JP2004238618A - ノボラック型アルキルフェノール樹脂の製造方法 - Google Patents
ノボラック型アルキルフェノール樹脂の製造方法 Download PDFInfo
- Publication number
- JP2004238618A JP2004238618A JP2004006301A JP2004006301A JP2004238618A JP 2004238618 A JP2004238618 A JP 2004238618A JP 2004006301 A JP2004006301 A JP 2004006301A JP 2004006301 A JP2004006301 A JP 2004006301A JP 2004238618 A JP2004238618 A JP 2004238618A
- Authority
- JP
- Japan
- Prior art keywords
- reaction
- novolak
- water
- resin
- alkylphenol resin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images

Landscapes
- Phenolic Resins Or Amino Resins (AREA)
Abstract
【課題】 未反応アルキルフェノールモノマー類が少なく、かつ、分子量分布が狭いノボラック型アルキルフェノール樹脂を高収率に製造する方法を提供する。
【解決手段】 ノボラック型アルキルフェノール樹脂の製造方法において、アルキルフェノール類とアルデヒド類とを、有機ホスホン酸を触媒として、非極性溶媒の存在下で反応させる工程を有することを特徴とするノボラック型アルキルフェノール樹脂の製造方法、及び、アルキルフェノール類とアルデヒド類とを、リン酸を触媒として、非極性溶媒の存在下で反応させる工程を有することを特徴とするノボラック型アルキルフェノール樹脂の製造方法。
【選択図】 なし
【解決手段】 ノボラック型アルキルフェノール樹脂の製造方法において、アルキルフェノール類とアルデヒド類とを、有機ホスホン酸を触媒として、非極性溶媒の存在下で反応させる工程を有することを特徴とするノボラック型アルキルフェノール樹脂の製造方法、及び、アルキルフェノール類とアルデヒド類とを、リン酸を触媒として、非極性溶媒の存在下で反応させる工程を有することを特徴とするノボラック型アルキルフェノール樹脂の製造方法。
【選択図】 なし
Description
本発明は、ノボラック型アルキルフェノール樹脂の製造方法に関するものである。
ノボラック型アルキルフェノール樹脂は、通常、アルキルフェノール類とアルデヒド類とを塩酸、硫酸、蓚酸、p−トルエンスルホン酸といった少量の無機酸、有機酸を触媒として反応させることで得られる。
ノボラック型アルキルフェノール樹脂の分子量は、アルキルフェノール類とアルデヒド類との仕込比率等で調整するのが一般的であるが、分子量の小さいノボラック型アルキルフェノール樹脂は、分子量分布が広くなりやすい。分子量分布を狭くする一般的手段としては、有機溶媒中で反応させる方法、水蒸気蒸留あるいは溶剤洗浄により低分子量成分を除去する方法などがあるが、前者の場合は低分子量のノボラック型アルキルフェノール樹脂は得られず、後者の場合は収率が大きく低下してしまうという問題があった。
ノボラック型アルキルフェノール樹脂の分子量は、アルキルフェノール類とアルデヒド類との仕込比率等で調整するのが一般的であるが、分子量の小さいノボラック型アルキルフェノール樹脂は、分子量分布が広くなりやすい。分子量分布を狭くする一般的手段としては、有機溶媒中で反応させる方法、水蒸気蒸留あるいは溶剤洗浄により低分子量成分を除去する方法などがあるが、前者の場合は低分子量のノボラック型アルキルフェノール樹脂は得られず、後者の場合は収率が大きく低下してしまうという問題があった。
また、近年アルキルフェノール、特にノニルフェノールモノマーやパラターシャリーオクチルフェノールモノマーは内分泌撹乱物質、いわゆる環境ホルモンとしてその危険性が指摘されている。このため、これらの化合物の使用を制限したり、含有量を少なくしたりする試みが多くなされている。これらの問題を解決する目的で、ノボラック型フェノール樹脂を合成する際に有機ホスホン酸触媒を用いる方法がある(例えば、特許文献1参照。)。
しかし、この方法でアルキルフェノール類を反応させ、その後反応系から分離しようとすると、アルキルフェノール樹脂が分子中のアルキル基を核にしてミセル構造を形成し、水酸基をその外側に配向させるようになるため、アルキルフェノール樹脂分子に親水性が発現し、水相からの分離が困難となるという問題があった。
しかし、この方法でアルキルフェノール類を反応させ、その後反応系から分離しようとすると、アルキルフェノール樹脂が分子中のアルキル基を核にしてミセル構造を形成し、水酸基をその外側に配向させるようになるため、アルキルフェノール樹脂分子に親水性が発現し、水相からの分離が困難となるという問題があった。
本発明は、未反応アルキルフェノールモノマー類の含有量が少なく、かつ、分子量分布が狭いノボラック型アルキルフェノール樹脂を高収率に製造する方法を提供するものである。
このような目的は、以下の本発明(1)〜(9)により達成される。
(1)ノボラック型アルキルフェノール樹脂の製造方法において、アルキルフェノール類とアルデヒド類とを、有機ホスホン酸を触媒として、非極性溶媒の存在下で反応させる工程を有することを特徴とする、ノボラック型アルキルフェノール樹脂の製造方法。
(2)前記工程において、反応系中の非極性溶媒量は20〜70重量%である上記(1)に記載のノボラック型アルキルフェノール樹脂の製造方法。
(3)前記工程における反応条件は、反応系中の水分量が30重量%以下、反応温度が110〜250℃である上記(1)又は(2)に記載のノボラック型アルキルフェノール樹脂の製造方法。
(4)前記有機ホスホン酸は、一般式(I)で示されるものである上記(1)ないし(3)のいずれかに記載のノボラック型アルキルフェノール樹脂の製造方法。
R−PO(OH)2 (I)
(Rは、炭素原子を含み、かつ、−COOH及び又は−PO(OH)2 を含む基である)
(5)ノボラック型アルキルフェノール樹脂の製造方法であって、アルキルフェノール類とアルデヒド類とを、リン酸類を触媒として、非極性溶媒の存在下で反応させる工程を有することを特徴とするノボラック型アルキルフェノール樹脂の製造方法。
(6)前記アルキルフェノール類1モルに対して、前記リン酸類0.2モル以上を用いる上記(5)に記載のノボラック型アルキルフェノール樹脂の製造方法。
(7)前記工程において、反応系中の非極性溶媒量は20〜70重量%である上記(5)又は(6)に記載のノボラック型アルキルフェノール樹脂の製造方法。
(8)前記工程における反応条件は、反応系中の水分量が1〜40重量%、反応温度が80〜180℃である上記(5)ないし(7)のいずれかに記載のノボラック型アルキルフェノール樹脂の製造方法。
(9)前記リン酸類は、リン酸である上記(5)ないし(8)のいずれかに記載のノボラック型アルキルフェノール樹脂の製造方法。
(1)ノボラック型アルキルフェノール樹脂の製造方法において、アルキルフェノール類とアルデヒド類とを、有機ホスホン酸を触媒として、非極性溶媒の存在下で反応させる工程を有することを特徴とする、ノボラック型アルキルフェノール樹脂の製造方法。
(2)前記工程において、反応系中の非極性溶媒量は20〜70重量%である上記(1)に記載のノボラック型アルキルフェノール樹脂の製造方法。
(3)前記工程における反応条件は、反応系中の水分量が30重量%以下、反応温度が110〜250℃である上記(1)又は(2)に記載のノボラック型アルキルフェノール樹脂の製造方法。
(4)前記有機ホスホン酸は、一般式(I)で示されるものである上記(1)ないし(3)のいずれかに記載のノボラック型アルキルフェノール樹脂の製造方法。
R−PO(OH)2 (I)
(Rは、炭素原子を含み、かつ、−COOH及び又は−PO(OH)2 を含む基である)
(5)ノボラック型アルキルフェノール樹脂の製造方法であって、アルキルフェノール類とアルデヒド類とを、リン酸類を触媒として、非極性溶媒の存在下で反応させる工程を有することを特徴とするノボラック型アルキルフェノール樹脂の製造方法。
(6)前記アルキルフェノール類1モルに対して、前記リン酸類0.2モル以上を用いる上記(5)に記載のノボラック型アルキルフェノール樹脂の製造方法。
(7)前記工程において、反応系中の非極性溶媒量は20〜70重量%である上記(5)又は(6)に記載のノボラック型アルキルフェノール樹脂の製造方法。
(8)前記工程における反応条件は、反応系中の水分量が1〜40重量%、反応温度が80〜180℃である上記(5)ないし(7)のいずれかに記載のノボラック型アルキルフェノール樹脂の製造方法。
(9)前記リン酸類は、リン酸である上記(5)ないし(8)のいずれかに記載のノボラック型アルキルフェノール樹脂の製造方法。
本発明は、アルキルフェノール類とアルデヒド類とを、有機ホスホン酸、あるいは、リン酸類を触媒として、非極性溶媒の存在下で反応させる工程を有することを特徴とするノボラック型アルキルフェノール樹脂の製造方法である。
本発明の製造方法により、未反応アルキルフェノール類の含有量が少なく、かつ、分子量分布が狭いノボラック型アルキルフェノール樹脂を高収率で得ることができる。
本発明の製造方法により、未反応アルキルフェノール類の含有量が少なく、かつ、分子量分布が狭いノボラック型アルキルフェノール樹脂を高収率で得ることができる。
以下に、本発明のノボラック型アルキルフェノール樹脂の製造方法について詳細に説明する。
まず、本発明のノボラック型アルキルフェノール樹脂の製造方法(以下、単に「製造方法」ということがある)の第一の形態は、アルキルフェノール類とアルデヒド類とを、有機ホスホン酸を触媒として、非極性溶媒の存在下で反応させる工程を有することを特徴とする。
まず、本発明のノボラック型アルキルフェノール樹脂の製造方法(以下、単に「製造方法」ということがある)の第一の形態は、アルキルフェノール類とアルデヒド類とを、有機ホスホン酸を触媒として、非極性溶媒の存在下で反応させる工程を有することを特徴とする。
本発明の製造方法で用いられるアルキルフェノール類としては特に限定されないが、フェノール環に結合したアルキル基の炭素数が3以上であるものが好ましい。このようなアルキルフェノール類としては例えば、パラターシャリーブチルフェノール、パラオクチルフェノール、パラフェニルフェノール、カルダノールなどが挙げられる。これらの中から選ばれた少なくとも1種以上のアルキルフェノール類を用いることができる。
本発明の製造方法で用いられるアルデヒド類は特に限定されないが、好ましくは、ホルムアルデヒド、アセトアルデヒド、ブチルアルデヒド、アクロレイン等あるいはこれらの混合物であり、これらのアルデヒド類の発生源となる物質、あるいは、これらのアルデヒド類の溶液を使用することも可能である。
上記アルキルフェノール類とアルデヒド類との反応モル比は特に限定されないが、アルキルフェノール類1.0モルに対して、アルデヒド類0.1〜3.0モルとすることが好ましく、さらに好ましくは0.7〜1.4モルである。これにより、軟化点が70℃〜120℃であり取り扱い作業性に優れたノボラック型アルキルフェノール樹脂を効率良く製造することができる。
また、アルキルフェノール類とアルデヒド類との反応方法としては特に限定されないが、例えば、
反応の開始時において、アルキルフェノール類とアルデヒド類とを全量一括して仕込み、ここへ有機ホスホン酸を添加し反応させてもよく、また、反応初期の発熱を抑えるため、アルキルフェノール類と有機ホスホン酸とを仕込んでからアルデヒド類を逐次添加して反応させてもよい。
また、アルキルフェノール類とアルデヒド類との反応方法としては特に限定されないが、例えば、
反応の開始時において、アルキルフェノール類とアルデヒド類とを全量一括して仕込み、ここへ有機ホスホン酸を添加し反応させてもよく、また、反応初期の発熱を抑えるため、アルキルフェノール類と有機ホスホン酸とを仕込んでからアルデヒド類を逐次添加して反応させてもよい。
本発明において触媒として使用する有機ホスホン酸は、ホスホン酸基[−PO(OH)2
]を含む有機化合物であり、いかなるものも使用可能であるが、下記一般式(I)で示されるホスホン酸が、未反応アルキルフェノール類の含有量が少なく、かつ、分子量分布が狭いノボラック型アルキルフェノール樹脂を高収率に得るために好ましい。
R−PO(OH)2 (I)
(Rは、炭素原子を含み、かつ、−COOH及び又は−PO(OH)2 を含む基である)
一般式(I)で示される有機ホスホン酸としては、アミノポリホスホン酸類であるエチレンジアミンテトラキスメチレンホスホン酸、エチレンジアミンビスメチレンホスホン酸、アミノトリスメチレンホスホン酸、β−アミノエチルホスホン酸N,N−ジ酢酸、アミノメチルホスホン酸N,N−ジ酢酸や、1−ヒドロキシエチリデン−1,1’−ジホスホン酸、2−ホスホノブタン−1,2,4−トリカルボン酸等がある。これらの中でも、工業的に大量生産され安価であるアミノトリスメチレンホスホン酸や、1−ヒドロキシエチリデン−1,1’−ジホスホン酸、2−ホスホノブタン−1,2,4−トリカルボン酸が好ましい。
]を含む有機化合物であり、いかなるものも使用可能であるが、下記一般式(I)で示されるホスホン酸が、未反応アルキルフェノール類の含有量が少なく、かつ、分子量分布が狭いノボラック型アルキルフェノール樹脂を高収率に得るために好ましい。
R−PO(OH)2 (I)
(Rは、炭素原子を含み、かつ、−COOH及び又は−PO(OH)2 を含む基である)
一般式(I)で示される有機ホスホン酸としては、アミノポリホスホン酸類であるエチレンジアミンテトラキスメチレンホスホン酸、エチレンジアミンビスメチレンホスホン酸、アミノトリスメチレンホスホン酸、β−アミノエチルホスホン酸N,N−ジ酢酸、アミノメチルホスホン酸N,N−ジ酢酸や、1−ヒドロキシエチリデン−1,1’−ジホスホン酸、2−ホスホノブタン−1,2,4−トリカルボン酸等がある。これらの中でも、工業的に大量生産され安価であるアミノトリスメチレンホスホン酸や、1−ヒドロキシエチリデン−1,1’−ジホスホン酸、2−ホスホノブタン−1,2,4−トリカルボン酸が好ましい。
上記有機ホスホン酸の添加量としては特に限定されないが、アルキルフェノール類1モルに対して0.001〜4.0モルとすることが好ましく、さらに好ましくは0.01〜0.5モルである。有機ホスホン酸の添加量が多いほど、未反応アルキルフェノール類の含有量が少なく、かつ、分子量分布が狭いノボラック型アルキルフェノール樹脂を高収率で得るという本発明の効果は大きいが、触媒添加量が4.0モルを越えるその効果が変わらなくなり、0.001モル未満では、触媒としての効果が実質的になくなる。
また、シュウ酸、硫酸、塩酸、p−トルエンスルホン酸など、通常のノボラック型アルキルフェノール樹脂の製造で使用する酸性触媒の併用も可能である。これらの酸性触媒の併用は、特に4核体以上の、高分子量の樹脂の反応促進に有効であり、分子量分布を制御する方法として有効な手段と言える。
また、シュウ酸、硫酸、塩酸、p−トルエンスルホン酸など、通常のノボラック型アルキルフェノール樹脂の製造で使用する酸性触媒の併用も可能である。これらの酸性触媒の併用は、特に4核体以上の、高分子量の樹脂の反応促進に有効であり、分子量分布を制御する方法として有効な手段と言える。
本発明の製造方法で使用する非極性溶媒とは特に限定されないが、例えば、ヘキサン、ヘプタン、オクタン、シクロヘキサンなどの水と混合し難い炭素系溶剤、ベンゼン、トルエン、キシレンなどの芳香族系溶剤、ジクロロメタン、クロロホルム、四塩化炭素などの塩素系溶剤、のほか、二硫化炭素などが挙げられる。これらを単独又は2種類以上を併用することができる。
これらの中でも、沸点111℃のトルエン、沸点約140℃のキシレンを用いることが好ましい。これにより、アルキルフェノール類とアルデヒド類との縮合反応を効率よく実施することができる。
これらの中でも、沸点111℃のトルエン、沸点約140℃のキシレンを用いることが好ましい。これにより、アルキルフェノール類とアルデヒド類との縮合反応を効率よく実施することができる。
本発明の製造方法における上記非極性溶媒の使用量としては特に限定されないが、反応系中において20〜70重量%とすることが好ましい。さらに好ましくは40〜60重量%である。これにより、ノボラック型アルキルフェノール樹脂を合成する反応を均一に安定して行うことができるとともに、生成したノボラック型アルキルフェノール樹脂を反応系から容易に分離精製することができる。
すなわち、この非極性溶媒は縮合反応温度を決定する還流溶媒として重要であり、また、生成したノボラック型アルキルフェノール樹脂を水相から分離するために十分な量配合されていることが重要である。
非極性溶媒の使用量が前記下限値より少ないと、アルキルフェノール類を溶解しきれず反応が充分に進行しないことがあり、また、生成したノボラック型アルキルフェノール樹脂を反応系から分離することが難しい場合がある。一方、前記上限値を超えると、有機ホスホン酸の触媒活性が低下し、反応時間が必要以上に多くかかったり、収率が低下したりすることがある。
また、縮合反応により発生した水分のみを系外に取り除きながら、溶剤還流反応を行うこ
ともできる。これにより、反応中の溶剤量を一定に保つことができ、反応系の温度を必要以上に低下させることなく、ノボラック型アルキルフェノール樹脂を効率的に得ることができる。
すなわち、この非極性溶媒は縮合反応温度を決定する還流溶媒として重要であり、また、生成したノボラック型アルキルフェノール樹脂を水相から分離するために十分な量配合されていることが重要である。
非極性溶媒の使用量が前記下限値より少ないと、アルキルフェノール類を溶解しきれず反応が充分に進行しないことがあり、また、生成したノボラック型アルキルフェノール樹脂を反応系から分離することが難しい場合がある。一方、前記上限値を超えると、有機ホスホン酸の触媒活性が低下し、反応時間が必要以上に多くかかったり、収率が低下したりすることがある。
また、縮合反応により発生した水分のみを系外に取り除きながら、溶剤還流反応を行うこ
ともできる。これにより、反応中の溶剤量を一定に保つことができ、反応系の温度を必要以上に低下させることなく、ノボラック型アルキルフェノール樹脂を効率的に得ることができる。
本発明の製造方法において、ノボラック型アルキルフェノール樹脂を合成する際の反応条件としては特に限定されないが、反応系中の水分を30重量%以下、反応温度を110〜250℃とすることが好ましい。かかる反応条件は未反応アルキルフェノール類のみならず、2核体や3核体といった低分子量領域のノボラック型アルキルフェノール樹脂が選択的に反応するのに有効で、分子量分布を効果的に狭くすることができる条件である。言い換えれば、未反応アルキルフェノール類の反応は、上記反応条件から外れた条件、即ち、水分が多く、低温下でも十分に行うことができるが、2核体、3核体等の低分子成分の選択的な反応に対しては、上記反応条件の範囲内とすることが効果的である。
本発明の製造方法において、反応系中の水分量とは、系内に存在するアルキルフェノール類、アルデヒド類、ノボラック型アルキルフェノール樹脂、有機ホスホン酸等全体に対する水分量である。また、ここで水分としては、仕込み時に添加した水分、添加するアルデヒド類に含まれる水分、添加する有機ホスホン酸に含まれる水分、有機ホスホン酸の結晶水等、仕込み原料に由来する水分、反応時に発生する縮合水などがある。これらの反応系中の水分量が30重量%以下であることが好ましく、さらに好ましくは1〜20重量%である。
反応系中の水分量の計算方法は、仕込み原料中の水分量と反応で生成する縮合水量との和を反応系中の水分量とし、仕込み全量で除した値である。また、水を蒸留して取り除きながら反応させる場合、上記仕込み原料中の水分量と反応で生成する縮合水量との和から、溜去した水分量を減じた水分量が反応系中の水分量である。
この水分量は少ないほど、未反応アルキルフェノール類が少なく、かつ、分子量分布が狭いノボラック型アルキルフェノール樹脂を高収率に製造できる効果が大きくなるので、20重量%以下であることが好ましい。しかし、水分が少なすぎると有機ホスホン酸が高粘度化もしくは固結するようになり、触媒作用が低下するため、結晶水を含む程度の水分量である1重量%以上であることが好ましい。水分量が30重量%を越えると、その効果がほとんど変わらないようになる。
反応系中の水分量の計算方法は、仕込み原料中の水分量と反応で生成する縮合水量との和を反応系中の水分量とし、仕込み全量で除した値である。また、水を蒸留して取り除きながら反応させる場合、上記仕込み原料中の水分量と反応で生成する縮合水量との和から、溜去した水分量を減じた水分量が反応系中の水分量である。
この水分量は少ないほど、未反応アルキルフェノール類が少なく、かつ、分子量分布が狭いノボラック型アルキルフェノール樹脂を高収率に製造できる効果が大きくなるので、20重量%以下であることが好ましい。しかし、水分が少なすぎると有機ホスホン酸が高粘度化もしくは固結するようになり、触媒作用が低下するため、結晶水を含む程度の水分量である1重量%以上であることが好ましい。水分量が30重量%を越えると、その効果がほとんど変わらないようになる。
また、本発明における反応温度は、110〜250℃が好ましい。110℃より低いと、上記のような水分の少ない条件下では、触媒である有機ホスホン酸が高粘度化もしくは固結するようになり、触媒作用が低下するようになる。一方、250℃を越えると、有機ホスホン酸の分解及びノボラック型アルキルフェノール樹脂の分解が起こるようになる。有機ホスホン酸、ノボラック型アルキルフェノール樹脂の分解は低温であるほうがより起こりにくく好ましいが、水分量1〜20重量%で有機ホスホン酸が高粘度化もしくは固結することなく触媒作用を十分に有した状態となるための温度範囲としては、さらに好ましくは110〜160℃である。
常圧下の反応であれば、用いる非極性溶媒の種類によっても異なるが、水分量が30重量%以下の範囲での還流温度は、150〜230℃に設定するのが好ましい。これにより、アルキルフェノール樹脂を効率よく合成することができる。
また、アルデヒド類を添加しながら、生成する縮合水を蒸留等で取り除く反応は、反応系中の水分量が一定となり好ましい条件である。しかし、この時、未反応のアルキルフェノール類が水分と一緒に取り除かれやすくなる欠点があるので、注意を要する。
この欠点を克服するため、未反応アルキルフェノール類が一定量以下となるまで、未反応のアルキルフェノール類が蒸留されないようにして反応を行い、次いで、蒸留により水分を取り除いた後あるいは取り除きながら、反応系中の水分を30重量%以下、反応温度を110〜250℃として反応を続けることができる。
また、アルデヒド類を添加しながら、生成する縮合水を蒸留等で取り除く反応は、反応系中の水分量が一定となり好ましい条件である。しかし、この時、未反応のアルキルフェノール類が水分と一緒に取り除かれやすくなる欠点があるので、注意を要する。
この欠点を克服するため、未反応アルキルフェノール類が一定量以下となるまで、未反応のアルキルフェノール類が蒸留されないようにして反応を行い、次いで、蒸留により水分を取り除いた後あるいは取り除きながら、反応系中の水分を30重量%以下、反応温度を110〜250℃として反応を続けることができる。
上記工程の反応終了後、触媒除去のために、中和や水洗を行ってもよい。また、必要により、水や有機溶剤、さらには未反応のアルキルフェノール類を除去するため、常圧蒸留や、減圧蒸留、水蒸気蒸留等を行うことも可能である。
また、本発明の製造方法の第二の形態は、アルキルフェノール類とアルデヒド類とを、リン酸類を触媒として、非極性溶媒の存在下で反応させる工程を有することを特徴とする。
ここで用いられるアルキルフェノール類、及び、アルデヒド類としては、上記第一の形態で記載したものと同じものを用いることができる。
上記アルキルフェノール類とアルデヒド類との反応モル比は特に限定されないが、アルキルフェノール類1.0モルに対して、アルデヒド類0.7〜1.4モルとすることが好ましく、さらに好ましくは0.9〜1.3モルである。これにより、軟化点が70℃〜120℃であり、取り扱い作業性に優れたアルキルフェノール樹脂を効率良く製造することができる。
また、アルキルフェノール類とアルデヒド類との反応方法としては特に限定されないが、例えば、
反応の開始時において、アルキルフェノール類とアルデヒド類とを全量一括して仕込み、ここへリン酸類を添加して反応させる方法、あるいは、反応初期の発熱を抑えるため、アルキルフェノール類とリン酸類とを仕込んでから、アルデヒド類を逐次添加して反応させる方法、などが挙げられる。
また、アルキルフェノール類とアルデヒド類との反応方法としては特に限定されないが、例えば、
反応の開始時において、アルキルフェノール類とアルデヒド類とを全量一括して仕込み、ここへリン酸類を添加して反応させる方法、あるいは、反応初期の発熱を抑えるため、アルキルフェノール類とリン酸類とを仕込んでから、アルデヒド類を逐次添加して反応させる方法、などが挙げられる。
ここでリン酸類としては、水に溶解してリン酸類水溶液となりうるリン酸系化合物を用いることができ、特に限定されないが、例えば、リン酸(オルトリン酸)、二リン酸、三リン酸などの直鎖状ポリリン酸、環状ポリリン酸、五酸化二リン、亜リン酸、次亜リン酸などのほか、各種リン酸エステル化合物が挙げられる。これらを単独または2種類以上組み合わせて使用することができる。
これらのリン酸類の中でも、リン酸が好ましい。リン酸は濃度調節を簡易に行うことができ、また、低コストで入手することができる。
リン酸類を水溶液の形態で用いる際のリン酸類の濃度としては特に限定されないが、20〜99重量%であることが好ましく、さらに好ましくは40〜99重量%である。リン酸類水溶液中のリン酸類の濃度を上記下限値以上とすることにより、アルキルフェノール類とアルデヒド類との反応を効率的に進行させることができる。
ここで用いられるリン酸類の量としては特に限定されないが、アルキルフェノール類1モルに対して、0.2モル以上であることが好ましい。これにより、アルキルフェノール類とアルデヒド類とを、リン酸類を用いて反応させる系において、アルキルフェノール類を主成分とする有機相と、リン酸類を含有する水相との分配を安定させることができる。
このリン酸類の量は、アルキルフェノール類1モルに対して、0.3〜1.0モルであることがさらに好ましく、0.4〜0.9モルであることが特に好ましい。これにより、未反応アルキルフェノール類の含有量が少ないアルキルフェノール樹脂を効率的に得ることができる。
このリン酸類の量は、アルキルフェノール類1モルに対して、0.3〜1.0モルであることがさらに好ましく、0.4〜0.9モルであることが特に好ましい。これにより、未反応アルキルフェノール類の含有量が少ないアルキルフェノール樹脂を効率的に得ることができる。
このリン酸類の量を多くすると、未反応アルキルフェノール類の含有量が少ないアルキルフェノール樹脂を高収率で得るという効果は大きくなる傾向があるが、アルキルフェノール類1モルに対して、1.0モルを越える量を用いても、この効果が実質的に変わらなくなるので経済的でないことがある。また、0.2モル未満では、有機相と水相とを安定
して分配するためには水相中のリン酸類濃度が低くなりすぎるので、反応速度が低下するようになる。
して分配するためには水相中のリン酸類濃度が低くなりすぎるので、反応速度が低下するようになる。
また、シュウ酸、硫酸、塩酸、p−トルエンスルホン酸など、通常のノボラック型アルキルフェノール樹脂の製造で使用する酸性触媒の併用も可能である。これらの酸性触媒の併用は、特に4核体以上の、高分子量の樹脂の反応促進に有効であり、分子量分布を制御する方法として有効な手段である。
第二の形態で使用する非極性溶媒としては特に限定されないが、例えば、ヘキサン、ヘプタン、オクタン、シクロヘキサンなどの水と混合し難い炭素系溶剤、ベンゼン、トルエン、キシレンなどの芳香族系溶剤、ジクロロメタン、クロロホルム、四塩化炭素などの塩素系溶剤、のほか、二硫化炭素などが挙げられる。これらを単独又は2種類以上を併用することができる。これらの中でも、沸点111℃のトルエン、沸点約140℃のキシレンを用いることが好ましい。これにより、アルキルフェノール類とアルデヒド類との縮合反応を効率よく実施することができる。
第二の形態における上記非極性溶媒の使用量としては特に限定されないが、反応系中において20〜70重量%とすることが好ましい。さらに好ましくは30〜60重量%である。
これにより、ノボラック型アルキルフェノール樹脂を合成する反応を均一に安定して行うことができるとともに、生成したノボラック型アルキルフェノール樹脂を反応系から容易に分離精製できる。すなわち、この非極性溶媒は縮合反応温度を決定する還流溶媒として重要であり、また、生成したノボラック型アルキルフェノール樹脂を水相から分離するためには、十分な量が使用されることが必要である。
これにより、ノボラック型アルキルフェノール樹脂を合成する反応を均一に安定して行うことができるとともに、生成したノボラック型アルキルフェノール樹脂を反応系から容易に分離精製できる。すなわち、この非極性溶媒は縮合反応温度を決定する還流溶媒として重要であり、また、生成したノボラック型アルキルフェノール樹脂を水相から分離するためには、十分な量が使用されることが必要である。
非極性溶媒の使用量が上記下限値より少ないと、アルキルフェノール類を溶解しきれず反応が充分に進行しないことがあり、また、生成したノボラック型アルキルフェノール樹脂を反応系から分離することが難しい場合がある。一方、上記上限値を超えると、リン酸類の触媒活性が低下し、反応時間が長くなったり、収率が低下したりすることがある。
また、縮合反応により発生した水分のみを系外に取り除きながら、溶剤還流反応を行うこともできる。これにより、反応中の溶剤量を一定に保つことができ、反応系の温度を必要以上に低下させることなく、ノボラック型アルキルフェノール樹脂を効率的に得ることができる。
また、縮合反応により発生した水分のみを系外に取り除きながら、溶剤還流反応を行うこともできる。これにより、反応中の溶剤量を一定に保つことができ、反応系の温度を必要以上に低下させることなく、ノボラック型アルキルフェノール樹脂を効率的に得ることができる。
第二の形態において、ノボラック型アルキルフェノール樹脂を合成する際の反応条件としては特に限定されないが、反応温度は80〜180℃とすることが好ましい。さらに好ましくは80〜150℃である。特に好ましくは100〜150℃である。
反応温度を上記下限値以上とすることにより、アルキルフェノール類とアルデヒド類との反応を促進させることができ、未反応アルキルフェノール類の含有量を低減させることができる。また、リン酸類水溶液を好ましい粘度にすることができ、触媒作用が低下するのを避けることができる。一方、上記上限値以下とすることにより、アルキルフェノール樹脂の分解を抑制することができる。
反応温度を上記下限値以上とすることにより、アルキルフェノール類とアルデヒド類との反応を促進させることができ、未反応アルキルフェノール類の含有量を低減させることができる。また、リン酸類水溶液を好ましい粘度にすることができ、触媒作用が低下するのを避けることができる。一方、上記上限値以下とすることにより、アルキルフェノール樹脂の分解を抑制することができる。
また、反応系中の水分を1〜40重量%とすることが好ましい。さらに好ましくは1〜30重量%である。
ここで、反応系中の水分含有率とは、反応系内に存在するアルキルフェノール類、アルデヒド類、リン酸類、非極性溶媒などの合計量に対する、反応系内に存在する水分の合計量の重量比率を指す。第二の形態において反応系内に存在する水分としては、リン酸類水溶液中の水分のほか、アルキルフェノール類とアルデヒド類との縮合によって生ずる縮合
水が挙げられる。
水が挙げられる。
反応系中の水分含有率は、反応系内に存在する水分量を、仕込み全量で除することで算出することができる。また、水を蒸留して取り除きながら反応させる場合、溜去した水分量を減じて反応系中の水分量とし、同様に算出することができる。
この水分含有率を、好ましくは上記の範囲内で反応を行うことにより、未反応アルキルフェノール類の含有量が少ないノボラック型アルキルフェノール樹脂を高収率で得ることができる。
反応系中の水分含有率を上記下限値以上とすることにより、リン酸類が高粘度化もしくは固結するのを抑えることができる。また、上記上限値以下とすることにより、反応速度の低下を抑制することができるので、フェノール類とアラルキルエーテル類との反応を効率的に進行させることができる。
反応系中の水分含有率を上記下限値以上とすることにより、リン酸類が高粘度化もしくは固結するのを抑えることができる。また、上記上限値以下とすることにより、反応速度の低下を抑制することができるので、フェノール類とアラルキルエーテル類との反応を効率的に進行させることができる。
上記第二の形態においては特に、反応温度を100〜150℃、反応系中の水分含有率を1〜30重量%とすることにより、未反応アルキルフェノール類だけでなく、2核体、3核体などの低分子量成分が選択的に反応しやすく、ノボラック型アルキルフェノール樹脂の分子量分布を効果的に狭くすることができる。
常圧下の反応であれば、用いる非極性溶媒の種類によっても異なるが、水分量が1〜30重量%の範囲での還流温度は、100〜150℃に設定するのが好ましい。これにより、ノボラック型アルキルフェノール樹脂を効率よく合成することができる。
また、アルデヒド類を添加しながら、生成する縮合水を蒸留等で取り除く反応は、反応系中の水分量が一定となり好ましい条件である。この時、未反応アルキルフェノール類が水分と一緒に取り除かれやすくなることがあるので、未反応アルキルフェノール類が一定量以下となるまで、未反応のアルキルフェノール類が蒸留されないようにして反応を行い、次いで、蒸留により水分を取り除いた後あるいは取り除きながら、反応系中の水分を1〜40重量%、反応温度を100〜150℃として反応を続けることができる。
また、アルデヒド類を添加しながら、生成する縮合水を蒸留等で取り除く反応は、反応系中の水分量が一定となり好ましい条件である。この時、未反応アルキルフェノール類が水分と一緒に取り除かれやすくなることがあるので、未反応アルキルフェノール類が一定量以下となるまで、未反応のアルキルフェノール類が蒸留されないようにして反応を行い、次いで、蒸留により水分を取り除いた後あるいは取り除きながら、反応系中の水分を1〜40重量%、反応温度を100〜150℃として反応を続けることができる。
上記工程の反応終了後、触媒除去のために、中和や水洗を行ってもよい。また、必要により、水や有機溶剤、さらには未反応のアルキルフェノール類を除去するため、常圧蒸留や、減圧蒸留、水蒸気蒸留等を行うことも可能である。
本発明の製造方法においては特に限定されないが、以上に説明した第二の形態によりノボラック型アルキルフェノール樹脂を合成した後、反応系の水洗を行い、ノボラック型アルキルフェノール樹脂中に含有されるリン酸類の濃度を3.0重量%以下にすることが好ましい。さらに好ましくは0.1重量%以下である。
これにより、水洗後、常圧蒸留もしくは減圧蒸留を行うとき、ノボラック型アルキルフェノール樹脂の分解を抑制することができる。
これにより、水洗後、常圧蒸留もしくは減圧蒸留を行うとき、ノボラック型アルキルフェノール樹脂の分解を抑制することができる。
ここで水洗を行う方法としては特に限定されないが、例えば、ノボラック型アルキルフェノール樹脂を含む有機相と、リン酸類水溶液を含む水相とを、遠心分離により分離する。次いで、得られた有機相を、純水やイオン交換水で水洗を行うことにより、ノボラック型アルキルフェノール樹脂中に含有されるリン酸類の濃度を3.0重量%以下とすることができる。また、この水洗を複数回数実施することにより、リン酸類の濃度を0.1重量%以下とすることができる。
また、さらに、リン酸類の濃度が上記上限値以下になるまで水洗を行った後、反応系中に残留しているリン酸類1当量に対して、アルカリ性物質0.8〜1.5当量を用いて中和することが好ましい。これにより、リン酸類の有する触媒活性を失活させることができ
るので、この後の工程で、高温で蒸留反応を行う場合でも、ノボラック型アルキルフェノール樹脂の分解を抑制することができる。
るので、この後の工程で、高温で蒸留反応を行う場合でも、ノボラック型アルキルフェノール樹脂の分解を抑制することができる。
ここで用いられるアルカリ性物質としては特に限定されないが、例えば、水酸化カルシウム、水酸化ナトリウム、トリエタノールアミンなどを用いることができる。アルカリ性物質の形態としては特に限定されないが、水溶液の形態で用いることが好ましい。
本発明の製造方法において、有機ホスホン酸、あるいは、リン酸類を触媒として用い、未反応アルキルフェノール類の含有量が少なく、分子量分布の狭いノボラック型アルキルフェノール樹脂を高収率で製造することができる理由は、以下のように考えられる。
本発明の製造方法に用いる有機ホスホン酸やリン酸類は、非常に水溶性が高い。しかし、アルキルフェノール類に対しては溶解性が小さく、ノボラック型アルキルフェノール樹脂に対しては、その分子量増大とともに更に溶解性が小さくなる性質を有している。このため反応時には、触媒である有機ホスホン酸やリン酸類を多量に含んだ水相と、アルキルフェノール類、ノボラック型アルキルフェノール樹脂、非極性溶媒からなる、触媒がほとんど存在しない有機相とに相分離した状態となる。
アルキルフェノール類、あるいは、ノボラック型アルキルフェノール樹脂の2核体等の低分子量成分は比較的水相に溶出しやすく、溶出した成分はアルデヒド類と反応する。反応して高分子量化したノボラック型アルキルフェノール樹脂は、速やかに有機相に抽出されて非極性溶媒に溶解し、その後水相にはほとんど溶出することがないので、それ以上は実質的に反応は進まなくなる。
このように、ノボラック型アルキルフェノール樹脂の低分子量成分と高分子量成分との間に反応速度差を生じるため、未反応アルキルフェノール類の含有量が少なく、かつ、分子量分布が狭いノボラック型アルキルフェノール樹脂を高収率に製造することが可能となる。
さらに、この反応系に非極性溶媒を用いることにより、有機相に抽出されて非極性溶媒に溶解したノボラック型アルキルフェノール樹脂を、反応終了後に反応系から容易に分離精製することができるという利点をも有する。
本発明の製造方法に用いる有機ホスホン酸やリン酸類は、非常に水溶性が高い。しかし、アルキルフェノール類に対しては溶解性が小さく、ノボラック型アルキルフェノール樹脂に対しては、その分子量増大とともに更に溶解性が小さくなる性質を有している。このため反応時には、触媒である有機ホスホン酸やリン酸類を多量に含んだ水相と、アルキルフェノール類、ノボラック型アルキルフェノール樹脂、非極性溶媒からなる、触媒がほとんど存在しない有機相とに相分離した状態となる。
アルキルフェノール類、あるいは、ノボラック型アルキルフェノール樹脂の2核体等の低分子量成分は比較的水相に溶出しやすく、溶出した成分はアルデヒド類と反応する。反応して高分子量化したノボラック型アルキルフェノール樹脂は、速やかに有機相に抽出されて非極性溶媒に溶解し、その後水相にはほとんど溶出することがないので、それ以上は実質的に反応は進まなくなる。
このように、ノボラック型アルキルフェノール樹脂の低分子量成分と高分子量成分との間に反応速度差を生じるため、未反応アルキルフェノール類の含有量が少なく、かつ、分子量分布が狭いノボラック型アルキルフェノール樹脂を高収率に製造することが可能となる。
さらに、この反応系に非極性溶媒を用いることにより、有機相に抽出されて非極性溶媒に溶解したノボラック型アルキルフェノール樹脂を、反応終了後に反応系から容易に分離精製することができるという利点をも有する。
また、上記第一の形態においては、ノボラック型アルキルフェノール樹脂を製造する条件として、反応系中の水分を30重量%以下、反応温度を110〜200℃とすることにより、以下の効果を得ることができる。
反応系の温度がこのように高温であることから、ノボラック型アルキルフェノール樹脂の2核体、3核体等の低分子量成分の成分が水相へ溶出されやすく、水相での反応を容易に進めることができる。そして、水分が少なく水相中のイオン濃度が高い状態で維持され、水相と有機相との界面が明確に分離するので、有機相側における反応を防止できる。また、有機ホスホン酸は高濃度であると粘度を高めたり固結したりする性質があるが、高温であるため溶融した状態となり触媒機能を失うことが防止できる。これらの効果から、未反応アルキルフェノール類の含有量が少なく、かつ、分子量分布が狭いノボラック型アルキルフェノール樹脂を高収率に得る効果を高めることができる。
反応系の温度がこのように高温であることから、ノボラック型アルキルフェノール樹脂の2核体、3核体等の低分子量成分の成分が水相へ溶出されやすく、水相での反応を容易に進めることができる。そして、水分が少なく水相中のイオン濃度が高い状態で維持され、水相と有機相との界面が明確に分離するので、有機相側における反応を防止できる。また、有機ホスホン酸は高濃度であると粘度を高めたり固結したりする性質があるが、高温であるため溶融した状態となり触媒機能を失うことが防止できる。これらの効果から、未反応アルキルフェノール類の含有量が少なく、かつ、分子量分布が狭いノボラック型アルキルフェノール樹脂を高収率に得る効果を高めることができる。
そいて、上記第二の形態において、ノボラック型アルキルフェノール樹脂を製造する条件として、反応系中の水分を1〜40重量%、反応温度を100〜150℃とすることにより、上記と同様の効果を得ることができる。
本発明の製造方法で得られたアルキルフェノール樹脂中の未反応アルキルフェノール類の含有量としては特に限定されないが、1.0重量%以下であることが好ましく、さらに好ましくは0.8重量%以下である。
本発明の製造方法においては、アルキルフェノール類とアルデヒド類とを、有機ホスホン酸、あるいは、リン酸類を触媒として用い、アルキルフェノール類や非極性溶媒を主成
分とする有機相と、有機ホスホン酸やリン酸類を含有する水相との間で液−液不均一反応を行うことにより、未反応アルキルフェノール類の含有量を上記上限値以下とすることができる。また、必要に応じて、未反応アルキルフェノール類を除去するために、常圧蒸留や、減圧蒸留、水蒸気蒸留等を併せて行うこともできる。
本発明の製造方法においては、アルキルフェノール類とアルデヒド類とを、有機ホスホン酸、あるいは、リン酸類を触媒として用い、アルキルフェノール類や非極性溶媒を主成
分とする有機相と、有機ホスホン酸やリン酸類を含有する水相との間で液−液不均一反応を行うことにより、未反応アルキルフェノール類の含有量を上記上限値以下とすることができる。また、必要に応じて、未反応アルキルフェノール類を除去するために、常圧蒸留や、減圧蒸留、水蒸気蒸留等を併せて行うこともできる。
なお、本発明の製造方法において、未反応アルキルフェノール類の含有量は、JIS K 0114に準拠し、ガスクロマトグラフィー法を用い、2,5−キシレノールを内部標準物質として内部標準法で測定した値である。
以下、本発明を実施例により詳細に説明する。ここで記載されている「部」は「重量部」、「%」は「重量%」を示す。
(実施例1)
5Lの三口フラスコ中に、パラターシャリーオクチルフェノール1000部、1−ヒドロキシエチリデン−1,1’−ジホスホン酸60%水溶液(フェリオックス115、ライオン社製)200部、キシレン1000部を添加した。これを100℃に昇温し、37%ホルムアルデヒド水溶液480部を30分間かけて逐次添加し、150℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応パラターシャリーオクチルフェノール量を測定した。その後、触媒相を分離して除去し、樹脂が溶解している溶剤相を300部の水で3回水洗分離を実施した。次いで、常圧蒸留を行い160℃まで昇温し、5000Paの減圧度で減圧蒸留を行ってキシレンを除去した後、180℃まで昇温し、ノボラック型パラターシャリーオクチルフェノール樹脂920部を得た。
5Lの三口フラスコ中に、パラターシャリーオクチルフェノール1000部、1−ヒドロキシエチリデン−1,1’−ジホスホン酸60%水溶液(フェリオックス115、ライオン社製)200部、キシレン1000部を添加した。これを100℃に昇温し、37%ホルムアルデヒド水溶液480部を30分間かけて逐次添加し、150℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応パラターシャリーオクチルフェノール量を測定した。その後、触媒相を分離して除去し、樹脂が溶解している溶剤相を300部の水で3回水洗分離を実施した。次いで、常圧蒸留を行い160℃まで昇温し、5000Paの減圧度で減圧蒸留を行ってキシレンを除去した後、180℃まで昇温し、ノボラック型パラターシャリーオクチルフェノール樹脂920部を得た。
(実施例2)
5Lの三口フラスコ中に、パラターシャリーオクチルフェノール1000部、アミノトリスメチレンホスホン酸50%水溶液(ディクエスト2000、ソルーシア・ジャパン社製)240部、キシレン1000部を添加した。これを100℃に昇温し、37%ホルムアルデヒド水溶液480部を30分間かけて逐次添加し、150℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応パラターシャリーオクチルフェノール量を測定した。その後、触媒相を分離して除去し、樹脂が溶解している溶剤相を300部の水で3回水洗分離を実施した。次いで、常圧蒸留を行い160℃まで昇温し、5000Paの減圧度で減圧蒸留を行って180℃まで昇温し、ノボラック型パラターシャリーオクチルフェノール樹脂890部を得た。
5Lの三口フラスコ中に、パラターシャリーオクチルフェノール1000部、アミノトリスメチレンホスホン酸50%水溶液(ディクエスト2000、ソルーシア・ジャパン社製)240部、キシレン1000部を添加した。これを100℃に昇温し、37%ホルムアルデヒド水溶液480部を30分間かけて逐次添加し、150℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応パラターシャリーオクチルフェノール量を測定した。その後、触媒相を分離して除去し、樹脂が溶解している溶剤相を300部の水で3回水洗分離を実施した。次いで、常圧蒸留を行い160℃まで昇温し、5000Paの減圧度で減圧蒸留を行って180℃まで昇温し、ノボラック型パラターシャリーオクチルフェノール樹脂890部を得た。
(実施例3)
5Lの三口フラスコ中に、ノニルフェノール1000部、1−ヒドロキシエチリデン−1,1’−ジホスホン酸60%水溶液(フェリオックス115、ライオン社製)200部、キシレン1000部を添加した。これを100℃に昇温し、37%ホルムアルデヒド水溶液440部を30分間かけて逐次添加し、150℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応ノニルフェノール量を測定した。その後、触媒相を分離して除去し、樹脂が溶解している溶剤相を300部の水で3回水洗分離を実施した。次いで、常圧蒸留を行い160℃まで昇温し、5000Paの減圧度で減圧蒸留を行って180℃まで昇温し、ノボラック型ノニルフェノール樹脂910部を得た。
5Lの三口フラスコ中に、ノニルフェノール1000部、1−ヒドロキシエチリデン−1,1’−ジホスホン酸60%水溶液(フェリオックス115、ライオン社製)200部、キシレン1000部を添加した。これを100℃に昇温し、37%ホルムアルデヒド水溶液440部を30分間かけて逐次添加し、150℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応ノニルフェノール量を測定した。その後、触媒相を分離して除去し、樹脂が溶解している溶剤相を300部の水で3回水洗分離を実施した。次いで、常圧蒸留を行い160℃まで昇温し、5000Paの減圧度で減圧蒸留を行って180℃まで昇温し、ノボラック型ノニルフェノール樹脂910部を得た。
(実施例4)
5Lの三口フラスコ中に、パラターシャリーオクチルフェノール1000部、1−ヒドロキシエチリデン−1,1’−ジホスホン酸60%水溶液(フェリオックス115、ライ
オン社製)200部、オクタン1000部を添加した。これを100℃に昇温し、37%ホルムアルデヒド水溶液480部を30分間かけて逐次添加し、140℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応パラターシャリーオクチルフェノール量を測定した。その後、触媒相を分離して除去し、樹脂が溶解している溶剤相を300部の水で3回水洗分離を実施した。次いで、常圧蒸留を行い160℃まで昇温し、5000Paの減圧度で減圧蒸留を行って180℃まで昇温し、ノボラック型パラターシャリーオクチルフェノール樹脂900部を得た。
5Lの三口フラスコ中に、パラターシャリーオクチルフェノール1000部、1−ヒドロキシエチリデン−1,1’−ジホスホン酸60%水溶液(フェリオックス115、ライ
オン社製)200部、オクタン1000部を添加した。これを100℃に昇温し、37%ホルムアルデヒド水溶液480部を30分間かけて逐次添加し、140℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応パラターシャリーオクチルフェノール量を測定した。その後、触媒相を分離して除去し、樹脂が溶解している溶剤相を300部の水で3回水洗分離を実施した。次いで、常圧蒸留を行い160℃まで昇温し、5000Paの減圧度で減圧蒸留を行って180℃まで昇温し、ノボラック型パラターシャリーオクチルフェノール樹脂900部を得た。
(実施例5)
5Lの三口フラスコ中に、パラターシャリーオクチルフェノール1000部、1−ヒドロキシエチリデン−1,1’−ジホスホン酸60%水溶液(フェリオックス115、ライオン社製)200部、トルエン1000部を添加した。これを100℃に昇温し、37%ホルムアルデヒド水溶液393部(反応モル比 1.00)を30分間かけて逐次添加し、140℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応パラターシャリーオクチルフェノール量を測定した。その後、触媒相を分離して除去し、樹脂が溶解している溶剤相を300部の水で3回水洗分離を実施した。次いで、常圧蒸留を行い160℃まで昇温し、5000Paの減圧度で減圧蒸留を行って180℃まで昇温し、ノボラック型パラターシャリーオクチルフェノール樹脂856部を得た。
5Lの三口フラスコ中に、パラターシャリーオクチルフェノール1000部、1−ヒドロキシエチリデン−1,1’−ジホスホン酸60%水溶液(フェリオックス115、ライオン社製)200部、トルエン1000部を添加した。これを100℃に昇温し、37%ホルムアルデヒド水溶液393部(反応モル比 1.00)を30分間かけて逐次添加し、140℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応パラターシャリーオクチルフェノール量を測定した。その後、触媒相を分離して除去し、樹脂が溶解している溶剤相を300部の水で3回水洗分離を実施した。次いで、常圧蒸留を行い160℃まで昇温し、5000Paの減圧度で減圧蒸留を行って180℃まで昇温し、ノボラック型パラターシャリーオクチルフェノール樹脂856部を得た。
(実施例6)
5Lの三口フラスコ中に、パラターシャリーオクチルフェノール1000部、85%リン酸水溶液448部(アルキルフェノール類1モルに対して0.8モル)、キシレン1000部を仕込んだ。
これを100℃に昇温して、37%ホルムアルデヒド水溶液480部(反応モル比 1.22)を30分間かけて逐次添加し、150℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応パラターシャリーオクチルフェノール量を測定した。
その後、純水300部を添加して混合し、樹脂と分離した水相を除去した。このような水洗工程を3回行った。水洗後のフェノール樹脂中に残存しているリン酸量を測定したところ、0.05部であった。ここへ、50%水酸化ナトリウム水溶液0.06部(上記残存リン酸1当量に対して1.5当量)を添加した。
その後、常圧蒸留を行い160℃まで昇温し、5000Paの減圧度で減圧蒸留を行ってキシレンを除去した後、180℃まで昇温し、ノボラック型パラターシャリーオクチルフェノール樹脂917部を得た。
5Lの三口フラスコ中に、パラターシャリーオクチルフェノール1000部、85%リン酸水溶液448部(アルキルフェノール類1モルに対して0.8モル)、キシレン1000部を仕込んだ。
これを100℃に昇温して、37%ホルムアルデヒド水溶液480部(反応モル比 1.22)を30分間かけて逐次添加し、150℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応パラターシャリーオクチルフェノール量を測定した。
その後、純水300部を添加して混合し、樹脂と分離した水相を除去した。このような水洗工程を3回行った。水洗後のフェノール樹脂中に残存しているリン酸量を測定したところ、0.05部であった。ここへ、50%水酸化ナトリウム水溶液0.06部(上記残存リン酸1当量に対して1.5当量)を添加した。
その後、常圧蒸留を行い160℃まで昇温し、5000Paの減圧度で減圧蒸留を行ってキシレンを除去した後、180℃まで昇温し、ノボラック型パラターシャリーオクチルフェノール樹脂917部を得た。
(実施例7)
5Lの三口フラスコ中に、パラターシャリーオクチルフェノール1000部、85%リン酸水溶液448部(アルキルフェノール類1モルに対して0.8モル)、オクタン1000部を仕込んだ。
これを100℃に昇温して、37%ホルムアルデヒド水溶液480部(反応モル比 1.22)を30分間かけて逐次添加し、140℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応パラターシャリーオクチルフェノール量を測定した。
その後、純水300部を添加して混合し、樹脂と分離した水相を除去した。このような水洗工程を3回行った。水洗後のフェノール樹脂中に残存しているリン酸量を測定したところ、0.05部であった。ここへ、50%水酸化ナトリウム水溶液0.06部(上記残存リン酸1当量に対して1.5当量)を添加した。
その後、常圧蒸留を行い160℃まで昇温し、5000Paの減圧度で減圧蒸留を行っ
てオクタンを除去した後、180℃まで昇温し、ノボラック型パラターシャリーオクチルフェノール樹脂893部を得た。
5Lの三口フラスコ中に、パラターシャリーオクチルフェノール1000部、85%リン酸水溶液448部(アルキルフェノール類1モルに対して0.8モル)、オクタン1000部を仕込んだ。
これを100℃に昇温して、37%ホルムアルデヒド水溶液480部(反応モル比 1.22)を30分間かけて逐次添加し、140℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応パラターシャリーオクチルフェノール量を測定した。
その後、純水300部を添加して混合し、樹脂と分離した水相を除去した。このような水洗工程を3回行った。水洗後のフェノール樹脂中に残存しているリン酸量を測定したところ、0.05部であった。ここへ、50%水酸化ナトリウム水溶液0.06部(上記残存リン酸1当量に対して1.5当量)を添加した。
その後、常圧蒸留を行い160℃まで昇温し、5000Paの減圧度で減圧蒸留を行っ
てオクタンを除去した後、180℃まで昇温し、ノボラック型パラターシャリーオクチルフェノール樹脂893部を得た。
(実施例8)
5Lの三口フラスコ中に、パラターシャリーオクチルフェノール1000部、85%リン酸水溶液196部(アルキルフェノール類1モルに対して0.35モル)、キシレン1000部を仕込んだ。
これを100℃に昇温して、37%ホルムアルデヒド水溶液480部(反応モル比 1.22)を30分間かけて逐次添加し、150℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応パラターシャリーオクチルフェノール量を測定した。
その後、純水500部を添加して混合し、樹脂と分離した水相を除去した。このような水洗工程を3回行った。水洗後のフェノール樹脂中に残存しているリン酸量を測定したところ、検出限界以下であった。
その後、常圧蒸留を行い160℃まで昇温し、5000Paの減圧度で減圧蒸留を行ってキシレンを除去した後、180℃まで昇温し、ノボラック型パラターシャリーオクチルフェノール樹脂873部を得た。
5Lの三口フラスコ中に、パラターシャリーオクチルフェノール1000部、85%リン酸水溶液196部(アルキルフェノール類1モルに対して0.35モル)、キシレン1000部を仕込んだ。
これを100℃に昇温して、37%ホルムアルデヒド水溶液480部(反応モル比 1.22)を30分間かけて逐次添加し、150℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応パラターシャリーオクチルフェノール量を測定した。
その後、純水500部を添加して混合し、樹脂と分離した水相を除去した。このような水洗工程を3回行った。水洗後のフェノール樹脂中に残存しているリン酸量を測定したところ、検出限界以下であった。
その後、常圧蒸留を行い160℃まで昇温し、5000Paの減圧度で減圧蒸留を行ってキシレンを除去した後、180℃まで昇温し、ノボラック型パラターシャリーオクチルフェノール樹脂873部を得た。
(実施例9)
5Lの三口フラスコ中に、ノニルフェノール1000部、85%リン酸水溶液419部(アルキルフェノール類1モルに対して0.8モル)、キシレン1000部を仕込んだ。
これを100℃に昇温して、37%ホルムアルデヒド水溶液450部(反応モル比 1.22)を30分間かけて逐次添加し、150℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応ノニルフェノール量を測定した。
その後、純水300部を添加して混合し、樹脂と分離した水相を除去した。このような水洗工程を3回行った。水洗後のフェノール樹脂中に残存しているリン酸量を測定したところ、0.05部であった。ここへ、50%水酸化ナトリウム水溶液0.06部(上記残存リン酸1当量に対して1.5当量)を添加した。
その後、常圧蒸留を行い160℃まで昇温し、5000Paの減圧度で減圧蒸留を行ってキシレンを除去した後、180℃まで昇温し、ノボラック型ノニルフェノール樹脂864部を得た。
5Lの三口フラスコ中に、ノニルフェノール1000部、85%リン酸水溶液419部(アルキルフェノール類1モルに対して0.8モル)、キシレン1000部を仕込んだ。
これを100℃に昇温して、37%ホルムアルデヒド水溶液450部(反応モル比 1.22)を30分間かけて逐次添加し、150℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応ノニルフェノール量を測定した。
その後、純水300部を添加して混合し、樹脂と分離した水相を除去した。このような水洗工程を3回行った。水洗後のフェノール樹脂中に残存しているリン酸量を測定したところ、0.05部であった。ここへ、50%水酸化ナトリウム水溶液0.06部(上記残存リン酸1当量に対して1.5当量)を添加した。
その後、常圧蒸留を行い160℃まで昇温し、5000Paの減圧度で減圧蒸留を行ってキシレンを除去した後、180℃まで昇温し、ノボラック型ノニルフェノール樹脂864部を得た。
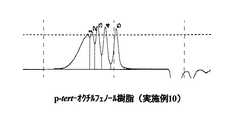
(実施例10)
5Lの三口フラスコ中に、パラターシャリーオクチルフェノール1000部、85%リン酸水溶液448部(アルキルフェノール類1モルに対して0.8モル)、トルエン1000部を仕込んだ。
これを100℃に昇温して、37%ホルムアルデヒド水溶液354部(反応モル比 0.90)を30分間かけて逐次添加し、150℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応パラターシャリーオクチルフェノール量を測定した。
その後、純水300部を添加して混合し、樹脂と分離した水相を除去した。このような水洗工程を3回行った。水洗後のフェノール樹脂中に残存しているリン酸量を測定したところ、0.05部であった。ここへ、50%水酸化ナトリウム水溶液0.06部(上記残存リン酸1当量に対して1.5当量)を添加した。
その後、常圧蒸留を行い160℃まで昇温し、5000Paの減圧度で減圧蒸留を行ってキシレンを除去した後、180℃まで昇温し、ノボラック型パラターシャリーオクチルフェノール樹脂812部を得た。
5Lの三口フラスコ中に、パラターシャリーオクチルフェノール1000部、85%リン酸水溶液448部(アルキルフェノール類1モルに対して0.8モル)、トルエン1000部を仕込んだ。
これを100℃に昇温して、37%ホルムアルデヒド水溶液354部(反応モル比 0.90)を30分間かけて逐次添加し、150℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応パラターシャリーオクチルフェノール量を測定した。
その後、純水300部を添加して混合し、樹脂と分離した水相を除去した。このような水洗工程を3回行った。水洗後のフェノール樹脂中に残存しているリン酸量を測定したところ、0.05部であった。ここへ、50%水酸化ナトリウム水溶液0.06部(上記残存リン酸1当量に対して1.5当量)を添加した。
その後、常圧蒸留を行い160℃まで昇温し、5000Paの減圧度で減圧蒸留を行ってキシレンを除去した後、180℃まで昇温し、ノボラック型パラターシャリーオクチルフェノール樹脂812部を得た。
(比較例1)
5Lの三口フラスコ中に、パラターシャリーオクチルフェノール1000部、シュウ酸10部を添加した。これを100℃に昇温し、37%ホルムアルデヒド水溶液480部を30分間かけて逐次添加し、100℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応パラターシャリーオクチルフェノール量を測定した。次いで、常圧蒸留を行い160℃まで昇温し、5000Paの減圧下で減圧蒸留を行って180℃まで昇温し、ノボラック型パラターシャリーオクチルフェノール樹脂860部を得た。
5Lの三口フラスコ中に、パラターシャリーオクチルフェノール1000部、シュウ酸10部を添加した。これを100℃に昇温し、37%ホルムアルデヒド水溶液480部を30分間かけて逐次添加し、100℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応パラターシャリーオクチルフェノール量を測定した。次いで、常圧蒸留を行い160℃まで昇温し、5000Paの減圧下で減圧蒸留を行って180℃まで昇温し、ノボラック型パラターシャリーオクチルフェノール樹脂860部を得た。
(比較例2)
5Lの三口フラスコ中に、パラターシャリーオクチルフェノール1000部、1−ヒドロキシエチリデン−1,1’−ジホスホン酸60%水溶液(フェリオックス115、ライオン社製)200部を添加した。これを100℃に昇温し、37%ホルムアルデヒド水溶液480部を30分間かけて逐次添加し、100℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応パラターシャリーオクチルフェノール量を測定した。その後、分離の目的で純水500部を添加し混合したが、樹脂と水相がうまく分離せず樹脂を単離できなかった。
5Lの三口フラスコ中に、パラターシャリーオクチルフェノール1000部、1−ヒドロキシエチリデン−1,1’−ジホスホン酸60%水溶液(フェリオックス115、ライオン社製)200部を添加した。これを100℃に昇温し、37%ホルムアルデヒド水溶液480部を30分間かけて逐次添加し、100℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応パラターシャリーオクチルフェノール量を測定した。その後、分離の目的で純水500部を添加し混合したが、樹脂と水相がうまく分離せず樹脂を単離できなかった。
実施例と比較例で得られたノボラック型アルキルフェノール樹脂の特性を表1に示す。

表の注:測定方法
(1)樹脂収得量:アルキルフェノール1000部に対するノボラック型アルキルフェノール樹脂の収得量で示した。
(2)数平均分子量、及び重量平均分子量:液体クロマトグラフィーで測定した。液体クロマトグラフィーは、東ソー製GPCカラム(G1000HXL:1本、G2000HXL:2本、G3000HXL:1本)を用い、流量1.0ml/分、溶出溶媒テトラヒドロフラン、カラム温度40℃の分析条件で示差屈折計を検出器として用いてGPC測定した。分子量は標準ポリスチレンにより換算した。
(3)未反応アルキルフェノール量:ガスクロマトグラフィーで測定した。ガスクロマトグラフィーは、JIS K 0114 に準拠し、2,5−キシレノールを内部標準として内部標準法で測定した。
(4)軟化点:JIS K 2531 に準拠して測定した。
(1)樹脂収得量:アルキルフェノール1000部に対するノボラック型アルキルフェノール樹脂の収得量で示した。
(2)数平均分子量、及び重量平均分子量:液体クロマトグラフィーで測定した。液体クロマトグラフィーは、東ソー製GPCカラム(G1000HXL:1本、G2000HXL:2本、G3000HXL:1本)を用い、流量1.0ml/分、溶出溶媒テトラヒドロフラン、カラム温度40℃の分析条件で示差屈折計を検出器として用いてGPC測定した。分子量は標準ポリスチレンにより換算した。
(3)未反応アルキルフェノール量:ガスクロマトグラフィーで測定した。ガスクロマトグラフィーは、JIS K 0114 に準拠し、2,5−キシレノールを内部標準として内部標準法で測定した。
(4)軟化点:JIS K 2531 に準拠して測定した。
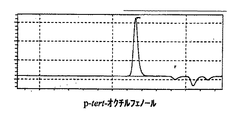
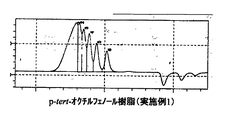
パラターシャリーオクチルフェノール、及び、実施例1と実施例10、及び比較例1で得られたノボラック型パラターシャリーオクチルフェノール樹脂の液体クロマトグラフィーチャートを図1〜図4に示す。
表1、及び図1〜4から明らかなように、実施例1〜5は、アルキルフェノール類とアルデヒド類とを、有機ホスホン酸を触媒として、非極性溶媒の存在下で反応させる本発明の製造方法で得られたノボラック型アルキルフェノール樹脂、実施例6〜10は、アルキルフェノール類とアルデヒド類とを、リン酸を触媒として、非極性溶媒の存在下で反応させる本発明の製造方法で得られたノボラック型アルキルフェノール樹脂であり、これらはいずれも分子量分布が狭く、未反応アルキルフェノールが少なく反応収量も高いものであった。
一方、比較例1は通常の酸性触媒を用いて反応を行ったが、未反応アルキルフェノールの含有量が多いものとなった。また、比較例2は有機ホスホン酸触媒を用いて反応を行ったが、非極性溶媒を用いなかったために、合成後のノボラック型アルキルフェノール樹脂の分離ができなかった。
一方、比較例1は通常の酸性触媒を用いて反応を行ったが、未反応アルキルフェノールの含有量が多いものとなった。また、比較例2は有機ホスホン酸触媒を用いて反応を行ったが、非極性溶媒を用いなかったために、合成後のノボラック型アルキルフェノール樹脂の分離ができなかった。
本発明は、アルキルフェノール類とアルデヒド類とを、有機ホスホン酸、あるいは、リン酸を触媒として、非極性溶媒の存在下で反応させる工程を有することを特徴とするノボラック型アルキルフェノール樹脂の製造方法である。本発明の製造方法により、未反応アルキルフェノール類が少なく、かつ、分子量分布が狭いノボラック型アルキルフェノール樹脂を高収率で得ることができる。
従って本発明は、ゴムなどに配合して相溶性、粘着性、粘弾性などを付与するノボラック型アルキルフェノール樹脂を製造する方法として好適に使用できるものである。
従って本発明は、ゴムなどに配合して相溶性、粘着性、粘弾性などを付与するノボラック型アルキルフェノール樹脂を製造する方法として好適に使用できるものである。
Claims (9)
- ノボラック型アルキルフェノール樹脂の製造方法において、アルキルフェノール類とアルデヒド類とを、有機ホスホン酸を触媒として、非極性溶媒の存在下で反応させる工程を有することを特徴とする、ノボラック型アルキルフェノール樹脂の製造方法。
- 前記工程において、反応系中の非極性溶媒量は20〜70重量%である請求項1に記載のノボラック型アルキルフェノール樹脂の製造方法。
- 前記工程における反応条件は、反応系中の水分量が30重量%以下、反応温度が110〜250℃である請求項1又は2に記載のノボラック型アルキルフェノール樹脂の製造方法。
- 前記有機ホスホン酸は、一般式(I)で示されるものである請求項1ないし3のいずれかに記載のノボラック型アルキルフェノール樹脂の製造方法。
R−PO(OH)2 (I)
(Rは、炭素原子を含み、かつ、−COOH及び又は−PO(OH)2 を含む基である) - ノボラック型アルキルフェノール樹脂の製造方法であって、アルキルフェノール類とアルデヒド類とを、リン酸類を触媒として、非極性溶媒の存在下で反応させる工程を有することを特徴とするノボラック型アルキルフェノール樹脂の製造方法。
- 前記アルキルフェノール類1モルに対して、前記リン酸類0.2モル以上を用いる請求項5に記載のノボラック型アルキルフェノール樹脂の製造方法。
- 前記工程において、反応系中の非極性溶媒量は20〜70重量%である請求項5又は6に記載のノボラック型アルキルフェノール樹脂の製造方法。
- 前記工程における反応条件は、反応系中の水分量が1〜40重量%、反応温度が80〜180℃である請求項5ないし7のいずれかに記載のノボラック型アルキルフェノール樹脂の製造方法。
- 前記リン酸類は、リン酸である請求項5ないし8のいずれかに記載のノボラック型アルキルフェノール樹脂の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004006301A JP2004238618A (ja) | 2003-01-16 | 2004-01-14 | ノボラック型アルキルフェノール樹脂の製造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003008250 | 2003-01-16 | ||
| JP2004006301A JP2004238618A (ja) | 2003-01-16 | 2004-01-14 | ノボラック型アルキルフェノール樹脂の製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2004238618A true JP2004238618A (ja) | 2004-08-26 |
Family
ID=32964780
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004006301A Pending JP2004238618A (ja) | 2003-01-16 | 2004-01-14 | ノボラック型アルキルフェノール樹脂の製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2004238618A (ja) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7488784B2 (en) * | 2003-10-03 | 2009-02-10 | Si Group, Inc. | Novolac alkylphenol resin formed with fatty acid |
| WO2011158751A1 (ja) * | 2010-06-14 | 2011-12-22 | 住友ベークライト株式会社 | ノボラック型フェノール樹脂の製造方法 |
| WO2018003513A1 (ja) * | 2016-06-29 | 2018-01-04 | Dic株式会社 | フェノールノボラック樹脂、硬化性樹脂組成物及びその硬化物 |
| US9932436B2 (en) | 2013-10-17 | 2018-04-03 | Si Group, Inc. | Modified alkylphenol-aldehyde resins stabilized by a salicylic acid |
| US9944744B2 (en) | 2013-10-17 | 2018-04-17 | Si Group, Inc. | In-situ alkylphenol-aldehyde resins |
| CN115216331A (zh) * | 2022-06-22 | 2022-10-21 | 中海油天津化工研究设计院有限公司 | 多烷基枝化酚醛树脂型油浆沉降剂及制备方法、使用方法 |
-
2004
- 2004-01-14 JP JP2004006301A patent/JP2004238618A/ja active Pending
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7488784B2 (en) * | 2003-10-03 | 2009-02-10 | Si Group, Inc. | Novolac alkylphenol resin formed with fatty acid |
| WO2011158751A1 (ja) * | 2010-06-14 | 2011-12-22 | 住友ベークライト株式会社 | ノボラック型フェノール樹脂の製造方法 |
| JP5252130B2 (ja) * | 2010-06-14 | 2013-07-31 | 住友ベークライト株式会社 | ノボラック型フェノール樹脂の製造方法 |
| US8822627B2 (en) | 2010-06-14 | 2014-09-02 | Sumitomo Bakelite Co., Ltd. | Method of manufacture of novolac-type phenol resin |
| US10647806B2 (en) | 2013-10-17 | 2020-05-12 | Si Group, Inc. | In-situ alkylphenol-aldehyde resins |
| US12012487B2 (en) | 2013-10-17 | 2024-06-18 | Si Group, Inc. | In-situ alkylphenol-aldehyde resins |
| US9932436B2 (en) | 2013-10-17 | 2018-04-03 | Si Group, Inc. | Modified alkylphenol-aldehyde resins stabilized by a salicylic acid |
| US9944744B2 (en) | 2013-10-17 | 2018-04-17 | Si Group, Inc. | In-situ alkylphenol-aldehyde resins |
| US11155667B2 (en) | 2013-10-17 | 2021-10-26 | Si Group, Inc. | In-situ alkylphenol-aldehyde resins |
| CN109415476A (zh) * | 2016-06-29 | 2019-03-01 | Dic株式会社 | 苯酚酚醛清漆树脂、固化性树脂组合物及其固化物 |
| US20190225758A1 (en) * | 2016-06-29 | 2019-07-25 | Dic Corporation | Phenol novolak resin, curable resin composition, and cured product thereof |
| KR20190025558A (ko) * | 2016-06-29 | 2019-03-11 | 디아이씨 가부시끼가이샤 | 페놀노볼락 수지, 경화성 수지 조성물 및 그 경화물 |
| US10808085B2 (en) | 2016-06-29 | 2020-10-20 | Dic Corporation | Phenol novolak resin, curable resin composition, and cured product thereof |
| CN109415476B (zh) * | 2016-06-29 | 2021-05-14 | Dic株式会社 | 苯酚酚醛清漆树脂、固化性树脂组合物及其固化物 |
| TWI731986B (zh) * | 2016-06-29 | 2021-07-01 | 日商迪愛生股份有限公司 | 苯酚酚醛清漆樹脂、硬化性樹脂組成物及其硬化物 |
| KR102283386B1 (ko) * | 2016-06-29 | 2021-07-30 | 디아이씨 가부시끼가이샤 | 페놀노볼락 수지, 경화성 수지 조성물 및 그 경화물 |
| JPWO2018003513A1 (ja) * | 2016-06-29 | 2018-07-05 | Dic株式会社 | フェノールノボラック樹脂、硬化性樹脂組成物及びその硬化物 |
| WO2018003513A1 (ja) * | 2016-06-29 | 2018-01-04 | Dic株式会社 | フェノールノボラック樹脂、硬化性樹脂組成物及びその硬化物 |
| CN115216331A (zh) * | 2022-06-22 | 2022-10-21 | 中海油天津化工研究设计院有限公司 | 多烷基枝化酚醛树脂型油浆沉降剂及制备方法、使用方法 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4998271B2 (ja) | フェノール樹脂及び樹脂組成物 | |
| JP2004238618A (ja) | ノボラック型アルキルフェノール樹脂の製造方法 | |
| JP3651843B2 (ja) | フェノール樹脂の製造方法 | |
| EP1108734B1 (en) | Process for producing phenol resin | |
| JP2005075938A (ja) | ハイオルソノボラック型フェノール樹脂の製造方法 | |
| JP4595751B2 (ja) | ビフェニルアラルキル変性フェノール樹脂及びその製造方法、これを含むエポキシ樹脂成形材料。 | |
| JP4399977B2 (ja) | 芳香族炭化水素変性フェノール樹脂の製造方法 | |
| JP2002179751A (ja) | 芳香族炭化水素フェノール樹脂の製造方法 | |
| JP2005075936A (ja) | ノボラック型フェノール樹脂およびその製造方法 | |
| JP2002128849A (ja) | フェノール樹脂の製造方法 | |
| JP2005179383A (ja) | アラルキル変性フェノール樹脂の製造方法 | |
| JP4720057B2 (ja) | エポキシ樹脂の製造方法 | |
| JP2005179382A (ja) | ノボラック型フェノール樹脂の製造方法 | |
| JP3874338B2 (ja) | ノボラック型フェノール樹脂の製造方法 | |
| JP4191019B2 (ja) | フェノールアラルキル樹脂の製造方法 | |
| JP2005206706A (ja) | エポキシ樹脂の製造方法 | |
| JP2005179448A (ja) | 芳香族炭化水素フェノール樹脂の製造方法 | |
| JP2001172348A (ja) | フェノール樹脂の製造方法 | |
| JP4206909B2 (ja) | トリアジン変性ノボラック型フェノール樹脂の製造方法 | |
| JP2002302525A (ja) | ノボラック型フェノール樹脂の製造方法 | |
| JP2002105157A (ja) | フェノール樹脂の製造方法 | |
| JP2005200489A (ja) | 熱可塑性樹脂変性ノボラック型フェノール樹脂の製造方法 | |
| JP2003212943A (ja) | ハイオルソノボラック型フェノール樹脂の製造方法 | |
| JP2003119233A (ja) | ノボラック型フェノール樹脂およびその製造方法 | |
| JP2005068395A (ja) | ノボラック型フェノール樹脂の製造方法 |