JP5060397B2 - 植物系繊維材料の糖化分離方法 - Google Patents
植物系繊維材料の糖化分離方法 Download PDFInfo
- Publication number
- JP5060397B2 JP5060397B2 JP2008145732A JP2008145732A JP5060397B2 JP 5060397 B2 JP5060397 B2 JP 5060397B2 JP 2008145732 A JP2008145732 A JP 2008145732A JP 2008145732 A JP2008145732 A JP 2008145732A JP 5060397 B2 JP5060397 B2 JP 5060397B2
- Authority
- JP
- Japan
- Prior art keywords
- fiber material
- cluster acid
- acid catalyst
- plant fiber
- cluster
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C13—SUGAR INDUSTRY
- C13K—SACCHARIDES OBTAINED FROM NATURAL SOURCES OR BY HYDROLYSIS OF NATURALLY OCCURRING DISACCHARIDES, OLIGOSACCHARIDES OR POLYSACCHARIDES
- C13K1/00—Glucose; Glucose-containing syrups
- C13K1/02—Glucose; Glucose-containing syrups obtained by saccharification of cellulosic materials
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Emergency Medicine (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- Organic Chemistry (AREA)
- Catalysts (AREA)
- Saccharide Compounds (AREA)
- Polysaccharides And Polysaccharide Derivatives (AREA)
Description
従来、セルロースやヘミセルロースを分解してグルコース等の糖を生成する種々の方法が提案されており(例えば、特許文献1〜4等)、一般的な方法としては、希硫酸や濃硫酸等の硫酸、塩酸を用いてセルロースを加水分解する方法(特許文献1等)が挙げられる。また、セルラーゼ酵素を用いる方法(特許文献2等)、活性炭やゼオライト等の固体触媒を用いる方法(特許文献3等)、加圧熱水を用いる方法(特許文献4等)もある。
また、濃硫酸を用いる場合には、長時間反応させると生成した糖の脱水反応が起こり、糖の収率が低下するという問題がある。そのため、加水分解反応中の反応系に植物系繊維材料を追加し、植物系繊維材料の加水分解処理量を増やそうとしても、植物系繊維材料に対する糖の収率が高まらない。
さらに、硫酸や塩酸等の酸は、分離、回収して再利用することが非常に困難である。そのため、これら酸をグルコース生成の触媒として用いることは、バイオエタノールのコストを引き上げる原因の一つとなっている。
また、上記特許出願において、擬融解状態のクラスター酸は、加水分解触媒として作用すると共に反応溶媒としても作用している。
前記加水分解工程において、
前記クラスター酸触媒と、
擬融解状態の該クラスター酸触媒に添加した時に該擬融解状態のクラスター酸触媒の粘度が上昇する量の植物系繊維材料(1)と、
を加熱混合し、
該加熱混合物の粘度の低下が生じた際に、さらに植物系繊維材料(2)を追加添加することを特徴とするものである。
このように、加水分解工程における反応混合物の粘度低下が生じた後に、新たに植物系繊維材料を追加投入することによって、糖の収率を確保しつつ、クラスター酸触媒の単位重量当りの植物系繊維材料の処理量を増加させることができる。その結果、クラスター酸触媒の使用量低減による糖生成のコスト削減、クラスター酸触媒を擬融解状態にするための加熱に要するエネルギーの削減が可能である。
前記加水分解工程において、前記クラスター酸触媒と、擬融解状態の該クラスター酸触媒に添加した時に、該擬融解状態のクラスター酸触媒の粘度が上昇する量(以下、高粘度添加量ということがある)の植物系繊維材料(1)と、を加熱混合し、該加熱混合物の粘度の低下が生じた際に、さらに植物系繊維材料(2)を追加添加することを特徴とするものである。
擬融解状態のクラスター酸触媒と、植物系繊維材料との加熱混合物は、加水分解反応当初は粘度が高いが、該植物系繊維材料の加水分解の進行に伴い、粘度が低下することがわかった。そして、該加熱混合物の粘度の低下により、該加熱混合物に新たに植物系繊維材料を投入しても、該加熱混合物を混合攪拌することが可能であり、糖収率を確保しながら、初期に投入した植物系繊維材料及び追加分の植物系繊維材料の加水分解が可能であることが見出された。
つまり、本発明によれば、従来と比較して、追加添加する植物系繊維材料分、植物系繊維材料の処理量を増加することができる。その結果、クラスター酸触媒単位重量当りの植物系繊維材料の処理量が増加し、クラスター酸触媒の使用量の削減による糖生成のコスト削減効果が得られる。また、擬融解状態のクラスター酸触媒を含有する加水分解工程中の加熱混合物に、植物系繊維材料を添加追加するため、クラスター酸触媒を擬融解状態とするのに必要な加熱に要するエネルギーを削減することができる。すなわち、エネルギー効率を向上させることができる。
尚、上記加熱混合物の粘度がどの程度が低下したときに追加で植物系繊維材料を添加するかは、追加で添加する植物系繊維材料の量に応じて適宜決めればよい。すなわち、少量の追加であれば粘度が多少低下した時点で追加可能であり、多量に追加するのであれば、充分加水分解反応が進んで粘度が大きく低下するまで添加を控える。いずれにしても最初に繊維材料を添加した際の粘度を越えないように追加添加することが好ましい。
まず、植物系繊維材料に含まれるセルロースを加水分解し、グルコースを主とする糖を生成させる加水分解工程について説明する。
尚、ここでは、主としてセルロースからグルコースを生成させる工程を中心に説明しているが、植物系繊維材料にはセルロース以外にヘミセルロースも含まれ、また、生成物もグルコース以外にキシロース等のその他の単糖もあり、これらの場合も本発明の範囲に含まれる。
これら繊維材料は、反応系における分散性の観点から、通常、粉末状のものを用いる。粉末状にする方法としては、一般的な方法に準じればよい。クラスター酸触媒との混合性、反応機会向上の観点から、植物系繊維材料は、数μm〜200μm程度の直径を有する粉末状であることが好ましい。
蒸解処理としては、例えば、NaOH、KOH、Ca(OH)2、Na2SO3、NaHCO3、NaHSO3、Mg(HSO3)2、Ca(HSO3)2などのアルカリや塩及びその水溶液、これらにさらにSO2溶液を混合したもの、NH3等のガスと、植物系繊維材料(例えば、数cm〜数mm)を、水蒸気下で接触させる方法が挙げられる。具体的な条件として、反応温度は120〜160℃、反応時間は数十分から1時間程度でよい。
尚、ここでは結晶状態のクラスター酸触媒、および、数分子のクラスター酸触媒で構成されるクラスター状態のクラスター酸触媒と水和又は配位する水を、一般的に使用される「結晶水」という用語で代用する。この結晶水にはクラスター酸触媒を構成するアニオンと水素結合したアニオン水、カチオンに配位した配位水、カチオン及びアニオンと配位しない格子水の他、OH基の形で含まれているものも含まれる。
また、クラスター状態のクラスター酸触媒とは、1〜数分子程度のクラスター酸から構成される集合体であり、結晶とは異なる。固体状態、擬融解状態、溶媒中に溶解(コロイド状)した状態でもクラスター状態とすることができる。
クラスター状態のクラスター酸触媒の調製方法は特に限定されない。具体的な方法については後述する。
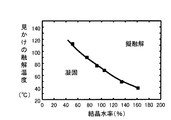
クラスター酸の擬融解状態は、温度と、クラスター酸触媒が含有する結晶水の量によって変わってくる(図2参照)。具体的には、クラスター酸であるリンタングステン酸は、含有する結晶水が多くなると擬融解状態を発現する温度が低下する。すなわち、結晶水を多く含むクラスター酸触媒は、相対的に結晶水量が少ないクラスター酸触媒よりも低い温度でセルロースの加水分解反応に対する触媒作用を発現する。つまり、加水分解工程の反応系におけるクラスター酸触媒が含有する結晶水の量をコントロールすることで、目的とする加水分解反応温度においてクラスター酸触媒を擬融解状態とすることができる。例えば、リンタグステン酸をクラスター酸触媒として用いる場合は、クラスター酸の結晶水量によって加水分解反応温度を110℃〜40℃の範囲内で制御可能である(図2参照)。
以上のように、クラスター酸の結晶水量は容易にコントロールが可能であり、結晶水量の制御によりセルロースの加水分解反応温度も容易に調整可能である。
さらに、硫酸等の酸を用いる従来のセルロースの加水分解法と異なり、クラスター酸を触媒として用いる本発明の方法は、糖と触媒の分離効率が高く、容易に分離可能である。クラスター酸は温度によっては固形状態となるため、生成物である糖類との分離が可能である。従って、分離したクラスター酸を回収し、再利用することも可能である。また、擬融解状態のクラスター酸触媒は、反応溶媒としても機能するため、従来の方法と比較して、反応溶媒としての溶剤量を大幅に減少させることができる。これは、クラスター酸と生成物である糖との分離、クラスター酸の回収の高効率化が可能であることを意味している。すなわち、クラスター酸をセルロースの加水分解触媒として利用する本発明は、低コストが可能であり、且つ、環境負荷も小さい。
通常、加水分解工程において使用するクラスター酸触媒に対する、植物系繊維材料(1)の体積比を60%以上とすると、擬融解状態の該クラスター酸触媒に植物系繊維材料(1)を添加した際に粘度が上昇する。特に、植物系繊維材料の処理効率の観点から、加水分解工程において使用するクラスター酸触媒に対する、植物系繊維材料(1)の体積比は、特に50%以上、さらに65%以上とすることが好ましい。
尚、クラスター酸触媒と植物系繊維材料(1)を投入した後、これらを加熱する場合には、加熱前に、クラスター酸触媒と植物系繊維材料(1)を、予め混合攪拌しておくことが好ましい。クラスター酸触媒が擬融解状態となる前にある程度混合しておくことによってクラスター酸と植物系繊維材料(1)との接触性を高めることができる。
しかしながら、加水分解工程においては、セルロースが加水分解されるための水が必要である。具体的には、n個のグルコースが重合したセルロースをn個のグルコースに分解するためには、(n−1)個の水分子が必要である。従って、反応系内に、クラスター酸触媒が反応温度において擬融解状態となるのに必要な結晶水量分の水分と、仕込まれたセルロース全量がグルコースに加水分解されるのに必要な水分の合計量が存在しない場合、クラスター酸触媒の結晶水がセルロースの加水分解に使用され、クラスター酸触媒の結晶水が減少し、クラスター酸が凝固状態となってしまう。すなわち、クラスター酸触媒と植物系繊維材料との接触性が低下するばかりか、植物系繊維材料とクラスター酸触媒の混合物の粘度が増加し、該混合物を充分に混合するのに時間がかかってしまう。
ここで、(B)の水量は、追加添加する植物系繊維材料(2)の追加添加前であれば、植物系繊維材料(1)に含有されるセルロースの全量がグルコースに加水分解されるのに必要な水分量(B1)であり、植物系繊維材料(2)の追加添加後であれば、植物系繊維材料(1)及び(2)に含有されるセルロースの全量がグルコースに加水分解されるのに必要な水分量(B1+B2)である。(B)の水分は、植物系繊維材料(2)の追加添加前に、全量(B1+B2)を添加してもよいし、植物系繊維材料(2)の追加添加に合わせてB1とB2を分割して添加してもよい。
また、加水分解工程において、加熱により反応系の相対湿度が低下しても、クラスター酸触媒の結晶水が所望量確保できるようにしておくことが好ましい。具体的には、予定の反応温度で反応系の雰囲気が飽和蒸気圧となるように、例えば、予め密閉された反応容器内で、加水分解反応温度で飽和蒸気圧状態を作り、密閉状態を保持したまま温度を下げて蒸気を凝縮させ、該凝縮水を植物系繊維材料及びクラスター酸触媒に添加する方法が挙げられる。
また、植物系繊維材料として、乾燥状態のものを用いる場合には、特に考慮する必要がないが、水分を含む植物系繊維材料を用いる場合には、反応系内に存在する水分量として、該植物系繊維材料が含有する水分量も考慮することが好ましい。
植物系繊維材料(2)を追加添加する際の加熱混合物の粘度は、加水分解工程の反応初期における擬融解状態のクラスター酸触媒と植物系繊維材料(1)を含む加熱混合物の粘度より低く、該加熱混合物に植物系繊維材料(2)を追加添加しても、該加熱混合物の混合が可能であれば、具体的な値に限定はない。追加で添加する植物系繊維材料の量に応じて適宜決めればよく、すなわち、少量の追加であれば粘度が多少低下した時点で追加可能であり、多量に追加するのであれば、充分加水分解反応が進んで粘度が大きく低下するまで添加を控える。いずれにしても最初に繊維材料を添加した際の反応初期の加熱混合物の粘度を越えないように追加添加することが好ましい。
通常は、加熱混合物の粘度が1500cp以下、特に1200cp以下、さらには1000cp以下になってから、植物系繊維材料(2)を追加添加することが好ましい。
植物系繊維材料(2)の追加添加は、複数行ってもよい。すなわち、第1回目の植物系繊維材料(2)の追加添加後、加熱混合物の粘度が低下したら、さらに植物系繊維材料(2)の追加添加を行うという工程を繰り返してよい。
具体的には、例えば、図3に示す固定反応装置(バッチ式)の場合、粘度センサ2及び液面センサ3により、反応槽1内の加熱混合物4の粘度が計測可能となっている。加熱混合物4の粘度を測定する粘度センサ2は、反応槽1の底面又は側面の底面に近い部分に設置することが好ましい。また、液面センサ3を、反応槽1の側面に複数設置することで、反応槽1内の加熱混合物4の液面変化を正確に計測することができる。このとき、液面センサ3と共に、温度センサ5を複数設けることで、攪拌翼の回転数を適切にフィードバック制御することができる(図3参照)。上記したように、粘度センサ2や液面センサ3により計測される加熱混合物4の粘度変化を、植物系繊維材料の投入機構9へフィードバックすることが好ましい。
尚、図3においては、反応槽1の底面に加熱ヒータ7及び温度センサ8が配置されており、反応槽1内の加熱混合物4の温度制御が可能となっている。また、固定反応装置(バッチ式)は、図3の形態に限定されず、例えば、上述したように、加熱混合物4の粘度を、攪拌翼6のトルクから間接的に計測されるようにしてもよい。
粘度センサ12と投入口11の設置位置は特に限定されないが、例えば、投入口11(1)については、下流側に設置された投入口11(2)の、上流側に隣接するように粘度センサ12(1)を設置することができる(図4参照)。投入口11(2)〜投入口11(3)と粘度センサ12(2)〜12(3)についても同様である。また、流通式反応装置の場合、リグニンを含む植物系繊維材料を用いる場合には、各投入口11の直前にヘテロポリ酸や生成した糖は除去しないが、リグニンを除去可能なフィルターを設置することで、上流側の残渣を除去することが好ましい。
従って、加水分解の反応温度は、セルロースの反応率とグルコース生成の選択性を左右する重要な要素であり、エネルギー効率の観点から加水分解反応の温度は低いことが好ましい旨を述べたが、セルロースの反応率やグルコース生成の選択性等も考慮して加水分解反応の温度を決定することが好ましい。
固液分離により得られる固形分は、蒸留水等の水を添加し、攪拌することで、糖が水に溶解するため、さらに固液分離することによって、糖水溶液と、残渣等を含む固形分とを分離することができる。
特に好ましくは、加水分解反応混合物中に含まれる全てのクラスター酸触媒の結晶水率が100%未満となるように、加水分解反応混合物の水分量を調節することが好ましい。クラスター酸触媒が多くの結晶水、典型的には、標準結晶水量以上の結晶水を有する場合、過剰な水分に生成物である糖が溶解し、クラスター酸有機溶媒溶液側に糖が混入することによって糖の回収率が低下してしまう。クラスター酸触媒の結晶水率を100%未満とすることで、このようにクラスター酸触媒に糖が混入することを抑制することができる。
クラスター酸触媒のクラスター化は、例えば、クラスター酸を、擬融解状態にして攪拌するか、又は、溶媒に溶解して加熱攪拌するか、又は、植物系繊維材料と加熱攪拌して加水分解触媒として使用することによって、促進される。
具体的なクラスター化促進処理の方法としては、以下の3つを挙げることができる。すなわち、(1)クラスター酸触媒と、該クラスター酸触媒を可溶な有機溶媒とを加熱攪拌する工程を備える方法、(2)植物系繊維材料をクラスター酸触媒を用いて加水分解する加水分解工程において、一回投入可能な量の植物系繊維材料の一部を、擬融解状態のクラスター酸触媒と加熱攪拌し、該植物系繊維材料の加水分解を行う方法、(3)クラスター酸触媒を擬融解状態で加熱攪拌する方法が挙げられる。以下、上記(1)〜(3)の方法について説明する。
クラスター酸触媒を可溶な有機溶媒としては、上記糖分離工程において使用可能な有機溶媒が挙げられる。中でも、クラスター酸の溶解度、沸点の観点から、エタノール、メタノールが好適である。
有機溶媒とクラスター酸触媒の混合割合は、特に限定されず、有機溶媒に対するクラスター酸触媒の溶解度等に応じて適宜選択することができる。また、加熱攪拌時間は、使用する有機溶媒に対するクラスター酸触媒の溶解度、加熱温度等に応じて、適宜決定すればよく、通常は、10分〜60分程度、又は30〜60分程度である。混合方法も特に限定されず、公知の方法を採用することができる。
また、上記糖分離工程において、加水分解反応混合物に有機溶媒を添加、攪拌した後、固液分離により得られるクラスター酸有機溶媒溶液を、加熱攪拌することで、再利用するクラスター酸触媒のクラスター化を促進することもできる。
具体的には、減圧蒸留、凍結乾燥等によって、有機溶媒を除去することが好ましい。加熱により有機溶媒を除去することも可能であるが、クラスター酸のクラスター状態を維持させるという観点からは、低温(具体的には、65℃以下)において、有機溶媒を除去することが好ましく、上記のような減圧蒸留、凍結乾燥等が好適であるといえる。
また、「一回投入可能な量の植物系繊維材料の一部」とは、上記「一回投入可能な量の植物系繊維材料」の一部であり、具体的な量に限定されないが、通常、擬融解状態のクラスター酸触媒に添加、攪拌しても、添加前の擬融解状態のクラスター酸触媒の粘度が維持される程度の微量である。加水分解工程において使用するクラスター酸触媒に対して、このような微量の植物系繊維材料を最初に添加することは、いわば、捨石による全体の反応効率の向上効果が期待できる。「一回投入可能な量の植物系繊維材料の一部」の具体的な量としては、一回投入可能な量の植物系繊維材料の10重量%以下が好ましく、特に5重量%以下が好ましい。
その他、反応温度、圧力などは、上記加水分解工程と同様にすることができる。
このようなクラスター酸を擬融解状態で加熱攪拌する方法は、擬融解状態のクラスター酸を加水分解触媒として使用する加水分解工程の前準備工程として、既存の工程に容易に組み込むことができる。また、未使用のクラスター酸試薬を用いる場合であっても、加水分解工程における単糖の脱水反応を抑制することができる。
尚、IR測定において、上記H2O分子に由来する吸収ピークは、強酸性の担体に結合したOH基に由来する吸収ピークの吸収に限らず、ブロードなピークとして観察されるのが一般的である。
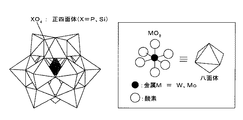
このような高波数シフトと低感度化は、クラスター酸触媒のクラスター化による以下の構造変化から起こる。WO6八面体はWのイオン半径が0.074nmと小さいため、図1のようにWOは非常にタイトになっている。クラスター化による表面エネルギー安定化が起こり、さらに球に近い形に歪むと、WO6の対称性は下がり、さらにWO6距離が短くなる。このため、感度低下と結合強度アップにより、散乱と高波数シフトが同時に起こることになる。この現象はリンタングステン酸だけでなく、他のクラスター酸でも同様に生じるため、ラマン分光測定によりクラスター酸触媒の構造変化を観察することで、クラスター酸触媒のクラスター状態を確認することができる。
密閉反応容器(バッチ式。図3参照)に、予め蒸留水を入れ、予定の反応温度(70℃)まで昇温し、容器内を飽和蒸気圧状態とし、容器内面に水蒸気を付着させた。
次に、繰り返し使用しているクラスター状態のヘテロポリ酸(予め結晶水量を測定済み。リンタングステン酸)1kg、ヘテロポリ酸の結晶水量を100%にするために必要な水分とセルロースが加水分解してグルコースになるのに必要な水分(55.6g)との合計量からの不足分(上記70℃での飽和蒸気圧分の水分は除く)の蒸留水(35g)を容器に投入して加熱攪拌し、容器内温度が70℃になってから、さらに10分間攪拌を続けた。その後、セルロース0.5kgを投入し、70℃で加熱混合した。加熱混合を開始して10分後、加熱混合物の粘度は3000cpであった。
1時間後、加熱混合物の粘度が700cpまで低下したため、さらに、セルロース0.5kgと、セルロースが加水分解してグルコースになるのに必要な水分(55.6g)を投入し、70℃で加熱混合を2時間続けた。
その後、加熱を停止し、容器の密閉を開放し余分な水蒸気を排出させながら、室温まで冷却した。
尚、ここで、単糖の収率は、以下のようにして算出した。
密閉反応容器(バッチ式。図3参照)に、予め蒸留水を入れ、予定の反応温度(70℃)まで昇温し、容器内を飽和蒸気圧状態とし、容器内面に水蒸気を付着させた。
次に、クラスター状態の繰り返し使用しているヘテロポリ酸(予め結晶水量を測定済み。リンタングステン酸)1.15kg、ヘテロポリ酸の結晶水量を100%にするために必要な水分とセルロースが加水分解してグルコースになるのに必要な水分(55.6g)との合計量からの不足分(上記70℃での飽和蒸気圧分の水分は除く)の蒸留水(35g)を投入して加熱攪拌し、70℃になってから、さらに10分間加熱混合を続けた。その後、リグニンを含む木材粉0.5kgを投入し、70℃で加熱混合した。加熱混合を開始して10分後、加熱混合物の粘度は3000cpであった。
3時間後、得られた加熱混合物から、焼結フィルタを用いてリグニンを取り除いた。リグニンを取り除いた加熱混合物の粘度は700cpまで低下したため、さらに、リグニンを含む木材粉0.5kgと、木材粉に含まれるセルロースが加水分解してグルコースになるのに必要な水分(35g)を投入し、70℃で加熱混合を3時間続けた。
尚、本実施例においては、上記焼結フィルタによるリグニン除去の際に、リグニンと共にリグニンに吸着したヘテロポリ酸が除去されるため、実施例1の1.15倍のヘテロポリ酸を使用した。
擬融解状態としたヘテロポリ酸(リンタングステン酸)の主流路に対して、加熱ライン(温度一定70℃)中で攪拌が可能な流通式反応装置(図4参照)を使用した。尚、ライン中の攪拌翼10は、反応槽100の内容物の搬送にはほとんど寄与せず、攪拌のみに有効な構造となっている。ゆえに、内容物の速度は、各成分(ヘテロポリ酸、植物系繊維材料)の投入速度の和となる。反応槽100は、擬融解状態のヘテロポリ酸の投入口13を最も上流側に備えており、該ヘテロポリ酸投入口13よりも下流側に、植物系繊維材料の投入口11を複数(第1〜第4の投入口)備えている。各植物系繊維材料の投入口11は、下流側(該投入口の下流側に設けられた投入口の直前又は反応器の下流壁直前)に粘度センサ12(第1〜第4の粘度センサ)が備えられており、該粘度センサ12による反応槽100内の内容物の粘度が、各植物系繊維材料の投入口11からの繊維材料の投入量にフィードバックされるようになっている。
反応器の下流から排出された反応混合物は、余分な水蒸気を除きながら、室温まで冷却した。尚、本実施例において、使用したヘテロポリ酸と木材粉の重量比は1:1.2(ヘテロポリ酸:木材粉)だった。
次に、実施例1と同様にして、加水分解反応混合物から糖とヘテロポリ酸を回収した。単糖収率は82.1%だった。このとき、除去したリグニンは、使用した木材分の30wt%として単糖収率を算出した。
密閉容器内(バッチ式)に、予め蒸留水を入れ、予定の反応温度(70℃)まで昇温し、容器内を飽和蒸気圧状態とし、容器内面に水蒸気を付着させた。次に、クラスター状態の繰り返し使用しているヘテロポリ酸(予め結晶水量を測定済み。リンタングステン酸)1kg、ヘテロポリ酸の結晶水量を100%にするために必要な水分とセルロースが加水分解してグルコースになるのに必要な水分(55.6g)との合計量からの不足分(上記70℃での飽和蒸気圧分の水分は除く)の蒸留水(35g)を容器に投入して加熱攪拌し、容器内温度が70℃になってから、さらに5分間攪拌を続けた。
続いて、容器内に0.5kgのセルロースを投入し、70℃で2時間攪拌を続けた。その後、加熱を停止し、容器の密閉を開放し余分な水蒸気を排出させながら、室温まで冷却した。
その後、実施例1と同様にして、加水分解反応混合物から糖とヘテロポリ酸を回収した。
単糖の収率は85.3%だった。
実施例1〜3及び比較例1について、単糖の収率及びヘテロポリ酸と繊維材料の重量比を表1に示す。

尚、工業的な反応装置では、反応系へ水の添加は、蒸気により導入することで、加熱と加水が同時に行うことができ、蒸留水による導入と比較してエネルギー的に有利である。
2…粘度センサ
3…液面センサ(温度センサ5付き)
4…加熱混合物
5…温度センサ
6…攪拌翼
7…加熱ヒータ
8…温度センサ
9…植物系繊維材料投入機構
10…攪拌翼
11(1)、11(2)、11(3)、11(4)…植物系繊維材料投入口
12(1)、12(2)、12(3)、12(4)…粘度センサ
13…擬融解状態クラスター酸触媒の投入口
Claims (3)
- 植物系繊維材料を加水分解し、グルコースを主とする糖を生成し、分離する植物系繊維材料の糖化分離方法であって、
擬融解状態のクラスター酸触媒を用いて、前記植物系繊維材料に含まれるセルロースを加水分解し、グルコースを生成させる加水分解工程を備えており、
前記加水分解工程において、
前記クラスター酸触媒と、
擬融解状態の該クラスター酸触媒に添加した時に該擬融解状態のクラスター酸触媒の粘度が上昇する量の植物系繊維材料(1)と、
を加熱混合し、
該加熱混合物の粘度の低下が生じた際に、さらに植物系繊維材料(2)を追加添加することを特徴とする、糖化分離方法。 - 前記植物系繊維材料(1)が、前記クラスター酸触媒に対して、体積比で60%以上である、請求項1に記載の糖化分離方法。
- 前記植物系繊維材料(2)が、前記クラスター酸触媒に対して、体積比で60%以上である、請求項1又は2に記載の糖化分離方法。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008145732A JP5060397B2 (ja) | 2008-06-03 | 2008-06-03 | 植物系繊維材料の糖化分離方法 |
| US12/995,809 US8460472B2 (en) | 2008-06-03 | 2009-06-02 | Method for glycosylating and separating plant fiber material |
| MYPI20105721 MY148089A (en) | 2008-06-03 | 2009-06-02 | Method for glycosylating and separating plant fiber material |
| RU2010149335/13A RU2461633C2 (ru) | 2008-06-03 | 2009-06-02 | Способ гидролиза растительного волокнистого материала для получения и выделения сахарида, включающего глюкозу |
| EP09757875A EP2300625B1 (en) | 2008-06-03 | 2009-06-02 | Method for glycosylating and separating plant fiber material |
| PCT/IB2009/005927 WO2009147522A1 (en) | 2008-06-03 | 2009-06-02 | Method for glycosylating and separating plant fiber material |
| BRPI0913405-0A BRPI0913405A2 (pt) | 2008-06-03 | 2009-06-02 | Método para glicosilar e separar material de fibra de planta |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008145732A JP5060397B2 (ja) | 2008-06-03 | 2008-06-03 | 植物系繊維材料の糖化分離方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2009291083A JP2009291083A (ja) | 2009-12-17 |
| JP5060397B2 true JP5060397B2 (ja) | 2012-10-31 |
Family
ID=40934882
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008145732A Expired - Fee Related JP5060397B2 (ja) | 2008-06-03 | 2008-06-03 | 植物系繊維材料の糖化分離方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US8460472B2 (ja) |
| EP (1) | EP2300625B1 (ja) |
| JP (1) | JP5060397B2 (ja) |
| BR (1) | BRPI0913405A2 (ja) |
| MY (1) | MY148089A (ja) |
| RU (1) | RU2461633C2 (ja) |
| WO (1) | WO2009147522A1 (ja) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5060397B2 (ja) * | 2008-06-03 | 2012-10-31 | トヨタ自動車株式会社 | 植物系繊維材料の糖化分離方法 |
| JP5114298B2 (ja) * | 2008-06-03 | 2013-01-09 | トヨタ自動車株式会社 | 植物系繊維材料の糖化分離方法 |
| EP2529036B1 (en) * | 2010-01-27 | 2017-11-22 | Council of Scientific & Industrial Research | A one pot and single step hydrolytic process for the conversion of lignocellulose into value added chemicals |
| JP5769165B2 (ja) * | 2010-04-01 | 2015-08-26 | 国立大学法人 宮崎大学 | セルロース系物質の分解方法 |
| JP2012005382A (ja) * | 2010-06-23 | 2012-01-12 | Equos Research Co Ltd | バイオマス加水分解反応装置 |
| FR2978156B1 (fr) | 2011-07-19 | 2014-11-07 | IFP Energies Nouvelles | Procede flexible de transformation de biomasse lignocellulosique avec etape de purification |
| FR2978157B1 (fr) | 2011-07-19 | 2014-09-12 | IFP Energies Nouvelles | Procede flexible de transformation de biomasse lignocellulosique en hydrocarbures en presence d'un acide |
| NZ629049A (en) * | 2014-04-22 | 2018-08-31 | Renmatix Inc | Method for mixed biomass hydrolysis |
| CN112218877B (zh) | 2018-08-27 | 2025-07-25 | 瑞泽恩制药公司 | 拉曼光谱在下游纯化中的应用 |
Family Cites Families (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE713610C (de) * | 1939-12-08 | 1941-11-11 | Sueddeutsche Holzverzuckerungs | Verfahren zum Verzuckern von Holz oder anderen cellulosehaltigen Stoffen mit Salzsaeure unter Zusatz von Katalysatoren |
| US2959500A (en) * | 1956-02-14 | 1960-11-08 | Schweizerische Eidgenossenschaft | Process for the saccharification of cellulose and cellulosic materials |
| US3652425A (en) * | 1969-02-18 | 1972-03-28 | Wilson Lab Inc | Process for the preparation of heteropoly acid complex compounds of metaphosphoric metasilic acid, metaphosphoric acid and phosphorous pentoxide |
| SU651652A3 (ru) * | 1976-02-16 | 1979-03-05 | V г, | Способ обработки грубых растительных кормов |
| US4237110A (en) * | 1979-04-30 | 1980-12-02 | The Dow Chemical Company | Process for separating and recovering concentrated hydrochloric acid from the crude product obtained from the acid hydrolysis of cellulose |
| JPS5630334A (en) | 1979-08-21 | 1981-03-26 | Kokusai Denshin Denwa Co Ltd <Kdd> | Light-relay supervising circuit |
| US4743669A (en) * | 1981-11-05 | 1988-05-10 | Union Oil Company Of California | Acid catalyzed organic reactions |
| JPS59124901A (ja) | 1982-12-29 | 1984-07-19 | Dai Ichi Seiyaku Co Ltd | 多糖誘導体 |
| JPS60206604A (ja) * | 1984-03-30 | 1985-10-18 | Ota Shoji | リグノセルロ−ス物質を再構成された複合物品に変換させる方法 |
| JPS61118420A (ja) | 1984-11-14 | 1986-06-05 | Asahi Chem Ind Co Ltd | ポリオキシテトラメチレングリコ−ル中のヘテロポリ酸の分離回収法 |
| JPH0393755A (ja) | 1989-09-05 | 1991-04-18 | Sumitomo Chem Co Ltd | α―アミノ酸の製造法 |
| JPH04226940A (ja) | 1990-06-11 | 1992-08-17 | Mitsui Toatsu Chem Inc | 酢酸シクロヘキシルの製造方法 |
| JPH04240138A (ja) | 1991-01-16 | 1992-08-27 | Sumitomo Electric Ind Ltd | 光ファイバの製造方法 |
| US5549789A (en) | 1992-08-28 | 1996-08-27 | The United States Of America As Represented By The Secretary Of Agriculture | Oxidation of lignin and polysaccharides mediated by polyoxometalate treatment of wood pulp |
| US5562777A (en) * | 1993-03-26 | 1996-10-08 | Arkenol, Inc. | Method of producing sugars using strong acid hydrolysis of cellulosic and hemicellulosic materials |
| JPH0741462A (ja) | 1993-07-28 | 1995-02-10 | Daicel Chem Ind Ltd | 2−ヒドロキシメチルメルカプト酪酸の製造方法 |
| US5380341A (en) * | 1993-09-27 | 1995-01-10 | Ventritex, Inc. | Solid state electrochemical capacitors and their preparation |
| JPH08299000A (ja) | 1995-05-10 | 1996-11-19 | Shokubutsu Kagaku Kenkyusho:Kk | 植物系繊維材料からグルコースを製造する方法 |
| JPH10137599A (ja) | 1996-11-12 | 1998-05-26 | Mitsubishi Gas Chem Co Inc | ヘテロポリ酸の回収方法 |
| JP3041380B2 (ja) | 1997-06-02 | 2000-05-15 | 工業技術院長 | 水溶性オリゴ糖類及び単糖類の製造方法 |
| JPH11240852A (ja) | 1998-02-24 | 1999-09-07 | Ube Ind Ltd | ヘテロポリ酸触媒の分離回収方法 |
| JPH11343301A (ja) * | 1998-06-01 | 1999-12-14 | Nisshin Oil Mills Ltd:The | 微粒子セルロースおよびその製造方法 |
| JP2000103758A (ja) | 1998-07-30 | 2000-04-11 | Toray Ind Inc | 芳香族ケトンの製造方法 |
| JP3802325B2 (ja) | 2000-08-23 | 2006-07-26 | 信行 林 | 植物系バイオマスの加圧熱水分解方法とそのシステム |
| JP2004241307A (ja) | 2003-02-07 | 2004-08-26 | Matsushita Electric Ind Co Ltd | ヘテロポリ酸を坦持した電極およびそれを用いた電気化学素子 |
| JP2004256370A (ja) | 2003-02-27 | 2004-09-16 | Toshihiro Yamase | ホウ素原子含有ヘテロポリ酸及びホウ素化合物 |
| EP1811038B1 (en) * | 2004-07-27 | 2012-02-15 | Asahi Kasei Chemicals Corporation | Process for producing cellooligosaccharide |
| JP4604194B2 (ja) | 2004-11-02 | 2010-12-22 | 国立大学法人広島大学 | 触媒を用いたセルロースの加水分解方法および触媒を用いたグルコースの生産方法 |
| JP2006149343A (ja) | 2004-11-26 | 2006-06-15 | Daitoo Fujitekku:Kk | 木質系バイオマスからのグルコース生成物とグルコース生成物製造方法 |
| JP4831666B2 (ja) | 2004-12-28 | 2011-12-07 | 独立行政法人産業技術総合研究所 | レブリン酸エステルの製造方法 |
| EP1860201A1 (en) * | 2006-05-25 | 2007-11-28 | BP p.l.c. | Conversion method |
| JP2010516296A (ja) * | 2007-01-30 | 2010-05-20 | ヴェレニウム コーポレイション | リグノセルロース性物質を処理する酵素、前記をコードする核酸並びに前記を製造及び使用する方法 |
| JP4877045B2 (ja) | 2007-04-25 | 2012-02-15 | トヨタ自動車株式会社 | 植物系繊維材料の分解方法 |
| JP4240138B1 (ja) * | 2007-09-05 | 2009-03-18 | トヨタ自動車株式会社 | 植物系繊維材料の糖化分離方法 |
| JP5114298B2 (ja) * | 2008-06-03 | 2013-01-09 | トヨタ自動車株式会社 | 植物系繊維材料の糖化分離方法 |
| JP4609526B2 (ja) * | 2008-06-03 | 2011-01-12 | トヨタ自動車株式会社 | 植物系繊維材料の糖化分離方法 |
| JP5060397B2 (ja) * | 2008-06-03 | 2012-10-31 | トヨタ自動車株式会社 | 植物系繊維材料の糖化分離方法 |
| JP4766130B2 (ja) * | 2009-03-06 | 2011-09-07 | トヨタ自動車株式会社 | 植物系繊維材料の糖化方法 |
-
2008
- 2008-06-03 JP JP2008145732A patent/JP5060397B2/ja not_active Expired - Fee Related
-
2009
- 2009-06-02 US US12/995,809 patent/US8460472B2/en not_active Expired - Fee Related
- 2009-06-02 RU RU2010149335/13A patent/RU2461633C2/ru active
- 2009-06-02 WO PCT/IB2009/005927 patent/WO2009147522A1/en not_active Ceased
- 2009-06-02 MY MYPI20105721 patent/MY148089A/en unknown
- 2009-06-02 EP EP09757875A patent/EP2300625B1/en not_active Not-in-force
- 2009-06-02 BR BRPI0913405-0A patent/BRPI0913405A2/pt not_active IP Right Cessation
Also Published As
| Publication number | Publication date |
|---|---|
| EP2300625A1 (en) | 2011-03-30 |
| JP2009291083A (ja) | 2009-12-17 |
| RU2461633C2 (ru) | 2012-09-20 |
| WO2009147522A8 (en) | 2010-11-25 |
| RU2010149335A (ru) | 2012-07-20 |
| WO2009147522A1 (en) | 2009-12-10 |
| US8460472B2 (en) | 2013-06-11 |
| BRPI0913405A2 (pt) | 2015-09-01 |
| US20110105744A1 (en) | 2011-05-05 |
| MY148089A (en) | 2013-02-28 |
| EP2300625B1 (en) | 2012-07-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5060397B2 (ja) | 植物系繊維材料の糖化分離方法 | |
| JP4877045B2 (ja) | 植物系繊維材料の分解方法 | |
| CN101778955B (zh) | 植物系纤维材料的糖化分离方法 | |
| JP5114298B2 (ja) | 植物系繊維材料の糖化分離方法 | |
| JP4766130B2 (ja) | 植物系繊維材料の糖化方法 | |
| JP4609526B2 (ja) | 植物系繊維材料の糖化分離方法 | |
| JP4983728B2 (ja) | 植物系繊維材料の糖化分離方法 | |
| JP5040001B2 (ja) | 植物系繊維材料の糖化分離方法 | |
| JP5463627B2 (ja) | 植物系繊維材料の糖化分離方法 | |
| JP2009291143A (ja) | 植物系繊維材料の糖化分離方法 | |
| BRPI0913405B1 (pt) | Method for glycosylating and separating plant fiber material |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20090908 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100119 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100319 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20100413 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20120703 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20120803 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20150810 Year of fee payment: 3 |
|
| R151 | Written notification of patent or utility model registration |
Ref document number: 5060397 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R151 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20150810 Year of fee payment: 3 |
|
| LAPS | Cancellation because of no payment of annual fees |