JP4184378B2 - ジペプチジルペプチダーゼivを阻害する化合物 - Google Patents
ジペプチジルペプチダーゼivを阻害する化合物 Download PDFInfo
- Publication number
- JP4184378B2 JP4184378B2 JP2005504761A JP2005504761A JP4184378B2 JP 4184378 B2 JP4184378 B2 JP 4184378B2 JP 2005504761 A JP2005504761 A JP 2005504761A JP 2005504761 A JP2005504761 A JP 2005504761A JP 4184378 B2 JP4184378 B2 JP 4184378B2
- Authority
- JP
- Japan
- Prior art keywords
- added
- group
- mixture
- acid
- reduced pressure
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 150000001875 compounds Chemical class 0.000 title claims description 187
- 101000930822 Giardia intestinalis Dipeptidyl-peptidase 4 Proteins 0.000 title claims description 4
- 102000016622 Dipeptidyl Peptidase 4 Human genes 0.000 title claims 3
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 26
- 150000003839 salts Chemical class 0.000 claims description 25
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 19
- 125000002618 bicyclic heterocycle group Chemical group 0.000 claims description 14
- 239000003112 inhibitor Substances 0.000 claims description 12
- 230000000694 effects Effects 0.000 claims description 10
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 9
- 125000005843 halogen group Chemical group 0.000 claims description 9
- 239000001257 hydrogen Substances 0.000 claims description 9
- 229910052739 hydrogen Inorganic materials 0.000 claims description 9
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 8
- 125000004432 carbon atom Chemical group C* 0.000 claims description 8
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 8
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 7
- 229910052799 carbon Inorganic materials 0.000 claims description 6
- 229910052757 nitrogen Inorganic materials 0.000 claims description 6
- 125000004043 oxo group Chemical group O=* 0.000 claims description 6
- 125000000041 C6-C10 aryl group Chemical group 0.000 claims description 5
- 206010012601 diabetes mellitus Diseases 0.000 claims description 5
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 5
- 239000004480 active ingredient Substances 0.000 claims description 4
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 4
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 3
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 2
- 239000008194 pharmaceutical composition Substances 0.000 claims description 2
- 125000001424 substituent group Chemical group 0.000 claims description 2
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 claims description 2
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 153
- 239000000203 mixture Substances 0.000 description 153
- 230000002829 reductive effect Effects 0.000 description 141
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 135
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 123
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 122
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 118
- 239000000243 solution Substances 0.000 description 94
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 90
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 84
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 82
- 238000005160 1H NMR spectroscopy Methods 0.000 description 79
- 239000013078 crystal Substances 0.000 description 71
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 69
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 60
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 54
- 238000001914 filtration Methods 0.000 description 54
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 52
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 51
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 45
- 238000006243 chemical reaction Methods 0.000 description 44
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 40
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 40
- 238000001816 cooling Methods 0.000 description 39
- 239000012074 organic phase Substances 0.000 description 36
- 102100025012 Dipeptidyl peptidase 4 Human genes 0.000 description 33
- 108010067722 Dipeptidyl Peptidase 4 Proteins 0.000 description 32
- 238000000034 method Methods 0.000 description 32
- -1 methoxy, ethoxy, propoxy, isopropoxy, cyclopropoxy, butoxy, isobutoxy, s-butoxy, t-butoxy, cyclobutoxy, pentyloxy, isopentyloxy, neopentyloxy Chemical group 0.000 description 32
- 238000001035 drying Methods 0.000 description 30
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 29
- 239000008346 aqueous phase Substances 0.000 description 27
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 26
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 25
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 24
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 24
- 238000004440 column chromatography Methods 0.000 description 24
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 21
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 20
- 235000017557 sodium bicarbonate Nutrition 0.000 description 20
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 20
- 239000012156 elution solvent Substances 0.000 description 19
- 238000003756 stirring Methods 0.000 description 19
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 18
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 16
- 239000002904 solvent Substances 0.000 description 16
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 15
- JYVHOGDBFNJNMR-UHFFFAOYSA-N hexane;hydrate Chemical compound O.CCCCCC JYVHOGDBFNJNMR-UHFFFAOYSA-N 0.000 description 15
- 239000012312 sodium hydride Substances 0.000 description 15
- 229910000104 sodium hydride Inorganic materials 0.000 description 15
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 14
- 239000000126 substance Substances 0.000 description 14
- 230000002401 inhibitory effect Effects 0.000 description 13
- 229920006395 saturated elastomer Polymers 0.000 description 13
- DTHNMHAUYICORS-KTKZVXAJSA-N Glucagon-like peptide 1 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 DTHNMHAUYICORS-KTKZVXAJSA-N 0.000 description 12
- 102400000322 Glucagon-like peptide 1 Human genes 0.000 description 12
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 12
- KMAKOBLIOCQGJP-UHFFFAOYSA-N indole-3-carboxylic acid Chemical compound C1=CC=C2C(C(=O)O)=CNC2=C1 KMAKOBLIOCQGJP-UHFFFAOYSA-N 0.000 description 12
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 12
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 12
- 101710198884 GATA-type zinc finger protein 1 Proteins 0.000 description 11
- 230000002378 acidificating effect Effects 0.000 description 11
- 239000000706 filtrate Substances 0.000 description 11
- 239000002253 acid Substances 0.000 description 10
- 239000003795 chemical substances by application Substances 0.000 description 10
- 229910000027 potassium carbonate Inorganic materials 0.000 description 10
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 9
- 239000008103 glucose Substances 0.000 description 9
- 239000002198 insoluble material Substances 0.000 description 9
- 239000012298 atmosphere Substances 0.000 description 8
- 125000002619 bicyclic group Chemical group 0.000 description 8
- 210000004369 blood Anatomy 0.000 description 8
- 239000008280 blood Substances 0.000 description 8
- 239000012141 concentrate Substances 0.000 description 8
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 8
- GWVMLCQWXVFZCN-UHFFFAOYSA-N isoindoline Chemical compound C1=CC=C2CNCC2=C1 GWVMLCQWXVFZCN-UHFFFAOYSA-N 0.000 description 8
- 239000002244 precipitate Substances 0.000 description 8
- 238000012360 testing method Methods 0.000 description 8
- JMTMSDXUXJISAY-UHFFFAOYSA-N 2H-benzotriazol-4-ol Chemical compound OC1=CC=CC2=C1N=NN2 JMTMSDXUXJISAY-UHFFFAOYSA-N 0.000 description 7
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 7
- 125000004093 cyano group Chemical group *C#N 0.000 description 7
- 235000019253 formic acid Nutrition 0.000 description 7
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 7
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 7
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 6
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 6
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 6
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 6
- 239000000654 additive Substances 0.000 description 6
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 6
- 229910052794 bromium Inorganic materials 0.000 description 6
- 201000010099 disease Diseases 0.000 description 6
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 6
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 6
- 238000002360 preparation method Methods 0.000 description 6
- 108090000790 Enzymes Proteins 0.000 description 5
- 102000004190 Enzymes Human genes 0.000 description 5
- TYHQDGAMZGNGCF-UHFFFAOYSA-N benzyl 1h-indole-5-carboxylate Chemical compound C=1C=C2NC=CC2=CC=1C(=O)OCC1=CC=CC=C1 TYHQDGAMZGNGCF-UHFFFAOYSA-N 0.000 description 5
- 238000010511 deprotection reaction Methods 0.000 description 5
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 5
- 230000008569 process Effects 0.000 description 5
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 4
- HZNVUJQVZSTENZ-UHFFFAOYSA-N 2,3-dichloro-5,6-dicyano-1,4-benzoquinone Chemical compound ClC1=C(Cl)C(=O)C(C#N)=C(C#N)C1=O HZNVUJQVZSTENZ-UHFFFAOYSA-N 0.000 description 4
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 4
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 4
- 208000030507 AIDS Diseases 0.000 description 4
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 4
- VGCXGMAHQTYDJK-UHFFFAOYSA-N Chloroacetyl chloride Chemical compound ClCC(Cl)=O VGCXGMAHQTYDJK-UHFFFAOYSA-N 0.000 description 4
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 4
- 102000004877 Insulin Human genes 0.000 description 4
- 108090001061 Insulin Proteins 0.000 description 4
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 4
- 208000008589 Obesity Diseases 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 4
- 230000009471 action Effects 0.000 description 4
- 150000001413 amino acids Chemical class 0.000 description 4
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 4
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 4
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 4
- 239000000460 chlorine Substances 0.000 description 4
- 229910052801 chlorine Inorganic materials 0.000 description 4
- 229910052731 fluorine Inorganic materials 0.000 description 4
- 239000011737 fluorine Substances 0.000 description 4
- 150000002430 hydrocarbons Chemical group 0.000 description 4
- 229940125396 insulin Drugs 0.000 description 4
- 230000003914 insulin secretion Effects 0.000 description 4
- 229910052740 iodine Inorganic materials 0.000 description 4
- DRYBMFJLYYEOBZ-UHFFFAOYSA-N methyl 1h-indole-5-carboxylate Chemical compound COC(=O)C1=CC=C2NC=CC2=C1 DRYBMFJLYYEOBZ-UHFFFAOYSA-N 0.000 description 4
- QXAUTQFAWKKNLM-UHFFFAOYSA-N methyl indole-3-carboxylate Chemical compound C1=CC=C2C(C(=O)OC)=CNC2=C1 QXAUTQFAWKKNLM-UHFFFAOYSA-N 0.000 description 4
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 4
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 4
- 235000020824 obesity Nutrition 0.000 description 4
- 230000002265 prevention Effects 0.000 description 4
- 108090000765 processed proteins & peptides Proteins 0.000 description 4
- ALSCEGDXFJIYES-UHFFFAOYSA-N pyrrolidine-2-carbonitrile Chemical class N#CC1CCCN1 ALSCEGDXFJIYES-UHFFFAOYSA-N 0.000 description 4
- JHJLBTNAGRQEKS-UHFFFAOYSA-M sodium bromide Chemical compound [Na+].[Br-] JHJLBTNAGRQEKS-UHFFFAOYSA-M 0.000 description 4
- 235000009518 sodium iodide Nutrition 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 4
- YCWRPKBYQZOLCD-LURJTMIESA-N (2s)-1-(2-chloroacetyl)pyrrolidine-2-carbonitrile Chemical compound ClCC(=O)N1CCC[C@H]1C#N YCWRPKBYQZOLCD-LURJTMIESA-N 0.000 description 3
- CJTZXIJETZZARD-UHFFFAOYSA-N 1-iodo-2,2-dimethylpropane Chemical compound CC(C)(C)CI CJTZXIJETZZARD-UHFFFAOYSA-N 0.000 description 3
- YHVGUXFUOSJCFJ-UHFFFAOYSA-N 2-methylpyrazolo[1,5-a]pyrimidine-6-carboxylic acid Chemical compound N1=CC(C(O)=O)=CN2N=C(C)C=C21 YHVGUXFUOSJCFJ-UHFFFAOYSA-N 0.000 description 3
- MHIJDDIBBUUVMD-UHFFFAOYSA-N 4-amino-3-sulfanylbenzoic acid Chemical compound NC1=CC=C(C(O)=O)C=C1S MHIJDDIBBUUVMD-UHFFFAOYSA-N 0.000 description 3
- 0 C*1CC*CC1 Chemical compound C*1CC*CC1 0.000 description 3
- 229940126062 Compound A Drugs 0.000 description 3
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 3
- 108010016626 Dipeptides Proteins 0.000 description 3
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 3
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 3
- 230000000996 additive effect Effects 0.000 description 3
- 150000001412 amines Chemical group 0.000 description 3
- 229940024606 amino acid Drugs 0.000 description 3
- 235000001014 amino acid Nutrition 0.000 description 3
- 125000003118 aryl group Chemical group 0.000 description 3
- 230000015556 catabolic process Effects 0.000 description 3
- 238000006731 degradation reaction Methods 0.000 description 3
- 238000006297 dehydration reaction Methods 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- QXRASANXLBTMJX-UHFFFAOYSA-N ethyl 2-(methoxymethyl)pyrazolo[1,5-a]pyrimidine-6-carboxylate Chemical compound C1=C(C(=O)OCC)C=NC2=CC(COC)=NN21 QXRASANXLBTMJX-UHFFFAOYSA-N 0.000 description 3
- YMCDYRGMTRCAPZ-UHFFFAOYSA-N ethyl 2-acetyl-3-oxobutanoate Chemical compound CCOC(=O)C(C(C)=O)C(C)=O YMCDYRGMTRCAPZ-UHFFFAOYSA-N 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 3
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 230000002503 metabolic effect Effects 0.000 description 3
- 210000000056 organ Anatomy 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- 125000006239 protecting group Chemical group 0.000 description 3
- 229910052979 sodium sulfide Inorganic materials 0.000 description 3
- GRVFOGOEDUUMBP-UHFFFAOYSA-N sodium sulfide (anhydrous) Chemical compound [Na+].[Na+].[S-2] GRVFOGOEDUUMBP-UHFFFAOYSA-N 0.000 description 3
- 125000004434 sulfur atom Chemical group 0.000 description 3
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 2
- PCVGSVPBMIEQQK-UHFFFAOYSA-N 1,2-bis(bromomethyl)-4-methoxybenzene Chemical compound COC1=CC=C(CBr)C(CBr)=C1 PCVGSVPBMIEQQK-UHFFFAOYSA-N 0.000 description 2
- AIOMGEMZFLRFJE-UHFFFAOYSA-N 1,3-thiazolidine-4-carboxamide Chemical compound NC(=O)C1CSCN1 AIOMGEMZFLRFJE-UHFFFAOYSA-N 0.000 description 2
- BTUGGGLMQBJCBN-UHFFFAOYSA-N 1-iodo-2-methylpropane Chemical compound CC(C)CI BTUGGGLMQBJCBN-UHFFFAOYSA-N 0.000 description 2
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 2
- DHXNGBMVPJHMBV-UHFFFAOYSA-N 3-(benzylazaniumyl)-3-methylbutanoate Chemical compound OC(=O)CC(C)(C)NCC1=CC=CC=C1 DHXNGBMVPJHMBV-UHFFFAOYSA-N 0.000 description 2
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 2
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 2
- QOYSPWLJTVJIAP-UHFFFAOYSA-N 4-amino-3-methoxy-5-sulfanylbenzoic acid Chemical compound COC1=CC(C(O)=O)=CC(S)=C1N QOYSPWLJTVJIAP-UHFFFAOYSA-N 0.000 description 2
- KQFLVUSDAWTRNT-UHFFFAOYSA-N 4-amino-3-thiocyanatobenzoic acid Chemical compound NC1=CC=C(C(O)=O)C=C1SC#N KQFLVUSDAWTRNT-UHFFFAOYSA-N 0.000 description 2
- YDPDODRZXXZMII-UHFFFAOYSA-N 4-methoxy-3-oxobutanenitrile Chemical compound COCC(=O)CC#N YDPDODRZXXZMII-UHFFFAOYSA-N 0.000 description 2
- KHKJLJHJTQRHSA-UHFFFAOYSA-N 4-methyl-4-nitropentanoic acid Chemical compound [O-][N+](=O)C(C)(C)CCC(O)=O KHKJLJHJTQRHSA-UHFFFAOYSA-N 0.000 description 2
- VFDNIDGPEBLDNH-UHFFFAOYSA-N 5-(methoxymethyl)-1h-pyrazol-3-amine Chemical compound COCC=1C=C(N)NN=1 VFDNIDGPEBLDNH-UHFFFAOYSA-N 0.000 description 2
- UKRUJPIJOJHCOB-UHFFFAOYSA-N 5-methylisoindole-1,3-dione Chemical compound CC1=CC=C2C(=O)NC(=O)C2=C1 UKRUJPIJOJHCOB-UHFFFAOYSA-N 0.000 description 2
- 208000023275 Autoimmune disease Diseases 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- XJUZRXYOEPSWMB-UHFFFAOYSA-N Chloromethyl methyl ether Chemical compound COCCl XJUZRXYOEPSWMB-UHFFFAOYSA-N 0.000 description 2
- 208000002249 Diabetes Complications Diseases 0.000 description 2
- 206010012655 Diabetic complications Diseases 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 2
- 208000001132 Osteoporosis Diseases 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 2
- 238000007112 amidation reaction Methods 0.000 description 2
- SOIFLUNRINLCBN-UHFFFAOYSA-N ammonium thiocyanate Chemical compound [NH4+].[S-]C#N SOIFLUNRINLCBN-UHFFFAOYSA-N 0.000 description 2
- 206010003246 arthritis Diseases 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- KKCWBKUOPJMUQV-UHFFFAOYSA-N azetidine-2-carboxamide Chemical compound NC(=O)C1CCN1 KKCWBKUOPJMUQV-UHFFFAOYSA-N 0.000 description 2
- IUBFCXDHYOWMAM-UHFFFAOYSA-N benzyl 1-(2,2-dimethylpropanoyl)indole-5-carboxylate Chemical compound C=1C=C2N(C(=O)C(C)(C)C)C=CC2=CC=1C(=O)OCC1=CC=CC=C1 IUBFCXDHYOWMAM-UHFFFAOYSA-N 0.000 description 2
- BHRDIGMZRQMPKX-UHFFFAOYSA-N benzyl 1-[2-[(2-methylpropan-2-yl)oxy]-2-oxoethyl]indole-5-carboxylate Chemical compound C=1C=C2N(CC(=O)OC(C)(C)C)C=CC2=CC=1C(=O)OCC1=CC=CC=C1 BHRDIGMZRQMPKX-UHFFFAOYSA-N 0.000 description 2
- DYVBOFXFWDECSX-UHFFFAOYSA-N benzyl 1-acetylindole-5-carboxylate Chemical compound C=1C=C2N(C(=O)C)C=CC2=CC=1C(=O)OCC1=CC=CC=C1 DYVBOFXFWDECSX-UHFFFAOYSA-N 0.000 description 2
- MMUZGGMJLZCFRK-UHFFFAOYSA-N benzyl 1-benzoylindole-5-carboxylate Chemical compound C=1C=C2N(C(=O)C=3C=CC=CC=3)C=CC2=CC=1C(=O)OCC1=CC=CC=C1 MMUZGGMJLZCFRK-UHFFFAOYSA-N 0.000 description 2
- 238000010241 blood sampling Methods 0.000 description 2
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical compound [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 230000001684 chronic effect Effects 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- SQAUUQRBOCJRCW-UHFFFAOYSA-N diethyl 2-acetylpropanedioate Chemical compound CCOC(=O)C(C(C)=O)C(=O)OCC SQAUUQRBOCJRCW-UHFFFAOYSA-N 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- 230000002708 enhancing effect Effects 0.000 description 2
- LQSOVGAUOHMPLK-UHFFFAOYSA-N ethyl 2-(dimethylaminomethylidene)-3-oxobutanoate Chemical compound CCOC(=O)C(C(C)=O)=CN(C)C LQSOVGAUOHMPLK-UHFFFAOYSA-N 0.000 description 2
- SXXOBMAAZHADHC-UHFFFAOYSA-N ethyl 2-(hydroxymethyl)pyrazolo[1,5-a]pyrimidine-6-carboxylate Chemical compound C1=C(C(=O)OCC)C=NC2=CC(CO)=NN21 SXXOBMAAZHADHC-UHFFFAOYSA-N 0.000 description 2
- KCBXCJFFWYBRFL-UHFFFAOYSA-N ethyl 2-methyl-5-oxo-4H-pyrazolo[1,5-a]pyrimidine-6-carboxylate Chemical compound N1=C(O)C(C(=O)OCC)=CN2N=C(C)C=C21 KCBXCJFFWYBRFL-UHFFFAOYSA-N 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine hydrate Chemical compound O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 2
- 201000001421 hyperglycemia Diseases 0.000 description 2
- 230000002218 hypoglycaemic effect Effects 0.000 description 2
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 2
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 2
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 2
- 125000002183 isoquinolinyl group Chemical class C1(=NC=CC2=CC=CC=C12)* 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- HBYFGEOBNHODCG-UHFFFAOYSA-N methyl 1-methylbenzimidazole-5-carboxylate Chemical compound COC(=O)C1=CC=C2N(C)C=NC2=C1 HBYFGEOBNHODCG-UHFFFAOYSA-N 0.000 description 2
- PLTXHAQWLYQPBN-UHFFFAOYSA-N methyl 3-(1,3-benzothiazole-6-carbonylamino)-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoate Chemical compound CC(C)(C)OC(=O)NC(C(=O)OC)CNC(=O)C1=CC=C2N=CSC2=C1 PLTXHAQWLYQPBN-UHFFFAOYSA-N 0.000 description 2
- UOQRESVEXATCGD-UHFFFAOYSA-N methyl 3-amino-4-(methylamino)benzoate Chemical compound CNC1=CC=C(C(=O)OC)C=C1N UOQRESVEXATCGD-UHFFFAOYSA-N 0.000 description 2
- RUJMPEWZLDVEAV-UHFFFAOYSA-N methyl 4-(methylamino)-3-nitrobenzoate Chemical compound CNC1=CC=C(C(=O)OC)C=C1[N+]([O-])=O RUJMPEWZLDVEAV-UHFFFAOYSA-N 0.000 description 2
- ODBFYEWPUQVAKY-UHFFFAOYSA-N methyl 5-methoxy-1h-indole-3-carboxylate Chemical compound C1=C(OC)C=C2C(C(=O)OC)=CNC2=C1 ODBFYEWPUQVAKY-UHFFFAOYSA-N 0.000 description 2
- HOCOQXZICBKRBF-JTQLQIEISA-N n-[2-[[2-[(2s)-2-cyanopyrrolidin-1-yl]-2-oxoethyl]amino]-2-methylpropyl]formamide Chemical compound O=CNCC(C)(C)NCC(=O)N1CCC[C@H]1C#N HOCOQXZICBKRBF-JTQLQIEISA-N 0.000 description 2
- YJFYIJUGKNWUQR-UHFFFAOYSA-N n-benzyl-4-methyl-4-nitropentan-1-amine Chemical compound [O-][N+](=O)C(C)(C)CCCNCC1=CC=CC=C1 YJFYIJUGKNWUQR-UHFFFAOYSA-N 0.000 description 2
- MATUPZQNPASCJO-UHFFFAOYSA-N n-benzyl-4-methyl-4-nitropentanamide Chemical compound [O-][N+](=O)C(C)(C)CCC(=O)NCC1=CC=CC=C1 MATUPZQNPASCJO-UHFFFAOYSA-N 0.000 description 2
- 125000001624 naphthyl group Chemical group 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 239000000825 pharmaceutical preparation Substances 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- 238000012746 preparative thin layer chromatography Methods 0.000 description 2
- 230000003449 preventive effect Effects 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 208000011580 syndromic disease Diseases 0.000 description 2
- OXGKKJVYQKKOIS-UHFFFAOYSA-N tert-butyl 4-carbamoyl-1,3-thiazolidine-3-carboxylate Chemical compound CC(C)(C)OC(=O)N1CSCC1C(N)=O OXGKKJVYQKKOIS-UHFFFAOYSA-N 0.000 description 2
- BDHHNWUPDIJMMW-ZDUSSCGKSA-N tert-butyl N-[2-[[(2S)-2-cyanopyrrolidin-1-yl]-(2-oxoethyl)amino]-2-methylpropyl]carbamate Chemical compound CC(C)(C)OC(=O)NCC(C)(C)N(CC=O)N1CCC[C@H]1C#N BDHHNWUPDIJMMW-ZDUSSCGKSA-N 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- KCBBEHBEAPOBSC-UHFFFAOYSA-N tert-butyl n-(2-amino-2-methylpropyl)carbamate Chemical compound CC(C)(C)OC(=O)NCC(C)(C)N KCBBEHBEAPOBSC-UHFFFAOYSA-N 0.000 description 2
- LMHRNNWMZKLHJQ-UHFFFAOYSA-N tert-butyl n-(3-indol-1-yl-3-oxopropyl)carbamate Chemical compound C1=CC=C2N(C(=O)CCNC(=O)OC(C)(C)C)C=CC2=C1 LMHRNNWMZKLHJQ-UHFFFAOYSA-N 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 2
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- VCGRFBXVSFAGGA-UHFFFAOYSA-N (1,1-dioxo-1,4-thiazinan-4-yl)-[6-[[3-(4-fluorophenyl)-5-methyl-1,2-oxazol-4-yl]methoxy]pyridin-3-yl]methanone Chemical compound CC=1ON=C(C=2C=CC(F)=CC=2)C=1COC(N=C1)=CC=C1C(=O)N1CCS(=O)(=O)CC1 VCGRFBXVSFAGGA-UHFFFAOYSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- YCWRPKBYQZOLCD-ZCFIWIBFSA-N (2r)-1-(2-chloroacetyl)pyrrolidine-2-carbonitrile Chemical compound ClCC(=O)N1CCC[C@@H]1C#N YCWRPKBYQZOLCD-ZCFIWIBFSA-N 0.000 description 1
- VLJNHYLEOZPXFW-SCSAIBSYSA-N (2r)-pyrrolidine-2-carboxamide Chemical compound NC(=O)[C@H]1CCCN1 VLJNHYLEOZPXFW-SCSAIBSYSA-N 0.000 description 1
- KQZRXFSDZVUWHV-YFKPBYRVSA-N (2s)-1-(2-chloroacetyl)azetidine-2-carbonitrile Chemical compound ClCC(=O)N1CC[C@H]1C#N KQZRXFSDZVUWHV-YFKPBYRVSA-N 0.000 description 1
- INESFTOSFRZYTN-VIFPVBQESA-N (2s)-1-[2-[(1-amino-2-methylpropan-2-yl)amino]acetyl]pyrrolidine-2-carbonitrile Chemical compound NCC(C)(C)NCC(=O)N1CCC[C@H]1C#N INESFTOSFRZYTN-VIFPVBQESA-N 0.000 description 1
- MPXHULGFMWRQFU-WWPIYYJJSA-N (2s)-1-[2-[(1-amino-2-methylpropan-2-yl)amino]acetyl]pyrrolidine-2-carbonitrile;dihydrochloride Chemical compound Cl.Cl.NCC(C)(C)NCC(=O)N1CCC[C@H]1C#N MPXHULGFMWRQFU-WWPIYYJJSA-N 0.000 description 1
- ALSCEGDXFJIYES-YFKPBYRVSA-N (2s)-pyrrolidine-2-carbonitrile Chemical compound N#C[C@@H]1CCCN1 ALSCEGDXFJIYES-YFKPBYRVSA-N 0.000 description 1
- QSJTUXCBPTVKQZ-JEDNCBNOSA-N (2s)-pyrrolidine-2-carbonitrile;hydrochloride Chemical compound Cl.N#C[C@@H]1CCCN1 QSJTUXCBPTVKQZ-JEDNCBNOSA-N 0.000 description 1
- 125000006274 (C1-C3)alkoxy group Chemical group 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- OPCJOXGBLDJWRM-UHFFFAOYSA-N 1,2-diamino-2-methylpropane Chemical compound CC(C)(N)CN OPCJOXGBLDJWRM-UHFFFAOYSA-N 0.000 description 1
- CHVFFSTYBJPSAM-UHFFFAOYSA-N 1,3-dimethylpyrazolo[3,4-b]pyridine Chemical compound C1=CC=C2C(C)=NN(C)C2=N1 CHVFFSTYBJPSAM-UHFFFAOYSA-N 0.000 description 1
- NMGFHFUUCGHRIZ-UHFFFAOYSA-N 1,3-dimethylpyrazolo[3,4-b]pyridine-5-carboxylic acid Chemical compound C1=C(C(O)=O)C=C2C(C)=NN(C)C2=N1 NMGFHFUUCGHRIZ-UHFFFAOYSA-N 0.000 description 1
- CVICYIYOWFQLFL-UHFFFAOYSA-N 1-(2,2-dimethylpropanoyloxymethyl)indole-3-carboxylic acid Chemical compound C1=CC=C2N(COC(=O)C(C)(C)C)C=C(C(O)=O)C2=C1 CVICYIYOWFQLFL-UHFFFAOYSA-N 0.000 description 1
- HOUHNDCEFIYGDU-UHFFFAOYSA-N 1-(2,2-dimethylpropyl)indole Chemical compound C1=CC=C2N(CC(C)(C)C)C=CC2=C1 HOUHNDCEFIYGDU-UHFFFAOYSA-N 0.000 description 1
- XNDDVADNOREXGM-UHFFFAOYSA-N 1-(2,2-dimethylpropyl)indole-3-carboxylic acid Chemical compound C1=CC=C2N(CC(C)(C)C)C=C(C(O)=O)C2=C1 XNDDVADNOREXGM-UHFFFAOYSA-N 0.000 description 1
- RNTCWULFNYNFGI-UHFFFAOYSA-N 1-(2,3-dihydroindol-1-yl)ethanone Chemical compound C1=CC=C2N(C(=O)C)CCC2=C1 RNTCWULFNYNFGI-UHFFFAOYSA-N 0.000 description 1
- ZOHDDNDXUPTLTK-UHFFFAOYSA-N 1-(2-methylpropyl)indole Chemical compound C1=CC=C2N(CC(C)C)C=CC2=C1 ZOHDDNDXUPTLTK-UHFFFAOYSA-N 0.000 description 1
- MUODAUUEASEJPI-UHFFFAOYSA-N 1-(acetyloxymethyl)indole-3-carboxylic acid Chemical compound C1=CC=C2N(COC(=O)C)C=C(C(O)=O)C2=C1 MUODAUUEASEJPI-UHFFFAOYSA-N 0.000 description 1
- YKGDAMWMFVRGDH-UHFFFAOYSA-N 1-(aminomethyl)cyclopentan-1-amine Chemical compound NCC1(N)CCCC1 YKGDAMWMFVRGDH-UHFFFAOYSA-N 0.000 description 1
- DIDZHWPGXZYJJH-UHFFFAOYSA-N 1-(methoxymethyl)indole Chemical compound C1=CC=C2N(COC)C=CC2=C1 DIDZHWPGXZYJJH-UHFFFAOYSA-N 0.000 description 1
- JGRXUJVCGCUHBS-UHFFFAOYSA-N 1-(methoxymethyl)indole-3-carboxylic acid Chemical compound C1=CC=C2N(COC)C=C(C(O)=O)C2=C1 JGRXUJVCGCUHBS-UHFFFAOYSA-N 0.000 description 1
- AKHSWAVGQJONPV-UHFFFAOYSA-N 1-(phenylmethoxymethyl)indole Chemical compound C1=CC2=CC=CC=C2N1COCC1=CC=CC=C1 AKHSWAVGQJONPV-UHFFFAOYSA-N 0.000 description 1
- YJWWHTNTPZAEEJ-UHFFFAOYSA-N 1-(phenylmethoxymethyl)indole-3-carboxylic acid Chemical compound C12=CC=CC=C2C(C(=O)O)=CN1COCC1=CC=CC=C1 YJWWHTNTPZAEEJ-UHFFFAOYSA-N 0.000 description 1
- VJLNUKOFIAYTTL-UHFFFAOYSA-N 1-(phenylmethoxymethyl)indole-5-carboxylic acid Chemical compound C1=CC2=CC(C(=O)O)=CC=C2N1COCC1=CC=CC=C1 VJLNUKOFIAYTTL-UHFFFAOYSA-N 0.000 description 1
- KRXHHESVJTVORG-UHFFFAOYSA-N 1-acetyl-2,3-dihydroindole-5-carboxylic acid Chemical compound OC(=O)C1=CC=C2N(C(=O)C)CCC2=C1 KRXHHESVJTVORG-UHFFFAOYSA-N 0.000 description 1
- GHLPNUSFQJHAPG-UHFFFAOYSA-N 1-acetylindole-3-carboxylic acid Chemical compound C1=CC=C2N(C(=O)C)C=C(C(O)=O)C2=C1 GHLPNUSFQJHAPG-UHFFFAOYSA-N 0.000 description 1
- DTRSVKLNWUNCQE-UHFFFAOYSA-N 1-indol-1-yl-2,2-dimethylpropan-1-one Chemical compound C1=CC=C2N(C(=O)C(C)(C)C)C=CC2=C1 DTRSVKLNWUNCQE-UHFFFAOYSA-N 0.000 description 1
- FIRXFHJQGIIJDB-UHFFFAOYSA-N 1-methyl-2,3-dihydroindole Chemical compound C1=CC=C2N(C)CCC2=C1 FIRXFHJQGIIJDB-UHFFFAOYSA-N 0.000 description 1
- FGYADSCZTQOAFK-UHFFFAOYSA-N 1-methylbenzimidazole Chemical compound C1=CC=C2N(C)C=NC2=C1 FGYADSCZTQOAFK-UHFFFAOYSA-N 0.000 description 1
- HVRCLXXJIQTXHC-UHFFFAOYSA-N 1-methylindole-3-carboxylic acid Chemical compound C1=CC=C2N(C)C=C(C(O)=O)C2=C1 HVRCLXXJIQTXHC-UHFFFAOYSA-N 0.000 description 1
- UHQAIJFIXCOBCN-UHFFFAOYSA-N 1-methylindole-5-carboxylic acid Chemical compound OC(=O)C1=CC=C2N(C)C=CC2=C1 UHQAIJFIXCOBCN-UHFFFAOYSA-N 0.000 description 1
- DWKIPLIDZYRILV-UHFFFAOYSA-N 1-methylindole-7-carboxylic acid Chemical compound C1=CC(C(O)=O)=C2N(C)C=CC2=C1 DWKIPLIDZYRILV-UHFFFAOYSA-N 0.000 description 1
- RTBFRGCFXZNCOE-UHFFFAOYSA-N 1-methylsulfonylpiperidin-4-one Chemical compound CS(=O)(=O)N1CCC(=O)CC1 RTBFRGCFXZNCOE-UHFFFAOYSA-N 0.000 description 1
- XHLHPRDBBAGVEG-UHFFFAOYSA-N 1-tetralone Chemical compound C1=CC=C2C(=O)CCCC2=C1 XHLHPRDBBAGVEG-UHFFFAOYSA-N 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- JVSFQJZRHXAUGT-UHFFFAOYSA-N 2,2-dimethylpropanoyl chloride Chemical compound CC(C)(C)C(Cl)=O JVSFQJZRHXAUGT-UHFFFAOYSA-N 0.000 description 1
- SIQBPWRTJNBBER-UHFFFAOYSA-N 2,3,4,5-tetrahydro-1h-2-benzazepine Chemical compound C1CCNCC2=CC=CC=C21 SIQBPWRTJNBBER-UHFFFAOYSA-N 0.000 description 1
- CACYYYIWDACOAQ-UHFFFAOYSA-N 2,7-dimethylpyrazolo[1,5-a]pyrimidine-6-carboxylic acid Chemical compound N1=CC(C(O)=O)=C(C)N2N=C(C)C=C21 CACYYYIWDACOAQ-UHFFFAOYSA-N 0.000 description 1
- CTEFMPLNGNDKLS-UHFFFAOYSA-N 2-(methoxymethyl)pyrazolo[1,5-a]pyrimidine-6-carboxylic acid Chemical compound N1=CC(C(O)=O)=CN2N=C(COC)C=C21 CTEFMPLNGNDKLS-UHFFFAOYSA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- IBCBHLQKAIHDIP-UHFFFAOYSA-N 2-methyl-7-oxo-1h-pyrazolo[1,5-a]pyrimidine-6-carboxylic acid Chemical compound C1=C(C(O)=O)C(=O)N2NC(C)=CC2=N1 IBCBHLQKAIHDIP-UHFFFAOYSA-N 0.000 description 1
- LDRXEOIHYVICNO-UHFFFAOYSA-N 2-methylpropane-1,2-diamine;hydrochloride Chemical compound Cl.CC(C)(N)CN LDRXEOIHYVICNO-UHFFFAOYSA-N 0.000 description 1
- WPHUUIODWRNJLO-UHFFFAOYSA-N 2-nitrobenzenesulfonyl chloride Chemical compound [O-][N+](=O)C1=CC=CC=C1S(Cl)(=O)=O WPHUUIODWRNJLO-UHFFFAOYSA-N 0.000 description 1
- SUBZYLZMYNBQLW-UHFFFAOYSA-N 2-phenyl-1,3-benzothiazole-6-carboxylic acid Chemical compound S1C2=CC(C(=O)O)=CC=C2N=C1C1=CC=CC=C1 SUBZYLZMYNBQLW-UHFFFAOYSA-N 0.000 description 1
- QIUJEIYANWQBLO-UHFFFAOYSA-N 2-phenyl-4h-isoquinoline-1,3-dione Chemical class O=C1CC2=CC=CC=C2C(=O)N1C1=CC=CC=C1 QIUJEIYANWQBLO-UHFFFAOYSA-N 0.000 description 1
- MFUPLJQNEXUUDW-UHFFFAOYSA-N 2-phenylisoindole-1,3-dione Chemical class O=C1C2=CC=CC=C2C(=O)N1C1=CC=CC=C1 MFUPLJQNEXUUDW-UHFFFAOYSA-N 0.000 description 1
- WAXSUZDXJVZCCD-UHFFFAOYSA-N 2-tert-butyl-7-methylpyrazolo[1,5-a]pyrimidine-6-carboxylic acid ethyl 3-oxobutanoate Chemical compound C(CC(=O)C)(=O)OCC.C(C)(C)(C)C1=NN2C(N=CC(=C2C)C(=O)O)=C1 WAXSUZDXJVZCCD-UHFFFAOYSA-N 0.000 description 1
- FJWNZTPXVSWUKF-UHFFFAOYSA-N 3-[(2-methylpropan-2-yl)oxycarbonyl]-1,3-thiazolidine-4-carboxylic acid Chemical compound CC(C)(C)OC(=O)N1CSCC1C(O)=O FJWNZTPXVSWUKF-UHFFFAOYSA-N 0.000 description 1
- WCFJUSRQHZPVKY-UHFFFAOYSA-N 3-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound CC(C)(C)OC(=O)NCCC(O)=O WCFJUSRQHZPVKY-UHFFFAOYSA-N 0.000 description 1
- AGXCRXUUNZLUGW-UHFFFAOYSA-N 3-amino-1-(1,3-dihydroisoindol-2-yl)-3-methylbutan-1-one Chemical compound C1=CC=C2CN(C(=O)CC(C)(N)C)CC2=C1 AGXCRXUUNZLUGW-UHFFFAOYSA-N 0.000 description 1
- NFQAIWOMJQWGSS-UHFFFAOYSA-N 3-amino-3-methylbutanoic acid Chemical compound CC(C)(N)CC(O)=O NFQAIWOMJQWGSS-UHFFFAOYSA-N 0.000 description 1
- OLJXRTRRJSMURJ-UHFFFAOYSA-N 4-amino-2-methoxybenzoic acid Chemical compound COC1=CC(N)=CC=C1C(O)=O OLJXRTRRJSMURJ-UHFFFAOYSA-N 0.000 description 1
- LVUBSVWMOWKPDJ-UHFFFAOYSA-N 4-methoxy-1,2-dimethylbenzene Chemical compound COC1=CC=C(C)C(C)=C1 LVUBSVWMOWKPDJ-UHFFFAOYSA-N 0.000 description 1
- WHLFXPIYRPOHGB-UHFFFAOYSA-N 4-methylpentane-1,4-diamine Chemical compound CC(C)(N)CCCN WHLFXPIYRPOHGB-UHFFFAOYSA-N 0.000 description 1
- FYTLHYRDGXRYEY-UHFFFAOYSA-N 5-Methyl-3-pyrazolamine Chemical compound CC=1C=C(N)NN=1 FYTLHYRDGXRYEY-UHFFFAOYSA-N 0.000 description 1
- ZOXBWJMCXHTKNU-UHFFFAOYSA-N 5-methyl-2-benzofuran-1,3-dione Chemical compound CC1=CC=C2C(=O)OC(=O)C2=C1 ZOXBWJMCXHTKNU-UHFFFAOYSA-N 0.000 description 1
- ZHBXGHWSVIBUCQ-UHFFFAOYSA-N 5-tert-butyl-1h-pyrazol-3-amine Chemical compound CC(C)(C)C1=CC(N)=NN1 ZHBXGHWSVIBUCQ-UHFFFAOYSA-N 0.000 description 1
- XOAZJKTUIFSUGD-UHFFFAOYSA-N 7-methoxy-1-methylindole Chemical compound COC1=CC=CC2=C1N(C)C=C2 XOAZJKTUIFSUGD-UHFFFAOYSA-N 0.000 description 1
- 230000035502 ADME Effects 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- VPBMIFXTTFPDBS-YLIVSKOQSA-N BrC(C(=O)O)C1=CC=CC=C1.BrC(C(=O)N1[C@@H](CCC1)C#N)C1=CC=CC=C1 Chemical compound BrC(C(=O)O)C1=CC=CC=C1.BrC(C(=O)N1[C@@H](CCC1)C#N)C1=CC=CC=C1 VPBMIFXTTFPDBS-YLIVSKOQSA-N 0.000 description 1
- MJKMAZRWFMINSA-UHFFFAOYSA-N C(C)#N.OCC1=NN2C(N=CC(=C2)C(=O)O)=C1 Chemical compound C(C)#N.OCC1=NN2C(N=CC(=C2)C(=O)O)=C1 MJKMAZRWFMINSA-UHFFFAOYSA-N 0.000 description 1
- PVWFEYYZMXHSCQ-UHFFFAOYSA-N CC(=CC(=O)O)C.NC(CC(=O)O)(C)C Chemical compound CC(=CC(=O)O)C.NC(CC(=O)O)(C)C PVWFEYYZMXHSCQ-UHFFFAOYSA-N 0.000 description 1
- GBGIASDIKMTNSK-UHFFFAOYSA-N CC(C)(C)OC(N(CC(O)=O)CC(O)=O)O Chemical compound CC(C)(C)OC(N(CC(O)=O)CC(O)=O)O GBGIASDIKMTNSK-UHFFFAOYSA-N 0.000 description 1
- HGALVEKTFYBMGE-UHFFFAOYSA-N CC1=NN2C(N=CC(=C2)C(=O)N)=C1.CC1=NN2C(N=CC(=C2)C(=O)O)=C1 Chemical compound CC1=NN2C(N=CC(=C2)C(=O)N)=C1.CC1=NN2C(N=CC(=C2)C(=O)O)=C1 HGALVEKTFYBMGE-UHFFFAOYSA-N 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- GHOKWGTUZJEAQD-UHFFFAOYSA-N Chick antidermatitis factor Natural products OCC(C)(C)C(O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-UHFFFAOYSA-N 0.000 description 1
- RJVJGCXMTDJWON-URWTVYCISA-N Cl.Cl.NCC(C)(C)NCC(=O)N1[C@@H](CCC1)C#N.CC(CNC)(C)NCC(=O)N1[C@@H](CCC1)C#N Chemical compound Cl.Cl.NCC(C)(C)NCC(=O)N1[C@@H](CCC1)C#N.CC(CNC)(C)NCC(=O)N1[C@@H](CCC1)C#N RJVJGCXMTDJWON-URWTVYCISA-N 0.000 description 1
- 102000001189 Cyclic Peptides Human genes 0.000 description 1
- 108010069514 Cyclic Peptides Proteins 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 1
- IYXGSMUGOJNHAZ-UHFFFAOYSA-N Ethyl malonate Chemical compound CCOC(=O)CC(=O)OCC IYXGSMUGOJNHAZ-UHFFFAOYSA-N 0.000 description 1
- 101800000224 Glucagon-like peptide 1 Proteins 0.000 description 1
- 208000002705 Glucose Intolerance Diseases 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 101000908391 Homo sapiens Dipeptidyl peptidase 4 Proteins 0.000 description 1
- 241000725303 Human immunodeficiency virus Species 0.000 description 1
- LELOWRISYMNNSU-UHFFFAOYSA-N Hydrocyanic acid Natural products N#C LELOWRISYMNNSU-UHFFFAOYSA-N 0.000 description 1
- 206010060378 Hyperinsulinaemia Diseases 0.000 description 1
- 206010020710 Hyperphagia Diseases 0.000 description 1
- 208000013016 Hypoglycemia Diseases 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- VLJNHYLEOZPXFW-BYPYZUCNSA-N L-prolinamide Chemical compound NC(=O)[C@@H]1CCCN1 VLJNHYLEOZPXFW-BYPYZUCNSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- QRMHDGWGLNLHMN-UHFFFAOYSA-N Methyl methoxyacetate Chemical compound COCC(=O)OC QRMHDGWGLNLHMN-UHFFFAOYSA-N 0.000 description 1
- ZSXGLVDWWRXATF-UHFFFAOYSA-N N,N-dimethylformamide dimethyl acetal Chemical compound COC(OC)N(C)C ZSXGLVDWWRXATF-UHFFFAOYSA-N 0.000 description 1
- UUCUQJHYUPXDHN-UHFFFAOYSA-N N-Acetylindole Chemical compound C1=CC=C2N(C(=O)C)C=CC2=C1 UUCUQJHYUPXDHN-UHFFFAOYSA-N 0.000 description 1
- AYCPARAPKDAOEN-LJQANCHMSA-N N-[(1S)-2-(dimethylamino)-1-phenylethyl]-6,6-dimethyl-3-[(2-methyl-4-thieno[3,2-d]pyrimidinyl)amino]-1,4-dihydropyrrolo[3,4-c]pyrazole-5-carboxamide Chemical compound C1([C@H](NC(=O)N2C(C=3NN=C(NC=4C=5SC=CC=5N=C(C)N=4)C=3C2)(C)C)CN(C)C)=CC=CC=C1 AYCPARAPKDAOEN-LJQANCHMSA-N 0.000 description 1
- OXNSELCAOXPPMX-KDDYFZQKSA-N N1[C@@H](CCC1)C(=O)N.N1[C@@H](CCC1)C#N Chemical compound N1[C@@H](CCC1)C(=O)N.N1[C@@H](CCC1)C#N OXNSELCAOXPPMX-KDDYFZQKSA-N 0.000 description 1
- XBVRJWDPXBKFEC-UHFFFAOYSA-N NC1=C(C=C(C(=O)O)C=C1)O.CC=1OC2=C(N1)C=CC(=C2)C(=O)O Chemical compound NC1=C(C=C(C(=O)O)C=C1)O.CC=1OC2=C(N1)C=CC(=C2)C(=O)O XBVRJWDPXBKFEC-UHFFFAOYSA-N 0.000 description 1
- ZZNZOYDVUKAYMC-UHFFFAOYSA-N NC1=C(C=C(C(=O)O)C=C1)S.O=C1SC2=C(N1)C=CC(=C2)C(=O)O Chemical compound NC1=C(C=C(C(=O)O)C=C1)S.O=C1SC2=C(N1)C=CC(=C2)C(=O)O ZZNZOYDVUKAYMC-UHFFFAOYSA-N 0.000 description 1
- FAJSTJGXKRYLCD-UHFFFAOYSA-N NC1=CC=C(C(=O)O)C=C1.CC=1SC2=C(N1)C=CC(=C2)C(=O)O Chemical compound NC1=CC=C(C(=O)O)C=C1.CC=1SC2=C(N1)C=CC(=C2)C(=O)O FAJSTJGXKRYLCD-UHFFFAOYSA-N 0.000 description 1
- RSBKAHFWAJDUAD-UHFFFAOYSA-N NC1=NNC(=C1)C(C)(C)C.C(C)(C)(C)C1=NN2C(N=C(C(=C2C)C(=O)O)C)=C1 Chemical compound NC1=NNC(=C1)C(C)(C)C.C(C)(C)(C)C1=NN2C(N=C(C(=C2C)C(=O)O)C)=C1 RSBKAHFWAJDUAD-UHFFFAOYSA-N 0.000 description 1
- ODEJIEMLGNZNJP-UHFFFAOYSA-N NC1=NNC(=C1)C.CC1=NN2C(N=C(C(=C2C)C(=O)O)C)=C1 Chemical compound NC1=NNC(=C1)C.CC1=NN2C(N=C(C(=C2C)C(=O)O)C)=C1 ODEJIEMLGNZNJP-UHFFFAOYSA-N 0.000 description 1
- QAGVVHUAFUNSEB-UHFFFAOYSA-N NC1=NNC(=C1)C.CC1=NN2C(N=CC(=C2C(F)(F)F)C(=O)O)=C1 Chemical compound NC1=NNC(=C1)C.CC1=NN2C(N=CC(=C2C(F)(F)F)C(=O)O)=C1 QAGVVHUAFUNSEB-UHFFFAOYSA-N 0.000 description 1
- YHCOMOVPNASPGS-UHFFFAOYSA-N NC1=NNC(=C1)C.COC1=C(C(=NC=2N1N=C(C2)C)C)C(=O)O Chemical compound NC1=NNC(=C1)C.COC1=C(C(=NC=2N1N=C(C2)C)C)C(=O)O YHCOMOVPNASPGS-UHFFFAOYSA-N 0.000 description 1
- FTBFXVULGIPZFW-UHFFFAOYSA-N NC1=NNC(=C1)C1=CC=CC=C1.CC1=C(C=NC=2N1N=C(C2)C2=CC=CC=C2)C(=O)O Chemical compound NC1=NNC(=C1)C1=CC=CC=C1.CC1=C(C=NC=2N1N=C(C2)C2=CC=CC=C2)C(=O)O FTBFXVULGIPZFW-UHFFFAOYSA-N 0.000 description 1
- LHARLHQRFQGCSK-UHFFFAOYSA-N NC1=NNC(=C1)C1=CC=CC=C1.CC1=NC=2N(C(=C1C(=O)O)C)N=C(C2)C2=CC=CC=C2 Chemical compound NC1=NNC(=C1)C1=CC=CC=C1.CC1=NC=2N(C(=C1C(=O)O)C)N=C(C2)C2=CC=CC=C2 LHARLHQRFQGCSK-UHFFFAOYSA-N 0.000 description 1
- OPYJJGDPVFCZBE-UHFFFAOYSA-N NCCN.OC1=CC=CC2=C1N=NN2 Chemical compound NCCN.OC1=CC=CC2=C1N=NN2 OPYJJGDPVFCZBE-UHFFFAOYSA-N 0.000 description 1
- 108090000189 Neuropeptides Proteins 0.000 description 1
- 102000003797 Neuropeptides Human genes 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- GBBQYVAPSDJJHG-UHFFFAOYSA-N OC1=C(C(=O)OCC)C(C)=NC2=CC(C)=NN21 Chemical compound OC1=C(C(=O)OCC)C(C)=NC2=CC(C)=NN21 GBBQYVAPSDJJHG-UHFFFAOYSA-N 0.000 description 1
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical class OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 1
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 208000025747 Rheumatic disease Diseases 0.000 description 1
- NPYYDRITBJTDJF-UHFFFAOYSA-N S1C=NC2=C1C=C(C=C2)C(=O)O.NC(C(=O)OC)CNC(=O)C2=CC1=C(N=CS1)C=C2 Chemical compound S1C=NC2=C1C=C(C=C2)C(=O)O.NC(C(=O)OC)CNC(=O)C2=CC1=C(N=CS1)C=C2 NPYYDRITBJTDJF-UHFFFAOYSA-N 0.000 description 1
- 108010022999 Serine Proteases Proteins 0.000 description 1
- 102000012479 Serine Proteases Human genes 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 230000006044 T cell activation Effects 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- QIOZLISABUUKJY-UHFFFAOYSA-N Thiobenzamide Chemical compound NC(=S)C1=CC=CC=C1 QIOZLISABUUKJY-UHFFFAOYSA-N 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- GLYOPNLBKCBTMI-UHFFFAOYSA-N [2-chloro-2-(1-chloro-2-phenylethoxy)ethyl]benzene Chemical compound C=1C=CC=CC=1CC(Cl)OC(Cl)CC1=CC=CC=C1 GLYOPNLBKCBTMI-UHFFFAOYSA-N 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 239000008351 acetate buffer Substances 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 239000001361 adipic acid Substances 0.000 description 1
- 235000011037 adipic acid Nutrition 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 125000002723 alicyclic group Chemical group 0.000 description 1
- 125000006848 alicyclic heterocyclic group Chemical group 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 125000000266 alpha-aminoacyl group Chemical group 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 1
- JFCQEDHGNNZCLN-UHFFFAOYSA-N anhydrous glutaric acid Natural products OC(=O)CCCC(O)=O JFCQEDHGNNZCLN-UHFFFAOYSA-N 0.000 description 1
- 230000001887 anti-feedant effect Effects 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000005870 benzindolyl group Chemical group 0.000 description 1
- 125000004604 benzisothiazolyl group Chemical group S1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000004603 benzisoxazolyl group Chemical group O1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 1
- KCXMKQUNVWSEMD-UHFFFAOYSA-N benzyl chloride Chemical compound ClCC1=CC=CC=C1 KCXMKQUNVWSEMD-UHFFFAOYSA-N 0.000 description 1
- 229940073608 benzyl chloride Drugs 0.000 description 1
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical class OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- NHYXMAKLBXBVEO-UHFFFAOYSA-N bromomethyl acetate Chemical compound CC(=O)OCBr NHYXMAKLBXBVEO-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 150000001718 carbodiimides Chemical class 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- OGEBRHQLRGFBNV-RZDIXWSQSA-N chembl2036808 Chemical compound C12=NC(NCCCC)=NC=C2C(C=2C=CC(F)=CC=2)=NN1C[C@H]1CC[C@H](N)CC1 OGEBRHQLRGFBNV-RZDIXWSQSA-N 0.000 description 1
- LADPCMZCENPFGV-UHFFFAOYSA-N chloromethoxymethylbenzene Chemical compound ClCOCC1=CC=CC=C1 LADPCMZCENPFGV-UHFFFAOYSA-N 0.000 description 1
- GGRHYQCXXYLUTL-UHFFFAOYSA-N chloromethyl 2,2-dimethylpropanoate Chemical compound CC(C)(C)C(=O)OCCl GGRHYQCXXYLUTL-UHFFFAOYSA-N 0.000 description 1
- 229940061627 chloromethyl methyl ether Drugs 0.000 description 1
- 125000004230 chromenyl group Chemical group O1C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000006165 cyclic alkyl group Chemical group 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000002933 cyclohexyloxy group Chemical group C1(CCCCC1)O* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 125000004652 decahydroisoquinolinyl group Chemical group C1(NCCC2CCCCC12)* 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- LTMHNWPUDSTBKD-UHFFFAOYSA-N diethyl 2-(ethoxymethylidene)propanedioate Chemical compound CCOC=C(C(=O)OCC)C(=O)OCC LTMHNWPUDSTBKD-UHFFFAOYSA-N 0.000 description 1
- RTXPQLJAMYBTCR-UHFFFAOYSA-N diethyl 2-[[3-methyl-5-(phenylmethoxycarbonylamino)pyrazol-1-yl]methylidene]propanedioate Chemical compound CCOC(=O)C(C(=O)OCC)=CN1N=C(C)C=C1NC(=O)OCC1=CC=CC=C1 RTXPQLJAMYBTCR-UHFFFAOYSA-N 0.000 description 1
- 125000001070 dihydroindolyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000004611 dihydroisoindolyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- VAYGXNSJCAHWJZ-UHFFFAOYSA-N dimethyl sulfate Chemical compound COS(=O)(=O)OC VAYGXNSJCAHWJZ-UHFFFAOYSA-N 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000000428 dust Substances 0.000 description 1
- 210000003038 endothelium Anatomy 0.000 description 1
- XNGGOXOLHQANRB-UHFFFAOYSA-N ethyl 2-(ethoxymethylidene)-4,4,4-trifluoro-3-oxobutanoate Chemical compound CCOC=C(C(=O)C(F)(F)F)C(=O)OCC XNGGOXOLHQANRB-UHFFFAOYSA-N 0.000 description 1
- HMFLBGNCDZYITR-UHFFFAOYSA-N ethyl 2-formyl-3-oxopropanoate Chemical compound CCOC(=O)C(C=O)C=O HMFLBGNCDZYITR-UHFFFAOYSA-N 0.000 description 1
- 125000000031 ethylamino group Chemical group [H]C([H])([H])C([H])([H])N([H])[*] 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 238000005755 formation reaction Methods 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 230000003451 hyperinsulinaemic effect Effects 0.000 description 1
- 201000008980 hyperinsulinism Diseases 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 125000004857 imidazopyridinyl group Chemical group N1C(=NC2=C1C=CC=N2)* 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- DIQDUFVJZPNLJQ-UHFFFAOYSA-N indol-1-yl(phenyl)methanone Chemical compound C1=CC2=CC=CC=C2N1C(=O)C1=CC=CC=C1 DIQDUFVJZPNLJQ-UHFFFAOYSA-N 0.000 description 1
- JZOFVAHGZLNPCV-UHFFFAOYSA-N indol-1-ylmethanol Chemical compound C1=CC=C2N(CO)C=CC2=C1 JZOFVAHGZLNPCV-UHFFFAOYSA-N 0.000 description 1
- IENZCGNHSIMFJE-UHFFFAOYSA-N indole-5-carboxylic acid Chemical compound OC(=O)C1=CC=C2NC=CC2=C1 IENZCGNHSIMFJE-UHFFFAOYSA-N 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000000741 isoleucyl group Chemical group [H]N([H])C(C(C([H])([H])[H])C([H])([H])C([H])([H])[H])C(=O)O* 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000003965 isoxazolidinyl group Chemical group 0.000 description 1
- 208000017169 kidney disease Diseases 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 230000037356 lipid metabolism Effects 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- UURRHMFRCYITCL-UHFFFAOYSA-N methyl 1-(2,2-dimethylpropyl)-5-methoxyindole-3-carboxylate Chemical compound C1=C(OC)C=C2C(C(=O)OC)=CN(CC(C)(C)C)C2=C1 UURRHMFRCYITCL-UHFFFAOYSA-N 0.000 description 1
- FTLOEULOTNVCGF-UHFFFAOYSA-N methyl 1h-indole-7-carboxylate Chemical compound COC(=O)C1=CC=CC2=C1NC=C2 FTLOEULOTNVCGF-UHFFFAOYSA-N 0.000 description 1
- SMCVPMKCDDNUCQ-UHFFFAOYSA-N methyl 3,3-dimethoxypropanoate Chemical compound COC(OC)CC(=O)OC SMCVPMKCDDNUCQ-UHFFFAOYSA-N 0.000 description 1
- HNGRJOCEIWGCTB-UHFFFAOYSA-N methyl 3-amino-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoate Chemical compound COC(=O)C(CN)NC(=O)OC(C)(C)C HNGRJOCEIWGCTB-UHFFFAOYSA-N 0.000 description 1
- HNTLUEZVPLRQEV-UHFFFAOYSA-N methyl 4-amino-3-nitrobenzoate Chemical compound COC(=O)C1=CC=C(N)C([N+]([O-])=O)=C1 HNTLUEZVPLRQEV-UHFFFAOYSA-N 0.000 description 1
- ZECAAQDHFZNFPB-UHFFFAOYSA-N methyl 5-methyl-1h-indole-3-carboxylate Chemical compound C1=C(C)C=C2C(C(=O)OC)=CNC2=C1 ZECAAQDHFZNFPB-UHFFFAOYSA-N 0.000 description 1
- JZMJDSHXVKJFKW-UHFFFAOYSA-N methyl sulfate Chemical compound COS(O)(=O)=O JZMJDSHXVKJFKW-UHFFFAOYSA-N 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 125000000896 monocarboxylic acid group Chemical group 0.000 description 1
- 230000007886 mutagenicity Effects 0.000 description 1
- 231100000299 mutagenicity Toxicity 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 201000001119 neuropathy Diseases 0.000 description 1
- 230000007823 neuropathy Effects 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000020830 overeating Nutrition 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 1
- 229940055726 pantothenic acid Drugs 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 239000000813 peptide hormone Substances 0.000 description 1
- 210000005259 peripheral blood Anatomy 0.000 description 1
- 239000011886 peripheral blood Substances 0.000 description 1
- 208000033808 peripheral neuropathy Diseases 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- YAVXLTMRALFZIS-UHFFFAOYSA-N piperidine-2-carbonitrile Chemical compound N#CC1CCCCN1 YAVXLTMRALFZIS-UHFFFAOYSA-N 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 201000009104 prediabetes syndrome Diseases 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000001226 reprecipitation Methods 0.000 description 1
- 230000000552 rheumatic effect Effects 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- BNWCETAHAJSBFG-UHFFFAOYSA-N tert-butyl 2-bromoacetate Chemical compound CC(C)(C)OC(=O)CBr BNWCETAHAJSBFG-UHFFFAOYSA-N 0.000 description 1
- NDEIKFCAPOYPHU-UHFFFAOYSA-N tert-butyl 2-carbamoylazetidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC1C(N)=O NDEIKFCAPOYPHU-UHFFFAOYSA-N 0.000 description 1
- XTFHTGRWEXUBRJ-UHFFFAOYSA-N tert-butyl 2-indol-1-ylacetate Chemical compound C1=CC=C2N(CC(=O)OC(C)(C)C)C=CC2=C1 XTFHTGRWEXUBRJ-UHFFFAOYSA-N 0.000 description 1
- QQWYQAQQADNEIC-RVDMUPIBSA-N tert-butyl [(z)-[cyano(phenyl)methylidene]amino] carbonate Chemical compound CC(C)(C)OC(=O)O\N=C(/C#N)C1=CC=CC=C1 QQWYQAQQADNEIC-RVDMUPIBSA-N 0.000 description 1
- LFKDJXLFVYVEFG-UHFFFAOYSA-N tert-butyl carbamate Chemical compound CC(C)(C)OC(N)=O LFKDJXLFVYVEFG-UHFFFAOYSA-N 0.000 description 1
- AOCSUUGBCMTKJH-UHFFFAOYSA-N tert-butyl n-(2-aminoethyl)carbamate Chemical compound CC(C)(C)OC(=O)NCCN AOCSUUGBCMTKJH-UHFFFAOYSA-N 0.000 description 1
- DUDVAAKHUZEZHZ-UHFFFAOYSA-N tert-butyl n-[2-[(1,3-dimethylpyrazolo[3,4-b]pyridine-5-carbonyl)amino]ethyl]carbamate Chemical compound C1=C(C(=O)NCCNC(=O)OC(C)(C)C)C=C2C(C)=NN(C)C2=N1 DUDVAAKHUZEZHZ-UHFFFAOYSA-N 0.000 description 1
- OARIFFHEWDGQQX-SFHVURJKSA-N tert-butyl n-[2-[(2s)-2-cyanopyrrolidin-1-yl]-2-oxoethyl]-n-[2-(1,3-dihydroisoindol-2-yl)-2-oxoethyl]carbamate Chemical compound C1C2=CC=CC=C2CN1C(=O)CN(C(=O)OC(C)(C)C)CC(=O)N1CCC[C@H]1C#N OARIFFHEWDGQQX-SFHVURJKSA-N 0.000 description 1
- MRQIYZGBLWVFDS-UHFFFAOYSA-N tert-butyl n-[3-(2,3-dihydro-1h-isoindol-1-yl)-3-oxopropyl]carbamate Chemical compound C1=CC=C2C(C(=O)CCNC(=O)OC(C)(C)C)NCC2=C1 MRQIYZGBLWVFDS-UHFFFAOYSA-N 0.000 description 1
- RWYGHCSYCRHHSP-UHFFFAOYSA-N tert-butyl n-[3-(2,3-dihydroindol-1-yl)-3-oxopropyl]carbamate Chemical compound C1=CC=C2N(C(=O)CCNC(=O)OC(C)(C)C)CCC2=C1 RWYGHCSYCRHHSP-UHFFFAOYSA-N 0.000 description 1
- 125000001973 tert-pentyl group Chemical group [H]C([H])([H])C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- JRMUNVKIHCOMHV-UHFFFAOYSA-M tetrabutylammonium bromide Chemical compound [Br-].CCCC[N+](CCCC)(CCCC)CCCC JRMUNVKIHCOMHV-UHFFFAOYSA-M 0.000 description 1
- 150000003526 tetrahydroisoquinolines Chemical class 0.000 description 1
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000004853 tetrahydropyridinyl group Chemical group N1(CCCC=C1)* 0.000 description 1
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 125000005306 thianaphthenyl group Chemical group 0.000 description 1
- YUKQRDCYNOVPGJ-UHFFFAOYSA-N thioacetamide Chemical compound CC(N)=S YUKQRDCYNOVPGJ-UHFFFAOYSA-N 0.000 description 1
- DLFVBJFMPXGRIB-UHFFFAOYSA-N thioacetamide Natural products CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 1
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/16—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/30—Indoles; Hydrogenated indoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to carbon atoms of the hetero ring
- C07D209/42—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/44—Iso-indoles; Hydrogenated iso-indoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/48—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
- C07D215/54—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen attached in position 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D217/00—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems
- C07D217/22—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the nitrogen-containing ring
- C07D217/26—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/60—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings condensed with carbocyclic rings or ring systems
- C07D277/62—Benzothiazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D311/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings
- C07D311/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D311/04—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring
- C07D311/06—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring with oxygen or sulfur atoms directly attached in position 2
- C07D311/08—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring with oxygen or sulfur atoms directly attached in position 2 not hydrogenated in the hetero ring
- C07D311/16—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring with oxygen or sulfur atoms directly attached in position 2 not hydrogenated in the hetero ring substituted in position 7
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/26—Heterocyclic compounds containing purine ring systems with an oxygen, sulphur, or nitrogen atom directly attached in position 2 or 6, but not in both
- C07D473/32—Nitrogen atom
- C07D473/34—Nitrogen atom attached in position 6, e.g. adenine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
Description
【0001】
本発明は、優れたジペプチジルペプチダーゼIV(以下DPP-IVと略す)の阻害作用を有し、2型糖尿病の治療又は予防、これに付随する合併症の治療又は予防、あるいはその他のDPP-IVが関与する病態の治療に有用な化合物又はその医薬的に許容される塩である化合物に関する。
【背景技術】
【0002】
DPP-IVは、ポリペプチド鎖のN末端から、Xaa-Pro又はXaa-Ala(Xaaはいかなるアミノ酸であってもよい)のジペプチドを特異的に加水分解するセリンプロテアーゼの1種である。DPP-IV(CD26とも称される)の生体内での役割、疾患との関係については、完全には解明されていないが、種々の報告がある。中でも、グルカゴン様ペプチド1(以下GLP-1と略す)の不活性化に関与する酵素としての役割が最近注目されている。
GLP-1は、単独ではインスリン分泌を誘導せずに、糖により引き起こされたインスリン分泌を強める働きをもつペプチドホルモンである。したがって、低血糖の危険性が低く、血糖の濃度に応じたインスリン分泌促進が期待できる。また、GLP-1が摂食抑制作用を有することを示唆する報告もある。しかし、GLP-1はDPP-IVにより速やかに分解されてしまうため、GLP-1そのものを薬剤とすることは難しい。そこで、GLP-1のペプチドアナログが検討されているが、いずれも注射剤等で経口製剤ではない。
【0003】
そこで考えられたのが、分解酵素であるDPP-IVを阻害することにより、GLP-1の分解を抑え、GLP-1の作用を強めることである。これは、DPP-IV阻害剤を経口投与することにより、GLP-1の体液中濃度を維持し、そのGLP-1の作用により、糖尿病等、特に2型糖尿病の予防又は治療を行なうものである。このような治療法はまた、損なわれた耐糖能によって誘発もしくは増悪されるその他の疾患、例えば、過血糖(食後過血糖)、高インスリン血症、糖尿病合併症(腎障害、神経障害等)、脂質代謝異常、肥満等の予防又は治療における効果も期待されている。さらにGLP-1が摂食抑制作用を増強することにより改善が見込まれる疾患、例えば、過食、肥満等の予防又は治療における効果も期待される。
一方、DPP-IVの作用としては、他に、神経ペプチドの分解、T細胞の活性化、転移性腫瘍細胞の内皮への接着、HIVウイルスのリンパ球への侵入等が報告されている。また、DPP-IVに関する知見としては、リウマチ患者における末梢血T細胞のDPP-IV陽性率の上昇や、腎炎患者の尿中でDPP-IV活性が高い事が知られている。よって、DPP-IVを阻害する物質には、自己免疫疾患(例えば、関節炎、慢性間接リウマチ)、骨粗鬆症、後天性免疫不全症候群(AIDS)、移植臓器・組織の拒絶反応等に対しての予防又は治療効果も期待されている。
【0004】
DPP-IV阻害薬に関する特許の出願も既になされている。WO02/51836、WO01/96295、US20020193390 、US6011155及び、特表平9-509921号には、2-シアノピロリジン誘導体が、WO97/40832には、アミノアシルチアゾリジド誘導体が開示されている。また、Annual Report in Medicinal Chemistry第36巻,第191-200頁,2001年には、上記化合物群のほか、ペプチド誘導体として、アミノアシルピロリジド誘導体、ジペプチドフォスフォネート誘導体、ジペプチド硼酸誘導体、テトラヒドロイソキノリン誘導体、環状ペプチド誘導体、又、非ペプチド誘導体としてN-フェニルフタルイミド誘導体、N-フェニルホモフタルイミド誘導体、イソキノリン誘導体が報告されている。
【発明の開示】
【発明が解決しようとする課題】
【0005】
現在までに多くのDPP-IV阻害薬が報告されているが、いずれの化合物も、阻害活性、安定性、安全性の面で十分とはいえず、医薬品として満足できるものではない。したがって、DPP-IV阻害作用による治療又は予防効果を有し、医薬品として十分に満足できる化合物の開発が望まれている。
本発明者らは、上記の点に鑑み新規DPP-IV阻害薬の開発を目的として鋭意検討を行った。その結果、本発明者らは、側鎖に適度な疎水性のニ環式環、特にニ環式複素環基を有した下記一般式で表される化合物が強力なDPP-IV阻害活性を有することを見出し、さらにその安定性を高める方向で化合物展開して本発明を完成した。
【課題を解決するための手段】
【0006】
すなわち、本発明によれば、以下の式
【化1】
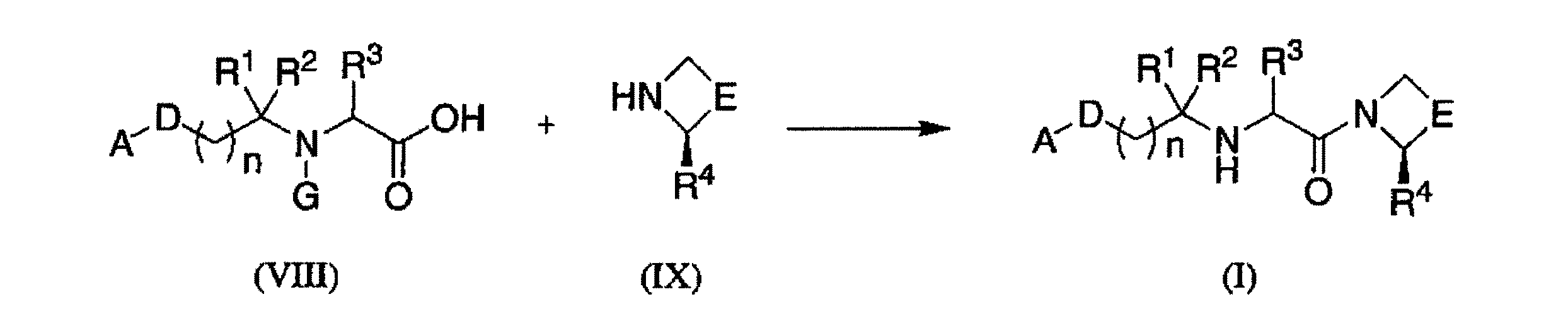
本発明はまた、前記本発明化合物を有効成分とするDPP-IV阻害剤をも提供する。当該DPP-IV阻害剤は、DPP-IV活性の阻害により病態の改善が見込まれる疾患、例えば、糖尿病(特に2型糖尿病)、糖尿病合併症等の予防又は治療剤となる。
【発明を実施するための最良の形態】
【0007】
本発明のDPP-IV阻害剤について、以下に更に詳細に説明する。本発明化合物は、次式、
【化2】
【0008】
置換されていてもよいC1-6アルキル基とは、C1-6アルキル基の任意の(「任意の」とは、複数の場合も含む。以後同じ。)水素原子がハロゲン原子(例えば、フッ素、塩素、臭素、ヨウ素原子)、オキソ基、ニトロ基、シアノ基、フェニル基、-OR14、-NR15R16、-OCOR17、NHCOR18、-NHS(O2)R19、-S(O2)NR20R21(ここでR14、R17、R18、R19は、水素原子、C1-6アルキル基、フェニル基、又はベンジル基を、R15、R16、R20、R21は、同時にあるいは別々に、水素原子、C1-6アルキル基、又はフェニル基を示すか、R15とR16、又はR20とR21とで3〜6員の脂環式環を形成していてもよい)等により置換されていてもよいことを意味する。C1-6アルキル基として具体的には、メチル、エチル、プロピル、イソプロピル、シクロプロピル、ブチル、イソブチル、s-ブチル、t-ブチル、シクロブチル、ペンチル、イソペンチル、ネオペンチル、t-ペンチル、シクロペンチル、ヘキシル、シクロヘキシル基等の直鎖又は分枝鎖状あるいは環状アルキル基が挙げられる。これらの中でも、C1-3アルキル基が好ましい。
【0009】
置換されていてもよいC1-6アルコキシ基とは、C1-6アルコキシ基の任意の水素原子がハロゲン原子(例えば、フッ素、塩素、臭素、ヨウ素原子)、オキソ基、ニトロ基、シアノ基、フェニル基、-OR14、-NR15R16、-OCOR17、NHCOR18、-NHS(O2)R19、-S(O2)NR20R21(ここでR14、R15、R16、R17、R18、R19、R20、R21は、前記に同じ)等により置換されていてもよいことを意味する。C1-6アルコキシ基として具体的には、メトキシ、エトキシ、プロポキシ、イソプロポキシ、シクロプロポキシ、ブトキシ、イソブトキシ、s-ブトキシ、t-ブトキシ、シクロブトキシ、ペンチルオキシ、イソペンチルオキシ、ネオペンチルオキシ、t-ペンチルオキシ、シクロペンチルオキシ、ヘキシルオキシ、シクロヘキシルオキシ基等の直鎖、又は分枝鎖状、あるいは環状アルコキシ基が挙げられる。これらの中でも、C1-3アルコキシ基が好ましい。
【0010】
置換されていてもよいC6-10アリール基とは、アリール基の環上の任意の水素原子がC1-6アルキル基、ハロゲン原子(例えば、フッ素、塩素、臭素、ヨウ素原子)、オキソ基、ニトロ基、シアノ基、フェニル基、-OR14、-NR15R16、-OCOR17、NHCOR18、-NHS(O2)R19、-S(O2)NR20R21(ここでR14、R15、R16、R17、R18、R19、R20、R21は、前記に同じ)等により置換されていることを意味する。アリール基としては、好ましくはフェニル、ナフチル、又は、6員環と、5、6、又は7員環が縮合したニ環式基で少なくともひとつの環が芳香環であるもの(例えばインダニル等)が挙げられる。
【0011】
置換されていてもよいニ環式複素環基とは、ニ環式複素環基の環上の任意の水素原子が置換されていてもよいC1-6アルキル基、置換されていてもよいC1-6アルコキシ基、ハロゲン原子(例えば、フッ素、塩素、臭素、ヨウ素原子)、オキソ基、ニトロ基、シアノ基、フェニル基、-OR14、-NR15R16、-OCOR17、NHCOR18、-NHS(O2)R19、-S(O2)NR20R21(ここでR14、R15、R16、R17、R18、R19、R20、R21は、前記に同じ)等により置換されていることを意味する。ニ環式複素環基としては、好ましくは炭素及び1〜4個のヘテロ原子(酸素、窒素、硫黄原子)を有する6員環と、5、6、又は7員環とが縮合したニ環式複素環基、特にベンズ誘導体、ピリジル誘導体、ピリミジル誘導体が挙げられる。例えば、インドリル、ベンゾチアゾリル、ベンゾイミダゾリル、ベンズオキサゾリル、ピラゾロピリジニル、イミダゾピリジニル、ピラゾロピリミジニル、トリアゾロピリミジニル、ベンゾトリアゾリル、ベンゾフラニル、イソベンゾフラニル、ベンゾチオフェニル、ベンズイソキサゾリル、ベンゾイソチアゾリル、トリアゾロピリミジニル、キノリニル、イソキノリニル、シンノリニル、クロメニル、ピリドピリミジニル、キナゾリニル、キノキサリニル、ナフチリジニル、チアナフテニル、イソチアナフテニル、ジヒドロインドリル、ジヒドロイソインドリル、ジヒドロプリニル、ジヒドロチアゾロピリミジニル、ジヒドロベンゾジオキサニル、イソインドリニル、インダゾリル、ピロロピリジニル、テトラヒドロキノリニル、デカヒドロキノリニル、テトラヒドロイソキノリニル、デカヒドロイソキノリニル、テトラヒドロナフチリジニル、テトラヒドロピリドアゼピニル等が挙げられる。
【0012】
置換されていてもよいニ環式炭化水素基とは、ニ環式炭化水素基上の任意の水素原子が、前述のニ環式複素環の場合と同様の置換基で置換されていてもよいことを意味する。例えば、ペンタレニル、インダニル、インデニル、ナフタレニル、テトラヒドロベンゾシクロヘプテニル、テトラヒドロナフタレニル等が挙げられる。
以下に、本発明化合物の中で、特に好ましい化合物について、更に詳細に説明する。
まず、R1、R2は、C1-6アルキル基が好ましく、更に好ましくはC1-3アルキル基であり、中でも、共にメチル基である化合物が化合物の安定性の面で特に好ましい。また、R3は水素原子が好ましく、R4 はDPP-IV阻害活性の面からシアノ基が好ましい。更に、Aは、窒素、酸素、硫黄原子のうち少なくとも1つのヘテロ原子を含む、置換されていてもよい6-5, 6-6, 6-7系のニ環式複素環基が好ましく、中でも、1〜3個の窒素原子を含む、置換されてもよい6-5系のニ環式複素環基が最適である。また更に、Dは-CONH-又は-CO-が好ましく、Eは-CH2CH2-が好ましく、nは1又は2が好ましい。
【0013】
次に、一般式(I)中の前記好ましい化合物において、Aについて、特に好ましいニ環式複素環基を更に詳細に説明する。
その1つのグループは、一般式(I)中のDが-CO-の場合で、Aが下式
【化3】
【0014】
また、別のグループは、一般式(I)中のDが-CONH-の場合で、Aが下式
【化4】
【化5】
【0015】
本発明化合物の製造方法を以下に反応工程式(1〜3)を挙げて説明する。
[反応工程式1]
【化6】
反応工程式1は、一般式(IV-1)で表される化合物と一般式(V)で表される化合物又はその塩を反応させて、一般式(I-1)で表される化合物を得る工程である。一般式(V)で表される化合物の塩としては、例えば、塩酸塩、トリフルオロ酢酸塩等が挙げられる。
【0016】
一般式(IV-1)で表される化合物と一般式(V)で表される化合物又はその塩との反応は、適当な溶媒(例えば、テトラヒドロフラン、ジクロロメタン、N,N-ジメチルホルムアミド等)中、脱酸剤(例えば、トリエチルアミン、4-ジメチルアミノピリジン等)の存在、又は、非存在下、一般式(IV-1)で表される化合物のカルボン酸を活性化する縮合剤(例えば、ジシクロヘキシルカルボジイミド、N-(3-ジメチルアミノプロピル)-N’-エチルカルボジイミド又は、その塩酸塩、N,N’-カルボニルジイミダゾール等)を単独で、又は、添加剤(N-ヒドロキシスクシンイミド、ヒドロキシベンゾトリアゾール等)を組み合わせて用い、-10〜80℃、とりわけ、0℃〜室温の温度条件下で、0.5時間〜3日間で好適に進行する。
【0017】
[反応工程式2]
【化7】
反応工程式2は、一般式(VI)で表される化合物又はその塩と一般式(VII)で表される化合物を反応させて、一般式(I)で表される化合物を得る工程である。一般式(VI)で表される化合物の塩としては、例えば、塩酸塩、トリフルオロ酢酸塩等が挙げられる。
一般式(VI)で表される化合物又はその塩と一般式(VII)で表される化合物との反応は、適当な溶媒(例えば、テトラヒドロフラン、ジクロロメタン、N,N-ジメチルホルムアミド、アセトン等)中、脱酸剤(例えば、トリエチルアミン、4-ジメチルアミノピリジン、炭酸カリウム等)、添加剤(例えば、臭化ナトリウム、ヨウ化ナトリウム、ヨウ化カリウム)の存在、又は、非存在下、-10〜80℃、とりわけ、0℃〜室温の温度条件下で、0.5時間〜3日間で好適に進行する。
【0018】
[反応工程式3]
【化8】
反応工程式3は、一般式(VIII)で表される化合物と一般式(IX)で表される化合物又はその塩を反応させて得られる化合物を脱保護させて、一般式(I)で表される化合物を得る工程である。一般式(IX)で表される化合物の塩としては、例えば、塩酸塩、トリフルオロ酢酸塩等が挙げられる。
アミド化反応は、適当な溶媒(例えば、テトラヒドロフラン、ジクロロメタン、N,N-ジメチルホルムアミド等)中、脱酸剤(例えば、トリエチルアミン、4-ジメチルアミノピリジン等)の存在、又は、非存在下、一般式(VIII)で表される化合物のカルボン酸を活性化する縮合剤(例えば、ジシクロヘキシルカルボジイミド、N-(3-ジメチルアミノプロピル)-N’-エチルカルボジイミド又は、その塩酸塩、N,N’-カルボニルジイミダゾール等)を単独で、又は、添加剤(N-ヒドロキシスクシンイミド、ヒドロキシベンゾトリアゾール等)を組み合わせて用い、-10〜80℃、とりわけ、0℃〜室温の温度条件下で、0.5時間〜3日間で好適に進行する。
脱保護反応は、例えば、保護基がBoc基の場合、適当な溶媒(例えば、1,4-ジオキサン、テトラヒドロフラン等)中、塩化水素、トリフルオロ酢酸等の酸を用いて、-10〜50℃、とりわけ、0℃〜室温の温度条件下で、10分〜24時間で好適に進行する。
【0019】
次に、原料の製造方法を以下に反応工程式(4〜7)を挙げて説明する。
[反応工程式4]
【化9】
反応工程式4は、一般式(X)で表される化合物と一般式(VII)で表される化合物を反応させた後、脱保護反応を行い化合物(V)を得る工程である。
一般式(X)で表される化合物と一般式(VII)で表される化合物との反応は、適当な溶媒(例えば、テトラヒドロフラン、ジクロロメタン、N,N-ジメチルホルムアミド、アセトン等)中、脱酸剤(例えば、トリエチルアミン、4-ジメチルアミノピリジン、炭酸カリウム等)、添加剤(例えば、臭化ナトリウム、ヨウ化ナトリウム、ヨウ化カリウム)の存在、又は、非存在下、-10〜80℃、とりわけ、0℃〜室温の温度条件下で、0.5時間〜3日間で好適に進行する。
脱保護反応は、例えば保護基がBoc基の場合、適当な溶媒(例えば、1,4-ジオキサン、テトラヒドロフラン等)中、塩化水素、トリフルオロ酢酸等の酸を用いて、-10〜50℃、とりわけ、0℃〜室温の温度条件下で、10分〜24時間で好適に進行する。
【0020】
[反応工程式5]
【化10】
反応工程式5は、一般式(IV-2)で表される化合物又はその塩(アミンの場合)と一般式(XI)で表される化合物又はその塩(アミンの場合)を反応させて(G1がアミノ酸の保護基の場合は、更に脱保護反応を行って)、一般式(VI)で表される化合物を得る工程である。一般式(IV-2)、(XI)で表される化合物の塩としては、例えば、塩酸塩、トリフルオロ酢酸塩等が挙げられる。
【0021】
アミド化反応は、適当な溶媒(例えば、テトラヒドロフラン、ジクロロメタン、N,N-ジメチルホルムアミド等)中、脱酸剤(例えば、トリエチルアミン、4-ジメチルアミノピリジン等)の存在、又は、非存在下、カルボン酸を活性化する縮合剤(例えば、ジシクロヘキシルカルボジイミド、N-(3-ジメチルアミノプロピル)-N’-エチルカルボジイミド又はその塩酸塩、N,N’-カルボニルジイミダゾール等)を単独で、又は、添加剤(N-ヒドロキシスクシンイミド、ヒドロキシベンゾトリアゾール等)を組み合わせて用い、-10〜80℃、とりわけ、0℃〜室温の温度条件下で、0.5時間〜3日間で好適に進行する。
又は、一般式(IV-2)で表される化合物がカルボン酸(R22が-COOH)の場合、以下のように反応させることも出来る。即ち、適当な溶媒(例えば、テトラヒドロフラン、ジクロロメタン、N,N-ジメチルホルムアミド等)中、オキサリルクロライド、チオニルクロライド等を用いて酸クロライド(R22を-COCl)とし、脱酸剤(例えば、トリエチルアミン、4-ジメチルアミノピリジン等)の存在、又は、非存在下、一般式(XI)で表される化合物(R23は、-NH2)又はその塩と、-10〜80℃、とりわけ、0℃〜室温の温度条件下で、0.5時間〜3日間で好適に進行する。
脱保護反応は、例えば、G1がBoc基の場合、適当な溶媒(例えば、1,4-ジオキサン、テトラヒドロフラン等)中、塩化水素、トリフルオロ酢酸等の酸を用いて、-10〜50℃、とりわけ、0℃〜室温の温度条件下で、10分〜24時間で好適に進行する。
【0022】
[反応工程式6]
【化11】
反応工程式6は、一般式(XII)で表される化合物と一般式(IX)で表される化合物又はその塩を反応させて一般式(VII)で表される化合物を得る工程である。
反応は、適当な溶媒(例えば、テトラヒドロフラン、ジクロロメタン、N,N-ジメチルホルムアミド等)中、脱酸剤(例えば、トリエチルアミン、4-ジメチルアミノピリジン等)の存在、又は、非存在下、一般式(XII)で表される化合物(Jが-OHの場合は例えばオキサリルクロライドやチオニルクロライド等で酸塩化物とした後)と一般式(IX)で表される化合物又はその塩とを、-10〜80℃、とりわけ、0℃〜室温の温度条件下で、0.5時間〜3日間反応させて一般式(VII)で表される化合物を得る。
【0023】
[反応工程式7]
【化12】
反応工程式7は、一般式(XIII)で表される化合物と一般式(IV-3)で表される化合物又はその塩を反応させて一般式(VIII)で表される化合物を得る工程である。
反応は、適当な溶媒(例えば、テトラヒドロフラン、ジクロロメタン、N,N-ジメチルホルムアミド等)中、脱酸剤(例えば、トリエチルアミン、4-ジメチルアミノピリジン等)の存在、又は、非存在下、カルボン酸を活性化する縮合剤(例えば、ジシクロヘキシルカルボジイミド、N-(3-ジメチルアミノプロピル)-N’-エチルカルボジイミド又はその塩酸塩、N,N’-カルボニルジイミダゾール等)を単独で、又は、添加剤(N-ヒドロキシスクシンイミド、ヒドロキシベンゾトリアゾール等)を組み合わせて用い、-10〜80℃、とりわけ、0℃〜室温の温度条件下で、0.5時間〜3日間で好適に進行する。
【0024】
上記それぞれの工程により得られる目的化合物は、通常の分離、精製手段により容易に単離することができる。該単離手段としては、一般に慣用される各種の手段のいずれをも採用することができ、その例としては、再結晶、再沈、溶媒抽出、カラムクロマトグラフィー等を例示できる。
本発明化合物は、多形(polymorphism)を示すことができ、また、複数の互変異性体として存在することができる。従って、本発明は、上記のようないかなる立体異性体、光学異性体、多形体、互変異性体、及びそれら任意の混合物等を含有するものである。
本発明化合物は、その医薬的に許容される塩を含む。医薬的に許容される塩としては、無機酸付加塩(例えば、塩酸、臭化水素酸、ヨウ化水素酸、硫酸、硝酸、リン酸等との塩)、有機酸付加塩(例えば、メタンスルホン酸、ベンゼンスルホン酸、p-トルエンスルホン酸、ギ酸、酢酸、トリフルオロ酢酸、シュウ酸、クエン酸、マロン酸、フマル酸、グルタル酸、アジピン酸、マレイン酸、酒石酸、コハク酸、マンデル酸、リンゴ酸、パントテン酸、メチル硫酸等との塩)、アミノ酸との塩(例えば、グルタミン酸、アスパラギン酸等との塩 )等が挙げられる。この酸付加塩の形成反応は、常法に従うことができる。
【0025】
本発明化合物は、DPP-IV阻害剤として提供することができる。即ち、本発明化合物は強力なDPP-IV阻害活性を示し、DPP-IV阻害作用により治療が可能な疾患、例えば、糖尿病(特に2型糖尿病)、これに付随する合併症、肥満、自己免疫疾患(例えば、関節炎、慢性間接リウマチ)、骨粗鬆症、後天性免疫不全症候群(AIDS)、移植臓器、組織の拒絶反応等の予防、治療に有用である。
本発明の化合物の投与形態としては、「日本薬局方」製剤総則記載の各種投与形態が目的に応じて選択できる。中でも、経口投与製剤とするのが好ましい。そのために、錠剤の形態に成形するに際しては、通例、当該分野で用いられる経口摂取可能な成分を選択すればよい。例えば、乳糖、結晶セルロース、白糖、リン酸カリウム等の賦形剤がそれにあたる。更に、所望により、結合剤、崩壊剤、滑沢剤、凝集防止剤等、通例製剤分野で常用される種々の添加剤を配合してもよい。
本発明製剤、即ち本発明の医薬組成物中に含有されるべき本発明化合物の量は、特に限定されず、広範囲より適宜選択される。有効成分化合物の投与量は、その用法、患者の年齢、性別その他の条件、疾患の程度により適宜選択されるが、通常、本発明化合物の量が、一日、体重1 kg当り約0.01〜500 mg程度と考えられる。尚、本発明製剤は、一日に1〜4回に分けて投与することもできる。
以下に実施例及び中間体実施例を挙げて本発明を更に詳しく説明するが、本発明はこれらによって何ら限定されるものではない。
【0026】
【中間体実施例1】
(S)-1-(2- クロロアセチル ) ピロリジン -2- カルボニトリル
特許(WO98/19998)記載の方法に従い、L-プロリンアミド(10.0 g)およびクロロアセチルクロライド(7.0 ml)を反応させた後、脱水反応させることにより、表記化合物(7.7 g, Y. 51%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 2.0-2.2 (4H, m), 3.4-3.5 (1H, m), 3.6-3.7 (1H, m), 4.4-4.5 (2H, m), 4.78 (1H, q).
ESI/MS(m/z) : 173 (M+H)+, 171 (M-H)-.
【中間体実施例2】
(R)-1-(2- クロロアセチル ) ピロリジン -2- カルボニトリル
中間体実施例1の方法を参考にD-プロリンアミド(3.2 g)およびクロロアセチルクロライド(2.5 ml)を反応させた後、脱水反応させることにより、表記化合物(3.2 g, Y. 66%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 2.1-2.4 (4H, m), 3.5-3.8 (2H, m), 4.0-4.2 (2H, m), 4.7-4.9 (1H, m).
ESI/MS(m/z) : 173 (M+H)+.
【0027】
【中間体実施例3】
(S)-3-(2- クロロアセチル ) チアゾリジン -4- カルボニトリル
チアゾリジン-3,4-ジカルボン酸-3-t-ブチルエステル(2.0 g)をテトラヒドロフラン(10 ml)に溶解し、氷冷下、N,N’-カルボニルジイミダゾール(1.4 g)を加えた。室温に戻し6時間撹拌した。1,4-ジオキサン(10 ml)を加え、氷冷した28%アンモニア水(40 ml)に滴下した。室温に戻し20時間撹拌した。酢酸エチル(60 ml)を加え抽出した。有機相を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。減圧下濃縮し、4-カルバモイルチアゾリジン-3-カルボン酸-t-ブチルエステル(1.6 g, Y. 81%)を得た。
上記で得られた4-カルバモイルチアゾリジン-3-カルボン酸-t-ブチルエステル(1.62 g)に4N塩酸/1,4-ジオキサン(3.5 ml)を加え一晩撹拌した。水と10%炭酸水素ナトリウム水溶液を加え中和し(pH7.5〜8)減圧下濃縮した。N,N-ジメチルホルムアミドを加え超音波にかけ、不溶物を濾去した。減圧下濃縮し、チアゾリジン-4-カルボン酸アミド(735 mg, Y. 80%)を得た。
中間体実施例1の方法を参考に、上記で得られたチアゾリジン-4-カルボン酸アミド(102 mg)およびクロロアセチルクロライド(105 mg)を反応させた後、脱水反応させることにより、表記化合物(87 mg, Y. 59%)を得た。
ESI/MS(m/z) : 191 (M+H)+.
【0028】
【中間体実施例4】
(S)-1-(2- クロロアセチル ) アゼチジン -2- カルボニトリル
2-カルバモイルアゼチジン-1-カルボン酸-t-ブチルエステル(500 mg)の1,4-ジオキサン(2.0 ml)溶液に氷浴下、4N塩酸/1,4-ジオキサン(2.5 ml)を加えた。室温で2時間撹拌した。5N水酸化ナトリウムを滴下し、反応溶液を中和した。減圧下濃縮し、N,N-ジメチルホルムアミドを加えて不溶物を濾去し、濾液を減圧下濃縮し、アゼチジン-2-カルボン酸アミド(161 mg, Y. 65%)を得た。
中間体実施例1の方法を参考に、上記で得られたアゼチジン-2-カルボン酸アミド(161 mg)およびクロロアセチルクロライド(200 mg)を反応させた後、脱水反応させることにより、表記化合物(112 mg, Y. 44%)を得た。
ESI/MS(m/z) : 159 (M+H)+.
【中間体実施例5】
(S)- 1-(2- ブロモ -2- フェニルアセチル ) ピロリジン -2- カルボニトリル
2-ブロモ-2-フェニル酢酸(500 mg)をジクロロメタン(30 ml)に溶解し、オキサリルクロライド(950 μl)、N,N-ジメチルホルムアミド(2滴)を加え室温で1時間撹拌した。減圧下濃縮後、ジクロロメタン(20 ml)に希釈した溶液を、(S)-ピロリジン-2-カルボニトリル(310 mg)とトリエチルアミン(650 μl)のジクロロメタン(30 ml)溶液に滴下し、室温で3時間撹拌した。10%クエン酸水溶液を加えて有機相を分取し、4%炭酸水素ナトリウム水溶液、飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥した。減圧下濃縮し、表記化合物(770 mg, Y. quant.)を得た。
ESI/MS(m/z) : 294 (M+H)+, 292 (M-H)-.
中間体実施例1〜5の方法を参考に、下記の反応式に従って化合物を合成した。合成した化合物とデータを表1に示した。(各記号は前記と同義である。)
【0029】
【化13】
【中間体実施例10】
(S)- ピロリジン -2- カルボニトリル
L-プロリンアミド(23 g)を テトラヒドロフラン(1200 ml)に溶解し、トリエチルアミン(22 g)を加え氷冷した。2-ニトロフェニルスルホニルクロライド(42 g)を加え室温で1時間撹拌した。酢酸エチル、水を加え有機相を分取し、無水硫酸ナトリウムで乾燥した。減圧下濃縮し、残渣にエーテルを加え析出した結晶を濾取し減圧下乾燥した。得られた結晶(45 g)をピリジン(890 ml)に溶解し、イミダゾール(23 g)を加え氷冷した。塩化ホスホリル(31 ml)を滴下し、室温で2時間撹拌した。氷(1000 g)、エーテル(2000 ml)を加え有機相を分取し、水で洗浄後、無水硫酸ナトリウムで乾燥した。減圧下濃縮し残渣にエーテル(4.1 l)を加えて溶解し、不溶物を濾去した。濾液に氷冷下、4N塩酸/1,4-ジオキサン(130 ml)を滴下し、室温で3時間撹拌した。析出した結晶を濾取し、エーテルで洗浄した。減圧下乾燥し、淡黄色結晶として表記化合物の塩酸塩(20 g, Y. 88%)を得た。
1H NMR; (CDCl3) δ (ppm) : 2.2-2.3 (2H, m), 2.3-2.4 (1H, m), 2.5-2.6 (1H, m), 3.5-3.7 (2H, m), 5.0 (1H, t).
【0031】
【中間体実施例11】
ピペリジン -2- カルボニトリル
中間体実施例3,10の方法を参考に、ピペリジン-2-カルボン酸(15 g)から表記化合物の塩酸塩(4.4 g, Y. 69%)を得た。
ESI/MS(m/z) : 111 (M+H)+.
【中間体実施例12】
(S)-1-[(2- アミノ -1,1- ジメチルエチル ) アミノアセチル ] ピロリジン -2- カルボニトリル 2 塩酸塩
2-メチルプロパン-1,2-ジアミン(5.0 g)をジクロロメタン(200 ml)に溶解し、0℃で15分間撹拌した。BOC-ON(15 g)のジクロロメタン(60 ml)溶液を滴下した後、室温で2時間撹拌した。クロロホルムで希釈して氷冷下、10%クエン酸水溶液で酸性とし有機相を分取した。水相は5N水酸化ナトリウム水溶液でアルカリ性にして酢酸エチルで抽出し、無水硫酸ナトリウムで乾燥した。減圧下濃縮して(2-アミノ-2-メチル-1-プロピル)カルバミン酸-t-ブチルエステル(7.9 g, Y. 74%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 0.9 (6H, s), 1.4 (9H, s), 2.8 (2H, d), 6.7 (1H, brt).
【0032】
上記で得られた(2-アミノ-2-メチル-1-プロピル)カルバミン酸-t-ブチルエステル(7.9 g)、よう化ナトリウム(8.7 g)、炭酸カリウム(8.0 g)をアセトン(230 ml)に懸濁した。氷冷下、(S)-1-(2-クロロアセチル)ピロリジン-2-カルボニトリル(10 g)のアセトン(80 ml)溶液を加えてそのまま30分間撹拌した。室温で15時間撹拌した後、減圧下濃縮した。残渣をクロロホルムに溶解し、不溶物を濾去した後、減圧下濃縮した。残渣をカラムクロマトグラフィー(流出溶媒;ジクロロメタン:メタノール 80:1 → 60:1 → 40:1)に供し(S)-{2-[(2-シアノピロリジン-1-イル)-2-オキソエチルアミノ]-2-メチル-1-プロピル}カルバミン酸-t-ブチルエステル(12 g, Y. 91%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 0.9 (6H, s), 1.4 (9H, s), 1.9-2.2 (4H, m), 2.9 (2H, d), 3.2-3.5 (4H, m), 3.5-3.7 (1H, m), 4.7-4.8 (1H, m), 6.6-6.7 (1H, brt).
ESI/MS(m/z) : 325 (M+H)+, 323 (M-H)-.
上記で得られた(S)-{2-[(2-シアノピロリジン-1-イル)-2-オキソエチルアミノ]-2-メチル-1-プロピル}カルバミン酸-t-ブチルエステル(4.8 g)をジクロロメタン(50 ml)に溶解した。氷冷下、4N塩酸/1,4-ジオキサン(50 ml)を加えて室温で1時間撹拌した。減圧下濃縮して表記化合物(4.2 g, Y. 96%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 1.4 (6H, s), 2.0-2.3 (4H, m), 3.2 (2H, brs), 3.5-3.6 (2H, m), 3.7-3.8 (1H, m), 4.0-4.2 (2H, m), 4.9 (1H, q), 8.5 (2H, brs), 9.4 (1H, brs), 9.5 (1H, brs).
ESI/MS(m/z) : 225 (M+H)+.
【0033】
【中間体実施例13】
(S)-1-[2-(1,1- ジメチル -2- メチルアミノエチルアミノ ) アセチル ] ピロリジン -2- カルボニトリル
(S)-1-[(2-アミノ-1,1-ジメチルエチル)アミノアセチル]ピロリジン-2-カルボニトリル2塩酸塩(1.48 g)をアセトニトリル(50 ml)に溶解し、ギ酸-4-ニトロフェニルエステル(1.00 g)、炭酸カリウム(1.37 g)を加え、室温で16時間撹拌した。減圧下濃縮し、残渣をカラムクロマトグラフィー(流出溶媒;ジクロロメタン:メタノール 5 :1)に供し(S)-N-{2-[2-(2-シアノピロリジン-1-イル)-2-オキソエチルアミノ]-2-メチル-1-プロピル}ホルムアミド(693 mg, Y. 55%)を得た。
ESI/MS(m/z) : 253 (M+H)+.
上記で得られた(S)-N-{2-[2-(2-シアノピロリジン-1-イル)-2-オキソエチルアミノ]-2-メチル-1-プロピル}ホルムアミド(690 mg)をMeOH(30 ml)に溶解した。シアノトリヒドロホウ酸ナトリウム(172 mg)を加え、室温にて6時間撹拌した。減圧下濃縮し、残渣をカラムクロマトグラフィー(流出溶媒;ジクロロメタン:メタノール 5:1 → 3:1)に供し表記化合物(455 mg, Y. 70%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 1.4 (6H, s), 2.0 (2H, brs), 2.0-2.3 (4H, m), 2.50 (3H, s), 3.2 (2H, brs), 3.5-3.6 (2H, m), 4.0-4.2 (2H, m), 4.9 (1H, q).
ESI/MS(m/z) : 225 (M+H)+.
【0034】
【中間体実施例14】
3- アミノ -3- メチルブタン酸
3-メチルクロトン酸(12.0 g)をピリジン(40 ml)に溶解し、ベンジルアミン(12.8 g)を加え、120℃で3時間撹拌した。室温に戻し、得られた懸濁溶液にアセトンを加え、結晶を濾取、洗浄した。減圧下乾燥し無色結晶として3-ベンジルアミノ-3-メチルブタン酸(10.3 g,Y.42%)を得た。
ESI/MS; 208 (M+H)+, 206 (M-H)-.
上記で得られた3-ベンジルアミノ-3-メチルブタン酸(6.0 g)のエタノール(90 ml)溶液に、6N塩酸(5.8 ml)を加えた。ここに、5%パラジウム-炭素(2.4 g)、酢酸(46 ml)を加え、水素雰囲気下、50℃で5時間撹拌した。不溶物を濾去し、濾液を減圧下濃縮した。析出した結晶をエーテルで洗浄後、減圧下乾燥し無色結晶として表記化合物(4.4 g, Y. quant.)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 1.4 (6H, s), 2.7 (2H, s), 8.3 (3H, brs).
ESI/MS(m/z) : 118 (M+H) +, 116 (M-H) -.
【中間体実施例15】
4- メチル -1,4- ペンタンジアミン
4-メチル-4-ニトロペンタン酸メチルエステル(5.00 g)をエタノール(25 ml)に溶解し、1N水酸化ナトリウム水溶液を加え1日間撹拌した。減圧下濃縮し、クロロホルム、水を加え、水相をクロロホルムで洗浄した。水相に2N塩酸(20 ml)を加え、クロロホルムで抽出、無水硫酸ナトリウムで乾燥した。減圧下濃縮し、白色結晶として4-メチル-4-ニトロペンタン酸(4.32 g, Y. 94%)を得た。
1H NMR; (CDCl3) δ (ppm) : 1.6 (6H, s), 2.2-2.3 (2H, m), 2.4-2.5 (2H, m), 10.8 (1H, brs).
上記で得られた4-メチル-4-ニトロペンタン酸(4.3 g)をジクロロメタンに溶解し、N-(3-ジメチルアミノプロピル)-N’-エチルカルボジイミド塩酸塩(6.1 g)、トリエチルアミン(4.5 ml)を加え1時間撹拌した。これにベンジルアミン(3.4 g)を加え、1日間撹拌した。水を加え、2N塩酸を加えて酸性とし、クロロホルムで抽出した。有機相を飽和炭酸水素ナトリウム水溶液、飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。減圧下濃縮し、残渣をカラムクロマトグラフィー (流出溶媒;酢酸エチル:n-ヘキサン 1:1.5)に供し、無色オイルとしてN-ベンジル-4-メチル-4-ニトロペンタン酸アミド(2.5 g, Y. 38%)を得た。
1H NMR; (CDCl3) δ (ppm) : 1.6 (6H, s), 2.1-2.2 (2H, m), 2.2-3.3 (2H, m), 4.4 (2H, d), 6.0 (1H, brs), 7.3-7.4 (5H, m).
【0035】
上記で得られたN-ベンジル-4-メチル-4-ニトロペンタン酸アミド(2.5 g)をテトラヒドロフラン(20 ml)に溶解し、0℃に冷却した。1Nボラン-テトラヒドロフラン錯体(13 ml)を滴下後、室温で一晩撹拌した。再び0℃に冷却し、2N塩酸(30 ml)を加え、50℃に昇温した。酢酸エチルを加え抽出した。水相に50%水酸化ナトリウム水溶液を加えアルカリ性とし、クロロホルムで抽出、飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥した。減圧下濃縮し、無色オイルとしてベンジル-4-メチル-4-ニトロペンチルアミン(1.7 g, Y. 73%)を得た。
1H NMR; (CDCl3) δ (ppm) : 1.4-1.5 (2H, m), 1.6 (6H, s), 2.0 (2H, dt), 2.6 (2H, t), 7.2-7.4 (5H, m).
上記で得られたベンジル-4-メチル-4-ニトロペンチルアミン(1.7 g)と10%パラジウム-炭素(500 mg)をエタノールに懸濁し、水素雰囲気下、60℃で1日撹拌した。室温に戻し、セライト濾過後、減圧下濃縮した。2N塩酸を加え酸性とし、エーテルで抽出した。水相に50%水酸化ナトリウム水溶液を加えアルカリ性とし、エーテルで抽出、無水硫酸ナトリウムで乾燥した。減圧下濃縮し、表記化合物(420 mg, Y. 50%)を得た。
1H NMR; (CDCl3) δ (ppm) : 1.2 (6H, s), 1.5-1.6 (4H, m), 2.7-2.8 (2H, m).
【0036】
【中間体実施例16】
2- メチルピラゾロ [1,5-a] ピリミジン -6- カルボン酸
2-メチルピラゾロ[1,5-a]ピリミジン-6-カルボン酸アミド(475 mg)をエタノール(5 ml)に溶解し、5N水酸化ナトリウム水溶液(2 ml)を加え70℃で1時間撹拌した。室温に冷却し水を加え、酢酸エチルで洗浄した。水相に2N塩酸を酸性になるまで加えて析出した結晶を濾取し、水、n-ヘキサンで洗浄した。減圧下乾燥し白色結晶として表記化合物(300 mg, Y. 63%)を得た。
ESI/MS; 178 (M+H) +, 176 (M-H) -.
【中間体実施例17】
2,5,7- トリメチルピラゾロ [1,5-a] ピリミジン -6- カルボン酸
3-アミノ-5-メチルピラゾール(970 mg)とジアセト酢酸エチル(1.7 g)を酢酸(5 ml)に溶解し120℃で3時間撹拌した。室温に冷却し、減圧下濃縮した。残渣にエタノール(5 ml)と5N水酸化ナトリウム水溶液(2 ml)を加え70℃で1時間撹拌した。室温に冷却し水を加え、酢酸エチルで洗浄した。水相に2N塩酸を酸性になるまで加えて析出した結晶を濾取し、水、n-ヘキサンで洗浄した。減圧下乾燥し白色結晶として表記化合物(1.6 g, Y. 80%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 2.4 (3H, s), 2.5 (3H, s), 2.8 (3H, s), 6.5 (1H, s), 13.8 (1H, brs).
ESI/MS(m/z) : 206(M-H)-.
【0037】
【中間体実施例18】
7- メトキシ -2,5- ジメチルピラゾロ [1,5-a] ピリミジン -6- カルボン酸
3-アミノ-5-メチルピラゾール(970 mg)とアセトマロン酸ジエチル(2.0 g)を酢酸(5 ml)に溶解し120℃で3時間撹拌した。室温に冷却し、減圧下濃縮して、残渣にエタノールを加え0℃に冷却した。析出した結晶を濾取し、冷エタノールで洗浄した。減圧下乾燥し白色結晶として7-ヒドロキシ-2,5-ジメチルピラゾロ[1,5-a]ピリミジン-6-カルボン酸エチルエステル(2.2 g, Y. 95%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 1.3 (3H, t), 2.3 (3H, s), 2.4 (3H, s), 4.2 (2H, q), 6.0 (1H, s), 12.6 (1H, brs).
ESI/MS(m/z) : 236(M+H)+, 234(M-H)-.
上記で得られた7-ヒドロキシ-2,5-ジメチルピラゾロ[1,5-a]ピリミジン-6-カルボン酸エチルエステル(235 mg)をアセトン(5 ml)に懸濁し、炭酸カリウム(138 mg)を加え室温で30分撹拌した。ヨウ化メチル(1.0 ml)を加え2時間還流した。室温に冷却し、水を加えクロロホルムで抽出、有機相を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥した。減圧下濃縮し、得られた結晶をエタノール(5 ml)に溶解した。5N水酸化ナトリウム水溶液(1 ml)を加え50℃で1時間撹拌した。室温に冷却し、水を加え、酢酸エチルにて洗浄した。水相に2N塩酸を酸性になるまで加えて析出した結晶を濾取し、水、n-ヘキサンで洗浄した。減圧下乾燥し白色結晶として表記化合物(162 mg, Y. 73%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 2.3 (3H, s), 2.7 (3H, s), 3.7 (3H, s), 6.4 (1H, s).ESI/MS(m/z) : 222(M+H)+.
【0038】
【中間体実施例19】
5,7- ジメチル -2- フェニルピラゾロ [1,5-a] ピリミジン -6- カルボン酸
3-アミノ-5-フェニルピラゾール(1.6 g)とジアセト酢酸エチル(1.7 g)を酢酸(5.0 ml)に溶解し120℃で3時間撹拌した。室温に冷却し、減圧下濃縮した。残渣にエタノール(10 ml)と5N水酸化ナトリウム水溶液(3 ml)を加え70℃で1時間撹拌した。室温に冷却し水を加え、酢酸エチルで洗浄した。水相に2N塩酸を酸性になるまで加えて析出した結晶を濾取し、水、n-ヘキサンで洗浄した。減圧下乾燥し白色結晶として表記化合物(2.1 g, Y. 78%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 2.6 (3H, s), 2.9 (3H, s), 7.2 (1H, s), 7.4 (1H, t), 7.5 (2H, t), 8.1 (1H, d), 13.9 (1H, brs).
ESI/MS(m/z) : 266(M-H)-.
【0039】
【中間体実施例20】
2- メチル -7- トリフルオロメチルピラゾロ [1,5-a] ピリミジン -6- カルボン酸
3-アミノ-5-メチルピラゾール(389 mg)と(エトキシメチリデン)トリフルオロアセト酢酸エチル(960 mg)をエタノール(10 ml)に溶解し70℃で1.5時間撹拌した。濃塩酸(1 ml)を加え、さらに1時間撹拌した。室温に冷却し、減圧下濃縮した。残渣にエタノール(10 ml)と5N水酸化ナトリウム水溶液(3 ml)を加え70℃で1時間撹拌した。室温に冷却し水を加え、酢酸エチルで洗浄した。水相に2N塩酸を酸性になるまで加えて析出した結晶を濾取し、水、n-ヘキサンで洗浄した。減圧下乾燥し白色結晶として表記化合物(102 mg, Y. 42%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 2.6 (3H, s), 2.9 (3H, s), 7.2 (1H, s), 7.4 (1H, t), 7.5 (2H, t), 8.1 (1H, d), 13.9 (1H, brs).
ESI/MS(m/z) : 244(M-H)-.
【中間体実施例21】
2-t- ブチル -5,7- ジメチルピラゾロ [1,5-a] ピリミジン -6- カルボン酸
3-アミノ-5-t-ブチルピラゾール(1.6 g)とジアセト酢酸エチル(1.7 g)を酢酸(5 ml)に溶解し120℃で3時間撹拌した。室温に冷却し、減圧下濃縮した。残渣にエタノール(10 ml)と5N水酸化ナトリウム水溶液(3 ml)を加え70℃で1時間撹拌した。室温に冷却し水を加え、酢酸エチルで洗浄した。水相に2N塩酸を酸性になるまで加えて析出した結晶を濾取し、水、n-ヘキサンで洗浄した。減圧下乾燥し白色結晶として表記化合物(2.1 g, Y. 78%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 2.6 (3H, s), 2.9 (3H, s), 7.2 (1H, s), 7.4 (1H, t), 7.5 (2H, t), 8.1 (1H, d), 13.9 (1H, brs).
ESI/MS(m/z) : 246(M-H)-.
【0040】
【中間体実施例22】
2-t- ブチル -7- メチルピラゾロ [1,5-a] ピリミジン -6- カルボン酸
アセト酢酸エチル(35.4 g)をアセトニトリル(200 ml)に溶解し、ジメチルホルムアミドジメチルアセタール(30.9 g)を加え、室温で一晩撹拌した。減圧下濃縮し、赤色オイルとして2-ジメチルアミノメチレンアセト酢酸エチル(50.4 g, Y. 99%)を得た。
1H NMR; (CDCl3-d6) δ (ppm) : 1.3 (3H, t), 2.3 (3H, s), 3.1 (6H, brs), 4.2 (2H, q), 7.7 (1H, s).
上記で得られた2-ジメチルアミノメチレンアセト酢酸エチル(556 mg)と3-アミノ-5-t-ブチルピラゾール(418 mg)をエタノール(10 ml)に溶解し70℃で1.5時間撹拌した。濃塩酸(1 ml)を加え、さらに1時間撹拌した。室温に冷却し、減圧下濃縮した。残渣にエタノール(10 ml)と5N水酸化ナトリウム水溶液を(3 ml)加え70℃で1時間撹拌した。室温に冷却し水を加え、酢酸エチルで洗浄した。水相に2N塩酸を酸性になるまで加え析出した結晶を濾取し、水、n-ヘキサンで洗浄した。減圧下乾燥し黄色結晶として表記化合物(396 mg, Y. 57%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 1.4 (9H, s), 3.1 (3H, s), 6.8 (1H, s), 8.8 (1H, s), 13.5 (1H, brs).
ESI/MS(m/z) : 232(M-H)-.
【0041】
【中間体実施例23】
7- メチル -2- フェニルピラゾロ [1,5-a] ピリミジン -6- カルボン酸
3-アミノ-5-フェニルピラゾール(477 mg)と2-N,N-ジメチルアミノメチレンアセト酢酸エチル(556 mg)をエタノール(10 ml)に溶解し70℃で1.5時間撹拌した。濃塩酸(1 ml)を加え、さらに1時間撹拌した。室温に冷却し、減圧下濃縮した。残渣にエタノール(10 ml)と5N水酸化ナトリウム水溶液を(3 ml)加え70℃で1時間撹拌した。室温に冷却し水を加え、酢酸エチルで洗浄した。水相に2N塩酸を酸性になるまで加え析出した結晶を濾取し、水、n-ヘキサンで洗浄した。減圧下乾燥し黄色結晶として表記化合物(463 mg, Y. 61%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 3.2 (3H, s), 7.4 (1H, s), 7.5 (3H, m), 8.1 (2H, d), 8.9 (1H, s), 13.6 (1H, brs).
ESI/MS(m/z) : 252(M-H)-.
【中間体実施例24】
7- メトキシ -5- メチル -2- フェニルピラゾロ [1,5-a] ピリミジン -6- カルボン酸
3-アミノ-5-フェニルピラゾール(1.56 mg)とアセトマロン酸ジエチル(2.00 g)を酢酸(5.0 ml)に溶解し120℃で3時間撹拌した。室温に冷却し、減圧下濃縮した。残渣にエタノールを加え0℃に冷却した。析出した結晶を濾取し、冷エタノールで洗浄した。減圧下乾燥し白色結晶として7-ヒドロキシ-5-メチル-2-フェニルピラゾロ[1,5-a]ピリミジン-6-カルボン酸エチルエステル(2.73 g, Y. 92%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 1.3 (3H, t), 2.4 (3H, s), 4.3 (2H, q), 6.7 (1H, s), 7.4 (2H, t), 7.5 (2H, t), 8.0 (1H, d).
上記で得られた7-ヒドロキシ-5-メチル-2-フェニルピラゾロ[1,5-a]ピリミジン-6-カルボン酸エチルエステル(297 mg)をアセトン(5 ml)に懸濁させ、炭酸カリウム(138 mg)を加え室温で30分撹拌した。ヨウ化メチル(1.0 ml)を加え2時間還流した。室温に冷却し、水を加えクロロホルムで抽出、有機相を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥した。減圧下濃縮し、得られた結晶をエタノール(5 ml)に溶解した。5N水酸化ナトリウム水溶液(1 ml)を加え50℃で1時間撹拌した。室温に冷却し、水を加え、酢酸エチルにて洗浄した。水相に2N塩酸を酸性になるまで加え析出した結晶を濾取し、水、n-ヘキサンで洗浄した。減圧下乾燥し白色結晶として表記化合物(121 mg, Y. 45%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 2.7 (3H, s), 3.8 (3H, s), 7.2 (1H, s), 7.5 (1H, t), 7.5 (2H, dd), 8.0 (2H, d), 13.5 (1H, brs).
【0042】
【中間体実施例25】
5- ヒドロキシ -2- メチルピラゾロ [1,5-a] ピリミジン -6- カルボン酸
3-アミノ-5-メチルピラゾール(971 mg)のクロロホルム(20 ml)溶液に0℃でトリエチルアミン(2.02 g)、ベンジルオキシカルボニルクロライド(1.71 g)を滴下し18時間撹拌した。減圧下濃縮し、残渣をカラムクロマトグラフィー(流出溶媒;n-ヘキサン:酢酸エチル 2:1)に供し5- メチル -2H- ピラゾール -3- イルカルバミン酸ベンジルエステル(1.65 g, Y. 67%)を得た。
上記で得られた5- メチル -2H- ピラゾール -3- イルカルバミン酸ベンジルエステル(600 mg)、エトキシメチレンマロン酸ジエチル(1.80 g)の混合液を100℃で18時間撹拌した。減圧下濃縮し、残渣をカラムクロマトグラフィー(流出溶媒;n-ヘキサン:酢酸エチル 3:1)に供し2-(5-ベンジルオキシカルボニルアミノ-3-メチルピラゾロ-1-イルメチレン)マロン酸ジエチルエステル(700 mg, Y. 67%)を得た。
上記で得られた2-(5-ベンジルオキシカルボニルアミノ-3-メチルピラゾール-1-イルメチレン)マロン酸ジエチルエステル(100 mg)に4N塩酸/1,4-ジオキサン(2 ml)を加え、22時間撹拌した。析出した結晶を濾取し、減圧下乾燥して5-ヒドロキシ-2-メチルピラゾロ[1,5-a]ピリミジン-6-カルボン酸エチルエステル(40 mg, Y. 73%)を得た。
中間体実施例24の方法を参考に、上記で得られた5-ヒドロキシ-2-メチルピラゾロ[1,5-a]ピリミジン-6-カルボン酸エチルエステル(154 mg)を加水分解して表記化合物(136 mg, Y. quant.)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 2.3 (3H, s), 6.3 (1H, s), 8.6 (1H, s).
【0043】
【中間体実施例26】
7- ヒドロキシ -2- メチルピラゾロ [1,5-a] ピリミジン -6- カルボン酸
中間体実施例24の方法を参考に、7-メトキシ-2-メチルピラゾロ[1,5-a]ピリミジン-6-カルボン酸エチルエステルを加水分解して表記化合物を得た。
1H NMR; (DMSO-d6) δ (ppm) : 2.3 (3H, s), 6.3 (1H, s), 8.8 (1H, s).
【中間体実施例27】
2- ヒドロキシメチルピラゾロ [1,5-a] ピリミジン -6- カルボン酸
ナトリウムメトキシド(1.40 g)のテトラヒドロフラン(50 ml)溶液にアセトニトリル(2.04 ml)を加え、1.5時間還流した。室温に戻し、メトキシ酢酸メチルエステル(2.57 ml)を加え一晩還流した。室温に戻し、水を加え1N塩酸でpH7にして、エーテルで抽出した。有機相を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。減圧下濃縮し、残渣をカラムクロマトグラフィー(流出溶媒;n-ヘキサン:酢酸エチル 2:1)に供し4-メトキシ-3-オキソブチロニトリル(1.14 g, Y. 39%)を得た。
上記で得られた4-メトキシ-3-オキソブチロニトリル(1.14 g)のエタノール(50 ml)溶液にヒドラジン一水和物(0.49 ml)を加え、17時間還流した。室温に戻し、減圧下濃縮した。残渣をカラムクロマトグラフィー(流出溶媒;ジクロロメタン:メタノール 50:1)に供し5-メトキシメチル-2H-ピラゾール-3-イルアミン(684 mg, Y. 53%)を得た。
【0044】
上記で得られた5-メトキシメチル-2H-ピラゾール-3-イルアミン(684 mg)のエタノール(50 ml)溶液に2-ホルミル-3-オキソプロピオン酸エチルエステル(775 mg)を加え、一晩撹拌した。減圧下濃縮し、残渣に飽和炭酸水素ナトリウム水溶液を加え酢酸エチルで抽出した。有機相を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。減圧下濃縮し、残渣をカラムクロマトグラフィー(流出溶媒;n-ヘキサン:酢酸エチル 4:1)に供し2-メトキシメチルピラゾロ[1,5-a]ピリミジン-6-カルボン酸エチルエステル(878 mg, Y. 69%)を得た。
上記で得られた2-メトキシメチルピラゾロ[1,5-a]ピリミジン-6-カルボン酸エチルエステル(20 mg)のジクロロメタン(2 ml)溶液に、-70℃で1M三臭化ホウ素/ジクロロメタン溶液(0.51 ml)を滴下した。撹拌しながら4.5時間かけて-70℃から-50℃に昇温した後、2時間かけて-50℃から室温に昇温した。0℃に冷却し、水を加えて酢酸エチルで抽出、無水硫酸ナトリウムで乾燥した。減圧下濃縮し、2-ヒドロキシメチルピラゾロ[1,5-a]ピリミジン-6-カルボン酸エチルエステル(19 mg, Y. quant.)を得た。
上記で得られた2-ヒドロキシメチルピラゾロ[1,5-a]ピリミジン-6-カルボン酸エチルエステル(19 mg)のテトラヒドロフラン(1 ml)溶液に5N水酸化ナトリウム水溶液(0.1 ml)を加え、室温で17時間撹拌した。水を加えて酢酸エチルで洗浄した。水相を2N塩酸で酸性にして酢酸エチルで抽出し、無水硫酸ナトリウムで乾燥した。減圧下濃縮し、残渣を熱酢酸エチルに溶解して濾過した。濾液を減圧下濃縮して表記化合物(11 mg, Y. 65%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 4.7 (2H, s), 6.8 (1H, s), 8.8 (1H, d), 9.3-9.4 (1H, m).
【0045】
【中間体実施例28】
2- メトキシメチルピラゾロ [1,5-a] ピリミジン -6- カルボン酸
中間体実施例27の中間体、2-メトキシメチルピラゾロ[1,5-a]ピリミジン-6-カルボン酸エチルエステルを中間体実施例27の方法を参考に、加水分解して表記化合物を得た。
1H NMR; (DMSO-d6) δ (ppm) : 3.4 (3H, s), 4.6 (2H, s), 6.8 (1H, s), 8.9 (1H, d), 9.4-9.5 (1H, m).
ESI/MS(m/z) : 206 (M-H)-.
【中間体実施例29】
1- メチル -1H- インドール -3- カルボン酸
1H-インドール-3-カルボン酸(960 mg)をN,N-ジメチルホルムアミド(15 ml)に溶解し0℃に冷却した。水素化ナトリウム(720 mg)を2回に分けて加え、室温に戻し1時間撹拌した。再び0℃に冷却しヨウ化メチル(0.67 ml)のN,N-ジメチルホルムアミド(5 ml)溶液をゆっくりと滴下し、室温に戻し2時間撹拌した。0℃に冷却し、反応液に氷を加え、さらに水(50 ml)を加え析出した結晶を濾取し、水、n-ヘキサンで洗浄した。減圧下乾燥し黄色結晶として表記化合物(910 mg, Y. 87%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 3.9 (3H, s), 7.2 (1H, dd), 7.3 (1H, dd), 7.5 (1H, d), 8.0 (1H, d), 8.1 (1H, s), 11.9 (1H, brs).
ESI/MS(m/z) : 174(M-H)-.
中間体実施例29の方法を参考に、下記の反応式に従って化合物を合成した。合成した化合物とデータを表2に示した。
【0046】
【化14】
【中間体実施例33】
1- メチル -1H- インドール -7- カルボン酸
1H-インドール-7-カルボン酸メチルエステル(546 mg)をN,N-ジメチルホルムアミド(8 ml)に溶解し0℃に冷却した。水素化ナトリウム(370 mg)を加え、そのまま30分間撹拌した。ヨウ化メチル(0.38 ml)をゆっくりと滴下し、室温に戻し2時間撹拌した。酢酸エチルで希釈して有機相を2N塩酸、飽和炭酸水素ナトリウム水溶液、飽和食塩水で洗浄した。無水硫酸ナトリウムで乾燥した後、減圧下濃縮した。
上記化合物に1,4-ジオキサン(14 ml)と1N水酸化ナトリウム水溶液(14 ml)を加えて40℃で17時間撹拌した。2N塩酸で酸性としてクロロホルムで抽出した。無水硫酸ナトリウムで乾燥した後、減圧下濃縮した。析出物を濾取し、n-ヘキサンで洗浄後、減圧下乾燥して表記化合物(296 mg, Y. 55%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 3.8 (1H, s), 6.5 (1H, d), 7.1 (1H, t), 7.4 (1H, d), 7.5 (1H, dd), 7.7 (1H, dd).
ESI/MS(m/z) : 176 (M+H)+, 174 (M-H) -.
【0048】
中間体実施例33の方法を参考に、下記の反応式に従って化合物を合成した。合成した化合物とデータを表3に示した。
【化15】
【中間体実施例38】
5- メトキシ -1- メチル -1H- インドール -3- カルボン酸
酢酸(8.0 ml)に4-メトキシフェニルヒドラジン塩酸塩(200 mg)、3,3-ジメトキシプロピオン酸メチルエステル(194 mg)を加えて70℃で4.5時間撹拌した。減圧下濃縮し、残渣をカラムクロマトグラフィー(流出溶媒;酢酸エチル:n-ヘキサン 1:5 → 1:3)に供し5-メトキシ-1H-インドール-3-カルボン酸メチルエステル(259 mg, Y. 97%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 3.8 (3H, s), 3.9 (3H, s), 6.8 (1H, dd), 7.4 (1H, d), 7.5 (1H, d), 8.0 (1H, s), 11.8 (1H, brs).
ESI/MS(m/z) : 204 (M-H) -.
上記で得られた5-メトキシ-1H-インドール-3-カルボン酸メチルエステル(121 mg)をN,N-ジメチルホルムアミド(1.5 ml)に溶解し0℃に冷却した。水素化ナトリウム(47 mg)を加え、そのまま30分間撹拌した。ヨウ化メチル(55 μl)を滴下し、室温に戻して1時間撹拌した。酢酸エチルで希釈し、有機相を2N塩酸、飽和炭酸水素ナトリウム水溶液、飽和食塩水で洗浄した。無水硫酸ナトリウムで乾燥した後、減圧下濃縮した。
上記化合物に1,4-ジオキサン(4 ml)と1N水酸化ナトリウム水溶液(4 ml)を加えて40℃で18時間撹拌した。2N塩酸で酸性として析出物を濾取し、水で洗浄後、減圧下乾燥して表記化合物(57 mg, Y. 52%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 3.7 (3H, s), 3.8 (3H, s), 6.8 (1H, dd), 7.4 (1H, d), 7.5 (1H, d), 7.9 (1H, s), 11.9 (1H, brs).
ESI/MS(m/z) : 206 (M+H)+, 204 (M-H) -.
【0050】
【中間体実施例39】
7- メトキシ -1- メチル -1H- インドール -5- カルボン酸
文献(J. Org. Chem., 1996, 61, 5804-5812)記載の方法に従い、3-メトキシ-4-アンスラニル酸メチルエステルより表記化合物を得た。
1H NMR; (DMSO-d6) δ (ppm) : 3.9 (3H, s), 4.0 (3H, s), 6.5 (1H, d), 7.2 (1H, s), 7.3 (1H, d), 7.9 (1H, s).
ESI/MS(m/z) : 206 (M+H)+, 204 (M-H) -.
【中間体実施例40】
1-(2,2- ジメチルプロピル )-1H- インドール -3- カルボン酸
1H-インドール-3-カルボン酸(208 mg)をN,N-ジメチルホルムアミド(10 ml)に溶解し、水素化ナトリウム(154 mg)を加え、室温で10分間撹拌した。反応液にヨウ化ネオペンチル(0.25 ml)を加え、80℃で15時間撹拌した。水を加え酢酸エチルで洗浄した。水相を1N塩酸でpH6にして酢酸エチルで抽出し、飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥した。減圧下濃縮し、残渣をカラムクロマトグラフィー(流出溶媒;n-ヘキサン:酢酸エチル 4:1)に供し表記化合物(264 mg, Y. 89%)を得た。
1H NMR; (CDCl3) δ (ppm) : 1.0 (9H, s), 3.9 (2H, s), 7.2-7.3 (2H, m), 7.3-7.4 (1H, m), 7.9 (1H, s), 8.2-8.3 (1H, m).
ESI/MS(m/z) : 232 (M+H)+, 230 (M-H)-.
【0051】
【中間体実施例41】
1- イソブイル -1H- インドール -3- カルボン酸
中間体実施例40の方法を参考に、1H-インドール-3-カルボン酸(251 mg)、ヨウ化イソブチルを用いて表記化合物(121 mg, Y. 36%)を得た。
1H NMR; (CDCl3) δ (ppm) : 0.9 (6H, d), 2.2-2.3 (1H, m), 3.9 (2H, d), 7.2-7.3 (2H, m), 7.3-7.4 (1H, m), 7.9 (1H, s), 8.2-8.3 (1H, m).
ESI/MS(m/z) : 218 (M+H)+, 216 (M-H)-.
【中間体実施例42】
1-(2,2- ジメチルプロピル )-1H- インドール -5- カルボン酸
中間体実施例40の方法を参考に、1H-インドール-5-カルボン酸メチルエステル(825 mg)、ヨウ化ネオペンチルを用いて表記化合物(473 mg, Y. 43%)を得た。
1H NMR; (CDCl3) δ (ppm) : 1.0 (9H, s), 3.9 (2H, s), 6.6 (1H, d), 7.1 (1H, d), 7.3 (1H, d), 7.9 (1H, dd), 8.4 (1H, s).
ESI/MS(m/z) : 232 (M+H)+, 230 (M-H)-.
【中間体実施例43】
1- イソブチル -1H- インドール -5- カルボン酸
中間体実施例40の方法を参考に、1H-インドール-5-カルボン酸メチルエステル(1.02 g)、ヨウ化イソブチルを用いて表記化合物(375 mg, Y. 30%)を得た。
1H NMR; (CDCl3) δ (ppm) : 0.9 (6H, d), 2.1-2.2 (1H, m), 3.9 (2H, d), 6.6 (1H, d), 7.1 (1H, d), 7.3 (1H, d), 7.9 (1H, dd), 8.4 (1H, s).
ESI/MS(m/z) : 218 (M+H)+, 216 (M-H)-.
【中間体実施例44】
1- ベンジルオキシメチル -1H- インドール -3- カルボン酸
1H-インドール-3-カルボン酸メチルエステル(1.00 g)をN,N-ジメチルホルムアミド(12 ml)に溶解し0℃に冷却した。水素化ナトリウム(0.46 g)を2回に分けて加え、そのまま30分間撹拌した。ベンジルオキシメチルクロライド(2.4 ml)をゆっくりと滴下し、室温に戻して2時間撹拌した。酢酸エチルで希釈して有機相を2N塩酸、飽和炭酸水素ナトリウム水溶液、飽和食塩水で洗浄した。無水硫酸ナトリウムで乾燥した後、減圧下濃縮した。
上記化合物に1,4-ジオキサン(20 ml)と1N水酸化ナトリウム水溶液(20 ml)を加えて40℃で18時間撹拌した。2N塩酸で酸性としてクロロホルムで抽出した。無水硫酸ナトリウムで乾燥した後、減圧下濃縮した。残渣はn-ヘキサンから結晶化を行い、減圧下乾燥して表記化合物(1.3g, Y. 83%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 5.7 (2H, s), 7.2-7.4 (7H, m), 7.6 (1H, d), 8.0 (1H, d), 8.2 (1H, s).
ESI/MS(m/z) : 282 (M+H)+, 280 (M-H) -.
【0052】
【中間体実施例45】
1- メトキシメチル -1H- インドール -3- カルボン酸
1H-インドール-3-カルボン酸メチルエステル(500 mg)をN,N-ジメチルホルムアミド(7.5 ml)に溶解し0℃に冷却した。水素化ナトリウム(340 mg)を加え、そのまま30分間撹拌した。メトキシメチルクロライド(0.43 ml)をゆっくりと滴下し、室温に戻し1時間撹拌した。酢酸エチルで希釈して有機相を2N塩酸、飽和炭酸水素ナトリウム水溶液、飽和食塩水で洗浄した。無水硫酸ナトリウムで乾燥した後、減圧下濃縮した。
上記化合物に1,4-ジオキサン(15 ml)と1N水酸化ナトリウム水溶液(15 ml)を加えて40℃で16時間撹拌した。2N塩酸で酸性としてクロロホルムで抽出した。無水硫酸ナトリウムで乾燥した後、減圧下濃縮した。析出物を濾取し、エーテルで洗浄後、減圧下乾燥して表記化合物(342 mg, Y. 58%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 3.1 (3H, s), 5.6 (2H, s), 7.2-7.3 (2H, m), 7.6 (1H, d), 8.0 (1H, d), 8.2 (1H, d).
ESI/MS(m/z) : 206 (M+H)+, 204 (M-H) -.
【中間体実施例46】
1- アセトキシメチル -1H- インドール -3- カルボン酸
1H-インドール-3-カルボン酸(400 mg)をN,N-ジメチルホルムアミド(6 ml)に溶解し0℃に冷却した。水素化ナトリウム(500 mg)を2回に分けて加え、そのまま30分間撹拌した。酢酸ブロモメチルエステル(0.32 ml)をゆっくりと滴下し0℃で15分間、室温に戻し45分間撹拌した。0℃に冷却して水を加えた後、2N塩酸で酸性として酢酸エチルで抽出した。無水硫酸ナトリウムで乾燥した後、減圧下濃縮した。残渣をカラムクロマトグラフィー(流出溶媒;ジクロロメタン:メタノール 50:1)に供し表記化合物(354 mg, Y. 61%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 2.0 (3H, s), 6.2 (2H, s), 7.2-7.4 (2H, m), 7.6 (1H, d), 7.9 (1H, s), 8.0 (1H, d).
ESI/MS(m/z) : 233 (M+H)+.
【0053】
【中間体実施例47】
1- ベンジルオキシメチル -1H- インドール -5- カルボン酸
1H-インドール-5-カルボン酸メチルエステル(500 mg)をN,N-ジメチルホルムアミド(6.0 ml)に溶解した。0℃に冷却して水素化ナトリウム(230 mg)を加え30分間撹拌した。ベンジルクロロメチルエーテル(1.2 ml)を加えて室温で2時間撹拌した。酢酸エチルで抽出し、有機相を2N塩酸、飽和炭酸水素ナトリウム水溶液、飽和食塩水で洗浄した。無水硫酸ナトリウムで乾燥後、減圧下濃縮した。残渣に1,4-ジオキサン(10 ml)と1N水酸化ナトリウム水溶液(5 ml)を加えて40℃で22時間撹拌した。2N塩酸で酸性とした後、クロロホルムで抽出した。無水硫酸ナトリウムで乾燥後、減圧下濃縮した。析出物を濾取し、n-ヘキサンで洗浄後、減圧下乾燥して表記化合物(740 mg, Y. 92%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 4.4 (2H, s), 5.7 (2H, s), 6.6 (1H, d), 7.2-7.4 (5H, m), 7.6-7.7 (3H, m), 7.8 (1H, d), 8.2 (1H, s).
ESI/MS(m/z) : 280 (M-H) -.
【中間体実施例48】
1- ヒドロキシメチル -1H- インドール -5- カルボン酸
中間体実施例47で得られた1-ベンジルオキシメチル-1H-インドール-5-カルボン酸(380 mg)をエタノール(6.5 ml)に懸濁した。10%パラジウム-炭素(190 mg)を加えて水素雰囲気下、60℃で47時間撹拌した。不溶物を濾去し、減圧下濃縮した。残渣をカラムクロマトグラフィー(流出溶媒;ジクロロメタン:メタノール 50:1)に供し表記化合物(120 mg, Y. 48%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 5.5 (2H, s), 6.5 (1H, d), 7.5 (1H, d), 7.6 (1H, d), 7.7 (1H, d), 8.2 (1H, s).
【0054】
【中間体実施例49】
1- メトキシメチル -1H- インドール -5- カルボン酸
1H-インドール-5-カルボン酸メチルエステル(500 mg)と、クロロメチルメチルエーテル(0.43 ml)から中間体実施例45の方法を参考に表記化合物(190 mg, Y. 70%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 5.5 (2H, s), 6.5 (1H, d), 7.5 (1H, d), 7.6 (1H, d), 7.7 (1H, d), 8.2 (1H, s).
【中間体実施例50】
1-(2,2- ジメチルプロピル) -5- メトキシ -1H- インドール -3- カルボン酸
5-メトキシ-1H-インドール-5-カルボン酸メチルエステル(357 mg)をN,N-ジメチルホルムアミド(17 ml)に溶解した。水素化ナトリウム(209 mg)を3回に分けて加え、そのまま15分間撹拌した。ヨウ化ネオペンチル(0.35 ml)を滴下し80℃で15時間撹拌した。酢酸エチルで希釈し、有機相を2N塩酸、飽和炭酸水素ナトリウム水溶液、飽和食塩水で洗浄した。無水硫酸ナトリウムで乾燥した後、減圧下濃縮した。残渣を分取用薄層クロマトグラフィー(展開溶媒;酢酸エチル:n-ヘキサン 1:3)に供し1-(2,2-ジメチルプロピル )-5-メトキシ-1H-インドール-3-カルボン酸ネオペンチルエステル(114 mg, Y. 20%)、1-(2,2-ジメチルプロピル )-5-メトキシ-1H-インドール-3-カルボン酸メチルエステル(130 mg, Y. 27%)を得た。
上記で得られた1-(2,2-ジメチルプロピル )-5-メトキシ-1H-インドール-3-カルボン酸ネオペンチルエステル(114 mg)に1,4-ジオキサン(2.5 ml)と1N水酸化ナトリウム水溶液(2.5 ml)を加えて40℃で15時間撹拌した。エタノール(3 ml)を加えて70℃で24時間撹拌した。2N塩酸で酸性としてクロロホルムで抽出した。無水硫酸ナトリウムで乾燥した後、減圧下濃縮して表記化合物(73 mg, Y. 81%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 0.9 (9H, s), 3.7 (3H, s), 4.0 (2H, s), 6.8 (1H, dd), 7.4 (1H, d), 7.5 (1H, d), 7.8 (1H, s).
ESI/MS(m/z) : 262 (M+H)+, 260 (M-H) -.
【0055】
【中間体実施例51】
1-(2,2- ジメチルプロピル )-5- メチル -1H- インドール -3- カルボン酸
中間体実施例50の方法と同様にして5-メチル-1H-インドール-3-カルボン酸メチルエステルより表記化合物を得た。
1H NMR; (DMSO-d6) δ (ppm) : 0.9 (9H, s), 2.4 (3H, s), 4.0 (2H, s), 7.0 (1H, d), 7.4 (1H, d), 7.8 (1H, s), 7.8 (1H, s).
ESI/MS(m/z) : 246 (M+H)+, 244 (M-H) -.
【中間体実施例52】
1-(2,2- ジメチルプロピル )-5- ヒドロキシ -1H- インドール -3- カルボン酸
1-(2,2-ジメチルプロピル )-5-メトキシ-1H-インドール-3-カルボン酸(102 mg)をジクロロメタン(3 ml)に溶解して-78℃に冷却した。1M 三臭化ホウ素/ジクロロメタン溶液(1.2 ml)をゆっくりと滴下して、-78℃から0℃に戻しながら1時間撹拌した。クロロホルムで希釈し、1N水酸化ナトリウム水溶液でアルカリ性とし、有機相を分取した。水相は2N塩酸で酸性としてクロロホルムで抽出し、無水硫酸ナトリウムで乾燥した。減圧下濃縮して表記化合物(78 mg, Y. 80%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 0.9 (9H, s), 3.9 (2H, s), 6.6 (1H, dd), 7.3-7.4 (2H, m), 7.8 (1H, s), 8.9 (1H, brs).
ESI/MS(m/z) : 248 (M+H)+, 246 (M-H) -.
【中間体実施例53】
1-(2,2- ジメチルプロピオニルオキシメチル )-1H- インドール -3- カルボン酸
1H-インドール-3-カルボン酸(400 mg)のN,N-ジメチルホルムアミド(4 ml)溶液を氷冷下、水素化ナトリウム(218 mg)を加え30分撹拌した。2,2-ジメチルプロピオン酸クロロメチルエステル(373 mg)を加え室温に戻し2時間撹拌した。水を加え水相をエーテルで洗浄した。水相を2N塩酸で酸性にし、エーテルで抽出した。有機相を飽和食塩水で洗浄し無水硫酸ナトリウムで乾燥した。減圧下濃縮し、橙色結晶として表記化合物(540 mg, Y. 79%)を得た。
ESI/MS(m/z) : 276 (M+H)+, 274 (M-H) -.
【0056】
【中間体実施例54】
1-t- ブトキシカルボニルメチル -1H- インドール -5- カルボン酸
1H-インドール-5-カルボン酸ベンジルエステル(600 mg)のN,N-ジメチルホルムアミド(2 ml)溶液に氷冷下、水素化ナトリウム(115 mg)を加え30分撹拌した。ブロモ酢酸-t-ブチルエステル(562 mg)を加えて2時間撹拌した。水を加えて、水相を中性にし、ジクロロメタンで抽出した。有機相を無水硫酸ナトリウムで乾燥した。減圧下濃縮し、1-t-ブトキシカルボニルメチル-1H-インドール-5-カルボン酸ベンジルエステル(944 mg, Y. quant.)を得た。
上記で得られた1-t-ブトキシカルボニルメチル-1H-インドール-5-カルボン酸ベンジルエステル(800 mg)をエタノールに溶解し、5%パラジウム-炭素(160 mg)を加え水素雰囲気下、室温で一晩撹拌した。不溶物を濾去し、濾液を減圧下濃縮して、表記化合物(670 mg, Y. quant.)を得た。
ESI/MS(m/z) : 276 (M+H)+, 274 (M-H) -.
【中間体実施例55】
1- メチル -2,3- ジヒドロ -1H- インドール -5- カルボン酸
1-メチル-1H-インドール-5-カルボン酸(100 mg)にジクロロメタン(2 ml)とトリエチルシラン(1 ml)を加えた。0℃に冷却してトリフルオロ酢酸(1 ml)を滴下し、室温に戻して2時間撹拌した。減圧下濃縮し、析出物を濾取した。エーテルで洗浄後、減圧下乾燥して表記化合物(66 mg, Y. 65%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 2.7 (3H, s), 2.9 (2H, t), 3.4 (2H, t), 6.4 (1H, d), 7.5 (1H, s), 7.6 (1H, d).
ESI/MS(m/z) : 178 (M+H)+, 176 (M-H) -.
【0057】
【中間体実施例56】
1- アセチル -1H- インドール -3- カルボン酸
1H-インドール-3-カルボン酸(400 mg)、酢酸ナトリウム(0.96 g)を無水酢酸(4.8 ml)に懸濁した。110℃で16時間撹拌した後、クロロホルムで抽出した。有機相を2N塩酸で洗浄し、無水硫酸ナトリウムで乾燥後、減圧下濃縮した。残渣をカラムクロマトグラフィー(流出溶媒;ジクロロメタン:メタノール 50:1)に供し表記化合物(170 mg, Y. 34%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 2.7 (3H, s), 7.3-7.4 (2H, m), 8.0-8.1 (1H, m), 8.3-8.4 (1H, m), 8.4-8.5 (1H, m).
ESI/MS(m/z) : 202 (M-H)-.
【中間体実施例57】
1- アセチル -2,3- ジヒドロ -1H- インドール -5- カルボン酸
1H-インドール-5-カルボン酸(2.0 g)をN,N-ジメチルホルムアミド(15 ml)に溶解した。ベンジルクロライド(1.53 ml)、炭酸カルシウム(3.4 g)を加えて室温で39時間撹拌した。酢酸エチルで希釈して有機相を2N塩酸、飽和炭酸水素ナトリウム水溶液、飽和食塩水で洗浄した。無水硫酸ナトリウムで乾燥後、減圧下濃縮した。析出してきた固体を濾取し、n-ヘキサンで洗浄後、減圧下乾燥して1H-インドール-5-カルボン酸ベンジルエステル(2.6 g, Y. 85%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 5.3 (2H, s), 6.6 (1H, s), 7.3-7.5 (7H, m), 7.7 (1H, d), 8.3 (1H, s), 11.5 (1H, brs).
ESI/MS(m/z) : 252(M+H)+, 250(M-H)-.
上記で得られた1H-インドール-5-カルボン酸ベンジルエステル(1.0 g)をN,N-ジメチルホルムアミド(10 ml)に溶解した。0℃に冷却して水素化ナトリウム(0.32 g)を加えて30分間撹拌した。アセチルクロライド(1.3 ml)を加えて室温で8時間撹拌した。酢酸エチルで希釈して有機相を2N塩酸、飽和炭酸水素ナトリウム水溶液、飽和食塩水で洗浄した。有機相は無水硫酸ナトリウムで乾燥後、減圧下濃縮した。残渣をカラムクロマトグラフィー(流出溶媒;酢酸エチル:n-ヘキサン 1:7 → 1:4)に供し1-アセチル-1H-インドール-5-カルボン酸ベンジルエステル(1.1 g, Y. 97%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 2.6 (3H, s), 5.3 (2H, s), 6.9 (1H, d), 7.3-7.5 (5H, m), 7.9 (1H, dd), 7.9 (1H, d), 8.3 (1H, d), 8.4 (1H, d).
ESI/MS(m/z) : 294(M+H)+, 292(M-H)-.
上記で得られた1-アセチル-1H-インドール-5-カルボン酸ベンジルエステル(550 mg)をエタノール(9 ml)に懸濁した。10%パラジウム-炭素を加え、水素雰囲気下、室温で16時間撹拌した。不溶物を濾去した後、減圧下濃縮した。析出した結晶を濾取し、エーテルで洗浄後、減圧下乾燥して表記化合物(180 mg, Y. 48%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 2.1 (3H, s), 3.1 (2H, t), 4.1 (2H, t), 7.7-7.8 (2H, m), 8.0 (1H, d).
ESI/MS(m/z) : 206(M+H)+, 204(M-H)-.
【0058】
【中間体実施例58】
1- アセチル -1H- インドール -5- カルボン酸
1-アセチル-2,3-ジヒドロ-1H-インドール-5-カルボン酸(100 mg)を1,4-ジオキサン(3 ml)に懸濁して2,3-ジクロロ-5,6-ジシアノ-p-ベンゾキノン(445 mg)を加えて110℃で16時間撹拌した。固形物を濾去した後、減圧下濃縮した。残渣を分取用薄層クロマトグラフィー(展開溶媒;ジクロロメタン:メタノール 20:1)に供し表記化合物(98 mg, Y. 99%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 2.6 (3H, s), 6.8 (1H, d), 7.9 (1H, d), 7.9 (1H, d), 8.2 (1H, s), 8.3 (1H, d).
ESI/MS(m/z) : 203 (M+H)+, 202 (M-H) -.
【中間体実施例59】
1- ベンゾイル -1H- インドール -5- カルボン酸
1H-インドール-5-カルボン酸ベンジルエステル(300 mg)のN,N-ジメチルホルムアミド(2 ml)溶液に氷冷下、水素化ナトリウム(58 mg)を加え30分撹拌した。ベンゾイルクロライド(202 mg)を加えて2時間撹拌した。ジクロロメタンで希釈して有機相を2N塩酸、飽和炭酸水素ナトリウム水溶液、飽和食塩水で洗浄した。有機相を無水硫酸ナトリウムで乾燥した。減圧下濃縮し淡橙色結晶として1-ベンゾイル-1H-インドール-5-カルボン酸ベンジルエステル(500 mg, Y. quant.)を得た。
上記で得られた1-ベンゾイル-1H-インドール-5-カルボン酸ベンジルエステル (100 mg)をエタノールに溶解し、5%パラジウム-炭素(20 mg)を加え水素雰囲気下、室温で一晩撹拌した。不溶物を濾去し、濾液を減圧下濃縮し、白色結晶として表記化合物(50 mg, Y. 66%)を得た。
ESI/MS(m/z) : 266 (M+H)+, 264 (M-H) -.
【0059】
【中間体実施例60】
1-(2,2- ジメチルプロピオニル )-1H- インドール -5- カルボン酸
1H-インドール-5-カルボン酸ベンジルエステル(276 mg)のN,N-ジメチルホルムアミド(2 ml)溶液に氷冷下、水素化ナトリウム(53 mg)を加え30分撹拌した。2,2-ジメチルプロピオニルクロライド(162 mg)を加えて2時間撹拌した。水を加えて、水相を中性にし、ジクロロメタンで抽出し、無水硫酸ナトリウムで乾燥した。減圧下濃縮し、淡橙色結晶として1-(2,2-ジメチルプロピオニル)-1H-インドール-5-カルボン酸ベンジルエステル(320 mg, Y. 87%)を得た。
上記で得られた1-(2,2-ジメチルプロピオニル)-1H-インドール-5-カルボン酸ベンジルエステル(220 mg)をエタノールに溶解し、5%パラジウム-炭素(44 mg)を加え水素雰囲気下、室温で一晩撹拌した。不溶物を濾去し、濾液を減圧下濃縮して、表記化合物(140 mg, Y. 86%)を得た。
ESI/MS(m/z) : 246 (M+H)+, 244 (M-H) -.
【中間体実施例61】
4- メトキシベンゾチアゾール -6- カルボン酸
4-アミノ-3-メトキシ安息香酸(1.0 g)とチオシアン酸アンモニウム(910 mg)をメタノール(15 ml)に溶解した。これに、0℃で臭素(0.30 ml)のメタノール(3.0 ml)溶液をゆっくり滴下した。その後、室温で2時間撹拌し、氷(50 g)を加えた。析出した結晶を濾取し、減圧下乾燥して得られた白色結晶(760 mg)を水(3.0 ml)とエタノール(3.0 ml)の混合溶媒中、硫化ナトリウム(1.6 g)と90℃で2時間撹拌した。冷却後、90%ギ酸で酸性にし、析出した結晶を濾取、減圧下乾燥し、黄色結晶として 4-アミノ-5-メルカプト-3-メトキシ安息香酸(670 mg, Y. 57%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 3.8 (3H, s), 7.1 (1H, brs), 7.4 (1H, brs).
ESI/MS(m/z) : 200 (M+H) +, 198 (M-H) -.
上記で得られた4-アミノ-5-メルカプト-3-メトキシ安息香酸(670 mg)を90%ギ酸(6.0 ml)中、50℃に加熱し、これに、亜鉛粉末(15 mg)を加えた。100℃で2時間撹拌し、室温に冷却後、析出した結晶を濾取し、水で洗浄後、減圧下乾燥し、白色結晶として表記化合物(470 mg, Y. 67%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 4.0 (3H, s), 7.5 (1H, d), 8.3 (1H, d), 9.4 (1H, s).
ESI/MS(m/z) : 210 (M+H) +, 208 (M-H) -.
【0060】
【中間体実施例62】
5- メトキシベンゾチアゾール -6- カルボン酸
中間体実施例61と同様の方法で、4-アミノ-2-メトキシ安息香酸(2.8 g)から表記化合物(1.3 g, Y. 38%)を得た。
ESI/MS(m/z) : 210 (M+H) +, 208 (M-H) -.
【中間体実施例63】
4- メトキシ -2- メチルベンゾチアゾール -6- カルボン酸
4-アミノ-3-メルカプト-5-メトキシ安息香酸(500 mg)をテトラヒドロフラン(15 ml)に溶解し-78℃に冷却した。無水酢酸(0.26 ml)を加え、30分かけて室温に戻し、3時間撹拌した。減圧下濃縮し、白色結晶として表記化合物(550 mg, Y. 99%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 2.8 (3H, s), 3.9 (3H, s), 7.4 (1H, s), 8.2 (1H, s).
ESI/MS(m/z) : 222 (M-H) -.
【中間体実施例64】
4- メトキシ -2- トリフルオロメチルベンゾチアゾール -6- カルボン酸
4-アミノ-3-メルカプト-5-メトキシ安息香酸(400 mg)をテトラヒドロフラン(15 ml)に溶解し-78℃に冷却した。無水トリフルオロ酢酸(0.31 ml)を加え、30分かけて室温に戻し、30分間撹拌した。減圧下濃縮し、白色結晶として表記化合物(550 mg, Y. 99%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 4.0 (3H, s), 7.6 (1H, s), 8.5 (1H, s).
【0061】
【中間体実施例65】
2- メチルベンゾチアゾール -6- カルボン酸
4-アミノ安息香酸(13 g)とチオシアン酸アンモニウム(6.9 g)をメタノール(200 ml)に懸濁し、氷浴中で-15℃に冷却した。臭素(4.7 ml)を含むメタノール溶液(40 ml)をゆっくりと滴下した。室温に戻し、2時間撹拌し、氷水(500 ml)を加え、析出した結晶を濾取し、水、n-ヘキサンで洗浄した。減圧下乾燥し白色結晶として4-アミノ-3-チオシアナト安息香酸(9.4 g, Y. 53%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 6.6 (2H, brs), 6.8 (1H, d), 7.7 (1H, dd), 7.9 (1H, d).
ESI/MS(m/z) : 193 (M-H) -.
硫化ナトリウム(25 g)を水(60 ml)とエタノール(60 ml)に懸濁し、40℃で、硫化ナトリウムが溶解したことを確認した後、上記で得られた4-アミノ-3-チオシアナト安息香酸(10 g)を加えた。90℃に昇温しそのまま2時間撹拌した。室温に戻し、90%ギ酸溶液を酸性になるまで加え、析出した結晶を濾取し、水、n-ヘキサンで洗浄した。減圧下乾燥し淡黄色結晶として4-アミノ-3-メルカプト安息香酸(8.8 g, Y. 96%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 6.6 (2H, brs), 6.8 (1H, d), 7.7 (1H, dd), 7.9 (1H, d).
ESI/MS(m/z) : 168 (M-H) -.
上記で得られた4-アミノ-3-メルカプト安息香酸(170 mg)とチオアセトアミド(83 mg)をエチレングリコール(1.5 ml)に懸濁した。濃塩酸(0.1 ml)を加え100℃で7時間撹拌した。室温に戻し、冷水を加え析出した結晶を濾取し、水、n-ヘキサンで洗浄した。減圧下乾燥し白色結晶として表記化合物(150 mg, Y. 78%)を得た。
ESI/MS(m/z) : 192 (M-H) -.
【0062】
【中間体実施例66】
4- メトキシ -2- フェニルベンゾチアゾール -6- カルボン酸
4-アミノ-3-メルカプト-5-メトキシ安息香酸(600 mg)とチオベンズアミド(450 mg)をエチレングリコール(10 ml)に懸濁した。濃塩酸(1.0 ml)を加え60℃で7時間撹拌した。室温に戻し、冷水を加え析出した結晶を濾取し、水、n-ヘキサンで洗浄した。減圧下乾燥し白色結晶として表記化合物(280 mg, Y. 32%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 4.0 (3H, s), 7.5 (1H, d), 7.5-7.6 (3H, m), 8.1-8.2 (2H, m), 8.3 (1H, d).
ESI/MS(m/z) : 284 (M-H) -.
【中間体実施例67】
2- フェニルベンゾチアゾール -6- カルボン酸
中間体実施例66と同様の方法で4-アミノ-3-メルカプト安息香酸(1.7 g)から表記化合物(1.9 g, Y. 74%)を得た。
ESI/MS(m/z) : 254 (M-H) -.
【中間体実施例68】
2- オキソ -2,3- ジヒドロベンゾチアゾール -6- カルボン酸
4-アミノ-3-メルカプト安息香酸(680 mg)をテトラヒドロフラン(20 ml)に溶解し炭酸カリウム(550 mg)を加え、室温で30分間撹拌した。-78℃に冷却し、トリホスゲン(400 mg)を加え1時間撹拌した。室温に戻し、減圧下溶媒を3分の1になるまで濃縮した。水(20 ml)とギ酸を酸性になるまで加え、析出した結晶を濾取し、水、n-ヘキサンで洗浄した。減圧下乾燥し白色結晶として表記化合物(740 mg, Y. 95%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 6.3 (1H, brs), 7.1 (1H, d), 7.8 (1H, d), 8.1 (1H, s).
ESI/MS(m/z) : 194 (M-H) -.
【0063】
【中間体実施例69】
1- メチル -1H- ベンゾイミダゾール -5- カルボン酸
4-アミノ-3-ニトロ安息香酸メチルエステル(7.0 g)、水酸化ナトリウム(5.7 g)、炭酸カリウム(4.9 g)、テトラブチルアンモニウムブロマイド(0.22 g)をトルエン(100 ml)に懸濁した。40℃で1時間撹拌した後、ジメチル硫酸(7.7 ml)を加えて2時間撹拌した。酢酸エチルで抽出し、水で洗浄後、無水硫酸ナトリウムで乾燥した。減圧下濃縮して4-メチルアミノ-3-ニトロ安息香酸メチルエステル(7.3 g, Y. 97%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 3.0 (d, 3H), 3.8 (s, 3H), 7.0 (d, 1H), 8.00 (dd, 1H), 8.5-8.7 (brs, 1H), 8.6 (d, 1H).
ESI/MS(m/z) : 325 (M+H) +, 323 (M-H) -.
上記で得られた4-メチルアミノ-3-ニトロ安息香酸メチルエステル(6.3 g)を1,4-ジオキサン(125 ml)に懸濁した。20%水酸化パラジウム(6.3 g)を加え、水素雰囲気下、室温で91時間撹拌した。不溶物を濾去した後、減圧下濃縮した。残渣をカラムクロマトグラフィー(流出溶媒;酢酸エチル:n-ヘキサン 1:4 → 2:3)に供し3-アミノ-4-メチルアミノ安息香酸メチルエステル(3.3 g, Y. 62%)を得た。
ESI/MS(m/z) : 181 (M+H) +, 179 (M-H) -.
上記で得られた3-アミノ-4-メチルアミノ安息香酸メチルエステル(3.3 g)をギ酸(96 ml)に溶解した。水(4 ml)を加えて90℃で3時間撹拌した。減圧下濃縮し、残渣に酢酸エチルを加えた。有機相を飽和炭酸水素ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥した。減圧下濃縮して1-メチル-1H-ベンゾイミダゾール-5-カルボン酸メチルエステル(3.4 g, Y. 97%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 3.8 (s, 3H), 3.8 (s, 3H), 7.6 (d, 1H), 7.9 (dd, 1H), 8.2 (d, 1H), 8.3 (s, 1H).
ESI/MS(m/z) : 191 (M+H) +.
上記で得られた1-メチル-1H-ベンゾイミダゾール-5-カルボン酸メチルエステル(500 mg)をメタノール(10 ml)に溶解した。1N水酸化ナトリウム水溶液(8 ml)を加えて室温で4時間撹拌した。水を加えた後、ギ酸で酸性にした。析出物を濾取し、減圧下乾燥して表記化合物(367 mg, Y. 79%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 3.8 (s, 3H), 7.6 (d, 1H), 7.8 (dd, 1H), 8.2 (d, 1H), 8.3 (s, 1H).
【0064】
【中間体実施例70】
2- メチルベンゾオキサゾール -6- カルボン酸
4-アミノ-3-ヒドロキシ安息香酸(4.9 g)を酢酸(250 ml)に加えて130℃で3日間撹拌した。減圧下濃縮し、析出物を濾取した。メタノールとクロロホルムに溶解した。減圧下濃縮し、析出物を濾取し、メタノールで洗浄し、減圧下乾燥して表記化合物(3.5 g, Y. 62%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 2.6 (s, 3H), 7.7 (d, 1H), 7.9 (dd, 1H), 8.1 (d, 1H).
ESI/MS(m/z) : 178 (M+H) +, 176 (M-H) -.
【中間体実施例71】
5- メチル -2,3- ジヒドロ -1H- イソインドール
4-メチルフタル酸無水物(3.0 g)、尿素(1.2 g)にキシレン(15 ml)を加え、150℃で一晩撹拌した。室温に冷却後、析出した結晶を濾取し、エタノール、水で洗浄した。減圧下乾燥し、白色結晶として4-メチルフタルイミド(2.4 g, Y. 82%)を得た。
1H NMR; (CDCl3) δ (ppm) : 2.5 (3H, s), 7.5 (1H, d), 7.6 (1H, s), 7.7 (1H, s).
上記で得られた4-メチルフタルイミド(1.8 g)をテトラヒドロフラン(3 ml)に懸濁し、1Nボラン-テトラヒドロフラン錯体(30 ml)を室温で加え、60℃で一晩撹拌した。0℃に冷却し、メタノール(2.8 ml)、6N塩酸(3.2 ml)を加え、1時間還流した。0℃に冷却し、6N水酸化ナトリウム水溶液を加え、酢酸エチルで抽出後、無水硫酸ナトリウムで乾燥した。減圧下濃縮し、残渣をカラムクロマトグラフィー(流出溶媒;ジクロロメタン→ジクロロメタン:メタノール 10:1 → 5:1)に供し表記化合物(400 mg, Y. 27%)を得た。
1H NMR; (CDCl3) δ (ppm) : 2.3 (3H, s), 2.7 (1H, brs), 7.0 (1H, d), 7.1 (1H, s), 7.2 (1H, d).
ESI/MS(m/z) : 134 (M+H) +.
【0065】
中間体実施例71の方法を参考に下記の反応式に従って化合物を合成した。合成した化合物とデータを表4に示した。(各記号は前記と同義である。)
【化16】
【表4】
【中間体実施例86】
5- メトキシ -2,3- ジヒドロ -1H- イソインドール
3,4-ジメチルアニソール(3.0 g)を四塩化炭素中、N-ブロモこはく酸イミド(7.9 g)、2,2'-アゾビスイソブチロニトリル(50 mg)を加え一晩還流した。室温に冷却後、不溶物を濾過し、濾液を減圧下濃縮した。残渣をカラムクロマトグラフィー(流出溶媒;n-ヘキサン:酢酸エチル 25:1 → 20:1)に供し、1,2-ビスブロモメチル-4-メトキシベンゼン(1.2 g, Y. 19%)を得た。
1H NMR; (CDCl3) δ (ppm) : 3.8 (3H, s), 4.6 (2H, s), 4.6 (2H, s), 6.8 (1H, dd), 6.9 (1H, d), 7.2 (1H, d).
水素化ナトリウム(0.35g)をN,N-ジメチルホルムアミド(1.2 ml)に懸濁し、p-トルエンスルホアミド(0.71 g)のN,N-ジメチルホルムアミド(2 ml)溶液を加え室温で30分間撹拌した。60℃で1時間撹拌後、上記で得られた1,2-ビスブロモメチル-4-メトキシベンゼン(1.2 g)のN,N-ジメチルホルムアミド(2 ml)溶液を60℃で加えた。室温で3時間撹拌後、酢酸エチルを加え水で洗浄した。有機相を無水硫酸ナトリウムで乾燥し、減圧下濃縮してスルホニル誘導体を得た。これをフェノール(0.54 g)、n-プロパノール(0.72 ml)、48%臭化水素酸(4.0 ml)と混合し、100℃で2時間撹拌した。冷却後、酢酸エチルで洗浄した。水相をアルカリ性にし、クロロホルムで抽出、無水硫酸ナトリウムで乾燥した。減圧下濃縮し、表記化合物(89 mg, Y. 14%)を得た。
1H NMR; (CDCl3) δ (ppm) : 3.8 (3H, s), 4.1-4.2 (4H, m), 6.7-6.8 (2H, m), 7.1-7.2 (1H, m).
【中間体実施例87】
4- メトキシ -2,3- ジヒドロ -1H- イソインドール
3,4-ジメチルアニソールから、中間体実施例86と同様の方法により、4-メトキシ-2,3-ジヒドロ-1H-イソインドールを合成した。
1H NMR; (CDCl3) δ (ppm) : 3.8 (3H, s), 4.2-4.3 (4H, m), 6.7-7.2 (3H, m).
【中間体実施例88】
2,3,4,5- テトラヒドロ -1H- ベンゾ [c] アゼピン
文献(Tetrahedoron, 1993, 49, 1807-1820)記載の方法に従い1-テトラロン(3.3 ml)から表記化合物(2.0 g, Y. 55%)を得た。
ESI/MS(m/z) : 148 (M+H) +.
【中間体実施例89】
3- アミノ -1-(1,3- ジヒドロイソインドール -2- イル )-3- メチルブタン -1- オン
2,3-ジヒドロ-1H-イソインドール(543 mg)と3-アミノ-3-メチルブタン酸(700 mg)をN,N-ジメチルホルムアミド(30 ml)に溶解した。これに0℃でN-(3-ジメチルアミノプロピル)-N’-エチルカルボジイミド塩酸塩(876 mg)とヒドロキシベンゾトリアゾール(698 mg)を加えた後、室温で一晩撹拌した。減圧下濃縮し、残渣に水と酢酸エチルを加えた。有機相を分離し、水相に飽和炭酸水素ナトリウム水溶液を加えpH9とし、酢酸エチルで抽出した。無水硫酸ナトリウムで乾燥後、減圧下濃縮し、褐色油状物として、表記化合物(0.60 g, Y. 60%) を得た。
1H NMR; (CDCl3) δ (ppm) : 1.2 (6H, s), 2.4 (2H, s), 4.7-4.8 (4H, m), 7.2-7.3 (4H, m).
ESI/MS(m/z) : 219 (M+H) +.
中間体実施例89の方法を参考に下記の反応式に従って化合物を合成した。合成した化合物とデータを表5及び表6に示した。(各記号は前記と同義である。)
【0068】
【化17】
【表6】
【中間体実施例111】
2- メチルピラゾロ [1,5-a] ピリミジン -6- カルボン酸 (2- アミノ -2- メチルプロピル ) アミド
2-メチルピラゾロ[1,5-a]ピリミジン-6-カルボン酸(0.18 g)をジクロロメタン(5 ml)に懸濁し、N,N-ジメチルホルムアミド(1 滴)を加えた。0℃に冷却し、オキサリルクロライド(10 μl)のジクロロメタン溶液(3 ml)を10分間で滴下し、そのまま0℃で1時間撹拌した。その後、室温で5時間撹拌し、酸クロライドを調整した。2-アミノ-2-メチルプロピルアミン(0.11 g)をジクロロメタンに溶解し、トリエチルアミン(0.33 ml)を加え、-78℃に冷却した。これに、調整した酸クロライド溶液を30分間で滴下し、そのまま30分間撹拌した。室温まで昇温し、室温で1時間撹拌した。水を加え2N塩酸で水相を酸性とした。クロロホルムで洗浄後、水相を5N水酸化ナトリウム水溶液でアルカリ性とし、クロロホルムで抽出した。有機相を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。減圧下濃縮し、黄色結晶として表記化合物(0.14 g, Y. 56%)を得た。
ESI/MS(m/z) : 248 (M+H) +.
【中間体実施例112】
2- メチルピラゾロ [1,5-a] ピリミジン -6- カルボン酸 (1- アミノシクロペンチルメチル ) アミド
2-メチルピラゾロ[1,5-a]ピリミジン-6-カルボン酸(0.31 g)をテトラヒドロフラン(7 ml)に懸濁し、N,N-ジメチルホルムアミド(0.04 ml)を加えた。これに、氷冷下、オキサリルクロライド(200 μl)のテトラヒドロフラン(0.8 ml)溶液を滴下し、同温で1時間撹拌後、室温で2時間撹拌した。-60℃以下で、炭酸カリウム(0.54 g)を加えた後、1-(アミノメチル)シクロペンチルアミン(0.22 g)のテトラヒドロフラン(0.8 ml)溶液を滴下した。-60℃以下で30分間撹拌後、室温で22時間撹拌した。氷浴中で水(6 ml)を加え、6N塩酸でpH2とした。クロロホルムで洗浄し、水相を5N水酸化ナトリウム水溶液でpH12とし、クロロホルムで抽出した。飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥した。減圧下濃縮し表記化合物(57 mg, Y. 12%)を得た。
1H NMR; (CDCl3) δ (ppm) : 1.4-1.8 (8H, m), 2.5 (3H, s), 3.2-3.3 (2H, m), 6.5, 8.8, 9.2 (3H, s).
ESI/MS(m/z) : 274(M+H)+, 272(M-H)-.
【0071】
【中間体実施例113】
2- メチルピラゾロ [1,5-a] ピリミジン -6- カルボン酸 (4- アミノ -4- メチルペンチル ) アミド
2-メチルピラゾロ[1,5-a]ピリミジン-6-カルボン酸(177 mg)をテトラヒドロフラン(5 ml)に懸濁し、N,N-ジメチルホルムアミド(1 滴)を加えた。氷冷下、オキサリルクロライド(100 μl)を加え、室温で30分撹拌した。再び、氷冷し、4-メチル-1,4-ペンタンジアミン(116 μl)とトリエチルアミン(0.21 ml)を加え室温で一晩撹拌した。水、2N塩酸を加え酸性とし、クロロホルムで洗浄した。水相を5N水酸化ナトリウム水溶液でアルカリ性とし、クロロホルムで抽出、無水硫酸ナトリウムで乾燥した。減圧下濃縮し、淡黄色結晶として表記化合物(151 mg, Y. 55%)を得た。
1H NMR; (CDCl3) δ (ppm) : 1.1 (6H, s), 1.7 (4H, m), 2.5 (3H, s), 3.4 (2H, dd), 6.5 (1H, s), 8.4 (1H, brs), 8.7 (1H, d), 9.1 (1H, d).
ESI/MS(m/z) : 276(M+H)+, 274(M-H)-.
【中間体実施例114】
2- アミノ -3-[( ベンゾチアゾール -6- カルボニル ) アミノ ] プロピオン酸メチルエステル
ベンゾチアゾール-6-カルボン酸(358 mg)、N-(3-ジメチルアミノプロピル)-N’-エチルカルボジイミド塩酸塩(382 mg)、ヒドロキシベンゾトリアゾール(306 mg)をN,N-ジメチルホルムアミド(10 ml)に溶解し、氷冷下、30分間撹拌した。3-アミノ-2-t-ブトキシカルボニルアミノプロピオン酸メチルエステル(560 mg)のN,N-ジメチルホルムアミド(8 ml)溶液を加え、氷冷〜室温で17時間撹拌した。減圧下濃縮後、水、酢酸エチルを加え有機相を抽出した。10%クエン酸水溶液、4%炭酸水素ナトリウム水溶液、水で洗浄後、無水硫酸ナトリウムで乾燥した。減圧下濃縮し3-[(ベンゾチアゾール-6-カルボニル)アミノ]-2-t-ブトキシカルボニルアミノプロピオン酸メチルエステル(750 mg, Y. 98.8%)を得た。
上記で得られた3-[(ベンゾチアゾール-6-カルボニル)アミノ]-2-t-ブトキシカルボニルアミノプロピオン酸メチルエステル(730 mg)を、氷冷したトリフルオロ酢酸(6 ml)に加え、1時間撹拌した。減圧下濃縮後、氷冷下、エーテルを加えて析出した結晶化を濾取し、減圧下乾燥し表記化合物(817 mg, Y. quant.)を得た。
ESI/MS(m/z) : 394(M+H)+.
【0072】
【中間体実施例115】
3- アミノ -1-(1,3- ジヒドロイソインドール -2- イル ) プロパン -1- オン
3-t-ブトキシカルボニルアミノプロピオン酸(1.90 g)のN,N-ジメチルホルムアミド溶液に、0℃でN-(3-ジメチルアミノプロピル)-N’-エチルカルボジイミド塩酸塩(1.94 g)を加えた。2,3-ジヒドロ-1H-イソインドール(1.00 g)のN,N-ジメチルホルムアミド溶液を加えた。室温に戻し、一晩撹拌した。減圧下濃縮し、残渣に水、ジクロロメタンを加えた。有機相を分取し、10%クエン酸水溶液、4%炭酸水素ナトリウム水溶液、飽和食塩水で洗浄した。無水硫酸ナトリウムで乾燥し、減圧下濃縮した。エーテルを加えて析出した結晶を濾取し、減圧下乾燥して淡橙色結晶として[3-(1,3-ジヒドロイソインドール)-3-オキソプロピル]カルバミン酸-t-ブチルエステル(1.33 g, Y. 55%)を得た。
上記で得られた[3-(1,3-ジヒドロイソインドリル)-3-オキソプロピル]カルバミン酸-t-ブチルエステル(1.33 g)を、氷冷したトリフルオロ酢酸(6 ml)に加え、そのまま30分間撹拌した。減圧下濃縮し、残渣にエーテルを加え、析出した結晶を濾取し、減圧下乾燥して、表記化合物(1.38 g, Y. 99%)を得た。
ESI/MS(m/z) : 191(M+H)+.
中間体実施例115の方法を参考に下記の反応式に従って化合物を合成した。合成した化合物とデータを表7に示した。(各記号は前記と同義である。)
【0073】
【化18】
【中間体実施例120】
3- アミノ -1- インドール -1- イルプロパン -1- オン
中間体実施例117の中間体として得られた[3-(2,3-ジヒドロインドール-1-イル)-3-オキソプロピル]カルバミン酸-t-ブチルエステル(290 mg)、 2,3-ジクロロ-5,6-ジシアノ-p-ベンゾキノン(510 mg)をクロロホルム(40 ml)に懸濁し、30時間還流した。室温に冷却後、不溶物を濾去し、濾液を水で洗浄後、有機相を無水硫酸ナトリウムで乾燥した。減圧下濃縮し、残渣をカラムクロマトグラフィー(流出溶媒;ジクロロメタン→ジクロロメタン:メタノール 10:1)に供し(3-インドール-1-イル-3-オキソプロピル)カルバミン酸-t-ブチルエステル(270 mg, Y. 95%)を得た。
ESI/MS(m/z) : 289 (M+H) +, 287 (M-H) -.
上記で得られた(3-インドール-1-イル-3-オキソプロピル)カルバミン酸-t-ブチルエステル(260 mg)を、氷冷したトリフルオロ酢酸(2.0 ml)に加え、そのまま1時間撹拌した。減圧下濃縮し、残渣にエーテルを加え析出した白色結晶を濾取した。減圧下乾燥して表記化合物のトリフルオロ酢酸塩(260 g, Y. 94%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 3.2-3.3 (2H, m), 3.4 (2H, t), 6.8 (1H, d), 7.2 (1H, t), 7.3 (1H, t), 7.6 (1H, d), 7.8 (3H, brs), 7.9 (1H, d), 8.3 (1H, d).
ESI/MS(m/z) : 189 (M+H) +, 187 (M-H) -.
【0075】
【中間体実施例121】
1,3- ジメチル -1H- ピラゾロ [3,4-b] ピリジン -5- カルボン酸 (2- アミノエチル ) アミド
1,3-ジメチル-1H-ピラゾロ[3,4-b]ピリジン-5-カルボン酸(4.00 g)のN,N-ジメチルホルムアミド(40 ml)溶液に氷冷下、ヒドロキシベンゾトリアゾール(3.55 g)、N-(3-ジメチルアミノプロピル)-N’-エチルカルボジイミド塩酸塩(4.45 g)を加えた。室温で30分間撹拌後、 (2-アミノエチル)カルバミン酸-t-ブチルエステル(3.65 ml)を加え室温で一晩撹拌した。減圧下濃縮し、残渣に酢酸エチル及び水を加えた。有機相を 10%クエン酸水溶液、4%炭酸水素ナトリウム水溶液、飽和食塩水で洗浄した。無水硫酸ナトリウムで乾燥後、減圧下濃縮し、無色結晶として{2-[(1,3-ジメチル-1H-ピラゾロ[3,4-b]ピリジン-5-カルボニル)アミノ]エチル}カルバミン酸-t-ブチルエステル(4.02 g, Y. 57%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 1.4 (9H, s), 2.5 (3H, s), 3.4-3.6 (4H, m), 4.1 (3H, s H), 5.0, 7.5 (2H, brs), 8.4 (1H, s), 9.0 (1H, s).
上記で得られた{2-[(1,3-ジメチル-1H-ピラゾロ[3,4-b]ピリジン-5-カルボニル)アミノ]エチル}カルバミン酸-t-ブチルエステル(4.02 g)を、氷冷したトリフルオロ酢酸(20 ml)に加え、そのまま2時間撹拌した。減圧下濃縮し、エーテルを加え析出した結晶を濾取した。減圧下乾燥し、淡黄色結晶として表記化合物(3.52 g, Y. 84%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 2.5 (3H, s), 3.0-3.1 (2H, m), 3.5-3.6 (2H, m), 4.0 (3H, s H), 7.8 (3H, brs), 8.7 (1H, s), 8.8 (1H, brt), 9.0 (1H, s).
中間体実施例121の方法を参考に下記の反応式に従って化合物を合成した。合成した化合物とデータを表8に示した。(各記号は前記と同義である。)
【0076】
【化19】
【中間体実施例138】
{t- ブトキシカルボニル -[2-(1,3- ジヒドロイソインドール -2- イル )-2- オキソエチル ] アミノ } 酢酸
Bocイミジン酢酸(580 mg)をN,N-ジメチルホルムアミド(3.5 ml)に溶解し、N-(3-ジメチルアミノプロピル)-N’-エチルカルボジイミド塩酸塩(480 mg)を加え室温で1時間撹拌した。2,3-ジヒドロ-1H-イソインドール(280 μl)を加えた後、室温で一晩撹拌した。減圧下濃縮し、残渣に10%クエン酸水溶液、酢酸エチルを加えた。有機相を分取し、4%炭酸水素ナトリウム水溶液、飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥した。減圧下濃縮し、残渣をカラムクロマトグラフィー(流出溶媒;ジクロロメタン:メタノール 20:1→10:1)に供し表記化合物(270 mg, Y. 33%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 1.4 (9H, s), 3.9 (2H, s), 4.2 (2H, s), 4.8 (4H, d), 7.2-7.3 (4H, m).
ESI/MS(m/z) : 335(M+H)+, 333(M-H) -.
中間体実施例138の方法を参考に下記の反応式に従って化合物を合成した。合成した化合物とデータを表9に示した。(各記号は前記と同義である。)
【0078】
【化20】
【実施例1】
(S)-2,7- ジメチルピラゾロ [1,5-a] ピリミジン -6- カルボン酸 {2-[(2- シアノピロリジン -1- イル )-2- オキソエチルアミノ ]-2- メチルプロピル } アミド
2,7-ジメチルピラゾロ[1,5-a]ピリミジン-6-カルボン酸(1.00 g)のテトラヒドロフラン(30 ml)溶液に、N,N’-カルボニルジイミダゾール(930 mg)を加え室温で4時間撹拌した。これを、氷冷下、(S)-1-[(2-アミノ-1,1-ジメチルエチル)アミノアセチル]ピロリジン-2-カルボニトリル2塩酸塩(1.56 g)、トリエチルアミン(3.6 ml)のテトラヒドロフラン(30 ml)溶液にゆっくり滴下した。室温に戻し、一晩撹拌した。減圧下濃縮し、残渣にジクロロメタンを加えた。不溶物を濾去し、濾液を減圧下濃縮した。残渣をカラムクロマトグラフィー(流出溶媒;ジクロロメタン:メタノール 50:1)に供し表記化合物(690 mg, Y. 33%)を得た。得られた化合物(690 mg)の1,4-ジオキサン(5.0 ml)溶液に4N塩酸/1,4-ジオキサン(0.50 ml)を10℃で加え、10分間撹拌した。エーテルを加え析出した結晶を濾取した。減圧下乾燥し、黄色結晶として 表記化合物の塩酸塩(670 mg, Y. 90%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 1.37 (6H, s), 2.05-2.31 (4H, m), 2.47 (3H, s), 2.87 (3H, s), 3.30-3.80 (4H, m) 4.10-4.30 (2H, m), 4.84-4.86 (1H, m), 6.60 (1H, s), 8.68 (1H, s), 8.93-8.97 (3H, m).
実施例1の方法を参考に、下記の反応式に従って化合物を合成した。合成した化合物とデータを表10〜表17に示した。
【0080】
【化21】
【表11】
【表12】
【表13】
【表14】
【表15】
【表16】
【表17】
【実施例60】
(S)-2- メチルピラゾロ [1,5-a] ピリミジン -6- カルボン酸 {2-[2-(2- シアノピロリジン -1- イル )-2- オキソエチルアミノ ]-2- メチルプロピル } メチルアミド
実施例1の方法を参考に、2-メチルピラゾロ[1,5-a]ピリミジン-6-カルボン酸(354 mg)と(S)-1-[2-(1,1-ジメチル-2-メチルアミノエチルアミノ)アセチル]ピロリジン-2-カルボニトリル(450 mg)から表記化合物(210 mg, Y. 28%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 1.36 (6H, s), 1.98 (1H, brs), 2.00-2.30 (4H, m), 2.50 (3H, s), 2.90 (3H, s), 3.30-3.80 (4H, m), 4.10-4.30 (2H, m), 4.80 (1H, m), 6.63 (1H, s), 8.80 (1H, s), 9.50 (1H, s).
ESI/MS(m/z) : 398(M+H)+, 396(M-H) -.
【0089】
【実施例61】
(S)-1-{2-[3-(1,3- ジヒドロイソインドール -2- イル )-1,1- ジメチル -3- オキソプロピルアミノ ] アセチル } ピロリジン -2- カルボニトリル
3-アミノ-1-(1,3-ジヒドロイソインドール-2-イル)-3-メチルブタン-1-オン(0.55 g)のアセトン溶液に、炭酸カリウム(370 mg)とヨウ化ナトリウム(200 mg)を加えた。これに、氷冷下、(S)-1-(2’-クロロアセチル)ピロリジン-2-カルボニトリル(467 mg)を加え室温で8時間撹拌した。ジクロロメタンを加え不溶物を濾去し、濾液を減圧下濃縮した。残渣をカラムクロマトグラフィー(流出溶媒;ジクロロメタン:メタノール 20:1)に供し、表記化合物(0.54 g, 61%)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 1.39, 1.40 (6H, 2s), 2.00-2.25 (4H, m), 2.85-2.95 (2H, m), 3.30-4.10 (4H, m), 4.71, 4.90 (4H, 2s), 4.85-4.90 (1H, m), 7.30-7.40 (4H, m).
ESI/MS(m/z) : 355(M+H)+.
実施例61の方法を参考に、下記の反応式に従って化合物を合成した。合成した化合物とデータを表18〜表22に示した。(各記号は前記と同義である。)
【化22】
【表18】
【表19】
【表20】
【表21】
【表22】
【実施例142】
(S)-1-{2-[2-(1,3- ジヒドロイソインドール -2- イル )-2- オキソエチルアミノ ] アセチル } ピロリジン -2- カルボニトリル
{t-ブトキシカルボニル-[2-(1,3-ジヒドロイソインドール-2-イル)-2-オキソエチル]アミノ}酢酸(260 mg)、N-(3-ジメチルアミノプロピル)-N’-エチルカルボジイミド塩酸塩(150 mg)、ヒドロキシベンゾトリアゾール(120 mg)をN,N-ジメチルホルムアミド(5.0 ml)に溶解した。これに、トリエチルアミン(110 μl)、(S)-ピロリジン-2-カルボニトリル塩酸塩(100 mg)を加えた後、室温で21時間撹拌した。減圧下濃縮後、残渣に酢酸エチル、10%クエン酸水溶液を加え有機相を分取した。有機相を4%炭酸水素ナトリウム水溶液、飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥した。減圧下濃縮し、残渣をカラムクロマトグラフィー(流出溶媒;ジクロロメタン:メタノール 20:1)に供し、(S)-[2-(シアノピロリジン-1-イル)-2-オキソエチル]-[2-(1,3-ジヒドロイソインドール-2-イル)-2-オキソエチル]カルバミン酸-t-ブチルエステル(290 mg, Y. 90%)を得た。
ESI/MS(m/z) : 413(M+H)+, 411(M-H)-.
上記で得られた(S)-[2-(シアノピロリジン-1-イル)-2-オキソエチル]-[2-(1,3-ジヒドロイソインドール-2-イル)-2-オキソエチル]カルバミン酸-t-ブチルエステル(280 mg)を1,4-ジオキサン(1.0 ml)に溶解し氷冷下、4N塩酸/1,4-ジオキサン(1.0 ml)を加え、30分間撹拌した。エーテルを加え析出した結晶を濾取し、減圧下乾燥して、表記化合物の塩酸塩(240 mg, Y. quant.)を得た。
1H NMR; (DMSO-d6) δ (ppm) : 2.03-2.19 (4H, m), 3.36-3.44 (2H, m), 3.57, 4.10 (4H, 2s), 4.74, 4.84 (4H, 2s), 4.86-4.88 (1H, m), 7.32-7.39 (4H, m).
ESI/MS(m/z) : 313(M+H)+, 311(M-H)-.
実施例142の方法を参考に、下記の反応式に従って化合物を合成した。合成した化合物とデータを表23〜表27に示した。
【0096】
【化23】
【表24】
【表25】
【表26】
【表27】
【薬理試験例1】
DPP-IV阻害剤のスクリーニングには、グリシル-プロリン-4-メチルクマリル-7-アミド(Gly-Pro-MCA)を基質とした以下の方法を用いた。
96ウエルマイクロタイタープレートに、測定緩衝液(塩化ナトリウム(140 mM)、塩化カルウム(10 mM)、1%ウシ血清アルブミンを含むトリス塩酸緩衝液(25 mM) pH7.4)に溶解した、種々の濃度の被検物質(40 μl)および150 μM Gly-Pro-MCA基質(40 μl)を加え、混和後室温に5分間静置した。その後、測定緩衝液で30倍希釈したヒト血漿(20 μl)を加えて撹拌し、暗所にて室温下30分間反応させた。1 M 酢酸緩衝液pH4.2を(100 μl)加えて反応を停止し、DPP-IV活性により遊離したMCAを360nmの励起で得られる465nmの蛍光を測定した。以下の式に従って算出されるDPP-IV活性をもとに、被検物質がDPP-IV活性を50%阻害する濃度(IC50)を求めた。結果を表28に示す。尚、比較薬剤としては、特許(WO97/40832)記載のイソロイシルチアゾリジド(化合物A)を用いた。
DPP-IV阻害活性=100×(1-(Fs-Fb)/F100-Fb)
F100:血漿での反応で得られる蛍光強度
Fb :反応停止液を加えて反応を行った場合のブランク蛍光強度
Fs :被検物質を加えて得られる蛍光強度
【0102】
【表28】
【0103】
【薬理試験例2】
Wistar/ST系雄性ラット((株)日本エスエルシー)を用い5日間以上馴化後(使用時8週令)、一夜絶食した。実施例1の化合物(3 mg/kg)、実施例61の化合物(1 mg/kg)及び化合物A(10 mg/kg)を(5 ml/kg)の割合で経口投与し、30分後に20%グルコース溶液(5 ml/kg)(グルコース(1 g/kg)に相当)で経口投与した。経時的に尾先端部より採血・血漿分離し、血中グルコースおよびインスリン濃度を測定した。血中グルコース濃度はグルテスト((株)三和化学研究所)で、血漿中インスリン濃度は市販のEAIキット((株)シバヤギ)を用いて測定した。
結果を表29に示す。ただし、血中グルコース濃度は、糖投与から60分後までの各採血時間の血糖値から曲線下面積(min・mg/dl)を算出したものを示し(ただし、0分の血糖値として試験開始前の採血で得られた試料の血糖値を代用した。)、血漿中インスリン濃度は化合物投与10分後の血漿中インスリン濃度(pg/ml)で示した。
【0104】
【表29】
以上に示してきたように、本発明化合物は強力なDPP-IV阻害活性を示し、化学的に安定であり、酵素選択性に優れた副作用等のない安全な化合物であるので、糖尿病(特に2型糖尿病)、これに付随する合併症、及び肥満等の治療に有用である。
Claims (9)
- 一般式(I)で表される化合物、又はその医薬的に許容される塩である化合物。
- 一般式(I)中の、R1、R2がメチル基である、請求項1に記載の化合物。
- 一般式(I)中のDが-CO-であり、Aが一般式( II )で表される6-5系のニ環式複素環基である、請求項1又は2に記載の化合物。
- 一般式(I)中のDが-CONH-であり、Aが一般式( III )で表される6-5系のニ環式複素環基である、請求項1又は2に記載の化合物。
- 式( III )中のyが窒素原子、z、v、wが炭素原子である、請求項4に記載の化合物。
- v、w、yが窒素原子、zが炭素原子である、請求項4に記載の化合物。
- 請求項1〜6のいずれかに記載の化合物を有効成分とする、ジペプチジルペプチダーゼIV活性の阻害剤。
- 糖尿病の治療のための、請求項7に記載のジペプチジルペプチダーゼIV活性の阻害剤。
- 請求項1〜6のいずれかに記載の化合物を有効成分として含有する医薬組成物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003023077 | 2003-01-31 | ||
| JP2003023077 | 2003-01-31 | ||
| PCT/JP2004/000886 WO2004067509A1 (ja) | 2003-01-31 | 2004-01-30 | ジペプチジルペプチダーゼivを阻害する化合物 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPWO2004067509A1 JPWO2004067509A1 (ja) | 2006-05-18 |
| JP4184378B2 true JP4184378B2 (ja) | 2008-11-19 |
Family
ID=32820710
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2005504761A Expired - Lifetime JP4184378B2 (ja) | 2003-01-31 | 2004-01-30 | ジペプチジルペプチダーゼivを阻害する化合物 |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US7345180B2 (ja) |
| EP (1) | EP1595866B1 (ja) |
| JP (1) | JP4184378B2 (ja) |
| KR (1) | KR100998796B1 (ja) |
| CN (1) | CN100434420C (ja) |
| AU (1) | AU2004207731B2 (ja) |
| CA (1) | CA2514191C (ja) |
| DK (1) | DK1595866T3 (ja) |
| ES (1) | ES2587885T3 (ja) |
| PT (1) | PT1595866T (ja) |
| WO (1) | WO2004067509A1 (ja) |
Families Citing this family (82)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2361862C2 (ru) * | 2003-12-29 | 2009-07-20 | Сепракор Инк. | Пиррольные и пиразольные ингибиторы daao |
| CN1942186B (zh) | 2004-03-09 | 2010-10-06 | 国家卫生研究院 | 吡咯烷化合物 |
| JP2008507541A (ja) | 2004-07-23 | 2008-03-13 | ロイヤルティ,スーザン・マリー | ペプチダーゼ阻害剤 |
| DOP2006000008A (es) | 2005-01-10 | 2006-08-31 | Arena Pharm Inc | Terapia combinada para el tratamiento de la diabetes y afecciones relacionadas y para el tratamiento de afecciones que mejoran mediante un incremento de la concentración sanguínea de glp-1 |
| CN101553217A (zh) | 2005-07-06 | 2009-10-07 | 塞普拉科公司 | 艾司佐匹克隆与反式4-(3,4-二氯苯基)-1,2,3,4-四氢-n-甲基-1-萘胺或反式4-(3,4-二氯苯基)-1,2,3,4-四氢-1-萘胺的组合以及治疗绝经期和心境障碍、焦虑症和认知障碍的方法 |
| WO2007033350A1 (en) | 2005-09-14 | 2007-03-22 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors for treating diabetes |
| EP1924567B1 (en) | 2005-09-16 | 2012-08-22 | Takeda Pharmaceutical Company Limited | Process for the preparation of pyrimidinedione derivatives |
| NZ569608A (en) * | 2006-01-06 | 2011-09-30 | Sepracor Inc | Tetralone-based monoamine reuptake inhibitors |
| AU2007205114B2 (en) * | 2006-01-06 | 2012-11-08 | Sunovion Pharmaceuticals Inc. | Cycloalkylamines as monoamine reuptake inhibitors |
| WO2007112347A1 (en) | 2006-03-28 | 2007-10-04 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| CN101421228B (zh) | 2006-03-31 | 2014-05-21 | 塞普拉柯公司 | 手性酰胺和胺的制备 |
| PE20071221A1 (es) | 2006-04-11 | 2007-12-14 | Arena Pharm Inc | Agonistas del receptor gpr119 en metodos para aumentar la masa osea y para tratar la osteoporosis y otras afecciones caracterizadas por masa osea baja, y la terapia combinada relacionada a estos agonistas |
| US7579370B2 (en) * | 2006-06-30 | 2009-08-25 | Sepracor Inc. | Fused heterocycles |
| US7884124B2 (en) * | 2006-06-30 | 2011-02-08 | Sepracor Inc. | Fluoro-substituted inhibitors of D-amino acid oxidase |
| US20080082066A1 (en) * | 2006-10-02 | 2008-04-03 | Weyerhaeuser Co. | Crosslinked carboxyalkyl cellulose fibers having non-permanent and temporary crosslinks |
| TW200838536A (en) | 2006-11-29 | 2008-10-01 | Takeda Pharmaceutical | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| US20090099248A1 (en) * | 2007-01-18 | 2009-04-16 | Sepracor Inc. | Inhibitors of d-amino acid oxidase |
| US7902252B2 (en) * | 2007-01-18 | 2011-03-08 | Sepracor, Inc. | Inhibitors of D-amino acid oxidase |
| EP2143443B1 (en) | 2007-04-03 | 2014-11-19 | Mitsubishi Tanabe Pharma Corporation | A combination of dipeptidyl peptidase iv inhibitor and sweetener for use in the treatment of obesity |
| KR101581289B1 (ko) | 2007-05-31 | 2015-12-31 | 세프라코 아이엔시. | 모노아민 재흡수 억제제로서 페닐 치환된 시클로알킬아민 |
| CL2008003653A1 (es) | 2008-01-17 | 2010-03-05 | Mitsubishi Tanabe Pharma Corp | Uso de un inhibidor de sglt derivado de glucopiranosilo y un inhibidor de dppiv seleccionado para tratar la diabetes; y composicion farmaceutica. |
| NZ587911A (en) | 2008-03-05 | 2012-02-24 | Nat Health Research Institutes | Pyrrolidine derivatives |
| EP2146210A1 (en) | 2008-04-07 | 2010-01-20 | Arena Pharmaceuticals, Inc. | Methods of using A G protein-coupled receptor to identify peptide YY (PYY) secretagogues and compounds useful in the treatment of conditions modulated by PYY |
| US8633199B2 (en) * | 2008-05-14 | 2014-01-21 | Sanwa Kagaku Kenkyusho Co., Ltd. | Medicine consisting of concomitant use or combination of DPP-IV inhibitor and other diabetic medicine |
| WO2010017418A1 (en) * | 2008-08-07 | 2010-02-11 | Sepracor Inc. | Prodrugs of fused heterocyclic inhibitors of d-amino acid oxidase |
| CZ2008512A3 (cs) * | 2008-08-26 | 2010-03-10 | Zentiva, A. S | Zpusob prípravy vysoce cistého vildagliptinu |
| WO2010047982A1 (en) | 2008-10-22 | 2010-04-29 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| CA2741672A1 (en) | 2008-10-31 | 2010-05-06 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| AR077642A1 (es) | 2009-07-09 | 2011-09-14 | Arena Pharm Inc | Moduladores del metabolismo y el tratamiento de trastornos relacionados con el mismo |
| WO2011017634A2 (en) * | 2009-08-07 | 2011-02-10 | Sepracore Inc. | Prodrugs of fused heterocyclic inhibitors of d-amino acid oxidase |
| JPWO2011055770A1 (ja) * | 2009-11-06 | 2013-03-28 | 武田薬品工業株式会社 | 縮合複素環化合物 |
| EP3646859A1 (en) | 2009-11-27 | 2020-05-06 | Boehringer Ingelheim International GmbH | Treatment of genotyped diabetic patients with dpp-iv inhibitors such as linagliptin |
| WO2011106273A1 (en) | 2010-02-25 | 2011-09-01 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| US20130109703A1 (en) | 2010-03-18 | 2013-05-02 | Boehringer Ingelheim International Gmbh | Combination of a GPR119 Agonist and the DPP-IV Inhibitor Linagliptin for Use in the Treatment of Diabetes and Related Conditions |
| CN102918027A (zh) | 2010-04-06 | 2013-02-06 | 艾尼纳制药公司 | Gpr119受体调节剂和对与所述受体有关的障碍的治疗 |
| AU2011249722B2 (en) | 2010-05-05 | 2015-09-17 | Boehringer Ingelheim International Gmbh | Combination therapy |
| BR112012032579B1 (pt) | 2010-06-24 | 2021-05-11 | Boehringer Ingelheim International Gmbh | uso de linagliptina e composição farmacêutica compreendendo linagliptina e insulina basal de longa duração |
| AU2011305525B2 (en) | 2010-09-22 | 2016-08-18 | Arena Pharmaceuticals, Inc. | Modulators of the GPR119 receptor and the treatment of disorders related thereto |
| CN103313968A (zh) | 2010-11-15 | 2013-09-18 | Abbvie公司 | Nampt和rock抑制剂 |
| EA025380B1 (ru) | 2011-02-25 | 2016-12-30 | Мерк Шарп Энд Домэ Корп. | Новые циклические производные азабензимидазола, используемые в качестве антидиабетических агентов |
| US20140018371A1 (en) | 2011-04-01 | 2014-01-16 | Arena Pharmaceuticals, Inc. | Modulators Of The GPR119 Receptor And The Treatment Of Disorders Related Thereto |
| US20140066369A1 (en) | 2011-04-19 | 2014-03-06 | Arena Pharmaceuticals, Inc. | Modulators Of The GPR119 Receptor And The Treatment Of Disorders Related Thereto |
| WO2012145603A1 (en) | 2011-04-22 | 2012-10-26 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| WO2012145604A1 (en) | 2011-04-22 | 2012-10-26 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| WO2012170554A1 (en) | 2011-06-06 | 2012-12-13 | Theodore Mark Kamenecka | N-biphenylmethylindole modulators of pparg |
| WO2012170561A1 (en) * | 2011-06-06 | 2012-12-13 | The Scripps Research Institute (T.S.R.I.) | N-benzylindole modulators of pparg |
| WO2012170702A1 (en) | 2011-06-08 | 2012-12-13 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| WO2013055910A1 (en) | 2011-10-12 | 2013-04-18 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| WO2013078240A1 (en) | 2011-11-22 | 2013-05-30 | Ripka Amy S | N-biphenylmethylbenzimidazole modulators of pparg |
| WO2013174767A1 (en) | 2012-05-24 | 2013-11-28 | Boehringer Ingelheim International Gmbh | A xanthine derivative as dpp -4 inhibitor for use in modifying food intake and regulating food preference |
| EP2865666B1 (en) | 2012-06-25 | 2017-05-31 | Sunshine Lake Pharma Co., Ltd. | Hexahydropentaleno derivatives, preparation method and use in medicine thereof |
| KR20150036245A (ko) | 2012-08-02 | 2015-04-07 | 머크 샤프 앤드 돔 코포레이션 | 항당뇨병 트리시클릭 화합물 |
| TW201412745A (zh) | 2012-08-28 | 2014-04-01 | Sanwa Kagaku Kenkyusho Co | N-[2-({2-[(2S)-2-氰基吡咯啶-1-基]-2-側氧乙基}胺基)-2-甲基丙基]-2-甲基吡唑并[1,5-a]嘧啶-6-甲醯胺之結晶 |
| WO2014034871A1 (ja) * | 2012-08-30 | 2014-03-06 | 株式会社 三和化学研究所 | 脂質異常症の予防又は治療薬 |
| JP6227352B2 (ja) * | 2012-09-27 | 2017-11-08 | 株式会社三和化学研究所 | アナグリプチン又はその塩を含有する固形製剤 |
| JP6309895B2 (ja) * | 2012-09-27 | 2018-04-11 | 株式会社三和化学研究所 | アナグリプチン含有製剤 |
| JP6283314B2 (ja) * | 2012-09-27 | 2018-02-21 | 株式会社三和化学研究所 | アナグリプチン含有固形製剤 |
| WO2014051024A1 (ja) * | 2012-09-27 | 2014-04-03 | 株式会社 三和化学研究所 | アナグリプチン含有医薬組成物 |
| JP6283316B2 (ja) * | 2012-10-26 | 2018-02-21 | 株式会社三和化学研究所 | アナグリプチン含有固形製剤 |
| WO2014074668A1 (en) | 2012-11-08 | 2014-05-15 | Arena Pharmaceuticals, Inc. | Modulators of gpr119 and the treatment of disorders related thereto |
| RU2015140066A (ru) | 2013-02-22 | 2017-03-30 | Мерк Шарп И Доум Корп. | Противодиабетические бициклические соединения |
| WO2014139388A1 (en) | 2013-03-14 | 2014-09-18 | Merck Sharp & Dohme Corp. | Novel indole derivatives useful as anti-diabetic agents |
| IN2013MU00848A (ja) * | 2013-03-19 | 2015-05-01 | Glenmark Generics Ltd | |
| WO2015051496A1 (en) | 2013-10-08 | 2015-04-16 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| CN103626775B (zh) * | 2013-12-02 | 2015-05-20 | 南京华威医药科技开发有限公司 | 具有二嗪结构的dpp-4抑制剂 |
| WO2015104602A2 (en) * | 2014-01-08 | 2015-07-16 | Wockhardt Limited | A process for the preparation of anagliptin and its intermediates thereof |
| IN2014MU01191A (ja) | 2014-03-29 | 2015-10-02 | Wockhardt Ltd | |
| WO2015152319A1 (ja) * | 2014-04-02 | 2015-10-08 | 株式会社 三和化学研究所 | 5,7-無置換-ピラゾロ[1,5-a]ピリミジン-6-カルボン酸の製造方法 |
| CN104974159A (zh) * | 2014-04-08 | 2015-10-14 | 辰欣药业股份有限公司 | 阿纳列汀的制备方法 |
| CN104974160B (zh) * | 2014-04-08 | 2017-12-05 | 辰欣药业股份有限公司 | 2‑甲基吡唑并[1,5‑a]嘧啶‑6‑羧酸衍生物及其应用 |
| US10016394B2 (en) | 2014-04-16 | 2018-07-10 | The Scripps Research Institute | PPARG modulators for treatment of osteoporosis |
| WO2016012927A1 (en) | 2014-07-23 | 2016-01-28 | Lupin Limited | A process for preparation of anagliptin hydrochloride |
| GB201415598D0 (en) | 2014-09-03 | 2014-10-15 | Univ Birmingham | Elavated Itercranial Pressure Treatment |
| CA2979033A1 (en) | 2015-03-09 | 2016-09-15 | Intekrin Therapeutics, Inc. | Methods for the treatment of nonalcoholic fatty liver disease and/or lipodystrophy |
| WO2016175092A1 (ja) * | 2015-04-27 | 2016-11-03 | 株式会社 三和化学研究所 | 肝細胞癌の予防又は治療のための医薬 |
| WO2016193988A1 (en) * | 2015-05-29 | 2016-12-08 | Harman Finochem Limited | An advantageous process for preparing anagliptin and its novel crystalline form-h |
| US11072602B2 (en) | 2016-12-06 | 2021-07-27 | Merck Sharp & Dohme Corp. | Antidiabetic heterocyclic compounds |
| US10968232B2 (en) | 2016-12-20 | 2021-04-06 | Merck Sharp & Dohme Corp. | Antidiabetic spirochroman compounds |
| CN110996951A (zh) | 2017-04-03 | 2020-04-10 | 科赫罗斯生物科学股份有限公司 | 治疗进行性核上性麻痹的PPARγ激动剂 |
| WO2021198981A1 (en) | 2020-04-01 | 2021-10-07 | Janssen Biopharma, Inc. | Antiviral compounds and uses thereof |
| IL303655A (en) | 2020-12-17 | 2023-08-01 | Astrazeneca Ab | N-(2-(4-cyanothiazolidin-3-yl)-2-oxoethyl)- quinoline-4-carboxamides |
| EP4359384A1 (en) | 2021-06-23 | 2024-05-01 | Orion Corporation | Process for the preparation of a cyp11a1 inhibitor and intermediates thereof |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IE54551B1 (en) * | 1982-01-22 | 1989-11-22 | Ici Plc | Amide derivatives |
| US5416093A (en) * | 1991-11-12 | 1995-05-16 | Eli Lilly And Company | Antithrombotic agents |
| IL111785A0 (en) * | 1993-12-03 | 1995-01-24 | Ferring Bv | Dp-iv inhibitors and pharmaceutical compositions containing them |
| DE122010000020I1 (de) | 1996-04-25 | 2010-07-08 | Prosidion Ltd | Verfahren zur Senkung des Blutglukosespiegels in Säugern |
| TW492957B (en) | 1996-11-07 | 2002-07-01 | Novartis Ag | N-substituted 2-cyanopyrrolidnes |
| US6011155A (en) * | 1996-11-07 | 2000-01-04 | Novartis Ag | N-(substituted glycyl)-2-cyanopyrrolidines, pharmaceutical compositions containing them and their use in inhibiting dipeptidyl peptidase-IV |
| TW583185B (en) | 2000-06-13 | 2004-04-11 | Novartis Ag | N-(substituted glycyl)-2-cyanopyrrolidines and pharmaceutical composition for inhibiting dipeptidyl peptidase-IV (DPP-IV) or for the prevention or treatment of diseases or conditions associated with elevated levels of DPP-IV comprising the same |
| US6432969B1 (en) * | 2000-06-13 | 2002-08-13 | Novartis Ag | N-(substituted glycyl)-2 cyanopyrrolidines, pharmaceutical compositions containing them and their use in inhibiting dipeptidyl peptidase-IV |
| JPWO2002051836A1 (ja) | 2000-12-27 | 2004-04-22 | 協和醗酵工業株式会社 | ジペプチジルペプチダーゼ−iv阻害剤 |
| US6861440B2 (en) * | 2001-10-26 | 2005-03-01 | Hoffmann-La Roche Inc. | DPP IV inhibitors |
-
2004
- 2004-01-30 US US10/541,108 patent/US7345180B2/en not_active Expired - Lifetime
- 2004-01-30 KR KR1020057013028A patent/KR100998796B1/ko active Protection Beyond IP Right Term
- 2004-01-30 WO PCT/JP2004/000886 patent/WO2004067509A1/ja active Application Filing
- 2004-01-30 ES ES04706796.2T patent/ES2587885T3/es not_active Expired - Lifetime
- 2004-01-30 EP EP04706796.2A patent/EP1595866B1/en not_active Expired - Lifetime
- 2004-01-30 JP JP2005504761A patent/JP4184378B2/ja not_active Expired - Lifetime
- 2004-01-30 CA CA2514191A patent/CA2514191C/en not_active Expired - Fee Related
- 2004-01-30 AU AU2004207731A patent/AU2004207731B2/en not_active Ceased
- 2004-01-30 PT PT47067962T patent/PT1595866T/pt unknown
- 2004-01-30 CN CNB2004800033423A patent/CN100434420C/zh not_active Expired - Fee Related
- 2004-01-30 DK DK04706796.2T patent/DK1595866T3/en active
Also Published As
| Publication number | Publication date |
|---|---|
| CA2514191C (en) | 2011-10-11 |
| WO2004067509A1 (ja) | 2004-08-12 |
| PT1595866T (pt) | 2016-09-14 |
| CA2514191A1 (en) | 2004-08-12 |
| US7345180B2 (en) | 2008-03-18 |
| JPWO2004067509A1 (ja) | 2006-05-18 |
| AU2004207731B2 (en) | 2009-08-13 |
| CN1745063A (zh) | 2006-03-08 |
| US20060229286A1 (en) | 2006-10-12 |
| EP1595866B1 (en) | 2016-06-08 |
| KR100998796B1 (ko) | 2010-12-06 |
| ES2587885T3 (es) | 2016-10-27 |
| EP1595866A1 (en) | 2005-11-16 |
| AU2004207731A1 (en) | 2004-08-12 |
| EP1595866A4 (en) | 2009-03-04 |
| CN100434420C (zh) | 2008-11-19 |
| KR20050094849A (ko) | 2005-09-28 |
| DK1595866T3 (en) | 2016-08-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4184378B2 (ja) | ジペプチジルペプチダーゼivを阻害する化合物 | |
| JP5424256B2 (ja) | アザビシクロオクタンの誘導体、その製造方法及びそのジペプチジルペプチダーゼivの阻害剤としての用途 | |
| US7393851B2 (en) | Azaindole derivatives as inhibitors of p38 kinase | |
| JP4329382B2 (ja) | 医薬組成物 | |
| JP4329291B2 (ja) | 含窒素五員環化合物 | |
| US20060041124A1 (en) | Process for preparing pyrrolotriazine kinase inhibitors | |
| CZ385098A3 (cs) | Nové heterocyklické sloučeniny, způsob jejich přípravy, farmaceutické kompozice s jejich obsahem a jejich použití v léčbě diabetu a příbuzných chorob | |
| JP2010522241A (ja) | 増殖性疾患、アレルギー性疾患、自己免疫疾患または炎症性疾患として有用な縮合ヘテロ環化合物 | |
| JPS6348272B2 (ja) | ||
| KR100488095B1 (ko) | 1H-피리도(3,4-b)인돌-4-카르복사미드 유도체, 그의 제법 및치료에의 응용 | |
| CZ94098A3 (cs) | Tertahydrochinolinové deriváty, způsob jejich výroby a farmaceutický prostředek s jejich obsahem | |
| JP2769578B2 (ja) | バソプレシン拮抗剤 | |
| JPH08208602A (ja) | インドロイルグアニジン誘導体 | |
| CN113754635A (zh) | 稠环类化合物及其制备方法和用途 | |
| WO1995021163A1 (fr) | Derive d'acide pyridonecarboxylique substitue par un groupe amino bicyclique, ester et sel de celui-ci, et amine bicyclique utilisee en tant qu'intermediaire pour celui-ci | |
| JPH0482147B2 (ja) | ||
| JP2005200427A (ja) | 含窒素五員環化合物 | |
| CZ414599A3 (cs) | Způsob a meziprodukty pro růstové hormonové sekretagogy |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20061222 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20080213 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20080318 |
|
| A871 | Explanation of circumstances concerning accelerated examination |
Free format text: JAPANESE INTERMEDIATE CODE: A871 Effective date: 20080318 |
|
| A975 | Report on accelerated examination |
Free format text: JAPANESE INTERMEDIATE CODE: A971005 Effective date: 20080501 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20080520 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20080618 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20080902 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20080903 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110912 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 4184378 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120912 Year of fee payment: 4 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120912 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130912 Year of fee payment: 5 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R153 | Grant of patent term extension |
Free format text: JAPANESE INTERMEDIATE CODE: R153 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R153 | Grant of patent term extension |
Free format text: JAPANESE INTERMEDIATE CODE: R153 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R153 | Grant of patent term extension |
Free format text: JAPANESE INTERMEDIATE CODE: R153 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |