JP2010077113A - 炭酸エステルの製造方法 - Google Patents
炭酸エステルの製造方法 Download PDFInfo
- Publication number
- JP2010077113A JP2010077113A JP2009168448A JP2009168448A JP2010077113A JP 2010077113 A JP2010077113 A JP 2010077113A JP 2009168448 A JP2009168448 A JP 2009168448A JP 2009168448 A JP2009168448 A JP 2009168448A JP 2010077113 A JP2010077113 A JP 2010077113A
- Authority
- JP
- Japan
- Prior art keywords
- reaction
- carbonate
- amount
- benzonitrile
- pressure
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/52—Improvements relating to the production of bulk chemicals using catalysts, e.g. selective catalysts
Landscapes
- Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
【解決手段】固体触媒とベンゾニトリルの存在下で、一価アルコールと二酸化炭素とを反応させて、炭酸エステルと水を生成すると共に、前記ベンゾニトリルと前記生成した水との水和反応によりベンズアミドを生成して前記生成した水を反応系から除去又は低減することで、前記炭酸エステルの生成を促進させる。
【選択図】なし
Description
(1)固体触媒とベンゾニトリルの存在下で、一価アルコールと二酸化炭素とを反応させて炭酸エステルを製造することを特徴とする炭酸エステルの製造方法である。また、
(2)固体触媒とベンゾニトリルの存在下で、一価アルコールと二酸化炭素とを反応させて、炭酸エステルと水を生成すると共に、前記ベンゾニトリルと前記生成した水との水和反応によりベンズアミドを生成させて、前記生成した水を反応系から除去又は低減することで、前記炭酸エステルの生成を促進させることを特徴とする炭酸エステルの製造方法である。また、
(3)前記固体触媒が、酸化セリウム、酸化ジルコニウム、及び酸化セリウムと酸化ジルコニウムとの化合物からなる群から選ばれた一種または二種以上からなることを特徴とする(1)又は(2)に記載の炭酸エステルの製造方法である。また、
(4)前記一価アルコールがメタノールであり、炭酸エステルとして炭酸ジメチルを製造することを特徴とする(1)〜(3)のいずれかに記載の炭酸エステルの製造方法である。また、
(5)前記反応時の圧力が5MPa以下であることを特徴とする(1)〜(4)のいずれかに記載の炭酸エステルの製造方法である。また、
(6)前記反応時の圧力が3MPa以下であることを特徴とする(1)〜(4)のいずれかに記載の炭酸エステルの製造方法である。また、
(7)前記反応時の圧力が0.1〜1MPaであることを特徴とする(1)〜(4)のいずれかに記載の炭酸エステルの製造方法である。
本発明の炭酸エステルの製造方法は、固体触媒とベンゾニトリルの存在下、一価アルコールと二酸化炭素とを直接反応させて炭酸エステルを生成するものである。下記反応式Iに示すように(反応式Iでは一価アルコールとしてメタノールの場合を例に説明する)、一価アルコールと二酸化炭素とを反応させると炭酸エステルの他に水も生成するが、ベンゾニトリルが存在することで、生成した水との水和反応によりベンズアミドを生成し、生成した水を反応系から除去又は低減することで、炭酸エステルの生成を促進させることが可能となる。
酸化セリウム(第一稀元素製:不純物濃度0.02%以下)を873Kで空気雰囲気下、3時間焼成し、粉末状の固体触媒を得た。そこで、190mlのオートクレーブ(反応器)に磁気攪拌子、上記固体触媒(1mmol)、メタノール(100mmol)及びベンゾニトリル(100mmol)を導入し、約5gのCO2でオートクレーブ内の空気を3回パージした後、表1に示した圧力になるようCO2を導入して、そのオートクレーブをバンドヒーター、ホットスターラーにより423Kまで攪拌しながら昇温し、目的の温度に達した時間を反応開始時間とした。そして、423Kで2時間反応させた後、オートクレーブを水冷し、室温まで冷えたら減圧して内部標準物質の2-プロパノールを加え、生成物を採取し、GC(ガスクロマトグラフィー)で分析した。このようにして、反応圧力(CO2の導入量)を変えて表1に示す試験No.1〜7の実験を行った。
ベンゾニトリル(BN)を下記表2に示すような量にする他は実施例1と同様にして、反応圧力が0.5MPaになるようにCO2量を40mmol導入した後、423Kで2hr反応を行った。その結果を表2に示す。
反応温度を下記表3に示し、メタノール(100mmol)及びベンゾニトリル(200mmol)を導入し、反応圧力が1MPaになるようにCO2量を80mmol導入する他は実施例1と同様にして、各反応温度で12hr反応を行った。その結果を表3に示す。
固体触媒の酸化セリウムを0.05g(0.3mmol)用い、反応時間を下記表4に示すようにする他は実施例3と同様にして、反応圧力が1MPaになるようにCO2量を80mmol導入した後、423Kで反応を行った。その結果を表4に示す。
固体触媒の酸化セリウムを0.17g(1mmol)用い、反応時間を下記表5に示すようにする他は実施例3と同様にして、反応圧力が1MPaになるようにCO2量を80mmol導入した後、423Kで反応を行った。その結果を表5に示す。
固体触媒の酸化セリウムを0.51g(3mmol)用い、反応時間を下記表6に示すようにする他は実施例3と同様にして、反応圧力が1MPaになるようにCO2量を80mmol導入した後、423Kで反応を行った。その結果を表6に示す。
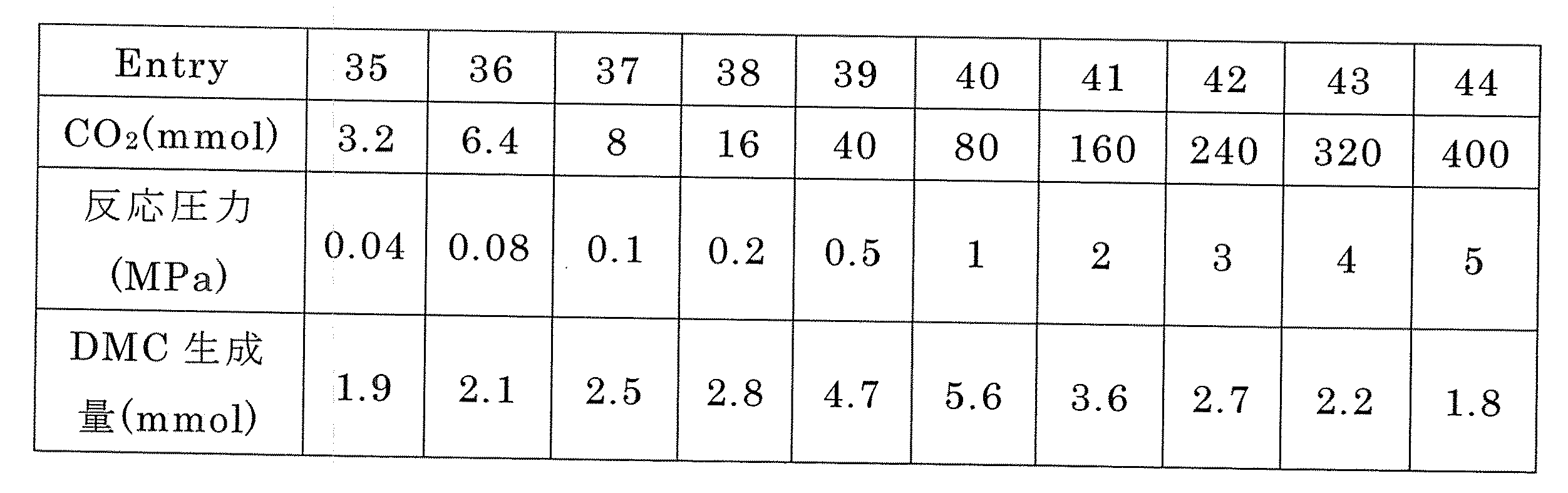
ベンゾニトリルを200mmol用いるほかは、実施例1と同様にして、CO2導入量及び反応圧力を変えて表7に示す試験No.35〜44の実験を行った。
固体触媒の酸化セリウムを0.17g(1mmol)用い、メタノール(200mmol)及びベンゾニトリル(400mmol)を導入し、反応圧力が0.2MPaになるようにCO2量を16mmol導入した後、423Kで下記表8に示すような各反応時間で反応を行った。その結果を表8に示す。
反応圧力が0.1MPaになるようにCO2量を8mmol導入する他は実施例8と同様にして、423Kで下記表9に示すような各反応時間で反応を行った。その結果を表9に示す。
酸化ジルコニウム(ナカライテスク製:不純物濃度0.08%以下)を673Kで空気雰囲気下、3時間焼成して得た粉末状の固体触媒を1mmol用いて、反応圧力及びCO2量を表10に示すようにした以外は、実施例1と同様にして実験した。尚、この場合は反応温度を443Kにて2時間反応させて行った。その結果を表10に示す。
硝酸セリウムと硝酸ジルコニウムをセリウムが20原子量%となるように溶解させた溶液に水酸化ナトリウムを導入して沈殿物を生成させた後、この沈殿物を濾過、水洗した後、1273Kで空気雰囲気下、3時間焼成し、粉末状の固体触媒を得た。そして、この固体触媒を1mmol用いて、反応圧力及びCO2量を表11に示すようにした以外は実施例1と同様にして実験した。尚、この場合、反応温度は443Kにて2時間反応させて行った。その結果を表11に示す。
固体触媒の酸化セリウムを0.17g(1mmol)用い、メタノールの代わりにエタノール(200mmol)を用いたほかは、実施例8と同様にして、反応圧力が0.2MPaになるようにCO2量を16mmol導入した後、423Kで下記表12に示すような各反応時間で反応を行った。その結果を表12に示す。
メタノールの代わりにプロパノール(100mmol)を用い、反応圧力及びCO2量を表13に示すようにした以外は実施例1と同様にして実験した。その結果を表13に示す。
メタノールの代わりにイソプロパノール(100mmol)を用い、反応圧力及びCO2量を表14に示すようにした以外は実施例1と同様にして実験した。その結果を表14に示す。
メタノールの代わりにt-ブチルアルコール(100mmol)を用い、反応圧力及びCO2量を表15に示すようにした以外は実施例1と同様にして実験した。その結果を表15に示す。
固体触媒の酸化セリウムを0.51g(3mmol)用い、且つベンゾニトリルを用いないようにして、反応圧力及びCO2量を表16に示すようにした以外は実施例1と同様にして実験した。その結果を表16に示す。
ベンゾニトリルの代わりに2,2-ジメトキシプロパンを30mmol用い、反応圧力及びCO2量を表17に示すようにした以外は実施例1と同様にして実験した。その結果を表17に示す。
Claims (7)
- 固体触媒とベンゾニトリルの存在下で、一価アルコールと二酸化炭素とを反応させて炭酸エステルを製造することを特徴とする炭酸エステルの製造方法。
- 固体触媒とベンゾニトリルの存在下で、一価アルコールと二酸化炭素とを反応させて、炭酸エステルと水を生成すると共に、前記ベンゾニトリルと前記生成した水との水和反応によりベンズアミドを生成させて、前記生成した水を反応系から除去又は低減することで、前記炭酸エステルの生成を促進させることを特徴とする炭酸エステルの製造方法。
- 前記固体触媒が、酸化セリウム、酸化ジルコニウム、及び酸化セリウムと酸化ジルコニウムとの化合物からなる群から選ばれた一種または二種以上からなることを特徴とする請求項1又は2に記載の炭酸エステルの製造方法。
- 前記一価アルコールがメタノールであり、炭酸エステルとして炭酸ジメチルを製造することを特徴とする請求項1〜3のいずれか1項に記載の炭酸エステルの製造方法。
- 前記反応時の圧力が5MPa以下であることを特徴とする請求項1〜4のいずれか1項に記載の炭酸エステルの製造方法。
- 前記反応時の圧力が3MPa以下であることを特徴とする請求項1〜4のいずれか1項に記載の炭酸エステルの製造方法。
- 前記反応時の圧力が0.1〜1MPaであることを特徴とする請求項1〜4のいずれか1項に記載の炭酸エステルの製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009168448A JP5458352B2 (ja) | 2008-08-27 | 2009-07-17 | 炭酸エステルの製造方法 |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008217524 | 2008-08-27 | ||
| JP2008217524 | 2008-08-27 | ||
| JP2009168448A JP5458352B2 (ja) | 2008-08-27 | 2009-07-17 | 炭酸エステルの製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2010077113A true JP2010077113A (ja) | 2010-04-08 |
| JP5458352B2 JP5458352B2 (ja) | 2014-04-02 |
Family
ID=42207979
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009168448A Active JP5458352B2 (ja) | 2008-08-27 | 2009-07-17 | 炭酸エステルの製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5458352B2 (ja) |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012162523A (ja) * | 2011-01-20 | 2012-08-30 | Nippon Steel Corp | 炭酸エステルの製造方法 |
| WO2015099053A1 (ja) * | 2013-12-27 | 2015-07-02 | 新日鐵住金株式会社 | シアノピリジンの製造方法、ベンゾニトリルの製造方法、炭酸エステルの製造方法、及び炭酸エステルの製造装置 |
| JP2016216363A (ja) * | 2014-09-09 | 2016-12-22 | 新日鐵住金株式会社 | 炭酸エステルの製造方法及び製造装置 |
| JP2016215101A (ja) * | 2015-05-18 | 2016-12-22 | 三菱重工業株式会社 | 炭酸ジブチル合成用触媒及びその製造方法 |
| JP2016216361A (ja) * | 2014-03-04 | 2016-12-22 | 新日鐵住金株式会社 | 炭酸エステルの製造方法及び製造装置 |
| WO2017221908A1 (ja) | 2016-06-22 | 2017-12-28 | 三菱瓦斯化学株式会社 | 芳香族ニトリル化合物の製造方法、及び炭酸エステルの製造方法 |
| WO2018116775A1 (ja) | 2016-12-21 | 2018-06-28 | 三菱瓦斯化学株式会社 | 芳香族ニトリル化合物の製造方法、及び炭酸エステルの製造方法 |
| WO2019065549A1 (ja) | 2017-09-29 | 2019-04-04 | 三菱瓦斯化学株式会社 | 触媒の再生方法、及び炭酸エステルの製造方法 |
| WO2019138993A1 (ja) | 2018-01-10 | 2019-07-18 | 三菱瓦斯化学株式会社 | 炭酸エステルの製造方法 |
| WO2020022416A1 (ja) | 2018-07-27 | 2020-01-30 | 三菱瓦斯化学株式会社 | 芳香族ニトリル化合物の製造方法、及び炭酸エステルの製造方法 |
| US10696619B2 (en) | 2017-12-28 | 2020-06-30 | Industrial Technology Research Institute | Method for preparing dialkyl carbonate |
| US11400440B2 (en) | 2019-12-16 | 2022-08-02 | Industrial Technology Research Institute | Catalyst and precursor thereof and method of forming dialkyl carbonate |
| WO2022181670A1 (ja) | 2021-02-25 | 2022-09-01 | 三菱瓦斯化学株式会社 | 2-フロニトリルの製造方法、及び炭酸エステルの製造方法 |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH05255214A (ja) * | 1992-03-11 | 1993-10-05 | Showa Denko Kk | ニトリル化合物の水和方法 |
| JPH0641020A (ja) * | 1992-06-01 | 1994-02-15 | Mitsubishi Gas Chem Co Inc | 芳香族炭酸エステルの製造方法 |
| JPH0656747A (ja) * | 1992-06-11 | 1994-03-01 | Enichem Sintesi Spa | 有機カーボネートの製法 |
| JPH06116221A (ja) * | 1991-05-22 | 1994-04-26 | Mitsubishi Gas Chem Co Inc | 芳香族カルボン酸アミドの製造法 |
| JP2003503472A (ja) * | 1999-06-30 | 2003-01-28 | ゼネラル・エレクトリック・カンパニイ | 触媒組成物及びニトリルを促進剤として用いるジアリールカーボネートの製造方法 |
| JP2005170821A (ja) * | 2003-12-10 | 2005-06-30 | Nippon Shokubai Co Ltd | ニトリル類の水和反応によるアミド化合物の製造方法 |
| WO2006109775A1 (ja) * | 2005-04-12 | 2006-10-19 | National Institute Of Advanced Industrial Science And Technology | 炭酸エステルの製造方法 |
| JP2006289157A (ja) * | 2005-04-05 | 2006-10-26 | Mitsubishi Heavy Ind Ltd | カーボネート合成触媒及びカーボネート製造方法 |
| JP2007099717A (ja) * | 2005-10-06 | 2007-04-19 | National Institute Of Advanced Industrial & Technology | 炭酸エステルの製造方法 |
-
2009
- 2009-07-17 JP JP2009168448A patent/JP5458352B2/ja active Active
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06116221A (ja) * | 1991-05-22 | 1994-04-26 | Mitsubishi Gas Chem Co Inc | 芳香族カルボン酸アミドの製造法 |
| JPH05255214A (ja) * | 1992-03-11 | 1993-10-05 | Showa Denko Kk | ニトリル化合物の水和方法 |
| JPH0641020A (ja) * | 1992-06-01 | 1994-02-15 | Mitsubishi Gas Chem Co Inc | 芳香族炭酸エステルの製造方法 |
| JPH0656747A (ja) * | 1992-06-11 | 1994-03-01 | Enichem Sintesi Spa | 有機カーボネートの製法 |
| JP2003503472A (ja) * | 1999-06-30 | 2003-01-28 | ゼネラル・エレクトリック・カンパニイ | 触媒組成物及びニトリルを促進剤として用いるジアリールカーボネートの製造方法 |
| JP2005170821A (ja) * | 2003-12-10 | 2005-06-30 | Nippon Shokubai Co Ltd | ニトリル類の水和反応によるアミド化合物の製造方法 |
| JP2006289157A (ja) * | 2005-04-05 | 2006-10-26 | Mitsubishi Heavy Ind Ltd | カーボネート合成触媒及びカーボネート製造方法 |
| WO2006109775A1 (ja) * | 2005-04-12 | 2006-10-19 | National Institute Of Advanced Industrial Science And Technology | 炭酸エステルの製造方法 |
| JP2007099717A (ja) * | 2005-10-06 | 2007-04-19 | National Institute Of Advanced Industrial & Technology | 炭酸エステルの製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| JPN6013045210; 鈴木 淳、他2名: 'アルコールと二酸化炭素からの有用有機カーボネート直接合成' 札幌大会(招待講演 第37回石油・石油化学討論会)(講演要旨) p.287-288, 20071025, 社団法人 石油学会 * |
Cited By (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012162523A (ja) * | 2011-01-20 | 2012-08-30 | Nippon Steel Corp | 炭酸エステルの製造方法 |
| TWI593676B (zh) * | 2013-12-27 | 2017-08-01 | Nippon Steel & Sumitomo Metal Corp | Process for producing benzonitrile, process for producing carbonate ester, and apparatus for producing carbonate ester |
| WO2015099053A1 (ja) * | 2013-12-27 | 2015-07-02 | 新日鐵住金株式会社 | シアノピリジンの製造方法、ベンゾニトリルの製造方法、炭酸エステルの製造方法、及び炭酸エステルの製造装置 |
| JP2016216361A (ja) * | 2014-03-04 | 2016-12-22 | 新日鐵住金株式会社 | 炭酸エステルの製造方法及び製造装置 |
| JP2016216363A (ja) * | 2014-09-09 | 2016-12-22 | 新日鐵住金株式会社 | 炭酸エステルの製造方法及び製造装置 |
| JP2016215101A (ja) * | 2015-05-18 | 2016-12-22 | 三菱重工業株式会社 | 炭酸ジブチル合成用触媒及びその製造方法 |
| WO2017221908A1 (ja) | 2016-06-22 | 2017-12-28 | 三菱瓦斯化学株式会社 | 芳香族ニトリル化合物の製造方法、及び炭酸エステルの製造方法 |
| KR20190018674A (ko) | 2016-06-22 | 2019-02-25 | 미츠비시 가스 가가쿠 가부시키가이샤 | 방향족 나이트릴 화합물의 제조 방법 및 탄산 에스터의 제조 방법 |
| US10584092B2 (en) | 2016-06-22 | 2020-03-10 | Mitsubishi Gas Chemical Company, Inc. | Method for producing aromatic nitrile compound and method for producing carbonate ester |
| WO2018116775A1 (ja) | 2016-12-21 | 2018-06-28 | 三菱瓦斯化学株式会社 | 芳香族ニトリル化合物の製造方法、及び炭酸エステルの製造方法 |
| US11161816B2 (en) | 2016-12-21 | 2021-11-02 | Mitsubishi Gas Chemical Company, Inc. | Method for producing aromatic nitrile compound and method for producing carbonic acid ester |
| KR20190094196A (ko) | 2016-12-21 | 2019-08-12 | 미츠비시 가스 가가쿠 가부시키가이샤 | 방향족 나이트릴 화합물의 제조 방법 및 탄산 에스터의 제조 방법 |
| US10793524B2 (en) | 2016-12-21 | 2020-10-06 | Mitsubishi Gas Chemical Company, Inc. | Method for producing aromatic nitrile compound and method for producing carbonic acid ester |
| JP7136112B2 (ja) | 2017-09-29 | 2022-09-13 | 三菱瓦斯化学株式会社 | 触媒の再生方法、及び炭酸エステルの製造方法 |
| WO2019065549A1 (ja) | 2017-09-29 | 2019-04-04 | 三菱瓦斯化学株式会社 | 触媒の再生方法、及び炭酸エステルの製造方法 |
| KR20200062250A (ko) | 2017-09-29 | 2020-06-03 | 미츠비시 가스 가가쿠 가부시키가이샤 | 촉매의 재생 방법 및 탄산 에스터의 제조 방법 |
| JPWO2019065549A1 (ja) * | 2017-09-29 | 2020-09-10 | 三菱瓦斯化学株式会社 | 触媒の再生方法、及び炭酸エステルの製造方法 |
| CN111201084A (zh) * | 2017-09-29 | 2020-05-26 | 三菱瓦斯化学株式会社 | 催化剂的再生方法和碳酸酯的制造方法 |
| KR102568451B1 (ko) | 2017-09-29 | 2023-08-18 | 미츠비시 가스 가가쿠 가부시키가이샤 | 촉매의 재생 방법 및 탄산 에스터의 제조 방법 |
| CN111201084B (zh) * | 2017-09-29 | 2023-06-06 | 三菱瓦斯化学株式会社 | 催化剂的再生方法和碳酸酯的制造方法 |
| US11596936B2 (en) | 2017-09-29 | 2023-03-07 | Mitsubishi Gas Chemical Company, Inc. | Method for regenerating catalyst and method for producing carbonate ester |
| US10696619B2 (en) | 2017-12-28 | 2020-06-30 | Industrial Technology Research Institute | Method for preparing dialkyl carbonate |
| KR20200107937A (ko) | 2018-01-10 | 2020-09-16 | 미츠비시 가스 가가쿠 가부시키가이샤 | 탄산에스테르의 제조 방법 |
| WO2019138993A1 (ja) | 2018-01-10 | 2019-07-18 | 三菱瓦斯化学株式会社 | 炭酸エステルの製造方法 |
| US11970442B2 (en) | 2018-01-10 | 2024-04-30 | Mitsubishi Gas Chemical Company, Inc. | Method for producing carbonic ester |
| KR20210040392A (ko) | 2018-07-27 | 2021-04-13 | 미쯔비시 가스 케미칼 컴파니, 인코포레이티드 | 방향족 나이트릴 화합물의 제조 방법, 및 탄산 에스터의 제조 방법 |
| US11673856B2 (en) | 2018-07-27 | 2023-06-13 | Mitsubishi Gas Chemical Company, Inc. | Method for producing aromatic nitrile compound and method for producing carbonate ester |
| EP4201925A1 (en) | 2018-07-27 | 2023-06-28 | Mitsubishi Gas Chemical Company, Inc. | Method for producing aromatic nitrile compound and method for producing carbonate ester |
| WO2020022416A1 (ja) | 2018-07-27 | 2020-01-30 | 三菱瓦斯化学株式会社 | 芳香族ニトリル化合物の製造方法、及び炭酸エステルの製造方法 |
| US11400440B2 (en) | 2019-12-16 | 2022-08-02 | Industrial Technology Research Institute | Catalyst and precursor thereof and method of forming dialkyl carbonate |
| US11565244B2 (en) | 2019-12-16 | 2023-01-31 | Industrial Technology Research Institute | Catalyst precursor |
| WO2022181670A1 (ja) | 2021-02-25 | 2022-09-01 | 三菱瓦斯化学株式会社 | 2-フロニトリルの製造方法、及び炭酸エステルの製造方法 |
| KR20230151985A (ko) | 2021-02-25 | 2023-11-02 | 미쯔비시 가스 케미칼 컴파니, 인코포레이티드 | 2-퓨로나이트릴의 제조 방법, 및 탄산 에스터의 제조방법 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP5458352B2 (ja) | 2014-04-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5458352B2 (ja) | 炭酸エステルの製造方法 | |
| JP5738206B2 (ja) | 炭酸エステルの製造方法 | |
| Shukla et al. | Diethyl carbonate: critical review of synthesis routes, catalysts used and engineering aspects | |
| WO2015099053A1 (ja) | シアノピリジンの製造方法、ベンゾニトリルの製造方法、炭酸エステルの製造方法、及び炭酸エステルの製造装置 | |
| JP5252424B2 (ja) | 炭酸エステルの製造方法 | |
| Raza et al. | Green synthesis of dimethyl carbonate from CO2 and methanol: new strategies and industrial perspective | |
| Ju et al. | Performance of ionic liquid as catalysts in the synthesis of dimethyl carbonate from ethylene carbonate and methanol | |
| JP7092305B2 (ja) | 芳香族ニトリル化合物の製造方法、及び炭酸エステルの製造方法 | |
| WO2017140800A1 (en) | Methanol production process | |
| CA2522183C (en) | Catalyst for dimethyl carbonate synthesis | |
| CN104072376B (zh) | 一种由co2和甲醇合成碳酸二甲酯的方法 | |
| JP6349787B2 (ja) | 炭酸エステルの製造方法 | |
| La et al. | Direct synthesis of dimethyl carbonate from CH 3 OH and CO 2 by H 3 PW 12 O 40/Ce x Ti 1-x O 2 catalyst | |
| JP2021151986A (ja) | 炭酸エステルの製造方法、および炭酸エステルの製造装置 | |
| WO2015132801A1 (en) | Process for the synthesis of dimethylcarbonate | |
| TWI770091B (zh) | 芳香族腈化合物之製造方法及碳酸酯之製造方法 | |
| JP6435728B2 (ja) | 炭酸エステルの製造方法及び製造装置 | |
| TWI783048B (zh) | 觸媒之再生方法,及碳酸酯之製造方法 | |
| JP7487664B2 (ja) | 炭酸ジアルキルの製造方法 | |
| WO2022181670A1 (ja) | 2-フロニトリルの製造方法、及び炭酸エステルの製造方法 | |
| GU | CO2 Transformation to Polycarbonates and Carbamates Using CeO2 as a Heterogeneous Catalyst | |
| JP2014224056A (ja) | 芳香族炭酸エステルの製造方法 | |
| 常陶 | Direct Synthesis of Organic Carbonates from Carbon Dioxide and Alcohols over Cerium Oxide Catalyst Assisted by Zeolites and Ketals | |
| Yabushita et al. | Thermodynamic and Catalytic Insights into Non‐Reductive Transformation of CO2 with Amines into Organic Urea Derivatives | |
| Razali et al. | Improving Product Yield in the Direct Carboxylation of Glycerol with CO2 through the Tailored Selection of Dehydrating Agents. Catalysts 2021, 11, 138 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20120628 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20120628 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20130807 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130910 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20131107 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20131203 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20131224 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5458352 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313117 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| S533 | Written request for registration of change of name |
Free format text: JAPANESE INTERMEDIATE CODE: R313533 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |