ES2793018T3 - Inmunomoduladores - Google Patents
Inmunomoduladores Download PDFInfo
- Publication number
- ES2793018T3 ES2793018T3 ES15781564T ES15781564T ES2793018T3 ES 2793018 T3 ES2793018 T3 ES 2793018T3 ES 15781564 T ES15781564 T ES 15781564T ES 15781564 T ES15781564 T ES 15781564T ES 2793018 T3 ES2793018 T3 ES 2793018T3
- Authority
- ES
- Spain
- Prior art keywords
- minutes
- methyl
- mobile phase
- hydrogen
- solution
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CCC(*)NOCN Chemical compound CCC(*)NOCN 0.000 description 23
- LUMGPUYAAZYVKH-ZZWQRSRUSA-N CCCC[C@@H](C(N[C@@H](C)C(N[C@@H](CCCNCC(N[C@@H](Cc(cc1)ccc1O)C(N(C)[C@@H](C)C(N[C@@H](CC(O)=O)C(N(CCC1)C1C(N[C@@H](Cc1cnc[nH]1)C(N[C@@H](CCC(N)=O)C(N(C[C@@H](C1)O)C1C(N[C@@H](Cc1c[nH]c2ccccc12)C(N[C@@H](CO)C(N[C@@H](Cc1c[n](CC(O)=O)c2ccccc12)C(N(C)[C@H]1CCCC)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)C(NCC(N)O)=O)=O)=O)N(C)C1=O Chemical compound CCCC[C@@H](C(N[C@@H](C)C(N[C@@H](CCCNCC(N[C@@H](Cc(cc1)ccc1O)C(N(C)[C@@H](C)C(N[C@@H](CC(O)=O)C(N(CCC1)C1C(N[C@@H](Cc1cnc[nH]1)C(N[C@@H](CCC(N)=O)C(N(C[C@@H](C1)O)C1C(N[C@@H](Cc1c[nH]c2ccccc12)C(N[C@@H](CO)C(N[C@@H](Cc1c[n](CC(O)=O)c2ccccc12)C(N(C)[C@H]1CCCC)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)C(NCC(N)O)=O)=O)=O)N(C)C1=O LUMGPUYAAZYVKH-ZZWQRSRUSA-N 0.000 description 1
- KOATWGUEPWYXLU-UBBXWSHDSA-N CCCC[C@@H](C(N[C@@H](CC(C)C)C(NCCCCC(N[C@@H](Cc(cc1)ccc1O)C(N(C)[C@@H](C)C(N[C@@H](CC(N)=O)C(N(CCC1)[C@@H]1C(N[C@@H](CN)C(N[C@@H](CC(C)C)C(N(C[C@@H](C1)O)[C@@H]1C(N[C@@H](Cc1c[nH]c2ccccc12)C(N[C@@H](CCN)C(N[C@@H](Cc1c[nH]c2ccccc12)C(N(C)[C@H]1CCCC)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)N(C)C1=O Chemical compound CCCC[C@@H](C(N[C@@H](CC(C)C)C(NCCCCC(N[C@@H](Cc(cc1)ccc1O)C(N(C)[C@@H](C)C(N[C@@H](CC(N)=O)C(N(CCC1)[C@@H]1C(N[C@@H](CN)C(N[C@@H](CC(C)C)C(N(C[C@@H](C1)O)[C@@H]1C(N[C@@H](Cc1c[nH]c2ccccc12)C(N[C@@H](CCN)C(N[C@@H](Cc1c[nH]c2ccccc12)C(N(C)[C@H]1CCCC)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)N(C)C1=O KOATWGUEPWYXLU-UBBXWSHDSA-N 0.000 description 1
- LDLPYQHWVSPUIE-ZWHVRFKPSA-N CCCC[C@@H](C(N[C@@H](CC(C)C)C(NCCCCC(N[C@@H](Cc(cc1)ccc1OC(C)(C)C)C(N(C)[C@@H](C)C(N[C@@H](CC(NC(c1ccccc1)(c1ccccc1)c1ccccc1)=O)C(N(CCC1)[C@@H]1C(N[C@@H](CN)C(N[C@@H](CC(C)C)C(N(C[C@@H](C1)OC(C)(C)C)[C@@H]1C(N[C@@H](Cc1c[n](C(OC(C)(C)C)=O)c2ccccc12)C(N[C@@H](CCNC(OC(C)(C)C)=O)C(N[C@@H](Cc1c[n](C(OC(C)(C)C)=O)c2ccccc12)C(N(C)[C@H]1CCCC)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)N(C)C1=O Chemical compound CCCC[C@@H](C(N[C@@H](CC(C)C)C(NCCCCC(N[C@@H](Cc(cc1)ccc1OC(C)(C)C)C(N(C)[C@@H](C)C(N[C@@H](CC(NC(c1ccccc1)(c1ccccc1)c1ccccc1)=O)C(N(CCC1)[C@@H]1C(N[C@@H](CN)C(N[C@@H](CC(C)C)C(N(C[C@@H](C1)OC(C)(C)C)[C@@H]1C(N[C@@H](Cc1c[n](C(OC(C)(C)C)=O)c2ccccc12)C(N[C@@H](CCNC(OC(C)(C)C)=O)C(N[C@@H](Cc1c[n](C(OC(C)(C)C)=O)c2ccccc12)C(N(C)[C@H]1CCCC)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)N(C)C1=O LDLPYQHWVSPUIE-ZWHVRFKPSA-N 0.000 description 1
- XMDGNMNNMJFYBV-DQEYMECFSA-N CN[C@@H](CC(NC(c1ccccc1)(c1ccccc1)c1ccccc1)=O)C(N(CCC1)[C@@H]1C(O)=O)=O Chemical compound CN[C@@H](CC(NC(c1ccccc1)(c1ccccc1)c1ccccc1)=O)C(N(CCC1)[C@@H]1C(O)=O)=O XMDGNMNNMJFYBV-DQEYMECFSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/64—Cyclic peptides containing only normal peptide links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/04—Linear peptides containing only normal peptide links
- C07K7/08—Linear peptides containing only normal peptide links having 12 to 20 amino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/12—Cyclic peptides, e.g. bacitracins; Polymyxins; Gramicidins S, C; Tyrocidins A, B or C
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/50—Cyclic peptides containing at least one abnormal peptide link
- C07K7/54—Cyclic peptides containing at least one abnormal peptide link with at least one abnormal peptide link in the ring
- C07K7/56—Cyclic peptides containing at least one abnormal peptide link with at least one abnormal peptide link in the ring the cyclisation not occurring through 2,4-diamino-butanoic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/10—Peptides having 12 to 20 amino acids
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/435—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans
- G01N2333/705—Assays involving receptors, cell surface antigens or cell surface determinants
- G01N2333/70503—Immunoglobulin superfamily, e.g. VCAMs, PECAM, LFA-3
- G01N2333/70532—B7 molecules, e.g. CD80, CD86
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2500/00—Screening for compounds of potential therapeutic value
- G01N2500/02—Screening involving studying the effect of compounds C on the interaction between interacting molecules A and B (e.g. A = enzyme and B = substrate for A, or A = receptor and B = ligand for the receptor)
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2800/00—Detection or diagnosis of diseases
- G01N2800/26—Infectious diseases, e.g. generalised sepsis
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/574—Immunoassay; Biospecific binding assay; Materials therefor for cancer
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Immunology (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Gastroenterology & Hepatology (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Un compuesto de fórmula (I) **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo, en la que: A se selecciona entre; **(Ver fórmula)** y en las que **(Ver fórmula)** representa el punto de unión al grupo carboxilo y **(Ver fórmula)** representa el punto de unión al átomo de nitrógeno; n es 0, 1 o 2; m es 1 o 2; p es 0, 1 o 2; R14 y R15 se seleccionan independientemente entre hidrógeno y metilo; Rz se selecciona entre hidrógeno y -C(O)NHR16; en donde R16 se selecciona entre hidrógeno, - CHR17C(O)NH2, -CHR17C(O)NHCHR18C(O)NH2 y -CHR17C(O)NHCHR18C(O)NHCH2C(O)NH2; en donde R17 se selecciona entre hidrógeno y -CH2OH y en donde R18 se selecciona entre hidrógeno y metilo; Rv es hidrógeno, metilo o una cadena lateral de aminoácido natural; Rc, Rf, Rh, Ri y Rm son hidrógeno; Rn es hidrógeno o metilo o Rv y Rn forman un anillo de pirrolidina; Ra, Re y Rj, se seleccionan cada uno independientemente entre hidrógeno y metilo; Rk es metilo; R1, R2, R3, R5, R6, R8, R9, R10, R11, R12 y R13 se seleccionan independientemente entre una cadena lateral de aminoácido natural y una cadena lateral de aminoácido no natural o R2 o R12 forman un anillo con el grupo R vecinal correspondiente como se describe más adelante; Rb es metilo o, Rb y R2, junto con los átomos a los que están unidos, forman un anillo seleccionado entre azetidina, pirrolidina, morfolina, piperidina, piperazina y tetrahidrotiazol; en donde cada anillo está opcionalmente sustituido con uno a cuatro grupos seleccionados independientemente entre amino, ciano, metilo, halo, e hidroxi; Rd y R4, junto con los átomos a los que están unidos, forman un anillo de pirrolidina; Rg y R7, junto con los átomos a los que están unidos, forman un anillo de pirrolidina que está opcionalmente sustituido con un grupo hidroxilo; y RL es metilo o, RL y R12, junto con los átomos a los que están unidos, forman un anillo seleccionado entre azetidina y pirrolidina, en donde cada anillo está opcionalmente sustituido con uno a cuatro grupos seleccionados independientemente entre amino, ciano, metilo, halo e hidroxi.
Description
DESCRIPCIÓN
Inmunomoduladores
La presente divulgación proporciona nuevos péptidos macrocíclicos que inhiben la interacción proteína/proteína PD-1/PD-L1 y CD80/PD-L1 y por tanto son útiles para la mejora de diversas enfermedades, incluyendo cáncer y enfermedades infecciosas.
La proteína Muerte programada 1 (PD-1) es un miembro inhibidor de la familia de receptores CD28, que también incluye CD28, CTLA-4, ICOS y BTLA. PD-1 se expresa en linfocitos B activados, linfocitos T y células mieloides (Agata et al., véase anteriormente; Okazaki et al., Curr. Opin. Immunol., 14:779-782 (2002); Bennett et al., J. Immunol., 170:711-718 (2003)).
La proteína PD-1 es una proteína transmembrana de tipo I de 55 kDa que es parte de la superfamilia de genes Ig (Agata et al., Int. Immunol., 8:765-772 (1996)). PD-1 contiene un motivo inhibidor de tirosina del inmunorreceptor proximal de membrana (ITIM) y un motivo de interruptor basado en tirosina distal de membrana (ITSM) (Thomas, M.L., J. Exp. Med., 181:1953-1956 (1995); Vivier, E. et al., Immunol. Today, 18:286-291 (1997)). Aunque estructuralmente similar a CTLA-4, PD-1 carece del motivo MYPPY que es crítico para unión a CD80 CD86 (B7-2). Se han identificado dos ligandos para PD-1, PD-L1 (B7-H1) y PD-L2 (b7-DC). La activación de linfocitos T que expresan PD-1 se ha mostrado que se regula negativamente tras interacción con células que expresan PD-L1 o PD-L2 (Freeman et al., J. Exp. Med., 192:1027-1034 (2000); Latchman et al., Nat. Immunol., 2:261-268 (2001); Carter et al., Eur. J. Immunol., 32:634-643 (2002)). Tanto PD-L1 como PD-L2 son miembros de la familia de proteínas B7 que se unen a PD-1, pero no se unen a otros miembros de la familia de CD28. El ligando PD-L1 es abundante en diversos cánceres humanos (Dong et al., Nat. Med., 8:787-789 (2002)). La interacción entre PD-1 y PD-L1 da como resultado una disminución de linfocitos infiltrantes de tumores, una disminución en la proliferación mediada por receptores de linfocitos T y evasión inmune por células cancerosas (Dong et al., J. Mol. Med., 81:281-287 (2003); Blank et al., Cancer Immunol. Immunother., 54:307-314 (2005); Konishi et al., Clin. Cancer Res., 10:5094-5100 (2004)). La supresión inmune se puede revertir inhibiendo la interacción local de PD-1 con PD-L1 y el efecto es aditivo cuando la interacción de PD-1 con PD-L2 también se bloquea (Iwai et al., Proc. Natl. Acad. Sci. USA, 99:12293-12297 (2002); Brown et al., J. Immunol., 170:1257-1266 (2003)).
También se ha mostrado que PD-L1 que interacciona con CD80 (Butte MJ et al., Immunity;27:111-122 (2007)). La interacción PD-L1/CD80 en expresión de células inmunes se ha mostrado que es inhibidora. El bloqueo de esta interacción se ha mostrado que elimina esta interacción inhibidora (Paterson Am , et al., J Immunol., 187:1097-1105 (2011); Yang J, et al. J Immunol. 1 de agosto;187(3):1113-9 (2011)).
Cuando los linfocitos T que expresan PD-1, entran en contacto con células que expresan sus ligandos, las actividades funcionales en respuesta a estímulos antigénicos, incluyendo la proliferación, secreción de citoquinas y citotoxicidad, se reducen. Las interacciones PD-1/PD-L1 o PD-L2 regulan negativamente las respuestas inmunitarias durante la resolución de una infección o tumor, o durante el desarrollo de autotolerancia (Keir, M.E. et al., Annu. Rev. Immunol., 26:Epub (2008)). La estimulación antigénica crónica, tal como la que se produce durante la enfermedad tumoral o infecciones crónicas, resulta en linfocitos T que expresan niveles elevados de PD-1 y son disfuncionales con respecto a la actividad hacia el antígeno crónico (revisado en Kim et al., Curr. Opin. I mm. (2010)). Esto se denomina "agotamiento de linfocitos T". Los linfocitos B también muestran la supresión y el "agotamiento" de PD-1/PD-ligando.
Se ha demostrado que el bloqueo de la ligadura PD-1/PD-L1 con anticuerpos contra PD-L1 restaura y aumenta la activación de los linfocitos T en muchos sistemas. Los pacientes con cáncer avanzado se benefician de la terapia con un anticuerpo monoclonal contra PD-L1 (Brahmer et al., New Engl. J. Med. (2012)). Modelos animales preclínicos de tumores e infecciones crónicas han demostrado que el bloqueo de la ruta PD-1/PD-L1 mediante anticuerpos monoclonales puede mejorar la respuesta inmunitaria y dar como resultado el rechazo del tumor o el control de la infección. La inmunoterapia antitumoral a través del bloqueo PD-1/PD-L1 puede aumentar la respuesta inmune terapéutica a varios tumores histológicamente distintos (Dong, H. et al., "B7-H1 pathway and its role in the evasion of tumor immunity", J. Mol. Med., 81(5):281-287 (2003); Dong, H. et al., "Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion", Nat. Med., 8(8):793-800 (2002)).
La interferencia con la interacción PD-1/PD-L1 provoca una mayor actividad de linfocitos T en sistemas con infección crónica. El bloqueo de PD-L1 causó un aclaramiento viral mejorado y una inmunidad restaurada en ratones con infección por virus de coriomeningitis linfocítica crónica (Barber, D.L. et al., "Restoring function in exhausted CD8 T cells during chronic viral infection", Nature, 439(7077):682-687 (2006)). Los ratones humanizados infectados con VIH-1 muestran una protección mejorada contra viremia y agotamiento viral de linfocitos T CD4+ (Palmer et al., J. Immunol. (2013)). El bloqueo de PD-1/PD-L1 a través de anticuerpos monoclonales contra PD-L1 puede restaurar la funcionalidad específica de antígeno in vitro en linfocitos T de pacientes con VIH (Day, Nature (2006); Petrovas, J. Exp. Med. (2006); Trautman, Nature Med. (2006); D'Souza, J. Immunol. (2007); Zhang, Blood (2007); Kaufmann, Nature Imm. (2007); Kasu, J. Immunol. (2010); Porichis, Blood (2011)), Pacientes con VHC (Golden-Mason, J. Virol. (2007); Jeung, J. Leuk. Biol. (2007); Urbani, J. Hepatol. (2008); Nakamoto, PLoSPath. (2009); Nakamoto,
Gastroenterology (2008)) y pacientes con VHB (Boni, J. Virol. (2007); Fisicaro, Gastro. (2010); Fisicaro et al., Gastroenterology (2012); Boni et al., Gastro. (2012); Penna et al., J. Hep. (2012); Raziorrough, Hepatology (2009); Liang, World J. Gastro. (2010); Zhang, Gastro. (2008)).
También se ha demostrado que el bloqueo de la interacción PD-L1/CD80 estimula la inmunidad (Yang J., et al., J Immunol. 1 de agosto;187(3):1113-9 (2011)). Se ha demostrado que la estimulación inmune resultante del bloqueo de la interacción PD-L1/CD80 mejora mediante un bloqueo combinado de otras interacciones PD-1/PD-L1 o PD-1/PD-L2.
Se supone que las alteraciones en los fenotipos de las células inmunes son un factor importante en choque séptico (Hotchkiss, et al., Nat Rev Immunol (2013)). Estas incluyen niveles incrementados de PD-1 y PD-L1 (Guignant, et al, Crit. Care (2011)), Las células de pacientes con choque séptico con niveles incrementados de PD-1 y PD-L1 exhiben un nivel incrementado de apoptosis de linfocitos T. Anticuerpos dirigidos a PD-L1, pueden reducir el nivel de apoptosis de células inmunes (Zhang et al, Crit. Care (2011)). Además, los ratones que carecen de expresión de PD-1 son más resistentes a los síntomas de choque séptico que los ratones de tipo natural. Yang J., et al. J Immunol. 1 de agosto;187(3):1113-9 (2011)). Los estudios han revelado que el bloqueo de interacciones de los anticuerpos contra PD-L1 puede suprimir las respuestas inmunitarias inapropiadas y mejorar los signos de la enfermedad.
Además de mejorar las respuestas inmunológicas a los antígenos crónicos, también se ha demostrado que el bloqueo de la ruta PD-1/PD-L1 mejora las respuestas a la vacunación, incluida la vacunación terapéutica en el contexto de infección crónica (Ha, S.J. et al., "Enhancing therapeutic vaccination by blocking PD-1-mediated inhibitory signals during chronic infection", J. Exp. Med., 205(3):543-555 (2008); Finnefrock, A.C. et al., "PD-1 blockade in rhesus macaques: impact on chronic infection and prophylactic vaccination", J. Immunol., 182(2):980-987 (2009); Song, M.-Y. et al., "Enhancement of vaccine-induced primary and memory CD8+ t-cell responses by soluble PD-1", J. Immunother., 34(3):297-306 (2011)).
El documento WO 2014/151634 desvela inhibidores macrocíclicos de la interacción proteína/proteína PD-1/PD-L1 y PD-1/CD-80.
Las moléculas descritas en este documento demuestran la capacidad de bloquear la interacción de PD-L1 con PD-1, tanto en sistemas bioquímicos como en sistemas experimentales basados en células. Estos resultados son consistentes con un potencial para la administración terapéutica para mejorar la inmunidad en cáncer o infección crónica, incluyendo la vacuna terapéutica.
Los péptidos macrocíclicos descritos en este documento son capaces de inhibir la interacción de PD-L1 con PD-1 y con CD80. Estos compuestos han demostrado una unión altamente eficaz a PD-L1, bloqueo de la interacción de PD-L1 con PD-1 o CD80 y son capaces de promover una mayor actividad funcional de linfocitos T, haciéndolos candidatos para formulaciones parenteral, oral, pulmonar, nasal, bucal y de liberación sostenida.
En un aspecto la presente divulgación proporciona un compuesto de fórmula (I)
A se selecciona entre;

en donde:

representa el punto de unión al grupo carboxilo y

representa el punto de unión al átomo de nitrógeno;
n es 0, 1 o 2;
m es 1 o 2;
p es 0, 1 o 2;
R14 y R15 se seleccionan independientemente entre hidrógeno y metilo:
Rz se selecciona entre hidrógeno y -C(O)NHR16; en donde R16 se selecciona entre hidrógeno, -CHR17C(O)NH2, -CHR17C(O)NHCHR18C(O)NH2 y -CHR17C(O)NHCHR18C(O)NHCH2C(O)NH2; en donde R17 se selecciona entre hidrógeno y -CH2OH y en donde R18 se selecciona entre hidrógeno y metilo;
Rv es hidrógeno, metilo, o una cadena lateral de aminoácido natural;
Rc, Rf, Rh, Ri y Rm son hidrógeno;
Rn es hidrógeno o metilo o Rv y Rn forman un anillo de pirrolidina;
Ra, Re y Rj, se seleccionan cada uno independientemente entre hidrógeno y metilo
Rk es metilo;
R1, R2, R3, R5, R6, R8, R9, R10, R R12 y R13 se seleccionan independientemente entre una cadena lateral de aminoácido natural y una cadena lateral de aminoácido no natural o forman un anillo con el grupo R vecinal correspondiente como se describe posteriormente;
Rb es metilo o, Rb y R2, junto con los átomos a los que están unidos, forman un anillo seleccionado entre azetidina, pirrolidina, morfolina, piperidina, piperazina y tetrahidrotiazol; en donde cada anillo está opcionalmente sustituido con uno a cuatro grupos seleccionados independientemente entre amino, ciano, metilo, halo, e hidroxi; Rd y R4, junto con los átomos a los que están unidos, pueden formar un anillo de pirrolidina;
Rg y R7, junto con los átomos a los que están unidos, forman un anillo de pirrolidina que está opcionalmente sustituido con un grupo hidroxi; y
Rl es metilo o, RL y R12, junto con los átomos a los que están unidos, forman un anillo seleccionado entre azetidina y pirrolidina, en donde cada anillo está opcionalmente sustituido con uno a cuatro grupos seleccionados independientemente entre amino, ciano, metilo, halo, e hidroxi.
En una realización del primer aspecto la presente divulgación proporciona un compuesto de fórmula (I) o una sal terapéuticamente aceptable del mismo, en donde:
Rd y R4, junto con los átomos a los que están unidos, forman un anillo de pirrolidina;
Rg y R7, junto con los átomos a los que están unidos, forman un anillo de pirrolidina, en donde dicho anillo está opcionalmente sustituido con un grupo hidroxi;
Rk es metilo
Ra, Re y Rj son hidrógeno;
Rb y R2 son cada uno metilo o, Rb y R2, junto con los átomos a los que
están unidos, forman un anillo de piperidina;
Rl es metilo;
Rn es hidrógeno, metilo, o Rn y Rv forman un anillo de pirrolidina;
R1 es fenilmetilo en donde el fenilo está sustituido con un grupo seleccionado entre halo, hidroxi, metoxi, o metilo; R3 se selecciona entre -CH2C(O)NH2 y -CH2CO2H;
R5 se selecciona entre hidrógeno, -CH2NH2, -CH2(imidazolilo) y -CH2C(O)NH2;
R6 se selecciona entre -CH2CH(CH3)2, -(CH2)4NH2, -(CH2)2CO2H y (CH2)2C(O)NH2;
R8 y R10 son -CH2(indolilo), en donde el indolilo está opcionalmente sustituido con -CH2CO2H;
R9 se selecciona entre hidrógeno, -(CH2)2NH2, -(CH2)4NH2, -CH2OH y -CH2C(O)NH2;
R11 y R12 son -(C^^CHa; y
R13 se selecciona entre metilo, -CH2OH, -CH2CH(CHa)2 y -(CH2)2CO2H.
En otra realización del primer aspecto la presente divulgación proporciona un compuesto de fórmula (I) o una sal terapéuticamente aceptable del mismo, en donde:
Rd y R4, junto con los átomos a los que están unidos, forman un anillo de pirrolidina;
Rg y R7, junto con los átomos a los que están unidos, forman un anillo de pirrolidina, en donde dicho anillo está opcionalmente sustituido con un grupo hidroxi;
Rk es metilo
Ra, Re y Rj hidrógeno;
Rb y R2 son cada uno metilo o, Rb y R2, junto con los átomos a los que están unidos, forman un anillo de piperidina;
Rl es metilo;
Rn es hidrógeno, metilo, o Rn y Rv forman un anillo de pirrolidina;
R1 es fenilmetilo en donde el fenilo está sustituido con un grupo seleccionado entre halo, hidroxi, metoxi, o metilo; R3 se selecciona entre -CH2C(O)NH2 y -CH2CO2H;
R5 se selecciona entre hidrógeno, -CH2NH2, -CH2(imidazolilo) y -CH2C(O)NH2;
R6 se selecciona entre -CH2CH(CH3)2, -(CH2)4NH2, -(CH2)2CO2H y (CH2)2C(O)NH2;
R8 y R10 son -CH2(indolilo), en donde el indolilo está opcionalmente sustituido con -CH2CO2H;
R9 se selecciona entre hidrógeno, -(CH2)2NH2, -(CH2)4NH2, -CH2OH y -CH2C(O)NH2;
R11 y R12 son -(CH2)3CH3; y
R13 se selecciona entre metilo, -CH2OH, -CH2CH(CH3)2 y -(CH2)2CO2H; y
A es

En otra realización del primer aspecto la presente divulgación proporciona un compuesto de fórmula (I), o una sal farmacéuticamente aceptable del mismo, en donde:
Rd y R4, junto con los átomos a los que están unidos, forman un anillo de pirrolidina;
Rg y R7, junto con los átomos a los que están unidos, forman un anillo de pirrolidina, en donde dicho anillo está opcionalmente sustituido con un grupo hidroxi;
Rk es metilo;
Ra, Re y Rj hidrógeno;
Rb y R2 son cada uno metilo o, Rb y R2, junto con los átomos a los que están unidos, forman un anillo de piperidina;
Rl es metilo;
Rn es hidrógeno, metilo, o Rn y Rv forman un anillo de pirrolidina;
R1 es fenilmetilo en donde el fenilo está sustituido con un grupo seleccionado entre halo, hidroxi, metoxi, o metilo; R3 se selecciona entre -CH2C(O)NH2 y -CH2CO2H;
R5 se selecciona entre hidrógeno, -CH2NH2, -CH2(imidazolilo) y -CH2C(O)NH2;
R6 se selecciona entre -CH2CH(CH3)2, -(CH2)4NH2, -(CH2)2CO2H y (CH2)2C(O)NH2;
R8 y R10 son -CH2(indolilo), en donde el indolilo está opcionalmente sustituido con -CH2CO2H;
R9 se selecciona entre hidrógeno, -(CH2)2NH2, -(CH2)4NH2, -CH2OH y -CH2C(O)NH2;
R11 y R12 son -(CH2)3CH3; y
R13 se selecciona entre metilo, -CH2OH, -CH2CH(CH3)2 y -(CH2)2CO2H; y
A es

En un segundo aspecto la presente divulgación proporciona un compuesto de fórmula (II),

en donde:

representa el punto de unión al grupo carboxilo y
representa el punto de unión al átomo de nitrógeno;
n es 0 o 1;
m es 1 o 2;
p es 0, 1 o 2;
R14 y R15 se seleccionan independientemente entre hidrógeno y metilo;y
Rz se selecciona entre hidrógeno y -C(O)NHR16; en donde R16 se selecciona entre hidrógeno, -CHR17C(O)NH2, -CHR17C(O)NHCHR18C(O)NH2 y -CHR17C(O)NHCHR18C(O)NHCH2C(O)NH2; en donde R17 se selecciona entre hidrógeno y -CH2OH y en donde R18 se selecciona entre hidrógeno y metilo;
Rv es hidrógeno o una cadena lateral de aminoácido natural;
Rc Rf, Rh, Ri y Rm son hidrógeno;
Rn es hidrógeno o metilo o Rv y Rn forman un anillo de pirrolidina;
Ra, Re y Rj, se seleccionan cada uno independientemente entre hidrógeno y metilo;
Rk es metilo;
R1, R2, R3, R5, R6, R8, R9, R10, R11, R12 y R13 se seleccionan independientemente entre una cadena lateral de aminoácido natural y una cadena lateral de aminoácido no natural o forman un anillo con el grupo R vecinal correspondiente como se describe posteriormente;
Re y Rk pueden formar cada uno un anillo con el correspondiente grupo vecinal R y los átomos a los que están unidos seleccionado entre azetidina, pirrolidina, morfolina, piperidina, piperazina y tetrahidrotiazol; en donde cada anillo está opcionalmente sustituido con uno a cuatro grupos seleccionados independientemente entre amino, ciano, metilo, halo, e hidroxi; Rb es metilo o, Rb y R2, junto con los átomos a los que están unidos, forman un anillo seleccionado entre azetidina, pirrolidina, morfolina, piperidina, piperazina y tetrahidrotiazol; en donde cada anillo está opcionalmente sustituido con uno a cuatro grupos seleccionados independientemente entre amino, ciano, metilo, halo, e hidroxi;
Rd y R4, junto con los átomos a los que están unidos, forman un anillo de pirrolidina;
Rg y R7, junto con los átomos a los que están unidos, forman un anillo de pirrolidina que está opcionalmente sustituido con un grupo hidroxi; y
Rl es metilo o, RL y R12, junto con los átomos a los que están unidos, forman un anillo seleccionado entre azetidina y pirrolidina, en donde cada anillo está opcionalmente sustituido con uno a cuatro grupos seleccionados independientemente entre amino, ciano, metilo, halo, e hidroxi.
En un tercer aspecto la presente divulgación proporciona un compuesto de fórmula (I) o una sal terapéuticamente aceptable del mismo para uso en mejorar, estimular y/o aumentar la respuesta inmune en un sujeto que lo necesita. En una primera realización, la invención comprende además administrar un agente adicional antes de, después, o simultáneamente con el compuesto de fórmula (I) o una sal terapéuticamente aceptable del mismo. En una segunda realización, el agente adicional es un agente antimicrobiano, un agente antivírico, un agente citotóxico y/o un modificador de la respuesta inmunitaria. En una tercera realización, el agente adicional es un inhibidor de HDAc . En una cuarta realización, el agente adicional es un agonista de TLR7 y/o TLR8.
En un cuarto aspecto, la presente divulgación proporciona un compuesto de fórmula (I) o una sal terapéuticamente aceptable del mismo para uso en inhibición de crecimiento, proliferación, o metástasis de células cancerosas en un sujeto que lo necesita. Debe entenderse que dicha inhibición puede ser directa o indirecta. En una primera realización, el cáncer se selecciona entre melanoma, carcinoma de células renales, cáncer de pulmón no microcítico escamoso (CPNM), CPNM no escamoso, cáncer colorrectal, cáncer de próstata resistente a la castración, cáncer de ovario, cáncer gástrico, carcinoma hepatocelular, carcinoma de páncreas, carcinoma de células escamosas de cabeza y cuello, carcinomas del esófago, del tracto gastrointestinal y de mama y neoplasia maligna hematológica. En un quinto aspecto, la presente documentación proporciona un compuesto de fórmula (I) o una sal terapéuticamente aceptable del mismo para su uso en el tratamiento de una enfermedad infecciosa en un sujeto que lo necesita. En una primera realización, la enfermedad infecciosa está causada por un virus. En una segunda realización, el virus se selecciona entre VIH, Hepatitis A, Hepatitis B, Hepatitis C, herpes virus, e influenza.
En un sexto aspecto, la presente divulgación proporciona uno o más péptidos macrocíclicos descritos en este documento para su uso en el tratamiento de choque séptico en un sujeto que lo necesita.
En un séptimo aspecto, la presente divulgación proporciona al menos un péptido macrocíclico descrito en este documento para su uso en el bloqueo de la interacción de PD-L1 con PD-1 y/o CD80 en un sujeto.
En compuestos de fórmula (I) y (II) donde las cadenas laterales R son parte de un anillo que está sustituido con metilo, se entiende que el grupo metilo puede estar en cualquier átomo de carbono sustituible en el anillo, incluido el carbono que forma parte de la estructura madre macrocíclica.
En compuestos de fórmula (I), cadenas laterales R1 preferentes son: fenilalanina, tirosina, 3-tien-2-ilo, 4-metilfenilalanina, 4-clorofenilalanina, 3-metoxifenilalanina, isotriptófano, 3-metilfenilalanina, 1-naftilalanina, 3,4-difluorofenilalanina, 4-fluorofenilalanina, 3,4-dimetoxifenilalanina, 3,4-diclorofenilalanina, 4-difluorometilfenilalanina, 2-metilfenilalanina, 2-naftilalanina, triptófano, 4-piridinilo, 4-bromofenilalanina, 3-piridinilo, 4-trifluorometilfenilalanina, 4-carboxifenilalanina, 4-metoxifenilalanina, bifenilalanina y 3-clorofenilalanina; y 2,4-diaminobutano.
En compuestos de fórmula (I) en donde R2 no es parte de un anillo, cadenas laterales R2 preferentes son: alanina, serina y glicina.
En compuestos de fórmula (I), cadenas laterales R3 preferentes son: asparagina, ácido aspártico, ácido glutámico, glutamina, serina, ornitina, lisina, histidina, treonina, leucina, alanina, 2,3-diaminopropano y 2,4-diaminobutano. En compuestos de fórmula (I), cadenas laterales R5 preferentes son: histidina, asparagina, 2,3-diaminopropano, serina, glicina, 2,4-diaminobutano, treonina, alanina, lisina, ácido aspártico, alanina y 3-tiazolilalanina.
En compuestos de fórmula (I), cadenas laterales R6 preferentes son: leucina, ácido aspártico, asparagina, ácido glutámico, glutamina, serina, lisina, 3-ciclohexano, treonina, omitina, 2,4-diaminobutano, alanina, arginina y ornitina (COCH3).
En compuestos de fórmula (I) las cadenas laterales R8 preferentes son triptófano y 1,2-benzoisotiazolinilalanina.
En compuestos de fórmula (I) las cadenas laterales R9 preferentes son: serina, histidina, lisina, ornitina, 2,4-dibutilamina, treonina, lisina, glicina, ácido glutámico, valina, 2,3-diaminopropano, arginina, ácido aspártico y tirosina. En compuestos de fórmula (I) las cadenas laterales R10 preferentes son: triptófano opcionalmente sustituido, benzoisotiazolilalanina, 1-naftilalanina y metionina.
En compuestos de fórmula (I) las cadenas laterales R11 preferentes son: norleucina, leucina, asparagina, fenilalanina, metionina, etoximetano, alanina, triptófano, isoleucina, fenilpropano, ácido glutámico, hexano y heptano. En compuestos de fórmula (I) en donde R12 no es parte de un anillo, cadenas laterales R12 preferentes son: norleucina, alanina, etoximetano, metionina, serina, fenilalanina, metoxietano, leucina, triptófano, isoleucina, ácido glutámico, hexano, heptano y glicina.
En compuestos de fórmula (I) las cadenas laterales R13 preferentes son: arginina, ornitina, alanina, 2,4-diaminobutano, 2,3-diaminopropano, leucina, ácido aspártico, ácido glutámico, serina, lisina, treonina, ciclopropilmetano, glicina, valina, isoleucina, histidina y 2-aminobutano.
Según la presente divulgación, hemos descubierto péptidos que se unen específicamente a PD-L1 y son capaces de inhibir la interacción de PD-L1 con PD-1 y CD80. Estos péptidos macrocíclicos exhiben eficacia inmunomoduladora in vitro, lo que los convierte en candidatos terapéuticos para el tratamiento de diversas enfermedades, incluidas el cáncer y las enfermedades infecciosas.
Los términos "unión específica" o "unión específica" se refieren a la interacción entre una proteína y una molécula de unión, tales como un compuesto o ligando. La interacción depende de la presencia de una estructura particular (es decir, un sitio de unión a enzimas, un determinante antigénico o epítopo) de la proteína que se reconoce por la molécula de unión. Por ejemplo, si un compuesto tiene una unión específica para el sitio de unión a proteínas "A", la presencia del compuesto en una reacción que contiene una proteína que incluye el sitio de unión A y un péptido marcado que se une específicamente al sitio de unión a la proteína A reducirá la cantidad de péptido marcado unido a la proteína. Por el contrario, la unión no específica de un compuesto a la proteína no da como resultado un desplazamiento dependiente de la concentración del péptido marcado de la proteína.
La presente divulgación pretende incluir todos los isótopos de átomos que se encuentran en los presentes compuestos. Los isótopos incluyen aquellos átomos que tienen el mismo número atómico pero diferentes números másicos. A modo de ejemplo general y sin limitación, los isótopos de hidrógeno incluyen deuterio y tritio. Los isótopos de carbono incluyen 13C y 14C. Los compuestos de la invención marcados isotópicamente se pueden preparar generalmente por técnicas convencionales conocidas por los expertos en la materia o por procedimientos análogos a los descritos en el presente documento, usando un reactivo marcado isotópicamente apropiado en lugar del reactivo no marcado empleado de otro modo. Dichos compuestos pueden tener diversos usos potenciales, por ejemplo, como patrones y reactivos para determinar la actividad biológica. En el caso de los isótopos estables, dichos compuestos pueden tener el potencial de modificar favorablemente las propiedades biológicas, farmacológicas o farmacocinéticas.
Un aspecto adicional de la materia objeto descrita en este documento es el uso de los péptidos descritos como ligandos radiomarcados para el desarrollo de ensayos de unión a ligandos o para el control de la adsorción in vivo, metabolismo, distribución, unión u ocupación de receptores, o disposición de compuesto. Por ejemplo, un péptido macrocíclico descrito en este documento puede prepararse usando el isótopo radiactivo 125I y el péptido radiomarcado resultante puede usarse para desarrollar un ensayo de unión para estudios de metabolismo. Como alternativa y para el mismo propósito, un péptido macrocíclico descrito en este documento puede convertirse en una forma radiomarcada mediante tritiación catalítica usando métodos conocidos por los expertos en la técnica.
Los péptidos macrocíclicos de la presente divulgación también se pueden usar como agentes de imagen de PET mediante la adición de un marcador radiactivo utilizando métodos conocidos por los expertos en la técnica.
Los péptidos preferentes incluyen al menos uno de los péptidos macrocíclicos proporcionados en este documento y estos péptidos pueden incluirse en composiciones y combinaciones farmacéuticas.
Las definiciones proporcionadas en este documento se aplican, sin limitación, a los términos usados en la presente memoria descriptiva, a no ser que se limite de otra forma en los casos específicos.
Los expertos en la técnica de la química de aminoácidos y péptidos son conscientes de que un aminoácido incluye un compuesto representado por la estructura general:

L- o S-a-aminoácido D- o R-a-aminoácido
(si R=H) (si R=H)
donde R y R' son como se discuten en este documento.
A menos que se indique otra cosa, el término "aminoácido" como se emplean este documento, solo o como parte de otro grupo, incluye, sin limitación, un grupo amino y un grupo carboxilo vinculado al mismo carbono, denominado carbono "a", donde R y/o R' pueden ser una cadena lateral natural o no natural, incluyendo hidrógeno. La configuración absoluta "S" en el carbono "a" se conoce comúnmente como la configuración "L" o "natural". En el caso en que los sustituyentes "R" y "R'" (prima) sean iguales a hidrógeno, el aminoácido es glicina y no es quiral.
Los términos "cadena lateral de aminoácido natural" y "cadena lateral de aminoácido de origen natural", como se usa en este documento, se refieren a la cadena lateral de cualquiera de los aminoácidos naturales (es decir, alanina, arginina, asparagina, ácido aspártico, cisteína, glutamina, ácido glutámico, glicina,-histidina, isoleucina, leucina, lisina, metionina, fenilalanina, prolina, serina, treonina, triptófano, tirosina y valina) habitualmente en la configuración S (es decir, el L-aminoácido).
Los términos "cadena lateral de aminoácido no natural" y "cadena lateral de aminoácido de origen no natural", como se usa en este documento, se refiere a una cadena lateral de cualquier aminoácido natural generalmente en la configuración R (es decir, el aminoácido D) o a un grupo distinto de una cadena lateral de aminoácido natural en la configuración R (o decir, el D-o L-aminoácido, respectivamente) seleccionado entre:
alquenilo C2-C7, alcoxi Ci-C3alquilo C1-C3, alcoxicarbonil Ci-C6alquilo C1-C3, alquilo Ci-C7, alquil C1-C3sulfanilalquilo C1-C3, alquil C1-C3 sulfonilalquilo C1-C3, amidoalquilo C1-C3, aminoalquilo C1-C3, azaindolilalquilo C1-C3, benzotiazolilalquilo C1-C3, benzotienilalquilo Ci-C3, benciloxialquilo C1-C3, carboxialquilo C1-C3, cianoalquilo C1-C3, cicloalquil C3-C6alquilo C1-C3, difenilmetilo, furanilalquilo C1-C3, haloalquilo C1-C3, hidroxialquilo C1-C3, imidazolilalquilo C1-C3, naftilalquilo C1-C3, piranilalquilo C1-C3, piridinilalquilo C1 -C3, tetrahidrofurilalquilo C1-C3, tiazolilalquilo C1-C3, tienilalquilo C1-C3;
bifenilalquilo C1-C3 en donde el bifenilo está opcionalmente sustituido con un grupo seleccionado entre alquilo C1-C3, amino, ciano, halo, hidroxi y nitro;
indolilalquilo C1-C3, en donde la parte indolilo está opcionalmente sustituida con un grupo seleccionado entre alquilo C1-C3, carboxialquilo C1-C3, ciano, halo, hidroxi, nitro y fenilo, en donde el fenilo está además opcionalmente sustituido con uno, dos, o tres grupos seleccionados independientemente entre alcoxi C1-C3, alquilo C1-C3 y halo;
NRaRb(alquilo C1-C7), en donde Ra y Rb se seleccionan independientemente entre hidrógeno, alqueniloxicarbonilo C2-C4, alcoxicarbonilo C1-C3, alquilo C1-C3, alquilcarbonilo C1-C3, cicloalquilcarbonilo C3-C6, furanilcarbonilo, fenilalquilo C1-C3, fenilcarbonilo, piranilcarbonilo, tetrahidrofurilcarbonilo y tienilcarbonilo. Cuando el conector alquilo contiene más de un carbono un grupo adicional NRaRb puede estar en la cadena.
NRcRdcarbonilalquilo C1-C3, en donde Rc y Rd se seleccionan independientemente entre hidrógeno, alquenilo C3-C4, alquilo C1-C3, fenilalquilo C1-C3 y trifenilmetilo;
fenilalquilo C1-C3 en donde la parte fenilo está opcionalmente sustituida con uno, dos, tres, cuatro, o cinco grupos seleccionados independientemente entre alcoxi C1-C4, alquilo C1-C4, alquilsulfonilamino Ci-C3, amido, amino, aminoalquilo C1-C3, aminosulfonilo, carboxi, ciano, halo, haloalquilo C1-C3, hidroxi, -NC(NH2)2, nitro y -OP(O)(OH)2; y
fenoxialquilo C1-C3 en donde el fenilo está opcionalmente sustituido con uno, dos, tres, cuatro, o cinco grupos seleccionados independientemente entre alcoxi C1-C4, alquilo C1-C4, alquilsulfonilamino C1-C3, amido, amino, aminoalquilo C1-C3, aminosulfonilo, carboxi, ciano, halo, haloalquilo C1-C3, hidroxi y nitro.
El término "alquenilo C2-C4", como se usa en este documento, se refiere a un grupo de cadena lineal o ramificada de dos a cuatro átomos de carbono que contiene al menos un doble enlace carbono-carbono.
El término "alquenilo C3-C4", como se usa en este documento, se refiere a un grupo de cadena lineal o ramificada de tres o cuatro átomos de carbono que contiene al menos un doble enlace carbono-carbono.
El término "alquenilo C2-C7", como se usa en este documento, se refiere a un grupo de cadena lineal o ramificada de dos a siete átomos de carbono que contiene al menos un doble enlace carbono-carbono.
El término "alqueniloxi C2-C4", como se usa en este documento, se refiere a un grupo alquenilo C2-C4 unido al resto molecular precursor a través de un átomo de oxígeno.
El término "alcoxi C1-C3", como se usa en este documento, se refiere a un grupo alquilo C1-C3 unido al resto molecular precursor a través de un átomo de oxígeno.
El término "alcoxi C1-C4", como se usa en este documento, se refiere a un grupo alquilo C1-C4 unido al resto molecular precursor a través de un átomo de oxígeno.
El término "alcoxi C1-C6", como se usa en este documento, se refiere a un grupo alquilo C1-C6 unido al resto molecular precursor a través de un átomo de oxígeno.
El término "alcoxi C1-C3alquilo C1-C3", como se usa en este documento, se refiere a un grupo alcoxi C1-C3 unido al resto molecular precursor a través de un grupo alquilo C1-C3.
El término "alcoxicarbonilo C1-C3", como se usa en este documento, se refiere a un grupo alcoxi C1-C3 unido al resto molecular precursor a través de un grupo carbonilo.
El término "alcoxicarbonilo C1-C6", como se usa en este documento, se refiere a un grupo alcoxi C1-C6 unido al resto molecular precursor a través de un grupo carbonilo.
El término "alcoxicarbonil C1-C6alquilo C1-C3", como se usa en este documento, se refiere a un grupo alcoxicarbonilo Ci-C6 unido al resto molecular precursor a través de un grupo alquilo C1-C3.
El término "alquilo C1-C3", como se usa en este documento, se refiere a un grupo obtenido a partir de un hidrocarburo saturado de cadena lineal o ramificada que contiene de uno a tres átomos de carbono.
El término "alquilo C1-C4", como se usa en este documento, se refiere a un grupo obtenido a partir de un hidrocarburo saturado de cadena lineal o ramificada que contiene de uno a cuatro átomos de carbono.
El término "alquilo CrC6", como se usa en este documento, se refiere a un grupo obtenido a partir de un hidrocarburo saturado de cadena lineal o ramificada que contiene de uno a seis átomos de carbono.
El término "alquilcarbonilo C1-C3", como se usa en este documento, se refiere a un grupo alquilo C1-C3 unido al resto molecular precursor a través de un grupo carbonilo.
El término "alquilsulfanilo C1-C3", como se usa en este documento, se refiere a un grupo alquilo C1-C3 unido al resto molecular parental a través de un átomo azufre.
El término "alquilsulfanil C1-C3alquilo C1-C3", como se usa en este documento, se refiere a un grupo alquilsulfanilo Ci-C3 unido al resto molecular precursor a través de un grupo alquilo C1-C3.
El término "alquilsulfonilo C1-C3", como se usa en este documento, se refiere a un grupo alquilo C1-C3 unido al resto molecular precursor a través de un grupo sulfonilo.
El término "alquilsulfonil C1-C3alquilo C1-C3", como se usa en este documento, se refiere a un grupo alquilsulfonilo Ci-C3 unido al resto molecular precursor a través de un grupo alquilo C1-C3.
El término "alquilsulfonilamino C1-C3", como se usa en este documento, se refiere a un grupo alquilsulfonilo Ci-C3 unido al resto molecular precursor a través de un grupo amino.
El término "amido", como se usa en este documento, se refiere a -C(O)NH2.
El término "amidoalquilo C1-C3", como se usa en este documento, se refiere a un grupo amido unido al resto molecular parental a través de un grupo alquilo C1-C3.
El término "amino", como se usa en este documento, se refiere a -NH2.
El término "aminoalquilo C1-C3", como se usa en este documento, se refiere a un grupo amino unido al resto molecular parental a través de un grupo alquilo C1-C3.
El término "aminosulfonilo", como se usa en este documento, se refiere a un grupo amino unido al resto molecular precursor a través de un grupo sulfonilo.
El término "azaindolilalquilo C1-C3", como se usa en este documento, se refiere a un grupo azaindolilo unido al resto molecular precursor a través de un grupo alquilo C1-C3. El grupo azaindolilo puede estar unido al resto alquilo a través de cualquier átomo sustituible en el grupo.
El término "benzotiazolilalquilo C1-C3", como se usa en este documento, se refiere a un grupo benzotiazolilo unido al resto molecular precursor a través de un grupo alquilo C1-C3. El grupo benzotiazolilo puede estar unido al resto alquilo a través de cualquier átomo sustituible en el grupo.
El término "benzotienilalquilo C1-C3", como se usa en este documento, se refiere a un grupo benzotienilo unido al resto molecular precursor a través de un grupo alquilo C1-C3. El grupo benzotienilo puede estar unido al resto alquilo a través de cualquier átomo sustituible en el grupo.
El término "benciloxi", como se usa en este documento, se refiere a un grupo bencilo unido al resto molecular precursor a través de un átomo de oxígeno.
El término "benciloxialquilo C1-C3", como se usa en este documento, se refiere a un grupo benciloxi unido al resto molecular precursor a través de un grupo alquilo C1-C3.
El término "bifenilalquilo C1-C3", como se usa en este documento, se refiere a un grupo bifenilo unido al resto molecular precursor a través de un grupo alquilo C1-C3. El grupo bifenilo puede estar unido al resto alquilo a través de cualquier átomo sustituible en el grupo.
El término "carbonilo", como se usa en este documento, se refiere a -C(O)-.
El término "carboxi", como se usa en este documento, se refiere a -CO2H.
El término "carboxialquilo C1-C3", como se usa en este documento, se refiere a un grupo carboxi unido al resto molecular parental a través de un grupo alquilo C1-C3.
El término "ciano", como se usa en este documento, se refiere a -CN.
El término "cianoalquilo C1-C3", como se usa en este documento, se refiere a un grupo ciano unido al resto molecular parental a través de un grupo alquilo C1-C3.
El término "cicloalquilo C3-C6", como se usa en este documento, se refiere a un sistema de anillo hidrocarbonado monocíclico saturado que tiene de tres a seis átomos de carbono y cero heteroátomos.
El término "cicloalquil C3-C6alquilo C1-C3", como se usa en este documento, se refiere a un grupo cicloalquilo C3-C6 unido al resto molecular precursor a través de un grupo alquilo C1-C3.
El término "cicloalquilcarbonilo C3-C6", como se usa en este documento, se refiere a un grupo cicloalquilo C3-C6 unido al resto molecular precursor a través de un grupo carbonilo.
El término "furanilalquilo C1-C3", como se usa en este documento, se refiere a un grupo furanilo unido al resto molecular precursor a través de un grupo alquilo C1-C3. El grupo furanilo puede estar unido al resto alquilo a través de cualquier átomo sustituible en el grupo.
El término "furanilcarbonilo", como se usa en este documento, se refiere a un grupo furanilo unido al resto molecular precursor a través de un grupo carbonilo.
Los términos "halo" y "halógeno", como se usa en este documento, se refieren a F, Cl, Br o I.
El término "haloalquilo C1-C3", como se usa en este documento, se refiere a un grupo alquilo C1-C3 sustituido con uno, dos o tres átomos de halógeno.
El término "halometilo", como se usa en este documento, se refiere a un grupo metilo sustituido con uno, dos o tres átomos de halógeno.
El término "hidroxi", como se usa en este documento, se refiere a -OH.
El término "hidroxialquilo C1-C3", como se usa en este documento, se refiere a un grupo hidroxi unido al resto molecular parental a través de un grupo alquilo C1-C3.
El término "imidazolilalquilo C1-C3", como se usa en este documento, se refiere a un grupo imidazolilo unido al resto molecular parental a través de un grupo alquilo C1-C3. El grupo imidazolilo puede estar unido al resto alquilo a través de cualquier átomo sustituible en el grupo.
El término "indolilalquilo C1-C3", como se usa en este documento, se refiere a un grupo indolilo unido al resto molecular precursor a través de un grupo alquilo C1-C3. El grupo indolilo puede estar unido al resto alquilo a través de cualquier átomo sustituible en el grupo.
El término "naftilalquilo C1-C3", como se usa en este documento, se refiere a un grupo naftilo unido al resto molecular precursor a través de un grupo alquilo C1-C3. El grupo naftilo puede estar unido al resto alquilo a través de cualquier átomo sustituible en el grupo.
El término "nitro", como se usa en este documento, se refiere a -NO2.
El término "NRaRb", como se usa en este documento, se refiere a dos grupos, Ra y Rb, que están unidos al resto molecular parental a través de un átomo de nitrógeno. Ra y Rb se seleccionan independientemente entre hidrógeno, alqueniloxicarbonilo C2-C4, alquilcarbonilo C1-C3, cicloalquilcarbonilo C3-C6, furanilcarbonilo y fenilcarbonilo.
El término "NRaRbalquilo (C1-C3)", como se usa en este documento, se refiere a un grupo NRaRb unido al resto molecular precursor a través de un grupo alquilo C1-C3.
El término "NRcRd", como se usa en este documento, se refiere a dos grupos, Rc y Rd, que están unidos al resto molecular parental a través de un átomo de nitrógeno. Rc y Rd se seleccionan independientemente entre hidrógeno, alquilo C1-C3 y trifenilmetilo.
El término "NRcRdcarbonilo", como se usa en este documento, se refiere a un grupo NRcRd unido al resto molecular precursor a través de un grupo carbonilo.
El término "NRcRdcarbonilalquilo C1-C3", como se usa en este documento, se refiere a un grupo NRcRdcarbonilo unido al resto molecular precursor a través de un grupo alquilo C1-C3.
El término "fenoxi", como se usa en este documento, se refiere a un grupo fenilo unido al resto molecular parental a través de un átomo de oxígeno.
El término "fenoxialquilo C1-C3", como se usa en este documento, se refiere a un grupo fenoxi unido al resto molecular precursor a través de un grupo alquilo C1-C3.
El término "fenilalquilo C1-C3", como se usa en este documento, se refiere a un grupo fenilo unido al resto molecular parental a través de un grupo alquilo C1-C3.
El término "fenilcarbonilo", como se usa en este documento, se refiere a un grupo fenilo unido al resto molecular parental a través de un grupo carbonilo.
El término "piranilalquilo C1-C3", como se usa en este documento, se refiere a un grupo piranilo unido al resto molecular precursor a través de un grupo alquilo C1-C3. El grupo piranilo puede estar unido al resto alquilo a través de cualquier átomo sustituible en el grupo.
El término "piranilcarbonilo", como se usa en este documento, se refiere a un grupo piranilo unido al resto molecular precursor a través de un grupo carbonilo.
El término "piridinilalquilo C1-C3", como se usa en este documento, se refiere a un grupo piridinilo unido al resto molecular precursor a través de un grupo alquilo C1-C3. El grupo piridinilo puede estar unido al resto alquilo a través de cualquier átomo sustituible en el grupo.
El término "sulfanilo", como se usa en este documento, se refiere a -S-.
El término "sulfonilo", como se usa en este documento, se refiere a -SO2-.
El término "tetrahidrofurilalquilo C1-C3", como se usa en este documento, se refiere a un grupo tetrahidrofurilo unido al resto molecular precursor a través de un grupo alquilo C1-C3. El grupo tetrahidrofurilo puede estar unido al resto alquilo a través de cualquier átomo sustituible en el grupo.
El término "tetrahidrofurilcarbonilo", como se usa en este documento, se refiere a un grupo tienilo unido al resto molecular precursor a través de un grupo carbonilo.
El término "tiazolilalquilo C1-C3", como se usa en este documento, se refiere a un grupo tiazolilo unido al resto molecular parental a través de un grupo alquilo C1-C3. El grupo tiazolilo puede estar unido al resto alquilo a través de cualquier átomo sustituible en el grupo.
El término "tienilalquilo C1-C3", como se usa en este documento, se refiere a un grupo tienilo unido al resto molecular precursor a través de un grupo alquilo C1-C3. El grupo tienilo puede estar unido al resto alquilo a través de cualquier átomo sustituible en el grupo.
El término "tienilcarbonilo", como se usa en este documento, se refiere a un grupo tienilo unido al resto molecular precursor a través de un grupo carbonilo.
El término "tratar" se refiere a: (i) prevenir que una enfermedad, trastorno, o afección que ocurre en un paciente que puede estar predispuesto a la enfermedad, trastorno y/o afección, pero al que aún no se le ha diagnosticado que la tenga; (ii) inhibir la enfermedad, trastorno o afección, es decir, detener su desarrollo; y (iii) aliviar la enfermedad, trastorno o afección, es decir, provocar la regresión de la enfermedad, trastorno y/o afección y/o síntomas asociados con la enfermedad, trastorno y/o afección.
La unión de péptidos macrocíclicos a PD-L1 puede medirse, por ejemplo, por métodos tales como fluorescencia homogénea de resolución temporal (HTRF), Resonancia de plasmón superficial (SPR), calorimetría de titulación isotérmica (ITC), espectroscopía de resonancia magnética nuclear (RMN) y similares. Además, la unión de los péptidos macrocíclicos a PD-L1 expresada en la superficie de las células puede medirse como se describe en este documento en ensayos de unión celular.
La administración de un agente terapéutico descrito en este documento incluye, sin limitación, administración de una cantidad terapéuticamente eficaz de agente terapéutico. El término "cantidad terapéuticamente eficaz" como se usa en este documento se refiere, sin limitación, a una cantidad de un agente terapéutico para tratar o prevenir una afección tratable mediante la administración de una composición de los inhibidores de unión a PD-1/PD-L1 descritos en este documento. Esta cantidad es la cantidad suficiente para presentar un efecto terapéutico o preventivo o mejorador. El efecto puede incluir, por ejemplo y sin limitación, el tratamiento o la prevención de las dolencias relacionadas en el presente documento. La cantidad eficaz precisa para un sujeto dependerá del tamaño y la salud del sujeto, la naturaleza y la extensión de la dolencia que se está tratando, recomendaciones del médico tratante y la terapéutica o combinación de la terapéutica seleccionada para la administración. Por tanto, no es útil especificar una cantidad efectiva exacta por adelantado.
En otro aspecto, la divulgación se refiere a los péptidos macrocíclicos de la presente divulgación para su uso en la inhibición del crecimiento de células tumorales en un sujeto. Tal como se demuestra en el presente documento, los péptidos macrocíclicos de la presente divulgación son capaces de unirse a PD-L1, interrumpiendo la interacción entre PD-L1 y PD-1, compitiendo con la unión de PD-L1 con anticuerpos monoclonales anti-PD-1 que se sabe que bloquean la interacción con PD-1, mejoran la secreción de IFNy de células T específicas de CMV y mejoran la secreción de IFNg de células T específicas de VIH. Como resultado, los péptidos macrocíclicos de la presente divulgación son útiles para modificar una respuesta inmune, tratar enfermedades tales como cáncer o enfermedades infecciosas, estimulando una respuesta autoinmune protectora para estimular respuestas inmunes específicas de antígeno (por ejemplo, mediante la administración conjunta de péptidos bloqueadores de PD-L1 con un antígeno de interés).
Con el fin de que se pueda entender más fácilmente la presente divulgación, se definen previamente determinados términos. Se establecen definiciones adicionales a lo largo de la descripción detallada.
Los términos "ligando de muerte programada 1", "ligando de muerte celular programada 1", "proteína PD-L1", "PD-L1", "PDL1", "PDCDL1", "hPD-L1", "hPD-LI", "CD274" y "B7-H1" se usan intercambiablemente, e incluyen variantes, isoformas, homólogos de especie de PD-L1 humana y análogos que tienen al menos un epítopo común con PD-L1. La secuencia completa de PD-L1 puede encontrarse en el n.° de acceso GENBANK® NP_054862.
Los términos "muerte programada 1", "muerte celular programada 1", "proteína PD-1", "PD-1", "PD1", "PDCD1", "hPD-1" y "hPD-I" se usan de forma indistinta e incluyen variantes, isoformas, homólogos de especie de PD-1 humana y análogos que tienen al menos un epítopo común con PD-1. La secuencia completa de PD-1 puede encontrarse en el n.° de acceso GENBANK® U64863.
Los términos "antígeno 4 asociado a linfocitos T citotóxicos", "CTLA-4", "CTLA4", "antígeno CTLA-4" y "CD152" (véase, por ejemplo, Murata, Am. J. Pathol., 155:453-460 (1999)) se usan intercambiablemente, e incluyen variantes, isoformas, homólogos de especie de CTLA-4 humano y análogos que tienen al menos un epítopo como un con CTLA-4 (véase, por ejemplo, Balzano, Int. J. Cancer Suppl., 7:28-32 (1992)). La secuencia de ácidos nucleicos completa de CTLA-4 puede encontrarse con el n.° de acceso GENBANK® L15006.
La expresión "respuesta inmunitaria" se refiere a la acción de, por ejemplo, linfocitos, células presentadoras de antígenos, células fagocíticas, granulocitos y macromoléculas solubles producidao por las células anteriores o el hígado (incluidos péptidos macrocíclicos, citoquinas y complemento) que da como resultado un daño selectivo sobre, la destrucción de o la eliminación en el cuerpo humano de patógenos invasores, células o tejidos infectados por patógenos, células cancerosas o, en los casos de autoinmunidad o inflamación patológica, células o tejidos humanos normales.
Un "evento adverso" (AE) como se usa en este documento es desfavorable y generalmente no intencionado, incluso indeseable, signo (incluido un hallazgo anormal de laboratorio), síntoma, enfermedad asociada con el uso de un tratamiento médico. Por ejemplo, un evento adverso puede estar asociado con la activación del sistema inmune o la
expansión de las células del sistema inmune (por ejemplo, linfocitos T) en respuesta a un tratamiento. Un tratamiento médico puede tener uno o más EA asociados y cada AE puede tener el mismo nivel de gravedad o diferente. La referencia a métodos capaces de "alterar los eventos adversos" significa un régimen de tratamiento que disminuye la incidencia y/o severidad de uno o más EA asociados con el uso de un régimen de tratamiento diferente.
Como se usa en este documento, la "enfermedad hiperproliferativa" se refiere a condiciones en las que el crecimiento celular aumenta a niveles normales. Por ejemplo, enfermedades hiperproliferativas o trastornos incluyen enfermedades malignas (por ejemplo, cáncer de esófago, cáncer de colon, cáncer biliar) y enfermedades no malignas (por ejemplo, ateroesclerosis, hiperplasia benigna, hipertrofia prostática benigna).
Como se usa en este documento, "aproximadamente" o "que comprende esencialmente de" significa dentro de un rango de error aceptable para el valor particular determinado por un experto en la técnica, que dependerá en parte de cómo se mida o determine el valor, es decir, de las limitaciones del sistema de medida. Por ejemplo, "aproximadamente" o "que comprende esencialmente de" puede significar dentro de una o más de una desviación estándar según la práctica en la técnica. Como alternativa, "aproximadamente" o "que comprende esencialmente de" puede significar un intervalo de hasta 20 %. Además, particularmente con respecto a los sistemas o procesos biológicos, las expresiones pueden significar hasta un orden de magnitud o hasta 5 veces de un valor. Cuando se proporcionan valores particulares en la solicitud y reivindicaciones, a menos que se indique lo contrario, se debe suponer que el significado de "aproximadamente" o "que comprende esencialmente de" está dentro de un rango de error aceptable para ese valor particular.
Como se describe en este documento, cualquier intervalo de concentración, intervalo de porcentaje, intervalo de relación o intervalo de número entero se entiende que incluye el valor de cualquier número entero dentro del intervalo citado y, cuando sea adecuado, fracciones del mismo (tal como una décima y una centésima de un número entero), salvo que se indique otra cosa.
Ensayos competitivos
La presente descripción también se refiere a péptidos macrocíclicos que son capaces de competir con la unión de un anticuerpo anti-PD-Ll de referencia (MDX-1105) en al menos aproximadamente 20 %, al menos aproximadamente 30 %, al menos aproximadamente 40 %, al menos aproximadamente 50 %, al menos aproximadamente 60 %, al menos aproximadamente 70 %, al menos aproximadamente 80 %, al menos aproximadamente 90 % y al menos aproximadamente 100 %. Dichos péptidos macrocíclicos pueden compartir homología estructural con uno o más péptidos macrocíclicos descritos en este documento, incluyendo mutante, sustitución conservadora, sustitución funcional y formas de deleción, siempre que se unan específicamente a PD-L1. Por ejemplo, si un péptido macrocíclico se une sustancialmente a la misma región de PD-L1 que un anticuerpo anti-PD-Ll de referencia, el péptido macrocíclico debe unirse a un epítopo de PD-L1 que al menos se superpone con el epítopo PD-L1 al que se une el anticuerpo monoclonal anti-PD-Ll. La región superpuesta puede variar desde un residuo de aminoácido hasta varios cientos de residuos de aminoácido. El péptido macrocíclico debe entonces competir con y/o bloquear la unión del anticuerpo monoclonal anti-PD-Ll a PD-L1 y así disminuir la unión del anticuerpo monoclonal anti-PD-Ll a PD-L1, probablemente en al menos 50 % en un ensayo de competición.
Los anticuerpos anti-PD-Ll que pueden usarse como anticuerpos de referencia para fines de ensayos de competición se conocen en la técnica. Por ejemplo, pueden usarse los siguientes anticuerpo representativos anti-PD-Ll: MDX-1105 (BMS); L01X-C (Serono), L1X3 (Serono), MSB-0010718C (Serono) y PD-L1 Probody (CytomX) y los anticuerpos PD-L1 desvelados en el documento del solicitante WO 2007/005874.
Os anticuerpos Anti-PD-1 que pueden usarse como anticuerpos de referencia para fines de ensayos de competición se conocen en la técnica. Por ejemplo, pueden usarse los siguientes anticuerpos representativos anti-PD-1: nivolumab (BMS); 17D8, 2D3, 4H1, 4A11, 7D3 y 5F4 desvelado cada uno en el documento de patente de Estados Unidos del propietario n.° 8.008.449 (BMS), MK-3475 (Merck, desvelado en el documento de patente de Estados Unidos n.° 8.168.757) y los anticuerpos desvelados en la patente de Estados Unidos n.° 7.488.802.
Composiciones farmacéuticas
En otro aspecto, la presente divulgación proporciona una composición, por ejemplo, una composición farmacéutica, que contiene uno o una combinación de péptidos macrocíclicos de la presente divulgación, formulado junto con un vehículo farmacéuticamente aceptable. Dichas composiciones pueden incluir uno o una combinación de (por ejemplo, dos o más diferentes) péptidos macrocíclicos, inmunoconjugados o moléculas biespecíficas de la divulgación. Por ejemplo, una composición farmacéutica de la divulgación puede comprender una combinación de péptidos macrocíclicos (inmunoconjugados o biespecíficos) que se unen a diferentes epítopos en el antígeno diana o que tienen actividades complementarias.
Las composiciones farmacéuticas de la divulgación también pueden administrarse en terapia de combinación, es decir, combinadas con otros agentes. Por ejemplo, la terapia combinada puede incluir un péptido macrocíclico combinado con al menos otro agente antiinflamatorio o inmunosupresor. Los ejemplos de agentes terapéuticos que
se pueden usar en la terapia de combinación se describen en mayor detalle a continuación en la sección sobre usos de los péptidos macrocíclicos de la divulgación.
Como se usa en este documento, "transportador farmacéuticamente aceptable" incluye todos y cada uno de los disolventes, medios de dispersión, recubrimientos, agentes antibacterianos y antifúngicos, agentes isotónicos y agentes retardantes de la absorción y similares, que sean fisiológicamente compatibles. Preferentemente, el vehículo es adecuado para intravenosa, intramuscular, subcutánea, parenteral, administración espinal o epidérmica (por ejemplo, por inyección o infusión). Dependiendo de la vía de administración, el compuesto activo, es decir, un péptido macrocíclico, inmunoconjugado o molécula biespecífica, se puede recubrir con un material para proteger el compuesto de la acción de los ácidos y de otras condiciones naturales que pueden inactivar el compuesto.
Los compuestos farmacéuticos de la divulgación pueden incluir una o más sales farmacéuticamente aceptables. Una "sal farmacéuticamente aceptable" o "sal terapéuticamente aceptable" se refiere a una sal que retiene la actividad biológica deseada del compuesto original y no imparte ningún efecto toxicológico no deseado (véase, por ejemplo, Berge, S.M. et al., J. Pharm. Sci., 66:1-19 (1977)). Los ejemplos de dichas sales incluyen sales de adición de ácido y sales de adición de bases. Las sales de adición de ácidos incluyen las derivadas de ácidos inorgánicos no tóxicos, tales como ácido clorhídrico, nítrico, fosfórico, sulfúrico, bromhídrico yodhídrico, fosforoso y similares, así como de ácidos orgánicos no tóxicos, tales como ácidos alifáticos monocarboxílicos y dicarboxílicos, ácidos alcanoicos sustituidos con fenilo, ácidos hidroxialcanoicos, ácidos aromáticos, ácidos sulfónicos alifáticos y aromáticos y similares. Las sales de adición de bases incluyen aquellas derivadas de metales alcalinotérreos, tales como sodio, potasio, magnesio, calcio y similares, así como de aminas orgánicas no tóxicas, tales como N,N'-dibenciletilendiamina, W-metilglucamina, cloroprocaína, colina, dietanolamina, etilendiamina, procaína y similares. Una composición farmacéutica de la divulgación también pueden incluir un antioxidante farmacéuticamente aceptable. Ejemplos de antioxidantes farmacéuticamente aceptables incluyen: (1) antioxidantes solubles en agua, tales como ácido ascórbico, clorhidrato de cisteína, bisulfato de sodio, metabisulfito de sodio, sulfito de sodio y similares; (2) antioxidantes solubles en aceite, tales como palmitato de ascorbilo, hidroxianisol butilado (BHA), hidroxitolueno butilado (BHT), lecitina, galato de propilo, alfa-tocoferol y similares; y (3) agentes quelantes de metales, tales como ácido cítrico, ácido etilendiaminotetraacético (EDTA), sorbitol, ácido tartárico, ácido fosfórico y similares.
Ejemplos de vehículos acuosos y no acuosos adecuados que pueden emplearse en las composiciones farmacéuticas de la divulgación incluyen agua, etanol, polioles (tales como glicerol, propilenglicol, polietilenglicol y similares) y mezclas adecuadas de los mismos, aceites vegetales, tales como aceite de oliva y ésteres orgánicos inyectables, tales como oleato de etilo. La fluidez adecuada se puede mantener, por ejemplo, mediante el uso de materiales de recubrimiento, tal como lecitina, mediante el mantenimiento del tamaño de partícula necesario en caso de dispersiones y mediante el uso de tensioactivos.
Estas composiciones también pueden contener adyuvantes tales como agentes conservantes, agentes humectantes, agentes emulsionantes y agentes dispersantes. La prevención de la presencia de microorganismos puede garantizarse mediante procedimientos de esterilización, véase anteriormente y mediante la inclusión de varios agentes antibacterianos y antifúngicos, por ejemplo, parabeno, clorobutanol, fenol de ácido sórbico y similares. También puede ser deseable incluir agentes isotónicos, tales como azúcares, cloruro sódico y similares en las composiciones. Además, la absorción prolongada de la forma inyectable del fármaco se puede llevar a cabo mediante la inclusión de agentes que retrasen la absorción, tales como monoestearato de aluminio y gelatina.
Los vehículos farmacéuticamente aceptables incluyen soluciones acuosas estériles o dispersiones y polvos estériles para la preparación extemporánea de soluciones estériles inyectables o dispersiones. El uso de dichos medios y agentes para sustancias farmacéuticamente activas es conocido en la materia. Excepto en el caso de que cualquier agente o medio convencional sea incompatible con el compuesto activo, se contempla el uso de los mismos en las composiciones farmacéuticas de la divulgación. También pueden incorporarse compuestos activos complementarios en las composiciones.
Normalmente, las composiciones terapéuticas deben ser estériles y estables en las condiciones de fabricación y almacenamiento. La composición puede formularse como una solución, microemulsión, liposoma u otra estructura ordenada adecuada para la alta concentración de fármaco. El vehículo puede ser un disolvente o medio de dispersión que contiene, por ejemplo, agua, etanol, poliol (por ejemplo, glicerol, propilenglicol y polietilenglicol líquido y similares) y mezclas adecuadas de los mismos. La fluidez apropiada puede mantenerse, por ejemplo, mediante el uso de un recubrimiento tal como lecitina, mediante el mantenimiento del tamaño de partícula necesario en el caso de una dispersión y mediante el uso de tensioactivos. En muchos casos, será preferible incluir agentes isotónicos, por ejemplo, azúcares, polialcoholes tales como manitol, sorbitol o cloruro sódico en la composición. Puede lograrse la absorción prolongada de las composiciones inyectables incluyendo en la composición un agente que retrase la absorción, por ejemplo, sales de monoestearato y gelatina.
Las soluciones inyectables estériles pueden prepararse incorporando el compuesto activo en la cantidad necesaria en un disolvente adecuado con uno o una combinación de los ingredientes enumerados anteriormente, según sea
necesario, seguido de esterilización por microfiltración. En general, las dispersiones se preparan mediante la incorporación del compuesto activo en un vehículo estéril que contiene un medio de dispersión básico y los otros ingredientes requeridos de los enumerados anteriormente. En el caso de polvos estériles para la preparación de soluciones inyectables estériles, los métodos de preparación preferidos son el secado al vacío y el secado en frío (liofilización) que producen un polvo del principio activo más cualquier ingrediente adicional deseado a partir una solución del mismo previamente esterilizada por filtración.
La cantidad de ingrediente activo que se puede combinar con un material vehículo para producir una sola forma de dosificación variará dependiendo del sujeto a tratar y el modo particular de administración. La cantidad de principio activo que se puede combinar con un material portador para producir una sola forma farmacéutica, en general, será la cantidad de la composición que produzca un efecto terapéutico. En general, de entre un cien por ciento, esta cantidad variará de aproximadamente 0,01 por ciento a aproximadamente noventa y nueve por ciento de ingrediente activo, aproximadamente de aproximadamente 0,1 por ciento a aproximadamente 70 por ciento, más convenientemente de aproximadamente 1 por ciento a aproximadamente 30 por ciento de ingrediente activo junto con un vehículo farmacéuticamente aceptable.
Los regímenes de dosificación se ajustan para proporcionar la respuesta óptima deseada (por ejemplo, una respuesta terapéutica). Por ejemplo, se puede administrar un único bolo, pueden administrarse varias dosis divididas con el tiempo o la dosis puede reducirse proporcionalmente o aumentarse según lo indiquen las exigencias de la situación terapéutica. Es especialmente ventajoso formular las composiciones parenterales en forma de dosis unitaria para facilitar la administración y la uniformidad de la dosis. La forma de dosificación unitaria usada en el presente documento se refiere a unidades físicamente discretas adecuadas como dosificaciones unitarias para los sujetos a tratar; cada unidad contiene una cantidad predeterminada de compuesto activo calculada para producir el efecto terapéutico deseado en asociación con el vehículo farmacéutico requerido. La memoria descriptiva para las formas de unidad de dosificación de la divulgación está dictada por y dependiente directamente de (a) las características únicas del compuesto activo y el efecto terapéutico particular que se va a lograr y (b) las limitaciones inherentes en la técnica de la composición de tales un compuesto activo para el tratamiento de sensibilidad en individuos.
Para la administración del péptido macrocíclico, la dosificación varía de aproximadamente 0,0001 a 100 mg/kg y más usualmente 0,01 a 5 mg/kg, del peso corporal del huésped. Por ejemplo las dosificaciones pueden ser 0,3 mg/kg de peso corporal, 1 mg/kg de peso corporal, 3 mg/kg de peso corporal, 5 mg/kg de peso corporal o 10 mg/kg de peso corporal o en el intervalo de 1-10 mg/kg. Un régimen de tratamiento a modo de ejemplo implica la administración una vez al día, dos veces al día, quincenalmente, tri-semanalmente, semanalmente, una vez cada dos semanas, una vez cada tres semanas, una vez cada cuatro semanas, una vez al mes, una vez cada 3 meses o una vez cada tres a 6 meses. Los regímenes de dosificación preferentes para un péptido macrocíclico de la descripción incluyen 1 mg/kg de peso corporal o 3 mg/kg de peso corporal mediante administración intravenosa, con el macrociclo que se administra usando uno de los siguientes horarios de dosificación: (i) cada cuatro semanas para seis dosis, entonces cada tres meses; (ii) cada tres semanas; (iii) 3 mg/kg de peso corporal una vez seguido de 1 mg/kg de peso corporal cada tres semanas.
Se pueden administrar dos o más péptidos macrocíclicos con diferentes especificidades de unión simultáneamente, en ese caso, la dosis de cada compuesto administrado cae dentro de los rangos indicados. Los compuestos generalmente se administran en múltiples ocasiones. Los intervalos entre dosis individuales pueden ser, por ejemplo, semanalmente, mensualmente, cada tres meses o anualmente. Los intervalos también pueden ser irregulares como se indica midiendo los niveles sanguíneos de péptido macrocíclico para el antígeno diana en el paciente. En algunas realizaciones, la dosificación se ajusta para conseguir una concentración plasmática de aproximadamente 1-1000.mu.g/ ml y en algunas realizaciones aproximadamente 25-300.mu.g/ ml.
Como alternativa, el péptido macrocíclico puede administrarse como una formulación de liberación sostenida, en cuyo caso se requiere una administración menos frecuente. La dosificación y la frecuencia de administración pueden variar dependiendo de si el tratamiento es profiláctico o terapéutico. En aplicaciones profilácticas, se administra una dosificación relativamente baja a intervalos relativamente infrecuentes durante un periodo de tiempo largo. Algunos pacientes continúan recibiendo tratamiento el resto de sus vidas. En aplicaciones terapéuticas, a veces se requiere una dosis relativamente alta a intervalos relativamente cortos hasta que la progresión de la enfermedad se reduzca o termine y hasta que el paciente muestre una mejoría parcial o completa de los síntomas de la enfermedad. Después de eso, al paciente se le puede administrar un régimen profiláctico.
Los niveles de dosificación reales de los ingredientes activos en las composiciones farmacéuticas de la presente divulgación pueden variar para obtener una cantidad del ingrediente activo que sea eficaz para lograr la respuesta terapéutica deseada para un paciente en particular, composición y modo de administración particular, sin que sean tóxicos para el paciente. El nivel de dosificación seleccionado dependerá de varios factores farmacocinéticos que incluyen la actividad de las composiciones particulares de la presente divulgación empleadas, el éster, la sal o la amida del mismo, la vía de administración, el tiempo de administración, la tasa de excreción del compuesto particular que se está empleando, la duración del tratamiento, otros fármacos, compuestos y/o materiales utilizados junto con las composiciones particulares empleadas, la edad, sexo, el peso, la afección, estado de salud general e historial
médico previo del paciente que se está tratando y factores similares bien conocidos en las técnicas médicas.
Una "dosificación terapéuticamente eficaz" de un péptido macrocíclico de la divulgación puede resultar en una disminución de la gravedad de los síntomas de la enfermedad, un aumento en la frecuencia y duración de los períodos libres de síntomas de la enfermedad, o una prevención del empeoramiento o discapacidad debido a la enfermedad. Por ejemplo, para el tratamiento de tumores, una "dosis terapéuticamente eficaz" inhibe el crecimiento celular o el crecimiento tumoral en al menos aproximadamente 20 %, más preferentemente en al menos aproximadamente 40 %, incluso más preferentemente en al menos aproximadamente el 60 % y aún más preferentemente en al menos aproximadamente el 80 % en relación con sujetos no tratados. La capacidad de un compuesto para inhibir el crecimiento tumoral y/o VIH se puede evaluar en un sistema modelo animal de eficacia predictiva en tumores humanos o eficacia viral. Como alternativa, esta propiedad de una composición puede evaluarse examinando la capacidad del compuesto para inhibir, dicha inhibición in vitro mediante ensayos conocidos por el profesional experto. Una cantidad terapéuticamente eficaz de un compuesto terapéutico puede disminuir el tamaño del tumor, disminuir la carga viral, o de lo contrario mejorar los síntomas en un sujeto. Una habilidad normal en la técnica sería capaz de determinar tales cantidades en función de factores tales como el tamaño del sujeto, la gravedad de los síntomas del sujeto y la composición particular de la ruta de administración seleccionada.
En otro aspecto, la presente divulgación proporciona un kit farmacéutico de partes que comprende un péptido macrocíclico y otro inmumodulador, como se describe en el presente documento. El kit también puede comprender además instrucciones para el uso en el tratamiento de una enfermedad hiperproliferativa (cáncer tal como se describe en este documento) y/o enfermedad antiviral.
Una composición de la presente divulgación se puede administrar a través de una o más rutas de administración utilizando uno o más de diversos métodos conocidos en la técnica. Como apreciarán lo expertos en la materia, la vía y/o el modo de administración variará dependiendo de los resultados deseados. Las rutas preferentes de administración para péptidos macrocíclicos de la divulgación incluyen rutas de administración intravenosa, intramuscular, intradérmica, intraperitoneal, subcutánea, espinal u otra ruta parenteral, por ejemplo, mediante inyección o infusión. La frase "administración parenteral" como se usa en el presente documento significa modos de administración diferentes de la administración enteral y tópica, habitualmente mediante inyección e incluyen, sin limitación, intravenosa, intramuscular, intraarterial, intratecal, intracapsular, intraorbital, intracardíaca, intradérmica, intraperitoneal, transtraqueal, subcutánea, subcuticular, intraarticular, subcapsular, subaracnoidea, intraespinal, epidural e inyección intraesternal e infusión.
Como alternativa, un péptido macrocíclico de la descripción se puede administrar a través de una ruta no parenteral, tal como una ruta de administración tópica, epidérmica o mucosa, por ejemplo, intranasalmente, por vía oral, vaginal, por vía rectal, sublingualmente o tópicamente.
Los compuestos activos pueden prepararse con vehículos que protegerán el compuesto contra la liberación rápida, tal como una formulación de liberación controlada, incluyendo implantes, parches transdérmicos y sistemas de suministro microencapsulados. Biodegradable, pueden usarse polímeros biocompatibles, tales como acetato de etilenvinilo, polianhídridos, ácido poliglicólico, colágeno, poliortoésteres y ácido poliláctico. Muchos métodos para la preparación de dichas formulaciones están patentados o se conocen generalmente por los expertos en la materia. Véase, por ejemplo, Robinson, J.R., ed., Sustained and Controlled Release Drug Delivery Systems, Marcel Dekker, Inc., Nueva York (1978).
Las composiciones terapéuticas pueden administrarse con dispositivos médicos conocidos en la técnica. Por ejemplo, en una realización preferente, una composición terapéutica de la divulgación se puede administrar con un dispositivo de inyección hipodérmica sin aguja, tales como los dispositivos desvelados en patentes de Estados Unidos n.° 5.399.163, 5.383.851, 5.312.335, 5.064.413, 4.941.880, 4.790.824, o 4.596.556. Ejemplos de implantes bien conocidos y módulos útiles en la presente divulgación incluyen: patente de Estados Unidos n.° 4.487.603, que desvela una bomba de microinfusión implantable para dispensar medicamento a una tasa controlada; patente de Estados Unidos n.° 4.486.194, que desvela un dispositivo terapéutico para administrar medicamentos a través de la piel; patente de Estados Unidos n.° 4.447.233, que desvela una bomba de infusión de medicamento para suministrar medicamento a una tasa de infusión precisa; patente de Estados Unidos n.° 4.447.224, que desvela un aparato de infusión implantable de flujo variable para la administración continua de fármaco; patente de Estados Unidos n.° 4.439.196, que desvela un sistema de administración osmótica de fármaco que tiene compartimentos multicámara; y patente de Estados Unidos n.° 4.475.196, que desvela un sistema de administración osmótica de fármaco. Muchos otros de estos implantes, sistemas de administración y módulos son conocidos por los expertos en la materia.
En ciertas realizaciones, los péptidos macrocíclicos de la divulgación pueden formularse para asegurar una distribución adecuada in vivo. Por ejemplo, la barrera hematoencefálica (BHM) excluye muchos compuestos altamente hidrofílicos. Para garantizar que los compuestos terapéuticos de la divulgación crucen la BBB (si se desea), se pueden formular, por ejemplo, en liposomas. Para los métodos de fabricación de liposomas, véase, por ejemplo, patente de Estados Unidos n.° 4.522.811, 5.374.548 y 5.399.331. Los liposomas pueden comprender uno o más grupos que se transportan de manera selectiva en células u órganos específicos, mejora así el suministro de
fármaco dirigido (véase, por ejemplo, Ranade, V.V., J. Clin. Pharmacol., 29:685 (1989)). Ejemplos de restos de direccionamiento incluyen folato o biotina (véase, por ejemplo, patente de Estados Unidos n.° 5.416.016 de Low et al.); manósidos (Umezawa et al., Biochem. Biophys. Res. Commun., 153:1038 (1988)); péptidos macrocíclicos (Bloeman, P.G. et al., FEBS Lett., 357:140 (1995); Owais, M. et al., Antimicrob. Agents Chemother., 39:180 (1995)); receptor A de proteína tensioactiva (Briscoe et al., Am. J. Physiol., 1233:134 (1995)); p120 (Schreier et al., J. Biol. Chem., 269:9090 (1994)); véase también Keinanen, K. et al., FEBS Lett., 346:123 (1994); Killion, J.J. et al., Immunomethods 4:273 (1994).
Usos de la divulgación
Los péptidos macrocíclicos, composiciones y realizaciones de la presente divulgación tienen numerosas utilidades in vitro e in vivo que implican, por ejemplo, detection de PD-L1 o mejora de la respuesta inmune por bloqueo de PD-L1. Por ejemplo, estas moléculas se pueden administrar a células en cultivo, in vitro o ex vivo, o a sujetos humanos, por ejemplo, in vivo, para mejorar inmunidad en diversas situaciones. Por tanto, en un aspecto, la divulgación proporciona el péptido macrocíclico de la divulgación para su uso en la modificación de una respuesta inmune en un sujeto. Preferentemente, la respuesta mejora, se estimula o se regula positivamente. En otros aspectos, el péptido macrocíclico puede tener unión anti-cino, anti-ratón y/o anti-marmota y actividad terapéutica.
Como se usa en este documento, el término "sujeto" pretende incluir seres humanos y animales no humanos. Animales no humanos incluyen todos los vertebrados, por ejemplo, mamíferos y no mamíferos, tales como primates no humanos, ovejas, perros, gatos, vacas, caballos, pollos, marmota, anfibios y reptiles, aunque se prefieren los mamíferos, tales como primates no humanos, ovejas, perros, gatos, vacas y caballos. Los sujetos preferentes incluyen pacientes humanos que necesitan una mejora de la respuesta inmune. Los pacientes humanos que tienen un trastorno pueden tratarse aumentando la respuesta inmune mediada por linfocitos T. En una realización particular, las realizaciones son particularmente adecuadas para el tratamiento de células cancerosas in vivo. Para lograr la mejora de la inmunidad específica de antígeno, los péptidos macrocíclicos se pueden administrar junto con un antígeno de interés. Cuando los péptidos macrocíclicos para PD-L1 se administran junto con otro agente, los dos pueden administrarse en cualquier orden o simultáneamente.
La divulgación describe además métodos para detectar la presencia de antígeno de PD-L1 humana, de ratón, marmota, cino y/o de ratón en una muestra, o medir la cantidad de antígeno PD-L1 humano, de marmota, cino y/o de ratón, que comprende poner en contacto la muestra y una muestra de control, con un péptido macrocíclico de referencia que se une específicamente a PD-L1 humana, de marmota, cino y/o ratón, en condiciones que permiten la formación de un complejo entre el macrociclo y PD-L1 humana, marmota, cino y/o ratón. Entonces, se detecta la formación de un complejo, en donde una diferencia de formación de complejo entre la muestra en comparación con la muestra de control es indicativa de la presencia de antígeno de PD-L1 humana, de marmota, cino y/o ratón en la muestra.
Dada la unión específica de los péptidos macrocíclicos de la divulgación para PD-L1, en comparación con CD28, ICOS y CTLA-4, los péptidos macrocíclicos de la divulgación pueden usarse para detectar específicamente la expresión de PD-L1 en la superficie de las células y, además, puede usarse para purificar PD-L1 mediante purificación de inmunoafinidad.
Cáncer
El bloqueo de PD-1 por péptidos macrocíclicos puede mejorar la respuesta inmune a las células cancerosas en el paciente. El ligando para p D-1, PD-L1, no se expresa en células humanas normales, pero es abundante en diversos cánceres humanos (Dong et al., Nat. Med., 8:787-789 (2002)). La interacción entre p D-1 y PD-L1 da como resultado una disminución de linfocitos infiltrantes de tumores, una disminución en la proliferación mediada por receptores de linfocitos T y evasión inmune por células cancerosas (Dong et al., J. Mol. Med., 81:281-287 (2003); Blank et al., Cancer Immunol. Immunother., 54:307-314 (2005); Konishi et al., Clin. Cancer Res., 10:5094-5100 (2004)). La supresión inmune puede revertirse inhibiendo la interacción local de PD-1 a PD-L1 y el efecto es aditivo cuando la interacción de PD-1 a PD-L2 también se bloquea (Iwai et al., Proc. Natl. Acad. Sci., 99:12293-12297 (2002); Brown et al., J. Immunol., 170:1257-1266 (2003)). Si bien los estudios anteriores han demostrado que la proliferación de linfocitos T puede restaurarse inhibiendo la interacción de PD-1 a PD-L1, no ha habido informes de un efecto directo sobre el crecimiento tumoral del cáncer in vivo al bloquear la interacción PD-1/PD-L1. En un aspecto, La presente divulgación se refiere a un tratamiento de un sujeto in vivo utilizando un péptido macrocíclico de manera que se inhibe el crecimiento de tumores cancerosos. Un péptido macrocíclico puede usarse solo para inhibir el crecimiento de tumores cancerosos. Como alternativa, se puede usar un péptido macrocíclico junto con otros agentes inmunogénicos, tratamientos estándar contra el cáncer, u otros péptidos macrocíclicos, como se describe a continuación.
Por tanto, en una realización, La divulgación proporciona un péptido macrocíclico para su uso en la inhibición del crecimiento de células tumorales en un sujeto.
Los cánceres preferentes cuyo crecimiento puede inhibirse mediante los péptidos macrocíclicos de la descripción
incluyen cánceres sensibles a la inmunoterapia. Los ejemplos no limitantes de cánceres preferentes para el tratamiento incluyen melanoma (por ejemplo, melanoma maligno metastásico), carcinoma de células renales (por ejemplo, carcinoma de células claras), cáncer de próstata (por ejemplo, adenocarcinoma de próstata resistente a hormonas y cáncer de próstata resistente a la castración), cáncer de mama, cáncer colorrectal y cáncer de pulmón (por ejemplo, cáncer de pulmón escamoso y no escamoso no microcítico). Además, la divulgación incluye tumores malignos recurrentes o refractarios cuyo crecimiento puede inhibirse usando los péptidos macrocíclicos de la divulgación.
Ejemplos de otros cánceres que pueden tratarse según la invención, incluyen cáncer de hueso, cáncer de páncreas, cáncer de piel, cáncer de cabeza o cuello, melanoma maligno cutáneo o intraocular, cáncer de útero, cáncer de ovario, cáncer de colon, cáncer de recto, cáncer de la región anal, cáncer de estómago/gástrico, cáncer de testículo, cáncer de útero, carcinoma de las trompas de Falopio, carcinoma del endometrio, carcinoma del cuello uterino, carcinoma de la vagina, carcinoma de la vulva, enfermedad de Hodgkin, linfoma no Hodgkin, cáncer del esófago, cáncer del intestino delgado, cáncer del sistema endocrino, cáncer de la glándula tiroides, cáncer de la glándula paratiroidea, cáncer de la glándula suprarrenal, sarcoma de tejidos blandos, cáncer de la uretra, cáncer del pene, leucemias crónicas o agudas, incluyendo leucemia mieloide aguda, leucemia mieloide crónica, leucemia linfoblástica aguda, leucemia linfocítica crónica, tumores sólidos de la infancia, linfoma linfocítico, cáncer de la vejiga, cáncer del riñón o del uréter, carcinoma de la pelvis renal, neoplasias del sistema nervioso central (SNC), linfoma primario del SNC, angiogénesis tumoral, tumor del eje espinal, glioma del tronco encefálico, adenoma hipofisario, sarcoma de Kaposi, cáncer epidermoide, cáncer de células escamosas, linfoma de linfocitos T, cánceres inducidos ambientalmente incluyendo aquellos inducidos por amianto y combinaciones de dichos cánceres. La presente divulgación también es útil para el tratamiento de cánceres metastásicos, especialmente cánceres metastásicos que expresan PD-L1 (Iwai et al., Int. Immunol., 17:133-144 (2005)).
Opcionalmente, péptidos macrocíclicos para PD-L1 pueden combinarse con un agente inmunogénico, tal como células cancerosas, antígenos tumorales purificados (incluyendo proteínas recombinantes, péptidos y moléculas de carbohidratos), células y células transfectadas con genes que codifican citoquinas estimulantes inmunes (He et al., J. Immunol., 173:4919-4928 (2004)). Los ejemplos no limitantes de vacunas tumorales que se pueden usar incluyen péptidos de antígenos de melanoma, tales como péptidos de gp100, antígenos MAGE, Trp-2, MART1 y/o tirosinasa, o células tumorales transfectadas para expresar la citoquina g M-CSF (discutido adicionalmente posteriormente).
En seres humanos, se ha demostrado que algunos tumores son inmunogénicos tales como melanomas. Se anticipa que al elevar el umbral de activación de linfocitos T por bloqueo de PD-L1, podemos esperar activar respuestas tumorales en el huésped.
El bloqueo de PD-L1 es probable que sea más eficaz cuando se combina con un protocolo de vacunación. Se han ideado muchas estrategias experimentales para la vacunación contra tumores (véase Rosenberg, S., Development of Cancer Vaccines, ASCO Educational Book Spring: 60-62 (2000); Logothetis, C., ASCO Educational Book Spring: 300-302 (2000); Khayat, D., ASCO Educational Book Spring: 414-428 (2000); Foon, K., ASCO Educational Book Spring: 730-738 (2000); véase también Restifo, N. et al., Cancer Vaccines, Capítulo 61, pág. 3023-3043, en DeVita, V. et al., eds., Cancer: Principles and Practice of Oncology, quinta edición (1997)). En una de estas estrategias, se prepara una vacuna usando células tumorales autólogas o alogénicas. Se ha demostrado que estas vacunas celulares son más eficaces cuando las células tumorales se transducen para expresar GM-CSF. GM-CSF ha demostrado ser un potente activador de presentación de antígenos para la vacunación tumoral (Dranoff et al., Proc. Natl. Acad. Sci. USA, 90: 3539-3543 (1993)).
El estudio de la expresión génica y los patrones de expresión génica a gran escala en varios tumores ha conducido a la definición de los llamados antígenos específicos de tumor (Rosenberg, S.A., Immunity, 10:281-287 (1999)). En muchos casos, estos antígenos específicos de tumor son antígenos diferenciados expresados en los tumores y en la célula de donde surgió el tumor, por ejemplo, antígenos de melanocitos gp100, antígenos MAGE y Trp-2. Más importante aún, se puede demostrar que muchos de estos antígenos son las dianas de linfocitos T específicos de tumor encontrados en el huésped. El bloqueo de PD-L1 se puede usar junto con una colección de proteínas recombinantes y péptidos expresados en un tumor para generar una respuesta inmune a estas proteínas. Estas proteínas son normalmente vistas por el sistema inmunitario como antígenos propios y por lo tanto, son tolerantes a ellos. El antígeno tumoral también puede incluir la proteína telomerasa, que se requiere para la síntesis de telómeros de cromosomas y que se expresa en más de 85 % de los cánceres humanos y solo en un número limitado de tejidos somáticos (Kim, N et al., Science, 266:2011-2013 (1994)). (Estos tejidos somáticos pueden protegerse del ataque inmunitario por diversos medios). El antígeno tumoral también puede ser "neoantígenos" expresados en células cancerosas debido a mutaciones somáticas que alteran la secuencia de proteínas o crean proteínas de fusión entre dos secuencias no relacionadas (es decir, bcr-abl en el cromosoma Filadelfia), o idiotipo de tumores de linfocitos B.
Otras vacunas de tumores pueden incluir las proteínas de virus implicados en cánceres humanos tales como virus del papiloma humano (HPV), Virus de la hepatitis (VHB y VHC) y Virus del sarcoma de herpes de Kaposi (KHSV). Otra forma de antígeno específico de tumor que se puede usar junto con el bloqueo de PD-L1 se purifica proteínas de choque térmico (HSP) aisladas del tejido tumoral. Estas proteínas de choque térmico contienen fragmentos de proteínas de las células tumorales y estas HSP son altamente eficaces en el suministro a las células presentadoras
de antígeno para inducir la inmunidad tumoral (Suot, R. et al., Science, 269:1585-1588 (1995); Tamura y. et al., Science, 278:117-120 (1997)).
Las células dendríticas (DC) son potentes células de presentación de antígeno que pueden usarse para preparar respuestas específicas de antígeno. DC pueden producirse ex vivo y cargarse con diversos antígenos de proteínas y péptidos, así como extractos de células tumorales (Nestle, F. et al., Nat. Med., 4:328-332 (1998)). Las Dc también pueden ser transducidas por medios genéticos para expresar también estos antígenos tumorales. DC también se han fusionado directamente a las células tumorales con fines de inmunización (Kugler, A. et al., Nat. Med., 6:332-336 (2000)). Como método de vacunación, La inmunización con DC puede combinarse eficazmente con el bloqueo de PD-L1 para activar respuestas antitumorales más potentes.
El bloqueo de PD-L1 también se puede combinar con tratamientos estándar contra el cáncer. El bloqueo de PD-L1 puede combinarse eficazmente con regímenes quimioterapéuticos. En estos casos, puede ser posible reducir la dosis de reactivo quimioterapéutico administrado (Mokyr, M. et al., Cancer Res., 58:5301-5304 (1998)). Un ejemplo de tal combinación es un péptido macrocíclico junto con la descarbazina para el tratamiento del melanoma. Otro ejemplo de tal combinación es un péptido macrocíclico junto con interleucina-2 (IL-2) para el tratamiento del melanoma. El fundamento científico detrás del uso combinado de bloqueo de PD-L1 y quimioterapia es que la muerte celular, que es una consecuencia de la acción citotóxica de la mayoría de los compuestos quimioterapéuticos, debería dar lugar a niveles aumentados de antígeno tumoral en la ruta de presentación del antígeno. Otras terapias combinadas que pueden dar como resultado una sinergia con el bloqueo de PD-L1 a través de la muerte celular son la radiación, cirugía y privación de hormonas. Cada uno de estos protocolos crea una fuente de antígeno tumoral en el huésped. Los inhibidores de la angiogénesis también pueden combinarse con el bloqueo de PD-L1. La inhibición de la angiogénesis conduce a la muerte de células tumorales que pueden alimentar el antígeno tumoral en las rutas de presentación del antígeno huésped.
Los péptidos macrocíclicos que bloquean PD-L1 también pueden usarse junto con péptidos macrocíclicos biespecíficos que dirigen células efectoras que expresan receptores de Fc alfa o Fc gamma a células tumorales (véase, por ejemplo, patente de Estados Unidos n.° 5.922.845 y 5.837.243). Se pueden usar péptidos macrocíclicos biespecíficos para dirigir dos antígenos separados. Por ejemplo, se han usado péptidos macrocíclicos biespecíficos de receptor anti-Fc/antígeno tumoral (por ejemplo, Her-2/neu) para dirigir macrófagos a sitios de tumor. Este direccionamiento puede activar más eficazmente las respuestas tumorales específicas. El brazo de linfocitos T de estas respuestas se vería aumentado por el uso del bloqueo de PD-L1. Como alternativa, el antígeno puede administrarse directamente a las DC mediante el uso de péptidos macrocíclicos biespecíficos que se unen al antígeno tumoral y un marcador de superficie celular específico de células dendríticas.
Los tumores evaden la vigilancia inmune del huésped por una gran diversidad de mecanismos. Muchos de estos mecanismos pueden ser superados por la inactivación de proteínas que son expresadas por los tumores y que son inmunosupresoras. Estos incluyen entre otros TGF-beta (Kehrl, J. et al., J. Exp. Med., 163:1037-1050 (1986)), IL-10 (Howard, M. et al., Immunology Today, 13:198-200 (1992)) y ligando Fas (Hahne, M. et al., Science, 274:1363-1365 (1996)). Los péptidos macrocíclicos para cada una de estas entidades pueden usarse junto con anti-PD-L1 para contrarrestar los efectos del agente inmunosupresor y favorecer las respuestas inmunes tumorales por parte del huésped.
Se pueden usar otros péptidos macrocíclicos que pueden activar la respuesta inmune del huésped junto con anti-PD-Ll. Estos incluyen moléculas en la superficie de las células dendríticas que activan la función DC y la presentación del antígeno. Los péptidos macrocíclicos anti-CD40 pueden sustituir eficazmente la actividad auxiliar de células T (Ridge, J. et al., Nature, 393:474-478 (1998)) y pueden usarse junto con anticuerpos PD-1 (Ito, N. et al., Immunobiology, 201(5):527-540 (2000)). Péptidos macrocíclicos activantes para moléculas coestimuladoras de linfocitos T tales como CTLA-4 (por ejemplo, patente de Estados Unidos n.° 5.811.097), OX-40 (Weinberg, A. et al., Immunol., 164:2160-2169 (2000)), 4-1BB (Melero, I. et al., Nat. Med., 3:682-685 (1997), e ICOS (Hutloff, A. et al., Nature, 397:262-266 (1999)) pueden proporcionar también niveles aumentados de activación de linfocitos T.
El trasplante de médula ósea se utiliza actualmente para tratar diversos tumores de origen hematopoyético. Mientras que la enfermedad del injerto contra huésped es una consecuencia de este tratamiento, el beneficio terapéutico puede obtenerse a partir de respuestas de injerto frente a tumor. El bloqueo de PD-L1 se puede usar para aumentar la eficacia de los linfocitos T específicos de tumor injertados de donante.
También hay varios protocolos de tratamiento experimental que involucran activación ex vivo y expansión de linfocitos T específicos de antígeno y transferencia adoptiva de estas células en receptores para linfocitos T específicos de antígeno contra tumor (Greenberg, R. et al., Science, 285:546-551 (1999)). Estos métodos también pueden usarse para activar las respuestas de los linfocitos T a agentes infecciosos tales como CMV. Se puede esperar que la activación ex vivo en presencia de péptidos macrocíclicos aumente la frecuencia y la actividad de los linfocitos T transferidos adoptivamente.
Enfermedades infecciosas
Otras realizaciones de la divulgación se usan para tratar pacientes que han sido expuestos a toxinas o agentes patógenos particulares. Por tanto, otro aspecto de la divulgación proporciona un péptido macrocíclico de la presente divulgación para su uso en el tratamiento de una enfermedad infecciosa en un sujeto.
Similar a su aplicación a los tumores como se ha analizado anteriormente, El bloqueo de PD-L1 se puede usar solo, o como adyuvante, en combinación con vacunas, para estimular la respuesta inmune a patógenos, toxinas y autoantígenos. Los ejemplos de agentes patógenos para los que este enfoque terapéutico puede ser particularmente útil, incluyen patógenos para los que actualmente no existe una vacuna eficaz, o patógenos para los cuales las vacunas convencionales son menos que completamente eficaces. Estos incluyen, pero sin limitación, VIH, Hepatitis (A, B y C), gripe, Herpes, Giardia, Malaria (Butler, N.S. et al., Nature Immunology 13, 188-195 (2012); Hafalla, J.C.R., et al. PLOS Pathogens; 2 de febrero, 2012)), Leishmania, Staphylococcus aureus, Pseudomonas Aeruginosa. El bloqueo de PD-L1 es particularmente útil contra infecciones establecidas por agentes tales como VIH que presentan antígenos alterados durante el curso de las infecciones. Estos nuevos epítopos se reconocen como extraños en el momento de la administración de PD-L1 anti-humano, provocando así una fuerte respuesta de linfocitos T que no se ve atenuada por señales negativas a través de PD-L1.
Algunos ejemplos de virus patógenos que causan infecciones tratables según la invención incluyen VIH, hepatitis (A, B o C), herpes virus (por ejemplo, VZV, HSV-1, HAV-6, HSV-II y CMV, virus Epstein Barr), adenovirus, virus de la gripe, flavivirus, ecovirus, rinovirus, virus coxsackie, cornovirus, virus sincitial respiratorio, virus de las paperas, rotavirus, virus del sarampión, virus de la rubeola, parvovirus, virus vaccinia, virus HTLV, virus del dengue, virus del papiloma, virus de molusco, poliovirus, virus de la rabia, virus JC y virus de la encefalitis arboviral.
Algunos ejemplos de bacterias patógenas que causan infecciones tratables según la invención incluyen clamidia, bacterias rickettsiales, micobacterias, estafilococos, estreptococos, neumonococos, meningococos y conococos, klebsiella, proteus, serratia, pseudomonas, legionella, difteria, salmonela, bacilos, cólera, tétanos, botulismo, ántrax, peste, leptospirosis y bacterias de enfermedad de Lyme.
Algunos ejemplos de hongos patógenos causantes de infecciones tratables según la invención incluyen Candida (albicans, krusei, glabrata, tropicalis, etc.), Cryptococcus neoformans, Aspergillus (fumigatus, niger, etc.), Genus Mucorales (mucor, absidia, rhizophus), Sporothrix schenkii, Blastomyces dermatitidis, Paracoccidioides brasiliensis, Coccidioides immitis e Histoplasma capsulatum.
Algunos ejemplos de parásitos patógenos que causan infecciones tratables según la invención incluyen Entamoeba histolytica, Balantidium coli, Naegleriafowleri, Acanthamoeba sp., Giardia lambia, Cryptosporidium sp., Pneumocystis carinii, Plasmodium vivax, Babesia microti, Trypanosoma brucei, Trypanosoma cruzi, Leishmania donovani, Toxoplasma gondi y Nippostrongylus brasiliensis.
En todas las realizaciones anteriores, el bloqueo de PD-L1 se puede combinar con otras formas de inmunoterapia tales como tratamiento con citoquinas (por ejemplo, interferones, agentes dirigidos a actividad de VEGF o receptores de VEGF, GM-CSF, G-CSF, IL-2) o terapia de anticuerpos biespecíficos, que proporciona presentación mejorada de antígenos tumorales (véase, por ejemplo, Holliger, Proc. Natl. Acad. Sci. USA, 90:6444-6448 (1993); Poljak, Structure, 2:1121-1123 (1994)).
Reacciones autoinmunes
Los péptidos macrocíclicos pueden provocar y amplificar respuestas autoinmunes. De hecho, la inducción de respuestas antitumorales usando vacunas de células tumorales y péptidos revela que muchas respuestas antitumorales implican anti-reactividades (despigmentación observada en el melanoma B16 modificado con anti-CTLA-4+GM-CSF en van Elsas et al. anteriormente; despigmentación en ratones vacunados con Trp-2 (Overwijk, W. et al., Proc. Natl. Acad. Sci. USA, 96:2982-2987 (1999)); prostatitis autoinmune provocada por vacunas de células tumorales TRAMP (Hurwitz, A., véase anteriormente (2000)), vacunación de antígeno de péptido de melanoma y vitíligo observada en ensayos clínicos en humanos (Rosenberg, S.A. et al., J. Immunother. Emphasis Tumor Immunol., 19(1):81-84 (1996)).
Por tanto, es posible considerar el uso del bloqueo anti-PD-Ll junto con varias proteínas propias para idear protocolos de vacunación para generar de manera eficaz respuestas inmunes contra estas proteínas propias para el tratamiento de la enfermedad. Por ejemplo, la enfermedad de Alzheimer implica una acumulación inapropiada de péptido A.beta. en depósitos amiloides en el cerebro; las respuestas de anticuerpos contra el amiloide son capaces de eliminar estos depósitos amiloides (Schenk et al., Nature, 400:173-177 (1999)).
También se pueden usar otras proteínas propias como dianas tales como IgE para el tratamiento de alergia y asma y TNF.alfa para artritis reumatoide. Finalmente, las respuestas de anticuerpos a diversas hormonas pueden ser inducidas mediante el uso de los macrociclos desvelados en este documento. Para la anticoncepción se pueden utilizar respuestas de anticuerpos neutralizantes a hormonas reproductivas. La respuesta de anticuerpos neutralizantes a hormonas y otros factores solubles que se requieren para el crecimiento de tumores particulares también pueden considerarse como posibles dianas de vacunación.
Las realizaciones análogas como se ha descrito previamente para el uso de macrociclos anti-PD-Ll pueden usarse para la inducción de respuestas autoinmunes terapéuticas para tratar pacientes que tienen una acumulación inapropiada de otros autoantígenos, tales como depósitos amiloides, incluyendo A-beta en la enfermedad de Alzheimer, citoquinas tales como TNF.alfa., e IgE.
Vacunas
Los péptidos macrocíclicos pueden usarse para estimular respuestas inmunitarias específicas de antígeno mediante la administración conjunta de un macrociclo anti-PD-1 con un antígeno de interés (por ejemplo, una vacuna). Por tanto, en otro aspecto la divulgación proporciona: (i) el antígeno; y (ii) un macrociclo anti-PD-1 para uso en mejorar una respuesta inmune a un antígeno en un sujeto. El antígeno puede ser, por ejemplo, un antígeno tumoral, un antígeno vírico, un antígeno bacteriano o un antígeno de un patógeno. Los ejemplos no limitativos de tales antígenos incluyen los analizados en las secciones anteriores, tales como los antígenos tumorales (o vacunas tumorales) analizados anteriormente, o antígenos de los virus, bacterias u otros patógenos descritos anteriormente.
Vías adecuadas para administrar las composiciones (por ejemplo, péptidos macrocíclicos, moléculas multiespecíficas y biespecíficas e inmunoconjugados) de la descripción in vivo e in vitro se conocen bien en la técnica y pueden seleccionarse por los expertos en la materia. Por ejemplo, las composiciones pueden administrarse por inyección (por ejemplo, intravenosa o subcutánea). Las dosis adecuadas de las moléculas utilizadas dependerán de la edad y el peso del sujeto y la concentración y/o formulación de la composición.
Como se describió previamente, los péptidos macrocíclicos de la descripción pueden administrarse conjuntamente con otro agente terapéutico más, por ejemplo, un agente citotóxico, un agente radiotóxico o un agente inmunosupresor. El péptido puede estar unido al agente (como un inmunocomplejo) o puede administrarse por separado del agente. En el último caso (administración separada), el péptido se puede administrar antes, después de o simultáneamente con el agente o puede administrarse conjuntamente con otras terapias conocidas, por ejemplo, una terapia anticancerosa, por ejemplo, radiación. Dichos agentes terapéuticos incluyen, entre otros, agentes antineoplásicos tales como doxorrubicina (adriamicina), cisplatino bleomicina sulfato, carmustina, clorambucilo, descarbazina y ciclofosfamida hidroxiurea que, por sí solos, son solamente eficaces a niveles que son tóxicos o subtóxicos para un paciente. El cisplatino se administra por vía intravenosa como una dosis de 100 mg/dosis una vez cada cuatro semanas y la adriamicina se administra por vía intravenosa como una dosis de 60-75 mg/ml una vez cada 21 días. La administración conjunta de los péptidos macrocíclicos de la presente divulgación con agentes quimioterapéuticos proporciona dos agentes anticancerígenos que operan a través de diferentes mecanismos que producen un efecto citotóxico para las células tumorales humanas. Dicha administración conjunta puede resolver problemas debido al desarrollo de resistencia a fármacos o un cambio en la antigenicidad de las células tumorales que las haría no reactivas con los péptidos.
Dentro del alcance de la presente divulgación también se encuentran kits que comprenden las composiciones de la divulgación (por ejemplo, péptidos macrocíclicos, biespecíficas o moléculas multiespecíficas, o inmunoconjugados) e instrucciones de uso. El kit puede contener además al menos un reactivo adicional, uno o más péptidos macrocíclicos adicionales de la descripción (por ejemplo, un anticuerpo humano que tiene una actividad complementaria que se une a un epítopo en el antígeno PD-L1 distinto del macrociclo). Los kits incluyen normalmente una etiqueta que indica el uso pretendido del contenido del kit. El término etiqueta incluye cualquier material escrito o grabado suministrado en o con el kit, o que de otro modo acompaña al kit.
Terapia combinada
La combinación de los péptidos macrocíclicos de la presente divulgación con otro antagonista de PD-L1 y otro inmunomodulador es útil para potenciar una respuesta inmune contra una enfermedad hiperproliferativa. Por ejemplo, estas moléculas se pueden administrar a células en cultivo, in vitro o ex vivo, o a sujetos humanos, por ejemplo, in vivo, para mejorar inmunidad en diversas situaciones. Por tanto, en un aspecto, la divulgación proporciona un péptido macrocíclico de la divulgación para su uso en la modificación de una respuesta inmune en un sujeto. Preferentemente, la respuesta mejora, se estimula o se regula positivamente. En otra realización, la presente divulgación proporciona un péptido macrocíclico de la presente divulgación y una dosis subterapéutica de otro inmunomodulador para uso que altera los eventos adversos asociados con el tratamiento de una enfermedad hiperproliferativa con un agente terapéutico inmunoestimulador.
El bloqueo de PD-L1 por péptidos macrocíclicos puede mejorar la respuesta inmune a las células cancerosas en el paciente. Los cánceres cuyo crecimiento puede inhibirse usando los péptidos macrocíclicos de la divulgación instantánea incluyen cánceres sensibles a la inmunoterapia. Los ejemplos representativos de cánceres para el tratamiento con la terapia combinada de la presente divulgación incluyen melanoma (por ejemplo, melanoma maligno metastásico), cáncer renal, cáncer de próstata, cáncer de mama, cáncer de colon y cáncer pulmonar. Ejemplos de otros cánceres que pueden tratarse según la invención, incluyen cáncer de hueso, cáncer de páncreas, cáncer de piel, cáncer de cabeza o cuello, melanoma maligno cutáneo o intraocular, cáncer de útero, cáncer de ovario, cáncer de recto, cáncer de la región anal, cáncer de estómago, cáncer de testículo, cáncer de útero,
carcinoma de las trompas de Falopio, carcinoma del endometrio, carcinoma del cuello uterino, carcinoma de la vagina, carcinoma de la vulva, enfermedad de Hodgkin, linfoma no Hodgkin, cáncer del esófago, cáncer del intestino delgado, cáncer del sistema endocrino, cáncer de la glándula tiroides, cáncer de la glándula paratiroidea, cáncer de la glándula suprarrenal, sarcoma de tejidos blandos, cáncer de la uretra, cáncer del pene, leucemias crónicas o agudas, incluyendo leucemia mieloide aguda, leucemia mieloide crónica, leucemia linfoblástica aguda, leucemia linfocítica crónica, tumores sólidos de la infancia, linfoma linfocítico, cáncer de la vejiga, cáncer del riñón o del uréter, carcinoma de la pelvis renal, neoplasias del sistema nervioso central (SNC), linfoma primario del SNC, angiogénesis tumoral, tumor del eje espinal, glioma del tronco encefálico, adenoma hipofisario, sarcoma de Kaposi, cáncer epidermoide, cáncer de células escamosas, linfoma de linfocitos T, cánceres inducidos ambientalmente incluyendo aquellos inducidos por amianto y combinaciones de dichos cánceres. La presente descripción también es útil para el tratamiento de cánceres metastásicos.
En ciertas realizaciones, la combinación de agentes terapéuticos que contienen al menos un péptido macrocíclico discutido en este documento puede administrarse simultáneamente como una composición única en un vehículo farmacéuticamente aceptable, o simultáneamente como composiciones separadas en donde cada agente puede administrarse secuencialmente. Por ejemplo, un segundo inmunomodulador y un péptido macrocíclico de la presente divulgación pueden administrarse secuencialmente, tal como el segundo inmunomodulador administrado primero y el péptido macrocíclico segundo, o el péptido macrocíclico administrado primero y el segundo inmunomodulador segundo. Además, si se administra secuencialmente más de una dosis de la terapia combinada, el orden de la administración secuencial se puede revertir o mantener en el mismo orden en cada momento de administración, Las administraciones secuenciales pueden combinarse con administraciones concurrentes, o cualquier combinación de las mismas. Por ejemplo, la primera administración de un segundo inmunomodulador y el péptido macrocíclico puede ser concurrente, la segunda administración puede ser secuencial con el segundo inmunomodulador primero y el péptido macrocíclico segundo y la tercera administración puede ser secuencial con el péptido macrocíclico primero y el segundo inmunomodulador segundo, etc. Otro esquema de dosificación representativo puede implicar una primera administración que es secuencial con el péptido macrocíclico primero y el segundo inmunomodulador segundo y las administraciones posteriores pueden ser concurrentes.
Opcionalmente, la combinación del péptido macrocíclico y un segundo inmunomodulador puede combinarse adicionalmente con un agente inmunogénico, tal como células cancerosas, antígenos tumorales purificados (incluyendo proteínas recombinantes, péptidos y moléculas de carbohidratos), células y células transfectadas con genes que codifican citoquinas estimulantes inmunes (He et al., J. Immunol., 173:4919-4928 (2004)). Los ejemplos no limitativos de vacunas tumorales que se pueden usar incluyen péptidos de antígenos de melanoma, tales como péptidos de gp100, antígenos MAGE, Trp-2, MART1 y/o tirosinasa, o células tumorales transfectadas para expresar la citoquina GM-CSF (discutido adicionalmente posteriormente).
Un péptido macrocíclico PD-L1 combinado y un segundo inmunomodulador pueden combinarse adicionalmente con un protocolo de vacunación. Se han ideado muchas estrategias experimentales para la vacunación contra tumores (véase Rosenberg, S., Development of Cancer Vaccines, ASCO Educational Book Spring: 60-62 (2000); Logothetis, C., ASCO Educational Book Spring: 300-302 (2000); Khayat, D., ASCO Educational Book Spring: 414-428 (2000); Foon, K., ASCO Educational Book Spring: 730-738 (2000); véase también Restifo et al., Cancer Vaccines, Capítulo 61, pág. 3023-3043 en DeVita et al., eds., Cancer: Principles and Practice of Oncology, quinta edición (1997)). En una de estas estrategias, se prepara una vacuna usando células tumorales autólogas o alogénicas. Se ha demostrado que estas vacunas celulares son más eficaces cuando las células tumorales se transducen para expresar GM-CSF. GM-CSF ha demostrado ser un potente activador de presentación de antígenos para la vacunación tumoral (Dranoff et al., Proc. Natl. Acad. Sci. USA, 90:3539-3543 (1993)).
El estudio de la expresión génica y los patrones de expresión génica a gran escala en varios tumores ha conducido a la definición de los llamados antígenos específicos de tumor (Rosenberg, Immunity, 10:281-287 (1999)). En muchos casos, estos antígenos específicos de tumor son antígenos de diferenciación expresados en los tumores y en la célula de la que se originó el tumor, por ejemplo, antígenos de melanocitos gp100, antígenos MAGE y Trp-2. Más importante aún, se puede demostrar que muchos de estos antígenos son las dianas de linfocitos T específicos de tumor encontrados en el huésped. En ciertas realizaciones, se puede usar un péptido macrocíclico PD-L1 combinado y un segundo inmunomodulador junto con una colección de proteínas recombinantes y/o péptidos expresados en un tumor para generar una respuesta inmune a estas proteínas. Estas proteínas son normalmente vistas por el sistema inmune como autoantígenos y son, por tanto, tolerantes a ello. El antígeno tumoral también puede incluir la proteína telomerasa, que se requiere para la síntesis de telómeros de cromosomas y que se expresa en más de 85 % de los cánceres humanos y solo en un número limitado de tejidos somáticos (Kim et al., Science, 266:2011-2013 (1994)). (Estos tejidos somáticos pueden protegerse del ataque inmunitario por diversos medios). El antígeno tumoral también puede ser "neoantígenos" expresados en células cancerosas debido a mutaciones somáticas que alteran la secuencia de proteínas o crean proteínas de fusión entre dos secuencias no relacionadas (es decir, bcr-abl en el cromosoma Filadelfia), o idiotipo de tumores de linfocitos B.
Otras vacunas de tumores pueden incluir las proteínas de virus implicados en cánceres humanos tales como virus del papiloma humano (HPV), Virus de la hepatitis (VHB y VHC) y Virus del sarcoma de herpes de Kaposi (KHSV). Otra forma de antígeno específico de tumor que se puede usar junto con el bloqueo del péptido macrocíclico PD-L1
es proteínas de choque térmico (HSP) purificadas aisladas del tejido tumoral. Estas proteínas de choque térmico contienen fragmentos de proteínas de las células tumorales y estas HSP son altamente eficaces en el suministro a las células presentadoras de antígeno para inducir inmunidad tumoral (Suot et al., Science, 269:1585-1588 (1995); Tamura et al., Science, 278:117-120 (1997)).
Las células dendríticas (DC) son potentes células de presentación de antígeno que pueden usarse para preparar respuestas específicas de antígeno. Las DC pueden producirse ex vivo y cargarse con diversos antígenos de proteínas y péptidos, así como extractos de células tumorales (Nestle et al., Nat. Med., 4:328-332 (1998)). Las DC también pueden ser transducidas por medios genéticos para expresar también estos antígenos tumorales. Las CD también se han fusionado directamente a las células tumorales con fines de inmunización (Kugler et al., Nat. Med., 6:332-336 (2000)). Como método de vacunación, La inmunización con DC puede combinarse eficazmente con un péptido macrocíclico anti-PD-Ll combinado y un segundo inmunomodulador para activar respuestas antitumorales más potentes.
Un péptido macrocíclico anti-PD-Ll combinado y un inmunomodulador adicional también se pueden combinar con los tratamientos estándar contra el cáncer. Por ejemplo, una combinación de un péptido macrocíclico y un segundo inmunomodulador puede combinarse eficazmente con regímenes quimioterapéuticos. En estos casos, como se observa con la combinación de un péptido macrocíclico y un segundo inmunomodulador, puede ser posible reducir la dosis de otro reactivo quimioterapéutico administrado con la combinación de la presente divulgación (Mokyr et al., Cancer Res., 58:5301-5304 (1998)). Un ejemplo de dicha combinación es una combinación de un péptido macrocíclico y un segundo inmunomodulador junto con la descarbina para el tratamiento del melanoma. Otro ejemplo es una combinación de un péptido macrocíclico y un segundo agente inmunomodulador además de interleucina-2 (IL-2) para el tratamiento del melanoma. El fundamento científico detrás del uso combinado del péptido macrocíclico PD-L1 y otro inmunomodulador con quimioterapia es que la muerte celular, que es una consecuencia de la acción citotóxica de la mayoría de los compuestos quimioterapéuticos, debería dar lugar a niveles aumentados de antígeno tumoral en la ruta de presentación del antígeno. Otras terapias combinadas que pueden dar como resultado una sinergia con un péptido macrocíclico combinado anti-PD-Ll y un inmunomodulador adicional a través de la muerte celular incluyen radiación, cirugía, o privación hormonal. Cada uno de estos protocolos crea una fuente de antígeno tumoral en el huésped. Los inhibidores de la angiogénesis también se pueden combinar con un PD-L1 y segundo inmunomodulador combinados. La inhibición de la angiogénesis conduce a la muerte de las células tumorales, que también puede ser una fuente de antígeno tumoral para ser alimentado en las vías de presentación del antígeno del huésped.
También se puede usar una combinación de PD-L1 y otro inmunomodulador junto con péptidos macrocíclicos biespecíficos que se dirigen a células efectoras que expresan receptores Fc.alfa. o Fc.gamma. a células tumorales (véase, por ejemplo, patente de Estados Unidos n.° 5.922.845 y 5.837.243). Se pueden usar péptidos macrocíclicos biespecíficos para dirigir dos antígenos separados. Por ejemplo, se han usado péptidos macrocíclicos biespecíficos de receptor anti-Fc/antígeno tumoral (por ejemplo, Her-2/neu) para dirigir macrófagos a sitios de tumor. Este direccionamiento puede activar más eficazmente las respuestas tumorales específicas. El brazo de linfocitos T de estas respuestas se incrementaría con el uso de un PD-L1 y un segundo inmunomodulador combinados. Como alternativa, el antígeno puede administrarse directamente a las DC mediante el uso de péptidos macrocíclicos biespecíficos que se unen al antígeno tumoral y un marcador de superficie celular específico de células dendríticas.
En otro ejemplo, se puede usar una combinación de un péptido macrocíclico y un segundo inmunomodulador junto con agentes macrocíclicos antineoplásicos, tales como RITUXAN® (rituximab), HERCEPTIN® (trastuzumab), BEXXAR® (tositumomab), ZEVAl In® (ibritumomab), CAMPATH® (alemtuzumab), Linfocida (eprtuzumab), AVASTIN® (bevacizumab) y TARCEVA® (erlotinib) y similares. A modo de ejemplo y sin el deseo de quedar vinculados a teoría alguna, el tratamiento con un anticuerpo anticancerígeno o un anticuerpo anticancerígeno conjugado con una toxina puede conducir a la muerte de células cancerosas (por ejemplo, células tumorales), lo que potenciaría una respuesta inmune mediada por la segunda diana de inmunomodulador o PD-L1. En una realización de ejemplo, un tratamiento de una enfermedad hiperproliferativa (por ejemplo, un tumor canceroso) puede incluir un anticuerpo anticancerígeno junto con un péptido macrocíclico y un segundo inmunomodulador simultáneamente o secuencialmente o cualquier combinación de los mismos, que puede potenciar una respuesta inmune antitumoral por parte del huésped.
Los tumores evaden la vigilancia inmune del huésped por una gran diversidad de mecanismos. Muchos de estos mecanismos pueden ser superados por la inactivación de proteínas, que se expresan por los tumores y que son inmunosupresoras. Estos incluyen, entre otros, TGF-.beta. (Kehrl, J. et al., J. Exp. Med., 163:1037-1050 (1986)), IL-10 (Howard, M. et al., Immunology Today, 13:198-200 (1992)) y ligando Fas (Hahne, M. et al., Science, 274:1363-1365 (1996)). En otro ejemplo, los anticuerpos para cada una de estas entidades pueden combinarse adicionalmente con un péptido macrocíclico y otro inmunomodulador para contrarrestar los efectos de los agentes inmunosupresores y favorecer las respuestas inmunitarias antitumorales por parte del huésped.
Otros agentes que pueden usarse para activar la respuesta inmune del huésped pueden usarse además junto con un péptido macrocíclico de la presente divulgación. Estos incluyen moléculas en la superficie de las células dendríticas que activan la función DC y la presentación del antígeno. Los péptidos macrocíclicos anti-CD40 pueden
sustituir eficazmente la actividad auxiliar de células T (Ridge, J. et al., Nature, 393:474-478 (1998)) y pueden usarse junto con los péptidos macrocíclicos de la presente divulgación, solos o junto con una combinación anti-CTLA-4 (Ito, N. et al., Immunobiology, 201(5):527-540 (2000)). Péptidos macrocíclicos activantes para moléculas coestimuladoras de linfocitos T, tales como OX-40 (Weinberg, A. et al., Immunol., 164:2160-2169 (2000)), 4-1BB (Melero, I. et al., Nat. Med., 3:682-685 (1997), e ICOS (Hutloff, A. et al., Nature, 397:262-266 (1999)) pueden proporcionar también niveles aumentados de activación de linfocitos T.
El trasplante de médula ósea se utiliza actualmente para tratar diversos tumores de origen hematopoyético. Mientras que la enfermedad del injerto contra huésped es una consecuencia de este tratamiento, el beneficio terapéutico puede obtenerse a partir de respuestas de injerto frente a tumor. Un péptido macrocíclico de la presente divulgación ya sea solo o junto con otro inmunomodulador, se puede usar para aumentar la eficacia de los linfocitos T específicos de tumor injertados en el donante.
También hay varios protocolos de tratamiento experimental que involucran activación ex vivo y expansión de linfocitos T específicos de antígeno y transferencia adoptiva de estas células en receptores para linfocitos T específicos de antígeno contra tumor (Greenberg, R. et al., Science, 285:546-551 (1999)). Estos métodos también pueden usarse para activar las respuestas de los linfocitos T a agentes infecciosos tales como CMV. Activación ex vivo en presencia de un péptido macrocíclico de la presente divulgación ya sea solo o junto con otro innumomodulador, cabe esperar que aumente la frecuencia y la actividad de los linfocitos T transferidos adoptivamente.
En ciertas realizaciones, la presente divulgación proporciona un péptido macrocíclico de la presente divulgación junto con una dosis subterapéutica de otro inmunomodulador para uso que altera un evento adverso asociado con el tratamiento de una enfermedad hiperproliferativa con un agente inmunoestimulador. Por ejemplo, Las realizaciones de la presente divulgación pueden reducir la incidencia de colitis o diarrea inducida por anticuerpos terapéuticos inmunoestimuladores mediante la administración de un esteroide no absorbible al paciente. Debido a que cualquier paciente que reciba un anticuerpo terapéutico inmunoestimulador está en riesgo de desarrollar colitis o diarrea inducida por dicho tratamiento, toda esta población de pacientes es adecuada para la terapia según las realizaciones de la presente divulgación. Aunque se han administrado esteroides para tratar la enfermedad inflamatoria del intestino (IBD) y prevenir las exacerbaciones de la IBD, no se han utilizado para prevenir (disminuir la incidencia de) IBD en pacientes que no han sido diagnosticados con IBD. Los efectos secundarios significativos asociados con esteroides, incluso esteroides no absorbibles, han desalentado el uso profiláctico.
En realizaciones adicionales, un péptido macrocíclico de la presente divulgación ya sea solo o junto con otro inmunomodulador, se puede combinar además con el uso de cualquier esteroide no absorbible. Como se usa en este documento, un "esteroide no absorbible" es un glucocorticoide que exhibe un extenso metabolismo de primer paso tal que, después del metabolismo en el hígado, la biodisponibilidad del esteroide es baja, es decir, menos de aproximadamente 20 %. En una realización de la divulgación, el esteroide no absorbible es budesonida. La budesonida es un glucocorticosteroide de acción local, que se metaboliza ampliamente, principalmente por el hígado, después de la administración oral. ENTOCORT® eC (Astra-Zeneca) es una formulación oral de budesonida dependiente del tiempo y del pH desarrollada para optimizar el suministro de fármacos al íleon y todo el colon. ENTOCORT® EC está aprobado en Estados Unidos para tratamiento de la enfermedad de Crohn leve a moderada que afecta al íleon y/o al colon ascendente. La dosis oral habitual de ENTOCORT® EC para el tratamiento de la enfermedad de Crohn es de 6 a 9 mg/día. ENTOCORT® EC se libera en los intestinos antes de ser absorbido y retenido en la mucosa intestinal. Una vez que pasa a través del tejido diana de la mucosa intestinal, ENTOCORT® EC se metaboliza ampliamente por el sistema del citocromo P450 en el hígado a metabolitos con una actividad glucocorticoide insignificante. Por tanto, La biodisponibilidad es baja (alrededor del 10 %). La baja biodisponibilidad de budesonida da como resultado una relación terapéutica mejorada en comparación con otros glucocorticoides con un metabolismo de primer paso menos extenso. Budesonida resulta en menos efectos adversos, incluyendo menos supresión hipotalámica-pituitaria, que los corticosteroides de acción sistémica. Sin embargo, la administración crónica de ENTOCORT® EC puede producir efectos glucocorticoides sistémicos tales como hipercorticismo y supresión suprarrenal. Véase Physicians' Desk Reference Supplement, 58a edición, 608-610 (2004).
En otras realizaciones adicionales, una combinación PD-L1 y otro inmunomodulador junto con un esteroide no absorbible pueden combinarse adicionalmente con un salicilato. Los salicilatos incluyen agentes 5-ASA tales como, por ejemplo: sulfasalazina (AZULFIDINE®, Pharmacia & Upjohn); olsalazina (DIPe Nt UM®, Pharmacia & Upjohn); balsalazida (COLAZAL®, Salix Pharmaceuticals, Inc.); y mesalamina (ASACOL®, Procter & Gamble Pharmaceuticals; PENTASA®, Shire US; CANASA®, Axcan Scandipharm, Inc.; ROWASA®, Solvay).
Dosificación y Formulación
Un péptido adecuado de Fórmula I, o más específicamente un péptido macrocíclico descrito en este documento, puede administrarse a pacientes para tratar diabetes y otras enfermedades relacionadas ya que el compuesto solo se mezcla con un vehículo aceptable en forma de formulaciones farmacéuticas. Los expertos en la técnica de tratamiento de diabetes pueden determinar fácilmente la dosis y la vía de administración del compuesto a mamíferos, incluyendo seres humanos, que necesitan dicho tratamiento. La ruta de administración puede incluir pero
sin limitación oral, intraoral, rectal, transdérmica, bucal, intranasal, pulmonar, subcutánea, intramuscular, intradérmica, sublingual, intracolónica, intraocular, intravenosa, o administración intestinal. El compuesto se formula según la ruta de administración basada en una práctica farmacéutica aceptable (Fingl et al., en The Pharmacological Basis de Therapeutics, Capítulo 1, pág. 1 (1975); Remington's Pharmaceutical Sciences, 18a edición, Mack Publishing Co., Easton, PA (1990)).
Las composiciones peptídicas farmacéuticamente aceptables descritas en el presente documento pueden administrarse en múltiples formas de dosificación tales como comprimidos, cápsulas (cada una de las cuales incluye formulaciones de liberación sostenida o de liberación programada), píldoras, polvos, gránulos, elixires, geles in situ, microesferas, complejos cristalinos, liposomas, microemulsiones, tinturas, suspensiones, jarabes, pulverizaciones de aerosol y emulsiones. Las composiciones descritas en el presente documento también se pueden administrar en forma oral, intravenosa (bolo o infusión), intraperitoneal, subcutánea, transdérmica o intramuscular, todas ellas utilizando formas de dosificación bien conocidas por los expertos en la técnica farmacéutica. Las composiciones pueden administrarse solas, pero generalmente se administrarán con un vehículo seleccionado dependiendo de la vía de administración escogida y de la práctica farmacéutica convencional.
El régimen de dosificación para las composiciones descritas en este documento, por supuesto, variará dependiendo de factores conocidos, tales como las características farmacodinámicas del agente particular y su modo y vía de administración; la especie, la edad, sexo, la salud, el estado médico y peso del receptor; la naturaleza y el alcance de los síntomas; la clase de tratamiento concurrente; la frecuencia del tratamiento; la vía de administración, la función renal y hepática del paciente y el efecto deseado. Un médico o un veterinario puede determinar y prescribir la cantidad eficaz del fármaco que es necesaria para prevenir, contrarrestar, o detener el progreso de la patología.
Como orientación general, la dosificación oral diaria del ingrediente activo, cuando se usan para los efectos indicados, variará entre aproximadamente 0,001 a 1000 mg/kg de peso corporal, preferentemente entre aproximadamente 0,01 a 100 mg/kg de peso corporal por día y lo más preferentemente entre aproximadamente 0,6 a 20 mg/kg/día. Por vía intravenosa, la dosis diaria del ingrediente activo cuando se usa para los efectos indicados oscilará entre 0,001 ng a 100,0 ng por minuto/por kg de peso corporal durante una infusión a velocidad constante. Dicha infusión intravenosa constante se puede administrar preferentemente a una velocidad de 0,01 ng a 50 ng por minuto por kg de peso corporal y lo más preferentemente de 0,01 ng a 10,0 mg por minuto por kg de peso corporal. Las composiciones descritas en este documento pueden administrarse en una dosis diaria única, o la dosis diaria total puede administrarse en dosis divididas de dos, tres o cuatro veces al día. Las composiciones descritas en este documento también pueden administrarse mediante una formulación de depósito que permitirá la liberación sostenida del fármaco durante un período de días/semanas/meses, según se desee.
Las composiciones descritas en el presente documento pueden administrarse en forma intranasal mediante el uso tópico de vehículos intranasales adecuados, o mediante rutas transdérmicas, usando parches cutáneos transdérmicos. Cuando se administra en forma de un sistema de suministro transdérmico, la administración de la dosis será, por supuesto, continua en lugar de intermitente a largo del régimen de dosificación.
Las composiciones están administradas en una mezcla con diluyentes farmacéuticos adecuados, excipientes o vehículos farmacéuticamente adecuados (denominados colectivamente en el presente documento como transportadores farmacéuticos) seleccionados adecuadamente con respecto a la forma de administración prevista, es decir, comprimidos orales, cápsulas, elixires, aerosoles generados con o sin propulsores y jarabes y consistentes con las prácticas farmacéuticas convencionales.
Por ejemplo, para la administración oral en forma de un comprimido o una cápsula, el componente de fármaco activo puede combinarse con un vehículo inerte oral, vehículo inerte, no tóxico, farmacéuticamente aceptable, tal como, sin limitación, lactosa, almidón, sacarosa, glucosa, metilcelulosa, estearato de magnesio, fosfato dicálcico, sulfato de calcio, manitol y sorbitol; para la administración oral en forma líquida, los componentes farmacológicos orales pueden combinarse con cualquier vehículo inerte oral, no tóxico, farmacéuticamente aceptable tal como, pero sin limitación, etanol, glicerol y agua. Además, cuando se desee o sea necesario, se pueden incorporar también aglutinantes, lubricantes, agentes disgregantes y agentes colorantes adecuados en la mezcla. Los aglutinantes adecuados incluyen, pero sin limitación, almidón, gelatina, azúcares naturales tales como, pero sin limitación, glucosa o beta-lactosa, edulcorantes de maíz, gomas naturales y sintéticas tales como goma arábiga, tragacanto o alginato de sodio, carboximetilcelulosa, polietilenglicol y ceras. Los lubricantes usados en estas formas de dosificación incluyen oleato sódico, estearato de sodio, estearato de magnesio, benzoato de sodio, acetato sódico y cloruro sódico. Los disgregantes incluyen, pero sin limitación, almidón, metilcelulosa, agar, bentonita y goma de xantano.
Las composiciones descritas en el presente documento también pueden administrarse en forma de sistemas mixtos de administración de micelas o liposomas, tales como vesículas unilamelares pequeñas, vesículas unilamelares grandes y vesículas multilamelares. Los liposomas se pueden formar a partir de varios fosfolípidos, tales como colesterol, estearilamina o fosfatidilcolinas. Se pueden agregar potenciadores de permeación para mejorar la absorción del fármaco.
Las composiciones descritas en el presente documento también se pueden acoplar con polímeros solubles como vehículos de fármacos dirigibles. Dichos polímeros pueden incluir polivinilpirrolidona, copolímero de pirano, polihidroxipropilmetacrilamida-fenol, polihidroxietilaspartamidafenol u óxido de polietilen-polilisina sustituido con restos palmitoílo. Además, las composiciones descritas en el presente documento pueden combinarse con una clase de polímeros biodegradables útiles para lograr la liberación controlada de un fármaco, por ejemplo, ácido poliláctico, ácido poliglicólico, copolímeros de ácido poliláctico y poliglicólico, poliépsilon caprolactona, ácido polihidroxibutírico, poliortoésteres, poliacetales, polihidropiranos, policianoacilatos y copolímeros de bloque de hidrogeles reticulados o anfipáticos.
Las formas de dosificación (composiciones farmacéuticas) adecuadas para la administración pueden contener de aproximadamente 0,01 miligramos a aproximadamente 500 miligramos de ingrediente activo por unidad de dosificación. En estas composiciones farmacéuticas, el ingrediente activo normalmente estará presente en una cantidad de aproximadamente 0,5-95 % en peso basado en el peso total de la composición.
Las cápsulas de gelatina pueden contener el principio activo y transportadores en polvo, tales como lactosa, almidón, derivado de celulosa, estearato de magnesio y ácido esteárico. Para elaborar comprimidos compactados pueden usarse diluyentes similares. Tanto los comprimidos como las cápsulas pueden fabricarse como productos de liberación sostenida para proporcionar la liberación continua de la medicación durante un periodo de horas. Los comprimidos pueden recubrirse con azúcar o con una película para ocultar cualquier sabor desagradable y proteger al comprimido de la atmósfera, o pueden tener un recubrimiento entérico para la disgregación selectiva en el tubo digestivo.
Las formas farmacéuticas líquidas para la administración oral pueden contener colorantes y saborizantes para aumentar la aceptación del paciente.
En general, agua, un aceite adecuado, solución salina, solución acuosa de dextrosa (glucosa) y soluciones de azúcares relacionados y glicoles tales como propilenglicol o polietilenglicoles son vehículos adecuados para las soluciones parenterales. La solución para la administración parenteral contiene una sal soluble en agua del ingrediente activo, agentes estabilizantes adecuados y, si es necesario, sustancias tamponantes. Los agentes antioxidantes tales como, bisulfito de sodio, sulfito de sodio o ácido ascórbico, solos o combinados, son agentes estabilizantes adecuados. También se usan ácido cítrico y sus sales y EDTA sódico. Además, las soluciones parenterales pueden contener conservantes, tales como cloruro de benzalconio, metil- o propil-parabeno y clorobutanol.
Se describen vehículos farmacéuticamente adecuados en Remington: The Science and Practice of Pharmacy, Decimonovena edición, Mack Publishing Company (1995), un texto de referencia en este campo.
Las formas de dosificación farmacéuticas útiles representativas para la administración de los compuestos descritas en el presente documento pueden ilustrarse como sigue:
Cápsulas
Se puede preparar una gran cantidad de cápsulas unitarias llenando cápsulas de gelatina dura estándar de dos piezas con 100 miligramos de ingrediente activo en polvo, 150 miligramos de lactosa, 50 miligramos de celulosa y 6 miligramos de estearato de magnesio.
Cápsulas de gelatina blanda
Una mezcla de ingrediente activo en un aceite digerible tal como aceite de soja, aceite de oliva o aceite de semilla de algodón puede prepararse e inyectarse por medio de una bomba de desplazamiento positivo en gelatina para formar cápsulas de gelatina blandas que contienen 100 miligramos del ingrediente activo. Las cápsulas deben lavarse y secarse.
Comprimidos
Pueden prepararse comprimidos mediante procedimientos específicos para que la unidad de dosificación, por ejemplo, sea 100 miligramos de ingrediente activo, 0,2 miligramos de dióxido de silicio coloidal, 5 miligramos de estearato de magnesio, 275 miligramos de celulosa microcristalina, 11 miligramos de almidón y 98,8 miligramos de lactosa. Se pueden aplicar revestimientos apropiados para aumentar la palatabilidad o retrasar la absorción.
Inyectable
Una formulación inyectable de una composición peptídica descrita en el presente documento puede no requerir el uso de excipientes tales como los que han sido aprobados por organismos reguladores. Estos excipientes incluyen, pero sin limitación, disolventes y codisolventes, solubilizante, agentes emulgentes o espesantes, agentes quelantes, antioxidantes y agentes reductores, conservantes antimicrobianos, tampones y agentes de ajuste de pH, agentes de
carga, protectores y ajustadores de tonicidad y aditivos especiales. Una formulación inyectable tiene que ser estéril, libre de pirógenos, en el caso de soluciones, libre de material particulado.
Una composición parenteral adecuada para administración por inyección puede prepararse agitando, por ejemplo, 1,5 % en peso de ingrediente activo en un tampón farmacéuticamente aceptable que puede no contener un codisolvente u otro excipiente. La solución debe hacerse isotónica con cloruro sódico y esterilizada.
Suspensión
Se puede preparar una suspensión acuosa para administración oral y/o parenteral de modo que, por ejemplo, cada 5 ml contenga 100 mg de ingrediente activo finamente dividido, 20 mg de carboximetil celulosa de sodio, 5 mg de benzoato de sodio, 1,0 g de solución de sorbitol, U.S.P. y 0,025 ml de vainillina u otro saborizante agradable al paladar.
Micropartículas Biodegradables
Se puede preparar una composición parenteral de liberación sostenida adecuada para la administración por inyección, por ejemplo, disolviendo un polímero biodegradable adecuado en un disolvente, añadiendo a la solución de polímero el agente activo a incorporar y eliminando el polímero de la matriz formando así la matriz del polímero con el agente activo distribuido por toda la matriz.
Síntesis de péptidos
Puede realizarse síntesis química de un péptido macrocíclico de la presente divulgación utilizando varios métodos reconocidos en la técnica, incluyendo síntesis en fase sólida por etapas, semi-síntesis a través de los fragmentos peptídicos de re-ligadura asistida conformacionalmente, ligadura enzimática clonada o segmentos de péptidos sintéticos y ligadura química. Un método preferente para sintetizar los péptidos macrocíclicos y sus análogos descritos en este documento es la síntesis química usando diversas técnicas de fase sólida tales como las descritas en Chan, W.C. et al., eds., Fmoc Solid Phase Synthesis, Oxford University Press, Oxford (2000); Barany, G. et al., The Peptides: Analysis, Synthesis, Biology, Vol. 2: "Special Methods in Peptide Synthesis, Parte A", pág. 3-284, Gross, E. et al., eds., Academic Press, Nueva York (1980); y en Stewart, J.M. et al., Solid-Phase Peptide Synthesis, 2a Edición, Pierce Chemical Co., Rockford, IL (1984). La estrategia preferente se basa en el grupo Fmoc (9-fluorenilmetil metiloxicarbonil) para la protección temporal del grupo a-amino, en combinación con el grupo tere-butilo para la protección temporal de las cadenas laterales de aminoácidos (véase por ejemplo Atherton, E. et al., "The Fluorenylmethoxycarbonyl Amino Protecting Group", en The Peptides: Analysis, Synthesis, Biology, Vol. 9: "Special Methods in Peptide Synthesis, Parte C", pág. 1-38, Undenfriend, S. et al., eds., Academic Press, San Diego (1987). Los péptidos pueden sintetizarse de manera gradual en un soporte de polímero insoluble (también denominado "resina") partiendo del extremo C-terminal del péptido. Se inicia una síntesis añadiendo el aminoácido C-terminal del péptido a la resina a través de la formación de un enlace amida o éster. Esto permite la liberación eventual del péptido resultante como una amida o ácido carboxílico C-terminal, respectivamente.
Se requiere que el aminoácido C-terminal y todos los demás aminoácidos utilizados en la síntesis tengan sus grupos a-amino y las funcionalidades de la cadena lateral (si están presentes) protegidas diferencialmente de manera que el protector del grupo a-amino pueda eliminarse selectivamente durante la síntesis. El acoplamiento de un aminoácido se realiza mediante la activación de su grupo carboxilo como cómo éster activo y reacción del mismo con el grupo aamino no bloqueado del aminoácido N-terminal unido a la resina. La secuencia de desprotección y acoplamiento del grupo a-amino se repite hasta que se ensambla toda la secuencia peptídica. El péptido se libera luego de la resina con la desprotección concomitante de las funcionalidades de la cadena lateral, generalmente en presencia de secuestradores apropiados para limitar las reacciones secundarias. El péptido resultante finalmente se purificó por HPLC de fase inversa.
La síntesis de las resinas de peptidilo requeridas como precursores de los péptidos finales utiliza resinas poliestireno-poliméricas reticuladas disponibles comercialmente (Novabiochem, San Diego, CA; Applied Biosystems, Foster City, CA). Los soportes sólidos preferentes son: resina 4-(2',4'-dimetoxifenil-Fmoc-aminometil)-fenoxiacetil-pmetil benzhidrilamina (resina Rink amida MBHA); resina 9-Fmoc-amino-xanten-3-iloxi-Merrifield (resina Sieber amida); resina 4-(9-Fmoc)aminometil-3,5-dimetoxifenoxi)valeril-aminometil-Merrifield (resina PAL), para carboxamidas C-terminales. El acoplamiento del primer y posteriores aminoácidos puede realizarse usando HOBt, 6-Cl-HOBt o HOAt ésteres activos producidos a partir de DIC/HOBt, HBTU/HOBt, BOP, PyBOP, o de DIC/6-Cl-HOBt, HCTU, DIC/HOAt o HATU, respectivamente. Los soportes sólidos preferentes son: resina cloruro de 2-Clorotritilo y resina 9-Fmoc-amino-xanten-3-iloxi-Merrifield (resina Sieber amida) para fragmentos peptídicos protegidos. La carga del primer aminoácido en la resina de cloruro de 2-clorotritilo se logra mejor haciendo reaccionar el aminoácido protegido con Fmoc con la resina en diclorometano y DIEA. Si fuera necesario, se puede agregar una pequeña cantidad de DMF para facilitar la disolución del aminoácido.
Las síntesis de los análogos de péptidos descritos en este documento pueden realizarse utilizando un sintetizador de
péptidos único o multicanal, tal como un sintetizador CEM Liberty Microwave, o Protein Technologies, Inc. Prelude (6 canales) o Sintetizador Symphony (12 canales).
Los precursores de resina de peptidilo para sus péptidos respectivos se pueden escindir y desproteger mediante cualquier procedimiento estándar (véase, por ejemplo, King, D.S. et al., Int. J. Peptide Protein Res., 36:255-266 (1990)). Un método deseado es el uso de TFA en presencia de agua y TIS como secuestradores. Normalmente, la resina de peptidilo se agita en TFA/agua/TIS (94:3:3, v:v:v; 1 ml/100 mg de resina de peptidilo) durante 2-6 horas a temperatura ambiente. La resina gastada se retiró por filtración después y la solución de TFA se concentra o seca a presión reducida. El péptido crudo resultante precipita y se lava con Et2Ü o se redisuelve directamente en DMSO o ácido acético acuoso al 50 % para purificación por HPLC preparativa.
Los péptidos con la pureza deseada se pueden obtener mediante purificación mediante HPLC preparativa, por ejemplo, en un Waters Modelo 4000 o en un cromatógrafo líquido Shimadzu Modelo LC-8A. La solución de péptido crudo se inyecta en una columna YMC S5 ODS (20X100 mm) y eluye con un gradiente lineal de MeCN en agua, ambos tamponados con 0,1 % de TFA, usando un caudal de 14-20 ml/min con monitorización de efluente por absorbancia UV a 220 nm. Las estructuras de los péptidos purificados se pueden confirmar mediante análisis MS por electronebulización.
Datos analíticos:
Espectrometría de Masas: "ESI-MS(+)" significa espectrometría de masas de ionización por electronebulización realizada en modo ion positivo; "ESI-MS(-)" significa espectrometría de masas de ionización por electronebulización realizada en modo ion negativo; "ESI-HRMS(+)" significa espectrometría de masas de ionización por electronebulización de alta resolución realizada en modo ion positivo; "ESI-HRMS(-)" significa espectrometría de masas de ionización por electronebulización de alta resolución realizada en modo ion negativo. Las masas detectadas se informan siguiendo las designación de unidades "m/z". los compuestos con masas exactas mayores de 1000 se detectaron a menudo como iones de doble carga o triple carga.
Datos analíticos:
Espectrometría de Masas: "ESI-MS(+)" significa espectrometría de masas de ionización por electronebulización realizada en modo ion positivo; "ESI-MS(-)" significa espectrometría de masas de ionización por electronebulización realizada en modo ion negativo; "ESI-HRMS(+)" significa espectrometría de masas de ionización por electronebulización de alta resolución realizada en modo ion positivo; "ESI-HRMS(-)" significa espectrometría de masas de ionización por electronebulización de alta resolución realizada en modo ion negativo. Las masas detectadas se informan siguiendo las designación de unidades " mlz". los compuestos con masas exactas mayores de 1000 se detectaron a menudo como iones de doble carga o triple carga.
Condiciones de análisis A:
Columna: Waters BEH C18, 2,0 x 50 mm, partículas de 1,7 pm; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 50 °C; Gradiente: 0% B, 0-100 % B durante 3 minutos, después mantener 0,5 minutos a 100 % B; Flujo: 1 ml/min; Detección: UV a 220 nm.
Condiciones de análisis B:
Columna: Waters BEH C18, 2,0 x 50 mm, partículas de 1,7 pm; Fase móvil A: 5:95 de metanol:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de metanol:agua con acetato amónico 10 mM; Temperatura: 50 °C; Gradiente: 0 % B, 0-100 % B durante 3 minutos, después mantener 0,5 minutos a 100 % B; Flujo: 0,5 ml/min; Detección: UV a 220 nm.
Condiciones de análisis C:
Columna: Waters Aquity BEH C18 2,1 X 50 mm partículas 1,7 pm; Fase móvil A: agua con TFA al 0,05 %; Fase móvil B: acetonitrilo con TFA al 0,05%; Temperatura: 40 °C; Gradiente: 0% B, 0-100 % B durante 3 minutos, después mantener 0,5 minutos a 100 % B; Flujo: 0,8 ml/min; Detección: UV a 220 nm.
Condiciones de análisis D:
Columna: Waters Aquity BEH C18 2,1 X 50 mm partículas 1,7 pm; Fase móvil A: agua con TFA al 0,05 %; Fase móvil B: metanol con t Fa al 0,05 %; Temperatura: 40 °C; Gradiente: 0 % B, 0-100 % B durante 3 minutos, después mantener 0,5 minutos a 100 % B; Flujo: 0,8 ml/min; Detección: UV a 220 nm.
Procedimientos generales:
Método Prelude A:
Todas las manipulaciones se realizaron con automatización en un sintetizador de péptidos Prelude (Protein Technologies). Todos los procedimientos, a menos que se indique lo contrario, se realizaron en un tubo de polipropileno de 10 ml equipado con una frita inferior; cuando la escala de reacción excedió de 0,100 mmol, se usó un tubo de polipropileno de 40 ml equipado con una frita inferior. El tubo se conecta al sintetizador de péptidos Prelude a través de la parte inferior y la parte superior del tubo. Pueden añadirse DMF y DCM a través de la parte superior del tubo, que lavan los lados del tubo por igual. Los reactivos restantes se agregan a través del fondo del tubo y pasan a través de la frita para contactar con la resina. Todas las soluciones se retiran a través del fondo del tubo. "Agitación periódica" describe un breve pulso de gas N2 a través de la frita inferior; el pulso dura aproximadamente 5 segundos y ocurre cada 30 segundos. Las soluciones de aminoácidos generalmente no se usaron más de tres semanas después de la preparación. Las soluciones HATU se utilizaron dentro de los 5 días de preparación. DMF = dimetilformamida; HATU = 3-óxido de hexafluorofosfato de 1-[Bis(dimetilamino)metileno]-1H-1,2,3-triazolo[4,5-b]piridinio; DIPEA = diisopropiletilamina; TIPS = triisopropilsilano; DTT=DL-ditiotreitol. La resina usada es resina cloruro de 2-Clorotritilo (1,42mmol/g carga). Los aminoácidos comunes utilizados se enumeran a continuación con los grupos protectores de cadena lateral indicados entre paréntesis: Fmoc-Ala-OH; Fmoc-Arg(Pbf)-OH; Fmoc-Asn(Trt)-OH; Fmoc-Asp(OtBu)-OH; Fmoc-Bzt-OH; Fmoc-Cys(Trt)-OH; Fmoc-Dab(Boc)-OH; Fmoc-Dap(Boc)-OH; Fmoc-Gln(Trt)-OH; Fmoc-Gly-OH; Fmoc-His(Trt)-OH; Fmoc-Hyp(tBu)-OH; Fmoc-Ile-OH; Fmoc-Leu-OH; Fmoc-Lys(Boc)-OH; Fmoc-Nle-OH; Fmoc-Met-OH; Fmoc-[N-Me]Ala-OH; Fmoc-[N-Me]Nle-OH; Fmoc-Phe-OH; Fmoc-Pro-OH; Fmoc-Sar-OH; Fmoc-Ser(tBu)-OH; Fmoc-Thr(tBu)-OH; Fmoc-Trp(Boc)-OH; Fmoc-Tyr(tBu)-OH; Fmoc-Val-OH.
Los procedimientos del "Método Prelude A" describen un experimento realizado a una escala de 0,100 mmol, donde la escala está determinada por la cantidad de aminoácido utilizado en el "Procedimiento de carga de resina" que se describe a continuación. En una escala de 0,100 mmol, se utilizan aproximadamente 100 mg de resina cloruro de 2-clorotritilo. Todos los procedimientos se pueden escalar más allá de la escala de 0,100 mmol ajustando los volúmenes descritos por el múltiplo de la escala.
Antes del acoplamiento de aminoácidos, todas las secuencias de síntesis de péptidos comenzaron con la carga del primer aminoácido en la resina, descrito a continuación como "Procedimiento de carga de resina". El acoplamiento de aminoácidos a una amina primaria N-terminal utilizó el "procedimiento de acoplamiento individual" que se describe a continuación. El acoplamiento de aminoácidos a una amina secundaria N-terminal usó el "procedimiento de doble acoplamiento" que se describe a continuación. procedimiento "detallado a continuación.
Procedimiento de carga de resina:
A un recipiente de reacción de fase sólida de polipropileno de 10 ml se añadió resina cloruro de 2-Clorotritilo (100 mg, 1,42mmol/g de carga). El recipiente de reacción se puso en el sintetizador de péptidos Prelude. Manualmente, al recipiente de reacción se añadió una solución del aminoácido C-terminal Fmoc-protegido (0,10 mmol) y diisopropiletilamina (0,65 mmol) en DCM (2,5 ml). Con automatización, la mezcla se agitó mediante burbujeo periódico de nitrógeno durante 60 minutos. Al recipiente de reacción se añadió metanol (0,20 ml). La mezcla se agitó mediante burbujeo periódico de nitrógeno durante 15 minutos, después el recipiente de reacción se drenó a través de la frita. La resina se lavó sucesivamente tres veces como sigue: para cada lavado, DCM (2,0 ml) se añadió a la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se lavó sucesivamente tres veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se lavó sucesivamente dos veces como sigue: para cada lavado, DCM (2,0 ml) se añadió a la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se lavó sucesivamente tres veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. Procedimiento de acoplamiento individual:
Al recipiente de reacción que contiene resina de la etapa previa se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 3 minutos y después la solución se drenó a través de la frita. Al recipiente de reacción se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 3 minutos y después la solución se drenó a través de la frita. La resina se lavó sucesivamente seis veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 30 segundos antes de drenar la solución a través de la frita. Al recipiente de reacción se añadió el aminoácido (0,2 M en DMF, 1,0 ml, 2 equiv.), después HATU (0,2 M en DMF, 1,0 ml, 2 equiv.) y finalmente DIPEA (0,8 M en DMF, 0,5 ml, 4 equiv.). La mezcla se agitó periódicamente durante 15 minutos, después la solución de reacción se drenó a través de la frita. La resina se lavó sucesivamente cuatro veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 30 segundos antes de drenar la solución a través de la frita. Se realiza una etapa de protección terminal como sigue: al recipiente de reacción se añadió DMF (0,65 ml), después DIPEA (0,8 M en DMF,
0,45 ml), después anhídrido acético (1,0 M en DMF, 1,45 ml). La mezcla se agitó periódicamente durante 10 minutos, después la solución se drenó a través de la frita. La resina se lavó sucesivamente cuatro veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina resultante se usó directamente en la siguiente etapa.
Procedimiento de acoplamiento doble:
Al recipiente de reacción que contiene resina de la etapa previa se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 3 minutos y después la solución se drenó a través de la frita. Al recipiente de reacción se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 3 minutos y después la solución se drenó a través de la frita. La resina se lavó sucesivamente seis veces como sigue: Para cada lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 30 segundos antes de drenar la solución a través de la frita. Al recipiente de reacción se añadió el aminoácido (0,2 M en DMF, 1,0 ml, 2 equiv.), después HATU (0,2 M en DMF, 1,0 ml, 2 equiv.) y finalmente DIPEA (0,8 M en DMF, 0,5 ml, 4 equiv.). La mezcla se agitó periódicamente durante 15 minutos, después la solución de reacción se drenó a través de la frita. La resina se lavó dos veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 30 segundos antes de drenar la solución a través de la frita. Al recipiente de reacción se añadió el aminoácido (0,2 M en DMF, 1,0 ml, 2 equiv.), después HATU (0,2 M en DMF, 1,0 ml, 2 equiv.) y finalmente DIPEA (0,8 M en DMF, 0,5 ml, 4 equiv.). La mezcla se agitó periódicamente durante 15 minutos, después la solución de reacción se drenó a través de la frita. La resina se lavó dos veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 30 segundos antes de drenar la solución a través de la frita. Se realiza una etapa de protección terminal como sigue: al recipiente de reacción se añadió DMF (0,65 ml), después DIPEA (0,8 M en DMF, 0,45 ml), después anhídrido acético (1,0 M en DMF, 1,45 ml). La mezcla se agitó periódicamente durante 10 minutos, después la solución se drenó a través de la frita. La resina se lavó sucesivamente cuatro veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina resultante se usó directamente en la siguiente etapa.
Método Symphony A:
Esta colección de procedimientos es idéntica al "Método Prelude A" excepto lo indicado. Para todos los procedimientos, se utiliza un sintetizador de péptidos Symphony X (Protein Technologies) en el lugar de un sintetizador de péptidos Prelude y todos los reactivos se agregan a través de la parte superior del recipiente de reacción.
Procedimiento de carga de resina:
Independientemente del sintetizador de péptidos usado en las etapas de acoplamiento de péptidos, la etapa de carga de resina se realizó siempre en el sintetizador Prelude siguiendo el "Método Prelude A: Procedimiento de carga de resina".
Procedimiento de acoplamiento individual:
Este procedimiento es idéntico al "Método Prelude A: Procedimiento de acoplamiento individual" excepto que la concentración de solución de DIPEA fue 0,4 M y 1,0 ml de esta solución se suministró a la reacción. Además, los reactivos usados en la etapa de protección terminal son anhídrido acético puro (1,0 ml) añadido a la resina suspendido en DIPEA (0,4 M, 1,0 ml).
Procedimiento de acoplamiento doble:
Este procedimiento es idéntico al "Método Prelude A: Procedimiento de acoplamiento doble" excepto que la concentración de solución de DIPEA fue 0,4 M y 1,0 ml de esta solución se suministró a la reacción. Además, los reactivos usados en la etapa de protección terminal son anhídrido acético puro (1,0 ml) añadido a la resina suspendido en DIPEA (0,4 M, 1,0 ml).
Secuencia de síntesis general A:
"Secuencia de síntesis general A" describe una secuencia general de procedimientos que se usaron para proporcionar los péptidos cíclicos descritos en este documento. Para los fines de este procedimiento general, los procedimientos de "Método Symphony A" son intercambiables con los de "Método Prelude A". El procedimiento se puede escalar más allá de la escala de 0,100 mmol ajustando los volúmenes descritos por el múltiplo de la escala. "Método Prelude A: procedimiento de carga de resina" se realizó usando 0,100 mmol de aminoácido. Luego se realizó una serie de acoplamientos de aminoácido secuencialmente en el Prelude siguiendo "Método Prelude A: procedimiento de acoplamiento individual" si el N-terminal del péptido unido a resina fue una amina primaria o
"Método Prelude A: procedimiento de acoplamiento doble" si el N-terminal del péptido unido a resina fue una amina secundaria.
Preparación de Ejemplo 5001

Preparación de Intermedio 5001A:
Se siguió "Secuencia de síntesis general A". Al recipiente de reacción que contiene resina de la secuencia automatizada se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. Al recipiente de reacción se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DCM (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se transfirió después inmediatamente usando DCM (8 ml) a un vial de 15 ml. A la solución se añadió hexafluoroisopropanol (2 ml). La resina se volvió inmediatamente rojo intenso; la solución permaneció incolora. La mezcla se agitó manualmente brevemente, después se dejó en reposo a t.a. durante 15 minutos, después se filtró. El filtrado se transfirió a un vial de 15 ml y se concentró bajo una corriente de N2 para proporcionar un residuo sólido, Intermedio 5001A.
Preparación de Intermedio 5001B:
A un vial de 15 ml cargado con la totalidad de Intermedio 5001A preparado anteriormente se añadió DMF (0,50 ml), después HATU (45,6 mg, 0,120 mmol) después DIPEA (0,114 ml, 0,650 mmol). La solución amarilla se agitó durante 30 minutos. La solución se sometió directamente a purificación por HPLC en las siguientes condiciones: Columna:
Luna C18, 30 x 100 mm, partículas de 5 |jm; Fase móvil A: 5:95 de acetonitrilo: agua con TFA al 0,1 %; Fase móvil B: 95:5 de acetonitrilo: agua con TFA al 0,1 %; Gradiente: 60-100 % B durante 10 minutos, después mantener 10 minutos a 100 % B; Flujo: 42 ml/min. Fracciones que contenían el producto deseado se combinaron y secaron por evaporación centrífuga para proporcionar un sólido de color blanco, Intermedio 5001B.
Preparación de Ejemplo 5001:
Una "solución de desprotección" se preparó combinando en un vial de vidrio de 40 ml ácido trifluoroacético (22 ml), fenol (1,325 g), agua (1,25 ml), TIPS (0,5 ml) y DTT (0,25 g). A un vial de 1 dram cargado con la totalidad de Intermedio 5001B preparado anteriormente se añadió la "solución de desprotección" (1,0 ml). La solución se mezcló durante 1,5h en un agitador funcionando a 500 rpm, después se vertió en un tubo de ensayo de 25 ml cargado con Et2O (20 ml). Precipitó una pequeña cantidad de sólido blanco. La mezcla se centrifugó; el líquido se decantó. Los sólidos se suspendieron en Et2O (10 ml). La mezcla se centrifugó, el líquido se decantó. El residuo resultante se disolvió en MeOH y el material crudo se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 jm ; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 15-55% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 0,5 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,49 min; ESI-MS(+) m/z 864,2 (M+2H)
Condiciones de análisis B: Tiempo de retención = 2,73 min; ESI-MS(+) m/z 863,8 (M+2H) ESI-HRMS(+) m/z: Calculado: 863,4774 Encontrado: 863,4768.
Preparación de Ejemplo 5002

Ejemplo 5002
Preparación de Intermedio 5002A:
Se siguió "Secuencia de síntesis general A". Al recipiente de reacción que contiene resina de la secuencia automatizada se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. Al recipiente de reacción se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DCM (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se transfirió después inmediatamente usando DCM (8 ml) a un vial de 15 ml. A la solución se añadió hexafluoroisopropanol (2 ml). La resina se volvió inmediatamente rojo intenso; la solución permaneció incolora. La mezcla se agitó manualmente brevemente, después se dejó en reposo a t.a. durante 15 minutos, después se filtró. El filtrado se transfirió a un vial de 15 ml y se concentró bajo una corriente de N2 para proporcionar un residuo sólido, Intermedio 5002A.
Preparación de Intermedio 5002B:
A un vial de 15 ml cargado con la totalidad de Intermedio 5002A preparado anteriormente se añadió DMF (0,50 ml), después HATU (45,6 mg, 0,120 mmol) después DIPEA (0,114 ml, 0,650 mmol). La solución amarilla se agitó durante 30 minutos. La solución se sometió directamente a purificación por HPLC en las siguientes condiciones: Columna: Luna C18, 30 x 100 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con TFA al 0,1 %; Fase móvil B: 95:5 de acetonitrilo: agua con TFA al 0,1 %; Gradiente: 60-100 % B durante 10 minutos, después mantener 10 minutos a 100 % B; Flujo: 42 ml/min. Fracciones que contenían el producto deseado se combinaron y secaron por evaporación centrífuga para proporcionar un sólido de color blanco, Intermedio 5002B.
Preparación de Ejemplo 5002:
Una "solución de desprotección" se preparó combinando en un vial de vidrio de 40 ml ácido trifluoroacético (22 ml), fenol (1,325 g), agua (1,25 ml), TIPS (0,5 ml) y DTT (0,25 g). A un vial de 1 dram cargado con la totalidad de Intermedio 5002B preparado anteriormente se añadió la "solución de desprotección" (1,0 ml). La solución se mezcló durante 1,0h en un agitador funcionando a 500 rpm, después se vertió en un tubo de ensayo de 25 ml cargado con Et2O (15 ml). Precipitó una pequeña cantidad de sólido blanco. La mezcla se centrifugó; el líquido se decantó. Los sólidos se suspendieron en Et2O (15 ml). La mezcla se centrifugó, el líquido se decantó. El residuo resultante se disolvió en MeOH y a la solución se añadió DIPEA (0,050 ml). El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de metanol: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de metanol: agua con acetato de amonio 10 mM; Gradiente: 45-85 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 1,7 mg y su pureza estimada por análisis de LCMS fue 95%. Condiciones de análisis A: Tiempo de retención = 1,69 min; ESI-MS(-) m/z 891,3 (M-2H) ESI-HRMS(+) m/z: Calculado: 892,4801 Encontrado: 892,4796.
Preparación de Ejemplo 5003 (no según la invención)

Ejemplo 5003
Preparación de Intermedio 5003A:
"Secuencia de síntesis general A" se siguió a una escala de 0,200 mmol usando un recipiente de reacción de 45 ml (RV). Al recipiente de reacción que contiene resina de la secuencia automatizada se añadió piperidina:DMF (20:80 v/v, 4,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. Al recipiente de reacción se añadió piperidina:DMF (20:80 v/v, 4,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DMF (4,0ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DCM (4,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. Al RV se añadió DCM (16 ml) seguido de hexafluoroisopropanol (4 ml). La mezcla se agitó manualmente brevemente, después se dejó en reposo a t.a. durante 15 minutos, después se filtró a través de la frita inferior del RV. El filtrado se transfirió a un ensayo de 25 ml y se concentró por evaporación centrífuga para proporcionar Intermedio 5003A.
Preparación de Intermedio 5003B:
Al tubo de ensayo de 25 ml cargado con la totalidad de Intermedio 5003A preparado anteriormente se añadió DMF (2,0 ml), después HATU (84 mg, 0,220 mmol) después DIPEA (0,350 ml, 2,00 mmol). El tubo de ensayo se puso en un agitador funcionando a 500 rpm durante 30 minutos. La solución se diluyó después con PhMe hasta un volumen de 20 ml y la solución se concentró después por evaporación centrífuga para proporcionar un residuo sólido, Intermedio crudo 5003B.
Preparación de Ejemplo 5003:
Una "solución de desprotección" se preparó combinando en un vial de vidrio de 40 ml ácido trifluoroacético (22 ml), fenol (1,325 g), agua (1,25 ml), TIPS (0,5 ml) y DTT (0,25 g). Al tubo de ensayo de 25 ml cargado con la totalidad de Intermedio crudo 5003B preparado anteriormente se añadió la "solución de desprotección" (2,0 ml). La solución se mezcló durante 1,0h en un agitador funcionando a 500 rpm, después se diluyó mediante la adición de Et2O (15 ml). Se formó un precipitado blanco a medida que se mezclaba bien la mezcla. La mezcla se centrifugó; el líquido se decantó. Los sólidos se suspendieron en Et2O (15ml). La mezcla se centrifugó, el líquido se decantó. El residuo
resultante se disolvió en agua: MeOH:MeCN (1 ml: 1 ml: 1 ml) y a la solución se añadió amonio bicarbonato (aprox.
25 mg). El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de metanol: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de metanol: agua con acetato de amonio 10 mM; Gradiente: 50-90% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60 % B durante 30 minutos, después mantener 3 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 1,0 mg y su pureza estimada por análisis de LCMS fue 100 %. Condiciones de análisis A: Tiempo de retención = 1,93 min; ESI-MS(+) m/z 846,7 (M+2H); ESI-HRMS(+) m/z: Calculado: 845,9588 Encontrado: 845,9576.
Preparación de Ejemplo 5004
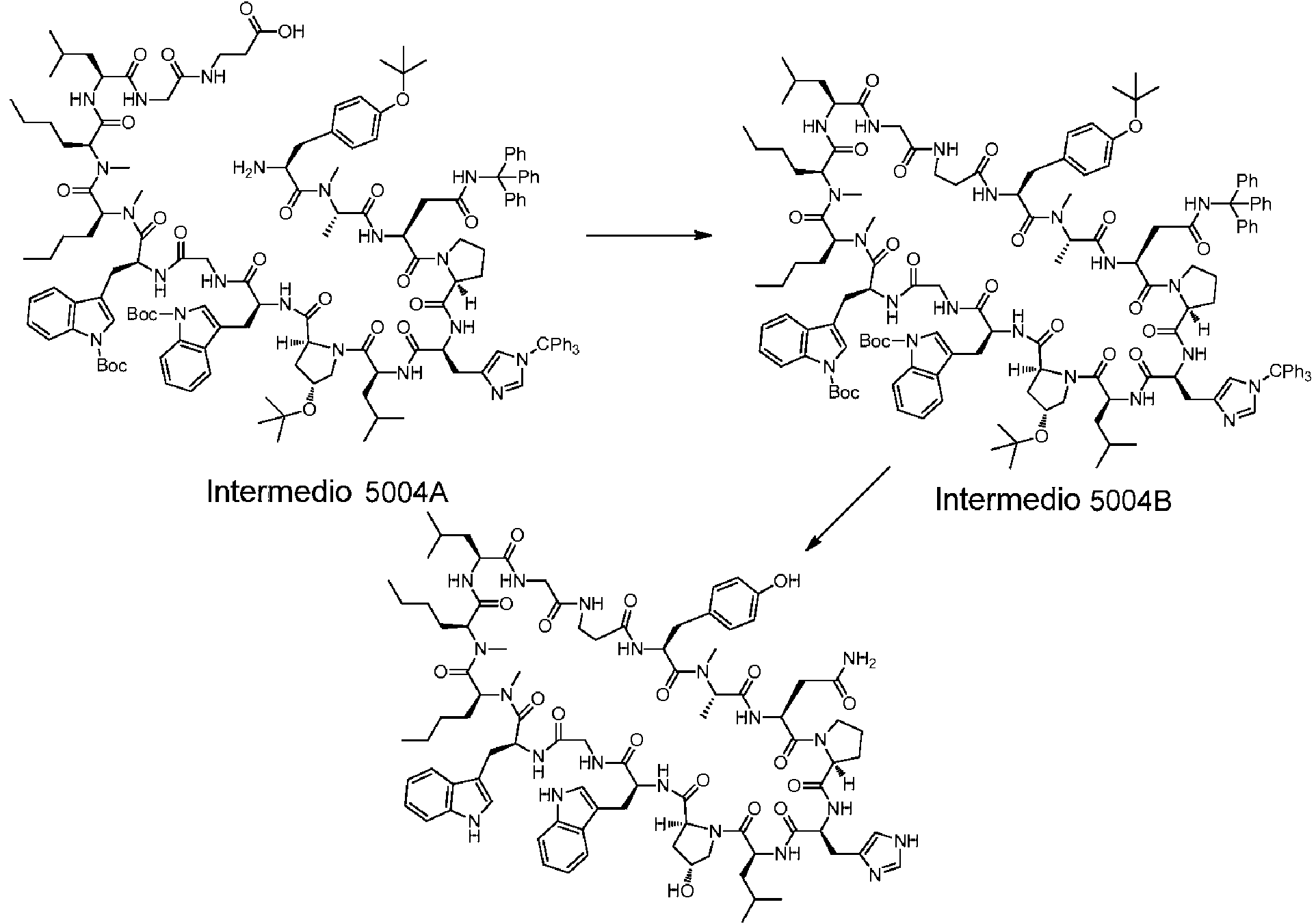
Ejemplo 5004
Preparación de Intermedio 5004A:
"Secuencia de síntesis general A" se siguió a una escala de 0,200 mmol usando un recipiente de reacción de 45 ml (RV). Al recipiente de reacción que contiene resina de la secuencia automatizada se añadió piperidina:DMF (20:80 v/v, 4,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. Al recipiente de reacción se añadió piperidina:DMF (20:80 v/v, 4,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DMF (4,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DCM (4,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. Al RV se añadió DCM (16 ml) seguido de hexafluoroisopropanol (4 ml). La mezcla se agitó manualmente brevemente, después se dejó en reposo a t.a. durante 15 minutos, después se filtró a través de la frita inferior del RV. El filtrado se transfirió a un ensayo de 25 ml y se concentró por evaporación centrífuga para proporcionar Intermedio 5004A.
Preparación de Intermedio 5004B:
Al tubo de ensayo de 25 ml cargado con la totalidad de Intermedio 5004A preparado anteriormente se añadió DMF (2,0 ml), después DIPEA (0,350 ml, 2,00 mmol), después una solución de HATU (84 mg, 0,220 mmol) en DMF (0,5 ml). El tubo de ensayo se puso en un agitador funcionando a 500 rpm durante 2 h. La solución después se diluyó con PhMe hasta un volumen de 20 ml y la solución se concentró después por evaporación centrífuga para proporcionar un residuo sólido, Intermedio crudo 5004B.
Preparación de Ejemplo 5004:
Una "solución de desprotección" se preparó combinando en un vial de vidrio de 40 ml ácido trifluoroacético (22 ml), fenol (1,325 g), agua (1,25 ml), TIPS (0,5 ml) y DTT (0,25 g). Al tubo de ensayo de 25 ml cargado con la totalidad de Intermedio crudo 5004B preparado anteriormente se añadió la "solución de desprotección" (2,0 ml). La solución se mezcló durante 1,0h en un agitador funcionando a 500 rpm, después se diluyó mediante la adición de Et2O (15 ml). Se formó un precipitado blanco a medida que se mezclaba bien la mezcla. La mezcla se centrifugó; el líquido se decantó. Los sólidos se suspendieron en Et2O (15ml). La mezcla se centrifugó, el líquido se decantó. El residuo resultante se disolvió en agua: MeOH:MeCN (1 ml: 1 ml: 1 ml) y a la solución se añadió amonio bicarbonato (aprox.
100mg). El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 15-55% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 5,5 mg y su pureza estimada por análisis de LCMS fue 100 %. Condiciones de análisis A: Tiempo de retención = 1,84 min; eSi-m S(+) m/z 874,4 (M+2H) ESI-HRMS(+) m/z: Calculado: 874,4696 Encontrado: 874,4679.
Preparación de Ejemplo 5005

Ejemplo 5005
Preparación de Intermedio 5005A:
"Secuencia de síntesis general A" se siguió a una escala de 0,200 mmol usando un recipiente de reacción de 45 ml (RV). Al recipiente de reacción que contiene resina de la secuencia automatizada se añadió piperidina:DMF (20:80 v/v, 4,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. Al recipiente de reacción se añadió piperidina:DMF (20:80 v/v, 4,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DMF (4,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DCM (4,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. Al RV se añadió DCM (16 ml) seguido de hexafluoroisopropanol (4 ml). La mezcla se agitó manualmente brevemente, después se dejó en reposo a t.a. durante 15 minutos, después se filtró a través de la frita inferior del RV. El filtrado se transfirió a un ensayo de 25 ml y se concentró por evaporación centrífuga para proporcionar Intermedio 5005A.
Preparación de Intermedio 5005B:
Al tubo de ensayo de 25 ml cargado con la totalidad de Intermedio 5005A preparado anteriormente se añadió DMF (2,0 ml), después DIPEA (0,350 ml, 2,00 mmol), después una solución de HATU (84 mg, 0,220 mmol) en DMF (0,5 ml). El tubo de ensayo se puso en un agitador funcionando a 500 rpm durante 2 h. La solución después se diluyó con PhMe hasta un volumen de 20 ml y la solución se concentró después por evaporación centrífuga para proporcionar un residuo sólido, Intermedio crudo 5005B.
Preparación de Ejemplo 5005:
Una "solución de desprotección" se preparó combinando en un vial de vidrio de 40 ml ácido trifluoroacético (22 ml), fenol (1,325 g), agua (1,25 ml), TIPS (0,5 ml) y DTT (0,25 g). Al tubo de ensayo de 25 ml cargado con la totalidad de Intermedio crudo 5005B preparado anteriormente se añadió la "solución de desprotección" (2,0 ml). La solución se mezcló durante 1,0h en un agitador funcionando a 500 rpm, después se diluyó mediante la adición de Et2O (15 ml). Se formó un precipitado blanco a medida que se mezclaba bien la mezcla. La mezcla se centrifugó; el líquido se decantó. Los sólidos se suspendieron en Et2O (15 ml). La mezcla se centrifugó, el líquido se decantó. El residuo resultante se disolvió en agua: MeOH:MeCN (1 ml: 1 ml: 1 ml) y a la solución se añadió amonio bicarbonato (aprox.
100 mg). El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de metanol: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de metanol: agua con acetato de amonio 10 mM; Gradiente: 50-90% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 4,5 mg y su pureza estimada por análisis de LCMS fue 95 %. Condiciones de análisis A: Tiempo de retención = 1,84 min; ESI-MS(+) m/z 881,5 (M+2H); ESI-HRMS(+) m/z: Calculado: 881,4774 Encontrado: 881,4761.
Preparación de Ejemplo 5006

Ejemplo 5006
Preparación de intermedio 5006A:
"Secuencia de síntesis general A" se siguió a una escala de 0,200 mmol usando un recipiente de reacción de 45 ml (RV). Al recipiente de reacción que contiene resina de la secuencia automatizada se añadió piperidina:DMF (20:80 v/v, 4,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. Al recipiente de reacción se añadió piperidina:DMF (20:80 v/v, 4,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DMF (4,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DCM (4,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. Al RV se añadió DCM (16 ml) seguido de hexafluoroisopropanol (4 ml). La mezcla se agitó manualmente brevemente, después se dejó en reposo a t.a. durante 15 minutos, después se filtró a través de la frita inferior del RV. El filtrado se reservó. Al RV que contenía la resina se añadió DCM (16 ml) seguido de hexafluoroisopropanol (4 ml). La mezcla se agitó manualmente brevemente, después se dejó en reposo a t.a. durante 15 minutos, después se filtró a través de la frita inferior del RV. Los filtrados se combinaron concentraron por evaporación centrífuga para proporcionar Intermedio 5006A.
Preparación de intermedio 5006B:
A un vial de 7 ml con la totalidad de Intermedio 5006A preparado anteriormente se añadió DMF (2,0 ml), después DIPEA (0,350 ml, 2,00 mmol), después a HATU (84 mg, 0,220 mmol). El tubo de ensayo se puso en un agitador funcionando a 500 rpm durante 1 h. La solución se concentró bajo una corriente de N2, después se concentró adicionalmente a alto vacío para proporcionar y sólido ámbar, Intermedio crudo 5006B.
Preparación de Ejemplo 5006:
Una "solución de desprotección" se preparó combinando en un vial de vidrio de 40 ml ácido trifluoroacético (22 ml), fenol (1,325 g), agua (1,25 ml), TIPS (0,5 ml) y DTT (0,25 g). Al vial cargado con la totalidad de Intermedio crudo 5006B preparado anteriormente se añadió la "solución de desprotección" (2,0 ml). La solución se mezcló durante 1,0h en un agitador funcionando a 500 rpm, después se diluyó mediante la adición de Et2O (15 ml). Se formó un precipitado blanco a medida que se mezclaba bien la mezcla. La mezcla se centrifugó; el líquido se decantó. Los
sólidos se suspendieron en Et2O (15ml). La mezcla se centrifugó, el líquido se decantó. El residuo resultante se disolvió en agua:MeOH:MeCN (1 ml: 1 ml: 1 ml) y a la solución se añadió amonio bicarbonato (aprox. 25 mg). El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de metanol: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de metanol: agua con acetato de amonio 10 mM; Gradiente: 45-85 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x 200mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 6,4 mg y su pureza estimada por análisis de LCMS fue 100 %. Condiciones de análisis A: Tiempo de retención = 1,77 min; ESI-MS(+) m/z 870,1 (M+2H); Condiciones de análisis C: Tiempo de retención = 1,44 min; ESI-MS(+) m/z 871,2 (M+2H).
Preparación de Ejemplo 5007

Preparación de Intermedio 5007A:
"Secuencia de síntesis general A" se siguió a una escala de 0,200 mmol usando un recipiente de reacción de 45 ml (RV). Al recipiente de reacción que contiene resina de la secuencia automatizada se añadió piperidina:DMF (20:80 v/v, 4,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. Al recipiente de reacción se añadió piperidina:DMF (20:80 v/v, 4,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DMF (4,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DCM (4,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. Al RV se añadió DCM (16 ml) seguido de hexafluoroisopropanol (4 ml). La mezcla se agitó manualmente brevemente, después se dejó en reposo a t.a. durante 15 minutos, después se filtró a través de la frita inferior del RV. El filtrado se reservó. Al RV que contenía la resina se añadió DCM (16 ml) seguido de hexafluoroisopropanol (4
ml). La mezcla se agitó manualmente brevemente, después se dejó en reposo a t.a. durante 15 minutos, después se filtró a través de la frita inferior del RV. Los filtrados se combinaron concentraron por evaporación centrífuga para proporcionar Intermedio 5007A.
Preparación de intermedio 5007B:
A un vial de 7 ml con la totalidad de Intermedio 5007A preparado anteriormente se añadió DMF (2,0 ml), después DIPEA (0,350 ml, 2,00 mmol), después a HATU (84 mg, 0,220 mmol). El tubo de ensayo se puso en un agitador funcionando a 500 rpm durante 1 h. La solución se concentró bajo una corriente de N2, después se concentró adicionalmente a alto vacío para proporcionar y sólido ámbar, Intermedio crudo 5007B.
Preparación de Ejemplo 5007:
Una "solución de desprotección" se preparó combinando en un vial de vidrio de 40 ml ácido trifluoroacético (22 ml), fenol (1,325 g), agua (1,25 ml), TIPS (0,5 ml) y DTT (0,25 g). Al vial cargado con la totalidad de Intermedio crudo 5007B preparado anteriormente se añadió la "solución de desprotección" (2,0 ml). La solución se mezcló durante 1,0h en un agitador funcionando a 500 rpm, después se diluyó mediante la adición de Et2O (15 ml). Se formó un precipitado blanco a medida que se mezclaba bien la mezcla. La mezcla se centrifugó; el líquido se decantó. Los sólidos se suspendieron en Et2O (15ml). La mezcla se centrifugó, el líquido se decantó. El residuo resultante se disolvió en agua:MeOH:MeCN (1 ml: 1 ml: 1 ml) y a la solución se añadió amonio bicarbonato (aprox. 25 mg). El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 15-55% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/e M preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 6,5 mg y su pureza estimada por análisis de LCMS fue 97 %. Condiciones de análisis A: Tiempo de retención = 1,70 min; ESI-MS(+) m/z 871,0 (M+2H).
Preparación de Ejemplo 5008

Ejemplo 5008
Preparación de Intermedio 5008A:
"Secuencia de síntesis general A" se siguió a una escala de 0,200 mmol usando un recipiente de reacción de 45 ml (RV). Al recipiente de reacción que contiene resina de la secuencia automatizada se añadió piperidina:DMF (20:80 v/v, 4,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. Al recipiente de reacción se añadió piperidina:DMF (20:80 v/v, 4,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DMF (4,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DCM (4,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. Al RV se añadió DCM (16 ml) seguido de hexafluoroisopropanol (4 ml). La mezcla se agitó manualmente brevemente, después se dejó en reposo a t.a. durante 15 minutos, después se filtró a través de la frita inferior del RV. El filtrado se reservó. Al RV que contenía la resina se añadió DCM (16 ml) seguido de hexafluoroisopropanol (4 ml). La mezcla se agitó manualmente brevemente, después se dejó en reposo a t.a. durante 15 minutos, después se filtró a través de la frita inferior del RV. Los filtrados se combinaron concentraron por evaporación centrífuga para proporcionar Intermedio 5008A.
Preparación de Intermedio 5008B:
A un vial de 7 ml con la totalidad de Intermedio 5008A preparado anteriormente se añadió DMF (2,0 ml), después DIPEA (0,350 ml, 2,00 mmol), después a HATU (84 mg, 0,220 mmol). El tubo de ensayo se puso en un agitador funcionando a 500 rpm durante 1 h. La solución se concentró bajo una corriente de N2, después se concentró adicionalmente a alto vacío para proporcionar y sólido ámbar, Intermedio crudo 5008B.
Preparación de Ejemplo 5008:
Una "solución de desprotección" se preparó combinando en un vial de vidrio de 40 ml ácido trifluoroacético (22 ml), fenol (1,325 g), agua (1,25 ml), TIPS (0,5 ml) y DTT (0,25 g). Al vial cargado con la totalidad de Intermedio crudo 5008B preparado anteriormente se añadió la "solución de desprotección" (2,0 ml). La solución se mezcló durante 1,0h en un agitador funcionando a 500 rpm, después se diluyó mediante la adición de Et2O (15 ml). Se formó un precipitado blanco a medida que se mezclaba bien la mezcla. La mezcla se centrifugó; el líquido se decantó. Los sólidos se suspendieron en Et2O (15 ml). La mezcla se centrifugó, el líquido se decantó. El residuo resultante se disolvió en agua:MeOH:MeCN (1 ml:1 ml:1 ml) y a la solución se añadió bicarbonato de amonio (aprox. 25 mg). El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 15-55% B durante 30 minutos, después mantener 4 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de metanol: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de metanol: agua con acetato de amonio 10 mM; Gradiente: 45-85 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 5,1 mg y su pureza estimada por análisis de LCMS fue 98%. Condiciones de análisis A: Tiempo de retención = 1,75 min; ESI-MS(+) m/z 883,7 (M+2H); Condiciones de análisis B: Tiempo de retención = 2,73 min; ESI-MS(+) m/z 884,3 (M+2H).
Preparación de Ejemplo 5009

Preparación de Intermedio 5009A:
"Secuencia de síntesis general A" se siguió a una escala de 0,200 mmol usando un recipiente de reacción de 45 ml (RV). Al recipiente de reacción que contiene resina de la secuencia automatizada se añadió piperidina:DMF (20:80 v/v, 4,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. Al recipiente de reacción se añadió piperidina:DMF (20:80 v/v, 4,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DMF (4,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DCM (4,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. Al RV se añadió DCM (16 ml) seguido de hexafluoroisopropanol (4 ml). La mezcla se agitó manualmente brevemente, después se dejó en reposo a t.a. durante 15 minutos, después se filtró a través de la frita inferior del RV. El filtrado se reservó. Al RV que contenía la resina se añadió DCM (16 ml) seguido de hexafluoroisopropanol (4 ml). La mezcla se agitó manualmente brevemente, después se dejó en reposo a t.a. durante 15 minutos, después se filtró a través de la frita inferior del RV. Los filtrados se combinaron concentraron por evaporación centrífuga para proporcionar Intermedio 5009A.
Preparación de Intermedio 5009B:
A un vial de 7 ml con la totalidad de Intermedio 5009A preparado anteriormente se añadió DMF (2,0 ml), después DIPEA (0,350 ml, 2,00 mmol), después a HATU (84 mg, 0,220 mmol). El tubo de ensayo se puso en un agitador funcionando a 500 rpm durante 1 h. La solución se concentró bajo una corriente de N2, después se concentró adicionalmente a alto vacío para proporcionar y sólido ámbar, Intermedio crudo 5009B.
Preparación de Ejemplo 5009:
Una "solución de desprotección" se preparó combinando en un vial de vidrio de 40 ml ácido trifluoroacético (22 ml), fenol (1,325 g), agua (1,25 ml), TIPS (0,5 ml) y DTT (0,25 g). Al vial cargado con la totalidad de Intermedio crudo
5009B preparado anteriormente se añadió la "solución de desprotección" (2,0 ml). La solución se mezcló durante 1,0h en un agitador funcionando a 500 rpm, después se diluyó mediante la adición de Et2O (15 ml). Se formó un precipitado blanco a medida que se mezclaba bien la mezcla. La mezcla se centrifugó; el líquido se decantó. Los
sólidos se suspendieron en Et2O (15 ml). La mezcla se centrifugó, el líquido se decantó. El residuo resultante se
disolvió en agua:MeOH:MeCN (1 ml: 1 ml: 1 ml) y a la solución se añadió amonio bicarbonato (aprox. 25 mg). El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19
x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de metanol: agua con acetato de amonio 10 mM; Fase móvil B:
95:5 de metanol: agua con acetato de amonio 10 mM; Gradiente: 45-85 % B durante 30 minutos, después mantener
5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x 200 mm, partículas de 5 pm; Fase
móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con
acetato de amonio 10 mM; Gradiente: 10-50 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo:
20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación
por centrifugación. El rendimiento del producto fue 6,2 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,72 min; ESI-MS(+) m/z 869,1 (M+2H); Condiciones de análisis
C: Tiempo de retención = 1,47 min; ESI-MS(+) m/z 1852,5 (M+TFA).
Procedimientos generales para Método Symphony X E/F/G
Procedimientos de acoplamiento generales:
Todas las manipulaciones se realizaron con automatización en un sintetizador de péptidos Symphony X (Protein Technologies). Todos los procedimientos, a menos que se indique lo contrario, se realizaron en un tubo de polipropileno de 10 ml equipado con una frita inferior; cuando la escala de reacción excedió de 0,100 mmol, se usó
un tubo de polipropileno de 40 ml equipado con una frita inferior. El tubo conecta al sintetizador de péptidos Symphony X a través tanto del fondo como de la parte superior del tubo. Pueden añadirse DMF y DCM a través de
la parte superior del tubo, que lavan los lados del tubo por igual. Los reactivos restantes se agregan a través de la
parte superior del tubo y pasan a través de la frita para contactar con la resina. Todas las soluciones se retiran a
través del fondo del tubo. "Agitación periódica" describe un breve pulso de gas N2 a través de la frita inferior; el pulso
dura aproximadamente 5 segundos y ocurre cada 30 segundos. Se usaron soluciones de cloruro de cloroacetilo en
DMF en 24h de preparación. Las soluciones de aminoácidos generalmente no se usaron más de tres semanas después de la preparación. Las soluciones HATU se utilizaron dentro de los 5 días de preparación. DMF = dimetilformamida; HATU = 3-óxido de hexafluorofosfato de 1-[Bis(dimetilamino)metileno]-1H-1,2,3-triazolo[4,5-b]piridinio; DIPEA = diisopropiletilamina; Resina Rink: 4-((2,4-dimetoxifenil)(Fmocamino) metil)fenoximetilpoliestireno.
Procedimiento de hinchamiento de resina:
A un recipiente de reacción de fase sólida de polipropileno de 10 ml se añadió resina (0,100 mmol). La resina se lavó (hinchó) tres veces como sigue: al recipiente de reacción se añadió DMF (2,0 ml), posteriormente la mezcla se agitó periódicamente durante 10 minutos antes de drenar el disolvente a través de la frita.
Procedimiento de acoplamiento individual:
Al recipiente de reacción que contiene resina de la etapa previa se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 3 minutos y después la solución se drenó a través de la frita. Al recipiente
de reacción se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 3 minutos y después la solución se drenó a través de la frita. La resina se lavó sucesivamente seis veces como sigue: para cada
lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 30 segundos antes de drenar la solución a través de la frita. Al recipiente de reacci añadió el aminoácido (0,2 M en DMF, 1,0 ml, 2 equiv.), después HATU (0,2 M en DMF, 1,0 ml, 2 equiv.) y finalmente
DIPEA (0,4 M en DMF, 1,0 ml, 4 equiv.). La mezcla se agitó periódicamente durante 15 minutos, después la solución
de reacción se drenó a través de la frita. La resina se lavó sucesivamente cuatro veces como sigue: para cada
lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 30 segundos antes de drenar la solución a través de la frita. Al recipiente de reacci añadió anhídrido acético (2,0 ml). La mezcla se agitó periódicamente durante 10 minutos, después la solución se
drenó a través de la frita. La resina se lavó sucesivamente cuatro veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina resultante se usó directamente en la siguiente
etapa.
Procedimiento de acoplamiento individual 1h:
Al recipiente de reacción que contiene resina de la etapa previa se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 3 minutos y después la solución se drenó a través de la frita. Al recipiente
de reacción se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 3 minutos y
después la solución se drenó a través de la frita. La resina se lavó sucesivamente seis veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 30 segundos antes de drenar la solución a través de la frita. Al recipiente de reacción se añadió el aminoácido (0,2 M en DMF, 1,0 ml, 2 equiv.), después HATU (0,2 M en DMF, 1,0 ml, 2 equiv.) y finalmente DIPEA (0,4 M en DMF, 1,0 ml, 4 equiv.). La mezcla se agitó periódicamente durante 60 minutos, después la solución de reacción se drenó a través de la frita. La resina se lavó sucesivamente cuatro veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 30 segundos antes de drenar la solución a través de la frita. Al recipiente de reacción se añadió anhídrido acético (2,0 ml). La mezcla se agitó periódicamente durante 10 minutos, después la solución se drenó a través de la frita. La resina se lavó sucesivamente cuatro veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina resultante se usó directamente en la siguiente etapa.
Procedimiento de acoplamiento doble:
Al recipiente de reacción que contiene resina de la etapa previa se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 3 minutos y después la solución se drenó a través de la frita. Al recipiente de reacción se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 3 minutos y después la solución se drenó a través de la frita. La resina se lavó sucesivamente seis veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 30 segundos antes de drenar la solución a través de la frita. Al recipiente de reacción se añadió el aminoácido (0,2 M en DMF, 1,0 ml, 2 equiv.), después HATU (0,2 M en DMF, 1,0 ml, 2 equiv.) y finalmente DIPEA (0,4 M en DMF, 1,0 ml, 4 equiv.). La mezcla se agitó periódicamente durante 15 minutos, después la solución de reacción se drenó a través de la frita. La resina se lavó dos veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 30 segundos antes de drenar la solución a través de la frita. Al recipiente de reacción se añadió el aminoácido (0,2 M en DMF, 1,0 ml, 2 equiv.), después HATU (0,2 M en DMF, 1,0 ml, 2 equiv.) y finalmente DIPEA (0,4 M en DMF, 1,0 ml, 4 equiv.). La mezcla se agitó periódicamente durante 15 minutos, después la solución de reacción se drenó a través de la frita. La resina se lavó dos veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 30 segundos antes de drenar la solución a través de la frita. Al recipiente de reacción se añadió anhídrido acético (2,0 ml). La mezcla se agitó periódicamente durante 10 minutos, después la solución se drenó a través de la frita. La resina se lavó sucesivamente cuatro veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina resultante se usó directamente en la siguiente etapa.
Procedimiento de acoplamiento de cloruro de cloroacetilo:
Al recipiente de reacción que contenía la resina de la etapa previa se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 3 minutos y después la solución se drenó a través de la frita. Al recipiente de reacción se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 3 minutos y después la solución se drenó a través de la frita. La resina se lavó sucesivamente seis veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 30 segundos antes de drenar la solución a través de la frita. Al recipiente de reacción se añadió DIPEA (0,8 M en DMF, 3,0 ml, 24 equiv.), después cloruro de cloroacetilo (0,8 M en DMF, 1,65 ml, 13,2 equiv.). La mezcla se agitó periódicamente durante 30 minutos, después la solución se drenó a través de la frita. La resina se lavó sucesivamente tres veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se lavó sucesivamente cuatro veces como sigue: para cada lavado, CH2Cl2 (2,0 ml) se añadió a la parte superior del recipiente y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina resultante se puso bajo una corriente de N2 durante 15 minutos.
Método de desprotección:
Todas las manipulaciones se realizaron manualmente a menos que se indique lo contrario. El procedimiento de "Método de desprotección" describe un experimento realizado en una escala de 0,100 mmol. El procedimiento se puede escalar más allá de la escala de 0,100 mmol ajustando los volúmenes descritos por el múltiplo de la escala. Una "solución de desprotección" se preparó combinando en un vial de vidrio de 40 ml ácido trifluoroacético (38 ml), DTT (1 g) y triisopropilsilano (1 ml). La resina se retiró del recipiente de reacción y transfirió a un vial de vidrio de 4 ml. Al vial se añadió la "solución de desprotección" (2,0 ml). La mezcla se mezcló vigorosamente en un agitador (1000 RPM durante 30-90 minutos). La mezcla se filtró a través de a filtro de jeringa de 0,2 micrómetros y los sólidos se extrajeron con la "solución de desprotección" (1,0 ml). Al tubo de ensayo de 24 ml cargado con los filtrados combinados se añadió Et2O (15 ml). La mezcla se mezcló vigorosamente hasta que precipitó una cantidad significativa de un sólido blanco. La mezcla se centrifugó durante 3 minutos, después la solución se decantó de los sólidos y descartó. Los sólidos se suspendieron en Et2O (20 ml); después la mezcla se centrifugó durante 3 minutos;
y la solución se decantó de los sólidos y descartó. Por última vez, los sólidos se suspendieron en Et2O (20 ml); la mezcla se centrifugó durante 3 minutos; y la solución se decantó de los sólidos y descartó para proporcionar el péptido crudo como sólido blanco a blanquecino.
Método de delación E:
Todas las manipulaciones se realizaron manualmente a menos que se indique lo contrario. El procedimiento de "Método de ciclación E" describe un experimento realizado en una escala de 0,100 mmol. El procedimiento se puede escalar más allá de la escala de 0,100 mmol ajustando los volúmenes descritos por el múltiplo de la escala. Los sólidos peptídicos crudos se disolvieron en 20 ml de DMF y a la solución se añadió 0,5 ml de DIEA después 50 mg de KI. La solución se calentó a 65 °C durante una noche. La solución de reacción se concentró y el residuo se sometió a purificación por HPLC de fase inversa para proporcionar el péptido cíclico deseado.
Procedimientos generales para Método Symphony X E:
"Procedimientos generales para Método Symphony X E" describe una secuencia general de procedimientos que se usaron para proporcionar los péptidos cíclicos descritos en este documento. A un recipiente de reacción de polipropileno de fase sólida de 10 ml se añadió resina Rink y el recipiente de reacción se puso en el sintetizador de péptidos Symphony X. "Procedimientos de acoplamiento generales E": Se siguió el procedimiento de hinchamiento de resina. Luego se realizó secuencialmente una serie de acoplamientos de aminoácidos en el sintetizador de péptidos Symphony X siguiendo el "Procedimiento de acoplamiento único" si el N-terminal del péptido unido a la resina era una amina primaria o "Procedimiento de doble acoplamiento" si el N-terminal el péptido unido a resina era una amina secundaria. El procedimiento "Acoplamiento único 1 h" se usó con el aminoácido ácido (S)-2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-(1-(2-(terc-butoxi)-2-oxoetil)-1H-indol-3-il)propanoico acoplado a la resina. Se siguió el procedimiento de acoplamiento de cloruro de cloroacetilo; después se siguió el Método de desprotección; después se siguió el Método de ciclación E.
Método de ciclación F:
Todas las manipulaciones se realizaron manualmente a menos que se indique lo contrario. El procedimiento "Método de ciclación F" describe un experimento realizado a una escala de 0,100 mmol. El procedimiento se puede escalar más allá de la escala de 0,100 mmol ajustando los volúmenes descritos por el múltiplo de la escala. Los sólidos peptídicos crudos se disolvieron en 20 ml de DMF y a la solución se añadió 0,2 ml de DIEA. La mezcla de reacción se agitó durante 5 minutos antes de enfriarse a 0 °C. Se añadieron 0,5 ml de CDI 0,2 M en solución de DMF a la mezcla de reacción gota a gota. La solución se agitó a ta durante una noche. La solución de reacción se concentró y el residuo se sometió a purificación por HPLC de fase inversa para proporcionar el péptido cíclico deseado.
Procedimientos generales para Método Symphony X F:
"Procedimientos generales para Método X F" describe una secuencia general de procedimientos que se usaron para proporcionar los péptidos cíclicos descritos en este documento. A un recipiente de reacción de polipropileno de fase sólida de 10 ml se añadió resina Rink y el recipiente de reacción se puso en el sintetizador de péptidos Symphony X. "Procedimientos generales de acoplamiento": Se siguió el procedimiento de hinchamiento de resina. Luego se realizó secuencialmente una serie de acoplamientos de aminoácidos en el sintetizador de péptidos Symphony X siguiendo el "Procedimiento de acoplamiento único" si el N-terminal del péptido unido a la resina era una amina primaria o "Procedimiento de doble acoplamiento" si el N-terminal el péptido unido a resina era una amina secundaria. El procedimiento "Acoplamiento único 1 h" se usó con el aminoácido ácido (S)-2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-(1-(2-(terc-butoxi)-2-oxoetil)-1H-indol-3-il)propanoico acoplado a la resina. Después de acoplar el último aminoácido en el péptido, al recipiente de reacción se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó durante 30 minutos en el agitador. La solución se drenó a través de la frita. La resina se lavó sucesivamente con DMF después DCM. Se siguió el Método de desprotección; después se siguió el Método de ciclación F.
Método de ciclación G:
Todas las manipulaciones se realizaron manualmente a menos que se indique lo contrario. El procedimiento de "Método de ciclación G" describe un experimento realizado en una escala de 0,100 mmol. El procedimiento se puede escalar más allá de la escala de 0,100 mmol ajustando los volúmenes descritos por el múltiplo de la escala. Los sólidos peptídicos crudos se disolvieron en 20 ml de THF y a la solución se añadió 0,2 ml de DIEA. La mezcla de reacción se agitó durante 5 minutos antes de enfriar a 0 °C. Se añadieron 0,5 ml de dicloruro de oxalilo 0,2 M en solución de THF a la mezcla de reacción gota a gota. La solución se agitó a ta durante una noche. La solución de reacción se concentró y el residuo se sometió a purificación por HPLC de fase inversa para proporcionar el péptido cíclico deseado.
Procedimientos generales para Método Symphony X G:
"Procedimientos generales para Método X G" describe una secuencia general de procedimientos que se usaron para proporcionar los péptidos cíclicos descritos en este documento. A un recipiente de reacción de polipropileno de fase sólida de 10 ml se añadió resina Rinky el recipiente de reacción se puso en el sintetizador de péptidos Symphony X. "Procedimientos generales de acoplamiento": Se siguió el procedimiento de hinchamiento de resina. Luego se realizó secuencialmente una serie de acoplamientos de aminoácidos en el sintetizador de péptidos Symphony X siguiendo el "Procedimiento de acoplamiento único" si el N-terminal del péptido unido a la resina era una amina primaria o "Procedimiento de doble acoplamiento" si el N-terminal el péptido unido a resina era una amina secundaria. El procedimiento "Acoplamiento único 1 h" se usó con el aminoácido ácido (S)-2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-(1-(2-(ferc-butoxi)-2-oxoetil)-1H-indol-3-il)propanoico acoplado a la resina. Después de acoplar el último aminoácido en el péptido, al recipiente de reacción se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó durante 30 minutos en el agitador. La solución se drenó a través de la frita. La resina se lavó sucesivamente con DMF después DCM. Se siguió el Método de desprotección; después se siguió el Método de ciclación G.
Ejemplo 10001-10028 se prepararon siguiendo los "Procedimientos generales para Método Symphony X E".
Preparación de Ejemplo 10001

Ejemplo 10001
El material en bruto del Ejemplo 10001 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 20,9 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,64 min; ESI-MS(+) m/z 927,55 (M+2H).
Condiciones de análisis B: Tiempo de retención = 3,14 min; ESI-MS(+) m/z 927,75 (M+2h ). ESI-HRMS(+) m/z: Calculado: 927,4885 (M+2H); Encontrado: 927,4877 (M+2H).
Preparación de Ejemplo 10002

Ejemplo 10002
El material en bruto del Ejemplo 10002 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 14,9 mg y su pureza estimada por análisis de LCMS fue 96 %.
Condiciones de análisis A: Tiempo de retención = 1,70 min; ESI-MS(+) m/z 919,75 (M+2H).
Condiciones de análisis B: Tiempo de retención = 3,19 min; ESI-MS(+) m/z 919,75 (M+2h ). ESI-HRMS(+) m/z: Calculado: 919,4910 (M+2H); Encontrado: 919,4897 (M+2H).
Preparación de Ejemplo 10003
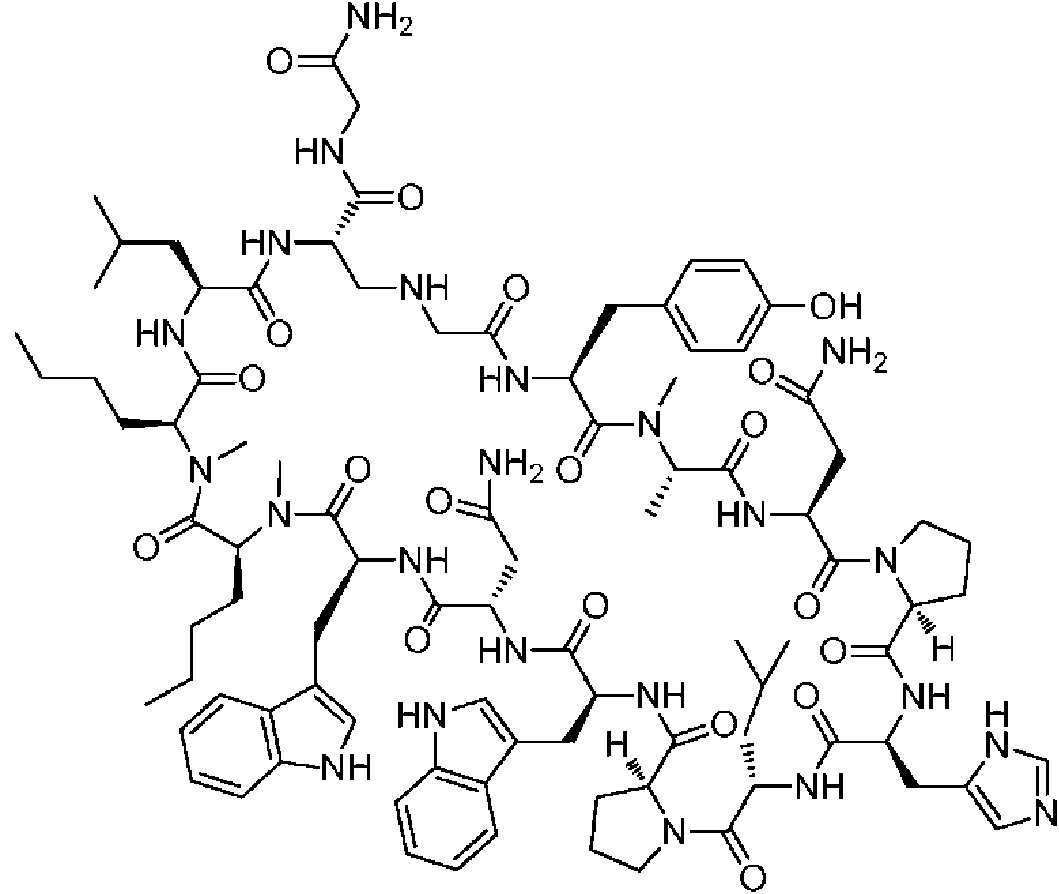
Ejemplo 10003
El material en bruto del Ejemplo 10003 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto
deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 14,2 mg y su pureza estimada por análisis de LCMS fue 98 %.
Condiciones de análisis A: Tiempo de retención = 1,71 min; ESI-MS(+) m/z 931,30 (M+2H).
Condiciones de análisis B: Tiempo de retención = 3,21 min; ESI-MS(+) m/z 931,30 (M+2h ). ESI-HRMS(+) m/z: Calculado: 930,9990 (M+2H); Encontrado: 930,9985 (M+2H).
Preparación de Ejemplo 10004

Ejemplo 10004
El material en bruto del Ejemplo 10004 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 2,7 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,64 min; ESI-MS(+) m/z 933,20 (M+2H).
Condiciones de análisis B: Tiempo de retención = 3,14 min; ESI-MS(+) m/z 933,25 (M+2h ). ESI-HRMS(+) m/z: Calculado: 932,9783 (M+2H); Encontrado: 932,9775 (M+2H).
Preparación de Ejemplo 10005
Ejemplo 10005
El material en bruto del Ejemplo 10005 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 20,3 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 2,46 min; ESI-MS(+) m/z 910,50 (M+2H).
Condiciones de análisis B: Tiempo de retención = 3,29 min; ESI-MS(+) m/z 910,50 (M+2h ). ESI-HRMS(+) m/z: Calculado: 910,0553 (M+2H); Encontrado: 910,0541 (M+2H).
Preparación de Ejemplo 10006

Ejemplo 10006
El material en bruto del Ejemplo 10006 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 8,4 mg y su pureza estimada por análisis de LCMS fue 98 %.
Condiciones de análisis A: Tiempo de retención = 1,44 min; ESI-MS(+) m/z 947,40 (M+2H).
Condiciones de análisis B: Tiempo de retención = 2,56 min; ESI-MS(+) m/z 946,80 (M+2h ). ESI-HRMS(+) m/z: Calculado: 946,4963 (M+2H); Encontrado: 946,4941 (M+2H).
Preparación de Ejemplo 10007

Ejemplo 10007
El material en bruto del Ejemplo 10007 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 |jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 1,5 mg y su pureza estimada por análisis de LCMS fue 97 %.
Condiciones de análisis A: Tiempo de retención = 1,38 min; ESI-MS(+) m/z 954,70 (M+2H).
Condiciones de análisis B: Tiempo de retención = 2,52 min; ESI-MS(+) m/z 954,40 (M+2h ). ESI-HRMS(+) m/z: Calculado: 954,4938 (M+2H); Encontrado: 954,4915 (M+2H).
Preparación de Ejemplo 10008

Ejemplo 10008
El material en bruto del Ejemplo 10008 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 |jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 5,6 mg y su pureza estimada por análisis de LCMS fue 97 %.
Condiciones de análisis A: Tiempo de retención = 1,40 min; ESI-MS(+) m/z 962,30 (M+2H).
Condiciones de análisis B: Tiempo de retención = 2,92 min; ESI-MS(+) m/z 962,25 (M+2h ). ESI-HRMS(+) m/z: Calculado: 961,9810 (M+2H); Encontrado: 961,9804 (M+2H).
Preparación de Ejemplo 10009
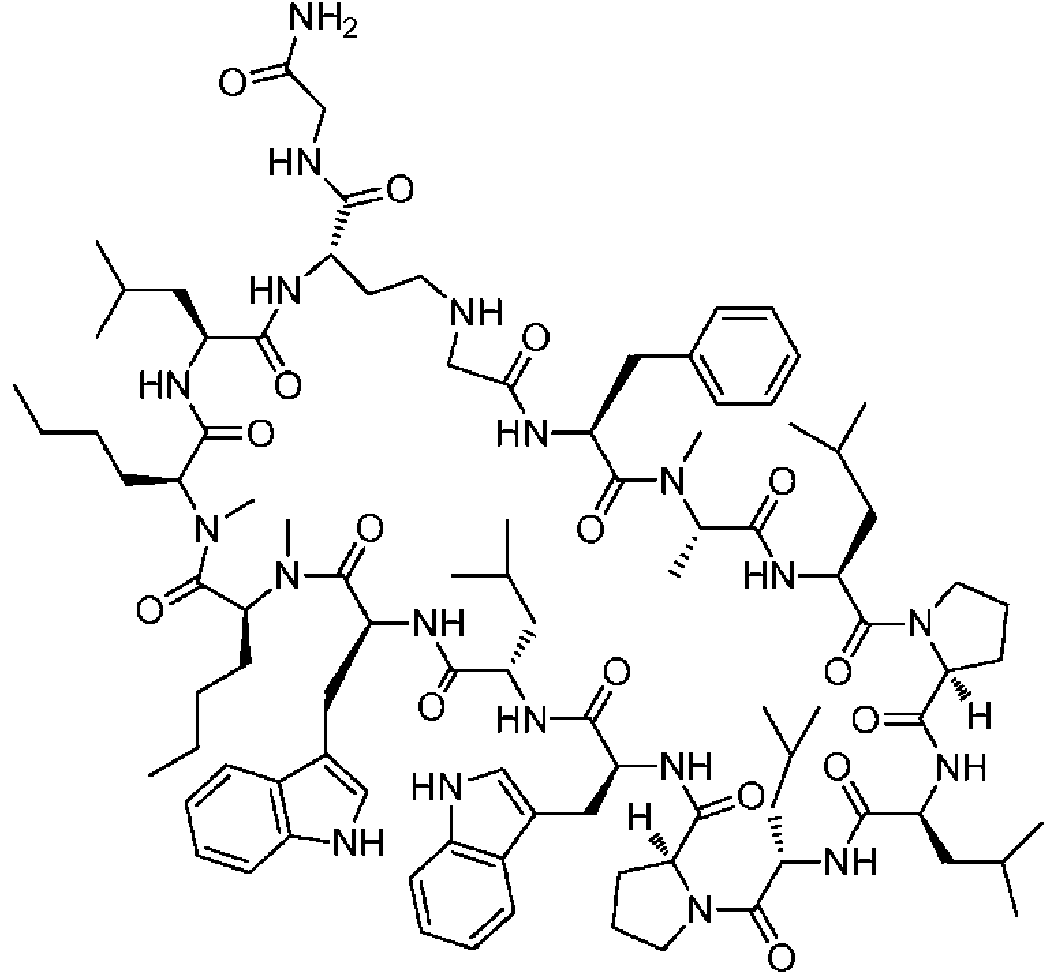
Ejemplo 10009
El material en bruto del Ejemplo 10009 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 jm ; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 35,8 mg y su pureza estimada por análisis de LCMS fue 100 %. Condiciones de análisis A: Tiempo de retención = 2,38 min; ESI-MS(+) m/z 917,50 (M+2H). ESI-HRMS(+) m/z: Calculado: 917,0631 (M+2H); Encontrado: 917,0618 (M+2H).
Preparación de Ejemplo 10010
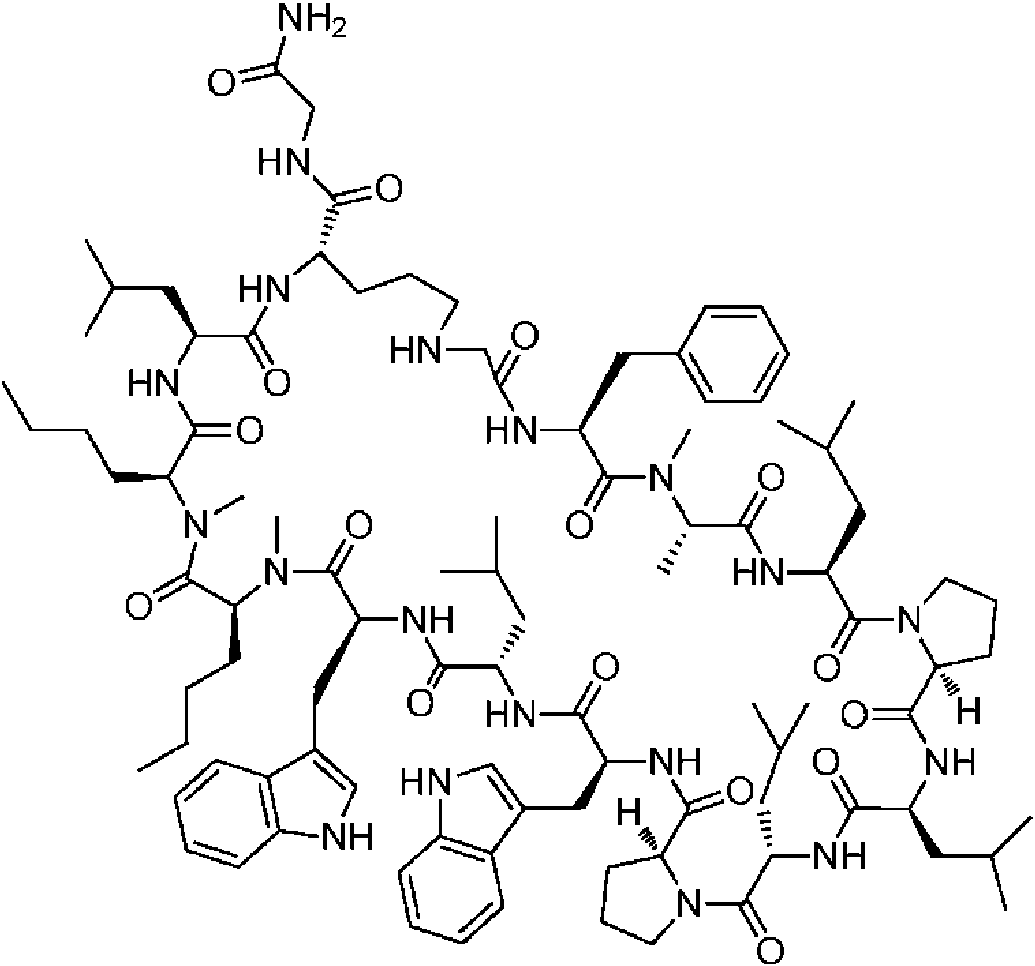
Ejemplo 10010
El material en bruto del Ejemplo 10010 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 24,5 mg y su pureza estimada por análisis de LCMS fue 100 %. Condiciones de análisis A: Tiempo de retención = 2,36 min; ESI-MS(+) m/z 924,55 (M+2H). ESI-HRMS(+) m/z: Calculado: 924,0709 (M+2H); Encontrado: 924,0700 (M+2H).
Preparación de Ejemplo 10011
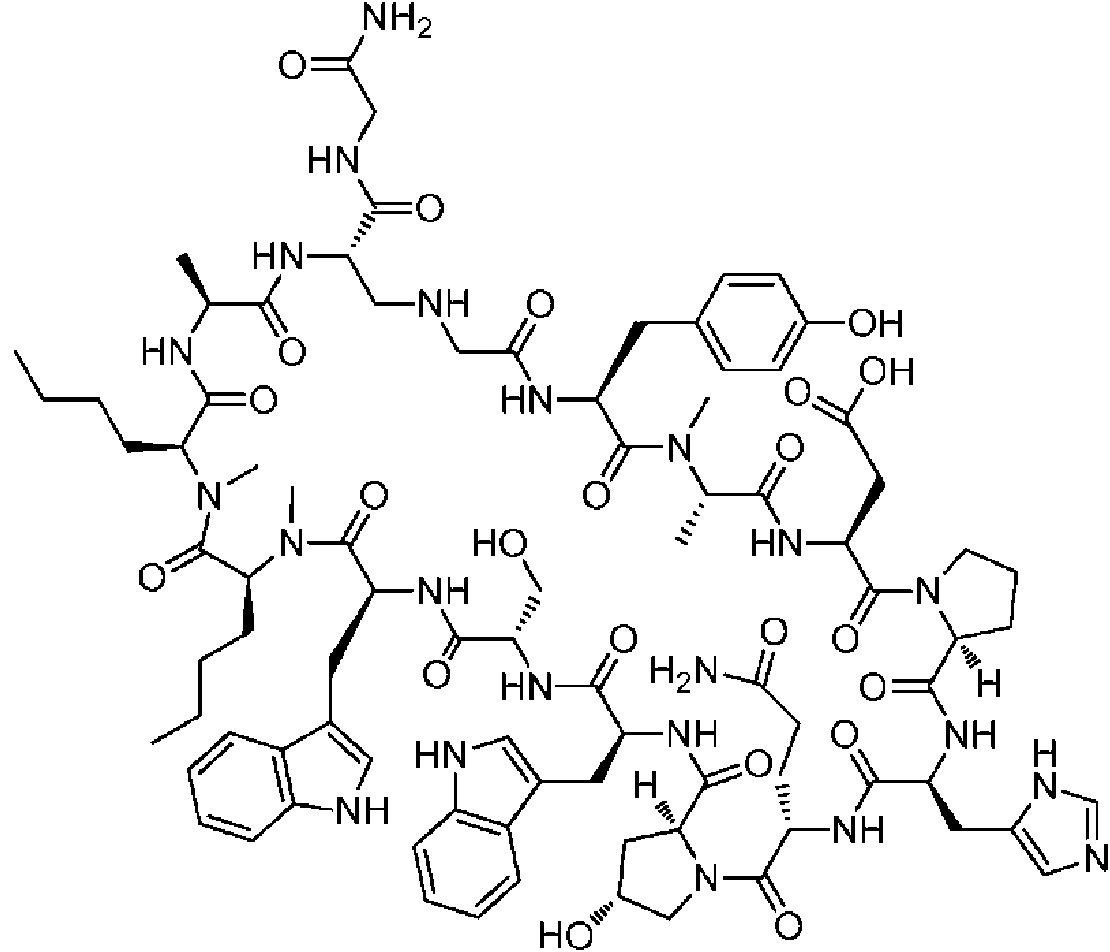
Ejemplo 10011
El material en bruto del Ejemplo 10011 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto
deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 2,4 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,33 min; ESI-MS(+) m/z 912,90 (M+2H).
Condiciones de análisis B: Tiempo de retención = 2,78 min; ESI-MS(+) m/z 912,85 (M+2h ). ESI-HRMS(+) m/z: Calculado: 912,4468 (M+2H); Encontrado: 912,4468 (M+2H).
Preparación de Ejemplo 10012

Ejemplo 10012
El material en bruto del Ejemplo 10012 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 11,7 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,50 min; ESI-MS(+) m/z 919,90 (M+2H).
Condiciones de análisis B: Tiempo de retención = 2,97 min; ESI-MS(+) m/z 919,90 (M+2h ). ESI-HRMS(+) m/z: Calculado: 919,4547 (M+2H); Encontrado: 919,4549 (M+2H).
Preparación de Ejemplo 10013
Ejemplo 10013
El material en bruto del Ejemplo 10013 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 6,4 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,38 min; ESI-MS(+) m/z 927,00 (M+2H).
Condiciones de análisis B: Tiempo de retención = 2,73 min; ESI-MS(+) m/z 926,95 (m 2h ).
Preparación de Ejemplo 10014

Ejemplo 10014
El material en bruto del Ejemplo 10014 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 1,1 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,28 min; ESI-MS(+) m/z 941,90 (M+2H).
Condiciones de análisis B: Tiempo de retención = 2,71 min; ESI-MS(+) m/z 941,90 (m 2H).
Preparación de Ejemplo 10015

Ejemplo 10015
El material en bruto del Ejemplo 10015 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 4,3 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,32 min; ESI-MS(+) m/z 948,90 (M+2H).
Condiciones de análisis B: Tiempo de retención = 2,80 min; ESI-MS(+) m/z 948,80 (m 2h ).
Preparación de Ejemplo 10016

Ejemplo 10016
El material en bruto del Ejemplo 10016 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue
5,3 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,31 min; ESI-MS(+) m/z 955,90 (M+2H).
Condiciones de análisis B: Tiempo de retención = 2,82 min; ESI-MS(+) m/z 955,90 (m 2h ).
Preparación de Ejemplo 10017

Ejemplo 10017
El material en bruto del Ejemplo 10017 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 28,9 mg y su pureza estimada por análisis de LCMS fue 98 %.
Condiciones de análisis A: Tiempo de retención = 1,64 min; ESI-MS(+) m/z 918,40 (M+2H).
Condiciones de análisis B: Tiempo de retención = 3,16 min; ESI-MS(+) m/z 918,40 (M+2h ). ESI-HRMS(+) m/z: Calculado: 917,9730 (M+2H); Encontrado: 917,9720 (M+2H).
Preparación de Ejemplo 10018
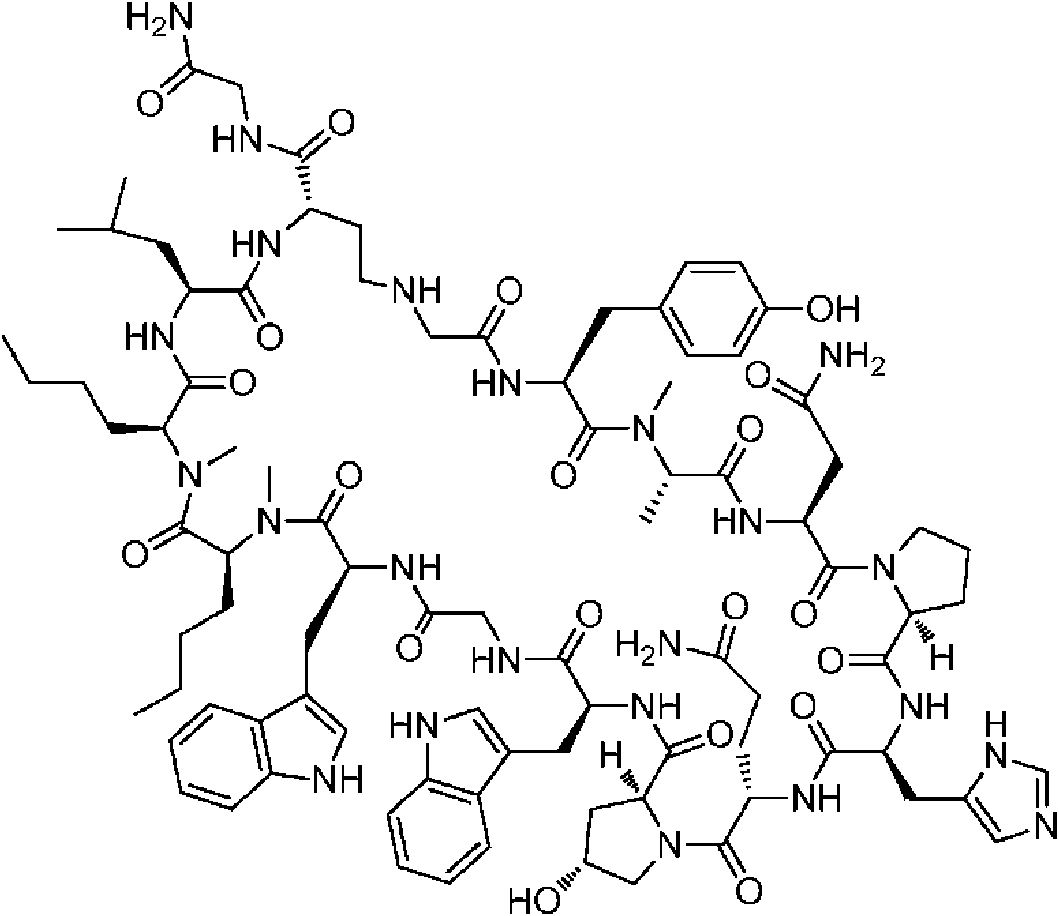
Ejemplo 10018
El material en bruto del Ejemplo 10018 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 31,3 mg y su pureza estimada por análisis de LCMS fue 98 %.
Condiciones de análisis A: Tiempo de retención = 1,61 min; ESI-MS(+) m/z 924,45 (M+2H).
Condiciones de análisis B: Tiempo de retención = 3,15 min; ESI-MS(+) m/z 925,40 (M+2h ). ESI-HRMS(+) m/z: Calculado: 924,9808 (M+2H); Encontrado: 924,9808 (M+2H).
Preparación de Ejemplo 10019

Ejemplo 10019
El material en bruto del Ejemplo 10019 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 15,0 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,61 min; ESI-MS(+) m/z 932,45 (M+2H).
Condiciones de análisis B: Tiempo de retención = 3,15 min; ESI-MS(+) m/z 932,40 (M+2h ). ESI-HRMS(+) m/z: Calculado: 931,9887 (M+2H); Encontrado: 931,9878 (M+2H).
Preparación de Ejemplo 10020

Ejemplo 10020
El material en bruto del Ejemplo 10020 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 11,1 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,42 min; ESI-MS(+) m/z 947,40 (M+2H).
Condiciones de análisis B: Tiempo de retención = 2,95 min; ESI-MS(+) m/z 947,40 (M+2h ). ESI-HRMS(+) m/z: Calculado: 946,9758 (M+2H); Encontrado: 946,9756 (M+2H).
Preparación de Ejemplo 10021

Ejemplo 10021
El material en bruto del Ejemplo 10021 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10
mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 11,1 mg y su pureza estimada por análisis de LCMS fue 99 %.
Condiciones de análisis A: Tiempo de retención = 1,44 min; ESI-MS(+) m/z 954,40 (M+2H).
Condiciones de análisis B: Tiempo de retención = 2,99 min; ESI-MS(+) m/z 954,35 (m 2h ).
Preparación de Ejemplo 10022

Ejemplo 10022
El material en bruto del Ejemplo 10022 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 5,0 mg y su pureza estimada por análisis de LCMS fue 95 %. Condiciones de análisis A: Tiempo de retención = 1,46 min; ESI-MS(+) m/z 961,45 (M+2H).
Preparación de Ejemplo 10023
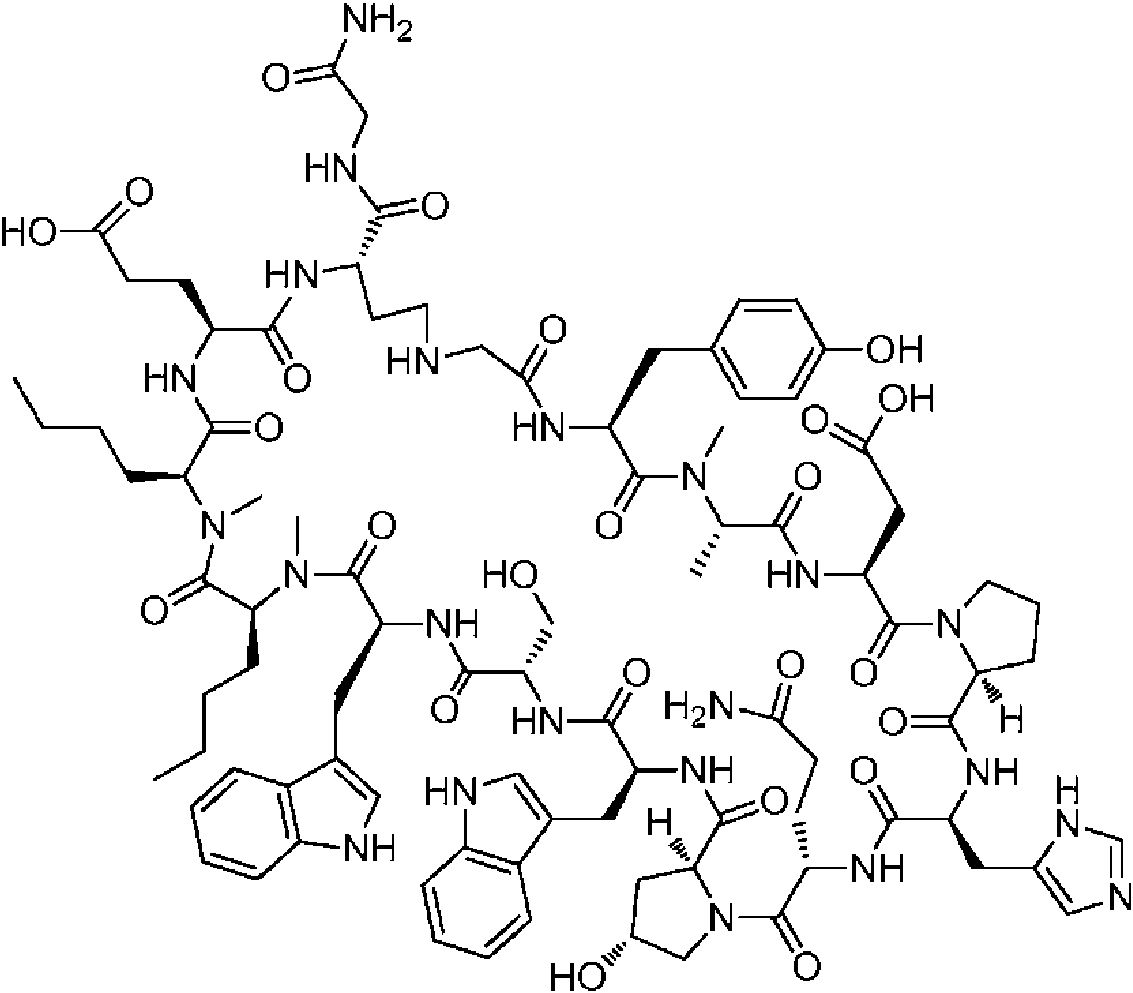
Ejemplo 10023
El material en bruto del Ejemplo 10023 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 3,5 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,52 min; ESI-MS(+) m/z 949,00 (M+2H).
Condiciones de análisis B: Tiempo de retención = 2,80 min; ESI-MS(+) m/z 948,85 (m +2H).
Preparación de Ejemplo 10024

Ejemplo 10024
El material en bruto del Ejemplo 10024 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10
mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 5,0 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,56 min; ESI-MS(+) m/z 920,30 (M+2H).
Condiciones de análisis B: Tiempo de retención = 3,05 min; ESI-MS(+) m/z 920,30 (m 2h ).
Preparación de Ejemplo 10025

Ejemplo 10025
El material en bruto del Ejemplo 10025 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 4,1 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,43 min; ESI-MS(+) m/z 934,60 (M+2H).
Condiciones de análisis B: Tiempo de retención = 2,97 min; ESI-MS(+) m/z 934,35 (m +2H).
Preparación de Ejemplo 10026

Ejemplo 10026
El material en bruto del Ejemplo 10026 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 19,7 mg y su pureza estimada por análisis de LCMS fue 97 %.
Condiciones de análisis A: Tiempo de retención = 1,69 min; ESI-MS(+) m/z 910,85 (M+2H).
Condiciones de análisis B: Tiempo de retención = 3,18 min; ESI-MS(+) m/z 910,85 (m 2h ).
Preparación de Ejemplo 10027

Ejemplo 10027
El material en bruto del Ejemplo 10027 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15
minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 21,0 mg y su pureza estimada por análisis de LCMS fue 99 %.
Condiciones de análisis A: Tiempo de retención = 1,66 min; ESI-MS(+) m/z 917,85 (M+2H).
Condiciones de análisis B: Tiempo de retención = 3,18 min; ESI-MS(+) m/z 917,90 (m +2h ).
Preparación de Ejemplo 10028

Ejemplo 10028
El material en bruto del Ejemplo 10028 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 20,1 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,65 min; ESI-MS(+) m/z 924,90 (M+2H).
Condiciones de análisis B: Tiempo de retención = 3,17 min; ESI-MS(+) m/z 924,90 (m +2h ).
Preparación de Ejemplo 10500 (no según la invención)

Preparación de Intermedio 10500:
Se siguió "Secuencia de síntesis general A". Se usó ácido 2-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)etoxi)acético en el "procedimiento de carga de resina". Al recipiente de reacción que contiene resina de la secuencia automatizada se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. Al recipiente de reacción se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DCM (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se transfirió después inmediatamente usando DCM (8 ml) a un vial de 15 ml. A la solución se añadió hexafluoroisopropanol (2 ml). La resina se volvió inmediatamente rojo intenso; la solución permaneció incolora. La mezcla se agitó manualmente brevemente, después se dejó en reposo a t.a. durante 15 minutos, después se filtró. El filtrado se transfirió a un vial de 15 ml y se concentró bajo una corriente de N2 para proporcionar un residuo sólido, Intermedio 10500.
Preparación de Ejemplo 10500:
Una "solución de desprotección" se preparó combinando en un vial de vidrio de 40 ml ácido trifluoroacético (23,75 ml), 1,4-Ditio-DL-treitol (625 mg), triisopropilsilano (0,625 ml). A un vial de 1 dram cargado con la totalidad de Intermedio 10500 preparado anteriormente se añadió la "solución de desprotección" (1,0 ml). La solución se mezcló durante 1,5h en un agitador funcionando a 500 rpm, después se vertió en un tubo de ensayo de 25 ml cargado con Et2O (20 ml). Precipitó una pequeña cantidad de sólido blanco. La mezcla se centrifugó; el líquido se decantó. Los sólidos se suspendieron en Et2O (10 ml). La mezcla se centrifugó, el líquido se decantó. El material bruto se purificó por medio de Cl/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 |jm;
Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 40-80 % B durante 20 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 7,3 mg y su pureza estimada por análisis de LCMS fue 100 %. Condiciones de análisis A: Tiempo de retención = 2,57 min; ESI-MS(+) m/z 764,9 (M+2H).
Condiciones de análisis E: Tiempo de retención = 2,57 min; ESI-MS(+) m/z 765,4 (M+2H). ESI-HRMS(+) m/z: Calculado: 765,4602 Encontrado: 765,4581.
Preparación de Ejemplo 10501 (no según la invención)

Ejemplo 10501
Preparación de Intermedio 10501A:
Se siguió "Secuencia de síntesis general A". Se usó ácido 2-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)etoxi)acético en el "procedimiento de carga de resina". Al recipiente de reacción que contiene resina de la secuencia automatizada se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. Al recipiente de reacción se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DCM (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se transfirió después inmediatamente usando DCM (8 ml) a un vial de 15 ml. A la solución se añadió hexafluoroisopropanol (2 ml). La resina se volvió inmediatamente rojo intenso; la solución permaneció incolora. La mezcla se agitó manualmente brevemente, después se dejó en reposo a t.a. durante 15 minutos, después se filtró. El filtrado se transfirió a un vial de 15 ml y se concentró bajo una corriente de N2 para proporcionar un residuo sólido, Intermedio 10501A.
Preparación de Intermedio 10501B:
A un vial de 15 ml cargado con la totalidad de Intermedio 10501A preparado anteriormente se añadió DCM (2 ml), después HATU (38mg, 0,10mmol) después DIPEA (0,114 ml, 0,650 mmol). La solución se agitó durante 2 h. La solución se secó al vacío para proporcionar Intermedio 10501B.
Preparación de Ejemplo 10501:
Una "solución de desprotección" se preparó combinando en un vial de vidrio de 40 ml ácido trifluoroacético
(23,75 ml), 1,4-Ditio-DL-treitol (625 mg), triisopropilsilano (0,625 ml). A un vial de 1 dram cargado con la totalidad de Intermedio 10501A preparado anteriormente se añadió la "solución de desprotección" (1,0 ml). La solución se mezcló durante 1,0h en un agitador funcionando a 500 rpm, después se vertió en un tubo de ensayo de 25 ml cargado con Et2O (15ml). Precipitó una pequeña cantidad de sólido blanco. La mezcla se centrifugó; el líquido se decantó. Los sólidos se suspendieron en Et2O (15ml). La mezcla se centrifugó, el líquido se decantó. El residuo resultante se disolvió en MeOH ya la solución se añadió DIPEA (0,050 ml). El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 15-55 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: XBridge C18, 19 x 200mm, partículas de 5 pm; Fase móvil A: 5:95 de metanol: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de metanol: agua con acetato de amonio 10 mM; Gradiente: 40-80 % B durante 30 minutos, después mantener 4 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 1,0 mg y su pureza estimada por análisis de LCMS fue 95 %.
Condiciones de análisis A: Tiempo de retención = 1,65 min; ESI-MS(-) m/z 856,8 (M+2H).
Condiciones de análisis B: Tiempo de retención = 2,72min; ESI-MS(-) m/z 857,2 (M+2H). ESI-HRMS(+) m/z: Calculado: 856,9798 Encontrado: 856,9790.
Preparación de Ejemplo 10502 (no según la invención)

Preparación de Intermedio 10502A:
Se siguió "Secuencia de síntesis general A". Se usó ácido 2-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)etoxi)acético
en el "procedimiento de carga de resina". Al recipiente de reacción que contiene resina de la secuencia automatizada se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. Al recipiente de reacción se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DCM (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se transfirió después inmediatamente usando DCM (8 ml) a un vial de 15 ml. A la solución se añadió hexafluoroisopropanol (2 ml). La resina se volvió inmediatamente rojo intenso; la solución permaneció incolora. La mezcla se agitó manualmente brevemente, después se dejó en reposo a t.a. durante 15 minutos, después se filtró. El filtrado se transfirió a un vial de 15 ml y se concentró bajo una corriente de N2 para proporcionar un residuo sólido, Intermedio 10502A.
Preparación de Intermedio 10502B:
A un vial de 15 ml cargado con la totalidad de Intermedio 10501A preparado anteriormente se añadió DCM (2 ml), después HATU (38 mg, 0,10 mmol) después DIPEA (0,114 ml, 0,650 mmol). La solución se agitó durante 2 h. La solución se secó al vacío para proporcionar redisuelto en MeOH después sometió purificación por HPLC en las siguientes condiciones: Columna: Waters XBridge C18, 30 x 100 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 55-100 % B durante 15 minutos, después mantener 15 minutos a 100 % B; Flujo: 30 ml/min. Fracciones que contenían el producto deseado se combinaron y secaron por evaporación centrífuga para proporcionar un sólido de color blanco, Intermedio 10502B.
Preparación de Ejemplo 10502:
Una "solución de desprotección" se preparó combinando en un vial de vidrio de 40 ml ácido trifluoroacético (23,75 ml), 1,4-Ditio-DL-treitol (625 mg), triisopropilsilano (0,625 ml). A un vial de 1 dram cargado con la totalidad de Intermedio 10501A preparado anteriormente se añadió la "solución de desprotección" (1,0 ml). La solución se mezcló durante 1,0h en un agitador funcionando a 500 rpm, después se vertió en un tubo de ensayo de 25 ml cargado con Et2O (15 ml). Precipitó una pequeña cantidad de sólido blanco. La mezcla se centrifugó; el líquido se decantó. Los sólidos se suspendieron en Et2O (15 ml). La mezcla se centrifugó, el líquido se decantó. El residuo resultante se disolvió en MeOH ya la solución se añadió DIPEA (0,050 ml). El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de metanol: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de metanol: agua con acetato de amonio 10 mM; Gradiente: 50-100 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 0,1 mg y su pureza estimada por análisis de LCMS fue 83 %. Condiciones de análisis A: Tiempo de retención = 1,79 min; ESI-MS(-) m/z 886,1 (M+2H).
Preparación de Ejemplo 10503 (no según la invención)

Ejemplo 10503
Preparación de Intermedio 10503A:
Se siguió "Secuencia de síntesis general A". Se usó ácido 2-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)etoxi)acético en el "procedimiento de carga de resina". Al recipiente de reacción que contiene resina de la secuencia automatizada se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. Al recipiente de reacción se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DCM (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se transfirió después inmediatamente usando DCM (8 ml) a un vial de 15 ml. A la solución se añadió hexafluoroisopropanol (2 ml). La resina se volvió inmediatamente rojo intenso; la solución permaneció incolora. La mezcla se agitó manualmente brevemente, después se dejó en reposo a t.a. durante 15 minutos, después se filtró. El filtrado se transfirió a un vial de 15 ml y se concentró bajo una corriente de N2 para proporcionar un residuo sólido, Intermedio 10503A.
Preparación de Intermedio 10503B:
A un vial de 15 ml cargado con la totalidad de Intermedio 10501A preparado anteriormente se añadió DCM (2 ml), después HATU (38 mg, 0,10 mmol) después DIPEA (0,114 ml, 0,650 mmol). La solución se agitó durante 2 h. La solución se secó al vacío para proporcionar Intermedio 10503B.
Preparación de Ejemplo 10503:
Una "solución de desprotección" se preparó combinando en un vial de vidrio de 40 ml ácido trifluoroacético (23,75 ml), 1,4-Ditio-DL-treitol (625 mg), triisopropilsilano (0,625 ml). A un vial de 1 dram cargado con la totalidad de Intermedio 10501A preparado anteriormente se añadió la "solución de desprotección" (1,0 ml). La solución se mezcló durante 1,0h en un agitador funcionando a 500 rpm, después se vertió en un tubo de ensayo de 25 ml cargado con Et2O (15 ml). Precipitó una pequeña cantidad de sólido blanco. La mezcla se centrifugó; el líquido se decantó. Los sólidos se suspendieron en Et2O (15 ml). La mezcla se centrifugó, el líquido se decantó. El residuo resultante se disolvió en MeOH ya la solución se añadió DIPEA (0,050 ml). El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de metanol: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de metanol: agua con acetato de amonio 10 mM; Gradiente: 40-90 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: waters CSH c-18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 10 50 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 0,8 mg y su pureza estimada por análisis de LCMS fue 99 %.
Condiciones de análisis A: Tiempo de retención = 1,81 min; ESI-MS(-) m/z 879,3 (M+2H).
Condiciones de análisis E: Tiempo de retención = 1,81 min; ESI-MS(-) m/z 1758,1 (M-H).
Preparación de Ejemplo 9005

Ejemplo 9005
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de metanol: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de metanol: agua con acetato de amonio 10 mM; Gradiente: 40-80 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50 % B durante 30 minutos, después mantener 3 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 4,5 mg y su pureza estimada por análisis de LCMS fue 100 %. ESI-MS(+) m/z 964,1 (M+2H).
Preparación de Ejemplo 9006

Ejemplo 9006
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 5-40% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 8,7 mg y su pureza estimada por análisis de LCMS fue 97 %. ESI-MS(+) m/z 964,5 (M+2H).
Preparación de Ejemplo 9007

Ejemplo 9007
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de metanol: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de metanol: agua con acetato de amonio 10 mM; Gradiente: 30-70 % B durante 30 minutos, después mantener
5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 5-40 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 2,7 mg y su pureza estimada por análisis de LCMS fue 100 %. ESI-MS(+) m/z 971,5 (M+2H).
Preparación de Ejemplo 9008
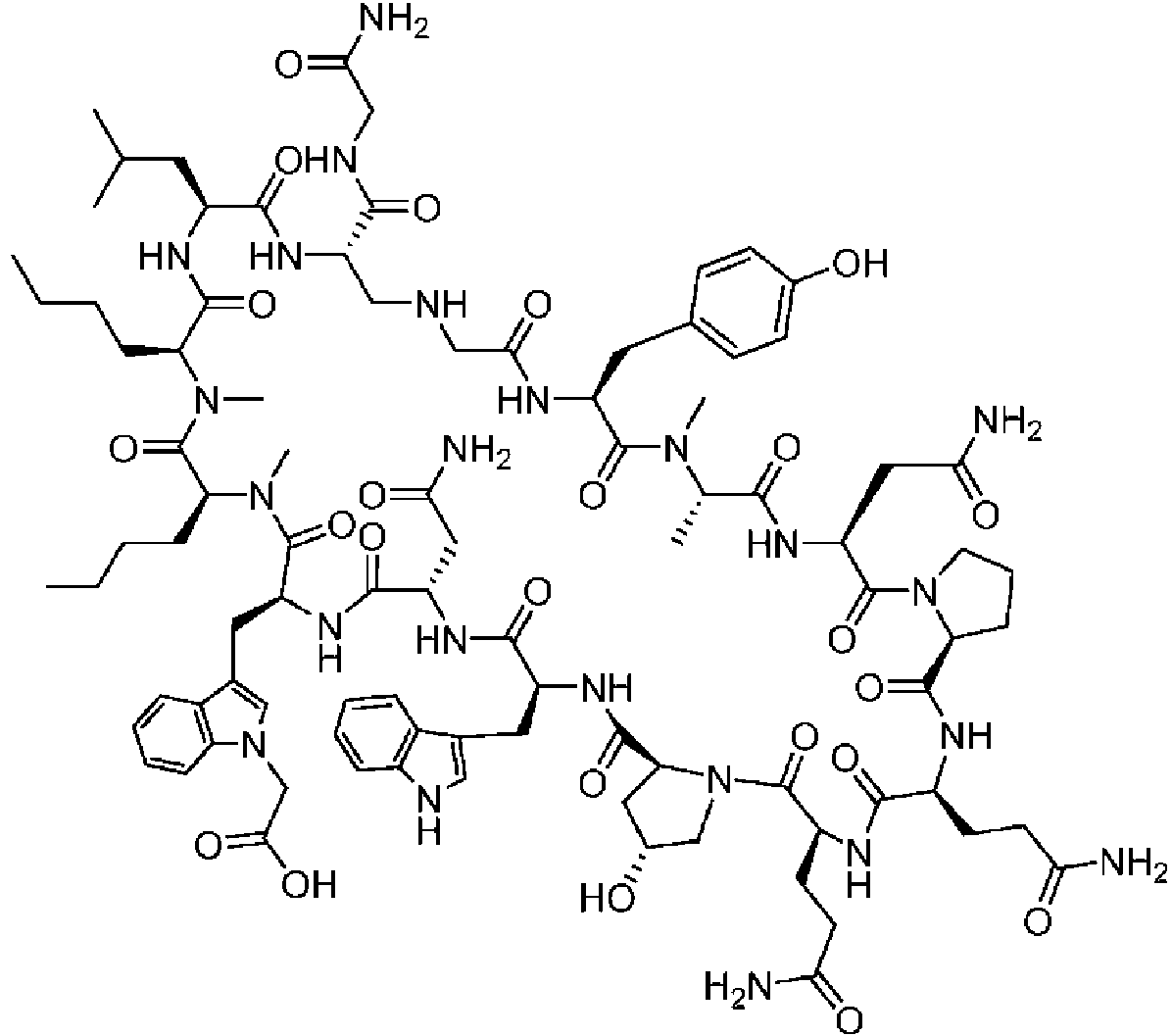
Ejemplo 9008
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de metanol: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de metanol: agua con acetato de amonio 10 mM; Gradiente: 40-80 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 15-55 % B durante 30 minutos, después mantener 3 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 5,7 mg y su pureza estimada por análisis de LCMS fue 100 %. ESI-MS(+) m/z 971,1 (M+2H).
Preparación de Ejemplo 9009

Ejemplo 9009
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 15-55% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/e M preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 15-55 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 9,5 mg y su pureza estimada por análisis de LCMS fue 97 %. ESI-MS(+) m/z 964,9 (M+2H).
Preparación de Ejemplo 9010

Ejemplo 9010
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 |jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 15-55% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/e M preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x 200 mm, partículas de 5 jm ; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 15-55 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 10,7 mg y su pureza estimada por análisis de LCMS fue 97 %. ESI-MS(+) m/z 966,1 (M+2H).
Preparación de Ejemplo 9011

Ejemplo 9011
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 jm ; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 20-60% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 15,6 mg y su pureza estimada por análisis de LCMS fue 82 %. ESI-MS(+) m/z 973,2 (M+2H).
Preparación de Ejemplo 9012

Ejemplo 9012
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 5-45% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/e M preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 5-45 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 7,3 mg y su pureza estimada por análisis de LCMS fue 96 %. ESI-MS(+) m/z 964,0 (M+2H).
Preparación de Ejemplo 9013

Ejemplo 9013
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 |jm; Fase móvil A: 5:95 de metanol: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de metanol: agua con acetato de amonio 10 mM; Gradiente: 30-70 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x 200 mm, partículas de 5 jm ; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 5-45 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 10,7 mg y su pureza estimada por análisis de LCMS fue 97 %. ESI-MS(+) m/z 965,1 (M+2H).
Preparación de Ejemplo 9014

El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 5-45% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/e M preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x 200 mm, partículas de 5 jm ; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 5-45 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: XBridge C18, 19 x 200 mm, partículas de 5 jm; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 10-50 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 2,4 mg y su pureza estimada por análisis de LCMS fue 97 %. ESI-MS(+) m/z 972,1 (M+2H).
Preparación de Ejemplo 9015

Ejemplo 9015
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 8,6 mg y su pureza estimada por análisis de LCMS fue 96 %. ESI-MS(+) m/z 966,2 (M+2H).
Preparación de Ejemplo 9016

Ejemplo 9016
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 2,1 mg y su pureza estimada
por análisis de LCMS fue 95 %. ESI-MS(+) m/z 973,2 (M+2H).
Preparación de Ejemplo 9017

Ejemplo 9017
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/e M preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 3,2 mg y su pureza estimada por análisis de LCMS fue 92 %. ESI-MS(+) m/z 973,1 (M+2H).
Preparación de Ejemplo 9018

Ejemplo 9018
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 |jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 5-45% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 4,5 mg y su pureza estimada por análisis de LCMS fue 100 %. ESI-MS(+) m/z 972,1 (M+2H).
Preparación de Ejemplo 9019

Ejemplo 9019
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 jm ; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 15-55% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 6,6 mg y su pureza estimada por análisis de LCMS fue 95 %. ESI-MS(+) m/z 972,4 (M+2H).
Preparación de Ejemplo 9020
Ejemplo 9020
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 |jm; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 10-50% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x 200 mm, partículas de 5 jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 5-45 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 4,5 mg y su pureza estimada por análisis de LCMS fue 94 %. ESI-MS(+) m/z 980,2 (M+2H).
Preparación de Ejemplo 9021

Ejemplo 9021
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 jm ; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 10-50% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x 200 mm, partículas de 5 jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 0-45 % B durante 50 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 4,7 mg y su pureza estimada por análisis de LCMS fue 97 %. ESI-MS(+) m/z 979,1 (M+2H).
Preparación de Ejemplo 9022

Ejemplo 9022
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50% B durante 33 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 2,1 mg y su pureza estimada por análisis de LCMS fue 100 %. ESI-MS(+) m/z 972,2 (M+2H).
Preparación de Ejemplo 9023

Ejemplo 9023
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 15-85% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se
combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 9,5 mg y su pureza estimada por análisis de LCMS fue 95 %. ESI-MS(+) m/z 974,0 (M+2H).
Preparación de Ejemplo 9024

Ejemplo 9024
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 5-45% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/e M preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 5-45 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 3,4 mg y su pureza estimada por análisis de LCMS fue 99 %. ESI-MS(+) m/z 980,6 (M+2H).
Preparación de Ejemplo 9025

Ejemplo 9025
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 5-45% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/e M preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 5-45 % B durante 30 minutos, después mantener 0 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 5 45 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 7,4 mg y su pureza estimada por análisis de LCMS fue 95 %. ESI-MS(+) m/z 980,0 (M+2H).
Preparación de Ejemplo 9026

Ejemplo 9026
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: waters xbridge c-18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 4,0 mg y su pureza estimada por análisis de LCMS fue 97 %. ESI-MS(+) m/z 948,2 (M+2H).
Preparación de Ejemplo 9027

Ejemplo 9027
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: waters CSH c-18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 10-50% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de
acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 5-45% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 3,7 mg y su pureza estimada por análisis de LCMS fue 100 %. ESI-MS(+) m/z 949,3 (M+2H).
Preparación de Ejemplo 9028

Ejemplo 9028
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: waters xbridge c-18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 3,9 mg y su pureza estimada por análisis de LCMS fue 90 %. ESI-MS(+) m/z 956,1 (M+2H).
Preparación de Ejemplo 9029
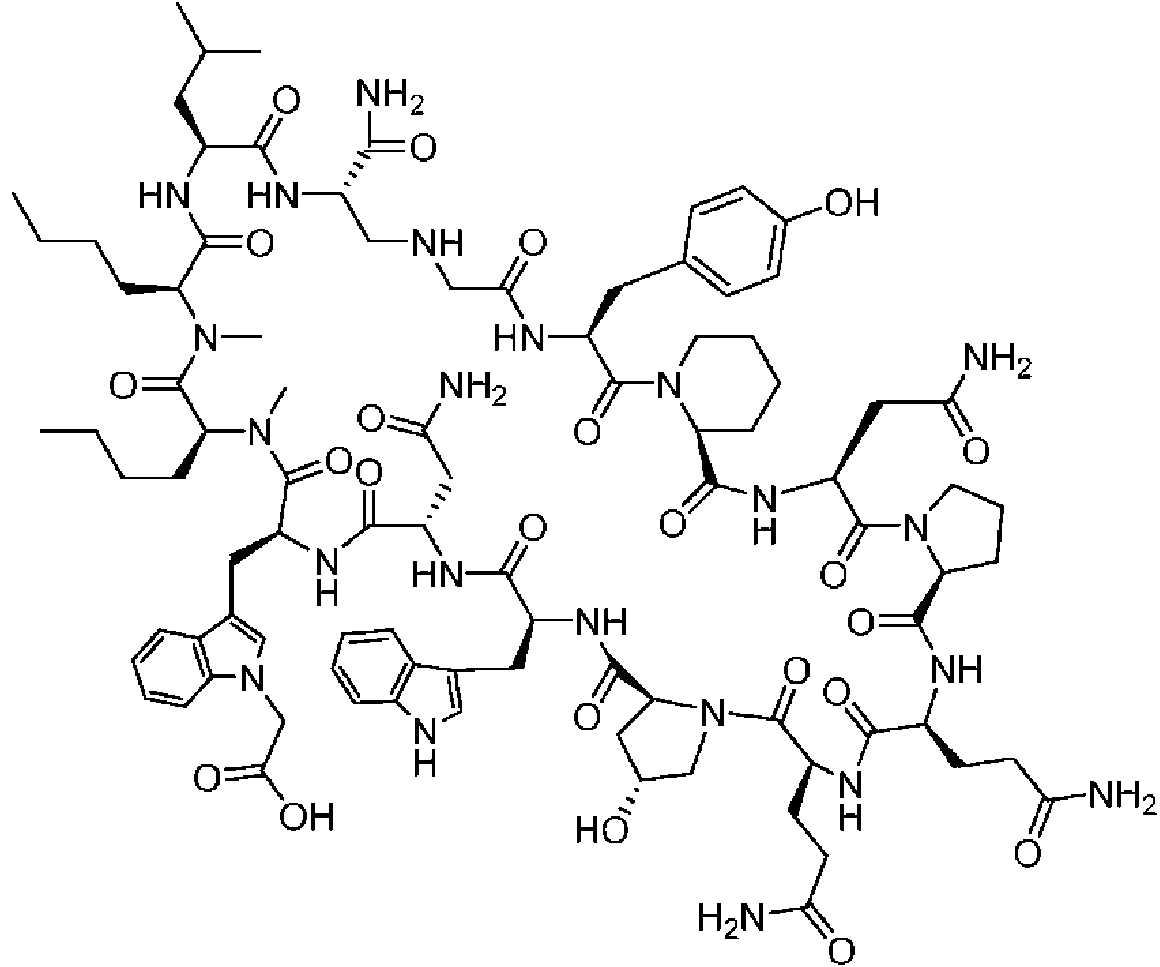
Ejemplo 9029
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: waters xbridge c-18, 19 x 200 mm, partículas de 5 |jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 4,3 mg y su pureza estimada por análisis de LCMS fue 99 %. ESI-MS(+) m/z 956,2 (M+2H).
Preparación de Ejemplo 9030

Ejemplo 9030
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 5,8 mg y su pureza estimada por análisis de LCMS fue 98 %. ESI-MS(+) m/z 949,2 (M+2H).
Preparación de Ejemplo 9031
Ejemplo 9031
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 15-55% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: XBridge C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-30 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 2,5 mg y su pureza estimada por análisis de LCMS fue 97 %. ESI-MS(+) m/z 950,1 (M+2H).
Preparación de Ejemplo 9032

Ejemplo 9032
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 10-50% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 15-55 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 5,9 mg y su pureza estimada por análisis de LCMS fue 82 %. ESI-MS(+) m/z 957,1 (M+2H).
Preparación de Ejemplo 9033

Ejemplo 9033
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 10-50 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/e M preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 15-55 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 7,0 mg y su pureza estimada por análisis de LCMS fue 99 %. ESI-MS(+) m/z 956,2 (M+2H).
Preparación de Ejemplo 9034

Ejemplo 9034
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18,
19 x mm, partículas de 5 |jm; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 15-55 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/e M preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x 200 mm, partículas de 5 jm ; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 15-55 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 3,3 mg y su pureza estimada por análisis de LCMS fue 100 %. ESI-MS(+) m/z 948,4 (M+2H).
Preparación de Ejemplo 9035

Ejemplo 9035
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 jm ; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 15-55% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: XBridge C18, 19 x 200 mm, partículas de 5 jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 5-45 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 2,1 mg y su pureza estimada por análisis de LCMS fue 100 %. ESI-MS(+) m/z 950,2 (M+2H).
Preparación de Ejemplo 9036

Ejemplo 9036
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 10-50% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 5-45 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 0,7 mg y su pureza estimada por análisis de LCMS fue 96 %. ESI-MS(+) m/z 956,3 (M+2H).
Preparación de Ejemplo 9037

Ejemplo 9037
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 |jm; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 15-55% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: XBridge C18, 19 x 200 mm, partículas de 5 jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 5-45 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 0,9 mg y su pureza estimada por análisis de LCMS fue 100 %. ESI-MS(+) m/z 956,2 (M+2H).
Preparación de Ejemplo 9038

Ejemplo 9038
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 jm ; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 20-60% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: XBridge C18, 19 x 200 mm, partículas de 5 jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 5-45 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 0,5 mg y su pureza estimada por análisis de LCMS fue 100 %. ESI-MS(+) m/z 949,2 (M+2H).
Preparación de Ejemplo 9039

Ejemplo 9039
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de metanol: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de metanol: agua con acetato de amonio 10 mM; Gradiente: 20-100 % B durante 20 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/e M preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 4,7 mg y su pureza estimada por análisis de LCMS fue 100 %. ESI-MS(+) m/z 951,1 (M+2H).
Preparación de Ejemplo 9040

Ejemplo 9040
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18,
19 x mm, partículas de 5 |jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/e M preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x mm, partículas de 5 jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 3,3 mg y su pureza estimada por análisis de LCMS fue 98 %. ESI-MS(+) m/z 957,4 (M+2H).
Preparación de Ejemplo 9041

Ejemplo 9041
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x mm, partículas de 5 jm; Fase móvil A: 5:95 de metanol: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de metanol: agua con acetato de amonio 10 mM; Gradiente: 25-65 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x mm, partículas de 5 jm ; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: XBridge C18, 19 x mm, partículas de 5 jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 1,7 mg y su pureza estimada por análisis de LCMS fue 100 %. ESI-MS(+) m/z 957,1 (M+2H).
Preparación de E jem plo 9042

Ejemplo 9042
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/e M preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 4,5 mg y su pureza estimada por análisis de LCMS fue 100 %. ESI-MS(+) m/z 956,2 (M+2H).
Preparación de Ejemplo 9043

Ejemplo 9043
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x mm, partículas de 5 |jm; Fase móvil A: 5:95 de metanol: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de metanol: agua con acetato de amonio 10 mM; Gradiente: 35-75 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x mm, partículas de 5 jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 3,3 mg y su pureza estimada por análisis de LCMS fue 100 %. ESI-MS(+) m/z 957,1 (M+2H).
Preparación de Ejemplo 9044

Ejemplo 9044
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x mm, partículas de 5 jm ; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/e M preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x mm, partículas de 5 jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 6,3 mg y su pureza estimada por análisis de LCMS fue 99 %. ESI-MS(+) m/z 964,1 (M+2H).
Preparación de E jem plo 9045

Ejemplo 9045
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de metanol: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de metanol: agua con acetato de amonio 10 mM; Gradiente: 35-75 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 5,5 mg y su pureza estimada por análisis de LCMS fue 99 %. ESI-MS(+) m/z 963,4 (M+2H).
Preparación de Ejemplo 9046

Ejemplo 9046
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de metanol: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de metanol: agua con acetato de amonio 10 mM; Gradiente: 35-75 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 5,2 mg y su pureza estimada por análisis de LCMS fue 98 %. ESI-MS(+) m/z 957,2 (M+2H).
Preparación de Ejemplo 9047

Ejemplo 9047
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de metanol: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de metanol: agua con acetato de amonio 10 mM; Gradiente: 35-75 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 4,9 mg y su pureza estimada por análisis de LCMS fue 100 %. ESI-MS(+) m/z 965,1 (M+2H).
Preparación de E jem plo 9048

Ejemplo 9048
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de metanol: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de metanol: agua con acetato de amonio 10 mM; Gradiente: 35-75 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 6,1 mg y su pureza estimada por análisis de LCMS fue 96 %. ESI-MS(+) m/z 965,1 (M+2H).
Preparación de Ejemplo 9049

Ejemplo 9049
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x mm, partículas de 5 |jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-100 % B durante 20 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/e M preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x mm, partículas de 5 jm; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 18-58% B durante 20 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 4,4 mg y su pureza estimada por análisis de LCMS fue 90 %. ESI-MS(+) m/z 932,1 (M+2H).
Preparación de Ejemplo 9050

Ejemplo 9050
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x mm, partículas de 5 jm ; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-100 % B durante 20 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/e M preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x mm, partículas de 5 jm; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 18-58% B durante 20 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 3,9 mg y su pureza estimada por análisis de LCMS fue 94 %. ESI-MS(+) m/z 938,7 (M+2H).
Preparación de E jem plo 9051

Ejemplo 9051
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-100 % B durante 20 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 18-58% B durante 20 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 3,9 mg y su pureza estimada por análisis de LCMS fue 97 %. ESI-MS(+) m/z 938,5 (M+2H).
Preparación de Ejemplo 9052

Ejemplo 9052
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x mm, partículas de 5 |jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-100 % B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/e M preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x mm, partículas de 5 jm ; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 20-60% B durante 20 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 5,9 mg y su pureza estimada por análisis de LCMS fue 86 %. ESI-MS(+) m/z 940,1 (M+2H).
Preparación de Ejemplo 9053
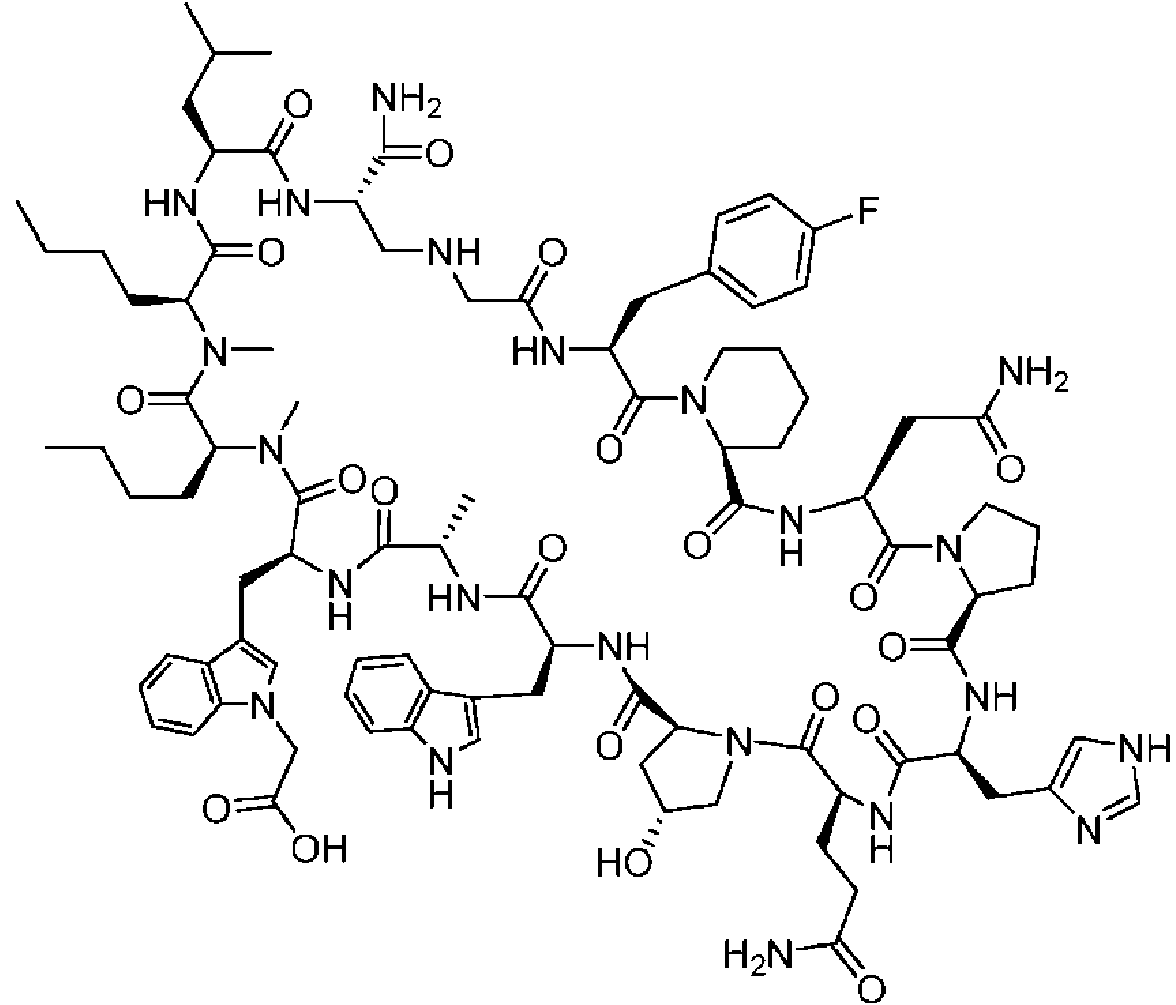
Ejemplo 9053
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x mm, partículas de 5 jm ; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-100 % B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x 200 mm, partículas de 5 jm ; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 20-60 % B durante 20 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 8,0 mg y su pureza estimada por análisis de LCMS fue 96 %. ESI-MS(+) m/z 940,4 (M+2H).
Preparación de Ejemplo 9054

Ejemplo 9054
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-100 % B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/e M preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 10-50% B durante 20 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 3,8 mg y su pureza estimada por análisis de LCMS fue 98 %. ESI-MS(+) m/z 940,2 (M+2H).
Preparación de Ejemplo 9055
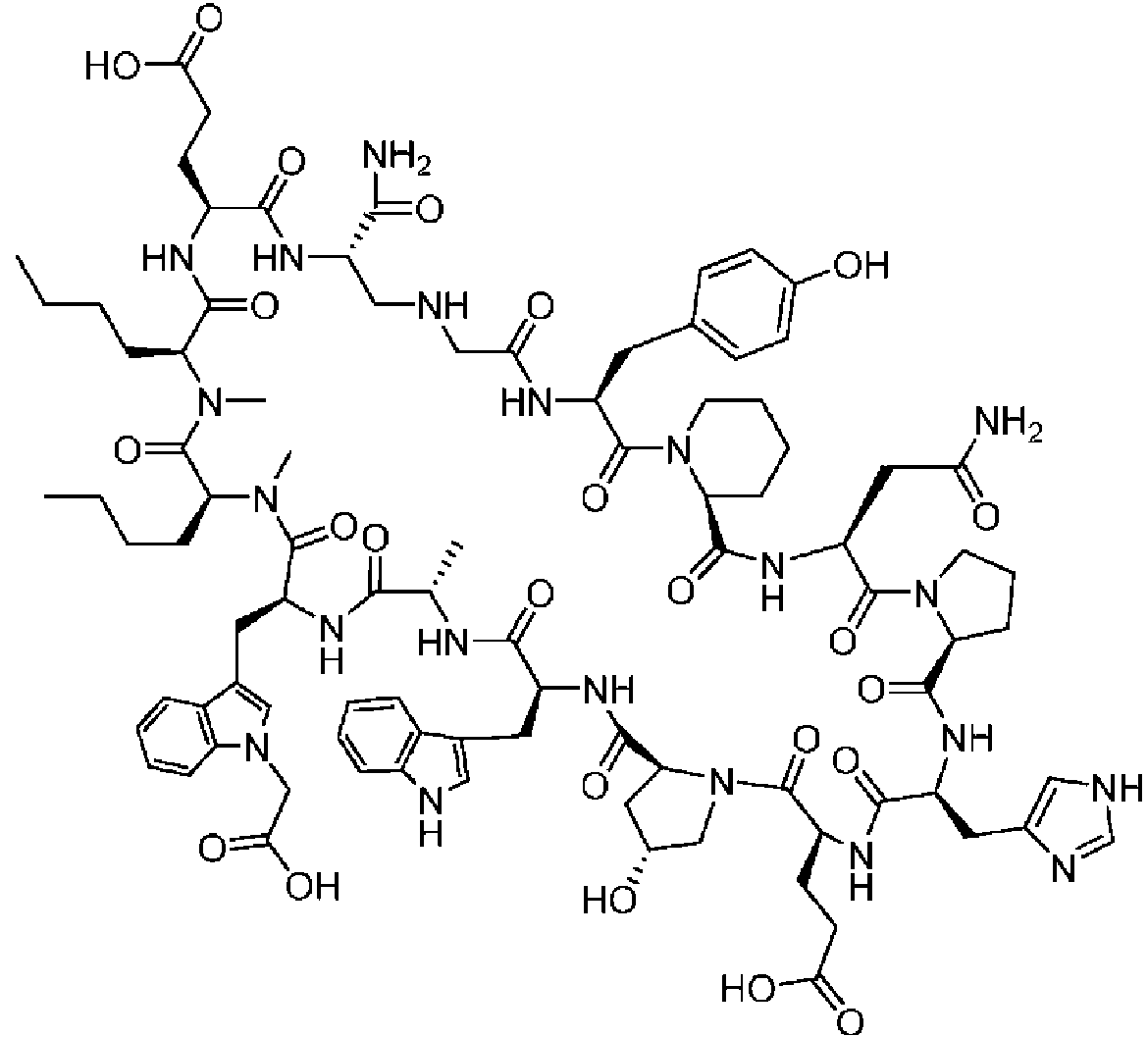
Ejemplo 9055
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-100 % B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 3,6 mg y su pureza estimada por análisis de LCMS fue 84 %. ESI-MS(+) m/z 947,2 (M+2H).
Preparación de Ejemplo 9056

Ejemplo 9056
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-100 % B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 4,0 mg y su pureza estimada por análisis de LCMS fue 90 %. ESI-MS(+) m/z 946,2 (M+2H).
Preparación de Ejemplo 9057

Ejemplo 9057
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de metanol: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de metanol: agua con acetato de amonio 10 mM; Gradiente: 30-100 % B durante 20 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/e M preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 10-50% B durante 20 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 4,1 mg y su pureza estimada por análisis de LCMS fue 92 %. ESI-MS(+) m/z 941,4 (M+2H).
Preparación de Ejemplo 9058
Ejemplo 9058
El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de metanol: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de metanol: agua con acetato de amonio 10 mM; Gradiente: 30-100 % B durante 20 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/e M preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 10-50% B durante 20 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 4,2 mg y su pureza estimada por análisis de LCMS fue 98 %. ESI-MS(+) m/z 947,6 (M+2H).
Preparación de Ejemplo 9059

Ejemplo 9059
Fase móvil A: 5:95 de metanol: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de metanol: agua con acetato de amonio 10 mM; Gradiente: 30-100 % B durante 20 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante c L/EM preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: 95:5 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Gradiente: 10-50 % B durante 20 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 3,5 mg y su pureza estimada por análisis de LCMS fue 98 %. ESI-MS(+) m/z 947,2 (M+2H).
Preparación de E jem plo 10029

Ejemplo 10029
El material en bruto del Ejemplo 10029 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 |jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 8,4 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,24 min; ESI-MS(+) m/z 907,2 (M+2H).
Condiciones de análisis B: Tiempo de retención = 1,67 min; ESI-Ms (+) m/z 906,9 (M+2H). ESI-HRMS(+) m/z: Calculado: 906,5014 (M+2H); Encontrado: 906,4994 (M+2H).
Preparación de Ejemplo 10030
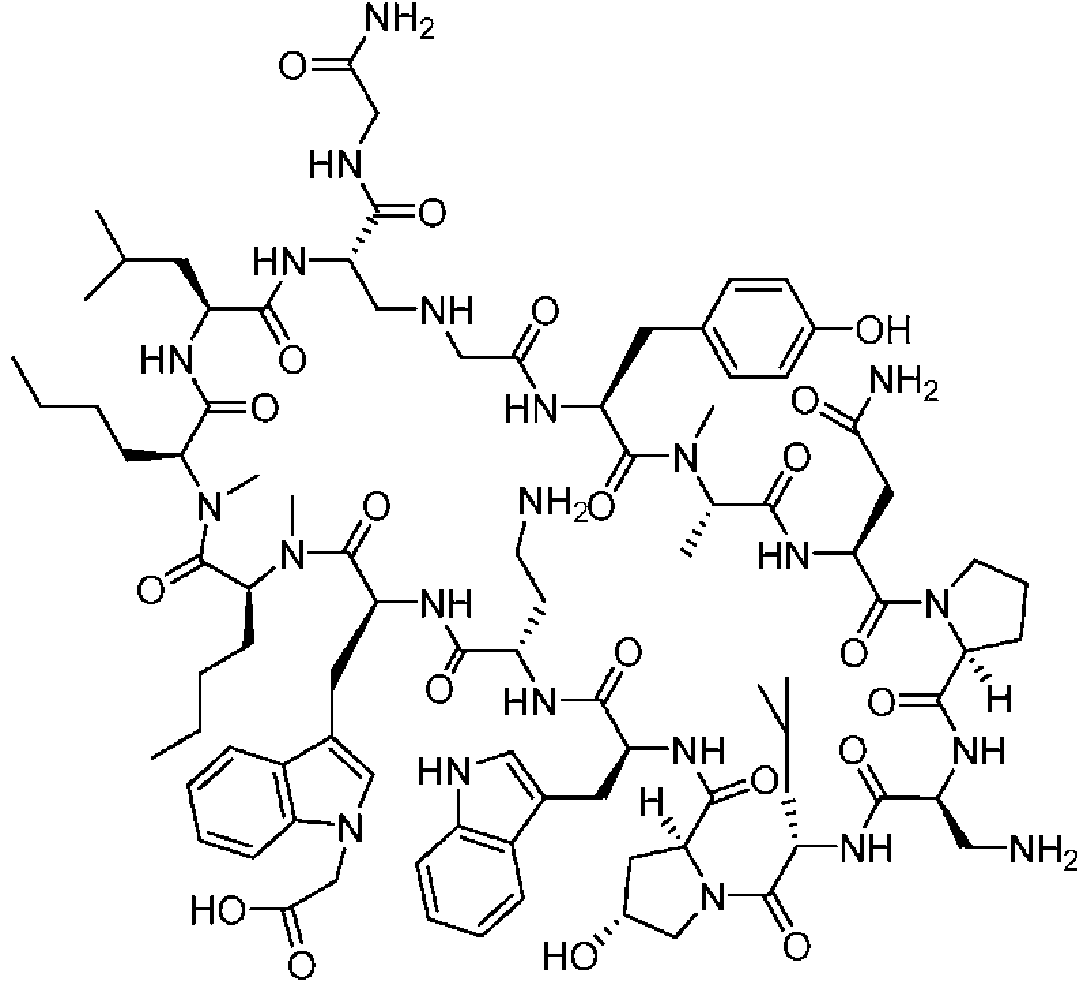
Ejemplo 10030
El material en bruto del Ejemplo 10030 se purificó por LC/MS preparativa con las siguientes condiciones: Columna:
XBridge C18, 19 x 200 mm, partículas de 5 |jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 8,4 mg y su pureza estimada por análisis de LCMS fue 97 %.
Condiciones de análisis A: Tiempo de retención = 1,65 min; ESI-MS(+) m/z 935,75 (M+2H).
Condiciones de análisis B: Tiempo de retención = 1,70 min; ESI-MS(+) m/z 935,80 (M+2h ). ESI-HRMS(+) m/z: Calculado: 935,5041 (M+2H); Encontrado: 935,5024 (M+2H).
Preparación de Ejemplo 10031

Ejemplo 10031
El material en bruto del Ejemplo 10031 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 jm ; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 3,0 mg y su pureza estimada por análisis de LCMS fue 95 %.
Condiciones de análisis A: Tiempo de retención = 1,14 min; ESI-MS(+) m/z 944,20 (M+2H).
Condiciones de análisis B: Tiempo de retención = 1,59 min; ESI-MS(+) m/z 94350 (M+2h ). ESI-HRMS(+) m/z: Calculado: 943,4834 (M+2H); Encontrado: 943,4808 (M+2H).
Preparación de Ejemplo 10032
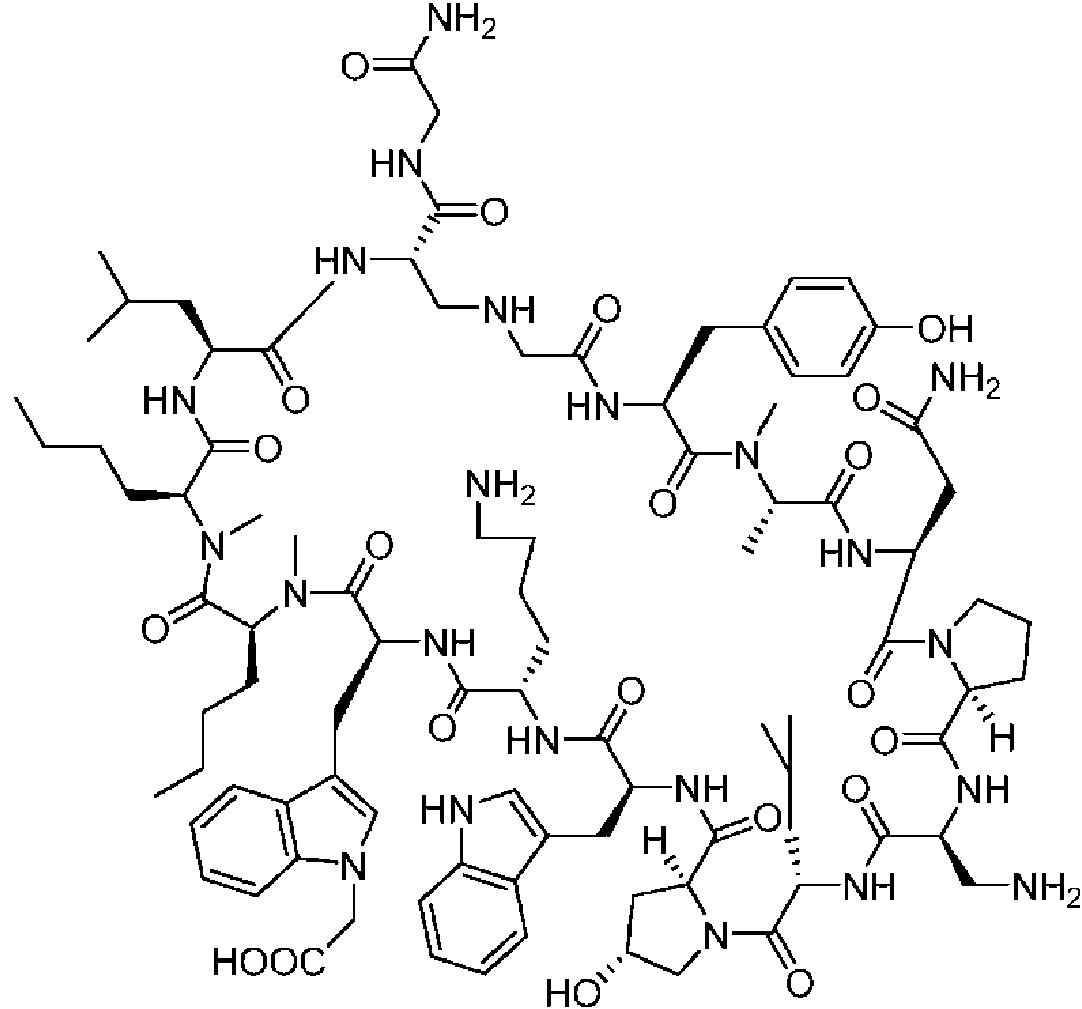
Ejemplo 10032
El material en bruto del Ejemplo 10032 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 3,9 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,15 min; ESI-MS(+) m/z 950,2 (M+2H).
Condiciones de análisis B: Tiempo de retención = 1,47 min; ESI-Ms (+) m/z 950,3 (M+2H). ESI-HRMS(+) m/z: Calculado: 949,5198 (M+2H); Encontrado: 949,5158 (M+2H).
Preparación de Ejemplo 10033

Ejemplo 10033
El material en bruto del Ejemplo 10033 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15
minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 1,8 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,17 min; ESI-MS(+) m/z 921,2 (M+2H).
Condiciones de análisis B: Tiempo de retención = 1,48 min; ESI-Ms (+) m/z 920,9 (M+2H). ESI-HRMS(+) m/z: Calculado: 921,0091 (M+2H); Encontrado: 921,0053 (M+2H).
Preparación de Ejemplo 10034

Ejemplo 10034
El material en bruto del Ejemplo 10034 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 0,6 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,17 min; ESI-MS(+) m/z 906,9 (M+2H).
Condiciones de análisis B: Tiempo de retención = 1,56 min; ESI-MS(+) m/z 907,1 (m 2h ).
Preparación de Ejemplo 10035

Ejemplo 10035
El material en bruto del Ejemplo 10035 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 |jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 0,3 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,15 min; ESI-MS(+) m/z 915,0 (M+2H).
Condiciones de análisis B: Tiempo de retención = 1,48 min; ESI-m S(+) m/z915,2 (M+2H). ESI-HRMS(+) m/z: 914,9724 (M+2H).
Preparación de Ejemplo 10036

Ejemplo 10036
El material en bruto del Ejemplo 10036 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 jm ; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 3,5 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,16 min; ESI-MS(+) m/z 943,0 (M+2H).
Condiciones de análisis B: Tiempo de retención = 1,51 min; ESI-Ms (+) m/z 942,6 (M+2H). ESI-HRMS(+) m/z: 942,5112 (M+2H).
Preparación de Ejemplo 10037

Ejemplo 10037
El material en bruto del Ejemplo 10037 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 20,8 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,17 min; ESI-MS(+) m/z 949,1 (M+2H).
Condiciones de análisis B: Tiempo de retención = 1,57 min; ESI-Ms (+) m/z 949,1 (M+2H). ESI-HRMS(+) m/z: 948,5098 (M+2H).
Preparación de Ejemplo 10038

Ejemplo 10038
El material en bruto del Ejemplo 10038 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 2,4 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,20 min; ESI-MS(+) m/z 920,1 (M+2H).
Condiciones de análisis B: Tiempo de retención = 1,60 min; ESI-Ms (+) m/z 921,0 (M+2H). ESI-HRMS(+) m/z: 919,9986 (M+2H).
Preparación de Ejemplo 10039

Ejemplo 10039
El material en bruto del Ejemplo 10039 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 8,4 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,24 min; ESI-MS(+) m/z 943,1 (M+2H).
Condiciones de análisis B: Tiempo de retención = 1,73 min; ESI-Ms (+) m/z 943,1 (M+2H). ESI-HRMS(+) m/z: 942,5096 (M+2H).
Preparación de Ejemplo 10040

Ejemplo 10040
El material en bruto del Ejemplo 10040 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 8,6 mg y su pureza estimada por análisis de LCMS fue 100 %.
Condiciones de análisis A: Tiempo de retención = 1,28 min; ESI-MS(+) m/z 956,1 (M+2H).
Condiciones de análisis B: Tiempo de retención = 1,80 min; ESI-Ms (+) m/z 956,1 (M+2H). ESI-HRMS(+) m/z: 955,5169 (M+2H).
Preparación de Ejemplo 10041

Ejemplo 10041
El material en bruto del Ejemplo 10041 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 2,6 mg y su pureza estimada por análisis de LCMS fue 97 %.
Condiciones de análisis A: Tiempo de retención = 1,23 min; ESI-MS(+) m/z 950,4 (M+2H).
Condiciones de análisis B: Tiempo de retención = 1,64 min; ESI-Ms (+) m/z 950,4 (M+2H). ESI-HRMS(+) m/z: 950,4883 (M+2H).
Preparación de Ejemplo 10042

Ejemplo 10042
El material en bruto del Ejemplo 10042 se purificó por LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 |jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 20-60% B durante 15 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 14,5 mg y su pureza estimada por análisis de LCMS fue 95 %.
Condiciones de análisis A: Tiempo de retención = 1,47 min; ESI-MS(+) m/z 926,9 (M+2H).
Condiciones de análisis B: Tiempo de retención = 1,83 min; ESI-Ms (+) m/z 926,2 (M+2H). ESI-HRMS(+) m/z: 927,0048 (M+2H).
Preparación de ácido (S)-2-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-((2-amino-2-oxoetil)amino)-3-oxopropoxi)acético

(S)-aziridina-1,2-dicarboxilato de 1-bencilo 2-metilo (5,105 g, 21,70 mmol) y 2-hidroxiacetato de bencilo (6,16 ml, 43,4 mmol) se disolvieron en DCM (43,4 ml) y enfriaron a 0 °C seguido de la adición de BF3.OEt2 (0,275 ml, 2,170 mmol). La reacción se agitó durante 2 h. TLC mostró que se había consumido el material de partida de aziridina. La reacción se agitó durante y 14 h adicionales. Se añadió solución saturada de bicarbonato sódico a la reacción y la mezcla bifásica se agitó vigorosamente durante 20 min. La reacción se diluyó con DCM y se separó de la fase acuosa. La capa orgánica se lavó con salmuera, se secó sobre sulfato sódico y se concentró al vacío. El
material crudo se purificó por cromatografía ultrarrápida usando 10-40 % de EtOAc/Hexanos. Las fracciones de producto se recogieron y el disolvente se retiró al vacío para dar O-(2-(benciloxi)-2-oxoetil)-N-((benciloxi)carbonil)-L-serinato de metilo, 3,0 (34 %). ESI-MS(+) m/z 402,1 (M+1). RMN 1H (400 MHz, CLOROFORMO-d) d 7,43 - 7,30 (m, 10H), 5,95 (d, J = 8,0 Hz, 1H), 5,18 (s, 2H), 5,15 (s, 2H), 4,50 (dt, J = 8,4, 3,1 Hz, 1H), 4,12 (d, J = 2,8 Hz, 2H), 4,10 -4,04 (m, 1H), 3,81 (dd, J = 9,4, 3,1 Hz, 1H), 3,77 (s, 3H).
Etapa 2:
(S)-3-(2-(benciloxi)-2-oxoetoxi)-2-(((benciloxi)carbonil)amino)propanoato de metilo (3 g, 7,47 mmol) se disolvió en MeOH (37,4 ml) y se puso en una atmósfera de N2. Pd-C (0,398 g, 0,374 mmol) se añadió a la solución con agitación vigorosa. La reacción se puso en una atmósfera de H2 y se agitó durante 16 h. La reacción se filtró a través de celite y se concentró al vacío para dar ácido (S)-2-(2-amino-3-metoxi-3-oxopropoxi)acético, 1,32 g, (100 %), que se usó en la etapa 3 sin purificación adicional. ESI-MS(+) m/z 178,1 (M+1).
Etapa 3:
Ácido (S)-2-(2-amino-3-metoxi-3-oxopropoxi)acético (1,323 g, 7,47 mmol) se disolvió en THF (29,9 ml) seguido de la adición de agua (29,9 ml). Después se añadió bicarbonato sódico (1,255 g, 14,94 mmol) seguido de la adición de carbonato de (9H-fluoren-9-il)metilo (2,5-dioxopirrolidin-1-ilo (2,52 g, 7,47 mmol). La reacción se agitó durante 2 h. La mayoría del THF se retiró al vacío después se añadió Et2O. La capa orgánica se descartó y la capa acuosa se lavó nuevamente con Et2O. La fase acuosa se recogió, acidificó con HCl 1 N y extrajo con EtOAc. La capa orgánica se recogió, se secó sobre sulfato sódico y concentró al vacío para dar ácido (S)-2-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-metoxi-3-oxopropoxi)acético, 2,76 g (93 %), que no se purificó adicionalmente. ESI-MS(+) m/z 400,1 (M+1).
Etapa 4:
A una solución de ácido (S)-2-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-metoxi-3-oxopropoxi)acético (3,78 g, 9,46 mmol) en acetato de etilo (40,6 ml) y hexanos (15,05 ml) se añadió 2,2,2-TRICLOROACETIMiDa TO DE TERC-BUTILO (1,692 ml, 9,46 mmol) gota a gota. Después de 15 min, BF3.OEt2 (0,060 ml, 0,473 mmol) se añadió y la reacción se agitó durante 1 h. Se añadió 2,2,2-t RiCLOROACETIMIDATO DE TERC-BUTILO (1,692 ml, 9,46 mmol) adicional seguido de BF3.OEt2 (0,060 ml, 0,473 mmol) y se agitó durante 1 h. El proceso se repitió una vez más, adición de 2,2,2-TRICLOROACETIMIDATO DE TERC-BUTILO (1,692 ml, 9,46 mmol) seguido de BF3.OEt2 (0,060 ml, 0,473 mmol) y agitación durante 1 h. Se añadió bicarbonato saturado a la reacción y se dejó agitar durante 15 min. La fase orgánica se recogió, se lavó con salmuera, se secó sobre sulfato sódico y concentró al vacío para dar el producto en bruto que se purificó adicionalmente por cromatografía ultrarrápida usando 20-30 % EtOAc/Hexanos como eluyente. Las fracciones de producto se recogieron y el disolvente se retiró al vacío para dar N-(((9H-fluoren-9-il)metoxi)carbonil)-O-(2-(terc-butoxi)-2-oxoetil)-L-serinato de metilo, 3,9 g, (90 %). ESI-m S(+) m/z 478,1 (M+Na).
Etapa 5:
(S)-2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-(2-(terc-butoxi)-2-oxoetoxi)propanoato de metilo (3,9 g, 8,56 mmol) se disolvió en DCE (42,8 ml) seguido de la adición de trimetilestannanol (3,10 g, 17,12 mmol). La reacción se calentó a 80 °C durante 5 h. El disolvente se retiró al vacío y el residuo se disolvió de nuevo en EtOAc y se lavó con HCl 1 N 3x, después salmuera. La capa orgánica se recogió, se secó sobre sulfato sódico y se concentró al vacío para dar el producto deseado. RMN mostró cierto estaño remanente. El material se disolvió de nuevo en EtOAc y se lavó con KHSO40,1 M, 3x seguido de salmuera. La capa orgánica se recogió, se secó sobre sulfato sódico y se concentró al vacío para dar N-(((9H-fluoren-9-il)metoxi)carbonil)-O-(2-(terc-butoxi)-2-oxoetil)-L-serina, 3,36 g (89 %). ESI-MS(+) m/z 464,0 (M+Na). RMN 1H (400 MHz, CLOROFORMO-d) d 7,78 (d, J = 7,5 Hz, 2H), 7,66 (t, J = 7,4 Hz, 2H), 7,45 -7,38 (m, 2H), 7,36 - 7,29 (m, 2H), 6,44 (d, J = 7,0 Hz, 1H), 4,55 - 4,33 (m, 3H), 4,26 (t, J = 7,3 Hz, 1H), 4,12 - 4,01 (m, 3H), 3,82 (dd, J = 9,7, 4,4 Hz, 1H), 1,52 (s, 9H).
Etapa 6:
Ácido (S)-2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-(terc-butoxi)-6-oxohexanoico (850 mg, 1,934 mmol), 2-aminoacetamida, HCl (278 mg, 2,51 mmol), se suspendieron en DCM (9670 pl) seguido de la adición de bases de Hunig (1013 pl, 5,80 mmol) después HATU (809 mg, 2,127 mmol). La reacción se agitó durante 2 h. La reacción se diluyó con DCM y se lavó con HCl 1 N, después salmuera. Después el orgánico se recogió, se secó sobre sulfato sódico y se concentró al vacío. El material en bruto se purificó por cromatografía ultrarrápida usando 20-60 % de Acetona/DCM. Las fracciones de producto se recogieron y el disolvente se retiró al vacío para dar el producto deseado., 789 mg (82 %). ESI-MS(+) m/z 498,1 (M+H).
Etapa 7:
(S)-2-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-((2-amino-2-oxoetil)amino)-3-oxopropoxi)acetato de terc-butilo
(1,25 g, 2,51 mmol) se disolvió en HCl (4 N en Dioxano) (15 ml, 60,0mmol). La reacción se agitó durante 4 h. El disolvente se retiró al vacío para dar ácido (S)-2-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-((2-amino-2-oxoetil)amino)-3-oxopropoxi)acético, 1,11 g (100 %). eS|-Ms (+) m/z 442,0 (M+H).
Preparación de ácido (S)-2-(2-((((9H-fíuoren-9-H)metoxi)carbonil)amino)-3-amino-3-oxopropoxi)acético

Etapa 1:
Ácido (S)-2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-(2-(terc-butoxi)-2-oxoetoxi)propanoico (1,875 g, 4,25 mmol)(síntesis descrita en otro lado), cloruro de amonio (0,568 g, 10,62 mmol), se suspendieron en DMF (21,24 ml) seguido de la adición de base de Hunig (2,225 ml, 12,74mmol) después HATU (1,776g, 4,67mmol). La reacción se agitó durante 2 h. La reacción se diluyó con DCM y se lavó con HCl 1 N, después salmuera. Después el orgánico se recogió, se secó sobre sulfato sódico y se concentró al vacío. El material en bruto se purificó por cromatografía ultrarrápida usando 20-40% de Acetona/DCM. Las fracciones de producto se recogieron y el disolvente se retiró al vacío para dar (S)-2-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-amino-3-oxopropoxi)acetato de terc-butilo, 1,43g (76%). ESI-MS(+) m/z 441,0 (M+1). RMN 1H (400 MHz, CLOROFORMO-d) d 7,78 (d, J = 7,5 Hz, 2H), 7,65 (d, J = 7,5 Hz, 2H), 7,46 -7,38 (m, 2H), 7,36 -7,30 (m, 2H), 6,99 (d, J = 17,3 Hz, 1H), 6,44 (s a, 1H), 5,46 (s a, 1H), 4,43 (d, J = 6,3 Hz, 2H), 4,31 (s a, 1H), 4,27 -4,21 (m, 1H), 4,16 -3,95 (m, 3H), 3,73 -3,64 (m, 1H), 1,51 (s, 9H).
Etapa 2:
2-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-amino-3-oxopropoxi)acetato de terc-butilo (1,432 g, 3,25mmol) se disolvió en HCl (4 N en Dioxano) (15 ml, 60,0 mmol). La reacción se agitó durante 4 h. El disolvente se retiró al vacío para dar ácido (S)-2-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-amino-3-oxopropoxi)acético, 1,25g, (100. ESI-MS(+) m/z 385,0 (M+1).
Preparación de ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-((2-amino-2-oxoetil)amino)-6-oxohexanoico

Etapa 1:
Ácido (S)-2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-(terc-butoxi)-6-oxohexanoico (850 mg, 1,934 mmol), 2-aminoacetamida, HCl (278 mg, 2,51 mmol), se suspendieron en DCM (9670 pl) seguido de la adición de bases de Hunig (1013 pl, 5,80 mmol) después HATU (809 mg, 2,127 mmol). La reacción se agitó durante 2 h. La reacción se diluyó con DCM y se lavó con HCl 1 N, después salmuera. Después el orgánico se recogió, se secó sobre sulfato sódico y se concentró al vacío. El material en bruto se purificó por cromatografía ultrarrápida usando 20-60 % de Acetona/DCM. Las fracciones de producto se recogieron y el disolvente se retiró al vacío para dar (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-((2-amino-2-oxoetil)amino)-6-oxohexanoato de terc-butilo, 789 mg (82 %). ESI-MS(+) m/z 496,1 (M+1). RMN 1H (400 MHz, CLOROFORMO-d) d 7,78 (d, J = 7,5 Hz, 2H), 7,60 (d, J = 7,5 Hz, 2H), 7,45 - 7,39 (m, 2H), 7,35 - 7,30 (m, 2H), 6,78 (s a, 1H), 6,42 (s a, 1H), 5,71 (d, J = 6,0 Hz, 1H), 5,45 (s a, 1H), 4,44
(d, J = 6,8 Hz, 2H), 4,25 - 4,19 (m, 1H), 4,10 (d, J = 6,0 Hz, 1H), 3,97 (d, J=5,5 Hz, 2H), 2,36 - 2,24 (m, 2H), 1,90 (s a, 1H), 1,76 - 1,55 (m, 3H), 1,46 (s, 9H)
Etapa 2:
(S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-((2-amino-2-oxoetil)amino)-6-oxohexanoato de tere-butilo (0,789 g, 1,593 mmol) se disolvió en HCl (4 N en Dioxano) (15 ml, 60,0 mmol). La reacción se agitó durante 6 h. El disolvente se retiró al vacío para dar ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-((2-amino-2-oxoetil)amino)-6-oxohexanoico, 700 mg (100%). ESI-MS(+) m/z 440,0 (M+1).
Preparaeión de áeido (S)-5-((((9H-fluoren-9-il)metoxi)earbonil)amino)-6-amino-6-oxohexanoieo
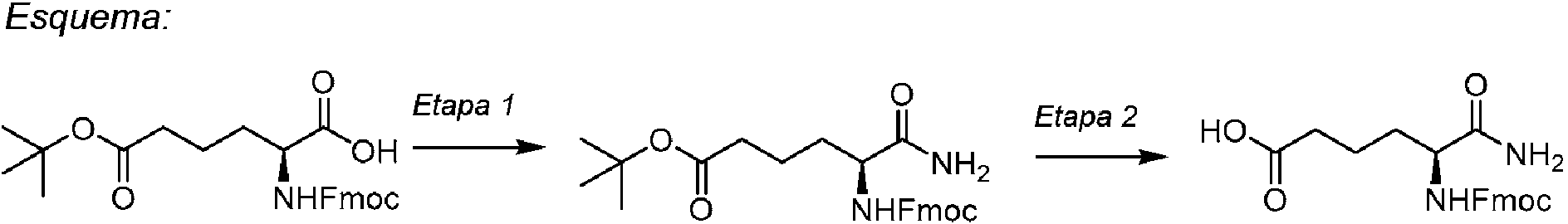
Ácido (S)-2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-(tere-butoxi)-6-oxohexanoico (2,5 g, 5,69 mmol), cloruro de amonio (0,761 g, 14,22 mmol), se suspendieron en DMF (28,4 ml) seguido de la adición de base de Hunig (2,98 ml, 17,06 mmol) después HATU (2,379 g, 6,26 mmol). La reacción se agitó durante 2 h. La reacción se diluyó con Et2O y se lavó con HCl 1 N, después salmuera. Después el orgánico se recogió, se secó sobre sulfato sódico y se concentró al vacío. El material en bruto se purificó por cromatografía ultrarrápida usando 0-20 % de Acetona/DCM. Las fracciones de producto se recogieron y el disolvente se retiró al vacío para dar (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-amino-6-oxohexanoato de tere-butilo, 1,89 g (76 %). ESI-MS(+) m/z 439,0 (M+1).
Etapa 2:
(S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-amino-6-oxohexanoato de tere-butilo (1,89 g, 4,31 mmol) se disolvió en HCl (4 N en Dioxano) (15 ml, 60,0 mmol). La reacción se agitó durante 4 h. El disolvente se retiró al vacío para dar ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-amino-6-oxohexanoico, 1,65 g, (100. ESI-MS(+) m/z 385,0 (M+1). RMN 1H (400 MHz, METANOL-d4) d 7,80 (d, J=7,5 Hz, 2H), 7,71 - 7,65 (m, 2H), 7,43 - 7,36 (m, 2H), 7,35 - 7,29 (m, 2H), 4,47 - 4,35 (m, 2H), 4,26 - 4,21 (m, 1H), 4,09 (d, J = 7,8 Hz, 1H), 2,32 (t, J = 6,4 Hz, 2H), 1,88 -1,57 (m, 4H).
Preparaeión de áeido (R)--5-((((9H-fluoren-9-il)metoxi)earbonil)amino)-2-((tere-butoxiearbonil)amino)pentanoieo Esquema

Etapa 1:
(R)-5-amino-2-((tere-butoxicarbonil)amino)pentanoico ácido (4,77 g, 20,54 mmol) se disolvió en THF (82 ml) seguido de la adición de agua (82 ml). Después se añadió bicarbonato sódico (3,45 g, 41,1 mmol) seguido de la adición de carbonato de (9H-fluoren-9-il)metilo (2,5-dioxopirrolidin-1-ilo (6,93 g, 20,54 mmol). La reacción se agitó durante 2 h. La mayoría del THF se retiró al vacío después se añadió Et2O. La capa orgánica se descartó y la capa acuosa se lavó nuevamente con Et2O. La fase acuosa se recogió, acidificó con HCl 1 N y extrajo con EtOAc. La capa orgánica se recogió, se secó sobre sulfato sódico y se concentró al vacío para dar ácido (R)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((tere-butoxicarbonil)amino)pentanoico, 8,76 g (94 %). ESI-MS(+) m/z 454,9 (M+1).
Preparaeión de Ejemplo 10504 (no según la inveneión)

Preparación de Intermedio 10504A:
Se siguió "Secuencia de síntesis general A". (S)-2-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-((2-amino-2-oxoetil)amino)-3-oxopropoxi)acético ácido se usó en el "Procedimiento de carga de resina". Al recipiente de reacción que contiene resina de la secuencia automatizada se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. Al recipiente de reacción se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DCM (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se transfirió después inmediatamente usando DCM (8 ml) a un vial de 15 ml. A la solución se añadió hexafluoroisopropanol (2 ml). La resina se volvió inmediatamente rojo intenso; la solución permaneció incolora. La mezcla se agitó manualmente brevemente, después se dejó en reposo a t.a. durante 15 minutos, después se filtró. El filtrado se transfirió a un vial de 15 ml y se concentró bajo una corriente de N2 para proporcionar un residuo sólido, Intermedio 10501A.
Preparación de Intermedio 10504B:
A un vial de 40 ml cargado con la totalidad de Intermedio 10504A preparado anteriormente se añadió DCM (20 ml), después HATU (38 mg, 0,10 mmol) después DIPEA (0,114 ml, 0,650 mmol). La solución se agitó durante 2 h. La solución se secó al vacío para proporcionar Intermedio 10504B.
Preparación de Ejemplo 10504:
Una "solución de desprotección" se preparó combinando en un vial de vidrio de 40 ml ácido trifluoroacético (23,75 ml), 1,4-Ditio-DL-treitol (625 mg), triisopropilsilano (0,625 ml). A un vial de 5 dram cargado con la totalidad de
Intermedio 10504A preparado anteriormente se añadió la "solución de desprotección" (2,0ml). La solución se mezcló durante 20 minutos en un agitador funcionando a 500 rpm, después se vertió en un tubo de ensayo de 25 ml cargado con Et2O (15 ml). Precipitó una pequeña cantidad de sólido blanco. La mezcla se centrifugó; el líquido se decantó. Los sólidos se suspendieron en Et2O (15 ml). La mezcla se centrifugó, el líquido se decantó. El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/EM preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 0-40 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 7,3 mg y su pureza estimada por análisis de LCMS fue 93 %. Condiciones de análisis A: Tiempo de retención = 1,58 min; ESI-MS(+) m/z 936,1 (M+2H).
Condiciones de análisis B: Tiempo de retención = 2,70 min; ESI-MS(+) m/z 935,9 (m +2h ).
Preparación de Ejemplo 10505 (no según ia invención)

Preparación de intermedio 10505A:
"Secuencia de síntesis general A" se siguió con la excepción de que la etapa de carga no fue necesaria dado que se usó resina Fmoc-L-Pro-2-Clorotritil precargada. Se usó ácido 5-((((9H-fluoren-9-il)metoxi)carbonil)amino)pentanoico en la 4a de formación de enlace amida. Al recipiente de reacción que contiene resina de la secuencia automatizada se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. Al recipiente de reacción se añadió piperidina:DMF (20:80 v/v, 2,0 ml). La mezcla se agitó periódicamente durante 4 minutos y después la solución se drenó a través de la frita. La resina se
lavó sucesivamente cinco veces como sigue: para cada lavado, DMF (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se lavó sucesivamente cinco veces como sigue: para cada lavado, DCM (2,0 ml) se añadió a través de la parte superior del matraz y la mezcla resultante se agitó periódicamente durante 90 segundos antes de drenar la solución a través de la frita. La resina se transfirió después inmediatamente usando DCM (8 ml) a un vial de 15 ml. A la solución se añadió hexafluoroisopropanol (2 ml). La resina se volvió inmediatamente rojo intenso; la solución permaneció incolora. La mezcla se agitó manualmente brevemente, después se dejó en reposo a t.a. durante 15 minutos y después se filtró. El filtrado se transfirió a un vial de 15 ml y se concentró bajo una corriente de N2 para proporcionar un residuo sólido, Intermedio 10501A.
Preparación de Intermedio 10505B:
A un vial de 40 ml cargado con la totalidad de Intermedio 10504A preparado anteriormente se añadió DCM (20 ml), después HATU (76 mg, 0,20 mmol) después DIPEA (0,052 ml, 0,30 mmol). La solución se agitó durante 2 h. La solución se secó al vacío para proporcionar Intermedio 10504B.
Preparación de Ejemplo 10505:
Una "solución de desprotección" se preparó combinando en un vial de vidrio de 40 ml ácido trifluoroacético (23,75 ml), 1,4-Ditio-DL-treitol (625 mg), triisopropilsilano (0,625 ml). A un vial de 5 dram cargado con la totalidad de Intermedio 10504A preparado anteriormente se añadió la "solución de desprotección" (2,0 ml). La solución se mezcló durante 20 minutos en un agitador funcionando a 500 rpm, después se vertió en un tubo de ensayo de 25 ml cargado con Et2O (15 ml). Precipitó una pequeña cantidad de sólido blanco. La mezcla se centrifugó; el líquido se decantó. Los sólidos se suspendieron en Et2O (15 ml). La mezcla se centrifugó, el líquido se decantó. El material bruto se purificó por medio de CL/EM preparativa con las siguientes condiciones: Columna: XBridge C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 10-50% B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El material se purificó adicionalmente mediante CL/e M preparativa con las condiciones siguientes: Columna: Waters CSH C18, 19 x 200 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: 0-40 % B durante 30 minutos, después mantener 5 minutos a 100 % B; Flujo: 20 ml/min. Las fracciones que contenían el producto deseado se combinaron y se secaron a través de evaporación por centrifugación. El rendimiento del producto fue 7,0 mg y su pureza estimada por análisis de LCMS fue 95 %.
Condiciones de análisis A: Tiempo de retención = 1,56 min; ESI-MS(+) m/z 927,0 (M+2H). Condiciones de análisis C: Tiempo de retención = 1,49 min; ESI-MS(+) m/z 927,1 (M+2H). acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 pm; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,52 minutos con detección ESI correspondiente de 922,9 m/z. acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 pm; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,52 minutos con detección ESI correspondiente de 922,9 m/z. acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 pm; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,52 minutos con detección ESI correspondiente de 922,9 m/z.
Preparación de Ejemplo 10506 (no según la invención)

Ejemplo 10506 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 para proporcionar 9,3 mg del producto con 98,6 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 pm; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 pm; Fase móvil A: acetonitrilo:agua 5:95 con 0,05 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,05% de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,43 minutos con detección ESI correspondiente de 1785,1 m/z.
Preparación de Ejemplo 10507 (no según la invención)

Ejemplo 10507 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-((2-amino-2-oxoetil)amino)-6-oxohexanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 2,3 mg del producto con 95,5 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 pm; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 pm; Fase móvil A: acetonitrilo:agua 5:95 con 0,05 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,05 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,36 minutos con detección ESI correspondiente de
1885,9 m/z.
Preparación de Ejemplo 10508 (no según la invención)

Ejemplo 10508 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-((2-amino-2-oxoetil)amino)-6-oxohexanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 5,2 mg del producto con 97,5 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 pm; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 pm; Fase móvil A: acetonitrilo:agua 5:95 con 0,05 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,05 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,38 minutos con detección ESI correspondiente de 956,2 m/z.
Preparación de Ejemplo 10509 (no según la invención)

Ejemplo 10509 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 para proporcionar 5,3 mg del producto con 98,2 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar
la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,05 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,05% de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,31 minutos con detección ESI correspondiente de 622 m/z.
Preparación de Ejemplo 10510 (no según la invención)

Ejemplo 10510 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-((2-amino-2-oxoetil)amino)-6-oxohexanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 7 mg del producto con 100% pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters Acquity UPLC BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 50 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 0,75 minutos a 100 % B; Flujo: 1,0 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters Acquity UPLC BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 50 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 0,75 minutos a 100 % B; Flujo: 1,0 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,65 minutos con detección ESI correspondiente de 946 m/z.
Preparación de Ejemplo 10511 (no según la invención)

Ejemplo 10511 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((ferc-butoxicarbonil)amino)pentanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 2,9 mg del producto con 95,1 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BeH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con 0,05 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,05% de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,73 minutos con detección ESI correspondiente de 1795 m/z.
Preparación de Ejemplo 10512 (no según la invención)

Ejemplo 10512 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 para proporcionar 8,6 mg del producto con 100 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters Acquity UPLC BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 50 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 0,75 minutos a 100 % B; Flujo: 1,0 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters Acquity UPLC BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 50 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 0,75 minutos a 100 % B; Flujo: 1,0 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,68 minutos con detección ESI
correspondiente de 898,2 m/z.
Preparación de Ejemplo 10513 (no según la invención)

Ejemplo 10513 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 para proporcionar 7,4 mg del producto con 100 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters Acquity UPLC BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 50 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 0,75 minutos a 100 % B; Flujo: 1,0 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2:
Columna: Waters Acquity UPLC BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético;
Temperatura: 50 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 0,75 minutos a 100 % B; Flujo:
1,0 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,6 minutos con detección ESI correspondiente de 906,2 m/z.
Preparación de Ejemplo 10514 (no según la invención)

Ejemplo 10514 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-((2-amino-2-oxoetil)amino)-6-oxohexanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 1,8 mg del producto con 100% pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters Acquity UPLC BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 50 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 0,75 minutos a 100 % B; Flujo: 1,0ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters Acquity UPLC BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 50 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 0,75 minutos a 100 % B; Flujo: 1,0 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,61 minutos con detección ESI correspondiente de 935,2 m/z.
Preparación de Ejemplo 10515 (no según la invención)

Ejemplo 10515 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5
((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-((2-amino-2-oxoetil)amino)-6-oxohexanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 0,3 mg del producto con 96,7 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters Acquity UPLC BEH C18, 2,1 x 50 mm, partículas de 1,7 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 50 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 0,75 minutos a 100 % B; Flujo: 1,0 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters Acquity UPLC BEH C18, 2,1 x 50 mm, partículas de 1,7 pm; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 50 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 0,75 minutos a 100 % B; Flujo: 1,0 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,65 minutos con detección ESI correspondiente de 948,3 m/z.
Preparación de Ejemplo 10516 (no según la invención)

Ejemplo 10516 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-((2-amino-2-oxoetil)amino)-6-oxohexanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 1,5 mg del producto con 100% pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 pm; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 pm; Fase móvil A: acetonitrilo:agua 5:95 con 0,05 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,05 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,27 minutos con detección ESI correspondiente de 983 m/z.
Preparación de Ejemplo 10517 (no según la invención)

Ejemplo 10517 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((ferc-butoxicarbonil)amino)pentanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 15,3 mg del producto con 97% pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,39 minutos con detección ESI correspondiente de 1809,7 m/z.
Preparación de Ejemplo 10518 (no según la invención)

Ejemplo 10518 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5
((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((ferc-butoxicarbonil)amino)pentanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 10,9 mg del producto con 97,5 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,79 minutos con detección ESI correspondiente de 1822,1 m/z.
Preparación de Ejemplo 10519 (no según la invención)

Ejemplo 10519 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((ferc-butoxicarbonil)amino)pentanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 8,5 mg del producto con 94,5 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,89 minutos con detección ESI correspondiente de 1807,1 m/z.
Preparación de Ejemplo 10520 (no según la invención)

Ejemplo 10520 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-amino-6-oxohexanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 7 mg del producto con 95,4 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,55 minutos con detección ESI correspondiente de 1853,1 m/z.
Preparación de Ejemplo 10521 (no según la invención)

Ejemplo 10521 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-amino-6-oxohexanoico usado en la 4a etapa de acoplamiento de amida)
para proporcionar 5,6 mg del producto con 100% pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,85 minutos con detección ESI correspondiente de 1853,3 m/z.
Preparación de Ejemplo 10522 (no según la invención)

Ejemplo 10522 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-amino-6-oxohexanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 5,9 mg del producto con 94,1 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,74 minutos con detección ESI correspondiente de 1867,1 m/z.
Preparación de Ejemplo 10525 (no según la invención)

Ejemplo 10525 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-amino-6-oxohexanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 2,8 mg del producto con 99,1 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C l8 , 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,72 minutos con detección ESI correspondiente de 926,8 m/z.
Preparación de Ejemplo 10526 (no según la invención)

Ejemplo 10526 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-amino-6-oxohexanoico usado en la 4a etapa de acoplamiento de amida)
para proporcionar 4,5 mg del producto con 99,2 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 |jm; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 jm ; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,72 minutos con detección ESI correspondiente de 920,9 m/z.
Preparación de Ejemplo 10527 (no según la invención)
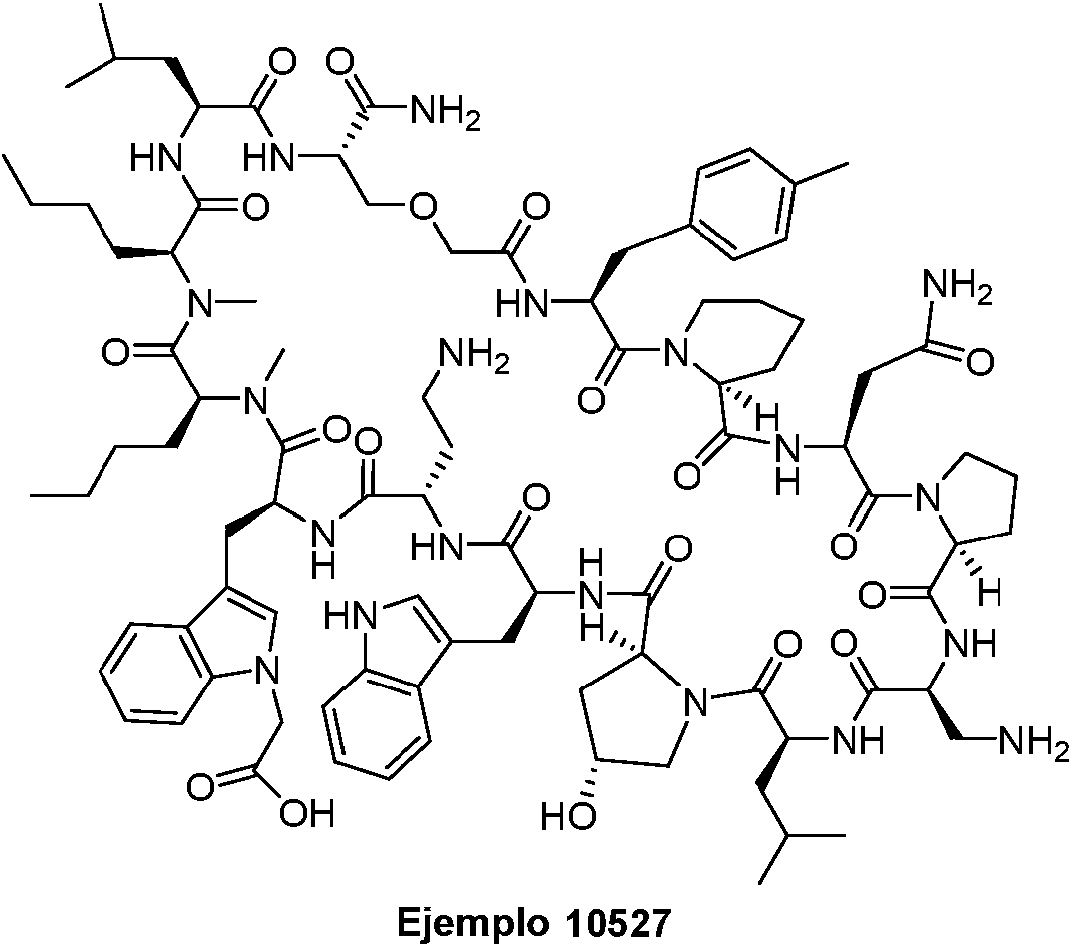
Ejemplo 10527 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-2-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-amino-3-oxopropoxi)acético usado en la 4a etapa de acoplamiento de amida) para proporcionar 2,8 mg del producto con 97,4 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 jm; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 jm ; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,97 minutos con detección ESI correspondiente de 1835,8 m/z.
Preparación de Ejemplo 10528 (no según la invención)

Ejemplo 10528 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-2-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-amino-3-oxopropoxi)acético usado en la 4a etapa de acoplamiento de amida) para proporcionar 3,4 mg del producto con 99 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,74 minutos con detección ESI correspondiente de 927,1 m/z.
Preparación de Ejemplo 10529 (no según la invención)

Ejemplo 10529 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-2-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-amino-3-oxopropoxi)acético usado en la 4a etapa de acoplamiento de
amida) para proporcionar 2,8 mg del producto con 98,9 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,89 minutos con detección ESI correspondiente de 1841,2 m/z.
Preparación de Ejemplo 10530 (no según la invención)

Ejemplo 10530 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-2-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-amino-3-oxopropoxi)acético usado en la 4a etapa de acoplamiento de amida) para proporcionar 4,5 mg del producto con 97,3 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,77 minutos con detección ESI correspondiente de 935,4 m/z.
Preparación de Ejemplo 10531 (no según la invención)

Ejemplo 10531 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-2-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-amino-3-oxopropoxi)acético usado en la 4a etapa de acoplamiento de amida) para proporcionar 5,8 mg del producto con 97,5 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,81 minutos con detección ESI correspondiente de 1859 m/z.
Preparación de Ejemplo 10532 (no según la invención)
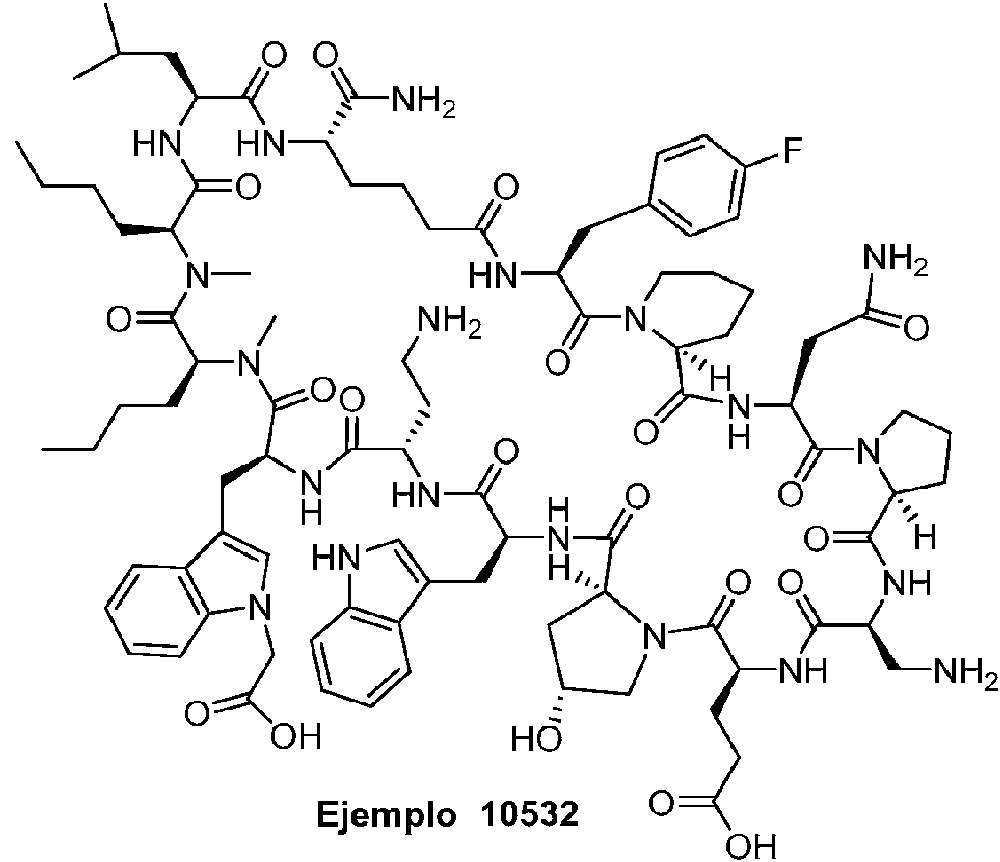
Ejemplo 10532 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-amino-6-oxohexanoico usado en la 4a etapa de acoplamiento de amida)
para proporcionar 3,7 mg del producto con 95,4 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,67 minutos con detección ESI correspondiente de 1854 m/z.
Preparación de Ejemplo 10533 (no según la invención)
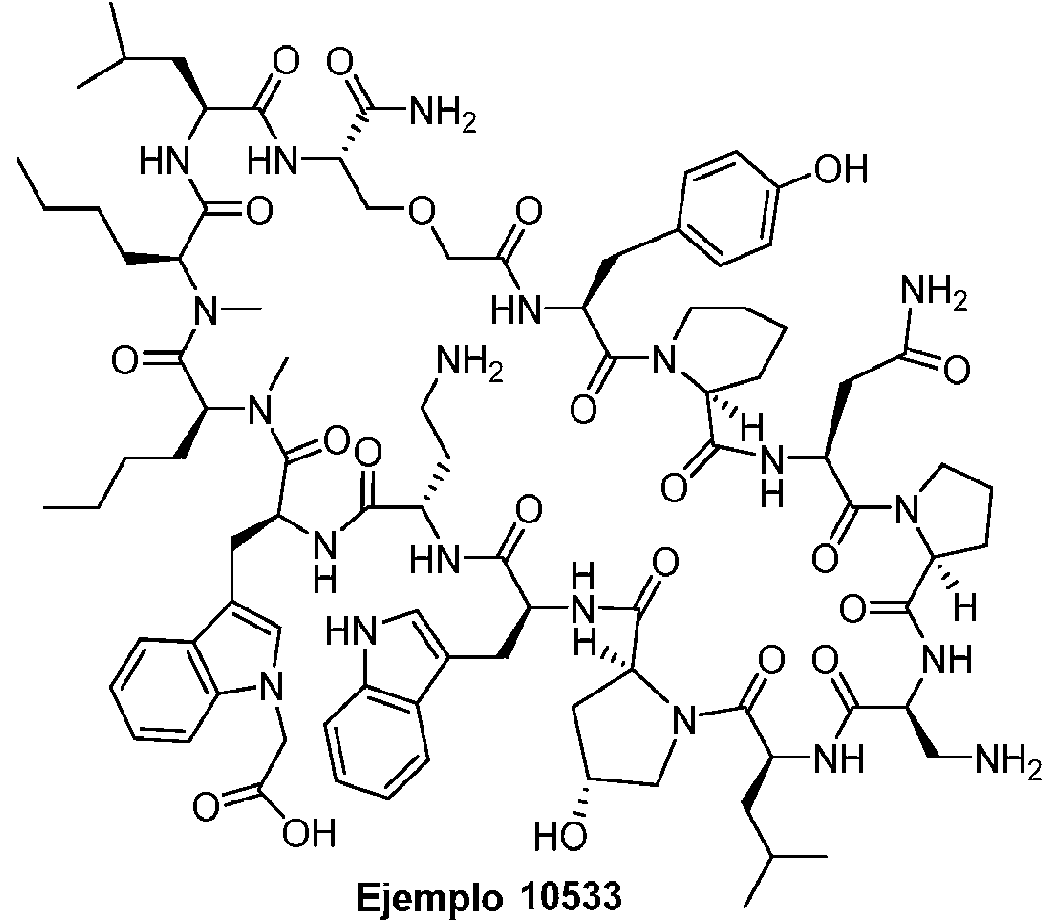
Ejemplo 10533 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-2-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-amino-3-oxopropoxi)acético usado en la 4a etapa de acoplamiento de amida) para proporcionar 3,5 mg del producto con 92,7 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,62 minutos con detección ESI correspondiente de 1839,9 m/z.
Preparación de Ejemplo 10534 (no según la invención)

Ejemplo 10534 se preparo siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (acido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-amino-6-oxohexanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 3,8 mg del producto con 100% pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters Acquity UPLC BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 50 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 0,75 minutos a 100 % B; Flujo: 1,0 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters Acquity UPLC BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de acido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de acido trifluoroacético; Temperatura: 50 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 0,75 minutos a 100 % B; Flujo: 1,0 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 2 minutos con detección ESI correspondiente de 929,2 m/z.
Preparación de Ejemplo 10535 (no según la invención)

Ejemplo 10535 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 para
proporcionar 9,3 mg del producto con 95,8 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,78 minutos con detección ESI correspondiente de 904,7 m/z.
Preparación de Ejemplo 10536 (no según la invención)

Ejemplo 10536 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 para proporcionar 11 ,6 mg del producto con 100 % pureza. Se usaron dos inyecciones de c L/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters Acquity UPLC BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 50 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 0,75 minutos a 100 % B; Flujo: 1,0 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters Acquity UPLC BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 50 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 0,75 minutos a 100 % B; Flujo: 1,0 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,9 minutos con detección ESI correspondiente de 899,1 m/z.
Preparación de Ejemplo 10537 (no según la invención)

Ejemplo 10537 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 para proporcionar 5,9 mg del producto con 100 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters Acquity UPLC BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 50 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 0,75 minutos a 100 % B; Flujo: 1,0ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters Acquity UPLC BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1% de ácido trifluoroacético; Temperatura: 50 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 0,75 minutos a 100 % B; Flujo: 1,0 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,94 minutos con detección ESI correspondiente de 605,1 m/z.
Preparación de Ejemplo 10538 (no según la invención)
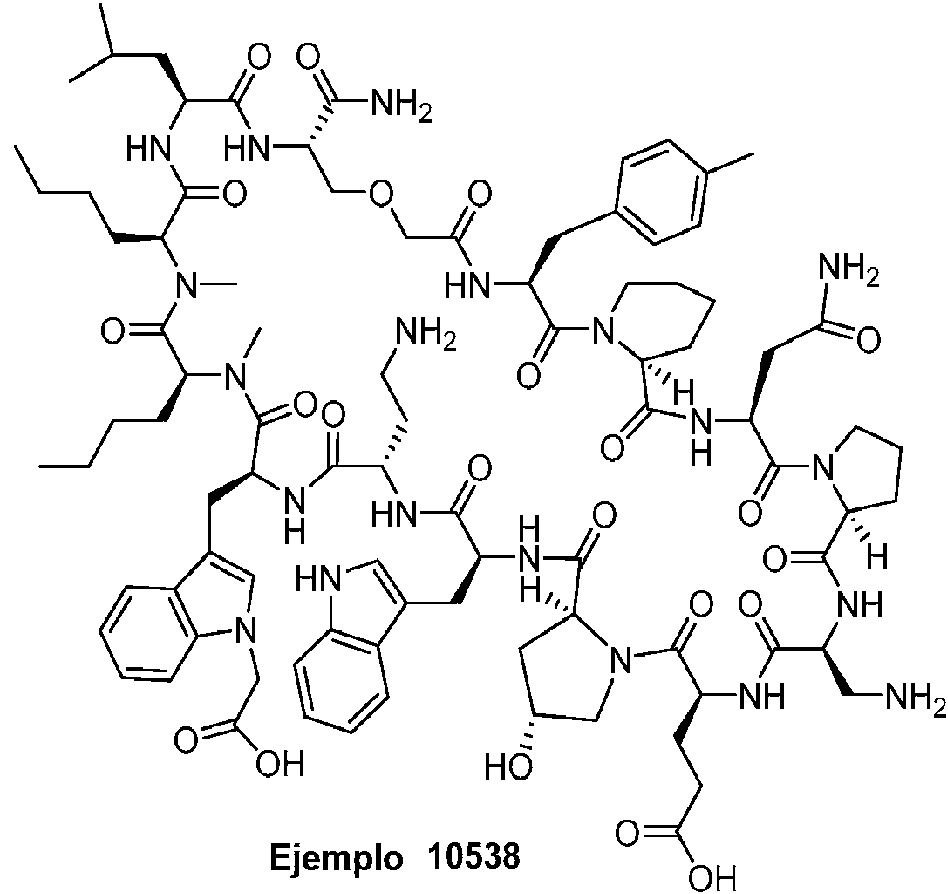
Ejemplo 10538 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-2-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-amino-3-oxopropoxi)acético usado en la 4a etapa de acoplamiento de
amida) para proporcionar 1,1 mg del producto con 99 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,78 minutos con detección ESI correspondiente de 1852,7 m/z.
Preparación de Ejemplo 10539 (no según la invención)

Ejemplo 10539 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-amino-6-oxohexanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 2,8 mg del producto con 97,8 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,64 minutos con detección ESI correspondiente de 923,0 m/z.
Preparación de Ejemplo 10540 (no según la invención)

Ejemplo 10540 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 para proporcionar 5,5 mg del producto con 95,7 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,96 minutos con detección ESI correspondiente de 1793,2 m/z.
Preparación de Ejemplo 10541 (no según la invención)

Ejemplo 10541 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 para proporcionar 9,2 mg del producto con 96,7 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase
móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 |jm; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,86 minutos con detección ESI correspondiente de 1810 m/z.
Preparación de Ejemplo 10542 (no según la invención)

Ejemplo 10542 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 para proporcionar 10,6 mg del producto con 94,5% pureza. Se usaron dos inyecciones de c L/e M analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 jm; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 jm; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,76 minutos con detección ESI correspondiente de 1824,8 m/z.
Preparación de Ejemplo 10543 (no según la invención)

Ejemplo 10543 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 para proporcionar 6,4 mg del producto con 95,4 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,79 minutos con detección ESI correspondiente de 1811,9 m/z.
Preparación de Ejemplo 10544 (no según la invención)

Ejemplo 10544 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 para proporcionar 6,6 mg del producto con 98,6 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase
móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,86 minutos con detección ESI correspondiente de 1829,9 m/z.
Preparación de Ejemplo 10545 (no según la invención)

Ejemplo 10545 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((ferc-butoxicarbonil)amino)pentanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 3 mg del producto con 94,6 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,91 minutos con detección ESI correspondiente de 1826,2 m/z.
Preparación de Ejemplo 10546 (no según la invención)

Ejemplo 10546 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((terc-butoxicarbonil)amino)pentanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 6,4 mg del producto con 100% pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,64 minutos con detección ESI correspondiente de [ERROR] m/z.
Preparación de Ejemplo 10547 (no según la invención)

Ejemplo 10547 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5
((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((ferc-butoxicarbonil)amino)pentanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 5,5 mg del producto con 97,6 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,89 minutos con detección ESI correspondiente de 1822,1 m/z.
Preparación de Ejemplo 10548 (no según la invención)

Ejemplo 10548 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((ferc-butoxicarbonil)amino)pentanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 5,2 mg del producto con 97,7 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,8 minutos con detección ESI correspondiente de 1828,7 m/z.
Preparación de Ejemplo 10549 (no según la invención)

Ejemplo 10549 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((ferc-butoxicarbonil)amino)pentanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 1,9 mg del producto con 98 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BeH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,39 minutos con detección ESI correspondiente de 1824,8 m/z.
Preparación de Ejemplo 10550 (no según la invención)

Ejemplo 10550 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((ferc-butoxicarbonil)amino)pentanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 1,8 mg del producto con 98,3 % pureza. Se usaron dos inyecciones de
CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,83 minutos con detección ESI correspondiente de 1810,6 m/z.
Preparación de Ejemplo 10551 (no según la invención)

Ejemplo 10551 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((ferc-butoxicarbonil)amino)pentanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 3,8 mg del producto con 95,5 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,79 minutos con detección ESI correspondiente de 1840,9 m/z.
Preparación de Ejemplo 10552 (no según la invención)

Ejemplo 10552 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-amino-6-oxohexanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 4,9 mg del producto con 100% pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,5 minutos con detección ESI correspondiente de 1837 m/z.
Preparación de Ejemplo 10553 (no según la invención)

Ejemplo 10553 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-amino-6-oxohexanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 4,2 mg del producto con 94,3 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con
acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,74 minutos con detección ESI correspondiente de 1834,9 m/z.
Preparación de Ejemplo 10554 (no según la invención)

Ejemplo 10554 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-amino-6-oxohexanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 5,1 mg del producto con 100% pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,79 minutos con detección ESI correspondiente de 1851,7 m/z.
Preparación de Ejemplo 10555 (no según la invención)

Ejemplo 10555 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-amino-6-oxohexanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 8,6 mg del producto con 98,8 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH Cl8, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,82 minutos con detección ESI correspondiente de 1841 m/z.
Preparación de Ejemplo 10556 (no según la invención)

Ejemplo 10556 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-amino-6-oxohexanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 4,3 mg del producto con 98,5 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18 , 2,1 x 50 mm, partículas de 1,7
^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,9 minutos con detección ESI correspondiente de 1855,6 m/z.
Preparación de Ejemplo 10557 (no según la invención)

Ejemplo 10557 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-amino-6-oxohexanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 2,2 mg del producto con 100% pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,69 minutos con detección ESI correspondiente de 1870,1 m/z.
Preparación de Ejemplo 10558 (no según la invención)

Ejemplo 10558 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (R)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((ferc-butoxicarbonil)amino)pentanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 1,4 mg del producto con 100 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BeH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,37 minutos con detección ESI correspondiente de 905,7 m/z.
Preparación de Ejemplo 10559 (no según la invención)

Ejemplo 10559 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (R)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((ferc-butoxicarbonil)amino)pentanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 1,9 mg del producto con 100 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BeH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B:
95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,91 minutos con detección ESI correspondiente de 1826,8 m/z.
Preparación de Ejemplo 10560 (no según la invención)

Ejemplo 10560 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (R)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((terc-butoxicarbonil)amino)pentanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 3,4 mg del producto con 95,9% pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,47 minutos con detección ESI correspondiente de 1823,1 m/z.
Preparación de Ejemplo 10561 (no según la invención)

Ejemplo 10561 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-amino-6-oxohexanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 3,1 mg del producto con 96,6% pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,37 minutos con detección ESI correspondiente de 1852,8 m/z.
Preparación de Ejemplo 10562 (no según la invención)

Ejemplo 10562 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-amino-6-oxohexanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 3,6 mg del producto con 96,9 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0
minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 pm; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,69 minutos con detección ESI correspondiente de 1851,3 m/z. Preparación de Ejemplo 10563 (no según la invención)
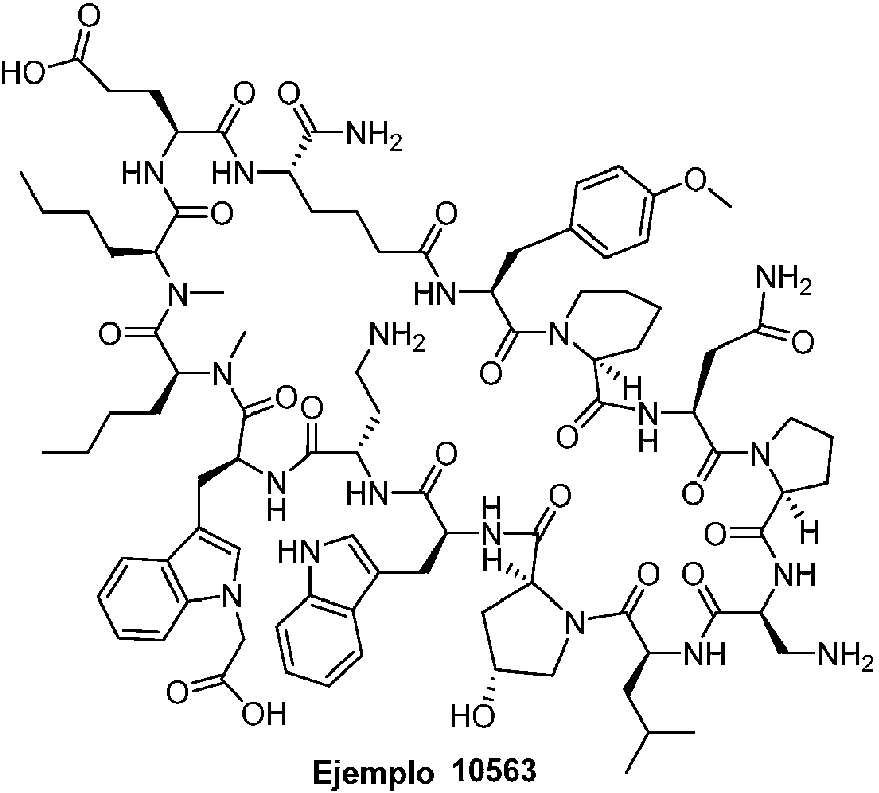
Ejemplo 10563 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-amino-6-oxohexanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 1,6 mg del producto con 97% pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 pm; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,6 minutos con detección ESI correspondiente de 1867,1 m/z. Preparación de Ejemplo 10564 (no según la invención)

Ejemplo 10564 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-6-amino-6-oxohexanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 3,2 mg del producto con 97,1 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,62 minutos con detección ESI correspondiente de 1854 m/z.
Preparación de Ejemplo 10566 (no según la invención)

Ejemplo 10566 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (R)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((ferc-butoxicarbonil)amino)pentanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 5,9 mg del producto con 96,6 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,47 minutos con detección ESI correspondiente de 1811,2 m/z.
Preparación de Ejemplo 10567 (no según la invención)

Ejemplo 10567 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (R)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((ferc-butoxicarbonil)amino)pentanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 4,3 mg del producto con 93,9% pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,52 minutos con detección ESI correspondiente de 1807,4 m/z.
Preparación de Ejemplo 10568 (no según la invención)

Ejemplo 10568 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (R)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((ferc-butoxicarbonil)amino)pentanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 4,5 mg del producto con 97,6% pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x
50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,6 minutos con detección ESI correspondiente de 1826,1 m/z.
Preparación de Ejemplo 10569 (no según la invención)

Ejemplo 10569 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (R)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((ferc-butoxicarbonil)amino)pentanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 6,1 mg del producto con 100 % pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,78 minutos con detección ESI correspondiente de 1838,1 m/z.
Preparación de Ejemplo 10570 (no según la invención)

Ejemplo 10570 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (R)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((ferc-butoxicarbonil)amino)pentanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 1,8 mg del producto con 97% pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,47 minutos con detección ESI correspondiente de 1827,2 m/z.
Preparación de Ejemplo 10571 (no según la invención)

Ejemplo 10571 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 (ácido (R)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((ferc-butoxicarbonil)amino)pentanoico usado en la 4a etapa de acoplamiento de amida) para proporcionar 0,7 mg del producto con 91,3% pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x
50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,52 minutos con detección ESI correspondiente de 922,9 m/z.
Preparación de Ejemplo 10572 (no según la invención)

Ejemplo 10572 se preparó siguiendo el procedimiento usado para la preparación del Ejemplo 10505 para proporcionar 21,7 mg del producto con 94,8% pureza. Se usaron dos inyecciones de CL/EM analítica para determinar la pureza final. Condiciones de inyección 1: Columna: Waters BEH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Condiciones de inyección 2: Columna: Waters CSH C18, 2,1 x 50 mm, partículas de 1,7 ^m; Fase móvil A: acetonitrilo:agua 5:95 con 0,1 % de ácido trifluoroacético; Fase móvil B: acetonitrilo:agua 95:5 con 0,1 % de ácido trifluoroacético; Temperatura: 70 °C; Gradiente: 0-100 % B durante 3 minutos, después mantener 2,0 minutos a 100 % B; Flujo: 0,75 ml/min; Detección: UV a 220 nm. Resultados del análisis: Tiempo de retención = 1,59 minutos con detección ESI correspondiente de 1810,8 m/z.
MÉTODOS PARA ENSAYAR LA CAPACIDAD DE LOS PÉPTIDOS MACROCÍCLICOS PARA COMPETIR POR LA UNIÓN DE PD-1 A PD-L1 UTILIZANDO ENSAYOS DE ENLACE DE FLUORESCENCIA HOMÓGENA RESUELTA EN TIEMPO (HTRF)
La capacidad de los péptidos macrocíclicos de la presente divulgación para unirse a PD-L1 se investigó mediante un ensayo de unión de fluorescencia homogénea resuelta en tiempo de PD-1/PD-L1 (HTRF).
Métodos
Ensayos de fluorescencia homogénea resuelta en tiempo (HTRF) de unión de PD-1 soluble a PD-L1 soluble. PD-1 soluble y PD-L1 soluble se refieren a proteínas con truncamientos del extremo carboxilo que eliminan las regiones transmembrana y se fusionan con secuencias heterólogas, específicamente la porción Fc de la secuencia G de inmunoglobulina humana (Ig) o la etiqueta del epítopo de hexahistidina (His). Todos los estudios de unión se realizaron en un tampón de ensayo HTRF que consiste en dPBS suplementado con albúmina de suero bovino al 0,1 % (p/v) y Tween-20 al 0,05 % (v/v). Para el ensayo de unión de PD-1-Ig/PD-L1-His, los inhibidores se incubaron previamente con PD-L1-His (10 nM final) durante 15 m en 4 ^l de tampón de ensayo, seguido de la adición de PD-1-Ig (final 20 nM) en 1 ^l de tampón de ensayo y una incubación adicional durante 15 m. Proteínas de fusión de PD-L1 humana, macacos cinomologos, ratón, u otras especies, se utilizaron. La detección de HTRF se logró usando el anticuerpo monoclonal anti-Ig marcado con criptato de europio (1 nM final) y el anticuerpo monoclonal anti-His marcado con aloficocianina (APC) (20 nM final). Los anticuerpos se diluyeron en tampón de detección de HTRF y se
dispensaron 5 |jl en la parte superior de la reacción de unión. La reacción se dejó equilibrar durante 30 minutos y la señal (relación 665 nm/620 nm) se comprobó utilizando un fluorómetro EnVision. Se establecieron ensayos de unión adicionales entre PD-1-Ig/PD-L2-His (20 y 5 nM, respectivamente), CD80-His/PD-L1-Ig (100 y 10 nM, respectivamente) y CD80-His/CTLA4-Ig (10 y 5 nM, respectivamente). Los estudios de unión/competición entre Compuesto n.° 71 biotinilado y PD-L1-His humano se realizaron como sigue. Los inhibidores de péptidos macrocíclicos se preincubaron con PD-L1-His (10 nM final) durante 60 minutos en 4 j l de tampón de ensayo seguido de la adición de Compuesto n.° 71 biotinilado (0,5 nM final) en 1 j l de tampón de ensayo. Se permitió que la unión se equilibrara durante 30 minutos seguido de la adición de estreptavidina marcada con criptato de europio (2,5 pM final) y anti-His marcado con APC (20 nM final) en 5 j l de tampón HTRF. La reacción se dejó equilibrar durante 30 m y se obtuvo una señal (relación 665 nm/620 nm) usando un fluorómetro EnVision.
Proteínas recombinantes. PD-1 humana truncada con carboxilo (aminoácidos 25-167) con una etiqueta de epítopo de Ig humana C-terminal [hPD-1 (25-167)-3S-IG] y PD-L1 humana (aminoácidos 18-239) con una etiqueta de epítopo His C-terminal [hPD-L1 (19-239)-sitio de escisión de proteasa del virus de moteado en venas del tabaco (TVMV)-His] se expresó en células HEK293T y se purificó secuencialmente por cromatografía de afinidad de Proteína A recombinante y cromatografía de exclusión por tamaño. PD-L2-His humana (Sino Biologicals), CD80-His (Sino Biologicals), CTLA4-Ig (RnD Systems) se obtuvieron todas de fuentes comerciales.
Secuencia de PD-1-Ig humana recombinante
hPDÍ (25-167)-3$~IG
l LDSPDRPWWP PTFSPALLW TEGDNATFTCSFSHTSESFV LMWYRM8PSK
51 QTDKLAAFPEDRSOPGQDCR FRVTQLPHGRDFHKSWRAR RIÍDSGTYIíCG
101 AISIAPKAQI KESLEAEIjRVTERKAEVPTAHPSPSPRPAGQPQGSPGGG3
151 GREPKSSDKTHTSPPSPAPELLGGSSVFLF PPKPKDTLMI SRTPEVTCW
201 VDVSHEDPEV KFKWYVDGVEVHNAKTKPRE SQYNSTYRW SVLTVLHQDW
251 LHGKSYKCKV SNKMiPAFIE KTXSKAKGQP REPQVYTLPP SRDELTKNQV
301 SLTCLVKGFY PSDIAVEWES NGQPENNYKTTPPVIiDSDGS FFLYSKLTVB
351 KSRWQQGNV'FSCSVMHEALHNHYTQKSLSL SFGK
(SEQ ID NO:1)
Secuencia de PD-L1-TVMV-His humana recombinante (PD-L1-His)
M*DLl(19-2»)-TVMV-eis
1 FTVTVPKDLY WEYGSNMTI ECKFFVEKQL DLAALIVYWE MEDKNIIQFV
51 HGEEDLKVQH SSYRQRARLL KDQLSLGKAALQITDVKLQD&GVYRCMXSY
101 GGADYKRITV KVNAPYNK1NQRILWDPVT SEHELTCQAE GYPKAEVIWT
151 SSDHQVLSGKTTTTNSKREE KLFNVTSTLR INTTTNEIFYCTFRRLDPEE
201 MHTAELVIPE LPLAHPPNERTGSSETVRFQGHHHHHH
(SEQ ID NO:2)
Los resultados se muestran en la Tabla 1. Como se muestra, los péptidos macrocíclicos de la presente divulgación demostraron una potente inhibición de la actividad de unión de PD-1-Ig a PD-L1-TVMV-His (PD-L1-His). Los intervalos son los siguientes: A = 0,10-10 jM; B = 0,01-0,099 jM; C = 0,001 - 0,0099 jM.
Tabla 1

(continuación)

(continuación)

(continuación)

(continuación)

Claims (17)
1. Un compuesto de fórmula (I)
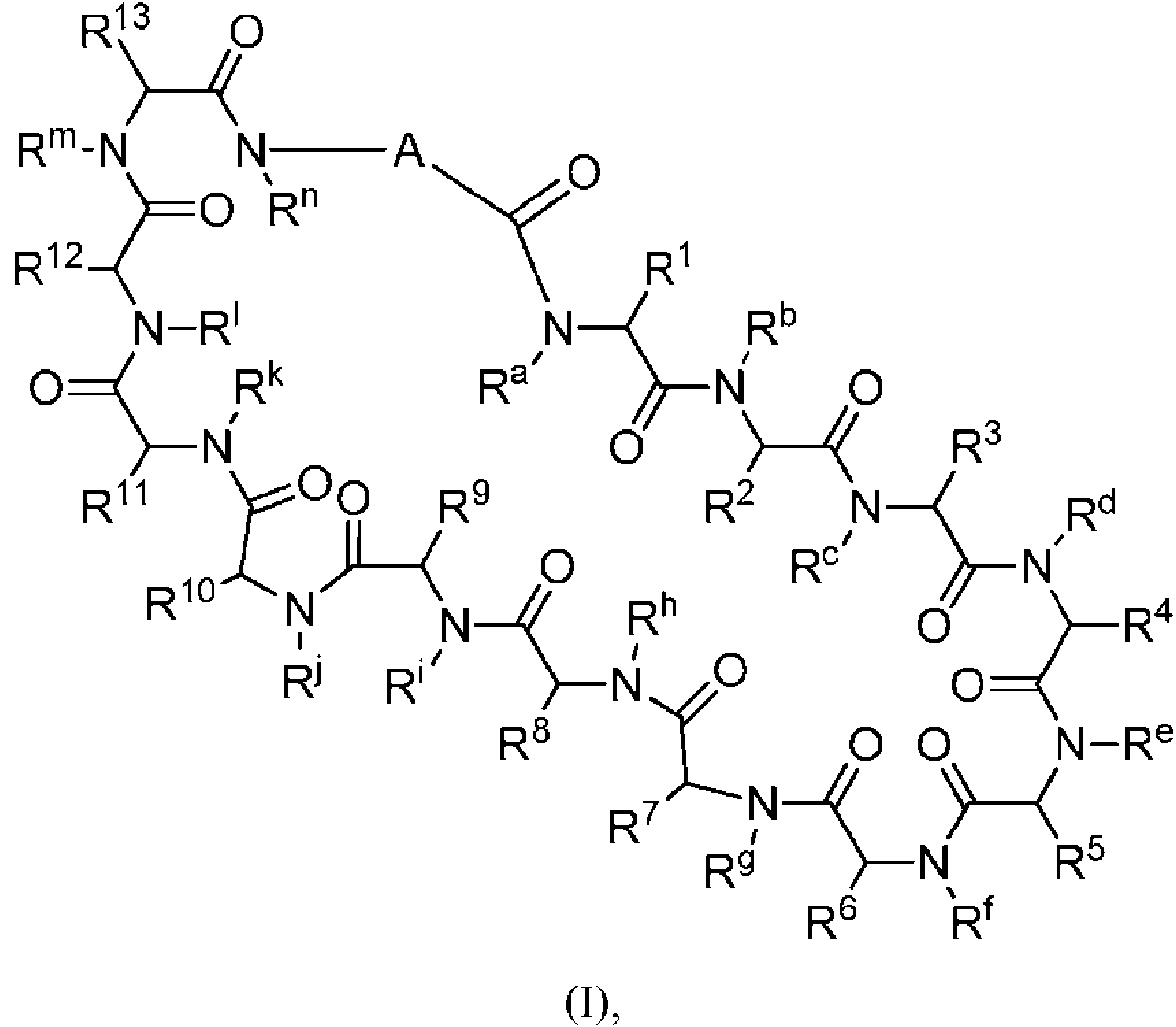
o una sal farmacéuticamente aceptable del mismo, en la que:
A se selecciona entre;

en las que:
representa el punto de unión al grupo carboxilo y
representa el punto de unión al átomo de nitrógeno;
n es 0, 1 o 2 ;
m es 1 o 2 ;
p es 0, 1 o 2 ;
R14 y R15 se seleccionan independientemente entre hidrógeno y metilo:
Rz se selecciona entre hidrógeno y -C(O)NHR16; en donde R16 se selecciona entre hidrógeno, -CHR17C(O)NH2, -CHR17C(O)NHCHR18C(O)NH2 y -CHR17C(O)NHCHR18C(O)NHCH2C(O)NH2; en donde R17 se selecciona entre hidrógeno y -CH2OH y en donde R18 se selecciona entre hidrógeno y metilo;
Rv es hidrógeno, metilo o una cadena lateral de aminoácido natural;
Rc, Rf, Rh, Ri y Rm son hidrógeno;
Rn es hidrógeno o metilo o Rv y Rn forman un anillo de pirrolidina;
Ra, Re y Rj, se seleccionan cada uno independientemente entre hidrógeno y metilo;
Rk es metilo;
R1, R2, R3, R5, R6, R8, R9, R10, R11, R12 y R13 se seleccionan independientemente entre una cadena lateral de aminoácido natural y una cadena lateral de aminoácido no natural o R2 o R12 forman un anillo con el grupo R vecinal correspondiente como se describe más adelante;
Rb es metilo o, Rb y R2, junto con los átomos a los que están unidos, forman un anillo seleccionado entre azetidina, pirrolidina, morfolina, piperidina, piperazina y tetrahidrotiazol; en donde cada anillo está opcionalmente sustituido con uno a cuatro grupos seleccionados independientemente entre amino, ciano, metilo, halo, e hidroxi; Rd y R4, junto con los átomos a los que están unidos, forman un anillo de pirrolidina;
Rg y R7, junto con los átomos a los que están unidos, forman un anillo de pirrolidina que está opcionalmente sustituido con un grupo hidroxilo; y
Rl es metilo o, RL y R12, junto con los átomos a los que están unidos, forman un anillo seleccionado entre azetidina y pirrolidina, en donde cada anillo está opcionalmente sustituido con uno a cuatro grupos seleccionados independientemente entre amino, ciano, metilo, halo e hidroxi.
2. Un compuesto de la reivindicación 1, o una sal terapéuticamente aceptable del mismo, en donde:
Ra, Rey Rj son hidrógeno;
Rb y R2 son cada uno metilo o, Rb y R2, junto con los átomos a los que
están unidos, forman un anillo de piperidina;
Rl es metilo;
Rn es hidrógeno, metilo, o Rn y Rv forman un anillo de pirrolidina;
R1 es fenilmetilo en donde el fenilo está sustituido con un grupo seleccionado entre halo, hidroxi, metoxi o metilo; R3 se selecciona entre -CH2C(O)NH2 y -CH2CO2H;
R5 se selecciona entre hidrógeno, -CH2NH2, -CH2(imidazolilo) y -CH2C(O)NH2;
R6 se selecciona entre -CH2CH(CH3)2, -(CH2)4NH2, -(CH2)2CO2H y (CH2)2C(O)NH2;
R8 y R10 son -CH2(indolilo), en donde el indolilo está opcionalmente sustituido con -CH2CO2H;
R9 se selecciona entre hidrógeno, -(CH2)2NH2, -(CH2)4NH2, -CH2OH y -CH2C(O)NH2;
R11 y R12 son -(CH2)3CH3; y
R13 se selecciona entre metilo, -CH2OH, -CH2CH(CH3)2 y -(CH2)2CO2H.
3. Un compuesto de la reivindicación 2, o una sal terapéuticamente aceptable del mismo, en donde A es

4. Un compuesto de la reivindicación 2, o una sal terapéuticamente aceptable del mismo, en donde A es

5. Un compuesto según la reivindicación 1 que se selecciona entre:

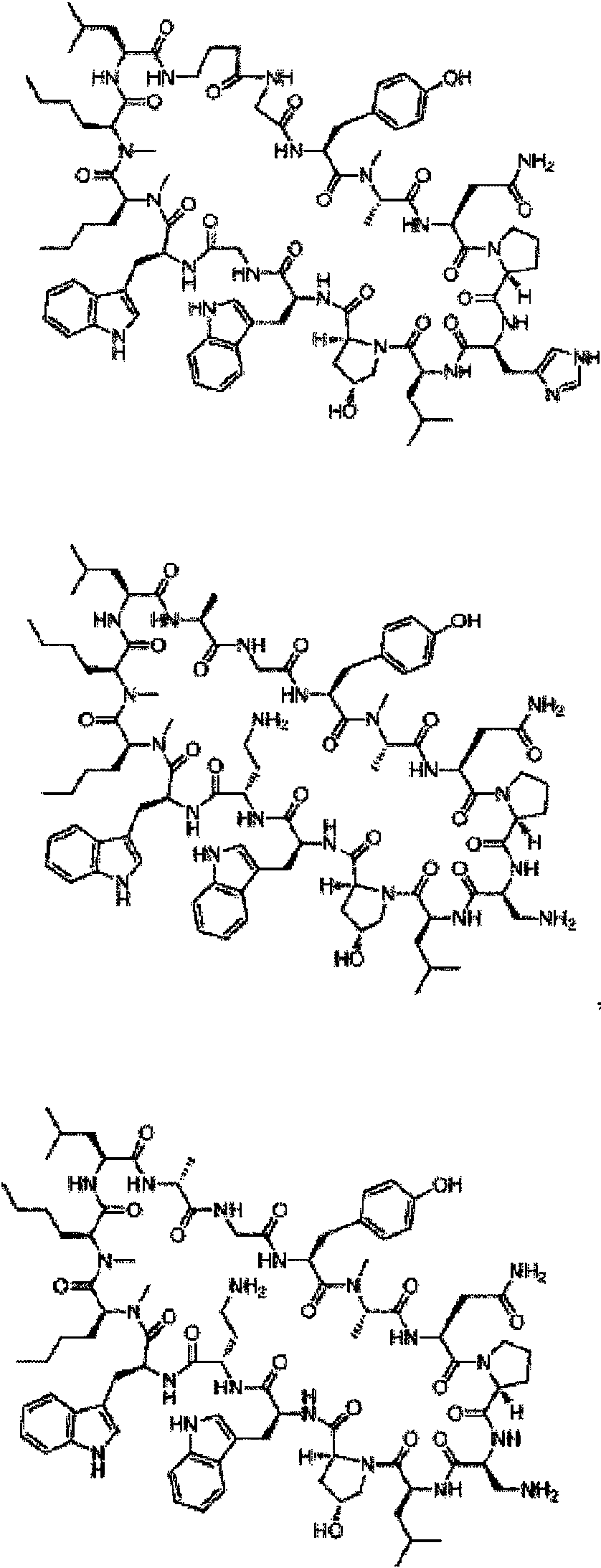





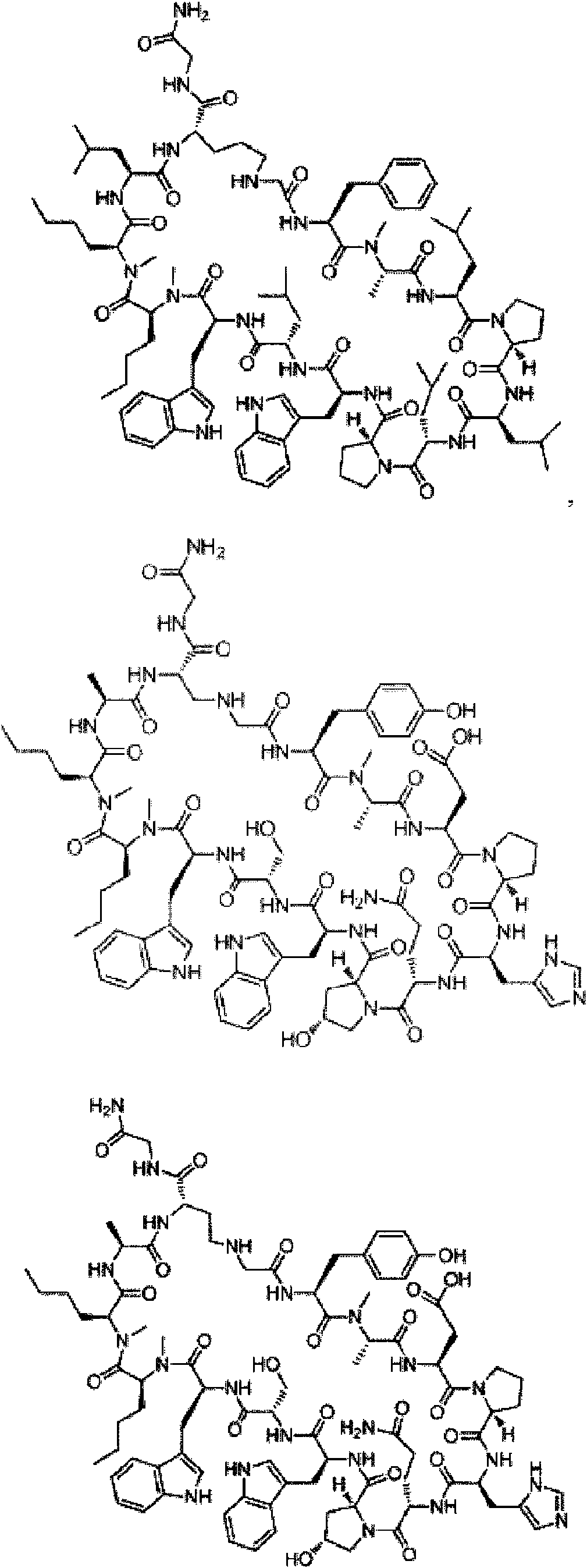




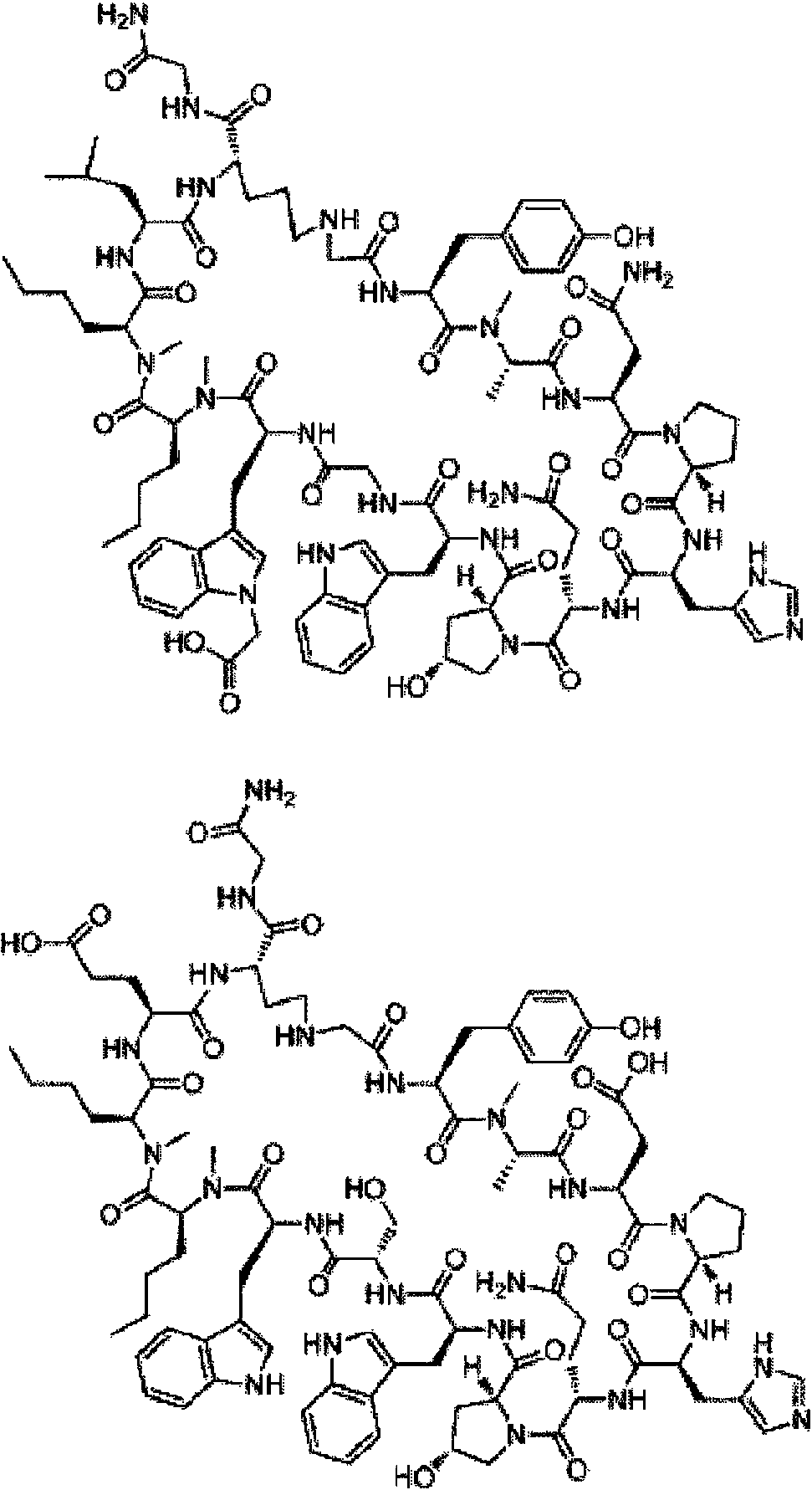


































o una sal farmacéuticamente aceptable del mismo.
6. Un compuesto de la reivindicación 1 o una sal terapéuticamente aceptable del mismo para uso en mejorar, estimular y/o aumentar la respuesta inmune en un sujeto que lo necesita.
7. Un compuesto o una sal terapéuticamente aceptable del mismo para uso según la reivindicación 6 que comprende además administrar un agente adicional antes de, después de, o simultáneamente con el compuesto de la reivindicación 1 o una sal terapéuticamente aceptable del mismo.
8. Un compuesto o una sal terapéuticamente aceptable del mismo para uso según la reivindicación 7 en donde el agente adicional es un agente antimicrobiano, un agente antivírico, un agente citotóxico y/o un modificador de la respuesta inmunitaria.
9. Un compuesto o una sal terapéuticamente aceptable del mismo para uso según la reivindicación 7 en donde el agente adicional es un inhibidor de HDAC.
10. Un compuesto o una sal terapéuticamente aceptable del mismo para uso según la reivindicación 7 en donde el agente adicional es un agonista de TLR7 y/o TLR8.
11. Un compuesto de la reivindicación 1 o una sal terapéuticamente aceptable del mismo para uso en inhibir crecimiento, proliferación, o metástasis de células cancerosas en un sujeto que lo necesita.
12. Un compuesto o una sal terapéuticamente aceptable del mismo para uso según la reivindicación 11 en donde el cáncer se selecciona entre melanoma, carcinoma de células renales, cáncer de pulmón no microcítico escamoso
(CPNM), CPNM no escamoso, cáncer colorrectal, cáncer de próstata resistente a la castración, cáncer de ovario, cáncer gástrico, carcinoma hepatocelular, carcinoma de páncreas, carcinoma de células escamosas de cabeza y cuello, carcinomas del esófago, tracto gastrointestinal y mama y tumores malignos hematológicos.
13. Un compuesto de la reivindicación 1 o una sal terapéuticamente aceptable del mismo para uso en el tratamiento de una enfermedad infecciosa en un sujeto que lo necesita.
14. Un compuesto o una sal terapéuticamente aceptable del mismo para uso según la reivindicación 13 en donde la enfermedad infecciosa está causada por un virus.
15. Un compuesto o una sal terapéuticamente aceptable del mismo para uso según la reivindicación 14 en donde el virus se selecciona entre VIH, Hepatitis A, Hepatitis B, Hepatitis C, herpes virus e influenza.
16. Un compuesto de la reivindicación 1 o una sal terapéuticamente aceptable del mismo para uso en el tratamiento de choque séptico en un sujeto que lo necesita.
17. Un compuesto de la reivindicación 1 o una sal terapéuticamente aceptable del mismo para uso en bloquear la interacción de PD-L1 con PD-1 y/o CD80 en un sujeto.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462062240P | 2014-10-10 | 2014-10-10 | |
| PCT/US2015/054407 WO2016057624A1 (en) | 2014-10-10 | 2015-10-07 | Immunomodulators |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2793018T3 true ES2793018T3 (es) | 2020-11-12 |
Family
ID=54330101
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15781564T Active ES2793018T3 (es) | 2014-10-10 | 2015-10-07 | Inmunomoduladores |
Country Status (20)
| Country | Link |
|---|---|
| US (1) | US9732119B2 (es) |
| EP (1) | EP3204403B1 (es) |
| JP (1) | JP6718439B2 (es) |
| KR (1) | KR102538436B1 (es) |
| CN (1) | CN107001424B (es) |
| AR (1) | AR102350A1 (es) |
| AU (1) | AU2015328136A1 (es) |
| BR (1) | BR112017006980A2 (es) |
| CA (1) | CA2963930A1 (es) |
| CL (1) | CL2017000855A1 (es) |
| CO (1) | CO2017004535A2 (es) |
| EA (1) | EA201790695A1 (es) |
| ES (1) | ES2793018T3 (es) |
| IL (1) | IL251589A0 (es) |
| MX (1) | MX2017004542A (es) |
| PE (1) | PE20170701A1 (es) |
| SG (1) | SG11201702792SA (es) |
| TW (1) | TW201629088A (es) |
| WO (1) | WO2016057624A1 (es) |
| ZA (1) | ZA201702484B (es) |
Families Citing this family (75)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9328116B2 (en) | 2012-12-07 | 2016-05-03 | Chemocentryx, Inc. | Diazole lactams |
| CA2960778C (en) | 2014-09-11 | 2023-03-07 | Bristol-Myers Squibb Company | Macrocyclic inhibitors of the pd-1/pd-l1 and cd80(b7-1)/pd-l1 protein/protein interactions |
| US9856292B2 (en) | 2014-11-14 | 2018-01-02 | Bristol-Myers Squibb Company | Immunomodulators |
| US9861680B2 (en) | 2014-12-18 | 2018-01-09 | Bristol-Myers Squibb Company | Immunomodulators |
| US9944678B2 (en) | 2014-12-19 | 2018-04-17 | Bristol-Myers Squibb Company | Immunomodulators |
| US11352642B2 (en) | 2015-01-09 | 2022-06-07 | Etubics Corporation | Methods and compositions for combination immunotherapy |
| US20160222060A1 (en) | 2015-02-04 | 2016-08-04 | Bristol-Myers Squibb Company | Immunomodulators |
| US9809625B2 (en) | 2015-03-18 | 2017-11-07 | Bristol-Myers Squibb Company | Immunomodulators |
| WO2016172249A1 (en) * | 2015-04-20 | 2016-10-27 | Etubics Corporation | Methods and compositions for combination immunotherapy |
| US10143746B2 (en) | 2016-03-04 | 2018-12-04 | Bristol-Myers Squibb Company | Immunomodulators |
| US10358463B2 (en) * | 2016-04-05 | 2019-07-23 | Bristol-Myers Squibb Company | Immunomodulators |
| CA3019391A1 (en) | 2016-04-07 | 2017-10-12 | Chemocentryx, Inc. | Reducing tumor burden by administering ccr1 antagonists in combination with pd-1 inhibitors or pd-l1 inhibitors |
| TWI794171B (zh) | 2016-05-11 | 2023-03-01 | 美商滬亞生物國際有限公司 | Hdac抑制劑與pd-l1抑制劑之組合治療 |
| TWI808055B (zh) | 2016-05-11 | 2023-07-11 | 美商滬亞生物國際有限公司 | Hdac 抑制劑與 pd-1 抑制劑之組合治療 |
| ES2864091T3 (es) | 2016-05-19 | 2021-10-13 | Bristol Myers Squibb Co | Inmunomoduladores de producción de imágenes de tomografía por emisión de positrones (PET) |
| EP3526323B1 (en) | 2016-10-14 | 2023-03-29 | Precision Biosciences, Inc. | Engineered meganucleases specific for recognition sequences in the hepatitis b virus genome |
| US10988507B2 (en) | 2016-11-07 | 2021-04-27 | Bristol-Myers Squibb Company | Immunomodulators |
| CN106963948A (zh) * | 2017-05-12 | 2017-07-21 | 顾艳宏 | 阿帕替尼与Anti‑PD‑1抗体联用在制备结肠癌药物中的应用 |
| US11066445B2 (en) * | 2017-06-23 | 2021-07-20 | Bristol-Myers Squibb Company | Immunomodulators acting as antagonists of PD-1 |
| KR20200110300A (ko) | 2017-09-25 | 2020-09-23 | 케모센트릭스, 인크. | 케모카인 수용체 2(ccr2) 길항제 및 pd-1/pd-l1 억제제를 사용하는 병용 요법 |
| CN111051332A (zh) | 2017-10-03 | 2020-04-21 | 百时美施贵宝公司 | 免疫调节剂 |
| CA3084582A1 (en) | 2017-12-20 | 2019-06-27 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3' cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| EP3728283B1 (en) | 2017-12-20 | 2023-11-22 | Institute of Organic Chemistry and Biochemistry ASCR, V.V.I. | 3'3' cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| US20190269664A1 (en) | 2018-01-08 | 2019-09-05 | Chemocentryx, Inc. | Methods of treating solid tumors with ccr2 antagonists |
| WO2019136368A2 (en) | 2018-01-08 | 2019-07-11 | Chemocentryx, Inc. | Methods of treating solid tumors with ccr2 antagonists |
| CN111788204B (zh) | 2018-02-26 | 2023-05-05 | 吉利德科学公司 | 作为hbv复制抑制剂的取代吡咯嗪化合物 |
| EP3774883A1 (en) | 2018-04-05 | 2021-02-17 | Gilead Sciences, Inc. | Antibodies and fragments thereof that bind hepatitis b virus protein x |
| TW202005654A (zh) | 2018-04-06 | 2020-02-01 | 捷克科學院有機化學與生物化學研究所 | 2,2,─環二核苷酸 |
| KR20200140867A (ko) | 2018-04-06 | 2020-12-16 | 인스티튜트 오브 오가닉 케미스트리 앤드 바이오케미스트리 에이에스 씨알 브이.브이.아이. | 3'3'-사이클릭 다이뉴클레오티드 |
| TWI818007B (zh) | 2018-04-06 | 2023-10-11 | 捷克科學院有機化學與生物化學研究所 | 2'3'-環二核苷酸 |
| TW201945388A (zh) | 2018-04-12 | 2019-12-01 | 美商精密生物科學公司 | 對b型肝炎病毒基因體中之識別序列具有特異性之最佳化之經工程化巨核酸酶 |
| US20190359645A1 (en) | 2018-05-03 | 2019-11-28 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotides comprising carbocyclic nucleotide |
| BR112020024404A2 (pt) | 2018-05-31 | 2021-03-02 | Ono Pharmaceutical Co., Ltd. | biomarcadores para determinar a eficácia de inibidores de checkpoint imunológico |
| WO2020028097A1 (en) | 2018-08-01 | 2020-02-06 | Gilead Sciences, Inc. | Solid forms of (r)-11-(methoxymethyl)-12-(3-methoxypropoxy)-3,3-dimethyl-8-0x0-2,3,8,13b-tetrahydro-1h-pyrido[2,1-a]pyrrolo[1,2-c] phthalazine-7-c arboxylic acid |
| TW202028212A (zh) | 2018-10-11 | 2020-08-01 | 日商小野藥品工業股份有限公司 | Sting促效化合物 |
| CN117105933A (zh) | 2018-10-31 | 2023-11-24 | 吉利德科学公司 | 具有hpk1抑制活性的取代的6-氮杂苯并咪唑化合物 |
| KR102650496B1 (ko) | 2018-10-31 | 2024-03-26 | 길리애드 사이언시즈, 인코포레이티드 | Hpk1 억제제로서의 치환된 6-아자벤즈이미다졸 화합물 |
| JP7350871B2 (ja) | 2019-03-07 | 2023-09-26 | インスティチュート オブ オーガニック ケミストリー アンド バイオケミストリー エーエスシーアール,ヴイ.ヴイ.アイ. | 2’3’-環状ジヌクレオチドおよびそのプロドラッグ |
| US11766447B2 (en) | 2019-03-07 | 2023-09-26 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3′3′-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide as sting modulator |
| EP3935066A1 (en) | 2019-03-07 | 2022-01-12 | Institute of Organic Chemistry and Biochemistry ASCR, V.V.I. | 3'3'-cyclic dinucleotides and prodrugs thereof |
| KR20210146348A (ko) | 2019-03-28 | 2021-12-03 | 브리스톨-마이어스 스큅 컴퍼니 | 종양을 치료하는 방법 |
| KR20210146349A (ko) | 2019-03-28 | 2021-12-03 | 브리스톨-마이어스 스큅 컴퍼니 | 종양을 치료하는 방법 |
| TW202210480A (zh) | 2019-04-17 | 2022-03-16 | 美商基利科學股份有限公司 | 類鐸受體調節劑之固體形式 |
| TW202212339A (zh) | 2019-04-17 | 2022-04-01 | 美商基利科學股份有限公司 | 類鐸受體調節劑之固體形式 |
| CN111909240B (zh) * | 2019-05-10 | 2023-10-24 | 中国药科大学 | 一种pd-1/pd-l1多肽类抑制剂及其医药用途 |
| KR20220010535A (ko) * | 2019-05-21 | 2022-01-25 | 브리스톨-마이어스 스큅 컴퍼니 | 면역조정제 |
| WO2020237025A1 (en) | 2019-05-23 | 2020-11-26 | Gilead Sciences, Inc. | Substituted exo-methylene-oxindoles which are hpk1/map4k1 inhibitors |
| US20220363760A1 (en) | 2019-05-30 | 2022-11-17 | Bristol-Myers Squibb Company | Multi-tumor gene signature for suitability to immuno-oncology therapy |
| KR20220016155A (ko) | 2019-05-30 | 2022-02-08 | 브리스톨-마이어스 스큅 컴퍼니 | 면역-종양학 (i-o) 요법에 적합한 대상체를 확인하는 방법 |
| CN114174537A (zh) | 2019-05-30 | 2022-03-11 | 百时美施贵宝公司 | 细胞定位特征和组合疗法 |
| EP4011391A4 (en) | 2019-08-05 | 2023-08-16 | ONO Pharmaceutical Co., Ltd. | BIOMARKER FOR ASSESSING THE EFFECTIVENESS OF AN IMMUNE CHECKPOINT INHIBITOR |
| WO2021034804A1 (en) | 2019-08-19 | 2021-02-25 | Gilead Sciences, Inc. | Pharmaceutical formulations of tenofovir alafenamide |
| KR20220066334A (ko) | 2019-09-22 | 2022-05-24 | 브리스톨-마이어스 스큅 컴퍼니 | Lag-3 길항제 요법에 대한 정량적 공간 프로파일링 |
| BR112022005687A2 (pt) | 2019-09-30 | 2022-06-21 | Gilead Sciences Inc | Vacinas contra o hbv e métodos para tratar o hbv |
| JP2022553851A (ja) | 2019-11-08 | 2022-12-26 | ブリストル-マイヤーズ スクイブ カンパニー | 黒色腫の処置のためのlag-3アンタゴニスト |
| US20230031465A1 (en) | 2019-12-06 | 2023-02-02 | Precision Biosciences, Inc. | Optimized engineered meganucleases having specificity for a recognition sequence in the hepatitis b virus genome |
| KR20220156884A (ko) | 2020-03-20 | 2022-11-28 | 길리애드 사이언시즈, 인코포레이티드 | 4'-c-치환된-2-할로-2'-데옥시아데노신 뉴클레오시드의 프로드러그 및 이의 제조 및 사용 방법 |
| US20230151024A1 (en) | 2020-04-10 | 2023-05-18 | Ono Pharmaceutical Co., Ltd. | Sting agonistic compound |
| JPWO2021206158A1 (es) | 2020-04-10 | 2021-10-14 | ||
| WO2022047189A1 (en) | 2020-08-28 | 2022-03-03 | Bristol-Myers Squibb Company | Lag-3 antagonist therapy for hepatocellular carcinoma |
| AU2021334361A1 (en) | 2020-08-31 | 2023-05-11 | Bristol-Myers Squibb Company | Cell localization signature and immunotherapy |
| AU2021347147A1 (en) | 2020-09-22 | 2023-05-18 | Barinthus Biotherapeutics North America, Inc. | Compositions and methods of manufacturing amphiphilic block copolymers that form nanoparticles in situ |
| CA3196496A1 (en) | 2020-10-23 | 2022-04-28 | Laurence David TOMS | Lag-3 antagonist therapy for lung cancer |
| WO2022120179A1 (en) | 2020-12-03 | 2022-06-09 | Bristol-Myers Squibb Company | Multi-tumor gene signatures and uses thereof |
| WO2022212400A1 (en) | 2021-03-29 | 2022-10-06 | Juno Therapeutics, Inc. | Methods for dosing and treatment with a combination of a checkpoint inhibitor therapy and a car t cell therapy |
| JP2024518558A (ja) | 2021-05-13 | 2024-05-01 | ギリアード サイエンシーズ, インコーポレイテッド | TLR8調節化合物と抗HBV siRNA治療薬との組合せ |
| CA3222752A1 (en) | 2021-06-11 | 2022-12-15 | Gilead Sciences, Inc. | Combination mcl-1 inhibitors with anti-body drug conjugates |
| CA3222269A1 (en) | 2021-06-11 | 2022-12-15 | Gilead Sciences, Inc. | Combination mcl-1 inhibitors with anti-cancer agents |
| EP4359411A1 (en) | 2021-06-23 | 2024-05-01 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| EP4359389A1 (en) | 2021-06-23 | 2024-05-01 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| CN117396478A (zh) | 2021-06-23 | 2024-01-12 | 吉利德科学公司 | 二酰基甘油激酶调节化合物 |
| CN117355531A (zh) | 2021-06-23 | 2024-01-05 | 吉利德科学公司 | 二酰基甘油激酶调节化合物 |
| WO2023077090A1 (en) | 2021-10-29 | 2023-05-04 | Bristol-Myers Squibb Company | Lag-3 antagonist therapy for hematological cancer |
| WO2023147371A1 (en) | 2022-01-26 | 2023-08-03 | Bristol-Myers Squibb Company | Combination therapy for hepatocellular carcinoma |
| US20230326022A1 (en) | 2022-04-08 | 2023-10-12 | Bristol-Myers Squibb Company | Machine Learning Identification, Classification, and Quantification of Tertiary Lymphoid Structures |
Family Cites Families (46)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4475196A (en) | 1981-03-06 | 1984-10-02 | Zor Clair G | Instrument for locating faults in aircraft passenger reading light and attendant call control system |
| US4447233A (en) | 1981-04-10 | 1984-05-08 | Parker-Hannifin Corporation | Medication infusion pump |
| US4439196A (en) | 1982-03-18 | 1984-03-27 | Merck & Co., Inc. | Osmotic drug delivery system |
| US4522811A (en) | 1982-07-08 | 1985-06-11 | Syntex (U.S.A.) Inc. | Serial injection of muramyldipeptides and liposomes enhances the anti-infective activity of muramyldipeptides |
| US4447224A (en) | 1982-09-20 | 1984-05-08 | Infusaid Corporation | Variable flow implantable infusion apparatus |
| US4487603A (en) | 1982-11-26 | 1984-12-11 | Cordis Corporation | Implantable microinfusion pump system |
| US4486194A (en) | 1983-06-08 | 1984-12-04 | James Ferrara | Therapeutic device for administering medicaments through the skin |
| US4596556A (en) | 1985-03-25 | 1986-06-24 | Bioject, Inc. | Hypodermic injection apparatus |
| US5374548A (en) | 1986-05-02 | 1994-12-20 | Genentech, Inc. | Methods and compositions for the attachment of proteins to liposomes using a glycophospholipid anchor |
| MX9203291A (es) | 1985-06-26 | 1992-08-01 | Liposome Co Inc | Metodo para acoplamiento de liposomas. |
| US4941880A (en) | 1987-06-19 | 1990-07-17 | Bioject, Inc. | Pre-filled ampule and non-invasive hypodermic injection device assembly |
| US4790824A (en) | 1987-06-19 | 1988-12-13 | Bioject, Inc. | Non-invasive hypodermic injection device |
| US5108921A (en) | 1989-04-03 | 1992-04-28 | Purdue Research Foundation | Method for enhanced transmembrane transport of exogenous molecules |
| US5312335A (en) | 1989-11-09 | 1994-05-17 | Bioject Inc. | Needleless hypodermic injection device |
| US5064413A (en) | 1989-11-09 | 1991-11-12 | Bioject, Inc. | Needleless hypodermic injection device |
| US5383851A (en) | 1992-07-24 | 1995-01-24 | Bioject Inc. | Needleless hypodermic injection device |
| US6410690B1 (en) | 1995-06-07 | 2002-06-25 | Medarex, Inc. | Therapeutic compounds comprised of anti-Fc receptor antibodies |
| US5869451A (en) | 1995-06-07 | 1999-02-09 | Glaxo Group Limited | Peptides and compounds that bind to a receptor |
| US5811097A (en) | 1995-07-25 | 1998-09-22 | The Regents Of The University Of California | Blockade of T lymphocyte down-regulation associated with CTLA-4 signaling |
| US5922845A (en) | 1996-07-11 | 1999-07-13 | Medarex, Inc. | Therapeutic multispecific compounds comprised of anti-Fcα receptor antibodies |
| US6277818B1 (en) | 1998-10-29 | 2001-08-21 | Angstrom Pharmaceuticals, Inc. | Cyclic peptide ligands that target urokinase plasminogen activator receptor |
| CA2508660C (en) | 2002-12-23 | 2013-08-20 | Wyeth | Antibodies against pd-1 and uses therefor |
| SI2439273T1 (sl) | 2005-05-09 | 2019-05-31 | Ono Pharmaceutical Co., Ltd. | Človeška monoklonska protitelesa za programirano smrt 1 (PD-1) in postopki za zdravljenje raka z uporabo protiteles proti PD-1 samostojno ali v kombinaciji z ostalimi imunoterapevtiki |
| CN101248089A (zh) | 2005-07-01 | 2008-08-20 | 米德列斯公司 | 抗程序性死亡配体1(pd-l1)的人单克隆抗体 |
| EP3012265B1 (en) | 2007-03-26 | 2017-06-28 | The University of Tokyo | Process for synthesizing cyclic peptide compound |
| EP2262837A4 (en) | 2008-03-12 | 2011-04-06 | Merck Sharp & Dohme | PD-1 BINDING PROTEINS |
| JP2012510429A (ja) | 2008-08-25 | 2012-05-10 | アンプリミューン、インコーポレーテッド | Pd−1アンタゴニストおよびその使用方法 |
| US8907053B2 (en) | 2010-06-25 | 2014-12-09 | Aurigene Discovery Technologies Limited | Immunosuppression modulating compounds |
| JP5818237B2 (ja) | 2010-09-09 | 2015-11-18 | 国立大学法人 東京大学 | N−メチルアミノ酸およびその他の特殊アミノ酸を含む特殊ペプチド化合物ライブラリーの翻訳構築と活性種探索法 |
| EP2647720B1 (en) | 2010-12-03 | 2019-06-19 | The University of Tokyo | Peptide library production method, peptide library, and screening method |
| EP2717895A1 (en) | 2011-06-08 | 2014-04-16 | Aurigene Discovery Technologies Limited | Therapeutic compounds for immunomodulation |
| AU2013239366A1 (en) | 2012-03-29 | 2014-10-16 | Aurigene Discovery Technologies Limited | Immunomodulating cyclic compounds from the BC loop of human PD1 |
| KR20150029678A (ko) | 2012-06-06 | 2015-03-18 | 바이오노르 이뮤노 에이에스 | 백신 |
| JP6047231B2 (ja) | 2012-06-06 | 2016-12-21 | ポリフォー・アクチェンゲゼルシャフトPolyphor Ag | β−ヘアピンペプチド模倣薬 |
| JP2013253842A (ja) | 2012-06-06 | 2013-12-19 | Univ Of Tokyo | pH依存的に標的分子に結合するペプチドのスクリーニング方法 |
| US9308236B2 (en) | 2013-03-15 | 2016-04-12 | Bristol-Myers Squibb Company | Macrocyclic inhibitors of the PD-1/PD-L1 and CD80(B7-1)/PD-L1 protein/protein interactions |
| KR102158924B1 (ko) | 2013-03-15 | 2020-09-22 | 제넨테크, 인크. | Pd-1 및 pd-l1 관련 상태를 치료하기 위한 바이오마커 및 방법 |
| EA031325B1 (ru) | 2013-09-06 | 2018-12-28 | Ауриген Дискавери Текнолоджиз Лимитед | Циклические пептидомиметические соединения в качестве иммуномодуляторов |
| WO2015044900A1 (en) | 2013-09-27 | 2015-04-02 | Aurigene Discovery Technologies Limited | Therapeutic immunomodulating compounds |
| CN103724290B (zh) * | 2013-11-15 | 2015-04-29 | 浙江大学 | 一种环肽化合物clavatustide A及其产生菌、制备方法和应用 |
| CA2960778C (en) | 2014-09-11 | 2023-03-07 | Bristol-Myers Squibb Company | Macrocyclic inhibitors of the pd-1/pd-l1 and cd80(b7-1)/pd-l1 protein/protein interactions |
| US9856292B2 (en) | 2014-11-14 | 2018-01-02 | Bristol-Myers Squibb Company | Immunomodulators |
| US9861680B2 (en) | 2014-12-18 | 2018-01-09 | Bristol-Myers Squibb Company | Immunomodulators |
| US9944678B2 (en) | 2014-12-19 | 2018-04-17 | Bristol-Myers Squibb Company | Immunomodulators |
| US20160222060A1 (en) | 2015-02-04 | 2016-08-04 | Bristol-Myers Squibb Company | Immunomodulators |
| US9809625B2 (en) | 2015-03-18 | 2017-11-07 | Bristol-Myers Squibb Company | Immunomodulators |
-
2015
- 2015-10-05 US US14/874,886 patent/US9732119B2/en active Active
- 2015-10-07 KR KR1020177012216A patent/KR102538436B1/ko active IP Right Grant
- 2015-10-07 ES ES15781564T patent/ES2793018T3/es active Active
- 2015-10-07 WO PCT/US2015/054407 patent/WO2016057624A1/en active Application Filing
- 2015-10-07 CA CA2963930A patent/CA2963930A1/en not_active Abandoned
- 2015-10-07 SG SG11201702792SA patent/SG11201702792SA/en unknown
- 2015-10-07 AU AU2015328136A patent/AU2015328136A1/en not_active Abandoned
- 2015-10-07 MX MX2017004542A patent/MX2017004542A/es unknown
- 2015-10-07 CN CN201580066937.1A patent/CN107001424B/zh active Active
- 2015-10-07 JP JP2017519313A patent/JP6718439B2/ja active Active
- 2015-10-07 BR BR112017006980A patent/BR112017006980A2/pt not_active Application Discontinuation
- 2015-10-07 EA EA201790695A patent/EA201790695A1/ru unknown
- 2015-10-07 PE PE2017000575A patent/PE20170701A1/es unknown
- 2015-10-07 EP EP15781564.8A patent/EP3204403B1/en active Active
- 2015-10-08 TW TW104133278A patent/TW201629088A/zh unknown
- 2015-10-09 AR ARP150103279A patent/AR102350A1/es unknown
-
2017
- 2017-04-05 IL IL251589A patent/IL251589A0/en unknown
- 2017-04-07 CL CL2017000855A patent/CL2017000855A1/es unknown
- 2017-04-07 ZA ZA2017/02484A patent/ZA201702484B/en unknown
- 2017-05-05 CO CONC2017/0004535A patent/CO2017004535A2/es unknown
Also Published As
| Publication number | Publication date |
|---|---|
| CN107001424A (zh) | 2017-08-01 |
| TW201629088A (zh) | 2016-08-16 |
| CN107001424B (zh) | 2021-03-05 |
| KR20170064548A (ko) | 2017-06-09 |
| IL251589A0 (en) | 2017-06-29 |
| JP2017536344A (ja) | 2017-12-07 |
| MX2017004542A (es) | 2017-06-23 |
| CO2017004535A2 (es) | 2017-08-31 |
| AR102350A1 (es) | 2017-02-22 |
| PE20170701A1 (es) | 2017-06-05 |
| KR102538436B1 (ko) | 2023-05-30 |
| AU2015328136A1 (en) | 2017-05-25 |
| US9732119B2 (en) | 2017-08-15 |
| ZA201702484B (en) | 2019-06-26 |
| EA201790695A1 (ru) | 2017-08-31 |
| SG11201702792SA (en) | 2017-05-30 |
| JP6718439B2 (ja) | 2020-07-08 |
| US20160102122A1 (en) | 2016-04-14 |
| BR112017006980A2 (pt) | 2017-12-12 |
| EP3204403B1 (en) | 2020-04-08 |
| WO2016057624A1 (en) | 2016-04-14 |
| CA2963930A1 (en) | 2016-04-14 |
| EP3204403A1 (en) | 2017-08-16 |
| CL2017000855A1 (es) | 2017-12-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2793018T3 (es) | Inmunomoduladores | |
| ES2910832T3 (es) | Inmunomoduladores | |
| US11952434B2 (en) | Immunomodulators | |
| ES2886635T3 (es) | Inmunomoduladores | |
| ES2910657T3 (es) | Inmunomoduladores | |
| ES2719135T3 (es) | Inmunomoduladores | |
| ES2940632T3 (es) | Inmunomoduladores | |
| ES2937286T3 (es) | Inmunomoduladores | |
| ES2768610T3 (es) | Inhibidores macrocíclicos de las interacciones proteína/proteína PD-1/PD-L1 y CD80(B7-1)/PD-L1 | |
| WO2019070643A1 (en) | IMMUNOMODULATORS |