-
BEREICH DER ERFINDUNG
-
Die
vorliegende Erfindung betrifft Somatostatinanaloga mit cyclischen
Gerüst
mit erzwungener Konformation, die über neue Bindungen cyclisiert
sind und pharmazeutische Zusammensetzungen, die dieselben enthalten.
-
HINTERGRUND DER ERFINDUNG
-
Somatostatinanaloga
-
Somatostatin
ist ein cyclisches Tetradecapeptid, das sowohl im zentralen Nervensystem
als auch in periphären
Geweben gefunden wird. Es wurde ursprünglich isoliert aus Sängerhypothalamus
und identifiziert als ein wichtiger Inhibitor der Wachstumshormonsekretion
des Hypophysenvorderlappens. Seine vielfachen biologischen Aktivitäten umfassen
Inhibierung der Sekretion von Glucagon und Insulin des Pankreas,
Regulation der meisten Darmhormone und Regulation der Freisetzung
anderer Neurotransmitter, die beteiligt sind an motorischen Aktivitäts- und
Wahrnehmungsprozessen im zentralen Nervensystem (siehe zur Übersicht
Lamberts, Endocrine Rev., 9:427, 1988). Zusätzlich sind Somatostatin und
seine Analoga potenziell geeignete Antiproliferationsmittel für die Behandlung
verschiedener Typen von Tumoren.
-
Natürliches
Somatostatin (ebenfalls bekannt als Somatotropin Release Inhibiting
Factor, SRIF (Somatotropinfreisetzungsinhibierungsfaktor)) mit der
folgenden Struktur:
H-Ala1-Gly2-Cys3-Lys4-Asn5-Phe6-Phe7-Trp8-Lys9-Thr10-Phe11-Thr12-Ser13-Cys14-OH
wurde
zum ersten mal isoliert von Guillemin und Kollegen (Bruzeau et al.
Science, 179:78, 1973). Es übt
seine Wirkungen durch Wechselwirkung mit einer Familie von Rezeptoren
aus. Kürzlich
sind fünft
Rezeptorsubtypen, bezeichnet als SST-R1 bis 5, identifiziert und
kloniert worden. Der exakte funktionale Unterschied zwischen diesen
Rezeptorsubtypen ist bis jetzt nicht vollständig aufgeklärt worden.
-
In
seiner natürlichen
Form hat Somatostatin eine begrenzte Anwendung als therapeutisches
Mittel, da es zwei unerwünschte
Eigenschaften zeigt: geringe Bioverfügbarkeit und kurze Wirkungsdauer.
Aus diesem Grund sind während
der letzten zwei Jahrzehnte große
Anstrengungen unternommen worden, um Somatostatinanaloga zu finden,
die überragend
sind sowohl hinsichtlich Potenz, Biostabilität, Wirkungsdauer als auch Selektivität im Hinblick
auf die Inhibierung der Freisetzung von Wachstumshormon, Insulin
oder Glucagon.
-
Untersuchungen
des Zusammenhangs zwischen Struktur und Aktivität, Spektroskopietechniken,
wie etwa Zirkulardichroismus und kernmagnetische Resonanz und Molekularmodellierung
ergeben das Folgende: die Konformation des cyclischen Teils von
natürlichem
Somatostatin ist wahrscheinlich ein antiparalleles β-Blatt; Phe6 und Phe11 spielen
eine wichtige Rolle beim Stabilisieren der Pharmakophorkonformation
durch hydrophobe Wechselwirkungen zwischen zwei aromatischen Ringen;
die vier Aminosäuren Phe7-Trp8-Lys9-Thr10, die um die β-Windung
in dem antiparallelen β-Blatt
verteilt sind, sind wesentlich für
das Pharmakophor; und (D)Trp8 ist bevorzugt
gegenüber
(L)Trp8 für die Wechselwirkungen mit
Somatostatinrezeptorsubtypen 2 bis 5.
-
Nichtsdestotrotz
ist ein Hexapeptid-Somatostatinanalog, das diese vier Aminosäuren, verankert
durch eine Disulfidbrücke:
nahezu inaktiv, sowohl in
vitro als auch in vivo, wenngleich es den Vorteil der kovalenten
Disulfidbrücke
zeigt, welche die Phe
6-Phe
11-hydrophoben
Wechselwirkungen in natürlichem
Somatostatin ersetzt.
-
Vier
Hauptansätze
sind unternommen worden, um die Aktivität dieses Hexapeptid-Somatostatinanalogs
zu erhöhen.
(1)
Ersetzen der Disulfidbrücke
durch eine Cyclisierung, die eine cis-Amidbindung unterstützt, oder Durchführen einer
zweiten Cyclisierung im Molekül,
was zu einem bicyclischen Analog führt. In beiden Fällen hat
das resultierende Analog eine verringerte Anzahl von konformativen
Freiheitsgraden. (2) Ersetzen der ursprünglichen Reste in der Sequenz
Phe7-(D)Trp8-Lys9-Thr10 mit anderen natürlichen oder nichtnatürlichen
Aminosäuren,
wie etwa Ersetzen von Phe7 durch Tyr7 und Thr10 mit Val10. (3) Einführen zusätzlicher funktioneller Gruppen von
natürlichem
Somatostatin mit dem Ziel, dass diese neuen Elemente zu der Wechselwirkung
mit dem Rezeptor beitragen werden. (4) Eliminieren einer der vier
Aminosäuren
Phe7-(D)Trp8-Lys9-Thr10 unter der Annahme, dass solche Analoga
selektiver sein würden.
Das Somatostatinanalog MK-678:
cyclo(N-Me-Ala6-Tyr7-(D)Trp8-Lys9-Val10-Phe)
ist
ein Beispiel eines hochpotenten Somatostatinanalogs, entwickelt
unter Verwendung der ersten drei obigen Ansätze (Veber, et al., Life Science,
34:371, 1984). In diesem Hexapeptidanalog ist eine cis-Amid-Bindung
lokalisiert zwischen N-Me-Ala und Phe11,
Tyr7 und Val10 ersetzen
Phe7 bzw. Thr10 und
Phe11 ist inkorporiert von natürlichem
Somatostatin.
-
Eine
andere Gruppe von Somatostatinanaloga (
U.S. Patente 4,310,518 und
4,235,886 ) umfassen Octreotid:
das erste
zugelassene Somatostatinanalog, das klinisch verfügbar ist.
-
Es
wurde entwickelt unter Verwendung des dritten oben beschriebenen
Ansatzes. Hier wird angenommen, dass (D)Phe5 und
das reduzierte C-terminale
Thr12-CH2OH einen
Teil des Konformationsraumes besetzen, der verfügbar ist für das natürliche Phe6 bzw.
Thr12.
-
Die
Verbindung TT-232:
ist eng
verwandt mit Octreotid und ist ein Beispiel der Ausführung des
vierten oben beschriebenen Ansatzes. Das Fehlen von Thr
10 ist
wahrscheinlich verantwortlich für
seine hohe funktionelle Selektivität im Hinblick auf Antitumoraktivität.
-
Diese
Beispiele von hochpotenten Somatostatinanaloga legen nahe, dass
die Phenylalanine in den Positionen 6 und 11 nicht nur eine wichtige
Rolle beim Stabilisieren der Pharmakophorkonformation spielen, sondern
auch eine funktionale Rolle bei der Wechselwirkung mit dem Rezeptor
spielen. Eine weitere offen Frage ist, ob ein Phenylalanin (entweder
Phe6 oder Phe11)
ausreichend ist für
die Wechselwirkung mit dem Rezeptor oder ob beide erforderlich sind.
-
Es
ist bekannt, dass die Somatostatinrezeptoren eine Familie von fünf verschiedenen
Rezeptorsubtypen bilden (Bell und Reisine, Trends Neurosci., 16,
34–38,
1993); die unterschieden werden können auf Basis ihrer Gewebespezifität und/oder
biologischen Aktivität.
-
Therapeutische Anwendungen von Somatostatinanaloga
-
Hinsichtlich
ihrer inhibitorischen pharmakologischen Eigenschaften können Somatostatinanaloga
verwendet werden zur Behandlung von Patienten mit Hormon-sekretierenden-
und Hormon-abhängigen
Tumoren. Derzeit werden Symptome, die verbunden sind mit metastatischen
karzinoiden Tumoren (Flush, Diarrhoe, Herzklappenerkrankung und
Abdominalschmerz) und vasoaktives intenstinales Peptid (VIP) sezernierende Adenome
(wässrige
Diarrhoe) mit Octreotid behandelt. Octreotid wurde ebenfalls zugelassen
zur Behandlung von schweren Gastrointestinalhämorrhagien und Acromegalie.
Zusätzlich
ermöglicht
die Fülle
von Hochaffinitäts-Somatostatinrezeptoren
in verschiedenen Tumoren die Verwendung von radioaktiv markierten
Somatostatinanaloga in vivo zur Sichtbarmachung dieser Tumore (Lamberts
et al. N. Engl. J. Med., 334:246, 1996). In neuroendokrinen Tumoren,
insbesondere Karzinoide und VIPoma, inhibiert Octreotid sowohl die
Sekretion als auch die Wirkung des aktiven Mittels. Daher verringern
Somatostatinanaloga in VIPoma, die gekennzeichnet sind durch Diarrhoe
mit übermäßiger Sekretion
durch die Inhibierung von VIP-Sekretion und durch direkte Wirkung
auf die Intestinalsekretion. Jedoch nimmt die Reaktion auf das Arzneimittel
mit der Zeit häufig
ab, möglicherweise
aufgrund der Hinabregulierung von Somatostatinrezeptoren auf Tumorzellen
oder aufgrund der Erzeugung von Rezeptornegativklon. Das Fehlen
von konsistenter antiproliferativer Wirkung kann verbunden sein
mit der geringen Affinität
von Octreotid für
einige der Somatostatinrezeptorsubtypen, die in diesen Tumoren gefunden
werden (Lamberts et al. ibid.).
-
Wie
berichtet, verbessern natives Somatostatin und Octreotid die Symptome
von sekretorischer Diarrhoe, im Unterschied zu denjenigen, die mit
Neuroendokrintumoren verbunden sind. Die Kontrolle von sekretorischer
Diarrhoe, die assoziiert ist mit dem Syndrom des kurzen Darms, Ileostomdiarrhoe,
idiopathische sekretorische Diarrhoe, Diarrhoe, die assoziiert ist
mit Amyloidose und diabetische Diarrhoe sind beschrieben worden.
Von beiden Verbindungen ist gezeigt worden, dass sie ebenfalls eine
gewisse Hoffnung geben bei der Handhabung von Refraktäriarrhoe,
die mit AIDS in Beziehung steht, im Besonderen bei Patienten ohne
identifizierbare Pathogene. Somatostatinanaloga, die in der Technik
bekannt sind, können
keine ausreichende Selektivität
oder Rezeptorsubtypselektivität
bereitstellen, insbesondere als antineoplastische Mittel (Reubi
und Laissue, TIPS, 16, 110–115,
1995).
-
Somatostatinanaloga,
die selektiv für
Typ 2- und Typ 5-Rezeptoren sind, die Wachstumshormon, jedoch nicht
Insulinfreisetzung inhibieren, können
potenziell verwendet werden zur Behandlung von nicht-Insulin abhängiger Diabetes
Mellitus (NIDDM). Geringere Potenz auf Glucagonfreisetzungsinhibierung
ist bevorzugt zur Reduktion der peripheren Resistenz gegenüber Insulin
und zur Verbesserung der glykämischen
Kontrolle.
-
Wachstumshormon
ist ein direkter Antagonist des Insulinrezeptors in der Peripherie
und Wachstumshormonüberproduktion
ist assoziiert mit Insulinperipherresistenz. Erhöhter IGF, was ein prinzipielles
biologisches Signal des Wachstumshormons ist, ist assoziiert mit
diabetischen Komplikationen, wie etwa Angiopathie, Retinopathie
und Nephropathie. Nephropathie ist eine der Hauptkomplikationen
von diabetischer Angiopathie und einer der Hauptgründe für das Endstadium
von Nierenversagen und Tod von Diabetespatienten. Der Nachweis der
signifikaten Beteiligung der GH-IGF-Achse
in diabetischen oder anderen Nephropathien kann bereitgestellt werden
durch mehrere Untersuchungen (Flyvbjerg A. Kidney Int. S12–S19, 1997).
Es wurde kürzlich
gefunden, dass erhöhte
Serumwachstumshormongehalte in den Non-Obese-Diabetic (NOD)-Mäusen ähnlich sind
zu den Änderungen,
die beschrieben sind bei Menschen (Landau et al. J. Am. Soc. Nephrol. 8:A2990,
1997). Diese Entdeckungen ermöglichen
die Erklärung
der Rolle der Wachstumshormon-IGF-Achse bei diabetischer Retinopathie
und das Testen von Somatostatinanaloga auf potenzielle therapeutische
Wirkung in diesen sekundären
mit Diabetes assoziierten Komplikationen.
-
Verbesserte Peptidanaloga
-
Es
wäre wünschenswert,
Peptidanaloga bereitzustellen mit einer größeren Spezifität für Rezeptorsubtypen,
wobei verbesserte klinische Serlektivität erreicht wird.
-
Als
ein Ergebnis der Hauptfortschritte in der organischen Chemie und
in der Molekularbiologie können viele
bioaktive Peptide nun hergestellt werden in Mengen, die ausreichend
sind für
pharmakologische und klinische Anwendungen. So sind in den letzten
Jahren neue Methoden entwickelt worden für die Behandlung und Therapie
von Krankheiten, worin Peptide impliziert waren. Jedoch ist die
Verwendung von Peptiden als Arzneimittel durch die folgenden Faktoren
begrenzt: a) ihre geringe Stoffwechselstabilität gegenüber Proteolyse im Gastrointestinaltrakt
und im Serum; b) ihre geringe Absorption nach oraler Aufnahme, insbesondere
aufgrund ihrer relativ hohen Molekülmasse oder des Fehlens spezifischer
Transportsysteme oder aufgrund von beidem; c) ihrer schnellen Ausscheidung
durch die Leber und Nieren; und d) ihrer ungewünschten Nebenwirkungen in nicht-Targetorgansystemen,
da Peptidrezeptoren weit in einem Organismus verteilt werden können.
-
Es
wäre am
vorteilhaftesten Peptidanaloga mit erzwungener Konformation zu erzeugen,
die die Nachteile von den nativen Peptidmolekülen beseitigen, wobei verbesserte
therapeutische Eigenschaften bereitgestellt werden.
-
Ein
neuer konzeptioneller Ansatz für
Peptide, deren Konformation erzwungen ist, wurde eingeleitet von
Gilon et al., (Biopolymers 31:745, 1991), der Gerüst-an-Gerüst-Zyklisierung
von Peptiden vorschlug. Die theoretischen Vorteile dieser Strategie
umfassen die Möglichkeit
zum Beeinflussen der Zyklisierung über die Kohlenstoffe oder Stickstoffe
des Peptigerüsts
ohne Wechselwirkung mit Seitenketten, die äußerst wichtig sein können für Wechselwirkung
mit dem spezifischen Rezeptor eines gegebenen Peptids. Während das
Konzept als anwendbar auf jedes lineare Peptid erachtet wurde, das
von Interesse ist, war tatsächlich
der limitierende Faktor des vorgeschlagenen Schemas die Verfügbarkeit
von geeigneten Baueinheiten, die verwendet werden müssen, um
die Aminosäuren
zu ersetzen, die verbunden werden müssen über überbrückende Gruppen. Die tatsächliche
Reduktion auf die Praxis dieses Konzepts der Gerüstzyklisierung wurde dadurch
verhindert, dass es nicht möglich
war ein praktisches Verfahren zum Herstellen von Baueinheiten von
Aminosäuren,
die verschieden von Glycin sind, zu erdenken (Gilon et al., J. Org.
Chem., 587:5687, 1992).
-
Weitere
Offenbarungen von Gilon und Mitarbeitern (
WO 95/33765 und
WO 97/09344 ) lieferten Verfahren zum
Herstellen von Bauinheiten, die erforderlich sind bei der Synthese
von Peptidanaloga mit zyklischem Gerüst. Kürzlich wurde auch die erfolgreiche
Verwendung dieser Verfahren zum Herstellen von Peptidanaloga zyklischem
Gerüst
mit Somatostatinaktivität
ebenfalls offenbart (
WO 98/04583 ).
-
Keine
der zugrundliegenden Techniken lehrt oder legt die Somastotatinanaloga,
die hier offenbart sind, nahe, die verbesserte therapeutische Selektivität aufweisen.
-
ZUSAMMENFASSUNG DER ERFINDUNG
-
Gemäß der vorliegenden
Erfindung werden neue Peptidanaloga bereitgestellt, die dadurch
gekennzeichnet sind, dass sie neue Baueinheiten mit überbrückenden
Gruppen umfassen, die gebunden sind an die alpha-Stickstoffe von alpha-Aminosäuren. Im
Speziellen sind diese Verbindungen Somatostatinanaloga mit zyklischem
Gerüst,
umfassend eine Peptidsequenz aus vier bis vierundzwanzig Aminosäuren, wobei
jedes Analog mindestens eine Baueinheit umfasst, wobei die Baueinheit
ein Stickstoffatom des Peptidgrundgerüsts, verbunden mit einer überbrückenden
Gruppe bzw. Überbrückungsgruppe
enthält,
umfassend ein Amid, Thioether, Thioester oder Disulfid, worin die
mindestens eine Baueinheit verbunden ist über die Überbrückungsgruppe, um eine zyklische
Struktur zu bilden mit einer Gruppe, ausgewählt aus der Gruppe, bestehend
aus einer zweiten Baueinheit, der Seitenkette eines Aminosäurerestes
der Sequenz oder dem N-terminalen Aminosäurerest.
-
Bisher
hatten Somatostatinanaloga mit cyclischem Gerüst mit erzwungener Konformation
vorherrschend Selektivität
für Rezeptorsubtyp
5. Diese Analoga hatten eine begrenzte therapeutische oder diagnostische
Anwendbarkeit.
-
Die
vorliegende Erfindung betrifft Verbindungen der Formel Nr. 7:
wie unten definiert.
-
Ein
derzeit am meisten bevorzugtes Analog der vorliegenden Erfindung
ist PTR 3173 mit verbesserter Selektivität der Bindung an SST-Rezeptorsubtyp
SST-R2 und SST-R5.
-
Für zusätzliche
am meisten bevorzugte Analoga, die offenbart sind, ist die Brücke verbunden
zwischen dem Nα-ω-funktionalisiertem
Derivat einer Aminosäure
und dem N-Terminus der Peptidsequenz. Für andere bevorzugte Analoga
der vorliegenden Erfindung ist die Brücke verbunden zwischen einer
Baueinheit, umfassend ein Nα-ω-funktionalisiertes Derivat
mit einer terminalen Thiogruppe und einem anderen solchen Derivat einer
Aminosäure,
oder an der Seitenkette eines Cys-Restes, mit einer Mercaptoenthaltenden
Säure oder
einem anderen SH-enthaltenden Rest, um eine Disulfidbrücke zu bilden.
-
Die
am meisten bevorzugten Somatostatinanaloga mit cyclischen Gerüst gemäß der Erfindung
sind beschrieben in Tabelle 1: Tabelle 1. Die am meisten bevorzugten
Analoga der Erfindung.
| PTR | Sequenz | SST-R |
| 3173 | GABA*-Phe-Trp-(D)Trp-Lys-Thr-Phe-Gly(C3)-X | 2,5 |
| 3229 | Galactose-Dab*-Phe-Trp-(D)Trp-Lys-Thr-Phe-Gly(C3)-X | |
worin X-NH
2 oder -OH ist
und die überbrückende Gruppe
sich zwischen den zwei Baueinheiten oder wie unten angegeben erstreckt:
Für PTR 3173
gibt das Sternchen an, dass die überbrückende Gruppe
verbunden ist zwischen dem N
α-ω-funktionalisierten Derivat
von einer Aminosäure
und dem N-Terminus des Peptids.
-
SST-R
bezeichnet die Somatostatinrezeptorsubtypen, für welche jedes Analog selektiv
ist.
-
Diese
Somatostatinpeptidanaloga mit cyclischem Gerüst werden hergestellt durch
Aufnehmen mindestens eines N
α-ω-funktionalisierten Derivats
einer Aminosäure
in eine Peptidsequenz und nachfolgendes selektives Cyclisieren der
funktionellen Gruppe mit einer der beiden Seitenketten der Aminosäuren in
der Peptidsequenz oder mit einem anderen ω-funktionalisierten Aminosäuresderivat.
Das N
α-ω-funktionalisierte
Derivat von Aminosäuren
hat vorzugsweise die folgende Formel:
worin X eine Abstandsgruppe
ist, ausgewählt
aus der Gruppe, bestehend aus Alkylen, substituiertem Alkylen, Arylen,
Cycloalkylen und substituiertem Cycloalkylen; R' eine Aminosäureseitenkette ist, optional
verbunden mit einer spezifischen Schutzgruppe; B eine Schutzgruppe
ist, ausgewählt
aus der Gruppe, bestehend aus Alkyloxy, substituiertem Alkyloxy
oder Arylcarbonylen; und G eine funktionelle Gruppe ist, ausgewählt aus
der Gruppe, bestehend aus Aminen, Thiolen, Alkoholen, Carbonsäuren und
Astern, Aldehyden, Alkoholen und Alkylhalogeniden; und A eine spezifische
Schutzgruppe von G ist.
-
Bevorzugte
Baueinheiten sind ω-funktionalisierte
Aminosäurederivate,
worin X Alkylen ist; G eine Thiolgruppe, eine Aminogruppe oder eine
Carboxylgruppe ist; und R' die
Seitenkette einer Aminosäure
ist. Weiter bevorzugt sind ω-funktionalisierte
Aminosäurederivate,
worin R' geschützt ist
mit einer spezifischen Schutzgruppe.
-
Mehr
bevorzugt sind ω-funktionalisierte
Aminosäurederivate,
worin G eine Aminogruppe, eine Carboxylgruppe oder eine Thiolgruppe
mit den folgenden Formeln ist:
worin
X, R' und B wie
oben definiert sind.
-
Die
am meisten herausragenden Vorteile dieser Verfahren sind:
- 1) Cyclisieren der Peptidsequenz wird erreicht
ohne Beeinträchtigen
einer der Seitenketten des Peptids, wobei die Wahrscheinlichkeiten
der Zerstörung
funktioneller Gruppen, die wesentliche für biologische Erkennung und
biologische Funktion sind, abnimmt.
- 2) Optimierung der Peptidkonformation wird erreicht indem Permutation
der Brückenlänge, -richtung
und des Bindungstyps (z.B. Amid, Disulfid, Thioether, Thioester
usw.) und der Position der Bindung im Ring zugelassen wird.
- 3) Bei Anwendung auf die Cyclisierung von linearen Peptiden
mit bekannter Aktivität
kann die Brücke
auf eine solche Art ausgestaltet werden, um Wechselwirkung mit der
aktiven Region des Peptids und seinem verwandten Rezeptor zu minimieren.
Dies erniedrigt die Möglichkeiten
des Cyclisierungsarms zum Stören der
Erkennung und Funktion und bildet ebenfalls eine Stelle, die geeignet
ist zur Bindung von Markierungen, wie etwa radioaktive Tracer, cytotoxische
Arzneimittel, Lichteinfangsubstanzen oder einem anderen gewünschten
Markierungsmittel.
-
Analoga
mit cyclischem Gerüst
der vorliegenden Erfindung können
verwendet werden als pharmazeutische Zusammensetzungen und in Verfahren
zur Behandlung von Erkrankungen, einschließlich: Krebs (einschließlich Karzinoidsyndrom),
endokrine Störungen
(einschließlich
Akromegalie und NIDDM), Diabetes-assoziierte Komplikationen (einschließlich diabetische
Nephropathie, diabetische Angiopathie und diabetische Retinopathie),
Magen-Darm-Störungen,
Pankreatitits, Autoimmunerkrankungen (einschließlich rheumatische Arthritis
und Psoriasis), Arteriosklerose, Retenose, post-operativer Schmerz
und Entzündungskrankheiten.
Zusätzlich
werden Somatostatinanaloga gemäß der Erfindung
geeignet sein bei der Verhinderung von Arteriosklerose und Restenose
durch Inhibieren von Wachstumfaktoren, die an diesen Störungen beteiligt
sind.
-
Die
bevorzugten Analoga, die in der vorliegenden Erfindung offenbart
sind, besitzen einzigartige Merkmale der Stoffwechselstabilität, Selektivität ihrer
in vivo-Aktivitäten
und Sicherheit. Das am meisten bevorzugte offenbarte Analog (PTR
3173) bietet einen Arzneimittelkandidaten mit einem klaren therapeutischen
Potenzial zur Behandlung von Karzinoidtumoren, Akromegalie und Diabetes-assoziierten
Komplikationen. Dieses am meisten bevorzugte Analog hat signifikante
Vorteile gegenüber
jedem anderen Somatostatinanalog, das derzeit verfügbar ist,
dahingehend, dass es äquipotent
ist wie verfügbare
Somatostatinanaloga bei der Wachstumshormoninhibierung, ohne nennenswerte
Wirkungen auf Insulin oder Glucagon.
-
Die
pharmazeutischen Zusammensetzungen, die pharmazeutisch aktive Somatostatinagonisten
oder -antagonisten mit cyclischem Gerüst und einen pharmazeutisch
verträglichen
Träger
oder ein pharmazeutisch verträgliches
Verdünnungsmittel
umfassen, stellen eine andere Ausführungsform der Erfindung dar,
wie auch die Verfahren zur Behandlung von Krebs, endokrinen Störungen,
Magen-Darm-Erkrankungen, Diabetes-assoziierten Komplikationen, Pankreatitis,
Autoimmunerkrankungen und Entzündungserkrankungen,
Arteriosklerose und Restenose unter Verwendung solcher Zusammensetzungen.
Die pharmazeutischen Zusammensetzungen gemäß der vorliegenden Erfindung
umfassen vorteilhafterweise mindestens ein Peptidanalog mit cyclischem
Gerüst,
das selektiv ist für
einen oder zwei Somatostatinrezeptorsubtypen. Diese pharmazeutischen Zusammensetzungen
können über jeden
geeigneten Verabreichungsweg verabreicht werden, einschließlich oral,
topisch oder systemisch. Bevorzugte Arten zur Verabreichung umfassen,
ohne darauf begrenzt zu sein, parenterale Wege, wie etwa intravenöse und intramuskuläre Injektionen
als auch nasale oder orale Aufnahme.
-
Analoga
mit cyclischen Gerüst
der vorliegenden Erfindung können
ebenfalls verwendet werden als pharmazeutische Zusammensetzungen
in Verfahren zur Diagnose von Krebs und Bilddarstellung des Vorliegens
von Tumoren oder ihren Metastasen. Die Verfahren zur Diagnose von
Krebs umfassen das Verabreichen an einen Säuger, einschließlich einen
humanen Patienten, eines Analogs oder von Analoga mit cyclischem Gerüst, markiert
mit einer nachweisbaren bzw. feststellbaren Sonde, ausgewählt aus
der Gruppe, bestehend aus einem radioaktiven Isotop und einem nichtradioaktivem
Tracer. Die Verfahren zur Diagnose oder Bilddarstellung von Krebs
unter Verwendung solcher Zusammensetzungen stellen eine andere Ausführungsform
der Erfindung dar.
-
KURZE BESCHREIBUNG DER ZEICHNUNGEN
-
1 ist
ein Graph, der die prozentuale Inhibierung von SRIF-Bindung an 5 humane
klonierte Somatostatinrezeptoren durch PTR-3173 zeigt.
-
2 ist
ein Graph, der die nichtspezifische Bindung von Somatostatinanaloga
(getestet bei einer Konzentration von 100 nM) an verschiedene G-Protein-gekoppelte
Rezeptoren zeigt.
-
3 ist ein Graph, der die Wirkung von Somatostatinanaloga
gemäß der Erfindung
auf die Freisetzung von Wachstumshormon im Vergleich mit Octreotid
zeigt.
-
4 ist
ein Graph, der die Dosisreaktionswirkung des Somatostatinanalogs
gemäß der vorliegenden Erfindung
auf die Freisetzung von Glucagon zeigt.
-
Die 5a und 5b sind
Graphen, die die Wirkung von Somatostatinanaloga gemäß der Erfindung
auf die Freisetzung von Insulin im Vergleich mit Octreotid in drei
verschiedenen Versuchen zeigen.
-
DETAILLIERTE BESCHREIBUNG
DER ERFINDUNG
-
Die
hier beschriebenen Verbindungen können asymmetrische Zentren
aufweisen. Alle chiralen, diastereomeren und racemischen Formen
sind von der vorliegenden Erfindung eingeschlossen. Viele geometrische
Isomere von Doppelbindungen und dgl. können ebenfalls in den Verbindungen,
die hier beschrieben sind, vorliegen, und alle solche stabilen Isomere
sind in der vorliegenden Erfindung vorgesehen.
-
Unter „stabile
Verbindung" oder „stabile
Struktur" versteht
sich hier eine Verbindung, die ausreichend stabil ist, um eine Isolierung
bis zu einem geeigneten Reinheitsgrad aus einem Reaktionsgemisch
und die Formulierung in einem wirkungsvollen therapeutischen Mittel
zu überstehen.
-
Wie
hier und in den Ansprüchen
verwendet, soll „Alkyl" oder „Alkylenyl" sowohl verzweigte
als auch geradkettige gesättigte
aliphatische Kohlenwasserstoffgruppen mit einem bis zehn Kohlenstoffatomen
umfassen; „Alkenyl" soll Kohlenwasserstoffketten
mit entweder einer geradkettigen oder verzweigten Konfiguration umfassen,
wobei zwei bis zehn Kohlenstoffatome und ein oder mehrere ungesättigte Kohlenstoff-Kohlenstoff-Bindungen
vorliegen können,
die an jedem stabilen Punkt entlang der Kette auftreten können, wie
etwa Ethenyl, Propenyl und dgl.; und „Alkinyl" soll Kohlenwasserstoffketten mit entweder
einer geradkettigten oder verzweigten Konfiguration umfassen, mit
von zwei bis zehn Kohlenstoffatomen und einer oder mehreren Kohlenstoff-Kohlenstoff-Dreifachbindungen,
die an jedem stabilen Punkt entlang der Kette auftreten können, wie etwa
Ethinyl, Propinyl und dgl.
-
Wie
hier und in den Ansprüchen
verwendet, soll „Aryl" einen beliebigen
stabilen 5- bis 7-gliedrigen monocyclischen oder bicyclischen oder
7- bis 14-gliedrigen
bicyclischen oder tricyclischen Kohlenstoffring umfassen, wobei jeder
von diesen gesättigt,
teilweise ungesättigt
oder aromatisch sein kann, z.B. Phenyl, Naphthyl, Indanyl oder Tetrahydronaphthyl
usw.
-
Wie
hier und in den Ansprüchen
verwendet, soll „Alkylhalogenid" sowohl verzweigte
als auch geradkettige gesättigte
aliphatische Kohlenwasserstoffgruppen mit einem bis zehn Kohlenstoffatomen,
worin 1 bis 3 Wasserstoffatome ersetzt worden sind durch ein Halogenatom,
wie etwa Cl, F, Br und I, umfassen.
-
Wie
hier und in den Ansprüchen
verwendet, bedeutet der Ausdruck „therapeutisch wirksame Menge" die Menge eines
neuen Peptidanalogs mit cyclischem Gerüst oder Zusammensetzungen,
die dasselbe umfassen, die zu verabreichen ist an einen Wirt, um
die gewünschten
Ergebnisse für
die hier beschriebenen Indikationen zu erreichen, wie etwa, ohne
darauf begrenzt zu sein, Entzündungskrankheiten,
Krebs, endokrine Störungen
und Magen-Darm-Erkrankungen.
-
Der
Ausdruck „substituiert" bedeutet, wie er
hier und in den Ansprüchen
verwendet wird, dass beliebig ein oder mehrere Wasserstoffatome
auf dem gekennzeichneten Atom ersetzt werden mit einer Auswahl aus der
angegebenen Gruppe, vorausgesetzt, dass die normale Valenz des angegebenen
Atoms nicht überschritten
wird und dass die Substitution zu einer stabilen Verbindung führt.
-
Wenn
eine Variable (z.B. R, X, Z usw.) mehr als einmal in einem beliebigen
Konstituenten oder in einer hier genannten Formel auftritt, ist
ihre Definition bei jedem Auftreten unabhängig von ihrer Definition bei
jedem anderen Auftreten. Ebenfalls sind Kombinationen von Substituenten
und/oder Variablen nur dann erlaubt, wenn solche Kombinationen zu
stabilen Verbindungen führen.
-
Wie
hier verwendet, gibt „Peptid" eine Sequenz von
Aminosäuren
an, die durch Peptidbindungen verbunden sind. Die Somatostatinpeptidanaloga
dieser Erfindung umfassen eine Sequenz von Aminosäuren, wobei
jeder Rest dadurch gekennzeichnet ist, dass er einen Amino- und
einen Carboxyterminus aufweist.
-
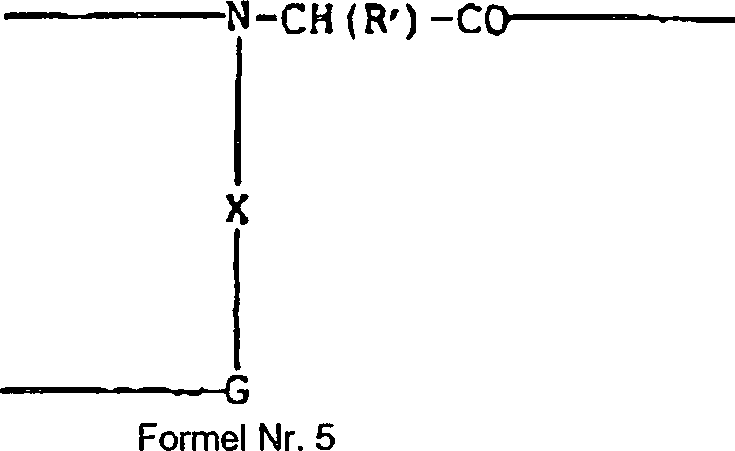
Eine „Baueinheit" gibt eine N
α-derivatisierte α-Aminosäure der
allgemeinen Formel Nr. 5:
an, worin X eine Abstandsgruppe
ist, ausgewählt
aus der Gruppe, bestehend aus Alkylen, substituiertem Alkylen, Arylen,
Cycloalkylen und substituiertem Cycloalkylen; R' eine Aminosäureseitenkette ist, die optional verbunden
ist mit einer spezifischen Schutzgruppe; und G eine funktionelle
Gruppe ist, ausgewählt
aus der Gruppe, bestehend aus Aminen, Thiolen, Alkoholen, Carbonsäuren und
Estern und Alkylhalogeniden; die eingebaut wird in die Peptidsequenz
und nachfolgend selektiv cyclisiert wird über die funktionelle Gruppe
G mit einer der Seitenketten der Aminosäuren in der Peptidsequenz oder
mit einem anderen ω-funktionalisiertem Aminosäurederivat.
-
Die
Methodologie zum Herstellen der Baueinheiten ist beschrieben in
den internationalen Patentanmeldungen, die veröffentlicht sind als
WO 95/33765 und
WO 98/04583 und in den
US Patenten 5,770,687 und
5,883,293 .
-
Die
Baueinheiten werden abgekürzt
durch den Dreibuchstabencode der entsprechend modifizierten Aminosäure, gefolgt
vom Typ der reaktiven Gruppe (N für Amin, C für Carboxyl) und eine Angabe
der Anzahl von Methylenabstandsgruppen. Zum Beispiel beschreibt
Gly-C2 einen modifizierten Gly-Rest mit einer reaktiven Carboxylgruppe
und einem Abstandhalter mit zwei Methylenkohlenstoffen und Phe-N3
bezeichnet eine modifizierte Phenylalaningruppe mit einer Amino-reaktiven
Gruppe und einem Methylen-Abstandshalter mit drei Kohlenstoffen.
-
In
einer generischen Formel werden die Baueinheiten abgekürzt als
R mit einer Hochstellung, die der Position in der Sequenz entspricht,
gefolgt von dem Buchstaben N als ein Hinweis, dass der Gerüststickstoff an
dieser Position der Bindungspunkt der überbrückenden Gruppe ist, die in
der Formel angegeben ist.
-
Wie
hier verwendet, bezeichnet „Peptid
mit cyclischem Gerüst" ein Analog eines
linearen Peptids, das mindestens eine Baueinheit enthält, die
verbunden worden ist, um eine Brücke über den
alpha-Stickstoff des Peptidgerüstes
an eine andere Baueinheit oder eine andere Aminosäure in der
Sequenz zu bilden.
-
Bestimmte
Abkürzungen
werden hier verwendet, um diese Erfindung und die Art zu ihrer Herstellung und
Verwendung zu beschreiben. Zum Beispiel betrifft AcOH Essigsäure, Alloc
betrifft Allyloxycarbonyl, Boc betrifft den t-Butyloxycarbonylrest, BOP betrifft Benzotriazol-1-yloxy-tris-(dimethylamino)phosphoniumhexafluorphosphat,
DCC betrifft Dicyclohexylcarbodiimid, DCM betrifft Dichlormethan,
DIEA betrifft Diisopropylethylamin, DMF betrifft Dimethylformamid,
EDT betrifft Ethandithiol, Fmoc betrifft den Fluorenylmethoxycarbonylrest, GH
betrifft das Wachstumshormon, HBTU betrifft 1-Hydroxybenztriazolyltetramethyluroniumhexafluorphosphat,
HF betrifft Fluorwasserstoffsäure,
HOBT betrifft 1-Hydroxybenzotriazol, HPLC betrifft Hochdruckflüssigchromatographie,
IGF betrifft Insulinwachstumsfaktor, MS betrifft Massenspektrometrie,
NIDDM betrifft Nicht-Insulin-abhängige-Diabetes-Mellitus,
NMM betrifft N-Methylmorpholin, NMP betrifft 1-Methyl-2- pyrrolidon, PyBOP
betrifft Benzotriazol-1-yl-oxy-tris-pyrrolidinophosphoniumhexafluorphosphat,
PyBrOP betrifft Brom-tris-pyrrolidinophosphoniumhexafluorphosphat,
rt betrifft Raumtemperatur, SRIF betrifft Somatotropin Release Inhibitory
Factor (Somatotropinfreisetzungsinhibierungsfaktor), TBTU betrifft
2-(1H-Benzotriazol-1-yl)-1,1,3,3-tetramethyluroniumtetrafluorborat,
t-Bu betrifft den tertiären
Butylrest, TFA betrifft Trifluoressigsäure, VIP betrifft vasoaktives
intestinales Peptid.
-
Die
Aminosäuren,
die in dieser Erfindung verwendet werden, sind diejenigen, die kommerziell
erhältlich
sind oder durch Routinesyntheseverfahren erhältlich sind. Bestimmte Reste
können
spezielle Verfahren zum Einbau in das Peptid erfordern und entweder
sequenzielle oder divergente und konvergente Syntheseansätze für die Peptidsequenz
sind in dieser Erfindung geeignet. Natürlich codierte Aminosäuren und
ihre Derivate werden durch den Dreibuchstabencode gemäß den IUPAC-Konventionen
dargestellt. Wenn es keine Angabe gibt, wurde das L-Isomer verwendet.
Die D-Isomere werden angezeigt durch „D" vor der Abkürzung des Restes. Liste der
nichtcodieren Aminosäuren:
Abu betrifft 2-Aminobuttersäure, β-Ala betrifft β-Alanin, Dab betrifft
Diaminobuttersäure,
GABA betrifft gamma-Aminobuttersäure, 1Nal
betrifft 1-Naphthylalanin, 2Nal betrifft 2-Naphthylalanin.
-
Die
Verbindung und Konjugation von Mono- und Disaccharidresten am Aminoterminus
zum Erhöhen oraler
Bioverfügbarkeit
(Nelson-Piercy et al., J. Clin. Endocrinol. And Metab. 78:329, 1994)
oder andere solche Substitutionen können orale Bioverfügbarkeit,
Penetration in das zentrale Nervensystem, das Zielrichten auf (Targeting)
spezifische Zellpopulationen und dgl. verbessern.
-
Synthetische Ansätze
-
Peptidanaloga
werden cyclisiert durch überbrückende Gruppen,
die an die alpha-Stickstoffe von Aminosäuren gebunden sind, die neue
Nicht-Peptidbindungen
erlauben. Im Allgemeinen beruhen die Verfahren, die verwendet werden
zum Aufbau solcher Peptidanaloga aus ihren Baueinheiten auf den
bekannten Prinzipien der Peptidsynthese; am üblichsten können die Verfahren durchgeführt werden
gemäß den bekannten
Prinzipien der Festphasenpeptidsynthese. Die Innovation erfordert
Ersatz von einer oder mehreren der Aminosäuren in einer Peptidsequenz
durch neue Baueinheiten der allgemeinen Formel:

worin R die Seitenkette einer
Aminosäure
ist, X eine Abstandsgruppe ist und G die funktionelle Endgruppe
ist, durch welche Cyclisierung bewirkt werden wird. Die Seitenkette
R ist die Seitenkette einer beliebigen natürlichen oder synthetischen
Aminosäure,
die ausgewählt
wird, um in die Peptidsequenz der Wahl eingebaut zu werden. X ist
eine Abstandsgruppe, die ausgewählt
wird, um einen größeren oder
geringeren Flexibilitätsgrad bereitzustellen,
um die geeigneten konformativen Zwänge des Peptidanalogs zu erreichen.
Solche Abstandsgruppen umfassen Alkylenketten, substituierte, verzweigte
und ungesättigte
Alkylene, Arylene, Cycloalkylene und ungesättigte und substituierte Cycloalkylene.
Darüber
hinaus können
X und R kombiniert werden, um eine heterocyclische Struktur zu bilden.
Die terminalen (ω)
funktionalen Gruppen, die zur Cyclisierung des Peptidanalogs zu
verwenden sind, umfassen, ohne darauf begrenzt zu sein:
- a. Amine zur Reaktion mit Elektrophilen, wie etwa aktivierte
Carboxylgruppen, Aldehyde und Ketone (mit oder ohne nachfolgende
Reduktion) und Alkyl- oder substituierte Alkylhalogenide.
- b. Alkohole zur Reaktion mit Elektrophilen, wie etwa aktivierte
Carboxylgruppen. c. Thiole zur Bildung von Disulfidbindungen und
Reaktion mit Elektrophilen, wie etwa aktivierte Carboxylgruppen
und Alkyl- oder substituierte Alkylhalogenide
- d. 1,2- und 1,3-Diole zur Bildung von Acetalen und Ketalen.
- e. Alkine und substituierte Alkine zur Reaktion mit Nukleophilen,
wie etwa Amine, Thiole und Carbanionen; freie Radikale; Elektrophile,
wie etwa Aldehyde und Ketone und Alkyl- oder substituierte Alkylhalogenide; oder
organometallische Komplexe.
- f. Carbonsäuren
und -ester zur Reaktion mit Nukleophilen (mit oder ohne vorherige
Aktivierung), wie etwa Amine, Alkohole und Thiole.
- g. Alkyl- oder substituierte Alkylhalogenide oder Ester zur
Reaktion mit Nukleophilen, wie etwa Amine, Alkohole, Thiole und
Carbanionen (von aktiven Methylengruppen, wie etwa Acetoacetaten
oder Malonaten); und Bildung von freien Radikalen zur nachfolgenden
Reaktion mit Alkenen oder substituierten Alkenen und Alkinen oder
substituierten Alkinen.
- h. Alkyl- oder Arylaldehyde und -ketone zur Reaktion mit Nukleophilen,
wie etwa Amine (mit oder ohne nachfolgende Reduktion), Carbanionen
(von aktiven Methylengruppen, wie etwa Acetoacetaten oder Malonaten),
Diole (zur Bildung von Acetalen und Ketalen).
- i. Alkene oder substituierte Alkene zur Reaktion mit Nukleophilen,
wie etwa Amine, Thiole, Carbanionen, freie Radikale oder organometallische
Komplexe.
- j. Aktive Methylengruppen, wie etwa Malonatester, Acetoacetatester
und andere zur Reaktion mit Elektrophilen, wie etwa Aldehyde und
Ketone, Alkyl- oder substituierte Alkylhalogenide.
-
Man
wird einzuschätzen
wissen, dass während
der Synthese des Peptides diese reaktiven Endgruppen als auch sämtliche
reaktiven Seitenketten durch geeignete Schutzgruppen geschützt werden
müssen.
Geeignete Schutzgruppen für
Amine sind Alkyloxy-, substituierte Alkyloxy- und Arlyoxycarbonyle,
einschließlich, ohne
darauf begrenzt zu sein, tert-Butyloxycarbonyl
(Boc), Fluorenylmethyloxycarbonyl (Fmoc), Allyloxycarbonyl (Alloc)
und Benzyloxycarbonyl (Z). Carboxylendgruppen zur Cyclisierung können geschützt werden
als ihre Alkyl- oder substituierte Alkylester oder Thioester oder
Aryl- oder substituierte Arylester oder Thioester. Beispiele umfassen,
ohne darauf begrenzt zu sein, Tertiärbutylester, Allylester, Benzylester,
2-(Trimethylsilyl)ethylester
und 9-Methylfluorenyl.
-
Thiolgruppen
für Cyclisierungen
können
geschützt
werden als ihre Alkyl- oder
substituierte Alkylthioether oder Disulfide oder Aryl- oder substituierte
Arylthioether oder Disulfide. Beispiele solcher Gruppen umfassen,
ohne darauf begrenzt zu sein, Tertiärbutyl, Trityl(triphenylmethyl),
Benzyl-2-(Trimethylsily)ethyl,
Pixyl(9-phenylxanthen-9-yl), Acetamidomethyl, Carboxymethyl, 2-Thio-4-nitropyridyl.
-
Es
wird weiter durch den Fachmann in der Technik einzuschätzen sein,
dass verschiedene reaktive Reste geschützt werden durch verschiedene
Schutzgruppen, um ihre selektive Entfernung zu erlauben. So wird
eine spezielle Aminosäure
gekoppelt an ihren Nachbarn in der Peptidsequenz wenn das Nα geschützt ist durch,
z.B. Schutzgruppe A. Wenn ein Amin als eine Endgruppe für die Cyclisierung
in dem Reaktionschema zu verwenden ist, wird das Nω geschützt werden
durch die Schutzgruppe B oder eine ε-Aminogruppe eines Lysins in der Sequenz
wird geschützt
werden durch die Schutzgruppe C usw.
-
Die
Kopplung der Aminosäuren
aneinander wird durchgeführt
als eine Reihe von Reaktionen, wie in der Technik der Peptidsynthese
bekannt. Neue Baueinheiten der Erfindung, nämlich die Nα-ω-funktionalisierte Aminosäuresderivate,
werden in die Peptidsequenz eingebaut, um eine oder mehrere der
Aminosäuren
zu ersetzen. Wenn nur ein solches Nα-ω-funktionalisiertes
Aminosäurederivat
ausgewählt
wird, wird es cyclisiert an eine Seitenkette von einer anderen Aminosäure in der
Sequenz oder an jede der beiden terminalen Aminosäuren der
Peptidsequenz. Zum Beispiel: (a) kann eine Nα-(ω-Aminoalkylen)-Aminosäure gebunden
werden an die Carboxylgruppe eines Asparagin- oder Glutaminsäurerestes;
(b) eine Nα-(ω-Carboxylalkylen)-Aminosäure kann
an die ε-Aminogruppe
eines Lysinrestes gebunden werden; (c) eine Nα-(ω-Thioalkylen)-Aminosäure kann an
die Thiolgruppe eines Cysteinrestes gebunden werden; usw. Eine mehr
bevorzugte Ausführungsform
der Erfindung umfasst zwei solche Nα-(ω-funktionalisierten
Aminosäurederivate,
die aneinander gebunden sein können,
um cyclische Peptidanaloga mit N-Gerüst an N-Gerüst zu bilden. Drei oder mehr
solcher Baueinheiten können
in eine Peptidsequenz eingebaut werden, um bicyclische Peptidanaloga
zu bilden, wie unten ausgeführt
wird.
-
So
können
Peptidanaloga konstruiert werden mit zwei oder mehr Cyclisierungen,
einschließlich
N-Gerüst-an-N-Gerüst als auch
Gerüst-an-Seitenkette oder
einer anderen Peptidcyclisierung.
-
Wie
oben angegeben, beruhen die Verfahren, die verwendet werden zum
Aufbau von Somatostatinanaloga der vorliegenden Erfindung aus neuen
Baueinheiten im Allgemeinen auf den bekannten Prinzipien der Peptidsynthese.
Jedoch wird es einzuschätzen
sein, dass eine Anpassung der Verfahren auf die voluminöseren Baueinheiten
der vorliegenden Erfindung erforderlich sein kann. Koppeln der Aminosäuren in
der Festphasepeptidchemie kann erreicht werden mittels eines Kopplungsmittels,
wie etwa, ohne darauf begrenzt zu sein, Dicyclohexylcarbodiimid
(DCC), bis(2-Oxo-3-oxazolidinyl)phosphinchlorid (BOP-Cl), Benzotriazolyl-N-oxytrisdimethylaminophosphoniumhexafluorphosphat
(3OP), 1-Oxo-1- chlorphospholan
(Cpt-Cl), Hydroxybenzotriazol (HOBT) oder Gemische davon.
-
Es
wurde nun gefunden, dass das Koppeln der nachfolgenden Aminosäure an die
voluminösen
Baueinheiten der vorliegenden Erfindung die Verwendung von zusätzlichen
Kopplungsmitteln erfordern kann, einschließlich, ohne darauf begrenzt
zu sein: Kopplungsmittel, wie etwa PyBOP (Benzotriazol-1-yl-oxytrispyrrolidinophosphoniumhexafluorphosphat),
PyBrOP (Brom-trispyrrolidinophosphoniumhexafluorphosphat), HBTU (2-(1H-Benzotriazol-1-yl)-1,1,3,3-tetramethyluroniumhexafluorphosphat),
TBTU (2-(1H-Benzotriazol-1-yl)-1,1,3,3-tetramethyluroniumtetrafluorborat).
-
Neue
Kopplungschemie kann verwendet werden, wie etwa vorgeformte Urethan-geschützte N-Carboxyanhydride
(UNCAs), vorgeformte Acylhalogenide, am meisten bevorzugt Acylchloride.
-
Vorteilhafterweise
ist es ebenfalls möglich
in situ erzeugte Aminosäurechloride
zu verwenden. Die Aminosäurechloride
könnten
erzeugt werden durch Verwenden von Reagenzien, wie etwa z.B. Bis(trichlormethyl)carbonat,
allgemein bekannt als Triphosgen.
-
Eine
solche Kopplung kann bei Raumtemperatur und ebenfalls bei erhöhten Temperaturen
stattfinden, in Lösungsmitteln,
wie etwa Toluol, DCM (Dichlormethan), DMF (Dimethylformamid), DMA
(Dimethylacetamid), NMP (N-Methylpyrrolidinon), Dioxan, Tetrahydrofuran,
Diglyme und 1,3-Dichlorpropan
oder Gemischen der obigen.
-
Die
Somatostatinanaloga mit cyclischem Gerüst der vorliegenden Erfindung
werden nun beschrieben.
-
Die
Somatostatinanaloga der vorliegenden Erfindung mit cyclischem Gerüst weisen
die allgemeine Formel auf:
worin
n 1 bis 5 ist;
X
eine terminale Carbonsäure-,
Amid- oder Alkoholgruppe bezeichnet;
Q Wasserstoff oder ein
Mono- oder Disaccharid ist;
R
5 gamma-Aminobuttersäure, Diaminobuttersäure,
Gly, β-Ala, 5-Aminopentansäure oder
Aminohexansäure
ist;
R
6 (D)- oder (L)-Phe oder Tyr
ist;
R
7 (D)- oder (L)-Trp, (D)- oder
(L)-Phe, (D)- oder (L)-1Nal oder (D)- oder (L)-2Nal oder Tyr ist;
R
8 (D)-
oder (L)-Trp ist;
R
9 (D)- oder (L)-Lys
ist;
R
10 Thr, Gly, Abu, Ser, Cys, Val,
(D)- oder (L)-Ala oder (D)- oder (L)-Phe ist;
R
11 (D)-
oder (L)-Phe, (D)- oder (L)-Ala, Nie oder Cys ist;
R
12 Gly, Val, Leu, (D)- oder (L)-Phe oder
1 Nal oder 2Nal ist.
-
Eine
am meisten bevorzugte Verbindung gemäß dieser Ausführungsform
wird als PTR 3173 bezeichnet, worin die Reste wie folgt sind:
Q
ist Wasserstoff;
R5 ist GABA;
R6 ist Phe;
R7 ist
Trp;
R8 ist (D)Trp;
R9 ist
Lys;
R10 ist Thr;
R11 ist
Phe;
R12 ist Gly;
n ist 3; und
X
ist ein Amid.
-
Eine
andere bevorzugte Verbindung gemäß dieser
Ausführungsform
wird als PTR 3229 bezeichnet, worin die Reste wie folgt sind: Q
ist Galactose;
R5 ist Dab;
R6 ist Phe;
R7 ist
(L)-Trp;
R8 ist (D)-Trp;
R9 ist Lys;
R10 ist
Thr;
R11 ist Phe;
R12 ist
Gly;
n ist 3; und
X ist Amid.
-
Die
am meisten bevorzugten Somatostatinanaloga mit cyclischem Gerüst gemäß der vorliegenden
Erfindung sind in Tabelle 3 beschrieben: Tabelle 3: Die am meisten bevorzugten
Analoga.
| PTR | Sequenz |
| 3173 | GABA*-Phe-Trp-(D)Trp-Lys-Thr-Phe-Gly(C3)-X |
| 3229 | Galactose-Dab*-Phe-Trp-(D)Trp-Lys-Thr-Phe-Gly(C3)-X |
worin X -NH
2 oder -OH
ist und die überbrückende Gruppe
sich zwischen zwei Baueinheiten oder wie unten angegeben erstreckt:
Für PTR 3173
gibt das Sternchen an, dass die überbrückende Gruppe
verbunden ist zwischen dem N
α-ω-funktionalisierten Derivat
einer Aminosäure
und dem N-Terminus des Peptids.
-
Somatostatin
ist ein Tetradecapeptidhormon, dessen zahlreiche regulatorischen
Funktionen vermittelt werden durch eine Familie aus fünf Rezeptoren,
wobei die Expression gewebeabhängig
ist. Es wird angenommen, dass rezeptorspezifische Analoga von Somatostatin
wertvolle therapeutische Mittel bei der Behandlung verschiedener
Krankheiten sind. Ansätze
zum Entwickeln kleiner Peptidanaloga mit dieser Selektivität sind nicht
von besonderem Erfolg gewesen. Es ist nun unerwarteterweise gefunden
worden, dass die Somatostatinanaloga mit cyclischem Gerüst mit erzwungener
Konformation der vorliegenden Erfindung hochselektiv für SST-Rezeptorsubtypen
sind.
-
Die
Peptide mit cyclischem Gerüst
dieser Erfindung sind neue selektive Analoga und binden vorzugsweise
mit höherer
Affinität
an einen einzelnen Rezeptor der Somatostatinrezeptorfamilie.
-
Die
Aminosäuresequenz
der entsprechenden Analoga mit hexacyclischem Grundgerüst (PTRs
3113, 3123 und 3171, nicht in der vorliegenden Erfindung enthalten)
basiert auf den angenommen am meisten wichtigen Aminosäuren, die
von dem nativen SRIF-14 abgeleitet sind. Aus den Daten in der Literatur
(Bauer et al., Life Sciences, 31:1133, 1982) wurde geschlossen,
dass die Aminosäuren
des nativen SRIF-14 in mindestens den Positionen 7 bis 10, nämlich Phe7, Trp8, Lys9 und Thr10, und
vorzugsweise den Positionen 6 bis 10, nämlich Phe8,
Phe7, Trp8, Lys9 und Thr10, wesentlich
sind für
das Pharmakophor des Hormons.
-
Die
vorliegenden innovativen Gerüstanaloga
umfassen vorzugsweise 8 Aminosäuren
mit speziellen Aminsäuremodifikationen.
Für bestimmte
bevorzugte Analoga wurde die Aminsäure Asn substituiert durch
die Gerüst-Phe-Baueinheit an
Position 5. Die Konfigurationssubstitution des nativen L-Trp an Position 8
durch D-Trp wurde durchgeführt
zum Verbessern der Stabilität
des Analogs. Der Thr-Rest an Position 10 wurde deletiert und die
Sequenz vervollständigt
durch die entsprechende Gerüst-ehe-Baueinheit.
Die einzigartige Konfigurationssubstitution an Position 9 von L-Lys
durch D-Lys, wie
in den PTRs 3123 und 3171 im Vergleich zu PTR 3113 gezeigt, führt eher
zu verbesserter Selektivität
der Bindung an SST-Rezeptorsubty SST-R3 als an SST-R5 (nicht in
der vorliegenden Erfindung enthalten).
-
Zusätzlich wurden
mehrere weitere Modifizierungen von Analoga von Aminosäuren durchgeführt. Zum Beispiel
Substitution von Phe-Resten mit Tyr zur Erleichterung der Iodierung.
Substitution von Phe-Resten mit N-Methyl-Phe-Rest (z.B. Substitution von
Phe6 in PTR 3173, um PTR 3223 zu ergeben
und Substitution von Phe6 und Phe11 in PTR 3173, um PTR 3225 zu ergeben) zum
Erhöhen
der Bioverfügbarkeit
der Verbindung (nicht in der vorliegenden Erfindung enthalten).
Addition von Mono- und Disaccharidresten am Amino-Terminus von bestimmten
Verbindungen wird durchgeführt
zum Erhöhen
der oralen Bioverfügbarkeit
(Nelson-Piercy et al., ibid.). Zum Beispiel wurde Galactose konjugiert
an den N-Terminus einer Verbindung, die ähnlich PTR 3173 ist, um ein
Analog zu ergeben, das die Sequenz aufweist:
Galactose-Dab-Phe-Trp-(D)Trp-Lys-Thr-Phe-Gly(C3)-NH2, hier bezeichnet als PTR 3229.
-
In
einem bestimmten am meisten bevorzugten Analog (PTR 3173 zum Beispiel)
ist die Brücke
verbunden zwischen dem Nα-ω-funktionalisierten Derivat
einer Aminosäure
und dem N-Terminus der Peptidsequenz. Für andere bevorzugte Analoga
der vorliegenden Erfindung ist die Brücke verbunden zwischen einer
Baueinheit, umfassend ein Nα-ω-funktionalisiertes Derivat
mit einer terminalen Thiogruppe, und einem anderen, wie etwa einem
Derivat einer Aminosäure
oder mit der Seitenkette eines Cys-Restes, an eine Mercapto-enthaltende
Säure oder
einen anderen beliebigen SH-enthaltenden Rest, um eine Disulfidbrücke zu bilden.
-
Die
vorliegenden neuen Analoga stellen eine zusätzliche Dimension für die Neuheit
der Gerüstcyclisierungstechnologie
dar, bei der Verwendung einer verkürzten Gerüstbrücke (d.h. nur ein bis drei
Methylene neben der Peptidbindung). Dieser Ansatz ermöglicht das
Erreichen einer viel größeren Steifheit
des Peptides und weiteres Erzwingen der gewünschten Konformation des nativen
Pharmakophors.
-
Ein
zusätzlicher
Vorteil der Hexapeptidanaloga der vorliegenden Erfindung ist verbunden
mit ihrem retativ geringen Molekutargewicht (wobei ihre Sequenz
nur aus sechs Aminosäuren
besteht), bis zu nur 1000 Dalton, im Vergleich zu den üblichsten
synthetischen Somatostatinanaloga, die üblicherweise Hepta- oder Octapeptide
sind.
-
Somatostatinanaloga
der vorliegenden Erfindung mit cyclischem Gerüst (z.B. PTR 3173) erwiesen sich
so, dass sie beachtenswerte Stoffwechselbiostabilität gegenüber Abbau
durch Enzyme aufweisen. Dieses Attribut könnte eine potenziell lange
Dauer der Aktivität
im Körper
nahelegen.
-
Die
Stabilität
der Analoga mit cyclischem Gerüst
war vergleichbar mit der von Stoffwechsel-stabilem Arzneimittel
Octreotid, unter Verwendung von experimentellen Stabilitätsmessungen,
basierend auf dem Abbau durch verschiedene Enzymgemische (z.B. Nierenhomogenat,
Rattenleberhomogenat und Humanserum). Alle getesteten Verbindungen
zeigten signifikant höhere
Biostabilität
als das native Hormon SRIF-14. In einigen der entsprechenden nichtcyclischen
Peptide wurde ein gewisser Abbau zwei Stunden nach Inkubation beobachtet,
was anzeigte, dass die Cyclisierung bemerkenswert zur Stabilität des Peptides
beitrug. Es wurde erwartet, dass der Einbau von N-alkylierten Aminosäuren, die
verwendet wurden für
die Gerüstcyclisierung,
die Stoffwechselbiostabilität
dieser Peptide verleiht.
-
Analoga
mit cyclischem Gerüst
der vorliegenden Erfindung binden in vitro mit hoher Affinität an einen definierten
Subsatz der humanen Somatostatinrezeptoren. Dieser Rezeptor zeigt
selektiv seine potenzielle physiologische Selektivität in vivo.
-
Übereinstimmend
mit der in vitro-Rezeptorbindung beeinflussen die Analoga mit cyclischem
Gerüst der
vorliegenden Erfindung selektiv ein definiertes System im Körper, während sie
andere bekannte physiologische Aktivitäten des nativen Hormons Somatostatin
nicht beeinflussen. Zum Beispiel zeigt PTR 3173 signifikante Inhibierung
mit ausgedehnter Wirkungsdauer auf der Wachstumshormon-IGF-1-Achse
mit einer ähnlichen
Größenordnung
wie das Arzneimittel Octreotid, jedoch fehlen die Nachteile von
Octreotid, wie etwa Inhibierung der Insulinsekretion. PTR 3173 zeigt
auch eine beachtenswert geringere Beeinträchtigung der Freisetzung von
Glucagon als Octreotid, es hat daher den Vorteil, dass es keine
Hyperglykämie
bewirkt, was es zu einer sehr attraktiven Verbindung zur Behandlung
von Diabetes Typ 2 (NIDDM) macht.
-
Wie
in Tabelle 4 zusammengefasst, besitzt PTR 3173 wesentliche physiologische
Selektivität
gegenüber
dem Arzneimittel Octreotid. PTR 3173 ist ein potenter Inhibitor
des Wachstumshormons, weist jedoch weniger Aktivität auf Glucagon
und keine nennenswerte Wirkung auf Insulin auf. Tabelle 4: Physiologische Selektivität von PTR
3173 im Vergleich mit Octreotid.
| Analog | GH
ID50 μg/kg | Glucagon
ID50 μg/kg | Insulin
ID50 μg/kg | GH/Insulin | GH
Glucagon |
| Octreotid | 0,08 | 0,65 | 26 | 309 | 8 |
| PTR
3173 | 0,1 | > 100 | > 1000 | > 10.000 | > 1000 |
-
PTR
3173 zeigt eine signifikante Wachstumsinhibierung von CHO-Zellen,
die kloniertes humanes SST-R5 exprimieren, wodurch eine potenzielle
Rolle bei der Behandlung von SST-R5-exprimierenden Tumoren (z.B.
Karzinoide, Hypophysentumore) gezeigt wird. Dieses Analog inhibiert
ebenfalls Chromogranin A-Freisetzung von der humanen Karzinoidzelllinie,
wodurch eine Antitumorwirkung angezeigt wird (Beispiel 5).
-
Das
einzigartige pharmakokinetische Profil von PTR 3173 ist, wie es
beurteilt wird in Tieren, übereinstimmend
mit seiner Stoffwechselbiostabilität, wie in vitro beurteilt.
Dieses Somatostatinanalog mit cyclischem Gerüst zeigt flip flog (eine langsame
Freisetzungskinetik)-Pharmakokinetiken. Nachfolgend auf subkutane
Verabreichung resultiert die scheinbare-Halbwertszeit im Organismus
aus seiner Absorptionsrate, jedoch nicht aus seiner Eliminierungsrate.
Nachfolgend auf subkutane Verabreichung an Ratten hatte PTR 3173
eine Halbwertszeit im Kreislauf von etwa 3 Stunden. Diese Aktivität überschreitet
wesentlich die des lange wirkenden Arzneimittels Octreotid, das
eine Halbwertszeit im Kreislauf von nur 40 Minuten aufweist.
-
Die
pharmakokinetischen Hauptparameter von PTR 3173 gegenüber Octreotid
sind in Tabelle 5 zusammengefasst. Tabelle 5: Pharmakokinetische Hauptparameter
von PTR 3173 gegenüber
Octreotid nachfolgend auf IV & SC-Verabreichung
an Wistar-Ratten, die bei Bewusstsein sind.
| Verabreichungsweg | Arzneimittel | F
(%) | Vs
(ml/kg) | T1/2 β (min) | E
% | Clearance (ml/min/kg) |
| IV | PTR
3173 | – | 653 | 31 | 10,3 | 13,0 |
| Octreotid* | – | 602 | 49 | 21,3 | 17,6 |
| SC | PTR
3173 | 99,6 | – | 170 | 15,9 | 13,3 |
| Octreotid* | 103 | – | 40 | 23,0 | 17,1 |
- * von Sandostatin (Octreotidacetat), Übersicht
und klinische Zusammenfassung. Sandoz Pharmaceutical Corporation,
1992.
- F – Bioverfügbarkeit,
- Vs. – Verteilungsvolumen,
- T1/2 – Halbwertszeit im Kreislauf
- E – extrahiert
aus Urin
-
Das
Somatostatinanalog mit cyclischem Gerüst PTR 3173 ist selektiv für Somatostatinrezeptoren
und bindet signifikant weniger andere G-Protein gekoppelte Rezeptoren
als Octreotid, wie gefunden wird durch Screenen beider Analoga und
SRIF auf Bindung an mehrere solche Rezeptoren (Beispiel 6). Dieses
Charakteristikum ist von großem
Vorteil, da Bindung an nicht Somatostatinrezeptoren potenziell nachteilige
Wirkungen im Körper
bewirken könnte.
-
Es
erwies sich weiterhin, dass PTR 3173 nicht mitogen ist für humane
Lymphozyten in humanen peripheren Blutlymphozyten (PBL)-Proliferationsassays.
-
PTR
3173 wurde in verschiedenen Spezies auf seine Grundsicherheitseigenschaften
getestet. In Hinblick auf die Pharmacopoeia-Europaea-Anforderungen für Sicherheitstesten
wurde es als sicherer Arzneimittelkandidat in diesem Entwicklungsstudium
deklariert. Keine Toxizitäthinweise
in Nagern oder Hunden waren ersichtlich bei Injektion einer Dosis,
die 10.000-fach höher
ist als die wirksame Dosis zum Inhibieren von Wachstumshormonfreisetzung.
-
Allgemeines Verfahren zur Synthese, Reinigung
und Charakterisierung von Peptiden mit cyclischem Gerüst
-
Synthese:
-
- Harz: 1 g Rink-Amid oder Tenta-Gelharz mit einer Beladung
von 0,2–0,7
mmol/g.
- Fmoc-Entschützung:
Mit 7 ml 20 % Piperidin in NMP. Zweimal für 15 Minuten, nachfolgend 5
Waschungen mit 10 ml NMP für
2 Minuten unter Schütteln.
- Kopplungen:
-
- 1. Reguläre
Kopplungen (Kopplung an einfache Aminosäuren): mit einer Lösung, enthaltend
3 Äquivalente Aminosäure, 3 Äquivalente
PyBroP und 6 Äquivalente
DIEA in 7 ml NMP. Für
0,5–2
Stunden unter Schütteln.
Das Koppeln wird beobachtet durch Ninhydrintest und wiederholt bis
die Ninhydrinlösung
gelb wird.
- 2. Koppeln von His und Asn mit einer Lösung, enthaltend 5 Äquivalente
DIC und 5 Äquivalente
HOBT in 10 ml DMF.
- 3. Koppeln an Gly-Baueinheiten: mit einer Lösung, enthaltend 3 Äquivalente
Aminosäure,
3 Äquivalente
PyBroP und 6 Äquivalente
DIEA in 7 ml NMP. Zweimal für
1–4 Stunden
unter Schütteln.
- 4. Koppeln an Baueinheiten, die nicht Gly sind: mit einer Lösung, enthaltend
5 Äquivalente
Aminosäure,
1,5 Äquivalente
Trisphosgen und 13 Äquivalente
Collidin in 15 ml Dioxan oder THF. Zweimal für 0,5 bis 2 Stunden bei 50°C unter Schütteln.
-
- Entfernen der Allyl- und Alloc-Schutzgruppen von den Baueinheiten:
mit 1,5 Äquivalenten
pro Peptid Pd(PPh3)4 in 30 ml DCM, enthaltend 5 % Essigsäure und
2,5 % NMM. Für
1–4 Stunden
unter Schütteln.
- Cyclisierung: mit einer Lösung,
enthaltend 3 Äquivalente
PyBOP und 6 Äquivalente
DIEA in 7 ml NMP. Für 0,5–2 Stunden
unter Schütteln.
Cyclisieren wird beobachtet durch einen Ninhydrintest und erforderlichenfalls wiederholt.
- Spaltung: mit 82 %–95
% TFA, supplementiert mit Einfängern:
1–15 %
H2O, 1–5
% TIS und 1–5
% EDT.
- Reinigung:
Ein individuelles Reinigungsverfahren für jedes
Peptid mit cyclischem Gerüst
wird entwickelt auf einer analytischen HPLC, um die maximale Isolierung
des cyclischen Peptids von anderen Rohkomponenten zu erreichen. Das
analytische Verfahren wird üblicherweise
durchgeführt
unter Verwendung einer C-18 Vydac Säule 250 × 4,6 mm als die stationäre Phase
und eines Wasser/ACN-Gemisch-Gradienten mit 0,1 % TFA.
-
Das
präparative
Verfahren wird entwickelt durch Einbeziehen des analytischen Trennverfahrens
in dem präparativen
2 Zoll-C-18 Vydac-Verfahren.
Während
des Reinigungsverfahrens wird das Material, das dem Signal für das cyclische
Peptid entspricht, gesammelt, unter Verwendung eines halbautomatischen
Fraktionssammlers. Die gesammelten Fraktionen werden zur Reinheitsüberprüfung in
die analytische HPLC injiziert. Die reinen Fraktionen werden vereinigt
und lyophylisiert.
-
Charakterisierung:
-
Das
vereinigte reine lyophylisierte Material wird durch HPLC, MS und
Kapillarelektrophorese analysiert und durch Aminosäureanalyse
auf den Peptidgehalt und zur Bestimmung des Aminosäureverhältnisses analysiert.
-
Allgemeines Screening von Somatostatinanaloga.
-
Somatostatinanaloga
mit cyclischem Grundgerüst
werden gescreent indem sie in vitro getestet werden auf ihre Inhibierung
der natürlichen
Peptid (SRIF-14)-Bindung
an seine G-Protein-gekoppelten Rezeptoren (Beispiel 3). Analoga,
die mit hoher Affinität
binden, werden dann auf ihren Einfluss auf zweite Messenger, wie
etwa cyclisches Adenosinmonophosphat (cAMP)-Gehalte, Typrosinphosphataseaktivität, Wachstumshormon-
und Chromogranin A-Sekretion, und auf Zellwachstum getestet.
-
Aktive
Analoga werden darüber
hinaus in vivo getestet auf Inhibierung von Hormonen und Enzymsekretion,
in speziellen relevanten Modellsystemen, basierend auf Literaturdaten,
die anzeigen, dass SST-R2 und SST-R5 die meisten endokrinen Wirkungen
von Somatostatin vermittelen, inhibierend sind für Freisetzung von Wachstumshormon
und Amylase, Magensäure,
Insulin- und Glucagonsekretion, die auf den bekannten endokrinen
Aktivitäten
des nativen Hormons SRIF und des Somatostatinanalogs, Octreotid,
basieren.
-
Das
am meisten bevorzugte Somatostatinanalog mit cyclischem Gerüst: PTR-3173,
das Rezeptorspezifität
für SST-R2
+ SST-R5 besitzt, wurde verwendet zum Aufklären der physiologischen Rolle
von jedem Somatostatinrezeptor auf die Endokrinprofile, zusätzlich zum
Auffinden ihrer Potenziale als Arzneimittelkandidaten.
-
Somatostatinanaloga
mit erzwungener Konformation, aufgebaut zum Teil basierend auf den
Sequenzen einer Vielzahl bekannter biologisch aktiver Peptide oder
basierend auf früher
nicht bekannten neuen Sequenzen, werden in den Beispielen unten
dargestellt. Die folgenden Beispiele sollen veranschaulichen, wie
die Verbindungen herzustellen und zu verwenden sind und die Verfahren
dieser Erfindung anzuwenden sind und sollen in keiner Art begrenzend
ausgelegt werden.
-
BEISPIELE
-
Beispiel 1. Detaillierte Synthese von
PTR 3173.
-
Fünf Gramm
Rink-Amidharz (NOVA) (0,56 mmol/g) wurden in N-Methylpyrrolidon (NMP) in einem Reaktionsbehältnis gequollen,
das ausgestattet war mit einem Sinterglasboden, und auf einem Schüttler angeordnet.
Die Fmoc-Schutzgruppe wurde von dem Harz entfernt durch Reaktion
mit 20 % Piperidin in NMP (2 mal 10 Minuten, jeweils 25 ml). Die
Fmoc-Entfernung wurde beobachtet durch Ultraviolettabsorptionsmessung bei
290 nm. Ein Kopplungszyklus wurde durchgeführt mit Fmoc-Gly-C3(Allyl)
(3 Äquivalente),
PyBrop (3 Äquivalente),
DIEA (6 Äquivalente)
in NMP (20 ml) für
1 Stunde bei Raumtemperatur. Reaktionsabschluss wurde beobachtet
durch den qualitativen Ninhydrintest (Kaiser Test). Nachfolgend
auf das Koppeln wurde das Peptidharz gewaschen mit NMP (7 mal mit
25 ml NMP, jeweils 2 Minuten). Ein Capping wurde durchgeführt durch Reaktion
des Peptidharzes mit Essigsäureanhydrid
(Capping-Gemisch: HOBt 400 mg, NMP 20 ml, Essigsäureanhydrid 10 ml, DIEA 4,4
ml) für
0,5 Stunden bei Raumtemperatur. Nach dem Capping wurden NMP-Waschungen
wie oben durchgeführt
(7 mal, jeweils 2 Minuten). Fmoc-Entfernung wurde wie oben durchgeführt. Fmoc-Phe-OH
wurde auf dieselbe Art gekoppelt und die Fmoc-Gruppe wie oben entfernt. Das Peptidharz
wurde umgesetzt mit Fmoc-Thr(OtBu)-OH:
Kopplungsbedingungen sind wie oben. Fmoc-Entfernung wurde wie oben
durchgeführt.
Fmoc-Lys(Boc)-OH wurde an das Peptidharz gekoppelt unter den die
gleichen Kopplungsbedingungen. Der Abschluss der Kopplung wurde
beobachtet durch den Fmoc-Test (eine Probe des Peptidharzes wurde
genommen und gewogen, das Fmoc wurde wie oben entfernt und die Ultraviolettabsorption
wurde gemessen). Fmoc-D-Trp-OH wurde gekoppelt an das Peptidharz
mit PyBrop, wie oben beschrieben. Nachfolgend auf Fmoc-Entfernung
wurde Fmoc-Trp-OH auf dieselbe Art gekoppelt. Nachfolgend auf Fmoc-Entfernung
wurde Fmoc-Phe-OH auf dieselbe Art gekoppelt. Nachfolgend auf Fmoc-Entfernung
wurde Fmoc-GABA-OH
auf dieselbe Art gekoppelt. Die Allylschutzgruppe wurde entfernt
durch Reaktion mit Pd(PPh3)4 und
Essigsäure
5 %, Morpholin 2,5 % in Chloroform unter Argon, für 2 Stunden
bei Raumtemperatur. Das Peptidharz wurde wie oben mit NMP gewaschen.
Die Fmoc-Schutzgruppe wurde von dem Peptid entfernt durch Reaktion mit
20 % Piperidin in NMP (2 mal 10 Minuten, jeweils 25 ml). Cyclisierung
wurde durchgeführt
mit 3 Äquivalenten
PyBOP, 6 Äquivalenten
DIEA in NMP, bei Raumtemperatur für 2 h. Das Peptidharz wurde
gewaschen und getrocknet. Das Peptid wurde von dem Harz abgespalten
durch Reaktion mit TFA 94 %, Wasser 2,5 %, EDT 2,5 %, TIS (Triisopropylsilan)
1 %, bei 0°C
für 15
Minuten und 2 Stunden bei Raumtemperatur unter Argon. Das Gemisch
wurde filtriert in kalten Ether (30 ml, 0°C) und das Harz wurde mit einem
kleinen Volumen TFA gewaschen. Das Filtrat wurde in einem Rotationsverdampfer
angeordnet und alle flüchtigen
Komponenten wurden entfernt. Ein öliges Produkt wurde erhalten.
Es wurde dreimal mit Ether verrieben und der Ether dekantiert. Ein
weißes
Pulver wurde erhalten. Dieses Rohprodukt wurde getrocknet. Das Gewicht
des Rohproduktes war 4 g.
-
Nach
Reinigung durch HPLC wurde ein einzelnes Signal erhalten, entsprechend
100 % Reinheit, gemäß Nachweis
durch analytische HPLC und Kapillarelektrophorese. Die erwartete
Masse von 1123 Dalton wurde durch Massenspektroskopie nachgewiesen.
-
Beispiel 3: Beständigkeit gegenüber Bioabbau.
-
Die
in vitro-Biostabilität
von SST-cyclischen Peptidanaloga; PTRs 3113
*,
3123
*, 3171
* und
3173, wurde gemessen in Nierenhomogenat und es erfolgte ein Vergleich
mit Octreotid (Sandostatin) und mit nativem Somatostatin (SRIF-14).
Die Ergebnisse sind unten in Tabelle 6 gezeigt. In diesem Assay
waren die Peptidanaloga mit cyclischem Gerüst der vorliegenden Erfindung
so stabil wie Octreotid und waren viel stabiler als SRIF. Der Assay
basierte auf einer HPLC-Bestimmung des Peptidabbaus als eine Funktion
der Zeit in Nierenhomogenat bei 37°C. Tabelle 6: Prozent intaktes Molekül nach Inkubieren
in Nierenhomogenat.
| Zeit
(h) | SRIF | Octreotid | PTR-3113* | PTR-3123* | PTR-3171* | PTR-3173 |
| 0 | 100 | 100 | 100 | 100 | 100 | 100 |
| 1 | 5 | 100 | 100 | 100 | 100 | 100 |
| 3 | 0 | 100 | 100 | 100 | 100 | 100 |
| 24 | 0 | 100 | 100 | 100 | 100 | 100 |
- *kein Bestandteil
der vorliegenden Erfindung
-
Beispiel 4: Bindung von Analoga an Somatostatinrezeptoren.
-
Die
Somatostatinanaloga wurden getestet auf ihre Potenz bei der Inhibierung
der Bindung von 125I-Tyr11-SRIF
(basierend auf dem von Raynor et al., Molecular Pharmacology 43:
838, 1993, beschriebenen Verfahren) an Membranpräparate, die die Transmembran-Somatostatinrezeptoren
exprimieren (SST-R1, 2, 3, 4 oder 5). Die Rezeptorpräparate,
die für
diese Tests verwendet wurden, waren entweder von den klonierten humanen
Rezeptoren, die selektiv und stabil exprimiert werden in Chinesischem
Hamster-Ovar (CHO)-Zellen oder von Zelllinien, die natürlich die
SST-Rs exprimieren. Typischerweise wurden Zellmembranen in Tris-Puffer in
der Gegenwart von Proteaseinhibitoren homogenisiert und inkubiert
für 30–40 Minuten
mit 125I-Tyr11-SRIF bei
verschiedenen Konzentrationen der getesteten Probe. Die Bindungsreaktionen
wurden filtriert, die Filter wurden gewaschen und die gebundene
Radioaktivität
wurde in einem Gammazähler
gezählt.
Nicht-spezifische Bindung wurde definiert als die Radioaktivität, die gebunden
bleibt in der Gegenwart von 1 μM
unmarkiertem SRIF-14.
-
Zum
Validieren positiver Signale der Bindungstests und zum Eliminieren
nichtspezifischer Signale wurden Proben irrelevanter Peptide, wie
etwa GnRH, die synthetisiert und gehandhabt wurden unter Verwendung
der gleichen Verfahren, in den gleichen Assays als negative Kontrollproben
getestet. Diese Proben hatten keine Bindungsaktivität in einem
der Assays. Ergebnisse sind unten in den Tabellen 8 und 9 und in
1 gezeigt. Tabelle 8: Prozentuale Inhibierung von
SRIF-14-Bindung an klonierte humane Somatostatinrezeptoren durch PTR
3173.
| Rezeptor-Subtyp | Konzentration
(M) |
| | 10–11 | 10–10 | 10–9 | 10–8 | 10–7 | 10–6 |
| SST-R1 | 0 | 0 | 0 | 0 | 5 | 15 |
| SST-R2 | 15 | 30 | 42 | 80 | 95 | 96 |
| SST-R3 | 2 | 1 | 1 | 4 | 50 | 89 |
| SST-R4 | 0 | 0 | 0 | 0 | 5 | 5 |
| SST-R5 | 20 | 48 | 63 | 82 | 95 | 95 |
Tabelle 9: Konzentration (nM) von Somatostatinanaloga
zum inhibieren von SRIF-Bindung an jeden der humanen klonierten
Somatostatinrezeptoren um 50 %.
| PTR | IC 50 (nM) |
| | SST-R1 | SST-R2 | SST-R3 | SST-R4 |
| 3173 | > 10–6 | 10–9 | 10–7 | 10–9 |
-
Beispiel 5 In vitro Bio-Reaktion von bevorzugten
Somatostatinanaloga mit cyclischem Gerüst.
-
A. Inhibierung von cAMP in humanen Karzinoid-BON-1-Zellen
durch das Somatostatinanalog mit cyclischem Gerüst PTR 3173:
-
Die
Aktivierung von SST-R5 führt
zur Verringerung der Adenylat-Cyclase-Aktivität. Somatostatinrezeptoren,
einschließlich
Typ 5-Rezeptoren, werden in dem humanen Karzinoid exprimiert, das
von der Zelllinie BON-1 abgeleitet ist. Diese humane Zellkultur
diente als ein in vitro Forschungsassay für neue Karzinoidtherapeutika.
Wechselwirkung von Somatostatinanaloga mit Somatostatinrezeptoren,
die in diesem System exprimiert werden, beeinträchtigt folglich die zelluläre Funktion
von BON-1. Es wurde gefunden, dass bevorzugte Analoga mit cyclischem
Gerüst
der vorliegenden Erfindung cAMP-Produktion nachfolgend auf Forskolin-Stimulierung
inhibieren. In diesem Signal-Absonderungsweg ist PTR 3173 äquipotent
wie das klinisch verwendete Arzneimittel Octreotid.
-
B. In vitro-Zellwachstumsinhibierung durch
das Somatostatinanalog mit cyclischem Gerüst PTR 3173:
-
Eine
pharmakologische Beurteilung der Wachstumsinhibierung wurde durchgeführt unter
Verwendung von CHO-Zellen, die humanes kloniertes SST-R5 exprimieren.
PTR 3173-Erkennung von SST-R5 bei zellulärem Gehalt war verbunden mit
beachtenswert höherer
Potenz der Wachstumsinhibierung im Vergleich mit dem nativen Hormon
und dem Arzneimittel Octreotid.
-
C. Inhibierung von Chromogranin A-Freisetzung
durch das Somatostatinanalog mit cyclischem Gerüst PTR 3173:
-
Eine
Abschätzung
der Chromogranin A-Freisetzung von BON-1 ist eine wichtige Untersuchung
mit dem Ziel zur Identifizierung potenzieller Antikarzinoid-Arzneimittel.
Chromogranin A ist einer der Hauptmediatoren bei der Degranulierung
von Tumorgranalien, die übermäßige Menge
vasoaktive Substanzen von Karzinoidtumoren sekretieren. PTR 3173
besitzt eine signifikante Antifreisetzungswirkung in diesem Stoffwechselweg.
Eine der faszinierendsten Entdeckungen hinsichtlich des Analogs
mit cyclischem Gerüst
im humanen BON-1-Assay ist seine äquivalente Potenz wie die des
nativen Hormons Somatostatin, wodurch eine potenzielle vorteilhafte
Wirkung. beim Karzinoidsyndrom angezeigt wird.
-
Beispiel 6: Vergleich von PTR 3173, Octreotid
und SRIF zur Bindung an nicht-Somatostatin G-gekoppelte Rezeptoren.
-
Somatostatinrezeptoren
gehören
zu der seven Transmembran-G-Protein gekoppelte Rezeptoren-Superfamilie.
G-Protein-gekoppelte Rezeptoren sind weit verbreitet im Körper und
vermitteln physiologische Aktivitäten verschiedener Hormone,
wie etwa Adrenalin, Acetylcholin, Opiaten, Neurokininen, Gastrin
und vielen anderen Hormonen. Ein Arzneimittelkandidat könnte entdeckt
werden durch einen definierten Subtyp von intrafamiliären Rezeptoren.
Jedoch könnten
potenzielle nachteilige Wirkungen im Körper bewirkt werden, aufgrund
der Erkennung anderer Rezeptoren, die verschieden von dieser Familie
sind. Diese Betrachtung erhöhte die
Wichtigkeit von inter- versus intra-Rezeptorselektivität im Zusammenhang
mit der Entwicklung physiologisch selektiver Arzneimittel.
-
NovaScreen
(Hannover, MD) führten
eine Beurteilung der nichtspezifischen Bindung an verschiedene G-Protein-gekoppelte
Rezeptorfamilien durch. Bindungsstudien an Neurokinin-, Opiat- und
Muscarin-Rezeptoren basierten auf einem Vergleich zwischen dem nativen
Hormon Somatostatin, Octreotid und PTR 3173.
-
In
einer Screeninguntersuchung, die durchgeführt wurde durch NovaScreen,
wurde eine signifikant hohe Affinität von Octreotid für Opiatrezeptoren
gefunden, während
unter den gleichen experimentellen Bedingungen PTR 3173 und das
native Hormon Somatostatin nicht an diese Rezeptoren banden (2).
Signifikant höhere
Affinität
von Octreotid über
PTR 3173 und dem nativen Hormon wurde ebenfalls für den Muscarin-2-Rezeptor
gefunden.
-
Die
Signifikanz der kreuzreaktiven Bindung von Octreotid an die Opiatrezeptoren
wurde weiter untersucht in dem Meerschweinchen-Ileum. Vorhergehende
Ergebnisse bestätigen
die Wirkung von Octreotid als ein Opiatantagonist, während unter
den gleichen experimentellen Bedingungen PTR 3173 die durch met-Enkephalin
hervorgerufene Zuckkontraktion nicht beeinflusst.
-
Beispiel 7: Die in vivo-Wirkung von Rezeptor-spezifischen
Somatostatinanaloga mit cyclischem Gerüst auf Wachstumshormonfreisetzung
-
Verfahren:
-
Inhibierung
von Wachstumshormon (GH)-Freisetzung als ein Ergebnis der Peptidverabreichung
wurde gemessen in männlichen
Wistar-Ratten. Die analoge Aktivität wurde in dieser Studie verglichen
mit SRIF oder Octreotid, unter Verwendung von 4 Ratten in jeder
Gruppe. Erwachsene männliche
Wistar-Ratten mit einem Gewicht von 200–250 g wurden bei einem konstanten
Licht-Dunkel-Zyklus (Licht von 8:00 bis 20:00 h), konstanter Temperatur
(21 ± 3°C) und relativer
Feuchte (55 ± 10%)
gehalten. Futterwasser und Leitungswasser waren ad libitum verfügbar. Am
Tag des Versuches wurden die Ratten anästhesiert mit Nembutal (IP,
60 mg/kg). Zehn Minuten nach Anästhesie
wurden Arzneimittel s.c. in einer Dosis von 0,01–100 Mikrogramm/kg verabreicht.
Stimulierung von GH wurde durch i.v.-Verabreichung von 0,5 g/kg
L-Arginin über
die Femoralvene durchgeführt.
Die Beprobung wurde wie folgt nachfolgend auf 5 Minuten Stimulierung
durchgeführt,
15 oder 30 Minuten nach Peptidverabreichung. Blutproben wurden aus
der abdominalen vena-cava gesammelt in Röhrchen, die Heparin enthielten(15
Einheiten pro ml Blut), und unmittelbar zentrifugiert. Plasma wurde
abgetrennt und bei –20°C gefrorengehalten
bis es untersucht wurde. Rattenwachstumshormon (rGH) [125I]-Gehalte wurden
bestimmt mittels eines Radioimmunoassaykits (Amersham). Der Standard
in diesem Kit ist kalibriert worden gegen eine Referenzstandardpräparation
(NIH-RP2), die erhalten wurde vom National Institute of Diabetes
and Digestive and Kidney Diseases. Alle Proben wurden doppelt gemessen.
Die Ergebnisse dieser Versuche sind in Figur 3 gezeigt.
-
Ergebnisse:
-
Wachstumshormonfreisetzung
wurde in Ratten stimuliert unter Verwendung von intravenöser (i.v.)
Bolus-Verabreichung von L-Arginin unter Nembutal-Anästhesie.
Der angegebene ED50 für
Octreotid (Bauer et al., ibid.) ist in diesem Modell ungefähr 0,1 Mikrogramm
pro Kilogramm. Folglich wurden Octreotid und die getesteten Rezeptor-spezifischen
Analoga mit cyclischem Gerüst
verabreicht in einer relativ hohen Dosis von 100 Mikrogramm pro
Kilogramm. Unter diesen experimentellen Bedingungen waren PTR 3205,
nicht enthalten in der vorliegenden Erfindung, und PTR 3173 äquipotente
Inhibitoren der Wachstumshormonfreisetzung im Vergleich mit Octreotid
(3). Faszinierende Ergebnisse wurden
erhalten mit PTR 3201, nicht in der vorliegenden Erfindung enthalten,
das ein Rezeptor 5-spezifisches Analog ist. Dieses selektive Analog
beeinflusste nicht die Wachstumshormonfreisetzung, wodurch gezeigt
wird, dass Wachstumshormoninhibierung nicht durch den Somatostatinrezeptor
Subtyp 5 vermittelt wird. Auf der anderen Seite zeigt die signifikante
Inhibierung, die gefunden wird mit PTR 3205, nicht in der vorliegenden
Erfindung enthalten, das selektiv ist für Rezeptorsubtyp 2, dass dieser
der prinzipielle Rezeptor ist, der Wachstumshormoninhibierung vermittelt.
Daher können
wir schließen,
dass die Wirkung auf das Wachstumshormon, die mit dem Arzneimittel
Octreotid oder PTR 3173 gefunden wird, auf ihre Erkennung von Rezeptorsubtyp
2 zurückzuführen ist.
-
Beispiel 8: Die in vivo Wirkung von Rezeptor-spezifischen
Somatostatinanaloga mit cyclischem Gerüst auf Glucagonfreisetzung.
-
In
vivo-Bestimmung der Freisetzung von Glucagon als ein Ergebnis von
Peptidverabreichung wurde gemessen in männlichen Wistar-Ratten. Die
analoge Aktivität
wurde in dieser Studie verglichen mit SRIF oder Octreotid, unter
Verwendung von 4 Ratten in jeder Gruppe. Zeitverlaufsprofile für Glucagonfreisetzung
unter konstanten experimentellen Bedingungen wurden gemessen.
-
Männliche
Wistar-Ratten ließ man über Nacht
fasten. Die Tiere wurden mit Nembutal anästhesiert (i.p., 60 mg/kg).
Zehn Minuten nach Anästhesie
wurden Arzneimittel s.c. in einer Dosis von 0,01–100 Mikrogramm/kg verabreicht.
Die Stimulierung der Glucagonsekretion wurde durch i.v.-Verabreichung von
L-Arginin, 0,5 g/kg, 5 Minuten vor dem Sammeln von Blut aus der
Portalvene durchgeführt.
Die Hormonkonzentration wurde durch RIA gemessen.
-
Der
einzig statistisch signifikante Unterschied der Glucagongehalte
im Vergleich mit der Kontrolle wurde erhalten mit der hohen Dosis
von 100 Mikrogramm pro Kilogramm PTR 3173 (4), einer
1000-fach höheren
Dosis im Vergleich zu der ED50 von PTR 3173 auf Wachstumshormonfreisetzung.
Diese Ergebnisse heben hervor, dass dieses Analog mit cyclischem
Gerüst
signifikante physiologische Selektivität im Vergleich mit Octreotid
aufweist, wie oben in Tabelle 4 zusammengefasst.
-
Beispiel 9: Die in vivo Wirkung von Rezeptor-spezifischen
Somatostatinanaloga mit cyclischem Gerüst auf Insulinfreisetzung.
-
Die
Inhibierung der Insulinfreisetzung durch Somatostatinanaloga ist
gut dokumentiert in der Literatur (Bauer et al., ibid., Lamberts
et al., 1996, ibid.). Jedoch sind synthetische Somatostatinanaloga
mit einer langen physiologischen Aktivitätsdauer beschrieben worden
als weniger aktiv für
Insulin im Vergleich mit ihrer potenten Inhibierung der Wachstumshormon- oder Glucagonfreisetzung
(Bauer et al., ibid., Lamberts et al., 1996, ibid.). Sandoz beansprucht,
dass eine physiologische Selektivität von Octreotid für das Wachstumshormon
gegenüber
Insulin besteht. Jedoch supprimiert in Typ 2 Diabetes das langwirkende
analoge Octeotid Insulin und Glucagonfreisetzung, wodurch Glucosegehalte
entweder unverändert
bleiben oder etwas erhöht
werden.
-
Andere
klinische Versuche haben gezeigt, dass das Versagen von Octreotid
beim Absenken glykämischer
Werte bei Typ 2-Diabetes trotz seiner Fähigkeit zum Erniedrigen von
Glucagon und Wachstumshormon möglicherweise
abhängig
war von einer temporären
Blockade von residualer endogener Insulinsekretion, die durch seine
Verabreichung induziert wird. In gesunden Individuen führte die
Verabreichung von Octreotid zur Entwicklung von schwacher Nüchtern-Hyperglykämie und
deutlicher Nüchtern-Hypoinsulinämie. Darüber hinaus
ist Octreotid beschrieben für
die Behandlung von Nesidioblastose, ein Syndrom, das verbunden ist
mit übermäßiger Freisetzung
von Insulin aus dem Pankreas, was die physiologisch nichtspezifische
Wirkung von Octreotid auf Insulin hervorhebt (Kane et al., J. Clin.
Inves., 100:1888, 1997).
-
Zum
Beurteilen der physiologischen Wirkungen von Rezeptor-spezifischen
Somatostatinanaloga mit cyclischem Gerüst auf Insulinfreisetzung wurde
das gleiche experimentelle Protokoll, das von Sandoz zur Beurteilung
von Octreotid verwendet wurde, durchgeführt. Insulinstimulierung wurde
induziert durch i.v.-Bolus-Verabreichung von D-Glucose an Ratten,
die man über
Nacht fasten ließ.
-
Verfahren:
-
Eine
in vivo-Bestimmung der Insulinfreisetzung als ein Ergebnis von Peptidverabreichung
wurde gemessen in männlichen
Wistar-Ratten. Die analoge Aktivität wurde verglichen in dieser
Studie mit SRIF oder Octreotid, unter Verwendung von 4 Ratten in
jeder Gruppe. Zeitverlaufsprofile für GH- Freisetzung unter konstanten experimentellen
Bedingungen wurden gemessen.
-
Männliche
Winstar-Ratten wurden über
Nacht fasten gelassen. Die Tiere wurden anästhesiert mit Nembutal (i.p.,
60 mg/kg). Zehn Minuten nach Anästhesie
wurden Arzneimittel s.c. in einer Dosis von 0,01–100 Mikrogramm/kg verabreicht,
30 Minuten vor Stimulierung der Insulinsekretion, ausgeführt durch
i.v.-Verabreichung von 0,5 g/kg D-Glucose, 5 Minuten vor Sammeln von Blut
aus der abdominalen vena-cava. Hormongehalte wurden durch RIA gemessen.
-
Ergebnisse:
-
PTR
3205, nicht in der vorliegenden Erfindung enthalten, und Octreotid
waren beide aktive Inhibitoren der Insulinfreisetzung (5a). Der ED50 von Octreotid war nachfolgend
auf subkutane Injektion zwischen 10 bis 100 Mikrogramm pro Kilogramm,
gemäß dem ED50,
der von Sandoz angegeben wird – 26
Mikrogramm pro Kilogramm. Die signifikante Wirkung, die gefunden
wird mit PTR-3205, nicht in der vorliegenden Erfindung enthalten,
zeigt, dass Somatostatinrezeptorsubtyp 2 die Wirkung auf Wachstumshormon
und auch auf Insulin vermittelt. Diese Rezeptor-Effektor-Beziehung wurde korreliert
mit früher
veröffentlichten
Daten, die angaben, dass Somatostatin β-Zellsekretion über Rezeptorsubtyp
2 in isoliertem perfundiertem humanem Pankreas inhibiert. Im Gegensatz
zu der signifikanten Wirkung, die gefunden wird für PTR 3205,
nicht in der vorliegenden Erfindung enthalten, und Octreotid, waren
hohe Dosen (100 Mikrogramm pro Kilogramm) PTR 3201, nicht in der
vorliegenden Erfindung enthalten, und PTR 3173 inaktiv für Insulin.
Es sollte festgehalten werden, dass PTR 3173 in einer ähnlichen
Dosis eine signifikante Wirkung auf die Freisetzung von Wachstumshormon
hatte. Diese faszinierende physiologische Selektivität von PTR
3173 führte
uns zur Wiederholung dieses Versuches mit einer viel höheren Dosis
von bis zu 1 Milligramm pro Kilogramm. Unter diesen experimentellen
Bedingungen wurde PTR 3173 definiert als ein physiologisch selektives
Somatostatinanalog mit keiner nennenswerten Wirkung auf Insulin
im Vergleich mit dem Arzneimittel Octreotid (5b).
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-