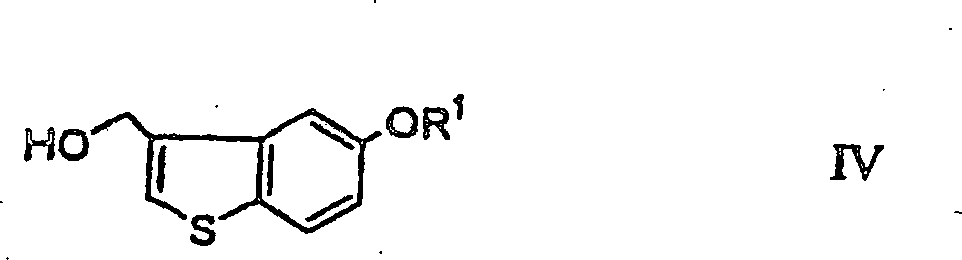-
Technisches Gebiet
-
Die
vorliegende Erfindung betrifft 5-Hydroxybenzo[b]thiophen-3-carbonsäurederivate,
die Schlüsselausgangsmaterialien
zur Herstellung von Verbindungen sind, die auf dem Gebiet der Pharmazeutika
nützlich sind. Stand
der Technik Hydroxybenzo[b]thiophen-3-carbonsäurederivate
der allgemeinen Formel (I):
wobei R ein Wasserstoffatom oder eine Hydroxyschutzgruppe
ist, sind wichtige Ausgangsmaterialien zur Synthese von pharmakologisch
wirksamen Verbindungen. Zum Beispiel ist eine Verbindung der Formel
(I) bei der Synthese von Benzothiophencarboxamidderivaten der allgemeinen
Formel (VI) essentiell:
wobei R die vorstehend angegebene
Bedeutung hat und X ein Wasserstoffatom oder ein Alkylrest ist.
Die Benzothiophencarboxamidderivate sind spezifische Antagonisten
von PGD
2 und es ist bekannt, daß sie als
Arzneimittel bei der Behandlung verschiedener Krankheiten, die mit
einer Funktionsstörung
der Mastzelle verbunden sind, die durch übermäßige Produktion von PGD
2 hervorgerufen wird, zum Beispiel systemische
Mastozytose, Störung
der systemischen Mastzellaktivierung, Luftröhrenkontraktion, Asthma, allergische
Rhinitis, allergische Conjunctivitis, Urtikaria, Verletzung durch
ischämische
Reperfusion, Entzündung
und atopische Dermatitis (WO 97/00853, PCT/JP 97/04527 (WO 98/25919))
nützlich
sind. Unter den Verbindungen der Formel (VI) hat eine Verbindung,
wobei OR eine 5-Hydroxylgruppe ist und X ein Wasserstoffatom ist
(nachfolgend als "Verbindung
A bezeichnet") eine
besonders hohe antagonistische Wirkung auf PGD
2,
wobei sie eine exzellente Wirkung gegen Nasenverstopfung zeigt und
wird als vielversprechendes Arzneimittel zur Behandlung nasaler Verstopfung
ins Auge gefaßt.
-
WO
97/02269 offenbart kondensierte Thiazolderivate mit 5-HT-Rezeptoraffinität, die zur
Behandlung zentraler Nervenstörungen
nützlich
sind.
-
Offenbarung
der Erfindung
-
Ein
Verfahren zur Herstellung der vorstehend genannten Verbindungen
wird durch das folgende Reaktionsschema dargestellt (WO 98/25919):
-
Um
die Verbindung A weitverbreitet klinisch einzusetzen ist es wesentlich,
ein Verfahren zur Herstellung eines Ausgangsmaterials, die Verbindung
(I), zu etablieren, wobei das Verfahren sicher, effizient und industriell
anwendbar ist.
-
Allerdings
ist es schwierig, Benzothiophenderivate mit einer 5-Hydroxylgruppe,
wie die Verbindung (I), zu synthetisieren und es gab bislang keine
industriell anwendbare Verfahren. Die bestehenden Verfahren schließen verschiedene
komplizierte Verfahren ein, sind ineffizient und von geringer Ausbeute.
Zum Beispiel gab es Verfahren, wo 5-Acetoxybenzo[b]thiophen bromiert wird
um 3-Brom-5-acetoxybenzo[b]thiophen zu ergeben, das wiederum an
der 5-Acetoxygruppe mit einer Benzylgruppe geschützt wird, um 3-Brom-5-benzyloxybenzo[b]thiophen
zu ergeben, gefolgt von einer Umwandlung in die metallische Form
mit Magnesium, einer Einführung
von Kohlendioxid und der Entfernung der Benzylgruppe (J. Chem. Soc.
(C) 1967, 1899–1905);
oder es wird 5-Brombenzo[b]thiophen
einer Friedel-Crafts-Reaktion unterworfen, um 3-Acetyl-5-Brombenzo[b]thiophen
zu ergeben, gefolgt von einer Oxidation mit Natriumhypochlorit,
um 5-Brombenzo[b]thiophen-3-carbonsäure zu ergeben (Nippon-Kagaku
Zasshi vol. 86, Nr. 10, 1067–1072
(1965), J. Chem. Soc (C). 1967, 2084–2089). 5-Hydroxybenzo[b]thiophen-3-carbonsäure oder
5-Acetoxybenzo[b]thiophen-3-carbonsäure werden
dann, ausgehend von den vorstehenden Umsetzungsprodukten sythetisiert.
Allerdings ist ein Ausgangsmaterial wie 5-Hydroxybenzo[b]thiophen
oder 5-Brombenzo[b]thiophen
nicht im Handel erhältlich
und mußte
in allen Fällen
aus einem geeigenten Reagenz synthetisiert werden (z. B., J. Am.
Chem. Soc., 57, 1611, (1935), J. Heterocyclic Chem, 25,1271 (1988)),
was das Syntheseverfahren länger
und komplexer machte.
-
Die
vorliegende Erfindung löst
die Probleme der bestehenden Verfahren und stellt ein Verfahren
zur Herstellung der Verbindungen der Formel (I) bereit, das industriell
anwendbar, effizient und sicher ist.
-
Folglich
stellt die vorliegende Erfindung ein Verfahren zur Herstellung einer
Verbindung der Formel (I) bereit:
wobei R für Wasserstoff oder eine Hydroxyschutzgruppe
steht, oder eines reaktiven Derivats davon, umfassend Aussetzen
von 4-Mercaptophenol Reaktionen zum Einführen einer Propargylgruppe
und Schützen
der Hydroxylgruppe, um eine Verbindung der Formel (II) zu erhalten:
wobei R
1 für eine Hydroxyschutzgruppe
steht; Oxidieren der Verbindung (II), um eine Verbindung der Formel (III)
zu erhalten:
wobei R
1 für eine Hydroxyschutzgruppe
steht; Aussetzen der Verbindung (III) einer thermischen Umlagerungsreaktion,
um eine Verbindung der Formel (IV) zu erhalten:
wobei R
1 wie
vorstehend definiert ist; und Aussetzen der Verbindung (IV) einer
schrittweisen Oxidierung der Hydroxymethylgruppe und gegebenenfalls
Entfernen der Schutzgruppe.
-
Die beste Ausführungsform
zur Anwendung der Erfindung
-
Die
hierin verwendeten Begriffe werden nachstehend definiert.
-
Der
Begriff "Hydroxyschutzgruppe" bedeutet einen Alkyl-,
Alkoxyalkyl-, Acyl-, Aralkyl-, Alkylsulfonyl-, Arylsulfonyl-, Alkyl-substituierten
Silyl-, Alkoxycarbonyl-, Aryloxycarbonyl-, Aralkyloxycarbonyl- oder
Tetrahydropyranylrest.
-
Der
Begriff "Alkyl" bedeutet einen C1-20 linearen oder verzweigten Alkylrest,
insbesondere eine Methyl-, Ethyl-, n-Propyl-, Isopropyl-, n-Butyl-,
Isobutyl-, sec-Butyl-, tert-Butyl-, n-Pentyl-, Isopentyl-, Neopentyl-, tert-Pentyl-,
Hexyl-, Heptyl-, Octyl-, Nonyl-, Decyl-, Undecyl-, Dodecyl-, Tridecyl-,
Tetradecyl-, Pentadecyl-, Hexadecyl-, Heptadecyl-, Octadecyl-, Nonadecyl-
und Eicosylgruppe, wobei ein C1-C6 Alkylrest bevorzugt ist.
-
Der
Begriff "Alkoxy" bedeutet einen C1 bis C6 linearen
oder verzweigten Alkoxyrest, insbesondere eine Methoxy-, Ethoxy-,
n-Propoxy-, i-Propoxy-, n-Butoxy-, i-Butoxy-, s-Butoxy-, t-Butoxy-, n-Pentyloxy-, i-Pentyloxy-,
Neopentyloxy-, s-Pentyloxy-, t-Pentyloxy-,
n-Hexyloxy-, Neohexyloxy-, i-Hexyloxy-, s-Hexyloxy- und t-Hexyloxygruppe, wobei
ein C1-C3 Alkoxyrest
bevorzugt ist.
-
Der
Begriff "Alkoxyalkyl" bedeutet einen mit
einem Alkyoxyrest substituiertem Alkylrest, einschließlich einer
Methoxymethyl-, Ethoxymethyl-, Methoxyethoxymethyl-, Ethoxyethyl-
und Methoxypropylgruppe.
-
Der
Begriff "Acyl" bedeutet einen C1-C12 Acylrest, der
sich von einer aliphatischen Carbonsäure oder einer aromatischen
Carbonsäure
ableitet. Beispiele für
einen von einer aliphatischen Carbonsäure abgeleiteten Acylrest schließen eine
Acetyl-, Chloracetyl-, Trichloracetyl-, Propionyl-, Butyryl- und
Valerylgruppe ein, und Beispiele für einen von einer aromatischen
Carbonsäure
abgeleiteten Acylrest schließen
eine Benzoyl-, p-Nitrobenzoyl-,
p-Methoxybenzoyl-, p-Brombenzoyl-, Toluoyl- und Naphthoylgruppe
ein.
-
Der
Begriff "Aryl" bedeutet z. B. eine
Phenyl-, Naphthylgruppe oder einen polycyclischen aromatischen Kohlenwasserstoffrest.
Zusätzlich
kann ein Arylrest mit den folgenden Substituenten substituiert sein:
Beispiele
für Substituenten
schließen
Alkylreste ein wie eine Methyl-, Ethyl-, n-Propyl-, Isopropyl-,
Isobutyl-, sec-Butyl-, tert-Butyl-, n-Peetyl-, Isopentyl-, Neopentyl-
oder tert-Pentylgruppe,
niedere Alkoxyreste wie eine Methoxy- oder Ethoxygruppe, Halogene
wie Fluor, Chlor, Brom oder Iod, eine Nitrogruppe, eine Hydroxygruppe,
eine Carboxygruppe, eine Cyanogruppe, eine Sulfonylgruppe, eine
Aminogruppe, niedere Alkylaminreste wie eine Methylamino-, Dimethylamino-,
Ethylmethylamino- oder Diethylaminogruppe ein. Der Arylrest kann einen
oder mehrere Substitutenten an jeder möglichen Position besitzen.
Spezielle Beispiele für
Arylreste schließen
eine 2-Methylphenyl-,
3-Methylphenyl-, 4-Methylphenyl-, 2-Ethylphenyl-, 3-Ethylphenyl-,
4-Ethylphenyl-,
4-Pentylphenyl-, 4-Carboxyphenyl-, 4-Acetylphenyl-, 4-(N,N-Dimethylamino)phenyl-,
4-Nitrophenyl-, 4-Hydroxyphenyl-, 4 Methoxyphenyl-, 4-Fluorphenyl-, 4-Chlorphenyl-
und 4-Iodphenylgruppe ein.
-
Der
Arylrest in den "Aralkyl-" "Arylsulfonyl-", "Aryloxycarbonyl-" oder "Aralkyloxycarbonykesten", die nachstehend
beschrieben sind, kann ähnliche
Substituenten wie vorstehend definiert haben.
-
Der
Begriff "Aralkyl" bedeutet einen Alkylrest,
der mit einen Arylrest substituiert ist und schließt eine Benzyl-,
4-Methylbenzyl-, 4-Methoxybenzyl-, 3,4-Dimethoxybenzyl-, Naphthylmethyl-,
und Phenylethylgruppe ein.
-
Der
Begriff "Alkylsulfonyl" bedeutet eine Sulfonylgruppe,
die mit einem Alkylrest substituiert ist und schließt eine
Methansulfonyl- und Ethansulfonylgruppe ein.
-
Der
Begriff "Arylsulfonyl" bedeutet eine Sulfonylgruppe,
die mit einem Arylrest substituiert ist und schließt eine
Benzolsulfonyl- und p-Toluolsulfonylgruppe ein.
-
Der
Begriff "Alkyl-substituiertes
Silyl" bedeutet
eine Mono-, Di- oder Tri-Alkyl-substituierte
Silylgruppe, zum Beispiel eine Methylsilyl-, Dimethylsilyl-, Trimethylsilyl- und t-Butyldimethylsilylgruppe.
-
Der
Begriff "Alkoxycarbonyl" bedeutet z.B. eine
Methoxycarbonyl-, Isopropoxycarbonyl-, und t-Butoxycarbonylgruppe.
-
Der
Begriff "Aryloxycarbonyl" bedeutet z. B. eine
Phenoxycarbonylgruppe.
-
Der
Begriff "Aralkyloxycarbonyl" bedeutet z. B. eine
Benzyloxycarbonylgruppe.
-
Obwohl
alle vorstehend genannten Hydroxyschutzgruppen als Hydroxyschutzgruppen,
wie durch R1 oder R in den entsprechenden
vorstehenden Formeln gezeigt, bevorzugt sind, wird ein Arylsulfonylrest
stärker bevorzugt
und unter ihnen ist eine Benzolsulfonylgruppe besonders bevorzugt.
-
Das
Verfahren der vorliegenden Erfindung wird nachstehend genauer beschrieben.
Falls ein Substituent oder Substituenten anwesend ist/sind, der/die
möglicherweise
mit der Umsetzung interferiert/en, kann/können er/sie zu einem gewünschten
Zeitpunkt angemessen geschützt
und dann entschützt
werden. Derartiges Schützen
oder Entschützen
kann durch ein im Fachgebiet bekanntes Verfahren erreicht werden.
-
-
Wobei
R und R1 wie vorstehend definiert sind.
-
Schritt 1
-
Dieser
Schritt betrifft die Einführung
einer Propargylgruppe an die Mercaptogruppe des 4-Mercaptophenols
(1) und das Schützen
der Hydroxylgruppe.
-
Die
Einführung
einer Propargylgruppe wird unter Verwendung eines Propargylhalogenids
wie Propargylbromid oder Propargylchlorid in Gegenwart eines basischen
Mittels durchgeführt.
Die Umsetzung kann innerhalb einiger zehn Minuten bis einiger Stunden
bei Raumtemperatur unter Anwendung einer anorganischen Base als
ein basisches Mittel wie Kaliumcarbonat oder Natriumcarbonat, oder
einer organischen Base wie Triethylamin, Pyridin oder 4-Dimethylaminopyridin
in einem Lösungsmittel
wie Aceton, Ethylacetat, Tetrahydrofuran oder Acetonitril, durchgeführt werden.
-
Wenn
eine starke Base wie Kaliumhydroxid oder Natriumhydroxid verwendet
wird, kann sie auch in einem Zwei-Schichten-Lösungssystem wie Toluol-Wasser
oder Xylol-Wasser durchgeführt
werden.
-
Das
Schützen
der Hydroxylgruppe kann unter Verwendung einer herkömmlichen
Hydroxyschutzgruppe in einer herkömmlichen Art und Weise durchgeführt werden.
Bevorzugte Schutzgruppen, die im vorliegenden Verfahren verwendet
werden, sind solche, die während
den oxidativen Umsetzungen im zweiten und vierten Schritt des vorliegenden
Verfahrens und im zweiten Schritt des nachstehenden Verfahrens II,
zur Herstellung der Verbindung der Formel (VI) und auch während der
Wittig-Reaktion des dritten Schrittes dieses Verfahrens keinen Veränderungen
unterliegen und die im vierten Schritt leicht entschützt werden
können,
um Abgangsgruppen zu ergeben, die von, zum Beispiel, Verbindung
A zu deren Reinigung einfach abzutrennen sind, was der Verbindung
der Formel VI entspricht, wobei OR eine 5-Hydroxygruppe ist, X ein
Wasserstoffatom ist und die Doppelbindung in der Z-Konfiguration
ist. Beispiele für
deratige Hydroxyschutzgruppen schließen einen Alkyl-, Alkoxyalkyl-,
Acyl-, Aralkyl-, Alkylsulfonyl-, Arylsulfonyl-, Alkyl-substituierten
Silyl-, Alkoxycarbonyl-, Aryloxycarbonyl, Aralkyloxycarbonyl- oder
Tetrahydropyranylrest ein.
-
Wenn
man die Voraussetzungen bedenkt, daß eine Schutzgruppe während der
Wittig-Reaktion
unter stark basischen Bedinungen überleben sollte, daß sie einfach
zu entschützen
sein soll, zum Beispiel im vierten Schritt bei der Herstellung von
Verbindung A, und daß sie
von Verbindung A abtrennbar sein soll, dann ist ein Arylsulfonylrest
stäker
bevorzugt und eine Benzolsulfonylgruppe besonders bevorzugt.
-
Die
Benzolsulfonylgruppe ist in wasserfreien Lösungsmittel gegenüber Basen
relativ stabil und gibt bei Entschützung Benzolsulfonsäure, die
wasserlöslich
ist und leicht vom Endprodukt der Formel (VI) abgetrennt werden
kann. Das Schützen
und Entschützen kann
durch auf dem Fachgebiet bekannte Verfahren ausgeführt werden.
Zum Beispiel wird im Fall der Benzolsulfonylgruppe die Einführung der
Benzolsulfonylgruppe in einer Weise ausgeführt, die zur Einführung einer
Propargylgruppe, unter Verwendung von Benzolsulfonylchlorid ähnlich ist.
-
Schritt 2
-
Dieser
Schritt betrifft die Oxidation der Verbindung (II). Es sind Oxidationsverfahren
bekannt, wie zum Beispiel wässrige
Wasserstoffperoxid-Essigsäure
(J. Am, Chem. Soc., 87, 1109–1114
(1965)), wässrige
Wasserstoffperoxid-Titan III-chlorid (Synthesis 1981, 204–206), m-Chlorperbenzoesäure (Org.
Synth., 64, 157–163 (1985)),
oder Natrium-Metaperiodat
(J. Org. Chem., 27, 282–284
(1962)) verwenden. Im vorliegenden Schritt ist es bevorzugt, einen
leichten Überschuß an 30%-igem
wässrigem
Wasserstoffperoxid in einem alkoholischen Lösungsmittel wie Ethanol, Methanol,
Isopropanol oder tert-Butanollösung, enthaltend
Ameisensäure zu
verwenden. Die Umsetzung wird innerhalb einiger zehn Minuten bis
zu einigen Stunden unter Kühlung
oder bei Raumtemperatur durchgeführt.
-
Schritt 3
-
Dieser
Schritt betrifft die Umwandlung der Verbindung (III) in die Hydroxymethylverbindung
(IV) durch eine thermische Umlagerungsreaktion. Die thermische Umlagerunsgsreaktion
in diesem Schritt wird nach dem in J. C. S. Chem. Communication.,
1974, 848–849
beschriebenen Verfahren ausgeführt.
Beispiele für
bevorzugte Lösungsmittel
für diese
Umsetzung schließen
Dioxan, 1,2-Dimethoxyethan, Propylacetat und 3-Pentanon ein. Die
Umsetzung wird unter Rückfluß in einem
Lösungsmittel über einige
Stunden hinweg, gefolgt von der Zugabe einer Säure (z. B. p-Toluolsulfonsäure, Methansulfonsäure oder
Schwefelsäure)
zum entstehenden Intermediat, durchgeführt.
-
Schritt 4
-
Dieser
Schritt betrifft die Oxidation der Verbindung (IV), um eine Carbonsäure (I)
zu ergeben. Die Oxidation kann entweder direkt oder schrittweise
duchgeführt
werden. Beispiele für
ein Oxidationsmittel zur Umsetzung eines aromatischen primären Alkohols
zur korrespondierenden Carbonsäure
schließen
Chromsäuren (Synthesis.
1986, 285–288),
Kaliumpermanganat (J. Org. Chem., 18, 806–809 (1953)) und Rutheniumoxide (J.
C. S. Chem. Communication., 1979, 58–59)) ein. Diese Verfahren
haben jedoch nicht nur hinsichtlich der Ausbeute sondern auch hinsichtlich
der folgenden Punkte Nachteile. Zum Beispiel ist die Umsetzungszeit
lang, es ist eine Entgiftungsbehandlung des Oxidationsmittels nach
der Umsetzung nötig,
die Reagenzien sind instabil und/oder sie beinhalten komplizierte
Arbeitsschritte.
-
Im
Gegensatz dazu kann in manchen Fällen
eine schrittweise Oxidation, wo ein primärer Alkohol zu einem Aldehyd
oxidiert wird und dann zu einer Carbonsäure, in Hinblick auf die Ausbeute
von Vorteil sein. Im Allgemeinen wurde die Oxidation eines Alkohols
zu einem Aldeyd unter Verwendung eines Oxidationsmittels aus der
Chromsäureserie,
zum Beispiel den Jones Reagenz (J. Org. Chem, 40, 1664–1665 (1975))
dem Collins Reagenz (J. C. S. Chem. Communication., 1972 1126))
oder, Pyridiniumchlorchromat (Tetrahedron Lett., 2647–2650 (1975))
ausgeführt.
Es gibt auch bekannte Verfahren, die Mangandioxyd (Helv. Chim. Acta.,
39, 858–862
(1956)) oder Dimethylsulfoxid (Swern oxidation, J. Org. Chem, 43,
2480–2482
(1978)) verwenden. Allerdings haben diese bestehenden Verfahren
Nachteile. Zum Beispiel sind Chromsäuren für den menschlichen Körper giftig
und müssen
nach Gebrauch entgiftet werden. Ferner ist die Swernoxidation unter
Verwendung von Dimethylsulfoxid-oxalylchlorid nicht für die Herstellung
im großen
Maßstab
geeignet, weil sie von der Entstehung von Kohlenmonoxid, das für die Arbeiter
schädlich
ist, und von schwefligem Geruch begleitet ist und auch bei niederen
Temperaturen ausgeführt
werden muß,
zum Beispiel zwischen –50 °C und –78 °C.
-
Alkohol
(IV) kann fast quantitativ in Aldehyd (IV) umgewandelt werden durch
ein Verfahren, wo ein Alkohol (IV) mit einem Oxidationsmittel, wie
einer Halogen-Oxo- Säure in Gegenwart
von z. B. 2, 2, 6, 6-Tetramethylpiperidin-1-oxyl ("TEMPOs" genannt), gemäß der Beschreibung
in der Literatur (z. B., J. Org. Chem., 52, 2559–2562 (1987)), oxidiert wird,
wobei die Probleme der bekannten Verfahren gelöst werden. Beispiele geeigneter
TEMPOs schließen
2,2,6,6-Tetramethylpiperidin-1-oxyl, 4-Methoxy-2,2,6,6-tetramethylpiperidin-1-oxyl,
4-Acetylamino-2,2,6,6-tetramethylpiperidin-1-oxyl,
4-Benzoyloxy-2,2,6,6-tetramethylpiperidin-1-oxyl und 4-Cyano-2,2,6,6-tetramethylpiperidin-1-oxyl
ein. Beispiele geeigneter Halogen-Oxo-Säuren schließen Natriumhypochlorit,
Natriumhypobromit, Natriumbromit und höhere Bleichpulver mit ein.
Eine Lösung
eines Oxidationsmittels kann zum Beispiel auf pH 8,5 bis 9,5 mit
einer Mineralsäure
wie Natriumhydrogencarbonat, Salzsäure oder Schwefelsäure eingestellt
werden. In einer anderen Ausführungsform
kann eine Lösung
eines Oxidationsmittels in Gegenwart von Natriumhydrogencarbonat
zugegeben werden. Die Umsetzung kann innerhalb einiger Minuten bis
zu einiger zehn Minuten bei einer Temperatur von Eiskühlung bis
zu Raumtemperatur in einem Lösungsmittel
wie Ethylacetat, Acetonitril oder Dichlormethan durchgeführt werden.
-
Wenn
die Umsetzungslösung,
die das entstandene Aldehyd (IV) enthält, angesäuert wird und Natriumchlorit
und wässriges
Wasserstoffperoxid zugegeben werden, wird das Aldehyd unter Eiskühlung innerhalb von
einigen zehn Minuten bis zu einigen Stunden in die Carbonsäure umgewandelt.
-
Wenn
gewünscht,
kann das Produkt weiter einer Entschützung der 5-Hydroxyschutzgruppe und/oder einer Umwandlung
in reaktive Derivate an der 3-Carboxylgruppe
unterworfen werden. Derartige "reaktive
Derivate" schließen die
entsprechenden Säurehalogenide
(z. B. Chlor, Brom, Jod), Säureanhydride
(z. B. gemischte Säureanhydride
mit Ameisensäure
oder Essigsäure),
aktivierte Ester (z. B. Succinimidester) ein und schließen acylierende
Agentien, die im allgemeinen zur Acylierung von Amiogruppen verwendet
werden, ein. Zum Beispiel wird, um Säurehalogenide zu erhalten,
eine Carbonsäure
mit Thionylhalogenid (z. B. Thionylchlorid), Phosphorhalogenid (z.
B. Phosphortrichlorid, Phosphorpentachlorid), Oxalylhalogenid (z.
B. Oxalylchlorid) gemäß bekannter
Verfahren, (z. B., Shin-jikken Kagaku Koza, vol. 14, 5.1787 (1978);
Synthesis 852–854
(1986); Shin-jikken Kagaku Koza vol. 22, S. 115 (1992)) umgesetzt.
-
Das
vorstehend erwähnte
Verfahren II wird nachstehend veranschaulicht. Verfahren II ist
kein Verfahren der vorliegenden Erfindung, aber es benötigt die
Verbindung der Formel I als ein Ausgangsmaterial. Verfahren
II
-
Wobei
R und X wie vorstehend definiert sind und die Doppelbindung eine
E- oder Z-Konfiguration
darstellt.
-
Schritt 1
-
Dieser
Schritt betrifft die Herstellung einer Verbindung der Formel (IX)
durch Umsetzung einer Verbindung der Formel (I) oder eines reaktiven
Derivats davon mit einer Verbindung der Formel (V') oder eines Salzes davon
in einer Weise, die ähnlich
ist zu der, die in Verfahren III vorstehend beschrieben ist. Die
Herstellung von einigen Verbindungen der Formel (V') ist in Chem. Pharm.
Bull. Vol. 37, No. 6 1524–1533
(1989) beschrieben.
-
Schritt 2
-
Dieser
Schritt betrifft die Herstellung eines Aldehyds der Formel (X) durch
Oxidieren einer Verbindung der Formel (IX). Die Umsetzung kann über einige
Stunden hinweg unter Kühlung
oder bei Raumtemperatur ausgeführt
werden, indem man ein Oxidationsmittel verwendet, das ausgewählt ist
aus der Chromsäureserie wie
z. B. dem Jones Reagenz, Collins Reagenz, Pyridiniumchlorchromat,
Pyridiniumdichromat oder Dimethylsulfoxid-Oxalylchlorid, in einem
Lösungsmittel
wie einem chlorierten Kohlenwasserstoff (z. B. Chloroform oder Dichlormethan),
einem Ether (z. B. Ethylether oder Tetrahydrofuran), Aceton oder
Benzol.
-
Schritt 3
-
Dieser
Schritt betrifft die Bildung einer Doppelbindung durch Umsetzen
einer Verbindung der Formel (X) mit einem Ylid (Ph3P=CH(CH2)3COOH). Die Umsetzung
zum Einführen
einer Doppelbindung kann in einer für die Wittig-Reaktion herkömmlichen
Weise durchgeführt
werden. Die Ylide, die bei der Umsetzung verwendet werden, können in
Anwesenheit einer Base synthetisiert werden, indem man ein Phosphoniumsalz,
das aus Triphenylphosphin und einem Halogenalkan mit einer gewünschten
zu kondensierenden Alkylgruppe, zum Beispiel 5-Brompentansäure, hergestellt
wurde behandelt. Beispiele für
Basen schließen
Dimsyl-Natrium, Dimsyl-Kalium,
Natriumhydrid, n-Butyllithium, Kalium-t-butoxid und Lithiumdüsopropylamid ein.
Die Umsetzung wird innerhalb einiger Stunden bei Raumtemperatur
in einem Lösungsmittel
wie Ether, Tetrahydrofuran, n-Hexan, 1,2-Dimethoxyethan oder Dimethylsulfoxid
erreicht.
-
Schritt 4
-
In
diesem Schritt wird eine Verbindung (VI), wobei R eine Hydroxyschutzgruppe
ist, gegebenenfalls entschützt,
um eine Verbindung (VI-1) zu ergeben. Die Umsetzung kann auf herkömmliche
Weise durchgeführt werden,
indem man einen Katalysator wie Salzsäure, Schwefelsäure, Natriumhydroxid,
Kaliumhydroxid oder Bariumhydroxid verwendet. Die Umsetzung wird
innerhalb einiger zehn Minuten bis einiger Stunden bei Erwärmen in
einem Lösungsmittel
wie Methanol-Wasser, Ethanol-Wasser, Aceton-Wasser oder Acetonitril-Wasser, vorzugsweise
Dimethylsulfoxyd-Wasser erreicht.
-
Die
folgenden Beispiele werden bereitgestellt, um die vorliegende Erfindung
detaillierter weiter zu veranschaulichen und sollen nicht dahingehend
ausgelegt werden, daß sie
ihren Schutzbereich begrenzen. Die in den Beispielen verwendeten
Abkürzungen
haben folgende Bedeutung:
- Ph:
- Phenyl
- Ac:
- Acetyl
- TEMPO:
- 2,2,6,6-Tetramethylpiperidin-1-oxyl
-
Beispiel 1
-
4-(2-Propyn-1-ylthio)phenylbenzolsulfonat
(2) (1)
Schritt 1:
-
4-Mercaptophenol
(1) (37,85 g, 300 mmol) und Propargylbromid (42,82 g, 360 mmol)
wurden in Ethylacetat (757 ml) gelöst. Zu der Lösung wurde über 25 Minuten
hinweg unter Rühren
und Eiskühlung
tropfenweise Triethylamin (42,5g, 420 mmol) zugegeben. Nach Rühren für weitere
1,5 Stunden bei der gleichen Temperatur wurde Triethylamin (42,5
g, 420 mmol) auf einmal zugegeben, Benzolsulfonylchlorid (63,58
g, 360 mmol) wurde tropfenweise über
20 Minuten hinweg zugegeben. Nachdem man eine Stunde lang die gleiche Temperatur
beibehalten hatte, wurde das Kühlbad
entfernt und das Gemisch über
weitere 30 Minuten hinweg bei Raumtemperatur gerührt und durch Zugabe von Eiswasser
(500 ml) und 2 N Salzsäure
(110 ml) in zwei Schichten aufgeteilt. Die wässrige Schicht wurde mit Ethylacetat
(200 ml) extrahiert. Die vereinigte organische Schicht wurde mit
Wasser gewaschen, über
wasserfreiem Magnesiumsulfat getrocknet und dann wurde das Lösungsmittel
unter reduziertem Druck abdestilliert, was 100,04 g der Titelverbindung
(2) als ein Öl
ergab. Rohausbeute: 109 %.
IR (CHCl
3);
3306, 3071, 3031, 3019, 3009, 1585, 1486,1449, 1378 cm
–1 1H NMR δ (CDCl
3), 300 MHz; 2,23 (1H, t, J = 2,7 Hz), 3,56
(2H, d, J = 2,7 Hz), 6,94 und 7,34 (jeweils 2H, jeweils d, J = 8,7
Hz), 7,51-7,56 (2H, m), 7,68 (1H, m), 7,8-7,85 (2H, m) 4-(2-Propyn-1-yltio)phenylbenzolsulfonat
(3) (2)
Schritt 2:
-
Die
Verbindung (2) (60,8 g, 183 mmol), die vorstehend in Schritt (1)
hergestellt wurde, wurde in Ameisensäure (30,4 ml) und Methanol
(122 ml) gelöst,
und dann wurden 31 % wässriges
Wasserstoffperoxid (26,29 g, 240 mmol) zugegeben. Nach 3,5 Stunden
wurde Eiswasser (240 ml) zugegeben und das Gemisch mit Ethylacetat
(2 × 300
ml) extrahiert. Die vereinigte organische Schicht wurde mit 5 %
wässrigem
Natriumcarbonat und Wasser gewaschen, über wasserfreiem Magnesiumsulfat
getrocknet und das Lösungsmittel
wurde dann unter reduziertem Druck abdestilliert, was 65,47 g der
Titelverbindung (3) als ein Öl
ergab. Rohausbeute: 117 %.
IR (CHCl
3);
3305, 3066, 3032, 3012, 1586, 1486, 1449, 1382 cm
–1 1H NMR 6(CDCl
3),
300 MHz; 2,34 (1 H, t, J = 3,9 Hz), 3,58 und 3,68 (jeweils 1H, jeweils
dd, J = 3,9 und 23,7 Hz), 7,18 und 7,67 (jeweils 2H, jeweils d,
J = 9,9 Hz), 7,51-7,59
(2H, m), 7,66 (1H, m), 7,82-7,87 (2H, m) 5-Benzolsulfonyloxy-3-hydroxymethylbenzo[b]thiophen
(4) (3)
Schritt 3:
-
Die
Verbindung (3) (65,47 g, 183 mmol), die vorstehend in (2) erhalten
wurde, wurde in 1,2-Dimethoxyethan (1,6 l) gelöst und die Lösung wurde über 4 Stunden
hinweg unter Rückfluß erhitzt.
Zu der Lösung
wurden Wasser (64 ml) und p-Toluolsulfonsäuremonohydrat
(19,2 g, 100 mmol) zugegeben und das Erhitzen unter Rückfluß wurde
2 Stunden fortgesetzt. Das Umsetzungsgemisch wurde unter vermindertem
Druck aufkonzentriert. Nachdem Wasser (200 ml) zu dem erhaltenen Öl zugegeben
worden war, wurde das Gemisch mit Ethylacetat (300 ml) extrahiert.
Die organische Schicht wurde mit wässrigem Natriumhydrogencarbonat
und Wasser gewaschen, über
wasserfreiem Magnesiumsulfat getrocknet und dann wurde das Lösungsmittel
unter reduziertem Druck abdestilliert, was 60,81 g der Titelverbindung
(4) als ein Öl
ergab.
Rohausbeute 103 %.
IR (CHCl
3);
3609, 3067, 3033, 3013, 2935, 2878, 1589, 1566, 1449, 1435, 1376
cm
–1 1H NMR δ (CDCl
3), 300 MHz; 4,78 (2H, d, J = 0,9 Hz), 6,98
(1H, dd, J = 2,4 und 8,7 Hz), 7,26 (1H, s), 7,43-7,45 (2H, m), 7,50-7,55
(2H, m), 7,66 (1H, m), 7,73 (1H, d, J = 8,7 Hz), 7,83-7,86 (2H,
m) 5-Benzolsulfonyloxybenzo[b]thiophen-3-carbonsäure (6) (4)
Schritt 4:
-
Die
Verbindung (4) (51,26 g 155 mmol), die vorstehend in (3) hergestellt
wurde, wurde in Acetonitril (1,54 L) gelöst, und TEMPO (2,2,6,6-Tetramethylpiperidin-1-oxyl,
250 mg, 0,01 eq.) hinzugefügt.
Zu der Mischung wurde tropfenweise 0,81 N wässrige Natriumhypochloritlösung zugegeben,
die durch Verdünnung
von 1,63 N wässriger
Natriumhypochloritlösung
(150 ml) mit Wasser (75 ml) hergestellt worden war, wobei der pH-Wert
mit 1 N Schwefelsäure
auf pH 8,6 eingestellt wurde und das Gesamtvolumen über 15 Minuten
hinweg auf 300 ml gebracht wurde, während die innere Temperatur
zwischen –1 °C und 8 °C gehalten
wurde. Nach Rühren
für 25
Minuten bei dieser Temperatur wurde 1 N wässrige Natriumsulfitlösung (32
ml) zugegeben. Anschließend
wurde 79 %-iges Natriumchlorit (27,48 g, 240 mmol) und 31 %-ige
wässrige
Wasserstoffperoxidlösung
(23,26 g, 212 mmol) unter Eiskühlung
zugegeben. Das Kühlbad
wurde entfernt und das Gemisch 2 Stunden lang gerührt. Das
Umsetzungsgemisch wurde mit Wasser (1,5 l) verdünnt, der pH-Wert mit 1 N Salzsäure auf
pH 3 eingestellt und die abgelagerten Kristalle wurden gefiltert
und zweimal mit Wasser (200 ml) und Acetonitril (50 ml) gewaschen,
um 32,4 g Rohkristalle zu ergeben. Die Rohkristalle (32,4 g) wurden
in Acetonitril (224 ml) suspendiert, über 15 Minuten hinweg unter
Rückfluß erhitzt
und auf Eis gekühlt.
Die Kristalle wurden gefiltert und mit Acetonitril (65 ml) gewaschen,
um 26,79 g der Titelverbindung (6) zu ergeben. Ausbeute: 51,7 %,
Schmp. 202–203 °C.
IR
(Nujol): 3102, 2925, 2854, 2744, 2640, 2577, 1672, 1599, 1558, 1500,
1460, 1451 cm–1
NMR δ (CDCl3), 300 MHz; 7,16 (1H, dd, J = 2,7 und 9,0
Hz), 7,55-7,61 (2H, m), 7,73 (1H, m), 7,81 (1H, d, J = 9,0 Hz),
7,90-7,94 (2H, m), 8,16 (1H, d, J = 2,7 Hz), 8,60 (1H, s)
Elementaranalyse
für C15H10O5S2
Berechnet (%): C, 53,88; H, 3,01;
S, 19,18
Erhalten (%): C, 53,73; H, 3,24; S, 19,09
 wobei R die vorstehend angegebene Bedeutung hat und X ein Wasserstoffatom oder ein Alkylrest ist. Die Benzothiophencarboxamidderivate sind spezifische Antagonisten von PGD2 und es ist bekannt, daß sie als Arzneimittel bei der Behandlung verschiedener Krankheiten, die mit einer Funktionsstörung der Mastzelle verbunden sind, die durch übermäßige Produktion von PGD2 hervorgerufen wird, zum Beispiel systemische Mastozytose, Störung der systemischen Mastzellaktivierung, Luftröhrenkontraktion, Asthma, allergische Rhinitis, allergische Conjunctivitis, Urtikaria, Verletzung durch ischämische Reperfusion, Entzündung und atopische Dermatitis (WO 97/00853, PCT/JP 97/04527 (WO 98/25919)) nützlich sind. Unter den Verbindungen der Formel (VI) hat eine Verbindung, wobei OR eine 5-Hydroxylgruppe ist und X ein Wasserstoffatom ist (nachfolgend als "Verbindung A bezeichnet") eine besonders hohe antagonistische Wirkung auf PGD2, wobei sie eine exzellente Wirkung gegen Nasenverstopfung zeigt und wird als vielversprechendes Arzneimittel zur Behandlung nasaler Verstopfung ins Auge gefaßt.
wobei R die vorstehend angegebene Bedeutung hat und X ein Wasserstoffatom oder ein Alkylrest ist. Die Benzothiophencarboxamidderivate sind spezifische Antagonisten von PGD2 und es ist bekannt, daß sie als Arzneimittel bei der Behandlung verschiedener Krankheiten, die mit einer Funktionsstörung der Mastzelle verbunden sind, die durch übermäßige Produktion von PGD2 hervorgerufen wird, zum Beispiel systemische Mastozytose, Störung der systemischen Mastzellaktivierung, Luftröhrenkontraktion, Asthma, allergische Rhinitis, allergische Conjunctivitis, Urtikaria, Verletzung durch ischämische Reperfusion, Entzündung und atopische Dermatitis (WO 97/00853, PCT/JP 97/04527 (WO 98/25919)) nützlich sind. Unter den Verbindungen der Formel (VI) hat eine Verbindung, wobei OR eine 5-Hydroxylgruppe ist und X ein Wasserstoffatom ist (nachfolgend als "Verbindung A bezeichnet") eine besonders hohe antagonistische Wirkung auf PGD2, wobei sie eine exzellente Wirkung gegen Nasenverstopfung zeigt und wird als vielversprechendes Arzneimittel zur Behandlung nasaler Verstopfung ins Auge gefaßt.

 wobei R1 für eine Hydroxyschutzgruppe steht; Oxidieren der Verbindung (II), um eine Verbindung der Formel (III) zu erhalten:
wobei R1 für eine Hydroxyschutzgruppe steht; Oxidieren der Verbindung (II), um eine Verbindung der Formel (III) zu erhalten: wobei R1 für eine Hydroxyschutzgruppe steht; Aussetzen der Verbindung (III) einer thermischen Umlagerungsreaktion, um eine Verbindung der Formel (IV) zu erhalten:
wobei R1 für eine Hydroxyschutzgruppe steht; Aussetzen der Verbindung (III) einer thermischen Umlagerungsreaktion, um eine Verbindung der Formel (IV) zu erhalten: wobei R1 wie vorstehend definiert ist; und Aussetzen der Verbindung (IV) einer schrittweisen Oxidierung der Hydroxymethylgruppe und gegebenenfalls Entfernen der Schutzgruppe.
wobei R1 wie vorstehend definiert ist; und Aussetzen der Verbindung (IV) einer schrittweisen Oxidierung der Hydroxymethylgruppe und gegebenenfalls Entfernen der Schutzgruppe.






 wobei R1 für eine Hydroxyschutzgruppe steht; Oxidieren der Verbindung (II), um eine Verbindung der Formel (III) zu erhalten:
wobei R1 für eine Hydroxyschutzgruppe steht; Oxidieren der Verbindung (II), um eine Verbindung der Formel (III) zu erhalten: wobei R1 für eine Hydroxyschutzgruppe steht; Aussetzen der Verbindung (III einer thermischen Umlagerungsreaktion, um eine Verbindung der Formel (IV) zu erhalten:
wobei R1 für eine Hydroxyschutzgruppe steht; Aussetzen der Verbindung (III einer thermischen Umlagerungsreaktion, um eine Verbindung der Formel (IV) zu erhalten: wobei R1wie vorstehend definiert ist; und Aussetzen der Verbindung (IV) schrittweiser Oxidierung der Hydroxymethylgruppe und gegebenenfalls Entfernen der Schutzgruppe.
wobei R1wie vorstehend definiert ist; und Aussetzen der Verbindung (IV) schrittweiser Oxidierung der Hydroxymethylgruppe und gegebenenfalls Entfernen der Schutzgruppe.