DE69918148T2 - Trizyclische delta3-pyridine als arzneimittel - Google Patents
Trizyclische delta3-pyridine als arzneimittel Download PDFInfo
- Publication number
- DE69918148T2 DE69918148T2 DE69918148T DE69918148T DE69918148T2 DE 69918148 T2 DE69918148 T2 DE 69918148T2 DE 69918148 T DE69918148 T DE 69918148T DE 69918148 T DE69918148 T DE 69918148T DE 69918148 T2 DE69918148 T2 DE 69918148T2
- Authority
- DE
- Germany
- Prior art keywords
- formula
- compounds
- mixture
- compound
- mol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 239000003814 drug Substances 0.000 title claims description 5
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 title description 4
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 title description 2
- 102100036466 Delta-like protein 3 Human genes 0.000 title 1
- 101710112748 Delta-like protein 3 Proteins 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims description 85
- 239000000203 mixture Substances 0.000 claims description 58
- 239000000543 intermediate Substances 0.000 claims description 45
- 239000000460 chlorine Substances 0.000 claims description 29
- 150000003839 salts Chemical class 0.000 claims description 28
- -1 nitro, hydroxy Chemical group 0.000 claims description 22
- 238000006243 chemical reaction Methods 0.000 claims description 15
- 238000002360 preparation method Methods 0.000 claims description 14
- 239000002253 acid Substances 0.000 claims description 13
- 239000003054 catalyst Substances 0.000 claims description 13
- 229910052739 hydrogen Inorganic materials 0.000 claims description 12
- 239000001257 hydrogen Substances 0.000 claims description 12
- 238000000034 method Methods 0.000 claims description 11
- 125000000217 alkyl group Chemical group 0.000 claims description 10
- 239000002585 base Substances 0.000 claims description 10
- 150000003254 radicals Chemical class 0.000 claims description 10
- 238000011282 treatment Methods 0.000 claims description 10
- 208000018737 Parkinson disease Diseases 0.000 claims description 7
- 229910052736 halogen Inorganic materials 0.000 claims description 7
- 150000002367 halogens Chemical class 0.000 claims description 7
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 6
- 238000007126 N-alkylation reaction Methods 0.000 claims description 6
- 150000002431 hydrogen Chemical class 0.000 claims description 6
- 239000004480 active ingredient Substances 0.000 claims description 5
- 239000003937 drug carrier Substances 0.000 claims description 5
- 239000012442 inert solvent Substances 0.000 claims description 5
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 5
- 230000002829 reductive effect Effects 0.000 claims description 5
- CUJPFPXNDSIBPG-UHFFFAOYSA-N 1,3-propanediyl Chemical group [CH2]C[CH2] CUJPFPXNDSIBPG-UHFFFAOYSA-N 0.000 claims description 4
- OMIVCRYZSXDGAB-UHFFFAOYSA-N 1,4-butanediyl Chemical group [CH2]CC[CH2] OMIVCRYZSXDGAB-UHFFFAOYSA-N 0.000 claims description 4
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 4
- 229910052801 chlorine Inorganic materials 0.000 claims description 4
- 238000004519 manufacturing process Methods 0.000 claims description 4
- 150000001204 N-oxides Chemical group 0.000 claims description 3
- 239000003513 alkali Substances 0.000 claims description 3
- 239000012458 free base Substances 0.000 claims description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 3
- 231100000252 nontoxic Toxicity 0.000 claims description 3
- 230000003000 nontoxic effect Effects 0.000 claims description 3
- 239000000376 reactant Substances 0.000 claims description 3
- 208000020401 Depressive disease Diseases 0.000 claims description 2
- 125000003545 alkoxy group Chemical group 0.000 claims description 2
- 239000002168 alkylating agent Substances 0.000 claims description 2
- 229940100198 alkylating agent Drugs 0.000 claims description 2
- 239000011872 intimate mixture Substances 0.000 claims description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 2
- 238000000844 transformation Methods 0.000 claims description 2
- 230000009466 transformation Effects 0.000 claims description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims 1
- 229910052731 fluorine Inorganic materials 0.000 claims 1
- 239000011737 fluorine Substances 0.000 claims 1
- 125000002485 formyl group Chemical class [H]C(*)=O 0.000 claims 1
- 125000001475 halogen functional group Chemical group 0.000 claims 1
- 239000002904 solvent Substances 0.000 description 34
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 30
- 239000000243 solution Substances 0.000 description 24
- 102000005962 receptors Human genes 0.000 description 23
- 108020003175 receptors Proteins 0.000 description 23
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 21
- 239000011541 reaction mixture Substances 0.000 description 20
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 15
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 14
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 13
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 12
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 12
- 239000012044 organic layer Substances 0.000 description 12
- 239000003480 eluent Substances 0.000 description 10
- 239000002244 precipitate Substances 0.000 description 10
- 230000027455 binding Effects 0.000 description 9
- 238000010992 reflux Methods 0.000 description 9
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 8
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 8
- 238000003756 stirring Methods 0.000 description 8
- 239000003826 tablet Substances 0.000 description 8
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 7
- 238000010898 silica gel chromatography Methods 0.000 description 7
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- 125000000815 N-oxide group Chemical group 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 239000000706 filtrate Substances 0.000 description 6
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 6
- 239000002287 radioligand Substances 0.000 description 6
- 239000011734 sodium Substances 0.000 description 6
- SFLSHLFXELFNJZ-QMMMGPOBSA-N (-)-norepinephrine Chemical compound NC[C@H](O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-QMMMGPOBSA-N 0.000 description 5
- 210000003169 central nervous system Anatomy 0.000 description 5
- 238000002347 injection Methods 0.000 description 5
- 239000007924 injection Substances 0.000 description 5
- 229960002748 norepinephrine Drugs 0.000 description 5
- SFLSHLFXELFNJZ-UHFFFAOYSA-N norepinephrine Natural products NCC(O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-UHFFFAOYSA-N 0.000 description 5
- 239000003921 oil Substances 0.000 description 5
- 235000019198 oils Nutrition 0.000 description 5
- 230000001225 therapeutic effect Effects 0.000 description 5
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 4
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 description 4
- 150000007513 acids Chemical class 0.000 description 4
- 239000002775 capsule Substances 0.000 description 4
- 125000004432 carbon atom Chemical group C* 0.000 description 4
- 239000008194 pharmaceutical composition Substances 0.000 description 4
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 4
- 229930192474 thiophene Natural products 0.000 description 4
- STIFURPZTKYNJV-UHFFFAOYSA-N 1,2,3,4-tetrahydro-[1]benzothiolo[3,2-c]pyridine Chemical compound S1C2=CC=CC=C2C2=C1CCNC2 STIFURPZTKYNJV-UHFFFAOYSA-N 0.000 description 3
- IHGPOPRPFXEICO-UHFFFAOYSA-N 4-(1,2-benzoxazol-3-ylamino)butan-1-ol Chemical compound C1=CC=C2C(NCCCCO)=NOC2=C1 IHGPOPRPFXEICO-UHFFFAOYSA-N 0.000 description 3
- QWLHJVDRPZNVBS-UHFFFAOYSA-N 4-phenoxybenzaldehyde Chemical compound C1=CC(C=O)=CC=C1OC1=CC=CC=C1 QWLHJVDRPZNVBS-UHFFFAOYSA-N 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical class OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 3
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 239000000654 additive Substances 0.000 description 3
- 239000000670 adrenergic alpha-2 receptor antagonist Substances 0.000 description 3
- SIKJAQJRHWYJAI-UHFFFAOYSA-N benzopyrrole Natural products C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 3
- 230000033228 biological regulation Effects 0.000 description 3
- 239000007795 chemical reaction product Substances 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 3
- 235000011056 potassium acetate Nutrition 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 239000012453 solvate Substances 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 0 *c(cc1)ccc1C(Nc1cc(CCC(CC2)Cc3c2[o]c2c3cccc2)ccc1)=* Chemical compound *c(cc1)ccc1C(Nc1cc(CCC(CC2)Cc3c2[o]c2c3cccc2)ccc1)=* 0.000 description 2
- ZQCBPTXJGPZNCQ-UHFFFAOYSA-N 1,2,3,4-tetrahydro-[1]benzofuro[3,2-c]pyridine;hydrochloride Chemical compound Cl.O1C2=CC=CC=C2C2=C1CCNC2 ZQCBPTXJGPZNCQ-UHFFFAOYSA-N 0.000 description 2
- CFFZDZCDUFSOFZ-UHFFFAOYSA-N 3,4-Dihydroxy-phenylacetic acid Chemical compound OC(=O)CC1=CC=C(O)C(O)=C1 CFFZDZCDUFSOFZ-UHFFFAOYSA-N 0.000 description 2
- 208000024827 Alzheimer disease Diseases 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 2
- 208000010228 Erectile Dysfunction Diseases 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- MFESCIUQSIBMSM-UHFFFAOYSA-N I-BCP Chemical compound ClCCCBr MFESCIUQSIBMSM-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- KFSLWBXXFJQRDL-UHFFFAOYSA-N Peracetic acid Chemical compound CC(=O)OO KFSLWBXXFJQRDL-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- 201000001880 Sexual dysfunction Diseases 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 230000000903 blocking effect Effects 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 2
- 208000010877 cognitive disease Diseases 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 206010012601 diabetes mellitus Diseases 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 108700006189 dopamine beta hydroxylase deficiency Proteins 0.000 description 2
- 208000009308 dopamine beta-hydroxylase deficiency Diseases 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 201000001881 impotence Diseases 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 230000004410 intraocular pressure Effects 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 238000004811 liquid chromatography Methods 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 239000012299 nitrogen atmosphere Substances 0.000 description 2
- 239000006186 oral dosage form Substances 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 210000001428 peripheral nervous system Anatomy 0.000 description 2
- 150000002978 peroxides Chemical class 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- NROKBHXJSPEDAR-UHFFFAOYSA-M potassium fluoride Chemical compound [F-].[K+] NROKBHXJSPEDAR-UHFFFAOYSA-M 0.000 description 2
- XXQBEVHPUKOQEO-UHFFFAOYSA-N potassium superoxide Chemical compound [K+].[K+].[O-][O-] XXQBEVHPUKOQEO-UHFFFAOYSA-N 0.000 description 2
- 230000003518 presynaptic effect Effects 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 238000010956 selective crystallization Methods 0.000 description 2
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 2
- 231100000872 sexual dysfunction Toxicity 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- CIHOLLKRGTVIJN-UHFFFAOYSA-N tert‐butyl hydroperoxide Chemical compound CC(C)(C)OO CIHOLLKRGTVIJN-UHFFFAOYSA-N 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 1
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 1
- DNXIKVLOVZVMQF-UHFFFAOYSA-N (3beta,16beta,17alpha,18beta,20alpha)-17-hydroxy-11-methoxy-18-[(3,4,5-trimethoxybenzoyl)oxy]-yohimban-16-carboxylic acid, methyl ester Natural products C1C2CN3CCC(C4=CC=C(OC)C=C4N4)=C4C3CC2C(C(=O)OC)C(O)C1OC(=O)C1=CC(OC)=C(OC)C(OC)=C1 DNXIKVLOVZVMQF-UHFFFAOYSA-N 0.000 description 1
- AGCPUUJGIKDYRM-WLHGVMLRSA-N (E)-but-2-enedioic acid N-[4-(3,4-dihydro-1H-[1]benzothiolo[3,2-c]pyridin-2-yl)butyl]-1,2-benzoxazol-3-amine Chemical compound OC(=O)\C=C\C(O)=O.c1cccc2c(CN(CC3)CCCCNc4noc5c4cccc5)c3sc21 AGCPUUJGIKDYRM-WLHGVMLRSA-N 0.000 description 1
- OMMBAUBTBDUGTQ-WLHGVMLRSA-N (e)-but-2-enedioic acid;1-[(4-phenoxyphenyl)methyl]-1,2,3,4-tetrahydro-[1]benzothiolo[3,2-c]pyridine Chemical compound OC(=O)\C=C\C(O)=O.N1CCC=2SC3=CC=CC=C3C=2C1CC(C=C1)=CC=C1OC1=CC=CC=C1 OMMBAUBTBDUGTQ-WLHGVMLRSA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- YGQZVYGZSCVMMN-UHFFFAOYSA-N 1,3,5,7-tetrazatricyclo[5.1.1.13,5]decane Chemical compound C1N2CN1CN(C1)CN1C2 YGQZVYGZSCVMMN-UHFFFAOYSA-N 0.000 description 1
- PFDVTBLQZCQHSF-UHFFFAOYSA-N 1-(3-chloropropyl)indole Chemical compound C1=CC=C2N(CCCCl)C=CC2=C1 PFDVTBLQZCQHSF-UHFFFAOYSA-N 0.000 description 1
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 1
- KWIBGLAHGINXJJ-UHFFFAOYSA-N 2-(2-iodoethyl)-6-methoxy-1-benzothiophene Chemical compound COC1=CC=C2C=C(CCI)SC2=C1 KWIBGLAHGINXJJ-UHFFFAOYSA-N 0.000 description 1
- TWADWNJKWBGRGU-UHFFFAOYSA-N 2-(3-indol-1-ylpropyl)-3,4-dihydro-1h-[1]benzofuro[3,2-c]pyridine Chemical compound C1=CC2=CC=CC=C2N1CCCN1CC(C2=CC=CC=C2O2)=C2CC1 TWADWNJKWBGRGU-UHFFFAOYSA-N 0.000 description 1
- JUKITXMBZQOPJN-UHFFFAOYSA-N 2-(4-chlorophenyl)-3-(2-hydroxyethyl)quinazolin-4-one Chemical compound N=1C2=CC=CC=C2C(=O)N(CCO)C=1C1=CC=C(Cl)C=C1 JUKITXMBZQOPJN-UHFFFAOYSA-N 0.000 description 1
- SOPGXZUWHVDQSW-UHFFFAOYSA-N 2-(6-methoxy-1-benzothiophen-2-yl)ethanol Chemical compound COC1=CC=C2C=C(CCO)SC2=C1 SOPGXZUWHVDQSW-UHFFFAOYSA-N 0.000 description 1
- MKPWXACOKOQLPM-UHFFFAOYSA-N 2-(6-methoxy-1-benzothiophen-2-yl)ethyl methanesulfonate Chemical compound COC1=CC=C2C=C(CCOS(C)(=O)=O)SC2=C1 MKPWXACOKOQLPM-UHFFFAOYSA-N 0.000 description 1
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 1
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 1
- IUVCFHHAEHNCFT-INIZCTEOSA-N 2-[(1s)-1-[4-amino-3-(3-fluoro-4-propan-2-yloxyphenyl)pyrazolo[3,4-d]pyrimidin-1-yl]ethyl]-6-fluoro-3-(3-fluorophenyl)chromen-4-one Chemical compound C1=C(F)C(OC(C)C)=CC=C1C(C1=C(N)N=CN=C11)=NN1[C@@H](C)C1=C(C=2C=C(F)C=CC=2)C(=O)C2=CC(F)=CC=C2O1 IUVCFHHAEHNCFT-INIZCTEOSA-N 0.000 description 1
- CCLBCKQPMVAVPS-UHFFFAOYSA-N 2-[(4-nitrophenyl)methyl]-3,4-dihydro-1h-[1]benzofuro[3,2-c]pyridine Chemical compound C1=CC([N+](=O)[O-])=CC=C1CN1CC(C2=CC=CC=C2O2)=C2CC1 CCLBCKQPMVAVPS-UHFFFAOYSA-N 0.000 description 1
- PPTROHGCWIQVRY-UHFFFAOYSA-N 2-[(4-nitrophenyl)methyl]-3,4-dihydro-1h-[1]benzofuro[3,2-c]pyridine;hydrochloride Chemical compound Cl.C1=CC([N+](=O)[O-])=CC=C1CN1CC(C2=CC=CC=C2O2)=C2CC1 PPTROHGCWIQVRY-UHFFFAOYSA-N 0.000 description 1
- XUUUGSDOVDBHKN-UHFFFAOYSA-N 2-[2-(1h-indol-3-yl)ethyl]-3,4-dihydro-1h-[1]benzothiolo[3,2-c]pyridine Chemical compound S1C2=CC=CC=C2C(C2)=C1CCN2CCC1=CNC2=CC=CC=C12 XUUUGSDOVDBHKN-UHFFFAOYSA-N 0.000 description 1
- OWHRXGOUYLKIPH-UHFFFAOYSA-N 2-cyclohexa-1,5-dien-1-ylacetic acid Chemical compound OC(=O)CC1=CCCC=C1 OWHRXGOUYLKIPH-UHFFFAOYSA-N 0.000 description 1
- NTLAICDKHHQUGC-UHFFFAOYSA-N 3-(2-bromoethyl)-1h-indole Chemical compound C1=CC=C2C(CCBr)=CNC2=C1 NTLAICDKHHQUGC-UHFFFAOYSA-N 0.000 description 1
- JNYKTYKWJMUQDT-UHFFFAOYSA-N 3-(2-bromoethyl)-2-(4-chlorophenyl)quinazolin-4-one;hydrobromide Chemical compound Br.C1=CC(Cl)=CC=C1C1=NC2=CC=CC=C2C(=O)N1CCBr JNYKTYKWJMUQDT-UHFFFAOYSA-N 0.000 description 1
- MHSKXVFALQYNNW-UHFFFAOYSA-N 3-(5-chloropentyl)-6-fluoro-1,2-benzoxazole Chemical compound FC1=CC=C2C(CCCCCCl)=NOC2=C1 MHSKXVFALQYNNW-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- INWUFXPCLZRSBH-UHFFFAOYSA-N 3-chloro-1,2-benzoxazole Chemical compound C1=CC=C2C(Cl)=NOC2=C1 INWUFXPCLZRSBH-UHFFFAOYSA-N 0.000 description 1
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 1
- SJTBRFHBXDZMPS-UHFFFAOYSA-N 3-fluorophenol Chemical compound OC1=CC=CC(F)=C1 SJTBRFHBXDZMPS-UHFFFAOYSA-N 0.000 description 1
- ZJQJXKGEXQWLRI-UHFFFAOYSA-N 4-(3,4-dihydro-1h-[1]benzofuro[3,2-c]pyridin-2-ylmethyl)aniline;hydrochloride Chemical compound Cl.C1=CC(N)=CC=C1CN1CC(C2=CC=CC=C2O2)=C2CC1 ZJQJXKGEXQWLRI-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- BLFRQYKZFKYQLO-UHFFFAOYSA-N 4-aminobutan-1-ol Chemical compound NCCCCO BLFRQYKZFKYQLO-UHFFFAOYSA-N 0.000 description 1
- WUBBRNOQWQTFEX-UHFFFAOYSA-N 4-aminosalicylic acid Chemical compound NC1=CC=C(C(O)=O)C(O)=C1 WUBBRNOQWQTFEX-UHFFFAOYSA-N 0.000 description 1
- CZKLEJHVLCMVQR-UHFFFAOYSA-N 4-fluorobenzoyl chloride Chemical compound FC1=CC=C(C(Cl)=O)C=C1 CZKLEJHVLCMVQR-UHFFFAOYSA-N 0.000 description 1
- JYMSHJGVOYWRHY-UHFFFAOYSA-N 6-chloro-1-(4-fluoro-2-hydroxyphenyl)hexan-1-one Chemical compound OC1=CC(F)=CC=C1C(=O)CCCCCCl JYMSHJGVOYWRHY-UHFFFAOYSA-N 0.000 description 1
- WZILXAPNPKMOSA-UHFFFAOYSA-N 6-chlorohexanoyl chloride Chemical compound ClCCCCCC(Cl)=O WZILXAPNPKMOSA-UHFFFAOYSA-N 0.000 description 1
- WGDVDMKNSDCNGB-UHFFFAOYSA-N 6-methoxy-1-benzothiophene Chemical compound COC1=CC=C2C=CSC2=C1 WGDVDMKNSDCNGB-UHFFFAOYSA-N 0.000 description 1
- MDYVKSNGFGVOCH-UHFFFAOYSA-N 7-methoxy-1,2,3,4-tetrahydro-[1]benzothiolo[3,2-c]pyridine Chemical compound C1NCCC2=C1C1=CC=C(OC)C=C1S2 MDYVKSNGFGVOCH-UHFFFAOYSA-N 0.000 description 1
- NLTAVZMAGZNIJZ-UHFFFAOYSA-N 7-methoxy-2-[(4-phenoxyphenyl)methyl]-3,4-dihydro-1h-[1]benzothiolo[3,2-c]pyridine Chemical compound C1CC=2SC3=CC(OC)=CC=C3C=2CN1CC(C=C1)=CC=C1OC1=CC=CC=C1 NLTAVZMAGZNIJZ-UHFFFAOYSA-N 0.000 description 1
- WTWMWHSXCSPWHH-UHFFFAOYSA-N 8-chloro-1,2,3,4-tetrahydro-[1]benzothiolo[3,2-c]pyridine;hydrochloride Chemical compound Cl.C1CNCC2=C1SC1=CC=C(Cl)C=C12 WTWMWHSXCSPWHH-UHFFFAOYSA-N 0.000 description 1
- RWEZBQWHEZPMHL-UHFFFAOYSA-N 8-methyl-1,2,3,4-tetrahydro-[1]benzothiolo[3,2-c]pyridine;hydrochloride Chemical compound Cl.C1CNCC2=C1SC1=CC=C(C)C=C12 RWEZBQWHEZPMHL-UHFFFAOYSA-N 0.000 description 1
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 1
- 108060003345 Adrenergic Receptor Proteins 0.000 description 1
- 102000017910 Adrenergic receptor Human genes 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- XZUMEXKQGXYIHK-UHFFFAOYSA-N C1=CC(=C(C=C1F)O)C(=NO)CCCCCCl Chemical compound C1=CC(=C(C=C1F)O)C(=NO)CCCCCCl XZUMEXKQGXYIHK-UHFFFAOYSA-N 0.000 description 1
- MUZYAOWTMODANQ-UHFFFAOYSA-N Cl.CC=1C=CC2=C(C1)C=1CN(CCC1S2)CC2=CC=C(C=C2)OC2=CC=CC=C2 Chemical compound Cl.CC=1C=CC2=C(C1)C=1CN(CCC1S2)CC2=CC=C(C=C2)OC2=CC=CC=C2 MUZYAOWTMODANQ-UHFFFAOYSA-N 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- UUNFQUAWHHNYJO-UHFFFAOYSA-N O1C2=CC=CC=C2C(C2)=C1CCN2CCCCCC1=NC2=CC=C(F)C=C2O1 Chemical compound O1C2=CC=CC=C2C(C2)=C1CCN2CCCCCC1=NC2=CC=C(F)C=C2O1 UUNFQUAWHHNYJO-UHFFFAOYSA-N 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- LCQMZZCPPSWADO-UHFFFAOYSA-N Reserpilin Natural products COC(=O)C1COCC2CN3CCc4c([nH]c5cc(OC)c(OC)cc45)C3CC12 LCQMZZCPPSWADO-UHFFFAOYSA-N 0.000 description 1
- QEVHRUUCFGRFIF-SFWBKIHZSA-N Reserpine Natural products O=C(OC)[C@@H]1[C@H](OC)[C@H](OC(=O)c2cc(OC)c(OC)c(OC)c2)C[C@H]2[C@@H]1C[C@H]1N(C2)CCc2c3c([nH]c12)cc(OC)cc3 QEVHRUUCFGRFIF-SFWBKIHZSA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 206010044565 Tremor Diseases 0.000 description 1
- OIPILFWXSMYKGL-UHFFFAOYSA-N acetylcholine Chemical compound CC(=O)OCC[N+](C)(C)C OIPILFWXSMYKGL-UHFFFAOYSA-N 0.000 description 1
- 229960004373 acetylcholine Drugs 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 150000004973 alkali metal peroxides Chemical class 0.000 description 1
- 150000001447 alkali salts Chemical class 0.000 description 1
- 150000004974 alkaline earth metal peroxides Chemical class 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 150000001414 amino alcohols Chemical class 0.000 description 1
- 229960004909 aminosalicylic acid Drugs 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 230000008485 antagonism Effects 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- HTZCNXWZYVXIMZ-UHFFFAOYSA-M benzyl(triethyl)azanium;chloride Chemical compound [Cl-].CC[N+](CC)(CC)CC1=CC=CC=C1 HTZCNXWZYVXIMZ-UHFFFAOYSA-M 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 1
- 210000004027 cell Anatomy 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 230000009137 competitive binding Effects 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 229940126543 compound 14 Drugs 0.000 description 1
- 229940126142 compound 16 Drugs 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 150000001907 coumarones Chemical class 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- MXZMFOJEOGTLJD-UHFFFAOYSA-N decane hydroiodide Chemical compound I.CCCCCCCCCC MXZMFOJEOGTLJD-UHFFFAOYSA-N 0.000 description 1
- 229940075894 denatured ethanol Drugs 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical class NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- 125000000816 ethylene group Chemical group [H]C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 150000002314 glycerols Chemical class 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- VKYKSIONXSXAKP-UHFFFAOYSA-N hexamethylene tetramine Natural products C1N(C2)CN3CN1CN2C3 VKYKSIONXSXAKP-UHFFFAOYSA-N 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical class C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 239000008172 hydrogenated vegetable oil Substances 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 1
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 150000004668 long chain fatty acids Chemical class 0.000 description 1
- 230000005923 long-lasting effect Effects 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229940126601 medicinal product Drugs 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- 125000005948 methanesulfonyloxy group Chemical group 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- BYKHVMAISBDKIR-UHFFFAOYSA-N n-(4-chlorobutyl)-1,2-benzoxazol-3-amine Chemical compound C1=CC=C2C(NCCCCCl)=NOC2=C1 BYKHVMAISBDKIR-UHFFFAOYSA-N 0.000 description 1
- MLHZJSUBZRCYFV-UHFFFAOYSA-N n-[4-(3,4-dihydro-1h-[1]benzofuro[3,2-c]pyridin-2-ylmethyl)phenyl]-4-fluorobenzamide Chemical compound C1=CC(F)=CC=C1C(=O)NC(C=C1)=CC=C1CN1CC(C2=CC=CC=C2O2)=C2CC1 MLHZJSUBZRCYFV-UHFFFAOYSA-N 0.000 description 1
- BBIBBVXOOIVZNF-UHFFFAOYSA-N n-[4-(8-methyl-3,4-dihydro-1h-[1]benzothiolo[3,2-c]pyridin-2-yl)butyl]-1,2-benzoxazol-3-amine;hydrochloride Chemical compound Cl.C1=CC=C2C(NCCCCN3CCC=4SC5=CC=C(C=C5C=4C3)C)=NOC2=C1 BBIBBVXOOIVZNF-UHFFFAOYSA-N 0.000 description 1
- 210000000653 nervous system Anatomy 0.000 description 1
- 210000002569 neuron Anatomy 0.000 description 1
- 230000003957 neurotransmitter release Effects 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 230000002474 noradrenergic effect Effects 0.000 description 1
- DBTXKJJSFWZJNS-UHFFFAOYSA-N o-phenylhydroxylamine;hydrochloride Chemical compound Cl.NOC1=CC=CC=C1 DBTXKJJSFWZJNS-UHFFFAOYSA-N 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000001451 organic peroxides Chemical class 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- KGCNHWXDPDPSBV-UHFFFAOYSA-N p-nitrobenzyl chloride Chemical compound [O-][N+](=O)C1=CC=C(CCl)C=C1 KGCNHWXDPDPSBV-UHFFFAOYSA-N 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- WLJNZVDCPSBLRP-UHFFFAOYSA-N pamoic acid Chemical compound C1=CC=C2C(CC=3C4=CC=CC=C4C=C(C=3O)C(=O)O)=C(O)C(C(O)=O)=CC2=C1 WLJNZVDCPSBLRP-UHFFFAOYSA-N 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 230000000149 penetrating effect Effects 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 230000002572 peristaltic effect Effects 0.000 description 1
- 150000004965 peroxy acids Chemical class 0.000 description 1
- XCRBXWCUXJNEFX-UHFFFAOYSA-N peroxybenzoic acid Chemical class OOC(=O)C1=CC=CC=C1 XCRBXWCUXJNEFX-UHFFFAOYSA-N 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 238000000053 physical method Methods 0.000 description 1
- BQOLKFJNJCOALF-UHFFFAOYSA-N piperidin-1-ium-4,4-diol;chloride Chemical compound Cl.OC1(O)CCNCC1 BQOLKFJNJCOALF-UHFFFAOYSA-N 0.000 description 1
- 239000002574 poison Substances 0.000 description 1
- 231100000614 poison Toxicity 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011698 potassium fluoride Substances 0.000 description 1
- 235000003270 potassium fluoride Nutrition 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- JADFCQKRKICRKI-UHFFFAOYSA-N quinoline;sulfane Chemical compound S.N1=CC=CC2=CC=CC=C21 JADFCQKRKICRKI-UHFFFAOYSA-N 0.000 description 1
- 239000012429 reaction media Substances 0.000 description 1
- BJOIZNZVOZKDIG-MDEJGZGSSA-N reserpine Chemical compound O([C@H]1[C@@H]([C@H]([C@H]2C[C@@H]3C4=C([C]5C=CC(OC)=CC5=N4)CCN3C[C@H]2C1)C(=O)OC)OC)C(=O)C1=CC(OC)=C(OC)C(OC)=C1 BJOIZNZVOZKDIG-MDEJGZGSSA-N 0.000 description 1
- 229960003147 reserpine Drugs 0.000 description 1
- 230000028527 righting reflex Effects 0.000 description 1
- MDMGHDFNKNZPAU-UHFFFAOYSA-N roserpine Natural products C1C2CN3CCC(C4=CC=C(OC)C=C4N4)=C4C3CC2C(OC(C)=O)C(OC)C1OC(=O)C1=CC(OC)=C(OC)C(OC)=C1 MDMGHDFNKNZPAU-UHFFFAOYSA-N 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 229940076279 serotonin Drugs 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- PFUVRDFDKPNGAV-UHFFFAOYSA-N sodium peroxide Chemical compound [Na+].[Na+].[O-][O-] PFUVRDFDKPNGAV-UHFFFAOYSA-N 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 239000012258 stirred mixture Substances 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000001665 trituration Methods 0.000 description 1
- 235000012431 wafers Nutrition 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- BPICBUSOMSTKRF-UHFFFAOYSA-N xylazine Chemical compound CC1=CC=CC(C)=C1NC1=NCCCS1 BPICBUSOMSTKRF-UHFFFAOYSA-N 0.000 description 1
- 229960001600 xylazine Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
- C07D491/044—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
- C07D491/048—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring the oxygen-containing ring being five-membered
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/10—Drugs for genital or sexual disorders; Contraceptives for impotence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Diabetes (AREA)
- Endocrinology (AREA)
- Ophthalmology & Optometry (AREA)
- Psychiatry (AREA)
- Reproductive Health (AREA)
- Pain & Pain Management (AREA)
- Hospice & Palliative Care (AREA)
- Obesity (AREA)
- Psychology (AREA)
- Hematology (AREA)
- Emergency Medicine (AREA)
- Gynecology & Obstetrics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Description
- Die vorliegende Erfindung betrifft tricyclische Δ3-Piperidine mit zentraler α2-Adrenozeptor-Antagonistenwirkung sowie deren Herstellung, diese Verbindungen enthaltende Zusammensetzungen und ihre Verwendung als Arzneimittel.
- Zentrale α2-Adrenozeptor-Antagonisten erhöhen bekanntlich die Noradrenalin-Ausschüttung, indem sie präsynaptische α2-Rezeptoren, die auf die Ausschüttung des Neurotransmitters inhibierend wirken, blockieren. Durch Erhöhung der Noradrenalinkonzentrationen können α2-Antagonisten zur Behandlung oder Prophylaxe von Depression, kognitiven Störungen, Morbus Parkinson, Diabetes mellitus, sexueller Dysfunktion und Impotenz, erhöhtem Augeninnendruck und mit gestörter Peristaltik in Zusammenhang stehenden Krankheiten im klinischen Bereich verwendet werden, da alle diese Beschwerden mit einem Noradrenalinmangel im zentralen oder peripheren Nervensystem verbunden sind.
- In der EP-A-206225 werden Verbindungen offenbart, die auf das zentrale Nervensystem wirken und eine an eine Indoleinheit gebundene tricyclische Piperidineinheit enthalten.
- Die erfindungsgemäßen Verbindungen sind neu und haben eine spezifische und selektive Bindungsaffinität zu den verschiedenen bekannten Subtypen der α2-Adrenozeptoren, d. h. dem α2A- , α2B- und α2C-Adrenozeptor.
- Die vorliegende Erfindung betrifft die Verbindungen der Formelund deren N-Oxidformen, pharmazeutisch unbedenkliche Additionssalze und stereochemisch isomere Formen, wobei:

Alk für C1–6-Alkandiyl steht;
n für 1 oder 2 steht;
X für -O-, -5-, -S(=O)- oder -S(=O)2- steht;
die Reste R1 jeweils unabhängig voneinander für Wasserstoff, Halogen, C1–6-Alkyl, Nitro, Hydroxy oder
C1–4-Alkyloxy stehen;
D für einen Rest der Formelsteht, wobei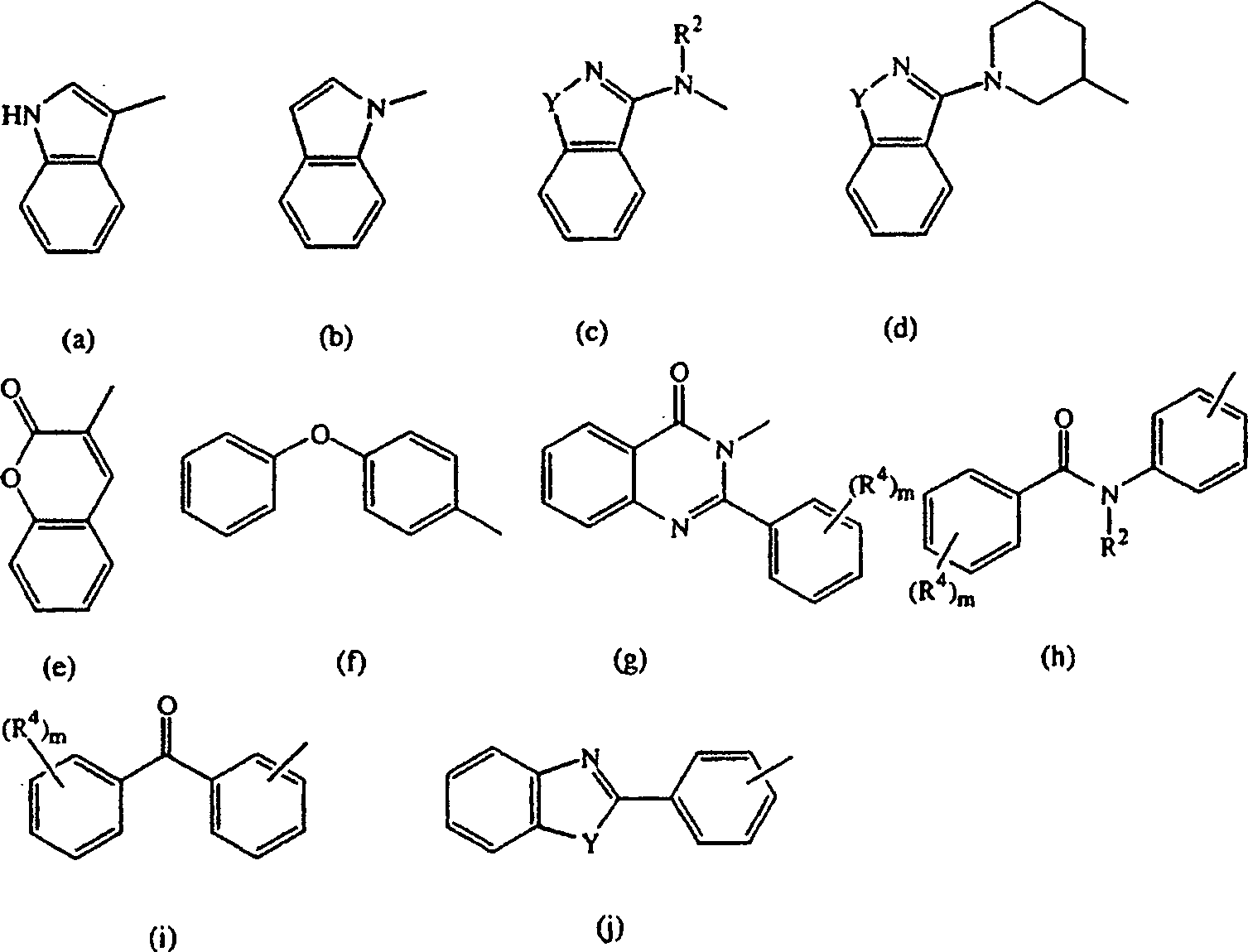
die Indices m jeweils unabhängig voneinander für 0, 1 oder 2 stehen;
die Reste Y jeweils unabhängig voneinander für -CH2-, -O-, -S- oder -NR3- stehen;
R2 und R3 jeweils unabhängig voneinander für Wasserstoff oder C1–6-Alkyl stehen; und
die Reste R4 jeweils unabhängig voneinander für Halogen oder C1–6-Alkyl stehen. - In den vorhergehenden Definitionen steht der Ausdruck „Halogen" allgemein für Fluor, Chlor, Brom und Iod. Der Ausdruck „C1–4-Alkyl" definiert geradkettige und verzweigte gesättigte Kohlenwasserstoffe mit 1 bis 4 Kohlenstoffatomen wie beispielsweise Methyl, Ethyl, Propyl, Butyl, 1-Methylethyl, 1,1-Dimethylethyl, 2-Methylpropyl und dergleichen. Der Ausdruck „C1–6-Alkyl" soll C1–4-Alkylreste und deren höhere Homologe mit 5 oder 6 Kohlenstoffatomen wie beispielsweise Pentyl, Hexyl und dergleichen umfassen. Der Ausdruck „C1–5-Alkandiyl" definiert zweiwertige geradkettige und verzweigte Alkandiylreste mit 1 bis 5 Kohlenstoffatomen wie beispielsweise Methylen, 1,2-Ethandiyl, 1,3-Propandiyl, 1,4-Butandiyl, 1,5-Pentandiyl und dergleichen. Der Ausdruck „C1–6-Alkandiyl" soll C1–5-Alkandiyl und dessen höhere Homologe mit 6 Kohlenstoffatomen wie beispielsweise 1,6-Hexandiyl umfassen.
- Unter den hier angesprochenen Additionssalzen versteht man die therapeutisch wirksamen Additionssalzformen, die die Verbindungen der Formel (I) mit geeigneten Säuren zu bilden vermögen, wie z.B. mit anorganischen Säuren, beispielsweise Halogenwasserstoffsäuren, z.B. Chlorwasserstoff- oder Bromwasserstoffsäure, Schwefelsäure, Salpetersäure, Phosphorsäure und dergleichen, oder mit organischen Säuren, wie zum Beispiel Essigsäure, Propansäure, Hydroxyessigsäure, Milchsäure, Brenztraubensäure, Oxalsäure, Malonsäure, Bernsteinsäure, Maleinsäure, Fumarsäure, Äpfelsäure, Weinsäure, Citronensäure, Methansulfonsäure, Ethansulfonsäure, Benzolsulfonsäure, p-Toluolsulfonsäure, Cyclaminsäure, Salicylsäure, p-Aminosalicylsäure, Pamoasäure und dergleichen.
- Unter den oben angesprochenen pharmazeutisch unbedenklichen Additionssalzen versteht man auch die therapeutisch wirksamen nichttoxischen Basen- und insbesondere Metall- oder Amin-Additionssalzformen, die die Verbindungen der Formel (I) zu bilden vermögen.
- Diese Salze erhält man zweckmäßigerweise durch Behandeln der Verbindungen der Formel (I), die saure Wasserstoffatome enthalten, mit geeigneten organischen und anorganischen Basen, wie beispielsweise Ammoniumsalzen, Alkali- und Erdalkalisalzen, z.B. Lithium-, Natrium-, Kalium-, Magnesium-, Calciumsalzen und dergleichen, Salzen mit organischen Basen, z.B. Benzathin-, N-Methyl-D-glucamin- und Hydrabaminsalze sowie Salze mit Aminosäuren, wie z.B. Arginin, Lysin und dergleichen.
- Umgekehrt lassen sich die Salzformen durch Behandlung mit einer geeigneten Base bzw. Säure in die freie Säure- oder Basenform überführen.
- Der Begriff „Additionssalz" umfaßt im Sinne der obigen Verwendung auch die Solvate, die die Verbindungen der Formel (I) zu bilden vermögen. Diese Solvate sollen in den Schutzbereich der vorliegenden Erfindung fallen. Beispiele für derartige Solvate sind Hydrate, Alkoholate und dergleichen.
- Die N-Oxidformen der Verbindungen der Formel (I) sollen diejenigen Verbindungen der Formel (I) umfassen, in denen ein oder mehrere Stickstoffatome zum sogenannten N-Oxid oxidiert sind.
- Unter dem hier verwendeten Begriff „stereochemisch isomere Formen" sind alle möglichen isomeren Formen zu verstehen, in denen die Verbindungen der Formel (I) auftreten können. Sofern nichts Anderes angegeben ist, bezeichnet die chemische Bezeichnung der Verbindungen das Gemisch aller möglichen stereochemisch isomeren Formen, wobei diese Gemische alle Diastereomere und Enantiomere der zugrundeliegenden Molekülstruktur enthalten.
- Einige der Verbindungen der Formel (I) können auch in ihrer tautomeren Form existieren. Derartige Formen sind zwar in der obigen Formel nicht explizit angegeben, sollen aber zum Schutzbereich der vorliegenden Erfindung gehören.
- Der Begriff "Verbindungen der Formel (I)" soll bei jeder nachfolgenden Erwähnung auch die N-Oxidformen, die pharmazeutisch unbedenklichen Additionssalze und alle stereoisomeren Formen mit einschließen.
- D steht geeigneterweise für einen Rest der Formel (a), (b), (c), (d), (e), (f) oder (g), wobei m für 0 steht; die Reste Y unabhängig voneinander jeweils für -CH2-, -O-, -S- oder -NR3- stehen; und R2 und R3 jeweils unabhängig voneinander für Wasserstoff oder C1–6-Alkyl stehen.
- Unten kommt bei Bezugnahme auf die Stellung des Substituenten R1 das folgende Numerierungssystem zur Anwendung:
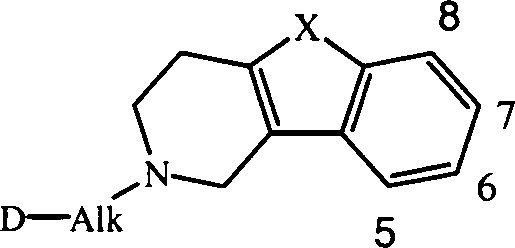
- Eine interessante Gruppe von Verbindungen sind die Verbindungen der Formel (I), in denen n für 1 steht und R1 für Wasserstoff, Chlor, Fluor, Methyl, Methoxy oder Nitro steht; R1 steht insbesondere für Wasserstoff, Chlor, Methyl oder Methoxy. Ebenfalls interessant sind die Verbindungen der Formel (I), in denen n für 2 steht und beide R1 für Methoxy stehen.
- Steht R1 nicht für Wasserstoff, dann ist R1 geeigneterweise in der 6- oder 7-Stellung an das tricyclische Ringsystem gebunden.
- Eine andere interessante Gruppe von Verbindungen sind die Verbindungen der Formel (I), in denen Alk für Methylen, 1,2-Ethandiyl, 1,3-Propandiyl, 1,4-Butandiyl oder 1,5-Pentandiyl, insbesondere Methylen, 1,2-Ethandiyl, 1,3-Propandiyl, 1,4-Butandiyl und ganz insbesondere 1,2-Ethandiyl steht.
- Noch eine interessante Gruppe von Verbindungen sind die Verbindungen der Formel (I), in denen D für einen Rest der Formel (a), (b), (c), (e), (f), (g), (h), (i) oder (j), insbesondere (a), (c), (j), (h), (i) oder (j), steht.
- Verbindungen der Formel (I), in denen D nicht für (a) oder (b) steht, sind ebenfalls von besonderem Interesse.
- Besondere Verbindungen sind die Verbindungen der Formel (I), in denen X für -O- oder -S-, insbesondere für -O-, steht.
- Andere besondere Verbindungen sind die Verbindungen der Formel (I), in denen Y für -O- oder -S- steht.
- Bevorzugte Verbindungen sind die Verbindungen der Formel (I), in denen n für 1 steht, R1 für Wasserstoff, Chlor, Methyl oder Methoxy steht und X für -O- oder -Ssteht.
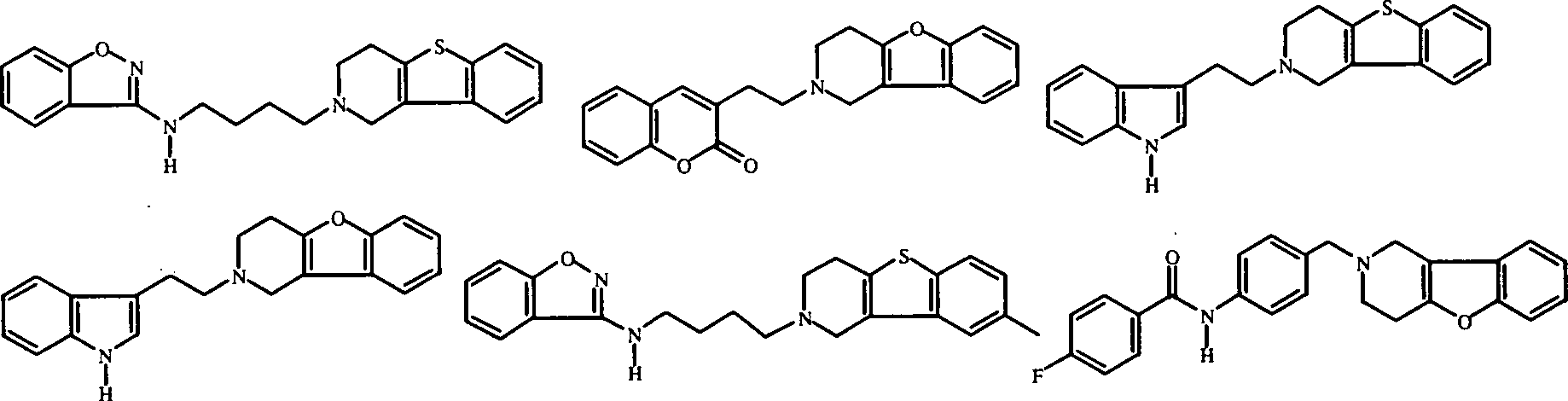
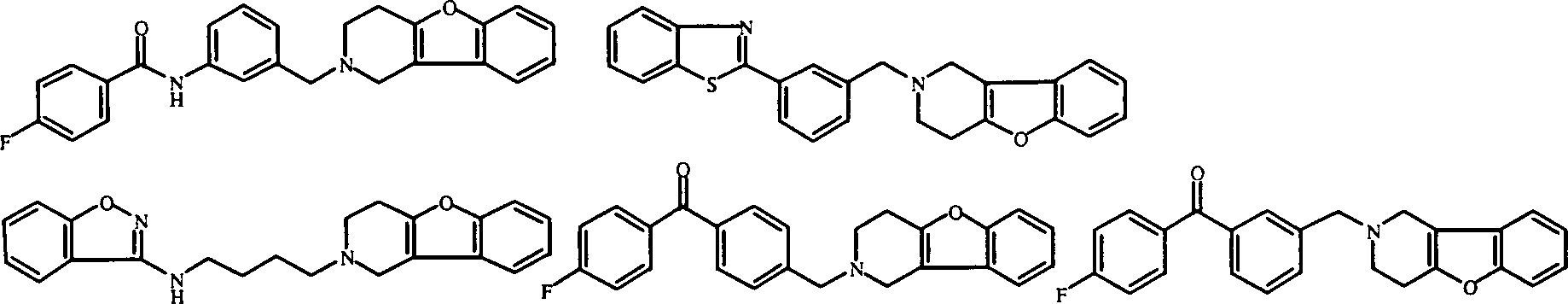
- Am meisten bevorzugte Verbindungen sind die unten gezeigten Verbindungen bzw. deren N-Oxidformen und die pharmazeutisch unbedenklichen Additionssalze und die stereochemisch isomeren Formen davon:
- Die Verbindungen der Formel (I) können im allgemeinen durch N-Alkylierung eines Zwischenprodukts der Formel (II) mit einem Alkylierungsmittel der Formel (III) nach der in EP-A-0,037,265, EP-A-0,070,053, EP-A-0,196,132 und EP-A-0,378,255 beschriebenen Verfahrensweise hergestellt werden. Insbesondere kann man die N-Alkylierung in einem unter den Reaktionsbedingungen inerten Lösungsmittel, wie Methylisobutylketon, N,N-Dimethylformamid oder N,N-Dimethylacetamid, in Gegenwart einer Base, beispielsweise Triethylamin, Natriumcarbonat oder Natriumhydrogencarbonat, und gegebenenfalls in Gegenwart eines Katalysators, wie Kaliumiodid, durchführen.
-
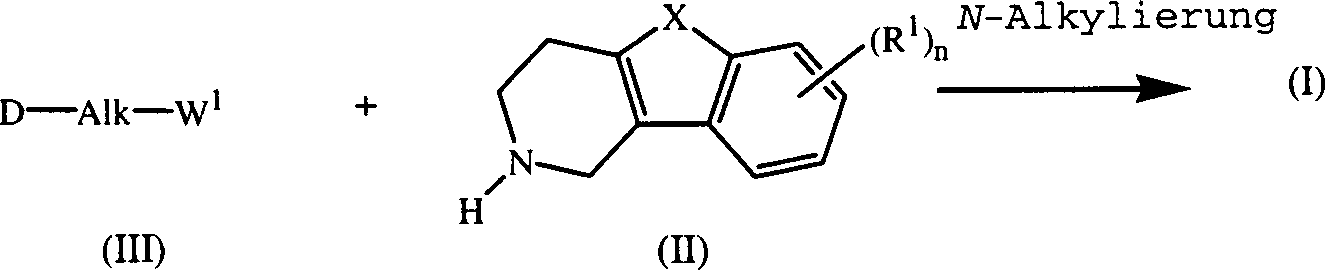
- Unter ähnlichen Reaktionsbedingungen lassen sich durch die Formel (I-h) wiedergegebene Verbindungen der Formel (I), in denen D für einen Rest der Formel (h) steht, durch Umsetzen eines Zwischenprodukts der Formel (VII) mit einem Zwischenprodukt der Formel (VIII) darstellen.
-
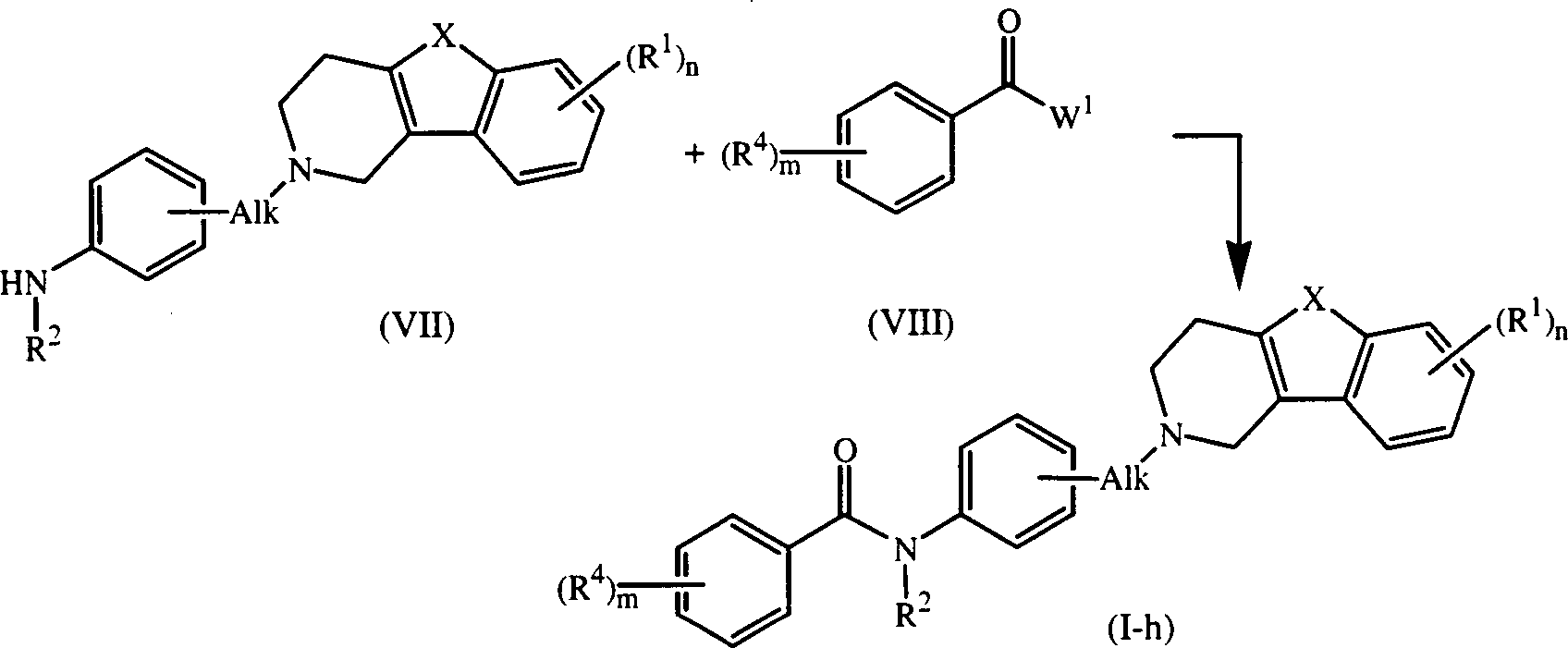
- Im Zwischenprodukt (III) und Zwischenprodukt (VIII) steht W1 für eine geeignete reaktive Abgangsgruppe, wie Halogen, z.B. Chlor, Brom oder Iod, oder Sulfonyloxy, z.B. Methansulfonyloxy oder 4-Methylbenzolsulfonyloxy.
- Bei dieser und den folgenden Umsetzungen können die Reaktionsprodukte nach an sich bekannten Verfahrensweisen aus dem Reaktionsmedium isoliert und gegebenenfalls weiter gereinigt werden, beispielsweise durch Extraktion, Kristallisation, Triturieren und Chromatographie.
- Ein spezieller Weg zur Darstellung von durch die Formel (I-g) wiedergegebenen Verbindungen der Formel (I), in denen D für einen Rest der Formel (c) oder (g) steht und Alk für -(Alk')p-CH2- steht, wobei Alk' für C1–5-Alkandiyl und p für 0 oder 1 steht, beinhaltet die reduktive N-Alkylierung des Zwischenprodukts der Formel (II) mit einem Aldehydderivat der Formel (IV-c) beziehungsweise (IV-g).
-

- Diese reduktive N-Alkylierungsreaktion läßt sich bequem durchführen, indem man eine Mischung der Reaktionspartner in einem geeigneten reaktionsinerten Lösungsmittel nach im Stand der Technik bekannten Vorschriften zur reduktiven N-Alkylierung reduziert. Insbesondere kann die Reaktionsmischung zur Erhöhung der Reaktionsgeschwindigkeit gerührt und/oder erhitzt werden. Als Lösungsmittel eignen sich beispielsweise Wasser; Methanol, Ethanol, 2-Propanol und dergleichen. Die Umsetzung erfolgt zweckmäßigerweise entweder mit Natriumcyanoborhydrid, Natriumborhydrid, Ameisensäure oder einem Salz davon und ähnlichen Reduktionsmitteln, oder alternativ dazu unter einer Wasserstoffatmosphäre, gegebenenfalls bei erhöhter Temperatur und/oder erhöhtem Druck, in Gegenwart eines geeigneten Katalysators wie beispielsweise Palladium-auf-Aktivkohle, Platin-auf-Aktivkohle und dergleichen. Um die unerwünschte weitere Hydrierung bestimmter funktioneller Gruppen in den Reaktionspartnern und den Reaktionsprodukten zu verhindern, kann es von Vorteil sein, die Reaktionsmischung mit einem geeigneten Katalysatorgift, beispielsweise Thiophen, Chinolin-Schwefel und dergleichen, zu versetzen. In einigen Fällen kann es weiterhin vorteilhaft sein, ein Alkalisalz wie beispielsweise Kaliumfluorid, Kalium acetat und ähnliche Salze zur Reaktionsmischung zu geben.
- Die Verbindungen der Formel (I) können durch an sich bekannte Transformationen funktioneller Gruppen ineinander umgewandelt werden.
- Die Verbindungen der Formel (I) lassen sich nach an sich bekannten Methoden zur Umwandlung eines dreiwertigen Stickstoffs in seine N-Oxidform auch in die entsprechenden N-Oxidformen überführen. Diese N-Oxidation kann im allgemeinen so durchgeführt werden, daß man das Edukt der Formel (I) mit einem geeigneten organischen oder anorganischen Peroxid umsetzt. Als anorganische Peroxide eignen sich beispielsweise Wasserstoffperoxid, Alkali- oder Erdalkalimetallperoxide, z.B. Natriumperoxid, Kaliumperoxid; als organische Peroxide eignen sich beispielsweise Peroxysäuren, wie zum Beispiel gegebenenfalls halogensubstituierte Benzolcarboperoxosäure, z.B. 3-Chlorbenzolcarboperoxosäure, Peroxoalkansäuren, z.B. Peroxoessigsäure, Alkylhydroperoxide, z.B. tert.-Butylhydroperoxid. Als Lösungsmittel eignen sich beispielsweise Wasser, niedere Alkanole, z.B. Ethanol und dergleichen, Kohlenwasserstoffe, z.B. Toluol, Ketone, z.B. 2-Butanon, halogenierte Kohlenwasserstoffe, z.B. Dichlormethan, sowie Gemische derartiger Lösungsmittel.
- Eine Reihe von Zwischenprodukten und Edukten sind im Handel erhältlich oder bekannt und nach an sich bekannten Verfahrensweisen zugänglich.
- So werden beispielsweise einige der Zwischenprodukte der Formel (III) und ihre Herstellung in EP-A-0,037,265, EP-A-0,070,053, EP-A-0,196,132 und EP-A-0,378,255 beschrieben.
- Zwischenprodukte der Formel (II), in denen X für 0 steht, lassen sich analog der in Cattanach C. et al. (J. Chem. Soc (C), 1971, S. 53–60); Kartashova T. (Khim. Geterotsikl. Soedin., 1979 (9), S. 1178–1180) und Zakusov V. et al. (Izobreteniya, 1992 (15) S. 247) beschriebenen Vorschriften darstellen. Zwischenprodukte der Formel (II), in denen X für S steht, lassen sich analog der in Capps et al. (J. Am. Chem. Soc., 1953, S. 697) oder US-3,752,820 beschriebenen Vorschrift darstellen.
- Eine besondere Syntheseroute zur Darstellung von Zwischenprodukten der Formel (II) ist in Schema 1 gezeigt.
- Schema 1
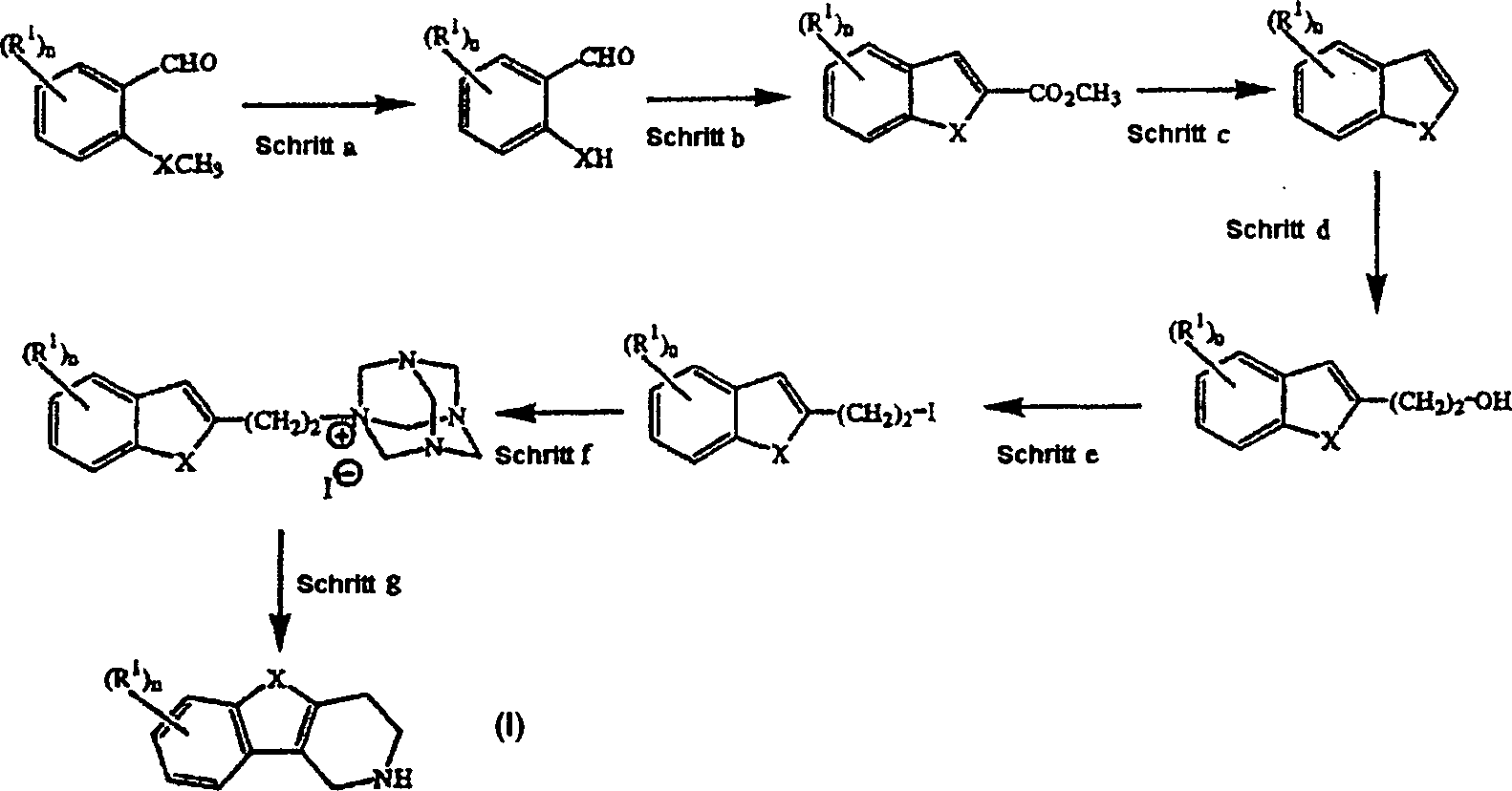
- Schritt a läßt sich analog der in Tetrahedron (1981), 37, S. 979–982 beschriebenen Vorschrift durchführen. Die in Schritt c erhaltenen Benzofurane wurden in
US 4,210,655 als Zwischenprodukte verwendet. Die weiteren Reaktionsschritte sind analog der inUS 3,752,820 beschriebenen Reaktionsvorschriften. - Alternativ dazu lassen sich Zwischenprodukte der Formel (II) darstellen, indem man die in Schema 2 gezeigten Reaktionsschritte anwendet.
- Schema 2
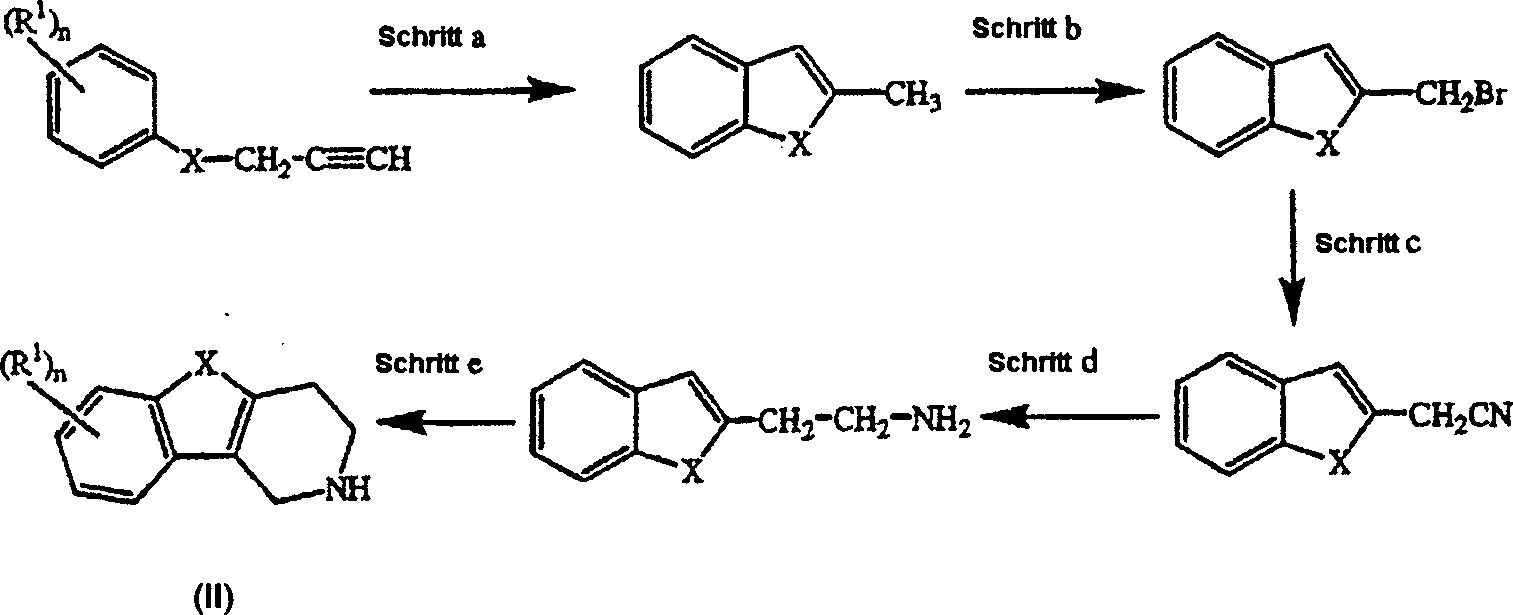
- Schritt a läßt sich analog der in Heterocycles (1994), 39 (1), S. 371–380 beschriebenen Vorschrift durchführen. Schritt b kann analog der in J. Med. Chem. (1986), 29 (9), S. 1643–1650 beschriebenen Vorschriften durchgeführt werden. Weitere Reaktionsschritte lassen sich analog der in J. Heterocycl. Chem. (1979), 16, S. 1321 beschriebenen Reaktionsschritte durchführen.
- Durch die Formel (III-c) wiedergegebene Zwischenprodukte der Formel (III), in denen D für einen Rest der Formel (c) steht, lassen sich darstellen, indem man ein Zwischenprodukt der Formel (V), in dem W2 für eine geeignete Abgangsgruppe wie beispielsweise ein Halogen steht, mit einem Aminoalkoholderivat der Formel (VI) in Gegenwart eines Katalysators wie beispielsweise Kaliumiodid umsetzt. Zweckmäßigerweise rührt man die Reaktionsmischung bei erhöhten Temperaturen. Anschließend kann man unter Anwendung von im Stand der Technik bekannten Verfahren wie beispielsweise der Umsetzung des Alkohols mit Thionylchlorid in einem Lösungsmittel wie Chloroform eine geeignete Abgangsgruppe wie beispielsweise ein Halogen, z.B. Chlor, einführen.
-

- Einige der Verbindungen der Formel (I) und einige der Zwischenprodukte bei der vorliegenden Erfindung enthalten mindestens ein asymmetrisches Kohlenstoffatom. Reine stereochemisch isomere Formen der Verbindungen und Zwischenprodukte sind nach an sich bekannten Verfahren erhältlich. So können beispielsweise Diastereoisomere durch physikalische Methoden, wie selektive Kristallisation, oder chromatographische Methoden, z.B. Gegenstromverteilung, Flüssigkeitschromatographie und dergleichen, getrennt werden. Enantiomere können aus racemischen Gemischen erhalten werden, indem man die racemischen Gemische zunächst mit geeigneten Trennreagenzien, wie beispielsweise chiralen Säuren, in Gemische aus diastereomeren Salzen oder Verbindungen überführt und diese Gemische aus diastereomeren Salzen oder Verbindungen dann beispielsweise durch selektive Kristallisation oder chromatographische Methoden, z.B. durch Flüssigkeitschromatographie oder dergleichen, physikalisch trennt und schließlich die getrennten diastereomeren Salze oder Verbindungen in die entsprechenden Enantiomere überführt.
- Reine stereochemisch isomere Formen der Verbindungen der Formel (I) lassen sich auch aus den reinen stereochemisch isomeren Formen der entsprechenden Zwischenprodukte und Edukte erhalten, vorausgesetzt, daß die dazwischen stattfindenden Umsetzungen stereospezifisch verlaufen. Die reinen und gemischten stereochemisch isomeren Formen der Verbindungen der Formel (I) sollen in den Schutzbereich der vorliegenden Erfindung fallen.
- Die Verbindungen der Formel (I), die N-Oxidformen, pharmazeutisch unbedenklichen Additionssalze und stereochemisch isomeren Formen davon blockieren die präsynaptischen α2-Rezeptoren an zentralen noradrenergen Neuronen und erhöhen somit die Noradrenalin-Ausschüttung. Die Blockierung der Rezeptoren wird verschiedene Symptome unterdrücken oder erleichtern, die mit einem Mangel an Noradrenalin im zentralen oder peripheren Nervensystem verbunden sind. Therapeutische Indikationen zur Verwendung der erfindungsgemäßen Verbindungen sind Depression, kognitive Störungen, Morbus Parkinson, Diabetes mellitus, sexuelle Dysfunktion und Impotenz sowie erhöhter Augeninnendruck.
- Es wurde weiterhin nachgewiesen, daß die Blockade von α2-Rezeptoren im Zentralnervensystem auch die Ausschüttung von Serotonin erhöht, was die therapeutische Wirkung bei Depression verstärken kann (Maura et al., 1992, Naunyn-Schmiedberg's Arch. Pharmacol., 345:410–416).
- Es wurde außerdem nachgewiesen, daß durch die Blockade von α2-Rezeptoren eine Erhöhung von extrazellulärem DOPAC (3,4-Dihydrophenylessigsäure), einem Metaboliten von Dopamin und Noradrenalin, induziert werden kann.
- Im Hinblick auf die Brauchbarkeit der erfindungsgemäßen Verbindungen bei der Behandlung von Krankeiten, die mit einem Noradrenalinmangel im Zentralnervensystem verbunden sind, insbesondere Depression und Morbus Parkinson, stellt die vorliegende Erfindung ein Verfahren zur Behandlung von Warmblütern, die an derartigen Krankheiten, insbesondere Depression und Morbus Parkinson, leiden, bereit, bei dem man eine therapeutisch wirksame Menge einer Verbindung der Formel (I) oder eines pharmazeutisch unbedenklichen Additionssalzes davon systemisch verabreicht.
- Die erfindungsgemäßen Verbindungen sind potentiell auch zur Verwendung bei der Behandlung von Alzheimer-Krankheit und Demenz geeignet, da α2-Antagonisten bekanntlich die Ausschüttung von Acetylcholin fördern (Tellez et al. 1997, J. Neurochem. 68:778–785).
- Im allgemeinen wird angenommen, daß eine wirksame therapeutische Tagesmenge etwa 0,01 mg/kg bis etwa 4 mg/kg Körpergewicht beträgt.
- Gegenstand der vorliegenden Erfindung sind auch Verbindungen der Formel (I) gemäß obiger Definition zur Verwendung als Arzneimittel. Des weiteren betrifft die Erfindung auch die Verwendung einer Verbindung der Formel (I) zur Herstellung eines Arzneimittels zur Behandlung von Depression oder Morbus Parkinson.
- Zur Beurteilung des α2-Adrenozeptor-Antagonismus der erfindungsgemäßen Verbindungen kann man ex-vivo- sowie in-vitro-Rezeptorsignalübertragungs- und -Rezeptorbindungsstudien heranziehen. Als Indices der Blockade zentraler α2-Adrenozeptoren in vivo kann man sich der Umkehr des Verlusts des Stellreflexes bei Ratten nach intravenöser Injektion von Xylazin und der Inhibierung des durch Reserpin induzierten Zitterns bei Ratten bedienen.
- Die erfindungsgemäßen Verbindungen haben auch die Fähigkeit, schnell in das Zentralnervensystem einzudringen.
- Zur Verabreichung können die erfindungsgemäßen Verbindungen als verschiedene pharmazeutische Zusammensetzungen formuliert werden, die einen pharmazeutisch unbedenklichen Träger und als Wirkstoff eine therapeutisch wirksame Menge einer Verbindung der Formel (I) enthalten. Zur Herstellung der erfindungsgemäßen pharmazeutischen Zusammensetzungen vereinigt man eine wirksame Menge der jeweiligen Verbindung in Additionssalzform oder in Form der freien Säure oder Base als Wirkstoff in Form einer innigen Mischung mit einem pharmazeutisch unbedenklichen Träger, der je nach der zur Verabreichung gewünschten Darreichungsform verschiedenste Formen annehmen kann. Diese pharmazeutischen Zusammensetzungen liegen wünschenswerterweise in Einheitsdosisform vor, die sich vorzugsweise zur oralen, oder perkutanen Verabreichung oder zur parenteralen Injektion eignet. Bei der Herstellung der Zusammensetzungen in oraler Dosisform können beispielsweise alle üblichen pharmazeutischen Medien verwendet werden, wie beispielsweise Wasser, Glykole, Öle, Alkohole und dergleichen bei oralen Flüssigpräparaten wie Suspensionen, Sirupen, Elixiren und Lösungen, oder feste Träger wie Stärken, Zucker, Kaolin, Gleitmittel, Bindemittel, Sprengmittel und dergleichen bei Pulvern, Pillen, Kapseln und Tabletten. Aufgrund ihrer leichten Verabreichbarkeit stellen Tabletten und Kapseln die vorteilhafteste orale Einzeldosisform dar, wobei man natürlich feste pharmazeutische Träger verwendet. Bei Zusammensetzungen zur parenteralen Applikation besteht der Träger in der Regel zumindest größtenteils aus sterilem Wasser, wenngleich auch andere Bestandteile, z.B. zur Förderung der Löslichkeit, vorhanden sein können. Es lassen sich beispielsweise Injektionslösungen herstellen, bei denen der Träger aus Kochsalzlösung, Glucoselösung oder einer Mischung von Kochsalz- und Glucoselösung besteht. Injektionslösungen, die Verbindungen der Formel (I) enthalten, können zwecks langanhaltender Wirkung in einem Öl formuliert werden. Als Öle für diesen Zweck eignen sich beispielsweise Erdnußöl, Sesamöl, Baumwollsamenöl, Maisöl, Sojabohnenöl, synthetische Glycerinester langkettiger Fettsäuren und Gemische aus diesen und anderen Ölen. Ferner lassen sich auch Injektionssuspensionen herstellen, wobei geeignete flüssige Träger, Suspendiermittel und dergleichen verwendet werden können. Bei den zur perkutanen Verabreichung geeigneten Zusammensetzungen enthält der Träger gegebenenfalls ein Penetriermittel und/oder ein geeignetes Netzmittel, gegebenenfalls in Kombination mit kleineren Mengen geeigneter Zusatzstoffe jeglicher Art, wobei diese Zusatzstoffe keine wesentliche negative Wirkung auf die Haut ausüben. Derartige Zusatzstoffe können die Aufbringung auf die Haut erleichtern und/oder für die Herstellung der gewünschten Zusammensetzungen von Nutzen sein. Diese Zusammensetzungen können auf verschiedenen Wegen verabreicht werden, z.B. als transdermales Pflaster, Direktauftrag oder Salbe. Additionssalze von (I) sind aufgrund ihrer gegenüber der entsprechenden freien Basen- oder Säureform erhöhten Wasserlöslichkeit offensichtlich besser für die Herstellung von wäßrigen Zusammensetzungen geeignet.
- Zwecks einfacher Verabreichung und einheitlicher Dosierung ist es besonders vorteilhaft, die obengenannten pharmazeutischen Zusammensetzungen in Einzeldosisform zu formulieren. Unter dem Begriff Einzeldosisform sind in der Beschreibung und in den Ansprüchen physikalisch diskrete Einheiten zu verstehen, die sich als Einheitsdosen eignen, wobei jede Einheit eine vorbestimmte Menge des Wirkstoffs enthält, die so berechnet ist, daß in Verbindung mit dem erforderlichen pharmazeutischen Träger die gewünschte therapeutische Wirkung erzielt wird. Beispiele für solche Einzeldosisformen sind Tabletten (darunter Tabletten mit Bruchrille und Dragees), Kapseln, Pillen, Pulverbeutel, Oblaten, Injektionslösungen, Injektionssuspensionen, Teelöffelvoll, Eßlöffelvoll und dergleichen sowie deren getrennte Vielfache.
- Die folgenden Beispiele sollen die vorliegende Erfindung näher erläutern.
- Experimenteller Teil
- A. Darstellung der Zwischenprodukte
- Beispiel A1
- Eine Mischung von O-Phenylhydroxylamin-hydrochlorid (1:1) (0,625 mol) und 4,4-Piperidindiol-hydrochlorid (1:1) (0,682 mol) in 2-Propanol (615 ml) wurde bei 20°C gerührt. HCl (353 ml) wurde bei 20°C zugetropft. Die Reaktionsmischung wurde vorsichtig auf Rückflußtemperatur erhöht. Die Reaktionsmischung wurde unter Rühren 3 Stunden lang auf Rückfluß erhitzt und dann auf Raumtemperatur abgekühlt. Der Niederschlag wurde abfiltriert, mit Diisopropylether gewaschen und getrocknet. Diese Fraktion wurde aus Wasser (1600 ml) kristallisiert. Die gewünschte Verbindung wurde unter Rühren auskristallisieren gelassen. Der Niederschlag wurde abfiltriert, mit 2-Propanol und Diisopropylether gewaschen und dann getrocknet, wodurch man 84 g (64%) 1,2,3,4-Tetrahydrobenzofuro[3,2-c]pyridin-hydrochlorid (1:1) (Zwischenpr. 1) erhielt.
- Beispiel A2
-
- a) Butyllithium (0,27 mol einer 2, 5 M Lösung) wurde zu 6-Methoxybenzo[b]thiophen [dargestellt analog der in J. Med. Chem. 1989, 32(12), 2548–2554 beschriebenen Vorschrift] (0,25 mol) in Tetrahydrofuran (1000 ml), das bei –30°C gerührt wurde, getropft. Die Mischung wurde 10 Minuten lang bei –30°C gerührt. Ethylenoxid (0,38 mol in 100 ml Tetrahydrofuran) wurde bei –30°C tropfenweise zugegeben. Die Mischung wurde auf Raumtemperatur erwärmen gelassen und 3 Stunden lang gerührt. Die Mischung wurde mit verdünnter HCl-Lösung angesäuert. Das Lösungsmittel wurde abgedampft. Der Rückstand wurde mit Wasser verdünnt, und die Mischung wurde mit CH2Cl2 extrahiert. Die abgetrennte organische Phase wurde getrocknet und filtriert, und das Lösungsmittel wurde abgedampft. Der Rückstand wurde in Hexan gerührt, abfiltriert und getrocknet, wodurch man 41,3 g 6-Methoxybenzo[b]thiophen-2-ethanol (Zwischen produkt 2) erhielt.
- b) Eine bei 0°C gerührte Mischung von Zwischenprodukt (2) (0,19 mol) und Triethylamin (0,21 mol) in CH2Cl2 (1000 ml) wurde mit Methansulfonylchlorid (0,21 mol) versetzt. Die Reaktionsmischung wurde 4 Stunden lang bei Raumtemperatur gerührt und dann in Wasser gegossen. Die abgetrennte organische Phase wurde getrocknet und filtriert, und das Lösungsmittel wurde abgedampft. Der Rückstand wurde mit Diisopropylether verrieben, abfiltriert und getrocknet, wodurch man 50,5 g (94%) 6-Methoxybenzo[b]thiophen-2-ethanol-methansulfonat(ester) (Zwischenprodukt 3) erhielt.
- c) Eine Mischung von Zwischenprodukt (3) (0,18 mol) und NaI (0,45 mol) in 2-Propanon (1000 ml) wurde 9 Stunden lang unter Rühren auf Rückfluß erhitzt und anschließend auf Raumtemperatur abgekühlt, und das Lösungsmittel wurde abgedampft. Der Rückstand wurde mit Wasser gewaschen und mit CH2Cl2 extrahiert. Die abgetrennte organische Phase wurde getrocknet und filtriert und das Lösungsmittel wurde abgedampft, wodurch man 57 g 2-(2-Iodethyl)-6-methoxybenzo(b]thiophen (Zwischenprodukt 4) erhielt.
- d) Zwischenprodukt (4) (0,18 mol) wurde portionsweise zu einer Mischung von 1,3,5,7-Tetraazatricyclo[5.1.1.13,5]decan (0,45 mol) in CHCl3 (600 ml) gegeben. Das Reaktionsgemisch wurde über Nacht unter Rühren auf Rückfluß erhitzt und anschließend auf Raumtemperatur abgekühlt. Der Niederschlag wurde abfiltriert und getrocknet, wodurch man 54,2 g 1-[2-(6-Methoxybenzo[b]thiophen-2-yl)ethyl]-1,3,5,7-tetraazatricyclo[5.1.1.15,7]decaniumiodid (Zwischenprodukt 5) erhielt.
- e) Eine Mischung von Zwischenprodukt (5) (0,12 mol) und HCl (0,50 mol) in Ethanol (171 ml) wurde 2 Tage lang bei Raumtemperatur gerührt. Mehr HCl (10 ml) und Ethanol (40 ml) wurden zugegeben, und das Reaktions gemisch wurde 1 Stunde lang unter Rühren auf Rückfluß erhitzt und anschließend auf Raumtemperatur abgekühlt. Das Lösungsmittel wurde abgedampft. Der Rückstand wurde in 2-Propanol gerührt und dann abfiltriert. Der Feststoff wurde getrocknet, und der Rückstand wurde mit 20% NaOH zurück in die freie Base umgewandelt. Die abgetrennte organische Phase wurde getrocknet und filtriert, und das Lösungsmittel wurde abgedampft. Der Rückstand wurde in 2-Propanol gelöst und mit HCl/2-Propanol in das Salzsäuresalz (1:1) überführt. Der Niederschlag wurde abfiltriert und getrocknet, wodurch man 13,1 g (50%) 1,2,3,4-Tetrahydro-7-methoxy[1]benzothieno[3,2-c]pyridin (Zwischenprodukt 6) erhielt.
- Auf analoge Weise wurde auch die folgende Verbindung dargestellt:
1,2,3,4-Tetrahydro-8-methyl-[1]benzothieno[3,2-c]pyridin-hydrochlorid (Zwischenprodukt 18). - Beispiel A3
-
- a) Eine Mischung von 3-Chlor-l,2-benzisoxazol (0,08 mol), 4-Amino-1-butanol (0,24 mol) und Kaliumiodid (1 g) wurde 4 Tage lang bei 80°C gerührt. Das Reaktionsgemisch wurde abgekühlt, in CH2Cl2 gelöst und durch Kieselgel-Säulenchromatographie (Laufmittel: CH2Cl2/CH3OH 95/5) aufgereinigt. Die reinen Fraktionen wurden gesammelt und das Lösungsmittel wurde abgedampft, wodurch man 15,4 g (93%) 4-(1,2-Benzisoxazol-3-ylamino)-1-butanol (Zwischenprodukt 7) erhielt.
- b) SOCl2 (0,048 mol) wurde auf 0°C abgekühlt. Eine Lösung von Zwischenprodukt 7 (0,048 mol) in CHCl3 (20 ml) wurde tropfenweise zugesetzt, und das Reaktionsgemisch wurde über Nacht bei Raumtemperatur gerührt. Das Lösungsmittel wurde abgedampft. Der Rückstand wurde mit Wasser gewaschen. Das Reaktionsgemisch wurde mit CH2Cl2 extrahiert. Die abgetrennte organische Phase wurde getrocknet und filtriert und das Lösungsmittel abgedampft, wodurch man 10,4 g N-(4-Chlorbutyl)-1,2-benzisoxazol-3-amin (Zwischenprodukt 8) erhielt.
- c) Umsetzung unter einer N2-Atmosphäre. Eine Lösung von Ethandioyldichlorid (0,026 mol) in CH2Cl2 (60 ml) wurde bei –60°C gerührt. Dimethylsulfoxid (3,8 ml) wurde bei –60°C zugetropft, und die Mischung wurde 10 Minuten lang gerührt. Eine Lösung von 4-(1,2-Benzisoxazol-3-ylamino)-1-butanol (0,024 mol) in CH2Cl2, (120 ml) wurde bei –60°C tropfenweise zugegeben, und die Mischung wurde eine Stunde lang bei –60°C gerührt. N,N-Diethylethanamin (13,7 ml) wurde zugetropft, und das Reaktionsgemisch wurde 10 Minuten lang bei –60°C gerührt und anschließend auf Raumtemperatur erwärmen gelassen. Die Mischung wurde in Wasser (250 ml) gegossen. Die Mischung wurde 10 Minuten lang gerührt. Die abgetrennte organische Phase wurde getrocknet und filtriert und das Lösungsmittel wurde abgedampft. Der Rückstand wurde mit Hexan verrieben, abfiltriert und getrocknet, wodurch man 3,9 g 4-(1,2-Benzisoxazol-3-ylamino)butanol (80%) (Zwischenprodukt 9) erhielt.
- Beispiel A4
- Eine gerührte Mischung von 58,5 g 1H-Indol, 107,5 ml 1-Brom-3-chlorpropan, 15 mg N,N,N-Triethylbenzolmethanaminiumchlorid und 450 ml Benzol wurde im Verlauf von 30 Minuten bei 40°C tropfenweise zu 250 ml einer 60%igen Natriumhydriddispersion gegeben. Nach Ende der Umsetzung wurde noch 1 weitere Stunde lang bei 40°C gerührt. Weitere 15 ml 1-Brom-3-chlorpropan wurden zugefügt, und es wurde 1 weitere Stunde lang bei 50°C gerührt. Nach dem Abkühlen wurde die Reaktionsmischung in Wasser gegossen. Das Produkt wurde mit Benzol extrahiert. Der Extrakt wurde abgetrennt, filtriert und eingedampft. Der Rückstand wurde destilliert, wodurch man 80 ml (83%) 1-(3-Chlorpropyl)-1H-indol (Zwischenprodukt 10); Sdp. 120–125°C erhielt.
- Beispiel A5
-
- a) Eine Mischung von 1,2,3,4-Tetrahydrobenzofuro[3,2-c]pyridin-hydrochlorid (1:1), (0,05 mol), 1-(Chlormethyl)-4-nitrobenzol (0,05 mol), Na2CO3 (7 g) und Kaliumiodid (0,1 g) in 4-Methyl-2-pentanon (250 ml) wurde unter Rühren 8 Stunden lang auf Rückfluß erhitzt. Die Mischung wurde auf Raumtemperatur abgekühlt. Das Reaktionsgemisch wurde filtriert und das Filtrat wurde eingedampft. Der ölige Rückstand wurde in CH3CN/Diisopropylether gelöst und gerührt. Der Niederschlag wurde abfiltriert und getrocknet, was 8 g 1,2,3,4-Tetrahydro-2-[(4-nitrophenyl)methyl]benzofuro[3,2-c]pyridin (Zwischenprodukt 11) lieferte. Das Filtrat wurde mit HCl/2-Propanol gerührt. Der Niederschlag wurde abfiltriert und getrocknet, wodurch man 9 g 1,2,3,4-Tetrahydro-2-[(4-nitrophenyl)methyl]benzofuro[3,2-c]pyridinhydrochlorid (.HCl) (Zwischenprodukt 12) erhielt.
- b) Eine Mischung von Zwischenprodukt (11) (0,027 mol) in 2-Methoxyethanol (300 ml) wurde bei Raumtemperatur mit 5% Platin-auf-Aktivkohle (2 g) als Katalysator in Gegenwart einer Thiophenlösung (2 ml) hydriert. Nach Ende der H2-Aufnahme (3 Äquivalente) wurde der Katalysator abfiltriert und das Filtrat eingedampft. Der Rückstand wurde in Diisopropylether + einer kleinen Menge CH3CN gerührt und mit HCl/2-Propanol behandelt. Das Salzsäuresalz (1:2) wurde abfiltriert und getrocknet, wodurch man 8,5 g 4-[(3,4-Dihydrobenzofuro[3,2-c]pyridin-2(1H)-yl)methyl]benzolamin-monohydrochlorid (Zwischenprodukt 13) erhielt.
- Beispiel A6
-
- a) Umsetzung unter einer N2-Atmosphäre. BF3·Et2O (215 ml) wurde auf 0°C abgekühlt. 3-Fluorphenol (0,25 mol) wurde zugegeben. 6-Chlorhexanoylchlorid (0,51 mol) wurde zugesetzt, und die so erhaltene Reaktionsmischung wurde 15 Minuten lang bei 0°C gerührt und anschließend auf Raumtemperatur erwärmt. Das Reaktionsgemisch wurde über Nacht bei 130°C gerührt. Die Mischung wurde auf Raumtemperatur abgekühlt. Während des Abkühlens wurde mit Wasser versetzt. Die Mischung wurde zweimal mit 2,2'-Oxybispropan extrahiert. Die abgetrennte organische Phase wurde getrocknet und filtriert, und das Lösungsmittel wurde abgedampft. Der Rückstand wurde durch Kieselgel-Säulenchromatographie (Laufmittel: CH2Cl2/Hexan 50/50) gefolgt von HPLC (Laufmittel: CH2Cl2/Hexan 50/50) aufgereinigt. Die reinen Fraktionen wurden gesammelt und das Lösungsmittel wurde abgedampft, wodurch man 52,2 g 6-Chlor-1-(4-fluor-2-hydroxyphenyl)-1-hexanon (Zwischenprodukt 14) erhielt.
- b) Eine Mischung von Zwischenprodukt (14) (0,21 mol) und Hydroxylamin (0,25 mol) in Pyridin (100 ml) wurde 2 Tage lang bei Raumtemperatur gerührt und anschließend in 1N HCl (450 ml) gegossen. Diese Mischung wurde 10 Minuten lang gerührt, und dann mit Essigsäureethylester extrahiert. Die abgetrennte organische Phase wurde getrocknet und filtriert, und das Lösungsmittel wurde abgedampft. Der Rückstand wurde durch Kieselgel-Säulenchromatographie (Laufmittel: CH2Cl2/CH3OH 99/1) aufgereinigt. Die gewünschten Fraktionen wurden gesammelt und das Lösungsmittel wurde abgedampft, wodurch man 22 g 6-Chlor-1-(4-fluor-2-hydroxyphenyl)-1-hexanon-oxim (Zwischenprodukt 15) erhielt.
- c) Das Zwischenprodukt (15) (0,077 mol) wurde in Tetrahydrofuran (200 ml) auf 60°C erhitzt. Eine Lösung von 1,1'-Carbonylbis[1H-imidazol] (0,16 mol) in Tetrahydrofuran (600 ml) wurde zugetropft, und das so erhaltene Reaktionsgemisch wurde 2 Stunden lang unter Rühren auf Rückfluß erhitzt. Das Reaktionsgemisch wurde auf Raumtemperatur abgekühlt, und das Lösungsmittel abgedampft. Der Rückstand wurde mit Wasser gewaschen und anschließend mit HCl angesäuert. Diese Mischung wurde mit CH2Cl2 extrahiert. Die abgetrennte organische Phase wurde getrocknet und filtriert, und das Lösungsmittel wurde abgedampft. Der Rückstand wurde durch Kieselgel-Säulenchromatographie (Laufmittel: CH2Cl2 100%) aufgereinigt. Zwei gewünschte Fraktionen wurden gesammelt und das Lösungsmittel wurde abgedampft, wodurch man 6,4 g 3-(5-Chlorpentyl)-6-fluor-1,2-benzisoxazol (Zwischenprodukt 16) und 11,1 g 2-(5-Chlorpentyl)-6-fluorobenzoxazol (Zwischenprodukt 17) erhielt.
- Beispiel A7
- Eine Mischung von 2-(4-Chlorphenyl)-3-(2-hydroxyethyl)-4(3H)-chinazolinon (0,068 mol) und 46%iger HBr in Wasser (200 ml) wurde 90 Minuten lang unter Rühren auf Rückfluß erhitzt. 300 ml Wasser wurden zugegeben. Das kristallisierte Produkt wurde abfiltriert und getrocknet (Fraktion 1). Das Filtrat (Öl) wurde verfestigt (Fraktion 2). Beide Fraktionen wurden vereinigt, wodurch man 23,5 g (78%) 3-(2-Bromethyl)-2-(4-chlorphenyl)-4(3H)-chinazolinon-monohydrobromid; Schmp. 214,0°C (Zwischenprodukt 19) erhielt.
- B. Darstellung der Endprodukte
- Beispiel B1
-
- a) Eine Mischung von 1,2,3,4-Tetrahydrobenzothieno[3,2-c]pyridin [dargestellt analog der in J. Am. Chem. Soc., 1953, S. 697 beschriebenen Vorschrift] (0,012 mol) und 4-Phenoxybenzaldehyd (0,012 mol) in Methanol (100 ml) wurde mit Palladium-auf-Aktivkohle (1 g) als Katalysator in Gegenwart einer Thiophenlösung (1 ml einer 4%igen Lösung) hydriert. Nach Ende der H2-Aufnahme (1 Äquivalent) wurde der Katalysator abfiltriert und das Filtrat eingedampft. Der Rückstand wurde in das (E)-2-Butendisäuresalz (1:1) überführt, abfiltriert und getrocknet, wodurch man 4,1 g (84%) 1,2,3,4-Tetrahydro- 1-[(4-phenoxyphenyl)methyl]-[1]-benzothieno[3,2-c]pyridin-(E)-2-butendioat (1:1) (Verbindung 1) erhielt.
- b) Eine Mischung von Zwischenprodukt (6) (0,0059 mol) und 4-Phenoxybenzaldehyd (0,0076 mol) und Kaliumacetat (1 g) in Methanol (150 ml) wurde bei 50°C mit Platin-auf-Aktivkohle (1 g) als Katalysator in Gegenwart einer Thiophenlösung (1 ml einer 5%igen Lösung) hydriert. Nach Ende der H2-Aufnahme (1 Äquivalent) wurde der Katalysator abfiltriert und das Lösungsmittel abgedampft. Der Rückstand wurde mit Wasser gewaschen und mit CH2Cl2 extrahiert. Die abgetrennte organische Phase wurde getrocknet und filtriert, und das Lösungsmittel wurde abgedampft. Der Rückstand wurde aus 2-Propanol kristallisiert, abfiltriert und getrocknet, wodurch man 1,2 g (50%) 1,2,3,4-Tetrahydro-7-methoxy-2-[(4-phenoxyphenyl)methyl]-[1]-benzothieno[3,2-c]pyridin (Verbindung 2) erhielt.
- c) Eine Mischung von 8-Chlor-1,2,3,4-tetrahydrobenzothieno[3,2-c]pyridin-hydrochlorid (1:1) (0,01 mol), 4-Phenoxybenzaldehyd (0,01 mol) und Kaliumacetat (1 g) in Methanol (150 ml) wurde bei 50°C hydriert. Nach Ende der H2-Aufnahme (1 Äquivalent) wurde der Katalysator abfiltriert und das Filtrat eingedampft. Der Rückstand wurde mit Wasser gewaschen und diese Mischung mit CH2Cl2 extrahiert. Die abgetrennte organische Phase wurde getrocknet und filtriert, und das Lösungsmittel wurde abgedampft. Der Rückstand wurde in das Salzsäuresalz (1:1) überführt, abfiltriert und getrocknet, wodurch man 2,9 g 1,2,3,4-Tetrahydro-8-methyl-2-[(4-phenoxyphenyl)methyl]-[1]-benzothieno[3,2-c]pyridin-hydrochlorid (69%) (Verbindung 10) erhielt.
- Beispiel B2
-
- a) Zwischenprodukt (10) (0,100 g) wurde zu einer Lösung von Zwischenprodukt (1) (0,00048 mol) und Na2CO3 (0.100 g) in N,N-Dimethylacetamid (1 ml) gegeben, und das so erhaltene Reaktionsgemisch wurde über Nacht bei 80°C gerührt. Die gewünschte Verbindung wurde isoliert und durch HPLC an nicht derivatisiertem Kromasil Spherical-Kieselgel (Laufmittel: CH2Cl2/(CH2Cl2/CH3OH 90/10)/CH3OH (0 Minuten) 100/0/0, (10,50 Minuten) 0/100/0, (12,50 Minuten) 50/0/50, (14,00 Minuten) 0/0/100, (15,01–20,00 Minuten) 100/0/0) aufgereinigt. Die reinen Fraktionen wurden gesammelt und das Lösungsmittel wurde abgedampft, wodurch man 0,045 g 1,2,3,4-Tetrahydro-2-[3-(1H-indol-1-yl)propyl]benzofuro[3,2-c]pyridin (Verbindung 4) erhielt.
- b) Eine Mischung von 1,2,3,4-Tetrahydrobenzothieno[3,2-c]pyridin [dargestellt analog der in J. Am. Chem. Soc., 1953, S. 697 beschriebenen Vorschrift] (0,01 mol), Zwischenprodukt (8) (0,02 mol) und Triethylamin (0,03 mol) in N,N-Dimethylacetamid (50 ml) wurde über Nacht bei 70°C gerührt und anschließend auf Raumtemperatur abgekühlt, und das Lösungsmittel wurde abgedampft. Der Rückstand wurde mit Wasser gewaschen und mit CH2Cl2 extrahiert. Die abgetrennte organische Phase wurde getrocknet und filtriert, und das Lösungsmittel wurde abgedampft. Der Rückstand wurde durch Kieselgel-Säulenchromatographie (Laufmittel: CH2Cl2/CH3OH 90/10) aufgereinigt. Die gewünschten Fraktionen wurden gesammelt, und das Lösungsmittel wurde abgedampft. Der Rückstand wurde in das (E)-2-Butendisäuresalz (2:1) überführt. Der Niederschlag wurde abfiltriert und getrocknet, wodurch man 0,38 g (9%) N-(1,2-Benzisoxazol-3-yl)-1,2,3,4-tetrahydro[1]benzothieno[3,2-c]pyridin-2-butanamin-(E)-2-butendioat (2:1) (Verbindung 7) erhielt.
- c) Na2CO3 (0,100 g) wurde zu einer Lösung von 1,2,3,4-Tetrahydrobenzothieno[3,2-c]pyridin [dargestellt analog der in J. Am. Chem. Soc., 1953, S. 697 beschriebenen Vorschrift] (0,00044 mol) und 3-(2-Bromethyl)-1H-indol (0,100 g) in Methylisobutylketon (2 ml) gegeben, und die so erhaltene Mischung wurde bei 100°C über Nacht gerührt. Die gewünschte Verbindung wurde isoliert und durch HPLC an nicht derivatisiertem Kromasil Spherical-Kieselgel (Laufmittel: CH2Cl2/(CH2Cl2/CH3OH 90/10)/CH3OH (0 Minuten) 100/0/0, (10,50 Minuten) 0/100/0, (12,50 Minuten) 50/0/50, (14,00 Minuten) 0/0/100, (15,01–20,00 Minuten) 100/0/0) aufgereinigt. Die reinen Fraktionen wurden gesammelt und das Lösungsmittel wurde abgedampft, wodurch man 0,045 g 1,2,3,4-Tetrahydro-2-[2-(1H-indol-3-yl)ethyl]-[1]-benzothieno[3,2-c]pyridin (Verbindung 8) erhielt.
- d) Eine Mischung von Zwischenprodukt (1) (0,01 mol), Zwischenprodukt (17) (0,012 mol), Na2CO3 (3 g) und KI in 4-Methyl-2-pentanon (200 ml) wurde über Nacht unter Rühren auf Rückfluß erhitzt und anschließend auf Raumtemperatur abgekühlt. Das Lösungsmittel wurde abgedampft. Der Rückstand wurde mit H2O gewaschen, und die Mischung wurde mit CH2Cl2 extrahiert. Die abgetrennte organische Phase wurde getrocknet und filtriert, und das Lösungsmittel wurde abgedampft. Der Rückstand wurde durch Kieselgel-Säulenchromatographie (Laufmittel: CH2Cl2/CH3OH 90/10) aufgereinigt. Die reinen Fraktionen wurden gesammelt, und die Lösung wurde eingedampft. Der Rückstand wurde in das (E)-2-Butendisäuresalz (1:1) überführt. Der Niederschlag wurde abfiltriert und aus CH3CN/2-Propanol umkristallisiert. Der Niederschlag wurde abfiltriert und getrocknet, wodurch man 2,0 g 2-[5-(6-Fluor-2-benzoxazolyl)pentyl]-1,2,3,4-tetrahydrobenzofuro[3,2-c]pyridin-(E)-2-butenedioat (1:1) (40%) (Verbindung 16) erhielt.
- Beispiel B3
- Essigsäure (0,0049 mol) wurde zum Zwischenprodukt (18) (0,0049 mol) in 1,2-Dichlorethan (50 ml) gegeben. Zwischenprodukt (9) (0,0049 mol) wurde zugesetzt, und die Mischung wurde gerührt, bis alles in Lösung gegangen war. NaHB(OAc)3 (0,0049 mol) wurde zugefügt, und das Reaktionsgemisch wurde über Nacht bei Raumtemperatur gerührt. Das Reaktionsgemisch wurde mit einer 10%igen Natronlauge (50 ml) gewaschen. Die Phasen wurden getrennt. Die wäßrige Phase wurde mit CH2Cl2 extrahiert. Die abgetrennte organische Phase wurde getrocknet und filtriert, und das Lösungsmittel wurde abgedampft. Der Rückstand wurde durch Kieselgel-Säulenchromatographie (Laufmittel: CH2Cl2/CH3OH 95/5) aufgereinigt. Die reinen Fraktionen wurden gesammelt und das Lösungmittel wurde abgedampft. Der Rückstand wurde in das Salzsäuresalz (1:1) überführt, abfiltriert und getrocknet, wodurch man 1,3 g N-(1,2-Benzisoxazol-3-yl)-1,2,3,4-tetrahydro-8-methyl-[1]-benzothieno[3,2-c]pyridin-2-butanamin-hydrochlorid (1:1) (57%) (Verbindung 11) erhielt.
- Beispiel B4
- Eine Mischung von 4-Fluorbenzoylchlorid (0,01 mol), Zwischenprodukt (13) (0,01 mol) und Na2CO3 (4 g) in CHCl3 (100 ml) wurde 30 Minuten lang unter Rückfluß gerührt. Die Mischung wurde auf Raumtemperatur abkühlen gelassen. Der Niederschlag wurde abgesaugt und der Filterkuchen wurde mit Wasser verrührt, dann abfiltriert und mit CH3CN verrührt, abfiltriert, mit Diisopropylether gewaschen und getrocknet, wodurch man 2,4 g N-[4-[(3,4-Dihydrobenzofuro[3,2-c]pyridin-2(1H)-yl)methyl]phenyl]-4-fluorbenzamid (Verbindung 14) erhielt.
- In Tabelle 1 sind Verbindungen der Formel (I) aufgeführt, die gemäß einem der obigen Beispiele dargestellt wurden.
- Tabelle 1
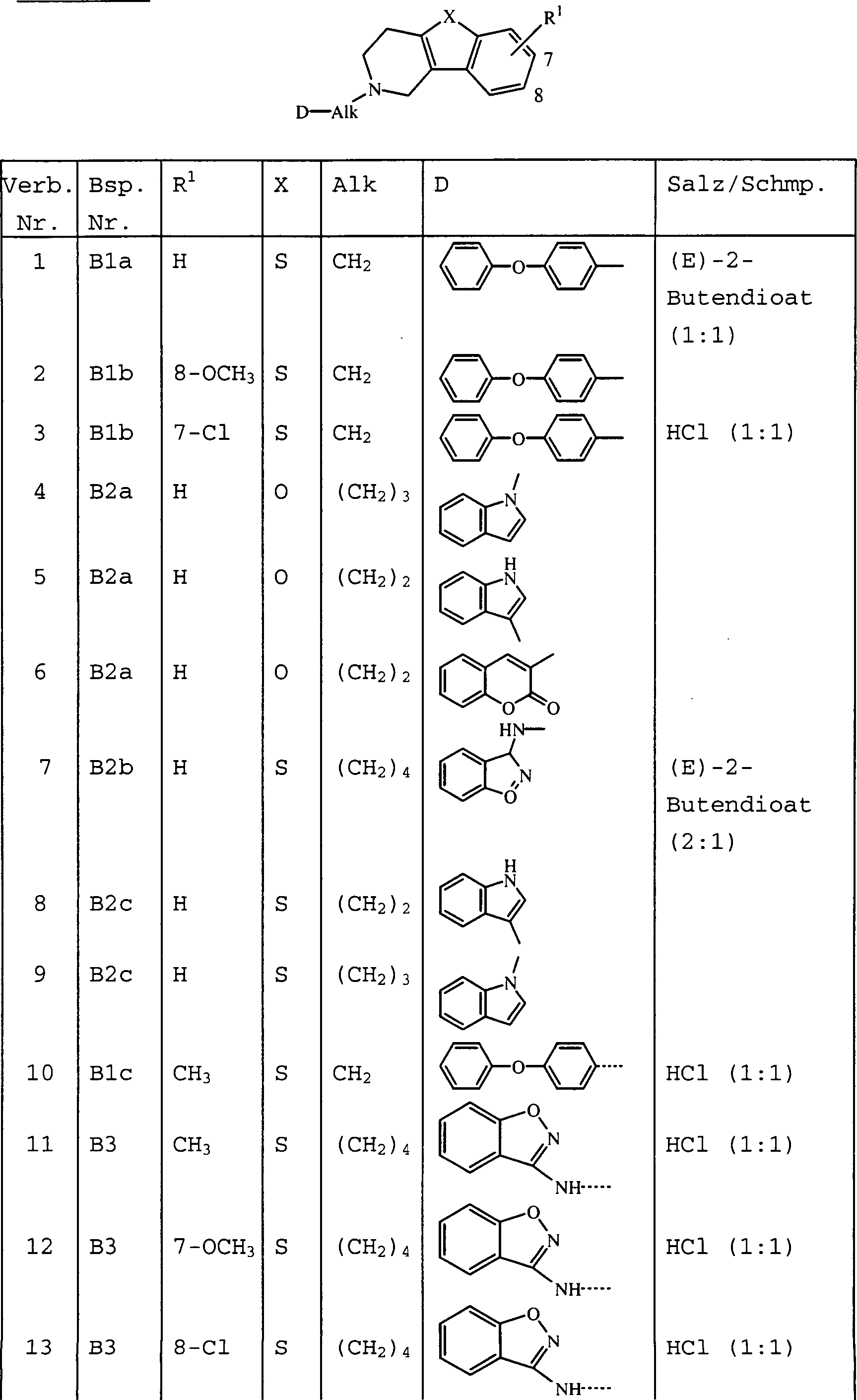
-
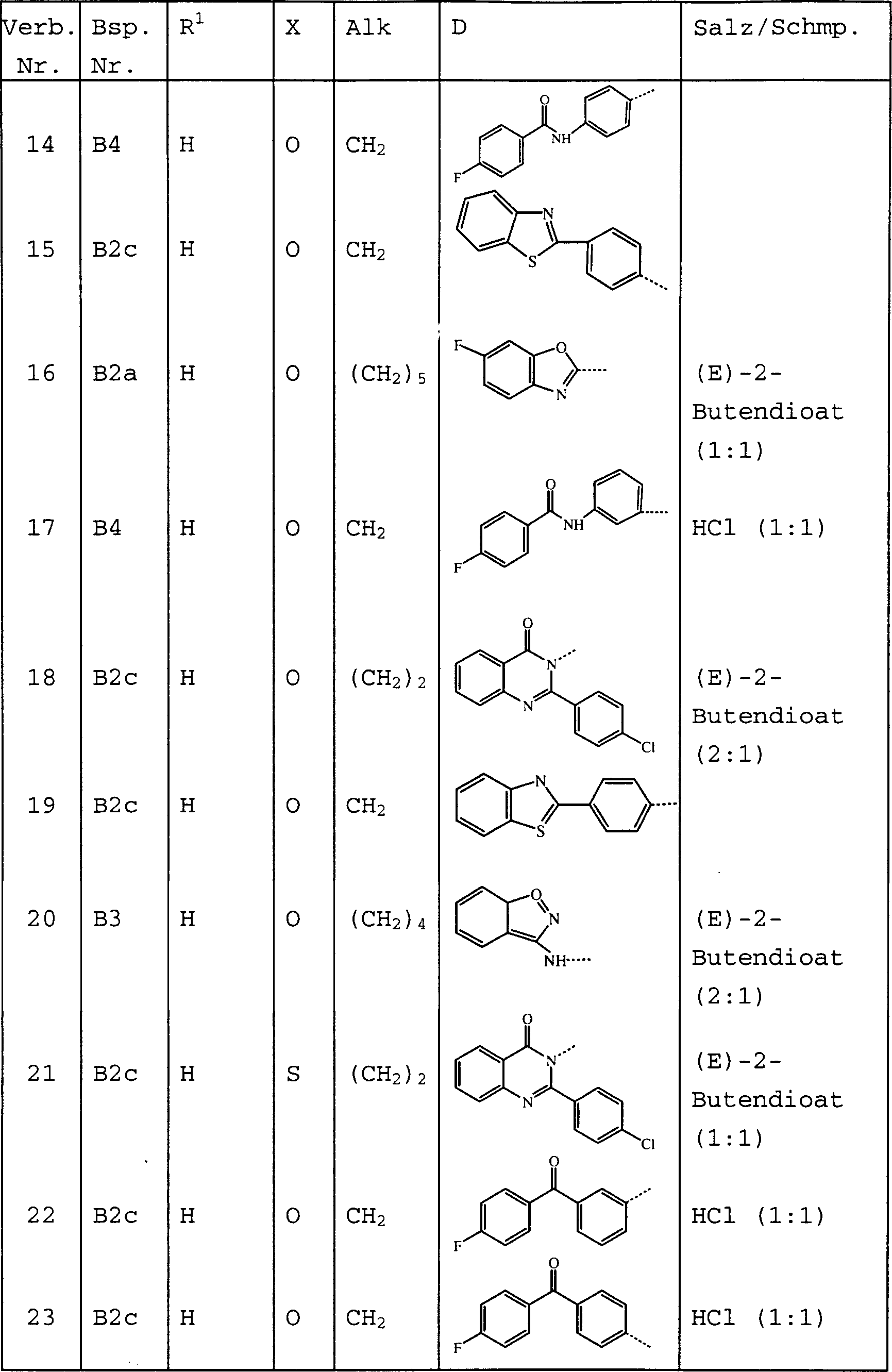
- C. Pharmakologische Beispiele
- Beispiel C.1: In-vitro-Bindungsaffinität zu α2-Rezeptoren
- Die Wechselwirkung der Verbindungen der Formel (I) mit α2-Rezeptoren wurde mit Hilfe von in-vitro-Radioligandenbindungsexperimenten beurteilt.
- Dazu wird im allgemeinen eine geringe Konzentration eines Radioliganden mit hoher Bindungsaffinität zu dem Rezeptor mit einer mit einem speziellen Rezeptor angereicherten Probe einer Gewebepräparation oder mit einer Präparation von klonierte Humanrezeptoren exprimierenden Zellen in einem gepufferten Medium inkubiert. Bei der Inkubation bindet der Radioligand an den Rezeptor. Nach Erreichen von Bindungsgleichgewicht wird die rezeptorgebundene Radioaktivität von der nicht gebundenen Radioaktivität getrennt und die rezeptorgebundene Aktivität gezählt. Die Beurteilung der Wechselwirkung der Testverbindungen mit dem Rezeptor erfolgt in kompetitiven Bindungsexperimenten. Der die Rezeptorpräparation und den Radioliganden enthaltenden Inkubationsmischung werden verschiedene Konzentrationen der Testverbindung zugesetzt. Die Bindung des Radioliganden wird durch die Testverbindung proportional zu seiner Bindungsaffinität und seiner Konzentration inhibiert.
- Als Radioligand für die α2A-, α2B- und α2C-Rezeptorbindung dient 3H-Rauwolscin und als Rezeptorpräparation die klonierte humane α2A-, α2B- und α2C-Rezeptoren exprimierende CHO-Zelle (Ovarzelle des Chinesischen Hamsters).
- Der IC50-Wert (die Konzentration, bei der 50% der Rezeptoren inhibiert sind) für die Verbindungen mit den Nummern 1 und 5 bis 23 für die drei Rezeptoren betrug wenigstens 10-6 M. Die anderen Verbindungen hatten für die drei Rezeptoren einen IC50-Wert von wenigstens 10-5 M.
- D. Beispiele für Zusammensetzungen
- Der in diesen Beispielen verwendete Begriff "aktive Substanz" (A.S.) bezieht sich auf eine Verbindung der Formel (I), ein pharmazeutisch unbedenkliches Additionssalz oder eine stereochemisch isomere Form davon.
- Beispiel D.1: Kapseln
- 20 g A.S., 6 g Natriumlaurylsulfat, 56 g Stärke, 56 g Lactose, 0,8 g kolloidales Siliciumdioxid und 1,2 g Magnesiumstearat werden kräftig miteinander verrührt. Die erhaltene Mischung wird dann in 1000 geeignete Hartgelatinekapseln gefüllt, die jeweils 20 mg A.S. enthalten.
- Beispiel D.2: Lacktabletten
- Herstellung des Tablettenkerns
- Eine Mischung aus 100 g A.S., 570 g Lactose und 200 g Stärke wird gut vermischt und anschließend mit einer Lösung aus 5 g Natriumdodecylsulfat und 10 g Polyvinylpyrrolidon in etwa 200 ml Wasser befeuchtet. Die feuchte Pulvermischung wird gesiebt, getrocknet und nochmals gesiebt. Dann werden 100 g mikrokristalline Cellulose und 15 g hydriertes Pflanzenöl zugesetzt. Das Ganze wird gut vermischt und zu Tabletten verpreßt, was 10.000 Tabletten ergibt, die jeweils 10 mg der aktiven Substanz enthalten.
- Überzug
- Eine Lösung von 10 g Methylcellulose in 75 ml denaturiertem Ethanol wird mit einer Lösung von 5 g Ethylcellulose in 150 ml Dichlormethan versetzt. Anschließend werden 75 ml Dichlormethan und 2,5 ml 1,2,3-Propantriol zugesetzt. 10 g Polyethylenglykol werden geschmolzen und in 75 ml Dichlormethan gelöst.
Claims (11)
- Verbindungen der Formelund deren N-Oxidformen, pharmazeutisch unbedenkliche Additionssalze und stereochemisch isomere Formen, wobei: Alk für C1–6-Alkandiyl steht; n für 1 oder 2 steht; X für -O-, -S-, -S(=O)- oder -S(=O)2- steht; die Reste R1 jeweils unabhängig voneinander für Wasserstoff, Halogen, C1–6-Alkyl, Nitro, Hydroxy oder C1–4-Alkyloxy stehen; D für einen Rest der Formel
 steht, wobei die Indices m jeweils unabhängig voneinander für 0, 1 oder 2 stehen; die Reste Y jeweils unabhängig voneinander für -CH2-, -O-, -S- oder -NR3- stehen; R2 und R3 jeweils unabhängig voneinander für Wasserstoff oder C1–6-Alkyl stehen; und die Reste R4 jeweils unabhängig voneinander für Halogen oder C1–6-Alkyl stehen.
steht, wobei die Indices m jeweils unabhängig voneinander für 0, 1 oder 2 stehen; die Reste Y jeweils unabhängig voneinander für -CH2-, -O-, -S- oder -NR3- stehen; R2 und R3 jeweils unabhängig voneinander für Wasserstoff oder C1–6-Alkyl stehen; und die Reste R4 jeweils unabhängig voneinander für Halogen oder C1–6-Alkyl stehen.
- Verbindungen nach Anspruch 1, wobei D für ein Rest der Formel (a),(b),(c),(d),(e),(f) oder (g) steht, wobei m für 0 steht; die Reste Y jeweils unabhängig voneinander für -CH2-, -O-, -S-, oder -NR3- stehen; und R2 und R3 jeweils unabhängig voneinander für Wasserstoff oder C1–6-Alkyl stehen.
- Verbindungen nach Anspruch 1 oder 2, wobei n für 1 steht und R1 für Wasserstoff, Chlor, Fluor, Methyl, Methoxy oder Nitro steht.
- Verbindungen nach einem der Ansprüche 1 bis 3, wobei x für -O- oder -S- steht.
- Verbindungen nach einem der Ansprüche 1 bis 4, wobei Alk für Methylen, 1,2-Ethandiyl, 1,3-Propandiyl, 1,4-Butandiyl oder 1,5-Pentandiyl steht.
- Verbindungen nach Anspruch 1, bei denen es sich umhandelt.
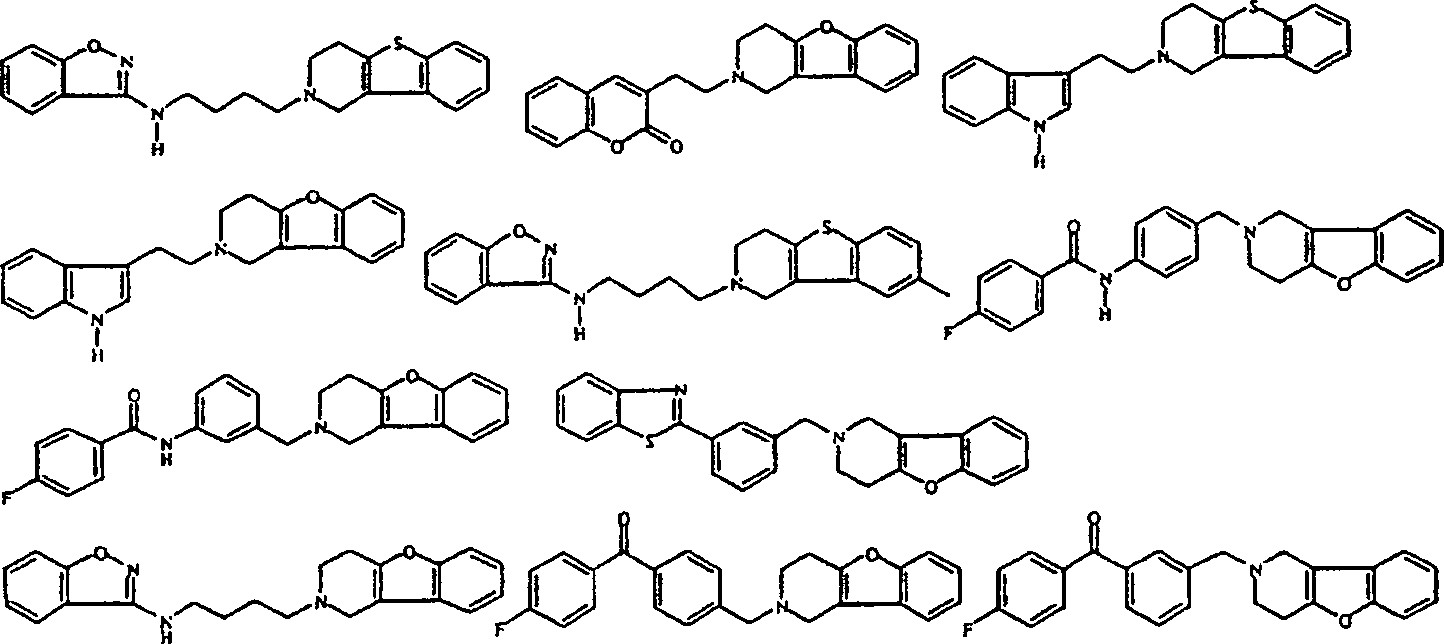
- Verbindungen nach einem der Ansprüche 1 bis 6 zur Verwendung als Medizin.
- Verwendung einer Verbindung nach einem der Ansprüche 1 bis 6 zur Herstellung eines Medikaments zur Behandlung von Depression oder Morbus Parkinson.
- Zusammensetzung, enthaltend einen pharmazeutisch unbedenklichen Träger und, als Wirkstoff, eine therapeutisch wirksame Menge einer Verbindung nach einem der Ansprüche 1 bis 6.
- Verfahren zur Herstellung einer Zusammensetzung nach Anspruch 9, bei dem man eine in einem der Ansprüche 1 bis 6 definierte Verbindung als Wirkstoff in einer innigen Mischung mit einem pharmazeutisch unbedenklichen Träger kombiniert.
- Verfahren zur Herstellung einer Verbindung nach Anspruch 1, dadurch gekennzeichnet, daß man a) ein Zwischenprodukt der Formel (II) mit einem Alkylierungsmittel der Formel (III)wobei W1 für eine geeignete Abgangsgruppe steht und D, Alk, X, n und R1 wie in Anspruch 1 definiert sind, in einem reaktionsinerten Lösungsmittel in Gegenwart einer Base und gegebenenfalls in Gegenwart eines Katalysators N-alkyliert; b) eines der Zwischenprodukte der Formel (VII) mit einem der Zwischenprodukte der Formel (VIII)
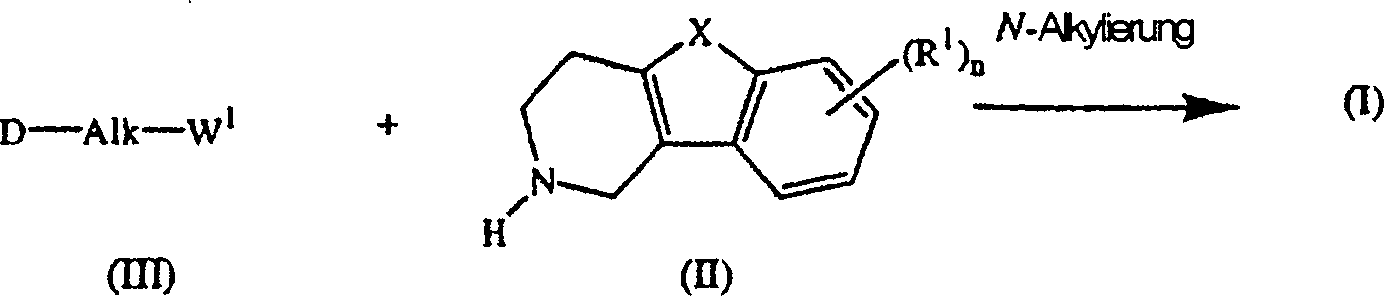 wobei W1 für eine geeignete Abgangsgruppe steht und Alk, X, n, m und R1, R2 und R4 wie in Anspruch 1 definiert sind, in einem reaktionsinerten Lösungsmittel in Gegenwart einer Base und gegebenenfalls in Gegenwart eines Katalysators umsetzt und so eine Verbindung der Formel (I-h) darstellt; c) ein Zwischenprodukt der Formel (II) reduktiv mit einem Aldehydderivat der Formel (IV-c) oder (IV-g)
wobei W1 für eine geeignete Abgangsgruppe steht und Alk, X, n, m und R1, R2 und R4 wie in Anspruch 1 definiert sind, in einem reaktionsinerten Lösungsmittel in Gegenwart einer Base und gegebenenfalls in Gegenwart eines Katalysators umsetzt und so eine Verbindung der Formel (I-h) darstellt; c) ein Zwischenprodukt der Formel (II) reduktiv mit einem Aldehydderivat der Formel (IV-c) oder (IV-g) wobei Alk' für C1–5-Alkandiyl steht, p für 0 oder 1 steht und X, Y, n und R1 wie in Anspruch 1 definiert sind, durch Reduktion einer Mischung der Reaktionspartner in einem geeigneten reaktionsinerten Lösungsmittel nach dem Stand der Technik bekannten Vorschriften zur reduktiven N-Alkylierung alkyliert und eine Verbindung der Formel (I-c) oder (I-g) bildet; d) und, falls gewünscht, Verbindungen der Formel (I) nach im Stand der Technik bekannten Transformationen ineinander umwandelt und weiterhin, falls gewünscht, die Verbindungen der Formel (I) durch Behandeln mit einer Säure in ein therapeutisch wirksames, nichttoxisches Säureadditionssalz umwandelt oder durch Behandeln mit einer Base in ein therapeutisch wirksames, nichttoxisches Basenadditionssalz umwandelt oder umgekehrt die Säureadditionssalzform durch Behandeln mit Alkali in die freie Base umwandelt oder das Basenadditionssalz durch Behandeln mit Säure in die freie Säure umwandelt; und, falls gewünscht, stereochemisch isomere Formen oder N-Oxide davon herstellt.
wobei Alk' für C1–5-Alkandiyl steht, p für 0 oder 1 steht und X, Y, n und R1 wie in Anspruch 1 definiert sind, durch Reduktion einer Mischung der Reaktionspartner in einem geeigneten reaktionsinerten Lösungsmittel nach dem Stand der Technik bekannten Vorschriften zur reduktiven N-Alkylierung alkyliert und eine Verbindung der Formel (I-c) oder (I-g) bildet; d) und, falls gewünscht, Verbindungen der Formel (I) nach im Stand der Technik bekannten Transformationen ineinander umwandelt und weiterhin, falls gewünscht, die Verbindungen der Formel (I) durch Behandeln mit einer Säure in ein therapeutisch wirksames, nichttoxisches Säureadditionssalz umwandelt oder durch Behandeln mit einer Base in ein therapeutisch wirksames, nichttoxisches Basenadditionssalz umwandelt oder umgekehrt die Säureadditionssalzform durch Behandeln mit Alkali in die freie Base umwandelt oder das Basenadditionssalz durch Behandeln mit Säure in die freie Säure umwandelt; und, falls gewünscht, stereochemisch isomere Formen oder N-Oxide davon herstellt.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP98203371 | 1998-10-06 | ||
| EP98203371 | 1998-10-06 | ||
| PCT/EP1999/007420 WO2000020423A1 (en) | 1998-10-06 | 1999-10-01 | Tricyclic δ3-piperidines as pharmaceuticals |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE69918148D1 DE69918148D1 (de) | 2004-07-22 |
| DE69918148T2 true DE69918148T2 (de) | 2005-07-28 |
Family
ID=8234192
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE69918148T Expired - Lifetime DE69918148T2 (de) | 1998-10-06 | 1999-10-01 | Trizyclische delta3-pyridine als arzneimittel |
Country Status (27)
| Country | Link |
|---|---|
| US (1) | US6352999B1 (de) |
| EP (1) | EP1119570B1 (de) |
| JP (1) | JP4590102B2 (de) |
| KR (1) | KR20010071844A (de) |
| CN (1) | CN1131868C (de) |
| AR (1) | AR023679A1 (de) |
| AT (1) | ATE269337T1 (de) |
| AU (1) | AU759199B2 (de) |
| BG (1) | BG64871B1 (de) |
| BR (1) | BR9913108A (de) |
| CA (1) | CA2346082C (de) |
| CZ (1) | CZ20011105A3 (de) |
| DE (1) | DE69918148T2 (de) |
| EE (1) | EE04680B1 (de) |
| ES (1) | ES2222735T3 (de) |
| HU (1) | HUP0103749A3 (de) |
| ID (1) | ID28451A (de) |
| IL (2) | IL142444A0 (de) |
| NO (1) | NO20011402D0 (de) |
| NZ (1) | NZ510116A (de) |
| PL (1) | PL347179A1 (de) |
| RU (1) | RU2226194C2 (de) |
| SK (1) | SK285386B6 (de) |
| TR (1) | TR200100953T2 (de) |
| TW (1) | TWI244485B (de) |
| UA (1) | UA70961C2 (de) |
| WO (1) | WO2000020423A1 (de) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| UA69420C2 (uk) * | 1998-10-06 | 2004-09-15 | Янссен Фармацевтика Н.В. | ТРИЦИКЛІЧНІ <font face="Symbol">D3</font>-ПІПЕРИДИНИ ЯК <font face="Symbol">a<sub></font>2</sub>-АНТАГОНІСТИ |
| EP1404679A2 (de) * | 2001-07-05 | 2004-04-07 | PHARMACIA & UPJOHN COMPANY | (hetero)aryl substituierte benzofuranen als 5-ht liganden |
| ATE521593T1 (de) * | 2002-04-03 | 2011-09-15 | Orion Corp | Verwendung eines alfa2-adrenorezeptorantagonisten bei mit dem zns zusammenhängenden erkrankungen |
| WO2005100355A1 (ja) | 2004-04-12 | 2005-10-27 | Taisho Pharmaceutical Co., Ltd. | 環状アミン化合物 |
| JP2008530229A (ja) | 2005-02-17 | 2008-08-07 | ワイス | シクロアルキル縮合インドール、ベンゾチオフェン、ベンゾフランおよびインデン誘導体 |
| DE102005038947A1 (de) * | 2005-05-18 | 2006-11-30 | Grünenthal GmbH | Substituierte Benzo[d]isoxazol-3-yl-amin-Verbindungen und deren Verwendung in Arzneimitteln |
| ES2897946T3 (es) * | 2013-07-25 | 2022-03-03 | Dong A St Co Ltd | Método para preparar un derivado de benzamida, intermedio novedoso utilizado en la preparación de la benzamida y método para preparar un intermedio novedoso |
| CN103755715B (zh) * | 2013-12-26 | 2015-12-09 | 武汉理工大学 | 苯并呋喃并[2,3-c]吡啶化合物及其合成方法 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SU926914A1 (ru) * | 1980-05-26 | 1992-04-23 | Научно-исследовательский институт фармакологии АМН СССР | Производные 1,2,3,4-тетрагидробензофуро[3,2-с]-пиридина или хлоргидрат, обладающие р дом свойств анальгетиков и антагонистов морфина |
| DE3522959A1 (de) * | 1985-06-27 | 1987-01-08 | Merck Patent Gmbh | Indolderivate |
| US4673680A (en) * | 1985-09-18 | 1987-06-16 | Pendleton Robert G | α2 -adrenergic receptor antagonists as modifiers of gastrointestinal motility |
| US4985422A (en) * | 1988-04-27 | 1991-01-15 | Glaxo Group Limited | Lactam derivatives |
| US4971974A (en) * | 1988-06-16 | 1990-11-20 | Neurex Corporation | Benzothiophenes as appetite suppressants |
| US5100891A (en) * | 1991-01-18 | 1992-03-31 | Hoechst-Roussel Pharmaceuticals Inc. | Substituted 1,2,3,4-tetrahydrocyclopent[b]indoles, 1,2,3,3a,4,8a-hexahydrocyclopent[B]indoles and related compounds |
| AU735764B2 (en) * | 1996-12-06 | 2001-07-12 | Abbott Laboratories | Benzopyranopyrrole and benzopyranopyridine alpha-1 adrenergic compounds |
| UA52681C2 (uk) * | 1997-04-08 | 2003-01-15 | Янссен Фармацевтика Н.В. | Похідні 1,2,3,4-тетрагідро-бензофуро[3,2,-c]піридину, спосіб їх одержання та фармацевтична композиція на їх основі |
| UA69420C2 (uk) * | 1998-10-06 | 2004-09-15 | Янссен Фармацевтика Н.В. | ТРИЦИКЛІЧНІ <font face="Symbol">D3</font>-ПІПЕРИДИНИ ЯК <font face="Symbol">a<sub></font>2</sub>-АНТАГОНІСТИ |
-
1999
- 1999-10-01 ID IDW20010779A patent/ID28451A/id unknown
- 1999-10-01 CN CN998116688A patent/CN1131868C/zh not_active Expired - Fee Related
- 1999-10-01 RU RU2001111811/04A patent/RU2226194C2/ru not_active IP Right Cessation
- 1999-10-01 PL PL99347179A patent/PL347179A1/xx not_active Application Discontinuation
- 1999-10-01 IL IL14244499A patent/IL142444A0/xx active IP Right Grant
- 1999-10-01 KR KR1020017000443A patent/KR20010071844A/ko not_active Ceased
- 1999-10-01 TW TW088116897A patent/TWI244485B/zh not_active IP Right Cessation
- 1999-10-01 WO PCT/EP1999/007420 patent/WO2000020423A1/en not_active Ceased
- 1999-10-01 AU AU63342/99A patent/AU759199B2/en not_active Ceased
- 1999-10-01 CA CA2346082A patent/CA2346082C/en not_active Expired - Fee Related
- 1999-10-01 JP JP2000574535A patent/JP4590102B2/ja not_active Expired - Fee Related
- 1999-10-01 EP EP99950628A patent/EP1119570B1/de not_active Expired - Lifetime
- 1999-10-01 EE EEP200100210A patent/EE04680B1/xx not_active IP Right Cessation
- 1999-10-01 HU HU0103749A patent/HUP0103749A3/hu unknown
- 1999-10-01 TR TR2001/00953T patent/TR200100953T2/xx unknown
- 1999-10-01 UA UA2001032000A patent/UA70961C2/uk unknown
- 1999-10-01 SK SK433-2001A patent/SK285386B6/sk unknown
- 1999-10-01 ES ES99950628T patent/ES2222735T3/es not_active Expired - Lifetime
- 1999-10-01 DE DE69918148T patent/DE69918148T2/de not_active Expired - Lifetime
- 1999-10-01 CZ CZ20011105A patent/CZ20011105A3/cs unknown
- 1999-10-01 BR BR9913108-0A patent/BR9913108A/pt not_active Application Discontinuation
- 1999-10-01 US US09/806,587 patent/US6352999B1/en not_active Expired - Lifetime
- 1999-10-01 NZ NZ510116A patent/NZ510116A/xx unknown
- 1999-10-01 AT AT99950628T patent/ATE269337T1/de not_active IP Right Cessation
- 1999-10-05 AR ARP990105041A patent/AR023679A1/es active IP Right Grant
-
2001
- 2001-03-12 BG BG105334A patent/BG64871B1/bg unknown
- 2001-03-20 NO NO20011402A patent/NO20011402D0/no not_active Application Discontinuation
- 2001-04-04 IL IL142444A patent/IL142444A/en not_active IP Right Cessation
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE69516867T2 (de) | Piperazinderivate als 5 ht1a antagonisten | |
| DE68926976T2 (de) | Kondensierte polyzyklische Verbindungen, Zusammenstellungen, Verfahren zur Herstellung und deren Anwendung als PAF-antagonistische, antihistaminische und/oder anti-inflammatorische Agenzien | |
| DE69716062T2 (de) | Benzofuran-derivate und deren verwendung | |
| DE3781173T2 (de) | N-heterozyklische-4-piperidinamine enthaltende anti-histaminische zusammensetzungen. | |
| DE3736664A1 (de) | Tetrahydro-furo- und -thieno(2,3-c)pyridine, ihre verwendung als arzneimittel und verfahren zu ihrer herstellung | |
| DE3502508A1 (de) | Heterocyclische verbindungen | |
| DE60206652T2 (de) | Benzothiazolderivate als adenosin-rezeptor-liganden | |
| DE69434888T2 (de) | 5-ht4-rezeptor-antagonisten | |
| DE69730962T2 (de) | Alkylaminobenzothiazole und -benzoxazole derivate | |
| DE69815700T2 (de) | Tetrahydro gamma-carboline | |
| DE69824872T2 (de) | 1,2,3,4-tetrahydro-benzofuro[3,2-c]pyridin derivate | |
| DE69720715T2 (de) | Hexahydro-pyrido (4,3-b)indolderivate als antipsychotische arzneimittel | |
| DE69629454T2 (de) | Antipsychotische 4-(1h-indolyl-1-yl)-1-substituierte piperidine derivate | |
| DE69918148T2 (de) | Trizyclische delta3-pyridine als arzneimittel | |
| DE69007905T2 (de) | 1-Oxa-2-oxo-8-azaspiro[4,5]decan-Derivate, Verfahren zu ihrer Herstellung und pharmazeutische Zusammensetzungen daraus. | |
| DE69905478T2 (de) | Tricyclische delta3-piperidine als alpha2-antagonisten | |
| DE69637412T2 (de) | Prokinetisch wirksame oxadiazole | |
| DE69007900T2 (de) | 4,4-Disubstituierte Piperidinderivate, Verfahren zu ihrer Herstellung und diese enthaltende pharmazeutische Zubereitungen. | |
| DE69910568T2 (de) | Furopyridinderivate und ihre therapeutische verwendung | |
| DE69406425T2 (de) | Trizyklische heterozyklische verbindungen als 5-ht4 rezeptorantagonisten | |
| DE69610104T2 (de) | Kondensierte thiazol-derivate, mit 5-ht-rezeptor affinität | |
| DE69911995T2 (de) | Benzisoxazole und phenone als alpha-2-antagonisten | |
| DE19602505A1 (de) | 1-(Pyrazol-4-Indol-3-yl)-Piperidine | |
| DE69919575T2 (de) | Benzoxazolderivate und medikamente die diese als aktiven wirkstoff enthalten | |
| DE68905363T2 (de) | Heterotetracyclische laktamderivate, verfahren zu ihrer herstellung und pharmazeutische zubereitungen, die sie enthalten. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition |