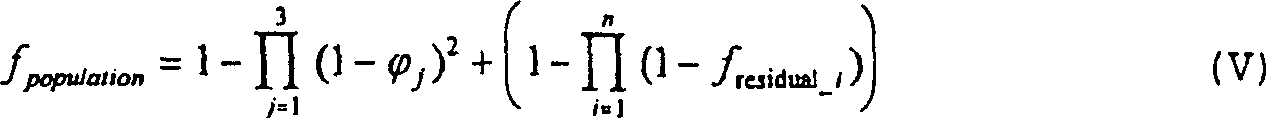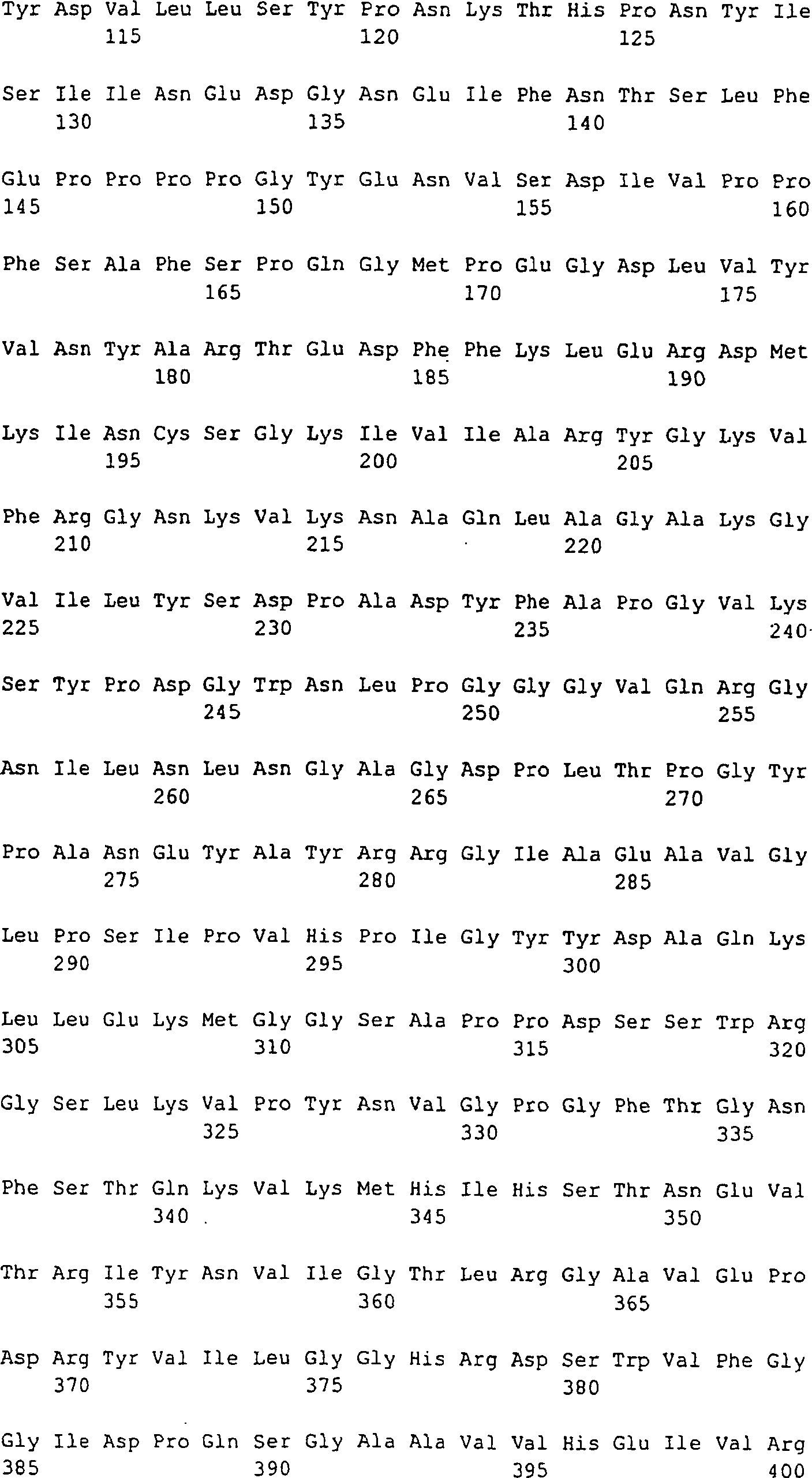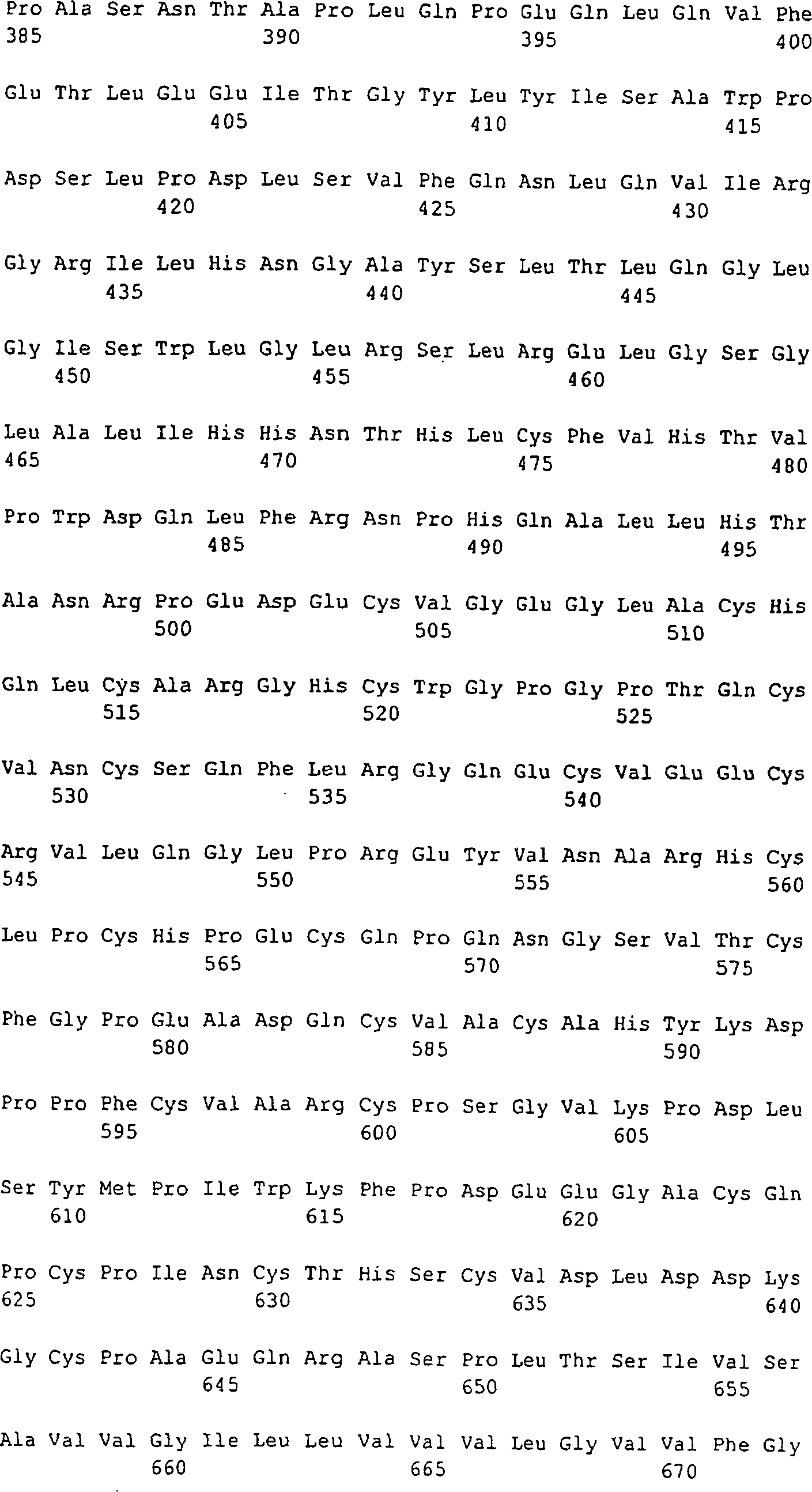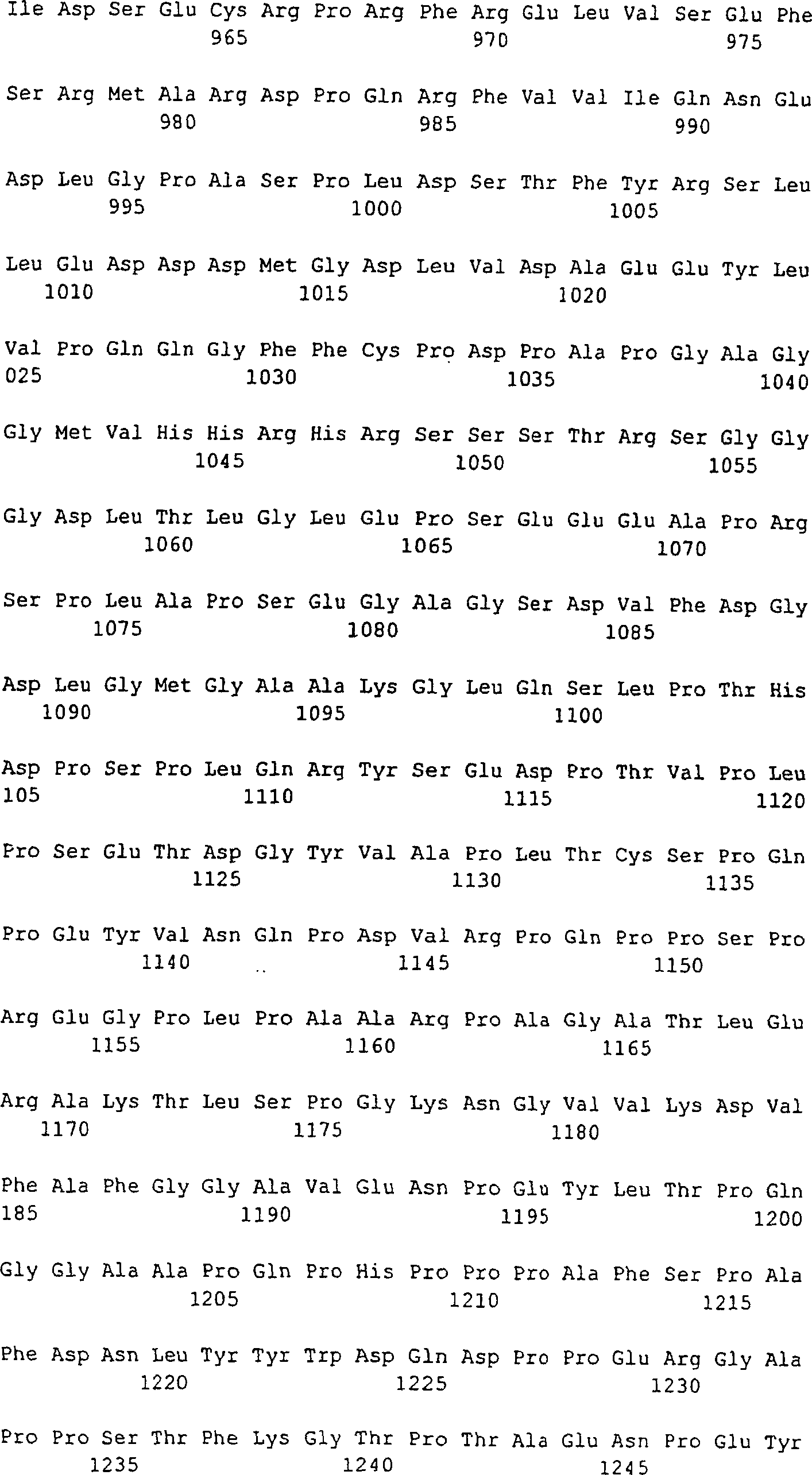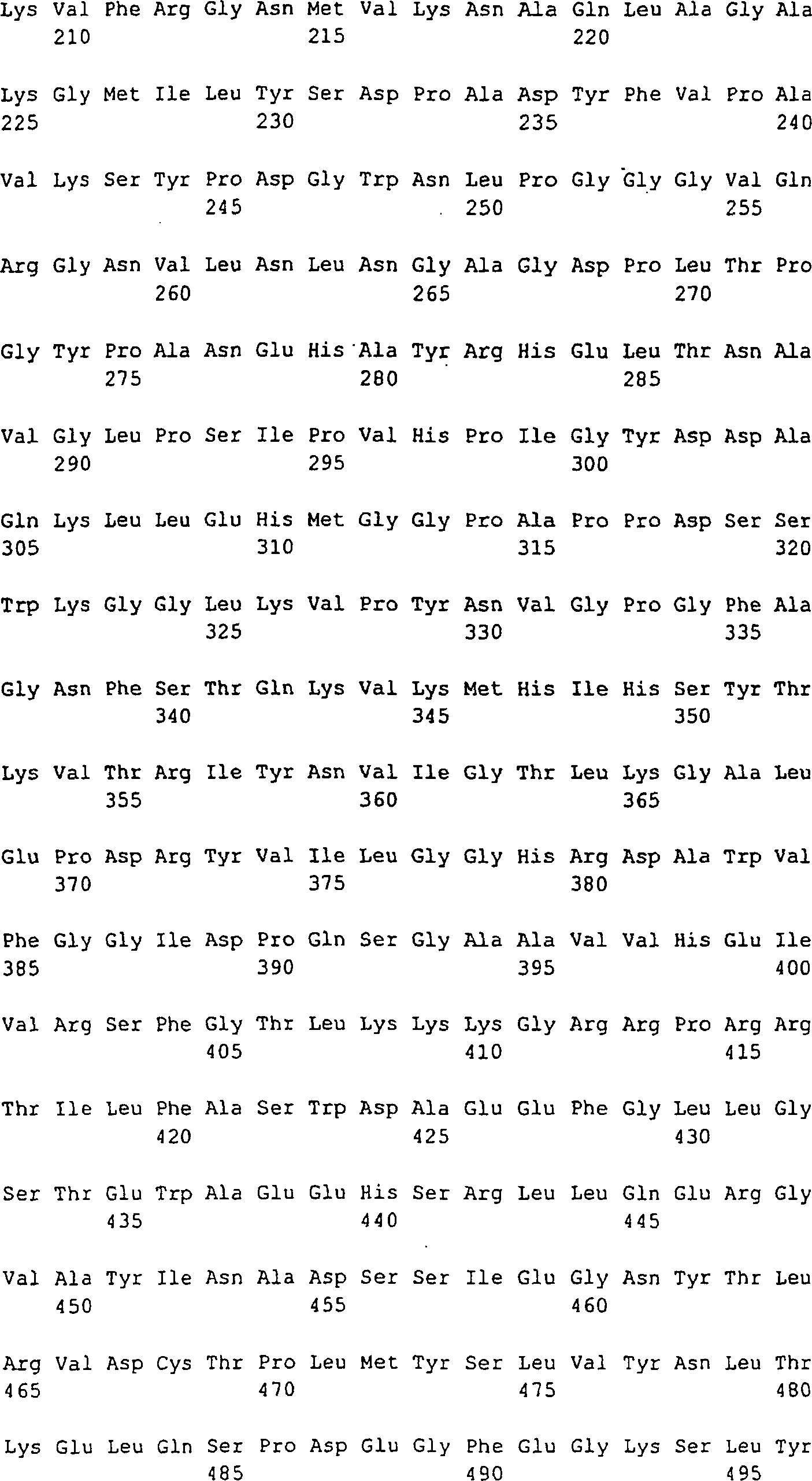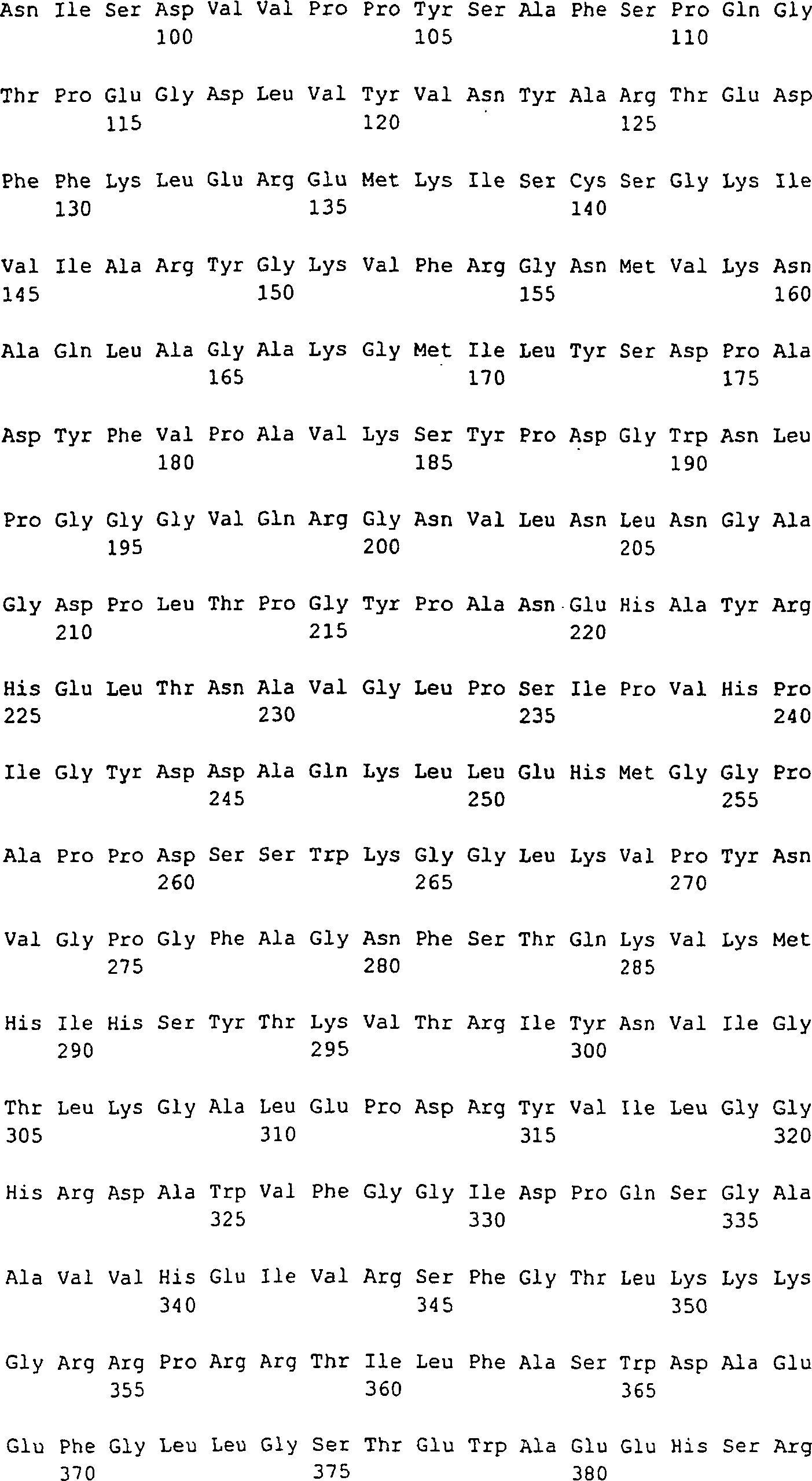-
GEBIET DER
ERFINDUNG
-
Die
vorliegende Erfindung bezieht sich auf neue Verfahren zur Bekämpfung von
Krankheiten, wie Krebs, welche durch das Vorhandensein von Zell-gebundenen
Genexpressionsprodukten, welche nicht-immunogen oder schwach immunogen
sind, gekennzeichnet sind. Insbesondere bezieht sich die vorliegende
Erfindung auf Verfahren zum Induzieren einer Immunantwort, die durch
cytotoxische T-Lymphozyten (CTLs) ausgeführt wird, wobei Zellen, die
Epitope von den Genexpressionsprodukten tragen, von den CTLs angegriffen und
getötet
werden. Die Erfindung bezieht sich auch auf ein Verfahren zur Herstellung
immunogener, modifizierter Polypeptid-Antigene, welche von schwach
immunogenen Antigenen abgeleitet sind.
-
Die
Erfindung bezieht sich weiterhin auf eine Reihe von Anwendungen
der AutoVac-Technologie
der Anmelder (welche der Gegenstand der WO 95/05849 ist) auf dem
Gebiet der therapeutischen Impfung gegen Krebs.
-
HINTERGRUND
DER ERFINDUNG
-
Die
Idee der Impfung gegen Krebs existiert seit über 100 Jahren und hat wiederholte
Ausbrüche
der Aktivität
erfahren, insbesondere seit der letzten Jahrhundertwende.
-
Jedoch
hat sich das Verständnis
der grundlegenden molekularen Mechanismen der Immunantwort während der
letzten 10 Jahre wesentlich verbessert. Unter den wichtigsten Meilensteinen,
die während
dieser Phase erreicht wurden, fanden sich auch die Entdeckung der
noch immer wachsenden Liste der Cytokine und Wachstumsfaktoren,
das Verstehen des Mechanismus der Interaktion zwischen T- und B-Zellen
sowie die Feststellung der zellulären Antigen-Prozessierungswege, einschließlich der
Rolle und Struktur der MHC-Klasse I- und II-Moleküle bei der
Antigenpräsentation.
Wichtige Entdeckungen in Bezug auf die Krebsimmunologie – obwohl
diese noch immer nicht vollständig
verstanden ist – betrafen
auch die Erklärung
der Mechanismen, die der Induktion der immunologischen Toleranz
in einem Wirt zugrunde liegen. Alle diese Forschungsunternehmen
haben zu einer großen
Anzahl von Versuchen geführt,
neue Behandlungen für
humanen Krebs zu entwickeln.
-
Abhängig davon,
wie Tumorimmunität
beim Patienten erreicht wird, können
Immuntherapiebehandlungen entweder als passive oder aktive Behandlungen
kategorisiert werden. Bei passiven Immuntherapiebehandlungen erhält der Patient
passiv Immunkomponenten, wie Cytokine, Antikörper, cytotoxische T-Zellen oder
aktivierte Killer-Lymphozytenzellen (LAK). Im Gegensatz dazu umfassen
aktive spezifische Immuntherapie-Protokolle das Induzieren der Tumorimmunität durch
Vakzinieren/Impfung mit der Tumorzelle oder seinen antigenischen
Komponenten. Letztere Form der Behandlung ist bevorzugt, da die
Immunität
länger
anhaltend ist.
-
Passive
und aktive Krebs-Impfstoffe konzentrierten sich entweder auf das
Induzieren von humoralen oder zellulären Immunantworten. In Bezug
auf aktive Impfstoffe besteht die wohletablierte Meinung, dass die Induktion
von CD4-positiven T-Helferzellen notwendig ist um sekundär entweder
Antikörper
oder cytotoxische CD8-positive T-Zellen zu induzieren.
-
Passive Vakzinierung mit
Antikörpern
-
Seit
der Entdeckung der monoklonalen Antikörper-Technologie in der Mitte
der siebziger Jahre ist eine große Anzahl therapeutischer monoklonaler
Antikörper
entwickelt worden, die gegen Tumor-spezifische oder Tumor-gebundene
Antigene gerichtet sind. Die monoklonale Antikörpertherapie verursacht jedoch
mehrere schwere Probleme:
- – Die Injektion dieser fremden
Substanzen induziert eine Immunantwort in dem Patienten gegen die
injizierten Antikörper,
welche zu einer weniger effizienten Behandlung sowie zu schweren
allergischen Nebenwirkungen bei den Patienten führen kann.
- – Monoklonale
Antikörper
müssen
für gewöhnlich in
großen
Mengen verabreicht werden. Dies ist ein Problem, da die Produktionskosten
für monoklonale
Antikörper
gewaltig sind.
- – Monoklonale
Antikörper
müssen über den
parenteralen Weg verabreicht werden und aufgrund der relativ großen erforderlichen
Mengen müssen
die Patienten häufig
während
der Behandlung ins Krankenhaus eingewiesen werden.
- – Die
Injektionen von monoklonalen Antikörpern müssen innerhalb relativ kurzer
Intervalle (Wochen) wiederholt werden um eine therapeutische Wirkung
aufrecht zu erhalten.
- – Monoklonale
Antikörper
sind für
gewöhnlich
nicht in der Lage, sekundäre
Effektorsysteme, wie beispielsweise das Abtöten von Tumorzellen durch Komplement,
NK-Zellen oder Makrophagen,
zu aktivieren.
-
Der
letztgenannte Nachteil ist bei der Krebstherapie von besonderer
Wichtigkeit und kann ein wichtiger Grund sein, warum die monoklonale
Antikörpertherapie
von Krebs in zahlreichen Fällen
nicht besonders erfolgreich war. Die sogenannten humanisierten monoklonalen
Antikörper,
die jetzt von vielen Unternehmen verwendet werden, sind weniger
immunogen, aber leider sind sie noch weniger in der Lage, die sekundären Immuneffektorsysteme
zu aktivieren. Außerdem
wurden Beispiele für
sekundäres
Wachstum von Tumoren, denen das ursprüngliche Tumor-Antigen fehlt,
beobachtet, da diese Antikörper
keine Wirkungen auf "unschuldige Zuschauer" ("innocent bystander" effects) auf Tumorzellen,
die das Tumor-Antigen nicht tragen, ausüben.
-
Die
nur schwache Effektor-Befähigung
der monoklonalen Antikörper
hat zur Entwicklung monoklonaler Antikörper, die chemisch mit verschiedenen
Toxinen und Radioisotopen verbunden sind, geführt. Z.B. Pharmacia Upjohn
AB hat eine Verknüpfung
von einem monoklonalen, Tumor-spezifischen Antikörper und dem Staphylococcus
aureus-Toxin A mit dem Zweck entwickelt, T-Zellen im Tumor zu aktivieren.
Medarex Inc. hat bispezifische monoklonale Antikörper entwickelt, die ein Tumor-spezifisches
Fab-Fragment sowie ein spezifisches Fc-Rezeptor-Antikörperfragment
enthalten, mit dem Zweck entwickelt, das Töten von Tumorzellen durch Makrophagen
zu aktivieren. Beide Konstrukte sind effektiver als der monoklonale Antikörper alleine, aber
sie sind auch teurer und stärker
immunogen. Antikörper,
die mit Radioisotopen verbunden sind, sind ebenfalls sowohl teuer
als auch immunogen, und es werden andere allgemeine toxische Nebenwirkungen
beobachtet.
-
Das
Erscheinen der monoklonalen Antikörpertechnologie war ein großer Schritt
vorwärts,
welcher die Herstellung von wohldefinierten Bindemolekülen mit
hoher Affinität
ermöglichte.
Da diese Antikörper
jedoch monoklonal sind, reagieren sie nur mit einem einzelnen Epitop-Typus
auf einem Tumor-Antigen. Dies ist der hauptsächliche Grund dafür, dass
sie für
gewöhnlich
nicht in der Lage sind, das Komplementsystem zu aktivieren oder
an die Fc-Rezeptoren
von NK-Zellen und Makrophagen zu binden. Diese sehr wirkungsvollen
Effektorsysteme erfordern für
gewöhnlich
die gleichzeitige Lokalisierung multipler Fc-Antikörperfragmente, die aus dem
Antigen herausragen.
-
Andere
Forscher haben daher versucht, zwei monoklonale Antikörper in
Kombination zu verwenden, und dies hat zu einer verbesserten Wirkung
geführt.
Statt dessen scheint es sehr sinnvoll zu sein, Tumorzellen mit hochspezifischen
polyklonalen Antikörpern,
die gegen ein Tumor-spezifisches oder gegen (überexprimierte) Tumor-assoziierte
Antigene oder Wachstumsfaktorrezeptoren gerichtet sind, anzugreifen.
Solche Antikörper
wären vollständig in
der Lage, die sekundären
Effektorsysteme, die oben genannt sind, zu aktivieren. Außerdem ist
es wahrscheinlich, dass die lokale Entzündungsreaktion, die von diesen
Effektorsystemen induziert wird, zu sekundären Wirkungen auf Zellen, die
sogenannte "unschuldige
Zuschauer" sind
und das fragliche Tumor-Antigen nicht exprimieren, führt, sowie
zur Aktivierung von Tumor-spezifischen
TILs (Tumor-infiltrierenden Lymphozyten) im Tumorgewebe. Solche
Wirkungen wurden von Medarex Inc. bei der Verwendung ihrer bispezifischen
monoklonalen Antikörperkonjugate
beobachtet.
-
Seit
der Entdeckung der monoklonalen Antikörpertechnologie wurde die mögliche Verwendung
von polyklonalen Antikörpern
zur Krebstherapie nicht sehr stark untersucht (mit der Ausnahme
der unten beschriebenen Antigene). Einer der Hauptgründe ist
der, dass wohldefinierte Tumor-spezifische oder Tumor-assoziierte
Oberflächenantigene
erst in den letzten Jahren charakterisiert worden sind, wobei sich
aber – was
noch wichtiger ist – viele
von diesen als Selbstantigene, und daher als nicht-immunogen entpuppt
haben. Dementsprechend hätten
xenogenische polyklonale Antikörper
verwendet werden müssen
um diese Wirkungen zu untersuchen. Solche Antikörper induzieren jedoch eine
energische Immunantwort gegen die injizierten fremden polyklonalen
Antikörper,
welche die therapeutischen Wirkungen schnell zerstört.
-
Aktive Impfung
zur Induktion von Antikörpern
-
Jüngste Versuche,
therapeutische polyklonale Autoantikörper bei Krebspatienten durch
aktive Impfung/Vakzinierung zu induzieren, waren erfolgreich. Es
wurden Vakzine/Impfstoffe gegen Membran-gebundene Kohlenhydrat-Selbstantigene
(wie die O-gebundenen, fehlexprimierten Tn- und sTn-Antigene und
die Gangliosidliposaccharide GM2 und GD3) entwickelt. Diese kleinen
Kohlenhydratstrukturen sind jedoch sehr schwache Antigene, so dass
Konjugate dieser Moleküle
mit Trägermolekülen, wie
dem Napfschneckenhaemocyanin ("keyhole
limpet haemocyanin, KLH) oder Schafmucinen (die Tn- und sTn enthalten)
verwendet werden müssen.
Bei Melanom-Patienten ging die Induktion von Anti-GM2-Antikörpern nach
einer Mindestnachfolgetherapie von 51 Monaten mit einem längeren krankheitsfreien
Zeitraum und längerer
Gesamtüberlebensrate einher.
Es wurden auch randomisierte Phase-II-Studien bei Brustkrebs-Patientinnen durchgeführt, bei
denen ein Konjugat von sTn und KLH in DETOX-B-Adjuvans (BIOMIRA
Inc.) verwendet wurde, die zeigten, dass sTn-immune Patienten eine
signifikant längere
mittlere Überlebensrate
im Vergleich mit Kontrollen besaßen. Ein weiteres Beispiel
der aktiven Induktion von polyklonalen Antikörpern bei Krebs ist die Verwendung
von Idiotyp-spezifischer
Impfung gegen B-Zell-Lymphome, welche – obwohl sie sehr vielversprechend
war – auf
diese Krebsart beschränkt
ist.
-
Schließlich hat
das US-Unternehmen Aphton Inc. aktive Konjugat-Impfstoffe gegen
Gonadotropin-Freisetzungshormon (GnRH) und Gastrin entwickelt. Es
wurde gezeigt, dass dieser Impfstoff in der Lage ist, die biologische
Aktivität
dieser Hormone zu kontrollieren, welche ebenfalls als autokrine
Wachstumsfaktoren für
gewisse Tumorzellen dienen können.
Erfolgreiche klinische Phase-II-Studien wurden bei Magen-Darm-Krebs-Patienten
durchgeführt,
und klinische Phase-III-Studien laufen noch.
-
Cytotoxische
T-Zellen
-
Es
wurde von mehreren Gruppen eindeutig gezeigt, dass Tumor-spezifische,
cytotoxische T-Zellen (CTLs) bei vielen Tumoren vorhanden sind.
Diese CTLs werden Tumor-infiltrierende
Lymphozyten (TILs) genannt. Jedoch werden diese Zellen mittels mehrerer
verschiedener möglicher
Mechanismen, einschließlich
der Sezernierung immunsuppressiver Cytokine durch die Tumorzellen,
Mangel bzw. Fehlen von co-stimulatorischen Signalen, negative Regulation
von MHC-Klasse I-Molekülen,
etc., nicht-reaktiv oder anerg.
-
Es
wurden viele Versuche unternommen, die Tumor-spezifischen HLA-Klasse
I-gebundenen Peptide, die
von TILs erkannt werden, zu isolieren, und in einigen Fällen war
dies auch erfolgreich (z.B. Peptide der Melanom-assoziierten Antigene).
Solche Peptide wurden verwendet um eine Tumor-spezifische Immunantwort
bei dem Wirt zu induzieren, aber die praktische Verwendung von Tumor-spezifischen
Peptiden in Impfstoffen ist aufgrund der engen Spezifität der HLA-Klasse
I-Bindung der Peptide auf einen eingeschränkten Teil der Bevölkerung
beschränkt.
Außerdem
ist es für
gewöhnlich
relativ schwierig, eine CTL-Antwort in vivo unter Verwendung synthetischer
Peptide hervorzurufen, und zwar aufgrund der niedrigen biologischen
Halbwertszeit dieser Substanzen sowie aufgrund der Schwierigkeiten
der exogenen Aktivierung bzw. des Primings von MHC-Klasse I-Molekülen.
-
Viele
andere Ansätze
wurden ausprobiert, mit dem Ziel, eine Tumor-spezifische CTL-Antwort hervorzurufen,
einschließlich
der Verwendung von Cytokinen (z.B. IL-2, IFN-γ, IL-6, IL-4, IL-10 oder GM-CSF)
oder co-stimulatorischer Moleküle
(B7), entweder in löslicher
Form oder exprimiert von der transfizierten Tumorzelle. Außerdem wurden
Immunisierungen mit allogenischen oder autologen vollständigen Zellen
oder mit Tumor-Antigenen, die in spezialisierten Adjuvantien präpariert
wurden, die so gestaltet waren, dass das Antigen über den
MHC-Klasse I-Antigen-Präsentationsweg
präsentiert
wurde, oder mit Tumor-Antigenen, die z.B. in Vaccinia-Vektoren exprimiert
wurden, etc., mit wechselndem Erfolg verwendet. Die allgemeine Meinung
unter Tumorimmunologen ist daher noch immer, dass einer der besten
Wege, Tumoren zu eliminieren, der ist, eine starke spezifische Antitumor-CTL-Antwort
zu induzieren.
-
Ungeachtet
der Tatsache, dass diese Behandlungen für gewöhnlich sehr teuer und schwierig
zu wiederholen sind, hat es sich auch herausgestellt, dass es schwierig
ist, eine gute Immunantwort gegen den Tumor hervorzurufen, da viele
der Tumor-assoziierten Antigene echte Selbstproteine sind, gegenüber welchen die
meistens T-Zellen tolerant zu sein scheinen. Daher scheint es notwendig,
eine kontrollierte zelluläre
Autoimmun-Situation im Patienten hervorzurufen.
-
AUFGABE DER
ERFINDUNG
-
Es
ist eine Aufgabe der vorliegenden Erfindung, verbesserte Verfahren
und Mittel zum Induzieren von Immunantworten in Wirtsorganismen
gegen unerwünschte
Antigene, z.B. Tumor-Antigene, zu induzieren. Es ist eine weitere
Aufgabe, ein Verfahren zum Herstellen von Polypeptid-Analoga solcher
unerwünschter
Antigene bereitzustellen, und zwar von Analoga, welche in der Lage
sind, eine effektive Immunantwort gegen das unerwünschte Antigen
hervorzurufen.
-
ZUSAMMENFASSUNG
DER ERFINDUNG
-
Die
Präsentation
von Antigenen wurde dogmatisch in zwei unabhängigen Wegen betrachtet, einem exogenen
Klasse II-Weg und einem endogenen Klasse I-Weg.
-
Kurz,
ein fremdes Protein außerhalb
der Zelle oder von der Zellmembran wird von der APC als Endosom
aufgenommen, welches mit einem intrazellulären Kompartiment fusioniert,
welches proteolytische Enzyme und MHC-Klasse II-Moleküle enthält. Einige
der produzierten Peptide binden an Klasse II, welche dann zur Zellmembran
verbracht werden.
-
Der
endogene Klasse I-Weg ist durch die vorherrschende Präsentation
cytosolischer Proteine gekennzeichnet. Es wird vermutet, dass diese über Proteasomen-vermittelten
Verdau, gefolgt vom Transport der Peptide in das endoplasmatische
Retikulum (ER) über
TAP-Moleküle, die
sich in der Membran des ER befinden, geschieht. Im ER binden die
Peptide an Klasse I, gefolgt vom Transport zur Plasmamembran.
-
Diese
zwei Wege sind jedoch nicht vollständig unabhängig. Es ist z.B. bekannt,
dass dendritische Zellen, und in gewissem Ausmaß auch Makrophagen, in der
Lage sind, extrazelluläre
Proteine durch Endocytose (Pinocytose) aufzunehmen und anschließend in
Verbindung mit MHC-Klasse I zu präsentieren. Es wurde ebenfalls
zuvor gezeigt, dass exogene Antigene bei der Verwendung von spezialisierten
Verabreichungswegen, z.B. durch Koppeln an Eisenoxidkügelchen,
in der Lage sind, in den Klasse I-Weg einzutreten (Rock, 1996).
Aufgrund der Wichtigkeit einer gleichzeitigen Expression von sowohl
Klasse I als auch Klasse II auf dersel ben APC zum Hervorbringen
eines Clusters des Drei-Zell-Typs, scheint dieser Mechanismus zentral
zu sein. Dieser Wechselwirkungs-Cluster vom Drei-Zell-Typ wurde
von Mitchison (1987) und später
von anderen Autoren vorhergesagt. Sie zeigten die Wichtigkeit der
gleichzeitigen Präsentation
von Klasse I- und Klasse II-Epitopen auf derselben APC. Entsprechend
dem kürzlich
beschriebenen Mechanismus der CTL-Aktivierung (vgl. Lanzavecchia,
1998, Nature 393: 413, Matzinger, 1999, Nature Med. 5: 616, Ridge
et al., 1998, Nature 393: 474, Bennett et al., 1998, Nature 393:
478, Schoenberger et al., 1998, Nature 393: 480, Ossendrop et al., 1998,
J. Exp. Med 187: 693, und Mackey et al., 1998, J. Immunol 161: 2094)
werden professionelle APCs, die Antigen auf MHC-Klasse II präsentieren,
von T-Helferzellen erkannt. Dies führt zu einer Aktivierung der
APC (vermittelt durch Wechselwirkung von CD40L auf der T-Helferzelle
und CD40 auf der APC). Dies ermöglicht es
der APC, CTLs direkt zu stimulieren, welche dadurch aktiviert werden.
Vgl. auch 2.
-
Es
wurde zuvor gezeigt, dass die Insertion eines Fremd-MHC-Klasse II-restringierten
T-Helferzell-Epitops
in ein Selbst-Antigen zur Bereitstellung eines Antigens führt, das
in der Lage ist, eine starke kreuzreaktive Antikörperreaktion zu induzieren,
die gegen das nicht-modifizierte Selbst-Antigen gerichtet ist (vgl.
WO 95/05849, ebenfalls vom Anmelder). Es wurde gezeigt, dass die
Autoantikörperinduktion
mittels spezifischer T-Zell-Hilfe verursacht wird, die durch das
insertierte Fremdepitop induziert wird.
-
In
Dalum et al., 1997, Molecular Immunology 34, 16-17, S. 1113-1120,
wurde ebenfalls gezeigt, dass eine Autoantikörper-Induktion durch ein insertiertes
Fremdepitop induziert werden konnte, und es wurde gezeigt, dass
die Feinspezifität
und möglicherweise
der Isotyp der Antikörper
als Folge der Position des insertierten Epitops manipuliert werden
kann.
-
Wir
sind jedoch zu der Schlussfolgerung gelangt, dass modifizierte Selbst-Antigene – mit der
Hilfe geeigneter Adjuvantien – ebenfalls
in der Lage sein sollten, starke CTL-Reaktionen gegen MHC-Klasse I-restringierte
Selbst-Epitope hervorzurufen, und daher kann die in der WO 95/05849
beschriebene Technologie ebenfalls so angepasst werden, dass eine
Vakzinierung gegen intrazelluläre
und andere Zell-gebundene Antigene, welche Epitope aufweisen, die
in Verbindung mit MHC-Klasse I präsentiert werden, bereitzustellen.
-
Die
in der WO 95/05849 beschriebene Autovakzin-Technologie bewirkt,
dass spezifische T-Zell-Hilfe für
selbst-reaktive B-Zellen bereitgestellt wird, wenn ein modifiziertes
Selbst-Antigen zur
Aufnahme in den MHC-Klasse II-Antigen-Prozessierungsweg verabreicht
wird (vgl. 1, und Dalum I et al., 1996,
J. Immunol. 157: 4796-4804, als auch Dalum I et al., 1999, Nature
Biotechnol. 17: 666-669). Es wurde gezeigt, dass potentiell selbst-reaktive
B-Lymphozyten, die
Selbst-Proteine erkennen, physiologisch bei normalen Individuen vorkommen.
Damit jedoch diese B-Lymphozyten induziert werden, tatsächlich Antikörper zu
produzieren, welche mit den relevanten Selbst-Proteinen reagieren,
ist die Mithilfe von Cytokin-produzierenden
T-Helferzellen (TH-Zellen oder TH-Lymphozyten) erforderlich. Normalerweise
wird diese Mithilfe nicht bereitgestellt, da T-Lymphozyten im Allgemeinen
keine T-Zell- Epitope
erkennen, die von Selbst-Proteinen abgeleitet sind, wenn sie von
Antigen-präsentierenden
Zellen (APCs) präsentiert
werden. Indem jedoch ein Element der "Fremdartigkeit" in einem Selbst-Protein zur Verfügung gestellt
wird (d.h. durch Einführen
einer immunologisch signifikanten Modifikation), werden T-Zellen,
die das fremde Element erkennen, auf das Erkennen des fremden Epitops auf
einer APC (wie beispielsweise, anfänglich, einer mononukleären Zelle)
aktiviert. Polyklonale B-Lymphozyten (welche T-Zell-Epitope präsentieren),
die in der Lage sind, Selbst-Epitope auf dem modifizierten Selbst-Protein
zu erkennen, nehmen das Antigen ebenfalls auf und präsentieren
anschließend
deren fremde T-Zell-Epitope bzw. deren fremdes T-Zell-Epitop, und
die aktivierten T-Lymphozyten stellen daraufhin diesen selbstreaktiven
polyklonalen B-Lymphozyten Cytokin-Hilfe zur Verfügung. Da
die Antikörper,
die von diesen polyklonalen B-Lymphozyten produziert werden, mit
verschiedenen Epitopen auf dem modifizierten Polypeptid reagieren,
schließen
sie jene mit ein, welche auch im nativen Polypeptid vorkommen, es
wird ein Antikörper induziert,
der mit dem nicht-modifizierten Selbst-Protein kreuzreaktiv ist. Im Ergebnis
können
die T-Lymphozyten dazu verleitet werden, so zu agieren, als ob die
Population der polyklonalen B-Lymphozyten ein gänzlich fremdes Antigen erkannt
hätten,
wobei tatsächlich
nur das eingefügte
Epitop bzw. die eingefügten
Epitope für den
Wirt fremd ist bzw. sind. Auf diese Weise werden Antikörper induziert,
die in der Lage sind, mit nicht-modifizierten Selbst-Antigenen eine
Kreuzreaktion einzugehen.
-
Wie
oben erwähnt,
benötigen
auch CTLs spezifische T-Zell-Hilfe, obwohl der Mechanismus für diese noch
nicht klar ist. Wir haben die vorliegende Erfindung auf unsere neue
Theorie aufgebaut, dass die Selbst-Proteine, die fremde MHC-Klasse
II-Epitope enthalten, nach exogener Aufnahme, Zugang zu dem Prozessierungsweg
für das
MHC-Klasse I-Antigen von z.B. Makrophagen und dendritischen Zellen
erlangen können.
Auf diese Weise könnte
eine starke CTL-Antwort gegen subdominante Epitope in dem Selbst-Protein
induziert werden. Alternativ dazu könnten Gene, die für modifizierte
Tumor-Antigene codieren, als Nukleinsäurevakzine verabreicht werden,
die gegebenenfalls auch sowohl zu MHC-Klasse II, als auch zu MHC-Klasse I-vermittelten
Immunantworten führen.
-
Tumorzellen
sind sehr schwache Antigen-präsentierende
Zellen, und zwar aufgrund ungenügender MHC-Klasse
I-Expression, Mangel an co-stimulatorischen Molekülen oder
der Sekretion von immunsuppressiven Cytokinen, etc. Unter Verwendung
der Autovakzin-Konstrukte
und der Vakzinierungsprotokolle, die oben erwähnt sind, könnte das modifizierte Tumor-Antigen
sowohl mittels MHC-Klasse I-, als auch durch MHC-Klasse II-Moleküle, auf
professionellen Antigen-präsentierenden
Zellen präsentiert
werden. Die Co-Präsentation subdominanter
Selbst-Epitope auf MHC-Klasse I-Molekülen und immundominanter Fremd-Epitope auf MHC-Klasse
II-Molekülen
würde eine
direkte Cytokin-Hilfe von aktivierten MHC-Klasse II-restringierten T-Helferzellen
für MHC-Klasse
I-restringierte CTLs vermitteln (2). Dies
wird unserer Meinung nach zu einem spezifischen Durchbrechen der
T-Zell-Autotoleranz gegenüber
dem Tumor-Antigen führen,
und dies ist exakt das, was bei der Krebs-Immuntherapie gewünscht ist.
-
Im
Ergebnis wird ein Vakzin, das unter Verwendung der oben dargestellten
Technologie konstruiert wird, eine humorale Autoantikörper-Antwort
mit sekundärer
Aktivierung von Komplement und Antikörper-abhängiger zellulärer Cytotoxizitäts-Aktivität (ADCC)
induzieren. Es wird außerdem
erwartet, dass dies eine cytotoxische T-Zell-Antwort, die gegen
z.B. ein Tumor-spezifisches
Membran-Antigen gerichtet ist, induzieren wird.
-
Daher
bezieht sich die vorliegende Erfindung in ihrem breitesten und allgemeinsten
Umfang auf die Verwendung von
1) mindestens einem CTL-Epitop,
das von einem Zell-gebundenen Polypeptid-Antigen abgeleitet ist, das in einem
Tier schwach immunogen oder nicht-immunogen ist, und 2) mindestens
einem ersten T-Helfer-Lymphozyten (TH)-Epitop,
welches für
das Tier fremd ist, oder von 1) mindestens einem Nukleinsäurefragment,
das für ein
CTL-Epitop, das von einem Zell-gebundenen Polypeptid-Antigen, das
in einem Tier schwach immunogen oder nicht-immunogen ist, abgeleitet ist, und 2)
mindestens einem ersten Nukleinsäurefragment,
das für
ein T-Helfer-Lymphozyten (TH)-Epitop codiert,
welches für
das Tier fremd ist, oder von einem nicht-pathogenen Mikroorganismus
oder Virus, welcher ein Nukleinsäurefragment
trägt,
welches für
1) mindestens ein CTL-Epitop, das von einem Zell-gebundenen Polypeptid-Antigen
abgeleitet ist, das in einem Tier schwach immunogen oder nicht-immunogen
ist, und 2) mindestens ein erstes T-Helfer-Lymphozyten (TH)-Epitop, welches für das Tier fremd ist, codiert
und diese exprimiert, zur Herstellung einer immunogenen Zusammensetzung
zur Behandlung eines pathologischen Prozesses, ausgewählt aus
einem Tumor, einer viralen Infektion und einer Infektion, die durch
einen intrazellulären
Parasiten oder ein Bakterium verursacht wird, indem in dem Tier
die simultane Präsentation
von dem mindestens einen CTL-Epitop und dem mindestens einen ersten
TH-Epitop auf einer geeigneten Antigen-präsentierenden
Zelle (APC) bewirkt wird und dabei eine spezifische cytotoxische
T-Lymphozyten (CTL)-Reaktion in dem Tier gegen Zellen induziert
wird, die das Zell-gebundene Polypeptid-Antigen auf ihrer Oberfläche tragen
oder das Zell-gebundene Antigen in ihrem intrazellulären Compartiment
beherbergen, wobei das Zell-gebundene Polypeptid-Antigen ausgewählt ist
aus einem Tumor-gebundenen Polypeptid-Antigen, einem Selbst-Protein,
einem viralen Polypeptid-Antigen und einem Polypeptid-Antigen, das von
einem intrazellulären
Parasiten oder Bakterium abgeleitet ist.
-
Ebenso
ist die neue Strategie zur Herstellung eines immunogenen Mittels
Teil der Erfindung. Diese neue Strategie umfasst die Selektion und
Produktion von Analoga von schwachen Zell-gebundenen Antigenen,
wobei das Bewahren eines wesentlichen Teils der bekannten und vorhergesagten
CTL-Epitope angestrebt wird, während
gleichzeitig mindestens ein fremdes TH-Epitop eingefügt wird.
-
Außerdem bezieht
sich die Erfindung auf bestimmte spezifische immunogene Konstrukte,
die auf bekannten Tumor-assoziierten Antigenen basieren, sowie auf
Zusammensetzungen, die diese Konstrukte enthalten. Letztlich bezieht
sich die Erfindung auf Nukleinsäurefragmente,
Vektoren, transformierte Zellen und andere Werkzeuge, die bei molekularbiologischen
Verfahren zur Herstellung der Analoga der Tumor-assoziierten Antigene
nützlich
sind.
-
ERKLÄRUNG DER
FIGUREN
-
1:
Das traditionelle AutoVac-Konzept. A: Tolerodominante Selbst-Epitope,
die auf MHC-Klasse II auf einer Antigen-präsentierenden Zelle (APC) präsentiert
werden, werden aufgrund der Depletion des T-Helfer-Zell (TH)-Repertoires ignoriert (T-Helferzelle mit
gepunkteten Linien angedeutet). Eingefügte fremde, immundominante
T-Zell-Epitope, die auf MHC-Klasse II präsentiert werden, aktivieren
T-Helferzellen, und B-Zellen (B), die spezifisch für native
Teile des Selbst-Proteins sind und die fremde immundominante T-Zell-Epitope auf
MHC-Klasse II präsentieren,
werden durch die Cytokin-Hilfe, die von der T-Helferzelle bereitgestellt
wird, aktiviert.
-
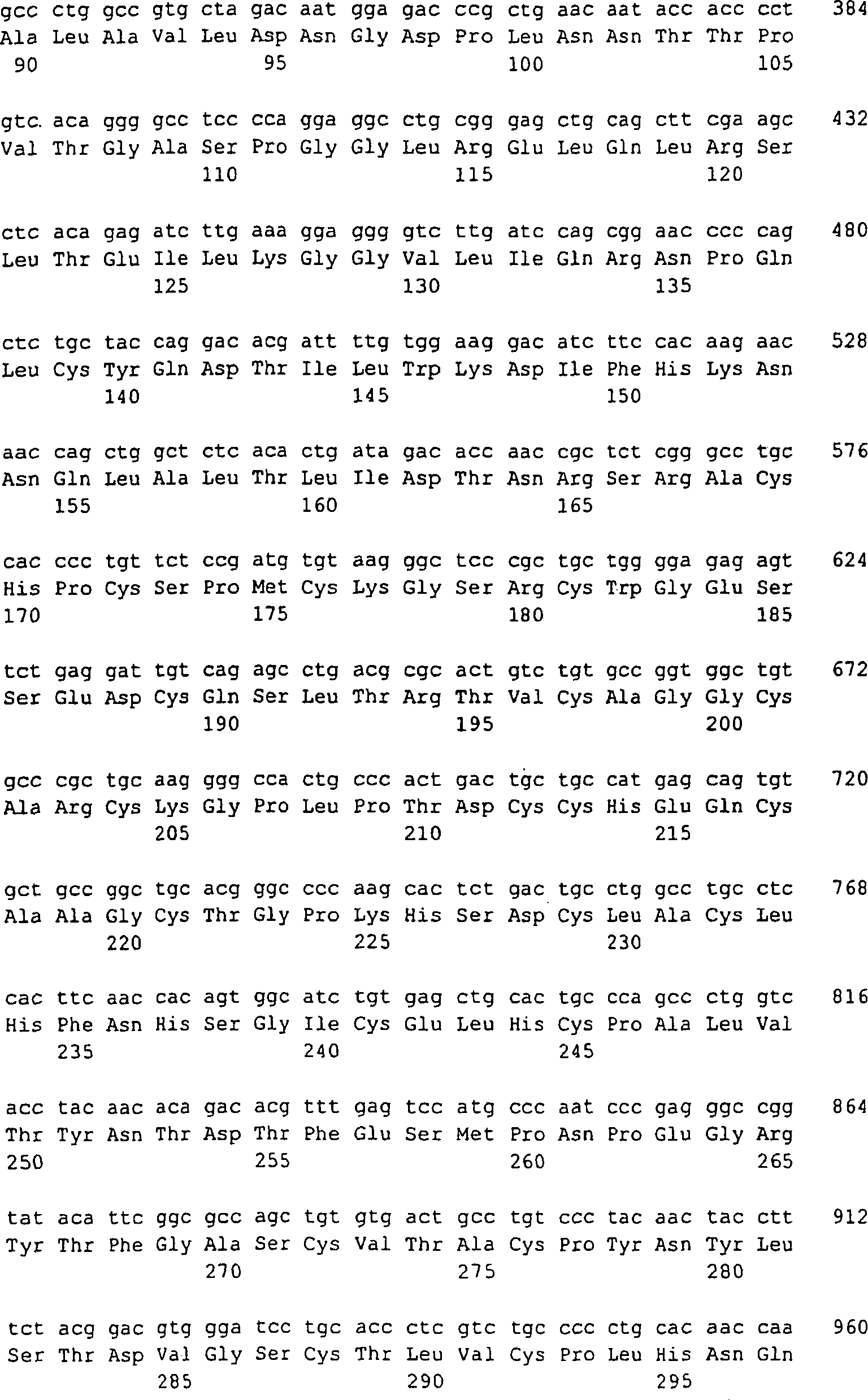
2:
Das AutoVac-Konzept zur Induzierung einer CTL-Antwort. Eingefügte, fremde,
immundominante T-Zell-Epitope, die auf MHC-Klasse II präsentiert
werden, aktivieren T-Helferzellen.
CTLs, die subdominante Selbst-Epitope erkennen, die auf MHC-Klasse
I präsentiert
werden, werden von den benachbarten aktivierten T-Helferzellen aktiviert.
-
3:
Eine schematische Wiedergabe des Her2-Polypeptids mit Angaben der
Epitop-Regionen
und N-Glykosylierungsstellen. Die 4 extrazellulären Domänen, die Transmembran (TM)-Domäne und die
2 intrazellulären
Domänen
sind mit den Angaben der Stellen mit unterschiedlichen Graden an
Homologie und der Stellen, die vermutete/bestimmte CTL-Epitope enthalten,
dargestellt.
-
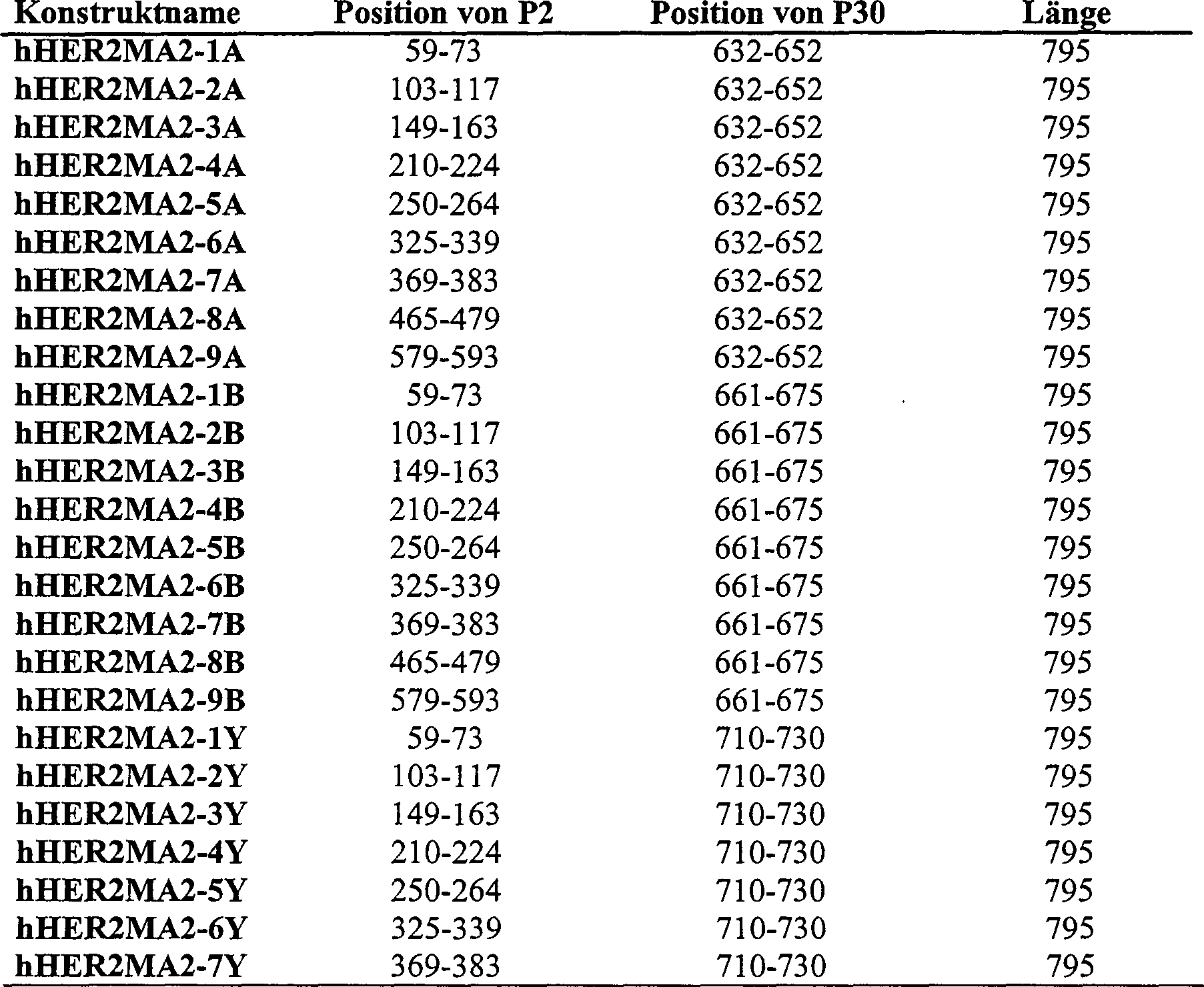
4:
Eine schematische Darstellung des humanen PSM-Polypeptids mit Angaben
der Insertionsregionen für
die P2- und P30-Epitope.
-
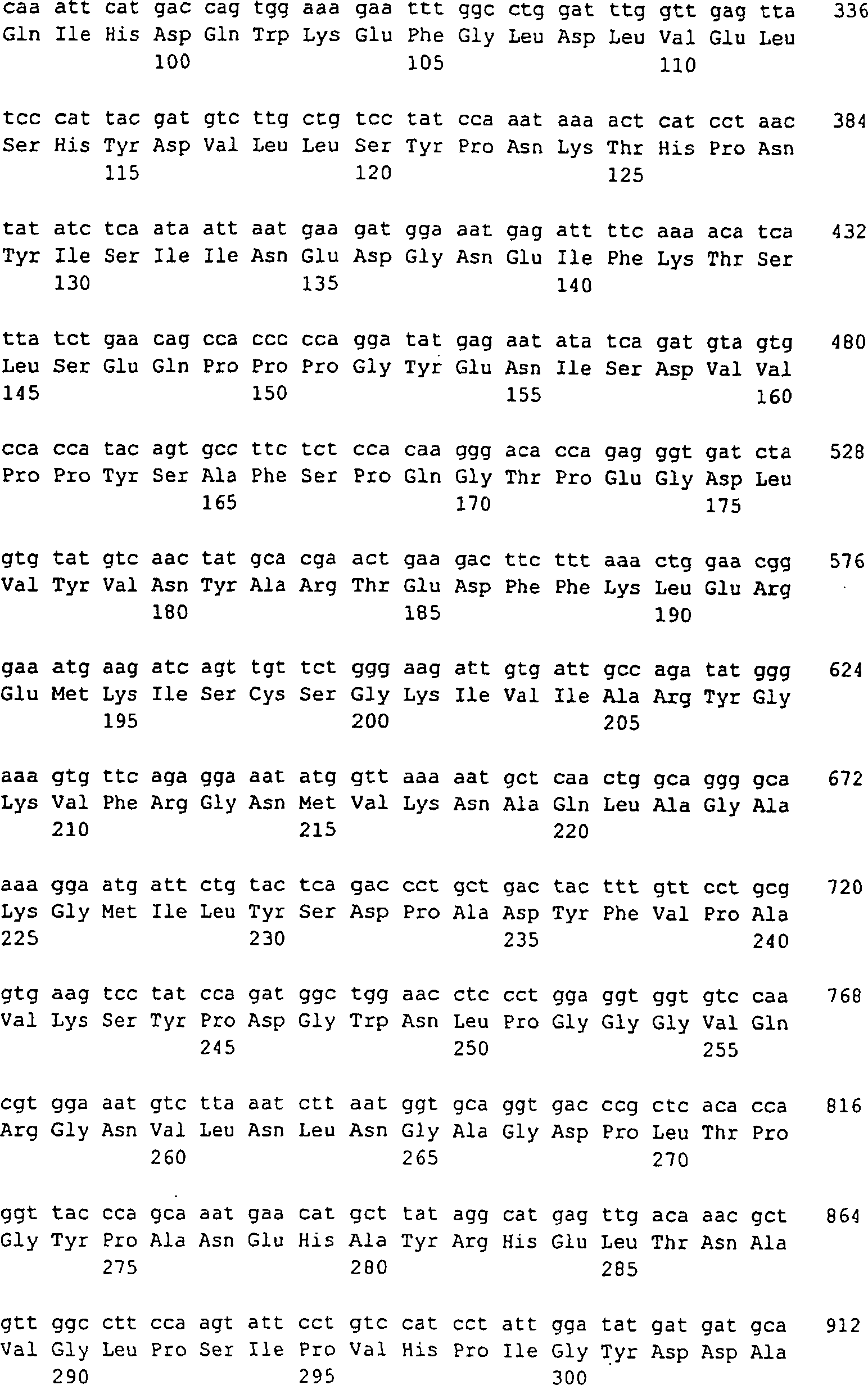
5: Die FGF-Gene und -Proteine. A: Exon-Intron-Struktur
der humanen und der Maus-FGF8-Gene. Darunter sind die acht unterschiedlichen
Splice-Formen dargestellt (nach Gemel 1996). B: Aminosäuresequenz
der unterschiedlichen FGF8-Isoformen. Die Polypeptidketten, die
jeweils auf FGF8b, FGF8f und FGF8e beschränkt sind, sind durch fette
und kursive oder unterstrichene Typen angedeutet. FGF8a ist die
kürzeste Variante,
die keine dieser hervorgehobenen Sequenzen enthält. Man erwartet, dass das
Signalpeptid am C-Terminus von Ala22 abgeschnitten wird. Die zwei
Cystein-Reste, die man beim reifen FGF8 (bei allen Isoformen) findet,
sind durch eine dicke Unterstreichung hervorgehoben. Die zwei möglichen
N-Glykosylierungsstellen
von FGF8b sind durch N ~ angezeigt. Die Nummerierung entspricht FGF8b.
-
6:
Darstellung der vier unterschiedlichen Varianten von FGF8b, die
zur Autovakzinierung gestaltet wurden. Oberes Feld: Theoretische
Modelle der Insertionspunkte der Epitope unter Verwendung der FGF2-Kristallstruktur
als Vorlage. Unteres Feld: Aminosäuresequenzen des Wildtyps-FGF8b
(WT) und der vier Varianten F30N, F2I, F30I und F2C. Das Signalpeptid
ist mit einer einzelnen Unterstreichung markiert. Die eingefügten Peptide
sind mit doppelter Unterstreichung markiert. Die N-terminale Sequenz
(MetAla) aller Varianten liegt in der Erzeugung einer Kozak-Sequenz
(Kozak 1991) zur besseren Translation in eukaryotischen Systemen
begründet.
-
DETAILLIERTE
OFFENBARUNG DER ERFINDUNG
-
Definitionen
-
Im
Folgenden wird eine Reihe von Begriffen, die in der vorliegenden
Beschreibung und den Ansprüchen
verwendet werden, definiert und im Detail erläutert um Maß und Ziel der Erfindung klarzustellen.
-
Es
ist beabsichtigt, dass ein "Zell-gebundenes
Polypeptid-Antigen" in
der vorliegenden Beschreibung und in den Ansprüchen ein Polypeptid bezeichnet,
welches an eine Zelle gebunden ist, welche in irgendeiner Weise
mit einem pathologischen Prozess in Verbindung steht. Des weiteren
präsentiert
die Zelle CTL-Epitope des Polypeptid-Antigens, gebunden an MHC-Klasse I-Moleküle auf ihrer
Oberfläche.
Zell-gebundene Polypeptid-Antigene können daher echte intrazelluläre Antigene
sein (und damit für
eine humorale Immunantwort unerreichbar), oder Antigene, die an
die Oberfläche
der Zellen gebunden sind. Das Zell-gebundene Antigen kann das Produkt
der zelleigenen Genexpression sein, das eines intrazellulären Parasiten,
eines Virus oder einer anderen Zelle. Im letzten Fall wird das Polypeptid-Antigen
anschließend
mit der Zelle, welche in den pathologischen Prozess involviert ist,
in Verbindung gebracht bzw. daran gebunden.
-
Die
Begriffe "T-Lymphozyt" und "T-Zelle" werden austauschbar
für Lymphozyten
verwendet, deren Ursprung im Thymus liegt, welche für verschiedene
Zell-vermittelte Immunantworten sowie für Effektorfurilctionen, wie
Helfer-Aktivität
in der humoralen Immunantwort, verantwortlich sind. Ebenso werden
die Ausdrücke "B-Lymphozyten" und "B-Zelle" austauschbar für Antikörper-produzierende
Lymphozyt verwendet.
-
Eine "Antigen-präsentierende
Zelle" (APC) ist
eine Zelle, welche Epitope gegenüber
T-Zellen präsentiert.
Typische Antigen-präsentierende
Zellen sind Makrophagen, dendritische Zellen und andere phagozytierende
und pinozytierende Zellen. Es sollte erwähnt werden, dass B-Zellen ebenfalls
als APCs fungieren, indem sie TH-Epitope
gebunden an MHC-Klasse II-Moleküle gegenüber TH-Zellen präsentieren, aber wenn der Ausdruck
APC im Allgemeinen in der vorliegenden Beschreibung und in den Ansprüchen verwendet
wird, wird beabsichtigt, sich auf die oben erwähnten phagozytierenden und
pinozytierenden Zellen zu beziehen.
-
"Helfer-T-Lymphozyten" oder "TH-Zellen" bezeichnen CD4-positive
T-Zellen, welche B-Zellen und cytotoxischen T-Zellen über die
Erkennung von TH-Epitopen, die an MHC-Klasse
II-Moleküle
auf Antigen-präsentierenden
Zellen gebunden sind, Hilfe bereitstellen.
-
Der
Ausdruck "cytotoxischer
T-Lymphozyt" (CTL)
wird für
CD8-positive T-Zellen verwendet, welche die Mithilfe von TH-Zellen benötigen um aktiviert zu werden.
-
Es
ist beabsichtigt, dass eine "spezifische" Immunantwort im
vorliegenden Zusammenhang eine polyklonale Immunantwort bezeichnet,
die sich vornehmlich gegen ein Molekül oder eine Gruppe von quasi
identischen Molekülen
oder, alternativ dazu, gegen Zellen richtet, welche CTL-Epitope
des Moleküls
oder der Gruppe von quasi identischen Molekülen präsentieren.
-
Es
ist beabsichtigt, dass "schwaches
oder nicht-immunogenes Polypeptid-Antigen" Polypeptide bezeichnet, die die Aminosäuresequenz
des schwachen Zell-gebundenen Protein-Antigens, das sich von dem fraglichen
Tier (z.B. einem Menschen) ableitet, aufweisen, aber ebenso sind
Polypeptide von dem Ausdruck umfasst, die die Aminosäuresequenz
aufweisen, die identisch zu Analoga solcher Proteine, die von anderen Arten
isoliert worden sind, identisch sind. Ebenso sind Formen der Polypeptide,
die aufgrund ihrer Herstellung in heterologen Systemen (z.B. Hefen
oder anderen eukaryontischen Nicht-Säuger-Expressionssystemen oder sogar
prokaryotischen Systemen) andere Glykosylierungsmuster aufweisen,
von den Grenzen des Ausdrucks umfasst. Es sollte jedoch erwähnt werden,
dass, wenn der Ausdruck verwendet wird, es beabsichtigt ist, dass das
fragliche Polypeptid normalerweise nicht-immunogen oder nur schwach
immunogen, jeweils in seiner natürlichen
Umgebung in dem zu behandelnden Tier, ist.
-
Der
Ausdruck "Polypeptid" soll im vorliegenden
Zusammenhang sowohl kurze Peptide von 2 bis 10 Aminosäureresten
Länge,
Oligopeptide von 11 bis 100 Aminosäureresten Länge und Polypeptide von mehr
als 100 Aminosäureresten
Länge umfassen.
Außerdem
ist beabsichtigt, dass der Ausdruck Proteine umfasst, d.h. funktionelle
Biomoleküle,
die mindestens ein Polypeptid umfassen; wenn diese mindestens zwei
Polypeptide umfassen, können
sie Komplexe bilden, kovalent verbunden sein, oder nicht-kovalent
verbunden sein. Das Polypeptid bzw. die Polypeptide in einem Protein
kann/können
glykosyliert und/oder an Lipide gebunden sein und/oder prosthetische
Gruppen umfassen.
-
Der
Ausdruck "Untersequenz" meint eine fortlaufende
Kette von mindestens 3 Aminosäuren
oder, wenn dies passend ist, von mindestens 3 Nukleotiden, die direkt
von einer natürlich
vorkommenden Aminosäuresequenz
bzw. Nukleinsäuresequenz
abgeleitet sind.
-
Der
Ausdruck "Tier" soll in dem vorliegenden
Zusammenhang im Allgemeinen eine Tierart (bevorzugt Säugerart)
bezeichnen, wie beispielsweise Homo sapiens, Canis domesticus, etc.,
und nicht nur ein einzelnes Tier. Der Ausdruck bezeichnet jedoch
auch eine Population von solch einer Tierart, da es wichtig ist,
dass die Individuen, die erfindungsgemäß immunisiert werden, alle
im Wesentlichen dasselbe schwache Zell-gebundene Polypeptid-Antigen
beherbergen, was die Immunisierung der Tiere mit demselben Immunogen
bzw. denselben Immunogenen erlaubt. Wenn z.B. genetische Varianten
der Polypeptide in unterschiedlichen humanen Populationen existieren,
kann es nötig
sein, unterschiedliche Immunogene in diesen unterschiedlichen Poplulationen
zu verwenden um in der Lage zu sein, die Autotoleranz gegen das
schwache Zell-gebundene Polypeptid-Antigen in jeder Population zu
durchbrechen.
-
Mit
dem Ausdruck "Herabregulierung
eines Zell-gebundenen Polypeptid-Antigens" ist hierin eine Verminderung der Menge
und/oder Aktivität
des fraglichen Antigens im lebenden Organismus gemeint. Die Herabregulierung
kann mittels verschiedener Mechanismen erreicht werden. Von diesen
ist die einfache Störung der
aktiven Stelle des Antigens durch Antikörper bindung die einfachste.
Es liegt jedoch ebenso im Umfang der vorliegenden Erfindung, dass
die Antikörperbindung
zum Entfernen des Polypeptids durch Fresszellen (wie beispielsweise
Makrophagen und andere phagozytierende Zellen) führt, und sogar noch wichtiger,
dass Zellen, die das Antigen tragen oder beherbergen, von CTLs im
Tier getötet
werden.
-
Der
Ausdruck "Bewirken
der simultanen Präsentation
durch eine geeignete APC" soll
ausdrücken, dass
das Immunsystem des Tieres auf kontrollierte Weise einer immunogenen
Herausforderung ausgesetzt wird, welche zur simultanen Präsentation
der fraglichen Epitope durch APCs führt. Solch eine Herausforderung des
Immunsystems kann, wie aus der untenstehenden Offenbarung hervorgehen
wird, auf eine Vielzahl von Wegen bewirkt werden, von denen die
wichtigsten die Vakzinierung mit Polypeptiden, die "Pharmaccine" enthalten (d.h.
einem Vakzin, welches verabreicht wird um eine andauernde Krankheit
zu behandeln oder zu verbessern), oder die Nukleinsäure-"Pharmaccin"-Vakzinierung sind.
Die dabei wichtige zu erreichende Folge ist, dass immun-kompetente
Zellen im Tier mit APCs konfrontiert werden, die die relevanten
Epitope auf immunologisch wirksame Weise aufzeigen.
-
Der
Ausdruck "immunologisch
wirksame Menge" hat
dabei seine im Stand der Technik übliche Bedeutung, d.h. eine
Menge an Immunogen, welche in der Lage ist, eine Immunantwort zu
induzieren, welche pathogene Agentien, welche immunologische Eigenschaften
mit dem Immunogen gemein haben, signifikant erfasst.
-
Bei
der Verwendung des Ausdrucks, dass die schwachen Zell-gebundenen
Polypeptid-Antigene
einer "Modifikation" unterzogen wurden,
ist hierin eine chemische Modifikation des Polypeptids gemeint,
welches das Rückgrat
des fraglichen Polypeptids darstellt. Solch eine Modifikation kann
z.B. eine Derivatisierung (z.B. Alkylierung) der Aminosäurereste
in der Aminosäuresequenz
sein, aber die bevorzugten Modifikationen umfassen Veränderungen
der Primärstruktur
der Aminosäuresequenz,
wie aufgrund der untenstehenden Offenbarung zugestanden werden wird.
-
Bei
der Diskussion von "Toleranz" und "Autotoleranz" versteht es sich,
dass normale Individuen in der Population keine Immunantwort gegen
das Polypeptid aufbauen, da die Polypeptide, welche die Ziele des
vorliegenden erfinderischen Verfahrens sind, Selbst-Proteine in
der zu vakzinierenden Population oder Proteine, welche nicht zu
Induktion einer effektiven Immunantwort führen, sind. Es kann dennoch
nicht ausgeschlossen werden, dass gelegentlich Individuen in einer
Tierpopulation in der Lage sein können, Antikörper gegen das native Polypeptid-Antigen
herzustellen, z.B. als Teil einer Autoimmunerkrankung. In jedem
Fall wird ein Tier normalerweise gegen seine eigenen Polypeptid-Antigene
autotolerant sein, aber es kann nicht ausgeschlossen werden, dass
Analoga, die von einer anderen Tierart oder von einer Population
mit einem anderen Phenotyp abgeleitet sind, ebenfalls von diesem
Tier toleriert werden würden.
-
Ein "fremdes T-Zell-Epitop" ist ein Peptid,
welches in der Lage ist, an ein MHC-Molekül zu binden und T-Zellen in
einer Tierart zu stimulieren. Bevorzugte Fremdepitope sind "gemischte" Epitope, d.h. Epitope,
welche an einen wesentlichen Teil von MHC-Klasse II-Molekülen in einer
Tierart oder -population binden. Nur eine sehr beschränkte Anzahl
solcher gemischter 7-Zell-Epitope ist bekannt, und diese werden
im Detail unten diskutiert. Es sollte erwähnt werden, dass es nötig sein
kann, 1) mehrere fremde T-Zell-Epitope in dasselbe Analogon einzufügen, oder
2) verschiedene Analoga herzustellen, bei denen in jedes Analogon
ein anderes gemischtes Epitop eingefügt wurde, damit die Immunogene,
welche erfindungsgemäß verwendet
werden, in einer möglichst
großen
Fraktion einer Tierpopulation wirksam sind. Es sollte erwähnt werden,
dass das Konzept der fremden T-Zell-Epitope ebenfalls die Verwendung
kryptischer T-Zell-Epitope umfasst, d.h. von Epitopen, welche von
einem Selbst-Protein abgeleitet sind und welche nur ein immunogenes
Verhalten entwickeln, wenn sie in isolierter Form vorliegen, ohne
ein Teil des fraglichen Selbst-Proteins zu sein.
-
Ein "fremdes T-Helfer-Lymphozyten-Epitop" (ein fremdes TH-Epitop) ist ein fremdes T-Zell-Epitop, welches
an ein MHC-Klasse II-Molekül
bindet und auf der Oberfläche
einer Antigen-präsentierenden
Zelle (APC), gebunden an das MHC-Klasse II-Molekül, präsentiert werden kann.
-
Ein "CTL"-Epitop ist ein Peptid,
welches in der Lage ist, an ein MHC-Klasse I-Molekül zu binden.
-
Ein "funktioneller Teil" eines (Bio-)Moleküls soll
im vorliegenden Zusammenhang den Teil des Moleküls bezeichnen, welcher für mindestens
eine der biochemischen oder physiologischen Wirkungen, die von dem Molekül bewirkt
werden, verantwortlich ist. Es ist wohlbekannt im Stand der Technik,
dass viele Enzyme und andere Effektormoleküle eine aktive Stelle aufweisen,
welche für
die Wirkungen, die von dem fraglichen Molekül ausgeübt werden, verantwortlich ist.
Andere Teile des Moleküls
können
einem stabilisierenden oder Löslichkeitverstärkenden
Zweck dienen und daher ausgelassen werden, wenn diese Zwecke im
Zusammenhang mit einer bestimmten Ausführungsform der vorliegenden
Erfindung nicht von Bedeutung sind. Z.B. ist es möglich, bestimmte
Cytokine als eine modifizierende Gruppierung im Analogon zu verwenden
(vgl. die detaillierte unterstehende Diskussion), und in einem solchen
Fall kann der Punkt der Stabilität
irrelevant sein, da die Kopplung an das Analogon die notwendige
Stabilität
bereitstellt.
-
Der
Ausdruck "Adjuvans" besitzt seine übliche Bedeutung
im Stand der Technik der Vakzinierungstechnologie, d.h. eine Substanz
oder eine Zusammensetzung, welche 1) nicht selbst in der Lage ist,
eine spezifische Immunantwort gegen das Immunogen des Vakzins hervorzurufen,
aber welche 2) nichtsdestotrotz in der Lage ist, die Immunantwort
gegen das Immunogen zu verstärken.
Oder, in anderen Worten, die Vakzinierung mit dem Adjuvans alleine
bewirkt keine Immunantwort gegen das Immunogen, die Vakzinierung
mit dem Immunogen kann eine Immunantwort gegen das Immunogen hervorrufen
oder nicht, aber die kombinierte Vakzinierung mit Immunogen und
Adjuvans induziert eine Immunantwort gegen das Immunogen, welche
stärker ist
als die, die von dem Immunogen alleine induziert wird.
-
"Targeting" eines Moleküls soll
im vorliegenden Zusammenhang die Situation bezeichnen, in der ein Molekül nach dem
Einführen
in das Tier bevorzugt in einem bestimmten Gewebe bzw. in bestimmten
Geweben auftauchen wird oder bevorzugt mit bestimmten Zellen oder
Zelltypen assoziiert sein wird. Die Wirkung kann auf einer Vielzahl
von Wegen erreicht werden, einschließlich der Formulierung des
Moleküls
in einer Zusammensetzung, die das Targeting erleichtert, oder durch
die Einführung
von Gruppen in das Molekül,
welche das Targeting erleichtern. Diese Punkte werden im Detail
unten diskutiert.
-
"Stimulation des Immunsystems" bedeutet, dass eine
Substanz oder eine Zusammensetzung eine allgemeine, nicht-spezifische
immunstimulatorische Wirkung zeigt. Eine Vielzahl von Adjuvantien
und vermutlichen Adjuvantien (wie beispielsweise bestimmte Cytokine)
besitzen die Fähigkeit,
das Immunsystem zu stimulieren. Das Ergebnis der Verwendung eines
immunstimulatorischen Mittels ist ein erhöhter "Alarmzustand" des Immunsystems, was bedeutet, dass
die simultane oder darauffolgende Immunisierung mit einem Immunogen eine
signifikant effektivere Immunantwort im Vergleich zur isolierten
Verwendung des Immunogens induziert.
-
Bevorzugte Ausführungsformen
-
Um
eine CTL-Antwort gegen eine Zelle zu induzieren, welche auf ihrer
Oberfläche
Epitope präsentiert, die
von dem Polypeptid-Antigen abgeleitet sind, ist es normalerweise
notwendig, dass mindestens ein CTL-Epitop mit einem MHC-Klasse I-Molekül auf der
Oberfläche
der APC assoziiert ist, wenn es präsentiert wird. Weiterhin ist
es bevorzugt, dass das mindestens eine erste fremde TH-Epitop
mit einem MHC-Klasse II-Molekül
auf der Oberfläche
der APC gebunden ist, wenn es präsentiert
wird.
-
Bevorzugte
APCs, die die Epitope präsentieren,
sind dendritische Zellen und Makrophagen, aber jede pino- oder phagozytierende
APC, welche in der Lage ist, simultan 1) CTL-Epitope, gebunden an MHC-Klasse I-Moleküle, und
2) TH-Epitope, gebunden an MHC-Klasse II-Moleküle, zu präsentieren,
ist eine bevorzugte APC entsprechend der Erfindung.
-
Erfindungsgemäß wird das
Zell-gebundene Polypeptid-Antigen aus Tumor-gebundenen Antigenen und
anderen Selbst-Proteinen, welche mit pathologischen Prozessen in
Verbindung stehen, ausgewählt,
aber auch aus viralen Antigenen und Antigenen, die von einem intrazellulären Parasiten
oder einem Bakterium abgeleitet sind. Es ist wohlbekannt im Stand
der Technik, dass solche Pathogen-gebundenen Antigene oft relativ schwache
Immunogene sind (z.B. Antigene von Mycobakterien, wie Mycobacterium
tuberculosis und Mycobacterium leprae, aber auch von Protozoen,
wie Plasmodium spp.). Es wird davon ausgegangen, dass die Erfindung,
unabhängig
davon, dass sie die Produktion von Antikörper- und CTL-Antworten gegen
echte Selbst-Protein-Antigene ermöglicht, in der Lage ist, die
oft unzureichende Immunantwort, die von dem Organismus gegen solche
intrazellulären
Antigene hervorgerufen wird, zu verstärken.
-
Normalerweise
wird es vorteilhaft sein, das Immunsystem mit einer großen Fraktion
der Aminosäuresequenz
des Polypeptid-Antigens, welches das Vakzinierungsziel ist, zu konfrontieren.
Daher wird die Präsentation
des CTL-Epitops und des ersten fremden TH-Epitops
durch die APC in einer bevorzugten Ausführungsform bewirkt, indem dem
Immunsystem des Tieres mindestens ein erstes Analogon des Zell-gebundenen
Polypeptid-Antigens präsentiert
wird, wobei dieses erste Analogon eine Variation der Aminosäuresequenz
des Zell-gebundenen Polypeptid-Antigens
umfasst, wobei diese Variation mindestens das CTL-Epitop und das
erste fremde TH-Epitop enthält. Dies ist ein Gegensatz
z.B. zu einer DNA-Vakzinierungs-Strategie, bei der die CTL- und
TH-Epitope von derselben Zelle exprimiert
werden, aber als Teile von getrennten Polypeptiden; solch eine DNA-Vakzinierungs-Strategie
ist ebenfalls eine Ausführungsform
der Erfindung, aber es wird davon ausgegangen, dass das Vorliegen
der zwei Epitope als Teil desselben Polypeptids normalerweise die
Immunantwort verstärken
wird, und in jedem Fall wird nur die Bereitstellung eines Expressionsproduktes
notwendig sein.
-
Um
die Chancen des Auslösens
einer effektiven Immunantwort zu maximieren, ist es bevorzugt, dass das
oben erwähnte
erste Analogon einen wesentlichen Teil von bekannten und vorhergesagten
CTL-Epitopen des Zell-gebundenen Polypeptid-Antigens enthält, d.h.,
einen Teil der bekannten und vorhergesagten CTL-Epitope, welche
an einen ausreichenden Teil von MHC-Klasse I-Molekülen in einer
Population binden. Es ist z.B. bevorzugt, dass der wesentliche Teil
von bekannten und vorhergesagten CTL-Epitopen in der Aminosäuresequenz
des Analogons von mindestens 50% der MHC-I-Haplotypen erkannt wird,
welche alle bekannten und vorhergesagten CTL-Epitope im Zell-gebundenen
Polypeptid-Antigen erkennen, aber höhere Prozentzahlen sind bevorzugt,
wie beispielsweise mindestens 60, mindestens 70, mindestens 80 und
mindestens 90%. Besonders bevorzugt ist die Verwendung von Analoga,
welche sicherstellt, dass im Wesentlichen alle bekannten CTL-Epitope
des Zell-gebundenen Polypeptid-Antigens
im Analogon vorhanden sind, d.h. nahezu 100% der bekannten CTL-Epitope.
Dementsprechend ist es ebenfalls besonders bevorzugt, dass im Wesentlichen
alle vorhergesagten CTL-Epitope des Zell-gebundenen Polypeptid-Antigens
zumindest in dem ersten Analogon vorhanden sind.
-
Verfahren
zur Vorhersage der Anwesenheit von CTL-Epitopen sind im Stand der
Technik wohlbekannt, vgl. z.B. Rothbard et al., EMBO J. 7: 93-100
(1988).
-
Wie
aus der vorliegenden Beschreibung und aus den Ansprüchen hervorgeht,
wird erwartet, dass das hierin beschriebene erfinderische Verfahren
die effektive Induzierung von CTL-Antworten gegen Zell-gebundene
Polypeptid-Antigene möglich
machen wird.
-
In
solchen Fällen,
in denen das Zell-gebundene Polypeptid-Antigen ein echtes intrazelluläres Antigen ist,
ist die Induzierung einer CTL-Antwort gegen Zellen, die das Antigen
beherbergen, der einzige Weg, seine Herabregulierung durch spezifische
immunologische Mittel zu erreichen. Im Falle von Membran-gebundenen Antigenen
ist es jedoch vorteilhaft, eine Antikörper-Antwort gegen das schwache
Zell-gebundene Polypeptid-Antigen zu induzieren. Wenn eine humorale
Immunantwort gegen ein schwaches Zell-gebundenes Antigen hervorgerufen
wird, ist es jedoch bevorzugt, die Antikörper-Antwort im Wesentlichen
auf die Wechselwirkung mit den Teilen des Antigens zu beschränken, welche
normalerweise zur möglichen
Wechsel wirkung mit Antikörpern
exponiert vorliegen. Ansonsten wäre
das Ergebnis höchstwahrscheinlich
die Induktion einer Antikörper-Antwort
gegen Teile des Antigens, welche normalerweise nicht das humorale
Immunsystem erfassen, und dieses wird umgekehrt das Risiko der Induzierung
einer Kreuzreaktivität
mit Antigenen erhöhen,
die zu keiner pathologischen Situation eine Beziehung aufweisen.
Ein eleganter Weg, diese Beschränkung
zu erreichen, ist es, eine Nukleinsäure-Vakzinierung mit einem
Analogon des schwachen Zell-gebundenen Antigens durchzuführen, wobei
dessen extrazellulärer
Teil entweder unverändert
ist oder ein TH-Epitop umfasst, welches
die 3D-Struktur des extrazellulären
Teils des Antigens nicht wesentlich verändert. Als eine mögliche Alternative kann
eine Immunisierung, sowohl mit einem CTL-gerichteten Immunogen als auch mit einem
B-Zell-gerichteten Immunogen, durchgeführt werden, wobei das B-Zell-gerichtete
Immunogen im Wesentlichen unfähig
ist, eine Immunisierung gegen den intrazellulären Teil des Ziel-Antigens
zu bewirken (das B-Zell-gerichtete Immunogen könnte z.B. keinerlei nicht-extrazelluläres Material
des Antigens aufweisen).
-
Die
Induzierung von Antikörper-Antworten
kann auf eine Vielzahl von Wegen erreicht werden, die dem Fachmann
bekannt sind, z.B. kann das mindestens eine erste Analogon einen
Teil umfassen, der aus einer Modifikation der Struktur des Zell-gebundenen
Polypeptid-Antigen
besteht, wobei die Modifikation zur Folge hat, dass die Immunisierung
des Tieres mit dem ersten Analogon die Herstellung von Antikörpern in
dem Tier gegen das Zell-gebundene Polypeptid-Antigen induziert – diese
Variante eignet sich, wie oben erwähnt, insbesondere zur Nukleinsäure-Vakzinierung.
Alternativ dazu kann das erfindungsgemäße Verfahren es umfassen, die
Präsentation
einer immunogenisch wirksamen Menge von mindestens einem zweiten
Analogon des Zell-gebundenen Polypeptid-Antigens, welches solch
eine Modifikation enthält,
gegenüber
dem Immunsystem des Tieres zu bewirken. Ein bequemer Weg zum Erreichen,
dass die Modifikation den gewünschten
Antikörper-induzierenden
Effekt besitzt, ist es, mindestens ein zweites fremdes TH-Epitop in das zweite Analogon mit einzuschließen, d.h.
eine Strategie, die der ähnlich
ist, die für
das erste Analogon verwendet wurde.
-
In
den Fällen,
in denen es erwünscht
ist, auch eine wirksame humorale Immunantwort hervorzurufen, ist
es vorteilhaft, dass das erste und/oder das zweite Analogon einen
wesentlichen Teil der B-Zell-Epitope des Zell-gebundenen Polypeptid-Antigens
umfasst bzw. umfassen, insbesondere einen wesentlichen Teil solcher B-Zell-Epitope,
welche in der natürlich
vorkommenden Form des Antigens in dem jeweiligen Tier extrazellulär vorliegen.
-
Die
oben diskutierten Variationen und Modifikationen des schwachen,
Zell-gebundenen Polypeptid-Antigens können verschiedene Formen einnehmen.
Es ist bevorzugt, dass die Variation und/oder Modifikation Aminosäuresubstitution
und/oder -deletion und/oder -insertion und/oder -addition umfasst.
Diese grundlegenden Operationen, die sich auf die Manipulation einer
Aminosäuresequenz
beziehen, sollen sowohl einzelne Aminosäureaustausche als auch Operationen
abdecken, die Ketten von Aminosäuren
umfassen (u.a. das Austauschen von Aminosäureketten innerhalb des Polypeptid-Antigens;
dies ist besonders interessant, wenn das An tigen ein echtes intrazelluläres Antigen
ist, da nur Erwägungen,
die das Bewahren von CTL-Epitopen betreffen,
relevant sind). Es versteht sich, dass die Einführung von z.B. einer einzelnen
Aminosäureinsertion oder
-deletion das Auftreten eines fremden TH-Epitops
in der Sequenz des Analogons hervorrufen kann, d.h. das Auftreten
einer MHC-Klasse II-Molekül-bindenden
Sequenz. In den meisten Situationen ist es jedoch bevorzugt (und
sogar notwendig), ein bekanntes fremdes TH-Epitop
einzufügen,
und solch eine Operation wird die Säuresubstitution und/oder -insertion
(oder manchmal der Addition in Form einer Konjugation mit einem Trägerprotein
oder der Bereitstellung eines Fusions-Polypeptids mittels molekularbiologischer
Verfahren) erfordern wird. Es ist bevorzugt, dass die Anzahl der
Aminosäureinsertionen,
-deletionen, -substitutionen oder -additionen mindestens 2 ist,
wie beispielsweise 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15,
16, 17, 18, 19, 20 und 25 Insertionen, Substitutionen, Additionen
oder Deletionen. Es ist außerdem
bevorzugt, dass die Anzahl an Aminosäuresubstitutionen 150 nicht übersteigt,
wie beispielsweise höchstens
100, höchstens
90, höchstens 80
und höchstens
70. Es ist besonders bevorzugt, dass die Anzahl der Substitutionen,
Insertionen, Deletionen oder Additionen 60 nicht überschreitet,
und insbesondere sollte die Anzahl 50 oder sogar 40 nicht überschreiten.
Besonders bevorzugt ist eine Anzahl von nicht mehr als 30.
-
Bevorzugte
Ausführungsformen
der Erfindung umfassen die Modifikationen durch Einführen von
mindestens einem fremden immundominanten TH-Epitop.
Es versteht sich, dass die Frage der Immundominanz eines T-Zell-Epitops
von der fraglichen Tierart abhängt.
Wie hier verwendet, bezieht sich der Ausdruck "Immundominanz" einfach auf Epitope, welche in dem
vakzinierten Tier/der Population eine signifikante Immunantwort
hervorrufen, aber es ist eine wohlbekannte Tatsache, dass ein T-Zell-Epitop,
welches immundominant in einem Individuum ist, nicht notwendigerweise
immundominant in anderen Individuen derselben Art ist, sogar obwohl
es in der Lage sein könnte,
MHC-II-Moleküle
in letzterem Individuum zu binden. Echte immundominante TH-Epitope sind jene, welche unabhängig von
dem Polypeptid, in dem sie eine Untersequenz bilden, die Aktivierung
von TH-Zellen hervorrufen – in anderen
Worten, einige TH-Epitope besitzen als intrinsische Eigenschaft
den Charakter, im Wesentlichen niemals kryptisch zu sein, da sie
im Wesentlichen immer von APCs prozessiert und in der Umgebung eines
MHC-II-Moleküls
auf der Oberfläche
der APC präsentiert
werden.
-
Ein
weiterer wichtiger Punkt ist die Frage der MHC-Restriktion von T-Zell-Epitopen.
Im Allgemeinen sind natürlich
vorkommende T-Zell-Epitope MHC-restringiert, d.h., ein bestimmtes
Peptid, das ein T-Zell-Epitop darstellt, wird lediglich an eine
Untergruppe von MHC-Klasse
II-Molekülen
effektiv binden. Dies hat umgekehrt zur Folge, dass in den meisten
Fällen
die Verwendung von einem bestimmten spezifischen T-Zell-Epitop zu einer
Vakzin-Komponente
führt,
welche lediglich in einem Teil der Population wirksam ist, und abhängig von der
Größe dieses
Teils kann es notwendig sein, mehr T-Zell-Epitope in dasselbe Molekül einzubauen,
oder alternativ dazu ein Multi-Komponenten-Vakzin herzustellen,
in dem die Komponenten Varianten des Antigens sind, welche sich
von den anderen durch die Natur des eingefügten T-Zell-Epitops unterscheiden.
-
Wenn
die MHC-Restriktion der verwendeten T-Zellen vollständig unbekannt
ist (z.B. in einer Situation, in der das vakzinierte Tier eine kaum
definierte MHC-Zusammensetzung aufweist), kann der Anteil der Population,
der von einer spezifischen Vakzin-Zusammensetzung abgedeckt wird,
mittels der folgenden Formel bestimmt werden
- – wobei
pi die Häufigkeit
von reagierenden Individuen bzw. Respondern in der Population gegen
das i-te Fremd-T-Zell-Epitop, das in der Vakzinzusammensetzung vorhanden
ist, und n die Gesamtzahl fremder T-Zell-Epitope in der Vakzinzusammensetzung
ist. Somit würde
eine Vakzinzusammensetzung, die drei fremde T-Zell-Epitope mit Reaktionshäufigkeiten
in der Population von 0,8 bzw. 0,7 bzw. 0,6 enthält, 1 – 0,2 × 0,3 × 0,4 =
0,976ergeben,
- – d.h.
97,6% der Population werden statistisch gesehen eine MHC-II-vermittelte
Antwort auf das Vakzin entwickeln.
-
Die
obige Formel ist in Situationen nicht anwendbar, in denen ein mehr
oder weniger präzises MHC-Restriktionsmuster
der verwendeten Peptide bekannt ist. Wenn z.B. ein bestimmtes Peptid
nur an die humanen MHC-II-Moleküle
bindet, das von den HLA-DR-Allelen DR1, DR3, DR5 und DR7 codiert
wird, dann wird die Verwendung dieses Peptids zusammen mit anderen
Peptiden, welche an die übrigen
MHC-II-Moleküle,
die von HLA-DR-Allelen codiert werden, binden, 100% Abdeckung in
der fraglichen Population erreichen. Wenn das zweite Peptid gleichermaßen nur
DR3 und DR5 bindet, wird die Zugabe dieses Peptids die Abdeckung überhaupt
nicht erhöhen.
Wenn man die Berechnung der Populationsantwort lediglich auf die
MHC-Restriktion von T-Zell-Epitopen im Vakzin beschränkt, kann
der Anteil der Population, der von einer spezifischen Vakzinzusammensetzung
abgedeckt wird, mittels der folgenden Formel bestimmt werden
– wobei φ
j die
Summe der Häufigkeiten
in der Population von allelischen Haplotypen ist, die für MHC-Moleküle codieren,
welche irgendeines der T-Zell-Epitope im Vakzin binden und welche
zum j-sten der drei bekannten HLA-Loci (DP, DR und DQ gehören; in
der Praxis wird zuerst bestimmt, welche MHC-Moleküle jedes T-Zell-Epitop
im Vakzin erkennen, und danach werden diese mittels des Typus (DP,
DR und DQ aufgelistet – dann
werden die individuellen Häufigkeiten
der unterschiedlichen aufgelisteten allelischen Haplotypen für jeden
Typus summiert, wodurch man φ
1, φ
2 und φ
3 erhält.
-
Es
kann vorkommen, dass der Wert p
i in der
Formel II den entsprechenden theoretischen Wert π
i übersteigt:
– wobei v
j die
Summe der Häufigkeiten
in der Population des allelischen Haplotyps ist, der für MHC-Moleküle codiert,
welche das i-te T-Zell-Epitop im Vakzin binden und welche zu dem
j-sten der drei bekannten HLA-Loci (DP, DR und DQ) gehören. Dies
bedeutet, dass πi
der Population eine Häufigkeit
von Respondern von f
residual_i = (p
i-π
i)/(1-π
i) ist. Daher kann Formel III unter Erhalt
der Formel V angepasst werden:
– wobei der Ausdruck 1-f
residual_i auf 0 gesetzt wird, wenn er negativ
ist. Es sollte erwähnt
werden, dass Formel V es erfordert, dass alle Epitope gegen identische
Haplotyp-Sätze
kartiert wurden.
-
Wenn
man daher T-Zell-Epitope auswählt,
die in das Analogon eingefügt
werden sollen, ist es wichtig, alles Wissen über die Epitope, welches verfügbar ist,
mit einzubringen: 1) die Häufigkeit
von Respondern in der Population gegenüber jedem Epitop, 2) die MHC-Restriktionsdaten,
und 3) die Häufigkeit
der relevanten Haplotypen in der Population.
-
Es
existiert eine Reihe von natürlich
vorkommenden "gemischten" T-Zell-Epitopen,
welche in einem großen
Teil der Individuen einer Tierart oder einer Tierpopulation aktiv
sind, und diese werden bevorzugt in das Vakzin eingefügt, wobei
das Erfordernis einer großen
Anzahl unterschiedlicher Analoga in demselben Vakzin verringert
wird.
-
Das
gemischte Epitop kann erfindungsgemäße ein natürlich vorkommendes humanes
T-Zell-Epitop sein,
wie beispielsweise Epitope von Tetanustoxoid (z.B. die P2- und P30-Epitope),
Diphterietoxoid, Grippevirus-Hämagluttinin
(HA) und P. falciparum CS-Antigen.
-
Über die
Jahre wurde eine große
Anzahl anderer gemischter T-Zell-Epitope identifiziert. Insbesondere Peptide,
die in der Lage sind, eine große
Anzahl von HLA-DR-Molekülen,
die von den unterschiedlichen HLA-DR-Allelen codiert werden, wurde
identifiziert, und diese sind alle mögliche T-Zell-Epitope, die
in Analoga eingefügt
werden können,
die erfindungsgemäß verwendet
werden. Vgl. ebenfalls die Epitope, die in den folgenden Zitatstellen
diskutiert werden: WO 98/23635 (Frazer IH et al., The University
of Queensland); Southwood S et al., 1998, J. Immunol. 160: 3363-3373;
Sinigaglia F et al., 1988, Nature 336: 778-780; Rammensee HG et
al., 1995, Immunogenetics 41: 4 178-228; Chicz RM et al., 1993,
J. Exp. Med 178: 27-47; Hammer J et al., 1993, Cell 74: 197-203;
und Falk K et al., 1994, Immunogenetics 39: 230-242. Die letzte
Bezugsstelle beschäftigt
sich auch mit HLA-DQ- und -DP-Liganden. Alle in diesen 5 Bezugsstellen
aufgelisteten Epitope sind als Kandidaten für natürliche Epitope, die in der
vorliegenden Erfindung zu verwenden sind, relevant, da sie Epitope
sind, welche gemeinsame Motive aufweisen.
-
Alternativ
dazu kann das Epitop jedes künstliche
T-Zell-Epitop sein, welches in der Lage ist, einen großen Anteil
von Haplotypen zu binden. In diesem Zusammenhang sind die pan-DR-Epitop-Peptide
("PADRE"), die in der WO
95/07707 und in dem dazugehörigen
wissenschaftlichen Artikel Alexander J et al., 1994, Immunity 1:
751-761, beschrieben sind, interessante Kandidaten für Epitope,
die erfindungsgemäß zu verwenden sind.
Es sollte erwähnt
werden, dass die effektivsten PADRE-Peptide, die in diesen Artikeln
veröffentlicht
sind, D-Aminosäuren an
den C- und N-Termini tragen um die Stabilität zu erhöhen, wenn diese verabreicht
werden. Die vorliegende Erfindung zielt jedoch in erster Linie darauf
ab, die relevanten Epitope als Teil des modifizierten Antigens einzubauen,
welches dann anschließend
innerhalb des lysosomalen Kompartiments der APCs enzymatisch abgebaut
wird um die anschließende
Präsentation
im Zusammenhang mit einem MHC-II-Molekül zu ermöglichen, und daher ist es nicht
zwingend erforderlich, D-Aminosäuren
in die Epitope einzubauen, die in der vorliegenden Erfindung verwendet
werden.
-
Ein
besonders bevorzugtes PADRE-Peptid ist das, das die Aminosäuresequenz
AKFVAAWTLKAAA besitzt, oder eine immunologisch wirksame Untersequenz
davon. Dieses, und andere Epitope, die ebenfalls keine MHC-Restriktion
aufweisen, sind bevorzugte T-Zell-Epitope, welche in dem Analogon, das
in dem erfinderischen Verfahren verwendet wird, vorhanden sein sollten.
Solche super-gemischten Epitope werden die einfachsten Ausführungsformen
der Erfindung ermöglichen,
wobei nur ein einzelnes aller Analogon dem Immunsystem des vakzinierten
Tiers präsentiert
wird.
-
Das
Wesen der oben beschriebenen Variation/Modifikation umfasst im Wesentlichen,
dass
- – mindestens
eine erste Gruppierung in dem ersten und/oder zweiten Analogon umfasst
ist, wobei die Gruppierung das Targeting des Analogons an eine Antigen-präsentierende
Zelle (APC) bewirkt, und/oder
- – mindestens
eine zweite Gruppierung in das erste und/oder das zweite Analogon
eingefügt
wird, wobei die zweite Gruppierung das Immunsystem stimuliert, und/oder
- – mindestens
eine dritte Gruppierung in das erste und/oder das zweite Analogon
eingefügt
wird, wobei diese dritte Gruppierung die Präsentation des Analogons gegenüber dem
Immunsystem optimiert.
-
Die
funktionellen und strukturellen Eigenschaften, die zu diesen ersten,
zweiten und dritten Gruppierungen gehören, werden im Folgenden diskutiert:
Sie
können
in Form von Seitengruppen vorliegen, die kovalent oder nicht-kovalent
an geeignete chemische Gruppen in der Aminosäuresequenz des Zell-gebundenen
Polypeptid-Antigens
oder einer Untersequenz davon angehängt werden. Dies soll bedeuten,
dass Ketten von Aminosäureresten,
die sich von dem Polypeptid-Antigen ableiten, derivatisiert werden,
ohne die primäre
Aminosäuresequenz
zu verändern,
oder zumindest ohne Veränderungen
in den Peptidbindungen zwischen den einzelnen Aminosäuren in
der Kette einzuführen.
-
Die
Gruppierungen können
auch in Form von Fusionspartnern mit der Aminosäurese quenz, die von dem Zell-gebundenen
Polypeptid-Antigen abgeleitet ist, vorliegen. In diesem Zusammenhang
sollte erwähnt werden,
dass beide Möglichkeiten
die Option umfassen, die Aminosäuresequenz
an einen Träger
zu binden, vgl. die unterstehende Diskussion von diesen. In anderen
Worten, in dem vorliegenden Zusammenhang ist der Ausdruck "Fusionsprotein" nicht nur auf ein
Fusionskonstrukt beschränkt,
das mittels der Expression eines DNA-Fragments hergestellt wird,
welches das Konstrukt codiert, sondern er umfasst auch eine Verbindung
von zwei Proteinen, welche mittels einer Peptidbindung in einer
anschließenden
chemischen Reaktion verbunden werden.
-
Wie
oben erwähnt,
kann das Analogon auch das Eizufügen
einer ersten Gruppierung umfassen, welche das Analogon zielgerichtet
zu einer APC oder zu einem B-Lymphozyten leitet. Zum Beispiel kann
die erste Gruppierung ein spezifischer Bindungspartner für ein B-Lymphozyten-spezifisches
Oberflächen-Antigen
oder für
ein APC-spezifisches Oberflächen-Antigen sein. Viele
solcher spezifischen Oberflächen-Antigene
sind im Stand der Technik bekannt. Z.B. kann die Gruppierung ein
Kohlenhydrat sein, für
das ein Rezeptor auf dem B-Lymphozyten
oder der APC existiert (z.B. Mannan oder Mannose). Alternativ dazu
kann die zweite Gruppierung ein Hapten sein. Auch ein Antikörperfragment,
welches spezifisch ein Oberflächenmolekül auf APCs
oder Lymphozyten erkennt, kann als erste Gruppierung verwendet werden
(das Oberflächenmolekül kann z.B.
ein FCγ-Rezeptor
von Makrophagen und Monozyten sein, wie beispielsweise FCγRI oder,
alternativ dazu, jeder andere spezifische Oberflächenmarker, wie beispielsweise
CD40 oder CTLA-4). Es sollte erwähnt
werden, dass alle diese beispielhaften Targeting-Moleküle als Teil
eines Adjuvans verwendet werden können, vgl. unten. CD40-Ligand,
Antikörper
gegen CD40 oder Varianten davon, welche CD40 binden, werden das
Analogon zielgerichtet zu dendritischen Zellen leiten. Gleichzeitig
haben jüngste
Ergebnisse gezeigt, dass die Wechselwirkung mit dem CD40-Molekül das essentielle
Vorhandensein von TH-Zellen zum Erhalten
einer CTL-Antwort beseitigt. Es wird somit vorgeschlagen, dass die
allgemeine Verwendung von CD40-bindenden Molekülen als erste Gruppierung (oder
als Adjuvantien, vgl. unten) die CTL-Antwort beachtlich erhöhen wird;
tatsächlich
wird die Verwendung solcher CD40-bindenden Moleküle als Adjuvantien und "erste Gruppierungen" im Sinne der vorliegenden
Erfindung als eigenständig
erfinderisch erachtet.
-
Als
Alternative oder Ersatz zum Targeting der Analoga zu einem bestimmten
Zelltypus zum Erzielen einer erhöhten
Immunantwort ist es möglich,
den Grad der Reaktivität
des Immunsystems zu erhöhen,
indem die oben beschriebene zweite Gruppierung eingefügt wird,
welche das Immunsystem stimuliert. Typische Beispiele solcher zweiten
Gruppierungen sind Cytokine, Hitzeschock-Proteine und Hormone, sowie
deren wirksame Teile.
-
Geeignete
Cytokine, die erfindungsgemäß zu verwenden
sind, sind jene, welche normalerweise auch als Adjuvantien in einer
Vakzinzusammensetzung fungieren, z.B. Interferon γ (IFNγ), Flt3-Ligand
(F1t3L), Interleukin 1 (IL-1), Interleukin 2 (IL-2), Interleukin
4 (IL-4), In terleukin 6 (IL-6), Interleukin 12 (IL-12), Interleukin 13
(IL-13), Interleukin 15 (IL-15), und Granulozyten-Makrophagen-Kolonie-stimulierender
Faktor (GM-CSF); alternativ dazu kann der funktionelle Teil des
Cytokinmoleküls
als zweite Gruppierung ausreichend sein. Im Hinblick auf die Verwendung
solcher Cytokine als Adjuvans-Substanzen vgl. die untenstehende
Diskussion.
-
Alternativ
dazu kann die zweite Gruppierung ein Toxin sein, wie beispielsweise
Listeriolycin (LLO), Lipid A und Hitze-labiles Enterotoxin. Eine
Reihe von mycobakteriellen Derivaten, wie MDP (Muramyldipeptid), CFA
(vollständiges
Freund'sches Adjuvans)
und die Trehalose-Diester TDM und TDE, sind ebenfalls interessante
Möglichkeiten.
-
Geeignete
erfindungsgemäße Hitzeschock-Proteine,
die als zweite Gruppierung verwendet werden können, können HSP70, HSP90, HSC70, GRP94
und Calreticulin (CRT) sein.
-
Auch
die Möglichkeit
der Einführung
einer dritten Gruppierung, welche die Präsentation des Analogons gegenüber dem
Immunsystem verstärkt,
ist eine wichtige Ausführungsform
der Erfindung. Der Stand der Technik hat mehrere Beispiele dieses
Prinzips aufgezeigt. Es ist z.B. bekannt, dass der Palmitoyl-Lipidanker in
dem OspA-Protein von Borrelia burgdorferi verwendet werden kann
um Polypeptide bereitzustellen, die sich selbst als Adjuvans dienen
(vgl. z.B. WO 96/40718). Es scheint, dass die Lipidproteine Micellen-ähnliche
Strukturen ausbilden, mit einem Kern, der aus den Lipidankerteilen
der Polypeptide besteht, wobei die übrigen Teile des Moleküls daraus
hervorragen, was zu multiplen Präsentationen
der antigenischen Determinanten führt. Die Anwendung von diesem
und von verwandten Ansätzen
unter Verwendung unterschiedlicher Lipidanker (z.B. einer Myristylgruppe,
einer Farnesylgruppe, einer Geranyl-Geranyl-Gruppe, eines GPI-Ankers und
einer N-Acyldiglyceridgruppe) stellt daher bevorzugte Ausführungsformen
der Erfindung dar, insbesondere da die Bereitstellung eines solchen
Lipidankers in einem rekombinant hergestellten Protein sehr einfach
ist und lediglich die Verwendung z.B. einer natürlich vorkommenden Signalsequenz
als Fusionspartner für
das Analogon erfordert. Eine andere Möglichkeit ist die Verwendung
des C3d-Fragmentes des Komplementfaktors C3 oder von C3 selbst (vgl.
Dempsey et al., 1996, Science 271, 348-350, und Lou & Kohler, 1998,
Nature Biotechnology 16, 458-462).
-
Es
ist wichtig zu erwähnen,
dass es insbesondere bevorzugt ist, dass das erste und/oder zweite
Analogon im Wesentlichen die vollständige Tertiärstruktur des Zell-gebundenen
Polypeptid-Antigens aufweist bzw. aufweisen, wenn versucht wird,
das erfindungsgemäße Verfahren
z.B. gegen Membran-gebundene Polypeptid-Antigene zu verwenden, welche
dem extrazellulären
Kompartiment ausgesetzt sind. In der vorliegenden Beschreibung und
in den Ansprüchen
soll dies bedeuten, dass die Gesamttertiärstruktur des Polypeptid-Antigens,
welches extrazellulär
exponiert ist, bewahrt bleibt, da, wie oben erwähnt, die Tertiärstruktur
der obligat intrazellulären
Polypeptide das humorale Immunsystem nicht mit einbezieht. Tatsächlich ist
es als Teil der Vakzinierungsstrategie oft erwünscht, das Aussetzen von putativen
B-Zell-Epitopen, die von dem intrazellulären Teil des Polypeptid-Antigens
abgeleitet sind, gegenüber
dem extra zellulären
Kompartiment zu vermeiden; auf diese Weise können potentiell nachteilige
Wirkungen, die durch eine Kreuzreaktivität mit anderen Antigenen verursacht
werden, minimiert werden.
-
Zum
Zwecke der vorliegenden Erfindung reicht es jedoch aus, wenn die
Variation/Modifikation (sei es eine Insertion, Addition, Deletion
oder Substitution) ein fremdes T-Zell-Epitop
hervorbringt und gleichzeitig eine wesentliche Anzahl der CTL-Epitope
in dem Polypeptid-Antigen bewahrt (und manchmal ebenfalls eine wesentliche
Anzahl von B-Zell-Epitopen).
-
Die
folgende Formel beschreibt die Konstrukte, die im Allgemeinen von
der Erfindung umfasst sind: (MOD1)s1(PAGe1)n1(MOD2)s2(PAGe2)n2 ....(MODx)sx(PAGex)nx (I)– wobei
PAGe1-PAGex x CTL-
und/oder B-Zell-Epitope sind, die Untersequenzen des relevanten
Polypeptid-Antigens enthalten, welche unabhängig voneinander identisch
oder nicht-identisch sind, und welche fremde Seitengruppen enthalten
können
oder nicht, x eine ganze Zahl ≥ 3
ist, n1-nx x ganze Zahlen ≥ 0
sind (mindestens eine ist ≥ 1),
MOD1-MODx x Modifikationen
sind, die in den bewahrten Epitopen eingefügt sind, und s1-sx x ganze
Zahlen ≥ 0
sind (mindestens eine ist ≥ 1,
wenn keine Seitengruppen in die Sequenzen eingefügt sind). Daher erlaubt die
Erfindung bei den gegebenen allgemeinen funktionellen Beschränkungen
der Immunogenizität
der Konstrukte, alle Arten von Permutationen der Original-Antigensequenz,
und alle Arten von Modifikationen darin. Somit sind von der Erfindung
Analoga erfasst, die durch Auslassen von Teilen der Polypeptid-Antigensequenz
erhalten werden, welche z.B. in vivo nachteilige Wirkungen aufweisen,
oder die Auslassung von Teilen, welche normalerweise intrazellulär vorliegen
und daher unerwünschte
immunologische Reaktionen hervorrufen könnten, vgl. die detaillierte
Diskussion unten.
-
Eine
weitere Ausarbeitung der obigen Prinzipien umfasst die Verwendung
von CTL- und/oder B-Zell-Epitopen
von mehr als einem Antigen mit pathologischem Bezug. Z.B. gibt es
mehrere Krebs-bezogene Antigene, die ihre onkogenen Wirkungen nur
ausüben,
wenn sie in mutierter Form vorliegen – Beispiele sind mutiertes
k-ras und P53, welche beide bei der normalen Zellzyklusregulation
notwendige Proteine sind und welche beide in den meisten normalen
Zellen Expressionsprodukte sind. In einigen Fällen wurde gezeigt, dass CTLs
mutierte Peptide von diesen Antigenen erkennen. Es ist daher wichtig,
dass das Immunsystem nur auf die mutierten Peptide reagiert und
nicht auf die unmutierten Teile, wenn eine Antigen-spezifische Immuntherapie
in Gang gesetzt wird.
-
Wir
haben eine Strategie entworfen, bei der Sequenzen von 8-25 Aminosäuren solcher
Krankheits-bezogenen Proteine als weitere Epitope in einem AutoVac-Konstrukt
verwendet werden können – in bevorzugten Ausführungsformen
würden
die eingefügten
Epitope gleichzeitig für
das Auftreten von TH-Epitopen im Endkonstrukt
sorgen, vgl. die obige Diskussion. Die Epitope, die für diesen
Zweck verwendet werden, würden
jene sein, welche die mutierte Region des Krankheits-bezogenen Proteins
umfassen. Indem solch ein Ansatz angewandt wird, wäre es möglich, CTLs
(und wo dies möglich
ist möglicherweise
Antikörper)
zu erzeugen, und zwar nur gegen die mutierte Form des Krankheits-bezogenen
Antigens. In den Fällen,
in denen das Krankheits-bezogene Antigen für das Auftreten eines TH-Epitops sorgt, könnte die Verwendung eines echten
fremden TH-Epitops vollständig ausgelassen
werden. Eine Ausführungsform
dieses Prinzips könnte
z.B. die Vakzinierung mit einem Nukleinsäure-Vakzin sein, welches für ein Analogon
eines Polypeptid-Antigens (z.B. Her2 oder PSM) codiert, in das mindestens
ein TH-Epitop
und mindestens ein Peptid, das von einem anderen Krankheits-bezogenen
Antigen (z.B. einem Peptid von dem mutierten Teil eines onkogenischen
Proteins eingefügt wurden).
In einer bevorzugten Ausführungsform
wird das mindestens eine TH-Epitop als Folge
des Einfügens des
Peptids eingefügt.
-
Es
ist außerdem
bevorzugt, dass die Variation und/oder Modifikation, wenn dies anwendbar
ist, eine Duplikation des mindestens einen B-Zell-Epitops, oder
des mindestens einen CTL-Epitops des Zell-gebundenen Polypeptid-Antigens
umfasst. Diese Strategie führt
zu dem Ergebnis, dass multiple Kopien bevorzugter Epitop-Regionen
dem Immunsystem präsentiert
werden und daher die Wahrscheinlichkeit einer effektiven Immunantwort
maximieren. Daher macht sich diese Ausführungsform der Erfindung multiple
Präsentationen
der Epitope, die von dem Polypeptid-Antigen abgeleitet sind, zunutze
(d.h. Formel I, bei der mindestens ein B-Zell-Epitop an zwei Positionen vorhanden
ist).
-
Diese
Wirkung kann auf verschiedene Weisen erreicht werden, z.B. indem
einfach Fusions-Polypeptide präpariert
werden, die die Struktur (PAG)m umfassen,
wobei m eine ganze Zahl ≥ 2
ist, und dann die hierin diskutierten Modifikationen in mindestens
einer der Polypeptid-Antigen-Sequenzen
eingefügt
werden.
-
Eine
alternative Ausführungsform
der Erfindung, welche ebenfalls zu der bevorzugten Präsentation von
multiplen (z.B. mindestens zwei) Kopien der wichtigen Epitop-Regionen
des Antigens gegenüber
dem Immunsystem führt,
ist das kovalente Koppeln des Antigens, einer Untersequenz oder
Varianten davon mit bestimmten Molekülen. Es können z.B. Polymere verwendet
werden, z.B. Kohlenhydrate, wie Dextran, vgl. z.B. Lees A et al.,
1994, Vaccine 12: 1160-1166; Lees A et al., 1990, J Immunol. 145:
3594-3600, aber ebenso sind Mannose und Mannan nützliche Alternativen. Integrale
Membranproteine, z.B. von E. coli und anderen Bakterien, sind ebenfalls
nützliche
Konjugationspartner. Die traditionellen Trägermoleküle wie Napfschnecken-Hämocyanin
(KLH, "keyhole limpet
haemocyanin"), Tetanustoxoid,
Diphterietoxoid und Rinderserumalbumin (BSA) sind ebenfalls bevorzugte
und nützliche
Konjugationspartner.
-
Die
Aufrechterhaltung der manchmal vorteilhaften wesentlichen Fraktion
der B-Zell-Epitope
oder sogar der gesamten Tertiärstruktur
eines Proteins, welches einer wie hierin beschriebenen Modifikation
unterworfen wurde, kann auf verschiedene Weise erreicht werden.
Eine ist es, einfach ein polyklonales Antiserum, das gegen das Polypeptid-Antigen
gerichtet ist (z.B. ein Antiserum, das in einem Kaninchen präpariert
wurde) zu präparieren,
und danach dieses Antiserum als Testreagens (z.B. in einem kompetitiven
ELISA) gegen die modifizierten Proteine, welche produziert werden,
zu verwenden. Modifizierte Versionen (Analoga), welche in demselben
Ausmaß mit
dem Antiserum reagieren, wie das Polypeptid-Antigen, müssen als
solche angesehen werden, die dieselbe Gesamttertiärstruktur,
wie das Polypeptid-Antigen aufweisen, während Analoga, die eine begrenzte
(aber noch immer signifikante und spezifische) Reaktivität mit solch
einem Antiserum zeigen, als solche angesehen werden, die einen wesentlichen
Teil der Original-B-Zell-Epitope behalten haben.
-
Alternativ
dazu kann eine Auswahl monoklonaler Antikörper, die mit unterschiedlichen
Epitopen auf dem Polypeptid-Antigen reagieren, präpariert
werden und als Testreihe verwendet werden. Dieser Ansatz hat den
Vorteil, dass er 1) eine Epitop-Kartierung des fraglichen Polypeptid-Antigens
erlaubt, und 2) eine Kartierung der Epitope, welche in den präparierten
Analoga aufrecht erhalten werden, erlaubt.
-
Natürlich wäre ein dritter
Ansatz, die 3-dimensionale Struktur des Polypeptid-Antigens oder
eines biologisch aktiven Teils davon (vgl. oben) aufzulösen, und
diese mit der aufgelösten
3-dimensionalen Struktur der präparierten
Analoga zu vergleichen. Eine dreidimensionale Struktur kann mit
der Hilfe von Röntgenbeugungsuntersuchungen
und der NMR-Spektroskopie aufgelöst
werden. Weitere Informationen, die sich auf die Tertiärstruktur
beziehen, können
zu einem gewissen Ausmaß aus
Zirkulardichroismus-Studien erhalten werden, welche den Vorteil
haben, dass sie lediglich die Polypeptide in reiner Form erfordern
(während
die Röntgenbeugung
die Bereitstellung von kristallisierten Polypeptiden erfordert und
NMR die Bereitstellung von Isotopvarianten des Polypeptids erfordert)
um nützliche
Informationen über
die Tertiärstruktur
eines gegebenen Moleküls
bereitzustellen. Letztlich sind jedoch Röntgenbeugung und/oder NMR erforderlich
um abschließende Daten
zu erhalten, da der Zirkulardichroismus lediglich einen indirekten
Beweis für
eine korrekte 3-dimensionale Struktur ermöglicht, und zwar über Informationen über Sekundärstrukturelemente.
-
Im
Wesentlichen gibt es zur Zeit drei ausführbare Wege, die Präsentation
der relevanten Epitope gegenüber
dem Immunsystem zu erreichen: Traditionelle Untereinheiten-Vakzinierung
mit Polypeptid-Antigenen, die Verabreichung von genetisch modifizierten
Lebendvakzinen und die Nukleinsäure-Vakzinierung.
Diese drei Möglichkeiten
werden getrennt im Folgenden diskutiert:
-
Polypeptid-Vakzinierung
-
Diese
bringt die Verabreichung einer immunogen wirksamen Menge des mindestens
einen ersten Analogons, und, wenn dies relevant ist, die Verabreichung
einer immunologisch wirksamen Menge des mindestens einen zweiten
Analogons, an das fragliche Tier mit sich. Bevorzugt wird/werden
das mindestens eine erste und/oder zweite Analogon mit einem pharmazeutisch
und immunologisch annehmbaren Träger
und/oder Vehikel und wahlweise mit einem Adjuvans formuliert.
-
Wenn
die Präsentation
des Analogons gegenüber
dem Immunsystem eines Tieres mittels dessen Verabreichung an das
Tier bewirkt wird, folgt die Formulierung des Polypeptids den im
Stand der Technik allgemein anerkannten Prinzipien.
-
Die
Herstellung von Vakzinen, welche Peptidsequenzen als aktive Inhaltsstoffe
enthalten, ist im Stand der Technik wohlbekannt, wie in den US-Patenten
4 608 251; 4 601 903; 4 599 231; 4 599 230; 4 596 792; und 4 578
770 beispielhaft dargestellt. Typischerweise werden solche Vakzine
entweder als Injektionen, entweder als flüssige Lösungen oder Suspensionen, hergestellt;
feste Formen, die sich zur Lösung
in oder zur Suspension in Flüssigkeit
vor der Injektion eigenen, können
ebenfalls hergestellt werden. Die Präparation kann außerdem emulgiert
werden. Der aktive immunogene Inhaltsstoff wird oft mit Exzipientien
gemischt, welche pharmazeutisch annehmbar sind, und mit dem aktiven
Inhaltsstoff kompatibel. Solche Exzipientien sind z.B. Wasser, Kochsalzlösung, Dextrose,
Glycerin, Ethanol oder ähnliches,
und Kombinationen davon. Zusätzlich
kann das Vakzin, wenn dies erwünscht
ist, geringe Mengen von Hilfssubstanzen, wie Benetzungsmittel oder
Emulgatoren, pH-Puffer oder Adjuvantien, welche die Wirksamkeit
der Vakzine erhöhen,
enthalten; vgl. die detaillierte Diskussion von Adjuvantien unten.
-
Die
Vakzine werden konventionell parenteral verabreicht, beispielsweise
durch Injektion, entweder subkutan, intradermal, subdermal oder
intramuskulär.
Zusätzliche
Formulierungen, welche für
andere Arten der Verabreichung geeignet sind, umfassen Zäpfchen und
in einigen Fallen orale, bukkale, sublinguale, intraperitoneale,
intravaginale, anale und intrakraniale Formulierungen. Für Zäpfchen können traditionelle
Bindemittel und Trägerstoffe,
z.B. Polyalkalenglykole oder Triglyceride, umfassen; solche Zäpfchen können aus
Gemischen gebildet sein, die den Inhaltsstoff im Bereich von 0,5%
bis 10%, bevorzugt 1-2%, enthalten. Orale Formulierungen umfassen
solche normalerweise verwendeten Exzipientien, wie beispielsweise
pharmazeutische Grade Mannit, Lactose, Stärke, Magnesiumstearat, Natriumsaccharin,
Cellulose, Magnesiumcarbonat und ähnliche. Diese Zusammensetzungen
haben die Form von Lösungen,
Suspensionen, Tabletten, Pillen, Kapseln, Retardpräparaten
oder Pudern, und enthalten 10-95%
des aktiven Inhaltsstoffes, bevorzugt 25-70%. Für orale Formulierungen ist
Choleratoxin ein interessanter Formulierungspartner (und ebenso
ein möglicher
Konjugationspartner).
-
Die
Polypeptide können
neutral oder in den Salzformen in dem Vakzin formuliert werden.
Pharmazeutisch annehmbare Salze umfassen Säureadditionssalze (gebildet
mit den freien Aminogruppen des Peptids), welche mit anorganischen
Säuren
gebildet werden, wie beispielsweise Salz- oder Phosphorsäuren, oder
beispielsweise organischen Säuren,
wie Essigsäure,
Oxalsäure,
Weinsäure,
Mandelsäure
und ähnlichen.
Salze, die mit den freien Carboxylgruppen gebildet werden, können ebenso
von anorganischen Basen, wie beispielsweise Natrium-, Kalium-, Ammonium-,
Calcium- oder Eisenhydroxiden, abgeleitet werden, und von solchen
organischen Basen, wie Isopropylamin, Trimethylamin, 2-Ethylaminoethanol,
Histidin, Procain und ähnlichen.
-
Die
Vakzine werden auf eine Weise verabreicht, welche mit der Dosisformulierung
kompatibel ist, und in einer solchen Menge, wie sie therapeutisch
wirksam und immunogen sein wird. Die zu verabreichende Menge hängt von
dem zu behandelnden Patienten ab, einschließlich z.B. des Zustandes des
Immunsystems des Individuums, das eine Immunantwort hervorbringen
soll, und des Grades an Schutz, der erwünscht ist. Geeignete Dosierungsbereiche
liegen im Bereich von mehreren hundert Mikrogramm aktiver Inhaltsstoff
pro Vakzinierung, mit einem bevorzugten Bereich von ungefähr 0,1 μg bis 2000 μg (obwohl
sogar höhere
Mengen im Bereich von 1 bis 10 mg vorgeschlagen werden), beispielsweise
im Bereich von ungefähr
0,5 μg bis
1000 μg, bevorzugt
im Bereich von 1 μg
bis 500 μg,
und insbesondere im Bereich von ungefähr 10 μg bis 100 μg. Geeignete Dosierungskurven
zur anfänglichen
Verabreichung und Booster sind ebenfalls variabel, aber sie werden
beispielhaft durch eine anfängliche
Verabreichung, gefolgt von nachfolgenden Impfungen oder anderen Verabreichungen,
vertreten.
-
Die
Art der Anwendung kann in weiten Grenzen variieren. Jede der konventionellen
Verfahren zur Verabreichung eines Vakzins ist anwendbar. Diese umfassen
die orale Anwendung auf einer festen, physiologisch annehmbaren
Basis oder in einer physiologisch annehmbaren Dispersion, parenteral,
durch Injektion oder ähnlich.
Die Dosierung des Vakzins hängt
vom Wege der Verabreichung ab und variiert je nach Alter der zu vakzinierenden
Person und der Formulierung des Antigens.
-
Einige
der Polypeptide des Vakzins sind in einem Vakzin ausreichend immunogen,
aber für
einige der anderen wird die Immunantwort erhöht, wenn das Vakzin außerdem eine
Adjuvanssubstanz umfasst. Es ist besonders bevorzugt, ein Adjuvans
zu verwenden, von dem gezeigt werden kann, dass es die Durchbrechung der
Autotoleranz gegen Autoantigene erleichtert.
-
Verschiedene
Verfahren zum Erreichen einer Adjuvanswirkung für das Vakzin sind bekannt.
Allgemeine Prinzipien und Verfahren sind in "The Theory and Practical Application
of Adjuvants", 1995,
Duncan E.S. Stewart-Tull (Hrsg.), John Wiley & Sons Ltd, ISBN 0-471-95170-6, und ebenso
in "Vaccines: New
Generation Immunological Adjuvants", 1995, Gregoriadis G et al. (Hrsg.),
Plenum Press, New York, ISBN 0-306-45283-9, beispielhaft dargestellt.
-
Bevorzugte
Adjuvantien erleichtern die Aufnahme der Vakzinmoleküle durch
APCs, wie beispielsweise dendritische Zellen, und aktivieren diese.
Nicht-beschränkende
Beispiele werden ausgewählt
aus der Gruppe, bestehend aus einem Immuntargeting-Adjuvans, einem
immunmodulierenden Adjuvans, wie beispielsweise einem Toxin, einem
Cytokin und einem mycobakteriellen Derivat; einer Ölformulierung;
einem Polymer; einem Micellen-bildenden Adjuvans; einem Saponin;
einer immunstimulierenden Komplexmatrix (ISCOM-Matrix); einem Partikel;
DDA; Aluminium-Adjuvantien; DNA-Adjuvantien; γ-Inulin; und einem einkapselnden
Adjuvans. Im Allgemeinen sollte erwähnt werden, dass die obigen
Offenbarungen, welche sich auf Verbindungen und Agentien beziehen,
welche als erste, zweite und dritte Gruppierung in den Analoga nützlich sind,
sich ebenso mutatis mutandis auf deren Verwendung im Adjuvans eines
Vakzins der Erfindung beziehen.
-
Die
Anwendung von Adjuvantien umfasst die Verwendung von Agentien, wie
beispielseweise Aluminiumhydroxid oder -phosphat (Alum), welches
für gewöhnlich als
0,05 bis 0,1%ige Lösung
in gepufferter Kochsalzlösung
verwendet wird, als Gemisch mit synthetischen Polymeren von Zuckern
(z.B. Carbopol®,
das als 0,25%ige Lösung
verwendet wird, die Verwendung einer Aggregation des Proteins in
dem Vakzin durch Hitzebehandlung mit Temperaturen im Bereich zwischen
70° und
101°C für 30 Sekunden
bis zu 2 Minuten, und auch die Aggregation mittels verknüpfender
Agentien ist möglich.
Die Aggregation durch Reaktivierung mit Pepsin-behandelten Antikörpern (Fab-Fragmente)
mit Albumin, das Mischen mit bakteriellen Zellen, wie C. parvum oder
Endotoxinen oder Lipopolysaccharid-Komponenten von gramnegativen
Bakterien, eine Emulsion in physiologisch annehmbaren Öl-Vehikeln,
wie Mannit-Mono-Oleat
(Aracel A) oder die Emulsion mit einer 20%igen Lösung Perfluorcarbon (Fluosol-DA), welche als Blocksubstitut
verwendet wird, kann ebenso verwendet werden. Die Mischung mit Ölen, wie
Squalen und IFA ist ebenso bevorzugt.
-
Entsprechend
der Erfindung ist DDA (Dimethyldioctadecylammoniumbromid) ein interessanter
Kandidat für
ein Adjuvans, genauso wie DNA und γ-Inulin, aber auch Freund'sches vollständiges und
unvollständiges
Adjuvans, sowie Quillajasaponine, wie QuilA und QS21, sind interessant.
Weitere Möglichkeiten
sind Monophosphoryl-Lipid A (MPL) und die oben erwähnten C3
und C3d.
-
Von
Liposom-Formulierungen ist ebenfalls bekannt, dass sie Adjuvans-Wirkungen
ausüben,
und daher sind erfindungsgemäße Liposom-Adjuvantien
bevorzugt.
-
Ebenso
sind Adjuvantien vom immunstimulatorischen Komplexmatrix-Typ (ISCOM®-Matrix) bevorzugte
Auswahlen entsprechend der Erfindung, insbesondere da es gezeigt
wurde, dass dieser Typ von Adjuvantien in der Lage ist, die MHC-Klasse
II-Expression bei APCs hochzuregulieren. Eine ISCOM®-Matrix
besteht aus (wahlweise fraktionierten) Saponinen (Triterpenoiden)
von Quillaja saponaria, Cholesterin und Phospholipid. Gemischt mit
dem immunogenen Protein ist die daraus hervorgehende partikuläre Formulierung
das, was als ISCOM-Partikel
bekannt ist, wobei das Saponin 60-70% Gew./Gew., das Cholesterin
und Phospholipid 10-15% Gew./Gew., und das Protein 10-15% Gew./Gew.
ausmacht. Details, die sich auf die Zusammensetzung und die Verwendung
von immunstimulatorischen Komplexen beziehen, findet man z.B. in
den oben erwähnten
Lehrbüchern,
die Adjuvantien behandeln, aber auch bei Marein B et al., 1995,
Clin. Immunother. 3: 461-475, sowie bei Barr IG und Mitchell GF,
1995, Immunol. and Cell Biol. 74: 8-25, welche nützliche Anleitungen für die Herstellung
von vollständigen
immunstimulatorischen Komplexen bereitstellen.
-
Eine
andere hochinteressante (und daher bevorzugte) Möglichkeit zum Erreichen einer
Adjuvanswirkung ist es, die Technik zu verwenden, die in Gosselin
et al., 1992, beschrieben ist. Kurz, die Präsentation von relevanten Antigenen,
wie einem erfindungsgemäßen Antigen
kann verstärkt
werden, indem das Antigen an Antikörper konjugiert wird (oder
an Antigen-bindende Antikörperfragmente),
und zwar gegen die Fcγ-Rezeptoren
auf Monozyten/Makrophagen. Ins besondere Konjugate von Antigen und
anti-FcγRI
zeigten, dass sie die Immunogenität zum Zwecke der Vakzinierung
erhöhen.
-
Andere
Möglichkeiten
umfassen die Verwendung von Targeting- und immunmodulierenden Substanzen
(unter anderem Cytokinen), die oben als Kandidaten für die ersten
und zweiten Gruppierungen in den modifizierten Analoga erwähnt sind.
In diesem Zusammenhang sind auch synthetische Induktoren von Cytokinen, wie
Poly-I:C eine Möglichkeit.
-
Geeignete
mycobakterielle Derivate werden aus der Gruppe ausgewählt, bestehend
aus Muramyldipeptid, vollständigem
Freund'schem Adjuvans,
RIBI und einem Diester aus Trehalose, wie TDM und TDE.
-
Geeignete
Immuntargeting-Adjuvantien werden aus der Gruppe ausgewählt, bestehend
aus CD40-Ligand und CD40-Antikörpern
oder deren spezifisch bindenden Fragmenten (vgl. die obige Diskussion),
Mannose, einem Fab-Fragment und CTLA-4.
-
Geeigeete
Polymer-Adjuvantien werden aus der Gruppe ausgewählt, bestehend aus Kohlenhydraten, wie
Dextran, PEG, Stärke,
Mannan und Mannose; einem Kunststoffpolymer; und Latex, wie beispielsweise
Latexkügelchen.
-
Noch
ein weiterer interessanter Weg zum Modulieren einer Immunantwort
ist es, das Immunogen (wahlweise zusammen mit den Adjuvantien und
pharmazeutisch annehmbaren Trägern
und Vehikeln) in einen "virtuellen
Lymphknoten" ("virtual lymph node", VLN) einzuschließen (ein
geschütztes
medizinisches Gerät, das
von ImmunoTherapy, Inc., 360 Lexington Avenue, New York, NY 10017-6501,
entwickelt wurde). Der VLN (ein dünnes röhrenartiges Gerät) ahmt
die Struktur und Funktion eines Lymphknotens nach. Das Einbringen eines
VLN unter die Haut ruft eine Stelle einer sterilen Entzündung mit
einem Anstieg von Cytokinen und Chemokinen hervor. T- und B-Zellen
sowie APCs reagieren sehr schnell auf die Gefahrensignale, wandern
in die entzündete
Stelle ein und reichern sich innerhalb der porösen Matrix des VLN an. Es wurde
gezeigt, dass die Antigendosis, die notwendig ist um eine Immunantwort
gegen ein Antigen hervorzurufen, vermindert ist, wenn der VLN verwendet
wird, und dass der Immunschutz, der durch Vakzinierung unter Verwendung
eines VLN verliehen wird, konventionelle Immunisierung unter Verwendung
von RIBI als Adjuvans übertrifft.
Die Technologie ist unter anderem kurz in Gelber C et al., 1998, "Elicitation of Robust
Cellular and Humoral Immune Responses to Small Amounts of Immunogens
Using a Novel Medical Device Designated the Virtual Lymph Node", in: "From the Laboratory
to the Clinic, Book of Abstracts, 12.-15. Oktober 1998, Seascape
Resort, Aptos, California" beschreiben.
-
Jüngste Ergebnisse
haben gezeigt, dass die gleichzeitige Verabreichung von H2-Agonisten das Überleben
von natürlichen
Killerzellen und CTLs innerhalb des Tumors erhöht. Daher wird auch vorgeschlagen, H2-Agonisten
als Adjuvantien in die erfindungsgemäßen Verfahren einzuführen.
-
Es
wird erwartet, dass das Vakzin mindestens einmal pro Jahr verabreicht
werden sollte, wie beispielsweise mindestens 1, 2, 3, 4, 5, 6 und
12 Mal pro Jahr. Insbesondere 1-12 Mal pro Jahr wird erwartet, wie
beispielsweise 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 oder 12 Mal pro
Jahr, an ein Individuum, das dies benötigt. Es wurde zuvor gezeigt,
dass die Gedächtnisimmunität, die durch
Verwendung der bevorzugten erfindungsgemäßen Autovakzine induziert wird,
nicht bleibend ist, und daher muss das Immunsystem periodisch mit
den Analoga konfrontiert werden.
-
Aufgrund
der genetischen Variation können
Individuen mit Immunantworten unterschiedlicher Stärke auf
dasselbe Polypeptid reagieren. Daher kann das erfindungsgemäße Vakzin
mehrere unterschiedliche Polypeptide umfassen um die Immunantwort
zu erhöhen,
vgl. ebenso die obige Diskussion, die die Auswahl der Einführungen
fremder T-Zell-Epitope betrifft. Das Vakzin kann zwei oder mehr
Polypeptide umfassen, wobei alle dieser Polypeptide solche sind,
wie sie oben beschrieben sind.
-
Das
Vakzin kann dementsprechend 3-20 unterschiedliche modifizierte oder
unmodifizierte Polypeptide umfassen, wie 3-10 unterschiedliche Polypeptide.
Jedoch wird normalerweise versucht, die Anzahl der Peptide auf einem
Minimum zu halten, wie 1 oder 2 Peptide.
-
Lebend-Vakzine
-
Die
zweite Alternative zum Bewirken der Präsentation gegenüber dem
Immunsystem ist die Anwendung der Lebend-Vakzintechnologie. Bei
der Lebend-Vakzinierung wird die Präsentation gegenüber dem
Immunsystem bewirkt, indem dem Tier ein nicht-pathogener Mikroorganismus
verabreicht wird, welcher mit einem Nukleinsäurefragment transformiert wurde,
das die notwendigen Epitop-Regionen oder ein vollständiges erstes
und/oder zweites Analogon codiert. Alternativ dazu wird der Mikroorganismus
mit einem Vektor transformiert, der ein solches Nukleinsäurefragment
umfasst. Der nicht-pathogene Mikroorganismus kann jeder geeignete
attenuierte Bakterienstamm sein (attenuiert mittels der Passagierung
oder mittels des Entfernens pathogener Expressionsprodukte durch
rekombinante DNA-Technologie), z.B. Mycobacterium bovis BCG., nicht-pathogene
Streptococcus spp., E. coli, Salmonella spp., Vibrio cholerae, Shigella,
etc. Übersichtsartikel, die
die Herstellung von Lebend-Vakzinen des Standes der Technik behandeln,
findet man z.B. in Saliou P, 1995, Rev. Prat. 45: 1492-1496, und
Walker PD, 1992, Vaccine 10: 977-990. Für Details über die Nukleinsäurefragmente
und Vektoren, die in solchen Lebend-Vakzinen verwendet werden, vgl.
die entenstehende Diskussion.
-
Wie
bei dem Polypeptid-Vakzin kann das TH-Epitop
und/oder das erste und/oder die erste und/oder zweite und/oder dritte
Gruppierungen, wenn sie vorhanden sind, in Form von Fusionspartnern
an die Aminosäuresequenz,
die sich von dem Zell-gebundenen Polypeptid-Antigen ableitet, vorliegen.
-
Als
Alternative zu bakteriellen Lebend-Vakzinen kann das unten diskutierte
erfindungsgemäße Nukleinsäurefragment
in einen nicht-virulenten viralen Vakzinvektor eingebaut sein. Eine
Möglichkeit
ist ein Pockenvirus, wie beispielsweise ein Vaccinia-Virus, MVA
(modifizierter Vaccinia-Virus), Kanarienpockenvirus, Vogelpockenvirus
und Hühnerpockenvirus,
etc. Alternativ dazu kann eine Herpes simplex-Virus-Variante verwendet werden.
-
Normalerweise
wird der nicht-pathogene Mikroorganismus oder Virus lediglich einmal
an das Tier verabreicht, aber in einigen Fällen kann es notwendig sein,
den Mikroorganismus mehr als einmal in einer Lebensdauer zu verabreichen.
-
Der
Mikroorganismus kann auch mit einer Nukleinsäure bzw. mit Nukleinsäuren transformiert
sein, die Regionen enthält
bzw. enthalten, die für
die 1., 2. und/oder 3. Gruppierungen) codiert/codieren, z.B. in
Form der oben beschriebenen immunmodulierenden Substanzen, wie den
Cytokinen, die als nützliche
Adjuvantien diskutiert wurden. Eine bevorzugte Version dieser Ausführungsform
umfasst das Vorhandensein der codierenden Region für das Analogon
und der codierenden Region für
den Immunmodulator in unterschiedlichen offenen Leserahmen oder
zumindest unter der Kontrolle unteschiedlicher Promotoren. Dadurch
wird es vermieden, dass das Analogon oder die Epitope als Fusionspartner
mit dem Immunmodulator hergestellt werden. Alternativ dazu können zwei
unterschiedliche Nukleotidfragmente als transformierende Agentien
verwendet werden.
-
Nukleinsäure-Vakzinierung
-
Als
Alternative zur klassischen Verabreichung eines Peptid-basierten
Vakzins bietet die Technologie der Nukleinsäure-Vakzinierung (auch als "Nukleinsäure-Immunisierung", "genetische Immunisierung", "Gen-Immunisierung" und "DNA-Vakzinierung" bekannt) eine Vielzahl
attraktiver Eigenschaften.
-
Als
erstes erfordert die Nukleinsäure-Vakzinierung,
im Gegensatz zum traditionellen Vakzinierungsansatz, keine Rohstoff
verbrauchende Produktion des immunogenen Mittels im großen Maßstab (z.B.
in Form der industriellen Fermentation von Mikroorganismen, die
die Analoga produzieren, was bei der Polypeptid-Vakzinierung erforderlich
ist). Außerdem
besteht kein Erfordernis, Reinigungs- und Faltungs-Schemata für das Immunogen
zu entwickeln. Und da schließlich
die Nukleinsäure-Vakzinierung
auf dem biochemischen Apparat des vakzinierten Individuums beruht
um das Expressionsprodukt der eingeführten Nukleinsäure zu produzieren,
wird erwartet, dass eine optimale posttranslationale Prozessierung
des Expressionsprodukts geschieht; dies ist besonders wichtig im
Falle der Autovakzinierung, da, wie oben erwähnt, ein signifikanter Teil
der Original-B-Zell-Epitope in den Analoga bewahrt werden soll,
die sich von extrazellulär
ausgesetzten Polypeptidsequenzen ableiten, und da B-Zell-Epitope
sich im Prinzip aus Teilen von irgendwelchen (Bio-)Molekülen (z.B. Kohlenhydraten,
Lipid, Protein, etc.) zusammensetzen können. Daher können native
Glykosylierungs- und Lipidmuster des Immunogens sehr wohl für die Gesamtimmunogenität von Bedeutung
sein, und diese werden am besten sichergestellt, indem man den Wirt
dazu veranlasst, das Immunogen herzustellen.
-
Daher
umfasst es eine wichtige Ausführungsform
des erfindungsgemäßen Verfahrens,
dass die Präsentation
bewirkt wird, indem in vivo mindestens ein Nukleinsäurefragment,
welches für
das mindestens eine CTL-Epitop und/oder das mindestens eine B-Zell-Epitop
und das mindestens eine erste fremde TH-Epitop
codiert und diese exprimiert, in die APC eingeführt wird (eine Alternative
umfasst die Verabreichung von mindestens zwei unterschiedlichen
Nuk leinsäurefragmenten,
wobei eine für
das mindestens eine CTL-Epitop und das andere für das mindestens eine fremde
TH-Epitop codiert). Bevorzugt wird dies
unternommen, indem ein Nukleinsäurefragment
verwendet wird, welches das oben diskutierte erste Analogon codiert.
Wenn das erste Analogon mit den oben detailliert ausgeführten TH-Epitopen und/oder ersten und/oder zweiten
und/oder dritten Gruppierungen ausgestattet ist, dann liegen diese
in Form von Fusionspartnern mit der Aminosäuresequenz vor, die sich von
dem Zell-gebundenen Polypeptid-Antigen
ableitet, wobei das Fusionskonstrukt von dem Nukleinsäurefragment
codiert wird.
-
Wie
bei dem traditionellen Vakzinierungsansatz kann die Nukleinsäure-Vakzinierung
mit der Einführung
in vivo von mindestens einem Nukleinsäurefragment, das das zweite
Analogon codiert und exprimiert in die APC, kombiniert werden. Die Überlegungen,
die die ersten, zweiten und dritten Gruppierungen und TH-Epitope
betreffen, treffen hier ebenfalls zu.
-
In
dieser Ausführungsform
ist die eingeführte
Nukleinsäure
bevorzugt DNA, welche in Form von nackter DNA, DNA formuliert mit
geladenen oder ungeladenen Lipiden, DNA formuliert in Liposomen,
DNA umfasst von einem viralen Vektor, DNA formuliert mit einem Transfektions-erleichternden
Protein oder Polypeptid, DNA formuliert mit einem Targeting-Protein oder -Polypeptid,
DNA formuliert mit Calcium-präzipitierenden
Agentien, DNA gekoppelt an ein inertes Trägermolekül und DNA formuliert mit einem
Adjuvans vorliegen kann. In diesem Zusammenhang wird festgestellt,
dass praktisch alle Überlegungen,
die die Verwendung von Adjuvantien in traditionellen Vakzinformulierungen
betreffen, für
die Formulierung von DNA-Vakzinen zutreffen. Daher treffen alle
hierin vorhandenen Offenbarungen, welche die Verwendung von Adjuvantien
im Zusammenhang mit Polypeptid-basierten Vakzinen betreffen, mutatis
mutandis auf deren Verwendung bei der Nukleinsäure-Vakzinierungstechnologie
zu. Gleiches gilt für
andere Überlegungen,
die sich auf die Formulierung und die Art und den Weg der Verabreichung
beziehen, und daher treffen auch diese Überlegungen, die im Zusammenhang
mit einem traditionellen Vakzin oben diskutiert werden, mutatis
mutandis auf deren Verwendung bei der Nukleinsäure-Vakzinierungstechnologie
zu.
-
Eine
besonders bevorzugte Art der Formulierung von Nukleinsäure-Vakzinen
sind Mikropartikel, die die DNA enthalten. Geeignete Mikropartikel
sind z.B. in der WO 98/31398 beschrieben.
-
Außerdem kann
bzw. können
die Nukleinsäure(n),
die als Immunisierungsmittel verwendet wird/werden, Regionen enthalten,
die für
die ersten, zweiten und/oder dritten Gruppierungen codieren, z.B.
in Form der immunmodulierenden Substanzen, die oben beschrieben
sind, wie die Cytokine, die als nützliche Adjuvantien diskutiert
werden. Eine bevorzugte Version dieser Ausführungsform umfasst es, dass
die codierende Region für
das Analogon und die codierende Region für den Immunmodulator in unterschiedlichen
offenen Leserahmen oder zumindest unter der Kontrolle unterschiedlicher
Promotoren enthalten sind. Dabei wird es vermieden, dass das Analogon
oder Epitop als Fusionspartner des Immunmodulators hergestellt wird.
Alternativ dazu können
zwei unterschiedliche Nukleotidfragmente verwendet werden, aber
dieses ist aufgrund des Vorteils der sicheren Co-Expression, wenn
beide codierenden Regionen umfasst von demselben Molekül vorliegen,
weniger bevorzugt.
-
Unter
normalen Umständen
wird die Nukleinsäure
des Vakzins in Form eines Vektors eingeführt, wobei die Expression unter
der Kontrolle eines viralen Promotors steht. Für eine detailliertere Diskussion
der erfindungsgemäßen Vektoren
vgl. die entenstehende Diskussion. Ebenso sind detaillierte Offenbarungen
in Bezug auf die Formulierung und Verwendung von Nukleinsäure-Vakzinen
verfügbar,
vgl. Donnelly JJ et al., 1997, Annu. Rev. Immunol. 15: 617-648, und Donnelly
JJ et al., 1997, Life Sciences 60: 163-172. Auf diese beiden Bezugsstellen
wird expressis verbis Bezug genommen.
-
Ein
wichtiger Teil der Erfindung betrifft ein neues Verfahren zum Selektieren
eines geeigneten immunogenen Analogons eines Zell-gebundenen Polypeptid-Antigens,
welches in einem Tier schwach immunogen oder nicht-immunogen ist,
wobei dieses immunogene Analogon in der Lage ist, eine CTL-Antwort
in dem Tier gegen Zellen zu induzieren, die ein Epitop, das von
dem Zell-gebundenen Polypeptid-Antigen abgeleitet ist, gebunden
an ein MHC-Klasse I-Molekül
aufweisen. Dieses Verfahren umfasst die Schritte
- a)
Identifizieren von mindestens einer Untersequenz der Aminosäuresequenz
des Zell-gebundenen
Polypeptid-Antigens, welches keine bekannten oder vorhergesagten
CTL-Epitope enthält,
- b) Herstellen von mindestens einem mutmaßlich immunogenen Analogon
des Zell-gebundenen
Polypeptid-Antigens, indem in die Aminosäuresequenz des Zell-gebundenen Polypeptid-Antigens
mindestens ein TH-Epitop, das für das Tier
fremd ist, an einer Position innerhalb der mindestens einen in Schritt
a) identifizierten Untersequenz eingefügt wird, und
- c) Auswahl des Analogons/der Analoga, die in Schritt b) hergestellt
wurden, welche verifizierbar in der Lage sind, eine CTL-Reaktion
in dem Tier zu induzieren.
-
Alternativ
dazu umfasst das Auswahlverfahren die Herstellung eines Nukleinsäurefragments
zum Zwecke der Nukleinsäurevakzinierung.
In dieser Situation ist es erforderlich, dass das codierte Peptid
mindestens ein TH-Epitop umfasst.
-
Wenn
das Analogon von einem Antigen abgeleitet ist, welches dem Extrazellulärraum ausgesetzt
ist, ist es bevorzugt, dass die in Schritt a) identifizierte Untersequenz
außerdem
keine Cystein-Reste enthält,
oder, alternativ dazu, dass das TH-Epitop,
das in Schritt b) eingefügt
wird, das Muster der Cystein-Reste nicht wesentlich verändert. Dieser
Ansatz erleichtert das Bewahren von Teil-B-Zell-Epitopen in dem
daraus hervorgehenden Konstrukt, welche den B-Zell-Epitopen in dem schwachen, Zell-gebundenen
Polypeptid-Antigen ähnlich
sind.
-
Aus
denselben Gründen
ist es bevorzugt, dass die in Schritt a) identifizierte Untersequenz
außerdem keine
bekannten oder vorhergesagten Glykosylierungsstellen enthält, oder,
alternativ dazu, eine Ausführungsform,
in der das TH-Epitop, das in Schritt b)
eingefügt
wird, im Wesentlichen das Glykosylierungsmuster nicht verändert.
-
Einige
der schwachen, Zell-gebundenen Polypeptid-Antigene üben ungewünschte Wirkungen
aus, indem sie eine pathophysiologische Rolle spielen. Es ist wünschenswert,
dass diese Wirkungen von den Vakzinierungskonstrukten nicht ausgeübt werden,
und daher ist es bevorzugt, dass die in Schritt a) identifizierte
Untersequenz signifikant zu einer pathophysiologischen Wirkung beiträgt, die
von dem Zell-gebundenen Polypeptid-Antigen ausgeübt wird, und dass die Einführung des
fremden TH-Epitops in Schritt b) diese pathophysiologische
Wirkung reduziert oder ausschaltet. Ein Beispiel für diesen
Ansatz ist es, die aktive Stelle in einem Enzym, Hormon oder Cytokin
zu entfernen und diese gegen das fremde TH-Epitop
auszutauschen.
-
Eine
andere wichtige Überlegung
betrifft die Frage der immunologischen Kreuzreaktivität des Polypeptidprodukts
des Vakzins mit anderen Selbst-Proteinen, welche mit pathologischen
Prozessen nicht in Verbindung stehen. Solch eine Kreuzreaktivität sollte
bevorzugt vermieden werden, und daher ist eine wichtige Ausführungsform
des erfindungsgemäßen Verfahrens
eine solche, bei der die Untersequenz, die in Schritt a) identifiziert
wird, zu einer Aminosäuresequenz
eines anderen Protein-Antigens des Tieres homolog ist, und bei der die
Einführung
des TH-Epitops in Schritt b) im Wesentlichen
die Homologie entfernt.
-
Diese
Ausführungsform
ist einer Ausführungsform ähnlich,
bei der jedwede Aminosäuresequenzen, welche
1) der extrazellulären
Phase nicht ausgesetzt sind, und 2) welche B-Zell-Epitope des schwachen, Zell-assoziierten
Polypeptid-Antigens darstellen könnten,
nicht bewahrt werden. Dies kann erreicht werden, indem solche Aminosäuresequenzen
mit TH-Epitopen,
welche keine B-Zell-Epitope darstellen, ausgetauscht werden, indem
man sie vollständig
entfernt, oder indem man sie teilweise entfernt.
-
Andererseits
ist es bevorzugt, dass alle "echten" B-Zell-Epitope des
schwachen Zell-gebundenen
Polypeptid-Antigens zu einem hohen Grad bewahrt werden, und daher
umfasst eine wichtige Ausführungsform des
erfindungsgemäßen Auswahlverfahrens,
das das Einführen
des fremden TH-Epitops in Schritt B zu dem Erhalt
eines wesentlichen Teils der B-Zell-Epitope des Zell-gebundenen
Polypeptid-Antigens führt.
Es ist insbesondere bevorzugt, dass das Analogon die Gesamttertiärstruktur
des Zell-gebundenen Polypeptid-Antigens behält.
-
Die
Präparation
in Schritt b) wird bevorzugt durch molekularbiologische Methoden
oder mittels Festphasen- oder Flüssigphasen-Peptidsynthese
erreicht. Kurze Peptide werden bevorzugt mittels der wohlbekannten
Techniken der Festphasen- oder Flüssigphasen-Peptidsynthese präpariert.
Jüngste
Fortschritte in dieser Technologie haben jedoch die Herstellung
von Volllängen-Polypeptiden
und Proteinen mittels dieser Verfahren möglich gemacht, und daher liegt
es ebenso im Umfang der vorliegenden Erfindung, die langen Konstrukte
durch synthetische Verfahren herzustellen.
-
Nachdem
die nützlichen
Analoga entsprechend dem oben diskutierten Verfahren identifiziert
wurden, ist es notwendig, die Analoga in größerem Maßstab herzustellen. Die Polypeptide
werden nach Verfahren hergestellt, die im Stand der Technik wohlbekannt
sind.
-
Dies
kann durch molekularbiologische Verfahren erreicht werden, die es
in einem ersten Schritt umfassen, eine transformierte Zelle zu präparieren,
indem eine Nukleinsäuresequenz,
die für
ein Analogon codiert, welches entsprechend dem Verfahren ausgewählt wurde,
in einen Vektor eingefügt
wird, und indem ein geeigneter Wirt mit dem Vektor transformiert
wird. Der nächste
Schritt ist es, die transformierte Zelle unter Bedingungen zu kultivieren,
welche die Expression des Nukleinsäurefragments, das für das Analogon
des Zell-gebundenen Antigens codiert, erleichtern, und anschließend das
Analogon aus dem Kulturüberstand
oder direkt aus den Zellen wiederzugewinnen, z.B. in Form eines
Lysats. Alternativ dazu kann das Analogon im großen Maßstab mittels Festphasen- oder
Flüssigphasen-Peptidsynthese
hergestellt werden, vgl. oben.
-
Letztlich
kann das Produkt, abhängig
von der als Wirt ausgesuchten Zelle oder dem verwendeten Syntheseverfahren,
künstlichen
posttranslationalen Modifikationen unterzogen werden. Diese können Rückfaltungsschemata
sein, die im Stand der Technik bekannt sind, Behandlung mit Enzymen
(um die Glykosylierung zu erreichen oder ungewünschte Fusionspartner zu entfernen),
chemische Modifikationen (wiederum ist Glykosylierung eine Möglichkeit)
und Konjugation, z.B. an traditionelle Trägermoleküle.
-
Es
sollte erwähnt
werden, dass erfindungsgemäß bevorzugte
Analoga (und auch die relevanten Analoga, die in den erfindungsgemäßen Verfahren
verwendet werden), Modifikationen umfassen, welche zu einem Polypeptid
führen,
das eine Sequenzidentität
von mindestens 70% mit dem Polypeptid-Antigen oder mit einer Teilsequenz
davon von mindestens 10 Aminosäuren
Länge aufweist.
Höhere
Sequenzidentitäten
sind bevorzugt, z.B. mindestens 75% oder sogar mindestens 80% oder
85%. Die Sequenzidentität
für Proteine
und Nukleinsäuren
können
errechnet werden als (Nref-(Ndif)·100/Nref, wobei Ndif die
Gesamtanzahl der nicht-identischen Reste in den zwei Sequenzen ist,
wenn sie aneinander angelagert werden, und wobei Nref die
Anzahl der Reste in einer der Sequenzen ist. Somit wird die DNA-Sequenz
AGTCAGTC eine Sequenzidentität
von 75% mit der Sequenz AATCAATC aufweisen (Ndif =
2 und Nref = 8).
-
Spezifische
beispielhafte Zielobjekte für
die Erfindung
-
Wie
oben diskutiert, sind bevorzugte schwache, Zell-gebundene Polypeptid-Antigene
Tumor-assoziierte Antigene. Eine nicht-einschränkende Liste von diesen ist
in der folgenden Tabelle angegeben.
| Antigen | Literaturstelle |
| 5 alpha
reductase | Délos 5,
Carsol JL, Fina F, Raynaud JP, Martin PM. 5alpha-reductase and 17beta-hydroxysteroid
dehydrogenase expression in epithelial cells from hyperplastic and
malignant human prostate. Int J Cancer 1998 Mar 16 75:6 890-6 |
| α-fetoprotein | Esteban
C, Terrier P, Frayssinet C, Uriel J. Expression of the alpha-fetoprotein
gene in human breast cancer. Tumour Biol 1996 17:5 299-305 |
| AM-1 | Harada
Y, Ohuchi N, Masuko T, Funaki Y, Mori S, Satomi S, Hashimoto Y.
Characterization of a new breast cancer-associated antigen and its
relationship to MUC1 and TAG-72 antigens. Tohoku J Exp Med 1996 Nov
180:3 273-88 |
| APC | Dihlmann
S, Amler LC, Schwab M, Wenzal A. Variations in the expression of
the adenomatous polyposis coli (APC) tumor suppressor gene in human
cancer cell lines of different tissue origin. Oncol Res 1997 9:3 119-27 |
| APRIL | LE,
Sordat B, Rimoldi D, Tschopp J. APRIL, a new ligand of the tumor
necrosis factor family, stimulates tumor cell growth. J Exp Med
1998 Sep 21 188:6 1185-90 |
| BAGE | Böel P, Wildmann
C, Sensi ML, Brasseur R, Renauld J-C, Coulie P, Hoon T, and Van
der Hruggen P. BAGE: a new gene encoding an antigen recognized on
human melanomas by cytolytic lymphocytes. Immunity 1995, 2: 167-175. |
| β-catenin | Hugh
TJ, Dillon SA, O'Dowd
G, Getty B, Pignatelli M, Poston GJ, Kinsella AR. beta-catenin expression
in primary and metastatic colorectal carcinoma. Int J Cancer 1999
Aug 12 82:4 509-11 |
| Bc12 | Koty
PP, Zhang N, Levitt ML. Antisense bcl-2 treatment increases programmed
cell death in non-small cell lung cancer cell lines. Lung Cancer
1999 Feb 23:2 115-27 |
| bcr-abl
(b3a2) | Verfaillie
CM, Bhatia R, Miller W, Mortari F, Roy V, Burger S, McCullough J,
Stieglbauer K, Dewald G, Heimfeld S, Miller JS, McGlave PH. BCR/ABL-negative
primitive progenitors suitable for transplantation can be selected
from the marrow of most early-chronic phase but not accelerated-phase
chronic myelogenous leukemia patients. Blood 1996 Jun 1 87:11 9770-9 |
| Antigen | Literaturstelle |
| CA-125 | Bast
RC Jr, Xu FJ, Yu YH, Barnhill S, Zhang 2, Mills GB. CA 125: the
past and the future. Int J Biol Markers 1998 Oct-Dec 13:4 179-87 |
| CASP-8/FLICE | Mandruzzato
S, Brasseur F, Andry G, Boon T, van der Bruggen P., A CASP-8 mutation
recognized by cytolytic T lymphocytes on a human head and neck carcinoma.
J Exp Med 1997 Aug 29 186:5 785-93. |
| Cathepsins | Thomssen
C, Schmitt M, Goretzki L, Oppelt P, Pache L, Dettmar P, Jänicke F,
Graeff H. Prognostic value of the cysteine proteases cathepsins
B and cathepsin L in human breast cancer. Clin Cancer Res 1995 Jul
1:7 791-6 |
| CD19 | Scheuermann
RH, Racila F. CD19 antigen in leukemia and lymphoma diagnosis and
immunotherapy. Leuk Lymphoma 1995 Aug 18:5-6 385-97 |
| CD20 | Knox
SJ, Goris ML, Trisler K, Negrin R, Davis T, Liles TM, Grillo-López A,
Chinn P, Varns C, Ning SC, Fowler S, Deb N, Becker M, Marquez C,
Levy R. Yttrium-90-labeled
anti-CD20 monoclonal antibody therapy of recurrent B-cell lymphoma.
Clin Cancer Res 1996 Mar 2:3 957-70 |
| CD21 | Shubinsky
G, Schlesinger M, Polliack A, Rabinowitz R. Pathways controlling
the expression of surface CD21 (CR2) and CD23 (Fc(epsilon)IIR) proteins
in human malignant B cells. Leuk Lymphoma 1997 May 25:5-6 521-30 |
| CD23 | Shubinsky
G, Schlesinger M, Polliack A, Rabinowitz R. Pathways controlling
the expression of surface CD21 (CR2) and CD23 (Fc(epsilon)IIR) proteins
in human malignant B cells. Leuk Lymphoma 1997 May 25: 5-6 521-30 |
| CD22 | French
RR, Penney CA, Browning AC, Stirpe F, George AJ, Glennie MJ. Delivery
of the ribosome-inactivating protein, gelonin, to lymphoma cells
via CD22 and CD38 using bispecific antibodies. Br J Cancer 1995 May
71:5 986-94 |
| CD33 | Nakase
K, Kita K, Shiku N, Tanaka I, Nasu K, Dohy N, Kyo T, Tsutani H,
Kamada N. Myeloid antigen, CD13, CD19, and/or CD33 expression is
restricted to certain lymphoid neoplasms. Am J Clin Pathol 1996
Jun 105:6 761-8 |
| CD35 | Yamakawa
M, Yamada K, Tsuge T, Ohrui H, Ogata T, Dobashi M, Imai Y. Protection
of thyroid cancer cells by complement-regulatory factors. Cancer
1994 Jun 1 73:11 2808-17 |
| Antigen | Literaturstelle |
| CD44 | Naot
D, Sionov RV, Ish-Shalom D. CD44: structure, function, and association
with the malignant process. Adv Cancer Res 1997 71: 241-319 |
| CD45 | Buzzi
M, Lu L, Lombardi AJ Jr, Posner MR, Brautigan DL, Fast LD, Frackelton
AR Jr. Differentiation-induced changes in protein-tyrosine phosphatase
activity and commensurate expression of CD45 in human leukemia cell
lines. Cancer Res 1992 Jul 15 52:19 4027-35 |
| CD46 | Yamakawa
M, Yamada K, Tsuge T, Ohrui H, Ogata T, Dobashi M, Imai Y. Protection
of thyroid cancer cells by complement-regulatory factors. Cancer
1999 Jun 1 73:11 2808–17 |
| CD5 | Stein
R, Witz IP, Ovadia J, Goldenberg DM, Yron I. CD5+ B cells and naturally
occurring autoantibodies in cancer patients. Clin Exp Immunol 1991
Sep 85:3 418-23 |
| CD52 | Ginaldi
L, De Martinis M, Matutes E, Farahat N, Morilla R, Dyer MJ, Catovsky
D. Levels of expression of CD52 in normal and leukemic B and T cells:
correlation with in vivo therapeutic responses to Campath-1H. Leuk
Res 1998 Feb 22:2 185-91 |
| CD55
(791Tgp72) | Spendlove,
I, L. Li, J. Carmichael, & L.
G. Durrant. Decay accelerating factor (CD55): A target for cancer vaccine?
(1999) Cancer Research 59: 2282-2286. |
| CD59 | Jarvis
GA, Li J, Hakulinen J, Brady KA, Nordling S, Dahiya R, Meri S. Expression
and function of the complement membrane attack complex inhibitor
protectin (CD59) in human prostate cancer. Int J Cancer 1997 Jun
11 71: 6 1049-55 |
| CDC27 | Wang
RF, Wang X, Atwood AC, Topalian SL, Rosenberg SA. Cloning genes
encoding MHC class II-restricted antigens: mutated CDC27 as a tumor
antigen. Science 1999 May 21 289: 5918 1351-9 |
| CDK4 | Wölfel, T.,
Hauer, M., Schneider, J., Serrano, M, Wölfel, C., Klehmann-Hieb, E.,
de Plaen, E., Hankeln, T., Meyer zum Büüschenfelde, K, and Beach, D.
A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes
in a human melanoma. Science 1995 Sep 1 269: 5228 1281-9 |
| CEA | Kass
E, Schlom J, Thompson J, Guadagni F, Graziano P, Greiner JW. Induction
of protective host immunity to carcinoembryonic antigen (CEA), a
self-antigen in CEA transgenic mice, by immunizing with a recombinant
vaccinia-CEA virus. Cancer Res 1999 Feb 1 59: 3 676-83 |
| c-myc | Watson
PH, Pon RT, Shiu RP. Inhibition of c-myc expres-sion by phosphorothioate antisense oligonucleotide
identifies a critical role for c-myc in the growth of human breast
cancer. Cancer Res 1991 Aug 1 51: 15 3996-9000 |
| Antigen | Literaturstelle |
| Cox-2 | Tsujii
M et al, Cycloxygenase regulates angiogenesis induced by colon cancer
cells. Cell 1998; 93: 705-716 |
| DCC | Gotley
DC, Reeder JA, Fawcett J, Walsh MD, Bates P, Simmons DL, Antalis
TM. The deleted in colon cancer (DCC) gene is consistently expressed
in colorectal cancers and metastases. Oncogene 1996 Aug 15 13: 9
787-95 |
| DcR3 | Genomic
ampliphication of a decoy receptor for Fas ligand in lung and colon
cancer. Pitti R et al, Nature 396, 699-703. 1998. |
| E6/E7 | Steller
MA, Zou Z, Schiller JT, Baserga R. Transformation by human papillomavirus
16 E6 and E7: role of the insulin-like growth factor 1 receptor.
Cancer Res 1996 Nov 1 56: 21 5087-91 |
| EGFR | Yang
XD, Jia XC, Corvalan JR, Wang P, Davis CG, Jakobovits A: Eradication
of established tumors by a fully human monoclonal antibody to the
epidermal growth factor receptor without concomitant chemotherapy. Cancer
Res 1999, 59(6): 1236-43. |
| EMBP | Clinical
study on estramustine binding protein (EMBP) in human prostate.
Shiina H, Igawa M, Ishibe T. Prostate 1996 Sep 29: 3 169-76. |
| Ena78 | D.A.
Arenberg et. al., Epithelial-neutrophil activating peptide (FNA-78)
is an important angiogenic factor in non-small cell lung cancer.
J. Clin. Invest. (1998) 102; 965-472. |
| farsyl
transferase FGF8b and FGF8a | Dorkin
TJ, Robinson MC, Marsh C, Bjartell A, Neal DE, Leung HY. FGF8 over-expression
in prostate cancer is associated with decreased patient survival
and persists in androgen independent disease. Oncogene 1999 Apr
29 18: 17 2755-61 |
| FLK-1/KDR | T.
Annie T. Fong et al, SU5416 is a potent and selective inhibitor
of the vascular endothelial growth factor receptor (Flk-1/KDR) that
inhibits tyrosine kinase catalysis, tumor vacularization and growth
of multiple tumor type. Cancer Res, 59, 99-106, 1999 |
| Folic
Acid Receptor | Dixon
KH, Mulligan T, Chung KN, Elwood PC, Cowan KH. Effects of folate
receptor expression following stable transfection into wild type
and methotrexate transportdeficient ZR-75-1 human breast cancer
cells. J Biol Chem 1992 Nov 25 267:33 29140-7 |
| Antigen | Literaturstelle |
| G250 | Divgi
CR, Bander NH, Scott AM, O'Donoghue
JA, Sgouros G, Welt S, Finn RD, Morrissey F, Capitelli P, Williams
JM, Deland D, Nakhre A, Oosterwijk E, Gulec S, Graham MC, Larson
SM, Old LJ. Phase I/II radioimmunotherapy trial with iodine-131-labeled
monoclonal antibody G250 in metastatic renal cell carcinoma. Clin
Cancer Res 1998 Nov 4:11 2729-39 |
| GAGE-Family | De
Backer O, 10 others, Boon T, van der Hruggen P. Characterization
of the GAGE genes that are expressed in various human cancers and
in normal testis. Cancer Res 1999 Jul 1;59(13): 3157-3165. |
| gastrin
17 | Watson
SA, Michaeli D, Grimes S, Morris TM, Crosbee D, Wilkinson M, Robinson
G, Robertson JF, Steele RJ, Hardcastle JD. Anti-gastrin antibodies
raised by gastrimmune inhibit growth of the human colorectal tumour
AP5. Int J Cancer 1995 Apr 10 61:2 233-40 |
| Gastrin-releasing
hormone (Bombesin) | Wang
QJ, Knezetic JA, Schally AV, Pour PM, Adrian TE. Bombesin may stimulate
proliferation of human pancreatic cancer cells through an autocrine
pathway. Int J Cancer 1996 Nov 15 68: 9 528-34 |
| GD2/GD3/GM2 | Wiesner
DA, Sweeley CC. Circulating gangliosides of breast-cancer patients.
Int J Cancer 1995 Jan 27 60:3 294-9 |
| GnRH | Bahk
JY, Hyun JS, Lee H, Kirn MO, Cho GJ, Lee BH, Choi WS. Expression
of gonadotropin-releasing hormone (GnRH) and GnRH receptor mRNA
in prostate cancer cells and effect of GnRH on the proliferation
of prostate cancer cells. Urol Res 1998 26: 4 259-69 |
| GnTV | Hengstler
JG, Arand M, Herrero ME, Oesch F. Polymorphisms of N-acetyltransferases,
glutathione S-transferases,
microsomal epoxide hydrolase and sulfotransferases: influence on
cancer susceptibility. Recent Results Cancer Res 1998 154: 47-85 |
| GP1
gp100/Pmel 17 | Wagner
SN, Wagner C, Schultewolter T, Goos M. Analysis of Pme117/gp100
expression in primary human tissue specimens: implications for melanoma
immuno- and genetherapy. Cancer Immunol Immunother 1997 Jun 44:
4 239-47 |
| gp-100-in4 | Kirkin
AF, Dzhandzhugazyan K, Zeuthen J. Melanomaassociated antigens recognized
by cytotoxic T lymphocytes. APMIS 1998 Jul 106:7 665-79 |
| Antigen | Literaturstelle |
| gp15 | Maeurer
MJ, et al. New treatment options for patients with melanoma: review
of melanoma-derived T-cell epitope-based peptide vaccines. Melanoma
Res. 1996 Feb; 6(1): 11-24. |
| gp75/TRP-1 | Lewis
JJ, Houghton AN. Definition of tumor antigens suitable for vaccine
construction. Semin Cancer Biol 1995 Dec 6:6 321-7 |
| hCG | Hoermann
R, Gerbes AL, Spoettl G, Jüüngst D,
Mann K. Immunoreactive human chorionic gonadotropin and its free
beta subunit in serum and ascites of patients with malignant tumors.
Cancer Res 1992 Mar 15 52:6 1520-4 |
| Heparanase | Vlodavsky
I, Friedmann Y, Elkin M, Aingorn H, Atzmon R, Ishai-Michaeli R,
Bitan M, Pappo O, Peretz T, Michal I, Spector L, Pecker I. Mammalian
heparanase: gene cloning, expression and function in tumor progression
and metastasis [see comments). Nat Med 1999 Jul 5:7 793-802 |
| Her2/neu | Lewis
JJ, Houghton AN. Definition of tumor antigens suitable for vaccine
construction. Semin Cancer Biol 1995 Dec 6:6 321-7 |
| HMTV | Kahl
LP, Carroll AR, Rhodes P, Wood J, Read NG. An evaluation of the
putative human mammary tumour retrovirus associated with peripheral
blood monocytes. Br J Cancer 1991 Apr 63: 4 534-40 |
| Hsp70 | Jaattela
M, et al. Hsp70 exerts its anti-apoptotic function downstream of
caspase-3-like proteases. EMBO J. 1998 Nov 2; 17(21): 6124-34. |
| hTERT
(telomerase) | Vonderheide
RH, Hahn W.C, Schultze JL, Nadler LM. The telomerase catalytic subunit
is a widely expressed tumor-associated antigen recognized by cytotoxic
T lymphocytes. Immunity June; 10: 673-679. 1999. |
| IGFR1 | M.J.
Ellis et. al., Insulin-like growth factors in human breast cancer.
Breast Cancer Res. Treat. (1998) 52; 175-184. |
| IL-13R | Murata
T, Obiri NI, Debinski W, Puri RK. Structure of IL-13 receptor: analysis
of subunit composition in cancer and immune cells. Biochem Biophys
Res Commun 1997 Sep 8 238:1 90-4 |
| iNOS | Klotz
T, Bloch W, Volberg C, Engelmann U, Addicks K. Selective expression
of inducible nitric oxide synthase in human prostate carcinoma.
Cancer 1998 May 15 82: 10 1897-903 |
| Ki
67 | Gerdes,
J., U. Schwab, H. Lemke, and H. Stein. Production of a mouse monoclonal
antibody reactive with a human nuclear antigen associated with cell
prolifera-tion.
Int J Cancer 31, 13-20, 1983 |
| Antigen | Literaturstelle |
| KIAA0205 | Gueguen
M, Patard JJ, Gaugler B, Hrasseur F, Renauld JC, Van Cangh PJ, Boon
T, Van den Eynde BJ. An antigen recognized by autologous CTLs on
a human bladder carcinoma. J Immunol 1998 Jun 15 160:12 6188-94 |
| K-ras,
H-ras, N-ras | Abrams
SI, Hand PH, Tsang KY, Schlom J. Mutant ras epitopes as targets
for cancer vaccines. Semin Oncol 1996 Feb 23:1 118-34 |
| KSA
(C017-1A) | Zhang
S, Zhang HS, Reuter VE, Slovin SF, Scher HI, Livingston PO. Expression
of potential target antigens for immunotherapy on primary and metastatic
prostate cancers. Clin Cancer Res 1998 Feb 4:2 295-302 |
| LDLR-FUT | Caruso
MG, Osella AR, Notarnicola M, Berloco P, Leo S, Bonfiglio C, Di
Leo A. Prognostic value of low density lipoprotein receptor expression
in colorectal carcinoma. Oncol Rep 1998 Jul-Aug 5:4 927-30 |
| MAGE
Family (MAGE1, MAGE3) | Marchand
M, et al., Tumor regressions observed in patients with metastatic
melanoma treated with an antigenic peptide encoded by gene MAGE-3
and presented by HLA-A1. Int J Cancer 1999 Jan 18;80(2): 219-30. |
| Mammaglobin | Watson
MA, Dintzis S, Darrow CM, Voss LE, DiPersio J, Jensen R, Fleming
TP, Mammaglobin expression in primary, metastatic, and occult breast
cancer. Cancer Res 1999 Jul 1 59:13 3028-31. |
| MAP17 | Kocher
O, Cheresh P, Lee SW. Identification and partial characterization
of a novel membrane-associated protein (MAP17) up-regulated in human
carcinomas and modulating cell replication and tumor growth. Am J
Pathol 1996 Aug 149:2 493-500 |
| Melan-A/MART-1 | Lewis
JJ, Houghton AN. Definition of tumor antigens suitable for vaccine
construction. Semin Cancer Biol 1995 Dec 6:6 321-7 |
| mesothelin | Chang
K, et al. Molecular cloning of mesothelin, a differentiation antigen
present on mesothelium, mesotheliomas, and ovarian cancers. Proc
Natl Acad Sci U S A. 1996 Jan 9;93(1): 136-40 |
| MIC
A/B | Groh
V, Steinle A, Bauer S, and Spies T. Recognition of stress-induced
MHC molecules by intestinal epithelial gammadelta T cells. Science
1998, 279: 1737-1740. |
| MT-MMP's, such as MMP2,
MMP3, MMP7, and MMP9 | Sato
H and Seiki M., Membrane-type matrix metalloproteinases (MT-MMPs)
in tumormetastasis., J Biochem (Tokyo) 1996 Feb; 119(2): 209-15 |
| Mox1 | Candia
AF, Hu J, Crosby J, Lalley PA, Noden D, Nadeau JH, Wright CV. Mox-1
and Mox-2 define a novel homeobox gene subfamily and are differentially
expressed during early mesodermal patterning in mouse embryos. Development
1992 Dec 116:9 1123-36 |
| Antigen | Literaturstelle |
| Mucin,
such as MUC-1, MUC-2,
MUC-3, and MUC-9 | Lewis
JJ, Houghton AN. Definition of tumor antigens suitable for vaccine
construction. Semin Cancer Biol 1995 Dec 6:6 321-7 |
| MUM-1 | Kirkin
AF, Dzhandzhugazyan K, Zeuthen J. Melanomaassociated antigens recognized
by cytotoxic T lymphocytes. APMIS 1998 Jul 106:7 665-79 |
| NY-ESO-1 | Jager,
E. et. al. Simultaneous humoral and cellular immune response against
cancer-testis antigen NY-ESO-1: definition of human histocompatibility
leukocyte antigen (HLA)-A2-binding peptide epitopes. J. Exp. Med.
1998, 187: 265-270. |
| Osteonectin | Graham
JD, Balleine RL, Milliken JS, Bilous AM, Clarke CL. Expression of
osteonectin mRNA in human breast tumours is inversely correlated
with oestrogen receptor content. Eur J Cancer 1997 Sep 33:10 1659-60 |
| p15 | Yoshida
S, Todoroki T, Ichikawa Y, Hanai S, Suzuki H, Hori M, Fukao K, Miwa
M, Uchida K. Mutations of p16Ink4/CDKN2 and p15Ink4B/MTS2 genes
in biliary tract cancers. Cancer Res 1995 Jul 1 55:13 2756-60 |
| P170/MDR1 | Trock
BJ, Leonessa F, Clarke R. Multidrug resistance in breast cancer:
a meta-analysis of MDR1/gp170 expression and its possible functional
significance. J Natl Cancer Inst 1997 Jul 2 89:13 917-31 |
| p53 | Roth
J, Dittmer D, Rea D, Tartaglia J, Paoletti E, Levine AJ. p53 as
a target for cancer vaccines: recombinant canarypox virus vectors
expressing p53 protect mice against lethal tumor cell challenge.
Proc Natl Acad Sci U S A 1996 May 19 93:10 4781-6. |
| p97/melanotrans-ferrin | Furukawa
KS, Furukawa K, Real FX, Old LJ, Lloyd KO. A unique antigenic epitope
of human melanoma is carried on the common melanoma glycoprotein gp95/p97.
J Exp Med 1989 Feb 1 169:2 585-90 |
| PAI-1 | Grondahl-Hansen
J, Christensen IJ, Rosenquist C, Brünner N, Mouridsen HT, Danø K, Blichert-Toft
M. High levels of urokinase-type plasminogen activator and its inhibitor
PAI-1 in cytosolic extracts of breast carcinomas are associated
with poor prognosis. Cancer Res 1993 Jun 1 53:11 2513-21 |
| PDGF | Vassbotn
FS, Andersson M, Westermark H, Heldin CH, Ostman A. Reversion of
autocrine transformation by a dominant negative platelet-derived
growth factor mutant. Mol Cell 8io1 1993 Jul 13:7 9066-76 |
| Antigen | Literaturstelle |
| Plasminogen
(uPA) | Naitoh
H, Eguchi Y, Ueyama H, Kodama M, Hattori T. Localization of urokinase-type
plasminogen activator, plasminogen activator inhibitor-1, 2 and
plasminogen in colon cancer. Jpn J Cancer Res 1995 Jan 86:1 98-56 |
| PRAME | Kirkin
AF, Dzhandzhugazyan K, Zeuthen J. Melanomaassociated antigens recognized
by cytotoxic T lymphocytes. APMIS 1998 Jul 106:7 665-79 |
| Probasin | Matuo
Y, Nishi N, Muguruma Y, Yoshitake Y, Kurata N, Wada F. Localization
of prostatic basic protein ("probasin") in the rat prostates
by use of monoclonal antibody. Biochem Biophys Res Commun 1985 Jul
16 130:1 293-300 |
| Progenipoietin
PSA | Sanda
MG, Smith DC, Charles LG, Hwang C, Pienta KJ, Schlom J, Milenic
D, Panicali D, Montie JE. Recombinant vaccinia-PSA (PROSTVAC) can
induce a prostate-specific immune response in androgen-modulated
human prostate cancer. Urology 1999 Feb 53:2 260-6. |
| PSM | Kawakami
M, Nakayama J. Enhanced expression of prostate-specific membrane
antigen gene in prostate cancer as revealed by in situ hybridization.
Cancer Res 1997 Jun 15 57:12 2321-4 |
| RAGE-1 | Gaugler
B, 7 others, Van den Eynde BJ. A new gene coding for an antigen
recognized by autologous cytolytic T lymphocytes on a human renal
carcinoma. Immunogenetics 1996; 99(5): 323-330. |
| Rb | Dosaka-Akita
H, Hu SX, Fujino M, Harada M, Kinoshita I, Xu HJ, Kuzumaki N, Kawakami
Y, Benedict WF. Altered retinoblastoma protein expression in nonsmall cell
lung cancer: its synergistic effects with altered ras and p53 protein
status on prognosis. Cancer 1997 Apr 1 79:7 1329-37 |
| RCAS1 | Sonoda
K, Nakashima M, Kaku T, Kamura T, Nakano N, Watanabe T. A novel
tumor-associated antigen expressed in human uterine and ovarian
carcinomas. Cancer 1996 Apr 15; 77(8): 1501-1509. |
| SART-1 | Kikuchi
M, Nakao M, Inoue Y, Matsunaga K, Shichijo S, Yamana H, Itoh K.
Identification of a SART-1-derived peptide capable of inducing HLA-A24-restricted and
tumor-specific cytotoxic T lymphocytes. Int J Cancer 1999 May 5
81:3 459-66 |
| SSX
gene family | Gure
AO, Tüüreci O,
Sahin U, Tsang S, Scanlan MJ, Jäger
E, Knuth A, Pfreundschuh M, Old LJ, Chen YT. SSX: a multigene family
with several members transcribed in normal testis and human cancer.
Int J Cancer 1997 Sep 17 72:6 965-71 |
| Antigen | Literaturstelle |
| STAT3 | Bromberg
JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C,
Darnell JE Jr. Stat3 as an oncogene. Cell 1999 Aug 6;98(3): 295-303 |
| STn
(mucin assoc.) | Sandmaier
BM, Oparin DV, Holmberg LA, Reddish MA, MacLean GD, Longenecker
BM. Evidence of a cellular immune response against sialyl-Tn in
breast and ovarian cancer patients after high-dose chemotherapy,
stem cell rescue, and immunization with Theratope STn-KLH cancer
vaccine. J Immunother 1999 Jan 22:1 59-66 |
| TAG-72 | Kuroki
M, Fernsten PD, Wunderlich D, Colcher D, Simpson JF, Poole DJ, Schlom
J. Serological mapping of the TAG-72 tumor-associated antigen using 19 distinct
monoclonal antibodies. Cancer Res 1990 Aug 15 50:16 9872-9 |
| TGF-α | Imanishi
K, Yamaguchi K, Suzuki M, Honda S, Yanaihara N, Abe K. Production
of transforming growth factor-alpha
in human tumour cell lines. Br J Cancer 1989 May 59:5 761-5 |
| TGF-β | Picon
A, Gold LI, Wang J, Cohen A, Friedman F. A subset of metastatic
human colon cancers expresses elevated levels of transforming growth
factor betal. Cancer Epidemiol Biomarkers Prev 1998 Jun 7:6 997-504 |
| Thymosin β 15 | Bao,
L., Loda, M., Janmey, P. A., Stewart, R., Anand-Apte, B., and Zetter, B. R. Thymosin
beta 15: a novel regulator of tumor cell motility upregulated in metastatic
prostate cancer. Nature Medicine. 2 (12), 1322-1328. 1996 |
| TNF-α | Moradi
MM, Carson LF, Weinberg B, Haney AF, Twiggs LB, Ramakrishnan S.
Serum and ascitic fluid levels of interleukin-1, interleukin-6,
and tumor necrosis factor-alpha in patients with ovarian epithelial
cancer. Cancer 1993 Oct 15 72:8 2933-40 |
| TPA | Maulard
C, Toubert ME, Chretien Y, Delanian S, Dufour B, Housset M. Serum
tissue polypeptide antigen (S-TPA) in bladder cancer as a tumor
marker. A prospective study. Cancer 1994 Jan 15 73:2 394-8 |
| TPI | Nishida
Y, Sumi H, Mihara H. A thiol protease inhibitor released from cultured
human malignant melanoma cells. Cancer Res 1989 Aug 44:8 3329-9 |
| TRP-2 | Parkhurst
MR, Fitzgerald EB, Southwood S, Sette A, Rosenberg SA, Kawakami
Y. Identification of a shared HLA-A*0201-restricted T-cell epitope
from the melanoma antigen tyrosinase-related protein 2 (TRP2). Cancer
Res 1998 Nov 1 58:21 4895-901 |
| Antigen | Literaturstelle |
| Tyrosinase | Kirkin
AF, Dzhandzhugazyan K, Zeuthen J. Melanomaassociated antigens recognized
by cytotoxic T lymphocytes. APMIS 1998 Jul 106:7 665-79 |
| VEGF | Hyodo
I, Doi T, Endo H, Hosokawa Y, Nishikawa Y, Tanimizu M, Jinno K,
Kotani Y. Clinical significance of plasma vascular endothelial growth
factor in gastrointestinal cancer. Eur J Cancer 1998 Dec 39:13 2041-5 |
| ZAG | Sanchez
LM, Chirino AJ, Bjorkman Pj. 1999, Crystal structure of human ZAG,
a fat-depleting factor related to MHC molecules. Science 19;283(5409):
1914-9 |
| p16INK4 | Quelle
DE, Ashmun RA, Hannon GJ, Rehberger PA, Trono D, Richter KH, Walker
C, Beach D, Sherr CJ, Serrano M. Cloning and characterization of
murine p16INK4a and p15INK4b genes. Oncogene 1995 Aug 17;11(4):
635-95 |
| Glutathione
S-transferase | Hengstler
JG, Arand M, Herrero ME, Oesch F. Polymorphisms of N-acetyltransferases,
glutathione S-transferases, microsomal epoxide hydrolase and sulfotransferases:
influence on cancer susceptibility. Recent Results Cancer Res 1998
154: 47-85 |
-
Im
Folgenden wird eine Reihe von spezifischen Tumor-assoziierten Antigenen
im Detail diskutiert.
-
Prostata-spezifisches
Membran Antigen (PSM)
-
In
den USA ist Prostata-Krebs die zweithäufigste Ursache für Krebstod
(ungefähr
40.000 pro Jahr), und 200.000 Patienten pro Jahr werden untersucht
(Boring 1993). Ungefähr
1 von 11 Männern
wird eventuell einen Prostata-Krebs entwickeln. Außerdem entwickeln
ungefähr
40-60% der Prostata-Krebs-Patienten Ausweitungen von dieser Erkrankung
außerhalb
der Prostata (Babaian 1994). Die Hauptstrategie der vorliegenden Erfindung
ist es, ein therapeutisches Vakzin als zusätzliche Therapie zur Prostatektomie
zu verwenden um restliches Tumorgewebe und Metastasen auszumerzen.
-
Mehrere
pathologische Zustände
werden der Prostatadrüse
zugeordnet, einschließlich
des gutartigen Wachstums (BPH), der Infektion (Prostatitis) und
der Neoplasie (Prostata-Krebs).
-
Die
biologische Aggressivität
von Prostata-Krebs ist unterschiedlich. In einigen Patienten bleibt
der aufgespürte
Tumor ein latenter histologischer Tumor und wird niemals klinisch
signifikant. In anderen Patienten schreitet das Tumorwachstum schnell
vorwärts,
der Tumor metastasiert und tötet
den Patienten in einer relativ kurzen Zeitspanne (2–5 Jahren).
-
Die
gängige
primäre
Behandlung von Prostata-Krebs ist die Prostatektomie. Aufgrund der
starken Ausbreitung von Prostata-Krebszellen werden die meisten
Patienten durch lokale chirurgische Behandlung jedoch nicht geheilt.
Patienten mit nicht beendeter Erkrankung erhalten gegebenenfalls
eine systemische Androgen-Herabsetzungstherapie, aber die jährliche
Todesrate von Prostata-Krebs wurde über die letzten 50 Jahre, seit
Androgen-Herabsetzungstherapie eine Standardtherapie für metastasierende
Erkrankungen wurde, überhaupt
nicht herabgesetzt.
-
PSM
ist ein Membranprotein, welches für Prostatagewebe hochspezifisch
ist, sowohl für
gutartige als auch bösartige,
obwohl auch die Expression von PSM in anderen Geweben, wie Nierengewebe
und Nierentumor, Dünndarm,
Gehirn und Tumor-Gefäßneuwachstum,
beobachtet wurde. Wenn eine Operation erfolgreich war, sollten daher
Krebs-Patienten, die eine Prostatektomie durchgemacht haben, theoretisch
PSM auf zurückbleibenden
bösartigen
Prostatatumorgeweben oder Metastasen, die von dem Tumor herrühren, exprimieren.
Indem eine starke CTL-Reaktion und/oder eine starke polyklonale
Antikörperantwort
gegen PSM induziert wird, wird erwartet, dass das restliche Tumorgewebe
entfernt werden kann.
-
Interessanterweise
wird eine Hochregulierung der PSM-Expression infolge der Androgen-Mangel-Therapie
von Prostata-Krebs-Patienten beobachtet (Wright 1996). Dieses würde eine
PSM-gerichtete Behandlung als Folgebehandlung für die traditionelle Androgen-Mangel-Therapie äußerst geeignet
machen.
-
PSM
wurde zuerst 1987 identifiziert, und zwar als Ergebnis der Erzeugung
eines monoklonalen Antikörpers,
7E11-C5.3, der gegen eine isolierte humane Prostata-Krebszelle,
LNCaP (Horoszewicz 1987) gezogen wurde. Der Antikörper erkennt
sowohl normales als auch bösartiges
Prostataepithel, und er wurde 1993 verwendet um das PSM-Protein
zu reinigen und dessen Aminosäuresequenz
zu bestimmen und schließlich das
Gen zu klonen (Israeli 1993).
-
PSM
ist ein Typ II-Transmembran-Glykoprotein mit einem Molekulargewicht
von 84 kD, vorhergesagt aus der Nukleinsäuresequenz, wobei die glykosylierte
Version ein beobachtetes Molekulargewicht von 100-120 kD aufweist.
Die Sequenzierung des Gens, das für PSM codiert, deckte eine
mutmaßliche
Membran-überspannende
Region in Verbindung mit drei cytosolischen Argenin-Ankerresten
auf. Der extrazelluläre Teil
von PSM macht 707 der insgesamt 750 Aminosäuren des Proteins aus, während für die cytoplasmatische Domäne vorhergesagt
wird, dass sie 19 Aminosäuren
lang ist (Israeli 1993). PSM-spezifische mRNA wurde in Prostatatumorgewebe
detektiert (Israeli 1994), was anzeigt, dass das Tumorantigen kein
falsch glykosyliertes Protein ist, was z.B. bei den Tn- oder sTn-Tumor-Antigenen
der Fall ist.
-
Die
Volllängen-PSM-cDNA
wurde in eine PSM-negative humane Prostata-Krebs-Zelllinie transfiziert und darin exprimiert,
und zwar in PC-3 (Kahn 1994). Außerdem wurde die Volllängen-cDNA
(2,65 Kilobasen) in vitro transkribiert und translatiert (Kahn 1994).
-
Es
wurde kürzlich
gezeigt, dass PSM hydrolytische Aktivität besitzt, die der N-acetylierten α-gebundenen
sauren Dipeptidase (NAALADase) ähnelt – tatsächlich wurde
gezeigt, dass die zwei Proteine identisch sind. NAALADase ist eine
Membran-gebundene Hydrolase des Nervensystems, welche das Neuropeptid N-Acetylaspartylglutamat
(NAAG) verstoff wechselt um glutamatergische Signalprozesse zu beeinflussen.
Es ist noch immer nicht bekannt, ob diese Aktivität von PSM
irgendeine relevante biologische Funktion hat.
-
Es
ist von gewisser Wichtigkeit, vorherzusagen, ob ungewünschte Kreuzreaktivität mit anderen
Proteinen, die für
CTLs oder Antikörper
zugänglich
sind, infolge der Behandlung mit einem Autovakzin, das eine PSM-spezifische
Immunantwort hervorruft, zu erwarten wäre. Es wurde gezeigt, dass
ein Teil der codierenden Region des PSM-Gens (Aminosäure-Positionen
418–567)
54% Homologie zum humanen Transferinrezeptor aufweist (Israeli 1993).
Ebenso wurde die vollständige
Sequenzidentität
mit dem NAALADase-Enzym aufgefunden, vgl. oben. Keine Identifizierung
von funktionell relevanten Ähnlichkeiten
mit anderen bekannten Peptidasen konnte erfolgen.
-
Die
Homologie mit dem Transferinrezeptor ist sehr niedrig und wird bevorzugt
in einigen der erfindungsgemäßen Konstrukte
zerstört.
Von der beobachteten Sequenzidentität mit der humanen NAALADase wird
nicht erwartet, dass sie ein Hindernis für ein PSM-Vakzin ist, teilweise
aufgrund der geringen Eignung von Antikörpern und CTLs zum Durchdringen
der Blut-Hirn-Schranke.
-
Insgesamt
wird auch mit dem PSM-ähnlichen
Konstrukt nicht erwartet, dass hinderliche Kreuzreaktivität mit anderen
Proteinen in den Patienten auftritt.
-
Aus
früheren
Studien ist klar, dass PSM in den meisten Prostatakrebs-Zellen und
aus der Prostata herrührenden
Metastasen, die getestet wurden, exprimiert wird. Außerdem wurde
nachgewiesen, dass die meisten anderen Krebsarten, wie Karzinome,
Sarkome und Melanome unterschiedlicher Gewebe, sowie eine ganze
Reihe von humanen Nicht-Prostatakrebszelllinien PSM-negativ sind.
-
Zusätzlich dazu
hat man herausgefunden, dass eine große Anzahl anderer Gewebe PSM-negativ ist. Diese
umfassen den Darm, die Brust, die Lunge, die Eierstöcke, die
Leber, die Blase, den Uterus, die Bronchien, die Milz, den Pankreas,
die Zunge, die Speiseröhre,
den Magen, den Thymus, den Parathymus, Nieren, Lymphknoten, die
Aorta, die Vena cava, die Haut, Milchdrüsen und die Placenta. RT-PCR
hat jedoch die Existenz von PSM-mRNA in einigen dieser Gewebe aufgedeckt.
-
Obwohl
PSM vornehmlich als Membran-gebundenes Molekül auf Prostatagewebe gefunden
wird, können
geringe Mengen PSM auch im Serum von normalen Individuen und mit
erhöhten
Spiegeln bei Prostatakrebs-Patienten aufgefunden werden (Rochon
1994, Murphy 1995). Der Spiegel an zirkulierendem PSM in diesen
Patienten erlaubt daher eine serologische Beobachtung der Wirksamkeit
eines PSM-Vakzins.
-
Im
Ergebnis, basierend auf dem vollständigen Umfang der zur Zeit
verfügbaren
Daten, ist PSM ein Antigen mit einer hohen Spezifität für humane
Prostatagewebe und Tumore, die aus diesen herrühren. Dies bedeutet, dass bei
Patienten, die eine Prostatektomie durchgemacht haben, PSM ein für den Tumor
quasi-spezifisches Selbst-Antigen ist. Es ist daher wahrschein lich,
dass ein effektives PSM-Vakzin hauptsächlich auf Prostatagewebe oder
von der Prostata herrührendes
Metastasengewebe abzielen wird.
-
Wie
aus Beispiel 1 deutlich werden wird, umfasst die Erfindung bevorzugt,
dass ein fremdes TH-Zell-Epitop in einen
Teil der PSM-Aminosäuresequenz
eingefügt
wird, der in der SEQ ID NO:2 durch Positionen 16-52 und/oder 87-108
und/oder 210-230 und/oder 269-289 und/oder 298-324 und/oder 442-465 und/oder
488-514 und/oder 598-630 und/oder 643-662 und/oder 672-699 definiert
ist. Außerdem
ist ein modifiziertes PSM-Molekül,
in das an diesen Positionen ein fremdes TH-Epitop
eingefügt
wurde, ebenso ein Teil der Erfindung.
-
Dementsprechend
betrifft die Erfindung auch ein Analogon von humanem PSM, welches
in Menschen immunogen ist, wobei das Analogon einen wesentlichen
Teil aller bekannten und vorhergesagten CTL- und B-Zell-Epitope
von PSM umfasst, und mindestens ein fremdes TH-Epitop
umfasst, wie hierin diskutiert. Bevorzugte PSM-Analoga sind jene,
bei denen das mindestens eine fremde TH-Epitop
als Insertion in die PSM-Aminosäuresequenz
oder als Substitution eines Teils der PSM-Aminosäuresequenz oder als Ergebnis
der Deletion eines Teils der PSM-Aminosäuresequenz vorliegt, und die
am meisten bevorzugten Analoga sind jene, in denen ein fremdes TH-Zell-Epitop in einen Teil der PSM-Aminosäuresequenz
eingefügt
ist, der durch SEQ ID NO:2 an den Positionen 16-52 und/oder 87-108
und/oder 210-230 und/oder 269-289
und/oder 298-324 und/oder 442-465 und/oder 488-514 und/oder 598-630
und/oder 643-662 und/oder 672-699 definiert wird.
-
Humanes Chorion-Gonadotropin
(HCG)
-
Der
Zusammenhang zwischen embryonalen Markern und bösartigen Phenotypen wird schon über viele
Jahrzehnte diskutiert. Eine wachsende Menge an Daten legt nahe,
dass mindestens ein solcher Marker, humanes Chorion-Gonadotropin
Beta (HCGβ)
regelmäßig auf
Krebszellen vieler unterschiedlicher histologischer Herkunft detektiert
wird, und dass die Expression dieses Proteins oft mit erhöhten metastasierenden
Eigenschaften korreliert. Eine humorale Immunantwort, die gegen
dieses lösliche
Protein gerichtet ist, könnte
die Chancen eines Tumors, sich auszubreiten, reduzieren und/oder
das Wiederauftreten von neuen primären Wachstumsschüben nach
der chirurgischen Behandlung hemmen.
-
Humanes
Chorion-Gonadotropin gehört
zu einer Familien von Glykoproteinhormonen, die das Follikel-stimulierende
Hormon (FSH), Thymus-stimulierende Hormon (TSH) und luteinisierendes
Hormon (LH) umfasst, wobei alle diese wichtige Regulatoren der reproduktiven
Expression und des fötalen Überlebens
sind. Die Mitglieder dieser Familie von Hormonen sind Heterodimere,
welche eine gemeinsame α-Kette
gemein haben. Die β-Kette
ist für
jedes Hormon einzigartig und stellt die Spezifität zur Verfügung, wobei die β-Kette von LH
die stärkste
Sequenzhomologie mit hCGβ aufweist
(ungefähr
80%). Das apparente Molekulargewicht von hCG-Holo ist 37 kD, zu
dem ein Drittel durch Kohlenhydrate beigetragen wird. Die posttranslationalen
Zuckermodifikationen umfassen sowohl N-gebundene als auch O-gebundene
Kohlen hydrate. Sialylsäurereste
sind zahlreich vorhanden und verleihen dem Protein eine große negative
Ladung. Die Kristallstruktur von hCG-Holo wurde aufgelöst (Lapthorn
et al., 1994).
-
Ausgehend
von der Kristallstruktur hat man herausgefunden, dass hCG eine Homologie
mit einer Familie von Wachstumsfaktoren aufweist, einschließlich PDGF
und TGFβ (Lapthorn
et al., 1994). Dies legt nahe, dass hCG-Expression bei der Regulation
des Krebszellwachstums helfen könnte.
-
Humanes
Chorion-Gonadotropin ist ein Glykoproteinhormon, welches von den
placentalen Syncytiotrophoblasten kurz nach der Empfängnis hergestellt
wird, und es ist für
die erfolgeiche Schwangerschaft der schwangeren Frau essentiell.
-
Die
pathophysiologische Rolle von embryonalen Markern für die Entwicklung
oder die Aufrechterhaltung einer Krebsmasse ist nicht bekannt. Es
ist jedoch von Interesse; zu bemerken, dass Trophoblasten (in denen
diese Proteine normalerweise hergestellt werden) sowohl angiogenische
als auch invasive Eigenschaften aufweisen, welche beide ebenso notwendige
Eigenschaften einer Krebszelle sind. Außerdem wurde vorgeschlagen,
dass hCG (oder seine Untereinheiten) die maternale zelluläre Immunantwort
gegen das fötale
Gewebe inhibieren. Studien haben z.B. gezeigt, dass hCG direkt T-Zell-Antworten
supprimiert (Jacoby et al., 1984), und es wurde vorgeschlagen, dass
eine geeignetere Umgebung zum Etablieren von metastasierenden Tumoren
resultieren könnte,
weil die Lyrnphknoten, die (in diesem Fall) einen primären Melanomtumor
herausfiltern (sollten), immunsupprimiert sind. Im Ergebnis kann
die Expression von hCGβ Krebszellen
helfen, sich in die sekundären
Lymphorgane auszubreiten. Schließlich wurde, wie oben erwähnt, eine
Strukturhomologie zwischen hCG und einer Reihe von Wachstumsfaktoren
gezeigt. Eine andere Möglichkeit
ist es daher, dass das Sezernieren von hCG durch Krebszellen dem
Tumor einen Wachstumsvorteil geben könnte.
-
Die
Expression von hCGβ wurde
in vielen unterschiedlichen Arten von Krebs gezeigt, beispielsweise: a)
Prostata-Adenokarzinom: ein positiver Nachweis für hCGβ in Gewebesektionen wurde bei
Patienten mit schwacher Prognose beobachtet, unabhängig von
dem histologischen Grad des Tumors (Sheaff et al., 1996), b) unterschiedliche
Arten von Lungen-Karzinomen;
Plattenepithel (SQCC), Adenokarzinom (AC) und goßzelliges Karzinom (LCC) zeigten
alle einen hohen Prozentsatz an Reaktivität für hCGβ an (Boucher et al., 1995), c)
Pankreas-Adenokarzinom (Syrigos et al., 1998), d) Neuroblastome,
Hirnkrebsarten und Retinoblastome (Acevedo et al., 1997), e) malignes
Melanom (Doi et al., 1996), f) Blasen-Karzinome (Lazar et al., 1995). Eine neuere
Veröffentlichung
beschreibt einen DNA-Ansatz, bei dem Mäuse mit einem hCGβ-Expressionskonstrukt immunisiert
wurden (Geissler et al., 1997). Bei diesem in vivo-Modell stand
die Inhibierung des Tumorwachstums in starker Beziehung zu der CTL-Aktivität, es wurden
jedoch auch höhere
Titer von Antikörpern
(welche die biologische Wirkung von intaktem hCG auf seinem zellulären Rezeptor
neutralisieren) detektiert.
-
Die
Verwendung von hCG als Immunogen wurde in mehreren Veröffentlichungen
beschrieben, welche sich auf ihre Verwendung als Empfängnis-verhütendes Vakzin
konzentrieren (Talwar et al., 1976, und Talwar et al., 1994). Ein
hoher Grad der Wirksamkeit und Sicherheit wurde beobachtet bei einer
klinischen Studie zur Antifertilität, die ein Vakzin gegen hCG-Holo
verwendete (Talwar et al., 1994). Klinische Studien der Phase I bei
Krebs-Patienten mit einem Vakzin gegen ein synthetisches C-terminales
Peptid von hCGβ,
das mit Diphterietoxoid konjugiert war, wurden ebenfalls durchgeführt (Triozzi
et al., 1994), und klinische Studien der Phase II dauern noch an.
Ungeachtet der Tatsache, dass die Idee, hCGβ als Krebs-Vakzin-Zielobjekt
zu verwenden, für
einige Zeit die Runde macht, wurde es nicht in Verbindung mit der
AutoVac-Technologie
untersucht.
-
Es
ist bekannt, dass Zellen nicht-embryonischer Gewebe oder gutartige
Neoplasmen kein hCGβ exprimieren.
Daher sollten keine möglichen
Nebenwirkungen aus der Vakzinierung gegen dieses Molekül (neben der
Wirkung auf die Schwangerschaft) entstehen. Da es auf so vielen
unterschiedlichen Arten von Krebs exprimiert wird, wurde dieses
Molekül
als "definitiver
Krebs-Biomarker" vorgeschlagen
(Acevedo et al., 1995, und Regelson W., 1995) und wäre als solches
ein attraktives Zielobjekt, dem man nachgehen sollte.
-
Geeignete
Tiermodelle für
weitere Studien der Wirksamkeit eines hCG-basierten Vakzins findet
man bei Acevedo et al., Cancer Det. and Prev. Suppl. (1987) 1: 477-486,
und in Kellen et al., Cancer Immunol. Immun. Ther. (1982) 13: 2-4.
-
Her2
-
Man
glaubt, dass die Tyrosinkinase-Rezeptoren Her2 und EGFr eine wesentliche
Rolle bei der malignen Transformation von normalen Zellen und bei
dem andauernden Wachstum von Krebszellen spielen. Eine Überexpression
ist für
gewöhnlich
mit einer sehr schwachen Prognose verknüpft. Während der letzten Jahre gab
es viele Berichte, die die Verwendung von Antikörpern gegen diese Rezeptoren
als Therapie gegen Krebsarten, die entweder einen oder beide Rezeptoren überexprimieren,
betrafen. Genentech Inc. hat mehrere erfolgreiche klinische Studien
bei Brustkrebs-Patientinnen unter Verwendung eines monoklonalen
Antikörpers gegen
Her2 beendet und hat vor kurzem eine FDA-Zulassung für die Vermarktung
der Präparation
des monoklonalen Antikörpers
anti-Her2, Herceptin®, erhalten.
-
Die
Autovakzinierungs-Technologie, die hierin offenbart wird, würde angewandt
auf das Her2-Molekül polyklonale
Antikörper
bewirken, die vornehmlich mit Her2 reagieren würden. Man erwartet, dass solche
Antikörper
Tumorzellen angreifen und eliminieren, sowie dass sie Metastasenzellen
daran hindern, sich zu Metastasen zu entwickeln. Der Wirkmechanismus
dieser Anti-Tumor-Wirkung würde über Komplement
und Antikörper-abhängige zelluläre Cytotoxizität vermittelt.
-
Abhängig von
der Auswahl der Konstrukte könnten
die induzierten Autoantikörper
auch Krebszellwachstum inhibieren, und zwar über die Inhibierung der Wachstumsfaktor-abhängigen Oligo-Dimerisierung und
Internalisierung der Rezeptoren. Und, was am wichtigsten ist, man erwartet,
dass die Her2-Analoga in der Lage sind, CTL-Antworten direkt gegen
bekannte und/oder vorhergesagte Her2-Epitope, die von den Tumorzellen
angezeigt werden, zu induzieren.
-
Her2
ist ein Mitglied der Familie der Epidermis-Wachstumsfaktor-Rezeptoren
(c-erbB), welche derzeit aus vier unterschiedlichen Rezeptoren besteht:
c-erbB-1 (EGFr), c-erbB-2 (Her2, c-Neu), c-erbB-3 und c-erbB-4 (Salomon
et al., 1995). C-erbB-3 und c-erbB-4 sind weniger gut charakterisiert
als EGFr und Her2. Her2 ist ein integrales Glykoprotein. Das reife
Protein besitzt ein Molekulargewicht von 185 kD mit strukturellen Eigenschaften,
die sehr stark dem EGFr-Rezeptor ähneln (Prigent
et al., 1992). EGFr ist auch ein integraler Membranrezeptor, der
aus einer Untereinheit besteht. Er besitzt ein apparentes Molekulargewicht
von 170 kD, und er besteht aus einer Liganden-bindenden Oberflächendomäne von 621
Aminosäuren,
einer einzelnen hydrophoben Transmembrandomäne aus 23 Aminosäuren und
einer hoch-konservierten cytoplasmatischen Tyrosinkinase-Domäne aus 542
Aminosäuren.
Das Protein ist N-glykosyliert (Prigent et al., 1994).
-
Alle
Proteine in dieser Familie sind Tyrosinkinasen. Die Interaktion
mit dem Liganden führt
zur Rezeptordimerisierung, welche die katalytische Wirkung der Tyrosinkinase
erhöht
(Bernard 1995, Chantry 1995). Die Proteine innerhalb der Familie
sind in der Lage, zu homo- und
zu heterodimerisieren, was für
ihre Aktivität wichtig
ist. Der EGFr vermittelt Wachstumsverstärkende Wirkungen und stimuliert
die Aufnahme von Glucose und Aminosäuren durch Zellen (Prigent
et al., 1992). Her2 vermittelt ebenso Wachstums-verstärkende Signale. Nur
EGFr bindet EGF und TGF-α.
Diese Liganden binden nicht an die anderen Rezeptoren in der Familie
(Prigent et al., 1992). Die Liganden für Her2 sind nicht vollständig bestimmt.
Es wurde jedoch gezeigt, dass Heregulin die Phosphorylierung durch
Aktivierung von Her2 induziert. Dies scheint nicht in einer direkten
Bindung an den Rezeptor begründet
zu sein, aber man glaubt, dass Heregulin ein Ligand für erbB-3
und erbB-4 ist, welches dann Her2 durch Oligo-Dimerisierung aktiviert (Solomon et
al., 1995).
-
Die
Homologie zwischen den Proteinen der EGF-Rezeptor-Familie ist in
der Tyrosinkinase-Domäne am
cytoplasmatischen Teil des Moleküls
am deutlichsten (82% zwischen EGFr und Her2). Die Homologie ist im
extrazellulären
Teil geringer – von
41% bis 46% in unterschiedlichen Domänen (Prigent et al., 1992).
-
Der
epidermale Wachstumsfaktor-Rezeptor wird auf normalen Geweben in
geringen Mengen exprimiert, aber er ist in vielen Krebsarten überexprimiert.
EGFr ist in Brustkrebs-Arten überexprimiert
(Earp et al., 1993, Eppenberger 1994), in Glioma (Schlegel et al.,
1994), in Magen-Krebs (Tkunaga et al., 1995), in Plattenepithel-Karzinom
(Fujii 1995), in Eierstock-Krebs
(van Dam et al., 1994), und anderen. Her2 ist ebenfalls auf wenigen
normalen humanen Geweben in geringen Mengen exprimiert, am charakteristischsten
auf sekretorischen Epithelien. Die Überexpression von Her2 tritt
bei ungefähr
30% der Brustkrebs-, Magenkrebs-, Pankreaskrebs-, Blasenkrebs- und
Eierstockkrebs-Arten auf.
-
Die
Expression dieser Rezeptoren variiert in Abhängigkeit von dem Grad der Differenzierung
des Tumors und des Krebstyps, z.B. bei Brustkrebs überexprimieren
primäre
Tumoren beide Rezeptoren, während bei
Magenkrebs die Überexpression
zu einem späteren
Stadium in metastasierenden Tumoren auftritt (Salomon et al., 1995).
Die Anzahl an überexprimierten
Rezeptoren auf Karzinomzellen ist größer als 106/Zelle
bei verschiedenen Kopf- und Nacken-Krebsarten, bei Vulva-, Brust- und Eierstock-Krebslinien,
die von Patientinnen isoliert wurden (Dean et al., 1994).
-
Es
gibt mehrere Gründe,
warum die EGFr-Familie von Rezeptoren geeignete Zielobjekte für die Tumor-Immuntherapie
darstellt. Zuerst sind sie in vielen Arten von Krebs überexprimiert,
was die Immunantwort auf den Tumor ausrichten sollte: Zweitens exprimieren
oder überexprimieren
die Tumoren häufig
die Liganden für
diese Familie von Rezeptoren, und einige sind hypersensitiv gegenüber der
proliferativen Wirkung, die von den Liganden vermittelt wird, drittens
haben Patienten mit Tumoren, die Wachstumsfaktor-Rezeptoren überexprimieren,
oft schlechte Aussichten. Die Überexpression
wurde insbesondere bei Brustkrebs, Lungenkrebs und Blasenkrebs (2)
stark mit der schlechten Prognose in Verbindung gebracht und geht
anscheinend mit invasiven/metastasierenden Phenotypen einher, welche
gegenüber
konventionellen Therapien recht unsensitiv sind (Eccles et al.,
1994).
-
Die Überexpression
von Her2 ist in einigen Fällen
ein Ergebnis der Amplifikation des Gens, und in anderen Fällen ein
Ergebnis erhöhter
Transkription und Translation. Die Überexpression von Her2 geht
mit einer schlechten Prognose bei Brustkrebs, Eierstockkrebs, Magenkrebs,
Blasenkrebs und möglicherweise
Lungenkrebs-Arten mit nicht-kleinen Zellen einher (Solomon et al.,
1995).
-
Klinische
Phase I-Studien wurden mit einem bispezifischen Antikörper bei
Patientinnen mit fortgeschrittenem Brust- und Eierstockkrebs durchgeführt. Der
Antikörper
war bispezifisch für
Her2 und FcγRI
(Weiner et al., 1995). Eine effiziente Lysis von Her2-überexprimierenden
Tumorzellen wurde bei einem bispezifischen Antikörper gegen Her2 und CD3 beobachtet
(Zhu et al., 1995).
-
Die
Behandlung von SCID-Mäusen,
auf die ein humaner Magenkrebs durch Xenotransplantation übertragen
wurde mit einem monoklonalen anti-Her2-Antikörper, verlängerte die Überlebensrate der Mäuse (Ohniski
et al., 1995). Die Anti-Tumor-Aktivitäten von monoklonalen Antikörpern gegen
Her2, in vitro und in vivo, beruhen nicht auf einem identischen
Mechanismus; diese können
als unvollständige
Liganden-Agonisten wirken, Her2-Rezeptor-Umsetzung und deren Phosphorylierung
verändern
oder die Dimerisierung beeinflussen (Lupu et al., 1995).
-
Ganz ähnlich wurde
gezeigt, dass Antikörper
gegen EGFr auch Wachstumsfaktor-Wechselwirkungen stören können (Baselga
et al., 1994, Modjahedi et al., 1993a, Wu et al., 1995, Modjahedi
et al., 1993b, Tosi et al., 1995, Dean et al., 1994, Bier et al.,
1995, Modjahedi et al., 1996, Valone 1995).
-
Daher
ist eine wichtige Ausführungsform
der erfindungsgemäßen Verfahren
eine solche, bei der das fremde T-Zell-Epitop in einen Teil der
Her2-Aminosäuresequenz
eingefügt
wird, der durch die Aminosäure-Nummerierung
in SEQ ID NO:3 durch die Positionen 5-25 und/oder 59-73 und/oder
103-117 und/oder 149-163 und/oder 210-224 und/oder 250-264 und/oder
325-339 und/oder 369-383 und/oder 465-479 und/oder 579-593 und/oder
632-652 und/oder 653–667
und/oder 661-675 und/oder 695-709 und/oder 710-730 definiert wird,
vgl. die Beispiele.
-
Dementsprechend
bezieht sich die Erfindung auch auf ein Analogon von humanem Her2,
welches in Menschen immunogen ist, wobei das Analogon einen wesentlichen
Teil aller bekannten und vorhergesagten CTL- und B-Zell-Epitope
von Her2 umfasst, und mindestens ein fremdes TH-Epitop
umfasst, wie hierin diskutiert ist. Es ist bevorzugt, dass das mindestens
eine fremde TH-Epitop als Insertion in der
Her2-Aminosäuresequenz
oder als Substitution eines Teils der Her2-Aminosäuresequenz
oder als Ergebnis einer Deletion eines Teils der Her2-Aminosäuresequenz
vorliegt. Insbesondere bevorzugte Analoga sind jene oben definierten,
d.h. jene, in denen das fremde T-Zell-Epitop in einem Teil der Her2-Aminosäuresequenz
eingefügt
ist, der durch die Positionen in SEQ ID NO:3 5-25 und/oder 59-73
und/oder 103-117 und/oder 149-163 und/oder 210-224 und/oder 250-264
und/oder 325-339 und/oder 369-383 und/oder 465-479 und/oder 579-593
und/oder 632-652 und/oder 653-667 und/oder 661-675 und/oder 695-709
und/oder 710-730 definiert wird.
-
FGF8b
-
Es
wurde von mehreren Forschern gezeigt, dass FGF8b die Proliferation,
Transformation, Differenzierung von Säugerzellen und -geweben induziert,
und in einigen Fällen
die Tumorigenizität
derselben stark erhöht
(Tanaka 1992, Kouhara 1994, Lorenzi 1995, MacArthur 1995a, Crossley
1996a, 1996b, Ghosh 1996, Ohuchi 1997a, Rudra-Ganguly 1998). Diese
Wirkungen werden vornehmlich durch die Bindung von FGF8b an Mitglieder
der Fibroblasten-Wachstumsfaktor-Rezeptoren
FGFR2, FGFR3 und FGFR4 vermittelt (MacArthur 1995b, Blunt 1997,
Tanaka 1998). Daher wurde gezeigt, dass Zellen, die einen dieser
Rezeptoren und FGF8(b) exprimieren, eine autokrine Wachstums-Signallcaskade
bereitstellen, die zur Proliferation führt.
-
Die
biologische Wirkung von FGF8b wird höchstwahrscheinlich zum Teil über den
JAK/STAT3-Weg vermittelt, da wir und andere beobachtet haben, dass
die Zugabe von FGF8b zum Wachstumsmedium bestimmter Zellen die Phosphorylierung
von STAT3 fördert,
ein Merkmal, von dem man vermutet, dass es Zellen resistent gegenüber der
Apoptose macht (Catlett-Falcone 1999).
-
Zusätzlich zu
den Beobachtungen in vitro, die oben erwähnt sind, wurde kürzlich gezeigt,
dass die Expression von FGF8(b) in sowohl Prostata- als auch Brustkrebs-Arten
hochreguliert ist (Marsh 1999, Dorkin 1999). Wir glauben daher,
dass ein Autovakzin gegen FGF8(b) eine sehr effiziente Methode der
Behandlung einer Reihe von FGF8-exprimierenden Tumoren ist, oder
vielleicht deren Sensitivität
gegenüber
Apoptose-induzierenden Mitteln erhöht.
-
Prostatakrebs
-
Die
biologische Aggressivität
von Prostatakrebs variiert. In einigen Patienten bleibt der Tumor
ein latenter histologischer Tumor und wird klinisch niemals signifikant.
In anderen Patienten schreitet das Tumorwachstum schnell fort, der
Tumor metastasiert und tötet
den Patienten in einer relativ kurzen Zeit (2-5 Jahre).
-
Zum
Zwecke der Diagnose und um die Reaktion auf eine Prostatakrebs-Therapie
zu verfolgen, wurde vornehmlich die Bestimmung des Spiegels im Blutkreislauf
von zwei Proteinen verwendet: PAP ("prostatic acid phosphatase") und PSA ("prostate specific
antigen") (Nguyen,
1990, Henttu 1989). Aufgrund der Zerstörung der normalen Architektur
der Prostatadrüse
in Reaktion auf die Krebsentwicklung werden diese Proteine in den Blutkreislauf
freigesetzt, wo sie als Marker, z.B. für metastatische Ausbreitung,
detektiert werden können.
-
Die
derzeitige primäre
Behandlung von Prostatakrebs ist die Prostatektomie. Aufgrund der
starken Ausbreitung von Prostatakrebszellen wird die Mehrheit der
Prostatakrebs-Patienten durch die lokale chirurgische Behandlung
nicht geheilt. Patienten mit einer nicht-beendeten Erkrankung erhalten
eine systemische Androgen-Herabsetzungstherapie, aber die jährliche
Todesrate von Prostatakrebs hat sich über die 50 Jahre, seit die
Androgen-Herabsetzung die Standardtherapie für metastatische Erkrankungen
wurde, überhaupt
nicht verringert.
-
RT-PCR-Analyse
hat gezeigt, dass FGF8-mRNA von den humanen Prostataepithel-Tumorzelllinien LNCaP,
PC-3, ALVA-31 und DU145 produziert wird, wobei FGF8b die am meisten
vorherrschende Isoform ist (Tanaka 1995, Ghosh 1996). Das Wachstum
der Androgenreaktiven LNCaP-Zellen wird durch Zugabe von rekombinantem
FGF8b stimuliert (Tanaka 1995), während DU145-Zellen durch die
Transfektion mit Viren, die Anti-Sense FGF8b exprimieren, im Wachstum
inhibiert werden konnten (Rudra-Ganguly 1998). Dieses, zusammen
mit den unten diskutierten Hinweisen aus Entwicklungsstudien, deutet
auf eine Rolle für
FGF8b bei der Aufrechterhaltung des Krebsstatus dieser Zelllinien.
-
Unter
Verwendung von FGF8a-cDNA für
in situ-Hybridisierungs-Experimente haben Leung und Mitarbeiter
gezeigt, dass ein hoher Anteil (80% (n = 106) und 71% (n = 31))
an Prostatakrebs FGF8-mRNA produziert und dass die Menge von FGF8-mRNA
mit der Schwere der Tumoren korreliert (P < 0,0016 bzw. P < 0,05) (Leung 1996, Dorkin 1999). Unter
Verwendung eines monoklonalen anti-FGF8b-Antikörpers konnte gezeigt werden,
dass diese Isoform für
die Überexpression
von FGF8b verantwortlich ist (Dorkin 1999). Zusätzlich zeigen Männer mit
Tumoren, welche hohe Spiegel an FGF8 exprimieren, eine schlechtere Überlebensrate
(P = 0,034), und die FGF8-Expression dauert in Androgen-unabhängigen Prostatakrebsarten
an (Dorkin 1999). Entsprechend der Daten, die von Dorkin und Mitarbeitern
dargestellt werden, kann die Expression von FGF8b bei Prostatakrebs
das Überleben
des Patienten vorhersagen.
-
Immunhistochemische
Analyse unter Verwendung eines monoklonalen Antikörpers gegen
FGF8 hat das Protein in 93% (n = 43) von humanen Prostatakrebsarten
detektiert (Tanaka 1998). Normales Prostatagewebe oder gutartige
Prostatahyperplasie bringt niedrige Spiegel an FGF8-mRNA hervor
und enthält
keine detektierbaren Mengen von FGF8-Protein (Leung 1996, Yoshimura
1996, Ghosh 1996, Tanaka 1998, Dorkin 1999).
-
Diese
Ergebnisse deuten darauf hin, dass ein Autovakzin gegen FGF8(b)
gegen Prostatakrebsgewebe reagieren würde und daher äußerst wertvoll
bei der Behandlung von Prostatakrebs wäre.
-
Brustkrebs
-
Die
derzeitige Behandlung von Brustkrebs ist die chirurgische Operation.
Aufgrund der starken Ausbreitung von Brustkrebszellen wird ein großer Teil
der Brustkrebs-Patientinnen durch die lokale chirurgische Behandlung
nicht geheilt. Patientinnen mit nicht-beendeter Krankheit erhalten
gegebenenfalls eine Androgen-Herabsetzungstherapie, eine Chemotherapie
oder eine Strahlungstherapie. Die jährliche Todesrate von Brustkrebs
ist jedoch immer noch relativ hoch.
-
FGF8
wurde ursprünglich
aus einer Maus-Mammakarzinom-Zelllinie (SC-3) isoliert, bei der
die Expression durch Zugabe von Androgen zum Medium induziert werden
konnte (Nonomura 1990). Von dem Protein ist auch bekannt, dass es
die Proliferation von diesen sowie von anderen Säugerzellen induziert. Jüngst wurde
gezeigt, dass FGF8b-mRNA in acht (n = 8) humanen Brustkrebs-Zelllinien
vorhanden ist (MDA-MB-231, MDA-MB-415, ZR 75-1, T-47-D, SK-BR-III, PMC-42,
HBL-100 und MCF-7) (Tanaka 1995, Payson 1996, Wu 1997, Marsh 1998).
-
Wnt-1-transgenische
Mäuse,
die mit einem Maus-Mammatumor-Virus (MMTV) infiziert wurden, entwickeln
Mammatumoren. FGF8-Transkription wird in 50% dieser Tumoren aktiviert
(MacArthur 1995c, Kapoun 1997).
-
Transgenische
Mäuse,
die die FGF8b-cDNA unter Kontrolle des sehr spezifischen MMTV-Promotors tragen,
entwickeln spontan FGF8b-exprimierende Mammatumoren (Coombes, persönliche Mitteilung).
-
Jüngste Daten
zeigen, dass FGF8(b)-Expression in Brustkrebs hochreguliert ist
(Tanaka 1998, Marsh 1999). Tanaka und Mitarbeiter verwendeten einen
neuen monoklonalen FGF8-Antikörper in
immunhistochemischen Untersuchungen. Sie zeigten, dass FGF8 in 67%
(n = 12) der Brustkrebs-Arten vorhanden war, und dass Androgen-Rezeptoren
in 89% von FGF8-positiven
Brustkrebserkrankungen vorhanden waren (Hyperplasie, Fibroadenom,
intraduktale Papillome und Krebsarten), was es ermöglichen
würde,
dass die autokrine Wachstumsfördernde
Schleife in das Fortschreiten von Brustkrebs-Arten verwickelt wäre (Tanaka
1998). Unter Verwendung eines semi-quantitativen RT-PCR-Verfahrens
wurde gezeigt, dass erhöhte
Spiegel an FGF8-mRNA in malignen Brustgeweben aufgefunden wurden,
verglichen mit nicht-malignen
Brustgeweben. Signifikant mehr maligne Gewebe exprimierten FGF8
(p = 0,019) mit signifikant höheren
Spiegeln (p = 0,031) (68 Brustkrebs-Arten und 24 nicht-maligne Brustgewebe)
(Marsh 1999).
-
Es
wurde noch nicht vollständig
dargelegt, dass FGF8(b) als autokriner Wachstumsfaktor fungiert.
Die Tatsache, dass eine große
Anzahl von Tumoren FGF8b überexprimiert,
spricht jedoch stark dafür,
dass ein Autovakzin gegen FGF8b wirksam gegen eine große Prozentzahl
von Brust- und Prostatakrebs-Arten sein könnte. Die Daten, über die
Marsh, Dorkin und Tanaka berichten, deuten darauf hin, dass ein
Autovakzin gegen FGF8(b) für
die Behandlung von sowohl Brust- als auch Prostatakrebs-Arten nützlich sein
könnte,
und die recht ungenauen Daten, die von Dorkin et al. dargestellt
werden, sind eine weitere Stütze
für die
Ansicht, dass FGF8 in die Proliferation von humanen Krebszellen
verwickelt ist.
-
Beschreibung von FGF8b
-
FGF8
gehört
zu der Familie der Fibroblasten-Wachstumsfaktoren (FGFs). Diese
Wachstumsregulierenden Proteine sind kleine 200 Aminosäurereste
lange Proteine, die alle in die Induktion der Proliferation und der
Differenzierung einer großen
Spanne von Zellen involviert sind. Für einen jüngeren Übersichtsartikel über die
Verstrickung der Fibroblasten-Wachstumsfaktoren in die Entwicklung
von Gliedern bei Vertebraten siehe Johnson 1997. Die FGF-Familienmitglieder
sind evolutionär
verwandt und weisen eine Aminosäure-Sequenzidentität von 20-50%
auf.
-
FGF8b
ist eine Splice-Variante von FGF8, die ursprünglich als Androgen-induzierender
Wachstumsfaktor (AIGF) bezeichnet wurde. AIGF wurde als erstes als
ein Protein identifiziert, das von einer Zelllinie, die von einem
Maus-Mamma-Karzinom abgeleitet ist (SC-3), nach Stimulation mit
Androgen sezerniert wird (Nonomura 1990). Das Maus-FGF8-Gen enthält 6 Exons,
die potentiell für
8 unterschiedliche FGF8-Isoformen (FGF8a-h) codieren, welche sich
nur im N-terminalen Teil der Moleküle unterscheiden (Crossley
1995, MacArthur 1995b). Humanes FGF8 besitzt dieselbe Genstruktur
wie das Maus-Gen. Aufgrund eines Stop-Kodons im Exon 1B kann humanes
FGF8 nur in vier unterschiedlichen Isoformen vorliegen, nämlich FGF8a,
FGF8b, FGF8e und FGF8f (Gemel 1996). Die Genstrukturen und die Aminosäuresequenzen
der vier humanen Isoformen sind in 5 dargestellt.
-
Reifes
FGF8b enthält
193 Aminosäure-Reste
und hat ein errechnetes Molekulargewicht von 22,5 kDa. Das stark
basische Protein enthält
21 Arginin- und 14 Lysin-Reste, was zu einem errechneten isoelektrischen Punkt
von 10,84 und einer errechneten positiven Ladung von 19,8 bei pH
7,0 führt.
Es enthält
zwei Cystein-Reste und zwei potentielle N-Glykosylierungsstellen.
Aufgrund der Eigenheiten der durchgeführten Untersuchungen, die FGF8b
einbeziehen, ist sehr wenig über
die FGF8b-Proteingruppierung bekannt. Es wurde jedoch heterolog
in Bakterien exprimiert, unter Verwendung eines C-terminalen Hexa-Histidin-Tags
gereinigt und in vitro bis zu einem löslichen und biologisch aktiven
Stadium rückgefaltet
(MacArthur 1995a, Blunt 1997).
-
Biologische Aktivität von FGF8b
-
Wie
oben erwähnt,
wurde FGF8(b) zuerst als ein Faktor isoliert, der von einer Androgen-abhängigen Maus-Mammatumor-Zelllinie
ausgeschüttet
wurde, und es wurde gezeigt, dass dieses Protein die Proliferation dieser
Zellen induzieren kann. Die morphologischen Veränderungen ähneln jenen, die durch Testosteron
induziert werden, von dem auch bekannt ist, dass es die Synthese
von FGF8(b)-mRNA induziert (Tanaka 1992). Die Proliferation kann
durch FGF8(b)-Antisense-Oligos inhibiert werden (Nonomura 1990,
Tanaka 1992 und Yamanishi 1994). Tatsächlich konnte eine humane Prostatakrebs-Zelllinie
DU145 durch Transfektion mit Viren, die Anti-Sense-FGF8b exprimieren,
im Wachstum behindert werden (Rudra-Ganguly 1998). Jüngste Daten zeigen,
dass FGF8b die Phosphorylierung von STAT3 induziert – einem
Protein, von dem man vermutet, dass es in die Resistenz gegen Apoptose
verwickelt ist (Catlett-Falcone
1999, Johnston, C.L., unveröffentlichte
Ergebnisse).
-
Es
wurde von mehreren Forschern gezeigt, dass FGF8b sehr effizient
beim Induzieren der Transformation von NIH3T3- oder SC115-Zellen
ist (Miyashita 1994, Kouhara 1994, Lorenzi 1995, MacArthur 1995a). Indem
rekombinant exprimierte Proteine verwendet wurden, wurde auch gezeigt,
dass diese Induktion von morphologischen Veränderungen sehr viel effizienter
mit FGF8b ist, im Gegensatz zur Verwendung von FGF8a oder FGF8c
(MacArthur 1995a, Ghosh 1996). Interessanterweise wurde gezeigt,
dass die N-terminale Hälfte des
FGF8b-Moleküls alleine
zur Transformation von NIH3T3-Zellen ausreicht, und sogar das kleine FGF8b-spezifische
Peptid (QVTVQSSPNFT) konnte es den Zellen ermöglichen, 2-3 Mal länger als
normal in 0,1% Serum zu wachsen (Rudra-Ganguly 1998). Außerdem wurde
berichtet, dass NIH3T3-Zellen, die stabil mit einem Expressionsvektor,
der für
FGF8b codiert, transfiziert waren, sehr tumorigen waren, wenn sie
intraokular in Nacktmäuse
injiziert wurden (Kouhara 1994, Ghosh 1996).
-
Es
ist bekannt, dass FGF8b in vivo in bestimmen Stadien der Entwicklung
in Vertebraten exprimiert wird. Eine Zusammenfassung der biologischen
Rollen, die FGF8(b) zugeschrieben werden, ist in Tabelle 1 gezeigt.
Für Übersichtsartikel über die
Beteiligung von FGFB bei der Vertebraten-Entwicklung siehe Goldfarb 1999,
Johnson 1997. Tabelle:
Verschiedene Stellen/Gewebe, die bekannt sind, FGF8 herzustellen,
und die vorgeschlagene biologische Rolle(n).
| Wirkung/Mechanismus/Vorhandensein
(Art) | Literaturstellen |
| Vorhanden
bei der Entwicklung von Extremitätenanlagen
(Maus) | Heikinheimo
1994, Ohuchi 1994 |
| Wachstum
von Extremitätenanlagen
(Huhn) | Kuwana
1997, Xu 1990 |
| Induktion
der ektopischen Gliedmaßenbildung
aus dem Mesoderm (Huhn) | Crossley
1996b |
| Induktion
der Mittelhirnbildung aus dem caudalen Diencephalon (Huhn) | Crossley
1996a |
| Initiation
des Flügelwachstums
bei einer flügellosen Mutante
(Huhn) | Ohuchi
1997a |
| Rolle
bei der dorsoventralen Musterung der Gastrula (Zebrafisch) | Fürthauer
1997 |
| Erforderlich
während
der Gastrulation, der Herz-, craniofacialen, Vorderhin-, Mittelhirn-
und Cerebellum-Entwicklung (Gewebe-spezifische knock-out-Mäuse) | Meyers
1998 |
| Rolle
bei der Zahnmorphogenese (Maus) | Kettunen
1998 |
-
Man
glaubt, dass FGF8(b) seine Wirkung über die Bindung an die Fibroblasten-Wachstumsfaktor-Rezeptoren
(FGFRs) vermittelt. Es ist bekannt, dass FGF8b in der Lage ist,
spezifisch FGFR2c, FGFR3c, FGFR4c und in gewissem Ausmaß auch FGFR1c
zu aktivieren, aber nicht FGFR1b, -2b oder -3b (MacArthur 1995b,
Blunt 1997). Im Falle der Induktion des Wachstums von ektopischen
Huhn-Gliedmaßen
ist impliziert, dass FGF10, FGFR2 und FGF8 interagieren, und dass
dies für
das Auswachsen ausreichend sein könnte (Kuwana 1997, Xu 1998).
-
Diese
Ergebnisse stützen
die Hypothese, dass FGF8(b) auf auto- und parakrine Weise wirken
kann, was zum normalen Wachsen und der normalen Musterbildung bei
mehreren anatomischen Strukturen während der Vertebraten-Entwicklung
führt.
Es ist wichtig, dass FGF8 "total-knock-out"-Mäuse nicht überleben, höchstwahrscheinlich
aufgrund der umfangreichen Beteiligung des Proteins bei der Entwicklung
des Embryos.
-
Homologie
zu anderen Proteinen
-
Es
ist von erhöhter
Wichtigkeit vorherzusagen, ob unerwünschte Kreuzreaktivität mit anderen
Proteinen, die für
Antikörper
zugänglich
sind, nach der Behandlung mit einem Autovakzin, das spezifische
Autoantikörper
gegen FGF8b induziert, zu erwarten wäre. Aufgrund des hohen Grades
der Sequenzidentität
zwischen FGF8b und den anderen FGF8-Molekülen wird erwartet, dass ein
Autovakzin mit diesen Proteinen kreuzreagieren würde. Dies wird jedoch vermutlich
vorteilhaft sein, da von keinem dieser Proteine berichtet wird,
dass sie in Geweben oder von Zelllinien exprimiert werden, die nicht
schon FGF8b exprimieren.
-
Die
Aminosäure-Reste
55-175 von FGF8b zeigen ein relativ niedriges Ausmaß, aber
signifikantes Ausmaß an
Sequenzidentität
mit den anderen FGFs. Es ist allgemein anerkannt (und mehrere Male
bewiesen), dass ein signifikanter Grad der Sequenzidentität zwischen
zwei Proteindomänen
sich ebenfalls in einem hohen Ausmaß an Tertiärstruktur-Ähnlichkeit niederschlägt. Daher
wird erwartet, dass die FGF-Familienmitglieder im Allgemeinen strukturell ähnlich sind.
Die 3-dimensionale Struktur von FGF2 wurde von Kristallen sowie
in Lösung
aufgelöst
(Ago 1991, Zhang 1991, Zhu 1991, Eriksson 1993, Blaber 1996, Moy
1996). FGF2 setzt sich insgesamt aus einer Beta-Faltblatt-Struktur
zusammen, die eine dreifache Wiederholung eines viersträngigen,
antiparallelen Beta-Meanders umfasst. Diese beta-Fass-Struktur ist
vollständig
konserviert bei Interleukin 1, FGF2 (oder basischem FGF), und FGF1
(oder saurem FGF). Kernspinresonanzanalysen von FGF2 in Lösung haben
gezeigt, dass der N-terminale Teil des Moleküls eine relativ flexible Struktur
bildet. Der übrige
Teil von FGF8b (Aminosäure-Reste 1-54 und 176-215)
zeigt nur ein geringes Ausmaß an
Sequenzidentität
mit bekannten Proteinen.
-
Ausgehend
von den strukturellen und Anlagerungsdaten wird allgemein angenommen,
dass der dreidimensionale strukturelle Kern der anderen Fibroblasten-Wachstumsfaktoren
jenem von FGF1 und FGF2 sehr stark ähnelt. Diese strukturellen
Abwägungen
sind wichtige Faktoren bei unserer Gestaltung der mutierten FGF8b-Autovakzin-Moleküle.
-
Es
ist äußerst wichtig,
dass aufgrund des relativ niedrigen Grads der Sequenzidentität zwischen
FGF8 und irgendwelchen anderen Mitgliedern der FGF-Familie die Oberfläche von
FGF8 sich von der anderer FGFs sehr stark unterscheidet, was die
Kreuzreaktivität
von Antikörpern,
die durch ein FGF8b-Autovakzin hervorgerufen werden, mit anderen
FGF-Familienmitgliedern minimiert. Aufgrund des sehr niedrigen Grads
der Homologie zu anderen Proteinen als den Fibroblasten-Wachstumsfaktoren
erwarten wir nicht, dass ein Autovakzin gegen FGF8b mit irgendwelchen
anderen Proteinen kreuzreagiert.
-
Es
sollte jedoch betont werden, dass ein Autovakzin gegen FGF8b vermutlich
mit allen Isoformen von FGF8 kreuzreagieren würde. Dies wird jedoch vermutlich
kein Problem sein, da man von keiner der FGF8-Isoformen erwartet,
dass sie in signifikantem Ausmaß in
dem Erwachsenen exprimiert wird. Es ist sogar möglich, dass diese Kreuzreaktivität bei der
Behandlung von Krebs vorteilhaft ist, da gezeigt wurde, dass mindestens einige
Krebs-Zelllinien andere Isoformen zusätzlich zu FGF8b exprimieren.
-
Gewebeverteilung von FGF8b
-
Idealerweise
sollten die induzierten Autoantikörper und die nachfolgenden
Wirkmechanismen sowie die erwartete CTL-Reaktion, die durch die
Autovakzinierung hervorgerufen wird, nur gegen Gewebe gerichtet sein,
die aus dem Patienten zu eliminieren sind. Daher ist die Gewebeverteilung
des Antigens, welches von einem Autovakzin anvisiert wird, eine
Frage von sehr hoher Wichtigkeit in Bezug auf die Sicherheit des
Vakzins. Tabelle:
Expression von FGF8b in verschiedenen Geweben und Zellen Mensch
| Brustkrebs-Zelllinien | ((RT-PCR)
Tanaka 1995, Payson 1996, |
| (MDA-MB-231,
MDA-MB-415, ZR 75-1, T-47-D, SK-BR-III, PMC-42, HBL-100 und MCF-7) | Wu
1997, Marsh 1999) |
| Brusttumoren | ((mAb)
Tanaka 1998, (RT-PCR) Marsh 1999) |
| Normales
Brustgewebe | ((RT-PCR)
Wu 1997, Marsh 1999 (mAb) Tanaka 1998) |
| Prostatakrebs
(93%) | ((in
situ hyb.) Leung 1996, Dorkin 1999, (mAb) Tanaka (1998) |
| Brusterkrankungen | ((mAb)
Tanaka (1998) |
| Prostata-Tumorzellen
(LNCaP, | ((RT-PCR)
Tanaka 1995, Ghosh |
| PC-3,
DU145 und ALVA-31) | 1996,
Schmitt 1996) |
| fötale Niere | ((Northern
blot) Ghosh 1996) |
| Adulte
Prostata, Hoden, Nieren, | ((RT-PCR)
Ghosh 1996, Wu |
| Neuronen | 1997,
Dorkin 1999) |
| Teratokarzinomzellen
(Tera-2) | ((RT-PCR)
Wu 1997) |
Maus
| Brustkrebs-Zelllinien
(SC-115, RENCA) | ((RT-PCR)
Yoshimura 1996) |
| Hypothalamus,
Hoden | ((RT-PCR)
Yoshimura 1996) |
| Mammatumoren
(Wnt-1-Transgen) | ((Northern
blot) MacArthur 1995c) |
| Embryonales
Gehirn | ((in
situ hyb.) Crossley 1995, Heikinheimo 1994, Ohuchi 1994, Shimamura
1997, (RT-PCR) Blunt 1997) |
| Eierstöcke, Hoden | ((Northern
blot) Valve 1997) |
| Das
sich entwickelnde Gesicht und | ((pAb)
MacArthur 1995b (in |
| Extremitätenanlagen | situ
hyb.) Heikinheimo 1994, Ohuchi 1994, Crossley 1995) |
| Gastrula | ((in
situ hyb.) Crossley 1995) |
Huhn
| Embryonales
Gehirn | ((in
situ hyb.) Crossley 1996a) |
| Sich
entwickelnde Extremitätenanlagen | ((in
situ hyb.) Ohuchi 1997a,b) |
Ratte
| Prostata
und Hoden | (RT-PCR)
Scmitt 1996 |
-
Die
obige Tabelle zeigt eine breite Auswahl der Gewebeverteilung und
Zelllinien-Daten der FGF8b-Expression. Wie aus der Tabelle ersichtlich,
wurden die meisten Daten, die die Gewebeverteilung betreffen, unter Verwendung
der sensitiven RT-PCR-Methode erzeugt. Dies liegt daran, dass Northern-Blot-Analyse
nicht jede FGF8b-mRNA in allen normalen Geweben detektiert, mit
der Ausnahme von der fötalen
Niere. Aus diesen Daten wird im Allgemeinen angenommen, dass die
Expression von FGF8b-mRNA im Erwachsenen sehr beschränkt ist
und daher ein Autovakzin gegen FGF8b vermutlich nicht gegen normale
Gewebe reagieren würde. Aufgrund
der Tatsache, dass geringe Mengen von FGF8b mit unbekannten Systemen
im Erwachsenen Wechselwirken könnten,
erfordert die Gewebeverteilung des Proteins eine weitere Analyse.
Es gibt jedoch unserer Meinung nach keine Anzeichen dafür, dass
ein Autovakzin gegen FGF8b zu ernsthaften ungewollten Wirkungen
bei den Patienten führen
würde.
-
Wirkungen von Antikörpern gegen
FGF8b
-
Bisher
wurden keine Versuche publiziert, Prostatakrebs unter Verwendung
von monoklonalen Antikörpern
zu behandeln. Klinische Untersuchungen mit monoklonalen Antikör pern in
Brustkrebs-Therapie-Studien dauern jedoch an. Antikörper gegen
FGF8b werden vermutlich die Wechselwirkung zwischen FGF8b und seinen
Rezeptoren blockieren, was das Ausdehnen der Zellmembran und die
Zellproliferation inhibieren wird, und sehr wahrscheinlich die Beweglichkeit
und die invasiven Eigenschaften der Krebszellen herabsetzen wird.
-
Daher
bezieht sich die Erfindung auch auf Ausführungsformen der hierin beschriebenen
Verfahren, in denen das fremde T-Zell-Epitop in einen Teil der FGF8b-Aminosäuresequenz
der in SEQ ID NO:6, der durch die Positionen 1-54 und/oder 178-215
und/oder 55-58 und/oder 63-68 und/oder 72-76 und/oder 85-91 und/oder
95-102 und/oder 106-111 und/oder 115-120 und/oder 128-134 und/oder
138-144 und/oder 149-154 und/oder 158-162 und/oder 173-177 definiert
wird, eingefügt
wird. Es sollte erwähnt
werden, dass es insbesondere bevorzugt ist, keine Variationen oder
Modifikationen an den Positionen 26-45 und im C-Terminus beginnend
von Aminosäuren
186-215 einzufügen,
da diese Bereiche zumindest eine Homologie mit einem jüngst entdeckten
Protein, FGF18, welches in einer Vielzahl von Nicht-Tumorgeweben
zu exprimiert sein scheint, aufweisen.
-
Dementsprechend
betrifft die Erfindung auch ein Analogon von humanem/Maus-FGF8b, welches in Menschen
immunogen ist, wobei das Analogon einen wesentlichen Teil aller
bekannten und vorhergesagten CTL- und B-Zell-Epitope von FGF8b umfasst
und mindestens ein fremdes TH-Epitop umfasst,
wie hierin diskutiert. Es ist bevorzugt, dass das mindestens eine
fremde TH-Epitop als Insertion in die FGF8b-Aminosäuresequenz
oder als Substitution eines Teils der FGF8b-Aminosäuresequenz
oder als Ergebnis der Deletion eines Teils der FGF8b-Aminosäuresequenz
vorliegt. Die insbesondere bevorzugten Analoga dieser Ausführungsform
sind jene, bei denen das fremde T-Zell-Epitop in einem Teil der
FGF8b-Aminosäuresequenz
eingefügt
ist, der durch die SEQ ID NO:6-Positionen 1-54 und/oder 178-215
und/oder 55-58 und/oder 63-68 und/oder 72-76 und/oder 85-91 und/oder
95-102 und/oder 106-111 und/oder 115-120 und/oder 128-134 und/oder
138-144 und/oder 149-154 und/oder 158-162 und/oder 173-177 definiert
ist.
-
Mucine
-
Die
Erfindung betrifft auch erfindungsgemäße Verfahren, die spezifisch
modifizierte Versionen der humanen Mucine verwenden, insbesondere
irgendeines der Mucine MUC-1 bis MUC-4, bevorzugt MUC-1. Die Analoga
umfassen die folgende Struktur TR(-S-P-S-(TR)m)n – wobei
TR ein Tandem-Repeat ist, der von dem natürlich vorkommenden Mucin abgeleitet
ist, P ein fremdes TH-Epitop ist, wie hierin
diskutiert, S ein inertes Platzhalter-Peptid ist, das 0 bis 15 Aminosäure-Reste
aufweist, bevorzugt zwischen 0 und 10 Aminosäure-Resten, und n eine ganze
Zahl von 1 bis 30 ist und m eine ganze Zahl von 1 bis 10, bevorzugt
von 3 bis 5, ist.
-
Wenn
solch ein Mucin-Analogon z.B. in einer humanen Zelllinie oder durch
Aufreinigung aus einem Gewebe produziert wird, wird das direkte
Ergebnis normalerweise nicht das gewünschte Glykosylierungsmuster
aufweisen, d.h. ein falsches Glykosylierungsmuster, das jenes eines
Tumor-abgeleiteten Mucins widerspiegelt. Es ist jedoch möglich, das
Analogon rekombinant, z.B. in E. coli, oder mittels synthetischer
Verfahren herzustellen und anschließend das Produkt enzymatisch
zu glykosylieren, so dass ein Tn- oder S-Tn-Glykosylierungsmuster
erreicht wird, das spezifisch für
MUC-1 ist, welches in Tumoren exprimiert wird. Alternativ dazu kann
das Polypeptid in einer Säuger-Zelllinie
oder in einer Insekten-Zelllinie präpariert werden, z.B. in Drosophila-Zellen,
welchen das relevante Enzym fehlt, oder indem das Protein intrazellulär exprimiert
wird (indem ein Sezernierungs-Signalpeptid ausgelassen wird), wobei
eine Glykosylierung nicht auftritt.
-
Erfindungsgemäße Nukleinsäurefragmente
und Vektoren
-
Es
ist aus der obigen Offenbarung anzunehmen, dass die Analoga mittels
rekombinanter Gentechnologie, aber auch mittels chemischer Synthese
oder Semisynthese, präpariert
werden können;
die letzten zwei Optionen sind besonders relevant, wenn die Modifikation
aus der Kopplung an Protein-Trägerstoffe
besteht (wie beispielsweise KLH, Diphterietoxoid, Tetanustoxoid
und BSA), und nicht-proteinöse
Moleküle,
wie Kohlenhydratpolymere, und natürlich auch, wenn die Modifikation
die Addition von Seitenketten oder Seitengruppen an eine Polypeptid-abgeleitete
Peptidkette umfasst.
-
Zum
Zwecke der rekombinanten Gentechnologie, und natürlich auch zum Zwecke der Nukleinsäure-Immunisierung,
sind Nukleinsäurefragmente,
die die notwendigen Epitop-Regionen
und Analoga codieren, wichtige chemische Produkte. Ein wichtiger
Teil der Erfindung betrifft daher ein Nukleinsäurefragment, welches ein oben
beschriebenes Analogon von irgendeinem der relevanten Tumor-spezifischen
Polypeptide codiert, bevorzugt ein Polypeptid, in das ein fremdes
TH-Zell-Epitop mittels der Insertion und/oder
Addition, bevorzugt mittels einer Substitution und/oder Deletion,
eingefügt
wurde. Die Nukleinsäurefragmente
der vorliegenden Erfindung sind entweder DNA- oder RNA-Fragmente.
-
Die
erfindungsgemäßen Nukleinsäurefragmente
werden normalerweise in geeignete Vektoren eingefügt um Klonierungs-
oder Expressions-Vektoren zu bilden, die die erfindungsgemäßen Nukleinsäurefragmente
tragen; solche neuen Vektoren sind ebenfalls Teil der Erfindung.
Details, die die Konstruktion dieser erfindungsgemäßen Vektoren
betreffen, werden im Zusammenhang mit transformierten Zellen und
Mikroorganismen unten diskutiert. Die Vektoren können, abhängig von Zweck und Art der
Anwendung, in Form von Plasmiden, Phagen, Cosmiden, Minichromosomen
oder Viren vorliegen, aber auch nackte DNA, welche nur transient
in bestimmten Zellen exprimiert wird, ist ein wichtiger Vektor.
Bevorzugte erfindungsgemäße Klonierungs- und
Expressions-Vektoren sind in der Lage, autonom zu replizieren, dabei
hohe Kopienzahlen zum Zwecke der Expression im großen Umfang
oder Replikation im großen
Umfang für
anschließende
Klonierung zu ermöglichen.
-
Die
allgemeine Beschreibung eines erfindungsgemäßen Vektors umfasst die folgenden
Eigenschaften in 5'→3'-Richtung und funktionell
verknüpft:
einen Promotor zum Betreiben der Expression des erfindungsgemäßen Nukleinsäurefragments,
wahlweise eine Nukleinsäurese quenz,
die für
ein Leitpeptid codiert, was die Sekretion des Polypeptidfragments
oder dessen Integration in die Membran ermöglicht, das erfindungsgemäße Nukleinsäurefragment
und eine Nukleinsäuresequenz,
die für
einen Terminator codiert. Wenn mit Expressions-Vektoren in Produktionsstämmen oder
-Zelllinien gearbeitet wird, ist es zum Zwecke der genetischen Stabilität der transformierten
Zellen bevorzugt, dass der Vektor, wenn er in eine Wirtszelle eingebracht
wird, in das Wirtszellgenom integriert wird. Im Gegensatz dazu,
wenn mit Vektoren gearbeitet wird, die verwendet werden um die Expression
in vivo in einem Tier zu bewirken, ist es (d.h., wenn der Vektor
bei der DNA-Vakzinierung verwendet wird) aus Gründen der Sicherheit bevorzugt,
dass der Vektor nicht in der Lage ist, in ein Wirtszellgenom integriert
zu werden; typischerweise werden nackte DNA oder nicht-integrierende
virale Vektoren verwendet, deren Auswahl dem Fachmann wohlbekannt
ist.
-
Die
erfindungsgemäßen Vektoren
werden verwendet um Wirtszellen zu transformieren um das erfindungsgemäße Analogon
zu produzieren. Solche transformierten Zellen, welche ebenfalls
Teil der Erfindung sind, können
kultivierte Zellen oder Zelllinien sein, die für die Vermehrung der Nukleinsäurefragmente
und -Vektoren der vorliegenden Erfindung verwendet werden, oder
sie können
zur rekombinanten Produktion des erfindungsgemäßen Analogous verwendet werden.
Alternativ dazu können
die transformierten Zellen geeignete Lebend-Vakzin-Stämme sein,
bei denen das Nukleinsäurefragrnent
(eine einzelne oder multiple Kopien) so eingefügt wurden, dass sie die Sezernierung
des Analogons oder dessen Integration in die bakterielle Membran
oder Zellwand bewirken.
-
Bevorzugte
transformierte erfindungsgemäße Zellen
sind Mikroorganismen, wie Bakterien (wie die Arten Escherichia [z.B.
E. coli], Bacillus [z.B. Bacillus subtillis], Salmonella oder Mycobakterium
[bevorzugt nicht-pathogene, z.B. M. bovis BCG]), Hefen (wie Saccharomyces
cerevisiae), und Protozoen. Alternativ dazu sind die transformierten
Zellen von einem multizellulären
Organismus, wie einem Pilz, einer Insektenzelle, einer Pflanzenzelle
oder einer Säugerzelle
abgeleitet. Besonders bevorzugt sind Zellen, die von einem Menschen
abgeleitet sind, vgl. die Diskussion von Zelllinien und Vektoren
unten.
-
Zum
Zwecke der Klonierung und/oder optimierten Expression ist es bevorzugt,
dass die transformierte Zelle in der Lage ist, das erfindungsgemäße Nukleinsäurefragment
zu replizieren. Zellen, die das Nukleinsäurefragment exprimieren, sind
bevorzugte nützliche
Ausführungsformen
der Erfindung; sie können
zur Präparation
des Analogous in kleinem oder großem Maßstab verwendet werden, oder,
im Falle nicht-pathogener Bakterien, als Vakzin-Bestandteile in
einem Lebend-Vakzin.
-
Bei
der Herstellung des erfindungsgemäßen Analogons mittels transformierter
Zellen ist es üblich, wenn
auch in keinem Fall wesentlich, dass das Expressionsprodukt entweder
in das Kulturmedium hinausexportiert wird oder auf der Oberfläche der
transformierten Zellen getragen wird.
-
Wenn
eine effektive Produktionszelle identifiziert wurde, ist es bevorzugt,
auf dieser Basis eine stabile Zelllinie zu etablieren, welche den
erfindungsgemäßen Vektor
trägt und
welche das Nukleinsäurefragment,
das für
das Analogon codiert, exprimiert. Bevorzugt sezerniert diese stabile
Zelllinie das erfindungsgemäße Analogon
oder trägt
dieses Analogon, was dessen Reinigung erleichtert.
-
Im
Allgemeinen werden Plasmid-Vektoren, die ein Replikon und Kontrollsequenzen
enthalten, welche von Arten abgeleitet sind, die mit der Wirtszelle
kompatibel sind, in Zusammenhang mit den Wirten verwendet. Der Vektor
besitzt für
gewöhnlich
eine Replikationsstelle sowie Markersequenzen, welche in der Lage
sind, eine phenotypische Selektion in transformierten Zellen zu
ermöglichen.
Z.B. wird E. coli typischerweise unter Verwendung von pBR322 transformiert,
einem Plasmid, das von einer E. coli-Art abgeleitet ist (siehe z.B.
Bolivar et al., 1977). Das pBR322-Plasmid enthält Gene für Ampicillin- und Tetrazyklin-Resistenz
und stellt daher einfache Mittel zur Identifikation von transformierten
Zellen bereit. Das pBR-Plasmid, oder ein anderes mikrobielles Plasmid
oder ein Phage, muss auch Promotoren enthalten oder so modifiziert
sein, dass es Promotoren enthält,
welche von dem prokaryotischen Mikroorganismus zur Expression verwendet
werden können.
-
Jene
Promotoren, die für
gewöhnlich
bei der Konstruktion rekombinanter DNA verwendet werden, umfassen
die B-Lactamase (Penicillinase)- und Lactose-Promotorsysteme (Chang
et al., 1978; Itakura et al., 1977; Goeddel et al., 1979) und ein
Tryptophan (trp)-Promotorsystem
(Goeddel et al., 1979;
EP-A-0
036 776 ). Während
diese die am meisten verwendeten sind, wurden andere mikrobielle
Promotoren diskutiert und verwendet, und Details, die deren Nukleotidsequenzen
betreffen, wurden veröffentlicht,
was es einem Fachmann ermöglicht,
sie funktionell mit Plasmid-Vektoren zu ligieren (Siebwenlist et
al., 1980). Bestimmte Gene von Prokaryonten können effizient in E. coli von
ihren eigenen Promotorsequenzen exprimiert werden, was das Erfordernis
der künstlichen
Zugabe eines anderen Promotors ausschließt.
-
Zusätzlich zu
Prokaryonten können
auch eukaryontische Mikroben, wie Hefekulturen, verwendet werden,
und hier sollte der Promotor in der Lage sein, die Expression anzutreiben.
Saccharomyces cerevisiae, oder die gewöhnliche Bäckerhefe, ist unter den eukaryontischen
Mikroorganismen der am allgemeinsten verwendete, obwohl eine Vielzahl
anderer Stämme
allgemein verfügbar
ist, wie beispielsweise Pichia pastoris. Zur Expression in Saccharomyces
wird für
gewöhnlich
z.B. das Plasmid YRp7 verwendet (Stinchcomb et al., 1979; Kingsman
et al., 1979; Tschemper et al., 1980). Dieses Plasmid enthält schon
das trp1-Gen, welches einen Selektionsmarker für einen mutierten Hefestamm
bereitstellt, dem die Fähigkeit,
auf Tryptophan zu wachsen, fehlt, beispielsweise ATCC No. 44076
oder PEP4-1 (Jones, 1977). Das Vorhandensein der trp1-Läsion als Charakteristikum
des Hefezell-Genoms bietet dann eine wirksame Umgebung zum Detektieren
der Transformation durch Wachstum in Abwesenheit von Tryptophan.
-
Geeignete
Promotorsequenzen in Hefe-Vektoren umfassen die Promotoren für 3-Phosphoglyceratkinase
(Hitzman et al., 1980) oder andere glykolytische Enzyme (Hess et
al., 1968; Holland et al., 1978), wie beispielsweise Enolase, Glycerinaldehyd-3-phosphat-Dehydrogenase, Hexokinase,
Pyruvatdecarboxylase, Phosphofructokinase, Glucose-6-phosphat-Isomerase, 3-Phosphoglycerat-Mutase,
Pyruvatkinase, Triosephosphat-Isomerase, Phosphoglucose-Isomerase
und Glucokinase. Bei der Konstruktion geeigneter Expressionsplasmide
werden die Terminationssequenzen, die diesen Genen zugeordnet sind,
ebenfalls 3' von
der gewünschten
Sequenz gelegen, die exprimiert werden soll, in den Expressions-Vektor
ligiert um die Polyadenylierung der mRNA und dessen Termination
sicherzustellen.
-
Andere
Promotoren, welche den zusätzlichen
Vorteil der kontrollierten Transkription durch Wachstumsbedingungen
aufweisen, sind die Promotorregion für Alkohol-Dehydrogenase 2,
Isocytochrom C, saure Phosphatase, degradierende Enzyme, die mit
dem Stickstoffstoffwechsel in Verbindung stehen, und die zuvor genannte
Glycerinaldehyd-3-phosphat-Dehydrogenase,
und Enzyme, die für
Maltose- und Galactose-Nutzung verantwortlich sind. Jeder Plasmid-Vektor,
der einen Hefe-kompatiblen Promotor, einen Replikationsursprung und
Terminationssequenzen enthält,
ist geeignet.
-
Zusätzlich zu
Mikroorganismen können
auch Kulturen von Zellen als Wirte verwendet werden, die von multizellulären Organismen
abgeleitet sind. Im Prinzip kann jede solche Zellkultur verwendet
werden, ob sie nun von Vertebraten- oder Invertebraten-Kultur stammt.
Das Interesse war jedoch am größten bei
Vertebratenzellen, und die Vermehrung von Vertebratenzellen in Kultur
(Gewebekultur) wurde ein Routineverfahren in den jüngsten Jahren
(Gewebekultur, 1973). Beispiele solcher nützlichen Wirtszelllinien sind
VERO und HeLa-Zellen, CHO ("Chinese
hamster ovary")-Zelllinien
und W138-, BHK-, COS-7-, 293- und MDCK-Zelllinien.
-
Expressions-Vektoren
für solche
Zellen umfassen für
gewöhnlich
(wenn dies notwendig ist) einen Replikationsursprung, einen Promotor,
der sich vor dem zu exprimierenden Gen befindet, zusammen mit irgendwelchen
notwendigen Ribosomen-Bindestellen, RNA-Splicestellen, Polyadenylierungsstelle
und transkriptionelle Terminatorsequenzen.
-
Zur
Verwendung in Säugerzellen
werden die Kontrollfunktionen auf den Expressions-Vektoren oft von viralem
Material bereitgestellt. Allgemein verwendete Promotoren sind z.B.
von Polyoma-, Adenovirus 2 und am häufigsten Affenvirus 40 ("Simian Virus 40", SV40) abgeleitet.
Die frühen
und späten
Promotoren des SV40-Virus sind besonders nützlich, da beide recht einfach
als Fragment aus dem Virus gewonnen werden können, welches auch den viralen
Replikationsursprung von SV40 enthält (Fiers et al., 1978). Kleinere
oder größere SV40-Fragmente können ebenfalls
verwendet werden, vorausgesetzt, die ungefähr 250 by lange Sequenz, die
sich von der HindIII-Schnittstelle bis zur BglI-Schnittstelle erstreckt,
die im viralen Replikationsursprung liegt, ist davon umfasst. Weiterhin
ist es auch möglich,
und oft wünschenswert,
Promotor- oder Kontrollsequenzen zu verwenden, die normalerweise
der ge wünschten
Gensequenz zugeordnet sind, vorausgesetzt solche Kontrollsequenzen
sind mit den Wirtszellsystemen kompatibel.
-
Ein
Replikationsursprung kann bereitgestellt werden entweder, indem
der Vektor so konstruiert ist, dass er einen exogenen Replikationsursprung
enthält,
wie er von SV40 oder anderen Viren abgeleitet sein kann (z.B. Polyoma,
Adeno, VSV, BPV), oder er kann von dem chromosomalen Replikationsmechanismus
der Wirtszelle bereitgestellt werden. Wenn der Vektor in das Wirtszellchromosom
integriert wird, ist Letzteres oft ausreichend.
-
Zusammensetzungen
der Erfindung
-
Die
Erfindung bezieht sich auch auf eine immunogene Zusammensetzung,
welche als wirksames immunogenes Mittel mindestens eines der hierin
beschriebenen Analoga umfasst, im Gemisch mit einem pharmazeutisch
und immunologisch annehmbaren Träger,
Vehikel, Lösungsmittel
oder Exzipiens, und wahlweise einem Adjuvans, vgl. auch die Diskussion
dieser Dinge in der obigen Beschreibung der Erfindung.
-
Außerdem bezieht
sich die Erfindung auch auf eine Zusammensetzung zum Induzieren
der Produktion von Antikörpern
gegen irgendeines der oben diskutierten Tumorantigene, wobei die
Zusammensetzung
- – ein erfindungsgemäßes Nukleinsäurefragment
oder einen erfindungsgemäßen Vektor
und
- – ein
pharmazeutisch und immunologisch annehmbares Lösungsmittel und/oder Vehikle
und/oder Trägerstoff
und/oder Exzipiens und/oder Adjuvans
umfasst.
-
Die
Formulierung und andere spezifische Einzelheiten, die solche Zusammensetzungen
betreffen, sind in dem relevanten Teil oben diskutiert, der die
Nukleinsäure-Immunisierung
betrifft.
-
BEISPIEL 1
-
Vakzinierung gegen PSM
-
Im
Folgenden wird beschrieben, wie ein humanes Autovakzin gegen PSM
durch Modifikation des Moleküls
durch Insertion von einer oder mehreren gemischten fremden T-Zell-Epitopen entwickelt
werden kann um eine Reihe von immununogenisierten PSM-Molekülen hervorzubringen.
-
Die
Konstrukte werden auf ihre Fähigkeit
getestet, spezifische CTL-Antworten gegen PSM-enthaltende Tumorzellen
zu induzieren. Außerdem
werden die Konstrukte auf ihre Fähigkeit
getestet, Antikörper
zu induzieren, welche mit den nativen Teilen des PSM-Moleküls kreuzreagieren.
Anschließend
wird in mehreren in vitro-Assays und in vivo-Tiermodellen die Wirksamkeit
der unterschiedlichen Konstrukte ermittelt. Die induzierten Antikörper werden
auf ihre Fähigkeit
getestet, Komplement über
den klassischen Weg zu induzieren und ADCC über Fc-Rezeptoren zu initiieren.
Schließlich
werden die unterschiedlichen Moleküle in Tiermodellen für humanen
Prostatakrebs getestet.
-
Strategie
beim Gestalten eines PSM-Autovakzins
-
Kurz,
der PSM-Vakzinierungsplan umfasst die folgenden experimentellen
Aufgaben
-
Gestaltung und Herstellung
einer Reihe von humanen PSM-Mutanten
-
- – Klonierung
der PSM-Sequenzen aus Menschen und Ratten/Mäusen.
- – Mutationsarbeit
zum Erzeugen immunogenisierter hPSM-Moleküle.
- – Expression
von Wildtyp- und immunogenisierten hPSM-Molekülen in E. coli und/oder Pichia
pastoris und/oder Säugerzellen
und/oder Insektenzellen (wie der S2-Zelllinie).
- – Reinigung,
Rückfaltung
und Charakterisierung der immunogenisierten hPSM-Moleküle.
-
DNA-Vakzinierung gegen
PSM
-
- – Erzeugung
von hPSM-DNA-Vakzinierungs-Vektoren, die für immunogenisierte hPSM-Moleküle codieren.
- – Testen
der hPSM-Vakzinierungs-Vektoren in in vitro- und in vivo-Experimenten.
-
Auswahl von hPSM-Kandidaten
-
- – Immunisierung
von Mäusen/Kaninchen.
- – ELISA.
- – FACS-Analyse.
- – Im
Falle einer Antikörper-Antwort:
Tumorzell-Proliferations-Assay.
- – T-Zell-Assays.
-
Testen der hPSM-Mutanten
in vivo
-
- – Tumor/Metastasen-Modell
in Mäusen
-
Konzeptstudie: CTL-Induktion
durch Autovakzinierung
-
- – Konstruktion
von immunogenisierten Mäusen-/Ratten-PSM,
entsprechend den ausgewählten
hPSM-Kandidaten (z.B. in Form von DNA-Vakzinen).
- – Testen
der Immunantwort, die bei Mäusen/Ratten-PSM-Mutanten
in in vitro-Assays hervorgerufen wird: Immunchemische Assays, ELISA,
FACS-Analyse, zelluläre
Assays, Komplementlyse von PSM-tragenden Zellen, ADCC-Assay, CTL-Aktivitäts-Assay, Tumorzell-Proliferations-Assay,
T-Zell-Präsentations-Assays.
- – Testen
der mPSM-Mutanten in vivo in einem Tumor/Metastase-Modell in Mäusen.
-
Nomenklatur der hPSM-Konstrukte
-
PSM
ist ein Typ II-Membranprotein von 750 Aminosäuren Länge, vgl. SEQ ID NO:2, welches
die Wildtyp-Sequenz widergibt, mit der Ausnahme, dass Trp an Position
2 aufgrund der Einführung
einer NcoI-Schnittstelle und einer Kozak-Sequenz in SEQ ID NO:1
durch Gly ersetzt ist, wobei tgg an Positionen 4-6 durch ggt ersetzt
ist. Natives PSM (d.h. PSM mit einem Trp an Position 2) wurde jedoch
ebenfalls für
einige humane PSM-basierte Autovakzin-Konstrukte verwendet.
-
In
PSM macht der extrazelluläre
Teil die 707 C-terminalen Aminosäuren
aus, während
vorausgesagt ist, dass die cytoplasmatische Domäne 19 Aminosäuren lang
ist und der Transmembranteil des Proteins aus 24 Aminosäuren besteht
(As 20-43).
-
Als
Ausgangspunkt für
die Konstrukte wurde die Splicevariante PSM' verwendet. Unsere Version dieser Splicevariante
besitzt die Aminosäuresequenz,
die den Resten 58-750 in SEQ ID NO:2 entspricht. Zur Erleichterung
der Nomenklatur wurden die Regionen in PSM' entsprechend der Nummerierung in PSM
nummeriert (was bedeutet, das z.B. Region 2 aus Aminosäuren 87-108
in PSM und Aminosäuren
30-51 in PSM' besteht),
vgl. auch die untenstehende Diskussion der Regionen.
-
Alle
genetischen Konstrukte von humanem PSM sind als hPSM_._ bezeichnet
(oder hPSM'_._,
wenn PSM' als Ausgangspunkt
verwendet wurde), wobei der erste _ die Insertionsregion ist, die
zur Insertion von P2 verwendet wurde, und der zweite _ die Insertionsregion
ist, die für
P30 verwendet wurde. Wenn P2 oder P30 nicht im Protein vorhanden
sind, ist die Zahl 0 (null) angegeben. Der Volllängen-Wildtyp von hPSM wird
als hPSM0.0 bezeichnet, und das Wildtyp-hPSM, dem die cytoplasmatischen
und Transmembran-Teile fehlen, wird hPSM÷0.0 bezeichnet. Die 13 geplanten
immunogenisierten hPSM-Gene, welche alle ein P2-Epitop und ein P30-Epitop
enthalten, werden hPSM1.1, hPSM6.1, hPSM8.1, hPSM10.1, hPSM1.6,
hPSM1.8, hPSM1.10, hPSMl.2, hPSM1.3, hPSM1.5, hPSM2.1, hPSM3.1,
hPSM10.3, hPSM6.3, hPSM'10.3,
hPSM'6.3, hPSM8.3, hPSM'8.3 und hPSM5.1 genannt,
vgl. die untenstehenden Details.
-
In
hPSM1.1 sind beide Epitope, P2 und P30, aufeinanderfolgend in der
Insertionsregion No. 1 (der Membran-überspannenden Region) eingefügt. Theoretisch
kann diese Mutante, hPSM1.1, als ein sehr attraktiver Vakzin-Kandidat
zur Induktion der Antikörperproduktion
angesehen werden, da die vollständige
extrazelluläre
Domäne
dieses Moleküls
intakt ist. Zur Induktion von CTL-Antworten unter Verwendung der
Nukleinsäure-Immunisierung
werden Konstrukte, wie hPSM10.3 und hPSM6.3 für nützlich gehalten.
-
Um
die Klonierungs- und Mutagenese-Verfahren zu erleichtern, wird viel
von der molekularen Konstruktionsarbeit durchgeführt, indem entweder der N-terminale
(Aminosäuren
1-436) oder der
C-terminale (Aminosäuren
437-750) Teil des hPSM-Gens als Ausgangsmaterial verwendet wird.
Diese zwei Teile des hPSM-Gens werden hPSMI_._ und hPSMII_._ genannt,
wobei der erste _ die Insertionsregion ist, die für P2 verwendet
wurde, und der zweite _ die Insertionsregion ist, die für P30 verwendet
wurde. Wiederum ist, wenn P2 oder P30 nicht im Protein vorhanden
sind, die Nummer 0 (null) angegeben, und die Wildtypen werden hPSMI0.0
und hPSMII0.0 genannt. Eine besondere Variante von hPSMI0.0 ohne
den cytoplasmatischen Teil von hPSM wird hPSMI÷0.0 genannt.
-
In
der Praxis wird die meiste Mutagenesearbeit durchgeführt, indem
hPSMI0.0 und hPSMII0.0 als Ausgangsmaterial verwendet werden.
-
Die
exprimierten mutierten hPSM-Proteine werden PROS_._ genannt, wobei
der erste _ die Insertionsregion ist, die für die Insertion von P2 verwendet
wurde, und der zweite _ die Insertionsregion ist, die für P30 verwendet
wurde. Wenn P2 oder P30 nicht in dem Protein vorkommen, ist die
Nummer 0 (null) angegeben. Das Wildtyp-hPSM wird PROS0.0 genannt.
-
PROSII0.0
ist das Proteinprodukt mit den hPSM-Aminosäuren 437-750. HIS-getaggte
Proteine werden HIS-PROS_._ genannt. Als HIS-tags wurden SEQ ID
NO:21 zur Expression in Hefen und Bakterien verwendet, während SEQ
ID NO:23 zur Expression in Säugerzellen
verwendet wurde.
-
Klonierung
der humanen PSM-Sequenz
-
Die
LNCaP-Zelllinie, welche von einer metastatischen Läsion eines
humanen Prostata-Adenokarzinoms
stammt, wurde von der American Type Culture Collection (ATCC) bezogen.
mRNA wurde von dieser Zelllinie isoliert und unter Verwendung eines
Standard-Kits revers transkribiert um eine cDNA zu erhalten, die die
humane PSM-Sequenz codiert. Unter Verwendung von unterschiedlichen
Sätzen
von hPSM-spezifischen Primern wurden PCR-Produkte, die für PSM (437-750)
codieren, erhalten, und weiter in pUC19 kloniert (Plasmid Nummer
pMR300), und durch DNA-Sequenzierung verifiziert. Dieser C-terminale
Teil des Wildtyps-PSM wird
hPSM Teil II (hPSMII0.0) genannt. Ganz ähnlich wurde der Wildtyp-hPSM-Teil
I (hPSMI0.0) in pUC19 kloniert, und zwar unter Verwendung von Primern
zur Amplifikation von Teil I, sowohl mit (hPSMI0.0) und ohne (hPSMI÷0.0),
die Transmembran und cytoplasmatische Domänen. Die Klone wurden zur Kontrolle
sequenziert und hPSMI0.0 und hPSMII0.0 wurden unter Verwendung der
Ligation an der EcoRI-Schnittstelle fusioniert. Die daraus hervorgehenden
Klone von hPSM0.0 (SEQ ID NO:2) und hPSM÷0.0 wurden in eine Reihe
von pro- und eukaryontischen Expressions-Vektoren subkloniert und
wiederum die Sequenz verifiziert. Die intrazellulär exprimierte
Form von humanem PSM (als hPSM' bezeichnet – Aminosäuren 58-750
von SEQ ID NO:2) wurde ebenfalls konstruiert, indem die cDNA als
Ausgangsmaterial verwendet wurde. Diese Sequenz wurde ebenfalls
in Säuger-Expressions-Vektoren
subkloniert und wurde als Ausgangsmaterial für einige hPSM-Autovakzin-Konstrukte
verwendet, z.B. hPSM'10.3
und hPSM'6.3.
-
Klonierung
der Ratten- und Maus-PSM-Sequenzen
-
Zwei
EST ("expressed
sequence tag")-Klone,
die Maus-PSM-cDNA-Sequenzen (aus fötaler Mäuseniere bzw. Maus-Makrophagen)
enthielten, wurden von der American Type Culture Collection (ATCC)
bezogen. Zusammen deckten diese ESTs die Maus-PSM-cDNA-Sequenz ab, und daher
wurden sowohl Volllängen-Maus-PSM
(SEQ ID NO:7 und 8) als auch Maus-PSM' (SEQ ID NO:9 und 10) in bakterielle
Vektoren und Säuger-Expressions-Vektoren
subkloniert. Maus-PSM-AutoVac-Konstrukte wurden ebenfalls durch
Insertion von P30 in die Maus-PSM-cDNA erzeugt.
-
Expression von Wildtyp-hPSM
in E. coli
-
Der
C-terminale Teil (Aminosäuren
437-750) von hPSM, hPSMII0.0, wurde in den bakteriellen Expressions-Vektor
pET28b kloniert um ein Produkt mit einem N-terminalen Poly-Histidin (HIS)-Schwanz
zu erhalten, welcher eine einfache Reinigung in großem Maßstab und
die Identifikation mit anti-Poly-HIS-Antikörpern erleichtert. Das Proteinprodukt
von Poly-HIS getaggtem hPSMII0.0 (das Proteinprodukt wurde als HIS-PROSII0.0
bezeichnet) wurde in E. coli exprimiert.
-
Die
DNA, die für
das verkürzte
humane Wildtyp-PSM hPSM÷cyt0.0
codiert, wurde ebenfalls von pET28b im E. coli-Stamm BL21 (DE3)
exprimiert, wobei das Expressionsprodukt sich in Einschlusskörperchen befindet.
Die SDS-PAGE-Analyse von bakteriellem Lysat zeigte ein Produkt mit
der erwarteten Wanderung und die Western-Blot-Analyse mit Kaninchen-anti-HIS-PROSII0.0
ergab ebenfalls die erwartete Bande. Außerdem ergab die N-terminale Sequenzierung
von fünf
Aminosäuren
dieses Produktes, welches aus einem SDS-PAGE-Gel eluiert wurde, die erwartete
Aminosäuresequenz.
-
Die
Wildtyp-hPSM-Konstrukte hPSM0.0, hPSM÷0.0 (sowie zwei hPSM-Mutanten,
hPSM1.1 und hPSM6.1, siehe unten) wurden in verschiedene E. coli-Expressions-Vektoren
kloniert um eine möglichst
effiziente Expression und zu gewissem Ausmaß die Faltung der rekombinanten
Proteine in E. coli zu ermöglichen. Die
ausgewählten
Expressionssysteme sind:
pMCT6, welches N-terminal His-getaggte
Versionen der exprimierten rekombinanten Proteine erzeugt,
pGH433,
welches die rekombinanten Proteine in Verbindung mit einer 22 Aminosäuren langen
pe1B-Leitsequenz exprimiert, welche das Protein in den periplasmatischen
Raum der E. coli-Bakterien leiten soll, und
pMa1-p2, in dem
rekombinante Proteine als C-terminale Fusionen mit Maltosebindendem
Protein (MBP) exprimiert werden, die die natürliche periplasmatische MBP-Leitsequenz enthalten.
Antikörper
gegen MBP können
zur Detektion der Fusionsproteine verwendet werden, und eine Kohlenhydrat-gekoppelte
Säule kann
zur Affinitätsaufreinigung
des Produkts verwendet werden.
-
Die
E. coli-Expressionsexperimente der Wildtyp-hPSM-Proteine von diesen
Vektoren zeigten jedoch nur einen geeigneten Expressionslevel bei
pMCT6. Die Probleme bei dem Erhalten einer periplasmatischen Expression
der Wildtyp-hPSM-Proteine sind derzeit noch immer nicht gelöst.
-
Expression
von Wildtyp-hPSM in Pichia pastoris
-
Aufgrund
des relativ hohen Molekulargewichts des PSM-Proteins und seinem
relativ hohen Grad der Glykosylierung (ungefähr 16% des Molekulargewichts),
und um die Reinigung durch Auslassen des Rückfaltungsschritts zu erleichtern,
wurde entschieden, eine alternative Technologie zur eukaryontischen
Expression der rekombinanten Proteine zu entwickeln. Mehrere gut
charakterisierte eurkaryontische Expressionssysteme wurden untersucht,
und für
das anfängliche
Screening von hPSM-Mutanten wurde die Hefe Pichia pastoris als Alternative
zur E. coli-Expression ausgewählt.
-
Ein
Expressionssystem, das auf der Hefe Pichia pastoris basiert, wurde
an die PSM-Konstrukte
angepasst. Das Glykosylierungsmuster von rekombinanten Proteinen,
die in diesem Organismus exprimiert werden, spiegelt der Erwartung
nach das Säuger-Glykosylierungsmuster besser
als z.B. in Saccharomyces cerevisiae wider, und zwar aufgrund einer
geringeren verzweigten Mannosylierung des rekombinanten Proteins. Es
wurde gezeigt, dass die Mannose-Rezeptor-vermittelte
Aufnahme von Antigenen durch dendritische Zellen zu einer ungefähr 100fach
effizienteren Präsentation
gegenüber
T-Zellen führt,
verglichen mit der Endozytose-Aufnahme
aus der Flüssigphase.
Daher kann die Mannosylierung eine Rolle bei der Antigenizität der hPSM-Mutanten
und anderer Antigene, gegen welche eine CTL-Antwort erwünscht ist
(und insbesondere in Bezug auf die Fähigkeit, eine CTL-Antwort zu
induzieren) spielen.
-
Ein
Stamm von Pichia pastoris sowie zwei unterschiedliche Expressions-Vektoren
wurden von Invitrogen bezogen. Der Vektor pPICZαA trägt einen Methanol-induzierbaren
Promotor stromaufwärts
der Polyklonierungsstelle, während
der pGAPZααA-Vektor
Proteine konstitutiv exprimiert. Beide Vektoren codieren für das Sekretionssignal
des α-Faktors
um die rekombinanten Proteine in das Medium zu exportieren. Das
Selektionssystem dieser Vektoren ist Zeozin-Resistenz. Die Sequenzen,
die für
hPSM0.0 und hPSM÷0.0
(sowie eine hPSM-Mutante, hPSM1.1, vgl. unten) codieren, wurden
in diese Vektoren subkloniert (im Leserahmen mit einem C-terminalen
c-myc-Epitop zur Identifikation, SEQ ID NO:27).
-
Vier
Pichia pastoris-Stämme
(X-33, SMD1168, GS115 und KM71), die z.B. im Hinblick auf ihre Wachstumserfordernisse
unterschiedlich sind, wurden mit jedem von diesen (linearisierten)
Plasmiden unter Verwendung der Elektroporation transformiert. Das
Transformationsverfahren wurde mehrere Male wiederholt mit geringen
Veränderungen
um eine große
Anzahl von Zeozin-resistenten Klonen zu erhalten. Es wurde die Expression
von WildtyphPSM÷0.0
(sowie von hPSM1.1, siehe unten) in dem Pichia pastoris-System erreicht.
Die exprimierten Produkte konnten in Western-Blots von Lysaten von
Pichia pastoris-Transformanden
detektiert werden, sowohl unter Verwendung eines monoklonalen anti-hPSM-Antikörpers als
auch eines monoklonalen anti-c-myc-Antikörpers als primärem Antikörper. Die
rekombinanten Produkte wurden jedoch intrazellulär detektiert.
-
Expression
von Wildtyp-hPSM in Säugerzellen
-
Ein
Expressionssystem unter Verwendung von CHO ("chinese hamster ovary")-Zellen wird ebenfalls für das finale
Testen und die Produktion ausgewählter
Moleküle
implementiert.
-
Bisher
wurden CHO-Zellen mit Wildtyp-hPSM und hPSM1.1 mit/ohne im Leserahmen
gelegene Leitsequenz im Säuger-Expressions-Vektor
pcDNA3.1 transfiziert. Geneticin-resistente Zellen wurden erhalten.
In COS-Zellen, die transient mit denselben Konstrukten transfiziert
wurden, wurde sowohl hPSM0.0 als auch hPSM1.I im Western-Blot von
Zellpellets detektiert, und zwar unter Verwendung von monoklonalem
anti-hPSM-Antikörper.
-
Gewebeverteilung
von hPSM
-
Ein
kommerzieller Kit wurde bezogen um zu bestimmen, ob die hPSM-Expression
in verschiedenen humanen Geweben detektiert werden kann, einschließlich Prostata,
Blut und Gehirn. Dieses Verfahren basiert auf einer dot-blot-Detektion
von polyA-enthaltender mRNA, die aus 50 unterschiedlichen humanen
Geweben extrahiert wurde. Vorläufige
Ergebnisse zeigen keine Hyperexpression von hPSM in Geweben, wie
Blut oder Gehirn, an. Nach dem Prioritätstag der vorliegenden Anmeldung
haben andere jedoch die Anwesenheit von PSM in anderen Geweben nachgewiesen,
vgl. oben.
-
Gestaltung
der hPSM-Mutanten
-
Beim
Gestalten der Mutationsarbeit von PSM ist es wichtig, einige Regionen
des Proteins in den modifizierten Konstrukten zu erhalten, z.B.
potentielle und identifizierte T-Zell-Epitope, B-Zell-Epitope und Disulfidbrücken-Cystein-Reste.
Daher wurden solche "verbotenen" Regionen innerhalb
der PSM-Sequenz identifiziert, was eine begrenzte Anzahl von "offenen" Regionen von 20
Aminosäuren
oder mehr übriglässt, die
zum Austausch mit den fremden T-Helfer-Epitopen P2 und/oder P30
zur Verfügung
stellen. Per definitionem wird die Transmembranregion ebenfalls
als "offene" Region angesehen,
da Autoantikörper,
die gegen diese Region gerichtet sind, irrelevant sind, und man
glaubt, dass das Entfernen dieser Sequenz die Löslichkeit der mutierten PSM-Proteine
erhöht,
aber es kann nicht ausgeschlossen werden, dass diese Region wichtige
CTL-Epitope enthält,
daher das Bewahren der Transmembranregion, z.B. in hPSM10.3.
-
Entsprechend
unserer Erwartung, dass das Autovakzin eine CTL-Antwort hervorrufen
wird, wäre
es wichtig, möglicherweise
subdominante CTL-Epitope in den Konstrukten zu identifizieren und
zu bewahren. Zwei solcher Epitope sind schon aus der Literatur bekannt:
1) das Peptid, das die PSM-Aminosäuren 4-12 (LLHETDSAV)
umfasst, kann auf dem humanen MHC-Klasse I-Molekül HLA-A2.1 präsentiert
werden (Tjoa 1996) und 2) das PSM (711719) (ALFDIESKV) bindet ebenfalls
an HLA-A2.1 (Ref. 25). Wir haben auch die PSM-Aminosäuresequenz
durchgesucht um primäre
Ankersequenzen von HLA-Klasse I-Bindemotiven zu identifizieren, wie
von Rammensee et al. (Rammensee, 1995) für die häufigsten HLA-Klasse I-Typen beschrieben
(HLA-A1, HLA-A2, HLA-A3, HLA-A23, HLA-A24 und HLA-A28), die zusammen
80% der HLA-A-Typen der humanen Population ausmachen. Gleichermaßen wurden
potentielle HLA-B- und HLA-C-Epitope identifiziert und als "verbotene" Bereiche ausgezeichnet.
-
Die
anfängliche
Intention war es, C57/black x SJL F1-Hybridmäuse in dem Falle zu benutzen,
dass entschieden wurde, transgenische Mäuse zum Testen der PSM-Autovakzin-Konstrukte zu benutzen.
Aufgrund dieser Intention wurden bestimmte potentielle Maus-H-2b- und
H-2s-T-Helfer-Epitope identifiziert und
als "verbotene" Regionen angesehen
(Rammensee 1995). Es ist auch wichtig, bestimmte Antikörper-Binde-Regionen in
dem PSM-Molekül
zu bewahren, da sie bei der Induktion spezifischer Anti-PSM-Autoantikörper wichtig
sein könnten.
Fünf solcher
Regionen wurden schon beschrieben: PSM(63-68), PSM(132-137), PSM(482-487)
(WO 94/09820), PSM(716-723) und PSM(1-7) (Murphy, 1996). Unter Verwendung
des Computer-basierten Verfahrens von Hopp und Woods für die Vorhersage
von Antigen-Determinanten
wurden fünf
Regionen vorhergesagt: PSM(63-69), PSM(183-191), PSM(404-414), PSM(479-486)
und PSM(716-723) (Hopp 1983), wobei einige von diesen mit den expe rimentell
gefundenen B-Zell-Epitopen überlappen.
Diese Regionen werden ebenfalls in den PSM-Vakzin-Kandidatenmolekülen bewahrt.
-
Das
PSM-Protein enthält
4 Cystein-Reste (Aminosäure-Positionen
22, 196, 466 und 597), welche in den immunogenisierten Konstrukten
aufgrund ihrer potentiellen Wichtigkeit bei der Bildung von Disulfidbrücken bewahrt
werden.
-
Basierend
auf den oben erwähnten "verbotenen" und "offenen" Regionen in dem
hPSM-Protein wurden
10 Regionen identifiziert, die sich zur Insertion fremder T-Helfer-Epitope
eignen:
Insertionsregionen in hPSMI (von der Initiationsstelle
bis zur EcoRI-Schnittstelle, As. 1-437):
Region 1: As. 16-52
in PSM (4 As. vor TM, TM (24 As.) und 9 As. nach TM = 37 As.)
Region
2: As. 87-108 in PSM, As. 30-51 in PSM' (22 As.)
Region 3: As. 210-230
in PSM, As. 153-173 in PSM' (21
As.)
Region 4: As. 269-289 in PSM, As. 212-232 in PSM' (21 As.)
Region
5: As. 298-324 in PSM, As. 241-267 in PSM' (27 As.)
-
Insertionsregionen
in hPSMII (von der EcoRI-Schnittstelle bis zur Terminationsstelle,
As. 437-750):
Region 6: As. 442-465 in PSM, As. 385-408 in
PSM' (24 As.)
Region
7: As. 488-514 in PSM, As. 431-457 in PSM' (27 As.)
Region 8: As. 598-630
in PSM, As. 541-573 in PSM' (33
As.)
Region 9: As. 643-662 in PSM, As. 586-605 in PSM' (20 As.)
Region
10: As. 672-699 in PSM, As. 615-642 in PSM' (28 As.)
-
Die
Insertionsregionen sowie die "verbotenen" Regionen sind graphisch
in 4 wiedergegeben.
-
Eine
Reihe unterschiedlicher immunogenisierter PSM-Konstrukte wird durch
Substitution eines Segmentes von Aminosäuren aus zwei der oben augeführten Insertionsregionen
durch P2 oder P30 hergestellt. Jedes mutierte Protein wird daher
sowohl P2 als auch P30 enthalten, obwohl solche Konstruktionen nur
beispielhaft wiedergegeben sind – einzelne Mutanten liegen
ebenso im Umfang der vorliegenden Erfindung. Experimentell werden
die Mutationen in Klonen von hPSMI- bzw. hPSMII-cDNA vorgenommen,
und die zwei mutierten Teile werden anschließend durch Ligation kombiniert
(an der EcoRI-Schnittstelle). Die P2- und P30-Epitope wurden zuvor
in die Insertionsregionen 1, 2, 3, 5, 6, 8 und 10 insertiert um
diese Mutanten zu kreieren.
-
Die
Sequenzen von P2 und P30 sind:
P2: QYIKANSKFIGITEL (SEQ ID
NO:12, 15 As.), in diesem Fall codiert von der Nukleotidsequenz
(SEQ ID NO:11, 45 Nukleotide),
wobei die fettgedruckte Sequenz eine SacI-Schnittstelle ist. Es
kann auch eine andere Ko donauswahl auftreten, abhängig von
der Wahl des Klonierungsvektors und des Expressionssystems.
P30:
FNNFTVSFWLRVPKVSASHLE (SEQ ID NO:14, 21 As.), in diesem Fall codiert
von der Nukleotidsequenz
(SEQ ID
NO:13, 63 Nukleotide), wobei Fettdruck eine HindIII-Schnittstelle,
einzelne Unterstreichung eine Eco47III-Schnittstelle, Großbuchstaben
eine BstNI-Schnittstelle anzeigen, und die doppelte Unterstreichung eine
NheI-Schnittstelle anzeigt.
-
Die
folgende Tabelle fasst die humanen PSM-Konstrukte, die hierin verwendet
wurden, zusammen:
-
Molekulare
Konstruktionen der hPSM-Mutanten
-
Mutationen
zum Einfügen
der codierenden Sequenzen von P2 und P30 wurden vorgenommen, indem sowohl
hPSMI0.0 als auch hPSMII0.0 als Ausgangsmaterial verwendet wurden.
-
Um
die Mehrheit der hPSM-Mutanten zu erzeugen, wurden P2 und P30 anfänglich in
hPSMI0.0 an der Insertionsposition 1 eingefügt. Das daraus hervorgehende
Material (hPSMI1.0 und hPSMI0.1) wurde anschließend als Ausgangsmaterial für die Mutagenese
zum Einfügen
von P2 und P30 an Positionen 2, 3 und 5 und zur Ligation mit dem
Epitop-mutierten hPSMII verwendet. hPSMI1.0 wurde konstruiert, indem
SOE ("single overlap
extension")-PCR
verwendet wurde und anschließend
die Sequenz verifiziert wurde. hPSMI0.1 wurde konstruiert, indem
die "Quick Change"-Technik verwendet
wurde, und die Sequenz anschließend
verifiziert wurde.
-
Das
P2-Epitop wurde an Positionen 2, 3 und 5 von hPSMI1.0 unter Verwendung
von SOE-PCR zum Kreieren von hPSMI1.2, hPSMI1.3 und hPSMI1.5 insertiert.
Diese Konstruktionen wurden mit hPSMII0.0 zum Kreieren von hPSM1.2,
hPSM1.3 und hPSM1.5 kombiniert.
-
hPSMI2.1,
hPSMI3.1 und hPSMI5.1 wurden durch SOE-PCR unter Verwendung von
hPSMI0.1 als Ausgangsmaterial konstruiert. Dieses Material wurde
durch Ligation an der Eco-RI-Schnittstelle
mit hPSMII0.0 zusammengesetzt um die Volllängen-Mutanten hPSM2.1, hPSM3.1
und hPSM5.1 zu kreieren.
-
Das
P2-Epitop wurde an drei unterschiedlichen Positionen von hPSMII0.0
insertiert um hPSMII6.0, hPSMII8.0 und hPSMII10.0 zu kreieren, und
zwar unter Verwendung der "Quick
Change"-Technik,
und die Sequenz dieser Klone wurde anschließend verifiziert.
-
Anschließend wurde
hPSMI0.l mit hPSMII6.0, hPSMII8.0, hPSMII10.0 unter Erhalt von hPSM6.1, hPSM8.1
und hPSM10.1 ligiert, und die Sequenz der Klone wurde verifziert.
-
Die
Insertion des P30-Epitops in hPSMII wird derzeit vorgenommen um
hPSMII0.6, hPSMII0.8 und hPSMII0.10 unter Verwendung der SOE-PCR
zu erzeugen.
-
hPSM1.1
wurde konstruiert, indem zwei Zwei-Schritt-PCR-Mutationen verwendet
wurden, gefolgt von der Ligation an einer HindIII-Schnittstelle
innerhalb der Epitopsequenz. Die Sequenz des Volllängenkonstrukts wurde
verifiziert.
-
hPSM10.3,
hPSM'10.3 und hPSM6.3
wurden konstruiert unter Verwendung der SOE-PCR. Mehrere andere
hPSM-Varianten mit sowohl P2 als auch P30, die im extrazellulären Teil
von hPSM eingefügt
wurden, werden derzeit konstruiert (hPSM'6.3, hPSM8.3, hPSM'8.3).
-
Zusätzlich zu
den ursprünglich
angedachten Mutanten, von denen jede sowohl P2 als auch P30 enthält, wurden
vier Mutanten konstruiert, welche lediglich ein einzelnes fremdes
Epitop enthalten, und deren Sequenz wurde verifiziert: hPSM1.0,
hPSM8.0, hPSM10.0 und hPSM0.1.
-
Expression von hPSM-Mutanten
in E. coli
-
In
Experimenten im kleinen Maßstab
wurden sieben hPSM-Mutanten, hPSM1.1, hPSM6.1, hPSM8.1, hPSM10.1,
hPSM2.1, hPSM3.1 und hPSM5.1 von pET28b in dem E. coli-Stamm BL21(DE3) exprimiert,
und IPTG-induzierbare Produkte der erwarteten Größe wurden in Coomassie Blau-gefärbten SDS-PAGE-Gelen identifiziert.
Ein Produkt von hPSM1.1 war jedoch nicht detektierbar. Die Expressionsspiegel
dieser hPSM-Mutanten waren, verglichen mit dem Produkt des Wildtyp-Konstruktes
hPSM÷0.0,
sehr gering. An dieser Stelle scheint ein vernünftiger Grad an Expression
der hPSM-Mutanten unter Verwendung des pET-Systems in E. coli unmöglich zu
sein, und die Verwendung von anderen E. coli-Expressionssystemen
und/oder anderen Wirtsorganismen ist daher notwendig.
-
Wie
oben erwähnt,
wurden hPSM6.1 und hPSM1.1 in unterschiedliche E. coli-Expressions-Vektoren subkloniert
um Folgendes zu erzeugen:
- – N-terminal His-getaggte Versionen
der exprimierten rekombinanten Proteine unter Verwendung des Vektors
pMCT6,
- – Versionen
der Proteine, die mit der pelB-Leitsequenz zusammen exprimiert werden,
welche das Protein in den periplasmatischen Raum der E. coli-Bakterien
leitet, und zwar unter Verwendung des Vektors pGH433, und
- – Versionen
der rekombinanten Proteine, die als C-terminales Fusionsprotein
mit Maltose-Bindungsprotein (MBP)
exprimiert werden, und zwar unter Verwendung des Vektors pMal-p2.
-
Bisher
wurde kein ausreichender Expressionsspiegel von irgendeinem dieser
Konstrukte erreicht.
-
Da
hPSM0.0 einigermaßen
in E. coli exprimiert wird, während
ein ähnlicher
Expressionsspiegel des Volllängen-hPSM0.0
oder der hPSM-Mutanten nicht beobachtet wurden, ist es möglich, dass
die Anwesenheit des cytoplasmatischen Teils des hPSM-Moleküls die Expression
der Volllängen-hPSM-Konstrukte
in E. coli irgendwie "inhibiert". Um diese Hypothese
zu testen, haben wir zunächst
zwei genetische Konstrukte von hPSM1.1 und hPSM6.1 ohne cytoplasmatische
Domänen
erstellt. In E. coli-Expressionsexperimenten ergab sich jedoch lediglich
eine schwache Expression dieser ÷cyt-Genprodukte.
-
Expression
von hPSM-Mutanten in Pichia pastoris
-
Um
das mutierte hPSM1.1-Protein in der Hefe Pichia pastoris zu exprimieren,
wurde die hPSM1.1-Sequenz (im Leserahmen mit einem C-terminalen
c-myc-Indentifikations-Epitop, SEQ ID NO:27) in die zwei unterschiedlichen
Expressionsvektoren pPICZαA
und pGAPZαA
subkloniert, und deren Sequenzen wurden verifiziert. hPSM1.1-Expression
(sowie die von hPSM÷0.0,
siehe oben) wurde intrazellulär
in den Pichia pastoris-Transformanden detektiert.
-
Expression
von hPSM-Mutanten in Säugerzellen
-
Wie
oben erwähnt,
wurde hPSM1.1 in die Säuger-Expressions-Vektoren
pcDNA3.1(+) und pZeoSV2 subkloniert, und diese Konstrukte (und andere)
können
für die
Expression in z.B. CHO-Zellen verwendet werden. Die transiente Expression
von hPSM1.1 sowie hPSM0.0 wurde in COS-Zellen erreicht, verifiziert
durch Western-Blot.
-
DNA-Vakzinierung
-
Die
DNA-Vakzinierung wäre,
wenn sie wirksam ist, für
das PSM-Autovakzin sehr geeignet – insbesondere, da gezeigt
wurde, dass diese Verabreichungsform sowohl CTL-vermittelte Immunantwort als auch Antikörperproduktion
fördert.
Daher bestand die Absicht, eine Parallelstudie mit dem Ziel durchzuführen, die
Auswirkung der DNA-Vakzinierung von Mäusen mit geeigneten Vektoren,
die für
immunogenisiertes Maus-Ubiquitin und/oder Maus-TNFα codieren,
zu untersuchen. Die DNA-Vakzinierung mit nackter DNA, die hPSM (und/oder
Mutanten) codiert, wird ebenfalls durchgeführt.
-
Machbarkeits-Studie
unter Verwendung von immunogenisiertem Ubiquitin zur DNA-Vakzinierung
-
Eine
Machbarkeits-Studie, die die Auswirkung der DNA-Vakzinierung mit
einem immunogenisierten Selbst-Protein feststellt, wurde unter Verwendung
von Ubiquitin mit einem eingefügten
T-Helfer-Epitop von Ovalbumin (UbiOVA) als Modellprotein durchgeführt. Die
Sequenzen, die für
UbiOVA sowie für
Wildtyp-Ubiquitin codieren, wurden in den Mäuse-Expressions-Vektor pcDNA3.1(-) subkloniert.
-
Gruppen
von 5 BALB/c-Mäusen
wurden mit 40 μg
pcDNA-UbiOVA oder pcDNA-Ubiquitin-Konstrukten, entweder
intramuskulär
in den Quadrizepsmuskel oder intradermal, immunisiert. Eine Kontrollgruppe
erhielt UbiOVA-Protein in vollständigem
Freund'schem Adjuvans.
Drei und sechs Wochen später
wurden die Immunisierungen wiederholt, mit dem einzigen Unterschied,
dass das UbiOVA-Protein in unvollständigem Freund'schem Adjuvans emulgiert
war.
-
Den
Mäusen
wurde gewöhnlich
Blut entnommen und die Titer von Anti-Ubiquitin-Antikörper wurden bestimmt. In den
DNA-vakzinierten UbiOVA-Gruppen wurden nur sehr schwache Anti-Ubiqutin-Antikörpertiter erzielt.
Anschließend
wurden allen Gruppen Booster-Impfungen
mit UbiOVA-Protein in unvollständigem Freund'schem Adjuvans verabreicht,
und es wurde Blut entnommen um zu bestimmen, ob die DNA-Vakzinierung
mit UbiOVA (und nicht Ubiquitin) die Antikörperantwort gegen das UbiOVA-Protein
verstärken
konnte. Die Ergebnisse dieses Experiments zeigten, dass es keinen
signifikanten Unterschied zwischen den UbiOVA-Gruppen und den Kontrollgruppen
gab, alle Mäuse
entwickelten nach diesem einzelnen UbiOVA/FIA-Boost starke Anti-Ubiquitin-Antikörper.
-
DNA-Vakzinierung
unter Verwendung von hPSM-Konstrukten
-
Derzeit
laufen verschiedene DNA-Vakzinierungsexperimente unter Verwendung
von hPSM-Konstrukten. Verschiedene humane PSM-Wildtyp- und -AutoVac-Konstrukte
(wie beispielsweise hPSM0.0, hPSM÷0.0, hPSM'0.0, hPSM1.1, hPSM10.3) wurden in DNA-Vakzinierungsvektoren
subkloniert (wie beispielsweise pcDNA3.1(+), pcDNA3.1(-), pVAX und
pZeoSV2). Bei einigen dieser Konstrukte wurden andere Leitsequenzen (wie
die CD11a-, tPA- und IL-5-Leitsequenzen; SEQ ID NO:29, 25 bzw. 31)
direkt N-terminal gelegen und innerhalb des Leserahmens eingefügt um die
Sezernierung der exprimierten hPSM-Proteine in vivo zu ermöglichen.
Alle Konstruktionen in DNA-Vakzinierungs-Vektoren wurden mittels
DNA-Sequenzierung und in vitro-Translation verifiziert.
-
Mäusen unterschiedlicher
Stämme
(wie Balb/cA, Balb/cJ, DBA/2 und A/J) wurden die oben beschriebenen
hPSM-DNA-Vakzine entweder intradermal oder intramuskulär injiziert,
und ihnen wurden mehrere Male unter Verwendung derselben Konstrukte
Booster-Impfungen verabreicht.
-
Es
wurden Serumproben während
der Immunisierungsphase entnommen und bei –20°C gelagert. Diese Proben werden
auf Anwesenheit von Antikörpern,
die mit dem Wildtyp-hPSM reagieren, analysiert.
-
Ebenso
werden Assays etabliert um bei diesen Mäusen proliferative CTL- und
T-Helfer-Reaktionen
zu beobachten.
-
Vorläufige Ergebnisse
deuten darauf hin, dass die Induktion von sowohl CTL- als auch Antikörper-Antworten
gegen PSM erreicht werden kann.
-
Reinigung/Charakterisierung
von HIS-getaggtem hPSM(437-750) (HIS--PROSII0.0)
-
HIS-getaggtes
Wildtyp-hPSMII (HIS-PROSII0.0) wurde von pET28b exprimiert, und
löslich
gemachte Einschlusskörperchen
wurden auf eine Gelfiltrations-FPLC-Säule aufgebracht und in einem
Puffer eluiert, der 8 M Harnstoff enthielt. Die Fraktionen, die
vornehmlich hPSMII enthielten, wurden verschiedenen Rückfaltungs-Bedingungen
unterworfen um das Verfahren zu optimieren. Das löslich gemachte
Produkt, das gegen Tris-Puffer dialysiert wurde, schätzte man
auf mehr als 90% rein, und zwar unter Verwendung von Silber-gefärbtem SDS-PAGE.
-
Kaninchen
wurden mit dem gereinigten HIS-PROSII0.0 immunisiert um das daraus
hervorgehende Antiserum zur späteren
Detektion und möglicherweise
Affinitätsaufreinigung
der hPSM-Mutanten zu verwenden.
-
Reinigung/Charakterisierung
von löslichem
hPSM (PROS÷0.0)
-
Wildtyp-hPSM,
dem die cytoplasmatischen und Transmembran-Teile fehlen, PROS÷0.0, wurde
in dem E. coli-Stamm BL21(DE3) nach der Induktion mit IPTG exprimiert
und konnte in Einschlusskörperchen
detektiert werden. SDS-PAGE dieses bakteriellen Lysats, gefolgt
von Western-Blot mit einem Kaninchen-anti-HIS-PROSII0.0-Antikörper, zeigte
ein Produkt mit den erwarteten Wanderungseigenschaften. Die N-terminale
Sequenzierung der ersten fünf
Aminosäuren
dieses Produkts, das aus einem SDS-PAGE-Gel eluiert wurde, zeigte
die erwartete Sequenz, die humanem PSM entspricht. Das Produkt wurde
einer großen
Reihe von Lösungs-
und Rückfaltungs-Experimenten
unterworfen. Ein Produkt, das in Lösung bleibt, kann in einem
Tris-Puffer ohne denaturierende oder reduzierende Mittel erhalten
werden, aber eine SDS-PAGE-Analyse zeigt, dass das Material vermutlich
große
Aggregate bildet. Mäuse
und Kaninchen wurden mit diesem Material immunisiert um Antikörper gegen
hPSM, z.B. zu analytischen Zwecken, zu erhalten – die Antiseren reagierten
nicht mit LNCap-hPSM.
-
Eine
Charge gewaschener E. coli-Einschlusskörperchen von PROS÷0.0 wurde
zur Immunisierung von Kaninchen zum Erzeugen eines polyklonalen
Antiserums gegen PSM präpariert.
Ungefähr
50% des Proteingehalts im nassen, pelletierten Material war PROS÷0.0. Die
Antiseren reagierten nicht mit LNCap-hPSM im Western-Blot.
-
Präparation
von KLH-konjugierten hPSM-Peptiden zur Immunisierung
-
Drei
15mer-Peptide wurden synthetisiert um ein immunogenes Konjugat bekannter
hPSM-B-Zell-Epitope mit einem Trägermolekül herzustellen
um ein polyklonales Antiserum zu erhalten, welches in der Lage ist, hPSM
zu erkennen. Diese Peptide enthalten das PSM-B-Zell-Epitop plus 5-6 flankierende
PSM-Aminosäuren an
jedem Ende.
-
Die
Peptide wurden mittels automatischer Synthese hergestellt, HPLC-gereinigt
und unter Verwendung des Edman-Abbaus zur Kontrolle sequenziert.
-
Ein
chemisch verknüpftes
Konjugat wurde durch Kreuzvernetzen des B-Zell-Epitops, das hPSM-Peptide
enthält,
mit dem KLH präpariert,
und zwar unter Verwendung eines standardmäßigen Ein-Schritt-Verfahrens
mit Glutaraldehyd als Kreuzvernetzungsmittel.
-
Synthese von P2- und P30-Peptiden
mit flankierenden hPSM-Sequenzen
-
Es
wurden sechs Peptide gestaltet, welche den P2- und P30-Epitopen
mit 5 flankierenden hPSM-Aminosäuren
an jedem Ende entsprachen. Die flankierenden Aminosäuren entsprachen
den Epitop-Insertionsstellen 6, 8 und 10. Die Peptide werden z.B.
in T-Zell-Proliferations-Assays
verwendet, aber auch für
andere Zwecke, wie ELISA oder andere in vitro-Assays. Die Peptidsequenzen
sind:
-
Die
P2- und P30-Epitope sind unterstrichen. Die Peptide wurden durch
automatische Synthese hergestellt und dem Verfahren der HPLC-Reinigung,
gefolgt von einer Kontrollsequenzierung unter Verwendung des Edman-Abbaus,
unterzogen.
-
Immunogenizitäts-Assays
-
Unterschiedliche
Experimentelle Aufbauten wurden begonnen um Material herzustellen
und die Immunogenizitäts-Assays
für zukünftige Tests
und Auswahl der mutierten PSM-Konstrukte
zu etablieren.
-
Erzeugung polyklonaler
Anti-HIS-PRO5II0.0- und Anti-KLH-PSM-Peptid-Konjugat-Antisera
-
Zwei
Kaninchen wurden mit gereinigtem HIS-PROSII0.0, dem HIS-getaggten
C-terminalen Teil
des hPSM-Proteins (Aminosäuren
437-750), emulgiert 1:1 in vollständigem Freund'schem Adjuvans, immunisiert und
mit demselben Antigen, emulgiert in unvollständigem Freund'schem Adjuvans, zweimal
eine Booster-Impfung (an Tagen 28 und 55) vorgenommen.
-
Zwei
Kaninchen wurden mit einem Cocktail, der aus dem KLH-PSM-Peptid-Konjugat
plus jedes der drei freien Peptide bestand, immunisiert. Diese drei
Peptide enthalten jeweils ein zuvor definiertes B-Zell-Epitop von
hPSM. Der Cocktail wurde in vollständigem Freund'schem Adjuvans 1:1
emulgiert. Den Kaninchen wurde zweimal (an Tagen 28 und 55) mit
demselben Antigen, emulgiert in unvollständigem Freund'schem Adjuvans, Booster-Impfungen
verabreicht.
-
Die
Kreuzreaktivität
zwischen Anti-HIS-PROSII0.0 und PSMpep005 einerseits und die Kreuzreaktivität zwischen
Anti-KLH-PSM-Peptid-Konjugat plus Peptide und HIS-PROSII0.0 andererseits
wurden in ELISA-Assays gezeigt.
-
Der
Anti-HIS-PROSII0.0-Antikörper
hat die Fähigkeit,
natives hPSM in Lysaten von LNCaP-Zellen in Western-Blots zu erkennen.
-
Immunisierung von Mäusen mit
retroviral exprimiertem hPSM0.0
-
In
diesem Stadium des PSM-Projekts ist ein großes Hindernis noch immer der
Mangel an Antikörpern, welche
in der Lage sind, korrekt gefaltetes natives hPSM zu erkennen. Daher
wurde ein Immunisierungsexperiment unter Verwendung von retroviral
exprimiertem hPSM0.0 durchgeführt.
-
Sechs
Gruppen von Balb/c-Mäusen
wurden jeweils immunisiert mit: 1) Mitomycin-C-behandelten BALB/c-Fibrosarkomzellen
(79.24.H8), transduziert mit hPSM0.0-cDNA (CMV-Koz-hPSM), 2) Mitomycin-C-behandelten
BALB/c-Fibrosarkomzellen (79.24.H8), transduziert mit leerem Vektor
(CMVBipep), 3) Verpackungszellen (BOSC), transfiziert mit hPSM0.0-cDNA (CMV-Koz-hPSM),
4) Verpackungszellen (BOSC), transfiziert mit leerem Vektor (CMVBipep),
5) Retrovirenstamm, der hPSM0.0-cDNA exprimiert (CMV-Koz-hPSM),
oder 6) Retrovirenstamm, leerer Vektor (CMVBipep).
-
Zu
verschiedenen Zeitpunkten wurde den Mäusen Blut entnommen, und die
erhaltenen Seren wurden im Hinblick auf die Reaktivität im ELISA
auf Reaktivität
gegen HIS-PROSII0.0 getestet. Leider entwickelte keine der Mäuse Antikörper, die
spezifisch die HIS-PROSII0.0-Präparation
erkennen.
-
Etablieren
eines Anti-hPSM-ELISA
-
Gereinigtes
HIS-PROSII0.0 wurde zum Beschichten von Polystyrol-Mikrotiterplatten
(Maxisorp) zum Zwecke des Etablierens eines ELISA-Assays verwendet,
und zwar zum Testen von z.B. Hybridom-Überständen oder Maus- und Kaninchen-Antiseren.
Seren von BALB/c-Mäusen, die
mit derselben Präparation
von HIS-PROSII0.0 immunisiert worden waren, zeigten Reaktivität gegen
das immobilisierte HIS-PROSII0.0 bei 0,5 μg pro Vertiefung, unter Verwendung
eines mit Meerrettich-Peroxidase markierten Kaninchen-anti-Maus-Ig als
sekundärem
Antikörper.
-
Wie
oben erwähnt,
wurde die Fähigkeit
eines Antiserums, das in Kaninchen gegen KLH-PSMpep004-PSMpep005-PSMpep006-Konjugats,
gemischt mit den freien Peptiden, gezogen wurde, mit immobilisierten
HIS-PROSII0.0 zu reagieren, unter Verwendung dieses ELISA-Assays
gezeigt.
-
Unter
Verwendung von AquaBind®-Mikrotiterplatten (vgl.
die Offenbarung in WO 94/03530, die unter anderem Mikrotiteroberflächen beschreibt,
die mit Tresyl-aktiviertem Dextran beschichtet sind, welche nun
unter der registrierten Marke AquaBind vermarktet werden) wurde
ein ELISA entwickelt, der immobilisierte PSM-Peptide (PSMpep004,
PSMpep005, PSMpep006) verwendet.
-
AquaBind®-Platten,
die mit diesen Peptiden beschichtet sind, können ein Kaninchen-Antiserum, das gegen
dieselbe Präparation
von Antigen gezogen wurde, erkennen. Wie oben erwähnt, konnte
Kaninchen-anti-HIS-PROSII0.0 auf AquaBind®-Platten,
die mit PSMpep005 beschichtet waren, detektiert werden.
-
Etablieren eines Anti-hPSM-Western-Blots
unter Verwendung von LNCaP-Zellen und dem monoklonalen Antikörper 7E11C5
-
7E11C5-B-Zell-Hybridome,
welche monoklonalen Maus-IgG2a-Antikörper gegen ein intrazelluläres Epitop
von humanem PSM sezernieren, wurden von ATCC bezogen. Kulturüberstände von
zu ungefähr
90% toten Zellen wurden gesammelt und im Western-Blot zur Detektion
von humanem PSM in sowohl Membran-angereicherten Präparationen
von LNCaP-Zellen
als auch in LNCaP-Zelllysaten verwendet. Dieser Antikörper wurde
unter Verwendung von Protein-G-Säulen
gereinigt, und seine Reaktivität
mit LNCaP in Western-Blots verifiziert.
-
Etablieren
einer FACS-Methode zum Detektieren von hPSM auf LNCaP-Zellen
-
Wir
haben zwei voneinander unabhängige
FACS-Methoden zum Detektieren von hPSM auf LNCaP-Zellen etabliert.
Mehrere Probleme werden angesprochen: Die LNCaP-Zellen wachsen sehr
langsam und in unregelmäßigen Klumpen,
und daher sollte das Verfahren, Einzelzellsuspensionen herzustellen,
optimiert werden. Zweitens wurde in der Literatur definiert, dass
das Epitop, das von dem mAb 7E11C5 erkannt wird, sich in der eytoplasmatischen
Domäne
von hPSM befindet. Daher wurde das Verfahren entwickelt um die Zellen
zu fixieren und zu permeabilisieren. Zu diesem Zweck wurde Protein-G-gereinigter
7E11C5-Antikörper FITC- konjugiert, und dieser
konnte daher ohne sekundären
Antikörper
in der FACS-Analyse verwendet werden.
-
Ebenso
wurde ein FACS-Verfahren etabliert, das den monoklonalen Anti-hPSM-Antikörper J591
verwendet, welcher ein Epitop auf dem extrazellulären Teil
von hPSM erkennt. Der Antikörper
wurde von BZL Biologicals bezogen und mit FITC konjugiert und anschließend zur
FACS-Analyse und -Sortierung von z.B. LNCaP-Zellen und hPSM-Transfektanden
verwendet (siehe unten).
-
Etablieren
eines Cytotoxizitäts-Assays
-
Ein
Verfahren zum Reinigen von dendritischen Zellen aus Maus-Knochenmark
wurde implementiert. Unter Verwendung von Modellproteinen wurde
die Immunisierung von Mäusen
mit dendritischen Zellen optimiert, welche in einem Pulsverfahren
mit Modell Klasse-I-Peptiden
und -Protein konfrontiert wurden. Mäuse wurden auch mit einem Modell-Protein
(β-Galactosidase), das
in der Form von ISCOMS formuliert war, immunisiert. T-Zellen, die
aus immunisierten Mäusen
gereinigt wurden, wurden in vitro mit unterschiedlichen Formen des
entsprechenden Antigens restimuliert. Die Fähigkeit dieser restimulierten
CTLs zum Lysieren unterschiedlicher Arten von Zielzellen (einschließlich der
Puls-markierten dendritischen Zellen, sowie Transfektanden, die retroviral-exprimiertes,
cytosolisches Klasse-I-Peptid exprimierten) wurde anschließend gemessen.
Zwei unterschidliche in vitro-Assays, die CTL-Aktivität messen,
wurden etabliert, entweder unter Verwendung der Chrom-Ausschüttung oder/und
der DNA-Fragmentierung
(JAM-Verfahren) als Maßstab
der Cytotoxizität.
Sehr schöne
Ergebnisse erhielt man mit dem β-Galactosidase-Modellprotein
und mit verschiedenen Kombinationen von MHC-Klasse I und -Klasse II-bindenden Modellpeptiden.
-
Etablierung von Werkzeugen
zum Studieren des Durchbrechens der Autotoleranz gegen Maus-PSM
in Mäusen
-
Es
besteht die Absicht, zu untersuchen, ob die Autotoleranz gegen Maus-PSM
in Mäusen
durch Immunisierung und/oder DNA-Vakzinierung gegen Maus-PSM unter
Verwendung von Maus-PSM-AutoVac-Molekülen durchbrochen werden kann.
-
Wie
oben erwähnt,
wurde eine cDNA, die für
Maus-PSM (mPSM) codiert, erhalten und dessen DNA sequenziert. Es
werden vier mPSM-Varianten-Moleküle
durch Insertion von P30 an wohldefinierten Stellen, entweder im
Volllängen-mPSM
oder mPSM', erzeugt.
Die Konstrukte sind die folgenden:
-
Zunächst werden
die mPSM-Wildtyp- und -Analogon-Moleküle in DNA-Vakzinierungs-Vektoren subkloniert und
zur DNA-Vakzinierung von Mäusen
verwendet.
-
Es
ist beabsichtigt, die Immunantworten, wie CTL-Reaktionen und Tumorelimination,
in den Mäusen zu
untersuchen. Dazu werden Maus-Tumorzelllinien mit Wildtyp-Maus-PSM
(im Leserahmen fusioniert mit einem Identifikations-tag, z.B. dem
c-myc-Epitop, SEQ ID NO:27, zu Detektionszwecken) transfiziert.
-
in vivo-PSM-Tumormodelle
-
Maus-T-Zell-Proliferations-Assay
mit P2 und P30
-
Eine
Reihe von T-Zell-Proliferationsexperimenten wurde durchgeführt um die
T-Zell-Immunogenität von P2-
und P30-Peptiden in verschiedenen Maus-Stämmen (BALB/cA (H-2d),
C3H/Hen (H-2k, DBA/1 (H-2q) und
C57BL/6 (H-2b)) festzustellen. Es ist wohlbekannt,
dass diese Epitope in Menschen gemischte Epitope sind, aber der
gemischte Charakter der T-Zelle musste auch in Mäusen unter Verwendung von einem
experimentellen Aufbau von M&Es
bestätigt
werden. Es wurde daher gezeigt, dass P30 T-Zell-immunogen in den BALB/cA-
und C57BL/6-Stämmen
ist, während
weder P2 noch P30 T-Zell-immunogen in dem C3H/Hen-Stamm ist. In DBA/1
konnten T-Zellen gegen P2 vermehrt werden.
-
Erzeugung
von hPSM-exprimierenden Maus-Tumorzellen
-
Zur
Anwendung in einem hPSM-spezifischen Tumormodell in Mäusen sowie
zur Anwendung in Tumorzell-Proliferations-Assays wurde eine Reihe
von hPSM-exprimierenden Maus-Tumorzellen etabliert. Ein Ansatz ist
es, diese Zelllinien zu erzeugen, indem die Maus-Tumorzelllinien mit retroviralen Vektoren
transduziert werden, die die Volllängen-WildtyphPSM0.0-cDNA codieren.
-
Drei
unterschiedliche Konstrukte wurden konstruiert, die die Volllängen-Wildtyp-cDNA, die humanes PSM
eingefügt
in die Polyklonierungsstelle des retroviralen Vektors CMVBipep codieren,
wobei zwei von diesen eine kurze Kozak-Sequenz, stromaufwärts vom
Startkodon gelegen, enthalten.
-
Diese
Konstrukte wurden in drei unterschiedliche Maus-Tumorzelllinien
transduziert: P815 (Mastocytom, H-2d), B16-F10
(Melanom, H-2b) und 79.24.H8 (Fibrosarkom,
H-2d), und zwar unter Verwendung der BOSC-Verpackungs-Zelllinie.
Man erhielt Geneticin-resistente Klone bei allen drei Zelltypen,
und es wurde in einer PCR-Analyse auf genomischer DNA bes tätigt, dass
die retroviralen Konstrukte in die Wirtszellen integriert waren.
Es war noch nicht möglich,
ein exprimiertes PSM-Genprodukt im Western-Blot oder in der FACS-Analyse
unter der Verwendung des monoklonalen 7E11C5-Antikörpers zu
detektieren.
-
Zwei
stabile Maus-Tumorzelllinien, die Membran-gebundenes humanes Wildtyp-PSM
enthalten, wurden durch Transfektion etabliert. Dies wurde erreicht,
indem hPSM0.0-cDNA in den Säuger-Expressionsvektor pcDNA3.1(÷) unter
der Kontrolle des CMV-Promotors und mit einer Kozak-Sequenz stromaufwärts des
Startkodons gelegen subkloniert wurde.
-
Das
daraus hervorgehende Plasmid wurde in zwei unterschiedliche Maus-Tumorzelllinien transfiziert: 79.24.H8
(Fibrosarkom, Balb/c-abgeleitet) und Sa1N (Fibrosarkom, A/J-abgeleitet).
Geneticin-resistente Kulturen wurden erhalten und einer Western-Blot- und FACS-Analyse
unterzogen, und zwar unter Verwendung der monoklonalen J591- und
7E11C5-anti-hPSM-Antikörper.
Unter Verwendung des J591-Antikörpers
wurden die Zellen über
mehrere Runden FACS-sortiert, bis eine hPSM-positive Population
erhalten wurde. Die hPSM-Expression wurde wiederum mittels intrazellulärer FACS-Färbung unter
Verwendung des 7E11C5-Antikörpers
verifiziert. Es wurde auch mittels FACS-Analyse überprüft, dass die MHC-Klasse I-Expressionsspiegel
dieselben Spiegel waren wie die Spiegel der parentalen Zelllinien.
-
Kulturen
von 79.24.H8 und SalN-Zelllinien, die hPSM exprimieren, wurden durch
limitierende Verdünnung
kloniert. Mehrere Klone wurden erhalten und mittels FACS-Analyse
unter Verwendung des monoklonalen Anti-hPSM-Antikörpers J591
auf unterschiedliche Expressionslevel von hPSM getestet.
-
79.24.H8-Zellen,
die hPSM exprimieren, wurden mit dem Gen, das für B7.1 codiert, zur Verwendung z.B.
in in vitro-Assays zum Beobachten der hPSM-spezifischen CTL-Antworten
und/oder der Interferon-gamma-Ausschüttung transfiziert. Die Zellen
wurden einmal unter Verwendung eines Anti-B7.1-Antiserums mittels FACS
sortiert.
-
Etablieren eines hP5M-spezifischen
Tumormodells in Mäusen
-
Es
wurde der Entschluss gefasst, mindestens zwei in vivo-Tumormodelle
in immunkompetenten Mäusen
zu etablieren um die Anti-Tumor-Wirkung von Antikörpern zu
bestimmen, die in Mäusen
gegen die immunogenisierten hPSM-Moleküle gezogen wurden. Dies wird
hoffentlich durch Injizieren in syngenische Maus-Tumorzelllinien
geschehen, die so modifiziert wurden, dass Wildtyp-hPSM auf der
Oberflächenmembran
exprimiert wird. Es werden Zellen verwendet werden, welche feste
Tumoren bilden, und/oder Zellen, von denen bekannt ist, dass sie
metastasieren. Es werden Zelllinien in diesem Modell verwendet,
welche in syngenische Mäuse
implantiert werden können,
ohne aufgrund der Anwesenheit des fremden hPSM-Moleküls abgestoßen zu werden. Die Fähigkeit
der hPSM-Vakzine, solche Tumorzellen zu eliminieren, wird für die Auswahl
der hPSM-Vakzin-Kandidaten verwendet werden.
-
Um
das Wachstum von SalN-Zellen, die mit dem humanen Volllängen-PSM
transfiziert waren, zu ermitteln, wurden unterschiedliche Dosen
(2 × 106 und 5 × 106) der mit hPSM transfi zierten Sa1N-Zellen
(S-PSM, 5 Mal sortiert) subkutan an der rechten unteren Flanke von
Gruppen von A/J-Mäusen
injiziert. Es entwickelten sich jedoch keine festen Tumoren. Anschließend wurden
drei Klone von S-PSM-Zellen mit unterschiedlichen Expressionsleveln
von hPSM subkutan in drei Gruppen von A/J-Mäusen mit einer Dosis von 107 Zellen/Maus injiziert. Die Größen der
etablierten Tumore wurden mittels einer Kaliberlehre gemessen, wobei
zwei unterschiedliche Durchmesser gemessen wurden, die multipliziert
wurden um die Tumorgröße in mm2 zu ergeben. Diese Werte wurden bei den
drei Gruppen verglichen. Innerhalb von 3-6 Tagen entwickelten alle
Mäuse eine feste
Tumor-ähnliche
Struktur, welche ungefähr
am Tag 15 wieder verschwand. Dies liegt wahrscheinlich in der Anwesenheit
von humanem hPSM auf den Tumorzell-Oberflächen begründet, obwohl es nicht verifiziert
wurde. SalN-Zellen, die nur mit dem pcDNA3.1-Vektor transfiziert
waren, setzten ihr Wachstum als fester Tumor in Mäusen fort.
-
Ein ähnliches
Bild wurde bei Mäusen
beobachtet, denen 106, 5 × 106 oder 107 79.24.H8-Zellen injiziert wurden,
welche mit hPSM transfiziert und über mehrere Runden im Hinblick
auf hPSM-Expression sortiert worden waren. Diese Zellen (79-PSM
genannt) etablierten sich ebenfalls nicht als Tumore in Balb/c-
oder DBA/2-Mäusen.
Wenn jedoch ein Klon hPSM-transfizierte
79.24.H8-Zellen, 79-PSM.3, in Balb/c- oder DBA/2-Mäuse injiziert
wurde, entwickelten die Mäuse
feste Tumor-ähnliche
Strukturen, welche wiederum um Tag 10-20 herum verschwanden. Lediglich
mit Vektor transfizierte 79.24.H8 setzten ihr Wachstum in Balb/c-Mäusen fort.
-
Es
muss noch immer bestimmt werden, ob diese "Tumore" behandelbar sind, oder ob ein besseres Tumormodell,
basierend auf den beschriebenen S-PSM- und 79-PSM-Zelllinien und
-Klonen etabliert werden kann.
-
Schlussfolgerungen
-
Bei
der molekularen Konstruktionsarbeit waren wir erfolgreich bei der
Klonierung des humanen PSM-Gens und beim Erhalten der Maus-PSM-cDNA.
Eine Reihe von vollständig
sequenzierten, immunogenisierten hPSM-Autovakzin-Konstrukten wurde
hervorgebracht. Die hPSM-Mutanten sowie unterschiedliche Wildtyp-hPSM-Moleküle wurden
in E. coli exprimiert, und man fand heraus und verifizierte, dass
die Expressionslevel in E. coli sehr niedrig sind. Polyklonale Antikörper gegen
die C-terminale Hälfte
von hPSM wurden in Kaninchen induziert. Es wurden Anstrengungen
unternommen, unterschiedliche Expressions-tags (HIS-tag und Maltose-Bindeprotein-Fusion)
sowie Expressionssysteme alternativ zu E. coli-Einschlusskörperchen
zu implementieren. Rekombinantes Wildtyp- und/oder Autovakzin-hPSM
wurde in transfiziertem Pichia pastoris und in Säugerzellen detektiert. Nützliche
Erwägungen,
die die DNA-Vakzintechnologie
betreffen, wurden vorgenommen, und eine vorläufige Machbarkeitsstudie wurde
durchgeführt.
DNA-Vakzinierungsexperimente mit hPSM-Autovakzin-Molekülen dauern
an und zeigen vielversprechende vorläufige Ergebnisse. Unterschiedliche
in vitro-Assays, die zum Testen und zur Auswahl der mutierten PSM-Konstrukte
nützlich
sind, sind etabliert, einschließlich
immunchemischer Assays und der FACS-Analyse. Maus-Tumorzellen wurden
stabil mit Volllängen-Wildtyp-hPSM
transfiziert und mittels FACS im Hinblick auf hPSM-Oberflächenexpression
sortiert. Man hat Klone dieser Zelllinien erhalten. Xenogenische
in vivo-Tumor-Modelle
in Mäusen
werden derzeit untersucht unter Verwendung dieser hPSM-enthaltenden syngenischen
Maus-Tumorzellen. Eine Reihe von T-Zell-Proliferations-Assays wurde
durchgeführt
um die Maus-Stämme
für die
Tumormodelle zu selektieren. CTL-Assays werden optimiert, und überzeugende
Ergebnisse mit Modell-Antigenen wurden erhalten, indem unterschiedliche
Immunisierungsmethoden und Assay-Bedingungen verwendet wurden. Außerdem werden Werkzeuge
etabliert, die notwendig sind um das Durchbrechen der Toleranz gegen
Maus-PSM durch Immunisierung gegen Maus-PSM-Autovakzine zu untersuchen.
-
BEISPIEL 2
-
Herstellung eines Her2
Autovakzins
-
Ein
humanes Autovakzin gegen Her2 kann durch Modifikation des Moleküls durch
Insertion von einem oder mehreren gemischten, fremden T-Zell-Epitopen
unter Hervorbringen einer Reihe von immunogenisierten Her2-Molekülen entwickelt
werden. Diese modifizierten Proteine werden auf ihre Fähigkeit
hin getestet, Antikörper
zu induzieren, welche mit den nativen Teilen des Her2-Moleküls kreuzreagieren.
Anschließend
wird in mehreren in vitro-Assays und in vivo-Tiermodellen die Wirksamkeit
der unterschiedlichen Konstrukte (wie das im Falle der DNA-Vakzinierung
der Fall sein kann) und der modifizierten Proteine ermittelt. Die
Induktion von spezifischen CTL-Antworten gegen Her2-enthaltende
Tumorzellen wird analysiert. Die induzierten Antikörper werden
auch auf ihre Fähigkeit
hin getestet, Komplement über
den klassischen Weg zu aktivieren und eine ADCC über Fc-Rezeptoren zu initiieren.
Schließlich
werden die unterschiedlichen modifizierten Moleküle in Tiermodellen von humanem
Brustkrebs getestet um ihre Wirkungen bei der Behandlung von Tumoren
zu untersuchen.
-
Immunogenische
Ratten und humane Moleküle
werden mit gemischten T-Zell-Epitopen an unterschiedlichen Positionen
in dem Molekül
konstruiert.
-
Während der
Vakzinierung gegen die gesamte extrazelluläre Domäne von Her2 besteht eine Möglichkeit
eines gewissen Grades an Kreuzreaktion der Antikörper mit anderen EGFr-Rezeptoren, da einige
dieser Rezeptoren mit bis zu 40-46% in den extrazellulären Domänen homolog
sind. Daher ist es geplant, dass die konservierten Regionen von
Her2 durch Einfügen
von fremden T-Zell-Epitopen mindestens in einigen der modifizierten
Proteine zerstört
sind (siehe unten für
Details).
-
Regionen
von Her2, die möglicherweise
CTL- oder B-Zell-Epitope sein können,
werden beim Gestalten von Konstrukten vermieden, wie in 3 ersichtlich.
Die Richtschnur zur Verwendung dieser Positionen ist die folgende:
Die
humane Her2-Sequenz kann in eine Reihe von Domänen geteilt werden, was vollständig auf
der Primärstruktur
des Proteins basiert.
-
Extrazellulärer (Rezeptor-)
Teil:
-
- 1-173: Domäne
I (N-terminale Domäne
des reifen Polypeptids).
- 174-323: Domäne
II (Cystein-reiche Domäne,
24 Cystein-Reste).
- 324-483: Domäne
III (Liganden-bindende Domäne
i des homologen EGF-Rezeptors).
- 484-623: Domäne
IV (Cystein-reiche Domäne,
20 Cystein-Reste).
- 624-654: Transmembran-Domäne
(TM-Reste von 654-675).
-
Intrazellulärer (Kinase-Teil:
-
- 655-1010: Tyrosinkinase-Domäne (Kern-TK-Domäne von 725-992).
- 1011-1235: C-terminale Domäne.
-
Die
Selektion der zu ersetzenden Stellen in der Aminosäuresequenz
von Her2 durch entweder die humane P2- oder die P30-T-Helfer-Epitope
wurde unter Beachtung der folgenden Parameter vorgenommen (schwach
priorisiert):
- 1. Bekannt und vorhergesagte
CTL-Epitope
- 2. Homologie gegenüber
verwandten Rezeptoren (EGFR insbesondere)
- 3. Konservierung von Cystein-Resten.
- 4. Vorhergesagte Haarnadel, α-Helix-
und β-Faltblatt-Strukturen
- 5. Potentielle N-Glykosylierungsstellen
- 6. Vorhersage exponierter und verborgener Aminosäurereste
- 7. Domänenorganisation
-
Die
CTL-Epitope scheinen in Domäne
I, Domäne
III, der TM-Domäne
und in zwei oder drei "hot
spots" in der TK-Domäne im Cluster
vorzuliegen. Entsprechend der Erfindung sollten diese weitestgehend
konserviert werden.
-
Regionen
mit einem hohen Grad an Homologie mit anderen Rezeptoren sind sehr
wahrscheinlich strukturell wichtig für die "Gesamt"tertiärstruktur von Her2, und daher
für die
Antikörpererkennung,
während Regionen
mit niedriger Homologie möglicherweise
ausgetauscht werden können,
mit der Folge von lediglich lokalen Änderungen der Struktur.
-
Cystein-Reste
sind häufig
in intramolekulare Disulfidbrücken-Bildung
involviert und daher kritisch für die
Tertiärstruktur,
und sie sollten nicht verändert
werden.
-
Regionen,
von denen vorhergesagt wurde, dass sie α-Helix- oder β-Faltblatt-Strukturen
bilden, sollten bevorzugt als Insertionspunkte fremder Epitope vermieden
werden, da diese Regionen vermutlich für die Faltung des Proteins
wichtig sind.
-
Potentielle
N-Glykosylierungsstellen sollten bevorzugt auch konserviert werden,
weil eine Mannosylierung des Proteins (beispielsweise durch Expression
in Hefe) erwünscht
ist, vgl. die Anwesenheit von Mannose-Rezeptoren auf APCs.
-
Regionen,
von denen vorhergesagt ist (aufgrund ihrer hydrophoben Eigenschaften),
dass sie im Inneren des Moleküls
vorliegen, sollten konserviert werden, da diese in die Faltung involviert
sein könnten.
Im Gegensatz dazu können
Regionen, die dem Lösungsmittel
ausgesetzt sind, als Kandidaten-Positionen zur Insertion der Modell-TH-Epitope P2 und P30 dienen.
-
Schließlich wurde
auch die Domänenorganisation
des Proteins aufgrund ihrer Relevanz für Protein-Struktur und -Funktion
in Erwägung
gezogen.
-
Der
Hauptpunkt der Strategie war es, die Struktur des extrazellulären Teils
von Her2 soviel wie möglich zu
konservieren, weil dies der Teil des Proteins ist, welcher als Zielobjekt
für neutralisierende
Antikörper
relevant ist. Im Gegensatz dazu ist der intrazelluläre Teil
von nativem Membran-gebundenen Her2 auf der Oberfläche von
Krebszellen für
das humorale Immunsystem unzugänglich.
-
Daher
gibt lediglich das Vorhandensein von CTL-Epitopen Grund dazu, diesen
Teil in ein Vakzin mit einzubeziehen. Es ist daher offensichtlich,
hier ein oder mehrere Epitope zu plazieren. Wenn sich herausstellt, dass
es unmöglich
ist, das Volllängen-Her22-Molekül in E.
coli oder in Hefe zu exprimieren, könnte der intrazelluläre Teil
nach dem ersten CTL-Epitop-"hot
spot" (um Position
800 herum) verkürzt
werden. Zusätzliche CTL-Epitope
können
danach an das C-terminale Ende des verkürzten Moleküls angefügt werden.
-
Die
Transmembran-Region ist vermutlich eine unabhängige Faltungseinheit, und
das Ersetzen von dieser durch TH-Epitope,
wie P2 oder P30, wird vermutlich die HER2-Struktur und -Faltung
nicht beeinflussen. Zusätzlich
könnte
die TM-Domäne
größere Probleme
für die
Expression in Hefe und E. coli verursachen und sollte in jedem Fall
ersetzt werden. Daher sollte ein Epitop in allen Konstruktionen
bevorzugt in dieser Domäne plaziert
werden (wobei sie vielleicht in einer Konstruktion intakt belassen
wird, da sie mehrere CTL-Epitope enthält und weil sie manchmal irgendwie
bei der Transmission von Signalen nach Liganden-Bindung involviert
ist).
-
Die
extrazelluläre
Domäne
könnte
im Prinzip intakt belassen werden, indem man P2 und P30 in der intrazellulären und
der Transmembran-Domäne
plaziert, wodurch die Anzahl möglicher
B-Zell-Epitope maximiert wird und die Struktur so wenig wie möglich gestört wird.
Der hohe Grad an Homologie mit EGFR und Her-3 und Her-4 erzeugt
ein Risiko der Kreuzreaktivität
mit diesen Rezeptoren, welches unerwünscht sein kann (oder auch
nicht). Zusätzlich
wurden einige monoklonale Antikörper
beschrieben, welche als Agonisten für den Rezeptor fungieren (vielleicht
durch Stimulieren der Heterodimerisierung oder der Liganden-Bindung) und
welche die Tumorgröße, in vivo,
erhöhen.
Positionen in der extrazellulären
Domäne
wurden daher ausgewählt,
welche diese Risiken hoffentlich dadurch reduzieren werden.
-
Diese
Auswahl hat alle der zuvor genannten Parameter mit einbezogen, und
sie wurde auf zwei unterschiedlichen Annahmen gefußt: (i)
Eine Insertion in nicht-konservierte (im Hinblick auf verwandte
Rezeptoren) Regionen hilft dabei, die Tertiärstruktur aufrecht zu erhalten
und könnte
ungewollte Aktivierung durch Antikörper reduzieren. (ii) Die Insertion
in die wohl-konservierten
Regionen könnte
die Struktur ändern,
aber könnte gleichzeitig
das Risiko der Kreuzreaktivität
verringern, indem die am stärksten
verwandten Sequenzen zerstört werden.
Beide Annahmen sind spekulativ, aber da es sehr schwierig ist, die
Wirkung der Platzierung eines Epitops in jeder Position des Proteins
vorherzusagen, wurden einige dieser Positionen in die Konstruktionen
mit einbezogen.
-
Es
wurde spekuliert, dass es ein Vorteil sein könnte, die zwei Cystein-reichen
Domänen
vollständig
zu entfernen. Von diesen ist vorhergesagt, dass sie lösliche,
exponierte Schleifenstrukturen bilden und unabhängige Faltungseinheiten bilden
können,
die vielleicht in die Dimerisierung involviert sind (angedeutet
durch die vielen Cysteine, die dazu dienen könnten, eine feste Struktur
aufrecht zu erhalten, die für
eine Dimerisierungs-Domäne
notwendig ist). Das Zerstören
dieser Strukturen könnte
daher das Risiko der Aktivierung durch Antikörper ausschalten sowie die
Anzahl kreuzreagierender Antikörper
reduzieren, da diese Domänen
die am besten konservierten Domänen
des extrazellulären
Teils des Proteins sind. Zusätzlich
könnte
es problematisch sein, solche Cystein-reichen Domänen in E.
coli oder Hefezellen herzustellen.
-
Die
Details der Konstrukte sind die folgenden, unter Verwendung der
P2- und P30-Epitope
als nicht-einschränkende
Beispiele: das P2-Epitop wird in erster Linie in der extrazellulären Domäne von Her2
plaziert, in Kombination mit dem P30-Epitop, das einen Teil von
der oder die gesamte Membran-überspannende Region
ersetzt. Das P2-Epitop wird, basierend auf den oben diskutierten
Kriterien, in den Regionen plaziert. Die bevorzugten Konstrukte
besitzen die folgenden Strukturen:
-
Die
Position des Epitops zeigt die erste und die letzte Aminosäure-Position
des Epitops in Bezug auf den Startpunkt des reifen Her2 an. Die
Länge ist
die Länge
in Aminosäuren
von dem vollständigen
Konstrukt. In allen Konstrukten, mit Ausnahme jener, in denen die
Position mit einem * angegeben ist, ersetzt das Epitop einen Aminosäurestrang
derselben Länge. "*" deutet darauf hin, dass das Epitop
in Her2 eingefügt
wird und nicht substituiert. Alle oben aufgelisteten Konstrukte
sind daher Verkürzungen
des reifen Her2, wobei der ausgelassene Teil vom C-Terminus stammt.
-
Die
meisten Konstruktionen liegen in unterschiedlichen Versionen vor,
z.B. in einem pcDNA3.1+-Vektor, in Fusion mit der natürlichen
HER2-Signal-Peptidsequenz, in dem Vektor pMT/BiP/VS-His-A als Fusion
mit dem BiP-Leitpeptid zur Expression in Drosophilazellen, und ohne
Leitsequenz in dem pET28b-Vektor zur Expression in E. coli-Zellen.
-
Unten
sind die Modelle beschrieben, die zur Verwendung beim Screening
und der Selektion modifizierter Her2-Proteine vorgesehen sind.
- 1. Die Induktion von Antikörpern gegen Maus-Her2 in transgenischen
Ratten, und in Kaninchen gegen Ratten- und humanes Her2 wird mittels
konventioneller ELISA-Technologie
nach mindestens drei Immunisierungen untersucht. Kommerziell erhältliche
Antikörper
gegen menschliches und Ratten-Her2 werden als Positivkontrollen
verwendet.
- 2. Diese Kaninchen-Antikörper
werden verwendet um die vermutete Inhibierung des Wachstums von
humanen und transgenischen Maus-Tumorzellen, die Her2 überexprimieren,
in einem in vitro-Modell zu untersuchen.
- 3. Die T-Zell-Proliferation von PBL von Tetanus-immunisierten
Patienten gegen ausgewählte
humane Her2-Moleküle
wird mittels herkömmlicher
Verfahren untersucht.
- 4. Die Fähigkeit
von modifizierten Ratten-Her2-Molekülen, CTL-Antworten in Ratten-Her2-transgenischen Mäusen zu
induzieren, wird unter Verwendung von Tumoren aus diesen Mäusen als
Zielobjekten untersucht.
- 5. Es ist beabsichtigt, eine ausgewählte Reihe von Peptiden in
der Transmembranregion von humanem Her2 zu synthetisieren, die P2-
und P30-Epitope umfassen. Diese Peptide werden auf Proliferation
von PBL von Menschen, die zuvor mit Tetanus-Toxoid immunisiert wurden,
getestet um zu bestimmen, ob P2- und P30-Epitope aus den Her2-Sequenzen effizient
herausprozessiert werden können
und gegenüber
T-Zellen präsentiert
werden können.
- 6. Es ist recht gut möglich,
dass ausgewählte
humane, modifizierte Her2-Proteine auf die Bildung neutralisierender
Antikörper
in einer Maus getestet werden, welche genetisch so konstruiert wurde,
dass sie nur humane VDJ-Gene exprimiert. Solch eine Maus ist von
Abgenix, Fremont, CA, USA, in Zusammenarbeit erhältlich.
-
Vier
gut charakterisierte transgenische Maus-Modelle für Brustkrebs
wurden beschrieben, die ein Ratten-Her2-Onkogen enthalten. Die ersten
drei transgenischen Mäuse
exprimierert aktiviertes Her2-Onkogen, während das vierte Modell ein
inaktiviertes Her2 verwendet. Alle Modelle verwenden einen MMTV-Promotor um
die Expression in Milchdrüsen
anzutreiben.
-
Wir
haben entschieden, zwei transgenische Maus-Modelle zu verwenden:
1) ein aggressiveres Tumormodell, das von Muller et al. beschrieben
wurde, das aktiviertes Her2-Onkogen verwendet (Muller et al., 1989), und
2) ein weniger aggressives Tumormodell, bei dem inaktiviertes Her2
verwendet wird um fokale Mammatumoren mit einer langen Latenzzeit
zu bilden (Guy et al., 1992). Beide transgenischen Mäuse werden
von Jackson und/oder Charles Rivers Laboratories bezogen.
-
In
den anfänglichen
Experimenten wird es diesen Mäusen
ermöglicht,
Antikörper
und CTL-Antworten hervorzubringen, und zwar indem mit modifizierten
Ratten-Her2-Proteinen immunisiert wird und Booster-Immunisierungen
verabreicht werden. Das Auftreten von Tumoren wird dann, wie von
anderen beschrieben, untersucht (Muller et al., 1998; Guy et al.,
1992; Katsumata et al., 1995). Die Antikörper-Spiegel werden mittels eines
ELISA-Assays gemessen. Die CTL-Aktivität wurde bestimmt, indem Zielzellen
erzeugt werden, die Ratten-Her2 exprimieren, wie oben erwähnt.
-
Alternativ
dazu kann das Nacktmäuse-Xenotransplantation-Karzinom-Modell
für passive
Vakzinierungsexperimente verwendet werden. Nacktmäuse können mit
humanen Tumoren transplantiert werden, und die Inhibierung von Tumoren
könnte
durch passiven Transfer von Serum von normalen oder humanisierten Mäusen, die
mit modifzierten Her2-Proteinen immunisiert werden, erreicht werden.
Während
dies nützlich wäre um die
Rolle von Antikörpern
bei der Unterdrückung
von Tumoren zu untersuchen, kann die CTL-Aktivität nicht direkt in diesem System
gemessen werden.
-
In
dem zweiten in vivo-Modell würden
auch Tumoren in Mäusen
erzeugt, indem Zelllinien von Tumoren transgenischer Mäuse, die
oben beschrieben sind, transplantiert würden. Zelllinien, die aus diesen
Mäusen
erzeugt würden,
würden
in relevante Maus-Stämme übertragen,
und deren Lokalisation würde
unter Verwendung von Standardprotokollen festgestellt.
-
Die Übertragung
von Maus-Tumorzellen, die Ratten-Her2 überexprimieren: In diesem System
werden Zellen mit Rattengenen transfiziert und in MHC-kompatible
Mäuse übertragen.
Die Inhibition des Tumorwachstums würde erreicht, indem Anti-Her2-Antworten erzeugt
würden.
-
In
diesen Systemen würden
modifizierte Her2-Proteine als Vakzin in Adjuvantien zum Erzeugen
von Antikörpern
und CTL-Reaktionen verwendet.
-
Die
DNA-Vakzinierung wurde erfolgreich in mehreren Systemen zum Hervorrufen
einer wirksamen Immunantwort verwendet. Wir untersuchen derzeit
Mittel der DNA-Übertragung
unter Verwendung von modifizierten Selbst-Proteinen. Es ist unsere
Absicht, den DNA-Vakzinierungs-Ansatz
zu benutzen um die Wirkungen von modifizierten Her2-Konstrukten
bei der Inhibierung von Tumoren in transgenischen Maus-Modellen für Brustkrebs
zu bestimmen. Ein ähnlicher
Ansatz kann dann möglicherweise
in Menschen für
die Behandlung dieser Krankheit angewandt werden.
-
BEISPIEL 3
-
Herstellung eines Anti-FGF8b-Vakzins
-
Im
Folgenden wird beschrieben, wie ein humanes Autovakzin gegen FGF8b über die
Modifikation des Moleküls
durch Insertion von einem oder mehreren gemischten fremden T-Zell-Epitopen entwickelt
werden könnte
um eine Reihe immunogenisierter "FGF8b"-Moleküle hervorzubringen.
Die Konstrukte werden auf ihre Fähigkeit
getestet, Antikörper
zu induzieren, die mit den authentischen Teilen des FGF8b-Moleküls kreuzreagieren.
Anschließend
wird in mehreren in vitro-Assays und in vivo-Tiermodellen die Wirksamkeit
der unterschiedlichen Konstrukte ermittelt. Die induzierten Antikörper werden
auf ihre Fähigkeit
getestet, Komplement über
den klassischen Weg zu aktivieren und ADCC über Fc-Rezeptoren zu initiieren.
Schließlich
werden die unterschiedlichen Moleküle in Tiermodellen für humanen
Prostata- und Brustkrebs getestet.
-
Konstruktion eines Autovakzins
gegen FGF8b
-
Aufgrund
der vollständigen
Identität
der Maus- und Mensch-FGF8b-Polypeptide können alle Konstrukte für Experimente
in sowohl Menschen als auch Mäusen
verwendet werden.
-
Die
gemischten Tetanus-Toxoid-T-Helfer-Zell-Epitope P2 und P30, die
erfolgreich in dem humanen TNFa-Vakzin verwendet wurden, werden
in das FGF8b-Polypeptid eingefügt.
Aufgrund der geringen Größe von FGF8b
werden Konstrukte hergestellt mit einem Epitop pro Molekül. Andere
gemischte T-Helfer-Zell-Epitope, wie das Influenza-Haemagglutinin-Epitop
HA(307-319) und andere T-Zell-Epitope, die hierin diskutiert werden,
könnten
ebenfalls in Erwägung
gezogen werden (O'Sullivan
1991).
-
4
unterschiedliche immunogenisierte FGF8b-Konstrukte wurden hergestellt,
wobei die Epitope über das
Molekül
verteilt waren. Diese vier Konstrukte werden auf der Basis multipler
und paarweiser Aneinanderanlagerungen der FGF-Familie von Proteinen
erstellt. Eine paarweise Gegenüberstellung
von FGF2 und FGF8b wird als Grundlage zur Analyse der vermuteten Sekundärstruktur
entlang des FGF8b-Moleküls
(d.h. der beta-Faltblatt-Verteilung) verwendet. Die Reste, die bei
FGF2 und FGF8b konserviert sind, bilden nirgendwo auf der dreidimensionalen
Struktur Cluster, was anzeigt, dass es keine bestimmten Regionen
des Moleküls gibt,
die nicht ersetzt werden könnten,
ohne zerstörerische
Wirkungen auf die Faltungsfähigkeiten
zu haben. Die Aminosäure-Reste
in FGF2, die den Cystein-Resten in FGF8b gegenüberstehen, liegen dreidimensional sehr
nah beieinander, was anzeigt, dass sie in FGF8b eine Disulfidbrücke bilden,
und dass die Gegenüberstellung
korrekt ist. Die Flexibilität
des N-terminalen Teils von FGF2 wurde ebenfalls beachtet.
-
Die
Variante von FGF8b mit dem P30-Epitop am N-Terminus (F30N) wurde
auf Grundlage einer lückenlosen
Gegenüberstellung
der Aminosäure-Reste
des FGF8b-Proteins und des P30-Epitops (SEQ ID NO:14) gestaltet,
und indem den unterschiedlichen Positionen im Hinblick auf ihre
chemischen Eigenschaften jedes Aminosäure-Restes Werte zugewiesen
wurden. Nur die N-terminal der vorhergesagten beta-Fass-Struktur
gelegene Region wurde in Erwägung
gezogen. Im Falle von F30N gibt es 9 ähnliche von 21 Resten. Unter Verwendung
dieses Pseudo-Algorithmus erwartet man, dass die Substitutionen
zu minimalen Gesamt-Strukturveränderungen
führen.
Die Sequenzen der vier unterschiedlichen Konstrukte, sowie dreidimensionale
Darstellungen der ersetzten Aminosäuren, sind in 6 gezeigt.
-
Die
Variante von FGF8b mit dem P2-Epitop (SEQ ID NO:12) am C-Terminus
(F2C) wurde ursprünglich wie
F30N gestaltet. Es wird jedoch ein gutes Kd-Epitop an Positionen
195-203 vorhergesagt.
Daher ist das P2-Epitop genau C-terminal von diesem Epitop eingefügt. Wiederum
wurde die Region C-terminal vom vorhergesagten Beta-Fass betrachtet.
-
Die
internen Varianten von FGF8b (F30I und F2I) wurden konstruiert,
indem externe Schleifen in der FGF2-Struktur mit den Epitopen P2
bzw. P30 ersetzt wurden, wobei das Rückgrat der Beta-Fass-Struktur
der FGF-Struktur wahrscheinlich unverändert bleibt. Die immunogenisierten
FGF8b-Moleküle
wurden in Escherichia coli exprimiert, was die Herstellung dieser
Proteine bei minimalen Kosten im großen Maßstab erleichtert. Obwohl FGF8b
zwei potentielle N-Glykosylierungsstellen (Asn31 und Asn177) enthält, wurde
gezeigt, dass bakteriell exprimiertes, rekombinantes FGF8b biologisch
aktiv ist (MacArthur 1995a, Blunt 1997). Um die Reinigung und Rückfaltung
zu erleichtern, wurden die FGF8b-Varianten in einer His-getaggten
Version hergestellt, was gleichzeitig das Koppeln an Nickel-beladene
Säulen
ermöglicht.
-
Die
Reinigung der Moleküle
wurde unter Ausnutzung der hohen positiven Ladung der Proteinmoleküle oder
des His-tags durchgeführt,
und die Rückfaltung
wird unter Anwendung von Standardverfahren durchgeführt, wobei
die Bildung der Disulfidbrücke
beachtet wird.
-
Die
vier immunogenisierten Moleküle
wurden auch zusammen mit der Wildtyp-FGF8b-cDNA in DNA-Vakzinierungsvektoren insertiert.
-
Screening und Auswahl
der modifizierten FGF8b-Moleküle
-
Die
vier immunogenisierten FGF8b-Moleküle wurden in Bakterien exprimiert
und anschließend
aus Einschlusskörperchen
aufgereinigt. Parallel werden die Konstrukte als DNA- Vakzine verwendet.
Die unterschiedlichen Konstrukte werden dann im Hinblick auf ihre
Fähigkeit,
verschiedene Wirkungen zu induzieren, verglichen, welche bei der
Behandlung von Prostata- und Brustkrebs-Patienten erwünscht sind.
Solche Untersuchungen werden unter Verwendung von mehreren verschiedenen
in vitro- und in vivo-Assays durchgeführt. Schließlich bilden die Ergebnisse
der Experimente die Basis für
die Endauswahl von ein oder zwei Kandidaten für ein FGF8b-Vakzin in Menschen.
-
in vitro-ModeUe
Analysen im Maussystem
-
Mäuse unterschiedlicher
Haplo-Typen sowie Kaninchen werden mit den unterschiedlichen FGF8b-Konstrukten
in vollständigem
Freund'schem Adjuvans
immunisiert und anschließend
eine Boost-Immunisierung mindestens zweimal mit denselben Antigenen,
emulgiert in unvollständigem
Freund'schem Adjuvans,
durchgeführt.
So kann die Fähigkeit
unterschiedlicher Konstrukte, die B-Zell-Toleranz zu durchbrechen, verglichen
werden. Die DNA-Vakzinierung
wird bei anderen Tieren durchgeführt,
indem gereinigte DNA in vollständigem
Freund'schem Adjuvans/unvollständigem Freund'schem Adjuvans intramuskulär innerhalb
von 14-tägigen
Intervallen injiziert wird.
-
Serumproben
erhält
man zu unterschiedlichen Zeitpunkten während des Immunisierungsplans,
und die Fähigkeit
der verschiedenen Konstrukte, FGF8b-spezifische Antikörper zu
induzieren, wird unter Verwendung eines konventionellen ELISA-Verfahrens
bestimmt (Rochon 1994). Ein kommerzielles polyklonales Antiserum,
sowie ein kommerzieller monoklonaler Antikörper, der gegen FGF8b gezogen
wurde (R&D),
werden für
Positivkontrollen verwendet. Das FGF8b-Protein ist kommerziell erhältlich (R&D), aber er wird
auch produziert werden zusammen mit den anderen FGF8b-Konstrukten,
und anschließend
gereinigt/rückgefaltet.
Dieses Produkt kann dann zur Beschichtung von Platten in einem direkten
ELISA zum Testen der Seren von Mäusen/Kaninchen
verwendet werden, die mit FGF8b-Varianten-Proteinen immunisiert
wurden.
-
Ein
wertvolles Werkzeug zum Untersuchen der Auswirkungen der Vakzinierung
gegen FGF8b wird eine FGF8b-abhängige
Krebszelllinie sein. Mehrere FGF8b-positive Krebszelllinien, z.B.
MCF-7 oder SC-3, sind in der Literatur beschrieben. Solch eine FGF8b-abhängige Maus-Krebszelllinie
wird identifiziert, indem eine quantitative RT-PCR, Zellproliferationsexperimente
und STAT-3-Phosphorylierungs-Assays verwendet werden.
-
Die
Anwesenheit von FGF8b, ligiert an einen FGF-Rezeptor auf der Zelloberfläche, wird
mit FGF8b-spezifischen Antikörpern
in einer FACS- oder ELISA-Analyse detektiert. Antikörper, die
gegen mehrere der unterschiedlichen FGF-Rezeptoren gerichtet sind,
sind kommerziell erhältlich
(R&D).
-
Die
Konstrukte werden im Hinblick auf ihre Fähigkeit, Antikörper zu
induzieren, die in der Lage sind, die Komplement-Lyse von FGF8b-produzierenden/-enthaltenden
Zellen zu induzieren, verglichen. Dies kann mit einer der Maus-Tumorzelllinien,
die FGF8b exprimieren, die zuvor beschrieben wurden, oder alternativ dazu
unter Verwendung osmotischer FGF8b-beladener Zellen bestimmt werden. Seren
von normalen oder transgenischen Mäusen (siehe unten), die mit
den humanen FGF8b-Konstrukten immunisiert wurden, werden mit der
Zelllinie inkubiert und anschließend mit frischem Meerschweinchen-Komplement
inkubiert. Die Antikörpervermittelte
Komplement-Lyse der Zellen kann durch Standardverfahren bestimmt
werden.
-
Die
Fähigkeit
der induzierten Antikörper,
eine ADCC zu vermitteln, kann untersucht werden, indem die 51 Cr-Ausschüttung aus
markierten FGF8b-exprimierenden Zellen gemessen wird. Die Effektorzellen
werden PBMC von syngenischen Mäusen
sein. Zum Etablieren des Assays kann es geeignet sein, eine Maus-Zelllinie zu
verwenden, die in der Lage ist, ADCC zu vermitteln (positiv für Fc(-Rezeptoren)
als Effektorzelle mit einem Antikörper gegen humanes FGF8b.
-
Um
zu zeigen, dass die FGF8b-Kandidaten-Vakzine nicht irgendwie Autoantikörperinduziertes
Tumorwachstum fördern,
werden wir auch einen Tumor-Proliferations-Assay durchgeführen. Serumproben
von immunisierten Mäusen
werden mit FGF8b-exprimierenden Tumorzellen inkubiert. Die Proliferation
der Tumorzellen kann dann aufgrund ihrer Fähigkeit, 3H-Thymidin einzubauen, welches anschließend zu
den Zellen gegeben wird, detektiert werden.
-
Da
von FGF8b bekannt ist, dass es die Proliferation einer Reihe von
Säugerzellen
induziert, wird es auch notwendig sein, die Wachstums-fördernden
Wirkungen der Varianten-Proteine
zu untersuchen. Dies kann vorgenommen werden, indem Zellproliferations-Assays
verwendet werden, wie der von Marsh 1999 verwendete.
-
Die
biologische Wirkung von FGF8b auf Säugerzellen sollte durch die
Autoantikörper
neutralisiert werden. Dies kann gezeigt werden, indem rekombinantes
FGF8b und z.B. NIH3T3 in Zellproliferations- (und Morphologieveränderungs-)
Studien verwendet werden. Die Zugabe der Autoantikörper sollte
die transformierende Aktivität
von FGF8b aufheben.
-
Immunisierungsprotokoll
-
Die
Anzahl von Tieren, die in ein FGF8b-AutoVac-Immunisierungsexperiment
eingehen müssen, hängt von
der erwarteten Penetranz der Erkrankung in dem Modell ab, und daher
von den Anzahlen, die notwendig sind um statistisch signifikante
Informationen zu erhalten. Das Immunisierungsprotokoll muss auf
der Erfahrung beruhen, die wir aus dem TNFa-AutoVac-Projekt gezogen haben. Verschiedene
Immunisierungsprotokolle wurden zur Immunisierung von Mäusen mit
den unterschiedlichen TNFa-Analoga für spezifische Zwecke verwendet,
aber die meisten Experimente wurden unter Verwendung des folgenden
Protokolls durchgeführt:
- 1. Die Mäuse
sollten entweder mittels Ohrmarken oder Transpondern individuell
markiert werden, 10 Tiere in jedem Käfig. Vermutlich müssen Männchen und
Weibchen getrennt untersucht werden, aber in jedem Fall sollten
nicht beide Geschlechter in demselben Käfig vorkommen. Den Tieren sollten
mindestens 3 Tage nach Transport und Markierung Ruhe zukommen.
- 2. Antigen (1 mg/ml in PBS-Puffer) wurde mit einem entsprechenden
Volumen Freundschem vollständigen Antigen
(CFA) (Difco oder Sigma) emulgiert. Die Emulsion wird überprüft, indem
ein Tropfen der Emulsion auf einer Wasseroberfläche plaziert wird und beobachtet
wird, ob der Tropfen zusammenhält
oder dispergiert. Die Mischung wird fortgesetzt, bis der Tropfen
nicht mehr dispergiert.
- 3. Die Standard-Immunisierungsdosis ist 100 μg Antigen in 100 μl Volumen
+ 100 μl
Adjuvans. Daher beträgt
das Gesamt-Immunisierungsvolumen 200 μl, verabreicht s.c. (subcutan) über den
Rücken
des Tieres.
- 4. Booster-Immunisierungen werden 2-3 Wochen nach der primären Immunisierung
vorgenommen und anschließend
mit 2-3-wöchigen
Intervallen. Das Booster-/Immunisierungsmaterial wird genauso präpariert und
verabreicht wie das Immunisierungsmaterial, aber es wird Freund'sches unvollständiges Adjuvans
verwendet. Drei Booster-Immunisierungen werden vermutlich den maximalen
Titer induzieren. Daher werden die höchsten Titer 6-9 Wochen nach
der ersten Immunisierung erzielt.
- 5. Die Blutentnahme ist eine orbitale Blutentnahme von 50-100 μl, die gewöhnlich vor
der ersten Immunisierung und eine Woche nach der Booster-Immunisierung
entnommen wird. Es können
auch Schwanzblutungen verwendet werden, und 10-20 μl können zur
Bestimmung des Titers ausreichend sein.
-
Der
Initiationspunkt des Immunisierungsprogramms hängt von der Entwicklung der
Krankheit ab und der Strategie, der wir folgen wollen. Wir schlagen
vor, dass anfänglich
versucht wird, die maximale Immunität so bald wie möglich zu
erzeugen, aber es ist schwierig, die Immunisierungen eher als bei
einem Alter von 5 Wochen zu beginnen. Danach sollten hohe Titer
aufrecht erhalten werden, indem in 6-8-wöchigen Intervallen nach den
drei anfänglichen
Booster-Impfungen Booster-Impfungen verabreicht werden. Es gibt
ein potentielles Problem, wenn FGF8b für die normale Entwicklung der
jungen Mäuse
notwendig ist, und man könnte
daher dafürsprechen,
die Immunisierung später
in der adulten Maus zu beginnen.
-
Analysen im
humanen System
-
Bei
der Selektion der unterschiedlichen FGF8b-Konstrukte wird die Fähigkeit
von humanen Antigen-präsentierenden
Zellen untersucht, die eingefügten
immunogenen T-Zell-Epitope
humanen T-Zellen zu präsentieren.
Dies wird vorgenommen, indem dieselben in vitro-Prozessierungs-Assays für die P2-
und P30-Präsentation
verwendet werden, die für
die TNFa-Vakzine
verwendet wurden. Humane T-Zelllinien, welche für P2 und P30 spezifisch sind,
werden von Donoren etabliert, die gegen Tetanus geimpft sind. Antigen-präsentierende
Zellen (PBMCs) von denselben Donoren werden mit den unterschiedlichen
Konstrukten inkubiert, und T-Zelllinien werden hinzugefügt. Der
Spiegel der Präsentation
der eingefügten
T-Zell-Epitope kann dann verglichen werden, indem die Stimulation
der T-Zelllinien gemessen wird.
-
in vivo-Tiermodelle
-
Mindestens
drei unterschiedliche Systeme können
verwendet werden um zu überwachen,
ob die induzierten FGF8b-Antikörper
in der Lage sind, eine FGF8b-abhängige
in vivo-Wirkung
zu kontrollieren.
-
Mäuse werden
mit Maus-Tumorzellen, die FGF8 exprimieren, transplantiert, und
die Inhibition des Fortschreitens des Tumors wird mit der Autovakzinierung
zusammen untersucht, indem die modifizierten FGF8b-Proteine oder
FGF8b-DNA-Vakzine verwendet werden. Das ideale System umfasst die
Verwendung von Zellen, die von Maus-Tumoren isoliert wurden. Alternativ
dazu werden wir andere Maus-Zelllinien (z.B. Balb/3T3) verwenden,
die stabil mit der FGF8b-cDNA in einem Expressions-Vektor transfiziert
sind.
-
Das
Maus-Xenotransplantations-Karzinom-Modell wird für passive Vakzinierungsexperimente
verwendet. Nacktmäuse
werden mit humanen Tumoren transplantiert, und die Inhibierung der
Tumoren würde
mit der Übertragung
von Serum von normalen oder humanisierten Mäusen, die mit modifizierten
FGF8b-Proteinen oder FGF8b-DNA-Vakzinen immunisiert wurden, versucht.
Dies wäre
sehr nützlich
zum Untersuchen der Fähigkeit
der gebildeten Antikörper,
Tumoren zu unterdrücken.
-
Ein
weiterer Ansatz um einen Beweis für das Konzept zu erhalten,
umfasst die Verwendung von Mäusen,
die für
FGF8b transgen sind. Diese Mäuse,
die die FGF8b-cDNA unter der Kontrolle des sehr spezifischen Mäuse-Mammatumor-Virus
(MMTV)-Promotors tragen, entwickeln nachgewiesenerweise spontan
FGF8b-exprimierende Mammatumoren (Coombes, persönliche Mitteilung). Die Autovakzinierung
dieser Mäuse
mit den FGF8b-Varianten-Proteinen
oder FGF8b-DNA-Vakzinen würde
uns in die Lage versetzen, zu zeigen, ob die Autovakzine es den
Mäusen
ermöglichen,
die Tumoren zu unterdrücken
oder abzustoßen.
-
Ein
möglicher
Ansatz um einen Beweis des Konzepts zu erhalten, wäre die Verwendung
der transgenischen Wnt-1-Mäuse
(MacArthur 1995c). Von der Induktion von Brustkrebs-Arten durch
MMTV-Virus wird berichtet, dass sie die FGF8-Expression in mehr
als der Hälfte
der Mäuse
aktivieren, die Tumoren entwickeln. Die FGF8b-Abhängigkeit
der Tumoren könnte
bestätigt
werden, wenn unser Autovakzin/unsere Autovakzine das Auftreten oder
die Wachstumsrate der Tumoren unterdrücken könnten.
-
Die
Tatsache, dass transgenische Mäuse
oft nicht-physiologische immunologische Toleranz-Muster zeigen,
wird dieses Projekt sehr wahrscheinlich nicht beeinflussen, da die
FGF8b-Polypeptide
bei Mensch und Maus identisch sind.
-
Wenn
eine hilfreiche Wirkung der FGF8b-Immunisierungen tatsächlich im
Maus-Modell gezeigt
wurde und geeignete humane Vakzin-Kandidaten ausgewählt wurden,
wird es möglich
sein, eine begrenzte Anzahl toxikologischer Studien durchzuführen. Anschließend können Vakzin-Studien
bei Brust- und Prostatakrebs-Patienten durchgeführt werden um einen endgültigen Beweis
des Konzepts zu erhalten.
-
Es
ist wichtig, dass mehr Mutanten auf der Basis der verfügbaren Daten
konstruiert werden, wenn die Experimente unter Verwendung von in
vivo-Modellen ein positives Ergebnis haben.
-
BEISPIEL 4
-
Herstellung eines MUC-1-Analogons
-
Es
wurde nur ein MUC-1-AutoVac-Molekül hergestellt. Dieses umfasst
insgesamt neun Mucin-Repeats, von denen jedes die Sequenz SEQ ID
NO:33 besitzt. Die Konstruktion beginnt mit drei solcher Sequenzen,
gefolgt von einem P2-Epitop, gefolgt von drei weiteren Mucin-Sequenzen, gefolgt
von einem P30-Epitop, und schließlich von drei Mucin-Sequenzen.
-
Die
Konstruktion wurde mit und ohne ein N-terminales UNI-his-tag erstellt
(SEQ ID NO:23). Beide Varianten wurden in E. coli exprimiert. Die
Identität
des exprimierten Proteins wurde sowohl durch Western-Blot als auch
N-terminale Sequenzierung bestätigt.
Das Protein wird in löslicher
Form exprimiert, aber als Dimer, was einigermaßen überraschend ist.
-
Die
HIS-getaggten MUC-1-Moleküle
wurden mittels Metall-Affinitäts-Chromatographie
gereinigt. Die Menge an reinem Protein und die Reinheit ist derzeit
unbekannt.
-
BEISPIEL 5
-
Durchbrechen
der Autotoleranz in einem Maus-Modellsystem
-
CTL-Experimente,
in denen Mäuse
mit dendritischen Zellen immunisiert wurden, die sowohl mit einem Klasse
I- als auch einem Klasse II-Epitop pulsmarkiert wurden, haben zuvor
eine verstärkte
CTL-Induktion aufgezeigt, wenn sowohl die Immunisierung als auch
die Restimulierung in vitro mit sowohl einem Klasse I- als auch
einem Klasse II-Peptid durchgeführt
wurden, im Gegensatz zu einer Immunisierung und Restimulierung lediglich
mit einem Klasse I-Epitop. Diese Situation ist vergleichbar mit
der Immunisierung mit einem Autovakzin, bei der ein fremdes Klasse
II-Epitop in einem Selbst-Protein eingefügt ist. Die Aufnahme und Prozessierung
dieser Moleküle
mittels professioneller Antigen-präsentierender Zellen, wie dendritischen
Zellen, führt
zur Präsentation
des fremden Klasse II-Epitops zusammen mit einigen Selbst-Klasse
I-Epitopen. Es ist bekannt, dass es möglich ist, autoreaktive CTLs
hervorzubringen, aber die Anwesenheit eines fremden Klasse II-Helfer-Epitops
sollte diese CTL-Induktion sehr wahrscheinlich verstärken.
-
Der
potentielle Vorteil der vorliegenden Erfindung zur Induktion von
selbstreaktiven CTLs wird derzeit in Ovalbumin-transgenen Mäusen untersucht.
Es existieren vier unterschiedliche transgenische Linien mit unterschiedlichen
Ovalbumin-Expressionsleveln und Toleranzzuständen, vgl. Kurts C et al.,
1997, J. Exp. Med. 186: 239-245, die die RIP-mOVA-transgenischen Mäuse offenbaren
(die Ovalbumin in Pankreas, Niere und Thymus exprimieren und einen
hohen Grad der Toleranz aufweisen), und Kurts C et al., 1998, J.
Exp. Med. 188: 409-414, welche die RIP-OVAlow-
und RIP-OVAhigh-transgenischen Mäuse offenbaren,
welche niedrige und hohe Expressionsspiegel von Ovalbumin aufweisen.
Die letzte Linie, RIP-OVAint, welche Ovalbumin
mit einem mittleren Spiegel exprimiert, wurde von Dr. William R.
Heath erhalten, einem Co-Autor der zwei oben erwähnten Literaturstellen.
-
Im
Körper
gibt es verschiedene Stufen der Toleranz gegen unterschiedliche
Antigene. Die mindeste Toleranzstufe findet man bei zirkulierenden
Antigenen in großen
Mengen. Diese Antigene treten alle in den Thymus ein, wo selbst-reaktive
T-Zellen zerstört
werden. Diese Antigene stehen unter "zentraler Toleranz". Gewebe-spezifische Antigene treten
andererseits nicht direkt in den Thymus ein, und sie stehen im Allgemeinen unter "peripherer Tol1eranz", die z.B. durch
T-Zell-Anergie ausgeübt
wird.
-
Es
wurden zwei Ovalbumin-AutoVac-Konstrukte produziert. Beide beziehen
sich auf die Sequenz mit der Zugangs-Nr.: J00845 bei EMBL, in die
die Sequenz von P30 (SEQ ID NO:14) an zwei verschiedenen Stellen
eingefügt
wurde.
-
In
dem Konstrukt "OVA
3.1" ist P30 an
der Position eingefügt,
die Aminosäuren
272-292 im Ovalbumin entspricht.
In dem Konstrukt "OVA
3.2" ist P30 an
der Position eingefügt,
die Aminosäuren
321-341 im Ovalbumin entspricht. Diese Konstrukte wurden in den
Vektor pVax1 eingefügt
und zur DNA-Immunisierung verwendet.
-
Die
Mäuse wurden
intradermal einmal mit 100 μg
von jeder DNA immunisiert. Drei Wochen nach dieser Immunisierung
wurden die Milzen entfernt und ein CTL-Assay unter Verwendung von
Zielzellen, die das dominante Ovalbumin-Epitop SIINFEKL und das
zusammengewürfelte
FILKSINE-Peptid als Kontrolle exprimierten. Die Immunisierung führte zu
einer klaren CTL-Induktion in Wildtyp-C57BL/6-Mäusen – wie erwartet, da sowohl Ovalbumin
als auch P30 in diesen Mäusen
fremd ist.
-
Wir
beabsichtigen nun, die 4 Linien der Ovalbumin-transgenen Mäuse mit
diesen AutoVac-Konstrukten zu immunisieren. RIP-OVAlow,
RIP-OVAint und RIP-OVAhigh exprimieren
ansteigende Mengen von Ovalbumin und besitzen unterschiedliche Stufen
der Toleranz, und, wie oben erwähnt,
auch RIP-mOVA besitzt einen hohen Grad an Toleranz.
-
In
diesen 4 Linien der transgenen Mäuse
wird nur das P30 fremd sein. Ovalbumin ist ein Selbst-Antigen in
diesen Mäusen,
und diese Situation stellt daher eine echte Autovakzinierung für die CTL-Induktion
gegen Ovalbumin dar.
-
Vorläufige Ergebnisse,
die in RIP-OVAlow-Mäusen erhalten wurden, welche
den niedrigsten Grad an "peripherer
Toleranz" gegen
Ovalbumin besitzen, zeigten, dass sowohl das Ovalbumin mit dem eingefügten P30
als auch die natürlich
vorkommenden Ovalbumin-Moleküle in der
Lage waren, CTL-Antworten hervorzurufen – es wird erwartet, dass transgenische
Mäuse mit
einem höheren
Grad der Toleranz lediglich in der Lage sein werden, eine CTL-Antwort gegen die
modifizierten Ovalbumin-Moleküle
hervorzurufen, und nicht gegen die natürlich vorkommende Form.
-
Liste der
Literaturstellen
-
- Ago H et al.,(1991). J Biochem (Tokyo), 110,
360-3.
- Abdel-Nabi H et al. (1992). Sem. Urol. 10, 45-54.
- Acevedo et al. (1995), Cancer 76; 1467-1475.
- Acevedo et al. (1997), Cancer Detect. Prev. 21; 295-303.
- Adelstein S et al. (1991), Science 251: 1223-1225.
- Babaian RJ et al. (1999), J. Urol 152, 1952-1955.
- Barnard JA et al. (1995) Gastroenterol. 108(2): 564-580.
- Bier H et al. (1995), Eur. Arch. Otorhinolaryngol. 252(7): 433-9.
- Bouchard L et al. (1989), Cell 57(6): 931-36.
- Blaber M et al. (1996), Biochemistry 35, 2086-94.
- Blunt AG et al. (1997), J Biol Chem 272, 3733-8.
- Boring CC et al. (1993), CA Cancer J. Clin. 43: 7-26.
- Boucher et al. (1995), Hum. Pathol. 26; 1201-1206.
- Callahan R (1996), Breast Cancer Res Treat, 39, 33-44.
- Carter RE (1996), Proc. Natl. Acad. Sci. U.S.A., 93, 749-753.
- Chang et al., (1981), Fertil. Steril. 36; 659-663.
- Chantry A, 1995, J.Biol. Chem. 270(7): 3068-73.
- Crossley PH et al. (1996b), Cell, 84, 127-36.
- Crossley PH et al. (1995), Development, 121, 439-51.
- Crossley PH, Martinez, S. and Martin, G.R. (1996a) Midbrain
development induced by FGF8 in the chick embryo. Nature, 380, 66-8.
- Dean C et al. (1994), Int. J. Cancer, suppl 8: 103-107.
- Dillioglugil Ö et
al. (1995), Eur. Urol. 28, 85-101.
- Doi et al. (1996), Int. J. Cancer 65; 459-459.
- Douglas TH et al. (1995), J. Surg. Oncol. 59(4), 246-250.
- Earp HS et al. (1995), Breast Cancer Res. Treat. 35(1):115-32.
- Eccles SA (1994), Invasion Metastasis 14 (1-6): 337-48.
- Eppenberger U et al. (1994),. J. Neurooncol. 22(3): 299-54.
- Dorkin TJ et al. (1999), Oncogene 18: 2755-2761.
- Eriksson AE et al. (1993), Protein. Sci. 2: 1274-84.
- Fernandez-Teran M. et al. (1997), Dev. Biol. 189, 246-55.
- Fujii K et al. (1995), Fxp. Cell. Res. 216(1): 261-72.
- Furst J et al. (1994), Urol. Res. 22, 107-113.
- Furthauer M et al. (1997), Development 129: 9253-64.
- Geissler et al. (1997), Lab. Invest. 76: 859-871.
- Gemel J et al. (1996), Genomics 35: 253-7.
- Ghosh AK et al. (1996), Cell Growth Differ 7: 1925-34.
- Goldfarb M et al. (1996), Cytokine Growth Factor Rev 7: 311-25.
- Goodnow CC et al. (1988), Nature 334: 676-682.
- Goodnow CC et al. (1991), Nature 352: 532-536.
- Greenberg NM et al. (1995), Proc. Natl. Acad. Sci. USA 92: 3439-3443.
- Guy CT et al. (1992), Proc. Natl. Acad. Sci, USA 89: 10578-10582.
- Heikinheimo M et al. (1994),. Mech Dev, 48: 129-38.
- Henttu P and Vihko P (1989), Bioch. Biophys. Res. Comm. 160:
903-910.
- Hopp TP and Woods KR (1983), Mol. Immunol. 20: 483-9.
- Horoszewicz JSH et al. (1983), Cancer Res. 43: 1803-1818.
- Horoszewicz JSH et al. (1987), Anticancer Res. 7: 927-936.
- Hoshikawa M et al. (1998), Biochem Biophys Res Commun 299: 187-91.
- Husmann I et al. (1996), Cytokine Growth Factor Rev 7: 249-58.
- Israeli RS et al. (1999), Cancer Res. 59: 1807-1811.
- Israeli RS et al. (1993), Cancer Res. 53: 227-230.
- Israeli RS et al. (1994), Cancer Res. 59: 6306-6310.
- Jacoby et al. (1984), Adv. Immunol. 35: 157-208.
- Johnson RL and Tabin CJ (1997), Cell 90: 979-90.
- Kahn D et al. (1999), J. Urol. 152: 1490-1995.
- Kapoun AM and Shackleford GM (1997), Oncogene 14: 2985-9.
- Kettunen P and Thesleff 2 (1998), Dev Dyn 211: 256-68.
- Koga M et al. (1995), J Steroid Biochem Mol Biol 54: 1-6.
- Kouhara H et al., (1994), Oncogene 9: 455-62.
- Kozak M (1991), J Cell Biol 115: 887-903.
- Lapthorn et al. (1994), Nature 369: 455-461.
- Lazar et al. (1995), Cancer Res. 55: 3735-3738.
- Leek J et al. (1995), British Journal of Cancer 72: 583-588.
- Leung HY et al. (1996), Oncogene 12: 1833-5.
- Lopes AD (1990), Cancer Res. 50: 6423-6429.
- Loric S et al. (1995), C1in. Chem. 41(12): 1698-1704.
- Lupu R et al. (1995), Semin. Cancer. Biol. 6: 135-145.
- MacArthur CA et al. (1995a), Cell Growth Differ 6: 817-25.
- MacArthur CA et al. (1995b), Development 121: 3603-13.
- MacArthur CA et al. (1995c), J Virol 69: 2501-7.
- Manabe et al. (1985), Gastroenterology 89: 1319-1325.
- Marsh SK et al. (1999), Oncogene 18, 1053-1060.
- Martin L et al. (1993), J. Immunol. 150(49): 1234-43.
- Mattei MG et al. (1995), Mamm Genome 6: 196-7.
- McDonnell WM and Askari FD (1996), New Engl. J. Med 334: 42-45.
- Meyers EN et al. (1998), Nat Genet 18: 136-41.
- Milich DR et al. (1994), J. Immunol. 153(1): 429-935.
- Milner PG et al. (1989), Biochem Biophys Res Commun 165: 1096-103.
- Miyashita Y et al. (1994), Jpn J Cancer Res 85: 1117-23.
- Modjtahedi H et al. (1993a), Br. J. Cancer 67(2): 254-261.
- Modjtahedi H et al. (1993b), Cell. Biophys. 22(1-3): 129-46.
- Modjtahedi H et al. (1996), Br. J. Cancer 73(2): 228-35.
- Moy FJ et al. (1996), Biochemistry, 35: 13552-13561.
- Muller WJ et al. (1988), Cell 54(1): 105-115.
- Murphy GP et al. (1996), Prostate 28: 266-271.
- Murphy GP et al. (1995), Prostate 26: 164-168.
- Murphy GP et al. (1995), Anticancer Research 15(4): 1473-1379.
- Nguyen L et al. (1990), Clin. Chem. 35: 1450-1955.
- Nonomura N et al. (1990), Cancer Res 50: 2316-21.
- O'Sullivan D
et al. (1991), J. Immunology 147: 2663-9.
- Ohnishi Y et al. (1995), Br. J. Cancer 71(5): 969-73.
- Ohuchi N et al. (1997a), Development 124: 2235-44.
- Ohuchi H et al. (1997b), Mech Dev 62: 3-13.
- Ohuchi N et al. (1994), Biochem Biophys Res Commun 204: 882-8.
- Payson RA et al. (1996), Oncogene 13: 47-53.
- Pillai et al. (1996), FEBS Lett. 387: 23-26.
- Pollard M and Luckert PH (1994), Anticancer Res. 14: 901-903.
- Prigent SA and Lemoine NR (1992), Progress in Growth Factor
Research 4: 1-24.
- R&D focus,
Drug News, (1996) 5, 21, 9.
- Rammensee H-G. et al. (1995), Immunogenetics 41: 178-228.
- Regelson W (1995), Cancer 76: 1299-1301.
- Ries LAG et al. (1996), SEER Cancer Statistics Review, 1973-1993:
Tables and Graphs, National Cancer Institute, Bethesda, MD.
- Rinker-Schaeffer CW et al. (1995), Genomics, 30(1): 105-108.
- Rochon YP et al. (1994), Prostate 25: 219-223.
- Rock KL et al. (1996), Vaccine 19: 1560-1568.
- Rock KL and Clark K (1996), J. Immunol. 156: 3721-3726.
- Rudra-Ganguly N et al. (1998), Oncogene 16: 1487-92.
- Salomon DS et al. (1995), Critical reviews in Oncology/Hematology
19: 183-232.
- Sato B et al. (1993), J Steroid Biochem Mol Biol 97: 91-8.
- Schlegel J et al. (1994), J. Neurooncol. 22(3): 249-54.
- Schmitt JF et al. (1996), J Steroid Biochem Mol Biol 57: 173-8.
- Sheaff et al. (1996), J. C1in. Pathol. 49: 329-332.
- Sherman L et al. (199B), Genes Dev 12: 1058-71.
- Shimamura K and Rubenstein JL (1997), Development 129: 2709-18.
- Shirai A and Klinman DM (1993), AIDS Res. Hum. Retroviruses
9, 979-983.
- Sokoloff M et al. (1996), Cancer 77(9): 1862-1872.
- Steinhoff U et al. (1994), Eur. J. Immunol. 24(3): 773-76.
- Stevens VC (1986), Ciba Found. Symp. 119: 200-225.
- Su SL et al. (1995), Cancer Res. 55: 1441-1443.
- Syrigos et al. (1998), Gut 42: 88-91.
- Talwar et al. (1976), PNAS 73: 218-222.
- Talwar et al. (1994), PNAS 91; 8532-8536.
- Tanaka A et al. (1992), Proc Natl Acad Sci USA 89: 8928-32.
- Tanaka A et al. (1998), Cancer Research 58: 2053-56.
- Tanaka A et al. (1995), FEBS Lett 363: 226-30.
- Thomas H et al. (1996), Br. J. Cancer, 73(1): 65-72.
- Tjoa B et al. (1996), Prostate 28: 65-69.
- Tjoa B et al. (1995), Prostate 27: 63-69.
- Tokunaga A et al., (1995), Cancer 75(6 suppl.): 1918-25.
- Tosi E et al. (1995), Int. J. Cancer 62(5): 643-50.
- Triozzi et al. (1994), Int. J. Onc. 5: 1997-1453.
- Troyer JK et al. (1995), Int. J. Cancer 62: 552-558.
- Valone FH et al. (1995), J. Clin. Oncol. 13(9): 2281-92.
- Valve E et al. (1997), Biochem Biophys Res Commun 232: 173-7.
- van Dam PA et al. J. Clin. Pathol. 1994 47(10): 919-19.
- Weiner LM et al. (1995), Cancer Res. 55(20): 4586-4593.
- Wright GL Jr et al. (1996), Urology 98: 326-334.
- Wu J et al. (1997), J Steroid Biochem Mol Biol 62: 1-10.
- Wu X et al., Fan, Z., Masui, H., Rosen, N., Mendelsohn, J. Apoptosis
induced by an anti-epidermal growth factor receptor monoclonal antibody
in a human colorectal carcinoma cell line and its delay by insulin.
- Wynant GE et al. (1991), Prostate 18: 229-291.
- Xu X et al. (1998), Development 125: 753-65.
- Yamanishi H et al. (1995), J Steroid Biochem Mol Biol 52: 99-53.
- Yogeeswaran and Salk (1981), Science 212: 1514-1516.
- Yokoyama N et al. (1998), Dev Biol 196: 1-10.
- Yoshimura K et al. (1996), Cancer Lett 103: 91-7.
- Yoshiura K et al. (1997), Am J Med Genet 72: 354-62.
- Young RA and Davis RW (1983), Proc. Natl. Acad. Sci. U.S.A.
80: 1194-1198.
- Yule TD (1993), J. Immunol. 151(6): 3057-3069.
- Zhang JD et al. (1991), [published erratum appears in Proc Natl
Acad Sci USA 88(12): 5477], Proc Natl Acad Sci USA 88, 3446-50.
- Zhu Z et al. (1995), Int. J. Cancer 62(3): 319-324.
- Zhu X et al. (1991), Science 251: 90-3.
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
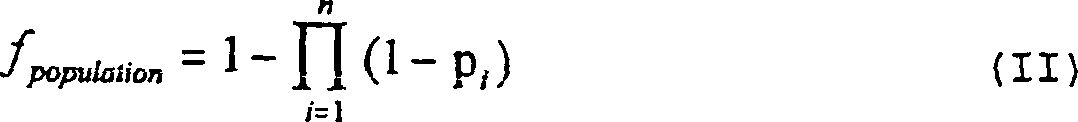
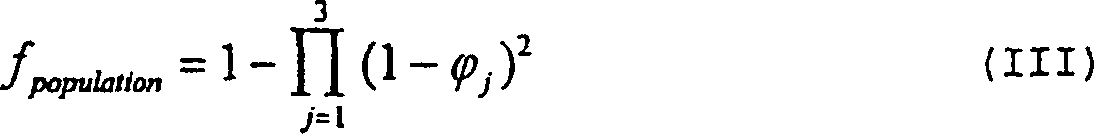
 – wobei der Ausdruck 1-fresidual_i auf 0 gesetzt wird, wenn er negativ ist. Es sollte erwähnt werden, dass Formel V es erfordert, dass alle Epitope gegen identische Haplotyp-Sätze kartiert wurden.
– wobei der Ausdruck 1-fresidual_i auf 0 gesetzt wird, wenn er negativ ist. Es sollte erwähnt werden, dass Formel V es erfordert, dass alle Epitope gegen identische Haplotyp-Sätze kartiert wurden.