CN1119329C - 6-氯-α-甲基-咔唑-2-乙酸的咔唑酯前体的纯化方法 - Google Patents
6-氯-α-甲基-咔唑-2-乙酸的咔唑酯前体的纯化方法 Download PDFInfo
- Publication number
- CN1119329C CN1119329C CN99108607A CN99108607A CN1119329C CN 1119329 C CN1119329 C CN 1119329C CN 99108607 A CN99108607 A CN 99108607A CN 99108607 A CN99108607 A CN 99108607A CN 1119329 C CN1119329 C CN 1119329C
- Authority
- CN
- China
- Prior art keywords
- impurity
- ester
- carbazole
- formula
- preparation
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/56—Ring systems containing three or more rings
- C07D209/80—[b, c]- or [b, d]-condensed
- C07D209/82—Carbazoles; Hydrogenated carbazoles
- C07D209/84—Separation, e.g. from tar; Purification
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/56—Ring systems containing three or more rings
- C07D209/80—[b, c]- or [b, d]-condensed
- C07D209/82—Carbazoles; Hydrogenated carbazoles
- C07D209/86—Carbazoles; Hydrogenated carbazoles with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to carbon atoms of the ring system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/56—Ring systems containing three or more rings
- C07D209/80—[b, c]- or [b, d]-condensed
- C07D209/82—Carbazoles; Hydrogenated carbazoles
- C07D209/88—Carbazoles; Hydrogenated carbazoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the ring system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/56—Ring systems containing three or more rings
- C07D209/96—Spiro-condensed ring systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Indole Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
一种纯化式(I)的(6-氯-2-咔唑基)甲基-丙二酸二(C1-C6烷基)酯的方法,式(I)中Ra和Rb必须相同并选自C1-C6烷基;该方法包括将一种或多种杂质从所述咔唑酯中至少进行一次析相作用,其中用于析相的溶剂为乙酸。在优选方案中,所述乙酸为保存在约30℃-约110℃温度下的冰乙酸,并且所述咔唑酯以至少99.90%(重量计)的纯度获得,这样存在于其中的杂质的量为0.10%或更少(重量计)。在更为优选的具体实施方案中,所述温度为约50℃-约70℃,并且仅进行一次析相。
Description
本发明涉及有机化合物的纯化方法领域,其中包括但不局限于由有机化学合成方法制备的终产物和中间体,尤其是后者的那些有机化合物。具体地说,这些方法是为了纯化羧酸有机化合物的烷基酯。本发明涉及一种改进的通过析相作用纯化(6-氯-2-咔唑基)甲基-丙二酸二(C1-C6烷基)酯,尤其是二乙酯的方法,在下文该化合物有时被称为“咔唑酯”,虽然该术语还通常用来指包括在本发明方法中的所有二(C1-C6烷基)酯。
咔唑酯是制备卡布洛芬过程中的起始物,在美国卡布洛芬是由食品和药物管理局、兽药委员会(FDA/CVM)批准用于狗的高效COX-2选择性抗炎药。业已知咔唑酯起始物可能含有至少一种在相关制备过程的一个步骤中产生的杂质,它可能含有为咔唑酯起始物重量的0.9%的杂质。该杂质的组成在下面将进一步详细讨论,但设想本发明的纯化方法在其范围内致力于不仅包括该杂质而且还包括其它杂质。为了获得足够纯形式的以用作动物药品的卡布洛芬终产物,必须将所有这些杂质减至最少。
Zwahlen的US4264500公开了一种制备6-氯-α-甲基-咔唑-2-乙酸的方法。终产物的最后的中间体为(6-氯-2-咔唑基)甲基丙二酸二乙酯,其中根据Zwahlen的描述,它通过水解作用和脱羧作用转化而来。另一方面,据报道转化步骤就地进行或在用已知方法分离所述倒数第二个中间体后进行,如通过结晶。然而,Zwahlen没有提出如由本发明提供的中间体的纯化方法或根据本发明方法产生的令人惊奇的高产率。
根据本发明的最广方面,提供了一种纯化式(I)(6-氯-2-咔唑基)甲基-丙二酸二(C1-C6烷基)酯的方法:其中Ra和Rb必须相同并选自C1-C6烷基,优选C1-C4烷基;该方法包括从所述酯中将一种或多种杂质至少进行一次析相作用,其中用来进行所述析相作用的溶剂为乙酸。
根据本发明,进一步提供了上述式(I)所述酯的纯化方法,其中所述酯以至少99.80%(重量计)的纯度获得,这样存在于其中的杂质的量为0.20%或更低(重量计);并且进一步其中所述乙酸为被保存在约30℃-约110℃的温度下的冰乙酸;并且进一步其中所述析相作用被任选进行两次或多次。
另外,根据本发明,提供上述式(I)所述酯的纯化方法,其中所述酯为二乙酯;并且进一步其中式(I)所述酯以至少99.90%(重量计)的纯度获得,这样存在于其中的杂质的量为0.10%或更低(重量计);并且进一步其中所述乙酸为被保存在约40℃-约90℃的温度下的冰乙酸,较优选约45℃-约75℃,最优选约50℃-约70℃;并且进一步其中所述析相作用仅被进行一次。
根据本发明较窄但仍然优选的具体实施方案,被纯化的式(I)所述(6-氯-2-咔唑基)甲基-丙二酸二(C1-C6烷基)酯以非晶体或晶体的分散固体的形式存在,其中它在其冰乙酸溶液中主要形成淤浆。
另外,提出了所述杂质可能在所述酯的制备方法过程中直接或间接产生,并可能含有任一种或多种起始物、合成中间体、反应体、反应副产物、降解产物、所述制备方法的各种反应步骤在其中进行的溶剂或具有与式(I)所述酯密切相关的化学结构的不希望的类似物。特别提出了所述杂质可能作为不适当或在次优的基础上进行所述方法的结果从所述制备方法间接产生。
还提供了所述杂质可能偶然来自没有被包括的在式(I)所述酯的所述制备方法中直接或间接产生的来源,例如来自在其中进行所述制备方法的设备的污染,来自所述制备方法所采用的起始物、溶剂或合成助剂的污染,来自周围空气中的污染物,即被吸收在所述方法中的围绕所述制备方法的环境,或来自其制备后的贮存或处理时的式(I)所述酯的污染。
在本发明纯化方法的一个特别优选的具体实施方案中,待纯化的中间体为咔唑(二乙)酯且待除去的杂质为式(IV)二聚体:
将要根据本发明的方法进行纯化的上述式(I)(6-氯-2-咔唑基)甲基-丙二酸二(C1-C6烷基)酯为卡布洛芬合成中的最后中间体: 其中Ra和Rb必须相同并选自C1-C6烷基。如已描述的,卡布洛芬为已被批准的尤其可用于治疗狗疼痛和炎症的抗炎药。
其中Ra和Rb必须相同并选自C1-C6烷基。如已描述的,卡布洛芬为已被批准的尤其可用于治疗狗疼痛和炎症的抗炎药。
要求Ra和Rb相同并且它们选自C1-C6烷基,优选C1-C4烷基。如果Ra和Rb被允许代表不同的烷基基团,例如甲基和乙基,则导致混合的二酯,那么丙二酸的碳将成为手性中心,产生式(I)酯的(S)和(R)对映体。该结果将进一步使令人满意的式(I)酯前体的纯化复杂化并可能完全受挫。例如,对于从外消旋混合物中析相分离所形成的非对映异构体,需要使用已知的通过与光学纯的分子,例如酒石酸和其衍生物混合的方法。
尽管它们所代表的部分必须都相同,但在本文Ra和Rb被使用作为不同的取代基鉴别基团。这种使鉴定不同化的目的是强调式(I)酯必须从中分离的可能的杂质包括可由制备方法的不适当操作或由一些其它未知或以前未预见的情况产生的混合酯。Ra和Rb优选地选自可为直链或支链的C1-C4烷基,包括甲基、乙基、正丙基、异丙基、正丁基、仲丁基和叔丁基。在这些代表种类中,优选甲基和乙基,尤其优选乙基。
从式(I)酯前体制备的卡布洛芬,即6-氯-α-甲基-9H-咔唑-2-乙酸可由式(II)表示:
注意到由于被水解和单脱羧,式(II)活性剂卡布洛芬不同于式(I)酯前体。在制备卡布洛芬的一个优选方法中,式(I)咔唑酯前体具有进一步如下式(III)所示的其自身的中间体。式(I)咔唑酯前体本身不同于式(III)中间体,其中它由于通过将两个另外的双键引入与α-甲基-乙酸部分相连的苯环中被芳化而优于式(III)中间体。从如下式(III)中间体的描述中将很容易理解这一点:
在根据上面提及的Zwahlen US4264500中描述的合成步骤进行的制备卡布洛芬的一个优选方法中进行式(I)咔唑酯前体和其前面式(III)中间体的上述修饰。
Zwahlen合成的第一个步骤是通过将其用氯气处理芳化式(III)中间体。该步骤优选在升高的温度,如反应混合物的回流温度下,在非质子传递溶剂,如甲苯、二氯甲烷或二氯乙烷中,同时将氯气缓慢加入到所述混合物中来进行。氯气的加入优选在2-8小时期间进行。在完成该步骤的一个典型的方式中,甲苯被用作溶剂且反应在75℃下进行4小时。产生的被芳化化合物为由式(I)代表的咔唑酯前体:
式(III)酯的芳化产生由上面式(I)代表的咔唑酯中间体,其中它接着被进行水解和脱羧以产生卡布洛芬终产物。在进行最后提及的合成步骤的一个优选方式中,根据包括用酸,如冰乙酸和盐酸的混合物处理的已知方法将式(I)中间体水解和脱羧。
上述合成转化可按照下面反应线路方案一起表示:
根据本发明广泛设想从式(I)酯前体分离的该杂质或多种杂质在性质上可能明显不同并可能来自不同来源。固有地,在本发明中进行的纯化通常依赖于纯化方法的基本性质,其中作为优选的具体实施方案,该纯化方法为析相作用法。这些已知方法可获得甚至在结构上密切相关的化合物的极高水平的分离,这在下面将被进一步详细说明。本发明纯化方法的参数以这样的方式选择,即所述方法不是由于待分离的杂质的结构才具有它的可操作性和优良的选择性。因此,设想本发明决不受这些杂质特性的限制。
对于式(I)咔唑酯前体遇到的一种较复杂的杂质进行了研究。该杂质随时间在所述酯前体的溶液以及式(II)卡布洛芬终产物的溶液中以沉淀的形式出现。杂质用X-射线晶体学和为咔唑酯前体的螺羟吲哚二聚体形式的其它分析数据来鉴定,其中该咔唑酯前体的螺羟吲哚在如上面合成线路中所描述的包含式(III)中间体的氯化的芳化步骤中产生。螺羟吲哚二聚体杂质的结构可用式(IV)表示:
二聚体杂质具有对常规纯化方法提出挑战的晶体性质,其可通过共沉淀来消除。使用常规的溶剂体系以达到通过本发明的纯化方法产生的所需纯化水平的最初努力没有成功。由于这些溶剂体系的上述动力学结晶作用,丙酮、乙腈、乙醇、丙醇、丁醇、乙酸乙酯、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、甲基异丁基酮以及这些溶剂体系的混合物导致二聚体杂质水平增加。使用甲苯/甲磺酸和甲苯/丁醇溶剂体系获得较能接受的结果。使用甲苯/甲磺酸体系获得的产率(75-85%)和产物质量(<0.1%二聚体杂质)是令人满意的,而甲苯/丁醇体系的产率较低。接着在打算模仿实际制备中将遇到的那些应力条件下实施甲苯/甲磺酸体系。将纯化在60-65℃升高的温度下进行大于2小时的延长时间。这些应力条件产生不能被分离和除去的降解产物。
最初用提高产率和能更好地去除杂质的乙醇/异丙基醚重晶体也获得令人满意的结果。然而,当该纯化体系使用延长的成粒时间进行应力试验时,产生的结果是不可接受的。事实上,结晶作用与随着产物的首先结晶以及随后在1小时内的二聚体杂质的结晶而动态变化。其中二聚体杂质也被结晶出来的该时间间隔对于商业规模的生产则太短。
成功以及作为本发明基础的溶剂体系是涉及温乙酸的溶剂体系。该温乙酸体系再浆化物,即析相作用的淤浆在由延长的成粒时间(大于36小时),延长的加热时间(大于12小时)和过度加热(大于70℃)构成的应力试验中仍然有效。因此,对于40kg量的生产,涉及循环时间以及设备变化的乙酸溶剂体系随之按比例增加。生产试验极其成功,通过HPLC分析测定仅产生0.02%的二聚体杂质。
除了特定的上述螺羟吲哚二聚体杂质外,明显存在许多其它可能的杂质。这些杂质可能直接或间接产生于所述式(I)的咔唑酯前体的制备方法中,并可能包含任一种或多种起始物、合成中间体、反应剂、反应副产物、降解产物、所述制备方法的各个反应步骤在其中进行的溶剂或不希望的具有与所述式(I)咔唑酯极其相关的化学结构的类似物。所述杂质最常在包括在采用的特定制备方法中的普通步骤中产生,因此,在本文中称为与所述制备方法“直接”相关。
然而,通常是这样的情况,即对于其基本的化学操作制备方法设计不恰当使用不适宜的起始物、反应剂或溶剂或需要不适宜的方法参数,如进行反应的时间和温度。另一方面,制备方法可基于完全适宜的化学操作,但在其执行过程中产生一些偶然的错误。例如,可能使用错误的起始物或不适量的反应剂;或反应进行的温度可能太高或太低。这些操作的错误还可能随着所需终产物一起产生杂质。这些类型的杂质在包括在所采用的制备方法的步骤范围之外产生,因此,在本文中被称为与所述制备方法“间接”相关。
还可能的是杂质可能不与制备方法直接或间接相关。相反,这些杂质可能偶然产生于不同的来源,来自例如完成制备方法的设备的污染、来自用于制备方法中的起始物、溶剂或合成助剂的污染、来自周围空气,即围绕制备方法的环境中的污染物。来自这些来源的杂质可能被吸收至制备方法的步骤中。在完成制备方法后,必须分离终产物并接着以某些预备根据已知方法将其配制成药物组合物的方式处理或贮存它。因此,杂质可作为在制备后的贮存或后序处理时,由于与所述杂质的来源接触而产生的所述式(I)酯被污染的结果而出现。
本发明的纯化方法提供足够高的式(I)咔唑酯前体的产率,以致所述咔唑酯前体终产物的纯度至少为99.80%(重量计),这样其中杂质的重量为0.20%或更低(重量计)。所述重量百分比基于终产物中的酯前体的重量除以所述终产物的重量×100。然而,从确定存在的杂质的量对终产物的定量分析结果计算纯度百分比通常更为方便,接着从中计算纯度百分比。这些定量分析方法是熟知的,其中任一或多种可适宜于本文描述的方法的需要。
在本发明的一个优选的具体实施方案中,所述式(I)咔唑酯前体为二乙酯,并且所述咔唑酯前体以至少99.90%(重量计)的纯度获得,这样其中存在的杂质的量为0.10%或更低(重量计)。在本发明一个更为优选的具体实施方案中,式(I)所述咔唑酯前体为二乙酯,并且所述咔唑酯前体以至少99.95%(重量计)的纯度获得,这样其中存在的杂质的量为0.05%或更低(重量计)。
采用的乙酸可为高度浓缩的非水溶液的形式,其中乙酸为非常主要的成分。然而,该乙酸非水溶液通常与式(I)终产物的咔唑酯前体的较低水平的纯度相关。因此,在本发明的一个优选具体实施方案中,所述乙酸为冰乙酸。
在其一个优选的具体实施方案中,本发明的纯化方法采用热乙酸作为溶剂,其被用于包含式(I)咔唑酯前体和其中所含杂质的固体产物中。待除去的杂质可高度溶于这种热的乙酸溶剂中,但是终产物咔唑酯前体在热的乙酸溶剂中具有极低的溶解度。式(I)咔唑酯前体在热乙酸溶剂中的不溶性水平为约85%的数量级(重量计),即仅约15%的咔唑酯前体溶解在热乙酸中。残留的咔唑酯前体被分散在热乙酸溶剂中,并因此可被确切地描述为以淤浆或浆的固体形式存在。在尽可能多的咔唑酯前体从热乙酸溶剂中沉淀出来后,它和已经分散的未溶解在溶剂中咔唑酯前体被从溶剂中分离。这种分离构成了析相作用,其中固相咔唑酯前体被从溶解杂质的液相中分离。
将乙酸溶剂保存在约30℃-约110℃的温度下;优选约35℃-约90℃的温度下,较优选约40℃-约75℃下,最优选约45℃-约70℃。可将沉淀过程,即包括为淤浆形式的咔唑酯前体材料的析相作用过程如所需地进行多次。虽然每一次析相作用产生一个较纯的产物,但这将以额外消耗能量的代价来实现,因此效率降低。然而,本发明一个令人惊奇的优点是从一次析相作用可获得高达至少99.90%(重量计),以及99.95%(重量计)或更高,甚至包括99.98%(重量计)的纯度。将析相作用过程进行两次通常是获得具有商业上作为动物健康药物所需高纯度终产物所需要的。
进一步设想根据待纯化的式(I)咔唑酯前体的特性和生产过程,可用许多不同的具体方案来完成本发明的纯化方法。例如,设想可以固体的形式分离所述酯前体物作为来自如上面详细描述的制备方法的中间体。所述咔唑酯前体物可以固体形式分离以允许它能贮存而用于在相同制备部位的后处理或为了其在不同生产设备中的加工的运送。该分离的固体中间体意味着一个能方便地除去存在的杂质的极好的机会,因为根据本发明的咔唑酯前体的加工将完全与正使用的步骤的制备合成顺序相适应。可直接用本发明方法的热乙酸析相作用溶剂处理所述分离的固体咔唑酯前体中间体。在一个较不优选的具体实施方案中,可首先将所述固体中间体咔唑酯前体溶解在一些与随后加入的乙酸相容的非水溶剂中。
不仅根据本文所公开的,而且根据本领域熟知的纯化方法的原则,尤其是析相作用方法来完成本发明的纯化方法。这些原则将在下面简要描述以总结最常在本领域普通技术人员作出的对本发明纯化方法的修改中发挥作用的那些条件。这些原则的总结还起到强调析相作用方法通常具有的结果以及尤其是本发明方法的未预料的成功的不可预测特性。
因此,例如根据本发明的析相作用的纯化不仅包括为分散的淤浆形式的酯前体的存在,而且还有酯前体的沉淀,其中该沉淀作用在杂质以溶解状态保留在乙酸溶剂中时必定发生。沉淀作用通常被认为主要由通过其中的物理或化学变化从以前清亮的溶液中分离固体颗粒的方法构成。这将与从本发明纯化方法开始的为分散状态的酯前体的存在区分开来。析相作用的一个最主要的用途是固体的纯化,其中它通常可被称为沉淀作用。
在其最简单的方面,析相作用包括在升高的温度下溶解在适宜溶剂中的不纯固体,以及冷却时,杂质材料仍被溶解,而同时沉淀产物被从中分离并纯化。在式(I)酯前体的情况下,甚至在高温,在存在乙酸溶剂时,产物具有低的溶解性,导致淤浆的最初形成。如果需要,可将本发明的析相分离作用重复数次并可在各种温度下使用乙酸溶剂。
为本发明的析相作用方法的纯化产物的式(I)固体酯前体可为无定型或晶体的形式,或为两种形式。如果为无定型形式,固体终产物可包含多种不同形状和大小中的任一种,并且这些无定型颗粒还可被附聚或絮集在一起以形成较大的块。如果为晶体形式,固体终产物可包含一种以上的晶体形式,并且它还可以混合物的形式出现。晶体颗粒的大小可在宽的大小范围内变化。
在更具体的术语中,析相作用或结晶指从多成分流体相中产生固体、单组分、无定型或晶体相,在本发明的情况下,所述流体相为不希望的杂质溶解在其中的乙酸溶液。在析相作用或结晶的目的是制备纯的干燥固体时,其中本发明的一些具体实施方案就是这种情况,则必须从所述流体相中分离固体,这通常通过离心或过滤,接着干燥来完成。这样的干燥固体无定型或晶体产物的有利性质包括易于处理、稳定性、良好的流动性和吸引人的外观。通常地,在套层或搅拌容器中进行析相作用或结晶,并且必须通过实验来确定获得适宜纯度、产率和可能的晶体形式所必需的条件。
在析相作用包括分散的晶体颗粒或来自溶液中的结晶时,它将按三个基本阶段来进行:过饱和的诱导、核的形成和晶体的生长。在给定的温度和浓度下,可通过冷却或除去溶剂使溶液饱和。还可加入降低溶解物溶解度的第三种成分或在其中产生的产物具有低溶解度的溶剂中进行化学反应。随着进一步冷却或浓缩,进入过饱和亚稳态范围。低水平的过饱和不可能导致晶核的自发形成,但可通过加入晶种启动晶体的生长。在落入限制亚稳态范围的曲线中的较低的温度或较高的浓度下,自发的成核作用实际上是肯定的并且在这些条件下也出现晶体的生长。
当超过亚稳态范围的界限时,成核作用的速率迅速增加,结晶过程变得不受控制。因此,需要将溶液的状态保持在亚稳态范围中。亚稳态范围曲线下的区域的宽度最显著地受搅拌、冷却速率、可溶性添加物的存在、溶剂以及具体溶液的热性质的影响。
成核作用引起小核的形成,晶体在小核周围生长。因此,没有成核作用,晶体生长则不能发生。当物质从溶液中晶体时,成核作用和晶体生长在一个宽的中间体温度范围内同时发生。成核作用依赖于过冷的程度,而低水平的过冷导致很少或没有成核作用。然而,成核作用的速率升至最大,接着下降,这样过度的冷却可通过限制形成的核的数量来抑制结晶速率。当足够的低动能分子达到它们的相互吸引足以克服它们的各自的动量的范围时,则发生自发的成核作用。一旦达到一定的大小,在普遍情况下核变得稳定,并且随着温度降低,出现更多的低能分子,成核作用的速率增加。这些情况部分说明了上面推理的在如上所述的式(I)酯前体的溶液中尤其麻烦的二聚体杂质的形成的特点。
晶核的形成或成核作用还是一个决定产物晶体大小的过程,并进一步在决定所述晶体的许多物理性质中起着重要的作用,并且更重要的是在本发明的情况下对它们的纯度起着重要作用。
对于晶体生长,在较高温度下,分子被过度激发而不能在晶格中保持被捕获状态,而在较低的温度下,较多的分子被保留,并且生长速率增大。然而,最终在仍然较低的温度下扩散并且在晶体表面的定向变得被抑制。在晶体表面的沉积导致紧邻部位的分子缺乏。因此,晶体生长的驱动力由从溶液的过饱和至晶面较低浓度的浓度梯度框架来提供。因此,高水平的过饱和促进高速率的晶体生长。
根据晶格的正确定位和适宜的定向导致所参与分子的动能损失。涉及结晶热的聚集必须进行,即被从整个溶液转移至一些表面,因此,晶体生长的速率受热转移速率和在所述表面进行的变化的影响。例如,已熟知体系的搅拌通过减少晶体附近的液体层的热阻来增加热转移直至在晶面的变化变为被有效控制。起初,搅拌通过降低该边界层的厚度以及扩散阻力迅速增加生长速率。然而,随着搅拌加强,达到通过表面反应动力学确定的限制值。
在晶体生长期间生长单位或前体经历的一些阶段显示了其它的关键因素,例如从大量溶液转移至对于晶体生长位点为非必需的撞击位点、在前体脱离溶剂分子并且溶剂被放回至溶液中的撞击处的吸附、前体从撞击部位到生长部位的扩散以及在去溶剂化后结合至晶格中,其中对于溶剂还可在逃逸到溶液中之前被吸附。所有这些过程取决于界面区的形态学。
在现有技术中已采用各种晶体生长的方式以确定晶面的生长机理以及随后的界面过程。例如,采用整体扩散和表面扩散模型以及二维成核作用和螺状生长方式。而且,根据现有技术的不同方法测定整个生长速率,但是从晶体生长理论的角度来看,晶体平面的线性生长速率是最常用的。另外,通过不同方法完成成核速率和成核动力学的测定。其中之一是诱导期的测定,其中它为研究体系在获得过饱和与出现固相之间所经历的时间。诱导期被认为与成核速率成反比。在结晶器中,成核作用和晶体生长与过饱和竞争,并均有助于终产物大小分布。
为了获得高组成均一性和由此高纯度的晶体,将线性生长速率在整个前进界面保持恒定非常重要,即在生长期间晶体形状保持不变。
通过结晶将终产物沉淀从中分离的可溶性杂质可增加或降低成核作用的速率。例如,不溶物可起到核的作用并从而促进结晶作用。杂质还可影响晶体形状。由于这些杂质的存在,固体沉淀的组成不同于结晶期间共存的流体的组成。这一现象被称为离析,并且由于各种原因,它对于晶体的生长非常重要,在每种情况下的中心问题是结晶组成在多大程度上反应它从中生长的养分的组成。
取决于它们对晶体Gibbs(吉布斯)自由能的影响,杂质被前进的界面部分排斥或优先吸收。因此,根据杂质的界面转移来定义离析系数。而且,已知杂质-溶剂的相互作用和复合物的形成导致离析系数的复杂浓度依赖性。对于晶体生长动力学本身而言,离析也是非常重要的,因为杂质可强烈地影响生长动力学。当晶体从不纯溶液中生长时,如果在晶体中比在溶液中更加不溶,则它将总体上排斥杂质。随着界面移动,杂质可能比可通过扩散被带走更为迅速地被排斥进入溶液中。结果,固体中的杂质浓度将由富集扩散层中的杂质浓度而不由溶液中的平均浓度决定。因此,以控制方式进行的离析可被有利地用于物质的纯化。
现有技术中已熟知可获得的最大的过饱和与晶体成核速率的大的差异可来自适宜的溶剂-溶质体系的选择。另外,当溶剂从极性变为非极性时,可获得的最大过饱和存在显著的差异,ΔCmax,并在ΔCmax与溶解度之间存在明显的相关性。溶解度越高,发生成核作用的过饱和度越低;因此,当溶液更加浓缩时,成核作用较为容易。溶剂的选择还对晶体的生长具有明显的作用。通过与生长界面性质相关的两个因素确定晶体从溶液中生长的生长动力学:分子粗糙度和溶剂在表面的吸附性质。
当根据上面讨论的原则和如本文所述地选择析相作用方法的所需参数,并用于本发明的方法中时,则将在用于获得所需结果的适宜的设备中进行所产生的纯化方法的具体实例。析相作用或结晶过程的目的本身是在最佳基础上产生所需形状大小分布,纯度和产率的无定型或晶体颗粒。当涉及结晶作用时,这通过保持一定的过饱和度来实现,其中在这种过饱和度下成核作用和晶体生长以适宜速率进行。除了溶质的溶解性和温度外,其它重要的因素包括溶质的热稳定性、存在的杂质的性质以及所需的水合程度。
在步骤一开始,本发明方法中的酯前体溶质在热乙酸溶剂中就极为不溶。然而,在此阶段溶解的酯前体将基本上随着温度增加而增加,通常在适宜的结晶器装置中通过冷却热的浓缩的溶液产生大部分溶质的过饱和和沉积。因此,可冷却蒸发晶体后的母液以产生另一批晶体。另一方面,可使用采用闪蒸的结晶装置。在该装置中,将热溶液通过其中发生蒸发和冷却的真空室。最佳地,采用的结晶器应产生平均大小的晶体,这便于母液的除去和洗涤。如果大量液体吸留在晶体块中,干燥将产生根据本发明为不可接受的不纯产物。另一个优点是在贮存时平均大小的晶体不易成块。
使用其中发生缓慢控制或完全自然冷却的搅拌反应容器可实现大而均匀的晶体的分批式生产。随着结晶作用发生,过饱和的程度和溶质的浓度降低,最后达到生长停止的饱和状态。在不存在自然成核作用时可通过人工引晶过饱和的溶液来获得对该过程的密切控制。可通过使用Oslo或Krystal结晶器实现大而均匀的晶体的连续生产,其中亚稳态,过饱和溶液被释放到溶质沉积在其中的生长晶体部分的底部。通过溶液的循环和分类使晶体成为流体,即该区域的分层作用使得足够大的晶体从结晶器的底部分离。
通常根据溶液过饱和的方式分类结晶器,例如冷却结晶器或蒸发结晶器。真空结晶器伴有这两个过程。在冷却结晶器中的分批式结晶在被搅拌器搅拌的密封罐中进行,其中溶液的比热和晶体热通过再循环冷却水流过的套管或旋管被除去。搅拌对于防止这些罐中的温度梯度、抑制沉淀和在罐底的不规则晶体生长以及对便于晶体生长非常重要。
当需要在连续的基础上进行结晶过程时,结晶器装置可采用如上述对于罐所描述的相同方式冷却的槽的形式。溶液在一端进入,晶体和液体在另一端被弃置。在该装置中的搅拌可通过使用缓慢移动的旋管来实现,其中该旋管在溶液中工作,并将晶体从冷却表面升高以分配它们通过溶液并缓慢将它们通过槽运送。可将整个槽的摇动与增加溶液在槽中的保留时间的挡板结合使用。这两种类型的结晶器特征在于低的热转移系数,更迅速的热交换可通过使用双管排列来实现,其中晶体流体在中心管中运送,冷却剂的逆流流动位于管之间的环隙中。这类装置中的搅拌通常通过使用在中心管旋转并携带有刮削热转移表面的叶片的轴来实现,这使得能获得高的热转移系数。
蒸发结晶器可为简单的盘状装置或搅拌反应容器。为了更高的生产水平,在加热和下水系统中采用排式管,其必须足够大以容纳悬液流动,其中通常装有一个叶轮,并伴有强制循环以增加对蒸发液体的热转移。可使用通过蒸发使溶液饱和的Oslo结晶器进行连续过程,其中对晶体产物大小的严格控制是非常重要的。在真空结晶器中,通常将热的浓缩溶液进料至保持在低压下的搅拌结晶室中。溶液沸腾并绝热冷却至对应于结晶器操作压力的沸点。结晶作用接在浓缩之后,并将产物从容器底部除去。
为了进一步说明它们的目的,紧接在下面提供本发明具体实施方案的实例,但决不限制本文权利要求所针对的本发明的范围。
实施例1咔唑酯前体的纯化
向反应容器中加入30.0g咔唑酯前体的具体产物,(6-氯-2-咔唑基)甲基-丙二酸二乙酯,已被确定含有0.6%(重量计)具有下面结构的螺羟基吲哚二聚体杂质:
将咔唑酯前体物与90ml冰乙酸混合并搅拌加热到50-55℃。产生在该温度下搅拌约2.5小时的稀的淤浆。接着将淤浆缓慢冷却到20-25℃,再搅拌2小时,随后过滤并干燥。获得的咔唑酯终产物的产率为23.14g(77%),其中它含有0.028%(重量计)的螺羟基吲哚二聚体杂质。
Claims (10)
1.一种纯化式(I)的(6-氯-2-咔唑基)甲基-丙二酸二(C1-C6烷基)酯的方法: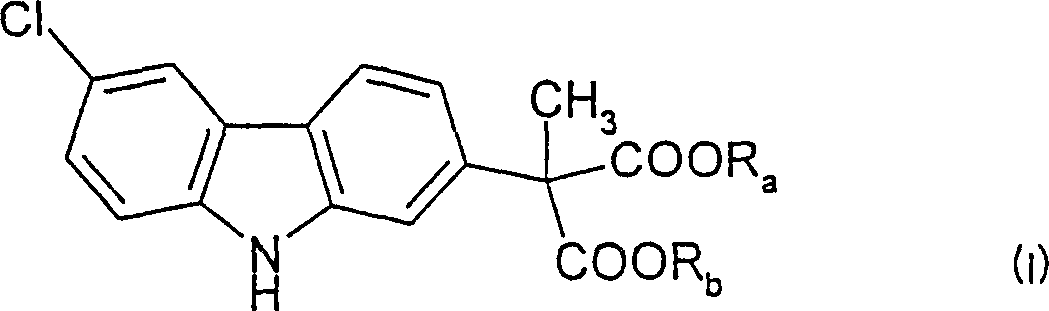 其中Ra和Rb必须相同并选自C1-C6烷基;该方法包括将一种或多种杂质从所述咔唑酯中至少进行一次析相作用,其中用来进行所述析相作用的溶剂为乙酸。
其中Ra和Rb必须相同并选自C1-C6烷基;该方法包括将一种或多种杂质从所述咔唑酯中至少进行一次析相作用,其中用来进行所述析相作用的溶剂为乙酸。
2.根据权利要求1的方法,其中所述乙酸为保存在30℃-110℃温度下的冰乙酸。
3.根据权利要求2的方法,其中所述温度为50℃-70℃,并且仅进行一次所述析相作用。
4.根据权利要求1的方法,其中所述式(I)咔唑酯为二乙酯。
5.根据权利要求4的方法,其中所述咔唑酯以至少99.95%重量计的纯度获得,这样存在于其中的杂质的量为0.05%重量计或更少。
6.根据权利要求5的方法,其中待纯化的所述式(I)咔唑酯以分离的晶体固体的形式存在。
7.根据权利要求1的方法,其中所述一种或多种杂质在所述酯的制备方法过程中直接或间接产生,并含有一种或多种起始物、合成中间体、反应体、反应副产物、降解产物、所述制备方法的各种反应步骤在其中进行的溶剂。
8.根据权利要求7的方法,其中所述一种或多种杂质作为不适当或在次佳基础上进行的结果而间接地从所述制备方法产生。
9.根据权利要求1的方法,其中所述一种或多种杂质偶然来自式(I)所述咔唑酯的制备方法在其中进行的设备的污染,来自所述制备方法中采用的起始物、溶剂或合成助剂的污染,来自所述制备方法周围的环境中的被吸收至所述方法中的污染物,或来自通过所述制备方法制备后的贮存或处理时的所述式(I)咔唑酯的污染。
10.根据权利要求1的方法,其中所述一种或多种杂质包括式(IV)螺羟吲哚二聚体:
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US8948098P | 1998-06-16 | 1998-06-16 | |
| US089480 | 1998-06-16 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1242362A CN1242362A (zh) | 2000-01-26 |
| CN1119329C true CN1119329C (zh) | 2003-08-27 |
Family
ID=22217887
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN99108607A Expired - Fee Related CN1119329C (zh) | 1998-06-16 | 1999-06-15 | 6-氯-α-甲基-咔唑-2-乙酸的咔唑酯前体的纯化方法 |
Country Status (26)
| Country | Link |
|---|---|
| US (1) | US6013808A (zh) |
| EP (1) | EP0965586B1 (zh) |
| JP (1) | JP3449602B2 (zh) |
| KR (1) | KR100342148B1 (zh) |
| CN (1) | CN1119329C (zh) |
| AR (1) | AR016726A1 (zh) |
| AT (1) | ATE237588T1 (zh) |
| AU (1) | AU745058C (zh) |
| BR (1) | BR9902279A (zh) |
| CA (1) | CA2274355C (zh) |
| CZ (1) | CZ293399B6 (zh) |
| DE (1) | DE69906876T2 (zh) |
| DK (1) | DK0965586T3 (zh) |
| ES (1) | ES2196723T3 (zh) |
| HK (1) | HK1023999A1 (zh) |
| HU (1) | HUP9901989A3 (zh) |
| ID (1) | ID23297A (zh) |
| IL (1) | IL130428A (zh) |
| PL (1) | PL333741A1 (zh) |
| PT (1) | PT965586E (zh) |
| RU (1) | RU2182148C2 (zh) |
| SG (1) | SG76627A1 (zh) |
| TR (1) | TR199901330A2 (zh) |
| TW (1) | TW491839B (zh) |
| YU (1) | YU26999A (zh) |
| ZA (1) | ZA993974B (zh) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20020049283A (ko) * | 2000-12-19 | 2002-06-26 | 이계안 | 하이브리드 전기 자동차용 토크 제어 시동방법 |
| JP4467880B2 (ja) | 2002-12-09 | 2010-05-26 | 株式会社日立製作所 | プロジェクトの評価システムおよび方法 |
| TWI692598B (zh) | 2019-05-03 | 2020-05-01 | 愛烙達股份有限公司 | 可變形之燭芯及設置該燭芯之燃燒裝置 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4193923A (en) * | 1978-08-21 | 1980-03-18 | Hoffmann-La Roche Inc. | Tetrahydro 2-carbazolyl methyl malonate derivatives |
| CH637117A5 (de) * | 1979-03-02 | 1983-07-15 | Hoffmann La Roche | Verfahren zur herstellung eines carbazolderivates. |
| JPS62263153A (ja) * | 1986-05-28 | 1987-11-16 | エフ・ホフマン―ラ ロシユ アーゲー | カルバゾ−ル誘導体 |
| DE3814887C1 (zh) * | 1988-05-02 | 1989-09-21 | Medice Chem.-Pharm. Fabrik Puetter Gmbh & Co Kg, 5860 Iserlohn, De |
-
1999
- 1999-04-15 US US09/292,138 patent/US6013808A/en not_active Expired - Fee Related
- 1999-05-05 PT PT99303509T patent/PT965586E/pt unknown
- 1999-05-05 DK DK99303509T patent/DK0965586T3/da active
- 1999-05-05 ES ES99303509T patent/ES2196723T3/es not_active Expired - Lifetime
- 1999-05-05 AT AT99303509T patent/ATE237588T1/de not_active IP Right Cessation
- 1999-05-05 EP EP99303509A patent/EP0965586B1/en not_active Expired - Lifetime
- 1999-05-05 DE DE69906876T patent/DE69906876T2/de not_active Expired - Fee Related
- 1999-06-02 JP JP15520099A patent/JP3449602B2/ja not_active Expired - Fee Related
- 1999-06-10 IL IL13042899A patent/IL130428A/xx not_active IP Right Cessation
- 1999-06-10 TW TW088109709A patent/TW491839B/zh not_active IP Right Cessation
- 1999-06-11 SG SG1999002908A patent/SG76627A1/en unknown
- 1999-06-11 AR ARP990102821A patent/AR016726A1/es not_active Application Discontinuation
- 1999-06-14 CA CA002274355A patent/CA2274355C/en not_active Expired - Fee Related
- 1999-06-14 YU YU26999A patent/YU26999A/sh unknown
- 1999-06-15 ID IDP990580A patent/ID23297A/id unknown
- 1999-06-15 KR KR1019990022158A patent/KR100342148B1/ko not_active IP Right Cessation
- 1999-06-15 TR TR1999/01330A patent/TR199901330A2/xx unknown
- 1999-06-15 ZA ZA9903974A patent/ZA993974B/xx unknown
- 1999-06-15 RU RU99113024/04A patent/RU2182148C2/ru not_active IP Right Cessation
- 1999-06-15 AU AU35038/99A patent/AU745058C/en not_active Ceased
- 1999-06-15 CZ CZ19992142A patent/CZ293399B6/cs not_active IP Right Cessation
- 1999-06-15 PL PL99333741A patent/PL333741A1/xx not_active IP Right Cessation
- 1999-06-15 HU HU9901989A patent/HUP9901989A3/hu unknown
- 1999-06-15 CN CN99108607A patent/CN1119329C/zh not_active Expired - Fee Related
- 1999-06-16 BR BR9902279-6A patent/BR9902279A/pt not_active IP Right Cessation
-
2000
- 2000-05-30 HK HK00103190A patent/HK1023999A1/xx not_active IP Right Cessation
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR100382122B1 (ko) | 고순도 테레프탈산의 제조방법 | |
| CN1058256C (zh) | 丙烯酸纯化方法和设备 | |
| CN102459138A (zh) | 分离发酵液成分的方法 | |
| SE533074C2 (sv) | Fraktioneringsprocess och kristalliserare för ätbara oljor och fetter genom smältning och/eller upplösning, kylning samt omrörning | |
| CN101050171A (zh) | 制备不饱和脂肪酸的方法 | |
| CN102329212A (zh) | 长链二元酸的精制方法 | |
| CN1119329C (zh) | 6-氯-α-甲基-咔唑-2-乙酸的咔唑酯前体的纯化方法 | |
| CN1029842C (zh) | 一种精制长链二元酸的方法 | |
| EP1173417A1 (en) | Process and apparatus for making ultra-pure dmso | |
| CN110386867A (zh) | 一种乙基香兰素的连续化纯化方法 | |
| CN1850768A (zh) | 高纯度结晶山梨酸的制备工艺 | |
| CN110642887B (zh) | 一种羟基亚乙基二膦酸晶体的连续化生产方法 | |
| JPS6010761B2 (ja) | 晶析装置 | |
| CN1171890C (zh) | 高纯桉叶素的制备方法及其装置 | |
| CN113248365A (zh) | 一种医用级l-乳酸生产方法 | |
| CN109071402B (zh) | 甲基丙烯酸的精制方法和制造方法 | |
| MXPA99005563A (en) | Method of purifying carbazole ester precursors of 6-chloro-alpha-methyl-carbazole-2-acetic acid | |
| JP5569108B2 (ja) | (メタ)アクリル酸の精製方法 | |
| CN115340462B (zh) | 一种尼龙56盐连续结晶纯化方法 | |
| CN203989946U (zh) | 一种双管式交叉混流结晶装置 | |
| CN110818593B (zh) | 一种邻羟基苯甲腈的精制方法 | |
| KR100463743B1 (ko) | 불포화지방산의 분리방법 및 분리된 지방산을 이용하여제조한 디글리세라이드 고함유 유지 조성물 | |
| JP6214156B2 (ja) | メタクリル酸の精製方法 | |
| CN115784900A (zh) | 草甘膦生产过程三乙胺纯化方法 | |
| EP4149918A1 (en) | Process of synthesizing and purifying (3r)-hydroxybutyl (3r)-hydroxybutanoate |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C06 | Publication | ||
| PB01 | Publication | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: GR Ref document number: 1022024 Country of ref document: HK |