CN1056373C - 含氮杂环类化合物的氟代烷氧基苄氨基衍生物的制备方法 - Google Patents
含氮杂环类化合物的氟代烷氧基苄氨基衍生物的制备方法 Download PDFInfo
- Publication number
- CN1056373C CN1056373C CN92104778A CN92104778A CN1056373C CN 1056373 C CN1056373 C CN 1056373C CN 92104778 A CN92104778 A CN 92104778A CN 92104778 A CN92104778 A CN 92104778A CN 1056373 C CN1056373 C CN 1056373C
- Authority
- CN
- China
- Prior art keywords
- formula
- compound
- group
- phenyl
- trifluoro
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D453/00—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids
- C07D453/02—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids containing not further condensed quinuclidine ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D205/00—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom
- C07D205/02—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/26—Psychostimulants, e.g. nicotine, cocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/44—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reduction and hydrolysis of nitriles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/51—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by pyrolysis, rearrangement or decomposition
- C07C45/511—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by pyrolysis, rearrangement or decomposition involving transformation of singly bound oxygen functional groups to >C = O groups
- C07C45/513—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by pyrolysis, rearrangement or decomposition involving transformation of singly bound oxygen functional groups to >C = O groups the singly bound functional group being an etherified hydroxyl group
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/67—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton
- C07C45/673—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by change of size of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/67—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton
- C07C45/68—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms
- C07C45/70—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms by reaction with functional groups containing oxygen only in singly bound form
- C07C45/71—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms by reaction with functional groups containing oxygen only in singly bound form being hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C47/00—Compounds having —CHO groups
- C07C47/52—Compounds having —CHO groups bound to carbon atoms of six—membered aromatic rings
- C07C47/575—Compounds having —CHO groups bound to carbon atoms of six—membered aromatic rings containing ether groups, groups, groups, or groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/56—Nitrogen atoms
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Psychiatry (AREA)
- Physical Education & Sports Medicine (AREA)
- Pain & Pain Management (AREA)
- Pulmonology (AREA)
- Immunology (AREA)
- Rheumatology (AREA)
- Anesthesiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Hydrogenated Pyridines (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Indole Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pyridine Compounds (AREA)
- Other In-Based Heterocyclic Compounds (AREA)
- Pyrrole Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Quinoline Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
本发明是关于新的含氮杂环化合物的氟烷氧基苄氨基衍生物,具体地说,本发明涉及下式I化合物,式中Q、X1、X2和X3的定义如说明书中所述。本发明新的化合物可用于治疗炎症和中枢神经系统疾病以及其他的病症。
Description
本发明涉及新的含氮杂环类化合物的氟代烷氧基苄氨基衍生物,包含这类化合物的药物组合物,这类化合物在治疗和预防炎症和中枢神经系统疾病,以及多种其他疾病中的应用。本发明的药物活性化合物是P物质受体拮抗剂。本发明还涉及用于合成这类P物质受体抗拮剂的新的中间体。
P物质是属于肽类中速激肽家族的天然存在的十一肽;速激肽的命名源于它们对平滑肌组织的迅速刺激作用。更具体地讲,P物质是一种具有药理活性的神经肽,它产生于哺乳动物体内(最初从肠中分离得到),并有特征的氨基酸顺序,该顺序由D.F.Veber等人描述于美国专利4,680,283号中。在本技术领域已充分证实了P物质及其他速激肽与许多疾病的病理生理学有着广泛的联系。例如,最近证实P物质参与疼痛或偏头痛的介导〔参见:B.E.B.Sandberg et al.,Journal of MedicinalChemistry,Vol.25,P.1009(1982)〕,以及中枢神经系统疾病(如:焦虑和精神分裂症)、呼吸道疾病和炎症(如:哮喘和类风湿性关节炎)、风湿病(如:肌风湿病),以及胃肠功能紊乱和胃肠道疾病(如:溃疡性结肠炎和克罗恩氏病)等〔参见:D.Regoli in“Trends in Oluster Headache,”F.Sicuteri等编,Elsevier Scientific Publishers,Amsterdam,1987,PP.85-95〕。
为了更有效地治疗上述疾病,最近人们已做出一些努力以提供P物质和其他速激肽的拮抗剂。因此,到目前为止所叙述的几种这类拮抗剂一般都具有类似于肽的性质,因此,从代谢的观点来看,在将其具体用作治疗疾病的治疗剂时它们过于不稳定了。换言之,本发明的非肽类拮抗剂不具有这种缺点,从代谢的观点来看它们远比上述药物稳定。
呈现P物质受体拮抗剂活性的奎宁环衍生物和有关化合物归类于1989年11月20日提交的PCT专利申请PCT/US 89/05338和1990年7月23日提交的美国专利申请557,442,上述两个申请与本申请一起转让。类似的化合物归类于1991年4月25日提交的PCT专利申请PCT/US 91/02853,和1991年5月14日提交的PCT专利申请PCT/US 91/03369。这两个申请也与本申请一起转让。
用作P物质拮抗剂的哌啶衍生物和有关含氮杂环化合物归类于1990年11月28日提交的美国专利申请619,361和1990年9月28日提交的美国专利申请590,423,这两个申请与本申请一起转让。
本发明涉及下式I化合物: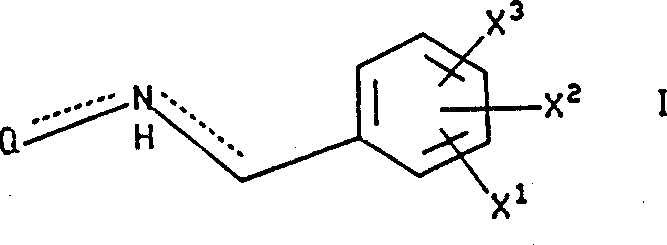 式中X1是氢,由1至3个氟原子任意取代的(C1-10)烷氧基或由1至3个氟原子任意取代的(C1-10)烷基;
式中X1是氢,由1至3个氟原子任意取代的(C1-10)烷氧基或由1至3个氟原子任意取代的(C1-10)烷基;
X2和X3独立地选自氢,卤素,硝基,由1至3个氟原子任意取代的(C1-10)烷基,由1至3个氟原子任意取代的(C1-10)烷氧基,三氟甲基,羟基,苯基,氰基,氨基,(C1-6)烷基氨基,二-(C1-C6)烷基氨基,
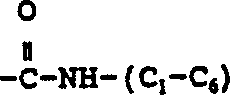 烷基,(C1-C6)烷基
烷基,(C1-C6)烷基
 烷基,羟基(C1-C4)烷基,(C1-C4)烷氧基(C1-C4)烷基,
烷基,羟基(C1-C4)烷基,(C1-C4)烷氧基(C1-C4)烷基,
 和
烷基;Q是下式基团:
和
烷基;Q是下式基团: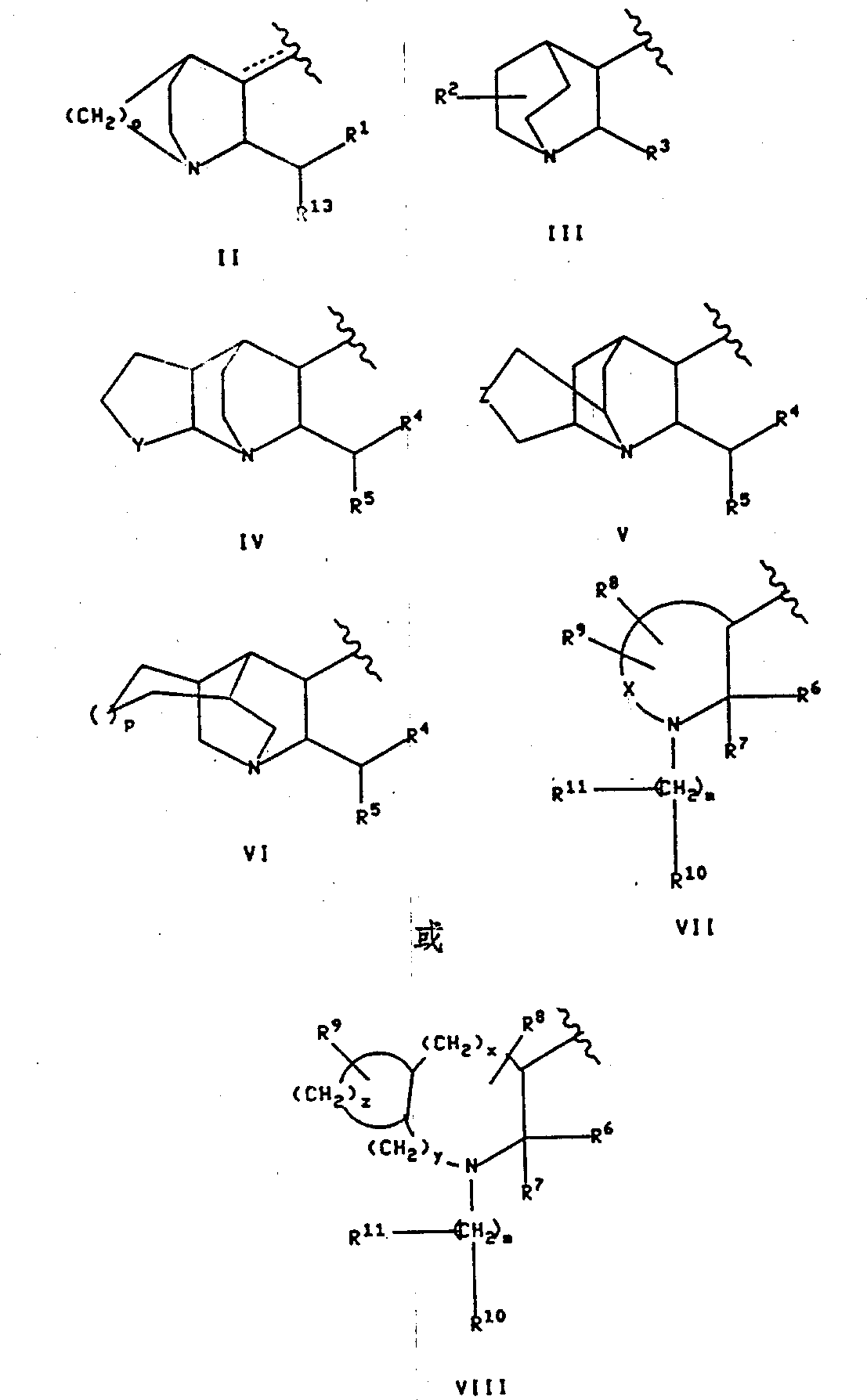 式中R1是选自呋喃基,噻吩基,吡啶基,吲哚基,联苯基和苯基的基团,这些基团可任意地被1或2个取代基取代,所述取代基独立地选自卤素,由1至3个氟原子任意取代的(C1-10)烷基,由1至3个氟原子任意取代的(C1-10)烷氧基,羧基,苄氧羰基和(C1-3)烷氧基羰基;
式中R1是选自呋喃基,噻吩基,吡啶基,吲哚基,联苯基和苯基的基团,这些基团可任意地被1或2个取代基取代,所述取代基独立地选自卤素,由1至3个氟原子任意取代的(C1-10)烷基,由1至3个氟原子任意取代的(C1-10)烷氧基,羧基,苄氧羰基和(C1-3)烷氧基羰基;
R13选自(C3-4)支链烷基,(C5-6)支链烯基,(C5-7)环烷基,和前述R1的定义中列举的基团;
R2是氢或(C1-6)烷基;
R3是苯基,联苯基,萘基,吡啶基,二苯甲基,噻吩基或呋喃基,并且R3可以由1至3个取代基任意地取代,所述取代基独立地选自卤素,由1至3个氟原子任意取代的(C1-10)烷基和由1至3个氟原子任意取代的(C1-10)烷氧基;
Y是(CH2)l,其中l是1至3的整数,或者Y是下式基团
Z是氧,硫,氨基,(C1-3)烷基氨基或(CH2)n,其中n是0,1或2;
O是2或3;
P是0或1;
R4是呋喃基,噻吩基,吡啶基,吲哚基,联苯基,或苯基,它们可由1或2个取代基任意地取代,所述取代基独立地选自卤素,由1至3个氟原子任意地取代的(C1-10)烷基,由1至3个氟原子任意地取代的(C1-10)烷氧基,羧基,(C1-3)烷氧羰基和苄氧羰基;
R5是由1或2个取代基任意取代的噻吩基,联苯基或苯基,所述取代基独立地选自卤素,由1至3个氟原子任意地取代的(C1-10)烷基和由1至3个氟原子任意地取代的(C1-10)烷氧基;
式1中的两个虚线中的每一个和式II中的虚线代表一个任意的双键,当Q是式II基团时该双键可任意地存在;
X是(OH2)q,其中q是1至6的整数,并且,其中在(CH2)q中的任何一个碳-碳单键可任意地由碳-碳双键替代,其中,所说(CH2)q中的任何一个碳原子可任意地由R8取代,其中,所说(CH2)q中的任何一个碳原子可任意地由R9取代;
m是0至8的整数,(CH2)m中的任何一个碳-碳单键可任意地由碳-碳双键或碳-碳叁键替代,所说(CH2)m中的任何一个碳原子可由R11任意取代;
R6是选自氢,(C1-6)直链或支链烷基,其中一个碳原子可任意地由氮、氧或硫替代的(C3-7)环烷基;选自联苯基,苯基,二氢化茚基和萘基的芳基;选自噻吩基,呋喃基,吡啶基,噻唑基,异噻唑基,噁唑基,异噁唑基,三唑基,四唑基和喹啉基的杂芳基;苯基(C2-6)烷基,二苯甲基和苄基,其中,每个所述芳基和杂芳基,以及所说苄基,苯基(C2-6)烷基和二苯甲基中的苯基部分可任意地由1个或多个取代基取代,所述取代基独立地选自卤素,硝基,由1至3个氟原子任意取代的(C1-10)烷基,由1至3个氟原子任意取代的(C1-10)烷氧基,氨基,羟基-(C1-6)烷基,(C1-6)烷氧基-(C1-6)烷基,(C1-C6)-烷氧基,(C1-C6)烷基
 (C1-C6)烷基
(C1-C6)烷基
 (C1-C6)烷基,(C1-C6)烷基
(C1-C6)烷基,(C1-C6)烷基
 (C1-C6)烷基
(C1-C6)烷基
 (C1-C6)烷基-O-,(C1-C6)烷基
(C1-C6)烷基-O-,(C1-C6)烷基
 烷基
烷基
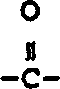 (C1-C6)烷基-,二-(C1-C6)烷基氨基,
(C1-C6)烷基-,二-(C1-C6)烷基氨基,
 烷基,(C1-C6)-烷基
烷基,(C1-C6)-烷基
 烷基,
和
烷基,
和
 烷基;其中,所述二苯甲基中的一个苯基可任意地由萘基,噻吩基,呋喃基或吡啶基替代;
烷基;其中,所述二苯甲基中的一个苯基可任意地由萘基,噻吩基,呋喃基或吡啶基替代;
R7是氢,苯基或(C1-6)烷基;
或者R6和R7与和它们相连接的碳一起形成含有3至7个碳原子的饱和碳环,其中,所说碳原子之一可任意地由氧,氮或硫替代;
R8和R9各自独立地选自氢,羟基,卤素,氨基,氧代(=O),氰基,羟基-(C1-6)烷基,(C1-6)烷氧基-(C1-6)烷基,(C1-6)烷基氨基,二-(C1-6)烷基氨基,(C1-6)烷氧基,(C1-C6)烷基
 ,(C1-C6)烷基
烷基,(C1-C6)烷基
,(C1-C6)烷基
烷基,(C1-C6)烷基
 ,(C1-C6)烷基
,(C1-C6)烷基
 烷基-O-,(C1-C6)烷基
,(C1-C6)烷基
烷基-,和前文在R6的定义中给出的基团;R10是
烷基-O-,(C1-C6)烷基
,(C1-C6)烷基
烷基-,和前文在R6的定义中给出的基团;R10是
 ,NHOH2R12,NHSO2R12或者前文在R6,R8和R9任意一个的定义中给出的基团之一;
,NHOH2R12,NHSO2R12或者前文在R6,R8和R9任意一个的定义中给出的基团之一;
R11是肟基(=NOH)或者前文在R6,R8和R9任意一个的定义中给出的基团之一;
R12是(C1-6)烷基,氢,苯基(C1-6)烷基,或由(C1-6)烷基任意取代的苯基;
上述定义的前提是:(a)当m为0时,R11不存在,(b)R8,R9,R10或R11均不可以与它所连接的碳原子一起与R7形成环,(c)当Q是式VIII基团时,R8和R9不可以连接在同一碳原子上,(d)当R8和R9连接在同一碳原子上时,那么或者R8和R9各自独立地选自氢,氟,(C1-6)烷基,羟基-(C1-6)烷基和(C1-6)烷氧基-(C1-6)烷基,或者R8和R9与和它们所连接的碳一起形成(C3-6)饱和碳环,该环与它们所连接的含氮环形成螺环化合物,(e)式I中的氮不可以同时通过双链与和它所连接的Q和取代苄基连接,(f)当Q是式VII基团,q是2,R8和R9中随便哪一个是5-羟基-(C1-6)烷基或5-(C1-6)烷氧基-(C1-6)烷基时,那么R8和R9中的另一个则是5-(C1-3)烷基或氢;(g)当Q是式VII基团,q是2时,R8或R9均不是4-羟基(C1-6)烷基或4-(C1-6)烷氧基-(C1-6)烷基,(h)如果X1,X2和X3均不是氟代烷氧基,那么,R1,R3,R4,R5,R6,R7和R13中至少有一个是由氟代烷氧基取代的芳基。
本发明还涉及式I化合物的可药用酸加成盐和碱盐。用于制备本发明前述碱性化合物的可药用酸加成盐的酸是能形成无毒酸加成盐的那些酸,所谓无毒酸加成盐即为含有药理学可接受阴离子的盐,例如,盐酸盐,氢溴酸盐,氢碘酸盐,硝酸盐,硫酸盐,硫酸氢盐,磷酸盐,酸式磷酸盐,乙酸盐,乳酸盐,柠檬酸盐,柠檬酸氢盐,酒石酸盐,酒石酸氢盐,琥珀酸盐,马来酸盐,富马酸盐,葡萄糖酸盐,糖二酸盐,苯甲酸盐,甲磺酸盐,乙磺酸盐,苯磺酸盐,对甲苯磺酸盐和pamoate〔即,1,1′-亚甲基二-(2-羟基-3-萘酸)盐〕。
本文采用的术语“卤素”,除特别指明外,包括氯,氟,溴和碘。
本文所采用的术语“烷基”,除特别指明外,包括具有直链,支链或环化部分或其组合的饱和一价烃基。
本文采用的术语“1个或多个取代基”,以可成键位置的数目计算,包括1个至最大可能数目的取代基。
在优选的式I化合物中,R1,R4,R5和R7是苯基,R2是氢;R3是由氯、氟、由1至3个氟原子任意取代的(C1-6)烷基或由1至3个氟原子任意取代的(C1-6)烷氧基任意取代的苯基,m是0,n是3或4。
具体的优选式I化合物是:
(2S,3S)-3-(5-叔丁基-2-甲氧基苄基)氨基-2-(3-三氟甲氧基苯基)哌啶;
(2S,3S)-3-(2-异丙氧基-5-三氟甲氧基苄基)氨基-2-苯基哌啶;
(2S,3S)-3-(2-乙氧基-5-三氟甲氧基苄基)氨基-2-苯基哌啶;
(2S,3S)-3-(2-甲氧基-5-三氟甲氧基苄基)氨基-2-苯基哌啶;
(2S,3S)-3-(5-叔丁基-2-三氟甲氧基苄基)氨基-2-苯基哌啶;
2-(二苯甲基)-N-(2-甲氧基-5-三氟甲氧基苯基)甲基-1-氮杂双环〔2.2.2.〕辛烷-3-胺;
(2S,3S)-3-〔5-氯-2-(2,2,2-三氟乙氧基)苄基〕氨基-2-苯基哌啶;
(2S,3S)-3-(5-叔丁基-2-三氟甲氧基苄基)氨基-2-苯基哌啶;
(2S,3S)-3-(2-异丙氧基-5-三氟甲氧基苄基)氨基-2-苯基哌啶;
(2S,3S)-3-(2-二氟甲氧基-5-三氟甲氧基苄基)氨基-2-苯基哌啶;
(2S,3S)-2-苯基-3-2-(2,2,2-三氟乙氧基苄基)氨基哌啶;
(2S,3S)-2-苯基-3-(2-三氟甲氧基苄基)〕氨基哌啶。
其他式I化合物是:
3-〔N-(2-甲氧基-5-三氟甲氧基苄基)氨基〕-5,5-二甲基-2-苯基吡咯烷;
3-〔N-(2-甲氧基-5-三氟甲氧基苄基)氨基〕-4,5-二甲基-2-苯基吡咯烷;
3-(2-环丙氧基-5-三氟甲氧基苄基)氨基-2-苯基哌啶;
3-(2-环丙基甲氧基-5-三氟甲氧基苄基)氨基-2-苯基哌啶;
3-(2-二氟甲氧基-5-苯基苄基)氨基-2-苯基哌啶;
3-(5-环丙基甲氧基-2-二氟甲氧基苄基)氨基-2-苯基哌啶;
3-(2-甲氧基苄基)氨基-2-(3-三氟甲氧基苯基)哌啶;
3-(2-甲氧基-5-三氟甲氧基苄基)氨基-2-(3-三氟甲氧基苯基)哌啶;
2-苯基-3-(5-正丙基-2-三氟甲氧基苄基)氨基哌啶;
3-(5-异丙基-2-三氟甲氧基苄基)氨基-2-苯基哌啶;
3-(5-乙基-2-三氟甲氧基苄基)氨基-2-苯基哌啶;
3-(5-仲丁基-2-三氟甲氧基苄基)氢基-2-苯基哌啶;
3-(5-二氟甲氧基-2-甲氧基苄基)氨基-2-苯基哌啶;
3-(2-甲氧基-5-三氟甲氧基苄基)氨基-2-苯基吡咯烷;
3-(2-甲氧基-5-三氟甲氧基苄基)氨基-2-苯基高哌啶;
2-二苯甲基-3-(2-甲氧基-5-三氟甲氧基苄基)氨基吡咯烷;
2-二苯甲基-3-(2-甲氧基-5-三氟甲氧基苄基)氨基高哌啶;
3-〔2,5-二-(2,2,2-三氟乙氧基)苄基〕氨基-2-苯基哌啶;
2-苯基-3-(3-三氟甲氧基苄基)氨基哌啶;
2-二苯甲基-3-(2-甲氧基-5-三氟甲氧基苄基)氨基哌啶;
1-(5,6-二氟己基)-3-(2-甲氧基-5-三氟甲氧基苄基)氨基-2-苯基哌啶;
1-(6-羟基己基)-3-(2-甲氧基-5-三氟甲氧基苄基)氨基-2-苯基哌啶;
3-苯基-4-(2-甲氧基-5-三氟甲氧基苄基)氨基-2-氮杂双环〔3.3.0〕辛烷;
4-二苯甲基-5-(2-甲氧基-5-三氟甲氧基苄基)氨基-3-氮杂双环〔4.1.0〕庚烷;
4-(2-甲氧基-5-三氟甲氧基苄基)氨基-3-苯基-2-氮杂双环〔4.4.0〕癸烷;
2-苯基-3-(2-甲氧基-5-三氟甲氧基苄基)氨基奎宁环;
8-二苯甲基-N-(2-甲氧基-5-三氟甲氧基苄基)-9-氮杂三环〔4.3.1.04,9〕癸烷-7-胺;
9-二苯甲基-N-(2-甲氧基-5-三氟甲氧基苄基)-10-氮杂三环〔4.4.1.05,10〕十一烷-8-胺;
9-二苯甲基-N-(2-甲氧基-5-三氟甲氧基苄基)-3-硫杂-10-氮杂三环〔4.4.1.05,10〕十一烷-8-胺;
8-二苯甲基-N-(2-甲氧基-5-三氟甲氧基苄基)-9-氮杂三环〔4.3.1.04,9〕癸烷-7-胺;
5,6-五亚甲基-2-二苯甲基-3-(2-甲氧基-5-三氟甲氧基苄基)氨基奎宁环;
5,6-三亚甲基-2-二苯甲基-3-(2-甲氧基-5-三氟甲氧基苄基)氨基奎宁环;
9-二苯甲基-N-((2-甲氧基-5-三氟甲氧基苯基)甲基)-3-氧杂-10-氮杂三环〔4.4.1.05,10〕十一烷-3-胺;
8-二苯甲基-N-((2-甲氧基-5-三氟甲氧基苯基)甲基)-7-氮杂三环〔4.4.1.05,10〕十一烷-9-胺;和
2-二苯甲基-N-((2-甲氧基-5-三氟甲氧基苯基)甲基)-1-氮杂双环〔3.2.2〕壬烷-3-胺。
本发明还涉及下式化合物 式中R14是三氟甲氧基或二氟甲氧基,R15是(C1-4)烷基,R16是二氟甲氧基或(C1-4)烷基,R17是三氟甲氧基,二氟甲氧基,(C1-4)烷基或(C1-4)烷氧基。
式中R14是三氟甲氧基或二氟甲氧基,R15是(C1-4)烷基,R16是二氟甲氧基或(C1-4)烷基,R17是三氟甲氧基,二氟甲氧基,(C1-4)烷基或(C1-4)烷氧基。
本发明还涉及治疗或预防哺乳动物(包括人)的下述疾病的药物组合物,这些疾病包括:炎症(如关节炎、牛皮癣、哮喘和炎症性肠病)、焦虑、抑郁或心境恶劣、结肠炎、精神病、疼痛、胃食管反流病、变应反应(如湿疹和鼻炎)、慢性阻塞性气管疾病、高敏性疾病(如毒葛)、血管痉挛性疾病(如绞痛、偏头痛和Reynaud氏病)、纤维化和胶原病(如硬皮病和嗜酸性片吸虫病)、反射性交感神经营养不良(如肩手综合症)、癖嗜病(如酒精中毒)、与应激反应有关的躯体疾病,外周神经病、神经痛、神经病理性疾病(如早老性痴呆、与爱滋病有关的痴呆、糖尿病性神经病和多发性硬化)、与免疫增强或抑制有关的疾病(如系统性红斑狼疮)、和风湿病(如肌风湿病),所述组合物包含治疗或预防这类疾病有效量的式I化合物或其可药用的盐及可药用的载体。
本发明还涉及治疗或预防哺乳动物(包括人)的下述疾病的方法,这些疾病包括:炎症(如关节炎、牛皮癖、哮喘和炎症性肠病)、焦虑、抑郁或心境恶劣,结肠炎、精神病、疼痛、胃食管反流病,变应反应(如湿疹和鼻炎)、慢性阻塞性气管疾病、高敏性疾病(如毒葛)、血管痉挛性疾病(如绞痛、偏头痛和Reynaud氏病)、纤维化和胶原病(如硬皮病和嗜酸性片吸虫病)、反射性交感神经营养不良(如肩手综合症)、癖嗜病(如酒精中毒)、与应激反应有关的躯体疾病、外周神经病、神经痛、神经病理性疾病(如早老性痴呆、与爱滋病有关的痴呆、糖尿病性神经病和多发性硬化)、与免疫增强或抑制有关的疾病(如系统性红斑狼疮)、和风湿病(如肌风湿病)。所述方法包括:将治疗或预防这类疾病有效量的式I化合物或其可药用的盐给予所述哺乳动物。
本发明还涉及拮抗哺乳动物(包括人)体内P物质的作用的药物组合物,该组合物包含拮抗P物质有效量的式I化合物或其可药用的盐及可药用的载体。
本发明还涉及拮抗哺乳动物(包括人)体内P物质的作用的方法,该方法包括将拮抗P物质有效量的式I化合物或其可药用的盐给予所述哺乳动物。
本发明还涉及用于治疗或预防由于P物质过量而引起的哺乳动物(包括人)疾病的药物组合物,该组合物包含拮抗P物质有效量的式I化合物或其可药用的盐及可药用的载体。
本发明还涉及治疗或预防由于P物质过量引起的哺乳动物(包括人)疾病的方法,该方法包括给所述哺乳动物服用拮抗P物质有效量的式I化合物或其可药用的盐。
本发明还涉及治疗或预防哺乳动物(包括人)的下述疾病的药物组合物,这些疾病包括:炎症(如关节炎、牛皮癣、哮喘和炎症性肠病)、焦虑、抑郁或心境恶劣、结肠炎、精神病、疼痛、胃食管反流病,变应反应(如湿疹和鼻炎)、慢性阻塞性气管疾病、高敏性疾病(如毒葛)、血管痉挛性疾病(如绞痛、偏头痛和Reynaud氏病)、纤维化和胶原病(如硬皮病和嗜酸性片吸虫病)、反射性交感神经营养不良(如肩手综合症)、癖嗜病(如酒精中毒)、与应激反应有关的躯体疾病,外周神经痛、神经病、神经病理性疾病(如早老性痴呆、与爱滋病有关的痴呆、糖尿病性神经病和多发性硬化)、与免疫增强或抑制有关的疾病(如系统性红斑狼疮)、和风湿病(如肌风湿病),所述药物组合物包含在P物质受体部位拮抗P物质作用有效量的式I化合物或其可药用的盐及可药用的载体。
本发明还涉及治疗或预防哺乳动物(包括人)的下述疾病的方法,这些疾病包括:炎症(如关节炎、牛皮癣、哮喘和炎症性肠病)、焦虑、抑郁或心境恶劣,结肠炎、精神病、疼痛、胃食管反流病、变应反应(如湿疹和鼻炎)、慢性阻塞性气管疾病、高敏性疾病(如毒葛)、血管痉挛性疾病(如绞痛、偏头痛和Reynaud氏病)、纤维化和胶原病(如硬皮病和嗜酸性片吸虫病)、反射性交感神经营养不良(如肩手综合症)、癖嗜病(如酒精中毒)、与应激反应有关的躯体疾病、外周神经病、神经痛、神经病理性疾病(如早老性痴呆、与爱滋病有关的痴呆、糖尿病性神经病和多发性硬化)、与免疫增强或抑制有关的疾病(如系统性红斑狼疮)、和风湿病(如肌风湿病)。所述方法包括:将在P物质受体部位拮抗P物质作用有效量的式I化合物或其可药用的盐给予所述哺乳动物。
本发明还涉及治疗或预防哺乳动物(包括人)疾病的药物组合物,所述疾病的治疗或预防是通过降低P物质介导的神经传导而发生作用或增强效果的,所述组合物包含能在P物质受体部位有效地拮抗P物质作用的一定量式I化合物或其可药用的盐及可药用的载体。
本发明还涉及治疗或预防哺乳动物(包括人)疾病的方法,所述疾病的治疗或预防是通过降低P物质介导的神经传导而发生作用或增强效果的,所述方法包括将能在P物质受体部位有效地拮抗P物质作用的一定量式I化合物或其可药用的盐给予所述哺乳动物。
本发明还涉及用于治疗或预防哺乳动物(包括人)疾病的药物组合物,所述疾病的治疗或预防是通过降低P物质介导的神经传导而发生作用或增强效果的,该组合物包含治疗或预防所述疾病有效量的式I化合物或其可药用的盐以及可药用的载体。
本发明还涉及治疗或预防哺乳动物(包括人)疾病的方法,所述疾病的治疗或预防是通过降低P物质介导的神经传导而起作用或增强效果的,该方法包括给予所述哺乳动物治疗或预防该疾病有效量的式I化合物或其可药用的盐。
本发明化合物具有手性中心,因此,可以以不同的对映异构体形式存在。本发明涉及式I化合物的所有光学异构体和所有的立体异构体,以及它们的混合物。
上述式I化合物包括与前文所述相同,但是由其放射性同位素置换其中一个或多个氢原子或碳原子的那些化合物。这些同位素标记的化合物可在代谢动力学研究及结合试验中用作研究和诊断的工具。在这些研究中的具体应用包括:放射配体结合测定试验,放射自显影研究和体内结合研究;而在诊断领域的具体应用包括:对人脑中的P物质受体在炎症相关组织中的体内结合作用研究,这些组织的例子包括免疫型细胞或与炎症性肠病直接有关的细胞等等。在式I化合物的放射性标记物中包括它们的氚和C14标记化合物。
按照下文反应式和讨论中描述的方法可以制得式I化合物。除特别指明外,下文反应式及讨论中的R1,R2,R3,R4,R5,R6,R7,R8,R9,R10,R11,R12,R13,X,Z,Q,Y,m,n,o,p,q,x,y和z如前文所限定。反应式1 反应式2
反应式2 反应式3
反应式3
式I化合物可以按反应式1和2所示方法制备。
参见反应式1,应用强的无机酸,如盐酸、氢溴酸或氢碘酸,于约室温至酸的回流温度使式X化合物水解脱去甲氧苄基。最好用氢溴酸于回流温度下进行该反应。得到相应的式XI化合物的上述反应通常进行约2小时。
最好在含有金属的催化剂(如铂或钯)存在下,将其中Q为式VII或VIII基团的式X化合物用氢处理以除去甲氧基苄基。该反应通常在对反应呈惰性的溶剂(如乙酸或低级醇)中,于约0℃~50℃进行(另外,还可以将上述化合物于约-30℃~-78℃用溶解在氨中的金属如锂或钠处理,或者在钯存在下用甲酸盐处理,或者在钯存在下用环己烷处理)。最好在钯-炭存在下,于甲醇/乙醇的水混合液或含有盐酸的甲醇/乙醇混合液中,于约25℃将上述化合物用氢处理。
通过与合适的式XII化合物反应,可以将得到的式XI化合物转变成相应的式I化合物(见反应式1)。该反应一般在还原剂(如氰基硼氢化钠、三乙酰氧基硼氢化钠、硼氢化钠、氢以及金属催化剂锌和盐酸、甲硼烷甲硫醚或甲酸)存在下,于约-60℃~50℃进行。该反应合适的反应惰性溶剂包括低级醇(如甲醇、乙醇和异丙醇)、乙酸和四氢呋喃(THF)。优选的溶剂为乙酸,温度为约25℃,还原剂为三乙酰氧基硼氢化钠。
另外,式XI化合物与式XII化合物的反应可以在干燥剂存在下进行,或在反应中应用经共沸除去生成水的仪器,结果得到下式的亚胺, 然后于约室温下使其与上述还原剂(最好是三乙酰氧基硼氢化钠)反应。上述亚胺的制备通常在对反应呈惰性的溶剂(如苯、二甲苯或甲苯,最好为甲苯)中,于约25~110℃(最好于溶剂的回流温度)下进行。合适的干燥剂/溶剂体系包括四氯化钛/二氯甲烷、异丙氧基钛/二氯甲烷及分子筛/THF。最好的是四氯化钛/二氯甲烷。
然后于约室温下使其与上述还原剂(最好是三乙酰氧基硼氢化钠)反应。上述亚胺的制备通常在对反应呈惰性的溶剂(如苯、二甲苯或甲苯,最好为甲苯)中,于约25~110℃(最好于溶剂的回流温度)下进行。合适的干燥剂/溶剂体系包括四氯化钛/二氯甲烷、异丙氧基钛/二氯甲烷及分子筛/THF。最好的是四氯化钛/二氯甲烷。
使式XI化合物与合适的下式化合物(XII′)反应,也可以将式XI化合物转变成相应的式I化合物,其中L为离去基团(如氯、溴、碘、甲苯磺酸酯或甲磺酸酯)。该反应通常在对反应呈惰性的溶剂(如二氯甲烷或THF,最好为二氯甲烷)中,于约0℃~60℃(最好于约25℃)进行。
使式XI化合物与合适的下式化合物反应,然后还原生成的酰胺,也可以将式XI化合物转变成相应的式I化合物,其中L的定义同上,或者为咪唑。该反应通常在惰性溶剂(如THF或二氯甲烷)中,于约-20℃~60℃进行,最好在二氯甲烷中于约0℃进行。生成的酰胺的还原反应是用还原剂(如甲硼烷甲硫醚配合物、氢化铝锂或氢化二异丁基铝)于惰性溶剂(如乙醚或THF)中完成的。反应温度的范围为约0℃至溶剂的回流温度。还原反应最好是用甲硼烷甲硫醚配合物于THF中在约60℃完成。
当Q为式II基团时,式X起始原料可以按美国专利申请系列号566,338(提交日期1990年7月20日,并转让给Pfizer公司)中所述方法进行制备。将该申请收编在本发明中作为参考。
当Q为式III基团时,式X起始原料可以按美国专利申请系列号532,525(提交日期1990年6月1日)和要求将其作为优先权的PCT专利申请(提交日期为1991年4月25日,其题目为“3-氨基-2-芳基奎宁环类”)中所述方法进行制备。上述两项专利申请均转让给Pfizer公司,并且将其收编在本发明中作为参考。
当Q为式IV、V或VI基团时,式X起始原料可以按美国专利申请系列号557,442(提交日期1990年7月23日)和要求将其作为优先权的PCT专利申请(提交日期为1991年5月15日,其题目为“奎宁环类衍生物”)中所述方法进行制备。上述二项申请均转让给Pfizer公司,并将它们收编在本发明中作为参考。
当Q为式VII基团时,式X起始原料可以按下述专利申请中所述方法进行制备,所有的申请均已转让给Pfizer公司:美国专利申请系列号:619,361(提交日期1990年11月28日)、675,244(提交日期1991年3月26日)、800,667(提交日期1991年11月27日);PCT专利申请系列号PCT/US92/00065(指定美国,提交日期1992年1月4日)。上面四项申请收编在本发明中作为参考。
当Q为式VII基团时,式X起始原料可以按美国专利申请系列号590,423(提交日期1990年9月28日,并转让给Pfizer公司)中所述方法进行制备。将该申请收编在本发明中作为参考。
反应式2列示了制备其中Q为式VII基团的式I化合物的另一方法。
如反应式2所示,用氰基硼氢化钠或三乙酰氧基硼氢化钠和式XIII化合物使式XII化合物进行还原性胺化作用,得到式XIV化合物。该反应通常在极性溶剂(如乙酸或低级链烷醇)中于约0~50℃进行。甲醇是优选的溶剂,并且温度优选为约25℃。反应混合物的pH值为约4~5也是优选的。
还原式XIV化合物得到其中Q为式VII基团,m为零的式I化合物。合适的还原剂包括甲硼烷甲硫醚(于THF中)、氢化铝锂、甲硼烷(于THF中)和硼氢化钠-氯化钛(IV)。应用甲硼烷甲硫醚(于THF中)得到了最好的结果。该反应可以在室温~约150℃进行,最好在溶剂的回流温度下进行。
可以将得到的式I化合物转变为其中Q为式VII基团,m不是零并具有相同立体化学结构的式I化合物,其方法是:使其与合适的式R10-(CH2)m-L′化合物反应,其中L′为卤素、甲磺酸酯或甲苯磺酸酯,并且其中(CH2)m的一个碳-碳单键可任意地由碳-碳双键或碳-碳叁键替代,以及其中该(CH2)m的一个碳可以任意地用R11取代。该反应一般在碱(如三乙胺或叔丁醇钾)存在下,于极性溶剂(如二氯甲烷或二氯乙烷)中,在室温~约150℃进行。最好使反应在二氯甲烷回流温度下,于三乙胺存在下进行。
式XIII起始原料可以按美国专利申请系列号619,361(提交日期1990年11月28日,并转让给Pfizer公司)。将该申请收编在本发明中作为参考。
反应式3列示了制备其中Q为式VIII基团的式I化合物的另一方法。
如反应式3所示,在式XV化合物存在下将式XII化合物进行还原性胺化,得到式XVI化合物。可以应用的还原剂的例子有金属催化剂存在下的氢、硼氢化钠、氰基硼氢化钠和三乙酰氧基硼氢化钠。该反应通常在极性溶剂(如乙酸或低级链烷醇)中,于脱水剂(如分子筛)存在下,在约0℃~50℃进行。甲醇是优选的溶剂,25℃是优选的温度。反应混合物的pH值优选为约4~5。
另外,用下式化合物使式XV化合物酰化,然后还原生成的酰胺,可以制得式XVI化合物,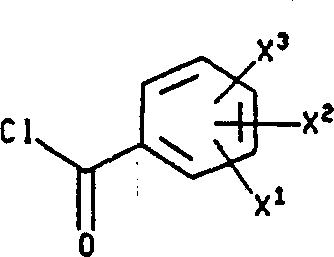 酰化反应通常在极性溶剂(如二氯甲烷、THF或乙醚)中,于约0℃~60℃进行。优选的溶剂是二氯甲烷,优选的温度为约25℃。可以应用的还原该酰胺的还原剂的例子有氢化铝锂和甲硼烷甲硫醚。还原反应一般在极性溶剂(如乙醚、THF或DMF)中,于约0℃~溶剂的回流温度(最好为大约室温)下进行。
酰化反应通常在极性溶剂(如二氯甲烷、THF或乙醚)中,于约0℃~60℃进行。优选的溶剂是二氯甲烷,优选的温度为约25℃。可以应用的还原该酰胺的还原剂的例子有氢化铝锂和甲硼烷甲硫醚。还原反应一般在极性溶剂(如乙醚、THF或DMF)中,于约0℃~溶剂的回流温度(最好为大约室温)下进行。
于钯-炭(如10%钯-炭)存在下,使式XVI化合物与甲酸铵反应,可以将式XVI化合物转变成相应的其中Q为式VIII基团,m为零的式I化合物。通常应用极性溶剂如乙酸乙酯或低级链烷醇,并且该反应于约室温~150℃进行约0.5~24小时。最好使该反应于乙醇中在室温下进行约3~24小时。
应用上述制备其中Q为式VII基团,m不为零的式I化合物的方法,可以将用前述方法制得的式I化合物转变成相同的但m不为零的化合物。
式XV起始原料可以按美国专利申请系列号590,423(提交日期1990年9月28日,并转让给Pfizer公司)中所述方法制备。将该申请收编在本发明中作为参考。
使Q=0(式II的Q)与合适的苄胺缩合,可以制得其中Q为式II基团,并且在Q和相邻氮之间有一双键的式I化合物(如下所示)。缩合反应通常在无羟基的溶剂(如苯、甲苯或THF)中,应用酸(如甲磺酸或对甲苯磺酸),于约20~溶剂的回流温度下进行。该反应优选在甲苯中,应用樟脑磺酸于回流温度下进行。
在前面实验部分未具体叙述的其他式I化合物的制备可以应用对本技术领域专业人员是显而易见的上述各反应的结合来完成。
在以上反应式1~3所示各反应中,压力要求不严格,除非另有说明。约0.5~5个大气压通常是合适的,并且为了方便,通常优选常压(即1个大气压)。
式I的新化合物及其可药用的盐可用作为P物质拮抗剂,即它们具有拮抗哺乳动物受体部位的P物质作用的能力,因此它们能够作为治疗患有上述疾病的哺乳动物的治疗剂。
在性质上为碱性的式I化合物能够与各种无机酸或有机酸生成各种不同的盐。虽然该盐必须是给动物服用的可药用的盐,但是实际上通常最好是首先作为药学上不适用的盐从反应混合物中分离出式I化合物,然后用碱性试剂处理,将该药学上不适用的盐转回成游离碱化合物,接着再将游离碱转变成可药用的酸加成盐。本发明碱基化合物的酸加成盐可以按下法容易地制得:在含水溶剂或合适的有机溶剂(如甲醇或乙醇)中,将碱基化合物用实质上等当量的经选择的无机酸或有机酸处理。小心地蒸去溶剂,容易地得到所需的盐。
在性质上呈酸性的式I化合物(例如其中R1为羧基苯基的式I化合物)可以与各种药理学上可以接受的阳离子形成碱盐。这样的盐的例子包括碱金属或碱土金属盐,尤其是钠盐和钾盐。上述盐均可以用一般的技术制得。用作制备本发明可药用的碱盐的试剂是与酸性式I化合物一起生成无毒碱盐的化学碱。所述无毒的碱盐包括由上述药理学上可以接受的阳离子(如钠、钾、钙、镁等)所得到的碱盐。这些盐可以按下法容易地制备:将相应的酸性化合物用含有所需药理学上可以接受的阳离子的水溶液处理,然后使所得溶液蒸发至干(最好在减压下)。另外,也可以按下法制备:将酸性化合物的低级链烷醇溶液与所需碱金属醇盐混合,然后按上述相同的方法将所得溶液蒸发至干。在任何一种情况下,优选应用化学计算量的试剂,以便确保反应完全和所需最终产物的产率最高。
式I化合物及其可药用的盐具有结合P物质受体的活性,因此在治疗和预防各种临床疾病中是有价值的,它们可以降低P物质介导的神经传递,因此在治疗和预防上是有效的或可以促进痊愈。所述疾病包括炎症(如关节炎、牛皮癣、哮喘和炎症性肠病)、焦虑、抑郁或心境恶劣、结肠炎、精神病、疼痛、胃食管反流疾病、变应反应(如湿疹和鼻炎)、慢性阻塞性气管疾病、高敏性疾病(如毒葛)、血管痉挛性疾病(如绞痛、偏头痛和Reynaud氏疾病)、纤维化和胶原疾病(如硬皮病和嗜酸性片吸虫病)、反射性交感神经营养不良(如肩手综合症)、癖嗜病(如酒精中毒)、与应激反应有关的躯体疾病、外周神经疾病、神经痛、神经病理性疾病(如早老性痴呆、与爱滋病有关的痴呆、糖尿病性神经病和多发性硬化)、与免疫增强或抑制有关的疾病(如全身性红斑狼疮)、以及风湿病(如肌风湿病)。因此,可以采用本发明化合物作为P物质拮抗剂应用进行治疗,以控制和/或治疗哺乳动物(包括人)的任一上述临床疾病。
本发明式I化合物及其可药用的盐可以经口服、非经胃肠道或局部途径给药。一般来讲,虽然根据体重、需治疗疾病的情况和所选择的具体给药途径改变剂量是一定会发生的,但是本发明化合物最好按每天约5.0毫克直到约1500毫克的剂量范围给药。但是应用的剂量水平为每天每千克体重按约0.07毫克~21毫克是最理想的。根据需治疗的动物的种类、对所用药物的个体反应以及所选用的药物剂型和给药间隔的时间,改变剂量仍然是会发生的。在一些情况下,剂量低于上述剂量范围的下限更为合适,而在另一些情况下,可以应用更大的剂量而不会引起任何有害的副作用,条件是在整个一天中这些大剂量首先要分成几个小剂量服用。
本发明化合物可以按以前所述三条途径之一单独地服用,或者与药学上适用的载体或稀释剂一起组成组合物服用,并且可以按单次剂量或按多次剂量给药。尤其是本发明新的治疗剂可以用多种不同的剂型给药,即本发明化合物可以与各种药学上适用的惰性载体组成片剂、胶囊剂、糖锭、锭剂、硬糖果剂、粉剂、喷雾剂、霜剂、油膏剂、栓剂、凝胶剂、明胶剂、糊剂、洗剂、软膏剂、水混悬剂、注射溶液剂、驰剂、糖浆剂等。所述载体包括固体稀释剂或填充剂、无菌含水介质和各种无毒的有机溶剂等。此外,口服药物组合物可以被适当地增甜和/或芳香化。一般来讲,本发明治疗上有效的化合物在上述剂型中的浓度范围约为5.0%~70%(重量)。
对于口服给药,片剂含有各种赋形剂如微晶纤维素、柠檬酸钠、碳酸钙、磷酸二钙和甘氨酸,各种崩解剂如淀粉(最好是玉米淀粉、马铃薯淀粉或木薯淀粉)、藻酸和某些复合的硅酸盐,以及颗粒粘合剂如聚乙烯吡咯烷酮、蔗糖、明胶和阿拉伯树胶。此外,对于压片来讲,润滑剂(如硬脂酸镁、十二烷基硫酸钠和滑石)通常是十分有用的。在明胶胶囊剂中,也可以应用相同类型的固体组合物作为填充剂;在这方面较好的物质还包括乳糖以及高分子量的聚乙二醇。当口服给药要求应用水混悬剂和/或驰剂时,可以将有效成分与各种甜味剂或芳香剂、有色物质或染料并且如需要,还有乳化剂和/或悬浮剂,以及这样的稀释剂如水、乙醇、丙二醇、甘油和其组合物混合。
对于非经胃肠道给药,可以应用具有治疗作用的本发明化合物在芝麻油、花生油或在含水丙二醇中的溶液剂。如果需要,水溶液剂应该用合适的缓冲剂进行缓冲,并且首先使液体稀释剂等渗。所述水溶液剂对于静脉注射是合适的。油溶液剂适合于关节内、肌内和皮下注射。按照本技术领域专业人员熟知的一般制药技术,在无菌条件下可以容易地完成上述所有溶液剂的制备。
此外,当治疗皮肤的炎症疾病时,还可以局部使用本发明化合物,并且最好使用按照一般制药方法制得的霜剂、凝胶剂、明胶剂、糊剂、软膏剂等。
本发明化合物作为P物质拮抗剂的活性可以通过自动射线照相的方法,应用放射性配位体使速激肽受体显影测得的抑制牛尾组织中受体部位P物质的结合能力来确证。这里所述化合物对P物质的拮抗活性可以应用M.A.Cascieri等(Journal of BiologicalChemistry,Vol.258,P.5158(1983))所述的一般试验方法进行测定。该方法主要包括测定在所分离的牛组织的受体部位减少50%放射性同位素标记的P物质配位体所需各个化合物的浓度,因此得到了表明各个受试化合物特征的IC50值。
在所述方法中,将牛尾组织从-70℃冷冻箱中取出,并置于50倍(w/v)冰冷的50mM Tris(即三羟甲氨基甲烷,2-氨基-2-羟甲基-1,3-丙二醇)盐酸盐缓冲液(pH 7.7)中进行匀浆。匀浆物以30,000×G转速离心20分钟。将沉降物重新悬浮在50倍体积的Tris缓冲液中,再次匀浆,然后再以30,000×G转速离心20分钟。然后将沉降物重新悬浮在冰冷却的40倍体积的含有2mM氯化钙、2mM氯化镁、40g/ml杆菌肽、4μg/ml亮肽素、2μg抑糜蛋白酶素和200g/ml小牛血清白蛋白的50MM Tris缓冲液(pH 7.7)中。该步骤完成了组织标本的制备。
然后按下述方法进行放射性配位体结合步骤,即通过加入100μl 1μM浓度的受试化合物激发反应,接着加入100μl最终浓度为0.5mM的放射性配位体,最后再加入800μl上述制备的组织标本。因此,最终体积为1.0ml,下一步将反应混合物旋转,并置于室温(约20℃)温育20分钟。在过滤步骤之前,将滤器预先浸泡2小时,各试管用细胞收集器过滤,玻璃纤维滤器(WhatmanGF/B)用50mM Tris缓冲液(pH 7.7)洗涤4次。然后用β计数器以53%计数效率测定放射性,并应用标准的统计学方法计算IC50值。
通过研究本发明化合物抑制P物质诱导的或抑制P物质激动剂诱导的豚鼠运动过强的能力,可以测定本发明化合物作为精神抑制药控制各种精神病的抗精神病作用。这样一种研究可以按下法进行:首先给豚鼠对照化合物或本发明合适的受试化合物,然后经插管通过大脑内给豚鼠注射P物质或P物质激动剂,其后测量它们对该刺激的各个运动反应。
通过下述实施例说明本发明。但是应该明白,本发明不限于这些实施例的具体细节。
实施例1
2-(二苯基甲基)-N-((2-二氟甲氧基)苯基)甲基-1-氮杂双环〔2.2.2〕辛-3-胺
A.2-(二氟甲氧基)苯甲醛
向装配有冷凝管和气体入口管的500ml三颈圆底烧瓶中加入5.0克(40.98毫摩尔)水杨醛、150ml二噁烷和150ml(164毫摩尔)1.1N氢氧化钠水溶液。加热至60℃,将氯代二氟甲烷气体鼓入反应混合物,并将反应混合物于该温度下搅拌2小时。然后冷却并用乙醚萃取。有机层用硫酸钠干燥,过滤和蒸发。残余物经硅胶柱层析,用己烷/乙酸乙酯作洗脱剂,得到1.63克(23%)淡黄色油状物。
1H NMR(δ,CDCl3):6.64(t,J=72.7(H-F),1H),7.16(d,J=7,1H),7.24(t,J=7,1H),7.53(m,1H),7.81(m,1H),10.29(s,1H)。
13C-NMR(CDCl3):112.2,115.6,115.645,115.7,119.1,119.2,119.5,125.6,125.7,125.8,125.9,127.5,128.8,128.9,135.7,152.71,152.73,188.4。
IR(cm-1,净态):1700(C=O)。
MS(%):172(100,M),171(48),122(45),121(82),120(69),104(37),95(40),92(55),91(49),76(39),65(49),63(76),51(81)。元素分析,C8H6F2O2·I/4 H2O:计算值:C 54.50,H 3.71测定值:C 54.68,H 3.33
B.2-(二苯基甲基)-N-((2-二氟甲氧基)苯基)甲基-1-氮杂双环〔2.2.2〕辛-3-胺
向装配有氮气入口管的25ml圆底烧瓶中加入500毫克(1.71毫摩尔)2-二苯基甲基-1-氮杂双环〔2.2.2〕辛-3-胺(按Warawa等的方法制备,J.Med.Chem.,17,497(1974))、8.5ml甲醇、383毫克(2.23毫摩尔)2-(二氟甲氧基)苯甲醛和216毫克(3.42毫摩尔)氰基硼氢化钠。反应液于室温下搅拌30小时,在乙酸乙酯和水之间进行分配。分出有机层,用盐水洗涤,经硫酸钠干燥并蒸发。为了除去最后未反应的微量胺,在室温下将混合物用三乙酰氧基硼氢化钠的乙酸溶液处理16小时,然后用氢氧化钠水溶液和二氯甲烷处理。残余物用异丙醇结晶,得到206毫克(27%)白色固体,m.p.144~147℃。
1H NMR(δ,CDCl3):1.27(m,1H),1.4-1.8(m,2H),1.90(m,1H),2.05(m,1H),2.63(m,1H),2.78(m,2H),2.88(m,1H),3.19(m,1H),3.45(ABq,JAB=13.5,Δν=105.5,2H),3.72(dd,J=8,12,1H),4.43(d,J=12,1H),6.31(t,J=74(H-F),1H),6.55 and 7.0-7.4(m,14H)。
13C-NMR(CDCl3):20.0,24.9,25.4,42.0,45.8,49.4,49.5,55.0,61.8,116.3,119.0,125.4,126.0,126.5,127.5,127.8,127.9,128.0,128.4,128.5,128.6,129.1,129.2,130.0,131.6,143.2,145.2,149.3。
IR(cm-1,净态):2940(C-H),1599(C=C)。
MS(%):449(<1,M+1),291(51),281(100),84(66),49(69)。元素分析,C28H30F2N2O:计算值:C 74.98,H 6.74,N 6.25测定值:C 74.72,H 6.70,N 6.23
实施例2
(2S,3S)-N-(2-甲氧基-5-三氟甲氧基苯基)甲基-2-二苯基甲基-1-氮杂双环〔2.2.2〕辛-3-胺甲磺酸盐
按类似实施例1所述的方法,用2-甲氧基-5-三氟甲氧基苯甲醛代替步骤B中的2-(三氟甲氧基)苯甲醛制备标题化合物。
M.p.135℃。
1H NMR(CDCl3)δ1.8-2.3(m,2H),2.2-2.8(m,6H),2.66(s,6H),3.56(s,3H),3.3-3.7(m,3H),3.90(m,3H),4.16(m,2H),5.06(m,1H),5.20(br,1H),5.50(m,1H),5.60(br,1H),6.77(d,1H,J=9.2),7.02(m,1H),7.2-7.8(m,11H),8.00(br,1H),10.8(br,1H).
IR(cm-1,KBr):3180,3140,3000,1500,1200,1062,782.
实施例3
(2S,3S)-2-苯基-3-〔2-(2,2,2-三氟乙氧基)苄基〕氨基哌啶盐酸盐
A.2-(2,2,2-三氟乙氧基)苯甲醛
在氮气氛下向装配有回流冷凝管的圆底烧瓶中加入0.2克(1毫摩尔)2-(2,2,2-三氟乙氧基)苄腈(J.Org.Chem.,377(1983))和5ml甲酸。向该溶液中加入约0.2克阮内镍,将混合物加热回流90分钟。混合物通过硅藻土过滤,并用水和氯仿(CHCl3)洗涤滤饼。分出有机层,水相用氯仿萃取3次。合并的有机相用饱和的碳酸氢钠水溶液和水洗涤,经硫酸钠(Na2SO4)干燥并浓缩(旋转蒸发器),得到176毫克标题化合物,为黄色固体,m.p.33~34℃。
B.(2S,3S)-2-苯基-3-〔2-(2,2,2-三氟乙氧基)苄基〕氨基哌啶盐酸盐
在氮气氛下向圆底烧瓶中加入112毫克(0.63毫摩尔)(2S,3S)-3-氨基-2-苯基哌啶、155毫克(0.76毫摩尔)由上面步骤A制得的醛和约2ml乙酸,溶液于室温下搅拌1小时。向该体系中分次加入294毫克(1.39毫摩尔)三乙酰氧基硼氢化钠,混合物于室温下搅拌过夜。用旋转蒸发器浓缩,并在1M氢氧化钠(NaOH)水溶液和二氯甲烷(CH2Cl2)间进行分配。分出有机层,水相用CH2Cl2萃取3次。合并的有机相用2N盐酸水溶液萃取3次,萃取液用2N NaOH水溶液碱化,混合物用CH2Cl2萃取4次。CH2Cl2萃取液经Na2SO4干燥并浓缩。将所得的油状物溶于约2ml乙酸乙酯中,用饱和氯化氢(HCl)乙醚溶液处理。收集产生的白色固体(73毫克,m.p.>275℃)。该物质在1NNaOH水溶液和CH2Cl2间进行分配使其转变成游离碱。游离碱(58毫克)经闪式柱层析纯化,依次用氯仿(CHCl3)和1∶19甲醇/CHCl3溶液洗脱,得到32毫克油状物。按上述方法将游离碱转变成相应的盐酸盐,得到17毫克标题化合物,m.p.>275℃。
1H NMR(游离碱,CDCl3)δ1.44(m,1H),1.63(m,1H),1.88(m,1H),2.1(m,1H),2.80(m,2H),3.26(m,1H),3.38(d,1H,J=15),3.66(d,1H,J=15),3.88(s,1H),4.08(m,2H),6.68(d,1H,J=6),6.90(m,1H),6.98(d,1H,J=6),7.16(m,1H),7.26(m,5H).
HRMS,C20H24F3N2O(M+1):计算值:365.1835。测定值:365.1980。元素分析,C20H23F3N2O·2HCl·1/3H2O:计算值:C 54.19,H 5.84,N 6.32测定值:C 54.22,H 5.57,N 6.28
实施例4
(2S,3S)-3-(2-甲氧基-5-三氟甲氧基苄基)氨基-2-苯基哌啶盐酸盐
A.2-甲氧基-5-三氟甲氧基苯甲醛
在氮气氛下向圆底烧瓶中加入3.63ml(28毫摩尔)4-三氟甲氧基苯酚和25ml丙酮。向该搅拌液中加入7.75克(56毫摩尔)碳酸钾和3.48ml(56毫摩尔)甲基碘,反应混合物于室温下搅拌过夜。抽滤分出固体,滤饼用丙酮洗涤。将滤液浓缩,得到6.5克固体/油混合物。该混合物用CHCl3稀释并过滤,浓缩滤液,得到5.5克1-甲氧基-4-三氟甲氧基苯,为黄色油状物。
1H NMR(CDCl3)δ3.78(s,3H),6.83(d,1H,J=12),7.10(d,1H,J=12).
MS m/z:192(M)
在氮气氛下向圆底烧瓶中加入1-甲氧基-4-三氟甲氧基苯(5.5克,29毫摩尔)和110ml CH2Cl2。在冰/丙酮浴冷却下,于约1分钟内向该体系中加入3.77ml(34毫摩尔)四氯化钛(TiCl4)。将反应混合物搅拌30分钟,向其中加入5.69ml(63毫摩尔)α,α-二氯甲基甲基醚。移去冰浴,混合物于室温下搅拌过夜。小心地将该混合物倒入水中,并用CH2Cl2萃取3次。合并的萃取液用水和盐水洗涤,经Na2SO4干燥并浓缩,得到6.06克油状物。该粗品经闪式柱层析(250克硅胶)纯化,用1∶9乙酸乙酯/己烷作为洗脱剂,得到920毫克带少量杂质的标题化合物,以及3.27克纯的标题化合物。
1H NMR(CDCl3)δ3.94(s,3H),7.00(d,1H,J=9),7.38(dd,1H,J=3,9),7.66(d,1H,J=3),10.4(s,1H)。质谱m/z:220(母峰)。
B.(2S,3S)-3-(2-甲氧基-5-三氟甲氧基苄基)氨基-2-苯基哌啶盐酸盐
在氮气氛下向圆底烧瓶中加入525毫克(2.4毫摩尔)2-甲氧基-5-三氟甲氧基苯甲醛、350毫克(2.0毫摩尔)(2S,3S)-3-氨基-2-苯基哌啶和5ml乙酸。反应混合物于室温下搅拌3天,并用旋转蒸发器浓缩。残余物在1N氢氧化钠水溶液和氯仿(CHCl3)间进行分配,混合物用氯仿萃取3次。合并的氯仿萃取液用1N盐酸水溶液萃取3次。合并的盐酸萃取液用浓氢氧化钠水溶液碱化,并用氯仿萃取4次。氯仿萃取液经Na2SO4干燥,用旋转蒸发器浓缩,得到760毫克油状物。将该油状物溶于乙酸乙酯中,向溶液中加入饱和氯化氢(HCl)乙醚溶液。抽滤收集产生的白色固体,用乙醚洗涤,得到600毫克标题化合物,m.p.>250℃。
1H NMR(游离碱,CDCl3)δ1.36(s,1H),1.54(m,1H),1.86(m,1H),2.06(m,1H),2.76(m,2H),3.22(m,1H),3.32(d,1H,J=15),3.48(s,3H),3.58(d,1H,J=15),3.85(d,1H,J=3),6.57(d,1H,J=9),6.80(d,1H,J=3),6.92(dd,1H,J=3,9),7.22(m,5H)。
HRMS,C20H23F3N2O2:计算值:380.1711,测定值:380.1704。
元素分析,C20H23F3N2O2·2HCl·0.2H2O:
计算值:C 52.57,H 5.60,N 6.13
测定值:C 52.58,H 5.40,N 5.97
实施例5
(2S,3S)-1-(5,6-二甲氧基己基)-3-(2-甲氧基-5-三氟甲氧基苄基)氨基-2-苯基哌啶盐酸盐
在氮气氛下向圆底烧瓶中加入250毫克(0.66毫摩尔)(2S,3S)-3-(2-甲氧基-5-三氟甲氧基苄基)氨基-2-苯基哌啶、2ml四氢呋喃(THF)和0.28ml(2.0毫摩尔)三乙胺。向该体系中加入475毫克(2.0毫摩尔)5,6-二甲氧基-1-甲磺酰氧基己烷(用1,5,6-己三醇制备,相继通过形成丙酮化合物(丙酮,对甲苯磺酸),乙酰化作用(乙酰氯,三乙胺,THF),丙酮化合物裂解(60%乙酸/水),二甲基化作用(氢化钠,甲基碘,THF),脱乙酰基作用(甲醇钠,甲醇)和形成甲磺酸酯(甲磺酰氯,三乙胺,THF)),混合物于50~60℃加热4天。反应混合物在CHCl3和饱和碳酸氢钠水溶液间进行分配,并用CHCl3萃取3次。合并的有机层经Na2SO4干燥、过滤和浓缩,得到853毫克橙色油状物。粗品经闪式柱层析(35克硅胶)纯化,用1∶19甲醇/氯仿作为洗脱剂,得到185毫克黄色油状物。将该油状物溶于乙酸乙酯中,向溶液中加入饱和氯化氢乙醚溶液。浓缩混合物,残余物和乙醚一起研磨,得到190毫克标题化合物。
1H NMR(游离碱,CDCl3)δ1.15(m,2H),1.38(m,6H),1.76(m,2H),1.96(m,3H),2.50(m,2H),3.16(m,2H),3.26(m,9H),3.46(s,3H),3.58(d,1H,J=15),6.52(d,1H,J=9),6.69(m,1H),6.86(m,1H),7.22(m,5H)。
HRMS,C28H39F3N2O4:计算值:524.28616,测定值:524.28634。元素分析,C28H39F3N2O4·2HCl·0.75H2O:计算值:C 55.03,H 7.00,N 4.58测定值:C 55.04,H 7.12,N 4.51
实施例6
(2S,3S)-2-苯基-3-(2-三氟甲氧基苄基)氨基哌啶盐酸盐
在氮气氛下向圆底烧瓶中加入3.0ml(23毫摩尔)三氟甲氧基苯和25ml苯。将其置于冰/丙酮浴中冷却,向搅拌溶液中加入4.1ml(45毫摩尔)α,α-二氯甲基甲基醚。向该体系中分次加入6.13克(46毫摩尔)三氯化铝(AlCl3)。加完后,将反应混合物逐渐温热至室温,并于室温下搅拌过夜。反应混合物缓慢地倒入水中,并用二氯甲烷萃取3次。合并的有机层用水洗涤,经Na2SO4干燥并用旋转蒸发器浓缩,得到3.7克油状物。该物质含有4-和2-三氟甲氧基苯甲醛的混合物,经闪式柱层析(160克硅胶)纯化,用1∶49乙酸乙酯/己烷作为洗脱剂,得到500毫克富集2-三氟甲氧基-苯甲醛的产物。
在氮气氛下向圆底烧瓶中加入155毫克(0.88毫摩尔)(2S,3S)-3-氨基-2-苯基哌啶,上面制得的醛和2ml乙酸。向其中加入370毫克(1.8毫摩尔)三乙酰氧基硼氢化钠,混合物于室温搅拌过夜。将混合物浓缩,残余物在1N氢氧化钠水溶液和二氯甲烷间进行分配,并用二氯甲烷萃取3次。合并的有机层用1N HCl萃取3次。酸萃取液用1N NaOH水溶液碱化,并用二氯甲烷萃取3次。二氯甲烷萃取液经干燥和浓缩,得到190毫克油状物,该油状物经闪式柱层析(5克硅胶)纯化,用1∶9甲醇/氯仿作为洗脱剂,得到95毫克标题化合物的游离碱。将游离碱溶于乙酸乙酯中,向溶液中加入饱和氯化氢乙醚溶液。抽滤收集生成的白色固体,用乙醚洗涤,得到72毫克标题化合物,m.p.231~233℃。
1H NMR(游离碱,CDCl3)δ1.40(m,1H),1.60(m,1H),1.84(m,1H),2.05(m,1H),2.78(m,2H),3.22(m,1H),3.42(d,1H,J=15),3.56(d,1H,J=15),3.86(d,1H,J=3),7.08(m,4H),7.24(m,5H)。 质谱:m/z 350(母峰).元素分析,C19H21F3N2O·2HCl·0.25 H2O:计算值:C 53.34,H 5.54,N 6.54测定值:C 53.19,H 5.40,N 6.54
实施例7
(2S,3S)-3-(2-羟基-5-三氟甲氧基苄基)氨基-2-苯基哌啶盐酸盐
A.2-羟基-5-三氟甲氧基苯甲醛
在氮气氛下向圆底烧瓶中加入300毫克(1.4毫摩尔)2-甲氧基-5-三氟甲氧基苯甲醛和30ml二氯甲烷。置于干冰丙酮浴中冷却,在约1分钟内向其中加入0.26ml(2.7毫摩尔)三溴化硼(BBr3)。将反应混合物搅拌1小时,用冰浴代替干冰/丙酮浴,混合物再搅拌1小时。向其中缓慢地加入10ml饱和碳酸氢钠水溶液,然后加入10ml水,将混合物温热至室温。混合物用二氯甲烷萃取2次,萃取液经Na2SO4干燥并浓缩。生成的油状物(280毫克)溶于CH2Cl2中,该溶液用1M NaOH水溶液萃取2次。合并的水萃取液用2M HCl水溶液酸化,并用二氯甲烷萃取3次。二氯甲烷萃取液经Na2SO4干燥并浓缩,得到200毫克标题化合物。
1H NMR(CDCl3)δ6.96(d,1H,J=9),7.36(m,2H),9.84(s,1H),10.9(s,1H)。
B.(2S,3S)-3-(2-羟基-5-三氟甲氧基苄基)氨基-2-苯基哌啶盐酸盐
按类似实施例4化合物的制备方法,用2-羟基-5-三氟甲氧基苯甲醛代替2-甲氧基-5-三氟甲氧基苯甲醛制备标题化合物。
1H NMR(游离碱,CDCl3)δ1.60(m,3H),2.04(m,1H),2.76(m,1H),2.88(m,1H),3.18(m,1H),3.42(s,2H),3.90(m,1H),6.52(m,1H),6.64(d,1H,J=9),6.89(m,1H),7.30(m,5H)。
HRMS,C19H21F3N2O2:计算值:366.1545,测定值:366.1562。元素分析,C19H21F3N2O2·2HCl·1/3 H2O:计算值:C 51.25,H 4.90,N 6.29测定值:C 51.30,H 4.75,N 6.22
实施例8
(2S,3S)-3-(5-氯-2-〔2,2,2-三氟乙氧基〕苄基)-氨基-2-苯基哌啶盐酸盐
A.5-氯-2-(2,2,2-三氟乙氧基)苯甲醛
在氮气氛下向圆底烧瓶中加入880毫克(22毫摩尔)60%氢化钠(NaH)和12ml N,N-二甲基甲酰胺。在15分钟内通过注射管向其中加入2.9ml(4克,40毫摩尔)2,2,2-三氟乙醇,混合物于室温搅拌20分钟。向该体系中加入1.72克(10毫摩尔)2,5-二氯苄腈,混合物于90℃加热3天。将混合物冷却至室温,并倒入50ml 2M HCl水溶液中,用乙醚萃取3次。合并的有机层经Na2SO4干燥并浓缩,得到2.5克固体。粗品经闪式柱层析纯化,用1∶49乙酸乙酯/己烷作为洗脱剂,得到1.4克5-氯-2-(2,2,2-三氟乙氧基)苄腈,为白色固体。m.p.61~62℃。
在氮气氛下向装配有回流冷凝管的圆底烧瓶中加入400毫克(1.7毫摩尔)上面制得的腈和10ml甲酸。向其中加入约500毫克阮内镍,将混合物加热回流6小时,并于室温下搅拌过夜。混合物通过硅藻土垫过滤,垫用水和CHCl3洗涤。分出有机相,水相用CHCl3萃取3次。合并的有机相经干燥和浓缩,得到270毫克标题化合物。
1H NMR(CDCl3)δ4.42(m,2H),6.86(d,1H,J=10),7.46(m,1H),7.80(d,1H,J=3),10.3(s,1H)。
质谱:m/z 238(M)。
B.(2S,3S)-3-(5-氯-2-〔2,2,2-三氟乙氧基〕苄基)氨基-2-苯基哌啶盐酸盐
按类似实施例4所述的方法,用5-氯-2-(2,2,2-三氟乙氧基)苯甲醛代替2-甲氧基-5-三氟甲氧基苯甲醛制备标题化合物。
M.p.267-269℃。
1H NMR(游离碱,CDCl3)δ1.4(m,1H),1.6(m,1H),1.82(m,1H),2.02(m,1H),2.78(m,2H),3.2(m,1H),3.3(d,1H,J=15),3.54(d,1H,J=15),3.84(d,1H,J=3),4.0(m,2H),6.54(d,1H,J=10),6.92(d,1H,J=3),7.04(m,1H),7.24(m,5H)。
元素分析,C20H22ClF3N2O·2HCl:
计算值:C 50.91,H 5.13,N 5.94
测定值:C 50.89,H 4.84,N 5.93
实施例9
(2S,3S)-2-苯基-3-(3-三氟甲氧基苄基)氨基哌啶盐酸盐
按类似实施例4所述的方法,用3-三氟甲氧基苯甲醛代替2-甲氧基-5-三氟甲氧基苯甲醛制备标题化合物。
M.p.>275℃。
1H NMR(游离碱,CDCl3)δ1.4(m,1H),1.56(m,1H),1.78(m,1H),1.96(m,1H),2.76(m,2H),3.18(m,1H),3.30(d,1H,J=15),3.46(d,1H,J=15),3.84(d,1H,J=3),6.79(s,1H),6.85(d,1H,J=6),6.94(m,1H),7.12(m,1H),7.24(m,5H).元素分析,C19H21F3N2O·2HCl:计算值:C 53.91,H 5.48,N 6.62测定值:C 53.84,H 5.07,N 6.59
按类似实施例4所述的方法,用合适的醛代替2-甲氧基-5-三氟甲氧基苯甲醛制备实施例10~23和26的标题化合物。
制备所需醛的反应结果列于下表1中。
| 表 1 | ||
| 制备式XII化合物 | ||
| -C6H2X1X2X3 | 起始原料 | 反应所需试剂* |
| 2-(2,2,2-三氟乙氧基)苯基 | 2-氯苄腈 | d,e |
| 2-羟基-5-三氟甲氧基苯基 | 2-甲氧基-5-三氟甲氧基苯甲醛 | f |
| 3-三氟甲氧基苯基 | - | 商品化的 |
| 5-氯-2-(2,2,2-三氟乙氧基)苯基 | 2,5-二氯苄腈 | d,e |
| 5-叔丁基-2-三氟甲氧基苯基 | 三氟甲氧基苯 | g,h |
| 2-乙氧基-5-三氟甲氧基苯基 | 4-三氟甲氧基苯酚 | i,a |
| 2-二氟甲氧基-5-三氟甲氧基苯基 | 2-羟基-5-三氟甲氧基苯甲醛 | j |
| 5-异丙基-2-(2,2,2-三氟乙氧基)苯基 | 4-异丙基-碘代苯 | d,a |
| 2-异丙氧基-5-三氟甲氧基苯基 | 4-三氟甲氧基苯酚 | k,a |
| 表 1 | ||
| 制备式XII化合物 | ||
| -C6H2X1X2X3 | 起始原料 | 反应所需试剂* |
| 5-叔丁基-2-二氟甲氧基苯基 | 4-叔丁基苯酚 | a,j |
| 2,5-双(二氟甲氧基)苯基 | 2,5-二羟基苯甲醛 | j |
| 2-二氟甲氧基-5-二甲氨基苯基 | 5-氨基-2-羟基苯甲醛 | l,j |
| 2-二氟甲氧基-5-异丙基苯基 | 4-异丙基苯酚 | a,j |
| 2-二氟甲氧基-5-硝基苯基 | 2-羟基-5-硝基苯甲醛 | j |
| 5-二甲氨基-2-(2,2,2-三氟乙氧基)苯基 | 2-氯-5-硝基苄腈 | d,l,e |
| 5-乙酰氨基-2-(2,2,2-三氟乙氧基)苯基 | 5-硝基-2-(2,2,2-三氟乙氧基)苄腈 | m,c,e |
| 2-二氟甲氧基-5-乙基苯基 | 4-乙基-甲氧基苯 | a,f,j |
| 表 1 | ||
| 制备式XII化合物 | ||
| -C6H2X1X2X3 | 起始原料 | 反应所需试剂* |
| 5-氯-2-二氟甲氧基苯基 | 5-氯-2-羟基苯甲醛 | j |
| 2-三氟甲氧基苯基 | - | 商品化的 |
| 2-甲氧基-5-三氟甲氧基苯基 | 4-三氟甲氧基苯酚 | b,a |
| 2-二氟甲氧基-5-甲基苯基 | 5-甲基-2-甲氧基苯甲醛 | f,j |
*用标准方法制备式XIV化合物所需的试剂。
a) Cl2CHOCH3,TiCl4
b) 甲基碘
c) 乙酰氯
d) NaOCH2CF3
e) 阮内镍 ,HCO2H
f) BBr3
g) 叔丁基氯/AlCl3
h) Cl2CHOCH3/AlC13
i) 乙基碘
j) ClF2CH
k) 异丙基溴
l) H2,Pd/C,HCHO
m) H2-Pd/BaSO4
实施例10
(2S,3S)-3-〔5-氯-2-(2,2,2-三氟乙氧基)苄基〕氨基-2-苯基哌啶盐酸盐
M.P.267-269℃。
1H NMR(游离碱;CDCl3)δ1.40(m,1H),1.60(m,1H),1.82(m,1H),2.02(m,1H),2.76(m,2H),3.20(m,1H),3.28(d,1H,J=15),3.52(d,1H,J=15),3.84(d,1H,J=3),4.00(m,2H),6.54(d,1H,J=10),6.92(d,1H,J=3),7.04.(m,1H),7.24(m,5H).
HRMS,C20H22ClF3N2O:计算值:398.1368,测定值:398.1352。元素分析,C20H22ClF3N2O·2HCl:计算值:C 50.91,H 5.13,N 5.94测定值:C 50.89,H 4.84,N 5.93
实施例11
(2S,3S)-3-(5-叔丁基-2-三氟甲氧基苄基)氨基-2-苯基哌啶盐酸盐
M.P.262-264℃。
1H NMR(游离碱;CDCl3)δ1.20(s,9H),1.40(m,1H),1.52(m,1H),1.84(m,1H),2.06(m,1H),2.80(m,2H),3.22(m,1H),3.38(d,1H,J=15),3.58(d,1H,J=15),3.86(d,1H,J=3),6.98(m,1H),7.12(m,2H),7.26(m,5H).
HRMS,C23H29F3N2O:计算值:406.2225,测定值:406.2271。元素分析,C23H29F3N2O·2HCl·1/3 H2O:计算值:C 56.92,H 6.56,N 5.77测定值:C 56.99,H 6.41,N 6.03
实施例12
(2S,3S)-3-〔5-异丙基-2-(2,2,2-三氟乙氧基)苄基〕氨基-2-苯基哌啶盐酸盐
M.P.>280℃。
1H NMR(游离碱;CDCl3)δ1.12(m,6H),1.4(m,1H),1.62(m,1H),1.82(m,1H),2.08(m,1H),2.76(m,3H),3.22(m,1H),3.30(d,1H,J=15),3.38(d,1H,J=15),3.82(d,1H,J=3),4.02(m,2H),6.56(d,1H,J=10),6.78(d,1H,J=3),6.94(m,1H),7.24(m,5H).
HRMS,C23H30F3N2O(M+1):计算值:407.2303,测定值:407.2287。元素分析,C22H29F3N2O·2HCl·1/2 H2O:计算值:C 56.55,H 6.60,N 5.70测定值:C 56.17,H 6.39,N 5.77
实施例13
(2S,3S)-3-〔5-二甲氨基-2-(2,2,2-三氟乙氧基)苄基〕氨基-2-苯基哌啶盐酸盐
M.P.250-252℃。
1H NMR(游离碱;CDCl3)δ1.40(m,1H),1.60(m,1H),1.86(m,1H),2.10(m,1H),2.82(m,8H),3.22(m,1H),3.34(d,1H,J=15),3.58(d,1H,J=15),3.88(d,1H,J=3),4.00(m,2H),6.42(d,1H,J=3),6.50(m,1H),6.64(d,1H,J=10),7.30(m,5H).
HRMS,C22H28F3N3O:计算值:407.2178,测定值:407.2179。
实施例14
(2S,3S)-3-(2-二氟甲氧基-5-N,N-二甲氨基苄基)氨基-2-苯基哌啶盐酸盐
M.P.243-245℃(分解)
1H NMR(游离碱:CDCl3)δ1.44(m,1H),1.72(m,2H),2.10(m,1H),2.84(m,8H),3.21(m,1H),3.28(d,1H,J=15),3.55(d,1H,J=15),3.88(d,1H,J=3),6.08(t,1H,J=72),6.36(d,1H,J=3),6.46(dd,1H,J=3,9),6.86(d,1H,J=9),7.28(m,5H).
HRMS,C21H27F2N3O:计算值:375.2122,测定值:375.2138。元素分析,C21H27F2N3O·3HCl·1/2H2O:计算值:C 51.07,H 6.44,N 8.51测定值:C 50.17,H 6.08,N 8.28
实施例15
(2S,3S)-3-〔2,5-双(二氟甲氧基)苄基〕氨基-2-苯基哌啶盐酸盐
M.P.238-239℃。
1H NMR(游离碱;CDCl3)δ1.64(m,3H),2.04(m,1H),2.76(m,2H),3.18(m,1H),3.28(d,1H,J=12),3.52(d,1H,J=12),3.84(d,1H,J=3),6.12(t,1H,J=75),6.40(t,1H,J=75),6.75(m,2H),6.94(d,1H,J=9),7.24(m,5H).
HRMS,C20H22F4N2O2:计算值:398.1612,测定值:
398.1591。
实施例16
(2S,3S)-3-(5-叔丁基-2-二氟甲氧基苄基)氨基-2-苯基哌啶盐酸盐
M.P.263-264℃(分解)。
1H NMR(游离碱;CDCl3)δ1.24(s,9H),1.42(m,1H),1.62(m,1H),1.80(m,1H),2.10(m,1H),2.80(m,2H),3.24(m,2H),3.58(d,1H,J=12),3.87(brs,1H),6.18(t,1H,J=72),6.86(d,1H,J=6),7.00(brs,1H),7.12(m,1H),7.24(m,5H).
HRMS,C23H30F2N2O:计算值:388.2321,测定值:
388.2336。
实施例17
(2S,3S)-3-(2-异丙氧基-5-三氟甲氧基苄基)氨基-2-苯基哌啶盐酸盐
M.P.245-246℃(分解)。
1H NMR(游离碱:CDCl3)δ1.08(d,3H,J=6),1.12(d,3H,J=6),1.40(m,1H),1.64(m,1H),1.87(m,1H),2.08(m,1H),2.78(m,2H),3.02(m,1H),3.34(d,1H,J=15),3.51(d,1H,J=15),3.85(d,1H,J=2),4.28(m,1H),6.01(d,1H,J=9),6.82(m,1H),6.91(m,1H),7.24(m,5H).
HRMS,C22H27F3N2O2:计算值:408.2024,测定值:408.2019。元素分析,C22H27F3N2O2·2HCl:计算值:C 54.89,H 6.07,N 5.82测定值:C 54.50,H 6.24,N 5.78
实施例18
(2S,3S)-3-(2-二氟甲氧基-5-三氟甲氧基苄基)氨基-2-苯基哌啶盐酸盐
M.P.257-259℃(分解)。
1H NMR(游离碱;CDCl3)δ1.44(m,1H),1.58(m,1H),1.78(m,1H),2.03(m,1H),2.78(m,2H),3.20(m,1H),3.32(d,1H,J=15),3.54(d,1H,J=15),3.87(d,1H,J=2),6.15(t,1H,J=72),6.94(m,3H),7.26(m,5H).
HRMS,C20H21F5N2O2:计算值:416.1523,测定值:416.1501。元素分析,C20H21F5N2O2·2HCl·1/3H2O:计算值:C 48.50,H 4.81,N 5.65测定值:C 48.45,H 4.57,N 5.66
实施例19
(2S,3S)-3-(2-乙氧基-5-三氟甲氧基苄基)氨基-2-苯基哌啶盐酸盐
M.P.>275℃(分解)。
1H NMR(游离碱;CDCl3)δ1.13(t,3H,J=6),1.38(m,1H),1.70(m,2H),2.06(m,1H),2.74(m,2H),3.22(m,1H),3.30(d,1H,J=15),3.68(m,3H),3.84(br s,1H),6.55(d,1H,J=9),6.79(br s,1H),6.90(m,1H),7.2(m,5H).
HRMS,C21H25F3N2O2:计算值:394.1868,测定值:394.1875。元素分析,C21H25F3N2O2·2HCl:计算值:C 53.97,H 5.82,N 6.00测定值:C 53.85,H 5.79,N 5.95
实施例20
(2S,3S)-3-(2-二氟甲氧基-5-硝基苄基)氨基-2-苯基哌啶盐酸盐
1H NMR(游离碱;CDCl3)δ1.50(m,1H),1.66(m,1H),1.98(m,2H),2.82(m,2H),3.28(m,1H),3.42(d,1H,J=15),3.64(d,1H,J=15),3.95(d,1H,J=2),6.30(t,1H,J=72),7.08(d,1H,J=8),7.30(m,5H),8.04(m,2H).FAB HRMS,C19H21F2N3O3(M+1):计算值:378.1629,测定值:378.1597。
实施例21
(2S,3S)-3-(2-二氟甲氧基-5-异丙基苄基)氨基-2-苯基哌啶盐酸盐
M.P.245-247℃(分解)。
1H NMR(游离碱;CDCl3)δ1.19(2d,6H,J=7),1.50(m,1H),1.75(m,2H),2.12(m,1H),2.83(m,3H),3.25(m,1H),3.35(d,1H,J=14),3.60(d,1H,J=14),3.90(d,1H,J=3),6.20(t,1H,J=75),6.90(m,2H),7.00(m,1H),7.30(m,5H).
HRMS,C22H28F2N2O:计算值:374.2170,测定值:374.2207。元素分析,C22H28F2N2O·2HCl·1/3H2O:计算值:C 58.28,H 6.67,N 6.18测定值:C 58.17,H 6.52,N 6.17
实施例22
(2S,3S)-3-〔5-乙酰氨基-2-(2,2,2-三氟乙氧基)-苄基〕氨基-2-苯基哌啶盐酸盐
M.P.>270℃。
1H NMR(游离碱;CDCl3)δ1.46(m,1H),1.82(m,1H),2.08(m,1H),2.12(s,3H),2.76(m,2H),3.20(m,1H),3.48(d,1H,J=15),3.58(d,1H,J=15),3.82(m,1H),4.08(m,2H),6.44(m,1H),6.58(d,1H,J=10),6.78(m,1H),7.26(m,5H),7.58(m,1H).
实施例23
(2S,3S)-3-(2-二氟甲氧基-5-乙基苄基)氨基-2-苯基哌啶盐酸盐
M.P.254-255℃。
1H NMR(游离碱;CDCl3)δ1.12(t,3H,J=10),1.36(m,1H),1.44(m,1H),1.82(m,1H),2.10(m,1H),2.48(q,2H,J=10),2.8(m,1H),3.10(m,1H),3.34(d,1H,J=15),3.58(d,1H,J=15),3.9(d,1H,J=3),6.12(t,1H,J=85),6.78(s,1H),6.90(m,2H),7.28(m,5H).元素分析,C21H26F2N2O·2HCl:计算值:C 58.19,H 6.51,N 6.47测定值:C 57.90,H 6.52,N 6.64
实施例24
顺式-3-(5-叔丁基-2-甲氧基苄基)氨基-2-(3-三氟甲氧基苯基)哌啶盐酸盐
A.顺式-5-硝基-6-(三氟甲氧基苯基)哌啶-2-酮
在氮气氛下向圆底烧瓶中加入15克(79毫摩尔)3-三氟甲氧基苯甲醛、80ml乙醇、11克(0.26摩尔)乙酸铵和12.6ml(79毫摩尔)4-硝基丁酸甲酯,将混合物加热回流6小时。冷却至室温后,浓缩混合物。残余物与约200ml CHCl3一起搅拌30分钟,过滤并浓缩。残余物经闪式柱层析纯化,用1∶49甲醇/氯仿洗脱,再用1∶19甲醇/氯仿洗脱,得到24克5-硝基-6-(3-三氟甲氧基苯基)哌啶-2-酮。
向圆底烧瓶中加入20克(66毫摩尔)上面制得的产物、13克KOH和100ml乙醇,混合物于室温搅拌90分钟。向其中加入约35ml 33%硫酸/乙醇溶液。将混合物倒入150ml水中,用100ml CHCl3萃取3次。合并的萃取液用水洗涤,经Na2SO4干燥并浓缩。粗品经柱层析(300克硅胶)纯化,依次用乙酸乙酯和1∶99甲醇/乙酸乙酯作为洗脱剂,得到5.8克顺式-5-硝基-6-(3-三氟甲氧基苯基)哌啶-2-酮,含约12%相应的反式异构体。该物经第2次层析纯化。得到4.6克顺式产物。
B.顺式-5-氨基-6-(3-三氟甲氧基苯基)哌啶-2-酮
在氮气氛下向装配有温度计和机械搅拌器的三颈圆底烧瓶中加入上述顺式物质以及THF(200ml)、甲醇(50ml)和水(5ml)的混合液。向该搅拌溶液中加入铝汞齐(通过用乙醚洗涤4.1克铝箔条,并浸渍在2%HgCl2水溶液中30~45秒,再用乙醚洗涤而制得),混合物于室温下搅拌过夜。混合物通过硅藻土垫过滤,垫用THF洗涤。浓缩滤液,溶于乙酸乙酯中,并用30ml饱和氯化氢乙醚溶液处理。经浓缩得到3.7克顺式-5-氨基-6-(3-三氟甲氧基苯基)哌啶-2-酮粗品,为蜡状固体,m、p.126~130℃。
C.顺式-3-(5-叔丁基-2-甲氧基苄基)氨基-2-(3-三氟甲氧基苯基)哌啶盐酸盐
在氮气氛下向圆底烧瓶中加入0.38克(1.4毫摩尔)上面制得的胺、6ml乙酸和0.32克(1.66毫摩尔)5-叔丁基-2-甲氧基苯甲醛。混合物搅拌45分钟。向其中分次加入0.65克(3.0毫摩尔)三乙酰氧基氢化钠,混合物于室温搅拌过夜。浓缩混合物,并在氯仿和水之间进行分配,用1N NaOH水溶液碱化。分出有机相,水相用CHCl3萃取2次。合并的有机相用水洗涤,干燥和浓缩。粗产物经闪式柱层析纯化,得到0.4克顺式-5-(5-叔丁基-2-甲氧基苄基)氨基-6-(3-三氟甲氧基苯基)哌啶-2-酮。
在氮气氛下向圆底烧瓶中加入0.4克(0.9毫摩尔)上面制得的产物和10ml THF。向其中加入2.2ml(4.4毫摩尔)2M甲硼烷甲硫醚配合物的THF溶液,将混合物逐步加热并使回流4小时。混合物冷却至室温,向其中加入2ml甲醇,浓缩混合物。向该体系中加入5ml乙醇和2.45 K2CO3,将混合物加热回流8小时,并于室温下搅拌过夜。浓缩混合物并在水和CH2Cl2间进行分配。分离有机相,水相用CHCl3萃取3次。合并的有机相经干燥并浓缩,得到油状物。将该油状物溶于乙酸乙酯中,溶液用饱和氯化氢乙醚溶液处理。浓缩后得到70毫克标题化合物,为蜡状固体。
M.P.247℃-249℃。
1H NMR(游离碱,CDCl3)δ1.26(s,9H),1.6(m,1H),1.90(m,2H),2.12(m,1H),2.80(m,2H),3.24(m,1H),3.35(d,1H,J=15),3.48(s,3H),3.64(d,1H,J=15),3.86(m,1H),6.60(d,1H,J=10),7.18(m,6H).
HRMS,C24H31N2O2F3:计算值:436.2330,测定值:436.2326。
实施例25
顺式-2-(3,5-二溴苯基)-3-(2-甲氧基-5-三氟甲氧基苄基)氨基哌啶
按类似实施例24所述的方法制备标题化合物,但是起始反应产物〔6-(3,5-二溴苯基)-5-硝基哌啶-2-酮〕的硝基取代基通过连续的氧化裂解(O3,KO+Bu)、形成肟(H2NOH)并用阮内镍进行催化还原转变成氨基。最终产物可通过在异丙醇中用(R)-(-)-扁桃酸处理进行拆分。从该方法分离出的固体重结晶2次(异丙醇),随后用饱和碳酸氢钠水溶液处理,得到(2S,3S)-对映体;〔α〕D(扁桃酸盐):+4.11°(MeOH,c=0.51)。
1H NMR(CDCl3)δ1.36(m,1H),1.50(m,1H),1.80(m,1H),2.04(m,1H),2.70(m,2H),3.18(m,1H),3.30(d,1H,J=18),3.57(s,3H),3.66(d,1H,J=18),3.75(m,1H),6.63(d,1H,J=9),6.86(d,1H,J=3),6.97(dd,1H,J=6,9),7.32(m,2H),7.48(s,1H).
实施例26
(2S,3S)-3-(2-二氟甲氧基-5-甲基苄基)氨基-2-苯基哌啶盐酸盐
按类似实施例4所述的方法制备标题化合物。
M.P.>275℃。
1H NMR(游离碱,CDCl3)δ1.44(m,1H),1.6(m,1H),1.84(m,1H),2.10(m,1H),2.20(s,3H),2.80(m,2H),3.22(m,1H),3.34(d,1H,J=15),3.58(d,1H,J=15),3.90(d,1H,J=3),6.10(t,1H,J=72),6.84(m,2H),7.26(m,5H).
HRMS,C20H24F2N2O:计算值:347.1929(M+1),测定值:347.1911。元素分析,C20H24F2N2O·2HCl·0.25H2O:计算值:C 56.67,H 6.30,N 6.61测定值:C 56.81,H 6.16,N 6.50
Claims (7)
1.制备式I化合物或其可药用的盐的方法, 式中:
式中:
各个
键代表任意选择的双键或单键,X1是氢,由1至3个氟原子任意选择地取代的C1-C10烷氧基或由1至3个氟原子任意选择地取代的C1-C10烷基;
X2和X3各自独立地选自氢,卤素,硝基,由1至3个氟原子任意选择地取代的C1-C10烷基,由1至3个氟原子任意选择地取代的C1-C10烷氧基,羟基,二-(C1-C6)烷基氨基,
O-NH-C-(C1-C6)烷基;以及
Q是下式基团:或
式中:键的定义如上所述;
R1是苯基;
R13是苯基;
o是2;
X是(CH2)q,其中q是3;
m是0至8的整数,而上式中的(CH2)m中的任何一个碳原子可任意选择地由R11取代;
R6可任意选择地被一个或多个取代基取代的C3-C7环烷基或苯基,该取代基独立地选自卤素和任意选择地被1至3个氟原子取代的C1-C10烷氧基;
R7是氢;
R8和R9各自独立地是氢;
R10是氢或C1-C6烷氧基;
R11是氢或C1-C6烷氧基;
上述定义的前提是:(a)当m为0时,R10和R11之一不存在而另一个是氢;(b)当X1、X2、X3没有一个是氟化的烷氧基时,R6是被氟化的烷氧基取代的苯基;以及(c)当Q是式II基团时,至少一个X1、X2或X3是氟化的烷氧基,该方法包括在还原剂存在下使式中Q的定义如上所述的式Q-NH2化合物与式XII化合物反应, 式中X1、X2和X3的定义如上所述。
式中X1、X2和X3的定义如上所述。
2.根据权利要求1所述的方法,其中所述式Q-NH2化合物通过下式化合物水解除去甲氧基苄基而得到,式中:各
 键的定义和Q的定义如权利要求1中所述。
键的定义和Q的定义如权利要求1中所述。
3.制备下式化合物的方法,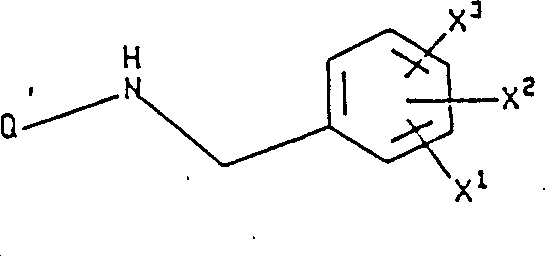
式中Q为在权利要求1中所定义的m为零的式VII基团,而X1、X2和X3的定义如权利要求1中所述,
该方法包括在钯-炭存在下使下式化合物与甲酸铵反应, 式中X1、X2、X3和R6的定义如权利要求1中所述,CBz是氮原子保护基团苄氧羰基。
式中X1、X2、X3和R6的定义如权利要求1中所述,CBz是氮原子保护基团苄氧羰基。
4.制备下式化合物的方法,
式中:Q为在权利要求1中所定义的式VII基团,而在式VII基团中m是权利要求1所述的整数但零除外,而X1、X2和X3的定义如权利要求1中所述,该方法包括使其中m为零的相应化合物与式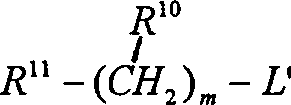 化合物反应,式中L′为卤素、甲磺酸酯或甲苯磺酸酯,R10、R11和m的定义如权利要求1中所述,上述(CH2)m的一个碳-碳单键可以任意选择地由碳-碳双键替代,并且该(CH2)m的一个碳原子可以任意选择地由R11取代。
化合物反应,式中L′为卤素、甲磺酸酯或甲苯磺酸酯,R10、R11和m的定义如权利要求1中所述,上述(CH2)m的一个碳-碳单键可以任意选择地由碳-碳双键替代,并且该(CH2)m的一个碳原子可以任意选择地由R11取代。
5.制备下式化合物的方法,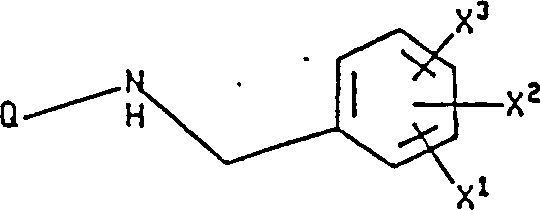
式中Q为在权利要求1中所定义的式VII基团,并且其中X1、X2和X3的定义如权利要求1中所述。该方法包括使下式化合物与还原剂反应,
式中X1、X2、X3、R6、R7和q的定义如权利要求1中所述。
6.制备下式化合物的方法,
式中:
键、Q、X1、X2和X3的定义如在权利要求1中所述,该方法包括使式中Q的定义如权利要求1所述的式Q-NH2化合物与下式化合物反应,式中L为离去基团,X1、X2和X3的定义如在权利要求1中所述。
7.制备权利要求1所定义的式I化合物的方法,其中Q为式II基团,并且在Q和相邻的氮之间有一双键,该方法包括使式中Q的定义如上所述的式Q=O化合物与下式合适的苄胺反应,
式中X1、X2和X3的定义如在权利要求1中所述。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US71794391A | 1991-06-20 | 1991-06-20 | |
| US717,943 | 1991-06-20 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1067655A CN1067655A (zh) | 1993-01-06 |
| CN1056373C true CN1056373C (zh) | 2000-09-13 |
Family
ID=24884155
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN92104778A Expired - Fee Related CN1056373C (zh) | 1991-06-20 | 1992-06-19 | 含氮杂环类化合物的氟代烷氧基苄氨基衍生物的制备方法 |
Country Status (31)
| Country | Link |
|---|---|
| US (3) | US5773450A (zh) |
| EP (1) | EP0589924B1 (zh) |
| JP (1) | JPH07110850B2 (zh) |
| KR (1) | KR0154882B1 (zh) |
| CN (1) | CN1056373C (zh) |
| AT (1) | ATE142199T1 (zh) |
| AU (1) | AU657967B2 (zh) |
| BR (2) | BR9206161A (zh) |
| CA (1) | CA2109613C (zh) |
| CZ (1) | CZ290475B6 (zh) |
| DE (2) | DE9290083U1 (zh) |
| DK (1) | DK0589924T3 (zh) |
| EG (1) | EG20280A (zh) |
| ES (1) | ES2092113T3 (zh) |
| FI (3) | FI990419A7 (zh) |
| GR (1) | GR3021411T3 (zh) |
| HU (2) | HU224443B1 (zh) |
| IE (1) | IE921986A1 (zh) |
| IL (1) | IL102188A (zh) |
| MX (1) | MX9203018A (zh) |
| NO (1) | NO180715C (zh) |
| NZ (1) | NZ243230A (zh) |
| PL (2) | PL172054B1 (zh) |
| PT (1) | PT100606B (zh) |
| RU (1) | RU2114848C1 (zh) |
| SK (1) | SK282203B6 (zh) |
| TW (1) | TW201301B (zh) |
| UA (1) | UA39168C2 (zh) |
| WO (1) | WO1993000331A1 (zh) |
| YU (1) | YU48997B (zh) |
| ZA (1) | ZA924528B (zh) |
Families Citing this family (89)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5364943A (en) * | 1991-11-27 | 1994-11-15 | Pfizer Inc. | Preparation of substituted piperidines |
| FI990419A7 (fi) * | 1991-06-20 | 1999-02-26 | Pfizer | Typpeä sisältävien hetrosyklisten yhdisteiden fluorialkoksibentsyyliaminojohdannaiset |
| DE69231395T3 (de) | 1991-09-20 | 2005-07-21 | Glaxo Group Ltd., Greenford | Neue medizinische Indikation für Tachykinin-Antagonisten |
| EP0641328B1 (en) * | 1992-05-18 | 2001-11-21 | Pfizer Inc. | Bridged aza-bicyclic derivatives as substance p antagonists |
| AU4396193A (en) * | 1992-08-04 | 1994-03-03 | Pfizer Inc. | 3-benzylamino-2-phenyl-piperidine derivatives as substance p receptor antagonists |
| ATE208376T1 (de) * | 1992-08-19 | 2001-11-15 | Pfizer | Substituierte benzylamin-stickstoff enthaltende nichtaromatische heterocyclen |
| FR2700472B1 (fr) | 1993-01-19 | 1995-02-17 | Rhone Poulenc Rorer Sa | Association synergisante ayant un effet antagoniste des récepteurs NK1 et NK2. |
| GB9305718D0 (en) * | 1993-03-19 | 1993-05-05 | Glaxo Group Ltd | Medicaments |
| IL109646A0 (en) * | 1993-05-19 | 1994-08-26 | Pfizer | Heteroatom substituted alkyl benzylamino-quinuclidines |
| JP2822274B2 (ja) * | 1993-05-19 | 1998-11-11 | ファイザー製薬株式会社 | P物質拮抗剤としてのヘテロ原子置換アルキルベンジルアミノキヌクリジン類 |
| DK0700384T3 (da) * | 1993-05-28 | 1997-12-08 | Pfizer | fremgangsmåde til fremstilling og opløsning af 2-phenyl-3-aminopiperidin |
| US5393762A (en) * | 1993-06-04 | 1995-02-28 | Pfizer Inc. | Pharmaceutical agents for treatment of emesis |
| WO1995007886A1 (en) * | 1993-09-17 | 1995-03-23 | Pfizer Inc. | 3-amino-5-carboxy-substituted piperidines and 3-amino-4-carboxy-substituted pyrrolidines as tachykinin antagonists |
| AU7082194A (en) * | 1993-09-17 | 1995-04-03 | Pfizer Inc. | Heteroarylamino and heteroarylsulfonamido substituted 3-benzylaminomethyl piperidines and related compounds |
| US6083943A (en) * | 1993-09-17 | 2000-07-04 | Pfizer Inc | Substituted azaheterocyclecarboxylic acid |
| IS4208A (is) * | 1993-09-22 | 1995-03-23 | Glaxo Group Limited | 3-(tetrazólýl-benzyl)amínó-piperadidín afleiður |
| EP0653208A3 (en) * | 1993-11-17 | 1995-10-11 | Pfizer | Substance P antagonists for the treatment or prevention of sunburn. |
| EP0659409A3 (en) * | 1993-11-23 | 1995-08-09 | Pfizer | Substance P antagonists to inhibit angiogenesis. |
| EP0655246A1 (en) * | 1993-11-30 | 1995-05-31 | Pfizer Inc. | Substance P antagonists for the treatment of disorders caused by helicobacter pylori or other spiral urease-positive gram-negative bacteria |
| FR2728169A1 (fr) | 1994-12-19 | 1996-06-21 | Oreal | Utilisation d'un antagoniste de substance p pour le traitement des prurits et des dysesthesies oculaires ou palpebrales |
| FR2728165A1 (fr) | 1994-12-19 | 1996-06-21 | Oreal | Utilisation d'un antagoniste de substance p pour le traitement des rougeurs cutanees d'origine neurogene |
| FR2728166A1 (fr) | 1994-12-19 | 1996-06-21 | Oreal | Composition topique contenant un antagoniste de substance p |
| EP0802912B1 (en) * | 1995-01-12 | 2004-10-13 | Glaxo Group Limited | Piperidine derivatives having tachykinin antagonist activity |
| GB9505692D0 (en) * | 1995-03-21 | 1995-05-10 | Glaxo Group Ltd | Chemical compounds |
| TW340842B (en) * | 1995-08-24 | 1998-09-21 | Pfizer | Substituted benzylaminopiperidine compounds |
| ATE182788T1 (de) | 1995-10-10 | 1999-08-15 | Pfizer | Nk-1 rezeptor antagonisten in neurogener entzündung in gen-therapie |
| TW458774B (en) | 1995-10-20 | 2001-10-11 | Pfizer | Antiemetic pharmaceutical compositions |
| FR2741262B1 (fr) | 1995-11-20 | 1999-03-05 | Oreal | Utilisation d'un antagoniste de tnf-alpha pour le traitement des rougeurs cutanees d'origine neurogene |
| PT780375E (pt) | 1995-12-21 | 2002-12-31 | Pfizer | 3-¬(benzilo substituido em 5)amino|-2-phenilpiperidinas como antagonistas da substancia p |
| US6329396B1 (en) | 1996-06-10 | 2001-12-11 | Pfizer Inc. | Substituted benzylaminopiperidine compounds |
| MX9706196A (es) * | 1996-08-14 | 1998-02-28 | Pfizer | Compuestos triciclicos de piperidinilamino como antagonistas de la sustancia p. |
| US5750549A (en) * | 1996-10-15 | 1998-05-12 | Merck & Co., Inc. | Cycloalkyl tachykinin receptor antagonists |
| NZ329807A (en) * | 1997-04-23 | 2000-07-28 | Pfizer | NK-1 receptor antagonists and P receptor antagonists 2-Diarylmethyl-3-amino-1-azabicyclo[2.2.2]octane derivatives and amino substituted N-containing rings as agents for treating irritable bowel syndrome |
| KR19990030614A (ko) * | 1997-10-02 | 1999-05-06 | 성재갑 | 스쿠알렌 합성 효소 억제활성을 갖는 신규 화합물, 그의 제조방법 및 그를 포함하는 조성물 |
| RS49964B (sr) | 1999-05-17 | 2008-09-29 | Pfizer Products Inc., | Postupak za dobijanje 2-fenil-3-aminopiridina,njegovih supstituisanih fenil derivata, i njegovih soli |
| JP2001172178A (ja) * | 1999-10-25 | 2001-06-26 | Pfizer Prod Inc | 偏頭痛治療用のnk−1レセプターアンタゴニスト及びエレトリプタン |
| MXPA02004330A (es) | 1999-11-03 | 2004-07-30 | Albany Molecular Res Inc | Tetrahidroisoquinolinas aril-y heteroaril-sustituidas y uso de las mismas para bloquear la recaptacion de norepinefrina, dopamina y serotonina.. |
| US7163949B1 (en) | 1999-11-03 | 2007-01-16 | Amr Technology, Inc. | 4-phenyl substituted tetrahydroisoquinolines and use thereof |
| KR100821410B1 (ko) | 2000-07-11 | 2008-04-10 | 에이엠알 테크놀로지, 인크. | 4-페닐 치환된 테트라하이드로이소퀴놀린 및 이의치료학적 용도 |
| US20020049211A1 (en) * | 2000-09-06 | 2002-04-25 | Sobolov-Jaynes Susan Beth | Combination treatment for depression and anxiety |
| EP1192952A3 (en) * | 2000-09-28 | 2003-03-26 | Pfizer Products Inc. | Combination, for treating depression and anxiety, containing an NK-3 receptor antagonist and a CNS penetrant NK-1 receptor antagonist |
| SK252004A3 (sk) * | 2001-07-20 | 2005-03-04 | Pfizer Products Inc. | Použitie antagonistov receptora NK-1 na výrobu liečiva na liečenie abnormálneho úzkostného správania domácich zvierat a spôsob screeningu skúšanej zlúčeniny s cieľom stanoviť anxiolytickú účinnosť u psov |
| US6686507B2 (en) | 2002-03-06 | 2004-02-03 | Pfizer Inc | Purification of 2-methoxy-5-trifluoromethoxybenzaldehyde |
| US6911544B2 (en) | 2002-10-23 | 2005-06-28 | Pfizer Inc. | Process for the preparation of (S,S)-cis-2-phenyl-3-aminopiperidine |
| US20040204455A1 (en) * | 2003-04-10 | 2004-10-14 | Cody Wayne Livingston | Piperidine derivative rennin inhibitors |
| US7671072B2 (en) * | 2003-11-26 | 2010-03-02 | Pfizer Inc. | Aminopyrazole derivatives as GSK-3 inhibitors |
| NZ552397A (en) | 2004-07-15 | 2011-04-29 | Amr Technology Inc | Aryl-and heteroaryl-substituted tetrahydroisoquinolines and use thereof to block reuptake of norepinephrine, dopamine, and serotonin |
| ES2382814T3 (es) | 2005-05-17 | 2012-06-13 | Merck Sharp & Dohme Ltd. | Ácido cis-4-[(4-clorofenil)sulfonil]-4-(2,5-difluorofenil)ciclohexanopropanoico para el tratamiento del cáncer |
| KR101594898B1 (ko) | 2005-07-15 | 2016-02-18 | 알바니 몰레큘라 리써치, 인크. | 아릴- 및 헤테로아릴-치환된 테트라히드로벤자제핀, 및 노르에피네프린, 도파민 및 세로토닌의 재흡수를 차단하기 위한 용도 |
| EP1940842B1 (en) | 2005-09-29 | 2012-05-30 | Merck Sharp & Dohme Corp. | Acylated spiropiperidine derivatives as melanocortin-4 receptor modulators |
| GB0603041D0 (en) | 2006-02-15 | 2006-03-29 | Angeletti P Ist Richerche Bio | Therapeutic compounds |
| US8173629B2 (en) | 2006-09-22 | 2012-05-08 | Merck Sharp & Dohme Corp. | Method of treatment using fatty acid synthesis inhibitors |
| US20110218176A1 (en) | 2006-11-01 | 2011-09-08 | Barbara Brooke Jennings-Spring | Compounds, methods, and treatments for abnormal signaling pathways for prenatal and postnatal development |
| JP4611444B2 (ja) | 2007-01-10 | 2011-01-12 | イステイチユート・デイ・リチエルケ・デイ・ビオロジア・モレコラーレ・ピ・アンジエレツテイ・エツセ・ピー・アー | ポリ(adp−リボース)ポリメラーゼ(parp)阻害剤としてのアミド置換インダゾール |
| EP2117538A1 (en) | 2007-01-24 | 2009-11-18 | Glaxo Group Limited | Pharmaceutical compositions comprising 2-methoxy-5- (5-trifluoromethyl-tetrazol-i-yl-benzyl) - (2s-phenyl-piperidin-3s-yl-) |
| JP5319518B2 (ja) | 2007-04-02 | 2013-10-16 | Msd株式会社 | インドールジオン誘導体 |
| AU2008269154B2 (en) | 2007-06-27 | 2014-06-12 | Merck Sharp & Dohme Llc | 4-carboxybenzylamino derivatives as histone deacetylase inhibitors |
| TWI410650B (zh) * | 2007-11-09 | 2013-10-01 | Hon Hai Prec Ind Co Ltd | 轉接板 |
| AU2009222122A1 (en) | 2008-03-03 | 2009-09-11 | Tiger Pharmatech | Tyrosine kinase inhibitors |
| US9156812B2 (en) | 2008-06-04 | 2015-10-13 | Bristol-Myers Squibb Company | Crystalline form of 6-[(4S)-2-methyl-4-(2-naphthyl)-1,2,3,4-tetrahydroisoquinolin-7-yl]pyridazin-3-amine |
| UA105182C2 (ru) | 2008-07-03 | 2014-04-25 | Ньюрексон, Інк. | Бензоксазины, бензотиазины и родственные соединения, которые имеют ингибирующую nos активность |
| WO2010114780A1 (en) | 2009-04-01 | 2010-10-07 | Merck Sharp & Dohme Corp. | Inhibitors of akt activity |
| AU2010247735B2 (en) | 2009-05-12 | 2015-07-16 | Albany Molecular Research, Inc. | Crystalline forms of (S)-7-([1,2,4]triazolo[1,5-a]pyridin-6-yl)-4-(3,4-dichlorophenyl)- 1,2,3,4-tetrahydroisoquinoline and use thereof |
| AU2010247763B2 (en) | 2009-05-12 | 2015-12-24 | Albany Molecular Research, Inc. | 7-([1,2,4,]triazolo[1,5,-a]pyridin-6-yl)-4-(3,4-dichlorophenyl)-1,2,3,4- tetrahydroisoquinoline and use thereof |
| WO2010132437A1 (en) | 2009-05-12 | 2010-11-18 | Albany Molecular Research, Inc. | Aryl, heteroaryl, and heterocycle substituted tetrahydroisoquinolines and use thereof |
| PE20121172A1 (es) | 2009-10-14 | 2012-09-05 | Merck Sharp & Dohme | Piperidinas sustituidas con actividad en la hdm2 |
| WO2011163330A1 (en) | 2010-06-24 | 2011-12-29 | Merck Sharp & Dohme Corp. | Novel heterocyclic compounds as erk inhibitors |
| EP3330377A1 (en) | 2010-08-02 | 2018-06-06 | Sirna Therapeutics, Inc. | Rna interference mediated inhibition of catenin (cadherin-associated protein), beta 1 (ctnnb1) gene expression using short interfering nucleic acid (sina) |
| US9029341B2 (en) | 2010-08-17 | 2015-05-12 | Sirna Therapeutics, Inc. | RNA interference mediated inhibition of hepatitis B virus (HBV) gene expression using short interfering nucleic acid (siNA) |
| US8883801B2 (en) | 2010-08-23 | 2014-11-11 | Merck Sharp & Dohme Corp. | Substituted pyrazolo[1,5-a]pyrimidines as mTOR inhibitors |
| EP2613782B1 (en) | 2010-09-01 | 2016-11-02 | Merck Sharp & Dohme Corp. | Indazole derivatives useful as erk inhibitors |
| US9242981B2 (en) | 2010-09-16 | 2016-01-26 | Merck Sharp & Dohme Corp. | Fused pyrazole derivatives as novel ERK inhibitors |
| WO2012058210A1 (en) | 2010-10-29 | 2012-05-03 | Merck Sharp & Dohme Corp. | RNA INTERFERENCE MEDIATED INHIBITION OF GENE EXPRESSION USING SHORT INTERFERING NUCLEIC ACIDS (siNA) |
| WO2012087772A1 (en) | 2010-12-21 | 2012-06-28 | Schering Corporation | Indazole derivatives useful as erk inhibitors |
| AU2012245971A1 (en) | 2011-04-21 | 2013-10-17 | Piramal Enterprises Limited | A crystalline form of a salt of a morpholino sulfonyl indole derivative and a process for its preparation |
| EP2770987B1 (en) | 2011-10-27 | 2018-04-04 | Merck Sharp & Dohme Corp. | Novel compounds that are erk inhibitors |
| US20150299696A1 (en) | 2012-05-02 | 2015-10-22 | Sirna Therapeutics, Inc. | SHORT INTERFERING NUCLEIC ACID (siNA) COMPOSITIONS |
| RU2660429C2 (ru) | 2012-09-28 | 2018-07-06 | Мерк Шарп И Доум Корп. | Новые соединения, которые являются ингибиторами erk |
| US20140206667A1 (en) | 2012-11-14 | 2014-07-24 | Michela Gallagher | Methods and compositions for treating schizophrenia |
| RS56680B1 (sr) | 2012-11-28 | 2018-03-30 | Merck Sharp & Dohme | Kompozicije i postupci za lečenje kancera |
| KR102196882B1 (ko) | 2012-12-20 | 2020-12-30 | 머크 샤프 앤드 돔 코포레이션 | Hdm2 억제제로서의 치환된 이미다조피리딘 |
| WO2014120748A1 (en) | 2013-01-30 | 2014-08-07 | Merck Sharp & Dohme Corp. | 2,6,7,8 substituted purines as hdm2 inhibitors |
| WO2015034925A1 (en) | 2013-09-03 | 2015-03-12 | Moderna Therapeutics, Inc. | Circular polynucleotides |
| EP3525785B1 (en) | 2016-10-12 | 2025-08-27 | Merck Sharp & Dohme LLC | Kdm5 inhibitors |
| US10947234B2 (en) | 2017-11-08 | 2021-03-16 | Merck Sharp & Dohme Corp. | PRMT5 inhibitors |
| EP3706747B1 (en) | 2017-11-08 | 2025-09-03 | Merck Sharp & Dohme LLC | Prmt5 inhibitors |
| US12173026B2 (en) | 2018-08-07 | 2024-12-24 | Merck Sharp & Dohme Llc | PRMT5 inhibitors |
| EP3833668B1 (en) | 2018-08-07 | 2025-03-19 | Merck Sharp & Dohme LLC | Prmt5 inhibitors |
| US11981701B2 (en) | 2018-08-07 | 2024-05-14 | Merck Sharp & Dohme Llc | PRMT5 inhibitors |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1990005729A1 (en) * | 1988-11-23 | 1990-05-31 | Pfizer Inc. | Quinuclidine therapeutic agents |
| WO1991018899A1 (en) * | 1990-06-01 | 1991-12-12 | Pfizer Inc. | 3-amino-2-aryl quinuclidines, process for their preparation and pharmaceutical compositions containing them |
| WO1992001688A1 (en) * | 1990-07-23 | 1992-02-06 | Pfizer Inc. | Quinuclidine derivatives |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3452026A (en) * | 1966-03-15 | 1969-06-24 | Bristol Myers Co | Substituted 1,2,3,4-tetrahydroquinolines |
| US3560510A (en) * | 1969-03-05 | 1971-02-02 | Aldrich Chem Co Inc | 2-benzhydrylquinuclidines |
| US5232929A (en) * | 1990-11-28 | 1993-08-03 | Pfizer Inc. | 3-aminopiperidine derivatives and related nitrogen containing heterocycles and pharmaceutical compositions and use |
| UA41251C2 (uk) * | 1990-01-04 | 2001-09-17 | Пфайзер, Інк. | Гідровані азотвмісні гетероциклічні сполуки, похідні піперидину, фармацевтична композиція та спосіб пригнічення активності речовини р в організмі |
| HUT68667A (en) * | 1990-09-28 | 1995-07-28 | Pfizer | Fused ring analogs of nitrogen containing nonaromatic heterocycles |
| EP0552197B1 (en) * | 1990-10-03 | 1999-01-13 | Commonwealth Scientific And Industrial Research Organisation | Epoxy resins based on diaminobisimide compounds |
| US5138060A (en) * | 1991-01-03 | 1992-08-11 | Pfizer Inc. | Process and intermediates for preparing azabicyclo(2.2.2)octan-3-imines |
| SK284565B6 (sk) * | 1991-03-26 | 2005-06-02 | Pfizer Inc. | Spôsob prípravy substituovaných piperidínov |
| PL171921B1 (pl) * | 1991-05-22 | 1997-06-30 | Pfizer | Sposób wytwarzania nowych pochodnych podstawionej 3-aminochinuklidyny PL PL PL PL PL |
| UA27776C2 (uk) * | 1991-05-31 | 2000-10-16 | Пфайзер Інк. | Похідні хінуклідину та їх фармацевтично прийнятні солі, що є антагоністами речовини р у ссавців, фармацевтична композиція, що має антагоністичну дію на речовину р у ссавців |
| FI990419A7 (fi) * | 1991-06-20 | 1999-02-26 | Pfizer | Typpeä sisältävien hetrosyklisten yhdisteiden fluorialkoksibentsyyliaminojohdannaiset |
| AU4396193A (en) * | 1992-08-04 | 1994-03-03 | Pfizer Inc. | 3-benzylamino-2-phenyl-piperidine derivatives as substance p receptor antagonists |
| US5393762A (en) * | 1993-06-04 | 1995-02-28 | Pfizer Inc. | Pharmaceutical agents for treatment of emesis |
| US5576317A (en) * | 1994-12-09 | 1996-11-19 | Pfizer Inc. | NK-1 receptor antagonists and 5HT3 receptor antagonists for the treatment of emesis |
-
1992
- 1992-05-05 FI FI990419A patent/FI990419A7/fi unknown
- 1992-05-05 PL PL92301884A patent/PL172054B1/pl not_active IP Right Cessation
- 1992-05-05 AT AT92911210T patent/ATE142199T1/de not_active IP Right Cessation
- 1992-05-05 WO PCT/US1992/003571 patent/WO1993000331A1/en not_active Ceased
- 1992-05-05 US US08/167,881 patent/US5773450A/en not_active Expired - Fee Related
- 1992-05-05 PL PL92310851A patent/PL170516B1/pl not_active IP Right Cessation
- 1992-05-05 JP JP4510950A patent/JPH07110850B2/ja not_active Expired - Fee Related
- 1992-05-05 DE DE9290083U patent/DE9290083U1/de not_active Expired - Lifetime
- 1992-05-05 HU HU9500836A patent/HU224443B1/hu not_active IP Right Cessation
- 1992-05-05 RU RU93058531A patent/RU2114848C1/ru not_active IP Right Cessation
- 1992-05-05 DK DK92911210.0T patent/DK0589924T3/da active
- 1992-05-05 ES ES92911210T patent/ES2092113T3/es not_active Expired - Lifetime
- 1992-05-05 CA CA002109613A patent/CA2109613C/en not_active Expired - Fee Related
- 1992-05-05 SK SK3908-92A patent/SK282203B6/sk not_active IP Right Cessation
- 1992-05-05 KR KR1019930703935A patent/KR0154882B1/ko not_active Expired - Fee Related
- 1992-05-05 EP EP92911210A patent/EP0589924B1/en not_active Expired - Lifetime
- 1992-05-05 UA UA93004396A patent/UA39168C2/uk unknown
- 1992-05-05 CZ CS19923908A patent/CZ290475B6/cs not_active IP Right Cessation
- 1992-05-05 HU HU9303668A patent/HU221634B1/hu not_active IP Right Cessation
- 1992-05-05 DE DE69213451T patent/DE69213451T2/de not_active Expired - Fee Related
- 1992-05-05 AU AU18893/92A patent/AU657967B2/en not_active Ceased
- 1992-05-05 BR BR9206161A patent/BR9206161A/pt not_active Application Discontinuation
- 1992-05-12 TW TW081103685A patent/TW201301B/zh active
- 1992-06-12 IL IL10218892A patent/IL102188A/en not_active IP Right Cessation
- 1992-06-17 EG EG31892A patent/EG20280A/xx active
- 1992-06-19 YU YU64092A patent/YU48997B/sh unknown
- 1992-06-19 PT PT100606A patent/PT100606B/pt not_active IP Right Cessation
- 1992-06-19 ZA ZA924528A patent/ZA924528B/xx unknown
- 1992-06-19 NZ NZ243230A patent/NZ243230A/en not_active IP Right Cessation
- 1992-06-19 MX MX9203018A patent/MX9203018A/es not_active IP Right Cessation
- 1992-06-19 CN CN92104778A patent/CN1056373C/zh not_active Expired - Fee Related
- 1992-07-01 IE IE198692A patent/IE921986A1/en not_active IP Right Cessation
-
1993
- 1993-12-17 FI FI935701A patent/FI108794B/fi active
- 1993-12-17 NO NO934691A patent/NO180715C/no unknown
-
1995
- 1995-05-22 US US08/443,418 patent/US5744480A/en not_active Expired - Fee Related
-
1996
- 1996-10-23 GR GR960402782T patent/GR3021411T3/el unknown
- 1996-11-18 BR BR1100086-4A patent/BR1100086A/pt active IP Right Grant
-
1999
- 1999-02-26 FI FI990418A patent/FI110686B/fi active
-
2003
- 2003-03-04 US US10/379,198 patent/US20030199540A1/en not_active Abandoned
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1990005729A1 (en) * | 1988-11-23 | 1990-05-31 | Pfizer Inc. | Quinuclidine therapeutic agents |
| WO1991018899A1 (en) * | 1990-06-01 | 1991-12-12 | Pfizer Inc. | 3-amino-2-aryl quinuclidines, process for their preparation and pharmaceutical compositions containing them |
| WO1992001688A1 (en) * | 1990-07-23 | 1992-02-06 | Pfizer Inc. | Quinuclidine derivatives |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1056373C (zh) | 含氮杂环类化合物的氟代烷氧基苄氨基衍生物的制备方法 | |
| CN1036652C (zh) | 碱式季酰胺,其制备方法及其药物组合物 | |
| CN1090188C (zh) | 新的苯并萘啶类化合物 | |
| CN1064356C (zh) | 新哌嗪、哌啶与1,2,5,6-四氢吡啶化合物,其制法及其药物组合物 | |
| CN1060285A (zh) | 含氮非芳香杂环的稠环类似物 | |
| CN1191862A (zh) | 8-取代的1,3,8-三氮杂螺[4.5]癸烷-4-酮衍生物 | |
| CN1092771A (zh) | 氨基亚甲基取代的非芳香杂环化合物 | |
| CN1294577A (zh) | 钾通道抑制剂 | |
| CN1058405A (zh) | 奎宁环衍生物 | |
| CN1018614B (zh) | 制备含n-杂环基-4-哌啶胺类的抗组胺组合物的方法 | |
| CN1042133C (zh) | N-取代的氮杂双环庚烷衍生物及其用途 | |
| CN1222521A (zh) | 1,3,8-三氮杂螺[4,5]癸-4-酮衍生物 | |
| CN1226825A (zh) | 胺化合物用于制备阻止肿瘤细胞增殖的药物 | |
| CN1071755C (zh) | 杂环甲酰胺衍生物及其作为治疗剂的用途 | |
| CN100338074C (zh) | 杂环化合物和以其为有效成分的脑机能改善剂 | |
| CN1049658C (zh) | 吡唑并[4,3-c]吡啶,它们的制备方法及其作为5-羟色胺再摄入抑制剂的用途 | |
| CN1166833A (zh) | 嘧啶基吡唑衍生物 | |
| CN1024663C (zh) | 治疗气喘用的取代的四氢化萘类化合物的制备方法 | |
| CN1041088C (zh) | 光学活性咪唑烷酮衍生物和抗痴呆药物组合物 | |
| CN1179962C (zh) | 氮杂吲哚嗪酮衍生物和以其为有效成分的脑功能改善剂 | |
| CN87104641A (zh) | 4-(芳酰氨基)哌啶丁酰氨衍生物 | |
| CN1281607C (zh) | 具有镇痛作用的新型单哌嗪季铵盐类化合物 | |
| CN1135215A (zh) | 咪唑啉酮衍生物、其酸的加成盐及老年痴呆症的治疗药物 | |
| CN1028531C (zh) | 1-氧杂-2-氧代-8-氮杂螺[4,5]癸烷衍生物及其盐的制备方法 | |
| CN87106267A (zh) | 新化合物2,3,4,5,6,7-六氢-2,7-亚甲基-1,5-苯并唑宁和-1,4-苯并唑宁,其制备方法及其药物组合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C06 | Publication | ||
| PB01 | Publication | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C17 | Cessation of patent right | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20000913 |