CN100372536C - 在口腔中迅速崩解的片剂 - Google Patents
在口腔中迅速崩解的片剂 Download PDFInfo
- Publication number
- CN100372536C CN100372536C CNB028050703A CN02805070A CN100372536C CN 100372536 C CN100372536 C CN 100372536C CN B028050703 A CNB028050703 A CN B028050703A CN 02805070 A CN02805070 A CN 02805070A CN 100372536 C CN100372536 C CN 100372536C
- Authority
- CN
- China
- Prior art keywords
- tablet
- oral cavity
- water
- component
- disintegrate
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images

Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
- A61K9/0056—Mouth soluble or dispersible forms; Suckable, eatable, chewable coherent forms; Forms rapidly disintegrating in the mouth; Lozenges; Lollipops; Bite capsules; Baked products; Baits or other oral forms for animals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2072—Pills, tablets, discs, rods characterised by shape, structure or size; Tablets with holes, special break lines or identification marks; Partially coated tablets; Disintegrating flat shaped forms
- A61K9/2077—Tablets comprising drug-containing microparticles in a substantial amount of supporting matrix; Multiparticulate tablets
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/10—Drugs for genital or sexual disorders; Contraceptives for impotence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1617—Organic compounds, e.g. phospholipids, fats
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1635—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Zoology (AREA)
- Nutrition Science (AREA)
- Physiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Gynecology & Obstetrics (AREA)
- Endocrinology (AREA)
- Reproductive Health (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Cosmetics (AREA)
Abstract
本发明涉及在服用时在口腔中迅速崩解、而不会给出令人不愉快的味道、并且在消化道内可被快速吸收从而使药物起效的片剂。这些片剂包含在中性或碱性条件下在水中几乎不溶、并且在酸性条件下水溶性很高、但有令人不愉快的味道的药物。可以通过将该药物与一种水溶性的酸性物质进行混合,用一种在醇溶剂中不溶的水溶性包衣剂对药物和酸性物质中的一种或两种进行包衣,进一步向其中加入在醇溶剂中可溶解的水溶性粘合剂和一种或多种水溶性的糖,然后在低压力下将混合物进行塑造,然后用醇对其进行处理,来制备该片剂。
Description
技术领域
本发明涉及在口腔中可迅速/快速崩解的片剂。更具体地,本发明涉及包含在中性或碱性条件下几乎不溶于水、并且在酸性条件下水溶性很高、但有令人不愉快味道的药用物质(药物)的在口中迅速崩解的片剂,该制剂包含水溶性的酸性物质并且用一种水溶性的包衣剂对该药用物质和酸性物质中的至少一种进行了包衣,该包衣剂在醇溶剂中不溶,由此,所述的制剂在被摄取后迅速起效而不会由于在口腔崩解过程中的药物而出现令人不愉快的味道。
背景技术
一般而言,本身为游离化合物形式的碱性药物在中性或碱性条件下在水中表现出低溶解度。因此,当刚吃完饭或患有酸缺乏的人服用该类药物时,由于所摄取的药物不能很好的溶解,所以会造成包括起效延迟和/或生物利用度(BA)降低等的问题。
在该类情况中,目前是将在中性或碱性条件下在水中具有低溶解度的药物转变成酸加成盐以使药物快速溶解、改善其生物利用度并使其及早起效。
此外,目前已经能获得起效更快并容易服用的在口腔中快速/迅速崩解的片剂或颗粒。但是,在这些制剂中,药物甚至在口腔中就开始溶解,因此,尽管所述的酸加成盐形式的药物的水溶性很高,但如果所述的药物具有令人不愉快的味道,则该类制剂本身很难以在口中迅速崩解的片剂或颗粒的形式给药于患者。
已经提出了一种掩味技术,例如该技术包含予先用在低pH下可溶的聚合物对包含酸加成盐的颗粒进行包衣。例如,WO98/30209公开了先用羟丙基甲基纤维素然后用EudragitE100(一种在胃酸中可溶的聚合物)进行包衣的枸橼酸西地那非(sildenafil citrate)的颗粒状制剂。但是,因为所述的制剂使用了一种在胃酸中可溶解的作为包衣剂的聚合物,所以药物的释放和生物利用度将受到不利影响并且当刚刚进食或患有胃酸缺乏症的患者使用该制剂时药物的起效将受到妨碍。
已知有许多药物需要在给药后立即起效。例如,为了改善生命质量(QOL),用一种环GMP(cGMP)-特异性的磷酸二酯酶(PDE)抑制剂,尤其是PDE5抑制剂作为治疗勃起功能障碍的药物。该类药物优选地在服药后立即起效以根据需要对性反应进行控制。
但是,在临床上用作PDE5抑制剂的枸橼酸西地那非(Viagra片)快速释放片剂的情况中,已经报道了使用者应当在希望其起效前1小时服用该制剂(W000/24383,第2页)。此外,还已知可以通过将包含枸橼酸西地那非和可结合的崩解剂的混合物进行湿法制粒,然后用压缩模塑法将该湿颗粒制备成一种在口腔中迅速崩解的片剂(JP-A 10-298062(1998))。但是,所述制剂不能有效地掩盖药物的苦味并且因此不能进行商业化制造。
发明内容
一直需要研制一种包含在中性或碱性条件下在水中几乎不溶、并且在酸性条件下水溶性很高、但在该酸性条件下有令人不愉快的味道的药物的药物制剂,该制剂在口腔中可以迅速崩解而不会出现不愉快的味道,该制剂在消化道中表现出药物的快速释放和吸收,并且能及早起效。
本发明提供了一种在口腔中迅速崩解的片剂,其包含有在中性或碱性条件下在水中几乎不溶、并且在酸性条件下水溶性很高、但有令人不愉快的味道的药物,以及制备该片剂的方法。
附图说明
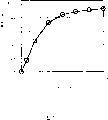
图1表示了用本发明实施例1的在口腔中迅速崩解的片剂进行的溶出试验的结果。
图2表示了用参考实施例6的片剂进行的溶出试验的结果。
图3表示了在口服了实施例1或参考实施例6的在口中迅速崩解的片剂的狗体内的药物血浆浓度。
实施本发明的最佳方式
本发明的发明者在他们的研究中发现通过将药物与一种酸性物质进行混合,并且将该药物和酸性物质中的至少一种用一种在醇溶剂中不溶的水溶性包衣剂进行包衣,可以获得一种在口腔中迅速崩解的片剂,其包含有在中性或碱性条件在水中几乎不溶、并且在酸性条件下水溶性很高、但在酸性条件下有令人不愉快的味道的药物,虽然该片剂在口腔中迅速崩解,但是在口腔中并出现该物质的令人不愉快的味道,并且还可以促进该药物在消化道中的释放和吸收。
因此,本发明提供来一种硬度20N或更高、并且孔隙率为25%至50%的在口腔中迅速崩解的片剂,其包含如下的组分:
i) 在碱性或中性条件下在水中几乎不溶、并且在酸性条件下水溶性很高、但在酸性条件下有令人不愉快的味道的药物;
ii) 水溶性的酸性物质;
iii) 可溶于醇溶剂的水溶性粘合剂;和
iv) 水溶性的糖,
其中用一种在醇溶剂中不溶的水溶性包衣剂对组分(i)和(ii)中的至少一种物质进行包衣。
本发明还提供了一种制备上述在口中迅速崩解的片剂的方法,该方法包含如下的步骤(A)至(C):
(A) 制备包含该在口中迅速崩解片剂所必需的组分的基本上均匀的混合物;
(B) 将由上述步骤(A)所获得的混合物在低压缩压力下进行压制;和
(C) 用醇对步骤(B)中通过压制得到的片剂进行处理,然后进行干燥。
本发明包含在中性或碱性条件下在水中几乎不溶、并且在酸性条件下水溶性很高、但在酸性条件下有令人不愉快的味道的药物(i)的在口中迅速崩解的片剂,其特征在于其包含一种酸性物质(ii),尤其是当将其给药于患有胃酸缺乏症或希望在进食后立即使用该制剂的患者时,其可增加该药物溶解度的水溶性;一种水溶性粘合剂(iii),其能溶解于在压制后进行醇溶剂处理所需的醇溶剂,用以提高片剂的硬度;和一种水溶性的糖(vi),其为该片剂提供良好的溶解度和甜味;并且用一种在醇溶剂中不溶的水溶性包衣剂对药物(i)和该水溶性的酸性物质(ii)中的至少一种进行包衣。用所述包衣剂进行处理,可以避免在整个所述在口中迅速崩解片剂的制备过程中药物和酸性物质的接触,其会引起不愉快的味道,其中所述所述在口中迅速崩解片剂的制备过程不仅不扩混合步骤(A)和压制模塑步骤(B),而且包括通过粘合剂的溶解和固化所进行的增强硬度的步骤(C)。
此外,虽然本发明在口中可迅速崩解的片剂可以在口腔中迅速崩解,但是其在药物与酸性物质进行接触前可以通过口腔,因此其在口腔中几乎不存在令人不愉快的味道。此外,本发明的制剂具有如下的优点,例如因为其在口腔中少量的唾液中就可崩解,所以在服用时不需要服用水,并且,在服用后,其会使药物在消化道的上部(从食道到胃)迅速溶解并且在消化道中药物浓度增加,从而改善了药物吸收并减少了药物的起效时间。
在本发明制剂中所用的水溶性的酸性物质的实例包括其pKa值不高于5,优选地不高于4,并且在25℃下在水中的溶解度不低于5mg/mL的物质。优选的实例包括有机酸,特别是富马酸、酒石酸、琥珀酸、苹果酸、抗坏血酸和天冬氨酸。该酸性物质可以单独使用或两种或多种联合使用。
该在醇溶剂中可溶解的水溶性粘合剂的实例包括那些在25℃下在水和醇溶剂中的溶解度都不低于20mg/mL,优选地不低于50mg/mL的物质。粘合剂的具体实例包括聚乙烯衍生物如聚乙烯吡咯烷酮(PVP)等等和纤维素衍生物如羟丙基纤维素(HPC)等等。可以使用一种或多种粘合剂。在本发明中,使用具有1-100μm,优选20-20μm的平均粒径的粘合剂。
水溶性糖的实例包括那些在25℃下在水中的溶解度不低于20mg/mL,优选不低于50mg/mL的物质。具体的实例包括甘露醇、木糖醇、赤藓醇、麦芽糖醇、山梨醇和乳糖,可以使用一种或多种糖。在本发明中,使用具有1-100μm,优选20-80μm,更优选40-60μm的平均粒径的水溶性糖。
用来对药物和水溶性酸性物质中的至少一种进行包衣的水溶性包衣剂的实例包括那些在25℃下在水中的溶解度不低于20mg/mL、并且在25℃下在醇溶剂中的溶解度不高于1mg/mL的物质,如纤维素衍生物(羟丙基甲基纤维素(HPMC)、甲基纤维素(MC)、羟乙基纤维素(HEC)等等)、多糖(葡聚糖、支链淀粉、精氨酸钠、琼脂等等)、聚环氧烷、聚乙烯衍生物(聚乙烯醇(PVA)等等)和肽衍生物(明胶等等)。本发明可以使用其中的一种或多种包衣剂。
优选地用该类包衣剂进行包衣的药物或水溶性酸性物质的平均粒径在30至500μm的范围内,优选地为50至300μm,更优选地为100至200μm。
可用于本发明的药物包括这些在中性或碱性条件下几乎不溶、并且在酸性条件下水溶性很好、但在酸性条件下有令人不愉快的味道的物质。该类药物的实例包括那些在25℃下在pH等于或高于7时在水中的溶解度不高于0.1mg/mL、并在pH等于或低于2时在水中的溶解度不低于1mg/mL的物质,优选地是那些在pH为1.2时在水中(25℃)的溶解度不低于在pH为7时在水中(25℃)溶解度的10倍,优选地不低于100倍的物质。
此外,可用于本发明的药物是那些在酸性条件下,例如pH等于或小于5,尤其是等于或小于4,具体地是在本发明所用的水溶性的酸性物质(例如具有等于或低于5的pKa,尤其是等于或低于4的酸)所造成的酸性条件下,有令人不愉快的味道如苦味、涩味、辛辣等等味道的物质。酸的实例包括有机酸如富马酸、酒石酸、琥珀酸、苹果酸、抗坏血酸、天冬氨酸等等。
本发明所优选的药物的实例尤其包括那些在给药后需要在1小时内的短时间内发挥其功效的物质,如环GMP-特异性的磷酸二酯酶抑制剂。
环GMP-特异性的磷酸二酯酶抑制剂的实例包括例如在JP-A 09-512835(1997),JP-A 09-503996(1997),JP-A 2000-128883(2000),JP-A 2000-128884(2000),JP-A 11-505236(1999),JP-A 11-505539(1999),JP-A 2000-507256(2000),JP-A 2000-503996(2000),JP-A 2000-95759(2000),JP-A 10-298164(1998),JP-A 2000-72675(2000),JP-A 2000-72751(2000),JP-A 09-124648(1997),JP-A 08-231545(1996),JP-A 08-231546(1996),JP-A 08-253457(2000),JP-A 11-503445(1999),WO97/45427,JP-A 11-509221(1999), JP-A-11-509517(1999)和JP-A 11-509535(1999),WO00/20033和W000/39099中所公开的PDE5抑制剂。
优选的PDE5抑制剂的实例包括式(I)的含氮的芳香族6-元环状化合物

其中环A是任选取代的含氮杂环基,R1是式:-NH-Q-R3(其中R3是任选取代的含氮杂环基,并且Q是低级亚烷基或单键)或式:-NH-R4(其中R4是任选取代的环烷基);R2是任选取代的芳基,Y和Z中的一个是式:=CH-的基团,并且另一个是式:=N-的基团。见WO01/19802(专利申请号为277652/2000的日本专利申请)。
环A“任选取代的含氮杂环基”中含氮杂环基的实例包括5-至10-元单环或双环含氮杂环基,更特别地是5-或6-元含氮的单环杂环基或8-至10-元含氮的双环杂环基,并且最特别地是如下式所示的5-至6-元含氮的单环杂环基:
或
或其中任何一种上述的5-或6-元含氮的单杂环与5-或6-元环基进行稠合的如下式所示的含氮的双环杂环基:



R3“任选取代的含氮杂环基”中含氮杂环基的实例包括含氮的单杂环或双杂环,特别是如下式所示的非芳香族的含氮杂环:

或如下式所示的芳香族的含氮杂环:
环A“任选取代的含氮杂环基”中取代基的实例包括选自如下的基团:(1)低级烷基,(2)羟基取代的低级烷基,(3)甲酰基,(4)氧代基,(5)氨基,(6)羟基,(7)低级烷氧基羰基,和(8)选自(i)和(ii)的基团,其中:(i)是被卤素原子和低级烷氧基所取代苄基氨基,和(ii)是被羟基取代的环烷基氨基甲酰基所取代的嘧啶基。
R1是式:-NH-Q-R3或式:-NH-R4的基团。R3“任选取代的含氮杂环基”中取代基的实例包括低级烷基、羟基取代的低级烷基、氧代基、氨基、二-(低级烷基)氨基、低级烷酰基和氰基取代的低级烷基。
R2“任选取代的芳基”中芳基的实例包括5-至10-元单环或双环的芳香烃基,特别地是苯基、萘基等等。
R2“任选取代的芳基”中取代基的实例包括低级烷氧基、卤素原子、氰基等等。
R4“任选取代的环烷基”中取代基的实例包括低级烷氧基、羟基、吗啉基等等。
在本说明书和权利要求中,“低级烷基”指的是具有1至6个碳原子的直链或支链烷基,如甲基、乙基、丙基、异丙基、丁基、异丁基、叔-丁基等等。“低级烷氧基“指的是具有1至6个碳原子的直链或支链烷氧基,如甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基、异丁氧基、叔-丁氧基等等。“环烷基”指的是具有3至8个碳原子的环烷基,如环丙基、环丁基、环戊基、环己基,环庚基等等。“低级亚烷基”指的是具有1至6个碳原子的直链或支链亚烷基,如亚甲基、亚乙基、1,3-亚丙基等等。
本发明在口中迅速崩解的片剂优选地用于游离形式的药物,当该药物被转化成酸加成盐时会表现出味道、稳定性或可操作性上的缺点。
在这里所用的PDE5抑制剂中,尤其优选的实例包括如下的化合物。
(S)-2-(2-羟基甲基-1-吡咯烷基)-4-(3-氯-4-甲氧基苄基氨基)-5-[N-(2-嘧啶基甲基)-氨基甲酰基]嘧啶;
(S)-2-(2-羟基甲基-1-吡咯烷基)-4-(3-氯-4-甲氧基苄基氨基)-5-[N-(2-吗啉代乙基)-氨基甲酰基]嘧啶;
2-(5,6,7,8-四氢咪唑并[1,2-a]吡嗪-7-基)-4-(3-氯-4-甲氧基苄基氨基)-5-[N-(2-嘧啶基甲基)-5-氨基甲酰基]嘧啶;
2-(5,6,7,8-四氢-1,7-二氮杂萘-7-基)-4-(3-氯-4-甲氧基苄基氨基)-5-[N-(2-吗啉代乙基)氨基甲酰基]-嘧啶;
(S)-2-(2-羟基甲基-1-吡咯烷基)-4-(3-氯-4-甲氧基苄基氨基)-5-[N-(5-嘧啶基甲基)氨基甲酰基]-嘧啶;
(S)-2-[N-(2-嘧啶基甲基)氨基甲酰基]-3-(3-氯-4-甲氧基苄基氨基)-5-[2-羟基甲基-1-吡咯烷基]-吡嗪;
(S)-2-[N-(2-吗啉代乙基)氨基甲酰基]-3-(3-氯-4-甲氧基苄基氨基)-5-(2-羟基甲基-1-吡咯烷基)-吡嗪;和
(S)-2-(2-羟基甲基-1-吡咯烷基)-4-(3-氯-4-甲氧基苄基氨基)-5-[N-(1,3,5-三甲基-4-吡唑基)-氨基甲酰基]嘧啶。
本发明在口中迅速崩解的片剂可以包含固体药物制剂中常用的添加剂。该添加剂的实例包括赋形剂如柠檬酸钙、磷酸钙、结晶纤维素和硅酸镁铝等等;崩解剂如玉米淀粉、马铃薯淀粉、羧甲基淀粉钠、部分预胶化的淀粉、羧甲基纤维素钙、羧甲基纤维素、低取代的羟基丙基纤维素、交联羧甲基纤维素钠、交联的聚乙烯吡咯烷酮等等;润滑剂如硬脂酸镁、硬脂酸钙、滑石、轻质无水硅酸、无水二氧化硅等等;矫味剂如橙油、小茴香油、肉桂油、丁香油、松节油、薄荷油、桉树油等等;着色剂如2号和3号食用染料红、4号和5号食用染料黄、3号食用染料绿、1号和2号使用染料蓝、以及这些食用染料的铝色淀、三氧化二铁、三氧化二铁黄等等;甜味剂如糖精、阿司帕坦、甜菊甙(Stevia)等等;调味剂如氯化钠等等;增溶剂如环糊精、精氨酸、赖氨酸、三氨基甲烷等等,等物质。当使用时,优选地加入少量的润滑剂。
本发明在口内迅速崩解片剂中,各组分的配方量是用各组分占片剂总重量的含量(w/w%)来表示,即用如下的

来表示,其中“%”指的是“w/w%”。
药物:1-70%,优选5-50%,更优选10-30%。
水溶性酸性物质:0.1-50%,优选1-30%。
水溶性糖:5-95%,优选10-85%,更优选30-80%。
水溶性粘合剂:0.1-30%,优选1-10%。
水溶性包衣剂:0.02-10%,优选0.1-6%。
本发明在口腔内迅速崩解的片剂具有如下的性质。
在口腔中的崩解时间:在60秒内,优选在45秒内,并且更优选在30秒内崩解。硬度:等于或大于20N(牛顿),优选等于或大于30N,其中20N是将片剂从水泡眼包装(通过包装压:PTP)中推出所必需的硬度。
孔隙率(%):当根据如下的方程进行测定时为5-50%,优选30-45%:

V:片剂体积(mL)
ρ:排除孔之后片剂的真密度(g/mL)
M:片重(g)
药物溶出速率:当用溶出试验方法2(桨法,第XII版日本药典)在50rpm的桨转速的条件下在37℃的水中进行测定时,在最初30分钟内等于和高于70%,优选等于和高于80%。
本发明在口中迅速崩解的片剂可以用上述包含步骤(A)、(B)和(C)的方法来进行制备。在下面对该方法进行详细的举例说明。
步骤(A):组分的均匀混合物的制备
所述混合物的制备包括在将组分连续混合后进行制粒或同时进行制粒,从而产生压片用颗粒的过程。可以用任何药学领域公知的常规方法来完成该混合过程,例如可通过double-com混合器(由Yashima Chemical EngineeringCo.,Ltd.制造的Double-Com混合器)、流化床造粒机(由Powrex Corporation制造的Multiplex;由Freund Co.,Ltd.制造的Spirer Flow)、高速剪切混合器造粒机(由Fukae Powtech Co.制造的高速混合器;由Powrex Corporation制造的Vertical造粒机),或振动筛(由Dalton制造的振动筛)等等来进行混合。
在本步骤中,将组分(i)-(iv)依次进行混合。在混合前,用在醇溶剂中不溶的水溶性包衣剂对组分(i)和(ii)中的任何一种或两种进行包衣,然后
(a)当将组分(i)和(ii)中的一种用水溶性包衣剂进行包衣时,将未进行包衣的组分(i)或(ii)与组分(iv)进行混合,在加入组分(iii)或其水溶液的同时将该混合物进行制粒,将所得颗粒进行干燥,如果需要的话,将其与包衣的组分(i)或(ii)进行混合;或
(b)当用水溶性包衣剂对组分(i)和(ii)两种进行包衣时,在加入组分(iii)或其水溶液的同时将组分(iv)进行制粒,将所得颗粒进行干燥,如果需要的话,将进行了包衣的组分(i)和(ii)与所得的颗粒进行混合。
可以用任何一种常规的制粒技术如湿法制粒、分层制粒或干法制粒来完成该制粒。
通过在加入组分(iii)的水溶液的同时,将要制粒的组分(iv)(和组分(i)和(ii)的未包衣组分)进行搅拌,并用混合造粒机或高速剪切混合造粒机等进行制粒,可以进行湿法制粒。备选地,将要进行制粒的组分与一种粘合剂的水溶液进行混合,揉合,用挤出造粒机进行制粒然后进行整粒。在另一种备选的方法中,将一种粘合剂的水溶液喷洒到组分的混合物上,用流化床造粒机、旋转的流化床造粒机等进行制粒。
分层制粒的进行可以通过用离心的流化床造粒机等将要进行制粒的组分(iv)(和组分(i)和(ii)的未包衣组分)添加到旋转的惰性载体中,同时喷洒组分(iii)的水溶液,以制备粘附到该载体上的组分的混合物。在本方法中所用的惰性载体的实例包括结晶的糖或无机盐如结晶乳糖、结晶纤维素、结晶氯化钠等等,以及球形颗粒如精制的白色糖的球形颗粒(商品名:Nonpareil-103,Freund Co.,Ltd.)、乳糖和预胶化淀粉的球形颗粒等等。
可以通过将要进行制粒的组分(iii)与组分(iv)(和组分(i)和(ii)的未包衣组分)进行混合,然后用旋转压片机、旋转造粒机等等进行制粒来进行干法制粒。
用在醇溶剂中不溶的水溶性包衣剂对组分(i)和/或(ii)进行包衣是通过将所述组分(i)和(ii)进行制粒,同时加入在醇溶剂中不溶的水溶性包衣剂的水溶液或水-醇溶液,从而用所述的水溶性包衣剂对组分(i)和/或(ii)的颗粒表面进行包衣,并且如果需要的话,可以喷洒一种在醇溶剂中不溶的所述水溶性包衣的水溶液。在这种方法中,优选地加入一种抗粘剂如滑石粉以防止包衣颗粒的聚集,并调节该水溶性包衣剂在口腔中的溶出速率。
步骤(B):在低压力下对步骤(A)中所得混合物进行的压缩
可以用常规的压片机在5-40MPa,优选7-20MPa的条件下对步骤(A)中得到的混合物进行压制,所述的压片机例如为三层旋转压片机(RT3L-14;Kikusui Seisakusyo,Ltd.)或双重压片机(Correct D65RC;Kikusui Seisakusyo,Ltd.)等等。当压片时,为了防止可能出现的诸如粘着的问题,可以使用一种通过将润滑剂(如硬脂酸镁、硬脂酸钙)和流化剂(例如结晶纤维素)的混合物、以及在步骤(A)中所得的混合物依次进行压制成形,从而在压制中使用润滑剂的方法(JP-A 10-29806(1998))。备选地,还可以使用其中压制是通过向压片机的冲上喷洒一种润滑剂粉末来进行的方法(JP-B 41-1273(1966),JP-B 48-20103(1973)),或其中压制是通过一种防粘连的聚合物膜来进行的方法(JP-A08-19589(1996))。
步骤(C):步骤(B)中压制得到的片剂的醇溶剂处理和干燥
用醇溶剂进行处理例如可以通过使该片剂在醇溶剂的气氛下进行放置或通过向该片剂上喷洒该醇溶剂的蒸汽来进行。可以通过在加热下喷洒醇溶剂可以产生醇溶剂的蒸汽。考虑到药物的稳定性,优选地将蒸汽维持在等于或低于60℃的温度上,更优选地将其维持在等于或低于40℃的温度上。
因为当用醇溶剂的蒸汽进行处理时,包含在片剂中的粘合剂会发生溶解,所以在一定限度内,通过延长处理时间可以增加粘合剂的溶解量,其通过下一步中的干燥处理中增强了硬度。在不能获得所需硬度或需要延长崩解时间的情况下,应当根据所需的硬度和/或所需的崩解时间来对粘合剂的量、处理时间、所喷洒醇蒸汽的量等进行适宜的选择。
通过从上述处理所得的产物中除去醇溶剂可以获得所需的在口中迅速崩解的片剂。可以通过将该产品在大气压或减压的条件下在室温下进行放置来除去该醇溶剂,同时可进行加热或通风以加速除去该醇溶剂。
可用的醇溶剂包括这些沸点不高于85℃的那些,例如低级醇,如甲醇、乙醇、异丙醇等等。
本发明在口腔中迅速崩解的片剂可以被塑造成片形、椭圆形、球形或方形的形状,并且其体积为0.05至1mL/单位,优选0.1至0.5mL。
在本发明中用作优选药物(PDE5抑制剂)的化合物(I)可以通过如下的方法来进行制备:将式(II)的化合物

其中X1是卤素原子,R5是羧基的保护基,并且其它符号具有如上所定义的含义,
与式(III)的化合物进行反应,
R-CH2-NH2(III)
其中符号具有如上所定义的含义,从而制备得到式(IV)的化合物:

其中各符号具有如上所述的含义;
氧化产物(IV),以制备式(V)的甲基磺酰基(或甲基亚磺酰基)化合物:

其中n是1或2的整数,并且其它符号具有如上所定义的含义;
将式(V)的化合物与式(VI)的化合物或其盐进行反应:

其中该符号具有如上所定义的含义,从而制得一种式(VII)的化合物:

其中的符号具有如上所定义的含义;
除去羧基保护基R5,制得式(VIII)的化合物:

其中的符号具有如上所定义的含义;
然后将式(VIII)的化合物与式(IX-a)的化合物进行反应:
R1-H (IX-a)
其中R1具有如上所定义的含义。
此外,化合物(I)可以用如下的方法来进行制备:
a)将化合物(VIII)卤化,从而制备出式(X)的化合物:

其中X2是卤素原子,并且其它符号具有如上所定义的含义,然后
b)将化合物(X)与化合物(IX-a)进行反应。
同时,还可以用如下的方法来制备化合物(VII):用二氧化碳处理式(XI)的二卤化的化合物:

其中X3和X4是卤素原子,并且其它符号具有如上所定义的含义,从而制备出式(XII)的化合物:

其中的符号具有如上所定义的含义,保护化合物(XII)的羧基,从而制备出式(XIII)的化合物:
其中的符号具有如上所定义的含义,将化合物(XIII)与化合物(III)进行反应,从而制备出式(XIV)的化合物:

其中的符号具有如上所定义的含义,然后将化合物(XIV)与化合物(VI)进行反应。
此外,化合物(XIV)还可以通过如下的方法制备:将化合物(V)水解,制备式(XV)的化合物:
其中的符号具有如上所定义的含义,
然后卤化式(XV)的化合物。
化合物(II)与(III)的反应是在溶剂中在存在或不存在一种酸清除剂的条件下进行的。例如该酸清除剂包括有机碱如N,N-二异丙基乙基胺、N-甲基吗啉、三乙胺和吡啶,以及无机碱如氢化钠、碳酸钠、碳酸钾和碳酸氢钠。该溶剂可以是不会干扰该反应的任何溶剂,例如N,N-二甲基甲酰胺、四氢呋喃、甲苯、乙酸乙酯、氯仿、二甲氧基乙烷、二甲苯或二甲基亚砜。该反应是在-10℃至室温,优选约0℃至室温的温度下进行的。
将化合物(IV)氧化以获得甲基磺酰基(或甲基亚磺酰基)化合物(V)的反应是在一种溶剂中在存在氧化剂的条件下进行的。例如,该氧化剂包括过酸如间-氯过苯甲酸或过乙酸或无机氧化剂如二氧化锰、高碘酸钠、过氧化氢、酸性硝酸盐、四氧化二氮、卤素、N-卤代化合物、氢过氧化物、醋酸碘苯、叔-丁基次氯酸盐、磺酰氯或过一硫酸钾。该溶剂可以是不干扰该反应的任何溶剂,例如氯仿、二氯甲烷、二氯乙烷和醋酸。该反应可以在-78℃至50℃,优选-10℃至10℃的温度下进行。
化合物(V)与化合物(VI)或其盐的反应可以在溶剂中在存在和不存在酸清除剂的条件下进行。例如,该酸清除剂包括一种有机碱如N,N-二异丙基乙基胺、N-甲基吗啉、三乙胺和吡啶、以及一种无机碱如氢化钠、碳酸钠、碳酸钾和碳酸氢钠。化合物(VI)的盐优选地是一种碱金属盐如钠盐、钾盐。该溶剂可以是不干扰该反应的任何溶剂,例如,N,N二甲基甲酰胺、四氢呋喃、二甲氧基乙烷或二甲基亚砜。该反应可以在0℃至150℃,优选室温至60℃的温度下进行。
除去化合物(VII)的羧基保护基R5以得到化合物(VIII)的反应可以用常规的方法来进行,其中所述的常规方法如水解或催化还原,可以根据所要除去的羧基保护基的类型来对所用方法进行选择。当通过水解来除去该羧基保护基时,水解是例如在溶剂中在存在或不存在碱的情况下进行的。该碱优选地是例如碱金属氢氧化物如氢氧化钠、氢氧化钾或氢氧化锂、碱金属碳酸盐如碳酸钠或碳酸钾。该溶剂可以是水或水和甲醇、乙醇、四氢呋喃、二噁烷、N,N-二甲基甲酰胺或二甲基亚砜的混合物。该反应是在0℃至80℃,优选5℃至60℃的温度下进行的。由R5所代表的羧基保护基可以是常规的羧基保护基如低级烷基或苄基。
化合物(VIII)与化合物(IX-a)的反应可以在适宜的溶剂中在存在或不存在缩合剂、碱或活化剂的条件下进行的。该缩合剂包括例如二环己基碳二亚胺、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺、二苯基膦酰基叠氮化物或二乙基氰基磷酸酯,其通常被用于肽的合成。该碱包括,例如有机碱如三乙胺或N-甲基吗啉,并且该活化剂包括例如1-羟基苯并三唑。该溶剂可以是不干扰该反应的任何溶剂,例如二氯甲烷、四氢呋喃、N,N-二甲基甲酰胺、乙腈、N,N-二甲基乙酰胺或乙酸乙酯。该反应是在-30℃至50℃,优选-10℃至10℃的温度下进行的。
作为备选的将化合物(VIII)转化成进一步与化合物(IX-a)进行反应的化合物(X)的方法,可以首先通过常规方法将化合物(VIII)与卤化剂在存在或不存在活化剂的条件下反应来进行,然后将所得化合物(X)与化合物(IX-a)进行反应。化合物(VIII)与卤化剂的反应是在溶剂中进行的。这种卤化剂优选地是亚硫酰氯、草酰氯或五氯化磷。该活化剂优选地是酰胺化合物,如N,N-二甲基甲酰胺。该溶剂可以是不干扰该反应的任何溶剂,例如二氯甲烷、氯仿、四氢呋喃、苯、甲苯或二噁烷。该反应是在-30℃至100℃,优选-5℃至10℃的温度下进行的。
随后的化合物(X)与化合物(IX-a)的反应可以在溶剂中在存在或不存在一种酸清除剂的条件下进行。该酸清除剂包括例如有机碱,如N,N-二异丙基乙基胺、N-甲基吗啉、三乙胺、吡啶、二甲基氨基吡啶、和无机碱如氢化钠、碳酸钠、碳酸钾、碳酸氢钠。该溶剂可以是不干扰该反应的任何溶剂,例如四氢呋喃、二氯甲烷、氯仿、甲苯、苯、二噁烷、乙酸乙酯。该反应是在-30℃至100℃,优选-5℃至10℃的温度下进行的。
用二氧化碳对二卤代化合物(XI)进行处理来得到化合物(XII)的处理可以在溶剂中在存在碱的条件下进行。该碱包括例如有机碱的碱金属盐如二异丙基酰胺锂、2,2,6,6-四甲基胡椒脂锂。该溶剂可以是不干扰该反应的任何溶剂,例如四氢呋喃、1,2-二甲氧基乙烷、二乙醚。该反应是在-100℃至30℃,优选-100℃至-70℃的温度下进行的。
将化合物(XII)的羧基保护起来以得到化合物(XIII)的反应可以用常规的方法来完成,例如当该保护基是一种低级烷基时,可以在溶剂中在存在碱的条件通过将其与一种烷化剂进行反应来完成。该烷化剂优选地是一种低级烷基卤化物如碘代甲烷。该碱优选地是一种碱金属碳酸氢盐,并且该溶剂可以是不干扰该反应的任何溶剂,例如N,N-二甲基甲酰胺、四氢呋喃。该反应是在0℃至100℃,优选室温至70℃的温度下进行的。
化合物(XIII)与化合物(III)反应得到化合物(XIV)的反应可以在与化合物(II)与化合物(III)的反应条件相同的条件下进行。
化合物(XIV)与化合物(VI)反应得到化合物(VII)的反应可以在与化合物(V)与化合物(VI)的反应条件相同的条件下进行反应。
由化合物(V)至给出化合物(XV)的水解可以在存在碱的情况下在溶剂中进行。该碱包括,例如碱金属氢氧化物如氢氧化钠、氢氧化钾、氢氧化钾或氢氧化锂、以及碱金属碳酸盐如碳酸钠或碳酸钾。该溶剂优选地是水、或水和甲醇的混合物、乙醇、四氢呋喃、二噁烷、N,N-二甲基甲酰胺或二甲基甲酰胺。该反应是在-20℃至80℃,优选-5℃至60℃的温度下进行的。
将化合物(XV)进行卤化以得到化合物(XIV)的反应可以在与用一种卤化剂将化合物(VIII)进行卤化来制备化合物(X)的条件相同的条件下来进行。
实施例1
(1)将具有如下结构式的具有PDE5抑制剂活性的化合物(平均粒度:10μm;50g)和甘露醇(日本药典级,其后被称为“JP级”,平均粒径:50μm;217.1g;Nikken Chemical Industry)置于流化床造粒机(Multiplex MP-01/03;PowrexCorporation)中,以5g/分钟的速度向其喷洒一种5w/w%PVP(JP级,PVP-k30;BASF)的水溶液(90g)18分钟,同时将进气温度控制在50℃。在喷洒后,将进气温度调节至70℃,并且将所得颗粒进行干燥直至其温度变为40℃,从而获得活性组分的颗粒。

(2)将富马酸(日本药典赋形剂规格,平均粒径:10μm;200g;FusoChemical Industry)置于流化床造粒机(Multiplex MP-01/03;Powrex Corporation)中,以5g/分钟的速度向其喷洒一种6w/w%HPMC(JP级,HPMC TC-5EW;Shin-Etsu Chemical,Co.Ltd.)水溶液20分钟,同时将进气温度控制在50℃。在喷洒完该水溶液后,以5g/分钟的速度喷洒一种HPMC(40g)、滑石(JP级,8g;Nippon Talc)、乙醇(526.4g)和水(225.6g)的混合物160分钟,同时将进气温度控制在60℃。在完成该喷洒后,将进气温度调节至70℃,并将所得颗粒进行干燥直至其温度变为40℃,得到富马酸的HPMC包衣颗粒。
(3)将得自(1)的活性物质颗粒(244.44g)、得自(2)的富马酸颗粒(22.86g)和阿司帕坦(日本药典赋形剂规格,化学名:N-L-α-天冬氨酰基-L-苯基丙氨酸,2.7g,平均粒径:10μm;Ajinomoto,co.,Ltd.)置于double com混合器中(容积:2升;Yashima Chemical Engineering Co.,Ltd.),并将其在55rpm下混合3分钟,得到压片所需的颗粒。
(4)在第一次预压缩部分中将单独制备的硬脂酸镁(9份)和结晶纤维素(1份)的混合物用一种三层旋转压片机(RT3L-14,Kikusui Seisakusyo Ltd.)进行压制,以使得硬脂酸镁粘附到冲(直径:10mm;球面曲率半径:13mm)上。在将该片剂排出后,在主压缩部分以10MPa的压缩压力对得自(3)的压片用颗粒进行压制,得到片剂(300mg/片)。
(5)将(4)所制备的片剂在25℃下在充满乙醇蒸汽的干燥器放置12小时,然后将其在箱式干燥器中在45℃下干燥5小时以除去乙醇,得到在口中迅速崩解的片剂(300mg/片)。
实施例2
(1)将实施例1(1)中所述的化合物(50g)和甘露醇(JP级,平均粒径:50μm,213.45g,Nikken Chemical)用高速剪切混合造粒机(ultra-slim High-speed混合器;Fukae20 Powtex)进行制粒,同时向其中加入18w/w%HPC(24.5g,HPC SL;Nippon Soda)的水溶液。在制粒后,将该颗粒在箱式干燥器、45℃下干燥8小时,通过22号筛(710μm)进行整粒和筛分,得到活性组分的颗粒。
(2)将(1)所得的活性组分颗粒(214.36g)、在实施例1(2)中所得的富马酸颗粒(15.24g)和阿司帕坦(食品添加剂的日本标准,化学名:N-L-α-天冬氨酰基-L-苯基丙氨酸,2.4g,平均粒径:10μm;Ajinomoto,Co.Ltd.)置于一种doublecom混合器(容积:2升;Yashima Chemical Engineering Co.,Ltd.)中,并将其在55rpm下混合3分钟,得到压片用颗粒。
(3)在第一次预压缩部分将单独制备的硬脂酸镁(9份)和结晶纤维素(1份)的混合物用一种三层旋转压片机(RT-3L-14,Kikusui Seisakusyo Ltd.)进行压制,以使得硬脂酸镁粘附到冲(直径:10mm;球面曲率半径:13mm)上。在将该片剂排出后,在主压缩部分以10MPa的压缩压力对得自(2)的压片用颗粒进行压制,得到片剂(290mg/片)。
(4)将在(3)中所制备的片剂在充满乙醇蒸汽的干燥器中、25℃下放置12小时,然后将将其在一种箱式干燥器中、45℃下放置5小时以除去乙醇,得到在口腔中迅速崩解的片剂(290mg/片)。
实施例3
(1)将实施例1(1)中所述的化合物(50g)和赤藓醇(日本药典赋形剂规格,平均粒径:60μm;217.1g;Nikken Chemical Industry)置于一种流化床造粒机(Multiplex MP-01/03;Powrex Corporation)中,然后以5g/分钟的速度向其喷洒一种5w/w%PVP(JP级,PVP-k30;BASF)的水溶液(90g)18分钟,同时将进气温度控制在50℃。在喷洒后,将进气温度调节至70℃,并将所得颗粒进行干燥直至其温度变为40℃,从而获得活性组分的颗粒。
(2)将(1)所得的活性组分颗粒(244.44g)、得自实施例1(2)的富马酸颗粒(22.86g)和阿司帕坦(食品添加剂的日本标准,化学名:N-L-α-天冬氨酰基-L-苯基丙氨酸,2.7g,平均粒径:10μm;Ajinomoto,co.,Ltd.)放在一种double corn混合器(容积:2升;Yashima Chemical Engineering Co.,Ltd.)中,并将其在55rpm下混合3分钟,得到压片用颗粒。
(3)在第一次预压缩部分将单独制备的硬脂酸镁(9份)和结晶纤维素(1份)的混合物用一种三层旋转压片机(RT3L-14,Kikusui Seisakusyo Ltd.)进行压制,以使得硬脂酸镁粘附到冲(直径:10mm;球面曲率半径:13mm)上。在将该片剂排出后,在主压缩部分以10MPa的压缩压力对得自(2)的压片用颗粒进行压制,得到片剂(300mg/片)。
(4)将得自(3)的片剂在25℃下在充满乙醇的蒸汽的干燥器中放置12小时,然后将其在一种箱式干燥器中、45℃下放置5小时以除去乙醇,得到在口腔中迅速崩解的片剂(300mg/片)。
实施例4
(1)将实施例1(1)中所述的化合物(50g)和赤藓醇(日本药典赋形剂规格,平均粒径:60μm;212.8g;Nikken Chemical Industry)置于一种流化床造粒机(Multiplex MP-01/03;Powrex Corporation)中,然后以5g/分钟的速度喷洒一种5w/w%PVP(JP级,PVP-k30;BASF)的水溶液(90g)18分钟,同时将进气温度控制在50℃。在进行喷洒后,将进气温度调节至70℃,然后将所得颗粒进行干燥直至其温度变成40℃,得到活性组分的颗粒。
(2)将得自(1)的活性组分颗粒(240.57g)和得自实施例1(2)的富马酸颗粒(11.43g)置于一种double corn混合器(容积:2升;Yashima ChemicalEngineering Co.,Ltd.)中,并将其在55rpm下混合3分钟,得到压片用颗粒。
(3)在第一次预压缩部分将单独制备的硬脂酸镁(9份)和结晶纤维素(1份)的混合物用一种三次旋转压片机(RT3L-14,Kikusui Seisakusyo Ltd.)进行压制,以使得硬脂酸镁粘附到冲(直径:8mm;球面曲率半径:10mm)上。在将该片剂排出后,在主压缩部分以10MPa的压缩压力将(2)所得的压片用颗粒进行压制,得到片剂(140mg/片)。
(4)将在(3)中所制备的片剂在25℃下在充满乙醇蒸汽的干燥器中放置12小时,然后将其在箱式干燥器中、45℃下干燥5小时以除去乙醇,得到在口腔中迅速崩解的片剂(140mg/片)。
实施例5
(1)将实施例1(1)中所述的化合物(平均粒径:10μm,100g)和甘露醇(JP级,平均粒径:50μm,403.8g,Nikken Chemical)置于一种流化床造粒机(Multiplex MP-01;Powrex Corporation)中,然后以9g/分钟的速度喷洒一种5w/w%PVP(JP级,PVP-k30;BASE)的水溶液(180g)20分钟,同时将进气温度控制在50℃。在进行喷洒后,将进气温度调节至70℃,并将所得颗粒进行干燥直至其温度变成40℃。得到活性组分的颗粒。
(2)将富马酸(日本药典赋形剂规格,平均粒径:80μm;400g;FusoChemical Industry)置于一种流化床造粒机(Multiplex MP-Ol;PowrexCorporation)中,然后以8g/分钟的速度喷洒一种6w/w%HPMC(JP级,HPMCTC-5EW;Shin-Etsu Chemical,Co.Ltd.)的水溶液(200g)25分钟,同时将进气温度控制在50℃。在完成喷洒后,将进气温度调节至70℃,并将所得颗粒进行干燥直至其温度变成40℃,得到富马酸的HPMC包衣颗粒。
(3)将得自(1)的活性组分颗粒(410.24g)、得自(2)的富马酸颗粒(32.96g)和阿司帕坦(食品添加剂的日本标准,化学名:N-L-α-天冬氨酰基-L-苯基丙氨酸,4.8g,平均粒径:10μm;Ajinomoto,co.,Ltd.)置于一种double corn混合器(容积:2升;Yashima Chemical Engineering Co.,Ltd.)中,并将其在55rpm下混合3分钟,得到压片用颗粒。
(4)在第一次预压缩部分将单独制备的硬脂酸镁(9份)和结晶纤维素(1份)的混合物用一种三层旋转压片机(RT3L-14,Kikusui Seisakusyo Ltd.)进行压制,以使得硬脂酸镁粘附到冲(直径:10mm;球面曲率半径:13mm)上。在将该片剂排出后,在主要压缩部位以10MPa的压缩压力将得自(3)的压片用颗粒进行压制,得到片剂(280mg/片)。
(5)将(4)中所制备的片剂在25℃下在充满乙醇蒸气的干燥器中放置12小时,然后将其在一种箱式干燥器中、45℃下放置5小时以除去乙醇,得到在口腔中迅速崩解的片剂(280mg/片)。
实施例6
用与实施例1相同的方法制备在口中迅速崩解的片剂,只是用2-(5,6,7,8-四氢咪唑并[1,2-a]吡嗪-7-基)-4-(3-氯-4-甲氧基苄基氨基)-5-[N-(2-嘧啶基甲基)氨基甲酰基]嘧啶作为实施例1(1)中的PDE5抑制剂。
实施例7
用与实施例1相同的方法来制备在口中迅速崩解的片剂,只是用2-(5,6,7,8-四氢-1,7-二氮杂萘-7-基)-4-(3-氯-4-甲氧基苄基氨基)-5-[N-(2-吗啉代(molphorino)乙基)-氨基甲酰基]嘧啶作为实施例1(1)中的PDE5抑制剂。
实施例8
用与实施例1相同的方法来制备在口中迅速崩解的片剂,只是用(S)-2-(2-羟基甲基-1-吡咯烷基(pyrolidinyl))-4-(3-氯-4-甲氧基苄基氨基)-5-[N-(5-嘧啶基甲基)-氨基甲酰基]嘧啶作为实施例1(1)中的PDE5抑制剂。
参考实施例1:没有用醇溶剂进行处理的制剂
用与实施例(1)-(4)相同的方法制备片剂(300mg/片)(省略实施例1(5)的乙醇-处理)。
参考实施例2:在改善了硬度的条件下压制的制剂
在第一次预压缩部分将单独制备的硬脂酸镁(9份)和结晶纤维素(1份)的混合物用一种三层旋转压片机(RT-3L-14,Kikusui Seisakusyo Ltd.)进行压制,以使硬脂酸镁粘附到冲(直径:10mm;球面曲率半径:13mm)上。在将该片剂排出后,在主要压缩部分将得自实施例1(3)的压片用颗粒以50MPa的压缩压力进行压制,得到片剂(300mg/片)。
参考实施例3:使用未包衣富马酸的制剂
(1)将实施例1(1)所述的化合物(平均粒径:10μm;75g)、甘露醇(JP级,平均粒径:50μm,338.25g,Nikken Chemical)和富马酸(日本药典赋形剂规格,平均粒径:10μm;30g;Fuso ChemicalIndustry)加入到一种高速剪切混合器造粒机(ultra-slim高速混合器;Fukae-Powtex)中,并在加入15w/w%PVP(JP级,PVP-k30;BASF)的水溶液(45g)的同时进行制粒。在制粒后,将颗粒在一种箱式干燥器中在45℃下干燥6小时,用22号筛(710μm)进行整粒和筛分,得到压片用颗粒。
(2)在第一次的预压缩部分将单独制备的硬脂酸镁(9份)和结晶纤维素(1份)的混合物用一种三层旋转压片机(RT3L-14,Kikusui Seisakusyo Ltd.)进行压制,以使得硬脂酸镁粘附到冲(直径:10mm;球面曲率半径:13mm)上。在将该片剂排出后,在主要压缩部分以10MPa的压缩压力将(1)中所得的压片用颗粒进行压制,得到(300mg/片)。
参考实施例4:带有未包衣的富马酸并用乙醇进行了处理的制剂
将参考实施例3(2)中所制备的片剂在25℃下在充满乙醇蒸气的干燥器中放置12小时,然后将其在一种箱式干燥器中、45℃下放置5小时以除去乙醇,得到在口腔中迅速崩解的片剂(300mg/片)。
参考实施例5:不含富马酸的制剂
(1)将实施例1(1)中所描述的化合物(平均粒径:10μm;50g)和甘露醇(JP级,平均粒径:50μm,247g,Nikken Chemical)置于一种流化床造粒机(MultiplexMP-01/03;Powrex Corporation)中,然后以5g/分钟的速度喷洒一种5w/w%PVP(JP级,PVP-30 k30;BASF)的水溶液(60g)12分钟,同时将进气温度控制在50℃。在进行喷洒后,将进气温度调节至70℃,并将所得颗粒进行干燥直至其温度变成40℃,得到活性组分的颗粒。
(2)在第一次预压缩部分将单独制备的硬脂酸镁(9份)和结晶纤维素(1份)的混合物用一种三层旋转压片机(RT-3L-14,Kikusui Seisakusyo Ltd.)进行压制,以使得硬脂酸镁粘附到冲(直径:10mm;球面曲率半径:13mm)上。在将该片剂排出后,在主压缩部分以10MPa的压缩压力将(1)中所得的压片用颗粒进行压制,得到片剂(300mg/片)。
参考实施例6:不含富马酸、用乙醇进行处理的制剂
将在参考实施例5(2)中所制备的片剂在25℃下在充满乙醇蒸气的干燥器中放置12小时,然后将其在一种箱式干燥器中、45℃下干燥5小时以除去乙醇,得到在口腔中迅速崩解的片剂(300mg/片)。
实验1:硬度的测定
用片剂硬度检测器(Tablet Tester 6D;Schleuniger)对实施例1至5和参考实施例1至6所得片剂的硬度进行了测定,其中硬度是破碎片剂所需的力。测定进行5轮,计算均值。结果如下面的表1所示。
表1
| 测定样品,得自如下实施例的片剂: | 硬度(N) |
| 实施例1 | 45 |
| 实施例2 | 40 |
| 实施例3 | 45 |
| 实施例4 | 38 |
| 实施例5 | 46 |
| 参考实施例1 | 11 |
| 参考实施例2 | 43 |
| 参考实施例3 | 10 |
| 参考实施例4 | 45 |
| 参考实施例5 | 10 |
| 参考实施例6 | 50 |
实验2:孔隙率的测定
测定得自实施例1至5和参考实施例1至6的各片剂的重量(M)。此外,用测微计(Digitrix II;NSK)测定各片剂的体积(V),用比重瓶(Autopycnometer 320型;Micrometric)测定除空隙空间外的密度(ρ)。用如下的方程计算各片剂的孔隙率。

表2
| 测定样品,得自如下实施例的片剂: | 孔隙率(%) |
| 实施例1 | 35 |
| 实施例2 | 34 |
| 实施例3 | 33 |
| 实施例4 | 31 |
| 实施例5 | 34 |
| 参考实施例1 | 35 |
| 参考实施例2 | 22 |
| 参考实施例3 | 35 |
| 参考实施例4 | 35 |
| 参考实施例5 | 33 |
| 参考实施例6 | 33 |
实验3:在口腔中崩解时间的评估
使健康的男性志愿者(志愿者们)(1-4名男性)将得自实施例1至5和实施例1至6的片剂放在舌头上并不施加任何作用地将其保留在舌头上,例如不咬破或不轻咬等。测定片剂完全崩解时所需的时间,获得该时间的均值。所得结果如下面的表3所示。
表3
| 测定样品,得自如下实施例的片剂: | 崩解时间(秒) |
| 实施例1 | 17 |
| 实施例2 | 20 |
| 实施例3 | 15 |
| 实施例4 | 15 |
| 实施例5 | 17 |
| 参考实施例1 | 16 |
| 参考实施例2 | 130 |
| 参考实施例3 | 20 |
| 参考实施例4 | 50 |
| 参考实施例5 | 8 |
| 参考实施例6 | 9 |
从实验1、2和3的结果可以清楚地看出实施例1-5所制备的本发明在口腔中迅速崩解的片剂具有足够的硬度、孔隙率和短时间内在口腔中崩解的能力。另一方面,尽管参考实施例1、3和5的片剂符合孔隙率的要求,并且具有在口腔中崩解的能力,但是其不够硬,因此其作为片剂是不合适的。用高压缩压力进行了压缩的参考实施例2的片剂虽然符合硬度要求,但是其表现出低孔隙率并且在口腔中的崩解力很差。参考实施例4的片剂具有足够的硬度和孔隙率;但是,当用如下面实验4所述的苦味试验进行检查时,其味道远远低于本发明在口腔内迅速崩解的片剂。参考实施例6的片剂表现出足够的硬度、孔隙率和在口腔中崩解的能力;但是,它们表现出如下面的实验5所示的极差的药物溶出曲线和如下面实验6所示的极低的生物利用度。
实验4:苦味实验
使健康的男性志愿者(4名男性)将实施例1的片剂和实施例4的片剂放在其舌头上并不施加任何作用地将其保留在舌头上,例如不咬破或不轻咬等,然后,在崩解后,用如表4所示的四等级评分来对味道进行评估。该结果的平均得分如表5所示。
表4
| 得分 | 评价 |
| 1 | 很好 |
| 2 | 好 |
| 3 | 有可以忍受的苦味 |
| 4 | 有不能忍受的苦味 |
表5
| 所试验的片剂 | 得分 |
| 实施例1的快速崩解的片剂 | 1 |
| 参考实施例4的片剂 | 3.5 |
实验5:溶出试验
根据溶出试验方法2(桨法,第XII版日本药典)在桨转速为50rpm、37℃下对实施例1所制备的在口腔中迅速崩解的片剂和参考实施例6中所制备的片剂在水中的药物溶出速率进行评估。
在溶出开始后在第2、5、10、15、20、25和30分钟从溶出液中取样并在295nm和450nm下测定吸收度。用单独制备的在吸收度和药物浓度之间表现出相关性的标准曲线来确定样品中的药物浓度。由所述药物的浓度和溶出液的体积来计算所溶出药物的量。
结果如图1和2所示。如图1和2所示,本发明实施例1的在口腔中迅速崩解的片剂在短时间内就表现出很高的溶出速率,而参考实施例6的片剂表现出很差的溶出曲线。
实验6:狗体内血浆浓度的测定
用将一种得自本发明实施例1或参考实施例6的在口腔中迅速崩解的片剂以及水(50mL)给禁食了一夜的雄性狗(1-2岁;每组6只狗)进行口服给药。在给药后第15、30、45、60、120、180、300和420分钟从每只狗的前肢血管上采集血样(2mL)。将样品离心以分离出血浆,然后将其用高效液相色谱(HPLC)进行分析。根据用包含已知浓度药物的样品所单独制备的标准曲线来估算血浆中的药物量。根据由此所获得的药物量和所采集血液的体积来计算药物浓度。结果如图3所示。
正如图3所示的那样,本发明实施例1的在口腔中迅速崩解的片剂在短时间内就表现出很高的血药浓度和高生物利用度,而参考实施例6的片剂表现出极低的血药浓度并且没有表现出足够的生物利用度。
制备1
(1)在用冰进行冷却的条件下,向4-氯-5-乙氧基羰基-2-甲硫基嘧啶(25.33g)在N,N-二甲基甲酰胺(85ml)中的溶液中加入3-氯-4-甲氧基苄基胺(19.62g)N,N-二甲基甲酰胺(15ml)和三乙胺(16.7ml)中的溶液。将该混合物在室温下搅拌20分钟,然后向其中加入3-氯-4-甲氧基苄基胺(940mg),并将该混合物再搅拌15分钟。再向该混合物中加入所述的胺(940mg),然后将该混合物搅拌15分钟。将反应混合物倒入到冰水和柠檬酸的混合物中,然后用乙酸乙酯进行萃取。将萃取液用10%柠檬酸水溶液、水和盐水连续进行洗涤,然后用无水硫酸钠进行干燥。在真空中蒸发掉溶剂并将残余物用正-己烷进行洗涤,得到4-(3-氯-4-甲氧基苄基氨基)-5-乙氧基羰基-2-甲硫基嘧啶(38.34g)。
(2)在用冰进行冷却的条件下向上面(1)中所得的化合物(5.00g)在氯仿(50ml)中的溶液中加入一种间-氯过苯甲酸(4.00g)在氯仿(50ml)中的溶液,并将该混合物搅拌2小时。用饱和的碳酸氢钠水溶液和盐水对该反应混合物进行洗涤,然后将有机层用无水硫酸钠进行干燥,然后在真空中蒸发掉溶剂,得到粗制的4-(3-氯-4-甲氧基苄基氨基)-5-乙氧基羰基-2-甲基亚磺酰基嘧啶。
(3)将在(2)中所得的粗品溶解于四氢呋喃(40ml)中,然后在室温下向其中加入L-脯氨醇(prolinol)(1.50g)和三乙胺(1.60g)在四氢呋喃(10ml)中的溶液。将该混合物搅拌过夜,然后将该反应混合物用乙酸乙酯进行稀释,然后用碳酸氢钠水溶液和盐水对其进行洗涤。将有机层用无水硫酸钠进行干燥,然后在真空中蒸发掉溶剂。用在硅胶上的柱色谱对残余物进行纯化(溶剂:氯仿),然后用乙醚和正-己烷的混合物进行结晶,得到(S)-4-(3-氯-4-甲氧基苄基氨基)-5-乙氧基羰基-2-(2-羟基甲基-1-吡咯烷基)嘧啶(4.72g)。
(4)将在上面(3)中所获得的化合物(3.4g)、10%氢氧化钠水溶液(23ml)、和二甲基亚砜(34ml)的混合物在室温下搅拌15小时。将该反应混合物倒入10%柠檬酸水溶液中,并用四氢呋喃和乙醚的混合物对沉淀进行结晶,得到(S)-4-(3-氯-4-甲氧基苄基氨基)-5-羧基-2-(2-羟基甲基-1-吡咯烷基)嘧啶(2.52g)。
(5)将在上面(4)中所得的化合物(600mg)、2-氨基甲基嘧啶(217mg)、1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐(323mg)、1-羟基苯并三唑一水合物(227mg)和N,N-二甲基甲酰胺(12ml)的混合物在室温下搅拌8小时,然后将该反应混合物倒入到碳酸氢钠水溶液中。将该混合物用乙酸乙酯进行萃取,用盐水进行洗涤,用无水硫酸钠进行干燥。在真空中蒸发掉溶剂,用使用硅胶的柱色谱(溶剂∶氯仿∶甲醇=50∶1)对残余物进行纯化,得到(S)-2-(2-羟基甲基-1-吡咯烷基)-4-(3-氯-4-甲氧基苄基氨基)-5-[N-(2-嘧啶基甲基)氨基甲酰基]嘧啶(610mg)。
制备2-4
用与制备1中所述方法相同的方法对相关起始材料进行处理,得到如下面的表6所列的化合物。
表6


工业实用性
根据本发明在口腔中迅速崩解的片剂,其包含一种在中性或碱性条件下在水中几乎不溶、并且在酸性条件下水溶性很高、但在酸性条件下有令人不愉快的味道的药物,通过混入一种水溶性的酸性物质可以增加药物的溶出曲线,从而获得快速崩解的能力并改善其生物利用度;并且通过用一种在醇溶剂中不溶的水溶性包衣剂对药物和水溶性酸性物质中的至少一种进行包衣,而避免了药物与酸性物质的最初接触,并且从而防止在口腔中出现令人不愉快的味道。此外,根据本发明,将其与混有的在醇溶剂中可溶解的水溶性粘合剂和水溶性糖一起在低压力下进行压片,并且然后用醇溶剂进行处理,从而所得的在口腔中迅速崩解的片剂符合硬度和孔隙率的要求,在口腔中迅速崩解,表现出高溶出速率、良好的生物利用度并且可以及早起效。因此,本发明尤其优选地适用于希望在服用后不久即起效的药物,例如在服用1小时内即起效的药物,如具有cGMP-特异性的磷酸二酯酶抑制活性的物质。
Claims (9)
1.一种具有20N或更高的硬度和25%至50%的孔隙率的在口腔中迅速崩解的片剂,其包含如下的组分:
i)(S)-2-(2-羟基甲基-1-吡咯烷基)-4-(3-氯-4-甲氧基苄氨基)-5-[N-(2-嘧啶基甲基)氨基甲酰基]嘧啶;
ii)富马酸;
iii)在醇溶剂中可溶解的水溶性粘合剂,其选自聚乙烯吡咯烷酮和羟丙基纤维素;和
iv)水溶性的糖,其选自甘露醇、赤藓醇,
其中用羟丙基甲基纤维素对组分(ii)进行包衣,
所述片剂是通过将包含组分(i)、(ii)、(iii)和(iv)的混合物进行压缩、用沸点不高于85℃的醇处理压缩的产品和去除该醇而获得的。
2.按照权利要求1所述的在口腔中迅速崩解的片剂,其在口腔中的崩解时间在60秒之内。
3.按照权利要求1所述的在口腔中迅速崩解的片剂,当用日本药典所定义的溶出试验方法2进行测定时,其在溶出开始后的最初30分钟内表现出在水中不低于70%的药物溶出速率,所述药典为第XII版,所述试验方法2为浆旋转,50rpm;37℃。
4.按照权利要求1所述的在口腔中迅速崩解的片剂,其包含1-70w/w%的组分(i),0.1-50w/w%的组分(ii),0.02-10w/w%的包衣组分(ii)的羟丙基甲基纤维素,0.1-30w/w%的组分(iii),以及5-95w/w%的组分(iv)。
5.按照权利要求1所述的在口腔中迅速崩解的片剂,其还进一步包含一种或多种用于药物制剂的可药用添加剂。
6.按照权利要求5所述的在口腔中迅速崩解的片剂,其中所述用于药物制剂的可药用添加剂是选自硬脂酸镁、硬脂酸钙、滑石粉、轻质无水硅酸和无水二氧化硅的润滑剂和/或选自糖精、阿司帕坦和甜菊甙的甜味剂。
7.一种制备在口腔中迅速崩解的片剂的方法,所述片剂具有20 N或更高的硬度、25%至50%的孔隙率并在服用时味道得到了改善,其包含如下的步骤(A)-(C):
(A)制备一种包含如下组分的基本上均匀的混合物:
i)(S)-2-(2-羟基甲基-1-吡咯烷基)-4-(3-氯-4-甲氧基苄氨基)-5-[N-(2-嘧啶基甲基)氨基甲酰基]嘧啶;
ii)富马酸;
iii)在醇溶剂中可溶解的水溶性粘合剂,其选自聚乙烯吡咯烷酮和羟丙基纤维素;和
iv)水溶性的糖,其选自甘露醇、赤藓醇,
其中用羟丙基甲基纤维素对组分(ii)进行包衣,
(B)将得自上面步骤(A)的混合物在压缩压力下进行压制,所述压缩压力是5至30MPa,从而得到硬度为5至20 N的片剂,和
(C)用沸点不高于85℃的醇对在步骤(B)中压制得到的片剂进行处理,然后对其进行干燥。
8.按照权利要求7所述的方法,其中所述的在步骤(B)中的压制是用如下方法中的任意一种方法来进行的:
(b-1)其中依次用含润滑剂和流化剂的混合物和在步骤(A)中得到的混合物进行压片的方法;
(b-2)其中在喷洒润滑剂粉末的同时进行压片的方法,和
(b-3)其中通过聚合物膜进行压制的方法。
9.按照权利要求7所述的方法,其中所述的醇溶剂是乙醇。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2001038343 | 2001-02-15 | ||
| JP038343/01 | 2001-02-15 | ||
| JP038343/2001 | 2001-02-15 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1527701A CN1527701A (zh) | 2004-09-08 |
| CN100372536C true CN100372536C (zh) | 2008-03-05 |
Family
ID=18901333
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB028050703A Expired - Fee Related CN100372536C (zh) | 2001-02-15 | 2002-02-12 | 在口腔中迅速崩解的片剂 |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US7927623B2 (zh) |
| EP (1) | EP1366760B1 (zh) |
| JP (1) | JP4179784B2 (zh) |
| KR (1) | KR100750554B1 (zh) |
| CN (1) | CN100372536C (zh) |
| AT (1) | ATE472319T1 (zh) |
| AU (1) | AU2002230217B2 (zh) |
| CA (1) | CA2437754C (zh) |
| DE (1) | DE60236850D1 (zh) |
| MX (1) | MXPA03007283A (zh) |
| NZ (1) | NZ527585A (zh) |
| WO (1) | WO2002064119A1 (zh) |
Families Citing this family (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2449163C (en) * | 2001-05-09 | 2010-07-13 | Bayer Healthcare Ag | New use of 2-phenyl-substituted imidazotriazinones |
| MY140561A (en) | 2002-02-20 | 2009-12-31 | Nycomed Gmbh | Dosage form containing pde 4 inhibitor as active ingredient |
| TWI286942B (en) * | 2002-06-10 | 2007-09-21 | Dainippon Sumitomo Pharma Co | Rapidly disintegrating tablet and preparing method thereof |
| DE10232113A1 (de) * | 2002-07-16 | 2004-01-29 | Bayer Ag | Vardenafil Hydrochlorid Trihydrat enthaltende Arzneimittel |
| ME00524B (me) | 2003-03-10 | 2011-10-10 | Astrazeneca Ab | Novi postupak za dobijanje roflumilasta |
| GB0317663D0 (en) * | 2003-07-29 | 2003-09-03 | Astrazeneca Ab | Pharmaceutical composition |
| JP4740740B2 (ja) | 2003-12-09 | 2011-08-03 | 大日本住友製薬株式会社 | 薬物含有粒子および該粒子を含む固形製剤 |
| KR20070030178A (ko) * | 2004-02-17 | 2007-03-15 | 트랜스오랄 파마슈티칼스, 인코포레이티드 | 구강 점막을 가로지르는 수면제 전달용 조성물 및 이의사용 방법 |
| DE102004023069A1 (de) * | 2004-05-11 | 2005-12-08 | Bayer Healthcare Ag | Neue Darreichungsformen des PDE 5-Inhibitors Vardenafil |
| DE102005001989A1 (de) * | 2005-01-15 | 2006-07-20 | Bayer Healthcare Ag | Intravenöse Formulierungen von PDE-Inhibitoren |
| DE102005009241A1 (de) * | 2005-03-01 | 2006-09-07 | Bayer Healthcare Ag | Arzneiformen mit kontrollierter Bioverfügbarkeit |
| DE102005009240A1 (de) * | 2005-03-01 | 2006-09-07 | Bayer Healthcare Ag | Arzneiformen mit verbesserten pharmakokinetischen Eigenschaften |
| CA2601250C (en) * | 2005-03-16 | 2014-10-28 | Nycomed Gmbh | Taste masked dosage form containing roflumilast |
| US20060276501A1 (en) * | 2005-05-25 | 2006-12-07 | Transoral Pharmaceuticals, Inc. | Solid compositions for treating middle-of-the-night insomnia |
| US20070225322A1 (en) * | 2005-05-25 | 2007-09-27 | Transoral Pharmaceuticals, Inc. | Compositions and methods for treating middle-of-the night insomnia |
| US20070287740A1 (en) * | 2005-05-25 | 2007-12-13 | Transcept Pharmaceuticals, Inc. | Compositions and methods of treating middle-of-the night insomnia |
| US7834201B2 (en) | 2005-06-22 | 2010-11-16 | H. Lundbeck A/S | Crystalline base of escitalopram and orodispersible tablets comprising escitalopram base |
| KR101011278B1 (ko) * | 2005-07-22 | 2011-01-28 | 미쓰비시 타나베 파마 코퍼레이션 | 구강내 속붕괴성 정제 |
| AU2006299232A1 (en) * | 2005-09-29 | 2007-04-12 | Bayer Schering Pharma Aktiengesellschaft | PDE inhibitors and combinations thereof for the treatment of urological disorders |
| US8497258B2 (en) | 2005-11-12 | 2013-07-30 | The Regents Of The University Of California | Viscous budesonide for the treatment of inflammatory diseases of the gastrointestinal tract |
| US20080305168A1 (en) * | 2006-02-10 | 2008-12-11 | Kyo-Tae Moon | In-Situ Melting and Gelling Tablet Composition For Oral Care |
| US20080069891A1 (en) * | 2006-09-15 | 2008-03-20 | Cima Labs, Inc. | Abuse resistant drug formulation |
| US8765178B2 (en) | 2006-07-19 | 2014-07-01 | Watson Laboratories, Inc. | Controlled release formulations and associated methods |
| WO2008079343A2 (en) * | 2006-12-21 | 2008-07-03 | Mallinckrodt Inc. | Composition of and method for preparing orally disintegrating tablets containing a high dose of pharmaceutically active ingredients |
| JP5366558B2 (ja) * | 2006-12-28 | 2013-12-11 | 武田薬品工業株式会社 | 口腔内崩壊性固形製剤 |
| WO2008151734A1 (en) * | 2007-06-13 | 2008-12-18 | Bayer Schering Pharma Aktiengesellschaft | Pde inhibitors for the treatment of hearing impairment |
| AU2009225869B2 (en) * | 2008-03-18 | 2014-01-23 | Merck Sharp & Dohme Llc | Substituted 4-hydroxypyrimidine-5-carboxamides |
| WO2010106936A1 (ja) * | 2009-03-16 | 2010-09-23 | ニプロ株式会社 | 口腔内崩壊錠 |
| JP2012520731A (ja) * | 2009-03-20 | 2012-09-10 | インキューブ ラブズ, エルエルシー | 固形薬物送達装置、処方物および使用法 |
| CA2765033C (en) | 2009-06-12 | 2020-07-14 | Meritage Pharma, Inc. | Methods for treating gastrointestinal disorders |
| WO2012005359A1 (ja) | 2010-07-09 | 2012-01-12 | 帝人ファーマ株式会社 | 口腔内崩壊錠剤 |
| US9011912B2 (en) | 2010-10-07 | 2015-04-21 | Abon Pharmaceuticals, Llc | Extended-release oral dosage forms for poorly soluble amine drugs |
| JP6245786B2 (ja) * | 2011-10-17 | 2017-12-13 | 大同化成工業株式会社 | 医薬用結合剤及び該結合剤を用いた製剤 |
| ES2895918T3 (es) | 2012-02-21 | 2022-02-23 | Towa Pharmaceutical Europe S L | Composiciones farmacéuticas orales de dabigatrán etexilato |
| JP5292520B1 (ja) * | 2013-02-13 | 2013-09-18 | アピ株式会社 | ローヤルゼリー含有錠剤及びその製造方法 |
| EP3410861B1 (en) * | 2016-02-05 | 2020-11-11 | Tiense Suikerraffinaderij N.V. | Pearl sugar; process for preparing pearl sugar |
| CN117529324A (zh) * | 2021-06-11 | 2024-02-06 | 奥夫艾治疗公司 | 稳定的阿匹莫德组合物和其用途 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH10298062A (ja) * | 1997-04-24 | 1998-11-10 | Pfizer Pharmaceut Co Ltd | 口腔内速溶型錠剤 |
| WO1999059544A2 (en) * | 1998-05-18 | 1999-11-25 | Takeda Chemical Industries, Ltd. | Orally disintegrable tablets |
| WO2000006126A1 (en) * | 1998-07-28 | 2000-02-10 | Takeda Chemical Industries, Ltd. | Rapidly disintegrable solid preparation |
| WO2000024383A1 (en) * | 1998-10-23 | 2000-05-04 | Pfizer Research And Development Company, N.V./S.A. | Controlled-release pharmaceutical formulations containing a cgmp pde-5 inhibitor |
| WO2000024379A1 (fr) * | 1998-10-26 | 2000-05-04 | Tanabe Seiyaku Co., Ltd. | Procede de production de fines particules spheriques contenant un medicament |
| CN1257422A (zh) * | 1997-05-27 | 2000-06-21 | 武田药品工业株式会社 | 固体药物制剂 |
| WO2000057857A1 (en) * | 1999-03-25 | 2000-10-05 | Yuhan Corporation | Rapidly disintegrable tablet for oral administration |
| US6149938A (en) * | 1997-07-25 | 2000-11-21 | Elan Pharma International Limited | Process for the preparation of a granulate suitable to the preparation of rapidly disintegrable mouth-soluble tablets and compositions obtained thereby |
Family Cites Families (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS4820103B1 (zh) | 1968-05-06 | 1973-06-19 | ||
| DE3822885C2 (de) * | 1987-07-06 | 1994-06-01 | Asahi Optical Co Ltd | Optisches Kabel und Verfahren zu dessen Herstellung |
| DE3827214A1 (de) * | 1988-08-11 | 1990-02-15 | Roehm Gmbh | Retardierte arzneiform und verfahren zu ihrer herstellung |
| JP3069458B2 (ja) * | 1992-01-29 | 2000-07-24 | 武田薬品工業株式会社 | 口腔内崩壊型錠剤およびその製造法 |
| US5254355A (en) * | 1992-05-29 | 1993-10-19 | Kraft General Foods, Inc. | Process for beverage tablets and products therefrom |
| GB9311920D0 (en) | 1993-06-09 | 1993-07-28 | Pfizer Ltd | Therapeutic agents |
| GB9514465D0 (en) * | 1995-07-14 | 1995-09-13 | Glaxo Lab Sa | Chemical compounds |
| JP3187657B2 (ja) | 1994-07-08 | 2001-07-11 | 株式会社三共製作所 | 錠剤製造方法およびその装置 |
| JP3067125B2 (ja) * | 1994-11-22 | 2000-07-17 | 田辺製薬株式会社 | 易服用性の有核錠型製剤 |
| GB9423911D0 (en) | 1994-11-26 | 1995-01-11 | Pfizer Ltd | Therapeutic agents |
| DE19501480A1 (de) * | 1995-01-19 | 1996-07-25 | Bayer Ag | 9-substituierte 2-(2-n-Alkoxyphenyl)-purin-6-one |
| DE19501482A1 (de) * | 1995-01-19 | 1996-07-25 | Bayer Ag | 2,9-disubstituierte Purin-6-one |
| DE19501481A1 (de) | 1995-01-19 | 1996-07-25 | Bayer Ag | 2,8-Disubstituierte Chinazolinone |
| EP0820441B1 (en) * | 1995-04-10 | 2002-06-26 | Fujisawa Pharmaceutical Co., Ltd. | INDOLE DERIVATIVES AS cGMP-PDE INHIBITORS |
| US6080782A (en) * | 1995-05-18 | 2000-06-27 | Byk Gulden Lomberg Chemische Fabrik Gmbh | Cyclohexyl dihydrobenzofuranes |
| PT828728E (pt) * | 1995-05-18 | 2003-06-30 | Altana Pharma Ag | Di-hidrobenzofuranos de fenilo |
| GB9514464D0 (en) * | 1995-07-14 | 1995-09-13 | Glaxo Lab Sa | Medicaments |
| GB9514473D0 (en) * | 1995-07-14 | 1995-09-13 | Glaxo Lab Sa | Chemical compounds |
| JPH0948726A (ja) * | 1995-08-07 | 1997-02-18 | Tanabe Seiyaku Co Ltd | 口腔内速崩壊性製剤およびその製法 |
| DE19541264A1 (de) * | 1995-11-06 | 1997-05-07 | Bayer Ag | Purin-6-on-derivate |
| SI0882021T1 (en) * | 1996-01-31 | 2003-10-31 | Altana Pharma Ag | New phenanthridines |
| US6127378A (en) * | 1996-03-26 | 2000-10-03 | Byk Gulden Lomberg Chemische Fabrik Gmbh | Phenanthridines substituted in the 6 position |
| US6087311A (en) * | 1996-12-06 | 2000-07-11 | The Proctor & Gamble Company | Coated detergent tablet |
| AU6230098A (en) | 1997-02-27 | 1998-09-18 | Tanabe Seiyaku Co., Ltd. | Isoquinolinone derivatives, process for preparing the same, and their use as phosphodiesterase inhibitors |
| JP3296412B2 (ja) | 1997-04-25 | 2002-07-02 | 田辺製薬株式会社 | 成型製剤及びその製法 |
| JP4939680B2 (ja) * | 1997-05-27 | 2012-05-30 | 武田薬品工業株式会社 | 固形製剤 |
| JP3591801B2 (ja) | 1997-06-19 | 2004-11-24 | 田辺製薬株式会社 | 口腔内速崩壊性製剤の製法 |
| US6037346A (en) * | 1997-10-28 | 2000-03-14 | Vivus, Inc. | Local administration of phosphodiesterase inhibitors for the treatment of erectile dysfunction |
| JP2000095759A (ja) | 1998-07-21 | 2000-04-04 | Takeda Chem Ind Ltd | 三環性化合物、その製造法および剤 |
| JP2000072675A (ja) | 1998-08-26 | 2000-03-07 | Tanabe Seiyaku Co Ltd | 医薬組成物 |
| JP2000072751A (ja) | 1998-08-26 | 2000-03-07 | Tanabe Seiyaku Co Ltd | イソキノリノン誘導体 |
| GB9823103D0 (en) | 1998-10-23 | 1998-12-16 | Pfizer Ltd | Pharmaceutically active compounds |
| GB9823101D0 (en) | 1998-10-23 | 1998-12-16 | Pfizer Ltd | Pharmaceutically active compounds |
| MY123528A (en) | 1999-09-16 | 2006-05-31 | Mitsubihsi Tanabe Pharma Corp | Aromatic nitrogen-containing 6-membered cyclic compounds. |
-
2002
- 2002-02-12 CA CA2437754A patent/CA2437754C/en not_active Expired - Fee Related
- 2002-02-12 AU AU2002230217A patent/AU2002230217B2/en not_active Ceased
- 2002-02-12 WO PCT/JP2002/001140 patent/WO2002064119A1/ja active IP Right Grant
- 2002-02-12 AT AT02711460T patent/ATE472319T1/de not_active IP Right Cessation
- 2002-02-12 EP EP02711460A patent/EP1366760B1/en not_active Expired - Lifetime
- 2002-02-12 MX MXPA03007283A patent/MXPA03007283A/es active IP Right Grant
- 2002-02-12 US US10/468,208 patent/US7927623B2/en not_active Expired - Fee Related
- 2002-02-12 NZ NZ527585A patent/NZ527585A/xx not_active IP Right Cessation
- 2002-02-12 DE DE60236850T patent/DE60236850D1/de not_active Expired - Lifetime
- 2002-02-12 CN CNB028050703A patent/CN100372536C/zh not_active Expired - Fee Related
- 2002-02-12 KR KR1020037010747A patent/KR100750554B1/ko not_active IP Right Cessation
- 2002-02-12 JP JP2002033547A patent/JP4179784B2/ja not_active Expired - Fee Related
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH10298062A (ja) * | 1997-04-24 | 1998-11-10 | Pfizer Pharmaceut Co Ltd | 口腔内速溶型錠剤 |
| CN1257422A (zh) * | 1997-05-27 | 2000-06-21 | 武田药品工业株式会社 | 固体药物制剂 |
| US6149938A (en) * | 1997-07-25 | 2000-11-21 | Elan Pharma International Limited | Process for the preparation of a granulate suitable to the preparation of rapidly disintegrable mouth-soluble tablets and compositions obtained thereby |
| WO1999059544A2 (en) * | 1998-05-18 | 1999-11-25 | Takeda Chemical Industries, Ltd. | Orally disintegrable tablets |
| WO2000006126A1 (en) * | 1998-07-28 | 2000-02-10 | Takeda Chemical Industries, Ltd. | Rapidly disintegrable solid preparation |
| WO2000024383A1 (en) * | 1998-10-23 | 2000-05-04 | Pfizer Research And Development Company, N.V./S.A. | Controlled-release pharmaceutical formulations containing a cgmp pde-5 inhibitor |
| WO2000024379A1 (fr) * | 1998-10-26 | 2000-05-04 | Tanabe Seiyaku Co., Ltd. | Procede de production de fines particules spheriques contenant un medicament |
| WO2000057857A1 (en) * | 1999-03-25 | 2000-10-05 | Yuhan Corporation | Rapidly disintegrable tablet for oral administration |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2002230217B2 (en) | 2005-02-17 |
| EP1366760A1 (en) | 2003-12-03 |
| US7927623B2 (en) | 2011-04-19 |
| MXPA03007283A (es) | 2003-12-04 |
| CN1527701A (zh) | 2004-09-08 |
| KR100750554B1 (ko) | 2007-08-20 |
| DE60236850D1 (de) | 2010-08-12 |
| WO2002064119A1 (fr) | 2002-08-22 |
| CA2437754A1 (en) | 2002-08-22 |
| EP1366760B1 (en) | 2010-06-30 |
| ATE472319T1 (de) | 2010-07-15 |
| EP1366760A4 (en) | 2005-09-21 |
| JP2002316923A (ja) | 2002-10-31 |
| KR20030086262A (ko) | 2003-11-07 |
| NZ527585A (en) | 2005-04-29 |
| CA2437754C (en) | 2010-05-18 |
| US20040109890A1 (en) | 2004-06-10 |
| JP4179784B2 (ja) | 2008-11-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN100372536C (zh) | 在口腔中迅速崩解的片剂 | |
| KR100534504B1 (ko) | 속붕괴성 의약 조성물 | |
| JP4084309B2 (ja) | 単一の結晶形を含有する固形製剤 | |
| AU2006271314B2 (en) | Gastroretentive formulations and manufacturing process thereof | |
| CA2426263C (en) | Nateglinide-containing preparation | |
| JPH11349483A (ja) | 医薬配合物 | |
| CN101227894A (zh) | 口腔速崩片 | |
| EP1316316B1 (en) | Preparations for oral administration | |
| JP3518601B2 (ja) | エバスタイムまたはその類似体に基づく医薬組成物 | |
| JPWO2002032403A1 (ja) | 口腔内速崩壊性医薬組成物およびその製造方法 | |
| KR20150015500A (ko) | 엔테카비어의 약제학적 조성물 및 제조 방법 | |
| JP2010202579A (ja) | アカルボースを含有する口腔内崩壊剤 | |
| JP2020169143A (ja) | アジルサルタンを含有する錠剤 | |
| JP2000191518A (ja) | 溶解性の改善された口腔内速崩壊性錠剤 | |
| JP2003221326A (ja) | 分岐鎖アミノ酸を含有するチュアブル剤 | |
| JP2000516601A (ja) | 水溶性化合物及びセルロースを含有する粒状物 | |
| WO1989006959A1 (en) | Pharmaceutical composition having improved releasability | |
| JP3470096B2 (ja) | ニルバジピン含有易溶性固形製剤およびその製造法 | |
| WO2023238929A1 (ja) | ピミテスピブを含有する医薬組成物 | |
| JP3815301B2 (ja) | 成型製剤及びその製法 | |
| WO2001085134A1 (en) | Pharmaceutical solid compositions and process for the production of mouth dissolving tablets | |
| KR910004481B1 (ko) | 용출성이 개량된 제제 조성물 | |
| JP2022023704A (ja) | プラスグレルを有効成分とする錠剤及び口腔内崩壊錠の製造方法、並びに医薬製剤 | |
| JP2003146878A (ja) | ニルバジピン含有易溶性固形製剤およびその製造法 | |
| JP2004143141A (ja) | ヨウ化イソプロパミド含有製剤 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C17 | Cessation of patent right | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20080305 Termination date: 20140212 |