WO2023012579A1 - リチウムイオン電池 - Google Patents
リチウムイオン電池 Download PDFInfo
- Publication number
- WO2023012579A1 WO2023012579A1 PCT/IB2022/056865 IB2022056865W WO2023012579A1 WO 2023012579 A1 WO2023012579 A1 WO 2023012579A1 IB 2022056865 W IB2022056865 W IB 2022056865W WO 2023012579 A1 WO2023012579 A1 WO 2023012579A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- positive electrode
- active material
- electrode active
- lithium
- crystal structure
- Prior art date
Links
- HBBGRARXTFLTSG-UHFFFAOYSA-N Lithium ion Chemical compound [Li+] HBBGRARXTFLTSG-UHFFFAOYSA-N 0.000 title claims abstract description 120
- 229910001416 lithium ion Inorganic materials 0.000 title claims abstract description 120
- 239000007774 positive electrode material Substances 0.000 claims abstract description 406
- 238000007600 charging Methods 0.000 claims abstract description 196
- 239000003792 electrolyte Substances 0.000 claims abstract description 74
- 238000007599 discharging Methods 0.000 claims abstract description 72
- 239000007773 negative electrode material Substances 0.000 claims abstract description 26
- 239000003575 carbonaceous material Substances 0.000 claims abstract description 12
- 239000013078 crystal Substances 0.000 claims description 292
- 229910052744 lithium Inorganic materials 0.000 claims description 176
- 235000002639 sodium chloride Nutrition 0.000 claims description 98
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 claims description 97
- 239000011780 sodium chloride Substances 0.000 claims description 96
- 229910018871 CoO 2 Inorganic materials 0.000 claims description 67
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 45
- 229910000625 lithium cobalt oxide Inorganic materials 0.000 claims description 42
- BFZPBUKRYWOWDV-UHFFFAOYSA-N lithium;oxido(oxo)cobalt Chemical compound [Li+].[O-][Co]=O BFZPBUKRYWOWDV-UHFFFAOYSA-N 0.000 claims description 42
- KMTRUDSVKNLOMY-UHFFFAOYSA-N Ethylene carbonate Chemical compound O=C1OCCO1 KMTRUDSVKNLOMY-UHFFFAOYSA-N 0.000 claims description 27
- IEJIGPNLZYLLBP-UHFFFAOYSA-N dimethyl carbonate Chemical compound COC(=O)OC IEJIGPNLZYLLBP-UHFFFAOYSA-N 0.000 claims description 21
- JBTWLSYIZRCDFO-UHFFFAOYSA-N ethyl methyl carbonate Chemical compound CCOC(=O)OC JBTWLSYIZRCDFO-UHFFFAOYSA-N 0.000 claims description 21
- 229910002804 graphite Inorganic materials 0.000 claims description 20
- 239000010439 graphite Substances 0.000 claims description 20
- 238000012360 testing method Methods 0.000 claims description 14
- 238000010280 constant potential charging Methods 0.000 claims description 13
- 150000002500 ions Chemical class 0.000 claims description 8
- 229910052799 carbon Inorganic materials 0.000 claims description 5
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 3
- CXHHBNMLPJOKQD-UHFFFAOYSA-M methyl carbonate Chemical compound COC([O-])=O CXHHBNMLPJOKQD-UHFFFAOYSA-M 0.000 claims description 2
- 230000008014 freezing Effects 0.000 abstract description 30
- 238000007710 freezing Methods 0.000 abstract description 30
- 239000000654 additive Substances 0.000 description 180
- 230000000996 additive effect Effects 0.000 description 180
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 163
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 159
- 239000011777 magnesium Substances 0.000 description 120
- 239000002344 surface layer Substances 0.000 description 112
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 105
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 102
- 229910052749 magnesium Inorganic materials 0.000 description 101
- 238000010438 heat treatment Methods 0.000 description 89
- 239000000463 material Substances 0.000 description 83
- 239000010941 cobalt Substances 0.000 description 77
- 229910017052 cobalt Inorganic materials 0.000 description 77
- 229910052759 nickel Inorganic materials 0.000 description 77
- 229910052731 fluorine Inorganic materials 0.000 description 75
- 239000010410 layer Substances 0.000 description 74
- 239000011737 fluorine Substances 0.000 description 73
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 71
- 238000003860 storage Methods 0.000 description 70
- 229910052782 aluminium Inorganic materials 0.000 description 68
- PQXKHYXIUOZZFA-UHFFFAOYSA-M lithium fluoride Chemical compound [Li+].[F-] PQXKHYXIUOZZFA-UHFFFAOYSA-M 0.000 description 68
- 239000000523 sample Substances 0.000 description 67
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 63
- 238000000034 method Methods 0.000 description 50
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 49
- 238000004519 manufacturing process Methods 0.000 description 49
- 239000001301 oxygen Substances 0.000 description 49
- 229910052760 oxygen Inorganic materials 0.000 description 49
- 239000000203 mixture Substances 0.000 description 48
- 230000006870 function Effects 0.000 description 42
- 238000002441 X-ray diffraction Methods 0.000 description 38
- 239000002131 composite material Substances 0.000 description 38
- 239000008151 electrolyte solution Substances 0.000 description 38
- 230000008859 change Effects 0.000 description 36
- 239000010408 film Substances 0.000 description 34
- 238000009826 distribution Methods 0.000 description 33
- 229910012851 LiCoO 2 Inorganic materials 0.000 description 32
- 238000004458 analytical method Methods 0.000 description 32
- 238000005259 measurement Methods 0.000 description 32
- 239000011230 binding agent Substances 0.000 description 30
- 238000010586 diagram Methods 0.000 description 28
- 229910052723 transition metal Inorganic materials 0.000 description 28
- 150000003624 transition metals Chemical class 0.000 description 28
- 239000011149 active material Substances 0.000 description 27
- 230000000694 effects Effects 0.000 description 26
- 239000012298 atmosphere Substances 0.000 description 24
- 238000010277 constant-current charging Methods 0.000 description 24
- -1 LiSCN Inorganic materials 0.000 description 22
- 239000002245 particle Substances 0.000 description 22
- 239000004065 semiconductor Substances 0.000 description 22
- 238000002156 mixing Methods 0.000 description 21
- 230000007547 defect Effects 0.000 description 20
- 102100027368 Histone H1.3 Human genes 0.000 description 19
- 101001009450 Homo sapiens Histone H1.3 Proteins 0.000 description 19
- 238000004833 X-ray photoelectron spectroscopy Methods 0.000 description 19
- 238000006243 chemical reaction Methods 0.000 description 19
- 229910052751 metal Inorganic materials 0.000 description 18
- 230000002829 reductive effect Effects 0.000 description 18
- 150000001450 anions Chemical class 0.000 description 17
- 238000002149 energy-dispersive X-ray emission spectroscopy Methods 0.000 description 17
- 239000002184 metal Substances 0.000 description 17
- 230000005540 biological transmission Effects 0.000 description 16
- 239000006229 carbon black Substances 0.000 description 16
- 238000010894 electron beam technology Methods 0.000 description 16
- 230000001965 increasing effect Effects 0.000 description 16
- 150000001768 cations Chemical class 0.000 description 15
- 150000001875 compounds Chemical class 0.000 description 15
- CPLXHLVBOLITMK-UHFFFAOYSA-N Magnesium oxide Chemical compound [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 14
- 125000004429 atom Chemical group 0.000 description 14
- 230000007423 decrease Effects 0.000 description 14
- 238000009792 diffusion process Methods 0.000 description 14
- 229910021389 graphene Inorganic materials 0.000 description 14
- 239000003960 organic solvent Substances 0.000 description 14
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 13
- PWHULOQIROXLJO-UHFFFAOYSA-N Manganese Chemical compound [Mn] PWHULOQIROXLJO-UHFFFAOYSA-N 0.000 description 13
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 13
- 230000006866 deterioration Effects 0.000 description 13
- 229910052748 manganese Inorganic materials 0.000 description 13
- 239000011572 manganese Substances 0.000 description 13
- 229910052698 phosphorus Inorganic materials 0.000 description 13
- 239000011574 phosphorus Substances 0.000 description 13
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 12
- 239000004020 conductor Substances 0.000 description 12
- 238000001514 detection method Methods 0.000 description 12
- 239000012535 impurity Substances 0.000 description 12
- 238000005211 surface analysis Methods 0.000 description 12
- 239000010936 titanium Substances 0.000 description 12
- 229910052719 titanium Inorganic materials 0.000 description 12
- 238000000354 decomposition reaction Methods 0.000 description 11
- 230000001976 improved effect Effects 0.000 description 11
- TWNQGVIAIRXVLR-UHFFFAOYSA-N oxo(oxoalumanyloxy)alumane Chemical compound O=[Al]O[Al]=O TWNQGVIAIRXVLR-UHFFFAOYSA-N 0.000 description 11
- 239000012071 phase Substances 0.000 description 11
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 10
- 239000002033 PVDF binder Substances 0.000 description 10
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 10
- 238000000576 coating method Methods 0.000 description 10
- 238000011156 evaluation Methods 0.000 description 10
- ORUIBWPALBXDOA-UHFFFAOYSA-L magnesium fluoride Chemical compound [F-].[F-].[Mg+2] ORUIBWPALBXDOA-UHFFFAOYSA-L 0.000 description 10
- 229910001635 magnesium fluoride Inorganic materials 0.000 description 10
- 238000002844 melting Methods 0.000 description 10
- 230000008018 melting Effects 0.000 description 10
- 229920002981 polyvinylidene fluoride Polymers 0.000 description 10
- 238000000851 scanning transmission electron micrograph Methods 0.000 description 10
- 229910052710 silicon Inorganic materials 0.000 description 10
- 239000010703 silicon Substances 0.000 description 10
- LIVNPJMFVYWSIS-UHFFFAOYSA-N silicon monoxide Chemical compound [Si-]#[O+] LIVNPJMFVYWSIS-UHFFFAOYSA-N 0.000 description 10
- 239000002002 slurry Substances 0.000 description 10
- 238000004364 calculation method Methods 0.000 description 9
- 239000011248 coating agent Substances 0.000 description 9
- 230000007797 corrosion Effects 0.000 description 9
- 238000005260 corrosion Methods 0.000 description 9
- 238000006073 displacement reaction Methods 0.000 description 9
- 238000004453 electron probe microanalysis Methods 0.000 description 9
- 238000000605 extraction Methods 0.000 description 9
- 238000001095 inductively coupled plasma mass spectrometry Methods 0.000 description 9
- 230000008569 process Effects 0.000 description 9
- 239000002904 solvent Substances 0.000 description 9
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 8
- 239000004743 Polypropylene Substances 0.000 description 8
- 238000003917 TEM image Methods 0.000 description 8
- 230000032683 aging Effects 0.000 description 8
- 239000000956 alloy Substances 0.000 description 8
- 239000000395 magnesium oxide Substances 0.000 description 8
- RVTZCBVAJQQJTK-UHFFFAOYSA-N oxygen(2-);zirconium(4+) Chemical compound [O-2].[O-2].[Zr+4] RVTZCBVAJQQJTK-UHFFFAOYSA-N 0.000 description 8
- 229920001155 polypropylene Polymers 0.000 description 8
- 239000011701 zinc Substances 0.000 description 8
- 229910001928 zirconium oxide Inorganic materials 0.000 description 8
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 7
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 7
- 229920000049 Carbon (fiber) Polymers 0.000 description 7
- 230000005536 Jahn Teller effect Effects 0.000 description 7
- 229910045601 alloy Inorganic materials 0.000 description 7
- 229910052796 boron Inorganic materials 0.000 description 7
- 239000004917 carbon fiber Substances 0.000 description 7
- 229920002678 cellulose Chemical class 0.000 description 7
- 239000001913 cellulose Chemical class 0.000 description 7
- 238000001816 cooling Methods 0.000 description 7
- 239000011888 foil Substances 0.000 description 7
- 238000001036 glow-discharge mass spectrometry Methods 0.000 description 7
- 238000001878 scanning electron micrograph Methods 0.000 description 7
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical compound [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 description 6
- 239000006230 acetylene black Substances 0.000 description 6
- 230000002411 adverse Effects 0.000 description 6
- 229920003235 aromatic polyamide Polymers 0.000 description 6
- 230000004888 barrier function Effects 0.000 description 6
- 230000015572 biosynthetic process Effects 0.000 description 6
- 238000004140 cleaning Methods 0.000 description 6
- 238000004891 communication Methods 0.000 description 6
- 230000000052 comparative effect Effects 0.000 description 6
- 238000003795 desorption Methods 0.000 description 6
- 238000010828 elution Methods 0.000 description 6
- 238000003780 insertion Methods 0.000 description 6
- 230000037431 insertion Effects 0.000 description 6
- 238000009830 intercalation Methods 0.000 description 6
- 230000002687 intercalation Effects 0.000 description 6
- 229920002647 polyamide Polymers 0.000 description 6
- 229920005989 resin Polymers 0.000 description 6
- 239000011347 resin Substances 0.000 description 6
- 229910001220 stainless steel Inorganic materials 0.000 description 6
- 239000010935 stainless steel Substances 0.000 description 6
- 230000003746 surface roughness Effects 0.000 description 6
- 229920003169 water-soluble polymer Polymers 0.000 description 6
- 229910052726 zirconium Inorganic materials 0.000 description 6
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 5
- 238000004435 EPR spectroscopy Methods 0.000 description 5
- 239000004760 aramid Substances 0.000 description 5
- 229910021383 artificial graphite Inorganic materials 0.000 description 5
- 230000008901 benefit Effects 0.000 description 5
- 239000003990 capacitor Substances 0.000 description 5
- 239000011651 chromium Substances 0.000 description 5
- 239000010949 copper Substances 0.000 description 5
- 239000011267 electrode slurry Substances 0.000 description 5
- 230000004907 flux Effects 0.000 description 5
- 229910000040 hydrogen fluoride Inorganic materials 0.000 description 5
- 230000014759 maintenance of location Effects 0.000 description 5
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 description 5
- 230000007246 mechanism Effects 0.000 description 5
- 239000007769 metal material Substances 0.000 description 5
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 5
- 239000004570 mortar (masonry) Substances 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 238000010926 purge Methods 0.000 description 5
- 239000007787 solid Substances 0.000 description 5
- 230000035882 stress Effects 0.000 description 5
- 230000002195 synergetic effect Effects 0.000 description 5
- 229910052720 vanadium Inorganic materials 0.000 description 5
- GPPXJZIENCGNKB-UHFFFAOYSA-N vanadium Chemical compound [V]#[V] GPPXJZIENCGNKB-UHFFFAOYSA-N 0.000 description 5
- VAYTZRYEBVHVLE-UHFFFAOYSA-N 1,3-dioxol-2-one Chemical compound O=C1OC=CO1 VAYTZRYEBVHVLE-UHFFFAOYSA-N 0.000 description 4
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 4
- OIFBSDVPJOWBCH-UHFFFAOYSA-N Diethyl carbonate Chemical compound CCOC(=O)OCC OIFBSDVPJOWBCH-UHFFFAOYSA-N 0.000 description 4
- UQSXHKLRYXJYBZ-UHFFFAOYSA-N Iron oxide Chemical compound [Fe]=O UQSXHKLRYXJYBZ-UHFFFAOYSA-N 0.000 description 4
- 229910013870 LiPF 6 Inorganic materials 0.000 description 4
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 4
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical group [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 4
- 239000012300 argon atmosphere Substances 0.000 description 4
- 229910052790 beryllium Inorganic materials 0.000 description 4
- ATBAMAFKBVZNFJ-UHFFFAOYSA-N beryllium atom Chemical compound [Be] ATBAMAFKBVZNFJ-UHFFFAOYSA-N 0.000 description 4
- 235000013339 cereals Nutrition 0.000 description 4
- 229910052804 chromium Inorganic materials 0.000 description 4
- 229910052802 copper Inorganic materials 0.000 description 4
- 238000009831 deintercalation Methods 0.000 description 4
- 229920001971 elastomer Polymers 0.000 description 4
- 230000005684 electric field Effects 0.000 description 4
- 238000000921 elemental analysis Methods 0.000 description 4
- 238000000227 grinding Methods 0.000 description 4
- 229910052742 iron Inorganic materials 0.000 description 4
- 229910003002 lithium salt Inorganic materials 0.000 description 4
- 159000000002 lithium salts Chemical class 0.000 description 4
- 239000002931 mesocarbon microbead Substances 0.000 description 4
- 150000002739 metals Chemical class 0.000 description 4
- 229910021421 monocrystalline silicon Inorganic materials 0.000 description 4
- 229910000480 nickel oxide Inorganic materials 0.000 description 4
- 229910052758 niobium Inorganic materials 0.000 description 4
- 239000010955 niobium Substances 0.000 description 4
- GUCVJGMIXFAOAE-UHFFFAOYSA-N niobium atom Chemical compound [Nb] GUCVJGMIXFAOAE-UHFFFAOYSA-N 0.000 description 4
- 150000004767 nitrides Chemical class 0.000 description 4
- 230000002093 peripheral effect Effects 0.000 description 4
- 239000002994 raw material Substances 0.000 description 4
- 239000005060 rubber Substances 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- 229910052717 sulfur Inorganic materials 0.000 description 4
- 239000011593 sulfur Substances 0.000 description 4
- YTZKOQUCBOVLHL-UHFFFAOYSA-N tert-butylbenzene Chemical compound CC(C)(C)C1=CC=CC=C1 YTZKOQUCBOVLHL-UHFFFAOYSA-N 0.000 description 4
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 4
- 229910052725 zinc Inorganic materials 0.000 description 4
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 3
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 3
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 3
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 description 3
- 239000004677 Nylon Substances 0.000 description 3
- 239000004952 Polyamide Substances 0.000 description 3
- 239000004698 Polyethylene Substances 0.000 description 3
- 239000004372 Polyvinyl alcohol Substances 0.000 description 3
- 238000003991 Rietveld refinement Methods 0.000 description 3
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 3
- 230000005856 abnormality Effects 0.000 description 3
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 3
- 229910052785 arsenic Inorganic materials 0.000 description 3
- RQNWIZPPADIBDY-UHFFFAOYSA-N arsenic atom Chemical compound [As] RQNWIZPPADIBDY-UHFFFAOYSA-N 0.000 description 3
- 239000011324 bead Substances 0.000 description 3
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 3
- 229910052794 bromium Inorganic materials 0.000 description 3
- 239000001768 carboxy methyl cellulose Substances 0.000 description 3
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 3
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 3
- 239000006182 cathode active material Substances 0.000 description 3
- 229910010293 ceramic material Inorganic materials 0.000 description 3
- 239000006258 conductive agent Substances 0.000 description 3
- 230000008602 contraction Effects 0.000 description 3
- 238000012937 correction Methods 0.000 description 3
- 238000005336 cracking Methods 0.000 description 3
- 238000001035 drying Methods 0.000 description 3
- 239000000428 dust Substances 0.000 description 3
- 230000005611 electricity Effects 0.000 description 3
- 230000002349 favourable effect Effects 0.000 description 3
- 150000002222 fluorine compounds Chemical class 0.000 description 3
- 125000001153 fluoro group Chemical group F* 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- 229910052732 germanium Inorganic materials 0.000 description 3
- 150000004676 glycans Chemical class 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- 239000011810 insulating material Substances 0.000 description 3
- 239000012212 insulator Substances 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 239000011244 liquid electrolyte Substances 0.000 description 3
- 150000002642 lithium compounds Chemical class 0.000 description 3
- 229910044991 metal oxide Inorganic materials 0.000 description 3
- 150000004706 metal oxides Chemical class 0.000 description 3
- 229910052750 molybdenum Inorganic materials 0.000 description 3
- 239000011733 molybdenum Substances 0.000 description 3
- 229910021382 natural graphite Inorganic materials 0.000 description 3
- BFDHFSHZJLFAMC-UHFFFAOYSA-L nickel(ii) hydroxide Chemical compound [OH-].[OH-].[Ni+2] BFDHFSHZJLFAMC-UHFFFAOYSA-L 0.000 description 3
- ZKATWMILCYLAPD-UHFFFAOYSA-N niobium pentoxide Inorganic materials O=[Nb](=O)O[Nb](=O)=O ZKATWMILCYLAPD-UHFFFAOYSA-N 0.000 description 3
- 229920001778 nylon Polymers 0.000 description 3
- 125000004430 oxygen atom Chemical group O* 0.000 description 3
- 229920003229 poly(methyl methacrylate) Polymers 0.000 description 3
- 229920000573 polyethylene Polymers 0.000 description 3
- 239000004926 polymethyl methacrylate Substances 0.000 description 3
- 229920001282 polysaccharide Polymers 0.000 description 3
- 239000005017 polysaccharide Substances 0.000 description 3
- 229920002451 polyvinyl alcohol Polymers 0.000 description 3
- 230000002265 prevention Effects 0.000 description 3
- 238000012545 processing Methods 0.000 description 3
- 230000001681 protective effect Effects 0.000 description 3
- 238000010298 pulverizing process Methods 0.000 description 3
- 230000003014 reinforcing effect Effects 0.000 description 3
- 125000006850 spacer group Chemical group 0.000 description 3
- 230000006641 stabilisation Effects 0.000 description 3
- 238000011105 stabilization Methods 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 230000008093 supporting effect Effects 0.000 description 3
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 description 3
- 229910052721 tungsten Inorganic materials 0.000 description 3
- 239000010937 tungsten Substances 0.000 description 3
- 238000003466 welding Methods 0.000 description 3
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 2
- HEZMWWAKWCSUCB-PHDIDXHHSA-N (3R,4R)-3,4-dihydroxycyclohexa-1,5-diene-1-carboxylic acid Chemical compound O[C@@H]1C=CC(C(O)=O)=C[C@H]1O HEZMWWAKWCSUCB-PHDIDXHHSA-N 0.000 description 2
- SBLRHMKNNHXPHG-UHFFFAOYSA-N 4-fluoro-1,3-dioxolan-2-one Chemical compound FC1COC(=O)O1 SBLRHMKNNHXPHG-UHFFFAOYSA-N 0.000 description 2
- 229910000838 Al alloy Inorganic materials 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 2
- QPLDLSVMHZLSFG-UHFFFAOYSA-N Copper oxide Chemical compound [Cu]=O QPLDLSVMHZLSFG-UHFFFAOYSA-N 0.000 description 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- 239000001856 Ethyl cellulose Substances 0.000 description 2
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 2
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 2
- 229910002601 GaN Inorganic materials 0.000 description 2
- GYHNNYVSQQEPJS-UHFFFAOYSA-N Gallium Chemical compound [Ga] GYHNNYVSQQEPJS-UHFFFAOYSA-N 0.000 description 2
- JMASRVWKEDWRBT-UHFFFAOYSA-N Gallium nitride Chemical compound [Ga]#N JMASRVWKEDWRBT-UHFFFAOYSA-N 0.000 description 2
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 229910032387 LiCoO2 Inorganic materials 0.000 description 2
- 229910013290 LiNiO 2 Inorganic materials 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- 229910052779 Neodymium Inorganic materials 0.000 description 2
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 2
- 239000004793 Polystyrene Substances 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- 238000005275 alloying Methods 0.000 description 2
- 229910052787 antimony Inorganic materials 0.000 description 2
- 229910001632 barium fluoride Inorganic materials 0.000 description 2
- YKYOUMDCQGMQQO-UHFFFAOYSA-L cadmium dichloride Chemical class Cl[Cd]Cl YKYOUMDCQGMQQO-UHFFFAOYSA-L 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910002091 carbon monoxide Inorganic materials 0.000 description 2
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 2
- 239000000919 ceramic Substances 0.000 description 2
- 150000005678 chain carbonates Chemical class 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 229910001429 cobalt ion Inorganic materials 0.000 description 2
- 229910000428 cobalt oxide Inorganic materials 0.000 description 2
- XLJKHNWPARRRJB-UHFFFAOYSA-N cobalt(2+) Chemical compound [Co+2] XLJKHNWPARRRJB-UHFFFAOYSA-N 0.000 description 2
- IVMYJDGYRUAWML-UHFFFAOYSA-N cobalt(ii) oxide Chemical compound [Co]=O IVMYJDGYRUAWML-UHFFFAOYSA-N 0.000 description 2
- 239000012141 concentrate Substances 0.000 description 2
- 239000000470 constituent Substances 0.000 description 2
- 230000008878 coupling Effects 0.000 description 2
- 238000010168 coupling process Methods 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- 238000002788 crimping Methods 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 229920005994 diacetyl cellulose Polymers 0.000 description 2
- QDOXWKRWXJOMAK-UHFFFAOYSA-N dichromium trioxide Chemical compound O=[Cr]O[Cr]=O QDOXWKRWXJOMAK-UHFFFAOYSA-N 0.000 description 2
- 230000005672 electromagnetic field Effects 0.000 description 2
- 238000002524 electron diffraction data Methods 0.000 description 2
- 230000008030 elimination Effects 0.000 description 2
- 238000003379 elimination reaction Methods 0.000 description 2
- 229920001249 ethyl cellulose Polymers 0.000 description 2
- 235000019325 ethyl cellulose Nutrition 0.000 description 2
- 239000000835 fiber Substances 0.000 description 2
- 239000000446 fuel Substances 0.000 description 2
- 229910052733 gallium Inorganic materials 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- GNPVGFCGXDBREM-UHFFFAOYSA-N germanium atom Chemical compound [Ge] GNPVGFCGXDBREM-UHFFFAOYSA-N 0.000 description 2
- 229910052735 hafnium Inorganic materials 0.000 description 2
- VBJZVLUMGGDVMO-UHFFFAOYSA-N hafnium atom Chemical compound [Hf] VBJZVLUMGGDVMO-UHFFFAOYSA-N 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 2
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 2
- 229910052738 indium Inorganic materials 0.000 description 2
- APFVFJFRJDLVQX-UHFFFAOYSA-N indium atom Chemical compound [In] APFVFJFRJDLVQX-UHFFFAOYSA-N 0.000 description 2
- 238000005304 joining Methods 0.000 description 2
- AMXOYNBUYSYVKV-UHFFFAOYSA-M lithium bromide Chemical compound [Li+].[Br-] AMXOYNBUYSYVKV-UHFFFAOYSA-M 0.000 description 2
- XGZVUEUWXADBQD-UHFFFAOYSA-L lithium carbonate Chemical compound [Li+].[Li+].[O-]C([O-])=O XGZVUEUWXADBQD-UHFFFAOYSA-L 0.000 description 2
- 229910052808 lithium carbonate Inorganic materials 0.000 description 2
- IIPYXGDZVMZOAP-UHFFFAOYSA-N lithium nitrate Chemical compound [Li+].[O-][N+]([O-])=O IIPYXGDZVMZOAP-UHFFFAOYSA-N 0.000 description 2
- INHCSSUBVCNVSK-UHFFFAOYSA-L lithium sulfate Chemical compound [Li+].[Li+].[O-]S([O-])(=O)=O INHCSSUBVCNVSK-UHFFFAOYSA-L 0.000 description 2
- 238000011068 loading method Methods 0.000 description 2
- AXZKOIWUVFPNLO-UHFFFAOYSA-N magnesium;oxygen(2-) Chemical compound [O-2].[Mg+2] AXZKOIWUVFPNLO-UHFFFAOYSA-N 0.000 description 2
- 229920000609 methyl cellulose Polymers 0.000 description 2
- 239000001923 methylcellulose Substances 0.000 description 2
- 235000010981 methylcellulose Nutrition 0.000 description 2
- QEFYFXOXNSNQGX-UHFFFAOYSA-N neodymium atom Chemical compound [Nd] QEFYFXOXNSNQGX-UHFFFAOYSA-N 0.000 description 2
- 238000001683 neutron diffraction Methods 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 239000011255 nonaqueous electrolyte Substances 0.000 description 2
- BTNXBLUGMAMSSH-UHFFFAOYSA-N octanedinitrile Chemical compound N#CCCCCCCC#N BTNXBLUGMAMSSH-UHFFFAOYSA-N 0.000 description 2
- 230000033116 oxidation-reduction process Effects 0.000 description 2
- GNRSAWUEBMWBQH-UHFFFAOYSA-N oxonickel Chemical compound [Ni]=O GNRSAWUEBMWBQH-UHFFFAOYSA-N 0.000 description 2
- UJMWVICAENGCRF-UHFFFAOYSA-N oxygen difluoride Chemical compound FOF UJMWVICAENGCRF-UHFFFAOYSA-N 0.000 description 2
- 238000012856 packing Methods 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 230000000704 physical effect Effects 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 229920002239 polyacrylonitrile Polymers 0.000 description 2
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 2
- 239000004810 polytetrafluoroethylene Substances 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 239000004627 regenerated cellulose Substances 0.000 description 2
- 239000011435 rock Substances 0.000 description 2
- WOCIAKWEIIZHES-UHFFFAOYSA-N ruthenium(iv) oxide Chemical compound O=[Ru]=O WOCIAKWEIIZHES-UHFFFAOYSA-N 0.000 description 2
- 150000003839 salts Chemical class 0.000 description 2
- 238000007086 side reaction Methods 0.000 description 2
- 229910021332 silicide Inorganic materials 0.000 description 2
- FVBUAEGBCNSCDD-UHFFFAOYSA-N silicide(4-) Chemical compound [Si-4] FVBUAEGBCNSCDD-UHFFFAOYSA-N 0.000 description 2
- HBMJWWWQQXIZIP-UHFFFAOYSA-N silicon carbide Chemical compound [Si+]#[C-] HBMJWWWQQXIZIP-UHFFFAOYSA-N 0.000 description 2
- 229910010271 silicon carbide Inorganic materials 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 239000007784 solid electrolyte Substances 0.000 description 2
- 239000006104 solid solution Substances 0.000 description 2
- 238000001179 sorption measurement Methods 0.000 description 2
- 230000000087 stabilizing effect Effects 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 229920003048 styrene butadiene rubber Polymers 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 229910052715 tantalum Inorganic materials 0.000 description 2
- GUVRBAGPIYLISA-UHFFFAOYSA-N tantalum atom Chemical compound [Ta] GUVRBAGPIYLISA-UHFFFAOYSA-N 0.000 description 2
- RBYFNZOIUUXJQD-UHFFFAOYSA-J tetralithium oxalate Chemical compound [Li+].[Li+].[Li+].[Li+].[O-]C(=O)C([O-])=O.[O-]C(=O)C([O-])=O RBYFNZOIUUXJQD-UHFFFAOYSA-J 0.000 description 2
- 239000010409 thin film Substances 0.000 description 2
- XOLBLPGZBRYERU-UHFFFAOYSA-N tin dioxide Chemical compound O=[Sn]=O XOLBLPGZBRYERU-UHFFFAOYSA-N 0.000 description 2
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 2
- PFNQVRZLDWYSCW-UHFFFAOYSA-N (fluoren-9-ylideneamino) n-naphthalen-1-ylcarbamate Chemical compound C12=CC=CC=C2C2=CC=CC=C2C1=NOC(=O)NC1=CC=CC2=CC=CC=C12 PFNQVRZLDWYSCW-UHFFFAOYSA-N 0.000 description 1
- FSSPGSAQUIYDCN-UHFFFAOYSA-N 1,3-Propane sultone Chemical compound O=S1(=O)CCCO1 FSSPGSAQUIYDCN-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- WKBPZYKAUNRMKP-UHFFFAOYSA-N 1-[2-(2,4-dichlorophenyl)pentyl]1,2,4-triazole Chemical compound C=1C=C(Cl)C=C(Cl)C=1C(CCC)CN1C=NC=N1 WKBPZYKAUNRMKP-UHFFFAOYSA-N 0.000 description 1
- VSKJLJHPAFKHBX-UHFFFAOYSA-N 2-methylbuta-1,3-diene;styrene Chemical compound CC(=C)C=C.C=CC1=CC=CC=C1.C=CC1=CC=CC=C1 VSKJLJHPAFKHBX-UHFFFAOYSA-N 0.000 description 1
- 206010000117 Abnormal behaviour Diseases 0.000 description 1
- 102100031786 Adiponectin Human genes 0.000 description 1
- 229910017692 Ag3Sn Inorganic materials 0.000 description 1
- KLZUFWVZNOTSEM-UHFFFAOYSA-K Aluminium flouride Chemical compound F[Al](F)F KLZUFWVZNOTSEM-UHFFFAOYSA-K 0.000 description 1
- 229910000967 As alloy Inorganic materials 0.000 description 1
- 229910020187 CeF3 Inorganic materials 0.000 description 1
- 229910020186 CeF4 Inorganic materials 0.000 description 1
- 229910052684 Cerium Inorganic materials 0.000 description 1
- 229910018992 CoS0.89 Inorganic materials 0.000 description 1
- 229910018989 CoSb Inorganic materials 0.000 description 1
- 229910019050 CoSn2 Inorganic materials 0.000 description 1
- 229910021503 Cobalt(II) hydroxide Inorganic materials 0.000 description 1
- 229910021583 Cobalt(III) fluoride Inorganic materials 0.000 description 1
- 229910018471 Cu6Sn5 Inorganic materials 0.000 description 1
- 229910005391 FeSn2 Inorganic materials 0.000 description 1
- 235000015842 Hesperis Nutrition 0.000 description 1
- 101000775469 Homo sapiens Adiponectin Proteins 0.000 description 1
- 235000012633 Iberis amara Nutrition 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 229910011939 Li2.6 Co0.4 N Inorganic materials 0.000 description 1
- 229910010820 Li2B10Cl10 Inorganic materials 0.000 description 1
- 229910010903 Li2B12Cl12 Inorganic materials 0.000 description 1
- 229910002986 Li4Ti5O12 Inorganic materials 0.000 description 1
- 229910000552 LiCF3SO3 Inorganic materials 0.000 description 1
- 229910012820 LiCoO Inorganic materials 0.000 description 1
- 229910015643 LiMn 2 O 4 Inorganic materials 0.000 description 1
- 229910001290 LiPF6 Inorganic materials 0.000 description 1
- 229910018688 LixC6 Inorganic materials 0.000 description 1
- 229910001091 LixCoO2 Inorganic materials 0.000 description 1
- JLVVSXFLKOJNIY-UHFFFAOYSA-N Magnesium ion Chemical compound [Mg+2] JLVVSXFLKOJNIY-UHFFFAOYSA-N 0.000 description 1
- 229910021569 Manganese fluoride Inorganic materials 0.000 description 1
- ZSBXGIUJOOQZMP-JLNYLFASSA-N Matrine Chemical compound C1CC[C@H]2CN3C(=O)CCC[C@@H]3[C@@H]3[C@H]2N1CCC3 ZSBXGIUJOOQZMP-JLNYLFASSA-N 0.000 description 1
- 229910019688 Mg2Ge Inorganic materials 0.000 description 1
- 229910019752 Mg2Si Inorganic materials 0.000 description 1
- 229910019743 Mg2Sn Inorganic materials 0.000 description 1
- 229910016964 MnSb Inorganic materials 0.000 description 1
- 229910005099 Ni3Sn2 Inorganic materials 0.000 description 1
- VEQPNABPJHWNSG-UHFFFAOYSA-N Nickel(2+) Chemical compound [Ni+2] VEQPNABPJHWNSG-UHFFFAOYSA-N 0.000 description 1
- 229920000459 Nitrile rubber Polymers 0.000 description 1
- 239000000020 Nitrocellulose Substances 0.000 description 1
- 240000007594 Oryza sativa Species 0.000 description 1
- 235000007164 Oryza sativa Nutrition 0.000 description 1
- 229920002319 Poly(methyl acrylate) Polymers 0.000 description 1
- 239000005062 Polybutadiene Substances 0.000 description 1
- 239000004642 Polyimide Substances 0.000 description 1
- 229920002367 Polyisobutene Polymers 0.000 description 1
- 229910018320 SbSn Inorganic materials 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- BQCADISMDOOEFD-UHFFFAOYSA-N Silver Chemical compound [Ag] BQCADISMDOOEFD-UHFFFAOYSA-N 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- 229920002978 Vinylon Polymers 0.000 description 1
- 229910007379 Zn3N2 Inorganic materials 0.000 description 1
- RUFZJUYWZZUTJE-UHFFFAOYSA-J [F-].[F-].[F-].[F-].F.F.[Na+].[Al+3] Chemical compound [F-].[F-].[F-].[F-].F.F.[Na+].[Al+3] RUFZJUYWZZUTJE-UHFFFAOYSA-J 0.000 description 1
- JFBZPFYRPYOZCQ-UHFFFAOYSA-N [Li].[Al] Chemical compound [Li].[Al] JFBZPFYRPYOZCQ-UHFFFAOYSA-N 0.000 description 1
- FDLZQPXZHIFURF-UHFFFAOYSA-N [O-2].[Ti+4].[Li+] Chemical compound [O-2].[Ti+4].[Li+] FDLZQPXZHIFURF-UHFFFAOYSA-N 0.000 description 1
- 230000001133 acceleration Effects 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- BTGRAWJCKBQKAO-UHFFFAOYSA-N adiponitrile Chemical compound N#CCCCCC#N BTGRAWJCKBQKAO-UHFFFAOYSA-N 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- WATWJIUSRGPENY-UHFFFAOYSA-N antimony atom Chemical compound [Sb] WATWJIUSRGPENY-UHFFFAOYSA-N 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 238000003491 array Methods 0.000 description 1
- 230000002238 attenuated effect Effects 0.000 description 1
- OYLGJCQECKOTOL-UHFFFAOYSA-L barium fluoride Chemical compound [F-].[F-].[Ba+2] OYLGJCQECKOTOL-UHFFFAOYSA-L 0.000 description 1
- JRPBQTZRNDNNOP-UHFFFAOYSA-N barium titanate Chemical compound [Ba+2].[Ba+2].[O-][Ti]([O-])([O-])[O-] JRPBQTZRNDNNOP-UHFFFAOYSA-N 0.000 description 1
- 229910002113 barium titanate Inorganic materials 0.000 description 1
- 229910052797 bismuth Inorganic materials 0.000 description 1
- JCXGWMGPZLAOME-UHFFFAOYSA-N bismuth atom Chemical compound [Bi] JCXGWMGPZLAOME-UHFFFAOYSA-N 0.000 description 1
- 230000036772 blood pressure Effects 0.000 description 1
- 230000036760 body temperature Effects 0.000 description 1
- 229910052793 cadmium Inorganic materials 0.000 description 1
- BDOSMKKIYDKNTQ-UHFFFAOYSA-N cadmium atom Chemical compound [Cd] BDOSMKKIYDKNTQ-UHFFFAOYSA-N 0.000 description 1
- WUKWITHWXAAZEY-UHFFFAOYSA-L calcium difluoride Chemical compound [F-].[F-].[Ca+2] WUKWITHWXAAZEY-UHFFFAOYSA-L 0.000 description 1
- 239000002041 carbon nanotube Substances 0.000 description 1
- 229910021393 carbon nanotube Inorganic materials 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- ZMIGMASIKSOYAM-UHFFFAOYSA-N cerium Chemical group [Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce] ZMIGMASIKSOYAM-UHFFFAOYSA-N 0.000 description 1
- QCCDYNYSHILRDG-UHFFFAOYSA-K cerium(3+);trifluoride Chemical compound [F-].[F-].[F-].[Ce+3] QCCDYNYSHILRDG-UHFFFAOYSA-K 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 229910021563 chromium fluoride Inorganic materials 0.000 description 1
- 238000000975 co-precipitation Methods 0.000 description 1
- YCYBZKSMUPTWEE-UHFFFAOYSA-L cobalt(ii) fluoride Chemical compound F[Co]F YCYBZKSMUPTWEE-UHFFFAOYSA-L 0.000 description 1
- ASKVAEGIVYSGNY-UHFFFAOYSA-L cobalt(ii) hydroxide Chemical compound [OH-].[OH-].[Co+2] ASKVAEGIVYSGNY-UHFFFAOYSA-L 0.000 description 1
- 239000000571 coke Substances 0.000 description 1
- 238000011109 contamination Methods 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- BERDEBHAJNAUOM-UHFFFAOYSA-N copper(I) oxide Inorganic materials [Cu]O[Cu] BERDEBHAJNAUOM-UHFFFAOYSA-N 0.000 description 1
- 229910052955 covellite Inorganic materials 0.000 description 1
- KRFJLUBVMFXRPN-UHFFFAOYSA-N cuprous oxide Chemical compound [O-2].[Cu+].[Cu+] KRFJLUBVMFXRPN-UHFFFAOYSA-N 0.000 description 1
- 150000005676 cyclic carbonates Chemical class 0.000 description 1
- 238000013481 data capture Methods 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 238000000151 deposition Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 238000000113 differential scanning calorimetry Methods 0.000 description 1
- 238000002050 diffraction method Methods 0.000 description 1
- CTNMMTCXUUFYAP-UHFFFAOYSA-L difluoromanganese Chemical compound F[Mn]F CTNMMTCXUUFYAP-UHFFFAOYSA-L 0.000 description 1
- AJNVQOSZGJRYEI-UHFFFAOYSA-N digallium;oxygen(2-) Chemical compound [O-2].[O-2].[O-2].[Ga+3].[Ga+3] AJNVQOSZGJRYEI-UHFFFAOYSA-N 0.000 description 1
- SXWUDUINABFBMK-UHFFFAOYSA-L dilithium;fluoro-dioxido-oxo-$l^{5}-phosphane Chemical compound [Li+].[Li+].[O-]P([O-])(F)=O SXWUDUINABFBMK-UHFFFAOYSA-L 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- QXYJCZRRLLQGCR-UHFFFAOYSA-N dioxomolybdenum Chemical compound O=[Mo]=O QXYJCZRRLLQGCR-UHFFFAOYSA-N 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 238000007580 dry-mixing Methods 0.000 description 1
- 230000005674 electromagnetic induction Effects 0.000 description 1
- 238000002003 electron diffraction Methods 0.000 description 1
- 238000005430 electron energy loss spectroscopy Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 238000000407 epitaxy Methods 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 230000005496 eutectics Effects 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000004880 explosion Methods 0.000 description 1
- 230000005669 field effect Effects 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 229920001973 fluoroelastomer Polymers 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 239000006232 furnace black Substances 0.000 description 1
- 229910001195 gallium oxide Inorganic materials 0.000 description 1
- 238000002290 gas chromatography-mass spectrometry Methods 0.000 description 1
- 238000001879 gelation Methods 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000003365 glass fiber Substances 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- 229910021469 graphitizable carbon Inorganic materials 0.000 description 1
- 230000005484 gravity Effects 0.000 description 1
- 238000009499 grossing Methods 0.000 description 1
- 229910021385 hard carbon Inorganic materials 0.000 description 1
- 230000020169 heat generation Effects 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 125000004435 hydrogen atom Chemical class [H]* 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 238000001027 hydrothermal synthesis Methods 0.000 description 1
- 238000010191 image analysis Methods 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- WPYVAWXEWQSOGY-UHFFFAOYSA-N indium antimonide Chemical compound [Sb]#[In] WPYVAWXEWQSOGY-UHFFFAOYSA-N 0.000 description 1
- 229910052809 inorganic oxide Inorganic materials 0.000 description 1
- 238000009413 insulation Methods 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 238000010884 ion-beam technique Methods 0.000 description 1
- 229920000554 ionomer Polymers 0.000 description 1
- JEIPFZHSYJVQDO-UHFFFAOYSA-N iron(III) oxide Inorganic materials O=[Fe]O[Fe]=O JEIPFZHSYJVQDO-UHFFFAOYSA-N 0.000 description 1
- SHXXPRJOPFJRHA-UHFFFAOYSA-K iron(iii) fluoride Chemical compound F[Fe](F)F SHXXPRJOPFJRHA-UHFFFAOYSA-K 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 229910052746 lanthanum Inorganic materials 0.000 description 1
- FZLIPJUXYLNCLC-UHFFFAOYSA-N lanthanum atom Chemical group [La] FZLIPJUXYLNCLC-UHFFFAOYSA-N 0.000 description 1
- 238000007561 laser diffraction method Methods 0.000 description 1
- 229910001540 lithium hexafluoroarsenate(V) Inorganic materials 0.000 description 1
- HSZCZNFXUDYRKD-UHFFFAOYSA-M lithium iodide Inorganic materials [Li+].[I-] HSZCZNFXUDYRKD-UHFFFAOYSA-M 0.000 description 1
- IDBFBDSKYCUNPW-UHFFFAOYSA-N lithium nitride Chemical group [Li]N([Li])[Li] IDBFBDSKYCUNPW-UHFFFAOYSA-N 0.000 description 1
- MHCFAGZWMAWTNR-UHFFFAOYSA-M lithium perchlorate Chemical compound [Li+].[O-]Cl(=O)(=O)=O MHCFAGZWMAWTNR-UHFFFAOYSA-M 0.000 description 1
- 229910001486 lithium perchlorate Inorganic materials 0.000 description 1
- 229910001537 lithium tetrachloroaluminate Inorganic materials 0.000 description 1
- 229910001496 lithium tetrafluoroborate Inorganic materials 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 150000002681 magnesium compounds Chemical class 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 239000000347 magnesium hydroxide Substances 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- 229910001425 magnesium ion Inorganic materials 0.000 description 1
- 239000000696 magnetic material Substances 0.000 description 1
- 238000001646 magnetic resonance method Methods 0.000 description 1
- 238000013507 mapping Methods 0.000 description 1
- 238000004452 microanalysis Methods 0.000 description 1
- 229910052953 millerite Inorganic materials 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 238000000329 molecular dynamics simulation Methods 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 229910001453 nickel ion Inorganic materials 0.000 description 1
- JDRCAGKFDGHRNQ-UHFFFAOYSA-N nickel(3+) Chemical compound [Ni+3] JDRCAGKFDGHRNQ-UHFFFAOYSA-N 0.000 description 1
- DBJLJFTWODWSOF-UHFFFAOYSA-L nickel(ii) fluoride Chemical compound F[Ni]F DBJLJFTWODWSOF-UHFFFAOYSA-L 0.000 description 1
- URLJKFSTXLNXLG-UHFFFAOYSA-N niobium(5+);oxygen(2-) Chemical compound [O-2].[O-2].[O-2].[O-2].[O-2].[Nb+5].[Nb+5] URLJKFSTXLNXLG-UHFFFAOYSA-N 0.000 description 1
- 229920001220 nitrocellulos Polymers 0.000 description 1
- 229910021470 non-graphitizable carbon Inorganic materials 0.000 description 1
- 238000009828 non-uniform distribution Methods 0.000 description 1
- 239000004745 nonwoven fabric Substances 0.000 description 1
- 239000011368 organic material Substances 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 239000000123 paper Substances 0.000 description 1
- 239000013618 particulate matter Substances 0.000 description 1
- 238000002161 passivation Methods 0.000 description 1
- AOLPZAHRYHXPLR-UHFFFAOYSA-I pentafluoroniobium Chemical compound F[Nb](F)(F)(F)F AOLPZAHRYHXPLR-UHFFFAOYSA-I 0.000 description 1
- NFVUDQKTAWONMJ-UHFFFAOYSA-I pentafluorovanadium Chemical compound [F-].[F-].[F-].[F-].[F-].[V+5] NFVUDQKTAWONMJ-UHFFFAOYSA-I 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 125000004437 phosphorous atom Chemical group 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 229920001495 poly(sodium acrylate) polymer Polymers 0.000 description 1
- 229920002857 polybutadiene Polymers 0.000 description 1
- 229920000515 polycarbonate Polymers 0.000 description 1
- 239000004417 polycarbonate Substances 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920001225 polyester resin Polymers 0.000 description 1
- 229920000139 polyethylene terephthalate Polymers 0.000 description 1
- 239000005020 polyethylene terephthalate Substances 0.000 description 1
- 229920001721 polyimide Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 239000002861 polymer material Substances 0.000 description 1
- 229920000098 polyolefin Polymers 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 229920002635 polyurethane Polymers 0.000 description 1
- 239000004814 polyurethane Substances 0.000 description 1
- 229920002689 polyvinyl acetate Polymers 0.000 description 1
- 239000011118 polyvinyl acetate Substances 0.000 description 1
- 239000004800 polyvinyl chloride Substances 0.000 description 1
- 229920000915 polyvinyl chloride Polymers 0.000 description 1
- NROKBHXJSPEDAR-UHFFFAOYSA-M potassium fluoride Chemical compound [F-].[K+] NROKBHXJSPEDAR-UHFFFAOYSA-M 0.000 description 1
- 238000001144 powder X-ray diffraction data Methods 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 238000003825 pressing Methods 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 239000003223 protective agent Substances 0.000 description 1
- 238000004080 punching Methods 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000006479 redox reaction Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 230000011514 reflex Effects 0.000 description 1
- 235000009566 rice Nutrition 0.000 description 1
- 229910052706 scandium Inorganic materials 0.000 description 1
- SIXSYDAISGFNSX-UHFFFAOYSA-N scandium atom Chemical compound [Sc] SIXSYDAISGFNSX-UHFFFAOYSA-N 0.000 description 1
- 238000001350 scanning transmission electron microscopy Methods 0.000 description 1
- 238000000790 scattering method Methods 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 238000005204 segregation Methods 0.000 description 1
- SBIBMFFZSBJNJF-UHFFFAOYSA-N selenium;zinc Chemical compound [Se]=[Zn] SBIBMFFZSBJNJF-UHFFFAOYSA-N 0.000 description 1
- 238000007873 sieving Methods 0.000 description 1
- 229910052814 silicon oxide Inorganic materials 0.000 description 1
- 229910052709 silver Inorganic materials 0.000 description 1
- 239000004332 silver Substances 0.000 description 1
- 239000002356 single layer Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- PUZPDOWCWNUUKD-UHFFFAOYSA-M sodium fluoride Chemical compound [F-].[Na+] PUZPDOWCWNUUKD-UHFFFAOYSA-M 0.000 description 1
- NNMHYFLPFNGQFZ-UHFFFAOYSA-M sodium polyacrylate Chemical compound [Na+].[O-]C(=O)C=C NNMHYFLPFNGQFZ-UHFFFAOYSA-M 0.000 description 1
- 229910021384 soft carbon Inorganic materials 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 238000010532 solid phase synthesis reaction Methods 0.000 description 1
- 239000000243 solution Substances 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- IAHFWCOBPZCAEA-UHFFFAOYSA-N succinonitrile Chemical compound N#CCCC#N IAHFWCOBPZCAEA-UHFFFAOYSA-N 0.000 description 1
- 230000019635 sulfation Effects 0.000 description 1
- 238000005670 sulfation reaction Methods 0.000 description 1
- 150000004763 sulfides Chemical class 0.000 description 1
- SFZCNBIFKDRMGX-UHFFFAOYSA-N sulfur hexafluoride Chemical compound FS(F)(F)(F)(F)F SFZCNBIFKDRMGX-UHFFFAOYSA-N 0.000 description 1
- 229960000909 sulfur hexafluoride Drugs 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 229920002994 synthetic fiber Polymers 0.000 description 1
- 239000012209 synthetic fiber Substances 0.000 description 1
- 229920003002 synthetic resin Polymers 0.000 description 1
- 239000000057 synthetic resin Substances 0.000 description 1
- TXEYQDLBPFQVAA-UHFFFAOYSA-N tetrafluoromethane Chemical compound FC(F)(F)F TXEYQDLBPFQVAA-UHFFFAOYSA-N 0.000 description 1
- 229910052718 tin Inorganic materials 0.000 description 1
- QHGNHLZPVBIIPX-UHFFFAOYSA-N tin(II) oxide Inorganic materials [Sn]=O QHGNHLZPVBIIPX-UHFFFAOYSA-N 0.000 description 1
- XROWMBWRMNHXMF-UHFFFAOYSA-J titanium tetrafluoride Chemical compound [F-].[F-].[F-].[F-].[Ti+4] XROWMBWRMNHXMF-UHFFFAOYSA-J 0.000 description 1
- 238000012876 topography Methods 0.000 description 1
- 238000004919 topotaxy Methods 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 150000003623 transition metal compounds Chemical class 0.000 description 1
- 229910000314 transition metal oxide Inorganic materials 0.000 description 1
- FTBATIJJKIIOTP-UHFFFAOYSA-K trifluorochromium Chemical compound F[Cr](F)F FTBATIJJKIIOTP-UHFFFAOYSA-K 0.000 description 1
- BYMUNNMMXKDFEZ-UHFFFAOYSA-K trifluorolanthanum Chemical compound F[La](F)F BYMUNNMMXKDFEZ-UHFFFAOYSA-K 0.000 description 1
- 239000004034 viscosity adjusting agent Substances 0.000 description 1
- 239000011800 void material Substances 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 239000013585 weight reducing agent Substances 0.000 description 1
- 229910009207 xMxN Inorganic materials 0.000 description 1
- 229910052727 yttrium Inorganic materials 0.000 description 1
- VWQVUPCCIRVNHF-UHFFFAOYSA-N yttrium atom Chemical group [Y] VWQVUPCCIRVNHF-UHFFFAOYSA-N 0.000 description 1
- BHHYHSUAOQUXJK-UHFFFAOYSA-L zinc fluoride Chemical compound F[Zn]F BHHYHSUAOQUXJK-UHFFFAOYSA-L 0.000 description 1
- OMQSJNWFFJOIMO-UHFFFAOYSA-J zirconium tetrafluoride Chemical compound F[Zr](F)(F)F OMQSJNWFFJOIMO-UHFFFAOYSA-J 0.000 description 1
Images
































































Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
- H01M10/0525—Rocking-chair batteries, i.e. batteries with lithium insertion or intercalation in both electrodes; Lithium-ion batteries
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/056—Accumulators with non-aqueous electrolyte characterised by the materials used as electrolytes, e.g. mixed inorganic/organic electrolytes
- H01M10/0564—Accumulators with non-aqueous electrolyte characterised by the materials used as electrolytes, e.g. mixed inorganic/organic electrolytes the electrolyte being constituted of organic materials only
- H01M10/0566—Liquid materials
- H01M10/0569—Liquid materials characterised by the solvents
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/13—Electrodes for accumulators with non-aqueous electrolyte, e.g. for lithium-accumulators; Processes of manufacture thereof
- H01M4/131—Electrodes based on mixed oxides or hydroxides, or on mixtures of oxides or hydroxides, e.g. LiCoOx
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/13—Electrodes for accumulators with non-aqueous electrolyte, e.g. for lithium-accumulators; Processes of manufacture thereof
- H01M4/133—Electrodes based on carbonaceous material, e.g. graphite-intercalation compounds or CFx
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/52—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of nickel, cobalt or iron
- H01M4/525—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of nickel, cobalt or iron of mixed oxides or hydroxides containing iron, cobalt or nickel for inserting or intercalating light metals, e.g. LiNiO2, LiCoO2 or LiCoOxFy
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/58—Selection of substances as active materials, active masses, active liquids of inorganic compounds other than oxides or hydroxides, e.g. sulfides, selenides, tellurides, halogenides or LiCoFy; of polyanionic structures, e.g. phosphates, silicates or borates
- H01M4/583—Carbonaceous material, e.g. graphite-intercalation compounds or CFx
- H01M4/587—Carbonaceous material, e.g. graphite-intercalation compounds or CFx for inserting or intercalating light metals
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
Definitions
- the invention disclosed in this specification etc. (hereinafter sometimes referred to as the "present invention" in this specification etc.) relates to a power storage device, a secondary battery and the like. In particular, it relates to lithium ion batteries.
- the present invention relates to an article, method, or manufacturing method.
- the invention relates to a process, machine, manufacture, or composition of matter.
- the present invention relates to a semiconductor device, a display device, a light-emitting device, a power storage device, a lighting device, an electronic device, or manufacturing methods thereof.
- lithium-ion batteries which have high output and high energy density
- portable information terminals such as mobile phones, smartphones, or notebook computers, portable music players, digital cameras, medical equipment, hybrid vehicles (HV), and electric vehicles.
- EV or clean energy vehicles
- PSV plug-in hybrid vehicles
- Lithium-ion batteries vary in charge characteristics and/or discharge characteristics depending on the battery charging environment and/or the battery discharging environment. For example, it is known that the discharge capacity of a lithium ion battery changes depending on the temperature during discharge.
- Patent Document 1 describes that a lithium ion battery that can operate even in a low-temperature environment can be realized by using the non-aqueous solvent described in Patent Document 1.
- the lithium ion battery described in Patent Document 1 cannot be said to have a large discharge capacity when discharged at a temperature of 0° C. or lower (also referred to as “below freezing”) at the time of filing, and further improvement is desired. It is rare.
- An object of one aspect of the present invention is to provide a lithium-ion battery that has excellent discharge characteristics even at subzero temperatures.
- another object is to provide a lithium-ion battery that has excellent charging characteristics even at subzero temperatures.
- An object is to provide a lithium ion battery that has a large discharge capacity and/or a large discharge energy density even when discharged at high temperatures.
- An object of the present invention is to provide a lithium ion battery in which the rate of decrease in discharge capacity and/or discharge energy density is small compared to the value of discharge capacity and/or discharge energy density when discharged at 25°C.
- a temperature below freezing e.g., 0°C or lower, -20°C or lower, preferably -30°C or lower, more preferably -40°C or lower, further preferably -50°C or lower, most preferably -60°C or lower.
- An object is to provide a lithium-ion battery whose rate of decrease in charge capacity is smaller than that in the case of charging at 25°C.
- one of the challenges is to provide a secondary battery with a high charging voltage. Another object is to provide a secondary battery with high safety or reliability. Another object is to provide a secondary battery that is less likely to deteriorate. Another object is to provide a long-life secondary battery. Another object is to provide a novel secondary battery.
- Another object is to provide a novel substance, active material, power storage device, or manufacturing method thereof.
- one aspect of the present invention has the following configuration.
- One aspect of the present invention is a lithium ion battery including a positive electrode having a positive electrode active material, an electrolyte, and a negative electrode having a carbon negative electrode active material.
- the electrolyte contains ethylene carbonate, ethyl methyl carbonate, and dimethyl carbonate.
- the volume ratio of methyl carbonate and the dimethyl carbonate is x:y:100-xy (where 5 ⁇ x ⁇ 35 and 0 ⁇ y ⁇ 65).
- the carbon material is graphite.
- one embodiment of the present invention is a lithium ion battery that includes a positive electrode having a positive electrode active material, an electrolyte, and a negative electrode, and is operable in a temperature range of at least -40°C to 25°C.
- one embodiment of the present invention is a lithium ion battery including a positive electrode having a positive electrode active material, an electrolyte, and a negative electrode.
- the positive electrode active material is used as a positive electrode and contains ethylene carbonate, ethyl methyl carbonate, and dimethyl carbonate, and the total content of the ethylene carbonate, the ethyl methyl carbonate, and the dimethyl carbonate is 100 vol%
- the an electrolyte in which the volume ratio of ethylene carbonate, the ethylmethyl carbonate, and the dimethyl carbonate is x:y:100-xy (where 5 ⁇ x ⁇ 35 and 0 ⁇ y ⁇ 65);
- below freezing e.g., 0°C or lower, -20°C or lower, preferably -30°C or lower, more preferably -40°C or lower, even more preferably -50°C or lower, most preferably -60°C or lower
- a lithium ion battery having a large discharge capacity and/or a large discharge energy density can be provided.
- discharge at a temperature below freezing e.g., 0°C or lower, -20°C or lower, preferably -30°C or lower, more preferably -40°C or lower, even more preferably -50°C or lower, most preferably -60°C or lower
- a temperature below freezing e.g., 0°C or lower, -20°C or lower, preferably -30°C or lower, more preferably -40°C or lower, even more preferably -50°C or lower, most preferably -60°C or lower
- a temperature below freezing e.g., 0°C or lower, -20°C or lower, preferably -30°C or lower, more preferably -40°C or lower, further preferably -50°C or lower, most preferably -60°C or lower
- a temperature below freezing e.g., 0°C or lower, -20°C or lower, preferably -30°C or lower, more preferably -40°C or lower, further preferably -50°C or lower, most preferably -60°C or lower
- a temperature below freezing e.g., 0°C or lower, -20°C or lower, preferably -30°C or lower, more preferably -40°C or lower, further preferably -50°C or lower, most preferably -60°C or lower
- a temperature below freezing e.g., 0°C or lower, -20°C or lower, preferably -30°C or lower, more preferably -40°C or lower, further preferably -50°C or lower, most preferably -60°C or lower
- a secondary battery with high charging voltage can be provided.
- a secondary battery with high safety or reliability can be provided.
- a secondary battery with little deterioration can be provided.
- a long-life secondary battery can be provided.
- a novel secondary battery can be provided.
- a novel substance, an active material, a power storage device, or a manufacturing method thereof can be provided.
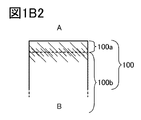
- FIGS. 1B1 and 1B2 are part of the cross-sectional views of the positive electrode active material.
- FIG. 2 is an example of a TEM image in which the crystal orientations are approximately the same.
- FIG. 3A is an example of an STEM image in which the crystal orientations are approximately matched.
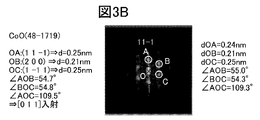
- FIG. 3B is an FFT pattern of the rocksalt crystal RS area
- FIG. 3C is an FFT pattern of the layered rocksalt crystal LRS area.
- FIG. 4 is a diagram for explaining the crystal structure of the positive electrode active material.
- FIG. 5 is a diagram for explaining the crystal structure of a conventional positive electrode active material.
- 6A1 and 6A2 are part of cross-sectional views of the positive electrode active material.
- 6B1 to 6C show the results of calculations for the crystal planes of lithium cobaltate and the distribution of magnesium.
- 7A and 7B are cross-sectional views of the positive electrode active material, and FIGS. 7C1 and 7C2 are part of the cross-sectional views of the positive electrode active material.
- FIG. 8 shows an XRD pattern calculated from the crystal structure.
- FIG. 9 shows an XRD pattern calculated from the crystal structure.
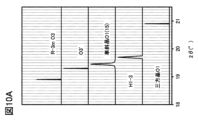
- 10A and 10B are diagrams showing XRD patterns calculated from the crystal structure.
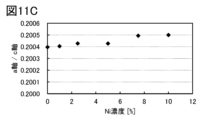
- 11A to 11C are lattice constants calculated from XRD.
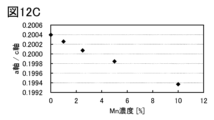
- 12A to 12C are lattice constants calculated from XRD.
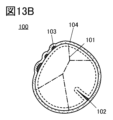
- FIG. 13A and 13B are cross-sectional views of positive electrode active materials.
- FIG. 14 is a cross-sectional view of a positive electrode active material.
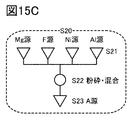
- 15A to 15C are diagrams illustrating a method for manufacturing a positive electrode active material.
- FIG. 16 is a diagram for explaining a method for producing a positive electrode active material.
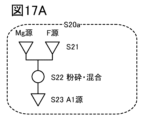
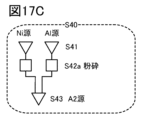
- 17A to 17C are diagrams illustrating a method for manufacturing a positive electrode active material.
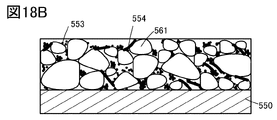
- 18A to 18D are cross-sectional views illustrating examples of positive electrodes of secondary batteries.
- 19A is an exploded perspective view of a coin-type secondary battery
- FIG. 19B is a perspective view of the coin-type secondary battery
- FIG. 19C is a cross-sectional perspective view thereof.
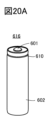
- FIG. 20A shows an example of a cylindrical secondary battery.
- FIG. 20A shows an example of a cylindrical secondary battery.
- FIG. 20A shows an example of a cylindrical secondary battery.
- FIG. 20A shows an example of a cylindrical secondary battery.
- FIG. 20B shows an example of a cylindrical secondary battery.
- FIG. 20C shows an example of a plurality of cylindrical secondary batteries.
- FIG. 20D shows an example of a power storage system having a plurality of cylindrical secondary batteries.
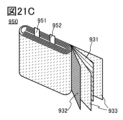
- 21A and 21B are diagrams for explaining an example of a secondary battery, and FIG. 21C is a diagram showing the state inside the secondary battery.
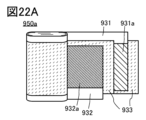
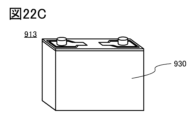
- 22A to 22C are diagrams illustrating examples of secondary batteries.
- 23A and 23B are diagrams showing the appearance of a secondary battery.
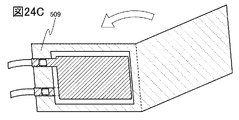
- 24A to 24C are diagrams illustrating a method for manufacturing a secondary battery.
- 25A shows a configuration example of a battery pack
- FIG. 25B shows a configuration example of a battery pack
- FIG. 25C shows a configuration example of a battery pack.
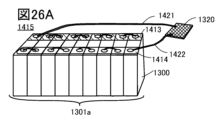
- FIG. 26A is a perspective view of a battery pack showing one embodiment of the present invention
- FIG. 26B is a block diagram of the battery pack
- FIG. 26C is a block diagram of a vehicle having a motor
- 27A to 27D are diagrams illustrating an example of a transportation vehicle.
- FIG. 27E is a diagram illustrating an example of an artificial satellite;
- 28A and 28B are diagrams illustrating a power storage device according to one embodiment of the present invention.
- 29A is a diagram showing an electric bicycle
- FIG. 29B is a diagram showing a secondary battery of the electric bicycle
- FIG. 29C is a diagram explaining an electric motorcycle.
- 30A to 30D are diagrams illustrating examples of electronic devices.
- FIG. 31A shows an example of a wearable device
- FIG. 31A shows an example of a wearable device
- FIG. 31A shows an example of a wearable device
- FIG. 31A shows an example of a wearable device
- FIG. 31A shows an example of
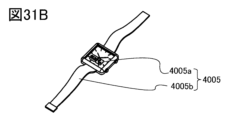
- FIG. 31B shows a perspective view of a wristwatch-type device
- FIG. 31C is a diagram explaining a side view of the wristwatch-type device.
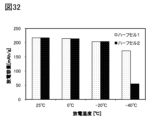
- 32 is a graph showing the discharge capacity of the secondary battery with respect to each temperature during discharge, as described in Example 1.
- FIG. 33 is a graph showing the charge capacity of the secondary battery with respect to each temperature during charging, described in Example 1.
- FIG. 34A and 34B are graphs showing discharge curves of the secondary battery with respect to each temperature, described in Example 1.
- FIG. 35A and 35B are graphs showing discharge curves of the secondary battery with respect to each temperature, described in Example 1.
- FIG. 36A and 36B are graphs showing cycle characteristics of the secondary battery described in Example 2.
- electro-optical devices having a power storage device
- information terminal devices having a power storage device
- the like are all electronic devices.
- power storage device refers to elements and devices in general that have a power storage function. Examples include power storage devices such as lithium ion batteries (also referred to as “secondary batteries”), lithium ion capacitors, electric double layer capacitors, and the like.
- lithium ion batteries also referred to as “secondary batteries”
- lithium ion capacitors lithium ion capacitors
- electric double layer capacitors and the like.
- space groups are expressed using Short notation in international notation (or Hermann-Mauguin notation).
- crystal planes and crystal orientations are expressed using Miller indexes.
- Individual planes indicating crystal planes are indicated using ( ).
- Space groups, crystal planes, and crystal orientations are indicated by a superscript bar on the number from the standpoint of crystallography. - (minus sign) may be attached to and expressed.
- individual orientations that indicate directions within the crystal are [ ]
- collective orientations that indicate all equivalent directions are ⁇ >
- individual planes that indicate crystal planes are ( )
- collective planes that have equivalent symmetry are ⁇ ⁇ to express each.
- the trigonal crystal represented by the space group R-3m is generally represented by a composite hexagonal lattice of hexagonal crystals for ease of understanding of the structure, and not only (hkl) but also (hkil) is used as the Miller index. Sometimes. where i is -(h+k).
- the theoretical capacity of a positive electrode active material refers to the amount of electricity when all the lithium that can be inserted and detached included in the positive electrode active material is desorbed.
- LiCoO 2 has a theoretical capacity of 274 mAh/g
- LiNiO 2 has a theoretical capacity of 275 mAh/g
- LiMn 2 O 4 has a theoretical capacity of 148 mAh/g.
- x in the composition formula for example, x in Li x CoO 2 (the occupancy rate of Li at the lithium site).
- the state in which x in Li x CoO 2 is small is, for example, x ⁇ 0.24, and considering the practical range when used as a lithium ion battery, for example, 0.1 ⁇ x ⁇ 0.24.
- LiCoO 2 and x 1.
- the charge capacity and/or discharge capacity used to calculate x in Li x CoO 2 is preferably measured under conditions where there is no or little influence of short circuit and/or decomposition of the electrolyte. For example, it is not preferable to use the data of a secondary battery in which a sudden change in capacity has occurred due to a short circuit for calculating x.
- the space group of the crystal structure is identified by XRD, electron diffraction, neutron diffraction, and the like. Therefore, in this specification and the like, belonging to a certain space group, belonging to a certain space group, or being in a certain space group can be rephrased as being identified by a certain space group.
- the anions do not have to form a strictly cubic lattice.
- the analysis results do not necessarily match the theory.
- FFT Fast Fourier Transform
- spots may appear at positions slightly different from their theoretical positions. For example, if the orientation with respect to the theoretical position is 5° or less, or 2.5° or less, it can be said that a cubic close-packed structure is obtained.
- the term “layered rock salt type crystal structure possessed by a composite oxide containing lithium and a transition metal” means a rock salt type ion arrangement in which cations and anions are alternately arranged, and a transition metal and A crystal structure in which lithium can diffuse two-dimensionally because lithium is arranged regularly to form a two-dimensional plane. In addition, it may have defects such as lack of cations or anions. Strictly speaking, the layered rock salt type crystal structure may be a structure in which the lattice of the rock salt type crystal is distorted.
- rock salt crystal structure refers to a structure in which cations and anions are arranged alternately.
- homogeneous refers to a phenomenon in which a certain element (eg, A) is distributed in a specific region with similar characteristics in a solid composed of multiple elements (eg, A, B, and C). say.
- concentrations of the elements in the specific regions may be substantially the same.
- difference in element concentration between specific regions may be within 10%.
- Specific regions include, for example, a surface layer portion, surface, convex portion, concave portion, inner portion, and the like.
- separation refers to a phenomenon in which an element (eg, B) is spatially unevenly distributed in a solid composed of multiple elements (eg, A, B, and C). Or, it means that the concentration of an element is different from others. It is synonymous with maldistribution, precipitation, non-uniformity, unevenness, or a mixture of high-concentration locations and low-concentration locations.
- the “surface portion” of a particle such as an active material is, for example, a region within 50 nm, more preferably within 35 nm, still more preferably within 20 nm, and most preferably within 10 nm from the surface toward the inside. be. Surfaces caused by cracks or cracks can also be considered surfaces. In addition, in this specification and the like, a region deeper than the surface layer may be called "inside".
- the term “grain boundary” refers to, for example, a portion where grains are stuck together, a portion where the crystal orientation changes inside the grain (including the central portion), a portion containing many defects, and a portion where the crystal structure is disturbed.
- the grain boundary can be said to be one of plane defects.
- the term “near the grain boundary” refers to a region within 20 nm, preferably within 10 nm, from the grain boundary.
- the term “particle” is not limited to indicating only a spherical shape (having a circular cross-sectional shape). Square, asymmetrical, etc. may be mentioned, and individual particles may be amorphous.
- a lithium ion battery of one embodiment of the present invention includes a positive electrode, a negative electrode, and an electrolyte.
- the electrolyte contains an electrolytic solution, it has a separator between the positive electrode and the negative electrode.
- the lithium-ion battery of one embodiment of the present invention may also have an exterior body that covers at least part of the positive electrode, the negative electrode, and the electrolyte.
- below freezing e.g., 0°C or lower, -20°C or lower, preferably -30°C or lower, more preferably -40°C or lower, further preferably -50°C or lower, most preferably -60°C or lower
- the positive electrode active material contained in the positive electrode and the electrolyte will be mainly described.
- the details of the configuration of the lithium ion battery other than the positive electrode active material and the electrolyte will be described in a third embodiment.
- the positive electrode has a positive electrode active material layer and a positive electrode current collector.
- the positive electrode active material layer contains a positive electrode active material and may further contain at least one of a conductive aid and a binder.
- the positive electrode active material has a function of taking in and/or releasing lithium ions during charging and discharging.
- the positive electrode active material used in one embodiment of the present invention can be charged and/or discharged (hereinafter also referred to as “charge/discharge”) under freezing temperature conditions even at a high charging voltage (hereinafter also referred to as "high charging voltage”).
- charge/discharge a high charging voltage
- a material that causes less deterioration (or a material that causes less increase in resistance) can be used.
- the “charging voltage” is represented based on the potential of lithium metal.
- high charging voltage is, for example, a charging voltage of 4.6 V or higher, preferably 4.65 V or higher, 4.7 V or higher, 4.75 V or higher, or 4.8 V or higher.
- Two or more kinds of materials having different particle diameters and/or compositions may be used as the positive electrode active material as long as they are less likely to deteriorate with charging and discharging even at a high charging voltage.
- composition is different means that the composition of the elements contained in the material is different, and even if the composition of the elements contained in the material is the same, the ratio of the contained elements is different. shall also include
- high charging voltage is 4.6 V or more based on the potential when the negative electrode is lithium metal, but the negative electrode is a carbon material (e.g., graphite).
- a voltage of 4.5 V or higher is called a "high charging voltage”.
- a charging voltage of 4.6 V or more is called a high charging voltage
- a charging voltage of 4.6 V or more is called a high charging voltage
- a charging voltage of .5 V or higher shall be referred to as a high charging voltage.
- any temperature below freezing e.g., 0° C., ⁇ 20° C., preferably ⁇ 30° C., more preferably ⁇ 40° C., further preferably ⁇ 50° C., most preferably ⁇ 60° C.
- a material that causes less deterioration or a material that causes less increase in resistance
- charge capacity and/or discharge at any temperature below freezing e.g., 0°C, -20°C, preferably -30°C, more preferably -40°C, even more preferably -50°C, most preferably -60°C
- a lithium ion battery having a capacity value of 50% or more (preferably 60% or more, more preferably 70% or more, and most preferably 80% or more) of the charge capacity and/or discharge capacity value at 25°C. can be realized.
- the value of the discharge capacity at an arbitrary temperature below freezing and the value of the discharge capacity at 25 ° C. are the temperature during discharge (hereinafter sometimes referred to as "discharge temperature" in this specification etc.). shall be the same.
- the discharge energy density is A large lithium-ion battery can be realized.
- the value of the discharge energy density at an arbitrary temperature below freezing for example, 0 ° C., -20 ° C., preferably -30 ° C., more preferably -40 ° C., further preferably -50 ° C., most preferably -60 ° C.
- 50% or more preferably 60% or more, more preferably 70% or more, and most preferably 80% or more of the discharge energy density at 25°C.
- the discharge energy density at an arbitrary temperature below freezing and the discharge energy density at 25° C. are measured under the same measurement conditions except for the temperature during discharge.
- the charge capacity value at an arbitrary temperature below freezing is A lithium ion battery having a charge capacity of 50% or more (preferably 60% or more, more preferably 70% or more, and most preferably 80% or more) of the value of charge capacity at 25° C. can be realized. Note that the value of charge capacity at an arbitrary temperature below freezing and the value of charge capacity at 25° C. are the same under the same measurement conditions except for the temperature during charging.
- the temperature during charging or discharging described in this specification etc. refers to the temperature of the lithium-ion battery.
- a thermostat that is stable at a desired temperature is used, and the battery to be measured (e.g., test battery or half cell) is placed in the thermostat, and then the test cell is The measurement can be started after a sufficient period of time (for example, 1 hour or longer) until the temperature reaches the temperature of the constant temperature bath, but the method is not necessarily limited to this method.
- the electrolyte used in one embodiment of the present invention can be used at any temperature below freezing (e.g., 0°C, -20°C, preferably -30°C, more preferably -40°C, even more preferably -50°C, most preferably -60°C). ) even at any temperature below freezing (e.g., 0 ° C., -20 ° C., preferably -30 ° C., more preferably -40 ° C., further preferably -50 ° C., most preferably -60 ° C.)
- a material having excellent lithium ion conductivity can be used.
- electrolyte An example of an electrolyte is described below. Note that the electrolyte described in this embodiment as an example is obtained by dissolving an electrolyte (lithium salt) in an organic solvent and can be called an electrolytic solution, but the electrolyte is a liquid electrolyte ( It is also possible to use a solid electrolyte without being limited to the electrolytic solution). Alternatively, an electrolyte (semi-solid electrolyte) containing both a liquid electrolyte that is liquid at room temperature and a liquid electrolyte that is solid at room temperature can be used.
- Examples of the organic solvent described in this embodiment include ethylene carbonate (EC), ethylmethyl carbonate (EMC), and dimethyl carbonate (DMC), and the ethylene carbonate, the ethylmethyl carbonate, and the dimethyl
- EC ethylene carbonate
- EMC ethylmethyl carbonate
- DMC dimethyl carbonate
- the volume ratio of the ethylene carbonate, the ethylmethyl carbonate, and the dimethyl carbonate is x: y: 100-x-y (where 5 ⁇ x ⁇ 35, 0 ⁇ y ⁇ 65.) can be used.
- the above volume ratio may be the volume ratio before the electrolyte is mixed, and the outside air may be room temperature (typically 25° C.) when the electrolyte is mixed.
- EC is a cyclic carbonate and has a high dielectric constant, so it has the effect of promoting the dissociation of lithium salts.
- the organic solvent specifically described as one aspect of the present invention further includes EMC and DMC instead of EC alone.
- EMC is a chain carbonate, has the effect of lowering the viscosity of the electrolytic solution, and has a freezing point of -54°C.
- DMC is also a chain carbonate and has the effect of lowering the viscosity of the electrolytic solution.
- EC, EMC, and DMC having such physical properties are used in a volume ratio of x: y: 100-x-y (where 5 ⁇ x ⁇ 35), with the total content of these three organic solvents being 100 vol%. , and 0 ⁇ y ⁇ 65.)
- the electrolyte produced using the mixed organic solvent has a freezing point of ⁇ 40° C. or lower.
- a lithium salt can be used as the electrolyte dissolved in the above solvent.
- the electrolytic solution has a low content of particulate matter or elements other than constituent elements of the electrolytic solution (hereinafter also simply referred to as "impurities") and is highly purified.
- the weight ratio of impurities to the electrolytic solution is preferably 1% or less, preferably 0.1% or less, and more preferably 0.01% or less.
- VC vinylene carbonate
- PS propane sultone
- TB tert-butylbenzene
- FEC fluoroethylene carbonate
- LiBOB lithium bis(oxalate)borate
- dinitrile compounds of succinonitrile or adiponitrile may be added.
- concentration of the additive may be, for example, 0.1 wt % or more and 5 wt % or less with respect to the solvent.
- an example of an electrolyte that can be used in the lithium ion battery of one embodiment of the present invention has been described.
- Other materials can also be used as long as they have excellent lithium ion conductivity even when charging and discharging at subzero temperatures (for example, ⁇ 20° C., preferably ⁇ 40° C.).
- a lithium ion battery of one embodiment of the present invention includes at least the positive electrode active material and the electrolyte described above, so that the lithium ion battery has excellent discharge characteristics even at sub-zero temperatures and/or lithium has excellent charge characteristics even at sub-zero temperatures.
- FIGS. 1 to 14 are used to describe a positive electrode active material that can be used in a lithium ion battery that is one embodiment of the present invention (hereinafter referred to as “a positive electrode active material that can be used as one embodiment of the present invention. ) and its manufacturing method will be described.
- the positive electrode active material that can be used for the lithium-ion battery that is one embodiment of the present invention is a material that is less likely to deteriorate due to charging and discharging even at a high charging voltage (high charging voltage). Anything can be used.
- the positive electrode active material that can be used for the lithium ion battery disclosed in this specification and the like does not need to be construed as being limited to the specific materials described in the present embodiment and the like, and at the time of filing the present application, a high charging voltage (for example, a known material can be used as a material that hardly deteriorates due to charging and discharging even when the voltage is 4.6 V or higher.
- Example of positive electrode active material An example of a positive electrode active material that can be used as one embodiment of the present invention is described below.
- a positive electrode active material 100 that can be used as one embodiment of the present invention will be described with reference to FIGS.
- FIGS. 1B1 and 1B2 are cross-sectional views of a positive electrode active material 100 that is one embodiment of the present invention.
- FIGS. 1B1 and 1B2 show enlarged views of the vicinity of AB in FIG. 1A1.
- the positive electrode active material 100 has a surface layer portion 100a and an inner portion 100b.
- the dashed line indicates the boundary between the surface layer portion 100a and the inner portion 100b.
- part of the grain boundary 101 is indicated by a dashed line in FIG. 1A2.
- the surface layer portion 100a of the positive electrode active material 100 is, for example, within 50 nm from the surface toward the inside, more preferably within 35 nm from the surface toward the inside, and still more preferably within 20 nm from the surface toward the inside. It refers to a region within 10 nm, most preferably within 10 nm from the surface toward the inside.
- a surface caused by cracks and/or cracks may also be referred to as a surface.
- Surface layer 100a is synonymous with near-surface, near-surface region, or shell.
- a region deeper than the surface layer portion 100a of the positive electrode active material is called an inner portion 100b.
- Interior 100b is synonymous with interior region or core.
- the surface of the positive electrode active material 100 means the surface of the composite oxide including the surface layer portion 100a, the inner portion 100b, the convex portion 103, and the like. Therefore, it is assumed that the positive electrode active material 100 does not contain carbonates, hydroxyl groups, and the like chemically adsorbed after production. Also, the electrolyte, binder, conductive material, and compounds derived from these attached to the positive electrode active material 100 are not included.
- the surface of the positive electrode active material 100 in a cross-sectional STEM (scanning transmission electron microscope) image or the like is the boundary between the region where the electron beam coupling image is observed and the region where it is not observed, and has an atomic number larger than that of lithium.
- EELS electron energy loss spectroscopy
- the grain boundary 101 is, for example, a portion where particles of the positive electrode active material 100 are fixed to each other, a portion where the crystal orientation changes inside the positive electrode active material 100, that is, a discontinuous repetition of bright lines and dark lines in an STEM image or the like. This refers to a portion that has become rough, a portion that contains many crystal defects, a portion where the crystal structure is disordered, etc.
- a crystal defect means a defect observable in a cross-sectional TEM (transmission electron microscope) image, a cross-sectional STEM image, or the like, that is, a structure in which another atom enters between lattices, a void, or the like.
- the grain boundary 101 can be said to be one of planar defects.
- the vicinity of the grain boundary 101 means a region within 20 nm (preferably within 15 nm, more preferably within 10 nm) from the grain boundary 101 .
- the positive electrode active material 100 contains lithium, cobalt, oxygen, and additive elements.
- the positive electrode active material 100 may be lithium cobaltate (LiCoO 2 ) to which additive elements are added.
- LiCoO 2 lithium cobaltate
- the positive electrode active material for lithium-ion batteries must contain transition metals that can be oxidized and reduced in order to maintain charge neutrality even when lithium ions are intercalated and deintercalated.
- the positive electrode active material 100 of the lithium-ion battery which is one embodiment of the present invention, preferably contains cobalt as a transition metal responsible for oxidation-reduction reaction. Also, in addition to cobalt, at least one of nickel and manganese may be included. When cobalt accounts for 75 atomic % or more, preferably 90 atomic % or more, and more preferably 95 atomic % or more of the transition metals included in the positive electrode active material 100, synthesis is relatively easy, handling is easy, and excellent cycle characteristics can be achieved. It is preferable in that it has
- nickel such as lithium nickelate (LiNiO 2 ) is the transition metal.
- LiNiO 2 lithium nickelate
- x is small in Li x CoO 2 , the stability is superior compared to composite oxides in which x is the majority. This is probably because cobalt is less affected by strain due to the Jahn-Teller effect than nickel.
- the Jahn-Teller effect in transition metal compounds varies in strength depending on the number of electrons in the d-orbital of the transition metal.
- Additive elements included in the positive electrode active material 100 include magnesium, fluorine, nickel, aluminum, titanium, zirconium, vanadium, iron, manganese, chromium, niobium, arsenic, zinc, silicon, sulfur, phosphorus, boron, bromine, and beryllium. It is preferable to use one or two or more selected.
- the transition metal if two or more are used, the total is preferably less than 25 atomic%, more preferably less than 10 atomic%, and further less than 5 atomic%. preferable.
- positive electrode active material 100 examples include lithium cobaltate to which magnesium and fluorine are added, magnesium, lithium cobaltate to which fluorine and titanium are added, magnesium, lithium cobaltate to which fluorine and aluminum are added, magnesium, and fluorine. and lithium cobaltate doped with nickel, lithium cobaltate doped with magnesium, fluorine, nickel and aluminum, and the like.
- the additive element may be a mixture or a part of the raw material.
- the additive elements may not necessarily contain magnesium, fluorine, nickel, aluminum, titanium, zirconium, vanadium, iron, manganese, chromium, niobium, arsenic, zinc, silicon, sulfur, phosphorus, boron, bromine, or beryllium. .
- the positive electrode active material 100 substantially does not contain manganese, the above advantages of being relatively easy to synthesize, easy to handle, and having excellent cycle characteristics are further enhanced.
- the weight of manganese contained in the positive electrode active material 100 is preferably, for example, 600 ppm or less, more preferably 100 ppm or less.
- “substantially does not contain” means that when measured using an analytical means, it is below the detection limit, or even if it is contained in the detection limit, there is no action or effect. It refers to the case where it is included within a range that does not affect it.
- the layered rock salt type composite oxide has a high discharge capacity, has a two-dimensional lithium ion diffusion path, is suitable for lithium ion insertion/extraction reactions, and is excellent as a positive electrode active material for secondary batteries. Therefore, it is particularly preferable that the inside 100b, which occupies most of the volume of the positive electrode active material 100, has a layered rock salt crystal structure.
- FIG. 4 shows the layered rock salt type crystal structure with R-3m(O3).
- the surface layer portion 100a of the positive electrode active material 100 that can be used as one aspect of the present invention, even if lithium is released from the positive electrode active material 100 by charging, the layered structure of the inner portion 100b consisting of octahedrons of cobalt and oxygen is not broken. It is preferable to have a reinforcing function.
- the surface layer portion 100 a preferably functions as a barrier film for the positive electrode active material 100 .
- Reinforcing here means suppressing structural changes of the surface layer portion 100a and the inner portion 100b of the positive electrode active material 100, such as desorption of oxygen, and/or the electrolyte is oxidatively decomposed on the surface of the positive electrode active material 100. It means to suppress things.
- the surface layer portion 100a preferably has a crystal structure different from that of the inner portion 100b. Moreover, the surface layer portion 100a preferably has a more stable composition and crystal structure at room temperature (25° C.) than the inner portion 100b.
- at least part of the surface layer portion 100a of the positive electrode active material 100 that can be used as one embodiment of the present invention preferably has a rock salt crystal structure.
- the surface layer portion 100a preferably has both a layered rock salt type crystal structure and a rock salt type crystal structure.
- the surface layer portion 100a preferably has characteristics of both a layered rock salt type crystal structure and a rock salt type crystal structure.
- the surface layer portion 100a is a region where lithium ions are first desorbed during charging, and is a region where the lithium concentration tends to be lower than in the inner portion 100b. It can also be said that the atoms on the surface of the positive electrode active material 100 included in the surface layer portion 100a are in a state in which some of the bonds are cut. Therefore, the surface layer portion 100a is likely to be unstable, and can be said to be a region where deterioration of the crystal structure is likely to occur.
- the layered structure of cobalt and oxygen octahedrons in the inner portion 100b can be made difficult to break even when x in Li x CoO 2 is small, for example, x is 0.24 or less. can be done. Further, it is possible to suppress the displacement of the layer composed of octahedrons of cobalt and oxygen in the interior 100b.
- the surface layer portion 100a preferably contains an additive element, and more preferably contains a plurality of additive elements. Further, it is preferable that the surface layer portion 100a has a higher concentration of one or more selected from the additive elements than the inner portion 100b. In addition, it is preferable that one or more of the additional elements included in the positive electrode active material 100 have a concentration gradient. Further, it is more preferable that the distribution of the positive electrode active material 100 differs depending on the additive element. For example, it is more preferable that the depth from the surface of the concentration peak differs depending on the additive element.
- the concentration peak as used herein means the maximum value of the concentration at 50 nm or less from the surface layer portion 100a or the surface.
- additive elements X For example, magnesium, fluorine, nickel, titanium, silicon, phosphorus, boron, calcium, etc., as some of the additive elements, have a concentration gradient that increases from the interior 100b toward the surface, as shown by the gradation in FIG. 1B1. is preferred. In this specification and the like, these additive elements are referred to as additive elements X. As shown in FIG. 1B1.
- additive elements such as aluminum, manganese, etc. may have a concentration gradient and/or have a concentration peak in a region deeper than the additive element X, as indicated by hatched densities in FIG. 1B2. preferable.
- the concentration peak may exist in the surface layer portion 100a or may be deeper than the surface layer portion 100a. For example, it preferably has a peak in a region of 5 nm or more and 50 nm or less from the surface toward the inside. In this specification and the like, these additive elements are referred to as additive elements Y. As shown in FIG.
- magnesium which is one of the additive elements X, is divalent, and magnesium ions are more stable in the lithium site than in the cobalt site in the layered rock salt type crystal structure, so they easily enter the lithium site.
- the layered rock salt crystal structure can be easily maintained. It is presumed that this is because the magnesium present in the lithium sites functions as a pillar supporting the CoO 2 layers.
- the presence of magnesium can suppress desorption of oxygen around magnesium when x in Li x CoO 2 is, for example, 0.24 or less.
- the density of the positive electrode active material 100 increases due to the presence of magnesium.
- the magnesium concentration of the surface layer portion 100a is high, it can be expected that corrosion resistance to hydrofluoric acid generated by decomposition of the electrolytic solution is improved.
- magnesium does not adversely affect the insertion and extraction of lithium during charging and discharging, and the above benefits can be enjoyed.
- excess magnesium can adversely affect lithium insertion and extraction.
- the effect of stabilizing the crystal structure may be reduced. It is considered that this is because magnesium enters the cobalt site in addition to the lithium site.
- unnecessary magnesium compounds oxides, fluorides, etc.
- the discharge capacity of the positive electrode active material may decrease. This is probably because too much magnesium enters the lithium sites and the amount of lithium that contributes to charging and discharging decreases.
- the amount of magnesium contained in the entire positive electrode active material 100 is appropriate.
- the number of atoms of magnesium is preferably 0.001 to 0.1 times the number of cobalt atoms, more preferably more than 0.01 times and less than 0.04 times, and still more preferably about 0.02 times.
- the amount of magnesium contained in the entire positive electrode active material 100 as used herein refers to the amount of magnesium in the entire positive electrode active material 100 using, for example, GD-MS (glow discharge mass spectrometry), ICP-MS (inductively coupled plasma mass spectrometry), or the like. It may be a value obtained by performing an elemental analysis, or it may be a value based on a blending value of raw materials in the process of producing the positive electrode active material 100 .
- nickel which is one of the additive elements X, can exist on both the cobalt site and the lithium site. When it exists in the cobalt site, the oxidation-reduction potential becomes lower than that of cobalt, which leads to an increase in discharge capacity, which is preferable.
- the shift of the layered structure composed of cobalt and oxygen octahedrons can be suppressed.
- the change in volume due to charge/discharge is suppressed.
- the elastic modulus increases, that is, the material becomes hard. It is presumed that this is because the nickel present in the lithium sites also functions as a pillar supporting the CoO 2 layers.
- the crystal structure can be expected to become more stable in a charged state at a high temperature, for example, 45° C. or higher.
- the amount of nickel contained in the entire positive electrode active material 100 is preferably an appropriate amount.
- the number of nickel atoms in the positive electrode active material 100 is preferably more than 0% and less than 7.5% of the number of cobalt atoms, preferably 0.05% or more and 4% or less, and 0.1% or more and 2% or less. is preferred, and 0.2% or more and 1% or less is more preferred.
- it is preferably more than 0% and 4% or less.
- it is preferably more than 0% and 2% or less.
- 0.05% or more and less than 7.5% is preferable.
- 0.05% or more and 2% or less is preferable.
- it is preferably 0.1% or more and less than 7.5%.
- the amount of nickel shown here may be a value obtained by elemental analysis of the entire positive electrode active material using, for example, GD-MS, ICP-MS, etc. may be based on the value of
- aluminum which is one of the additive elements Y, can exist in cobalt sites in the layered rock salt crystal structure. Since aluminum is a trivalent typical element and does not change its valence, lithium around aluminum does not easily move during charging and discharging. Therefore, aluminum and lithium around it function as pillars and can suppress changes in the crystal structure. In addition, aluminum has the effect of suppressing the elution of surrounding cobalt and improving the continuous charge resistance. In addition, since the Al--O bond is stronger than the Co--O bond, detachment of oxygen around aluminum can be suppressed. These effects improve thermal stability. Therefore, if aluminum is included as an additive element, safety can be improved when the positive electrode active material 100 is used in a secondary battery.
- the positive electrode active material 100 whose crystal structure does not easily collapse even after repeated charging and discharging can be obtained.
- the aluminum exists at a position slightly deeper than the surface (specifically, the peak of the concentration of aluminum is present in a region deeper than the peak of the concentration of the additional element X).
- a region deeper than the deepest region where the presence of the additive element X is confirmed exists, where the presence of aluminum is confirmed and the deepest region is present.
- the amount of aluminum contained in the entire positive electrode active material 100 is appropriate.
- the number of aluminum atoms contained in the entire positive electrode active material 100 is preferably 0.05% or more and 4% or less, preferably 0.1% or more and 2% or less, or 0.3% or more and 1.5% or more. % or less is more preferable. Alternatively, 0.05% or more and 2% or less is preferable. Alternatively, 0.1% or more and 4% or less is preferable.
- the amount of the entire positive electrode active material 100 referred to here may be, for example, a value obtained by elemental analysis of the entire positive electrode active material 100 using GD-MS, ICP-MS, or the like. 100 may be based on the values of the raw material formulations during the fabrication process.
- Furine which is one of the additional elements X, is a monovalent anion, and if part of the oxygen in the surface layer portion 100a is replaced with fluorine, the lithium desorption energy is reduced. This is because the change in the valence of cobalt ions due to desorption of lithium changes from trivalent to tetravalent when fluorine is not present, and from divalent to trivalent when fluorine is present, resulting in different oxidation-reduction potentials. Therefore, when a part of oxygen is replaced with fluorine in the surface layer portion 100a of the positive electrode active material 100, it can be said that desorption and insertion of lithium ions in the vicinity of fluorine easily occur.
- the positive electrode active material 100 when used in a secondary battery, charge/discharge characteristics, large current characteristics, and the like can be improved. Further, the presence of fluorine in the surface layer portion 100a having the surface which is the portion in contact with the electrolytic solution can effectively improve the corrosion resistance to hydrofluoric acid.
- a fluxing agent also referred to as a flux agent that lowers the melting point of the other additive element sources is used. ).
- titanium oxide which is one of the additive elements X
- titanium oxide is known to have superhydrophilicity. Therefore, by using the positive electrode active material 100 including titanium oxide in the surface layer portion 100a, wettability to a highly polar solvent may be improved. When used as a secondary battery, the interface between the positive electrode active material 100 and the highly polar electrolyte solution is in good contact, and an increase in internal resistance may be suppressed.
- phosphorus which is one of the additive elements X
- it may suppress short circuits when the state of x in Li x CoO 2 is kept small.
- it preferably exists in the surface layer portion 100a as a compound containing phosphorus and oxygen.
- the hydrogen fluoride generated by decomposition of the electrolyte reacts with phosphorus, which is preferable because the concentration of hydrogen fluoride in the electrolyte can be reduced.
- the electrolyte has LiPF 6
- hydrolysis can generate hydrogen fluoride.
- hydrogen fluoride may be generated due to the reaction between polyvinylidene fluoride (PVDF), which is used as a component of the positive electrode, and alkali.
- PVDF polyvinylidene fluoride
- By reducing the concentration of hydrogen fluoride in the electrolyte corrosion of the current collector and/or peeling of the coating 104 can be suppressed in some cases.
- the positive electrode active material 100 contains phosphorus together with magnesium, because the stability is extremely high in a state where x in Li x CoO 2 is small.
- the number of phosphorus atoms is preferably 1% or more and 20% or less, more preferably 2% or more and 10% or less, and even more preferably 3% or more and 8% or less of the number of cobalt atoms.
- it is preferably 1% or more and 10% or less.
- it is preferably 1% or more and 8% or less.
- it is preferably 2% or more and 20% or less.
- it is preferably 2% or more and 8% or less.
- the number of atoms of magnesium is preferably 0.1% or more and 10% or less, more preferably 0.5% or more and 5% or less, and more preferably 0.7% or more and 4% or less of the number of cobalt atoms.
- 0.1% or more and 5% or less is preferable.
- 0.1% or more and 4% or less is preferable.
- 0.5% or more and 10% or less is preferable.
- 0.5% or more and 4% or less is preferable.
- the concentrations of phosphorus and magnesium shown here may be, for example, values obtained by elemental analysis of the entire positive electrode active material 100 using GC-MS, ICP-MS, or the like, or It may be based on the value of the blend of raw materials in
- the positive electrode active material 100 has cracks
- the presence of phosphorus, more specifically, for example, a compound containing phosphorus and oxygen inside the positive electrode active material with the cracks on the surface, for example, the embedded portion 102 causes the cracks to form. Progression can be inhibited.
- additive elements with different distributions such as additive element X and additive element Y
- the crystal structure of a wider region can be stabilized.
- the positive electrode active material 100 contains both magnesium and nickel, which are part of the additional element X, and aluminum, which is one of the additional elements Y
- the amount of the positive electrode active material 100 is higher than when only one of the additional elements X and Y is contained.
- the crystal structure of a wide region can be stabilized.
- the additive element X such as magnesium and nickel can sufficiently stabilize the surface.
- aluminum it is preferable for aluminum to be widely distributed in a deep region, for example, a region having a depth of 5 nm or more and 50 nm or less from the surface, because the crystal structure of a wider region can be stabilized.
- the effects of the respective additive elements are synergistic and can contribute to further stabilization of the surface layer portion 100a.
- the effect of making the composition and crystal structure stable is high, which is preferable.
- the surface layer portion 100a is occupied only by the compound of the additive element and oxygen, it becomes difficult to intercalate and deintercalate lithium, which is not preferable.
- the surface layer portion 100a is occupied only by a structure in which MgO, MgO and NiO(II) are in solid solution, and/or a structure in which MgO and CoO(II) are in solid solution. Therefore, the surface layer portion 100a preferably contains at least cobalt, also contains lithium in a discharged state, and has a lithium intercalation/deintercalation path.
- the concentration of cobalt in the surface layer portion 100a is preferably higher than that of magnesium.
- the ratio Mg/Co between the number Mg of magnesium atoms and the number Co of cobalt atoms is preferably 0.62 or less.
- the concentration of cobalt in the surface layer portion 100a is higher than that of nickel.
- the concentration of cobalt in the surface layer portion 100a is higher than that of aluminum.
- the concentration of cobalt in the surface layer portion 100a is higher than that of fluorine.
- the surface layer portion 100a preferably has a higher concentration of magnesium than nickel.
- the number of atoms of nickel is preferably 1/6 or less of the number of atoms of magnesium.
- Some of the additive elements particularly magnesium, nickel and aluminum, preferably have a higher concentration in the surface layer portion 100a than in the inner portion 100b, but preferably also exist randomly and sparsely in the inner portion 100b.
- magnesium and aluminum are present at appropriate concentrations in the lithium sites in the interior 100b, there is an effect that the layered rock salt type crystal structure can be easily maintained in the same manner as described above.
- nickel is present in the inside 100b at an appropriate concentration, it is possible to suppress the shift of the layered structure composed of octahedrons of cobalt and oxygen in the same manner as described above.
- both magnesium and nickel are contained, a synergistic effect of suppressing the elution of magnesium can be expected similarly to the above.
- the crystal structure changes continuously from the inside 100b toward the surface.
- the crystal orientations of the surface layer portion 100a and the inner portion 100b substantially match.
- the surface layer part 100a and the inner part 100b are topotaxy.
- topotaxis means having three-dimensional structural similarity such that the orientation of the crystals roughly matches, or having the same crystallographic orientation.
- epitaxy refers to the structural similarity of two-dimensional interfaces.
- a pit means a hole formed as a defect progresses in the positive electrode active material.
- the crystal structure changes continuously from the layered rock salt type interior 100b toward the rock salt type or the surface and surface layer portion 100a having characteristics of both the rock salt type and the layered rock salt type.
- the crystal orientation of the surface layer portion 100a having characteristics of the rock salt type or both of the rock salt type and the layered rock salt type and the layered rock salt type inside 100b substantially match.
- the layered rock salt type crystal structure belonging to the space group R-3m which is possessed by a composite oxide containing a transition metal such as lithium and cobalt, refers to a structure in which cations and anions are alternately arranged. It is a crystal structure that has a rock salt-type ion arrangement that allows two-dimensional diffusion of lithium because the transition metal and lithium are regularly arranged to form a two-dimensional plane. In addition, there may be defects such as lack of cations or anions. Strictly speaking, the layered rock salt type crystal structure may be a structure in which the lattice of the rock salt type crystal is distorted.
- rock salt type crystal structure refers to a structure that has a cubic crystal structure including space group Fm-3m, in which cations and anions are arranged alternately. In addition, there may be a lack of cations or anions.
- the rocksalt type has no distinction in the cation sites, but the layered rocksalt type has two types of cation sites in the crystal structure, one of which is occupied mostly by lithium and the other is occupied by a transition metal.
- the layered structure in which the two-dimensional planes of cations and two-dimensional planes of anions are alternately arranged is the same for both the rock salt type and the layered rock salt type.
- the bright spots of the electron beam diffraction pattern corresponding to the crystal plane forming this two-dimensional plane when the central spot (transmission spot) is set to the origin 000, the bright spot closest to the central spot is ideal.
- the rock salt type has the (111) plane
- the layered rock salt type has the (003) plane, for example.
- the bright spots on the (003) plane of LiCoO2 are about half the distance between the bright spots on the (111) plane of MgO. is observed at the position of Therefore, when the analysis region has two phases, for example, rocksalt-type MgO and layered rocksalt-type LiCoO, in the electron beam diffraction pattern, there is a plane orientation in which bright spots with strong brightness and bright spots with weak brightness are alternately arranged. do. Bright spots common to the rocksalt type and layered rocksalt type exhibit high brightness, and bright spots occurring only in the layered rocksalt type exhibit weak brightness.
- the anions of layered rock salt crystals and rock salt crystals have a cubic close-packed structure (face-centered cubic lattice structure).
- the O3' type and monoclinic O1(15) crystals which will be described later, are also presumed to have a cubic close-packed structure of anions. Therefore, when the layered rock-salt crystal and the rock-salt crystal are in contact with each other, there exists a crystal plane in which the direction of the cubic close-packed structure composed of anions is aligned.
- the anions in the ⁇ 111 ⁇ planes of the cubic crystal structure have a triangular lattice.
- the layered rocksalt type has a space group R-3m and has a rhombohedral structure, but is generally represented by a compound hexagonal lattice to facilitate understanding of the structure, and the (0001) plane of the layered rocksalt type has a hexagonal lattice.
- the triangular lattice of the cubic ⁇ 111 ⁇ planes has a similar atomic arrangement to the hexagonal lattice of the (0001) planes of the layered rocksalt type. It can be said that the orientation of the cubic close-packed structure is aligned when both lattices are consistent.
- the space group of layered rocksalt crystals and O3′ crystals is R-3m, which is different from the space group of rocksalt crystals Fm-3m (the space group of general rocksalt crystals).
- the Miller indices of the crystal planes to be filled are different between the layered rocksalt type crystal and the O3′ type crystal, and the rocksalt type crystal.
- TEM Transmission Electron Microscope, transmission electron microscope
- STEM Sccanning Transmission Electron Microscope, scanning transmission electron microscope
- HAADF-STEM High-angle Annular Dark Field Scanning TEM, high-angle scattering annular dark-field scanning transmission electron microscope
- ABF-STEM Annular Bright-Field Scanning Transmission Electron Microscope, annular bright-field scanning transmission electron microscope
- FIG. 2 shows an example of a TEM image in which the orientations of the layered rock salt crystal LRS and the rock salt crystal RS are approximately the same.
- a TEM image, an STEM image, an HAADF-STEM image, an ABF-STEM image, or the like provides an image that reflects the crystal structure.
- a contrast derived from a crystal plane can be obtained.
- the contrast derived from the (0003) plane is bright (bright strip) and dark (dark strip) ) is obtained as a repetition of Therefore, repetition of bright lines and dark lines is observed in the TEM image, and when the angle between the bright lines (for example, L RS and L LRS shown in FIG. 2) is 5° or less, or 2.5° or less, the crystal plane is roughly It can be determined that they match, that is, that the crystal orientations roughly match. Similarly, when the angle between the dark lines is 5° or less, or 2.5° or less, it can be determined that the crystal orientations are approximately the same.
- FIG. 3A shows an example of an STEM image in which the orientations of the layered rock salt crystal LRS and the rock salt crystal RS are approximately the same.
- FIG. 3B shows the FFT pattern of the rocksalt crystal RS region
- FIG. 3C shows the FFT pattern of the layered rocksalt crystal LRS region.
- Compositions, JCPDS card numbers, and d values and angles calculated therefrom are shown on the left of FIGS. 3B and 3C. Measured values are shown on the right.
- the spots marked with an O are the 0th diffraction order.
- the spots marked with A in FIG. 3B are derived from the cubic 11-1 reflection.
- the spots labeled A in FIG. 3C are derived from layered rock salt type 0003 reflections. From FIGS. 3B and 3C, it can be seen that the orientation of the cubic crystal 11-1 reflection and the orientation of the layered rock salt type 0003 reflection approximately match. That is, it can be seen that the straight line passing through AO in FIG. 3B and the straight line passing through AO in FIG. 3C are substantially parallel. As used herein, “substantially coincident” and “substantially parallel” mean that the angle is 5° or less or 2.5° or less.
- these reciprocal lattice points are spot-like, that is, not continuous with other reciprocal lattice points.
- the fact that the reciprocal lattice points are spot-like and are not continuous with other reciprocal lattice points means that the crystallinity is high.
- the orientation of the 0003 reflection of the layered rocksalt type may vary depending on the incident direction of the electron beam. Spots not derived from layered rocksalt-type 0003 reflection may be observed on a reciprocal lattice space with a different orientation.
- the spot labeled B in FIG. 3C originates from the layered rock salt type 1014 reflection. This is an angle of 52° or more and 56° or less from the orientation of the reciprocal lattice point (A in FIG.
- ⁇ AOB is 52° or more and 56° or less
- d is sometimes observed at a location of 0.19 nm or more and 0.21 nm or less.
- this index is an example, and does not necessarily have to match this index. For example, they may be equivalent reciprocal lattice points.
- a spot not derived from the cubic 11-1 reflection may be observed on a reciprocal lattice space different from the orientation in which the cubic 11-1 reflection is observed.
- the spot labeled B in FIG. 3B is from the cubic 200 reflection. This is a diffraction spot at an angle of 54° or more and 56° or less (that is, ⁇ AOB is 54° or more and 56° or less) from the orientation of the reflection derived from cubic crystal 11-1 (A in FIG. 3B). is sometimes observed.
- this index is an example, and does not necessarily have to match this index. For example, they may be equivalent reciprocal lattice points.
- the (0003) plane and its equivalent planes and the (10-14) plane and its equivalent planes tend to appear as crystal planes.
- the observation sample is prepared with an FIB or the like so that the (0003) plane can be easily observed, for example, the electron beam is [12-10] incident in the TEM or the like. Thin section processing is possible.
- it is preferable to thin the crystal so that the (0003) plane of the layered rock salt type can be easily observed.
- the positive electrode active material 100 that can be used as one aspect of the present invention has the above-described additive element distribution and/or crystal structure in a discharged state, so that x in Li x CoO 2 is small.
- the crystal structure of is different from that of conventional positive electrode active materials.
- "x is small” means 0.1 ⁇ x ⁇ 0.24.
- FIG. 5 shows changes in the crystal structure of conventional positive electrode active materials.
- the conventional positive electrode active material shown in FIG. 5 is lithium cobaltate (LiCoO 2 ) with no additional element.
- LiCoO 2 lithium cobaltate
- not having any additional elements means that when measured using an analytical means, it is below the detection limit, or even if it is contained at about the detection limit, the presence or absence of an effect It refers to the case where it is contained within a range that does not affect the
- lithium occupies octahedral sites and there are three CoO 2 layers in the unit cell. Therefore, this crystal structure is sometimes called an O3 type crystal structure.
- the CoO 2 layer is a structure in which an octahedral structure in which six oxygen atoms are coordinated to cobalt continues in the planar direction in a state of edge sharing. This is sometimes referred to as a layer composed of octahedrons of cobalt and oxygen.
- This structure has one CoO 2 layer in the unit cell. Therefore, it is sometimes called O1 type or monoclinic O1 type.
- This structure can also be said to be a structure in which a CoO 2 structure such as a trigonal O1 type and a LiCoO 2 structure such as R-3m(O3) are alternately laminated. Therefore, this crystal structure is sometimes called an H1-3 type crystal structure.
- the H1-3 type crystal structure has twice the number of cobalt atoms per unit cell as other structures.
- the c-axis of the H1-3 type crystal structure is shown with half the unit cell in order to facilitate comparison with other crystal structures.
- the coordinates of cobalt and oxygen in the unit cell are Co (0,0,0.42150 ⁇ 0.00016), O1 (0,0,0.27671 ⁇ 0.00045) , O2(0,0,0.11535 ⁇ 0.00045).
- O1 and O2 are each oxygen atoms.
- Which unit cell should be used to express the crystal structure of the positive electrode active material can be determined, for example, by Rietveld analysis of an XRD pattern. In this case, a unit cell with a small GOF (goodness of fit) value should be adopted.
- conventional lithium cobalt oxide has an H1-3 crystal structure and a discharged R-3m (O3) structure. , the change in crystal structure (that is, non-equilibrium phase change) is repeated.
- these two crystal structures have a large difference in volume.
- the volume difference between the H1-3 type crystal structure and the R-3m(O3) type crystal structure in the discharged state exceeds 3.5%, typically 3.9% or more. is.
- the positive electrode active material 100 that can be used as one embodiment of the present invention shown in FIG . of the positive electrode active material More specifically, the shift between the CoO 2 layer when x is 1 and when x is 0.24 or less can be reduced. In addition, the change in volume when compared per cobalt atom can be reduced. Therefore, in the positive electrode active material 100 that can be used as one embodiment of the present invention, even when charging and discharging are repeated such that x becomes 0.24 or less, the crystal structure does not easily collapse, and excellent cycle characteristics can be achieved. In addition, the positive electrode active material 100 that can be used as one embodiment of the present invention can have a more stable crystal structure than a conventional positive electrode active material when x in Li x CoO 2 is 0.24 or less. Therefore, in the positive electrode active material 100 that can be used as one embodiment of the present invention, short-circuiting is unlikely to occur when x in Li x CoO 2 is maintained at 0.24 or less, so the safety of the lithium ion battery is improved. do.
- FIG. 4 shows the crystal structure of the inside 100b of the positive electrode active material 100 when x in Li x CoO 2 is about 1, 0.2, and about 0.15. Since the inside 100b occupies most of the volume of the positive electrode active material 100 and is a portion that greatly contributes to charging and discharging, it can be said that displacement of the CoO 2 layer and volume change are the most problematic portions.
- the positive electrode active material 100 has the same R-3m(O3) crystal structure as conventional lithium cobaltate.
- the positive electrode active material 100 has a crystal structure belonging to the trigonal space group R-3m. This is the same symmetry of CoO2 layer as O3. Therefore, this crystal structure is called an O3' type crystal structure. This crystal structure is shown in FIG. 4 with R-3m(O3)'.
- the crystal structure of the O3′ type has the coordinates of cobalt and oxygen in the unit cell as Co (0, 0, 0.5), O (0, 0, x), within the range of 0.20 ⁇ x ⁇ 0.25 can be shown as
- the positive electrode active material 100 that can be used as one embodiment of the present invention has a crystal structure belonging to the monoclinic space group P2/m. There is one CoO 2 layer in the unit cell. Further, lithium present in the positive electrode active material 100 at this time is about 15 atomic % of the discharged state. Therefore, in this specification and the like, this crystal structure is referred to as a "monoclinic O1(15) type crystal structure". This crystal structure is shown in FIG. 4 labeled P2/m monoclinic O1 (15).
- the crystal structure of the monoclinic O1(15) type has the coordinates of cobalt and oxygen in the unit cell as Co1(0.5,0,0.5), Co2(0,0.5,0.5), O1(X O1 , 0, Z O1 ), 0.23 ⁇ X O1 ⁇ 0.24, 0.61 ⁇ Z O1 ⁇ 0.65, O2(X O2 , 0.5, Z O2 ), 0.75 ⁇ X 02 ⁇ 0.78 and 0.68 ⁇ Z 02 ⁇ 0.71.
- ions such as cobalt, nickel, and magnesium occupy 6 oxygen coordination positions.
- light elements such as lithium may occupy 4-coordinated positions of oxygen in some cases.
- the difference in volume per cobalt atom of the same number in the R-3m(O3) in the discharged state and the O3′ type crystal structure is 2.5% or less, more specifically 2.2% or less, typically 1 0.8%.
- the difference in volume per cobalt atom of the same number of R-3m (O3) in the discharged state and the monoclinic O1 (15) type crystal structure is 3.3% or less, more specifically 3.0% or less, representative Typically it is 2.5%.
- Table 1 shows the difference in volume per cobalt atom between R-3m(O3) in the discharged state, O3', monoclinic O1(15), H1-3 type, and trigonal O1.
- the lattice constant of each crystal structure used for the calculation in Table 1 is the literature value (ICSD coll.code.172909 and 88721) and non Reference can be made to Patent Literature 1.
- O3′ and monoclinic O1(15) can be calculated from XRD experimental values.
- the change in crystal structure is similar to that of the conventional positive electrode active material. More restrained than matter. Also, the change in volume when compared per the same number of cobalt atoms is suppressed. Therefore, even if the positive electrode active material 100 is repeatedly charged and discharged so that x becomes 0.24 or less, the crystal structure does not easily collapse, and a decrease in charge/discharge capacity during charge/discharge cycles is suppressed.
- the positive electrode active material 100 since more lithium can be stably used than the conventional positive electrode active material, the positive electrode active material 100 has a large discharge capacity per weight and per volume. Therefore, by using the positive electrode active material 100, a secondary battery with high discharge capacity per weight and per volume can be manufactured.
- the positive electrode active material 100 sometimes has an O3′ type crystal structure when x in Li x CoO 2 is 0.15 or more and 0.24 or less, and x exceeds 0.24. It is presumed to have an O3' type crystal structure even when the E is 0.27 or less.
- x in Li x CoO 2 exceeds 0.1 and is 0.2 or less, typically x is 0.15 or more and 0.17 or less, it has a monoclinic O1(15) type crystal structure. It has been confirmed that there are cases.
- the crystal structure is affected not only by x in Li x CoO 2 but also by the number of charge/discharge cycles, charge/discharge current, temperature, electrolyte, etc., x is not necessarily limited to the above range.
- the positive electrode active material 100 may have only the O3′ type or only the monoclinic O1(15) type. or both crystal structures. Further, not all the particles in the interior 100b of the positive electrode active material 100 may have the crystal structure of the O3′ type and/or the monoclinic O1(15) type. It may contain other crystal structures, or may be partially amorphous.
- the state in which x in Li x CoO 2 is small can be rephrased as the state of being charged at a high charging voltage.
- CC/CV constant current/constant voltage
- the H1-3 type crystal structure appears in the conventional cathode active material. Therefore, a charging voltage of 4.6 V or more based on the potential of lithium metal can be said to be a high charging voltage.
- the charging voltage is expressed based on the potential of lithium metal.
- the positive electrode active material 100 that can be used as one embodiment of the present invention has a crystal structure having R-3m(O3) symmetry even when charged at a high charging voltage, for example, a voltage of 4.6 V or higher at 25°C. can be held.
- it can be said that it is preferable because it can have an O3' type crystal structure when charged at a higher charging voltage, for example, a voltage of 4.65 V or more and 4.7 V or less at 25°C.
- the monoclinic O1(15) type crystal structure can be obtained when charged at a higher charging voltage, for example, a voltage exceeding 4.7 V and not more than 4.8 V at 25°C.
- the H1-3 type crystal structure may be observed only when the charging voltage is further increased.
- the crystal structure is affected by the number of charge-discharge cycles, charge-discharge current, temperature, electrolyte, etc. Therefore, when the charge voltage is lower, for example, even if the charge voltage is 4.5 V or more and less than 4.6 V at 25 ° C. , the positive electrode active material 100 that can be used as one embodiment of the present invention may have an O3′ crystal structure.
- a monoclinic O1(15) type crystal structure may be obtained.
- the voltage of the secondary battery is lowered by the potential of the graphite.
- the potential of graphite is about 0.05 V to 0.2 V with respect to the potential of lithium metal. Therefore, in the case of a secondary battery using graphite as a negative electrode active material, it has a similar crystal structure at a voltage obtained by subtracting the potential of graphite from the above voltage.
- lithium is shown to be present at all lithium sites with equal probability, but the present invention is not limited to this. It may exist disproportionately at some lithium sites, or may have symmetry such as monoclinic O1 (Li 0.5 CoO 2 ) shown in FIG. 5, for example.
- the lithium distribution can be analyzed, for example, by neutron diffraction.
- the O3′ and monoclinic O1(15) type crystal structures are similar to the CdCl 2 type crystal structure, although they have lithium randomly between the layers.
- the crystal structure similar to this CdCl2 type is close to the crystal structure when lithium nickelate is charged to Li0.06NiO2 , but pure lithium cobalt oxide or a layered rock salt type positive electrode active material containing a large amount of cobalt is used. It is known that CdCl 2 -type crystal structure is not usually taken.
- the concentration gradient of the additive element is preferably the same at multiple locations on the surface layer portion 100 a of the positive electrode active material 100 .
- the barrier film derived from the additive element is homogeneously present on the surface layer portion 100a. Even if there is a barrier film on a part of the surface layer portion 100a, if there is a portion without the barrier film, stress may concentrate on the portion without the barrier film. If the stress concentrates on a portion of the positive electrode active material 100, defects such as cracks may occur there, leading to cracking of the positive electrode active material and a decrease in discharge capacity.
- FIGS. 6A1 and 6A2 show enlarged views of the vicinity of CD in FIG. 1A1.
- FIG. 6A1 shows an example of the distribution of the additional element X near C-D in FIG. 1A1
- FIG. 6A2 shows an example of the distribution of the additional element Y near C-D.
- the vicinity of C-D has a layered rock salt type crystal structure of R-3m, and the surface is (001) oriented.
- the (001) oriented surface may have a different distribution of additive elements than other surfaces.
- the (001) oriented surface and its surface layer portion 100a have a distribution of one or more concentration peaks selected from the additive element X and the additive element Y, which is higher than that of the surface other than the (001) oriented surface. It may be limited to a shallow portion.
- the (001) oriented surface and its surface layer portion 100a may have a lower concentration of one or more elements selected from the additive element X and the additive element Y than the surface other than the (001) oriented surface.
- the (001)-oriented surface and its surface layer portion 100a may have a concentration of one or more selected from the additive element X and the additive element Y below the detection limit.
- the CoO 2 layer is relatively stable, it is more stable for the surface of the positive electrode active material 100 to be (001) oriented. The main diffusion paths of lithium ions during charging and discharging are not exposed on the (001) plane.
- the surface other than the (001) orientation and the surface layer portion 100a are important regions for maintaining the diffusion path of lithium ions, and at the same time, they are the regions where lithium ions are first desorbed, so they tend to be unstable. Therefore, reinforcing the surface other than the (001) orientation and the surface layer portion 100a is extremely important for maintaining the crystal structure of the positive electrode active material 100 as a whole.
- the distribution of the additive element on the surface other than the (001)-oriented surface and the surface layer portion 100a is as shown in FIGS. 1B1 and 1B2.
- the (001)-oriented surface and its surface layer portion 100a may have a low or no additive element concentration as described above.
- the additive element spreads mainly through the diffusion path of lithium ions, which will be described later. Therefore, the distribution of the additive element on the surface other than the (001)-oriented surface and the surface layer portion 100a can be easily controlled within a preferable range.
- FIG. 6B1 shows the results of calculations for the (104)-oriented surface and its surface layer portion 100a. It was calculated by classical molecular dynamics method. LiCoO 2 (LCO) was placed in the lower part of the system, and LiF and MgF 2 as magnesium source, lithium source and fluorine source were placed in the upper part of the system.
- the ensemble is NVT (canonical ensemble), the density of the initial structure is 1.8 g/cm 3 , the temperature of the system is 2000 K, the elapsed time is 100 psec, the potential is optimized with the LCO crystal structure, and the other atoms are UFF. (Universal Force Field), the number of atoms in the system is about 10,000, and the charge of the system is neutral. In order to simplify the drawing, only Co atoms and Mg atoms are shown.
- Fig. 6B2 is the result of calculation up to 200psec
- Fig. 6B3 is up to 1200psec.
- magnesium diffuses in the following process.
- Lithium is desorbed from LCO by heat.
- Magnesium enters the lithium layer of the LCO and diffuses inside.
- Lithium derived from LiF enters the lithium layer of LCO and supplements the lithium desorbed in (1).
- FIG. 6C is the result of calculation similar to FIG. 6B1, except for the (001) orientation. In FIG. 6C, it can be seen that the magnesium atoms remain on the surface of the LCO.
- the surface of the positive electrode active material 100 is smooth and has few irregularities, but not necessarily the entire surface of the positive electrode active material 100 .
- a composite oxide having an R-3m layered rocksalt type crystal structure is prone to slip on a plane parallel to the (001) plane, for example, a plane in which lithium is arranged.
- a slip is also called a stacking fault, and refers to a state in which LiCoO 2 is deformed along the lattice pattern direction (ab plane direction) by pressing. Deformation includes shifting the checkered fringes back and forth. When the lattice fringes are shifted back and forth, a step occurs on the surface in the direction perpendicular to the lattice fringes (c-axis direction). For example, as shown in FIG. 7A, when the (001) plane exists, there is a case where slip occurs parallel to the (001) plane as indicated by the arrow in FIG. be.
- FIGS. 7C1 and 7C2 show enlarged views of the vicinity of E-F.
- the additional element X and the additional element Y are not distributed.
- At least part of the additive element included in the positive electrode active material 100 that can be used as one embodiment of the present invention is more preferably unevenly distributed in the grain boundary 101 and its vicinity.
- the magnesium concentration in the grain boundary 101 of the positive electrode active material 100 and its vicinity may be higher than in other regions of the interior 100b.
- the fluorine concentration in the grain boundary 101 and its vicinity is preferably higher than that in other regions of the interior 100b.
- the nickel concentration in the grain boundary 101 and its vicinity is preferably higher than that in other regions of the interior 100b.
- the aluminum concentration in the grain boundary 101 and its vicinity is higher than that in other regions of the interior 100b.
- the grain boundary 101 is one of planar defects, it tends to be unstable like the surface, and the crystal structure tends to start changing. Therefore, by increasing the concentration of the additive element at and near the grain boundary 101, such a change in the crystal structure can be more effectively suppressed.
- the magnesium concentration and the fluorine concentration at and near the grain boundaries 101 are high, even if cracks are generated along the grain boundaries 101 of the positive electrode active material 100 that can be used as one embodiment of the present invention, the cracks are generated. Magnesium concentration and fluorine concentration are high in the vicinity of the surface. Therefore, the corrosion resistance to hydrofluoric acid can be improved even in the positive electrode active material after cracks have occurred.
- the median diameter (D50) is preferably 1 ⁇ m or more and 100 ⁇ m or less, more preferably 2 ⁇ m or more and 40 ⁇ m or less, and even more preferably 5 ⁇ m or more and 30 ⁇ m or less. Alternatively, it is preferably 1 ⁇ m or more and 40 ⁇ m or less.
- it is preferably 1 ⁇ m or more and 30 ⁇ m or less. Alternatively, it is preferably 2 ⁇ m or more and 100 ⁇ m or less. Alternatively, it is preferably 2 ⁇ m or more and 30 ⁇ m or less. Alternatively, it is preferably 5 ⁇ m or more and 100 ⁇ m or less. Alternatively, it is preferably 5 ⁇ m or more and 40 ⁇ m or less.
- the positive electrode active material 100 that can be used as one embodiment of the present invention has an O3′ type and/or a monoclinic O1(15) type crystal structure when x in Li x CoO 2 is small in a certain positive electrode active material. Whether or not the positive electrode having a positive electrode active material in which x in Li x CoO 2 is small is analyzed using XRD, electron beam diffraction, neutron beam diffraction, electron spin resonance (ESR), nuclear magnetic resonance (NMR), etc. can be determined by doing
- XRD can analyze the symmetry of transition metals such as cobalt in the positive electrode active material with high resolution, can compare the crystallinity level and crystal orientation, and can analyze the periodic strain and crystallite size of the lattice. It is preferable in that sufficient accuracy can be obtained even if the positive electrode obtained by disassembling the secondary battery is measured as it is.
- XRDs in powder XRD, a diffraction peak reflecting the crystal structure of the inside 100b of the positive electrode active material 100, which occupies most of the volume of the positive electrode active material 100, is obtained.
- the positive electrode active material 100 that can be used as one embodiment of the present invention is characterized by little change in crystal structure when x in Li x CoO 2 is 1 and when x is 0.24 or less. is.
- a material in which the crystal structure occupies 50% or more of which the change in crystal structure is large when charged at a high voltage (for example, 4.6 V) is not preferable because it cannot withstand charging and discharging at a high voltage.
- the crystal structure of O3′ type or monoclinic O1(15) type may not be obtained only by adding an additive element.
- lithium cobalt oxide with magnesium and fluorine, or lithium cobalt oxide with magnesium and aluminum, x in Li x CoO 2 is 0.24 depending on the concentration and distribution of the additive element.
- the O3′ type and/or monoclinic O1(15) type crystal structure accounts for 60% or more, and cases where the H1-3 type crystal structure accounts for 50% or more.
- the positive electrode active material 100 that can be used as one embodiment of the present invention, when x is too small, such as 0.1 or less, or under conditions where the charging voltage exceeds 4.9 V, H1-3 type or three-way Crystal O1-type crystal structures may also occur. Therefore, in order to determine whether the positive electrode active material 100 can be used as one aspect of the present invention, analysis of the crystal structure such as XRD and information such as charge capacity or charge voltage are required. .
- the positive electrode active material with small x may undergo a change in crystal structure when exposed to the atmosphere.
- the O3' and monoclinic O1(15) crystal structures may change to the H1-3 crystal structure. Therefore, all samples to be analyzed for crystal structure are preferably handled in an inert atmosphere such as an argon atmosphere.
- whether or not the distribution of additive elements possessed by a positive electrode active material is in the state described above can be determined, for example, by XPS, energy dispersive X-ray spectroscopy (EDX), EPMA (electron probe microanalysis) or the like can be used for determination.
- XPS energy dispersive X-ray spectroscopy
- EPMA electron probe microanalysis
- the crystal structure of the surface layer portion 100a, the crystal grain boundary 101, and the like can be analyzed by electron beam diffraction or the like of the cross section of the positive electrode active material 100.
- Charging for determining whether a certain composite oxide is the positive electrode active material 100 that can be used as one aspect of the present invention is performed by, for example, using a coin cell (CR2032 type, diameter 20 mm height 3.2 mm) and charge it.
- the charging method described below is a condition for confirming physical properties of the positive electrode active material 100 that can be used as one embodiment of the present invention. Therefore, the electrolyte and the like described below for the structure other than the positive electrode active material are different from the structure of the lithium ion battery which is one embodiment of the present invention.
- a slurry obtained by mixing a positive electrode active material, a conductive material, and a binder can be applied to a positive current collector made of aluminum foil.
- Lithium metal can be used as an example of the negative electrode (counter electrode).
- the potential of the secondary battery and the potential of the positive electrode are different. Voltage and potential in this specification and the like are the potential of the positive electrode when the counter electrode is lithium metal, unless otherwise specified.
- LiPF 6 lithium fluorophosphate
- a polypropylene porous film with a thickness of 25 ⁇ m can be used as an example of the separator.
- the positive electrode can and the negative electrode can, those made of stainless steel (SUS) can be used.
- SUS stainless steel
- Constant current charging also referred to as CC charging
- the temperature should be 25°C or 45°C.
- XRD XRD can be performed in a sealed container with an argon atmosphere.
- the charging and discharging conditions for the multiple times may be different from the above charging conditions.
- charging is performed by constant current charging at a current value of 100 mA/g to an arbitrary voltage (for example, 4.6 V, 4.65 V, 4.7 V, 4.75 V or 4.8 V) until the current value reaches 10 mA/g. It can be charged at a constant voltage and discharged at a constant current of 2.5 V and 100 mA/g.
- constant current discharge can be performed, for example, at 2.5 V and a current value of 100 mA/g.
- XRD XRD
- the device and conditions for XRD measurement are not particularly limited. For example, it can be measured using the following apparatus and conditions.
- XRD device D8 ADVANCE manufactured by Bruker AXS X-ray source: CuK ⁇ 1 -line output: 40 kV, 40 mA Slit width: Div. Slit, 0.5° Detector: LynxEye Scanning method: 2 ⁇ / ⁇ continuous scan Measurement range (2 ⁇ ): 15° to 90° Step width (2 ⁇ ): 0.01° setting Counting time: 1 second/step Sample table rotation: 15 rpm
- the measurement sample is powder, it can be set by placing it in a glass sample holder or by sprinkling the sample on a greased silicone non-reflective plate.
- the sample to be measured is a positive electrode
- the positive electrode can be attached to the substrate with a double-sided tape, and the positive electrode active material layer can be set according to the measurement surface required by the device.
- FIGS. 10A and 10B show the ideal powder XRD patterns by CuK ⁇ 1 line calculated from models of the O3′ type crystal structure, the monoclinic O1(15) type crystal structure, and the H1-3 type crystal structure.
- 10A and 10B show the XRD patterns of the O3′ type crystal structure, the monoclinic O1(15) type crystal structure, and the H1-3 type crystal structure.
- FIG. 10B is an enlarged view of the range of 2 ⁇ from 42° to 46°.
- the patterns of LiCoO 2 (O3) and CoO 2 (O1) were created using Reflex Powder Diffraction, which is one of the modules of Materials Studio (BIOVIA) from crystal structure information obtained from ICSD (Inorganic Crystal Structure Database). bottom.
- the pattern of the H1-3 type crystal structure was created similarly from the crystal structure information described in Non-Patent Document 1.
- the crystal structure patterns of the O3′ type and the monoclinic O1(15) type are estimated from the XRD pattern of a positive electrode active material that can be used as one embodiment of the present invention, and TOPAS ver. 3 (Crystal structure analysis software manufactured by Bruker) was used for fitting, and an XRD pattern was created in the same manner as the others.
- the positive electrode active material 100 that can be used as one embodiment of the present invention has an O3′-type and/or monoclinic O1(15)-type crystal structure when x in Li x CoO 2 is small. may not be the O3′ type and/or the monoclinic O1(15) type crystal structure. It may contain other crystal structures, or may be partially amorphous. However, when the XRD pattern is subjected to Rietveld analysis, the crystal structure of O3′ type and/or monoclinic O1(15) type is preferably 50% or more, more preferably 60% or more, It is more preferably 66% or more.
- a positive electrode active material with sufficiently excellent cycle characteristics has a crystal structure of O3′ type and/or monoclinic O1(15) type of 50% or more, more preferably 60% or more, and still more preferably 66% or more. be able to.
- the crystal structure of O3' type and / or monoclinic O1 (15) type is preferably 35% or more, It is more preferably 40% or more, and even more preferably 43% or more.
- each diffraction peak after charging is sharp, that is, the half width is narrow.
- the crystallite size of the O3′ type and monoclinic O1(15) crystal structure of the positive electrode active material 100 is reduced to only about 1/20 of LiCoO 2 (O3) in the discharged state. Therefore, even under the same XRD measurement conditions as for the positive electrode before charge/discharge, when x in Li x CoO 2 is small, distinct O3′-type and monoclinic O1(15) crystal structure peaks can be observed.
- conventional LiCoO 2 has a smaller crystallite size and a broader and smaller peak, even if a part of it can have a crystal structure similar to the O3′ type and monoclinic O1(15). The crystallite size can be obtained from the half width of the XRD peak.
- the Jahn-Teller effect has little influence as described above.
- transition metals such as nickel and manganese may be added as long as the influence of the Jahn-Teller effect is small.
- FIG. 11 shows the lattice constants of the a-axis and c-axis calculated using XRD when the positive electrode active material 100 that can be used as one embodiment of the present invention has a layered rock salt crystal structure and contains cobalt and nickel. The results are shown.
- FIG. 11A shows the results for the a-axis
- FIG. 11B shows the results for the c-axis.
- the XRD pattern used for these calculations is the powder after synthesizing the positive electrode active material and before incorporating it into the positive electrode.
- the nickel concentration on the horizontal axis indicates the concentration of nickel when the sum of the number of atoms of cobalt and nickel is 100%.
- the positive electrode active material was produced according to the production method of FIGS. 15A and 15C, except that the aluminum source was not used.
- FIG. 12 shows the a-axis and c-axis lattice constants estimated by XRD in the case where a positive electrode active material that can be used as one embodiment of the present invention has a layered rock salt crystal structure and contains cobalt and manganese.
- the results are shown.
- FIG. 12A shows the results for the a-axis
- FIG. 12B shows the results for the c-axis.
- the lattice constant shown in FIG. 12 is the powder after synthesis of the positive electrode active material, and is obtained by XRD measured before incorporating into the positive electrode.
- the manganese concentration on the horizontal axis indicates the concentration of manganese when the sum of the number of atoms of cobalt and manganese is taken as 100%.
- the positive electrode active material was produced according to the production method of FIGS. 15A and 15C except that a manganese source was used instead of a nickel source and an aluminum source was not used.
- FIG. 11C shows the value obtained by dividing the lattice constant of the a-axis by the lattice constant of the c-axis (a-axis/c-axis) for the positive electrode active materials whose lattice constant results are shown in FIGS. 11A and 11B.
- FIG. 12C shows the value obtained by dividing the lattice constant of the a-axis by the lattice constant of the c-axis (a-axis/c-axis) for the positive electrode active materials whose lattice constant results are shown in FIGS. 12A and 12B.
- the concentration of manganese is preferably 4% or less, for example.
- nickel concentration and manganese concentration ranges described above do not necessarily apply to the surface layer portion 100a. That is, in the surface layer portion 100a, the concentration may be higher than the concentration described above.
- the positive electrode active material 100 in a state in which charging and discharging are not performed, or in a discharged state, which can be estimated from the XRD pattern
- the a-axis lattice constant is greater than 2.814 ⁇ 10 -10 m and less than 2.817 ⁇ 10 -10 m
- the c-axis lattice constant is 14.05 ⁇ 10 -10 m. It has been found to be preferably greater than 10 m and less than 14.07 ⁇ 10 ⁇ 10 m.
- the state in which charging and discharging are not performed may be, for example, the state of powder before manufacturing the positive electrode of the secondary battery.
- the value obtained by dividing the lattice constant of the a-axis by the lattice constant of the c-axis is It is preferably greater than 0.20000 and less than 0.20049.
- XRD analysis shows a first peak at 2 ⁇ of 18.50° to 19.30°. is observed, and a second peak may be observed at 2 ⁇ of 38.00° or more and 38.80° or less.
- XPS X-ray photoelectron spectroscopy
- inorganic oxides it is possible to analyze a region from the surface to a depth of about 2 nm to 8 nm (usually 5 nm or less) when monochromatic aluminum K ⁇ rays are used as the X-ray source. It is possible to quantitatively analyze the concentration of each element. Also, the bonding state of elements can be analyzed by narrow scan analysis. In most cases, the quantitative accuracy of XPS is about ⁇ 1 atomic %, and the detection limit is about 1 atomic % although it depends on the element.
- the concentration of one or more elements selected from the additive elements is higher in the surface layer portion 100a than in the inner portion 100b.
- concentration of one or more selected additive elements in the surface layer portion 100 a is preferably higher than the average additive element concentration of the entire positive electrode active material 100 . Therefore, for example, the concentration of one or two or more selected from the additive elements in the surface layer portion 100a measured by XPS or the like is measured by ICP-MS (inductively coupled plasma mass spectrometry), GD-MS (glow discharge mass spectrometry), or the like. It can be said that it is preferably higher than the average additive element concentration of the entire positive electrode active material 100 measured by .
- the concentration of magnesium in at least a portion of the surface layer portion 100 a measured by XPS or the like is higher than the concentration of magnesium in the entire positive electrode active material 100 .
- the concentration of nickel in at least part of the surface layer portion 100 a is higher than the nickel concentration in the entire positive electrode active material 100 .
- the concentration of aluminum in at least part of the surface layer portion 100 a is higher than the concentration of aluminum in the entire positive electrode active material 100 .
- the concentration of fluorine in at least a portion of the surface layer portion 100 a is higher than the concentration of fluorine in the entire positive electrode active material 100 .
- the surface and surface layer portion 100a of the positive electrode active material 100 that can be used as one embodiment of the present invention do not contain carbonates, hydroxyl groups, and the like chemically adsorbed after the positive electrode active material 100 is manufactured.
- the electrolyte, binder, conductive material, and compounds derived from these adhered to the surface of the positive electrode active material 100 are not included. Therefore, when quantifying the elements contained in the positive electrode active material, correction may be made to exclude carbon, hydrogen, excess oxygen, excess fluorine, etc. that can be detected by surface analysis such as XPS. For example, in XPS, it is possible to separate the types of bonds by analysis, and correction may be performed to exclude binder-derived C—F bonds.
- the samples such as the positive electrode active material and the positive electrode active material layer are washed in order to remove the electrolytic solution, binder, conductive material, or compounds derived from these adhered to the surface of the positive electrode active material. may be performed. At this time, lithium may dissolve into the solvent or the like used for washing, but even in such a case, since the additive element is difficult to dissolve, the atomic number ratio of the additive element is not affected.
- the concentration of the additive element may be compared in terms of the ratio with cobalt. It is preferable to use the ratio with cobalt, because it is possible to reduce the influence of the chemically adsorbed carbonate and the like after the production of the positive electrode active material for comparison.
- the atomic ratio Mg/Co of magnesium and cobalt determined by XPS analysis of the surface or surface layer portion of the positive electrode active material is preferably 0.4 or more and 1.5 or less.
- Mg/Co is preferably 0.001 or more and 0.06 or less by ICP-MS analysis for the entire positive electrode active material.
- the surface layer portion 100a of the positive electrode active material 100 has a higher concentration of lithium and cobalt than each additive element in order to sufficiently secure the lithium intercalation and deintercalation paths.
- concentration of lithium and cobalt in the surface layer portion 100a is preferably higher than the concentrations of one or more additive elements selected from the additive elements possessed by the surface layer portion 100a measured by XPS or the like. can be done.
- concentration of cobalt in at least a portion of the surface layer portion 100a measured by XPS or the like is preferably higher than the concentration of magnesium in at least a portion of the surface layer portion 100a measured by XPS or the like.
- the concentration of lithium is higher than the concentration of magnesium.
- the concentration of cobalt is higher than the concentration of nickel.
- the lithium concentration be higher than the nickel concentration.
- the concentration of cobalt is preferably higher than that of aluminum.
- the lithium concentration be higher than the aluminum concentration.
- the concentration of cobalt is preferably higher than that of fluorine. Similarly, a higher concentration of lithium than fluorine is preferred.
- the additive element Y including aluminum be distributed widely in a deep region, for example, a region with a depth of 5 nm or more and 50 nm or less from the surface. Therefore, although the additive element Y including aluminum is detected in the analysis of the entire positive electrode active material 100 using ICP-MS, GD-MS, etc., it is more preferable that this is below the detection limit in XPS or the like.
- the number of magnesium atoms is 0.4 times or more and 1.2 times or less with respect to the number of cobalt atoms. is preferable, and 0.65 times or more and 1.0 times or less is more preferable.
- the number of nickel atoms is preferably 0.15 times or less, more preferably 0.03 to 0.13 times the number of cobalt atoms.
- the number of aluminum atoms is preferably 0.12 times or less, more preferably 0.09 times or less, relative to the number of cobalt atoms.
- the number of fluorine atoms is preferably 0.3 to 0.9 times, more preferably 0.1 to 1.1 times, the number of cobalt atoms.
- monochromatic aluminum K ⁇ rays can be used as the X-ray source.
- the extraction angle may be set to 45°, for example.
- it can be measured using the following apparatus and conditions.
- Measurement spectrum wide scan, narrow scan for each detected element
- the peak indicating the binding energy between fluorine and another element is preferably 682 eV or more and less than 685 eV. More preferably, it is about 3 eV. This value is different from both 685 eV, which is the binding energy of lithium fluoride, and 686 eV, which is the binding energy of magnesium fluoride. That is, in the case where the positive electrode active material 100 that can be used as one embodiment of the present invention contains fluorine, it is preferably a bond other than lithium fluoride and magnesium fluoride.
- the peak indicating the binding energy between magnesium and another element is preferably 1302 eV or more and less than 1304 eV, and 1303 eV. It is more preferable that the degree is This value is different from 1305 eV, which is the binding energy of magnesium fluoride, and is close to the binding energy of magnesium oxide. That is, in the case where the positive electrode active material 100 that can be used as one embodiment of the present invention contains magnesium, a bond other than magnesium fluoride is preferable.
- EDX It is preferable that one or two or more elements selected from additive elements contained in the positive electrode active material 100 have a concentration gradient. Further, it is more preferable that the positive electrode active material 100 has different depths from the surface of the concentration peak depending on the additive element.
- the concentration gradient of the additive element is obtained by, for example, exposing a cross section of the positive electrode active material 100 by FIB (Focused Ion Beam) or the like, and subjecting the cross section to energy dispersive X-ray spectroscopy (EDX), EPMA (electron It can be evaluated by analyzing using a probe microanalysis) or the like.
- EDX surface analysis measuring while scanning the area and evaluating the area two-dimensionally.
- line analysis measuring while linearly scanning to evaluate the distribution of the atomic concentration in the positive electrode active material.
- line analysis data of a linear region extracted from EDX surface analysis is sometimes called line analysis.
- point analysis measuring a certain area without scanning is called point analysis.
- EDX surface analysis for example, elemental mapping
- concentration distribution and maximum value of additive elements can be analyzed by EDX-ray analysis.
- analysis of thinning a sample like STEM-EDX can analyze the concentration distribution in the depth direction from the surface to the center of the positive electrode active material in a specific region without being affected by the distribution in the depth direction. , is more preferred.
- the concentration of each additive element, particularly the additive element X, in the surface layer portion 100a is higher than that in the inner portion 100b. is preferred.
- the magnesium concentration in the surface layer portion 100a is higher than that in the inner portion 100b.
- the magnesium concentration peak of the surface layer portion 100a preferably exists at a depth of 3 nm from the surface toward the center of the positive electrode active material 100, and may exist at a depth of 1 nm. More preferably, it exists up to a depth of 0.5 nm.
- the concentration of magnesium attenuates to 60% or less of the peak at a point 1 nm deep from the peak top.
- the peak is attenuated to 30% or less at a point 2 nm deep from the peak top.
- the density peak means the maximum value of the density.
- the distribution of fluorine preferably overlaps with the distribution of magnesium.
- the difference in the depth direction between the fluorine concentration peak and the magnesium concentration peak is preferably within 10 nm, more preferably within 3 nm, and even more preferably within 1 nm.
- the peak of the fluorine concentration in the surface layer portion 100a preferably exists at a depth of 3 nm from the surface toward the center of the positive electrode active material 100, and may exist at a depth of 1 nm. More preferably, it exists up to a depth of 0.5 nm. Further, it is preferable that the peak of the fluorine concentration is located slightly closer to the surface side than the peak of the magnesium concentration, because the resistance to hydrofluoric acid increases. For example, the fluorine concentration peak is more preferably 0.5 nm or more closer to the surface than the magnesium concentration peak, and more preferably 1.5 nm or more closer to the surface.
- the nickel concentration peak of the surface layer portion 100a preferably exists at a depth of 3 nm from the surface toward the center of the positive electrode active material 100, and up to a depth of 1 nm. It is more preferable to exist at a depth of 0.5 nm.
- the distribution of nickel preferably overlaps with the distribution of magnesium.
- the difference in the depth direction between the nickel concentration peak and the magnesium concentration peak is preferably within 10 nm, more preferably within 3 nm, and even more preferably within 1 nm.
- the concentration peak of magnesium, nickel, or fluorine is closer to the surface than the aluminum concentration peak of the surface layer portion 100a in EDX-ray analysis.
- the peak of the aluminum concentration preferably exists at a depth of 0.5 nm or more and 50 nm or less, more preferably 5 nm or more and 50 nm or less, from the surface toward the center of the positive electrode active material 100 .
- the atomic ratio (Mg/Co) between magnesium Mg and cobalt Co at the magnesium concentration peak is 0.05 or more. 0.6 or less is preferable, and 0.1 or more and 0.4 or less is more preferable.
- the atomic ratio (Al/Co) of aluminum Al and cobalt Co at the aluminum concentration peak is preferably 0.05 or more and 0.6 or less, more preferably 0.1 or more and 0.45 or less.
- the atomic number ratio (Ni/Co) of nickel Ni and cobalt Co at the nickel concentration peak is preferably 0 or more and 0.2 or less, more preferably 0.01 or more and 0.1 or less.
- the atomic ratio (F/Co) of fluorine F to cobalt Co at the fluorine concentration peak is preferably 0 or more and 1.6 or less, more preferably 0.1 or more and 1.4 or less.
- the surface of the positive electrode active material 100 in the EDX-ray analysis results can be estimated, for example, as follows. For an element such as oxygen or cobalt uniformly present in the interior 100b of the positive electrode active material 100, the point at which the amount detected in the interior 100b is 1/2 is defined as the surface.
- the surface can be estimated using the detected amount of oxygen. Specifically, first, the average value O ave of the oxygen concentration is obtained from the region where the detected amount of oxygen in the interior 100b is stable. At this time, if oxygen O background , which is considered to be due to chemisorption or background, is detected in a region that can be clearly determined to be outside the surface, subtract O background from the measured value and then take the average oxygen concentration O ave . be able to. It can be estimated that the measurement point showing the value of 1/2 of this average value O ave , that is, the measurement value closest to 1/2 O ave , is the surface of the positive electrode active material.
- the surface can be estimated in the same way as above using the detected amount of cobalt.
- it can be similarly estimated using the sum of detected amounts of a plurality of transition metals. Detected amounts of transition metals such as cobalt are suitable for surface estimation because they are less susceptible to chemisorption.
- the ratio (A/Co) between the additive element A and cobalt Co in the vicinity of the grain boundary 101 is 0.020 or more and 0.50 or less. It is preferably 0.025 or more and 0.30 or less, and still more preferably 0.030 or more and 0.20 or less. It should be noted that these upper and lower limits can be freely combined unless otherwise specified in this specification.
- the additive element is magnesium
- the atomic ratio (Mg/Co) of magnesium and cobalt in the vicinity of the grain boundary 101 is 0 when the surface or surface layer of the positive electrode active material 100 is subjected to line analysis or surface analysis.
- 0.020 or more and 0.50 or less are preferable, 0.025 or more and 0.30 or less are more preferable, and 0.030 or more and 0.20 or less are still more preferable.
- ⁇ EPMA ⁇ EPMA electron probe microanalysis
- Surface analysis can analyze the distribution of each element.
- one or more elements selected from the additive elements have a concentration gradient, similar to the EDX analysis results. is preferred. Further, it is more preferable that the depth from the surface of the concentration peak differs depending on the additive element. The preferred range of the concentration peak of each additive element is also the same as in the case of EDX.
- EPMA analyzes the area from the surface to a depth of about 1 ⁇ m. Therefore, the quantitative value of each element may differ from the measurement results obtained using other analytical methods. For example, when the surface analysis of the positive electrode active material 100 is performed by EPMA, the concentration of each additive element present in the surface layer portion 100a may be lower than the result of XPS.
- the positive electrode active material 100 that can be used as one embodiment of the present invention exhibits a characteristic voltage change during charging in some cases.
- a change in voltage can be read from a dQ/dVvsV curve obtained by differentiating the capacity (Q) of the charge curve by the voltage (V) (dQ/dV).
- Q capacity of the charge curve by the voltage (V)
- V voltage
- a non-equilibrium phase change means a phenomenon that causes a nonlinear change in physical quantity.
- the positive electrode active material 100 may have a broad peak near 4.55 V in the dQ/dVvsV curve.
- the peak around 4.55 V reflects the change in voltage during the phase change from the O3 type to the O3' type. Therefore, the broadness of this peak means less change in the energy required for lithium to be abstracted, ie less change in the crystal structure, than when the peak is sharp. The smaller these changes are, the less the effect of displacement and volume change of the CoO 2 layer is, which is preferable.
- the half width of the first peak is 0.10 V or more. and is sufficiently broad, which is preferable.
- the half width of the first peak is defined as the first peak and the first
- the average value HWHM 1 between the minimum value of , and the first peak and the second minimum value when the minimum value of the dQ/dV value appearing between 4.6 V and 4.8 V is the second minimum value
- the charging when obtaining the dQ/dVvsV curve can be constant current charging at 10 mA/g up to 4.9 V, for example. Moreover, when obtaining the dQ/dV of the initial charge, it is preferable to discharge the battery to 2.5 V at 100 mA/g before measurement, and then start the charging.
- the setting of the data capture interval during charging can be set to capture the voltage and current at intervals of 1 second or when the voltage fluctuates by 1 mV, for example.
- the charge capacity is the sum of the current value and time.
- the difference between the n-th and n+1-th data of the charge capacity data be the n-th value of the capacity change dQ.
- the difference between the n-th and (n+1)-th data of the voltage data is taken as the n-th value of the voltage change dV.
- dQ/dV may be obtained from a moving average of a certain number of intervals for the difference in voltage and charge capacity.
- the number of sections can be 500, for example.
- the average value of dQ from the nth to the n+500th is calculated, and similarly the average of the dV from the nth to the n+500th is calculated.
- dQ (average of 500)/dV (average of 500) can be defined as dQ/dV.
- moving average values of 500 sections can be used.
- the charging and discharging conditions for the multiple times may be different from the above charging conditions.
- charging is performed at an arbitrary voltage (eg, 4.6 V, 4.65 V, 4.7 V, 4.75 V or 4.8 V), constant current charging at 100 mA/g, and constant voltage charging until the current value reaches 10 mA/g.
- the discharge can be constant current discharge at 2.5 V and 100 mA/g.
- the phase changes from the O3 type to the O3' type, and the O3 type at this time is about 0.3 in x in Li x CoO 2 . It has the same symmetry as the O3 type with x 1 described in FIG. 5, but the distance between the CoO 2 layers is slightly different.
- the positive electrode active material 100 that can be used as one embodiment of the present invention preferably contains cobalt and nickel and magnesium as additive elements.
- some Co 3+ is preferably replaced by Ni 3+ and some Li + is replaced by Mg 2+ .
- the Ni 3+ may be reduced to Ni 2+ .
- part of Li + may be replaced with Mg 2+ , and along with this, Co 3+ near Mg 2+ may be reduced to Co 2+ .
- part of Co 3+ may be replaced with Mg 2+ , and along with this, Co 3+ in the vicinity of Mg 2+ may be oxidized to become Co 4+ .
- the positive electrode active material 100 preferably contains at least one of Ni 2+ , Ni 3+ , Co 2+ and Co 4+ .
- the spin density due to at least one of Ni 2+ , Ni 3+ , Co 2+ , and Co 4+ per weight of the positive electrode active material 100 is 2.0 ⁇ 10 17 spins/g or more and 1.0 ⁇ 10 21 spins. /g or less.
- the crystal structure becomes stable especially in a charged state, which is preferable. Note that if the magnesium concentration is too high, the spin density due to one or more of Ni 2+ , Ni 3+ , Co 2+ and Co 4+ may decrease.
- the spin density in the positive electrode active material can be analyzed, for example, using an electron spin resonance method (ESR: Electron Spin Resonance).
- ESR Electron Spin Resonance
- the positive electrode active material 100 that can be used as one embodiment of the present invention preferably has a smooth surface with few unevenness.
- the fact that the surface is smooth and has little unevenness indicates that the effect of the flux, which will be described later, is sufficiently exhibited, and the surface of the additive element source and the lithium cobaltate are melted (solid dissolved). Therefore, this is one factor indicating that the distribution of the additive element in the surface layer portion 100a is good.
- the fact that the surface is smooth and has little unevenness can be determined from, for example, a cross-sectional SEM image or a cross-sectional TEM image of the positive electrode active material 100, the specific surface area of the positive electrode active material 100, or the like.
- the smoothness of the surface can be quantified from the cross-sectional SEM image of the positive electrode active material 100 as follows.
- the positive electrode active material 100 is processed by FIB or the like to expose the cross section. At this time, it is preferable to cover the positive electrode active material 100 with a protective film, a protective agent, or the like.
- the surface roughness of the positive electrode active material is the surface roughness of at least 400 nm of the outer circumference of the particle.
- the root mean square (RMS) surface roughness which is an index of roughness, is less than 3 nm, preferably less than 1 nm, more preferably less than 0.5 nm ( RMS) surface roughness.
- the image processing software for noise processing, interface extraction, etc. is not particularly limited, but for example, "ImageJ" can be used.
- the spreadsheet software is not particularly limited, but for example, Microsoft Office Excel can be used.
- the smoothness of the surface of the positive electrode active material 100 can also be quantified from the ratio between the actual specific surface area S R measured by the constant volume gas adsorption method and the ideal specific surface area Si . can be done.
- the ideal specific surface area Si is obtained by calculation assuming that all the particles of the positive electrode active material have the same diameter as D50, the same weight, and an ideal sphere shape.
- the median diameter D50 can be measured with a particle size distribution meter or the like using a laser diffraction/scattering method.
- the specific surface area can be measured by, for example, a specific surface area measuring device using a gas adsorption method based on a constant volume method.
- the ratio S R / S i between the ideal specific surface area S i obtained from the median diameter D50 and the actual specific surface area S R is 2.1 or less. is preferred.
- the smoothness of the surface can be quantified from the cross-sectional SEM image of the positive electrode active material 100 by the following method.
- a surface SEM image of the positive electrode active material 100 is obtained.
- a conductive coating may be applied as a pretreatment for observation.
- the viewing plane is preferably perpendicular to the electron beam.
- an image (this is called a grayscale image) is obtained by converting the above SEM image into, for example, 8 bits using image processing software (eg, "ImageJ").
- a grayscale image contains luminance (brightness information).
- a dark part has a low number of gradations, and a bright part has a high number of gradations.
- the brightness change can be quantified in association with the number of gradations.
- Such numerical values are called grayscale values.
- a histogram is a three-dimensional representation of the gradation distribution in a target area, and is also called a luminance histogram. Acquiring the luminance histogram makes it possible to visually understand and evaluate the unevenness of the positive electrode active material.
- the difference between the maximum value and the minimum value of the grayscale value is preferably 120 or less, more preferably 115 or less, and 70 or more and 115 or less. is more preferable.
- the standard deviation of gray scale values is preferably 11 or less, more preferably 8 or less, and even more preferably 4 or more and 8 or less.
- the cathode active material 100 may have depressions, cracks, depressions, V-shaped cross-sections, and the like. These are one of the defects, and repeated charging and discharging may result in elution of cobalt, collapse of the crystal structure, cracking of the main body, desorption of oxygen, and the like. Therefore, by providing a buried portion 102 containing an additive element as shown in FIG. 1A2, the elution of cobalt can be suppressed. Therefore, the positive electrode active material 100 can have excellent reliability and cycle characteristics.
- the positive electrode active material 100 may have a convex portion 103 as a region where the additive element is unevenly distributed.
- the additive element contained in the positive electrode active material 100 may adversely affect the insertion and extraction of lithium.
- the additive element when used as a secondary battery, there is a risk of causing an increase in internal resistance, a decrease in charge/discharge capacity, and the like.
- the additive element if the additive element is insufficient, it may not be distributed over the entire surface layer portion 100a, and the effect of suppressing deterioration of the crystal structure may be insufficient.
- the additive element needs to have an appropriate concentration in the positive electrode active material 100, but the adjustment is not easy.
- the positive electrode active material 100 has a region where the additive element is unevenly distributed, part of the excess additive element is removed from the inside 100b of the positive electrode active material 100, and the additive element concentration is made appropriate in the inside 100b. be able to.
- This makes it possible to suppress an increase in internal resistance, a decrease in charge/discharge capacity, and the like when used as a secondary battery.
- the ability to suppress an increase in the internal resistance of a secondary battery is an extremely favorable characteristic particularly in charging and discharging at a large current, for example, charging and discharging at 400 mA/g or more.
- the positive electrode active material 100 having a region in which the additive element is unevenly distributed it is allowed to mix the additive element excessively to some extent in the manufacturing process, which is preferable because the production margin is widened.
- the positive electrode active material 100 may have a film on at least part of the surface.
- 13A and 13B show examples of cathode active materials 100 having coatings 104.
- FIG. 13A and 13B show examples of cathode active materials 100 having coatings 104.
- the film 104 is preferably formed by depositing decomposition products of the electrolytic solution due to charging and discharging, for example.
- Coating 104 preferably comprises carbon, oxygen and fluorine, for example.
- the coating 104 containing one or more selected from boron, nitrogen, sulfur and fluorine may be a good coating and is therefore preferable.
- the film 104 does not have to cover all of the positive electrode active material 100 .
- the positive electrode active material when the positive electrode active material is charged at 4.5 V or higher, or charged and discharged at a high temperature, such as 45 ° C. or higher, progressive defects that progress deep from the surface to the inside occur.
- a phenomenon in which defects progress to form holes in the positive electrode active material can also be called pitting corrosion, and holes generated by this phenomenon are also called pits in this specification and the like.
- FIG. 14 shows a cross-sectional schematic diagram of the positive electrode active material 51 having pits. A crystal plane 55 parallel to the arrangement of cations is also shown. Since FIG. 14 is a cross-sectional view, the pits 54 and 58 are shown as holes, but the shape of these openings is deep and groove-like rather than circular. Moreover, as shown by pits 54 and 58, unlike the recesses 52, they tend to occur parallel to the alignment of the lithium ions.
- 53 and 56 indicate surface layer portions of the positive electrode active material 51 where the additive element exists.
- the added element is less than 53 and 56 or below the detection limit, and it is presumed that the function of the barrier film is reduced.
- the crystal structure of lithium cobalt oxide collapses in the vicinity of the formation of pits, resulting in a crystal structure different from that of the layered rock salt type. Since the collapse of the crystal structure hinders the diffusion and release of lithium ions, which are carrier ions, pits are considered to be a factor in deterioration of cycle characteristics.
- the source of pits may be point defects. It is thought that the point defects of the positive electrode active material change with repeated charging and discharging and are chemically or electrochemically eroded by the surrounding electrolyte or the like, or the material is deteriorated and pits are generated. This deterioration does not occur uniformly on the surface of the positive electrode active material, but occurs locally intensively.
- cracks 57 in FIG. 14 defects such as cracks (also called fissures) may occur due to expansion and contraction of the positive electrode active material due to charging and discharging.
- cracks and pits are different. Immediately after the production of the positive electrode active material, there are cracks but no pits.
- a pit can be said to be a hole from which several layers of cobalt and oxygen have escaped, or a place where cobalt has been eluted, by charging/discharging under a high voltage condition of 4.5 V or higher or at a high temperature (45° C. or higher), for example.
- a crack refers to a crack caused by a new surface or a crystal grain boundary 101 caused by, for example, physical pressure being applied. Cracks may occur due to expansion and contraction of the positive electrode active material due to charging and discharging.
- cracks and/or pits may be generated from cavities inside the positive electrode active material.
- Example 1 of method for producing positive electrode active material An example of a method for manufacturing a positive electrode active material (Example 1 of a method for manufacturing a positive electrode active material) that can be used as one embodiment of the present invention will be described with reference to FIGS. 15A to 15C. Note that the manufacturing method described here is an example of a method for manufacturing the positive electrode active material 100 having the features described above in this embodiment.
- Step S11 In step S11 shown in FIG. 15A, a lithium source (Li source) and a cobalt source (Co source) are prepared as starting materials of lithium and transition metal materials, respectively.
- a lithium source Li source
- a cobalt source Co source
- the lithium source it is preferable to use a compound containing lithium.
- a compound containing lithium for example, lithium carbonate, lithium hydroxide, lithium nitrate, or lithium fluoride can be used.
- the lithium source preferably has a high purity, and for example, a material with a purity of 99.99% or higher is preferably used.
- cobalt source it is preferable to use a compound containing cobalt.
- cobalt oxide, cobalt hydroxide, and the like can be used.
- the cobalt source preferably has a high purity, for example, a purity of 3N (99.9%) or higher, preferably 4N (99.99%) or higher, more preferably 4N5 (99.995%) or higher, further preferably 5N (99%) or higher. .999%) or higher.
- Impurities in the positive electrode active material can be controlled by using a high-purity material. As a result, the capacity of the secondary battery is increased and/or the reliability of the secondary battery is improved.
- the cobalt source has high crystallinity, for example, it should have single crystal grains.
- TEM transmission electron microscope
- STEM scanning transmission electron microscope
- HAADF-STEM high angle scattering annular dark field scanning transmission electron microscope
- ABF-STEM annular dark field scanning transmission electron microscope
- XRD X-ray diffraction
- the method for evaluating the crystallinity described above can be applied not only to the transition metal source but also to the evaluation of other crystallinity.
- Step S12 the lithium source and the cobalt source are pulverized and mixed to produce a mixed material. Grinding and mixing can be dry or wet. The wet method is preferred because it can be pulverized into smaller pieces.
- the lithium source and the transition metal source are mixed with dehydrated acetone with a purity of 99.5% or more and with a water content of 10 ppm or less, followed by pulverization and mixing.
- dehydrated acetone with the above purity, possible impurities can be reduced.
- a ball mill, bead mill, or the like can be used as means for mixing.
- a ball mill it is preferable to use aluminum oxide balls or zirconium oxide balls as grinding media. Zirconium oxide balls are preferable because they emit less impurities.
- the peripheral speed should be 100 mm/s or more and 2000 mm/s or less in order to suppress contamination from the media. In this embodiment, the peripheral speed is 838 mm/s (rotational speed: 400 rpm, ball mill diameter: 40 mm).
- Step S13 the mixed material is heated. Heating is preferably performed at 800° C. or higher and 1100° C. or lower, more preferably 900° C. or higher and 1000° C. or lower, and even more preferably about 950° C. or lower and 1000° C. or lower. If the temperature is too low, decomposition and melting of the lithium source and transition metal source may be insufficient. On the other hand, if the temperature is too high, defects may occur, such as by evaporation of lithium from the lithium source and/or excessive reduction of cobalt. For example, cobalt changes from trivalent to divalent and may induce oxygen defects and the like.
- the heating time may be 1 hour or more and 100 hours or less, more preferably 2 hours or more and 20 hours or less.
- the rate of temperature increase depends on the temperature reached by the heating temperature, but is preferably 80°C/h or more and 250°C/h or less. For example, when heating at 1000° C. for 10 hours, the heating rate is preferably 200° C./h.
- Heating is preferably carried out in an atmosphere with little water such as dry air, for example, an atmosphere with a dew point of -50°C or lower, more preferably -80°C or lower. In this embodiment mode, heating is performed in an atmosphere with a dew point of -93°C. Further, in order to suppress impurities that may be mixed into the material, the concentrations of impurities such as CH 4 , CO, CO 2 and H 2 in the heating atmosphere should each be 5 ppb (parts per billion) or less.
- An atmosphere containing oxygen is preferable as the heating atmosphere.
- the heating atmosphere there is a method of continuously introducing dry air into the reaction chamber.
- the flow rate of dry air is preferably 10 L/min.
- the process by which oxygen continues to be introduced into the reaction chamber and is flowing through the reaction chamber is referred to as flow.
- the heating atmosphere is an atmosphere containing oxygen
- a method that does not flow may be used.
- the reaction chamber may be decompressed and then filled with oxygen to prevent the oxygen from entering or exiting the reaction chamber. This is called purging.
- the reaction chamber may be evacuated to -970 hPa and then filled with oxygen to 50 hPa.
- Cooling after heating may be natural cooling, but it is preferable if the cooling time from the specified temperature to room temperature is within 10 hours or more and 50 hours or less. However, cooling to room temperature is not necessarily required, and cooling to a temperature that the next step allows is sufficient.
- Heating in this process may be performed by a rotary kiln or a roller hearth kiln. Heating by a rotary kiln can be performed while stirring in either a continuous system or a batch system.
- the crucible used for heating is preferably a crucible made of aluminum oxide.
- a crucible made of aluminum oxide is a material that is less likely to be contaminated with impurities.
- an aluminum oxide crucible with a purity of 99.9% is used.
- step S13 After the heating is over, it may be pulverized and sieved as necessary. When recovering the material after heating, it may be recovered after being moved from the crucible to a mortar. Moreover, it is preferable to use an aluminum oxide mortar as the mortar.
- a mortar made of aluminum oxide is a material that does not easily get mixed with impurities. Specifically, a mortar made of aluminum oxide with a purity of 90% or higher, preferably 99% or higher is used. Note that the same heating conditions as in step S13 can be applied to the later-described heating process other than step S13.
- Step S14 Through the above steps, lithium cobaltate (LiCoO 2 ) shown in step S14 shown in FIG. 15A can be synthesized.
- a composite oxide by a solid-phase method as in steps S11 to S14 has been shown, but the composite oxide may be produced by a coprecipitation method. Alternatively, the composite oxide may be produced by a hydrothermal method.
- Step S15 lithium cobaltate is heated in step S15 shown in FIG. 15A.
- the heating in step S15 may be called initial heating because it is the first heating for lithium cobaltate.
- the heating since the heating is performed before step S20 described below, it may be called preheating or pretreatment.
- the lithium source and/or the cobalt source prepared in step S11 or the like may contain impurities. It is possible to reduce impurities from the lithium cobalt oxide completed in step S14 by the initial heating. Note that the effect of enhancing the crystallinity of the interior 100b is the effect of alleviating strain, displacement, etc., caused by the difference in contraction, etc., of the lithium cobalt oxide produced in step S13.
- the initial heating has the effect of smoothing the surface of the lithium cobalt oxide.
- smooth surface means that the lithium cobaltate has little unevenness, and the lithium cobaltate is generally rounded, and the corners are rounded.
- the state in which there are few foreign substances adhering to the surface is also called smooth. Foreign matter is considered to be a cause of unevenness, and it is preferable not to allow foreign matter to adhere to the surface.
- the heating time in this process is too short, a sufficient effect cannot be obtained, but if it is too long, productivity will decrease.
- it can be implemented by selecting from the heating conditions described in step S13.
- the heating temperature in step S15 is preferably lower than the temperature in step S13 in order to maintain the crystal structure of the composite oxide.
- the heating time in step S15 is preferably shorter than the time in step S13 in order to maintain the crystal structure of the composite oxide. For example, heating may be performed at a temperature of 700° C. to 1000° C. for 2 hours to 20 hours.
- a temperature difference may occur between the surface and the inside of the lithium cobalt oxide due to the heating in step S13. Differences in temperature can induce differential shrinkage. It is also considered that the difference in shrinkage occurs due to the difference in fluidity between the surface and the inside due to the temperature difference.
- the energy associated with differential shrinkage imparts internal stress differentials to lithium cobaltate.
- the difference in internal stress is also called strain, and the energy is sometimes called strain energy.
- strain energy is homogenized by the initial heating in step S15.
- the strain energy is homogenized, the strain of lithium cobaltate is relaxed. Along with this, the surface of lithium cobaltate may become smooth. It is also called surface-improved. In other words, after step S15, it is thought that the difference in shrinkage caused in the lithium cobalt oxide is relaxed and the surface of the composite oxide becomes smooth.
- step S15 may be performed. After step S15, it is possible to homogenize the displacement of the composite oxide (relax the displacement of crystals or the like occurring in the composite oxide, or align the crystal grains). As a result, the surface of the composite oxide may become smooth.
- lithium cobalt oxide with a smooth surface When lithium cobalt oxide with a smooth surface is used as a positive electrode active material, deterioration during charging and discharging as a secondary battery is reduced, and cracking of the positive electrode active material can be prevented.
- lithium cobaltate synthesized in advance may be used as step S14.
- steps S11 to S13 can be omitted.
- step S15 By performing step S15 on previously synthesized lithium cobalt oxide, lithium cobalt oxide with a smooth surface can be obtained.
- Step S20 Next, as shown in steps S20 to S33, it is preferable to add an additive element A as an A source to the lithium cobalt oxide that has undergone the initial heating.
- the additive element A When the additive element A is added to lithium cobalt oxide that has undergone initial heating, the additive element A can be added evenly. Therefore, it is preferable to add the additive element A after the initial heating (step S15), rather than adding the additive element A and then performing the initial heating (step S15).
- step S20 the details of step S20 of preparing the additive element A as the A source will be described with reference to FIGS. 15B and 15C.
- Step S21 prepares an additive element A.
- the additive element A the additive element described in the previous embodiment, for example, the additive element X and the additive element Y can be used.
- one or more selected from magnesium, fluorine, nickel, aluminum, titanium, zirconium, vanadium, iron, manganese, chromium, niobium, arsenic, zinc, silicon, sulfur, phosphorus and boron can be used.
- One or more selected from bromine and beryllium can also be used.
- FIG. 15B illustrates a case where a magnesium source and a fluorine source are prepared.
- a lithium source may be prepared separately.
- the additive element source can be called the magnesium source.
- magnesium source magnesium fluoride, magnesium oxide, magnesium hydroxide, magnesium carbonate, or the like can be used. Multiple sources of magnesium may be used.
- the additive element source can be called a fluorine source.
- the fluorine source include lithium fluoride (LiF), magnesium fluoride (MgF 2 ), aluminum fluoride (AlF 3 ), titanium fluoride (TiF 4 ), cobalt fluoride (CoF 2 , CoF 3 ) and fluorine.
- lithium fluoride is preferable because it has a relatively low melting point of 848° C. and is easily melted in a heating step to be described later.
- Magnesium fluoride can be used as both a fluorine source and a magnesium source. Lithium fluoride can also be used as a lithium source. Other lithium sources used in step S21 include lithium carbonate.
- the fluorine source may also be gaseous, such as fluorine ( F2 ), carbon fluoride, sulfur fluoride, or oxygen fluoride ( OF2 , O2F2 , O3F2 , O4F2 , O5F 2 , O 6 F 2 , O 2 F) or the like may be used and mixed in the atmosphere in the heating step described later. Multiple fluorine sources may be used.
- lithium fluoride (LiF) is prepared as a fluorine source
- magnesium fluoride (MgF 2 ) is prepared as a fluorine source and a magnesium source.
- LiF:MgF 2 65:35 (molar ratio)
- the effect of lowering the melting point is maximized.
- the amount of lithium fluoride increases, there is a concern that the amount of lithium becomes excessive and the cycle characteristics deteriorate.
- the neighborhood is a value that is more than 0.9 times and less than 1.1 times that value.
- step S22 shown in FIG. 15B the magnesium source and the fluorine source are pulverized and mixed. This step can be performed by selecting from the pulverization and mixing conditions described in step S12.
- a heating process may be performed after step S22, if necessary.
- the heating process can be performed by selecting from the heating conditions described in step S13.
- the heating time is preferably 2 hours or longer, and the heating temperature is preferably 800° C. or higher and 1100° C. or lower.
- step S23 shown in FIG. 15B the pulverized and mixed material can be recovered to obtain the additive element source (A source).
- the additive element source (A source) shown in step S23 has a plurality of starting materials and can also be called a mixture.
- D50 (median diameter) is preferably 600 nm or more and 20 ⁇ m or less, more preferably 1 ⁇ m or more and 10 ⁇ m or less. Even when one type of material is used as the additive element source, the D50 (median diameter) is preferably 600 nm or more and 20 ⁇ m or less, more preferably 1 ⁇ m or more and 10 ⁇ m or less.
- Such a pulverized mixture (including the case where one additive element is added) is easy to uniformly adhere to the surface of lithium cobaltate when mixed with lithium cobaltate in a later step. It is preferable that the mixture is uniformly adhered to the surface of the lithium cobalt oxide, because the additive element is easily distributed or diffused uniformly in the surface layer portion 100a of the composite oxide after heating.
- Step S21> A process different from that in FIG. 15B will be described with reference to FIG. 15C.
- Step S20 shown in FIG. 15C has steps S21 to S23.
- step S21 shown in FIG. 15C four types of additive element sources to be added to lithium cobaltate are prepared. That is, FIG. 15C differs from FIG. 15B in the type of additive element source. Also, in addition to the additive element source, a lithium source may be prepared separately.
- a magnesium source (Mg source), a fluorine source (F source), a nickel source (Ni source), and an aluminum source (Al source) are prepared as four types of additive element sources. Note that the magnesium source and the fluorine source can be selected from the compounds and the like described with reference to FIG. 15B.
- a nickel source nickel oxide, nickel hydroxide, or the like can be used.
- Aluminum oxide, aluminum hydroxide, and the like can be used as the aluminum source.
- Step S22 and Step S23 are the same as the steps described in FIG. 15B.
- step S31 shown in FIG. 15A lithium cobalt oxide and an additive element source (A source) are mixed.
- the mixing in step S31 is preferably performed under milder conditions than the mixing in step S12.
- the number of revolutions is smaller than that of the mixing in step S12, or that the time is short.
- the conditions of the dry method are milder than those of the wet method.
- a ball mill, bead mill, or the like can be used.
- zirconium oxide balls it is preferable to use, for example, zirconium oxide balls as media.
- a ball mill using zirconium oxide balls with a diameter of 1 mm is used for dry mixing at 150 rpm for 1 hour.
- the mixing is performed in a dry room with a dew point of -100°C or higher and -10°C or lower.
- step S32 of FIG. 15A the mixed materials are recovered to obtain a mixture 903.
- FIGS. 15A to 15C describe a manufacturing method in which an additive element is added only after initial heating, but the present invention is not limited to the above method.
- the additive element may be added at other timings, or may be added multiple times. Also, the timing may be changed depending on the element.
- the additive element may be added to the lithium source and the transition metal source at the stage of step S11, that is, at the stage of the starting material of the composite oxide. After that, in step S13, lithium cobaltate having the additive element can be obtained. In this case, there is no need to separate the steps S11 to S14 from the steps S21 to S23. It can be said that it is a simple and highly productive method.
- lithium cobaltate having a part of the additive element in advance may be used.
- part of steps S11 to S14 and step S20 can be omitted. It can be said that it is a simple and highly productive method.
- lithium cobaltate to which magnesium and fluorine are added in advance is heated in step S15, and then, as in step S20, a magnesium source and a fluorine source, or a magnesium source, a fluorine source, a nickel source, and an aluminum source. may be added.
- step S33 shown in FIG. 15A the mixture 903 is heated.
- the heating conditions described in step S13 can be selected and implemented.
- the heating time is preferably 2 hours or more.
- the lower limit of the heating temperature in step S33 must be higher than or equal to the temperature at which the reaction between the lithium cobalt oxide and the additive element source proceeds.
- the temperature at which the reaction proceeds may be any temperature at which interdiffusion of the elements of the lithium cobalt oxide and the additive element source occurs, and may be lower than the melting temperature of these materials. Taking an oxide as an example, solid-phase diffusion occurs from 0.757 times the melting temperature T m (Tamman temperature T d ). Therefore, the heating temperature in step S33 may be 500° C. or higher.
- the reaction proceeds more easily when the temperature is higher than or equal to the temperature at which one or more selected from the materials included in the mixture 903 melt.
- the eutectic point of LiF and MgF2 is around 742°C, so the lower limit of the heating temperature in step S33 is preferably 742°C or higher.
- a mixture 903 obtained by mixing LiCoO 2 :LiF:MgF 2 100:0.33:1 (molar ratio) has an endothermic peak near 830° C. in differential scanning calorimetry (DSC measurement). is observed. Therefore, the lower limit of the heating temperature is more preferably 830° C. or higher.
- the upper limit of the heating temperature should be less than the decomposition temperature of lithium cobaltate (1130°C). At temperatures in the vicinity of the decomposition temperature, there is concern that lithium cobaltate will decompose, albeit in a very small amount. Therefore, it is preferably 1000° C. or lower, more preferably 950° C. or lower, and even more preferably 900° C. or lower.
- the heating temperature in step S33 is preferably 500° C. or higher and 1130° C. or lower, more preferably 500° C. or higher and 1000° C. or lower, even more preferably 500° C. or higher and 950° C. or lower, and further preferably 500° C. or higher and 900° C. or lower. preferable.
- the temperature is preferably 742°C or higher and 1130°C or lower, more preferably 742°C or higher and 1000°C or lower, even more preferably 742°C or higher and 950°C or lower, and even more preferably 742°C or higher and 900°C or lower.
- the temperature is preferably 800° C. to 1100° C., preferably 830° C.
- step S33 is preferably higher than that in step S13.
- some materials such as LiF, which is a fluorine source, may function as a flux.
- the heating temperature can be lowered to below the decomposition temperature of lithium cobalt oxide, for example, 742° C. or higher and 950° C. or lower, and additional elements such as magnesium are distributed in the surface layer portion to produce a positive electrode active material with good characteristics. can.
- LiF has a lower specific gravity in a gaseous state than oxygen
- LiF may volatilize due to heating, and the volatilization reduces LiF in the mixture 903 .
- the function as a flux is weakened. Therefore, it is necessary to heat while suppressing volatilization of LiF.
- LiF is not used as a fluorine source or the like, there is a possibility that Li on the surface of LiCoO 2 reacts with F in the fluorine source to generate LiF and volatilize. Therefore, even if a fluoride having a higher melting point than LiF is used, it is necessary to similarly suppress volatilization.
- the mixture 903 in an atmosphere containing LiF, that is, to heat the mixture 903 in a state where the partial pressure of LiF in the heating furnace is high. Such heating can suppress volatilization of LiF in the mixture 903 .
- the heating in this step is preferably performed so that the particles of the mixture 903 do not adhere to each other. If the particles of the mixture 903 adhere to each other during heating, the contact area with oxygen in the atmosphere is reduced, and the diffusion path of the additive element (eg, fluorine) is inhibited. fluorine) distribution may deteriorate.
- the additive element eg, fluorine
- the additive element for example, fluorine
- a positive electrode active material that is smooth and has few irregularities can be obtained. Therefore, in order to maintain or smoothen the surface after the heating in step S15 in this step, it is preferable that the particles of the mixture 903 do not adhere to each other.
- the flow rate of the oxygen-containing atmosphere in the kiln when heating with a rotary kiln, it is preferable to control the flow rate of the oxygen-containing atmosphere in the kiln. For example, it is preferable to reduce the flow rate of the oxygen-containing atmosphere, or to stop the flow of the atmosphere after first purging the atmosphere and introducing the oxygen atmosphere into the kiln.
- Flowing oxygen may evaporate the fluorine source, which is not preferable for maintaining smoothness of the surface.
- the mixture 903 can be heated in an atmosphere containing LiF, for example, by placing a lid on the container containing the mixture 903 .
- the heating temperature is preferably 600° C. or higher and 950° C. or lower, for example.
- the heating time is, for example, preferably 3 hours or longer, more preferably 10 hours or longer, and even more preferably 60 hours or longer.
- the cooling time after heating is, for example, 10 hours or more and 50 hours or less.
- the heating temperature is preferably 600° C. or higher and 950° C. or lower, for example.
- the heating time is, for example, preferably 1 hour or more and 10 hours or less, more preferably about 2 hours.
- the cooling time after heating is, for example, 10 hours or more and 50 hours or less.
- step S34 shown in FIG. 15A the heated material is collected and, if necessary, pulverized to obtain the positive electrode active material 100.
- FIG. At this time, it is preferable to further screen the recovered positive electrode active material 100 .
- the positive electrode active material 100 having the features described in this embodiment can be manufactured.
- Example 2 of method for producing positive electrode active material Another example of a method for manufacturing a positive electrode active material (Example 2 of a method for manufacturing a positive electrode active material) that can be used as one embodiment of the present invention will be described with reference to FIGS.
- Example 2 of the method for producing a positive electrode active material differs from Example 1 of the method for producing a positive electrode active material described above in terms of the number of times the additive element is added and the mixing method. can be applied.
- steps S11 to S15 are performed in the same manner as in FIG. 15A to prepare lithium cobalt oxide that has undergone initial heating.
- Step S20a is a step of preparing a first additive element source (A1 source) used for adding the additive element A1, and will be described with reference to FIG. 17A.
- a first additive element source (A1 source) is prepared.
- the additional element A1 it is possible to select and use from the additional elements A described in step S21 shown in FIG. 15B.
- the additive element A1 one or more selected from magnesium, fluorine, and calcium can be used.
- FIG. 17A illustrates a case where a magnesium source (Mg source) and a fluorine source (F source) are pulverized and mixed and used as the A1 source.
- Steps S21 to S23 shown in FIG. 17A can be performed under the same conditions as steps S21 to S23 shown in FIG. 15B.
- a first additive element source (A1 source) can be obtained in step S23.
- steps S31 to S33 shown in FIG. 16 can be performed under the same conditions as steps S31 to S33 shown in FIG. 15A.
- Step S34a Next, the material heated in step S33 is recovered, and lithium cobaltate having the additive element A1 is produced.
- the composite oxide (first composite oxide) in step S14 it is also called a second composite oxide.
- Step S40 is a step of preparing a second additive element source (A2 source) used for adding the additive element A2, which will be described with reference to FIGS. 17B and 17C.
- a second additive element source (A2 source) is prepared.
- A2 source As the additional element A2, it is possible to select and use from the additional elements A described in step S21 shown in FIG. 15B.
- the additive element A2 any one or more selected from nickel, titanium, boron, zirconium, and aluminum can be suitably used.
- FIG. 17B exemplifies a case where a nickel source and an aluminum source are pulverized and mixed and used as the A2 source.
- Steps S41 to S43 shown in FIG. 17B can be manufactured under the same conditions as steps S21 to S23 shown in FIG. 15B. As a result, a second additive element source (A2 source) can be obtained in step S43.
- A2 source a second additive element source
- Steps S41 to S43 shown in FIG. 17C are a modification of FIG. 17B.
- a nickel source (Ni source) and an aluminum source (Al source) are prepared in step S41 shown in FIG. 17C, and pulverized independently in step S42a.
- a plurality of second additive element sources (A2 sources) are prepared in step S43.
- the step of FIG. 17C differs from that of FIG. 17B in that the additive elements are independently pulverized in step S42a.
- Steps S51 to S53 shown in FIG. 16 can be performed under the same conditions as steps S31 to S33 shown in FIG. 15A.
- the conditions of step S53 regarding the heating process may be a lower temperature and a shorter time than those of step S33.
- step S54 shown in FIG. 16 the heated material is recovered and, if necessary, pulverized to obtain the positive electrode active material 100.
- FIG. Through the above steps, the positive electrode active material 100 having the features described in this embodiment can be manufactured.
- the additive element to lithium cobaltate is introduced separately into the first additive element A1 and the second additive element A2.
- the profile of each additive element in the depth direction can be changed. For example, it is possible to profile the first additive element so that the concentration is higher in the surface layer than in the inside, and to profile the second additive element so that the concentration is higher inside than in the surface layer. .
- the positive electrode has a positive electrode active material layer and a positive electrode current collector.
- the positive electrode active material layer contains a positive electrode active material and may further contain at least one of a conductive aid and a binder.
- the positive electrode active material described in Embodiment 1 can be used.
- FIG. 18A shows an example of a schematic diagram of the cross section of the positive electrode.
- a metal foil for example, can be used for the current collector 550 .
- the positive electrode can be formed by applying a slurry onto a metal foil and drying it. In addition, you may add a press after drying.
- the positive electrode is obtained by forming an active material layer on the current collector 550 .
- a slurry is a material liquid used to form an active material layer on the current collector 550, and refers to a liquid containing an active material, a binder, and a solvent, preferably further mixed with a conductive aid.
- the slurry is sometimes called an electrode slurry or an active material slurry, and is called a positive electrode slurry when forming a positive electrode active material layer, and is called a negative electrode slurry when forming a negative electrode active material layer.
- the positive electrode active material 561 has a function of taking in and/or releasing lithium ions during charging and discharging.
- a material that is less likely to deteriorate due to charge/discharge even at high charging voltage can be used.
- the charge voltage is represented based on the potential of lithium metal.
- a high charging voltage is, for example, a charging voltage of 4.6 V or higher, preferably 4.65 V or higher, more preferably 4.7 V or higher, still more preferably 4.75 V or higher, and most preferably 4.75 V or higher. is 4.8V or higher.
- any material can be used as long as it is less likely to deteriorate due to charging and discharging even at a high charging voltage, and the materials described in Embodiment 1 or 2 can be used. can be used. Note that two or more kinds of materials having different particle sizes can be used for the positive electrode active material 561 as long as the material is less deteriorated due to charging and discharging even at a high charging voltage.
- the conductive aid is also called a conductive agent or a conductive material, and a carbon material can be used.
- a conductive aid By adhering the conductive aid between the plurality of active materials, the plurality of active materials are electrically connected to each other, and the conductivity is increased.
- adheresion does not only refer to the fact that the active material and the conductive aid are in close physical contact. The concept includes the case where the conductive aid covers part of the surface of the active material, the case where the conductive aid is stuck in the unevenness of the surface of the active material, and the case where they are electrically connected even if they are not in contact with each other.
- FIG. 18A illustrates carbon black 553 as a conductive aid.
- a binder may be mixed to fix the current collector 550 such as a metal foil and the active material as the positive electrode of the secondary battery.
- a binder is also called a binding agent.
- the binder is a polymer material, and if the binder is contained in a large amount, the ratio of the active material in the positive electrode is lowered, and the discharge capacity of the secondary battery is reduced. Therefore, it is preferable to mix the amount of binder to a minimum.
- regions not filled with the positive electrode active material 561, the second active material 562, and the carbon black 553 indicate voids or binders.
- FIG. 18A shows an example in which the positive electrode active material 561 is spherical, it is not particularly limited.
- the cross-sectional shape of the positive electrode active material 561 may be oval, rectangular, trapezoidal, triangular, polygonal with rounded corners, or asymmetrical.
- FIG. 18B shows an example in which the positive electrode active material 561 has a polygonal shape with rounded corners.
- graphene 554 is used as a carbon material used as a conductive aid.
- 18B forms a positive electrode active material layer including a positive electrode active material 561, graphene 554, and carbon black 553 on a current collector 550.
- the weight of the carbon black to be mixed is 1.5 to 20 times, preferably 2 to 9.5 times the weight of the graphene. preferably.
- the carbon black 553 has excellent dispersion stability during preparation of the slurry, and agglomerates are less likely to occur.
- the electrode density can be higher than that of the positive electrode in which only the carbon black 553 is used as the conductive aid. By increasing the electrode density, the capacity per unit weight can be increased. Specifically, the density of the positive electrode active material layer by gravimetric measurement can be 3.5 g/cc or more.
- the electrode density is lower than that of a positive electrode that uses only graphene as a conductive agent, by setting the mixture of the first carbon material (graphene) and the second carbon material (acetylene black) in the above range, rapid charging can be achieved. can correspond to Therefore, it is particularly effective when used as a vehicle-mounted secondary battery.
- FIG. 18C illustrates an example of a positive electrode using carbon fiber 555 instead of graphene.
- FIG. 18C shows an example different from FIG. 18B.
- Using the carbon fiber 555 can prevent the aggregation of the carbon black 553 and improve the dispersibility.
- regions not filled with the positive electrode active material 561, the carbon fibers 555, and the carbon black 553 refer to voids or binders.
- FIG. 18D is illustrated as another example of the positive electrode.
- FIG. 18D shows an example using carbon fiber 555 in addition to graphene 554 .
- Using both the graphene 554 and the carbon fiber 555 can prevent carbon black such as the carbon black 553 from agglomerating and further improve the dispersibility.
- regions not filled with the positive electrode active material 561, the carbon fibers 555, the graphene 554, and the carbon black 553 refer to voids or binders.
- a separator is stacked on the positive electrode, and the stack is placed in a container (such as an outer package, a metal can, etc.) that houses the laminate in which the negative electrode is stacked on the separator, and the electrolyte solution is placed in the container.
- a secondary battery can be produced by filling the
- ⁇ Binder> As the binder, it is preferable to use rubber materials such as styrene-butadiene rubber (SBR), styrene-isoprene-styrene rubber, acrylonitrile-butadiene rubber, butadiene rubber, and ethylene-propylene-diene copolymer. Fluororubber can also be used as the binder.
- SBR styrene-butadiene rubber
- styrene-isoprene-styrene rubber acrylonitrile-butadiene rubber
- butadiene rubber butadiene rubber
- Fluororubber can also be used as the binder.
- the binder it is preferable to use, for example, a water-soluble polymer.
- Polysaccharides for example, can be used as the water-soluble polymer.
- cellulose derivatives such as carboxymethyl cellulose (CMC), methyl cellulose, ethyl cellulose, hydroxypropyl cellulose, diacetyl cellulose, regenerated cellulose, starch, and the like can be used. Further, it is more preferable to use these water-soluble polymers in combination with the aforementioned rubber material.
- Binders may be used in combination with more than one of the above.
- a material having a particularly excellent viscosity adjusting effect may be used in combination with another material.
- rubber materials and the like are excellent in adhesive strength and elasticity, it may be difficult to adjust the viscosity when they are mixed with a solvent. In such a case, for example, it is preferable to mix with a material having a particularly excellent viscosity-adjusting effect.
- a water-soluble polymer may be used as a material having a particularly excellent viscosity-adjusting effect.
- the aforementioned polysaccharides such as carboxymethyl cellulose (CMC), methyl cellulose, ethyl cellulose, hydroxypropyl cellulose and diacetyl cellulose, cellulose derivatives such as regenerated cellulose, or starch are used. be able to.
- cellulose derivatives such as carboxymethyl cellulose
- solubility of cellulose derivatives is increased by making them into salts such as sodium or ammonium salts of carboxymethyl cellulose, making it easier to exert its effect as a viscosity modifier.
- the higher solubility also allows for better dispersibility with the active material or other constituents when preparing the electrode slurry.
- cellulose and cellulose derivatives used as binders for electrodes also include salts thereof.
- the water-soluble polymer stabilizes the viscosity by dissolving it in water, and can stably disperse the active material and other materials combined as a binder, such as styrene-butadiene rubber, in the aqueous solution.
- a binder such as styrene-butadiene rubber
- it since it has a functional group, it is expected to be stably adsorbed on the surface of the active material.
- many cellulose derivatives such as carboxymethyl cellulose are materials having functional groups such as hydroxyl groups or carboxyl groups, and due to the presence of functional groups, the macromolecules interact with each other, and the surface of the active material can be widely covered. Be expected.
- the binder that covers or contacts the surface of the active material forms a film
- it is expected to play a role as a passive film and suppress the decomposition of the electrolyte.
- the "passive film” is a film with no electrical conductivity or a film with extremely low electrical conductivity.
- WHEREIN The decomposition
- the positive electrode current collector highly conductive materials such as metals such as stainless steel, gold, platinum, aluminum and titanium, and alloys thereof can be used. Moreover, it is preferable that the material used for the positive electrode current collector does not elute at the potential of the positive electrode.
- an aluminum alloy added with an element that improves heat resistance such as silicon, titanium, neodymium, scandium, or molybdenum, can be used.
- a metal element that forms silicide by reacting with silicon may be used.
- Metal elements that react with silicon to form silicide include zirconium, titanium, hafnium, vanadium, niobium, tantalum, chromium, molybdenum, tungsten, cobalt, and nickel.
- the shape of the positive electrode current collector can be appropriately used such as foil, plate, sheet, mesh, punching metal, expanded metal, and the like.
- a positive electrode current collector having a thickness of 5 ⁇ m or more and 30 ⁇ m or less is preferably used.
- the negative electrode has a negative electrode active material layer and a negative electrode current collector. Moreover, the negative electrode active material layer contains a negative electrode active material, and may further contain a conductive aid and a binder.
- Niobium electrode active material for example, an alloy material or a carbon material can be used.
- the negative electrode active material can use an element capable of undergoing charge/discharge reaction by alloying/dealloying reaction with lithium.
- an element capable of undergoing charge/discharge reaction by alloying/dealloying reaction with lithium for example, materials containing at least one of silicon, tin, gallium, aluminum, germanium, lead, antimony, bismuth, silver, zinc, cadmium, indium, etc. can be used.
- Such an element has a larger capacity than carbon, and silicon in particular has a high theoretical capacity of 4200 mAh/g. Therefore, it is preferable to use silicon for the negative electrode active material. Compounds containing these elements may also be used.
- elements capable of undergoing charge/discharge reactions by alloying/dealloying reactions with lithium, compounds containing such elements, and the like are sometimes referred to as alloy-based materials.
- SiO refers to silicon monoxide, for example.
- SiO can be represented as SiO x .
- x preferably has a value of 1 or close to 1.
- x is preferably 0.2 or more and 1.5 or less, and preferably 0.3 or more and 1.2 or less.
- Graphite, graphitizable carbon (soft carbon), non-graphitizable carbon (hard carbon), carbon fiber (carbon nanotube), graphene, carbon black, etc. may be used as the carbon material.
- Graphite includes artificial graphite and natural graphite.
- artificial graphite include mesocarbon microbeads (MCMB), coke-based artificial graphite, and pitch-based artificial graphite.
- Spherical graphite having a spherical shape can be used here as the artificial graphite.
- MCMB may have a spherical shape and are preferred.
- MCMB is also relatively easy to reduce its surface area and may be preferred.
- natural graphite include flake graphite and spherical natural graphite.
- Graphite exhibits a potential as low as that of lithium metal when lithium ions are inserted into graphite (at the time of formation of a lithium-graphite intercalation compound) (0.05 V or more and 0.3 V or less vs. Li/Li + ). Accordingly, a lithium-ion battery using graphite can exhibit a high operating voltage. Furthermore, graphite is preferable because it has advantages such as relatively high capacity per unit volume, relatively small volume expansion, low cost, and high safety compared to lithium metal.
- titanium dioxide TiO2
- lithium titanium oxide Li4Ti5O12
- lithium -graphite intercalation compound LixC6
- niobium pentoxide Nb2O5
- oxide Oxides such as tungsten (WO 2 ) and molybdenum oxide (MoO 2 ) can be used.
- Li 2.6 Co 0.4 N 3 exhibits a large discharge capacity (900 mAh/g, 1890 mAh/cm 3 ) and is preferred.
- lithium ions are contained in the negative electrode active material, so that it can be combined with materials such as V 2 O 5 and Cr 3 O 8 that do not contain lithium ions as the positive electrode active material, which is preferable.
- materials such as V 2 O 5 and Cr 3 O 8 that do not contain lithium ions as the positive electrode active material, which is preferable.
- a composite nitride of lithium and a transition metal can be used as the negative electrode active material by preliminarily desorbing the lithium ions contained in the positive electrode active material.
- a material that causes a conversion reaction can also be used as the negative electrode active material.
- transition metal oxides such as cobalt oxide (CoO), nickel oxide (NiO), and iron oxide (FeO) that do not form an alloy with lithium may be used as the negative electrode active material.
- oxides such as Fe2O3 , CuO, Cu2O , RuO2 and Cr2O3 , sulfides such as CoS0.89 , NiS and CuS, and Zn3N2 , Cu 3 N, Ge 3 N 4 and other nitrides, NiP 2 , FeP 2 and CoP 3 and other phosphides, and FeF 3 and BiF 3 and other fluorides.
- the same materials as the conductive aid and binder that the positive electrode active material layer can have can be used.
- ⁇ Negative electrode current collector> copper or the like can be used in addition to the same material as the positive electrode current collector.
- the negative electrode current collector it is preferable to use a material that does not alloy with carrier ions such as lithium.
- Electrolyte The electrolyte described in Embodiment 1 can be used.
- separator When the electrolyte includes an electrolytic solution, a separator is placed between the positive and negative electrodes.
- separators include fibers containing cellulose such as paper, non-woven fabrics, glass fibers, ceramics, synthetic fibers using nylon (polyamide), vinylon (polyvinyl alcohol-based fiber), polyester, acrylic, polyolefin, and polyurethane. can be used. It is preferable that the separator be processed into a bag shape and arranged so as to enclose either the positive electrode or the negative electrode.
- the separator may have a multilayer structure.
- an organic material film such as polypropylene or polyethylene can be coated with a ceramic material, a fluorine material, a polyamide material, or a mixture thereof.
- the ceramic material for example, aluminum oxide particles, silicon oxide particles, or the like can be used.
- PVDF, polytetrafluoroethylene, or the like can be used as the fluorine-based material.
- the polyamide-based material for example, nylon, aramid (meta-aramid, para-aramid) and the like can be used.
- Coating with a ceramic material improves oxidation resistance, so it is possible to suppress deterioration of the separator during high-voltage charging and discharging and improve the reliability of the secondary battery.
- the separator and the electrode are more likely to adhere to each other, and the output characteristics can be improved.
- Coating with a polyamide-based material, particularly aramid improves the heat resistance, so that the safety of the secondary battery can be improved.
- both sides of a polypropylene film may be coated with a mixed material of aluminum oxide and aramid.
- a polypropylene film may be coated with a mixed material of aluminum oxide and aramid on the surface thereof in contact with the positive electrode, and coated with a fluorine-based material on the surface thereof in contact with the negative electrode.
- the safety of the secondary battery can be maintained even if the overall thickness of the separator is thin, so the capacity per unit volume of the secondary battery can be increased.
- a metal material such as aluminum or a resin material can be used for the exterior body of the secondary battery.
- a film-like exterior body can also be used.
- a film for example, a film made of a material such as polyethylene, polypropylene, polycarbonate, ionomer, polyamide, etc. is provided with a highly flexible metal thin film such as aluminum, stainless steel, copper, nickel, etc., and an exterior is provided on the metal thin film.
- a film having a three-layer structure provided with an insulating synthetic resin film such as a polyamide-based resin or a polyester-based resin can be used as the outer surface of the body.
- FIG. 19A is an exploded perspective view of a coin-type (single-layer flat type) secondary battery
- FIG. 19B is an external view
- FIG. 19C is a cross-sectional view thereof.
- Coin-type secondary batteries are mainly used in small electronic devices.
- FIG. 19A is a schematic diagram so that the overlapping of members (vertical relationship and positional relationship) can be understood for the sake of clarity. Therefore, FIG. 19A and FIG. 19B do not correspond to each other completely.
- the positive electrode 304, separator 310, negative electrode 307, spacer 322, and washer 312 are stacked. These are sealed with a negative electrode can 302 and a positive electrode can 301 .
- a gasket for sealing is not shown in FIG. 19A.
- the spacer 322 and the washer 312 are used to protect the inside or fix the position inside the can when the positive electrode can 301 and the negative electrode can 302 are pressure-bonded. Spacers 322 and washers 312 are made of stainless steel or an insulating material.
- a positive electrode 304 has a laminated structure in which a positive electrode active material layer 306 is formed on a positive electrode current collector 305 .
- the separator 310 is arranged so as to cover the upper surface of the positive electrode 304 .
- the separator 310 has a larger planar area than the positive electrode 304 .
- FIG. 19B is a perspective view of a completed coin-shaped secondary battery.
- a positive electrode can 301 that also serves as a positive electrode terminal and a negative electrode can 302 that also serves as a negative electrode terminal are insulated and sealed with a gasket 303 made of polypropylene or the like.
- the positive electrode 304 is formed of a positive electrode current collector 305 and a positive electrode active material layer 306 provided so as to be in contact therewith.
- the negative electrode 307 is formed of a negative electrode current collector 308 and a negative electrode active material layer 309 provided so as to be in contact therewith.
- the negative electrode 307 is not limited to a laminated structure, and may be a lithium metal foil or a lithium-aluminum alloy foil.
- the positive electrode 304 and the negative electrode 307 used in the coin-shaped secondary battery 300 may each have an active material layer formed on only one side.
- the positive electrode can 301 and the negative electrode can 302 may be made of a metal such as nickel, aluminum, or titanium that is corrosion resistant to the electrolyte, an alloy thereof, or an alloy of these metals with another metal (for example, stainless steel). can. In addition, it is preferable to coat with nickel, aluminum, or the like in order to prevent corrosion due to an electrolytic solution or the like.
- the positive electrode can 301 and the negative electrode can 302 are electrically connected to the positive electrode 304 and the negative electrode 307, respectively.
- the negative electrode 307, the positive electrode 304, and the separator 310 are immersed in an electrolytic solution, and as shown in FIG. 301 and a negative electrode can 302 are crimped via a gasket 303 to manufacture a coin-shaped secondary battery 300 .
- the coin-type secondary battery 300 having high capacity, high discharge capacity, and excellent cycle characteristics can be obtained.
- a cylindrical secondary battery 616 has a positive electrode cap (battery lid) 601 on its top surface and battery cans (armor cans) 602 on its side and bottom surfaces.
- the positive electrode cap 601 and the battery can (outer can) 602 are insulated by a gasket (insulating packing) 610 .
- FIG. 20B is a diagram schematically showing a cross section of a cylindrical secondary battery.
- the cylindrical secondary battery shown in FIG. 20B has a positive electrode cap (battery lid) 601 on the top surface and battery cans (armor cans) 602 on the side and bottom surfaces.
- the positive electrode cap 601 and the battery can (outer can) 602 are insulated by a gasket (insulating packing) 610 .
- a battery element in which a strip-shaped positive electrode 604 and a strip-shaped negative electrode 606 are wound with a separator 605 interposed therebetween is provided inside a hollow cylindrical battery can 602 .
- the battery element is wound around the central axis.
- Battery can 602 is closed at one end and open at the other end.
- the battery can 602 can be made of metals such as nickel, aluminum, titanium, etc., which are corrosion-resistant to the electrolyte, alloys thereof, and alloys of these and other metals (for example, stainless steel). .
- the battery element in which the positive electrode, the negative electrode and the separator are wound is sandwiched between a pair of insulating plates 608 and 609 facing each other.
- a non-aqueous electrolyte (not shown) is filled inside the battery can 602 in which the battery element is provided. The same non-aqueous electrolyte as used in coin-type secondary batteries can be used.
- the positive electrode and negative electrode used in a cylindrical storage battery are wound, it is preferable to form active materials on both sides of the current collector.
- a positive electrode terminal (positive collector lead) 603 is connected to the positive electrode 604
- a negative electrode terminal (negative collector lead) 607 is connected to the negative electrode 606 .
- a metal material such as aluminum can be used for both the positive electrode terminal 603 and the negative electrode terminal 607 .
- the positive electrode terminal 603 and the negative electrode terminal 607 are resistance welded to the safety valve mechanism 613 and the bottom of the battery can 602, respectively.
- the safety valve mechanism 613 is electrically connected to the positive electrode cap 601 via a PTC element (Positive Temperature Coefficient) 611 .
- the safety valve mechanism 613 disconnects the electrical connection between the positive electrode cap 601 and the positive electrode 604 when the increase in internal pressure of the battery exceeds a predetermined threshold.
- the PTC element 611 is a thermal resistance element whose resistance increases when the temperature rises, and the increase in resistance limits the amount of current to prevent abnormal heat generation.
- Barium titanate (BaTiO 3 ) semiconductor ceramics or the like can be used for the PTC element.
- FIG. 20C shows an example of the power storage system 615.
- a power storage system 615 includes a plurality of secondary batteries 616 .
- the positive electrode of each secondary battery contacts and is electrically connected to a conductor 624 separated by an insulator 625 .
- Conductor 624 is electrically connected to control circuit 620 via wiring 623 .
- a negative electrode of each secondary battery is electrically connected to the control circuit 620 through a wiring 626 .
- As the control circuit 620 a charge/discharge control circuit that performs charge/discharge or a protection circuit that prevents overcharge and/or overdischarge can be applied.
- FIG. 20D shows an example of the power storage system 615.
- FIG. A power storage system 615 includes a plurality of secondary batteries 616 that are sandwiched between a conductive plate 628 and a conductive plate 614 .
- the plurality of secondary batteries 616 are electrically connected to the conductive plates 628 and 614 by wirings 627 .
- the plurality of secondary batteries 616 may be connected in parallel, may be connected in series, or may be connected in series after being connected in parallel.
- a plurality of secondary batteries 616 may be connected in series after being connected in parallel.
- a temperature control device may be provided between the plurality of secondary batteries 616 .
- the secondary battery 616 When the secondary battery 616 is overheated, it can be cooled by the temperature control device, and when the secondary battery 616 is too cold, it can be heated by the temperature control device. Therefore, the performance of power storage system 615 is less likely to be affected by the outside air temperature.
- the power storage system 615 is electrically connected to the control circuit 620 via wiring 621 and wiring 622 .
- the wiring 621 is electrically connected to the positive electrodes of the plurality of secondary batteries 616 through the conductive plate 628
- the wiring 622 is electrically connected to the negative electrodes of the plurality of secondary batteries 616 through the conductive plate 614 .
- FIG. 21 A structural example of a secondary battery is described with reference to FIGS. 21 and 22.
- FIG. 21 A structural example of a secondary battery is described with reference to FIGS. 21 and 22.
- a secondary battery 913 shown in FIG. 21A has a wound body 950 provided with terminals 951 and 952 inside a housing 930 .
- the wound body 950 is immersed in the electrolytic solution inside the housing 930 .
- the terminal 952 is in contact with the housing 930, and the terminal 951 is not in contact with the housing 930 by using an insulating material or the like.
- the housing 930 is shown separately for the sake of convenience. exist.
- a metal material such as aluminum
- a resin material can be used as the housing 930.
- the housing 930 shown in FIG. 21A may be made of a plurality of materials.
- a housing 930a and a housing 930b are attached together, and a wound body 950 is provided in a region surrounded by the housings 930a and 930b.
- An insulating material such as organic resin can be used as the housing 930a.
- a material such as an organic resin for the surface on which the antenna is formed shielding of the electric field by the secondary battery 913 can be suppressed.
- an antenna may be provided inside the housing 930a.
- a metal material, for example, can be used as the housing 930b.
- a wound body 950 has a negative electrode 931 , a positive electrode 932 , and a separator 933 .
- the wound body 950 is a wound body in which the negative electrode 931 and the positive electrode 932 are laminated with the separator 933 interposed therebetween, and the laminated sheet is wound. Note that the negative electrode 931, the positive electrode 932, and the separator 933 may be stacked more than once.
- a secondary battery 913 having a wound body 950a as shown in FIG. 22 may be used.
- a wound body 950 a illustrated in FIG. 22A includes a negative electrode 931 , a positive electrode 932 , and a separator 933 .
- the negative electrode 931 has a negative electrode active material layer 931a.
- the positive electrode 932 has a positive electrode active material layer 932a.
- the secondary battery 913 having high capacity, high discharge capacity, and excellent cycle characteristics can be obtained.
- the separator 933 has a wider width than the negative electrode active material layer 931a and the positive electrode active material layer 932a, and is wound so as to overlap with the negative electrode active material layer 931a and the positive electrode active material layer 932a.
- the width of the negative electrode active material layer 931a is wider than that of the positive electrode active material layer 932a.
- the wound body 950a having such a shape is preferable because of its good safety and productivity.
- the negative electrode 931 is electrically connected to the terminal 951 by ultrasonic bonding, welding, or crimping.
- Terminal 951 is electrically connected to terminal 911a.
- the positive electrode 932 is electrically connected to the terminal 952 by ultrasonic bonding, welding, or crimping.
- Terminal 952 is electrically connected to terminal 911b.
- the casing 930 covers the wound body 950a and the electrolytic solution to form a secondary battery 913.
- the housing 930 is preferably provided with a safety valve, an overcurrent protection element, and the like.
- the safety valve is a valve that opens the interior of housing 930 at a predetermined internal pressure in order to prevent battery explosion.
- the secondary battery 913 may have a plurality of wound bodies 950a. By using a plurality of wound bodies 950a, the secondary battery 913 can have a higher discharge capacity.
- the description of the secondary battery 913 illustrated in FIGS. 21A to 21C can be referred to.
- FIGS. 23A and 23B show an example of an external view of an example of a laminated secondary battery.
- 23A and 23B have a positive electrode 503, a negative electrode 506, a separator 507, an outer casing 509, a positive electrode lead electrode 510, and a negative electrode lead electrode 511.
- FIG. 23A and 23B have a positive electrode 503, a negative electrode 506, a separator 507, an outer casing 509, a positive electrode lead electrode 510, and a negative electrode lead electrode 511.
- the positive electrode 503 has a positive electrode current collector 501 , and the positive electrode active material layer 502 is formed on the surface of the positive electrode current collector 501 .
- the positive electrode 503 has a region where the positive electrode current collector 501 is partially exposed (hereinafter referred to as a tab region).
- the negative electrode 506 has a negative electrode current collector 504 , and the negative electrode active material layer 505 is formed on the surface of the negative electrode current collector 504 .
- the negative electrode 506 has a region where the negative electrode current collector 504 is partially exposed, that is, a tab region.
- the area or shape of the tab regions of the positive and negative electrodes is not limited to the example shown in FIG. 24A.
- FIG. 24B shows the negative electrode 506, separator 507 and positive electrode 503 stacked.
- an example is shown in which five sets of negative electrodes and four sets of positive electrodes are used. It can also be called a laminate consisting of a negative electrode, a separator, and a positive electrode.
- the tab regions of the positive electrode 503 are joined together, and the positive electrode lead electrode 510 is joined to the tab region of the outermost positive electrode.
- For joining for example, ultrasonic welding or the like may be used.
- bonding between the tab regions of the negative electrode 506 and bonding of the negative electrode lead electrode 511 to the tab region of the outermost negative electrode are performed.
- the negative electrode 506 , the separator 507 and the positive electrode 503 are arranged on the outer package 509 .
- the exterior body 509 is bent at the portion indicated by the dashed line. After that, the outer peripheral portion of the exterior body 509 is joined. For example, thermocompression bonding or the like may be used for joining. At this time, a region (hereinafter referred to as an introduction port) that is not joined is provided in a part (or one side) of the exterior body 509 so that the electrolytic solution can be introduced later.
- an introduction port a region (hereinafter referred to as an introduction port) that is not joined is provided in a part (or one side) of the exterior body 509 so that the electrolytic solution can be introduced later.
- the electrolytic solution is introduced into the exterior body 509 through an inlet provided in the exterior body 509 . It is preferable to introduce the electrolytic solution under a reduced pressure atmosphere or an inert atmosphere. And finally, the inlet is joined. In this manner, a laminated secondary battery 500 can be manufactured.
- the secondary battery 500 having high capacity, high discharge capacity, and excellent cycle characteristics can be obtained.
- Battery pack example An example of a secondary battery pack of one embodiment of the present invention that can be wirelessly charged using an antenna will be described with reference to FIGS.
- FIG. 25A is a diagram showing the appearance of the secondary battery pack 531, which has a thin rectangular parallelepiped shape (also called a thick flat plate shape).
- FIG. 25B is a diagram illustrating the configuration of the secondary battery pack 531. As shown in FIG.
- the secondary battery pack 531 has a circuit board 540 and a secondary battery 513 .
- a label 529 is attached to the secondary battery 513 .
- Circuit board 540 is secured by seal 515 .
- the secondary battery pack 531 has an antenna 517 .
- the inside of the secondary battery 513 may have a structure having a wound body or a structure having a laminated body.
- the secondary battery pack 531 has a control circuit 590 on a circuit board 540, as shown in FIG. 25B, for example. Also, the circuit board 540 is electrically connected to the terminals 514 . In addition, the circuit board 540 is electrically connected to the antenna 517 , one of the positive and negative leads 551 and the other of the positive and negative leads 552 of the secondary battery 513 .
- FIG. 25C it may have a circuit system 590 a provided on the circuit board 540 and a circuit system 590 b electrically connected to the circuit board 540 via the terminals 514 .
- antenna 517 is not limited to a coil shape, and may be linear or plate-shaped, for example. Further, antennas such as planar antennas, aperture antennas, traveling wave antennas, EH antennas, magnetic field antennas, and dielectric antennas may be used. Alternatively, antenna 517 may be a planar conductor. This flat conductor can function as one of conductors for electric field coupling. That is, the antenna 517 may function as one of the two conductors of the capacitor. As a result, electric power can be exchanged not only by electromagnetic fields and magnetic fields, but also by electric fields.
- the secondary battery pack 531 has a layer 519 between the antenna 517 and the secondary battery 513 .
- the layer 519 has a function of shielding an electromagnetic field generated by the secondary battery 513, for example.
- a magnetic material for example, can be used as the layer 519 .
- FIG. 26C shows an example of application to an electric vehicle (EV).
- EV electric vehicle
- the electric vehicle is equipped with first batteries 1301a and 1301b as secondary batteries for main driving, and a second battery 1311 that supplies power to an inverter 1312 that starts the motor 1304.
- the second battery 1311 is also called cranking battery (also called starter battery).
- the second battery 1311 only needs to have a high output and does not need a large capacity so much, and the capacity of the second battery 1311 is smaller than that of the first batteries 1301a and 1301b.
- the internal structure of the first battery 1301a may be the wound type shown in FIG. 21C or 22A, or the laminated type shown in FIG. 23A or 23B. Further, the all-solid-state battery of Embodiment 6 may be used as the first battery 1301a. By using the all-solid-state battery of Embodiment 6 for the first battery 1301a, the capacity can be increased, the safety can be improved, and the size and weight can be reduced.
- first batteries 1301a and 1301b are connected in parallel
- three or more batteries may be connected in parallel.
- the first battery 1301a can store sufficient electric power
- the first battery 1301b may be omitted.
- a large amount of electric power can be extracted by forming a battery pack including a plurality of secondary batteries.
- a plurality of secondary batteries may be connected in parallel, may be connected in series, or may be connected in series after being connected in parallel.
- a plurality of secondary batteries is also called an assembled battery.
- a secondary battery for vehicle has a service plug or a circuit breaker that can cut off high voltage without using a tool in order to cut off power from a plurality of secondary batteries.
- the power of the first batteries 1301a and 1301b is mainly used to rotate the motor 1304, but is also used to supply 42V in-vehicle components (electric power steering 1307, heater 1308, defogger 1309, etc.) via the DCDC circuit 1306. to power the The first battery 1301a is also used to rotate the rear motor 1317 when the rear wheel has the rear motor 1317 .
- the second battery 1311 supplies power to 14V vehicle-mounted components (audio 1313, power window 1314, lamps 1315, etc.) via the DCDC circuit 1310.
- FIG. 26A shows an example in which nine prismatic secondary batteries 1300 are used as one battery pack 1415 .
- Nine square secondary batteries 1300 are connected in series, one electrode is fixed by a fixing portion 1413 made of an insulator, and the other electrode is fixed by a fixing portion 1414 made of an insulator.
- an example of fixing by fixing portions 1413 and 1414 is shown; Since it is assumed that the vehicle is subjected to vibration or shaking from the outside (such as the road surface), the fixed portions 1413 and 1414 are used. It is preferable to fix a plurality of secondary batteries with a battery housing box or the like.
- One electrode is electrically connected to the control circuit portion 1320 through a wiring 1421 .
- the other electrode is electrically connected to the control circuit section 1320 by wiring 1422 .
- control circuit portion 1320 may use a memory circuit including a transistor using an oxide semiconductor.
- a charge control circuit or a battery control system including a memory circuit including a transistor using an oxide semiconductor is sometimes called a BTOS (battery operating system or battery oxide semiconductor).
- oxides include In-M-Zn oxide (element M is aluminum, gallium, yttrium, copper, vanadium, beryllium, boron, titanium, iron, nickel, germanium, zirconium, molybdenum, lanthanum, cerium, neodymium, A metal oxide such as one selected from hafnium, tantalum, tungsten, magnesium, or the like, or a plurality of types thereof may be used.
- element M is aluminum, gallium, yttrium, copper, vanadium, beryllium, boron, titanium, iron, nickel, germanium, zirconium, molybdenum, lanthanum, cerium, neodymium
- a metal oxide such as one selected from hafnium, tantalum, tungsten, magnesium, or the like, or a plurality of types thereof may be used.
- In-M-Zn oxides that can be applied as oxides are preferably CAAC-OS (C-Axis Aligned Crystal Oxide Semiconductor) and CAC-OS (Cloud-Aligned Composite Oxide Semiconductor).
- CAAC-OS is an oxide semiconductor that includes a plurality of crystal regions, and the c-axes of the plurality of crystal regions are oriented in a specific direction. Note that the specific direction is the thickness direction of the CAAC-OS film, the normal direction to the formation surface of the CAAC-OS film, or the normal direction to the surface of the CAAC-OS film.
- a crystalline region is a region having periodicity in atomic arrangement. If the atomic arrangement is regarded as a lattice arrangement, the crystalline region is also a region with a uniform lattice arrangement.
- CAC-OS has a mosaic structure in which the material is separated into the first region and the second region, and the first region is distributed in the film (hereinafter referred to as a cloud-like structure). It is also called.). That is, CAC-OS is a composite metal oxide in which the first region and the second region are mixed. However, it may be difficult to observe a clear boundary between the first area and the second area.
- a region containing In as the main component (first 1 region) and a region containing Ga as a main component (second region) are unevenly distributed and can be confirmed to have a mixed structure.
- the conductivity attributed to the first region and the insulation attributed to the second region complementarily act to provide a switching function (on/off function).
- a switching function on/off function
- CAC-OS a part of the material has a conductive function
- a part of the material has an insulating function
- the whole material has a semiconductor function.
- Oxide semiconductors have a variety of structures, each with different characteristics.
- An oxide semiconductor of one embodiment of the present invention includes two or more of an amorphous oxide semiconductor, a polycrystalline oxide semiconductor, an a-like OS, a CAC-OS, an nc-OS, and a CAAC-OS. may
- the control circuit portion 1320 may be formed using unipolar transistors.
- a transistor using an oxide semiconductor for a semiconductor layer has an operating ambient temperature of ⁇ 40° C. or more and 150° C. or less, which is wider than that of single crystal Si, and changes in characteristics are smaller than those of a single crystal even when the secondary battery is heated.
- the off-state current of a transistor using an oxide semiconductor is below the lower limit of measurement regardless of the temperature even at 150° C.
- the off-state current characteristics of a single crystal Si transistor are highly dependent on temperature.
- a single crystal Si transistor has an increased off-current and does not have a sufficiently large current on/off ratio.
- the control circuitry 1320 can improve safety. Further, by combining the positive electrode active material 100 obtained in Embodiments 1, 2, and the like with a secondary battery using the positive electrode for the positive electrode, a synergistic effect regarding safety can be obtained.
- the secondary battery in which the positive electrode active material 100 obtained in Embodiments 1, 2, and the like is used for the positive electrode and the control circuit portion 1320 can greatly contribute to the elimination of accidents such as fire caused by the secondary battery.
- the control circuit unit 1320 using a memory circuit including a transistor using an oxide semiconductor can also function as an automatic control device for a secondary battery against the cause of instability such as a micro-short.
- Functions that eliminate the causes of secondary battery instability include overcharge prevention, overcurrent prevention, overheat control during charging, cell balance in the assembled battery, overdischarge prevention, fuel gauge, temperature-dependent Automatic control of charging voltage and current amount, control of charging current amount according to the degree of deterioration, detection of micro-short abnormal behavior, prediction of abnormality related to micro-short, etc., among which the control circuit section 1320 has at least one function.
- micro short refers to a minute short circuit inside a secondary battery. It refers to a phenomenon in which a small amount of short-circuit current flows at a short-circuited portion. Since a large voltage change occurs in a relatively short time and even at a small location, the abnormal voltage value may affect subsequent estimation.
- micro-shorts One of the causes of micro-shorts is that the non-uniform distribution of the positive electrode active material caused by repeated charging and discharging causes localized concentration of current in a portion of the positive electrode and a portion of the negative electrode, resulting in a separator failure. It is said that a micro short-circuit occurs due to the generation of a portion where a part fails or the generation of a side reaction product due to a side reaction.
- control circuit unit 1320 not only detects micro-shorts, but also detects the terminal voltage of the secondary battery and manages the charging/discharging state of the secondary battery. For example, both the output transistor of the charging circuit and the cut-off switch can be turned off almost simultaneously to prevent overcharging.
- FIG. 26B shows an example of a block diagram of the battery pack 1415 shown in FIG. 26A.
- the control circuit unit 1320 includes a switch unit 1324 including at least a switch for preventing overcharge and a switch for preventing overdischarge, a control circuit 1322 for controlling the switch unit 1324, a voltage measurement unit for the first battery 1301a, have
- the control circuit unit 1320 is set with an upper limit voltage and a lower limit voltage of the secondary battery to be used, and limits the upper limit of the current from the outside or the upper limit of the output current to the outside.
- the range from the lower limit voltage to the upper limit voltage of the secondary battery is within the voltage range recommended for use.
- the control circuit unit 1320 controls the switch unit 1324 to prevent over-discharging and/or over-charging, it can also be called a protection circuit.
- control circuit 1322 detects a voltage that is likely to cause overcharging
- the switch of the switch section 1324 is turned off to cut off the current.
- a PTC element may be provided in the charging/discharging path to provide a function of interrupting the current according to the temperature rise.
- the control circuit section 1320 also has an external terminal 1325 (+IN) and an external terminal 1326 (-IN).
- the switch section 1324 can be configured by combining n-channel transistors or p-channel transistors.
- the switch unit 1324 is not limited to a switch having a Si transistor using single crystal silicon. indium), SiC (silicon carbide), ZnSe (zinc selenide), GaN (gallium nitride), GaOx (gallium oxide; x is a real number greater than 0), or the like.
- a memory element using an OS transistor can be freely arranged by stacking it on a circuit using a Si transistor or the like, integration can be easily performed.
- an OS transistor can be manufactured using a manufacturing apparatus similar to that of a Si transistor, it can be manufactured at low cost. That is, the control circuit portion 1320 using an OS transistor can be stacked on the switch portion 1324 and integrated into one chip. Since the volume occupied by the control circuit section 1320 can be reduced, miniaturization is possible.
- the first batteries 1301a and 1301b mainly supply power to 42V system (high voltage system) in-vehicle equipment, and the second battery 1311 supplies power to 14V system (low voltage system) in-vehicle equipment.
- the second battery 1311 is often adopted as a lead-acid battery because of its cost advantage.
- a lead-acid battery has a larger self-discharge than a lithium-ion battery, and has the disadvantage of being easily deteriorated due to a phenomenon called sulfation.
- Using a lithium-ion battery as the second battery 1311 has the advantage of being maintenance-free, but if it is used for a long period of time, for example, three years or more, there is a risk that an abnormality that is difficult to determine may occur during manufacturing.
- the second battery 1311 that starts the inverter becomes inoperable, the second battery 1311 is lead-free in order to prevent the motor from being unable to start even if the first batteries 1301a and 1301b have remaining capacity.
- power is supplied from the first battery to the second battery and charged so as to always maintain a fully charged state.
- the second battery 1311 may use a lead-acid battery, an all-solid battery, or an electric double layer capacitor.
- the all-solid-state battery of Embodiment 6 may be used.
- the capacity can be increased, and the size and weight can be reduced.
- regenerated energy from the rotation of the tire 1316 is sent to the motor 1304 via the gear 1305 and charged to the second battery 1311 via the control circuit section 1321 from the motor controller 1303 or the battery controller 1302 .
- the battery controller 1302 charges the first battery 1301 a through the control circuit unit 1320 .
- the battery controller 1302 charges the first battery 1301 b through the control circuit unit 1320 . In order to efficiently charge the regenerated energy, it is desirable that the first batteries 1301a and 1301b be capable of rapid charging.
- the battery controller 1302 can set the charging voltage and charging current of the first batteries 1301a and 1301b.
- the battery controller 1302 can set charging conditions according to the charging characteristics of the secondary battery to be used and perform rapid charging.
- the outlet of the charger or the connection cable of the charger is electrically connected to the battery controller 1302 .
- Electric power supplied from an external charger charges the first batteries 1301 a and 1301 b via the battery controller 1302 .
- Some chargers are provided with a control circuit and do not use the function of the battery controller 1302. In order to prevent overcharging, the first batteries 1301a and 1301b are charged via the control circuit unit 1320. is preferred.
- the connection cable or the connection cable of the charger is provided with the control circuit.
- the control circuit section 1320 is sometimes called an ECU (Electronic Control Unit).
- the ECU is connected to a CAN (Controller Area Network) provided in the electric vehicle.
- CAN is one of serial communication standards used as an in-vehicle LAN.
- the ECU includes a microcomputer.
- the ECU uses a CPU or a GPU.
- External chargers installed at charging stands and the like include 100V outlets and 200V outlets, or 3-phase 200V and 50kW. Also, the battery can be charged by receiving power supply from an external charging facility by a non-contact power supply method or the like.
- the operating voltage of the secondary battery can be increased by using the positive electrode active material 100 described in Embodiments 1 and 2, and as the charging voltage increases, , can increase the available capacity.
- the positive electrode active material 100 described in Embodiments 1 and 2 for the positive electrode it is possible to provide a vehicle secondary battery having excellent cycle characteristics.
- next-generation clean energy such as a hybrid vehicle (HV), an electric vehicle (EV), or a plug-in hybrid vehicle (PHV) can be used.
- HV hybrid vehicle
- EV electric vehicle
- PSV plug-in hybrid vehicle
- a car can be realized.
- secondary batteries are used in agricultural machinery, motorized bicycles including electrically assisted bicycles, motorcycles, electric wheelchairs, electric carts, ships, submarines, aircraft, rockets, artificial satellites, space probes, planetary probes, or spacecraft. It can also be installed.
- the secondary battery of one embodiment of the present invention can be a high-capacity secondary battery. Therefore, the secondary battery of one embodiment of the present invention is suitable for miniaturization and weight reduction, and can be suitably used for transportation vehicles.
- a vehicle 2001 shown in FIG. 27A is an electric vehicle that uses an electric motor as a power source for running. Alternatively, it is a hybrid vehicle in which an electric motor and an engine can be appropriately selected and used as power sources for running.
- a secondary battery is mounted in a vehicle, an example of the secondary battery described in Embodiment 4 is installed at one or more places.
- a car 2001 shown in FIG. 27A has a battery pack 2200, and the battery pack has a secondary battery module to which a plurality of secondary batteries are connected. Furthermore, it is preferable to have a charging control device electrically connected to the secondary battery module.
- the vehicle 2001 can be charged by receiving power from an external charging facility by a plug-in system or a contactless power supply system to the secondary battery of the vehicle 2001 .
- the charging method or the standard of the connector may be appropriately performed by a predetermined method such as CHAdeMO (registered trademark) or Combo.
- the secondary battery may be a charging station provided in a commercial facility, or may be a household power source.
- plug-in technology can charge a power storage device mounted on the automobile 2001 by power supply from the outside. Charging can be performed by converting AC power into DC power via a conversion device such as an ACDC converter.
- a power receiving device can be mounted on a vehicle, and power can be supplied from a power transmission device on the ground in a contactless manner for charging.
- this non-contact power supply system it is possible to charge the vehicle not only while the vehicle is stopped but also while the vehicle is running by installing a power transmission device on the road or the outer wall.
- power may be transmitted and received between two vehicles.
- a solar battery may be provided on the exterior of the vehicle, and the secondary battery may be charged while the vehicle is stopped or running.
- An electromagnetic induction method or a magnetic resonance method can be used for such contactless power supply.
- FIG. 27B shows a large transport vehicle 2002 with electrically controlled motors as an example of a transport vehicle.
- the secondary battery module of the transportation vehicle 2002 has a maximum voltage of 170 V, for example, a four-cell unit of secondary batteries having a nominal voltage of 3.0 V or more and 5.0 V or less, and 48 cells connected in series. Except for the number of secondary batteries forming the secondary battery module of the battery pack 2201, the function is the same as that of FIG. 27A, so the description is omitted.
- FIG. 27C shows, as an example, a large transport vehicle 2003 with electrically controlled motors.
- the secondary battery module of the transportation vehicle 2003 has a maximum voltage of 600 V, for example, a hundred or more secondary batteries with a nominal voltage of 3.0 V or more and 5.0 V or less connected in series. Therefore, a secondary battery with small variations in characteristics is required.
- a secondary battery having stable battery characteristics can be manufactured at low cost from the viewpoint of yield. Mass production is possible. 16A except that the number of secondary batteries constituting the secondary battery module of the battery pack 2202 is different, description thereof is omitted.
- FIG. 27D shows an aircraft 2004 with an engine that burns fuel as an example. Since the aircraft 2004 shown in FIG. 27D has wheels for takeoff and landing, it can be said to be a type of transport vehicle, and a secondary battery module is configured by connecting a plurality of secondary batteries, and the secondary battery module and charging control are performed. It has a battery pack 2203 containing a device.
- the secondary battery module of aircraft 2004 has a maximum voltage of 32V, for example, eight 4V secondary batteries connected in series. Except for the number of secondary batteries forming the secondary battery module of the battery pack 2203, the function is the same as that of FIG. 27A, so the description is omitted.
- FIG. 27E shows a satellite 2005 with a secondary battery 2204 as an example. Since the artificial satellite 2005 is used in extremely cold outer space, it preferably includes the secondary battery 2204 which is one embodiment of the present invention and has excellent low-temperature resistance. Moreover, it is more preferable that the secondary battery 2204 is mounted inside the artificial satellite 2005 while being covered with a heat insulating member.
- the house illustrated in FIG. 28A includes a power storage device 2612 including a secondary battery that is one embodiment of the present invention and a solar panel 2610.
- the power storage device 2612 is electrically connected to the solar panel 2610 through a wiring 2611 or the like. Alternatively, the power storage device 2612 and the ground-mounted charging device 2604 may be electrically connected.
- a power storage device 2612 can be charged with power obtained from the solar panel 2610 . Electric power stored in power storage device 2612 can be used to charge a secondary battery of vehicle 2603 via charging device 2604 .
- Power storage device 2612 is preferably installed in the underfloor space. By installing in the space under the floor, the space above the floor can be effectively used. Alternatively, power storage device 2612 may be installed on the floor.
- the power stored in the power storage device 2612 can also supply power to other electronic devices in the house. Therefore, the use of the power storage device 2612 according to one embodiment of the present invention as an uninterruptible power supply makes it possible to use the electronic device even when power cannot be supplied from a commercial power supply due to a power failure or the like.
- FIG. 28B illustrates an example of a power storage device according to one embodiment of the present invention.
- a power storage device 791 according to one embodiment of the present invention is installed in an underfloor space 796 of a building 799.
- the power storage device 791 may be provided with the control circuit described in Embodiment 7, and the power storage device 791 may be a secondary battery whose positive electrode is the positive electrode active material 100 obtained in Embodiments 1, 2, or the like.
- a synergistic effect on safety can be obtained with The control circuit described in Embodiment 7 and the secondary battery using the positive electrode active material 100 described in Embodiments 1, 2, etc. for the positive electrode are greatly effective in eliminating accidents such as fire caused by the power storage device 791 having the secondary battery. can contribute.
- a control device 790 is installed in the power storage device 791, and the control device 790 is connected to the distribution board 703, the power storage controller 705 (also referred to as a control device), the display 706, and the router 709 by wiring. electrically connected.
- Electric power is sent from the commercial power source 701 to the distribution board 703 via the service wire attachment portion 710 . Electric power is sent to the distribution board 703 from the power storage device 791 and the commercial power supply 701, and the distribution board 703 distributes the sent power to the general load via an outlet (not shown). 707 and power storage system load 708 .
- a general load 707 is, for example, an electrical device such as a television or a personal computer
- a power storage system load 708 is, for example, an electrical device such as a microwave oven, refrigerator, or air conditioner.
- the power storage controller 705 has a measurement unit 711, a prediction unit 712, and a planning unit 713.
- the measuring unit 711 has a function of measuring the amount of electric power consumed by the general load 707 and the power storage system load 708 during a day (for example, from 00:00 to 24:00).
- the measurement unit 711 may also have a function of measuring the amount of power in the power storage device 791 and the amount of power supplied from the commercial power source 701 .
- the prediction unit 712 predicts the demand to be consumed by the general load 707 and the storage system load 708 during the next day based on the amount of power consumed by the general load 707 and the storage system load 708 during the day. It has a function of predicting power consumption.
- the planning unit 713 also has a function of planning charging and discharging of the power storage device 791 based on the amount of power demand predicted by the prediction unit 712 .
- the amount of power consumed by the general load 707 and the power storage system load 708 measured by the measurement unit 711 can be confirmed by the display 706 . Also, it can be checked on an electric device such as a television or a personal computer via the router 709 . In addition, it can be confirmed by a mobile electronic terminal such as a smart phone or a tablet via the router 709 . In addition, it is possible to check the amount of power demand for each time period (or for each hour) predicted by the prediction unit 712 by using the display 706, the electric device, and the portable electronic terminal.
- FIG. 29A illustrates an example of an electric bicycle using the power storage device of one embodiment of the present invention.
- the power storage device of one embodiment of the present invention can be applied to the electric bicycle 8700 illustrated in FIG. 29A.
- a power storage device of one embodiment of the present invention includes, for example, a plurality of storage batteries and a protection circuit.
- the electric bicycle 8700 includes a power storage device 8702.
- the power storage device 8702 can supply electricity to a motor that assists the driver.
- the power storage device 8702 is portable, and is shown removed from the bicycle in FIG. 29B.
- the power storage device 8702 includes a plurality of storage batteries 8701 included in the power storage device of one embodiment of the present invention, and the remaining battery power and the like can be displayed on a display portion 8703 .
- the power storage device 8702 also includes a control circuit 8704 capable of controlling charging of the secondary battery or detecting an abnormality, one example of which is shown in Embodiment 7.
- the control circuit 8704 is electrically connected to the positive and negative electrodes of the storage battery 8701 .
- the positive electrode active material 100 obtained in Embodiments 1, 2, etc. with a secondary battery using the positive electrode for the positive electrode, a synergistic effect regarding safety can be obtained.
- the secondary battery in which the positive electrode active material 100 obtained in Embodiments 1, 2, and the like is used for the positive electrode and the control circuit 8704 can greatly contribute to the elimination of accidents such as fire caused by the secondary battery.
- FIG. 29C is an example of a motorcycle using the power storage device of one embodiment of the present invention.
- the power storage device 8602 can supply electricity to the turn signal lights 8603 .
- the power storage device 8602 containing a plurality of secondary batteries using the positive electrode active material 100 obtained in Embodiments 1, 2, and the like for positive electrodes can have a high capacity and can contribute to miniaturization.
- the power storage device 8602 can be stored in the storage space 8604 under the seat.
- the power storage device 8602 can be stored in the underseat storage 8604 even if the underseat storage 8604 is small.
- a secondary battery which is one embodiment of the present invention, in an electronic device
- electronic devices that implement secondary batteries include television devices (also referred to as televisions or television receivers), monitors for computers, digital cameras, digital video cameras, digital photo frames, mobile phones (mobile phones, Also referred to as a mobile phone device), a portable game machine, a personal digital assistant, a sound reproducing device, a large game machine such as a pachinko machine, and the like.
- Portable information terminals include notebook personal computers, tablet terminals, electronic book terminals, mobile phones, and the like.
- FIG. 30A shows an example of a mobile phone.
- a mobile phone 2100 includes a display unit 2102 incorporated in a housing 2101, operation buttons 2103, an external connection port 2104, a speaker 2105, a microphone 2106, and the like.
- the mobile phone 2100 has a secondary battery 2107 .
- the secondary battery 2107 By including the secondary battery 2107 in which the positive electrode active material 100 described in Embodiments 1, 2, and the like is used for the positive electrode, the capacity can be increased, and a structure that can cope with the space saving associated with the downsizing of the housing is provided. can be realized.
- the mobile phone 2100 can execute various applications such as mobile phone, e-mail, reading and creating text, playing music, Internet communication, and computer games.
- the operation button 2103 can have various functions such as time setting, power on/off operation, wireless communication on/off operation, manner mode execution/cancellation, and power saving mode execution/cancellation.
- the operating system installed in the mobile phone 2100 can freely set the functions of the operation buttons 2103 .
- the mobile phone 2100 is capable of performing standardized short-range wireless communication. For example, by intercommunicating with a headset capable of wireless communication, hands-free communication is also possible.
- the mobile phone 2100 has an external connection port 2104, and can directly exchange data with other information terminals via connectors. Also, charging can be performed via the external connection port 2104 . Note that the charging operation may be performed by wireless power supply without using the external connection port 2104 .
- the mobile phone 2100 preferably has a sensor.
- a sensor for example, a fingerprint sensor, a pulse sensor, a body sensor such as a body temperature sensor, a touch sensor, a pressure sensor, an acceleration sensor, or the like is preferably mounted.
- FIG. 30B is an unmanned aerial vehicle 2300 having multiple rotors 2302 .
- Unmanned aerial vehicle 2300 may also be referred to as a drone.
- Unmanned aerial vehicle 2300 has a secondary battery 2301 that is one embodiment of the present invention, a camera 2303, and an antenna (not shown).
- Unmanned aerial vehicle 2300 can be remotely operated via an antenna.
- a secondary battery using the positive electrode active material 100 obtained in Embodiments 1, 2, etc. as a positive electrode has a high energy density and is highly safe. It is suitable as a secondary battery to be mounted on aircraft 2300 .
- FIG. 30C shows an example of a robot.
- a robot 6400 shown in FIG. 30C includes a secondary battery 6409, an illuminance sensor 6401, a microphone 6402, an upper camera 6403, a speaker 6404, a display unit 6405, a lower camera 6406 and an obstacle sensor 6407, a moving mechanism 6408, an arithmetic device, and the like.
- the microphone 6402 has a function of detecting the user's speech and environmental sounds. Also, the speaker 6404 has a function of emitting sound. Robot 6400 can communicate with a user using microphone 6402 and speaker 6404 .
- the display unit 6405 has a function of displaying various information.
- the robot 6400 can display information desired by the user on the display unit 6405 .
- the display portion 6405 may include a touch panel. Further, the display unit 6405 may be a detachable information terminal, and by installing it at a fixed position of the robot 6400, charging and data transfer are possible.
- the upper camera 6403 and the lower camera 6406 have the function of imaging the surroundings of the robot 6400.
- the obstacle sensor 6407 can detect the presence or absence of an obstacle in the direction in which the robot 6400 moves forward using the movement mechanism 6408 .
- the robot 6400 uses an upper camera 6403, a lower camera 6406, and an obstacle sensor 6407 to recognize the surrounding environment and can move safely.
- a robot 6400 includes a secondary battery 6409 according to one embodiment of the present invention and a semiconductor device or an electronic component in its internal region.
- a secondary battery using the positive electrode active material 100 obtained in Embodiments 1 and 2 as a positive electrode has a high energy density and is highly safe. It is suitable as the secondary battery 6409 mounted on the 6400.
- FIG. 30D shows an example of a cleaning robot.
- the cleaning robot 6300 has a display unit 6302 arranged on the top surface of a housing 6301, a plurality of cameras 6303 arranged on the side surfaces, a brush 6304, an operation button 6305, a secondary battery 6306, various sensors, and the like.
- the cleaning robot 6300 is provided with tires, a suction port, and the like.
- the cleaning robot 6300 can run by itself, detect dust 6310, and suck the dust from a suction port provided on the bottom surface.
- the cleaning robot 6300 can analyze the image captured by the camera 6303 and determine the presence or absence of obstacles such as walls, furniture, or steps. Further, when an object such as wiring that is likely to get entangled in the brush 6304 is detected by image analysis, the rotation of the brush 6304 can be stopped.
- Cleaning robot 6300 includes a secondary battery 6306 according to one embodiment of the present invention and a semiconductor device or an electronic component in its internal region.
- a secondary battery using the positive electrode active material 100 obtained in Embodiments 1 and 2 as a positive electrode has a high energy density and is highly safe. It is suitable as the secondary battery 6306 mounted on the robot 6300 .
- FIG. 31A shows an example of a wearable device.
- a wearable device uses a secondary battery as a power source.
- wearable devices that can be charged not only by wires with exposed connectors but also by wireless charging are being developed. Desired.
- the secondary battery which is one embodiment of the present invention can be mounted in a spectacles-type device 4000 as shown in FIG. 31A.
- the glasses-type device 4000 has a frame 4000a and a display section 4000b.
- the spectacles-type device 4000 that is lightweight, has a good weight balance, and can be used continuously for a long time can be obtained.
- a secondary battery in which the positive electrode active material 100 obtained in Embodiments 1, 2, and the like is used for the positive electrode has a high energy density, and can realize a structure that can cope with space saving due to downsizing of the housing.
- a secondary battery that is one embodiment of the present invention can be mounted in the headset device 4001 .
- the headset type device 4001 has at least a microphone section 4001a, a flexible pipe 4001b, and an earphone section 4001c.
- a secondary battery can be provided in the flexible pipe 4001b or the earphone part 4001c.
- a secondary battery in which the positive electrode active material 100 obtained in Embodiments 1, 2, and the like is used for the positive electrode has a high energy density, and can realize a structure that can cope with space saving due to downsizing of the housing.
- the device 4002 that can be attached directly to the body can be equipped with the secondary battery that is one embodiment of the present invention.
- a secondary battery 4002b can be provided in a thin housing 4002a of the device 4002 .
- a secondary battery in which the positive electrode active material 100 obtained in Embodiments 1, 2, and the like is used for the positive electrode has a high energy density, and can realize a structure that can cope with space saving due to downsizing of the housing.
- the device 4003 that can be attached to clothes can be equipped with a secondary battery that is one embodiment of the present invention.
- a secondary battery 4003b can be provided in a thin housing 4003a of the device 4003 .
- a secondary battery in which the positive electrode active material 100 obtained in Embodiments 1, 2, and the like is used for the positive electrode has a high energy density, and can realize a structure that can cope with space saving due to downsizing of the housing.
- a secondary battery that is one embodiment of the present invention can be mounted in the belt-type device 4006 .
- the belt-type device 4006 has a belt portion 4006a and a wireless power supply receiving portion 4006b, and a secondary battery can be mounted in the inner region of the belt portion 4006a.
- a secondary battery in which the positive electrode active material 100 obtained in Embodiments 1, 2, and the like is used for the positive electrode has a high energy density, and can realize a structure that can cope with space saving due to downsizing of the housing.
- a secondary battery that is one embodiment of the present invention can be mounted in the wristwatch-type device 4005 .
- a wristwatch-type device 4005 has a display portion 4005a and a belt portion 4005b, and a secondary battery can be provided in the display portion 4005a or the belt portion 4005b.
- a secondary battery in which the positive electrode active material 100 obtained in Embodiments 1, 2, and the like is used for the positive electrode has a high energy density, and can realize a structure that can cope with space saving due to downsizing of the housing.
- the display unit 4005a can display not only the time but also various information such as incoming e-mails or phone calls.
- the wristwatch-type device 4005 is a type of wearable device that is directly wrapped around the arm, it may be equipped with a sensor that measures the user's pulse, blood pressure, and the like. It is possible to accumulate data on the amount of exercise and health of the user and manage the health.
- FIG. 31B shows a perspective view of the wristwatch-type device 4005 removed from the arm.
- FIG. 31C shows a state in which a secondary battery 913 is incorporated in the inner region.
- a secondary battery 913 is the secondary battery described in Embodiment 4.
- the secondary battery 913 is provided so as to overlap with the display portion 4005a, can have high density and high capacity, and is small and lightweight.
- the wristwatch-type device 4005 is required to be small and lightweight, by using the positive electrode active material 100 obtained in Embodiments 1 and 2 for the positive electrode of the secondary battery 913, high energy density, In addition, the secondary battery 913 can be small.
- lithium cobalt oxide (Cellseed C-10N, manufactured by Nippon Kagaku Kogyo Co., Ltd.) having no particular additive element was prepared as the lithium cobalt oxide (LiCoO 2 ) in step S14 of FIG. 16 .
- this lithium cobalt oxide was placed in a crucible, which was covered with a lid and then heated at 850° C. for 2 hours in a muffle furnace. After an oxygen atmosphere was created in the muffle furnace, no flow occurred ( O2 purge). When the amount recovered after the initial heating was confirmed, it was found that the weight had slightly decreased. The weight loss may have been due to the removal of impurities from the lithium cobaltate.
- step S21 shown in FIG. 17A LiF was prepared as the F source, and MgF 2 was prepared as the Mg source. LiF:MgF 2 was weighed to be 1:3 (molar ratio). Next, LiF and MgF 2 were mixed in dehydrated acetone and stirred at a rotational speed of 400 rpm for 12 hours to prepare an additive element source (A1 source). A ball mill was used for mixing, and zirconium oxide balls were used as grinding media.
- a total of about 9 g of F source and Mg source was added to 45 mL of the mixing ball mill container together with 20 mL of dehydrated acetone and 22 g of zirconium oxide balls (1 mm ⁇ ) and mixed. After that, it was sieved through a sieve having a mesh of 300 ⁇ m to obtain an A1 source having a uniform particle size.
- step S31 the number of magnesium atoms in the A1 source is weighed so that the number of cobalt atoms in the lithium cobalt oxide is 1 atomic %. mixed with. At this time, the mixture was stirred for 1 hour at a rotation speed of 150 rpm. This is a gentler condition than the stirring when obtaining the A1 source. Finally, the mixture was sieved through a sieve having a mesh size of 300 ⁇ m to obtain a mixture 903 having a uniform particle size (step S32).
- step S33 the mixture 903 was heated.
- the heating conditions were 900° C. and 20 hours.
- a lid was placed over the crucible containing mixture 903 during heating.
- the inside of the crucible was made into an atmosphere containing oxygen, and the entry and exit of the oxygen was shut off (purge).
- a composite oxide containing Mg and F was obtained by heating (step S34a).
- step S51 the composite oxide and the additive element source (A2 source) were mixed.
- nickel hydroxide was prepared as a Ni source
- aluminum hydroxide was prepared as an Al source.
- the number of nickel atoms in the nickel hydroxide is 0.5 atomic % with respect to the number of cobalt atoms in the composite oxide
- the number of aluminum atoms in the aluminum hydroxide is equal to the number of aluminum atoms in the composite oxide. It was weighed so as to be 0.5 atomic % with respect to the number of atoms of cobalt, and mixed with the composite oxide in a dry process. At this time, the mixture was stirred for 1 hour at a rotational speed of 150 rpm.
- a ball mill was used for mixing, and zirconium oxide balls were used as grinding media.
- a total of about 7.5 g of Ni source and Al source was put into a 45 mL container of a mixing ball mill and mixed together with 22 g of zirconium oxide balls (1 mm ⁇ ). This is a gentler condition than the stirring when obtaining the A1 source.
- the mixture was sieved through a sieve having a mesh of 300 ⁇ m to obtain a mixture 904 having a uniform particle size (step S52).
- step S53 the mixture 904 was heated.
- the heating conditions were 850° C. and 10 hours.
- a lid was placed over the crucible containing mixture 904 during heating.
- the inside of the crucible was made into an atmosphere containing oxygen, and the entry and exit of the oxygen was shut off (purge).
- lithium cobaltate containing Mg, F, Ni, and Al was obtained (step S54).
- the positive electrode active material (composite oxide) thus obtained was used as sample 1-1.
- the positive electrode active material of Sample 1-1 obtained in this example was manufactured according to the method for manufacturing the positive electrode active material 100 specifically described in Embodiment 2, and the manufactured positive electrode The characteristics of the active material 100 also have the characteristics of the positive electrode active material 100 specifically described in the second embodiment.
- a half-cell (half-cell 1) was produced for the purpose of evaluating a lithium-ion battery having excellent discharge characteristics and/or charge characteristics even at subzero temperatures. The conditions for manufacturing the half-cell will be described.
- NMP N-methyl-2-pyrrolidone
- the solvent was volatilized, and then press treatment was performed using a roll press device.
- the temperature of both the upper roll and the lower roll was set to 120° C., and the pressure (linear pressure) was set to 210 kN ⁇ m.
- a positive electrode was obtained through the above steps.
- the amount of active material supported on the positive electrode was about 7 mg/cm 2 .
- the electrolyte used for the half cell 1 contains an organic solvent.
- a polypropylene porous film was used as the separator. Lithium metal was used for the negative electrode (counter electrode). Using these, a coin-shaped half cell (half cell 1) was produced. Note that the half cell 1 can be called a test battery.
- a half cell (half cell 2) was produced for the purpose of evaluating a lithium ion battery having standard discharge characteristics and/or charge characteristics below freezing. Note that, like the half cell 1, the half cell 2 can be called a test battery.
- the half cell 2 was manufactured under the same conditions as the half cell 1 except that the electrolyte was different, so the electrolyte used for the half cell 2 will be described here.
- the electrolyte used in the half cell 2 contains an organic solvent.
- Lithium hexafluorophosphate (LiPF 6 ) was dissolved in this organic solvent so as to have a concentration of 1 mol/L, and 2 wt % of vinylene carbonate (VC) was added as an additive to prepare an electrolytic solution.
- VC vinylene carbonate
- a coin-shaped half cell (half cell 2) was fabricated using this electrolytic solution and the like. This electrolytic solution will be referred to as electrolytic solution B hereinafter.
- half-cell 1 and half-cell 2 the discharge capacity was measured under each of a plurality of temperature conditions.
- the temperature during discharge was set to four conditions of 25°C, 0°C, -20°C, and -40°C, and charging was performed under the same conditions at 25°C before the discharge test at each temperature.
- the temperature during charging or discharging described in the examples of this specification was set at the temperature of the constant temperature bath in which the half-cell was left for a certain period of time.
- FIG. 32 shows the discharge capacity for each temperature during discharge.
- both half cell 1 and half cell 2 have high discharge capacities, and even when compared with the 25° C. condition, they have equivalent discharge capacities.
- the discharge capacities of half-cell 1 and half-cell 2 differ significantly.
- the half cell 2 has a discharge capacity of about 50 mAh/g under the condition of -40°C, which is only about 25% of the discharge capacity obtained under the condition of -40°C.
- the discharge capacity of the half cell 1 under the conditions of 1 was about 170 mAh/g, which is at least 70% or more of the discharge capacity obtained under the conditions of 25°C.
- the battery was charged at a constant current of 0.2 C until the voltage reached 4.6 V under an environment of 4.6 V, and then charged at a constant voltage of 4.6 V until the current reached 0.02 C. It is possible to realize a lithium ion battery that is at least 50% or more (or 70% or more) of the discharge capacity value obtained by constant current discharge at a discharge rate of 0.1 C until the voltage reaches 5 V. Proven.
- the lithium ion battery including the positive electrode active material and the electrolyte described in Embodiment 1 and the like can operate at least in the temperature range of -40°C or higher and 25°C or lower. rice field.
- the lithium ion battery including the positive electrode active material and the electrolyte described in Embodiment 1 and the like achieves a discharge capacity of 100 mAh/g or more (specifically, about 170 mAh/g) at -40°C. It became clear that
- Fig. 33 shows the charging capacity for each temperature during charging.
- the charging temperature in FIG. 33 refers to the temperature during charging.
- the charge capacity of the half cell 1 at ⁇ 20° C. was about 175 mAh/g, which was more than 80% of the discharge capacity at 25° C.
- the charge capacity of the half cell 1 under the condition of -40°C was about 120 mAh/g, which was about 60% of the charge capacity under the condition of 25°C.
- both the charge capacity at -20°C and the charge capacity at -40°C were approximately 0 mAh/g.
- the reason why the charge capacity was not obtained is that the electrolyte used in the half cell 2 has a high freezing point, the viscosity of the electrolyte at ⁇ 20° C. and ⁇ 40° C. or the bulk resistance of the positive electrode active material is high, and the lithium ion conductivity is low. is presumed to be due to the extremely low As described above, the lithium-ion battery including at least the positive electrode active material and the electrolyte described in Embodiment 1 and the like can perform at 25°C even in a very low temperature environment of -20°C or -40°C. It was demonstrated that the charging capacity is not so inferior even when compared with the charging capacity.
- Sample 21-1 A total of four samples (Sample 21-1, Sample 21-2, Sample 22-1, and Sample 22-2) were prepared for measuring the discharge capacity. All samples had a loading of approximately 7 mg/cm 2 . Samples 21-2, 22-1, and 22-2 are comparative examples.
- constant current discharging was performed at a cutoff voltage of 2.5 V. The temperature during discharge was measured under four conditions of 25°C, 0°C, -20°C, and -40°C.
- Sample 21-2 uses half cell 2 prepared in Example 1 (that is, half cell 1 having lithium cobaltate containing Mg, F, Ni, and Al as the positive electrode active material and electrolyte solution A as the electrolyte). It has the same structure as Sample 21-1.
- Sample 22-1 is obtained by changing the positive electrode active material used for the half cell compared to Sample 21-1.
- the positive electrode active material of Sample 22-1 is a commercially available cobalt acid Lithium (Cellseed C-10N, manufactured by Nippon Kagaku Kogyo Co., Ltd.).
- Cellseed C-10N manufactured by Nippon Kagaku Kogyo Co., Ltd.
- a series of operations of constant current discharging at 25° C., cutoff voltage of 2.5 V, and 0.1 C were performed twice.
- constant current discharging at a cutoff voltage of 2.5 V and 0.1 C was performed. The temperature during discharge was measured under two conditions of 25°C and -40°C.
- the sample 22-2 has the same conditions as the sample 22-1 except for the value of the charging voltage. In addition, it can be said that Sample 22-2 is different from Sample 21-2 only in the positive electrode active material used in the half cell. Specifically, the positive electrode active material of Sample 22-2 is a commercially available lithium cobaltate (Cellseed C-10N, manufactured by Nippon Kagaku Kogyo Co., Ltd.) that does not have any additive element. After the initial aging, evaluation of low temperature properties was carried out. The aging conditions are the same as for sample 22-1.
- constant current discharging at a cutoff voltage of 2.5 V and 0.1 C was performed. The temperature during discharge was measured under four conditions of 25°C, 0°C, -20°C, and -40°C.
- FIGS. 34 and 35 show the discharge curves of each sample at each temperature during discharge.
- FIG. 34A shows the discharge curve for sample 21-1
- FIG. 34B shows the discharge curve for sample 21-2
- FIG. 35A shows the discharge curve for sample 22-1
- FIG. 35B shows the discharge curve for sample 22-2.
- the dotted line indicates the result when the temperature during discharge is 25°C
- the dashed line indicates the result when the temperature during discharge is 0°C
- the dashed line indicates the result when the temperature during discharge is -0°C.
- the results at 20°C are shown
- the solid line shows the results at a temperature of -40°C during discharge.
- Table 2 shows the measurement results of discharge capacity, average discharge voltage, and discharge energy density at each temperature during discharge.
- the ratio of the discharge capacity, average discharge voltage, and discharge energy density normalized by dividing the values of discharge capacity, average discharge voltage, and discharge energy density at each temperature during discharge by the value at a temperature of 25 ° C. during discharge (Unit: %) is shown in Table 3.
- the discharge capacity (unit: mAh/g) in Table 2 is a value calculated per weight of the positive electrode active material.
- the discharge energy density (unit: mWh/g) in Table 2 is a value calculated by multiplying the discharge capacity by the average discharge voltage (unit: V). Also, "-" in Tables 2 and 3 indicates that it has not been acquired.
- Sample 21-1 had a very high discharge capacity of about 200 mAh/g even at a discharge temperature of -20°C. From another point of view, a result was obtained in which the discharge capacity at -20°C discharge was about 90% of the discharge capacity at 25°C discharge. At a discharge temperature of -40°C, a discharge capacity of about 150 mAh/g was obtained. From another point of view, at a discharge temperature of -40°C, a high discharge energy density of about 480 mWh/g was obtained. From another point of view, a result was obtained in which the discharge energy density in discharge at -40°C was about 50% of the discharge energy density in discharge at 25°C.
- the discharge capacity at -40°C discharge was about 70% of the discharge capacity at 25°C discharge.
- the lithium ion battery of this example has a discharge capacity of 200 mAh/g or more at a discharge temperature of -20°C, and the discharge capacity at -20°C discharge is higher than the discharge capacity at 25°C discharge. 90% or more, the discharge capacity at a discharge temperature of -40 ° C. is 150 mAh / g or more, and the discharge capacity at -40 ° C.
- the discharge energy density at a discharge temperature of -40°C is about 475 mAh/g or more, and the discharge energy density at -40°C discharge is 50% or more of the discharge energy density at 25°C discharge. was gotten.
- the sample 21-2 with a charging voltage of 4.3 V uses a half cell having the same structure as the sample 21-1.
- a low discharge capacity was obtained.
- the result was obtained that the discharge capacity at -40°C was only about 30% of the discharge capacity at 25°C.
- Sample 22-1 which differs from Sample 21-1 only in the positive electrode active material, had a very low discharge capacity at -40°C. From this result, it was found that when the positive electrode active material used in Sample 22-1, the commercially available lithium cobalt oxide containing no additional element, was charged at a high voltage, structural changes occurred in the surface layer or inside of the lithium cobalt oxide. In addition, it is presumed that the discharge capacity was extremely low due to the formation of a film with high resistance due to the decomposition of the electrolytic solution.
- sample 22-2 which differs from sample 22-1 only in the charging voltage conditions, has a discharge capacity of about 95 mAh/g when discharged at -40°C, and can be said to have a larger discharge capacity than sample 22-1. .
- Sample 21-1 which is one embodiment of the present invention, only about 60% of the discharge capacity was obtained. Also, only about 285 mAh/g was obtained for the discharge energy density in the discharge at -40°C.
- Sample 21-1 which is one embodiment of the present invention, is a positive electrode active material that is less likely to deteriorate due to charging and discharging under sub-zero temperature conditions even at a high charging voltage, and charging and discharging at sub-zero temperatures. It was demonstrated that a lithium ion battery having excellent discharge characteristics even at below freezing point ( ⁇ 40° C.) can be obtained by having at least both the electrolyte and the electrolyte, which is a material having excellent lithium ion conductivity.
- Sample 31-1 A total of four samples (Sample 31-1, Sample 31-2, Sample 32-1, and Sample 32-2) were prepared for measuring cycle characteristics. All samples had a loading of approximately 7 mg/cm 2 . Samples 32-1 and 32-2 are comparative examples.
- Samples 31-1 and 31-2 are half cells having the same structure as Samples 21-1 and 21-2 described in Example 2, and lithium cobalt oxide containing Mg, F, Ni, and Al is used as a positive electrode. It has as an active material, and has the electrolytic solution A as an electrolyte.
- Samples 32-1 and 32-2 which are comparative examples, are half cells having the same structure as Samples 22-1 and 22-2 described in Example 2, and are commercially available cells having no particular additive element. It has lithium cobalt oxide as a positive electrode active material, and electrolyte solution A as an electrolyte.
- FIG. 36B shows the relationship between the number of cycles and the discharge capacity obtained by repeating such charging and discharging 50 cycles.
- sample 31-1 is indicated by a solid line and sample 32-1 is indicated by a dotted line.
- the sample 31-2 As shown in FIG. 36B, the sample 31-2, whose cycle characteristics were evaluated under the condition of 4.3 V charging, had a discharge capacity value at the 50th cycle that was the maximum discharge capacity in the discharge capacity from the 1st to the 50th cycles. The values did not change much when compared to each other. Specifically, as a result of calculating the discharge capacity retention rate by (discharge capacity after 50 cycles/maximum discharge capacity) x 100 (unit: %), the discharge capacity retention rate of sample 31-2 was 99.1%. , resulting in very little deterioration. On the other hand, the discharge capacity retention rate of sample 32-2 was 88.4%, which was ten times or more that of sample 31-2.
- a positive electrode active material that can be used in a lithium ion battery that is one embodiment of the present invention and an electrolyte (electrolyte solution A) that is a material that has excellent lithium ion conductivity even when charged and discharged at subzero temperatures. It was found that a lithium-ion battery having at least both has potential to be excellent in both discharge capacity and cycle characteristics.
- 100 positive electrode active material
- 100a surface layer portion
- 100b inside
- 101 grain boundary
- 102 embedded portion
- 103 convex portion
- 104 coating
Abstract
氷点下においても優れた放電特性を有するリチウムイオン電池を提供する。 正極活物質を有する正極と、電解質と、炭素材料の負極活物質を有する負極と、を備えたリチウムイオン電池を、25℃環境下で4.5Vの電圧になるまで0.1C(ただし、1C=200mA/gとする)の充電レートで定電流充電し、電流値が0.01Cとなるまで4.5Vでの定電圧充電をした後、-40℃環境下で2.5Vの電圧になるまで0.1Cの放電レートで定電流放電することで求められた放電容量の値が、前記リチウムイオン電池を25℃環境下で4.5Vの電圧になるまで0.1C(ただし、1C=200mA/gとする)の充電レートで定電流充電し、電流値が0.01Cとなるまで4.5Vでの定電圧充電をした後、25℃環境下で2.5Vの電圧になるまで0.1Cの放電レートで定電流放電することで求められた放電容量の値に比して50%以上である。
Description
本明細書等に開示する発明(以下、本明細書等において「本発明」と表記することがある。)は、蓄電装置、二次電池等に関する。特に、リチウムイオン電池に関する。
または、本発明は、物、方法、もしくは製造方法に関する。または、本発明は、プロセス、マシン、マニュファクチャ、もしくは組成物(コンポジション・オブ・マター)に関する。または、本発明は、半導体装置、表示装置、発光装置、蓄電装置、照明装置、電子機器、もしくはそれらの製造方法に関する。
近年、リチウムイオン電池、リチウムイオンキャパシタ、空気電池等、種々の蓄電装置の開発が盛んに行われている。特に、高出力、高エネルギー密度であるリチウムイオン電池は、携帯電話機、スマートフォン、もしくはノート型コンピュータ等の携帯情報端末、携帯音楽プレーヤ、デジタルカメラ、医療機器、又は、ハイブリッド車(HV)、電気自動車(EV)、もしくはプラグインハイブリッド車(PHV)等のクリーンエネルギー自動車など、半導体産業の発展と併せて急速にその需要が拡大し、繰り返し充電可能なエネルギーの供給源として現代の情報化社会に不可欠なものとなっている。
リチウムイオン電池は、電池の充電環境および/または電池の放電環境に依存して、充電特性および/または放電特性が変動する。例えば、リチウムイオン電池は、放電時の温度によって放電容量が変化することが知られている。
そのため、低温環境下であっても優れた電池特性を有するリチウムイオン電池が求められている。(例えば、特許文献1参照)。
Zhaohui Chen et al,"Staging Phase Transitions in Li▲x▼CoO▲2▼",Journal of The Electrochemical Society,2002,149(12)A1604−A1609
特許文献1に記載の非水溶媒を用いることにより、低温環境下でも動作可能なリチウムイオン電池を実現できたことが特許文献1に記載されている。しかしながら、特許文献1に記載のリチウムイオン電池であっても、0℃以下(「氷点下」ともいう。)の温度で放電した際の放電容量は本出願時では大きいと言えず、さらなる改善が望まれている。
本発明の一態様は、氷点下においても優れた放電特性を有するリチウムイオン電池の提供を課題の一とする。または、氷点下においても優れた充電特性を有するリチウムイオン電池の提供を課題の一とする。
具体的には、氷点下(例えば、0℃以下、−20℃以下、好ましくは−30℃以下、より好ましくは−40℃以下、さらに好ましくは−50℃以下、最も好ましくは−60℃以下)の温度で放電しても放電容量および/または放電エネルギー密度の大きいリチウムイオン電池の提供を課題の一とする。または、氷点下(例えば、0℃以下、−20℃以下、好ましくは−30℃以下、より好ましくは−40℃以下、さらに好ましくは−50℃以下、最も好ましくは−60℃以下)の温度で放電しても、25℃で放電した場合の放電容量および/または放電エネルギー密度の値に比して減少率の少ないリチウムイオン電池の提供を課題の一とする。または、氷点下(例えば、0℃以下、−20℃以下、好ましくは−30℃以下、より好ましくは−40℃以下、さらに好ましくは−50℃以下、最も好ましくは−60℃以下)の温度で充電しても充電容量の大きいリチウムイオン電池の提供を課題の一とする。または、氷点下(例えば、0℃以下、−20℃以下、好ましくは−30℃以下、より好ましくは−40℃以下、さらに好ましくは−50℃以下、最も好ましくは−60℃以下)の温度で充電しても、25℃で充電した場合の充電容量の値に比して減少率の少ないリチウムイオン電池の提供を課題の一とする。
または、充電電圧の高い二次電池を提供することを課題の一とする。または、安全性もしくは信頼性の高い二次電池を提供することを課題の一とする。または、劣化が少ない二次電池を提供することを課題の一とする。または、長寿命の二次電池を提供することを課題の一とする。または、新規の二次電池を提供することを課題の一とする。
または、新規の物質、活物質、蓄電装置、もしくはそれらの作製方法を提供することを課題の一とする。
なお、これらの課題の記載は、他の課題の存在を妨げるものではない。また、本発明の一態様は、これらの課題の全てを解決する必要はないものとする。また、本明細書、図面、請求項等の記載から、これら以外の課題を抽出することも可能である。
上記の課題等を解決するため、本発明の一態様は、以下の構成を有する。
本発明の一態様は、正極活物質を有する正極と、電解質と、炭素材料の負極活物質を有する負極と、を備えたリチウムイオン電池である。前記電解質は、エチレンカーボネートと、エチルメチルカーボネートと、ジメチルカーボネートと、を含み、前記エチレンカーボネート、前記エチルメチルカーボネート、及び前記ジメチルカーボネートの全含有量を100vol%としたとき、前記エチレンカーボネート、前記エチルメチルカーボネート、及び前記ジメチルカーボネートの体積比が、x:y:100−x−y(ただし、5≦x≦35であり、0<y<65である。)である。前記リチウムイオン電池を25℃環境下で4.5Vの電圧になるまで0.1C(ただし、1C=200mA/gとする)の充電レートで定電流充電し、電流値が0.01Cとなるまで4.5Vでの定電圧充電をした後、−40℃環境下で2.5Vの電圧になるまで0.1Cの放電レートで定電流放電することで求められた放電容量の値が、前記リチウムイオン電池を25℃環境下で4.5Vの電圧になるまで0.1C(ただし、1C=200mA/gとする)の充電レートで定電流充電し、電流値が0.01Cとなるまで4.5Vでの定電圧充電をした後、25℃環境下で2.5Vの電圧になるまで0.1Cの放電レートで定電流放電することで求められた放電容量の値に比して50%以上である。
または、本発明の一態様において、前記炭素材料は黒鉛である。
または、本発明の一態様は、正極活物質を有する正極と、電解質と、負極と、を備え、少なくとも−40℃以上25℃以下の温度範囲で動作可能である、リチウムイオン電池である。
または、本発明の一態様は、正極活物質を有する正極と、電解質と、負極と、を備えたリチウムイオン電池である。前記正極活物質を正極として用い、エチレンカーボネートと、エチルメチルカーボネートと、ジメチルカーボネートと、を含み、前記エチレンカーボネート、前記エチルメチルカーボネート、及び前記ジメチルカーボネートの全含有量を100vol%としたとき、前記エチレンカーボネート、前記エチルメチルカーボネート、及び前記ジメチルカーボネートの体積比が、x:y:100−x−y(ただし、5≦x≦35であり、0<y<65である。)である電解質を用い、リチウム金属を負極として用いて試験用電池とした際に、前記試験用電池を25℃で4.6Vの電圧になるまで0.1C(ただし、1C=200mA/gとする)の充電レートで定電流充電し、電流値が0.01Cとなるまで4.6Vでの定電圧充電をした後、−40℃で2.5Vの電圧になるまで0.1Cの放電レートで定電流放電することで求められた放電容量の値が、前記試験用電池を25℃で4.6Vの電圧になるまで0.1C(ただし、1C=200mA/gとする)の充電レートで定電流充電し、電流値が0.01Cとなるまで4.6Vでの定電圧充電をした後、25℃で2.5Vの電圧になるまで0.1Cの放電レートで定電流放電することで求められた放電容量の値に比して50%以上である。
または、本発明の一態様において、前記正極活物質は、LixCoO2(ただし、0<x≦1である。)で表されるコバルト酸リチウムを有し、前記LixCoO2中のxが1のとき、空間群R−3mの層状岩塩型の結晶構造を有し、前記LixCoO2中のxが0.1を超えて0.24以下の充電状態のとき、空間群P2/m、格子定数a=4.88±0.01(×10−1nm)、格子定数b=2.82±0.01(×10−1nm)、格子定数c=4.84±0.01(×10−1nm)、α=90°、β=109.58±0.01°、γ=90°の結晶構造を有する。
または、本発明の一態様において、前記正極活物質は、LixCoO2(ただし、0<x≦1である。)で表されるコバルト酸リチウムを有し、前記LixCoO2中のxが1のとき、空間群R−3mの層状岩塩型の結晶構造を有し、前記LixCoO2中のxが0.1を超えて0.24以下の充電状態のとき、粉末X線回折で分析すると、回折パターンは、2θ=19.37°以上19.57°以下と、2θ=45.57°以上45.67°以下と、に少なくともピークを有する。
本発明の一態様により、氷点下(例えば、0℃以下、−20℃以下、好ましくは−30℃以下、より好ましくは−40℃以下、さらに好ましくは−50℃以下、最も好ましくは−60℃以下)の温度で放電しても放電容量および/または放電エネルギー密度の大きいリチウムイオン電池を提供することができる。または、氷点下(例えば、0℃以下、−20℃以下、好ましくは−30℃以下、より好ましくは−40℃以下、さらに好ましくは−50℃以下、最も好ましくは−60℃以下)の温度で放電しても、25℃で放電した場合の放電容量および/または放電エネルギー密度の値に比して減少率の少ないリチウムイオン電池を提供することができる。または、氷点下(例えば、0℃以下、−20℃以下、好ましくは−30℃以下、より好ましくは−40℃以下、さらに好ましくは−50℃以下、最も好ましくは−60℃以下)の温度で充電しても充電容量の大きいリチウムイオン電池を提供することができる。または、氷点下(例えば、0℃以下、−20℃以下、好ましくは−30℃以下、より好ましくは−40℃以下、さらに好ましくは−50℃以下、最も好ましくは−60℃以下)の温度で充電しても、25℃で充電した場合の充電容量の値に比して減少率の少ないリチウムイオン電池を提供することができる。
または、本発明の一態様により、充電電圧の高い二次電池を提供することができる。または、安全性もしくは信頼性の高い二次電池を提供することができる。または、劣化が少ない二次電池を提供することができる。または、長寿命の二次電池を提供することができる。または、新規の二次電池を提供することができる。
または、本発明の一態様により、新規の物質、活物質、蓄電装置、もしくはそれらの作製方法を提供することができる。
図1A1及び図1A2は、正極活物質の断面図、図1B1及び図1B2は正極活物質の断面図の一部である。
図2は、結晶の配向が概略一致しているTEM像の例である。
図3Aは、結晶の配向が概略一致しているSTEM像の例である。図3Bは、岩塩型結晶RSの領域のFFTパターン、図3Cは、層状岩塩型結晶LRSの領域のFFTパターンである。
図4は、正極活物質の結晶構造を説明する図である。
図5は、従来の正極活物質の結晶構造を説明する図である。
図6A1及び図6A2は、正極活物質の断面図の一部である。図6B1乃至図6Cは、コバルト酸リチウムの結晶面とマグネシウムの分布について計算した結果である。
図7A及び図7Bは、正極活物質の断面図、図7C1及び図7C2は正極活物質の断面図の一部である。
図8は、結晶構造から計算されるXRDパターンを示す図である。
図9は、結晶構造から計算されるXRDパターンを示す図である。
図10A及び図10Bは、結晶構造から計算されるXRDパターンを示す図である。
図11A乃至図11Cは、XRDから算出される格子定数である。
図12A乃至図12Cは、XRDから算出される格子定数である。
図13A及び図13Bは、正極活物質の断面図である。
図14は、正極活物質の断面図である。
図15A乃至図15Cは正極活物質の作製方法を説明する図である。
図16は正極活物質の作製方法を説明する図である。
図17A乃至図17Cは正極活物質の作製方法を説明する図である。
図18A乃至図18Dは、二次電池の正極の例を説明する断面図である。
図19Aはコイン型二次電池の分解斜視図であり、図19Bはコイン型二次電池の斜視図であり、図19Cはその断面斜視図である。
図20Aは、円筒型の二次電池の例を示す。図20Bは、円筒型の二次電池の例を示す。図20Cは、複数の円筒型の二次電池の例を示す。図20Dは、複数の円筒型の二次電池を有する蓄電システムの例を示す。
図21A及び図21Bは、二次電池の例を説明する図であり、図21Cは、二次電池の内部の様子を示す図である。
図22A乃至図22Cは、二次電池の例を説明する図である。
図23A、及び図23Bは、二次電池の外観を示す図である。
図24A乃至図24Cは、二次電池の作製方法を説明する図である。
図25Aは、電池パックの構成例を示し、図25Bは、電池パックの構成例を示し、図25Cは、電池パックの構成例を示す。
図26Aは、本発明の一態様を示す電池パックの斜視図であり、図26Bは、電池パックのブロック図であり、図26Cは、モータを有する車両のブロック図である。
図27A乃至図27Dは、輸送用車両の一例を説明する図である。図27Eは、人工衛星の一例を説明する図である。
図28A、及び図28Bは、本発明の一態様に係る蓄電装置を説明する図である。
図29Aは、電動自転車を示す図であり、図29Bは、電動自転車の二次電池を示す図であり、図29Cは、電動バイクを説明する図である。
図30A乃至図30Dは、電子機器の一例を説明する図である。
図31Aは、ウェアラブルデバイスの例を示しており、図31Bは、腕時計型デバイスの斜視図を示しており、図31Cは、腕時計型デバイスの側面を説明する図である。
図32は、実施例1で説明した、放電時の各温度に対する二次電池の放電容量を示すグラフである。
図33は、実施例1で説明した、充電時の各温度に対する二次電池の充電容量を示すグラフである。
図34A、図34Bは、実施例1で説明した、各温度に対する二次電池の放電曲線を示すグラフである。
図35A、図35Bは、実施例1で説明した、各温度に対する二次電池の放電曲線を示すグラフである。
図36A、図36Bは、実施例2で説明した二次電池のサイクル特性を示すグラフである。
図2は、結晶の配向が概略一致しているTEM像の例である。
図3Aは、結晶の配向が概略一致しているSTEM像の例である。図3Bは、岩塩型結晶RSの領域のFFTパターン、図3Cは、層状岩塩型結晶LRSの領域のFFTパターンである。
図4は、正極活物質の結晶構造を説明する図である。
図5は、従来の正極活物質の結晶構造を説明する図である。
図6A1及び図6A2は、正極活物質の断面図の一部である。図6B1乃至図6Cは、コバルト酸リチウムの結晶面とマグネシウムの分布について計算した結果である。
図7A及び図7Bは、正極活物質の断面図、図7C1及び図7C2は正極活物質の断面図の一部である。
図8は、結晶構造から計算されるXRDパターンを示す図である。
図9は、結晶構造から計算されるXRDパターンを示す図である。
図10A及び図10Bは、結晶構造から計算されるXRDパターンを示す図である。
図11A乃至図11Cは、XRDから算出される格子定数である。
図12A乃至図12Cは、XRDから算出される格子定数である。
図13A及び図13Bは、正極活物質の断面図である。
図14は、正極活物質の断面図である。
図15A乃至図15Cは正極活物質の作製方法を説明する図である。
図16は正極活物質の作製方法を説明する図である。
図17A乃至図17Cは正極活物質の作製方法を説明する図である。
図18A乃至図18Dは、二次電池の正極の例を説明する断面図である。
図19Aはコイン型二次電池の分解斜視図であり、図19Bはコイン型二次電池の斜視図であり、図19Cはその断面斜視図である。
図20Aは、円筒型の二次電池の例を示す。図20Bは、円筒型の二次電池の例を示す。図20Cは、複数の円筒型の二次電池の例を示す。図20Dは、複数の円筒型の二次電池を有する蓄電システムの例を示す。
図21A及び図21Bは、二次電池の例を説明する図であり、図21Cは、二次電池の内部の様子を示す図である。
図22A乃至図22Cは、二次電池の例を説明する図である。
図23A、及び図23Bは、二次電池の外観を示す図である。
図24A乃至図24Cは、二次電池の作製方法を説明する図である。
図25Aは、電池パックの構成例を示し、図25Bは、電池パックの構成例を示し、図25Cは、電池パックの構成例を示す。
図26Aは、本発明の一態様を示す電池パックの斜視図であり、図26Bは、電池パックのブロック図であり、図26Cは、モータを有する車両のブロック図である。
図27A乃至図27Dは、輸送用車両の一例を説明する図である。図27Eは、人工衛星の一例を説明する図である。
図28A、及び図28Bは、本発明の一態様に係る蓄電装置を説明する図である。
図29Aは、電動自転車を示す図であり、図29Bは、電動自転車の二次電池を示す図であり、図29Cは、電動バイクを説明する図である。
図30A乃至図30Dは、電子機器の一例を説明する図である。
図31Aは、ウェアラブルデバイスの例を示しており、図31Bは、腕時計型デバイスの斜視図を示しており、図31Cは、腕時計型デバイスの側面を説明する図である。
図32は、実施例1で説明した、放電時の各温度に対する二次電池の放電容量を示すグラフである。
図33は、実施例1で説明した、充電時の各温度に対する二次電池の充電容量を示すグラフである。
図34A、図34Bは、実施例1で説明した、各温度に対する二次電池の放電曲線を示すグラフである。
図35A、図35Bは、実施例1で説明した、各温度に対する二次電池の放電曲線を示すグラフである。
図36A、図36Bは、実施例2で説明した二次電池のサイクル特性を示すグラフである。
本発明の実施の形態について、図面を適宜用いながら説明する。但し、本発明は以下の説明に限定されず、本発明の趣旨及びその範囲から逸脱することなくその形態及び詳細を様々に変更しうることは当業者であれば容易に理解される。従って、本発明は以下に示す実施の形態において、同じ物を指し示す符号は異なる図面において共通とする。
また、以下に説明する実施の形態及び実施例それぞれにおいて、特に断りがない限り、本明細書等に記載されている実施形態及び実施例等を適宜組み合わせて実施することが可能である。
本明細書等において「電子機器」とは、蓄電装置を有する装置全般を指し、蓄電装置を有する電気光学装置、蓄電装置を有する情報端末装置などは全て電子機器である。
本明細書等において、「蓄電装置」とは、蓄電機能を有する素子及び装置全般を指すものである。例えば、リチウムイオン電池などの蓄電装置(「二次電池」ともいう)、リチウムイオンキャパシタ、及び電気二重層キャパシタなどを含む。
本明細書等において、空間群は国際表記(またはHermann−Mauguin記号)のShort notationを用いて表記する。また、ミラー指数を用いて結晶面及び結晶方向を表記する。結晶面を示す個別面は( )を用いて表記する。空間群、結晶面、および結晶方向の表記は、結晶学上、数字に上付きのバーを付すが、本明細書等では書式の制約上、数字の上にバーを付す代わりに、数字の前に−(マイナス符号)を付して表現する場合がある。また、結晶内の方向を示す個別方位は[ ]で、等価な方向全てを示す集合方位は< >で、結晶面を示す個別面は( )で、等価な対称性を有する集合面は{ }でそれぞれ表現する。また、空間群R−3mで表される三方晶は、構造の理解のしやすさのため、一般に六方晶の複合六方格子で表され、ミラー指数として(hkl)だけでなく(hkil)を用いることがある。ここでiは−(h+k)である。
本明細書等において、正極活物質の理論容量とは、正極活物質が有する挿入脱離可能なリチウムが全て脱離した場合の電気量をいう。例えば、LiCoO2の理論容量は274mAh/g、LiNiO2の理論容量は275mAh/g、LiMn2O4の理論容量は148mAh/gである。
また、正極活物質中に挿入脱離可能なリチウムがどの程度残っているかを、組成式中のx、例えばLixCoO2中のx(リチウムサイトのLiの占有率)で示すことが可能である。二次電池の有する正極活物質の場合、x=充電容量/理論容量とすることができる。例えば、LiCoO2を正極活物質に用いた二次電池を219.2mAh/g充電した場合、Li0.2CoO2またはx=0.2ということができる。LixCoO2中のxが小さい状態とは、例えばx≦0.24であり、リチウムイオン電池として用いる際の実用的な範囲を考慮すると、例えば0.1<x≦0.24であるものとする。
コバルト酸リチウムが化学量論比をおよそ満たす場合、LiCoO2であり、x=1である。また、放電が終了した二次電池に含まれるコバルト酸リチウムも、LiCoO2であり、x=1といってよい。また、一般的にLiCoO2を用いたリチウムイオン電池では、放電電圧が2.5Vになるまでに放電電圧が急激に降下する。このため、本明細書等においては、例えば100mA/g以下の電流で、電圧が2.5V(対極はリチウム)となった状態を、放電が終了した状態と見なし、x=1と見なす。したがって、例えばx=0.2のときのコバルト酸リチウムとするためには、放電が終了した状態から219.2mAh/g充電すればよい。
LixCoO2中のxの算出に用いる充電容量および/または放電容量は、短絡および/または電解質の分解の影響がないか、少ない条件で計測することが好ましい。例えば、短絡とみられる急激な容量の変化が生じた二次電池のデータは、xの算出に使用するのは好ましくない。
また、結晶構造の空間群は、XRD、電子線回折、中性子線回折等によって同定されるものである。そのため、本明細書等において、ある空間群に帰属する、ある空間群に属する、またはある空間群であるとは、ある空間群に同定されると言い換えることができる。
また、陰イオンがABCABCのように3層が互いにずれて積み重なる構造であれば、「立方最密充填構造」と呼ぶこととする。そのため、陰イオンは厳密に立方格子でなくてもよい。同時に現実の結晶は必ず欠陥を有するため、分析結果が必ずしも理論通りでなくてもよい。例えば電子線回折パターンまたはTEM像等のFFT(高速フーリエ変換)パターンにおいて、理論上の位置と若干異なる位置にスポットが現れてもよい。例えば理論上の位置との方位が5°以下、または2.5°以下であれば立方最密充填構造を取るといってよい。
本明細書等において、「リチウムと遷移金属を含む複合酸化物が有する層状岩塩型の結晶構造」とは、陽イオンと陰イオンが交互に配列する岩塩型のイオン配列を有し、遷移金属とリチウムが規則配列して二次元平面を形成するため、リチウムの二次元的拡散が可能である結晶構造をいう。なお、陽イオンまたは陰イオンの欠損等の欠陥を有していてもよい。また、層状岩塩型結晶構造は、厳密に言えば、岩塩型結晶の格子が歪んだ構造となっている場合がある。
本明細書等において、「岩塩型の結晶構造」とは、陽イオンと陰イオンが交互に配列している構造をいう。なお、陽イオンまたは陰イオンの欠損があってもよい。
本明細書等において、「均質」とは、複数の元素(例えばA,B,C)からなる固体において、ある元素(例えばA)が特定の領域に同様の特徴を有して分布する現象をいう。具体的には、特定の領域同士の元素の濃度が実質的に同一であればよい。例えば、特定領域同士の元素濃度の差が10%以内であればよい。特定の領域としては、例えば表層部、表面、凸部、凹部、内部などが挙げられる。
本明細書等において、「偏析」とは、複数の元素(例えばA,B,C)からなる固体において、ある元素(例えばB)が空間的に不均一に分布する現象をいう。または、ある元素の濃度が他と異なることをいう。偏在、析出、不均一、偏り、または濃度が高い箇所と濃度が低い箇所が混在する、と同義である。
本明細書等において、活物質等の粒子の「表層部」とは、例えば、表面から内部に向かって50nm以内、より好ましくは35nm以内、さらに好ましくは20nm以内、最も好ましくは10nm以内の領域である。また、ひび又はクラックにより生じた面は、表面と見なすことができる。また、本明細書等において、表層部より深い領域を「内部」と呼ぶことがある。また、本明細書等において、「粒界」とは、例えば粒子同士が固着している部分、粒子内部(中央部を含む)で結晶方位が変わる部分、欠陥を多く含む部分、結晶構造が乱れている部分等をいう。また、粒界は、面欠陥の一つとも言える。また、「粒界の近傍」とは、粒界から20nm以内、好ましくは10nm以内の領域をいうこととする。また、本明細書等において、「粒子」とは、球形(断面形状が円)のみを指すことに限定されず、個々の粒子の断面形状が楕円形、長方形、台形、三角形、角が丸まった四角形、非対称の形状などが挙げられ、さらに個々の粒子は不定形であってもよい。
(実施の形態1)
[リチウムイオン電池]
本発明の一態様のリチウムイオン電池は、正極と、負極と、電解質と、を有する。電解質が電解液を含む場合は、正極と負極との間にセパレータを有する。本発明の一態様のリチウムイオン電池は、また、正極、負極、及び電解質の周囲の少なくとも一部を覆う外装体を有していてもよい。
[リチウムイオン電池]
本発明の一態様のリチウムイオン電池は、正極と、負極と、電解質と、を有する。電解質が電解液を含む場合は、正極と負極との間にセパレータを有する。本発明の一態様のリチウムイオン電池は、また、正極、負極、及び電解質の周囲の少なくとも一部を覆う外装体を有していてもよい。
本実施の形態では、氷点下(例えば、0℃以下、−20℃以下、好ましくは−30℃以下、より好ましくは−40℃以下、さらに好ましくは−50℃以下、最も好ましくは−60℃以下)においても優れた放電特性を有するリチウムイオン電池、および/または氷点下においても優れた充電特性を有するリチウムイオン電池を実現するために必要とされるリチウムイオン電池の構成に焦点を当てて説明する。具体的には、正極に含まれる正極活物質と、電解質を中心に説明する。リチウムイオン電池の有する正極活物質と電解質以外の構成の詳細については、実施の形態3で説明する。
[正極]
正極は、正極活物質層及び正極集電体を有する。正極活物質層は正極活物質を有し、さらに導電助剤及びバインダの少なくとも一を有していてもよい。
正極は、正極活物質層及び正極集電体を有する。正極活物質層は正極活物質を有し、さらに導電助剤及びバインダの少なくとも一を有していてもよい。
<正極活物質>
正極活物質は、充放電に伴い、リチウムイオンを取り込む、および/または放出する機能を有する。本発明の一態様として用いる正極活物質は、高い充電電圧(以下、「高充電電圧」とも記す)としても、氷点下の温度条件における充電および/または放電(以下、「充放電」とも呼ぶ。)に伴う劣化の少ない材料(または抵抗の増加の少ない材料)を用いることができる。なお、本明細書等において特に言及しない場合、「充電電圧」はリチウム金属の電位を基準として表すものとする。また、本明細書等において、「高充電電圧」とは、例えば4.6V以上の充電電圧とし、好ましくは4.65V以上、4.7V以上、4.75V以上、または4.8V以上とする。なお、正極活物質は、高充電電圧としても充放電に伴う劣化の少ない材料であれば、粒径および/または組成が異なる2種類以上の材料を用いることも可能である。本明細書等において、「組成が異なる」とは、材料に含まれる元素の構成が異なる場合に加えて、材料に含まれる元素の構成が同じであっても、含まれる元素の割合が異なる場合も含むものとする。
正極活物質は、充放電に伴い、リチウムイオンを取り込む、および/または放出する機能を有する。本発明の一態様として用いる正極活物質は、高い充電電圧(以下、「高充電電圧」とも記す)としても、氷点下の温度条件における充電および/または放電(以下、「充放電」とも呼ぶ。)に伴う劣化の少ない材料(または抵抗の増加の少ない材料)を用いることができる。なお、本明細書等において特に言及しない場合、「充電電圧」はリチウム金属の電位を基準として表すものとする。また、本明細書等において、「高充電電圧」とは、例えば4.6V以上の充電電圧とし、好ましくは4.65V以上、4.7V以上、4.75V以上、または4.8V以上とする。なお、正極活物質は、高充電電圧としても充放電に伴う劣化の少ない材料であれば、粒径および/または組成が異なる2種類以上の材料を用いることも可能である。本明細書等において、「組成が異なる」とは、材料に含まれる元素の構成が異なる場合に加えて、材料に含まれる元素の構成が同じであっても、含まれる元素の割合が異なる場合も含むものとする。
なお、前述したとおり、本明細書等において、「高充電電圧」とは、負極がリチウム金属である場合の電位を基準として4.6V以上としたが、負極が炭素材料(例えば、黒鉛)である場合の電位を基準とした場合は、4.5V以上を「高充電電圧」と呼ぶものとする。端的には、負極としてリチウム金属が用いられるハーフセルの場合においては、4.6V以上の充電電圧を高充電電圧と呼び、負極として炭素材料(例えば、黒鉛)が用いられるフルセルの場合においては、4.5V以上の充電電圧を高充電電圧と呼ぶものとする。
高い充電電圧としても、氷点下の任意温度(例えば、0℃、−20℃、好ましくは−30℃、より好ましくは−40℃、さらに好ましくは−50℃、最も好ましくは−60℃)における充放電に伴う劣化の少ない材料(または抵抗の増加の少ない材料)を正極活物質として用いることにより、氷点下の温度においても充電容量および/または放電容量が大きいリチウムイオン電池を実現できる。または、氷点下の任意温度(例えば、0℃、−20℃、好ましくは−30℃、より好ましくは−40℃、さらに好ましくは−50℃、最も好ましくは−60℃)における充電容量および/または放電容量の値が、25℃における充電容量および/または放電容量の値に比して50%以上(好ましくは60%以上、より好ましくは70%以上、最も好ましくは80%以上)であるリチウムイオン電池を実現できる。なお、氷点下の任意温度における放電容量の値と、25℃における放電容量の値は、放電時の温度(以下、本明細書等において「放電温度」と呼ぶことがある。)以外の測定条件は同じものとする。
または、氷点下の任意温度(例えば、0℃、−20℃、好ましくは−30℃、より好ましくは−40℃、さらに好ましくは−50℃、最も好ましくは−60℃)においても、放電エネルギー密度が大きいリチウムイオン電池を実現できる。または、氷点下の任意温度(例えば、0℃、−20℃、好ましくは−30℃、より好ましくは−40℃、さらに好ましくは−50℃、最も好ましくは−60℃)における放電エネルギー密度の値が、25℃における放電エネルギー密度の値に比して50%以上(好ましくは60%以上、より好ましくは70%以上、最も好ましくは80%以上)であるリチウムイオン電池を実現できる。なお、氷点下の任意温度における放電エネルギー密度の値と、25℃における放電エネルギー密度の値は、放電時の温度以外の測定条件は同じものとする。
または、氷点下の任意温度(例えば、0℃、−20℃、好ましくは−30℃、より好ましくは−40℃、さらに好ましくは−50℃、最も好ましくは−60℃)における充電容量の値が、25℃における充電容量の値に比して50%以上(好ましくは60%以上、より好ましくは70%以上、最も好ましくは80%以上)であるリチウムイオン電池を実現できる。なお、氷点下の任意温度における充電容量の値と、25℃における充電容量の値は、充電時の温度以外の測定条件は同じものとする。
本明細書等に記載した、充電時または放電時の温度とは、リチウムイオン電池の温度のことをいう。種々の温度での電池特性の測定においては、一例として所望の温度で安定した恒温槽を用い、測定対象の電池(例えば、試験用電池またはハーフセル)を当該恒温槽内に設置後、試験セルが恒温槽の温度と同程度になるまで十分な時間(例えば、1時間以上)をおいてから測定を開始することができるが、必ずしもこの方法に限定されるものではない。
<電解質>
本発明の一態様として用いる電解質は、氷点下の任意温度(例えば、0℃、−20℃、好ましくは−30℃、より好ましくは−40℃、さらに好ましくは−50℃、最も好ましくは−60℃)における充放電であっても氷点下の任意温度(例えば、0℃、−20℃、好ましくは−30℃、より好ましくは−40℃、さらに好ましくは−50℃、最も好ましくは−60℃)におけるリチウムイオン伝導性に優れた材料を用いることができる。
本発明の一態様として用いる電解質は、氷点下の任意温度(例えば、0℃、−20℃、好ましくは−30℃、より好ましくは−40℃、さらに好ましくは−50℃、最も好ましくは−60℃)における充放電であっても氷点下の任意温度(例えば、0℃、−20℃、好ましくは−30℃、より好ましくは−40℃、さらに好ましくは−50℃、最も好ましくは−60℃)におけるリチウムイオン伝導性に優れた材料を用いることができる。
電解質の一例について、以下に説明する。なお、一例として本実施の形態で説明する電解質は、有機溶媒に電解質(リチウム塩)が溶解されたものであり、電解液と呼ぶこともできるが、電解質は、常温で液体である液体電解質(電解液)に限定されず、固体電解質を用いることも可能である。または、常温で液体である液体電解質と、常温で固体である液体電解質の双方を含む電解質(半固体の電界質)を用いることも可能である。
一例として本実施の形態で説明する有機溶媒は、エチレンカーボネート(EC)と、エチルメチルカーボネート(EMC)と、ジメチルカーボネート(DMC)と、を含み、前記エチレンカーボネート、前記エチルメチルカーボネート、及び前記ジメチルカーボネートの全含有量を100vol%としたとき、前記エチレンカーボネート、前記エチルメチルカーボネート、及び前記ジメチルカーボネートの体積比が、x:y:100−x−y(ただし、5≦x≦35であり、0<y<65である。)であるものを用いることができる。より具体的には、ECと、EMCと、DMCと、を、EC:EMC:DMC=30:35:35(体積比)で含んだ有機溶媒を用いることができる。なお、上記の体積比は、電解液の混合前における体積比であってもよく、電解液を混合する際の外気は室温(代表的には、25℃)であってもよい。
ECは、環状カーボネートであり、高い比誘電率を有するため、リチウム塩の解離を促進させる効果を有する。一方で、ECは、粘度が高く、凝固点(融点)が38℃と高いので、有機溶媒としてEC単体を用いた場合、低温環境下での使用が難しい。そこで、本発明の一態様として具体的に説明する有機溶媒は、EC単体ではなく、EMCとDMCを更に含む。EMCは、鎖状カーボネートであり、電界液の粘度を下げる効果を有する上に、凝固点が−54℃である。また、DMCも、鎖状カーボネートであり、電界液の粘度を下げる効果を有する。このような物性を有するEC、EMC、及びDMCを、これら3つの有機溶媒の全含有量を100vol%として、体積比が、x:y:100−x−y(ただし、5≦x≦35であり、0<y<65である。)となるように混合した有機溶媒を用いて作製された電解質は、凝固点が−40℃以下という特徴を有する。
リチウムイオン電池に用いられている一般的な電解質は、低くても−20℃程度で凝固してしまうため、−40℃で充放電できる電池を作製することは困難である。本実施の形態において一例として説明した電解質は、凝固点が−40℃以下であるため、−40℃という極低温環境下においても充放電可能なリチウムイオン電池を実現できる。
また、上記の溶媒に溶解させる電解質は、リチウム塩を用いることが可能である。例えば、LiPF6、LiClO4、LiAsF6、LiBF4、LiAlCl4、LiSCN、LiBr、LiI、Li2SO4、Li2B10Cl10、Li2B12Cl12、LiCF3SO3、LiC4F9SO3、LiC(CF3SO2)3、LiC(C2F5SO2)3、LiN(CF3SO2)2、LiN(C4F9SO2)(CF3SO2)、LiN(C2F5SO2)2、リチウムビス(オキサレート)ボレート(LiBOB)のうち少なくとも一種のリチウム塩を任意の組み合わせ及び比率で用いることが可能である。
また、電解液は、粒状のごみ、または電解液の構成元素以外の元素(以下、単に「不純物」ともいう。)の含有量が少なく、高純度化されていることが好ましい。具体的には、電解液に対する不純物の重量比を1%以下、好ましくは0.1%以下、より好ましくは0.01%以下とすることが好ましい。
また、安全性向上等を目的として、電極(活物質層)と電解液との界面に被膜(Solid Electrolyte Interphase膜)を形成するため、電解液に対し、ビニレンカーボネート(VC)、プロパンスルトン(PS)、tert−ブチルベンゼン(TBB)、フルオロエチレンカーボネート(FEC)、リチウムビス(オキサレート)ボレート(LiBOB)、またはスクシノニトリルもしくはアジポニトリルのジニトリル化合物の添加剤を添加してもよい。添加剤の濃度は、例えば溶媒に対して0.1wt%以上5wt%以下とすればよい。
以上のとおり、本発明の一態様のリチウムイオン電池に用いることが可能な電解質の一例について説明したが、本発明の一態様のリチウムイオン電池に用いることが可能な電解質は、この一例に限定解釈されるものではない。氷点下(例えば−20℃、好ましくは−40℃)における充放電であってもリチウムイオン伝導性に優れた材料であれば、他の材料を用いることも可能である。
本発明の一態様のリチウムイオン電池は、上述した正極活物質と電解質を少なくとも含むことにより、氷点下においても優れた放電特性を有するリチウムイオン電池、および/または氷点下においても優れた充電特性を有するリチウムイオン電池を実現することができる。より具体的には、上述した正極活物質と電解質を少なくとも含み、リチウム金属を負極として用いて試験用電池とした際に、試験用電池を25℃環境下において4.6Vの電圧になるまで0.1Cまたは0.2C(ただし、1C=200mA/gとする)の充電レートで定電流充電した後、−40℃環境下において2.5Vの電圧になるまで0.1Cの放電レートで定電流放電することで求められた放電容量の値が、前記試験用電池を25℃環境下において4.6Vの電圧になるまで0.1Cまたは0.2C(ただし、1C=200mA/gとする)の充電レートで定電流充電した後、25℃環境下において2.5Vの電圧になるまで0.1Cの放電レートで定電流放電することで求められた放電容量の値に比して50%以上であるリチウムイオン電池を実現することができる。本明細書等において、25℃環境下における放電容量と比較して、T℃(Tは任意の温度(℃)とする。)環境下における放電容量が50%以上を実現できる場合、そのリチウムイオン電池はT℃で動作可能であると表現することとする。
(実施の形態2)
本実施の形態では、図1乃至図14を用いて、本発明の一態様であるリチウムイオン電池に用いることが可能な正極活物質(以下、「本発明の一態様として利用可能な正極活物質」と呼ぶことがある。)、及びその作製方法について説明する。なお、実施の形態1で上述したとおり、本発明の一態様であるリチウムイオン電池に用いることが可能な正極活物質は、高い充電電圧(高充電電圧)としても充放電に伴う劣化の少ない材料であれば何でも用いることが可能である。したがって、本明細書等で開示するリチウムイオン電池に使用可能な正極活物質は、本実施の形態等で説明する具体的な材料に限定解釈される必要はなく、本願出願時において高い充電電圧(例えば、4.6V以上)としても充放電に伴う劣化の少ない材料として公知の材料も使用可能である。
本実施の形態では、図1乃至図14を用いて、本発明の一態様であるリチウムイオン電池に用いることが可能な正極活物質(以下、「本発明の一態様として利用可能な正極活物質」と呼ぶことがある。)、及びその作製方法について説明する。なお、実施の形態1で上述したとおり、本発明の一態様であるリチウムイオン電池に用いることが可能な正極活物質は、高い充電電圧(高充電電圧)としても充放電に伴う劣化の少ない材料であれば何でも用いることが可能である。したがって、本明細書等で開示するリチウムイオン電池に使用可能な正極活物質は、本実施の形態等で説明する具体的な材料に限定解釈される必要はなく、本願出願時において高い充電電圧(例えば、4.6V以上)としても充放電に伴う劣化の少ない材料として公知の材料も使用可能である。
<正極活物質の一例>
本発明の一態様として利用可能な正極活物質の一例を、以下に説明する。
本発明の一態様として利用可能な正極活物質の一例を、以下に説明する。
本実施の形態では、図1乃至図14を用いて本発明の一態様として利用可能な正極活物質100について説明する。
図1A1及び図1A2は本発明の一態様である正極活物質100の断面図である。図1A1中のA−B付近を拡大した図を図1B1及び図1B2に示す。
図1A1、図1B1、図1B2に示すように、正極活物質100は、表層部100aと、内部100bを有する。これらの図中に破線で表層部100aと内部100bの境界を示す。また図1A2に一点破線で結晶粒界101の一部を示す。
本明細書等において、正極活物質100の表層部100aとは、例えば、表面から内部に向かって50nm以内、より好ましくは表面から内部に向かって35nm以内、さらに好ましくは表面から内部に向かって20nm以内、最も好ましくは表面から内部に向かって10nm以内の領域をいう。本明細書等において、ひび及び/またはクラックにより生じた面も表面といってもよい。表層部100aは、表面近傍、表面近傍領域またはシェルと同義である。
また、正極活物質の表層部100aより深い領域を、内部100bと呼ぶ。内部100bは、内部領域またはコアと同義である。
また、正極活物質100の表面とは、表層部100a、内部100b、及び凸部103等を含む複合酸化物の表面をいうこととする。そのため、正極活物質100は、作製後に化学吸着した炭酸塩、ヒドロキシ基等は含まないとする。また、正極活物質100に付着した電解質、バインダ、導電材、またはこれら由来の化合物も含まないとする。また、断面STEM(走査型透過電子顕微鏡)像等における正極活物質100の表面とは、電子線の結合像が観察される領域と、観察されない領域の境界であって、リチウムより原子番号の大きな金属元素の原子核に由来する輝点が確認される領域の最も外側とする。断面STEM像等における表面は、より空間分解能の高い分析、例えば電子エネルギー損失分光法(Electron Energy Loss Spectroscopy,EELS)等の分析結果と併せて判断してもよい。
また、結晶粒界101とは、例えば正極活物質100の粒子同士が固着している部分、正極活物質100内部で結晶方位が変わる部分、つまりSTEM像等における明線と暗線の繰り返しが不連続になった部分、結晶欠陥を多く含む部分、結晶構造が乱れている部分等をいう。また、結晶欠陥とは断面TEM(透過型電子顕微鏡)像、断面STEM像等で観察可能な欠陥、つまり格子間に他の原子が入り込んだ構造、空洞(ボイド)等をいうこととする。結晶粒界101は、面欠陥の一つといえる。また、結晶粒界101の近傍とは、結晶粒界101から20nm以内(好ましくは15nm以内、さらに好ましくは10nm以内)の領域をいうこととする。
<含有元素>
正極活物質100は、リチウムと、コバルトと、酸素と、添加元素と、を有する。または、正極活物質100は、コバルト酸リチウム(LiCoO2)に添加元素が加えられたものでもよい。ただし、本実施の形態で説明する正極活物質100は、後述する結晶構造を有していればよい。そのため、コバルト酸リチウムの組成が厳密にLi:Co:O=1:1:2に限定されるものではない。
正極活物質100は、リチウムと、コバルトと、酸素と、添加元素と、を有する。または、正極活物質100は、コバルト酸リチウム(LiCoO2)に添加元素が加えられたものでもよい。ただし、本実施の形態で説明する正極活物質100は、後述する結晶構造を有していればよい。そのため、コバルト酸リチウムの組成が厳密にLi:Co:O=1:1:2に限定されるものではない。
リチウムイオン電池の正極活物質は、リチウムイオンが挿入脱離しても電荷中性を保つために、酸化還元が可能な遷移金属を有する必要がある。本発明の一態様であるリチウムイオン電池の正極活物質100は、酸化還元反応を担う遷移金属としてコバルトを有することが好ましい。また、コバルトに加えて、ニッケル及びマンガンの少なくとも一以上を有していてもよい。正極活物質100の有する遷移金属のうち、コバルトが75原子%以上、好ましくは90原子%以上、さらに好ましくは95原子%以上であると、合成が比較的容易で取り扱いやすく、優れたサイクル特性を有している点で好ましい。
また、正極活物質100の遷移金属のうち、コバルトが75原子%以上、好ましくは90原子%以上、さらに好ましくは95原子%以上であると、ニッケル酸リチウム(LiNiO2)等のニッケルが遷移金属の過半を占めるような複合酸化物と比較して、LixCoO2中のxが小さいときの安定性がより優れる。これは、ニッケルよりもコバルトの方が、ヤーン・テラー効果による歪みの影響が小さいためと考えられる。遷移金属化合物におけるヤーン・テラー効果は、遷移金属のd軌道の電子の数により、その効果の強さが異なる。ニッケル酸リチウム等の8面体配位の低スピンニッケル(III)が遷移金属の過半を占めるような層状岩塩型の複合酸化物は、ヤーン・テラー効果の影響が大きく、ニッケルと酸素の8面体からなる層に歪みが生じやすい。そのため充放電サイクルにおいて結晶構造の崩れが生じる懸念が高まる。またニッケルイオンはコバルトイオンと比較して大きく、リチウムイオンの大きさに近い。そのため、ニッケル酸リチウムのようにニッケルが遷移金属の過半を占めるような層状岩塩型の複合酸化物は、ニッケルとリチウムのカチオンミキシングが生じやすいという課題がある。
正極活物質100が有する添加元素としては、マグネシウム、フッ素、ニッケル、アルミニウム、チタン、ジルコニウム、バナジウム、鉄、マンガン、クロム、ニオブ、ヒ素、亜鉛、ケイ素、硫黄、リン、ホウ素、臭素、及びベリリウムから選ばれた一または二以上を用いることが好ましい。なお、添加元素として一または二以上の遷移金属を用いる場合、遷移金属(二以上を用いる場合は、合計)は25原子%未満が好ましく、10原子%未満がより好ましく、5原子%未満がさらに好ましい。
正極活物質100の具体例としては、マグネシウム及びフッ素が添加されたコバルト酸リチウム、マグネシウム、フッ素及びチタンが添加されたコバルト酸リチウム、マグネシウム、フッ素及びアルミニウムが添加されたコバルト酸リチウム、マグネシウム、フッ素及びニッケルが添加されたコバルト酸リチウム、マグネシウム、フッ素、ニッケル及びアルミニウムが添加されたコバルト酸リチウム、等を有することができる。
これらの添加元素を有することにより、後述するように正極活物質100が有する結晶構造をより安定化させる効果を奏する。なお、本明細書等において、添加元素は混合物、または原料の一部であってもよい。
なお添加元素として、必ずしもマグネシウム、フッ素、ニッケル、アルミニウム、チタン、ジルコニウム、バナジウム、鉄、マンガン、クロム、ニオブ、ヒ素、亜鉛、ケイ素、硫黄、リン、ホウ素、臭素、またはベリリウムを含まなくてもよい。
例えば、マンガンを実質的に含まない正極活物質100とすると、合成が比較的容易で取り扱いやすく、優れたサイクル特性を有するといった上記の利点がより大きくなる。正極活物質100に含まれるマンガンの重量は例えば600ppm以下、より好ましくは100ppm以下であることが好ましい。なお、本明細書等において、「実質的に含まない」とは、分析手段を用いて測定した際に検出下限以下の場合、または検出下限程度に含んでいたとしても、作用効果の有無には影響しない程度の範囲で含まれている場合のことを指すものとする。
<結晶構造>
≪LixCoO2中のxが1のとき≫
本発明の一態様として利用可能な正極活物質100は、放電状態、つまりLixCoO2中のx=1の場合に、空間群R−3mに帰属する層状岩塩型の結晶構造を有することが好ましい。層状岩塩型の複合酸化物は、放電容量が高く、二次元的なリチウムイオンの拡散経路を有し、リチウムイオンの挿入/脱離反応に適しており、二次電池の正極活物質として優れる。そのため、特に正極活物質100の体積の大半を占める内部100bが層状岩塩型の結晶構造を有することが好ましい。図4に層状岩塩型の結晶構造をR−3m(O3)を付して示す。
≪LixCoO2中のxが1のとき≫
本発明の一態様として利用可能な正極活物質100は、放電状態、つまりLixCoO2中のx=1の場合に、空間群R−3mに帰属する層状岩塩型の結晶構造を有することが好ましい。層状岩塩型の複合酸化物は、放電容量が高く、二次元的なリチウムイオンの拡散経路を有し、リチウムイオンの挿入/脱離反応に適しており、二次電池の正極活物質として優れる。そのため、特に正極活物質100の体積の大半を占める内部100bが層状岩塩型の結晶構造を有することが好ましい。図4に層状岩塩型の結晶構造をR−3m(O3)を付して示す。
一方、本発明の一態様として利用可能な正極活物質100の表層部100aは、充電により正極活物質100からリチウムが抜けても、内部100bのコバルトと酸素の8面体からなる層状構造が壊れないよう補強する機能を有することが好ましい。または、表層部100aが正極活物質100のバリア膜として機能することが好ましい。または、正極活物質100の外周部である表層部100aが正極活物質100を補強することが好ましい。ここでいう補強とは、酸素の脱離をはじめとする正極活物質100の表層部100a及び内部100bの構造変化を抑制すること、及び/または電解質が正極活物質100の表面で酸化分解されることを抑制することをいう。
そのため、表層部100aは、内部100bと異なる結晶構造を有していることが好ましい。また表層部100aは、内部100bよりも室温(25℃)で安定な組成及び結晶構造であることが好ましい。例えば、本発明の一態様として利用可能な正極活物質100の表層部100aの少なくとも一部が、岩塩型の結晶構造を有することが好ましい。または表層部100aは、層状岩塩型と岩塩型の結晶構造の両方の結晶構造を有していることが好ましい。または表層部100aは、層状岩塩型と岩塩型の結晶構造の両方の特徴を有することが好ましい。
表層部100aは充電時にリチウムイオンが最初に脱離する領域であり、内部100bよりもリチウム濃度が低くなりやすい領域である。また表層部100aが有する正極活物質100の表面の原子は、一部の結合が切断された状態ともいえる。そのため、表層部100aは不安定になりやすく、結晶構造の劣化が始まりやすい領域といえる。一方で表層部100aを十分に安定にできれば、LixCoO2中のxが小さいとき、例えばxが0.24以下においても内部100bのコバルトと酸素の8面体からなる層状構造を壊れにくくすることができる。さらに、内部100bのコバルトと酸素の8面体からなる層のずれを抑制することができる。
表層部100aを安定な組成及び結晶構造とするために、表層部100aは添加元素を有することが好ましく、添加元素を複数有することがより好ましい。また、表層部100aは内部100bよりも添加元素から選ばれた一または二以上の濃度が高いことが好ましい。また、正極活物質100が有する添加元素から選ばれた一または二以上は濃度勾配を有していることが好ましい。また、正極活物質100は添加元素によって分布が異なっていることがより好ましい。例えば、添加元素によって濃度ピークの表面からの深さが異なっていることがより好ましい。ここでいう濃度ピークとは、表層部100aまたは表面から50nm以下における濃度の極大値をいうこととする。
例えば、添加元素の一部として、マグネシウム、フッ素、ニッケル、チタン、ケイ素、リン、ホウ素、カルシウム等は、図1B1にグラデーションで示すように、内部100bから表面に向かって高くなる濃度勾配を有することが好ましい。本明細書等において、これらの添加元素を添加元素Xと呼ぶこととする。
また、別の添加元素、例えばアルミニウム、マンガン等は、図1B2にハッチの濃さで示すように、濃度勾配を有し、及び/または添加元素Xよりも深い領域に濃度のピークを有することが好ましい。濃度のピークは表層部100aに存在してもよいし、表層部100aより深くてもよい。例えば、表面から内部に向かって5nm以上50nm以下の領域にピークを有することが好ましい。本明細書等において、これらの添加元素を添加元素Yと呼ぶこととする。
例えば、添加元素Xの一つであるマグネシウムは2価で、マグネシウムイオンは層状岩塩型の結晶構造におけるコバルトサイトよりもリチウムサイトに存在する方が安定であるため、リチウムサイトに入りやすい。マグネシウムが表層部100aのリチウムサイトに適切な濃度で存在することで、層状岩塩型の結晶構造を保持しやすくできる。これはリチウムサイトに存在するマグネシウムが、CoO2層同士を支える柱として機能するためと推測される。また、マグネシウムが存在することで、LixCoO2中のxが例えば0.24以下の状態においてマグネシウムの周囲の酸素の脱離を抑制することができる。また、マグネシウムが存在することで正極活物質100の密度が高くなることが期待できる。また表層部100aのマグネシウム濃度が高いと、電解液が分解して生じたフッ酸に対する耐食性が向上することも期待できる。
マグネシウムは、適切な濃度であれば充放電に伴うリチウムの挿入及び脱離に悪影響を及ぼさず、上記のメリットを享受できる。しかしながら、マグネシウムが過剰であるとリチウムの挿入及び脱離に悪影響が出る恐れがある。さらに結晶構造の安定化への効果が小さくなってしまう場合がある。これはマグネシウムが、リチウムサイトに加えてコバルトサイトにも入るようになるためと考えられる。加えて、リチウムサイトにもコバルトサイトにも置換しない、不要なマグネシウム化合物(酸化物、フッ化物等)が正極活物質の表面等に偏析し、二次電池の抵抗成分となる恐れがある。また、正極活物質のマグネシウム濃度が高くなるのに伴って正極活物質の放電容量が減少することがある。これはリチウムサイトにマグネシウムが入りすぎ、充放電に寄与するリチウム量が減少するためと考えられる。
そのため、正極活物質100全体が有するマグネシウムが適切な量であることが好ましい。例えばマグネシウムの原子数はコバルトの原子数の0.001倍以上0.1倍以下が好ましく、0.01倍より大きく0.04倍未満がより好ましく、0.02倍程度がさらに好ましい。ここでいう正極活物質100全体が有するマグネシウムの量とは、例えばGD−MS(グロー放電質量分析法)、ICP−MS(誘導結合プラズマ質量分析法)等を用いて正極活物質100の全体の元素分析を行った値であってもよいし、正極活物質100の作製の過程における原料の配合の値に基づいたものであってもよい。
また、添加元素Xの一つであるニッケルは、コバルトサイトとリチウムサイトのどちらにも存在しうる。コバルトサイトに存在する場合、コバルトと比較して酸化還元電位が低くなるため、放電容量の増加に繋がり好ましい。
また、ニッケルがリチウムサイトに存在する場合、コバルトと酸素の8面体からなる層状構造のずれが抑制されうる。また、充放電に伴う体積の変化が抑制される。また、弾性係数が大きくなる、つまり硬くなる。これはリチウムサイトに存在するニッケルも、CoO2層同士を支える柱として機能するためと推測される。特に高温、例えば45℃以上での充電状態において結晶構造がより安定になることが期待できるため、好ましい。
一方で、ニッケルが過剰であるとヤーン・テラー効果による歪みの影響が強まり好ましくない。またニッケルが過剰であるとリチウムの挿入及び脱離に悪影響が出る恐れがある。
そのため、正極活物質100全体が有するニッケルは、適切な量であることが好ましい。例えば正極活物質100が有するニッケルの原子数は、コバルトの原子数の0%を超えて7.5%未満が好ましく、0.05%以上4%以下が好ましく、0.1%以上2%以下が好ましく、0.2%以上1%以下がより好ましい。または0%を超えて4%以下が好ましい。または0%を超えて2%以下が好ましい。または0.05%以上7.5%未満が好ましい。または0.05%以上2%以下が好ましい。または0.1%以上7.5%未満が好ましい。または0.1%以上4%以下が好ましい。ここで示すニッケルの量は、例えばGD−MS、ICP−MS等を用いて正極活物質の全体の元素分析を行った値であってもよいし、正極活物質の作製の過程における原料の配合の値に基づいてもよい。
また、添加元素Yの一つであるアルミニウムは、層状岩塩型の結晶構造におけるコバルトサイトに存在しうる。アルミニウムは3価の典型元素であり価数が変化しないため、充放電の際もアルミニウム周辺のリチウムは移動しにくい。そのためアルミニウムとその周辺のリチウムが柱として機能し、結晶構造の変化を抑制しうる。また、アルミニウムは周囲のコバルトの溶出を抑制し、連続充電耐性を向上する効果がある。また、Al−Oの結合はCo−O結合よりも強いため、アルミニウムの周囲の酸素の脱離を抑制することができる。これらの効果により、熱安定性が向上する。そのため、添加元素としてアルミニウムを有すると、二次電池に正極活物質100を用いたときの安全性を向上できる。また、充放電を繰り返しても結晶構造が崩れにくい正極活物質100とすることができる。また、アルミニウムは再表面よりも少し深い位置に存在すること(具体的には、添加元素Xの濃度のピークよりも深い領域にアルミニウムの濃度のピークを有すること)が好ましい。または、添加元素Xの存在が確認される、再表面より最も深い領域よりも深い領域に、アルミニウムの存在が確認され、再表面より最も深い領域が存在することが好ましい。これは、アルミニウムがリチウムサイトに置換された場合、アルミニウムが置換されたリチウムサイトの近傍に存在するリチウムまで固定されてしまうため、アルミニウムが再表面にあった場合、添加元素Xよりもリチウムの拡散経路を阻害してしまうおそれがあるためである。
一方でアルミニウムが過剰であると、リチウムの挿入及び脱離に悪影響が出る恐れがある。
そのため、正極活物質100全体が有するアルミニウムが適切な量であることが好ましい。例えば正極活物質100の全体が有するアルミニウムの原子数は、コバルトの原子数の0.05%以上4%以下が好ましく、0.1%以上2%以下が好ましく、0.3%以上1.5%以下がより好ましい。または0.05%以上2%以下が好ましい。または0.1%以上4%以下が好ましい。ここでいう正極活物質100全体が有する量とは、例えば、GD−MS、ICP−MS等を用いて正極活物質100の全体の元素分析を行った値であってもよいし、正極活物質100の作製の過程における原料の配合の値に基づいてもよい。
また、添加元素Xの一つであるフッ素は1価の陰イオンであり、表層部100aにおいて酸素の一部がフッ素に置換されていると、リチウム脱離エネルギーが小さくなる。これは、リチウム脱離に伴うコバルトイオンの価数の変化が、フッ素を有さない場合は3価から4価、フッ素を有する場合は2価から3価となり、酸化還元電位が異なることによる。そのため正極活物質100の表層部100aにおいて酸素の一部がフッ素に置換されていると、フッ素近傍のリチウムイオンの脱離及び挿入がスムースに起きやすいと言える。そのため二次電池に正極活物質100を用いたときに充放電特性、大電流特性等を向上させることができる。また電解液に接する部分である表面を有する表層部100aにフッ素が存在することで、フッ酸に対する耐食性を効果的に向上させることができる。また後の実施の形態で述べるが、フッ化リチウムをはじめとするフッ化物の融点が、他の添加元素源の融点より低い場合、その他の添加元素源の融点を下げる融剤(フラックス剤ともいう)として機能しうる。
また、添加元素Xの一つであるチタンの酸化物は超親水性を有することが知られている。そのため、表層部100aにチタン酸化物を有する正極活物質100とすることで、極性の高い溶媒に対して濡れ性がよくなる可能性がある。二次電池としたときに正極活物質100と、極性の高い電解液との界面の接触が良好となり、内部抵抗の上昇を抑制できる可能性がある。
また、添加元素Xの一つであるリンを表層部100aに有すると、LixCoO2中のxが小さい状態を保持した場合において、ショートを抑制できる場合があり好ましい。例えばリンと酸素を含む化合物として表層部100aに存在することが好ましい。
正極活物質100がリンを有する場合には、電解質の分解により発生したフッ化水素とリンが反応し、電解質中のフッ化水素濃度を低下できる可能性があり好ましい。
電解質がLiPF6を有する場合、加水分解により、フッ化水素が発生する恐れがある。また、正極の構成要素として用いられるポリフッ化ビニリデン(PVDF)とアルカリとの反応によりフッ化水素が発生する恐れもある。電解質中のフッ化水素濃度が低下することにより、集電体の腐食及び/または被膜104のはがれを抑制できる場合がある。また、PVDFのゲル化及び/または不溶化による接着性の低下を抑制できる場合がある。
また、正極活物質100がマグネシウムと共にリンを有すると、LixCoO2中のxが小さい状態における安定性が極めて高くなり好ましい。正極活物質100がリンを有する場合、リンの原子数は、コバルトの原子数の1%以上20%以下が好ましく、2%以上10%以下がより好ましく、3%以上8%以下がさらに好ましい。または1%以上10%以下が好ましい。または1%以上8%以下が好ましい。または2%以上20%以下が好ましい。または2%以上8%以下が好ましい。または3%以上20%以下が好ましい。または3%以上10%以下が好ましい。加えてマグネシウムの原子数は、コバルトの原子数の0.1%以上10%以下が好ましく、0.5%以上5%以下がより好ましく、0.7%以上4%以下がより好ましい。または0.1%以上5%以下が好ましい。または0.1%以上4%以下が好ましい。または0.5%以上10%以下が好ましい。または0.5%以上4%以下が好ましい。または0.7%以上10%以下が好ましい。または0.7%以上5%以下が好ましい。ここで示すリン及びマグネシウムの濃度は例えば、GC−MS、ICP−MS等を用いて正極活物質100の全体の元素分析を行った値であってもよいし、正極活物質100の作製の過程における原料の配合の値に基づいてもよい。
また、正極活物質100がクラックを有する場合、クラックを表面とした正極活物質の内部、例えば埋め込み部102にリン、より具体的には例えばリンと酸素を含む化合物が存在することにより、クラックの進行が抑制されうる。
また、表層部100aにマグネシウムとニッケルを併せて有する場合、2価のニッケルの近くでは2価のマグネシウムがより安定に存在できる可能性がある。そのためLixCoO2中のxが小さい状態でもマグネシウムの溶出が抑制されうる。そのため表層部100aの安定化に寄与しうる。
また、添加元素Xと添加元素Yのように分布が異なる添加元素を併せて有すると、より広い領域の結晶構造を安定化でき好ましい。例えば正極活物質100は添加元素Xの一部であるマグネシウム及びニッケルと、添加元素Yの一であるアルミニウムと、を共に有すると、添加元素Xと添加元素Yの一方しか有さない場合よりも広い領域の結晶構造を安定化できる。このように正極活物質100が添加元素Xと添加元素Yを併せて有する場合は、表面の安定化はマグネシウム、ニッケル等の添加元素Xによって十分に果たせるため、アルミニウムなどの添加元素Yは表面に必須ではない。むしろアルミニウムは深い領域、例えば表面からの深さが5nm以上50nm以内の領域に広く分布する方が、より広い領域の結晶構造を安定化でき好ましい。
上記のように複数の添加元素を有すると、それぞれの添加元素の効果が相乗し表層部100aのさらなる安定化に寄与しうる。特にマグネシウム、ニッケル及びアルミニウムを有すると安定な組成及び結晶構造とする効果が高く好ましい。
ただし、表層部100aが添加元素と酸素の化合物のみで占められると、リチウムの挿入脱離が難しくなってしまうため、好ましくない。例えば表層部100aが、MgO、MgOとNiO(II)が固溶した構造、及び/またはMgOとCoO(II)が固溶した構造のみで占められるのは好ましくない。そのため、表層部100aは少なくともコバルトを有し、放電状態においてはリチウムも有し、リチウムの挿入脱離の経路を有していることが好ましい。
また、十分にリチウムの挿入脱離の経路を確保するために、表層部100aはマグネシウムよりもコバルトの濃度が高いことが好ましい。例えば、マグネシウムの原子数Mgとコバルトの原子数Coの比Mg/Coは0.62以下であることが好ましい。また、表層部100aはニッケルよりもコバルトの濃度が高いことが好ましい。また、表層部100aはアルミニウムよりもコバルトの濃度が高いことが好ましい。また、表層部100aはフッ素よりもコバルトの濃度が高いことが好ましい。
さらに、ニッケルが多すぎるとリチウムの拡散を阻害する恐れがあるため、表層部100aはニッケルよりもマグネシウムの濃度が高いことが好ましい。例えばニッケルの原子数はマグネシウムの原子数の1/6以下であることが好ましい。
また、添加元素の一部、特にマグネシウム、ニッケル及びアルミニウムは、内部100bよりも表層部100aの濃度が高いことが好ましいものの、内部100bにもランダムかつ希薄に存在することが好ましい。マグネシウム及びアルミニウムが内部100bのリチウムサイトに適切な濃度で存在すると、上記と同様に層状岩塩型の結晶構造を保持しやすくできるといった効果がある。またニッケルが内部100bに適切な濃度で存在すると、上記と同様にコバルトと酸素の8面体からなる層状構造のずれが抑制されうる。またマグネシウムとニッケルを併せて有する場合も上記と同様にマグネシウムの溶出を抑制する相乗効果が期待できる。
また、上述のような添加元素の濃度勾配に起因して、内部100bから、表面に向かって結晶構造が連続的に変化することが好ましい。または、表層部100aと内部100bの結晶の配向が概略一致していることが好ましい。または、表層部100aと内部100bがトポタキシ(topotaxy)であることが好ましい。
本明細書等において、トポタキシとは、結晶の配向が概略一致するような三次元的な構造上の類似性を有すること、または結晶学的に同じ配向であることをいう。なお、エピタキシとは二次元界面の構造上の類似性をいう。
表層部100aと内部100bとがトポタキシであることで、結晶構造の歪み、および/または原子配列のずれを減少させることができる。これにより、ピットの原因を抑制することができる。本明細書等において、ピットとは、正極活物質において欠陥が進行して形成される穴のことをいう。
また、層状岩塩型の内部100bから、岩塩型、または岩塩型と層状岩塩型の両方の特徴を有する表面及び表層部100aに向かって結晶構造が連続的に変化することが好ましい。または、岩塩型、または岩塩型と層状岩塩型の両方の特徴を有する表層部100aと、層状岩塩型の内部100bの結晶の配向が概略一致していることが好ましい。
なお、本明細書等において、リチウムとコバルトをはじめとする遷移金属を含む複合酸化物が有する、空間群R−3mに帰属する層状岩塩型の結晶構造とは、陽イオンと陰イオンが交互に配列する岩塩型のイオン配列を有し、遷移金属とリチウムが規則配列して二次元平面を形成するため、リチウムの二次元的拡散が可能である結晶構造をいう。なお陽イオンまたは陰イオンの欠損等の欠陥があってもよい。また、層状岩塩型結晶構造は、厳密に言えば、岩塩型結晶の格子が歪んだ構造となっている場合がある。
また、岩塩型の結晶構造とは、空間群Fm−3mをはじめとする立方晶系の結晶構造を有し、陽イオンと陰イオンが交互に配列している構造をいう。なお陽イオンまたは陰イオンの欠損があってもよい。
また、層状岩塩型と岩塩型の結晶構造の特徴の両方を有することは、電子線回折パターン、TEM像、断面STEM像等によって判断することができる。
岩塩型は陽イオンのサイトに区別がないが、層状岩塩型は結晶構造の陽イオンのサイトが2種あり、1つはリチウムが大半を占有し、もう1つは遷移金属が占有する。陽イオンの二次元平面と陰イオンの二次元平面とが交互に配列する積層構造は、岩塩型も層状岩塩型も同じである。この二次元平面を形成する結晶面に対応する電子線回折パターンの輝点の中で、中心のスポット(透過斑点)を原点000とした際、中心のスポットに最も近い輝点は、理想的な状態の岩塩型では例えば(111)面、層状岩塩型では例えば(003)面になる。例えば岩塩型MgOと層状岩塩型LiCoO2の電子線回折パターンを比較する場合、LiCoO2の(003)面の輝点は、MgOの(111)面の輝点間の距離のおよそ半分程度の距離の位置に観察される。そのため分析領域に、例えば岩塩型MgOと層状岩塩型LiCoO2の2相を有する場合、電子線回折パターンでは、強い輝度の輝点と、弱い輝度の輝点とが交互に配列する面方位が存在する。岩塩型と層状岩塩型で共通する輝点は強い輝度となり、層状岩塩型のみで生じる輝点は弱い輝度となる。
また、断面STEM像等では、層状岩塩型の結晶構造をc軸に垂直な方向から観察したとき、強い輝度で観察される層と、弱い輝度で観察される層が交互に観察される。岩塩型は陽イオンのサイトに区別がないためこのような特徴はみられない。岩塩型と層状岩塩型の両方の特徴を有する結晶構造の場合、特定の結晶方位から観察すると、断面STEM像等では強い輝度で観察される層と、弱い輝度で観察される層が交互に観察され、さらに弱い輝度の層、すなわちリチウム層の一部にリチウムより原子番号の大きい金属が存在する。
層状岩塩型結晶、及び岩塩型結晶の陰イオンは立方最密充填構造(面心立方格子構造)をとる。後述するO3’型及び単斜晶O1(15)結晶も、陰イオンは立方最密充填構造をとると推定される。そのため層状岩塩型結晶と岩塩型結晶が接するとき、陰イオンにより構成される立方最密充填構造の向きが揃う結晶面が存在する。
または、以下のように説明することもできる。立方晶の結晶構造の{111}面における陰イオンは三角格子を有する。層状岩塩型は空間群R−3mであって、菱面体構造であるが、構造の理解を容易にするため一般に複合六方格子で表現され、層状岩塩型の(0001)面は六角格子を有する。立方晶{111}面の三角格子は、層状岩塩型の(0001)面の六角格子と同様の原子配列を有する。両者の格子が整合性を持つことを、立方最密充填構造の向きが揃うということができる。
ただし、層状岩塩型結晶及びO3’型結晶の空間群はR−3mであり、岩塩型結晶の空間群Fm−3m(一般的な岩塩型結晶の空間群)とは異なるため、上記の条件を満たす結晶面のミラー指数は層状岩塩型結晶及びO3’型結晶と、岩塩型結晶では異なる。本明細書等では、層状岩塩型結晶、O3’型及び岩塩型結晶において、陰イオンにより構成される立方最密充填構造の向きが揃うとき、結晶の配向が概略一致する、と言う場合がある。
二つの領域の結晶の配向が概略一致することは、TEM(Transmission Electron Microscope、透過電子顕微鏡)像、STEM(Scanning Transmission Electron Microscope、走査透過電子顕微鏡)像、HAADF−STEM(High−angle Annular Dark Field Scanning TEM、高角散乱環状暗視野走査透過電子顕微鏡)像、ABF−STEM(Annular Bright−Field Scanning Transmission Electron Microscope、環状明視野走査透過電子顕微鏡)像、電子線回折パターン、TEM像及びSTEM像等のFFTパターン等から判断することができる。XRD(X−ray Diffraction、X線回折)、電子線回折、中性子線回折等も判断の材料にすることができる。
図2に、層状岩塩型結晶LRSと岩塩型結晶RSの配向が概略一致しているTEM像の例を示す。TEM像、STEM像、HAADF−STEM像、ABF−STEM像等では、結晶構造を反映した像が得られる。
例えばTEMの高分解能像等では、結晶面に由来するコントラストが得られる。電子線の回折及び干渉によって、例えば層状岩塩型の複合六方格子のc軸と垂直に電子線が入射した場合、(0003)面に由来するコントラストが明るい帯(明るいストリップ)と暗い帯(暗いストリップ)の繰り返しとして得られる。そのためTEM像において明線と暗線の繰り返しが観察され、明線同士(例えば図2に示すLRSとLLRS)の角度が5°以下、または2.5°以下である場合、結晶面が概略一致している、すなわち結晶の配向が概略一致していると判断することができる。同様に、暗線同士の角度が5°以下、または2.5°以下である場合も、結晶の配向が概略一致していると判断することができる。
また、HAADF−STEM像では、原子番号に比例したコントラストが得られ、原子番号が大きい元素ほど明るく観察される。例えば空間群R−3mに属する層状岩塩型のコバルト酸リチウムの場合、コバルト(原子番号27)が最も原子番号が大きいため、コバルト原子の位置で電子線が強く散乱され、コバルト原子の配列が明線もしくは強い輝度の点の配列として観察される。そのため層状岩塩型の結晶構造を有するコバルト酸リチウムをc軸と垂直に観察した場合、c軸と垂直にコバルト原子の配列が明線もしくは強い輝度の点の配列として観察され、リチウム原子、酸素原子の配列は暗線もしくは輝度の低い領域として観察される。コバルト酸リチウムの添加元素としてフッ素(原子番号9)及びマグネシウム(原子番号12)を有する場合も同様である。
そのため、HAADF−STEM像において、結晶構造の異なる二つの領域で明線と暗線の繰り返しが観察され、明線同士の角度が5°以下、または2.5°以下である場合、原子の配列が概略一致している、すなわち結晶の配向が概略一致していると判断することができる。同様に、暗線同士の角度が5°以下、または2.5°以下である場合も、結晶の配向が概略一致していると判断することができる。
なお、ABF−STEMでは原子番号が小さい元素ほど明るく観察されるが、原子番号に応じたコントラストが得られる点ではHAADF−STEMと同様であるため、HAADF−STEM像と同様に結晶の配向を判断することができる。
図3Aに層状岩塩型結晶LRSと岩塩型結晶RSの配向が概略一致しているSTEM像の例を示す。岩塩型結晶RSの領域のFFTパターンを図3Bに、層状岩塩型結晶LRSの領域のFFTパターンを図3Cに示す。図3B及び図3Cの左に組成、JCPDSのカードナンバー、及びこれから計算されるd値及び角度を示す。右に実測値を示す。Oを付したスポットは0次回折である。
図3BでAを付したスポットは立方晶の11−1反射に由来するものである。図3CでAを付したスポットは層状岩塩型の0003反射に由来するものである。図3B及び図3Cから、立方晶の11−1反射の方位と、層状岩塩型の0003反射の方位と、が概略一致していることがわかる。すなわち図3BのAOを通る直線と、図3CのAOを通る直線と、が概略平行であることがわかる。ここでいう概略一致及び概略平行とは、角度が5°以下、または2.5°以下であることをいう。
このように、FFTパターン及び電子線回折パターンでは、層状岩塩型結晶と岩塩型結晶の配向が概略一致していると、層状岩塩型の〈0003〉方位と、岩塩型の〈11−1〉方位と、が概略一致する場合がある。このとき、これらの逆格子点はスポット状であること、つまり他の逆格子点と連続していないことが好ましい。逆格子点がスポット状で、他の逆格子点と連続していないことは、結晶性が高いことを意味する。
また、上述のように立方晶の11−1反射の方位と、層状岩塩型の0003反射の方位と、が概略一致している場合、電子線の入射方位によっては、層状岩塩型の0003反射の方位とは異なる逆格子空間上に、層状岩塩型の0003反射由来ではないスポットが観測されることがある。例えば図3CでBを付したスポットは、層状岩塩型の1014反射に由来するものである。これは、層状岩塩型の0003反射由来の逆格子点(図3CのA)の方位から、52°以上56°以下の角度であり(すなわち∠AOBが52°以上56°以下であり)、dが0.19nm以上0.21nm以下の箇所に観測されることがある。なおこの指数は一例であり、必ずしもこれに一致している必要は無い。例えば、それぞれにおける等価な逆格子点でも良い。
同様に、立方晶の11−1反射が観測された方位とは別の逆格子空間上に、立方晶の11−1反射由来ではないスポットが観測されることがある。例えば、図3BでBを付したスポットは、立方晶の200反射に由来するものである。これは、立方晶の11−1由来の反射(図3BのA)の方位から、54°以上56°以下の角度である(すなわち∠AOBが54°以上56°以下である)箇所に回折スポットが観測されることがある。なおこの指数は一例であり、必ずしもこれに一致している必要は無い。例えば、それぞれにおける等価な逆格子点でも良い。
なお、コバルト酸リチウムをはじめとする層状岩塩型の正極活物質は、(0003)面及びこれと等価な面、並びに(10−14)面及びこれと等価な面が結晶面として現れやすいことが知られている。そのため正極活物質の形状をSEM等でよく観察することで、(0003)面が観察しやすいように、例えばTEM等において電子線が[12−10]入射となるように観察サンプルをFIB等で薄片加工することが可能である。結晶の配向の一致について判断したいときは、層状岩塩型の(0003)面が観察しやすいよう薄片化することが好ましい。
≪LixCoO2中のxが小さい状態≫
本発明の一態様として利用可能な正極活物質100は、放電状態において上述のような添加元素の分布及び/または結晶構造を有することに起因して、LixCoO2中のxが小さい状態での結晶構造が、従来の正極活物質と異なる。なお、本明細書等において、「xが小さい」とは、0.1<x≦0.24をいうこととする。
本発明の一態様として利用可能な正極活物質100は、放電状態において上述のような添加元素の分布及び/または結晶構造を有することに起因して、LixCoO2中のxが小さい状態での結晶構造が、従来の正極活物質と異なる。なお、本明細書等において、「xが小さい」とは、0.1<x≦0.24をいうこととする。
図4乃至図8を用いて、LixCoO2中のxの変化に伴う結晶構造の変化について、従来の正極活物質と本発明の一態様として利用可能な正極活物質100を比較しながら説明する。
従来の正極活物質の結晶構造の変化を図5に示す。図5に示す従来の正極活物質は、特に添加元素を有さないコバルト酸リチウム(LiCoO2)である。なお、本明細書等において、「特に添加元素を有さない」とは、分析手段を用いて測定した際に検出下限以下の場合、または検出下限程度に含んでいたとしても、作用効果の有無には影響しない程度の範囲で含まれている場合のことを指すものとする。
図5にR−3m(O3)を付してLixCoO2中のx=1のコバルト酸リチウムが有する結晶構造を示す。この結晶構造はリチウムが8面体(Octahedral)サイトを占有し、ユニットセル中にCoO2層が3層存在する。そのためこの結晶構造をO3型結晶構造と呼ぶ場合がある。なお、CoO2層とはコバルトに酸素が6配位した8面体構造が、稜共有の状態で平面方向に連続した構造をいうこととする。これをコバルトと酸素の8面体からなる層、という場合もある。
また従来のコバルト酸リチウムは、x=0.5程度のときリチウムの対称性が高まり、単斜晶系の空間群P2/mに帰属する結晶構造を有することが知られている。この構造は、ユニットセル中にCoO2層が1層存在する。そのためO1型、または単斜晶O1型と呼ぶ場合がある。
また、x=0のときの正極活物質は、三方晶系の空間群P−3m1の結晶構造を有し、やはりユニットセル中にCoO2層が1層存在する。そのためこの結晶構造を、O1型、または三方晶O1型と呼ぶ場合がある。また三方晶を複合六方格子に変換し、六方晶O1型と呼ぶ場合もある。
またx=0.12程度のときの従来のコバルト酸リチウムは、空間群R−3mの結晶構造を有する。この構造は、三方晶O1型のようなCoO2の構造と、R−3m(O3)のようなLiCoO2の構造と、が交互に積層された構造ともいえる。そのため、この結晶構造をH1−3型結晶構造と呼ぶ場合がある。なお、実際にはH1−3型結晶構造は、ユニットセルあたりのコバルト原子の数が他の構造の2倍となっている。しかし図5をはじめ本明細書等では、他の結晶構造と比較しやすくするためH1−3型結晶構造のc軸をユニットセルの1/2にした図で示すこととする。
H1−3型結晶構造は、一例として、ユニットセルにおけるコバルトと酸素の座標を、Co(0,0,0.42150±0.00016)、O1(0,0,0.27671±0.00045)、O2(0,0,0.11535±0.00045)と表すことができる。O1及びO2は、それぞれ酸素原子である。正極活物質が有する結晶構造をいずれのユニットセルを用いて表すべきかは、例えばXRDパターンのリートベルト解析により判断することができる。この場合はGOF(goodness of fit)の値が小さくなるユニットセルを採用すればよい。
LixCoO2中のxが0.12以下になるような充電と、放電とを繰り返すと、従来のコバルト酸リチウムはH1−3型結晶構造と、放電状態のR−3m(O3)の構造と、の間で結晶構造の変化(つまり非平衡な相変化)を繰り返すことになる。
しかしながら、これらの2つの結晶構造は、CoO2層のずれが大きい。図5に点線及び矢印で示すように、H1−3型結晶構造では、CoO2層が放電状態のR−3m(O3)から大きくずれている。このようなダイナミックな構造変化は、結晶構造の安定性に悪影響を与えうる。
さらにこれらの2つの結晶構造は体積の差も大きい。同数のコバルト原子あたりで比較した場合、H1−3型結晶構造と放電状態のR−3m(O3)型結晶構造の体積の差は3.5%を超え、代表的には3.9%以上である。
加えて、H1−3型結晶構造が有する、三方晶O1型のようにCoO2層が連続した構造は不安定である可能性が高い。
そのため、xが0.12以下になるような充放電を繰り返すと従来のコバルト酸リチウムの結晶構造は崩れていく。結晶構造の崩れが、サイクル特性の悪化を引き起こす。これは、結晶構造が崩れることで、リチウムが安定して存在できるサイトが減少し、またリチウムの挿入脱離が難しくなるためである。なお、xが0.12以下になるような充放電を繰り返す場合のときだけでなく、実際にはxが0.24以下であっても結晶構造の崩れが多く発生し、サイクル特性の悪化を引き起こす。このため、従来のコバルト酸リチウムは、実用上、xが0.24を超える範囲で充放電を繰り返すような制御が行われている。
一方、図4に示す本発明の一態様として利用可能な正極活物質100は、LixCoO2中のxが1の放電状態と、xが0.24以下の状態における結晶構造の変化が従来の正極活物質よりも少ない。より具体的には、xが1の状態と、xが0.24以下の状態におけるCoO2層のずれを小さくすることができる。また、コバルト原子あたりで比較した場合の体積の変化を小さくすることができる。したがって、本発明の一態様として利用可能な正極活物質100は、xが0.24以下になるような充放電を繰り返しても結晶構造が崩れにくく、優れたサイクル特性を実現することができる。また、本発明の一態様として利用可能な正極活物質100は、LixCoO2中のxが0.24以下の状態において従来の正極活物質よりも安定な結晶構造を取り得る。したがって、本発明の一態様として利用可能な正極活物質100は、LixCoO2中のxが0.24以下の状態を保持した場合においてショートが生じづらいため、リチウムイオン電池の安全性が向上する。
LixCoO2中のxが1、0.2程度、及び0.15程度のときに正極活物質100の内部100bが有する結晶構造を図4に示す。内部100bは正極活物質100の体積の大半を占め、充放電に大きく寄与する部分であるため、CoO2層のずれ及び体積の変化が最も問題となる部分といえる。
正極活物質100は、x=1のとき、従来のコバルト酸リチウムと同じR−3m(O3)の結晶構造を有する。
一方、正極活物質100は、x=0.24以下、例えば0.2程度及び0.15程度のとき、従来のコバルト酸リチウムのH1−3型結晶構造とは異なる結晶構造を有する。
具体的には、x=0.2程度のときの正極活物質100は、三方晶系の空間群R−3mに帰属される結晶構造を有する。これは、CoO2層の対称性がO3と同じである。よって、この結晶構造をO3’型結晶構造と呼ぶこととする。図4にR−3m(O3)’を付してこの結晶構造を示す。
O3’型の結晶構造は、ユニットセルにおけるコバルトと酸素の座標を、Co(0,0,0.5)、O(0,0,x)、0.20≦x≦0.25の範囲内で示すことができる。またユニットセルの格子定数は、a軸は2.797≦a≦2.837(×10−1nm)が好ましく、2.807≦a≦2.827(×10−1nm)がより好ましく、代表的にはa=2.817(×10−1nm)である。c軸は13.68≦c≦13.88(×10−1nm)が好ましく、13.75≦c≦13.81がより好ましく、代表的にはc=13.78(×10−1nm)である。
また、x=0.15程度のときの本発明の一態様として利用可能な正極活物質100は、単斜晶系の空間群P2/mに帰属される結晶構造を有する。これは、ユニットセル中にCoO2層が1層存在する。また、このとき正極活物質100中に存在するリチウムは放電状態の15原子%程度である。このため、本明細書等においては、この結晶構造を「単斜晶O1(15)型結晶構造」と呼ぶこととする。図4にP2/m 単斜晶O1(15)を付して、この結晶構造を示す。
単斜晶O1(15)型の結晶構造は、ユニットセルにおけるコバルトと酸素の座標を、
Co1(0.5,0,0.5)、
Co2(0,0.5,0.5)、
O1(XO1,0,ZO1)、
0.23≦XO1≦0.24、0.61≦ZO1≦0.65、
O2(XO2,0.5,ZO2)、
0.75≦XO2≦0.78、0.68≦ZO2≦0.71、の範囲内で示すことができる。またユニットセルの格子定数は、−
a=4.880±0.05(×10−1nm)、
b=2.817±0.05(×10−1nm)、
c=4.839±0.05(×10−1nm)、
α=90°、
β=109.6±0.1°、
γ=90°である。
Co1(0.5,0,0.5)、
Co2(0,0.5,0.5)、
O1(XO1,0,ZO1)、
0.23≦XO1≦0.24、0.61≦ZO1≦0.65、
O2(XO2,0.5,ZO2)、
0.75≦XO2≦0.78、0.68≦ZO2≦0.71、の範囲内で示すことができる。またユニットセルの格子定数は、−
a=4.880±0.05(×10−1nm)、
b=2.817±0.05(×10−1nm)、
c=4.839±0.05(×10−1nm)、
α=90°、
β=109.6±0.1°、
γ=90°である。
なお、この結晶構造は、ある程度の誤差を許容すれば空間群R−3mでもフィッティング可能である。この場合のユニットセルにおけるコバルトと酸素の座標は、
Co(0,0,0.5)、
O(0,0,ZO)、
0.21≦ZO≦0.23、の範囲内で示すことができる。
またユニットセルの格子定数は、
a=2.817±0.02(×10−1nm)、
c=13.68±0.1(×10−1nm)である。
Co(0,0,0.5)、
O(0,0,ZO)、
0.21≦ZO≦0.23、の範囲内で示すことができる。
またユニットセルの格子定数は、
a=2.817±0.02(×10−1nm)、
c=13.68±0.1(×10−1nm)である。
O3’型及び単斜晶O1(15)型結晶構造は、いずれもコバルト、ニッケル、マグネシウム等のイオンが酸素6配位位置を占める。なお、リチウムなどの軽元素は酸素4配位位置を占める場合がありうる。
図4中に点線で示すように、放電状態のR−3m(O3)と、O3’及び単斜晶O1(15)型結晶構造とでは、CoO2層のずれがほとんどない。
また、放電状態のR−3m(O3)と、O3’型結晶構造の同数のコバルト原子あたりの体積の差は2.5%以下、より詳細には2.2%以下、代表的には1.8%である。
また放電状態のR−3m(O3)と、単斜晶O1(15)型結晶構造の同数のコバルト原子あたりの体積の差は3.3%以下、より詳細には3.0%以下、代表的には2.5%である。
表1に、放電状態のR−3m(O3)と、O3’、単斜晶O1(15)、H1−3型、及び三方晶O1のコバルト原子1つあたりの体積の差を示す。表1の算出に用いた各結晶構造の格子定数は、放電状態のR−3m(O3)、三方晶O1、及びH1−3型については文献値(ICSD coll.code.172909および88721)及び非特許文献1を参照することができる。O3’、単斜晶O1(15)についてはXRDの実験値から算出することができる。

このように、本発明の一態様として利用可能な正極活物質100は、LixCoO2中のxが小さいとき、つまり多くのリチウムが脱離したときの結晶構造の変化が、従来の正極活物質よりも抑制されている。また同数のコバルト原子あたりで比較した場合の体積の変化も抑制されている。このため、正極活物質100は、xが0.24以下になるような充放電を繰り返しても結晶構造が崩れにくく、充放電サイクルにおける充放電容量の低下が抑制される。また、従来の正極活物質よりも多くのリチウムを安定して利用できるため、正極活物質100は重量あたり及び体積あたりの放電容量が大きい。そのため正極活物質100を用いることで、重量あたり及び体積あたりの放電容量の高い二次電池を作製できる。
なお、正極活物質100は、LixCoO2中のxが0.15以上0.24以下のとき、O3’型の結晶構造を有する場合があることが確認され、xが0.24を超えて0.27以下のときであってもO3’型の結晶構造を有すると推定されている。また、LixCoO2中のxが0.1を超えて0.2以下、代表的にはxが0.15以上0.17以下のとき単斜晶O1(15)型の結晶構造を有する場合があることが確認されている。しかし、結晶構造は、LixCoO2中のxだけでなく充放電サイクル数、充放電電流、温度、電解質等の影響を受けるため、必ずしも上記のxの範囲に限定されない。
このため、正極活物質100はLixCoO2中のxが0.1を超えて0.24以下のとき、O3’型のみを有してもよいし、単斜晶O1(15)型のみを有してもよいし、両方の結晶構造を有してもよい。また正極活物質100の内部100bの粒子の全てがO3’型及び/または単斜晶O1(15)型の結晶構造でなくてもよい。他の結晶構造を含んでいてもよいし、一部が非晶質であってもよい。
また、LixCoO2中のxが小さい状態にするには、一般的には高い充電電圧で充電する必要がある。そのため、LixCoO2中のxが小さい状態を、高い充電電圧で充電した状態と言い換えることができる。例えば、リチウム金属の電位を基準として4.6V以上の電圧で、25℃の環境でCC/CV(定電流/定電圧)充電すると、従来の正極活物質ではH1−3型結晶構造が現れる。そのためリチウム金属の電位を基準として4.6V以上の充電電圧は高い充電電圧ということができる。また本明細書等において、特に言及しない場合、充電電圧はリチウム金属の電位を基準として表すとする。
そのため、本発明の一態様として利用可能な正極活物質100は、高い充電電圧、例えば25℃において4.6V以上の電圧で充電しても、R−3m(O3)の対称性を有する結晶構造を保持できるため好ましい、と言い換えることができる。またより高い充電電圧、例えば25℃において4.65V以上4.7V以下の電圧で充電したときO3’型の結晶構造を取り得るため好ましい、と言い換えることができる。さらに高い充電電圧、例えば25℃において4.7Vを超えて4.8V以下の電圧で充電したとき単斜晶O1(15)型の結晶構造を取り得るため好ましい、と言い換えることができる。
正極活物質100でもさらに充電電圧を高めるとようやく、H1−3型結晶構造が観測される場合がある。また上述したように結晶構造は充放電サイクル数、充放電電流、温度、電解質等の影響を受けるため、充電電圧がより低い場合、例えば充電電圧が25℃において4.5V以上4.6V未満でも、本発明の一態様として利用可能な正極活物質100はO3’型結晶構造を取り得る場合が有る。同様に25℃において4.65V以上4.7V以下の電圧で充電したときに単斜晶O1(15)型の結晶構造を取り得る場合がある。
なお、二次電池において例えば負極活物質として黒鉛を用いる場合、上記よりも黒鉛の電位の分だけ二次電池の電圧が低下する。黒鉛の電位はリチウム金属の電位を基準として0.05V乃至0.2V程度である。そのため負極活物質として黒鉛を用いた二次電池の場合は、上記の電圧から黒鉛の電位を差し引いた電圧のとき同様の結晶構造を有する。
また図4のO3’及び単斜晶O1(15)ではリチウムが全てのリチウムサイトに等しい確率で存在するように示したが、これに限らない。一部のリチウムサイトに偏って存在していてもよいし、例えば図5に示す単斜晶O1(Li0.5CoO2)のような対称性を有していてもよい。リチウムの分布は、例えば中性子線回折により分析することができる。
またO3’及び単斜晶O1(15)型の結晶構造は、層間にランダムにリチウムを有するもののCdCl2型の結晶構造に類似する結晶構造であるということもできる。このCdCl2型に類似した結晶構造は、ニッケル酸リチウムをLi0.06NiO2まで充電したときの結晶構造と近いが、純粋なコバルト酸リチウム、またはコバルトを多く含む層状岩塩型の正極活物質では通常CdCl2型の結晶構造を取らないことが知られている。
また添加元素の濃度勾配は、正極活物質100の表層部100aの複数個所において同じような勾配であることが好ましい。つまり添加元素に由来するバリア膜が表層部100aに均質に存在することが好ましい。表層部100aの一部にバリア膜があっても、バリア膜のない部分が存在すれば、ない部分に応力が集中する恐れがある。正極活物質100の一部に応力が集中すると、そこからクラック等の欠陥が生じ、正極活物質の割れ及び放電容量の低下につながる恐れがある。
ただし必ずしも、正極活物質100の表層部100a全てにおいて添加元素が同じような濃度勾配を有していなくてもよい。図1A1中のC−D付近を拡大した図を図6A1及び図6A2に示す。図1A1のC−D付近の添加元素Xの分布の例を図6A1に、C−D付近の添加元素Yの分布の例を図6A2に示す。
ここで、C−D付近はR−3mの層状岩塩型の結晶構造を有し、表面は(001)配向であるとする。(001)配向した表面は、その他の表面と添加元素の分布が異なっていてもよい。例えば、(001)配向した表面とその表層部100aは、添加元素X及び添加元素Yから選ばれた一または二以上の濃度ピークの分布が、(001)配向以外の表面と比較して表面から浅い部分に限定されていてもよい。または、(001)配向した表面とその表層部100aは、(001)配向以外の表面と比較して添加元素X及び添加元素Yから選ばれた一または二以上の濃度が低くてもよい。または、(001)配向した表面とその表層部100aは、添加元素X及び添加元素Yから選ばれた一または二以上の濃度が検出下限以下であってもよい。
R−3mの層状岩塩型の結晶構造では、(001)面に平行に陽イオンが配列している。これはCoO2層と、リチウム層と、が(001)面と平行に交互に積層した構造であるということができる。そのためリチウムイオンの拡散経路も(001)面に平行に存在する。
CoO2層は比較的安定であるため、正極活物質100の表面は(001)配向である方が安定である。(001)面には充放電におけるリチウムイオンの主な拡散経路は露出していない。
一方、(001)配向以外の表面ではリチウムイオンの拡散経路が露出している。そのため(001)配向以外の表面及び表層部100aは、リチウムイオンの拡散経路を保つために重要な領域であると同時に、リチウムイオンが最初に脱離する領域であるため不安定になりやすい。そのため(001)配向以外の表面及び表層部100aを補強することが、正極活物質100全体の結晶構造を保つために極めて重要である。
そのため、本発明の別の一態様の正極活物質100では、(001)配向以外の表面及びその表層部100aの添加元素の分布が図1B1及び図1B2に示すような分布となっていることが好ましい。一方、(001)配向した表面及びその表層部100aでは上述のように添加元素の濃度は低くてもよいし、またはなくてもよい。
後の実施の形態で説明する、純度の高いLiCoO2を作製した後に、添加元素を後から混合して加熱する作製方法は、主にリチウムイオンの拡散経路を介して添加元素が広がる。そのため(001)配向以外の表面及びその表層部100aの添加元素の分布を好ましい範囲にしやすい。
図6B1乃至図6Cを用いて、LiCoO2を作製した後に、添加元素を混合して加熱した場合の添加元素の分布について計算した結果を説明する。
図6B1は、(104)配向している表面及びその表層部100aについて計算した結果である。古典的分子動力学法で計算した。系の下部にLiCoO2(LCO)を、系の上部にマグネシウム源、リチウム源及びフッ素源としてLiFとMgF2を配した。アンサンブルはNVT(正準集団:カノニカルアンサンブル)、初期構造の密度は1.8g/cm3、系の温度は2000K、経過時間は100psec、ポテンシャルはLCO結晶構造にて最適化し、その他の原子はUFF(Universal Force Field)との混合、系の原子数は約1万個、系の電荷は中性とした。図を簡潔にするため、Co原子とMg原子を抜粋して示す。
図6B2は同様に200psec、図6B3は1200psecまで計算した結果である。
以上の計算から、下記のような過程でマグネシウムが拡散している様子が推察される。(1)熱でリチウムがLCOから脱離する。(2)マグネシウムがLCOのリチウム層に入り、内部へ拡散する。(3)LiF由来のリチウムがLCOのリチウム層に入り、(1)で脱離したリチウムが補完される。
100psec経過した図6B1からマグネシウム原子がLCO内に拡散している様子が明らかである。コバルト原子の配列に沿ってマグネシウム原子が拡散していき、1200psec経過した図6B3では、系の上部に用意したマグネシウム原子がほぼ全てLCOに取り込まれる。
図6Cは、(001)配向とした他は図6B1と同様に計算した結果である。図6Cでは、マグネシウム原子はLCOの表面にとどまっている様子がわかる。
このように純度の高いLiCoO2を作製した後に、添加元素を混合して加熱する作製方法により、(001)面よりも、(001)配向以外の表面及びその表層部100aの添加元素を好ましい分布にすることができる。
また後述する初期加熱を経る作製方法では、初期加熱によりLiCoO2の表面に意図せず残っているリチウム化合物などが脱離することが期待できる。そのため、マグネシウムをはじめとする添加元素を高濃度に表層部に分布させやすくなる。
また、正極活物質100の表面はなめらかで凹凸が少ないことが好ましいが、必ずしも、正極活物質100が有する表面の全てがそうでなくてもよい。R−3mの層状岩塩型の結晶構造を有する複合酸化物は、(001)面に平行な面、例えばリチウムが配列した面においてスリップが生じやすい。ここで、スリップとは、積層欠陥とも呼び、プレスによってLiCoO2が格子縞方向(ab面方向)に沿って変形した状態をいう。変形には、格子縞同士が前後にずれることが含まれる。格子縞同士が前後にずれると、格子縞に対して垂直方向(c軸方向)の表面には、段差が生じる。例えば図7Aのように、(001)面が存在する場合は、プレス等の工程を経ることで図7B中に矢印で示したように(001)面と平行にスリップが起こり、変形する場合がある。
この場合、スリップした結果新たに生じた表面及びその表層部100aには、添加元素が存在しないか、検出下限以下である場合がある。図7B中のE−Fはスリップした結果新たに生じた表面及びその表層部100aの例である。E−F付近を拡大した図を図7C1及び図7C2に示す。図7C1及び図7C2では、図1B1、図1B2、図6A1、図6A2と異なり、添加元素X及び添加元素Yが分布しない。
しかしスリップは(001)面に平行に生じやすいため、新たに生じた表面及びその表層部100aは(001)配向となりやすい。この場合リチウムイオンの拡散経路が露出せず、比較的安定であるため、添加元素が存在しないか、検出下限以下であっても問題がほとんどない。
なお上述のように、組成がLiCoO2、結晶構造がR−3mの層状岩塩型を有する複合酸化物では、(001)面と平行にコバルト原子が配列する。またHAADF−STEM像では、LiCoO2のうち原子番号の最も大きいコバルトの輝度が最も高くなる。そのためHAADF−STEM像において、輝度の高い原子の配列はコバルト原子の配列と考えてよい。この輝度の高い配列の繰り返しは、結晶縞または格子縞と同義である。
≪結晶粒界≫
本発明の一態様として利用可能な正極活物質100が有する添加元素は、上記のような分布に加え、少なくとも一部は結晶粒界101及びその近傍に偏在していることがより好ましい。
本発明の一態様として利用可能な正極活物質100が有する添加元素は、上記のような分布に加え、少なくとも一部は結晶粒界101及びその近傍に偏在していることがより好ましい。
例えば、正極活物質100の結晶粒界101及びその近傍(例えば、結晶粒界101を中心として、数nm離れた領域の範囲内)のマグネシウム濃度は、内部100bの他の領域よりも高いことが好ましい。また、結晶粒界101及びその近傍のフッ素濃度も内部100bの他の領域より高いことが好ましい。また、結晶粒界101及びその近傍のニッケル濃度も、内部100bの他の領域より高いことが好ましい。また、結晶粒界101及びその近傍のアルミニウム濃度も、内部100bの他の領域より高いことが好ましい。
結晶粒界101は、面欠陥の一つであるため、表面と同様不安定になりやすく、結晶構造の変化が始まりやすい。そこで結晶粒界101及びその近傍の添加元素の濃度を高くすることにより、このような結晶構造の変化をより効果的に抑制することができる。
また、結晶粒界101及びその近傍のマグネシウム濃度及びフッ素濃度が高い場合、本発明の一態様として利用可能な正極活物質100の結晶粒界101に沿ってクラックが生じた場合でも、クラックにより生じた表面の近傍でマグネシウム濃度及びフッ素濃度が高くなる。そのため、クラックが生じた後の正極活物質においてもフッ酸に対する耐食性を高めることができる。
<粒径>
本発明の一態様として利用可能な正極活物質100の粒径は、大きすぎるとリチウムの拡散が難しくなる、集電体に塗工したときに活物質層の表面が粗くなりすぎる、等の問題がある。一方、小さすぎると、集電体への塗工時に活物質層を担持しにくくなる、電解質(電解液)との反応が過剰に進む等の問題点も生じる。そのため、メディアン径(D50)が、1μm以上100μm以下が好ましく、2μm以上40μm以下であることがより好ましく、5μm以上30μm以下がさらに好ましい。または1μm以上40μm以下が好ましい。または1μm以上30μm以下が好ましい。または2μm以上100μm以下が好ましい。または2μm以上30μm以下が好ましい。または5μm以上100μm以下が好ましい。または5μm以上40μm以下が好ましい。
本発明の一態様として利用可能な正極活物質100の粒径は、大きすぎるとリチウムの拡散が難しくなる、集電体に塗工したときに活物質層の表面が粗くなりすぎる、等の問題がある。一方、小さすぎると、集電体への塗工時に活物質層を担持しにくくなる、電解質(電解液)との反応が過剰に進む等の問題点も生じる。そのため、メディアン径(D50)が、1μm以上100μm以下が好ましく、2μm以上40μm以下であることがより好ましく、5μm以上30μm以下がさらに好ましい。または1μm以上40μm以下が好ましい。または1μm以上30μm以下が好ましい。または2μm以上100μm以下が好ましい。または2μm以上30μm以下が好ましい。または5μm以上100μm以下が好ましい。または5μm以上40μm以下が好ましい。
<分析方法>
ある正極活物質においてLixCoO2中のxが小さいとき、O3’型及び/または単斜晶O1(15)型の結晶構造を有する本発明の一態様として利用可能な正極活物質100であるか否かは、LixCoO2中のxが小さい正極活物質を有する正極を、XRD、電子線回折、中性子線回折、電子スピン共鳴(ESR)、核磁気共鳴(NMR)等を用いて解析することで判断できる。
ある正極活物質においてLixCoO2中のxが小さいとき、O3’型及び/または単斜晶O1(15)型の結晶構造を有する本発明の一態様として利用可能な正極活物質100であるか否かは、LixCoO2中のxが小さい正極活物質を有する正極を、XRD、電子線回折、中性子線回折、電子スピン共鳴(ESR)、核磁気共鳴(NMR)等を用いて解析することで判断できる。
特にXRDは、正極活物質が有するコバルト等の遷移金属の対称性を高分解能で解析できる、結晶性の高さ及び結晶の配向性を比較できる、格子の周期性歪み及び結晶子サイズの解析ができる、二次電池を解体して得た正極をそのまま測定しても十分な精度を得られる、等の点で好ましい。XRDの中でも粉体XRDでは、正極活物質100の体積の大半を占める正極活物質100の内部100bの結晶構造を反映した回折ピークが得られる。
本発明の一態様として利用可能な正極活物質100は、これまで述べたようにLixCoO2中のxが1のときと、0.24以下のときで結晶構造の変化が少ないことが特徴である。高電圧(例えば、4.6V)で充電したとき、結晶構造の変化が大きな結晶構造が50%以上を占める材料は、高電圧の充放電に耐えられないため好ましくない。
ただし、添加元素を添加するだけではO3’型または単斜晶O1(15)型の結晶構造を取らない場合があることに注意が必要である。例えば、マグネシウム及びフッ素を有するコバルト酸リチウム、またはマグネシウム及びアルミニウムを有するコバルト酸リチウム、という点で共通していても、添加元素の濃度及び分布次第で、LixCoO2中のxが0.24以下でO3’型及び/または単斜晶O1(15)型の結晶構造が60%以上になる場合と、H1−3型結晶構造が50%以上を占める場合と、がある。
また、本発明の一態様として利用可能な正極活物質100であっても、xが0.1以下など小さすぎる場合、または充電電圧が4.9Vを超えるような条件ではH1−3型または三方晶O1型の結晶構造が生じる場合もある。そのため、本発明の一態様として利用可能な正極活物質100であるか否かを判断するには、XRDをはじめとする結晶構造についての解析と、充電容量または充電電圧等の情報が必要である。
また、xが小さい状態の正極活物質は、大気に触れると結晶構造の変化を起こす場合がある。例えば、O3’型及び単斜晶O1(15)型の結晶構造からH1−3型結晶構造に変化する場合がある。そのため、結晶構造の分析に供するサンプルは、全てアルゴン雰囲気等の不活性雰囲気でハンドリングすることが好ましい。
また、ある正極活物質が有する添加元素の分布が、上記で説明したような状態であるか否かは、例えばXPS、エネルギー分散型X線分光法(EDX:Energy Dispersive X−ray Spectroscopy)、EPMA(電子プローブ微小分析)等を用いて解析することで判断できる。
また、表層部100a、結晶粒界101等の結晶構造は、正極活物質100の断面の電子線回折等で分析することができる。
≪充電方法≫
ある複合酸化物が、本発明の一態様として利用可能な正極活物質100であるか否かを判断するための充電は、例えば対極(この場合、負極)をリチウム金属でコインセル(CR2032タイプ、直径20mm高さ3.2mm)を作製して充電する方法が挙げられる。なお、以下に述べる充電方法は、本発明の一態様として利用可能な正極活物質100の物性を確認するための条件である。そのため、正極活物質以外の構成について以下に述べる電解質等は、本発明の一態様であるリチウムイオン電池の構成とは異なる。
ある複合酸化物が、本発明の一態様として利用可能な正極活物質100であるか否かを判断するための充電は、例えば対極(この場合、負極)をリチウム金属でコインセル(CR2032タイプ、直径20mm高さ3.2mm)を作製して充電する方法が挙げられる。なお、以下に述べる充電方法は、本発明の一態様として利用可能な正極活物質100の物性を確認するための条件である。そのため、正極活物質以外の構成について以下に述べる電解質等は、本発明の一態様であるリチウムイオン電池の構成とは異なる。
より具体的には、正極の一例として、正極活物質、導電材及びバインダを混合したスラリーを、アルミニウム箔の正極集電体に塗工したものを用いることができる。
負極(対極)の一例として、リチウム金属を用いることができる。なお対極にリチウム金属以外の材料を用いたときは、二次電池の電位と正極の電位が異なる。本明細書等における電圧及び電位は、特に言及しない限り、対極をリチウム金属とした場合の正極の電位である。
電解質の一例として、エチレンカーボネート(EC)とジエチルカーボネート(DEC)がEC:DEC=3:7(体積比)、ビニレンカーボネート(VC)が2wt%で混合された有機溶媒に、1mol/Lの六フッ化リン酸リチウム(LiPF6)が溶解されたものを用いることができる。
セパレータの一例として、厚さ25μmのポリプロピレンの多孔質フィルムを用いることができる。
正極缶及び負極缶の一例として、ステンレス(SUS)で形成されているものを用いることができる。
上記条件で作製したコインセルを、任意の電圧(例えば、4.5V、4.55V、4.6V、4.65V、4.7V、4.75Vまたは4.8V)まで、電流値10mA/g(1C=200mA/gとした場合、0.05Cに相当)で定電流充電(CC充電とも呼ぶ)する。正極活物質の相変化を観測するためには、このような小さい電流値で充電を行うことが望ましい。温度は25℃または45℃とする。このような条件で充電した後、コインセルをアルゴン雰囲気のグローブボックス中で解体して正極を取り出すことで、任意の充電容量の正極活物質が得られる。この後に各種分析を行う際、外界成分との反応を抑制するため、アルゴン雰囲気で密封することが好ましい。例えば、XRDは、アルゴン雰囲気の密閉容器内に封入して行うことができる。また充電完了後、速やかに正極を取り出し分析に供することが好ましい。具体的には充電完了後1時間以内が好ましく、30分以内がより好ましい。
また、複数回充放電した後の充電状態の結晶構造を分析する場合、該複数回の充放電条件は上記の充電条件と異なっていてもよい。例えば充電は任意の電圧(例えば4.6V、4.65V、4.7V、4.75Vまたは4.8V)まで、電流値100mA/gで定電流充電し、電流値が10mA/gとなるまで定電圧充電し、放電は2.5V、100mA/gで定電流放電とすることができる。
さらに複数回充放電した後の放電状態の結晶構造を分析する場合も、例えば2.5V、電流値100mA/gで定電流放電とすることができる。
≪XRD≫
XRD測定の装置及び条件は、特に限定されない。例えば、下記のような装置及び条件で測定することができる。
XRD装置 :Bruker AXS社製、D8 ADVANCE
X線源 :CuKα1線
出力 :40kV、40mA
スリット幅 :Div.Slit、0.5°
検出器:LynxEye
スキャン方式 :2θ/θ連続スキャン
測定範囲(2θ) :15°以上90°以下
ステップ幅(2θ) :0.01°設定
計数時間 :1秒間/ステップ
試料台回転 :15rpm
XRD測定の装置及び条件は、特に限定されない。例えば、下記のような装置及び条件で測定することができる。
XRD装置 :Bruker AXS社製、D8 ADVANCE
X線源 :CuKα1線
出力 :40kV、40mA
スリット幅 :Div.Slit、0.5°
検出器:LynxEye
スキャン方式 :2θ/θ連続スキャン
測定範囲(2θ) :15°以上90°以下
ステップ幅(2θ) :0.01°設定
計数時間 :1秒間/ステップ
試料台回転 :15rpm
測定サンプルが粉末の場合は、ガラスのサンプルホルダーに入れる、グリースを塗ったシリコン無反射板にサンプルを振りかける、等の手法でセッティングすることができる。測定サンプルが正極の場合は、正極を基板に両面テープで貼り付け、正極活物質層を装置の要求する測定面に合わせてセッティングすることができる。
O3’型の結晶構造と、単斜晶O1(15)型の結晶構造と、H1−3型結晶構造のモデルから計算される、CuKα1線による理想的な粉末XRDパターンを図8、図9、図10A及び図10Bに示す。また比較のため、LixCoO2中のx=1のLiCoO2 O3と、x=0の三方晶O1の結晶構造から計算される理想的なXRDパターンも示す。図10A及び図10Bは、O3’型の結晶構造、単斜晶O1(15)型の結晶構造とH1−3型の結晶構造のXRDパターンを併記したものであり、図10Aは2θの範囲が18°以上21°以下の領域、図10Bは2θの範囲が42°以上46°以下の領域について拡大したものである。なお、LiCoO2(O3)及びCoO2(O1)のパターンはICSD(Inorganic Crystal Structure Database)より入手した結晶構造情報からMaterials Studio(BIOVIA)のモジュールの一つである、Reflex Powder Diffractionを用いて作成した。2θの範囲は15°から75°とし、Step size=0.01、波長λ1=1.540562×10−10m、λ2は設定なし、Monochromatorはsingleとした。H1−3型結晶構造のパターンは非特許文献1に記載の結晶構造情報から同様に作成した。O3’型及び単斜晶O1(15)型の結晶構造のパターンは本発明の一態様として利用可能な正極活物質のXRDパターンから結晶構造を推定し、TOPAS ver.3(Bruker社製結晶構造解析ソフトウェア)を用いてフィッティングし、他と同様にXRDパターンを作成した。
図8、図10A及び図10Bに示すように、O3’型の結晶構造では、2θ=19.25±0.12°(19.13°以上19.37°未満)、及び2θ=45.47±0.10°(45.37°以上45.57°未満)に回折ピークが出現する。
また、単斜晶O1(15)型の結晶構造では、2θ=19.47±0.10°(19.37°以上19.57°以下)、及び2θ=45.62±0.05°(45.57°以上45.67°以下)に回折ピークが出現する。
一方で、図9、図10A及び図10Bに示すように、H1−3型結晶構造及び三方晶O1ではこれらの位置にピークは出現しない。このため、LixCoO2中のxが小さい状態で19.13以上19.37未満及び/または19.37°以上19.57°以下、並びに45.37°以上45.57°未満及び/または45.57°以上45.67°以下にピークが出現することは、本発明の一態様として利用可能な正極活物質100の特徴であるといえる。
また、x=1と、x≦0.24の結晶構造で、XRDの回折ピークが出現する位置が近いということもできる。より具体的には、x=1と、x≦0.24の結晶構造の主な回折ピークのうち2θが42°以上46°以下に出現するピークについて、2θの差が、0.7°以下、より好ましくは0.5°以下であるということができる。
なお、本発明の一態様として利用可能な正極活物質100はLixCoO2中のxが小さいときO3’型及び/または単斜晶O1(15)型の結晶構造を有するが、粒子の全てがO3’型及び/または単斜晶O1(15)型の結晶構造でなくてもよい。他の結晶構造を含んでいてもよいし、一部が非晶質であってもよい。ただし、XRDパターンについてリートベルト解析を行ったとき、O3’型及び/または単斜晶O1(15)型の結晶構造が50%以上であることが好ましく、60%以上であることがより好ましく、66%以上であることがさらに好ましい。O3’型及び/または単斜晶O1(15)型の結晶構造が50%以上、より好ましくは60%以上、さらに好ましくは66%以上あれば、十分にサイクル特性に優れた正極活物質とすることができる。
また、測定開始から100サイクル以上の充放電を経ても、リートベルト解析を行ったとき、O3’型及び/または単斜晶O1(15)型の結晶構造が35%以上であることが好ましく、40%以上であることがより好ましく、43%以上であることがさらに好ましい。
また、XRDパターンにおける回折ピークの鋭さは、結晶性の高さを示す。そのため、充電後の各回折ピークは鋭い、すなわち半値幅が狭い方が好ましい。半値幅は、同じ結晶相から生じたピークでも、XRDの測定条件または2θの値によっても異なる。上述した測定条件の場合は、2θ=43°以上46°以下に観測されるピークにおいて、半値幅は例えば0.2°以下が好ましく、0.15°以下がより好ましく、0.12°以下がさらに好ましい。なお、必ずしも全てのピークがこの要件を満たしていなくてもよい。一部のピークがこの要件を満たせば、その結晶相の結晶性が高いことがいえる。そのため、十分に充電後の結晶構造の安定化に寄与する。
また、正極活物質100が有するO3’型及び単斜晶O1(15)の結晶構造の結晶子サイズは、放電状態のLiCoO2(O3)の1/20程度までしか低下しない。そのため、充放電前の正極と同じXRDの測定条件であっても、LixCoO2中のxが小さいとき明瞭なO3’型及び単斜晶O1(15)の結晶構造のピークが確認できる。一方、従来のLiCoO2では、一部がO3’型及び単斜晶O1(15)の結晶構造に似た構造を取り得たとしても、結晶子サイズが小さくなり、ピークはブロードで小さくなる。結晶子サイズは、XRDピークの半値幅から求めることができる。
本発明の一態様として利用可能な正極活物質100においては、前述の通りヤーン・テラー効果の影響が小さいことが好ましい。ヤーン・テラー効果の影響が小さい範囲であれば、コバルトの他に添加元素としてニッケル、マンガン等の遷移金属を有してもよい。
正極活物質において、XRD分析を用いて、ヤーン・テラー効果の影響が小さいと推測されるニッケル及びマンガンの割合及び格子定数の範囲について考察する。
図11は、本発明の一態様として利用可能な正極活物質100が層状岩塩型の結晶構造を有し、コバルトとニッケルを有する場合において、XRDを用いてa軸及びc軸の格子定数を算出した結果を示す。図11Aがa軸、図11Bがc軸の結果である。なお、これらの算出に用いたXRDパターンは、正極活物質の合成を行った後の粉体であり、正極に組み込む前のものである。横軸のニッケル濃度は、コバルトとニッケルの原子数の和を100%とした場合のニッケルの濃度を示す。正極活物質は、アルミニウム源を用いない他は、図15A及び図15Cの作製方法に準じて作製した。
図12には、本発明の一態様として利用可能な正極活物質が層状岩塩型の結晶構造を有し、コバルトとマンガンを有する場合において、XRDを用いてa軸及びc軸の格子定数を見積もった結果を示す。図12Aがa軸、図12Bがc軸の結果である。なお、図12に示す格子定数は、正極活物質の合成を行った後の粉体であり、正極に組み込む前に測定したXRDによるものである。横軸のマンガン濃度は、コバルトとマンガンの原子数の和を100%とした場合のマンガンの濃度を示す。正極活物質は、ニッケル源に代えてマンガン源を用い、さらにアルミニウム源を用いない他は、図15A及び図15Cの作製方法に準じて作製した。
図11Cには、図11A及び図11Bに格子定数の結果を示した正極活物質について、a軸の格子定数をc軸の格子定数で割った値(a軸/c軸)を示す。図12Cには、図12A及び図12Bに格子定数の結果を示した正極活物質について、a軸の格子定数をc軸の格子定数で割った値(a軸/c軸)を示す。
図11Cより、ニッケル濃度が5%と7.5%ではa軸/c軸が顕著に変化する傾向がみられ、ニッケル濃度7.5%ではa軸の歪みが大きくなっている。この歪みはヤーン・テラー歪みである可能性がある。ニッケル濃度が7.5%未満において、ヤーン・テラー歪みの小さい、優れた正極活物質が得られることが示唆される。
次に、図12Aより、マンガン濃度が5%以上においては、格子定数の変化の挙動が異なり、ベガード則に従わないことが示唆される。よって、マンガン濃度が5%以上では結晶構造が異なることが示唆される。よって、マンガンの濃度は例えば、4%以下が好ましい。
なお、上記のニッケル濃度及びマンガン濃度の範囲は、表層部100aにおいては必ずしもあてはまらない。すなわち、表層部100aにおいては、上記の濃度より高くてもよい。
以上より、格子定数の好ましい範囲について考察を行ったところ、本発明の一態様として利用可能な正極活物質において、XRDパターンから推定できる、充放電を行わない状態、または放電状態の正極活物質100が有する層状岩塩型の結晶構造において、a軸の格子定数が2.814×10−10mより大きく2.817×10−10mより小さく、かつc軸の格子定数が14.05×10−10mより大きく14.07×10−10mより小さいことが好ましいことがわかった。充放電を行わない状態とは、例えば二次電池の正極を作製する前の粉体の状態であってもよい。
または、充放電を行わない状態、あるいは放電状態の正極活物質100が有する層状岩塩型の結晶構造において、a軸の格子定数をc軸の格子定数で割った値(a軸/c軸)が0.20000より大きく0.20049より小さいことが好ましい。
または、充放電を行わない状態、あるいは放電状態の正極活物質100が有する層状岩塩型の結晶構造において、XRD分析をしたとき、2θが18.50°以上19.30°以下に第1のピークが観測され、かつ2θが38.00°以上38.80°以下に第2のピークが観測される場合がある。
≪XPS≫
X線光電子分光(XPS)では、無機酸化物の場合で、X線源として単色アルミニウムのKα線を用いると、表面から2nm乃至8nm程度(通常5nm以下)の深さまでの領域の分析が可能であり、各元素の濃度を定量的に分析することができる。また、ナロースキャン分析をすれば元素の結合状態を分析することができる。なお、XPSの定量精度は多くの場合±1原子%程度、検出下限は元素にもよるが約1原子%である。
X線光電子分光(XPS)では、無機酸化物の場合で、X線源として単色アルミニウムのKα線を用いると、表面から2nm乃至8nm程度(通常5nm以下)の深さまでの領域の分析が可能であり、各元素の濃度を定量的に分析することができる。また、ナロースキャン分析をすれば元素の結合状態を分析することができる。なお、XPSの定量精度は多くの場合±1原子%程度、検出下限は元素にもよるが約1原子%である。
本発明の一態様として利用可能な正極活物質100は、添加元素から選ばれた一または二以上の濃度が内部100bよりも表層部100aにおいて高いことが好ましい。これは、表層部100aにおける添加元素から選ばれた一または二以上の濃度が、正極活物質100全体の平均の添加元素の濃度よりも高いことが好ましい、と同義である。そのため、例えばXPS等で測定される表層部100aにおける添加元素から選ばれた一または二以上の濃度が、ICP−MS(誘導結合プラズマ質量分析)、あるいはGD−MS(グロー放電質量分析法)等で測定される正極活物質100全体の平均の添加元素の濃度よりも高いことが好ましい、ということができる。例えば、XPS等で測定される表層部100aの少なくとも一部のマグネシウムの濃度が、正極活物質100全体のマグネシウム濃度よりも高いことが好ましい。また表層部100aの少なくとも一部のニッケルの濃度が、正極活物質100全体のニッケル濃度よりも高いことが好ましい。また、表層部100aの少なくとも一部のアルミニウムの濃度が、正極活物質100全体のアルミニウム濃度よりも高いことが好ましい。また、表層部100aの少なくとも一部のフッ素の濃度が、正極活物質100全体のフッ素濃度よりも高いことが好ましい。
なお、本発明の一態様として利用可能な正極活物質100の表面及び表層部100aには、正極活物質100作製後に化学吸着した炭酸塩、ヒドロキシ基等は含まないとする。また、正極活物質100の表面に付着した電解液、バインダ、導電材、またはこれら由来の化合物も含まないとする。そのため、正極活物質が有する元素を定量するときは、XPSをはじめとする表面分析で検出されうる炭素、水素、過剰な酸素、過剰なフッ素等を除外する補正をしてもよい。例えば、XPSでは結合の種類を解析で分離することが可能であり、バインダ由来のC−F結合を除外する補正をおこなってもよい。
さらに各種分析に供する前に、正極活物質の表面に付着した電解液、バインダ、導電材、またはこれら由来の化合物を除くために、正極活物質及び正極活物質層等の試料に対して洗浄等を行ってもよい。このとき洗浄に用いる溶媒等にリチウムが溶け出す場合があるが、たとえその場合であっても、添加元素は溶け出しにくいため、添加元素の原子数比に影響があるものではない。
また、添加元素の濃度は、コバルトとの比で比較してもよい。コバルトとの比を用いることにより、正極活物質を作製後に化学吸着した炭酸塩等の影響を減じて比較することができるため、好ましい。例えば正極活物質の表面または表層部に対するXPSの分析によるマグネシウムとコバルトの原子数の比Mg/Coは、0.4以上1.5以下であることが好ましい。一方、正極活物質全体に対するICP−MSの分析によるMg/Coは0.001以上0.06以下であることが好ましい。
同様に、正極活物質100は、十分にリチウムの挿入脱離の経路を確保するために、表層部100aにおいて各添加元素よりもリチウム及びコバルトの濃度が高いことが好ましい。これは、XPS等で測定される表層部100aが有する添加元素から選ばれた一または二以上の各添加元素の濃度よりも、表層部100aのリチウム及びコバルトの濃度が高いことが好ましい、ということができる。例えば、XPS等で測定される表層部100aの少なくとも一部のマグネシウムの濃度よりも、XPS等で測定される表層部100aの少なくとも一部のコバルトの濃度が高いことが好ましい。同様に、マグネシウムの濃度よりもリチウムの濃度が高いことが好ましい。また、ニッケルの濃度よりもコバルトの濃度が高いことが好ましい。同様に、ニッケルの濃度よりもリチウムの濃度が高いことが好ましい。また、アルミニウムよりもコバルトの濃度が高いことが好ましい。同様に、アルミニウムの濃度よりもリチウムの濃度が高いことが好ましい。また、フッ素よりもコバルトの濃度が高いことが好ましい。同様に、フッ素よりもリチウムの濃度が高いことが好ましい。
さらに、アルミニウムをはじめとする添加元素Yは、深い領域、例えば表面からの深さが5nm以上50nm以内の領域に広く分布する方がより好ましい。そのため、ICP−MS、GD−MS等を用いた正極活物質100全体の分析ではアルミニウムをはじめとする添加元素Yが検出されるものの、XPS等ではこれが検出下限以下であると、より好ましい。
さらに、本発明の一態様として利用可能な正極活物質100の表面または表層部についてXPS分析をしたとき、コバルトの原子数に対して、マグネシウムの原子数は0.4倍以上1.2倍以下が好ましく、0.65倍以上1.0倍以下がより好ましい。またコバルトの原子数に対して、ニッケルの原子数は0.15倍以下が好ましく、0.03倍以上0.13倍以下がより好ましい。またコバルトの原子数に対して、アルミニウムの原子数は0.12倍以下が好ましく、0.09倍以下がより好ましい。またコバルトの原子数に対して、フッ素の原子数は0.3倍以上0.9倍以下が好ましく、0.1倍以上1.1倍以下がより好ましい。
XPS分析を行う場合には、例えば、X線源として単色化アルミニウムKα 線を用いることができる。また、取出角は例えば45°とすればよい。例えば下記の装置及び条件で測定することができる。
測定装置 :PHI 社製QuanteraII
X線源 :単色化Al Kα(1486.6eV)
検出領域 :100μmφ
検出深さ :約4~5nm(取出角45°)
測定スペクトル :ワイドスキャン,各検出元素のナロースキャン
測定装置 :PHI 社製QuanteraII
X線源 :単色化Al Kα(1486.6eV)
検出領域 :100μmφ
検出深さ :約4~5nm(取出角45°)
測定スペクトル :ワイドスキャン,各検出元素のナロースキャン
また、本発明の一態様として利用可能な正極活物質100の表面または表層部についてXPS分析したとき、フッ素と他の元素の結合エネルギーを示すピークは682eV以上685eV未満であることが好ましく、684.3eV程度であることがさらに好ましい。これは、フッ化リチウムの結合エネルギーである685eV、及びフッ化マグネシウムの結合エネルギーである686eVのいずれとも異なる値である。つまり、本発明の一態様として利用可能な正極活物質100がフッ素を有する場合、フッ化リチウム及びフッ化マグネシウム以外の結合であることが好ましい。
さらに、本発明の一態様として利用可能な正極活物質100の表面または表層部についてXPS分析したとき、マグネシウムと他の元素の結合エネルギーを示すピークは、1302eV以上1304eV未満であることが好ましく、1303eV程度であることがさらに好ましい。これは、フッ化マグネシウムの結合エネルギーである1305eVと異なる値であり、酸化マグネシウムの結合エネルギーに近い値である。つまり、本発明の一態様として利用可能な正極活物質100がマグネシウムを有する場合、フッ化マグネシウム以外の結合であることが好ましい。
≪EDX≫
正極活物質100が有する添加元素から選ばれた一または二以上は濃度勾配を有していることが好ましい。また、正極活物質100は添加元素によって、濃度ピークの表面からの深さが異なっていることがより好ましい。添加元素の濃度勾配は、例えばFIB(Focused Ion Beam)等により正極活物質100の断面を露出させ、その断面をエネルギー分散型X線分光法(EDX:Energy Dispersive X−ray Spectroscopy)、EPMA(電子プローブ微小分析)等を用いて分析することで評価できる。
正極活物質100が有する添加元素から選ばれた一または二以上は濃度勾配を有していることが好ましい。また、正極活物質100は添加元素によって、濃度ピークの表面からの深さが異なっていることがより好ましい。添加元素の濃度勾配は、例えばFIB(Focused Ion Beam)等により正極活物質100の断面を露出させ、その断面をエネルギー分散型X線分光法(EDX:Energy Dispersive X−ray Spectroscopy)、EPMA(電子プローブ微小分析)等を用いて分析することで評価できる。
EDX測定のうち、領域内を走査しながら測定し、領域内を2次元に評価することをEDX面分析と呼ぶ。また線状に走査しながら測定し、原子濃度について正極活物質内の分布を評価することを線分析と呼ぶ。さらに、EDXの面分析から線状の領域のデータを抽出したものを線分析と呼ぶ場合もある。また、ある領域について走査せずに測定することを点分析と呼ぶ。
EDX面分析(例えば元素マッピング)により、正極活物質100の表層部100a、内部100b及び結晶粒界101近傍等における、添加元素の濃度を定量的に分析することができる。また、EDX線分析により、添加元素の濃度分布及び最大値を分析することができる。また、STEM−EDXのようにサンプルを薄片化する分析は、奥行き方向の分布の影響を受けずに、特定の領域における正極活物質の表面から中心に向かった深さ方向の濃度分布を分析でき、より好適である。
そのため、本発明の一態様として利用可能な正極活物質100についてEDX面分析またはEDX点分析したとき、表層部100aの各添加元素、特に添加元素Xの濃度が、内部100bのそれよりも高いことが好ましい。
例えば、添加元素としてマグネシウムを有する正極活物質100についてEDX面分析またはEDX点分析したとき、表層部100aのマグネシウム濃度が、内部100bのマグネシウム濃度よりも高いことが好ましい。また、EDX線分析をしたとき、表層部100aのマグネシウム濃度のピークは、正極活物質100の表面から中心に向かった深さ3nmまでに存在することが好ましく、深さ1nmまでに存在することがより好ましく、深さ0.5nmまでに存在することがさらに好ましい。また、マグネシウムの濃度はピークトップから深さ1nmの点でピークの60%以下に減衰することが好ましい。またピークトップから深さ2nmの点でピークの30%以下に減衰することが好ましい。なお、ここでいう濃度のピークとは、濃度の極大値をいうこととする。
また、添加元素としてマグネシウム及びフッ素を有する正極活物質100では、フッ素の分布は、マグネシウムの分布と重畳することが好ましい。例えばフッ素濃度のピークと、マグネシウム濃度のピークの深さ方向の差が10nm以内であると好ましく、3nm以内であるとより好ましく、1nm以内であるとさらに好ましい。
また、EDX線分析をしたとき、表層部100aのフッ素濃度のピークは、正極活物質100の表面から中心に向かった深さ3nmまでに存在することが好ましく、深さ1nmまでに存在することがより好ましく、深さ0.5nmまでに存在することがさらに好ましい。またフッ素濃度のピークはマグネシウムの濃度のピークよりもわずかに表面側に存在すると、フッ酸への耐性が増してより好ましい。例えばフッ素濃度のピークはマグネシウムの濃度のピークよりも0.5nm以上表面側であるとより好ましく、1.5nm以上表面側であるとさらに好ましい。
また、添加元素としてニッケルを有する正極活物質100では、表層部100aのニッケル濃度のピークは、正極活物質100の表面から中心に向かった深さ3nmまでに存在することが好ましく、深さ1nmまでに存在することがより好ましく、深さ0.5nmまでに存在することがさらに好ましい。またマグネシウム及びニッケルを有する正極活物質100では、ニッケルの分布は、マグネシウムの分布と重畳することが好ましい。例えばニッケル濃度のピークと、マグネシウム濃度のピークの深さ方向の差が10nm以内であると好ましく、3nm以内であるとより好ましく、1nm以内であるとさらに好ましい。
また、正極活物質100が添加元素としてアルミニウムを有する場合は、EDX線分析をしたとき、表層部100aのアルミニウム濃度のピークよりも、マグネシウム、ニッケルまたはフッ素の濃度のピークが表面に近いことが好ましい。例えばアルミニウム濃度のピークは正極活物質100の表面から中心に向かった深さ0.5nm以上50nm以下に存在することが好ましく、深さ5nm以上50nm以下に存在することがより好ましい。
また、正極活物質100の表面または表層部についてEDX線分析、面分析または点分析をしたとき、マグネシウム濃度のピークにおけるマグネシウムMgとコバルトCoの原子数の比(Mg/Co)は0.05以上0.6以下が好ましく、0.1以上0.4以下がより好ましい。アルミニウム濃度のピークにおけるアルミニウムAlとコバルトCoの原子数の比(Al/Co)は0.05以上0.6以下が好ましく、0.1以上0.45以下がより好ましい。ニッケル濃度のピークにおけるニッケルNiとコバルトCoの原子数の比(Ni/Co)は0以上0.2以下が好ましく、0.01以上0.1以下がより好ましい。フッ素濃度のピークにおけるフッ素FとコバルトCoの原子数の比(F/Co)は0以上1.6以下が好ましく、0.1以上1.4以下がより好ましい。
なお、EDX線分析結果における正極活物質100の表面は、例えば以下のように推定することができる。正極活物質100の内部100bにおいて均一に存在する元素、例えば酸素またはコバルトについて、内部100bの検出量の1/2となった点を表面とする。
正極活物質100は複合酸化物であるので、酸素の検出量を用いて表面を推定することができる。具体的には、まず内部100bの酸素の検出量が安定している領域から酸素濃度の平均値Oaveを求める。このとき明らかに表面より外と判断できる領域に化学吸着またはバックグラウンドによると考えられる酸素Obackgroundが検出される場合は、測定値からObackgroundを減じてから、酸素濃度の平均値Oaveとすることができる。この平均値Oaveの1/2の値、つまり1/2Oaveに最も近い測定値を示した測定点を、正極活物質の表面であると推定することができる。
また、コバルトの検出量を用いても上記と同様に表面を推定することができる。または複数の遷移金属の検出量の和を用いて同様に推定することもできる。コバルトをはじめとする遷移金属の検出量は、化学吸着の影響を受けにくい点で、表面の推定に好適である。
また、正極活物質100の表面または表層部について線分析または面分析をしたとき、結晶粒界101近傍における添加元素AとコバルトCoの比(A/Co)は0.020以上0.50以下が好ましく、0.025以上0.30以下がより好ましく、0.030以上0.20以下がさらに好ましい。なお、これらの上限と下限の値は、本明細書において断りが無い限り、自由に組み合わせが可能である。
例えば、添加元素がマグネシウムのとき、正極活物質100の表面または表層部について線分析または面分析をしたとき、結晶粒界101近傍におけるマグネシウムとコバルトの原子数の比(Mg/Co)は、0.020以上0.50以下が好ましく、0.025以上0.30以下がより好ましく、0.030以上0.20以下がさらに好ましい。
≪EPMA≫
EPMA(電子プローブ微小分析)も元素の定量が可能である。面分析ならば各元素の分布を分析することができる。
EPMA(電子プローブ微小分析)も元素の定量が可能である。面分析ならば各元素の分布を分析することができる。
本発明の一態様として利用可能な正極活物質100の断面についてEPMA面分析をしたとき、EDXの分析結果と同様に、添加元素から選ばれた一または二以上は濃度勾配を有していることが好ましい。また添加元素によって、濃度ピークの表面からの深さが異なっていることがより好ましい。各添加元素の濃度ピークの好ましい範囲も、EDXの場合と同様である。
ただし、EPMAでは表面から1μm程度の深さまでの領域を分析する。そのため、各元素の定量値が他の分析法を用いた測定結果と異なる場合がある。例えば、正極活物質100の表面分析をEPMAで行ったとき、表層部100aに存在する各添加元素の濃度が、XPSの結果より低くなる場合がある。
≪充電曲線と、電圧Vに対するdQ/dV曲線≫
本発明の一態様として利用可能な正極活物質100は、充電していくとき特徴的な電圧の変化が表れることがある。電圧の変化は、充電曲線の容量(Q)を電圧(V)で微分(dQ/dV)することで得られるdQ/dVvsV曲線から読み取ることができる。例えばdQ/dVvsV曲線におけるピークの前後では、非平衡な相変化が起き、結晶構造が大きく変わっていると考えられる。なお、本明細書等において、非平衡な相変化とは、物理量の非線形変化を起こす現象をいうこととする。
本発明の一態様として利用可能な正極活物質100は、充電していくとき特徴的な電圧の変化が表れることがある。電圧の変化は、充電曲線の容量(Q)を電圧(V)で微分(dQ/dV)することで得られるdQ/dVvsV曲線から読み取ることができる。例えばdQ/dVvsV曲線におけるピークの前後では、非平衡な相変化が起き、結晶構造が大きく変わっていると考えられる。なお、本明細書等において、非平衡な相変化とは、物理量の非線形変化を起こす現象をいうこととする。
本発明の一態様として利用可能な正極活物質100は、dQ/dVvsV曲線において、4.55V付近にブロードなピークを有する場合がある。4.55V付近のピークは、O3型からO3’型へと相変化する際の電圧の変化を反映している。そのため、このピークがブロードであることは、ピークが鋭い場合よりもリチウムが引き抜かれるのに必要なエネルギーの変化が少ない、すなわち結晶構造の変化が少ないことを意味する。これらの変化は少ない方が、CoO2層のずれ及び体積の変化の影響が少なく、好ましい。
より具体的には、充電曲線のdQ/dVvsV曲線において、4.5V以上4.6V以下に現れる最大値を第1のピークとしたとき、第1のピークの半値幅が0.10V以上であると、十分にブロードであるといえ、好ましい。本明細書等において、第1のピークの半値幅は、4.3V以上4.5V以下に現れるdQ/dV値の最小値を第1の最小値としたときの、第1のピークと第1の最小値との平均値HWHM1と、4.6V以上4.8V以下に現れるdQ/dV値の最小値を第2の最小値としたときの第1のピークと第2の最小値との平均値HWHM2と、の差とする。
dQ/dVvsV曲線を取得する際の充電は、例えば4.9Vまで10mA/gで定電流充電とすることができる。また、初回充電のdQ/dVを取得するときは、測定前に100mA/gで2.5Vまで放電したのちに上記充電を開始することが好ましい。
充電時のデータ取り込み間隔の設定は、例えば1秒間隔または1mVの電圧変動があったときの電圧及び電流を取り込む設定とすることができる。電流値と時間を積算した値を充電容量とする。
上記充電容量のデータの、n番目とn+1番目データの差分を、容量の変化dQのn番目の値とする。同様に上記電圧データの、n番目とn+1番目データの差分を、電圧の変化dVのn番目の値とする。
ただし、上記のデータを用いると微細なノイズの影響が大きいため、電圧及び充電容量の差分について、ある区間数の移動平均からdQ/dVを求めてもよい。区間数は例えば500とすることができる。
具体的には、dQのn番目からn+500番目までの平均値を算出し、同様にdVのn番目からn+500番目までの平均値を算出する。dQ(500個平均)/dV(500個平均)を、dQ/dVとすることができる。dQ/dVvsVグラフにおける横軸の電圧も、同じように区間数500の移動平均の値を用いることができる。なお上記のような区間数500の移動平均を用いる場合は、501番目以降のデータはノイズの影響が大きくなるため、dQ/dVvsVグラフには用いないことが好ましい。
また、複数回充放電した後のdQ/dVvsV曲線を分析する場合、該複数回の充放電条件は上記の充電条件と異なっていてもよい。例えば充電は任意の電圧(例えば4.6V、4.65V、4.7V、4.75Vまたは4.8V)、100mA/gで定電流充電し、電流値が10mA/gとなるまで定電圧充電し、放電は2.5V、100mA/gで定電流放電とすることができる。
なお、4.55V付近においてO3型からO3’型へと相変化するが、このときのO3型はLixCoO2中のxが0.3程度である。これは図5で説明したx=1のO3型と同じ対称性を有するが、CoO2層間の距離は若干異なる。本明細書等において、xの大きさの異なるO3型を区別する場合、x=1のO3型をO3(2θ=18.85)、x=0.3程度のO3型をO3(2θ=18.57)ということとする。これは、XRD測定において2θが19°付近に現れるピークの位置が、CoO2層間距離と対応するためである。
≪放電曲線とdQ/dVvsV曲線≫
また、本発明の一態様として利用可能な正極活物質100は、高電圧で充電した後、例えば40mA/g以下の低い電流で放電すると、放電終了間近に特徴的な電圧の変化が表れることがある。この変化は、放電曲線から求めたdQ/dVvsV曲線において、3.9V前後に出現するピークよりも低電圧で、3.5Vまでの範囲に、少なくとも1つのピークが存在することで明瞭に確かめることができる。
また、本発明の一態様として利用可能な正極活物質100は、高電圧で充電した後、例えば40mA/g以下の低い電流で放電すると、放電終了間近に特徴的な電圧の変化が表れることがある。この変化は、放電曲線から求めたdQ/dVvsV曲線において、3.9V前後に出現するピークよりも低電圧で、3.5Vまでの範囲に、少なくとも1つのピークが存在することで明瞭に確かめることができる。
≪ESR≫
本発明の一態様として利用可能な正極活物質100は、コバルトを有し、添加元素としてニッケル及びマグネシウムを有することが好ましい。その結果、一部のCo3+がNi3+に置換され、また一部のLi+がMg2+に置換されることが好ましい。Li+がMg2+に置換されることに伴い、当該Ni3+は還元されて、Ni2+になることがある。また、一部のLi+がMg2+に置換され、それに伴いMg2+近傍のCo3+が還元されてCo2+になる場合がある。また、一部のCo3+がMg2+に置換され、それに伴いMg2+近傍のCo3+が酸化されてCo4+になる場合がある。
本発明の一態様として利用可能な正極活物質100は、コバルトを有し、添加元素としてニッケル及びマグネシウムを有することが好ましい。その結果、一部のCo3+がNi3+に置換され、また一部のLi+がMg2+に置換されることが好ましい。Li+がMg2+に置換されることに伴い、当該Ni3+は還元されて、Ni2+になることがある。また、一部のLi+がMg2+に置換され、それに伴いMg2+近傍のCo3+が還元されてCo2+になる場合がある。また、一部のCo3+がMg2+に置換され、それに伴いMg2+近傍のCo3+が酸化されてCo4+になる場合がある。
したがって正極活物質100は、Ni2+、Ni3+、Co2+及びCo4+のいずれか一以上を有することが好ましい。また、正極活物質100の重量当たりのNi2+、Ni3+、Co2+及びCo4+のいずれか一以上に起因するスピン密度が、2.0×1017spins/g以上1.0×1021spins/g以下であることが好ましい。前述のスピン密度を有する正極活物質100とすることで、特に充電状態での結晶構造が安定となり好ましい。なお、マグネシウム濃度が高すぎると、Ni2+、Ni3+、Co2+及びCo4+のいずれか一以上に起因するスピン密度が低くなる場合がある。
正極活物質中のスピン密度は、例えば、電子スピン共鳴法(ESR:Electron Spin Resonance)などを用いて分析することができる。
≪表面粗さと比表面積≫
本発明の一態様として利用可能な正極活物質100は、表面がなめらかで凹凸が少ないことが好ましい。表面がなめらかで凹凸が少ないことは、後述する融剤の効果が十分に発揮されて、添加元素源とコバルト酸リチウムの表面が溶融(固溶)したことを示す。そのため表層部100aにおける添加元素の分布が良好であることを示す一つの要素である。
本発明の一態様として利用可能な正極活物質100は、表面がなめらかで凹凸が少ないことが好ましい。表面がなめらかで凹凸が少ないことは、後述する融剤の効果が十分に発揮されて、添加元素源とコバルト酸リチウムの表面が溶融(固溶)したことを示す。そのため表層部100aにおける添加元素の分布が良好であることを示す一つの要素である。
表面がなめらかで凹凸が少ないことは、例えば正極活物質100の断面SEM像または断面TEM像、正極活物質100の比表面積等から判断することができる。
例えば、以下のように正極活物質100の断面SEM像から表面のなめらかさを数値化することができる。
まず、正極活物質100をFIB等により加工して断面を露出させる。このとき保護膜、保護剤等で正極活物質100を覆うことが好ましい。次に保護膜等と正極活物質100との界面のSEM像を撮影する。該SEM像に画像処理ソフトでノイズ処理を行う。例えばガウスぼかし(σ=2)を行った後、二値化を行う。さらに画像処理ソフトで界面抽出を行う。さらに自動選択ツール等で保護膜等と正極活物質100との界面ラインを選択し、データを表計算ソフト等に抽出する。表計算ソフト等の機能を用いて、回帰曲線(二次回帰)から補正を行い、傾き補正後データからラフネス算出用パラメータを求め、標準偏差を算出した二乗平均平方根表面粗さ(RMS)を求める。また、この表面粗さは、正極活物質は少なくとも粒子外周の400nmにおける表面粗さである。
本実施の形態の正極活物質100の粒子表面においては、ラフネスの指標である二乗平均平方根(RMS)表面粗さは3nm未満、好ましくは1nm未満、さらに好ましくは0.5nm未満の二乗平均平方根(RMS)表面粗さであることが好ましい。
なお、ノイズ処理、界面抽出等を行う画像処理ソフトについては特に限定されないが、例えば「ImageJ」を用いることができる。また表計算ソフト等についても特に限定されないが、例えばMicrosoft Office Excelを用いることができる。
また、例えば、定容法によるガス吸着法にて測定した実際の比表面積SRと、理想的な比表面積Siとの比からも、正極活物質100の表面のなめらかさを数値化することができる。
理想的な比表面積Siは、全ての正極活物質の粒子の直径がD50と同じであり、重量が同じであり、形状は理想的な球であるとして計算して求める。
メディアン径D50は、レーザ回折・散乱法を用いた粒度分布計等によって測定することができる。比表面積は、例えば定容法によるガス吸着法を用いた比表面積測定装置等によって測定することができる。
本発明の一態様として利用可能な正極活物質100は、メディアン径D50から求めた理想的な比表面積Siと、実際の比表面積SRの比SR/Siが2.1以下であることが好ましい。
または、下記のような方法によっても正極活物質100の断面SEM像から表面のなめらかさを数値化することができる。
まず、正極活物質100の表面SEM像を取得する。このとき観察前処理として導電性コーティングを施してもよい。観察面は電子線と垂直であることが好ましい。複数のサンプルを比較する場合は測定条件及び観察面積を同じとする。
次に、画像処理ソフト(例えば「ImageJ」)を用いて上記のSEM像を例えば8ビットに変換した画像(これをグレースケール画像と呼ぶ)を取得する。グレースケール画像は輝度(明るさ情報)を含んでいる。例えば8ビットのグレースケール画像では、輝度を2の8乗=256階調で表すことができる。暗い部分は階調数が低くなり、明るい部分は階調数が高くなる。階調数と関連付けて輝度変化を数値化することができる。当該数値をグレースケール値と呼ぶ。グレースケール値を取得することで正極活物質の凹凸を数値として評価することが可能となる。
さらに、対象領域の輝度変化をヒストグラムで表すことも可能となる。ヒストグラムとは対象領域における階調分布を立体的に示したもので、輝度ヒストグラムとも呼ぶ。輝度ヒストグラムを取得することで正極活物質の凹凸を視覚的にわかりやすく、評価することが可能となる。
本発明の一態様として利用可能な正極活物質100は、上記グレースケール値の最大値と最小値との差が120以下であることが好ましく、115以下であることがより好ましく、70以上115以下であることがさらに好ましい。またグレースケール値の標準偏差は、11以下となることが好ましく、8以下であることがより好ましく、4以上8以下であることがさらに好ましい。
<その他の特徴>
正極活物質100は凹部、クラック、窪み、断面V字形などを有する場合がある。これらは欠陥の一つであり、充放電を繰り返すとこれらからコバルトの溶出、結晶構造の崩れ、本体の割れ、酸素の脱離などが生じるおそれがある。そこで、図1A2に示すような添加元素を含む埋め込み部102を設けることで、コバルトの溶出などを抑制できる。そのため、信頼性及びサイクル特性の優れた正極活物質100とすることができる。
正極活物質100は凹部、クラック、窪み、断面V字形などを有する場合がある。これらは欠陥の一つであり、充放電を繰り返すとこれらからコバルトの溶出、結晶構造の崩れ、本体の割れ、酸素の脱離などが生じるおそれがある。そこで、図1A2に示すような添加元素を含む埋め込み部102を設けることで、コバルトの溶出などを抑制できる。そのため、信頼性及びサイクル特性の優れた正極活物質100とすることができる。
また、正極活物質100は添加元素が偏在する領域として凸部103を有していてもよい。
上述したように、正極活物質100が有する添加元素は、過剰であるとリチウムの挿入及び脱離に悪影響が出る恐れがある。また二次電池としたときに内部抵抗の上昇、充放電容量の低下等を招く恐れもある。一方、添加元素が不足していると表層部100a全体に分布せず、結晶構造の劣化を抑制する効果が不十分になる恐れがある。このように添加元素は、正極活物質100において適切な濃度である必要があるが、その調整は容易ではない。
そのため、正極活物質100が、添加元素が偏在する領域を有していると、過剰な添加元素の一部が正極活物質100の内部100bから除かれ、内部100bにおいて適切な添加元素濃度とすることができる。これにより、二次電池としたときの内部抵抗の上昇、充放電容量の低下等を抑制することができる。二次電池の内部抵抗の上昇を抑制できることは、特に大電流での充放電、例えば400mA/g以上での充放電において極めて好ましい特性である。
また、添加元素が偏在している領域を有する正極活物質100では、作製工程においてある程度過剰に添加元素を混合することが許容され、生産におけるマージンが広くなり好ましい。
また、正極活物質100は、表面の少なくとも一部に被膜を有していてもよい。図13A及び図13Bに被膜104を有する正極活物質100の例を示す。
被膜104は、例えば充放電に伴い電解液の分解物が堆積して形成されたものであることが好ましい。特にLixCoO2中のxが0.24以下となるような充電を繰り返す場合、正極活物質100の表面に電解液由来の被膜を有することで、充放電サイクル特性が向上することが期待される。これは正極活物質表面のインピーダンスの上昇を抑制する、またはコバルトの溶出を抑制する、等の理由による。被膜104は、例えば炭素、酸素及びフッ素を有することが好ましい。さらに電解液の一部にLiBOB、及び/またはSUN(スベロニトリル)を用いた場合などは良質な被膜を得られやすい。そのため、ホウ素、窒素、硫黄及びフッ素から選ばれた一または二以上を有する被膜104は良質な被膜である場合があり好ましい。また被膜104は正極活物質100の全てを覆っていなくてもよい。
また、正極活物質は、4.5V以上で充電するような条件、または高温、例えば45℃以上の環境で充放電することにより、表面から内部に向かって深くまで進む進行性の欠陥が生じる場合がある。正極活物質において欠陥が進行して穴を形成する現象を孔食(Pitting Corrosion)とも呼ぶことができ、この現象で発生した穴を本明細書等ではピットとも呼ぶ。
図14にピットを有する正極活物質51の断面模式図を示す。陽イオンの配列と平行な結晶面55を併せて示した。図14は断面図であるためピット54及びピット58を穴として示しているが、これらの開口形状は円ではなく奥行きがあり溝のような形状を有する。また、ピット54及びピット58に示すように、凹部52と異なりリチウムイオンの配列と平行に生じやすい。
また、正極活物質51のうち添加元素の存在する表層部を53及び56で示す。ピットが生じた表層部は添加元素が53及び56よりも少ないか検出下限以下であり、バリア膜の機能が減じていると予想される。またピットができる近傍ではコバルト酸リチウムの結晶構造が崩れ、層状岩塩型とは異なった結晶構造になると考えられる。結晶構造が崩れるとキャリアイオンであるリチウムイオンの拡散及び放出を阻害するため、ピットはサイクル特性劣化の要因と考えられる。
ピットの発生源は、点欠陥の可能性がある。正極活物質が有する点欠陥が充放電を繰り返すことで変化し、周囲の電解質等によって化学的または電気化学的に侵食されるか、または材質が劣化してピットが生じると考えられる。この劣化は、正極活物質の表面で均一に発生するのではなく、局部的に集中して生じる。
また、図14のクラック57に示すように、充放電による正極活物質の膨張及び収縮により、クラック(割れ目とも呼ぶ)などの欠陥が発生する場合もある。本明細書等において、クラックとピットは異なる。正極活物質の作製直後にクラックは存在してもピットは存在しない。ピットは、例えば4.5V以上の高電圧条件または高温(45℃以上)下で充放電することにより、コバルト及び酸素が何層分か抜けた穴とも言え、コバルトが溶出した箇所ともいえる。クラックは例えば物理的な圧力が加えられることで生じる新たな面、或いは結晶粒界101が起因となって生じた割れ目を指す。充放電による正極活物質の膨張及び収縮によりクラックが発生する場合もある。また、クラック及び/または正極活物質内部の空洞からピットが発生する場合もある。
<正極活物質の作製方法の例1>
図15A乃至図15Cを用いて、本発明の一態様として利用可能な正極活物質の作製方法の一例(正極活物質の作製方法の例1)について説明する。なお、ここで説明する作製方法は、本実施の形態で先に説明した特徴を有する正極活物質100の作製方法の一例である。
図15A乃至図15Cを用いて、本発明の一態様として利用可能な正極活物質の作製方法の一例(正極活物質の作製方法の例1)について説明する。なお、ここで説明する作製方法は、本実施の形態で先に説明した特徴を有する正極活物質100の作製方法の一例である。
<ステップS11>
図15Aに示すステップS11では、出発材料であるリチウム及び遷移金属の材料として、それぞれリチウム源(Li源)及びコバルト源(Co源)を準備する。
図15Aに示すステップS11では、出発材料であるリチウム及び遷移金属の材料として、それぞれリチウム源(Li源)及びコバルト源(Co源)を準備する。
リチウム源としては、リチウムを有する化合物を用いると好ましく、例えば炭酸リチウム、水酸化リチウム、硝酸リチウム、又はフッ化リチウム等を用いることができる。リチウム源は純度が高いと好ましく、例えば純度が99.99%以上の材料を用いるとよい。
コバルト源としては、コバルトを有する化合物を用いると好ましく、例えば酸化コバルト、水酸化コバルト等を用いることができる。コバルト源は純度が高いと好ましく、例えば純度が3N(99.9%)以上、好ましくは4N(99.99%)以上、より好ましくは4N5(99.995%)以上、さらに好ましくは5N(99.999%)以上の材料を用いるとよい。高純度の材料を用いることで、正極活物質の不純物を制御することができる。その結果、二次電池の容量が高まり、及び/または二次電池の信頼性が向上する。
加えて、コバルト源は結晶性が高いと好ましく、例えば単結晶粒を有するとよい。遷移金属源の結晶性の評価としては、TEM(透過電子顕微鏡)像、STEM(走査透過電子顕微鏡)像、HAADF−STEM(高角散乱環状暗視野走査透過電子顕微鏡)像、ABF−STEM(環状明視野走査透過電子顕微鏡)像等による判断、またはX線回折(XRD)、電子線回折、中性子線回折等の判断がある。なお、上記の結晶性の評価に関する手法は、遷移金属源だけではなく、その他の結晶性の評価にも適用することができる。
<ステップS12>
次に、図15Aに示すステップS12として、リチウム源及びコバルト源を粉砕及び混合して、混合材料を作製する。粉砕及び混合は、乾式または湿式で行うことができる。湿式はより小さく解砕することができるため好ましい。湿式で行う場合は、溶媒を準備する。溶媒としてはアセトン等のケトン、エタノール及びイソプロパノール等のアルコール、エーテル、ジオキサン、アセトニトリル、N−メチル−2−ピロリドン(NMP)等を用いることができるが、リチウムと反応が起こりにくい、非プロトン性溶媒を用いることが好ましい。本実施の形態では、純度が99.5%以上の脱水アセトンを用いることとする。水分含有量を10ppm以下まで抑えた、純度が99.5%以上の脱水アセトンにリチウム源及び遷移金属源を混合して、粉砕及び混合を行うと好適である。上記のような純度の脱水アセトンを用いることで、混入しうる不純物を低減できる。
次に、図15Aに示すステップS12として、リチウム源及びコバルト源を粉砕及び混合して、混合材料を作製する。粉砕及び混合は、乾式または湿式で行うことができる。湿式はより小さく解砕することができるため好ましい。湿式で行う場合は、溶媒を準備する。溶媒としてはアセトン等のケトン、エタノール及びイソプロパノール等のアルコール、エーテル、ジオキサン、アセトニトリル、N−メチル−2−ピロリドン(NMP)等を用いることができるが、リチウムと反応が起こりにくい、非プロトン性溶媒を用いることが好ましい。本実施の形態では、純度が99.5%以上の脱水アセトンを用いることとする。水分含有量を10ppm以下まで抑えた、純度が99.5%以上の脱水アセトンにリチウム源及び遷移金属源を混合して、粉砕及び混合を行うと好適である。上記のような純度の脱水アセトンを用いることで、混入しうる不純物を低減できる。
混合等の手段には、ボールミルまたはビーズミル等を用いることができる。ボールミルを用いる場合は、粉砕メディアとして酸化アルミニウムボール又は酸化ジルコニウムボールを用いるとよい。酸化ジルコニウムボールは、不純物の排出が少なく好ましい。また、ボールミルまたはビーズミル等を用いる場合、メディアからのコンタミネーションを抑制するために、周速を100mm/s以上2000mm/s以下とするとよい。本実施の形態では、周速838mm/s(回転数400rpm、ボールミルの直径40mm)として実施する。
<ステップS13>
次に、図15Aに示すステップS13として、上記の混合材料を加熱する。加熱は、800℃以上1100℃以下で行うことが好ましく、900℃以上1000℃以下で行うことがより好ましく、950℃程度1000℃以下がさらに好ましい。温度が低すぎると、リチウム源及び遷移金属源の分解及び溶融が不十分となるおそれがある。一方、温度が高すぎると、リチウム源からリチウムが蒸散する、及び/またはコバルトが過剰に還元される、などが原因となり、欠陥が生じるおそれがある。例えばコバルトが3価から2価へ変化し、酸素欠陥などを誘発することがある。
次に、図15Aに示すステップS13として、上記の混合材料を加熱する。加熱は、800℃以上1100℃以下で行うことが好ましく、900℃以上1000℃以下で行うことがより好ましく、950℃程度1000℃以下がさらに好ましい。温度が低すぎると、リチウム源及び遷移金属源の分解及び溶融が不十分となるおそれがある。一方、温度が高すぎると、リチウム源からリチウムが蒸散する、及び/またはコバルトが過剰に還元される、などが原因となり、欠陥が生じるおそれがある。例えばコバルトが3価から2価へ変化し、酸素欠陥などを誘発することがある。
加熱時間は短すぎるとコバルト酸リチウムが合成されないが、長すぎると生産性が低下する。このため、加熱時間は1時間以上100時間以下とすればよく、2時間以上20時間以下とすることがさらに好ましい。
昇温レートは、加熱温度の到達温度によるが、80℃/h以上250℃/h以下がよい。例えば1000℃で10時間加熱する場合、昇温レートは200℃/hとするとよい。
加熱は、乾燥空気等の水が少ない雰囲気で行うことが好ましく、例えば露点が−50℃以下、より好ましくは露点が−80℃以下の雰囲気がよい。本実施の形態においては、露点−93℃の雰囲気にて、加熱を行うこととする。また材料中に混入しうる不純物を抑制するためには、加熱雰囲気におけるCH4、CO、CO2、及びH2等の不純物濃度が、それぞれ5ppb(parts per billion)以下にするとよい。
加熱雰囲気として、酸素を有する雰囲気が好ましい。例えば反応室に乾燥空気を導入し続ける方法がある。この場合、乾燥空気の流量は10L/minとすることが好ましい。酸素を反応室へ導入し続け、酸素が反応室内を流れている方法をフローと呼ぶ。
加熱雰囲気を、酸素を有する雰囲気とする場合、フローさせないやり方でもよい。例えば反応室を減圧してから酸素を充填し、当該酸素が反応室から出入りしないようにする方法でもよく、これをパージと呼ぶ。例えば反応室を−970hPaまで減圧してから、50hPaまで酸素を充填すればよい。
加熱後の冷却は自然放冷でよいが、規定温度から室温までの降温時間が10時間以上50時間以下に収まると好ましい。ただし、必ずしも室温までの冷却は要せず、次のステップが許容する温度まで冷却されればよい。
本工程の加熱は、ロータリーキルン又はローラーハースキルンによる加熱を行ってもよい。ロータリーキルンによる加熱は、連続式、バッチ式いずれの場合でも攪拌しながら加熱することができる。
加熱の際に用いる、るつぼは酸化アルミニウム製のるつぼが好ましい。酸化アルミニウム製のるつぼは、不純物が混入しにくい材質である。本実施の形態においては、純度が99.9%の酸化アルミニウムのるつぼを用いる。また、るつぼは蓋を配してから加熱すると、材料の揮発を防ぐことができるため、好ましい。
加熱が終わった後、必要に応じて粉砕し、さらにふるいを実施してもよい。加熱後の材料を回収する際に、るつぼから乳鉢へ移動させたのち回収してもよい。また、当該乳鉢は酸化アルミニウムの乳鉢を用いると好適である。酸化アルミニウムの乳鉢は不純物が混入しにくい材質である。具体的には、純度が90%以上、好ましくは純度が99%以上の酸化アルミニウムの乳鉢を用いる。なお、ステップS13以外の後述の加熱の工程においても、ステップS13と同等の加熱条件を適用できる。
<ステップS14>
以上の工程により、図15Aに示すステップS14で示すコバルト酸リチウム(LiCoO2)を合成することができる。
以上の工程により、図15Aに示すステップS14で示すコバルト酸リチウム(LiCoO2)を合成することができる。
ステップS11乃至ステップS14のように固相法で複合酸化物を作製する例を示したが、共沈法で複合酸化物を作製してもよい。また、水熱法で複合酸化物を作製してもよい。
<ステップS15>
次に、図15Aに示すステップS15としてコバルト酸リチウムを加熱する。コバルト酸リチウムに対する最初の加熱のため、ステップS15の加熱を初期加熱と呼ぶことがある。または、以下に示すステップS20の前に加熱するものであるため、予備加熱又は前処理と呼ぶことがある。
次に、図15Aに示すステップS15としてコバルト酸リチウムを加熱する。コバルト酸リチウムに対する最初の加熱のため、ステップS15の加熱を初期加熱と呼ぶことがある。または、以下に示すステップS20の前に加熱するものであるため、予備加熱又は前処理と呼ぶことがある。
初期加熱により、コバルト酸リチウムの表面に意図せず残っているリチウム化合物などが脱離する。また内部100bの結晶性を高める効果が期待できる。またステップS11等で準備したリチウム源及び/またはコバルト源には、不純物が混入していることがある。ステップS14で完成したコバルト酸リチウムから不純物を低減させることが、初期加熱によって可能である。なお、内部100bの結晶性を高める効果とは、例えばステップS13で作製したコバルト酸リチウムが有する収縮差等に由来する歪み、ずれ等を緩和する効果である。
また、初期加熱を経ることで、コバルト酸リチウムの表面がなめらかになる効果がある。表面がなめらかとは、凹凸が少なく、コバルト酸リチウムが全体的に丸みを帯び、さらに角部が丸みを帯びる様子をいう。または、表面に付着した異物が少ない状態もなめらかと呼ぶ。異物は凹凸の要因になると考えられ、表面に付着させない方が好ましい。
なお、この初期加熱にあたり、リチウム化合物源、添加元素源、または融剤として機能する材料を別途用意することは不要である。
本工程の加熱時間は、短すぎると十分な効果が得られないが、長すぎると生産性が低下する。例えば、ステップS13で説明した加熱条件から選択して実施できる。なお、ステップS15の加熱温度は、複合酸化物の結晶構造を維持するため、ステップS13の温度より低くするとよい。また、ステップS15の加熱時間は、複合酸化物の結晶構造を維持するため、ステップS13の時間より短くするとよい。例えば700℃以上1000℃以下の温度で、2時間以上20時間以下の加熱を行うとよい。
コバルト酸リチウムは、ステップS13の加熱によって、コバルト酸リチウムの表面と内部に温度差が生じることがある。温度差が生じると収縮差が誘発されることがある。温度差により、表面と内部の流動性が異なるため収縮差が生じるとも考えられる。収縮差に関連するエネルギーは、コバルト酸リチウムに内部応力の差を与えてしまう。内部応力の差は歪みとも称され、当該エネルギーを歪みエネルギーと呼ぶことがある。内部応力はステップS15の初期加熱により除去され、別言すると歪みエネルギーはステップS15の初期加熱により均質化されると考えられる。歪みエネルギーが均質化されるとコバルト酸リチウムの歪みが緩和される。これに伴い、コバルト酸リチウムの表面がなめらかになる可能性がある。表面が改善されたとも称する。別言すると、ステップS15を経るとコバルト酸リチウムに生じた収縮差が緩和され、複合酸化物の表面がなめらかになると考えられる。
また、収縮差は上記コバルト酸リチウムにミクロなずれ、例えば結晶のずれを生じさせることがある。当該ずれを低減するためにも、ステップS15を実施するとよい。ステップS15を経ると、上記複合酸化物のずれを均一化させる(複合酸化物に生じた結晶等のずれを緩和させる、または結晶粒の整列が行われる)ことが可能である。この結果、複合酸化物の表面がなめらかになる可能性がある。
表面がなめらかなコバルト酸リチウムを正極活物質として用いると、二次電池として充放電した際の劣化が少なくなり、正極活物質の割れを防ぐことができる。
なお、ステップS14として、予め合成されたコバルト酸リチウムを用いてもよい。この場合、ステップS11乃至ステップS13を省略することができる。予め合成されたコバルト酸リチウムに対してステップS15を実施することで、表面がなめらかなコバルト酸リチウムを得ることができる。
<ステップS20>
次に、ステップS20乃至ステップS33に示すように、初期加熱を経たコバルト酸リチウムに対し、A源として添加元素Aを加えることが好ましい。初期加熱を経たコバルト酸リチウムに添加元素Aを加えると、添加元素Aをムラなく添加することができる。このため、添加元素Aを添加した後に初期加熱(ステップS15)する順ではなく、初期加熱(ステップS15)後に添加元素Aを添加する順が好ましい。次に、A源として添加元素Aを用意するステップS20の詳細について、図15B、及び図15Cを用いて説明する。
次に、ステップS20乃至ステップS33に示すように、初期加熱を経たコバルト酸リチウムに対し、A源として添加元素Aを加えることが好ましい。初期加熱を経たコバルト酸リチウムに添加元素Aを加えると、添加元素Aをムラなく添加することができる。このため、添加元素Aを添加した後に初期加熱(ステップS15)する順ではなく、初期加熱(ステップS15)後に添加元素Aを添加する順が好ましい。次に、A源として添加元素Aを用意するステップS20の詳細について、図15B、及び図15Cを用いて説明する。
<ステップS21>
図15Bに示すステップS20は、ステップS21乃至ステップS23を有する。ステップS21は、添加元素Aを準備する。添加元素Aとしては、先の実施の形態で説明した添加元素、例えば添加元素X及び添加元素Yを用いることができる。具体的にはマグネシウム、フッ素、ニッケル、アルミニウム、チタン、ジルコニウム、バナジウム、鉄、マンガン、クロム、ニオブ、ヒ素、亜鉛、ケイ素、硫黄、リン及びホウ素から選ばれた一または二以上を用いることができる。また臭素、及びベリリウムから選ばれた一または二以上用いることもできる。図15Bにおいては、マグネシウム源及びフッ素源を用意した場合を例示している。なお、ステップS21において、添加元素Aに加えて、リチウム源を別途準備してもよい。
図15Bに示すステップS20は、ステップS21乃至ステップS23を有する。ステップS21は、添加元素Aを準備する。添加元素Aとしては、先の実施の形態で説明した添加元素、例えば添加元素X及び添加元素Yを用いることができる。具体的にはマグネシウム、フッ素、ニッケル、アルミニウム、チタン、ジルコニウム、バナジウム、鉄、マンガン、クロム、ニオブ、ヒ素、亜鉛、ケイ素、硫黄、リン及びホウ素から選ばれた一または二以上を用いることができる。また臭素、及びベリリウムから選ばれた一または二以上用いることもできる。図15Bにおいては、マグネシウム源及びフッ素源を用意した場合を例示している。なお、ステップS21において、添加元素Aに加えて、リチウム源を別途準備してもよい。
添加元素Aとしてマグネシウムを選んだとき、添加元素源はマグネシウム源と呼ぶことができる。マグネシウム源としては、フッ化マグネシウム、酸化マグネシウム、水酸化マグネシウム、又は炭酸マグネシウム等を用いることができる。マグネシウム源は複数用いてもよい。
添加元素Aとしてフッ素を選んだとき、添加元素源はフッ素源と呼ぶことができる。当該フッ素源としては、例えばフッ化リチウム(LiF)、フッ化マグネシウム(MgF2)、フッ化アルミニウム(AlF3)、フッ化チタン(TiF4)、フッ化コバルト(CoF2、CoF3)、フッ化ニッケル(NiF2)、フッ化ジルコニウム(ZrF4)、フッ化バナジウム(VF5)、フッ化マンガン、フッ化鉄、フッ化クロム、フッ化ニオブ、フッ化亜鉛(ZnF2)、フッ化カルシウム(CaF2)、フッ化ナトリウム(NaF)、フッ化カリウム(KF)、フッ化バリウム(BaF2)、フッ化セリウム(CeF3、CeF4)、フッ化ランタン(LaF3)、又は六フッ化アルミニウムナトリウム(Na3AlF6)等を用いることができる。なかでも、フッ化リチウムは融点が848℃と比較的低く、後述する加熱工程で溶融しやすいため好ましい。
フッ化マグネシウムは、フッ素源としてもマグネシウム源としても用いることができる。また、フッ化リチウムはリチウム源としても用いることができる。ステップS21に用いられるその他のリチウム源としては、炭酸リチウムが挙げられる。
また、フッ素源は、気体でもよく、フッ素(F2)、フッ化炭素、フッ化硫黄、又はフッ化酸素(OF2、O2F2、O3F2、O4F2、O5F2、O6F2、O2F)等を用い、後述する加熱工程において雰囲気中に混合させてもよい。フッ素源は複数用いてもよい。
本実施の形態では、フッ素源としてフッ化リチウム(LiF)を準備し、フッ素源及びマグネシウム源としてフッ化マグネシウム(MgF2)を準備する。フッ化リチウムとフッ化マグネシウムは、LiF:MgF2=65:35(モル比)程度で混合すると融点を下げる効果が最も高くなる。一方、フッ化リチウムが多くなると、リチウムが過剰になりすぎサイクル特性が悪化する懸念がある。そのため、フッ化リチウムとフッ化マグネシウムのモル比は、LiF:MgF2=x:1(0≦x≦1.9)であることが好ましく、LiF:MgF2=x:1(0.1≦x≦0.5)がより好ましく、LiF:MgF2=x:1(x=0.33近傍)がさらに好ましい。なお本明細書等において、近傍とは、特に断りがない限り、その値の0.9倍より大きく1.1倍より小さい値とする。
<ステップS22>
次に、図15Bに示すステップS22では、マグネシウム源及びフッ素源を粉砕及び混合する。本工程は、ステップS12で説明した粉砕及び混合の条件から選択して実施することができる。
次に、図15Bに示すステップS22では、マグネシウム源及びフッ素源を粉砕及び混合する。本工程は、ステップS12で説明した粉砕及び混合の条件から選択して実施することができる。
ここで、必要に応じてステップS22の後に加熱工程を行ってもよい。加熱工程はステップS13で説明した加熱条件から選択して実施することができる。加熱時間は2時間以上が好ましく、加熱温度は800℃以上1100℃以下が好ましい。
<ステップS23>
次に、図15Bに示すステップS23では、上記で粉砕、混合した材料を回収して、添加元素源(A源)を得ることができる。なお、ステップS23に示す添加元素源(A源)は、複数の出発材料を有するものであり、混合物と呼ぶこともできる。
次に、図15Bに示すステップS23では、上記で粉砕、混合した材料を回収して、添加元素源(A源)を得ることができる。なお、ステップS23に示す添加元素源(A源)は、複数の出発材料を有するものであり、混合物と呼ぶこともできる。
上記混合物の粒径は、D50(メディアン径)が600nm以上20μm以下であることが好ましく、1μm以上10μm以下であることがより好ましい。添加元素源として、一種の材料を用いた場合においても、D50(メディアン径)が600nm以上20μm以下であることが好ましく、1μm以上10μm以下であることがより好ましい。
このような微粉化された混合物(添加元素が1種の場合も含む)は、後の工程でコバルト酸リチウムと混合したときに、コバルト酸リチウムの表面に混合物を均一に付着させやすい。コバルト酸リチウムの表面に混合物が均一に付着していると、加熱後に複合酸化物の表層部100aに均一に添加元素を分布又は拡散させやすいため、好ましい。
<ステップS21>
図15Bとは異なる工程について図15Cを用いて説明する。 図15Cに示すステップS20は、ステップS21乃至ステップS23を有する。
図15Bとは異なる工程について図15Cを用いて説明する。 図15Cに示すステップS20は、ステップS21乃至ステップS23を有する。
図15Cに示すステップS21では、コバルト酸リチウムに添加する添加元素源を4種用意する。すなわち、図15Cは図15Bと添加元素源の種類が異なる。また、添加元素源に加えて、リチウム源を別途準備してもよい。
4種の添加元素源として、マグネシウム源(Mg源)、フッ素源(F源)、ニッケル源(Ni源)、及びアルミニウム源(Al源)を準備する。なお、マグネシウム源及びフッ素源は図15Bで説明した化合物等から選択することができる。ニッケル源としては、酸化ニッケル、水酸化ニッケル等を用いることができる。アルミニウム源としては、酸化アルミニウム、水酸化アルミニウム、等を用いることができる。
<ステップS22及びステップS23>
次に、図15Cに示すステップS22及びステップS23は、図15Bで説明したステップと同様である。
次に、図15Cに示すステップS22及びステップS23は、図15Bで説明したステップと同様である。
<ステップS31>
次に、図15Aに示すステップS31では、コバルト酸リチウムと、添加元素源(A源)とを混合する。コバルト酸リチウム中のコバルトの原子数Coと、添加元素源(A源)が有するマグネシウムの原子数Mgとの比は、Co:Mg=100:y(0.1≦y≦6)であることが好ましく、M:Mg=100:y(0.3≦y≦3)であることがより好ましい。
次に、図15Aに示すステップS31では、コバルト酸リチウムと、添加元素源(A源)とを混合する。コバルト酸リチウム中のコバルトの原子数Coと、添加元素源(A源)が有するマグネシウムの原子数Mgとの比は、Co:Mg=100:y(0.1≦y≦6)であることが好ましく、M:Mg=100:y(0.3≦y≦3)であることがより好ましい。
ステップS31の混合は、コバルト酸リチウムの形状を破壊させないために、ステップS12の混合よりも穏やかな条件とすることが好ましい。例えば、ステップS12の混合よりも回転数が少ない、または短時間の条件とすることが好ましい。また、湿式よりも乾式の方が穏やかな条件であると言える。混合には、例えばボールミル、ビーズミル等を用いることができる。ボールミルを用いる場合は、例えばメディアとして酸化ジルコニウムボールを用いることが好ましい。
本実施の形態では、直径1mmの酸化ジルコニウムボールを用いたボールミルで、150rpm、1時間、乾式で混合することとする。また該混合は、露点が−100℃以上−10℃以下のドライルームで行うこととする。
<ステップS32>
次に、図15AのステップS32において、上記で混合した材料を回収し、混合物903を得る。回収の際、必要に応じて解砕した後にふるいを実施してもよい。
次に、図15AのステップS32において、上記で混合した材料を回収し、混合物903を得る。回収の際、必要に応じて解砕した後にふるいを実施してもよい。
なお、図15A乃至図15Cでは、初期加熱を経た後にのみ添加元素を加える作製方法について説明しているが、本発明は上記方法に限定されない。添加元素は他のタイミングで加えてもよいし、複数回にわたって加えてもよい。また、元素によってタイミングを変えてもよい。
例えば、ステップS11の段階、つまり複合酸化物の出発材料の段階で添加元素をリチウム源及び遷移金属源に添加してもよい。その後ステップS13で添加元素を有するコバルト酸リチウムを得ることができる。この場合は、ステップS11乃至ステップS14の工程と、ステップS21乃至ステップS23の工程を分ける必要がない。簡便で生産性が高い方法であるといえる。
また、あらかじめ添加元素の一部を有するコバルト酸リチウムを用いてもよい。例えばマグネシウム及びフッ素が添加されたコバルト酸リチウムを用いれば、ステップS11乃至ステップS14、及びステップS20の一部の工程を省略することができる。簡便で生産性が高い方法であるといえる。
また、あらかじめマグネシウム及びフッ素が添加されたコバルト酸リチウムに対して、ステップS15の加熱を行った後、ステップS20のようにマグネシウム源及びフッ素源、又はマグネシウム源、フッ素源、ニッケル源、及びアルミニウム源を添加してもよい。
<ステップS33>
次に、図15Aに示すステップS33では、混合物903を加熱する。ステップS13で説明した加熱条件から選択して実施することができる。加熱時間は2時間以上が好ましい。ステップS33の加熱温度の下限は、コバルト酸リチウムと添加元素源との反応が進む温度以上である必要がある。反応が進む温度とは、コバルト酸リチウムと添加元素源との有する元素の相互拡散が生じる温度であればよく、これらの材料の溶融温度よりも低くてもよい。酸化物を例にして説明するが、溶融温度Tmの0.757倍(タンマン温度Td)から固相拡散が生じる。このため、ステップS33における加熱温度としては、500℃以上であればよい。
次に、図15Aに示すステップS33では、混合物903を加熱する。ステップS13で説明した加熱条件から選択して実施することができる。加熱時間は2時間以上が好ましい。ステップS33の加熱温度の下限は、コバルト酸リチウムと添加元素源との反応が進む温度以上である必要がある。反応が進む温度とは、コバルト酸リチウムと添加元素源との有する元素の相互拡散が生じる温度であればよく、これらの材料の溶融温度よりも低くてもよい。酸化物を例にして説明するが、溶融温度Tmの0.757倍(タンマン温度Td)から固相拡散が生じる。このため、ステップS33における加熱温度としては、500℃以上であればよい。
なお、混合物903が有する材料から選ばれた一または二以上が溶融する温度以上であると、より反応が進みやすい。例えば、添加元素源として、LiF及びMgF2を有する場合、LiFとMgF2の共融点は742℃付近であるため、ステップS33の加熱温度の下限は742℃以上とすると好ましい。
また、LiCoO2:LiF:MgF2=100:0.33:1(モル比)となるように混合して得られた混合物903は、示差走査熱量測定(DSC測定)において830℃付近に吸熱ピークが観測される。よって、加熱温度の下限は830℃以上がより好ましい。
加熱温度は高い方が反応が進みやすく、加熱時間が短く済み、生産性が高く好ましい。
加熱温度の上限は、コバルト酸リチウムの分解温度(1130℃)未満とする。分解温度の近傍の温度では、微量ではあるがコバルト酸リチウムの分解が懸念される。そのため、1000℃以下であると好ましく、950℃以下であるとより好ましく、900℃以下であるとさらに好ましい。
これらを踏まえると、ステップS33における加熱温度としては、500℃以上1130℃以下が好ましく、500℃以上1000℃以下がより好ましく、500℃以上950℃以下がさらに好ましく、500℃以上900℃以下がさらに好ましい。また、742℃以上1130℃以下が好ましく、742℃以上1000℃以下がより好ましく、742℃以上950℃以下がさらに好ましく、742℃以上900℃以下がさらに好ましい。また、800℃以上1100℃以下、830℃以上1130℃以下が好ましく、830℃以上1000℃以下がより好ましく、830℃以上950℃以下がさらに好ましく、830℃以上900℃以下がさらに好ましい。なおステップS33における加熱温度は、ステップS13よりも高いとよい。
さらに、混合物903を加熱する際、フッ素源等に起因するフッ素またはフッ化物の分圧を適切な範囲に制御することが好ましい。
本実施の形態で説明する作製方法では、一部の材料、例えばフッ素源であるLiFが融剤として機能する場合がある。この機能により加熱温度をコバルト酸リチウムの分解温度未満、例えば742℃以上950℃以下にまで低温化でき、表層部にマグネシウムをはじめとする添加元素を分布させ、良好な特性の正極活物質を作製できる。
しかし、LiFは酸素よりも気体状態での比重が軽いため、加熱によりLiFが揮発する可能性があり、揮発すると混合物903中のLiFが減少してしまう。すると融剤としての機能が弱くなってしまう。したがって、LiFの揮発を抑制しつつ、加熱する必要がある。なお、フッ素源等としてLiFを用いなかったとしても、LiCoO2表面のLiとフッ素源のFが反応して、LiFが生じ、揮発する可能性もある。そのため、LiFより融点が高いフッ化物を用いたとしても、同じように揮発の抑制が必要である。
そこで、LiFを含む雰囲気で混合物903を加熱すること、すなわち、加熱炉内のLiFの分圧が高い状態で混合物903を加熱することが好ましい。このような加熱により混合物903中のLiFの揮発を抑制することができる。
また、本工程の加熱は、混合物903の粒子同士が固着しないように加熱すると好ましい。加熱中に混合物903の粒子同士が固着すると、雰囲気中の酸素との接触面積が減る、及び添加元素(例えばフッ素)が拡散する経路を阻害することにより、表層部への添加元素(例えばマグネシウム及びフッ素)の分布が悪化する可能性がある。
また、添加元素(例えばフッ素)が表層部に均一に分布すると、なめらかで凹凸が少ない正極活物質を得られると考えられている。そのため、本工程でステップS15の加熱を経た、表面がなめらかな状態を維持する又はより一層なめらかになるためには、混合物903の粒子同士が固着しない方がよい。
また、ロータリーキルンによって加熱する場合は、キルン内の酸素を含む雰囲気の流量を制御して加熱することが好ましい。例えば酸素を含む雰囲気の流量を少なくする、最初に雰囲気をパージしキルン内に酸素雰囲気を導入した後は雰囲気のフローはしない、等が好ましい。酸素をフローするとフッ素源が蒸散する可能性があり、表面のなめらかさを維持するためには好ましくない。
ローラーハースキルンによって加熱する場合は、例えば混合物903の入った容器に蓋を配することでLiFを含む雰囲気で混合物903を加熱することができる。
図15AのステップS14のコバルト酸リチウムのメディアン径(D50)が12μm程度の場合、加熱温度は、例えば600℃以上950℃以下が好ましい。加熱時間は例えば3時間以上が好ましく、10時間以上がより好ましく、60時間以上がさらに好ましい。なお、加熱後の降温時間は、例えば10時間以上50時間以下とすることが好ましい。
また、ステップS14のコバルト酸リチウムのメディアン径(D50)が5μm程度の場合、加熱温度は例えば600℃以上950℃以下が好ましい。加熱時間は例えば1時間以上10時間以下が好ましく、2時間程度がより好ましい。なお、加熱後の降温時間は、例えば10時間以上50時間以下とすることが好ましい。
<ステップS34>
次に、図15Aに示すステップS34では、加熱した材料を回収し、必要に応じて解砕して、正極活物質100を得る。このとき、回収された正極活物質100を、さらにふるいにかけると好ましい。以上の工程により、本実施の形態で説明した特徴を有する正極活物質100を作製することができる。
次に、図15Aに示すステップS34では、加熱した材料を回収し、必要に応じて解砕して、正極活物質100を得る。このとき、回収された正極活物質100を、さらにふるいにかけると好ましい。以上の工程により、本実施の形態で説明した特徴を有する正極活物質100を作製することができる。
<正極活物質の作製方法の例2>
図16乃至図17を用いて、本発明の一態様として利用可能な正極活物質の作製方法の別の一例(正極活物質の作製方法の例2)について説明する。正極活物質の作製方法の例2は、添加元素を加える回数及び混合方法が先に述べた正極活物質の作製方法の例1と異なるが、その他の記載は正極活物質の作製方法の例1の記載を適用することができる。
図16乃至図17を用いて、本発明の一態様として利用可能な正極活物質の作製方法の別の一例(正極活物質の作製方法の例2)について説明する。正極活物質の作製方法の例2は、添加元素を加える回数及び混合方法が先に述べた正極活物質の作製方法の例1と異なるが、その他の記載は正極活物質の作製方法の例1の記載を適用することができる。
図16において、図15Aと同様にステップS11乃至S15までを行い、初期加熱を経たコバルト酸リチウムを準備する。
<ステップS20a>
次に、ステップS20a乃至ステップS33に示すように、初期加熱を経たコバルト酸リチウムに添加元素A1を加える。ステップS20aは、添加元素A1を加えるために用いる第1の添加元素源(A1源)を準備するステップであり、図17Aを参照しながら説明する。
次に、ステップS20a乃至ステップS33に示すように、初期加熱を経たコバルト酸リチウムに添加元素A1を加える。ステップS20aは、添加元素A1を加えるために用いる第1の添加元素源(A1源)を準備するステップであり、図17Aを参照しながら説明する。
<ステップS21>
図17Aに示すステップS21乃至ステップS23では、第1の添加元素源(A1源)を準備する。添加元素A1としては、図15Bに示すステップS21で説明した添加元素Aの中から選択して用いることができる。例えば、添加元素A1としては、マグネシウム、フッ素、及びカルシウムの中から選ばれるいずれか一または複数を用いることができる。図17Aでは、A1源として、マグネシウム源(Mg源)、及びフッ素源(F源)を、粉砕及び混合して用いる場合を例示している。
図17Aに示すステップS21乃至ステップS23では、第1の添加元素源(A1源)を準備する。添加元素A1としては、図15Bに示すステップS21で説明した添加元素Aの中から選択して用いることができる。例えば、添加元素A1としては、マグネシウム、フッ素、及びカルシウムの中から選ばれるいずれか一または複数を用いることができる。図17Aでは、A1源として、マグネシウム源(Mg源)、及びフッ素源(F源)を、粉砕及び混合して用いる場合を例示している。
図17Aに示すステップS21乃至ステップS23は、図15Bに示すステップS21乃至ステップS23と同様の条件で行うことができる。その結果、ステップS23で第1の添加元素源(A1源)を得ることができる。
また、図16に示すステップS31乃至S33については、図15Aに示すステップS31乃至S33と同様の条件で行うことができる。
<ステップS34a>
次に、ステップS33で加熱した材料を回収し、添加元素A1を有するコバルト酸リチウムを作製する。ここでは、ステップS14の複合酸化物(第1の複合酸化物)と区別するため、第2の複合酸化物とも呼ぶ。
次に、ステップS33で加熱した材料を回収し、添加元素A1を有するコバルト酸リチウムを作製する。ここでは、ステップS14の複合酸化物(第1の複合酸化物)と区別するため、第2の複合酸化物とも呼ぶ。
<ステップS40>
図16に示すステップS40乃至ステップS53では、第2の複合酸化物に添加元素A2を添加する。ステップS40は、添加元素A2を加えるために用いる第2の添加元素源(A2源)を準備するステップであり、図17B及び図17Cを参照しながら説明する。
図16に示すステップS40乃至ステップS53では、第2の複合酸化物に添加元素A2を添加する。ステップS40は、添加元素A2を加えるために用いる第2の添加元素源(A2源)を準備するステップであり、図17B及び図17Cを参照しながら説明する。
<ステップS41>
図17Bに示すステップS41乃至ステップS43では、第2の添加元素源(A2源)を準備する。添加元素A2としては、図15Bに示すステップS21で説明した添加元素Aの中から選択して用いることができる。例えば、添加元素A2としては、ニッケル、チタン、ホウ素、ジルコニウム、及びアルミニウムの中から選ばれるいずれか一または複数を好適に用いることができる。図17BではA2源として、ニッケル源、及びアルミニウム源を、粉砕及び混合して用いる場合を例示している。
図17Bに示すステップS41乃至ステップS43では、第2の添加元素源(A2源)を準備する。添加元素A2としては、図15Bに示すステップS21で説明した添加元素Aの中から選択して用いることができる。例えば、添加元素A2としては、ニッケル、チタン、ホウ素、ジルコニウム、及びアルミニウムの中から選ばれるいずれか一または複数を好適に用いることができる。図17BではA2源として、ニッケル源、及びアルミニウム源を、粉砕及び混合して用いる場合を例示している。
図17Bに示すステップS41乃至ステップS43は、図15Bに示すステップS21乃至ステップS23と同様の条件で作製することができる。その結果、ステップS43で第2の添加元素源(A2源)を得ることができる。
図17Cに示すステップS41乃至ステップS43は、図17Bの変形例である。図17Cに示すステップS41ではニッケル源(Ni源)、及びアルミニウム源(Al源)を準備し、ステップS42aではそれぞれ独立に粉砕する。その結果、ステップS43では、複数の第2の添加元素源(A2源)を準備することとなる。図17Cのステップは、ステップS42aにて添加元素を独立に粉砕している点で図17Bと異なる。
<ステップS51乃至ステップS53>
次に、図16に示すステップS51乃至ステップS53は、図15Aに示すステップS31乃至ステップS33と同様の条件で行うことができる。加熱工程に関するステップS53の条件は、ステップS33より低い温度且つ短時間でよい。
次に、図16に示すステップS51乃至ステップS53は、図15Aに示すステップS31乃至ステップS33と同様の条件で行うことができる。加熱工程に関するステップS53の条件は、ステップS33より低い温度且つ短時間でよい。
<ステップS54>
次に、図16に示すステップS54では、加熱した材料を回収し、必要に応じて解砕して、正極活物質100を得る。以上の工程により、本実施の形態で説明した特徴を有する正極活物質100を作製することができる。
次に、図16に示すステップS54では、加熱した材料を回収し、必要に応じて解砕して、正極活物質100を得る。以上の工程により、本実施の形態で説明した特徴を有する正極活物質100を作製することができる。
図16及び図17に示すように、作製方法2では、コバルト酸リチウムへの添加元素を第1の添加元素A1と、第2の添加元素A2とに分けて導入する。分けて導入することにより、各添加元素の深さ方向のプロファイルを変えることができる。例えば、第1の添加元素を内部に比べて表層部で高い濃度となるようにプロファイルし、第2の添加元素を表層部に比べて内部で高い濃度となるようにプロファイルすることも可能である。
(実施の形態3)
本実施の形態では、リチウムイオン電池に含まれる正極活物質と電解質以外の構成について説明する。
本実施の形態では、リチウムイオン電池に含まれる正極活物質と電解質以外の構成について説明する。
[正極]
正極は、正極活物質層及び正極集電体を有する。正極活物質層は正極活物質を有し、さらに導電助剤及びバインダの少なくとも一を有していてもよい。正極活物質は、実施の形態1で説明したものを用いることができる。
正極は、正極活物質層及び正極集電体を有する。正極活物質層は正極活物質を有し、さらに導電助剤及びバインダの少なくとも一を有していてもよい。正極活物質は、実施の形態1で説明したものを用いることができる。
図18Aは、正極の断面の模式図の一例を示している。
集電体550は、例えば金属箔を用いることができる。正極は、金属箔上にスラリーを塗布して乾燥させることによって形成することができる。なお、乾燥後にプレスを加えてもよい。正極は、集電体550上に活物質層を形成したものである。
スラリーとは、集電体550上に活物質層を形成するために用いる材料液であり、活物質とバインダと溶媒を含有し、好ましくはさらに導電助剤を混合させたものを指している。なお、スラリーは、電極用スラリーまたは活物質スラリーと呼ばれることもあり、正極活物質層を形成する場合には正極用スラリーを用い、負極活物質層を形成する場合には負極用スラリーと呼ばれることもある。
正極活物質561は、充放電に伴い、リチウムイオンを取り込む、および/または放出する機能を有する。本発明の一態様として用いる正極活物質561は、高い充電電圧としても充放電に伴う劣化の少ない材料を用いることができる。なお、本明細書等において、特に言及しない場合、充電電圧はリチウム金属の電位を基準として表すものとする。また、本明細書等において、高い充電電圧とは、例えば4.6V以上の充電電圧とし、好ましくは4.65V以上、さらに好ましくは4.7V以上、よりさらに好ましくは4.75V以上、最も好ましくは4.8V以上とする。
本発明の一態様として用いる正極活物質561は、高い充電電圧としても充放電に伴う劣化の少ない材料であれば何でも用いることが可能であり、実施の形態1または実施の形態2で説明したものを用いることができる。なお、正極活物質561は、高い充電電圧としても充放電に伴う劣化の少ない材料であれば、粒径が異なる2種類以上の材料を用いることができる。
導電助剤は、導電付与剤、導電材とも呼ばれ、炭素材料を用いることができる。複数の活物質の間に導電助剤を付着させることで複数の活物質同士が電気的に接続され、導電性が高まる。なお、本明細書等において「付着」とは、活物質と導電助剤が物理的に密着していることのみを指しているのではなく、共有結合が生じる場合、ファンデルワールス力により結合する場合、活物質の表面の一部を導電助剤が覆う場合、活物質の表面凹凸に導電助剤がはまりこむ場合、互いに接していなくとも電気的に接続される場合などを含む概念とする。
導電助剤として用いることができる炭素材料の具体例は、カーボンブラック(ファーネスブラック、アセチレンブラック、黒鉛など)が挙げられる。
図18Aは、導電助剤としてカーボンブラック553を図示している。
二次電池の正極として、金属箔などの集電体550と、活物質と、を固着させるために、バインダ(樹脂)を混合してもよい。バインダは結着剤とも呼ばれる。バインダは高分子材料であり、バインダを多く含ませると正極における活物質の割合が低下して、二次電池の放電容量が小さくなる。そのため、バインダの量は最小限に混合させることが好ましい。図18Aにおいて、正極活物質561、第2の活物質562、カーボンブラック553で埋まっていない領域は、空隙またはバインダを指している。
なお、図18Aでは正極活物質561を球形として図示した例を示しているが、特に限定されない。例えば、正極活物質561の断面形状は楕円形、長方形、台形、三角形、角が丸まった多角形、非対称の形状であってもよい。例えば、図18Bでは、正極活物質561が角が丸まった多角形の形状を有する例を示している。
また、図18Bの正極では、導電助剤として用いられる炭素材料として、グラフェン554を用いている。図18Bは、集電体550上に正極活物質561、グラフェン554、カーボンブラック553を有する正極活物質層を形成している。
なお、グラフェン554、カーボンブラック553を混合し、電極スラリーを得る工程において、混合するカーボンブラックの重量はグラフェンの1.5倍以上20倍以下、好ましくは2倍以上9.5倍以下の重量とすることが好ましい。
また、グラフェン554とカーボンブラック553の混合を上記範囲とすると、スラリー調製時に、カーボンブラック553の分散安定性に優れ、凝集部が生じにくい。また、グラフェン554とカーボンブラック553の混合を上記範囲とすると、カーボンブラック553のみを導電助剤に用いる正極よりも高い電極密度とすることができる。電極密度を高くすることで、単位重量当たりの容量を大きくすることができる。具体的には、重量測定による正極活物質層の密度は、3.5g/cc以上とすることができる。
また、グラフェンのみを導電助剤に用いる正極に比べると電極密度は低いが、第1の炭素材料(グラフェン)と第2の炭素材料(アセチレンブラック)の混合を上記範囲とすることで、急速充電に対応することができる。このため、車載用の二次電池として用いる場合に特に有効である。
図18Cでは、グラフェンに代えて炭素繊維555を用いる正極の例を図示している。図18Cは、図18Bと異なる例を示している。炭素繊維555を用いるとカーボンブラック553の凝集を防ぎ、分散性を高めることができる。
なお、図18Cにおいて、正極活物質561、炭素繊維555、カーボンブラック553で埋まっていない領域は、空隙またはバインダを指している。
また、他の正極の例として、図18Dを図示している。図18Dでは、グラフェン554に加えて炭素繊維555を用いる例を示している。グラフェン554及び炭素繊維555の両方を用いると、カーボンブラック553などのカーボンブラックの凝集を防ぎ、分散性をより高めることができる。
なお、図18Dにおいて、正極活物質561、炭素繊維555、グラフェン554、カーボンブラック553で埋まっていない領域は、空隙またはバインダを指している。
図18A乃至図18Dのいずれか一の正極を用い、正極上にセパレータを重ね、セパレータ上に負極を重ねた積層体を収容する容器(外装体、金属缶など)などに入れ、容器に電解液を充填させることで二次電池を作製することができる。
<バインダ>
バインダとしては、例えば、スチレン−ブタジエンゴム(SBR)、スチレン−イソプレン−スチレンゴム、アクリロニトリル−ブタジエンゴム、ブタジエンゴム、エチレン−プロピレン−ジエン共重合体などのゴム材料を用いることが好ましい。またバインダとして、フッ素ゴムを用いることができる。
バインダとしては、例えば、スチレン−ブタジエンゴム(SBR)、スチレン−イソプレン−スチレンゴム、アクリロニトリル−ブタジエンゴム、ブタジエンゴム、エチレン−プロピレン−ジエン共重合体などのゴム材料を用いることが好ましい。またバインダとして、フッ素ゴムを用いることができる。
また、バインダとしては、例えば水溶性の高分子を用いることが好ましい。水溶性の高分子としては、例えば多糖類などを用いることができる。多糖類としては、カルボキシメチルセルロース(CMC)、メチルセルロース、エチルセルロース、ヒドロキシプロピルセルロース、ジアセチルセルロース、再生セルロースなどのセルロース誘導体、または澱粉などを用いることができる。また、これらの水溶性の高分子を、前述のゴム材料と併用して用いると、さらに好ましい。
または、バインダとしては、ポリスチレン、ポリアクリル酸メチル、ポリメタクリル酸メチル(ポリメチルメタクリレート、PMMA)、ポリアクリル酸ナトリウム、ポリビニルアルコール(PVA)、ポリエチレンオキシド(PEO)、ポリプロピレンオキシド、ポリイミド、ポリ塩化ビニル、ポリテトラフルオロエチレン、ポリエチレン、ポリプロピレン、ポリイソブチレン、ポリエチレンテレフタレート、ナイロン、ポリフッ化ビニリデン(PVDF)、ポリアクリロニトリル(PAN)、エチレンプロピレンジエンポリマー、ポリ酢酸ビニル、ニトロセルロース等の材料を用いることが好ましい。
バインダは上記のうち複数を組み合わせて使用してもよい。
例えば粘度調整効果の特に優れた材料と、他の材料とを組み合わせて使用してもよい。例えばゴム材料等は接着力及び弾性力に優れる反面、溶媒に混合した場合に粘度調整が難しい場合がある。このような場合には例えば、粘度調整効果の特に優れた材料と混合することが好ましい。粘度調整効果の特に優れた材料としては、例えば水溶性高分子を用いるとよい。また、粘度調整効果に特に優れた水溶性高分子としては、前述の多糖類、例えばカルボキシメチルセルロース(CMC)、メチルセルロース、エチルセルロース、ヒドロキシプロピルセルロース及びジアセチルセルロース、再生セルロースなどのセルロース誘導体、または澱粉を用いることができる。
なお、カルボキシメチルセルロースなどのセルロース誘導体は、例えばカルボキシメチルセルロースのナトリウム塩またはアンモニウム塩などの塩とすることにより溶解度が上がり、粘度調整剤としての効果を発揮しやすくなる。溶解度が高くなることにより電極のスラリーを作製する際に活物質または他の構成要素との分散性を高めることもできる。本明細書等においては、電極のバインダとして使用するセルロース及びセルロース誘導体としては、それらの塩も含むものとする。
水溶性高分子は水に溶解することにより粘度を安定化させ、活物質及びバインダとして組み合わせる他の材料、例えばスチレンブタジエンゴムを水溶液中に安定して分散させることができる。また、官能基を有するために活物質表面に安定に吸着しやすいことが期待される。また、例えばカルボキシメチルセルロースなどのセルロース誘導体は、水酸基またはカルボキシル基などの官能基を有する材料が多く、官能基を有するために高分子同士が相互作用し、活物質表面を広く覆って存在することが期待される。
活物質表面を覆う、または表面に接するバインダが膜を形成する場合には、不動態膜としての役割を果たして電解液の分解を抑える効果も期待される。ここで、「不動態膜」とは、電気の電導性のない膜、または電気電導性の極めて低い膜であり、例えば活物質の表面に不動態膜が形成された場合には、電池反応電位において、電解液の分解を抑制することができる。また、不動態膜は、電気の電導性を抑えるとともに、リチウムイオンは伝導できるとさらに望ましい。
<正極集電体>
正極集電体としては、ステンレス、金、白金、アルミニウム、チタン等の金属、及びこれらの合金など、導電性が高い材料を用いることができる。また正極集電体に用いる材料は、正極の電位で溶出しないことが好ましい。また、シリコン、チタン、ネオジム、スカンジウム、モリブデンなどの耐熱性を向上させる元素が添加されたアルミニウム合金を用いることができる。また、シリコンと反応してシリサイドを形成する金属元素で形成してもよい。シリコンと反応してシリサイドを形成する金属元素としては、ジルコニウム、チタン、ハフニウム、バナジウム、ニオブ、タンタル、クロム、モリブデン、タングステン、コバルト、ニッケル等がある。正極集電体は、箔状、板状、シート状、網状、パンチングメタル状、エキスパンドメタル状等の形状を適宜用いることができる。正極集電体は、厚みが5μm以上30μm以下のものを用いるとよい。
正極集電体としては、ステンレス、金、白金、アルミニウム、チタン等の金属、及びこれらの合金など、導電性が高い材料を用いることができる。また正極集電体に用いる材料は、正極の電位で溶出しないことが好ましい。また、シリコン、チタン、ネオジム、スカンジウム、モリブデンなどの耐熱性を向上させる元素が添加されたアルミニウム合金を用いることができる。また、シリコンと反応してシリサイドを形成する金属元素で形成してもよい。シリコンと反応してシリサイドを形成する金属元素としては、ジルコニウム、チタン、ハフニウム、バナジウム、ニオブ、タンタル、クロム、モリブデン、タングステン、コバルト、ニッケル等がある。正極集電体は、箔状、板状、シート状、網状、パンチングメタル状、エキスパンドメタル状等の形状を適宜用いることができる。正極集電体は、厚みが5μm以上30μm以下のものを用いるとよい。
[負極]
負極は、負極活物質層及び負極集電体を有する。また、負極活物質層は負極活物質を有し、さらに導電助剤及びバインダを有していてもよい。
負極は、負極活物質層及び負極集電体を有する。また、負極活物質層は負極活物質を有し、さらに導電助剤及びバインダを有していてもよい。
<負極活物質>
負極活物質としては、例えば合金系材料または炭素材料を用いることができる。
負極活物質としては、例えば合金系材料または炭素材料を用いることができる。
また、負極活物質は、リチウムとの合金化・脱合金化反応により充放電反応を行うことが可能な元素を用いることができる。例えば、シリコン、スズ、ガリウム、アルミニウム、ゲルマニウム、鉛、アンチモン、ビスマス、銀、亜鉛、カドミウム、インジウム等のうち少なくとも一つを含む材料を用いることができる。このような元素は炭素と比べて容量が大きく、特にシリコンは理論容量が4200mAh/gと高い。このため、負極活物質にシリコンを用いることが好ましい。また、これらの元素を有する化合物を用いてもよい。例えば、SiO、Mg2Si、Mg2Ge、SnO、SnO2、Mg2Sn、SnS2、V2Sn3、FeSn2、CoSn2、Ni3Sn2、Cu6Sn5、Ag3Sn、Ag3Sb、Ni2MnSb、CeSb3、LaSn3、La3Co2Sn7、CoSb3、InSb、SbSn等がある。ここで、リチウムとの合金化・脱合金化反応により充放電反応を行うことが可能な元素、及び該元素を有する化合物等を合金系材料と呼ぶ場合がある。
本明細書等において、「SiO」は例えば一酸化シリコンを指す。あるいはSiOは、SiOxと表すこともできる。ここでxは1または1近傍の値を有することが好ましい。例えばxは、0.2以上1.5以下が好ましく、0.3以上1.2以下が好ましい。
炭素材料は、黒鉛、易黒鉛化性炭素(ソフトカーボン)、難黒鉛化性炭素(ハードカーボン)、炭素繊維(カーボンナノチューブ)、グラフェン、カーボンブラック等を用いればよい。
黒鉛は、人造黒鉛または天然黒鉛等が挙げられる。人造黒鉛としては例えば、メソカーボンマイクロビーズ(MCMB)、コークス系人造黒鉛、ピッチ系人造黒鉛等が挙げられる。ここで人造黒鉛として、球状の形状を有する球状黒鉛を用いることができる。例えば、MCMBは球状の形状を有する場合があり、好ましい。また、MCMBはその表面積を小さくすることが比較的容易であり、好ましい場合がある。天然黒鉛としては、例えば、鱗片状黒鉛、球状化天然黒鉛等が挙げられる。
黒鉛は、リチウムイオンが黒鉛に挿入されたとき(リチウム−黒鉛層間化合物の生成時)にリチウム金属と同程度に低い電位を示す(0.05V以上0.3V以下 vs.Li/Li+)。これにより、黒鉛を用いたリチウムイオン電池は高い作動電圧を示すことができる。さらに、黒鉛は、単位体積当たりの容量が比較的高い、体積膨張が比較的小さい、安価である、リチウム金属に比べて安全性が高い等の利点を有するため、好ましい。
また、負極活物質として、二酸化チタン(TiO2)、リチウムチタン酸化物(Li4Ti5O12)、リチウム−黒鉛層間化合物(LixC6)、五酸化ニオブ(Nb2O5)、酸化タングステン(WO2)、酸化モリブデン(MoO2)等の酸化物を用いることができる。
また、負極活物質として、リチウムと遷移金属の複窒化物である、Li3N型構造をもつLi3−xMxN(M=Co、Ni、Cu)を用いることができる。例えば、Li2.6Co0.4N3は大きな放電容量(900mAh/g、1890mAh/cm3)を示し好ましい。
リチウムと遷移金属の複窒化物を用いると、負極活物質中にリチウムイオンを含むため、正極活物質としてリチウムイオンを含まないV2O5、Cr3O8等の材料と組み合わせることができ好ましい。なお、正極活物質にリチウムイオンを含む材料を用いる場合でも、あらかじめ正極活物質に含まれるリチウムイオンを脱離させることで、負極活物質としてリチウムと遷移金属の複窒化物を用いることができる。
また、コンバージョン反応が生じる材料を負極活物質として用いることもできる。例えば、酸化コバルト(CoO)、酸化ニッケル(NiO)、酸化鉄(FeO)等の、リチウムとの合金を作らない遷移金属酸化物を負極活物質に用いてもよい。コンバージョン反応が生じる材料としては、さらに、Fe2O3、CuO、Cu2O、RuO2、Cr2O3等の酸化物、CoS0.89、NiS、CuS等の硫化物、Zn3N2、Cu3N、Ge3N4等の窒化物、NiP2、FeP2、CoP3等のリン化物、FeF3、BiF3等のフッ化物でも起こる。
負極活物質層が有することのできる導電助剤及びバインダとしては、正極活物質層が有することのできる導電助剤及びバインダと同様の材料を用いることができる。
<負極集電体>
負極集電体には、正極集電体と同様の材料に加え、銅なども用いることができる。なお負極集電体は、リチウム等のキャリアイオンと合金化しない材料を用いることが好ましい。
負極集電体には、正極集電体と同様の材料に加え、銅なども用いることができる。なお負極集電体は、リチウム等のキャリアイオンと合金化しない材料を用いることが好ましい。
[電解質]
電解質は、実施の形態1で説明したものを用いることができる。
電解質は、実施の形態1で説明したものを用いることができる。
[セパレータ]
電解質が電解液を含む場合、正極と負極の間にセパレータを配置する。セパレータとしては、例えば、紙をはじめとするセルロースを有する繊維、不織布、ガラス繊維、セラミックス、或いはナイロン(ポリアミド)、ビニロン(ポリビニルアルコール系繊維)、ポリエステル、アクリル、ポリオレフィン、ポリウレタンを用いた合成繊維等で形成されたものを用いることができる。セパレータは袋状に加工し、正極または負極のいずれか一方を包むように配置することが好ましい。
電解質が電解液を含む場合、正極と負極の間にセパレータを配置する。セパレータとしては、例えば、紙をはじめとするセルロースを有する繊維、不織布、ガラス繊維、セラミックス、或いはナイロン(ポリアミド)、ビニロン(ポリビニルアルコール系繊維)、ポリエステル、アクリル、ポリオレフィン、ポリウレタンを用いた合成繊維等で形成されたものを用いることができる。セパレータは袋状に加工し、正極または負極のいずれか一方を包むように配置することが好ましい。
セパレータは多層構造であってもよい。例えばポリプロピレン、ポリエチレン等の有機材料フィルムに、セラミック系材料、フッ素系材料、ポリアミド系材料、またはこれらを混合したもの等をコートすることができる。セラミック系材料としては、例えば酸化アルミニウム粒子、酸化シリコン粒子等を用いることができる。フッ素系材料としては、例えばPVDF、ポリテトラフルオロエチレン等を用いることができる。ポリアミド系材料としては、例えばナイロン、アラミド(メタ系アラミド、パラ系アラミド)等を用いることができる。
セラミック系材料をコートすると耐酸化性が向上するため、高電圧充放電の際のセパレータの劣化を抑制し、二次電池の信頼性を向上させることができる。またフッ素系材料をコートするとセパレータと電極が密着しやすくなり、出力特性を向上させることができる。ポリアミド系材料、特にアラミドをコートすると、耐熱性が向上するため、二次電池の安全性を向上させることができる。
例えば、ポリプロピレンのフィルムの両面に酸化アルミニウムとアラミドの混合材料をコートしてもよい。また、ポリプロピレンのフィルムの、正極と接する面に酸化アルミニウムとアラミドの混合材料をコートし、負極と接する面にフッ素系材料をコートしてもよい。
多層構造のセパレータを用いると、セパレータ全体の厚さが薄くても二次電池の安全性を保つことができるため、二次電池の体積あたりの容量を大きくすることができる。
[外装体]
二次電池が有する外装体としては、例えばアルミニウムなどの金属材料または樹脂材料を用いることができる。また、フィルム状の外装体を用いることもできる。フィルムとしては、例えばポリエチレン、ポリプロピレン、ポリカーボネート、アイオノマー、ポリアミド等の材料からなる膜上に、アルミニウム、ステンレス、銅、ニッケル等の可撓性に優れた金属薄膜を設け、さらに該金属薄膜上に外装体の外面としてポリアミド系樹脂、ポリエステル系樹脂等の絶縁性合成樹脂膜を設けた三層構造のフィルムを用いることができる。
二次電池が有する外装体としては、例えばアルミニウムなどの金属材料または樹脂材料を用いることができる。また、フィルム状の外装体を用いることもできる。フィルムとしては、例えばポリエチレン、ポリプロピレン、ポリカーボネート、アイオノマー、ポリアミド等の材料からなる膜上に、アルミニウム、ステンレス、銅、ニッケル等の可撓性に優れた金属薄膜を設け、さらに該金属薄膜上に外装体の外面としてポリアミド系樹脂、ポリエステル系樹脂等の絶縁性合成樹脂膜を設けた三層構造のフィルムを用いることができる。
(実施の形態4)
本実施の形態では、先の実施の形態で説明した作製方法によって作製された正極または負極を有する二次電池の複数種類の形状の例について説明する。
本実施の形態では、先の実施の形態で説明した作製方法によって作製された正極または負極を有する二次電池の複数種類の形状の例について説明する。
[コイン型二次電池]
コイン型の二次電池の一例について説明する。図19Aはコイン型(単層偏平型)の二次電池の分解斜視図であり、図19Bは、外観図であり、図19Cは、その断面図である。コイン型の二次電池は主に小型の電子機器に用いられる。
コイン型の二次電池の一例について説明する。図19Aはコイン型(単層偏平型)の二次電池の分解斜視図であり、図19Bは、外観図であり、図19Cは、その断面図である。コイン型の二次電池は主に小型の電子機器に用いられる。
なお、図19Aでは、わかりやすくするために部材の重なり(上下関係、及び位置関係)がわかるように模式図としている。従って図19Aと図19Bは完全に一致する対応図とはしていない。
図19Aでは、正極304、セパレータ310、負極307、スペーサ322、ワッシャー312を重ねている。これらを負極缶302と正極缶301で封止している。なお、図19Aにおいて、封止のためのガスケットは図示していない。スペーサ322、ワッシャー312は、正極缶301と負極缶302を圧着する際に、内部を保護または缶内の位置を固定するために用いられている。スペーサ322、ワッシャー312はステンレスまたは絶縁材料を用いる。
正極集電体305上に正極活物質層306が形成された積層構造を正極304としている。
正極と負極の短絡を防ぐため、セパレータ310を正極304の上面を覆うように配置する。セパレータ310は、正極304よりも広い平面面積を有している。
図19Bは、完成したコイン型の二次電池の斜視図である。
コイン型の二次電池300は、正極端子を兼ねた正極缶301と負極端子を兼ねた負極缶302とが、ポリプロピレン等で形成されたガスケット303で絶縁シールされている。正極304は、正極集電体305と、これと接するように設けられた正極活物質層306により形成される。また、負極307は、負極集電体308と、これに接するように設けられた負極活物質層309により形成される。また、負極307は、積層構造に限定されず、リチウム金属箔またはリチウムとアルミニウムの合金箔を用いてもよい。
なお、コイン型の二次電池300に用いる正極304及び負極307は、それぞれ活物質層は片面のみに形成すればよい。
正極缶301、負極缶302には、電解液に対して耐食性のあるニッケル、アルミニウム、チタン等の金属、若しくはこれらの合金又はこれらと他の金属との合金(例えばステンレス鋼等)を用いることができる。また、電解液などによる腐食を防ぐため、ニッケルまたはアルミニウム等を被覆することが好ましい。正極缶301は正極304と、負極缶302は負極307とそれぞれ電気的に接続する。
これら負極307、正極304及びセパレータ310を電解液に浸し、図19Cに示すように、正極缶301を下にして正極304、セパレータ310、負極307、負極缶302をこの順で積層し、正極缶301と負極缶302とをガスケット303を介して圧着してコイン形の二次電池300を製造する。
上記の構成を有することで、高容量、且つ、放電容量が高く、且つ、サイクル特性に優れたコイン型の二次電池300とすることができる。
[円筒型二次電池]
円筒型の二次電池の例について図20Aを参照して説明する。円筒型の二次電池616は、図20Aに示すように、上面に正極キャップ(電池蓋)601を有し、側面及び底面に電池缶(外装缶)602を有している。これら正極キャップ601と電池缶(外装缶)602とは、ガスケット(絶縁パッキン)610によって絶縁されている。
円筒型の二次電池の例について図20Aを参照して説明する。円筒型の二次電池616は、図20Aに示すように、上面に正極キャップ(電池蓋)601を有し、側面及び底面に電池缶(外装缶)602を有している。これら正極キャップ601と電池缶(外装缶)602とは、ガスケット(絶縁パッキン)610によって絶縁されている。
図20Bは、円筒型の二次電池の断面を模式的に示した図である。図20Bに示す円筒型の二次電池は、上面に正極キャップ(電池蓋)601を有し、側面及び底面に電池缶(外装缶)602を有している。これら正極キャップ601と電池缶(外装缶)602とは、ガスケット(絶縁パッキン)610によって絶縁されている。
中空円柱状の電池缶602の内側には、帯状の正極604と負極606とがセパレータ605を間に挟んで捲回された電池素子が設けられている。図示しないが、電池素子は中心軸を中心に捲回されている。電池缶602は、一端が閉じられ、他端が開いている。電池缶602には、電解液に対して耐腐食性のあるニッケル、アルミニウム、チタン等の金属、又はこれらの合金、これらと他の金属との合金(例えば、ステンレス鋼等)を用いることができる。また、電解液による腐食を防ぐため、ニッケル及びアルミニウム等を電池缶602に被覆することが好ましい。電池缶602の内側において、正極、負極及びセパレータが捲回された電池素子は、対向する一対の絶縁板608、609により挟まれている。また、電池素子が設けられた電池缶602の内部は、非水電解液(図示せず)が注入されている。非水電解液は、コイン型の二次電池と同様のものを用いることができる。
円筒型の蓄電池に用いる正極及び負極は捲回するため、集電体の両面に活物質を形成することが好ましい。
実施の形態1、2等で得られる正極活物質100を正極604に用いることで、高容量、且つ、放電容量が高く、且つ、サイクル特性に優れた円筒型の二次電池616とすることができる。
正極604には正極端子(正極集電リード)603が接続され、負極606には負極端子(負極集電リード)607が接続される。正極端子603及び負極端子607は、ともにアルミニウムなどの金属材料を用いることができる。正極端子603は安全弁機構613に、負極端子607は電池缶602の底にそれぞれ抵抗溶接される。安全弁機構613は、PTC素子(Positive Temperature Coefficient)611を介して正極キャップ601と電気的に接続されている。安全弁機構613は電池の内圧の上昇が所定の閾値を超えた場合に、正極キャップ601と正極604との電気的な接続を切断するものである。また、PTC素子611は温度が上昇した場合に抵抗が増大する熱感抵抗素子であり、抵抗の増大により電流量を制限して異常発熱を防止するものである。PTC素子には、チタン酸バリウム(BaTiO3)系半導体セラミックス等を用いることができる。
図20Cは蓄電システム615の一例を示す。蓄電システム615は複数の二次電池616を有する。それぞれの二次電池の正極は、絶縁体625で分離された導電体624に接触し、電気的に接続されている。導電体624は配線623を介して、制御回路620に電気的に接続されている。また、それぞれの二次電池の負極は、配線626を介して制御回路620に電気的に接続されている。制御回路620として、充放電などを行う充放電制御回路、または過充電もしくは/及び過放電を防止する保護回路を適用することができる。
図20Dは、蓄電システム615の一例を示す。蓄電システム615は複数の二次電池616を有し、複数の二次電池616は、導電板628及び導電板614の間に挟まれている。複数の二次電池616は、配線627により導電板628及び導電板614と電気的に接続される。複数の二次電池616は、並列接続されていてもよいし、直列接続されていてもよいし、並列に接続された後さらに直列に接続されていてもよい。複数の二次電池616を有する蓄電システム615を構成することで、大きな電力を取り出すことができる。
複数の二次電池616が、並列に接続された後、さらに直列に接続されてもよい。
また、複数の二次電池616の間に温度制御装置を有していてもよい。二次電池616が過熱されたときは、温度制御装置により冷却し、二次電池616が冷えすぎているときは温度制御装置により加熱することができる。そのため蓄電システム615の性能が外気温に影響されにくくなる。
また、図20Dにおいて、蓄電システム615は制御回路620に配線621及び配線622を介して電気的に接続されている。配線621は導電板628を介して複数の二次電池616の正極に、配線622は導電板614を介して複数の二次電池616の負極に、それぞれ電気的に接続される。
[二次電池の他の構造例]
二次電池の構造例について図21及び図22を用いて説明する。
二次電池の構造例について図21及び図22を用いて説明する。
図21Aに示す二次電池913は、筐体930の内部に端子951と端子952が設けられた捲回体950を有する。捲回体950は、筐体930の内部で電解液中に浸される。端子952は、筐体930に接し、端子951は、絶縁材などを用いることにより筐体930に接していない。なお、図21Aでは、便宜のため、筐体930を分離して図示しているが、実際は、捲回体950が筐体930に覆われ、端子951及び端子952が筐体930の外に延在している。筐体930としては、金属材料(例えばアルミニウムなど)又は樹脂材料を用いることができる。
なお、図21Bに示すように、図21Aに示す筐体930を複数の材料によって形成してもよい。例えば、図21Bに示す二次電池913は、筐体930aと筐体930bが貼り合わされており、筐体930a及び筐体930bで囲まれた領域に捲回体950が設けられている。
筐体930aとしては、有機樹脂など、絶縁材料を用いることができる。特に、アンテナが形成される面に有機樹脂などの材料を用いることにより、二次電池913による電界の遮蔽を抑制できる。なお、筐体930aによる電界の遮蔽が小さければ、筐体930aの内部にアンテナを設けてもよい。筐体930bとしては、例えば金属材料を用いることができる。
さらに、捲回体950の構造について図21Cに示す。捲回体950は、負極931と、正極932と、セパレータ933と、を有する。捲回体950は、セパレータ933を挟んで負極931と、正極932が重なり合って積層され、該積層シートを捲回させた捲回体である。なお、負極931と、正極932と、セパレータ933と、の積層を、さらに複数重ねてもよい。
また、図22に示すような捲回体950aを有する二次電池913としてもよい。図22Aに示す捲回体950aは、負極931と、正極932と、セパレータ933と、を有する。負極931は負極活物質層931aを有する。正極932は正極活物質層932aを有する。
実施の形態1、2等で得られる正極活物質100を正極932に用いることで、高容量、且つ、放電容量が高く、且つ、サイクル特性に優れた二次電池913とすることができる。
セパレータ933は、負極活物質層931a及び正極活物質層932aよりも広い幅を有し、負極活物質層931a及び正極活物質層932aと重畳するように捲回されている。また正極活物質層932aよりも負極活物質層931aの幅が広いことが安全性の点で好ましい。またこのような形状の捲回体950aは安全性及び生産性がよく好ましい。
図22Bに示すように、負極931は、超音波接合、溶接、または圧着により端子951と電気的に接続される。端子951は端子911aと電気的に接続される。また正極932は、超音波接合、溶接、または圧着により端子952と電気的に接続される。端子952は端子911bと電気的に接続される。
図22Cに示すように、筐体930により捲回体950a及び電解液が覆われ、二次電池913となる。筐体930には安全弁、過電流保護素子等を設けることが好ましい。安全弁は、電池破裂を防止するため、筐体930の内部が所定の内圧で開放する弁である。
図22Bに示すように二次電池913は複数の捲回体950aを有していてもよい。複数の捲回体950aを用いることで、より放電容量の大きい二次電池913とすることができる。図22A及び図22Bに示す二次電池913の他の要素は、図21A乃至図21Cに示す二次電池913の記載を参酌することができる。
<ラミネート型二次電池>
次に、ラミネート型の二次電池の例について、外観図の一例を図23A及び図23Bに示す。図23A及び図23Bは、正極503、負極506、セパレータ507、外装体509、正極リード電極510、及び負極リード電極511を有する。
次に、ラミネート型の二次電池の例について、外観図の一例を図23A及び図23Bに示す。図23A及び図23Bは、正極503、負極506、セパレータ507、外装体509、正極リード電極510、及び負極リード電極511を有する。
図24Aは正極503及び負極506の外観図を示す。正極503は正極集電体501を有し、正極活物質層502は正極集電体501の表面に形成されている。また、正極503は正極集電体501が一部露出する領域(以下、タブ領域という)を有する。負極506は負極集電体504を有し、負極活物質層505は負極集電体504の表面に形成されている。また、負極506は負極集電体504が一部露出する領域、すなわちタブ領域を有する。なお、正極及び負極が有するタブ領域の面積または形状は、図24Aに示す例に限られない。
<ラミネート型二次電池の作製方法>
図23Aに外観図を示すラミネート型二次電池の作製方法の一例について、図24B及び図24Cを用いて説明する。
図23Aに外観図を示すラミネート型二次電池の作製方法の一例について、図24B及び図24Cを用いて説明する。
まず、負極506、セパレータ507及び正極503を積層する。図24Bに積層された負極506、セパレータ507及び正極503を示す。ここでは負極を5組、正極を4組使用する例を示す。負極とセパレータと正極からなる積層体とも呼べる。次に、正極503のタブ領域同士の接合と、最表面の正極のタブ領域への正極リード電極510の接合を行う。接合には、例えば超音波溶接等を用いればよい。同様に、負極506のタブ領域同士の接合と、最表面の負極のタブ領域への負極リード電極511の接合を行う。
次に、外装体509上に、負極506、セパレータ507及び正極503を配置する。
次に、図24Cに示すように、外装体509を破線で示した部分で折り曲げる。その後、外装体509の外周部を接合する。接合には例えば熱圧着等を用いればよい。この時、後に電解液を入れることができるように、外装体509の一部(または一辺)に接合されない領域(以下、導入口という)を設ける。
次に、外装体509に設けられた導入口から、電解液を外装体509の内側へ導入する。電解液の導入は、減圧雰囲気下、或いは不活性雰囲気下で行うことが好ましい。そして最後に、導入口を接合する。このようにして、ラミネート型の二次電池500を作製することができる。
実施の形態1、2等で得られる正極活物質100を正極503に用いることで、高容量、且つ、放電容量が高く、且つ、サイクル特性に優れた二次電池500とすることができる。
[電池パックの例]
アンテナを用いて無線充電が可能な本発明の一態様の二次電池パックの例について、図25を用いて説明する。
アンテナを用いて無線充電が可能な本発明の一態様の二次電池パックの例について、図25を用いて説明する。
図25Aは、二次電池パック531の外観を示す図であり、厚さの薄い直方体形状(厚さのある平板形状とも呼べる)である。図25Bは、二次電池パック531の構成を説明する図である。二次電池パック531は、回路基板540と、二次電池513と、を有する。二次電池513には、ラベル529が貼られている。回路基板540は、シール515により固定されている。また、二次電池パック531は、アンテナ517を有する。
二次電池513の内部は、捲回体を有する構造にしてもよいし、積層体を有する構造にしてもよい。
二次電池パック531において、例えば図25Bに示すように、回路基板540上に制御回路590を有する。また、回路基板540は、端子514と電気的に接続されている。また回路基板540は、アンテナ517、二次電池513の正極リード及び負極リードの一方551、正極リード及び負極リードの他方552と電気的に接続される。
または、図25Cに示すように、回路基板540上に設けられる回路システム590aと、端子514を介して回路基板540に電気的に接続される回路システム590bと、を有してもよい。
なお、アンテナ517はコイル状に限定されず、例えば線状、板状であってもよい。また、平面アンテナ、開口面アンテナ、進行波アンテナ、EHアンテナ、磁界アンテナ、誘電体アンテナ等のアンテナを用いてもよい。又は、アンテナ517は、平板状の導体でもよい。この平板状の導体は、電界結合用の導体の一つとして機能することができる。つまり、コンデンサの有する2つの導体のうちの一つの導体として、アンテナ517を機能させてもよい。これにより、電磁界、磁界だけでなく、電界で電力のやり取りを行うこともできる。
二次電池パック531は、アンテナ517と、二次電池513との間に層519を有する。層519は、例えば二次電池513による電磁界を遮蔽することができる機能を有する。層519としては、例えば磁性体を用いることができる。
(実施の形態5)
本実施の形態では、円筒型の二次電池である図20Dとは異なる例である。図26Cを用いて電気自動車(EV)に適用する例を示す。
本実施の形態では、円筒型の二次電池である図20Dとは異なる例である。図26Cを用いて電気自動車(EV)に適用する例を示す。
電気自動車には、メインの駆動用の二次電池として第1のバッテリ1301a、1301bと、モータ1304を始動させるインバータ1312に電力を供給する第2のバッテリ1311が設置されている。第2のバッテリ1311はクランキングバッテリー(スターターバッテリーとも呼ばれる)とも呼ばれる。第2のバッテリ1311は高出力できればよく、大容量はそれほど必要とされず、第2のバッテリ1311の容量は第1のバッテリ1301a、1301bと比較して小さい。
第1のバッテリ1301aの内部構造は、図21Cまたは図22Aに示した捲回型であってもよいし、図23Aまたは図23Bに示した積層型であってもよい。また、第1のバッテリ1301aは、実施の形態6の全固体電池を用いてもよい。第1のバッテリ1301aに実施の形態6の全固体電池を用いることで高容量とすることができ、安全性が向上し、小型化、軽量化することができる。
本実施の形態では、第1のバッテリ1301a、1301bを2つ並列に接続させている例を示しているが3つ以上並列に接続させてもよい。また、第1のバッテリ1301aで十分な電力を貯蔵できるのであれば、第1のバッテリ1301bはなくてもよい。複数の二次電池を有する電池パックを構成することで、大きな電力を取り出すことができる。複数の二次電池は、並列接続されていてもよいし、直列接続されていてもよいし、並列に接続された後、さらに直列に接続されていてもよい。複数の二次電池を組電池とも呼ぶ。
また、車載用の二次電池において、複数の二次電池からの電力を遮断するため、工具を使わずに高電圧を遮断できるサービスプラグまたはサーキットブレーカを有しており、第1のバッテリ1301aに設けられる。
また、第1のバッテリ1301a、1301bの電力は、主にモータ1304を回転させることに使用されるが、DCDC回路1306を介して42V系の車載部品(電動パワステ1307、ヒーター1308、デフォッガ1309など)に電力を供給する。後輪にリアモータ1317を有している場合にも、第1のバッテリ1301aがリアモータ1317を回転させることに使用される。
また、第2のバッテリ1311は、DCDC回路1310を介して14V系の車載部品(オーディオ1313、パワーウィンドウ1314、ランプ類1315など)に電力を供給する。
次に、第1のバッテリ1301aについて、図26Aを用いて説明する。
図26Aでは9個の角型二次電池1300を一つの電池パック1415としている例を示している。また、9個の角型二次電池1300を直列接続し、一方の電極を絶縁体からなる固定部1413で固定し、もう一方の電極を絶縁体からなる固定部1414で固定している。本実施の形態では固定部1413、1414で固定する例を示しているが電池収容ボックス(筐体とも呼ぶ)に収納させる構成としてもよい。車両は外部(路面など)から振動または揺れが加えられることを想定されているため、固定部1413、1414や。電池収容ボックスなどで複数の二次電池を固定することが好ましい。また、一方の電極は配線1421によって制御回路部1320に電気的に接続されている。またもう一方の電極は配線1422によって制御回路部1320に電気的に接続されている。
また、制御回路部1320は、酸化物半導体を用いたトランジスタを含むメモリ回路を用いてもよい。酸化物半導体を用いたトランジスタを含むメモリ回路を有する充電制御回路、又は電池制御システムを、BTOS(Battery operating system、又はBattery oxide semiconductor)と呼称する場合がある。
酸化物半導体として機能する金属酸化物を用いることが好ましい。例えば、酸化物として、In−M−Zn酸化物(元素Mは、アルミニウム、ガリウム、イットリウム、銅、バナジウム、ベリリウム、ホウ素、チタン、鉄、ニッケル、ゲルマニウム、ジルコニウム、モリブデン、ランタン、セリウム、ネオジム、ハフニウム、タンタル、タングステン、又はマグネシウム等から選ばれた一種、又は複数種)等の金属酸化物を用いるとよい。特に、酸化物として適用できるIn−M−Zn酸化物は、CAAC−OS(C−Axis Aligned Crystal Oxide Semiconductor)、CAC−OS(Cloud−Aligned Composite Oxide Semiconductor)であることが好ましい。また、酸化物として、In−Ga酸化物、In−Zn酸化物を用いてもよい。CAAC−OSは、複数の結晶領域を有し、当該複数の結晶領域はc軸が特定の方向に配向している酸化物半導体である。なお、特定の方向とは、CAAC−OS膜の厚さ方向、CAAC−OS膜の被形成面の法線方向、またはCAAC−OS膜の表面の法線方向である。また、結晶領域とは、原子配列に周期性を有する領域である。なお、原子配列を格子配列とみなすと、結晶領域とは、格子配列の揃った領域でもある。
なお、「CAC−OS」は、第1の領域と、第2の領域と、に材料が分離することでモザイク状となり、当該第1の領域が、膜中に分布した構成(以下、クラウド状ともいう。)である。つまり、CAC−OSは、当該第1の領域と、当該第2の領域とが、混合している構成を有する複合金属酸化物である。ただし、第1の領域と第2の領域は、明確な境界が観察困難な場合がある。
例えば、In−Ga−Zn酸化物におけるCAC−OSでは、エネルギー分散型X線分光法(EDX:Energy Dispersive X−ray spectroscopy)を用いて取得したEDXマッピングにより、Inを主成分とする領域(第1の領域)と、Gaを主成分とする領域(第2の領域)とが、偏在し、混合している構造を有することが確認できる。
CAC−OSをトランジスタに用いる場合、第1の領域に起因する導電性と、第2の領域に起因する絶縁性とが、相補的に作用することにより、スイッチングさせる機能(On/Offさせる機能)をCAC−OSに付与することができる。つまり、CAC−OSとは、材料の一部では導電性の機能と、材料の一部では絶縁性の機能とを有し、材料の全体では半導体としての機能を有する。導電性の機能と絶縁性の機能とを分離させることで、双方の機能を最大限に高めることができる。よって、CAC−OSをトランジスタに用いることで、高いオン電流(Ion)、高い電界効果移動度(μ)、及び良好なスイッチング動作を実現することができる。
酸化物半導体は、多様な構造をとり、それぞれが異なる特性を有する。本発明の一態様の酸化物半導体は、非晶質酸化物半導体、多結晶酸化物半導体、a−like OS、CAC−OS、nc−OS、CAAC−OSのうち、二種以上を有していてもよい。
また、高温環境下で使用可能であるため、制御回路部1320は酸化物半導体を用いるトランジスタを用いることが好ましい。プロセスを簡略なものとするため、制御回路部1320は単極性のトランジスタを用いて形成してもよい。半導体層に酸化物半導体を用いるトランジスタは、動作周囲温度が単結晶Siよりも広く−40℃以上150℃以下であり、二次電池が加熱しても特性変化が単結晶に比べて小さい。酸化物半導体を用いるトランジスタのオフ電流は、150℃であっても温度によらず測定下限以下であるが、単結晶Siトランジスタのオフ電流特性は、温度依存性が大きい。例えば、150℃では、単結晶Siトランジスタはオフ電流が上昇し、電流オン/オフ比が十分に大きくならない。制御回路部1320は、安全性を向上することができる。また、実施の形態1、2等で得られる正極活物質100を正極に用いた二次電池と組み合わせることで安全性についての相乗効果が得られる。実施の形態1、2等で得られる正極活物質100を正極に用いた二次電池及び制御回路部1320は、二次電池による火災等の事故撲滅に大きく寄与することができる。
酸化物半導体を用いたトランジスタを含むメモリ回路を用いた制御回路部1320は、マイクロショート等の不安定性の原因に対し、二次電池の自動制御装置として機能させることもできる。二次電池の不安定性の原因を解消する機能としては、過充電の防止、過電流の防止、充電時過熱制御、組電池でのセルバランス、過放電の防止、残量計、温度に応じた充電電圧及び電流量自動制御、劣化度に応じた充電電流量制御、マイクロショート異常挙動検知、マイクロショートに関する異常予測などが挙げられ、そのうちの少なくとも一つの機能を制御回路部1320が有する。また、二次電池の自動制御装置の超小型化が可能である。
また、「マイクロショート」とは、二次電池の内部の微小な短絡のことを指しており、二次電池の正極と負極が短絡して充放電不可能の状態になるというほどではなく、微小な短絡部でわずかに短絡電流が流れてしまう現象を指している。比較的短時間、且つ、わずかな箇所であっても大きな電圧変化が生じるため、その異常な電圧値がその後の推定に影響を与える恐れがある。
マイクロショートの原因の一つは、充放電が複数回行われることによって、正極活物質の不均一な分布により、正極の一部と負極の一部で局所的な電流の集中が生じ、セパレータの一部が機能しなくなる箇所が発生、または副反応による副反応物の発生によりミクロな短絡が生じていると言われている。
また、マイクロショートの検知だけでなく、制御回路部1320は、二次電池の端子電圧を検知し、二次電池の充放電状態を管理するとも言える。例えば、過充電を防ぐために充電回路の出力トランジスタと遮断用スイッチの両方をほぼ同時にオフ状態とすることができる。
次に、図26Aに示す電池パック1415のブロック図の一例を図26Bに示す。
制御回路部1320は、少なくとも過充電を防止するスイッチと、過放電を防止するスイッチを含むスイッチ部1324と、スイッチ部1324を制御する制御回路1322と、第1のバッテリ1301aの電圧測定部と、を有する。制御回路部1320は、使用する二次電池の上限電圧と下限電圧が設定されており、外部からの電流上限、または外部への出力電流の上限などを制限している。二次電池の下限電圧以上上限電圧以下の範囲内は、使用が推奨されている電圧範囲内であり、その範囲外となるとスイッチ部1324が作動し、保護回路として機能する。また、制御回路部1320は、スイッチ部1324を制御して過放電および/または過充電を防止するため、保護回路とも呼べる。例えば、過充電となりそうな電圧を制御回路1322で検知した場合にスイッチ部1324のスイッチをオフ状態とすることで電流を遮断する。さらに充放電経路中にPTC素子を設けて温度の上昇に応じて電流を遮断する機能を設けてもよい。また、制御回路部1320は、外部端子1325(+IN)と、外部端子1326(−IN)とを有している。
スイッチ部1324は、nチャネル型のトランジスタまたはpチャネル型のトランジスタを組み合わせて構成することができる。スイッチ部1324は、単結晶シリコンを用いるSiトランジスタを有するスイッチに限定されず、例えば、Ge(ゲルマニウム)、SiGe(シリコンゲルマニウム)、GaAs(ガリウムヒ素)、GaAlAs(ガリウムアルミニウムヒ素)、InP(リン化インジウム)、SiC(シリコンカーバイド)、ZnSe(セレン化亜鉛)、GaN(窒化ガリウム)、GaOx(酸化ガリウム;xは0より大きい実数)などを有するパワートランジスタでスイッチ部1324を形成してもよい。また、OSトランジスタを用いた記憶素子は、Siトランジスタを用いた回路上などに積層することで自由に配置可能であるため、集積化を容易に行うことができる。またOSトランジスタは、Siトランジスタと同様の製造装置を用いて作製することが可能であるため、低コストで作製可能である。即ち、スイッチ部1324上にOSトランジスタを用いた制御回路部1320を積層し、集積化することで1チップとすることもできる。制御回路部1320の占有体積を小さくすることができるため、小型化が可能となる。
第1のバッテリ1301a、1301bは、主に42V系(高電圧系)の車載機器に電力を供給し、第2のバッテリ1311は14V系(低電圧系)の車載機器に電力を供給する。第2のバッテリ1311は鉛蓄電池がコスト上有利のため採用されることが多い。鉛蓄電池はリチウムイオン電池と比べて自己放電が大きく、サルフェーションとよばれる現象により劣化しやすい欠点がある。第2のバッテリ1311をリチウムイオン電池とすることでメンテナンスフリーとするメリットがあるが、長期間の使用、例えば3年以上となると、製造時には判別困難な異常発生が生じる恐れがある。特にインバータを起動する第2のバッテリ1311が動作不能となると、第1のバッテリ1301a、1301bに残容量があってもモータを起動させることができなくなることを防ぐため、第2のバッテリ1311が鉛蓄電池の場合は、第1のバッテリから第2のバッテリに電力を供給し、常に満充電状態を維持するように充電されている。
本実施の形態では、第1のバッテリ1301aと第2のバッテリ1311の両方にリチウムイオン電池を用いる一例を示す。第2のバッテリ1311は、鉛蓄電池、全固体電池、または電気二重層キャパシタを用いてもよい。例えば、実施の形態6の全固体電池を用いてもよい。第2のバッテリ1311に実施の形態6の全固体電池を用いることで高容量とすることができ、小型化、軽量化することができる。
また、タイヤ1316の回転による回生エネルギーは、ギア1305を介してモータ1304に送られ、モータコントローラ1303、またはバッテリーコントローラ1302から制御回路部1321を介して第2のバッテリ1311に充電される。またはバッテリーコントローラ1302から制御回路部1320を介して第1のバッテリ1301aに充電される。またはバッテリーコントローラ1302から制御回路部1320を介して第1のバッテリ1301bに充電される。回生エネルギーを効率よく充電するためには、第1のバッテリ1301a、1301bが急速充電可能であることが望ましい。
バッテリーコントローラ1302は第1のバッテリ1301a、1301bの充電電圧及び充電電流などを設定することができる。バッテリーコントローラ1302は、用いる二次電池の充電特性に合わせて充電条件を設定し、急速充電することができる。
また、図示していないが、外部の充電器と接続させる場合、充電器のコンセントまたは充電器の接続ケーブルは、バッテリーコントローラ1302に電気的に接続される。外部の充電器から供給された電力はバッテリーコントローラ1302を介して第1のバッテリ1301a、1301bに充電する。また、充電器によっては、制御回路が設けられており、バッテリーコントローラ1302の機能を用いない場合もあるが、過充電を防ぐため制御回路部1320を介して第1のバッテリ1301a、1301bを充電することが好ましい。また、接続ケーブルまたは充電器の接続ケーブルに制御回路を備えている場合もある。制御回路部1320は、ECU(Electronic Control Unit)と呼ばれることもある。ECUは、電動車両に設けられたCAN(Controller Area Network)に接続される。CANは、車内LANとして用いられるシリアル通信規格の一つである。また、ECUは、マイクロコンピュータを含む。また、ECUは、CPUまたはGPUを用いる。
充電スタンドなどに設置されている外部の充電器は、100Vコンセント−200Vコンセント、または3相200V且つ50kWなどがある。また、非接触給電方式等により外部の充電設備から電力供給を受けて、充電することもできる。
急速充電を行う場合、短時間での充電を行うためには、高電圧での充電に耐えうる二次電池が望まれている。
また、導電助剤としてグラフェンを用い、電極層を厚くして担持量を高くしても容量低下を抑え、高容量を維持することが相乗効果として大幅に電気特性が向上された二次電池を実現できる。特に車両に用いる二次電池に有効であり、車両全重量に対する二次電池の重量の割合を増加させることなく、航続距離が長い、具体的には一充電走行距離が500km以上の車両を提供することができる。
特に上述した本実施の形態の二次電池は、実施の形態1、2等で説明した正極活物質100を用いることで二次電池の動作電圧を高くすることができ、充電電圧の増加に伴い、使用できる容量を増加させることができる。また、実施の形態1、2等で説明した正極活物質100を正極に用いることでサイクル特性に優れた車両用の二次電池を提供することができる。
次に、本発明の一態様である二次電池を車両、代表的には輸送用車両に実装する例について説明する。
図20D、図22C、図26Aのいずれか一に示した二次電池を車両に搭載すると、ハイブリッド車(HV)、電気自動車(EV)、又はプラグインハイブリッド車(PHV)等の次世代クリーンエネルギー自動車を実現できる。また、農業機械、電動アシスト自転車を含む原動機付自転車、自動二輪車、電動車椅子、電動カート、船舶、潜水艦、航空機、ロケット、人工衛星、宇宙探査機、惑星探査機、または宇宙船に二次電池を搭載することもできる。本発明の一態様の二次電池は高容量の二次電池とすることができる。そのため本発明の一態様の二次電池は、小型化、軽量化に適しており、輸送用車両に好適に用いることができる。
図27A乃至図27Dにおいて、本発明の一態様を用いた輸送用車両を例示する。図27Aに示す自動車2001は、走行のための動力源として電気モータを用いる電気自動車である。または、走行のための動力源として電気モータとエンジンを適宜選択して用いることが可能なハイブリッド自動車である。二次電池を車両に搭載する場合、実施の形態4で示した二次電池の一例を一箇所または複数個所に設置する。図27Aに示す自動車2001は、電池パック2200を有し、電池パックは、複数の二次電池を接続させた二次電池モジュールを有する。さらに二次電池モジュールに電気的に接続する充電制御装置を有すると好ましい。
また、自動車2001は、自動車2001が有する二次電池にプラグイン方式または非接触給電方式等により外部の充電設備から電力供給を受けて、充電することができる。充電に際しては、充電方法またはコネクタの規格等はCHAdeMO(登録商標)またはコンボ等の所定の方式で適宜行えばよい。二次電池は、商用施設に設けられた充電ステーションでもよく、また家庭の電源であってもよい。例えば、プラグイン技術によって、外部からの電力供給により自動車2001に搭載された蓄電装置を充電することができる。充電は、ACDCコンバータ等の変換装置を介して、交流電力を直流電力に変換して行うことができる。
また、図示しないが、受電装置を車両に搭載し、地上の送電装置から電力を非接触で供給して充電することもできる。この非接触給電方式の場合には、道路または外壁に送電装置を組み込むことで、停車中に限らず走行中に充電を行うこともできる。また、この非接触給電の方式を利用して、2台の車両どうしで電力の送受電を行ってもよい。さらに、車両の外装部に太陽電池を設け、停車時または走行時に二次電池の充電を行ってもよい。このような非接触での電力の供給には、電磁誘導方式または磁界共鳴方式を用いることができる。
図27Bは、輸送用車両の一例として電気により制御するモータを有した大型の輸送車2002を示している。輸送車2002の二次電池モジュールは、例えば公称電圧3.0V以上5.0V以下の二次電池を4個セルユニットとし、48セルを直列に接続した170Vの最大電圧とする。電池パック2201の二次電池モジュールを構成する二次電池の数などが違う以外は、図27Aと同様な機能を備えているので説明は省略する。
図27Cは、一例として電気により制御するモータを有した大型の輸送車両2003を示している。輸送車両2003の二次電池モジュールは、例えば公称電圧3.0V以上5.0V以下の二次電池を百個以上直列に接続した600Vの最大電圧とする。従って、特性バラツキの小さい二次電池が求められる。実施の形態1、2等で説明した正極活物質100を正極に用いた二次電池を用いることで、安定した電池特性を有する二次電池を製造することができ、歩留まりの観点から低コストで大量生産が可能である。また、電池パック2202の二次電池モジュールを構成する二次電池の数などが違う以外は、図16Aと同様な機能を備えているので説明は省略する。
図27Dは、一例として燃料を燃焼するエンジンを有した航空機2004を示している。図27Dに示す航空機2004は、離着陸用の車輪を有しているため、輸送車両の一種とも言え、複数の二次電池を接続させて二次電池モジュールを構成し、二次電池モジュールと充電制御装置とを含む電池パック2203を有している。
航空機2004の二次電池モジュールは、例えば4Vの二次電池を8個直列に接続した32Vの最大電圧とする。電池パック2203の二次電池モジュールを構成する二次電池の数などが異なる以外は、図27Aと同様な機能を備えているので説明は省略する。
図27Eは、一例として二次電池2204を備えた人工衛星2005を示している。人工衛星2005は極低温の宇宙空間で使用されるため、低温耐性に優れた本発明の一態様である二次電池2204を備えることが好ましい。また、人工衛星2005の内部において、保温部材に覆われた状態で二次電池2204が搭載されることがさらに好ましい。
(実施の形態6)
本実施の形態では、本発明の一態様である二次電池を建築物に実装する例について図28A及び図28Bを用いて説明する。
本実施の形態では、本発明の一態様である二次電池を建築物に実装する例について図28A及び図28Bを用いて説明する。
図28Aに示す住宅は、本発明の一態様である二次電池を有する蓄電装置2612と、ソーラーパネル2610を有する。蓄電装置2612は、ソーラーパネル2610と配線2611等を介して電気的に接続されている。また蓄電装置2612と地上設置型の充電装置2604が電気的に接続されていてもよい。ソーラーパネル2610で得た電力は、蓄電装置2612に充電することができる。また蓄電装置2612に蓄えられた電力は、充電装置2604を介して車両2603が有する二次電池に充電することができる。蓄電装置2612は、床下空間部に設置されることが好ましい。床下空間部に設置することにより、床上の空間を有効的に利用することができる。あるいは、蓄電装置2612は床上に設置されてもよい。
蓄電装置2612に蓄えられた電力は、住宅内の他の電子機器にも電力を供給することができる。よって、停電などにより商用電源から電力の供給が受けられない時でも、本発明の一態様に係る蓄電装置2612を無停電電源として用いることで、電子機器の利用が可能となる。
図28Bに、本発明の一態様に係る蓄電装置の一例を示す。図28Bに示すように、建物799の床下空間部796には、本発明の一態様に係る蓄電装置791が設置されている。また、蓄電装置791に実施の形態7に説明した制御回路を設けてもよく、実施の形態1、2等で得られる正極活物質100を正極に用いた二次電池を蓄電装置791に用いることで安全性についての相乗効果が得られる。実施の形態7に説明した制御回路及び実施の形態1、2等で説明した正極活物質100を正極に用いた二次電池は、二次電池を有する蓄電装置791による火災等の事故撲滅に大きく寄与することができる。
蓄電装置791には、制御装置790が設置されており、制御装置790は、配線によって、分電盤703と、蓄電コントローラ705(制御装置ともいう)と、表示器706と、ルータ709と、に電気的に接続されている。
商業用電源701から、引込線取付部710を介して、電力が分電盤703に送られる。また、分電盤703には、蓄電装置791と、商業用電源701と、から電力が送られ、分電盤703は、送られた電力を、コンセント(図示せず)を介して、一般負荷707及び蓄電系負荷708に供給する。
一般負荷707は、例えばテレビまたはパーソナルコンピュータなどの電気機器であり、蓄電系負荷708は、例えば、電子レンジ、冷蔵庫、空調機などの電気機器である。
蓄電コントローラ705は、計測部711と、予測部712と、計画部713と、を有する。計測部711は、一日(例えば、0時から24時)の間に、一般負荷707、蓄電系負荷708で消費された電力量を計測する機能を有する。また、計測部711は、蓄電装置791の電力量と、商業用電源701から供給された電力量と、を計測する機能を有していてもよい。また、予測部712は、一日の間に一般負荷707及び蓄電系負荷708で消費された電力量に基づいて、次の一日の間に一般負荷707及び蓄電系負荷708で消費される需要電力量を予測する機能を有する。また、計画部713は、予測部712が予測した需要電力量に基づいて、蓄電装置791の充放電の計画を立てる機能を有する。
計測部711によって計測された一般負荷707及び蓄電系負荷708で消費された電力量は、表示器706によって確認することができる。また、ルータ709を介して、テレビまたはパーソナルコンピュータなどの電気機器において、確認することもできる。さらに、ルータ709を介して、スマートフォンまたはタブレットなどの携帯電子端末によっても確認することができる。また、表示器706、電気機器、携帯電子端末によって、予測部712が予測した時間帯ごと(または一時間ごと)の需要電力量なども確認することができる。
(実施の形態7)
本実施の形態では、二輪車、自転車に本発明の一態様である蓄電装置を搭載する例を示す。
本実施の形態では、二輪車、自転車に本発明の一態様である蓄電装置を搭載する例を示す。
図29Aは、本発明の一態様の蓄電装置を用いた電動自転車の一例である。図29Aに示す電動自転車8700に、本発明の一態様の蓄電装置を適用することができる。本発明の一態様の蓄電装置は例えば、複数の蓄電池と、保護回路と、を有する。
電動自転車8700は、蓄電装置8702を備える。蓄電装置8702は、運転者をアシストするモータに電気を供給することができる。また、蓄電装置8702は、持ち運びができ、図29Bに自転車から取り外した状態を示している。また、蓄電装置8702は、本発明の一態様の蓄電装置が有する蓄電池8701が複数内蔵されており、そのバッテリ残量などを表示部8703で表示できるようにしている。また蓄電装置8702は、実施の形態7に一例を示した二次電池の充電制御または異常検知が可能な制御回路8704を有する。制御回路8704は、蓄電池8701の正極及び負極と電気的に接続されている。また、実施の形態1、2等で得られる正極活物質100を正極に用いた二次電池と組み合わせることで、安全性についての相乗効果が得られる。実施の形態1、2等で得られる正極活物質100を正極に用いた二次電池及び制御回路8704は、二次電池による火災等の事故撲滅に大きく寄与することができる。
図29Cは、本発明の一態様の蓄電装置を用いた二輪車の一例である。図29Cに示すスクータ8600は、蓄電装置8602、サイドミラー8601、方向指示灯8603を備える。蓄電装置8602は、方向指示灯8603に電気を供給することができる。また、実施の形態1、2等で得られる正極活物質100を正極に用いた二次電池を複数収納された蓄電装置8602は高容量とすることができ、小型化に寄与することができる。
また、図29Cに示すスクータ8600は、座席下収納8604に、蓄電装置8602を収納することができる。蓄電装置8602は、座席下収納8604が小型であっても、座席下収納8604に収納することができる。
(実施の形態8)
本実施の形態では、本発明の一態様である二次電池を電子機器に実装する例について説明する。二次電池を実装する電子機器として、例えば、テレビジョン装置(テレビ、又はテレビジョン受信機ともいう)、コンピュータ用などのモニタ、デジタルカメラ、デジタルビデオカメラ、デジタルフォトフレーム、携帯電話機(携帯電話、携帯電話装置ともいう)、携帯型ゲーム機、携帯情報端末、音響再生装置、パチンコ機などの大型ゲーム機などが挙げられる。携帯情報端末としてはノート型パーソナルコンピュータ、タブレット型端末、電子書籍端末、携帯電話機などがある。
本実施の形態では、本発明の一態様である二次電池を電子機器に実装する例について説明する。二次電池を実装する電子機器として、例えば、テレビジョン装置(テレビ、又はテレビジョン受信機ともいう)、コンピュータ用などのモニタ、デジタルカメラ、デジタルビデオカメラ、デジタルフォトフレーム、携帯電話機(携帯電話、携帯電話装置ともいう)、携帯型ゲーム機、携帯情報端末、音響再生装置、パチンコ機などの大型ゲーム機などが挙げられる。携帯情報端末としてはノート型パーソナルコンピュータ、タブレット型端末、電子書籍端末、携帯電話機などがある。
図30Aは、携帯電話機の一例を示している。携帯電話機2100は、筐体2101に組み込まれた表示部2102の他、操作ボタン2103、外部接続ポート2104、スピーカ2105、マイク2106などを備えている。なお、携帯電話機2100は、二次電池2107を有している。実施の形態1、2等で説明した正極活物質100を正極に用いた二次電池2107を備えることで高容量とすることができ、筐体の小型化に伴う省スペース化に対応できる構成を実現することができる。
携帯電話機2100は、移動電話、電子メール、文章閲覧及び作成、音楽再生、インターネット通信、コンピュータゲームなどの種々のアプリケーションを実行することができる。
操作ボタン2103は、時刻設定のほか、電源のオン、オフ動作、無線通信のオン、オフ動作、マナーモードの実行及び解除、省電力モードの実行及び解除など、様々な機能を持たせることができる。例えば、携帯電話機2100に組み込まれたオペレーティングシステムにより、操作ボタン2103の機能を自由に設定することもできる。
また、携帯電話機2100は、通信規格された近距離無線通信を実行することが可能である。例えば無線通信可能なヘッドセットと相互通信することによって、ハンズフリーで通話することもできる。
また、携帯電話機2100は、外部接続ポート2104を備え、他の情報端末とコネクタを介して直接データのやりとりを行うことができる。また外部接続ポート2104を介して充電を行うこともできる。なお、充電動作は外部接続ポート2104を介さずに無線給電により行ってもよい。
また、携帯電話機2100は、センサを有することが好ましい。センサとしては、例えば、指紋センサ、脈拍センサ、体温センサ等の人体センサ、タッチセンサ、加圧センサ、または加速度センサ等が搭載されることが好ましい。
図30Bは、複数のローター2302を有する無人航空機2300である。無人航空機2300はドローンと呼ばれることもある。無人航空機2300は、本発明の一態様である二次電池2301と、カメラ2303と、アンテナ(図示しない)を有する。無人航空機2300はアンテナを介して遠隔操作することができる。実施の形態1、2等で得られる正極活物質100を正極に用いた二次電池は高エネルギー密度であり、安全性が高いため、長期間に渡って長時間の安全な使用ができ、無人航空機2300に搭載する二次電池として好適である。
図30Cは、ロボットの一例を示している。図30Cに示すロボット6400は、二次電池6409、照度センサ6401、マイクロフォン6402、上部カメラ6403、スピーカ6404、表示部6405、下部カメラ6406及び障害物センサ6407、移動機構6408、演算装置等を備える。
マイクロフォン6402は、使用者の話し声及び環境音等を検知する機能を有する。また、スピーカ6404は、音声を発する機能を有する。ロボット6400は、マイクロフォン6402及びスピーカ6404を用いて、使用者とコミュニケーションをとることが可能である。
表示部6405は、種々の情報の表示を行う機能を有する。ロボット6400は、使用者の望みの情報を表示部6405に表示することが可能である。表示部6405は、タッチパネルを搭載していてもよい。また、表示部6405は取り外しのできる情報端末であっても良く、ロボット6400の定位置に設置することで、充電及びデータの受け渡しを可能とする。
上部カメラ6403及び下部カメラ6406は、ロボット6400の周囲を撮像する機能を有する。また、障害物センサ6407は、移動機構6408を用いてロボット6400が前進する際の進行方向における障害物の有無を察知することができる。ロボット6400は、上部カメラ6403、下部カメラ6406及び障害物センサ6407を用いて、周囲の環境を認識し、安全に移動することが可能である。
ロボット6400は、その内部領域に本発明の一態様に係る二次電池6409と、半導体装置または電子部品を備える。実施の形態1、2等で得られる正極活物質100を正極に用いた二次電池は高エネルギー密度であり、安全性が高いため、長期間に渡って長時間の安全な使用ができ、ロボット6400に搭載する二次電池6409として好適である。
図30Dは、掃除ロボットの一例を示している。掃除ロボット6300は、筐体6301上面に配置された表示部6302、側面に配置された複数のカメラ6303、ブラシ6304、操作ボタン6305、二次電池6306、各種センサなどを有する。図示されていないが、掃除ロボット6300には、タイヤ、吸い込み口等が備えられている。掃除ロボット6300は自走し、ゴミ6310を検知し、下面に設けられた吸い込み口からゴミを吸引することができる。
掃除ロボット6300は、カメラ6303が撮影した画像を解析し、壁、家具または段差などの障害物の有無を判断することができる。また、画像解析により、配線などブラシ6304に絡まりそうな物体を検知した場合は、ブラシ6304の回転を止めることができる。掃除ロボット6300は、その内部領域に本発明の一態様に係る二次電池6306と、半導体装置または電子部品を備える。実施の形態1、2等で得られる正極活物質100を正極に用いた二次電池は高エネルギー密度であり、安全性が高いため、長期間に渡って長時間の安全な使用ができ、掃除ロボット6300に搭載する二次電池6306として好適である。
図31Aは、ウェアラブルデバイスの例を示している。ウェアラブルデバイスは、電源として二次電池を用いる。また、使用者が生活または屋外で使用する場合において、防沫性能、耐水性能または防塵性能を高めるため、接続するコネクタ部分が露出している有線による充電だけでなく、無線充電も行えるウェアラブルデバイスが望まれている。
例えば、図31Aに示すような眼鏡型デバイス4000に本発明の一態様である二次電池を搭載することができる。眼鏡型デバイス4000は、フレーム4000aと、表示部4000bを有する。湾曲を有するフレーム4000aのテンプル部に二次電池を搭載することで、軽量であり、且つ、重量バランスがよく継続使用時間の長い眼鏡型デバイス4000とすることができる。実施の形態1、2等で得られる正極活物質100を正極に用いた二次電池は高エネルギー密度であり、筐体の小型化に伴う省スペース化に対応できる構成を実現することができる。
また、ヘッドセット型デバイス4001に本発明の一態様である二次電池を搭載することができる。ヘッドセット型デバイス4001は、少なくともマイク部4001aと、フレキシブルパイプ4001bと、イヤフォン部4001cを有する。フレキシブルパイプ4001b内またはイヤフォン部4001c内に二次電池を設けることができる。実施の形態1、2等で得られる正極活物質100を正極に用いた二次電池は高エネルギー密度であり、筐体の小型化に伴う省スペース化に対応できる構成を実現することができる。
また、身体に直接取り付け可能なデバイス4002に本発明の一態様である二次電池を搭載することができる。デバイス4002の薄型の筐体4002aの中に、二次電池4002bを設けることができる。実施の形態1、2等で得られる正極活物質100を正極に用いた二次電池は高エネルギー密度であり、筐体の小型化に伴う省スペース化に対応できる構成を実現することができる。
また、衣服に取り付け可能なデバイス4003に本発明の一態様である二次電池を搭載することができる。デバイス4003の薄型の筐体4003aの中に、二次電池4003bを設けることができる。実施の形態1、2等で得られる正極活物質100を正極に用いた二次電池は高エネルギー密度であり、筐体の小型化に伴う省スペース化に対応できる構成を実現することができる。
また、ベルト型デバイス4006に本発明の一態様である二次電池を搭載することができる。ベルト型デバイス4006は、ベルト部4006a及びワイヤレス給電受電部4006bを有し、ベルト部4006aの内部領域に、二次電池を搭載することができる。実施の形態1、2等で得られる正極活物質100を正極に用いた二次電池は高エネルギー密度であり、筐体の小型化に伴う省スペース化に対応できる構成を実現することができる。
また、腕時計型デバイス4005に本発明の一態様である二次電池を搭載することができる。腕時計型デバイス4005は表示部4005a及びベルト部4005bを有し、表示部4005aまたはベルト部4005bに、二次電池を設けることができる。実施の形態1、2等で得られる正極活物質100を正極に用いた二次電池は高エネルギー密度であり、筐体の小型化に伴う省スペース化に対応できる構成を実現することができる。
表示部4005aには、時刻だけでなく、メールまたは電話の着信等、様々な情報を表示することができる。
また、腕時計型デバイス4005は、腕に直接巻きつけるタイプのウェアラブルデバイスであるため、使用者の脈拍、血圧等を測定するセンサを搭載してもよい。使用者の運動量及び健康に関するデータを蓄積し、健康を管理することができる。
図31Bに腕から取り外した腕時計型デバイス4005の斜視図を示す。
また、側面図を図31Cに示す。図31Cには、内部領域に二次電池913を内蔵している様子を示している。二次電池913は実施の形態4に示した二次電池である。二次電池913は表示部4005aと重なる位置に設けられており、高密度、且つ、高容量とすることができ、小型、且つ、軽量である。
腕時計型デバイス4005においては、小型、且つ、軽量であることが求められるため、実施の形態1、2等で得られる正極活物質100を二次電池913の正極に用いることで、高エネルギー密度、且つ、小型の二次電池913とすることができる。
本実施例では、実施の形態等で説明したリチウムイオン電池を作製し、電池特性を取得した結果を示す。
<正極活物質の作製>
リチウムイオン電池に用いた正極活物質を説明する。まず、図16及び図17に示す作製方法を参照しながら、正極活物質の作製工程を詳述する。なお、本実施例で用いた正極活物質の作製方法は、実施の形態2で具体的に説明した正極活物質の作製方法に準じたものである。
リチウムイオン電池に用いた正極活物質を説明する。まず、図16及び図17に示す作製方法を参照しながら、正極活物質の作製工程を詳述する。なお、本実施例で用いた正極活物質の作製方法は、実施の形態2で具体的に説明した正極活物質の作製方法に準じたものである。
図16のステップS14のコバルト酸リチウム(LiCoO2)として、添加元素を特に有さない市販のコバルト酸リチウム(日本化学工業株式会社製、セルシードC−10N)を用意した。ステップS15の加熱として、このコバルト酸リチウムをるつぼに入れ、蓋をした後、850℃、2時間、マッフル炉にて加熱した。マッフル炉内は酸素雰囲気とした後、フローしなかった(O2パージ)。初期加熱後の回収量を確認すると重量がやや減少していることがわかった。コバルト酸リチウムから不純物が除去されたため、重量が減少した可能性がある。
次に、図17A及び図17Bで示したステップS21及びステップS41に従って、添加元素としてMg及びFと、Ni及びAlを、2回に分けて添加した。まず、図17Aで示したステップS21に従って、F源としてLiFを用意し、Mg源としてMgF2を用意した。LiF:MgF2を1:3(モル比)となるように秤量した。次に、脱水アセトン中にLiF,及びMgF2を混合して、400rpmの回転速度で12時間攪拌して添加元素源(A1源)を作製した。混合にはボールミルを用い、粉砕メディアとして酸化ジルコニウムボールを用いた。混合用ボールミルの容器の容量45mLに対し、脱水アセトン20mL、酸化ジルコニウムボール(1mmφ)22gと共に合計約9gのF源及びMg源を入れて混合した。その後300μmの目を有するふるいでふるい、粒径の揃ったA1源を得た。
次に、ステップS31として、A1源が有するマグネシウムの原子数が、コバルト酸リチウムが有するコバルトの原子数に対して、1原子%となるように秤量して、初期加熱後のコバルト酸リチウムと乾式で混合した。このとき、150rpmの回転速度で1時間攪拌した。これはA1源を得るときの攪拌より緩やかな条件である。最後に300μmの目を有するふるいでふるい、粒径の揃った混合物903を得た(ステップS32)。
次に、ステップS33として、混合物903を加熱した。加熱条件は、900℃及び20時間とした。加熱の際、混合物903を入れたるつぼに蓋を配した。るつぼ内は酸素を有する雰囲気とし、当該酸素の出入りは遮断した(パージ)。加熱により、Mg及びFを有する複合酸化物を得た(ステップS34a)。
次に、ステップS51として、複合酸化物と添加元素源(A2源)を混合した。図17Bで示したステップS41に従って、Ni源としてニッケル水酸化物を用意し、Al源としてアルミニウム水酸化物を用意した。ニッケル水酸化物が有するニッケルの原子数が、複合酸化物が有するコバルトの原子数に対して0.5原子%となり、また、アルミニウム水酸化物が有するアルミニウムの原子数が、複合酸化物が有するコバルトの原子数に対して0.5原子%となるように秤量して、複合酸化物と乾式で混合した。このとき150rpmの回転速度で1時間攪拌した。混合にはボールミルを用い、粉砕メディアとして酸化ジルコニウムボールを用いた。混合用ボールミルの容器の容量45mLに対し、酸化ジルコニウムボール(1mmφ)22gと共に合計約7.5gのNi源及びAl源を入れて混合した。これはA1源を得るときの攪拌より緩やかな条件である。最後に300μmの目を有するふるいでふるい、粒径の揃った混合物904を得た(ステップS52)。
次に、ステップS53として、混合物904を加熱した。加熱条件は、850℃及び10時間とした。加熱の際、混合物904をいれたるつぼに蓋を配した。るつぼ内は酸素を有する雰囲気とし、当該酸素の出入りは遮断した(パージ)。加熱により、Mg、F、Ni、及びAlを有するコバルト酸リチウムを得た(ステップS54)。このようにして得られた正極活物質(複合酸化物)をサンプル1−1とした。なお、本実施例で得られたサンプル1−1の正極活物質は、実施の形態2で具体的に説明した正極活物質100の作製方法に準じて作製されたものであり、作製された正極活物質100の特徴も、実施の形態2で具体的に説明した正極活物質100の特徴を有している。
<ハーフセル1>
次に、氷点下においても優れた放電特性および/又は充電特性を有するリチウムイオン電池の評価を目的としたハーフセル(ハーフセル1)を作製した。ハーフセルの作製条件を説明する。
次に、氷点下においても優れた放電特性および/又は充電特性を有するリチウムイオン電池の評価を目的としたハーフセル(ハーフセル1)を作製した。ハーフセルの作製条件を説明する。
正極活物質としてサンプル1−1を用意し、導電助剤としてアセチレンブラック(AB)を用意し、結着剤としてポリフッ化ビニリデン(PVDF)を用意した。PVDFはあらかじめN−メチル−2−ピロリドン(NMP)に対して重量比で5%の割合で溶解したものを用意した。次に、正極活物質:AB:PVDF=95:3:2(重量比)で混合してスラリーを作製し、該スラリーをアルミニウムの正極集電体に塗工した。スラリーの溶媒としてNMPを用いた。
正極集電体にスラリーを塗工した後、溶媒を揮発させた後、ロールプレス装置を用いてプレス処理を行った。プレス処理の条件として、上ロール及び下ロールの温度をともに120℃とし、圧力(線圧)を210kN・mとした。以上の工程により、正極を得た。正極の活物質担持量はおよそ7mg/cm2とした。
ハーフセル1に用いた電解液は、有機溶媒を含む。有機溶媒は、エチレンカーボネート(EC)と、エチルメチルカーボネート(EMC)と、ジメチルカーボネート(DMC)と、を含み、EC、EMC、及びDMCの全含有量を100vol%としたとき、EC、EMC、及びDMCの体積比が、x:y:100−x−y(ただし、5≦x≦35であり、0<y<65である。)であるものを用いた。より具体的には、ECと、EMCと、DMCとをEC:EMC:DMC=30:35:35(体積比)で含む有機溶媒を用意した。この有機溶媒に対し、1mol/Lとなるように六フッ化リン酸リチウム(LiPF6)を溶解したものを電解液として用いた。以下、この電解液を電解液Aと呼ぶ。
リチウムイオン電池に用いられている一般的な電解液は、低くても−20℃程度で凝固してしまうため、−40℃で充放電できる電池を作製することは困難である。本実施例で用いる電解液は、凝固点が−40℃以下であるため、−40℃という極低温環境下においても充放電可能なリチウムイオン電池を実現できる必要条件の一つとなる。
セパレータは、ポリプロピレンの多孔質フィルムを用いた。また、負極(対極)はリチウム金属を用いた。これらを用いて、コイン型のハーフセル(ハーフセル1)を作製した。なお、ハーフセル1は、試験用電池と呼ぶことができる。
<ハーフセル2>
次に、比較例として、氷点下において標準的な放電特性および/又は充電特性を有するリチウムイオン電池の評価を目的としたハーフセル(ハーフセル2)を作製した。なお、ハーフセル1と同様に、ハーフセル2は、試験用電池と呼ぶことができる。ハーフセル2は、電解液が異なる点以外はハーフセル1と同様の条件で作製したので、ここではハーフセル2に使用した電解液について説明する。
次に、比較例として、氷点下において標準的な放電特性および/又は充電特性を有するリチウムイオン電池の評価を目的としたハーフセル(ハーフセル2)を作製した。なお、ハーフセル1と同様に、ハーフセル2は、試験用電池と呼ぶことができる。ハーフセル2は、電解液が異なる点以外はハーフセル1と同様の条件で作製したので、ここではハーフセル2に使用した電解液について説明する。
ハーフセル2に用いた電解液は、有機溶媒を含む。有機溶媒は、エチレンカーボネート(EC)とジエチルカーボネート(DEC)と、を含み、ECとDECをEC:DEC=3:7(体積比)で混合したものを用いた。この有機溶媒に対し、1mol/Lとなるように六フッ化リン酸リチウム(LiPF6)を溶解し、さらに添加剤としてビニレンカーボネート(VC)を2wt%加えたものを電解液として用いた。この電解液等を用いて、コイン型のハーフセル(ハーフセル2)を作製した。以下、この電解液を電解液Bと呼ぶ。
<放電容量の温度特性>
次に、作製したハーフセル1及びハーフセル2を用いて、充電容量及び放電容量を測定した。なお、ハーフセル1及びハーフセル2の測定条件は同じである。
次に、作製したハーフセル1及びハーフセル2を用いて、充電容量及び放電容量を測定した。なお、ハーフセル1及びハーフセル2の測定条件は同じである。
ハーフセル1及びハーフセル2を用いて、複数の温度条件のそれぞれにおいて放電容量を測定した。放電時の温度は25℃、0℃、−20℃、−40℃の4条件とし、それぞれの温度での放電試験の前に、25℃において同一の条件で充電を行った。充電は、4.60Vの電圧になるまで0.2C(1C=200mA/gとした)の充電電流で定電流充電を行い、続けて4.60Vでの定電圧充電を、充電電流が0.02C以下になるまで行った。放電時の条件は、温度以外全て同一としており、2.5V(カットオフ電圧)になるまで0.1C(ただし、1C=200mA/gとする)の放電レートで定電流放電する条件とした。また、充電時の条件は、全て同一としており、25℃環境下において4.6Vの電圧になるまで0.2C(ただし、1C=200mA/gとする)の充電レートで定電流充電する条件とした。なお、本明細書の実施例において記載した、充電時または放電時の温度は、ハーフセルを一定時間放置した恒温槽の温度で設定した。
図32に、放電時の各温度に対する放電容量を示す。図32に示されるとおり、−20℃の条件では、ハーフセル1及びハーフセル2の双方ともに高い放電容量を有し、25℃の条件と比較しても同等の放電容量を有している。一方、−40℃の条件では、ハーフセル1及びハーフセル2の放電容量に顕著な差を有することが明らかになった。具体的には、−40℃の条件におけるハーフセル2の放電容量は約50mAh/gであり、25℃の条件と比較して約25%の放電容量しか得られていないのに対し、−40℃の条件におけるハーフセル1の放電容量は約170mAh/gであり、25℃の条件と比較して少なくとも70%以上の放電容量が得られた。このように、実施の形態1等に記載の正極活物質及び電解質を備えたリチウムイオン電池は、−20℃または−40℃という非常に低温な環境下であっても、優れた放電容量を示すことが明らかとなった。具体的には、25℃で4.6Vの電圧になるまで0.2C(ただし、1C=200mA/gとする)の充電レートで定電流充電し、電流値が0.02Cとなるまで4.6Vでの定電圧充電をした後、−40℃で2.5Vの電圧になるまで0.1Cの放電レートで定電流放電することで求められた放電容量の値が、前記リチウムイオン電池を25℃環境下で4.6Vの電圧になるまで0.2Cの充電レートで定電流充電し、電流値が0.02Cとなるまで4.6Vでの定電圧充電をした後、25℃で2.5Vの電圧になるまで0.1Cの放電レートで定電流放電することで求められた放電容量の値に比して、少なくとも50%以上(または70%以上)であるリチウムイオン電池を実現できることが実証された。
また、図32の結果から、実施の形態1等に記載の正極活物質及び電解質を備えたリチウムイオン電池は、少なくとも−40℃以上25℃以下の温度範囲で動作可能であることが明らかとなった。また、実施の形態1等に記載の正極活物質及び電解質を備えたリチウムイオン電池は、−40℃の条件における放電容量が100mAh/g以上(具体的には、約170mAh/g)を実現していることが明らかとなった。
<充電容量の温度特性>
ハーフセル1及びハーフセル2を用いて、複数の温度条件のそれぞれにおいて充電容量を測定した。まず、各サンプルで充電前の状態を均一にするため、充電開始前に25℃、2.5Vになるまで0.2Cで定電流放電した充電時の温度は、25℃、0℃、−20℃、−40℃の4条件とした。充電時の条件は、温度以外全て同一としており、4.6Vになるまで0.1C(ただし、1C=200mA/gとする)の充電レートで定電流充電する条件とした。
ハーフセル1及びハーフセル2を用いて、複数の温度条件のそれぞれにおいて充電容量を測定した。まず、各サンプルで充電前の状態を均一にするため、充電開始前に25℃、2.5Vになるまで0.2Cで定電流放電した充電時の温度は、25℃、0℃、−20℃、−40℃の4条件とした。充電時の条件は、温度以外全て同一としており、4.6Vになるまで0.1C(ただし、1C=200mA/gとする)の充電レートで定電流充電する条件とした。
図33に、充電時の各温度に対する充電容量を示す。図33における充電温度とは、充電時の温度を指す。図33に示されるとおり、−20℃の条件におけるハーフセル1の充電容量は約175mAh/gであり、25℃の条件における放電容量に比して80%超の充電容量が得られた。−40℃の条件におけるハーフセル1の充電容量は約120mAh/gであり、25℃の条件における充電容量に比して約60%の充電容量が得られた。これに対し、比較例となるハーフセル2は、−20℃の充電容量、及び−40℃の充電容量のいずれも充電容量がほぼ0mAh/gであった。充電容量が得られなかった理由は、ハーフセル2に用いた電解液は凝固点が高く、−20℃、及び−40℃における電解液の粘度または正極活物質のバルク抵抗が高く、リチウムイオンの伝導率が非常に低いことが原因であると推定される。このように、実施の形態1等に記載の正極活物質及び電解質を少なくとも備えたリチウムイオン電池は、−20℃または−40℃という非常に低温な環境下であっても、25℃の条件における充電容量と比較してもあまり見劣りしない程度の充電容量を示すことが実証された。具体的には、25℃で2.5Vの電圧になるまで0.1Cの放電レートで定電流放電した後、−40℃で4.6Vの電圧になるまで0.1C(ただし、1C=200mA/gとする)の充電レートで定電流充電することで求められた充電容量の値が、25℃で2.5Vの電圧になるまで0.1Cの放電レートで定電流放電した後、25℃で4.6Vの電圧になるまで0.1Cの充電レートで定電流充電することで求められた充電容量の値に比して、50%以上(または60%以上)であるリチウムイオン電池を実現できることが実証された。
本実施例では、正極活物質と充電電圧を変化させた場合の放電容量について説明する。
放電容量を測定するサンプルは、計4つ(サンプル21−1、サンプル21−2、サンプル22−1、サンプル22−2)を準備した。いずれのサンプルも、担持量はおよそ7mg/cm2とした。なお、サンプル21−2、サンプル22−1、サンプル22−2は比較例である。
サンプル21−1は、実施例1で作製したハーフセル1(すなわち、正極活物質としてMg、F、Ni、及びAlを有するコバルト酸リチウムを有し、電解質として電解液Aを有するハーフセル1)を用いたものである。最初にエージングを行った後、低温特性の評価を行った。エージングの条件として、25℃で充電電圧4.6Vになるまで電流値0.1C(ただし、1C=200mA/gとする。)で定電流充電し、電流値が0.01Cとなるまで定電圧充電した後、25℃、カットオフ電圧2.5V、0.1Cで定電流放電する、という一連の操作を2回行った。また、低温特性の評価として、25℃で充電電圧4.6Vになるまで電流値0.1C(ただし、1C=200mA/gとする。)で定電流充電し、電流値が0.01Cとなるまで4.6Vでの定電圧充電をした後、カットオフ電圧2.5V、0.1Cで定電流放電する条件とした。放電時の温度は、25℃、0℃、−20℃、−40℃の4条件で測定した。
サンプル21−2は、実施例1で作製したハーフセル2(すなわち、正極活物質としてMg、F、Ni、及びAlを有するコバルト酸リチウムを有し、電解質として電解液Aを有するハーフセル1)を用いたものであり、サンプル21−1と同じ構造を有するものである。最初にエージングを行った後、低温特性の評価を行った。エージングの条件として、25℃で充電電圧4.3Vになるまで電流値0.1C(ただし、1C=200mA/gとする。)で定電流充電し、電流値が0.01Cとなるまで定電圧充電した後、25℃、カットオフ電圧2.5V、0.1Cで定電流放電する、という一連の操作を2回行った。低温特性の評価として、25℃で充電電圧4.3Vになるまで電流値0.1Cで定電流充電し、電流値が10mA/gとなるまで4.3Vでの定電圧充電をした後、2.5V、0.1Cで定電流放電する条件とした。放電時の温度は、25℃、0℃、−20℃、−40℃の4条件で測定した。すなわち、低温特性の評価を行う充電電圧の値以外の条件は、サンプル21−1とサンプル21−2で同じである。
サンプル22−1は、サンプル21−1と比較して、ハーフセルに用いた正極活物質を変更したものであり、サンプル22−1の正極活物質は、添加元素を特に有さない市販のコバルト酸リチウム(日本化学工業株式会社製、セルシードC−10N)である。最初にエージングを行った後、低温特性の評価を行った。エージングの条件として、25℃で充電電圧4.3Vになるまで電流値0.1C(ただし、1C=200mA/gとする。)で定電流充電し、電流値が0.01Cとなるまで定電圧充電した後、25℃、カットオフ電圧2.5V、0.1Cで定電流放電する、という一連の操作を2回行った。低温特性の評価として、25℃で充電電圧4.6Vになるまで電流値0.1C(ただし、1C=200mA/gとする。)で定電流充電し、電流値が0.01Cとなるまで4.6Vでの定電圧充電をした後、カットオフ電圧2.5V、0.1Cで定電流放電する条件とした。放電時の温度は、25℃、−40℃の2条件で測定した。
サンプル22−2は、充電電圧の値以外の条件がサンプル22−1と共通である。また、サンプル22−2は、サンプル21−2と比較して、ハーフセルに用いた正極活物質のみを変更したものであるとも言える。具体的には、サンプル22−2の正極活物質は、添加元素を特に有さない市販のコバルト酸リチウム(日本化学工業株式会社製、セルシードC−10N)である。最初にエージングを行った後、低温特性の評価を行った。エージングの条件は、サンプル22−1と同じである。低温特性の評価として、25℃で充電電圧4.3Vになるまで電流値0.1C(ただし、1C=200mA/gとする。)で定電流充電し、電流値が0.01Cとなるまで4.3Vでの定電圧充電をした後、カットオフ電圧2.5V、0.1Cで定電流放電する条件とした。放電時の温度は、25℃、0℃、−20℃、−40℃の4条件で測定した。
図34、図35は、放電時の各温度における各サンプルの放電曲線を示すものである。詳細には、図34Aはサンプル21−1の放電曲線を示し、図34Bはサンプル21−2の放電曲線を示し、図35Aはサンプル22−1の放電曲線を示し、図35Bはサンプル22−2の放電曲線を示すものである。また、図34、図35の各放電曲線において、点線は放電時の温度が25℃の結果を示し、一点鎖線は放電時の温度が0℃の結果を示し、破線は放電時の温度が−20℃の結果を示し、実線は放電時の温度が−40℃の結果を示すものである。また、放電時の各温度における放電容量、平均放電電圧、放電エネルギー密度の測定結果を表2に示す。また、放電時の各温度における放電容量、平均放電電圧、放電エネルギー密度の値を、放電時の温度が25℃における値で除して規格化した放電容量、平均放電電圧、放電エネルギー密度の比(単位:%)を表3に示す。なお、表2における放電容量(単位:mAh/g)は、正極活物質の重量あたりで算出した値である。また、表2における放電エネルギー密度(単位:mWh/g)は、放電容量に平均放電電圧(単位:V)を掛けて算出した値である。また、表2、表3における「−」は、未取得であることを示す。


図34、図35、表2、表3に示されるとおり、サンプル21−1は、放電温度が−20℃であっても約200mAh/gという非常に高い放電容量が得られた。別の視点では、−20℃の放電における放電容量が、25℃の放電における放電容量に比して約90%である結果が得られた。放電温度が−40℃では、約150mAh/gという放電容量が得られた。別の視点では、放電温度が−40℃では、約480mWh/gという、高い放電エネルギー密度が得られた。別の視点では、−40℃の放電における放電エネルギー密度が、25℃の放電における放電エネルギー密度に比して約50%である結果が得られた。別の視点では、−40℃の放電における放電容量が、25℃の放電における放電容量に比して約70%である結果が得られた。このように、本実施例におけるリチウムイオン電池は、放電温度が−20℃における放電容量が200mAh/g以上であり、−20℃の放電における放電容量が25℃の放電における放電容量に比して90%以上であり、放電温度が−40℃における放電容量が150mAh/g以上であり、−40℃の放電における放電容量が25℃の放電における放電容量に比して約70%以上であり、放電温度が−40℃における放電エネルギー密度が約475mAh/g以上であり、−40℃の放電における放電エネルギー密度が25℃の放電における放電エネルギー密度に比して50%以上を実現している結果が得られた。
一方で、充電電圧を4.3Vとしたサンプル21−2は、サンプル21−1と同じ構造を有するハーフセルを用いているにも関わらず、氷点下(特に、−20℃、−40℃)の放電における放電容量は低い結果となった。特に、−40℃の放電における放電容量は、25℃の放電における放電容量に比して約30%程度しか得られない結果が得られた。
また、正極活物質のみがサンプル21−1と異なるサンプル22−1は、−40℃の放電における放電容量が非常に低い結果となった。この結果より、サンプル22−1に用いた正極活物質である、添加元素を特に有さない市販のコバルト酸リチウムは、高い電圧で充電すると、コバルト酸リチウムの表層部または内部の構造変化が生じたり、電解液の分解による抵抗の高い被膜が生成されたりするため、放電容量が非常に低い結果になったと推察される。
また、充電電圧の条件のみがサンプル22−1と異なるサンプル22−2は、−40℃の放電における放電容量が約95mAh/gであり、サンプル22−1に比較すれば放電容量が大きいと言える。しかしながら、本発明の一態様であるサンプル21−1に比較すると、約60%の放電容量しか得られなかった。また、−40℃の放電における放電エネルギー密度についても約285mAh/gしか得られなかった。
以上の結果より、本発明の一態様であるサンプル21−1は、高い充電電圧としても氷点下の温度条件における充放電に伴う劣化の少ない材料である正極活物質と、氷点下における充放電であってもリチウムイオン伝導性に優れた材料である電解質の双方を少なくとも有することにより、氷点下(−40℃)においても優れた放電特性を有するリチウムイオン電池が得られることが実証された。
本実施例では、実施例1または実施例2で説明したリチウムイオン電池のサイクル特性について説明する。
サイクル特性を測定するサンプルは、計4つ(サンプル31−1、サンプル31−2、サンプル32−1、サンプル32−2)を準備した。いずれのサンプルも、担持量はおよそ7mg/cm2とした。なお、サンプル32−1、サンプル32−2は比較例である。
サンプル31−1、サンプル31−2は、実施例2で説明したサンプル21−1、サンプル21−2と同じ構造を有するハーフセルであり、Mg、F、Ni、及びAlを有するコバルト酸リチウムを正極活物質として有し、電解質として電解液Aを有するものである。一方、比較例であるサンプル32−1、サンプル32−2は、実施例2で説明したサンプル22−1、サンプル22−2と同じ構造を有するハーフセルであり、添加元素を特に有さない市販のコバルト酸リチウムを正極活物質として有し、電解質として電解液Aを有するものである。
<サイクル特性の測定条件>
サンプル31−1、サンプル32−1におけるサイクル特性の測定条件は同じものを用いた。具体的には、25℃で4.6Vの電圧になるまで0.5C(ただし、1C=200mA/gとする)の充電レートで定電流充電し、0.05Cになるまで4.6Vで定電圧充電し、25℃で2.5Vの電圧になるまで0.5Cで定電流放電したときの放電容量を測定した。このような充電と放電を50サイクル繰り返して得られた、サイクル回数と放電容量との関係を図36Aに示す。図36Aにおいて、サンプル31−1は実線で示し、サンプル32−1は点線で示す。
サンプル31−1、サンプル32−1におけるサイクル特性の測定条件は同じものを用いた。具体的には、25℃で4.6Vの電圧になるまで0.5C(ただし、1C=200mA/gとする)の充電レートで定電流充電し、0.05Cになるまで4.6Vで定電圧充電し、25℃で2.5Vの電圧になるまで0.5Cで定電流放電したときの放電容量を測定した。このような充電と放電を50サイクル繰り返して得られた、サイクル回数と放電容量との関係を図36Aに示す。図36Aにおいて、サンプル31−1は実線で示し、サンプル32−1は点線で示す。
サンプル31−2、サンプル32−2におけるサイクル特性の測定条件は同じものを用いた。具体的には、25℃で4.3Vの電圧になるまで0.5C(ただし、1C=200mA/gとする)の充電レートで定電流充電し、0.05Cになるまで4.3Vで定電圧充電し、25℃で2.5Vの電圧になるまで0.5Cで定電流放電したときの放電容量を測定した。このような充電と放電を50サイクル繰り返して得られた、サイクル回数と放電容量との関係を図36Bに示す。図36Bにおいて、サンプル31−1は実線で示し、サンプル32−1は点線で示す。
図36Bに示されるとおり、4.3V充電の条件でサイクル特性を評価したサンプル31−2は、サイクル50回目の放電容量の値が、サイクル1回目乃至50回目までの放電容量における最大放電容量の値と比較しても殆ど変わらなかった。具体的には、放電容量維持率を(50サイクル後の放電容量/最大放電容量)×100(単位:%)で計算した結果、サンプル31−2の放電容量維持率は99.1%であり、非常に劣化が少ない結果となった。一方、サンプル32−2の放電容量維持率は88.4%であり、サンプル31−2に比べると10倍以上劣化している結果となった。
図36Aに示されるとおり、4.6V充電の条件でサイクル特性を評価した場合、本実施例のサンプル(サンプル31−1)と、比較例のサンプル(サンプル32−1)との差は、より顕著なものとなった。具体的には、サンプル31−1の放電容量維持率は97.5%であり、サイクル50回目の放電容量でさえ200mAh/g以上を達成していた。一方、サンプル32−1の放電容量維持率は35.5%であり、サンプル31−1に比べると25倍以上劣化している結果となった。このように、本発明の一態様であるリチウムイオン電池に用いることが可能な正極活物質と、氷点下における充放電であってもリチウムイオン伝導性に優れた材料である電解質(電解液A)の双方を少なくとも有するリチウムイオン電池は、放電容量、サイクル特性の双方共に優れたポテンシャルを有していることが明らかとなった。
100:正極活物質、100a:表層部、100b:内部、101:結晶粒界、102:埋め込み部、103:凸部、104:被膜
Claims (6)
- 正極活物質を有する正極と、電解質と、炭素材料の負極活物質を有する負極と、を備えたリチウムイオン電池であって、
前記電解質は、エチレンカーボネートと、エチルメチルカーボネートと、ジメチルカーボネートと、を含み、前記エチレンカーボネート、前記エチルメチルカーボネート、及び前記ジメチルカーボネートの全含有量を100vol%としたとき、前記エチレンカーボネート、前記エチルメチルカーボネート、及び前記ジメチルカーボネートの体積比が、x:y:100−x−y(ただし、5≦x≦35であり、0<y<65である。)であり、
前記リチウムイオン電池を25℃環境下で4.5Vの電圧になるまで0.1C(ただし、1C=200mA/gとする)の充電レートで定電流充電し、電流値が0.01Cとなるまで4.5Vでの定電圧充電をした後、−40℃環境下で2.5Vの電圧になるまで0.1Cの放電レートで定電流放電することで求められた放電容量の値が、前記リチウムイオン電池を25℃環境下で4.5Vの電圧になるまで0.1C(ただし、1C=200mA/gとする)の充電レートで定電流充電し、電流値が0.01Cとなるまで4.5Vでの定電圧充電をした後、25℃環境下で2.5Vの電圧になるまで0.1Cの放電レートで定電流放電することで求められた放電容量の値に比して50%以上である、リチウムイオン電池。 - 請求項1において、前記炭素材料は黒鉛である、リチウムイオン電池。
- 正極活物質を有する正極と、電解質と、負極と、を備え、
少なくとも−40℃以上25℃以下の温度範囲で動作可能である、リチウムイオン電池。 - 正極活物質を有する正極と、電解質と、負極と、を備えたリチウムイオン電池であって、
前記正極活物質を正極として用い、
エチレンカーボネートと、エチルメチルカーボネートと、ジメチルカーボネートと、を含み、前記エチレンカーボネート、前記エチルメチルカーボネート、及び前記ジメチルカーボネートの全含有量を100vol%としたとき、前記エチレンカーボネート、前記エチルメチルカーボネート、及び前記ジメチルカーボネートの体積比が、x:y:100−x−y(ただし、5≦x≦35であり、0<y<65である。)である電解質を用い、
リチウム金属を負極として用いて試験用電池とした際に、
前記試験用電池を25℃環境下で4.6Vの電圧になるまで0.1C(ただし、1C=200mA/gとする)の充電レートで定電流充電し、電流値が0.01Cとなるまで4.6Vでの定電圧充電をした後、−40℃環境下で2.5Vの電圧になるまで0.1Cの放電レートで定電流放電することで求められた放電容量の値が、前記試験用電池を25℃環境下で4.6Vの電圧になるまで0.1C(ただし、1C=200mA/gとする)の充電レートで定電流充電し、電流値が0.01Cとなるまで4.6Vでの定電圧充電をした後、25℃環境下で2.5Vの電圧になるまで0.1Cの放電レートで定電流放電することで求められた放電容量の値に比して50%以上である、リチウムイオン電池。 - 請求項1乃至4のいずれか一において、
前記正極活物質は、LixCoO2(ただし、0<x≦1である。)で表されるコバルト酸リチウムを有し、
前記LixCoO2中のxが1のとき、空間群R−3mの層状岩塩型の結晶構造を有し、
前記LixCoO2中のxが0.1を超えて0.24以下の充電状態のとき、
空間群P2/m、
格子定数a=4.88±0.01(×10−1nm)、
格子定数b=2.82±0.01(×10−1nm)、
格子定数c=4.84±0.01(×10−1nm)、
α=90°、
β=109.58±0.01°、
γ=90°の結晶構造を有する、リチウムイオン電池。 - 請求項1乃至4のいずれか一において、
前記正極活物質は、LixCoO2(ただし、0<x≦1である。)で表されるコバルト酸リチウムを有し、
前記LixCoO2中のxが1のとき、空間群R−3mの層状岩塩型の結晶構造を有し、
前記LixCoO2中のxが0.1を超えて0.24以下の充電状態のとき、粉末X線回折で分析すると、回折パターンは、
2θ=19.37°以上19.57°以下と、
2θ=45.57°以上45.67°以下と、に少なくともピークを有する、リチウムイオン電池。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020247003700A KR20240042425A (ko) | 2021-08-06 | 2022-07-26 | 리튬 이온 전지 |
| JP2023539217A JPWO2023012579A1 (ja) | 2021-08-06 | 2022-07-26 | |
| CN202280054033.7A CN117795731A (zh) | 2021-08-06 | 2022-07-26 | 锂离子电池 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021-130258 | 2021-08-06 | ||
| JP2021130258 | 2021-08-06 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023012579A1 true WO2023012579A1 (ja) | 2023-02-09 |
Family
ID=85154306
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2022/056865 WO2023012579A1 (ja) | 2021-08-06 | 2022-07-26 | リチウムイオン電池 |
Country Status (4)
| Country | Link |
|---|---|
| JP (1) | JPWO2023012579A1 (ja) |
| KR (1) | KR20240042425A (ja) |
| CN (1) | CN117795731A (ja) |
| WO (1) | WO2023012579A1 (ja) |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004342626A (ja) * | 2004-08-05 | 2004-12-02 | Sumitomo Chem Co Ltd | 非水電解液リチウム二次電池の低温放電特性向上方法 |
| JP2015162406A (ja) * | 2014-02-28 | 2015-09-07 | 三洋電機株式会社 | 円筒形非水電解液二次電池 |
| JP2018067444A (ja) * | 2016-10-19 | 2018-04-26 | トヨタ自動車株式会社 | 非水電解液二次電池の製造方法 |
| JP2021093356A (ja) * | 2019-11-28 | 2021-06-17 | 株式会社半導体エネルギー研究所 | 正極活物質、二次電池、電子機器 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6342230B2 (ja) | 2013-06-21 | 2018-06-13 | 株式会社半導体エネルギー研究所 | 非水溶媒、非水電解質および蓄電装置 |
-
2022
- 2022-07-26 JP JP2023539217A patent/JPWO2023012579A1/ja active Pending
- 2022-07-26 CN CN202280054033.7A patent/CN117795731A/zh active Pending
- 2022-07-26 KR KR1020247003700A patent/KR20240042425A/ko unknown
- 2022-07-26 WO PCT/IB2022/056865 patent/WO2023012579A1/ja active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004342626A (ja) * | 2004-08-05 | 2004-12-02 | Sumitomo Chem Co Ltd | 非水電解液リチウム二次電池の低温放電特性向上方法 |
| JP2015162406A (ja) * | 2014-02-28 | 2015-09-07 | 三洋電機株式会社 | 円筒形非水電解液二次電池 |
| JP2018067444A (ja) * | 2016-10-19 | 2018-04-26 | トヨタ自動車株式会社 | 非水電解液二次電池の製造方法 |
| JP2021093356A (ja) * | 2019-11-28 | 2021-06-17 | 株式会社半導体エネルギー研究所 | 正極活物質、二次電池、電子機器 |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2023012579A1 (ja) | 2023-02-09 |
| CN117795731A (zh) | 2024-03-29 |
| KR20240042425A (ko) | 2024-04-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20230387394A1 (en) | Method for forming positive electrode active material, positive electrode, secondary battery, electronic device, power storage system, and vehicle | |
| US20240021862A1 (en) | Secondary battery, power storage system, vehicle, and method for fabricating positive electrode | |
| KR20230027121A (ko) | 이차 전지 및 전자 기기 | |
| US20230343952A1 (en) | Secondary battery, manufacturing method of secondary battery, electronic device, and vehicle | |
| JP2022045353A (ja) | 二次電池の作製方法、および二次電池 | |
| WO2022248968A1 (ja) | 電池、電子機器、蓄電システムおよび移動体 | |
| WO2023281346A1 (ja) | 正極活物質 | |
| WO2022254284A1 (ja) | 二次電池、電子機器及び飛行体 | |
| JP2022173119A (ja) | 正極活物質 | |
| WO2023012579A1 (ja) | リチウムイオン電池 | |
| WO2023031729A1 (ja) | 正極および正極の作製方法 | |
| WO2023209474A1 (ja) | 正極活物質、リチウムイオン電池、電子機器、および車両 | |
| WO2023047234A1 (ja) | 複合酸化物の作製方法、及びリチウムイオン電池の作製方法 | |
| WO2023073480A1 (ja) | リチウムイオン電池 | |
| WO2023209475A1 (ja) | 正極活物質、正極、二次電池、電子機器および車両 | |
| WO2024074938A1 (ja) | 二次電池 | |
| WO2022243782A1 (ja) | 正極活物質の作製方法、正極、リチウムイオン二次電池、移動体、蓄電システム、及び電子機器 | |
| WO2024052785A1 (ja) | 電池、電子機器、及び車両 | |
| WO2022189889A1 (ja) | 複合酸化物の作製方法、正極、リチウムイオン二次電池、電子機器、蓄電システム、及び移動体 | |
| WO2022167885A1 (ja) | 正極活物質の製造方法、二次電池および車両 | |
| WO2022038451A1 (ja) | 正極活物質の作製方法、および二次電池の作製方法 | |
| WO2023242669A1 (ja) | リチウムイオン二次電池 | |
| WO2022038454A1 (ja) | 正極活物質の作製方法 | |
| US20240030413A1 (en) | Positive electrode, method for forming positive electrode, secondary battery, electronic device, power storage system, and vehicle | |
| WO2022200908A1 (ja) | 電池、電子機器および車両 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22852420 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023539217 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |