WO2016152937A1 - 電子写真感光体、及び画像形成装置 - Google Patents
電子写真感光体、及び画像形成装置 Download PDFInfo
- Publication number
- WO2016152937A1 WO2016152937A1 PCT/JP2016/059263 JP2016059263W WO2016152937A1 WO 2016152937 A1 WO2016152937 A1 WO 2016152937A1 JP 2016059263 W JP2016059263 W JP 2016059263W WO 2016152937 A1 WO2016152937 A1 WO 2016152937A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- polyester resin
- formula
- divalent
- dichloromethane
- Prior art date
Links
- 0 CC1(C2C1C2)C1=C(C)CCC1* Chemical compound CC1(C2C1C2)C1=C(C)CCC1* 0.000 description 6
- SOBTXHZWIHOYAG-UHFFFAOYSA-N CCC(C)C(C)(C)c1ccc(C)cc1C Chemical compound CCC(C)C(C)(C)c1ccc(C)cc1C SOBTXHZWIHOYAG-UHFFFAOYSA-N 0.000 description 1
- UFWIBTONFRDIAS-UHFFFAOYSA-N c1ccc(cccc2)c2c1 Chemical compound c1ccc(cccc2)c2c1 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 1
Images

Classifications
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G5/00—Recording members for original recording by exposure, e.g. to light, to heat, to electrons; Manufacture thereof; Selection of materials therefor
- G03G5/02—Charge-receiving layers
- G03G5/04—Photoconductive layers; Charge-generation layers or charge-transporting layers; Additives therefor; Binders therefor
- G03G5/05—Organic bonding materials; Methods for coating a substrate with a photoconductive layer; Inert supplements for use in photoconductive layers
- G03G5/0528—Macromolecular bonding materials
- G03G5/0557—Macromolecular bonding materials obtained otherwise than by reactions only involving carbon-to-carbon unsatured bonds
- G03G5/0567—Other polycondensates comprising oxygen atoms in the main chain; Phenol resins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/02—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds
- C08G63/12—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds derived from polycarboxylic acids and polyhydroxy compounds
- C08G63/16—Dicarboxylic acids and dihydroxy compounds
- C08G63/18—Dicarboxylic acids and dihydroxy compounds the acids or hydroxy compounds containing carbocyclic rings
- C08G63/19—Hydroxy compounds containing aromatic rings
- C08G63/193—Hydroxy compounds containing aromatic rings containing two or more aromatic rings
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G5/00—Recording members for original recording by exposure, e.g. to light, to heat, to electrons; Manufacture thereof; Selection of materials therefor
- G03G5/02—Charge-receiving layers
- G03G5/04—Photoconductive layers; Charge-generation layers or charge-transporting layers; Additives therefor; Binders therefor
- G03G5/05—Organic bonding materials; Methods for coating a substrate with a photoconductive layer; Inert supplements for use in photoconductive layers
- G03G5/0528—Macromolecular bonding materials
- G03G5/0557—Macromolecular bonding materials obtained otherwise than by reactions only involving carbon-to-carbon unsatured bonds
- G03G5/056—Polyesters
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G5/00—Recording members for original recording by exposure, e.g. to light, to heat, to electrons; Manufacture thereof; Selection of materials therefor
- G03G5/02—Charge-receiving layers
- G03G5/04—Photoconductive layers; Charge-generation layers or charge-transporting layers; Additives therefor; Binders therefor
- G03G5/05—Organic bonding materials; Methods for coating a substrate with a photoconductive layer; Inert supplements for use in photoconductive layers
- G03G5/0528—Macromolecular bonding materials
- G03G5/0557—Macromolecular bonding materials obtained otherwise than by reactions only involving carbon-to-carbon unsatured bonds
- G03G5/0564—Polycarbonates
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G5/00—Recording members for original recording by exposure, e.g. to light, to heat, to electrons; Manufacture thereof; Selection of materials therefor
- G03G5/14—Inert intermediate or cover layers for charge-receiving layers
- G03G5/147—Cover layers
- G03G5/14708—Cover layers comprising organic material
- G03G5/14713—Macromolecular material
- G03G5/14747—Macromolecular material obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds
- G03G5/14752—Polyesters
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G5/00—Recording members for original recording by exposure, e.g. to light, to heat, to electrons; Manufacture thereof; Selection of materials therefor
- G03G5/14—Inert intermediate or cover layers for charge-receiving layers
- G03G5/147—Cover layers
- G03G5/14708—Cover layers comprising organic material
- G03G5/14713—Macromolecular material
- G03G5/14747—Macromolecular material obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds
- G03G5/14756—Polycarbonates
Definitions
- the present invention relates to an electrophotographic photosensitive member, and more particularly to an electrophotographic photosensitive member having good wear resistance and electrical characteristics, and an image forming apparatus. Furthermore, the present invention relates to a resin used for the electrophotographic photoreceptor and a method for producing the same.
- the electrophotographic photosensitive member is repeatedly used in an electrophotographic process, that is, a cycle of charging, exposure, development, transfer, cleaning, static elimination, and the like, and therefore deteriorates due to various stresses during that period.
- Such deterioration includes, for example, strongly oxidative ozone generated from a corona charger normally used as a charger, chemical damage caused by NOx to the photosensitive layer, and carrier (current) generated by image exposure.
- chemical and electrical degradations such as decomposition of the photosensitive layer composition caused by flowing inside or static elimination light and external light.
- the photosensitive layer is particularly susceptible to such a load.
- the photosensitive layer is usually composed of a binder resin and a photoconductive material, and it is the binder resin that substantially determines the strength, but since the amount of the photoconductive material doped is considerably large, sufficient mechanical strength is provided. Has not reached.
- a polyester resin excellent in sensitivity and wear resistance has been used as a binder resin for a photosensitive layer (see Patent Documents 1 to 8).
- an object of the present invention is to provide an electrophotographic photoreceptor excellent in abrasion resistance and electrical characteristics against a practical load by including a polyester resin excellent in solubility, abrasion resistance, and electrical characteristics in the photosensitive layer. It is to be.
- the present invention provides a polyester resin excellent in solubility, wear resistance, and electrical properties, and a method for producing the same.
- the present inventors have excellent mechanical properties by including a polyester resin having a specific chemical structure in the photosensitive layer. Based on this finding, it has been found that an electrophotographic photoreceptor having high solubility and excellent coating solution stability with respect to the solvent used in the coating solution for forming the photosensitive layer can be obtained, and having excellent electrical characteristics.
- the present invention has been completed.
- the present inventors have found that when a polyester resin is produced, the remaining carboxylic acid chloride in the ester oligomer passes through the ester oligomer.
- the gist of the invention resides in the following ⁇ 1> to ⁇ 8>.
- the photosensitive layer contains a polyester resin composed of a divalent phenol residue and a divalent carboxylic acid residue, and the divalent phenol residue.
- An electrophotographic photoreceptor comprising the electrophotographic photoreceptor.
- R 1 represents any one of a hydrogen atom, an alkyl group which may have a substituent having 1 to 20 carbon atoms, an alkoxy group, an aromatic group which may be substituted, and a halogen group.
- N 1 is an integer from 0 to 4.
- R 2 represents any one of a hydrogen atom, an alkyl group optionally having a substituent having 1 to 20 carbon atoms, an alkoxy group, an optionally substituted aromatic group, and a halogen group.
- N 2 is an integer of 0 to 6.
- Ar 1 and Ar 2 each independently represent an arylene group optionally having a substituent having 6 to 16 carbon atoms.
- Z represents an arylene group optionally having a substituent.
- s is an integer of 0 or 1.
- R 3 to R 6 are each independently a hydrogen atom, an alkyl group optionally having a substituent having 1 to 20 carbon atoms, an alkoxy group, an optionally substituted aromatic group
- X represents a single bond, —CR 7 R 8 —, O, CO, or S.
- R 7 and R 8 each independently represents a hydrogen atom or a carbon atom having 1 to 10 carbon atoms.
- An alkyl group or R 7 and R 8 represent a C 5-10 cycloalkylidene group formed by combining R 7 and R 8.
- At least one divalent phenol residue selected from the divalent phenol residue group represented by the above formulas (1) to (3) is 5 with respect to all divalent phenol residues of the polyester resin.
- ⁇ 3> The electrophotographic photosensitive member according to ⁇ 1> or ⁇ 2>, wherein the divalent carboxylic acid residue of the polyester resin is a divalent carboxylic acid residue represented by the following formula (5).
- Ar 3 and Ar 4 each independently represent an arylene group which may have a substituent.
- Y represents a single bond, an oxygen atom, a sulfur atom, or Formula (6).
- R 9 and R 10 in formula (6) are each independently a hydrogen atom, an alkyl group, or an aryl group.
- R 11 is an alkylene group, an arylene group, or a group represented by Formula (8).
- R 12 and R 13 each independently represents an alkylene group
- Ar 5 represents an arylene group
- k represents an integer of 0 to 5.
- a method for producing a polyester resin comprising a repeating unit represented by the following formula (1a) and a repeating unit represented by the following formula (2a), wherein the polyester resin is represented by the following formula (4a 1 ).
- the manufacturing method of the polyester resin characterized by satisfy
- X 1a is a divalent group represented by the following formula (3a), and Y 1a is a divalent group.
- Y 1a is a divalent group.
- X 2a is X 1a. Is a group having a divalent aromatic that is not the same.
- Y 2a is a divalent group.
- R 1a and R 2a are each independently a hydrogen atom, an alkyl group which may have a substituent having 1 to 20 carbon atoms, an alkoxy group, or an aromatic group which may be substituted.
- N 1a and n 2a are each an integer of 0 to 4.
- R 3a and R 4a each independently have a hydrogen atom, an alkyl group having 1 to 20 carbon atoms, or a substituent. Or an aromatic group or a group in which R 3a and R 4a are bonded to each other to form a ring which may have a substituent.
- ⁇ 5> A method for producing a polyester resin, wherein at least one ester oligomer is used as a raw material and polymerized by an interfacial polymerization method.
- the polyester resin is obtained by polymerizing a divalent phenol residue and a divalent carboxylic acid residue, and the divalent phenol residue is represented by the above formulas (1) to (3).
- the at least one dihydric phenol residue selected from the dihydric phenol residue group and the dihydric phenol residue represented by the above formula (4), any one of the above ⁇ 4> to ⁇ 6> A method for producing a polyester resin.
- R 1 represents any one of a hydrogen atom, an alkyl group which may have a substituent having 1 to 20 carbon atoms, an alkoxy group, an aromatic group which may be substituted, and a halogen group.
- N 1 is an integer from 0 to 4.
- R 2 represents any one of a hydrogen atom, an alkyl group optionally having a substituent having 1 to 20 carbon atoms, an alkoxy group, an optionally substituted aromatic group, and a halogen group.
- N 2 is an integer of 0 to 6.
- Ar 1 and Ar 2 each independently represent an arylene group optionally having a substituent having 6 to 16 carbon atoms.
- Z represents an arylene group optionally having a substituent.
- s is an integer of 0 or 1.
- R 3 to R 6 are each independently a hydrogen atom, an alkyl group optionally having a substituent having 1 to 20 carbon atoms, an alkoxy group, an optionally substituted aromatic group, X represents a single bond, —CR 7 R 8 —, O, CO, or S.
- R 7 and R 8 each independently represents a hydrogen atom or a carbon atom having 1 to 10 carbon atoms.
- An alkyl group or R 7 and R 8 represent a C 5-10 cycloalkylidene group formed by combining R 7 and R 8.
- an electrophotographic photoreceptor excellent in wear resistance and electrical characteristics can be obtained.
- a polyester resin particularly excellent in solubility and wear resistance can be obtained.
- FIG. 1 is a conceptual diagram showing an embodiment of an image forming apparatus using the electrophotographic photosensitive member of the present invention.
- the photosensitive layer of the electrophotographic photoreceptor to which the exemplary embodiment is applied includes a polyester resin containing at least one divalent phenol residue selected from divalent phenol residues represented by the following formulas (1) to (3) Containing.
- R 1 is a hydrogen atom, an alkyl group which may have a substituent having 1 to 20 carbon atoms, an alkoxy group which may have a substituent having 1 to 20 carbon atoms, a carbon number It represents either an aromatic group optionally having 1 to 20 substituents or a halogen group, and n 1 is an integer of 0 to 4.
- the number of carbons is the number of carbons in the entire group including the substituent.
- substituent for R 1 include an alkyl group, an alkoxy group, a halogen atom, and the like, and preferably has no substituent.
- alkyl group which may have a substituent having 1 to 20 carbon atoms include methyl group, ethyl group, propyl group, isopropyl group, butyl group, tert-butyl group, isobutyl group, cyclohexyl group, chloro group
- alkyl group which may have a substituent having 1 to 20 carbon atoms include methyl group, ethyl group, propyl group, isopropyl group, butyl group, tert-butyl group, isobutyl group, cyclohexyl group, chloro group
- examples thereof include a methyl group, a fluorinated alkyl group, a trifluoromethyl group, and a perfluoroalkyl group.
- alkoxy group which may have a substituent having 1 to 20 carbon atoms include a methoxy group, an ethoxy group, a propoxy group, and a cyclohexoxy group.
- aromatic group which may have a substituent having 1 to 20 carbon atoms include a phenyl group, a methylphenyl group, a dimethylphenyl group, a halogenated phenyl group, and a naphthyl group.
- halogen group examples include a fluorine group, a chloro group, and a bromo group.
- an alkyl group having 1 to 6 carbon atoms, an alkoxy group having 1 to 6 carbon atoms and an unsubstituted aromatic group are preferable, and a methyl group and an ethyl group are particularly preferable.
- dihydric phenol compound that derives the dihydric phenol residue represented by the formula (1) include, for example, hydroquinone, methylhydroquinone, chlorohydroquinone, tert-butylhydroquinone, bromohydroquinone, and 2,3-dimethylhydroquinone.
- 2,5-dimethylhydroquinone trimethylhydroquinone, phenylhydroquinone, resorcinol, 2-methylresorcinol, 5-methylresorcinol, 5-bromoresorcinol, catechol, 4-methylcatechol and the like.
- hydroquinone, methylhydroquinone, 2,3-dimethylhydroquinone, 2,5-dimethylhydroquinone, trimethylhydroquinone, and resorcinol are particularly preferable from the viewpoint of ease of production and wear resistance.
- R 2 is a hydrogen atom, an alkyl group which may have a substituent having 1 to 20 carbon atoms, an alkoxy group which may have a substituent having 1 to 20 carbon atoms, a carbon number It represents either an aromatic group optionally having 1 to 20 substituents or a halogen group, and n 2 is an integer of 0 to 6.
- the number of carbons is the number of carbons in the entire group including the substituent.
- the substituent for R 2 include an alkyl group, an alkoxy group, a halogen atom, and the like, and preferably has no substituent. Specific examples of each group can be those exemplified for R 1 . From the viewpoint of wear resistance, an alkyl group having 1 to 6 carbon atoms and an alkoxy group having 1 to 6 carbon atoms are preferable, and a methyl group and an ethyl group are particularly preferable.
- dihydric phenol compound for deriving the dihydric phenol residue represented by the formula (2) include, for example, 1,2-dihydroxynaphthalene, 1,3-dihydroxynaphthalene, 1,5-dihydroxynaphthalene, , 6-dihydroxynaphthalene, 1,7-dihydroxynaphthalene, 2,3-dihydroxynaphthalene, 2,6-dihydroxynaphthalene, 2,7-dihydroxynaphthalene and the like.
- 1,6-dihydroxynaphthalene, 1,7-dihydroxynaphthalene, 2,6-dihydroxynaphthalene, and 2,7-dihydroxynaphthalene are preferable from the viewpoint of ease of production and solubility, and wear resistance. From the viewpoint, 2,6-dihydroxynaphthalene and 2,7-dihydroxynaphthalene are particularly preferable.
- Ar 1 and Ar 2 each independently represent an arylene group optionally having a substituent having 6 to 16 carbon atoms.
- Z is an arylene group which may have a substituent, and s is an integer of 0 or 1.
- arylene group which may have a substituent of Ar 1 and Ar 2 include a phenylene group, a naphthylene group, an anthrylene group, a phenanthrylene group, a pyrenylene group, and a biphenylene group.
- a phenylene group is preferred from the standpoint of production convenience.
- substituents examples include an alkyl group having 1 to 10 carbon atoms, an alkoxy group having 1 to 10 carbon atoms, a halogenated alkyl group, a halogen group, and a benzyl group.
- alkyl group examples include a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, a tert-butyl group, an isobutyl group, and a cyclohexyl group.
- alkoxy group examples include methoxy group, ethoxy group, propoxy group, cyclohexoxy group and the like.
- halogenated alkyl group examples include a chloromethyl group and a fluorinated alkyl group.
- halogen group examples include a fluorine group, a chloro group, and a bromo group. From the viewpoint of wear resistance, an alkyl group having 1 to 6 carbon atoms and an alkoxy group having 1 to 6 carbon atoms are preferable, and a methyl group and an ethyl group are particularly preferable.
- arylene group which may have a Z substituent include a p-phenylene group, an m-phenylene group, and a structure represented by the following formula (11). From the viewpoint of simplicity in production, a p-phenylene group or a structure represented by the following formula (11) is preferable.
- R 16 to R 19 are each independently a hydrogen atom or an alkyl group having 1 to 10 carbon atoms.
- alkyl group in the formula (11) examples include a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, a tert-butyl group, an isobutyl group, and a cyclohexyl group.
- R 16 to R 19 are preferably a hydrogen atom or a methyl group from the viewpoints of solubility, wear resistance, and ease of production.
- S is preferably an integer of 0 or 1 from the viewpoint of wear resistance, and more preferably 0 from the viewpoint of solubility. It represents below as a specific example of the dihydric phenol compound which induces
- polyester resin in the present invention contains a divalent phenol residue represented by the following formula (4).
- R 3 to R 6 each independently have a hydrogen atom, an alkyl group which may have a substituent having 1 to 20 carbon atoms, or a substituent having 1 to 20 carbon atoms. Or an aromatic group which may have a substituent having 1 to 20 carbon atoms, or a halogen group.
- X represents a single bond, —CR 7 R 8 —, O, CO, or S.
- R 7 and R 8 each independently represent a hydrogen atom, an alkyl group having 1 to 10 carbon atoms, or a cycloalkylidene group having 5 to 10 carbon atoms formed by combining R 7 and R 8 .
- each group of R 3 to R 6 includes those exemplified for R 1 .
- an alkyl group having 1 to 6 carbon atoms and an alkoxy group having 1 to 6 carbon atoms are preferable, and a methyl group is particularly preferable.
- alkyl group having 1 to 10 carbon atoms of R 7 and R 8 include a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, and an isobutyl group.
- a methyl group and an ethyl group are preferable from the viewpoint of solubility, wear resistance, and ease of production.
- the cycloalkylidene group having 5 to 10 carbon atoms formed by combining R 7 and R 8 include a cyclopentylidene group, a cyclohexylidene group, a cycloheptylidene group, and the like.
- bisphenol residue represented by the formula (4) include bis- (4-hydroxyphenyl) methane, bis- (4-hydroxy-3-methylphenyl) methane, and bis- (3,5-dimethyl).
- -4-hydroxyphenyl) methane 1,1-bis- (4-hydroxyphenyl) ethane, 1,1-bis- (4-hydroxy-3-methylphenyl) ethane, 1,1-bis- (3,5 -Dimethyl-4-hydroxyphenyl) ethane
- 1,1-bis- (4-hydroxyphenyl) propane 1,1-bis- (4-hydroxy-3-methylphenyl) propane, 1,1-bis- (3 5-dimethyl-4-hydroxyphenyl) propane, 2,2-bis- (4-hydroxyphenyl) propane, 2,2-bis- (4-hydrokey 3-methylphenyl) Lopan, 2,2-bis- (3,5-dimethyl-4-hydroxyphenyl) propane, 1,1-bis- (4-hydroxyphenyl) cyclohexane, 1,1-bis- (4-bis
- bis- (4-hydroxyphenyl) methane bis- (4-hydroxy-3-methylphenyl) methane, bis- ( 3,5-dimethyl-4-hydroxyphenyl) methane, 1,1-bis- (4-hydroxyphenyl) ethane, 1,1-bis- (4-hydroxy-3-methylphenyl) ethane, 2,2-bis -(4-hydroxyphenyl) propane, 2,2-bis- (4-hydrokey 3-methylphenyl) propane, 2,2-bis- (3,5-dimethyl-4-hydroxyphenyl) propane, 1,1- Bis- (4-hydroxyphenyl) cyclohexane, 4,4'-biphenol, 3,3'-dimethyl-4,4'-biphenol, 3,3 ', 5,5'-tetramethyl -4,4'-biphenol and 4,4'-dihydroxydiphenyl ether are preferred.
- bis- (4-hydroxyphenyl) methane bis- (4-hydroxy-3-methylphenyl) methane, 1,1-bis- (4-hydroxyphenyl) ethane, 1,1- Bis- (4-hydroxy-3-methylphenyl) ethane, 2,2-bis- (4-hydrokey 3-methylphenyl) propane, 4,4'-biphenol, 1,1-bis- (4-hydroxyphenyl) More preferred is cyclohexane.
- the content of the divalent phenol residues represented by the above formulas (1) to (3) is preferably 5 mol% or more and 80 mol% or less with respect to the total amount of all divalent phenol residues in the polyester resin. Further, from the viewpoint of solubility, it is more preferably 60 mol% or less, still more preferably 50 mol% or less, and particularly preferably 40 mol% or less. Further, from the viewpoint of wear resistance, it is more preferably 8 mol% or more, and further preferably 10 mol% or more.
- the content of the divalent phenol residue represented by the formula (4) is preferably 20 mol% or more with respect to the total amount of all divalent phenol residues, and from the viewpoint of solubility, it is 40 mol% or more. More preferably, 50 mol% or more is further more preferable, and 60 mol% or more is particularly preferable. Moreover, 95 mol% or less is preferable, and from a viewpoint of abrasion resistance, 92 mol% or less is more preferable, and 90 mol% or less is more preferable.
- the divalent phenol residues exemplified as the formulas (1) to (4) can be used in combination with a plurality of compounds as necessary. It is also possible to combine with a dihydric alcohol other than the formulas (1) to (4).
- Specific examples of the dihydric alcohol other than the formulas (1) to (4) include, for example, ethylene glycol, 1,3-propanediol, 1,4-butanediol, 1,8-octanediol, polyester diol, and polycarbonate. Examples include diol, polytetramethylene glycol, polysiloxane-containing diol, and the like.
- the divalent carboxylic acid residue of the polyester resin is preferably a divalent carboxylic acid residue represented by the following formula (5).
- Ar 3 and Ar 4 each independently represent an arylene group which may have a substituent.
- Y represents a divalent organic residue having a single bond, an oxygen atom, a sulfur atom, a structure represented by the formula (6), or a structure represented by the formula (7).
- R 9 and R 10 in Formula (6) each independently represent a hydrogen atom, an alkyl group, or an aryl group, or a cycloalkylidene group formed by combining R 9 and R 10 .
- R 11 is an alkylene group, an arylene group, or a group represented by Formula (8).
- R 12 and R 13 are each independently an alkylene group.
- Ar 5 represents an arylene group.
- k represents an integer of 0 to 5.
- Ar 3 and Ar 4 are preferably an arylene group having 6 to 20 carbon atoms, and examples thereof include a phenylene group, a naphthylene group, an anthrylene group, a phenanthrylene group, and a pyrenylene group. Among these, from the viewpoint of production cost, a phenylene group, a naphthylene group, or a biphenylene group is more preferable.
- the substituent that the arylene group may have independently include a hydrogen atom, an alkyl group, an alkoxy group, an aryl group, a condensed polycyclic group, and a halogen group.
- the aryl group is preferably a phenyl group or a naphthyl group
- the halogen group is a fluorine group, a chloro group, a bromo group or an iodo group.
- the alkoxy group is preferably a methoxy group, an ethoxy group, or a butoxy group.
- the alkyl group is preferably an alkyl group having 1 to 10 carbon atoms, more preferably an alkyl group having 1 to 8 carbon atoms, and 1 to 2 carbon atoms.
- a methyl group is particularly preferable.
- the number of substituents for each of Ar 3 and Ar 4 is not particularly limited, but is preferably 3 or less, more preferably 2 or less, and particularly preferably 1 or less. From the viewpoint of electrical characteristics and solubility, Ar 3 and Ar 4 are preferably the same arylene group having the same substituent, and more preferably an unsubstituted phenylene group.
- Y represents a divalent organic residue having a single bond, an oxygen atom, a sulfur atom, a structure represented by formula (6), or a structure represented by formula (7).
- R 9 and R 10 each independently represent a hydrogen atom, an alkyl group, or an aryl group, or a cycloalkylidene group formed by combining R 9 and R 10 .
- Examples of the alkyl group for R 9 and R 10 include a methyl group, an ethyl group, a propyl group, and a butyl group, and examples of the aryl group include a phenyl group and a naphthyl group.
- examples of the cycloalkylidene group formed by combining R 9 and R 10 in Formula (6) include a cyclopentylidene group, a cyclohexylidene group, and a cycloheptylidene group.
- R 11 is an alkylene group, an arylene group, or a group represented by Formula (8).
- R 12 and R 13 each independently represent an alkylene group.
- Ar 5 represents an arylene group.
- Examples of the alkylene group for R 11 in formula (7) include a methylene group, an ethylene group, and a propylene group.
- Examples of the arylene group for R 11 in formula (7) include a phenylene group and a terphenylene group. It is done.
- Specific examples of the group represented by the formula (8) include a group represented by the following formula (9).
- Y is preferably an oxygen atom from the viewpoint of wear resistance.
- k is an integer of 0 to 5, preferably 0 or 1, and particularly preferably 1 from the viewpoint of wear resistance.
- specific examples of the divalent carboxylic acid compound from which the divalent carboxylic acid residue is derived include terephthalic acid and isophthalic acid.
- the formula (5) is particularly preferably the following general formula (10).
- R 14 and R 15 each independently represent a hydrogen atom, an alkyl group, an aryl group, a halogen group, or an alkoxy group, and n 3 and n 4 are each independently an integer of 0 to 4 is there.
- R 14 and R 15 are, for example, a hydrogen atom; an alkyl group having 1 to 6 carbon atoms such as a methyl group, an ethyl group or an isopropyl group; an aryl group such as a phenyl group or a naphthyl group; a fluorine atom And halogen groups such as chlorine atom, bromine atom and iodine atom; alkoxy groups such as methoxy group, ethoxy group and butoxy group.
- R 14 and R 15 are particularly preferably a hydrogen atom or a methyl group.
- divalent carboxylic acid compound for deriving the divalent carboxylic acid residue represented by the formula (10) include, for example, diphenyl ether-2,2′-dicarboxylic acid, diphenyl ether-2,4′-dicarboxylic acid, Examples include diphenyl ether-4,4′-dicarboxylic acid. Among these, diphenyl ether-4,4′-dicarboxylic acid is particularly preferable in view of the convenience in production.
- the compound exemplified as the formula (5) can be used in combination with a plurality of compounds as necessary.
- Specific examples of the divalent carboxylic acid compound that may be combined include, for example, adipic acid, suberic acid, sebacic acid, phthalic acid, isophthalic acid, terephthalic acid, toluene-2,5-dicarboxylic acid, p-xylene-2,5 -Dicarboxylic acid, pyridine-2,3-dicarboxylic acid, pyridine-2,4-dicarboxylic acid, pyridine-2,5-dicarboxylic acid, pyridine-2,6-dicarboxylic acid, pyridine-3,4-dicarboxylic acid, pyridine -3,5-dicarboxylic acid, naphthalene-1,4-dicarboxylic acid, naphthalene-2,3-dicarboxylic acid, naphthalene-2,6-dicarboxylic acid,
- the total content of divalent carboxylic acid residues represented by the formula (5) is preferably 70 mol% or more based on the total divalent carboxylic acid component. From the viewpoint of wear resistance, it is preferably 90 mol% or more, particularly preferably 100 mol%.
- the viscosity average molecular weight (Mv) of the polyester resin is usually 10,000 or more, preferably 25,000 or more, more preferably 35,000 or more from the viewpoint of mechanical strength. Further, it is usually 200,000 or less, and preferably 150,000 or less from the viewpoint of applicability.
- the amount of carboxylic acid chloride group present at the terminal of the polyester resin is usually 0.1 ⁇ equivalent / g or less, preferably 0.05 ⁇ equivalent / g or less.
- the amount of the terminal carboxylic acid chloride group exceeds the above range, the storage stability when the polyester resin is used as a coating solution for an electrophotographic photoreceptor tends to decrease.
- the carboxylic acid value of the polyester resin is preferably 300 ⁇ equivalent / g or less, more preferably 150 ⁇ equivalent / g or less.
- the carboxylic acid value exceeds 150 ⁇ equivalent / g, the electrical characteristics of the photoreceptor tend to deteriorate, and further, the storage stability when the resin is dissolved in a solvent to form a coating solution tends to be lowered. .
- the amount of OH groups present at the terminal of the polyester resin is usually 100 ⁇ equivalent / g or less, preferably 50 ⁇ equivalent / g or less. If the amount of terminal OH groups exceeds the above range, the electrical characteristics when the polyester resin is used as an electrophotographic photoreceptor may be deteriorated.
- the total nitrogen amount (TN amount) contained in the polyester resin is preferably 3000 ppm or less, more preferably 1500 ppm or less, further preferably 1000 ppm or less, particularly preferably 500 pp or less, and most preferably 300 ppm or less. preferable. If the total nitrogen amount exceeds 3000 ppm, the electrical characteristics may deteriorate.
- the amount of the free divalent carboxylic acid contained in the polyester resin is not particularly limited, but is preferably 50 ppm or less, and more preferably 10 ppm or less.
- the content of the free divalent carboxylic acid exceeds 50 ppm, the electrical characteristics of the photoreceptor may be deteriorated or it may become a foreign substance appearing in image evaluation.
- the amount of the free divalent phenol contained in the polyester resin is not particularly limited, but is preferably 100 ppm or less, more preferably 50 ppm or less. If it exceeds 100 ppm, foreign matters may be produced depending on the deterioration of electrical characteristics, coloring, and the solvent used. From the viewpoint of the electrical characteristics of the photoreceptor, the less free divalent phenol is better, but from the viewpoint of ease of production, it is preferably 0.001 ppm or more, and particularly preferably 0.01 ppm or more.
- Polyester resin of the second embodiment has a repeating unit represented by the following formula (1a) and a repeating unit represented by the following formula (2a).
- X 1a is a divalent group represented by the following formula (3a), and Y 1a is a divalent group.
- X 2a is a group containing a divalent aromatic.
- Y 2a is a divalent group.
- X 2a is not the same as X 1a .
- R 1a and R 2a each independently have a hydrogen atom, an alkyl group which may have a substituent having 1 to 20 carbon atoms, or a substituent having 1 to 20 carbon atoms.
- the number of carbons is the number of carbons in the entire group including the substituent.
- substituents for R 1a and R 2a include an alkyl group, an alkoxy group, a halogen atom, and the like, and preferably no substituent.
- alkyl group examples include a methyl group, an ethyl group, an n-propyl group, an isopropyl group, an n-butyl group, an isobutyl group, a tert-butyl group, and a cyclohexyl group.
- alkoxy group examples include a methoxy group, an ethoxy group, an n-propoxy group, and an n-butoxy group.
- aromatic group that may have a substituent include a phenyl group, a naphthyl group, and a 4-methylphenyl group. Among these, a methyl group and an ethyl group are preferable from the viewpoint of solubility, wear resistance, and ease of production.
- n 1a and n 2a are each an integer of 0 to 4. From the viewpoint of wear resistance, it is preferably an integer of 0 to 2, particularly preferably 0 or 1.
- R 3a and R 4a each independently represents a hydrogen atom, an alkyl group having 1 to 20 carbon atoms, or an aromatic group optionally having a substituent having 1 to 20 carbon atoms. However, R 3a and R 4a may be bonded to each other to form a ring. The number of carbons is the number of carbons in the entire group including the substituent.
- substituents for R 3a and R 4a those exemplified as the substituent for R 1a and R 2a can be applied.
- the alkyl group include a methyl group, an ethyl group, an n-propyl group, an isopropyl group, an n-butyl group, and an isobutyl group.
- the aromatic group that may have a substituent include a phenyl group, a naphthyl group, and a biphenyl group.
- R 3a and R 4a are bonded to each other to form a ring
- a cyclopentylidene group a cyclohexylidene group, a cyclohexylidene group substituted with 1 to 3 methyl groups, and the like.
- R 3a and R 4a are preferably a hydrogen atom, a methyl group, an ethyl group, or a cyclohexylidene group in view of solubility, wear resistance, and ease of production.
- bisphenol as the base of the divalent group represented by the formula (3a) include bis- (4-hydroxyphenyl) methane, bis- (4-hydroxy-3-methylphenyl) methane, bis- (3,5-dimethyl-4-hydroxyphenyl) methane, 1,1-bis- (4-hydroxyphenyl) ethane, 1,1-bis- (4-hydroxy-3-methylphenyl) ethane, 1,1- Bis- (3,5-dimethyl-4-hydroxyphenyl) ethane, 1,1-bis- (4-hydroxyphenyl) propane, 1,1-bis- (4-hydrokey 3-methylphenyl) propane, 1,1 -Bis- (3,5-dimethyl-4-hydroxyphenyl) propane, 2,2-bis- (4-hydroxyphenyl) propane, 2,2-bis- (4-hydrokey 3- Tilphenyl) propane, 2,2-bis- (3,5-dimethyl-4-hydroxyphenyl) propane, 1,1-bis- (4-hydroxyphenyl) cyclohex
- bis- (4-hydroxyphenyl) methane, bis- (4-hydroxy-3-methylphenyl) methane, bis- (3,5-dimethyl) are considered in consideration of the ease of production, solubility, and electrical characteristics.
- bis- (4-hydroxyphenyl) methane bis- (4-hydroxy-3-methylphenyl) methane, 1,1-bis- (4-hydroxyphenyl) ethane, 1,1- Bis- (4-hydroxy-3-methylphenyl) ethane, 2,2-bis- (4-hydrokey 3-methylphenyl) propane, and 1,1-bis- (4-hydroxyphenyl) cyclohexane are more preferred.
- X 2a in the formula (2a) is a group containing a divalent aromatic that is not the same as X 1a .
- the group containing a divalent aromatic include a divalent group such as the above formula (3a) and a divalent group represented by the following formulas (7a) to (10a). From the viewpoint of printing durability, a group containing at least one divalent aromatic selected from the following formulas (7a) to (10a) is preferable.
- R 5a is a hydrogen atom, an alkyl group which may have a substituent having 1 to 20 carbon atoms, an alkoxy group which may have a substituent having 1 to 20 carbon atoms, a carbon number It represents an aromatic group or halogen group which may have 1 to 20 substituents.
- the number of carbons is the number of carbons in the entire group including the substituent.
- Specific examples of the alkyl group include a methyl group, an ethyl group, an n-propyl group, an isopropyl group, an n-butyl group, an isobutyl group, a tert-butyl group, and a cyclohexyl group.
- alkoxy group examples include a methoxy group, an ethoxy group, an n-propoxy group, and an n-butoxy group.
- aromatic group that may have a substituent include a phenyl group, a naphthyl group, and a methyl-substituted phenyl group.
- halogen group examples include a fluoro group, a chloro group, and a bromo group.
- a methyl group, a phenyl group, a fluoro group, and a chloro group are preferable from the viewpoint of solubility, wear resistance, and ease of production.
- n 3a is an integer of 0 to 4.
- an integer of 0 to 2 is preferable, and 0 is particularly preferable.
- Specific examples of the divalent phenol that is the base of the divalent group represented by the formula (7a) include hydroquinone, methylhydroquinone, chlorohydroquinone, fluorohydroquinone, resorcinol, and catechol. From the viewpoint of printing durability, hydroquinone is preferred.
- R 6a is a hydrogen atom, an alkyl group which may have a substituent having 1 to 20 carbon atoms, an alkoxy group which may have a substituent having 1 to 20 carbon atoms, a carbon number It represents an aromatic group or halogen group which may have 1 to 20 substituents.
- R 6a include those equivalent to R 5a .
- n 4a is an integer of 0-6. From the viewpoint of printing durability, it is preferably an integer of 0 to 2, particularly preferably 0.
- dihydric phenol that is the base of the divalent group represented by the formula (8a) include 1,6-dihydroxynaphthalene, 1,7-dihydroxynaphthalene, 1,4-dihydroxynaphthalene, 2, Examples thereof include 6-dihydroxynaphthalene and 2,7-dihydroxynaphthalene. From the viewpoint of printing durability, 2,6-dihydroxynaphthalene and 2,7-dihydroxynaphthalene are preferable.
- R 7a and R 8a are each independently an alkyl group which may have a substituent having 1 to 20 carbon atoms, or an alkoxy which may have a substituent having 1 to 20 carbon atoms.
- Specific examples of R 7a and R 8a include those equivalent to R 5a .
- n 5a and n 6a are integers of 0 to 4. From the viewpoint of printing durability, an integer of 0 to 2 is preferable, and 0 or 1 is particularly preferable.
- W 1 represents a single bond, oxygen, or sulfur.
- dihydric phenol that is the base of the divalent group represented by the formula (9a) include 4,4′-biphenol, 4,4′-dihydroxy-3,3′-dimethylbiphenyl, 4, 4'-dihydroxy-3,3 ', 5,5'-tetramethylbiphenyl, 4,4'-dihydroxy-2,2', 3,3 ', 5,5'-hexamethylbiphenyl, 4,4'- Dihydroxydiphenyl ether, 4,4′-dihydroxy-3,3′-dimethyldiphenyl ether, 4,4′-dihydroxy-3,3 ′, 5,5′-tetramethyldiphenyl ether, 4,4′-dihydroxydiphenyl sulfide, 4, 4'-dihydroxy-3,3'-dimethyldiphenyl sulfide, 4,4'-dihydroxy-3,3 ', 5,5'-tetramethyldiphenyl sulfide, etc.
- Ar 1a and Ar 2a each independently represent an arylene group optionally having a substituent having 6 to 16 carbon atoms.
- Z is an optionally substituted arylene group, or have a substituent represents an even alkylene group, s a is an integer of 0 or 1.
- arylene group in Ar 1a and Ar 2a examples include a phenylene group, a naphthylene group, an anthrylene group, a phenanthrylene group, a pyrenylene group, and a biphenylene group.
- a phenylene group is preferred from the standpoint of production convenience.
- Examples of the substituent for the arylene group include an alkyl group having 1 to 10 carbon atoms, an alkoxy group having 1 to 10 carbon atoms, a halogenated alkyl group, a halogen group, and a benzyl group.
- Specific examples of the alkyl group include a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, a tert-butyl group, an isobutyl group, and a cyclohexyl group.
- Specific examples of the alkoxy group include a methoxy group, an ethoxy group, a propoxy group, and a cyclohexoxy group.
- halogenated alkyl group examples include a chloromethyl group and a fluorinated alkyl group.
- examples of the halogen group include a fluorine group, a chloro group, and a bromo group. From the viewpoint of wear resistance, an alkyl group having 1 to 6 carbon atoms and an alkoxy group having 1 to 6 carbon atoms are preferable, and a methyl group and an ethyl group are particularly preferable.
- arylene group which may have a substituent in Z include a p-phenylene group, an m-phenylene group, and a structure represented by the following formula (11).
- alkylene group which may have a substituent include an ethylene group, a propylene group, a cyclohexylene group, and a 1,4-dimethylcyclohexane group.
- a p-phenylene group or a structure represented by the following formula (11) described in the first embodiment and ethylidene are preferable from the standpoint of production convenience.
- R 16 to R 19 each independently represent a hydrogen atom or an alkyl group having 1 to 10 carbon atoms.
- the alkyl group in the formula (11) include a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, a tert-butyl group, an isobutyl group, and a cyclohexyl group.
- R 16 to R 19 are preferably a hydrogen atom or a methyl group from the viewpoints of solubility, wear resistance, and ease of production.
- s a is an integer of 0 or 1 is preferred from the viewpoint of wear resistance, it is 0 in view of solubility more preferred. It represents below as a specific example of the dihydric phenol used as the origin of the bivalent group represented by Formula (10a).
- Y 1a and Y 2a in the formulas (1a) and (2a) are preferably divalent groups represented by the following formula (11a). Y 1a and Y 2a are preferably the same.
- Ar 3a and Ar 4a each independently represent an arylene group which may have a substituent.
- W 2 represents a single bond, an oxygen atom, a sulfur atom, a divalent organic residue having a structure represented by formula (12a), or a divalent organic residue having a structure represented by formula (13a).
- R 9a and R 10a in formula (12a) each independently represent a hydrogen atom, an alkyl group, or an aryl group, or a cycloalkylidene group formed by combining R 9a and R 10a .
- R 11a represents an alkylene group, an arylene group, or a group represented by formula (14a).
- R 12a and R 13a each independently represent an alkylene group
- Ar 5a represents an arylene group.
- k a represents an integer of 0 to 5.
- Ar 3a and Ar 4a are preferably an arylene group having 6 to 20 carbon atoms, and examples thereof include a phenylene group, a naphthylene group, a biphenylene group, an anthrylene group, a phenanthrylene group, and a pyrenylene group.
- a phenylene group, a naphthylene group, or a biphenylene group is more preferable.
- the substituent that the arylene group may have independently include an alkyl group, an alkoxy group, an aryl group, a condensed polycyclic group, and a halogen group.
- the aryl group is preferably a phenyl group or a naphthyl group
- the halogen group is a fluorine group, a chloro group, a bromo group or an iodo group.
- the alkoxy group is preferably a methoxy group, an ethoxy group, or a butoxy group.
- the alkyl group is preferably an alkyl group having 1 to 10 carbon atoms, more preferably an alkyl group having 1 to 8 carbon atoms, and 1 to 2 carbon atoms.
- a methyl group is particularly preferable.
- the number of substituents for Ar 3a and Ar 4a is not particularly limited, but is preferably 3 or less, more preferably 2 or less, and particularly preferably 1 or less. From the viewpoint of wear resistance and ease of production, Ar 3a and Ar 4a are preferably the same arylene group having the same substituent, and more preferably an unsubstituted phenylene group.
- R 9a and R 10a each independently represent a hydrogen atom, an alkyl group, or an aryl group, or a cycloalkylidene group formed by combining R 9a and R 10a .
- alkyl group of R 9a and R 10a include a methyl group, an ethyl group, a propyl group, and a butyl group
- aryl group include a phenyl group and a naphthyl group.
- Examples of the cycloalkylidene group formed by combining R 9a and R 10a in formula (12a) include a cyclopentylidene group, a cyclohexylidene group, and a cycloheptylidene group.
- R 11a is an alkylene group, an arylene group, or a group represented by Formula (14a)
- R 12a and R 13a each independently represent an alkylene group.
- Ar 5a represents an arylene group.
- Examples of the alkylene group represented by R 11a in formula (13a) include a methylene group, an ethylene group, and a propylene group.
- Examples of the arylene group represented by R 11a include a phenylene group and a terphenylene group.
- Specific examples of the group represented by the formula (14a) include a group represented by the following formula (15a).
- W 2 is preferably an oxygen atom from the viewpoint of wear resistance.
- ka is an integer of 0 to 5. From the viewpoint of wear resistance, it is preferably an integer of 0 or 1, and is particularly preferably 1.
- specific examples of the divalent carboxylic acid that is the base of the divalent group represented by the formula (11a) include terephthalic acid and isophthalic acid.
- the formula (11a) is particularly preferably the following general formula (16a).
- R 18a and R 19a each independently represent a hydrogen atom, an alkyl group, an aryl group, a halogen group, or an alkoxy group, and n 7 and m are each independently an integer of 0 to 4. .
- examples of R 18a and R 19a include a hydrogen atom; an alkyl group having 1 to 6 carbon atoms such as a methyl group, an ethyl group, and an isopropyl group; an aryl group such as a phenyl group and a naphthyl group; a fluorine atom And halogen groups such as chlorine atom, bromine atom and iodine atom; alkoxy groups such as methoxy group, ethoxy group and butoxy group.
- R 18a and R 19a are particularly preferably a hydrogen atom or a methyl group.
- Specific examples of the divalent carboxylic acid compound derived from the divalent group represented by the formula (16a) include, for example, diphenyl ether-2,2′-dicarboxylic acid, diphenyl ether-2,4′-dicarboxylic acid, diphenyl ether- 4,4'-dicarboxylic acid and the like. Among these, diphenyl ether-4,4′-dicarboxylic acid is particularly preferable in view of the convenience in production.
- the dicarboxylic acid residue can be used in combination of a plurality of compounds as required.
- Specific examples of the divalent carboxylic acid compound that may be combined include, for example, adipic acid, suberic acid, sebacic acid, phthalic acid, isophthalic acid, terephthalic acid, toluene-2,5-dicarboxylic acid, p-xylene-2,5 -Dicarboxylic acid, pyridine-2,3-dicarboxylic acid, pyridine-2,4-dicarboxylic acid, pyridine-2,5-dicarboxylic acid, pyridine-2,6-dicarboxylic acid, pyridine-3,4-dicarboxylic acid, pyridine -3,5-dicarboxylic acid, naphthalene-1,4-dicarboxylic acid, naphthalene-2,3-dicarboxylic acid, naphthalene-2,6-dicarboxylic acid, biphenyl
- the content of the repeating unit represented by the formula (2a) in the polyester resin is 0.45 ⁇ (2a) / ((1a) + () in terms of a molar ratio with the repeating unit represented by the formula (1a). 2a)) ⁇ 0.05 is preferred. Further, from the viewpoint of the solubility and wear resistance of the polyester resin, preferably 0.40 ⁇ (2a) / ((1a) + (2a)) ⁇ 0.08, and more preferably 0.35. ⁇ (2a) / ((1a) + (2a)) ⁇ 0.10.
- the viscosity average molecular weight (Mv) of the polyester resin is usually 10,000 or more, and preferably 20,000 or more from the viewpoint of mechanical strength. Further, it is usually 200,000 or less, and preferably 150,000 or less from the viewpoint of applicability.
- the amount of carboxylic acid chloride group present at the terminal of the polyester resin is usually 0.1 ⁇ equivalent / g or less, preferably 0.05 ⁇ equivalent / g or less.
- the amount of the terminal carboxylic acid chloride group exceeds the above range, the storage stability when the polyester resin is used as a coating solution for an electrophotographic photoreceptor tends to decrease.
- the carboxylic acid value of the polyester resin is preferably 300 ⁇ equivalent / g or less, more preferably 150 ⁇ equivalent / g or less.
- the carboxylic acid value exceeds 300 ⁇ equivalent / g, the electrical characteristics of the photoreceptor tend to deteriorate, and further, the storage stability when the resin is dissolved in a solvent to form a coating solution tends to decrease. .
- the amount of OH groups present at the end of the polyester resin is usually 200 ⁇ eq / g or less, preferably 100 ⁇ eq / g or less.
- the amount of terminal OH groups exceeds the above range, the electrical characteristics when the polyester resin is used as an electrophotographic photoreceptor may be deteriorated.
- the total nitrogen amount (TN amount) contained in the polyester resin is preferably 500 ppm or less, more preferably 300 ppm or less, and particularly preferably 150 ppm or less. If the total nitrogen amount exceeds 500 ppm, the electrical characteristics may deteriorate.
- the amount of the free divalent carboxylic acid contained in the polyester resin is not particularly limited, but is preferably 50 ppm or less, and more preferably 10 ppm or less.
- the content of the free divalent carboxylic acid exceeds 50 ppm, the electrical characteristics of the photoreceptor may be deteriorated or it may become a foreign substance appearing in image evaluation.
- the amount of free divalent phenol contained in the polyester resin is not particularly limited, but is preferably 100 ppm or less, and more preferably 50 ppm or less. If it exceeds 100 ppm, foreign matters may be produced depending on the deterioration of electrical characteristics, coloring, and the solvent used. From the viewpoint of the electrical characteristics of the photoreceptor, the less free divalent phenol is better, but from the viewpoint of ease of production, it is preferably 0.001 ppm or more, and particularly preferably 0.01 ppm or more.
- the structure of the polyester resin produced according to the present invention is not particularly limited, but a polyester resin having the following structure is preferable from the viewpoint of solubility, mechanical properties, electrical characteristics, and the like.
- a polyester resin having the following structure is preferable from the viewpoint of solubility, mechanical properties, electrical characteristics, and the like.
- the above method for producing a polyester resin is effective.
- the types referred to here are distinguished by molecular structure, and positional isomers, stereoisomers and the like are also distinguished to be one type.
- the polyester resin preferably has at least one divalent carboxylic acid residue represented by the following formulas (1b) to (4b).
- each R 1b independently represent an alkyl group having 1 to 10 carbon atoms, an alkoxy group having 1 to 10 carbon atoms, a halogenated alkyl group, a halogen group, or a benzyl group, n b is 0 It is an integer of ⁇ 4.
- alkyl group examples include a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, a tert-butyl group, an isobutyl group, and a cyclohexyl group.
- alkoxy group examples include a methoxy group, an ethoxy group, a propoxy group, and a cyclohexoxy group.
- halogenated alkyl group examples include a chloromethyl group and a fluorinated alkyl group.
- examples of the halogen group include a fluorine group, a chloro group, and a bromo group. From the viewpoint of wear resistance, an alkyl group having 1 to 6 carbon atoms and an alkoxy group having 1 to 6 carbon atoms are preferable, and a methyl group and an ethyl group are particularly preferable.
- Specific examples of the divalent carboxylic acid compound that derives the divalent carboxylic acid residue represented by the formula (1b) include orthophthalic acid, isophthalic acid, terephthalic acid, and the like. Among these, isophthalic acid and terephthalic acid are particularly preferable from the viewpoint of wear resistance.
- R 2b and R 3b each independently represent an alkyl group having 1 to 10 carbon atoms, an alkoxy group having 1 to 10 carbon atoms, a halogenated alkyl group, a halogen group, or a benzyl group, mb, lb is an integer of 0 to 4, and Xb represents a single bond or an oxygen atom.
- the alkyl group include a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, a tert-butyl group, an isobutyl group, and a cyclohexyl group.
- alkoxy group examples include methoxy group, ethoxy group, propoxy group, cyclohexoxy group and the like.
- halogenated alkyl group examples include a chloromethyl group and a fluorinated alkyl group.
- examples of the halogen group include a fluorine group, a chloro group, and a bromo group.
- an alkyl group having 1 to 6 carbon atoms and an alkoxy group having 1 to 6 carbon atoms are preferable, and a methyl group and an ethyl group are particularly preferable.
- divalent carboxylic acid compound for deriving the divalent carboxylic acid residue represented by the formula (2b) include, for example, diphenyl ether-2,2′-dicarboxylic acid, diphenyl ether-2,4′-dicarboxylic acid, Examples thereof include diphenyl ether-4,4′-dicarboxylic acid, 4,4′-diphenyldicarboxylic acid, 2,2′-diphenyldicarboxylic acid, 2,4′-diphenyldicarboxylic acid and the like. Of these, diphenyl ether-4,4′-dicarboxylic acid and 4,4′-diphenyldicarboxylic acid are particularly preferable from the viewpoint of ease of production and wear resistance.
- R 4b independently represents an alkyl group having 1 to 10 carbon atoms.
- the alkyl group include a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, a tert-butyl group, an isobutyl group, and a cyclohexyl group.
- an alkyl group having 1 to 6 carbon atoms is preferable, and a methyl group and an ethyl group are particularly preferable.
- divalent carboxylic acid compound for deriving the divalent carboxylic acid residue represented by the formula (3b) include, for example, trans-1,4-cyclohexanedicarboxylic acid, cis-1,4-cyclohexanedicarboxylic acid, Examples thereof include trans-1,3-cyclohexanedicarboxylic acid and cis-1,3-cyclohexanedicarboxylic acid.
- trans-1,4-cyclohexanedicarboxylic acid and cis-1,4-cyclohexanedicarboxylic acid are particularly preferable from the viewpoints of production convenience and wear resistance.
- Y b represents a single bond, an alkylene group having 1 to 14 carbon atoms, p- phenylene, m- phenylene, or 4,4'-biphenyl group.
- alkylene group include methylene group, ethylene group, propylene group, butylene group, 2,2-dimethylpropylene group, cyclohexylene group, and cyclohexanedimethyl group.
- divalent carboxylic acid compound for deriving the divalent carboxylic acid residue represented by the formula (4b) include malonic acid, succinic acid, 2,2-dimethylsuccinic acid, glutamic acid, azelaic acid, 1, Examples include 3-adamantane dicarboxylic acid, sebacic acid, 1,10-decanedicarboxylic acid, 1,4-phenylenediacetic acid and the like.
- polyester resin contains a dicarboxylic acid residue of the formula (3b) or formula (4b)
- dicarboxylic acid residue of the above formula (1b) or (2b) and Copolymerization is preferred.
- the polyester resin preferably has at least one dihydric phenol and / or dihydric alcohol represented by the following formulas (5b) to (12b).
- each R 5b independently represents an alkyl group having 1 to 10 carbon atoms, an alkoxy group having 1 to 10 carbon atoms, a halogenated alkyl group, a halogen group, or a benzyl group, and q b is 0 It is an integer of ⁇ 4.
- alkyl group examples include a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, a tert-butyl group, an isobutyl group, and a cyclohexyl group.
- alkoxy group examples include a methoxy group, an ethoxy group, a propoxy group, and a cyclohexoxy group.
- halogenated alkyl group examples include a chloromethyl group and a fluorinated alkyl group.
- the halogen group examples include a fluorine group, a chloro group, and a bromo group.
- an alkyl group having 1 to 6 carbon atoms and an alkoxy group having 1 to 6 carbon atoms are preferable, and a methyl group and an ethyl group are particularly preferable.
- dihydric phenol compound for deriving the dihydric phenol residue represented by the formula (5b) include, for example, hydroquinone, methylhydroquinone, bromohydroquinone, 2,3-dimethylhydroquinone, trimethylhydroquinone, resorcinol, 2- Examples thereof include methylresorcinol, 5-methylresorcinol, 5-bromoresorcinol, catechol, 4-methylcatechol and the like. Among these, hydroquinone, methylhydroquinone, and resorcinol are particularly preferable from the viewpoints of production convenience and wear resistance.
- each R 6b independently represent an alkyl group having 1 to 10 carbon atoms, an alkoxy group having 1 to 10 carbon atoms, a halogenated alkyl group, a halogen group, or a benzyl group, r b is 0 An integer of ⁇ 6.
- alkyl group examples include a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, a tert-butyl group, an isobutyl group, and a cyclohexyl group.
- alkoxy group examples include a methoxy group, an ethoxy group, a propoxy group, and a cyclohexoxy group.
- halogenated alkyl group examples include a chloromethyl group and a fluorinated alkyl group.
- examples of the halogen group include a fluorine group, a chloro group, and a bromo group.
- an alkyl group having 1 to 6 carbon atoms and an alkoxy group having 1 to 6 carbon atoms are preferable, and a methyl group and an ethyl group are particularly preferable.
- dihydric phenol compound for deriving the dihydric phenol residue represented by the formula (6b) include, for example, 1,2-dihydroxynaphthalene, 1,3-dihydroxynaphthalene, 1,5-dihydroxynaphthalene, , 6-dihydroxynaphthalene, 1,7-dihydroxynaphthalene, 2,3-dihydroxynaphthalene, 2,6-dihydroxynaphthalene, 2,7-dihydroxynaphthalene and the like.
- 1,6-dihydroxynaphthalene, 1,7-dihydroxynaphthalene, 2,6-dihydroxynaphthalene, and 2,7-dihydroxynaphthalene are particularly preferable from the viewpoint of ease of production, solubility, and wear resistance. .
- Ar 1b, the Ar 2b each independently, p- phenylene, m- phenylene group, a divalent naphthalene radical, or a divalent biphenyl group
- Z b is an alkylene group having 1 to 10 carbon atoms
- s b is an integer of 0 or 1.
- R 15b to R 18b each independently represents a hydrogen atom or an alkyl group having 1 to 10 carbon atoms.
- alkylene group in the formula (7b) examples include a methylene group, an ethylene group, a propylene group, a butylene group, a 2,2-dimethylpropylene group, a cyclohexylene group, and a cyclohexanedimethyl group.
- an alkyl group having 1 to 8 carbon atoms is preferable, and an ethylene group, a cyclohexylene group, and a cyclohexanedimethyl group are particularly preferable.
- alkyl group in the formula (12b) examples include methyl group, ethyl group, propyl group, isopropyl group, butyl group, tert-butyl group, isobutyl group, cyclohexyl group and the like.
- a methyl group is preferred from the viewpoint of solubility, abrasion resistance, and ease of production.
- dihydric phenol compound for deriving the dihydric phenol residue represented by the formula (7b) are shown below.
- W b represents a single bond, an alkylene group having 1 to 16 carbon atoms, a perfluoroalkylene group having 3 to 10 carbon atoms, an m-phenylene group, a p-phenylene group, a divalent biphenyl group, or Represents a divalent naphthalene group.
- alkylene group examples include methylene group, ethylene group, propylene group, butylene group, hexylene group, cyclohexylene group, octylene group and the like.
- perfluoroalkylene group examples include a hexafluoro-1,3-propylene group, an octafluoro-1,4-butylene group, a dodecafluoro-1,6-hexylene group, and a hexadecafluoro-1,8-octylene group.
- Etc From the viewpoint of solubility and abrasion resistance, an alkylene group having 1 to 6 carbon atoms and a perfluoroalkylene group having 3 to 8 carbon atoms are preferable.
- dihydric alcohol compound derived from the dihydric alcohol residue represented by the formula (8b) include, for example, ethylene glycol, 1,3-propylene glycol, 1,4-butanediol, 1,5-pentane.
- ethylene glycol, 1,4-butanediol, 1,4-cyclohexanedimethanol, 1,8-octanediol, and 1,4-benzene are preferred from the viewpoint of ease of production, solubility, and wear resistance.
- Dimethanol and 4,4′-biphenyldimethanol are particularly preferred.
- R 7b are each R 8b independently represent an alkyl group, or an alkoxy group having 1 to 10 carbon atoms having 1 to 10 carbon atoms, t b, u b each represent an integer of 0 to 10 It is.
- a b represents a single bond, —CR 9b R 10b —.
- R 9b and R 10b each independently represent a hydrogen atom, an alkyl group having 1 to 20 carbon atoms, or a group that forms a ring in which R 9b and R 10b are bonded to each other and may have a substituent.
- v b is an integer of 0 or 1.
- alkyl group examples include a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, a tert-butyl group, an isobutyl group, and a cyclohexyl group.
- alkoxy group examples include a methoxy group, an ethoxy group, a propoxy group, and a cyclohexoxy group. From the viewpoint of wear resistance, an alkyl group having 1 to 6 carbon atoms and an alkoxy group having 1 to 6 carbon atoms are preferable, and a methyl group and an ethyl group are particularly preferable.
- dihydric alcohol compound from which the dihydric alcohol residue represented by the formula (9b) is derived include 1,4-cyclohexanediol, 2,2-bis (4-hydroxycyclohexyl) propane, 4, 4-bicyclohexanol and the like can be mentioned.
- dihydric alcohol compound that induces the dihydric alcohol residue represented by the formula (10b) include isomannide, isosorbide, and the like.
- R 11b to R 14b each independently represent a hydrogen atom or a methyl group.
- V b represents a single bond, —CR 15b R 16b —, O, CO, or S.
- R 15b and R 16b are each independently a hydrogen atom, a methyl group, or an ethyl group, or R 15b and R 16b are a cyclopentyl group or a cyclohexylidene group formed by combining R 15b and R 16b. Represents.
- bisphenol residue of the polyester resin represented by the formula (11b) include bis- (4-hydroxyphenyl) methane, bis- (4-hydroxy-3-methylphenyl) methane, bis- (3, 5-dimethyl-4-hydroxyphenyl) methane, 1,1-bis- (4-hydroxyphenyl) ethane, 1,1-bis- (4-hydroxy-3-methylphenyl) ethane, 1,1-bis- ( 3,5-dimethyl-4-hydroxyphenyl) ethane, 1,1-bis- (4-hydroxyphenyl) propane, 1,1-bis- (4-hydroxy 3-methylphenyl) propane, 1,1-bis- (3,5-dimethyl-4-hydroxyphenyl) propane, 2,2-bis- (4-hydroxyphenyl) propane, 2,2-bis- (4-hydroxy 3-methylphenyl) propane, 2,2-bis- (3,5-dimethyl-4-hydroxyphenyl) propane, 1,1-bis- (4-hydroxyphenyl) cyclohexane, 1,1-bis- (4- Hydro
- bis- (4-hydroxyphenyl) methane bis- (4-hydroxy-3-methylphenyl) methane, bis- ( 3,5-dimethyl-4-hydroxyphenyl) methane, 1,1-bis- (4-hydroxyphenyl) ethane, 1,1-bis- (4-hydroxy-3-methylphenyl) ethane, 2,2-bis -(4-hydroxyphenyl) propane, 2,2-bis- (4-hydrokey 3-methylphenyl) propane, 2,2-bis- (3,5-dimethyl-4-hydroxyphenyl) propane, 1,1- Bis- (4-hydroxyphenyl) cyclohexane, 4,4'-biphenol, 3,3'-dimethyl-4,4'-biphenol, 3,3 ', 5,5'-tetramethyl -4,4'-biphenol and 4,4'-dihydroxydiphenyl ether are preferred.
- bis- (4-hydroxyphenyl) methane bis- (4-hydroxy-3-methylphenyl) methane, 1,1-bis- (4-hydroxyphenyl) ethane, 1,1- Bis- (4-hydroxy-3-methylphenyl) ethane, 2,2-bis- (4-hydrokey 3-methylphenyl) propane, 4,4'-biphenol, 3,3'-dimethyl-4,4'- Biphenol, 3,3 ′, 5,5′-tetramethyl-4,4′-biphenol is more preferred.
- the polyester resin of the present invention when one of the dihydric phenols or dihydric alcohols of the above formulas (5b) to (10b) is used, from the viewpoint of solubility, molecular weight of the resin, and mechanical properties, It is preferable to copolymerize with the dihydric phenol of (11b).
- the content of the formula (11b) is not particularly limited, but is preferably 5 mol% or more, more preferably 30 mol% or more, further preferably 50 mol% or more of the total dihydric phenol and dihydric alcohol.
- Polyester polycarbonate resin of the third embodiment >> About the polyester site
- the formula (13b) is preferable.
- R 19b to R 22b each independently represents a hydrogen atom or a methyl group.
- Q b represents a single bond, —CR 23b R 24b —, O, CO, or S.
- R 23b and R 24b each independently represent a hydrogen atom, a methyl group, or an ethyl group, or R 23b and R 24b are a cyclopentyl group or a cyclohexyl group formed by combining R 23b and R 24b.
- R 23b and R 24b are each preferably a hydrogen atom or a methyl group.
- the viscosity average molecular weight (Mv) of the polyester resin and polyester polycarbonate resin produced by the production method of the present invention is usually 15,000 or more, preferably 20,000 or more. Moreover, from a viewpoint of applicability
- the amount of the carboxylic acid chloride group present at the terminal of the polyester resin in the present invention is usually 0.1 ⁇ equivalent / g or less, preferably 0.05 ⁇ equivalent / g or less.
- the amount of the terminal carboxylic acid chloride group exceeds the above range, the storage stability when the polyester resin is used as a coating solution for an electrophotographic photoreceptor tends to decrease.
- the amount of OH groups present at the end of the polyester resin in the present invention is usually 100 ⁇ equivalent / g or less, preferably 50 ⁇ equivalent / g or less. If the amount of terminal OH groups exceeds the above range, the electrical characteristics when the polyester resin is used as an electrophotographic photoreceptor may be deteriorated.
- the amount of the carboxylic acid group present at the terminal of the polyester resin in the present invention is usually 200 ⁇ eq / g or less, preferably 100 ⁇ eq / g or less. If the amount of the terminal carboxylic acid group exceeds the above range, the electrical characteristics when the polyester resin is used as an electrophotographic photoreceptor may be deteriorated.
- the total nitrogen amount (TN amount) contained in the polyester resin is preferably 3000 ppm or less, more preferably 1500 ppm or less, and particularly preferably 1000 ppm or less. If the total nitrogen amount exceeds 3000 ppm, the electrical characteristics may deteriorate.
- the polyester resin production method in the present invention is preferably a solution polymerization method or a polymerization method in which a solution polymerization method and an interfacial polymerization method are combined.
- the polyester resin of the present invention when the polyester resin of the present invention is produced, a dihydric phenol having an aromatic ester bond is used. Therefore, in the interfacial polymerization method, when the divalent phenol that has become an alkali salt is dissolved in the aqueous layer, hydroxylation is performed. It is rapidly hydrolyzed by nucleophiles such as product ions, and polymerization does not proceed. Therefore, the method for producing a polyester resin in the present invention is preferably a solution polymerization method or a polymerization method in which a solution polymerization method and an interfacial polymerization method are combined. An example of a method for producing a polyester resin will be described below.
- the polymerization can be performed by dissolving a dihydric phenol compound and a divalent carboxylic acid chloride compound in a solvent and adding a base such as triethylamine.
- the polymerization temperature is preferably in the range of ⁇ 10 ° C. to 40 ° C.
- the polymerization time is preferably in the range of 0.5 hour to 10 hours from the viewpoint of productivity.
- Examples of the base used in the solution polymerization method include triethylamine, tripropylamine, tributylamine, N, N-diisopropylethylamine, N, N-dipropylethylamine, N, N-diethylmethylamine, N, N-dimethylethylamine.
- tertiary amines such as octane, pyridines such as pyridine and 4-methylpyridine, and organic bases such as 1,8-diazabicyclo [5.4.0] -7-undecene.
- the base is not particularly limited as long as it is a base such as phosphazene base and inorganic base used for esterification reaction.
- bases triethylamine, N, N-dipropylethylamine, N, N-diethylmethylamine, and pyridine are preferable from the viewpoint of the reactivity of the esterification reaction and availability, and the acid chloride decomposition is inhibited and washed.
- Triethylamine is particularly preferable from the viewpoint of ease of removal.
- the amount of base used is preferably in the range of 1.01 to 2 equivalents of the carboxylic acid chloride group contained in the reaction system.
- solvent examples include halogenated hydrocarbon compounds such as dichloromethane, chloroform, 1,2-dichloroethane, trichloroethane, tetrachloroethane, chlorobenzene and dichlorobenzene, aromatic hydrocarbon compounds such as toluene, anisole and xylene, cyclohexane, methylcyclohexane and the like.
- halogenated hydrocarbon compounds such as dichloromethane, chloroform, 1,2-dichloroethane, trichloroethane, tetrachloroethane, chlorobenzene and dichlorobenzene
- aromatic hydrocarbon compounds such as toluene, anisole and xylene, cyclohexane, methylcyclohexane and the like.
- Hydrocarbon compounds such as tetrahydrofuran, tetrahydropyran, 1,4-dioxane, 1,3-dioxolane, ester compounds such as ethyl acetate, methyl benzoate, benzyl acetate, N, N-dimethylformamide, N, N- Examples thereof include amide compounds such as dimethylacetamide.
- pyridine may be used as a base and a solvent.
- dichloromethane, chloroform, 1,2-dichloroethane, tetrahydrofuran, N, N-dimethylformamide, and pyridine are preferable from the viewpoints of solubility of monomers and oligomers to be formed and reactivity of esterification reaction. Further, dichloromethane is particularly preferable from the viewpoint of washing efficiency and electrical characteristics.
- the ratio of the dihydric phenol and the divalent carboxylic acid chloride is not particularly limited. However, from the viewpoint of producing a high molecular weight polyester resin and controlling the end group, divalent phenol: 2
- the monovalent carboxylic acid chloride is preferably in a molar ratio of 1: 0.95 to 1: 1.05, more preferably 1: 0.99 to 1: 1.01, particularly preferably 1: 0.995 to 1: 1.005. It is.
- the dihydric phenol when the dihydric phenol has poor solubility in a solvent, it may precipitate when reacting with the divalent carboxylic acid chloride, resulting in the insoluble content remaining in the produced polyester resin and the deterioration of electrical characteristics. .
- the dihydric phenol when the dihydric phenol has poor solubility in the solvent, it may precipitate when reacting with the divalent carboxylic acid chloride, resulting in white turbidity of the reaction solution or inhibition of oligomerization. Therefore, when divalent phenol having poor solubility is introduced into the polyester resin, it is preferable to copolymerize with divalent phenol having good solubility.
- a divalent phenol and a divalent carboxylic acid chloride with good solubility are reacted in advance to form an oligomer in the reaction system, and then a divalent phenol with poor solubility is added and reacted to make a polyester. It is more preferable to produce a resin.
- a molecular weight regulator When producing a polyester resin, a molecular weight regulator can be used.
- the molecular weight regulator include phenol, o, m, p-cresol, o, m, p-ethylphenol, o, m, p-propylphenol, o, m, p- (tert-butyl) phenol, and pentyl.
- Alkylphenols such as phenol, hexylphenol, octylphenol, nonylphenol, 2,6-dimethylphenol derivatives, 2-methylphenol derivatives; monofunctional phenols such as o, m, p-phenylphenol; acetic acid chloride, butyric acid chloride, octyl Examples thereof include monofunctional acid halides such as acid chloride, benzoyl chloride, benzenesulfonyl chloride, benzenesulfinyl chloride, sulfinyl chloride, benzenephosphonyl chloride and substituted products thereof.
- monofunctional acid halides such as acid chloride, benzoyl chloride, benzenesulfonyl chloride, benzenesulfinyl chloride, sulfinyl chloride, benzenephosphonyl chloride and substituted products thereof.
- monofunctional aliphatic alcohols such as methanol, ethanol and propanol
- monofunctional alcohols having acrylics such as 2-hydroxyethyl acrylate, 4-hydroxybutyl acrylate and 2-hydroxy methacrylate, 1H, 1H, 2H, 2H-
- monofunctional alcohols having perfluoroalkyl such as tridecafluoro-1-n-octanol, 1H, 1H, 2H, 2H-heptadecafluoro-1-decanol, and monofunctional alcohols having siloxane.
- o, m, p- (tert-butyl) phenol, 2,6-dimethylphenol derivatives, 2-methylphenol are preferred because of their high molecular weight controllability and solution stability.
- Particularly preferred are p- (tert-butyl) phenol, 2,3,6-trimethylphenol, and 2,3,5-trimethylphenol.
- an antioxidant can be added during the polymerization reaction or in the cleaning liquid.
- the antioxidant include sodium sulfite, hydrosulfite (sodium hyposulfite), sulfur dioxide, potassium sulfite, sodium hydrogen sulfite and the like.
- hydrosulfite is particularly preferable from the viewpoint of the antioxidant effect and reduction of environmental load.
- As the usage-amount of antioxidant 0.01 mass% or more and 10.0 mass% or less are preferable with respect to all the bivalent phenols. More preferably, it is 0.1 mass% or more and 5 mass% or less.
- the polyester resin solution is an alkaline aqueous solution such as sodium hydroxide or potassium hydroxide; hydrochloric acid, An aqueous solution of acid such as nitric acid and phosphoric acid; a method of separating by standing separation, centrifugation, etc. after washing with water or the like.
- the polyester resin solution after washing is precipitated in water, alcohol or other organic solvent in which the polyester resin is insoluble, or the polyester resin solution is distilled off in warm water or in a dispersion medium in which the polyester resin is insoluble, or heated, It may be taken out by distilling off the solvent under reduced pressure or the like, and when taken out in the form of a slurry, the solid can be taken out by a centrifugal separator or a filter.
- the polyester resin is usually dried at a temperature equal to or lower than the decomposition temperature of the polyester resin, preferably 20 ° C. or higher and lower than the melting temperature of the polyester resin. At this time, it is preferable to dry under reduced pressure. Preferably, the drying time is longer than the time until the purity of impurities such as the residual solvent becomes below a certain level.
- the residual solvent is usually 1000 ppm or lower, preferably 300 ppm or lower, more preferably 100 ppm or lower. dry.
- the polyester resin in the present invention uses a dihydric phenol having an aromatic ester bond in the first stage to produce an ester oligomer by a solution polymerization method, or a carbonate oligomer or a polycarbonate resin by a melt polymerization method. It can be produced by an interfacial polymerization method using an ester oligomer, a carbonate oligomer or a polycarbonate resin.
- a dihydric phenol residue having an aromatic ester bond into the oligomer produced in the first stage, the oligomer is not dissolved in the aqueous layer during the interfacial polymerization in the second stage.
- the aromatic ester bond does not hydrolyze and a polyester resin can be obtained.
- Ester oligomer production method examples include solution polymerization, melt polymerization, and interfacial polymerization. Among these, solution polymerization and melt polymerization are preferable from the viewpoints of antioxidation of bisphenol, reactivity of dihydric alcohol, stability of ester bond in the monomer, and the like. Furthermore, solution polymerization is particularly preferred from the standpoint of production.
- a dihydric phenol having an ester bond When a dihydric phenol having an ester bond is used, it is preferably used at the time of producing the ester oligomer.
- a nucleophilic agent such as hydroxide ion. Is hydrolyzed and polymerization does not proceed. Therefore, by introducing a dihydric phenol having an ester bond into the ester oligomer by a solution polymerization method or a melt polymerization method, it becomes difficult to dissolve in an aqueous solution and can be used for the following polyester production without hydrolysis. It becomes.
- dihydric phenol that is easily oxidized
- it is preferably used at the time of producing the ester oligomer.
- Divalent phenols that are easily oxidized are particularly easily oxidized when they become anions. Therefore, in the interfacial polymerization method, the strong alkali aqueous solution becomes an anion and is rapidly oxidized to prevent the polymerization from proceeding. Therefore, by introducing a dihydric phenol that is easily oxidized into an ester oligomer in advance by a solution polymerization method or a melt polymerization method that does not form an anion, it can be used for the following polyester production.
- a dihydric phenol compound, a dihydric alcohol compound, or a dicarboxylic acid chloride compound can be dissolved in a solvent, and polymerization can be performed by adding a base such as triethylamine.
- the polymerization temperature is preferably in the range of ⁇ 10 ° C. to 40 ° C.
- the polymerization time is preferably in the range of 0.5 hour to 10 hours from the viewpoint of productivity.
- the base, solvent, terminator, and antioxidant used in the solution polymerization method are preferably under the same conditions as the polyester resin obtained by the solution polymerization.
- Examples of the base used in the solution polymerization method include triethylamine, tripropylamine, tributylamine, N, N-diisopropylethylamine, N, N-dipropylethylamine, N, N-diethylmethylamine, N, N-dimethylethylamine.
- Examples include tertiary amines such as octane, pyridines such as pyridine and 4-methylpyridine, and organic bases such as 1,8-diazabicyclo [5.4.0] -7-undecene.
- the base is not particularly limited as long as it is a base used for esterification reaction such as phosphazene base and inorganic base.
- a base used for esterification reaction such as phosphazene base and inorganic base.
- triethylamine, N, N-dipropylethylamine, N, N-diethylmethylamine, and pyridine are preferable from the viewpoint of the reactivity of the esterification reaction and availability, and the acid chloride decomposition is inhibited and washed.
- Triethylamine is particularly preferable from the viewpoint of ease of removal.
- the amount of base used is preferably in the range of 1.01 to 2 equivalents of the carboxylic acid chloride group contained in the reaction system.
- solvent examples include halogenated hydrocarbon compounds such as dichloromethane, chloroform, 1,2-dichloroethane, trichloroethane, tetrachloroethane, chlorobenzene and dichlorobenzene, aromatic hydrocarbon compounds such as toluene, anisole and xylene, cyclohexane, methylcyclohexane and the like.
- halogenated hydrocarbon compounds such as dichloromethane, chloroform, 1,2-dichloroethane, trichloroethane, tetrachloroethane, chlorobenzene and dichlorobenzene
- aromatic hydrocarbon compounds such as toluene, anisole and xylene, cyclohexane, methylcyclohexane and the like.
- Hydrocarbon compounds such as tetrahydrofuran, tetrahydropyran, 1,4-dioxane, 1,3-dioxolane, ester compounds such as ethyl acetate, methyl benzoate, benzyl acetate, N, N-dimethylformamide, N, N- Examples thereof include amide compounds such as dimethylacetamide. Pyridine may be used as a base and a solvent.
- dichloromethane chloroform, 1,2-dichloroethane, tetrahydrofuran, N, N-dimethylformamide and pyridine are preferred from the viewpoints of solubility of monomers and oligomers to be formed and reactivity of esterification reaction. Further, dichloromethane is particularly preferable from the viewpoint of washing efficiency and electrical characteristics.
- the ratio of dihydric phenol and / or dihydric alcohol to dicarboxylic acid chloride is not particularly limited, but one of them must be excessive for use in the production of polyester shown below. There is. Since the phenol end is higher in the air and in the solution than the carboxylic acid chloride end, the dihydric phenol and / or the dihydric alcohol is preferably used in excess of the dicarboxylic acid chloride. When the dihydric phenol and / or dihydric alcohol is in excess of the dicarboxylic acid chloride, the dihydric phenol and / or dihydric alcohol: dicarboxylic acid chloride ratio is 10: 1 to 1.1: in molar ratio. 1 is preferable.
- a dihydric alcohol for the production of an ester oligomer
- the hydroxyl reactivity thereof is low, and therefore it may not be used in the polyester production method shown below. Therefore, in order to introduce a divalent alcohol residue into the structure of the ester oligomer, it is necessary to copolymerize with a divalent phenol.
- a dihydric alcohol and a dicarboxylic acid chloride are reacted in advance, and then a divalent phenol is added and reacted to obtain a copolymer oligomer having a terminal hydroxyl group.
- the ratio of dihydric alcohol to dicarboxylic acid chloride is preferably 1:10 to 1: 1.1 in molar ratio, and the dicarboxylic acid chloride is preferably used in excess.
- ester oligomers when using a dihydric phenol having poor solubility in a solvent, it may precipitate when reacting with a divalent carboxylic acid chloride, resulting in clouding of the reaction solution or inhibition of oligomerization. . Therefore, when dihydric phenol having poor solubility is introduced into the ester oligomer, it is preferable to use a copolymer with divalent phenol and / or dihydric alcohol having good solubility.
- a dihydric phenol and / or dihydric alcohol and a divalent dicarboxylic acid chloride which have good solubility in advance, and then add and react a dihydric phenol having poor solubility.
- the poorly soluble monomer include rigid aromatic compounds such as hydroquinone, 4,4′-biphenol, and 4-hydroxyphenyl 4-hydroxybenzoate.
- a molecular weight regulator can be used for the production of the ester oligomer. It is also possible to use it at the time of polyester production shown below.
- the molecular weight regulator include phenol, o, m, p-cresol, o, m, p-ethylphenol, o, m, p-propylphenol, o, m, p- (tert-butyl) phenol, and pentyl.
- Alkylphenols such as phenol, hexylphenol, octylphenol, nonylphenol, 2,6-dimethylphenol derivatives, 2-methylphenol derivatives; monofunctional phenols such as o, m, p-phenylphenol; acetic acid chloride, butyric acid chloride, octyl Examples thereof include monofunctional acid halides such as acid chloride, benzoyl chloride, benzenesulfonyl chloride, benzenesulfinyl chloride, sulfinyl chloride, benzenephosphonyl chloride and substituted products thereof.
- monofunctional acid halides such as acid chloride, benzoyl chloride, benzenesulfonyl chloride, benzenesulfinyl chloride, sulfinyl chloride, benzenephosphonyl chloride and substituted products thereof.
- monofunctional aliphatic alcohols such as methanol, ethanol and propanol
- monofunctional alcohols having acrylics such as 2-hydroxyethyl acrylate, 4-hydroxybutyl acrylate and 2-hydroxy methacrylate, 1H, 1H, 2H, 2H-
- monofunctional alcohols having perfluoroalkyl such as tridecafluoro-1-n-octanol, 1H, 1H, 2H, 2H-heptadecafluoro-1-decanol, and monofunctional alcohols having siloxane.
- o, m, p- (tert-butyl) phenol, 2,6-dimethylphenol derivatives, and 2-methylphenol derivatives are preferred because of their high molecular weight controllability and solution stability. It is. Particularly preferred are p- (tert-butyl) phenol, 2,3,6-trimethylphenol, and 2,3,5-trimethylphenol.
- the viscosity average molecular weight (Mv) of the ester oligomer is usually 800 or more. Moreover, it is 20,000 or less normally, Preferably it is 15,000 or less. When the viscosity average molecular weight is less than 800, it may be dissolved in an alkaline aqueous solution at the time of producing a polyester by the interfacial polymerization described below and the ester bond may be decomposed. If it exceeds 20,000, it may be difficult to adjust the viscosity-average molecular weight as intended during the production of the polyester as shown below, and the production efficiency is reduced.
- an antioxidant can be added during the polymerization reaction or in the cleaning liquid.
- the antioxidant include sodium sulfite, hydrosulfite (sodium hyposulfite), sulfur dioxide, potassium sulfite, sodium hydrogen sulfite and the like.
- hydrosulfite is particularly preferable from the viewpoint of the antioxidant effect and reduction of environmental load.
- the amount of the antioxidant used is usually 0.01% by mass or more based on the total dihydric phenol, and preferably 0.1% by mass or more from the viewpoint of antioxidant. Usually, it is 10.0 mass% or less, and 5 mass% or less is preferable from a viewpoint of an electrical property.
- a solution of the ester oligomer is an alkaline aqueous solution such as sodium hydroxide or potassium hydroxide; hydrochloric acid, An aqueous solution of acid such as nitric acid and phosphoric acid; a method of separating by standing separation, centrifugation, etc. after washing with water or the like.
- the ester oligomer after washing is precipitated in water, alcohol or other organic solvent in which the ester oligomer is insoluble, or the solvent of the ester oligomer is distilled off in warm water or in a dispersion medium in which the polyester resin is insoluble, or heated and reduced in pressure.
- the solvent may be removed by distilling off the solvent, and when the slurry is taken out, the solid can be taken out by a centrifuge or a filter.
- the ester oligomer may not be taken out after washing, and may be used as it is for the production of the following polyester resin as a solution.
- the ester oligomer When the ester oligomer is taken out, it is usually dried at a temperature not higher than the decomposition temperature of the ester oligomer, but can be preferably dried at 20 ° C. or higher and not higher than the melting temperature of the ester oligomer. At this time, it is preferable to dry under reduced pressure. Preferably, the drying time is longer than the time until the purity of impurities such as the residual solvent becomes below a certain level. Specifically, the residual solvent is usually 1000 ppm or lower, preferably 300 ppm or lower, more preferably 100 ppm or lower. dry.
- the residual amount of the base is usually 50,000 ppm or less, preferably 10 1,000 ppm or less, more preferably 5,000 ppm or less.
- the carbonate oligomer or polycarbonate resin used in the present invention is produced by melt polymerization. Since the carbonate oligomer or polycarbonate produced by melt polymerization has many phenol ends, it can react with a carboxylic acid chloride during the production of the polyester shown below, and a copolymer can be easily obtained.
- the amount of OH groups present at the ends of the carbonate oligomer and the polycarbonate is usually 30 ⁇ eq / g or more, preferably 50 ⁇ eq / g or more. If the amount of terminal OH groups does not exceed the above range, there is a possibility that the copolymer will not be obtained due to insufficient reaction during the production of the following polyester resin.
- an alkaline aqueous solution and a halogenated hydrocarbon or aromatic hydrocarbon solution in which the ester oligomer and the aromatic dicarboxylic acid chloride compound are dissolved are mixed.
- a quaternary ammonium salt or a quaternary phosphonium salt can be present as a catalyst.
- the polymerization temperature is preferably in the range of 0 ° C. to 40 ° C., and the polymerization time is preferably in the range of 2 hours to 20 hours from the viewpoint of productivity.
- the water phase and the organic phase are separated, and the polymer dissolved in the organic phase is washed and recovered by a known method to obtain the intended resin.
- a dihydric phenol it is also possible to add a dihydric phenol separately to the alkaline aqueous solution.
- alkali component used in the interfacial polymerization method examples include alkali metal hydroxides such as sodium hydroxide and potassium hydroxide.
- the amount of the alkali component used is preferably in the range of 1.01 to 3 equivalents of the phenolic hydroxyl group contained in the reaction system.
- halogenated hydrocarbon examples include dichloromethane, chloroform, 1,2-dichloroethane, trichloroethane, tetrachloroethane, dichlorobenzene and the like.
- aromatic hydrocarbon examples include toluene, xylene, benzene and the like.
- Examples of the quaternary ammonium salt or quaternary phosphonium salt used as the catalyst include, for example, salts of tertiary alkylamines such as tributylamine and trioctylamine such as hydrochloric acid, bromic acid and iodic acid; benzyltriethylammonium chloride, benzyltrimethylammonium Examples include chloride, benzyltributylammonium chloride, tetraethylammonium chloride, tetrabutylammonium chloride, tetrabutylammonium bromide, trioctylmethylammonium chloride, tetrabutylphosphonium bromide, triethyloctadecylphosphonium bromide, N-laurylpyridinium chloride, laurylpicolinium chloride It is done.
- tertiary alkylamines such as tributylamine and trioctyl
- a molecular weight regulator can be used.
- the molecular weight regulator include phenol, o, m, p-cresol, o, m, p-ethylphenol, o, m, p-propylphenol, o, m, p- (tert-butyl) phenol, and pentyl.
- Alkylphenols such as phenol, hexylphenol, octylphenol, nonylphenol, 2,6-dimethylphenol derivatives, 2-methylphenol derivatives; monofunctional phenols such as o, m, p-phenylphenol; acetic acid chloride, butyric acid chloride, octyl Examples thereof include monofunctional acid halides such as acid chloride, benzoyl chloride, benzenesulfonyl chloride, benzenesulfinyl chloride, sulfinyl chloride, benzenephosphonyl chloride and substituted products thereof.
- monofunctional acid halides such as acid chloride, benzoyl chloride, benzenesulfonyl chloride, benzenesulfinyl chloride, sulfinyl chloride, benzenephosphonyl chloride and substituted products thereof.
- o, m, p- (tert-butyl) phenol, 2,6-dimethylphenol derivatives, and 2-methylphenol derivatives are preferred because of their high molecular weight controllability and solution stability. It is. Particularly preferred are p- (tert-butyl) phenol, 2,3,6-trimethylphenol, and 2,3,5-trimethylphenol.
- An antioxidant can be added in order not to oxidize the dihydric phenol in the alkaline solution.
- the antioxidant include sodium sulfite, hydrosulfite (sodium hyposulfite), sulfur dioxide, potassium sulfite, sodium hydrogen sulfite and the like.
- hydrosulfite is particularly preferable from the viewpoint of the antioxidant effect and reduction of environmental load.
- antioxidant 0.01 mass% or more and 10.0 mass% or less are preferable with respect to all the bivalent phenols. More preferably, it is 0.1 mass% or more and 5 mass% or less. If the content is too small, the antioxidant effect may be insufficient. If the content is too large, it may remain in the polyester and adversely affect electrical characteristics.
- the polyester resin solution is an alkaline aqueous solution such as sodium hydroxide or potassium hydroxide; hydrochloric acid, An aqueous solution of acid such as nitric acid and phosphoric acid; a method of separating by standing separation, centrifugation, etc. after washing with water or the like.
- Other purification methods include, for example, a method in which the produced polyester resin solution is precipitated in a solvent in which the polyester resin is insoluble, a method in which the polyester resin solution is dispersed in warm water, and the solvent is distilled off, or polyester You may refine
- the polyester resin after purification is precipitated in water, alcohol or other organic solvent in which the polyester resin is insoluble, or the solvent of the polyester resin is distilled off in warm water or in a dispersion medium in which the polyester resin is insoluble, or heating and decompression are performed.
- the solvent may be removed by distilling off the solvent, and when the slurry is taken out, the solid can be taken out by a centrifuge or a filter.
- the obtained polyester resin is usually dried at a temperature equal to or lower than the decomposition temperature of the polyester resin, but can be preferably dried at a temperature not lower than 20 ° C. and not higher than the melting temperature of the resin. At this time, it is preferable to dry under reduced pressure.
- the drying time is longer than the time until the purity of impurities such as the residual solvent becomes below a certain level.
- the residual solvent is usually 1000 ppm or lower, preferably 300 ppm or lower, more preferably 100 ppm or lower. dry.
- the manufacturing method of the polyester resin of 2nd Embodiment includes a step of reacting the dihydric phenol represented by the formula (4a 1 ) with the dicarboxylic acid chloride represented by the formula (5a) to obtain an ester oligomer, and the ester oligomer and the formula ( 4a 2 ) and the step of reacting the ester oligomer with the ester oligomer to obtain a polyester resin.
- the method for producing a polyester resin include a solution polymerization method, a polymerization method in which a solution polymerization method and an interfacial polymerization method are combined, or an interfacial polymerization method.
- a polyester for an electrophotographic photoreceptor When producing a polyester for an electrophotographic photoreceptor, it is generally produced by an interfacial polymerization method.
- a monomer that easily oxidizes in an alkaline aqueous solution such as hydroquinone, or a dihydric phenol that has an ester bond and is easily hydrolyzed is used as the monomer
- the interfacial polymerization method the monomer is alkaline in the aqueous solution.
- a solution polymerization method or a solution polymerization method A polymerization method combined with an interfacial polymerization method is preferred.
- the solution polymerization method is particularly preferable from the viewpoint of ease of production. Below, an example of the manufacturing method of the polyester resin by a solution polymerization method is demonstrated.
- a divalent phenol compound and a divalent carboxylic acid chloride compound that derive a divalent group represented by X 1a in the formula (1a) are dissolved in a solvent, and triethylamine or the like is dissolved.
- a divalent phenol compound for inducing a divalent group represented by X 2a in the formula (2a) is added to the same reaction vessel, and further a base is added. By adding it, a polyester resin can be obtained in one pot.
- the polymerization temperature is preferably in the range of ⁇ 10 ° C. to 40 ° C.
- the polymerization time is preferably in the range of 0.5 to 10 hours from the viewpoint of productivity.
- the target polyester resin is obtained by washing and collecting the polyester resin dissolved in the organic phase.
- Y 1a and Y 2a may be contained in the ester oligomer, or after the production of the ester oligomer containing Y 1a , the dicarboxylic acid chloride monomer corresponding to Y 2a and the above formula It may be contained in the polyester resin by adding a divalent phenol compound that induces a divalent group represented by X 2a in (2a).
- ⁇ Production method combining solution polymerization and interfacial polymerization> for example, a divalent phenol compound and a divalent carboxylic acid chloride compound that derive a divalent group represented by X 1a in the formula (1a) are used. After dissolving in a solvent and adding a base such as triethylamine to obtain an ester oligomer (solution polymerization), a divalent phenol compound for inducing a divalent group represented by X 2a in the formula (2a) is added to an aqueous alkaline solution. And a solution of halogenated hydrocarbon and aromatic hydrocarbon in which ester oligomer and dicarboxylic acid chloride compound are dissolved are mixed.
- a quaternary ammonium salt or a quaternary phosphonium salt can be present as a catalyst.
- the polymerization temperature is preferably in the range of 0 ° C. to 40 ° C., and the polymerization time is preferably in the range of 2 hours to 20 hours from the viewpoint of productivity.
- the divalent phenol compound for inducing the divalent group represented by X 1a in the formula (1a) is excellent in solubility, it is combined with a poorly soluble divalent phenol by passing through an ester oligomer composed thereof.
- a polyester resin having high solubility and excellent transparency is produced, and the obtained polyester resin can be applied to a coating film.
- dicarboxylic acid having poor solubility without passing through an ester oligomer and divalent phenol for inducing the divalent group represented by X 1a in the above (1a) are mixed with dicarboxylic acid chloride in the same reaction vessel from the beginning.
- divalent phenol and dicarboxylic acid chloride reactant having poor solubility are precipitated in the reaction solution, so that insoluble matter remains in the obtained polyester resin. Therefore, the polyester resin coating solution may become cloudy, and the resulting coating film may also become cloudy, or the coating film surface may become non-uniform.
- the amount of residual dicarboxylic acid chloride monomer in the ester oligomer satisfies the condition of the following formula (6a).
- the residual dicarboxylic acid chloride monomer represents the amount of dicarboxylic acid chloride monomer remaining in the ester oligomer.
- the charged dicarboxylic acid chloride monomer represents the amount of dicarboxylic acid chloride monomer initially charged in order to produce an ester oligomer.
- the said Formula (6a) is satisfy
- the amount of the residual dicarboxylic acid chloride monomer in the ester oligomer satisfies the condition of the above formula (6a), but from the viewpoint of the solubility of the polyester resin to be produced, the amount of the residual dicarboxylic acid chloride monomer is 0.001 in the above formula (6a). 18 or less is preferable, and 0.16 or less is more preferable. These values can be measured by the method described in Example [Measurement of residual acid chloride monomer in ester oligomer]. Moreover, the residual dicarboxylic acid chloride monomer amount in the ester oligomer can be set to a specific range, for example, by adjusting the amount of the base used to a range described later.
- the charge ratio of the divalent phenol compound and divalent carboxylic acid chloride that induces the divalent group represented by X 1a in (1a) is represented by the formula (4a 1 ). It is preferable that the charged molar ratio of the divalent phenol and the dicarboxylic acid chloride represented by the formula (5a) satisfy the following formula.
- the molar ratio of divalent carboxylic acid chloride: divalent phenol is 1.05: 1 to 1.8: 1. From the viewpoint of wear resistance when contained in the photoreceptor, 1.08 or more is more preferable, and 1.1 or more is still more preferable. From the viewpoint of the solubility of the resin, 1.6 or less is more preferable, and 1.55 or less is even more preferable.
- Examples of the base used in the solution polymerization method include triethylamine, tripropylamine, tributylamine, N, N-diisopropylethylamine, N, N-dipropylethylamine, N, N-diethylmethylamine, N, N-dimethylethylamine.
- tertiary amines such as octane, pyridines such as pyridine and 4-methylpyridine, and organic bases such as 1,8-diazabicyclo [5.4.0] -7-undecene.
- the base is not particularly limited as long as it is a base such as phosphazene base and inorganic base used for esterification reaction.
- a base such as phosphazene base and inorganic base used for esterification reaction.
- triethylamine, N, N-dipropylethylamine, N, N-diethylmethylamine, and pyridine are preferable from the viewpoint of the reactivity of the esterification reaction and availability, and the acid chloride decomposition is inhibited and washed.
- Triethylamine is particularly preferable from the viewpoint of ease of removal.
- the amount of the base used is usually 0.50 equivalents or more, preferably 0.60 equivalents or more, relative to the carboxylic acid chloride group used for producing the ester oligomer when the ester oligomer is produced. On the other hand, it is usually 0.95 times equivalent or less, preferably 0.90 times equivalent or less. In addition, in order to prevent unnecessary decomposition of the acid chloride, 1.1 equivalents or less is preferable with respect to the phenolic hydroxyl group used.
- the amount is usually 1.01 times equivalent or more with respect to the carboxylic acid chloride group used for the reaction, and more preferably 1.05 times equivalent or more in order to rapidly advance the polymerization reaction. Further, it is usually 2 times equivalent or less, and 1.8 times equivalent or less is preferable in order to prevent decomposition of the formed ester bond or to reduce the residual base.
- Examples of the solvent used in the solution polymerization method include halogenated hydrocarbon compounds such as dichloromethane, chloroform, 1,2-dichloroethane, trichloroethane, tetrachloroethane, chlorobenzene and dichlorobenzene, aromatic hydrocarbon compounds such as toluene, anisole and xylene, Hydrocarbon compounds such as cyclohexane and methylcyclohexane, ether compounds such as tetrahydrofuran, tetrahydropyran, 1,4-dioxane and 1,3-dioxolane, ester compounds such as ethyl acetate, methyl benzoate and benzyl acetate, N, N-dimethyl Examples thereof include amide compounds such as formamide and N, N-dimethylacetamide.
- halogenated hydrocarbon compounds such as dichloromethane, chloroform, 1,2-dichloroethane, trichloroe
- pyridine may be used as a base and a solvent.
- dichloromethane, chloroform, 1,2-dichloroethane, tetrahydrofuran, N, N-dimethylformamide, and pyridine are preferable from the viewpoints of solubility of monomers and oligomers to be formed and reactivity of esterification reaction. Further, dichloromethane is particularly preferred from the viewpoint of washing efficiency and electrical characteristics.
- the ratio of the total divalent phenol to the total divalent carboxylic acid chloride is determined from the viewpoint of the production of the high molecular weight polyester resin and the control of the end groups.
- the carboxylic acid chloride is preferably in a molar ratio of 1: 0.95 to 1: 1.05, more preferably 1: 0.99 to 1: 1.01, and particularly preferably 1: 0.992 to 1: 1.008. is there.
- a molecular weight regulator When producing a polyester resin, a molecular weight regulator can be used.
- the molecular weight regulator include phenol, o, m, p-cresol, o, m, p-ethylphenol, o, m, p-propylphenol, o, m, p- (tert-butyl) phenol, and pentyl.
- Alkylphenols such as phenol, hexylphenol, octylphenol, nonylphenol, 2,6-dimethylphenol derivatives, 2-methylphenol derivatives; monofunctional phenols such as o, m, p-phenylphenol; acetic acid chloride, butyric acid chloride, octyl Examples thereof include monofunctional acid halides such as acid chloride, benzoyl chloride, benzenesulfonyl chloride, benzenesulfinyl chloride, sulfinyl chloride, benzenephosphonyl chloride and substituted products thereof.
- monofunctional acid halides such as acid chloride, benzoyl chloride, benzenesulfonyl chloride, benzenesulfinyl chloride, sulfinyl chloride, benzenephosphonyl chloride and substituted products thereof.
- monofunctional aliphatic alcohols such as methanol, ethanol, and propanol
- monofunctional alcohols having acrylics such as 2-hydroxyethyl acrylate, 4-hydroxybutyl acrylate, and 2-hydroxy methacrylate, 1H, 1H, 2H, and 2H-
- monofunctional alcohols having perfluoroalkyl such as tridecafluoro-1-n-octanol, 1H, 1H, 2H, 2H-heptadecafluoro-1-decanol, and monofunctional alcohols having siloxane.
- o, m, p- (tert-butyl) phenol, 2,6-dimethylphenol derivatives, 2-methylphenol are preferred because of their high molecular weight controllability and solution stability.
- Particularly preferred are p- (tert-butyl) phenol, 2,3,6-trimethylphenol, and 2,3,5-trimethylphenol.
- an antioxidant can be added during the polymerization reaction or in the cleaning liquid.
- the antioxidant include sodium sulfite, hydrosulfite (sodium hyposulfite), sulfur dioxide, potassium sulfite, sodium hydrogen sulfite and the like.
- hydrosulfite is particularly preferable from the viewpoint of the antioxidant effect and reduction of environmental load.
- As the usage-amount of antioxidant 0.01 mass% or more and 10.0 mass% or less are preferable with respect to all the bivalent phenols. More preferably, it is 0.1 mass% or more and 5 mass% or less.
- the method for washing after polymerization of the polyester resin is, for example, washing the polyester resin solution with an alkali aqueous solution such as sodium hydroxide or potassium hydroxide; an acid aqueous solution such as hydrochloric acid, nitric acid or phosphoric acid; And a method of liquid separation by centrifugation or the like.
- an alkali aqueous solution such as sodium hydroxide or potassium hydroxide
- an acid aqueous solution such as hydrochloric acid, nitric acid or phosphoric acid
- a method of liquid separation by centrifugation or the like is, for example, washing the polyester resin solution with an alkali aqueous solution such as sodium hydroxide or potassium hydroxide; an acid aqueous solution such as hydrochloric acid, nitric acid or phosphoric acid; And a method of liquid separation by centrifugation or the like.
- the polyester resin solution after washing is precipitated in water, alcohol or other organic solvent in which the polyester resin is insoluble, or the polyester resin solution is distilled off in warm water or in a dispersion medium in which the polyester resin is insoluble, or heated, It may be taken out by distilling off the solvent under reduced pressure or the like, and when taken out in the form of a slurry, the solid can be taken out by a centrifugal separator or a filter.
- the polyester resin is usually dried at a temperature equal to or lower than the decomposition temperature of the polyester resin, preferably 20 ° C. or higher and lower than the melting temperature of the polyester resin. At this time, it is preferable to dry under reduced pressure. It is preferable to perform the drying time for at least the time until the purity of impurities such as the residual solvent becomes a certain level or less. Specifically, the drying time is usually 1000 ppm or less, preferably 300 ppm or less, more preferably 100 ppm or less. dry.
- the amount of residual dicarboxylic acid chloride monomer in the ester oligomer only needs to satisfy the condition of the above formula (6a), and a polymerization method or an interfacial polymerization method combining the solution polymerization method and the interfacial polymerization method.
- a polymerization method or an interfacial polymerization method combining the solution polymerization method and the interfacial polymerization method.
- the electrophotographic photosensitive member to which this embodiment is applied has a photosensitive layer provided on a conductive support, and the photosensitive layer contains the polyester resin.
- the photosensitive layer for example, a laminate in which a charge generation layer mainly composed of a charge generation material and a charge transport layer mainly composed of a charge transport material and a binder resin are stacked on a conductive support.
- the polyester resin is usually used for a layer containing a charge transport material, and preferably used for a charge transport layer of a multilayer photoreceptor.
- the photosensitive layer used in the electrophotographic photosensitive member to which the exemplary embodiment is applied for example, in the case of a laminated type photosensitive member, it contains a charge transport material and a binder resin, and maintains an electrostatic charge.
- a charge transfer material and a charge generation material are dispersed in a binder resin in the photosensitive layer.
- ⁇ Conductive support> There is no particular limitation on the conductive support, but for example, metal materials such as aluminum, aluminum alloy, stainless steel, copper, and nickel, and conductive powders such as metal, carbon, and tin oxide are added to impart conductivity.
- metal materials such as aluminum, aluminum alloy, stainless steel, copper, and nickel
- conductive powders such as metal, carbon, and tin oxide are added to impart conductivity.
- a resin material, a resin, glass, paper, or the like on which a conductive material such as aluminum, nickel, or ITO (indium tin oxide) is deposited or applied on the surface is mainly used.
- a form of the conductive support a drum form, a sheet form, a belt form or the like is used. Furthermore, a conductive material having an appropriate resistance value may be used on a conductive support made of a metal material in order to control conductivity and surface properties and to cover defects.
- a metal material such as an aluminum alloy
- it may be used after an anodized film is applied.
- an anodized film it is preferable to perform a sealing treatment by a known method.
- the support surface may be smooth, or may be roughened by using a special cutting method or performing a polishing treatment. Further, it may be roughened by mixing particles having an appropriate particle diameter with the material constituting the support. In order to reduce the cost, it is possible to use the drawing tube as it is without performing the cutting process.
- An undercoat layer may be provided between the conductive support and the photosensitive layer described later for improving adhesion and blocking properties.
- As the undercoat layer a resin and a resin in which particles such as a metal oxide are dispersed are used.
- the undercoat layer may be a single layer or a plurality of layers.
- the undercoat layer may contain a known antioxidant, pigment particles, resin particles, or the like.
- the film thickness is usually 0.01 ⁇ m or more, preferably 0.1 ⁇ m or more, from the viewpoint of improving the electrical characteristics, strong exposure characteristics, image characteristics, repeat characteristics, and coating properties during production of the electrophotographic photoreceptor. Usually, it is 30 ⁇ m or less, preferably 20 ⁇ m or less.
- metal oxide particles used for the undercoat layer include metal oxide particles containing one metal element such as titanium oxide, aluminum oxide, silicon oxide, zirconium oxide, zinc oxide, iron oxide, calcium titanate, titanium Examples thereof include metal oxide particles containing a plurality of metal elements such as strontium acid and barium titanate. One kind of these particles may be used alone, or a plurality of kinds of particles may be mixed and used.
- titanium oxide and aluminum oxide are preferable, and titanium oxide is particularly preferable.
- the surface of the titanium oxide particles may be treated with an inorganic substance such as tin oxide, aluminum oxide, antimony oxide, zirconium oxide, or silicon oxide, or an organic substance such as stearic acid, polyol, or silicon.
- an inorganic substance such as tin oxide, aluminum oxide, antimony oxide, zirconium oxide, or silicon oxide, or an organic substance such as stearic acid, polyol, or silicon.
- any of rutile, anatase, brookite, and amorphous can be used.
- the thing of the several crystal state may be contained.
- the average primary particle size is preferably 10 nm or more and 100 nm or less, and particularly 10 nm or more and 50 nm or less. preferable. This average primary particle size can be obtained from a TEM photograph or the like.
- the undercoat layer is preferably formed in a form in which metal oxide particles are dispersed in a binder resin.
- the binder resin used for the undercoat layer is epoxy resin, polyethylene resin, polypropylene resin, acrylic resin, methacrylic resin, polyamide resin, vinyl chloride resin, vinyl acetate resin, phenol resin, polycarbonate resin, polyurethane resin, polyimide resin, chloride resin.
- the organic zirconium compound alkoxide compounds, titanyl chelate compounds, organic titanyl compounds such as titanium alkoxide compounds include known binder resins such as a silane coupling agent.
- the binder resin is usually 10% by mass or more, It is preferable to use in the range of 500 mass% or less.
- Photosensitive layer As a type of the photosensitive layer, a charge generation material and a charge transport material are present in the same layer and are dispersed in a binder resin, a charge generation layer in which a charge generation material is dispersed in a binder resin, and a charge.
- a functional separation type (laminated type) composed of two layers of a charge transport layer in which a transport material is dispersed in a binder resin can be mentioned, but any type may be used.
- a charge generation layer and a charge transport layer are laminated in this order from the conductive support side, and a reverse lamination layer in which a charge transport layer and a charge generation layer are laminated in reverse order. Any one of them can be used, but a sequentially laminated photosensitive layer that can exhibit the most balanced photoconductivity is preferable.
- the charge generation layer is formed by binding a charge generation material with a binder resin.
- the film thickness is usually 0.1 ⁇ m or more, preferably 0.15 ⁇ m or more, and usually 10 ⁇ m or less, preferably 0.6 ⁇ m or less.
- Examples of the charge generation material include inorganic photoconductive materials such as selenium and its alloys, cadmium sulfide, and organic photoconductive materials such as organic pigments, but organic photoconductive materials are preferred, especially organic pigments. Is preferred.
- organic pigments include phthalocyanine pigments, azo pigments, dithioketopyrrolopyrrole pigments, squalene (squarylium) pigments, quinacridone pigments, indigo pigments, perylene pigments, polycyclic quinone pigments, anthanthrone pigments, and benzimidazole pigments. .
- phthalocyanine pigments or azo pigments are particularly preferable.
- organic pigments are used as the charge generating substance, usually, fine particles of these organic pigments are used in the form of a dispersion layer bound with various binder resins.
- a photosensitive member having a high sensitivity to a laser beam having a relatively long wavelength for example, a laser beam having a wavelength around 780 nm
- a photosensitive member having a high sensitivity to a laser beam having a relatively long wavelength for example, a laser beam having a wavelength around 780 nm
- an azo pigment such as diazo or trisazo
- a photosensitive member having a high sensitivity can be obtained.
- a phthalocyanine pigment or an azo pigment is particularly preferable.
- the phthalocyanine pigment provides a photosensitive material with high sensitivity to a laser beam having a relatively long wavelength, and the azo pigment has a sufficient sensitivity to white light and a laser beam having a relatively short wavelength. , Each is excellent.
- a phthalocyanine pigment as a charge generating material, specifically, metal-free phthalocyanine, copper, indium, gallium, tin, titanium, zinc, vanadium, silicon, germanium, aluminum or other metal or oxide thereof, halide, water Those having crystal forms of coordinated phthalocyanines such as oxides and alkoxides, and phthalocyanine dimers using oxygen atoms as bridging atoms are used.
- titanyl phthalocyanines also known as oxytitanium
- A-type also known as ⁇ -type
- B-type also known as ⁇ -type
- D-type also known as Y-type
- vanadyl phthalocyanine vanadyl phthalocyanine
- chloroindium phthalocyanine hydroxyindium phthalocyanine
- chlorogallium phthalocyanine such as type II
- hydroxygallium phthalocyanine such as type V
- ⁇ -oxo-gallium phthalocyanine dimer such as type G and type I, type II, etc.
- the ⁇ -oxo-aluminum phthalocyanine dimer is preferred.
- A-type (also known as ⁇ -type), B-type (also known as ⁇ -type), and powder X-ray diffraction angle 2 ⁇ ( ⁇ 0.2 °) are 27.1 ° or 27.3 °.
- D-type (Y-type) titanyl phthalocyanine, II-type chlorogallium phthalocyanine, V-type and 28.1 ° have the strongest peaks, and 26.2 ° have peaks Hydroxygallium phthalocyanine having a clear peak at 28.1 ° and a half width W of 25.9 ° of 0.1 ° ⁇ W ⁇ 0.4 °, G-type ⁇ -oxo -Gallium phthalocyanine dimer and the like are particularly preferred.
- D-type (Y-type) titanyl phthalocyanine is preferable because it exhibits good sensitivity.
- the phthalocyanine compound a single compound may be used, or several mixed or mixed crystals may be used.
- the mixed state that can be put in the phthalocyanine compound or crystal state here those obtained by mixing the respective constituent elements later may be used, or they may be mixed in the production / treatment process of the phthalocyanine compound such as synthesis, pigmentation, and crystallization. It may be the one that caused the condition.
- acid paste treatment, grinding treatment, solvent treatment and the like are known.
- two types of crystals are mixed, mechanically ground and made amorphous, and then a specific crystal state is obtained by solvent treatment. The method of converting into is mentioned.
- the binder resin used for the charge generation layer is not particularly limited, but examples include polyvinyl butyral resin, polyvinyl formal resin, polyvinyl acetal type such as partially acetalized polyvinyl butyral resin in which a part of butyral is modified with formal, acetal, or the like.
- the mixing ratio (mass) of the binder resin and the charge generation material is usually 10 parts by mass or more, preferably 30 parts by mass or more, and usually 1000 parts by mass with respect to 100 parts by mass of the binder resin. Part or less, preferably 500 parts by weight or less.
- the charge transport layer of the multilayer photoreceptor contains a charge transport material and usually contains a binder resin and other components used as necessary.
- the charge transport layer may be composed of a single layer, or may be a stack of a plurality of layers having different constituent components or composition ratios.
- the film thickness is usually 5 ⁇ m to 50 ⁇ m, preferably 10 ⁇ m to 45 ⁇ m.
- the charge transport material is not particularly limited, and any material can be used.
- charge transport materials include aromatic nitro compounds such as 2,4,7-trinitrofluorenone, cyano compounds such as tetracyanoquinodimethane, electron withdrawing materials such as quinone compounds such as diphenoquinone, carbazole derivatives, indoles Derivatives, imidazole derivatives, oxazole derivatives, pyrazole derivatives, thiadiazole derivatives, heterocyclic compounds such as benzofuran derivatives, aniline derivatives, hydrazone derivatives, aromatic amine derivatives, stilbene derivatives, butadiene derivatives, enamine derivatives and multiple types of these compounds bind Or an electron donating substance such as a polymer having a group consisting of these compounds in the main chain or side chain.
- carbazole derivatives aromatic amine derivatives, stilbene derivatives, butadiene derivatives, enamine derivatives, and those in which a plurality of these compounds are bonded are preferable.
- charge transport materials may be used alone or in combination. Specific examples of suitable structures of the charge transport material are shown below.
- the polyester resin is preferably used as a binder resin for a charge transport layer. It is also possible to use a mixture of the polyester resin and another resin.
- the resin having another structure mixed here include vinyl polymers such as polymethyl methacrylate, polystyrene, and polyvinyl chloride and copolymers thereof; polycarbonate resin, polyester resin, polyester polycarbonate resin, polysulfone resin, phenoxy
- thermoplastic resins such as resins, epoxy resins, and silicone resins, various thermosetting resins, and copolymers thereof.
- the mixing ratio of the resin having another structure to be mixed is not particularly limited, but usually it is preferably used in combination within the range not exceeding the ratio of the polyester resin, specifically, the other structure relative to the polyester resin.
- the content of the resin having a content of 50 parts by mass or less is preferably 30 parts by mass or less from the viewpoint of wear resistance.
- the ratio of the polyester resin to the charge transport material is 30 parts by mass or more of the charge transport material with respect to 100 parts by mass of the polyester resin. From the viewpoint of electrical characteristics, it is preferably 40 parts by mass or more. From the viewpoint of wear resistance, it is 200 parts by mass or less, preferably 150 parts by mass or less.
- the charge transport material is usually used at a ratio of 10 parts by mass or more with respect to 100 parts by mass of the binder resin in the same layer. Among them, 20 parts by mass or more is preferable from the viewpoint of reducing the residual potential, 30 parts by mass or more is more preferable, and 40 parts by mass or more is more preferable from the viewpoint of stability and charge mobility when repeatedly used.
- the charge transport material is usually used at a ratio of 150 parts by mass or less and 120 parts by mass or less from the viewpoint of the thermal stability of the photosensitive layer.
- 110 parts by mass or less is preferable, 100 parts by mass or less is more preferable, 80 parts by mass or less is further preferable from the viewpoint of wear resistance, and from the viewpoint of scratch resistance. 70 parts by mass or less is particularly preferable.
- the charge transport layer has well-known plasticizers, antioxidants, ultraviolet absorbers, and electron withdrawing properties in order to improve film forming properties, flexibility, coating properties, stain resistance, gas resistance, light resistance, etc.
- You may contain additives, such as a compound, dye, a pigment, and a leveling agent.
- the antioxidant include hindered phenol compounds and hindered amine compounds.
- dyes and pigments include various pigment compounds and azo compounds.
- the single-layer type photosensitive layer is formed using a binder resin in order to ensure film strength, in the same manner as the charge transport layer of the multilayer photoreceptor, in addition to the charge generation material and the charge transport material.
- a charge generation material, a charge transport material, and various binder resins are dissolved or dispersed in a solvent to prepare a coating solution, and on a conductive support (or an undercoat layer when an undercoat layer is provided). It can be obtained by coating and drying.
- the types of the charge transport material and the binder resin and the use ratios thereof are the same as those described for the charge transport layer of the multilayer photoreceptor.
- the charge generating material is further dispersed in the charge transport medium comprising these charge transport material and binder resin.
- the charge generation material the same materials as those described for the charge generation layer of the multilayer photoreceptor can be used. However, in the case of a photosensitive layer of a single-layer type photoreceptor, it is necessary to sufficiently reduce the particle size of the charge generating material. Specifically, the range is usually 1 ⁇ m or less, preferably 0.5 ⁇ m or less.
- the amount of the charge generating material dispersed in the single-layer type photosensitive layer is too small, sufficient sensitivity cannot be obtained, but if it is too large, there are adverse effects such as a decrease in chargeability and a decrease in sensitivity. It is used in the range of usually 0.5% by mass or more, preferably 1% by mass or more, and usually 50% by mass or less, preferably 20% by mass or less based on the whole layer-type photosensitive layer.
- the usage ratio of the binder resin and the charge generation material in the single-layer type photosensitive layer is such that the charge generation material is usually 0.1 parts by weight or more, preferably 1 part by weight or more, based on 100 parts by weight of the binder resin. 30 parts by mass or less, preferably 10 parts by mass or less.
- the film thickness of the single-layer type photosensitive layer is usually 5 ⁇ m or more, preferably 10 ⁇ m or more, and usually 100 ⁇ m or less, preferably 50 ⁇ m or less.
- a known plasticizer for improving film formability, flexibility, mechanical strength, an additive for suppressing residual potential, a dispersion aid for improving dispersion stability, and coating properties are provided.
- Leveling agents and surfactants for improvement, such as silicone oil, fluorine oil and other additives may be added.
- ⁇ Other functional layers> For the purpose of improving film-forming properties, flexibility, coating properties, stain resistance, gas resistance, light resistance, etc., in both the photosensitive layer and each layer constituting it, both in the multilayer type photosensitive member and the single layer type photosensitive member. Additives such as well-known antioxidants, plasticizers, ultraviolet absorbers, electron-withdrawing compounds, leveling agents, and visible light shielding agents may be contained.
- the surface layer is made of fluorine resin, silicon resin, polyethylene resin, etc. Particles made of the above resin or inorganic compound particles may be contained in the surface layer. Alternatively, a layer containing these resins and particles may be newly formed as a surface layer. Further, if necessary, it may have a layer for improving electrical properties and mechanical properties, such as an intermediate layer such as a barrier layer, an adhesive layer and a blocking layer, and a transparent insulating layer.
- Each layer constituting these photoreceptors is formed by immersing, coating, spraying, nozzle coating, bar coating, roll coating, blade coating, etc., on a support, a coating solution obtained by dissolving or dispersing a substance to be contained in a solvent. In this method, the coating and drying steps are sequentially repeated for each layer.
- solvent or dispersion medium used for the preparation of the coating solution, but specific examples include alcohols such as methanol, ethanol, propanol and 2-methoxyethanol, tetrahydrofuran, 1,4-dioxane, dimethoxyethane and the like.
- esters such as methyl formate and ethyl acetate, ketones such as acetone, methyl ethyl ketone and cyclohexanone, aromatic hydrocarbons such as benzene, toluene and xylene, dichloromethane, chloroform, 1,2-dichloroethane, 1,1, Chlorinated hydrocarbons such as 2-trichloroethane, 1,1,1-trichloroethane, tetrachloroethane, 1,2-dichloropropane, trichloroethylene, n-butylamine, isopropanolamine, diethylamine, triethanolamine, ethylenedia Emissions, nitrogen-containing compounds such as triethylenediamine, acetonitrile, N- methylpyrrolidone, N, N- dimethylformamide, aprotic polar solvents such as dimethyl sulfoxide and the like.
- aromatic hydrocarbons such as benzene,
- non-halogen solvents are preferable from the viewpoint of environmental considerations, and toluene, xylene, anisole, dimethoxyethane, tetrahydrofuran, and 1,4-dioxane are particularly preferable from the viewpoint of solubility. Any of these may be used alone or in combination of two or more.
- the amount of the solvent or dispersion medium used is not particularly limited, but considering the purpose of each layer and the properties of the selected solvent / dispersion medium, it is appropriate so that the physical properties such as solid content concentration and viscosity of the coating liquid are within a desired range It is preferable to adjust.
- the solid content concentration of the coating solution is usually 5% by mass or more, preferably 10% by mass or more, and usually 40% by mass.
- the range is preferably 35% by mass or less.
- the viscosity of the coating solution is usually in the range of 10 cps or more, preferably 50 cps or more, and usually 500 cps or less, preferably 400 cps or less.
- the solid content concentration of the coating solution is usually 0.1% by mass or more, preferably 1% by mass or more, and usually 15% by mass or less, preferably 10% by mass. % Or less.
- the viscosity of the coating solution is usually 0.01 cps or more, preferably 0.1 cps or more, and usually 20 cps or less, preferably 10 cps or less.
- Examples of the coating method include a dip coating method, a spray coating method, a spinner coating method, a bead coating method, a wire bar coating method, a blade coating method, a roller coating method, an air knife coating method, and a curtain coating method. Other known coating methods can also be used.
- the drying of the coating solution is preferably performed by drying at the room temperature, and then drying by heating in a temperature range of usually 30 ° C. or more and 200 ° C. or less for 1 minute to 2 hours while still or blowing.
- the heating temperature may be constant or the heating may be performed while changing the temperature during drying.
- the image forming apparatus includes an electrophotographic photosensitive member 1, a charging device 2, an exposure device 3, and a developing device 4, and further includes a transfer device 5, a cleaning device 6, and a fixing device as necessary.
- a device 7 is provided.
- the electrophotographic photoreceptor 1 is not particularly limited as long as it is the above-described electrophotographic photoreceptor of the present invention, but in FIG. 1, as an example, a drum in which the above-described photosensitive layer is formed on the surface of a cylindrical conductive support.
- the photoconductor is shown.
- a charging device 2, an exposure device 3, a developing device 4, a transfer device 5, and a cleaning device 6 are disposed along the outer peripheral surface of the electrophotographic photosensitive member 1.
- the charging device 2 charges the electrophotographic photoreceptor 1 and uniformly charges the surface of the electrophotographic photoreceptor 1 to a predetermined potential.
- the charging device include a corona charging device such as a corotron and a scorotron, a direct charging device (contact type charging device) that charges a direct charging member to which a voltage is applied by contacting the photosensitive member surface, and a contact type charging device such as a charging brush. Often used.
- Examples of direct charging means include contact chargers such as charging rollers and charging brushes.
- a roller-type charging device charging roller
- FIG. 1 a roller-type charging device (charging roller) is shown as an example of the charging device 2.
- the direct charging means either charging with air discharge or injection charging without air discharge is possible.
- a voltage applied at the time of charging it is possible to use only a direct current voltage or to superimpose an alternating current on a direct current.
- the type of exposure apparatus 3 is not particularly limited as long as it can expose the electrophotographic photoreceptor 1 to form an electrostatic latent image on the photosensitive surface of the electrophotographic photoreceptor 1.
- Specific examples include halogen lamps, fluorescent lamps, lasers such as semiconductor lasers and He—Ne lasers, LEDs, and the like. Further, exposure may be performed by a photoreceptor internal exposure method.
- the light used for the exposure is arbitrary.
- the exposure may be performed using monochromatic light having a wavelength of 780 nm, monochromatic light having a wavelength of 600 nm to 700 nm, slightly short wavelength, or monochromatic light having a wavelength of 380 nm to 500 nm. .
- monochromatic light having a wavelength of 780 nm monochromatic light having a wavelength of 780 nm
- monochromatic light having a wavelength of 600 nm to 700 nm slightly short wavelength
- monochromatic light having a wavelength of 380 nm to 500 nm it is preferable to use light having a short wavelength of 380 to 500 nm because the resolution is increased.
- 405 nm monochromatic light is preferable.
- the type of the developing device 4 is not particularly limited, and an arbitrary device such as a dry development method such as cascade development, one-component insulating toner development, one-component conductive toner development, or two-component magnetic brush development, or a wet development method is used. be able to.
- the developing device 4 includes a developing tank 41, an agitator 42, a supply roller 43, a developing roller 44, and a regulating member 45, and has a configuration in which toner T is stored inside the developing tank 41. .
- a replenishing device (not shown) for replenishing toner T may be attached to the developing device 4.
- the replenishing device is configured to be able to replenish toner T from a container such as a bottle or a cartridge.
- the type of the transfer device 5 is not particularly limited, and an apparatus using an arbitrary system such as an electrostatic transfer method such as corona transfer, roller transfer, or belt transfer, a pressure transfer method, or an adhesive transfer method can be used.
- the transfer device 5 includes a transfer charger, a transfer roller, a transfer belt, and the like that are disposed to face the electrophotographic photoreceptor 1.
- the transfer device 5 applies a predetermined voltage value (transfer voltage) having a polarity opposite to the charging potential of the toner T, and transfers the toner image formed on the electrophotographic photosensitive member 1 to a recording paper (paper, medium) P. Is.
- the cleaning device 6 is not particularly limited, and any cleaning device such as a brush cleaner, a magnetic brush cleaner, an electrostatic brush cleaner, a magnetic roller cleaner, or a blade cleaner can be used.
- the cleaning device 6 scrapes the residual toner adhering to the electrophotographic photosensitive member 1 with a cleaning member and collects the residual toner. However, when there is little or almost no toner remaining on the surface of the photoreceptor, the cleaning device 6 may be omitted.
- the toner transferred onto the recording paper P passes between the upper fixing member 71 and the lower fixing member 72 heated to a predetermined temperature, the toner is heated to a molten state and cooled after passing through the recording paper. Toner is fixed on P.
- an image is recorded as follows. That is, first, the surface (photosensitive surface) of the electrophotographic photosensitive member 1 is charged to a predetermined potential (for example, ⁇ 600 V) by the charging device 2. At this time, charging may be performed with a DC voltage, or charging may be performed by superimposing an AC voltage on the DC voltage.
- a predetermined potential for example, ⁇ 600 V
- the photosensitive surface of the charged electrophotographic photosensitive member 1 is exposed by the exposure device 3 according to the image to be recorded, and an electrostatic latent image is formed on the photosensitive surface. Then, development of the electrostatic latent image formed on the photosensitive surface of the electrophotographic photoreceptor 1 is performed by the developing device 4.
- the developing device 4 thins the toner T supplied by the supply roller 43 with a regulating member (developing blade) 45 and has a predetermined polarity (here, the same polarity as the charging potential of the electrophotographic photosensitive member 1). Negatively charged) and conveyed while being carried on the developing roller 44 and brought into contact with the surface of the electrophotographic photosensitive member 1.
- the final image can be obtained by passing the fixing device 7 and thermally fixing the toner image onto the recording paper P.
- the image forming apparatus may be configured to perform, for example, a static elimination process in addition to the above-described configuration.
- the neutralization step is a step of neutralizing the electrophotographic photosensitive member by exposing the electrophotographic photosensitive member, and a fluorescent lamp, an LED, or the like is used as the neutralizing device.
- the light used in the static elimination process is often light having an exposure energy that is at least three times that of the exposure light.
- the image forming apparatus may be further modified.
- the image forming apparatus may be configured to perform a pre-exposure process, an auxiliary charging process, or the like, or may be configured to perform offset printing.
- a full-color tandem system configuration using toner may be used.
- the electrophotographic photosensitive member 1 is combined with one or more of the charging device 2, the exposure device 3, the developing device 4, the transfer device 5, the cleaning device 6, and the fixing device 7 to form an integrated cartridge (
- the electrophotographic photosensitive member cartridge may be configured to be detachable from a main body of an electrophotographic apparatus such as a copying machine or a laser beam printer.
- the electrophotographic photosensitive member cartridge is removed from the main body of the image forming apparatus, and another new electrophotographic photosensitive member cartridge is mounted on the main body of the image forming apparatus. This facilitates maintenance and management of the image forming apparatus.
- Example 1-1 (Production of Polyester Resin (1)) Sodium hydroxide (1.57 g) and H 2 O (188 ml) were weighed in a 500 mL beaker and dissolved with stirring. 2,3,5-Trimethylphenol (0.21 g) and benzyltriethylammonium chloride (0.11 g) were added thereto and dissolved by stirring, and the aqueous alkaline solution was transferred to a 1 L reaction vessel. Separately, a mixed solution of the ester oligomer (1) synthesized in Production Example 1-1 (16.13 g) and diphenyl ether-4,4′-dicarboxylic acid chloride (4.87 g) and dichloromethane (107 mL) was placed in the dropping funnel.
- Example 1-2 (Production of polyester resin (2)) Sodium hydroxide (1.55 g) and H 2 O (188 ml) were weighed in a 500 mL beaker and dissolved with stirring. 2,3,5-Trimethylphenol (0.21 g) and benzyltriethylammonium chloride (0.11 g) were added thereto and dissolved by stirring, and the aqueous alkaline solution was transferred to a 1 L reaction vessel. Separately, a mixed solution of the ester oligomer (2) synthesized in Production Example 1-2 (16.18 g) and diphenyl ether-4,4′-dicarboxylic acid chloride (4.81 g) and dichloromethane (107 mL) was placed in the dropping funnel.
- Example 1-3 (Production of polyester resin (3)) Sodium hydroxide (1.87 g) and H 2 O (235 ml) were weighed in a 500 mL beaker and dissolved with stirring. 2,3,5-trimethylphenol (0.32 g) and benzyltriethylammonium chloride (0.14 g) were added thereto and dissolved by stirring, and then the alkaline aqueous solution was transferred to a 1 L reaction vessel. Separately, a mixed solution of the ester oligomer (3) (20.33 g) synthesized in Production Example 1-3 and diphenyl ether-4,4′-dicarboxylic acid chloride (5.78 g) and dichloromethane (134 mL) was placed in the dropping funnel.
- the resulting polyester resin had a viscosity average molecular weight (Mv) of 43,500.
- Mv viscosity average molecular weight
- Example 1-4 (Production of polyester resin (4)) Sodium hydroxide (1.52 g) and H 2 O (188 ml) were weighed in a 500 mL beaker and dissolved with stirring. 2,3,5-trimethylphenol (0.24 g) and benzyltriethylammonium chloride (0.11 g) were added thereto and dissolved by stirring, and then the alkaline aqueous solution was transferred to a 1 L reaction vessel.
- Example 1-5 (Production of polyester resin (5)) Sodium hydroxide (1.47 g) and H 2 O (188 ml) were weighed in a 500 mL beaker and dissolved with stirring. 2,3,5-trimethylphenol (0.25 g) and benzyltriethylammonium chloride (0.11 g) were added thereto and dissolved by stirring, and then the alkaline aqueous solution was transferred to a 1 L reaction vessel.
- Example 1-6 (Production of polyester resin (6)) Sodium hydroxide (1.78 g) and H 2 O (188 ml) were weighed in a 500 mL beaker and dissolved with stirring. 2,3,5-trimethylphenol (0.26 g) and benzyltriethylammonium chloride (0.08 g) were added thereto and dissolved by stirring, and then the alkaline aqueous solution was transferred to a 1 L reaction vessel.
- Example 1-7 (Production of polyester resin (7)) Sodium hydroxide (2.31 g) and H 2 O (188 ml) were weighed in a 500 mL beaker and dissolved with stirring. 2,3,5-Trimethylphenol (0.25 g) and benzyltriethylammonium chloride (0.17 g) were added thereto and dissolved by stirring, and then the alkaline aqueous solution was transferred to a 1 L reaction vessel.
- Example 1-8 (Production of polyester resin (8)) Sodium hydroxide (2.26 g) and H 2 O (188 ml) were weighed in a 500 mL beaker and dissolved with stirring. 2,3,5-Trimethylphenol (0.25 g) and benzyltriethylammonium chloride (0.17 g) were added thereto and dissolved by stirring, and then the alkaline aqueous solution was transferred to a 1 L reaction vessel.
- Example 1-9 (Production of polyester resin (9)) Sodium hydroxide (2.72 g) and H 2 O (188 ml) were weighed in a 500 mL beaker and dissolved with stirring. BP-a (4.64 g), 2,3,5-trimethylphenol (0.26 g), and benzyltriethylammonium chloride (0.20 g) were added thereto and dissolved by stirring. Moved to the tank. Separately, a mixed solution of the ester oligomer (9) (8.76 g) synthesized in Production Example 1-9 and diphenyl ether-4,4′-dicarboxylic acid chloride (8.43 g) and dichloromethane (107 mL) was placed in the dropping funnel. Moved.
- Example 1-10 (Production of polyester resin (10)) Sodium hydroxide (1.77 g) and H 2 O (188 ml) were weighed in a 500 mL beaker and dissolved with stirring. 2,3,5-trimethylphenol (0.25 g) and benzyltriethylammonium chloride (0.13 g) were added thereto and dissolved by stirring, and then the alkaline aqueous solution was transferred to a 1 L reaction vessel.
- This organic layer was washed with 0.1 N hydrochloric acid (190 mL) three times, and further washed with demineralized water (190 mL) twice.
- Dichloromethane (100 ml) was added to the washed organic layer for dilution, and the precipitate obtained by pouring into methanol (1800 ml) was collected by filtration and dried to obtain the desired polyester resin (10).
- the viscosity average molecular weight (Mv) of the obtained polyester resin was 22,500.
- the structural formula of the polyester resin (10) is shown below.
- Example 1-11 (Production of polyester resin (11)) Sodium hydroxide (1.45 g) and H 2 O (188 ml) were weighed in a 500 mL beaker and dissolved with stirring. 2,3,5-Trimethylphenol (0.20 g) and benzyltriethylammonium chloride (0.07 g) were added thereto and dissolved by stirring, and then the alkaline aqueous solution was transferred to a 1 L reaction vessel.
- This organic layer was washed with 0.1 N hydrochloric acid (190 mL) three times, and further washed with demineralized water (190 mL) twice.
- Dichloromethane (100 ml) was added to the washed organic layer for dilution, and the precipitate obtained by pouring into methanol (1800 ml) was collected by filtration and dried to obtain the desired polyester resin (11).
- the viscosity average molecular weight (Mv) of the obtained polyester resin was 40,200.
- the structural formula of the polyester resin (11) is shown below.
- Example 1-12 (Production of polyester polycarbonate resin (12)) Sodium hydroxide (4.16 g) and H 2 O (234 ml) were weighed in a 500 mL beaker and dissolved with stirring. 2,3,5-trimethylphenol (0.34 g), BP-a (10.15 g), and benzyltriethylammonium chloride (0.11 g) were added thereto and dissolved by stirring. Moved to the tank.
- a polycarbonate resin of the following formula (13) produced by the melt polymerization method described in Production Example 1-1 of Japanese Patent Application Laid-Open No.
- This organic layer was washed with 0.1N hydrochloric acid (235 mL) three times, and further washed twice with demineralized water (235 mL).
- Dichloromethane (150 ml) was added to the washed organic layer for dilution, and the precipitate obtained by pouring into methanol (2300 ml) was collected by filtration and dried to obtain the desired polyester polycarbonate resin (12).
- the viscosity average molecular weight (Mv) of the obtained polyester resin was 40,100.
- the structural formula of the polyester resin (12) is shown below.
- Comparative Example 1-1 (Production by Interfacial Polymerization of Polyester Resin (3)) Sodium hydroxide (4.23 g) and H 2 O (188 ml) were weighed in a 500 mL beaker and dissolved with stirring. 2,3,5-trimethylphenol (0.26 g), BP-a (10.15 g), hydroquinone (0.47 g), and benzyltriethylammonium chloride (0.12 g) were added and dissolved with stirring. The aqueous alkaline solution was transferred to a 1 L reaction vessel.
- Comparative Example 1-2 (Production by Interfacial Polymerization of Polyester Resin (6)) Sodium hydroxide (4.13 g) and H 2 O (188 ml) were weighed and dissolved in a 500 mL beaker while stirring. There, 2,3,5-trimethylphenol (0.26 g), BP-a (9.16 g), 4-hydroxybenzoic acid 4-hydroxyphenyl ether (0.97 g), and benzyltriethylammonium chloride (0.12 g) ) was added and dissolved by stirring, and the aqueous alkaline solution was transferred to a 1 L reaction vessel.
- 4-hydroxybenzoic acid 4-hydroxyphenyl ether is hydrolyzed during the polymerization, becomes hydroquinone, is oxidized, the water layer turns brown, and the divalent phenol is insufficient, so that a sufficiently elongated polymer is obtained. There wasn't.
- Comparative Example 1-3 Production by Interfacial Polymerization of Polyester Resin (11)
- Sodium hydroxide (4.14 g) and H 2 O (188 ml) were weighed in a 500 mL beaker and dissolved with stirring.
- 2,3,5-trimethylphenol (0.26 g), BP-a (6.93 g), 4,4′-biphenyldimethanol (3.02 g), and benzyltriethylammonium chloride (0.12 g) were added there. After adding and dissolving with stirring, the aqueous alkaline solution was transferred to a 1 L reaction vessel.
- Example 1-13 10 parts by mass of oxytitanium phthalocyanine and 150 parts by mass of 4-methoxy-4-methyl-2-pentanone were mixed and pulverized and dispersed in a sand grind mill to produce a pigment dispersion.
- Oxytitanium phthalocyanine has Bragg angles (2 ⁇ ⁇ 0.2) of 9.3 °, 10.6 °, 13.2 °, 15.1 °, 15.7 °, 16.5 ° in X-ray diffraction by CuK ⁇ ray. Strong diffraction peaks are shown at 1 °, 20.8 °, 23.3 °, 26.3 °, and 27.1 °.
- This charge generation layer forming coating solution was applied on a polyethylene terephthalate sheet having aluminum deposited on the surface so that the film thickness after drying was 0.4 ⁇ m and dried to provide a charge generation layer.
- 2002-080432 comprising a group of geometric isomers having the following structure as a main component as a charge transport material 50 parts by mass of (CTM-1), 100 parts by mass of the polyester resin (1) produced in Example 1-1, 8 parts by mass of an antioxidant (Irganox 1076), and 0.05 parts by mass of silicone oil as a leveling agent was mixed with 640 parts by mass of a mixed solvent of tetrahydrofuran and toluene (tetrahydrofuran 80% by mass, toluene 20% by mass) to prepare a coating solution for forming a charge transport layer.
- CTM-1 charge transport material 50 parts by mass of (CTM-1), 100 parts by mass of the polyester resin (1) produced in Example 1-1, 8 parts by mass of an antioxidant (Irganox 1076), and 0.05 parts by mass of silicone oil as a leveling agent was mixed with 640 parts by mass of a mixed solvent of tetrahydrofuran and toluene (tetrahydrofuran 80% by
- This charge transport layer forming coating solution is applied onto the above-described charge generation layer using an applicator so that the film thickness after drying is 25 ⁇ m, and dried at 125 ° C. for 20 minutes to form a charge transport layer.
- a photoreceptor sheet was prepared.
- Example 1-14 A photoreceptor sheet was produced in the same manner as in Example 1-1 except that the polyester resin (1) was changed to the polyester resin (2).
- Example 1-15 A photoreceptor sheet was produced in the same manner as in Example 1-1 except that the polyester resin (1) was changed to the polyester resin (3).
- Example 1-16 A photoreceptor sheet was produced in the same manner as in Example 1-1 except that the polyester resin (1) was changed to the polyester resin (4).
- Example 1-17 A photoreceptor sheet was produced in the same manner as in Example 1-1 except that the polyester resin (1) was changed to the polyester resin (5).
- Example 1-18 A photoreceptor sheet was produced in the same manner as in Example 1-1 except that the polyester resin (1) was changed to the polyester resin (6).
- Example 1-19 A photoreceptor sheet was produced in the same manner as in Example 1-1 except that the polyester resin (1) was changed to the polyester resin (7).
- Example 1-20 A photoreceptor sheet was produced in the same manner as in Example 1-1 except that the polyester resin (1) was changed to the polyester resin (8).
- Example 1-21 A photoreceptor sheet was produced in the same manner as in Example 1-1 except that the polyester resin (1) was changed to the polyester resin (9).
- Example 1-22 A photoreceptor sheet was produced in the same manner as in Example 1-1 except that the polyester resin (1) was changed to the polyester resin (10).
- Example 1-23 A photoreceptor sheet was produced in the same manner as in Example 1-1 except that the polyester resin (1) was changed to the polyester resin (11).
- Example 1-24 A photoreceptor sheet was produced in the same manner as in Example 1-1 except that the polyester resin (1) was changed to the polyester resin (12).
- the surface potential (VL) when the exposure light was irradiated by 2.4 ⁇ J / cm 2 was measured.
- the time required from the exposure to the potential measurement was 139 ms.
- the irradiation energy (half exposure energy: ⁇ J / cm 2 ) when the surface potential was half of the initial surface potential ( ⁇ 350 V) was measured as sensitivity (E 1/2 ).
- E 1/2 sensitivity
- the measurement environment was 25 ° C. and 50% relative humidity (N / N). The results are shown in Table 1-1.
- the ester oligomer (2-1) solution (1.25 g) was weighed into a 100 ml sample bottle containing morpholine (0.3 ml: manufactured by Tokyo Chemical Industry) and acetonitrile (50 ml: manufactured by Junsei Chemical Co., Ltd. for high speed liquid chromatography) Mixed. After standing at room temperature for 30 minutes to amidate the acid chloride site, 1 ml of this solution was weighed and mixed with 9 ml of mobile phase used in the HPLC analysis described below. The mixed solution was filtered through GL Chromatodisc 13P (GL Science Co., Ltd., pore size 0.45 ⁇ m) to remove solids, thereby preparing a sample solution.
- morpholine 0.3 ml: manufactured by Tokyo Chemical Industry
- acetonitrile 50 ml: manufactured by Junsei Chemical Co., Ltd. for high speed liquid chromatography
- Example 2-1 Synthesis of polyester resin (1-1) 1: Ester oligomer (2-1) formulation
- polyester resin (1-1) was produced in the same reaction vessel.
- the production method is shown below.
- BP-1 14.57 g
- AC-1 22.44 g
- polyester resin (1-1) Diluted dichloromethane (300 ml) was added to the organic layer after washing, and the resulting precipitate was poured into methanol (4000 ml), filtered out and dried to obtain the desired polyester resin (1-1).
- the viscosity average molecular weight (Mv) of the obtained polyester resin was 61,000.
- the structural formula of the polyester resin (1-1) group is shown below.
- the resulting polyester resin had a viscosity average molecular weight (Mv) of 45,000.
- the resulting polyester resin had a viscosity average molecular weight (Mv) of 46,500.
- Example 2-2 Synthesis of polyester resin (2-1): ester oligomer (2-2) formulation
- An ester oligomer was produced according to the same formulation as in Production Example 2-2, and then a polyester resin (2-1) was produced in the same reaction vessel.
- the production method is shown below. Weigh BP-1 (7.92 g), 2,3,5-trimethylphenol (0.064 g) and AC-1 (13.86 g) in a 500 mL four-necked reaction vessel purged with nitrogen and dissolve in dichloromethane (80 mL). I let you.
- Example 2-3 Synthesis of polyester resin (3-1) 1: Ester oligomer (2-2) formulation
- An ester oligomer was produced according to the same formulation as in Production Example 2-2, and then a polyester resin (3-1) was produced in the same reaction vessel.
- the production method is shown below.
- BP-1 13.89 g
- 2,3,5-trimethylphenol 0.067 g
- AC-1 24.33 g
- polyester resin (3-1) Diluted dichloromethane (300 ml) was added to the organic layer after washing, and the precipitate obtained by pouring into methanol (4000 ml) was taken out by filtration and dried to obtain the desired polyester resin (3-1).
- the resulting polyester resin had a viscosity average molecular weight (Mv) of 55,000.
- Mv viscosity average molecular weight
- the resulting polyester resin had a viscosity average molecular weight (Mv) of 39,000.
- Example 2-4 Synthesis of polyester resin (4-1): ester oligomer (2-3) formulation
- An ester oligomer was produced according to the same formulation as in Production Example 2-3, and then a polyester resin (4-1) was produced in the same reaction vessel.
- the production method is shown below.
- BP-1 (7.00 g), 2,3,5-trimethylphenol (0.065 g) and AC-1 (14.34 g) are weighed in a nitrogen-substituted 500 mL four-necked reaction vessel and dissolved in dichloromethane (80 mL). I let you.
- Example 2-5 Synthesis of polyester resin (5-1) 1: Ester oligomer (2-1) formulation
- An ester oligomer was produced according to the same formulation as in Production Example 2-1, and then a polyester resin (5-1) was produced in the same reaction vessel.
- the production method is shown below.
- BP-1 (8.44 g), 2,3,5-trimethylphenol (0.119 g) and AC-1 (13.02 g) are weighed in a nitrogen-substituted 500 mL four-necked reaction vessel and dissolved in dichloromethane (80 mL). I let you.
- Example 2-6 Synthesis of polyester resin (6-1): ester oligomer (2-3) formulation
- An ester oligomer was produced in the same formulation as in Production Example 2-3, and then a polyester resin (6-1) was produced in the same reaction vessel.
- the production method is shown below.
- BP-1 (6.36 g)
- AC-1 13.07 g
- BP-1 2,3,5-trimethylphenol
- AC-1 13.07 g
- Example 2-7 Synthesis of polyester resin (7-1)
- An ester oligomer was produced according to the same formulation as in Production Example 2-2, and then a polyester resin (7-1) was produced in the same reaction vessel.
- the production method is shown below. Weigh BP-1 (7.63 g), 2,3,5-trimethylphenol (0.092 g) and AC-1 (13.42 g) in a 500 mL four-necked reaction vessel purged with nitrogen, and dissolve in dichloromethane (80 mL). I let you. Subsequently, a mixed solution of triethylamine (6.42 g) and dichloromethane (20 mL) is dropped into a reaction vessel cooled to 5 to 15 ° C.
- the mixture was further diluted with dichloromethane (80 mL) and stirred for 4 hours. Thereafter, washing with demineralized water (200 mL) was performed, followed by washing with 0.2 N hydrochloric acid (200 mL) three times, and further washing with demineralized water (200 mL) twice.
- Dichloromethane (150 ml) was added to the washed organic layer for dilution, and the precipitate obtained by pouring into methanol (2000 ml) was collected by filtration and dried to obtain the desired polyester resin (7-1).
- the resulting polyester resin had a viscosity average molecular weight (Mv) of 40,700.
- Mv viscosity average molecular weight
- Example 2-8 (Synthesis of polyester resin (8-1))
- the polyester resin (8-1) was prepared in the same manner as in Example 2-7 except that 2,6-dihydroxynaphthalene in Example 2-7 was replaced with 2,7-dihydroxynaphthalene (hereinafter referred to as BP-7). Obtained.
- the resulting polyester resin had a viscosity average molecular weight (Mv) of 35,500.
- Mv viscosity average molecular weight
- the structural formula of the polyester resin (8-1) is shown below.
- Polyester resin (8-1) Polyester resin (8-1)
- Example 2-9 Synthesis of polyester resin (9-1)
- An ester oligomer was produced in the same formulation as in Production Example 2-1, and then a polyester resin (9-1) was produced in the same reaction vessel.
- the production method is shown below.
- a 500 mL four-necked reaction vessel purged with nitrogen was charged with bis (4-hydroxy-3-methylphenyl) methane (hereinafter referred to as BP-8) (9.03 g), 2,3,5-trimethylphenol (0.060 g), and AC- 1 (13.13 g) was weighed and dissolved in dichloromethane (75 mL).
- BP-8 bis (4-hydroxy-3-methylphenyl) methane
- AC- 1 13.13 g
- Example 2-10 Synthesis of polyester resin (10-1) Same as Example 2-10, except that bis (4-hydroxy-3-methylphenyl) methane in Example 2-10 was replaced with 1,1-bis (4-hydroxyphenyl) ethane (hereinafter referred to as BP-9). To obtain a polyester resin (10-1). The viscosity average molecular weight (Mv) of the obtained polyester resin was 56,200. The structural formula of the polyester resin (10-1) is shown below.
- Polyester resin (10-1) Polyester resin (10-1)
- ⁇ Preparation of photoreceptor sheet> 10 parts by mass of oxytitanium phthalocyanine and 150 parts by mass of 4-methoxy-4-methyl-2-pentanone were mixed and pulverized and dispersed in a sand grind mill to produce a pigment dispersion.
- Oxytitanium phthalocyanine has Bragg angles (2 ⁇ ⁇ 0.2) of 9.3 °, 10.6 °, 13.2 °, 15.1 °, 15.7 °, 16.5 ° in X-ray diffraction by CuK ⁇ ray. Strong diffraction peaks are represented at 1 °, 20.8 °, 23.3 °, 26.3 °, and 27.1 °.
- This charge generation layer forming coating solution was applied on a polyethylene terephthalate sheet having aluminum deposited on the surface so that the film thickness after drying was 0.4 ⁇ m and dried to provide a charge generation layer.
- a charge transport material a mixture (CTM) produced by the method described in Example 1 of Japanese Patent Application Laid-Open No.
- 2002-080432 which comprises a group of geometric isomers whose main component is the structure shown below -1) 50 parts by mass, polyester resin (1-1) produced in Example 2-1 100 parts by mass, antioxidant (Irganox 1076) 8 parts by mass, leveling agent 0.05 parts by mass of silicone oil was mixed with 640 parts by mass of a mixed solvent of tetrahydrofuran and toluene (tetrahydrofuran 80% by mass, toluene 20% by mass) to prepare a coating solution for forming a charge transport layer.
- This charge transport layer forming coating solution is applied onto the above-described charge generation layer using an applicator so that the film thickness after drying is 25 ⁇ m, and dried at 125 ° C. for 20 minutes to form a charge transport layer.
- a photoreceptor sheet was prepared.
- Example 2-12 A photoreceptor sheet was produced in the same manner as in Example 2-11 except that the polyester resin (1-1) was changed to the polyester resin (3-1) produced in Example 2-3.
- Example 2-13 A photoreceptor sheet was produced in the same manner as in Example 2-11 except that the polyester resin (1-1) was changed to the polyester resin (5-1) produced in Example 2-5.
- Example 2-14 A photoreceptor sheet was produced in the same manner as in Example 2-11 except that the polyester resin (1-1) was changed to the polyester resin (6-1) produced in Example 2-6.
- Example 2-15 A photoreceptor sheet was produced in the same manner as in Example 2-11 except that the polyester resin (1-1) was changed to the polyester resin (7-1) produced in Example 2-7.
- Example 2-16 A photoreceptor sheet was produced in the same manner as in Example 2-11 except that the polyester resin (1-1) was changed to the polyester resin (8-1) produced in Example 2-8.
- Example 2-17 A photoreceptor sheet was produced in the same manner as in Example 2-11 except that the polyester resin (1-1) was changed to the polyester resin (9-1) produced in Example 2-9.
- polyester resin (1-1) was changed to the polyester resin (11) (viscosity average molecular weight 36,200) having the following structure, which was produced by the method described in Example 6 of Japanese Patent Application Laid-Open No. 2006-53549.
- a photoreceptor sheet was produced in the same manner as in Example 2-1, except for the above.
- the potential holding ratio (DDR) after being charged to ⁇ 700 V and left for 5 seconds was measured (%).
- DDR potential holding ratio
- N / N 50% relative humidity
- Example 3-1 (Production of polyester resin (3-1) using ester oligomer (3-1))
- Sodium hydroxide (1.78 g) and H 2 O (188 ml) were weighed in a 500 mL beaker and dissolved with stirring.
- 2,3,5-trimethylphenol (0.26 g) and benzyltriethylammonium chloride (0.08 g) were added thereto and dissolved by stirring, and then the alkaline aqueous solution was transferred to a 1 L reaction vessel.
- Example 3-2 (Production of polyester resin (3-1) by solution polymerization method) 1,1-bis (4-hydroxy-3-methylphenyl) ethane (3.43 g) and diphenyl ether-4,4′-dicarboxylic acid chloride (8.35 g) were weighed in a nitrogen-substituted 500 mL four-necked reaction vessel. Dissolved in dichloromethane (80 mL). Subsequently, a mixed solution of triethylamine (3.01 g) and dichloromethane (15 mL) was dropped into a reaction vessel cooled to 5 to 15 ° C. over 20 minutes.
- Example 3-3 (Production of polyester resin (3-2) by solution polymerization method) 1,1-bis (4-hydroxy-3-methylphenyl) ethane (3.46 g) and diphenyl ether-4,4′-dicarboxylic acid chloride (8.43 g) were weighed in a nitrogen-substituted 500 mL four-necked reaction vessel. Dissolved in dichloromethane (80 mL). Subsequently, a mixed solution of triethylamine (3.03 g) and dichloromethane (15 mL) was dropped into the reaction vessel cooled to 5 to 15 ° C. over 20 minutes.
- the resulting polyester resin had a viscosity average molecular weight (Mv) of 79,700.
- Comparative Example 3-1 (Production of polyester resin (3-1) by interfacial polymerization method) Sodium hydroxide (4.13 g) and H 2 O (188 ml) were weighed and dissolved in a 500 mL beaker while stirring. There, 2,3,5-trimethylphenol (0.26 g), BP-a (9.16 g), 4-hydroxybenzoic acid 4-hydroxyphenyl ether (0.97 g), and benzyltriethylammonium chloride (0.12 g) ) was added and dissolved by stirring, and the aqueous alkaline solution was transferred to a 1 L reaction vessel.
- 4-hydroxybenzoic acid 4-hydroxyphenyl ether is hydrolyzed during the polymerization, becomes hydroquinone, is oxidized, the water layer turns brown, and the divalent phenol is insufficient, so that a sufficiently elongated polymer is obtained. There wasn't.
- Example 3-4 10 parts by mass of oxytitanium phthalocyanine and 150 parts by mass of 4-methoxy-4-methyl-2-pentanone were mixed and pulverized and dispersed in a sand grind mill to produce a pigment dispersion.
- Oxytitanium phthalocyanine has Bragg angles (2 ⁇ ⁇ 0.2) of 9.3 °, 10.6 °, 13.2 °, 15.1 °, 15.7 °, 16.5 ° in X-ray diffraction by CuK ⁇ ray. Strong diffraction peaks are represented at 1 °, 20.8 °, 23.3 °, 26.3 °, and 27.1 °.
- a charge transport material a mixture (CTM) produced by the method described in Example 1 of Japanese Patent Application Laid-Open No. 2002-080432, which comprises a group of geometric isomers whose main component is the structure shown below -1) 50 parts by mass, polyester resin (3-1) produced in Example 3-1 100 parts by mass, antioxidant (Irganox 1076) 8 parts by mass, leveling agent 0.05 parts by mass of silicone oil was mixed with 640 parts by mass of a mixed solvent of tetrahydrofuran and toluene (tetrahydrofuran 80% by mass, toluene 20% by mass) to prepare a coating solution for forming a charge transport layer.
- CTM mixture
- Example 1 of Japanese Patent Application Laid-Open No. 2002-080432 which comprises a group of geometric isomers whose main component is the structure shown below -1) 50 parts by mass
- polyester resin (3-1) produced in Example 3-1 100 parts by mass
- This charge transport layer forming coating solution is applied onto the above-described charge generation layer using an applicator so that the film thickness after drying is 25 ⁇ m, and dried at 125 ° C. for 20 minutes to form a charge transport layer.
- a photoreceptor sheet was prepared.
- Example 3-5 A photoreceptor sheet was produced in the same manner as in Example 3-1, except that the polyester resin (3-1) was changed to the polyester resin (3-1) produced in Example 3-2.
- Example 3-6 A photoreceptor sheet was produced in the same manner as in Example 3-1, except that the polyester resin (3-1) was changed to the polyester resin (3-2).
- Polyester resin (3-1) was produced by the method described in Example 6 of Japanese Patent Application Laid-Open No. 2006-53549, and polyester resin (3-3) (viscosity average molecular weight 36,200) having the following structure was prepared.
- a photoreceptor sheet was produced in the same manner as in Example 3-1, except that the changes were made.
- the surface potential (VL) when the exposure light was irradiated by 2.4 ⁇ J / cm 2 was measured.
- the time required from the exposure to the potential measurement was 139 ms.
- the irradiation energy (half exposure energy: ⁇ J / cm 2 ) when the surface potential was half of the initial surface potential ( ⁇ 350 V) was measured as sensitivity (E 1/2 ).
- E 1/2 sensitivity
- the measurement environment was 25 ° C. and 50% relative humidity (N / N). The results are shown in Table 3-1.
- the electrophotographic photosensitive member containing the specific polyester resin of the present invention is excellent not only in electrical characteristics but also in abrasion resistance.
- the polyester resin of the present invention uses a low molecular weight dihydric phenol having an ester bond as a raw material, but can be easily manufactured by combining a solution polymerization method or a solution polymerization method with an interfacial polymerization method, and an electron It was clarified that the characteristics of the photographic photoreceptor can be satisfied.
- Oligomer production example 4-2 (Production of ester oligomer (4-2)) Methylhydroquinone (3.00 g) (hereinafter referred to as BP-3), BP-2 (11.71 g) and AC-1 (14.26 g) were weighed in a 300 mL four-necked reaction vessel purged with nitrogen, and dissolved in dichloromethane (90 mL). Dissolved. Subsequently, a mixed solution of triethylamine (10.28 g) and dichloromethane (30 mL) was dropped into the reaction vessel cooled to 0 to 5 ° C. over 30 minutes. After stirring for 1 hour, demineralized water (100 mL) was added and stirred for 10 minutes.
- Oligomer Production Example 4-4 (Production of ester oligomer (4-4)) Weigh 1,5-dihydroxynaphthalene (1.50 g) (hereinafter BP-5), BP-2 (12.86 g) and AC-1 (12.28 g) in a 300 mL four-necked reaction vessel purged with nitrogen, (80 mL). Subsequently, a mixed solution of triethylamine (8.84 g) and dichloromethane (40 mL) was dropped into the reaction vessel cooled to 0 to 5 ° C. over 30 minutes. After stirring for 1 hour, demineralized water (100 mL) was added and stirred for 10 minutes.
- Polyester Production Example 4-1 (Production of Polyester Resin (4-1)) Sodium hydroxide (1.57 g) and H 2 O (188 ml) were weighed in a 500 mL beaker and dissolved with stirring. 2,3,5-Trimethylphenol (0.21 g) and benzyltriethylammonium chloride (0.11 g) were added thereto and dissolved by stirring, and the aqueous alkaline solution was transferred to a 1 L reaction vessel. Separately, a mixed solution of ester oligomer (4-1) (16.13 g) and AC-1 (4.87 g) synthesized in oligomer production example 4-1 and dichloromethane (107 mL) was transferred into a dropping funnel.
- polyester resin (4-1) Diluted dichloromethane (100 ml) was added to the organic layer after washing, and the resulting precipitate was poured into methanol (1800 ml), filtered out and dried to obtain the desired polyester resin (4-1).
- the viscosity average molecular weight (Mv) of the obtained polyester resin was 44,200.
- the structural formula of the polyester resin (4-1) is shown below.
- Polyester Production Example 4-2 (Production of Polyester Resin (4-2)) Sodium hydroxide (1.55 g) and H 2 O (188 ml) were weighed in a 500 mL beaker and dissolved with stirring. 2,3,5-Trimethylphenol (0.21 g) and benzyltriethylammonium chloride (0.11 g) were added thereto and dissolved by stirring, and the aqueous alkaline solution was transferred to a 1 L reaction vessel. Separately, a mixed solution of the ester oligomer (4-2) (16.18 g) synthesized in the oligomer production example 4-2 and AC-1 (4.81 g) and dichloromethane (107 mL) was transferred into the dropping funnel.
- Polyester Production Example 4-3 (Production of Polyester Resin (4-3)) Sodium hydroxide (1.52 g) and H 2 O (188 ml) were weighed in a 500 mL beaker and dissolved with stirring. 2,3,5-trimethylphenol (0.24 g) and benzyltriethylammonium chloride (0.11 g) were added thereto and dissolved by stirring, and then the alkaline aqueous solution was transferred to a 1 L reaction vessel. Separately, a mixed solution of ester oligomer (4-3) (16.22 g) and AC-1 (4.70 g) synthesized in oligomer production example 4-3 and dichloromethane (107 mL) was transferred into a dropping funnel.
- the resulting polyester resin had a viscosity average molecular weight (Mv) of 39,000.
- Mv viscosity average molecular weight
- Polyester Production Example 4-4 (Production of polyester resin (4-4)) Sodium hydroxide (1.47 g) and H 2 O (188 ml) were weighed in a 500 mL beaker and dissolved with stirring. 2,3,5-trimethylphenol (0.25 g) and benzyltriethylammonium chloride (0.11 g) were added thereto and dissolved by stirring, and then the alkaline aqueous solution was transferred to a 1 L reaction vessel. Separately, a mixed solution of ester oligomer (4-4) (16.32 g) and AC-1 (4.56 g) synthesized in oligomer production example 4-4 and dichloromethane (107 mL) was transferred into the dropping funnel.
- the resulting polyester resin had a viscosity average molecular weight (Mv) of 41,300.
- Mv viscosity average molecular weight
- Polyester Production Example 4-5 (Production of polyester resin (4-5)) BP-2 (13.89 g), 2,3,5-trimethylphenol (0.067 g) and AC-1 (24.33 g) were weighed in a nitrogen-substituted 1000 mL four-necked reaction vessel and dissolved in dichloromethane (120 mL). I let you. Subsequently, a mixed solution of triethylamine (11.60 g) and dichloromethane (30 mL) was dropped into a reaction vessel cooled to 5 to 15 ° C. over 20 minutes, and then stirring was continued for 10 minutes to form an ester oligomer. It was.
- hydroquinone (hereinafter referred to as BP-6) (2.71 g) was added into the reaction vessel. Thereafter, a mixed solution of triethylamine (6.13 g) and dichloromethane (50 mL) was dropped into the reaction vessel into the reaction vessel cooled to 5 to 15 ° C. over 20 minutes. Stirring was continued for 0.5 hours while maintaining the internal temperature of the reaction at 15 to 23 ° C., then diluted with dichloromethane (230 mL), and further stirred for 0.5 hours. Subsequently, the mixture was further diluted with dichloromethane (230 mL) and stirred for 4 hours.
- BP-6 hydroquinone
- polyester resin (4-5) had a viscosity average molecular weight (Mv) of 55,000.
- Mv viscosity average molecular weight
- Polyester Production Example 4-6 (Synthesis of polyester resin (4-6)) BP-2 (7.63 g), 2,3,5-trimethylphenol (0.092 g) and AC-1 (13.42 g) are weighed in a nitrogen-substituted 500 mL four-necked reaction vessel and dissolved in dichloromethane (80 mL). I let you. Subsequently, a mixed solution of triethylamine (6.42 g) and dichloromethane (20 mL) is dropped into a reaction vessel cooled to 5 to 15 ° C. over 20 minutes, and then stirring is continued for 10 minutes to form an ester oligomer. It was.
- BP-7 2,6-dihydroxynaphthalene (2.16 g) was added into the reaction vessel. Thereafter, a mixed solution of triethylamine (3.41 g) and dichloromethane (30 mL) was dropped into the reaction vessel, which was cooled to 5 to 15 ° C., over 20 minutes. Stirring was continued for 0.5 hours while maintaining the internal temperature of the reaction at 15 to 23 ° C., then diluted with dichloromethane (90 mL), and further stirred for 0.5 hours. Subsequently, the mixture was further diluted with dichloromethane (80 mL) and stirred for 4 hours.
- BP-7 2,6-dihydroxynaphthalene
- Polyester Production Example 4-7 (Synthesis of polyester resin (4-7)) The same procedure as in Polyester Production Example 4-6 was conducted, except that 2,6-dihydroxynaphthalene (BP-7) in Polyester Production Example 4-6 was replaced with 2,7-dihydroxynaphthalene (hereinafter referred to as BP-8). Resin (4-7) was obtained. The resulting polyester resin had a viscosity average molecular weight (Mv) of 35,500. The structural formula of the polyester resin (4-7) is shown below.
- Polyester Production Example 4-8 (Production by Interfacial Polymerization of Polyester Resin (4-8)) Sodium hydroxide (4.23 g) and H 2 O (188 ml) were weighed in a 500 mL beaker and dissolved with stirring. 2,3,5-trimethylphenol (0.26 g), BP-2 (10.15 g), BP-6 (0.47 g), and benzyltriethylammonium chloride (0.12 g) were added and dissolved with stirring. Then, this alkaline aqueous solution was transferred to a 1 L reaction vessel. Separately, a mixed solution of AC-1 (13.08 g) and dichloromethane (94 mL) was transferred into the dropping funnel.
- AC-1 13.08 g
- dichloromethane 94 mL
- Example 4-1 10 parts by mass of oxytitanium phthalocyanine and 150 parts by mass of 4-methoxy-4-methyl-2-pentanone were mixed and pulverized and dispersed in a sand grind mill to produce a pigment dispersion.
- Oxytitanium phthalocyanine has Bragg angles (2 ⁇ ⁇ 0.2) of 9.3 °, 10.6 °, 13.2 °, 15.1 °, 15.7 °, 16.5 ° in X-ray diffraction by CuK ⁇ ray. Strong diffraction peaks are represented at 1 °, 20.8 °, 23.3 °, 26.3 °, and 27.1 °.
- This charge generation layer forming coating solution was applied on a polyethylene terephthalate sheet having aluminum deposited on the surface so that the film thickness after drying was 0.4 ⁇ m and dried to provide a charge generation layer.
- a charge transport material a mixture (CTM) produced by the method described in Example 1 of Japanese Patent Application Laid-Open No.
- 2002-080432 which comprises a group of geometric isomers whose main component is the structure shown below -1) 50 parts by mass, polyester resin (4-1) 100 parts by mass, antioxidant (Irganox 1076) 8 parts by mass, silicone oil 0.05 parts by mass as a leveling agent, tetrahydrofuran and toluene mixed It mixed with 640 mass parts of solvents (tetrahydrofuran 80 mass%, toluene 20 mass%), and prepared the coating liquid for charge transport layer formation.
- This charge transport layer forming coating solution is applied onto the above-described charge generation layer using an applicator so that the film thickness after drying is 25 ⁇ m, and dried at 125 ° C. for 20 minutes to form a charge transport layer.
- a photoreceptor sheet was prepared.
- Example 4-2 A photoreceptor sheet was produced in the same manner as in Example 4-1, except that the polyester resin (4-1) was changed to the polyester resin (4-2).
- Example 4-3 A photoreceptor sheet was produced in the same manner as in Example 4-1, except that the polyester resin (4-1) was changed to the polyester resin (4-3).
- Example 4-4 A photoreceptor sheet was produced in the same manner as in Example 4-1, except that the polyester resin (4-1) was changed to the polyester resin (4-4).
- Example 4-5 A photoreceptor sheet was produced in the same manner as in Example 4-1, except that the polyester resin (4-1) was changed to the polyester resin (4-5).
- Example 4-6 A photoreceptor sheet was produced in the same manner as in Example 4-1, except that the polyester resin (4-1) was changed to the polyester resin (4-6).
- Example 4-7 A photoreceptor sheet was produced in the same manner as in Example 4-1, except that the polyester resin (4-1) was changed to the polyester resin (4-7).
- polyester resin (4-1) was produced by the method described in Example 6 of Japanese Patent Application Laid-Open No. 2006-53549, and the polyester resin (4-9) (viscosity average molecular weight 36,200) having the following structure was produced.
- a photoreceptor sheet was produced in the same manner as in Example 4-1, except that the change was made.
- the surface potential (VL) when the exposure light was irradiated by 2.4 ⁇ J / cm 2 was measured.
- the time required from the exposure to the potential measurement was 139 ms.
- the irradiation energy (half exposure energy: ⁇ J / cm 2 ) when the surface potential was half of the initial surface potential ( ⁇ 350 V) was measured as sensitivity (E 1/2 ).
- E 1/2 sensitivity
- the measurement environment was 25 ° C. and 50% relative humidity (N / N). The results are shown in Table 4-1.
- the electrophotographic photosensitive member containing the specific polyester resin of the present invention is excellent not only in electrical characteristics but also in abrasion resistance.
- the polyester resin of the present invention is produced using a dihydric phenol that is easily oxidized, but can be easily produced by combining a solution polymerization method or a solution polymerization method with an interfacial polymerization method. It was clarified that the characteristics can be satisfied.
- Electrophotographic photoreceptor Charging device (charging roller) DESCRIPTION OF SYMBOLS 3 Exposure apparatus 4 Developing apparatus 5 Transfer apparatus 6 Cleaning apparatus 7 Fixing apparatus 41 Developing tank 42 Agitator 43 Supply roller 44 Developing roller 45 Control member 71 Upper fixing member (pressure roller) 72 Lower fixing member (fixing roller) 73 Heating device T Toner P Recording paper
Abstract
本発明は、実用上の負荷に対する耐摩耗性及び電気特性に優れた電子写真感光体を提供する。本発明は、導電性支持体上に感光層を有する電子写真感光体において、前記感光層は、2価フェノール残基および2価カルボン酸残基からなるポリエステル樹脂を含有し、前記2価フェノール残基が、下記式(1)~式(3)で表される2価フェノール残基群より選ばれる少なくとも一つの2価フェノール残基、及び下記式(4)で表される2価フェノール残基を含むことを特徴とする電子写真感光体に関する。
Description
本発明は電子写真感光体に関し、より詳しくは、耐摩耗性及び電気特性が良好な電子写真感光体、及び画像形成装置に関する。さらに本発明は、その電子写真感光体に用いられる樹脂、及びその製造方法に関する。
電子写真感光体は、電子写真プロセス、即ち、帯電、露光、現像、転写、クリーニング、除電等のサイクルで繰り返し使用されるため、その間の様々なストレスを受けて劣化する。このような劣化としては、例えば、帯電器として普通用いられるコロナ帯電器から発生する強酸化性のオゾンやNOxが感光層に与える化学的なダメージ、像露光で生成したキャリアー(電流)が感光層内を流れること、又は除電光及び外部からの光に起因する感光層組成物の分解等の化学的、電気的劣化がある。さらに、クリーニングブレード、磁気ブラシ等の摺擦、現像剤、紙との接触等による感光層表面の摩耗、傷の発生、膜の剥がれ等の機械的劣化がある。このような機械的劣化による損傷は画像上に現れやすく、直接画像品質を損なうため感光体の寿命を制限する大きな要因となっている。
表面保護層等の機能層を設けない一般的な感光体の場合、特に感光層がこのような負荷を受け易い。感光層は、通常、バインダー樹脂と光導電性物質とからなり、実質的に強度を決めるのはバインダー樹脂であるが、光導電性物質のドープ量が相当多いため充分な機械強度を持たせるには至っていない。近年、感光層のバインダー樹脂として、感度や耐摩耗性に優れているポリエステル樹脂が使用されている(特許文献1~特許文献8参照)。
一方で、ポリエステル樹脂の製造方法は種々知られているが、分子量が高く、且つ着色が少なく、純度の高いポリエステル樹脂が得られる界面重合が広く利用されている。しかしながら、例えば、ヒドロキノンのような酸化されやすいモノマーを用いて界面重合法によりポリエステル樹脂を製造しようとすると、アルカリ水溶液中で簡単に酸化されキノン等の酸化体になってしまう。この酸化体は、ジカルボン酸クロライドと反応しないため想定量を樹脂中に組み込みことは難しく、また樹脂中に残存する酸化体により樹脂が着色する。このような重合中のモノマー酸化を防ぐ方法は検討されており、過剰量の酸化防止剤を入れる重合方法等が知られている(特許文献9参照)。
しかしながら、本発明者らの検討によると、前記特許文献1~8に記載の技術のポリエステル樹脂を使用した感光体では、100,000枚以上印刷するような高寿命・高速ハイエンド機種においては、機械強度は十分なものではなかった。また、特許文献6に記載の技術ではポリエステル樹脂の溶解性が十分ではなく、塗布液に溶解しない、または、塗布液が数日でゲル化するなど溶解性が十分なものではなかった。さらに、前記特許文献9に記載の技術により、酸化しやすいモノマーを使用しポリエステル樹脂の製造が可能になるが、酸化防止剤を多量に使用するため重合された樹脂中に不純物として残存し、電気特性が悪化することがある。また、重合中に厳密に水溶液中の酸素を除く必要があり、実際に製造する上では困難が伴う。
本発明は上記課題に鑑みてなされたものである。即ち、本発明の目的は、溶解性、耐摩耗性、電気特性に優れたポリエステル樹脂を感光層に含むことにより、実用上の負荷に対する耐摩耗性及び電気特性に優れた電子写真感光体を提供することである。あわせて、上記溶解性、耐摩耗性、電気特性に優れたポリエステル樹脂、及びその製造方法を提供するものである。
本発明者らは、上記の課題を解決しうる電子写真感光体について鋭意検討を行なった結果、感光層に特定の化学構造を有するポリエステル樹脂を含有させることにより、優れた機械的特性を有し、感光層形成用塗布液に用いる溶媒に対して高い溶解性及び優れた塗布液安定性を有し、且つ、電気特性に優れた電子写真感光体を得ることができることを見出し、かかる知見に基づき本発明を完成させるに至った。
また、本発明者らは、上記の課題を解決しうるポリエステル樹脂の製造方法について鋭意検討を行なった結果、ポリエステル樹脂の製造において、エステルオリゴマーを経由し、該エステルオリゴマー中の残存カルボン酸時クロライドモノマー量を制御することにより、電子写真感光体に含有した場合の電気特性、溶解性及び耐摩耗性に優れたポリエステル樹脂を簡易に製造できることを見出し、かかる知見に基づき本発明を完成させた。
即ち、発明の要旨は、以下<1>~<8>に存する。
<1>導電性支持体上に感光層を有する電子写真感光体において、前記感光層は、2価フェノール残基および2価カルボン酸残基からなるポリエステル樹脂を含有し、前記2価フェノール残基が、下記式(1)~式(3)で表される2価フェノール残基群より選ばれる少なくとも一つの2価フェノール残基、及び下記式(4)で表される2価フェノール残基を含むことを特徴とする電子写真感光体。
また、本発明者らは、上記の課題を解決しうるポリエステル樹脂の製造方法について鋭意検討を行なった結果、ポリエステル樹脂の製造において、エステルオリゴマーを経由し、該エステルオリゴマー中の残存カルボン酸時クロライドモノマー量を制御することにより、電子写真感光体に含有した場合の電気特性、溶解性及び耐摩耗性に優れたポリエステル樹脂を簡易に製造できることを見出し、かかる知見に基づき本発明を完成させた。
即ち、発明の要旨は、以下<1>~<8>に存する。
<1>導電性支持体上に感光層を有する電子写真感光体において、前記感光層は、2価フェノール残基および2価カルボン酸残基からなるポリエステル樹脂を含有し、前記2価フェノール残基が、下記式(1)~式(3)で表される2価フェノール残基群より選ばれる少なくとも一つの2価フェノール残基、及び下記式(4)で表される2価フェノール残基を含むことを特徴とする電子写真感光体。

(式(1)中、R1は水素原子、炭素数1~20の置換基を有していてもよいアルキル基、アルコキシ基、置換されてもよい芳香族基、ハロゲン基のいずれかを表し、n1は0~4の整数である。)

(式(2)中、R2は水素原子、炭素数1~20の置換基を有していてもよいアルキル基、アルコキシ基、置換されてもよい芳香族基、ハロゲン基のいずれかを表し、n2は0~6の整数である。)

(式(3)中、Ar1、Ar2は、それぞれ独立に炭素数6~16の置換基を有していてもよいアリーレン基を表す。Zは、置換基を有してもよいアリーレン基であり、sは0又は1の整数である。)

(式(4)中、R3~R6は、それぞれ独立に水素原子、炭素数1~20の置換基を有していてもよいアルキル基、アルコキシ基、置換されてもよい芳香族基、ハロゲン基のいずれかを表す。Xは、単結合、―CR7R8-、O、CO、又はSを表す。またR7、R8はそれぞれ独立に、水素原子、炭素数1~10のアルキル基、又は、R7及びR8は、R7とR8とが結合して形成される炭素数5~10のシクロアルキリデン基を表す。)
<2>前記ポリエステル樹脂の全2価フェノール残基に対して、上記式(1)~式(3)で表される2価フェノール残基群より選ばれる少なくとも一つの2価フェノール残基が5~80モル%である、前記<1>に記載の電子写真感光体。
<3>前記ポリエステル樹脂の2価カルボン酸残基が、下記式(5)で表される2価カルボン酸残基である、前記<1>又は<2>に記載の電子写真感光体。
<2>前記ポリエステル樹脂の全2価フェノール残基に対して、上記式(1)~式(3)で表される2価フェノール残基群より選ばれる少なくとも一つの2価フェノール残基が5~80モル%である、前記<1>に記載の電子写真感光体。
<3>前記ポリエステル樹脂の2価カルボン酸残基が、下記式(5)で表される2価カルボン酸残基である、前記<1>又は<2>に記載の電子写真感光体。

(式(5)中、Ar3、Ar4は、それぞれ独立に置換基を有していてもよいアリーレン基を表す。Yは、単結合、酸素原子、硫黄原子、式(6)で表される構造、又は式(7)で表される構造を有する2価の有機残基を表す。式(6)中のR9及びR10は、それぞれ独立に、水素原子、アルキル基、若しくはアリール基、又はR9とR10とが結合して形成されるシクロアルキリデン基を表す。さらに、式(7)中、R11は、アルキレン基、アリーレン基、又は式(8)で表される基であり、式(8)中、R12及びR13は、それぞれ独立にアルキレン基を表し、Ar5はアリーレン基を表す。kは0~5の整数を表す。)



<4>下記式(1a)で表される繰返し単位および下記式(2a)で表される繰返し単位を含むポリエステル樹脂の製造方法であって、該ポリエステル樹脂は下記式(4a1)で表される2価フェノールおよび下記式(5a)で表されるジカルボン酸クロライドから得られるエステルオリゴマーを重合させて得られ、該エステルオリゴマー中の下記式(5a)で表されるジカルボン酸クロライドモノマー残存量が下記式(6a)を満たすことを特徴としたポリエステル樹脂の製造方法。


(式(1a)中、X1aは、下記式(3a)で表される2価の基であり、Y1aは、2価の基である。式(2a)中、X2aは、X1aとは同一ではない2価の芳香族を有する基である。Y2aは、2価の基である。)

(式(3a)中、R1a、R2aは、それぞれ独立に、水素原子、炭素数1~20の置換基を有していてもよいアルキル基、アルコキシ基、置換されても良い芳香族基のいずれかを表し、n1a、n2aはそれぞれ0~4の整数である。R3a,R4aはそれぞれ独立に、水素原子、炭素数1~20のアルキル基、置換基を有していてもよい芳香族基、またはR3aとR4aが互いに結合して置換基を有していてもよい環を形成する基のいずれかを表す。)



<5>ポリエステル樹脂の製造方法であって、少なくとも1種類のエステルオリゴマーを原料とし、界面重合法により重合することを特徴とする、ポリエステル樹脂の製造方法。
<6>前記エステルオリゴマーが、溶液重合法又は溶融重合法により得られることを特徴とする、前記<5>に記載のポリエステル樹脂の製造方法。
<7>前記ポリエステル樹脂は、2価フェノール残基および2価カルボン酸残基を重合することにより得られ、前記2価フェノール残基が、上記式(1)~式(3)で表される2価フェノール残基群より選ばれる少なくとも一つの2価フェノール残基、及び上記式(4)で表される2価フェノール残基を含む前記<4>~<6>のいずれか1に記載のポリエステル樹脂の製造方法。
<8>2価フェノール残基および2価カルボン酸残基からなるポリエステル樹脂であって、前記2価フェノール残基が、下記式(1)~式(3)で表される2価フェノール残基群より選ばれる少なくとも一つの2価フェノール残基、及び下記式(4)で表される2価フェノール残基を含むことを特徴とするポリエステル樹脂。
<6>前記エステルオリゴマーが、溶液重合法又は溶融重合法により得られることを特徴とする、前記<5>に記載のポリエステル樹脂の製造方法。
<7>前記ポリエステル樹脂は、2価フェノール残基および2価カルボン酸残基を重合することにより得られ、前記2価フェノール残基が、上記式(1)~式(3)で表される2価フェノール残基群より選ばれる少なくとも一つの2価フェノール残基、及び上記式(4)で表される2価フェノール残基を含む前記<4>~<6>のいずれか1に記載のポリエステル樹脂の製造方法。
<8>2価フェノール残基および2価カルボン酸残基からなるポリエステル樹脂であって、前記2価フェノール残基が、下記式(1)~式(3)で表される2価フェノール残基群より選ばれる少なくとも一つの2価フェノール残基、及び下記式(4)で表される2価フェノール残基を含むことを特徴とするポリエステル樹脂。

(式(1)中、R1は水素原子、炭素数1~20の置換基を有していてもよいアルキル基、アルコキシ基、置換されてもよい芳香族基、ハロゲン基のいずれかを表し、n1は0~4の整数である。)

(式(2)中、R2は水素原子、炭素数1~20の置換基を有していてもよいアルキル基、アルコキシ基、置換されてもよい芳香族基、ハロゲン基のいずれかを表し、n2は0~6の整数である。)

(式(3)中、Ar1、Ar2は、それぞれ独立に炭素数6~16の置換基を有していてもよいアリーレン基を表す。Zは、置換基を有してもよいアリーレン基であり、sは0又は1の整数である。)

(式(4)中、R3~R6は、それぞれ独立に水素原子、炭素数1~20の置換基を有していてもよいアルキル基、アルコキシ基、置換されてもよい芳香族基、ハロゲン基のいずれかを表す。Xは、単結合、―CR7R8-、O、CO、又はSを表す。またR7、R8はそれぞれ独立に、水素原子、炭素数1~10のアルキル基、又は、R7及びR8は、R7とR8とが結合して形成される炭素数5~10のシクロアルキリデン基を表す。)
本発明によれば、耐摩耗性に特に優れ、且つ、電気特性に優れた電子写真感光体が得られる。また、溶解性、耐摩耗性に特に優れたポリエステル樹脂が得られる。
以下、本発明を実施するための形態について詳細に説明する。尚、本発明は、以下の実施の形態に限定されるものではなく、その要旨の範囲内で種々変形して実施することができる。
≪第1の実施形態のポリエステル樹脂≫
本実施の形態が適用される電子写真感光体の感光層は、下記式(1)~(3)で表される2価フェノール残基より選ばれる少なくとも1つの2価フェノール残基を含むポリエステル樹脂を含有する。
本実施の形態が適用される電子写真感光体の感光層は、下記式(1)~(3)で表される2価フェノール残基より選ばれる少なくとも1つの2価フェノール残基を含むポリエステル樹脂を含有する。

式(1)中、R1は水素原子、炭素数1~20の置換基を有していてもよいアルキル基、炭素数1~20の置換基を有していてもよいアルコキシ基、炭素数1~20の置換基を有していてもよい芳香族基、又はハロゲン基のいずれかを表し、n1は0~4の整数である。
炭素数は、置換基を含めた基全体の炭素数である。R1の置換基としては、アルキル基、アルコキシ基、ハロゲン原子等が挙げられ、置換基を有さないことが好ましい。
炭素数1~20の置換基を有していてもよいアルキル基の具体例としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、tert-ブチル基、イソブチル基、シクロヘキシル基、クロロメチル基、フッ化アルキル基、トリフルオロメチル基、パーフルオロアルキル基等が挙げられる。
炭素数1~20の置換基を有していてもよいアルコキシ基の具体例としては、メトキシ基、エトキシ基、プロポキシ基、シクロヘキソキシ基等が挙げられる。炭素数1~20の置換基を有していてもよい芳香族基としては、フェニル基、メチルフェニル基、ジメチルフェニル基、ハロゲン化フェニル基、ナフチル基等が挙げられる。
ハロゲン基としては、フッ素基、クロロ基、ブロモ基が挙げられる。耐摩耗性の観点から、好ましくは、炭素数1~6のアルキル基、炭素数1~6のアルコキシ基、無置換の芳香族基であり、特に好ましくはメチル基、エチル基である。n1は各々独立に、0~4の整数であり、製造上の簡便性の観点から、好ましくは、n1=0、1である。
式(1)で表される2価フェノール残基を誘導する2価フェノール化合物の具体例としては、例えば、ヒドロキノン、メチルヒドロキノン、クロロヒドロキノン、tert-ブチルヒドロキノン、ブロモヒドロキノン、2,3-ジメチルヒドロキノン、2,5-ジメチルヒドロキノントリメチルヒドロキノン、フェニルヒドロキノン、レソルシノール、2-メチルレソルシノール、5-メチルレソルシノール、5-ブロモレソルシノール、カテコール、4-メチルカテコール等が挙げられる。これらの中でも、製造上の簡便性及び耐摩耗性の観点から、ヒドロキノン、メチルヒドロキノン、2,3-ジメチルヒドロキノン、2,5-ジメチルヒドロキノン、トリメチルヒドロキノン、レソルシノールが特に好ましい。

式(2)中、R2は水素原子、炭素数1~20の置換基を有していてもよいアルキル基、炭素数1~20の置換基を有していてもよいアルコキシ基、炭素数1~20の置換基を有していてもよい芳香族基、又はハロゲン基のいずれかを表し、n2は0~6の整数である。
炭素数は、置換基を含めた基全体の炭素数である。R2の置換基としては、アルキル基、アルコキシ基、ハロゲン原子等が挙げられ、置換基を有さないことが好ましい。それぞれの基の具体例はR1で挙げたものが適用できる。耐摩耗性の観点から、好ましくは、炭素数1~6のアルキル基、炭素数1~6のアルコキシ基であり、特に好ましくはメチル基、エチル基である。n2は各々独立に、0~4の整数であり、製造上の簡便性の観点から、好ましくは、n2=0である。
式(2)で表される2価フェノール残基を誘導する2価フェノール化合物の具体例としては、例えば、1,2-ジヒドロキシナフタレン、1,3-ジヒドロキシナフタレン、1,5-ジヒドロキシナフタレン、1,6-ジヒドロキシナフタレン、1,7-ジヒドロキシナフタレン、2,3-ジヒドロキシナフタレン、2,6-ジヒドロキシナフタレン、2,7-ジヒドロキシナフタレン等が挙げられる。これらの中でも、製造上の簡便性、溶解性の観点から、1,6-ジヒドロキシナフタレン、1,7-ジヒドロキシナフタレン、2,6-ジヒドロキシナフタレン、2,7-ジヒドロキシナフタレンが好ましく、耐摩耗性の観点から、2,6-ジヒドロキシナフタレン、2,7-ジヒドロキシナフタレンが特に好ましい。

式(3)中、Ar1、Ar2はそれぞれ独立に、炭素数6~16の置換基を有していてもよいアリーレン基を表す。Zは、置換基を有してもよいアリーレン基であり、sは0又は1の整数である。
式(3)中、Ar1、Ar2の置換基を有していてもよいアリーレン基の具体例としては、フェニレン基、ナフチレン基、アントリレン基、フェナントリレン基、ピレニレン基、ビフェニレン基が挙げられる。製造上の簡便性から、フェニレン基が好ましい。
置換基としては、炭素数1~10のアルキル基、炭素数1~10のアルコキシ基、ハロゲン化アルキル基、ハロゲン基、ベンジル基等が挙げられる。
アルキル基の具体例としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、tert-ブチル基、イソブチル基、シクロヘキシル基等が挙げられる。
アルコキシ基の具体例としては、メトキシ基、エトキシ基、プロポキシ基、シクロヘキソキシ基等が挙げられる。
ハロゲン化アルキル基としては、クロロメチル基、フッ化アルキル基等が挙げられる。
ハロゲン基としては、フッ素基、クロロ基、ブロモ基等が挙げられる。耐摩耗性の観点から、好ましくは、炭素数1~6のアルキル基、炭素数1~6のアルコキシ基であり、特に好ましくはメチル基、エチル基である。
式(3)中、Zの置換基を有していてもよいアリーレン基の具体例としては、p-フェニレン基、m-フェニレン基、又は下記式(11)で表される構造が挙げられる。製造上の簡便性から、p-フェニレン基、又は下記式(11)で表される構造が好ましい。

(式(11)中、R16~R19は、それぞれ独立に水素原子、又は炭素数1~10のアルキル基である。)
式(11)中のアルキル基の具体例としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、tert-ブチル基、イソブチル基、シクロヘキシル基等が挙げられる。溶解性、耐摩耗性、製造上の簡便性から、R16~R19は、水素原子又はメチル基が好ましい。
sは、耐摩耗性の観点から0又は1の整数が好ましく、溶解性の観点から0がさらに好ましい。式(3)で表される2価フェノール残基を誘導する2価フェノール化合物の具体例として以下に表す。

さらに、本発明におけるポリエステル樹脂には下記式(4)で表される2価フェノール残基を含有する。

式(4)中、R3~R6は、それぞれ独立に水素原子、炭素数1~20の置換基を有していてもよいアルキル基、炭素数1~20の置換基を有していてもよいアルコキシ基、炭素数1~20の置換基を有していてもよい芳香族基、又はハロゲン基のいずれかを表す。Xは、単結合、―CR7R8-、O、CO、又はSを表す。R7、R8は、それぞれ独立に水素原子、炭素数1~10のアルキル基、又は、R7とR8とが結合して形成される炭素数5~10のシクロアルキリデン基を表す。
R3~R6のそれぞれの基の具体例はR1で挙げたものが適用できる。製造上の簡便性、耐摩耗性の観点から、好ましくは、炭素数1~6のアルキル基、炭素数1~6のアルコキシ基であり、特に好ましくはメチル基である。
R7、R8の炭素数1~10のアルキル基の具体例としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基等が挙げられる。溶解性、耐摩耗性、製造上の簡便性から、メチル基、エチル基が好ましい。また、R7とR8とが結合して形成される炭素数5~10のシクロアルキリデン基としては、シクロペンチリデン基、シクロヘキシリデン基、シクロへプチリデン基等が挙げられる。
式(4)で表されるビスフェノール残基として具体的に例示すると、ビス-(4-ヒドロキシフェニル)メタン、ビス-(4-ヒドロキシ-3-メチルフェニル)メタン、ビス-(3,5-ジメチル-4-ヒドロキシフェニル)メタン、1,1-ビス-(4-ヒドロキシフェニル)エタン、1,1-ビス-(4-ヒドロキシ-3-メチルフェニル)エタン、1,1-ビス-(3,5-ジメチル-4-ヒドロキシフェニル)エタン、1,1-ビス-(4-ヒドロキシフェニル)プロパン、1,1-ビス-(4-ヒドロキー3-メチルフェニル)プロパン、1,1-ビス-(3,5-ジメチル-4-ヒドロキシフェニル)プロパン、2,2-ビス-(4-ヒドロキシフェニル)プロパン、2,2-ビス-(4-ヒドロキー3-メチルフェニル)プロパン、2,2-ビス-(3,5-ジメチル-4-ヒドロキシフェニル)プロパン、1,1-ビス-(4-ヒドロキシフェニル)シクロヘキサン、1,1-ビス-(4-ヒドロキー3-メチルフェニル)シクロヘキサン、1,1-ビス-(3,5-ジメチル-4-ヒドロキシフェニル)シクロヘキサン、1,1-ビス-(4-ヒドロキシフェニル)-1-フェニルエタン、4,4´-ビフェノール、3,3´-ジメチル-4,4´-ビフェノール、3,3´,5,5´-テトラメチル-4,4´-ビフェノール、4,4´-ジヒドロキシジフェニルエーテル、3,3´-ジメチル-4,4´-ジヒドロキシジフェニルエーテル、ビス(4-ヒドロキシフェニル)スルフィド、4,4´-ジヒドロキシベンゾフェノン等が挙げられる。
この中でも、二価フェノール成分の製造の簡便性及び溶解性、電気特性を考慮すれば、ビス-(4-ヒドロキシフェニル)メタン、ビス-(4-ヒドロキシ-3-メチルフェニル)メタン、ビス-(3,5-ジメチル-4-ヒドロキシフェニル)メタン、1,1-ビス-(4-ヒドロキシフェニル)エタン、1,1-ビス-(4-ヒドロキシ-3-メチルフェニル)エタン、2,2-ビス-(4-ヒドロキシフェニル)プロパン、2,2-ビス-(4-ヒドロキー3-メチルフェニル)プロパン、2,2-ビス-(3,5-ジメチル-4-ヒドロキシフェニル)プロパン、1,1-ビス-(4-ヒドロキシフェニル)シクロヘキサン、4,4´-ビフェノール、3,3´-ジメチル-4,4´-ビフェノール、3,3´,5,5´-テトラメチル-4,4´-ビフェノール、4,4´-ジヒドロキシジフェニルエーテルが好ましい。
さらに機械物性を考慮すれば、ビス-(4-ヒドロキシフェニル)メタン、ビス-(4-ヒドロキシ-3-メチルフェニル)メタン、1,1-ビス-(4-ヒドロキシフェニル)エタン、1,1-ビス-(4-ヒドロキシ-3-メチルフェニル)エタン、2,2-ビス-(4-ヒドロキー3-メチルフェニル)プロパン、4,4´-ビフェノール、1,1-ビス-(4-ヒドロキシフェニル)シクロヘキサンがより好ましい。
前記式(1)~(3)で表される2価フェノール残基の含有量は、ポリエステル樹脂中の全2価フェノール残基の合計量に対し、5モル%以上80モル%以下が好ましい。さらに、溶解性の観点から、より好ましくは60モル%以下、さらに好ましくは50モル%以下、特に好ましくは40モル%以下である。また、耐摩耗性の観点から、より好ましくは8モル%以上であり、さらに好ましくは10モル%以上である。
また、式(4)で表される2価フェノール残基の含有量は、全2価フェノール残基の合計量に対し、20モル%以上が好ましく、溶解性の観点から、40モル%以上がより好ましく、50モル%以上がさらに好ましく、60モル%以上が特に好ましい。また、95モル%以下が好ましく、耐摩耗性の観点から、92モル%以下がより好ましく、90モル%以下がさらに好ましい。
式(1)~(4)として例示した2価フェノール残基は、必要に応じて複数の化合物を組み合わせて用いることも可能である。また、式(1)~(4)以外の、2価アルコールと組み合わせることも可能である。式(1)~(4)以外の、2価アルコールの具体例としては、例えば、エチレングリコール、1,3-プロパンジオール、1,4-ブタンジオール、1,8-オクタンジオール、ポリエステルジオール、ポリカーボネートジオール、ポリテトラメチレングリコール、ポリシロキサン含有ジオール等が挙げられる。
前記ポリエステル樹脂の2価カルボン酸残基は、下記式(5)で表される2価カルボン酸残基であることが好ましい。

式(5)中、Ar3、Ar4は、それぞれ独立に置換基を有していてもよいアリーレン基を表す。Yは、単結合、酸素原子、硫黄原子、式(6)で表される構造、又は式(7)で表される構造を有する2価の有機残基を表す。式(6)中のR9及びR10は、それぞれ独立に、水素原子、アルキル基、若しくはアリール基、又はR9とR10とが結合して形成されるシクロアルキリデン基を表す。さらに、式(7)中、R11は、アルキレン基、アリーレン基、又は式(8)で表される基であり、式(8)中、R12及びR13は、それぞれ独立にアルキレン基を表し、Ar5はアリーレン基を表す。kは0~5の整数を表す。

式(5)中、Ar3、Ar4は、炭素数6~20のアリーレン基が好ましく、例えば、フェニレン基、ナフチレン基、アントリレン基、フェナントリレン基、ピレニレン基が挙げられる。中でも、製造コストの面から、フェニレン基、ナフチレン基、又はビフェニレン基がより好ましい。
前記アリーレン基がそれぞれ独立に有していてもよい置換基としては、例えば、水素原子、アルキル基、アルコキシ基、アリール基、縮合多環基、ハロゲン基が挙げられる。感光層用バインダー樹脂としての機械的特性と感光層形成用塗布液に対する溶解性を勘案すれば、アリール基としてフェニル基、ナフチル基が好ましく、ハロゲン基としてフッ素基、クロロ基、ブロモ基、ヨード基が好ましく、アルコキシ基としてメトキシ基、エトキシ基、ブトキシ基が好ましく、アルキル基としては、炭素数1~10のアルキル基が好ましく、炭素数1~8のアルキル基がさらに好ましく、炭素数1~2のアルキル基が特に好ましく、具体的にはメチル基が特に好ましい。
Ar3、Ar4それぞれの置換基の数に特に制限は無いが、3個以下であることが好ましく、2個以下であることがより好ましく、1個以下であることが特に好ましい。電気特性、溶解性の観点から、Ar3とAr4は同じ置換基を有する同じアリーレン基であることが好ましく、無置換のフェニレン基であることがより好ましい。
式(5)中、Yは、単結合、酸素原子、硫黄原子、式(6)で表される構造、又は式(7)で表される構造を有する2価の有機残基を表す。式(6)中、R9及びR10は、それぞれ独立に水素原子、アルキル基、若しくはアリール基、又はR9とR10とが結合して形成されるシクロアルキリデン基を表す。
R9及びR10のアルキル基としては、メチル基、エチル基、プロピル基、ブチル基等が挙げられ、アリール基としては、フェニル基、ナフチル基等が挙げられる。また、式(6)中のR9とR10とが結合して形成されるシクロアルキリデン基としては、シクロペンチリデン基、シクロヘキシリデン基、シクロヘプチリデン基等が挙げられる。
式(7)中、R11は、アルキレン基、アリーレン基、又は式(8)で表される基であって、式(8)中、R12及びR13は、それぞれ独立にアルキレン基を表し、Ar5はアリーレン基を表す。
式(7)中のR11のアルキレン基としては、メチレン基、エチレン基、プロピレン基等が挙げられ、式(7)中のR11のアリーレン基としては、フェニレン基、テルフェニレン基等が挙げられる。式(8)で表される基としては、具体的には下記式(9)で表される基等が挙げられる。
Yは、耐摩耗性の観点から、酸素原子であることが好ましい。
Yは、耐摩耗性の観点から、酸素原子であることが好ましい。

式(5)中、kは0~5の整数であるが、好ましくは0、1の整数であり、耐摩耗性の観点から1であることが特に好ましい。kが0の場合、2価カルボン酸残基を誘導する2価カルボン酸化合物の具体例としては、テレフタル酸、イソフタル酸が挙げられる。kが1である場合、式(5)は、下記一般式(10)であることが特に好ましい。

式(10)中、R14、R15は、それぞれ独立に水素原子、アルキル基、アリール基、ハロゲン基、又はアルコキシ基を表し、n3,n4は、それぞれ独立に0~4の整数である。式(10)中、R14、R15としては、例えば、水素原子;メチル基、エチル基、イソプロピル基等の炭素数1~6のアルキル基;フェニル基、ナフチル基等のアリール基;フッ素原子、塩素原子、臭素原子、ヨウ素原子等のハロゲン基;メトキシ基、エトキシ基、ブトキシ基等のアルコキシ基等が挙げられる。
式(10)で表される2価カルボン酸残基を誘導する2価カルボン酸化合物の製造上の簡便性を考慮すれば、R14、R15は、水素原子、メチル基が特に好ましい。n3,n4は、それぞれ独立に、0~4の整数であり、特に好ましくは、n3=n4=0である。
式(10)で表される2価カルボン酸残基を誘導する2価カルボン酸化合物の具体例としては、例えば、ジフェニルエーテル-2,2´-ジカルボン酸、ジフェニルエーテル-2,4´-ジカルボン酸、ジフェニルエーテル-4,4´-ジカルボン酸等が挙げられる。これらの中でも、製造上の簡便性を考慮すれば、ジフェニルエーテル-4,4´-ジカルボン酸が特に好ましい。
式(5)として例示した化合物は、必要に応じて複数の化合物を組み合わせて用いることも可能である。組み合わせてよい2価カルボン酸化合物の具体例としては、例えば、アジピン酸、スベリン酸、セバシン酸、フタル酸、イソフタル酸、テレフタル酸、トルエン-2,5-ジカルボン酸、p-キシレン-2,5-ジカルボン酸、ピリジン-2,3-ジカルボン酸、ピリジン-2,4-ジカルボン酸、ピリジン-2,5-ジカルボン酸、ピリジン-2,6-ジカルボン酸、ピリジン-3,4-ジカルボン酸、ピリジン-3,5-ジカルボン酸、ナフタレン-1,4-ジカルボン酸、ナフタレン-2,3-ジカルボン酸、ナフタレン-2,6-ジカルボン酸、ビフェニル-2,2´-ジカルボン酸、ビフェニル-4,4´-ジカルボン酸、ジフェニルエーテル-2,2´-ジカルボン酸、ジフェニルエーテル-2,3´-ジカルボン酸、ジフェニルエーテル-2,4´-ジカルボン酸、ジフェニルエーテル-3,3´-ジカルボン酸、ジフェニルエーテル-3,4´-ジカルボン酸、ジフェニルエーテル-4,4´-ジカルボン酸が挙げられる。ジカルボン酸成分の製造の簡便性を考慮すれば、イソフタル酸、テレフタル酸、ジフェニルエーテル-4,4´-ジカルボン酸が特に好ましい。
前記ポリエステル樹脂において、式(5)で表される2価カルボン酸残基の合計含有量は全2価カルボン酸成分に対し70モル%以上であることが好ましい。耐摩耗性の観点から、好ましくは90モル%以上であり、特に好ましくは100モル%である。
前記ポリエステル樹脂の粘度平均分子量(Mv)は、通常、10,000以上、機械的強度の観点から、好ましくは25,000以上、さらに好ましくは35,000以上である。また、通常、200,000以下、塗布性の観点から、好ましくは、150,000以下である。
前記ポリエステル樹脂の末端に存在するカルボン酸クロライド基量は、通常0.1μ当量/g以下、好ましくは0.05μ当量/g以下である。末端カルボン酸クロライド基量が上記範囲を超えると、ポリエステル樹脂を電子写真感光体用塗布液とした際の保存安定性が低下する傾向がある。
前記ポリエステル樹脂のカルボキシル酸価は、300μ当量/g以下とすることが好ましく、より好ましくは150μ当量/g以下である。カルボキシル酸価が150μ当量/gを超えると、感光体の電気的特性が悪化する傾向があり、さらに、樹脂を溶媒に溶解して塗工液としたときの保存安定性が低下する傾向がある。
前記ポリエステル樹脂の末端に存在するOH基量は、通常100μ当量/g以下、好ましくは50μ当量/g以下である。末端OH基量が上記範囲を超えると、ポリエステル樹脂を電子写真感光体とした際の電気特性が悪化する可能性がある。
前記ポリエステル樹脂に含まれる全窒素量(T-N量)は、3000ppm以下が好ましく、1500ppm以下がより好ましく、1000ppm以下がさらに好ましく、500pp以下であることが特に好ましく、300ppm以下であることが最も好ましい。全窒素量が3000ppmを超えると電気特性が悪化する場合がある。
前記ポリエステル樹脂に含まれる遊離の2価カルボン酸量は、その量は特に限定されないが、50ppm以下であることが好ましく、10ppm以下であることがより好ましい。遊離の2価カルボン酸含有量が50ppmを超える場合、感光体の電気特性が悪化したり、画像評価に現れる異物となったりする場合がある。感光体の電気特性・画像特性の観点からは、遊離の2価カルボン酸量は少ないほどよいが、ポリエステル樹脂の安定性の観点からは、0.01ppm以上であることが好ましく、0.1ppm以上であることが特に好ましい。
前記ポリエステル樹脂に含有される遊離の2価フェノールについては、その量は特に限定されないが、100ppm以下が好ましい、50ppm以下が更に好ましい。100ppmを超えると電気特性の悪化や着色、使用する溶媒によっては異物が生じる場合がある。感光体の電気特性の観点からは、遊離の2価フェノールは少ないほどよいが、製造の簡便性からは、0.001ppm以上であることが好ましく、0.01ppm以上であることが特に好ましい。
≪第2の実施形態のポリエステル樹脂≫
本発明の製造方法が適用されるポリエステル樹脂は、下記式(1a)で表される繰返し単位及び下記式(2a)で表される繰返し単位を有している。
本発明の製造方法が適用されるポリエステル樹脂は、下記式(1a)で表される繰返し単位及び下記式(2a)で表される繰返し単位を有している。


式(1a)中、X1aは、下記式(3a)で表される2価の基であり、Y1aは、2価の基である。式(2a)中、X2aは、2価の芳香族を含む基である。Y2aは、2価の基である。但し、X2aはX1aとは同一ではない。

式(3a)中、R1a、R2aは、それぞれ独立に、水素原子、炭素数1~20の置換基を有していてもよいアルキル基、炭素数1~20の置換基を有していてもよいアルコキシ基、又は炭素数1~20の置換基を有していてもよい芳香族基を表す。炭素数は、置換基を含めた基全体の炭素数である。R1a、R2aの置換基としては、アルキル基、アルコキシ基、ハロゲン原子等が挙げられ、置換基を有さないことが好ましい。
アルキル基の具体例としては、メチル基、エチル基、n-プロピル基、イソプロピル基、n-ブチル基、イソブチル基,tert-ブチル基、シクロヘキシル基等が挙げられる。アルコキシ基の具体例としては、メトキシ基、エトキシ基、n-プロポキシ基、n-ブトキシ基等が挙げられる。置換基を有していてもよい芳香族基の具体例としては、フェニル基、ナフチル基、4-メチルフェニル基等が挙げられる。これらの中でも、溶解性、耐摩耗性、製造上の簡便性から、メチル基、エチル基が好ましい。
n1a、n2aは、それぞれ0~4の整数である。耐摩耗性の観点から、好ましくは、0~2の整数であり、特に好ましくは0又は1である。
R3a、R4aはそれぞれ独立に、水素原子、炭素数1~20のアルキル基、又は炭素数1~20の置換基を有していてもよい芳香族基を表す。但し、R3aとR4aが互いに結合して環を形成してもよい。炭素数は、置換基を含めた基全体の炭素数である。
R3a、R4aの置換基としては、R1a、R2aの置換基として挙げたものが適用できる。アルキル基の具体例としては、メチル基、エチル基、n-プロピル基、イソプロピル基、n-ブチル基、イソブチル基等が挙げられる。置換基を有していてもよい芳香族基の具体例として、フェニル基、ナフチル基、ビフェニル基等が挙げられる。R3aとR4aが互いに結合して環を形成する場合の具体例として、シクロペンチリデン基、シクロヘキシリデン基、1~3個のメチル基で置換されたシクロヘキシリデン基等が挙げられる。R3a、R4aは、溶解性、耐摩耗性、製造上の簡便性から、水素原子、メチル基、エチル基、又はシクロヘキシリデン基が好ましい。
式(3a)で表される2価の基の元となるビスフェノールとして具体的に例示すると、ビス-(4-ヒドロキシフェニル)メタン、ビス-(4-ヒドロキシ-3-メチルフェニル)メタン、ビス-(3,5-ジメチル-4-ヒドロキシフェニル)メタン、1,1-ビス-(4-ヒドロキシフェニル)エタン、1,1-ビス-(4-ヒドロキシ-3-メチルフェニル)エタン、1,1-ビス-(3,5-ジメチル-4-ヒドロキシフェニル)エタン、1,1-ビス-(4-ヒドロキシフェニル)プロパン、1,1-ビス-(4-ヒドロキー3-メチルフェニル)プロパン、1,1-ビス-(3,5-ジメチル-4-ヒドロキシフェニル)プロパン、2,2-ビス-(4-ヒドロキシフェニル)プロパン、2,2-ビス-(4-ヒドロキー3-メチルフェニル)プロパン、2,2-ビス-(3,5-ジメチル-4-ヒドロキシフェニル)プロパン、1,1-ビス-(4-ヒドロキシフェニル)シクロヘキサン、1,1-ビス-(4-ヒドロキー3-メチルフェニル)シクロヘキサン、1,1-ビス-(3,5-ジメチル-4-ヒドロキシフェニル)シクロヘキサン、1,1-ビス-(4-ヒドロキシフェニル)-1-フェニルエタン等が挙げられる。
この中でも、製造の簡便性及び溶解性、電気特性を考慮すれば、ビス-(4-ヒドロキシフェニル)メタン、ビス-(4-ヒドロキシ-3-メチルフェニル)メタン、ビス-(3,5-ジメチル-4-ヒドロキシフェニル)メタン、1,1-ビス-(4-ヒドロキシフェニル)エタン、1,1-ビス-(4-ヒドロキシ-3-メチルフェニル)エタン、2,2-ビス-(4-ヒドロキシフェニル)プロパン、2,2-ビス-(4-ヒドロキシ-3-メチルフェニル)プロパン、2,2-ビス-(3,5-ジメチル-4-ヒドロキシフェニル)プロパン、1,1-ビス-(4-ヒドロキシフェニル)シクロヘキサンが好ましい。更に機械物性を考慮すれば、ビス-(4-ヒドロキシフェニル)メタン、ビス-(4-ヒドロキシ-3-メチルフェニル)メタン、1,1-ビス-(4-ヒドロキシフェニル)エタン、1,1-ビス-(4-ヒドロキシ-3-メチルフェニル)エタン、2,2-ビス-(4-ヒドロキー3-メチルフェニル)プロパン、1,1-ビス-(4-ヒドロキシフェニル)シクロヘキサンがより好ましい。
前記式(2a)中のX2aは、X1aとは同一ではない2価の芳香族を含む基である。2価の芳香族を含む基としては、前記式(3a)のような2価の基や下記式(7a)~(10a)の2価の基等が挙げられる。耐刷性の観点から、下記式(7a)~(10a)から選ばれる少なくとも1種の2価の芳香族を含む基が好ましい。

式(7a)中、R5aは水素原子、炭素数1~20の置換基を有していてもよいアルキル基、炭素数1~20の置換基を有していてもよいアルコキシ基、炭素数1~20の置換基を有していてもよい芳香族基、又はハロゲン基を表す。炭素数は、置換基を含めた基全体の炭素数である。アルキル基の具体例としては、メチル基、エチル基、n-プロピル基、イソプロピル基、n-ブチル基、イソブチル基,tert-ブチル基、シクロヘキシル基等が挙げられる。
アルコキシ基の具体例としては、メトキシ基、エトキシ基、n-プロポキシ基、n-ブトキシ基等が挙げられる。置換基を有していてもよい芳香族基の具体例としては、フェニル基、ナフチル基、メチル置換されたフェニル基等が挙げられる。ハロゲン基としては、フルオロ基、クロロ基、ブロモ基が挙げられる。溶解性、耐摩耗性、製造上の簡便性から、メチル基、フェニル基、フルオロ基、クロロ基が好ましい。n3aは0~4の整数である。耐刷性の観点から、好ましくは、0~2の整数であり、特に好ましくは0である。式(7a)で表される2価の基の元となる2価フェノールとして具体的に例表すると、ハイドロキノン、メチルハイドロキノン、クロロハイドロキノン、フルオロハイドロキノン、レゾルシノール、カテコール等が挙げられる。耐刷性の観点から、ハイドロキノンが好ましい。

式(8a)中、R6aは水素原子、炭素数1~20の置換基を有していてもよいアルキル基、炭素数1~20の置換基を有していてもよいアルコキシ基、炭素数1~20の置換基を有していてもよい芳香族基、又はハロゲン基を表す。R6aの具体例としては、前記R5aと同等のものが挙げられる。
n4aは0~6の整数である。耐刷性の観点から、好ましくは、0~2の整数、特に好ましくは0である。
式(8a)で表される2価の基の元となる2価フェノールとして具体的に例表すると、1,6-ジヒドロキシナフタレン、1,7-ジヒドロキシナフタレン、1,4-ジヒドロキシナフタレン、2,6-ジヒドロキシナフタレン、2,7-ジヒドロキシナフタレン等が挙げられる。耐刷性の観点から、2,6-ジヒドロキシナフタレン、2,7-ジヒドロキシナフタレンが好ましい。

式(9a)中、R7a、R8aは、それぞれ独立に炭素数1~20の置換基を有していてもよいアルキル基、炭素数1~20の置換基を有していてもよいアルコキシ基、炭素数1~20の置換基を有していてもよい芳香族基、又はハロゲン基を表す。R7a、R8aの具体例としては、前記R5aと同等のものが挙げられる。
n5a、n6aは0~4の整数である。耐刷性の観点から、好ましくは、0~2の整数であり、特に好ましくは0又は1である。W1は、単結合、酸素、又は硫黄を表す。
式(9a)で表される2価の基の元となる2価フェノールとして具体的に例示すると、4,4´-ビフェノール、4,4´-ジヒドロキシ-3,3´-ジメチルビフェニル、4,4´-ジヒドロキシ-3,3´,5,5´-テトラメチルビフェニル、4,4´-ジヒドロキシ-2,2´,3,3´,5,5´-ヘキサメチルビフェニル、4,4´-ジヒドロキシジフェニルエーテル、4,4´-ジヒドロキシ-3,3´-ジメチルジフェニルエーテル、4,4´-ジヒドロキシ-3,3´,5,5´-テトラメチルジフェニルエーテル、4,4´-ジヒドロキシジフェニルスルフィド、4,4´-ジヒドロキシ-3,3´-ジメチルジフェニルスルフィド、4,4´-ジヒドロキシ-3,3´,5,5´-テトラメチルジフェニルスルフィド等が挙げられる。耐刷性及び入手容易性の観点から、4,4´-ビフェノール、4,4´-ジヒドロキシ-3,3´-ジメチルビフェニル、4,4´-ジヒドロキシ-3,3´,5,5´-テトラメチルビフェニル、4,4´-ジヒドロキシジフェニルエーテルが好ましい。

式(10a)中、Ar1a、Ar2aは、それぞれ独立に炭素数6~16の置換基を有していてもよいアリーレン基を表す。Zは、置換基を有していてもよいアリーレン基、又は置換基を有していてもよいアルキレン基を表し、saは0又は1の整数である。
式(10a)中、Ar1a、Ar2aにおけるアリーレン基の具体例としては、フェニレン基、ナフチレン基、アントリレン基、フェナントリレン基、ピレニレン基、ビフェニレン基等が挙げられる。製造上の簡便性から、フェニレン基が好ましい。
アリーレン基の置換基としては、炭素数1~10のアルキル基、炭素数1~10のアルコキシ基、ハロゲン化アルキル基、ハロゲン基、ベンジル基等が挙げられる。アルキル基の具体例としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、tert-ブチル基、イソブチル基、シクロヘキシル基等が挙げられる。アルコキシ基の具体例としては、メトキシ基、エトキシ基、プロポキシ基、シクロヘキソキシ基等が挙げられる。ハロゲン化アルキル基としては、クロロメチル基、フッ化アルキル基等が挙げられる。ハロゲン基としては、フッ素基、クロロ基、ブロモ基等が挙げられる。耐摩耗性の観点から、好ましくは、炭素数1~6のアルキル基、炭素数1~6のアルコキシ基であり、特に好ましくはメチル基、エチル基である。
式(10a)中、Zにおける置換基を有していてもよいアリーレン基の具体例としては、p-フェニレン基、m-フェニレン基、又は下記式(11)で表される構造等が挙げられる。置換基を有していてもよいアルキレン基の具体例としては、エチレン基、プロピレン基、シクロへキシレン基、1,4-ジメチルシクロヘキサン基等が挙げられる。製造上の簡便性から、p-フェニレン基、又は、第1の実施形態でも記した、下記式(11)で表される構造、エチリデンが好ましい。

式(11)中、R16~R19は、それぞれ独立に水素原子、又は炭素数1~10のアルキル基を表す。
式(11)中のアルキル基の具体例としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、tert-ブチル基、イソブチル基、シクロヘキシル基等が挙げられる。溶解性、耐摩耗性、製造上の簡便性から、R16~R19は、水素原子又はメチル基が好ましい。
式(11)中のアルキル基の具体例としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、tert-ブチル基、イソブチル基、シクロヘキシル基等が挙げられる。溶解性、耐摩耗性、製造上の簡便性から、R16~R19は、水素原子又はメチル基が好ましい。
式(10a)中、saは、耐摩耗性の観点から0又は1の整数が好ましく、溶解性の観点から0が更に好ましい。
式(10a)で表される2価の基の元となる2価フェノールの具体例として以下に表す。
式(10a)で表される2価の基の元となる2価フェノールの具体例として以下に表す。

また、下記式(4a1)及び式(4a2)は、式(1a)及び式(2a)に対応する2価フェノールでありX1a及びX2aは前述の通りである。


前記式(1a)及び(2a)中のY1a、Y2aは、下記式(11a)で表される2価の基であることが好ましい。Y1a、Y2aは同一であることが好ましい。

Ar3a、Ar4aは、それぞれ独立に置換基を有していてもよいアリーレン基を表す。W2は、単結合、酸素原子、硫黄原子、式(12a)で表される構造を有する2価の有機残基、又は式(13a)で表される構造を有する2価の有機残基を表す。式(12a)中のR9a及びR10aは、それぞれ独立に、水素原子、アルキル基、若しくはアリール基、又はR9aとR10aとが結合して形成されるシクロアルキリデン基を表す。更に、式(13a)中、R11aは、アルキレン基、アリーレン基、又は式(14a)で表される基を表す。式(14a)中、R12a及びR13aは、それぞれ独立にアルキレン基を表し、Ar5aはアリーレン基を表す。kaは0~5の整数を表す。


式(11a)中、Ar3a、Ar4aは、炭素数6~20のアリーレン基が好ましく、例えば、フェニレン基、ナフチレン基、ビフェニレン基、アントリレン基、フェナントリレン基、ピレニレン基が挙げられる。中でも、製造コストの面から、フェニレン基、ナフチレン基、又はビフェニレン基がより好ましい。
前記アリーレン基がそれぞれ独立に有していてもよい置換基としては、例えば、アルキル基、アルコキシ基、アリール基、縮合多環基、ハロゲン基が挙げられる。感光層用バインダー樹脂としての機械的特性と感光層形成用塗布液に対する溶解性を勘案すれば、アリール基としてフェニル基、ナフチル基が好ましく、ハロゲン基としてフッ素基、クロロ基、ブロモ基、ヨード基が好ましく、アルコキシ基としてメトキシ基、エトキシ基、ブトキシ基が好ましく、アルキル基としては、炭素数1~10のアルキル基が好ましく、炭素数1~8のアルキル基が更に好ましく、炭素数1~2のアルキル基が特に好ましく、具体的にはメチル基が特に好ましい。
Ar3a、Ar4aそれぞれの置換基の数に特に制限は無いが、3個以下であることが好ましく、2個以下であることがより好ましく、1個以下であることが特に好ましい。耐摩耗性、製造の簡便性の観点から、Ar3aとAr4aは同じ置換基を有する同じアリーレン基であることが好ましく、無置換のフェニレン基であることがより好ましい。
式(12a)中、R9a及びR10aは、それぞれ独立に水素原子、アルキル基、若しくはアリール基、又はR9aとR10aとが結合して形成されるシクロアルキリデン基を表す。R9a及びR10aのアルキル基としては、メチル基、エチル基、プロピル基、ブチル基等が挙げられ、アリール基としては、フェニル基、ナフチル基等が挙げられる。
また、式(12a)中のR9aとR10aとが結合して形成されるシクロアルキリデン基としては、シクロペンチリデン基、シクロヘキシリデン基、シクロヘプチリデン基等が挙げられる。式(13a)中、R11aは、アルキレン基、アリーレン基、又は式(14a)で表される基であって、式(14a)中、R12a及びR13aは、それぞれ独立にアルキレン基を表し、Ar5aはアリーレン基を表す。式(13a)中のR11aのアルキレン基としては、メチレン基、エチレン基、プロピレン基等が挙げられ、R11aのアリーレン基としては、フェニレン基、テルフェニレン基等が挙げられる。式(14a)で表される基としては、具体的には下記式(15a)で表される基等が挙げられる。W2は、耐摩耗性の観点から、酸素原子であることが好ましい。

式(11a)中、kaは0~5の整数である。耐摩耗性の観点から、好ましくは0又は1の整数であり、1であることが特に好ましい。kaが0の場合、式(11a)で表される2価の基の元となる2価カルボン酸の具体例としては、テレフタル酸、イソフタル酸等が挙げられる。kaが1である場合、式(11a)は、下記一般式(16a)であることが特に好ましい。

式(16a)中、R18a、R19aは、それぞれ独立に水素原子、アルキル基、アリール基、ハロゲン基、又はアルコキシ基を表し、n7,mは、それぞれ独立に0~4の整数である。式(16a)中、R18a、R19aとしては、例えば、水素原子;メチル基、エチル基、イソプロピル基等の炭素数1~6のアルキル基;フェニル基、ナフチル基等のアリール基;フッ素原子、塩素原子、臭素原子、ヨウ素原子等のハロゲン基;メトキシ基、エトキシ基、ブトキシ基等のアルコキシ基等が挙げられる。
式(16a)で表される2価の基を誘導する2価カルボン酸化合物の製造上の簡便性を考慮すれば、R18a、R19aは、水素原子、メチル基が特に好ましい。n7,mはそれぞれ独立に、0~4の整数であり、特に好ましくは、n7=m=0である。式(16a)で表される2価の基を誘導する2価カルボン酸化合物の具体例としては、例えば、ジフェニルエーテル-2,2´-ジカルボン酸、ジフェニルエーテル-2,4´-ジカルボン酸、ジフェニルエーテル-4,4´-ジカルボン酸等が挙げられる。これらの中でも、製造上の簡便性を考慮すれば、ジフェニルエーテル-4,4´-ジカルボン酸が特に好ましい。
ジカルボン酸残基は、必要に応じて複数の化合物を組み合わせて用いることも可能である。組み合わせてよい2価カルボン酸化合物の具体例としては、例えば、アジピン酸、スベリン酸、セバシン酸、フタル酸、イソフタル酸、テレフタル酸、トルエン-2,5-ジカルボン酸、p-キシレン-2,5-ジカルボン酸、ピリジン-2,3-ジカルボン酸、ピリジン-2,4-ジカルボン酸、ピリジン-2,5-ジカルボン酸、ピリジン-2,6-ジカルボン酸、ピリジン-3,4-ジカルボン酸、ピリジン-3,5-ジカルボン酸、ナフタレン-1,4-ジカルボン酸、ナフタレン-2,3-ジカルボン酸、ナフタレン-2,6-ジカルボン酸、ビフェニル-2,2´-ジカルボン酸、ビフェニル-4,4´-ジカルボン酸、ジフェニルエーテル-2,2´-ジカルボン酸、ジフェニルエーテル-2,3´-ジカルボン酸、ジフェニルエーテル-2,4´-ジカルボン酸、ジフェニルエーテル-3,3´-ジカルボン酸、ジフェニルエーテル-3,4´-ジカルボン酸、ジフェニルエーテル-4,4´-ジカルボン酸が挙げられる。ジカルボン酸成分の製造の簡便性を考慮すれば、イソフタル酸、テレフタル酸、ジフェニルエーテル-4,4´-ジカルボン酸が特に好ましい。
また、下記式(5a)は、Y1aに対応する2価カルボン酸化合物であり、Y1aは前述の通りである。

ポリエステル樹脂中の前記式(2a)で表される繰返し単位の含有量は、前記式(1a)で表される繰返し単位とのモル比率で0.45≧(2a)/((1a)+(2a))≧0.05であることが好ましい。更に、ポリエステル樹脂の溶解性の観点及び耐摩耗性の観点から、好ましくは0.40≧(2a)/((1a)+(2a))≧0.08であり、更に好ましくは、0.35≧(2a)/((1a)+(2a))≧0.10である。
ポリエステル樹脂の粘度平均分子量(Mv)は、通常、10,000以上、機械的強度の観点から、好ましくは20,000以上である。また、通常、200,000以下、塗布性の観点から、好ましくは、150,000以下である。
前記ポリエステル樹脂の末端に存在するカルボン酸クロライド基量は、通常0.1μ当量/g以下、好ましくは0.05μ当量/g以下である。末端カルボン酸クロライド基量が前記範囲を超えると、ポリエステル樹脂を電子写真感光体用塗布液とした際の保存安定性が低下する傾向がある。
前記ポリエステル樹脂のカルボキシル酸価は、300μ当量/g以下とすることが好ましく、より好ましくは150μ当量/g以下である。カルボキシル酸価が300μ当量/gを超えると、感光体の電気的特性が悪化する傾向があり、更に、樹脂を溶媒に溶解して塗工液としたときの保存安定性が低下する傾向がある。
前記ポリエステル樹脂の末端に存在するOH基量は、通常200μ当量/g以下、好ましくは100μ当量/g以下である。末端OH基量が前記範囲を超えると、ポリエステル樹脂を電子写真感光体とした際の電気特性が悪化する可能性がある。
前記ポリエステル樹脂に含まれる全窒素量(T-N量)は、500ppm以下が好ましく、300ppm以下であることが更に好ましく、150ppm以下であることが特に好ましい。全窒素量が500ppmを超えると電気特性が悪化する場合がある。
前記ポリエステル樹脂に含まれる遊離の2価カルボン酸量は、その量は特に限定されないが、50ppm以下であることが好ましく、10ppm以下であることがより好ましい。遊離の2価カルボン酸含有量が50ppmを超える場合、感光体の電気特性が悪化したり、画像評価に現れる異物となったりする場合がある。感光体の電気特性・画像特性の観点からは、遊離の2価カルボン酸量は少ないほどよいが、ポリエステル樹脂の安定性の観点からは、0.01ppm以上であることが好ましく、0.1ppm以上であることが特に好ましい。
前記ポリエステル樹脂に含有される遊離の2価フェノールについては、その量は特に限定されないが、100ppm以下が好ましく、50ppm以下が更に好ましい。100ppmを超えると電気特性の悪化や着色、使用する溶媒によっては異物が生じる場合がある。感光体の電気特性の観点からは、遊離の2価フェノールは少ないほどよいが、製造の簡便性からは、0.001ppm以上であることが好ましく、0.01ppm以上であることが特に好ましい。
≪第3の実施形態のポリエステル樹脂≫
本発明により製造されるポリエステル樹脂の構造は、特に限定されないが、溶解性、機械物性、電気特性等の観点から以下に示す構造を有するポリエステル樹脂が好ましい。少なくとも2種類のジカルボン酸クロライドを使用するか、少なくとも2種類の2価フェノール又は2価アルコールを使用する場合に、上記ポリエステル樹脂の製造方法が有効である。ここで言う種類とは、分子構造で区別するものであり、位置異性体、立体異性体等も区別して1種類とする。
本発明により製造されるポリエステル樹脂の構造は、特に限定されないが、溶解性、機械物性、電気特性等の観点から以下に示す構造を有するポリエステル樹脂が好ましい。少なくとも2種類のジカルボン酸クロライドを使用するか、少なくとも2種類の2価フェノール又は2価アルコールを使用する場合に、上記ポリエステル樹脂の製造方法が有効である。ここで言う種類とは、分子構造で区別するものであり、位置異性体、立体異性体等も区別して1種類とする。
(2価カルボン酸残基)
ポリエステル樹脂として下記式(1b)~(4b)で表される2価カルボン酸残基を少なくとも1つ有することが好ましい。
ポリエステル樹脂として下記式(1b)~(4b)で表される2価カルボン酸残基を少なくとも1つ有することが好ましい。

式(1b)中、R1bはそれぞれ独立に、炭素数1~10のアルキル基、炭素数1~10のアルコキシ基、ハロゲン化アルキル基、ハロゲン基、又はベンジル基を表し、nbは、0~4の整数である。
アルキル基の具体例としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、tert-ブチル基、イソブチル基、シクロヘキシル基等が挙げられる。アルコキシ基の具体例としては、メトキシ基、エトキシ基、プロポキシ基、シクロヘキソキシ基等が挙げられる。
ハロゲン化アルキル基としては、クロロメチル基、フッ化アルキル基等が挙げられる。ハロゲン基としては、フッ素基、クロロ基、ブロモ基等が挙げられる。耐摩耗性の観点から、好ましくは、炭素数1~6のアルキル基、炭素数1~6のアルコキシ基であり、特に好ましくはメチル基、エチル基である。
nbは各々独立に、0~4の整数であり、製造上の簡便性の観点から、好ましくは、nb=0である。式(1b)で表される2価カルボン酸残基を誘導する2価カルボン酸化合物の具体例としては、例えば、オルトフタル酸、イソフタル酸、テレフタル酸等が挙げられる。これらの中でも、耐摩耗性の観点から、イソフタル酸、テレフタル酸が特に好ましい。

式(2b)中、R2b、R3bはそれぞれ独立に、炭素数1~10のアルキル基、炭素数1~10のアルコキシ基、ハロゲン化アルキル基、ハロゲン基、又はベンジル基を表し、mb、lbは、0~4の整数であり、Xbは単結合又は酸素原子を表す。アルキル基の具体例としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、tert-ブチル基、イソブチル基、シクロヘキシル基等が挙げられる。
アルコキシ基の具体例としては、メトキシ基、エトキシ基、プロポキシ基、シクロヘキソキシ基等が挙げられる。ハロゲン化アルキル基としては、クロロメチル基、フッ化アルキル基等が挙げられる。ハロゲン基としては、フッ素基、クロロ基、ブロモ基が挙げられる。耐摩耗性の観点から、好ましくは、炭素数1~6のアルキル基、炭素数1~6のアルコキシ基であり、特に好ましくはメチル基、エチル基である。mb、lbは各々独立に、0~4の整数であり、製造上の簡便性の観点から、好ましくは、mb=lb=0である。
式(2b)で表される2価カルボン酸残基を誘導する2価カルボン酸化合物の具体例としては、例えば、ジフェニルエーテル-2,2´-ジカルボン酸、ジフェニルエーテル-2,4´-ジカルボン酸、ジフェニルエーテル-4,4´-ジカルボン酸、4,4´-ジフェニルジカルボン酸、2,2´-ジフェニルジカルボン酸、2,4´-ジフェニルジカルボン酸等が挙げられる。これらの中でも、製造上の簡便性及び耐摩耗性の観点から、ジフェニルエーテル-4,4´-ジカルボン酸、4,4´-ジフェニルジカルボン酸が特に好ましい。

式(3b)中、R4bはそれぞれ独立に、炭素数1~10のアルキル基を表す。アルキル基の具体例としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、tert-ブチル基、イソブチル基、シクロヘキシル基等が挙げられる。耐摩耗性の観点から、好ましくは、炭素数1~6のアルキル基であり、特に好ましくはメチル基、エチル基である。pbは各々独立に、0~10の整数であり、製造上の簡便性の観点から、好ましくは、pb=0である。
式(3b)で表される2価カルボン酸残基を誘導する2価カルボン酸化合物の具体例としては、例えば、trans-1,4-シクロヘキサンジカルボン酸、cis-1,4-シクロヘキサンジカルボン酸、trans-1,3-シクロヘキサンジカルボン酸、cis-1,3-シクロヘキサンジカルボン酸等が挙げられる。これらの中でも、製造上の簡便性及び耐摩耗性の観点から、trans-1,4-シクロヘキサンジカルボン酸、cis-1,4-シクロヘキサンジカルボン酸が特に好ましい。

式(4b)中、Ybは単結合、炭素数1~14のアルキレン基、p-フェニレン基、m-フェニレン基、又は4,4’-ビフェニル基を表す。アルキレン基の具体例としては、メチレン基、エチレン基、プロピレン基、ブチレン基、2,2-ジメチルプロピレン基、シクロヘキシレン基、シクロヘキサンジメチル基等が挙げられる。式(4b)で表される2価カルボン酸残基を誘導する2価カルボン酸化合物の具体例としては、マロン酸、こはく酸、2,2-ジメチルこはく酸、グルタン酸、アゼライン酸、1,3-アダマンタンジカルボン酸、セバシン酸、1,10-デカンジカルボン酸、1,4-フェニレンジ酢酸等が挙げられる。
必要に応じて上記ジカルボン酸残基を組み合わせて用いることも可能である。特に、式(3b)、式(4b)のジカルボン酸残基をポリエステル樹脂に含む場合は、耐摩耗性、溶解性等の観点から、上記式(1b)、(2b)のジカルボン酸残基と共重合することが好ましい。
(2価フェノール残基、2価脂肪族アルコール残基)
ポリエステル樹脂として下記式(5b)~(12b)で表される2価フェノール及び/又は2価アルコールを少なくとも1つ有することが好ましい。
ポリエステル樹脂として下記式(5b)~(12b)で表される2価フェノール及び/又は2価アルコールを少なくとも1つ有することが好ましい。

式(5b)中、R5bはそれぞれ独立に、炭素数1~10のアルキル基、炭素数1~10のアルコキシ基、ハロゲン化アルキル基、ハロゲン基、又はベンジル基を表し、qbは、0~4の整数である。
アルキル基の具体例としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、tert-ブチル基、イソブチル基、シクロヘキシル基等が挙げられる。アルコキシ基の具体例としては、メトキシ基、エトキシ基、プロポキシ基、シクロヘキソキシ基等が挙げられる。ハロゲン化アルキル基としては、クロロメチル基、フッ化アルキル基等が挙げられる。ハロゲン基としては、フッ素基、クロロ基、ブロモ基が挙げられる。
耐摩耗性の観点から、好ましくは、炭素数1~6のアルキル基、炭素数1~6のアルコキシ基であり、特に好ましくはメチル基、エチル基である。qbは各々独立に、0~4の整数であり、製造上の簡便性の観点から、好ましくは、qb=0、1である。
式(5b)で表される2価フェノール残基を誘導する2価フェノール化合物の具体例としては、例えば、ヒドロキノン、メチルヒドロキノン、ブロモヒドロキノン、2,3-ジメチルヒドロキノン、トリメチルヒドロキノン、レソルシノール、2-メチルレソルシノール、5-メチルレソルシノール、5-ブロモレソルシノール、カテコール、4-メチルカテコール等が挙げられる。これらの中でも、製造上の簡便性及び耐摩耗性の観点から、ヒドロキノン、メチルヒドロキノン、レソルシノールが特に好ましい。

式(6b)中、R6bはそれぞれ独立に、炭素数1~10のアルキル基、炭素数1~10のアルコキシ基、ハロゲン化アルキル基、ハロゲン基、又はベンジル基を表し、rbは、0~6の整数である。
アルキル基の具体例としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、tert-ブチル基、イソブチル基、シクロヘキシル基等が挙げられる。アルコキシ基の具体例としては、メトキシ基、エトキシ基、プロポキシ基、シクロヘキソキシ基等が挙げられる。
ハロゲン化アルキル基としては、クロロメチル基、フッ化アルキル基等が挙げられる。ハロゲン基としては、フッ素基、クロロ基、ブロモ基が挙げられる。耐摩耗性の観点から、好ましくは、炭素数1~6のアルキル基、炭素数1~6のアルコキシ基であり、特に好ましくはメチル基、エチル基である。rbは各々独立に、0~4の整数であり、製造上の簡便性の観点から、好ましくは、rb=0である。
式(6b)で表される2価フェノール残基を誘導する2価フェノール化合物の具体例としては、例えば、1,2-ジヒドロキシナフタレン、1,3-ジヒドロキシナフタレン、1,5-ジヒドロキシナフタレン、1,6-ジヒドロキシナフタレン、1,7-ジヒドロキシナフタレン、2,3-ジヒドロキシナフタレン、2,6-ジヒドロキシナフタレン、2,7-ジヒドロキシナフタレン等が挙げられる。
これらの中でも、製造上の簡便性、溶解性及び耐摩耗性の観点から、1,6-ジヒドロキシナフタレン、1,7-ジヒドロキシナフタレン、2,6-ジヒドロキシナフタレン、2,7-ジヒドロキシナフタレンが特に好ましい。

式(7b)中、Ar1b、Ar2bはそれぞれ独立に、p-フェニレン基、m-フェニレン基、2価のナフタレン基、又は2価のビフェニル基、Zbは炭素数1~10のアルキレン基、p-フェニレン基、又は下記式(12b)を表し、sbは0又は1の整数である。

式(12b)中、R15b~R18bはそれぞれ独立に、水素原子、又は炭素数1~10のアルキル基を表す。
式(7b)中のアルキレン基の具体例としては、メチレン基、エチレン基、プロピレン基、ブチレン基、2,2-ジメチルプロピレン基、シクロヘキシレン基、シクロヘキサンジメチル基等が挙げられる。溶解性、耐摩耗性の観点から、好ましくは、炭素数1~8のアルキル基であり、特に好ましくはエチレン基、シクロヘキシレン基、シクロヘキサンジメチル基である。
式(12b)中のアルキル基の具体例としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、tert-ブチル基、イソブチル基、シクロヘキシル基等が挙げられる。溶解性、耐摩耗性、製造上の簡便性から、メチル基が好ましい。
式(7b)で表される2価フェノール残基を誘導する2価フェノール化合物の具体例として以下に示す。
式(7b)で表される2価フェノール残基を誘導する2価フェノール化合物の具体例として以下に示す。


アルキレン基の具体例としては、メチレン基、エチレン基、プロピレン基、ブチレン基、へキシレン基、シクロへキシレン基、オクチレン基等が挙げられる。パーフルオロアルキレン基の具体例としては、ヘキサフルオロ-1,3-プロピレン基、オクタフルオロ-1,4-ブチレン基、ドデカフルオロ-1,6-ヘキシレン基、ヘキサデカフルオロ-1,8-オクチレン基等が挙げられる。溶解性耐摩耗性の観点から、好ましくは、炭素数1~6のアルキレン基、炭素数3~8のパーフルオロアルキレン基である。
式(8b)で表される2価アルコール残基を誘導する2価アルコール化合物の具体例としては、例えば、エチレングリコール、1,3-プロピレングリコール、1,4-ブタンジオール、1,5-ペンタンジオール、1,3-シクロペンタンジメタノール、1,6-ヘキサンジオール、1,4-シクロヘキサンジメタノール、1,8-オクタンジオール、1,10-デカンジオール、1,12-ドデカンジオール、1,16-ヘキサデカンジオール、1,4-ベンゼンジメタノール、4,4’-ビフェニルジメタノール、2,2,3,3,4,4-ヘキサフルオロ-1,5-ペンタンジオール、2,2,3,3,4,4,5,5,6,6,7,7-ドデカフルオロ-1,8-オクタンジオール、1H,1H,10H,10H-ヘキサデカフルオロ-1,10-デカンジオール等が挙げられる。
これらの中でも、製造上の簡便性、溶解性及び耐摩耗性の観点から、エチレングリコール、1,4-ブタンジオール、1,4-シクロヘキサンジメタノール、1,8-オクタンジオール、1,4-ベンゼンジメタノール、4,4’-ビフェニルジメタノールが特に好ましい。

式(9b)中、R7b、R8bはそれぞれ独立に、炭素数1~10のアルキル基、又は炭素数1~10のアルコキシ基を表し、tb、ubは、それぞれ0~10の整数である。
Abは、単結合、-CR9bR10b-を示す。R9b、R10bはそれぞれ独立に、水素原子、炭素数1~20のアルキル基、又はR9bとR10bが互いに結合して置換基を有していてもよい環を形成する基を表す。vbは、0又は1の整数である。
アルキル基の具体例としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、tert-ブチル基、イソブチル基、シクロヘキシル基等が挙げられる。アルコキシ基の具体例としては、メトキシ基、エトキシ基、プロポキシ基、シクロヘキソキシ基等が挙げられる。耐摩耗性の観点から、好ましくは、炭素数1~6のアルキル基、炭素数1~6のアルコキシ基であり、特に好ましくはメチル基、エチル基、である。tb、ubは各々独立に、0~10の整数であり、製造上の簡便性の観点から、好ましくは、tb=ub=0である。
式(9b)で表される2価アルコール残基を誘導する2価アルコール化合物の具体例としては、例えば、1,4-シクロヘキサンジオール、2,2-ビス(4-ヒドロキシシクロヘキシル)プロパン、4,4-ビシクロヘキサノール等が挙げられる。

式(10b)で表される2価アルコール残基を誘導する2価アルコール化合物の具体例としては、例えば、イソマンニド、イソソルバイド等が挙げられる。

式(11b)中、R11b~R14bは、各々独立に、水素原子、又はメチル基を表す。Vbは、単結合、―CR15bR16b-、O、CO、又はSを表す。またR15b、R16bは各々独立に、水素原子、メチル基、又はエチル基、もしくは、R15b及びR16bは、R15bとR16bとが結合して形成されるシクロペンチル基又はシクロヘキシリデン基を表す。
式(11b)で表されるポリエステル樹脂のビスフェノール残基として具体的に例示すると、ビス-(4-ヒドロキシフェニル)メタン、ビス-(4-ヒドロキシ-3-メチルフェニル)メタン、ビス-(3,5-ジメチル-4-ヒドロキシフェニル)メタン、1,1-ビス-(4-ヒドロキシフェニル)エタン、1,1-ビス-(4-ヒドロキシ-3-メチルフェニル)エタン、1,1-ビス-(3,5-ジメチル-4-ヒドロキシフェニル)エタン、1,1-ビス-(4-ヒドロキシフェニル)プロパン、1,1-ビス-(4-ヒドロキー3-メチルフェニル)プロパン、1,1-ビス-(3,5-ジメチル-4-ヒドロキシフェニル)プロパン、2,2-ビス-(4-ヒドロキシフェニル)プロパン、2,2-ビス-(4-ヒドロキー3-メチルフェニル)プロパン、2,2-ビス-(3,5-ジメチル-4-ヒドロキシフェニル)プロパン、1,1-ビス-(4-ヒドロキシフェニル)シクロヘキサン、1,1-ビス-(4-ヒドロキー3-メチルフェニル)シクロヘキサン、1,1-ビス-(3,5-ジメチル-4-ヒドロキシフェニル)シクロヘキサン、1,1-ビス-(4-ヒドロキシフェニル)-1-フェニルエタン、4,4´-ビフェノール、3,3´-ジメチル-4,4´-ビフェノール、3,3´,5,5´-テトラメチル-4,4´-ビフェノール、4,4´-ジヒドロキシジフェニルエーテル、3,3´-ジメチル-4,4´-ジヒドロキシジフェニルエーテル、ビス(4-ヒドロキシフェニル)スルフィド、4,4´-ジヒドロキシベンゾフェノン等が挙げられる。
この中でも、2価フェノール成分の製造の簡便性及び溶解性、電気特性を考慮すれば、ビス-(4-ヒドロキシフェニル)メタン、ビス-(4-ヒドロキシ-3-メチルフェニル)メタン、ビス-(3,5-ジメチル-4-ヒドロキシフェニル)メタン、1,1-ビス-(4-ヒドロキシフェニル)エタン、1,1-ビス-(4-ヒドロキシ-3-メチルフェニル)エタン、2,2-ビス-(4-ヒドロキシフェニル)プロパン、2,2-ビス-(4-ヒドロキー3-メチルフェニル)プロパン、2,2-ビス-(3,5-ジメチル-4-ヒドロキシフェニル)プロパン、1,1-ビス-(4-ヒドロキシフェニル)シクロヘキサン、4,4´-ビフェノール、3,3´-ジメチル-4,4´-ビフェノール、3,3´,5,5´-テトラメチル-4,4´-ビフェノール、4,4´-ジヒドロキシジフェニルエーテルが好ましい。
さらに機械物性を考慮すれば、ビス-(4-ヒドロキシフェニル)メタン、ビス-(4-ヒドロキシ-3-メチルフェニル)メタン、1,1-ビス-(4-ヒドロキシフェニル)エタン、1,1-ビス-(4-ヒドロキシ-3-メチルフェニル)エタン、2,2-ビス-(4-ヒドロキー3-メチルフェニル)プロパン、4,4´-ビフェノール、3,3´-ジメチル-4,4´-ビフェノール、3,3´,5,5´-テトラメチル-4,4´-ビフェノールがより好ましい。
本発明のポリエステル樹脂を製造する場合において、上記式(5b)~(10b)の2価フェノール又は2価アルコールを1つでも使用する場合、溶解性、樹脂の分子量、機械物性の観点から、式(11b)の2価フェノールと共重合することが好ましい。式(11b)の含有量は特に規定しないが、全2価フェノール及び2価アルコールの5モル%以上であることが好ましく、より好ましくは30モル%以上、さらに好ましくは50モル%以上である。
≪第3の実施形態のポリエステルポリカーボネート樹脂≫
ポリエステルポリカーボネート樹脂に含まれるポリエステル部位については、ジカルボン酸、2価フェノール及び/又は2価アルコールについては、上記ポリエステル樹脂と同等のものが好ましい。カーボネートオリゴマー及びポリカーボネート樹脂については式(13b)が好ましい。
ポリエステルポリカーボネート樹脂に含まれるポリエステル部位については、ジカルボン酸、2価フェノール及び/又は2価アルコールについては、上記ポリエステル樹脂と同等のものが好ましい。カーボネートオリゴマー及びポリカーボネート樹脂については式(13b)が好ましい。

式(13b)中、R19b~R22bは、各々独立に、水素原子、メチル基を表す。Qbは、単結合、―CR23bR24b-、O、CO、Sを表す。またR23b、R24bは各々独立に、水素原子、メチル基、もしくはエチル基を示し、又は、R23b及びR24bは、R23bとR24bとが結合して形成されるシクロペンチル基又はシクロヘキシリデン基を示す。耐摩耗性、製造上の簡便性から、R19b=R21b=水素原子、メチル基、R20b=R22b=水素原子であることが好ましい。また、R23b、R24bは、それぞれ水素原子、メチル基が好ましい。
≪第3の実施形態の樹脂の物性≫
本発明の製造方法により製造されるポリエステル樹脂、ポリエステルポリカーボネート樹脂の粘度平均分子量(Mv)は、機械的強度の観点から、通常15,000以上であり、好ましくは20,000以上である。また、塗布性の観点から通常150,000以下であり、好ましくは100,000以下である。
本発明の製造方法により製造されるポリエステル樹脂、ポリエステルポリカーボネート樹脂の粘度平均分子量(Mv)は、機械的強度の観点から、通常15,000以上であり、好ましくは20,000以上である。また、塗布性の観点から通常150,000以下であり、好ましくは100,000以下である。
また、本発明におけるポリエステル樹脂の末端に存在するカルボン酸クロライド基量は、通常0.1μ当量/g以下、好ましくは0.05μ当量/g以下である。末端カルボン酸クロライド基量が上記範囲を超えると、ポリエステル樹脂を電子写真感光体用塗布液とした際の保存安定性が低下する傾向がある。
本発明におけるポリエステル樹脂の末端に存在するOH基量は、通常100μ当量/g以下、好ましくは50μ当量/g以下である。末端OH基量が上記範囲を超えると、ポリエステル樹脂を電子写真感光体とした際の電気特性が悪化する可能性がある。
発明におけるポリエステル樹脂の末端に存在するカルボン酸基量は、通常200μ当量/g以下、好ましくは100μ当量/g以下である。末端カルボン酸基量が上記範囲を超えると、ポリエステル樹脂を電子写真感光体とした際の電気特性が悪化する可能性がある。
前記ポリエステル樹脂に含まれる全窒素量(T-N量)は、3000ppm以下が好ましく、1500ppm以下であることがさらに好ましく、1000ppm以下であることが特に好ましい。全窒素量が3000ppmを超えると電気特性が悪化する場合がある。
≪第1の実施形態のポリエステル樹脂の製造方法≫
次に、本実施の形態が適用される電子写真感光体に使用するポリエステル樹脂の製造方法について説明する。電子写真感光体用ポリエステルを製造する場合一般的には界面重合法で製造される。
次に、本実施の形態が適用される電子写真感光体に使用するポリエステル樹脂の製造方法について説明する。電子写真感光体用ポリエステルを製造する場合一般的には界面重合法で製造される。
しかしながら、本発明のポリエステル樹脂を製造する場合、ハイドロキノンのように容易にアルカリ水溶液中で酸化されてしまうモノマーであるため、界面重合法では、前記モノマーが水溶液中でアルカリ塩となった際に、キノンに速やかに酸化する。そこで酸化を防ぐ観点から本発明中のポリエステル樹脂の製造方法は、溶液重合法又は溶液重合法と界面重合法を組み合わせた重合方法であることが好ましい。
あるいは、本発明のポリエステル樹脂を製造する場合、芳香族エステル結合を有する2価フェノールを使用するため、界面重合法では、アルカリ塩となった2価フェノールが水層に溶解した際に、水酸化物イオン等の求核剤により速やかに加水分解され、重合が進行しない。そのため、本発明中のポリエステル樹脂の製造方法は、溶液重合法又は溶液重合法と界面重合法を組み合わせた重合方法であることが好ましい。
ポリエステル樹脂の製造法の一例を以下説明する。
ポリエステル樹脂の製造法の一例を以下説明する。
<溶液重合法による製造方法>
溶液重合法による製造の場合は、例えば、2価フェノール化合物及び2価カルボン酸クロライド化合物を溶媒に溶解させ、トリエチルアミン等の塩基を添加することで重合することができる。重合温度は-10℃~40℃の範囲、重合時間は0.5時間~10時間の範囲であるのが生産性の点で好ましい。重合終了後、有機相中に溶解しているポリエステル樹脂を、洗浄、回収することにより、目的とするポリエステル樹脂が得られる。
溶液重合法による製造の場合は、例えば、2価フェノール化合物及び2価カルボン酸クロライド化合物を溶媒に溶解させ、トリエチルアミン等の塩基を添加することで重合することができる。重合温度は-10℃~40℃の範囲、重合時間は0.5時間~10時間の範囲であるのが生産性の点で好ましい。重合終了後、有機相中に溶解しているポリエステル樹脂を、洗浄、回収することにより、目的とするポリエステル樹脂が得られる。
溶液重合法で用いられる塩基としては、例えば、トリエチルアミン、トリプロピルアミン、トリブチルアミン、N,N-ジイソプロピルエチルアミン、N,N-ジプロピルエチルアミン、N,N-ジエチルメチルアミン、N,N-ジメチルエチルアミン、N,N-ジメチルブチルアミン、N,N-ジメチルイソプロピルアミン、N,N-ジエチルイソプロピルアミン、N,N,N´,N´-テトラメチルジエチルアミン、1,4-ジアザビシクロ[2,2,2]オクタン等の3級アミンや、ピリジン、4-メチルピリジン等のピリジン類及び1,8-ジアザビシクロ[5.4.0]-7-ウンデセン等の有機塩基が挙げられる。
また、フォスファゼン塩基、無機塩基等エステル化反応に使用されるような塩基ならば特に制限されない。これらの塩基の中で、エステル化反応の反応性及び入手の簡便性の観点からトリエチルアミン、N,N-ジプロピルエチルアミン、N,N-ジエチルメチルアミン、ピリジンが好ましく、酸クロリドの分解抑制や洗浄における除去の容易さの観点からトリエチルアミンが特に好ましい。塩基の使用量としては、反応系中に含まれるカルボン酸クロライド基の1.01倍当量~2倍当量の範囲が好ましい。
溶媒としては、ジクロロメタン、クロロホルム、1,2-ジクロロエタン、トリクロロエタン、テトラクロロエタン、クロロベンゼン、ジクロロベンゼン等のハロゲン化炭化水素化合物、トルエン、アニソール、キシレン等の芳香族炭化水素化合物、シクロヘキサン、メチルシクロヘキサン等の炭化水素化合物、テトラヒドロフラン、テトラヒドロピラン、1,4-ジオキサン、1,3-ジオキソラン等のエーテル化合物、酢酸エチル、安息香酸メチル、酢酸ベンジル等のエステル化合物、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド化合物、等が挙げられる。
また、ピリジンを塩基かつ溶媒として使用してもよい。これらの中で、モノマーや生成するオリゴマーの溶解性及びエステル化反応の反応性の観点から、ジクロロメタン、クロロホルム、1,2-ジクロロエタン、テトラヒドロフラン、N,N-ジメチルホルムアミド、ピリジンが好ましい。さらに洗浄効率及び電気特性の観点からジクロロメタンが特に好ましい。
溶液重合法においてポリエステル樹脂を製造する場合、2価フェノールと2価カルボン酸クロライドの比率は特に決まりがないが、高分子量のポリエステル樹脂の製造及び末端基の制御の観点から、2価フェノール:2価カルボン酸クロライドはモル比率で1:0.95~1:1.05が好ましく、さらには1:0.99~1:1.01、特に好ましくは1:0.995~1:1.005である。
また、2価フェノールが溶媒に対する溶解性が悪い場合、2価のカルボン酸クロライドと反応した際に析出し、製造されるポリエステル樹脂に不溶分の残留及び電気特性等の悪化となる可能性がある。あるいは、2価フェノールが溶媒に対する溶解性が悪い場合、2価のカルボン酸クロライドと反応した際に析出し、反応溶液の白濁、オリゴマー化の阻害となる場合がある。そのため、溶解性の悪い2価フェノールをポリエステル樹脂に導入する場合は、溶解性の良い2価フェノールとの共重合させることが好ましい。また、溶解性を向上させるため、あらかじめ溶解性の良い2価フェノール及び2価カルボン酸クロライドを反応させ反応系中にオリゴマーを生成させた後に、溶解性の悪い2価フェノールを添加、反応させポリエステル樹脂を製造することがより好ましい。
ポリエステル樹脂を製造する際には、分子量調節剤を使用することができる。分子量調節剤としては、例えば、フェノール、o,m,p-クレゾール、o,m,p-エチルフェノール、o,m,p-プロピルフェノール、o,m,p-(tert-ブチル)フェノール、ペンチルフェノール、ヘキシルフェノール、オクチルフェノール、ノニルフェノール、2,6-ジメチルフェノール誘導体、2-メチルフェノール誘導体等のアルキルフェノール類;o,m,p-フェニルフェノール等の1官能性のフェノール;酢酸クロライド、酪酸クロライド、オクチル酸クロライド、塩化ベンゾイル、ベンゼンスルホニルクロライド、ベンゼンスルフィニルクロライド、スルフィニルクロライド、ベンゼンホスホニルクロライドやそれらの置換体等の1官能性酸ハロゲン化物等が挙げられる。
また、メタノール、エタノール、プロパノール等の1官能脂肪族アルコールや、2-ヒドロキシエチルアクリレート、4-ヒドロキシブチルアクリレート、2-ヒドロキシメタクリレート等のアクリル類を有する1官能アルコール、1H,1H,2H,2H-トリデカフルオロ-1-n-オクタノール、1H,1H,2H,2H-ヘプタデカフルオロ-1-デカノール等のパーフルオロアルキルを有する1官能アルコール、シロキサンを有する1官能アルコール等が挙げられる。これらの分子量調節剤の中でも、分子量調節能が高く、かつ溶液安定性の点で好ましいのは、o,m,p-(tert-ブチル)フェノール、2,6-ジメチルフェノール誘導体、2-メチルフェノール誘導体である。特に好ましくは、p-(tert-ブチル)フェノール、2,3,6-トリメチルフェノール、2,3,5-トリメチルフェノールである。
また、2価フェノールを酸化させないために、重合反応中や洗浄液中に酸化防止剤を添加することができる。酸化防止剤としては、例えば、亜硫酸ナトリウム、ハイドロサルファイト(次亜硫酸ナトリウム)、二酸化硫黄、亜硫酸カリウム、亜硫酸水素ナトリウム等が挙げられる。これらの中でも、酸化防止の効果及び環境負荷の低減からもハイドロサルファイトが特に好ましい。酸化防止剤の使用量としては、全2価フェノールに対して、0.01質量%以上、10.0質量%以下が好ましい。さらに好ましくは0.1質量%以上、5質量%以下である。
ポリエステル樹脂の重合後の洗浄方法は、本発明の効果を著しく損なわない限り任意の方法を用いることができるが、例えば、ポリエステル樹脂の溶液を水酸化ナトリウム、水酸化カリウム等のアルカリ水溶液;塩酸、硝酸、リン酸等の酸水溶液;水等で洗浄した後、静置分離、遠心分離等により分液する方法が挙げられる。
洗浄後のポリエステル樹脂溶液は、ポリエステル樹脂が不溶の水、アルコールその他有機溶媒中に析出させるか、ポリエステル樹脂の溶液を温水又はポリエステル樹脂が不溶の分散媒中で溶媒を留去するか、加熱、減圧等により溶媒を留去することにより取り出してもよいし、スラリー状で取り出した場合は遠心分離器、濾過器とうにより固体を取り出すこともできる。
ポリエステル樹脂の乾燥は、通常ポリエステル樹脂の分解温度以下の温度で乾燥するが、好ましくは20℃以上、ポリエステル樹脂の溶融温度以下で乾燥することができる。このとき減圧下で乾燥することが好ましい。乾燥時間は残存溶媒等の不純物の純度が一定以下になるまでの時間以上行うことが好ましく、具体的には、残存溶媒が通常1000ppm以下、好ましくは300ppm以下、さらに好ましくは100ppm以下になる時間以上乾燥する。
<溶液重合法及び界面重合法の組合せによる製造方法>
本発明中のポリエステル樹脂は、1段階目に芳香族エステル結合を有する2価フェノールを使用し溶液重合法によりエステルオリゴマーを製造し、または溶融重合法によりカーボネートオリゴマーもしくはポリカーボネート樹脂を製造し、2段階目にエステルオリゴマー、カーボネートオリゴマー又はポリカーボネート樹脂を使用した界面重合法により製造することができる。1段階目で製造されるオリゴマー中に芳香族エステル結合を有する2価フェノール残基を導入することにより、2段階目の界面重合の際には、オリゴマーは水層に溶解することがないため、芳香族エステル結合が加水分解せずポリエステル樹脂を得ることができる。
本発明中のポリエステル樹脂は、1段階目に芳香族エステル結合を有する2価フェノールを使用し溶液重合法によりエステルオリゴマーを製造し、または溶融重合法によりカーボネートオリゴマーもしくはポリカーボネート樹脂を製造し、2段階目にエステルオリゴマー、カーボネートオリゴマー又はポリカーボネート樹脂を使用した界面重合法により製造することができる。1段階目で製造されるオリゴマー中に芳香族エステル結合を有する2価フェノール残基を導入することにより、2段階目の界面重合の際には、オリゴマーは水層に溶解することがないため、芳香族エステル結合が加水分解せずポリエステル樹脂を得ることができる。
(1段階目:(1)エステルオリゴマーの製造方法)
エステルオリゴマーを製造する方法としては、溶液重合、溶融重合、界面重合が挙げられる。これらの中で、ビスフェノールの酸化防止、2価アルコールの反応性、モノマー中のエステル結合の安定性などの観点から、溶液重合及び溶融重合が好ましい。さらに、製造の簡便さから、溶液重合が特に好ましい。
エステルオリゴマーを製造する方法としては、溶液重合、溶融重合、界面重合が挙げられる。これらの中で、ビスフェノールの酸化防止、2価アルコールの反応性、モノマー中のエステル結合の安定性などの観点から、溶液重合及び溶融重合が好ましい。さらに、製造の簡便さから、溶液重合が特に好ましい。
エステル結合を有する2価フェノールを用いる場合、エステルオリゴマー製造時に用いることが好ましい。電子写真感光体用ポリエステルを製造する場合に一般的な界面重合法では、アルカリ塩となった2価フェノールが水層に溶解した際に、水酸化物イオン等の求核剤により速やかにエステル結合が加水分解され、重合が進行しない。そこで、溶液重合法や溶融重合法によりエステルオリゴマー中にエステル結合を有する2価フェノールを導入することにより、水溶液に溶解しづらくなり加水分解されることなく、下記のポリエステル製造に使用することが可能となる。
酸化されやすい2価フェノールを用いる場合、エステルオリゴマー製造時に用いることが好ましい。酸化されやすい2価フェノールは、アニオンになった際に、特に酸化されやすくなる。そのため、界面重合法では、強アルカリ水溶液によりアニオンとなり速やかに酸化され重合が進行しなくなる。そこで、アニオンを形成しない溶液重合法や溶融重合法により、エステルオリゴマー中に酸化されやすい2価フェノールをあらかじめ導入することにより、下記のポリエステル製造に使用することが可能となる。
溶液重合法によるエステルオリゴマーの製造では、例えば、2価フェノール化合物及び又は2価アルコール化合物、ジカルボン酸クロライド化合物を溶媒に溶解させ、トリエチルアミン等の塩基を添加することで重合することができる。重合温度は-10℃~40℃の範囲、重合時間は0.5時間~10時間の範囲であるのが生産性の点で好ましい。重合終了後、有機相中に溶解しているオリゴマーを、洗浄、回収することにより、目的とするオリゴマーが得られる。また、回収せずに洗浄したオリゴマー溶液を次のポリエステル重合にそのまま使用してもよい。エステルオリゴマー製造の際、溶液重合法で用いられる塩基、溶媒、停止剤、酸化防止剤は上記溶液重合によるポリエステル樹脂と同等の条件が好ましい。
溶液重合法で用いられる塩基としては、例えば、トリエチルアミン、トリプロピルアミン、トリブチルアミン、N,N-ジイソプロピルエチルアミン、N,N-ジプロピルエチルアミン、N,N-ジエチルメチルアミン、N,N-ジメチルエチルアミン、N,N-ジメチルブチルアミン、N,N-ジメチルイソプロピルアミン、N,N-ジエチルイソプロピルアミン、N,N,N´,N´-テトラメチルジエチルアミン、1,4-ジアザビシクロ[2,2,2]オクタンなどの3級アミンや、ピリジン、4-メチルピリジンなどのピリジン類及び1,8-ジアザビシクロ[5.4.0]-7-ウンデセンなどの有機塩基が挙げられる。
また、フォスファゼン塩基、無機塩基などエステル化反応に使用されるような塩基ならば特に制限されない。これらの塩基の中で、エステル化反応の反応性及び入手の簡便性の観点からトリエチルアミン、N,N-ジプロピルエチルアミン、N,N-ジエチルメチルアミン、ピリジンが好ましく、酸クロリドの分解抑制や洗浄における除去の容易さの観点からトリエチルアミンが特に好ましい。塩基の使用量としては、反応系中に含まれるカルボン酸クロライド基の1.01倍当量~2倍当量の範囲が好ましい。
溶媒としては、ジクロロメタン、クロロホルム、1,2-ジクロロエタン、トリクロロエタン、テトラクロロエタン、クロロベンゼン、ジクロロベンゼン等のハロゲン化炭化水素化合物、トルエン、アニソール、キシレン等の芳香族炭化水素化合物、シクロヘキサン、メチルシクロヘキサン等の炭化水素化合物、テトラヒドロフラン、テトラヒドロピラン、1,4-ジオキサン、1,3-ジオキソラン等のエーテル化合物、酢酸エチル、安息香酸メチル、酢酸ベンジル等のエステル化合物、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド化合物、などが挙げられる。また、ピリジンを塩基かつ溶媒として使用してもよい。
これらの中で、モノマーや生成するオリゴマーの溶解性及びエステル化反応の反応性の観点から、ジクロロメタン、クロロホルム、1,2-ジクロロエタン、テトラヒドロフラン、N,N-ジメチルホルムアミド、ピリジンが好ましい。さらに洗浄効率及び電気特性の観点からジクロロメタンが特に好ましい。
溶液重合法においてオリゴマーを製造する場合、2価フェノール及び/又は2価アルコールとジカルボン酸クロライドの比率は特に決まりがないが、以下に示すポリエステルの製造に使用するためにどちらかを過剰にする必要がある。カルボン酸クロライド末端よりもフェノール末端の方が、空気中及び溶液中の保存安定性が高いため、2価フェノール及び/又は2価アルコールがジカルボン酸クロライドよりも過剰にすることが好ましい。2価フェノール及び/又は2価アルコールがジカルボン酸クロライドよりも過剰とする場合は、2価フェノール及び/又は2価アルコール:ジカルボン酸クロライドの比率は、モル比率で、10:1~1.1:1であることが好ましい。
エステルオリゴマーの製造に2価アルコールを用いる場合、アルコールがオリゴマーの末端になると、その水酸基反応性は低いため、以下に示すポリエステルの製造方法には使用することができない場合がある。そのため、2価のアルコール残基をエステルオリゴマーの構造に導入するためには、2価フェノールと共重合する必要がある。本共重合方法としては、あらかじめ2価アルコールとジカルボン酸クロライドを反応させた後に、2価フェノールを添加、反応させることで、末端がフェノール水酸基である共重合体オリゴマーを得ることができる。2価アルコールとジカルボン酸クロライドの比率は、モル比率で1:10~1:1.1とジカルボン酸クロライドを過剰に使用することが好ましい。
また、エステルオリゴマーの製造において、溶媒に対する溶解性の悪い2価フェノールを使用する場合、2価のカルボン酸クロライドと反応した際に析出し、反応溶液の白濁、オリゴマー化の阻害となる場合がある。そのため、溶解性の悪い2価フェノールをエステルオリゴマーに導入する場合は、溶解性の良い2価フェノール及び/又は2価アルコールとの共重合体とすることが好ましい。さらに溶解性を向上させるため、あらかじめ溶解性の良い2価フェノール及び/又は2価アルコールと2価ジカルボン酸クロライドを反応させた後に、溶解性の悪い2価フェノールを添加、反応させることがより好ましい。上記溶解性の悪いモノマーとしては、例えば、ヒドロキノン、4,4‘-ビフェノール、4―ヒドロキシ安息香酸4―ヒドロキシフェニルなど剛直性の芳香族化合物等が挙げられる。
エステルオリゴマーの製造には、分子量調節剤を使用することができる。以下に示すポリエステル製造時に使用することも可能である。分子量調節剤としては、例えば、フェノール、o,m,p-クレゾール、o,m,p-エチルフェノール、o,m,p-プロピルフェノール、o,m,p-(tert-ブチル)フェノール、ペンチルフェノール、ヘキシルフェノール、オクチルフェノール、ノニルフェノール、2,6-ジメチルフェノール誘導体、2-メチルフェノール誘導体等のアルキルフェノール類;o,m,p-フェニルフェノール等の1官能性のフェノール;酢酸クロライド、酪酸クロライド、オクチル酸クロライド、塩化ベンゾイル、ベンゼンスルホニルクロライド、ベンゼンスルフィニルクロライド、スルフィニルクロライド、ベンゼンホスホニルクロライドやそれらの置換体等の1官能性酸ハロゲン化物等が挙げられる。
また、メタノール、エタノール、プロパノール等の1官能脂肪族アルコールや、2-ヒドロキシエチルアクリレート、4-ヒドロキシブチルアクリレート、2-ヒドロキシメタクリレート等のアクリル類を有する1官能アルコール、1H,1H,2H,2H-トリデカフルオロ-1-n-オクタノール、1H,1H,2H,2H-ヘプタデカフルオロ-1-デカノール等のパーフルオロアルキルを有する1官能アルコール、シロキサンを有する1官能アルコール等が挙げられる。
これら分子量調節剤の中でも、分子量調節能が高く、かつ溶液安定性の点で好ましいのは、o,m,p-(tert-ブチル)フェノール、2,6-ジメチルフェノール誘導体、2-メチルフェノール誘導体である。特に好ましくは、p-(tert-ブチル)フェノール、2,3,6-トリメチルフェノール、2,3,5-トリメチルフェノールである。
エステルオリゴマーの粘度平均分子量(Mv)は、通常800以上である。また、通常20,000以下であり、好ましくは15,000以下である。粘度平均分子量が800未満であると以下に示す界面重合によるポリエステル製造時にアルカリ水溶液中に溶解してしまいエステル結合が分解してしまう場合がある。20,000を超えると製造効率の低下や、以下に示すポリエステル製造時に粘度平均分子量を狙い通りに調整することが困難となる場合がある。
また、2価フェノールを酸化させないために、重合反応中や洗浄液中に酸化防止剤を添加することができる。酸化防止剤としては、例えば、亜硫酸ナトリウム、ハイドロサルファイト(次亜硫酸ナトリウム)、二酸化硫黄、亜硫酸カリウム、亜硫酸水素ナトリウム等が挙げられる。これらの中でも、酸化防止の効果及び環境負荷の低減からもハイドロサルファイトが特に好ましい。酸化防止剤の使用量としては、全2価フェノールに対して、通常0.01質量%以上、酸化防止の観点から、好ましくは0.1質量%以上である。通常10.0質量%以下であり、電気特性の観点から、5質量%以下が好ましい。
エステルオリゴマーの重合後の洗浄方法は、本発明の効果を著しく損なわない限り任意の方法を用いることができるが、例えば、エステルオリゴマーの溶液を水酸化ナトリウム、水酸化カリウム等のアルカリ水溶液;塩酸、硝酸、リン酸等の酸水溶液;水等で洗浄した後、静置分離、遠心分離等により分液する方法が挙げられる。
洗浄後のエステルオリゴマーは、エステルオリゴマーが不溶の水、アルコールその他有機溶媒中に析出させるか、エステルオリゴマーの溶液を温水又はポリエステル樹脂が不溶の分散媒中で溶媒を留去するか、加熱、減圧等により溶媒を留去することにより取り出してもよいし、スラリー状で取り出した場合は遠心分離器、濾過器とうにより固体を取り出すこともできる。また、エステルオリゴマーは洗浄後に取り出さず、溶液として以下のポリエステル樹脂製造にそのまま使用してもよい。
エステルオリゴマーを取り出した場合は、通常エステルオリゴマーの分解温度以下の温度で乾燥するが、好ましくは20℃以上、エステルオリゴマーの溶融温度以下で乾燥することができる。このとき減圧下で乾燥することが好ましい。乾燥時間は残存溶媒等の不純物の純度が一定以下になるまでの時間以上行うことが好ましく、具体的には、残存溶媒が通常1000ppm以下、好ましくは300ppm以下、さらに好ましくは100ppm以下になる時間以上乾燥する。
エステルオリゴマー製造時に使用した塩基は、以下に示すポリエステル製造に反応の阻害や、ジカルボン酸クロライドの分解などの悪影響を与える可能性があるため、塩基の残存量は通常50,000ppm以下、好ましくは10,000ppm以下、更に好ましくは5,000ppm以下である。
(1段階目:(2)カーボネートオリゴマー、ポリカーボネート樹脂の製造方法)
本発明で使用するカーボネートオリゴマー又はポリカーボネート樹脂は、溶融重合による製造のものである。溶融重合で製造されるカーボネートオリゴマー又はポリカーボネートは、多くのフェノール末端を有しているため、下記に示すポリエステルの製造時にカルボン酸クロライドと反応し、簡便に共重合体を得ることが可能である。カーボネートオリゴマー及びポリカーボネートの末端に存在するOH基量は、通常30μ当量/g以上、好ましくは50μ当量/g以上である。末端OH基量が上記範囲を超えないと、下記ポリエステル樹脂の製造時に十分反応せず、共重合体が得られない可能性がある。
本発明で使用するカーボネートオリゴマー又はポリカーボネート樹脂は、溶融重合による製造のものである。溶融重合で製造されるカーボネートオリゴマー又はポリカーボネートは、多くのフェノール末端を有しているため、下記に示すポリエステルの製造時にカルボン酸クロライドと反応し、簡便に共重合体を得ることが可能である。カーボネートオリゴマー及びポリカーボネートの末端に存在するOH基量は、通常30μ当量/g以上、好ましくは50μ当量/g以上である。末端OH基量が上記範囲を超えないと、下記ポリエステル樹脂の製造時に十分反応せず、共重合体が得られない可能性がある。
(2段階目: ポリエステルの製造方法)
前記エステルオリゴマー、カーボネートオリゴマー、ポリカーボネート樹脂の少なくとも1つを使用し、ポリエステル樹脂を製造する際、溶液重合法又は界面重合法が好ましい。特に、製造されるポリエステル樹脂の分子量調整の簡便さ及び電気特性の観点から、界面重合法が特に好ましい。
前記エステルオリゴマー、カーボネートオリゴマー、ポリカーボネート樹脂の少なくとも1つを使用し、ポリエステル樹脂を製造する際、溶液重合法又は界面重合法が好ましい。特に、製造されるポリエステル樹脂の分子量調整の簡便さ及び電気特性の観点から、界面重合法が特に好ましい。
界面重合法による製造の場合は、例えば、アルカリ水溶液と、前記エステルオリゴマーと芳香族ジカルボン酸クロライド化合物を溶解したハロゲン化炭化水素又は芳香族炭化水素の溶液とを混合する。この際、触媒として、4級アンモニウム塩もしくは4級ホスホニウム塩を存在させることも可能である。重合温度は0℃~40℃の範囲、重合時間は2時間~20時間の範囲であるのが生産性の点で好ましい。重合終了後、水相と有機相とを分離し、有機相中に溶解しているポリマーを公知の方法で、洗浄、回収することにより、目的とする樹脂が得られる。界面重合による製造の際、必要によっては、アルカリ水溶液に別途2価フェノールを添加することも可能である。
界面重合法で用いられるアルカリ成分としては、例えば、水酸化ナトリウム、水酸化カリウム等のアルカリ金属の水酸化物等を挙げることができる。アルカリ成分の使用量としては、反応系中に含まれるフェノール性水酸基の1.01倍当量~3倍当量の範囲が好ましい。
ハロゲン化炭化水素としては、例えば、ジクロロメタン、クロロホルム、1,2-ジクロロエタン、トリクロロエタン、テトラクロロエタン、ジクロロベンゼン等が挙げられる。芳香族炭化水素としては、例えば、トルエン、キシレン、ベンゼン等が挙げられる。
触媒として用いられる4級アンモニウム塩もしくは4級ホスホニウム塩としては、例えば、トリブチルアミンやトリオクチルアミン等の3級アルキルアミンの塩酸、臭素酸、ヨウ素酸等の塩;ベンジルトリエチルアンモニウムクロライド、ベンジルトリメチルアンモニウムクロライド、ベンジルトリブチルアンモニウムクロライド、テトラエチルアンモニウムクロライド、テトラブチルアンモニウムクロライド、テトラブチルアンモニウムブロマイド、トリオクチルメチルアンモニウムクロライド、テトラブチルホスホニウムブロマイド、トリエチルオクタデシルホスホニウムブロマイド、N-ラウリルピリジニウムクロライド、ラウリルピコリニウムクロライド等が挙げられる。
また、界面重合法では、分子量調節剤を使用することができる。分子量調節剤としては、例えば、フェノール、o,m,p-クレゾール、o,m,p-エチルフェノール、o,m,p-プロピルフェノール、o,m,p-(tert-ブチル)フェノール、ペンチルフェノール、ヘキシルフェノール、オクチルフェノール、ノニルフェノール、2,6-ジメチルフェノール誘導体、2-メチルフェノール誘導体等のアルキルフェノール類;o,m,p-フェニルフェノール等の1官能性のフェノール;酢酸クロライド、酪酸クロライド、オクチル酸クロライド、塩化ベンゾイル、ベンゼンスルホニルクロライド、ベンゼンスルフィニルクロライド、スルフィニルクロライド、ベンゼンホスホニルクロライドやそれらの置換体等の1官能性酸ハロゲン化物等が挙げられる。
これら分子量調節剤の中でも、分子量調節能が高く、かつ溶液安定性の点で好ましいのは、o,m,p-(tert-ブチル)フェノール、2,6-ジメチルフェノール誘導体、2-メチルフェノール誘導体である。特に好ましくは、p-(tert-ブチル)フェノール、2,3,6-トリメチルフェノール、2,3,5-トリメチルフェノールである。
2価フェノールをアルカリ溶液中で酸化させないために、酸化防止剤を添加することができる。酸化防止剤としては、例えば、亜硫酸ナトリウム、ハイドロサルファイト(次亜硫酸ナトリウム)、二酸化硫黄、亜硫酸カリウム、亜硫酸水素ナトリウム等が挙げられる。これらの中でも、酸化防止の効果及び環境負荷の低減からもハイドロサルファイトが特に好ましい。
酸化防止剤の使用量としては、全2価フェノールに対して、0.01質量%以上、10.0質量%以下が好ましい。さらに好ましくは0.1質量%以上、5質量%以下である。含有量が少なすぎると酸化防止効果が不十分の可能性があり、多すぎるとポリエステル中に残存してしまい電気特性に悪影響する場合がある。
ポリエステル樹脂の重合後の精製方法は、本発明の効果を著しく損なわない限り任意の方法を用いることができるが、例えば、ポリエステル樹脂の溶液を水酸化ナトリウム、水酸化カリウム等のアルカリ水溶液;塩酸、硝酸、リン酸等の酸水溶液;水等で洗浄した後、静置分離、遠心分離等により分液する方法が挙げられる。
また、他の精製方法としては、例えば、生成したポリエステル樹脂の溶液を、ポリエステル樹脂が不溶の溶媒中に析出させる方法、ポリエステル樹脂の溶液を温水中に分散させ溶媒を留去する方法、又はポリエステル樹脂溶液を吸着カラム等に流通させる方法等により精製してもよい。
精製後のポリエステル樹脂は、ポリエステル樹脂が不溶の水、アルコールその他有機溶媒中に析出させるか、ポリエステル樹脂の溶液を温水又はポリエステル樹脂が不溶の分散媒中で溶媒を留去するか、加熱、減圧等により溶媒を留去することにより取り出してもよいし、スラリー状で取り出した場合は遠心分離器、濾過器とうにより固体を取り出すこともできる。得られたポリエステル樹脂は、通常ポリエステル樹脂の分解温度以下の温度で乾燥するが、好ましくは20℃以上、樹脂の溶融温度以下で乾燥することができる。このとき減圧下で乾燥することが好ましい。
乾燥時間は残存溶媒等の不純物の純度が一定以下になるまでの時間以上行うことが好ましく、具体的には、残存溶媒が通常1000ppm以下、好ましくは300ppm以下、さらに好ましくは100ppm以下になる時間以上乾燥する。
なお、本発明中のポリエステルポリカーボネート樹脂の製造は、上記ポリエステル製造と同等の条件を適用できる。
≪第2の実施形態のポリエステル樹脂の製造方法≫
次に、第2の実施形態のポリエステル樹脂の製造方法について説明する。製造方法としては、前記式(4a1)で表される2価フェノールと前記式(5a)で表されるジカルボン酸クロライドとを反応させてエステルオリゴマーを得る工程、及び前記エステルオリゴマーと前記式(4a2)で表される2価フェノールと前記エステルオリゴマーとを反応させてポリエステル樹脂を得る工程を含むものである。ポリエステル樹脂の製造方法としては、溶液重合法、溶液重合法と界面重合法を組み合わせた重合法又は界面重合法が挙げられる。
次に、第2の実施形態のポリエステル樹脂の製造方法について説明する。製造方法としては、前記式(4a1)で表される2価フェノールと前記式(5a)で表されるジカルボン酸クロライドとを反応させてエステルオリゴマーを得る工程、及び前記エステルオリゴマーと前記式(4a2)で表される2価フェノールと前記エステルオリゴマーとを反応させてポリエステル樹脂を得る工程を含むものである。ポリエステル樹脂の製造方法としては、溶液重合法、溶液重合法と界面重合法を組み合わせた重合法又は界面重合法が挙げられる。
電子写真感光体用ポリエステルを製造する場合、一般的には界面重合法で製造される。しかしながら、ハイドロキノンのように容易にアルカリ水溶液中で酸化されてしまうモノマーや、エステル結合を有し加水分解されやすい2価フェノールをモノマーとして使用する場合、界面重合法では、前記モノマーが水溶液中でアルカリ塩となった際に、キノンに速やかに酸化したり、エステル結合が水酸化物イオン等の求核剤により速やかに加水分解されたりすることを防ぐ観点から、溶液重合法又は、溶液重合法と界面重合法とを組み合わせた重合法であることが好ましい。製造の簡便性から、特に溶液重合法が好ましい。以下に溶液重合法によるポリエステル樹脂の製造方法の一例を説明する。
<溶液重合法による製造方法>
溶液重合法による製造の場合は、例えば、前記式(1a)のX1aで表される2価の基を誘導する2価フェノール化合物及び2価カルボン酸クロライド化合物を溶媒に溶解させ、トリエチルアミン等の塩基を添加し、反応系中にエステルオリゴマーを形成した後、同一反応容器内へ前記式(2a)のX2aで表される2価の基を誘導する2価フェノール化合物を添加、更に塩基を添加することによりワンポットでポリエステル樹脂が得られる。
重合温度は-10℃~40℃の範囲、重合時間は0.5時間~10時間の範囲であるのが生産性の点から好ましい。重合終了後、有機相中に溶解しているポリエステル樹脂を、洗浄、回収することにより、目的とするポリエステル樹脂が得られる。Y1aとY2aが異なる場合には、Y1aとY2aがエステルオリゴマーに含有されていてもよいし、Y1aを含むエステルオリゴマー製造後に、更にY2aに対応するジカルボン酸クロライドモノマーと前記式(2a)のX2aで表される2価の基を誘導する2価フェノール化合物とを添加することでポリエステル樹脂に含んでいてもよい。
<溶液重合法と界面重合法を組み合わせた製造方法>
溶液重合法と界面重合法を組み合わせた重合法の場合には、例えば、前記式(1a)のX1aで表される2価の基を誘導する2価フェノール化合物及び2価カルボン酸クロライド化合物を溶媒に溶解させ、トリエチルアミン等の塩基を添加し、エステルオリゴマーを得た後(溶液重合)、前記式(2a)のX2aで表される2価の基を誘導する2価フェノール化合物をアルカリ水溶液に溶解した溶液と、エステルオリゴマー及びジカルボン酸クロライド化合物を溶解したハロゲン化炭化水素及び芳香族炭化水素の溶液とを混合する。
溶液重合法と界面重合法を組み合わせた重合法の場合には、例えば、前記式(1a)のX1aで表される2価の基を誘導する2価フェノール化合物及び2価カルボン酸クロライド化合物を溶媒に溶解させ、トリエチルアミン等の塩基を添加し、エステルオリゴマーを得た後(溶液重合)、前記式(2a)のX2aで表される2価の基を誘導する2価フェノール化合物をアルカリ水溶液に溶解した溶液と、エステルオリゴマー及びジカルボン酸クロライド化合物を溶解したハロゲン化炭化水素及び芳香族炭化水素の溶液とを混合する。
この際、触媒として、4級アンモニウム塩もしくは4級ホスホニウム塩を存在させることも可能である。重合温度は0℃~40℃の範囲、重合時間は2時間~20時間の範囲であるのが生産性の点で好ましい。重合終了後、水相と有機相とを分離し、有機相中に溶解しているポリマーを公知の方法で、洗浄、回収することにより、目的とする樹脂が得られる。
前記式(1a)のX1aで表される2価の基を誘導する2価フェノール化合物は溶解性に優れるためそれから成るエステルオリゴマーを経由することにより、溶解性の悪い2価フェノールと組み合わせた場合でも溶解性が高く透明性に優れたポリエステル樹脂が製造され、得られたポリエステル樹脂は塗膜への適用が可能となる。
一方で、エステルオリゴマーを経由せず溶解性の悪い2価フェノールと前記(1a)のX1aで表される2価の基を誘導する2価フェノールを最初から同一反応容器内でジカルボン酸クロライドと反応させた場合、溶解性の悪い2価フェノールとジカルボン酸クロライド反応物が反応溶液中で析出するため、得られるポリエステル樹脂中に不溶物が残る。そのため、そのポリエステル樹脂の塗布液は白濁し、得られる塗布膜も白濁したり、塗膜表面が不均一となったりする可能性がある。また、前記不溶物中には酸クロライド末端やフェノール末端が残るため、得られる塗布膜の電気特性が悪化する可能性もある。特に溶液重合では、この析出は顕著となる。界面重合では、溶解性の悪い2価フェノールでもアルカリ塩となり水溶液中に溶解するため、途中析出する可能性は低くなるが、溶液重合では2価フェノールのアルカリ塩を経由しないため析出する。
エステルオリゴマー中の残存ジカルボン酸クロライドモノマー量は、下記式(6a)の条件を満たす。

残存ジカルボン酸クロライドモノマーとは、エステルオリゴマー中に残存するジカルボン酸クロライドモノマー量を表す。仕込みジカルボン酸クロライドモノマーとは、エステルオリゴマーを製造するために、最初に仕込むジカルボン酸クロライドモノマー量を表す。尚、エステルオリゴマーを製造した後に、ジカルボン酸クロライドモノマーを添加する場合においても、前記式(6a)を満たせば本発明の範囲内である。即ち、前記残存ジカルボン酸クロライドモノマー、仕込みジカルボン酸クロライドモノマーにそのエステルオリゴマーを製造した後に更に添加した量を加えて計算する。
エステルオリゴマー中の残存ジカルボン酸クロライドモノマー量は、前記式(6a)の条件を満たすが、製造されるポリエステル樹脂の溶解性の観点から、残存ジカルボン酸クロライドモノマー量は前記式(6a)において0.18以下が好ましく、0.16以下がよりに好ましい。これらの値は、実施例[エステルオリゴマー中の残存酸クロライドモノマー測定]に記載の方法により測定できる。また、エステルオリゴマー中の残存ジカルボン酸クロライドモノマー量は、例えば塩基の使用量を後述する範囲に調節することにより特定の範囲とすることができる。
前記エステルオリゴマーを製造する場合、前記(1a)のX1aで表される2価の基を誘導する2価フェノール化合物と2価カルボン酸クロライドの仕込み比率は、前記式(4a1)で表される2価フェノール及び前記式(5a)で表されるジカルボン酸クロライドの仕込みモル比率が、下記式を満たすことが好ましい。

モル比率で2価カルボン酸クロライド:2価フェノールが1.05:1~1.8:1であること示す。感光体に含有した際の耐摩耗性の観点から、1.08以上がより好ましく、1.1以上が更に好ましい。樹脂の溶解性の観点から、1.6以下がより好ましく、1.55以下が更に好ましい。
溶液重合法で用いられる塩基としては、例えば、トリエチルアミン、トリプロピルアミン、トリブチルアミン、N,N-ジイソプロピルエチルアミン、N,N-ジプロピルエチルアミン、N,N-ジエチルメチルアミン、N,N-ジメチルエチルアミン、N,N-ジメチルブチルアミン、N,N-ジメチルイソプロピルアミン、N,N-ジエチルイソプロピルアミン、N,N,N´,N´-テトラメチルジエチルアミン、1,4-ジアザビシクロ[2,2,2]オクタン等の3級アミンや、ピリジン、4-メチルピリジン等のピリジン類及び1,8-ジアザビシクロ[5.4.0]-7-ウンデセン等の有機塩基が挙げられる。
また、フォスファゼン塩基、無機塩基等エステル化反応に使用されるような塩基ならば特に制限されない。これらの塩基の中で、エステル化反応の反応性及び入手の簡便性の観点からトリエチルアミン、N,N-ジプロピルエチルアミン、N,N-ジエチルメチルアミン、ピリジンが好ましく、酸クロリドの分解抑制や洗浄における除去の容易さの観点からトリエチルアミンが特に好ましい。
塩基の使用量としては、エステルオリゴマーを製造する場合は、エステルオリゴマー製造に使用するカルボン酸クロライド基に対して通常0.50倍当量以上、好ましくは0.60倍当量以上である。一方、通常0.95倍当量以下、好ましくは0.90倍当量以下である。また、酸クロライドの不要な分解を防ぐため、使用するフェノール性水酸基に対して1.1倍当量以下が好ましい。
一方、ポリエステル樹脂を製造する場合は、反応に使用するカルボン酸クロライド基に対して、通常1.01倍当量以上、重合反応を速やかに進行させるため、1.05倍当量以上が更に好ましい。また、通常2倍当量以下、形成したエステル結合の分解を防いだり、残存塩基を少なくしたりするため、1.8倍当量以下が好ましい。
溶液重合法で用いられる溶媒としては、ジクロロメタン、クロロホルム、1,2-ジクロロエタン、トリクロロエタン、テトラクロロエタン、クロロベンゼン、ジクロロベンゼン等のハロゲン化炭化水素化合物、トルエン、アニソール、キシレン等の芳香族炭化水素化合物、シクロヘキサン、メチルシクロヘキサン等の炭化水素化合物、テトラヒドロフラン、テトラヒドロピラン、1,4-ジオキサン、1,3-ジオキソラン等のエーテル化合物、酢酸エチル、安息香酸メチル、酢酸ベンジル等のエステル化合物、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド化合物、等が挙げられる。
また、ピリジンを塩基かつ溶媒として使用してもよい。これらの中で、モノマーや生成するオリゴマーの溶解性及びエステル化反応の反応性の観点から、ジクロロメタン、クロロホルム、1,2-ジクロロエタン、テトラヒドロフラン、N,N-ジメチルホルムアミド、ピリジンが好ましい。更に洗浄効率及び電気特性の観点からジクロロメタンが特に好ましい。
溶液重合法においてポリエステル樹脂を製造する場合、全2価フェノールと全2価カルボン酸クロライドの比率は、高分子量のポリエステル樹脂の製造及び末端基の制御の観点から、全2価フェノール:全2価カルボン酸クロライドはモル比率で1:0.95~1:1.05が好ましく、更には1:0.99~1:1.01、特に好ましくは1:0.992~1:1.008である。
ポリエステル樹脂を製造する際には、分子量調節剤を使用することができる。分子量調節剤としては、例えば、フェノール、o,m,p-クレゾール、o,m,p-エチルフェノール、o,m,p-プロピルフェノール、o,m,p-(tert-ブチル)フェノール、ペンチルフェノール、ヘキシルフェノール、オクチルフェノール、ノニルフェノール、2,6-ジメチルフェノール誘導体、2-メチルフェノール誘導体等のアルキルフェノール類;o,m,p-フェニルフェノール等の1官能性のフェノール;酢酸クロライド、酪酸クロライド、オクチル酸クロライド、塩化ベンゾイル、ベンゼンスルホニルクロライド、ベンゼンスルフィニルクロライド、スルフィニルクロライド、ベンゼンホスホニルクロライドやそれらの置換体等の1官能性酸ハロゲン化物等が挙げられる。
また、メタノール、エタノール、プロパノール等の1官能脂肪族アルコールや、2-ヒドロキシエチルアクリレート、4―ヒドロキシブチルアクリレート、2-ヒドロキシメタクリレート等のアクリル類を有する1官能アルコール、1H,1H,2H,2H-トリデカフルオロ-1-n-オクタノール、1H,1H,2H,2H-ヘプタデカフルオロ-1-デカノール等のパーフルオロアルキルを有する1官能アルコール、シロキサンを有する1官能アルコール等が挙げられる。
これらの分子量調節剤の中でも、分子量調節能が高く、かつ溶液安定性の点で好ましいのは、o,m,p-(tert-ブチル)フェノール、2,6-ジメチルフェノール誘導体、2-メチルフェノール誘導体である。特に好ましくは、p-(tert-ブチル)フェノール、2,3,6-トリメチルフェノール、2,3,5-トリメチルフェノールである。
また、2価フェノールを酸化させないために、重合反応中や洗浄液中に酸化防止剤を添加することができる。酸化防止剤としては、例えば、亜硫酸ナトリウム、ハイドロサルファイト(次亜硫酸ナトリウム)、二酸化硫黄、亜硫酸カリウム、亜硫酸水素ナトリウム等が挙げられる。これらの中でも、酸化防止の効果及び環境負荷の低減からもハイドロサルファイトが特に好ましい。酸化防止剤の使用量としては、全2価フェノールに対して、0.01質量%以上、10.0質量%以下が好ましい。更に好ましくは0.1質量%以上、5質量%以下である。
ポリエステル樹脂の重合後の洗浄方法は、例えば、ポリエステル樹脂の溶液を水酸化ナトリウム、水酸化カリウム等のアルカリ水溶液;塩酸、硝酸、リン酸等の酸水溶液;水等で洗浄した後、静置分離、遠心分離等により分液する方法が挙げられる。洗浄後のポリエステル樹脂溶液は、ポリエステル樹脂が不溶の水、アルコールその他有機溶媒中に析出させるか、ポリエステル樹脂の溶液を温水又はポリエステル樹脂が不溶の分散媒中で溶媒を留去するか、加熱、減圧等により溶媒を留去することにより取り出してもよいし、スラリー状で取り出した場合は遠心分離器、濾過器とうにより固体を取り出すこともできる。
ポリエステル樹脂の乾燥は、通常ポリエステル樹脂の分解温度以下の温度で乾燥するが、好ましくは20℃以上、ポリエステル樹脂の溶融温度以下で乾燥することができる。このとき減圧下で乾燥することが好ましい。乾燥時間は残存溶媒等の不純物の純度が一定以下になるまでの時間以上行うことが好ましく、具体的には、残存溶媒が通常1000ppm以下、好ましくは300ppm以下、更に好ましくは100ppm以下になる時間以上乾燥する。
溶液重合法以外の製造方法においても、エステルオリゴマー中の残存ジカルボン酸クロライドモノマー量が、前記式(6a)の条件を満たせばよく、溶液重合法と界面重合法を組み合わせた重合法又は界面重合法も本発明の範囲内である。
≪電子写真感光体≫
本実施の形態が適用される電子写真感光体は、導電性支持体上に設けた感光層を有し、感光層が、前記ポリエステル樹脂を含有するものである。感光層の具体的な構成としては、例えば、導電性支持体上に、電荷発生物質を主成分とする電荷発生層と電荷輸送物質及びバインダー樹脂を主成分とする電荷輸送層とを積層した積層型感光体;導電性支持体上に、電荷輸送物質及びバインダー樹脂を含有する層中に電荷発生物質を分散させた感光層を有する分散型(単層型)感光体等が挙げられる。前記ポリエステル樹脂は、通常、電荷輸送物質を含有する層に用いられ、好ましくは積層型感光体の電荷輸送層に用いられる。
本実施の形態が適用される電子写真感光体は、導電性支持体上に設けた感光層を有し、感光層が、前記ポリエステル樹脂を含有するものである。感光層の具体的な構成としては、例えば、導電性支持体上に、電荷発生物質を主成分とする電荷発生層と電荷輸送物質及びバインダー樹脂を主成分とする電荷輸送層とを積層した積層型感光体;導電性支持体上に、電荷輸送物質及びバインダー樹脂を含有する層中に電荷発生物質を分散させた感光層を有する分散型(単層型)感光体等が挙げられる。前記ポリエステル樹脂は、通常、電荷輸送物質を含有する層に用いられ、好ましくは積層型感光体の電荷輸送層に用いられる。
本実施の形態が適用される電子写真感光体に使用される感光層の具体的な構成としては、例えば、積層型感光体の場合は電荷輸送物質及びバインダー樹脂を含有し、静電荷を保持して露光により発生した電荷を輸送する電荷輸送層と、電荷発生物質を含有し、露光により電荷対を発生する電荷発生層と、を有する。また、その他にも必要に応じて、例えば、導電性支持体からの電荷注入を阻止する電荷阻止層、レーザー光等の光を拡散させて干渉縞の発生を防止する光拡散層等を有する場合がある。分散型(単層型)感光体の場合は、感光層は、電荷移動物質及び電荷発生物質がバインダー樹脂中に分散されている。
<導電性支持体>
導電性支持体について特に制限は無いが、例えばアルミニウム、アルミニウム合金、ステンレス鋼、銅、ニッケル等の金属材料や、金属、カーボン、酸化錫等の導電性粉体を添加して導電性を付与した樹脂材料や、アルミニウム、ニッケル、ITO(酸化インジウム酸化錫)等の導電性材料をその表面に蒸着又は塗布した樹脂、ガラス、紙等が主として使用される。
導電性支持体について特に制限は無いが、例えばアルミニウム、アルミニウム合金、ステンレス鋼、銅、ニッケル等の金属材料や、金属、カーボン、酸化錫等の導電性粉体を添加して導電性を付与した樹脂材料や、アルミニウム、ニッケル、ITO(酸化インジウム酸化錫)等の導電性材料をその表面に蒸着又は塗布した樹脂、ガラス、紙等が主として使用される。
これらは1種を単独で用いてもよく、2種以上を任意の組み合わせ及び比率で併用してもよい。導電性支持体の形態としては、ドラム状、シート状、ベルト状等のものが用いられる。更には、金属材料の導電性支持体の上に、導電性・表面性等の制御や欠陥被覆のために、適当な抵抗値を有する導電性材料を塗布したものを用いてもよい。
また、導電性支持体としてアルミニウム合金等の金属材料を用いた場合、陽極酸化被膜を施してから用いてもよい。陽極酸化被膜を施した場合には、公知の方法により封孔処理を施すのが好ましい。
支持体表面は、平滑であってもよいし、特別な切削方法を用いたり、研磨処理を施したりすることにより、粗面化されていてもよい。また、支持体を構成する材料に適当な粒径の粒子を混合することによって、粗面化されたものでもよい。また、安価化のためには、切削処理を施さず、引き抜き管をそのまま使用することも可能である。
<下引き層>
導電性支持体と後述する感光層との間には、接着性・ブロッキング性等の改善のため、下引き層を設けてもよい。下引き層としては、樹脂、及び樹脂に金属酸化物等の粒子を分散したもの等が用いられる。また、下引き層は、単一層からなるものであっても、複数層からなるものであってもよい。
導電性支持体と後述する感光層との間には、接着性・ブロッキング性等の改善のため、下引き層を設けてもよい。下引き層としては、樹脂、及び樹脂に金属酸化物等の粒子を分散したもの等が用いられる。また、下引き層は、単一層からなるものであっても、複数層からなるものであってもよい。
下引き層には、公知の酸化防止剤等、顔料粒子、樹脂粒子等を含有させて用いてもよい。その膜厚は、電子写真感光体の電気特性、強露光特性、画像特性、繰り返し特性、及び製造時の塗布性を向上させる観点から、通常は0.01μm以上、好ましくは0.1μm以上、また、通常30μm以下、好ましくは20μm以下である。
下引き層に用いる金属酸化物粒子の例としては、酸化チタン、酸化アルミニウム、酸化珪素、酸化ジルコニウム、酸化亜鉛、酸化鉄等の1種の金属元素を含む金属酸化物粒子、チタン酸カルシウム、チタン酸ストロンチウム、チタン酸バリウム等の複数の金属元素を含む金属酸化物粒子等が挙げられる。これらは一種類の粒子を単独で用いてもよいし、複数の種類の粒子を混合して用いてもよい。
これらの金属酸化物粒子の中で、酸化チタン及び酸化アルミニウムが好ましく、特に酸化チタンが好ましい。酸化チタン粒子は、その表面に、酸化錫、酸化アルミニウム、酸化アンチモン、酸化ジルコニウム、酸化珪素等の無機物、又はステアリン酸、ポリオール、シリコン等の有機物による処理を施されていてもよい。酸化チタン粒子の結晶型としては、ルチル、アナターゼ、ブルッカイト、アモルファスのいずれも用いることができる。また、複数の結晶状態のものが含まれていてもよい。
また、金属酸化物粒子の粒径としては種々のものが利用できるが、中でも特性及び液の安定性の点から、その平均一次粒径は、10nm以上100nm以下が好ましく、特に10nm以上50nm以下が好ましい。この平均一次粒径は、TEM写真等から得ることができる。
下引き層は、金属酸化物粒子をバインダー樹脂に分散した形で形成するのが好ましい。下引き層に用いられるバインダー樹脂としては、エポキシ樹脂、ポリエチレン樹脂、ポリプロピレン樹脂、アクリル樹脂、メタクリル樹脂、ポリアミド樹脂、塩化ビニル樹脂、酢酸ビニル樹脂、フェノール樹脂、ポリカーボネート樹脂、ポリウレタン樹脂、ポリイミド樹脂、塩化ビニリデン樹脂、ポリビニルアセタール樹脂、塩化ビニル-酢酸ビニル共重合体、ポリビニルアルコール樹脂、ポリウレタン樹脂、ポリアクリル樹脂、ポリアクリルアミド樹脂、ポリビニルピロリドン樹脂、ポリビニルピリジン樹脂、水溶性ポリエステル樹脂、ニトロセルロース等のセルロースエステル樹脂、セルロースエーテル樹脂、カゼイン、ゼラチン、ポリグルタミン酸、澱粉、スターチアセテート、アミノ澱粉、ジルコニウムキレート化合物、ジルコニウムアルコキシド化合物等の有機ジルコニウム化合物、チタニルキレート化合物、チタンアルコキシド化合物等の有機チタニル化合物、シランカップリング剤等の公知のバインダー樹脂が挙げられる。
これらは単独で用いてもよく、或いは2種以上を任意の組み合わせ及び比率で併用してもよい。また、硬化剤とともに硬化した形で使用してもよい。中でも、アルコール可溶性の共重合ポリアミド、変性ポリアミド等は、良好な分散性、塗布性を表すことから好ましい。下引き層に用いられるバインダー樹脂に対する無機粒子の使用比率は任意に選ぶことが可能であるが、分散液の安定性、塗布性の観点から、バインダー樹脂に対して、通常は10質量%以上、500質量%以下の範囲で使用することが好ましい。
<感光層>
感光層の形式としては、電荷発生物質と電荷輸送物質とが同一層に存在し、バインダー樹脂中に分散された単層型と、電荷発生物質がバインダー樹脂中に分散された電荷発生層及び電荷輸送物質がバインダー樹脂中に分散された電荷輸送層の二層からなる機能分離型(積層型)とが挙げられるが、何れの形式であってもよい。
感光層の形式としては、電荷発生物質と電荷輸送物質とが同一層に存在し、バインダー樹脂中に分散された単層型と、電荷発生物質がバインダー樹脂中に分散された電荷発生層及び電荷輸送物質がバインダー樹脂中に分散された電荷輸送層の二層からなる機能分離型(積層型)とが挙げられるが、何れの形式であってもよい。
積層型感光層としては、導電性支持体側から電荷発生層、電荷輸送層をこの順に積層して設ける順積層型感光層と、逆に電荷輸送層、電荷発生層の順に積層して設ける逆積層型感光層とがあり、いずれを採用することも可能であるが、最もバランスの取れた光導電性を発揮できる順積層型感光層が好ましい。
[電荷発生層-積層型]
積層型感光体(機能分離型感光体)の場合、電荷発生層は、電荷発生物質をバインダー樹脂で結着することにより形成される。その膜厚は通常0.1μm以上、好ましくは0.15μm以上、また、通常10μm以下、好ましくは0.6μm以下の範囲である。
積層型感光体(機能分離型感光体)の場合、電荷発生層は、電荷発生物質をバインダー樹脂で結着することにより形成される。その膜厚は通常0.1μm以上、好ましくは0.15μm以上、また、通常10μm以下、好ましくは0.6μm以下の範囲である。
電荷発生物質としては、セレニウム及びその合金、硫化カドミウム等の無機系光導電材料と、有機顔料等の有機系光導電材料とが挙げられるが、有機系光導電材料の方が好ましく、特に有機顔料が好ましい。有機顔料としては、例えば、フタロシアニン顔料、アゾ顔料、ジチオケトピロロピロール顔料、スクアレン(スクアリリウム)顔料、キナクリドン顔料、インジゴ顔料、ペリレン顔料、多環キノン顔料、アントアントロン顔料、ベンズイミダゾール顔料等が挙げられる。
これらの中でも、特にフタロシアニン顔料又はアゾ顔料が好ましい。電荷発生物質として有機顔料を使用する場合、通常はこれらの有機顔料の微粒子を、各種のバインダー樹脂で結着した分散層の形で使用する。
電荷発生物質として無金属フタロシアニン化合物、金属含有フタロシアニン化合物を用いた場合は比較的長波長のレーザー光、例えば780nm近辺の波長を有するレーザー光に対して高感度の感光体が得られ、またモノアゾ、ジアゾ、トリスアゾ等のアゾ顔料を用いた場合には、白色光、又は660nm近辺の波長を有するレーザー光、もしくは比較的短波長のレーザー光、例えば450nm、400nm近辺の波長を有するレーザーに対して十分な感度を有する感光体を得ることができる。
電荷発生物質として有機顔料を使用する場合、特にフタロシアニン顔料又はアゾ顔料が好ましい。フタロシアニン顔料は、比較的長波長のレーザー光に対して高感度の感光体が得られる点で、また、アゾ顔料は、白色光及び比較的短波長のレーザー光に対し十分な感度を持つ点で、それぞれ優れている。
電荷発生物質としてフタロシアニン顔料を使用する場合、具体的には無金属フタロシアニン、銅、インジウム、ガリウム、スズ、チタン、亜鉛、バナジウム、シリコン、ゲルマニウム、アルミニウム等の金属又はその酸化物、ハロゲン化物、水酸化物、アルコキシド等の配位したフタロシアニン類の各結晶型を持ったもの、酸素原子等を架橋原子として用いたフタロシアニンダイマー類等が使用される。
特に、感度の高い結晶型であるX型、τ型無金属フタロシアニン、A型(別称β型)、B型(別称α型)、D型(別称Y型)等のチタニルフタロシアニン(別称:オキシチタニウムフタロシアニン)、バナジルフタロシアニン、クロロインジウムフタロシアニン、ヒドロキシインジウムフタロシアニン、II型等のクロロガリウムフタロシアニン、V型等のヒドロキシガリウムフタロシアニン、G型、I型等のμ-オキソ-ガリウムフタロシアニン二量体、II型等のμ-オキソ-アルミニウムフタロシアニン二量体が好適である。
また、これらフタロシアニンの中でも、A型(別称β型)、B型(別称α型)、及び粉末X線回折の回折角2θ(±0.2゜)が27.1゜、もしくは27.3゜に明瞭なピークを表すことを特徴とするD型(Y型)チタニルフタロシアニン、II型クロロガリウムフタロシアニン、V型及び28.1゜に最も強いピークを有すること、また26.2゜にピークを持たず28.1゜に明瞭なピークを有し、かつ25.9゜の半値幅Wが0.1゜≦W≦0.4゜であることを特徴とするヒドロキシガリウムフタロシアニン、G型μ-オキソ-ガリウムフタロシアニン二量体等が特に好ましい。これらの中でも、D型(Y型)チタニルフタロシアニンが良好な感度を表すため好ましい。
フタロシアニン化合物は単一の化合物のものを用いてもよいし、幾つかの混合又は混晶状態のものを用いてもよい。ここでのフタロシアニン化合物ないしは結晶状態に置ける混合状態としては、それぞれの構成要素を後から混合したものを用いてもよいし、合成、顔料化、結晶化等のフタロシアニン化合物の製造・処理工程において混合状態を生じさせたものでもよい。このような処理としては、酸ペースト処理・磨砕処理・溶剤処理等が知られている。混晶状態を生じさせるためには、日本国特開平10-48859号公報記載のように、2種類の結晶を混合後に機械的に磨砕、不定形化した後に、溶剤処理によって特定の結晶状態に変換する方法が挙げられる。
電荷発生層に用いるバインダー樹脂は特に制限されないが、例としては、ポリビニルブチラール樹脂、ポリビニルホルマール樹脂、ブチラールの一部がホルマールや、アセタール等で変性された部分アセタール化ポリビニルブチラール樹脂等のポリビニルアセタール系樹脂、ポリアリレート樹脂、ポリカーボネート樹脂、ポリエステル樹脂、変性エーテル系ポリエステル樹脂、フェノキシ樹脂、ポリ塩化ビニル樹脂、ポリ塩化ビニリデン樹脂、ポリ酢酸ビニル樹脂、ポリスチレン樹脂、アクリル樹脂、メタクリル樹脂、ポリアクリルアミド樹脂、ポリアミド樹脂、ポリビニルピリジン樹脂、セルロース系樹脂、ポリウレタン樹脂、エポキシ樹脂、シリコン樹脂、ポリビニルアルコール樹脂、ポリビニルピロリドン樹脂、カゼインや、塩化ビニル-酢酸ビニル共重合体、ヒドロキシ変性塩化ビニル-酢酸ビニル共重合体、カルボキシル変性塩化ビニル-酢酸ビニル共重合体、塩化ビニル-酢酸ビニル-無水マレイン酸共重合体等の塩化ビニル-酢酸ビニル系共重合体、スチレン-ブタジエン共重合体、塩化ビニリデン-アクリロニトリル共重合体、スチレン-アルキッド樹脂、シリコン-アルキッド樹脂、フェノール-ホルムアルデヒド樹脂等の絶縁性樹脂や、ポリ-N-ビニルカルバゾール、ポリビニルアントラセン、ポリビニルペリレン等の有機光導電性ポリマー等が挙げられる。これらのバインダ樹脂は、何れか1種を単独で用いても良く、2種類以上を任意の組み合わせで混合して用いてもよい。
電荷発生層において、バインダー樹脂と電荷発生物質との配合比(質量)は、バインダー樹脂100質量部に対して電荷発生物質が通常10質量部以上、好ましくは30質量部以上、また、通常1000質量部以下、好ましくは500質量部以下の範囲である。
[電荷輸送層-積層型]
積層型感光体の電荷輸送層は、電荷輸送物質を含有するとともに、通常はバインダー樹脂と、必要に応じて使用されるその他の成分とを含有する。電荷輸送層は、単一の層から成ってもよいし、構成成分あるいは組成比の異なる複数の層を重ねたものでもよい。その膜厚は、通常、5μm~50μm、好ましくは10μm~45μmである。
積層型感光体の電荷輸送層は、電荷輸送物質を含有するとともに、通常はバインダー樹脂と、必要に応じて使用されるその他の成分とを含有する。電荷輸送層は、単一の層から成ってもよいし、構成成分あるいは組成比の異なる複数の層を重ねたものでもよい。その膜厚は、通常、5μm~50μm、好ましくは10μm~45μmである。
電荷輸送物質としては特に限定されず、任意の物質を用いることが可能である。電荷輸送物質の例としては、2,4,7-トリニトロフルオレノン等の芳香族ニトロ化合物、テトラシアノキノジメタン等のシアノ化合物、ジフェノキノン等のキノン化合物等の電子吸引性物質、カルバゾール誘導体、インドール誘導体、イミダゾール誘導体、オキサゾール誘導体、ピラゾール誘導体、チアジアゾール誘導体、ベンゾフラン誘導体等の複素環化合物、アニリン誘導体、ヒドラゾン誘導体、芳香族アミン誘導体、スチルベン誘導体、ブタジエン誘導体、エナミン誘導体及びこれらの化合物の複数種が結合したもの、あるいはこれらの化合物からなる基を主鎖又は側鎖に有する重合体等の電子供与性物質等が挙げられる。
これらの中でも、カルバゾール誘導体、芳香族アミン誘導体、スチルベン誘導体、ブタジエン誘導体、エナミン誘導体、及びこれらの化合物の複数種が結合したものが好ましい。これらの電荷輸送物質は単独で用いてもよいし、いくつかを混合してもよい。電荷輸送物質の好適な構造の具体例を以下に表す。



前記ポリエステル樹脂は、電荷輸送層のバインダー樹脂として用いられることが好ましい。前記ポリエステル樹脂と他の樹脂とを混合して用いることも可能である。ここで混合される他の構造を有する樹脂としては、例えば、ポリメチルメタクリレート、ポリスチレン、ポリ塩化ビニル等のビニル重合体及びその共重合体;ポリカーボネート樹脂、ポリエステル樹脂、ポリエステルポリカーボネート樹脂、ポリスルホン樹脂、フェノキシ樹脂、エポキシ樹脂、シリコーン樹脂等の熱可塑性樹脂及び種々の熱硬化性樹脂等及びこれらの共重合体が挙げられる。
これら樹脂のなかでもポリカーボネート樹脂、ポリエステル樹脂、ポリカーボネート樹脂とシリコーン樹脂の共重合体及びポリエステル樹脂とシリコーン樹脂の共重合体が好ましい。また、混合される他の構造を有する樹脂の混合割合は、特に限定されないが、通常、前記ポリエステル樹脂の割合を超えない範囲で併用することが好ましく、具体的には前記ポリエステル樹脂に対する他の構造を有する樹脂の含有量は、通常50質量部以下、耐摩耗性の観点から、30質量部以下が好ましい。
前記ポリエステル樹脂と電荷輸送物質との割合は、ポリエステル樹脂100質量部に対して電荷輸送物質を30質量部以上の比率で使用する。電気特性の観点から、好ましくは40質量部以上である。耐摩耗性の観点から、200質量部以下、好ましくは150質量部以下である。
前記他の構造を有する樹脂の好適な構造の具体例を以下に表す。これら具体例は例示のために示したものであり、本発明の趣旨に反しない限りはいかなるバインダー樹脂を混合して用いてもよい。


バインダー樹脂全体と電荷輸送物質との割合としては、通常同一層中のバインダー樹脂100質量部に対して電荷輸送物質を10質量部以上の比率で使用する。中でも、残留電位低減の観点から20質量部以上が好ましく、繰り返し使用した際の安定性や電荷移動度の観点から30質量部以上がより好ましく、40質量部以上が更に好ましい。
一方、通常電荷輸送物質を150質量部以下、感光層の熱安定性の観点から120質量部以下の比率で使用する。中でも、電荷輸送物質とバインダー樹脂との相溶性の観点から、110質量部以下が好ましく、100質量部以下がより好ましく、耐摩耗性の観点から80質量部以下がさらに好ましく、耐傷性の観点から70質量部以下が特に好ましい。
尚、電荷輸送層には成膜性、可撓性、塗布性、耐汚染性、耐ガス性、耐光性等を向上させるために周知の可塑剤、酸化防止剤、紫外線吸収剤、電子吸引性化合物、染料、顔料、レベリング剤等の添加剤を含有させてもよい。酸化防止剤の例としては、ヒンダードフェノール化合物、ヒンダードアミン化合物等が挙げられる。また染料、顔料の例としては、各種の色素化合物、アゾ化合物等が挙げられる。
<単層型感光層>
単層型感光層は、電荷発生物質と電荷輸送物質に加えて、積層型感光体の電荷輸送層と同様に、膜強度確保のためにバインダー樹脂を使用して形成する。具体的には、電荷発生物質と電荷輸送物質と各種バインダー樹脂とを溶剤に溶解又は分散して塗布液を作製し、導電性支持体上(下引き層を設ける場合は下引き層上)に塗布、乾燥して得ることができる。
単層型感光層は、電荷発生物質と電荷輸送物質に加えて、積層型感光体の電荷輸送層と同様に、膜強度確保のためにバインダー樹脂を使用して形成する。具体的には、電荷発生物質と電荷輸送物質と各種バインダー樹脂とを溶剤に溶解又は分散して塗布液を作製し、導電性支持体上(下引き層を設ける場合は下引き層上)に塗布、乾燥して得ることができる。
電荷輸送物質及びバインダー樹脂の種類並びにこれらの使用比率は、積層型感光体の電荷輸送層について説明したものと同様である。これらの電荷輸送物質及びバインダー樹脂からなる電荷輸送媒体中に、さらに電荷発生物質が分散される。
電荷発生物質は、積層型感光体の電荷発生層について説明したものと同様のものが使用できる。但し、単層型感光体の感光層の場合、電荷発生物質の粒子径を充分に小さくする必要がある。具体的には、通常1μm以下、好ましくは0.5μm以下の範囲とする。
単層型感光層内に分散される電荷発生物質の量は、少な過ぎると充分な感度が得られない一方で、多過ぎると帯電性の低下、感度の低下等の弊害があることから、単層型感光層全体に対して通常0.5質量%以上、好ましくは1質量%以上、また、通常50質量%以下、好ましくは20質量%以下の範囲で使用される。
また、単層型感光層におけるバインダー樹脂と電荷発生物質との使用比率は、バインダー樹脂100質量部に対して電荷発生物質が通常0.1質量部以上、好ましくは1質量部以上、また、通常30質量部以下、好ましくは10質量部以下である。
単層型感光層の膜厚は、通常5μm以上、好ましくは10μm以上、また、通常100μm以下、好ましくは50μm以下の範囲である。この場合にも成膜性、可とう性、機械的強度等を改良するための公知の可塑剤、残留電位を抑制するための添加剤、分散安定性向上のための分散補助剤、塗布性を改善するためのレベリング剤、界面活性剤、例えばシリコ-ンオイル、フッ素系オイルその他の添加剤が添加されていてもよい。
<その他の機能層>
積層型感光体、単層型感光体ともに、感光層又はそれを構成する各層には、成膜性、可撓性、塗布性、耐汚染性、耐ガス性、耐光性等を向上させる目的で、周知の酸化防止剤、可塑剤、紫外線吸収剤、電子吸引性化合物、レベリング剤、可視光遮光剤等の添加物を含有させてもよい。
積層型感光体、単層型感光体ともに、感光層又はそれを構成する各層には、成膜性、可撓性、塗布性、耐汚染性、耐ガス性、耐光性等を向上させる目的で、周知の酸化防止剤、可塑剤、紫外線吸収剤、電子吸引性化合物、レベリング剤、可視光遮光剤等の添加物を含有させてもよい。
また、感光体表面の摩擦抵抗や、摩耗を低減、トナーの感光体から転写ベルト、紙への転写効率を高める等の目的で、表面層にフッ素系樹脂、シリコン樹脂、ポリエチレン樹脂等、又はこれらの樹脂からなる粒子や無機化合物の粒子を、表面層に含有させてもよい。或いは、これらの樹脂や粒子を含む層を新たに表面層として形成してもよい。さらに必要に応じて、バリアー層、接着層、ブロッキング層等の中間層、透明絶縁層等、電気特性、機械特性の改良のための層を有していてもよい。
<各層の形成方法>
これらの感光体を構成する各層は、含有させる物質を溶剤に溶解又は分散させて得られた塗布液を、支持体上に浸漬塗布、スプレー塗布、ノズル塗布、バーコート、ロールコート、ブレード塗布等の公知の方法により、各層ごとに順次塗布・乾燥工程を繰り返すことにより形成される。
これらの感光体を構成する各層は、含有させる物質を溶剤に溶解又は分散させて得られた塗布液を、支持体上に浸漬塗布、スプレー塗布、ノズル塗布、バーコート、ロールコート、ブレード塗布等の公知の方法により、各層ごとに順次塗布・乾燥工程を繰り返すことにより形成される。
塗布液の作製に用いられる溶媒又は分散媒に特に制限は無いが、具体例としては、メタノール、エタノール、プロパノール、2-メトキシエタノール等のアルコール類、テトラヒドロフラン、1,4-ジオキサン、ジメトキシエタン等のエーテル類、ギ酸メチル、酢酸エチル等のエステル類、アセトン、メチルエチルケトン、シクロヘキサノン等のケトン類、ベンゼン、トルエン、キシレン等の芳香族炭化水素類、ジクロロメタン、クロロホルム、1,2-ジクロロエタン、1,1,2-トリクロロエタン、1,1,1-トリクロロエタン、テトラクロロエタン、1,2-ジクロロプロパン、トリクロロエチレン等の塩素化炭化水素類、n-ブチルアミン、イソプロパノールアミン、ジエチルアミン、トリエタノールアミン、エチレンジアミン、トリエチレンジアミン等の含窒素化合物類、アセトニトリル、N-メチルピロリドン、N,N-ジメチルホルムアミド、ジメチルスルホキシド等の非プロトン性極性溶剤類等が挙げられる。
これらの溶剤の中で、環境配慮の観点から非ハロゲン系溶剤が好ましく、溶解性の観点から、トルエン、キシレン、アニソール、ジメトキシエタン、テトラヒドロフラン、1,4-ジオキサンが特に好ましい。これらは何れか1種を単独で用いてもよく、2種以上を併用して用いてもよい。
溶媒又は分散媒の使用量は特に制限されないが、各層の目的や選択した溶媒・分散媒の性質を考慮して、塗布液の固形分濃度や粘度等の物性が所望の範囲となるように適宜調整するのが好ましい。
例えば、単層型感光体、及び機能分離型感光体の電荷輸送層層の場合には、塗布液の固形分濃度を通常5質量%以上、好ましくは10質量%以上、また、通常40質量%以下、好ましくは35質量%以下の範囲とする。また、塗布液の粘度を通常10cps以上、好ましくは50cps以上、また、通常500cps以下、好ましくは400cps以下の範囲とする。
また、積層型感光体の電荷発生層の場合には、塗布液の固形分濃度は、通常0.1質量%以上、好ましくは1質量%以上、また、通常15質量%以下、好ましくは10質量%以下の範囲とする。また、塗布液の粘度は、通常0.01cps以上、好ましくは0.1cps以上、また、通常20cps以下、好ましくは10cps以下の範囲とする。
塗布液の塗布方法としては、浸漬コーティング法、スプレーコーティング法、スピナーコーティング法、ビードコーティング法、ワイヤーバーコーティング法、ブレードコーティング法、ローラーコーティング法、エアーナイフコーティング法、カーテンコーティング法等が挙げられるが、他の公知のコーティング法を用いることも可能である。
塗布液の乾燥は、室温における指触乾燥後、通常30℃以上、200℃以下の温度範囲で、1分から2時間の間、静止又は送風下で加熱乾燥させることが好ましい。また、加熱温度は一定であってもよく、乾燥時に温度を変更させながら加熱を行なってもよい。
<画像形成装置>
次に、本発明の電子写真感光体を用いた画像形成装置(本発明の画像形成装置)の実施の形態について、装置の要部構成を表す図1を用いて説明する。但し、実施の形態は以下の説明に限定されるものではなく、本発明の要旨を逸脱しない限り任意に変形して実施することができる。
次に、本発明の電子写真感光体を用いた画像形成装置(本発明の画像形成装置)の実施の形態について、装置の要部構成を表す図1を用いて説明する。但し、実施の形態は以下の説明に限定されるものではなく、本発明の要旨を逸脱しない限り任意に変形して実施することができる。
図1に表すように、画像形成装置は、電子写真感光体1,帯電装置2,露光装置3及び現像装置4を備えて構成され、更に、必要に応じて転写装置5,クリーニング装置6及び定着装置7が設けられる。
電子写真感光体1は、上述した本発明の電子写真感光体であれば特に制限はないが、図1ではその一例として、円筒状の導電性支持体の表面に上述した感光層を形成したドラム状の感光体を示している。この電子写真感光体1の外周面に沿って、帯電装置2,露光装置3,現像装置4,転写装置5及びクリーニング装置6がそれぞれ配置されている。
帯電装置2は、電子写真感光体1を帯電させるもので、電子写真感光体1の表面を所定電位に均一帯電させる。帯電装置としては、コロトロンやスコロトロン等のコロナ帯電装置、電圧印加された直接帯電部材を感光体表面に接触させて帯電させる直接帯電装置(接触型帯電装置)帯電ブラシ等の接触型帯電装置などがよく用いられる。
直接帯電手段の例としては、帯電ローラ、帯電ブラシ等の接触帯電器などが挙げられる。なお、図1では、帯電装置2の一例としてローラ型の帯電装置(帯電ローラ)を示している。直接帯電手段として、気中放電を伴う帯電、あるいは気中放電を伴わない注入帯電いずれも可能である。また、帯電時に印可する電圧としては、直流電圧だけの場合、及び直流に交流を重畳させて用いることもできる。
露光装置3は、電子写真感光体1に露光を行なって電子写真感光体1の感光面に静電潜像を形成することができるものであれば、その種類に特に制限はない。具体例としては、ハロゲンランプ、蛍光灯、半導体レーザーやHe-Neレーザー等のレーザー、LEDなどが挙げられる。また、感光体内部露光方式によって露光を行なうようにしてもよい。
露光を行なう際の光は任意であるが、例えば波長が780nmの単色光、波長600nm~700nmのやや短波長寄りの単色光、波長380nm~500nmの短波長の単色光などで露光を行なえばよい。これらの中で380~500nmの短波長光を用いると解像度が高くなり好ましい。中でも405nmの単色光が好適である。
現像装置4は、その種類に特に制限はなく、カスケード現像、一成分絶縁トナー現像、一成分導電トナー現像、二成分磁気ブラシ現像などの乾式現像方式や、湿式現像方式などの任意の装置を用いることができる。図1では、現像装置4は、現像槽41、アジテータ42、供給ローラ43、現像ローラ44、及び、規制部材45からなり、現像槽41の内部にトナーTを貯留している構成となっている。
また、必要に応じ、トナーTを補給する補給装置(図示せず)を現像装置4に付帯させてもよい。この補給装置は、ボトル、カートリッジなどの容器からトナーTを補給することが可能に構成される。
転写装置5は、その種類に特に制限はなく、コロナ転写、ローラ転写、ベルト転写などの静電転写法、圧力転写法、粘着転写法など、任意の方式を用いた装置を使用することができる。ここでは、転写装置5が電子写真感光体1に対向して配置された転写チャージャー,転写ローラ,転写ベルト等から構成されるものとする。この転写装置5は、トナーTの帯電電位とは逆極性で所定電圧値(転写電圧)を印加し、電子写真感光体1に形成されたトナー像を記録紙(用紙,媒体)Pに転写するものである。
クリーニング装置6について特に制限はなく、ブラシクリーナー、磁気ブラシクリーナー、静電ブラシクリーナー、磁気ローラクリーナー、ブレードクリーナーなど、任意のクリーニング装置を用いることができる。クリーニング装置6は、電子写真感光体1に付着している残留トナーをクリーニング部材で掻き落とし、残留トナーを回収するものである。但し、感光体表面に残留するトナーが少ないか、殆ど無い場合には、クリーニング装置6は無くても構わない。
記録紙P上に転写されたトナーは、所定温度に加熱された上部定着部材71と下部定着部材72との間を通過する際、トナーが溶融状態まで熱加熱され、通過後冷却されて記録紙P上にトナーが定着される。
以上のように構成された電子写真装置では、次のようにして画像の記録が行なわれる。即ち、まず電子写真感光体1の表面(感光面)が、帯電装置2によって所定の電位(例えば-600V)に帯電される。この際、直流電圧により帯電させてもよく、直流電圧に交流電圧を重畳させて帯電させてもよい。
続いて、帯電された電子写真感光体1の感光面を、記録すべき画像に応じて露光装置3により露光し、感光面に静電潜像を形成する。そして、その電子写真感光体1の感光面に形成された静電潜像の現像を、現像装置4で行なう。
現像装置4は、供給ローラ43により供給されるトナーTを、規制部材(現像ブレード)45により薄層化するとともに、所定の極性(ここでは電子写真感光体1の帯電電位と同極性であり、負極性)に摩擦帯電させ、現像ローラ44に担持しながら搬送して、電子写真感光体1の表面に接触させる。
現像ローラ44に担持された帯電トナーTが電子写真感光体1の表面に接触すると、静電潜像に対応するトナー像が電子写真感光体1の感光面に形成される。そしてこのトナー像は、転写装置5によって記録紙Pに転写される。この後、転写されずに電子写真感光体1の感光面に残留しているトナーが、クリーニング装置6で除去される。
トナー像の記録紙P上への転写後、定着装置7を通過させてトナー像を記録紙P上へ熱定着することで、最終的な画像が得られる。
なお、画像形成装置は、上述した構成に加え、例えば除電工程を行なうことができる構成としてもよい。除電工程は、電子写真感光体に露光を行なうことで電子写真感光体の除電を行なう工程であり、除電装置としては、蛍光灯、LED等が使用される。また除電工程で用いる光は、強度としては露光光の3倍以上の露光エネルギーを有する光である場合が多い。
また、画像形成装置は更に変形して構成してもよく、例えば、前露光工程、補助帯電工程などの工程を行なうことができる構成としたり、オフセット印刷を行なう構成としたり、更には複数種のトナーを用いたフルカラータンデム方式の構成としてもよい。
なお、電子写真感光体1を、帯電装置2、露光装置3、現像装置4、転写装置5、クリーニング装置6、及び定着装置7のうち1つ又は2つ以上と組み合わせて、一体型のカートリッジ(以下適宜「電子写真感光体カートリッジ」という)として構成し、この電子写真感光体カートリッジを複写機やレーザービームプリンタ等の電子写真装置本体に対して着脱可能な構成にしてもよい。
この場合、例えば電子写真感光体1やその他の部材が劣化した場合に、この電子写真感光体カートリッジを画像形成装置本体から取り外し、別の新しい電子写真感光体カートリッジを画像形成装置本体に装着することにより、画像形成装置の保守・管理が容易となる。
以下に、本発明の具体的態様を実施例によりさらに詳細に説明するが、本発明はその要旨を越えない限り、これらの実施例によって限定されるものではない。
(試験例1)
[エステルオリゴマーの製造]
製造例1-1(エステルオリゴマー(1)の合成)
窒素置換した300mL4つ口反応容器にレゾルシノール(3.00g)、1,1-ビス(4-ヒドロキシ-3,メチルフェニル)エタン(13.20g)(以下、BP-a)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(16.08g)を秤取り、ジクロロメタン(90mL)に溶解させた。続いて、トリエチルアミン(11.58g)とジクロロメタン(45mL)との混合溶液を0~5℃へ冷却した反応容器内へ30分かけて滴下した。1時間撹拌を続けた後、脱塩水(100mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(100mL)にて洗浄を2回行い、さらに、脱塩水(100mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(1)を得た。
[エステルオリゴマーの製造]
製造例1-1(エステルオリゴマー(1)の合成)
窒素置換した300mL4つ口反応容器にレゾルシノール(3.00g)、1,1-ビス(4-ヒドロキシ-3,メチルフェニル)エタン(13.20g)(以下、BP-a)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(16.08g)を秤取り、ジクロロメタン(90mL)に溶解させた。続いて、トリエチルアミン(11.58g)とジクロロメタン(45mL)との混合溶液を0~5℃へ冷却した反応容器内へ30分かけて滴下した。1時間撹拌を続けた後、脱塩水(100mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(100mL)にて洗浄を2回行い、さらに、脱塩水(100mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(1)を得た。
製造例1-2(エステルオリゴマー(2)の合成)
窒素置換した300mL4つ口反応容器にメチルヒドロキノン(3.00g)、BP-a(11.71g)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(14.26g)を秤取り、ジクロロメタン(90mL)に溶解させた。続いて、トリエチルアミン(10.28g)とジクロロメタン(30mL)との混合溶液を0~5℃へ冷却した反応容器内へ30分かけて滴下した。1時間撹拌を続けた後、脱塩水(100mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(100mL)にて洗浄を2回行い、さらに、脱塩水(100mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(2)を得た。
窒素置換した300mL4つ口反応容器にメチルヒドロキノン(3.00g)、BP-a(11.71g)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(14.26g)を秤取り、ジクロロメタン(90mL)に溶解させた。続いて、トリエチルアミン(10.28g)とジクロロメタン(30mL)との混合溶液を0~5℃へ冷却した反応容器内へ30分かけて滴下した。1時間撹拌を続けた後、脱塩水(100mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(100mL)にて洗浄を2回行い、さらに、脱塩水(100mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(2)を得た。
製造例1-3(エステルオリゴマー(3)の合成)
窒素置換した300mL4つ口反応容器にBP-a(5.86g)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(14.29g)を秤取り、ジクロロメタン(60mL)に溶解させた。続いて、トリエチルアミン(5.14g)とジクロロメタン(40mL)との混合溶液を0~5℃へ冷却した反応容器内へ30分かけて滴下した。30分撹拌を続けた後、BP-a(9.10g)とヒドロキノン(1.20g)を反応容器へ加えた。その後、トリエチルアミン(5.14g)とジクロロメタン(20mL)との混合溶液を反応容器内へ30分かけて滴下した。1時間攪拌を続けた後、脱塩水(100mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(100mL)にて洗浄を2回行い、さらに、脱塩水(100mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(3)を得た。
窒素置換した300mL4つ口反応容器にBP-a(5.86g)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(14.29g)を秤取り、ジクロロメタン(60mL)に溶解させた。続いて、トリエチルアミン(5.14g)とジクロロメタン(40mL)との混合溶液を0~5℃へ冷却した反応容器内へ30分かけて滴下した。30分撹拌を続けた後、BP-a(9.10g)とヒドロキノン(1.20g)を反応容器へ加えた。その後、トリエチルアミン(5.14g)とジクロロメタン(20mL)との混合溶液を反応容器内へ30分かけて滴下した。1時間攪拌を続けた後、脱塩水(100mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(100mL)にて洗浄を2回行い、さらに、脱塩水(100mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(3)を得た。
製造例1-4(エステルオリゴマー(4)の合成)
窒素置換した300mL4つ口反応容器に1,6-ジヒドロキシナフタレン(4.00g)、BP-a(12.10g)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(14.74g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(10.62g)とジクロロメタン(45mL)との混合溶液を0~5℃へ冷却した反応容器内へ30分かけて滴下した。1時間撹拌を続けた後、脱塩水(100mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(100mL)にて洗浄を2回行い、さらに、脱塩水(100mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(4)を得た。
窒素置換した300mL4つ口反応容器に1,6-ジヒドロキシナフタレン(4.00g)、BP-a(12.10g)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(14.74g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(10.62g)とジクロロメタン(45mL)との混合溶液を0~5℃へ冷却した反応容器内へ30分かけて滴下した。1時間撹拌を続けた後、脱塩水(100mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(100mL)にて洗浄を2回行い、さらに、脱塩水(100mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(4)を得た。
製造例1-5(エステルオリゴマー(5)の合成)
窒素置換した300mL4つ口反応容器に1,5-ジヒドロキシナフタレン(1.50g)、BP-a(12.86g)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(12.28g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(8.84g)とジクロロメタン(40mL)との混合溶液を0~5℃へ冷却した反応容器内へ30分かけて滴下した。1時間撹拌を続けた後、脱塩水(100mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(100mL)にて洗浄を2回行い、さらに、脱塩水(100mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(5)を得た。
窒素置換した300mL4つ口反応容器に1,5-ジヒドロキシナフタレン(1.50g)、BP-a(12.86g)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(12.28g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(8.84g)とジクロロメタン(40mL)との混合溶液を0~5℃へ冷却した反応容器内へ30分かけて滴下した。1時間撹拌を続けた後、脱塩水(100mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(100mL)にて洗浄を2回行い、さらに、脱塩水(100mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(5)を得た。
製造例1-6(エステルオリゴマー(6)の合成)
窒素置換した500mL4つ口反応容器にBP-a(7.00g)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(17.05g)を秤取り、ジクロロメタン(160mL)に溶解させた。続いて、トリエチルアミン(6.14g)とジクロロメタン(20mL)との混合溶液を0~5℃へ冷却した反応容器内へ20分かけて滴下した。5分撹拌を続けた後、BP-a(11.90g)と4―ヒドロキシ安息香酸4―ヒドロキシフェニル(2.00g)を反応容器へ加えた。その後、トリエチルアミン(6.14g)とジクロロメタン(20mL)との混合溶液を反応容器内へ20分かけて滴下した。1時間攪拌を続けた後、0.1規定塩酸(170mL)を添加し、30分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(170mL)にて洗浄を2回行い、さらに、脱塩水(170mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(6)を得た。
窒素置換した500mL4つ口反応容器にBP-a(7.00g)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(17.05g)を秤取り、ジクロロメタン(160mL)に溶解させた。続いて、トリエチルアミン(6.14g)とジクロロメタン(20mL)との混合溶液を0~5℃へ冷却した反応容器内へ20分かけて滴下した。5分撹拌を続けた後、BP-a(11.90g)と4―ヒドロキシ安息香酸4―ヒドロキシフェニル(2.00g)を反応容器へ加えた。その後、トリエチルアミン(6.14g)とジクロロメタン(20mL)との混合溶液を反応容器内へ20分かけて滴下した。1時間攪拌を続けた後、0.1規定塩酸(170mL)を添加し、30分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(170mL)にて洗浄を2回行い、さらに、脱塩水(170mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(6)を得た。
製造例1-7(エステルオリゴマー(7)の合成)
窒素置換した500mL4つ口反応容器にBP-a(18.54g)とtrans-1,4-シクロヘキサンジカルボン酸クロライド(イハラニッケイ化学工業製)(8.00g)を秤取り、ジクロロメタン(160mL)に溶解させた。続いて、トリエチルアミン(8.52g)とジクロロメタン(30mL)との混合溶液を0~5℃へ冷却した反応容器内へ30分かけて滴下した。1時間撹拌を続けた後、脱塩水(150mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(150mL)にて洗浄を2回行い、さらに、脱塩水(150mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(7)を得た。
窒素置換した500mL4つ口反応容器にBP-a(18.54g)とtrans-1,4-シクロヘキサンジカルボン酸クロライド(イハラニッケイ化学工業製)(8.00g)を秤取り、ジクロロメタン(160mL)に溶解させた。続いて、トリエチルアミン(8.52g)とジクロロメタン(30mL)との混合溶液を0~5℃へ冷却した反応容器内へ30分かけて滴下した。1時間撹拌を続けた後、脱塩水(150mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(150mL)にて洗浄を2回行い、さらに、脱塩水(150mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(7)を得た。
製造例1-8(エステルオリゴマー(8)の合成)
窒素置換した500mL4つ口反応容器にBP-a(17.31g)と1,4-フェニレン二酢酸ジクロライド(イハラニッケイ化学工業製)(10.00g)を秤取り、ジクロロメタン(150mL)に溶解させた。続いて、トリエチルアミン(9.19g)とジクロロメタン(65mL)との混合溶液を0~5℃へ冷却した反応容器内へ1時間かけて滴下した。1時間撹拌を続けた後、脱塩水(150mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(150mL)にて洗浄を2回行い、さらに、脱塩水(150mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(8)を得た。
窒素置換した500mL4つ口反応容器にBP-a(17.31g)と1,4-フェニレン二酢酸ジクロライド(イハラニッケイ化学工業製)(10.00g)を秤取り、ジクロロメタン(150mL)に溶解させた。続いて、トリエチルアミン(9.19g)とジクロロメタン(65mL)との混合溶液を0~5℃へ冷却した反応容器内へ1時間かけて滴下した。1時間撹拌を続けた後、脱塩水(150mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(150mL)にて洗浄を2回行い、さらに、脱塩水(150mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(8)を得た。
製造例1-9(エステルオリゴマー(9)の合成)
窒素置換した500mL4つ口反応容器に1,4-ベンゼンジメタノール(3.00g)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(12.81g)を秤取り、ジクロロメタン(130mL)に溶解させた。続いて、トリエチルアミン(4.61g)とジクロロメタン(20mL)との混合溶液を0~5℃へ冷却した反応容器内へ30分かけて滴下した。2時間撹拌を続けた後、BP-a(10.52g)を反応容器へ加えた。その後、トリエチルアミン(4.61g)とジクロロメタン(20mL)との混合溶液を反応容器内へ30分かけて滴下した。1時間攪拌を続けた後、脱塩水(150mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(150mL)にて洗浄を2回行い、さらに、脱塩水(150mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(9)を得た。
窒素置換した500mL4つ口反応容器に1,4-ベンゼンジメタノール(3.00g)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(12.81g)を秤取り、ジクロロメタン(130mL)に溶解させた。続いて、トリエチルアミン(4.61g)とジクロロメタン(20mL)との混合溶液を0~5℃へ冷却した反応容器内へ30分かけて滴下した。2時間撹拌を続けた後、BP-a(10.52g)を反応容器へ加えた。その後、トリエチルアミン(4.61g)とジクロロメタン(20mL)との混合溶液を反応容器内へ30分かけて滴下した。1時間攪拌を続けた後、脱塩水(150mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(150mL)にて洗浄を2回行い、さらに、脱塩水(150mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(9)を得た。
製造例1-10(エステルオリゴマー(10)の合成)
窒素置換した300mL4つ口反応容器にイソソルビド(4.00g)とテレフタル酸クロライド(11.11g)を秤取り、ジクロロメタン(90mL)に溶解させた。続いて、トリエチルアミン(5.82g)とジクロロメタン(26mL)との混合溶液を0~5℃へ冷却した反応容器内へ30分かけて滴下した。3時間撹拌を続けた後、BP-a(13.26g)を反応容器へ加えた。その後、トリエチルアミン(5.82g)とジクロロメタン(20mL)との混合溶液を反応容器内へ30分かけて滴下した。1時間攪拌を続けた後、脱塩水(100mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(100mL)にて洗浄を2回行い、さらに、脱塩水(100mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(10)を得た。
窒素置換した300mL4つ口反応容器にイソソルビド(4.00g)とテレフタル酸クロライド(11.11g)を秤取り、ジクロロメタン(90mL)に溶解させた。続いて、トリエチルアミン(5.82g)とジクロロメタン(26mL)との混合溶液を0~5℃へ冷却した反応容器内へ30分かけて滴下した。3時間撹拌を続けた後、BP-a(13.26g)を反応容器へ加えた。その後、トリエチルアミン(5.82g)とジクロロメタン(20mL)との混合溶液を反応容器内へ30分かけて滴下した。1時間攪拌を続けた後、脱塩水(100mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(100mL)にて洗浄を2回行い、さらに、脱塩水(100mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(10)を得た。
製造例1-11(エステルオリゴマー(11)の合成)
窒素置換した300mL4つ口反応容器に4,4‘-ビフェニルジメタノール(4.50g)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(12.04g)を秤取り、ジクロロメタン(120mL)に溶解させた。続いて、トリエチルアミン(4.67g)とジクロロメタン(30mL)との混合溶液を0~5℃へ冷却した反応容器内へ20分かけて滴下した。反応容器を20℃へと昇温し、1時間撹拌を続けた後、BP-a(10.18g)を反応容器へ加えた。反応容器を0~5℃へ冷却した後、トリエチルアミン(4.67g)とジクロロメタン(30mL)との混合溶液を反応容器内へ20分かけて滴下した。1時間攪拌を続けた後、0.1規定塩酸(150mL)を添加し、30分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(150mL)にて洗浄を2回行い、さらに、脱塩水(150mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(11)を得た。
窒素置換した300mL4つ口反応容器に4,4‘-ビフェニルジメタノール(4.50g)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(12.04g)を秤取り、ジクロロメタン(120mL)に溶解させた。続いて、トリエチルアミン(4.67g)とジクロロメタン(30mL)との混合溶液を0~5℃へ冷却した反応容器内へ20分かけて滴下した。反応容器を20℃へと昇温し、1時間撹拌を続けた後、BP-a(10.18g)を反応容器へ加えた。反応容器を0~5℃へ冷却した後、トリエチルアミン(4.67g)とジクロロメタン(30mL)との混合溶液を反応容器内へ20分かけて滴下した。1時間攪拌を続けた後、0.1規定塩酸(150mL)を添加し、30分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(150mL)にて洗浄を2回行い、さらに、脱塩水(150mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(11)を得た。
実施例1-1(ポリエステル樹脂(1)の製造)
500mLビーカーに水酸化ナトリウム(1.57g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.21g)、及びベンジルトリエチルアンモニウムクロライド(0.11g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、製造例1-1で合成したエステルオリゴマー(1)(16.13g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(4.87g)とジクロロメタン(107mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(166mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(205mL)にて洗浄を3回行い、さらに、脱塩水(205mL)にて洗浄を2回行った。洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は44,200であった。ポリエステル樹脂(1)の構造式を以下に示す。
500mLビーカーに水酸化ナトリウム(1.57g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.21g)、及びベンジルトリエチルアンモニウムクロライド(0.11g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、製造例1-1で合成したエステルオリゴマー(1)(16.13g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(4.87g)とジクロロメタン(107mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(166mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(205mL)にて洗浄を3回行い、さらに、脱塩水(205mL)にて洗浄を2回行った。洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は44,200であった。ポリエステル樹脂(1)の構造式を以下に示す。

[粘度平均分子量(Mv)の測定]
ポリエステル樹脂をジクロロメタンに溶解し濃度Cが6.00g/Lの溶液を調製した。溶媒(ジクロロメタン)の流下時間t0が136.16秒のウベローデ型毛細管粘度計を用いて、20.0℃に設定した恒温水槽中で試料溶液の流下時間tを測定した。以下の式に従って粘度平均分子量(Mv)を算出した。
a=0.438×ηsp+1 ηsp=t/t0-1
b=100×ηsp/C C=6.00(g/L)
η=b/a
Mv=3207×η1.205
ポリエステル樹脂をジクロロメタンに溶解し濃度Cが6.00g/Lの溶液を調製した。溶媒(ジクロロメタン)の流下時間t0が136.16秒のウベローデ型毛細管粘度計を用いて、20.0℃に設定した恒温水槽中で試料溶液の流下時間tを測定した。以下の式に従って粘度平均分子量(Mv)を算出した。
a=0.438×ηsp+1 ηsp=t/t0-1
b=100×ηsp/C C=6.00(g/L)
η=b/a
Mv=3207×η1.205
実施例1-2(ポリエステル樹脂(2)の製造)
500mLビーカーに水酸化ナトリウム(1.55g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.21g)、及びベンジルトリエチルアンモニウムクロライド(0.11g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、製造例1-2で合成したエステルオリゴマー(2)(16.18g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(4.81g)とジクロロメタン(107mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(166mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(205mL)にて洗浄を3回行い、さらに、脱塩水(205mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(2)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は54,200であった。ポリエステル樹脂(2)の構造式を以下に示す。
500mLビーカーに水酸化ナトリウム(1.55g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.21g)、及びベンジルトリエチルアンモニウムクロライド(0.11g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、製造例1-2で合成したエステルオリゴマー(2)(16.18g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(4.81g)とジクロロメタン(107mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(166mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(205mL)にて洗浄を3回行い、さらに、脱塩水(205mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(2)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は54,200であった。ポリエステル樹脂(2)の構造式を以下に示す。

実施例1-3(ポリエステル樹脂(3)の製造)
500mLビーカーに水酸化ナトリウム(1.87g)とH2O(235ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.32g)、及びベンジルトリエチルアンモニウムクロライド(0.14g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、製造例1-3で合成したエステルオリゴマー(3)(20.33g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(5.78g)とジクロロメタン(134mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(200mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(250mL)にて洗浄を3回行い、さらに、脱塩水(250mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(150ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(3)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は43,500であった。ポリエステル樹脂(3)の構造式を以下に示す。
500mLビーカーに水酸化ナトリウム(1.87g)とH2O(235ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.32g)、及びベンジルトリエチルアンモニウムクロライド(0.14g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、製造例1-3で合成したエステルオリゴマー(3)(20.33g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(5.78g)とジクロロメタン(134mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(200mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(250mL)にて洗浄を3回行い、さらに、脱塩水(250mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(150ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(3)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は43,500であった。ポリエステル樹脂(3)の構造式を以下に示す。

実施例1-4(ポリエステル樹脂(4)の製造)
500mLビーカーに水酸化ナトリウム(1.52g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.24g)、及びベンジルトリエチルアンモニウムクロライド(0.11g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、製造例1-4で合成したエステルオリゴマー(4)(16.22g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(4.70g)とジクロロメタン(107mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(176mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(210mL)にて洗浄を3回行い、さらに、脱塩水(210mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(4)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は39,000であった。ポリエステル樹脂(4)の構造式を以下に示す。
500mLビーカーに水酸化ナトリウム(1.52g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.24g)、及びベンジルトリエチルアンモニウムクロライド(0.11g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、製造例1-4で合成したエステルオリゴマー(4)(16.22g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(4.70g)とジクロロメタン(107mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(176mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(210mL)にて洗浄を3回行い、さらに、脱塩水(210mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(4)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は39,000であった。ポリエステル樹脂(4)の構造式を以下に示す。

実施例1-5(ポリエステル樹脂(5)の製造)
500mLビーカーに水酸化ナトリウム(1.47g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.25g)、及びベンジルトリエチルアンモニウムクロライド(0.11g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、製造例1-5で合成したエステルオリゴマー(5)(16.32g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(4.56g)とジクロロメタン(107mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(176mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(210mL)にて洗浄を3回行い、さらに、脱塩水(210mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(5)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は41,300であった。ポリエステル樹脂(5)の構造式を以下に示す。
500mLビーカーに水酸化ナトリウム(1.47g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.25g)、及びベンジルトリエチルアンモニウムクロライド(0.11g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、製造例1-5で合成したエステルオリゴマー(5)(16.32g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(4.56g)とジクロロメタン(107mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(176mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(210mL)にて洗浄を3回行い、さらに、脱塩水(210mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(5)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は41,300であった。ポリエステル樹脂(5)の構造式を以下に示す。

実施例1-6(ポリエステル樹脂(6)の製造)
500mLビーカーに水酸化ナトリウム(1.78g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.26g)、及びベンジルトリエチルアンモニウムクロライド(0.08g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、製造例1-6で合成したエステルオリゴマー(6)(20.57g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(5.53g)とジクロロメタン(135mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(179mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(235mL)にて洗浄を3回行い、さらに、脱塩水(235mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(180ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(6)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は51,000であった。ポリエステル樹脂(6)の構造式を以下に示す。
500mLビーカーに水酸化ナトリウム(1.78g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.26g)、及びベンジルトリエチルアンモニウムクロライド(0.08g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、製造例1-6で合成したエステルオリゴマー(6)(20.57g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(5.53g)とジクロロメタン(135mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(179mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(235mL)にて洗浄を3回行い、さらに、脱塩水(235mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(180ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(6)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は51,000であった。ポリエステル樹脂(6)の構造式を以下に示す。

実施例1-7(ポリエステル樹脂(7)の製造)
500mLビーカーに水酸化ナトリウム(2.31g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.25g)、及びベンジルトリエチルアンモニウムクロライド(0.17g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、製造例1-7で合成したエステルオリゴマー(7)(14.37g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(7.16g)とジクロロメタン(107mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(160mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(200mL)にて洗浄を3回行い、さらに、脱塩水(200mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(7)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は35,300であった。ポリエステル樹脂(7)の構造式を以下に示す。
500mLビーカーに水酸化ナトリウム(2.31g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.25g)、及びベンジルトリエチルアンモニウムクロライド(0.17g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、製造例1-7で合成したエステルオリゴマー(7)(14.37g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(7.16g)とジクロロメタン(107mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(160mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(200mL)にて洗浄を3回行い、さらに、脱塩水(200mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(7)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は35,300であった。ポリエステル樹脂(7)の構造式を以下に示す。

実施例1-8(ポリエステル樹脂(8)の製造)
500mLビーカーに水酸化ナトリウム(2.26g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.25g)、及びベンジルトリエチルアンモニウムクロライド(0.17g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、製造例1-8で合成したエステルオリゴマー(8)(14.50g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(6.99g)とジクロロメタン(107mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(160mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(200mL)にて洗浄を3回行い、さらに、脱塩水(200mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(8)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は20,200であった。ポリエステル樹脂(8)の構造式を以下に示す。
500mLビーカーに水酸化ナトリウム(2.26g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.25g)、及びベンジルトリエチルアンモニウムクロライド(0.17g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、製造例1-8で合成したエステルオリゴマー(8)(14.50g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(6.99g)とジクロロメタン(107mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(160mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(200mL)にて洗浄を3回行い、さらに、脱塩水(200mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(8)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は20,200であった。ポリエステル樹脂(8)の構造式を以下に示す。

実施例1-9(ポリエステル樹脂(9)の製造)
500mLビーカーに水酸化ナトリウム(2.72g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこにBP-a(4.64g)、2,3,5-トリメチルフェノール(0.26g)、及びベンジルトリエチルアンモニウムクロライド(0.20g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、製造例1-9で合成したエステルオリゴマー(9)(8.76g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(8.43g)とジクロロメタン(107mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(160mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(200mL)にて洗浄を3回行い、さらに、脱塩水(200mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(9)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は33,200であった。ポリエステル樹脂(9)の構造式を以下に示す。
500mLビーカーに水酸化ナトリウム(2.72g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこにBP-a(4.64g)、2,3,5-トリメチルフェノール(0.26g)、及びベンジルトリエチルアンモニウムクロライド(0.20g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、製造例1-9で合成したエステルオリゴマー(9)(8.76g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(8.43g)とジクロロメタン(107mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(160mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(200mL)にて洗浄を3回行い、さらに、脱塩水(200mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(9)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は33,200であった。ポリエステル樹脂(9)の構造式を以下に示す。

実施例1-10(ポリエステル樹脂(10)の製造)
500mLビーカーに水酸化ナトリウム(1.77g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.25g)、及びベンジルトリエチルアンモニウムクロライド(0.13g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、製造例1-10で合成したエステルオリゴマー(10)(15.62g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(5.50g)とジクロロメタン(107mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(143mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(190mL)にて洗浄を3回行い、さらに、脱塩水(190mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(10)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は22,500であった。ポリエステル樹脂(10)の構造式を以下に示す。
500mLビーカーに水酸化ナトリウム(1.77g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.25g)、及びベンジルトリエチルアンモニウムクロライド(0.13g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、製造例1-10で合成したエステルオリゴマー(10)(15.62g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(5.50g)とジクロロメタン(107mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(143mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(190mL)にて洗浄を3回行い、さらに、脱塩水(190mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(10)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は22,500であった。ポリエステル樹脂(10)の構造式を以下に示す。

実施例1-11(ポリエステル樹脂(11)の製造)
500mLビーカーに水酸化ナトリウム(1.45g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.20g)、及びベンジルトリエチルアンモニウムクロライド(0.07g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、製造例1-11で合成したエステルオリゴマー(11)(16.42g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(4.62g)とジクロロメタン(94mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(156mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(190mL)にて洗浄を3回行い、さらに、脱塩水(190mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(11)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は40,200であった。ポリエステル樹脂(11)の構造式を以下に示す。
500mLビーカーに水酸化ナトリウム(1.45g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.20g)、及びベンジルトリエチルアンモニウムクロライド(0.07g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、製造例1-11で合成したエステルオリゴマー(11)(16.42g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(4.62g)とジクロロメタン(94mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(156mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(190mL)にて洗浄を3回行い、さらに、脱塩水(190mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(11)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は40,200であった。ポリエステル樹脂(11)の構造式を以下に示す。

実施例1-12(ポリエステルポリカーボネート樹脂(12)の製造)
500mLビーカーに水酸化ナトリウム(4.16g)とH2O(234ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.34g)、BP-a(10.15g)、及びベンジルトリエチルアンモニウムクロライド(0.11g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、日本国特開2012-185206号公報の製造例1-1に記載の溶融重合法で製造した下記式(13)のポリカーボネート樹脂(Mv26,000、末端OH量63.8μ当量/g)(4.81g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(12.86g)とジクロロメタン(117mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(195mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(235mL)にて洗浄を3回行い、さらに、脱塩水(235mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(150ml)を加えて希釈し、メタノール(2300ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステルポリカーボネート樹脂(12)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は40,100であった。ポリエステル樹脂(12)の構造式を以下に示す。
500mLビーカーに水酸化ナトリウム(4.16g)とH2O(234ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.34g)、BP-a(10.15g)、及びベンジルトリエチルアンモニウムクロライド(0.11g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、日本国特開2012-185206号公報の製造例1-1に記載の溶融重合法で製造した下記式(13)のポリカーボネート樹脂(Mv26,000、末端OH量63.8μ当量/g)(4.81g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(12.86g)とジクロロメタン(117mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(195mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(235mL)にて洗浄を3回行い、さらに、脱塩水(235mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(150ml)を加えて希釈し、メタノール(2300ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステルポリカーボネート樹脂(12)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は40,100であった。ポリエステル樹脂(12)の構造式を以下に示す。
ポリエステルポリカーボネート樹脂(12)

ポリカーボネート樹脂(13)

比較例1-1(ポリエステル樹脂(3)の界面重合による製造)
500mLビーカーに水酸化ナトリウム(4.23g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.26g)、BP-a(10.15g)、ヒドロキノン(0.47g)、及びベンジルトリエチルアンモニウムクロライド(0.12g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、ジフェニルエーテル-4,4´-ジカルボン酸クロライド(13.08g)とジクロロメタン(94mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(156mL)を加え、撹拌を9時間続けた。しかしながら、重合中にヒドロキノンが酸化され水層が茶色に変色し、2価のフェノールが足りなくなったため、十分に伸長した重合体が得られなかった。
500mLビーカーに水酸化ナトリウム(4.23g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.26g)、BP-a(10.15g)、ヒドロキノン(0.47g)、及びベンジルトリエチルアンモニウムクロライド(0.12g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、ジフェニルエーテル-4,4´-ジカルボン酸クロライド(13.08g)とジクロロメタン(94mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(156mL)を加え、撹拌を9時間続けた。しかしながら、重合中にヒドロキノンが酸化され水層が茶色に変色し、2価のフェノールが足りなくなったため、十分に伸長した重合体が得られなかった。
比較例1-2(ポリエステル樹脂(6)の界面重合による製造)
500mLビーカーに水酸化ナトリウム(4.13g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.26g)、BP-a(9.16g)、4―ヒドロキシ安息香酸4―ヒドロキシフェニルエーテル(0.97g)、及びベンジルトリエチルアンモニウムクロライド(0.12g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、ジフェニルエーテル-4,4´-ジカルボン酸クロライド(12.75g)とジクロロメタン(94mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(156mL)を加え、撹拌を9時間続けた。しかしながら、重合中に4―ヒドロキシ安息香酸4―ヒドロキシフェニルエーテルが加水分解され、ヒドロキノンとなり酸化され水層が茶色に変色し、2価のフェノールが足りなくなったため、十分に伸長した重合体が得られなかった。
500mLビーカーに水酸化ナトリウム(4.13g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.26g)、BP-a(9.16g)、4―ヒドロキシ安息香酸4―ヒドロキシフェニルエーテル(0.97g)、及びベンジルトリエチルアンモニウムクロライド(0.12g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、ジフェニルエーテル-4,4´-ジカルボン酸クロライド(12.75g)とジクロロメタン(94mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(156mL)を加え、撹拌を9時間続けた。しかしながら、重合中に4―ヒドロキシ安息香酸4―ヒドロキシフェニルエーテルが加水分解され、ヒドロキノンとなり酸化され水層が茶色に変色し、2価のフェノールが足りなくなったため、十分に伸長した重合体が得られなかった。
比較例1-3(ポリエステル樹脂(11)の界面重合による製造)
500mLビーカーに水酸化ナトリウム(4.14g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.26g)、BP-a(6.93g)、4,4‘-ビフェニルジメタノール(3.02g)、及びベンジルトリエチルアンモニウムクロライド(0.12g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、ジフェニルエーテル-4,4´-ジカルボン酸クロライド(12.97g)とジクロロメタン(94mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(156mL)を加え、撹拌を9時間続けた。しかしながら、4,4’-ビフェニルジメタノールの水酸基は、反応性が悪いため、十分に伸長した重合体が得られなかった。
500mLビーカーに水酸化ナトリウム(4.14g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.26g)、BP-a(6.93g)、4,4‘-ビフェニルジメタノール(3.02g)、及びベンジルトリエチルアンモニウムクロライド(0.12g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、ジフェニルエーテル-4,4´-ジカルボン酸クロライド(12.97g)とジクロロメタン(94mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(156mL)を加え、撹拌を9時間続けた。しかしながら、4,4’-ビフェニルジメタノールの水酸基は、反応性が悪いため、十分に伸長した重合体が得られなかった。
<感光体シートの作製>
[実施例1-13]
10質量部のオキシチタニウムフタロシアニンと、150質量部の4-メトキシ-4-メチル-2-ペンタノンとを混合し、サンドグラインドミルにて粉砕分散処理を行い顔料分散液を製造した。尚、オキシチタニウムフタロシアニンは、CuKα線によるX線回折においてブラッグ角(2θ±0.2)9.3゜、10.6゜、13.2゜、15.1゜、15.7゜、16.1゜、20.8゜、23.3゜、26.3゜、27.1゜に強い回折ピークを示す。
この顔料分散液に、ポリビニルブチラール(電気化学工業株式会社製、商品名デンカブチラール♯6000C)の5質量%1,2-ジメトキシエタン溶液を50質量部、フェノキシ樹脂(ユニオンカーバイド株式会社製、商品名PKHH)の5質量%1,2-ジメトキシエタン溶液を50質量部混合し、更に、適量の1,2-ジメトキシエタンを加え、固形分濃度4.0%の電荷発生層形成用塗布液を調製した。この電荷発生層形成用塗布液を、表面にアルミ蒸着したポリエチレンテレフタレートシート上に、乾燥後の膜厚が0.4μmになるように塗布、乾燥して電荷発生層を設けた。
次に、電荷輸送物質として、下記に示す構造を主成分とする、幾何異性体の化合物群からなる、日本国特開2002-080432号公報の実施例1-1に記載の方法で製造した混合物(CTM-1)を50質量部、実施例1-1で製造したポリエステル樹脂(1)を100質量部、酸化防止剤(イルガノックス1076)8質量部、レベリング剤としてシリコーンオイル0.05質量部を、テトラヒドロフランとトルエンとの混合溶媒(テトラヒドロフラン80質量%、トルエン20質量%)640質量部に混合し、電荷輸送層形成用塗布液を調製した。
[実施例1-13]
10質量部のオキシチタニウムフタロシアニンと、150質量部の4-メトキシ-4-メチル-2-ペンタノンとを混合し、サンドグラインドミルにて粉砕分散処理を行い顔料分散液を製造した。尚、オキシチタニウムフタロシアニンは、CuKα線によるX線回折においてブラッグ角(2θ±0.2)9.3゜、10.6゜、13.2゜、15.1゜、15.7゜、16.1゜、20.8゜、23.3゜、26.3゜、27.1゜に強い回折ピークを示す。
この顔料分散液に、ポリビニルブチラール(電気化学工業株式会社製、商品名デンカブチラール♯6000C)の5質量%1,2-ジメトキシエタン溶液を50質量部、フェノキシ樹脂(ユニオンカーバイド株式会社製、商品名PKHH)の5質量%1,2-ジメトキシエタン溶液を50質量部混合し、更に、適量の1,2-ジメトキシエタンを加え、固形分濃度4.0%の電荷発生層形成用塗布液を調製した。この電荷発生層形成用塗布液を、表面にアルミ蒸着したポリエチレンテレフタレートシート上に、乾燥後の膜厚が0.4μmになるように塗布、乾燥して電荷発生層を設けた。
次に、電荷輸送物質として、下記に示す構造を主成分とする、幾何異性体の化合物群からなる、日本国特開2002-080432号公報の実施例1-1に記載の方法で製造した混合物(CTM-1)を50質量部、実施例1-1で製造したポリエステル樹脂(1)を100質量部、酸化防止剤(イルガノックス1076)8質量部、レベリング剤としてシリコーンオイル0.05質量部を、テトラヒドロフランとトルエンとの混合溶媒(テトラヒドロフラン80質量%、トルエン20質量%)640質量部に混合し、電荷輸送層形成用塗布液を調製した。
電荷輸送材(CTM-1)

この電荷輸送層形成用塗布液を上述の電荷発生層上に、乾燥後の膜厚が25μmとなるようにアプリケーターを用いて塗布し、125℃で20分間乾燥して電荷輸送層を形成して、感光体シートを作製した。
[実施例1-14]
ポリエステル樹脂(1)をポリエステル樹脂(2)に変えた以外は実施例1-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(1)をポリエステル樹脂(2)に変えた以外は実施例1-1と同様にして、感光体シートを作製した。
[実施例1-15]
ポリエステル樹脂(1)をポリエステル樹脂(3)に変えた以外は実施例1-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(1)をポリエステル樹脂(3)に変えた以外は実施例1-1と同様にして、感光体シートを作製した。
[実施例1-16]
ポリエステル樹脂(1)をポリエステル樹脂(4)に変えた以外は実施例1-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(1)をポリエステル樹脂(4)に変えた以外は実施例1-1と同様にして、感光体シートを作製した。
[実施例1-17]
ポリエステル樹脂(1)をポリエステル樹脂(5)に変えた以外は実施例1-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(1)をポリエステル樹脂(5)に変えた以外は実施例1-1と同様にして、感光体シートを作製した。
[実施例1-18]
ポリエステル樹脂(1)をポリエステル樹脂(6)に変えた以外は実施例1-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(1)をポリエステル樹脂(6)に変えた以外は実施例1-1と同様にして、感光体シートを作製した。
[実施例1-19]
ポリエステル樹脂(1)をポリエステル樹脂(7)に変えた以外は実施例1-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(1)をポリエステル樹脂(7)に変えた以外は実施例1-1と同様にして、感光体シートを作製した。
[実施例1-20]
ポリエステル樹脂(1)をポリエステル樹脂(8)に変えた以外は実施例1-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(1)をポリエステル樹脂(8)に変えた以外は実施例1-1と同様にして、感光体シートを作製した。
[実施例1-21]
ポリエステル樹脂(1)をポリエステル樹脂(9)に変えた以外は実施例1-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(1)をポリエステル樹脂(9)に変えた以外は実施例1-1と同様にして、感光体シートを作製した。
[実施例1-22]
ポリエステル樹脂(1)をポリエステル樹脂(10)に変えた以外は実施例1-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(1)をポリエステル樹脂(10)に変えた以外は実施例1-1と同様にして、感光体シートを作製した。
[実施例1-23]
ポリエステル樹脂(1)をポリエステル樹脂(11)に変えた以外は実施例1-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(1)をポリエステル樹脂(11)に変えた以外は実施例1-1と同様にして、感光体シートを作製した。
[実施例1-24]
ポリエステル樹脂(1)をポリエステル樹脂(12)に変えた以外は実施例1-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(1)をポリエステル樹脂(12)に変えた以外は実施例1-1と同様にして、感光体シートを作製した。
[参考例1-1]
ポリエステル樹脂(1)を日本国特開2006-53549号公報の実施例6に記載の方法により製造した下記に示す構造を有するポリエステル樹脂(14)(粘度平均分子量36,200)に変えた以外は実施例1-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(1)を日本国特開2006-53549号公報の実施例6に記載の方法により製造した下記に示す構造を有するポリエステル樹脂(14)(粘度平均分子量36,200)に変えた以外は実施例1-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(14)

[電気特性評価]
電子写真学会測定標準に従って作製された電子写真特性評価装置(続電子写真技術の基礎と応用、電子写真学会編、コロナ社、404-405頁記載(1996年))を使用し、上記感光体をアルミニウム製ドラムに貼り付けて円筒状にし、アルミニウム製ドラムと感光体のアルミニウム基体との導通を取った上で、ドラムを一定回転数で回転させ、帯電、露光、電位測定、除電のサイクルによる電気特性評価試験を行った。その際、初期表面電位を-700Vとし、露光は780nm、除電は660nmの単色光を用い、露光光を2.4μJ/cm2照射した時点の表面電位(VL)を測定した。VL測定に際しては、露光から電位測定に要する時間を139msとした。また、表面電位が初期表面電位の半分(-350V)となる時の照射エネルギー(半減露光エネルギー:μJ/cm2)を感度(E1/2)として測定した。VLの値の絶対値が小さいほど電気特性が良好であることを示し、E1/2の値が小さいほど高感度であることを示す。測定環境は、温度25℃、相対湿度50%下(N/N)で行った。結果を表1-1に示す。
電子写真学会測定標準に従って作製された電子写真特性評価装置(続電子写真技術の基礎と応用、電子写真学会編、コロナ社、404-405頁記載(1996年))を使用し、上記感光体をアルミニウム製ドラムに貼り付けて円筒状にし、アルミニウム製ドラムと感光体のアルミニウム基体との導通を取った上で、ドラムを一定回転数で回転させ、帯電、露光、電位測定、除電のサイクルによる電気特性評価試験を行った。その際、初期表面電位を-700Vとし、露光は780nm、除電は660nmの単色光を用い、露光光を2.4μJ/cm2照射した時点の表面電位(VL)を測定した。VL測定に際しては、露光から電位測定に要する時間を139msとした。また、表面電位が初期表面電位の半分(-350V)となる時の照射エネルギー(半減露光エネルギー:μJ/cm2)を感度(E1/2)として測定した。VLの値の絶対値が小さいほど電気特性が良好であることを示し、E1/2の値が小さいほど高感度であることを示す。測定環境は、温度25℃、相対湿度50%下(N/N)で行った。結果を表1-1に示す。

[摩耗試験]
感光体フィルムを直径10cmの円状に切断しテーバー摩耗試験機(東洋精機社製)により、摩耗評価を行った。試験条件は、25℃、50%RHの雰囲気下、摩耗輪CS-10Fを用いて、荷重500gで1000回回転後の摩耗量を試験前後の質量を比較することにより測定した。結果を表1-2に示す。
感光体フィルムを直径10cmの円状に切断しテーバー摩耗試験機(東洋精機社製)により、摩耗評価を行った。試験条件は、25℃、50%RHの雰囲気下、摩耗輪CS-10Fを用いて、荷重500gで1000回回転後の摩耗量を試験前後の質量を比較することにより測定した。結果を表1-2に示す。

以上から、本発明により酸化されやすい、加水分解されやすいといった問題があった2価フェノールや、反応性の悪い2価アルコールを導入したポリエステル樹脂を製造できることを明らかとした。本方法により、感光体に求められる高分子量のポリエステル樹脂が製造できることも明らかとした。また、ポリエステルポリカーボネート樹脂においても簡便に製造できることを明らかとした。さらに、本発明により製造されたポリエステル樹脂から提供される感光体は、電気特性が良好であり、耐摩耗性等の特性も良好であることを明らかとした。
(試験例2)
[エステルオリゴマー中の残存酸クロライドモノマー測定]
[エステルオリゴマーの製造]
製造例2-1(エステルオリゴマー(2-1)の製造)
窒素置換した500mL4つ口反応容器に1,1-ビス(4-ヒドロキシ-3-メチルフェニル)エタン(以下、BP-1)(8.32g)、2,3,5-トリメチルフェノール(0.085g)及び以下、ジフェニルエーテル-4,4’-ジカルボン酸クロライド(以下、AC-1)(12.83g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(6.92g:東京化成製)とジクロロメタン(15mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることにより、反応容器内にエステルオリゴマー(2-1)を生成させた。
続いて、生成したエステルオリゴマー溶液に以下の酸クロライドモノマー測定用の内部標準として安息香酸メチル(6.41g)を添加した。
[エステルオリゴマー中の残存酸クロライドモノマー測定]
[エステルオリゴマーの製造]
製造例2-1(エステルオリゴマー(2-1)の製造)
窒素置換した500mL4つ口反応容器に1,1-ビス(4-ヒドロキシ-3-メチルフェニル)エタン(以下、BP-1)(8.32g)、2,3,5-トリメチルフェノール(0.085g)及び以下、ジフェニルエーテル-4,4’-ジカルボン酸クロライド(以下、AC-1)(12.83g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(6.92g:東京化成製)とジクロロメタン(15mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることにより、反応容器内にエステルオリゴマー(2-1)を生成させた。
続いて、生成したエステルオリゴマー溶液に以下の酸クロライドモノマー測定用の内部標準として安息香酸メチル(6.41g)を添加した。
[残存酸クロライドモノマー測定]
酸クロライドの分析は、酸クロライドをアミド化により安定化合物とした後に、HPLC分析により実施した。本特許ではアミド化物のHPLCピークを、酸クロライドモノマーのピークとして記載する。以下に実施方法を記載する。
酸クロライドの分析は、酸クロライドをアミド化により安定化合物とした後に、HPLC分析により実施した。本特許ではアミド化物のHPLCピークを、酸クロライドモノマーのピークとして記載する。以下に実施方法を記載する。
モルホリン(0.3ml:東京化成製)及びアセトニトリル(50ml:純正化学製、高速液クロ用)を入れた100mlサンプル瓶に、前記エステルオリゴマー(2-1)溶液(1.25g)を量りとり、混合させた。室温下で30分間放置し酸クロライド部位をアミド化した後、この溶液を1ml量りとり、下記HPLC分析で使用する移動相9mlと混合した。この混合溶液をGLクロマトディスク13P(ジーエルサイエンス(株)、孔径0.45μm)でろ過することにより固形分を除去し、サンプル溶液を調製した。
続いて、前記サンプル溶液を用いて下記の条件でHPLC分析を行い、酸クロライドモノマーピーク面積/安息香酸メチルピーク面積比を求め、下記検量線から酸クロライドモノマー量を求めた。エステルオリゴマー(2-1)に含まれる残存酸クロモノマー量を以下の表2-1に示す。
続いて、前記サンプル溶液を用いて下記の条件でHPLC分析を行い、酸クロライドモノマーピーク面積/安息香酸メチルピーク面積比を求め、下記検量線から酸クロライドモノマー量を求めた。エステルオリゴマー(2-1)に含まれる残存酸クロモノマー量を以下の表2-1に示す。
(HPLC測定条件)
カラム:Inertsil ODS-3 5μm 4.6mmX150mm(GLサイエンス製)
カラム温度:40℃
移動相:メタノール:純水:酢酸=60:40:0.05(vol%)
流速:1.0ml/min
測定時間:20min
検出波長:254nm
サンプル溶液注入量:10μl
カラム:Inertsil ODS-3 5μm 4.6mmX150mm(GLサイエンス製)
カラム温度:40℃
移動相:メタノール:純水:酢酸=60:40:0.05(vol%)
流速:1.0ml/min
測定時間:20min
検出波長:254nm
サンプル溶液注入量:10μl
(検量線の作成)
酸クロライドモノマーと安息香酸メチル比率を各100:50、50:50、20:50、10:50、5:50、3:50、1:50、0.5:50、0.1:50(mg)としたサンプルをそれぞれ前記のエステルオリゴマー溶液(2-1)と変更した以外は、前記残存酸クロライドモノマー測定と同様に、アミド化し、HPLC分析することにより、酸クロライドモノマーピーク面積/安息香酸メチルピーク面積比の検量線を作成した。
酸クロライドモノマーと安息香酸メチル比率を各100:50、50:50、20:50、10:50、5:50、3:50、1:50、0.5:50、0.1:50(mg)としたサンプルをそれぞれ前記のエステルオリゴマー溶液(2-1)と変更した以外は、前記残存酸クロライドモノマー測定と同様に、アミド化し、HPLC分析することにより、酸クロライドモノマーピーク面積/安息香酸メチルピーク面積比の検量線を作成した。
製造例2-2(エステルオリゴマー(2-2)の製造)
窒素置換した500mL4つ口反応容器にBP-1(7.27g)、2,3,5-トリメチルフェノール(0.090g)及び以下、AC-1(12.88g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(6.12g)とジクロロメタン(15mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることにより、反応容器内にエステルオリゴマー(2-2)を生成させた。続いて、生成したエステルオリゴマー溶液に安息香酸メチル(6.44g)を添加した。
製造例2-1と同様にエステルオリゴマー(2-2)に含まれる残存酸クロモノマー量の測定を行った。結果を以下の表2-1に示す。
窒素置換した500mL4つ口反応容器にBP-1(7.27g)、2,3,5-トリメチルフェノール(0.090g)及び以下、AC-1(12.88g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(6.12g)とジクロロメタン(15mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることにより、反応容器内にエステルオリゴマー(2-2)を生成させた。続いて、生成したエステルオリゴマー溶液に安息香酸メチル(6.44g)を添加した。
製造例2-1と同様にエステルオリゴマー(2-2)に含まれる残存酸クロモノマー量の測定を行った。結果を以下の表2-1に示す。
製造例2-3(エステルオリゴマー(2-3)の製造)
窒素置換した500mL4つ口反応容器にBP-1(6.27g)、2,3,5-トリメチルフェノール(0.090g)及び以下、AC-1(12.88g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(5.26g)とジクロロメタン(15mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることにより、反応容器内にエステルオリゴマー(2-3)を生成させた。続いて、生成したエステルオリゴマー溶液に安息香酸メチル(6.44g)を添加した。
製造例2-1と同様にエステルオリゴマー(2-3)に含まれる残存酸クロモノマー量の測定を行った。結果を以下の表2-1に示す。
窒素置換した500mL4つ口反応容器にBP-1(6.27g)、2,3,5-トリメチルフェノール(0.090g)及び以下、AC-1(12.88g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(5.26g)とジクロロメタン(15mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることにより、反応容器内にエステルオリゴマー(2-3)を生成させた。続いて、生成したエステルオリゴマー溶液に安息香酸メチル(6.44g)を添加した。
製造例2-1と同様にエステルオリゴマー(2-3)に含まれる残存酸クロモノマー量の測定を行った。結果を以下の表2-1に示す。
製造例2-4(エステルオリゴマー(2-4)の製造)
窒素置換した500mL4つ口反応容器にBP-1(8.32g)、2,3,5-トリメチルフェノール(0.085g)及び以下、AC-1(12.83g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(4.35g)とジクロロメタン(15mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることにより、反応容器内にエステルオリゴマー(2-4)を生成させた。続いて、生成したエステルオリゴマー溶液に安息香酸メチル(6.41g)を添加した。
製造例2-1と同様にエステルオリゴマー(2-4)に含まれる残存酸クロモノマー量の測定を行った。結果を以下の表2-1に示す。
窒素置換した500mL4つ口反応容器にBP-1(8.32g)、2,3,5-トリメチルフェノール(0.085g)及び以下、AC-1(12.83g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(4.35g)とジクロロメタン(15mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることにより、反応容器内にエステルオリゴマー(2-4)を生成させた。続いて、生成したエステルオリゴマー溶液に安息香酸メチル(6.41g)を添加した。
製造例2-1と同様にエステルオリゴマー(2-4)に含まれる残存酸クロモノマー量の測定を行った。結果を以下の表2-1に示す。
製造例2-5(エステルオリゴマー(2-5)の製造)
窒素置換した500mL4つ口反応容器にBP-1(5.21g)、2,3,5-トリメチルフェノール(0.090g)及び以下、AC-1(12.93g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(4.61g)とジクロロメタン(15mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることにより、反応容器内にエステルオリゴマー(2-5)を生成させた。続いて、生成したエステルオリゴマー溶液に安息香酸メチル(6.47g)を添加した。
製造例2-1と同様にエステルオリゴマー(2-5)に含まれる残存酸クロモノマー量の測定を行った。結果を以下の表2-1に示す。
窒素置換した500mL4つ口反応容器にBP-1(5.21g)、2,3,5-トリメチルフェノール(0.090g)及び以下、AC-1(12.93g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(4.61g)とジクロロメタン(15mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることにより、反応容器内にエステルオリゴマー(2-5)を生成させた。続いて、生成したエステルオリゴマー溶液に安息香酸メチル(6.47g)を添加した。
製造例2-1と同様にエステルオリゴマー(2-5)に含まれる残存酸クロモノマー量の測定を行った。結果を以下の表2-1に示す。

[ポリエステル樹脂の製造]
実施例2-1(ポリエステル樹脂(1-1)の合成1:エステルオリゴマー(2-1)処方)
前記製造例2-1と同様の処方でエステルオリゴマーを製造した後、同一反応容器内でポリエステル樹脂(1-1)の製造を実施した。以下に製法を示す。
窒素置換した1000mL4つ口反応容器にBP-1(14.57g)、2,3,5-トリメチルフェノール(0.102g)及びAC-1(22.44g)を秤取り、ジクロロメタン(140mL)に溶解させた。続いて、トリエチルアミン(12.17g)とジクロロメタン(35mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることによりエステルオリゴマーを生成させた。
続いて前記反応容器内へ、4-ヒドロキシ安息香酸4-ヒドロキシフェニル(以下、BP-2)(3.46g)を加えた。その後、トリエチルアミン(3.22g)とジクロロメタン(35mL)との混合溶液を反応容器内へ5~15℃へ冷却した反応容器内へ20分かけて滴下した。反応内温を15~23℃に保ちながら、0.5時間攪拌を続けた後、ジクロロメタン(230mL)で希釈した後、更に0.5時間撹拌を行った。続いてジクロロメタン(230mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(490mL)にて洗浄を行い、続いて0.2規定塩酸(490mL)にて洗浄を3回行い、更に、脱塩水(490mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(300ml)を加えて希釈し、メタノール(4000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(1-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は61,000であった。ポリエステル樹脂(1-1)群の構造式を以下に表す。
実施例2-1(ポリエステル樹脂(1-1)の合成1:エステルオリゴマー(2-1)処方)
前記製造例2-1と同様の処方でエステルオリゴマーを製造した後、同一反応容器内でポリエステル樹脂(1-1)の製造を実施した。以下に製法を示す。
窒素置換した1000mL4つ口反応容器にBP-1(14.57g)、2,3,5-トリメチルフェノール(0.102g)及びAC-1(22.44g)を秤取り、ジクロロメタン(140mL)に溶解させた。続いて、トリエチルアミン(12.17g)とジクロロメタン(35mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることによりエステルオリゴマーを生成させた。
続いて前記反応容器内へ、4-ヒドロキシ安息香酸4-ヒドロキシフェニル(以下、BP-2)(3.46g)を加えた。その後、トリエチルアミン(3.22g)とジクロロメタン(35mL)との混合溶液を反応容器内へ5~15℃へ冷却した反応容器内へ20分かけて滴下した。反応内温を15~23℃に保ちながら、0.5時間攪拌を続けた後、ジクロロメタン(230mL)で希釈した後、更に0.5時間撹拌を行った。続いてジクロロメタン(230mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(490mL)にて洗浄を行い、続いて0.2規定塩酸(490mL)にて洗浄を3回行い、更に、脱塩水(490mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(300ml)を加えて希釈し、メタノール(4000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(1-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は61,000であった。ポリエステル樹脂(1-1)群の構造式を以下に表す。
ポリエステル樹脂(1-1)

[粘度平均分子量(Mv)の測定]
ポリエステル樹脂をジクロロメタンに溶解し濃度Cが6.00g/Lの溶液を調製した。溶媒(ジクロロメタン)の流下時間t0が136.16秒のウベローデ型毛細管粘度計を用いて、20.0℃に設定した恒温水槽中で試料溶液の流下時間tを測定した。以下の式に従って粘度平均分子量(Mv)を算出した。
ポリエステル樹脂をジクロロメタンに溶解し濃度Cが6.00g/Lの溶液を調製した。溶媒(ジクロロメタン)の流下時間t0が136.16秒のウベローデ型毛細管粘度計を用いて、20.0℃に設定した恒温水槽中で試料溶液の流下時間tを測定した。以下の式に従って粘度平均分子量(Mv)を算出した。
a=0.438×ηsp+1 ηsp=t/t0-1
b=100×ηsp/C C=6.00(g/L)
η=b/a
Mv=3207×η1.205
b=100×ηsp/C C=6.00(g/L)
η=b/a
Mv=3207×η1.205
比較例2-1(ポリエステル樹脂(1-2)の合成2:エステルオリゴマー経由無)
窒素置換した500mL4つ口反応容器にBP-1(8.33g)、BP-2(1.98g)、2,3,5-トリメチルフェノール(0.059g)及びAC-1(12.83g)を秤取り、ジクロロメタン(100mL)に溶解させた。続いて、トリエチルアミン(9.37g)とジクロロメタン(35mL)との混合溶液を5~15℃へ冷却した反応容器内へ40分かけて滴下した。反応内温を15~23℃に保ちながら、0.1時間攪拌を続けた後、ジクロロメタン(120mL)で希釈し、0.5時間撹拌を行った。続いてジクロロメタン(120mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(280mL)にて洗浄を行い、続いて0.2規定塩酸(280mL)にて洗浄を3回行い、更に、脱塩水(280mL)にて洗浄を2回行った。洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥してポリエステル樹脂(1-2)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は36,800であった。
窒素置換した500mL4つ口反応容器にBP-1(8.33g)、BP-2(1.98g)、2,3,5-トリメチルフェノール(0.059g)及びAC-1(12.83g)を秤取り、ジクロロメタン(100mL)に溶解させた。続いて、トリエチルアミン(9.37g)とジクロロメタン(35mL)との混合溶液を5~15℃へ冷却した反応容器内へ40分かけて滴下した。反応内温を15~23℃に保ちながら、0.1時間攪拌を続けた後、ジクロロメタン(120mL)で希釈し、0.5時間撹拌を行った。続いてジクロロメタン(120mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(280mL)にて洗浄を行い、続いて0.2規定塩酸(280mL)にて洗浄を3回行い、更に、脱塩水(280mL)にて洗浄を2回行った。洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥してポリエステル樹脂(1-2)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は36,800であった。
比較例2-2(ポリエステル樹脂(1-3)の合成3:エステルオリゴマー(2-4)処方)
前記製造例2-4と同様の処方でエステルオリゴマーを製造した後、同一反応容器内でポリエステル樹脂(1-3)の製造を実施した。以下に製法を示す。
窒素置換した500mL4つ口反応容器にBP-1(8.33g)、2,3,5-トリメチルフェノール(0.059g)及びAC-1(12.83g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(4.35g)とジクロロメタン(20mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることによりエステルオリゴマーを生成させた。
続いて前記反応容器内へ、BP-2(1.98g)を加えた。その後、トリエチルアミン(5.03g)とジクロロメタン(35mL)との混合溶液を反応容器内へ5~15℃へ冷却した反応容器内へ20分かけて滴下した。反応内温を15~23℃に保ちながら、0.2時間攪拌を続けた後、ジクロロメタン(120mL)で希釈した後、更に0.5時間撹拌を行った。続いてジクロロメタン(120mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(280mL)にて洗浄を行い、続いて0.2規定塩酸(280mL)にて洗浄を3回行い、更に、脱塩水(280mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥してポリエステル樹脂(1-3)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は45,000であった。
前記製造例2-4と同様の処方でエステルオリゴマーを製造した後、同一反応容器内でポリエステル樹脂(1-3)の製造を実施した。以下に製法を示す。
窒素置換した500mL4つ口反応容器にBP-1(8.33g)、2,3,5-トリメチルフェノール(0.059g)及びAC-1(12.83g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(4.35g)とジクロロメタン(20mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることによりエステルオリゴマーを生成させた。
続いて前記反応容器内へ、BP-2(1.98g)を加えた。その後、トリエチルアミン(5.03g)とジクロロメタン(35mL)との混合溶液を反応容器内へ5~15℃へ冷却した反応容器内へ20分かけて滴下した。反応内温を15~23℃に保ちながら、0.2時間攪拌を続けた後、ジクロロメタン(120mL)で希釈した後、更に0.5時間撹拌を行った。続いてジクロロメタン(120mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(280mL)にて洗浄を行い、続いて0.2規定塩酸(280mL)にて洗浄を3回行い、更に、脱塩水(280mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥してポリエステル樹脂(1-3)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は45,000であった。
比較例2-3(ポリエステル樹脂(1-4)の合成4:エステルオリゴマー(2-5)処方)
前記製造例2-5と同様の処方でエステルオリゴマーを製造した後、同一反応容器内でポリエステル樹脂(1-4)の製造を実施した。以下に製法を示す。
窒素置換した500mL4つ口反応容器にBP-1(5.21g)、2,3,5-トリメチルフェノール(0.059g)及びAC-1(12.83g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(4.46g)とジクロロメタン(20mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることによりエステルオリゴマーを生成させた。
続いて前記反応容器内へ、BP-1(3.12g)及びBP-2(1.98g)を加えた。その後、トリエチルアミン(4.92g)とジクロロメタン(35mL)との混合溶液を反応容器内へ5~15℃へ冷却した反応容器内へ20分かけて滴下した。反応内温を15~23℃に保ちながら、0.2時間攪拌を続けた後、ジクロロメタン(120mL)で希釈した後、更に0.5時間撹拌を行った。続いてジクロロメタン(120mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(280mL)にて洗浄を行い、続いて0.2規定塩酸(280mL)にて洗浄を3回行い、更に、脱塩水(280mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥してポリエステル樹脂(1-4)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は46,500であった。
前記製造例2-5と同様の処方でエステルオリゴマーを製造した後、同一反応容器内でポリエステル樹脂(1-4)の製造を実施した。以下に製法を示す。
窒素置換した500mL4つ口反応容器にBP-1(5.21g)、2,3,5-トリメチルフェノール(0.059g)及びAC-1(12.83g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(4.46g)とジクロロメタン(20mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることによりエステルオリゴマーを生成させた。
続いて前記反応容器内へ、BP-1(3.12g)及びBP-2(1.98g)を加えた。その後、トリエチルアミン(4.92g)とジクロロメタン(35mL)との混合溶液を反応容器内へ5~15℃へ冷却した反応容器内へ20分かけて滴下した。反応内温を15~23℃に保ちながら、0.2時間攪拌を続けた後、ジクロロメタン(120mL)で希釈した後、更に0.5時間撹拌を行った。続いてジクロロメタン(120mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(280mL)にて洗浄を行い、続いて0.2規定塩酸(280mL)にて洗浄を3回行い、更に、脱塩水(280mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥してポリエステル樹脂(1-4)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は46,500であった。
実施例2-2(ポリエステル樹脂(2-1)の合成:エステルオリゴマー(2-2)処方)
前記製造例2-2と同様の処方でエステルオリゴマーを製造した後、同一反応容器内でポリエステル樹脂(2-1)の製造を実施した。以下に製法を示す。
窒素置換した500mL4つ口反応容器にBP-1(7.92g)、2,3,5-トリメチルフェノール(0.064g)及びAC-1(13.86g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(6.64g)とジクロロメタン(20mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることによりエステルオリゴマーを生成させた。
続いて前記反応容器内へ、BP-2(2.96g)を加えた。その後、トリエチルアミン(3.20g)とジクロロメタン(30mL)との混合溶液を反応容器内へ5~15℃へ冷却した反応容器内へ20分かけて滴下した。反応内温を15~23℃に保ちながら、0.5時間攪拌を続けた後、ジクロロメタン(120mL)で希釈した後、更に0.5時間撹拌を行った。続いてジクロロメタン(120mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(280mL)にて洗浄を行い、続いて0.2規定塩酸(280mL)にて洗浄を3回行い、更に、脱塩水(280mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(150ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(2-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は75,000であった。ポリエステル樹脂(2-1)群の構造式を以下に表す。
前記製造例2-2と同様の処方でエステルオリゴマーを製造した後、同一反応容器内でポリエステル樹脂(2-1)の製造を実施した。以下に製法を示す。
窒素置換した500mL4つ口反応容器にBP-1(7.92g)、2,3,5-トリメチルフェノール(0.064g)及びAC-1(13.86g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(6.64g)とジクロロメタン(20mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることによりエステルオリゴマーを生成させた。
続いて前記反応容器内へ、BP-2(2.96g)を加えた。その後、トリエチルアミン(3.20g)とジクロロメタン(30mL)との混合溶液を反応容器内へ5~15℃へ冷却した反応容器内へ20分かけて滴下した。反応内温を15~23℃に保ちながら、0.5時間攪拌を続けた後、ジクロロメタン(120mL)で希釈した後、更に0.5時間撹拌を行った。続いてジクロロメタン(120mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(280mL)にて洗浄を行い、続いて0.2規定塩酸(280mL)にて洗浄を3回行い、更に、脱塩水(280mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(150ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(2-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は75,000であった。ポリエステル樹脂(2-1)群の構造式を以下に表す。
ポリエステル樹脂(2-1)

実施例2-3(ポリエステル樹脂(3-1)の合成1:エステルオリゴマー(2-2)処方)
前記製造例2-2と同様の処方でエステルオリゴマーを製造した後、同一反応容器内でポリエステル樹脂(3-1)の製造を実施した。以下に製法を示す。
窒素置換した1000mL4つ口反応容器にBP-1(13.89g)、2,3,5-トリメチルフェノール(0.067g)及びAC-1(24.33g)を秤取り、ジクロロメタン(120mL)に溶解させた。続いて、トリエチルアミン(11.60g)とジクロロメタン(30mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることによりエステルオリゴマーを生成させた。
続いて前記反応容器内へ、ハイドロキノン(以下、BP-3)(2.71g)を加えた。その後、トリエチルアミン(6.13g)とジクロロメタン(50mL)との混合溶液を反応容器内へ5~15℃へ冷却した反応容器内へ20分かけて滴下した。反応内温を15~23℃に保ちながら、0.5時間攪拌を続けた後、ジクロロメタン(230mL)で希釈した後、更に0.5時間撹拌を行った。続いてジクロロメタン(230mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(490mL)にて洗浄を行い、続いて0.2規定塩酸(490mL)にて洗浄を3回行い、更に、脱塩水(490mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(300ml)を加えて希釈し、メタノール(4000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(3-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は55,000であった。ポリエステル樹脂(3-1)群の構造式を以下に表す。
前記製造例2-2と同様の処方でエステルオリゴマーを製造した後、同一反応容器内でポリエステル樹脂(3-1)の製造を実施した。以下に製法を示す。
窒素置換した1000mL4つ口反応容器にBP-1(13.89g)、2,3,5-トリメチルフェノール(0.067g)及びAC-1(24.33g)を秤取り、ジクロロメタン(120mL)に溶解させた。続いて、トリエチルアミン(11.60g)とジクロロメタン(30mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることによりエステルオリゴマーを生成させた。
続いて前記反応容器内へ、ハイドロキノン(以下、BP-3)(2.71g)を加えた。その後、トリエチルアミン(6.13g)とジクロロメタン(50mL)との混合溶液を反応容器内へ5~15℃へ冷却した反応容器内へ20分かけて滴下した。反応内温を15~23℃に保ちながら、0.5時間攪拌を続けた後、ジクロロメタン(230mL)で希釈した後、更に0.5時間撹拌を行った。続いてジクロロメタン(230mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(490mL)にて洗浄を行い、続いて0.2規定塩酸(490mL)にて洗浄を3回行い、更に、脱塩水(490mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(300ml)を加えて希釈し、メタノール(4000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(3-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は55,000であった。ポリエステル樹脂(3-1)群の構造式を以下に表す。
ポリエステル樹脂(3-1)

比較例2-4(ポリエステル樹脂(3-2)の合成2:エステルオリゴマー経由無)
窒素置換した500mL4つ口反応容器にBP-1(6.95g)、BP-3(1.35g)、2,3,5-トリメチルフェノール(0.034g)及びAC-1(12.17g)を秤取り、ジクロロメタン(100mL)に溶解させた。続いて、トリエチルアミン(8.87g)とジクロロメタン(35mL)との混合溶液を5~15℃へ冷却した反応容器内へ40分かけて滴下した。反応内温を15~23℃に保ちながら、0.1時間攪拌を続けた後、ジクロロメタン(120mL)で希釈し、0.5時間撹拌を行った。続いてジクロロメタン(120mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(280mL)にて洗浄を行い、続いて0.2規定塩酸(280mL)にて洗浄を3回行い、更に、脱塩水(280mL)にて洗浄を2回行った。洗浄後の有機層にジクロロメタン(150ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥してポリエステル樹脂(3-2)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は39,000であった。
窒素置換した500mL4つ口反応容器にBP-1(6.95g)、BP-3(1.35g)、2,3,5-トリメチルフェノール(0.034g)及びAC-1(12.17g)を秤取り、ジクロロメタン(100mL)に溶解させた。続いて、トリエチルアミン(8.87g)とジクロロメタン(35mL)との混合溶液を5~15℃へ冷却した反応容器内へ40分かけて滴下した。反応内温を15~23℃に保ちながら、0.1時間攪拌を続けた後、ジクロロメタン(120mL)で希釈し、0.5時間撹拌を行った。続いてジクロロメタン(120mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(280mL)にて洗浄を行い、続いて0.2規定塩酸(280mL)にて洗浄を3回行い、更に、脱塩水(280mL)にて洗浄を2回行った。洗浄後の有機層にジクロロメタン(150ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥してポリエステル樹脂(3-2)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は39,000であった。
実施例2-4(ポリエステル樹脂(4-1)の合成:エステルオリゴマー(2-3)処方)
前記製造例2-3と同様の処方でエステルオリゴマーを製造した後、同一反応容器内でポリエステル樹脂(4-1)の製造を実施した。以下に製法を示す。
窒素置換した500mL4つ口反応容器にBP-1(7.00g)、2,3,5-トリメチルフェノール(0.065g)及びAC-1(14.34g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(5.87g)とジクロロメタン(20mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることによりエステルオリゴマーを生成させた。
続いて前記反応容器内へ、BP-3(2.12g)を加えた。その後、トリエチルアミン(4.56g)とジクロロメタン(30mL)との混合溶液を反応容器内へ5~15℃へ冷却した反応容器内へ20分かけて滴下した。反応内温を15~23℃に保ちながら、0.5時間攪拌を続けた後、ジクロロメタン(120mL)で希釈した後、更に0.5時間撹拌を行った。続いてジクロロメタン(120mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(280mL)にて洗浄を行い、続いて0.2規定塩酸(280mL)にて洗浄を3回行い、更に、脱塩水(280mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(150ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(4-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は57,500であった。ポリエステル樹脂(4-1)群の構造式を以下に表す。
前記製造例2-3と同様の処方でエステルオリゴマーを製造した後、同一反応容器内でポリエステル樹脂(4-1)の製造を実施した。以下に製法を示す。
窒素置換した500mL4つ口反応容器にBP-1(7.00g)、2,3,5-トリメチルフェノール(0.065g)及びAC-1(14.34g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(5.87g)とジクロロメタン(20mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることによりエステルオリゴマーを生成させた。
続いて前記反応容器内へ、BP-3(2.12g)を加えた。その後、トリエチルアミン(4.56g)とジクロロメタン(30mL)との混合溶液を反応容器内へ5~15℃へ冷却した反応容器内へ20分かけて滴下した。反応内温を15~23℃に保ちながら、0.5時間攪拌を続けた後、ジクロロメタン(120mL)で希釈した後、更に0.5時間撹拌を行った。続いてジクロロメタン(120mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(280mL)にて洗浄を行い、続いて0.2規定塩酸(280mL)にて洗浄を3回行い、更に、脱塩水(280mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(150ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(4-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は57,500であった。ポリエステル樹脂(4-1)群の構造式を以下に表す。
ポリエステル樹脂(4-1)

実施例2-5(ポリエステル樹脂(5-1)の合成1:エステルオリゴマー(2-1)処方)
前記製造例2-1と同様の処方でエステルオリゴマーを製造した後、同一反応容器内でポリエステル樹脂(5-1)の製造を実施した。以下に製法を示す。
窒素置換した500mL4つ口反応容器にBP-1(8.44g)、2,3,5-トリメチルフェノール(0.119g)及びAC-1(13.02g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(7.07g)とジクロロメタン(20mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることによりエステルオリゴマーを生成させた。
続いて前記反応容器内へ、4,4’-ビフェノール(以下、BP-4)(1.62g)を加えた。その後、トリエチルアミン(2.42g)とジクロロメタン(30mL)との混合溶液を反応容器内へ5~15℃へ冷却した反応容器内へ20分かけて滴下した。反応内温を15~23℃に保ちながら、0.5時間攪拌を続けた後、ジクロロメタン(90mL)で希釈した後、更に0.5時間撹拌を行った。続いてジクロロメタン(80mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(230mL)にて洗浄を行い、続いて0.2規定塩酸(230mL)にて洗浄を3回行い、更に、脱塩水(230mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(150ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(5-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は38,200であった。ポリエステル樹脂(5-1)の構造式を以下に表す。
前記製造例2-1と同様の処方でエステルオリゴマーを製造した後、同一反応容器内でポリエステル樹脂(5-1)の製造を実施した。以下に製法を示す。
窒素置換した500mL4つ口反応容器にBP-1(8.44g)、2,3,5-トリメチルフェノール(0.119g)及びAC-1(13.02g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(7.07g)とジクロロメタン(20mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることによりエステルオリゴマーを生成させた。
続いて前記反応容器内へ、4,4’-ビフェノール(以下、BP-4)(1.62g)を加えた。その後、トリエチルアミン(2.42g)とジクロロメタン(30mL)との混合溶液を反応容器内へ5~15℃へ冷却した反応容器内へ20分かけて滴下した。反応内温を15~23℃に保ちながら、0.5時間攪拌を続けた後、ジクロロメタン(90mL)で希釈した後、更に0.5時間撹拌を行った。続いてジクロロメタン(80mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(230mL)にて洗浄を行い、続いて0.2規定塩酸(230mL)にて洗浄を3回行い、更に、脱塩水(230mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(150ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(5-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は38,200であった。ポリエステル樹脂(5-1)の構造式を以下に表す。
ポリエステル樹脂(5-1)

比較例2-5(ポリエステル樹脂(5-2)の合成2:エステルオリゴマー経由無)
窒素置換した500mL4つ口反応容器にBP-1(8.44g)、BP-4(1.62g)、2,3,5-トリメチルフェノール(0.119g)及びAC-1(13.02g)を秤取り、ジクロロメタン(100mL)に溶解させた。続いて、トリエチルアミン(9.49g)とジクロロメタン(35mL)との混合溶液を5~15℃へ冷却した反応容器内へ40分かけて滴下した。反応内温を15~23℃に保ちながら、0.1時間攪拌を続けた後、ジクロロメタン(85mL)で希釈し、0.5時間撹拌を行った。続いてジクロロメタン(80mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(230mL)にて洗浄を行い、続いて0.2規定塩酸(230mL)にて洗浄を3回行い、更に、脱塩水(230mL)にて洗浄を2回行った。洗浄後の有機層にジクロロメタン(150ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥してポリエステル樹脂(5-2)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は25,000であった。
窒素置換した500mL4つ口反応容器にBP-1(8.44g)、BP-4(1.62g)、2,3,5-トリメチルフェノール(0.119g)及びAC-1(13.02g)を秤取り、ジクロロメタン(100mL)に溶解させた。続いて、トリエチルアミン(9.49g)とジクロロメタン(35mL)との混合溶液を5~15℃へ冷却した反応容器内へ40分かけて滴下した。反応内温を15~23℃に保ちながら、0.1時間攪拌を続けた後、ジクロロメタン(85mL)で希釈し、0.5時間撹拌を行った。続いてジクロロメタン(80mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(230mL)にて洗浄を行い、続いて0.2規定塩酸(230mL)にて洗浄を3回行い、更に、脱塩水(230mL)にて洗浄を2回行った。洗浄後の有機層にジクロロメタン(150ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥してポリエステル樹脂(5-2)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は25,000であった。
実施例2-6(ポリエステル樹脂(6-1)の合成:エステルオリゴマー(2-3)処方)
前記製造例2-3と同様の処方でエステルオリゴマーを製造した後、同一反応容器内でポリエステル樹脂(6-1)の製造を実施した。以下に製法を示す。
窒素置換した500mL4つ口反応容器にBP-1(6.36g)、2,3,5-トリメチルフェノール(0.060g)及びAC-1(13.07g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(5.42g)とジクロロメタン(20mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることによりエステルオリゴマーを生成させた。
続いて前記反応容器内へ、4,4’-ジヒドロキシ-3,3’-ジメチルビフェニル(以下、BP-5)(3.75g)を加えた。その後、トリエチルアミン(4.16g)とジクロロメタン(50mL)との混合溶液を反応容器内へ5~15℃へ冷却した反応容器内へ20分かけて滴下した。反応内温を15~23℃に保ちながら、0.5時間攪拌を続けた後、ジクロロメタン(75mL)で希釈した後、更に0.5時間撹拌を行った。続いてジクロロメタン(75mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(230mL)にて洗浄を行い、続いて0.2規定塩酸(230mL)にて洗浄を3回行い、更に、脱塩水(230mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(150ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(6-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は55,200であった。ポリエステル樹脂(6-1)の構造式を以下に表す。
前記製造例2-3と同様の処方でエステルオリゴマーを製造した後、同一反応容器内でポリエステル樹脂(6-1)の製造を実施した。以下に製法を示す。
窒素置換した500mL4つ口反応容器にBP-1(6.36g)、2,3,5-トリメチルフェノール(0.060g)及びAC-1(13.07g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(5.42g)とジクロロメタン(20mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることによりエステルオリゴマーを生成させた。
続いて前記反応容器内へ、4,4’-ジヒドロキシ-3,3’-ジメチルビフェニル(以下、BP-5)(3.75g)を加えた。その後、トリエチルアミン(4.16g)とジクロロメタン(50mL)との混合溶液を反応容器内へ5~15℃へ冷却した反応容器内へ20分かけて滴下した。反応内温を15~23℃に保ちながら、0.5時間攪拌を続けた後、ジクロロメタン(75mL)で希釈した後、更に0.5時間撹拌を行った。続いてジクロロメタン(75mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(230mL)にて洗浄を行い、続いて0.2規定塩酸(230mL)にて洗浄を3回行い、更に、脱塩水(230mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(150ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(6-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は55,200であった。ポリエステル樹脂(6-1)の構造式を以下に表す。
ポリエステル樹脂(6-1)

実施例2-7(ポリエステル樹脂(7-1)の合成)
前記製造例2-2と同様の処方でエステルオリゴマーを製造した後、同一反応容器内でポリエステル樹脂(7-1)の製造を実施した。以下に製法を示す。
窒素置換した500mL4つ口反応容器にBP-1(7.63g)、2,3,5-トリメチルフェノール(0.092g)及びAC-1(13.42g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(6.42g)とジクロロメタン(20mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることによりエステルオリゴマーを生成させた。
続いて前記反応容器内へ、2,6-ジヒドロキシナフタレン(以下、BP-6)(2.16g)を加えた。その後、トリエチルアミン(3.41g)とジクロロメタン(30mL)との混合溶液を反応容器内へ5~15℃へ冷却した反応容器内へ20分かけて滴下した。反応内温を15~23℃に保ちながら、0.5時間攪拌を続けた後、ジクロロメタン(90mL)で希釈した後、更に0.5時間撹拌を行った。続いてジクロロメタン(80mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(200mL)にて洗浄を行い、続いて0.2規定塩酸(200mL)にて洗浄を3回行い、更に、脱塩水(200mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(150ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(7-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は40,700であった。ポリエステル樹脂(7-1)の構造式を以下に表す。
前記製造例2-2と同様の処方でエステルオリゴマーを製造した後、同一反応容器内でポリエステル樹脂(7-1)の製造を実施した。以下に製法を示す。
窒素置換した500mL4つ口反応容器にBP-1(7.63g)、2,3,5-トリメチルフェノール(0.092g)及びAC-1(13.42g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(6.42g)とジクロロメタン(20mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることによりエステルオリゴマーを生成させた。
続いて前記反応容器内へ、2,6-ジヒドロキシナフタレン(以下、BP-6)(2.16g)を加えた。その後、トリエチルアミン(3.41g)とジクロロメタン(30mL)との混合溶液を反応容器内へ5~15℃へ冷却した反応容器内へ20分かけて滴下した。反応内温を15~23℃に保ちながら、0.5時間攪拌を続けた後、ジクロロメタン(90mL)で希釈した後、更に0.5時間撹拌を行った。続いてジクロロメタン(80mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(200mL)にて洗浄を行い、続いて0.2規定塩酸(200mL)にて洗浄を3回行い、更に、脱塩水(200mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(150ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(7-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は40,700であった。ポリエステル樹脂(7-1)の構造式を以下に表す。
ポリエステル樹脂(7-1)

実施例2-8(ポリエステル樹脂(8-1)の合成)
実施例2-7における2,6-ジヒドロキシナフタレンを2,7-ジヒドロキシナフタレン(以下、BP-7)に代えた以外は実施例2-7と同様に実施し、ポリエステル樹脂(8-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は35,500であった。ポリエステル樹脂(8-1)の構造式を以下に表す。
実施例2-7における2,6-ジヒドロキシナフタレンを2,7-ジヒドロキシナフタレン(以下、BP-7)に代えた以外は実施例2-7と同様に実施し、ポリエステル樹脂(8-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は35,500であった。ポリエステル樹脂(8-1)の構造式を以下に表す。
ポリエステル樹脂(8-1)
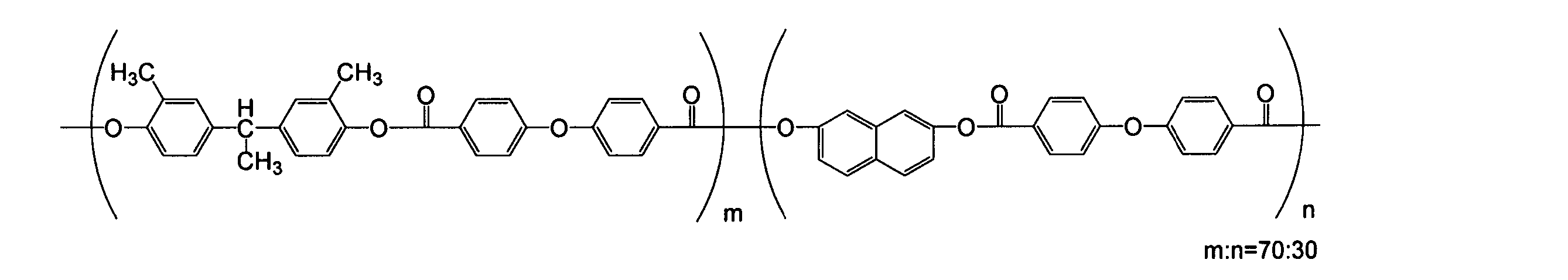
実施例2-9(ポリエステル樹脂(9-1)の合成)
前記製造例2-1と同様の処方でエステルオリゴマーを製造した後、同一反応容器内でポリエステル樹脂(9-1)の製造を実施した。以下に製法を示す。
窒素置換した500mL4つ口反応容器にビス(4-ヒドロキシ-3-メチルフェニル)メタン(以下、BP-8)(9.03g)、2,3,5-トリメチルフェノール(0.060g)及びAC-1(13.13g)を秤取り、ジクロロメタン(75mL)に溶解させた。続いて、トリエチルアミン(7.20g)とジクロロメタン(20mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることによりエステルオリゴマーを生成させた。
続いて前記反応容器内へ、BP-1(1.01g)を加えた。その後、トリエチルアミン(2.41g)とジクロロメタン(80mL)との混合溶液を反応容器内へ5~15℃へ冷却した反応容器内へ20分かけて滴下した。反応内温を15~23℃に保ちながら、0.2時間攪拌を続けた後、ジクロロメタン(50mL)で希釈した後、更に0.5時間撹拌を行った。続いてジクロロメタン(70mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(210mL)にて洗浄を行い、続いて0.2規定塩酸(210mL)にて洗浄を3回行い、更に、脱塩水(210mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(150ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(9-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は60,100であった。ポリエステル樹脂(9-1)の構造式を以下に表す。
前記製造例2-1と同様の処方でエステルオリゴマーを製造した後、同一反応容器内でポリエステル樹脂(9-1)の製造を実施した。以下に製法を示す。
窒素置換した500mL4つ口反応容器にビス(4-ヒドロキシ-3-メチルフェニル)メタン(以下、BP-8)(9.03g)、2,3,5-トリメチルフェノール(0.060g)及びAC-1(13.13g)を秤取り、ジクロロメタン(75mL)に溶解させた。続いて、トリエチルアミン(7.20g)とジクロロメタン(20mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることによりエステルオリゴマーを生成させた。
続いて前記反応容器内へ、BP-1(1.01g)を加えた。その後、トリエチルアミン(2.41g)とジクロロメタン(80mL)との混合溶液を反応容器内へ5~15℃へ冷却した反応容器内へ20分かけて滴下した。反応内温を15~23℃に保ちながら、0.2時間攪拌を続けた後、ジクロロメタン(50mL)で希釈した後、更に0.5時間撹拌を行った。続いてジクロロメタン(70mL)で更に希釈した後、4時間撹拌を行った。その後、脱塩水(210mL)にて洗浄を行い、続いて0.2規定塩酸(210mL)にて洗浄を3回行い、更に、脱塩水(210mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(150ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(9-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は60,100であった。ポリエステル樹脂(9-1)の構造式を以下に表す。
ポリエステル樹脂(9-1)

実施例2-10(ポリエステル樹脂(10-1)の合成)
実施例2-10におけるビス(4-ヒドロキシ-3-メチルフェニル)メタンを1,1-ビス(4-ヒドロキシフェニル)エタン(以下、BP-9)に代えた以外は実施例2-10と同様に実施し、ポリエステル樹脂(10-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は56,200であった。ポリエステル樹脂(10-1)の構造式を以下に表す。
実施例2-10におけるビス(4-ヒドロキシ-3-メチルフェニル)メタンを1,1-ビス(4-ヒドロキシフェニル)エタン(以下、BP-9)に代えた以外は実施例2-10と同様に実施し、ポリエステル樹脂(10-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は56,200であった。ポリエステル樹脂(10-1)の構造式を以下に表す。
ポリエステル樹脂(10-1)

<ポリエステルTHF溶液の外観>
実施例2-1~2-10、比較例2-1~2-5で製造したポリエステル樹脂100質量部を透明サンプル瓶に入れた後、テトラヒドロフラン溶媒660質量部で溶解させ、溶液の外観を確認した。○は透明な溶液を表し、×は白濁した溶液を表す。結果を以下の表2-2に示す。
実施例2-1~2-10、比較例2-1~2-5で製造したポリエステル樹脂100質量部を透明サンプル瓶に入れた後、テトラヒドロフラン溶媒660質量部で溶解させ、溶液の外観を確認した。○は透明な溶液を表し、×は白濁した溶液を表す。結果を以下の表2-2に示す。

表2-2の結果より、エステルオリゴマーを経由し、更にエステルオリゴマー中の残存酸クロライドモノマー量を本特許の範囲内にすることにより、溶解性に優れたポリエステル樹脂が得られることが分かる。一方で、エステルオリゴマーを経由しない場合又は、本発明中の範囲よりもエステルオリゴマー中の残存酸クロライドモノマー量が多い場合は不溶性のオリゴマーが形成されるため溶液が白濁する。
<感光体シートの作製>
[実施例2-11]
10質量部のオキシチタニウムフタロシアニンと、150質量部の4-メトキシ-4-メチル-2-ペンタノンとを混合し、サンドグラインドミルにて粉砕分散処理を行い顔料分散液を製造した。尚、オキシチタニウムフタロシアニンは、CuKα線によるX線回折においてブラッグ角(2θ±0.2)9.3゜、10.6゜、13.2゜、15.1゜、15.7゜、16.1゜、20.8゜、23.3゜、26.3゜、27.1゜に強い回折ピークを表す。
この顔料分散液に、ポリビニルブチラール(電気化学工業株式会社製、商品名デンカブチラール♯6000C)の5質量%1,2-ジメトキシエタン溶液を50質量部、フェノキシ樹脂(ユニオンカーバイド株式会社製、商品名PKHH)の5質量%1,2-ジメトキシエタン溶液を50質量部混合し、更に、適量の1,2-ジメトキシエタンを加え、固形分濃度4.0%の電荷発生層形成用塗布液を調製した。この電荷発生層形成用塗布液を、表面にアルミ蒸着したポリエチレンテレフタレートシート上に、乾燥後の膜厚が0.4μmになるように塗布、乾燥して電荷発生層を設けた。
次に、電荷輸送物質として、下記に表す構造を主成分とする、幾何異性体の化合物群からなる、日本国特開2002-080432号公報の実施例1に記載の方法で製造した混合物(CTM-1)を50質量部、実施例2-1で製造したポリエステル樹脂(1-1)を100質量部、酸化防止剤(イルガノックス1076)8質量部、レベリング剤としてシリコーンオイル0.05質量部を、テトラヒドロフランとトルエンとの混合溶媒(テトラヒドロフラン80質量%、トルエン20質量%)640質量部に混合し、電荷輸送層形成用塗布液を調製した。
[実施例2-11]
10質量部のオキシチタニウムフタロシアニンと、150質量部の4-メトキシ-4-メチル-2-ペンタノンとを混合し、サンドグラインドミルにて粉砕分散処理を行い顔料分散液を製造した。尚、オキシチタニウムフタロシアニンは、CuKα線によるX線回折においてブラッグ角(2θ±0.2)9.3゜、10.6゜、13.2゜、15.1゜、15.7゜、16.1゜、20.8゜、23.3゜、26.3゜、27.1゜に強い回折ピークを表す。
この顔料分散液に、ポリビニルブチラール(電気化学工業株式会社製、商品名デンカブチラール♯6000C)の5質量%1,2-ジメトキシエタン溶液を50質量部、フェノキシ樹脂(ユニオンカーバイド株式会社製、商品名PKHH)の5質量%1,2-ジメトキシエタン溶液を50質量部混合し、更に、適量の1,2-ジメトキシエタンを加え、固形分濃度4.0%の電荷発生層形成用塗布液を調製した。この電荷発生層形成用塗布液を、表面にアルミ蒸着したポリエチレンテレフタレートシート上に、乾燥後の膜厚が0.4μmになるように塗布、乾燥して電荷発生層を設けた。
次に、電荷輸送物質として、下記に表す構造を主成分とする、幾何異性体の化合物群からなる、日本国特開2002-080432号公報の実施例1に記載の方法で製造した混合物(CTM-1)を50質量部、実施例2-1で製造したポリエステル樹脂(1-1)を100質量部、酸化防止剤(イルガノックス1076)8質量部、レベリング剤としてシリコーンオイル0.05質量部を、テトラヒドロフランとトルエンとの混合溶媒(テトラヒドロフラン80質量%、トルエン20質量%)640質量部に混合し、電荷輸送層形成用塗布液を調製した。
電荷輸送材(CTM-1)

この電荷輸送層形成用塗布液を上述の電荷発生層上に、乾燥後の膜厚が25μmとなるようにアプリケーターを用いて塗布し、125℃で20分間乾燥して電荷輸送層を形成して、感光体シートを作製した。
[実施例2-12]
ポリエステル樹脂(1-1)を実施例2-3で製造したポリエステル樹脂(3-1)に変えた以外は実施例2-11と同様にして、感光体シートを作製した。
ポリエステル樹脂(1-1)を実施例2-3で製造したポリエステル樹脂(3-1)に変えた以外は実施例2-11と同様にして、感光体シートを作製した。
[実施例2-13]
ポリエステル樹脂(1-1)を実施例2-5で製造したポリエステル樹脂(5-1)に変えた以外は実施例2-11と同様にして、感光体シートを作製した。
ポリエステル樹脂(1-1)を実施例2-5で製造したポリエステル樹脂(5-1)に変えた以外は実施例2-11と同様にして、感光体シートを作製した。
[実施例2-14]
ポリエステル樹脂(1-1)を実施例2-6で製造したポリエステル樹脂(6-1)に変えた以外は実施例2-11と同様にして、感光体シートを作製した。
ポリエステル樹脂(1-1)を実施例2-6で製造したポリエステル樹脂(6-1)に変えた以外は実施例2-11と同様にして、感光体シートを作製した。
[実施例2-15]
ポリエステル樹脂(1-1)を実施例2-7で製造したポリエステル樹脂(7-1)に変えた以外は実施例2-11と同様にして、感光体シートを作製した。
ポリエステル樹脂(1-1)を実施例2-7で製造したポリエステル樹脂(7-1)に変えた以外は実施例2-11と同様にして、感光体シートを作製した。
[実施例2-16]
ポリエステル樹脂(1-1)を実施例2-8で製造したポリエステル樹脂(8-1)に変えた以外は実施例2-11と同様にして、感光体シートを作製した。
ポリエステル樹脂(1-1)を実施例2-8で製造したポリエステル樹脂(8-1)に変えた以外は実施例2-11と同様にして、感光体シートを作製した。
[実施例2-17]
ポリエステル樹脂(1-1)を実施例2-9で製造したポリエステル樹脂(9-1)に変えた以外は実施例2-11と同様にして、感光体シートを作製した。
ポリエステル樹脂(1-1)を実施例2-9で製造したポリエステル樹脂(9-1)に変えた以外は実施例2-11と同様にして、感光体シートを作製した。
[比較例2-6]
ポリエステル樹脂(1-1)を比較例2-1で製造したポリエステル樹脂(1-2)に変えた以外は実施例2-11と同様にして、感光体シートを作製した。
ポリエステル樹脂(1-1)を比較例2-1で製造したポリエステル樹脂(1-2)に変えた以外は実施例2-11と同様にして、感光体シートを作製した。
[比較例2-7]
ポリエステル樹脂(1-1)を比較例2-2で製造したポリエステル樹脂(1-3)に変えた以外は実施例2-11と同様にして、感光体シートを作製した。
ポリエステル樹脂(1-1)を比較例2-2で製造したポリエステル樹脂(1-3)に変えた以外は実施例2-11と同様にして、感光体シートを作製した。
[参考例2-1]
ポリエステル樹脂(1-1)を日本国特開2006-53549号公報の実施例6に記載の方法により製造した以下に表す構造を有するポリエステル樹脂(11)(粘度平均分子量36,200)に変えた以外は実施例2-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(1-1)を日本国特開2006-53549号公報の実施例6に記載の方法により製造した以下に表す構造を有するポリエステル樹脂(11)(粘度平均分子量36,200)に変えた以外は実施例2-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(11)

[電荷輸送層の表面状態]
実施例2-11~2-17、比較例2-6~2-7、参考例2-1で作製した感光体シートの表面状態を目視で確認した。目視で表面に凹凸がない場合を○、表面に凹凸がある場合を×とした。目視で分かる凹凸がある場合、印刷した際に画像欠陥となる。
実施例2-11~2-17、比較例2-6~2-7、参考例2-1で作製した感光体シートの表面状態を目視で確認した。目視で表面に凹凸がない場合を○、表面に凹凸がある場合を×とした。目視で分かる凹凸がある場合、印刷した際に画像欠陥となる。
[電気特性評価]
実施例2-11~2-17、比較例2-6~2-7、参考例2-1で作製した感光体シートを用いて測定を行った。電子写真学会測定標準に従って作製された電子写真特性評価装置(続電子写真技術の基礎と応用、電子写真学会編、コロナ社、404-405頁記載(1996年))を使用し、前記感光体シートをアルミニウム製ドラムに貼り付けて円筒状にし、アルミニウム製ドラムと感光体のアルミニウム基体との導通を取った上で実施した。なお本評価装置は、ドラムを一定回転数で回転させ、帯電、露光、電位測定、除電のサイクルができる。電気特性評価として-700Vに帯電して5秒放置後の電位保持率(DDR)を測定した(%)。電位保持率は高い方が感光体として安定であり、かぶり等の画像欠陥が発生しづらい。測定環境は、温度25℃、相対湿度50%下(N/N)で行った。結果を表2-3に表す。
実施例2-11~2-17、比較例2-6~2-7、参考例2-1で作製した感光体シートを用いて測定を行った。電子写真学会測定標準に従って作製された電子写真特性評価装置(続電子写真技術の基礎と応用、電子写真学会編、コロナ社、404-405頁記載(1996年))を使用し、前記感光体シートをアルミニウム製ドラムに貼り付けて円筒状にし、アルミニウム製ドラムと感光体のアルミニウム基体との導通を取った上で実施した。なお本評価装置は、ドラムを一定回転数で回転させ、帯電、露光、電位測定、除電のサイクルができる。電気特性評価として-700Vに帯電して5秒放置後の電位保持率(DDR)を測定した(%)。電位保持率は高い方が感光体として安定であり、かぶり等の画像欠陥が発生しづらい。測定環境は、温度25℃、相対湿度50%下(N/N)で行った。結果を表2-3に表す。

[摩耗試験]
感光体フィルムを直径10cmの円状に切断しテーバー摩耗試験機(東洋精機社製)により、摩耗評価を行った。試験条件は、25℃、50%RHの雰囲気下、摩耗輪CS-10Fを用いて、荷重500gで1000回回転後の摩耗量を試験前後の質量を比較することにより測定した。値が小さい方が耐摩耗性に優れる。結果を表2-4に表す。
感光体フィルムを直径10cmの円状に切断しテーバー摩耗試験機(東洋精機社製)により、摩耗評価を行った。試験条件は、25℃、50%RHの雰囲気下、摩耗輪CS-10Fを用いて、荷重500gで1000回回転後の摩耗量を試験前後の質量を比較することにより測定した。値が小さい方が耐摩耗性に優れる。結果を表2-4に表す。

以上から、本発明により製造されたポリエステル樹脂を用いることにより、欠陥がないきれいな塗布膜が得られ、電気特性及び耐摩耗性に優れた電子写真感光体が提供できることを明らかとした。
(試験例3)
[エステルオリゴマーの製造]
製造例3-1(エステルオリゴマー(3-1)の合成)
窒素置換した500mL4つ口反応容器に1,1-ビス(4-ヒドロキシ-3-メチルフェニル)エタン(7.00g)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(17.05g)を秤取り、ジクロロメタン(160mL)に溶解させた。続いて、トリエチルアミン(6.14g)とジクロロメタン(20mL)との混合溶液を0~5℃へ冷却した反応容器内へ20分かけて滴下した。5分撹拌を続けた後、1,1-ビス(4-ヒドロキシ-3-メチルフェニル)エタン(11.90g)と4―ヒドロキシ安息香酸4―ヒドロキシフェニル(2.00g)を反応容器へ加えた。その後、トリエチルアミン(6.14g)とジクロロメタン(20mL)との混合溶液を反応容器内へ20分かけて滴下した。1時間攪拌を続けた後、0.1規定塩酸(170mL)を添加し、30分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(170mL)にて洗浄を2回行い、さらに、脱塩水(170mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(3-1)を得た。
[エステルオリゴマーの製造]
製造例3-1(エステルオリゴマー(3-1)の合成)
窒素置換した500mL4つ口反応容器に1,1-ビス(4-ヒドロキシ-3-メチルフェニル)エタン(7.00g)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(17.05g)を秤取り、ジクロロメタン(160mL)に溶解させた。続いて、トリエチルアミン(6.14g)とジクロロメタン(20mL)との混合溶液を0~5℃へ冷却した反応容器内へ20分かけて滴下した。5分撹拌を続けた後、1,1-ビス(4-ヒドロキシ-3-メチルフェニル)エタン(11.90g)と4―ヒドロキシ安息香酸4―ヒドロキシフェニル(2.00g)を反応容器へ加えた。その後、トリエチルアミン(6.14g)とジクロロメタン(20mL)との混合溶液を反応容器内へ20分かけて滴下した。1時間攪拌を続けた後、0.1規定塩酸(170mL)を添加し、30分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(170mL)にて洗浄を2回行い、さらに、脱塩水(170mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(3-1)を得た。
実施例3-1(エステルオリゴマー(3-1)を使用したポリエステル樹脂(3-1)の製造)
500mLビーカーに水酸化ナトリウム(1.78g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.26g)、及びベンジルトリエチルアンモニウムクロライド(0.08g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、製造例3-1で合成したエステルオリゴマー(3-1)(20.57g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(5.53g)とジクロロメタン(135mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(179mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(235mL)にて洗浄を3回行い、さらに、脱塩水(235mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(180ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(3-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は51,000であった。ポリエステル樹脂(3-1)の構造式を以下に表す。
500mLビーカーに水酸化ナトリウム(1.78g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.26g)、及びベンジルトリエチルアンモニウムクロライド(0.08g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、製造例3-1で合成したエステルオリゴマー(3-1)(20.57g)及びジフェニルエーテル-4,4´-ジカルボン酸クロライド(5.53g)とジクロロメタン(135mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(179mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(235mL)にて洗浄を3回行い、さらに、脱塩水(235mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(180ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(3-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は51,000であった。ポリエステル樹脂(3-1)の構造式を以下に表す。
ポリエステル樹脂(3-1)

[粘度平均分子量(Mv)の測定]
ポリエステル樹脂をジクロロメタンに溶解し濃度Cが6.00g/Lの溶液を調製した。溶媒(ジクロロメタン)の流下時間t0が136.16秒のウベローデ型毛細管粘度計を用いて、20.0℃に設定した恒温水槽中で試料溶液の流下時間tを測定した。以下の式に従って粘度平均分子量(Mv)を算出した。
ポリエステル樹脂をジクロロメタンに溶解し濃度Cが6.00g/Lの溶液を調製した。溶媒(ジクロロメタン)の流下時間t0が136.16秒のウベローデ型毛細管粘度計を用いて、20.0℃に設定した恒温水槽中で試料溶液の流下時間tを測定した。以下の式に従って粘度平均分子量(Mv)を算出した。
a=0.438×ηsp+1 ηsp=t/t0-1
b=100×ηsp/C C=6.00(g/L)
η=b/a
Mv=3207×η1.205
b=100×ηsp/C C=6.00(g/L)
η=b/a
Mv=3207×η1.205
実施例3-2(溶液重合法によるポリエステル樹脂(3-1)の製造)
窒素置換した500mL4つ口反応容器に1,1-ビス(4-ヒドロキシ-3-メチルフェニル)エタン(3.43g)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(8.35g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(3.01g)とジクロロメタン(15mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下した。5分撹拌を続けた後、1,1-ビス(4-ヒドロキシ-3-メチルフェニル)エタン(5.83g)と4―ヒドロキシ安息香酸4―ヒドロキシフェニル(0.98g)を反応容器へ加えた。その後、トリエチルアミン(3.29g)とジクロロメタン(15mL)との混合溶液を10~20℃へ冷却した反応容器内へ20分かけて滴下した。10分攪拌を続けた後、2,3,5-トリメチルフェノール(0.10g)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(4.40g)を反応容器へ加えた。その後、トリエチルアミン(3.72g)とジクロロメタン(50mL)との混合溶液を0~10℃へ冷却した反応容器内へ30分かけて滴下した。室温へと昇温し30分攪拌を続けた後、ジクロロメタン(179mL)を加え、撹拌を3時間続けた。
その後、0.1規定水酸化ナトリウム水溶液(280mL)でアルカリ洗浄し、続いて、0.1規定塩酸水溶液(280mL)にて酸洗浄を3回行い、さらに、脱塩水(280mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(3-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は36,800であった。
窒素置換した500mL4つ口反応容器に1,1-ビス(4-ヒドロキシ-3-メチルフェニル)エタン(3.43g)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(8.35g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(3.01g)とジクロロメタン(15mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下した。5分撹拌を続けた後、1,1-ビス(4-ヒドロキシ-3-メチルフェニル)エタン(5.83g)と4―ヒドロキシ安息香酸4―ヒドロキシフェニル(0.98g)を反応容器へ加えた。その後、トリエチルアミン(3.29g)とジクロロメタン(15mL)との混合溶液を10~20℃へ冷却した反応容器内へ20分かけて滴下した。10分攪拌を続けた後、2,3,5-トリメチルフェノール(0.10g)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(4.40g)を反応容器へ加えた。その後、トリエチルアミン(3.72g)とジクロロメタン(50mL)との混合溶液を0~10℃へ冷却した反応容器内へ30分かけて滴下した。室温へと昇温し30分攪拌を続けた後、ジクロロメタン(179mL)を加え、撹拌を3時間続けた。
その後、0.1規定水酸化ナトリウム水溶液(280mL)でアルカリ洗浄し、続いて、0.1規定塩酸水溶液(280mL)にて酸洗浄を3回行い、さらに、脱塩水(280mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(3-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は36,800であった。
実施例3-3(溶液重合法によるポリエステル樹脂(3-2)の製造)
窒素置換した500mL4つ口反応容器に1,1-ビス(4-ヒドロキシ-3-メチルフェニル)エタン(3.46g)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(8.43g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(3.03g)とジクロロメタン(15mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下した。5分撹拌を続けた後、1,1-ビス(4-ヒドロキシ-3-メチルフェニル)エタン(3.29g)、1,1-ビス(4―ヒドロキシフェニル)メタン(2.14g)と4―ヒドロキシ安息香酸4―ヒドロキシフェニル(0.99g)を反応容器へ加えた。その後、トリエチルアミン(3.32g)とジクロロメタン(15mL)との混合溶液を10~20℃へ冷却した反応容器内へ20分かけて滴下した。10分攪拌を続けた後、2,3,5-トリメチルフェノール(0.03g)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(4.35g)を反応容器へ加えた。その後、トリエチルアミン(3.76g)とジクロロメタン(50mL)との混合溶液を0~10℃へ冷却した反応容器内へ30分かけて滴下した。室温へと昇温し30分攪拌を続けた後、ジクロロメタン(220mL)を加え、撹拌を3時間続けた。
その後、0.1規定水酸化ナトリウム水溶液(280mL)でアルカリ洗浄し、続いて、0.1規定塩酸水溶液(280mL)にて酸洗浄を3回行い、さらに、脱塩水(280mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(150ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(3-2)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は79,700であった。
窒素置換した500mL4つ口反応容器に1,1-ビス(4-ヒドロキシ-3-メチルフェニル)エタン(3.46g)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(8.43g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(3.03g)とジクロロメタン(15mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下した。5分撹拌を続けた後、1,1-ビス(4-ヒドロキシ-3-メチルフェニル)エタン(3.29g)、1,1-ビス(4―ヒドロキシフェニル)メタン(2.14g)と4―ヒドロキシ安息香酸4―ヒドロキシフェニル(0.99g)を反応容器へ加えた。その後、トリエチルアミン(3.32g)とジクロロメタン(15mL)との混合溶液を10~20℃へ冷却した反応容器内へ20分かけて滴下した。10分攪拌を続けた後、2,3,5-トリメチルフェノール(0.03g)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(4.35g)を反応容器へ加えた。その後、トリエチルアミン(3.76g)とジクロロメタン(50mL)との混合溶液を0~10℃へ冷却した反応容器内へ30分かけて滴下した。室温へと昇温し30分攪拌を続けた後、ジクロロメタン(220mL)を加え、撹拌を3時間続けた。
その後、0.1規定水酸化ナトリウム水溶液(280mL)でアルカリ洗浄し、続いて、0.1規定塩酸水溶液(280mL)にて酸洗浄を3回行い、さらに、脱塩水(280mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(150ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(3-2)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は79,700であった。
ポリエステル樹脂(3-2)

比較例3-1(界面重合法によるポリエステル樹脂(3-1)の製造)
500mLビーカーに水酸化ナトリウム(4.13g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.26g)、BP-a(9.16g)、4―ヒドロキシ安息香酸4―ヒドロキシフェニルエーテル(0.97g)、及びベンジルトリエチルアンモニウムクロライド(0.12g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、ジフェニルエーテル-4,4´-ジカルボン酸クロライド(12.75g)とジクロロメタン(94mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(156mL)を加え、撹拌を9時間続けた。しかしながら、重合中に4―ヒドロキシ安息香酸4―ヒドロキシフェニルエーテルが加水分解され、ヒドロキノンとなり酸化され水層が茶色に変色し、2価のフェノールが足りなくなったため、十分に伸長した重合体が得られなかった。
500mLビーカーに水酸化ナトリウム(4.13g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.26g)、BP-a(9.16g)、4―ヒドロキシ安息香酸4―ヒドロキシフェニルエーテル(0.97g)、及びベンジルトリエチルアンモニウムクロライド(0.12g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、ジフェニルエーテル-4,4´-ジカルボン酸クロライド(12.75g)とジクロロメタン(94mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(156mL)を加え、撹拌を9時間続けた。しかしながら、重合中に4―ヒドロキシ安息香酸4―ヒドロキシフェニルエーテルが加水分解され、ヒドロキノンとなり酸化され水層が茶色に変色し、2価のフェノールが足りなくなったため、十分に伸長した重合体が得られなかった。
<感光体シートの作製>
[実施例3-4]
10質量部のオキシチタニウムフタロシアニンと、150質量部の4-メトキシ-4-メチル-2-ペンタノンとを混合し、サンドグラインドミルにて粉砕分散処理を行い顔料分散液を製造した。尚、オキシチタニウムフタロシアニンは、CuKα線によるX線回折においてブラッグ角(2θ±0.2)9.3゜、10.6゜、13.2゜、15.1゜、15.7゜、16.1゜、20.8゜、23.3゜、26.3゜、27.1゜に強い回折ピークを表す。この顔料分散液に、ポリビニルブチラール(電気化学工業株式会社製、商品名デンカブチラール♯6000C)の5質量%1,2-ジメトキシエタン溶液を50質量部、フェノキシ樹脂(ユニオンカーバイド株式会社製、商品名PKHH)の5質量%1,2-ジメトキシエタン溶液を50質量部混合し、更に、適量の1,2-ジメトキシエタンを加え、固形分濃度4.0%の電荷発生層形成用塗布液を調製した。この電荷発生層形成用塗布液を、表面にアルミ蒸着したポリエチレンテレフタレートシート上に、乾燥後の膜厚が0.4μmになるように塗布、乾燥して電荷発生層を設けた。
[実施例3-4]
10質量部のオキシチタニウムフタロシアニンと、150質量部の4-メトキシ-4-メチル-2-ペンタノンとを混合し、サンドグラインドミルにて粉砕分散処理を行い顔料分散液を製造した。尚、オキシチタニウムフタロシアニンは、CuKα線によるX線回折においてブラッグ角(2θ±0.2)9.3゜、10.6゜、13.2゜、15.1゜、15.7゜、16.1゜、20.8゜、23.3゜、26.3゜、27.1゜に強い回折ピークを表す。この顔料分散液に、ポリビニルブチラール(電気化学工業株式会社製、商品名デンカブチラール♯6000C)の5質量%1,2-ジメトキシエタン溶液を50質量部、フェノキシ樹脂(ユニオンカーバイド株式会社製、商品名PKHH)の5質量%1,2-ジメトキシエタン溶液を50質量部混合し、更に、適量の1,2-ジメトキシエタンを加え、固形分濃度4.0%の電荷発生層形成用塗布液を調製した。この電荷発生層形成用塗布液を、表面にアルミ蒸着したポリエチレンテレフタレートシート上に、乾燥後の膜厚が0.4μmになるように塗布、乾燥して電荷発生層を設けた。
次に、電荷輸送物質として、下記に表す構造を主成分とする、幾何異性体の化合物群からなる、日本国特開2002-080432号公報の実施例1に記載の方法で製造した混合物(CTM-1)を50質量部、実施例3-1で製造したポリエステル樹脂(3-1)を100質量部、酸化防止剤(イルガノックス1076)8質量部、レベリング剤としてシリコーンオイル0.05質量部を、テトラヒドロフランとトルエンとの混合溶媒(テトラヒドロフラン80質量%、トルエン20質量%)640質量部に混合し、電荷輸送層形成用塗布液を調製した。
電荷輸送材(CTM-1)

この電荷輸送層形成用塗布液を上述の電荷発生層上に、乾燥後の膜厚が25μmとなるようにアプリケーターを用いて塗布し、125℃で20分間乾燥して電荷輸送層を形成して、感光体シートを作製した。
[実施例3-5]
ポリエステル樹脂(3-1)を実施例3-2で製造したポリエステル樹脂(3-1)に変えた以外は実施例3-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(3-1)を実施例3-2で製造したポリエステル樹脂(3-1)に変えた以外は実施例3-1と同様にして、感光体シートを作製した。
[実施例3-6]
ポリエステル樹脂(3-1)をポリエステル樹脂(3-2)に変えた以外は実施例3-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(3-1)をポリエステル樹脂(3-2)に変えた以外は実施例3-1と同様にして、感光体シートを作製した。
[比較例3-1]
ポリエステル樹脂(3-1)を日本国特開2006-53549号公報の実施例6に記載の方法により製造した以下に表す構造を有するポリエステル樹脂(3-3)(粘度平均分子量36,200)に変えた以外は実施例3-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(3-1)を日本国特開2006-53549号公報の実施例6に記載の方法により製造した以下に表す構造を有するポリエステル樹脂(3-3)(粘度平均分子量36,200)に変えた以外は実施例3-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(3-3)

[電気特性評価]
電子写真学会測定標準に従って作製された電子写真特性評価装置(続電子写真技術の基礎と応用、電子写真学会編、コロナ社、404-405頁記載(1996年))を使用し、上記感光体をアルミニウム製ドラムに貼り付けて円筒状にし、アルミニウム製ドラムと感光体のアルミニウム基体との導通を取った上で、ドラムを一定回転数で回転させ、帯電、露光、電位測定、除電のサイクルによる電気特性評価試験を行った。その際、初期表面電位を-700Vとし、露光は780nm、除電は660nmの単色光を用い、露光光を2.4μJ/cm2照射した時点の表面電位(VL)を測定した。VL測定に際しては、露光から電位測定に要する時間を139msとした。また、表面電位が初期表面電位の半分(-350V)となる時の照射エネルギー(半減露光エネルギー:μJ/cm2)を感度(E1/2)として測定した。VLの値の絶対値が小さいほど電気特性が良好であることを示し、E1/2の値が小さいほど高感度であることを表す。測定環境は、温度25℃、相対湿度50%下(N/N)で行った。結果を表3-1に表す。
電子写真学会測定標準に従って作製された電子写真特性評価装置(続電子写真技術の基礎と応用、電子写真学会編、コロナ社、404-405頁記載(1996年))を使用し、上記感光体をアルミニウム製ドラムに貼り付けて円筒状にし、アルミニウム製ドラムと感光体のアルミニウム基体との導通を取った上で、ドラムを一定回転数で回転させ、帯電、露光、電位測定、除電のサイクルによる電気特性評価試験を行った。その際、初期表面電位を-700Vとし、露光は780nm、除電は660nmの単色光を用い、露光光を2.4μJ/cm2照射した時点の表面電位(VL)を測定した。VL測定に際しては、露光から電位測定に要する時間を139msとした。また、表面電位が初期表面電位の半分(-350V)となる時の照射エネルギー(半減露光エネルギー:μJ/cm2)を感度(E1/2)として測定した。VLの値の絶対値が小さいほど電気特性が良好であることを示し、E1/2の値が小さいほど高感度であることを表す。測定環境は、温度25℃、相対湿度50%下(N/N)で行った。結果を表3-1に表す。
[摩耗試験]
感光体フィルムを直径10cmの円状に切断しテーバー摩耗試験機(東洋精機社製)により、摩耗評価を行った。試験条件は、25℃、50%RHの雰囲気下、摩耗輪CS-10Fを用いて、荷重500gで1000回回転後の摩耗量を試験前後の質量を比較することにより測定した。値が小さい方が耐摩耗性に優れる。結果を表3-1に表す。
感光体フィルムを直径10cmの円状に切断しテーバー摩耗試験機(東洋精機社製)により、摩耗評価を行った。試験条件は、25℃、50%RHの雰囲気下、摩耗輪CS-10Fを用いて、荷重500gで1000回回転後の摩耗量を試験前後の質量を比較することにより測定した。値が小さい方が耐摩耗性に優れる。結果を表3-1に表す。

以上から、本発明の特定のポリエステル樹脂を含有する電子写真感光体は、電気特性に優れるだけでなく、耐摩耗性に非常に優れることを明らかとした。また、本発明のポリエステル樹脂は原料にエステル結合を有する低分子量の2価フェノールを使用しているが、溶液重合法や溶液重合法と界面重合法とを組み合わせることにより、簡便に製造可能かつ電子写真感光体の特性を満たすことができることを明らかとした。
(試験例4)
[エステルオリゴマーの製造]
オリゴマー製造例4-1(エステルオリゴマー(4-1)の製造)
窒素置換した300mL4つ口反応容器にレゾルシノール(3.00g)(以下、BP-1)、1,1-ビス(4-ヒドロキシ-3,メチルフェニル)エタン(13.20g)(以下、BP-2)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(以下、AC-1)(16.08g)を秤取り、ジクロロメタン(90mL)に溶解させた。続いて、トリエチルアミン(11.58g)とジクロロメタン(45mL)との混合溶液を0~5℃へ冷却した反応容器内へ30分かけて滴下した。1時間撹拌を続けた後、脱塩水(100mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(100mL)にて洗浄を2回行い、さらに、脱塩水(100mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(4-1)を得た。
[エステルオリゴマーの製造]
オリゴマー製造例4-1(エステルオリゴマー(4-1)の製造)
窒素置換した300mL4つ口反応容器にレゾルシノール(3.00g)(以下、BP-1)、1,1-ビス(4-ヒドロキシ-3,メチルフェニル)エタン(13.20g)(以下、BP-2)とジフェニルエーテル-4,4’-ジカルボン酸クロライド(以下、AC-1)(16.08g)を秤取り、ジクロロメタン(90mL)に溶解させた。続いて、トリエチルアミン(11.58g)とジクロロメタン(45mL)との混合溶液を0~5℃へ冷却した反応容器内へ30分かけて滴下した。1時間撹拌を続けた後、脱塩水(100mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(100mL)にて洗浄を2回行い、さらに、脱塩水(100mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(4-1)を得た。
オリゴマー製造例4-2(エステルオリゴマー(4-2)の製造)
窒素置換した300mL4つ口反応容器にメチルヒドロキノン(3.00g)(以下、BP-3)、BP-2(11.71g)とAC-1(14.26g)を秤取り、ジクロロメタン(90mL)に溶解させた。続いて、トリエチルアミン(10.28g)とジクロロメタン(30mL)との混合溶液を0~5℃へ冷却した反応容器内へ30分かけて滴下した。1時間撹拌を続けた後、脱塩水(100mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(100mL)にて洗浄を2回行い、さらに、脱塩水(100mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(4-2)を得た。
窒素置換した300mL4つ口反応容器にメチルヒドロキノン(3.00g)(以下、BP-3)、BP-2(11.71g)とAC-1(14.26g)を秤取り、ジクロロメタン(90mL)に溶解させた。続いて、トリエチルアミン(10.28g)とジクロロメタン(30mL)との混合溶液を0~5℃へ冷却した反応容器内へ30分かけて滴下した。1時間撹拌を続けた後、脱塩水(100mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(100mL)にて洗浄を2回行い、さらに、脱塩水(100mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(4-2)を得た。
オリゴマー製造例4-3(エステルオリゴマー(4-3)の合成)
窒素置換した300mL4つ口反応容器に1,6-ジヒドロキシナフタレン(4.00g)(以下、BP-4)、BP-2(12.10g)とAC-1(14.74g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(10.62g)とジクロロメタン(45mL)との混合溶液を0~5℃へ冷却した反応容器内へ30分かけて滴下した。1時間撹拌を続けた後、脱塩水(100mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(100mL)にて洗浄を2回行い、さらに、脱塩水(100mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(4-3)を得た。
窒素置換した300mL4つ口反応容器に1,6-ジヒドロキシナフタレン(4.00g)(以下、BP-4)、BP-2(12.10g)とAC-1(14.74g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(10.62g)とジクロロメタン(45mL)との混合溶液を0~5℃へ冷却した反応容器内へ30分かけて滴下した。1時間撹拌を続けた後、脱塩水(100mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(100mL)にて洗浄を2回行い、さらに、脱塩水(100mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(4-3)を得た。
オリゴマー製造例4-4(エステルオリゴマー(4-4)の製造)
窒素置換した300mL4つ口反応容器に1,5-ジヒドロキシナフタレン(1.50g)(以下、BP-5)、BP-2(12.86g)とAC-1(12.28g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(8.84g)とジクロロメタン(40mL)との混合溶液を0~5℃へ冷却した反応容器内へ30分かけて滴下した。1時間撹拌を続けた後、脱塩水(100mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(100mL)にて洗浄を2回行い、さらに、脱塩水(100mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(4-4)を得た。
窒素置換した300mL4つ口反応容器に1,5-ジヒドロキシナフタレン(1.50g)(以下、BP-5)、BP-2(12.86g)とAC-1(12.28g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(8.84g)とジクロロメタン(40mL)との混合溶液を0~5℃へ冷却した反応容器内へ30分かけて滴下した。1時間撹拌を続けた後、脱塩水(100mL)を添加し、10分撹拌した。撹拌後、有機層を分離し、この有機層を0.1規定塩酸(100mL)にて洗浄を2回行い、さらに、脱塩水(100mL)にて洗浄を2回行った。その後、濃縮、乾燥して分子末端にフェノール基を有するエステルオリゴマー(4-4)を得た。
[ポリエステル樹脂の製造]
ポリエステル製造例4-1(ポリエステル樹脂(4-1)の製造)
500mLビーカーに水酸化ナトリウム(1.57g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.21g)、及びベンジルトリエチルアンモニウムクロライド(0.11g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、オリゴマー製造例4-1で合成したエステルオリゴマー(4-1)(16.13g)及びAC-1(4.87g)とジクロロメタン(107mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(166mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(205mL)にて洗浄を3回行い、さらに、脱塩水(205mL)にて洗浄を2回行った。洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(4-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は44,200であった。ポリエステル樹脂(4-1)の構造式を以下に示す。
ポリエステル製造例4-1(ポリエステル樹脂(4-1)の製造)
500mLビーカーに水酸化ナトリウム(1.57g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.21g)、及びベンジルトリエチルアンモニウムクロライド(0.11g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、オリゴマー製造例4-1で合成したエステルオリゴマー(4-1)(16.13g)及びAC-1(4.87g)とジクロロメタン(107mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(166mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(205mL)にて洗浄を3回行い、さらに、脱塩水(205mL)にて洗浄を2回行った。洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(4-1)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は44,200であった。ポリエステル樹脂(4-1)の構造式を以下に示す。
ポリエステル樹脂(4-1)

[粘度平均分子量(Mv)の測定]
ポリエステル樹脂をジクロロメタンに溶解し濃度Cが6.00g/Lの溶液を調製した。溶媒(ジクロロメタン)の流下時間t0が136.16秒のウベローデ型毛細管粘度計を用いて、20.0℃に設定した恒温水槽中で試料溶液の流下時間tを測定した。以下の式に従って粘度平均分子量(Mv)を算出した。
ポリエステル樹脂をジクロロメタンに溶解し濃度Cが6.00g/Lの溶液を調製した。溶媒(ジクロロメタン)の流下時間t0が136.16秒のウベローデ型毛細管粘度計を用いて、20.0℃に設定した恒温水槽中で試料溶液の流下時間tを測定した。以下の式に従って粘度平均分子量(Mv)を算出した。
a=0.438×ηsp+1 ηsp=t/t0-1
b=100×ηsp/C C=6.00(g/L)
η=b/a
Mv=3207×η1.205
b=100×ηsp/C C=6.00(g/L)
η=b/a
Mv=3207×η1.205
ポリエステル製造例4-2(ポリエステル樹脂(4-2)の製造)
500mLビーカーに水酸化ナトリウム(1.55g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.21g)、及びベンジルトリエチルアンモニウムクロライド(0.11g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、オリゴマー製造例4-2で合成したエステルオリゴマー(4-2)(16.18g)及びAC-1(4.81g)とジクロロメタン(107mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(166mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(205mL)にて洗浄を3回行い、さらに、脱塩水(205mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(4-2)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は54,200であった。ポリエステル樹脂(4-2)の構造式を以下に示す。
500mLビーカーに水酸化ナトリウム(1.55g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.21g)、及びベンジルトリエチルアンモニウムクロライド(0.11g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、オリゴマー製造例4-2で合成したエステルオリゴマー(4-2)(16.18g)及びAC-1(4.81g)とジクロロメタン(107mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(166mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(205mL)にて洗浄を3回行い、さらに、脱塩水(205mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(4-2)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は54,200であった。ポリエステル樹脂(4-2)の構造式を以下に示す。
ポリエステル樹脂(4-2)

ポリエステル製造例4-3(ポリエステル樹脂(4-3)の製造)
500mLビーカーに水酸化ナトリウム(1.52g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.24g)、及びベンジルトリエチルアンモニウムクロライド(0.11g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、オリゴマー製造例4-3で合成したエステルオリゴマー(4-3)(16.22g)及びAC-1(4.70g)とジクロロメタン(107mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(176mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(210mL)にて洗浄を3回行い、さらに、脱塩水(210mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(4-3)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は39,000であった。ポリエステル樹脂(4-3)の構造式を以下に示す。
500mLビーカーに水酸化ナトリウム(1.52g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.24g)、及びベンジルトリエチルアンモニウムクロライド(0.11g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、オリゴマー製造例4-3で合成したエステルオリゴマー(4-3)(16.22g)及びAC-1(4.70g)とジクロロメタン(107mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(176mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(210mL)にて洗浄を3回行い、さらに、脱塩水(210mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(4-3)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は39,000であった。ポリエステル樹脂(4-3)の構造式を以下に示す。
ポリエステル樹脂(4-3)

ポリエステル製造例4-4(ポリエステル樹脂(4-4)の製造)
500mLビーカーに水酸化ナトリウム(1.47g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.25g)、及びベンジルトリエチルアンモニウムクロライド(0.11g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、オリゴマー製造例4-4で合成したエステルオリゴマー(4-4)(16.32g)及びAC-1(4.56g)とジクロロメタン(107mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(176mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(210mL)にて洗浄を3回行い、さらに、脱塩水(210mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(4-4)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は41,300であった。ポリエステル樹脂(4-4)の構造式を以下に示す。
500mLビーカーに水酸化ナトリウム(1.47g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.25g)、及びベンジルトリエチルアンモニウムクロライド(0.11g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、オリゴマー製造例4-4で合成したエステルオリゴマー(4-4)(16.32g)及びAC-1(4.56g)とジクロロメタン(107mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(176mL)を加え、撹拌を9時間続けた。その後、撹拌を停止し30分間静置した後に有機層を分離した。この有機層を0.1規定塩酸(210mL)にて洗浄を3回行い、さらに、脱塩水(210mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(100ml)を加えて希釈し、メタノール(1800ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(4-4)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は41,300であった。ポリエステル樹脂(4-4)の構造式を以下に示す。
ポリエステル樹脂(4-4)

ポリエステル製造例4-5(ポリエステル樹脂(4-5)の製造)
窒素置換した1000mL4つ口反応容器にBP-2(13.89g)、2,3,5-トリメチルフェノール(0.067g)及びAC-1(24.33g)を秤取り、ジクロロメタン(120mL)に溶解させた。続いて、トリエチルアミン(11.60g)とジクロロメタン(30mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることによりエステルオリゴマーを生成させた。
続いて上記反応容器内へ、ハイドロキノン(以下、BP-6)(2.71g)を加えた。その後、トリエチルアミン(6.13g)とジクロロメタン(50mL)との混合溶液を反応容器内へ5~15℃へ冷却した反応容器内へ20分かけて滴下した。反応内温を15~23℃に保ちながら、0.5時間攪拌を続けた後、ジクロロメタン(230mL)で希釈した後、さらに0.5時間撹拌を行った。続いてジクロロメタン(230mL)でさらに希釈した後、4時間撹拌を行った。その後、脱塩水(490mL)にて洗浄を行い、続いて0.2規定塩酸(490mL)にて洗浄を3回行い、さらに、脱塩水(490mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(300ml)を加えて希釈し、メタノール(4000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(4-5)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は55,000であった。ポリエステル樹脂(4-5)の構造式を以下に表す。
窒素置換した1000mL4つ口反応容器にBP-2(13.89g)、2,3,5-トリメチルフェノール(0.067g)及びAC-1(24.33g)を秤取り、ジクロロメタン(120mL)に溶解させた。続いて、トリエチルアミン(11.60g)とジクロロメタン(30mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることによりエステルオリゴマーを生成させた。
続いて上記反応容器内へ、ハイドロキノン(以下、BP-6)(2.71g)を加えた。その後、トリエチルアミン(6.13g)とジクロロメタン(50mL)との混合溶液を反応容器内へ5~15℃へ冷却した反応容器内へ20分かけて滴下した。反応内温を15~23℃に保ちながら、0.5時間攪拌を続けた後、ジクロロメタン(230mL)で希釈した後、さらに0.5時間撹拌を行った。続いてジクロロメタン(230mL)でさらに希釈した後、4時間撹拌を行った。その後、脱塩水(490mL)にて洗浄を行い、続いて0.2規定塩酸(490mL)にて洗浄を3回行い、さらに、脱塩水(490mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(300ml)を加えて希釈し、メタノール(4000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(4-5)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は55,000であった。ポリエステル樹脂(4-5)の構造式を以下に表す。
ポリエステル樹脂(4-5)

ポリエステル製造例4-6(ポリエステル樹脂(4-6)の合成)
窒素置換した500mL4つ口反応容器にBP-2(7.63g)、2,3,5-トリメチルフェノール(0.092g)及びAC-1(13.42g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(6.42g)とジクロロメタン(20mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることによりエステルオリゴマーを生成させた。
続いて上記反応容器内へ、2,6-ジヒドロキシナフタレン(以下、BP-7)(2.16g)を加えた。その後、トリエチルアミン(3.41g)とジクロロメタン(30mL)との混合溶液を反応容器内へ5~15℃へ冷却した反応容器内へ20分かけて滴下した。反応内温を15~23℃に保ちながら、0.5時間攪拌を続けた後、ジクロロメタン(90mL)で希釈した後、さらに0.5時間撹拌を行った。続いてジクロロメタン(80mL)でさらに希釈した後、4時間撹拌を行った。その後、脱塩水(200mL)にて洗浄を行い、続いて0.2規定塩酸(200mL)にて洗浄を3回行い、さらに、脱塩水(200mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(150ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(4-6)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は40,700であった。ポリエステル樹脂(4-6)の構造式を以下に表す。
窒素置換した500mL4つ口反応容器にBP-2(7.63g)、2,3,5-トリメチルフェノール(0.092g)及びAC-1(13.42g)を秤取り、ジクロロメタン(80mL)に溶解させた。続いて、トリエチルアミン(6.42g)とジクロロメタン(20mL)との混合溶液を5~15℃へ冷却した反応容器内へ20分かけて滴下し、その後10分撹拌を続けることによりエステルオリゴマーを生成させた。
続いて上記反応容器内へ、2,6-ジヒドロキシナフタレン(以下、BP-7)(2.16g)を加えた。その後、トリエチルアミン(3.41g)とジクロロメタン(30mL)との混合溶液を反応容器内へ5~15℃へ冷却した反応容器内へ20分かけて滴下した。反応内温を15~23℃に保ちながら、0.5時間攪拌を続けた後、ジクロロメタン(90mL)で希釈した後、さらに0.5時間撹拌を行った。続いてジクロロメタン(80mL)でさらに希釈した後、4時間撹拌を行った。その後、脱塩水(200mL)にて洗浄を行い、続いて0.2規定塩酸(200mL)にて洗浄を3回行い、さらに、脱塩水(200mL)にて洗浄を2回行った。
洗浄後の有機層にジクロロメタン(150ml)を加えて希釈し、メタノール(2000ml)に注いで得られた沈殿物を濾過にて取り出し、乾燥して目的のポリエステル樹脂(4-6)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は40,700であった。ポリエステル樹脂(4-6)の構造式を以下に表す。
ポリエステル樹脂(4-6)

ポリエステル製造例4-7(ポリエステル樹脂(4-7)の合成)
ポリエステル製造例4-6における2,6-ジヒドロキシナフタレン(BP-7)を2,7-ジヒドロキシナフタレン(以下、BP-8)に代えた以外はポリエステル製造例4-6と同様に実施し、ポリエステル樹脂(4-7)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は35,500であった。ポリエステル樹脂(4-7)の構造式を以下に表す。
ポリエステル製造例4-6における2,6-ジヒドロキシナフタレン(BP-7)を2,7-ジヒドロキシナフタレン(以下、BP-8)に代えた以外はポリエステル製造例4-6と同様に実施し、ポリエステル樹脂(4-7)を得た。得られたポリエステル樹脂の粘度平均分子量(Mv)は35,500であった。ポリエステル樹脂(4-7)の構造式を以下に表す。
ポリエステル樹脂(4-7)

ポリエステル製造例4-8(ポリエステル樹脂(4-8)の界面重合による製造)
500mLビーカーに水酸化ナトリウム(4.23g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.26g)、BP-2(10.15g)、BP-6(0.47g)、及びベンジルトリエチルアンモニウムクロライド(0.12g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、AC-1(13.08g)とジクロロメタン(94mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(156mL)を加え、撹拌を9時間続けた。しかしながら、重合中にヒドロキノンが酸化され水層が茶色に変色し、2価のフェノールが足りなくなったため、十分に伸長した重合体が得られなかった。
500mLビーカーに水酸化ナトリウム(4.23g)とH2O(188ml)を秤取り、攪拌しながら溶解させた。そこに2,3,5-トリメチルフェノール(0.26g)、BP-2(10.15g)、BP-6(0.47g)、及びベンジルトリエチルアンモニウムクロライド(0.12g)を添加し、攪拌溶解した後、このアルカリ水溶液を1L反応槽に移した。
別途、AC-1(13.08g)とジクロロメタン(94mL)との混合溶液を滴下ロート内に移した。
反応槽の外温を20℃に保ち、反応槽内のアルカリ水溶液を撹拌しながら、滴下ロートよりジクロロメタン溶液を1時間かけて滴下した。さらに1時間撹拌を続けた後、ジクロロメタン(156mL)を加え、撹拌を9時間続けた。しかしながら、重合中にヒドロキノンが酸化され水層が茶色に変色し、2価のフェノールが足りなくなったため、十分に伸長した重合体が得られなかった。
<感光体シートの作製>
[実施例4-1]
10質量部のオキシチタニウムフタロシアニンと、150質量部の4-メトキシ-4-メチル-2-ペンタノンとを混合し、サンドグラインドミルにて粉砕分散処理を行い顔料分散液を製造した。尚、オキシチタニウムフタロシアニンは、CuKα線によるX線回折においてブラッグ角(2θ±0.2)9.3゜、10.6゜、13.2゜、15.1゜、15.7゜、16.1゜、20.8゜、23.3゜、26.3゜、27.1゜に強い回折ピークを表す。
この顔料分散液に、ポリビニルブチラール(電気化学工業株式会社製、商品名デンカブチラール♯6000C)の5質量%1,2-ジメトキシエタン溶液を50質量部、フェノキシ樹脂(ユニオンカーバイド株式会社製、商品名PKHH)の5質量%1,2-ジメトキシエタン溶液を50質量部混合し、更に、適量の1,2-ジメトキシエタンを加え、固形分濃度4.0%の電荷発生層形成用塗布液を調製した。この電荷発生層形成用塗布液を、表面にアルミ蒸着したポリエチレンテレフタレートシート上に、乾燥後の膜厚が0.4μmになるように塗布、乾燥して電荷発生層を設けた。
次に、電荷輸送物質として、下記に表す構造を主成分とする、幾何異性体の化合物群からなる、日本国特開2002-080432号公報の実施例1に記載の方法で製造した混合物(CTM-1)を50質量部、ポリエステル樹脂(4-1)を100質量部、酸化防止剤(イルガノックス1076)8質量部、レベリング剤としてシリコーンオイル0.05質量部を、テトラヒドロフランとトルエンとの混合溶媒(テトラヒドロフラン80質量%、トルエン20質量%)640質量部に混合し、電荷輸送層形成用塗布液を調製した。
[実施例4-1]
10質量部のオキシチタニウムフタロシアニンと、150質量部の4-メトキシ-4-メチル-2-ペンタノンとを混合し、サンドグラインドミルにて粉砕分散処理を行い顔料分散液を製造した。尚、オキシチタニウムフタロシアニンは、CuKα線によるX線回折においてブラッグ角(2θ±0.2)9.3゜、10.6゜、13.2゜、15.1゜、15.7゜、16.1゜、20.8゜、23.3゜、26.3゜、27.1゜に強い回折ピークを表す。
この顔料分散液に、ポリビニルブチラール(電気化学工業株式会社製、商品名デンカブチラール♯6000C)の5質量%1,2-ジメトキシエタン溶液を50質量部、フェノキシ樹脂(ユニオンカーバイド株式会社製、商品名PKHH)の5質量%1,2-ジメトキシエタン溶液を50質量部混合し、更に、適量の1,2-ジメトキシエタンを加え、固形分濃度4.0%の電荷発生層形成用塗布液を調製した。この電荷発生層形成用塗布液を、表面にアルミ蒸着したポリエチレンテレフタレートシート上に、乾燥後の膜厚が0.4μmになるように塗布、乾燥して電荷発生層を設けた。
次に、電荷輸送物質として、下記に表す構造を主成分とする、幾何異性体の化合物群からなる、日本国特開2002-080432号公報の実施例1に記載の方法で製造した混合物(CTM-1)を50質量部、ポリエステル樹脂(4-1)を100質量部、酸化防止剤(イルガノックス1076)8質量部、レベリング剤としてシリコーンオイル0.05質量部を、テトラヒドロフランとトルエンとの混合溶媒(テトラヒドロフラン80質量%、トルエン20質量%)640質量部に混合し、電荷輸送層形成用塗布液を調製した。
電荷輸送材(CTM-1)

この電荷輸送層形成用塗布液を上述の電荷発生層上に、乾燥後の膜厚が25μmとなるようにアプリケーターを用いて塗布し、125℃で20分間乾燥して電荷輸送層を形成して、感光体シートを作製した。
[実施例4-2]
ポリエステル樹脂(4-1)をポリエステル樹脂(4-2)に変えた以外は実施例4-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(4-1)をポリエステル樹脂(4-2)に変えた以外は実施例4-1と同様にして、感光体シートを作製した。
[実施例4-3]
ポリエステル樹脂(4-1)をポリエステル樹脂(4-3)に変えた以外は実施例4-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(4-1)をポリエステル樹脂(4-3)に変えた以外は実施例4-1と同様にして、感光体シートを作製した。
[実施例4-4]
ポリエステル樹脂(4-1)をポリエステル樹脂(4-4)に変えた以外は実施例4-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(4-1)をポリエステル樹脂(4-4)に変えた以外は実施例4-1と同様にして、感光体シートを作製した。
[実施例4-5]
ポリエステル樹脂(4-1)をポリエステル樹脂(4-5)に変えた以外は実施例4-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(4-1)をポリエステル樹脂(4-5)に変えた以外は実施例4-1と同様にして、感光体シートを作製した。
[実施例4-6]
ポリエステル樹脂(4-1)をポリエステル樹脂(4-6)に変えた以外は実施例4-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(4-1)をポリエステル樹脂(4-6)に変えた以外は実施例4-1と同様にして、感光体シートを作製した。
[実施例4-7]
ポリエステル樹脂(4-1)をポリエステル樹脂(4-7)に変えた以外は実施例4-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(4-1)をポリエステル樹脂(4-7)に変えた以外は実施例4-1と同様にして、感光体シートを作製した。
[比較例4-1]
ポリエステル樹脂(4-1)を日本国特開2006-53549号公報の実施例6に記載の方法により製造した以下に表す構造を有するポリエステル樹脂(4-9)(粘度平均分子量36,200)に変えた以外は実施例4-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(4-1)を日本国特開2006-53549号公報の実施例6に記載の方法により製造した以下に表す構造を有するポリエステル樹脂(4-9)(粘度平均分子量36,200)に変えた以外は実施例4-1と同様にして、感光体シートを作製した。
ポリエステル樹脂(4-9)

[電気特性評価]
電子写真学会測定標準に従って作製された電子写真特性評価装置(続電子写真技術の基礎と応用、電子写真学会編、コロナ社、404-405頁記載(1996年))を使用し、上記感光体をアルミニウム製ドラムに貼り付けて円筒状にし、アルミニウム製ドラムと感光体のアルミニウム基体との導通を取った上で、ドラムを一定回転数で回転させ、帯電、露光、電位測定、除電のサイクルによる電気特性評価試験を行った。その際、初期表面電位を-700Vとし、露光は780nm、除電は660nmの単色光を用い、露光光を2.4μJ/cm2照射した時点の表面電位(VL)を測定した。VL測定に際しては、露光から電位測定に要する時間を139msとした。また、表面電位が初期表面電位の半分(-350V)となる時の照射エネルギー(半減露光エネルギー:μJ/cm2)を感度(E1/2)として測定した。VLの値の絶対値が小さいほど電気特性が良好であることを示し、E1/2の値が小さいほど高感度であることを示す。測定環境は、温度25℃、相対湿度50%下(N/N)で行った。結果を表4-1に示す。
電子写真学会測定標準に従って作製された電子写真特性評価装置(続電子写真技術の基礎と応用、電子写真学会編、コロナ社、404-405頁記載(1996年))を使用し、上記感光体をアルミニウム製ドラムに貼り付けて円筒状にし、アルミニウム製ドラムと感光体のアルミニウム基体との導通を取った上で、ドラムを一定回転数で回転させ、帯電、露光、電位測定、除電のサイクルによる電気特性評価試験を行った。その際、初期表面電位を-700Vとし、露光は780nm、除電は660nmの単色光を用い、露光光を2.4μJ/cm2照射した時点の表面電位(VL)を測定した。VL測定に際しては、露光から電位測定に要する時間を139msとした。また、表面電位が初期表面電位の半分(-350V)となる時の照射エネルギー(半減露光エネルギー:μJ/cm2)を感度(E1/2)として測定した。VLの値の絶対値が小さいほど電気特性が良好であることを示し、E1/2の値が小さいほど高感度であることを示す。測定環境は、温度25℃、相対湿度50%下(N/N)で行った。結果を表4-1に示す。

[摩耗試験]
感光体フィルムを直径10cmの円状に切断しテーバー摩耗試験機(東洋精機社製)により、摩耗評価を行った。試験条件は、25℃、50%RHの雰囲気下、摩耗輪CS-10Fを用いて、荷重500gで1000回転後の摩耗量を試験前後の質量を比較することにより測定した。値が小さい方が耐摩耗性に優れる。結果を表4-2に表す。
感光体フィルムを直径10cmの円状に切断しテーバー摩耗試験機(東洋精機社製)により、摩耗評価を行った。試験条件は、25℃、50%RHの雰囲気下、摩耗輪CS-10Fを用いて、荷重500gで1000回転後の摩耗量を試験前後の質量を比較することにより測定した。値が小さい方が耐摩耗性に優れる。結果を表4-2に表す。

以上から、本発明の特定のポリエステル樹脂を含有する電子写真感光体は、電気特性に優れるだけでなく、耐摩耗性に非常に優れることを明らかとした。また、本発明のポリエステル樹脂は酸化しやすい2価フェノールを使用し製造しているが、溶液重合法または溶液重合法と界面重合法とを組み合わせることにより、簡便に製造可能かつ電子写真感光体の特性を満たすことができることを明らかとした。
本発明を特定の態様を用いて詳細に説明したが、本発明の意図と範囲を離れることなく様々な変更および変形が可能であることは、当業者にとって明らかである。なお本出願は、2015年3月23日付で出願された日本特許出願(特願2015-060011)、2015年3月26日付で出願された日本特許出願(特願2015-064865)、2015年10月28日付で出願された日本特許出願(特願2015-212163)、および2015年11月30日付で出願された日本特許出願(特願2015-233304)に基づいており、その全体が引用により援用される。
1 電子写真感光体
2 帯電装置(帯電ローラ)
3 露光装置
4 現像装置
5 転写装置
6 クリーニング装置
7 定着装置
41 現像槽
42 アジテータ
43 供給ローラ
44 現像ローラ
45 規制部材
71 上部定着部材(加圧ローラ)
72 下部定着部材(定着ローラ)
73 加熱装置
T トナー
P 記録紙
2 帯電装置(帯電ローラ)
3 露光装置
4 現像装置
5 転写装置
6 クリーニング装置
7 定着装置
41 現像槽
42 アジテータ
43 供給ローラ
44 現像ローラ
45 規制部材
71 上部定着部材(加圧ローラ)
72 下部定着部材(定着ローラ)
73 加熱装置
T トナー
P 記録紙
Claims (8)
- 導電性支持体上に感光層を有する電子写真感光体において、前記感光層は、2価フェノール残基および2価カルボン酸残基からなるポリエステル樹脂を含有し、前記2価フェノール残基が、下記式(1)~式(3)で表される2価フェノール残基群より選ばれる少なくとも一つの2価フェノール残基、及び下記式(4)で表される2価フェノール残基を含むことを特徴とする電子写真感光体。




- 前記ポリエステル樹脂の全2価フェノール残基に対して、前記式(1)~式(3)で表される2価フェノール残基群より選ばれる少なくとも一つの2価フェノール残基が5~80モル%である、請求項1に記載の電子写真感光体。
- 前記ポリエステル樹脂の2価カルボン酸残基が、下記式(5)で表される2価カルボン酸残基である、請求項1又は2に記載の電子写真感光体。




- 下記式(1a)で表される繰返し単位および下記式(2a)で表される繰返し単位を含むポリエステル樹脂の製造方法であって、該ポリエステル樹脂は下記式(4a1)で表される2価フェノールおよび下記式(5a)で表されるジカルボン酸クロライドから得られるエステルオリゴマーを重合させて得られ、該エステルオリゴマー中の下記式(5a)で表されるジカルボン酸クロライドモノマー残存量が下記式(6a)を満たすことを特徴としたポリエステル樹脂の製造方法。






- ポリエステル樹脂の製造方法であって、少なくとも1種類のエステルオリゴマーを原料とし、界面重合法により重合することを特徴とする、ポリエステル樹脂の製造方法。
- 前記エステルオリゴマーが、溶液重合法又は溶融重合法により得られることを特徴とする、請求項5に記載のポリエステル樹脂の製造方法。
- 前記ポリエステル樹脂は、2価フェノール残基および2価カルボン酸残基を重合することにより得られ、前記2価フェノール残基が、前記式(1)~式(3)で表される2価フェノール残基群より選ばれる少なくとも一つの2価フェノール残基、及び前記式(4)で表される2価フェノール残基を含む請求項4~6のいずれか1項に記載のポリエステル樹脂の製造方法。
- 2価フェノール残基および2価カルボン酸残基からなるポリエステル樹脂であって、前記2価フェノール残基が、下記式(1)~式(3)で表される2価フェノール残基群より選ばれる少なくとも一つの2価フェノール残基、及び下記式(4)で表される2価フェノール残基を含むことを特徴とするポリエステル樹脂。




Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201680017656.1A CN107428922B (zh) | 2015-03-23 | 2016-03-23 | 电子照相感光体及图像形成装置 |
| US15/713,126 US10725391B2 (en) | 2015-03-23 | 2017-09-22 | Electrophotographic photoreceptor and image forming apparatus |
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015-060011 | 2015-03-23 | ||
| JP2015060011A JP6609951B2 (ja) | 2015-03-23 | 2015-03-23 | ポリエステル樹脂の製造方法及びそれを用いた電子写真感光体 |
| JP2015-064865 | 2015-03-26 | ||
| JP2015064865A JP6418026B2 (ja) | 2015-03-26 | 2015-03-26 | 電子写真感光体 |
| JP2015212163A JP6648486B2 (ja) | 2015-10-28 | 2015-10-28 | ポリエステル樹脂の製造方法、及び電子写真感光体の製造方法 |
| JP2015-212163 | 2015-10-28 | ||
| JP2015233304A JP6657860B2 (ja) | 2015-11-30 | 2015-11-30 | 電子写真感光体 |
| JP2015-233304 | 2015-11-30 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/713,126 Continuation US10725391B2 (en) | 2015-03-23 | 2017-09-22 | Electrophotographic photoreceptor and image forming apparatus |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016152937A1 true WO2016152937A1 (ja) | 2016-09-29 |
Family
ID=56978908
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/059263 WO2016152937A1 (ja) | 2015-03-23 | 2016-03-23 | 電子写真感光体、及び画像形成装置 |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US10725391B2 (ja) |
| CN (1) | CN107428922B (ja) |
| WO (1) | WO2016152937A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018180244A (ja) * | 2017-04-12 | 2018-11-15 | 京セラドキュメントソリューションズ株式会社 | 電子写真感光体、プロセスカートリッジ、及び画像形成装置 |
| JP2018180245A (ja) * | 2017-04-12 | 2018-11-15 | 京セラドキュメントソリューションズ株式会社 | 電子写真感光体、プロセスカートリッジ及び画像形成装置 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002311608A (ja) * | 2001-04-17 | 2002-10-23 | Mitsubishi Chemicals Corp | 電子写真感光体 |
| JP2003119262A (ja) * | 2001-10-05 | 2003-04-23 | Bayer Ag | ポリエステルカーボネートの製造 |
| JP2007084650A (ja) * | 2005-09-21 | 2007-04-05 | Fujifilm Corp | 高耐熱ポリマー前駆体フィルム、光学フィルムおよびその製造方法、並びに、これを用いた画像表示装置 |
| WO2009128325A1 (ja) * | 2008-04-16 | 2009-10-22 | 日東電工株式会社 | 複屈折性フィルム、偏光板、画像表示装置、及び芳香族ポリエステル |
| JP2010083986A (ja) * | 2008-09-30 | 2010-04-15 | Unitika Ltd | ポリアリレートおよびその製造方法 |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IT1116721B (it) | 1976-04-02 | 1986-02-10 | Allied Chem | Copolimero bisfenolo a tereftalato carbonato lavorabili in massa fusa |
| US4156069A (en) | 1976-04-02 | 1979-05-22 | Allied Chemical Corporation | Bisphenol-A/terephthalate/carbonate melt processable copolymers |
| JPS5614526A (en) | 1979-07-17 | 1981-02-12 | Mitsubishi Gas Chem Co Inc | Production of low-molecular aromatic dihydroxy ester |
| JPH036567A (ja) | 1989-06-02 | 1991-01-14 | Kanegafuchi Chem Ind Co Ltd | 電子写真感光体 |
| JP2680530B2 (ja) | 1993-09-27 | 1997-11-19 | 三田工業株式会社 | 電子写真感光体 |
| JPH08143655A (ja) | 1994-11-17 | 1996-06-04 | Idemitsu Kosan Co Ltd | ポリエステルカーボネートの製造方法 |
| JP3602890B2 (ja) | 1995-07-04 | 2004-12-15 | ユニチカ株式会社 | 電子写真感光体 |
| JP3537065B2 (ja) | 1996-07-01 | 2004-06-14 | キヤノン株式会社 | 電子写真感光体、プロセスカートリッジ及び電子写真装置 |
| JP3785019B2 (ja) | 2000-03-22 | 2006-06-14 | 三菱化学株式会社 | 電子写真感光体 |
| JP2004294750A (ja) | 2003-03-27 | 2004-10-21 | Mitsubishi Chemicals Corp | 電子写真感光体 |
| JP2005024852A (ja) | 2003-07-01 | 2005-01-27 | Fuji Xerox Co Ltd | 電子写真感光体、電子写真プロセスカートリッジ及び画像形成装置 |
| US7175955B2 (en) | 2003-06-30 | 2007-02-13 | Fuji Xerox Co., Ltd. | Electrophotographic photoreceptor, electrophotographic process cartridge and image forming apparatus |
| JP4517964B2 (ja) | 2004-07-16 | 2010-08-04 | 三菱化学株式会社 | 電子写真感光体 |
| EP2154575B1 (en) | 2004-07-16 | 2014-12-24 | Mitsubishi Chemical Corporation | Electrophotographic photoreceptor |
| JP4631349B2 (ja) | 2004-08-10 | 2011-02-16 | 三菱化学株式会社 | 芳香族ポリエステルポリカーボネート樹脂及びその製造方法 |
| JP2008293006A (ja) | 2007-04-27 | 2008-12-04 | Mitsubishi Chemicals Corp | 電子写真感光体 |
| JP5481829B2 (ja) | 2008-10-16 | 2014-04-23 | 三菱化学株式会社 | 電子写真感光体、並びにそれを備えたカートリッジ及び画像形成装置 |
| JP6123338B2 (ja) | 2013-02-18 | 2017-05-10 | 三菱化学株式会社 | 電子写真感光体、電子写真カートリッジ、及び画像形成装置 |
-
2016
- 2016-03-23 CN CN201680017656.1A patent/CN107428922B/zh active Active
- 2016-03-23 WO PCT/JP2016/059263 patent/WO2016152937A1/ja active Application Filing
-
2017
- 2017-09-22 US US15/713,126 patent/US10725391B2/en active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002311608A (ja) * | 2001-04-17 | 2002-10-23 | Mitsubishi Chemicals Corp | 電子写真感光体 |
| JP2003119262A (ja) * | 2001-10-05 | 2003-04-23 | Bayer Ag | ポリエステルカーボネートの製造 |
| JP2007084650A (ja) * | 2005-09-21 | 2007-04-05 | Fujifilm Corp | 高耐熱ポリマー前駆体フィルム、光学フィルムおよびその製造方法、並びに、これを用いた画像表示装置 |
| WO2009128325A1 (ja) * | 2008-04-16 | 2009-10-22 | 日東電工株式会社 | 複屈折性フィルム、偏光板、画像表示装置、及び芳香族ポリエステル |
| JP2010083986A (ja) * | 2008-09-30 | 2010-04-15 | Unitika Ltd | ポリアリレートおよびその製造方法 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018180244A (ja) * | 2017-04-12 | 2018-11-15 | 京セラドキュメントソリューションズ株式会社 | 電子写真感光体、プロセスカートリッジ、及び画像形成装置 |
| JP2018180245A (ja) * | 2017-04-12 | 2018-11-15 | 京セラドキュメントソリューションズ株式会社 | 電子写真感光体、プロセスカートリッジ及び画像形成装置 |
Also Published As
| Publication number | Publication date |
|---|---|
| US10725391B2 (en) | 2020-07-28 |
| US20180011412A1 (en) | 2018-01-11 |
| CN107428922A (zh) | 2017-12-01 |
| CN107428922B (zh) | 2021-06-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2007179038A (ja) | 電子写真感光体、および画像形成装置 | |
| JP5365164B2 (ja) | 電子写真感光体、ポリエステル樹脂、樹脂組成物、及びポリエステル樹脂の製造方法 | |
| JP6380124B2 (ja) | 電子写真感光体、画像形成装置、及びポリエステル樹脂 | |
| WO2016152937A1 (ja) | 電子写真感光体、及び画像形成装置 | |
| JP6123338B2 (ja) | 電子写真感光体、電子写真カートリッジ、及び画像形成装置 | |
| JP6592943B2 (ja) | 電子写真感光体、及び画像形成装置 | |
| JP5741180B2 (ja) | 電子写真感光体、電子写真カートリッジ、及び画像形成装置 | |
| JP6609951B2 (ja) | ポリエステル樹脂の製造方法及びそれを用いた電子写真感光体 | |
| JP5593818B2 (ja) | 電子写真感光体、電子写真カートリッジ、及び画像形成装置 | |
| WO2018056326A1 (ja) | 電子写真感光体並びにそれを含有する電子写真カートリッジ及び画像形成装置 | |
| JP6070261B2 (ja) | 電子写真感光体、電子写真カートリッジ、及び画像形成装置 | |
| JP5593817B2 (ja) | 電子写真感光体、電子写真カートリッジ、及び画像形成装置 | |
| JP6648486B2 (ja) | ポリエステル樹脂の製造方法、及び電子写真感光体の製造方法 | |
| JP6657860B2 (ja) | 電子写真感光体 | |
| JP6418026B2 (ja) | 電子写真感光体 | |
| JP6798215B2 (ja) | 電子写真感光体、電子写真カートリッジおよび画像形成装置 | |
| JP4298190B2 (ja) | 電子写真感光体 | |
| JP6953694B2 (ja) | 電子写真感光体、電子写真カートリッジおよび画像形成装置 | |
| JP2018159087A (ja) | ポリアリレート樹脂、及びそれを用いた電子写真感光体 | |
| JP4487997B2 (ja) | 電子写真感光体 | |
| JP4107135B2 (ja) | 電子写真感光体 | |
| JP6569394B2 (ja) | 電子写真感光体、及びポリアリレート樹脂の製造方法 | |
| JP2005227527A (ja) | 電子写真感光体 | |
| JP2015017224A (ja) | ポリアリレート樹脂、及びそれを用いた電子写真感光体 | |
| JP2008158553A (ja) | 電子写真感光体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16768840 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 16768840 Country of ref document: EP Kind code of ref document: A1 |