WO2015156228A1 - 高分子電解質組成物ならびにそれを用いた高分子電解質膜、膜電極複合体および固体高分子形燃料電池 - Google Patents
高分子電解質組成物ならびにそれを用いた高分子電解質膜、膜電極複合体および固体高分子形燃料電池 Download PDFInfo
- Publication number
- WO2015156228A1 WO2015156228A1 PCT/JP2015/060630 JP2015060630W WO2015156228A1 WO 2015156228 A1 WO2015156228 A1 WO 2015156228A1 JP 2015060630 W JP2015060630 W JP 2015060630W WO 2015156228 A1 WO2015156228 A1 WO 2015156228A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- polymer
- polymer electrolyte
- bis
- electrolyte membrane
- Prior art date
Links
- DNZSHSJERXNJGX-BCWCAWDXSA-N c1c(/C(/c2ccncc2)=C(/C=C2)\N=C2/C(/c2ccncc2)=C(/C=C2)\N/C2=C(\C(C=C2)=N/C2=C2/c3ccncc3)/c3ccncc3)[nH]c2c1 Chemical compound c1c(/C(/c2ccncc2)=C(/C=C2)\N=C2/C(/c2ccncc2)=C(/C=C2)\N/C2=C(\C(C=C2)=N/C2=C2/c3ccncc3)/c3ccncc3)[nH]c2c1 DNZSHSJERXNJGX-BCWCAWDXSA-N 0.000 description 1
Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M8/00—Fuel cells; Manufacture thereof
- H01M8/10—Fuel cells with solid electrolytes
- H01M8/1016—Fuel cells with solid electrolytes characterised by the electrolyte material
- H01M8/1018—Polymeric electrolyte materials
- H01M8/1041—Polymer electrolyte composites, mixtures or blends
- H01M8/1046—Mixtures of at least one polymer and at least one additive
- H01M8/1051—Non-ion-conducting additives, e.g. stabilisers, SiO2 or ZrO2
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M8/00—Fuel cells; Manufacture thereof
- H01M8/10—Fuel cells with solid electrolytes
- H01M8/1016—Fuel cells with solid electrolytes characterised by the electrolyte material
- H01M8/1018—Polymeric electrolyte materials
- H01M8/1069—Polymeric electrolyte materials characterised by the manufacturing processes
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M8/00—Fuel cells; Manufacture thereof
- H01M8/10—Fuel cells with solid electrolytes
- H01M8/1004—Fuel cells with solid electrolytes characterised by membrane-electrode assemblies [MEA]
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M8/00—Fuel cells; Manufacture thereof
- H01M8/10—Fuel cells with solid electrolytes
- H01M8/1016—Fuel cells with solid electrolytes characterised by the electrolyte material
- H01M8/1018—Polymeric electrolyte materials
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M8/00—Fuel cells; Manufacture thereof
- H01M8/10—Fuel cells with solid electrolytes
- H01M8/1016—Fuel cells with solid electrolytes characterised by the electrolyte material
- H01M8/1018—Polymeric electrolyte materials
- H01M8/102—Polymeric electrolyte materials characterised by the chemical structure of the main chain of the ion-conducting polymer
- H01M8/1025—Polymeric electrolyte materials characterised by the chemical structure of the main chain of the ion-conducting polymer having only carbon and oxygen, e.g. polyethers, sulfonated polyetheretherketones [S-PEEK], sulfonated polysaccharides, sulfonated celluloses or sulfonated polyesters
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M8/00—Fuel cells; Manufacture thereof
- H01M8/10—Fuel cells with solid electrolytes
- H01M8/1016—Fuel cells with solid electrolytes characterised by the electrolyte material
- H01M8/1018—Polymeric electrolyte materials
- H01M8/102—Polymeric electrolyte materials characterised by the chemical structure of the main chain of the ion-conducting polymer
- H01M8/103—Polymeric electrolyte materials characterised by the chemical structure of the main chain of the ion-conducting polymer having nitrogen, e.g. sulfonated polybenzimidazoles [S-PBI], polybenzimidazoles with phosphoric acid, sulfonated polyamides [S-PA] or sulfonated polyphosphazenes [S-PPh]
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M8/00—Fuel cells; Manufacture thereof
- H01M8/10—Fuel cells with solid electrolytes
- H01M8/1016—Fuel cells with solid electrolytes characterised by the electrolyte material
- H01M8/1018—Polymeric electrolyte materials
- H01M8/1041—Polymer electrolyte composites, mixtures or blends
- H01M8/1044—Mixtures of polymers, of which at least one is ionically conductive
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M8/00—Fuel cells; Manufacture thereof
- H01M8/10—Fuel cells with solid electrolytes
- H01M8/1016—Fuel cells with solid electrolytes characterised by the electrolyte material
- H01M8/1018—Polymeric electrolyte materials
- H01M8/1041—Polymer electrolyte composites, mixtures or blends
- H01M8/1046—Mixtures of at least one polymer and at least one additive
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M8/00—Fuel cells; Manufacture thereof
- H01M8/10—Fuel cells with solid electrolytes
- H01M8/1016—Fuel cells with solid electrolytes characterised by the electrolyte material
- H01M8/1018—Polymeric electrolyte materials
- H01M8/1041—Polymer electrolyte composites, mixtures or blends
- H01M8/1046—Mixtures of at least one polymer and at least one additive
- H01M8/1048—Ion-conducting additives, e.g. ion-conducting particles, heteropolyacids, metal phosphate or polybenzimidazole with phosphoric acid
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M8/00—Fuel cells; Manufacture thereof
- H01M8/10—Fuel cells with solid electrolytes
- H01M2008/1095—Fuel cells with polymeric electrolytes
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M2300/00—Electrolytes
- H01M2300/0017—Non-aqueous electrolytes
- H01M2300/0065—Solid electrolytes
- H01M2300/0082—Organic polymers
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/30—Hydrogen technology
- Y02E60/50—Fuel cells
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P70/00—Climate change mitigation technologies in the production process for final industrial or consumer products
- Y02P70/50—Manufacturing or production processes characterised by the final manufactured product
Definitions
- the present invention relates to a polymer electrolyte composition, and in particular, has excellent chemical stability that can withstand a strong oxidizing atmosphere during fuel cell operation, and excellent proton conductivity under low humidification conditions, and excellent
- the present invention relates to a polymer electrolyte composition having excellent practicality capable of achieving mechanical strength and physical durability, and a polymer electrolyte membrane, a membrane electrode assembly and a polymer electrolyte fuel cell using the same.
- a fuel cell is a kind of power generation device that extracts electric energy by electrochemically oxidizing a fuel such as hydrogen or methanol, and has recently attracted attention as a clean energy supply source.
- the polymer electrolyte fuel cell has a standard operating temperature as low as around 100 ° C. and a high energy density, so that it is a relatively small-scale distributed power generation facility, a mobile power generator such as an automobile or a ship.
- a mobile power generator such as an automobile or a ship.
- secondary batteries such as nickel metal hydride batteries and lithium ion batteries.
- an anode electrode and a cathode electrode in which a reaction responsible for power generation occurs, and a polymer electrolyte membrane serving as a proton conductor between the anode and the cathode are sometimes referred to as a membrane electrode assembly (hereinafter, abbreviated as MEA).
- MEA membrane electrode assembly
- a cell in which this MEA is sandwiched between separators is configured as a unit.
- the main component of the polymer electrolyte membrane is an ionic group-containing polymer (polymer electrolyte material), but it is also possible to use a polymer electrolyte composition containing an additive or the like in order to enhance durability.
- the polymer electrolyte composition is also suitable as a binder for an electrode catalyst layer used in a particularly severe oxidizing atmosphere.
- the polymer electrolyte membrane and the polymer electrolyte composition As the required characteristics of the polymer electrolyte membrane and the polymer electrolyte composition, firstly, high proton conductivity is mentioned, and it is particularly necessary to have high proton conductivity even under high temperature and low humidification conditions. In addition, since the polymer electrolyte membrane and the polymer electrolyte composition serve as a barrier that prevents direct reaction between the fuel and oxygen, low fuel permeability is required. Other examples include chemical stability to withstand a strong oxidizing atmosphere during fuel cell operation, mechanical strength and physical durability to withstand repeated thinning and swelling and drying.
- Nafion registered trademark
- DuPont which is a perfluorosulfonic acid polymer
- Nafion registered trademark
- DuPont which is a perfluorosulfonic acid polymer
- fuel crossover amount of fuel permeation
- problems such as loss of mechanical strength and physical durability of the membrane due to swelling and drying, problems of low softening point and inability to use at high temperatures, problems of disposal treatment after use and difficulty in recycling materials are pointed out. It has been.
- development of hydrocarbon-based electrolyte membranes has recently been activated as an inexpensive polymer electrolyte membrane that can replace Nafion (registered trademark) and has excellent membrane characteristics.
- any of these polymer electrolyte membranes has a problem of insufficient chemical stability when used in a polymer electrolyte fuel cell.
- the mechanism related to chemical degradation has not yet been fully elucidated, but hydrogen peroxide, which has strong oxidizing power generated during power generation, and a trace amount of metal such as iron that can exist in the system reacts with hydrogen peroxide.
- the polymer main chain and side chain being cleaved by the generated hydroxy radical, etc.
- the polymer electrolyte membrane becomes thin, weakened, fuel permeation increases, hydrogen peroxide, hydroxy radical, etc. are further generated, It is believed that film degradation proceeds at an accelerated rate.
- the polymer electrolyte membrane that has become fragile is damaged while it repeatedly swells and contracts in accordance with changes in humidity, and there is a problem that power generation cannot be performed.
- Patent Documents 1 and 2 propose polymer electrolyte compositions to which a phosphorus-based antioxidant is added. Specifically, a polymer electrolyte composition in which a phosphite (phosphite) antioxidant is blended with a sulfonic acid group-containing polyethersulfone polymer, or a phosphonic acid group-containing polymer such as polyvinylphosphonic acid A polymer electrolyte composition blended with a sulfonic acid group-containing polyethersulfone polymer or a sulfonic acid group-containing polyetherketone polymer.
- Patent Documents 3 to 5 propose electrolyte compositions in which sulfur-based, amine-based, phenol-based antioxidants and the like are added in addition to phosphorus-based antioxidants. Specifically, antioxidants such as phosphites (phosphites), thioethers, hindered amines, and hindered phenols are blended into sulfonic acid group-containing polyethersulfone polymers and sulfonic acid group-containing polyarylene polymers. The polymer electrolyte composition.
- antioxidants such as phosphites (phosphites), thioethers, hindered amines, and hindered phenols are blended into sulfonic acid group-containing polyethersulfone polymers and sulfonic acid group-containing polyarylene polymers.
- Patent Document 6 proposes a polymer electrolyte composition in which cerium ions or manganese ions are blended with a perfluorosulfonic acid polymer or a sulfonic acid group-containing polyether ketone polymer.
- Patent Document 7 proposes a polymer electrolyte composition containing a phosphorus-containing additive selected from a phosphine compound and a phosphinite compound, and further a transition metal atom such as cerium or manganese.
- Patent Document 8 proposes a peroxide decomposition catalyst in which a nitrogen atom such as imidazole or pyridine is coordinated to a base metal atom such as manganese or iron.
- Patent Documents 9 and 10 propose polymer electrolyte compositions in which a perfluoro-based electrolyte membrane is mixed with a phenanthroline derivative or a complex of phenanthroline and cerium ions or manganese ions.
- 2,2′-bipyridyl and 1,10-phenanthroline described in Patent Document 9 are oxidized by hydrogen peroxide and hydroxy radicals generated during operation and may be eluted out of the membrane, so that there is still sufficient chemistry. It cannot be said that mechanical stability and durability have been obtained.
- Patent Document 6 since the sulfonic acid group is ion-exchanged with cerium ions or manganese ions, which are polyvalent metals, the proton conductivity of the polymer electrolyte composition is lowered. There was a problem that the film-forming property deteriorated and the film became brittle.
- the metal is coordinated (complex) with a phosphorus additive in Patent Document 7, 2,2-bipyridyl in Patent Document 8, 1,10-phenanthroline in Patent Document 10, and the like.
- a phosphorus additive in Patent Document 7
- 2,2-bipyridyl in Patent Document 8 1,10-phenanthroline in Patent Document 10
- the complex structure is relatively hydrophilic and may leach out of the membrane during operation, sufficient chemical stability and durability have not yet been obtained.
- the polymer electrolyte composition according to the prior art has insufficient economic efficiency, processability, proton conductivity, mechanical strength, chemical stability, and physical durability, and is an industrially useful polymer electrolyte composition. It wasn't be a thing.
- the present invention has excellent chemical stability that can withstand a strong oxidizing atmosphere during fuel cell operation, and also has excellent proton conductivity and excellent mechanical strength under low humidification conditions. It is intended to provide a polymer electrolyte composition excellent in practical use that can achieve physical durability and a polymer electrolyte membrane, a membrane electrode assembly and a solid polymer fuel cell using the same. is there.
- the present inventors have blended an ionic group-containing polymer (A) with an organophosphorus additive (C).
- an organophosphorus additive C
- a specific nitrogen-containing heteroaromatic ring additive D
- a polymer electrolyte composition particularly as a polymer electrolyte membrane for fuel cells
- proton conductivity including low humidification conditions and power generation characteristics
- Excellent performance in terms of workability such as film-forming properties, chemical durability such as oxidation resistance, radical resistance and hydrolysis resistance, mechanical strength of the membrane, and physical durability such as hot water resistance
- the present invention was completed by investigating that the problems could be solved all at once, and adding various studies.
- the polymer electrolyte composition of the present invention contains at least an ionic group-containing polymer (A), an organic phosphorus-based additive (C), and a nitrogen-containing heteroaromatic ring-based additive (D).
- the molecular electrolyte composition is characterized in that the nitrogen-containing heteroaromatic ring additive (D) contains at least three nitrogen-containing heteroaromatic rings in the molecule.
- the polymer electrolyte membrane, the membrane electrode assembly, and the solid polymer fuel cell of the present invention are configured using such a polymer electrolyte composition.
- the present invention has excellent chemical stability that can withstand a strong oxidizing atmosphere, and can also achieve excellent proton conductivity, excellent mechanical strength and physical durability under low humidification conditions. It is possible to provide a polymer electrolyte composition excellent in practicality, a polymer electrolyte membrane using the same, a membrane electrode assembly, and a solid polymer fuel cell.
- the additive means a compound other than the ionic group-containing polymer (A) contained in the polymer electrolyte composition and mixed with the ionic group-containing polymer.
- the additive in the present invention mainly serves as an antioxidant.
- a "radical chain initiation inhibitor (metal deactivator)” that inactivates metal ions (Fe 2+ , Cu 2+, etc.) that serve as catalysts for generating peroxide radicals and inhibits the initiation of chain reactions by radicals Function
- function as a "radical scavenger” that inactivates the generated hydroxyl radicals and peroxide radicals and suppresses chain reactions by hydroxy radicals and peroxide radicals, reaction that hydrogen peroxide decomposes and radicalizes It is a compound having at least one of the functions as a “peroxide decomposer” that inhibits It is preferred.
- the antioxidant may be either a low molecular type having a molecular weight of less than 2000 or a high molecular type having 2000 or more. From the viewpoint of elution resistance, a polymer type is more preferable and can be appropriately selected in consideration of cost.
- the antioxidant having such a function various compounds such as phosphite, thioether, hindered amine, hindered phenol have been reported.
- the organic phosphorus additive (C) It has been newly found that by adding a nitrogen-containing heteroaromatic ring additive (D) containing at least three nitrogen-containing heteroaromatic rings, it is possible to obtain synergistically superior chemical stability and durability. It was.
- Organophosphorous additive (C) has high functionality as a “peroxide decomposing agent” or “radical scavenger” that captures, decomposes, and detoxifies highly oxidizing hydroxyl radicals, peroxide radicals and hydrogen peroxide
- a nitrogen-containing heteroaromatic ring additive (D) containing at least three nitrogen-containing heteroaromatic rings in the molecule is present in a system that promotes the generation of hydroxy radicals and peroxide radicals. Separation mechanism that functions as a “metal deactivator” that coordinates strongly to and inactivates any metal.
- the organophosphorus additive (C) functions as a “metal deactivator” and the nitrogen-containing heteroaromatic ring additive (D) functions as a “peroxide decomposer” or “radical scavenger”. Separation mechanism.
- the reduced product of the organic phosphorus additive (C) generated by reduction with hydrogen during operation is reduced and returned to the original nitrogen-containing heteroaromatic compound, while the organophosphorus addition is inferior in elution resistance.
- the oxide of the agent (C) is reduced with the operating hydrogen and returns to the original organophosphorus additive (C). This prevents the dissolution of additive oxides that are inferior in elution resistance and hot water resistance, and improves the decomposition efficiency of hydrogen peroxide, hydroxy radicals, and peroxide radicals in cycles.
- organic phosphorus additive (C) represents an organic phosphorus compound containing a chemical bond between carbon and phosphorus.
- Organic phosphorus compounds include those containing trivalent phosphorus and those containing pentavalent phosphorus.
- a general formula O P (OR)
- a phosphate (phosphoric acid) compound having a group represented by 3 (R is an organic group).
- the organic phosphorus additive (C) of the present invention preferably contains at least two phosphorus atoms in the molecule as a constituent element of the compound from the viewpoint of durability and proton conductivity.
- the decomposition efficiency of hydrogen peroxide, hydroxy radicals, and peroxide radicals is improved and low.
- the added amount not only improves durability without impairing proton conductivity, but also has the ability to coordinate and inactivate metal impurities that promote the generation of hydroxy radicals and peroxide radicals by chelating effects. And the reduction efficiency of the oxidized nitrogen-containing heteroaromatic compound can be increased.
- the chelate effect means that when a ligand is coordinated to a metal, it is a multidentate ligand (coordinating ability) from a monodentate ligand (a ligand having one coordination ability atom in the molecule).
- a ligand having two or more atoms having a) in the molecule indicates an effect that can further stabilize (inactivate) the complex by forming a chelate ring.
- this effect makes it easier for the target substance such as peroxide and the active phosphorus atom in the compound to approach each other, thereby improving the peroxide resolution, radical scavenging ability, and reduction efficiency of the oxidized nitrogen-containing heteroaromatic compound. It is thought to bring about an effect.
- the organophosphorus additive (C) used in the polymer electrolyte composition of the present invention includes hydrogen peroxide, hydroxyl radical, peroxide radical decomposition efficiency, nitrogen-containing heteroaromatic compound oxidation product reduction efficiency and resistance. From the viewpoint of elution, a trivalent organophosphorus compound is preferable. By using a trivalent organophosphorus compound with a smaller oxidation number, oxygen acceptability and reducing ability (electron donating ability) from hydrogen peroxide, hydroxy radical, and peroxide radical, which have strong oxidizing power, are increased. In addition to improving the detoxification efficiency of the substance, it is considered that the reduction efficiency of the oxidized nitrogen-containing heteroaromatic compound can be increased. Further, trivalent organophosphorus compounds having lower hydrophilicity than pentavalent organophosphorus compounds are less likely to elute out of the system even during fuel cell operation, and can impart higher chemical stability to the electrolyte composition. it is conceivable that.
- organic phosphorus additive (C) used in the present invention an organic phosphorus compound (monodentate organic phosphorus compound) represented by the following general formula (C1), and a general formula (C2) shown below are used.
- Organophosphorus compounds containing two total phosphorus atoms (bidentate organophosphorus compounds), organophosphorus compounds containing three total phosphorus atoms represented by the following general formula (C3) (tridentate organophosphorus compounds),
- An organic phosphorus compound (tetradentate organic phosphorus compound) containing a total of 4 phosphorus atoms represented by the general formula (C4-1) or (C4-2) can be given.
- J 1 represents P or P ⁇ O
- R 1 to R 3 are each independently a straight chain represented by the general formula C m H n (m and n are integers).
- halogens such as fluorine, chlorine and bromine Represents a substituent selected from an atom or a hydrogen atom
- R 1 to R 3 may be arbitrarily bonded to form a ring structure as represented by the general formula (C1a).
- n is independent in each formula.
- J 2 and J 3 each independently represent P or P ⁇ O
- R 4 to R 7 are each independently a general formula C m H n (m and n are integers).
- Z 1 is a linear, cyclic, or branched group represented by the general formula C m H n (m and n are integers).
- R 4 ⁇ R 7, Z 1 is independently in each formula, good .
- R 4 ⁇ R 7, Z 1 may be the same or different, as in the general formula (C1a), optionally It may be bonded to form a ring structure.
- J 4 to J 6 each independently represents P or P ⁇ O
- R 8 to R 12 each independently represents a linear or cyclic group represented by the general formula C m H n.
- Z 2 and Z 3 are each independently a linear, cyclic or branched hydrocarbon group represented by the general formula C m H n (m and n are integers); Divalent substitution selected from a linear, cyclic or branched alkoxy group represented by the general formula OC m H n (m and n are integers) or OC m H n O (m and n are integers)
- m and n are independent in each formula.
- .R 8 ⁇ R 12, Z 2 , Z 3 is the good .R 8 ⁇ R 12, Z 2 , Z 3 be the same as or different from each other, as in the general formula (C1a), optionally It may be bonded to form a ring structure.)
- J 7 to J 10 each independently represents P or P ⁇ O
- R 13 to R 18 each independently represents the general formula C m H n.
- a linear, cyclic, or branched structure represented by the general formula OC m H n (m and n are integers) Represents an alkoxy group, a halogen atom such as fluorine, chlorine, bromine, or a hydrogen atom
- Z 4 to Z 6 are each independently represented by the general formula C m H n (m and n are integers).
- the .m and n represents the valence of the substituent is independently in each formula .
- R 13 ⁇ R 18, Z 4 ⁇ Z 6 MAY be the same or different .
- R 13 ⁇ R 18, Z 4 ⁇ Z 6 may be optionally bonded to form a ring structure as in the general formula (C1a).
- organic phosphorus additive (C) in which a part of R or Z is an alkoxy group include, as phosphinite and phosphinate compounds, compounds represented by the following general formulas (C1b), (C2a), and (C2b) As phosphonite and phosphonate compounds, the compounds represented by the following general formulas (C1c), (C2c) and (C2d) and the like, and the phosphites and phosphate compounds as represented by the following general formulas (C1d) and (C2e) Although the compound etc. can be mentioned, the phosphinite, the phosphinate compound, the phosphonite, the phosphonate compound, the phosphite and the phosphate compound of the present invention are not limited to these.
- J 1 to J 3 each independently represents P or P ⁇ O
- R 1 to R 7 and Z 1 are the general formulas C m H n (m and n Represents a linear, cyclic, or branched hydrocarbon group represented by the formula (II), a halogen atom such as fluorine, chlorine or bromine, or a hydrogen atom, wherein R 1 to R 7 and Z 1 are the same as each other.
- R 1 to R 7 and Z 1 may be optionally combined to form a ring structure as in the general formula (C1a).
- Organophosphorus compounds containing 2 or more (bidentate or more) of at least one selected from phosphine groups, phosphinite groups, phosphonite groups and phosphite groups are preferred as the organophosphorus additive (C). More preferred are tetradentate multidentate phosphine compounds, phosphinite compounds, phosphonite compounds, and phosphite compounds.
- bidentate phosphine compounds, phosphinite compounds, phosphonite compounds, and phosphite compounds are more preferable, and in terms of hot water resistance, bidentate phosphine compounds, phosphinite compounds, and phosphonite compounds are preferable, and are resistant to hydrolysis. In this respect, a bidentate phosphine compound is most preferable.
- the organophosphorus additive (C) is a polydentate organophosphorus compound, it is capable of coordinating to metal, detoxifying hydrogen peroxide, hydroxy radicals, peroxide radicals, nitrogen-containing heteroaromatic compounds
- the bonding group that connects the phosphorus atoms is preferably a linear hydrocarbon group represented by the general formula C m H n (m and n are integers). More preferably, it is an aliphatic hydrocarbon group having a structure.
- the linear hydrocarbon group ensures high metal inertness without hindering the coordination of phosphorus atoms to metals and the approach to oxidized peroxides, radicals, and nitrogen-containing heteroaromatic compounds.
- Phosphorus-containing compound can be imparted with the ability to oxidize, peroxide resolution, radical scavenging ability, reducing ability of nitrogen-containing heteroaromatic compound, and a linear aliphatic hydrocarbon group As a result of the flexibility, the effect can be further enhanced.
- m is preferably 20 or less, more preferably 10 or less, in terms of coordination ability to metal, Most preferably, it is 6 or less.
- the hydrocarbon group is 20 or more, as a result of the active phosphorus atoms being too far apart in the molecule, the metal coordination ability, peroxide resolution, radical scavenging ability, nitrogen-containing heteroaromatic ring compound oxide associated with the chelate effect May not have sufficient reducing ability.
- the organophosphorus additive (C) preferably contains at least one aromatic hydrocarbon group in the molecule in terms of chemical stability and production process.
- the organic phosphorus additive (C) can be stabilized by electronic interaction between the ⁇ electron and the phosphorus atom of the aromatic hydrocarbon group, and more A stable polymer electrolyte composition manufacturing process can be established.
- these trivalent organophosphorus compounds may be oxidized to the corresponding pentavalent phosphate-containing compounds during the fuel cell operation.
- organic phosphorus additives C
- phosphine compound represented by the general formula (C1) include trimethylphosphine, triethylphosphine, tripropylphosphine, triisopropylphosphine, tri-n-butylphosphine, tri-s-butylphosphine, tri-i -Butylphosphine, tri-t-butylphosphine, tripentylphosphine, trihexylphosphine, triheptylphosphine, trioctylphosphine, tris (ethylhexyl) phosphine, trinonylphosphine, tridecylphosphine, tris (hydroxymethyl) phosphine, tris ( 2-carboxyethyl) phosphine, dicyclohexylethylphosphine, di-t-butylneopentylphosphine, diadamantylpho
- bidentate phosphine compound represented by the general formula (C2) include bis (diphenylphosphino) methane, bis (diphenylphosphino) ethane, bis (diphenylphosphino) propane, and bis (diphenyl).
- Phosphino) butane bis (diphenylphosphino) pentane, bis (diphenylphosphino) hexane, bis (diphenylphosphino) pentane, bis (diphenylphosphino) octane, bis (diphenylphosphino) nonane, bis (diphenylphosphino) ) Decane, bis [bis (pentafluorophenyl) phosphino] ethane, bis (diphenylphosphino) ethylene, bis (diphenylphosphino) acetylene, bis [(phenylpropanesulfonic acid) phosphine] butane and its salts, ((diphenylphosphine 6) phenyl) diphenylphosphine, bis (dimethylphosphino) methane, bis (dimethylphosphino) ethane, bis (diethylphos
- tridentate phosphine compound represented by the general formula (C3) include bis (2-diphenylphosphinoethyl) phenylphosphine, bis (2-dicyclopentylphosphinoethyl) cyclopentylphosphine, bis (2- Dicyclohexylphosphinoethyl) cyclohexylphosphine, tris (diphenylphosphino-methyl) methane, tris (diphenylphosphino-ethyl) methane, tris (diphenylphosphino-methyl) ethane, tris (diphenylphosphino-ethyl) ethane, tris ( Examples thereof include diphenylphosphino-methyl) propane and tris (diphenylphosphino-ethyl) propane.
- tetradentate phosphine compound represented by the general formula (C4) include tris [2- (diphenylphosphino) ethyl] phosphine.
- phosphine compounds in terms of hot water resistance and cost, bis (diphenylphosphino) methane, bis (diphenylphosphino) ethane, bis (diphenylphosphino) propane, bis (diphenylphosphino) butane, bis (diphenylphosphine) Fino) pentane, bis (diphenylphosphino) hexane, bis (diphenylphosphino) pentane, bis (diphenylphosphino) octane, bis (diphenylphosphino) nonane, bis (diphenylphosphino) decane, bis [bis (pentafluoro Phenyl) phosphino] ethane, bis (diphenylphosphino) ethylene, bis (diphenylphosphino) acetylene, bis [(phenylpropanesulfonic acid) phosphine]
- Examples of the phosphinite compound include methoxydiphenylphosphine, ethoxydiphenylphosphine, butoxydiphenylphosphine, and compounds represented by the following structural formulas (C111) and (C112).
- Examples of the bidentate phosphinite compound include ethyl 3,5-bis [(diphenylphosphino) oxy] benzoate.
- Examples of the phosphonite compound include dimethoxyphenylphosphine, diethoxyphenylphosphine, and compounds represented by the following structural formulas (C113) to (C117).
- Examples of the bidentate phosphonite compound include 4,4 ′-(oxadi-2,1-phenylene) bis-dinaphthodioxaphosphine, tetrakis (2,4-di-t-butylphenyloxy) 4,4 ′.
- Examples of the phosphine oxide compound represented by the general formula (C1) include trimethylphosphine oxide, triethylphosphine oxide, tripropylphosphine oxide, triisopropylphosphine oxide, tri-n-butylphosphine oxide, tri-s-butylphosphine oxide, -I-butylphosphine oxide, tri-t-butylphosphine oxide, tripentylphosphine oxide, trihexylphosphine oxide, triheptylphosphine oxide, trioctylphosphine oxide, tris (ethylhexyl) phosphine oxide, trinonylphosphine oxide, tridecylphosphine Oxide, tris (hydroxymethyl) phosphine oxide, tris (2-carboxyethyl) phosphine oxide, disic Hexylethylphosphine oxide, di-t-butylneopen
- phosphine oxide compound represented by the general formula (C2) include bis (diphenylphosphino) methane monooxide, bis (diphenylphosphino) methane dioxide, and bis (diphenylphosphino) ethane monooxide.
- Preferred specific examples of the phosphine oxide compound represented by the general formula (C3) include phosphorylated form of bis (2-diphenylphosphinoethyl) phenylphosphine, phosphorous of bis (2-dicyclopentylphosphinoethyl) cyclopentylphosphine.
- Oxidized substance phosphorylated form of bis (2-dicyclohexylphosphinoethyl) cyclohexylphosphine, phosphorylated form of tris (diphenylphosphino-methyl) methane, phosphorylated form of tris (diphenylphosphino-ethyl) methane, tris (diphenyl) Phosphino-methyl) ethane phosphor, tris (diphenylphosphino-ethyl) ethane phosphor, tris (diphenylphosphino-methyl) propane phosphor, tris (diphenylphosphino-ethyl) propane phosphor Oxides etc. It is.
- phosphine oxide compound represented by the general formula (C4-1) or (C4-2) include a phosphorylated form of tris [2- (diphenylphosphino) ethyl] phosphine. .
- the phosphinate compounds include benzenephosphinic acid and its anhydride, methylphenylphosphinic acid and its anhydride, ethylphenylphosphinic acid and its anhydride, propylphenylphosphinic acid and its anhydride, isopropylphenylphosphinic acid and its anhydride, butyl Phenylphosphinic acid and its anhydride, i-butylphenylphosphinic acid and its anhydride, t-butylphenylphosphinic acid and its anhydride, pentylphenylphosphinic acid and its anhydride, hexylphenylphosphinic acid and its anhydride, heptylphenyl Phosphinic acid and its anhydride, octylphenylphosphinic acid and its anhydride, diphenylphosphinic acid and its anhydride, phenylvinylphosphinic
- Phosphonate compounds include dimethyl phosphonate, diethyl phosphonate, dipropyl phosphonate, diisopropyl phosphonate, dibutyl phosphonate, di-s-butyl phosphonate, di-t-butyl phosphonate, dipentyl phosphonate, dihexyl phosphonate, diheptyl phosphonate Dioctyl phosphonate, dioleyl phosphonate, diethyl vinyl phosphonate, diethyl allyl phosphonate, diethyl butyl phosphonate, diethyl decyl phosphonate, dimethyl allyl phosphonate, dimethyl decyl phosphonate, dimethyl methyl phosphonate, dimethyl vinyl phosphonate, vinyl phosphone Diethyl acid, diphenyl vinylphosphonate, dibutyl propylphosphonate, diethyl benzylphosphonate, diethy
- phosphate compound examples include methyl phosphate and its salt, ethyl phosphate and its salt, propyl phosphate and its salt, isopropyl phosphate and its salt, butyl phosphate and its salt, isobutyl phosphate and its salt, 2-butyl Phosphate and its salt, t-butyl phosphate and its salt, pentyl phosphate and its salt, cyclopentyl phosphate and its salt, hexyl phosphate and its salt, cyclohexyl phosphate and its salt, heptyl phosphate and its salt, phosphoric acid Cycloheptyl and its salt, octyl phosphate and its salt, cyclooctyl phosphate and its salt, nonyl phosphate and its salt, decyl phosphate and its salt, undecyl phosphate and its salt, dodecyl phosphate and its salt, phosphorus Tridecyl
- the nitrogen-containing heteroaromatic compound refers to a compound group consisting of a heteroaromatic ring in which a part of the CH bond constituting the aromatic ring is substituted with an N atom or a derivative thereof.
- the nitrogen-containing heteroaromatic ring additive (D) used in the present invention comprises a nitrogen-containing heteroaromatic ring compound having three or more nitrogen-containing heteroaromatic rings in the molecule.
- the decomposition efficiency of hydrogen peroxide, hydroxy radicals, and peroxide radicals is improved, and durability is improved with a small addition amount without impairing proton conductivity.
- the coordinating ability and inactivation ability with respect to metal impurities that promote the generation of hydroxy radicals and peroxide radicals can be remarkably enhanced by the stronger chelating effect.
- the molecular weight of the nitrogen-containing heteroaromatic additive (D) increases, the elution resistance and hot water resistance are improved, and the oxide of the nitrogen-containing heteroaromatic ring produced by reaction with hydrogen peroxide and radicals It is considered that it can more easily react with the organophosphorus additive (C) and return to the original nitrogen-containing heteroaromatic ring.
- the number of nitrogen-containing heteroaromatic rings is 2 or less, the chelating effect is not exhibited well, the inactivation ability of metal impurities is inferior, or it reacts with hydrogen peroxide, hydroxy radicals, peroxide radicals and is rendered harmless. Since the nitrogen-containing heteroaromatic ring is reduced, the detoxification efficiency of hydrogen peroxide, hydroxy radicals, and peroxide radicals may be reduced.
- the nitrogen-containing heteroaromatic ring constituting the nitrogen-containing heteroaromatic ring additive (D) is preferably a pyridine ring, pyridazine ring, pyrimidine ring, pyrazine ring, triazine ring, pyrrole ring, pyrazole ring, imidazole ring, 1 , 2,3-triazole ring, 1,2,4-triazole ring, tetrazole ring, oxazole ring, thiazole ring, thiadiazole ring, and pyridine ring, pyrazine ring, pyrimidine ring from the viewpoint of ease of synthesis and cost , A pyridazine ring, a pyrrole ring, and an imidazole ring are more preferable.
- the nitrogen-containing heteroaromatic ring constituting the nitrogen-containing heteroaromatic ring additive (D) contains two or more nitrogens in terms of metal deactivation ability, hydrogen peroxide, hydroxy radical, and peroxide radical resolution.
- the heteroaromatic ring preferably has a structure condensed to one aliphatic ring or an aromatic ring, and more preferably has a structure condensed to an aromatic ring. By condensing two or more nitrogen-containing heteroaromatic rings into one ring, the nitrogen-containing heteroaromatic ring has a more rigid structure, and the basicity and coordination ability of the N atom are improved, resulting in metal deactivation.
- nitrogen-containing heteroaromatic ring additive (D) used in the present invention a nitrogen-containing heteroaromatic ring compound represented by the following general formula (D1) or (D2) is preferably used.
- R 1 is a linear, cyclic, or branched hydrocarbon group, amino group, sulfide group, ketone group, sulfonyl group, sulfone group represented by the general formula C m H n (m and n are integers)
- Q 1 represents at least one substituent selected from the above general formulas (d1) to (d6)
- n 1 represents a positive integer whose product of n 3 to n 8 is 3 or more.
- E 1 -E 25 each independently represent C—H, N—H, N—R 3 (R 3 is an arbitrary organic group), N, and E 1 to E which are constituent atoms of the same aromatic ring 6 , at least one of E 7 to E 10 , E 11 to E 14 , E 15 to E 19 , E 20 to E 22 , E 23 to E 25 is N—H, N—R 3 or N Ar 1 to Ar 4 each represents an arbitrary aromatic group, a nitrogen-containing heteroaromatic ring and A Each of r 1 to Ar 4 may be substituted with an arbitrary substituent, * represents a monovalent or higher linking site, and W 1 to W 6 are a ketone group, a sulfone group, a direct bond, an amino group, a sulfide Represents a group, a sulfoxide group, an ether group or an arbitrary organic group, and W 1 to W 6 having a valence of 2 or more may optionally combine with R 1 to form a ring structure.
- an amino group represents a primary to tertiary amino group or a quaternary ammonium cation.
- the monovalent or higher binding site refers to having at least one site capable of binding to other structural units, and the bivalent or higher binding site refers to two sites capable of binding to other structural units. It means having the above.
- R 2 is a linear, cyclic, or branched hydrocarbon group, amino group, sulfide group, ketone group, sulfonyl group, sulfone group represented by the general formula C m H n (m and n are integers)
- E 1 to E 25 each independently represent C—H, N—H, N—R 4 (R 4 is an arbitrary organic group), or N, and are atoms constituting the same aromatic ring.
- At least one of E 1 to E 6 , E 7 to E 10 , E 11 to E 14 , E 15 to E 19 , E 20 to E 22 , E 23 to E 25 is N—H, N-R 4 or .
- Ar 1 ⁇ Ar 4 representing the N represents any aromatic group, Both-containing heteroaromatic ring and Ar 1 ⁇ Ar 4 may be substituted with any substituent.
- (D7) represents * monovalent or more binding sites in ⁇ (d12), the general formula (d13 ) To (d18) represents a divalent or higher bond site, W 7 to W 18 represent a ketone group, a sulfone group, a direct bond, an amino group, a sulfide group, a sulfoxide group, an ether group, or an arbitrary organic group.
- W 19 to W 24 represent an amino group or an arbitrary organic group.
- N—H, N—R (R is an arbitrary organic group), or N is preferably 1 to 3, and N—H, N are preferred in view of ease of synthesis and basicity. More preferably, -R or N is 1 or 2.
- the number of NH, NR, or N among atoms constituting one aromatic ring is 4 or more, the electron density of the nitrogen-containing heteroaromatic ring decreases, the stability of the ring decreases, Coordination / inactivation ability may be reduced.
- N—H, N—R (R is an arbitrary organic group) constituting the same aromatic ring, or N is not adjacent in the ring is more preferable.
- N—H, N—R, or adjacent to each other the stability of the ring may be reduced.
- those representing N and NH may be oxidized to an N oxide structure (N + —O ⁇ ) during operation of the fuel cell.
- Q 1 is (d3) to or a nitrogen-containing heteroaromatic ring compound represented by (d6) (D1)
- Q 2 is (d9) ⁇ (d12) or (d15) ⁇ nitrogen-containing heteroaromatic ring compound represented by (d18) More preferably (D2).
- These compounds have a rigid nitrogen-containing hetero-fused aromatic ring in which ⁇ electrons are more delocalized, resulting in improved basicity and coordination ability of the N atom, resulting in metal deactivation ability, hydrogen peroxide, It is considered that the hydroxy radical, peroxide radical resolution, and reactivity with the organophosphorus additive (C) are improved. Further, when n 3 to n 20 are 2 or more, these compounds are formed by condensing a plurality of nitrogen-containing heteroaromatic rings and fixing an aromatic ring containing an N atom having metal coordination ability. It is considered that a higher metal coordination ability as a kind of chelate effect is exhibited.
- the two condensed nitrogen-containing heteroaromatic rings in these compounds cannot revolve freely as described above, and there is no steric twist between the two aromatic rings, so that they resonate with the two aromatic rings. It is thought that there is an electronic interaction due to the effect. Thereby, the reducibility of the nitrogen-containing heteroaromatic ring compounds (D1) and (D2) is improved, and the oxidized form of the nitrogen-containing heteroaromatic ring compound produced by reacting with the peroxide and radical is It is considered that the organic phosphorus-based additive (C) can be more easily reduced to easily return to the original nitrogen-containing heteroaromatic compound.
- the contained heteroaromatic ring-based compound (D2) is considered to be inferior in metal coordination ability because two nitrogen-containing heteroaromatic rings can freely rotate.
- (D1) and (D2) can be appropriately selected from the viewpoints of solvent solubility, processability and elution resistance, and bleed-out resistance of the nitrogen-containing heteroaromatic compound (D). That is, by using the structure represented by (D1), packing between molecules can be suppressed, solvent solubility and workability can be improved, and by using the structure represented by (D2), the molecular weight By increasing the intermolecular packing, it is possible to achieve excellent elution resistance and bleed out resistance.
- the aromatic group represented by Ar 1 to Ar 4 is not particularly limited, but a phenyl group , Naphthyl group, anthracenyl group, triphenylenyl group, biphenyl group, terphenyl group, tetraphenyl group, fluorenyl group, triptycenyl group, phenalenyl group and other hydrocarbon-based arylene groups, thiophene group, selenophene group and other heteroarylene groups, etc. It is done. From the viewpoint of ease of production and cost, a phenyl group, a naphthyl group, and a biphenyl group are preferably used.
- the nitrogen-containing heteroaromatic ring containing E 1 to E 25 and the aromatic group represented by Ar 1 to Ar 4 are represented by the general formula C m H n (m and n are A linear, cyclic, or branched hydrocarbon group represented by an integer), a linear, cyclic, or branched alkoxy group represented by the general formula OC m H n (where m and n are integers), It may have a substituent such as a halogen atom such as fluorine, chlorine or bromine, a hydrogen atom, a carboxyl group, a carboxylic acid ester group, a sulfonic acid group, a sulfuric acid ester group, a hydroxyl group, an amino group, a cyano group or a nitro group.
- a halogen atom such as fluorine, chlorine or bromine
- a hydrogen atom such as fluorine, chlorine or bromine
- a hydrogen atom such as fluorine, chlorine or bromine
- a linear, cyclic, or branched hydrocarbon group represented by the general formula C m H n (m and n are integers), a general formula OC m H n (m and n n is an integer), a linear, cyclic, or branched alkoxy group, halogen atom, hydrogen atom, amino group, cyano group, or nitro group is preferred.
- the general formula C a linear, cyclic, or branched hydrocarbon group represented by m H n (m and n are integers), a straight chain, cyclic, represented by the general formula OC m H n (m and n are integers), Or an alkoxy group having a branched structure, a hydrogen atom, an amino group, a cyano group, or a nitro group is more preferable. Furthermore, it is more preferable that at least one of the substituents is an amino group from the viewpoint of suppressing bleed out.
- W 1 to W 18 represent a ketone group, a sulfone group, a direct bond, an amino group, a sulfide group, a sulfoxide group, an ether group, or an arbitrary organic group.
- a hydrocarbon having a direct bond, an ether group, an amino group, or a straight chain, cyclic, or branched structure represented by the general formula C m H n (m and n are integers) is more preferable. It is a group.
- W 19 to W 24 each represents an amino group or an arbitrary organic group, and more preferably an amino group or a linear, cyclic, or branched structure represented by a general formula C m H n (m and n are integers). It is a certain hydrocarbon group.
- the groups represented by R 1 and R 2 in the formulas (D1) and (D2) are linear, cyclic, or branched structures represented by the general formula C m H n (m and n are integers). It represents a certain hydrocarbon group, amino group, sulfide group, ketone group, sulfonyl group, sulfone group or ether group. From the viewpoints of elution resistance, bleed-out resistance, compound stability, and ease of synthesis, aromatic groups, amino groups, and ether groups are preferred.
- Phenyl group, naphthyl group, anthracenyl group, triphenylenyl group, biphenyl group, terphenyl Group, tetraphenyl group, fluorenyl group, triptycenyl group, phenalenyl group, amino group, and ether group are more preferable, and from the viewpoint of cost, a phenyl group, a naphthyl group, a biphenyl group, an amino group, and an ether group are more preferably used.
- R 1 , R 2 , Q 1 , and Q 2 in the formulas (D1) and (D2) are not limited to one type, but as a combination of a plurality of types in the chemical structure of the nitrogen-containing heteroaromatic compound. It may be contained. Moreover, also used preferably when a structure Q 2 in (D2) is represented by two or more valences (D1).
- nitrogen-containing heteroaromatic ring additive (D) those having a salt structure such as phenanthroline hydrochloride and bipyridine p-toluenesulfonate can be suitably used.
- Examples of the nitrogen-containing heteroaromatic additive represented by the formula (D1) include compounds represented by the following structural formulas (E1) to (E110).
- Examples of the nitrogen-containing heteroaromatic ring additive represented by the formula (D2) include compounds represented by the following structural formulas (F1) to (F21).
- any of these compounds when combined with the organophosphorus additive (C), exhibits an excellent durability improving effect.
- hydrogen peroxide, hydroxy radicals, peroxide radical resolution and the organophosphorus additive From the viewpoint of reactivity with the agent (C), (E1) to (E97), (E107) and (F1) to (F18) are preferable.
- (E1), (E4), (E12 ), (E89) to (E90), (E92) to (E93), (F12), (F14), (F16) to (F18) are more preferable, and (E1) and (E4) are preferable in terms of production cost.
- (E12), (E89) to (E90), (E92) to (E93), and (F16) are more preferable.
- the contents of the organic phosphorus additive (C) and nitrogen-containing heteroaromatic ring additive (D) used in the present invention can be appropriately selected in consideration of the balance between power generation characteristics and durability, and are limited.
- the total content of the organophosphorus additive (C) and the nitrogen-containing heteroaromatic ring additive (D) is 0.01% by weight or more, 15%, based on the whole polymer electrolyte composition. More preferably, it is less than or equal to weight percent. More preferably, they are 0.05 weight% or more and 3 weight% or less, Most preferably, it is 0.1 weight% or more and 2 weight% or less. If it is less than 0.01% by weight, the durability may be insufficient. On the other hand, if it exceeds 15% by weight, proton conductivity may be insufficient.
- the total content of the organic phosphorus additive (C) and the nitrogen-containing heteroaromatic additive (D) is 0. More preferably, it is 0.02 wt% or more and 35 wt% or less. More preferably, they are 0.1 weight% or more and 5 weight% or less, Most preferably, they are 0.5 weight% or more and 3 weight% or less. If it is less than 0.02% by weight, the durability may be insufficient, which is not preferable. On the other hand, if it exceeds 35% by weight, proton conductivity may be insufficient.
- the polymer electrolyte composition of the present invention includes at least one selected from Ce, Mn, Ti, Zr, V, Cr, Mo, W, Ru, Co, Rh, Ir, Ni, Pd, Pt, Ag, Au. It is also preferred to further contain a seed transition metal. These transition metals may be one or more selected from the group consisting of such transition metals, ions of such transition metals, salts containing such transition metal ions, and oxides of such transition metals.
- Ce, Mn, V, Mo, W, Ru, Co, Rh, Ir, Ni, Pd, Pt, Ag, and Au are used because of their high functions as radical scavengers and peroxide decomposers. More preferably, Ce, Mn, Ru, Co, Rh, Ir, Ni, Pd, Pt, Ag, Au, more preferably Ru, Co, Rh, Ir, Ni, Pd, Pt, Ag, Au Most preferably, they are Pt, Ru, Co, Rh, Ir, Ni, Pd, and Au.
- the content of the transition metal used in the present invention can be appropriately selected in consideration of the balance between power generation characteristics and durability, and is not limited, but as a transition metal equivalent (transition metal part), a polymer More preferably, the content is 0.01% by weight or more and 15% by weight or less of the entire electrolyte composition. More preferably, they are 0.05 weight% or more and 3 weight% or less, Most preferably, it is 0.1 weight% or more and 2 weight% or less.
- the content of the transition metal is 0.02 wt% or more and 35 wt% or less with respect to the entire polymer electrolyte membrane. More preferably, they are 0.1 weight% or more and 5 weight% or less, Most preferably, they are 0.5 weight% or more and 3 weight% or less.
- the content of the transition metal is defined by the content of the transition metal equivalent (only the transition metal portion) in the compound.
- the content ratio of the organophosphorus additive (C), nitrogen-containing heteroaromatic additive (D) and transition metal used in the present invention should be appropriately selected in consideration of the balance between power generation characteristics and durability.
- the phosphorus / transition metal molar ratio and the nitrogen / transition metal molar ratio in the aromatic ring be 0.01 or more and 100 or less.
- the molar ratio is more preferably 20 or less, and further preferably 10 or less.
- Examples of the salt containing a transition metal ion include a salt containing + 3-valent cerium ion, a salt containing + 4-valent cerium ion, a salt containing + 2-valent manganese ion, a salt containing + 3-valent manganese, and the like.
- Examples of the salt containing + trivalent cerium ions include cerium acetate, cerium chloride, cerium nitrate, cerium carbonate, and cerium sulfate.
- Examples of the salt containing +4 valent cerium ions include cerium sulfate and tetraammonium cerium sulfate.
- Examples of the salt containing +2 valent manganese ions include manganese acetate, manganese chloride, manganese nitrate, manganese carbonate, and manganese sulfate.
- Examples of the salt containing + trivalent manganese include manganese acetate. Of these, cerium nitrate and manganese nitrate are preferably used because they have a high effect of suppressing oxidative degradation.
- Such transition metal ions may exist alone or as a complex coordinated with an organic compound.
- organophosphorus compound (C) and the nitrogen-containing heteroaromatic compound (D) of the present invention elution of additives during the operation of the fuel cell and ion crosslinking of the polymer electrolyte composition, It is preferable from the viewpoint that gelation is suppressed (excellent in gel resistance), and further, when the organic phosphorus additive (C) is a polydentate compound having two or more phosphorus, or a nitrogen-containing heteroaromatic ring additive
- (D) has three or more imidazole rings or pyridine rings in the molecule, it is more preferable because the complex can be further improved in elution resistance and gel resistance by a strong chelating effect.
- organophosphorus compound is a phosphine compound, a phosphinite compound, or a phosphonite compound, it is most preferable because it can be a complex that is more excellent in
- transition metal oxides include cerium oxide, manganese oxide, ruthenium oxide, cobalt oxide, nickel oxide, chromium oxide, iridium oxide, and lead oxide. Of these, cerium oxide and manganese oxide are preferably used because they have a high effect of suppressing oxidative degradation.
- blending an organic phosphorus type additive (C) and a nitrogen-containing heteroaromatic ring type additive (D) with the ionic group containing polymer (A) in this invention is not specifically limited, For example, the following method is mentioned. Among these, it is more preferable to use the method (1) or (3) from the viewpoint of excellent mass productivity.
- the ionic group-containing polymer (A) used in the present invention may be either a perfluoro polymer or a hydrocarbon polymer as long as it can achieve both power generation characteristics and chemical stability.
- the perfluoro polymer means a polymer in which most or all of the hydrogen of the alkyl group and / or alkylene group in the polymer is substituted with fluorine atoms.
- Representative examples of perfluoro polymers having an ionic group include Nafion (registered trademark) (manufactured by DuPont), Flemion (registered trademark) (manufactured by Asahi Glass Co., Ltd.), and Aciplex (registered trademark) (manufactured by Asahi Kasei Co., Ltd.).
- Nafion registered trademark
- Flemion registered trademark
- Aciplex registered trademark
- the ionic group-containing polymer (A) used in the present invention is preferably a hydrocarbon polymer, and has an aromatic ring in the main chain. More preferably, it is a hydrocarbon-based polymer. Among these, polymers having sufficient mechanical strength and physical durability that are used as engineering plastics are preferable.
- the aromatic ring may include not only a hydrocarbon aromatic ring but also a hetero ring. Further, a part of the aliphatic units may constitute a polymer together with the aromatic ring unit.
- hydrocarbon polymer having an aromatic ring in the main chain examples include polysulfone, polyethersulfone, polyphenylene oxide, polyarylene ether polymer, polyphenylene sulfide, polyphenylene sulfide sulfone, polyparaphenylene, polyarylene polymer, polyarylene.
- examples include ketones, polyether ketones, polyarylene phosphine oxides, polyether phosphine oxides, polybenzoxazoles, polybenzthiazoles, polybenzimidazoles, aromatic polyamides, polyimides, polyether imides, and polyimide sulfones.
- aromatic polyether polymers are more preferable in terms of mechanical strength, physical durability, and manufacturing cost. In addition, it exhibits crystallinity due to good packing of the main chain skeleton structure and extremely strong intermolecular cohesion, has no property of dissolving in general solvents, and has excellent tensile strength and elongation, tear strength and fatigue resistance. Therefore, an aromatic polyether ketone polymer is particularly preferable.
- the aromatic polyether ketone polymer is a general term for polymers having at least an aromatic ring, an ether bond and a ketone bond in the main chain, and includes an aromatic polyether ketone, an aromatic polyether ketone ketone, an aromatic Aromatic polyether ether ketone, aromatic polyether ether ketone ketone, aromatic polyether ketone ether ketone ketone, aromatic polyether ketone sulfone, aromatic polyether ketone phosphine oxide, aromatic polyether ketone nitrile and the like.
- the ionic group of the ionic group-containing polymer (A) is preferably a negatively charged atomic group, and preferably has proton exchange ability.
- a functional group a sulfonic acid group, a sulfonimide group, a sulfuric acid group, a phosphonic acid group, a phosphoric acid group, and a carboxylic acid group are preferably used.
- the ionic group includes a case where it is a salt. Examples of cations that form such salts include arbitrary metal cations, NR 4 + (R is an arbitrary organic group), and the like. In the case of a metal cation, the valence and the like are not particularly limited and can be used.
- preferable metal cations include cations such as Li, Na, K, Rh, Mg, Ca, Sr, Ti, Al, Fe, Pt, Rh, Ru, Ir, and Pd.
- Na, K, and Li cations that are inexpensive and can be easily proton-substituted are preferably used.
- Examples of the method of introducing an ionic group include a method of polymerizing using a monomer having an ionic group and a method of introducing an ionic group by a polymer reaction.
- a monomer having an ionic group in a repeating unit may be used as a method for polymerizing using a monomer having an ionic group. Such a method is described, for example, in Journal of Membrane Science, 197, 2002, p. 231-242. This method is preferable because the ion exchange capacity of the polymer can be easily controlled.
- a method for introducing an ionic group by a polymer reaction for example, Polymer Preprints (Japan), 51, 2002, p. It is possible by the method described in 750 etc.
- introduction of a phosphate group into a hydrocarbon-based polymer having an aromatic ring in the main chain is by esterification of a polymer having a hydroxyl group.
- introduction of sulfate groups is possible, for example, by sulfate esterification of a polymer having hydroxyl groups.
- a method described in JP-A-2-16126 or JP-A-2-208322 can be used.
- a hydrocarbon polymer having an aromatic ring in the main chain is reacted with a sulfonating agent such as chlorosulfonic acid in a solvent such as chloroform, or in concentrated sulfuric acid or fuming sulfuric acid.
- a sulfonating agent such as chlorosulfonic acid in a solvent such as chloroform, or in concentrated sulfuric acid or fuming sulfuric acid.
- the sulfonating agent is not particularly limited as long as it sulfonates the polymer, and in addition to the above, sulfur trioxide and the like can be used.
- the degree of sulfonation can be controlled by the amount of sulfonating agent used, the reaction temperature and the reaction time.
- Introduction of a sulfonimide group into a hydrocarbon-based polymer having an aromatic ring in the main chain is possible by, for example, a method of reacting a sulfonic acid group and a sulfonamide group.
- the molecular weight of the ionic group-containing polymer (A) thus obtained is preferably from 10,000 to 5,000,000, more preferably from 10,000 to 500,000 in terms of polystyrene-converted weight average molecular weight. If it is less than 10,000, any of the mechanical strength, physical durability, and solvent resistance may be insufficient, such as cracking in the molded film. On the other hand, if it exceeds 5 million, the solubility becomes insufficient, the solution viscosity is high, and the processability may be poor.
- a segment (A1) containing an ionic group and a segment containing no ionic group from the viewpoint of proton conductivity and power generation characteristics under low humidification conditions ( More preferred is a block polymer having A2).
- the segment (A2) which does not contain an ionic group the said segment (A2) has an ionic group in the range which does not have a decisive bad influence on the performance as an electrolyte membrane. It is not excluded that a small amount is contained.
- a block polymer having a linker site for connecting the segments is more preferable. Due to the presence of the linker, different segments can be linked while effectively suppressing side reactions.
- the number average molecular weight of the segment (A1) containing an ionic group and the segment (A2) containing no ionic group is related to the domain size of the phase separation structure, and the proton conductivity and physical durability at low humidification From the balance, it is more preferably 50,000 or more, more preferably 10,000 or more, and most preferably 15,000 or more. Moreover, 50,000 or less is more preferable, More preferably, it is 40,000 or less, Most preferably, it is 30,000 or less.
- the segment (A1) containing an ionic group is represented by the following general formula (S1)
- the segment (A2) not containing an ionic group is represented by the following general formula (S2). More preferred are those containing a structural unit.
- Ar 1 to Ar 4 represent any divalent arylene group, and at least one of Ar 1 and Ar 2 has an ionic group as a substituent.
- Ar 3 and Ar 4 may or may not have an ionic group as a substituent,
- Ar 1 to Ar 4 may be optionally substituted with a group other than the ionic group, and
- Ar 1 to Ar 4 are structural units.
- R may be the same as or different from each other,
- R represents a ketone group or a protecting group that can be derived from the ketone group, and may be the same or different from each other. Represents the binding site.
- Ar 5 to Ar 8 each represents an arbitrary divalent arylene group, which may be optionally substituted, but does not have an ionic group.
- Ar 5 to Ar 8 are each a structural unit.
- R may represent a ketone group or a protecting group that can be derived from a ketone group, and may be the same or different from each other, and * may be the same as or different from the general formula (S2) or other structural units.
- Such protecting groups include, for example, Theodora W. Greene, “Protective Groups in Organic Synthesis”, United States, John Willy & Sons, Inc), 1981, which can be preferably used. It can be appropriately selected in consideration of the reactivity and yield of the protection reaction and deprotection reaction, the stability of the protecting group-containing state, the production cost, and the like.
- a method of protecting / deprotecting a ketone moiety with a ketal moiety and a method of protecting / deprotecting a ketone moiety with a heteroatom analog of the ketal moiety, such as a thioketal are preferably used.
- the structural unit containing a protecting group includes at least one selected from the following general formulas (U1) and (U2).
- Ar9 to Ar12 are any divalent arylene group
- R1 and R2 are at least one group selected from H and an alkyl group
- R3 is any alkylene group
- E is O or S, each of which may represent two or more groups, and the groups represented by formulas (U1) and (U2) may be optionally substituted.
- E is O in the general formulas (U1) and (U2) in terms of the odor, reactivity, stability, etc. of the compound, that is, a method for protecting / deprotecting a ketone moiety with a ketal moiety.
- E O in the general formulas (U1) and (U2) in terms of the odor, reactivity, stability, etc. of the compound
- R1 and R2 in the general formula (U1) are more preferably an alkyl group from the viewpoint of stability, more preferably an alkyl group having 1 to 6 carbon atoms, and most preferably an alkyl group having 1 to 3 carbon atoms. is there.
- R3 in the general formula (U2) is more preferably an alkylene group having 1 to 7 carbon atoms, and most preferably an alkylene group having 1 to 4 carbon atoms from the viewpoint of stability.
- R3 examples include —CH 2 CH 2 —, —CH (CH 3 ) CH 2 —, —CH (CH 3 ) CH (CH 3 ) —, —C (CH 3 ) 2 CH 2 —, —C (CH 3 ) 2 CH (CH 3 ) —, —C (CH 3 ) 2 O (CH 3 ) 2 —, —CH 2 CH 2 CH 2 —, —CH 2 C (CH 3 ) 2 CH 2 — and the like However, it is not limited to these.
- R3 in the general formula (U2) is preferably an alkylene group having 1 to 7 carbon atoms, that is, a group represented by C n1 H 2n1 (n1 is an integer of 1 to 7), Most preferred is at least one selected from —CH 2 CH 2 —, —CH (CH 3 ) CH 2 —, or —CH 2 CH 2 CH 2 — from the viewpoint of ease of synthesis.
- the deprotection reaction can be performed in the presence of water and acid under non-uniform or uniform conditions, but from the viewpoint of mechanical strength, physical durability, and solvent resistance, after being molded into a film or the like
- the acid treatment method is more preferable. Specifically, it is possible to deprotect the molded membrane by immersing it in an aqueous hydrochloric acid solution or an aqueous sulfuric acid solution, and the acid concentration and aqueous solution temperature can be appropriately selected.
- Ar 1 to Ar 8 include hydrocarbon-based arylene groups such as phenylene group, naphthylene group, biphenylene group, and fluorenediyl group, pyridinediyl, quinoxalinediyl, thiophenediyl, and the like
- a heterophenylene group preferably a phenylene group, and most preferably a p-phenylene group.
- segment (A1) containing an ionic group a structural unit that is chemically stable, has an increased acidity due to an electron withdrawing effect, and is introduced with a high density of ionic groups is more preferable.
- the segment (A2) not containing an ionic group is more preferably a structural unit that is chemically stable and exhibits crystallinity due to strong intermolecular cohesion.
- the structural unit represented by the general formula (S1) include a structural unit represented by the following general formula (P2) in terms of raw material availability.
- a structural unit represented by the following formula (P3) is more preferable, and a structural unit represented by the following formula (P4) is most preferable.
- M 1 to M 4 represent hydrogen, a metal cation, and an ammonium cation NR 4 + (R is an arbitrary organic group), and M 1 to M 4 are two or more types.
- R1 to r4 each independently represents an integer of 0 to 2
- r1 + r2 represents an integer of 1 to 8
- r1 to r4 may be different for each structural unit.
- * represents a binding site with the formula (P2) (P3) (P4) or other structural unit.
- the molar composition ratio (A1 / A2) of the segment (A1) containing an ionic group and the segment (A2) containing no ionic group is 0.2 or more. Is more preferably 0.33 or more, and most preferably 0.5 or more. Moreover, 5 or less is more preferable, 3 or less is more preferable, and 2 or less is the most preferable.
- the molar composition ratio A1 / A2 is less than 0.2 or exceeds 5, the proton conductivity under low humidification conditions tends to be insufficient, or the hot water resistance and physical durability tend to be insufficient. .
- the ion exchange capacity of the segment (A1) containing an ionic group is preferably 2.5 meq / g or more, more preferably 3 meq / g or more, and still more preferably from the viewpoint of proton conductivity under low humidification conditions. It is 3.5 meq / g or more. Moreover, from the point of hot water resistance and physical durability, 6.5 meq / g or less is more preferable, 5 meq / g or less is more preferable, and 4.5 meq / g or less is further more preferable.
- the ion exchange capacity of the segment (A2) not containing an ionic group is preferably 1 meq / g or less, more preferably 0.5 meq / g, from the viewpoint of hot water resistance, mechanical strength, dimensional stability, and physical durability. More preferably, it is 0.1 meq / g or less.
- the block polymer has a sulfonic acid group
- its ion exchange capacity is preferably 0.1 to 5 meq / g, more preferably 1.5 meq / g or more, and still more preferably, from the viewpoint of the balance between proton conductivity and water resistance. Is 2 meq / g or more. Moreover, 3.5 meq / g or less is more preferable, More preferably, it is 3 meq / g or less.
- the ion exchange capacity is less than 0.1 meq / g, proton conductivity may be insufficient, and when it is greater than 5 meq / g, water resistance may be insufficient.
- the ion exchange capacity is a value determined by a neutralization titration method.
- the neutralization titration method is performed as follows. In addition, a measurement shall be performed 3 times or more and the average value shall be taken.
- (2) Add 50 mL of 5 wt% sodium sulfate aqueous solution to the electrolyte and leave it for 12 hours for ion exchange.
- the generated sulfuric acid is titrated using 0.01 mol / L sodium hydroxide aqueous solution.
- a commercially available phenolphthalein solution for titration, 0.1 w / v%, is added as an indicator, and the point at which light reddish purple is obtained is the end point.
- the ion exchange capacity is determined by
- Ion exchange capacity (meq / g) [Concentration of sodium hydroxide aqueous solution (mmol / ml) ⁇ Drip amount (ml)] / Dry weight of sample (g)
- the method for synthesizing the oligomer constituting the segment (A1) containing an ionic group and the segment (A2) containing no ionic group is not particularly limited as long as it has a substantially sufficient molecular weight.
- the aromatic active dihalide compound used for the synthesis of the oligomer constituting the segment (A1) containing an ionic group using a compound in which an ionic group is introduced into the aromatic active dihalide compound as a monomer is chemically stable,
- the production cost and the amount of ionic groups are preferable from the viewpoint that precise control is possible.
- Specific examples of the monomer having a sulfonic acid group as an ionic group include 3,3′-disulfonate-4,4′-dichlorodiphenylsulfone and 3,3′-disulfonate-4,4′-difluorodiphenyl.
- 3,3′-disulfonate-4,4′-dichlorodiphenyl ketone and 3,3′-disulfonate-4,4′-difluorodiphenyl ketone are more preferable from the viewpoint of chemical stability and physical durability. From the viewpoint of polymerization activity, 3,3′-disulfonate-4,4′-difluorodiphenyl ketone is most preferable.
- aromatic active dihalide compound which does not have an ionic group used for the synthesis
- combination of the oligomer which comprises the segment (A2) which does not contain the oligomer which comprises the segment (A1) containing an ionic group, and an ionic group 4 , 4'-dichlorodiphenylsulfone, 4,4'-difluorodiphenylsulfone, 4,4'-dichlorodiphenylketone, 4,4'-difluorodiphenylketone, 4,4'-dichlorodiphenylphenylphosphine oxide, 4,4 ' -Difluorodiphenylphenylphosphine oxide, 2,6-dichlorobenzonitrile, 2,6-difluorobenzonitrile and the like.
- 4,4′-dichlorodiphenyl ketone and 4,4′-difluorodiphenyl ketone are more preferable in terms of imparting crystallinity, mechanical strength, physical durability and hot water resistance, and 4,4′-difluoro in terms of polymerization activity.
- Diphenyl ketone is most preferred.
- aromatic active dihalide compounds can be used alone, but a plurality of aromatic active dihalide compounds can also be used in combination.
- halogenated aromatic hydroxy compounds as monomers having no ionic group used for the synthesis of the oligomer constituting the segment (A1) containing an ionic group and the oligomer constituting the segment (A2) containing no ionic group can be mentioned.
- the said segment can synthesize
- the halogenated aromatic hydroxy compound is not particularly limited, but 4-hydroxy-4′-chlorobenzophenone, 4-hydroxy-4′-fluorobenzophenone, 4-hydroxy-4′-chlorodiphenylsulfone, 4-hydroxy -4′-fluorodiphenylsulfone, 4- (4′-hydroxybiphenyl) (4-chlorophenyl) sulfone, 4- (4′-hydroxybiphenyl) (4-fluorophenyl) sulfone, 4- (4′-hydroxybiphenyl) Examples include (4-chlorophenyl) ketone, 4- (4′-hydroxybiphenyl) (4-fluorophenyl) ketone, and the like.
- these halogenated aromatic hydroxy compounds may be reacted together to synthesize an aromatic polyether compound.
- the method for synthesizing the block polymer is not particularly limited as long as a substantially sufficient molecular weight can be obtained.
- the oligomer that constitutes the segment containing the ionic group and the ionic group are not contained. It can be synthesized by utilizing the aromatic nucleophilic substitution reaction of the oligomer constituting the segment.
- the monomer mixture or segment mixture is reacted in the presence of a basic compound.
- the polymerization can be carried out in a temperature range of 0 to 350 ° C., but a temperature of 50 to 250 ° C. is preferable. When the temperature is lower than 0 ° C., the reaction does not proceed sufficiently, and when the temperature is higher than 350 ° C., the polymer tends to be decomposed.
- the polymerization reaction can be carried out in the absence of a solvent, but is preferably carried out in a solvent.
- Solvents that can be used include N, N-dimethylacetamide, N, N-dimethylformamide, N-methyl-2-pyrrolidone, dimethyl sulfoxide, sulfolane, 1,3-dimethyl-2-imidazolidinone, hexamethylphosphontriamide, etc. However, it is not limited to these, and any solvent that can be used as a stable solvent in the aromatic nucleophilic substitution reaction may be used. These organic solvents may be used alone or as a mixture of two or more.
- Examples of the basic compound used for the aromatic nucleophilic substitution reaction include sodium hydroxide, potassium hydroxide, sodium carbonate, potassium carbonate, sodium hydrogen carbonate, potassium hydrogen carbonate, etc., but aromatic diols are active phenoxide structures. As long as it can be converted to, it is not limited to these and can be used. In order to increase the nucleophilicity of phenoxide, it is also preferable to add a crown ether such as 18-crown-6. These crown ethers may be preferably used because they may be coordinated to a sodium ion or potassium ion of a sulfonic acid group to improve the solubility in an organic solvent.
- water may be generated as a by-product.
- water can be removed from the system as an azeotrope by coexisting toluene or the like in the reaction system.
- a water-absorbing agent such as molecular sieve can also be used.
- Azeotropic agents used to remove reaction water or water introduced during the reaction generally do not substantially interfere with polymerization, co-distill with water and boil between about 25 ° C. and about 250 ° C. Any inert compound.
- Common azeotropic agents include benzene, toluene, xylene, chlorobenzene, methylene chloride, dichlorobenzene, trichlorobenzene, cyclohexane and the like. Of course, it is beneficial to select an azeotropic agent whose boiling point is lower than that of the dipolar solvent used.
- An azeotropic agent is commonly used, but it is not always necessary when high reaction temperatures, such as temperatures above 200 ° C., are used, especially when the reaction mixture is continuously sparged with inert gas. In general, it is desirable to carry out the reaction in an inert atmosphere and in the absence of oxygen.
- the aromatic nucleophilic substitution reaction is performed in a solvent, it is preferable to charge the monomer so that the polymer concentration obtained is 5 to 50% by weight.
- the polymer concentration obtained is less than 5% by weight, the degree of polymerization tends to be difficult to increase.
- it exceeds 50% by weight the viscosity of the reaction system becomes too high, and the post-treatment of the reaction product tends to be difficult.
- the desired polymer is obtained by removing the solvent from the reaction solution by evaporation and washing the residue as necessary.
- the reaction solution by adding the reaction solution to a solvent having low polymer solubility and high by-product inorganic salt solubility, the inorganic salt is removed, the polymer is precipitated as a solid, and the polymer is obtained by filtering the precipitate. You can also.
- the recovered polymer is optionally washed with water, alcohol or other solvent and dried.
- halide or phenoxide end groups can optionally be reacted by introducing a phenoxide or halide end-capping agent that forms a stable end group.
- the organophosphorus additive (C) and the nitrogen-containing heteroaromatic ring additive (D) are polar (hydrophilic). And (hydrophobic and hydrophobic) are selected as appropriate so that they are concentrated on the hydrophilic domain formed by the segment (A1) containing an ionic group and the hydrophobic domain formed by the segment (A2) containing no ionic group. It is possible to make it. Hydroxy radicals and hydrogen peroxide are usually highly hydrophilic and are considered to exist in the hydrophilic domain formed by the segment (A) containing an ionic group and cut the segment.
- the hydrophilic additive is effective for stabilizing the segment (A1) containing an ionic group.
- the hydrophobic domain formed by the segment (A2) that does not contain an ionic group is a component responsible for mechanical strength. Therefore, by placing a hydrophobic additive, there is an effect of improving physical durability. it is conceivable that. It is also suitable to use a hydrophilic additive and a hydrophobic additive in combination as necessary.
- a phase separation structure is observed when TEM observation is performed at a magnification of 50,000 times, and an average interlayer distance or an average interparticle distance measured by image processing is 5 nm or more. , 500 nm or less is preferable. Among them, the average interlaminar distance or the average interparticle distance is more preferably 10 nm or more and 50 nm or less, and most preferably 15 nm or more and 30 nm or less.
- the phase separation structure is not observed by a transmission electron microscope, or when the average interlayer distance or the average interparticle distance is less than 5 nm, the continuity of the ion channel may be insufficient and the conductivity may be insufficient. Further, when the interlayer distance exceeds 500 nm, the mechanical strength and dimensional stability may be poor.
- the block polymer used as the ionic group-containing polymer (A) preferably has crystallinity while having a phase separation structure. That is, it is preferable that crystallinity is recognized by differential scanning calorimetry (DSC) or wide-angle X-ray diffraction. Specifically, the crystallization calorific value measured by differential scanning calorimetry is 0.1 J / g or more, or The crystallinity measured by wide angle X-ray diffraction is preferably 0.5% or more. Note that “having crystallinity” means that the polymer can be crystallized when the temperature is raised, has a crystallizable property, or has already been crystallized.
- An amorphous polymer means a polymer that is not a crystalline polymer and that does not substantially proceed with crystallization. Therefore, even if it is a crystalline polymer, if the crystallization is not sufficiently advanced, the polymer may be in an amorphous state.
- the polymer electrolyte composition of the present invention is particularly suitably used as a molded polymer electrolyte.
- the polymer electrolyte molded body means a molded body containing the polymer electrolyte composition of the present invention.
- the polymer electrolyte molded body of the present invention includes membranes (including films and films), plates, fibers, hollow fibers, particles, lumps, micropores, coatings, and foams. It can take various forms depending on the intended use.
- the polymer can be applied to a wide range of applications because it can improve the design freedom of the polymer and improve various properties such as mechanical properties and solvent resistance. It is particularly suitable when the polymer electrolyte molded body is a membrane.
- the method for forming the polymer electrolyte composition of the present invention into a polymer electrolyte membrane is not particularly limited, but a method of forming a film from a solution state or a method of forming a film from a molten state is possible.
- the former for example, there is a method of forming a film by dissolving the polymer electrolyte material in a solvent such as N-methyl-2-pyrrolidone, casting the solution on a glass plate or the like, and removing the solvent. It can be illustrated.
- the solvent used for film formation is not particularly limited as long as the polymer electrolyte composition can be dissolved and then removed.
- N, N-dimethylacetamide, N, N-dimethylformamide, N-methyl-2- Aprotic polar solvents such as pyrrolidone, dimethyl sulfoxide, sulfolane, 1,3-dimethyl-2-imidazolidinone, hexamethylphosphontriamide, ester solvents such as ⁇ -butyrolactone, butyl acetate, ethylene carbonate, propylene carbonate, etc.
- Carbonate solvents ethylene glycol monomethyl ether, ethylene glycol monoethyl ether, propylene glycol monomethyl ether, alkylene glycol monoalkyl ethers such as propylene glycol monoethyl ether, or isopropanol.
- Alcohol solvents, water and mixtures thereof is preferably used, is preferred for high highest solubility aprotic polar solvent.
- a crown ether such as 18-crown-6.
- the filter medium used here is not particularly limited, but a glass filter or a metallic filter is suitable.
- the pore size of the minimum filter through which the polymer solution passes is preferably 1 ⁇ m or less.
- the obtained polymer electrolyte membrane is preferably heat-treated in the state of a metal salt at least part of the ionic group.
- the polymer electrolyte material to be used is polymerized in the form of a metal salt at the time of polymerization, it is preferable to perform film formation and heat treatment as it is.
- the metal of the metal salt may be any metal that can form a salt with an ionic group, but from the viewpoint of cost and environmental load, Li, Na, K, Rb, Cs, Mg, Ca, Sr, Ba, Ti, V , Mn, Fe, Co, Ni, Cu, Zn, Zr, Mo, W, and the like are preferable.
- the temperature of this heat treatment is preferably 80 to 350 ° C., more preferably 100 to 200 ° C., and particularly preferably 120 to 150 ° C.
- the heat treatment time is preferably 10 seconds to 12 hours, more preferably 30 seconds to 6 hours, and particularly preferably 1 minute to 1 hour. If the heat treatment temperature is too low, mechanical strength and physical durability may be insufficient. On the other hand, if it is too high, chemical decomposition of the film material may proceed. If the heat treatment time is less than 10 seconds, the heat treatment effect may be insufficient. On the other hand, when it exceeds 12 hours, the film material tends to deteriorate.
- the polymer electrolyte membrane obtained by the heat treatment can be proton-substituted by being immersed in an acidic aqueous solution as necessary.
- the polymer electrolyte membrane can achieve both a good balance between proton conductivity and physical durability.
- the film thickness of the polymer electrolyte membrane is preferably 1 to 2000 ⁇ m. In order to obtain the mechanical strength and physical durability of the membrane that can withstand practical use, it is more preferably thicker than 1 ⁇ m, and in order to reduce membrane resistance, that is, improve power generation performance, it is preferably thinner than 2000 ⁇ m. A more preferable range of the film thickness is 3 to 50 ⁇ m, and a particularly preferable range is 10 to 30 ⁇ m.
- the film thickness can be controlled by the solution concentration or the coating thickness on the substrate.
- additives such as a crystallization nucleating agent, a plasticizer, a stabilizer, an antioxidant, or a mold release agent used for ordinary polymer compounds are within the scope not departing from the object of the present invention. Can be further added.
- various polymers, elastomers, fillers, fine particles, various additives, etc. are included for the purpose of improving mechanical strength, thermal stability, processability, etc. within the range that does not adversely affect the above-mentioned various characteristics. Also good. Moreover, you may reinforce with a microporous film, a nonwoven fabric, a mesh, etc.
- the polymer electrolyte molded body of the present invention can be applied to various uses. For example, extracorporeal circulation column, medical use such as artificial skin, filtration use, ion exchange resin use such as chlorine-resistant reverse osmosis membrane, various structural materials use, electrochemical use, humidification membrane, anti-fogging membrane, antistatic membrane, Applicable to solar cell membranes and gas barrier materials. It is also suitable as an artificial muscle and actuator material.
- the polymer electrolyte molded body of the present invention can be preferably used for various electrochemical applications. Examples of the electrochemical application include a fuel cell, a redox flow battery, a water electrolysis device, a chloroalkali electrolysis device, and the like, and among them, it can be particularly preferably used for a fuel cell.
- a polymer electrolyte membrane, an electrode catalyst layer, and the like are preferable, and among them, the polymer electrolyte membrane is preferably used.
- the polymer electrolyte composition of the present invention has high chemical stability, and can be particularly suitably used for an electrode catalyst layer binder in which an electrochemical reaction takes place nearby.
- the polymer electrolyte fuel cell has a structure in which a catalyst layer, an electrode substrate, and a separator are sequentially laminated on both sides of a polymer electrolyte membrane.
- the catalyst layer laminated on both sides of the electrolyte membrane (that is, the catalyst layer / electrolyte membrane / catalyst layer configuration) is called an electrolyte membrane with a catalyst layer (CCM), and is formed on both sides of the electrolyte membrane.
- a catalyst layer and a gas diffusion base material laminated in sequence is an electrode-electrolyte membrane assembly (MEA). ).
- a coating method in which a catalyst layer paste composition for forming a catalyst layer is applied and dried on the electrolyte membrane surface and a catalyst layer alone are produced on a substrate, and the catalyst layer is transferred.
- a method (transfer method) in which the catalyst layer is laminated on the electrolyte membrane is generally performed.
- the MEA When the MEA is produced by pressing, a known method (for example, chemical plating method described in Electrochemistry, 1985, 53, p. 269, edited by Electrochemical Society (J. Electrochem. Soc.), Electrochemical Science and Technology (Electrochemical Science and Technology), 1988, 135, 9, p. 2209, etc. can be applied.
- the temperature and pressure during pressing may be appropriately selected depending on the thickness of the electrolyte membrane, the moisture content, the catalyst layer, and the electrode substrate. Further, in the present invention, it is possible to form a composite by pressing even when the electrolyte membrane is in a dry state or in a state of absorbing water.
- Specific pressing methods include a roll press that defines pressure and clearance, and a flat plate press that defines pressure.
- Ion exchange capacity It was measured by the neutralization titration method described in the following (i) to (iv). The measurement was performed 3 times and the average value was taken.
- Moisture on the membrane surface of the electrolyte membrane that had been proton-substituted and thoroughly washed with pure water was wiped off, and then vacuum-dried at 100 ° C. for 12 hours or more to determine the dry weight.
- Ii 50 mL of a 5 wt% aqueous sodium sulfate solution was added to the electrolyte, and the mixture was allowed to stand for 12 hours for ion exchange.
- Ion exchange capacity [Concentration of sodium hydroxide aqueous solution (mmol / mL) ⁇ Drip amount (mL)] / Dry weight of sample (g)] (2) Proton conductivity After immersing the membrane-like sample in pure water at 25 ° C for 24 hours, it is kept in a constant temperature and humidity chamber at 80 ° C and a relative humidity of 25 to 95% for 30 minutes at each step. Proton conductivity was measured by the AC impedance method.
- a Solartron electrochemical measurement system (Solartron 1287, Electrochemical Interface and Solartron 1255B Frequency Response Analyzer) was used to measure proton impedance by a two-terminal method.
- the AC amplitude was 50 mV.
- a sample having a width of 10 mm and a length of 50 mm was used.
- the measurement jig was made of phenol resin, and the measurement part was opened.
- As an electrode a platinum plate (thickness: 100 ⁇ m, 2 sheets) was used. The electrodes were arranged at a distance of 10 mm between the front and back sides of the sample film so as to be parallel to each other and perpendicular to the longitudinal direction of the sample film.
- N-Methyl is used as an integrated device of UV detector and differential refractometer, using HLC-8022GPC manufactured by Tosoh Corporation, and two TSK gel SuperHM-Hs manufactured by Tosoh Corporation (inner diameter 6.0 mm, length 15 cm) as GPC columns. Measured with a -2-pyrrolidone solvent (N-methyl-2-pyrrolidone solvent containing 10 mmol / L of lithium bromide) at a sample concentration of 0.1% by weight, a flow rate of 0.2 mL / min, and a temperature of 40 ° C. The number average molecular weight and the weight average molecular weight were determined by polystyrene conversion.
- the electrolyte membrane was cut out in a size of 5 cm ⁇ 5 cm, dried at 110 ° C. under reduced pressure for 2 hours, accurately weighed, and allowed to stand at 550 ° C. for 2 days. To obtain a liquid in which the additive was completely extracted. This solution was measured by ICP emission analysis, and the amount of phosphorus, nitrogen, and various metal elements was measured to quantify the additives.
- Hot water resistance of the additive was evaluated by measuring the residual rate after immersion in hot water at 95 ° C.
- the electrolyte membrane was cut into two strips having a length of about 5 cm and a width of about 10 cm, and the additive was eluted by immersing it in hot water at 95 ° C. for 8 hours.
- the electrolyte membrane before and after immersion in hot water was cut out in a size of 5 cm ⁇ 5 cm, the ICP emission analysis was performed to measure the additive content, and the hot water resistance was evaluated as the additive residual rate.
- This membrane electrode assembly was set in a JARI standard cell “Ex-1” (electrode area 25 cm 2 ) manufactured by Eiwa Co., Ltd., and kept at 80 ° C. while maintaining low humidity in hydrogen (70 mL / min, back pressure 0.1 MPaG). ) And air (174 mL / min, back pressure 0.05 MPaG) were introduced into the cell, and an accelerated deterioration test was performed in an open circuit. After operating the fuel cell under these conditions for 200 hours, the membrane-electrode assembly was taken out, put into a mixed solution of ethanol / water, and further subjected to ultrasonic treatment to remove the catalyst layer. Then, the molecular weight of the remaining polymer electrolyte membrane was measured and evaluated as a molecular weight retention rate.
- a membrane electrode assembly was prepared by the same method as described above, and set in an evaluation cell. Subsequently, a deterioration acceleration test in an open circuit was performed under the same conditions as described above. The time until the open circuit voltage decreased to 0.7 V or less was evaluated as the open circuit holding time.
- DHBP 4,4'-dihydroxybenzophenone
- ethylene glycol ethylene glycol
- 96.9 g trimethyl orthoformate
- p-toluenesulfonic acid monohydrate Charge 0.50 g and dissolve. Thereafter, the mixture was stirred at 78-82 ° C. for 2 hours. Further, the internal temperature was gradually raised to 120 ° C. and heated until the distillation of methyl formate, methanol and trimethyl orthoformate completely stopped.
- m represents a positive integer.
- a 1000 mL three-necked flask equipped with a stirrer, a nitrogen inlet tube, and a Dean-Stark trap 16.59 g of potassium carbonate (Aldrich reagent, 120 mmol), 25.8 g (100 mmol) of K-DHBP, and 4,4′-difluorobenzophenone 20. 3 g (Aldrich reagent, 93 mmol) was added, purged with nitrogen, dehydrated at 160 ° C.
- NMP N-methylpyrrolidone
- M in the OM group represents Na or K, and the following notation follows this). It was.
- the number average molecular weight was 10,000.
- M represents Na or K.
- M represents Na or K.
- a 1000 mL three-necked flask equipped with a stirrer, a nitrogen inlet tube, and a Dean-Stark trap 27.6 g of potassium carbonate (Aldrich reagent, 200 mmol), 12.9 g (50 mmol) of K-DHBP, and 9.3 g of 4,4′-biphenol were added.
- the block copolymer b1 contains 50 mol% of the structural unit represented by the general formula (S1) as a segment (A1) containing an ionic group and the general formula (S2) as a segment (A2) containing no ionic group. It contained 100 mol% of the structural unit represented by S2).
- the block copolymer b1 itself is a polymer electrolyte membrane
- the ion exchange capacity obtained from neutralization titration is 2.2 meq / g
- Synthesis Example 2 Synthesis of block copolymer b2 (Synthesis of polyethersulfone (PES) block copolymer precursor b2 ′ composed of a segment represented by the following formula (G6) and a segment represented by the following formula (G7)) 1.85 g of anhydrous nickel chloride and 17 mL of dimethyl sulfoxide were mixed and adjusted to 70 ° C. To this, 2.46 g of 2,2′-bipyridyl was added and stirred at the same temperature for 10 minutes to prepare a nickel-containing solution.
- PES polyethersulfone
- the reaction mixture was added to 70 mL of methanol, and then 70 mL of 6 mol / L hydrochloric acid was added and stirred for 1 hour.
- the precipitated solid was separated by filtration and dried to obtain 1.89 g of a block copolymer precursor b2 ′ (polyarylene precursor) containing a gray-white segment represented by the following formula (G6) and the following formula (G7). Obtained at 94%.
- the weight average molecular weight was 220,000.
- the separated solid was dried to obtain a block copolymer b2 composed of a gray-white segment represented by the formula (G7) and a segment represented by the following formula (G8).
- the resulting polyarylene had a weight average molecular weight of 210,000.
- the ion exchange capacity determined from neutralization titration was 2.03 meq / g.
- the resulting reaction solution was allowed to cool and then diluted by adding 100 mL of toluene.
- the precipitate of the inorganic compound produced as a by-product was removed by filtration, and the filtrate was put into 2 L of methanol.
- the precipitated product was separated by filtration, collected, dried, and then dissolved in 250 mL of tetrahydrofuran. This was reprecipitated in 2 L of methanol to obtain 103 g of the target oligomer a3.
- the number average molecular weight of the oligomer a3 was 7,200.
- the reaction system was heated with stirring (finally heated to 82 ° C.) and reacted for 3 hours. An increase in viscosity in the system was observed during the reaction.
- the polymerization reaction solution was diluted with 185 mL of DMAc, stirred for 30 minutes, and filtered using Celite as a filter aid. Add 36.1 g (301 mmol) of lithium bromide to the filtrate in three 1/3 portions at 1 hour intervals with a 1 L three-neck equipped with a stirrer, and react at 120 ° C. for 5 hours under a nitrogen atmosphere. I let you. After the reaction, the mixture was cooled to room temperature, poured into 4 L of acetone and solidified.
- the coagulum was collected by filtration, air-dried, pulverized with a mixer, and washed with 1500 mL of 1N sulfuric acid while stirring. After filtration, the product was washed with ion-exchanged water until the pH of the washing solution reached 5 or higher, and then dried at 80 ° C. overnight to obtain 40.0 g of the target block copolymer b3.
- the weight average molecular weight of this block copolymer was 190,000.
- the ion exchange capacity determined from neutralization titration was 2.22 meq / g.
- Example 1 (Synthesis of AD-1) 81 g of 8-amino-7-quinolinecarbaldehyde was reacted in 36 g of 1,3-diacetylbenzene (manufactured by Tokyo Chemical Industry Co., Ltd.), 80 g of 85% potassium hydroxide and 1440 mL of ethanol for 10 hours under reflux, followed by liquid separation extraction. 30 g of the product was reacted with 150 mL of phenyllithium (0.94M cyclohexane / diethyl ether solution) in 550 mL of toluene for 2.5 hours under ice-cooling and purified by recrystallization. The obtained product was reacted with 89 g of nitrobenzene at 110 ° C. for 3 hours and purified by recrystallization to obtain 16.2 g of compound AD-1 represented by the following structural formula.
- Example 2 Synthesis of AD-2) 43 g of 2,7-dihydroxynaphthalene (manufactured by Tokyo Chemical Industry Co., Ltd.) was dissolved in 544 mL of dichloromethane and 109 mL of pyridine, and 180 g of trifluoromethanesulfonic anhydride (manufactured by Tokyo Chemical Industry Co., Ltd.) was added dropwise at 0 ° C. The mixture was reacted at 5 ° C. for 2 hours and then at room temperature for 1 day, and then treated in a conventional manner to obtain 114 g of 2,7-bis (trifluoromethanesulfonyloxy) naphthalene.
- Example 3 Synthesis of AD-3 2 g of AD-1 and 30 mL of sulfuric acid were added and dissolved by stirring at room temperature. The solution temperature was ice-cooled to 1 ° C., and a solution prepared by dissolving 0.793 g of potassium nitrate in 30 mL of sulfuric acid was added. After reacting at 140 ° C. for 8 hours, the reaction solution was added dropwise to 2 L of a saturated sodium bicarbonate solution. The solid was filtered, washed with pure water and dried, and 2.0 g of the resulting compound was dissolved in 50 mL of NMP.
- Example 4 An electrolyte membrane was obtained in the same manner as in Example 1, except that 4 g of 1,2-bis (diphenylphosphino) ethane and 3 g of AD-1 were changed. Since the obtained film was insoluble in NMP and the molecular weight retention rate was not measurable, the open circuit retention time was measured as a durability test, but the evaluation was not completed within 5000 hours. The chemical durability of the electrolyte membrane was evaluated. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 5 An electrolyte membrane was obtained in the same manner as in Example 1, except that 1,2-bis (diphenylphosphino) ethane was changed to 2 mg and AD-1 was changed to 2 mg. Since the obtained film was insoluble in NMP and the molecular weight retention rate was not measurable, the open circuit retention time was measured as a durability test, but the evaluation was not completed within 5000 hours. The chemical durability of the electrolyte membrane was evaluated. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 6 An electrolyte membrane was obtained in the same manner as in Example 1 except that AD-1 was changed to 1,3,5-tri (4-pyridyl) benzene. Since the obtained film was insoluble in NMP and the molecular weight retention rate was not measurable, the open circuit retention time was measured as a durability test, but the evaluation was not completed within 5000 hours. The chemical durability of the electrolyte membrane was evaluated. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 7 The electrolyte membrane was prepared in the same manner as in Example 1 except that AD-1 was changed to 4 ′, 4 ′′ ′′-(1,4-phenylene) bis (2,2 ′: 6 ′, 2 ′′ -terpyridine). Since the obtained film was insoluble in NMP and the molecular weight retention rate was not measurable, the open circuit retention time was measured as a durability test, but the evaluation was not completed within 5000 hours. In addition, the chemical durability of the electrolyte membrane was evaluated as a voltage holding ratio, and the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity were measured. It is shown in 1.
- Example 8 An electrolyte membrane was obtained in the same manner as in Example 1 except that AD-1 was changed to AD-4 represented by the following structural formula. Since the obtained film was insoluble in NMP and the molecular weight retention rate was not measurable, the open circuit retention time was measured as a durability test, but the evaluation was not completed within 5000 hours. The chemical durability of the electrolyte membrane was evaluated. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 9 An electrolyte membrane was obtained in the same manner as in Example 1 except that AD-1 was changed to AD-5 represented by the following structural formula. Since the obtained film was insoluble in NMP and the molecular weight retention rate was not measurable, the open circuit retention time was measured as a durability test, but the evaluation was not completed within 5000 hours. The chemical durability of the electrolyte membrane was evaluated. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 10 An electrolyte membrane was obtained in the same manner as in Example 1 except that AD-1 was changed to AD-6 represented by the following structural formula. Since the obtained film was insoluble in NMP and the molecular weight retention rate was not measurable, the open circuit retention time was measured as a durability test, but the evaluation was not completed within 5000 hours. The chemical durability of the electrolyte membrane was evaluated. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 11 An electrolyte membrane was obtained in the same manner as in Example 1 except that AD-1 was changed to AD-7 represented by the following structural formula. Since the obtained film was insoluble in NMP and the molecular weight retention rate was not measurable, the open circuit retention time was measured as a durability test, but the evaluation was not completed within 5000 hours. The chemical durability of the electrolyte membrane was evaluated. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 12 (Synthesis of AD-8) 926 mg of cesium carbonate, 468 mg of 1,10-phenanthroline-5-amine, and 744 mg of 1,2-diiodoethane were dissolved in 8.5 mL of N, N-dimethylformamide and reacted at 80 ° C. for 12 hours. The solution after the reaction was reprecipitated in 100 mL of water and then 300 mL of IPA, and the filtrate was dried to obtain 1.01 g of AD-8 represented by the following structural formula. The number average molecular weight was 2000.
- Example 13 (Synthesis of AD-9) 1,7-dichloro-1,10-phenanthroline 1 g 4,7-dihydroxy-1,10-phenanthroline 0.85 g was dissolved in 50 mL of NMP, dehydrated in 25 mL of toluene at 160 ° C., heated to toluene Removal and polymerization were carried out at 180 ° C. for 1 hour. Purification was carried out by reprecipitation with a large amount of methanol to obtain 1.7 g of AD-9 represented by the following structural formula. The number average molecular weight was 2100.
- Example 14 An electrolyte membrane was obtained in the same manner as in Example 1 except that AD-1 was changed to AD-10 represented by the following structural formula (number average molecular weight 2500). Since the obtained film was insoluble in NMP and the molecular weight retention rate was not measurable, the open circuit retention time was measured as a durability test, but the evaluation was not completed within 5000 hours. The chemical durability of the electrolyte membrane was evaluated. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 15 An electrolyte membrane was obtained in the same manner as in Example 1 except that AD-1 was changed to AD-11 (number average molecular weight 3200) represented by the following structural formula. Since the obtained film was insoluble in NMP and the molecular weight retention rate was not measurable, the open circuit retention time was measured as a durability test, but the evaluation was not completed within 5000 hours. The chemical durability of the electrolyte membrane was evaluated. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 16 An electrolyte membrane was obtained in the same manner as in Example 1 except that 1,2-bis (diphenylphosphino) ethane was changed to triphenylphosphine. Since the obtained film was insoluble in NMP and the molecular weight retention rate was not measurable, the open circuit retention time was measured as a durability test, but the evaluation was not completed within 5000 hours. The chemical durability of the electrolyte membrane was evaluated. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 17 Example 1 except that 1,2-bis (diphenylphosphino) ethane was changed to 2,2′-bis (diphenylphosphino) -1,1′-binaphthyl and AD-1 was changed to AD-5.
- an electrolyte membrane was obtained. Since the obtained film was insoluble in NMP and the molecular weight retention rate was not measurable, the open circuit retention time was measured as a durability test, but the evaluation was not completed within 5000 hours. The chemical durability of the electrolyte membrane was evaluated. In addition, the ion exchange capacity, proton-resistant water and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 18 An electrolyte membrane was obtained in the same manner as in Example 1 except that 2-bis (diphenylphosphino) ethane was changed to the following general formula AD-12 and AD-1 was changed to AD-3. Since the obtained film was insoluble in NMP and the molecular weight retention rate was not measurable, the open circuit retention time was measured as a durability test, but the evaluation was not completed within 5000 hours. The chemical durability of the electrolyte membrane was evaluated. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 19 An electrolyte membrane was obtained in the same manner as in Example 1 except that 2-bis (diphenylphosphino) ethane was changed to diphenylmethoxyphosphine. Since the obtained film was insoluble in NMP and the molecular weight retention rate was not measurable, the open circuit retention time was measured as a durability test, but the evaluation was not completed within 5000 hours. The chemical durability of the electrolyte membrane was evaluated. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 20 An electrolyte membrane was obtained in the same manner as in Example 1 except that 2-bis (diphenylphosphino) ethane was changed to dimethoxyphenylphosphine. Since the obtained film was insoluble in NMP and the molecular weight retention rate was not measurable, the open circuit retention time was measured as a durability test, but the evaluation was not completed within 5000 hours. The chemical durability of the electrolyte membrane was evaluated. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 21 An electrolyte membrane was obtained in the same manner as in Example 1 except that 2-bis (diphenylphosphino) ethane was changed to triphenoxyphosphine. Since the obtained film was insoluble in NMP and the molecular weight retention rate could not be measured, the open circuit retention time was measured as a durability test. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 22 An electrolyte membrane was obtained in the same manner as in Example 1 except that the block polymer b1 was changed to Nafion (registered trademark) NRE211CS which is a fluorine-based electrolyte polymer. Since the obtained film was insoluble in NMP and the molecular weight retention rate could not be measured, the open circuit retention time was measured as a durability test. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 23 An electrolyte membrane was obtained in the same manner as in Example 1 except that the block polymer b1 was changed to the PES block copolymer b2. Since the obtained film was soluble in NMP, the molecular weight retention rate was measured as a durability test. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 24 An electrolyte membrane was obtained in the same manner as in Example 1 except that the block polymer b1 was changed to the polyarylene block copolymer b3. Since the obtained film was soluble in NMP, the molecular weight retention rate was measured as a durability test. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 26 2-bis (diphenylphosphino) ethane is converted to dichloro [(R)-(+)-2,2′-bis (diphenylphosphino) -1,1′-binaphthyl] ruthenium (II) (BINAP-Ru)
- An electrolyte membrane was obtained in the same manner as in Example 1 except that AD-1 was changed to AD-3. Since the obtained film was insoluble in NMP and the molecular weight retention rate was not measurable, the open circuit retention time was measured as a durability test, but the evaluation was not completed within 5000 hours. The chemical durability of the electrolyte membrane was evaluated. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 27 An electrolyte membrane was obtained in the same manner as in Example 1 except that 2-bis (diphenylphosphino) ethane was changed to a tetrakis (triphenylphosphine) platinum (0) complex and AD-1 was changed to AD-3. . Since the obtained film was insoluble in NMP and the molecular weight retention rate was not measurable, the open circuit retention time was measured as a durability test, but the evaluation was not completed within 5000 hours. The chemical durability of the electrolyte membrane was evaluated. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 28 22 g of the electrolyte membrane obtained in Example 1 was immersed for 72 hours in a 30 L aqueous solution in which 23.9 mg (0.138 mmol) of manganese acetate was dissolved in pure water, and manganese acetate was taken in to obtain a polymer electrolyte membrane. Since the obtained film was insoluble in NMP and the molecular weight retention rate was not measurable, the open circuit retention time was measured as a durability test, but the evaluation was not completed within 5000 hours. The chemical durability of the electrolyte membrane was evaluated. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 31 20 g of the electrolyte membrane obtained in Example 2 was further immersed in a 30 L aqueous solution in which 36.4 mg (0.125 mmol) of cobalt nitrate hexahydrate was dissolved in pure water for 72 hours to incorporate cobalt nitrate to form a polymer electrolyte. A membrane was obtained. Since the obtained film was insoluble in NMP and the molecular weight retention rate was not measurable, the open circuit retention time was measured as a durability test, but the evaluation was not completed within 5000 hours. The chemical durability of the electrolyte membrane was evaluated. In addition, the ion exchange capacity, proton-resistant water and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 32 20 g of the electrolyte membrane obtained in Example 7 was further immersed in a 30 L aqueous solution in which 32.7 mg (0.125 mmol) of ruthenium chloride trihydrate was dissolved in pure water for 72 hours to incorporate ruthenium chloride and polymer electrolyte. A membrane was obtained. Since the obtained film was insoluble in NMP and the molecular weight retention rate was not measurable, the open circuit retention time was measured as a durability test, but the evaluation was not completed within 5000 hours. The chemical durability of the electrolyte membrane was evaluated. In addition, the ion exchange capacity, proton-resistant water and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 1 An electrolyte membrane was obtained in the same manner as in Example 1 except that the amount of AD-1 added was changed to 200 mg and 2-bis (diphenylphosphino) ethane was not used. Since the obtained film was insoluble in NMP and the molecular weight retention rate could not be measured, the open circuit retention time was measured as a durability test. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 2 An electrolyte membrane was obtained in the same manner as in Example 1 except that the amount of 2-bis (diphenylphosphino) ethane added was changed to 400 mg and AD-1 was not used. Since the obtained film was insoluble in NMP and the molecular weight retention rate could not be measured, the open circuit retention time was measured as a durability test. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 3 An electrolyte membrane was obtained in the same manner as in Example 1 except that the amount of AD-1 added was changed to 400 mg and 2-bis (diphenylphosphino) ethane was not used. Since the obtained film was insoluble in NMP and the molecular weight retention rate could not be measured, the open circuit retention time was measured as a durability test. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 4 An electrolyte membrane was obtained in the same manner as in Example 1 except that the amount of 2-bis (diphenylphosphino) ethane added was changed to 800 mg and AD-1 was not used. Since the obtained film was insoluble in NMP and the molecular weight retention rate could not be measured, the open circuit retention time was measured as a durability test. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 5 An electrolyte membrane was obtained in the same manner as in Example 1 except that AD-1 was changed to 1,10-phenanthroline. Since the obtained film was insoluble in NMP and the molecular weight retention rate was not measurable, the open circuit retention time was measured as a durability test, but the evaluation was not completed within 5000 hours. The chemical durability of the electrolyte membrane was evaluated. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 7 An electrolyte membrane was obtained in the same manner as in Example 1 except that AD-1 was changed to dichloro (1,10-phenanthroline) platinum (II). Since the obtained film was insoluble in NMP and the molecular weight retention rate was not measurable, the open circuit retention time was measured as a durability test, but the evaluation was not completed within 5000 hours. The chemical durability of the electrolyte membrane was evaluated. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 8 An electrolyte membrane was obtained in the same manner as in Example 1 except that AD-1 was changed to 1,10-phenanthroline and 2-bis (diphenylphosphino) ethane was changed to triphenoxyphosphine. Since the obtained film was insoluble in NMP and the molecular weight retention rate could not be measured, the open circuit retention time was measured as a durability test. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 9 An electrolyte membrane was obtained by the method described in Example 1 except that AD-1 was changed to AD-13 represented by the following structural formula. Since the obtained film was insoluble in NMP and the molecular weight retention rate could not be measured, the open circuit retention time was measured as a durability test. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 10 An electrolyte membrane was obtained by the method described in Example 1 except that AD-1 was changed to 2,2′-bipyridyl. Since the obtained film was insoluble in NMP and the molecular weight retention rate could not be measured, the open circuit retention time was measured as a durability test. In addition, the ion exchange capacity, proton-resistant water and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 11 An electrolyte membrane was obtained in the same manner as in Example 1 except that 2-bis (diphenylphosphino) ethane and AD-1 were not used. Since the obtained film was insoluble in NMP and the molecular weight retention rate could not be measured, the open circuit retention time was measured as a durability test. In addition, the ion exchange capacity, proton-resistant water and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 12 Except that 2-bis (diphenylphosphino) ethane and AD-1 were not used and the block polymer b1 was changed to Nafion (registered trademark) NRE211CS (manufactured by DuPont), which is a fluorine-based electrolyte polymer, Example 1 and An electrolyte membrane was obtained in the same manner. Since the obtained film was insoluble in NMP and the molecular weight retention rate could not be measured, the open circuit retention time was measured as a durability test. In addition, the ion exchange capacity, hot water resistance, and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 13 An electrolyte membrane was obtained in the same manner as in Example 1, except that 2-bis (diphenylphosphino) ethane and AD-1 were not used and the block polymer b1 was changed to the PES block copolymer b2. Since the obtained film was soluble in NMP, the molecular weight retention rate was measured as a durability test. In addition, the ion exchange capacity, proton-resistant water and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 14 An electrolyte membrane was obtained in the same manner as in Example 1 except that 2-bis (diphenylphosphino) ethane and AD-1 were not used and the block polymer b1 was changed to the polyarylene block copolymer b3. Since the obtained film was soluble in NMP, the molecular weight retention rate was measured as a durability test. In addition, the ion exchange capacity, proton-resistant water and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 15 An electrolyte membrane was obtained in the same manner as in Example 22 except that AD-1 was not used. Since the obtained film was insoluble in NMP and the molecular weight retention rate could not be measured, the open circuit retention time was measured as a durability test. In addition, the ion exchange capacity, proton-resistant water and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 16 An electrolyte membrane was obtained in the same manner as in Example 22 except that DPPE was not used. Since the obtained film was insoluble in NMP and the molecular weight retention rate could not be measured, the open circuit retention time was measured as a durability test. In addition, the ion exchange capacity, proton-resistant water and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 17 An electrolyte membrane was obtained in the same manner as in Example 23 except that AD-1 was not used. Since the obtained film was soluble in NMP, the molecular weight retention rate was measured as a durability test. In addition, the ion exchange capacity, proton-resistant water and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 18 An electrolyte membrane was obtained in the same manner as in Example 23 except that DPPE was not used. Since the obtained film was soluble in NMP, the molecular weight retention rate was measured as a durability test. In addition, the ion exchange capacity, proton-resistant water and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 19 An electrolyte membrane was obtained in the same manner as in Example 24 except that AD-1 was not used. Since the obtained film was soluble in NMP, the molecular weight retention rate was measured as a durability test. In addition, the ion exchange capacity, proton-resistant water and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
- Example 20 An electrolyte membrane was obtained in the same manner as in Example 24 except that DPPE was not used. Since the obtained film was soluble in NMP, the molecular weight retention rate was measured as a durability test. In addition, the ion exchange capacity, proton-resistant water and proton conductivity at 80 ° C. and 25% relative humidity of the obtained electrolyte membrane were measured. The results are shown in Table 1.
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Manufacturing & Machinery (AREA)
- Sustainable Energy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Sustainable Development (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Composite Materials (AREA)
- Crystallography & Structural Chemistry (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Fuel Cell (AREA)
- Polyethers (AREA)
- Conductive Materials (AREA)
Abstract
Description
本発明において、有機リン系添加剤(C)とは、炭素とリンの化学結合を含む有機リン化合物を表す。



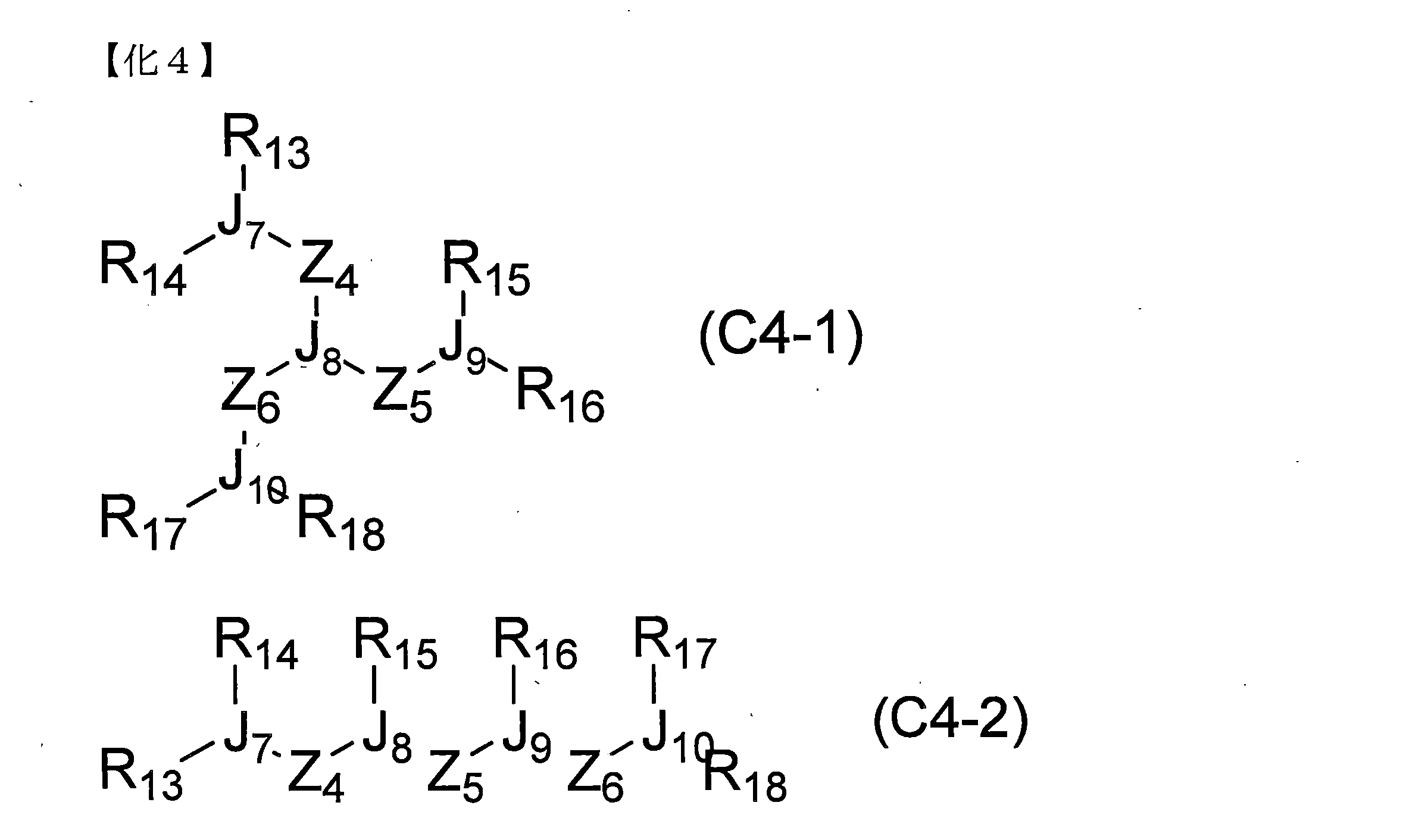
RやZの一部がアルコキシ基である有機リン系添加剤(C)の具体例としては、ホスフィナイト、ホスフィネート化合物として、下記一般式(C1b)、(C2a)、(C2b)で表される化合物など、ホスホナイト、ホスホネート化合物として、下記一般式(C1c)、(C2c)、(C2d)で表される化合物など、ホスファイト、ホスフェート化合物として、下記一般式(C1d)、(C2e)で表される化合物などを挙げることができるが、本発明のホスフィナイト、ホスフィネート化合物、ホスホナイト、ホスホネート化合物、ホスファイト、ホスフェート化合物はこれらに限定されるものではない。
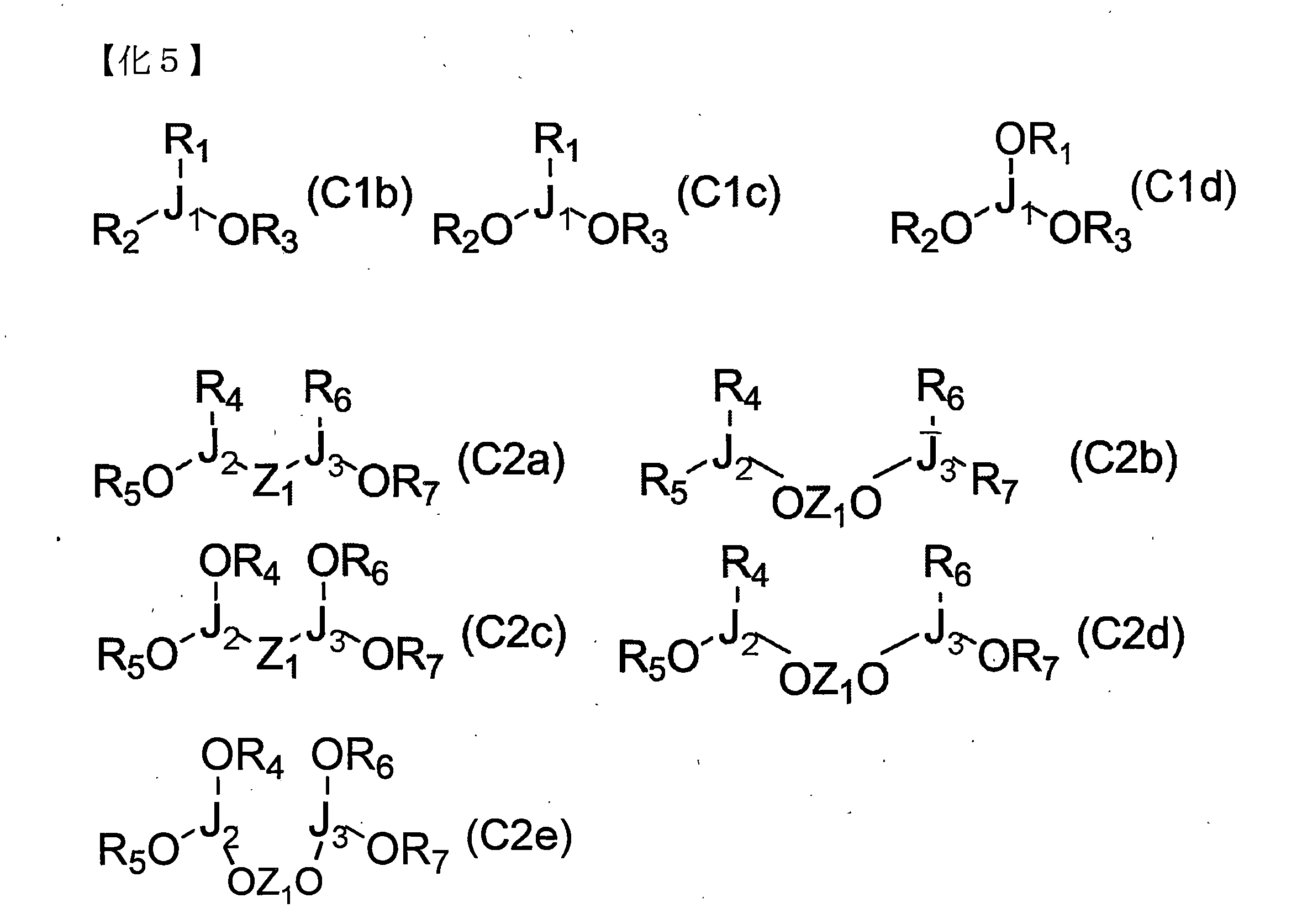
なかでも、耐熱水性により優れ、リンの電子供与性に起因する、より強いキレート効果により金属不活性化剤としての効果が大きく、窒素含有複素芳香環系化合物酸化体の還元能が大きい点で、ホスフィン基、ホスフィナイト基、ホスホナイト基、ホスファイト基から選ばれた少なくとも1種を合計2個以上含有する(2座以上の)有機リン系化合物が有機リン系添加剤(C)として好ましく、2~4座の多座ホスフィン化合物、ホスフィナイト化合物、ホスホナイト化合物、ホスファイト化合物がより好ましい。また、コストの点では、2座のホスフィン化合物、ホスフィナイト化合物、ホスホナイト化合物、ホスファイト化合物がさらに好ましく、耐熱水性の点で、2座のホスフィン化合物、ホスフィナイト化合物、ホスホナイト化合物が好ましく、耐加水分解性の点で、2座のホスフィン化合物が最も好ましい。






次に、本発明で使用する窒素含有複素芳香環系添加剤(D)について説明する。

なお、本明細書において、アミノ基とは一級~三級アミノ基、四級アンモニウムカチオンを表すものとする。また、1価以上の結合部位とは、他構造単位との結合可能な部位を1個以上有することを指し、2価以上の結合部位とは、他構造単位との結合可能な部位を2個以上有することを指す。

なかでも、コストの点で、同一芳香環の構成原子であるE1~E6、E7~E10、E11~E14、E15~E19、E20~E22、E23~E25のそれぞれのうち、N-H、N-R(Rは任意の有機基)、またはNが1~3個であることが好ましく、合成の容易さや塩基性の点で、N-H、N-R、またはNが1個または2個であることがさらに好ましい。一つの芳香環を構成する原子のうち、N-H、N-R、またはNが4個以上の場合、窒素含有複素芳香環の電子密度が低下し、環の安定性が低下したり、金属への配位・不活性化能が低下したりする場合がある。








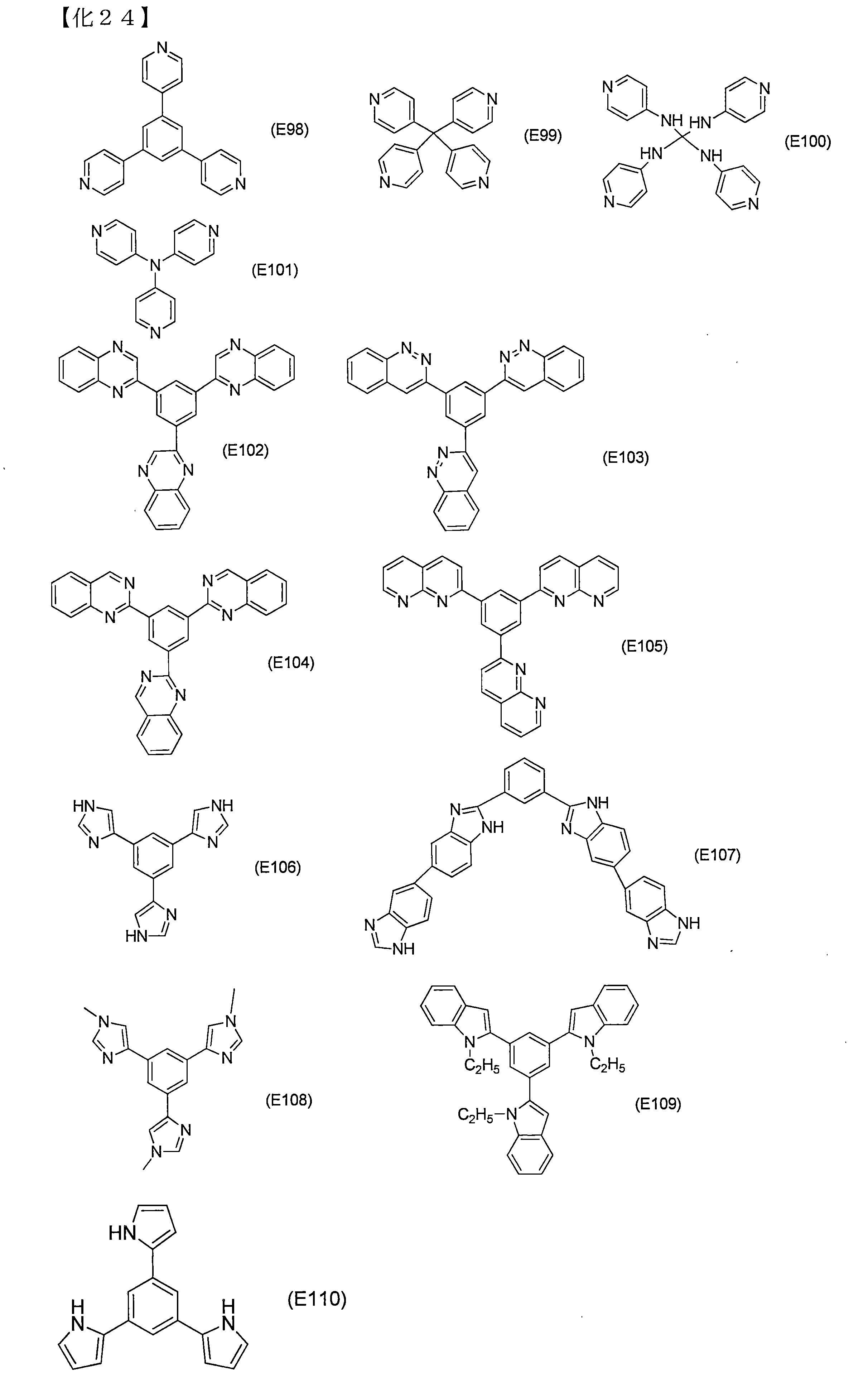
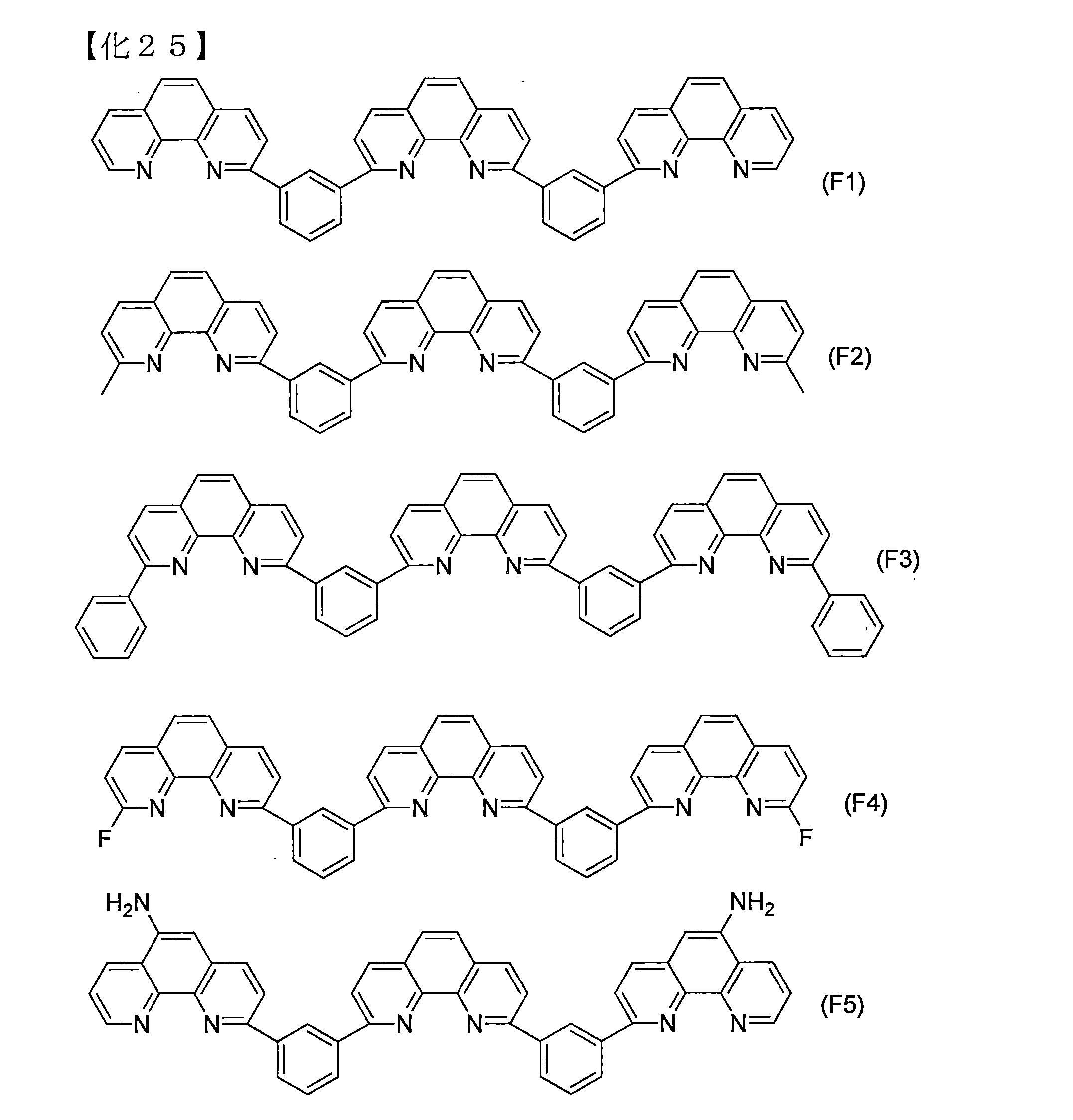

本発明の高分子電解質組成物としては、Ce、Mn、Ti、Zr、V、Cr、Mo、W、Ru、Co、Rh、Ir、Ni、Pd、Pt、Ag、Auから選ばれた少なくとも1種の遷移金属をさらに含有することも好ましい。これら遷移金属は、かかる遷移金属、かかる遷移金属のイオン、かかる遷移金属イオンを含む塩、かかる遷移金属の酸化物からなる群から選ばれる1種以上を用いることができる。
(1)イオン性基含有ポリマー(A)の溶液または分散液に、有機リン系添加剤(C)および/または窒素含有複素芳香環系添加剤(D)を溶解または分散させた後、得られた液を用いて製膜し、高分子電解質膜を作製する方法。
(2)有機リン系添加剤(C)および/または窒素含有複素芳香環系添加剤(D)を溶解させた液を、イオン性基含有ポリマー(A)からなる高分子電解質膜に塗布する方法。
(3)有機リン系添加剤(C)および/または窒素含有複素芳香環系添加剤(D)を溶解させた溶液に、イオン性基含有ポリマー(A)からなる高分子電解質膜を浸漬する方法。
次に、本発明に使用するイオン性基含有ポリマー(A)について説明する。

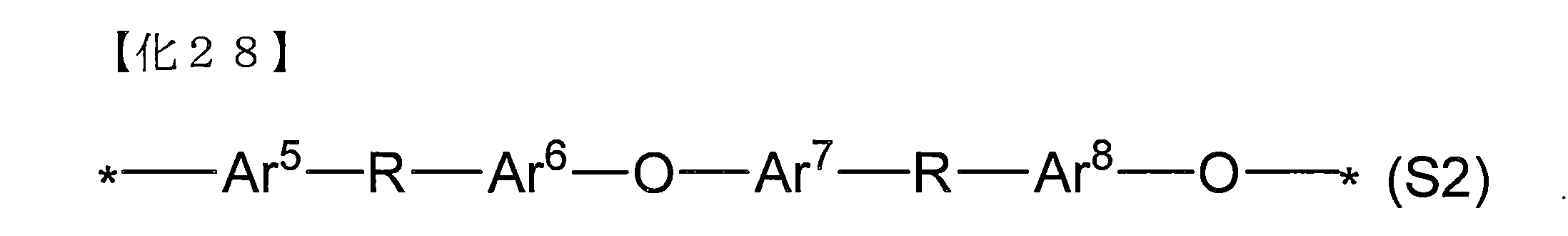
ケトン基に誘導されうる保護基の具体例としては、有機合成で一般的に用いられる保護基があげられ、後の段階で除去することを前提に、一時的に導入される置換基を表し、脱保護により元のケトン基に戻すことのできるものである。

なかでも、化合物の臭いや反応性、安定性等の点で、前記一般式(U1)および(U2)において、EがOである、すなわち、ケトン部位をケタール部位で保護/脱保護する方法が最も好ましい。
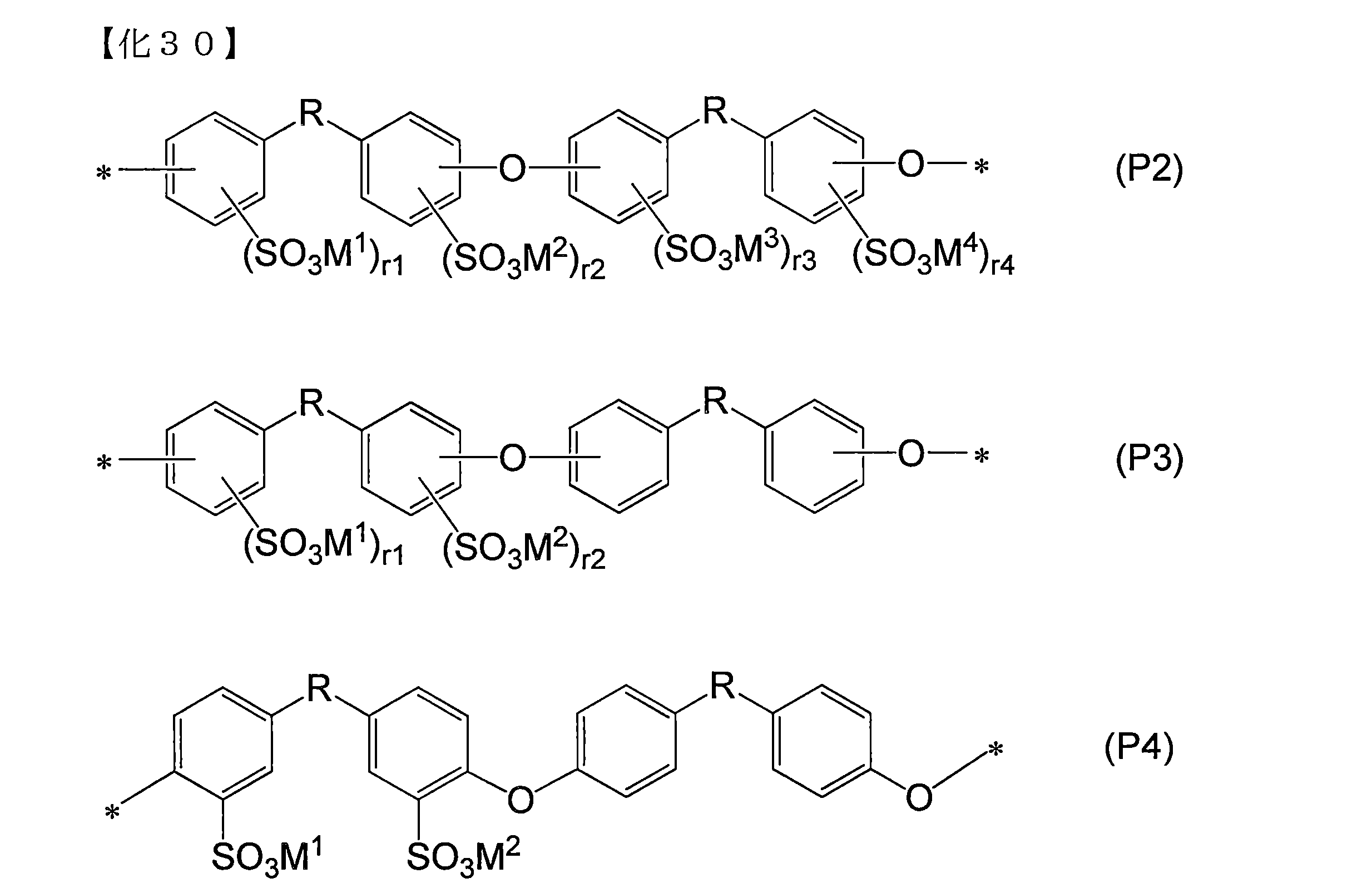
本発明で使用するブロックポリマーとしては、イオン性基を含有するセグメント(A1)と、イオン性基を含有しないセグメント(A2)のモル組成比(A1/A2)が、0.2以上であることがより好ましく、0.33以上がさらに好ましく、0.5以上が最も好ましい。また、5以下がより好ましく、3以下がさらに好ましく、2以下が最も好ましい。当該モル組成比A1/A2が、0.2未満あるいは5を越える場合には、低加湿条件下でのプロトン伝導性が不足したり、耐熱水性や物理的耐久性が不足したりする傾向がある。
(1)プロトン置換し、純水で十分に洗浄した電解質膜の膜表面の水分を拭き取った後、100℃にて12時間以上真空乾燥し、乾燥重量を求める。
(2)電解質に5wt%硫酸ナトリウム水溶液を50mL加え、12時間静置してイオン交換する。
(3)0.01mol/L水酸化ナトリウム水溶液を用いて、生じた硫酸を滴定する。指示薬として市販の滴定用フェノールフタレイン溶液0.1w/v% を加え、薄い赤紫色になった点を終点とする。
(4)下記式によりイオン交換容量を求める。
〔水酸化ナトリウム水溶液の濃度(mmol/ml)×滴下量(ml)〕/試料の乾燥重量(g)
イオン性基を含有するセグメント(A1)およびイオン性基を含有しないセグメント(A2)を構成するオリゴマーの合成方法は、実質的に十分な分子量が得られる方法であれば特に限定されるものではないが、例えば芳香族活性ジハライド化合物と2価フェノール化合物の芳香族求核置換反応、またはハロゲン化芳香族フェノール化合物の芳香族求核置換反応を利用して合成することができる。
本発明の高分子電解質組成物は、特に、高分子電解質成型体として好適に用いられる。本発明において高分子電解質成型体とは、本発明の高分子電解質組成物を含有する成型体を意味する。本発明の高分子電解質成型体としては、膜類(フィルムおよびフィルム状のものを含む)の他、板状、繊維状、中空糸状、粒子状、塊状、微多孔状、コーティング類、発泡体類など、使用用途によって様々な形態をとりうる。ポリマーの設計自由度の向上および機械特性や耐溶剤性等の各種特性の向上が図れることから、幅広い用途に適応可能である。特に高分子電解質成型体が膜類であるときに好適である。
本発明の高分子電解質組成物を固体高分子形燃料電池用として使用する際には、高分子電解質膜および電極触媒層などが好適であり、中でも高分子電解質膜に好適に用いられる。本発明の高分子電解質組成物は、高い化学的安定性を有しており、電気化学反応が近くで起こる電極触媒層バインダーにも特に好適に使用できる。
以下の(i)~(iv)に記載の中和滴定法により測定した。測定は3回行って、その平均値を取った。
(i)プロトン置換し、純水で十分に洗浄した電解質膜の膜表面の水分を拭き取った後、100℃にて12時間以上真空乾燥し、乾燥重量を求めた。
(ii)電解質に5重量%硫酸ナトリウム水溶液を50mL加え、12時間静置してイオン交換した。
(iii)0.01mol/L水酸化ナトリウム水溶液を用いて、生じた硫酸を滴定した。指示薬として市販の滴定用フェノールフタレイン溶液0.1w/v%を加え、薄い赤紫色になった点を終点とした。
(iv)イオン交換容量は下記の式により求めた。
〔水酸化ナトリウム水溶液の濃度(mmol/mL)×滴下量(mL)〕/試料の乾燥重量(g)〕
(2)プロトン伝導度
膜状の試料を25℃の純水に24時間浸漬した後、80℃、相対湿度25~95%の恒温恒湿槽中にそれぞれのステップで30分保持し、定電位交流インピーダンス法でプロトン伝導度を測定した。
ポリマーの数平均分子量、重量平均分子量をGPCにより測定した。紫外検出器と示差屈折計の一体型装置として東ソー社製HLC-8022GPCを、またGPCカラムとして東ソー社製TSK gel SuperHM-H(内径6.0mm、長さ15cm)2本を用い、N-メチル-2-ピロリドン溶媒(臭化リチウムを10mmol/L含有するN-メチル-2-ピロリドン溶媒)にて、サンプル濃度0.1重量%、流量0.2mL/min、温度40℃で測定し、標準ポリスチレン換算により数平均分子量、重量平均分子量を求めた。
ミツトヨ社製グラナイトコンパレータスタンドBSG-20にセットしたミツトヨ社製ID-C112型を用いて測定した。
下記条件のガスクロマトグラフィー(GC)により定量分析した。
カラム:DB-5(J&W社製) L=30m Φ=0.53mm D=1.50μm
キャリヤー:ヘリウム(線速度=35.0cm/sec)
分析条件
Inj.temp.; 300℃
Detct.temp.; 320℃
Oven; 50℃×1min
Rate; 10℃/min
Final; 300℃×15min
SP ratio; 50:1
(6)添加剤の添加量測定
電解質膜の添加剤添加量は、誘導結合プラズマ(ICP)発光分析により評価した。5cm×5cmの大きさで電解質膜を切り出し、110℃、減圧下で2時間乾燥した後、質量を精秤し、550℃で2日間静置して、残った灰分を0.1規定硝酸水溶液に溶解させ、添加剤を完全に抽出した液を得た。この液をICP発光分析にて測定し、リン、窒素及び各種金属元素量を測定することで、添加剤の定量を行った。
添加剤の耐熱水性は、95℃の熱水浸漬後の残存率を測定することにより評価した。電解質膜を長さ約5cm、幅約10cmの短冊2枚に切り取り、95℃の熱水中に8時間浸漬させることで添加剤を溶出させた。熱水浸漬前後の電解質膜を、5cm×5cmの大きさで切り出し、上記ICP発光分析を行うことで添加剤含量を測定し、添加剤残存率として耐熱水性を評価した。
下記の測定条件で、1H-NMRの測定を行い、構造確認、およびイオン性基を含有するセグメント(A1)とイオン性基を含有しないセグメント(A2)のモル組成比の定量を行った。該モル組成比は、8.2ppm(ジスルホネート-4,4’-ジフルオロベンゾフェノン由来)と6.5~8.0ppm(ジスルホネート-4,4’-ジフルオロベンゾフェノンを除く全芳香族プロトン由来)に認められるピークの積分値から算出した。
共鳴周波数 :270MHz(1H-NMR)
測定温度 :室温
溶解溶媒 :DMSO-d6
内部基準物質:TMS(0ppm)
積算回数 :16回
(9)化学的安定性
(A)分子量保持率
N-メチルピロリドン(NMP)に可溶な電解質膜については、以下の方法にて電解質膜を劣化させ、劣化試験前後の分子量を比較することで化学安定性を評価した。
NMPに溶解不可能な電解質膜については、以下の方法にて電解質膜を劣化させ、開回路電圧の保持時間を比較することで化学安定性を評価した。
上記(B)の開回路保持時間評価を行っても5000時間以上、0.7V以上を維持できる場合には、そこで評価を打ち切り初期電圧と5000時間後の電圧を比較し電圧保持率として化学耐久性を評価した。
(下記一般式(G1)で表される2,2-ビス(4-ヒドロキシフェニル)-1,3-ジオキソラン(K-DHBP)の合成)



かき混ぜ機、窒素導入管、Dean-Starkトラップを備えた1000mL三口フラスコに、炭酸カリウム16.59g(アルドリッチ試薬、120mmol)、K-DHBP 25.8g(100mmol)および4,4’-ジフルオロベンゾフェノン20.3g(アルドリッチ試薬、93mmol)を入れ、窒素置換後、N-メチルピロリドン(NMP)300mL、トルエン100mL中にて160℃で脱水後、昇温してトルエン除去、180℃で1時間重合を行った。多量のメタノールで再沈殿することで精製を行い、イオン性基を含有しないオリゴマーa1(末端OM基、なおOM基のMはNaまたはKを表し、これ以降の表記もこれに倣う。)を得た。数平均分子量は10000であった。
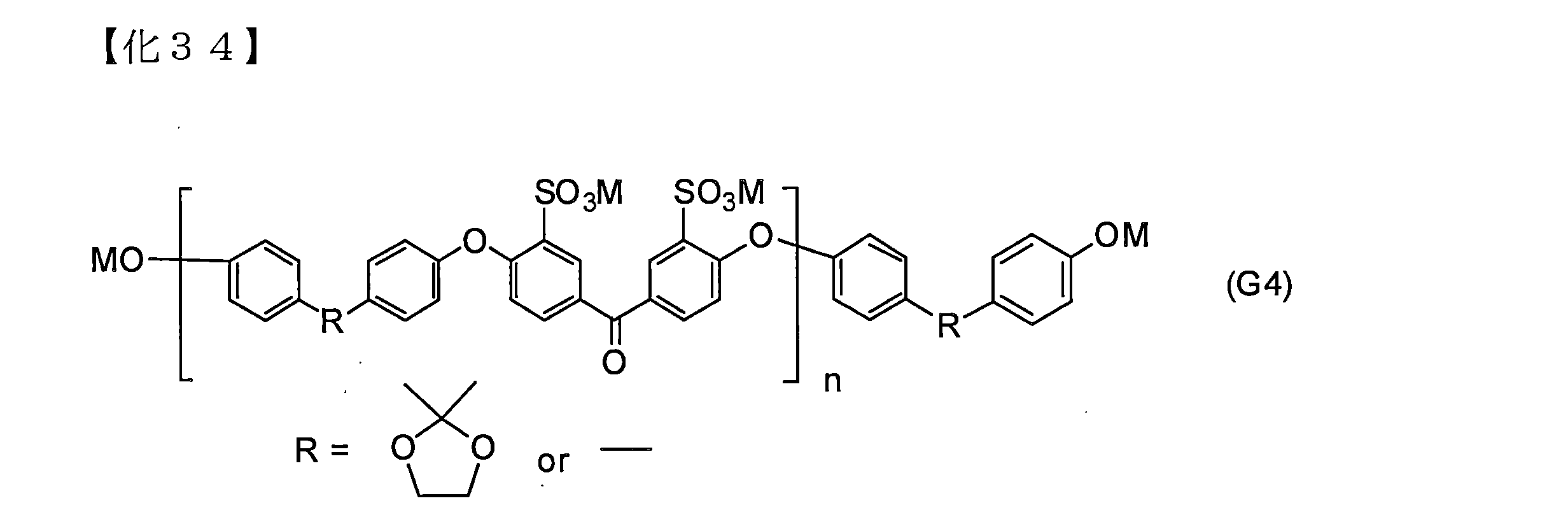
かき混ぜ機、窒素導入管、Dean-Starkトラップを備えた1000mL三口フラスコに、炭酸カリウム27.6g(アルドリッチ試薬、200mmol)、K-DHBP 12.9g(50mmol)および4,4’-ビフェノール9.3g(アルドリッチ試薬、50mmol)、ジソジウム 3,3’-ジスルホネート-4,4’-ジフルオロベンゾフェノン40.6g(96mmol)、および18-クラウン-6エーテル17.9g(和光純薬、82mmol)を入れ、窒素置換後、N-メチルピロリドン(NMP)300mL、トルエン100mL中にて170℃で脱水後、昇温してトルエン除去、180℃で1時間重合を行った。多量のイソプロピルアルコールで再沈殿することで精製を行い、前記式(G4)で示されるイオン性基を含有するオリゴマーa2(末端OM基)を得た。数平均分子量は29000であった。
かき混ぜ機、窒素導入管、Dean-Starkトラップを備えた500mL三口フラスコに、炭酸カリウム0.56g(アルドリッチ試薬、4mmol)、イオン性基を含有するオリゴマーa2(末端OM基)を29g(1mmol)を入れ、窒素置換後、N-メチルピロリドン(NMP)100mL、トルエン30mL中にて100℃で脱水後、昇温してトルエン除去し、イオン性基を含有しないオリゴマーa1’(末端フルオロ基)11g(1mmol)を入れ、105℃で24時間反応を行った。多量のイソプロピルアルコールで再沈殿することで精製を行い、ブロックコポリマーb1を得た。重量平均分子量は39万であった。
(下記式(G6)で表されるセグメントと下記式(G7)で表されるセグメントからなるポリエーテルスルホン(PES)系ブロックコポリマー前駆体b2’の合成)
無水塩化ニッケル1.85gとジメチルスルホキシド17mLとを混合し、70℃に調整した。これに、2,2’-ビピリジル2.46gを加え、同温度で10分撹拌し、ニッケル含有溶液を調製した。

ブロックコポリマー前駆体b2’0.3gを、臭化リチウム1水和物0.23gとN-メチル-2-ピロリドン10mLとの混合溶液に加え、120℃で24時間反応させた。反応混合物を、6mol/L塩酸100mL中に注ぎ込み、1時間撹拌した。析出した固体を濾過により分離した。分離した固体を乾燥し、灰白色の式(G7)で示されるセグメントと下記式(G8)で表されるセグメントからなるブロックコポリマーb2を得た。得られたポリアリーレンの重量平均分子量は21万であった。

(下記構造式(G9)で表される疎水性オリゴマーa3の合成)


(AD-1の合成)
8-アミノ- 7-キノリンカルボアルデヒド81gを1,3-ジアセチルベンゼン(東京化成工業(株)製)36g、85%水酸化カリウム80gとエタノール1440mL中で還流下10時間反応させ、分液抽出したもの30gをトルエン550mL中でフェニルリチウム(0.94M シクロヘキサン/ジエチルエーテル溶液)150mLと氷冷下で2.5時間反応させ、再結晶精製した。得られた生成物をニトロベンゼン89gと110℃で3時間反応させ、再結晶精製し、下記構造式で表される化合物AD-1を16.2g得た。
合成例1にて得た20gのブロックコポリマーb1を80gのNMPに溶解した。この溶液に、1,2-ビス(ジフェニルホスフィノ)エタン(DPPE、Aldrich製)を200mg、AD-1を100mg添加し、撹拌機で20,000rpm、3分間撹拌しポリマー濃度20質量%の透明な溶液を得た。ポリマーの溶解性は極めて良好であった。得られた溶液を、ガラス繊維フィルターを用いて加圧ろ過後、ガラス基板上に流延塗布し、100℃にて4時間乾燥後、窒素下150℃で10分間熱処理し、ポリケタールケトン膜(膜厚15μm)を得た。95℃、10重量%硫酸水溶液に24時間浸漬してプロトン置換、脱保護反応した後に、大過剰量の純水に24時間浸漬して充分洗浄し、高分子電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
(AD-2の合成)
2,7-ジヒドロキシナフタレン(東京化成工業(株)製)43gをジクロロメタン544mLピリジン109mLに溶解させ、0℃でトリフルオロメタンスルホン酸無水物(東京化成工業(株)製)180gを滴下した。5℃で2時間、次いで室温で1日間反応させた後、常法で処理し、2,7-ビス(トリフルオロメタンスルフォニルオキシ)ナフタレン114gを得た。この2,7-ビス(トリフルオロメタンスルフォニルオキシ)ナフタレン80gをn-ブチルビニルエーテル(東京化成工業(株)製)152mL、トリエチルアミン79mL、1,3-ビス(ジフェニルホスフィノ)プロパン(東京化成工業(株)製)1.94g、酢酸パラジウム(和光純薬工業(株)製)0.53g、ジメチルホルムアミド477mLと混合し、70~85℃で2日間反応させた。常法で処理し、2,7-ジアセチルナフタレン27gを得た。この2,7-ジアセチルナフタレン5g をエタノール238mL中60℃で8-アミノ-7-キノリンカルボアルデヒド8.52g、水酸化カリウム8.55gと反応させ、常法で処理し、下記構造式で表されるAD-2を4.7g得た。
AD-1の代わりに、AD-2を用いた以外は実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
(AD-3の合成)
AD-1 2g、硫酸30mLを入れ、室温で攪拌し溶解した。液温を1℃に氷冷し、硝酸カリウム0.793gを硫酸30mLに溶解した溶液を加えた。140℃で8時間反応させた後、2Lの炭酸水素ナトリウム飽和溶液に反応液を滴下した。固体を濾過、純水洗浄、乾燥させ、得られた化合物2.0gをNMP50mLに溶解させた。Pd/C10wt% 199mgを入れたオートクレーブを窒素置換し、前記NMP溶液50mLを投入した。水素圧0.5MPaで加圧して、21時間攪拌、反応させた。反応液を濾過した後、ろ液からNMPを留去、ジクロロメタン可溶部を抽出した。抽出物をジクロロメタン/メタノールで再結晶することで、下記構造式で表されるAD-3を1.02gを得た。

AD-1の代わりに、AD-3を用いた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
1,2-ビス(ジフェニルホスフィノ)エタンを4g、AD-1を3gに変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
1,2-ビス(ジフェニルホスフィノ)エタンを2mg、AD-1を2mgに変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
AD-1を1,3,5‐トリ(4‐ピリジル)ベンゼンに変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
AD-1を4′,4″″-(1,4-フェニレン)ビス(2,2′:6′,2″-テルピリジン)に変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
AD-1を下記構造式で表されるAD-4に変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。

AD-1を下記構造式で表されるAD-5に変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。

AD-1を下記構造式で表されるAD-6に変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。

AD-1を下記構造式で表されるAD-7に変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。

(AD-8の合成)
炭酸セシウム926mg、1,10-フェナントロリン-5-アミン 468mg、1,2-ジヨードエタン 744mgをN,N-ジメチルホルムアミド 8.5mLに溶解させ、80℃で12時間反応させた。反応後の溶液を水100mL、続いてIPA300mLに再沈殿し、濾物を乾燥させ、下記構造式で表されるAD-8を1.01g得た。数平均分子量は、2000であった。

AD-1を上記構造式で表されるAD-8に変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
(AD-9の合成)
4,7-ジクロロ-1,10-フェナントロリン 1g 4,7-ジヒドロキシ-1,10-フェナントロリン 0.85 gをNMP50mLに溶解性させ、トルエン25mL中にて160℃で脱水後、昇温してトルエン除去、180℃で1時間重合を行った。多量のメタノールで再沈殿することで精製を行い、下記構造式で表されるAD-9を1.7g得た。数平均分子量は、2100であった。

AD-1を上記構造式で表されるAD-9に変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
AD-1を下記構造式で表されるAD-10(数平均分子量2500)に変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。

AD-1を下記構造式で表されるAD-11(数平均分子量3200)に変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。

1,2-ビス(ジフェニルホスフィノ)エタンをトリフェニルホスフィンに変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
1,2-ビス(ジフェニルホスフィノ)エタンを2,2’-ビス(ジフェニルホスフィノ)-1,1’-ビナフチルに、AD-1をAD-5に変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
2-ビス(ジフェニルホスフィノ)エタンを下記一般式AD-12に、AD-1を
AD-3に変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。

2-ビス(ジフェニルホスフィノ)エタンをジフェニルメトキシホスフィンに変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
2-ビス(ジフェニルホスフィノ)エタンをジメトキシフェニルホスフィンに変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
2-ビス(ジフェニルホスフィノ)エタンをトリフェノキシホスフィンに変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
ブロックポリマーb1をフッ素系電解質ポリマーであるナフィオン(登録商標)NRE211CSに変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
ブロックポリマーb1をPES系ブロックコポリマーb2に変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに可溶であったため、耐久性試験として分子量保持率を測定した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
ブロックポリマーb1をポリアリーレン系ブロックコポリマーb3に変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに可溶であったため、耐久性試験として分子量保持率を測定した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
(2-ビス(ジフェニルホスフィノ)エタンと硝酸セリウム(III)との錯体の合成)
100mLのナスフラスコ中に、DPPE2.0g(5.02mmol)と硝酸セリウム六水和物544mg(1.26mmol)を加えた。そこに、エタノール40mLを注ぎ込み、25℃で24時間攪拌した。白色の懸濁液をロータリーエバポレーターにて濃縮し溶媒を除去した。得られた、白色の固体を精製せずにそのまま添加剤として使用した。
2-ビス(ジフェニルホスフィノ)エタンを前記2-ビス(ジフェニルホスフィノ)エタン-セリウム錯体に変えた以外は、実施例1と同様の方法で、電解質膜を得た。 得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
2-ビス(ジフェニルホスフィノ)エタンをジクロロ[(R)-(+)-2,2’-ビス(ジフェニルホスフィノ)-1,1’-ビナフチル]ルテニウム(II)(BINAP-Ru)に、AD-1をAD-3に変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
2-ビス(ジフェニルホスフィノ)エタンをテトラキス(トリフェニルホスフィン)白金(0)錯体に、AD-1をAD-3に変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
実施例1で得た電解質膜22gを酢酸マンガン23.9mg(0.138mmol)を純水に溶解させた30Lの水溶液に72時間浸漬し、酢酸マンガンを取り込ませ高分子電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
(AD-3と酢酸パラジウム(II)との錯体の合成)
100mナスフラスコ中に、AD-3 2,0g(3.08mmol)と酢酸パラジウム173mg(0.77mmol)を加えた。そこに、NMP40mLを注ぎ込み、25℃で24時間攪拌した。反応液を濃縮、溶媒除去し、得られた固体を精製せずに、そのまま添加剤として使用した。
AD-1を、合成したAD-3のパラジウム錯体に変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
(AD-3と塩化白金(II)との錯体の合成)
100mナスフラスコ中に、AD-3 2,0g(3.08mmol)と塩化白金205mg(0.77mmol)を加えた。そこに、NMP40mLを注ぎ込み、25℃で24時間攪拌した。反応液を濃縮、溶媒除去し、得られた固体を精製せずに、そのまま添加剤として使用した。
AD-1を、合成したAD-3の白金錯体に変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
実施例2で得た電解質膜20gをさらに、硝酸コバルト六水和物 36.4mg(0.125mmol)を純水に溶解させた30Lの水溶液に72時間浸漬し、硝酸コバルトを取り込ませ高分子電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
実施例7で得た電解質膜20gをさらに、塩化ルテニウム三水和物 32.7mg(0.125mmol)を純水に溶解させた30Lの水溶液に72時間浸漬し、塩化ルテニウムを取り込ませ高分子電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
AD-1の添加量を200mgに変え、2-ビス(ジフェニルホスフィノ)エタンを使用しない以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
2-ビス(ジフェニルホスフィノ)エタンの添加量を400mgに変え、AD-1を使用しない以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
AD-1の添加量を400mgに変え、2-ビス(ジフェニルホスフィノ)エタンを使用しない以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
2-ビス(ジフェニルホスフィノ)エタンの添加量を800mgに変え、AD-1を使用しない以外は実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
AD-1を1,10―フェナントロリンに変えた以外は実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
比較例5で得られた電解質膜18gを酢酸マンガン19.6mg(0.113mmol)を純水に溶解させた30Lの水溶液に72時間浸漬し、酢酸マンガンを取り込ませ高分子電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
AD-1をジクロロ(1,10-フェナントロリン)白金(II)に変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定したが、5000時間以内に評価が終了しなかったので、電圧保持率として電解質膜の化学耐久性を評価した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
AD-1を1,10―フェナントロリンに変え、2-ビス(ジフェニルホスフィノ)エタンをトリフェノキシホスフィン変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
AD-1を下記構造式で表されるAD-13に変えた以外は、実施例1に記載の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。

AD-1を2,2’-ビピリジルに変えた以外は、実施例1に記載の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
2-ビス(ジフェニルホスフィノ)エタンおよびAD-1を使用しない以外は実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
2-ビス(ジフェニルホスフィノ)エタンおよびAD-1を使用せず、かつブロックポリマーb1をフッ素系電解質ポリマーであるナフィオン(登録商標)NRE211CS(デュポン社製)に変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と、80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
2-ビス(ジフェニルホスフィノ)エタンおよびAD-1を使用せず、かつブロックポリマーb1をPES系ブロックコポリマーb2に変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに可溶であったため、耐久性試験として分子量保持率を測定した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
2-ビス(ジフェニルホスフィノ)エタンおよびAD-1を使用せず、かつブロックポリマーb1をポリアリーレン系ブロックコポリマーb3に変えた以外は、実施例1と同様の方法で、電解質膜を得た。得られた膜は、NMPに可溶であったため、耐久性試験として分子量保持率を測定した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
AD-1を使用しない以外は、実施例22と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
DPPEを使用しない以外は、実施例22と同様の方法で、電解質膜を得た。得られた膜は、NMPに不溶であり分子量保持率が測定不能であったため、耐久性試験として開回路保持時間を測定した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
AD-1を使用しない以外は、実施例23と同様の方法で、電解質膜を得た。得られた膜は、NMPに可溶であったため、耐久性試験として分子量保持率を測定した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
DPPEを使用しない以外は、実施例23と同様の方法で、電解質膜を得た。得られた膜は、NMPに可溶であったため、耐久性試験として分子量保持率を測定した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
AD-1を使用しない以外は、実施例24と同様の方法で、電解質膜を得た。得られた膜は、NMPに可溶であったため、耐久性試験として分子量保持率を測定した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
DPPEを使用しない以外は、実施例24と同様の方法で、電解質膜を得た。得られた膜は、NMPに可溶であったため、耐久性試験として分子量保持率を測定した。加えて、得られた電解質膜のイオン交換容量、耐熱水性と80℃、相対湿度25%におけるプロトン伝導度を測定した。その結果を表1に示す。
合成例1で得た20gのブロックコポリマーb1を80gのNMPに溶解し、さらに硝酸セリウム43mgを添加、20,000rpmで攪拌した。攪拌中に溶液粘度が増大、ゲル化し、ガラス繊維フィルターを用いた加圧ろ過は困難で、製膜に供することはできなかった。

Claims (15)
- 少なくともイオン性基含有ポリマー(A)と、有機リン系添加剤(C)と、さらに窒素含有複素芳香環系添加剤(D)とを含有する高分子電解質組成物であって、窒素含有複素芳香環系添加剤(D)が分子内に少なくとも窒素含有複素芳香環を3個以上含有する窒素含有複素芳香環系化合物であることを特徴とする高分子電解質組成物。
- 有機リン系添加剤(C)が構成元素として分子内に少なくともリンを2個以上含有する有機リン化合物である、請求項1に記載の高分子電解質組成物。
- 有機リン系添加剤(C)が3価の有機リン化合物である、請求項1または2に記載の高分子電解質組成物。
- 有機リン系添加剤(C)が、ホスフィン基、ホスフィナイト基、ホスホナイト基から選ばれた少なくとも1種を合計2個以上含有する有機リン系化合物である、請求項3に記載の高分子電解質組成物。
- 窒素含有複素芳香環系添加剤(D)が、窒素含有複素芳香環としてピリジン環またはイミダゾール環を含有する化合物である、請求項1~4のいずれかに記載の高分子電解質組成物。
- 有機リン系添加剤(C)および窒素含有複素芳香環系添加剤(D)の含有量の総和が、高分子電解質組成物全体の0.01重量%以上、15重量%以下である、請求項1~5のいずれかに記載の高分子電解質組成物。
- Ce、Mn、Ti、Zr、V、Cr、Mo、W、Ru、Co、Rh、Ir、Ni、Pd、Pt、Ag、Auから選ばれた少なくとも1種の遷移金属をさらに含有する、請求項1~6のいずれかに記載の高分子電解質組成物。
- イオン性基含有ポリマー(A)が、主鎖に芳香環を有する炭化水素系ポリマーである、請求項1~7のいずれかに記載の高分子電解質組成物。
- イオン性基含有ポリマー(A)が、イオン性基を含有するセグメント(A1)とイオン性基を含有しないセグメント(A2)を有するブロックポリマーである、請求項1~8のいずれかに記載の高分子電解質組成物。
- イオン性基含有ポリマー(A)が芳香族ポリエーテルケトン系ポリマーである、請求項8または9に記載の高分子電解質組成物。
- イオン性基を含有するセグメント(A1)およびイオン性基を含有しないセグメント(A2)が、それぞれ下記一般式(S1)および(S2)で表される構成単位を含有する、請求項9または10に記載の高分子電解質組成物。

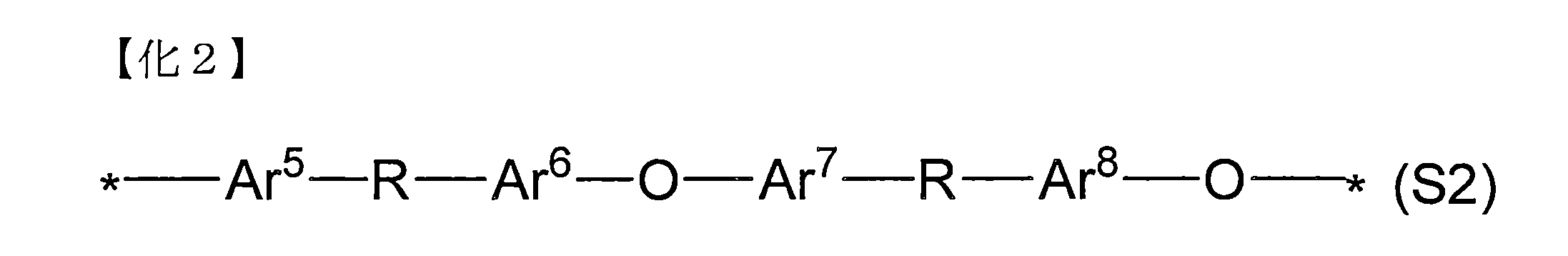
- 請求項1~11のいずれかに記載の高分子電解質組成物を用いて構成されることを特徴とする高分子電解質膜。
- 請求項1~11のいずれかに記載の高分子電解質組成物を用いて構成されることを特徴とする触媒層付き電解質膜。
- 請求項1~11のいずれかに記載の高分子電解質組成物を用いて構成されることを特徴とする膜電極複合体。
- 請求項1~11のいずれかに記載の高分子電解質組成物を用いて構成されることを特徴とする固体高分子形燃料電池。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2944439A CA2944439C (en) | 2014-04-07 | 2015-04-03 | Polymer electrolyte composition and polymer electrolyte membrane, polymer electrolyte membrane with catalyst layer, membrane electrode assembly, and polymer electrolyte fuel cell each using the same |
| CN201580018329.3A CN106165175B (zh) | 2014-04-07 | 2015-04-03 | 高分子电解质组合物以及使用其的高分子电解质膜、膜电极复合体及固体高分子型燃料电池 |
| JP2015529360A JP6341204B2 (ja) | 2014-04-07 | 2015-04-03 | 高分子電解質組成物ならびにそれを用いた高分子電解質膜、膜電極複合体および固体高分子形燃料電池 |
| US15/302,073 US10103401B2 (en) | 2014-04-07 | 2015-04-03 | Polymer electrolyte composition and polymer electrolyte membrane, polymer electrolyte membrane with catalyst layer, membrane electrode assembly, and polymer electrolyte fuel cell each using the same |
| KR1020167025432A KR102273035B1 (ko) | 2014-04-07 | 2015-04-03 | 고분자 전해질 조성물 및 그것을 사용한 고분자 전해질막, 막전극 복합체 및 고체 고분자형 연료 전지 |
| EP15776250.1A EP3131145B1 (en) | 2014-04-07 | 2015-04-03 | Polymer electrolyte composition and polymer electrolyte membrane, membrane-electrolyte assembly, and solid polymer fuel cell using same |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014078401 | 2014-04-07 | ||
| JP2014-078401 | 2014-04-07 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015156228A1 true WO2015156228A1 (ja) | 2015-10-15 |
Family
ID=54287804
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/060630 WO2015156228A1 (ja) | 2014-04-07 | 2015-04-03 | 高分子電解質組成物ならびにそれを用いた高分子電解質膜、膜電極複合体および固体高分子形燃料電池 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US10103401B2 (ja) |
| EP (1) | EP3131145B1 (ja) |
| JP (1) | JP6341204B2 (ja) |
| KR (1) | KR102273035B1 (ja) |
| CN (1) | CN106165175B (ja) |
| CA (1) | CA2944439C (ja) |
| TW (1) | TWI643394B (ja) |
| WO (1) | WO2015156228A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018060789A (ja) * | 2016-09-30 | 2018-04-12 | 東レ株式会社 | 高分子電解質組成物ならびにそれを用いた高分子電解質膜、触媒層付き電解質膜、膜電極複合体、固体高分子形燃料電池、電気化学式水素ポンプおよび水電解式水素発生装置 |
| KR20220157948A (ko) | 2020-03-26 | 2022-11-29 | 도레이 카부시키가이샤 | 페난트롤린 유도체의 결정 및 그 제조 방법, 그리고 그것을 사용한 발광 소자 |
| JP2023522655A (ja) * | 2020-11-12 | 2023-05-31 | コーロン インダストリーズ インク | 高分子電解質膜、その製造方法、及びそれを含む電気化学装置 |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107093750A (zh) * | 2017-04-27 | 2017-08-25 | 高延敏 | 一种组合物及其用途以及锌锰电池 |
| CN111868986B (zh) * | 2018-02-28 | 2024-02-23 | 可隆工业株式会社 | 离子交换膜和包括它的储能装置 |
| US11674011B2 (en) | 2018-08-31 | 2023-06-13 | Lg Chem, Ltd. | Cross-linked polyolefin separator and manufacturing method thereof |
| CN109929117B (zh) * | 2019-01-15 | 2021-04-30 | 浙江大学宁波理工学院 | 一种磷氮型刚性骨架多孔阻燃剂及其制备方法和应用 |
| CN109867795A (zh) * | 2019-03-15 | 2019-06-11 | 上海应用技术大学 | 一种含酚酞的磺化聚芳醚氧膦聚合物及其膜制备方法 |
| CN110204817B (zh) * | 2019-06-25 | 2021-12-17 | 苏州颢冉智能科技有限公司 | 一种无卤阻燃汽车线束用电缆料组合物的制备方法 |
| CN112436168A (zh) * | 2020-11-30 | 2021-03-02 | 山东东岳未来氢能材料股份有限公司 | 长寿命增强型全氟质子膜及其制备方法 |
| CN113477068B (zh) * | 2021-05-30 | 2023-05-16 | 中国人民解放军东部战区疾病预防控制中心 | 香烟烟气活性氧清除剂的制备方法及添加方法 |
| CN113724910B (zh) * | 2021-08-17 | 2023-11-17 | 广东风华高新科技股份有限公司 | 一种铜浆及其制备方法与应用 |
| CN116314986B (zh) * | 2023-01-16 | 2024-06-11 | 山东东岳未来氢能材料股份有限公司 | 一种液流电池膜的添加剂及高性能液流电池膜 |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000011756A (ja) * | 1998-06-22 | 2000-01-14 | Toyota Central Res & Dev Lab Inc | 高耐久性固体高分子電解質 |
| JP2003151346A (ja) * | 2001-08-30 | 2003-05-23 | Sumitomo Chem Co Ltd | 高分子電解質組成物及びその用途 |
| JP2005290318A (ja) * | 2004-04-05 | 2005-10-20 | Teijin Ltd | 固体高分子電解質 |
| WO2006006502A1 (ja) * | 2004-07-09 | 2006-01-19 | Nissan Motor Co., Ltd. | 燃料電池システム及び固体高分子電解質膜 |
| JP2009235260A (ja) * | 2008-03-27 | 2009-10-15 | Tokyo Institute Of Technology | スルホン化ピリジロキシ基またはスルホン酸塩を結合したピリジロキシ基を有する芳香環を構成単位として分子内に有する高分子有機化合物、およびスルホン化ピリジロキシ基またはスルホン酸塩を結合したピリジロキシ基を有する芳香環から成る有機化合物、それらを用いた医薬品、農薬、消毒剤あるいは抗菌剤、イオン交換体、電解質膜、触媒、膜電極接合体、燃料電池 |
| US20110111321A1 (en) * | 2009-11-10 | 2011-05-12 | Daimler Ag | Composite proton conducting membrane with low degradation and membrane electrode assembly for fuel cells |
| WO2014084138A1 (ja) * | 2012-11-27 | 2014-06-05 | 東レ株式会社 | 高分子電解質組成物、およびそれを用いた高分子電解質膜、膜電極複合体および固体高分子型燃料電池 |
Family Cites Families (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE58909860D1 (de) | 1988-04-30 | 1999-11-11 | Akzo Nobel Nv | Verfahren zur Sulfonierung von aromatischen Polyäthersulfonen |
| JPH02208322A (ja) | 1989-02-08 | 1990-08-17 | Kurita Water Ind Ltd | スルホン化樹脂の製造方法 |
| EP1222707A2 (en) * | 1999-09-09 | 2002-07-17 | Danish Power Systems APS | Polymer electrolyte membrane fuel cells |
| TW589760B (en) | 2001-08-09 | 2004-06-01 | Sumitomo Chemical Co | Polymer electrolyte composition and fuel cell |
| JP2003201403A (ja) | 2002-01-09 | 2003-07-18 | Jsr Corp | 高分子電解質組成物およびプロトン伝導膜 |
| DE10228657A1 (de) * | 2002-06-27 | 2004-01-15 | Celanese Ventures Gmbh | Protonenleitende Membran und deren Verwendung |
| US7537857B2 (en) * | 2003-12-17 | 2009-05-26 | Bdf Ip Holdings Ltd. | Reduced degradation of ion-exchange membranes in electrochemical fuel cells |
| JP4733352B2 (ja) | 2004-01-28 | 2011-07-27 | Jsr株式会社 | ポリアリーレン重合体組成物およびその用途 |
| US7259230B2 (en) * | 2004-06-07 | 2007-08-21 | Battelle Energy Alliance, Llc | Polybenzimidazole compounds, polymeric media, and methods of post-polymerization modifications |
| JP2006099999A (ja) | 2004-09-28 | 2006-04-13 | Asahi Glass Co Ltd | 固体高分子形燃料電池用電解質膜、その製造方法及び固体高分子形燃料電池用膜電極接合体 |
| KR101319764B1 (ko) * | 2005-02-15 | 2013-10-17 | 도레이 카부시키가이샤 | 고분자 전해질 성형체의 제조 방법, 고분자 전해질 재료,고분자 전해질막 및 고분자 전해질형 연료 전지 |
| JP4704283B2 (ja) | 2005-06-28 | 2011-06-15 | 住友化学株式会社 | 過酸化物分解触媒 |
| JP2007066882A (ja) | 2005-08-02 | 2007-03-15 | Toyobo Co Ltd | 高分子電解質膜/電極接合体及び燃料電池 |
| GB0700819D0 (en) * | 2006-01-19 | 2007-02-21 | Sumitomo Chemical Co | Polymer electrolyte composition and application thereof |
| CN101601156B (zh) * | 2007-01-10 | 2012-03-28 | 旭硝子株式会社 | 固体高分子电解质膜及固体高分子型燃料电池用膜电极接合体 |
| US20100167160A1 (en) * | 2007-04-25 | 2010-07-01 | Tomoyuki Takane | Method For Producing Polymer Electrolyte Membrane For Solid Polymer Fuel Cell, Membrane Eelctrode Assembly For Solid Polymer Fuel Cell, and Solid Polymer Fuel Cell |
| JP4864107B2 (ja) * | 2009-02-17 | 2012-02-01 | 株式会社日立製作所 | ブロック共重合体並びにこれを用いた燃料電池用固体高分子電解質、燃料電池用固体高分子電解質膜、膜電極接合体及び燃料電池 |
| KR20110020186A (ko) * | 2009-08-21 | 2011-03-02 | 한양대학교 산학협력단 | 고분자 전해질형 연료 전지용 고분자 전해질 막, 이의 제조 방법 및 이를 포함하는 고분자 전해질형 연료 전지 시스템 |
| JP5566645B2 (ja) | 2009-09-08 | 2014-08-06 | 株式会社クラレ | 泥水用分散剤 |
| JP2011057768A (ja) | 2009-09-08 | 2011-03-24 | Uchiyama Manufacturing Corp | アクリルゴム組成物及びその成形品 |
| EP2493890B1 (en) * | 2009-10-30 | 2018-03-21 | Sumitomo Chemical Company, Limited | Nitrogen-containing aromatic compounds and metal complexes |
| EP2499693B1 (en) | 2009-11-10 | 2015-09-09 | Daimler AG | Composite proton conducting electrolyte with improved additives and membrane electrode assembly for fuel cells |
| WO2011065483A1 (ja) * | 2009-11-29 | 2011-06-03 | 国立大学法人豊橋技術科学大学 | 電解質膜、燃料電池、及び電解質膜の製造方法 |
| US8461296B2 (en) * | 2011-03-24 | 2013-06-11 | Basf Se | Method for mechanical stabilization of nitrogen-containing polymers |
| CA2858343C (en) | 2011-12-20 | 2019-04-09 | Toray Industries, Inc. | Polymer electrolyte composition, and polymer electrolyte membrane, membrane electrode assembly, and polymer electrolyte fuel cell each using same |
| KR101325722B1 (ko) | 2012-02-16 | 2013-11-08 | 김태민 | 사용자 입력 노래에 대응한 악보 생성 장치와 그 방법 |
-
2015
- 2015-04-03 JP JP2015529360A patent/JP6341204B2/ja active Active
- 2015-04-03 EP EP15776250.1A patent/EP3131145B1/en active Active
- 2015-04-03 CN CN201580018329.3A patent/CN106165175B/zh active Active
- 2015-04-03 US US15/302,073 patent/US10103401B2/en active Active
- 2015-04-03 KR KR1020167025432A patent/KR102273035B1/ko active IP Right Grant
- 2015-04-03 WO PCT/JP2015/060630 patent/WO2015156228A1/ja active Application Filing
- 2015-04-03 CA CA2944439A patent/CA2944439C/en active Active
- 2015-04-07 TW TW104111109A patent/TWI643394B/zh active
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000011756A (ja) * | 1998-06-22 | 2000-01-14 | Toyota Central Res & Dev Lab Inc | 高耐久性固体高分子電解質 |
| JP2003151346A (ja) * | 2001-08-30 | 2003-05-23 | Sumitomo Chem Co Ltd | 高分子電解質組成物及びその用途 |
| JP2005290318A (ja) * | 2004-04-05 | 2005-10-20 | Teijin Ltd | 固体高分子電解質 |
| WO2006006502A1 (ja) * | 2004-07-09 | 2006-01-19 | Nissan Motor Co., Ltd. | 燃料電池システム及び固体高分子電解質膜 |
| JP2009235260A (ja) * | 2008-03-27 | 2009-10-15 | Tokyo Institute Of Technology | スルホン化ピリジロキシ基またはスルホン酸塩を結合したピリジロキシ基を有する芳香環を構成単位として分子内に有する高分子有機化合物、およびスルホン化ピリジロキシ基またはスルホン酸塩を結合したピリジロキシ基を有する芳香環から成る有機化合物、それらを用いた医薬品、農薬、消毒剤あるいは抗菌剤、イオン交換体、電解質膜、触媒、膜電極接合体、燃料電池 |
| US20110111321A1 (en) * | 2009-11-10 | 2011-05-12 | Daimler Ag | Composite proton conducting membrane with low degradation and membrane electrode assembly for fuel cells |
| WO2014084138A1 (ja) * | 2012-11-27 | 2014-06-05 | 東レ株式会社 | 高分子電解質組成物、およびそれを用いた高分子電解質膜、膜電極複合体および固体高分子型燃料電池 |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018060789A (ja) * | 2016-09-30 | 2018-04-12 | 東レ株式会社 | 高分子電解質組成物ならびにそれを用いた高分子電解質膜、触媒層付き電解質膜、膜電極複合体、固体高分子形燃料電池、電気化学式水素ポンプおよび水電解式水素発生装置 |
| JP7087315B2 (ja) | 2016-09-30 | 2022-06-21 | 東レ株式会社 | 高分子電解質組成物ならびにそれを用いた高分子電解質膜、触媒層付き電解質膜、膜電極複合体、固体高分子形燃料電池、電気化学式水素ポンプおよび水電解式水素発生装置 |
| KR20220157948A (ko) | 2020-03-26 | 2022-11-29 | 도레이 카부시키가이샤 | 페난트롤린 유도체의 결정 및 그 제조 방법, 그리고 그것을 사용한 발광 소자 |
| JP2023522655A (ja) * | 2020-11-12 | 2023-05-31 | コーロン インダストリーズ インク | 高分子電解質膜、その製造方法、及びそれを含む電気化学装置 |
| JP7433471B2 (ja) | 2020-11-12 | 2024-02-19 | コーロン インダストリーズ インク | 高分子電解質膜、その製造方法、及びそれを含む電気化学装置 |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2944439C (en) | 2021-10-26 |
| US10103401B2 (en) | 2018-10-16 |
| EP3131145A1 (en) | 2017-02-15 |
| JP6341204B2 (ja) | 2018-06-13 |
| EP3131145A4 (en) | 2017-10-04 |
| TWI643394B (zh) | 2018-12-01 |
| TW201603383A (zh) | 2016-01-16 |
| CN106165175B (zh) | 2019-11-08 |
| KR102273035B1 (ko) | 2021-07-06 |
| US20170125832A1 (en) | 2017-05-04 |
| EP3131145B1 (en) | 2018-09-05 |
| KR20160142289A (ko) | 2016-12-12 |
| JPWO2015156228A1 (ja) | 2017-04-13 |
| CA2944439A1 (en) | 2015-10-15 |
| CN106165175A (zh) | 2016-11-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6341204B2 (ja) | 高分子電解質組成物ならびにそれを用いた高分子電解質膜、膜電極複合体および固体高分子形燃料電池 | |
| JP6171342B2 (ja) | 高分子電解質組成物、およびそれを用いた高分子電解質膜、膜電極複合体および固体高分子型燃料電池 | |
| JP6156358B2 (ja) | 高分子電解質組成物、およびそれを用いた高分子電解質膜、膜電極複合体および固体高分子型燃料電池 | |
| JP6336601B2 (ja) | 高分子電解質組成物ならびにそれを用いた高分子電解質膜、触媒層付き電解質膜、膜電極複合体および固体高分子型燃料電池 | |
| JP7087315B2 (ja) | 高分子電解質組成物ならびにそれを用いた高分子電解質膜、触媒層付き電解質膜、膜電極複合体、固体高分子形燃料電池、電気化学式水素ポンプおよび水電解式水素発生装置 | |
| KR20240011175A (ko) | 설폰화 아릴 폴리머로부터의 새로운 포스폰화 비-플루오르화 및 부분적으로 플루오르화된 아릴 폴리머 및 퍼플루오로설폰산으로부터의 새로운 고분자 퍼플루오로포스폰산, 이들의 제조 과정 및 일렉트로멤브레인 용도에서의 사용 | |
| AU2022243650A1 (en) | Polymer electrolyte membrane, block copolymer, polymer electrolyte material, polymer electrolyte molded body, electrolyte membrane with catalyst layer, membrane electrode composite, solid polymer fuel cell, and water electrolytic hydrogen generator | |
| JP2009263634A (ja) | 芳香族系高分子の製造方法およびその原料化合物 | |
| Cabasso | Frederick Edward Johnson |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2015529360 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15776250 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20167025432 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2944439 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15302073 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015776250 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2015776250 Country of ref document: EP |