WO2015075939A1 - T細胞受容体およびb細胞受容体レパトアの解析システムならびにその治療および診断への利用 - Google Patents
T細胞受容体およびb細胞受容体レパトアの解析システムならびにその治療および診断への利用 Download PDFInfo
- Publication number
- WO2015075939A1 WO2015075939A1 PCT/JP2014/005849 JP2014005849W WO2015075939A1 WO 2015075939 A1 WO2015075939 A1 WO 2015075939A1 JP 2014005849 W JP2014005849 W JP 2014005849W WO 2015075939 A1 WO2015075939 A1 WO 2015075939A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- region
- tcr
- sequence
- bcr
- gene
- Prior art date
Links
Images





































































































Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6881—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for tissue or cell typing, e.g. human leukocyte antigen [HLA] probes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
- C12Q1/6886—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material for cancer
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B30/00—ICT specially adapted for sequence analysis involving nucleotides or amino acids
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B30/00—ICT specially adapted for sequence analysis involving nucleotides or amino acids
- G16B30/10—Sequence alignment; Homology search
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/156—Polymorphic or mutational markers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/158—Expression markers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/16—Primer sets for multiplex assays
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/172—Haplotypes
Definitions
- the present invention relates to a technique for amplifying a gene generated from a biological sample by gene reconstruction without bias, a system for analyzing the obtained genetic information, and treatment and diagnosis thereof.
- T cells and B cells do not react with their own cells or molecules, but can specifically recognize and attack foreign pathogens such as viruses and bacteria. Therefore, T cells and B cells have a mechanism capable of recognizing and distinguishing various antigens derived from other organisms together with self-antigens by receptor molecules expressed on the cell surface.
- T cell receptor TCR
- BCR receptor BCR
- Intracellular signals are transmitted by stimulation from these antigen receptors, production of inflammatory cytokines and chemokines is enhanced, cell proliferation is enhanced, and various immune responses are initiated.
- TCR recognizes a peptide (peptide-MHC complex, pMHC) bound to the peptide binding groove of a major histocompatibility complex (MHC) expressed on an antigen-presenting cell, and thereby recognizes self and non-self. It recognizes and recognizes an antigenic peptide (Non-patent Document 1).
- TCR is a heterodimeric receptor molecule composed of two TCR polypeptide chains, and there are an ⁇ type TCR expressed by normal T cells and a ⁇ type TCR having a special function.
- ⁇ and ⁇ chain TCR molecules form a complex with a plurality of CD3 molecules (CD3 ⁇ chain, CD3 ⁇ chain, CD3 ⁇ chain, CD3 ⁇ chain), transmit intracellular signals after antigen recognition, and initiate various immune responses.
- Endogenous antigens such as viral antigens that have proliferated in cells with viral infection and cancer antigens derived from cancer cells are presented as antigenic peptides on MHC class I molecules.
- an antigen derived from a foreign microorganism is taken up into an antigen-presenting cell by endocytosis, processed and then presented on an MHC class II molecule.
- These antigens are recognized by TCRs expressed by CD8 + T cells or CD4 + T cells, respectively.
- costimulatory molecules such as CD28, ICOS and OX40 molecules are important for stimulation via TCR molecules.
- the TCR gene has a large number of V regions (variable regions, V), J regions (joining region, J), D regions (diversity region, D) and constant regions C regions (constant regions) encoded in different regions on the genome. , C). In the T cell differentiation process, these gene fragments are rearranged in various combinations.
- the ⁇ -chain and ⁇ -chain TCR are VJ-C genes, and the ⁇ -chain and ⁇ -chain TCRs are VDJ-. A gene consisting of C is expressed.
- TCR V gene fragments there are 43 functional ⁇ chain TCR V gene fragments (TRAV), 50 TCR J gene fragments (TRAJ), and functional ⁇ chain TCR V gene fragments (TRBV).
- TRBD TCR D gene fragment
- TRBJ TCR J gene fragment
- TRGV functional ⁇ chain V gene fragment
- TRGJ TCR J gene fragment
- TRDV Three types of functional ⁇ chain V gene fragments (TRDV), three types of TCR D gene fragments (TRDD), and four types of TCR J gene fragments (TRDJ) are known (Non-patent Document 2). Reconstruction of these gene fragments creates diversity, and insertion or deletion of one or more bases between V and D or D and J gene fragments results in the formation of random amino acid sequences. More diverse TCR gene sequences have been created.
- the region where the TCR molecule and the pMHC complex surface directly bind is composed of the complementarity determining region (CDR) CDR1, CDR2 and CDR3 regions rich in diversity within the V region.
- CDR3 region includes a part of the V region, a VDJ region formed by a random sequence, and a part of the J region, and forms the most diverse antigen recognition site.
- the other region is called FR (framework region) and plays a role of forming a structure that becomes a skeleton of the TCR molecule.
- the ⁇ chain TCR is first reconstituted and associates with the pT ⁇ molecule to form a pre-TCR complex molecule.
- the ⁇ chain TCR is then reconstituted to form an ⁇ TCR molecule and, if no functional ⁇ TCR is formed, reconstitution occurs in the other ⁇ chain TCR gene allele. It is known that a positive or negative selection in the thymus is performed, and a TCR having an appropriate affinity is selected to acquire antigen specificity (Non-patent Document 3).
- BCR is known as immunoglobulin (Ig), and the membrane-bound type of Ig functions as an antigen receptor molecule as BCR, and its secreted protein is secreted extracellularly as an antibody.
- Antibodies are secreted in large quantities from plasma cells (plasma cells) from which B cells have finally differentiated, and bind to pathogen molecules such as viruses and bacteria, and then eliminate pathogens by immune reactions such as complement-binding reactions.
- plasma cells plasma cells
- pathogen molecules such as viruses and bacteria
- complement-binding reactions have a job.
- BCR is expressed on the surface of B cells and, after binding to an antigen, transmits intracellular signals to initiate various immune responses and cell proliferation. The specificity of BCR is borne by the diversity of amino acid sequences at the antigen binding site.
- variable region The sequence of the antigen binding site varies greatly between BCR molecules and is called the variable region (V region).
- the sequence of the constant region (C region) is highly conserved between BCR molecules or antibody molecules, and has an antibody effector function and a receptor signal transduction function.
- An Ig molecule consists of a polypeptide chain of two heavy chains (heavy chain, H chain) and two light chains (light chain, L chain). In one Ig molecule, two H chains, and one H chain and one L chain are linked by a disulfide bond.
- Ig has five different H chain classes (isotypes) called ⁇ chain, ⁇ chain, ⁇ chain, ⁇ chain, and ⁇ chain, which are called IgM, IgA, IgG, IgD, and IgE, respectively.
- IgG type antibodies such as IgA type antibodies involved in mucosal immunity and IgE type antibodies important for allergy, asthma, and atopic dermatitis. It is known that the roles are different. Furthermore, it is known that there are several subclasses of isotypes such as IgG1, IgG2, IgG3, and IgG4. There are two types of L chain, ⁇ chain (IgL) and ⁇ chain (IgK), which can bind to any class of H chains, and it is considered that there is no functional difference (Non-Patent Document 4). ).
- the BCR gene is formed by gene rearrangement that occurs in somatic cells, like the TCR gene.
- the variable region is encoded by being divided into several gene fragments on the genome, and they undergo somatic gene recombination during the differentiation process of cells.
- the gene sequence of the variable region of the H chain consists of a C region (constant region, C) that defines an isotype different from the V region, J region, and D region.
- C constant region
- each gene fragment exists in the genome, it is expressed as a series of VDJC genes by gene rearrangement.
- IgM is first produced by immature B cells. Naive B cells not exposed to antigen co-express IgM and IgD, and after activation upon stimulation with the antigen, the variable region sequences remain as they are, and the C region of IgM C region and the C region sequence of IgG.
- a class switch isotype switch that converts some C ⁇ occurs. Similarly, C ⁇ is converted to the C region (C ⁇ ) of IgA or the C region (C ⁇ ) of IgE to produce IgA and IgG. These class switch recombination will produce the type of antibody needed to eliminate the pathogen where needed.
- Non-Patent Document 5 In the process of proliferation of B cells that have undergone class switching, mutations frequently occur in variable regions of IgG, IgA, or IgE regions (somatic hypermutation, somatic hypermutation). As a result, B cells that have acquired higher specificity for the antigen are further stimulated and proliferated, and antibody-producing B cells having higher specificity are selected through this process (affinity maturation, affinity maturation). (Non-Patent Document 5).
- T cells or B cells produce one type of TCR or BCR with high specificity for a specific antigen. Since a large number of antigen-specific T cells and B cells are present in the living body, various TCR repertoires (repertoires) and BCR repertoires are formed, and can effectively function as a defense mechanism against various pathogens. Therefore, analysis of TCR and BCR repertoire, which are important indicators of immune cell specificity and diversity, is a useful analysis tool for analysis of monoclonality and immune abnormalities. If T cells or B cells proliferate in response to an antigen, it is observed that the ratio of specific TCR or BCR genes increases among various repertoires (increased clonality).
- Non-patent Document 6 Attempts have been made to detect tumorigenicity of lymphoid cells expressing TCR or BCR as an increase in clonality by TCR or BCR repertoire analysis. Further, it has been reported that the frequency of use of a specific V ⁇ chain increases when exposed to a molecule that selectively stimulates a TCR having a specific V ⁇ chain such as a superantigen (Non-patent Document 7). . For the purpose of investigating antigen-specific immune responses, it is also frequently used for analysis of intractable autoimmune diseases caused by immune abnormalities such as rheumatoid arthritis, systemic lupus erythematosus, Sjogren's syndrome, idiopathic thrombocytopenic purpura, etc. Sex has been shown.
- the conventional TCR repertoire analysis is an analysis method for examining how much each T cell in a sample uses each V chain.
- One is a method of analyzing the ratio of T cells expressing individual V ⁇ chains by flow cytometry using a specific V ⁇ chain specific antibody (FACS analysis).
- FACS analysis a specific V ⁇ chain specific antibody
- this method requires a relatively large number of cells, it is useful for analysis of peripheral blood containing a large amount of lymphocytes, but cannot be applied to a sample derived from tissue material.
- no antibody corresponding to all V chains is available at present, a comprehensive analysis cannot be performed.
- TCR repertoire analysis by molecular biological techniques has been devised based on TCR gene information obtained from human genome sequences.
- RNA is extracted from a cell sample, complementary DNA is synthesized, and then the TCR gene is PCR amplified and quantified.
- a method of designing many individual TCR V chain specific primers and individually quantifying them by a real-time PCR method or the like, or a method of simultaneously amplifying these specific primers (Multiple PCR) method has been used.
- Multiple PCR multiple PCR
- the multiple PCR method has a drawback that a difference in amplification efficiency between primers causes a bias during PCR amplification.
- next-generation sequence analysis technology In recent years, it has become possible to determine the base sequence of large-scale genes by the next-generation sequence analysis technology that has advanced rapidly.
- next-generation sequencing analysis technology By PCR amplification of TCR genes from human samples and using next-generation sequencing analysis technology, conventional TCR repertoire analysis, which obtains limited information such as V-chain usage frequency, more detailed gene information at the clone level
- the next generation TCR repertoire analysis method that can be obtained and analyzed can be realized. Under such circumstances, several next-generation TCR repertoire analysis methods have been developed (Patent Documents 1 and 2), and other attempts have been made (Patent Documents 3 to 11).
- the present invention includes (1) a technique for uniformly amplifying TCR or BCR gene sequences generated by gene rearrangement from gene fragments on a plurality of genomes without applying a bias (unbiased gene amplification technique), (2) A technique for analyzing the TCR repertoire and BCR repertoire by determining the base sequence of the TCR or BCR gene amplified by the non-biased gene amplification technique on a large scale by the next generation sequencing method and assigning the V, D, J, and C regions.
- a technique for uniformly amplifying TCR or BCR gene sequences generated by gene rearrangement from gene fragments on a plurality of genomes without applying a bias (unbiased gene amplification technique) (2) A technique for analyzing the TCR repertoire and BCR repertoire by determining the base sequence of the TCR or BCR gene amplified by the non-biased gene amplification technique on a large scale by the next generation sequencing method and assigning the V, D, J, and C regions.
- TCR and BCR create various gene sequences by gene rearrangement of gene fragments of multiple V, D, J, and C regions existing on the genome.
- a large number of primers specific to the V region or J region are created in the same reaction solution or in separate reactions.
- a technique for amplifying in liquid is widely used.
- PCR amplification in which a few genes are amplified exponentially, a difference in amplification efficiency between primers becomes a fatal problem.
- the primers set in the V region and the J region need to correspond to all known allele sequences.
- BCR point mutations are introduced with high frequency (about 20%) into the variable region of IgG, IgA or IgE by the mechanism of somatic hypermutation. Therefore, if a 20-base primer is set, about 4 bases are mismatched and it is difficult to achieve uniform gene amplification by the conventional method. That is, in the existing method of designing a V chain specific primer based on the genome sequence, mismatch with the actual BCR gene sequence cannot be avoided, and quantitative gene amplification is not guaranteed. Furthermore, BCR has isotypes and subclasses defined by C region sequences. It is necessary to develop a quantification method for each isotype or subclass using the difference in nucleotide sequence between these isotypes or subclasses.
- the inventors of the present invention used all the isotypes and a single set of primers consisting of one forward primer and one reverse primer.
- a TCR or BCR gene containing a subtype gene was amplified without changing the presence frequency, and a method for determining a base sequence using a next-generation sequence on a large scale was completed.
- a gene containing all V regions is amplified by adding an adapter sequence to its 5 ′ end without setting a primer in the highly diverse V region. .
- This adapter has an arbitrary length and sequence on the base sequence, and about 20 base pairs is optimal, but sequences of 10 to 100 bases can be used.
- the adapter added to the 3 'end is removed by a restriction enzyme and all TCR or BCR genes are amplified by amplifying with an adapter primer of the same sequence as the 20 base pair adapter and a reverse primer specific to the C region which is a common sequence. Amplify.
- Complementary strand DNA is synthesized from the TCR or BCR gene messenger RNA by reverse transcriptase, and then double-stranded complementary DNA is synthesized. Double-stranded complementary DNAs containing V regions of different lengths are synthesized by reverse transcription reaction or double-stranded synthesis reaction, and an adapter consisting of 20 base pairs and 10 base pairs at the 5 ′ end of these genes is subjected to DNA ligase reaction. Add by.
- a reverse primer can be set to amplify these genes.
- the reverse primers set in the C region correspond to the sequences of C ⁇ , C ⁇ , C ⁇ , and C ⁇ for TCR, match the sequences of C ⁇ , C ⁇ , C ⁇ , C ⁇ , and C ⁇ for BCR, and others.
- a primer having a mismatch that does not prime is set in the C region sequence.
- the reverse primer in the C region is optimally prepared taking into account the base sequence, base composition, DNA melting temperature (Tm), and the presence or absence of a self-complementary sequence so that amplification with the adapter primer is possible.
- IgG subtype ( ⁇ 1, ⁇ 2, ⁇ 3, ⁇ 4) and IgA subtype ( ⁇ 1, ⁇ 2) can be amplified with the same primers, and the subtype can be determined by sequencing.
- Alleles can be uniformly amplified by setting primers in the region excluding base sequences that differ between allele sequences in the C region sequence.
- the length (number of bases) of the candidate primer sequence is not particularly limited with respect to a sequence in which none of the primers includes a sequence that differs between allele sequences, but is 10 to 100 bases, preferably 15 to 50 bases. The number is more preferably 20 to 30 bases. Accordingly, the present invention also provides the following. ⁇ In silico> In one aspect, the present invention relates to a technique for analyzing a TCR or BCR repertoire based on a group of expressed TCR or BCR gene sequences derived from a biological sample.
- a sequence of nucleic acids of V (-D) -JC does not depend on the sequencer model. Classification is possible without being biased. Either the positive strand or the complementary strand can be input.
- reference sequences that serve as the basis for classification
- a method of setting a reference database for each of V, D, and J is also conceivable.
- V the difference from the reference sequence is increased due to random mutation, and the region for D and J is short.
- the possibility of oversight cannot be ignored in the homology search method.
- a method of translating the entire nucleic acid sequence to be analyzed into an amino acid sequence and classifying it as a material can be considered, but it is particularly vulnerable to sequencing / deleting sequencing errors. The correspondence becomes unclear, making it difficult to use known information.
- the reference database used in the present invention is prepared for each V, D, J (in addition to C in the case of BCR) gene region.
- nucleic acid sequence data sets for each region and allele published by IMGT are used, but not limited thereto, any data set in which a unique ID is assigned to each sequence can be used.
- the input sequence set used in the present invention is generally trimmed from an adapter sequence and a low quality region in advance, and only a sequence having a length sufficient for analysis is extracted to constitute a high quality set. This step is not necessary, but is used in the preferred embodiment. This is because the LQ sequence just becomes “classification impossible” even if it is as it is.
- the input sequence set used in the present invention performs homology search with a reference database for each gene region, and records the closest reference allele and its alignment with the sequence.
- an algorithm with high mismatch tolerance is used for the homology search.
- general BLAST is used as a homology search program
- settings such as a reduction in window size, a reduction in mismatch penalty, and a reduction in gap penalty are performed for each region.
- the homology score, alignment length, kernel length (length of consecutively matched base sequences) and the number of matching bases are used as indices, and these are applied according to a predetermined priority order.
- the CDR3 sequence is extracted with the CDR3 head on the reference V and the CDR3 end on the reference J as markers. By translating this into an amino acid sequence, it is used for classification of the D region.
- the reference database for the D region is prepared, a combination of the homology search result and the amino acid sequence translation result is set as the classification result.
- the present invention provides the following.
- a method for analyzing a TCR or BCR repertoire comprising the following steps: (1) Providing a reference database for each gene region including at least one of V region, D region, J region and optionally C region: (2) Step of providing an input sequence set obtained by performing trimming as necessary and extracting an appropriate length as necessary; (3) performing a homology search with the reference database for each gene region with respect to the input sequence set, and recording an alignment with the approximate reference allele and / or the sequence of the reference allele; (4) assigning a V region and a J region with respect to the input sequence set, and extracting a nucleic acid sequence of the D region based on the assignment result; (5) translating the nucleic acid sequence of the D region into an amino acid sequence, and classifying the D region using the amino acid sequence; (6) Based on the classification in (5), the TCR or BCR repertoire is calculated by calculating the appearance frequency of each of the V region, the D region, the J region, and if necessary, the C region, or a combination thereof.
- the gene region includes all of a V region, a D region, a J region, and, if necessary, a C region.
- the reference database is a database in which a unique ID is assigned to each sequence.
- the input array set is a non-biased array set.
- the array set is trimmed.
- ⁇ 6> The trimming deletes the low quality region from both ends of the lead; deletes the region matching the adapter sequence by 10 bp or more from both ends of the lead; and if the remaining length is 200 bp or more (TCR) or 300 bp or more (BCR) Item 6.
- ⁇ 7> The method according to item ⁇ 6>, wherein the low quality is a QV value having a 7 bp moving average of less than 30.
- ⁇ 8> The method according to any one of items ⁇ 1> to ⁇ 7>, wherein the approximate sequence is the closest sequence.
- the approximate sequence is as follows. 1. Number of matching bases 2.
- step (5) if there is a D region reference database, the combination of the homology search result with the CDR3 nucleic acid sequence and the amino acid sequence translation result is used as the classification result, items ⁇ 1> to ⁇ 13>
- ⁇ 15> The method according to any one of items ⁇ 1> to ⁇ 14>, wherein in step (5), when there is no D region reference database, classification is performed based only on the appearance frequency of the amino acid sequence. .
- step (5) when there is no D region reference database, classification is performed based only on the appearance frequency of the amino acid sequence. .
- ⁇ 16> The method according to any one of items ⁇ 1> to ⁇ 15>, wherein the appearance frequency is determined in gene name units and / or allyl units.
- the step (4) includes a step of assigning a V region and a J region to the input sequence set, and extracting a CDR3 sequence with the CDR3 head on the reference V region and the CDR3 end on the reference J as markers.
- the method according to any one of items ⁇ 1> to ⁇ 16>. ⁇ 18> Any of items ⁇ 1> to ⁇ 17>, wherein the step (5) includes a step of translating the CDR3 nucleic acid sequence into an amino acid sequence and classifying the D region using the amino acid sequence. 2. The method according to item 1.
- a system for analyzing a TCR or BCR repertoire comprising: (1) Means for providing a reference database for each gene region including at least one of a V region, a D region, a J region, and if necessary, a C region: (2) Means for providing an input sequence set obtained by performing trimming as necessary and extracting an appropriate length as necessary; (3) Means for performing homology search with the reference database for each gene region for the input sequence set and recording an alignment with the approximate reference allele and / or the sequence of the reference allele; (4) Means for assigning a V region and a J region for the input sequence set, and extracting a nucleic acid sequence of the D region based on the assignment result; (5) means for translating the nucleic acid sequence of the D region into an amino acid sequence and classifying the D region using the amino acid sequence; (6) Means for deriving a TCR or a BCR repertoire by calculating an appearance frequency of each of the V region, the D region, the J region, and if necessary, the C region
- ⁇ 19A> The system according to item ⁇ 19>, including any one or more of the items ⁇ 1> to ⁇ 18>.
- a recording medium storing a computer program that causes a computer to execute processing of a method of analyzing a TCR or BCR repertoire, the method including the following steps: (1) Providing a reference database for each gene region including at least one of V region, D region, J region and optionally C region: (2) Step of providing an input sequence set obtained by performing trimming as necessary and extracting an appropriate length as necessary; (3) performing a homology search with the reference database for each gene region with respect to the input sequence set, and recording an alignment with the approximate reference allele and / or the sequence of the reference allele; (4) assigning a V region and a J region with respect to the input sequence set, and extracting a nucleic acid sequence of the D region based on the assignment result; (5) translating the nucleic acid sequence of the D region into an amino acid sequence, and classifying the D region using the amino acid sequence; (6) Deriving a T
- the present invention provides (1) a technique (unbiased gene amplification technique) for uniformly amplifying a TCR or BCR gene sequence generated by gene rearrangement from gene fragments on a plurality of genomes without applying a bias. ), (2) TCR or BCR gene amplified by the non-biased gene amplification technique is determined on a large scale by next-generation sequencing, and V, D, J, and C regions are assigned to perform TCR repertoire and BCR It is a technique for analyzing repertoire.
- a technique unbiased gene amplification technique for uniformly amplifying a TCR or BCR gene sequence generated by gene rearrangement from gene fragments on a plurality of genomes without applying a bias.
- TCR or BCR gene amplified by the non-biased gene amplification technique is determined on a large scale by next-generation sequencing, and V, D, J, and C regions are assigned to perform TCR repertoire and BCR It is a technique for analyzing repertoire.
- TCR and BCR create various gene sequences by gene rearrangement of gene fragments of multiple V, D, J, and C regions existing on the genome.
- a large number of primers specific to the V region or J region are created in the same reaction solution or in separate reactions.
- a technique for amplifying in liquid is widely used.
- PCR amplification in which a few genes are amplified exponentially, a difference in amplification efficiency between primers becomes a fatal problem.
- the primers set in the V region and the J region need to correspond to all known allele sequences.
- BCR point mutations are introduced with high frequency (about 20%) into the variable region of IgG, IgA or IgE by the mechanism of somatic hypermutation. Therefore, if a 20-base primer is set, about 4 bases are mismatched and it is difficult to achieve uniform gene amplification by the conventional method. That is, in the existing method of designing a V chain specific primer based on the genome sequence, mismatch with the actual BCR gene sequence cannot be avoided, and quantitative gene amplification is not guaranteed. Furthermore, BCR has isotypes and subclasses defined by C region sequences. It is necessary to develop a quantification method for each isotype or subclass using the difference in nucleotide sequence between these isotypes or subclasses.
- the inventors of the present invention used all the isotypes and a single set of primers consisting of one forward primer and one reverse primer.
- a TCR or BCR gene containing a subtype gene was amplified without changing the presence frequency, and a method for determining a base sequence using a next-generation sequence on a large scale was completed.
- a gene containing all V regions is amplified by adding an adapter sequence to its 5 ′ end without setting a primer in the highly diverse V region. .
- This adapter has an arbitrary length and sequence on the base sequence, and about 20 base pairs is optimal, but sequences of 10 to 100 bases can be used.
- the adapter added to the 3 'end is removed by a restriction enzyme and all TCR or BCR genes are amplified by amplifying with an adapter primer of the same sequence as the 20 base pair adapter and a reverse primer specific to the C region which is a common sequence. Amplify.
- Complementary strand DNA is synthesized from the TCR or BCR gene messenger RNA by reverse transcriptase, and then double-stranded complementary DNA is synthesized. Double-stranded complementary DNAs containing V regions of different lengths are synthesized by reverse transcription reaction or double-stranded synthesis reaction, and an adapter consisting of 20 base pairs and 10 base pairs at the 5 ′ end of these genes is subjected to DNA ligase reaction. Add by.
- a reverse primer can be set to amplify these genes.
- the reverse primers set in the C region correspond to the sequences of C ⁇ , C ⁇ , C ⁇ , and C ⁇ for TCR, match the sequences of C ⁇ , C ⁇ , C ⁇ , C ⁇ , and C ⁇ for BCR, and others.
- a primer having a mismatch that does not prime is set in the C region sequence.
- the reverse primer in the C region is optimally prepared taking into account the base sequence, base composition, DNA melting temperature (Tm), and the presence or absence of a self-complementary sequence so that amplification with the adapter primer is possible.
- IgG subtype ( ⁇ 1, ⁇ 2, ⁇ 3, ⁇ 4) and IgA subtype ( ⁇ 1, ⁇ 2) can be amplified with the same primers, and the subtype can be determined by sequencing.
- Alleles can be uniformly amplified by setting primers in the region excluding base sequences that differ between allele sequences in the C region sequence.
- the length (number of bases) of the candidate primer sequence is not particularly limited with respect to a sequence in which none of the primers includes a sequence that differs between allele sequences, but is 10 to 100 bases, preferably 15 to 50 bases. The number is more preferably 20 to 30 bases. Accordingly, the present invention also provides the following.
- ⁇ A1> A method for preparing a sample for quantitative analysis of a repertoire of a variable region of a T cell receptor (TCR) or a B cell receptor (BCR) by gene sequence analysis using a database.
- a step of synthesizing complementary DNA using an RNA sample derived from a target cell as a template (2) a step of synthesizing a double-stranded complementary DNA using the complementary DNA as a template; (3) adding a common adapter primer sequence to the double-stranded complementary DNA to synthesize an adapter-added double-stranded complementary DNA; (4) A first PCR amplification reaction is performed using the adapter-added double-stranded complementary DNA, the common adapter primer comprising the common adapter primer sequence, and the first TCR or BCR C region-specific primer.
- the C region specific primer of the first TCR or BCR is It contains a sequence that is sufficiently specific for the target C region of the TCR or BCR, has no homology to other gene sequences, and contains a mismatched base between subtypes downstream when amplified.
- a step of performing a second PCR amplification reaction using the PCR amplification product of (4), the common adapter primer, and a second TCR or BCR C region-specific primer The TCR or BCR C region-specific primer has a sequence perfectly matched to the TCR or BCR C region in a sequence downstream from the sequence of the first TCR or BCR C region-specific primer, but other genes
- the PCR amplification product of (5), the additional common adapter primer containing the first additional adapter nucleic acid sequence in the nucleic acid sequence of the common adapter primer, the second additional adapter nucleic acid sequence and the molecular identification (MID Tag) sequence A third PCR amplification reaction using a third TCR or BCR C region-specific primer with an adapter to which is added to the C region specific sequence of the third TCR or BCR,
- the first additional adapter nucleic acid sequence is a sequence suitable for binding to a DNA capture bead and emPCR reaction;
- the second additional adapter nucleic acid sequence is a sequence suitable for an emPCR reaction;
- the molecular identification (MID Tag) sequence is a sequence for imparting uniqueness so that an amplification product can be identified; Including the method.
- the C region-specific primer contains a sequence perfectly matched to the target isotype C region selected from the group consisting of IgM, IgA, IgG, IgE and IgD, and other C If the region has no homology, and for IgA or IgG, it is a sequence that perfectly matches a subtype that is either IgG1, IgG2, IgG3 or IgG4, or either IgA1 or IgA2, or is a TCR
- the C region specific primer perfectly matches the C region of the target chain selected from the group consisting of ⁇ chain, ⁇ chain, ⁇ chain, and ⁇ chain, and has no homology to other C regions.
- the method according to item ⁇ A1> which is an array.
- ⁇ A3> The method according to item 1 or ⁇ A2>, wherein the C region-specific primer selects a sequence portion that completely matches all C region allele sequences of the same isotype in the database.
- the common adapter primer is designed so that homodimer and intramolecular hairpin structures are difficult to form and can form a double strand stably, and is not highly homologous to all TCR gene sequences in the database, and The method according to any one of items ⁇ A1> to ⁇ A3>, which is designed to have the same melting temperature (Tm) as that of the C region-specific primer.
- Tm melting temperature
- the common adapter primer is designed not to have a homodimer and an intramolecular hairpin structure, and is selected to have no homology to other genes including BCR or TCR.
- Method. ⁇ A6> The method according to item ⁇ A5>, wherein the common adapter primer is P20EA (SEQ ID NO: 2) and / or P10EA (SEQ ID NO: 3).
- the first, second and third TCR or BCR C region-specific primers are each independently for BCR repertoire analysis, and are IgM, IgG, IgA, IgD, or IgE.
- the common adapter primer sequence has a base length suitable for amplification, is difficult to form a homodimer and an intramolecular hairpin structure, can be stably formed into a double strand, and has all TCR gene sequences in the database.
- Each of the first, second and third TCR or BCR C region-specific primers is independently for TCR or BCR repertoire analysis, and each primer is one ⁇ chain (TRAC) Sequences that perfectly match two types of ⁇ chains (TRBC01 and TRBC02), two types of ⁇ chains (TRGC1 and TRGC2), and one type of ⁇ chain (TRDC1). Selected so as to include a mismatched base between subtypes downstream of the primer,
- the common adapter primer sequence has a base length suitable for amplification, is difficult to form a homodimer and an intramolecular hairpin structure, can be stably formed into a double strand, and has all TCR gene sequences in the database. Item 6.
- the method according to any one of items ⁇ A1> to ⁇ A7> which is designed so as not to have high homology and to have the same Tm as that of the C region-specific primer.
- the C region specific primer of the third TCR or BCR is set to a region of about 150 bases from the C region 5 ′ end side, and the first TCR or BCR C region specific primer and the second TCR or BCR C region specific primer.
- the first, second and third TCR or BCR C region-specific primers are each independently for quantitative analysis of BCR, Designed to have specific primers separately for the five isotype sequences, perfectly matched to the target sequence, and to ensure a mismatch of 5 bases or more for other isotypes, and similar IgG subtypes (IgG1 , IgG2, IgG3 and IgG4) or IgA subtypes (IgA1 and IgA2) are designed to perfectly match all subtypes so that they can be handled by one primer each, items ⁇ A1> to ⁇ A9 The method of any one of>.
- the primer design parameters are set to base sequence length 18-22 bases, melting temperature 54-66 ° C.,% GC (% guanine / cytosine content) 40-65%, items ⁇ A1> to ⁇ A10 The method of any one of>.
- the primer design parameters were set to base sequence length 18-22 bases, melting temperature 54-66 ° C,% GC (% guanine / cytosine content) 40-65%, self-annealing score 26, self-terminal annealing
- the method according to any one of items ⁇ A1> to ⁇ A11>, which is set to a score of 10 and a secondary structure score of 28.
- ⁇ A13> The following conditions 1.
- the C region specific primer of the first TCR or BCR is 41 to 300 bases based on the first base of the first codon of the C region sequence generated by splicing, and the C region specific of the second TCR or BCR.
- the primary primer is up to 21-300 bases based on the first base
- the third TCR or BCR C region specific primer is within 150 bases based on the first base
- subtype and / or allele mismatch The method according to any one of items ⁇ A1> to ⁇ A13>, which is set at a position including a part.
- the C region specific primer of the first TCR or BCR has the following structures: CM1 (SEQ ID NO: 5), CA1 (SEQ ID NO: 8), CG1 (SEQ ID NO: 11), CD1 (SEQ ID NO: 14), CE1
- CM1 SEQ ID NO: 5
- CA1 SEQ ID NO: 8
- CG1 SEQ ID NO: 11
- CD1 SEQ ID NO: 14
- CE1 CE1
- the second TCR or BCR C region-specific primer has the following structure: CM2 (SEQ ID NO: 6), CA2 (SEQ ID NO: 9), CG2 (SEQ ID NO: 12), CD2 (SEQ ID NO: 15), CE2
- CM2 SEQ ID NO: 6
- CA2 SEQ ID NO: 9
- CG2 SEQ ID NO: 12
- CD2 SEQ ID NO: 15
- CE2 CE2
- the third TCR or BCR C region-specific primer has the following structure: CM3-GS (SEQ ID NO: 7), CA3-GS (SEQ ID NO: 10), CG3-GS (SEQ ID NO: 13), CD3- The method according to any one of items ⁇ A1> to ⁇ A16>, which has GS (SEQ ID NO: 16) or CE3-GS (SEQ ID NO: 19).
- ⁇ A18> The TCR or BCR C region-specific primer according to any one of items ⁇ A1> to ⁇ A17>, which is provided in a set corresponding to all subclasses of TCR or BCR. Method.
- ⁇ A19> A method for performing gene analysis using the sample produced by the method according to any one of items ⁇ A1> to ⁇ A18>.
- ⁇ A20> The method according to item ⁇ A19>, wherein the gene analysis is a quantitative analysis of a repertoire of a variable region of a T cell receptor (TCR) or a B cell receptor (BCR).
- TCR T cell receptor
- BCR B cell receptor
- ⁇ Analysis system> A method of quantitatively analyzing a subject's T cell receptor (TCR) or B cell receptor (BCR) variable region repertoire using a ⁇ B1> database, the method comprising: (1) providing a nucleic acid sample comprising a nucleic acid sequence of a T cell receptor (TCR) or a B cell receptor (BCR), amplified non-biased from the subject; (2) determining the nucleic acid sequence contained in the nucleic acid sample; and (3) calculating the appearance frequency of each gene or a combination thereof based on the determined nucleic acid sequence, and deriving the TCR or BCR repertoire of the subject Including the method.
- TCR T cell receptor
- BCR B cell receptor
- the nucleic acid sample includes a plurality of types of nucleic acid sequences of T cell receptor (TCR) or B cell receptor (BCR), and the item (2) is a method in which the nucleic acid sequence is determined by single sequencing.
- TCR T cell receptor
- BCR B cell receptor
- the item (2) is a method in which the nucleic acid sequence is determined by single sequencing.
- ⁇ B3> In the single sequencing, in the amplification from the nucleic acid sample to the sequencing sample, at least one of the sequences used as a primer has the same sequence as the nucleic acid sequence encoding the C region or its complementary strand.
- ⁇ B4> The method according to item ⁇ B2> or ⁇ B3>, wherein the single sequencing is performed using a common adapter primer.
- ⁇ B5> The method according to any one of items ⁇ B1> to ⁇ B4>, wherein the non-biased amplification is not V-region specific amplification.
- ⁇ B6> The method according to any one of items ⁇ B1> to ⁇ B5>, wherein the repertoire is a repertoire of a BCR variable region, and the nucleic acid sequence is a BCR nucleic acid sequence.
- ⁇ B7> A method for analyzing the disease, disorder or condition of the subject based on the TCR or BCR repertoire derived based on any one of items ⁇ B1> to ⁇ B6>.
- ⁇ B8> The method according to item ⁇ B7>, wherein the subject's disease, disorder, or condition is selected from the group consisting of blood tumors and colorectal cancer.
- a method for treating or preventing a disease, disorder or condition in the subject comprising selecting a means for proper treatment or prevention.
- ⁇ B10> The method according to item ⁇ B9>, wherein the disease, disorder, or condition of the subject is selected from the group consisting of a blood tumor and a colorectal cancer.
- a system for quantitatively analyzing a subject's T cell receptor (TCR) or B cell receptor (BCR) variable region repertoire using a ⁇ B11> database comprising: (1) A kit for providing a nucleic acid sample containing a nucleic acid sequence of a T cell receptor (TCR) or a B cell receptor (BCR) amplified from the subject in a biased manner; (2) an apparatus for determining the nucleic acid sequence contained in the nucleic acid sample; and (3) An apparatus for calculating the appearance frequency of each gene or a combination thereof based on the determined nucleic acid sequence and deriving the TCR or BCR repertoire of the subject
- a system comprising: ⁇ B12>
- the nucleic acid sample includes the nucleic acid sequences of a plurality of types of T cell receptors (TCR) or B cell receptors (BCR), and (2) is determined by single sequencing.
- the system according to item ⁇ B11>. ⁇ B13> The item ⁇ B12> is characterized in that in the single sequencing, at least one of the sequences used as primers in the amplification from the nucleic acid sample to the sequencing sample has the same sequence as the C region.
- System. ⁇ B14> The system according to item ⁇ B12> or ⁇ B13>, wherein the single sequencing is performed using a common adapter primer.
- ⁇ B15> The system according to any one of items ⁇ B11> to ⁇ B14>, wherein the non-biased amplification is not V region specific amplification.
- ⁇ B16> The system according to any one of items ⁇ B11> to ⁇ B15>, wherein the repertoire is a repertoire of a BCR variable region, and the nucleic acid sequence is a BCR nucleic acid sequence.
- ⁇ B17> The system according to any one of items ⁇ B11> to ⁇ B16>, and means for analyzing the disease, disorder or condition of the subject based on the TCR or BCR repertoire derived based on the system; A system for analyzing a disease, disorder or condition of a subject.
- ⁇ B18> The system according to item ⁇ B17>, wherein the subject's disease, disorder, or condition is selected from the group consisting of a blood tumor and a colorectal cancer.
- ⁇ B19> Means for quantitatively associating the TCR or BCR repertoire with the disease, disorder or condition of the subject determined by the system according to item ⁇ B17> or ⁇ B18>, and the quantitative relationship,
- a means for treating or preventing a disease, disorder or condition in said subject comprising means for selecting a means for proper treatment or prevention.
- ⁇ B20> The system according to item ⁇ B19>, wherein the subject's disease, disorder, or condition is selected from the group consisting of blood tumors and colon cancer.
- T-LGL Monoclonal T cells that express T cell large granular lymphocytic leukemia
- TRAC ⁇ / TRAJ15 / CVVRATGTALIFG (SEQ ID NO: 1450) or a nucleic acid encoding the same in TCR ⁇ and / or TRBV29-1 / TRBJ2-7 / CSVERGGSLGEQYFG (SEQ ID NO: 1500) or a nucleic acid encoding the same in TCR ⁇ , Use as a diagnostic indicator of cellular large granular lymphocytic leukemia (T-LGL).
- T-LGL T-cell large granular lymphocytic leukemia
- TRAV10 / TRAJ15 / CVVRATGITALIFG (SEQ ID NO: 1450) in TCR ⁇ or a nucleic acid encoding the same, and / or TRBV29-1 / TRBJ2-7 / CSVERGGSLGEQYFG (SEQ ID NO: 1500) in TCR ⁇ or the nucleic acid encoding the same Detection agent.
- T-LGL T-cell large granular lymphocytic leukemia
- ⁇ B27> A peptide that is an indicator of mucosa-associated invariant T (MAIT) cells, comprising a sequence selected from the group consisting of SEQ ID NOs: 1648 to 1651, 1653 to 1654, 1666 to 1667, 1844 to 1848, and 1851.
- ⁇ B28> A nucleic acid encoding the peptide according to item ⁇ B27>.
- ⁇ B29> Use of the peptide according to item ⁇ B27> or ⁇ B28> or a nucleic acid encoding the peptide as a diagnostic index for colorectal cancer.
- ⁇ B30> A peptide that is an indicator of natural killer T cells (NKT), comprising the sequence shown in SEQ ID NO: 1668.
- ⁇ B31> A nucleic acid encoding the peptide according to item ⁇ B30>.
- ⁇ B32> Use of the peptide according to item ⁇ B30> or ⁇ B31> or a nucleic acid encoding the peptide as a diagnostic index for colorectal cancer.
- ⁇ B33> A peptide that is specific for colon cancer, comprising a sequence selected from the group consisting of SEQ ID NOs: 1652, 1655 to 1665, 1669 to 1843, 1849 to 1850, and 1852 to 1860.
- ⁇ B34> A nucleic acid encoding the peptide according to item ⁇ B33>.
- ⁇ B35> Use of the peptide according to item ⁇ B33> or ⁇ B34> or a nucleic acid encoding the peptide as a diagnostic index for colorectal cancer.
- ⁇ B36> a peptide that is specific for colorectal cancer, comprising a sequence selected from the group consisting of SEQ ID NOs: 1861 to 1865 and 1867 to 1909 ⁇ B37> A nucleic acid encoding the peptide according to item ⁇ B36>.
- ⁇ B38> Use of the peptide according to item ⁇ B36> or ⁇ B37> or a nucleic acid encoding the peptide as a diagnostic index for colorectal cancer.
- ⁇ B39> Cell population, T cell line, or recombination in which T cells having a peptide according to item ⁇ B33>, ⁇ B34>, ⁇ B36> or ⁇ B37> or a nucleic acid sequence encoding the peptide are induced at high frequency Expressed T cells.
- ⁇ B40> A therapeutic agent for colorectal cancer comprising the cell population, T cell line or T cell according to item ⁇ B39>.
- ⁇ B41> A method for treating or preventing colorectal cancer using the cell population, T cell line or T cell according to item ⁇ B39>.
- ⁇ B42> Using the method according to any one of items ⁇ B1> to ⁇ B10> or the system according to any one of items ⁇ B11> to ⁇ B20> to detect the frequency of use of the V gene how to.
- ⁇ B43> A method for detecting the frequency of use of the J gene using the method according to any one of items ⁇ B1> to ⁇ B10> or the system according to items ⁇ B11> to ⁇ B20>.
- ⁇ B44> Using the method described in any one of items ⁇ B1> to ⁇ B10> or the system described in items ⁇ B11> to ⁇ B20>, the frequency of subtype frequency analysis (BCR) is determined. How to detect.
- ⁇ B45> A method of analyzing a CDR3 sequence length pattern using the method according to any one of items ⁇ B1> to ⁇ B10> or the system according to items ⁇ B11> to ⁇ B20>.
- ⁇ B46> A method of analyzing the clonality of TCR or BCR using the method described in any one of items ⁇ B1> to ⁇ B10> or the system described in items ⁇ B11> to ⁇ B20>.
- ⁇ B47> A method for extracting duplicate leads using the method according to any one of items ⁇ B1> to ⁇ B10> or the system according to items ⁇ B11> to ⁇ B20>.
- ⁇ B48> A method for searching for a disease-specific TCR or BCR clone using the method according to any one of items ⁇ B1> to ⁇ B10> or the system according to items ⁇ B11> to ⁇ B20>.
- ⁇ B49> A method according to any one of items ⁇ B1> to ⁇ B10>, or a method of analyzing a target using a diversity index using the system according to items ⁇ B11> to ⁇ B20>.
- ⁇ B50> Use the method described in any one of items ⁇ B1> to ⁇ B10> or the system described in items ⁇ B11> to ⁇ B20> to support analysis of the target using the diversity index Method.
- ⁇ B51> In item ⁇ B49> or ⁇ B50>, wherein the diversity index is used as an index for measuring the degree of recovery of the immune system after bone marrow transplantation, or as an index for detecting abnormality of immune system cells accompanying hematopoietic tumor The method described.
- the diversity index is based on Shannon-Wiener diversity index (H ′), Simpson diversity index ( ⁇ , 1- ⁇ or 1 / ⁇ ), Pierou equality index (J ′), and Chao1 index.
- ⁇ B54> Use the method described in any one of items ⁇ B1> to ⁇ B10> or the system described in items ⁇ B11> to ⁇ B20> to support analysis of the target using the similarity index Method.
- ⁇ B55> Item ⁇ B53> or ⁇ B54>, wherein the similarity index is used as an evaluation of repertoire similarity between HLA-type matches or mismatches, and evaluation of repertoire similarity between recipient and donor after bone marrow transplantation The method described in 1.
- ⁇ B56> The method according to item ⁇ B53> or ⁇ B54>, wherein the similarity index is selected from the group consisting of a Morisita-Horn index, a Kimoto C ⁇ index, and a Pianka ⁇ index.
- ⁇ B57> (1) is the following process (1-1) a step of synthesizing complementary DNA using an RNA sample derived from a target cell as a template; (1-2) synthesizing double-stranded complementary DNA using the complementary DNA as a template; (1-3) adding a common adapter primer sequence to the double-stranded complementary DNA to synthesize an adapter-added double-stranded complementary DNA; (1-4) The first PCR amplification reaction using the adapter-added double-stranded complementary DNA, the common adapter primer comprising the common adapter primer sequence, and the first TCR or BCR C region specific primer A process of performing The first TCR or BCR C region specific primer comprises a sequence that is sufficiently specific for the C region of interest of the TCR or BCR and has no homology to other gene sequences; and Designed to contain mismatched bases between subtypes downstream when amplified (1-5) A step of performing a second PCR amplification reaction using the PCR amplification product of (1-4), the common adapter primer, and a
- the second TCR or BCR C region specific primer has a perfect match sequence to the TCR or BCR C region in a sequence downstream from the sequence of the first TCR or BCR C region specific primer.
- are designed to contain sequences that are not homologous to other gene sequences and, when amplified, contain mismatched bases between subtypes downstream; and (1-6) PCR amplification product of (1-5), additional common adapter primer containing the first additional adapter nucleic acid sequence in the nucleic acid sequence of the common adapter primer, second additional adapter nucleic acid sequence and molecular identification ( MID Tag) is a step of performing a third PCR amplification reaction using a third TCR or BCR C region specific primer with an adapter to which a sequence is added to the third TCR or BCR C region specific sequence.
- the third TCR or BCR C region-specific primer has a sequence perfectly matched to the TCR or BCR C region in a sequence downstream from the sequence of the second TCR or BCR C region specific primer.
- the first additional adapter nucleic acid sequence is a sequence suitable for binding to a DNA capture bead and emPCR reaction;
- the second additional adapter nucleic acid sequence is a sequence suitable for an emPCR reaction;
- the molecular identification (MID Tag) sequence is a sequence for imparting uniqueness so that an amplification product can be identified; Including The method according to item ⁇ B1>.
- the (1) kit is as follows: (1-1) Means for synthesizing complementary DNA using an RNA sample derived from a target cell as a template; (1-2) Means for synthesizing double-stranded complementary DNA using the complementary DNA as a template; (1-3) means for adding adapter-added double-stranded complementary DNA by adding a common adapter primer sequence to the double-stranded complementary DNA; (1-4) The first PCR amplification reaction using the adapter-added double-stranded complementary DNA, the common adapter primer comprising the common adapter primer sequence, and the first TCR or BCR C region specific primer Means for performing The first TCR or BCR C region specific primer comprises a sequence that is sufficiently specific for the C region of interest of the TCR or BCR and has no homology to other gene sequences; and Means designed to contain mismatched bases between subtypes downstream when amplified (1-5) Means for performing a second PCR amplification reaction using the PCR amplification product of (1-4), the common adapter primer,
- the second TCR or BCR C region specific primer has a perfect match sequence to the TCR or BCR C region in a sequence downstream from the sequence of the first TCR or BCR C region specific primer.
- Means that comprises a sequence that is not homologous to other gene sequences and is designed to contain mismatched bases between subtypes downstream when amplified; and (1-6) PCR amplification product of (1-5), additional common adapter primer containing the first additional adapter nucleic acid sequence in the nucleic acid sequence of the common adapter primer, second additional adapter nucleic acid sequence and molecular identification ( MID Tag) is a means for performing a third PCR amplification reaction using a third TCR or BCR C region-specific primer with an adapter in which a sequence is added to a third TCR or BCR C region specific sequence.
- the third TCR or BCR C region-specific primer has a sequence perfectly matched to the TCR or BCR C region in a sequence downstream from the sequence of the second TCR or BCR C region specific primer.
- the first additional adapter nucleic acid sequence is a sequence suitable for binding to a DNA capture bead and emPCR reaction;
- the second additional adapter nucleic acid sequence is a sequence suitable for an emPCR reaction;
- the molecular identification (MID Tag) sequence is a sequence for imparting uniqueness so that an amplification product can be identified; means; Including The system according to item ⁇ B11>.
- the C region-specific primer contains a sequence perfectly matched to the target isotype C region selected from the group consisting of IgM, IgA, IgG, IgE and IgD, and others Is a sequence that perfectly matches a subtype of either IgG1, IgG2, IgG3, or IgG4, or either IgA1 or IgA2, or has no homology in the C region of In some cases, the C region specific primer perfectly matches the C region of the target chain selected from the group consisting of ⁇ chain, ⁇ chain, ⁇ chain, and ⁇ chain, and has homology to other C regions.
- ⁇ B58-3> The method according to item ⁇ B57> or ⁇ B58-2>, wherein the C region-specific primer selects a sequence part that completely matches all C region allele sequences of the same isotype in the database Or the system according to ⁇ B58> or ⁇ B58-2>.
- the common adapter primer is designed so that it is difficult to form a homodimer and an intramolecular hairpin structure and can form a double strand stably, and is not highly homologous to all TCR gene sequences in the database.
- any one of items ⁇ B57> and ⁇ B58-2> to ⁇ B58-3> which is designed to have the same melting temperature (Tm) as the C region-specific primer or The system according to any one of ⁇ B58> to ⁇ B58-3>.
- Tm melting temperature
- ⁇ B58-5> The common adapter primer is selected not to have a homodimer and an intramolecular hairpin structure, and is selected to have no homology to other genes including BCR or TCR, ⁇ B57> and The method according to any one of ⁇ B58-2> to ⁇ B58-4> or the system according to any one of ⁇ B58> to ⁇ B58-4>.
- the common adapter primer is P20EA (SEQ ID NO: 2) and / or P10EA (SEQ ID NO: 3), any of items ⁇ B57> and ⁇ B58-2> to ⁇ B58-5> Or the system according to any one of ⁇ B58> to ⁇ B58-5>.
- the first, second and third TCR or BCR C region-specific primers are each independently for BCR repertoire analysis, and include IgM, IgG, IgA, IgD, Or a sequence that perfectly matches each isotype C region of IgE, and in the case of IgG and IgA, it is a sequence that also perfectly matches subtypes and has no homology to other sequences contained in the database, And selected such that a mismatched base is included between subtypes downstream of the primer,
- the common adapter primer sequence has a base length suitable for amplification, is difficult to form a homodimer and an intramolecular hairpin structure, can be stably formed into a double strand, and has all TCR gene sequences in the database.
- ⁇ B58-8> The first, second and third TCR or BCR C region-specific primers are each independently for TCR or BCR repertoire analysis.
- Alpha chain (TRAC), two ⁇ chains (TRBC01 and TRBC02), two ⁇ chains (TRGC1 and TRGC2), one sequence of ⁇ chains (TRDC1), and a perfect match, included in the database Selected so as to include a mismatched base between subtypes downstream of the primer,
- the common adapter primer sequence has a base length suitable for amplification, is difficult to form a homodimer and an intramolecular hairpin structure, can be stably formed into a double strand, and has all TCR gene sequences in the database.
- the method according to any one of items ⁇ B57> and ⁇ B58-2> to ⁇ B58-7>, which is designed not to have high homology and to have the same Tm as the C region-specific primer.
- the C region-specific primer of the third TCR or BCR is set to a region of about 150 bases from the C region 5 ′ end, and the first TCR or BCR C region-specific primer and 2 TCR or BCR C region-specific primers are set between C region 5 ′ end and about 300 bases, and any of items ⁇ B57> and ⁇ B58-2> to ⁇ B58-8> The method described in the above or the system described in ⁇ B58> to ⁇ B58-8>.
- the C region-specific primers of the first, second and third TCR or BCR are each independently for quantitative analysis of BCR, Designed to have specific primers separately for the five isotype sequences, perfectly matched to the target sequence, and to ensure a mismatch of 5 bases or more for other isotypes, and similar IgG subtypes (IgG1 , IgG2, IgG3 and IgG4) or IgA subtypes (IgA1 and IgA2), respectively, designed to perfectly match all subtypes so that one primer can correspond, items ⁇ B57> and ⁇ B58 -2> to ⁇ B58-9> or the system according to any of ⁇ B58> to ⁇ B58-9>.
- the primer design parameters are set to a base sequence length of 18-22 bases, a melting temperature of 54-66 ° C., and a% GC (% guanine / cytosine content) of 40-65%, items ⁇ B57> and The method according to any one of ⁇ B58-2> to ⁇ B58-10> or the system according to any one of ⁇ B58> to ⁇ B58-10>.
- ⁇ B58-12> The primer design parameters were set to base sequence length 18-22 bases, melting temperature 54-66 ° C,% GC (% guanine / cytosine content) 40-65%, self-annealing score 26, self The method according to any one of items ⁇ B57> and ⁇ B58-2> to ⁇ B58-11>, or any of ⁇ B58> to ⁇ B58-11> set in the terminal annealing score 10 and the secondary structure score 28 The system described in Crab. ⁇ B58-13> The following conditions 1. Incorporating a plurality of subtype sequences and / or allele sequences into a base sequence analysis software and aligning them; 2. Search for multiple primers that satisfy the parameter conditions in the C region using primer design software; 3.
- the C region-specific primer of the first TCR or BCR is a C region of the second TCR or BCR up to 41-300 bases based on the first base of the first codon of the C region sequence generated by splicing. Region-specific primers are up to 21-300 bases based on the first base, C region-specific primers of the third TCR or BCR are within 150 bases based on the first base, and subtypes and / or alleles.
- the C region specific primer of the first TCR or BCR has the following structure: CM1 (SEQ ID NO: 5), CA1 (SEQ ID NO: 8), CG1 (SEQ ID NO: 11), CD1 (SEQ ID NO: 14) , CE1 (SEQ ID NO: 17), CA1 (SEQ ID NO: 35), or CB1 (SEQ ID NO: 37), the method according to any of items ⁇ B57> and ⁇ B58-2> to ⁇ B58-14> or ⁇ The system according to any one of B58> to ⁇ B58-14>.
- the second TCR or BCR C region-specific primer has the following structure: CM2 (SEQ ID NO: 6), CA2 (SEQ ID NO: 9), CG2 (SEQ ID NO: 12), CD2 (SEQ ID NO: 15) , CE2 (SEQ ID NO: 18), CA2 (SEQ ID NO: 35), or CB2 (SEQ ID NO: 37), the method according to any of items ⁇ B57> and ⁇ B58-2> to ⁇ B58-15> or ⁇ The system according to any one of B58> to ⁇ B58-15>.
- the third TCR or BCR C region-specific primer has the following structure: CM3-GS (SEQ ID NO: 7), CA3-GS (SEQ ID NO: 10), CG3-GS (SEQ ID NO: 13), The method according to any of items ⁇ B57> and ⁇ B58-2> to ⁇ B58-16>, or ⁇ B58> to ⁇ B58, which has CD3-GS (SEQ ID NO: 16) or CE3-GS (SEQ ID NO: 19) The system according to any one of -16>.
- TCR or BCR C region-specific primers are provided in sets corresponding to all subclasses of TCR or BCR, items ⁇ B57> and ⁇ B58-2> to ⁇ B58- 17> or the system according to ⁇ B58> to ⁇ B58-17>.
- ⁇ B58-19> Item ⁇ B57> and a sample produced by the method according to any one of ⁇ B58-2> to ⁇ B58-18> or the system according to any one of ⁇ B58> to ⁇ B58-18>
- Method or system for performing gene analysis using ⁇ B58-20> The method according to any one of items ⁇ B58-19>, wherein the gene analysis is a quantitative analysis of a T cell receptor (TCR) or a repertoire of a variable region of a B cell receptor (BCR). Or system. ⁇ B59> (3) Derivation of the TCR or BCR repertoire is as follows.
- (3-1) Means for providing a reference database for each gene region including at least one of the V region, the D region, the J region, and, if necessary, the C region:
- (3-2) Means for providing an input sequence set obtained by performing trimming as necessary and extracting an appropriate length as necessary;
- (3-3) means for performing a homology search with the reference database for each gene region for the input sequence set and recording an alignment with the approximate reference allele and / or the sequence of the reference allele;
- (3-4) means for assigning a V region and a J region to the input sequence set, and extracting a nucleic acid sequence of the D region based on the assignment result;
- (3-5) Means for translating the nucleic acid sequence of the D region into an amino acid sequence and classifying the D region using the amino acid sequence;
- (3-6) Based on the classification in (3-5), by calculating the appearance frequency of each of the V region, the D region, the J region and, if necessary, the C region, or a combination thereof, the TCR Or means for
- the reference database is a database in which a unique ID is assigned to each sequence. Items ⁇ B57>, ⁇ B58-2> to ⁇ B58-20>, ⁇ B59>, and ⁇ B60-2> Or the system according to any one of ⁇ B58> to ⁇ B58-20>, ⁇ B59>, ⁇ B60> to ⁇ B60-2>.
- the input array set is an unbiased array set, items ⁇ B57>, ⁇ B58-2> to ⁇ B58-20>, ⁇ B59>, and ⁇ B60-2> to ⁇ B60-3> Or the system according to any one of ⁇ B58> to ⁇ B58-20>, ⁇ B59>, ⁇ B60> to ⁇ B60-3>.
- ⁇ B60-5> Any of items ⁇ B57>, ⁇ B58-2> to ⁇ B58-20>, ⁇ B59>, and ⁇ B60-2> to ⁇ B60-4>, wherein the array set is trimmed Or the system according to any one of ⁇ B58> to ⁇ B58-20>, ⁇ B59>, ⁇ B60> to ⁇ B60-4>.
- ⁇ B60-6> The trimming deletes the low quality region from both ends of the lead; deletes the region matching the adapter sequence by 10 bp or more from both ends of the lead; and the remaining length is 200 bp or more (TCR) or 300 bp or more (BCR ), Any of items ⁇ B57>, ⁇ B58-2> to ⁇ B58-20>, ⁇ B59>, and ⁇ B60-2> to ⁇ B60-5> achieved by the step used for analysis as high quality Or the system according to any one of ⁇ B58> to ⁇ B58-20>, ⁇ B59>, ⁇ B60> to ⁇ B60-5>.
- ⁇ B60-7> The low quality is such that the 7 bp moving average of the QV value is less than 30, items ⁇ B57>, ⁇ B58-2> to ⁇ B58-20>, ⁇ B59> and ⁇ B60-2> ⁇
- the approximate sequence is the closest sequence of items ⁇ B57>, ⁇ B58-2> to ⁇ B58-20>, ⁇ B59>, and ⁇ B60-2> to ⁇ B60-7>
- ⁇ B60-9> The approximate sequence is as follows. 1. Number of matching bases 2. kernel length; Score, 4.
- ⁇ B60-10> The homology search is performed under conditions that allow random mutations to be scattered throughout, items ⁇ B57>, ⁇ B58-2> to ⁇ B58-20>, ⁇ B59>, and The method according to any one of ⁇ B60-2> to ⁇ B60-9> or the system according to any one of ⁇ B58> to ⁇ B58-20>, ⁇ B59>, ⁇ B60> to ⁇ B60-9>.
- ⁇ B60-11> In the homology search, (1) the window size is reduced, (2) the mismatch penalty is reduced, (3) the gap penalty is reduced, and (4) the index priority is the top.
- ⁇ B60-13> The D region is classified according to the frequency of appearance of the amino acid sequence, items ⁇ B57>, ⁇ B58-2> to ⁇ B58-20>, ⁇ B59>, and ⁇ B60-2> to ⁇ B60-2>.
- step (5) if there is a D region reference database, the combination of the homology search result with the CDR3 nucleic acid sequence and the amino acid sequence translation result is used as the classification result, item ⁇ B57 >, ⁇ B58-2> to ⁇ B58-20>, ⁇ B59> and ⁇ B60-2> to ⁇ B60-13>, or ⁇ B58> to ⁇ B58-20>, ⁇ B59> ⁇ B60> to ⁇ B60-13>.
- ⁇ B60-15> In the step (5), when there is no D region reference database, classification is performed only by the appearance frequency of the amino acid sequence, items ⁇ B57>, ⁇ B58-2> to ⁇ B58-20 >, ⁇ B59> and ⁇ B60-2> to ⁇ B60-14>, or any of ⁇ B58> to ⁇ B58-20>, ⁇ B59>, ⁇ B60> to ⁇ B60-14> The system described in Crab.
- ⁇ B60-16> The appearance frequency is determined in gene name units and / or allyl units, items ⁇ B57>, ⁇ B58-2> to ⁇ B58-20>, ⁇ B59>, and ⁇ B60-2> to ⁇ B60-2> The method according to any one of B60-15> or the system according to any one of ⁇ B58> to ⁇ B58-20>, ⁇ B59>, ⁇ B60> to ⁇ B60-15>.
- ⁇ B60-17> In the step (4), a V region and a J region are assigned to the input sequence set, and a CDR3 sequence is extracted using the CDR3 head on the reference V region and the CDR3 end on the reference J as markers.
- the step (5) includes the steps of translating the nucleic acid sequence of CDR3 into an amino acid sequence and classifying the D region using the amino acid sequence, ⁇ B57>, ⁇ B58-2 > To ⁇ B58-20>, ⁇ B59> and ⁇ B60-2> to ⁇ B60-17>, or ⁇ B58> to ⁇ B58-20>, ⁇ B59>, ⁇ B60> to ⁇ B B60-17>.
- ⁇ B60-19> (3)
- the apparatus for deriving the TCR or BCR repertoire is as follows.
- (3-1) Means for providing a reference database for each gene region including at least one of the V region, the D region, the J region, and, if necessary, the C region:
- (3-2) Means for providing an input sequence set obtained by performing trimming as necessary and extracting an appropriate length as necessary;
- (3-3) means for performing a homology search with the reference database for each gene region for the input sequence set and recording an alignment with the approximate reference allele and / or the sequence of the reference allele;
- (3-4) means for assigning a V region and a J region to the input sequence set, and extracting a nucleic acid sequence of the D region based on the assignment result;
- (3-5) Means for translating the nucleic acid sequence of the D region into an amino acid sequence and classifying the D region using the amino acid sequence;
- (3-6) Means for deriving TCR or BCR repertoire by calculating the appearance frequency of each of the V region, the D region, the J region, and if necessary, the C region, or a combination thereof in the input
- ⁇ Application example of analysis> ⁇ C1> A method of providing cancer idiotype peptide-sensitized immune cell therapy to a subject, the method comprising: (1) The method according to any one of items ⁇ B1> to ⁇ B10>, ⁇ B57>, ⁇ B58> to ⁇ B58-20>, ⁇ B59>, or ⁇ B60> to ⁇ B60-21> and / or By the system according to any one of items ⁇ B11> to ⁇ B20>, ⁇ B58> to ⁇ B58-20>, ⁇ B59>, or ⁇ B60> to ⁇ B60-21>, the subject's T cell receptor (TCR ) Or analyzing a B cell receptor (BCR) repertoire; (2) A step of determining a TCR or BCR derived from the cancer cell of the subject based on the result of the analysis, wherein the determination is in the frequency ranking of the TCR or BCR gene derived from the cancer cell of the subject.
- TCR T cell receptor
- a step wherein the higher rank sequence is made by selecting as a TCR or BCR from the cancer cell; (3) A step of determining a candidate amino acid sequence of an HLA test peptide based on the determined TCR or BCR derived from the cancer, wherein the determination is a score calculated using an HLA-binding peptide prediction algorithm Made on the basis of a process; (4) synthesizing the determined peptide; A method comprising the step of (5) treating with a synthesized peptide as necessary.
- ⁇ C2> The method according to item ⁇ C1>, wherein the candidate for the HLA test peptide in the step (3) is determined using BIMAS, SYFPEITHI, RANKPEP, or NetMHC.
- ⁇ C3> ⁇ Improved CTL method> After the step (4), the peptide, the dendritic cell or antigen-presenting cell derived from the subject, and the CD8 + T cell derived from the subject are mixed and cultured, and the mixture after the culture is treated with a patient.
- ⁇ C4> ⁇ DC vaccine therapy> After the step (4), the steps comprising mixing the peptide and the dendritic cell derived from the subject and culturing, and administering the cultured mixture to a patient ⁇ C1> to ⁇ The method according to any one of C3>.
- the peptide, the dendritic cell or antigen-presenting cell derived from the subject, and the CD8 + T cell derived from the subject are mixed and cultured to obtain CD8 + T cell-dendritic cell /
- ⁇ D1> isolation of custom-made cancer-specific T cell receptor gene, isolation of cancer-specific TCR gene by in vitro antigen stimulation>
- A an antigen protein or antigen peptide derived from a subject or a lymphocyte derived from the subject or the determined peptide according to any one of items ⁇ C1> to ⁇ C5> and inactivation derived from the subject Mixing tumor cells and T lymphocytes derived from the subject and culturing to produce tumor-specific T cells;
- the TCR of the tumor-specific T cell is the item ⁇ B1> to ⁇ B10>, ⁇ B57>, ⁇ B58> to ⁇ B58-20>, ⁇ B59>, or ⁇ B60> to ⁇ B60-21> According to any of the methods and / or items ⁇ B11> to ⁇ B20>, ⁇ B58> to ⁇ B58-20>, ⁇ B59>, or ⁇ B60> to ⁇ B60-21>
- C a method for is
- ⁇ D1-1> The step comprises mixing and culturing an antigen protein or antigen peptide derived from a subject, inactivated cancer cells derived from the subject, and T lymphocytes derived from the subject, to produce tumor-specific T cells.
- the method according to item ⁇ D1> which is a production step.
- ⁇ D1-2> A process mixes and culture
- Step is a tumor-specific T cell obtained by mixing and culturing the determined peptide according to item C1, inactivated cancer cells derived from the subject, and T lymphocytes derived from the subject.
- the method according to any one of items ⁇ D1> to ⁇ D1-2>, wherein ⁇ D2> ⁇ Isolation of tailor-made cancer-specific T cell receptor gene, isolation of cancer-specific TCR gene by consensus sequence search> (A) isolating lymphocytes or cancer tissue from subjects having a common HLA; (B) analyzing the TCR of the tumor-specific T cell by the method according to item B1 for the lymphocyte or cancer tissue; and (C) a T cell having a sequence common to the tumor-specific T cell.
- a method for isolating a cancer-specific TCR gene by consensus sequence search comprising a step of isolating.
- ⁇ E1> ⁇ CPC> A) collecting T lymphocytes from the patient; B) After antigen stimulation of the T lymphocytes, items ⁇ B1> to ⁇ B10>, ⁇ B57>, ⁇ B58> to ⁇ B58-20>, ⁇ B59>, or ⁇ B60> to ⁇ B60-21> Any of the methods and / or items ⁇ B11> to ⁇ B20>, ⁇ B58> to ⁇ B58-20>, ⁇ B59>, or ⁇ B60> to ⁇ B60-21> Analyzing TCR based on the antigen, wherein the antigen stimulation is performed by an antigen protein or antigen peptide derived from the subject, an inactivated cancer cell derived from the subject, or a tumor-derived idiotype peptide ; C) selecting an optimal antigen and an optimal TCR in the analyzed TCR; D) producing
- ⁇ E1-1> The cell processing therapy according to Item ⁇ E1>, wherein the antigen stimulation is performed by an antigen protein or an antigen peptide derived from the subject.
- ⁇ E1-2> The cell processing therapy according to item ⁇ E1> or ⁇ E1-1>, wherein the antigen stimulation is performed by inactivated cancer cells derived from the subject.
- ⁇ E1-3> The cell processing therapy according to any one of items ⁇ E1> and ⁇ E1-1> to ⁇ E1-2>, wherein the antigen stimulation is performed by the tumor-derived idiotype peptide.
- Step C) is any one of items ⁇ E1>, ⁇ E1-1> to ⁇ E1-4>, comprising selecting an antigen that most strongly activates T cells in an antigen-specific lymphocyte stimulation test.
- Method. ⁇ E1-6> Step C) includes any of items ⁇ B1> to ⁇ B10>, ⁇ B57>, ⁇ B58> to ⁇ B58-20>, ⁇ B59>, or ⁇ B60> to ⁇ B60-21> before and after antigen stimulation.
- the method and / or item ⁇ B11> to ⁇ B20>, ⁇ B58> to ⁇ B58-20>, ⁇ B59>, or ⁇ B60> to ⁇ B60-21> The method according to any one of items ⁇ E1> and ⁇ E1-1> to ⁇ E1-5>, comprising selecting an antigen that most increases the frequency of a specific TCR from a repertoire analysis.
- ⁇ E2> ⁇ CPCRAC> A method for evaluating efficacy and / or safety by conducting an in vitro stimulation test using a cancer-specific TCR gene isolated by the method according to item ⁇ D2>.
- ⁇ CC1> A method of preparing a composition for use in cancer idiotype peptide-sensitized immune cell therapy in a subject, the method comprising: (1) The method according to any one of items ⁇ B1> to ⁇ B10>, ⁇ B57>, ⁇ B58> to ⁇ B58-20>, ⁇ B59>, or ⁇ B60> to ⁇ B60-21> and / or By the system according to any one of items ⁇ B11> to ⁇ B20>, ⁇ B58> to ⁇ B58-20>, ⁇ B59>, or ⁇ B60> to ⁇ B60-21>, the subject's T cell receptor (TCR ) Or analyzing a B cell receptor (BCR) repertoire; (2) A step of determining a TCR or BCR derived from the cancer cell of the subject based on the result of the analysis, wherein the determination is in the frequency ranking of the TCR or BCR gene derived from the cancer cell of the subject.
- TCR T cell receptor
- BCR
- a step wherein the higher rank sequence is made by selecting as a TCR or BCR from the cancer cell; (3) A step of determining a candidate amino acid sequence of an HLA test peptide based on the determined TCR or BCR derived from the cancer, wherein the determination is a score calculated using an HLA-binding peptide prediction algorithm And (4) synthesizing the determined peptide; Including the method.
- ⁇ CC2> The method according to item ⁇ CC1>, wherein the candidate for the HLA test peptide in the step (3) is determined using BIMAS, SYFPEEITHI, RANKPEEP, or NEETMHHC.
- ⁇ CC3> ⁇ Improved CTL method> Item ⁇ CC1>, comprising the step of mixing and culturing the peptide, dendritic cells or antigen-presenting cells derived from the subject, and CD8 + T cells derived from the subject after the step (4) The method according to ⁇ CC2>.
- ⁇ CC4> ⁇ DC vaccine therapy> The method according to any one of items ⁇ CC1> to ⁇ CC2>, comprising a step of mixing and culturing the peptide and the dendritic cell derived from the subject after the step (4).
- the peptide, the dendritic cell or antigen-presenting cell derived from the subject, and the CD8 + T cell derived from the subject are mixed and cultured to obtain CD8 + T cell-dendritic cell / Comprising the steps of producing an antigen-presenting cell-peptide mixture, and producing a dendritic cell-peptide mixture by mixing and culturing the peptide and dendritic cells derived from the subject.
- A An antigen protein or antigen peptide derived from a subject, a lymphocyte derived from the subject, or the determined peptide according to any one of items ⁇ CC1> to ⁇ CC5> and inactivation derived from the subject Mixing tumor cells and T lymphocytes derived from the subject and culturing to produce tumor-specific T cells;
- the TCR of the tumor-specific T cell is the item ⁇ B1> to ⁇ B10>, ⁇ B57>, ⁇ B58> to ⁇ B58-20>, ⁇ B59>, or ⁇ B60> to ⁇ B60-21> According to any of the methods and / or items ⁇ B11> to ⁇ B20>, ⁇ B58> to ⁇ B58-20>, ⁇ B59>, or ⁇ B60> to ⁇ B60-21>
- ⁇ DD1-1> The step comprises mixing and culturing an antigen protein or antigen peptide derived from a subject, inactivated cancer cells derived from the subject, and T lymphocytes derived from the subject, to produce tumor-specific T cells.
- the method according to item ⁇ DD1> which is a production step.
- ⁇ DD1-2> A process mixes and culture
- Step is a tumor-specific T cell obtained by mixing and culturing the determined peptide according to item CC1, inactivated cancer cells derived from the subject, and T lymphocytes derived from the subject.
- the method according to any one of items ⁇ DD1> to ⁇ DD1-2>, which is a step of producing ⁇ DD2> ⁇ Isolation of tailor-made cancer-specific T cell receptor gene, isolation of cancer-specific TCR gene by consensus sequence search>
- A providing lymphocytes or cancer tissue isolated from subjects having a common HLA
- B For the lymphocytes or cancer tissues, the TCR of the tumor-specific T cells is determined in items ⁇ B1> to ⁇ B10>, ⁇ B57>, ⁇ B58> to ⁇ B58-20>, ⁇ B59>, or ⁇ B59>
- ⁇ EE1> ⁇ CCPCC> A) providing T lymphocytes collected from a patient; B) After antigen stimulation of the T lymphocytes, items ⁇ B1> to ⁇ B10>, ⁇ B57>, ⁇ B58> to ⁇ B58-20>, ⁇ B59>, or ⁇ B60> to ⁇ B60-21> Any of the methods and / or items ⁇ B11> to ⁇ B20>, ⁇ B58> to ⁇ B58-20>, ⁇ B59>, or ⁇ B60> to ⁇ B60-21> TCCR analysis based on the step, wherein the antigen stimulation is performed by an antigen protein or antigen peptide derived from the subject, an inactivated cancer cell derived from the subject, or a tumor-derived idiotype peptide ; CC) selecting the optimal antigen and optimal TCR in the analyzed TCR; and DD) for cell processing therapy comprising the steps of producing a tumor-specific ⁇ and ⁇ TCR
- ⁇ EE1-1> The method according to item ⁇ EE1>, wherein the antigen stimulation is performed by an antigen protein or an antigen peptide derived from the subject.
- ⁇ EE1-2> The method according to item ⁇ EE1> or ⁇ EE1-1>, wherein the antigen stimulation is performed by inactivated cancer cells derived from the subject.
- ⁇ EE1-3> The method according to any one of items ⁇ EE1> to ⁇ EE1-2>, wherein the antigen stimulation is performed by the tumor-derived idiotype peptide.
- step C) comprises selecting an antigen highly expressed in the cancer tissue of the subject.
- Step C) includes any one of items ⁇ EE1> to ⁇ EE1-5> including selecting an antigen that most increases the frequency of a specific TCCR from a repertoire analysis performed based on item B1 before and after antigen stimulation.
- ⁇ EE2> ⁇ CCPCRACC> A method for evaluating efficacy and / or safety by conducting an in vitro stimulation test using a cancer-specific TCCR gene isolated by the method according to item ⁇ DD2>. Specific steps of this efficacy and / or safety assessment can be exemplified as follows.
- ⁇ Efficacy> For example, a T cell into which a cancer-specific TCR gene has been introduced and an antigen protein or antigen peptide derived from the subject described in ⁇ EE1-1>, an inactivity derived from the subject described in ⁇ EE1-2> Measuring the amount of cytokines (such as interferon ⁇ ) secreted extracellularly in response to activation of T cells after culturing with cancerous cells or the tumor-derived idiotype peptide according to ⁇ EE1-3> Efficacy can be evaluated by measuring the expression level of a specific gene that increases in response to cell activation, or by measuring cell surface molecules that are expressed or increased in response to T cell activation. .
- cytokines such as interferon ⁇
- Safety For example, when T cells derived from the subject into which a cancer-specific TCR gene has been introduced and normal cells derived from the subject are mixed, cytokines secreted in response to the activation of the T cells, Safety can be evaluated by measuring gene expression or expression of cell surface molecules and confirming that the TCR gene-introduced T cells are not activated by normal cells.
- the present invention has an effect that it can cope with a “large-scale” sequence as compared with the prior art.
- BCR since a large number of mutations are observed, it can be said that there is a particularly advantageous effect in that it can be amplified “unbiased” regardless of the mutation and an accurate determination can be made.
- conventional systems for amplification method and sequencing method using V-strand specific primer, 1. Be non-biased; 2. Therefore, it can be said that the quantitative property is excellent.
- technologies such as SMART PCR 1. “Non-bias degree” is remarkably improved. This is advantageous in that it does not have the inherent disadvantages of each technology. For example, SMART reports Repeated Template Switching as a problem, but this problem does not occur in this system. 3.
- the ability to perform comprehensive analysis including identification of isotypes and subtypes can also be cited as an advantageous effect.
- TCR derives TCR and BCR repertoire for ⁇ , ⁇ , ⁇ , and ⁇ chains
- BCR derives IgM, IgD, IgA, IgG, IgE heavy chain and IgL, IgL light chain
- Changes in the repertoire from various aspects can be detected.
- the position of the C region primer for sequencing is arranged at an appropriate position. Furthermore, the position of the primer is devised so that the type of isotype or subtype can be identified from the amplified gene sequence, and the gene associated with the disease can be easily identified.
- invariant TCR screening can be performed.
- invariant TCR can be screened by searching for leads that overlap in many samples regardless of HLA in the TCR ⁇ chain.
- MAIT-derived TCRs that recognize MR1, which is a non-classical MHC, could be detected.
- NKT and MAIT that express invariant TCR play an important role in immune responses such as infectious immunity, antitumor, and inflammation. It is expected that the novel invariant TCRs in various tissue samples can be screened and used for the purpose of searching for cells having unique functions.
- TCR ⁇ and TCR ⁇ are receptor molecules that form heterodimers.
- Antigen-specific T cells that proliferate in response to antigens consist of specific unique TCR ⁇ and TCR ⁇ chains.
- TCR repertoire analysis amplifies the TCR ⁇ or TCR ⁇ gene separately, it cannot know which TCR ⁇ and which TCR ⁇ form a pair. Therefore, by examining whether or not a combination of individuals in which specific TCR ⁇ chain reads overlap is coincident with a TCR ⁇ chain overlapping individual, a pair of TCR ⁇ and TCR ⁇ chain genes can be estimated (FIG. 44).
- a matching TCR ⁇ chain could be inferred using an individual with a specific TCR ⁇ chain as an index (Table 3-11). Although there is an example in which a plurality of reads are assigned, it is considered that this is a useful search method for identifying a paired TCR gene.
- a highly accurate and unbiased sample for large-scale gene analysis is provided, which is particularly useful in clinical applications where quantitative analysis is particularly required.
- the ability to identify a “low frequency” (1 / 10000-1 / 100,000 or less) gene leads to more accurate diagnosis and treatment of leukemia, for example. This was not possible due to the detection limit (about 1%) in the prior art (method of combining the plate method with the adapter, or method of combining the plate method with the SMART method).
- the conventional method takes one night, but this is advantageous because it takes only a few minutes in this case.
- the conventional method takes one night, but this is advantageous because it takes only a few minutes in this case.
- IMGT / High-V-QUEST there is no C region classification function, and the repertoire classification is "gene name unit""allylunit" (That is, (*) V (gene name) -D (gene name) -J (gene name) or V (allyl) -D (allyl) -J (allyl)).
- the CDR3 classification can be performed separately from the repertoire, but there is no degree of freedom.
- the C region can be classified.
- “gene name unit” and “allyl unit” can be selected for each region. It is also possible to use CDR3 instead of D.
- V gene name
- V gene name
- V gene name
- V gene name
- V gene name
- V (allyl) -J (allyl)
- V (allyl) -CDR3-J (allyl)
- CDR3 can be used as a part of the repertoire classification, and can be classified independently.
- IMGT / High-V-QUEST has 150,000 as the maximum number of sequences that can be processed in one batch, whereas the analysis method of the present invention is unlimited. The time required for processing the same data is approximately 1/10 in this system.
- the cancer idiotype peptide therapy of the present invention is used for patients who do not have a specific marker (molecular target) effective for treatment in the target cancer cells or are ineffective in treatment with existing specific molecular target drugs. It is valid. That is, since peptides are prepared based on genetic information of cancer cells derived from individual patients, they are effective against many tumors that express TCR or BCR. Lymphoma cells and leukemia cells have T-cell and B-cell tumors from their origin, and this technology can be applied to any tumor type and is useful for the treatment of many patients. In addition, when targeting tumorized B cell subpopulations, antibody drugs that target cell surface molecules expressed on most B cells, such as anti-CD20 antibodies, are used.
- Existing CTL therapy induces patient-specific killer T cells or tumor-specific DC by co-culturing patient lymphocytes and patient tumor cells, and existing DC therapy co-cultures patient DC cells and patient tumor cells.
- treatment targeting anti-tumor effects is performed by stimulating lymphocytes or DC cells using cancer antigens from the population and transferring them to patients.
- cancer antigen protein is more effective and has fewer side effects than the protein as the antigen that imparts specificity, rather than using the whole tumor cell.
- peptides also have the advantage that they can be easily chemically synthesized directly based on gene sequence information. Peptides do not use biomaterials such as cells, culture media, and infectious substances in the production process, so safety can be ensured.
- CTL cells are expected to work as cells that have already been stimulated and activated by an antigen and exert an early therapeutic effect.
- Tumor-specific DC cells induce CTL cells in the transferred patient and thus have a sustained anti-tumor effect, and a synergistic anti-tumor effect is expected when these different cells are used in combination.
- cancer-specific TCR gene therapy it is important that expression of the target antigen is limited to cancer cells.
- antigens that are localized in limited tissues such as cancer cells and testis tissue, such as cancer-testis antigens, are selected, but these antigens are also known to be expressed in some normal cells.
- the tailor-made cancer TCR gene therapy of the technology of the present invention identifies T cells that infiltrate patient tumor tissue and uses the gene sequence of the TCR. Therefore, a higher effect is expected because a functional TCR that is considered to actually work on an antitumor in a patient is used.
- it is a T cell present in a patient its action on normal cells is likely to be limited.
- TCR gene therapy is limited to patients with specific HLA and expressing the target cancer antigen. On the other hand, if it is made to order, it can be applied to patient HLA, TCR specific to the cancer antigen derived from the patient can be individually prepared, and treatment for a wider range of patients becomes possible. Isolation of a cancer-specific TCR gene by in vitro stimulation is performed by stimulating patient lymphocytes with antigen protein, antigen peptide, inactivated cancer cell, idiotype peptide, or the like. The TCR gene isolated through an experimental process for each patient is a TCR that matches the patient's HLA type, cancer cell type, cancer antigen type, and other genetic backgrounds, and is considered to be more effective for treatment.
- FIG. 1 shows the cross-reactivity of isotype specific primers.
- the left panel is an example for the second IgM sample, the left end (L) shows the lane of molecular weight markers.
- M, G, A, D and E show the results with specific primers for IgM, IgG, IgA, IgD and IgE, respectively.
- the middle panel shows the results with the second IgG sample on the left and the second IgA sample on the right and the right end (L) shows the lane of molecular weight markers.
- M, G, A, D and E show the results with specific primers for IgM, IgG, IgA, IgD and IgE, respectively.
- the right panel shows the second IgD sample on the left and the second IgE sample on the right.
- the left end (L) shows a lane for molecular weight markers.
- M, G, A, D and E show the results with specific primers for IgM, IgG, IgA, IgD and IgE, respectively.
- amplification with another isotype-specific primer together with the target immunoglobulin isotype-specific primer was performed to confirm the presence or absence of cross-reactivity. 10 ⁇ L of the GS-PCR amplification product was electrophoresed in 2% agarose gel in TAE buffer and evaluated by ethidium bromide staining.
- FIG. 2 shows the examination result of the optimum dilution concentration.
- the optimum conditions for GS-PCR in each isotype were examined.
- a 2-fold serial dilution series of 2nd PCR amplification products was prepared, and 20 cycles of GS-PCR were performed. From the left, the results of 1-fold, 2-fold, 4-fold, 8-fold and 16-fold dilutions of IgM, IgG, IgA, IgD and IgE are shown for the 2nd PCR amplification product.
- the leftmost L indicates a lane of molecular weight marker.
- FIG. 3 shows the examination result of the optimum number of cycles. PCR was performed for 10, 15, and 20 cycles using a 16-fold diluted 2nd PCR amplification product. The upper panel shows the results of 20 cycles, the middle panel shows the results of 15 cycles, and the lower panel shows the results of 10 cycles. In each panel, the leftmost L indicates a lane of molecular weight marker, and IgM, IgG, IgA, IgD and IgE are shown from the left. In IgM, IgG, IgA, and IgD, good amplification was confirmed in 10 cycles. Further, it was confirmed that 20 cycles is appropriate for IgE.
- FIG. 4 shows the read length of the next generation sequence.
- the graph shows the number of library reads (vertical axis), and the horizontal axis shows the analysis result of the lead length.
- the read length of the next generation sequence of the BCR gene is shown.
- the number of leads of raw data was 130,000, and the number of leads passed through Filter pass was 90,000 or more.
- Table 2 shows the number of leads derived from each isotype labeled.
- FIG. 5 shows the analysis result of the lead length by MID. IgM, IgG and IgA are shown from the upper panel left, and IgD and IgE are shown from the lower panel left.
- the vertical axis indicates the number of reads
- the horizontal axis indicates the read length (base length). The number of leads divided by MID and the distribution of lead lengths were also uniform.
- FIG. 6A shows the analysis results of the frequency of use of C region sequences by isotype. IgM, IgG and IgA are shown from the upper panel left, and IgD and IgE are shown from the lower panel left. In each graph, the vertical axis indicates% and the horizontal axis indicates the identified C region gene name. The obtained leads for each isotype were subjected to a homology search with immunoglobulin isotype C region sequences including subclasses.
- FIG. 6A shows the result of analysis by IMGT HighV-Quest. The result similar to FIG. 6A was analyzed with the improved software (Repertoire genesis). Similar results were obtained with this software, and a no-hit result indicating reads that were not classified into any isotype or subtype was also obtained.
- FIGS. 7A and 7B show the results of analysis of the V region repertoire by isotype. From the top, IgM, IgG, IgA, IgD and IgE are shown, respectively. The horizontal axis shows each isotype name.
- the repertoire (BCR V repertoire) of the V region sequence by isotype is shown.
- the BCR V repertoire was very similar among IgM, IgG, IgA, and IgD, but only the lead with IGHV3-30 was obtained for IgE. This is because the number of IgE positive cells in peripheral blood is very small compared to other classes, suggesting the possibility that a biased repertoire was detected.
- FIGS. 7A and 7B show the results of analysis by IMGT HighV-Quest.
- FIGS. 7A and 7B show the results of analysis of the V region repertoire by isotype. From the top, IgM, IgG, IgA, IgD and IgE are shown, respectively. The horizontal axis shows each isotype name.
- the repertoire (BCR V repertoire) of the V region sequence by isotype is shown.
- the BCR V repertoire was very similar among IgM, IgG, IgA, and IgD, but only the lead with IGHV3-30 was obtained for IgE. This is because the number of IgE positive cells in peripheral blood is very small compared to other classes, suggesting the possibility that a biased repertoire was detected.
- FIGS. 7A and 7B show the results of analysis by IMGT HighV-Quest. FIGS.
- FIGS. 7C and D show the results of performing the same analysis as FIGS. 7A and B with improved software (Repertoire genesis). Similar results were obtained with this software, and a no hit result was also obtained.
- FIGS. 7C and D show the results of performing the same analysis as FIGS. 7A and B with improved software (Repertoire genesis). Similar results were obtained with this software, and a no hit result was also obtained.
- 8A and B show the analysis results of the V region repertoire by subtype. IgA1, IgA2, IgG1 and IgG2 are shown from the top. The horizontal axis shows each isotype name of each subclass. Shown are BCR V repertoires by IgA and IgG subclass.
- IgA The subclasses of IgA differed in the frequency of several V chains between IgA1 and IgA2.
- IGHV1-18 and IGHV4-39 were higher in IgA1 than IgA2, while IGHV3-23 and IGHV3-74 were higher in IgA2 than IgA1.
- the frequency of IGHV3-23 and IGHV3-74 which showed an increase in IgA2, was higher in IgG2 compared to IgG1.
- the number of reads of IgG3 and IgG4 is small (10 reads).
- FIGS. 8A and 8B show the results of analysis by IMGT HighV-Quest.
- 8A and B show the analysis results of the V region repertoire by subtype. IgA1, IgA2, IgG1 and IgG2 are shown from the top. The horizontal axis shows each isotype name of each subclass. Shown are BCR V repertoires by IgA and IgG subclass.
- IgA The subclasses of IgA differed in the frequency of several V chains between IgA1 and IgA2.
- IGHV1-18 and IGHV4-39 were higher in IgA1 than IgA2, while IGHV3-23 and IGHV3-74 were higher in IgA2 than IgA1.
- the frequency of IGHV3-23 and IGHV3-74 which showed an increase in IgA2, was higher in IgG2 compared to IgG1.
- the number of reads of IgG3 and IgG4 is small (10 reads).
- FIGS. 8A and 8B show the results of analysis by IMGT HighV-Quest.
- 8C and D show the results of the same analysis as in FIGS. 8A and 8 performed by improved software (Repertoire genesis). Similar results were obtained with this software, and a no hit result was also obtained.
- 8C and D show the results of the same analysis as in FIGS. 8A and 8 performed by improved software (Repertoire genesis).
- FIG. 9A shows the analysis result of BCRJ repertoire by subclass.
- BCRJ repertoires by subclass are shown.
- the upper panel shows IgM, IgG, IgA, IgD and IgE, respectively, and the horizontal axis shows the name of each isotype.
- the lower panel is a display by subclass, showing IgA1, IgA2, IgG1 and IgG2 from the left.
- the horizontal axis indicates the name of each isotype in each subclass.
- IgM, IgG, IgA, and IgD IGHJ4 was used in about half of the leads while IGHJ2 was rarely used.
- IgE only IGHJ1 was used.
- FIG. 9A shows the result of analysis by IMGT HighV-Quest. The same analysis as that of FIG. 9A is performed with the improved software. The same result was obtained with the pending software (Repertoire genesis) together with this patent, and the result of no hit could also be obtained.
- FIG. 10 shows a schematic diagram of a TCR gene amplification method. The description about the primer pair illustrated in the Example is shown.
- FIG. 11 shows the results of electrophoresis of 10 ⁇ L of GS-PCR amplification products derived from 10 healthy subjects on a 2% agarose gel. Upper row: GS-PCR (TRA), TCR ⁇ chain amplification product, lower row: GS-PCR (TRB), TCR ⁇ chain amplification product. The number indicates the sample number.
- TRA GS-PCR
- TRB GS-PCR
- FIG. 12 shows parameter settings of TCR / BCR repertoire analysis software (Repertoire genesis).
- FIG. 13 (AD) shows the analysis results of TRAV repertoire in healthy individuals. The TRAV repertoire for each sample (see number) is shown. The horizontal axis indicates the name of each TRAV gene, and the vertical axis indicates the presence frequency. Mean represents an average. The TRBV repertoire and average values of 10 healthy subjects were shown. The presence frequency of TRAV9-2, 12 and 13 was high, TRAV20 in # 1, TRAV21 in # 5 was higher than other healthy individuals, and individual differences were also observed.
- FIG. 13 (AD) shows the analysis results of TRAV repertoire in healthy individuals. The TRAV repertoire for each sample (see number) is shown.
- the horizontal axis indicates the name of each TRAV gene, and the vertical axis indicates the presence frequency. Mean represents an average.
- the TRBV repertoire and average values of 10 healthy subjects were shown.
- the presence frequency of TRAV9-2, 12 and 13 was high, TRAV20 in # 1, TRAV21 in # 5 was higher than other healthy individuals, and individual differences were also observed.
- FIG. 13 (AD) shows the analysis results of TRAV repertoire in healthy individuals.
- the TRAV repertoire for each sample is shown.
- the horizontal axis indicates the name of each TRAV gene, and the vertical axis indicates the presence frequency.
- Mean represents an average.
- the TRBV repertoire and average values of 10 healthy subjects were shown.
- FIG. 13 shows the analysis results of TRAV repertoire in healthy individuals.
- the TRAV repertoire for each sample is shown.
- the horizontal axis indicates the name of each TRAV gene, and the vertical axis indicates the presence frequency.
- Mean represents an average.
- the TRBV repertoire and average values of 10 healthy subjects were shown.
- the presence frequency of TRAV9-2, 12 and 13 was high, TRAV20 in # 1, TRAV21 in # 5 was higher than other healthy individuals, and individual differences were also observed.
- FIG. 14 shows the analysis results of TRBV repertoire in healthy individuals.
- the TRBV repertoire for each sample (see number) is shown.
- the horizontal axis indicates the name of each TRBV gene, and the vertical axis indicates the presence frequency.
- Mean represents an average.
- the TRBV repertoire and average values of 10 healthy subjects were shown.
- the presence frequency of TRBV20-1, 28, and 29-1 was high.
- TRBV3-1 was higher than other healthy individuals, and individual differences were observed.
- FIG. 14 (AD) shows the analysis results of TRBV repertoire in healthy individuals.
- the TRBV repertoire for each sample (see number) is shown.
- the horizontal axis indicates the name of each TRBV gene, and the vertical axis indicates the presence frequency.
- Mean represents an average.
- the TRBV repertoire and average values of 10 healthy subjects were shown.
- the presence frequency of TRBV20-1, 28, and 29-1 was high.
- FIG. 14 (AD) shows the analysis results of TRBV repertoire in healthy individuals.
- the TRBV repertoire for each sample (see number) is shown.
- the horizontal axis indicates the name of each TRBV gene, and the vertical axis indicates the presence frequency.
- Mean represents an average.
- the TRBV repertoire and average values of 10 healthy subjects were shown.
- the presence frequency of TRBV20-1, 28, and 29-1 was high.
- TRBV3-1 was higher than other healthy individuals, and individual differences were observed.
- FIG. 14 (AD) shows the analysis results of TRBV repertoire in healthy individuals.
- the TRBV repertoire for each sample (see number) is shown.
- the horizontal axis indicates the name of each TRBV gene, and the vertical axis indicates the presence frequency.
- FIGS. 15A to 15D show the results of TRAJ repertoire analysis in healthy individuals. The horizontal axis indicates the name of each TRAJ gene, and the vertical axis indicates the presence frequency. Mean represents an average. TRAJ repertoire and average values of 10 healthy subjects were shown. TRAJ repertoires of healthy individuals show that within about 5% of all AJ families, TRAJ12 in # 1, TRAJ27 in # 4, TRAJ37 in # 5, TRAJ45 in # 8 are higher than other healthy individuals, and individual differences are observed It was. FIGS.
- FIGS. 15A to 15D show the results of TRAJ repertoire analysis in healthy individuals.
- the horizontal axis indicates the name of each TRAJ gene, and the vertical axis indicates the presence frequency.
- Mean represents an average.
- TRAJ repertoire and average values of 10 healthy subjects were shown.
- TRAJ repertoires of healthy individuals show that within about 5% of all AJ families, TRAJ12 in # 1, TRAJ27 in # 4, TRAJ37 in # 5, TRAJ45 in # 8 are higher than other healthy individuals, and individual differences are observed It was.
- FIGS. 15A to 15D show the results of TRAJ repertoire analysis in healthy individuals.
- the horizontal axis indicates the name of each TRAJ gene, and the vertical axis indicates the presence frequency.
- Mean represents an average. TRAJ repertoire and average values of 10 healthy subjects were shown.
- FIGS. 15A to 15D show the results of TRAJ repertoire analysis in healthy individuals.
- the horizontal axis indicates the name of each TRAJ gene, and the vertical axis indicates the presence frequency. Mean represents an average.
- TRAJ repertoire and average values of 10 healthy subjects were shown.
- TRAJ repertoires of healthy individuals show that within about 5% of all AJ families, TRAJ12 in # 1, TRAJ27 in # 4, TRAJ37 in # 5, TRAJ45 in # 8 are higher than other healthy individuals, and individual differences are observed It was.
- FIG. 16 shows the results of TRBJ repertoire analysis in healthy individuals.
- the horizontal axis indicates the name of each TRBJ gene, and the vertical axis indicates the presence frequency. Mean represents an average.
- the TRBJ repertoire and average values of 10 healthy subjects were shown. In the healthy person, TRBJ repertoire had a high frequency of TRBJ2-1, 2-3, and 2-7, and in # 8, TRBJ2-2 was high, showing individual differences.
- FIG. 17 shows the result of electrophoresing each 2 nd PCR amplification product synthesized in Preparation Example 3 on a 2% agarose gel, and confirming the amplification product of the desired size in visual observation.
- the target sequence is an artificially spliced functional TRAC exon region sequence. It consists of exons EX1, EX2, and EX3, and is a primer over the entire length. Setting is possible.
- the possible primer setting regions of TRBC are shown in the lower row (the target sequence is an exon region sequence of functionally spliced functional TRBC. It consists of exons EX1, EX2, EX3 and EX4, and allows primer setting over the entire length. is there.).
- the TRAC sequence to be the target sequence can be used in addition to the one shown in the figure (SEQ ID NO: 1376) or a variant thereof.
- the target TRBC sequence can be those shown in the figure (SEQ ID NO: 1377), as well as those of SEQ ID NOs: 1392, 1393, and other variants. 18 to 25, each setting sequence in this full-length sequence is merely an example, and the C region-specific primer of the first TCR or BCR should be set most on the 5 ′ end side of the complementary DNA. Once the first TCR or BCR C region specific primer is set, the second TCR or BCR C region specific primer can be set downstream. Further, once a third TCR or BCR C region specific primer is set, a third TCR or BCR C region specific primer can be set. FIG.
- the target sequence is an exon region sequence of functional TRGC artificially spliced. It consists of exons EX1, EX2 and EX3, and is a primer over the entire length. Setting is possible.
- the upper and lower tiers show possible TRDC primer setting regions (the target sequence is an artificially spliced functional TRDC exon region sequence. It consists of exons EX1, EX2, EX3 and EX4, and allows primer setting over the entire length.
- the TRGC sequence to be the target sequence can be those shown in the figure (SEQ ID NO: 1378), as well as those of SEQ ID NOS: 1394, 1395, 1396, 1397, 1398, 1399, and variants thereof. Is done. It is understood that the target TRDC sequence can be used in addition to the one shown in the figure (SEQ ID NO: 1379) or a variant thereof.
- FIG. 20 shows an example of possible primer setting regions of IGHM (the target sequence is an artificially spliced functional IGHM exon region sequence. The secreted forms are exons CH1, CH2, CH3, CH4 and CH ⁇ . S, membrane-bound type consists of CH1, CH2, CH3, CH4, M1 and M2.
- the figure shows an example of membrane-bound type.
- the target sequence IGHM sequence is the one shown in the figure (SEQ ID NO: 1380) It is understood that those of SEQ ID NOs: 1447, 1448, 1449, and variants thereof can also be used.
- FIG. 21 shows an example of a possible primer setting region of IGHA (the target sequence is an artificially spliced functional IGHA exon region sequence. Secretory forms are exons CH1, H, CH2, CH3 and CH ⁇ .
- S, membrane-bound type is composed of CH1, H, CH2, CH3, M1 and M2.
- FIG. 22 shows an example of a possible primer setting region of IGHG (the target sequence is an artificially spliced functional IGHG exon region sequence.
- Secretion types are exons CH1, H (H1, H2, H3, H4), CH2, CH3 and CH-S, and the membrane-bound type is composed of CH1, H (H1, H2, H3, H4), CH2, CH3, M1 and M2.
- the figure shows an example of the secreted type.
- FIG. 23 shows an example of a possible primer setting region of IGHD (the target sequence is an artificially spliced functional IGHD exon region sequence.
- Secretory forms are exons CH1, H1, H2, CH2, CH3 and CH-S, membrane-bound type is composed of CH1, H1, H2, CH2, CH3, M1 and M2.
- the figure shows an example of membrane-bound type.
- the target sequence IGHD sequence is the one shown in the figure (SEQ ID NO: 1383), it is understood that those of SEQ ID NOs: 1404 to 1406 and variants thereof can also be used.
- FIG. 24 shows an example of possible primer setting regions of IGHE (the target sequence is an artificially spliced functional IGHE exon region sequence. Secretory forms are exon CH1, exon CH2, exon CH3 and CH- S, membrane-bound type is composed of CH1, exon CH2, exon CH3, M1 and M2.
- FIG. 25 shows an example of a primer setting region capable of IGKC in the upper part (the target sequence is a functional IGKC CL sequence.
- the IGKC sequence serving as the target sequence is that shown in the figure (SEQ ID NO: 1379)).
- Primers can be set over the entire length.
- the possible primer setting region of IGLC is shown in the lower row (the target sequence is a functional IGLC CL sequence.
- FIG. 26 shows an RNA electrophoresis image by Agilent 2100 Bioanalyzer. Total RNA was extracted from the cell serial dilution, and the amount of RNA was measured using an Agilent bioanalyzer. RNA was separated using a microchip electrophoresis apparatus, and RNA quality was checked. In any sample, 28S (upper band) and 18S rRNA (lower band) were detected, indicating that RNA that had not undergone degradation was obtained.
- FIG. 27 shows TCR reads in Molt-4 cell serially diluted samples.
- TCR leads obtained from 10%, 1%, 0.1% and 0.01% of each Molt-4 serially diluted sample are indicated. Ranking in order of the number of leads, showing the top 40. Rankings 365 to 404 are shown for 0.01% samples.
- the TRBV, TRBJ and CDR3 amino acid sequences of each lead and the number of reads are shown.
- a functional TCR lead derived from Molt-4 (TRBV20-1 / TRBJ2-1 / CSARESTTSDPKNEQFFG) is shown in bold and gray background, and the other functional defect is suspected (TRBV10-3 / TRBJ2-5 / CAISEPTGIRRDPVLR) Is shown in bold.
- TCR reads in Molt-4 cell serially diluted samples (SEQ ID NOs: 1165-1324). TCR leads obtained from 10%, 1%, 0.1% and 0.01% of each Molt-4 serially diluted sample are indicated. Ranking in order of the number of leads, showing the top 40. Rankings 365 to 404 are shown for 0.01% samples. The TRBV, TRBJ and CDR3 amino acid sequences of each lead and the number of reads are shown.
- FIG. 27 shows TCR reads in Molt-4 cell serially diluted samples. (SEQ ID NOs: 1165-1324). TCR leads obtained from 10%, 1%, 0.1% and 0.01% of each Molt-4 serially diluted sample are indicated. Ranking in order of the number of leads, showing the top 40. Rankings 365 to 404 are shown for 0.01% samples.
- FIG. 27 shows TCR reads in Molt-4 cell serially diluted samples. (SEQ ID NOs: 1165-1324). TCR leads obtained from 10%, 1%, 0.1% and 0.01% of each Molt-4 serially diluted sample are indicated. Ranking in order of the number of leads, showing the top 40.
- Rankings 365 to 404 are shown for 0.01% samples.
- the TRBV, TRBJ and CDR3 amino acid sequences of each lead and the number of reads are shown.
- a functional TCR lead derived from Molt-4 (TRBV20-1 / TRBJ2-1 / CSARESTTSDPKNEQFFG) is shown in bold and gray background, and the other functional defect is suspected (TRBV10-3 / TRBJ2-5 / CAISEPTGIRRDPVLR) Is shown in bold.
- FIG. 28 shows the number of TCR reads and detection sensitivity in a Molt-4 cell serially diluted sample.
- FIG. 29 is a schematic diagram showing a flow of TCR data analysis.
- FIG. 30 is a schematic diagram showing a flow of BCR data analysis.
- FIG. 31 is a diagram showing the frequency of C for each class.
- FIG. 32 (A and B) is a diagram showing a comparison of V repertoires between classes. The vertical axis represents frequency (%), and the horizontal axis represents gene name. No-hit indicates the frequency that did not correspond to any of them.
- FIG. 32 (A and B) is a diagram showing a comparison of V repertoires between classes. The vertical axis represents frequency (%), and the horizontal axis represents gene name. No-hit indicates the frequency that did not correspond to any of them.
- FIG. 33 is a diagram showing a comparison of J repertoires between classes. The vertical axis represents frequency (%), and the horizontal axis represents gene name.
- FIG. 34 shows a comparison of V repertoires between subclasses.
- the vertical axis represents frequency (%), and the horizontal axis represents gene name. No-hit indicates the frequency that did not correspond to any of them.
- FIG. 34 (A and B) shows a comparison of V repertoires between subclasses. The vertical axis represents frequency (%), and the horizontal axis represents gene name. No-hit indicates the frequency that did not correspond to any of them.
- FIG. 35 is a diagram showing a comparison of J repertoires between subclasses. The vertical axis represents frequency (%), and the horizontal axis represents gene name. No-hit indicates the frequency that did not correspond to any of them.
- FIG. 34 shows a comparison of V repertoires between subclasses. The vertical axis represents frequency (%), and the horizontal axis represents gene name. No-hit indicates the frequency that did not correspond to any of them.
- FIG. 34 (A and B) shows a comparison of V repertoires between subclasses. The vertical axis
- FIG. 36 (A and B) shows a comparison of IgM V repertoires between specimens.
- the vertical axis represents frequency (%), and the horizontal axis represents gene name. No-hit indicates the frequency that did not correspond to any of them.
- FIG. 36 (A and B) shows a comparison of IgM V repertoires between specimens. The vertical axis represents frequency (%), and the horizontal axis represents gene name. No-hit indicates the frequency that did not correspond to any of them.
- FIG. 37 is a diagram showing a comparison of IgM J repertoire between samples. The vertical axis represents frequency (%), and the horizontal axis represents gene name. No-hit indicates the frequency that did not correspond to any of them.
- FIG. 38 (AD) shows a TRAV repertoire comparison between specimens.
- FIG. 38 (AD) shows a TRAV repertoire comparison between specimens. Vertical axis: frequency (%), horizontal axis: gene name. “Average” is the average of all specimens, and error bars indicate ⁇ standard deviation.
- FIG. 38 (AD) shows a TRAV repertoire comparison between specimens. Vertical axis: frequency (%), horizontal axis: gene name. “Average” is the average of all specimens, and error bars indicate ⁇ standard deviation.
- FIG. 38 (AD) shows a TRAV repertoire comparison between specimens. Vertical axis: frequency (%), horizontal axis: gene name. “Average” is the average of all specimens, and error bars indicate ⁇ standard deviation.
- FIG. 38 (AD) shows a TRAV repertoire comparison between specimens. Vertical axis: frequency (%), horizontal axis: gene name. “Average” is the average of all specimens, and error bars indicate ⁇ standard deviation.
- FIG. 38 (AD) shows a TRAV repertoire comparison between specimens.
- FIG. 39 shows a TRBV repertoire comparison between specimens. Vertical axis: frequency (%), horizontal axis: gene name. “Average” is the average of all specimens, and error bars indicate ⁇ standard deviation.
- FIG. 39 (AD) shows a TRBV repertoire comparison between specimens. Vertical axis: frequency (%), horizontal axis: gene name. “Average” is the average of all specimens, and error bars indicate ⁇ standard deviation.
- FIG. 39 (AD) shows a TRBV repertoire comparison between specimens. Vertical axis: frequency (%), horizontal axis: gene name. “Average” is the average of all specimens, and error bars indicate ⁇ standard deviation.
- FIG. 39 (AD) shows a TRBV repertoire comparison between specimens. Vertical axis: frequency (%), horizontal axis: gene name. “Average” is the average of all specimens, and error bars indicate ⁇ standard deviation.
- FIG. 39 (AD) shows a TRBV repertoire comparison between specimens. Vertical axis: frequency (%)
- FIG. 39 shows a TRBV repertoire comparison between specimens.
- Vertical axis frequency (%), horizontal axis: gene name. “Average” is the average of all specimens, and error bars indicate ⁇ standard deviation.
- FIG. 40 (AD) shows a TRAJ repertoire comparison between specimens. Vertical axis: frequency (%), horizontal axis: gene name. “Average” is the average of all specimens, and error bars indicate ⁇ standard deviation.
- FIG. 40 (AD) shows a TRAJ repertoire comparison between specimens. The vertical axis represents frequency (%), and the horizontal axis represents the name of the child. “Average” is the average of all specimens, and error bars indicate ⁇ standard deviation.
- FIG. 40 (AD) shows a TRAJ repertoire comparison between specimens.
- FIG. 40 shows a TRAJ repertoire comparison between specimens. Vertical axis: frequency (%), horizontal axis: gene name. “Average” is the average of all specimens, and error bars indicate ⁇ standard deviation.
- FIG. 41 shows a TRBJ repertoire comparison between specimens. Vertical axis: frequency (%), horizontal axis: gene name. “Average” is the average of all specimens, and error bars indicate ⁇ standard deviation.
- FIG. 42 shows a block diagram of the system of the present invention.
- FIG. 43 shows a flowchart for the processing of the present invention.
- FIG. 44 shows the distribution of unique reads in the TCR ⁇ and TCR ⁇ chain repertoire analysis.
- the distribution of the unique reads of all sequence reads was examined with the copy number as the horizontal axis. In TCR ⁇ , only one lead was detected (single), 73.3% (1250 reads), and 70.5% (6502 reads) in the TCR ⁇ chain.
- FIG. 45 shows TRAV and TRAJ repertoire. The frequency of use of each TRAV and TRAJ in all leads is shown.
- the horizontal axis shows the TRAV gene (upper graph) and the TRAJ gene (lower graph).
- the vertical axis shows the ratio (% Usage) in all leads.
- FIG. 46 shows a 3D plot of the TRA repertoire.
- FIG. 47 shows TRBV and TRBJ repertoire. The frequency of use of each TRBV and TRBJ in all leads is shown. The horizontal axis shows the TRBV gene (upper graph) and the TRBJ gene (lower graph). The vertical axis shows the ratio (% Usage) in all leads.
- FIG. 48 shows a 3D plot of the TRB repertoire.
- FIG. 49 is a schematic diagram of a TCR ⁇ pair read estimation method (see Example 3 of the analysis system).
- FIG. 50 is a schematic diagram of MiSeq Dual-indexed Paired-end Sequencing according to the fourth embodiment of the analysis system.
- FIG. 51 shows the use of TRAV and TRAJ in 20 healthy individuals. The number of TCR sequences with TRAV and TRAJ, respectively, was counted.
- the frequency percentage of 54 TRAV and 61 TRAJ was calculated and presented as a scatter plot. Each dot indicates the frequency percentage of TRAV or TRAJ in each individual. The horizontal line shows an average value of 20.
- P pseudogene
- ORF open reading frame.
- FIG. 52 shows the use of TRBV and TRBJ in 20 healthy individuals. The frequency percentage of 65 TRBV and 14 TRBJ is shown as a scatter plot. Each dot indicates the frequency percentage of TRBV or TRBJ in each individual. Red bars indicate average values.
- FIG. 53 shows the frequency of occurrence of genetic recombination in TRAV TRAJ in read data pooled from 20 healthy individuals.
- the number of TCR sequence reads having the respective gene recombination of TRAV and TRAJ was counted.
- the occurrence tendency of recombination is visualized by a heat map display of each recombination number.
- the color of each pixel indicates the number of each recombination.
- TRAV8-5 there are 8 pseudogenes (TRAV8-5, TRAV11, TRAV15, TRAV28, TRAV31, TRAV32, TRAV33 and TRAV37), one ORF (TRAV8-7, a gene that was not fully expressed (TRAV7, TRAV9- 1, TRAV18 and TRAV36) for TRAJ, 3 pseudogenes (TRAJ51, TRAJ55, and TRAJ60), 6 ORFs (TRAJ1, TRAJ2, TRAJ19, TRAJ25, TRAJ59, and TRAJ61), and fully Genes that were not expressed (TRAJ14 and TRAJ46) were excluded, including two ORFs that were found to be expressed (TRAJ35 and TRAJ48), and 2,050 recombination events (4 It shows a heat map display of individual TRAV ⁇ 50 pieces of TRAJ).
- FIG. 54 shows a 3D image of the TCR ⁇ repertoire.
- the number of TCR sequence reads having a given gene recombination in TRAJ of TRAV was counted.
- the average frequency percentage of 3,294 (54 TRAV ⁇ 61 TRAJ) in 20 healthy individuals is shown as a 3D bar graph.
- the X axis and Y axis indicate TRAV and TRAJ, respectively. Recombination of TRAV1-2 with TRAJ33 (AV1-2 / AJ33) was most expressed (0.99 ⁇ 0.85).
- P pseudogene
- (ORF) open reading frame.
- FIG. 55 shows a 3D image of the TCR ⁇ repertoire.
- FIG. 56 shows the digital CDR3 chain length distribution of TCR ⁇ and TCR ⁇ . CDR3 length was determined in 172,109 TCR ⁇ and 94,928 TCR ⁇ sequence reads obtained from pooled data of 20 individuals.
- the length of the nucleotide sequence from the conserved cysteine at position 104 (Cys104) (IMGT designation) to the conserved phenylalanine at position 118 (Phe118) was automatically calculated using RG software.
- the CDR3 chain length distribution in TCR ⁇ (top) and TCR ⁇ (bottom) is shown as a histogram.
- FIG. 57 shows the diversity of TCR ⁇ and TCR ⁇ repertoires in healthy individuals.
- the copy number (read number) of the unique sequence read (USR) was calculated.
- the average number of copies per unique sequence read in each individual is shown as a white circle (left).
- FIG. 58 shows the similarity of TCR ⁇ and TCR ⁇ repertoires in healthy individuals.
- the frequency of occurrence of TCR sequence reads shared among pairs of all 20 individuals was calculated (Tables 4-6 and 4-7).
- FIG. 59 shows that the public TCR had a CDR3 with a shorter chain length than the private TCR.
- the length of CDR3 was calculated with 7,237 USR (grey) for public TCR and 83,997 USR (black) for private TCR.
- the frequency percentage of USR at each CDR3 length was plotted as a bar graph.
- FIG. 60 shows the correlation of gene usage of TRAV, TRAJ, TRBV and TRBJ among healthy individuals.
- FIG. 61 shows coincidence correlation coefficients for TRAV, TRAJ, TRBV and TRBJ.
- the correlation coefficient between two samples from healthy individuals was calculated by Spearman's correlation test. Each dot indicates a correlation coefficient value between individual pairs.
- FIG. 62 shows an overview of cancer idiotype peptide-sensitized immune cell therapy.
- Lymphocytes are collected from the upper left patient and a repertoire analysis for TCR or BCR is performed to predict HLA-binding peptides. Thereafter, using the predicted HLA-binding peptide, tailored peptide-sensitized CTL therapy or tailored peptide-sensitized DC vaccine therapy is performed.
- tailored peptide-sensitized CTL therapy or tailored peptide-sensitized DC vaccine therapy is performed.
- FIG. 63 shows an overview of the improved CTL method.
- FIG. 64 shows an overview of DC vaccine therapy.
- Dendritic cells are isolated from the left patient and mixed with the antigenic peptide.
- DC vaccine therapy since individualized peptides are created based on sequence information obtained from patient-derived tumor cells, they do not act on normal cells but act more specifically on tumor cells, resulting in high therapeutic effects. I can expect. Since peptides are used as antigens, there is an advantage that they can be easily chemically synthesized unlike proteins.
- FIG. 65 shows an overview of patient autoimmune cell therapy. In improved CTL therapy (left), CD8 + T cells and dendritic cells are isolated from the peripheral blood of a patient, and co-culture stimulation is performed using an antigenic peptide. Both cytotoxic T cells and antigen presenting cells are transferred to the patient.
- FIG. 66 shows the outline of the isolation of a tailor-made cancer-specific T cell receptor gene and the isolation of a cancer-specific TCR gene by in vitro antigen stimulation.
- a tumor-specific TCR gene is obtained by co-culturing an antigenic peptide, a patient-derived inactivated cancer, and a patient-derived T cell.
- Preparation of an isolated cancer-specific TCR gene by in vitro antigen stimulation can be performed using any technique known in the art once the genetic information is obtained.
- FIG. 67 shows an outline of preparation of an isolated cancer-specific TCR gene by in vitro antigen stimulation. As shown, the resulting TCR ⁇ and TCR ⁇ genes are introduced into a TCR-expressing viral vector (middle) and transformed by infecting patient-derived T lymphocytes.
- FIG. 68 shows an overview of cell processing therapy. As shown, a tumor-specific TCR gene obtained by TCR repertoire analysis is introduced into a patient-derived T lymphocyte from a T lymphocyte isolated from the upper right patient, and the tumor-specific T lymphocyte is transferred to the patient. .
- FIG. 69 shows an outline of a method for evaluating efficacy and / or safety by performing a stimulation test in vitro. Evaluation of the efficacy and / or safety of tumor-specific TCR gene-introduced T lymphocytes is performed by an in vitro stimulation test (downward arrow). Based on these in vitro evaluations, T lymphocytes suitable for treatment are selected (upward arrow). Evaluation of efficacy is performed by co-culturing tumor-specific TCR gene-introduced lymphocytes and patient-derived cancer cells and testing the reactivity. When evaluating safety, the same test is performed using normal cells instead of cancer cells.
- the “database” refers to an arbitrary database relating to a gene, and particularly refers to a database including a T cell receptor and a B cell receptor repertoire in the present invention.
- databases include the IMGT (the international ImmunGeneTics information system, www.imgt.org) database, the Japan DNA Databank (DDBJ, DNA Data Bank of Japan, www.ddbj.jp. US Biotechnology Information Center, www.ncbi.nlm.nih.gov/genbank/) database, ENA (EMBL (European Institute for Molecular Biology), www.ebi.ac.uk/ena) database, etc. However, it is not limited to this.
- gene sequence analysis refers to analysis of nucleic acid sequence and / or amino acid sequence constituting a gene. Determination of base or residue, determination of homology, determination of domain, potential function, etc. Any analysis related to the gene is included.
- T cell receptor is also referred to as a T cell receptor, a T cell antigen receptor, or a T cell antigen receptor.
- the TCR consisting of the former combination is called ⁇ TCR
- the TCR consisting of the latter combination is called ⁇ TCR
- T cells having the respective TCRs are called ⁇ T cells and ⁇ T cells. It is structurally very similar to the Fab fragment of an antibody produced by B cells and recognizes antigen molecules bound to MHC molecules.
- TCR Since the TCR gene of a mature T cell has undergone gene rearrangement, one individual has a variety of TCRs and can recognize various antigens.
- the TCR further binds to an invariable CD3 molecule present in the cell membrane to form a complex.
- CD3 has an amino acid sequence called ITAM (immunoreceptor tyrosine-based activation motif) in the intracellular region, and this motif is considered to be involved in intracellular signal transduction.
- ITAM immunoimmunoreceptor tyrosine-based activation motif
- Each TCR chain is composed of a variable part (V) and a constant part (C), and the constant part penetrates through the cell membrane and has a short cytoplasmic part.
- the variable region exists outside the cell and binds to the antigen-MHC complex.
- the variable region has three regions called hypervariable regions or complementarity determining regions (CDRs), and these regions bind to the antigen-MHC complex.
- the three CDRs are called CDR1, CDR2 and CDR3, respectively.
- CDR1 and CDR2 bind to MHC
- CDR3 binds to an antigen.
- TCR gene rearrangement is similar to the process of the B cell receptor known as immunoglobulin. In the gene rearrangement of ⁇ TCR, first, VDJ rearrangement of ⁇ chain is performed, and then VJ rearrangement of ⁇ chain is performed.
- the ⁇ chain gene When the ⁇ chain is rearranged, the ⁇ chain gene is deleted from the chromosome, so that T cells having ⁇ TCR do not have ⁇ TCR at the same time.
- this TCR-mediated signal suppresses ⁇ -chain expression, so that T cells having ⁇ TCR do not have ⁇ TCR at the same time.
- B cell receptor is also called a B cell receptor, a B cell antigen receptor, or a B cell antigen receptor, and Ig ⁇ / Ig ⁇ associated with a membrane-bound immunoglobulin (mIg) molecule ( CD79a / CD79b) refers to those composed of heterodimers ( ⁇ / ⁇ ).
- the mIg subunit binds to the antigen and causes receptor aggregation, while the ⁇ / ⁇ subunit transmits a signal into the cell. Aggregation of BCR is said to rapidly activate Src family kinases Lyn, Blk, and Fyn, similar to tyrosine kinases Syk and Btk.
- the complexity of BCR signaling produces many different results, including survival, tolerance (anergy; lack of hypersensitivity to antigen) or apoptosis, cell division, differentiation into antibody-producing cells or memory B cells, etc. Is included.
- Hundreds of millions of T cells with different TCR variable region sequences are generated, and hundreds of millions of B cells with different BCR (or antibody) variable region sequences are generated.
- the antigen specificity of T cells and B cells can be determined by determining the TCR / BCR genomic sequence or mRNA (cDNA) sequence. You can get a clue.
- V region refers to the variable region (V) region of the variable region of the TCR chain or BCR chain.
- D region refers to the D region of the variable region of the TCR chain or BCR chain.
- J region refers to a J region region of a variable region of a TCR chain or a BCR chain.
- C region refers to a constant region (C) region of a TCR chain or a BCR chain.
- variable region repertoire refers to a set of V (D) J regions arbitrarily created by gene rearrangement by TCR or BCR. Although it is used in idioms such as TCR repertoire and BCR repertoire, these may be referred to as T cell repertoire, B cell repertoire and the like.
- T cell repertoire refers to a collection of lymphocytes characterized by expression of a T cell receptor (TCR) that plays an important role in antigen recognition. Because changes in T cell repertoires provide a significant indicator of immune status in physiological and disease states, T cell repertoire analysis identifies antigen-specific T cells involved in disease development and T lymphocyte abnormalities Has been done for diagnosis.
- variable regions by fluorescence activated cell sorter analysis using a larger panel of antibodies specific for TCR variable regions (van den Beemd Ret al. (2000) Cytometry 40: 336-345; MacIsaac C et al. ( 2003) J Immunol Methods283: 9-15; Tembhare P et al. (2011) Am J Clin Pathol 135: 890-900; Langerak AWet al. (2001) Blood 98: 165-173), polymerase chain using multiple primers Reaction (PCR) (Rebai N et al. (1994) Proc Natl Acad Sci U S A 91: 1529-1533) or PCR-based enzyme-linked immunosorbent assay (Matsutani T et al.
- quantitative analysis refers to quantitative analysis, and in the present invention, repertoire analysis refers to analysis in a format that reflects the amount originally present in each clone.
- sample can include components derived from the subject (body fluids such as blood), but is not limited thereto.
- complementary DNA refers to DNA that forms a complementary strand with respect to RNA contained in a target nucleic acid molecule, for example, an RNA sample derived from a target cell.
- double-stranded complementary DNA refers to DNAs that are complementary to each other and form a double strand.
- a complementary DNA that forms a complementary strand with respect to RNA contained in an RNA sample or the like derived from a target cell can be generated as a template.
- the “common adapter primer sequence” means any sequence in the adapter-added double-stranded complementary DNA used as a primer in the first PCR amplification reaction and the second PCR amplification reaction of the present invention. Also refers to the sequence of commonly added portions.
- adaptive-added double-stranded complementary DNA is DNA used as a primer in the first PCR of the present invention, and is a common adapter for various double-stranded complementary DNAs in a sample.
- a primer sequence is added. Used as a template in the first primer amplification reaction.
- the “common adapter primer” is DNA used as a primer in the first PCR reaction and the second PCR amplification reaction of the present invention, and a single common sequence is used for each reaction. Say what you can.
- the “first TCR or BCR C region-specific primer” is a primer used in the first PCR amplification reaction of the present invention, and is a sequence specific to the C region of TCR or BCR.
- FIG. 18 shows an example of a TRAC-possible primer setting region in the upper part (the target sequence is an artificially spliced functional TRAC exon region sequence. It consists of exons EX1, EX2, and EX3, and is a primer over the entire length. Setting is possible.)
- An example of a possible primer setting region of TRBC is shown in the lower row (the target sequence is an artificially spliced functional TRBC exon region sequence. It consists of exons EX1, EX2, EX3 and EX4.
- the C region specific primer of the first TCR or BCR can be set to the 5 ′ end side of the most complementary DNA, and once the C region specific primer of the first TCR or BCR is set, The C region specific primer of the second TCR or BCR can be set downstream. Further, once a third TCR or BCR C region specific primer is set, a third TCR or BCR C region specific primer can be set. That is, when the first setting is made, the second is downstream, and the third is further downstream. Theoretically, it is understood that it is only necessary to be downstream by the length of the primer.
- FIG. 19 shows an example of a possible primer setting region of TRGC in the upper row (the target sequence is an artificially spliced functional TRGC exon region sequence. It consists of exons EX1, EX2 and EX3 and covers the entire length. Primer setting is possible.)
- An example of a possible primer setting region of TRDC is shown in the bottom row (the target sequence is an exon region sequence of a functionally spliced functional TRDC. It consists of exons EX1, EX2 and EX3, and allows primer setting over the entire length.
- the first TCR or BCR C region-specific primer can be set at the 5 ′ end of the most complementary DNA, and once the first TCR or BCR C region-specific primer is set.
- the C region specific primer of the second TCR or BCR can be set downstream. Further, once a third TCR or BCR C region specific primer is set, a third TCR or BCR C region specific primer can be set. That is, when the first setting is made, the second is downstream, and the third is further downstream. Theoretically, it is understood that it is only necessary to be downstream by the length of the primer.
- FIG. 20 shows an example of a possible primer setting region of IGHM (the target sequence is an artificially spliced functional IGHM exon region sequence. It consists of exons CH1, CH2, CH3 and CH4. Primer setting is possible.)
- the C region specific primer of the first TCR or BCR can be set to the 5 ′ end side of the most complementary DNA, and once the C region specific primer of the first TCR or BCR is set, The C region specific primer of the second TCR or BCR can be set downstream. Further, once a third TCR or BCR C region specific primer is set, a third TCR or BCR C region specific primer can be set. That is, when the first setting is made, the second is downstream, and the third is further downstream. Theoretically, it is understood that it is only necessary to be downstream by the length of the primer.
- FIG. 21 shows an example of a possible primer setting region of IGHA (the target sequence is an artificially spliced functional IGHA exon region sequence.
- Secretion types are exons CH1, H, CH2, CH3 and CH.
- -S the membrane-bound type is composed of CH1, H, CH2, CH3, M1 and M2, all of which can be set with primers over the entire length.
- the C region specific primer of the first TCR or BCR can be set to the 5 ′ end side of the most complementary DNA, and once the C region specific primer of the first TCR or BCR is set, The C region specific primer of the second TCR or BCR can be set downstream.
- a third TCR or BCR C region specific primer can be set. That is, when the first setting is made, the second is downstream, and the third is further downstream. Theoretically, it is understood that it is only necessary to be downstream by the length of the primer.
- FIG. 22 shows an example of a possible primer setting region of IGHG (the target sequence is an artificially spliced functional IGHG exon region sequence.
- Secretion types are exons CH1, H (H1, H2, H3). , H4), CH2, CH3 and CH-S, and the membrane-bound type is CH1, H (H1, H2, H3, H4), CH2, CH3, M1 and M2, all of which can be set with primers over the entire length. ).
- the C region specific primer of the first TCR or BCR can be set to the 5 ′ end side of the most complementary DNA, and once the C region specific primer of the first TCR or BCR is set, The C region specific primer of the second TCR or BCR can be set downstream.
- a third TCR or BCR C region specific primer can be set. That is, when the first setting is made, the second is downstream, and the third is further downstream. Theoretically, it is understood that it is only necessary to be downstream by the length of the primer.
- FIG. 23 shows examples of possible primer setting regions of IGHD (the target sequence is an artificially spliced functional IGHD exon region sequence.
- Secretion types are exons CH1, H1, H2, CH2, CH3.
- CH—S the membrane-bound type is composed of CH1, H1, H2, CH2, CH3, M1 and M2, all of which can be set with primers over the entire length.
- the C region specific primer of the first TCR or BCR can be set to the 5 ′ end side of the most complementary DNA, and once the C region specific primer of the first TCR or BCR is set, The C region specific primer of the second TCR or BCR can be set downstream.
- a third TCR or BCR C region specific primer can be set. That is, when the first setting is made, the second is downstream, and the third is further downstream. Theoretically, it is understood that it is only necessary to be downstream by the length of the primer.
- FIG. 24 shows an example of a possible primer setting region of IGHE (the target sequence is an artificially spliced functional IGHE exon region sequence.
- Secretion types are exon CH1, exon CH2, exon CH3 and CH.
- -S the membrane-bound type consists of CH1, exon CH2, exon CH3, M1 and M2, all of which can be set as primers.
- the C region specific primer of the first TCR or BCR can be set to the 5 ′ end side of the most complementary DNA, and once the C region specific primer of the first TCR or BCR is set, The C region specific primer of the second TCR or BCR can be set downstream.
- a third TCR or BCR C region specific primer can be set. That is, when the first setting is made, the second is downstream, and the third is further downstream. Theoretically, it is understood that it is only necessary to be downstream by the length of the primer.
- FIG. 25 shows an example of a primer setting region capable of IGKC in the upper row (the target sequence is a functional IGKC CL sequence. Primer setting is possible over the entire length).
- An example of a primer setting region capable of IGLC is shown in the lower part (the target sequence is a functional IGLC CL sequence. Primer setting is possible over the entire length).
- the C region specific primer of the first TCR or BCR can be set to the 5 ′ end side of the most complementary DNA, and once the C region specific primer of the first TCR or BCR is set, The C region specific primer of the second TCR or BCR can be set downstream. Further, once a third TCR or BCR C region specific primer is set, a third TCR or BCR C region specific primer can be set. That is, when the first setting is made, the second is downstream, and the third is further downstream. Theoretically, it is understood that it is only necessary to be downstream by the length of the primer.
- the C region specific primer of the first TCR or BCR has the following structure: CM1 (SEQ ID NO: 5), CA1 (SEQ ID NO: 8), CG1 (SEQ ID NO: 11), CD1 for BCR Examples of (SEQ ID NO: 14), CE1 (SEQ ID NO: 17), and TCR include CA1 (SEQ ID NO: 35) and CB1 (SEQ ID NO: 37), but are not limited thereto.
- such primer sequences can be set within the following ranges, but are not limited thereto, and the first, second, and third ranges can be set over the entire range, but determined mutually. can do.
- TCR ⁇ sequence base number 213 to base number 235 of SEQ ID NO: 1376 (FIG. 18)
- TCR ⁇ sequence base number 278 to base number 300 of SEQ ID NO: 1377 (FIG. 18)
- TCR ⁇ sequence base number 184 to base number 201 of SEQ ID NO: 1378 (FIG. 19)
- TCR ⁇ sequence base number 231 to base number 249 of SEQ ID NO: 1379 (FIG. 19)
- BCR IgM heavy chain sequence base number 77 to base number 95 of SEQ ID NO: 1380 (FIG. 20)
- BCR IgA heavy chain sequence base number 189 to base number 208 of SEQ ID NO: 1381 (FIG.
- IgG heavy chain sequence of BCR base number 262 to base number 282 of SEQ ID NO: 1382
- BCR IgD heavy chain sequence base number 164 to base number 183 of SEQ ID NO: 1383
- BCR IgE heavy chain sequence base number 182 to base number 199 of SEQ ID NO: 1384
- BCR Ig ⁇ chain constant region sequence base number 230 to base number 248 of SEQ ID NO: 1385
- BCR Ig ⁇ chain sequence base number 273 to base number 291 of SEQ ID NO: 1386 (FIG. 25).
- specific binds to a sequence of interest, but binds to other sequences at least in all TCR or BCR sequences present, preferably in a pool of TCR or BCR of interest. Is low, preferably not bound.
- the specific sequence is preferably, but not necessarily limited to, perfectly complementary to the sequence of interest.
- “sufficiently specific (to the target C region)” means having sufficient specificity to perform a gene amplification reaction.
- the same sequence as the target C region is advantageous, but not necessarily limited thereto.
- the “first PCR amplification reaction” is a PCR amplification reaction performed in the first stage in the method for preparing a sample of the present invention.
- “having no homology with other gene sequences” means that the gene has no homology with a sequence other than the target sequence (for example, the target C region of TCR or BCR) to the extent that no gene amplification reaction occurs. Say low.
- “including a mismatched base downstream (between subtypes)” means that a mismatched base is included between subtypes downstream of a sequence when set as a primer.
- the “second TCR or BCR C region-specific primer” is a primer used in the second PCR amplification reaction of the present invention, and is a sequence specific to the C region of TCR or BCR.
- a primer containing The second TCR or BCR C region specific primer has a sequence perfectly matched to the C region of the TCR or BCR in a sequence downstream from the sequence of the first TCR or BCR C region specific primer. It is designed to include a sequence having no homology to the gene sequence of and to include mismatched bases between subtypes downstream when amplified.
- CM2 SEQ ID NO: 6
- CA2 SEQ ID NO: 9
- CG2 SEQ ID NO: 12
- CD2 SEQ ID NO: 15
- CE2 SEQ ID NO: 18
- CA2 sequence ID for TCR No. 36
- CB2 SEQ ID NO: 38
- primer sequences can be set within the following ranges, but are not limited thereto, and the first, second, and third ranges can be set over the entire range, but determined mutually. can do. That is, when the first setting is made, the second is downstream, the third is further downstream, and theoretically, it is only necessary to be downstream by the length of the primer.
- TCR ⁇ sequence base number 146 to base number 168 of SEQ ID NO: 1376 (FIG. 18)
- T ⁇ ⁇ sequence base number 205 to base number 227 of SEQ ID NO: 1377 (FIG. 18)
- T ⁇ gamma sequence SEQ ID NO: 1378 (FIG. 19) base number 141 to base number 160
- TCR ⁇ sequence base number 135 to base number 155 of SEQ ID NO: 1379
- BCR IgM heavy chain sequence base number 43 to base of SEQ ID NO: 1380 (Fig. 20) No. 62
- BCR IgA heavy chain sequence base number 141 to base number 161 of SEQ ID NO: 1381 (FIG.
- BCR IgG heavy chain sequence base number 163 to base number 183 of SEQ ID NO: 1382 (FIG. 22) IgD heavy of BCR Chain sequence: base number 125 to base number 142 of SEQ ID NO: 1383 (FIG. 23) BCR IgE heavy chain sequence: SEQ ID NO: 1384 (FIG. 24) base number 155 to base number 173 BCR Ig ⁇ chain constant region sequence: SEQ ID NO: 1385 (FIG. 25) base number 103 to base number 120 BCR Ig ⁇ chain sequence: SEQ ID NO: 1386 (FIG. 25) Base number 85 to base number 100.
- the “second PCR amplification reaction” refers to a PCR amplification reaction that is performed using the product as a template after the first PCR reaction in the sample production for the analysis of the present invention. ) A reaction that takes place in the form.
- a common adapter primer and a second TCR or BCR C region-specific primer are used.
- the C region specific primer of the second TCR or BCR has a perfect match sequence with the C region of the TCR or BCR in a sequence downstream from the sequence of the C region specific primer of the first TCR or BCR. Is designed to contain sequences that are not homologous to other gene sequences and, when amplified, contain mismatched bases between subtypes downstream.
- the “third PCR amplification reaction” is a PCR amplification reaction that is performed using the product as a template after the second nested PCR reaction in the sample production for the analysis of the present invention.
- the third PCR amplification reaction includes an additional common adapter primer containing the first additional adapter nucleic acid sequence in the nucleic acid sequence of the common adapter primer and a second additional adapter using the product after the second nested PCR reaction as a template. It is made using a third TCR or BCR C region-specific primer with an adapter in which a nucleic acid sequence and a molecular identification sequence (MID Tag sequence) are added to the C region specific sequence of the third TCR or BCR.
- MID Tag sequence molecular identification sequence
- the C region-specific primer of the third TCR or BCR with an adapter may contain a sequence for confirming the nucleic acid sequence position called a key sequence.
- Adapter B SEQ ID NO: 1375) -TAATACGACTCCGAATTCCC can be used
- a third TCR or BCR C region-specific primer with an adapter for example, Adapter A (SEQ ID NO: 39) -key (TCAG) -MID1- (SEQ ID NO: 40) AAAGGGTTGGGGCGGAGC (SEQ ID NO: 1387) (primer is SEQ ID NO: 7), Adapter A (SEQ ID NO: 39) -key (TCAG) -MID2 (sequence No.
- the “first additional adapter nucleic acid sequence” is a sequence added to the primer used in the third PCR amplification reaction of the present invention, and is used by adding to the nucleic acid sequence of the common adapter primer. Is an array.
- the first additional adapter nucleic acid sequence and the second additional adapter nucleic acid sequence may be different or the same.
- this nucleic acid sequence includes DNA capture bead binding and emPCR reaction (for example, Chee-Seng, Ku; En Yun, Loy; Yudi, Pawitan; and Kee-Seng, Chia. Next Generation Sequencing Technologies and Their Applications. In: Encyclopedia of Life Sciences (ELS). John Wiley & Sons, Ltd: Chichester.
- the “second additional adapter nucleic acid sequence” is a sequence added to a primer used in the third PCR amplification reaction of the present invention, and a molecular identification sequence (for example, ( MID Tag sequence)) and / or key sequence, and added to the third TCR or BCR C region specific sequence to construct a third TCR or BCR C region specific primer with adapter It is an array to do.
- the second additional adapter nucleic acid sequence and the second additional adapter nucleic acid sequence may be different or the same.
- the nucleic acid sequence is a sequence suitable for emPCR reaction, and any sequence may be used as long as it has such characteristics. Specifically, CCATCTCATCCCTGCGTGTCCGCAC (SEQ ID NO: 39) is used, but is not limited thereto.
- the “key sequence” used in the present specification is a sequence added to a primer used in the third PCR amplification reaction of the present invention, and a molecular identification sequence (for example, (MID Tag Sequence)), and is added to the third TCR or BCR C region-specific sequence to constitute a third TCR or BCR C region-specific primer with an adapter.
- a molecular identification sequence for example, (MID Tag Sequence)
- TCAG 4-base key sequence
- the “molecular identification (MID Tag) sequence is a sequence for imparting uniqueness so that an amplification product can be identified. Therefore, it is preferably different from the target sequence.
- examples of such sequences include, but are not limited to, those of SEQ ID NOS: 1325 to 1374. Criteria for identification sequences (tag sequences) and representative examples thereof That is, the tag sequence determination criteria will be described below.
- a tag sequence is a base sequence added to identify each sample when a plurality of samples are mixed and sequenced simultaneously.
- One tag sequence is associated with reads from one sample, and the acquired read sequence is assigned to which sample. It is an arbitrary sequence of four types of A, C, G, and T.
- the base sequence length is preferably 2 to 40 bases, more preferably 6 to 10 bases, including a continuous sequence (AA, CC, GG, TT).
- Typical tags that can be used herein are as follows: ACGAGTGGT (SEQ ID NO: 1325), ACGTCTCGACA (SEQ ID NO: 1326), AGAGCCGACTC (SEQ ID NO: 1327), AGCACTGTTAG (SEQ ID NO: 1) No.
- ACACAGTGCGGT SEQ ID NO: 1370
- ACGATCTGCGGT SEQ ID NO: 1371
- AGAGACGGAGT SEQ ID NO: 1372
- ACTCGTAGAGT SEQ ID NO: 1373
- ACGACGGGGAGT SEQ ID NO: 1374
- the “third TCR or BCR C region-specific sequence” is a sequence specific to the C region of the TCR or BCR, the first TCR or BCR C region specific sequence and the It is a sequence existing downstream of the C region specific sequence of 2 TCRs or BCRs. This is the sequence used to construct the third TCR or BCR C region specific primer.
- CM3-GS CM3-GS
- CA3-GS CA3-GS
- CD3-GS SEQ ID NO: 1390
- CE3-GS CE3-GS
- TCR TCR in Table 6 in HuVaF and HuVbF Specific sequences
- such a primer sequence can be specifically set in the following ranges, but is not limited thereto, the first, second, and third ranges can be set in the entire range, It can be determined mutually. That is, when the first setting is made, the second is downstream, the third is further downstream, and theoretically, it is only necessary to be downstream by the length of the primer.
- TCR ⁇ sequence of TCR base number 51 to base number 73 of SEQ ID NO: 1376 (FIG. 18)
- TCR ⁇ sequence base number 69 to base number 91 of SEQ ID NO: 1377 (FIG. 18)
- TCR ⁇ sequence base number 34 to base number 53 of SEQ ID NO: 1378 (FIG. 19)
- TCR ⁇ sequence base number 61 to base number 78 of SEQ ID NO: 1379 (FIG. 19)
- BCR IgM heavy chain sequence base number 7 to base number 25 of SEQ ID NO: 1380 (FIG. 20)
- BCR IgA heavy chain sequence base number 115 to base number 134 of SEQ ID NO: 1381 (FIG.
- IgG heavy chain sequence of BCR base number 109 to base number 129 of SEQ ID NO: 1382
- BCR IgD heavy chain sequence base number 78 to base number 96 of SEQ ID NO: 1383
- BCR IgE heavy chain sequence base number 45 to base number 64 of SEQ ID NO: 1384
- BCR Ig ⁇ chain constant region sequence base number 75 to base number 92 of SEQ ID NO: 1385
- BCR Ig ⁇ chain sequence base number 52 to base number 69 of SEQ ID NO: 1386 (FIG. 25) (this SEQ ID NO is also used for CM).
- third TCR or BCR C region-specific primer refers to a primer used in the third PCR amplification reaction of the present invention, which is specific to the second TCR or BCR C region. Including a sequence having a perfect match in the C region of the TCR or BCR in the sequence downstream from the sequence of the target primer but not homologous to other gene sequences, and when amplified, between subtypes downstream Designed to contain mismatched bases. Furthermore, an adapter sequence, a key sequence and an identification sequence are further included.
- CM3-GS SEQ ID NO: 7
- CA3-GS SEQ ID NO: 10
- CG3-GS SEQ ID NO: 13
- CD3-GS SEQ ID NO: 16
- CE3-GS SEQ ID NO: 19
- isotype refers to types that belong to the same type in IgM, IgA, IgG, IgE, IgD, etc., but have different sequences. Isotypes are displayed using various gene abbreviations and symbols.
- the “subtype” is a type within the types existing in IgA and IgG in the case of BCR, and IgG1, IgG2, IgG3 or IgG4 is present for IgG, and IgA1 or IgA2 is present for IgA.
- TCR is also known to exist in ⁇ and ⁇ chains, and TRBC1 and TRBC2 or TRGC1 and TRGC2 exist, respectively.
- perfect match means 100% identity when sequences are compared.
- “completely match all C region allele sequences of the same isotype” means that all sequences match when C region allele sequences belonging to the same isotype are aligned. Since the C region sequences are not all the same even in the same isotype, using a sequence that completely matches all the C region allele sequences of the same isotype, the isotype is immediately determined when the amplification product is sequenced. It is advantageous to determine.
- “difficult to form homodimer and intramolecular hairpin structure” means that a dimer is formed for pairing with a complementary strand or the like with respect to a state of a nucleic acid molecule, particularly a common adapter primer, A sequence that hardly forms a hairpin structure or the like due to pairing with a complementary strand.
- the term “difficult to remove” allows, for example, the degree to which homodimers and hairpins do not substantially affect the subsequent analysis. For example, 10% or less, 5% or less, 1% or less, 0.5% % Or less, 0.1% or less, 0.05% or less, or 0.01% or less.
- “does not take a homodimer and an intramolecular hairpin structure” means that a nucleic acid molecule state, particularly a common adapter primer, forms a dimer for pairing with a complementary strand, etc.
- Such sequences are known in the art (SantaLucia, J. Proc Natl Acad Sci US A, 95 (4): 1460-1465. (1998), Bombarito et al., Nucleic Acids Res, 28 (9 ): 1929-1934. (2000), SantaLucia, J. Proc Natl Acad Sci U S A, 95 (4): 1460-1465.
- a structure that “can stably form a double strand” refers to a structure in which a nucleic acid molecule, particularly a common adapter primer, forms a double strand with other nucleic acid molecules such as a template stably. Is formed. Such stability can be evaluated mainly by temperature, pH, melting temperature (Tm) calculated from the base composition, pHm, and structure stabilization energy ( ⁇ G 37 ° C. ). Such sequences are known in the art (Santa Lucia, J. Proc Natl Acad Sci USA, 95 (4): 1460-1465. (1998), Bommarito et al., Nucleic Acids Res, 28 (9) : 1929-1934. (2000), Santa Lucia, J.
- not having high homology refers to nucleic acid molecules, in particular, common adapter primers.
- characteristics such as not having high homology with all TCR gene sequences in the database. It means having.
- the degree of homology is, for example, 80% or less, 70% or less, 60% or less, 50% or less, 40% or less, 30% or less, 25% or less, 20% or less, 15 % Or less and preferably 10% or less.
- “same melting temperature (Tm)” means that the DNA melting temperature (Tm) of the primer or sequence used is substantially the same. This is a preferable condition for appropriately performing the PCR amplification reaction.
- “Same degree” means Tm within ⁇ 15 ° C, within ⁇ 14 ° C, within ⁇ 13 ° C, within ⁇ 12 ° C, within ⁇ 11 ° C, within ⁇ 10 ° C, within ⁇ 9 ° C, within ⁇ 8 ° C, ⁇ It can be within 7 ° C, within ⁇ 6 ° C, within ⁇ 5 ° C, within ⁇ 4 ° C, within ⁇ 3 ° C, within ⁇ 2 ° C, within ⁇ 1 ° C, within ⁇ 0.5 ° C.
- appropriate base length for amplification refers to a length appropriate for the amplification reaction for the primer or sequence used. Such a length can be obtained by, for example, a commercially available computer program or the like (CLC Main Workbench or Primer 3) used in the examples, and the following documents can be referred to: Santa Lucia, J. Proc Natl Acad Sci US A, 95 (4): 1460-1465. (1998), Bommarito et al., Nucleic Acids Res, 28 (9): 1929-1934. (2000), Santa Lucia, J. Proc Natl Acad Sci U S A, 95 (4): 1460-1465. (1998), and von Ahsen et al., Clin Chem, 47 (11): 1956-11961. (2001)).
- mismatch means that there are bases that are not identical to each other when gene sequences are aligned.
- % GC % guanine / cytosine content
- G guanine
- C cytosine
- T thymine
- U uracil
- a set corresponding to all subclasses of TCR or BCR means a known subclass of a target TCR or BCR (TRBC1, TRBC2, or TRGC1, TRGC2, etc. for TCR).
- TCR target TCR or BCR
- IgG IgG1, IgG2, IgG3 or IgG4
- IgA means IgA1 or IgA2, etc.
- protein protein
- polypeptide oligopeptide
- peptide refers to a polymer of amino acids having an arbitrary length.
- This polymer may be linear, branched, or cyclic.
- the amino acid may be natural or non-natural and may be a modified amino acid.
- the term can also encompass one assembled into a complex of multiple polypeptide chains.
- the term also encompasses natural or artificially modified amino acid polymers. Such modifications include, for example, disulfide bond formation, glycosylation, lipidation, acetylation, phosphorylation or any other manipulation or modification (eg, conjugation with a labeling component).
- This definition also includes, for example, polypeptides containing one or more analogs of amino acids (eg, including unnatural amino acids, etc.), peptide-like compounds (eg, peptoids) and other modifications known in the art. Is done.
- amino acid may be natural or non-natural as long as the object of the present invention is satisfied.
- polynucleotide As used herein, “polynucleotide”, “oligonucleotide”, and “nucleic acid” are used interchangeably herein and refer to a nucleotide polymer of any length. The term also includes “oligonucleotide derivatives” or “polynucleotide derivatives”. “Oligonucleotide derivatives” or “polynucleotide derivatives” refer to oligonucleotides or polynucleotides that include derivatives of nucleotides or that have unusual linkages between nucleotides, and are used interchangeably.
- oligonucleotide examples include, for example, 2′-O-methyl-ribonucleotide, an oligonucleotide derivative in which a phosphodiester bond in an oligonucleotide is converted to a phosphorothioate bond, and a phosphodiester bond in an oligonucleotide.
- oligonucleotide derivatives in which ribose and phosphodiester bond in oligonucleotide are converted to peptide nucleic acid bond uracil in oligonucleotide is C— Oligonucleotide derivatives substituted with 5-propynyluracil, oligonucleotide derivatives wherein uracil in the oligonucleotide is substituted with C-5 thiazole uracil, cytosine in the oligonucleotide is C-5 propynylcytosine Substituted oligonucleotide derivatives, oligonucleotide derivatives in which cytosine in the oligonucleotide is replaced with phenoxazine-modified cytosine, oligonucleotide derivatives in which the ribose in DNA is replaced with 2'-O-
- a particular nucleic acid sequence may also be conservatively modified (eg, degenerate codon substitutes) and complementary sequences, as well as those explicitly indicated. Is contemplated. Specifically, a degenerate codon substitute creates a sequence in which the third position of one or more selected (or all) codons is replaced with a mixed base and / or deoxyinosine residue. (Batzer et al., Nucleic Acid Res. 19: 5081 (1991); Ohtsuka et al., J. Biol. Chem. 260: 2605-2608 (1985); Rossolini et al., Mol. Cell .Probes 8: 91-98 (1994)).
- nucleic acid is also used interchangeably with gene, cDNA, mRNA, oligonucleotide, and polynucleotide.
- nucleotide may be natural or non-natural.
- gene refers to a factor that defines a genetic trait. Usually arranged in a certain order on the chromosome. A gene that defines the primary structure of a protein is called a structural gene, and a gene that affects its expression is called a regulatory gene. As used herein, “gene” may refer to “polynucleotide”, “oligonucleotide”, and “nucleic acid”. A “gene product” is a substance produced based on a gene and refers to a protein, mRNA, and the like.
- homology of a gene refers to the degree of identity of two or more gene sequences to each other, and generally “having homology” means that the degree of identity or similarity is high. Say. Therefore, the higher the homology between two genes, the higher the sequence identity or similarity. Whether two genes have homology can be examined by direct sequence comparison or, in the case of nucleic acids, hybridization methods under stringent conditions. When directly comparing two gene sequences, the DNA sequence between the gene sequences is typically at least 50% identical, preferably at least 70% identical, more preferably at least 80%, 90% , 95%, 96%, 97%, 98% or 99% are identical, the genes are homologous.
- a “homolog” or “homologous gene product” is a protein in another species, preferably a mammal, that performs the same biological function as the protein component of the complex further described herein. Means.
- Amino acids may be referred to herein by either their commonly known three letter symbols or by the one letter symbols recommended by the IUPAC-IUB Biochemical Nomenclature Commission. Nucleotides may also be referred to by a generally recognized one letter code.
- BLAST is a sequence analysis tool.
- the identity search can be performed using, for example, NCBI BLAST 2.2.9 (issued May 12, 2004).
- the identity value usually refers to a value when the above BLAST is used and aligned under default conditions. However, if a higher value is obtained by changing the parameter, the highest value is set as the identity value. When identity is evaluated in a plurality of areas, the highest value among them is set as the identity value. Similarity is a numerical value calculated for similar amino acids in addition to identity.
- polynucleotide hybridizing under stringent conditions refers to well-known conditions commonly used in the art.
- a polynucleotide can be obtained by using a colony hybridization method, a plaque hybridization method, a Southern blot hybridization method or the like using a polynucleotide selected from among the polynucleotides of the present invention as a probe.
- hybridization was performed at 65 ° C. in the presence of 0.7 to 1.0 M NaCl using a filter on which colony or plaque-derived DNA was immobilized, and then a 0.1 to 2-fold concentration was obtained.
- a polynucleotide that can be identified by washing the filter under conditions of 65 ° C using an SSC (saline-sodium citrate) solution (composition of a 1-fold concentration of SSC solution is 150 mM sodium chloride, 15 mM sodium citrate).
- SSC saline-sodium citrate
- composition of a 1-fold concentration of SSC solution is 150 mM sodium chloride, 15 mM sodium citrate.
- Hybridization is described in experimental documents such as Molecular Cloning 2nd ed., Current Protocols in Molecular Biology, Supplement 1-38, DNA Cloning 1: Core Techniques, A Prac1tical Approach, Second Edition, Oxford University Press (1995). It can be carried out according to the method.
- the sequence containing only the A sequence or only the T sequence is preferably excluded from the sequences that hybridize under stringent conditions.
- a polypeptide (for example, transthyretin) used in the present invention is a nucleic acid molecule that hybridizes under stringent conditions to a nucleic acid molecule encoding a polypeptide particularly described in the present invention.
- the polypeptide encoded by is also encompassed. These low stringency conditions are: 35% formamide, 5 ⁇ SSC, 50 mM Tris-HCl (pH 7.5), 5 mM EDTA, 0.02% polyvinylpyrrolidone (PVP), 0.02% BSA, 100 ⁇ g / ml denatured salmon sperm DNA , And 10% (weight / volume) dextran sulfate in buffer solution at 40 ° C.
- a “purified” substance or biological factor refers to a substance from which at least a part of the factor naturally associated with the biological factor has been removed.
- the purity of a biological agent in a purified biological agent is higher (ie, enriched) than the state in which the biological agent is normally present.
- the term “purified” as used herein is preferably at least 75% by weight, more preferably at least 85% by weight, even more preferably at least 95% by weight, and most preferably at least 98% by weight, Means the presence of the same type of biological agent.
- the materials used in the present invention are preferably “purified” materials.
- a “corresponding” amino acid or nucleic acid has or has the same action as a predetermined amino acid or nucleotide in a reference polypeptide or polynucleotide in a polypeptide molecule or polynucleotide molecule.
- a reference polypeptide or polynucleotide in a polypeptide molecule or polynucleotide molecule in particular, in the case of an enzyme molecule, it means an amino acid that is present at the same position in the active site and contributes similarly to the catalytic activity.
- an antisense molecule can be a similar part in an ortholog corresponding to a particular part of the antisense molecule.
- Corresponding amino acids are identified as, for example, cysteinylation, glutathioneation, SS bond formation, oxidation (eg, oxidation of methionine side chain), formylation, acetylation, phosphorylation, glycosylation, myristylation, etc.
- the corresponding amino acid can be an amino acid responsible for dimerization.
- Such “corresponding” amino acids or nucleic acids may be a range of regions or domains (eg, V region, D region, etc.). Thus, in such cases, it is referred to herein as a “corresponding” region or domain.
- fragment refers to a polypeptide or polynucleotide having a sequence length of 1 to n ⁇ 1 with respect to a full-length polypeptide or polynucleotide (length is n).
- the length of the fragment can be appropriately changed according to the purpose.
- the lower limit of the length is 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 40, 50 and more amino acids, and lengths expressed in integers not specifically listed here (eg 11 etc.) are also suitable as lower limits obtain.
- examples include 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 40, 50, 75, 100 and more nucleotides.
- Non-integer lengths may also be appropriate as a lower limit.
- a fragment falls within the scope of the present invention as long as the full-length fragment functions as a marker, as long as the fragment itself also functions as a marker.
- the term “activity” refers herein to the function of a molecule in the broadest sense. Activities are not intended to be limiting, but generally include the biological function, biochemical function, physical function, therapeutic activity, diagnostic activity or chemical function of a molecule. Activity activates, promotes, stabilizes, inhibits, suppresses, or destabilizes, for example, enzyme activity, the ability to interact with other molecules, and the function of other molecules Ability, stability, and ability to localize to specific intracellular locations. Where applicable, the term also relates to the function of the protein complex in the broadest sense.
- expression of a gene, polynucleotide, polypeptide or the like means that the gene or the like undergoes a certain action in vivo to take another form.
- a gene, polynucleotide or the like is transcribed and translated into a polypeptide form.
- transcription and production of mRNA can also be an aspect of expression.
- polypeptide forms may be post-translationally processed (derivatives as referred to herein).
- search refers to another nucleic acid having a specific function and / or property using a certain nucleobase sequence electronically or biologically or by other methods, preferably electronically. This refers to finding the base sequence.
- Electronic searches include BLAST (Altschul et al., J. Mol. Biol. 215: 403-410 (1990)), FASTA (Pearson & Lipman, Proc. Natl. Acad. Sci., USA 85: 2444- 2448 (1988)), Smith and Waterman method (Smith and Waterman, J. Mol. Biol.
- BLAST is typically used.
- Biological searches include stringent hybridization, macroarrays with genomic DNA affixed to nylon membranes, microarrays affixed to glass plates (microarray assays), PCR and in situ hybridization, etc. It is not limited to. In the present specification, it is intended that the gene used in the present invention should include a corresponding gene identified by such an electronic search or biological search.
- an amino acid sequence having one or more amino acid insertions, substitutions or deletions, or those added to one or both ends can be used.
- “insertion, substitution or deletion of one or a plurality of amino acids in the amino acid sequence, or addition to one or both ends thereof” means a well-known technical method such as site-directed mutagenesis.
- site-directed mutagenesis means that the amino acid has been altered by substitution of a plurality of amino acids to the extent that it can occur naturally.
- the modified amino acid sequence of the molecule is, for example, an insertion or substitution of 1 to 30, preferably 1 to 20, more preferably 1 to 9, more preferably 1 to 5, particularly preferably 1 to 2, amino acids. Alternatively, it can be deleted or added to one or both ends.
- the modified amino acid sequence is preferably an amino acid whose amino acid sequence has one or more (preferably one or several or 1, 2, 3, or 4) conservative substitutions in the amino acid sequence of a molecule such as CD98. It may be an array.
- conservative substitution means substitution of one or more amino acid residues with another chemically similar amino acid residue so as not to substantially alter the function of the protein. For example, when a certain hydrophobic residue is substituted by another hydrophobic residue, a certain polar residue is substituted by another polar residue having the same charge, and the like. Functionally similar amino acids that can make such substitutions are known in the art for each amino acid.
- non-polar (hydrophobic) amino acids such as alanine, valine, isoleucine, leucine, proline, tryptophan, phenylalanine, and methionine.
- polar (neutral) amino acids include glycine, serine, threonine, tyrosine, glutamine, asparagine, and cysteine.
- positively charged (basic) amino acids include arginine, histidine, and lysine.
- negatively charged (acidic) amino acids include aspartic acid and glutamic acid.
- marker refers to a certain state (eg, normal cell state, transformed state, disease state, disordered state, proliferative ability, differentiation state level, presence / absence, etc. ) Or a substance that serves as an indicator for tracking whether there is a danger or not.
- detection, diagnosis, preliminary detection, prediction or pre-diagnosis for a certain condition is a drug, agent, factor or means specific for the marker associated with the condition, or It can be realized by using a composition, kit or system containing them.
- a certain condition eg, a disease such as differentiation disorder
- gene product refers to a protein or mRNA encoded by a gene.
- the “subject” refers to a target (for example, a human or other organism or an organ or cell taken out from the organism) that is a target of diagnosis or detection of the present invention.
- sample refers to any substance obtained from a subject or the like, and includes, for example, eye cells. Those skilled in the art can appropriately select a preferable sample based on the description of the present specification.
- drug drug
- drug may also be a substance or other element (eg energy such as light, radioactivity, heat, electricity).
- Such substances include, for example, proteins, polypeptides, oligopeptides, peptides, polynucleotides, oligonucleotides, nucleotides, nucleic acids (eg, DNA such as cDNA, genomic DNA, RNA such as mRNA), poly Saccharides, oligosaccharides, lipids, small organic molecules (for example, hormones, ligands, signaling substances, small organic molecules, molecules synthesized by combinatorial chemistry, small molecules that can be used as pharmaceuticals (for example, small molecule ligands, etc.)) , These complex molecules are included, but not limited thereto.
- a polynucleotide having a certain sequence homology to the sequence of the polynucleotide (for example, 70% or more sequence identity) and complementarity examples include, but are not limited to, a polypeptide such as a transcription factor that binds to the promoter region.
- Factors specific for a polypeptide typically include an antibody specifically directed against the polypeptide or a derivative or analog thereof (eg, a single chain antibody), and the polypeptide is a receptor.
- specific ligands or receptors in the case of ligands, and substrates thereof when the polypeptide is an enzyme include, but are not limited to.
- detection agent refers to any drug that can detect a target object in a broad sense.
- diagnostic agent refers to any drug capable of diagnosing a target state (for example, a disease) in a broad sense.
- the detection agent of the present invention may be a complex or a complex molecule in which another substance (for example, a label or the like) is bound to a detectable moiety (for example, an antibody or the like).
- a detectable moiety for example, an antibody or the like.
- complex or “complex molecule” means any construct comprising two or more moieties.
- the other part may be a polypeptide or other substance (eg, sugar, lipid, nucleic acid, other hydrocarbon, etc.).
- two or more parts constituting the complex may be bonded by a covalent bond, or bonded by other bonds (for example, hydrogen bond, ionic bond, hydrophobic interaction, van der Waals force, etc.). May be.
- the “complex” includes a molecule formed by linking a plurality of molecules such as a polypeptide, a polynucleotide, a lipid, a sugar, and a small molecule.
- interaction refers to two substances. Force (for example, intermolecular force (van der Waals force), hydrogen bond, hydrophobic interaction between one substance and the other substance. Etc.). Usually, two interacting substances are in an associated or bound state.
- bond means a physical or chemical interaction between two substances or a combination thereof. Bonds include ionic bonds, non-ionic bonds, hydrogen bonds, van der Waals bonds, hydrophobic interactions, and the like.
- a physical interaction can be direct or indirect, where indirect is through or due to the effect of another protein or compound. Direct binding refers to an interaction that does not occur through or due to the effects of another protein or compound and does not involve other substantial chemical intermediates. By measuring the binding or interaction, the degree of expression of the marker of the present invention can be measured.
- a “factor” (or drug, detection agent, etc.) that interacts (or binds) “specifically” to a biological agent such as a polynucleotide or a polypeptide is defined as that
- the affinity for a biological agent such as a nucleotide or polypeptide thereof is typically equal or greater than the affinity for other unrelated (especially less than 30% identity) polynucleotides or polypeptides. Includes those that are high or preferably significantly (eg, statistically significant). Such affinity can be measured, for example, by hybridization assays, binding assays, and the like.
- a first substance or factor interacts (or binds) “specifically” to a second substance or factor means that the first substance or factor has a relationship to the second substance or factor. Interact (or bind) with a higher affinity than a substance or factor other than the second substance or factor (especially other substances or factors present in the sample containing the second substance or factor) That means. Specific interactions (or bindings) for a substance or factor involve both nucleic acids and proteins, for example, ligand-receptor reactions, hybridization in nucleic acids, antigen-antibody reactions in proteins, enzyme-substrate reactions, etc.
- Examples include, but are not limited to, protein-lipid interaction, nucleic acid-lipid interaction, and the like, such as a reaction between a transcription factor and a binding site of the transcription factor.
- the first substance or factor “specifically interacts” with the second substance or factor means that the first substance or factor has the second substance Or having at least a part of complementarity to the factor.
- both substances or factors are proteins
- the fact that the first substance or factor interacts (or binds) “specifically” to the second substance or factor is, for example, by antigen-antibody reaction Examples include, but are not limited to, interaction by receptor-ligand reaction, enzyme-substrate interaction, and the like.
- the first substance or factor interacts (or binds) “specifically” to the second substance or factor by the transcription factor and its Interaction (or binding) between the transcription factor and the binding region of the nucleic acid molecule of interest is included.
- “detection” or “quantification” of polynucleotide or polypeptide expression uses suitable methods, including, for example, mRNA measurement and immunoassay methods, including binding or interaction with marker detection agents. In the present invention, it can be measured by the amount of PCR product.
- molecular biological measurement methods include Northern blotting, dot blotting, and PCR.
- immunological measurement methods include ELISA using a microtiter plate, RIA, fluorescent antibody method, luminescence immunoassay (LIA), immunoprecipitation (IP), immunodiffusion method (SRID), immunization. Examples are turbidimetry (TIA), Western blotting, immunohistochemical staining, and the like.
- Examples of the quantitative method include an ELISA method and an RIA method. It can also be performed by a gene analysis method using an array (eg, DNA array, protein array).
- the DNA array is widely outlined in (edited by Shujunsha, separate volume of cell engineering "DNA microarray and latest PCR method”).
- Examples of gene expression analysis methods include, but are not limited to, RT-PCR, RACE method, SSCP method, immunoprecipitation method, two-hybrid system, in vitro translation and the like.
- expression level refers to the amount of polypeptide or mRNA expressed in a target cell, tissue or the like. Such expression level is evaluated by any appropriate method including immunoassay methods such as ELISA method, RIA method, fluorescent antibody method, Western blot method, and immunohistochemical staining method using the antibody of the present invention. The amount of expression of the polypeptide of the present invention at the protein level, or the polypeptide used in the present invention evaluated by any suitable method including molecular biological measurement methods such as Northern blotting, dot blotting, and PCR. The expression level of the peptide at the mRNA level can be mentioned.
- “Change in expression level” means expression at the protein level or mRNA level of the polypeptide used in the present invention evaluated by any appropriate method including the above immunological measurement method or molecular biological measurement method. Means that the amount increases or decreases. By measuring the expression level of a certain marker, various detection or diagnosis based on the marker can be performed.
- “decrease” or “suppression” or synonyms for activity, expression products (eg, proteins, transcripts (RNA, etc.)) or a synonym is a decrease in the quantity, quality or effect of a particular activity, transcript or protein. Or activity to decrease.
- an expression product eg, a protein, transcript (such as RNA)
- a synonym thereof refers to a quantity, quality or effect of a particular activity, transcript or protein.
- the activity of the immune system can be detected and screened using the ability of the marker of the present invention such as decrease, inhibition, increase or activation as an index.
- “means” refers to any tool that can achieve a certain purpose (for example, detection, diagnosis, treatment).
- a certain purpose for example, detection, diagnosis, treatment.
- “means for selective recognition (detection)” means capable of recognizing (detecting) a certain object differently from others.
- the present invention is useful as an index of the state of the immune system.
- an indicator of the state of the immune system can be identified and used to know the state of the disease.
- nucleic acid primer refers to a substance necessary for the initiation of a reaction of a polymer compound to be synthesized in a polymer synthase reaction.
- a nucleic acid molecule for example, DNA or RNA
- the primer can be used as a marker detection means.
- a nucleic acid sequence is preferably at least 12 contiguous nucleotides long, at least 9 contiguous nucleotides, more preferably at least 10 contiguous nucleotides, and even more preferably at least 11 contiguous nucleotides.
- Nucleic acid sequences used as probes are nucleic acid sequences that are at least 70% homologous, more preferably at least 80% homologous, more preferably at least 90% homologous, at least 95% homologous to the sequences described above. Is included.
- a sequence suitable as a primer may vary depending on the nature of the sequence intended for synthesis (amplification), but those skilled in the art can appropriately design a primer according to the intended sequence. Such primer design is well known in the art, and may be performed manually or using a computer program (eg, LASERGENE, PrimerSelect, DNAStar).
- the primer according to the present invention can also be used as a primer set composed of two or more kinds of the primers.
- the primer and primer set according to the present invention can be prepared according to conventional methods in known methods for detecting a target gene using a nucleic acid amplification method such as a PCR method, an RT-PCR method, a real-time PCR method, an in situ PCR method, or a LAMP method. It can be used as a primer and a primer set.
- a nucleic acid amplification method such as a PCR method, an RT-PCR method, a real-time PCR method, an in situ PCR method, or a LAMP method. It can be used as a primer and a primer set.
- the primer set according to the present invention can be selected so that the nucleotide sequence of a target protein such as a T cell receptor molecule can be amplified by a nucleic acid amplification method such as a PCR method.
- Nucleic acid amplification methods are well known, and selection of primer pairs in nucleic acid amplification methods is obvious to those skilled in the art.
- one of two primers (primer pair) is paired with a plus strand of a double-stranded DNA of a target protein of a T cell receptor molecule, and the other primer is a minus strand of a double-stranded DNA.
- the primer can be selected so that the other primer is paired with the extended strand that has been paired with the other and extended with one primer.
- the primer of the present invention can be chemically synthesized based on the nucleotide sequence disclosed herein. Preparation of the primer is well known, for example, "Molecular Cloning, A Laboratory Manual 2 nd ed. (Cold Spring Harbor Press (1989)), “Current Protocols in Molecular Biology” (John Wiley & Sons (1987-1997)).
- the term “probe” refers to a substance that serves as a search means used in biological experiments such as screening in vitro and / or in vivo.
- a nucleic acid molecule containing a specific base sequence or a specific Examples include, but are not limited to, peptides containing amino acid sequences, specific antibodies or fragments thereof.
- the probe is used as a marker detection means.
- the detection agent of the present invention may be labeled.
- the detection agent of the present invention may have a tag bound thereto.
- the “label” refers to a presence (for example, a substance, energy, electromagnetic wave, etc.) for distinguishing a target molecule or substance from others.
- a labeling method include RI (radioisotope) method, fluorescence method, biotin method, chemiluminescence method and the like.
- the labeling is performed with fluorescent substances having different fluorescence emission maximum wavelengths. The difference in the maximum fluorescence emission wavelength is preferably 10 nm or more.
- Alexa TM Fluor is preferred as the fluorescent substance.
- Alexa TM Fluor is a water-soluble fluorescent dye obtained by modifying coumarin, rhodamine, fluorescein, cyanine, etc., and is a series corresponding to a wide range of fluorescent wavelengths. Compared with other applicable fluorescent dyes, Stable, bright and low pH sensitive. Examples of combinations of fluorescent dyes having a fluorescence maximum wavelength of 10 nm or more include a combination of Alexa TM 555 and Alexa TM 633, a combination of Alexa TM 488 and Alexa TM 555, and the like. Any nucleic acid can be used as long as it can bind to its base moiety.
- a cyanine dye eg, CyDye TM series Cy3, Cy5, etc.
- rhodamine 6G reagent N-acetoxy-N2— Acetylaminofluorene (AAF), AAIF (iodine derivative of AAF) or the like
- the fluorescent substance having a difference in fluorescence emission maximum wavelength of 10 nm or more include a combination of Cy5 and rhodamine 6G reagent, a combination of Cy3 and fluorescein, a combination of rhodamine 6G reagent and fluorescein, and the like.
- the target object by using such a label, the target object can be modified so that it can be detected by the detection means used. Such modifications are known in the art, and those skilled in the art can appropriately carry out such methods depending on the label and the target object.
- a “tag” is a substance for sorting molecules by a specific recognition mechanism such as a receptor-ligand, more specifically, a binding for binding a specific substance.
- a substance that plays the role of a partner eg, having a relationship such as biotin-avidin, biotin-streptavidin
- label can be included in the category of “label”.
- a specific substance to which a tag is bound can be selected by bringing the substrate to which the binding partner of the tag sequence is bound into contact.
- tags or labels are well known in the art.
- Representative tag sequences include, but are not limited to, myc tag, His tag, HA, Avi tag and the like. Such a tag may be bound to the marker or marker detection agent of the present invention.
- test sample may be a sample that is considered to include a target cell or a substance derived from the target cell and capable of gene expression.
- diagnosis refers to identifying various parameters related to a disease, disorder, or condition in a subject and determining the current state or future of such a disease, disorder, or condition.
- conditions within the body can be examined, and such information can be used to formulate a disease, disorder, condition, treatment to be administered or prevention in a subject.
- various parameters such as methods can be selected.
- diagnosis in a narrow sense means diagnosis of the current state, but in a broad sense includes “early diagnosis”, “predictive diagnosis”, “preliminary diagnosis”, and the like.
- the diagnostic method of the present invention is industrially useful because, in principle, the diagnostic method of the present invention can be used from the body and can be performed away from the hands of medical personnel such as doctors.
- diagnosis, prior diagnosis or diagnosis may be referred to as “support”.
- the prescription procedure as a medicine such as the diagnostic agent of the present invention is known in the art, and is described in, for example, the Japanese Pharmacopoeia, the US Pharmacopoeia, and the pharmacopoeia of other countries. Accordingly, those skilled in the art can determine the amount to be used without undue experimentation as described herein.
- trimming means removing inappropriate parts in gene analysis. Trimming is performed by deleting a low quality region from both ends of the lead, a partial sequence of the artificial nucleic acid sequence added in the experimental procedure, or both. Trimming is performed using software and literature known in the art (eg, cutadapt http://journal.embnet.org/index.php/embnetjournal/article/view/200/(EMBnet.journal,2011); fastq-mcf Aronesty E., The Open BioinformaticsJournal (2013) 7, 1-8 (DOI: 10.2174 / 1875036201307010001); and fastx-toolkit http://hannonlab.cshl.edu/fastx_toolkit/ (2009)) .
- trimming removes the low quality region from both ends of the lead; removes the region that matches the adapter sequence by 10 bp or more from both ends of the lead; and the remaining length is 200 bp or more.
- TCR or more than 300 bp (BCR) is achieved by the step used for analysis as high quality.
- the “appropriate length” means a length suitable for analysis when performing analysis such as alignment in the gene analysis of the present invention. Such length can be determined, for example, from the sequence determination start position on the C region so as to be not less than a length including 100 bases near the D region on the V region.
- TCR it may be 200 nucleotides or more, preferably 250 nucleotides or more
- BCR it may be 300 nucleotides or more, preferably 350 nucleotides or more, but is not limited thereto.
- the “input sequence set” refers to a set of sequences to be analyzed for TCR or BCR repertoire in the gene analysis of the present invention.
- gene region refers to each region such as V region, D region, J region and C region. Such a gene region is known in the art and can be appropriately determined in consideration of a database or the like.
- homology of a gene refers to the degree of identity of two or more gene sequences to each other, and generally “having homology” means that the degree of identity or similarity is high. Say. Therefore, the higher the homology between two genes, the higher the sequence identity or similarity. Whether two genes have homology can be examined by direct sequence comparison or, in the case of nucleic acids, hybridization methods under stringent conditions.
- homoology search refers to homology search. Preferably, it can be performed in silico using a computer.
- “approximate” means that the degree of homology is high when homology search is performed.
- software that performs homology search BLAST, FASTA, etc.
- it is usually listed in the order of high homology, and therefore it can be approximated by appropriately selecting the ones with high rank.
- “closest” means that the degree of homology is highest when homology search is performed.
- homology search is performed by software, the one displayed in the first place is selected.
- reference allele refers to a reference allele that is hit in the reference database when homology search is performed.
- alignment is a bioinformatics that is arranged so that regions similar to the primary structure of biomolecules such as DNA, RNA, and protein can be identified, And to line up. Provides clues to know functional, structural, or evolutionary sequence relationships.
- assignment refers to assigning information such as a specific gene name, function, characteristic region (eg, V region, J region, etc.) to a certain sequence (eg, nucleic acid sequence, protein sequence, etc.). . Specifically, this can be achieved by inputting or linking specific information to a certain array.
- CDR3 refers to a third complementarity-determining region (CDR), where the CDR is a variable region, and the region directly contacting the antigen is particularly changed. Largely refers to this hypervariable region. There are three CDRs (CDR1 to CDR3) and four FRs (FR1 to FR4) surrounding the three CDRs in the light chain and heavy chain variable regions, respectively. Since the CDR3 region is said to exist across the V region, D region, and J region, it is said to hold the key to the variable region and is used as an analysis target.
- the top of the CDR3 on the reference V region refers to a sequence corresponding to the top of the CDR3 in the V region targeted by the present invention.
- end of CDR3 on reference J refers to a sequence corresponding to the end of CDR3 in the J region targeted by the present invention.
- condition that allow random mutations to be scattered throughout refers to any conditions that result in random mutations being scattered, for example, BLAST / FASTA optimal parameters. It is well expressed with the following conditions: allows up to 33% mismatch over the entire alignment length and allows up to 60% non-matching mismatches for any 30 bp therein.
- Functional equivalents such as isotypes of molecules such as IgG used in the present invention can be found by searching a database or the like.
- search refers to another nucleic acid having a specific function and / or property using a certain nucleobase sequence electronically or biologically or by other methods, preferably electronically. This refers to finding the base sequence.
- BLAST Altschul et al., J. Mol. Biol. 215: 403-410 (1990)
- FASTA Pearson & Lipman, Proc. Natl. Acad. Sci., USA 85: 2444- 2448 (1988)
- Smith and Waterman method Smith and Waterman, J. Mol. Biol. 147: 195-197 (1981)
- Needleman and Wunsch method Needleman and Wunsch, J. Mol. Biol. 48: 443). -453 (1970)
- BLAST is typically used.
- Bio searches include stringent hybridization, macroarrays with genomic DNA affixed to nylon membranes, microarrays affixed to glass plates (microarray assays), PCR and in situ hybridization, etc. It is not limited to. In the present specification, it is intended that the gene used in the present invention should include a corresponding gene identified by such an electronic search or biological search.
- the present invention can adjust samples for quantitative analysis of T cell receptor (TCR) or B cell receptor (BCR) variable region repertoire using next generation sequencing technology. . These sequencing techniques can obtain 1 million or more leads from a sample at a reasonable cost. Even genotypes present at a frequency of 1 / 1,000,000 or less can be detected in a specific and unbiased manner using these techniques. Unbiased amplification methods are achieved to amplify all different types of sequences of specific portions of genes or transcripts from samples derived from DNA such as blood or bone marrow.
- the present invention prepares a sample for quantitative analysis of T cell receptor (TCR) or B cell receptor (BCR) variable region repertoire by gene sequence analysis using a database.
- TCR T cell receptor
- BCR B cell receptor
- Providing a method for In this method (1) a step of synthesizing complementary DNA using an RNA sample derived from a target cell as a template; (2) a step of synthesizing double-stranded complementary DNA using the complementary DNA as a template; A step of adding a common adapter primer sequence to the double-stranded complementary DNA to synthesize an adapter-added double-stranded complementary DNA; (4) from the adapter-added double-stranded complementary DNA and the common adapter primer sequence; A first PCR amplification reaction using a common adapter primer and a first TCR or BCR C region specific primer, wherein the first TCR or BCR C region specific primer comprises: It contains a sequence that is sufficiently specific for the target C region of the TCR or BCR, has no homology to other gene sequences
- the second TCR or BCR C region specific primer comprises a sequence of the TCR or BCR in a sequence downstream of the first TCR or BCR C region specific primer.
- the third TCR or BCR C region specific primer is a sequence downstream of the second TCR or BCR C region specific primer in the TCR or BCR sequence.
- the first addition is designed to include a sequence having a perfect match in the C region of the gene but not homologous to other gene sequences, and to include a mismatched base between subtypes downstream when amplified.
- the adapter nucleic acid sequence is a sequence suitable for binding to the DNA capture bead and the emPCR reaction
- the second additional adapter nucleic acid sequence is emPC This is a sequence suitable for the R reaction
- the molecular identification (MID Tag) sequence includes a step for providing uniqueness so that an amplification product can be identified.
- the SMART PCR method uses the terminal transferase activity of the reverse transcriptase derived from Moloney MurineLeukemia Virus (MMLV). That is, when the reverse transcriptase reaches the 5 'end of the mRNA that is the template in the complementary strand DNA synthesis reaction, it uses a secondary reaction that mainly adds a C base to the 3' end of the newly synthesized complementary DNA. is doing.
- MMLV Moloney MurineLeukemia Virus
- TS oligo By using a primer (TS oligo) having a base sequence (GGG) complementary to the added base (CCC) at the 3 ′ end, the template is changed during the reverse transcription reaction and double-stranded synthesis occurs. Therefore, it is known that TS oligo addition reactions occur continuously and TS oligo concatamers are formed (Villanyi Z, Mai, A, Szabad J. Repeated template switching: Obstacles in cDNA libraries and ways to avoid them The open genomics journal, 2012, 5, 1-6).
- TS oligo inhibits the progression of polymerase, resulting in bias (Tang DT, Plessy C, Salimullah M , Suzuki AM, Calligaris R, Gustincich S, Carninci P. Suppression of artifacts and barcodes bias in high-throughput transcriptome analyses utilizing template switching.
- the C region-specific primer when performing quantitative analysis of the BCR variable region repertoire, is present in a target isotype C region selected from the group consisting of IgM, IgA, IgG, IgE, and IgD. It has a sequence that contains a perfect match and has no homology to other C regions.
- the C region specific primer is a sequence that, for IgA or IgG, perfectly matches a subtype that is either IgG1, IgG2, IgG3 or IgG4, or either IgA1 or IgA2.
- the C region-specific primer when performing quantitative analysis of the repertoire of the variable region of TCR, is a target strand selected from the group consisting of ⁇ chain, ⁇ chain, ⁇ chain, and ⁇ chain. A sequence that perfectly matches the C region and has no homology to other C regions.
- the C region-specific primer preferably selects a sequence portion that completely matches all C region allele sequences of the same isotype in the database. By selecting such a perfect match, a highly accurate analysis can be performed.
- the common adapter primer is designed so that homodimer and intramolecular hairpin structures are difficult to form and can form a stable double strand, and is not highly homologous to all TCR gene sequences in the database. And it is designed to have the same Tm as the C region specific primer.
- Examples of such common adapter primer sequences include TAATACGACTCCGAATTCCC (SEQ ID NO: 2), GGGAATTCGG (P10EA; SEQ ID NO: 3), and the like.
- the common adapter primer is selected so as not to have a homodimer and an intramolecular hairpin structure, and has no homology to other genes including BCR or TCR.
- Examples of such common adapter primer sequences include P20EA, P10EA, and the like.
- the common adapter primer is P20EA and / or P10EA, and the sequences thereof are TAATACGACTCCGAATTTCCC (P20EA; SEQ ID NO: 2), GGGATTTCGG (P10EA; SEQ ID NO: 3).
- the first, second and third TCR or BCR C region specific primers are each independently for BCR repertoire analysis, and include IgM, IgG, IgA, IgD, Or a sequence that perfectly matches each isotype C region of IgE, and in the case of IgG and IgA, it is a sequence that also perfectly matches subtypes and has no homology to other sequences contained in the database,
- the common adapter primer sequence is selected so as to contain mismatched bases between subtypes downstream of the primer, the base length is suitable for amplification, and it is difficult to take homodimer and intramolecular hairpin structures.
- sequences include, but are not limited to, P20EA (TAATACGACTCCGAATTCCC (SEQ ID NO: 2)) and P10EA (GGGAATTCGG (SEQ ID NO: 3)).
- the first, second and third TCR C region specific primers are each independently for TCR repertoire analysis, each primer comprising one alpha chain (TRAC).
- TRBC01, TRBC02 two types of ⁇ chains
- TRGC1, TRGC2 two types of ⁇ chains
- TRDC1 one type of ⁇ chain
- the common adapter primer sequence has a base length suitable for amplification, and is selected to include a mismatched base between subtypes downstream of the primer. Designed so that the inner hairpin structure is difficult to form and can form a stable double strand, homology with all TCR gene sequences in the database Not high, and is designed to be the Tm of the same extent as the C region-specific primers. Examples of such sequences include, but are not limited to, P20EA (TAATACGACTCCGAATTCCC (SEQ ID NO: 2)) and P10EA (GGGAATTCGG (SEQ ID NO: 3)).
- the third TCR or BCR C region-specific primer is set to a region of about 150 bases from the C region 5 ′ end, and the first TCR or BCR C region-specific primer and Two TCR or BCR C region-specific primers are set between the C region 5 ′ end and about 300 bases.
- the first, second and third TCR or BCR C region-specific primers are each independently for quantitative BCR analysis, and the five isotype sequences include Designed separately with specific primers, perfectly matched to the target sequence and designed to ensure a mismatch of 5 bases or more for other isotypes and similar IgG subtypes (IgG1, IgG2, IgG3, IgG4) Alternatively, for IgA subtypes (IgA1, IgA2), each subtype is designed to be a perfect match so that it can be handled with one primer.
- the primer design parameters are set to a base sequence length of 18-22 bases, a melting temperature of 54-66 ° C., and% GC (% guanine / cytosine content) of 40-65%.
- These preferable values such as the base sequence length may vary depending on the model, but those skilled in the art can appropriately set them depending on the model.
- the conditions of the method for determining the sequence of the C region specific primer of the first, second and third TCR or BCR include the following. 1. 1. Incorporate a plurality of subtype sequences and / or allele sequences into base sequence analysis software and align them; Search for multiple primers that satisfy the parameter conditions in the C region using the primer design software; select a primer in a region without mismatched bases in the alignment sequence of 3.1; according to 4.3 Confirm that there are multiple mismatched sequences for each subtype and / or allele downstream of the determined primer, and if not, search for the primer further upstream and repeat this as necessary.
- the C region specific primer of the first TCR or BCR is up to 41-300 bases relative to the first base of the first codon of the C region sequence resulting from splicing up to the C of the second TCR or BCR.
- Region-specific primers are up to 21-300 bases based on the first base
- C region-specific primers of the third TCR or BCR are within 150 bases based on the first base
- subtypes and / or alleles It is set at a position including the non-matching part.
- the first TCR or BCR C region specific primer has the following structure: CM1 (SEQ ID NO: 5), CA1 (SEQ ID NO: 8), CG1 (SEQ ID NO: 11), CD1 (SEQ ID NO: 14), CE1 (SEQ ID NO: 17), CA1 (SEQ ID NO: 35), CB1 (SEQ ID NO: 37) and the like, but are not limited thereto.
- the second TCR or BCR C region specific primer has the following structure: CM2 (SEQ ID NO: 6), CA2 (SEQ ID NO: 9), CG2 (SEQ ID NO: 12), CD2 (SEQ ID NO: 15), CE2 (SEQ ID NO: 18), CA2 (SEQ ID NO: 35), CB2 (SEQ ID NO: 37) and the like, but are not limited thereto.
- the third TCR or BCR C region specific primer has the following structure: CM3-GS (SEQ ID NO: 7), CA3-GS (SEQ ID NO: 10), CG3-GS (SEQ ID NO: 13 ), CD3-GS (SEQ ID NO: 16) or CE3-GS (SEQ ID NO: 19).
- all of the TCR or BCR C region specific primers are provided in sets corresponding to all subclasses of TCR or BCR.
- the specific sequences are as follows: CM1 (SEQ ID NO: 5), CA1 (SEQ ID NO: 8), CG1 (SEQ ID NO: 11), CD1 (SEQ ID NO: 14), CE1 (SEQ ID NO: 17), CM2 (SEQ ID NO: 6), CA2 (SEQ ID NO: 9), CG2 (SEQ ID NO: 12), CD2 (SEQ ID NO: 15), CE2 (SEQ ID NO: 18), CM3-GS (SEQ ID NO: 7), CA3-GS (SEQ ID NO: 10), CG3-GS ( SEQ ID NO: 13), CD3-GS (SEQ ID NO: 16) or CE3-GS (SEQ ID NO: 19), CA1 (SEQ ID NO: 35), CB1 (SEQ ID NO: 37), CA2 (SEQ ID NO: 35), CB2 (SEQ ID NO: 37)
- the present invention provides a method for performing gene analysis using a sample produced by the method of the present invention.
- the gene analysis can be performed using any analysis method.
- V, D, J obtained from the well-known IMGT (the international ImMgeneTics information system, http://www.imgt.org) database.
- IMGT the international ImMgeneTics information system, http://www.imgt.org
- new software Repetoire Genesis
- the gene analysis is a quantitative analysis of a repertoire of a variable region of a T cell receptor (TCR) or a B cell receptor (BCR).
- TCR T cell receptor
- BCR B cell receptor
- a method for determining the profile of recombinant DNA sequences in T cells and / or B cells. The method can include the steps of isolating a sample from the subject, including one or more rounds of nucleic acid amplification, spatially isolating individual nucleic acids, and sequencing the nucleic acids.
- a method for determining the correlation of one or more repertoires in a subject or individual is provided.
- a method is provided for developing an algorithm that can predict the correlation of one or more repertoires in any sample from a subject having a disease.
- an algorithm that can predict the correlation of one or more repertoires in any sample derived from a subject is used to discover the correlation of one or more repertoires of an individual Providing a method for
- a method is provided for creating an algorithm for calculating a disease activity score.
- a method for monitoring an individual's disease state is provided.
- the present invention provides bioinformatics for quantitative analysis of T cell receptor (TCR) or B cell receptor (BCR) variable region repertoire using next generation sequencing technology.
- TCR T cell receptor
- BCR B cell receptor
- the present invention is a method for analyzing TCR or BCR repertoire, which comprises the following steps: (1) a gene comprising at least one of a V region, a D region, a J region, and optionally a C region Step of providing a reference database for each area: (2) Step of providing an input sequence set obtained by performing trimming as necessary and extracting an appropriate length as necessary; (3) About the input sequence set Performing a homology search with the reference database for each gene region and recording an alignment with the approximate reference allele and the sequence of the reference allele; (4) assigning the V region and the J region to the input sequence set; And extracting the nucleic acid sequence of the D region based on the assignment result (preferably, the V region for the input sequence set).
- the J region are assigned, and the CDR3 sequence is extracted using the CDR3 beginning on the reference V region and the CDR3 end on the reference J as markers, and (5) the nucleic acid sequence of the D region is translated into an amino acid sequence;
- a step of classifying the D region using an amino acid sequence preferably, a step of translating the nucleic acid sequence of the CDR3 into an amino acid sequence and classifying the D region using the amino acid sequence;
- (6) in (5) The step of deriving a TCR or a BCR repertoire by calculating the appearance frequency of each of the V region, the D region, the J region, and, if necessary, the appearance frequency of the C region, or a combination thereof, based on the above classification is included.
- FIG. 43 is a flowchart showing a processing flow showing a method of analyzing TCR or BCR repertoire in the gene analysis system of the present invention.
- each symbol S1 to S6 in the figure corresponds to each step (1) to step (6) in the following description.
- the step of providing a reference database for each gene region including at least one of V region, D region, J region and optionally C region is, This can be achieved by appropriately selecting and providing a database including information on the V region.
- the step of (2) trimming as necessary and providing an input sequence set in which an appropriate length is extracted as necessary includes functions such as software as needed. This is achieved by providing trimming and, if necessary, selecting the length as appropriate and providing the extracted input sequence set.
- the input sequence can be, for example, a set of amplification products amplified by known methods, or a set of amplification products PCR amplified by an unbiased method as described in the application filed on the same day as this application.
- (3) performing a homology search with the reference database for each gene region for the input sequence set, and recording an alignment with the approximate reference allele and / or the sequence of the reference allele Uses a software for performing homology search as appropriate, and performs a homology search with the reference database for each gene region (for example, V region, etc.) on the input sequence set, and the resulting approximate reference allele and This is done by recording an alignment with the sequence of the reference allele.
- the “BLAST” or “BLAST analysis” box, the IMGT database below it, and the place where they are connected by a vertical double line are applicable.
- the step of (4) assigning the V region and J region to the input sequence set and extracting the nucleic acid sequence of the D region based on the assignment result is based on known information from the sequence alignment.
- an extraction which can be achieved by determining the V region and / or the J region, preferably the V region and the J region are assigned to the input sequence set, and the CDR3 head on the reference V region is assigned. It can be achieved by extracting the CDR3 sequence with the end of CDR3 on the reference J as a landmark.
- FIG. 29 and FIG. 30 using the horizontal arrow below V and the horizontal arrow below J as markers, it is possible to determine both ends of this region as indicated by the horizontal arrow below Dno.
- the step of translating the nucleic acid sequence of the D region into an amino acid sequence and classifying the D region using the amino acid sequence comprises translating into the amino acid using a method known in the art. This can be achieved by extracting the sequence corresponding to the D region by homology search or the like for the amino acid sequence.
- the CDR3 nucleic acid sequence can be translated into an amino acid sequence, and the D region can be classified using the amino acid sequence.
- the step of deriving the TCR or BCR repertoire can be calculated by, for example, arranging the appearance frequencies of the V region, D region, J region and / or C region calculated in the above steps in a list. Thereby, a TCR or a BCR repertoire can be derived.
- a reference database is provided. This may be stored in the external storage device 1405, but can usually be acquired as a publicly provided database through the communication device 1411. Alternatively, it may be input using the input device 1409 and recorded in the RAM 1403 or the external storage device 1405 as necessary.
- a database including a target area such as the V area is provided.
- an input array set is provided.
- a set of sequence information obtained from, for example, a set of amplification products amplified by a PCR amplification reaction is input using the input device 1409 or via the communication device 1411.
- a device for receiving the amplification product of the PCR amplification reaction and analyzing the gene sequence may be connected. Such a connection is made through the system bus 1420 or through the communication device 1411.
- trimming and / or extraction of an appropriate length can be performed as necessary.
- Such processing is performed by the CPU 1401.
- Programs for trimming and / or extracting can be provided via an external storage device, a communication device, or an input device, respectively.
- step (3) alignment is performed.
- the input sequence set is searched for homology with the reference database for each gene region.
- This homology search is performed with respect to the reference database obtained via the communication device 1411 or the like.
- Perform program processing This process is performed by the CPU 1401. Further, the obtained result is analyzed and approximated with a reference allele and / or a sequence of the reference allele. This process is also performed by the CPU 1401. Programs for executing these can be provided via an external storage device, a communication device, or an input device, respectively.
- step (4) the nucleic acid sequence of D information is detected.
- This process is also performed by the CPU 1401.
- Programs for executing these can be provided via an external storage device, a communication device, or an input device, respectively.
- the V region and the J region are assigned to the input array set.
- Assignment processing is also performed by the CPU 1401.
- the CPU 1401 also extracts the nucleic acid sequence of the D region based on the assignment result.
- Programs for assignment and extraction processing may also be provided via an external storage device or a communication device or an input device, respectively.
- it can be preferably achieved by determining the V region and / or the J region from the sequence alignment based on known information or the like.
- the result can be saved in the RAM 1403 or the external storage device 1405.
- Such extraction is preferably achieved by assigning a V region and a J region to the input sequence set, and extracting a CDR3 sequence with the CDR3 head on the reference V region and the CDR3 end on the reference J as markers. can do.
- Such processing can also be performed by the CPU 1401.
- Programs for this purpose can also be provided via an external storage device, a communication device, or an input device, respectively.
- Step (5) classifies the D region.
- the nucleic acid sequence of the D region is translated into an amino acid sequence, and the D region is classified using the amino acid sequence.
- a program for this processing can also be provided via an external storage device, a communication device, or an input device, respectively, by extracting a sequence corresponding to the D region by homology search etc. with respect to the obtained amino acid sequence.
- Such processing is also performed by the CPU 1401.
- a program for this processing can also be provided via an external storage device, a communication device, or an input device, respectively. It can be translated into an amino acid sequence, and the D region can be classified using the amino acid sequence. Made in 401. Program for this process it may also be respectively provided via an external storage device or communication device or the input device.
- step (6) by calculating the appearance frequency of each of the V region, D region, J region and, if necessary, the C region or a combination thereof based on the above classification, the TCR or BCR repertoire is calculated. Is derived. Processing for this calculation and derivation is also performed by the CPU 1401. A program for this processing can also be provided via an external storage device, a communication device, or an input device, respectively.
- the gene region used in the present invention includes all of the V region, D region, J region and optionally the C region.
- the reference database is a database in which a unique ID is assigned to each array. By uniquely assigning IDs, gene sequences can be analyzed based on a simple index called ID.
- the input array set is an unbiased array set.
- Unbiased sequence sets can be performed by PCR amplification by an unbiased method as described herein. If the accuracy of the non-bias method is not required, a “pseudo non-bias method” having a relatively low level such as the Smart method may be used. Therefore, in this specification, the term “unbiased” refers to an unbiased accuracy that can be achieved by the method of the present invention, and a “pseudo-unbiased method” when the level is not reached.
- non-bias methods as described herein may be referred to as “accurate unbias” when distinguished from each other. It is understood that this is the level achieved using the methods described in the specification.
- the array set is trimmed. By performing trimming, unnecessary or inappropriate nucleic acid sequences can be removed, and the efficiency of analysis can be increased.
- trimming removes low quality regions from both ends of the lead; removes regions that match the adapter sequence by 10 bp or more from both ends of the lead; and a remaining length of 200 bp or more (TCR) or 300 bp or more (BCR) If so, it is achieved by the steps used for analysis as high quality.
- the low quality is a QV value with a 7 bp moving average of less than 30.
- the approximate sequence is the closest sequence.
- the approximating array is: 1. Number of matching bases 2. kernel length; Score, 4. Determined by the rank of the alignment length.
- the homology search is performed under conditions that allow random mutations to be scattered throughout. Such conditions are well expressed, for example, for BLAST / FASTA optimal parameters: allow up to 33% mismatch over the entire alignment length and up to 60 for any 30 bp in it.
- the homology search includes (1) reduced window size, (2) reduced mismatch penalty, (3) reduced gap penalty, and ( 4)
- the top index priority includes at least one condition for the number of matching bases.
- This condition can be used, for example, in a situation where only a part of the region is classified using a shorter ( ⁇ 200 bp) sequence (a situation that deviates from the “preferred example”), and a situation where an Illumina sequencer is used. But it can be used. In this case, the possibility of using bwa or bowtie for the homology search is considered.
- the D region is classified based on the appearance frequency of the amino acid sequence.
- step (5) if there is a D region reference database, the combination of the homology search result with the CDR3 nucleic acid sequence and the amino acid sequence translation result is used as the classification result.
- the classification is made only by the appearance frequency of the amino acid sequence.
- the appearance frequency is made in gene name units and / or allyl units.
- the step (4) assigns a V region and a J region to the input sequence set, and extracts a CDR3 sequence with the CDR3 head on the reference V region and the CDR3 end on the reference J as markers. Is included.
- the step (5) includes translating the CDR3 nucleic acid sequence into an amino acid sequence and classifying the D region using the amino acid sequence.
- the present invention is a system for analyzing a TCR or BCR repertoire, the system comprising: (1) a gene region comprising at least one of a V region, a D region, a J region, and optionally a C region Means for providing a reference database for each: (2) Means for providing an input sequence set obtained by performing trimming as necessary and extracting an appropriate length as necessary; (3) About the input sequence set; Means for performing homology search with the reference database for each gene region and recording an alignment with the approximate reference allele and / or sequence of the reference allele; (4) V region and J region for the input sequence set; Means for assigning and extracting the nucleic acid sequence of the D region based on the assignment result; (5) translating the nucleic acid sequence of the D region into an amino acid sequence; Means for classifying the D region using the non-acid sequence; (6) Each appearance frequency of the V region, the D region, the J region and, if necessary, the C region based on the classification in (5), or a combination thereof A system is
- the present invention is a computer program that causes a computer to execute processing of a method of analyzing a TCR or BCR repertoire, the method comprising the following steps: (1) V region, D region, J region, and necessary A step of providing a reference database for each gene region including at least one of the C regions according to: (2) Trimming as necessary, and extracting an input sequence set having an appropriate length as necessary (3) performing a homology search with the reference database for each of the gene regions with respect to the input sequence set, and recording an alignment with the approximate reference allele and / or the sequence of the reference allele; 4) Assign V region and J region for the input sequence set, and based on the assignment result, nucleic acid of D region (5) translating the nucleic acid sequence of the D region into an amino acid sequence, and classifying the D region using the amino acid sequence; (6) based on the classification in (5), V Deriving a TCR or a BCR repertoire by calculating the appearance frequency of each region, region D, region J, and, if necessary, region
- the present invention is a recording medium storing a computer program that causes a computer to execute processing of a method for analyzing a TCR or BCR repertoire, the method comprising the following steps: (1) V region, D Step of providing a reference database for each gene region including at least one of region, J region, and C region as necessary: (2) Trimming as necessary, and having an appropriate length as necessary Providing the extracted input sequence set; (3) performing a homology search with the reference database for each gene region for the input sequence set, and aligning with the reference allele and / or the sequence of the reference allele (4) assigning V region and J region to the input array set, and assigning result And (5) translating the nucleic acid sequence of the D region into an amino acid sequence and classifying the D region using the amino acid sequence; (6) in (5) Deriving a TCR or BCR repertoire by calculating the appearance frequency of each of the V region, the D region, the J region, and, if necessary, the C region, or a combination thereof based on the classification of A recording
- the gene analysis system 1 includes a CPU 1401 built in a computer system via a system bus 1420, a RAM 1403, an external storage device 1405 such as a flash memory such as a ROM, HDD, magnetic disk, or USB memory, and an input / output interface (I). / F) 1425 is connected.
- An input device 1409 such as a keyboard and a mouse, an output device 1407 such as a display, and a communication device 1411 such as a modem are connected to the input / output I / F 1425, respectively.
- the external storage device 1405 includes an information database storage unit 1430 and a program storage unit 1440. Both are fixed storage areas secured in the external storage device 1405.
- the reference database In the database storage unit 1430, the reference database, the input sequence set, the generated classification data, TCR or BCR repertoire data, etc., or information acquired via the communication device 1411 or the like is written and updated as needed.
- the information belonging to the sample to be accumulated can be identified by the ID defined in each master table. It becomes possible to manage.
- the database storage unit 1430 stores sample provider ID, sample information, nucleic acid analysis results, known individual / physiology information, and TCR or BCR repertoire analysis results in association with the sample IDs as input sequence set entry information.
- the TCR or BCR repertoire analysis result is information obtained by processing the nucleic acid analysis result by the processing of the present invention.
- the computer program stored in the program storage unit 1440 configures the computer as the above-described processing system, for example, a system that performs processing such as trimming, extraction, alignment, assignment, classification, translation, and the like.
- Each of these functions is an independent computer program, its module, routine, etc., and is executed by the CPU 1401 to configure the computer as each system or device. In the following, it is assumed that each function in each system cooperates to constitute each system.
- the present invention provides a method for quantitatively analyzing a subject's T cell receptor (TCR) or B cell receptor (BCR) variable region repertoire using a database.
- the method comprises (1) providing a nucleic acid sample comprising a T cell receptor (TCR) or B cell receptor (BCR) nucleic acid sequence, amplified unbiased from the subject; (2) contained in the nucleic acid sample And (3) calculating the frequency of occurrence of each gene or a combination thereof based on the determined nucleic acid sequence, and deriving the TCR or BCR repertoire of the subject.
- This method and methods that include one or more additional features described herein are also referred to herein as “the repertoire analysis method of the present invention”.
- a system for realizing the repertoire analysis method of the present invention is also referred to as a “repertoire analysis system of the present invention”.
- (1) providing a nucleic acid sample comprising a T cell receptor (TCR) or B cell receptor (BCR) nucleic acid sequence amplified in a biased manner from the subject in the method of the present invention comprises determining the nucleic acid sequence; Any sample may be used as long as the sample is suitable.
- Reverse transcriptase-PCR in addition to the above-mentioned preferred amplification method of the present invention, Reverse transcriptase-PCR, real-time PCR, digital PCR, emulsion PCR, amplified fragment length polymorphism (AFLP) PCR, allele-specific PCR, assembled PCR, asymmetric PCR , Colony PCR, helicase-dependent amplification, hot start PCR, inverse PCR, in situ PCR nested PCR, Touchdown PCR, loop-mediated isometric PCR (LAMP), Nucleic acid sequence based amplification (a NASB) Reaction, Branch DNA Amplification, Rolling Circle Amplification, Circle to circle Amplification, SPIA amplification, Trget Amplification by Capture and Ligation (TACL), 5'-Rapid amplification of cDNA end (5'-RACE), 3'-Rapid amplification of cDNA End (3'-RACE), Switching Mechanism at 5'-endof the RNA Transscript (SMART
- any method may be used as long as the nucleic acid sequence can be determined.
- an automated large-scale sequencing method include sequencing using the Roche 454 sequencer (GS FLX +, GS Junior), sequencing using the ion torrent sequencer (Ion PGM TM Sequencer) method, Illumina's method (GenomeAnalyzer IIx, There is sequencing using Hiseq, Miseq).
- Other sequence methods include Helicescope TM Sequencer, Helicos True Single Molecule Sequencing (tSMA) (Harris. TD et al.
- the Roche 454 sequence creates a single-stranded DNA in which two types of adapters that specifically bind to the 3 ′ end and the 5 ′ end are bound.
- the single-stranded DNA binds to the bead via an adapter, and wraps in a water-in-oil emulsion to form a microreactor with the bead and the DNA fragment.
- emulsion PCR is performed in a water-in-oil emulsion to amplify the target gene.
- the beads are applied to a pico titer plate and sequenced.
- ATP is produced by sulfurylase using pyrophosphate generated when dNTP is taken into DNA by DNA polymerase (Pyrosequencing).
- Luciferase emits fluorescence using this ATP and Luciferin as substrates, and the base sequence is determined by detecting with a CCD camera.
- the ion torrent method after emulsion PCR is performed in the same manner as Roche, the beads are transferred to a microchip and a sequence reaction is performed on the microchip. In the detection, the hydrogen ion concentration released when DNA is extended by polymerase is detected on a semiconductor chip and converted into a base sequence.
- Illumina sequencing is a method in which the target DNA is amplified on a flow cell by the bridge PCR method and Sequencing-by-synthesis, and sequencing is performed while synthesis.
- the bridge PCR method creates a single-stranded DNA with different adapter sequences added to both ends.
- An adapter sequence on the 5 ′ end side is fixed in advance on the flow cell, and is fixed to the flow cell by performing an extension reaction.
- an adapter on the 3 ′ end side is fixed at an adjacent position, and is combined with the 3 ′ end of the synthesized DNA to synthesize a double-stranded DNA in a so-called bridge form.
- bridge binding ⁇ extension ⁇ denaturation a large number of single-stranded DNA fragments are locally amplified to form an accumulated cluster. Sequencing is performed using this single-stranded DNA as a template. Sequencing-by-synthesis performs a one-base synthesis reaction with 3 ′ end block fluorescent dNTPs by DNA polymerase after adding a sequence primer.
- a fluorescent substance bound to a base is excited by laser light, and light emission is recorded as a photograph by a fluorescence microscope.
- the base sequence is determined by removing the fluorescent substance and blocking, performing the next extension reaction, and proceeding with the step of detecting fluorescence.
- the nucleic acid sample includes a plurality of types of T cell receptor (TCR) or B cell receptor (BCR) nucleic acid sequences, and in the step of determining the sequence of (2), single sequencing is performed.
- TCR T cell receptor
- BCR B cell receptor
- single sequencing is performed.
- the method of the present invention can reduce or eliminate the bias that can arise from performing multiple types of sequencing by performing a single sequencing. Therefore, it is particularly useful for accurately detecting TCR or BCR leads that occur only infrequently.
- At least one of the sequences used as a primer in the single sequencing, in the amplification from the nucleic acid sample to the sample for sequencing, at least one of the sequences used as a primer is the same as the nucleic acid sequence encoding the C region or a complementary strand thereof. It has the arrangement.
- a primer having the same sequence as the nucleic acid sequence encoding the C region or its complementary strand similar amplification can be performed in any TCR or BCR, and unbiased can be achieved.
- the single sequencing is performed using a common adapter primer.
- the common adapter primer is of a base length suitable for amplification, is designed to be able to form stable duplexes with less difficulty in homodimers and intramolecular hairpin structures, and all TCRs in the database.
- the gene sequence is not highly homologous and / or designed to have the same melting temperature (T m ) as the C region specific primer.
- T m melting temperature
- common adapter primers are selected that do not have homodimer and intramolecular hairpin structures and are not homologous to other genes, including BCR or TCR.
- the common adapter primer is P20EA (SEQ ID NO: 2) and / or P10EA (SEQ ID NO: 3).
- the non-biased amplification includes not being V region specific amplification.
- the bias can be further reduced or eliminated as compared with the case where non-biased amplification is performed by devising a multiplex using a V-specific primer.
- the repertoire targeted by the present invention is a BCR variable region repertoire
- the nucleic acid sequence is a BCR nucleic acid sequence.
- BCR is likely to be mutated, and in particular, it is said that many mutations occur in the V region, and accurate analysis of BCR repetoa is difficult by the technique using V region specific amplification.
- the present invention provides a method for analyzing a disease, disorder or condition of the subject based on a TCR or BCR repertoire derived based on the repertoire analysis method of the present invention.
- a technique for analyzing the disease, disorder or condition of the subject based on the TCR or BCR repertoire derived based on the repertoire analysis method of the present invention is a disease, disorder or the like.
- Clinical information such as status, and derived lead type, number of leads, read frequency, V data, J data, C data, CDR3 data, etc., are linked, and a database is created using a spreadsheet such as EXCEL It starts from doing.
- a TCR having a known function such as NKT or MAIT is searched. 2. Search existing public database and collate with TCR or BCR whose functions such as antigen specificity are known. 3.
- the subject's disease, disorder or condition is a blood tumor, colon cancer, immune condition, rheumatoid arthritis, adult T-cell leukemia, T-cell large granular lymphocytic leukemia, Examples include idiopathic thrombocytopenic purpura, but are not limited thereto.
- the present invention provides a method for quantitatively associating a disease, disorder or condition of a subject determined by the method of the present invention with the TCR or BCR repertoire, Methods are provided for treating or preventing a disease, disorder or condition in the subject comprising selecting a means for treatment or prevention.
- the disease, disorder or condition of the subject targeted in the method for treatment or prevention of the present invention is a blood tumor, colon cancer, immune condition, rheumatoid arthritis, adult T cell leukemia, T cell.
- examples include, but are not limited to, large granular lymphocytic leukemia and idiopathic thrombocytopenic purpura.
- the present invention provides a system (analysis system) for quantitatively analyzing a subject's T cell receptor (TCR) or B cell receptor (BCR) variable region repertoire using a database.
- the system comprises: (1) a kit for providing a nucleic acid sample comprising a T cell receptor (TCR) or a B cell receptor (BCR) nucleic acid sequence, amplified unbiased from the subject; (2) the nucleic acid sample An apparatus for determining the nucleic acid sequence contained in the gene; and (3) calculating the appearance frequency of each gene or a combination thereof based on the determined nucleic acid sequence, and deriving the TCR or BCR repertoire of the subject
- a device for A system that includes such a system and one or more additional features described herein is referred to as a “repertoire analysis system of the present invention”.
- the repertoire analysis system of the present invention realizes the “repertoire analysis method of the present invention”.
- the nucleic acid sample comprises a plurality of types of T cell receptor (TCR) or B cell receptor (BCR) nucleic acid sequences
- the device of (2) uses a single sequencing to extract the nucleic acid sequences. Configured to be able to determine.
- the single sequencing is characterized in that at least one of the sequences used as primers in the amplification from the nucleic acid sample to the sequencing sample has the same sequence as the C region.
- the system of the present invention can reduce or eliminate the bias that can arise from performing multiple types of sequencing by performing a single sequencing.
- the system of the present invention is particularly useful for accurately detecting TCR or BCR leads that occur only infrequently.
- At least one of the sequences used as a primer in the single sequencing, in the amplification from the nucleic acid sample to the sample for sequencing, at least one of the sequences used as a primer is the same as the nucleic acid sequence encoding the C region or a complementary strand thereof. It has the arrangement.
- a primer may be provided in this apparatus, may be included in a kit, or may be provided separately.
- the single sequencing is performed using a common adapter primer.
- a common adapter primer may be provided in this apparatus, may be included in a kit, or may be provided separately.
- the common adapter primer is of a base length suitable for amplification, is designed to be able to form stable duplexes with less difficulty in homodimers and intramolecular hairpin structures, and all TCRs in the database. It is not homologous to the gene sequence and / or is designed to have the same melting temperature (Tm) as the C region specific primer. More preferably, common adapter primers are selected that do not have homodimer and intramolecular hairpin structures and are not homologous to other genes, including BCR or TCR.
- the common adapter primer is P20EA (SEQ ID NO: 2) and / or P10EA (SEQ ID NO: 3).
- the nucleic acid sequence contained in the nucleic acid sample provided by the kit of the present invention is subjected to the unbiased amplification, but the amplification is not V region specific amplification.
- the bias can be further reduced or eliminated as compared with the case where non-biased amplification is performed by devising a multiplex using a V region specific primer.
- the repertoire to be analyzed by the system of the present invention is a BCR variable region repertoire
- the nucleic acid sequence is a BCR nucleic acid sequence.
- BCR is likely to be mutated, and in particular, it is said that many mutations occur in the V region, and accurate analysis of BCR repetoa is difficult by the technique using V region specific amplification. By using the system of the present invention, it has become possible to accurately analyze BCR repeaters.
- the present invention comprises a subject's analysis system, and means for analyzing the subject's disease, disorder or condition based on a TCR or BCR repertoire derived based on the system.
- a system for analyzing a disease, disorder or condition is provided.
- the means for analyzing the disease, disorder or condition of the subject based on the TCR or BCR repertoire derived based on the analysis system of the present invention includes clinical information such as the disease, disorder or condition and the derived lead. It begins with connecting read data consisting of type, number of reads, read frequency, V region, J region, C region, CDR3 array, etc., and creating a database using a spreadsheet such as EXCEL.
- a TCR having a known function such as NKT or MAIT is searched. 2. Search existing public database and collate with TCR or BCR whose functions such as antigen specificity are known. 3. Search in the constructed database or existing public database, and relate to the disease, disorder or condition from the origin, characteristics or function of the common sample. Next, regarding the lead arrangement in the sample, 1. Determine if the frequency of a particular lead increases (increases clonality). 2. It is examined whether the frequency of use of a specific V chain or J chain increases or decreases depending on the onset of the disease and the state of the disorder. 3. It is examined whether the CDR3 sequence length in a specific V chain is increased or decreased depending on the onset of the disease and the state of the disorder. 4).
- composition and sequence of the CDR3 region that changes depending on the onset of the disease and the state of the disorder are examined. 5. Search for leads that appear or disappear depending on the onset of the disease or the state of the disorder. 6). Search for leads that increase or decrease depending on the onset of the disease and the state of the disorder. 7). Leads that appear or increase / decrease in response to the onset of the disease or the state of the disorder are searched in other samples and associated with the disease, disorder or condition. 8). Diversity index or similarity index is calculated using statistical analysis software such as ESTIMATES or R (vegan) using data such as number of samples, lead type and number of leads. 9. Changes in diversity index or similarity index can be associated with disease onset and disorder status.
- the disease, disorder or condition of the subject that can be analyzed by the analysis system of the present invention includes blood tumor, colon cancer, immune condition, rheumatoid arthritis, adult T cell leukemia, T cell large granules.
- examples thereof include, but are not limited to, lymphocytic leukemia and idiopathic thrombocytopenic purpura.
- the present invention provides a means for quantitatively associating a disease, disorder or condition of a subject determined by the analysis system of the present invention with the TCR or BCR repertoire,
- a system treatment system or prevention system for treating or preventing a disease, disorder or condition in the subject, comprising means for selecting a means for treatment or prevention.
- the means for associating the subject's disease, disorder or condition with the TCR or BCR repertoire quantitatively in the system of the present invention can be realized by the following configuration. That is, repertoire information derived by the analysis system of the present invention can be read, and this can be realized by reading information on a subject's disease, disorder or condition and associating them.
- the V region, the J region, and the C region are assigned from the collation operation with respect to the existing reference sequence, and the CDR3 sequence is determined.
- the corresponding reads are counted, and the number of reads detected in the sample and the ratio to the total number of reads for each unique lead (the other lead that does not have the same sequence) ( Frequency) is calculated.
- This information (read sequence, number of reads, read frequency, V region, J region, C region, CDR3 sequence) and clinical information of the subject (history, disease name, disease type, progression, severity, HLA type, immune status, etc.) )
- a spreadsheet such as EXCEL or software having a database creation function.
- the lead array in the sample is sorted by the number of reads and frequency, and ranking processing is performed.
- the number of reads is totaled for each V area or J area, and the V area usage frequency or J area usage frequency is calculated. Based on these information, 1. Determine if the frequency of a particular lead increases (increases clonality). 2. It is examined whether the frequency of use of a specific V chain or J chain increases or decreases depending on the onset of the disease and the state of the disorder. 3. It is examined whether the CDR3 sequence length in a specific V chain is increased or decreased depending on the onset of the disease and the state of the disorder. 4). The composition and sequence of the CDR3 region that changes depending on the onset of the disease and the state of the disorder are examined. 5. Search for leads that appear or disappear depending on the onset of the disease or the state of the disorder. 6).
- a means for selecting an appropriate treatment or prevention means from a quantitative relationship can be configured as follows. In other words, the selection of this means of selection is realized by associating data showing quantitativeness with past information on treatment, treatment or prevention, or information currently available, and realizing selection of those whose subsequent courses improve. Can do.
- the disease, disorder or condition of the subject is a blood tumor, colon cancer, immune condition, rheumatoid arthritis, adult T cell leukemia, T cell large granular lymphocytic leukemia, idiopathic thrombocytopenic purpura.
- a blood tumor colon cancer
- immune condition rheumatoid arthritis
- adult T cell leukemia T cell large granular lymphocytic leukemia
- idiopathic thrombocytopenic purpura idiopathic thrombocytopenic purpura.
- the present invention relates to TRCR10 / TRAJ15 / CVVRTAGTALIFG (SEQ ID NO: 1450) or TCR ⁇ containing a nucleic acid encoding the same, and / or TRBV29-1 / TRBJ2-7 / CSVERGGSLGEQYFG ( SEQ ID NO: 1500) or a monoclonal T cell for T cell large granular lymphocytic leukemia (T-LGL) that expresses TCR ⁇ comprising the nucleic acid encoding it.
- T-LGL T cell large granular lymphocytic leukemia
- T cell has various usefulness as shown in Examples and the like.
- TRAV10 / TRAJ15 / CVVRATGITALIFG SEQ ID NO: 1450
- a nucleic acid encoding the same in TCR ⁇ and / or TRBV29-1 / TRBJ2-7 / CSVERGGSLGEQYFG SEQ ID NO: 1500
- T-LGL large granular lymphocytic leukemia
- the “detection agent” broadly refers to any drug that can detect a target object (eg, peptide, nucleic acid, cell, etc.).
- a target object eg, peptide, nucleic acid, cell, etc.
- “detection” or “quantification” of polynucleotide or polypeptide expression includes, for example, measurement of mRNA and immunology, including binding or interaction with a marker detection agent. It can be achieved using any suitable method, including measurement methods. Examples of molecular biological measurement methods include Northern blotting, dot blotting, and PCR.
- immunological measurement methods include ELISA using a microtiter plate, RIA, fluorescent antibody method, luminescence immunoassay (LIA), immunoprecipitation (IP), immunodiffusion method (SRID), immunization. Examples are turbidimetry (TIA), Western blotting, immunohistochemical staining, and the like. Examples of the quantitative method include an ELISA method and an RIA method. It can also be performed by a gene analysis method using an array (eg, DNA array, protein array). The DNA array is widely outlined in (edited by Shujunsha, separate volume of cell engineering "DNA microarray and latest PCR method”). For protein arrays, see Nat Genet. 2002 Dec; 32 Suppl: 526-32.
- gene expression analysis methods include, but are not limited to, RT-PCR, RACE method, SSCP method, immunoprecipitation method, two-hybrid system, in vitro translation and the like.
- Such further analysis methods are described in, for example, Genome Analysis Experimental Method / Yusuke Nakamura Laboratory Manual, Editing / Yusuke Nakamura Yodosha (2002), etc., all of which are incorporated herein by reference. Is done.
- the “expression amount” refers to an amount in which a polypeptide or mRNA is expressed in a target cell, tissue or the like.
- Such expression level is evaluated by any appropriate method including immunoassay methods such as ELISA method, RIA method, fluorescent antibody method, Western blot method, and immunohistochemical staining method using the antibody of the present invention.
- the expression level of the peptide at the mRNA level can be mentioned.
- “Change in expression level” means expression at the protein level or mRNA level of the polypeptide used in the present invention evaluated by any appropriate method including the above immunological measurement method or molecular biological measurement method. Means that the amount increases or decreases.
- the present invention also provides an agent for detecting TRAV10 / TRAJ15 / CVVRATGTALIFG (SEQ ID NO: 1450) or a nucleic acid encoding the same in TCR ⁇ , and / or TRBV29-1 / TRBJ2-7 / CSVERGGSLGEQYFG (SEQ ID NO: 1500) in TCR ⁇ or encoding the same.
- a diagnostic agent for T-cell large granular lymphocytic leukemia (T-LGL) is provided.
- “decrease” or “suppression” or synonyms for activity, expression products (eg, proteins, transcripts (RNA, etc.)) or a synonym is a decrease in the quantity, quality or effect of a particular activity, transcript or protein. Or activity to decrease.
- an expression product eg, a protein, transcript (such as RNA)
- a synonym thereof refers to a quantity, quality or effect of a particular activity, transcript or protein.
- drugs having various activities can be detected and screened using the ability of the marker of the present invention to reduce, suppress, increase or activate as an index.
- drug drug
- drug may also be a substance or other element (eg energy such as light, radioactivity, heat, electricity).
- Such substances include, for example, proteins, polypeptides, oligopeptides, peptides, polynucleotides, oligonucleotides, nucleotides, nucleic acids (eg, DNA such as cDNA, genomic DNA, RNA such as mRNA), poly Saccharides, oligosaccharides, lipids, small organic molecules (for example, hormones, ligands, signaling substances, small organic molecules, molecules synthesized by combinatorial chemistry, small molecules that can be used as pharmaceuticals (for example, small molecule ligands, etc.)) , These complex molecules are included, but not limited thereto.
- a polynucleotide having a certain sequence homology to the sequence of the polynucleotide (for example, 70% or more sequence identity) and complementarity examples include, but are not limited to, a polypeptide such as a transcription factor that binds to the promoter region.
- Factors specific for a polypeptide typically include an antibody specifically directed against the polypeptide or a derivative or analog thereof (eg, a single chain antibody), and the polypeptide is a receptor.
- specific ligands or receptors in the case of ligands, and substrates thereof when the polypeptide is an enzyme include, but are not limited to.
- detecting agent broadly means that a target object (eg, a normal cell (eg, normal corneal endothelial cell) or a transformed cell (eg, transformed corneal endothelial cell)) is detected. Any drug that can be used.
- a target object eg, a normal cell (eg, normal corneal endothelial cell) or a transformed cell (eg, transformed corneal endothelial cell)
- diagnostic agent refers to any drug capable of diagnosing a target state (for example, a disease) in a broad sense.
- the detection agent of the present invention may be a complex or a complex molecule in which another substance (for example, a label or the like) is bound to a detectable moiety (for example, an antibody or the like).
- a detectable moiety for example, an antibody or the like.
- complex or “complex molecule” means any construct comprising two or more moieties.
- the other part may be a polypeptide or other substance (eg, sugar, lipid, nucleic acid, other hydrocarbon, etc.).
- two or more parts constituting the complex may be bonded by a covalent bond, or bonded by other bonds (for example, hydrogen bond, ionic bond, hydrophobic interaction, van der Waals force, etc.). May be.
- the “complex” includes a molecule formed by linking a plurality of molecules such as a polypeptide, a polynucleotide, a lipid, a sugar, and a small molecule.
- interaction refers to two substances. Force (for example, intermolecular force (van der Waals force), hydrogen bond, hydrophobic interaction between one substance and the other substance. Etc.). Usually, two interacting substances are in an associated or bound state.
- bond means a physical or chemical interaction between two substances or a combination thereof. Bonds include ionic bonds, non-ionic bonds, hydrogen bonds, van der Waals bonds, hydrophobic interactions, and the like.
- a physical interaction can be direct or indirect, where indirect is through or due to the effect of another protein or compound. Direct binding refers to an interaction that does not occur through or due to the effects of another protein or compound and does not involve other substantial chemical intermediates. By measuring the binding or interaction, the degree of expression of the marker of the present invention can be measured.
- a “factor” (or drug, detection agent, etc.) that interacts (or binds) “specifically” to a biological agent such as a polynucleotide or a polypeptide is defined as that
- the affinity for a biological agent such as a nucleotide or polypeptide thereof is typically equal or greater than the affinity for other unrelated (especially less than 30% identity) polynucleotides or polypeptides. Includes those that are high or preferably significantly (eg, statistically significant). Such affinity can be measured, for example, by hybridization assays, binding assays, and the like.
- a first substance or factor interacts (or binds) “specifically” to a second substance or factor means that the first substance or factor has a relationship to the second substance or factor. Interact (or bind) with a higher affinity than a substance or factor other than the second substance or factor (especially other substances or factors present in the sample containing the second substance or factor) That means. Specific interactions (or bindings) for a substance or factor involve both nucleic acids and proteins, for example, ligand-receptor reactions, hybridization in nucleic acids, antigen-antibody reactions in proteins, enzyme-substrate reactions, etc.
- Examples include, but are not limited to, protein-lipid interaction, nucleic acid-lipid interaction, and the like, such as a reaction between a transcription factor and a binding site of the transcription factor.
- the first substance or factor “specifically interacts” with the second substance or factor means that the first substance or factor has the second substance Or having at least a part of complementarity to the factor.
- both substances or factors are proteins
- the fact that the first substance or factor interacts (or binds) “specifically” to the second substance or factor is, for example, by antigen-antibody reaction Examples include, but are not limited to, interaction by receptor-ligand reaction, enzyme-substrate interaction, and the like.
- the first substance or factor interacts (or binds) “specifically” to the second substance or factor by the transcription factor and its Interaction (or binding) between the transcription factor and the binding region of the nucleic acid molecule of interest is included.
- antibody broadly refers to polyclonal antibodies, monoclonal antibodies, multispecific antibodies, chimeric antibodies, and anti-idiotype antibodies, and fragments thereof such as Fv fragments, Fab ′ fragments, F (ab ′). 2 and Fab fragments, and other recombinantly produced conjugates or functional equivalents (eg, chimeric antibodies, humanized antibodies, multifunctional antibodies, bispecific or oligospecific antibodies, single chains Antibody, scFV, diabody, sc (Fv) 2 (single chain (Fv) 2 ), scFv-Fc).
- antibodies may be covalently linked or recombinantly fused to enzymes such as alkaline phosphatase, horseradish peroxidase, alpha galactosidase, and the like.
- enzymes such as alkaline phosphatase, horseradish peroxidase, alpha galactosidase, and the like.
- the antibodies to various leads used in the present invention may be bound to specific various leads, respectively, and their origin, type, shape, etc. are not questioned.
- known antibodies such as non-human animal antibodies (eg, mouse antibodies, rat antibodies, camel antibodies), human antibodies, chimeric antibodies, and humanized antibodies can be used.
- monoclonal or polyclonal antibodies can be used as antibodies, but monoclonal antibodies are preferred.
- the binding of the antibody to a particular lead is preferably specific binding.
- antigen refers to any substrate that can be specifically bound by an antibody molecule.
- immunogen refers to an antigen capable of initiating lymphocyte activation that produces an antigen-specific immune response.
- epitope or “antigenic determinant” refers to a site in an antigen molecule to which an antibody or lymphocyte receptor binds. Methods for determining epitopes are well known in the art, and such epitopes can be determined by those skilled in the art using such well known techniques once the primary sequence of the nucleic acid or amino acid is provided.
- the “means” refers to anything that can be an arbitrary tool for achieving a certain purpose (for example, detection, diagnosis, treatment).
- the antibody used in the present invention may be a polyclonal antibody or a monoclonal antibody.
- the detection agent, diagnostic agent or other medicine of the present invention can take the form of probes and primers.
- the probes and primers of the present invention can specifically hybridize with specific leads.
- the expression of a particular lead is, for example, an indicator of whether it is colorectal cancer and is useful as an indicator of the degree of disease.
- nucleic acid primer refers to a substance necessary for the initiation of a reaction of a polymer compound to be synthesized in a polymer synthase reaction.
- a nucleic acid molecule for example, DNA or RNA
- the primer can be used as a marker detection means.
- a nucleic acid sequence is preferably at least 12 contiguous nucleotides long, at least 9 contiguous nucleotides, more preferably at least 10 contiguous nucleotides, and even more preferably at least 11 contiguous nucleotides.
- Nucleic acid sequences used as probes are nucleic acid sequences that are at least 70% homologous, more preferably at least 80% homologous, more preferably at least 90% homologous, at least 95% homologous to the sequences described above. Is included.
- a sequence suitable as a primer may vary depending on the nature of the sequence intended for synthesis (amplification), but those skilled in the art can appropriately design a primer according to the intended sequence. Such primer design is well known in the art, and may be performed manually or using a computer program (eg, LASERGENE, PrimerSelect, DNAStar).
- the primer according to the present invention can also be used as a primer set composed of two or more kinds of the primers.
- the primer and primer set according to the present invention can be prepared according to conventional methods in known methods for detecting a target gene using a nucleic acid amplification method such as a PCR method, an RT-PCR method, a real-time PCR method, an in situ PCR method, or a LAMP method. It can be used as a primer and a primer set.
- a nucleic acid amplification method such as a PCR method, an RT-PCR method, a real-time PCR method, an in situ PCR method, or a LAMP method. It can be used as a primer and a primer set.
- the term “probe” refers to a substance that serves as a search means used in biological experiments such as screening in vitro and / or in vivo.
- a nucleic acid molecule containing a specific base sequence or a specific Examples include, but are not limited to, peptides containing amino acid sequences, specific antibodies or fragments thereof.
- the probe is used as a marker detection means.
- the detection agent of the present invention may be labeled.
- the detection agent of the present invention may have a tag bound thereto.
- the “label” refers to a presence (for example, a substance, energy, electromagnetic wave, etc.) for distinguishing a target molecule or substance from others.
- a labeling method include RI (radioisotope) method, fluorescence method, biotin method, chemiluminescence method and the like.
- the labeling is performed with fluorescent substances having different fluorescence emission maximum wavelengths. The difference in the maximum fluorescence emission wavelength is preferably 10 nm or more.
- Alexa TM Fluor is preferred as the fluorescent substance.
- Alexa TM Fluor is a water-soluble fluorescent dye obtained by modifying coumarin, rhodamine, fluorescein, cyanine, etc., and is a series corresponding to a wide range of fluorescent wavelengths. Compared with other applicable fluorescent dyes, Stable, bright and low pH sensitive. Examples of combinations of fluorescent dyes having a fluorescence maximum wavelength of 10 nm or more include a combination of Alexa TM 555 and Alexa TM 633, a combination of Alexa TM 488 and Alexa TM 555, and the like. Any nucleic acid can be used as long as it can bind to its base moiety.
- a cyanine dye eg, CyDye TM series Cy3, Cy5, etc.
- rhodamine 6G reagent N-acetoxy-N2— Acetylaminofluorene (AAF), AAIF (iodine derivative of AAF) or the like
- the fluorescent substance having a difference in fluorescence emission maximum wavelength of 10 nm or more include a combination of Cy5 and rhodamine 6G reagent, a combination of Cy3 and fluorescein, a combination of rhodamine 6G reagent and fluorescein, and the like.
- the target object by using such a label, the target object can be modified so that it can be detected by the detection means used. Such modifications are known in the art, and those skilled in the art can appropriately carry out such methods depending on the label and the target object.
- a “tag” is a substance for sorting molecules by a specific recognition mechanism such as a receptor-ligand, more specifically, a binding for binding a specific substance.
- a substance that plays the role of a partner eg, having a relationship such as biotin-avidin, biotin-streptavidin
- label can be included in the category of “label”.
- a specific substance to which a tag is bound can be selected by bringing the substrate to which the binding partner of the tag sequence is bound into contact.
- tags or labels are well known in the art.
- Representative tag sequences include, but are not limited to, myc tag, His tag, HA, Avi tag and the like. Such a tag may be bound to the marker or marker detection agent of the present invention.
- the method of the present invention comprises contacting a target sample with the detection agent or diagnostic agent of the present invention, and whether the target target lead or the gene of these leads is present in the sample, or the level or amount thereof. It can be implemented by measuring.
- contacting refers to a polypeptide that can function as a marker, a detection agent, a diagnostic agent, a ligand, and the like of the present invention either directly or indirectly. It means physical proximity to the polynucleotide.
- the polypeptide or polynucleotide can be present in many buffers, salts, solutions, and the like. Contact includes placing the compound in, for example, a beaker, microtiter plate, cell culture flask or microarray (eg, gene chip) containing a polypeptide encoding a nucleic acid molecule or fragment thereof.
- the present invention provides a peptide that is a novel invariant TCR comprising any of the sequences shown in SEQ ID NOs: 1627 to 1647.
- a peptide can be used as an invariant and used as various indicators (for example, indicators such as diseases).
- the invention provides a mucosal associated invariant T (MAIT) comprising a sequence selected from the group consisting of SEQ ID NOs: 1648-1651, 1653-1654, 1666-1667, 1844-1848, and 1851.
- MAIT mucosal associated invariant T
- TCR peptides carried by cells and nucleic acids encoding these peptides are provided.
- Such peptides and nucleic acids can be used as mucosa-associated invariant T (MAIT) and used as various indicators (for example, indicators of diseases, etc.).
- peptides that are TCRs possessed by the mucosa-associated invariant T (MAIT) cells of the present invention, and nucleic acids encoding these peptides may be used as a diagnostic indicator for colorectal cancer. it can.
- the present invention provides a peptide that is a TCR possessed by natural killer T (NKT) cells, which contains the sequence shown in SEQ ID NO: 1668, and a nucleic acid encoding this peptide.
- NKT natural killer T
- peptides that are TCRs possessed by the NKT of the present invention and nucleic acids encoding these peptides can be used as a diagnostic indicator for colorectal cancer.
- the present invention provides peptides that are specific for colorectal cancer comprising a sequence selected from the group consisting of SEQ ID NOs: 1652, 1655 to 1665, 1669 to 1843, 1849 to 1850, and 1852 to 1860, and these A nucleic acid encoding is provided.
- these peptides and the nucleic acids that encode them can be used as a diagnostic indicator for colorectal cancer.
- the present invention provides a peptide that is specific for colorectal cancer, including a sequence selected from the group consisting of SEQ ID NOs: 1861 to 1865, and 1867 to 1909, and nucleic acids encoding them.
- these peptides and the nucleic acids that encode them can be used as a diagnostic indicator for colorectal cancer.
- the invention provides a sequence selected from the group consisting of SEQ ID NOs: 1652, 1655-1665, 1669-1843, 1849-1850, and 1852-1860, and SEQ ID NOs: 1861-1865, and 1867-1909.
- a cell population, a T cell line, or a recombinantly expressed T cell with high frequency induction of T cells having a peptide containing or a nucleic acid sequence encoding the peptide is provided.
- a peptide of a colorectal cancer-specific TCR or a nucleic acid sequence, cell, cell population, or cell line encoding the peptide is useful for diagnosis or treatment.
- colorectal cancer is found by examining that the sequence is found only in colorectal cancer patients, that the sequence is often found in colorectal cancer patients, or is accumulated in the cancer tissue of the same patient. Prognosis can be predicted.
- a cell population in which T cells having a colon cancer-specific sequence are frequently induced, a T cell line having a colon cancer-specific sequence, and a T cell in which a colon cancer-specific sequence is artificially expressed can be used for the treatment of colorectal cancer (reference: 1: Uttenthal BJ, Chua I, Morris EC, Stauss HJ. Challenges in T cell receptor gene therapy. J Gene Med. 2012 Jun; 14 (6): 386-99.
- the present invention provides a therapeutic or preventive agent for colorectal cancer comprising the above cell population, T cell line or T cell.
- the base sequence (read) of the TCR or BCR gene identified by a large-scale sequence and the appearance frequency thereof can be calculated on software, and a list, distribution, or graph can be drawn. Based on such information, the change of the repertoire is detected by using various indicators as follows. Based on the changes, the association with diseases and disorders can be clarified.
- the present invention provides a method for detecting the use frequency of a V gene using the analysis method or analysis system of the present invention.
- the V gene of each lead can be identified, and the proportion of each V gene in the total TCR or BCR gene can be calculated.
- the increase or decrease in the frequency of use of V related to the disease or condition can be clarified.
- the present invention provides a method for detecting the frequency of use of the J gene using the analysis method or analysis system of the present invention.
- the J gene of each lead can be identified, and the proportion of each J gene in the total TCR or BCR gene can be calculated.
- the increase or decrease in the frequency of use of J related to the disease or condition can be clarified.
- the present invention provides a method for detecting the frequency of subtype frequency analysis (BCR) using the analysis method or analysis system of the present invention. Based on the sequencing of the C region, the presence frequency of IgA1, IgA2, IgG1, IgG2, IgG3, and IgG4 subtypes can be calculated. Can reveal increases or decreases in specific subtypes associated with a disease or condition.
- BCR subtype frequency analysis
- the present invention provides a method for analyzing a CDR3 sequence length pattern using the analysis method or analysis system of the present invention.
- the CDR3 base sequence length of each read can be calculated to clarify the distribution.
- a normal TCR or BCR shows a normal distribution-like peak pattern, but a peak deviating from the normal distribution can be detected to clarify the relationship with a disease or disease state.
- the present invention provides a method for analyzing TCR or BCR clonality using the analysis method or analysis system of the present invention. Based on the V sequence, J sequence and CDR3 sequence of each lead, reads having the same sequence are classified, and the copy number is calculated. By calculating the ratio of the number of copies of each lead to the total number of reads, it is possible to clarify the presence of a lead that exists frequently. The degree of clonality is evaluated by sorting the descending order according to the appearance frequency, and comparing the number and ratio of leads that exist frequently with normal samples. Therefore, the change of TCR or BCR clonality related to the disease or pathological condition is examined. In particular, it can be used for detection of leukemia cells and the like.
- the present invention provides a method for extracting duplicate leads using the analysis method or analysis system of the present invention.
- the present invention provides a method for searching for a disease-specific TCR or BCR clone using the analysis method or analysis system of the present invention.
- Search for TCR or BCR leads associated with a specific disease or disorder condition in a test sample, revealing its appearance, disappearance, increase or decrease, and predicting the onset or progression of disease can do.
- the present invention provides a method for analyzing an object using a diversity index using the analysis method or analysis system of the present invention.
- the present invention provides a method for supporting analysis of an object using a diversity index using the analysis method or analysis system of the present invention.
- the number of read sequences identified based on the CDR3 sequence is counted, the number of read species and the number of individuals are calculated, and the diversity of TCR or BCR repertoire is indexed. Contrast with normal samples using Shannon-Wiener diversity index (H ′), Simpson diversity index ( ⁇ , 1- ⁇ or 1 / ⁇ ), Pierou uniformity index (J ′), Chao1 index, etc.
- H ′ Shannon-Wiener diversity index
- Simpson diversity index ⁇ , 1- ⁇ or 1 / ⁇
- Pierou uniformity index J ′
- Chao1 index etc.
- It can be used as an index for measuring the degree of recovery of the immune system after bone marrow transplantation. It can also be used as an indicator for detecting abnormalities in immune system cells associated
- the diversity in the method for analyzing a subject using a diversity index, is used as an index for measuring the degree of recovery of the immune system after bone marrow transplantation or detecting an abnormality of immune system cells associated with a hematopoietic tumor.
- Use sex index. Analysis using such diversity indexes has been difficult with conventional systems.
- various diversity indexes are obtained from the data of the number of samples, the type of lead, and the number of leads, EXCEL spreadsheet, ESTIMATES (Colwell, RK et al. Journal of Plant Ecology 5: 3-21.) Or R It can be calculated using software such as a package (vegan).
- the Shannon-Wiener diversity index (H ′), the Simpson diversity index ( ⁇ , 1- ⁇ or 1 / ⁇ ), the Pierou uniformity index (J ′), or the Chao1 index are obtained by the following formulas.
- N total number of leads
- n i number of leads in lead i Shannon-Weaver index H ′
- the present invention is a method of analyzing an object using a similarity index using the analysis method or analysis system of the present invention.
- the present invention provides a method for supporting analysis of an object using a similarity index using the analysis method or analysis system of the present invention.
- the number of species and individuals of the lead sequence identified based on the CDR3 sequence are calculated, and the similarity of TCR or BCR repertoire between samples to be compared is clarified.
- the Morisita-Horn index, Kimoto's C ⁇ index or Pianka's ⁇ index the degree of similarity between samples is clarified. It can be used for evaluation of repertoire similarity between HLA type agreement or mismatch, evaluation of repertoire similarity between recipient and donor after bone marrow transplantation, and the like.
- the similarity index is used as an assessment of repertoire similarity between HLA-type matches or mismatches, and an assessment of repertoire similarity between recipient and donor after bone marrow transplantation. Analysis using such a similarity index is difficult with a conventional system.
- Various similarity indices can be calculated using the following formulas using ESTIMATES (Colwell, R. K. et al. Journal of Plant Ecology 5: 3-21.) Or R package (vegan).
- the Morisita-Horn index, Kimoto's C ⁇ index, or Pianka's ⁇ index is obtained by the following equation.
- Morosita-Horn index X i number of leads i appearing in all X leads derived from one sample
- y i number of leads i appearing in all Y leads derived from the other sample S: number of unique reads
- the present invention can adjust samples for quantitative analysis of T cell receptor (TCR) or B cell receptor (BCR) variable region repertoire using next generation sequencing technology.
- TCR T cell receptor
- BCR B cell receptor
- These sequencing techniques can obtain 1 million or more leads from a sample at a reasonable cost. Even genotypes present at a frequency of 1 / 1,000,000 or less can be detected in a specific and unbiased manner using these techniques.
- Unbiased amplification methods are achieved to amplify all different types of sequences of specific portions of genes or transcripts from samples derived from DNA such as blood or bone marrow.
- the present invention provides a method of preparing a composition for use in cancer idiotype peptide-sensitized immune cell therapy in a subject.
- This method comprises (1) analyzing the T cell receptor (TCR) or B cell receptor (BCR) repertoire of the subject by the repertoire analysis method of the present invention or the repertoire analysis system of the present invention; (2) A step of determining a TCR or BCR derived from the cancer cell of the subject based on the result, wherein the determination is based on the highest ranking sequence in the frequency ranking of the TCR or BCR gene derived from the cancer cell of the subject.
- a step of selecting the TCR or BCR derived from the cancer cell (3) determining a candidate amino acid sequence of the HLA test peptide based on the determined TCR or BCR derived from the cancer The determination is based on a score calculated using an HLA-binding peptide prediction algorithm; And (4) comprising the step of combining the determined peptide.
- the peptide synthesized here can be used for cancer idiotype peptide-sensitized immune cell therapy. This method is sometimes referred to herein as "cancer idiotype peptide sensitized immune cell therapy".
- cancer idiotype peptide-sensitized immune cell therapy specifically, it can be performed clinically using the following procedure. For example, briefly, (1) peripheral blood cells of a cancer patient suffering from a blood tumor can be collected, lymphocyte cells can be isolated, and then the repertoire analysis method of the present invention can be performed, and this can be used. Cancer idiotype peptide sensitized immune cell therapy. In another embodiment, the repertoire analysis method of the present invention can be performed for a TCR in the case of a T cell tumor and for a BCR in the case of a B cell tumor.
- the higher rank sequence is selected as the TCR or BCR derived from the cancer cell, and the cancer separately determined from the sequence containing the CDR3 region of the TCR or BCR gene
- Peptides that bind to a patient's human leukocyte antigen (HLA) are predicted using an HLA-binding peptide prediction program (any known program can be used as described elsewhere herein).
- an HLA binding peptide is synthesize
- patient peripheral blood mononuclear cells are collected, added to the mixture of mononuclear cells or patient-derived antigen-presenting cells and CD8 + T cells, cultured, Stimulation with peptides can be performed.
- the peptide-stimulated lymphocyte cells can be transferred to a patient and subjected to CTL therapy.
- peptide-sensitized DC vaccine therapy after collecting peripheral blood mononuclear cells from patients, monocytes are isolated and induced to differentiate into dendritic cells (DC) in the presence of differentiation-inducing factors. Then, the peptide can be added and cultured, and the peptide-sensitized dendritic cells can be transferred to a patient and subjected to dendritic cell therapy.
- DC dendritic cells
- Cancer idiotype peptide sensitized immune cell therapy includes, for example, acute myeloid leukemia and related progenitor cell tumors, lymphoblastic leukemia / lymphoma, T lymphoblastic leukemia / lymphoma, chronic lymphocytic leukemia / small lymphocytic lymphoma B cell prolymphocytic leukemia, hairy cell leukemia, T cell prolymphocytic leukemia, T cell large granular lymphocytic leukemia, adult T cell leukemia / lymphoma and other hematological cancers, multiple myeloma, bone marrow Can be used for leukemia-related diseases such as dysplasia syndrome, rheumatoid arthritis, systemic lupus erythematosus, autoimmune diseases such as type I diabetes, patients with various infectious diseases, etc.
- leukemia-related diseases such as dysplasia syndrome, rheumatoid arthritis, systemic lup
- the candidate for the HLA test peptide in step (3) of the present invention is determined using BIMAS, SYFPEITHI, RANKPEP, or NetMHC.
- the present invention comprises, after step (4) of the present invention, mixing the peptide, the subject-derived dendritic cell or antigen-presenting cell, and the subject-derived CD8 + T cell. Including the step of culturing. This is also called an improved CTL method.
- the improved CTL method for example, unlike the widespread activation of T cells by existing anti-CD3 antibodies and IL-2, antigen specificity is imparted to CD8 + T cells using an antigen peptide, resulting in higher specificity. Treatment with fewer side effects can be expected. Moreover, since the individualized peptide created based on the information obtained from the tumor cells of the patient is used, a high therapeutic effect can be expected.
- Improved CTL methods include, for example, acute myeloid leukemia and related progenitor cell tumors, lymphoblastic leukemia / lymphoma, T lymphoblastic leukemia / lymphoma, chronic lymphocytic leukemia / small lymphocytic lymphoma, B cell prolymphocyte Leukemia, hairy cell leukemia, T-cell prolymphocytic leukemia, T-cell large granular lymphocytic leukemia, blood cancer such as adult T-cell leukemia / lymphoma, multiple myeloma, leukemia such as myelodysplastic syndrome Can be used for related diseases, rheumatoid arthritis, systemic lupus erythematosus, autoimmune diseases such as type I diabetes, patients with various infectious diseases, etc. Patients with end-stage cancer patients, refractory autoimmune diseases, severe infections Can be used.
- the present invention includes a step of mixing and culturing the peptide and the dendritic cell derived from the subject after the step (4) of the present invention. This is also called DC vaccine therapy.
- DC vaccine therapy for example, since individualized peptides are created based on sequence information obtained from tumor cells derived from the patient, they do not act on normal cells, but act more specifically on tumor cells and are high A therapeutic effect can be expected. Since a peptide is used as an antigen, there is an advantage that it can be easily chemically synthesized unlike a protein.
- DC vaccine therapy includes, for example, acute myeloid leukemia and related progenitor cell tumors, lymphoblastic leukemia / lymphoma, T lymphoblastic leukemia / lymphoma, chronic lymphocytic leukemia / small lymphocytic lymphoma, B cell prolymphocytic Leukemia such as leukemia, hairy cell leukemia, T-cell prolymphocytic leukemia, T-cell large granular lymphocytic leukemia, adult T-cell leukemia / lymphoma, etc., multiple myeloma, myelodysplastic syndrome Can be used for diseases, rheumatoid arthritis, systemic lupus erythematosus, autoimmune diseases such as type I diabetes, patients with various infectious diseases, etc. For patients with terminal cancer patients, refractory autoimmune diseases, severe infections Can be used).
- the present invention comprises, after step (4) of the present invention, mixing the peptide, the subject-derived dendritic cell or antigen-presenting cell, and the subject-derived CD8 + T cell. Culturing to produce a CD8 + T cell-dendritic cell / antigen-presenting cell-peptide mixture, and mixing the peptide and dendritic cells derived from the subject to culture, Including the process of producing. This is also called patient autoimmune cell therapy.
- Patient autoimmune cell therapy includes, for example, blood cancer (leukemia, etc.), acute myeloid leukemia and related progenitor cell tumors, lymphoblastic leukemia / lymphoma, T lymphoblastic leukemia / lymphoma, chronic lymphocytic leukemia / small lymphoma Hematopoietic lymphoma, B cell prolymphocytic leukemia, hairy cell leukemia, T cell prolymphocytic leukemia, T cell large granular lymphocytic leukemia, blood cancer such as adult T cell leukemia / lymphoma, multiple bone marrow It can be used for leukemia-related diseases such as myeloma, myelodysplastic syndrome, rheumatoid arthritis, systemic lupus erythematosus, autoimmune diseases such as type I diabetes, various infectious disease patients, etc., terminal cancer patients, refractory autoimmunity It can be used for patients with diseases and serious
- the present invention provides a method of giving cancer idiotype peptide-sensitized immune cell therapy to a subject.
- This method comprises (1) analyzing the T cell receptor (TCR) or B cell receptor (BCR) repertoire of the subject by the repertoire analysis method of the present invention or the repertoire analysis system of the present invention; (2) A step of determining a TCR or BCR derived from the cancer cell of the subject based on the result, wherein the determination is based on the highest ranking sequence in the frequency ranking of the TCR or BCR gene derived from the cancer cell of the subject.
- a step of selecting the TCR or BCR derived from the cancer cell (3) determining a candidate amino acid sequence of the HLA test peptide based on the determined TCR or BCR derived from the cancer The determination is based on a score calculated using an HLA-binding peptide prediction algorithm; Comprising the step of performing treated with optionally (5) synthesized peptide; 4) a step of synthesizing the determined peptide.
- This method includes both a method of producing a therapeutic agent and a method of performing the treatment itself. When excluding medical practice, it can be completed in the steps up to (5).
- the candidate for the HLA test peptide in the step (3) is determined using BIMAS, SYFPEITHI, RANKPEP, or NetMHC.
- BIMAS is an estimation program for HLA peptide binding provided at www-bimas.cit.nih.gov/.
- SYFPEITHI is a database and search engine for MHC ligands and peptide motifs provided at www.syfpeithi.de/.
- RANKPEP is a peptide bond prediction program for class I and class II MHC molecules provided at http://imed.med.ucm.es/Tools/rankpep.html.
- NetMHC is a program server that predicts the binding of peptides to many HLA alleles, which is provided at www.cbs.dtu.dk/services/NetMHC/.
- the present invention comprises, after the step (4), the peptide, the dendritic cell or antigen-presenting cell derived from the subject, and the CD8 + T cell derived from the subject.
- the present invention comprises a step of mixing and culturing the peptide and dendritic cells derived from the subject after the step (4), and treating the cultured mixture with a patient.
- the present invention comprises, after the step (4), the peptide, dendritic cells or antigen-presenting cells derived from the subject, and CD8 + T cells derived from the subject. Culturing the mixture to produce a CD8 + T cell-dendritic cell / antigen-presenting cell-peptide mixture, and mixing the peptide with the subject-derived dendritic cell and culturing the dendritic cell. Performing the step of producing a peptide mixture and administering the CD8 + T cell-dendritic cell / antigen presenting cell-peptide mixture, and the dendritic cell-peptide mixture to a patient.
- the present invention provides a technique for isolating a bespoke cancer-specific T cell receptor gene and a technique for isolating a cancer-specific TCR gene by in vitro antigen stimulation.
- the present invention relates to (A) an antigen protein or antigen peptide derived from a subject or a lymphocyte derived from the subject or a peptide determined in the “cancer idiotype peptide-sensitized immune cell therapy” of the present invention, Mixing inactivated cancer cells derived from a subject and T lymphocytes derived from the subject and culturing to produce tumor-specific T cells; (B) TCR of the tumor-specific T cells of the present invention Analyzing with the repertoire analysis method of the present invention or the repertoire analysis system of the present invention; and (C) isolating a desired tumor-specific T cell based on the result of the analysis, Methods are provided for preparing isolated cancer-specific TCR genes.
- Preparation of such an isolated cancer-specific TCR gene by in vitro antigen stimulation can be carried out using any technique known in the art once genetic information is obtained.
- Such isolated tailored cancer-specific T cell receptor genes and cancer-specific TCR genes can be used for the treatment and prevention of various cancers.
- Such an isolated tailor-made cancer-specific T cell receptor gene and cancer-specific TCR gene can be specifically carried out clinically using the following procedure.
- tumor cells are removed from a cancer patient.
- the patient-derived tumor cells are crushed, separated into single cells, and inactivated by chemical treatment such as mitomycin C or irradiation.
- Isolating peripheral blood cells from the whole blood of the cancer patient. (4) Extract RNA from cells using a part of peripheral blood cells as an untreated control sample. (5) Activate and proliferate tumor-specific T cells by mixing and culturing inactivated tumor cells and peripheral blood cells. .
- peripheral blood cells are collected, and RNA is extracted from the cells as a sample after stimulation.
- the repertoire analysis method of the present invention is performed from the RNA sample extracted in (4) and (6).
- TCR genes greatly increased in the stimulation sample as compared with the control sample, rank them, and then select the upper TCR ⁇ and TCR ⁇ genes.
- These full-length TCR ⁇ and TCR ⁇ genes are cloned and introduced into a retrovirus vector for gene expression.
- a virus for gene transfer is prepared from these TCR ⁇ and TCR ⁇ gene expression retroviral vectors.
- (11) Transform by infecting lymphocytes collected from the patient with TCR ⁇ and TCR ⁇ alone, or by creating a gene expression retroviral vector containing both TCR ⁇ and TCR ⁇ genes at the same time. Transform genes.
- (12) Confirm the expression of TCR ⁇ / TCR ⁇ heterodimer on the cell surface.
- (13) Using an isolated tailored cancer-specific T cell receptor gene and a cancer-specific TCR gene by transferring a tumor-specific patient lymphocyte expressing a target TCR ⁇ / TCR ⁇ into a patient. could be realized.
- TCR or BCR determined by the method described in “cancer idiotype peptide-sensitized immune cell therapy” is used as an antigen or peptide.
- Can do is used as an antigen or peptide.
- any cancer antigen or inactivated cancer tissue derived from a patient is assumed here, the following methods can be typically used: any antigen protein or any antigen peptide, T lymphocyte and antigen-presenting cell A method of mixing a subject-derived lymphocyte and a subject-derived inactivated cancer cell; a TCR or BCR-derived peptide determined by a repertoire analysis provided in “cancer idiotype peptide-sensitized immune cell therapy” A method of mixing T lymphocytes and antigen presenting cells is provided.
- the step (A) in the present invention comprises an antigen protein or antigen peptide derived from a subject, inactivated cancer cells derived from the subject, and T lymphocytes derived from the subject. Mixing and culturing to produce tumor-specific T cells.
- the step (A) in the present invention comprises mixing and culturing lymphocytes derived from the subject, inactivated cancer cells derived from the subject, and T lymphocytes derived from the subject. This is a process for producing tumor-specific T cells.
- step (A) in the present invention is derived from the peptide determined in “cancer idiotype peptide-sensitized immune cell therapy”, inactivated cancer cells derived from the subject, and the subject. This is a step of producing tumor-specific T cells by mixing and culturing with T lymphocytes.
- Treatments using such isolated tailored cancer-specific T cell receptor genes and cancer-specific TCR genes include, for example, adrenal cancer, anal cancer, bile duct cancer, bladder cancer, breast cancer, cervical cancer , Chronic lymphocytic leukemia, chronic myeloid leukemia, colon cancer, endometrial cancer, esophageal cancer, Ewing tumor, gallbladder cancer, Hodgkin's disease, hypopharyngeal cancer, laryngeal cancer, oral cavity cancer, liver cancer, non-small cell lung cancer , Non-Hodgkin lymphoma, melanoma, mesothelioma, multiple myeloma, ovarian cancer, pancreatic cancer, prostate cancer, gastric cancer, testicular cancer, thyroid cancer, etc. .
- the present invention provides isolation of a tailor-made cancer-specific T cell receptor gene and isolation of a cancer-specific TCR gene by consensus sequence search. Accordingly, the present invention provides (A) providing lymphocytes or cancer tissue isolated from subjects having a common HLA; (B) for the lymphocytes or cancer tissue, the tumor-specific T cells.
- a common sequence search comprising the steps of: analyzing the TCR of the subject by the repertoire analysis method of the present invention or the repertoire analysis system of the present invention; Provides a method for preparing an isolated cancer-specific TCR gene by Preparation of an isolated cancer-specific TCR gene by such a common sequence search can be performed using any technique known in the art once genetic information is obtained.
- Such a gene obtained by isolation of a tailor-made cancer-specific T cell receptor gene and isolation of a cancer-specific TCR gene by consensus sequence search can be used for the treatment and prevention of various cancers. it can.
- This method is also referred to as “isolation of a customized cancer-specific T cell receptor gene of the present invention and isolation of a cancer-specific TCR gene by consensus sequence search”.
- the gene obtained by the isolation of such a tailor-made cancer-specific T cell receptor gene and the isolation of a cancer-specific TCR gene by consensus sequence search is specifically as follows in clinical practice. Can be implemented.
- (2) Repertoire analysis is performed using tumor tissue or lymphocyte cells containing tumor cell infiltrating T cells.
- a tumor-specific T cell that ranks based on its presence frequency in each sample and shows a higher presence frequency in tumor cells than in peripheral blood cells is selected.
- a sequence common to a plurality of HLA-matched cancer patients is searched.
- a tumor-specific TCR gene common to most cancer patients is selected as a therapeutic tumor-specific TCR.
- TCR ⁇ and TCR ⁇ genes are cloned and introduced into a retrovirus vector for gene expression.
- a virus for gene transfer is prepared from these TCR ⁇ and TCR ⁇ gene expression retroviral vectors.
- Tumor-specific patient lymphocytes expressing the target TCR ⁇ / TCR ⁇ are transferred to a patient to isolate a custom-made cancer-specific T cell receptor gene, and a cancer-specific TCR by consensus sequence search Treatment using a gene obtained by gene isolation can be realized.
- the treatment using the gene obtained by the isolation of such a tailor-made cancer-specific T cell receptor gene and the isolation of the cancer-specific TCR gene by consensus sequence search includes, for example, adrenocortical cancer, anal cancer, Bile duct cancer, bladder cancer, breast cancer, cervical cancer, chronic lymphocytic leukemia, chronic myelogenous leukemia, colon cancer, endometrial cancer, esophageal cancer, Ewing tumor, gallbladder cancer, Hodgkin's disease, hypopharyngeal cancer, laryngeal cancer,
- adrenocortical cancer anal cancer
- Bile duct cancer bladder cancer
- breast cancer cervical cancer
- the present invention provides (A) an antigen protein or antigen peptide derived from a subject or a peptide determined in lymphocyte or cancer idiotype peptide-sensitized immune cell therapy derived from the subject, Mixing inactivated cancer cells derived from a subject and T lymphocytes derived from the subject and culturing to produce tumor-specific T cells; (B) TCR of the tumor-specific T cells of the present invention Analyzing by the repertoire analysis method of the present invention or the repertoire analysis system of the present invention; and (C) isolating a desired tumor-specific T cell based on the result of the analysis, by in vitro antigen stimulation.
- a method for isolating cancer-specific TCR genes is provided.
- Preparation of such an isolated cancer-specific TCR gene by in vitro antigen stimulation can be performed using any technique known in the art once the genetic information is obtained.
- Such isolated tailored cancer-specific T cell receptor genes and cancer-specific TCR genes can be used for the treatment and prevention of various cancers.
- step (A) in the present invention is derived from an antigen protein or antigen peptide derived from a subject and the subject.
- the step (A) in the present invention comprises mixing and culturing lymphocytes derived from the subject, inactivated cancer cells derived from the subject, and T lymphocytes derived from the subject. This is a process for producing tumor-specific T cells.
- step (A) in the present invention is derived from the peptide determined in “cancer idiotype peptide-sensitized immune cell therapy”, inactivated cancer cells derived from the subject, and the subject. This is a step of producing tumor-specific T cells by mixing and culturing with T lymphocytes.
- the present invention provides a technique for isolating a bespoke cancer-specific TCR gene by isolating a tailor-made cancer-specific T cell receptor gene and searching for a common sequence.
- B a step of analyzing a TCR of the tumor-specific T cell for the lymphocyte or cancer tissue by the repertoire analysis method of the present invention
- C A method for isolating a cancer-specific TCR gene by consensus sequence search, comprising the step of isolating T cells having a sequence common to tumor-specific T cells.
- Such isolated tailored cancer-specific T cell receptor genes and cancer-specific TCR genes can be used for the treatment and prevention of various cancers.
- the present invention provides cell processing therapy. Specifically, the present invention is based on A) the step of providing T lymphocytes collected from a patient; B) after the antigen stimulation of the T lymphocytes, and the repertoire analysis method of the present invention or the repertoire analysis system of the present invention.
- Analyzing the TCR wherein the antigen stimulation is performed by an antigen protein or antigen peptide derived from the subject, an inactivated cancer cell derived from the subject, or a tumor-derived idiotype peptide; C) selecting the optimal antigen and optimal TCR in the analyzed TCR; and D) for cell processing therapy comprising the steps of producing a tumor-specific alpha and beta TCR expressing viral vector of the TCR gene of the optimal TCR
- a method for preparing the tumor-specific TCR transgenic T lymphocytes for use in the present invention is provided. This cell processing therapy using tumor-specific TCR gene-transferred T lymphocytes can be used for the treatment and prevention of various cancers.
- the cell processing therapy using such tumor-specific TCR gene-introduced T lymphocytes can be specifically carried out clinically using the following procedure.
- ⁇ isolation of tailor-made cancer-specific T cell receptor gene, isolation of cancer-specific TCR gene by in vitro antigen stimulation> or ⁇ isolation of tailor-made cancer-specific T cell receptor gene, Tumor-specific TCR gene-introduced lymphocytes can be used by the method described in> Isolation of cancer-specific TCR gene by consensus sequence search.
- any cancer antigen or cancer peptide can be produced or synthesized as an antigen by production or synthesis, and the collected inactivated patient cancer cells can be used, or a tumor-derived idio Type peptides can be used.
- a selection method an antigen highly expressed in cancer tissue can be selected, or a peptide that binds to a patient HLA type can be selected as an antigen.
- an optimal antigen for example, (1) an antigen highly expressed in patient cancer tissue, (2) the T cell is most strongly activated in an antigen-specific lymphocyte stimulation test Antigen, (3) From the repertoire analysis before and after antigen stimulation, it can be envisaged to select an antigen that most increases the frequency of a specific TCR, but is not limited thereto.
- (3) a method of selecting the most increased TCR as the optimum TCR in an example in which the frequency of a specific TCR is most increased from repertoire analysis before and after antigen stimulation can be assumed.
- an optimal TCR candidate is artificially introduced into a patient lymphocyte, and the one that exhibits the highest reactivity with an actual patient cancer tissue can be selected as the optimal TCR.
- T lymphocytes include, for example, adrenocortical cancer, anal cancer, bile duct cancer, bladder cancer, breast cancer, cervical cancer, chronic lymphocytic leukemia, chronic myeloid leukemia Colorectal cancer, endometrial cancer, esophageal cancer, Ewing tumor, gallbladder cancer, Hodgkin's disease, hypopharyngeal cancer, laryngeal cancer, oral cavity cancer, liver cancer, non-small cell lung cancer, non-Hodgkin lymphoma, melanoma, mesothelioma It can be used for a wide range of cancer patients such as multiple myeloma, ovarian cancer, pancreatic cancer, prostate cancer, stomach cancer, testicular cancer, thyroid cancer, etc., but is not limited thereto.
- adrenocortical cancer anal cancer, bile duct cancer, bladder cancer, breast cancer, cervical cancer, chronic lymphocytic leukemia, chronic myeloid leukemia Colore
- the antigen stimulation of the method of the present invention is performed by an antigen protein or an antigen peptide derived from the subject.
- the antigen stimulation of the present invention is performed by inactivated cancer cells derived from the subject.
- the antigen stimulation of the method of the present invention is performed by the tumor-derived idiotype peptide.
- step C) of the method of the present invention comprises selecting an antigen that is highly expressed in the cancer tissue of the subject.
- step C) of the method of the invention comprises selecting the antigen that most strongly activates T cells in an antigen-specific lymphocyte stimulation test.
- step C) of the method of the present invention most increases the frequency of a specific TCR from a repertoire analysis performed based on the repertoire analysis method of the present invention or the repertoire analysis system of the present invention before and after antigen stimulation. Selecting an antigen.
- the present invention is isolated by “isolation of a customized cancer-specific T cell receptor gene of the present invention, a method for isolating a cancer-specific TCR gene by consensus sequence search”.
- the present invention provides a method for evaluating efficacy and / or safety by conducting an in vitro stimulation test using a cancer-specific TCR gene.
- the effectiveness is derived from a subject, for example, an antigenic protein or antigenic peptide derived from the subject that has been stimulated by an antigenic protein or antigenic peptide derived from the subject and a T cell into which a cancer-specific TCR gene has been introduced.
- cytokine interferon ⁇ , etc.
- Safety For example, when T cells derived from the subject into which a cancer-specific TCR gene has been introduced and normal cells derived from the subject are mixed, cytokines secreted in response to the activation of the T cells, Safety can be evaluated by measuring gene expression or expression of cell surface molecules and confirming that the TCR gene-introduced T cells are not activated by normal cells.
- the specific steps of efficacy and / or safety assessment can be realized as follows. That is, for example, (1) tumor-specific TCR ⁇ and TCR ⁇ gene-introduced T lymphocyte cells are produced using a retrovirus gene expression system. (2) When evaluating the effectiveness, cancer cells derived from a patient are removed and separated, immortalized, and then mixed and cultured with tumor-specific TCR gene-introduced T lymphocytes. (3) Using the above cultured cells, a cell proliferation test (such as a thymidine incorporation test, an MTT test, or an IL-2 production test) is performed to quantitatively evaluate the reactivity with tumor cells, and more strongly to tumor cells. A reactive TCR gene can be selected.
- a cell proliferation test such as a thymidine incorporation test, an MTT test, or an IL-2 production test
- the present invention is based on A) the step of collecting T lymphocytes from a patient; B) after the antigen stimulation of the T lymphocytes, and the repertoire analysis method of the present invention or the repertoire analysis system of the present invention. Analyzing the TCR, wherein the antigen stimulation is performed by an antigen protein or antigen peptide derived from the subject, an inactivated cancer cell derived from the subject, or a tumor-derived idiotype peptide; C) selecting an optimal antigen and optimal TCR in the analyzed TCR; D) producing a tumor-specific ⁇ and ⁇ TCR-expressing viral vector of the TCR gene of the optimal TCR; E) introducing the tumor-specific TCR gene Introducing a T lymphocyte into the patient.
- the method for carrying out the step of introducing the obtained tumor-specific TCR gene-introduced T lymphocytes into the patient is as follows: A) Step of producing tumor-specific TCR gene-introduced T lymphocytes; B) Tumor-specific TCR ⁇ and TCR ⁇ C) a step of instilling tumor-specific TCR gene-introduced T lymphocytes from a vein.
- the antigen stimulation in the cell processing therapy of the present invention is performed by an antigen protein or an antigen peptide derived from the subject.
- antigen stimulation in the cell processing therapy of the present invention is performed by inactivated cancer cells derived from the subject.
- antigen stimulation in the cell processing therapy of the present invention is performed by the tumor-derived idiotype peptide.
- step C) in the cell processing therapy of the present invention includes selecting an antigen that is highly expressed in the cancer tissue of the subject.
- step C) in the cell processing therapy of the present invention comprises selecting an antigen that most strongly activates T cells in an antigen-specific lymphocyte stimulation test.
- the step C) in the cell processing therapy of the present invention selects an antigen that most increases the frequency of a specific TCR from repertoire analysis performed based on the repertoire analysis method of the present invention before and after antigen stimulation. including.
- BCR gene repertoire analysis is performed using the repertoire analysis method of the present invention, and a human-type antibody specific for a target antigen can be rapidly obtained by the method described below (A) And a cell population containing antibody-producing B cells (eg, spleen, lymph nodes, peripheral blood cells) is isolated from the mouse, and immunoglobulin is analyzed by BCR repertoire analysis using the repertoire analysis method of the present invention.
- BCR gene repertoire analysis is performed using the repertoire analysis method of the present invention, and a human-type antibody specific for a target antigen can be rapidly obtained by the method described below (A)
- a cell population containing antibody-producing B cells eg, spleen, lymph nodes, peripheral blood cells
- immunoglobulin is analyzed by BCR repertoire analysis using the repertoire analysis method of the present invention.
- A1 Method A in which immunized mice are KM mice capable of producing fully human antibodies while maintaining antibody diversity
- the immunized mice are NOD / scid Humans in NOG (NOD / Shi-scid, IL-2R ⁇ null) mice with severe combined immunodeficiency created by mating IL-2 receptor ⁇ chain knockout mice to mice
- Method A of humanized mice produced by transplanting cells B) immunoglobulin heavy and light chain gene sequences obtained from samples from mice derived from control mice and immunized mice, or mice before and after antigen immunization Compare frequency (C) Identify immunoglobulin heavy and light chain genes that are strongly expressed in immunized mice or increase after immunization (D) Select immunoglobulin heavy and light chain genes selected from step C And inserting the immunoglobulin heavy chain and light chain gene expression vectors prepared in the step (E) D into a single antibody expression vector or by separately inserting them into two different antibody expression vectors.
- eukaryotic cells such as CHO (Chinese HamsterOvary), and perform cell culture (F) Isolate and purify antibody molecules produced and secreted by genetically modified cells, target
- eukaryotic cells such as CHO (Chinese HamsterOvary)
- F cell culture
- Isolate and purify antibody molecules produced and secreted by genetically modified cells target
- the antigen can be directly and rapidly changed without being changed into a chimeric antibody or humanized antibody with a human antibody.
- This is a method for obtaining a specific human antibody and can be used for the development and production of an antibody drug comprising a human antibody.
- NOD / SCID / gamma (c) (null) mouse an excellent recipient mouse model for engraftment of human. Nov 1; 100 (9): 3175-82.
- Jayapal KP Wlaschin KF, Hu WS, Yap MGS. Recombinant protein therapeutics from CHO cells-20 years and Count.Chem Eng Prog. 2007; 103: 40? 47.; Chusainow J, Yang YS, Yeo JH, Toh PC, Asvadi P, Wong NS, Yap MG.
- a study of monoclonal antigen-producing CHO cell lines whatwhat You can refer to stable high producer? Biotechnol Bioeng. 2009 Mar 1; 102 (4): 1182-96.
- a human-type antibody specific for a target antigen can be rapidly obtained by the method described below using the BCR gene repertoire analysis method.
- A Target antigen protein or antigen A method in which a mouse is immunized with a peptide, a cell population containing antibody-producing B cells (eg, spleen, lymph nodes, peripheral blood cells) is isolated from the mouse, and immunoglobulin heavy chain and light chain genes are analyzed by BCR repertoire analysis ( A1) Method A in which the immunized mouse is a KM mouse that can produce fully human antibodies while maintaining antibody diversity (A2) The immunized mouse is a NOD / scid mouse and an IL-2 receptor ⁇ chain knockout mouse A humanized mouse produced by transplanting human stem cells into NOG (NOD / Shi-scid, IL-2R ⁇ null) mice exhibiting severe combined immunodeficiency produced by mating Method (B) Compare the frequency of immunoglobulin heavy and light chain
- the immunoglobulin heavy and light chain genes that increase after immunization are identified (D)
- the immunoglobulin heavy and light chain genes selected from step C are selected and inserted in accordance with one antibody expression vector.
- the method of inserting each into the two antibody expression vectors separately (E)
- the immunoglobulin heavy chain and light chain gene expression vectors prepared in step D are introduced into eukaryotic cells such as CHO (Chinese HamsterOvary),
- F) Incubate F) Isolate and purify antibody molecules produced and secreted by genetically modified cells, and test their specificity for the target antibody protein or peptide
- mice are immunized with MyelinOligodendrocyte Glycoprotein (MOG35-55, MOG), an antigenic peptide of experimental autoimmune encephalomyelitis.
- MOG35-55 MyelinOligodendrocyte Glycoprotein
- An equal volume of 2 mg / mL MOG peptide and complete Freund's adjuvant are mixed to make an emulsion, 200 ⁇ g MOG is immunized subcutaneously, and 400 ng pertussis toxin is simultaneously immunized to the peritoneal cavity.
- Control mice are immunized with PBS and complete Freund's adjuvant.
- 400 ng of pertussis toxin is immunized.
- Next generation BCR repertoire analysis will be performed using the spleen of the affected and control mice.
- IgG immunoglobulin heavy chains and immunoglobulin light chains the frequency of occurrence of individual BCR sequences and the ranking are performed. 4).
- BCR sequences whose frequency of occurrence is greatly increased in the affected mice compared to the control mice are extracted and ranked. A combination of the highest ranking BCR sequences induced by administration of these antibodies is identified as a MOG-specific antibody gene. 5.
- the full-length human immune fibrillin sequence is cloned from the BCR gene amplification product amplified from the affected mouse by PCR-cloning method.
- Each of an IgG immunoglobulin heavy chain and an immunoglobulin light chain is cloned into an antibody expression vector.
- CHO Choinese Hamster Ovary
- the CHO cell culture medium is recovered, and the secreted antibody protein is recovered by purification with a Protein A affinity column and concentration by gel filtration. 8).
- the binding activity against MOG35-55 or MOG protein is measured by ELISA assay using the collected antibody, and the specificity of the antibody is verified. 9.
- an antibody expression stable expression cell line is obtained, and a human anti-MOG antibody is produced by a mass culture system.
- peptide of the present invention or the nucleic acid encoding it can be used for immunotherapy. This will be described below.
- Peptides provided by the present invention are derived from antigens associated with tumorigenesis, and bind sufficiently to MHC (HLA) class II molecules to human leukocyte immune responses, particularly lymphocytes, particularly T lymphocytes.
- HLA MHC
- it may be capable of initiating a TH1-type immune response provided by CD4-positive T lymphocytes, particularly CD4-positive T lymphocytes.
- protein protein
- polypeptide oligopeptide
- peptide refers to a polymer of amino acids having an arbitrary length.
- This polymer may be linear, branched, or cyclic.
- the amino acid may be natural or non-natural and may be a modified amino acid.
- the term can also encompass one assembled into a complex of multiple polypeptide chains.
- the term also encompasses natural or artificially modified amino acid polymers. Such modifications include, for example, disulfide bond formation, glycosylation, lipidation, acetylation, phosphorylation or any other manipulation or modification (eg, conjugation with a labeling component).
- amino acid may be natural or non-natural as long as it satisfies the object of the present invention.
- polynucleotide refers to a polymer of nucleotides of any length.
- the term also includes “oligonucleotide derivatives” or “polynucleotide derivatives”.
- Oligonucleotide derivatives or “polynucleotide derivatives” refer to oligonucleotides or polynucleotides that include derivatives of nucleotides or that have unusual linkages between nucleotides, and are used interchangeably.
- oligonucleotides include, for example, 2′-O-methyl-ribonucleotides, oligonucleotide derivatives in which phosphodiester bonds in oligonucleotides are converted to phosphorothioate bonds, and phosphodiester bonds in oligonucleotides.
- oligonucleotide derivative in which ribose and phosphodiester bond in oligonucleotide are converted to peptide nucleic acid bond uracil in oligonucleotide is C- Oligonucleotide derivatives substituted with 5-propynyluracil, oligonucleotide derivatives wherein uracil in the oligonucleotide is substituted with C-5 thiazole uracil, oligonucleotides in which cytosine in the oligonucleotide is substituted with C-5 propynylcytosine Leotide derivatives, oligonucleotide derivatives in which the cytosine in the oligonucleotide is replaced with phenoxazine-modified cytosine, oligonucleotide derivatives in which the ribose in DNA is replaced with 2
- a particular nucleic acid sequence may also be conservatively modified (eg, degenerate codon substitutes) and complementary sequences, as well as those explicitly indicated. Is contemplated. Specifically, a degenerate codon substitute creates a sequence in which the third position of one or more selected (or all) codons is replaced with a mixed base and / or deoxyinosine residue. (Batzeret al., Nucleic Acid Res. 19: 5081 (1991); Ohtsuka et al., J. Biol. Chem. 260: 2605-2608 (1985); Rossolinet al., Mol. Cell. Probes 8: 91-98 (1994)).
- nucleic acid is also used interchangeably with gene, cDNA, mRNA, oligonucleotide, and polynucleotide.
- nucleotide may be natural or non-natural.
- gene refers to a factor that defines a genetic trait
- gene may refer to “polynucleotide”, “oligonucleotide”, and “nucleic acid”.
- a variant thereof in addition to the identified peptide, a variant thereof may be used.
- Such variants include, but are not limited to those having homology to the identified peptide.
- homology of a gene refers to the degree of identity of two or more gene sequences to each other, and generally “having homology” means that the degree of identity or similarity is high. Say. Therefore, the higher the homology between two genes, the higher the sequence identity or similarity. Whether two genes have homology can be determined by direct sequence comparison or, in the case of nucleic acids, hybridization methods under stringent conditions. When directly comparing two gene sequences, the DNA sequence between the gene sequences is typically at least 50% identical, preferably at least 70% identical, more preferably at least 80%, 90% , 95%, 96%, 97%, 98% or 99% are identical, the genes have homology.
- a “homolog” or “homologous gene product” is a protein in another species, preferably a mammal, that performs the same biological function as the protein component of the complex further described herein. Means. Such homologues may also be referred to as “ortholog gene products”. It will be understood that such homologues, homologous gene products, orthologous gene products and the like can be used as long as they meet the objectives of the present invention.
- Amino acids may be referred to herein by either their commonly known three letter symbols or by the one letter symbols recommended by the IUPAC-IUB Biochemical Nomenclature Commission. Nucleotides can also be referred to by the generally recognized one letter code.
- comparison of similarity, identity and homology between amino acid sequences and base sequences is calculated using default parameters using BLAST, which is a sequence analysis tool.
- the identity search can be performed, for example, using NCBI's BLAST 2.2.28 (issued 2013.4.2).
- the identity value usually refers to a value when the above BLAST is used and aligned under default conditions. However, if a higher value is obtained by changing the parameter, the highest value is the identity value. When identity is evaluated in a plurality of areas, the highest value among them is set as the identity value. Similarity is a numerical value calculated for similar amino acids in addition to identity.
- “several” may be, for example, 10, 8, 6, 5, 4, 3, or 2, and may be any value or less.
- Polypeptides that have undergone deletion, addition, insertion, or substitution with other amino acids of one or several amino acid residues are known to maintain their biological activity (Market al., ProcNatl Acad). Sci U S A.1984 Sep; 81 (18): 5662-5666., Zoller et al., Nucleic Acids Res. 1982 Oct 25; 10 (20): 6487-6500., Wang et al., Science. 1984 Jun29 ; 224 (4656): 1431-1433.).
- Antibodies with deletions and the like can be prepared by, for example, site-specific mutagenesis, random mutagenesis, or biopanning using an antibody phage library.
- site-specific mutagenesis method for example, KOD-Plus-Mutagenesis Kit (TOYOBO CO., LTD.) Can be used. It is possible to select an antibody having the same activity as that of the wild type from mutant antibodies into which deletion or the like has been introduced by performing various characterizations such as FACS analysis and ELISA.
- “90% or more” may be, for example, 90, 95, 96, 97, 98, 99, or 100% or more, and is within the range of any two values thereof. Also good.
- the above-mentioned “homology” may be calculated according to a method known in the art, based on the ratio of the number of amino acids homologous in two or more amino acid sequences. Before calculating the ratio, the amino acid sequences of the group of amino acid sequences to be compared are aligned, and a gap is introduced into a part of the amino acid sequence when necessary to maximize the ratio of the same amino acids.
- polynucleotide hybridizing under stringent conditions refers to well-known conditions commonly used in the art.
- a polynucleotide can be obtained by using a colony hybridization method, a plaque hybridization method, a Southern blot hybridization method or the like using a polynucleotide selected from among the polynucleotides of the present invention as a probe.
- hybridization was performed at 65 ° C. in the presence of 0.7 to 1.0 M NaCl using a filter on which colony or plaque-derived DNA was immobilized, and then a 0.1 to 2-fold concentration was obtained.
- a polynucleotide that can be identified by washing the filter under conditions of 65 ° C using SSC (saline-sodium citrate) solution (composition of 1x concentrated SSC solution is 150 mM sodium chloride, 15 mM sodium citrate) .
- SSC saline-sodium citrate
- composition of 1x concentrated SSC solution is 150 mM sodium chloride, 15 mM sodium citrate
- stringent conditions for example, the following conditions can be adopted.
- Hybridization MolecularCloning 2nd. it can.
- a sequence that contains only the A sequence or only the T sequence is preferably excluded from the sequences that hybridize under stringent conditions.
- Moderate stringent conditions can be readily determined by those skilled in the art based on, for example, the length of the DNA, Sambrook et al., Molecular Cloning: A Laboratory Manual, 3rd edition, Vol.
- a polypeptide used in the present invention is encoded by a nucleic acid molecule that hybridizes under highly or moderately stringent conditions to a nucleic acid molecule encoding a polypeptide specifically described in the present invention. Are also included.
- a “purified” substance or biological factor refers to a substance from which at least a part of the factor naturally associated with the substance or biological factor has been removed. .
- the purity of a biological agent in a purified biological agent is higher (ie, enriched) than the state in which the biological agent is normally present.
- the term “purified” as used herein is preferably at least 75% by weight, more preferably at least 85% by weight, even more preferably at least 95% by weight, and most preferably at least 98% by weight, Means the presence of the same type of biological agent.
- the substance or biological agent used in the present invention is preferably a “purified” substance.
- an “isolated” substance or biological agent is substantially free of the factors that naturally accompany the substance or biological agent. Say things.
- the term “isolated” as used herein does not necessarily have to be expressed in purity, as it will vary depending on its purpose, but is preferably at least 75% by weight, more preferably if necessary. Means that there is at least 85%, more preferably at least 95%, and most preferably at least 98% by weight of the same type of biological agent.
- the materials used in the present invention are preferably “isolated” materials or biological agents.
- fragment refers to a polypeptide or polynucleotide having a sequence length of 1 to n ⁇ 1 with respect to a full-length polypeptide or polynucleotide (length is n).
- the length of the fragment can be appropriately changed according to the purpose.
- the lower limit of the length is 3, 4, 5, 6, 7, 8, 9, 10, 15, 20 , 25, 30, 40, 50 and more amino acids, and lengths expressed in integers not specifically listed here (eg, 11 etc.) may also be appropriate as lower limits.
- examples include 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 40, 50, 75, 100 and more nucleotides.
- Non-integer lengths may also be appropriate as a lower limit.
- such a fragment falls within the scope of the present invention as long as the full-length fragment functions as a marker or target molecule as long as the fragment itself also functions as a marker or target molecule. Is understood.
- the “functional equivalent” refers to any object having the same target function but different structure from the target original entity. Functional equivalents can be found by searching a database or the like.
- search refers to finding another nucleobase sequence having a specific function and / or property using a nucleobase sequence electronically or biologically or by other methods.
- Electronic searches include BLAST (Altschul et al., J. Mol. Biol. 215: 403-410 (1990)), FASTA (Pearson & Lipman, Proc. Natl. Acad. Sci., USA 85: 2444-2448 ( 1988), Smith and Waterman method (Smithand Waterman, J. Mol. Biol.
- Bio searches include stringent hybridization, macroarrays with genomic DNA affixed to nylon membranes, microarrays affixed to glass plates (microarray assays), PCR and in situ hybridization. It is not limited. In the present specification, it is intended that the gene used in the present invention should include a corresponding gene identified by such an electronic search or biological search.
- an amino acid sequence having one or more amino acid insertions, substitutions or deletions, or those added to one or both ends can be used.
- “insertion, substitution or deletion of one or a plurality of amino acids in the amino acid sequence, or addition to one or both ends thereof” is a well-known technical method such as site-directed mutagenesis. It means that a modification has been made by substitution of a plurality of amino acids to the extent that can occur naturally by a method or by natural mutation.
- the modified amino acid sequence has, for example, 1 to 30, preferably 1 to 20, more preferably 1 to 9, further preferably 1 to 5, particularly preferably 1 to 2 amino acid insertions, substitutions or deletions. Lost or added to one or both ends thereof.
- the modified amino acid sequence preferably has one or more (preferably 1 or several, or 1, 2, 3, or 4) conservative substitutions in the amino acid sequence such as CCL21, CXCR3, CCR7, etc.
- the amino acid sequence may have.
- conservative substitution means substitution of one or more amino acid residues with another chemically similar amino acid residue so as not to substantially alter the function of the protein. For example, when a certain hydrophobic residue is substituted by another hydrophobic residue, a certain polar residue is substituted by another polar residue having the same charge, and the like. Functionally similar amino acids that can make such substitutions are known in the art for each amino acid.
- non-polar (hydrophobic) amino acids such as alanine, valine, isoleucine, leucine, proline, tryptophan, phenylalanine, and methionine.
- polar (neutral) amino acids include glycine, serine, threonine, tyrosine, glutamine, asparagine, and cysteine.
- positively charged (basic) amino acids include arginine, histidine, and lysine.
- negatively charged (acidic) amino acids include aspartic acid and glutamic acid.
- the “subject (person)” refers to a subject of diagnosis or detection or treatment of the present invention.
- drug drug
- drug may also be a substance or other element (eg energy such as light, radioactivity, heat, electricity).
- Such substances include, for example, proteins, polypeptides, oligopeptides, peptides, polynucleotides, oligonucleotides, nucleotides, nucleic acids (eg, DNA such as cDNA, genomic DNA, RNA such as mRNA), poly Saccharides, oligosaccharides, lipids, small organic molecules (for example, hormones, ligands, signaling substances, small organic molecules, molecules synthesized by combinatorial chemistry, small molecules that can be used as pharmaceuticals (for example, small molecule ligands, etc.)) , These complex molecules are included, but not limited thereto.
- a factor specific for a polynucleotide is a polynucleotide having complementarity with a certain sequence homology to the sequence of the polynucleotide (eg, 70% or more sequence identity), Examples include, but are not limited to, a polypeptide such as a transcription factor that binds to the promoter region.
- Factors specific for a polypeptide typically include an antibody specifically directed against the polypeptide or a derivative or analog thereof (eg, a single chain antibody), and the polypeptide is a receptor.
- specific ligands or receptors in the case of ligands, and substrates thereof when the polypeptide is an enzyme include, but are not limited to.
- treatment refers to prevention of worsening of a disease or disorder when it becomes such a condition or disease (for example, cerebral malaria). Preferably, it refers to reduction, more preferably elimination, and includes the ability to exert a symptom improving effect or a preventive effect on one or more symptoms associated with a patient's disease or disease. Diagnosing in advance and performing appropriate treatment is referred to as “companion treatment”, and the diagnostic agent therefor is sometimes referred to as “companion diagnostic agent”.
- the term “therapeutic agent (agent)” broadly refers to any drug capable of treating a target condition (for example, a disease such as cerebral malaria), and an inhibitor (for example, provided by the present invention) Antibody).
- the “therapeutic agent” may be a pharmaceutical composition comprising an active ingredient and one or more pharmacologically acceptable carriers.
- the pharmaceutical composition can be produced by any method known in the technical field of pharmaceutics, for example, by mixing the active ingredient and the carrier.
- the form of use of the therapeutic agent is not limited as long as it is a substance used for treatment, and it may be an active ingredient alone or a mixture of an active ingredient and an arbitrary ingredient.
- the shape of the carrier is not particularly limited, and may be, for example, a solid or a liquid (for example, a buffer solution).
- prevention means that a certain disease or disorder (for example, cerebral malaria) is prevented from becoming such a state before it becomes such a state. Diagnosis can be performed using the drug of the present invention, and for example, cerebral malaria can be prevented or a preventive measure can be taken using the drug of the present invention as necessary.
- a certain disease or disorder for example, cerebral malaria
- prophylactic agent refers to any agent that can prevent a target condition (for example, a disease such as cerebral malaria) in a broad sense.
- the present invention relates to human leukocytes, in particular lymphocytes, in particular T lymphocytes, in particular CD8 positive cytotoxic T lymphocytes, derived from antigens associated with tumorigenesis and fully bound to MHC (HLA) class I molecules It also provides peptides that have the ability to trigger the immune response of, and also provides a combination of two peptides that are particularly useful as vaccinations for cancer patients.
- lymphocytes in particular T lymphocytes, in particular CD8 positive cytotoxic T lymphocytes, derived from antigens associated with tumorigenesis and fully bound to MHC (HLA) class I molecules
- HLA MHC
- the peptide of the present invention can be derived from a tumor-related antigen, particularly a tumor-related antigen having functions in proteolysis, angiogenesis, cell growth, cell cycle control, cell division, transcriptional regulation, tissue invasion and the like.
- the peptide can be chemically synthesized and can be used as an active pharmaceutical ingredient of a pharmaceutical
- the peptide provided by the present invention can be used for immunotherapy, preferably cancer immunotherapy.
- composition of the present invention further comprises additional peptides and / or excipients to increase the effect, as will be further described below.
- the pharmaceutical composition of the present invention may comprise a peptide identified in the present invention, the peptide comprising 8 to 100 amino acids, preferably 8 to 30 amino acids, most preferably 8 to 16 amino acids. Has as a full length.
- peptide or variant can be further modified to improve stability and / or binding to MHC molecules to elicit a stronger immune response.
- Methods for optimizing such peptide sequences are well known to those skilled in the art and include, for example, the introduction of reverse peptide bonds or non-peptide bonds.
- a pharmaceutical composition in which at least one peptide or variant comprises a non-peptide bond.
- U.S. Patent No. 4,897,445 non-peptide bonds in the polypeptide chain provides a method for the solid phase synthesis of (-CH 2 -NH), the polypeptide synthesized by standard procedures to this, the amino aldehyde It involves non-peptide bonds synthesized by reacting amino acids in the presence of NaCNBH 3 .
- Peptides having the sequences of the present invention can be synthesized with additional chemical groups at their amino terminus and / or carboxy terminus, for example, to enhance the stability, bioavailability, and / or affinity of the peptides.
- a hydrophobic group such as a carbobenzoxyl group, a dansyl group, or a t-butyloxycarbonyl group can be added to the amino terminus of the peptide.
- an acetyl group or a 9-fluorenylmethoxy-carbonyl group can be placed at the amino terminus of the peptide.
- the above-mentioned hydrophobic group t-butyloxycarbonyl group or amide group can be added to the carboxy terminus of the peptide.
- the peptides used in the present invention can be synthesized so as to change their configuration.
- the D-isomer of one or more amino acid residues of the peptide can be used in place of the normal L-isomer.
- at least one amino acid residue of the peptides of the invention can be replaced with one of the well-known unnatural amino acid residues. Such modifications can serve to increase the stability, bioavailability, and / or binding action of the peptides of the invention.
- the peptide or variant of the present invention can be chemically modified by reacting a specific amino acid before or after the synthesis of the peptide used in the present invention. Examples of such modifications are well known in the art and are summarized, for example, in R. Lundblad, ChemicalReagents for Protein Modification, 3rd ed. CRC Press, 2005, which is hereby incorporated by reference.
- Chemical modifications of amino acids include acylation, amidination, lysine pyridoxylation, reductive alkylation, trinitrobenzeneation of amino groups with 2, 4, 6-trinitrobenzenesulfonic acid TN (TNBS), amide modification of carboxyl groups and excess Sulfidyl modification with formic acid, oxidation of cysteine to cysteic acid, formation of mercury derivatives, formation of mixed disulfides with other thiol compounds, reaction with maleimide, carboxymethyl hatching with iodoacetic acid or iodoacetamide, and at alkaline pH Including, but not limited to, modification by carbamoylation of cyanide with cyanate.
- one skilled in the art can refer to a broader methodology for chemical modification of proteins by Current Protocols In Protein Science, Eds. Colligan et al. (John Wiley & Sons NY NY 1995-2000).
- modification of protein arginine residues is often the formation of adducts based on the reaction of adjacent dicarbonyl compounds such as phenylglyoxal, 2,3-butanedione, and 1,2-cyclohexanedione.
- Another example is the reaction of methylglycosal with an arginine residue.
- Cysteine can be modified without simultaneous modification of other nucleophilic sites such as lysine and histidine. Therefore, a large number of reagents can be used for cysteine modification. Specific reagent information is provided by Pierce Chemical Company, Sigma-Aldrich, and other websites.
- Disulfide bonds can be formed and oxidized during heat treatment of biopharmaceuticals.
- Woodward's ReagentK can be used to modify specific glutamate residues.
- N- (3- (dimethylamino) propyl) -N′-ethylcarbodiimide can be used to form intramolecular crosslinks between lysine residues and glutamic acid residues.
- diethyl pyrocarbonate is a reagent that modifies histidyl residues in proteins. Histidine can also be modified with 4-hydroxy-2-nonenal.
- lysine residues with other ⁇ -amino groups are useful, for example, for peptide-surface binding or protein / peptide cross-linking.
- Lysine is a site to which poly (ethylene) glycol is attached and is a major site of modification in protein glycation.
- the methionine residue of the protein can be modified with, for example, iodoacetamide, bromoethylamine, chloramine T. Tetranitromethane and N-acetylimidazole can be used to modify tyrosyl residues.
- Crosslinking due to the formation of dityrosine can be achieved by hydrogen peroxide / copper ions.
- peptides and variants used in the present invention are disclosed in Lu et al (1981) J. Org. Chem. 46, 3433 and references thereof, as disclosed therein.
- it can be synthesized using the Fmoc-polyamide form of solid phase peptide synthesis. Purification is one of techniques such as recrystallization, size exclusion chromatography, ion exchange chromatography, hydrophobic interaction chromatography, and (usually) reversed-phase high-performance liquid chromatography using acetonitrile / water gradient separation. Or it can carry out by the combination.
- Peptide analysis includes thin layer chromatography, electrophoresis, especially capillary electrophoresis, solid phase extraction (CSPE), reversed-phase high performance liquid chromatography, amino acid analysis after acid hydrolysis, fast atom bombardment (FAB) mass spectrometry , MALDI and ESI-Q-TOF mass spectrometry.
- CSPE solid phase extraction
- FAB fast atom bombardment
- Still another aspect of the present invention provides a nucleic acid (eg, polynucleotide) encoding the peptide of the present invention or a variant thereof.
- the polynucleotide include DNA, cDNA, PNA, CNA, RNA, single-stranded and / or double-stranded, or a natural or stable polynucleotide such as a polynucleotide having a phosphorothioate backbone, Intron inclusion is not essential as long as combinations are possible and the polynucleotide encodes the peptide.
- an expression vector capable of expressing a polypeptide according to the invention Expression vectors for different cell types are well known in the art and can be selected without special experimentation.
- DNA is inserted into an expression vector such as a plasmid in the correct orientation and with the correct reading frame for expression.
- the DNA can be linked to appropriate transcriptional and translational regulatory administrative nucleotide sequences recognized by the desired host, but such administrative functions are generally contained in expression vectors.
- the vector is then introduced into the host by standard techniques. For this, reference can be made to Sambrook1989et al (1989) Molecular Cloning, A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, ⁇ NY.
- the optimal amount of the nuclear peptide contained in the vaccine and the optimal dosage regimen can be determined by those skilled in the art without special experimentation.
- the peptide or variant thereof can be intravenous (iv) injection, subcutaneous (sc) injection, intradermal (id) injection, intraperitoneal (ip) injection, intramuscular. (Im) Can be prepared as an injection.
- the preferred route of administration for peptide injection is s. c. I. d. I. p. I. m. I. v. It is.
- the preferred route of administration for DNA injection is i. d. I. m. , S. c. I. p. I. v. It is.
- 1 to 500 mg, 50 ⁇ g to 1.5 mg, preferably 125 ⁇ g to 500 ⁇ g of peptide or DNA can be administered, and the dose depends on each peptide or DNA.
- Dose in this range has been used in previous trials and has been successfully used (Brunsvig PF, Aamdal S, Gjertsen MK, Kvalheim G, Markowski-Grimsrud CJ, Sve I, Dyrhaug M, Trachsel S, Muller M, Eriksen JA, Gaudernack G; Telomerase peptide vaccination: a phase I / II study in patients with non-small cell lung cancer; Cancer ImmunolImmunother. 2006; 55 (12): 1553-1564; M.
- the selection, number and / or amount of peptides present in the composition can be specific to tissue, cancer and / or patient.
- side effects can be avoided by guiding the correct selection of peptides by the protein expression pattern of a given patient tissue. This choice may depend on the type of cancer specific to the patient being treated and the disease state, previous treatment regimen, the patient's immune status, and, of course, the patient's HLA haplotype.
- the vaccine according to the invention can comprise individual components according to the personal needs of a particular patient. Examples are the amount of different peptides in a particular patient depending on the expression of the relevant TAA, the side effects of individual allergies or other treatments, and the adjustment of secondary treatment after a series of initial treatment regimens.
- Peptides in which the parent protein is expressed in high amounts in normal tissues are avoided or present in low amounts in the composition of the present invention.
- each pharmaceutical composition for the treatment of this cancer can be present in high amounts and / or this particular protein or pathway A plurality of peptides specific to can be included.
- Those skilled in the art will be able to determine in vitro T cell formation, their efficacy, and overall presentation, proliferation, affinity, expansion, and functionality of a particular T cell for a particular peptide, such as IFN- ⁇ . By testing by production analysis, preferred combinations of immunogenic peptides can be selected (see also the examples below). Usually, the most efficient peptides are then combined as a vaccine for the purposes described above.
- Suitable vaccines are preferably 1-20 peptides, more preferably 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18 , 19, or 20 different peptides, more preferably 6, 7, 8, 9, 10, 11, 12, 13, or 14 different peptides, most preferably 14 different peptides.
- the length of the peptide used as a cancer vaccine may be any suitable peptide. Specifically, a suitable 9-mer peptide or a suitable 7-mer or 8-mer or 10-mer or 11-mer peptide or 12-mer, 13-mer, 14-mer or 15-mer peptide It can be.
- the peptide of the present invention constitutes a tumor or cancer vaccine.
- the tumor or cancer vaccine is administered directly to the patient in its diseased organ or systemic, or is administered to the patient ex vivo applied to cells or human cell lines from the patient, or used in vitro A sub-population from the patient's immune cells can be selected and administered to the patient again.
- the peptide can be substantially pure or can be used in combination with an immunostimulatory adjuvant (see below), or in combination with an immunostimulatory cytokine, or administered with an appropriate delivery system (e.g., a liposome). .
- the peptide can also be conjugated to a suitable carrier such as keyhole limpet hemocyanin (KLH) or mannan (see WO95 / 18145 and Longgenecker et al (1993) Ann. NY Acad. Sci.690,276-291). ).
- KLH keyhole limpet hemocyanin
- mannan see WO95 / 18145 and Longgenecker et al (1993) Ann. NY Acad. Sci.690,276-291).
- the peptide can also be tagged, a fusion protein, or a hybrid molecule. Peptides whose sequences are given in the present invention are expected to stimulate CD4 or CD8CTL.
- the pharmaceutical composition of a preferred embodiment of the present invention further comprises at least one suitable adjuvant.
- the adjuvant used in the present invention is a substance that non-specifically enhances or promotes an immune response ⁇ to an antigen (e.g., an immune response mediated by CTL and helper T (TH) cells), and thus is considered useful for the agent of the present invention. It is done.
- an adjuvant such as Freund's incomplete or GM-CSF is preferred.
- An adjuvant such as Freund's incomplete or GM-CSF is preferred.
- Several immunological adjuvants specific to dendritic cells e.g. MF59
- their formulations have already been described (Dupuis M, Murphy TJ, Higgins D, Ugozzoli M, van Nest G, Ott G, McDonald DM; Dendriticcells internalize vaccine adjuvant after intramuscular injection; Cell Immunol.1998; 186 (1): 18-27; Allison AC; The mode of action of immunological adjuvants; Dev Biol Stand. 1998; 92: 3-11).
- cytokines e.g. MF59
- cytokines have been described as accelerating the process of dendritic cells to mature into antigen-presenting cells effective against T lymphocytes (e.g., GM-CSF, IL-1, IL-4) (U.S. Patent No. 5,849,589). This reference is incorporated in its entirety)) and also acts as an immunoadjuvant IL (e.g. IL-12) (Gabrilovich DI, Cunningham HT, Carbone DP; IL-12 and mutant P53 peptide-pulsed dendritic cells for the specific immuneotherapy of Cancer; J Immunother Emphasis Tumor Immunol. 1996 (6): 414-418), some directly linked to the effect on migration of dendritic cells to lymphocyte tissue (eg TNF- ⁇ ).
- TNF- ⁇ lymphocyte tissue
- CpG immunostimulatory oligonucleotides also enhance the effectiveness of adjuvants in vaccine settings.
- CpG oligonucleotides have the effect of activating the innate (non-adaptive) immune system via toll-like receptors (TLR) (mainly TLR9).
- TLR9 toll-like receptors
- CTL cytotoxic T lymphocyte
- the TH1 bias induced by stimulation of TLR9 is maintained even in the presence of vaccine adjuvants such as alum or Freund's incomplete adjuvant (IFA) that normally promote TH2 bias.
- CpG oligonucleotides exhibit higher adjuvant activity when co-formulated or co-administered with other adjuvants, or in microparticles, nanoparticles, lipid emulsions or similar formulations, for relatively weak antigens This is especially necessary to induce a strong response.
- adjuvants useful in the present invention include chemically modified CpGs (eg CpR, Idera), poly (I: C) (eg poly I: C12U), non-CpG bacterial DNA or RNA, And imidazoquinoline, cyclophosphamide, sunitinib, bivacizumab, celebrex, NCX-4016, sildenafil, tadalafil, vardenafil, sorafinib, XL-999, CP-547632, pazopanib, ZD2171, AZD2171, ipilimumab, tremelimumab, SC58175 They can act as therapeutic agents and / or adjuvants.
- adjuvants and additives useful in the context of the present invention can be readily determined by one skilled in the art without undue experimentation.
- Preferred adjuvants are dSLIM, BCG, OK432, imiquimod, PeviTer, and JuvImmune.
- the adjuvant is selected from the group comprising a colony stimulating factor such as granulocytic macrophage colony stimulating factor (GM-CSF, sargramostim).
- GM-CSF granulocytic macrophage colony stimulating factor
- the adjuvant is imiquimod.
- composition of the present invention is used for parenteral or oral administration such as subcutaneous, intradermal and intramuscular.
- the peptide and optionally other molecules for this purpose are dissolved or suspended in a pharmaceutically acceptable, preferably aqueous carrier.
- the composition can include buffers, binders, blasting agents, diluents, flavoring agents, excipients such as lubricants, and the like.
- the peptide can also be administered with an immunostimulatory substance such as a cytokine.
- An extensive list of excipients that can be used in such compositions is obtained, for example, from A. Kibbe, Handbook of Pharmaceutical Excipients, 3 Ed. 2000, American Pharmaceutical Association and pharmaceutical press.
- the composition can be used as a prophylaxis and / or therapy for tumors or cancer, preferably CRC.
- Cytotoxic T cells recognize peptide-shaped antigens bound to MHC molecules and not intact foreign antigens themselves.
- the MHC molecule itself is on the cell surface of the antigen-presenting cell. Therefore, CTL activation is only possible in the presence of peptide antigen trimer complex, MHC molecule and APC.
- the pharmaceutical composition of the invention additionally comprises at least one antigen presenting cell.
- Antigen presenting cells typically have MHC class I or II molecules on their surface, and in one embodiment, load themselves with the selected MHC class I or II molecule with the selected antigen. It is virtually impossible to do. As described in detail below, the MHC class I or II molecule can be easily loaded with the selected antigen in vitro.
- a pharmaceutical composition of the invention comprising a nucleic acid of the invention is administered in the same manner as a pharmaceutical composition comprising a peptide of the invention, i.e., intravenous, intraarterial, intraperitoneal, intramuscular, intradermal, It can be administered intratumorally, orally, transdermally, nasally, orally, rectally, vaginally, or by inhalation or topical administration.
- the pharmaceutical composition is administered in conjunction with a second anticancer agent.
- the second agent used in the present invention can be administered before, after, or simultaneously with the pharmaceutical composition of the present invention.
- Co-administration can be performed, for example, by mixing the pharmaceutical composition of the present invention with the second anticancer agent if the chemical properties are compatible.
- Another method of simultaneous administration is, for example, by injecting the pharmaceutical composition of the present invention and administering the second anticancer agent, for example, orally, so that the composition and the anticancer agent are administered on an independent route on the same day.
- the pharmaceutical composition and the second anticancer agent may be administered on different days within the same course of treatment and / or within separate courses of treatment.
- a method for treating or preventing cancer in a patient comprises administering to the patient any one of the pharmaceutical compositions of the invention in a therapeutically effective amount.
- a therapeutically effective amount is an amount sufficient to elicit an immune response, particularly to activate a subpopulation of CTLs.
- One skilled in the art can readily determine the effective amount using standard immunological methods, such as those provided in the Examples herein.
- Another way of monitoring the effect of a particular amount of the pharmaceutical composition of the invention is to observe the growth and / or recurrence of the treated tumor.
- the pharmaceutical composition is used as an anti-cancer vaccine.
- composition containing the peptide or peptide-encoding nucleic acid of the present invention can constitute a tumor or cancer vaccine.
- the tumor or cancer vaccine is administered directly to the patient in its diseased organ or systemically, or administered ex vivo to cells or human cell lines from the patient, or used in vitro A sub-population from the patient's immune cells can then be selected and administered to the patient again.
- the composition of the present invention can be used as a method for treating cancer or as a vaccine.
- the cancer includes oral cavity, pharyngeal cancer, gastrointestinal cancer, colon, rectum, anal cancer, airway cancer, breast cancer, uterus, vagina, vulvar cancer, uterine body, ovarian cancer, male reproductive tract cancer, urethral cancer, Bone and soft tissue cancer, Kaposi's sarcoma, cutaneous melanoma, ocular melanoma, non-melanoma eye cancer, brain, central nervous system cancer, thyroid and other endocrine cancers, Hodgkin lymphoma, non-Hodgkin lymphoma, myeloma, preferred Is kidney cancer, colorectal cancer, lung cancer, breast cancer, pancreatic cancer, prostate cancer, stomach cancer, brain cancer, GIST or glioblastoma.
- the preferred amount of peptide can vary between about 0.1-100 mg, preferably about 0.1-1 mg, most preferably about 300-800 ⁇ g in a 500 ⁇ l solution.
- the term “about” means +/ ⁇ 10 percent of a given value unless otherwise stated.
- One skilled in the art can adjust the actual amount of peptide used based on several factors, such as the individual's immune status and / or the amount of TUMAP presented in a particular type of cancer. I will.
- the peptide of the present invention may be provided in other suitable forms (such as a sterilized solution) other than the frozen lysate.
- the pharmaceutical composition of the present invention having a peptide and / or nucleic acid according to the present invention is administered to a patient with an adenoma or cancer disease associated with each corresponding peptide or antigen.
- a T cell-mediated immune response is triggered, preferably a pharmaceutical composition of the present invention, wherein the peptide composition (especially tumor-related peptide) or nucleic acid of the pharmaceutical composition of the present invention, or the
- the amount of the expression vector of the invention present in the composition is specific to the tissue, cancer, and / or patient.
- the vaccine of the invention is a nucleic acid vaccine. It is well known that vaccination with a nucleic acid vaccine, such as a DNA vaccine encoding a polypeptide, elicits a T cell response.
- a nucleic acid vaccine such as a DNA vaccine encoding a polypeptide
- the tumor or cancer vaccine can be administered directly to the patient in its diseased organ or systemically, or in vitro applied to cells or human cell lines from the patient, or in vitro
- the sub-population from the patient's immune cells can be selected and administered to the patient again.
- the nucleic acid is administered to cells in vitro, it may be useful to transfer the cells so as to co-express immunostimulatory cytokines such as interleukin-2 or GM-CSF.
- the nucleic acid can be substantially pure, or combined with an immunostimulatory adjuvant, used in combination with an immunostimulatory cytokine, or administered with an appropriate delivery system (eg, a liposome).
- the nucleic acid vaccine may be administered with an adjuvant as described for the peptide vaccine above. Preferably, the nucleic acid vaccine is administered without an adjuvant.
- the polynucleotide of the present invention may be substantially pure or contained in a suitable vector or delivery system.
- suitable vectors and delivery systems include those that are viral, such as adenoviruses, vaccinia viruses, retroviruses, herpesviruses, adeno-associated viruses, or hybrid-based systems that contain multiple viral elements . Included as non-viral delivery systems are cationic lipids and cationic polymers well known in the DNA delivery art. Physical delivery such as a “gene gun” can also be used.
- the peptide, or peptide encoded by the nucleic acid can be a fusion protein, for example, a fusion protein with an epitope from a tetanus toxoid that stimulates CD4 positive T cells.
- nucleic acid vaccine can have any nucleic acid delivery means.
- the nucleic acid, preferably DNA, can be delivered in liposomes or as part of a viral vector delivery system.
- Nucleic acid vaccines such as sputum DNA vaccines, are preferably administered intramuscularly, and peptide vaccines are preferably administered at s.c. or i.d. It is also preferred to administer the vaccine intradermally.
- Nucleic acid uptake by professional antigen-presenting cells such as dendritic cells and expression of the encoded polypeptide may be the priming mechanism of the immune response.
- dendritic cells may not be transferred, it is still important because they can take up the expressed peptide from the transferred cells in the tissue (“cross-priming”, eg Thomas : AM, Santarsiero LM).
- cross-priming eg Thomas : AM, Santarsiero LM
- Lutz ER Armstrong TD, Chen YC, Huang LQ, Laheru DA, Goggins M, Hruban RH, Jaffee EM.
- Mesothelin-specific CD8 (+) T cell responses provide creativic cross-primingpresentaccv cancerpatients. J Exp Med. 2004 Aug 2; 200 (3): 297-306).
- the target of the vaccine of the present invention is directed to a specific cell population, such as antigen presenting cells, by use of an injection site, targeting vector and delivery system, or selective purification of a specific cell population from a patient.
- a targeting vector can have a tissue or tumor specific promoter that directs expression of the antigen at the appropriate location.
- the vaccine of the present invention may depend on the type of cancer specific to the patient being treated and the disease state, previous treatment regimen, the patient's immune status, and, of course, the patient's HLA haplotype.
- the vaccine according to the invention can comprise individual components according to the personal needs of a particular patient. Examples are the amount of different peptides in a particular patient depending on the expression of the relevant TAA, the side effects of individual allergies or other treatments, and the adjustment of secondary treatment after a series of initial treatment regimens.
- the peptide of the present invention is useful not only for cancer treatment but also for diagnosis. Since these peptides are produced from glioblastoma, and it has been specified that these peptides are not present in normal tissues, the presence of cancer can be diagnosed using these peptides.
- Pathologists can aid in the diagnosis of cancer by the presence of the peptides of the present invention on tissue biopsy.
- a pathologist can know whether the tissue is malignant, inflamed or generally affected by detection of specific peptides of the invention, mass spectrometry, or other methods well known in the art using antibodies. it can.
- the presence of the group of peptides of the present invention allows the classification or subclassification of affected tissues.
- Detection of the peptides of the present invention in diseased tissue specimens allows decisions on the benefits of therapy involving the immune system, especially when T lymphocytes are known or expected to be involved in the mechanism of action. To do. Loss of MHC expression is a well-understood mechanism that allows malignant cells to escape immune surveillance. Thus, the presence of the peptides of the present invention indicates that this mechanism is not utilized by the analyzed cells.
- the peptide of the present invention can be used for analysis of lymphocyte response to the peptide of the present invention.
- a T cell response or antibody response to the peptide of the present invention or a peptide of the present invention which is a complex with an MHC molecule can be used.
- These lymphocyte responses can be used as prognostic markers to determine further treatment steps.
- These responses can also be used as surrogate markers in immunotherapy approaches that seek to elicit lymphocyte responses by different means such as, for example, vaccination of proteins, nucleic acids, self-substances, lymphocyte immune transfer.
- lymphocyte responses to the peptides of the invention can be taken into account in the evaluation of side effects. Monitoring of lymphocyte responses may be useful in follow-up testing after transplantation therapy, for example, in detecting graft-to-host and host-to-graft disease.
- the peptide of the present invention can be used for production and development of an antibody specific to the MHC / peptide complex. They can be used in therapy to target the affected tissue and apply a toxin or radioactive material. Another use of these antibodies is to target them against radionuclides against diseased tissue for imaging methods such as PET. This method of use can help detect small metastases or determine the size and exact location of diseased tissue.
- the peptides can be used for verification of cancer diagnosis based on biopsy specimens performed by a pathologist.
- kits are a unit provided with a portion to be provided (eg, a test agent, a diagnostic agent, a therapeutic agent, an antibody, a label, instructions, etc.) usually divided into two or more compartments.
- a portion to be provided eg, a test agent, a diagnostic agent, a therapeutic agent, an antibody, a label, instructions, etc.
- This kit form is preferred when it is intended to provide a composition that should not be provided in admixture for stability or the like, but preferably used in admixture immediately before use.
- kits preferably include instructions or instructions that describe how to use the provided parts (eg, test agents, diagnostic agents, therapeutic agents, or how the reagents should be processed).
- the kit when the kit is used as a reagent kit, the kit usually contains instructions including usage of test agents, diagnostic agents, therapeutic agents, antibodies, etc. Is included.
- the invention relates to a kit comprising: (a) a container containing the pharmaceutical composition of the invention in solution or lyophilized form; and (b) selected A second container containing a diluent or reconstitution liquid for the lyophilized formulation, and (c) optionally (i) use of the solution or (ii) reconstitution of the lyophilized formulation and And / or instructions for use.
- the kit further comprises one or more (iii) a buffer, (iv) a diluent, (v) a filter, (vi) a needle, or (v) a syringe.
- the container is preferably a bottle, vial, syringe, or test tube and may be a versatile container.
- the pharmaceutical composition is preferably dried and frozen.
- the kit of the present invention preferably has the dry frozen formulation of the present invention and instructions regarding its reconstitution and / or use in a suitable container.
- suitable containers include, for example, bottles, vials (eg, dual chamber vials), syringes (such as dual champ syringes), and test tubes.
- the container can be formed from a variety of materials such as glass or plastic.
- the kit and / or container includes instructions on how to reconstitute and / or use that are on or associated with the container.
- the label can indicate that the dried frozen formulation is reconstituted to the peptide concentration described above.
- the label can further indicate that the formulation is useful for or for subcutaneous injection.
- the container for the formulation may be a versatile vial that can be used for repeated administration (eg, 2-6 administrations).
- the kit can further include a second container having a suitable diluent (eg, a baking soda solution).
- the kit further includes other materials desirable from a commercial and user standpoint, including other buffers, diluents, filters, needles, syringes, and instructions inserted into the package. Can do.
- the kit of the present invention has a single container containing the formulation of the pharmaceutical composition of the present invention with or without other components (e.g., other compounds or pharmaceutical compositions of these other compounds). Or, each component can have a separate container.
- the kit of the invention comprises a second compound (adjuvant (eg GM-CSF), chemotherapeutic agent, natural product, hormone or antagonist, anti-angiogenesis agent or angiogenesis inhibitor, apoptosis inducer or chelate Etc.) or a formulation of the invention packaged for use in combination with the co-administration of the pharmaceutical composition.
- a second compound eg GM-CSF
- chemotherapeutic agent eg GM-CSF
- natural product e.g. GM-CSF
- hormone or antagonist e.g., anti-angiogenesis agent or angiogenesis inhibitor, apoptosis inducer or chelate Etc.
- a formulation of the invention packaged for use in combination with the co-administration of the pharmaceutical composition.
- the components of the kit can be pre-made as a complex, or each component can be in a separate container until administered to a patient.
- the kit components can be provided as one or more liquid solutions, preferably an aqueous solution, more preferably
- the container of the therapy kit can be a vial, test tube, flask, bottle, syringe, or any other means of sealing a solid or liquid.
- the kit includes a second vial or other container so that it can be dispensed separately.
- the kit can also include another container for a pharmaceutically acceptable liquid.
- the treatment kit includes a device (eg, one or more needles, syringes, eye drops, pipettes, etc.) that allows administration of an agent of the invention that is a component of the kit.
- the pharmaceutical composition of the present invention administers the peptide by any acceptable route such as oral (enteral), nasal, ocular, subcutaneous, intradermal, intramuscular, intravenous, or transdermal. It is suitable for. Preferably the administration is subcutaneous, most preferably intradermal. Administration can be performed by an infusion pump.
- the “instruction sheet” describes the method for using the present invention for a doctor or other user.
- This instruction manual includes a word indicating that the detection method of the present invention, how to use a diagnostic agent, or administration of a medicine or the like is given.
- the instructions may include a word indicating that the administration site is oral or esophageal administration (for example, by injection).
- This instruction is prepared in accordance with the format prescribed by the national supervisory authority (for example, the Ministry of Health, Labor and Welfare in Japan and the Food and Drug Administration (FDA) in the United States, etc.) It is clearly stated that it has been received.
- the instruction sheet is a so-called package insert and is usually provided in a paper medium, but is not limited thereto, and is in a form such as an electronic medium (for example, a homepage or an e-mail provided on the Internet). But it can be provided.
- RNA extraction 5 mL of whole blood was collected from a healthy human subject in a heparin-containing blood vessel, and peripheral blood mononuclear cells (PBMC) were separated by ficoll density gradient centrifugation. Isolated 5 ⁇ 10 6 RNeasy Lipid from PBMC cells Tissue Mini Kit (QIAGEN, Germany) was used to extract and purify total RNA. The obtained RNA was quantified by the absorbance of A260 using an absorptiometer. The concentration was 232 ng / ⁇ L at an eluate volume of 30 ⁇ L.
- E. coli in the following double-stranded DNA synthesis buffer solution was used.
- Coli DNA polymerase I E.I.
- Double-stranded complementary DNA was synthesized by incubating at 16 ° C. for 2 hours in the presence of E. coli DNA Ligase and RNase H. Further, T4 DNA polymerase was reacted at 16 ° C. for 5 minutes to perform a 5 ′ end blunting reaction.
- Double-stranded DNA was column purified by High Pure PCR Cleanup Micro Kit (Roche) and then in the following T4 ligase buffer at 16 ° C in the presence of P20EA / 10EA adapter (Table 1-1) and T4 ligase. The ligation reaction was carried out by incubating overnight.
- the adapter-added double-stranded DNA purified by the column as described above was digested with NotI restriction enzyme (50 U / ⁇ L, Takara) with the following composition in order to remove the adapter added to the 3 ′ end.
- NotI restriction enzyme 50 U / ⁇ L, Takara
- PCR Primary PCR amplification (1 st PCR) from double-stranded complementary DNA is performed using the common adapter primer P20EA and each immunoglobulin isotype C region specific primer (CM1, CA1, CG1, CD1, CE1). 20 cycles of 95 ° C. for 30 seconds, 55 ° C. for 30 seconds, and 72 ° C. for 1 minute were performed.
- the primer sequences used are shown in Table 1-1.
- PCR cycle was 20 cycles of 95 ° C. for 30 seconds, 55 ° C. for 30 seconds, 72 ° C. for 1 minute.
- the primer sequences used are shown in Table 1-1.
- the DNA amounts of the recovered amplification products derived from the respective isotypes were IgM (1611 ng / mL), IgG (955 ng / mL), IgA (796 ng / mL), IgD (258 ng / mL), IgE (871 ng / mL), respectively. there were. These isotype amplification products were mixed so that the amounts of DNA were uniform, 10 million DNA was used for emulsion PCR, and sequence analysis was performed using a next-generation sequence analyzer (GS Junior bench top system) manufactured by Roche.
- the crossover property of isotype-specific primers is shown in FIG.
- amplification with another isotype-specific primer together with the target immunoglobulin isotype-specific primer was performed to confirm the presence or absence of cross-reactivity.
- 10 ⁇ L of the GS-PCR amplification product was electrophoresed in 2% agarose gel in TAE buffer and evaluated by ethidium bromide staining.
- the 2nd PCR amplification product amplified with each isotype-specific primer was confirmed to have high primer specificity without being amplified with other isotype-specific GS-PCR primers.
- Fig. 2 shows the optimum dilution concentration.
- the optimum conditions for GS-PCR in each isotype were examined. A 2-fold serial dilution series of 2nd PCR amplification products was prepared, and 20 cycles of GS-PCR were performed. Good results were obtained at 16-fold dilution.
- Figure 3 shows the optimum number of cycles. PCR was performed for 10, 15, and 20 cycles using a 16-fold diluted 2nd PCR amplification product. In IgM, IgG, IgA, and IgD, good amplification was confirmed in 10 cycles. Further, it was confirmed that 20 cycles is appropriate for IgE.
- FIG. 4 shows the read length of the next generation sequence of the BCR gene.
- the number of leads of raw data was 130,000, and the number of leads passed through Filter pass was 90,000 or more.
- Table 1-2 shows the number of leads derived from each isotype labeled.
- the lead length by MID is shown in FIG.
- the number of leads divided by MID and the distribution of lead lengths were also uniform.
- the lead length having a length sufficient for the analysis of the V region is counted as 400 bp or more, it is considered that about half of the 10,000 leads are effective for the BCR repertoire analysis.
- the frequency of use of isotype-specific C region sequences is shown in FIG.
- the obtained leads for each isotype were subjected to a homology search with immunoglobulin isotype C region sequences including subclasses.
- the frequency of reads by subclass was 73% for IgA1 and 27% for IgA2 in the IgA subclass, 62% for IgG1 and 36% for IgG2, and almost no IgG3 and IgG4 reads were obtained. .
- the specificity of the primer could be reconfirmed at the sequence level.
- the V region repertoires by isotype are shown in Fig. 7 (AD).
- the repertoire (BCR V repertoire) of the V region sequence by isotype is shown.
- the BCR V repertoire was very similar among IgM, IgG, IgA, and IgD, but only the lead with IGHV3-30 was obtained for IgE. This is because the number of IgE positive cells in peripheral blood is very small compared to other classes, suggesting the possibility that a biased repertoire was detected.
- the V region repertoire by subtype is shown in Fig. 8 (AD).
- BCR V repertoires by IgA and IgG subclass are shown.
- the subclasses of IgA differed in the frequency of several V chains between IgA1 and IgA2.
- IGHV1-18 and IGHV4-39 were higher in IgA1 than IgA2, while IGHV3-23 and IGHV3-74 were higher in IgA2 than IgA1.
- the frequency of IGHV3-23 and IGHV3-74 which showed an increase in IgA2, was higher in IgG2 compared to IgG1.
- the number of reads of IgG3 and IgG4 is small (10 reads).
- IgG3 clones with IGHV4-59-IGHJ4-IGHD1-7 have a high clonality of 3/10, and IGHV3-23-IGHJ4-IGHD3- is also used for IgG4. Leads with 10 accounted for 5/10 (Table 1-3).
- Figure 9 shows BCRJ repertoires by subclass.
- IgM IgG, IgA, and IgD
- IGHJ4 was used in about half of the leads while IGHJ2 was rarely used.
- IgE only IGHJ1 was used.
- IGHJ repertoires in the IgM and IgA subclasses were also examined. Unlike the IGHV repertoire, there was no significant difference between subclasses.
- TCR repertoire was analyzed in peripheral blood of healthy individuals.
- RNA extraction From 10 healthy individuals, 5 mL of whole blood was collected in heparin-containing blood collection tubes, and peripheral blood mononuclear cells (PBMC) were separated by ficoll density gradient centrifugation. Total RNA was extracted and purified from the isolated PBMC using RNeasy Lipid Tissue Mini Kit (Qiagen, Germany). The obtained RNA was quantified using an Agilent 2100 bioanalyzer (Agilent). The obtained RNA amounts are shown in the following Table 1-4.
- Adapter-ligation PCR was performed using the extracted RNA sample. The method was carried out according to the method shown in Preparation Example 1. Specifically, BSL-18E primer (Table 1-5) and RNA were mixed and annealed, and then complementary strand DNA was synthesized using reverse transcriptase. Subsequently, double-stranded complementary DNA was synthesized, and a 5 ′ end blunting reaction was performed using T4 DNA polymerase. After column purification by High Pure PCR Cleanup Micro Kit (Roche), a P20EA / P10EA adapter was added by a ligation reaction. The adapter-added double-stranded complementary DNA purified by the column was digested with NotI restriction enzyme.
- PCR From the double-stranded complementary DNA, using the common adapter primer P20EA shown in Table 1-1 and the TCR ⁇ -chain or ⁇ -chain C region specific primer (CA1 or CB1), 1 st which is the product of the first PCR amplification reaction PCR amplification was performed. PCR had the following composition, and 20 cycles of 95 ° C. for 30 seconds, 55 ° C. for 30 seconds, and 72 ° C. for 1 minute were performed.
- 2nd PCR was performed using the P20EA primer and the TCR ⁇ chain or ⁇ chain C region specific primer (CA2 or CB2) with the following reaction composition.
- the PCR cycle was 20 cycles of 95 ° C. for 30 seconds, 55 ° C. for 30 seconds, and 72 ° C. for 1 minute.
- the primer was removed from the obtained 2nd PCR amplification product, which was the product of the second PCR amplification reaction, using High Pure PCR Cleanup Micro Kit (Roche), and further 10-fold diluted 2nd PCR amplification product was used as a template. Analysis was performed using a generation sequence analyzer (GS Junior Bench Top System).
- a B-P20EA primer obtained by adding an adapter B sequence to the P20EA adapter primer shown in FIG. 10, an adapter A sequence and a TCR ⁇ chain or ⁇ chain C region specific sequence, and each MID tag sequence (MID-1 to 26) HuVaF-01 to HuVaF10 ( ⁇ chain) and HuVbF-01 to HuVbF-10 ( ⁇ chain) to which was added were used.
- the primer sequences used are shown in Table 1-6.
- the PCR cycle was 10 cycles of 95 ° C. for 30 seconds, 55 ° C. for 30 seconds, 72 ° C. for 1 minute.
- 10 ⁇ L of the amplified product was amplified by 2% agarose gel electrophoresis (FIG. 11).
- a band containing an amplification product of about 600 bp was visually cut out and purified using a DNA purification kit (QIAEX II Gel Extraction Kit, Qiagen).
- the recovered PCR amplification product was measured for the amount of DNA by Quant-T TM PicoGreen (registered trademark) dsDNA Assay Kit (Invitrogen).
- Table 1-7 shows the amount of DNA recovered from 10 healthy individuals.
- the obtained sequence data (SFF file) was classified into read sequences for each MID Tag by using GS Junior attached software (sffile or sffinfo), and a Fasta format sequence file was generated.
- the average number of leads obtained was TRA: 17840 leads, TRB: 5122 leads, and the ratio of raw data of 200 bp or more was TRA: 34.9-63.7% (average 42.2%), TRB: 68.8- It was 78.7% (average 73.1%) (Table 1-8).
- the reference sequence of the IMGT the international ImmunGeneTics information system, www.imgt.org
- FIGS. 13 (A to D), 14 (A to D), 15 (A to D), and FIG. 16 show TRV and TRJ repertoire generated using leads obtained by Repertoire Genesis.
- FIG. 10 shows a method for amplifying the TCR gene. Amplification was performed using a B-P20EA primer with B-adaptor added to the P20EA adapter primer and a primer with A-adaptor and MID tag sequences (MID-1 to 26) added to the 3rd nested primer.
- the confirmation of the GS-PCR amplification product is shown in FIG.
- Ten ⁇ L of GS-PCR amplification products derived from 10 healthy subjects were electrophoresed on a 2% agarose gel.
- the upper row shows GS-PCR (TRA) (TCR ⁇ chain amplification product), and the lower row shows GS-PCR (TRB) (TCR ⁇ chain amplification product).
- TRA GS-PCR
- TRB GS-PCR
- the TRAV repertoire for healthy individuals is shown in FIG.
- the TRAV repertoire and average values of 10 healthy subjects were shown.
- the presence frequency of TRAV9-2, 12 and 13 was high, TRAV20 in # 1, TRAV21 in # 5 was higher than other healthy individuals, and individual differences were also observed.
- the TRBV repertoire for healthy individuals is shown in FIG. The TRBV repertoire and average values of 10 healthy subjects were shown. The presence frequency of TRBV20-1, 28, and 29-1 was high. In # 8, TRBV3-1 was higher than other healthy individuals, and individual differences were observed. *
- TRAJ repertoire in healthy individuals is shown in FIG. TRAJ repertoire and average values of 10 healthy subjects were shown. TRAJ repertoires of healthy individuals show that within about 5% of all AJ families, TRAJ12 in # 1, TRAJ27 in # 4, TRAJ37 in # 5, TRAJ45 in # 8 are higher than other healthy individuals, and individual differences are observed It was.
- FIG. 16 shows the TRBJ repertoire for healthy individuals.
- the TRBJ repertoire and average values of 10 healthy subjects were shown.
- TRBJ repertoire had a high frequency of TRBJ2-1, 2-3, and 2-7, and in # 8, TRBJ2-2 was high, showing individual differences.
- TCR and BCR gene amplification by non-biased Adapter-ligation PCR method TCR and BCR gene amplification by non-biased Adapter-ligation PCR method.
- RNA extraction From one healthy subject, 5 mL of whole blood was collected in a heparin-containing blood collection tube, and peripheral blood mononuclear cells (PBMC) were separated by Ficoll density gradient centrifugation. Isolated 5 ⁇ 10 6 RNeasy Lipid from PBMC cells Tissue Mini Kit (QIAGEN, Germany) was used to extract and purify total RNA.
- E. coli in the following double-stranded DNA synthesis buffer solution was used.
- Coli DNA polymerase I E.I.
- Double-stranded complementary DNA was synthesized by incubating at 16 ° C. for 2 hours in the presence of E. coli DNA Ligase and RNase H. Further, T4 DNA polymerase was reacted at 16 ° C. for 5 minutes to perform a 5 ′ end blunting reaction.
- Double-stranded DNA was purified by column using High Pure PCR Cleanup Micro Kit (Roche) and then in the following T4 ligase buffer at 16 ° C in the presence of P20EA / 10EA adapter (Table 1-1) and T4 ligase. The ligation reaction was carried out by incubating overnight.
- the adapter-added double-stranded DNA purified by the column as described above was digested with NotI restriction enzyme (50 U / ⁇ L, Takara) with the following composition in order to remove the adapter added to the 3 ′ end.
- NotI restriction enzyme 50 U / ⁇ L, Takara
- the 1st PCR from double-stranded complementary DNA was performed using P20EA and TCRC region specific primers (CA1, CB1, CG1, CD1) or immunoglobulin isotype C region specific primers (CM1, CA1, CG1, CD1). , CE1, CK1, CL1).
- Primers were set on the 3 ′ end, center or 5 ′ side of the C region so as to amplify the sequence including the full length of the C region. With the reaction composition shown, a cycle of 95 ° C. for 30 seconds, 55 ° C. for 30 seconds, 72 ° C. for 1 minute was performed 20 cycles.
- the primer sequences used are shown in Table 1-1.
- PCR cycle was 20 cycles of 95 ° C. for 30 seconds, 55 ° C. for 30 seconds, 72 ° C. for 1 minute.
- the primer sequences used are shown in Table 1-1.
- FIGS. The primer positions relative to the template are shown in FIGS. This indicates that a considerable range of regions are suitable as the PCR primer for the purpose of the present invention, and it is understood that a specific sequence can be determined as appropriate based on the principle of the present invention.
- Example Normal human peripheral blood mononuclear cells, MOLT-4 human acute lymphoblastic leukemia cell line (Method) (1. Culture of T cell leukemia cell line) As a T cell line expressing T cell receptor (TCR), human acute lymphoblastic leukemia cell line Molt-4 was used. The cells were cultured in RPMI-1640 medium containing 10% fetal bovine serum, 100 IU / ml penicillin, 100 ⁇ g / ml streptomycin, 2 mM L-glutamine at 37 ° C. under 5% CO 2 to give a total cell count of 1 ⁇ 10 7. Cells were collected. The cells were washed with RPMI-1640 medium, and the cells were suspended at 1 ⁇ 10 6 cells / mL.
- PBMC peripheral blood mononuclear cells
- RNA extraction and RNA amount measurement Total RNA was extracted and purified from the serially diluted cell suspension using RNeasy Lipid Tissue Mini Kit (QIAGEN, Germany). Elution was performed with 20 ⁇ L of the eluate, and the amount of RNA was quantified based on the absorbance of A260 using an Agilent 2100 bioanalyzer (Agilent). The RNA electrophoresis image is shown in FIG. 26, and the amount of RNA obtained from each sample is shown in Table 1-4B.
- RNAsin RNase inhibitor
- E. coli in the following double-stranded DNA synthesis buffer solution was used.
- Coli DNA polymerase I E.I.
- Double-stranded complementary DNA was synthesized by incubating at 16 ° C. for 2 hours in the presence of E. coli DNA Ligase and RNase H. Further, T4 DNA polymerase was reacted at 16 ° C. for 5 minutes to perform a 5 ′ end blunting reaction.
- Double-stranded DNA was purified by column using High Pure PCR Cleanup Micro Kit (Roche) and then in the following T4 ligase buffer at 16 ° C in the presence of P20EA / 10EA adapter (Table 1-4E) and T4 ligase. The ligation reaction was carried out by incubating overnight.
- the adapter-added double-stranded DNA purified by the column as described above was digested with NotI restriction enzyme (50 U / ⁇ L, Takara) with the following composition in order to remove the adapter added to the 3 ′ end.
- NotI restriction enzyme 50 U / ⁇ L, Takara
- PCR From double-stranded complementary DNA, using a common adapter primer P20EA and TCR ⁇ chain or ⁇ chain C region-specific primers shown in Table 1-1 (CB1), it was performed 1 st PCR amplification. PCR had the following composition, and 20 cycles of 95 ° C. for 30 seconds, 55 ° C. for 30 seconds, and 72 ° C. for 1 minute were performed.
- PCR cycle was 20 cycles of 95 ° C. for 30 seconds, 55 ° C. for 30 seconds, 72 ° C. for 1 minute.
- Primers were removed from the obtained 2nd PCR amplification product using High Pure PCR Cleanup Micro Kit (Roche), and further 10-fold diluted 2nd PCR amplification product was used as a template, Roche's next-generation sequencing analyzer (GS Junior bench top system) ) was conducted.
- a B-P20EA primer obtained by adding an adapter B sequence to a P20EA adapter primer and a HuVbF Primer obtained by adding an adapter A sequence and each MID tag sequence to a TCR ⁇ chain C region specific sequence were used.
- the PCR cycle was 10 cycles of 95 ° C. for 30 seconds, 55 ° C. for 30 seconds, 72 ° C. for 1 minute.
- the obtained sequence data (SFF file) was classified into read sequences according to MID Tag by GS Junior attached software (sfffile or sffinfo), and a Fasta format sequence file was generated. The number of effective leads obtained was 11,651.
- the repertoire analysis software (Repertoire Genesis)
- the reference sequence of the IMGT database was compared, and the assignment of the BV region and BJ region of each read and the determination of the CDR3 sequence were performed.
- an in-frame TCR lead (Read 1) having a functional sequence and a frame-shifted TCR lead (Read 2) were observed (Table 1-4J). All were detected with a similar frequency, and were assumed to be TCR genes derived from Molt-4 cells.
- RNA electrophoresis image by Agilent 2100 bioanalyzer is shown in FIG.
- Total RNA was extracted from the cell serial dilution, and the amount of RNA was measured using an Agilent bioanalyzer.
- RNA was separated using a microchip electrophoresis apparatus, and RNA quality was checked. In both samples, 28S (upper band) and 18S rRNA (lower band) were detected, indicating that RNA that had not undergone degradation was obtained.
- TCR reads in Molt-4 cell serially diluted samples are shown in FIGS. 27 (A to D) (SEQ ID NOS: 1165 to 1324).
- the TCR leads obtained from 10%, 1%, 0.1% and 0.01% of each Molt-4 serially diluted sample are listed in Table 1. Ranking in order of the number of leads, showing the top 40. Rankings 365-404 were shown for 0.01% samples.
- the TRBV, TRBJ and CDR3 amino acid sequences of each lead and the number of reads are shown.
- a functional TCR lead derived from Molt-4 (TRBV20-1 / TRBJ2-1 / CSARESTTSDPKNEQFFG (SEQ ID NO: 1166)) is shown in bold and background gray, and the other functional defect is predicted (TRBV10-3 / TRBJ2) ⁇ 5 / CAISEPTTGIRRDDPVLR (SEQ ID NO: 1165)) is shown in bold.
- FIG. 28 shows the number of TCR leads and detection sensitivity in the Molt-4 cell serially diluted sample.
- Two TCR reads were detected from Molt-4 cells ( ⁇ : TRBV20-1 / TRBJ2-1 / CSARESTSDPKNEQFFG (SEQ ID NO: 1166), ⁇ : TRBV10-3 / TRBJ2-5 / CAISEPTGIRRDPVLR (SEQ ID NO: 1165)).
- the percentage of Molt-4 derived TCR reads detected in TCR reads obtained from 10%, 1%, 0.1% and 0.01% of each Molt-4 serially diluted sample is shown.
- the detection limit (Detection limit) of each read was 0.1% ( ⁇ ) and 0.01% ( ⁇ ).
- a lead set was used in which BCR cDNA obtained in an unbiased manner from RNA obtained from peripheral blood mononuclear cells of one healthy subject was sequenced by Roche GS-Junior. It is a lead set for each class of IgM, IgG, IgA, IgD, and IgE.
- FIG. 30 An overall view of the method is shown in FIG. 30 (FIG. 29 shows a TCR analysis scheme).
- the previously reported allele nucleic acid sequence was obtained from IMGT and used. Although BLASTN was used for the homology search, the following parameters were set for each region.
- the indicators for selecting the closest reference allele were applied in the following order of priority:
- Table 2-3 and Table 2-4 show a comparison of D repertoires between classes. The number of appearance reads was described for each gene name and CDR3 amino acid sequence, and the gene name and amino acid sequence of lead number 1 were omitted.
- FIG. 32 (A and B) shows the V repertoire
- FIG. 33 shows the J repertoire.
- FIG. 34 (A and B) shows a comparison of V repertoires between subclasses.
- FIG. 35 shows a comparison of J repertoires between subclasses.
- the frequency is derived for the combination of the D gene name and the CDR3 amino acid sequence.
Landscapes
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Organic Chemistry (AREA)
- Analytical Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Physics & Mathematics (AREA)
- Immunology (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Biotechnology (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- General Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- Molecular Biology (AREA)
- Pathology (AREA)
- Bioinformatics & Computational Biology (AREA)
- Medical Informatics (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Evolutionary Biology (AREA)
- Theoretical Computer Science (AREA)
- Cell Biology (AREA)
- Hospice & Palliative Care (AREA)
- Oncology (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Apparatus Associated With Microorganisms And Enzymes (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
Abstract
Description
<インシリコ>
1つの局面では、本発明は、生体試料に由来する一群の発現TCRもしくはBCR遺伝子配列を元に、そのTCRもしくはBCRレパトアを解析する手法に関する。
<1>TCRもしくはBCRレパトアを解析する方法であって、以下のステップ:
(1)V領域、D領域、J領域および必要に応じてC領域の少なくとも1つを含む遺伝子領域ごとの参照データベースを提供するステップ:
(2)必要に応じてトリミングを行い、必要に応じて適切な長さのものを抽出した入力配列セットを提供するステップ;
(3)該入力配列セットについて、該遺伝子領域ごとに、該参照データベースと相同性検索を行い、近似する参照アリルおよび/または該参照アリルの配列とのアラインメントを記録するステップ;
(4)該入力配列セットについてV領域およびJ領域をアサインし、アサイン結果に基づいて、D領域の核酸配列を抽出するステップ;
(5)該D領域の核酸配列をアミノ酸配列に翻訳し、該アミノ酸配列を利用してD領域を分類するステップ;
(6)(5)での分類に基づいて、V領域、D領域、J領域および必要に応じてC領域の各出現頻度、もしくはその組合せの出現頻度を算出することで、TCRもしくはBCRレパトアを導出するステップ;
を包含する、方法。
<2>前記遺伝子領域は、V領域、D領域、J領域および必要に応じてC領域の全部を含む、項目<1>に記載の方法。
<3>前記参照データベースは、各配列に一意のIDが割り振られたデータベースである、項目<1>~<2>のいずれか1項に記載の方法。
<4>前記入力配列セットは、非バイアス配列セットである、項目<1>~<3>のいずれか1項に記載の方法。
<5>前記配列セットはトリミングされたものである、項目<1>~<4>のいずれか1項に記載の方法。
<6> 前記トリミングは、リード両端から低クオリティ領域を削除し;リード両端からアダプタ配列と10bp以上マッチする領域を削除し;および残った長さが200bp以上(TCR)もしくは300bp以上(BCR)なら高クオリティとして解析に使用するステップによって達成される、項目<1>~<5>のいずれか1項に記載の方法。
<7>前記低クオリティーは、QV値の7bp移動平均が30未満のものである、項目<6>に記載の方法。
<8>前記近似する配列は、最も近しい配列である、項目<1>~<7>のいずれか1項に記載の方法。
<9>前記近似する配列は、1. 一致塩基数、2. カーネル長、3. スコア、4. アラインメント長の順位によって決定される、項目<1>~<8>のいずれか1項に記載の方法。
<10>前記相同性検索は、ランダムな変異が全体的に散在することを許容する条件で行われる、項目<1>~<9>のいずれか1項に記載の方法。
<11>前記相同性検索は、デフォルト条件に比べて(1)ウィンドウサイズの短縮、(2)ミスマッチペナルティの低減、(3)ギャップペナルティの低減および(4)指標の優先順位のトップが一致塩基数の少なくとも1つの条件を含む、項目<1>~<10>のいずれか1項に記載の方法。
<12>前記相同性検索は、BLASTまたはFASTAにおいて以下の条件
V ミスマッチペナルティ=-1、最短アラインメント長=30、最短カーネル長=15
D ワード長=7(BLASTの場合)またはK-tup=3(FASTAの場合)、ミスマッチペナルティ=-1、ギャップペナルティ=0、
最短アラインメント長=11、最短カーネル長=8
J ミスマッチペナルティ=-1、最短ヒット長=18、最短カーネル長=10
C 最短ヒット長=30、最短カーネル長=15
で実施される、項目<1>~<11>のいずれか1項に記載の方法。
<13>前記D領域の分類は、前記アミノ酸配列の出現頻度によってなされる、項目<1>~<12>のいずれか1項に記載の方法。
<14>前記ステップ(5)において、D領域の参照データベースが存在する場合は、前記CDR3の核酸配列との相同性検索結果とアミノ酸配列翻訳結果の組合せを分類結果とする、項目<1>~<13>のいずれか1項に記載の方法。
<15>前記ステップ(5)において、D領域の参照データベースが存在しない場合、前記アミノ酸配列の出現頻度のみによって分類がなされる、項目<1>~<14>のいずれか1項に記載の方法。
<16>前記出現頻度は、遺伝子名単位および/またはアリル単位でなされる、項目<1>~<15>のいずれか1項に記載の方法。
<17>前記ステップ(4)は該入力配列セットについてV領域およびJ領域をアサインし、参照V領域上のCDR3先頭および参照J上のCDR3末尾を目印に、CDR3配列を抽出するステップを包含する、項目<1>~<16>のいずれか1項に記載の方法。
<18>前記ステップ(5)は、該CDR3の核酸配列をアミノ酸配列に翻訳し、該アミノ酸配列を利用してD領域を分類するステップを包含する、項目<1>~<17>のいずれか1項に記載の方法。
<19>TCRもしくはBCRレパトアを解析するシステムであって、該システムは:
(1)V領域、D領域、J領域および必要に応じてC領域の少なくとも1つを含む遺伝子領域ごとの参照データベースを提供する手段:
(2)必要に応じてトリミングを行い、必要に応じて適切な長さのものを抽出した入力配列セットを提供する手段;
(3)該入力配列セットについて、該遺伝子領域ごとに、該参照データベースと相同性検索を行い、近似する参照アリルおよび/または該参照アリルの配列とのアラインメントを記録する手段;
(4)該入力配列セットについてV領域およびJ領域をアサインし、アサイン結果に基づいて、D領域の核酸配列を抽出する手段;
(5)該D領域の核酸配列をアミノ酸配列に翻訳し、該アミノ酸配列を利用してD領域を分類する手段;
(6)入力配列セットにおいて、V領域、D領域、J領域および必要に応じてC領域の各出現頻度、もしくはその組合せの出現頻度を算出することで、TCRもしくはBCRレパトアを導出する手段;
を包含する、システム。
<19A>項目<1>~<18>のいずれか1項または複数の特徴を有する、項目<19>に記載のシステム。
<20>TCRもしくはBCRレパトアを解析する方法の処理をコンピュータに実行させるコンピュータプログラムであって、該方法は以下のステップ:
(1)V領域、D領域、J領域および必要に応じてC領域の少なくとも1つを含む遺伝子領域ごとの参照データベースを提供するステップ:
(2)必要に応じてトリミングを行い、必要に応じて適切な長さのものを抽出した入力配列セットを提供するステップ;
(3)該入力配列セットについて、該遺伝子領域ごとに、該参照データベースと相同性検索を行い、近似する参照アリルおよび/または該参照アリルの配列とのアラインメントを記録するステップ;
(4)該入力配列セットについてV領域およびJ領域をアサインし、アサイン結果に基づいて、D領域の核酸配列を抽出するステップ;
(5)該D領域の核酸配列をアミノ酸配列に翻訳し、該アミノ酸配列を利用してD領域を分類するステップ;
(6)入力配列セットにおいて、V領域、D領域、J領域および必要に応じてC領域の各出現頻度、もしくはその組合せの出現頻度を算出することで、TCRもしくはBCRレパトアを導出するステップ;
を包含する、プログラム。
<20A>項目<1>~<18>のいずれか1項または複数の特徴を有する、項目<20>に記載のプログラム。
<21>TCRもしくはBCRレパトアを解析する方法の処理をコンピュータに実行させるコンピュータプログラムを格納する記録媒体であって、該方法は以下のステップ:
(1)V領域、D領域、J領域および必要に応じてC領域の少なくとも1つを含む遺伝子領域ごとの参照データベースを提供するステップ:
(2)必要に応じてトリミングを行い、必要に応じて適切な長さのものを抽出した入力配列セットを提供するステップ;
(3)該入力配列セットについて、該遺伝子領域ごとに、該参照データベースと相同性検索を行い、近似する参照アリルおよび/または該参照アリルの配列とのアラインメントを記録するステップ;
(4)該入力配列セットについてV領域およびJ領域をアサインし、アサイン結果に基づいて、D領域の核酸配列を抽出するステップ;
(5)該D領域の核酸配列をアミノ酸配列に翻訳し、該アミノ酸配列を利用してD領域を分類するステップ;
(6)入力配列セットにおいて、V領域、D領域、J領域および必要に応じてC領域の各出現頻度、もしくはその組合せの出現頻度を算出することで、TCRもしくはBCRレパトアを導出するステップ;
を包含する、記録媒体。
<21A>項目<1>~<18>のいずれか1項または複数の特徴を有する、項目<21>に記載の記録媒体。
<ウェット>
別の局面では、本発明は、(1)複数のゲノム上の遺伝子断片から遺伝子再構成により生成されたTCRまたはBCR遺伝子配列を、バイアスをかけることなく均一に増幅する技術(非バイアス遺伝子増幅技術)、(2)その非バイアス遺伝子増幅技術により増幅したTCRまたはBCR遺伝子を次世代シーケンス法により大規模に塩基配列決定し、V、D、J、C領域のアサインメントを行ってTCRレパトアやBCRレパトアを解析する手法である。
<A1>データベースを用いた遺伝子配列解析により、T細胞レセプター(TCR)またはB細胞レセプター(BCR)の可変領域のレパトア(repertoire)の定量解析を行うための試料を調製するための方法であって、
(1)標的となる細胞に由来するRNA試料を鋳型として相補的DNAを合成する工程;
(2)該相補的DNAを鋳型として二本鎖相補的DNAを合成する工程;
(3)該二本鎖相補的DNAに共通アダプタープライマー配列を付加してアダプター付加二本鎖相補的DNAを合成する工程;
(4)該アダプター付加二本鎖相補的DNAと、該共通アダプタープライマー配列からなる共通アダプタープライマーと、第1のTCRまたはBCRのC領域特異的プライマーとを用いて第1のPCR増幅反応を行う工程であって、
該第1のTCRまたはBCRのC領域特異的プライマーは、
該TCRまたはBCRの目的とするC領域に十分に特異的であり、かつ、他の遺伝子配列に相同性のない配列を含み、かつ、増幅された場合に下流にサブタイプ間に不一致塩基を含むよう設計される、工程
(5)(4)のPCR増幅産物と、該共通アダプタープライマーと、第2のTCRまたはBCRのC領域特異的プライマーとを用いて第2のPCR増幅反応を行う工程であって、該第2のTCRまたはBCRのC領域特異的プライマーは、該第1のTCRまたはBCRのC領域特異的プライマーの配列より下流の配列において該TCRまたはBCRのC領域に完全マッチの配列を有するが他の遺伝子配列に相同性のない配列を含み、かつ、増幅された場合に下流にサブタイプ間に不一致塩基を含むよう設計される、工程;および
(6)(5)のPCR増幅産物と、該共通アダプタープライマーの核酸配列に第1の追加アダプター核酸配列を含む付加共通アダプタープライマーと、第2の追加アダプター核酸配列および分子同定(MID Tag)配列を第3のTCRまたはBCRのC領域特異的配列に付加したアダプター付の第3のTCRまたはBCRのC領域特異的プライマーとを用いて第3のPCR増幅反応を行う工程であって、
該第3のTCRまたはBCRのC領域特異的プライマーは、該第2のTCRまたはBCRのC領域特異的プライマーの配列より下流の配列において該TCRまたはBCRのC領域に完全マッチの配列を有するが他の遺伝子配列に相同性のない配列を含み、かつ、増幅された場合に下流にサブタイプ間に不一致塩基を含むよう設計され、
該第1の追加アダプター核酸配列は、DNA捕捉ビーズへの結合およびemPCR反応に適切な配列であり、
該第2の追加アダプター核酸配列は、emPCR反応に適切な配列であり、
該分子同定(MID Tag)配列は、増幅産物が同定できるように、ユニークさを付与するための配列である、工程;
を包含する、方法。
<A2>BCRである場合、前記C領域特異的プライマーは、IgM、IgA、IgG、IgEおよびIgDからなる群より選択される目的とするアイソタイプC領域に完全マッチの配列を含み、かつ他のC領域に相同性を持たず、IgAまたはIgGについては、IgG1、IgG2、IgG3もしくはIgG4のいずれか、またはIgA1もしくはIgA2のいずれかであるサブタイプに完全マッチする配列であるか、あるいはTCRである場合、前記C領域特異的プライマーは、α鎖、β鎖、γ鎖およびδ鎖からなる群より選択される目的とする鎖のC領域に完全マッチし、かつ他のC領域に相同性を持たない配列である、項目<A1>に記載の方法。
<A3>前記C領域特異的プライマーは、前記データベース中の同じアイソタイプのC領域アレル配列についてすべてに完全マッチする配列部分を選択する、項目1または<A2>に記載の方法。
<A4>前記共通アダプタープライマーは、ホモダイマーおよび分子内ヘアピン構造が取りにくく、安定して2本鎖を形成できるように設計され、該データベース内のすべてのTCR遺伝子配列と相同性が高くなく、かつ、該C領域特異的プライマーと同じ程度の融解温度(Tm)になるように設計される、項目<A1>~<A3>のいずれか1項に記載の方法。
<A5>前記共通アダプタープライマーは、ホモダイマーおよび分子内ヘアピン構造をとらないよう設計され、BCRまたはTCRを含む他の遺伝子に対して相同性がないものを選択される、項目<A4>に記載の方法。
<A6>前記共通アダプタープライマーは、P20EA(配列番号2)および/またはP10EA(配列番号3)である、項目<A5>に記載の方法。
<A7> 前記第1、第2および第3のTCRまたはBCRのC領域特異的プライマーは、それぞれ独立して、BCRのレパトア解析のためのものであり、IgM,IgG,IgA,IgD,またはIgEの各アイソタイプC領域に完全マッチする配列であって、該IgGおよびIgAの場合は、サブタイプについても完全マッチし、該データベースに含まれる他の配列に相同性を持たない配列であり、かつ、該プライマーの下流においてサブタイプ間に不一致塩基が含まれるように選択され、
該共通アダプタープライマー配列は、増幅に適切な塩基長であり、ホモダイマーおよび分子内ヘアピン構造が取りにくく、安定して2本鎖を形成できるように設計され、該データベース内のすべてのTCR遺伝子配列と相同性が高くなく、かつ、該C領域特異的プライマーと同じ程度のTmになるように設計される、項目<A1>~<A6>のいずれか1項に記載の方法。
<A8> 前記第1、第2および第3のTCRまたはBCRのC領域特異的プライマーは、それぞれ独立して、TCRまたはBCRのレパトア解析のためのものであり、各プライマーは1種のα鎖(TRAC)、2種のβ鎖(TRBC01およびTRBC02)、2種のγ鎖(TRGC1およびTRGC2)、1種のδ鎖(TRDC1)に対して完全マッチする配列であり、該データベースに含まれる他の配列に相同性を持たない配列であり、かつ、該プライマーの下流においてサブタイプ間に不一致塩基が含まれるように選択され、
該共通アダプタープライマー配列は、増幅に適切な塩基長であり、ホモダイマーおよび分子内ヘアピン構造が取りにくく、安定して2本鎖を形成できるように設計され、該データベース内のすべてのTCR遺伝子配列と相同性が高くなく、かつ、該C領域特異的プライマーと同じ程度のTmになるように設計される項目<A1>~<A7>のいずれか1項に記載の方法。
<A9>前記第3のTCRまたはBCRのC領域特異的プライマーはC領域5’末端側から約150塩基までの領域に設定され、第1のTCRまたはBCRのC領域特異的プライマーおよび第2のTCRまたはBCRのC領域特異的プライマーはC領域5’末端側から約300塩基までの間に設定される、項目<A1>~<A8>のいずれか1項に記載の方法。
<A10>前記第1、第2および第3のTCRまたはBCRのC領域特異的プライマーは、それぞれ独立して、BCRの定量解析を行うためのものであり、
5種のアイソタイプ配列には別個に特異的プライマーを設定し、標的配列には完全マッチして、かつ他のアイソタイプには5塩基以上のミスマッチを確保するよう設計され、類似するIgGサブタイプ(IgG1,IgG2,IgG3およびIgG4)あるいはIgAサブタイプ(IgA1およびIgA2)に対しては、それぞれ1種のプライマーで対応できるようすべてのサブタイプに完全マッチするよう設計される、項目<A1>~<A9>のいずれか1項に記載の方法。
<A11>プライマー設計におけるパラメータは、塩基配列長18―22塩基、融解温度54-66℃、%GC(%グアニン・シトシン含量)は40-65%に設定される、項目<A1>~<A10>のいずれか1項に記載の方法。
<A12>プライマー設計におけるパラメータは、塩基配列長18―22塩基、融解温度54-66℃、%GC(%グアニン・シトシン含量)は40-65%に設定し、自己アニーリングスコア26、自己末端アニーリングスコア10、二次構造スコア28に設定される、項目<A1>~<A11>のいずれか1項に記載の方法。
<A13>以下の条件
1.複数のサブタイプ配列および/またはアレル配列を塩基配列解析ソフトに取り込み、アライメントすること;
2.プライマーデザイン用のソフトウェアを用いて、C領域内にパラメータ条件を満たすプライマーを複数検索すること;
3.1のアライメント配列の中で不一致塩基のない領域にあるプライマーを選択すること;
4.3により決定されたプライマーの下流に各サブタイプおよび/またはアレル毎に不一致配列が複数あることを確認し、ない場合は、さらに上流にプライマーを検索し、必要に応じてさらにこれを反復すること
により、前記第1、第2および第3のTCRまたはBCRのC領域特異的プライマーの配列が決定される、項目<A1>~<A12>のいずれか1項に記載の方法。
<A14>第1のTCRまたはBCRのC領域特異的プライマーは、スプライシングにより生じるC領域配列の第一コドンの第一塩基を基準として41-300塩基まで、第2のTCRまたはBCRのC領域特異的プライマーは該第一塩基を基準として21-300塩基まで、第3のTCRまたはBCRのC領域特異的プライマーは該第一塩基を基準として150塩基以内で、かつサブタイプおよび/またはアレルの不一致部位を含む位置で設定される、項目<A1>~<A13>のいずれか1項に記載の方法。
<A15>第1のTCRまたはBCRのC領域特異的プライマーは、以下の構造:CM1(配列番号5)、CA1(配列番号8)、CG1(配列番号11)、CD1(配列番号14)、CE1(配列番号17)、CA1(配列番号35)、またはCB1(配列番号37)を有する、項目<A1>~<A14>のいずれか1項に記載の方法。
<A16>第2のTCRまたはBCRのC領域特異的プライマーは、以下の構造:CM2(配列番号6)、CA2(配列番号9)、CG2(配列番号12)、CD2(配列番号15)、CE2(配列番号18)、CA2(配列番号35)、またはCB2(配列番号37)を有する、項目<A1>~<A15>のいずれか1項に記載の方法。
<A17>第3のTCRまたはBCRのC領域特異的プライマーは、以下の構造:CM3-GS(配列番号7)、CA3-GS(配列番号10)、CG3-GS(配列番号13)、CD3-GS(配列番号16)またはCE3-GS(配列番号19)を有する、項目<A1>~<A16>のいずれか1項に記載の方法。
<A18>前記TCRまたはBCRのC領域特異的プライマーは、いずれも、TCRまたはBCRのすべてのサブクラスに対応するセットで提供される、項目<A1>~<A17>のいずれか1項に記載の方法。
<A19>項目<A1>~<A18>のいずれか1項に記載の方法で製造された試料を用いて遺伝子解析を行う方法。
<A20> 前記遺伝子解析は、T細胞レセプター(TCR)またはB細胞レセプター(BCR)の可変領域のレパトア(repertoire)の定量解析である、項目<A19>に記載の方法。
<解析システム>
<B1>データベースを用いて被験体のT細胞レセプター(TCR)またはB細胞レセプター(BCR)の可変領域のレパトア(repertoire)を定量的に解析する方法であって、該方法は、
(1)該被験者から非バイアス的に増幅した、T細胞レセプター(TCR)またはB細胞レセプター(BCR)の核酸配列を含む核酸試料を提供する工程;
(2)該核酸試料に含まれる該核酸配列を決定する工程;および
(3)決定された該核酸配列にもとづいて、各遺伝子の出現頻度またはその組み合わせを算出し、該被験体のTCRもしくはBCRレパトアを導出する工程
を包含する、方法。
<B2>前記核酸試料は、複数種類のT細胞レセプター(TCR)またはB細胞レセプター(BCR)の核酸配列を含み、前記(2)は単一の配列決定により前記核酸配列が決定される、項目<B1>に記載の方法。
<B3>前記単一の配列決定は、前記核酸試料から配列決定用の試料への増幅において、プライマーとして使用する配列は少なくとも一方がC領域をコードする核酸配列またはその相補鎖と同一配列を有することを特徴とする、項目<B2>に記載の方法。
<B4>前記単一の配列決定は、共通アダプタープライマーを用いて行われることを特徴とする、項目<B2>または<B3>に記載の方法。
<B5>前記非バイアス的な増幅が、V領域特異的な増幅ではない、項目<B1>~<B4>のいずれか1項に記載の方法。
<B6>前記レパトアはBCRの可変領域のレパトアであり、前記核酸配列はBCRの核酸配列である、項目<B1>~<B5>のいずれか1項に記載の方法。
<B7>項目<B1>~<B6>のいずれか1項に基づいて導出されたTCRもしくはBCRレパトアに基づいて前記被験者の疾患、障害または状態を分析する方法。
<B8>前記被験者の疾患、障害または状態は、血液腫瘍および大腸がんからなる群より選択される、項目<B7>に記載の方法。
<B9>項目<B7>または<B8>に記載の方法で決定された被験者の疾患、障害または状態と、前記TCRもしくはBCRレパトアとを定量的に関連付ける工程、および該定量的な関連から、適切な処置または予防のための手段を選択する工程を含む、該被験者の疾患、障害または状態を処置または予防するための方法。
<B10>前記被験者の疾患、障害または状態は、血液腫瘍および大腸がんからなる群より選択される、項目<B9>に記載の方法。
<B11>データベースを用いて被験体のT細胞レセプター(TCR)またはB細胞レセプター(BCR)の可変領域のレパトア(repertoire)を定量的に解析するためのシステムであって、該システムは、
(1)該被験者から非バイアス的に増幅した、T細胞レセプター(TCR)またはB細胞レセプター(BCR)の核酸配列を含む核酸試料を提供するためのキット;
(2)該核酸試料に含まれる該核酸配列を決定するための装置;および
(3)決定された該核酸配列にもとづいて、各遺伝子の出現頻度またはその組み合わせを算出し、該被験体のTCRもしくはBCRレパトアを導出するための装置
を備えるシステム。
<B12>前記核酸試料は、前記核酸配列を複数種類のT細胞レセプター(TCR)またはB細胞レセプター(BCR)の核酸配列を含み、前記(2)は単一の配列決定により前記核酸配列が決定される、項目<B11>に記載のシステム。
<B13>前記単一の配列決定は、前記核酸試料から配列決定用の試料への増幅において、プライマーとして使用する配列は少なくとも一方がC領域と同一配列を有することを特徴とする、項目<B12>に記載のシステム。
<B14>前記単一の配列決定は、共通アダプタープライマーを用いて行われることを特徴とする、項目<B12>または<B13>に記載のシステム。
<B15>前記非バイアス的な増幅が、V領域特異的な増幅ではない、項目<B11>~<B14>のいずれか1項に記載のシステム。
<B16>前記レパトアはBCRの可変領域のレパトアであり、前記核酸配列はBCRの核酸配列である、項目<B11>~<B15>のいずれか1項に記載のシステム。
<B17>項目<B11>~<B16>のいずれか1項に記載のシステムと、該システムに基づいて導出されたTCRもしくはBCRレパトアに基づいて前記被験者の疾患、障害または状態を分析する手段とを備える、被験者の疾患、障害または状態を分析するシステム。
<B18>前記被験者の疾患、障害または状態は、血液腫瘍および大腸がんからなる群より選択される、項目<B17>に記載のシステム。
<B19>項目<B17>または<B18>に記載のシステムで決定された被験者の疾患、障害または状態と、前記TCRもしくはBCRレパトアとを定量的に関連付ける手段、および該定量的な関連から、適切な処置または予防のための手段を選択する手段とを備える、該被験者の疾患、障害または状態を処置または予防するためのシステム。
<B20>前記被験者の疾患、障害または状態は、血液腫瘍および大腸がんからなる群より選択される、項目<B19>に記載のシステム。
<B21>TRAV10/TRAJ15/CVVRATGTALIFG(配列番号1450)もしくはこれをコードする核酸を含むTCRα、および/またはTRBV29-1/TRBJ2-7/CSVERGGSLGEQYFG(配列番号1500)もしくはこれをコードする核酸を含むTCRβを発現する、T細胞大顆粒リンパ球性白血病(T-LGL)に関するモノクローナルなT細胞。
<B22>TCRαにおいてTRAV10/TRAJ15/CVVRATGTALIFG(配列番号1450)もしくはこれをコードする核酸、および/またはTCRβにおいてTRBV29-1/TRBJ2-7/CSVERGGSLGEQYFG(配列番号1500)もしくはこれをコードする核酸の、T細胞大顆粒リンパ球性白血病(T-LGL)の診断指標としての使用。
<B23>TCRαにおいてTRAV10/TRAJ15/CVVRATGTALIFG(配列番号1450)もしくはこれをコードする核酸、および/またはTCRβにおいてTRBV29-1/TRBJ2-7/CSVERGGSLGEQYFG(配列番号1500)もしくはこれをコードする核酸を検出する工程を包含する、T細胞大顆粒リンパ球性白血病(T-LGL)の検出する方法。
<B24>TCRαにおけるTRAV10/TRAJ15/CVVRATGTALIFG(配列番号1450)またはこれをコードする核酸の検出剤、および/またはTCRβにおけるTRBV29-1/TRBJ2-7/CSVERGGSLGEQYFG(配列番号1500)またはこれをコードする核酸の検出剤。
<B25>TCRαにおけるTRAV10/TRAJ15/CVVRATGTALIFG(配列番号1450)またはこれをコードする核酸の検出剤、および/またはTCRβにおけるTRBV29-1/TRBJ2-7/CSVERGGSLGEQYFG(配列番号1500)またはこれをコードする核酸の検出剤を含む、T細胞大顆粒リンパ球性白血病(T-LGL)の診断薬。
<B26>配列番号1627~1647に示す配列のいずれかを含む、新規インバリアントTCRであるペプチド。
<B27>配列番号1648~1651、1653~1654、1666~1667、1844~1848、および1851からなる群より選択されるの配列を含む、粘膜関連インバリアントT(MAIT)細胞の指標であるペプチド。
<B28>項目<B27>に記載のペプチドをコードする核酸。
<B29>項目<B27>または<B28>に記載のペプチドまたは該ペプチドをコードする核酸の、大腸がんの診断指標としての使用。
<B30>配列番号1668に示す配列を含む、ナチュラルキラーT細胞(NKT)の指標であるペプチド。
<B31>項目<B30>に記載のペプチドをコードする核酸。
<B32>項目<B30>または<B31>に記載のペプチドまたは該ペプチドをコードする核酸の、大腸がんの診断指標としての使用。
<B33>配列番号1652、1655~1665、1669~1843、1849~1850、および1852~1860からなる群より選択される配列を含む、大腸がん特異的であるペプチド。
<B34>項目<B33>に記載のペプチドをコードする核酸。
<B35>項目<B33>または<B34>に記載のペプチドまたは該ペプチドをコードする核酸の、大腸がんの診断指標としての使用。
<B36>配列番号1861~1865、および1867~1909からなる群より選択される配列を含む、大腸がん特異的であるペプチド
<B37>項目<B36>に記載のペプチドをコードする核酸。
<B38>項目<B36>または<B37>に記載のペプチドまたは該ペプチドをコードする核酸の、大腸がんの診断指標としての使用。
<B39>項目<B33>、<B34>、<B36>または<B37>に記載のペプチドまたは該ペプチドをコードする核酸配列を有するT細胞を高頻度に誘導した細胞集団、T細胞株、または組み換え発現させたT細胞。
<B40>項目<B39>に記載の細胞集団、T細胞株またはT細胞を含む大腸がんの治療剤。
<B41>項目<B39>に記載の細胞集団、T細胞株またはT細胞を用いる大腸がんの治療または予防のための方法。
<B42>項目<B1>~<B10>のいずれか1項に記載の方法、または項目<B11>~<B20>のいずれか1項に記載のシステムを用いて、V遺伝子の使用頻度を検出する方法。
<B43>項目<B1>~<B10>のいずれか1項に記載の方法、または項目<B11>~<B20>に記載のシステムを用いて、J遺伝子の使用頻度を検出する方法。
<B44>項目<B1>~<B10>のいずれか1項に記載の方法、または項目<B11>~<B20>に記載のシステムを用いて、サブタイプの頻度解析(BCR)の使用頻度を検出する方法。
<B45>項目<B1>~<B10>のいずれか1項に記載の方法、または項目<B11>~<B20>に記載のシステムを用いて、CDR3配列長のパターンを分析する方法。
<B46>項目<B1>~<B10>のいずれか1項に記載の方法、または項目<B11>~<B20>に記載のシステムを用いて、TCRあるいはBCRのクローナリティを分析する方法。
<B47>項目<B1>~<B10>のいずれか1項に記載の方法、または項目<B11>~<B20>に記載のシステムを用いて、重複リードを抽出する方法。
<B48>項目<B1>~<B10>のいずれか1項に記載の方法、または項目<B11>~<B20>に記載のシステムを用いて、疾患特異的TCRあるいはBCRクローンを検索する方法。
<B49>項目<B1>~<B10>のいずれか1項に記載の方法、または項目<B11>~<B20>に記載のシステムを用いて、多様性指数を使って対象を分析する方法。
<B50>項目<B1>~<B10>のいずれか1項に記載の方法、または項目<B11>~<B20>に記載のシステムを用いて、多様性指数を使って対象を分析を支援する方法。
<B51>骨髄移植後の免疫系の回復度を測る指標、または、造血器腫瘍に伴う免疫系細胞の異常を検出する指標として前記多様性指数を使用する、項目<B49>または<B50>に記載の方法。
<B52>前記多様性指数は、Shannon-Wienerの多様性指数(H’)、Simpsonの多様性指数(λ、1-λあるいは1/λ)、Pielou均等度指数(J’)およびChao1指数からなる群より選択されるものである、項目<B49>または<B50>に記載の方法。
<B53>項目<B1>~<B10>のいずれか1項に記載の方法、または項目<B11>~<B20>に記載のシステムを用いて、類似性指数を使って対象を分析する方法。
<B54>項目<B1>~<B10>のいずれか1項に記載の方法、または項目<B11>~<B20>に記載のシステムを用いて、類似性指数を使って対象を分析を支援する方法。
<B55>HLA型一致あるいは不一致間のレパトアの類似度の評価、骨髄移植後のレシピエントとドナー間のレパトアの類似度の評価として前記類似性指数を使用する、項目<B53>または<B54>に記載の方法。
<B56>前記類似性指数は、Morisita―Horn指数、木元のCπ指数およびPiankaのα指数からなる群より選択されるものである、項目<B53>または<B54>に記載の方法。
<B57>前記(1)は、以下の工程
(1-1)標的となる細胞に由来するRNA試料を鋳型として相補的DNAを合成する工程;
(1-2)該相補的DNAを鋳型として二本鎖相補的DNAを合成する工程;
(1-3)該二本鎖相補的DNAに共通アダプタープライマー配列を付加してアダプター付加二本鎖相補的DNAを合成する工程;
(1-4)該アダプター付加二本鎖相補的DNAと、該共通アダプタープライマー配列からなる共通アダプタープライマーと、第1のTCRまたはBCRのC領域特異的プライマーとを用いて第1のPCR増幅反応を行う工程であって、
該第1のTCRまたはBCRのC領域特異的プライマーは、該TCRまたはBCRの目的とするC領域に十分に特異的であり、かつ、他の遺伝子配列に相同性のない配列を含み、かつ、増幅された場合に下流にサブタイプ間に不一致塩基を含むよう設計される、工程
(1-5)(1-4)のPCR増幅産物と、該共通アダプタープライマーと、第2のTCRまたはBCRのC領域特異的プライマーとを用いて第2のPCR増幅反応を行う工程であって、該第2のTCRまたはBCRのC領域特異的プライマーは、該第1のTCRまたはBCRのC領域特異的プライマーの配列より下流の配列において該TCRまたはBCRのC領域に完全マッチの配列を有するが他の遺伝子配列に相同性のない配列を含み、かつ、増幅された場合に下流にサブタイプ間に不一致塩基を含むよう設計される、工程;および
(1-6)(1-5)のPCR増幅産物と、該共通アダプタープライマーの核酸配列に第1の追加アダプター核酸配列を含む付加共通アダプタープライマーと、第2の追加アダプター核酸配列および分子同定(MID Tag)配列を第3のTCRまたはBCRのC領域特異的配列に付加したアダプター付の第3のTCRまたはBCRのC領域特異的プライマーとを用いて第3のPCR増幅反応を行う工程であって、
該第3のTCRまたはBCRのC領域特異的プライマーは、該第2のTCRまたはBCRのC領域特異的プライマーの配列より下流の配列において該TCRまたはBCRのC領域に完全マッチの配列を有するが他の遺伝子配列に相同性のない配列を含み、かつ、増幅された場合に下流にサブタイプ間に不一致塩基を含むよう設計され、
該第1の追加アダプター核酸配列は、DNA捕捉ビーズへの結合およびemPCR反応に適切な配列であり、
該第2の追加アダプター核酸配列は、emPCR反応に適切な配列であり、
該分子同定(MID Tag)配列は、増幅産物が同定できるように、ユニークさを付与するための配列である、工程;
を包含する、
項目<B1>に記載の方法。
<B58>前記(1)キットは、以下:
(1-1)標的となる細胞に由来するRNA試料を鋳型として相補的DNAを合成する手段;
(1-2)該相補的DNAを鋳型として二本鎖相補的DNAを合成する手段;
(1-3)該二本鎖相補的DNAに共通アダプタープライマー配列を付加してアダプター付加二本鎖相補的DNAを合成する手段;
(1-4)該アダプター付加二本鎖相補的DNAと、該共通アダプタープライマー配列からなる共通アダプタープライマーと、第1のTCRまたはBCRのC領域特異的プライマーとを用いて第1のPCR増幅反応を行う手段であって、
該第1のTCRまたはBCRのC領域特異的プライマーは、該TCRまたはBCRの目的とするC領域に十分に特異的であり、かつ、他の遺伝子配列に相同性のない配列を含み、かつ、増幅された場合に下流にサブタイプ間に不一致塩基を含むよう設計される、手段
(1-5)(1-4)のPCR増幅産物と、該共通アダプタープライマーと、第2のTCRまたはBCRのC領域特異的プライマーとを用いて第2のPCR増幅反応を行う手段であって、該第2のTCRまたはBCRのC領域特異的プライマーは、該第1のTCRまたはBCRのC領域特異的プライマーの配列より下流の配列において該TCRまたはBCRのC領域に完全マッチの配列を有するが他の遺伝子配列に相同性のない配列を含み、かつ、増幅された場合に下流にサブタイプ間に不一致塩基を含むよう設計される、手段;および
(1-6)(1-5)のPCR増幅産物と、該共通アダプタープライマーの核酸配列に第1の追加アダプター核酸配列を含む付加共通アダプタープライマーと、第2の追加アダプター核酸配列および分子同定(MID Tag)配列を第3のTCRまたはBCRのC領域特異的配列に付加したアダプター付の第3のTCRまたはBCRのC領域特異的プライマーとを用いて第3のPCR増幅反応を行う手段であって、
該第3のTCRまたはBCRのC領域特異的プライマーは、該第2のTCRまたはBCRのC領域特異的プライマーの配列より下流の配列において該TCRまたはBCRのC領域に完全マッチの配列を有するが他の遺伝子配列に相同性のない配列を含み、かつ、増幅された場合に下流にサブタイプ間に不一致塩基を含むよう設計され、
該第1の追加アダプター核酸配列は、DNA捕捉ビーズへの結合およびemPCR反応に適切な配列であり、
該第2の追加アダプター核酸配列は、emPCR反応に適切な配列であり、
該分子同定(MID Tag)配列は、増幅産物が同定できるように、ユニークさを付与するための配列である、手段;
を包含する、
項目<B11>に記載のシステム。
<B58-2>BCRである場合、前記C領域特異的プライマーは、IgM、IgA、IgG、IgEおよびIgDからなる群より選択される目的とするアイソタイプC領域に完全マッチの配列を含み、かつ他のC領域に相同性を持たず、IgAまたはIgGについては、IgG1、IgG2、IgG3もしくはIgG4のいずれか、またはIgA1もしくはIgA2のいずれかであるサブタイプに完全マッチする配列であるか、あるいはTCRである場合、前記C領域特異的プライマーは、α鎖、β鎖、γ鎖およびδ鎖からなる群より選択される目的とする鎖のC領域に完全マッチし、かつ他のC領域に相同性を持たない配列である、項目<B57>に記載の方法または<B58>に記載のシステム。
<B58-3>前記C領域特異的プライマーは、前記データベース中の同じアイソタイプのC領域アレル配列についてすべてに完全マッチする配列部分を選択する、項目<B57>もしくは<B58-2>に記載の方法または<B58>もしくは<B58-2>に記載のシステム。
<B58-4>前記共通アダプタープライマーは、ホモダイマーおよび分子内ヘアピン構造が取りにくく、安定して2本鎖を形成できるように設計され、該データベース内のすべてのTCR遺伝子配列と相同性が高くなく、かつ、該C領域特異的プライマーと同じ程度の融解温度(Tm)になるように設計される、項目<B57>および<B58-2>~<B58-3>のいずれかに記載の方法または<B58>~<B58-3>のいずれかに記載のシステム。
<B58-5>前記共通アダプタープライマーは、ホモダイマーおよび分子内ヘアピン構造をとらないよう設計され、BCRまたはTCRを含む他の遺伝子に対して相同性がないものを選択される、項目<B57>および<B58-2>~<B58-4>のいずれかに記載の方法または<B58>~<B58-4>のいずれかに記載のシステム。
<B58-6>前記共通アダプタープライマーは、P20EA(配列番号2)および/またはP10EA(配列番号3)である、項目<B57>および<B58-2>~<B58-5>のいずれかに記載の方法または<B58>~<B58-5>のいずれかに記載のシステム。
<B58-7> 前記第1、第2および第3のTCRまたはBCRのC領域特異的プライマーは、それぞれ独立して、BCRのレパトア解析のためのものであり、IgM,IgG,IgA,IgD,またはIgEの各アイソタイプC領域に完全マッチする配列であって、該IgGおよびIgAの場合は、サブタイプについても完全マッチし、該データベースに含まれる他の配列に相同性を持たない配列であり、かつ、該プライマーの下流においてサブタイプ間に不一致塩基が含まれるように選択され、
該共通アダプタープライマー配列は、増幅に適切な塩基長であり、ホモダイマーおよび分子内ヘアピン構造が取りにくく、安定して2本鎖を形成できるように設計され、該データベース内のすべてのTCR遺伝子配列と相同性が高くなく、かつ、該C領域特異的プライマーと同じ程度のTmになるように設計される、項目<B57>および<B58-2>~<B58-6>のいずれかに記載の方法または<B58>~<B58-6>のいずれかに記載のシステム。
<B58-8> 前記第1、第2および第3のTCRまたはBCRのC領域特異的プライマーは、それぞれ独立して、TCRまたはBCRのレパトア解析のためのものであり、各プライマーは1種のα鎖(TRAC)、2種のβ鎖(TRBC01およびTRBC02)、2種のγ鎖(TRGC1およびTRGC2)、1種のδ鎖(TRDC1)に対して完全マッチする配列であり、該データベースに含まれる他の配列に相同性を持たない配列であり、かつ、該プライマーの下流においてサブタイプ間に不一致塩基が含まれるように選択され、
該共通アダプタープライマー配列は、増幅に適切な塩基長であり、ホモダイマーおよび分子内ヘアピン構造が取りにくく、安定して2本鎖を形成できるように設計され、該データベース内のすべてのTCR遺伝子配列と相同性が高くなく、かつ、該C領域特異的プライマーと同じ程度のTmになるように設計される、項目<B57>および<B58-2>~<B58-7>のいずれかに記載の方法または<B58>~<B58-7>のいずれかに記載のシステム。
<B58-9>前記第3のTCRまたはBCRのC領域特異的プライマーはC領域5’末端側から約150塩基までの領域に設定され、第1のTCRまたはBCRのC領域特異的プライマーおよび第2のTCRまたはBCRのC領域特異的プライマーはC領域5’末端側から約300塩基までの間に設定される、項目<B57>および<B58-2>~<B58-8>のいずれかに記載の方法または<B58>~<B58-8>に記載のシステム。
<B58-10>前記第1、第2および第3のTCRまたはBCRのC領域特異的プライマーは、それぞれ独立して、BCRの定量解析を行うためのものであり、
5種のアイソタイプ配列には別個に特異的プライマーを設定し、標的配列には完全マッチして、かつ他のアイソタイプには5塩基以上のミスマッチを確保するよう設計され、類似するIgGサブタイプ(IgG1,IgG2,IgG3およびIgG4)あるいはIgAサブタイプ(IgA1およびIgA2)に対しては、それぞれ1種のプライマーで対応できるようすべてのサブタイプに完全マッチするよう設計される、項目<B57>および<B58-2>~<B58-9>のいずれかに記載の方法または<B58>~<B58-9>のいずれかに記載のシステム。
<B58-11>プライマー設計におけるパラメータは、塩基配列長18―22塩基、融解温度54-66℃、%GC(%グアニン・シトシン含量)は40-65%に設定される、項目<B57>および<B58-2>~<B58-10>のいずれかに記載の方法または<B58>~<B58-10>のいずれかに記載のシステム。
<B58-12>プライマー設計におけるパラメータは、塩基配列長18―22塩基、融解温度54-66℃、%GC(%グアニン・シトシン含量)は40-65%に設定し、自己アニーリングスコア26、自己末端アニーリングスコア10、二次構造スコア28に設定される、項目<B57>および<B58-2>~<B58-11>のいずれかに記載の方法または<B58>~<B58-11>のいずれかに記載のシステム。
<B58-13>以下の条件
1.複数のサブタイプ配列および/またはアレル配列を塩基配列解析ソフトに取り込み、アライメントすること;
2.プライマーデザイン用のソフトウェアを用いて、C領域内にパラメータ条件を満たすプライマーを複数検索すること;
3.1のアライメント配列の中で不一致塩基のない領域にあるプライマーを選択すること;
4.3により決定されたプライマーの下流に各サブタイプおよび/またはアレル毎に不一致配列が複数あることを確認し、ない場合は、さらに上流にプライマーを検索し、必要に応じてさらにこれを反復すること
により、前記第1、第2および第3のTCRまたはBCRのC領域特異的プライマーの配列が決定される、項目<B57>および<B58-2>~<B58-12>のいずれかに記載の方法または<B58>~<B58-12>のいずれかに記載のシステム。
<B58-14>第1のTCRまたはBCRのC領域特異的プライマーは、スプライシングにより生じるC領域配列の第一コドンの第一塩基を基準として41-300塩基まで、第2のTCRまたはBCRのC領域特異的プライマーは該第一塩基を基準として21-300塩基まで、第3のTCRまたはBCRのC領域特異的プライマーは該第一塩基を基準として150塩基以内で、かつサブタイプおよび/またはアレルの不一致部位を含む位置で設定される、項目<B57>および<B58-2>~<B58-13>のいずれかに記載の方法または<B58>~<B58-13>のいずれかに記載のシステム。
<B58-15>第1のTCRまたはBCRのC領域特異的プライマーは、以下の構造:CM1(配列番号5)、CA1(配列番号8)、CG1(配列番号11)、CD1(配列番号14)、CE1(配列番号17)、CA1(配列番号35)、またはCB1(配列番号37)を有する、項目<B57>および<B58-2>~<B58-14>のいずれかに記載の方法または<B58>~<B58-14>のいずれかに記載のシステム。
<B58-16>第2のTCRまたはBCRのC領域特異的プライマーは、以下の構造:CM2(配列番号6)、CA2(配列番号9)、CG2(配列番号12)、CD2(配列番号15)、CE2(配列番号18)、CA2(配列番号35)、またはCB2(配列番号37)を有する、項目<B57>および<B58-2>~<B58-15>のいずれかに記載の方法または<B58>~<B58-15>のいずれかに記載のシステム。
<B58-17>第3のTCRまたはBCRのC領域特異的プライマーは、以下の構造:CM3-GS(配列番号7)、CA3-GS(配列番号10)、CG3-GS(配列番号13)、CD3-GS(配列番号16)またはCE3-GS(配列番号19)を有する、項目<B57>および<B58-2>~<B58-16>のいずれかに記載の方法または<B58>~<B58-16>のいずれかに記載のシステム。
<B58-18>前記TCRまたはBCRのC領域特異的プライマーは、いずれも、TCRまたはBCRのすべてのサブクラスに対応するセットで提供される、項目<B57>および<B58-2>~<B58-17>のいずれかに記載の方法または<B58>~<B58-17>に記載のシステム。
<B58-19>項目<B57>および<B58-2>~<B58-18>のいずれかに記載の方法または<B58>~<B58-18>のいずれかに記載のシステムで製造された試料を用いて遺伝子解析を行う方法またはシステム。
<B58-20> 前記遺伝子解析は、T細胞レセプター(TCR)またはB細胞レセプター(BCR)の可変領域のレパトア(repertoire)の定量解析である、項目<B58-19>のいずれかに記載の方法またはシステム。
<B59>(3)前記TCRもしくはBCRレパトアの導出は以下の工程
(3-1)V領域、D領域、J領域および必要に応じてC領域の少なくとも1つを含む遺伝子領域ごとの参照データベースを提供する工程:
(3-2)必要に応じてトリミングを行い、必要に応じて適切な長さのものを抽出した入力配列セットを提供する工程;
(3-3)該入力配列セットについて、該遺伝子領域ごとに、該参照データベースと相同性検索を行い、近似する参照アリルおよび/または該参照アリルの配列とのアラインメントを記録する工程;
(3-4)該入力配列セットについてV領域およびJ領域をアサインし、アサイン結果に基づいて、D領域の核酸配列を抽出する工程;
(3-5)該D領域の核酸配列をアミノ酸配列に翻訳し、該アミノ酸配列を利用してD領域を分類する工程;
(3-6)(3-5)での分類に基づいて、V領域、D領域、J領域および必要に応じてC領域の各出現頻度、もしくはその組合せの出現頻度を算出することで、TCRもしくはBCRレパトアを導出する工程;
を包含する方法によって達成される、項目<B57>および<B58-2>~<B58-20>のいずれかに記載の方法または<B58>~<B58-20>のいずれかに記載のシステム。
<B60>(3)前記TCRもしくはBCRレパトアの導出のための装置は以下
(3-1)V領域、D領域、J領域および必要に応じてC領域の少なくとも1つを含む遺伝子領域ごとの参照データベースを提供する手段:
(3-2)必要に応じてトリミングを行い、必要に応じて適切な長さのものを抽出した入力配列セットを提供する手段;
(3-3)該入力配列セットについて、該遺伝子領域ごとに、該参照データベースと相同性検索を行い、近似する参照アリルおよび/または該参照アリルの配列とのアラインメントを記録する手段;
(3-4)該入力配列セットについてV領域およびJ領域をアサインし、アサイン結果に基づいて、D領域の核酸配列を抽出する手段;
(3-5)該D領域の核酸配列をアミノ酸配列に翻訳し、該アミノ酸配列を利用してD領域を分類する手段;
(3-6)(3-5)での分類に基づいて、V領域、D領域、J領域および必要に応じてC領域の各出現頻度、もしくはその組合せの出現頻度を算出することで、TCRもしくはBCRレパトアを導出する手段;
を備える、項目<B11>~<B20>、<B58>~<B58-20>または59に記載のシステム。
<B60-2>前記遺伝子領域は、V領域、D領域、J領域および必要に応じてC領域の全部を含む、項目<B57>、<B58-2>~<B58-20>および<B59>のいずれかに記載の方法または<B58>~<B58-20>、<B59>および<B60>のいずれかに記載のシステム。
<B60-3>前記参照データベースは、各配列に一意のIDが割り振られたデータベースである、項目<B57>、<B58-2>~<B58-20>、<B59>および<B60-2>のいずれかに記載の方法または<B58>~<B58-20>、<B59>、<B60>~<B60-2>のいずれかに記載のシステム。
<B60-4>前記入力配列セットは、非バイアス配列セットである、項目<B57>、<B58-2>~<B58-20>、<B59>および<B60-2>~<B60-3>のいずれかに記載の方法または<B58>~<B58-20>、<B59>、<B60>~<B60-3>のいずれかに記載のシステム。
<B60-5>前記配列セットはトリミングされたものである、項目<B57>、<B58-2>~<B58-20>、<B59>および<B60-2>~<B60-4>のいずれかに記載の方法または<B58>~<B58-20>、<B59>、<B60>~<B60-4>のいずれかに記載のシステム。
<B60-6> 前記トリミングは、リード両端から低クオリティ領域を削除し;リード両端からアダプタ配列と10bp以上マッチする領域を削除し;および残った長さが200bp以上(TCR)もしくは300bp以上(BCR)なら高クオリティとして解析に使用するステップによって達成される、項目<B57>、<B58-2>~<B58-20>、<B59>および<B60-2>~<B60-5>のいずれかに記載の方法または<B58>~<B58-20>、<B59>、<B60>~<B60-5>のいずれかに記載のシステム。
<B60-7>前記低クオリティーは、QV値の7bp移動平均が30未満のものである、項目<B57>、<B58-2>~<B58-20>、<B59>および<B60-2>~<B60-6>のいずれかに記載の方法または<B58>~<B58-20>、<B59>、<B60>~<B60-6>のいずれかに記載のシステム。
<B60-8>前記近似する配列は、最も近しい配列である、項目<B57>、<B58-2>~<B58-20>、<B59>および<B60-2>~<B60-7>のいずれかに記載の方法または<B58>~<B58-20>、<B59>、<B60>~<B60-7>のいずれかに記載のシステム。
<B60-9>前記近似する配列は、1. 一致塩基数、2. カーネル長、3. スコア、4. アラインメント長の順位によって決定される、項目<B57>、<B58-2>~<B58-20>、<B59>および<B60-2>~<B60-8>のいずれかに記載の方法または<B58>~<B58-20>、<B59>、<B60>~<B60-8>のいずれかに記載のシステム。
<B60-10>前記相同性検索は、ランダムな変異が全体的に散在することを許容する条件で行われる、項目<B57>、<B58-2>~<B58-20>、<B59>および<B60-2>~<B60-9>のいずれかに記載の方法または<B58>~<B58-20>、<B59>、<B60>~<B60-9>のいずれかに記載のシステム。
<B60-11>前記相同性検索は、デフォルト条件に比べて(1)ウィンドウサイズの短縮、(2)ミスマッチペナルティの低減、(3)ギャップペナルティの低減および(4)指標の優先順位のトップが一致塩基数の少なくとも1つの条件を含む、項目<B57>、<B58-2>~<B58-20>、<B59>および<B60-2>~<B60-10>のいずれかに記載の方法または<B58>~<B58-20>、<B59>、<B60>~<B60-10>のいずれかに記載のシステム。
<B60-12>前記相同性検索は、BLASTまたはFASTAにおいて以下の条件
V ミスマッチペナルティ=-1、最短アラインメント長=30、最短カーネル長=15
D ワード長=7(BLASTの場合)またはK-tup=3(FASTAの場合)、ミスマッチペナルティ=-1、ギャップペナルティ=0、
最短アラインメント長=11、最短カーネル長=8
J ミスマッチペナルティ=-1、最短ヒット長=18、最短カーネル長=10
C 最短ヒット長=30、最短カーネル長=15
で実施される、項目<B57>、<B58-2>~<B58-20>、<B59>および<B60-2>~<B60-11>のいずれかに記載の方法または<B58>~<B58-20>、<B59>、<B60>~<B60-11>のいずれかに記載のシステム。
<B60-13>前記D領域の分類は、前記アミノ酸配列の出現頻度によってなされる、項目<B57>、<B58-2>~<B58-20>、<B59>および<B60-2>~<B60-12>のいずれかに記載の方法または<B58>~<B58-20>、<B59>、<B60>~<B60-12>のいずれかに記載のシステム。
<B60-14>前記ステップ(5)において、D領域の参照データベースが存在する場合は、前記CDR3の核酸配列との相同性検索結果とアミノ酸配列翻訳結果の組合せを分類結果とする、項目<B57>、<B58-2>~<B58-20>、<B59>および<B60-2>~<B60-13>のいずれかに記載の方法または<B58>~<B58-20>、<B59>、<B60>~<B60-13>のいずれかに記載のシステム。
<B60-15>前記ステップ(5)において、D領域の参照データベースが存在しない場合、前記アミノ酸配列の出現頻度のみによって分類がなされる、項目<B57>、<B58-2>~<B58-20>、<B59>および<B60-2>~<B60-14>のいずれかに記載の方法または<B58>~<B58-20>、<B59>、<B60>~<B60-14>のいずれかに記載のシステム。
<B60-16>前記出現頻度は、遺伝子名単位および/またはアリル単位でなされる、項目<B57>、<B58-2>~<B58-20>、<B59>および<B60-2>~<B60-15>のいずれかに記載の方法または<B58>~<B58-20>、<B59>、<B60>~<B60-15>のいずれかに記載のシステム。
<B60-17>前記ステップ(4)は該入力配列セットについてV領域およびJ領域をアサインし、参照V領域上のCDR3先頭および参照J上のCDR3末尾を目印に、CDR3配列を抽出するステップを包含する、項目<B57>、<B58-2>~<B58-20>、<B59>および<B60-2>~<B60-16>のいずれかに記載の方法または<B58>~<B58-20>、<B59>、<B60>~<B60-16>のいずれかに記載のシステム。
<B60-18>前記ステップ(5)は、該CDR3の核酸配列をアミノ酸配列に翻訳し、該アミノ酸配列を利用してD領域を分類するステップを包含する、項目<B57>、<B58-2>~<B58-20>、<B59>および<B60-2>~<B60-17>のいずれかに記載の方法または<B58>~<B58-20>、<B59>、<B60>~<B60-17>のいずれかに記載のシステム。
<B60-19>(3)前記TCRもしくはBCRレパトアの導出のための装置は以下
(3-1)V領域、D領域、J領域および必要に応じてC領域の少なくとも1つを含む遺伝子領域ごとの参照データベースを提供する手段:
(3-2)必要に応じてトリミングを行い、必要に応じて適切な長さのものを抽出した入力配列セットを提供する手段;
(3-3)該入力配列セットについて、該遺伝子領域ごとに、該参照データベースと相同性検索を行い、近似する参照アリルおよび/または該参照アリルの配列とのアラインメントを記録する手段;
(3-4)該入力配列セットについてV領域およびJ領域をアサインし、アサイン結果に基づいて、D領域の核酸配列を抽出する手段;
(3-5)該D領域の核酸配列をアミノ酸配列に翻訳し、該アミノ酸配列を利用してD領域を分類する手段;
(3-6)入力配列セットにおいて、V領域、D領域、J領域および必要に応じてC領域の各出現頻度、もしくはその組合せの出現頻度を算出することで、TCRもしくはBCRレパトアを導出する手段;
を包含する、項目<B11>~<B20>、<B58>~<B58-20>または<B59>、<B60>~<B60-18>のいずれかにに記載のシステム。
<B60-20>
前記TCRもしくはBCRレパトアを解析する方法の処理は、以下のステップ:
(1)V領域、D領域、J領域および必要に応じてC領域の少なくとも1つを含む遺伝子領域ごとの参照データベースを提供するステップ:
(2)必要に応じてトリミングを行い、必要に応じて適切な長さのものを抽出した入力配列セットを提供するステップ;
(3)該入力配列セットについて、該遺伝子領域ごとに、該参照データベースと相同性検索を行い、近似する参照アリルおよび/または該参照アリルの配列とのアラインメントを記録するステップ;
(4)該入力配列セットについてV領域およびJ領域をアサインし、アサイン結果に基づいて、D領域の核酸配列を抽出するステップ;
(5)該D領域の核酸配列をアミノ酸配列に翻訳し、該アミノ酸配列を利用してD領域を分類するステップ;
(6)入力配列セットにおいて、V領域、D領域、J領域および必要に応じてC領域の各出現頻度、もしくはその組合せの出現頻度を算出することで、TCRもしくはBCRレパトアを導出するステップ;
を含むコンピュータに実行させるコンピュータプログラムによって実現される、
項目<B11>~<B20>、<B58>~<B58-20>、<B59>、<B60>~<B60-19>のいずれかに記載のシステム。
<B60-21>TCRもしくはBCRレパトアを解析する方法の処理をコンピュータに実行させる項目<B11>~<B20>、<B58>~<B58-20>、<B59>、<B60>~<B60-20>のいずれかに記載のシステムであって、該方法は以下のステップ:
(1)V領域、D領域、J領域および必要に応じてC領域の少なくとも1つを含む遺伝子領域ごとの参照データベースを提供するステップ:
(2)必要に応じてトリミングを行い、必要に応じて適切な長さのものを抽出した入力配列セットを提供するステップ;
(3)該入力配列セットについて、該遺伝子領域ごとに、該参照データベースと相同性検索を行い、近似する参照アリルおよび/または該参照アリルの配列とのアラインメントを記録するステップ;
(4)該入力配列セットについてV領域およびJ領域をアサインし、アサイン結果に基づいて、D領域の核酸配列を抽出するステップ;
(5)該D領域の核酸配列をアミノ酸配列に翻訳し、該アミノ酸配列を利用してD領域を分類するステップ;
(6)入力配列セットにおいて、V領域、D領域、J領域および必要に応じてC領域の各出現頻度、もしくはその組合せの出現頻度を算出することで、TCRもしくはBCRレパトアを導出するステップ;
を包含する、システム。
<C1>
被験者にがんイディオタイプペプチド感作免疫細胞療法を付与する方法であって、該方法は、
(1)項目<B1>~<B10>、<B57>、<B58>~<B58-20>、<B59>、または<B60>~<B60-21>のいずれかに記載の方法および/または項目<B11>~<B20>、<B58>~<B58-20>、<B59>、または<B60>~<B60-21>のいずれかに記載のシステムによって、該被験者のT細胞レセプター(TCR)またはB細胞レセプター(BCR)レパトアを解析する工程;
(2)該解析の結果に基づいて、該被験者のがん細胞由来のTCRまたはBCRを決定する工程であって、該決定は該被験者のがん細胞由来のTCRまたはBCR遺伝子の存在頻度ランキングにおいて、上位ランクの配列が、該がん細胞由来のTCRまたはBCRとして選択することによってなされる、工程;
(3)決定された該がん由来のTCRまたはBCRに基づいて、HLA検査ペプチドの候補のアミノ酸配列を決定する工程であって、該決定は、HLA結合ペプチド予測アルゴリズムを用いて算出されたスコアに基づきなされる、工程;
(4)決定されたペプチドを合成する工程;
必要に応じて(5)合成したペプチドを用いて治療を行う工程
を包含する、方法。
<C2>
前記(3)工程のHLA検査ペプチドの候補は、BIMAS、SYFPEITHI、RANKPEPまたはNetMHCを用いて決定される、項目<C1>に記載の方法。
<C3><改良型CTL法>
前記(4)工程の後に、前記ペプチドと、前記被験者由来の樹状細胞または抗原提示細胞と、前記被験者由来のCD8+T細胞とを混合して培養する工程、および該培養後の混合物を患者に投与する工程を包含する、項目<C1>または<C2>に記載の方法。
<C4><DCワクチン療法>
前記(4)工程の後に、前記ペプチドと、前記被験者由来の樹状細胞とを混合して培養する工程、および該培養された混合物を患者に投与する工程を包含する、項目<C1>~<C3>のいずれか1項に記載の方法。
<C5><患者自己免疫細胞療法>
前記(4)工程の後に、前記ペプチドと、前記被験者由来の樹状細胞または抗原提示細胞と、前記被験者由来のCD8+T細胞とを混合して培養してCD8+T細胞-樹状細胞/抗原提示細胞-ペプチド混合物を生産する工程、および前記ペプチドと、前記被験者由来の樹状細胞とを混合して培養して樹状細胞-ペプチド混合物を生産する工程を行い、該CD8+T細胞-樹状細胞/抗原提示細胞-ペプチド混合物、および該樹状細胞-ペプチド混合物を患者に投与する工程を包含する、項目<C1>~<C5>のいずれか1項に記載の方法。
<D1><オーダーメイドがん特異的T細胞受容体遺伝子の単離、in vitro抗原刺激によるがん特異的TCR遺伝子の単離>
(A)被験者に由来する抗原タンパク質または抗原ペプチドまたは該被験者に由来するリンパ球または項目<C1>~<C5>のいずれかに記載の前記決定されたペプチドと、該被験者に由来する不活化がん細胞と、該被験者に由来するTリンパ球とを混合し培養して腫瘍特異的T細胞を生産する工程;
(B)該腫瘍特異的T細胞のTCRを項目<B1>~<B10>、<B57>、<B58>~<B58-20>、<B59>、または<B60>~<B60-21>のいずれかに記載の方法および/または項目<B11>~<B20>、<B58>~<B58-20>、<B59>、または<B60>~<B60-21>のいずれかに記載のシステムによって解析する工程;および
(C)該解析の結果に基づいて、所望の腫瘍特異的T細胞を単離する工程
を包含する、in vitro抗原刺激によるがん特異的TCR遺伝子の単離方法。
<D1-1>
(A)工程は、被験者に由来する抗原タンパク質または抗原ペプチドと、該被験者に由来する不活化がん細胞と、該被験者に由来するTリンパ球とを混合し培養して腫瘍特異的T細胞を生産する工程である、項目<D1>に記載の方法。
<D1-2>
(A)工程は、前記被験者に由来するリンパ球と、該被験者に由来する不活化がん細胞と、該被験者に由来するTリンパ球とを混合し培養して腫瘍特異的T細胞を生産する工程である、項目<D1>~<D1-1>のいずれかに記載の方法。
<D1-3>
(A)工程は、項目C1に記載の前記決定されたペプチドと、該被験者に由来する不活化がん細胞と、該被験者に由来するTリンパ球とを混合し培養して腫瘍特異的T細胞を生産する工程である、項目<D1>~<D1-2>のいずれかに記載の方法。
<D2>
<オーダーメイドがん特異的T細胞受容体遺伝子の単離、共通配列検索によるがん特異的TCR遺伝子の単離>
(A)共通のHLAを有する被験体からリンパ球またはがん組織を単離する工程;
(B)該リンパ球またはがん組織について、該腫瘍特異的T細胞のTCRを項目B1に記載の方法によって解析する工程;および
(C)腫瘍特異的T細胞に共通する配列を有するT細胞を単離する工程
を包含する、共通配列検索によるがん特異的TCR遺伝子の単離する方法。
<E1><CPC>
A)患者からTリンパ球を採取する工程;
B)該Tリンパ球を抗原刺激した後に、項目<B1>~<B10>、<B57>、<B58>~<B58-20>、<B59>、または<B60>~<B60-21>のいずれかに記載の方法および/または項目<B11>~<B20>、<B58>~<B58-20>、<B59>、または<B60>~<B60-21>のいずれかに記載のシステムに基づいてTCRを解析する工程であって、該抗原刺激は、前記被験者に由来する抗原タンパク質または抗原ペプチド、該被験者に由来する不活性化がん細胞、または腫瘍由来イディオタイプペプチドによってなされる、工程;
C)解析された該TCRにおいて最適抗原および最適TCRを選択する工程;
D)該最適TCRのTCR遺伝子の腫瘍特異的αおよびβTCR発現ウイルスベクターを生産する工程;
E)該腫瘍特異的TCR遺伝子導入Tリンパ球を該患者に導入する工程;
を包含する細胞加工療法。
<E1-1>
前記抗原刺激は、前記被験者に由来する抗原タンパク質または抗原ペプチドによってなされる、項目<E1>に記載の細胞加工療法。
<E1-2>
前記抗原刺激は、前記被験者に由来する不活性化がん細胞によってなされる、項目<E1>または<E1-1>に記載の細胞加工療法。
<E1-3>
前記抗原刺激は、前記腫瘍由来イディオタイプペプチドによってなされる、項目<E1>、<E1-1>~<E1-2>のいずれかに記載の細胞加工療法。
<E1-4>
C)工程は、前記被験体のがん組織に高発現する抗原を選択することを含む、項目<E1>、<E1-1>~<E1-3>のいずれかに記載の方法。
<E1-5>
C)工程は、抗原特異的リンパ球刺激試験において最も強くT細胞を活性させる抗原を選択することを含む、項目<E1>、<E1-1>~<E1-4>のいずれかに記載の方法。
<E1-6>
C)工程は、抗原刺激前後において項目<B1>~<B10>、<B57>、<B58>~<B58-20>、<B59>、または<B60>~<B60-21>のいずれかに記載の方法および/または項目<B11>~<B20>、<B58>~<B58-20>、<B59>、または<B60>~<B60-21>のいずれかに記載のシステムに基づいて行ったレパトア解析から特定のTCRの頻度を最も増加させる抗原を選択することを含む、項目<E1>、<E1-1>~<E1-5>のいずれかに記載の方法。
<E2><CPCのRAC>
項目<D2>に記載の方法で単離されたがん特異的TCR遺伝子を用いてインビトロで刺激試験を行い有効性および/または安全性評価を行う方法。
<CC1>
被験者にがんイディオタイプペプチド感作免疫細胞療法に使用するための組成物を調製する方法であって、該方法は、
(1)項目<B1>~<B10>、<B57>、<B58>~<B58-20>、<B59>、または<B60>~<B60-21>のいずれかに記載の方法および/または項目<B11>~<B20>、<B58>~<B58-20>、<B59>、または<B60>~<B60-21>のいずれかに記載のシステムによって、該被験者のT細胞レセプター(TCR)またはB細胞レセプター(BCR)レパトアを解析する工程;
(2)該解析の結果に基づいて、該被験者のがん細胞由来のTCRまたはBCRを決定する工程であって、該決定は該被験者のがん細胞由来のTCRまたはBCR遺伝子の存在頻度ランキングにおいて、上位ランクの配列が、該がん細胞由来のTCRまたはBCRとして選択することによってなされる、工程;
(3)決定された該がん由来のTCRまたはBCRに基づいて、HLA検査ペプチドの候補のアミノ酸配列を決定する工程であって、該決定は、HLA結合ペプチド予測アルゴリズムを用いて算出されたスコアに基づきなされる、工程;および
(4)決定されたペプチドを合成する工程;
を包含する、方法。
<CC2>
前記(3)工程のHLA検査ペプチドの候補は、BIMAS、SYFPEEITHI、RANKPEEPまたはNEEtMHCCを用いて決定される、項目<CC1>に記載の方法。
<CC3><改良型CTL法>
前記(4)工程の後に、前記ペプチドと、前記被験者由来の樹状細胞または抗原提示細胞と、前記被験者由来のCD8+T細胞とを混合して培養する工程を包含する、項目<CC1>または<CC2>に記載の方法。
<CC4><DCワクチン療法>
前記(4)工程の後に、前記ペプチドと、前記被験者由来の樹状細胞とを混合して培養する工程を包含する、項目<CC1>~<CC2>のいずれかに記載の方法。
<CC5><患者自己免疫細胞療法>
前記(4)工程の後に、前記ペプチドと、前記被験者由来の樹状細胞または抗原提示細胞と、前記被験者由来のCD8+T細胞とを混合して培養してCD8+T細胞-樹状細胞/抗原提示細胞-ペプチド混合物を生産する工程、および前記ペプチドと、前記被験者由来の樹状細胞とを混合して培養して樹状細胞-ペプチド混合物を生産する工程を包含する、項目<CC1>~<CC4>のいずれかに記載の方法。
<DD1><オーダーメイドがん特異的T細胞受容体遺伝子の単離、in vitro抗原刺激によるがん特異的TCR遺伝子の単離>
(A)被験者に由来する抗原タンパク質または抗原ペプチドまたは該被験者に由来するリンパ球または項目<CC1>~<CC5>のいずれかに記載の前記決定されたペプチドと、該被験者に由来する不活化がん細胞と、該被験者に由来するTリンパ球とを混合し培養して腫瘍特異的T細胞を生産する工程;
(B)該腫瘍特異的T細胞のTCRを項目<B1>~<B10>、<B57>、<B58>~<B58-20>、<B59>、または<B60>~<B60-21>のいずれかに記載の方法および/または項目<B11>~<B20>、<B58>~<B58-20>、<B59>、または<B60>~<B60-21>のいずれかに記載のシステムによって解析する工程;および
(CC)該解析の結果に基づいて、所望の腫瘍特異的T細胞を単離する工程
を包含する、in vitro抗原刺激による単離されたがん特異的TCR遺伝子を調製する方法。
<DD1-1>
(A)工程は、被験者に由来する抗原タンパク質または抗原ペプチドと、該被験者に由来する不活化がん細胞と、該被験者に由来するTリンパ球とを混合し培養して腫瘍特異的T細胞を生産する工程である、項目<DD1>に記載の方法。
<DD1-2>
(A)工程は、前記被験者に由来するリンパ球と、該被験者に由来する不活化がん細胞と、該被験者に由来するTリンパ球とを混合し培養して腫瘍特異的T細胞を生産する工程である、項目<DD1>または<DD1-1>に記載の方法。
<DD1-3>
(A)工程は、項目CC1に記載の前記決定されたペプチドと、該被験者に由来する不活化がん細胞と、該被験者に由来するTリンパ球とを混合し培養して腫瘍特異的T細胞を生産する工程である、項目<DD1>~<DD1-2>のいずれかに記載の方法。
<DD2>
<オーダーメイドがん特異的T細胞受容体遺伝子の単離、共通配列検索によるがん特異的TCR遺伝子の単離>
(A)共通のHLAを有する被験体から単離されたリンパ球またはがん組織を提供する工程;
(B)該リンパ球またはがん組織について、該腫瘍特異的T細胞のTCRを項目<B1>~<B10>、<B57>、<B58>~<B58-20>、<B59>、または<B60>~<B60-21>のいずれかに記載の方法および/または項目<B11>~<B20>、<B58>~<B58-20>、<B59>、または<B60>~<B60-21>のいずれかに記載のシステムによって解析する工程;および
(C)腫瘍特異的T細胞に共通する配列を有するT細胞を単離する工程
を包含する、共通配列検索による単離されたがん特異的TCR遺伝子を調製する方法。
<EE1><CCPCC>
A)患者から採取されたTリンパ球を提供する工程;
B)該Tリンパ球を抗原刺激した後に、項目<B1>~<B10>、<B57>、<B58>~<B58-20>、<B59>、または<B60>~<B60-21>のいずれかに記載の方法および/または項目<B11>~<B20>、<B58>~<B58-20>、<B59>、または<B60>~<B60-21>のいずれかに記載のシステムに基づいてTCCRを解析する工程であって、該抗原刺激は、前記被験者に由来する抗原タンパク質または抗原ペプチド、該被験者に由来する不活性化がん細胞、または腫瘍由来イディオタイプペプチドによってなされる、工程;
CC)解析された該TCRにおいて最適抗原および最適TCRを選択する工程; および
DD)該最適TCRのTCCR遺伝子の腫瘍特異的αおよびβTCR発現ウイルスベクターを生産する工程
を包含する細胞加工療法に使用するための該腫瘍特異的TCR遺伝子導入Tリンパ球を調製する方法。
<EE1-1>
前記抗原刺激は、前記被験者に由来する抗原タンパク質または抗原ペプチドによってなされる、項目<EE1>に記載の方法。
<EE1-2>
前記抗原刺激は、前記被験者に由来する不活性化がん細胞によってなされる、項目<EE1>または<EE1-1>に記載の方法。
<EE1-3>
前記抗原刺激は、前記腫瘍由来イディオタイプペプチドによってなされる、項目<EE1>~<EE1-2>のいずれかに記載の方法。
<EE1-4>
C)工程は、前記被験体のがん組織に高発現する抗原を選択することを含む、項目<EE1>~<EE1-3>のいずれかに記載の方法。
<EE1-5>
C)工程は、抗原特異的リンパ球刺激試験において最も強くT細胞を活性させる抗原を選択することを含む、項目<EE1>~<EE1-4>のいずれかに記載の方法。
<EE1-6>
C)工程は、抗原刺激前後において項目B1に基づいて行ったレパトア解析から特定のTCCRの頻度を最も増加させる抗原を選択することを含む、項目<EE1>~<EE1-5>のいずれかに記載の方法。
<EE2><CCPCCのRACC>
項目<DD2>に記載の方法で単離されたがん特異的TCCR遺伝子を用いてインビトロで刺激試験を行い有効性および/または安全性評価を行う方法。この有効性および/または安全性評価の具体的なステップを以下を例示することができる。
精確度の高い非バイアスでの大規模遺伝子解析のための試料が提供され、定量的な分析が特に必要とされる臨床応用場面において特に有用である。また、本発明では、「低頻度」(1/10000-1/100000)遺伝子を同定できることで、たとえば白血病のより正確な診断、治療につながることになる。これは、従来技術(アダプターにプレート法を組み合わせる方法、またはSMART法にプレート法を組み合わせる方法)では検出限界(1%程度)のためできなかった。
従来汎用されるIMGT/High-V-QUESTとの大きな相違点は以下が挙げられる:IMGT/High-V-QUESTでは、C領域の分類機能は無く、レパトア分類は「遺伝子名単位」「アリル単位」どちらかである(すなわち、(*)V(遺伝子名)-D(遺伝子名)-J(遺伝子名)、もしくは、V(アリル)-D(アリル)-J(アリル)となる)。また、CDR3分類は、上記レパトアとは別個に行うことなら可能であるが、自由度がない。他方、本発明の解析方法ではC領域の分類が可能であり、レパトア分類では領域ごとに「遺伝子名単位」「アリル単位」を選択可能である。また、Dの代わりにCDR3を用いることも可能である。
標的となる癌細胞において治療に有効な特異的マーカー(分子標的)が存在しない、あるいは既存の特定の分子標的薬による治療では効果がない患者に対して、本発明のがんイディオタイプペプチド療法は有効である。すなわち、個別患者由来の癌細胞の遺伝子情報に基づいてペプチドが作製されるため、TCRもしくはBCRを発現する多くの腫瘍に対して効果を発揮する。リンパ腫細胞や白血病細胞はその起源からT細胞系腫瘍とB細胞系腫瘍が存在し、いずれの腫瘍型においても本技術が適応可能であり、多くの患者の治療に有用である。また、腫瘍化したB細胞亜集団を標的とする場合、抗CD20抗体のような大部分のB細胞に発現する細胞表面分子を標的とした抗体医薬が使用される。こららの抗体医薬は正常B細胞にも働くため、癌細胞のみならず正常細胞にも働き免疫能の低下などの副作用を引き起こす。他方、本発明のように癌細胞だけを標的とした治療は安全性も高い。癌ペプチドを利用する場合も、癌細胞に対するより特異性の高いペプチドを使用することにより、安全性の高い治療を実現することができる。また、既存のがんペプチドを使用した治療は、その結合する特定のHLAを有する患者に限定される。他方、本発明のように患者の遺伝情報に基づいてペプチドが設計されるため、HLA型に限定されず、広範な患者に適応できる利点がある。
TCRのβ配列:配列番号1377(図18)の塩基番号278~塩基番号300
TCRのγ配列:配列番号1378(図19)の塩基番号184~塩基番号201
TCRのδ配列:配列番号1379(図19)の塩基番号231~塩基番号249
BCRのIgM重鎖配列:配列番号1380(図20)の塩基番号77~塩基番号95
BCRのIgA重鎖配列:配列番号1381(図21)の塩基番号189~塩基番号208
BCRのIgG重鎖配列:配列番号1382(図22)の塩基番号262~塩基番号282
BCRのIgD重鎖配列:配列番号1383(図23)の塩基番号164~塩基番号183
BCRのIgE重鎖配列:配列番号1384(図24)の塩基番号182~塩基番号199
BCRのIgκ鎖定常領域配列:配列番号1385(図25)の塩基番号230~塩基番号248
BCRのIgλ鎖配列:配列番号1386(図25)の塩基番号273~塩基番号291。
TCRのβ配列:配列番号1377(図18)の塩基番号69~塩基番号91
TCRのγ配列:配列番号1378(図19)の塩基番号34~塩基番号53
TCRのδ配列:配列番号1379(図19)の塩基番号61~塩基番号78
BCRのIgM重鎖配列:配列番号1380(図20)の塩基番号7~塩基番号25
BCRのIgA重鎖配列:配列番号1381(図21)の塩基番号115~塩基番号134
BCRのIgG重鎖配列:配列番号1382(図22)の塩基番号109~塩基番号129
BCRのIgD重鎖配列:配列番号1383(図23)の塩基番号78~塩基番号96
BCRのIgE重鎖配列:配列番号1384(図24)の塩基番号45~塩基番号64
BCRのIgκ鎖定常領域配列:配列番号1385(図25)の塩基番号75~塩基番号92
BCRのIgλ鎖配列:配列番号1386(図25)の塩基番号52~塩基番号69(この配列番号はCMにも使っている。)。
以下に本発明の好ましい実施形態を説明する。以下に提供される実施形態は、本発明のよりよい理解のために提供されるものであり、本発明の範囲は以下の記載に限定されるべきでないことが理解される。従って、当業者は、本明細書中の記載を参酌して、本発明の範囲内で適宜改変を行うことができることは明らかである。これらの実施形態について、当業者は適宜、任意の実施形態を組み合わせ得る。
本発明は、次世代シークエンシング技術を使用して、T細胞レセプター(TCR)またはB細胞レセプター(BCR)の可変領域のレパトア(repertoire)の定量解析を行うための試料の調整を行うことができる。これらのシークエンシング技術は、妥当なコストで、試料から100万またはそれ以上のリードを得ることができる。1/1,000,000またはそれよりも低い頻度で存在する遺伝子型でさえ、これらの技術を用いて特異的様式で、しかもバイアスのかかっていない様式で検出することができる。血液または骨髄等のDNA由来の試料から、遺伝子または転写物の特定部分の配列の異なる型をすべて増幅するための非バイアス増幅方法が達成される。
別の局面において、本発明は、本発明の方法で製造された試料を用いて遺伝子解析を行う方法を提供する。
本発明は、次世代シークエンシング技術を使用して、T細胞レセプター(TCR)またはB細胞レセプター(BCR)の可変領域のレパトア(repertoire)の定量解析を行うためのバイオインフォマティクスを提供する。
V ミスマッチペナルティ=-1、最短アラインメント長=30、最短カーネル長=15
D ワード長=7(BLASTの場合)またはK-tup=3(FASTAの場合)、ミスマッチペナルティ=-1、ギャップペナルティ=0、最短アラインメント長=11、最短カーネル長=8
J ミスマッチペナルティ=-1、最短ヒット長=18、最短カーネル長=10
C 最短ヒット長=30、最短カーネル長=15で実施される。この条件は、例えば、より短い(~200bp)配列を用いて一部の領域だけでも分類する状況(「好ましい例」から外れる状況)であれば使用することができ、イルミナ製シーケンサを利用する状況でも使用することができる。この場合は、相同性検索にbwaもしくはbowtieを用いる可能性が考えられる。
次に、図42の機能ブロック図を参照して、本発明のシステム1の構成を説明する。なお、本図においては、単一のシステムで実現した場合を示している。
1つの局面において、本発明は、データベースを用いて被験体のT細胞レセプター(TCR)またはB細胞レセプター(BCR)の可変領域のレパトア(repertoire)を定量的に解析する方法を提供する。この方法は、(1)該被験者から非バイアス的に増幅した、T細胞レセプター(TCR)またはB細胞レセプター(BCR)の核酸配列を含む核酸試料を提供する工程;(2)該核酸試料に含まれる該核酸配列を決定する工程;および(3)決定された該核酸配列にもとづいて、各遺伝子の出現頻度またはその組み合わせを算出し、該被験体のTCRもしくはBCRレパトアを導出する工程を包含する。この方法および本明細書で説明される1つまたは複数の更なる特徴を含む方法を、本明細書において「本発明のレパトア解析法」とも呼ぶ。そして本発明のレパトア解析法を実現するシステムを「本発明のレパトア解析システム」ともいう。








1つの局面では、本発明は、被験者にがんイディオタイプペプチド感作免疫細胞療法に使用するための組成物を調製する方法を提供する。この方法は、(1)本発明のレパトア解析法または本発明のレパトア解析システムによって、該被験者のT細胞レセプター(TCR)またはB細胞レセプター(BCR)レパトアを解析する工程;(2)該解析の結果に基づいて、該被験者のがん細胞由来のTCRまたはBCRを決定する工程であって、該決定は該被験者のがん細胞由来のTCRまたはBCR遺伝子の存在頻度ランキングにおいて、上位ランクの配列が、該がん細胞由来のTCRまたはBCRとして選択することによってなされる、工程;(3)決定された該がん由来のTCRまたはBCRに基づいて、HLA検査ペプチドの候補のアミノ酸配列を決定する工程であって、該決定は、HLA結合ペプチド予測アルゴリズムを用いて算出されたスコアに基づきなされる、工程;および(4)決定されたペプチドを合成する工程を包含する。ここで合成されたペプチドは、がんイディオタイプペプチド感作免疫細胞療法に使用することができる。この方法は、本明細書において「がんイディオタイプペプチド感作免疫細胞療法」と呼ばれることがある。
別の局面において、本発明は、オーダーメイドがん特異的T細胞受容体遺伝子の単離、in vitro抗原刺激によるがん特異的TCR遺伝子の単離技術を提供する。したがって、本発明は、(A)被験者に由来する抗原タンパク質または抗原ペプチドまたは該被験者に由来するリンパ球または本発明の「がんイディオタイプペプチド感作免疫細胞療法」において決定されたペプチドと、該被験者に由来する不活化がん細胞と、該被験者に由来するTリンパ球とを混合し培養して腫瘍特異的T細胞を生産する工程;(B)該腫瘍特異的T細胞のTCRを本発明のレパトア解析法または本発明のレパトア解析システムによって解析する工程;および(C)該解析の結果に基づいて、所望の腫瘍特異的T細胞を単離する工程を包含する、in vitro抗原刺激による単離されたがん特異的TCR遺伝子を調製する方法を提供する。このようなin vitro抗原刺激による単離されたがん特異的TCR遺伝子を調製は、いったん遺伝子情報が得られたならば、当該分野で周知の任意の手法を用いて実施することができる。このような単離されたオーダーメイドがん特異的T細胞受容体遺伝子およびがん特異的TCR遺伝子を用いて、種々のがんの治療および予防に用いることができる。
さらなる局面において、本発明は、細胞加工療法を提供する。詳細には、本発明は、A)患者から採取されたTリンパ球を提供する工程;B)該Tリンパ球を抗原刺激した後に、本発明のレパトア解析法または本発明のレパトア解析システムに基づいてTCRを解析する工程であって、該抗原刺激は、前記被験者に由来する抗原タンパク質または抗原ペプチド、該被験者に由来する不活性化がん細胞、または腫瘍由来イディオタイプペプチドによってなされる、工程;C)解析された該TCRにおいて最適抗原および最適TCRを選択する工程; およびD)該最適TCRのTCR遺伝子の腫瘍特異的αおよびβTCR発現ウイルスベクターを生産する工程を包含する細胞加工療法に使用するための該腫瘍特異的TCR遺伝子導入Tリンパ球を調製する方法を提供する。この腫瘍特異的TCR遺伝子導入Tリンパ球を用いた細胞加工療法は、種々のがんの治療および予防に用いることができる。
一つの実施形態として、本発明のレパトア解析法を用いてBCR遺伝子レパトア解析を行い、以下に記載の方法で標的抗原に特異的なヒト型抗体を迅速に取得することができる
(A)標的となる抗原タンパクまたは抗原ペプチドをマウスに免疫し、該マウスより抗体産生B細胞を含む細胞集団(例えば脾臓、リンパ節、末梢血細胞)を分離し、本発明のレパトア解析法によるBCRレパトア解析により免疫グロブリン重鎖および軽鎖遺伝子を解析する方法
(A1)免疫するマウスが抗体多様性を維持したまま完全ヒト抗体を産生することができるKMマウスであるAの方法
(A2)免疫するマウスがNOD/scidマウスにIL-2レセプターγ鎖ノックアウトマウスを交配して作製された重度な複合型免疫不全を呈するNOG(NOD/Shi-scid, IL-2Rγnull)マウスにヒト幹細胞を移植して作製されたヒト化マウスであるAの方法
(B)対照マウスと免疫マウス、あるいは抗原免疫前と後のマウス由来の試料から得られた免疫グロブリン重鎖および軽鎖遺伝子配列とその頻度を比較する
(C)免疫マウスで強く発現する、あるいは免疫後に増加する免疫グロブリン重鎖および軽鎖遺伝子を同定する
(D)Cの工程から選択した免疫グロブリン重鎖と軽鎖遺伝子を選択し、1種の抗体発現用ベクターにあわせて挿入する、あるいは別個に2種の抗体発現用ベクターにそれぞれ挿入する方法
(E)Dの工程で作製した免疫グロブリン重鎖および軽鎖遺伝子発現ベクターをCHO(Chinese HamsterOvary)などの真核細胞に導入し、細胞培養を行う
(F)遺伝子組換え細胞により産生、分泌された抗体分子を分離・精製し、標的抗体タンパクあるいはペプチドに対する特異性を検証する
上記A-Fの工程により、動物由来の抗体遺伝子を取得した後にヒト抗体とのキメラ型抗体あるいはヒト化抗体に改変することなく、直接的かつ迅速に抗原特異的ヒト型抗体を取得する方法であり、ヒト型抗体から成る抗体医薬品の開発と製造に用いることができる。
一つの実施形態として、当該BCR遺伝子レパトア解析法を利用して、以下に記載の方法で標的抗原に特異的なヒト型抗体を迅速に取得することができる
(A)標的となる抗原タンパクまたは抗原ペプチドをマウスに免疫し、該マウスより抗体産生B細胞を含む細胞集団(例えば脾臓、リンパ節、末梢血細胞)を分離し、BCRレパトア解析法により免疫グロブリン重鎖および軽鎖遺伝子を解析する方法
(A1)免疫するマウスが抗体多様性を維持したまま完全ヒト抗体を産生することができるKMマウスであるAの方法
(A2)免疫するマウスがNOD/scidマウスにIL-2レセプターγ鎖ノックアウトマウスを交配して作製された重度な複合型免疫不全を呈するNOG(NOD/Shi-scid, IL-2Rγnull)マウスにヒト幹細胞を移植して作製されたヒト化マウスであるAの方法
(B)対照マウスと免疫マウス、あるいは抗原免疫前と後のマウス由来の試料から得られた免疫グロブリン重鎖および軽鎖遺伝子配列とその頻度を比較する
(C)免疫マウスで強く発現する、あるいは免疫後に増加する免疫グロブリン重鎖および軽鎖遺伝子を同定する
(D)Cの工程から選択した免疫グロブリン重鎖と軽鎖遺伝子を選択し、1種の抗体発現用ベクターにあわせて挿入する、あるいは別個に2種の抗体発現用ベクターにそれぞれ挿入する方法
(E)Dの工程で作製した免疫グロブリン重鎖および軽鎖遺伝子発現ベクターをCHO(Chinese HamsterOvary)などの真核細胞に導入し、細胞培養を行う
(F)遺伝子組換え細胞により産生、分泌された抗体分子を分離・精製し、標的抗体タンパクあるいはペプチドに対する特異性を検証する
上記A-Fの工程により、動物由来の抗体遺伝子を取得した後にヒト抗体とのキメラ型抗体あるいはヒト化抗体に改変することなく、直接的かつ迅速に抗原特異的ヒト型抗体を取得する方法であり、ヒト型抗体から成る抗体医薬品の開発と製造に用いることができる。
1.KMマウスに実験的自己免疫性脳脊髄炎の抗原ペプチドであるMyelinOligodendrocyte Glycoprotein(MOG35-55, MOG)を免疫する。等量の2mg/mL MOGペプチドと完全フロイントアジュバントを混合し、エマルジョンを作成し、200μgのMOGを皮下に免疫し、同時に400ngの百日咳毒素を腹腔に免疫する。対PBSと完全フロイントアジュバントによる免疫を行い、対照マウスとする。
2.初回免疫後2日目に400ngの百日咳毒素を免疫し、免疫後10日目に発症を確認した後、脳脊髄炎発症マウスより脾臓を摘出する。
3.発症マウスと対照マウスの脾臓を用いて、次世代BCRレパトア解析を実施する。IgG免疫グロブリン重鎖および免疫グロブリン軽鎖について、個々のBCR配列の出現頻度の集計とランキングを行う。
4.対照ウスに比し、発症マウスにおいて出現頻度が大きく増加したBCR配列を抽出し、ランキングする。これら抗体投与によって誘導されたBCR配列のランキング上位のものの組み合わせについて、MOG特異的抗体遺伝子として同定する。
5.発症マウスから増幅したBCR遺伝子増幅産物からPCR-cloning法により全長ヒト免疫フロブリン配列をクローニングする。抗体発現用ベクターにIgG免疫グロブリン重鎖および免疫グロブリン軽鎖のそれぞれをクローニングする。1種の抗体発現用ベクターにあわせて挿入する、あるいは別個に2種の抗体発現用ベクターにそれぞれ挿入する方法がある。
6.これら構築した発現ベクターでリポフェクタミン3000(Life Science)を用いてCHO(Chinese Hamster Ovary)細胞を形質転換し、IgG免疫グロブリン重鎖および免疫グロブリン軽鎖を導入する。
7.CHO細胞培養液を回収し、ProteinAアフィニティーカラムによる精製、ゲル濾過による濃縮により、分泌された抗体蛋白を回収する。
8.回収した抗体を用いたELISAアッセイにより、MOG35-55またはMOG蛋白に対する結合活性を測定し、抗体の特異性を検証する。
9.十分な特異性が得られたら、抗体発現安定発現細胞株を取得し、大量培養系によりヒト型抗MOG抗体を製造する。
本発明のペプチドまたはそれをコードする核酸は、免疫療法に用いることができる。以下に説明する。
本明細書において同じ意味で使用され、任意の長さのヌクレオチドのポリマーをいう。この用語はまた、「オリゴヌクレオチド誘導体」または「ポリヌクレオチド誘導体」を含む。「オリゴヌクレオチド誘導体」または「ポリヌクレオチド誘導体」とは、ヌクレオチドの誘導体を含むか、またはヌクレオチド間の結合が通常とは異なるオリゴヌクレオチドまたはポリヌクレオチドをいい、互換的に使用される。そのようなオリゴヌクレオチドとして具体的には、例えば、2’-O-メチル-リボヌクレオチド、オリゴヌクレオチド中のリン酸ジエステル結合がホスホロチオエート結合に変換されたオリゴヌクレオチド誘導体、オリゴヌクレオチド中のリン酸ジエステル結合がN3’-P5’ホスホロアミデート結合に変換されたオリゴヌクレオチド誘導体、オリゴヌクレオチド中のリボースとリン酸ジエステル結合とがペプチド核酸結合に変換されたオリゴヌクレオチド誘導体、オリゴヌクレオチド中のウラシルがC-5プロピニルウラシルで置換されたオリゴヌクレオチド誘導体、オリゴヌクレオチド中のウラシルがC-5チアゾールウラシルで置換されたオリゴヌクレオチド誘導体、オリゴヌクレオチド中のシトシンがC-5プロピニルシトシンで置換されたオリゴヌクレオチド誘導体、オリゴヌクレオチド中のシトシンがフェノキサジン修飾シトシン(phenoxazine-modified cytosine)で置換されたオリゴヌクレオチド誘導体、DNA中のリボースが2’-O-プロピルリボースで置換されたオリゴヌクレオチド誘導体およびオリゴヌクレオチド中のリボースが2’-メトキシエトキシリボースで置換されたオリゴヌクレオチド誘導体などが例示される。他にそうではないと示されなければ、特定の核酸配列はまた、明示的に示された配列と同様に、その保存的に改変された改変体(例えば、縮重コドン置換体)および相補配列を包含することが企図される。具体的には、縮重コドン置換体は、1またはそれ以上の選択された(または、すべての)コドンの3番目の位置が混合塩基および/またはデオキシイノシン残基で置換された配列を作成することにより達成され得る(Batzeret al.,Nucleic Acid Res.19:5081(1991);Ohtsuka et al.,J.Biol.Chem.260:2605-2608(1985);Rossoliniet al., Mol.Cell.Probes 8:91-98(1994))。本明細書に
おいて「核酸」はまた、遺伝子、cDNA、mRNA、オリゴヌクレオチド、およびポリヌクレオチドと互換可能に使用される。本明細書において「ヌクレオチド」は、天然のものでも非天然のものでもよい。
、25、30、40、50およびそれ以上のアミノ酸が挙げられ、ここの具体的に列挙していない整数で表される長さ(例えば、11など)もまた、下限として適切であり得る。また、ポリヌクレオチドの場合、5、6、7、8、9、10、15、20、25、30、40、50、75、100およびそれ以上のヌクレオチドが挙げられ、ここの具体的に列挙していない整数で表される長さ(例えば、11など)もまた、下限として適切であり得る。本明細書において、このようなフラグメントは、例えば、全長のものがマーカーまたは標的分子として機能する場合、そのフラグメント自体もまたマーカーまたは標的分子としての機能を有する限り、本発明の範囲内に入ることが理解される。
該製剤の容器は、繰り返し投与(例えば2~6回の投与)に使うことができる多用途バイアル瓶でもよい。該キットは、さらに、適切な希釈剤(例えば重曹溶液)を有する第2の容器を有することができる。
本明細書において用いられる分子生物学的手法、生化学的手法、微生物学的手法は、当該分野において周知であり慣用されるものであり、例えば、Sambrook J. et al.(1989).Molecular Cloning: A Laboratory Manual, Cold Spring Harborおよびその3rd Ed.(2001); Ausubel, F. M.(1987).Current Protocols in Molecular Biology, Greene Pub. Associates and Wiley-Interscience; Ausubel,F.M.(1989).Short Protocols in Molecular Biology: A Compendium of Methods from Current Protocols in Molecular Biology, Greene Pub. Associates and Wiley-Interscience; Innis, M. A. (1990). PCR Protocols: A Guide to Methods and Applications, Academic Press; Ausubel, F. M. (1992).Short Protocols in Molecular Biology: A Compendium of Methods from Current Protocols in Molecular Biology, Greene Pub. Associates; Ausubel, F. M.(1995).Short Protocols in Molecular Biology: A Compendium of Methods from Current Protocols in Molecular Biology, Greene Pub. Associates; Innis, M. A. et al.(1995). PCR Strategies, Academic Press; Ausubel, F. M. (1999). Short Protocols in Molecular Biology: A Compendium of Methods from Current Protocols in Molecular Biology, Wiley, and annual updates; Sninsky, J. J. et al.(1999).PCR Applications: Protocols for Functional Genomics, Academic Press, Gait, M. J. (1985). Oligonucleotide Synthesis: A Practical Approach, IRL Press; Gait, M. J. (1990). Oligonucleotide Synthesis: A Practical Approach, IRL Press; Eckstein, F.(1991). Oligonucleotides and Analogues: A Practical Approach, IRL Press; Adams, R. L. et al.(1992).The Biochemistry of the Nucleic Acids, Chapman & Hall; Shabarova, Z. et al.(1994). Advanced Organic Chemistry of Nucleic Acids, Weinheim; Blackburn, G. M. et al.(1996). Nucleic Acids in Chemistry and Biology, Oxford University Press; Hermanson, G. T. (I996). Bioconjugate Techniques, Academic Press、別冊実験医学「遺伝子導入&発現解析実験法」羊土社、1997などに記載されている。これらは本明細書において関連する部分(全部であり得る)が参考として援用される。
(調製実施例1:健常人末梢血におけるBCRレパトアの解析) 本実施例では、健常人末梢血におけるBCRレパトアの解析を行った。
試料:健常人末梢血単核球細胞
方法:
(1.RNA抽出)
例の健常人から全血5mLをヘパリン含採血管に採取し、フィコール密度勾配遠心分離により末梢血単核球細胞(peripheral blood mononuclear cells, PBMC)を分離した。単離した5×106細胞のPBMCからRNeasy Lipid Tissue Mini Kit (QIAGEN, Germany)を用いて、全RNAを抽出・精製した。取得されたRNAは、吸光度計を用いてA260の吸光度により定量された。溶出液量30μLにおいて濃度232ng/μLであった。
抽出されたRNA試料を用いて、adaptor-ligation PCRを実施した。最初に、相補的DNAを合成するため、BSL-18Eプライマー(表1-1)と 3.5 μL(812ng)のRNAを混和して、70℃で8分間アニーリングした。氷上で冷却後、下記の組成においてRNase阻害剤(RNAsin)の存在下で逆転写反応を行い、相補的DNAを合成した。

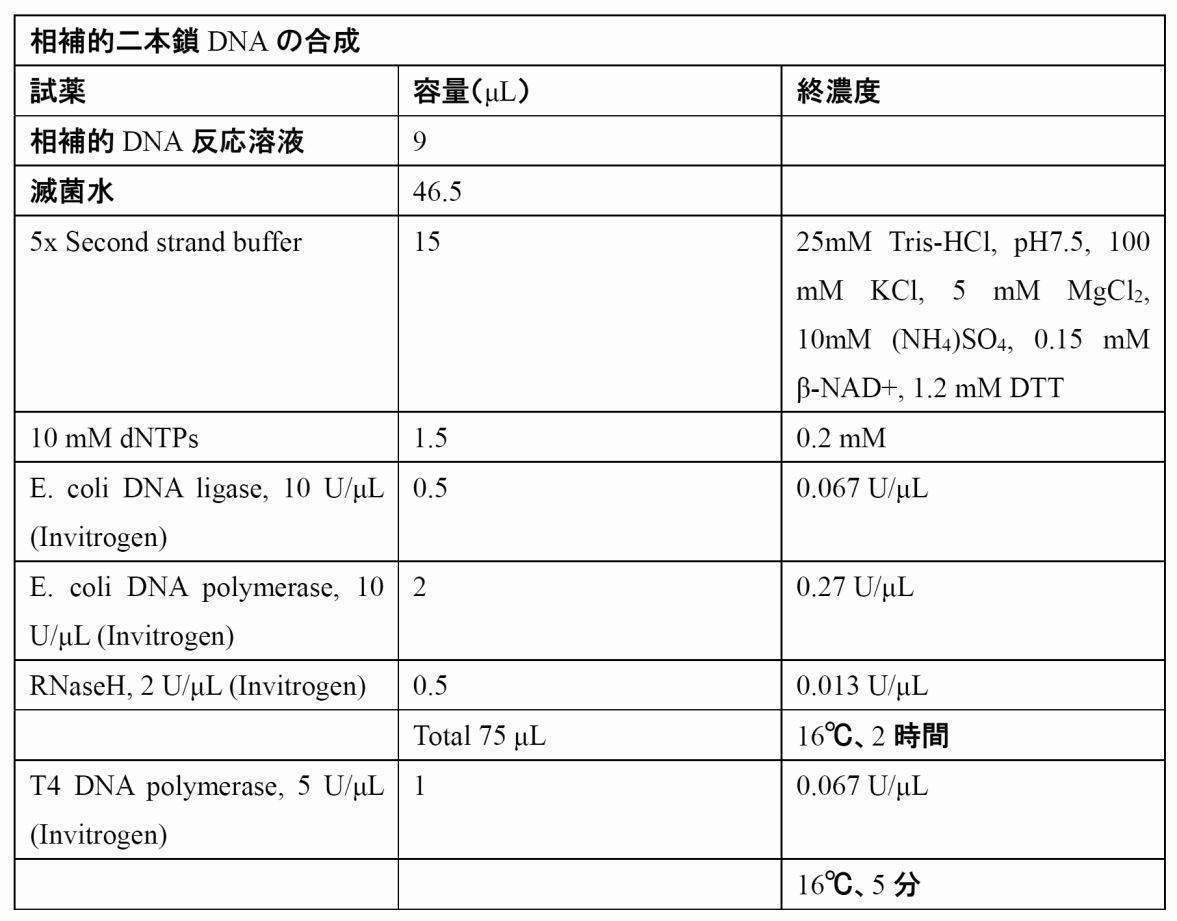



二本鎖相補的DNAからの一次PCR増幅(1st PCR)は、共通アダプタープライマーのP20EAと各々の免疫グロブリンアイソタイプC領域特異的プライマー(CM1、CA1、CG1、CD1、CE1)を用いて、次に示す反応組成で、95℃ 30秒、55℃ 30秒、72℃ 1分のサイクルを20サイクル行った。用いたプライマー配列は表1-1に示す。


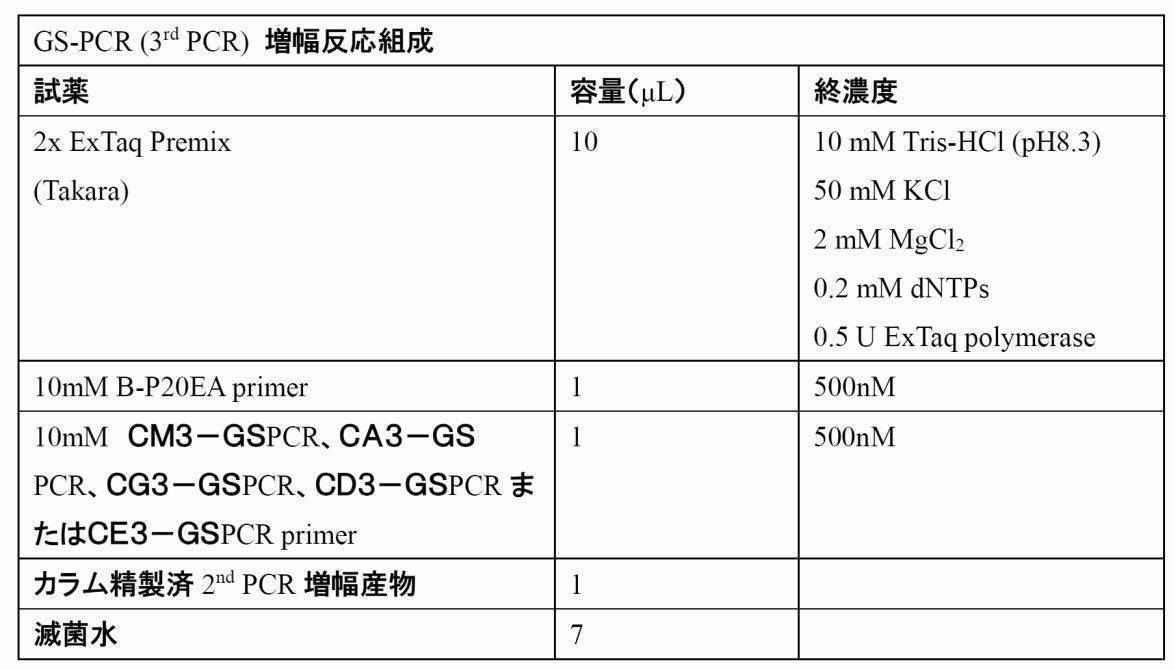
至適条件下におけるGS-PCR増幅の後、2%アガロースゲル電気泳動を実施し、目視化において目的のサイズ(500bp~700bp)のバンドを切り出し、DNA精製キット(QIAEX II Gel Extraction Kit、QIAGEN)を用いて精製した。回収されたDNAをQuant-iTTM PicoGreen(登録商標) dsDNA Assay Kit(Invitrogen)を用いてDNA量を測定した。回収された各アイソタイプ由来の増幅産物のDNA量は、それぞれIgM(1611ng/mL)、IgG(955ng/mL)、IgA(796ng/mL)、IgD(258ng/mL)、IgE(871ng/mL)であった。これらアイソタイプ増幅産物のDNA量が均等になるよう混和し、1000万DNAをエマルジョンPCRに用い、ロシュ社製次世代シーケンス解析装置(GS Juniorベンチトップシステム)によるシーケンス解析を行った。
シーケンスリードの解析は、IMGT (the international ImMunoGeneTics information system, http://www.imgt.org)データベースから入手されるV、D、J、C配列をリファレンス配列として各リード配列のV、D、J、C配列をアサインした。アサインメントにはIMGTのHighV-Questおよび新規に開発したレパトア解析ソフトウェア(Repertoire Genesis、同日付で出願される特許出願を参照。この内容は本明細書において参考として援用される)を用いた。




































たことが示された。
本実施例では、健常人末梢血におけるTCRレパトアの解析を行った。
(試料)
10例の健常人末梢血単核球細胞
(方法)
(1.RNA抽出)
10例の健常人から全血5mLをヘパリン含採血管に採取し、フィコール密度勾配遠心分離により末梢血単核球細胞(peripheral blood mononuclear cells, PBMC)を分離した。単離したPBMCからRNeasy Lipid Tissue Mini Kit (Qiagen, Germany)を用いて、全RNAを抽出・精製した。取得されたRNAは、Agilent 2100バイオアナライザ(Agilent)を用いて定量された。獲得されたRNA量を次の表1-4に示す。

抽出されたRNA試料を用いて、adaptor-ligation PCRを実施した。方法は調製実施例1に示した方法に従って実施した。すなわち、BSL-18Eプライマー(表1-5)とRNAを混和してアニーリング後、逆転写酵素を使って相補鎖DNAを合成した。続いて、二本鎖相補的DNAを合成し、T4 DNA polymeraseによる5’末端平滑化反応を行った。High Pure PCR Cleanup Micro Kit(Roche)によりカラム精製した後、P20EA/P10EAアダプターをLigation反応にて付加した。カラムにより精製されたアダプター付加二本鎖相補的DNAは、NotI制限酵素により消化された。

二本鎖相補的DNAから、表1-1に示す共通アダプタープライマーP20EAとTCRα鎖またはβ鎖C領域特異的プライマー(CA1またはCB1)を用いて、第1のPCR増幅反応の産物である1st PCR増幅を行った。PCRは次に示す組成で、95℃30秒、55℃30秒、72℃1分のサイクルを20サイクル行った。





ロシュ社製GS Juniorシーケンス解析装置による次世代シーケンスを実施した。具体的には、GS Junior Titanium emPCR Kit (Lib-L)を用いて、メーカーのプロトコールに従ってemPCRを実施した。ビーズとDNAの割合(copy per beads:cpb)0.5で実施した。emPCR後、ビーズエンリッチメントにて回収されたビーズは、シーケンスラン試薬であるGS Junior Titanium Sequencing KitおよびPicoTiterPlate Kitを用いて、メーカーのプロトコールに従ってシーケンスランを実施した。
得られたシーケンスデータ(SFFファイル)をGS Junior付属ソフトウェア(sfffileもしくはsffinfo)により、MID Tag別のリード配列に分類し、Fasta形式のシーケンスファイルを生成した。得られた平均リード数はTRA:17840リード、TRB:5122リードで、200bp以上のRaw dataの割合はTRA:34.9-63.7%(平均42.2%)、TRB:68.8-78.7%(平均73.1%)であった(表1-8)。次に、新規に開発したレパトア解析ソフトウェア(Repertoire Genesis、特許出願中)を用いて、IMGT(the international ImMunoGeneTics information system, www.imgt.org)データベースのリファレンス配列との照合を行い、各リードのV領域、D領域、J領域のアサインメントとCDR3配列の決定を行った。アサインされたリード数は表1-8に示した。また、同一リードの頻度解析、V、D、J鎖の使用頻度を調べた。Repertoire Genesisにより得られたリードを用いて生成したTRVおよびTRJレパトアについて図13(A~D)、14(A~D)、15(A~D)、図16に示す。
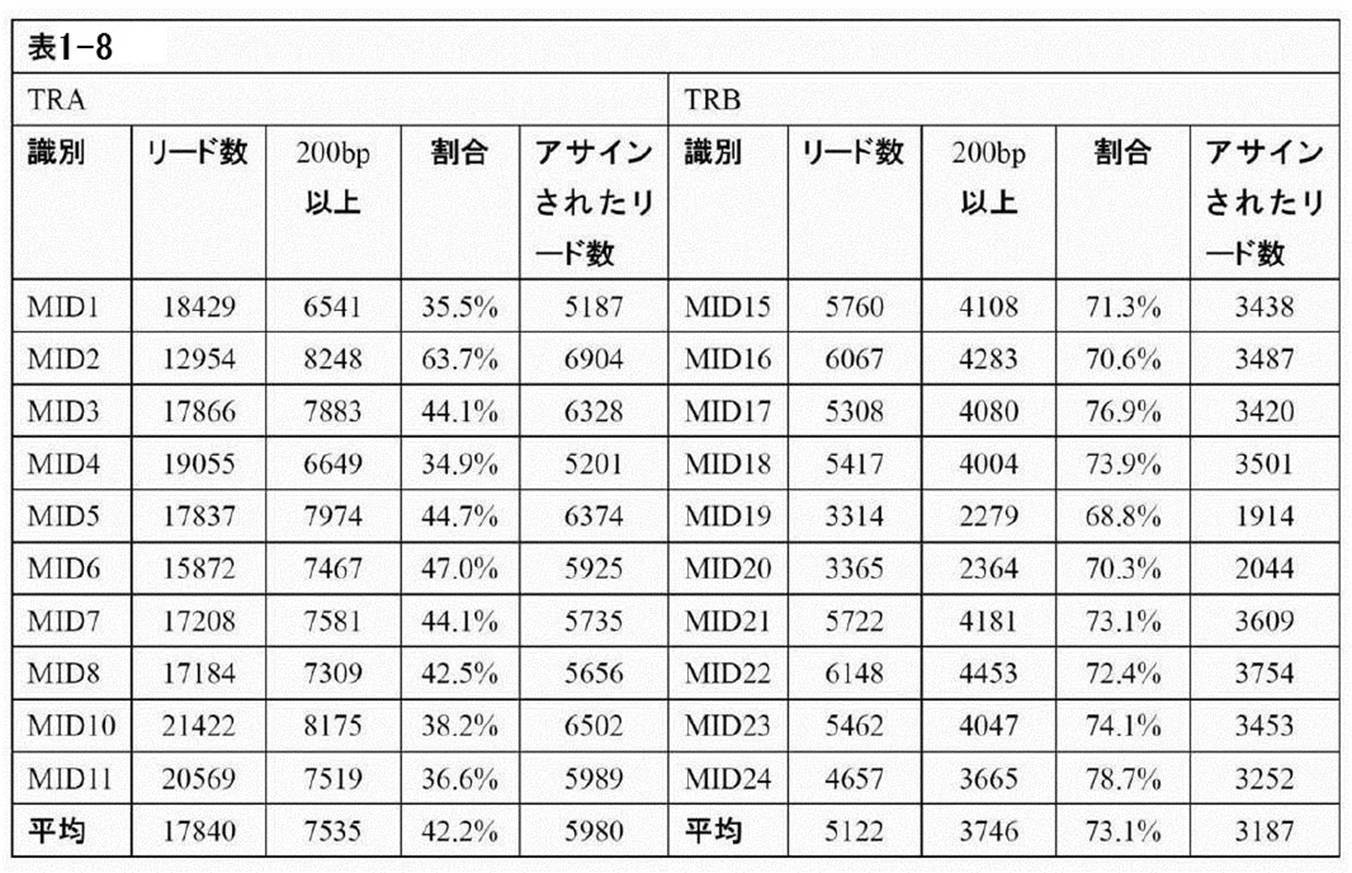
本実施例では、非バイアスAdaptor-ligation PCR法によるTCRおよびBCR遺伝子の増幅を行う。
(試料)
健常人末梢血単核球細胞
(方法)
(1.RNA抽出)
1例の健常人から全血5mLをヘパリン含採血管に採取し、フィコール密度勾配遠心分離により末梢血単核球細胞(peripheral blood mononuclear cells, PBMC)を分離した。単離した5×106細胞のPBMCからRNeasy Lipid Tissue Mini Kit (QIAGEN, Germany)を用いて、全RNAを抽出・精製した。
抽出されたRNA試料を用いて、adaptor-ligation PCRを実施した。最初に、相補的DNAを合成するため、BSL-18Eプライマー(表1-1)と 3.5 μL(812ng)のRNAを混和して、70℃で8分間アニーリングした。氷上で冷却後、下記の組成においてRNase阻害剤(RNAsin)の存在下で逆転写反応を行い、相補的DNAを合成した。




二本鎖相補的DNAからの1st PCRは、共通アダプタープライマーのP20EAとTCR C領域特異的プライマー(CA1、CB1、CG1、CD1)または免疫グロブリンアイソタイプC領域特異的プライマー(CM1、CA1、CG1、CD1、CE1、CK1、CL1)を用いて行った。C領域の全長を含む配列を増幅できるようC領域の3′末端側、中央部あるいは5′側にプライマーを設定した。示す反応組成で、95℃ 30秒、55℃ 30秒、72℃ 1分のサイクルを20サイクル行った。用いたプライマー配列は表1-1に示す。

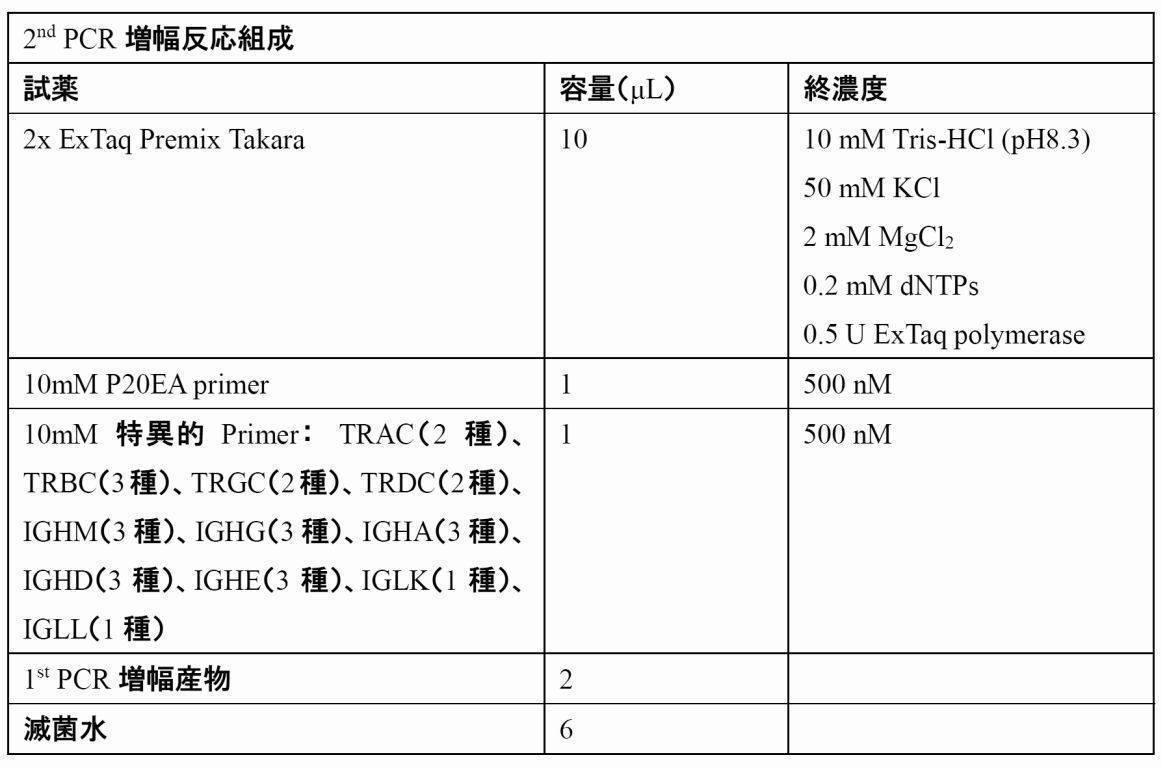
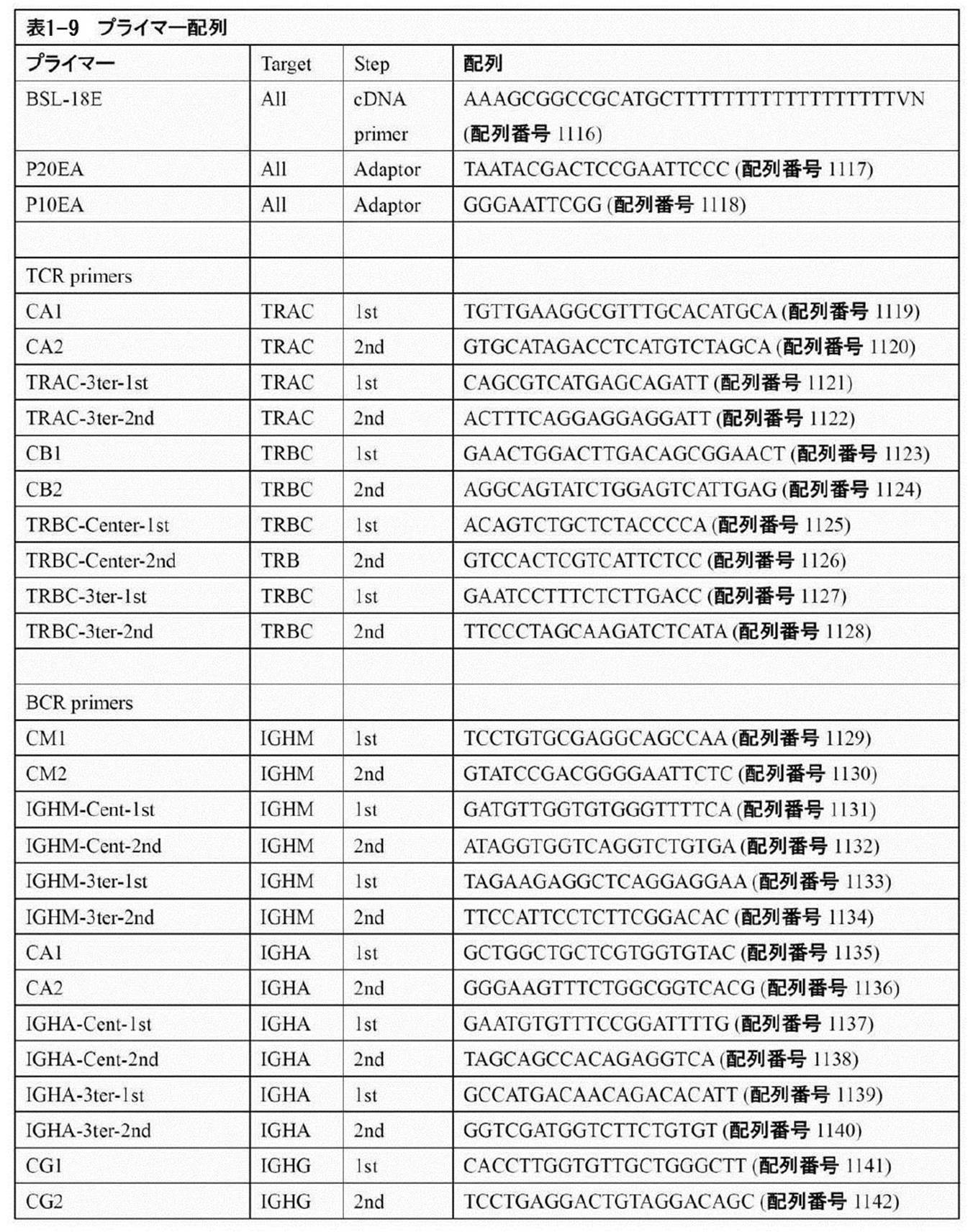
(試料)
健常人末梢血単核球細胞、MOLT-4ヒト急性リンパ芽球性白血病細胞株
(方法)
(1.T細胞系白血病細胞株の培養)
T細胞受容体(TCR)を発現するT細胞系細胞株として、ヒト急性リンパ芽球性白血病細胞株Molt-4を用いた。10%ウシ胎児血清、100 IU/mlペニシリン、100 μg/mlストレプトマイシン、2mM L-グルタミンを含むRPMI-1640培地で、37℃、5%CO2下において培養し、全細胞数として1 x 107細胞を回収した。RPMI-1640培地で洗浄し、1 x 106細胞/mLになるように細胞を懸濁した。
1例の健常人から全血5mLをヘパリン含採血管に採取し、フィコール密度勾配遠心分離により末梢血単核球細胞(peripheral blood mononuclear cells, PBMC)を分離した。RPMI1640培地で洗浄後、細胞数をカウントして1 x 106細胞/mLになるように懸濁した。
得られた1x106 細胞/mLのPBMCと1 x 106 細胞/mLのMolt-4細胞を下記の細胞数となるように混合し、Molt-4段階希釈細胞懸濁液を調製した。

段階希釈細胞懸濁液からRNeasy Lipid Tissue Mini Kit (QIAGEN, Germany)を用いて、全RNAを抽出・精製した。20 μLの溶出液で溶出し、Agilent 2100バイオアナライザ(Agilent)を用いてA260の吸光度によりRNA量を定量した。RNA電気泳動像を図26に示し、各試料から得られたRNA量を表1-4Bに示した。

抽出されたRNA試料を用いて、adaptor-ligation PCRを実施した。最初に、相補的DNAを合成するため、BSL-18Eプライマーと3.5 μLのRNAを混和して、70℃で8分間アニーリングした。氷上で冷却後、下記の組成においてRNase阻害剤(RNAsin)の存在下で逆転写反応を行い、相補的DNAを合成した。




二本鎖相補的DNAから、表1-1に示す共通アダプタープライマーP20EAとTCRα鎖またはβ鎖C領域特異的プライマー(CB1)を用いて、1st PCR増幅を行った。PCRは次に示す組成で、95℃30秒、55℃30秒、72℃1分のサイクルを20サイクル行った。



ロシュ社製GS Juniorシーケンス解析装置による次世代シーケンスを実施した。具体的には、GS Junior Titanium emPCR Kit (Lib-L)を用いて、メーカーのプロトコールに従ってemPCRを実施した。ビーズとDNAの割合(copy per beads:cpb)2で実施した。emPCR後、ビーズエンリッチメントにて回収されたビーズは、シーケンスラン試薬であるGS Junior Titanium Sequencing KitおよびPicoTiterPlate Kitを用いて、メーカーのプロトコールに従ってシーケンスランを実施した。


Agilent 2100 バイオアナライザによるRNA電気泳動像を図26に示す。細胞段階希釈液より全RNAを抽出して、Agilent社製バイオアナライザを用いてRNA量を測定した。マイクロチップ型電気泳動装置でRNAを分離し、RNAの品質チェックを行った。いずれの試料においても、28S(上バンド)および18S rRNA(下バンド)が検出され、分解を受けていないRNAが得られたことを示している。
(解析試験例1 健常者のBCRレパトア解析)
本実施例では、健常者のBCRレパトアの比較を行った。
(材料)
健常者1検体の末梢血単核球細胞より得られるRNAより、非バイアス的に得られたBCRのcDNAをRoche GS-Juniorで配列決定したリードセットを使用した。IgM、IgG、IgA、IgD、IgEのクラスごとのリードセットとなっている。
方法の全体像を図30(図29にはTCRの解析スキームを示す)に示す。
D ワード長=7、ミスマッチペナルティ=-1、ギャップペナルティ=0、 最短アラインメント長=11、最短カーネル長=8
J ミスマッチペナルティ=-1、最短ヒット長=18、最短カーネル長=10
C 最短ヒット長=30、最短カーネル長=15
最も近しい参照アリルを選択する際の指標は以下の優先順位で適用した。
IgM、IgG、IgA、IgD、IgEのリードセットごとにC遺伝子名の出現頻度を導出した結果を図31に示す。各クラスに対応する遺伝子名が専ら出現しており、また、no-hitがほとんど見られないことから、解析対象であるリードセットのクオリティは十分であることが示唆される。
(表2-3)クラス間のDレパトアの比較 縦軸:頻度(%)、横軸:遺伝子名




































本実施例では、検体間のBCRレパトアの比較を行った。
(材料)
解析実施例1と同様の手法で得られた5検体のリードセットで、4検体(No.1-4)が健常人、1検体(No.5)が白血病患者である。
解析実施例1と同様の方法で各検体について、クラスごと、領域ごとのレパトアを導出し、検体間で比較を行った。
結果の一例としてIgMにおけるVレパトアの比較結果を図36(AおよびB)に、Jレパトアの比較結果を図37に示す。No.5の検体のみ大きく異なることが示されている。
本実施例では、健常人のTCRレパトアの比較を行った。
(材料)
実施例1と同様の手法で得られた10検体のリードセットで、10検体(No.1-10)とも健常人である。
実施例1と同様の方法で各検体について、クラスごと、領域ごとのレパトアを導出し、検体間で比較を行った。
結果を図38~41に示す。図38(A~D)は、検体間のTRAVのレパトア比較の結果を示し、図39(A~D)は、検体間のTRBVのレパトア比較の結果を示し、図40(A~D)は、検体間のTRAJレパトア比較の結果を示し、図41は、検体間のTRBJレパトア比較の結果を示す。
(解析システムの実施例1:T細胞大顆粒リンパ球性白血病(T-LGL)の診断応用)
本実施例では、本発明のシステムのT細胞大顆粒リンパ球性白血病(T-LGL)の診断への応用を確認する実験を行った。
試料:T細胞大顆粒リンパ球性白血病由来末梢血単核球細胞
方法
(RNA抽出)
1例のT細胞大顆粒リンパ球性白血病を罹患した患者から全血7mLをヘパリン含採血管に採取し、フィコール密度勾配遠心分離により末梢血単核球細胞(peripheral blood mononuclear cells, PBMC)を分離した。単離した1.66x107細胞のPBMCからRNeasy Lipid Tissue Mini Kit (QIAGEN, Germany)を用いて、全RNAを抽出・精製した。取得されたRNAは、吸光度計を用いてA260の吸光度により定量され、全RNA量は15μgであった。
抽出されたRNA試料を用いて、adaptor-ligation PCRを実施した。最初に、相補的DNAを合成するため、BSL-18Eプライマー(表3-1A)と 3.5 μLのRNAを混和して、70℃で8分間アニーリングした。氷上で冷却後、下記の組成においてRNase阻害剤(RNAsin)の存在下で逆転写反応を行い、相補的DNAを合成した。






二本鎖相補的DNAから、共通アダプタープライマーP20EAとTCRα鎖またはβ鎖C領域特異的プライマー(CA1またはCB1)を用いて、1stPCR増幅を行った。PCRは次に示す組成で、95℃30秒、55℃30秒、72℃1分のサイクルを20サイクル行った。




Roche社製GSJuniorシーケンス解析装置による次世代シーケンスを実施した。具体的には、GS Junior Titanium emPCR Kit (Lib-L)を用いて、メーカーのプロトコールに従ってemPCRを実施した。ビーズとDNAの割合(copyper beads:cpb)0.5で実施した。emPCR後、ビーズエンリッチメントにて回収されたビーズは、シーケンスラン試薬であるGS JuniorTitanium Sequencing KitおよびPicoTiterPlateKitを用いて、メーカーのプロトコールに従ってシーケンスランを実施した。
得られたシーケンスデータ(SFFファイル)をGS Junior付属ソフトウェア(sfffileもしくはsffinfo)により、MID Tag別のリード配列に分類し、Fasta形式のシーケンスファイルを生成した。シーケンスリードの解析は、IMGT(the international ImMuno Gene Ticsinformation system, http://www.imgt.org)データベースから入手されるV、D、J、C配列をリファレンス配列として各リード配列のV、D、J、C配列をアサインした。アサインメントには新規開発ソフトウェア(RepertoireGenesis)を用いた。TCRαにおいては、22833のリードが得られ、16407リード(71.9%)がアサインされた。ユニークリード数は1705リードであった。TCRβにおいては、121080のリードが得られ、81542リード(67.3%)がアサインされた。ユニークリード数は9224リードであった。得られたリードについて、同一のTRAV遺伝子、TRAJ遺伝子およびCDR3配列をもつリードをユニークリードとして、その頻度を調べた(表3-1)。また、同様に、同一のTRBV遺伝子、TRBJ遺伝子およびCDR3配列をもつリードについて頻度を調べた(表3-2)。その結果、TRAレパトアでは、TRAV10、TRAJ15およびCVVRATGTALIFG(配列番号1450)をもつリードが1971リード(12.53%)を占め、特定のTCRを発現する細胞がクローナルに増加している可能性を示唆した。また、TRBレパトアでは、TRBV29-1、TRBJ2-7およびCSVERGGSLGEQYFG(配列番号1500)をもつリードが22568リード(28.57%)を占めた。これらの結果から、TRAV10およびTRAJ15を有するTCRαとTRBV29-1およびTRBJ2-7を有するTCRβからなるTCR分子を発現するT細胞がモノクローナルに増殖している可能性が示唆された。健常人10例とLGL患者の間で種々の多様度指数について比較した(表3-3)。多様度を示すシャノン・ウェーバー指数(H')、シンプソン指数(λ)、逆シンプソン指数(1/λ)およびPielouの指数(J)は健常人と比べ低い値を示し、多様性が減少していることが示された。
本LGL患者においては、薬物療法などの治療を施行した後、TRAV10/TRAJ15/CVVRATGTALIFG(配列番号1450)あるいはTRBV29-1/TRBJ2-7/CSVERGGSLGEQYFG(配列番号1500)を持つシーケンスリードを指標として、微少残存病変の検出ができると期待される。また、リード頻度を用いた定量的解析から白血病細胞に対する治療効果を測ることができると考えられる。また、種々の多様度指数を用いて、クローナリティの増加疾患の有無を推測できる可能性が示唆された。
(表3-1) TRAリード(上位50位)(配列番号1450-1499)






試料:外科手術にて摘出された大腸がん患者の腫瘍組織、健常人末梢血
方法
(大腸がん組織の採取と保存)
60例の大腸がん患者における腫瘍摘出手術において腫瘍組織を採取した。摘出臓器のがん病変部から大豆大の大きさに相当する100mgの組織を採材し、5mm四方に切断したものをただちにRNA安定化試薬(RNAlater(登録商標)、Ambion)に浸漬した。一晩、4℃で保管した後、RNAlater(登録商標)を除去した後-80℃で保管した。
コントロールとして、健常人末梢血細胞を用いた。10例の健常人から全血5mLをヘパリン含採血管に採取し、フィコール密度勾配遠心分離により末梢血単核球細胞(peripheral bloodmononuclear cells, PBMC)を分離した。単離した5×106細胞のPBMCからRNeasyLipid TissueMini Kit (QIAGEN, Germany)を用いて、全RNAを抽出・精製した。取得されたRNAは、吸光度計を用いてA260の吸光度により定量された(表3-3A)。表3-3A 健常人末梢血細胞の全RNA量


がん組織におけるHLAの発現およびHLAハプロタイプを同定するため、HLA-Aタイピングを実施した。RNAlater(登録商標)に浸漬したがん組織を一部取り出し、QIAampDNA Mini Kit(Qiagen, Germany)を用いてゲノムDNAを抽出した。次にWAKFlow HLAタイピング試薬 HLA-A(Wakunaga)を用いて増幅および標識を行い、Luminex(LuminexCorp.)で解析した。その結果、HLA-A2402遺伝子は60検体中25検体でホモもしくはヘテロで発現し
ていた(表3-4)。
表3-4 HLA-A2402を発現した大腸がん組織の一覧


HLA-A2402遺伝子を発現する25検体においてTCRレパトアを解析するため、RNAlater(登録商標)に浸漬した組織を一部取り出し、RNeasyLipidTissue Mini Kit (QIAGEN, Germany)を用いて、全RNAを抽出・精製した。カラムからの溶出はRNAaseフリーの滅菌水50 μLで実施した。各試料から得られたRNA量を表3-5に示した。
表3-5 大腸がん試料の全RNA量


抽出されたRNA試料を用いて、adaptor-ligationPCRを実施した。最初に、相補的DNAを合成するため、BSL-18Eプライマーと3.5 μLのRNAを混和して、70℃で8分間アニーリングした。氷上で冷却後、下記の組成においてRNase阻害剤(RNAsin)の存在下で逆転写反応を行い、相補的DNAを合成した。






二本鎖相補的DNAから、共通アダプタープライマーP20EAとTCRα鎖またはβ鎖C領域特異的プライマー(CB1)を用いて、1stPCR増幅を行った。PCRは次に示す組成で、95℃30秒、55℃30秒、72℃1分のサイクルを20サイクル行った。




Roche社製GSJuniorシーケンス解析装置による次世代シーケンスを実施した。具体的には、GS Junior Titanium emPCR Kit (Lib-L)を用いて、メーカーのプロトコールに従ってemPCRを実施した。ビーズとDNAの割合(copyper beads:cpb)2で実施した。emPCR後、ビーズエンリッチメントにて回収されたビーズは、シーケンスラン試薬であるGS JuniorTitanium Sequencing KitおよびPicoTiterPlateKitを用いて、メーカーのプロトコールに従ってシーケンスランを実施した。
得られたシーケンスデータ(SFFファイル)をGS Junior付属ソフトウェア(sfffileもしくはsffinfo)により、MID Tag別のリード配列に分類し、Fasta形式のシーケンスファイルを生成した。レパトア解析ソフトウェア(RepertoireGenesis)を用いて、IMGTデータベースのリファレンス配列との照合を行い、各リードのAV、BV領域およびAJ、BJ、領域のアサインメントとCDR3配列の決定を行った。
正常コントロールとして、10例の健常人末梢血単核球のTCRシーケンスを調べた。各健常人から得られたTCRαおよびTCRβシーケンスリードについて、V、JおよびCDR3配列を指標にして個体間で重複するリードを検索し、抽出した。TCRα鎖とTCRβ鎖の間で、重複するユニークリード数ならびにその重複ユニーリードをもつ個体数を調べた(表3-6)。TCRβ鎖に比べ、TCRα鎖において著しく重複ユニークリードの数が多く(809対39)、その割合も高かった(2.37%対0.19%)。また、TCRα鎖は最大10個体中8個体に重複するリードが存在し、一方でTCRβ鎖のすべての重複リードは2個体だけに重複していた。これらの結果は、TCRαレパトアは個体間でより類似していることを示唆している。表3-6 健常人における重複ユニークリード数

TCRβ鎖に対比し高い個体間での重複度を示すTCRα鎖について、重複リードの塩基配列を詳細に調べた。その結果、高い重複度を示すTCRリードの多くは、インバリアント鎖を発現するとして知られるナテュラルキラーT細胞(Natural killer T,NKT)あるいは粘膜関連インバリアントT細胞(Mucosal-associated invariant T、MAIT)由来のTCRα遺伝子であることが分かった(表3-7)。NKT細胞はTRAV10(Vα24)-TRAJ18を、MAITはTRAV1-2(Vα7.2)-TRAJ33からなるTCRを主に発現する。最近、MAITのTCRは、MR1分子によって提示された細菌のビタミンB代謝物を認識することが報告され、免疫監視機能における役割が注目されている(Nature. 2012 Nov 29;491(7426):717-23;JExp Med. 2013 Oct21;210(11):2305-20)。重複個体数が4以上の重複リードについて、既報のインバリアントTCRと照合した結果、その45%がインバリアントTCRで占められることが分かった(表3-7)。TCRα鎖において高頻度重複リードが存在するのとは対称的に、TCRβ鎖の重複個体数は最大2であった(表3-8)。従って、TCRαでの高い重複度はインバリアントTCRの存在によると推測される。4個体以上に重複する38種の高頻度重複リードのうち既報のインバリアントTCRと照合できなかったTCRαリードを21種類特定できた(表3-9)。これらは、新規インバリアントTCRである可能性が示唆される。
表3-7 健常人における重複TCRα鎖リード配列





がん患者のがん組織にはがん抗原特異的T細胞が存在し、抗腫瘍効果に重要な働きをすることが知られている。がん抗原特異的TCR遺伝子を同定するため、特定のHLAを有する患者を対象にTCRレパトアを解析して、特定の抗原に反応して増殖するTCR遺伝子を同定する。本実験では、25例の共通のHLA-A2402を有する大腸がん患者のがん組織を用いてTCRレパトア解析を実施し、がん患者サンプル間で重複して存在するユニークリードを検索した(表3-10)。その結果、TCRα鎖においては213リード(1.65%)、TCRβ鎖では49リード(0.11%)が複数の患者に重複して存在することが分かった。健常人と同様に、TCRα鎖においては最大25例中12例に存在する高頻度重複リードが存在する一方で、TCRβ鎖は最大2個体で重複するのみであった。TCRα鎖では、最大12検体の間で共通するリードが存在し、4検体以上のがん組織で重複する7リードの配列は1例を除きMAITに由来するTRAV1-2/TRAJ33を有するTCRα鎖であった(表3-11)。(表3-10 がん組織における重複ユニークリード数とがん特異的リード数)









高頻度に重複するTCRαリードはインバリアントTCRを多く含んでいる。これらの配列は正常コントロールである健常人にも存在し、腫瘍抗原に反応するTCRではない。がん特異的TCRを抽出する目的で、がん組織で重複するリードのうち健常人試料で検出されないものをがん特異的TCRとして分類した(表3-12)。健常人にも存在する重複リードはTCRα鎖では56リード、一方TCRβ鎖ではわずか1リードであった。重複個体数が4個体以上のリードはインバリアントTCRまたは健常人にも存在するリードであった。がん特異的リードは3個体間以内で重複し、TCRα鎖では157リード(1.22%)、TCRβ鎖では48リード(0.11%)検出された。(TCRαβペアリードの推定方法については図49も参照)。
表3-12 がん患者における重複TCRβリード配列とがん特異的TCRβ




抽出されたRNA試料を用いて、adaptor-ligation PCRを実施した。方法は実施例1に示した方法に従って実施した。すなわち、BSL-18Eプライマー(表3-14)とRNAを混和してアニーリング後、逆転写酵素を使って相補鎖DNAを合成した。続いて、二本鎖相補的DNAを合成し、T4 DNA polymeraseによる5’末端平滑化反応を行った。High Pure PCR Cleanup Micro Kit(Roche)によりカラム精製した後、P20EA/P10EAアダプターをLigation反応にて付加した。カラムにより精製されたアダプター付加二本鎖相補的DNAは、NotI制限酵素により消化された。

二本鎖相補的DNAから、表3-14に示す共通アダプタープライマーP20EAとTCRα鎖またはβ鎖C領域特異的プライマー(CA1またはCB1)を用いて、第1のPCR増幅反応の産物である1st PCR増幅を行った。PCRは次に示す組成で、95℃30秒、55℃30秒、72℃1分のサイクルを20サイクル行った。








Ion OneTouch ES(IonTorrent社)を用いてサンプルのエンリッチメントを行う。新しいチューブをチップローダーにセットし、チップアームを取り付ける。次に、下記のMelt-Off溶液を調整する。



本実施例では、Illumina MiSeqシステムを用いたTCRシーケンスでも本発明のシステムが実施可能かを実証する。
健常人から全血5mLをヘパリン含採血管に採取し、フィコール密度勾配遠心分離により末梢血単核球細胞(peripheral blood mononuclear cells, PBMC)を分離した。単離したPBMCからRNeasy Lipid Tissue Mini Kit (Qiagen, Germany)を用いて、全RNAを抽出・精製した。取得されたRNAは、Agilent 2100バイオアナライザ(Agilent)を用いて定量された。
抽出されたRNA試料を用いて、adaptor-ligation PCRを実施した。方法は実施例1に示した方法に従って実施した。すなわち、BSL-18Eプライマー(表3-21)とRNAを混和してアニーリング後、逆転写酵素を使って相補鎖DNAを合成した。続いて、二本鎖相補的DNAを合成し、T4 DNA polymeraseによる5’末端平滑化反応を行った。High Pure PCR Cleanup Micro Kit(Roche)によりカラム精製した後、P20EA/P10EAアダプターをLigation反応にて付加した。カラムにより精製されたアダプター付加二本鎖相補的DNAは、NotI制限酵素により消化された。

二本鎖相補的DNAから、表1に示す共通アダプタープライマーP20EAとTCRα鎖またはβ鎖C領域特異的プライマー(CA1またはCB1)を用いて、第1のPCR増幅反応の産物である1st PCR増幅を行った。PCRは次表3-22に示す組成で、95℃30秒、55℃30秒、72℃1分のサイクルを20サイクル行った。


10倍希釈した2nd PCR増幅産物を鋳型として、図50のようにP20EAアダプタープライマーにP5配列、R1 Seq Primer配列、Index2配列を付加したP5-P20EAプライマーとTCRα鎖またはβ鎖C領域特異的配列にP7配列、R2 Seq Primer配列、Index1配列を付加したP7-CA3またはP7-CB3を用いて、PCR増幅反応を行う。異なるIndex1配列とIndex2配列を使って増幅プライマーに標識し、複数の試料から増幅したTCR遺伝子増幅産物を識別する。使用するプライマー配列は、表3-24に示した。PCRサイクルは、95℃ 30秒、55℃ 30秒、72℃ 1分を10サイクル行う。




E-Gelアガロースゲル電気泳動システムを用いて、増幅したPCR産物を電気泳動する。高感度蛍光染色剤入りプレキャストゲルを電気泳動装置に設置し、2%アガロースゲルにウェル当たり20μLの試料を入れ、電気泳動を行う。500-600bpに相当する目的バンドが溶出された時点で、増幅産物を回収する。回収されたPCR増幅産物はQuant-TTMPicoGreen(登録商標) dsDNA AssayKit (Invitrogen)によりDNA量を測定する。得られたDNA量をもとに、複数の試料を等モル量ずつ混合し、シーケンス反応を行う。
MiSeq サンプルシートを作成する。PhiXコントロールを5-50%の範囲で加え、MiSeq Reagent Kit v.3 (600ccyle、イルミナ社)をインストールしたMiSEQシーケンス装置でシーケンスを開始する。約65時間後に、シーケンスデータを得ることができる。
解析システムの実施例1~4に記載の手法をまとめる実施例として以下に総合した実施例を記載する。
これまでに述べてきたように、近年では、次世代配列決定(NGS)として公知のハイスループット配列決定技術における進歩が急激に進み、ラージスケールのシーケンスデータの分析が可能となった(Shendure J etal. (2008) Nat Biotechnol 26: 1135-1145; Metzker ML et al. (2010) Nat Rev Genet11: 31-46)。いくつかのNGSに基づくTCRレパトア解析システムが、他の研究者らによって開発されてきたが、多くの増幅技術が、各可変領域に特異的な異なるプライマーを含むMultiple PCRに基づいている。そのため、異なる標的遺伝子に対する可変領域特異的プライマー間の示差的なハイブリダイゼーション動態に起因して、バイアスは最も一般的であるため、PCR増幅中のバイアスが回避できない。それゆえ、Multiple PCRアッセイを用いる場合、補正およびさらなる計算的な標準化法が、PCRバイアスを最小化するために必要とされる(Carlson CS etal. (2013) Nat Commun 4: 2680)。プライマーの単一セットの使用は、配列の5’末端が高度に多様である未知の変異体を含むすべてのTCR遺伝子の非バイアスで定量的な増幅を達成する好ましい方法である。T4 RNAリガーゼを含むcDNAの3’末端に対する一本鎖オリゴヌクレオチドアンカーライゲーション(Troutt AB et al.(1992) Proc Natl Acad Sci U S A 89: 9823-9825)、cDNAのホモポリマーテーリング、cDNA末端の5’急速増幅(RACE)(Frohman MA et al. (1988)Proc Natl Acad Sci U S A 85: 8998-9002)およびテンプレートスイッチングPCR(TS-PCRまたはSMART PCR)(Zhu YY et al.(2001) Biotechniques 30: 892-897)は、TCRレパトアを分析するために使用されてきた(Freeman JD etal. (2009) Genome Res 19: 1817-1824; Warren RL et al. (2011) Genome Res 21:790-797)。TS-PCRは、単純で便利だが、TSプライマーは、非特異的にRNAのランダムな領域にアニーリングするか、TSプライマーを繰り返し付加させるため、高いレベルのバックグラウンド増幅が生じる(Alon S et al.(2011) Genome Res 21: 1506-1511; Kapteyn J (2010) BMC Genomics 11: 413)。そこで、本明細書は、TCR転写物ならびにアダプタープライマーおよび定常領域特異的プライマーによるその後のPCR増幅物に由来する二本鎖相補的DNAの5’末端に対するアダプターの付加によって開発されたadaptor-ligation媒介PCR(Tsurutaら(Tsuruta Y et al.(1993) J Immunol Methods 161: 7-21; Tsuruta Y et al. (1994) J Immunol Methods169: 17-23)によって初めて報告された)を記載している。平滑末端化された二本鎖相補的DNAへのadaptor-ligationは、特定のcDNAの配列にほとんど影響されず、他方で、T4 RNAリガーゼを用いた5’adaptor-ligationの効率は、配列依存的である(Jayaprakash AD et al. (2011) Nucleic Acids Res 39:e141)。さらに、T4リガーゼを用いた二本鎖DNAのライゲーションは、ライゲーションアンカー(ligation anchored)PCR(LA-PCR)におけるT4 RNAリガーゼを用いたssDNAライゲーションよりも効率的である。したがって、この非バイアスAL-PCR法は、補正または標準化を必要としないTCRレパトアの正確な分析を可能とする。
(本実施例における実証)
T細胞レセプター(TCR)遺伝子のハイスループット配列決定は、抗原特異性、Tリンパ球のクローナリティおよび多様性の分析に強力なツールとなる。ここで、発明者らは、adaptor-ligation媒介ポリメラーゼ連鎖反応(PCR)と組み合わせた454DNA配列決定技術を用いた新規TCRレパトア解析法を開発した。この方法は、PCRに通常生じるバイアスをかけることなく、SMART PCR法で達成し得る擬似バイアス程度のレベルではなく、真の意味での非バイアスの様式ですべてのTCR遺伝子の増幅が可能となった。
末梢血単核球細胞およびRNA抽出物の単離
インフォームドコンセントを得た後、全血を20例の健常人から回収した。本研究は、独立行政法人国立病院機構相模原病院臨床研究センターの倫理委員会によって承認された。全血10mLをヘパリン処置したチューブに回収した。末梢血単核球細胞(PBMC)をFicoll-Paque PLUSTM(GE Healthcare Health Sciences、Uppsala、Sweden)の密度勾配遠心により単離し、リン酸緩衝食塩水(PBS)で洗浄した。細胞数をカウントして1 × 106細胞をRNA抽出に使用した。全RNAを単離して、RNeasy Lipid Tissue Mini Kit(Qiagen、Hilden、Germany)によりメーカーの説明書に従って精製した。RNA量および純度を、Agilent 2100バイオアナライザ(Agilent Technologies、Palo Alto、CA)を用いて測定した。
全RNA1μgを、Superscript III逆転写酵素(Invitrogen、Carlsbad、CA)を用いて相補的DNA(cDNA)に変換した。poly18およびNotI部位を含むBSL-18EプライマーをcDNA合成に使用した。cDNA合成後、二本鎖(ds)-cDNAを、E.Coli DNA polymerase I(Invitrogen)、E.coli DNAリガーゼ(Invitrogen)、およびRNase H(Invitrogen)を用いて合成した。ds-cDNAをT4 DNAポリメラーゼ(Invitrogen)を用いて平滑末端化した。P10EA/P20EAアダプターをds-cDNAの5’末端に連結させ、その後NotI制限酵素で切断した。MinElute Reaction Cleanup kit(Qiagen)を用いてアダプターおよびプライマーを除いた後、TCRα鎖定常領域特異的プライマー(CA1)またはTCRβ鎖定常領域特異的プライマー(CB1)のいずれかとP20EA(表4-1)とを使用して、PCRを行った。PCR条件は以下のとおりである:95℃(30秒)、55℃(30秒)、および72℃(1分)を20サイクル。2nd PCRを、同じPCR条件を使用して、CA2またはCB2のいずれかとP20EAプライマーとにより行った。

NGS用の増幅産物を、P20EAプライマーと融合タグプライマー(表4-1)とを使用した2nd PCR産物の増幅により調製した。アダプターA配列(CCATCTCATCCCTGCGTGTCTCCGAC)を含む融合タグプライマー、4塩基配列のkey(TCAG)、分子同定(MID)タグ配列(10ヌクレオチド)、およびTCR定常領域特異的配列を、メーカーの説明書に従って設計した。PCR増幅後、増幅産物を分離して、アガロースゲル電気泳動により評価した。得られた断片(約600bp)をゲルから取り除き、QIAEX IIゲル抽出キット(Qiagen)を用いて精製した。精製された増幅産物の量をQuant-iTTM PicoGreen(登録商標)dsDNA Assay Kit(Life Technologies、Carlsbad、CA)によって定量した。10例の健常人から異なる融合タグプライマーを用いて得られた各増幅産物を、等モル濃度で混合した。エマルションPCR(emPCR)を、GS Junior Titanium emPCR Lib-L kit(Roche 454 Life Sciences、Branford、CT)により、増幅産物混合物を使用して、メーカーの説明書に従って行った。
すべてのリード配列をMID Tag配列によって分類した。人工的に付加された配列(タグ、アダプター、およびkey)および低い品質スコアを有する配列を、454配列決定システム上にインストールされたソフトウェアを使用してリード配列の両末端から取り除いた。残りの配列を、TRAVおよびTRAJのTCRα配列に対するアサインメントならびにTRBVおよびTRBJのTCRβ配列に対するアサインメントに使用した。配列のアサインメントを、偽遺伝子を含む54個のTRAV、61個のTRAJ、65個のTRBVおよび14個のTRBJ遺伝子についてのリファレンス配列のデータセット、ならびにImMunoGeneTics information system(登録商標)(IMGT)データベース(http://www.imgt.org)から利用可能なオープンリーディングフレーム(ORF)リファレンス配列のデータセットにおける最も高い同一性を用いた配列決定により行った。データ処理、アサインメント、データ集積を、本発明者らによって独自に開発されたレパトア解析ソフトウェア(Repertoire Genesis、RG)を使用して自動的に行った。RGは、BLATN、自動集積プログラム、TRVおよびTRJの使用についてのグラフィックプログラム、ならびにCDR3の鎖長分布を用いた配列相同性検索についてのプログラムを実行した。クエリー配列とエントリー配列との間のヌクレオチドレベルでの配列相同性を自動的に計算した。感受性および精度を増加させたパラメータ(E値閾値、最小カーネル、ハイスコアセグメントペア(HSP)スコア)を、それぞれのレパトア解析に対して慎重に最適化した。
104位の保存されたシステイン(Cys104)(IMGTの命名)から118位の保存されたフェニルアラニン(Phe118)までの範囲のCDR3のヌクレオチド配列およびその次のグリシン(Gly119)を、推定アミノ酸配列に翻訳した。ユニークシーケンスリード(USR)を、TRV、TRJおよび他のシーケンスリードを含むCDR3の推定アミノ酸配列において、同一性を有さないシーケンスリードとして定義した。同一のUSRのコピー数を、各試料においてRGソフトウェアによって自動的にカウントし、その後、コピー数の順位でランク付けした。全シーケンスリードにおけるTRAV、TRAJ、TRBVおよびTRBJ遺伝子を含むシーケンスリードの発生頻度の割合を計算した。
試料間で共有された配列を検索するために、個人のUSRの「TRV遺伝子名」_「CDR3領域の推定アミノ酸配列」_「TRJ遺伝子名」の文字列(例えば、TRBV1_CASTRVVJFG_TRBJ2-5)を、TCR識別子として使用した。試料中のTCR識別子を、すべての他の試料からのリードデータセットにおいて検索した。
ディープシーケンスデータにおいてTCR多様性を推定するために、いくつかの多様性指数、シンプソン指数およびシャノン・ウェーバー指数を、Rプログラムにおけるveganパッケージの関数「多様性(diversity)」を使用して計算した。これらの指数を、生態学における生物学的多様性についての尺度として、試料当たりの種の数および試料当たりの個人の数に基づいて計算した。ディープシーケンスデータにおいて、USRおよびコピー数を、種および個人にそれぞれ使用した。シンプソン指数(1-λ)を以下:



統計学的有意性を、GraphPad Prismソフトウェア(version4.0、San Diego、CA)を使用して、ノンパラメトリックなMann-WhitneyのU検定によって試験した。p<0.05の値を統計学的に有意であるとみなす。
レパトア解析ソフトウェア
本研究において開発されたクラウドベースのソフトウェアプラットフォームであるRGは、TCRレパトア解析のための高速で、正確で、便利な計算システムである。RGは、(1)V、DおよびJセグメントのアサインメント、(2)配列同一性の計算、(3)CDR3配列の抽出、(4)同一リードのカウント、(5)アミノ酸翻訳、(6)フレーム解析(ストップおよびフレームシフト)、ならびに(7)CDR3の長さの分析についての統合されたソフトウェアパッケージを提供する。NGSシーケンサから配列決定データをアップロードした後、V、DおよびJセグメントを、最適化されたパラメータを用いてそれらの配列類似性に基づいて同定することができる。リード数を自動的に集約し、その後、加工されたデータ、集計表、およびグラフを容易にダウンロードすることができる。
本発明者らは、20例の健常人由来のPBMCにおけるTCRαおよびTCRβ遺伝子のハイスループット配列決定を行った。全部で172,109個および91,234個のシーケンスリードを、RGプログラムを使用して、それぞれ、TCRαおよびTCRβのレパトア解析に対してアサインした(表4-2および4-3)。




TCRシーケンスリードにおけるTRVおよびTRJ遺伝子の使用を決定するために、TRVまたはTRJそれぞれを有するUSRのコピー数(リード数)をカウントした。個々のUSRをコピー数の順番でランク付けし、TRVおよびTRJそれぞれの頻度パーセンテージを計算した(図51および図52)。TCRαレパトアに関して、8つの偽遺伝子(AV8-5、AV11、AV15、AV28、AV31、AV32、AV33およびAV37)は、健常人において発現されなかった。ORFとして分類されるAV8-7(IMGTによるスプライシング部位、組換えシグナルおよび/または制御エレメントにおける変更に基づいて定義される)は、わずかに発現した(20個体のうち11個体において43リード)。AV18およびAV36の発現(機能遺伝子として分類される)は、健常人において観察されなかった。さらに、機能遺伝子であるAV7およびAV9-1は、1個体(9リード)および2個体(3リード)において、それぞれ十分に発現されなかった。ORF遺伝子として分類される8つのAJ遺伝子(AJ1、AJ2、AJ19、AJ25、AJ35、AJ58およびAJ61)のうち、AJ35およびAJ58の発現は、20個体すべてにおいて観察された。これらのうち、AJ25およびAJ61は、3個体(21リード)および7個体(35リード)において、それぞれわずかに発現した。AJ1、AJ2、AJ19およびAJ59は、いずれの個体にも存在しなかった。いずれの個体においても3つの偽遺伝子、AJ51、AJ55およびAJ60の発現は存在しなかった。機能遺伝子AJ14を、3個体から3リードだけ検出した。
41個のTRAVの50個のTRAJでの遺伝子組換え(偽遺伝子、ORF、および十分に発現されない遺伝子を除く)は、総数2,050個のAV-AJ組換えを生成することが可能であり(図53)、そのうち1,969個のAV-AJ組換え(96.0%)を20個体において検出した。これは、ほとんどすべてのAV-AJ組換えが、制限されずにTCR転写物において使用されたことを示した。特に、AV1-1~AV6遺伝子を、AJ50~AJ58で優先的には組換えられず、同様に、AV35~AV41遺伝子のAJ3~AJ16での組換えは、ほとんど観察されなかった。これらの遺伝子セグメントの染色体上の位置を考慮すると、これらの結果は、AV-AJ組換えは、近位AV遺伝子と遠位AJ遺伝子との間および遠位AV遺伝子および近位AJ遺伝子との間で、ほとんど起こらないことを示した。
全TCR転写物におけるTRVおよびTRJの使用を明らかにするため、TRVまたはTRJそれぞれを有するUSRの発生頻度を計算した(図51および図52)。いくつかのTRAV遺伝子における優先的使用は、ハイブリダイゼーションに基づく定量アッセイを使用して得られた以前の結果(6)と類似していた。いくつかのTRBV遺伝子をTRBVレパトアにおいて多く使用した。上位3位のTRAV9-2(ArdenによるBV4S1)、TRBV20-1(BV2S1)およびTRBV28(BV3S1)は、全シーケンスリードの3分の1を占めた。これは、マイクロプレートハイブリダイゼーションアッセイを使用した本発明者らの以前の研究で得られた結果(6)と似ていた。遺伝子の使用は、TRBJ遺伝子の間でかなり変動した。TRBJ2-1およびTRBJ2-7は非常に発現され、他方で、TRBJ1-3、TRBJ1-4、TRBJ1-6、TRBJ2-4およびTRBJ2-6の発現は低かった。
TRV遺伝子とTRJ遺伝子との組み合わせを有するTCRの使用を可視化するために、本発明者らは、TCRレパトアの3D描写を作製した(図54および図55)。3Dイメージの利点は、TRV遺伝子とTRJ遺伝子との特定の組み合わせの優位およびTCRの多様性の程度を容易に観察し得ることである。TCRβについては、TRBV遺伝子とTRBJ遺伝子との間の組換えの優先的な使用はほとんど存在しなかった。各組換えの頻度は、TRBVまたはTRBJの使用に依存した。BV29-1/BJ2-7、BV29-1/BJ2-1、BV29-1/BJ2-3およびBV20-1/BJ2-7は、すべての組み合わせにおいて高い頻度で使用され、他方で、その他は低い頻度で発現した。対照的に、TCRαレパトアの3Dイメージングは、TRAVおよびTRAJの広範な分布で、低いレベルでの発現を示した。占有率は、すべての組み合わせについて1%未満だった。注目すべきことに、AV1-2およびAJ33を有するTCRリードは、すべての健常人において高度に発現した(平均±SD:0.99±0.85)。
CDR3サイズスペクトラタイピング(Yassai M et al.(2000) J Immunol 165: 3706-3712; Yassai M et al. (2002) J Immunol 168:3801-3807)またはイムノスコープ解析(Pannetier C etal. (1993) Proc Natl Acad Sci U S A 90: 4319-4323; Pannetier C et al. (1995)Immunol Today 16: 176-181)と呼ばれるCDR3の鎖長分布の分析を効率的に使用し、TCRレパトアの多様性を推定した。この技術は、ゲル電気泳動による、CDR3配列を含むPCR増幅産物の実際のピーク分布に基づいている。本研究において、保存されたCys104(IMGTの命名)から118位の保存されたフェニルアラニン(Phe118)の範囲のTCRの決定されたヌクレオチド配列の長さを自動的に計算した。これは、NGSデータを使用することによってTCRの多様性およびクローナリティを推定するための可視的に容易な方法を提供する。RGは、各V領域についてのデジタルCDR3鎖長分布を表す図を生成することができる。TCRαおよびTCRβ両方のCDR3鎖長分布は、通常の分布と似ていたが、完全に対称的というわけではなかった(図56)。CDR3の鎖長は、TCRαにおいてTCRβよりも短く(平均±SD:41.2±8.3対42.8±6.1)、TCRαは、TCRβよりも正の歪度を有しており(歪度指数:11.1対5.41)、TCRαにおける分布が左側に集中したことを示した。さらに、TCRαは、TCRβよりも正の尖度を有しており、TCRαにおいて高い尖度を示した(尖度指数:282.4対176.7)。
TCRレパトアの多様性を示すために、本発明者らは、USRの平均コピー数および多様性指数(シンプソン指数およびシャノン・ウェーバー指数など)を計算した(図57)。USRの平均コピー数は、TCRαとTCRβとの間で顕著に異なった(2.0±0.72対1.70±0.57)。さらに、TCRαとTCRβとの間のシンプソン指数の逆数(D)およびシャノン・ウェーバー指数(H)において顕著な違いはなかった(D:710.3±433.0対729.7±493.9、H:7.02±0.33対6.97±0.43)。これらの結果は、健常人におけるTCRαおよびTCRβについての免疫多様性に違いがないことを示した。
個人間の遺伝子の使用の相関を明らかにするために、TRVおよびTRJの各々の頻度パーセンテージを、散布図によってすべての個体のペアの間でプロットした(図60)。各ペア間のスピアマンの相関係数を計算した。一致相関係数は、TRAVにおいてTRBVよりも低く(平均±SD、TRAVにおいて0.86±0.059、TRBVにおいて0.89±0.038、p<0.001)、TRAJにおいてTRBJよりも低かった(TRAJにおいて0.74±0.095、TRBJにおいて0.91±0.063、p<0.001)。これらの結果は、健常人の間のTRVおよびTRJの発現レベルが、TCRαと比較して、TCRβにおいて個人間でより類似していたことを示した。


少数のTCR配列は、異なる健常人の間で共有され、それらの共有されたTCRはパブリックTCRと呼ばれる。対照的に、ほとんどのTCRは各健常人に対して特異的であった(プライベートTCR)。20例の健常人におけるパブリックTCR配列を同定するために、本発明者らは、2以上の健常人の間で共有されたTCRαリードおよびTCRβリードを検索した。20例の健常人において、3041個のパブリックTCRαおよび206個のパブリックTCRβ配列が、それぞれ90,643個および57,982個のUSRから得られた(表4-8)。

パブリックTCRαは、健常人由来のPBLにおいて高頻度で観察された。パブリックTCRαの起源を決定するために、本発明者らは、以前に報告されたパブリックTCRαのCDR3配列を調べた。興味深いことに、複数の個体に対して共通であったパブリックTCRα配列は、iNKT細胞またはMAIT細胞を示す高い割合のインバリアントTCRαを含んでいた(表4-9)。


ハイスループット配列決定技術は、幅広い種類のNGSプラットフォームの開発により大きな飛躍をしてきた。NGSは、莫大な量のシーケンスデータの取得を促進するが、未だに、全ゲノムまたは遺伝子ライブラリの代わりに、目的の配列遺伝子のPCR増幅または遺伝子エンリッチメントを必要とする。多くの遺伝子セグメントの再構成によって生成された不均一なTCRまたはBCR遺伝子については、多くの遺伝子特異的プライマーによるマルチプレックスPCRが広く使用されていた。しかし、複数のプライマーの使用により、それぞれの遺伝子の間で増幅バイアスが生じ、遺伝子頻度の正確な推定を妨害する。ここで、本発明者らは、NGSに基づくTCRレパトア解析のために、非バイアスPCR技術であるadaptor-ligation媒介PCRを使用した。この方法は、単一セットのプライマーを使用し、PCRバイアスをかけることなく、理論上すべてのTCR遺伝子の増幅を可能とする。したがって、この方法は、幅広い試料からのTCR遺伝子のそれぞれの存在量を正確に推定するのに最も適している。
本実施例では、実際に応用した具体的な態様としては、BCRレパトア解析を利用したヒト型抗体の単離例を行う。
ヒト化NOGマウスを用いたヒト型抗イディオタイプ抗体の取得
1.B細胞系白血病あるいは悪性リンパ腫患者においては腫瘍細胞に由来するモノクローナルBCRの高度な発現が観察される。
2.B細胞系白血病あるいは悪性リンパ腫患者の末梢血単核球細胞を採取し、本編記載のBCRレパトア解析を実施する。決定された数万リードの遺伝子配列からランキング最上位に位置し、優位に存在する腫瘍細胞由来の免疫グロブリンH鎖遺伝子を同定する。
3.決定された免疫グロブリンH鎖遺伝子配列を使って、多様性の高いCDR3領域のアミノ酸配列を推定し、その配列と同一のペプチドを合成する。
4.200μgの合成ペプチドを完全フロイントアジュバント(CFA,シグマ・アルドリッチ社)とよく混和し、シリンジを用いてヒト化NOGマウスの皮下に投与する(初回免疫)。同様に、対照マウスに対しPBSを投与する。さらに、初回投与後2週後に再度同量の抗原ペプチドを投与する。
5.初回免役から4週後に、マウスよりリンパ節または脾臓を摘出する。リン酸緩衝液(PBS、Invitrogen社)中で組織を細断し、セルストレイナー(0.75μm、BD社)でフィルターろ過し、シングルセルを調整する。
6.得られた細胞をTrizol溶液(Invitrogen社)に溶解し、本特許記載のBCRレパトア解析法により遺伝子配列を決定する。
7.得られた数万リードのBCR遺伝子配列について、その存在頻度(リード数)順にソートし、ランキング上位に位置する免疫グロブリンH鎖およびL鎖遺伝子配列を決定する。対照としてPBSを投与したマウスのリードランキングと比較して、存在頻度が有意に高い免疫グロブリンH鎖およびL鎖遺伝子配列を選択する。
8.得られた免疫グロブリンH鎖およびL鎖遺伝子配列について、P20EAアダプタープライマーとC末端primerを用いて全長免疫グロブリンH鎖および全長L鎖遺伝子をPCR増幅する。抗体発現ベクターであるpEHX1.1(抗体H鎖用、TOYOBO社)およびpELX2.2(抗体L鎖用、TOYOBO社)にLigation Kit(TAKARA社)を用いてLigation反応により、それぞれ全長遺伝子をベクター内のマルチクローニング部位に挿入する。大腸菌TOP10細胞株(One Shot(登録商標) TOP10 Chemically Competent E. coli、Invitrogen社)を形質転換し、H鎖発現プラスミドおよびL鎖発現プラスミドを得る。
9.両プラスミドについてBglIIおよびEcoRI制限酵素で二重消化し、次いでH鎖プラスミドのBglII-EcoRI切断部位に、L鎖プラスミドのBglII-EcoRI断片を挿入し、H鎖およびL鎖を共発現する抗体発現プラスミドを得る。
10.抗体発現プラスミドをQIAGEN Plasmid Mini kitを用いて大腸菌より抽出・精製し、TransIT(登録商標)-CHO Transfection Kit(TAKARA社)を用いてCHO細胞に導入する。
11.形質転換した抗体発現CHO細胞株を拡大培養し、培養上清を回収して、ProteinAアガロースアフィニティーカラム(HiTrap Protein A HP Columns、GEヘルスケア社)を用いて、使用方法に従い精製する。
12.得られた抗体蛋白量を吸光度計を用いて測定した後、抗原ペプチドとの結合反応性をELISA法で検証する。
本実施例では、本発明のレパトア解析を用いたがんイディオタイプペプチド感作免疫細胞療法での実証例を行う。以下にその手順を説明する(図62参照)。
(1)悪性リンパ腫患者の全血10mLを採血し、Ficoll-Paque比重遠心分離(GEヘルスケアバイオサイエンス、17-1440-02)により末梢血単核球(PBMC)を分離する。
(2)患者PBMCよりTrizol Reagent(Invitrogen社)を用いて全RNAを抽出する。
(3)RNAから逆転写酵素(Superscript II, Invitrogen, 18064-014)によりcDNA合成し、その後、DNA Polymerase(Invitrogen, 18010-017)、E.coli Ligase(Invitrogen, 18052-019)、RNase H(Invitrogen, 18021071)によりdsDNA合成し、さらにT4 DNA Polymerase(Invitrogen, 18005-025)により末端の平滑化を行う。T4 ligase(Invitrogen, 15224-025)によりP20EA/P10EA adaptorのライゲーション反応(調製実施例2等を参照)を行なった後、NotI(TaKaRa, 1166A)で消化を行なう。
(4)BCR遺伝子のIgMのC領域特異的プライマー(CM1(配列番号5))とP20EAアダプター(配列番号2)により1st PCRを、CM2(配列番号6)とP20EAプライマー(配列番号2)により2nd PCRを実施する。PCR反応は、95℃ 30秒、55℃ 30秒、72℃ 1分のサイクルをそれぞれ20サイクル行う。
(5)2nd PCR産物からプライマーを除去するため、High Pure PCR Cleanup Micro Kit(Roche)によりカラム精製を行った後、P20EAプライマー(配列番号2)にアダプターB配列(配列番号1375)を付加したB-P20EAプライマー(配列番号4)とIgM C領域特異的プライマー(CM3にアダプターA配列(配列番号39)および同定配列であるMID Tag配列(表1-6を参照)を付加したGS-PCRプライマー(配列情報は表1-1を参照)を用いてPCRを行う。
(6)GS-PCR増幅の後、2%アガロースゲル電気泳動を実施し、目視化において目的のサイズ(500bp~700bp)のバンドを切り出し、QIAEX II Gel Extraction Kit(QIAGEN)を用いて精製した。回収されたDNAをQuant-iTTM PicoGreen(登録商標)dsDNA Assay Kit(Invitrogen)を用いてDNA量を測定した。1000万DNAをエマルジョンPCRに用い、Roche社製次世代シーケンス解析装置(GS Juniorベンチトップシステム)によるシーケンス解析を行う。
(7)得られた配列データについて本発明において新規に開発したTCR/BCRレパトア解析ソフトウェア(Repertoire Genesis、本明細書解析試験例、解析実施例1~5等参照)を用いて、V及びJ配列のアサイメント並びにCDR3領域の推定アミノ酸配列を決定する。同時に、同一の塩基配列についてそのコピー数を集計し、出現頻度によるランキングを行なう。
(8)ランキング最上位のBCR遺伝子を決定し、そのBCRのリード数が全体の10%以上を占めることを著明に高いとして確認し、該BCR遺伝子を腫瘍由来BCR遺伝子として同定する。
(9)該腫瘍由来BCR遺伝子の推定アミノ酸配列について、HLA結合ペプチド予測プログラムであるBIMAS(www-bimas.cit.nih.gov/)を用いて、HLA結合ぺプチドを予測する。特に条件指定がなければデフォルトの条件を用いる。該BCRアミノ酸配列と患者HLA型をBIMASに入力し、CDR3アミノ酸配列内のペプチド、もしくはCDR3アミノ酸配列の一部を含むペプチドの中で、最も高いスコアを示した推定HLA結合ペプチドを決定する。
(10)ハイスコアHLA結合ペプチドを個別化がんペプチドとして用いて、細胞傷害性T細胞(CTL)療法または樹状細胞(DC)ワクチン療法を実施する。ここでは、DCワクチン療法を実施する。
(11)個別化がんペプチド配列を、全自動ペプチド合成装置(Protein Technologies, Inc.社)を用いて化学合成する。ペプチドは収量1mg以上、純度95%以上のものを獲得する。ペプチドは50%DMSOに溶解し、-20℃で保存する。
(12)成分採血装置(テルモアフェレーシス装置AC-555)を実施し、がん患者より単球細胞を分離する。単球を含む細胞をAIM-V培地(Invitrogen, 12055091)で洗浄した後、細胞数を計測する。
(13)プラスティックプレート上で付着しなかった細胞を除去した後、2000U/mLの顆粒球単球コロニー刺激因子(GM-CSF、和光純薬)及び400U/mLのインターロイキン-4(IL-4、ペトロテック)を含むAIM-V培地で約1週間培養し、樹状細胞(DC)に分化誘導させる。
(14)MHCクラスI&II分子、CD40,CD80またはCD86の発現をFACS解析を用いて調べることでDCに分化したことを確認した後、2×106 細胞に対して20μg/mLの個別化がんペプチドを添加し、刺激因子(ピシバニール(OK-432), ピシバニール注射用0.5KE, 中外製薬)とともにAIM-V培地(上記同様)でさらに1日培養する。
(15)ペプチド刺激したDC細胞を回収し、生理食塩水で洗浄した後、該がん患者に点滴による静脈注入を行なう。
本実施例により、以下が達成される。
(1)悪性リンパ腫患者の末梢血においる次世代BCRレパトア解析によって、全体のBCRリードのうち50%以上を占める1種のIgM免疫グロブリン重鎖及び1種のIgM免疫グロブリン軽鎖が同定される。
(2)これら免疫グロブリン遺伝子のCDR3領域をRepertoire Genesisプログラムで特定される。
(3)BIMASプログラムに患者HLA型(例:HLA-A*02)及びIgM免疫グロブリン重鎖CDR3アミノ酸配列を入力し、結合スコアが最もハイスコアを示すペプチド配列が選択される。
(4)このペプチドを個別化がんペプチドとして、全自動ペプチド合成装置にて化学合成し、患者由来のDCをペプチドがin vitroで刺激活性化される。
(5)個別化ペプチド刺激DC細胞を患者に静脈移入し、腫瘍細胞数の減少、臨床症状の改善を見ることができる。
(考察)
本実施例から、本発明により以下のような効果が達成されることが理解される。
(1)患者癌細胞由来のBCR配列を使って、治療用個別化がんぺプチドを作製することができ、HLA型や抗原の発現に関係なく幅広い患者にDC療法やCTL療法を行なうことができる。
(2)患者のHLAに適合したペプチドを用いるため、より患者に適合し、癌細胞に特異性の高い、効果的なDC療法やCTL療法が実現できる。
(3)BCR解析から得られた遺伝子配列から直接抗原ペプチドを化学合成できるため、
安全性が高く、抗原同定などの必要がない。
本実施例では、本発明のレパトア解析を用いた改良型CTL法での実証例を行う。以下にその手順を説明する(図63参照)。
(1)応用実施例2(1)~(9)に記載の方法でがんイディオタイプペプチドを同定する。
(2)既存のがんペプチド(NY-ESO-1ペプチド)またはがんイディオタイプペプチド(1)により同定されたペプチド)について、全自動ペプチド合成装置(Protein Technologies,Inc.社)を用いて化学合成する。ペプチドは収量1mg以上、純度95%以上のものを獲得する。ペプチドは50%DMSOに溶解し、-20℃で保存する。
(3)該がん患者より20mLの末梢血を採血し、Ficoll-Paque比重遠心分離(応用実施例2を参照)により末梢血単核球(PBMC)を分離する。
(4)CD8+T細胞分離用磁気ビーズ(Miltenyi Biotech)もしくはフローサイトメトリー装置(FACS Aria II, Beckton Dickinson)を用いて、CD8+T細胞を分離する。
(5)成分採血装置(テルモアフェレーシス装置AC-555)またはPBMCより分離した単球細胞を培養プレート(100mm ディッシュ、コーニング、353003)で培養し、非付着系の細胞を除去する。
(6)付着した単球細胞を、2000U/mLの顆粒球単球コロニー刺激因子(GM-CSF、和光純薬)及び400U/mLのインターロイキン-4(IL-4、ペトロテック)を含むAIM-V培地(応用実施例2と同じ)で約1週間培養し、樹状細胞(DC)に分化誘導させる。
(7)DCに分化したことを確認した後、2×106細胞に対して20μg/mLのペプチド(応用実施例2の「最も高いスコアを示した推定HLA結合ペプチド」)を添加し、刺激因子(ピシバニール(OK-432), ピシバニール注射用0.5KE, 中外製薬)とともにAIM-V培地でさらに1日培養する。
(8)さらに、DC培養液に対し、20μg/mLの合成ペプチド(応用実施例2の「最も高いスコアを示した推定HLA結合ペプチド」)、及び上記(3)で分離した2×106/mLのCD8+T細胞とAIM-V培地(応用実施例2等を参照)にて刺激培養する。
(9)抗原刺激により増殖したCD8+T細胞をプラスティック培養プレート(100mm ディッシュ、コーニング、353003)((5)のプレートと同様)に付着したDCと分離した後に、さらに5μg/mLの抗CD3抗体(OKT3、オルソクローンOKT3、ヤンセンファーマ)及び200U/mLのインターロイキン-2(IL-2)(Roche Applied Science、10799068001)の存在下で拡大培養する。
(9)活性化CD8+T細胞をCTL細胞として回収した後、生理食塩水にて洗浄した後、該がん患者に点滴による静脈注入を行なう。
本実施例により、以下が達成される。
(1)悪性リンパ腫患者の腫瘍細胞由来BCR遺伝子のCDR3領域からHLA結合ペプチドが同定される。
(2)患者末梢血からCD8+T細胞分離用磁気ビーズにより2x106細胞のCD8陽性細胞を回収した。純度は98%である。
(3)患者単球由来DC細胞、CD8+細胞、ペプチドとの混合培養で抗原刺激を行い、さらに、抗CD3抗体、IL-2の存在下での拡大培養で、CD8+CTL細胞を50倍まで増殖することができる。
(4)培養CTL細胞を患者に静脈移入し、腫瘍細胞数の減少、臨床症状の改善を見ることができる。
本実施例から、本発明により以下のような効果が達成されることが理解される。
(1)患者癌細胞由来のBCR配列を使って、治療用個別化がんぺプチドを作製することができ、HLA型や抗原の発現に関係なく幅広い患者にCTL療法を行なうことができる。
(2)患者のHLAに適合したペプチドを用いるため、より患者に適合し、癌細胞に特異性の高い、効果的なCTL療法が実現できる。
(3)BCR解析から得られた遺伝子配列から直接抗原ペプチドを化学合成できるため、
安全性が高く、抗原同定などの必要がない。
本実施例では、本発明のレパトア解析を用いたDCワクチン療法の実証例を行う。以下にその手順を説明する(図64参照)。
(1)応用実施例2(1)~(9)に記載の方法でがんイディオタイプペプチドを同定する。
(2)既存のがんペプチド(NY-ESO-1ペプチド)またはがんイディオタイプペプチド(1)により同定されたペプチド)について、全自動ペプチド合成装置(Protein Technologies,Inc.社)を用いて化学合成する。ペプチドは収量1mg以上、純度95%以上のものを獲得する。ペプチドは50%DMSOに溶解し、-20℃で保存する。がん患者より成分採血(アフェレーシス)により単球細胞を分離する。
(3)成分採血装置(テルモアフェレーシス装置AC-555)を実施し、該がん患者より単球細胞を分離する。単球を含む細胞をAIM-V培地(応用実施例2等を参照)で洗浄した後、細胞数を計測した。
(4)プラスティックプレート(100mm ディッシュ、コーニング、353003)上で付着しなかった細胞を除去した後、2000U/mLの顆粒球単球コロニー刺激因子(GM-CSF、和光純薬)及び400U/mLのインターロイキン-4(IL-4、ペトロテック)を含むAIM-V培地(応用実施例2参照)で約1週間培養し、樹状細胞(DC)に分化誘導させる。
(5)MHCクラスI&II分子、CD40,CD80またはCD86の発現をFACSにて調べることでDCに分化したことを確認した後、2×106細胞に対して20μg/mLのペプチド((2)で合成したペプチド)を添加し、刺激因子(ピシバニール(OK-432), ピシバニール注射用0.5KE, 中外製薬)とともにAIM-V培地(応用実施例2等を参照)でさらに1日培養する。
(6)ペプチド刺激したDC細胞を回収し、生理食塩水で洗浄した後、該がん患者に点滴による静脈注入(テルフュージョン輸液システム、テルモ)を行なう。
本実施例により、以下が達成される。
(1)悪性リンパ腫患者の腫瘍細胞由来BCR遺伝子のCDR3領域からHLA結合ペプチドを同定する。
(2)患者末梢血から単球を分離し、分化培養培地で培養することで、MHC DR+, CD40+,CD80/CD86+細胞を検出し、単球からDCへの分化が確認する。
(3)ペプチド刺激DC細胞を患者に静脈移入し、腫瘍細胞数の減少、臨床症状の改善を見ることができる。
本実施例から、本発明により以下のような効果が達成されることが理解される。
(1)患者癌細胞由来のBCR配列を使って、治療用個別化がんぺプチドを作製することができ、HLA型や抗原の発現に関係なく幅広い患者にDC療法を行なうことができる。
(2)患者のHLAに適合したペプチドを用いるため、より患者に適合し、癌細胞に特異性の高い、効果的なDC療法が実現できる。
(3)BCR解析から得られた遺伝子配列から直接抗原ペプチドを化学合成できるため、安全性が高く、抗原同定などの必要がない。
本実施例では、本発明のレパトア解析を用いた患者自己免疫細胞療法の実証例を行う。以下にその手順を説明する(図65参照)。
(1)応用実施例2(1)~(9)に記載の方法でがんイディオタイプペプチドを同定する。
(2)既存のがんペプチドまたはがんイディオタイプペプチド(1)により同定されたペプチド)(について、全自動ペプチド合成装置(Protein Technologies,Inc.社)を用いて化学合成する。ペプチドは収量1mg以上、純度95%以上のものを獲得する。ペプチドは50%DMSOに溶解し、-20℃で保存する。
(3)該がん患者より20mLの末梢血を採血し、Ficoll-Paque比重遠心分離により末梢血単核球(PBMC)を分離する。
(4)CD8+T細胞分離用磁気ビーズ(Miltenyi Biotech)もしくはフローサイトメトリー装置(FACS Aria II, Beckton Dickinson)を用いて、CD8+T細胞を分離する。
(5)成分採血装置(テルモアフェレーシス装置AC-555)またはPBMCより分離した単球細胞を培養プレート(100mm ディッシュ、コーニング、353003)で培養し、非付着系の細胞を除去する。
(6)付着した単球細胞を、2000U/mLの顆粒球単球コロニー刺激因子(GM-CSF、和光純薬)及び400U/mLのインターロイキン-4(IL-4、ペトロテック)を含むAIM-V培地(応用実施例2等と同じ)で約1週間培養し、樹状細胞(DC)に分化誘導させる。
(7)DCに分化したことを確認した後、2×106細胞に対して20μg/mLのペプチド((2)で合成したペプチド)を添加し、刺激因子とともにAIM-V培地でさらに1日培養する。
(8)さらに、DC培養液に対し、20μg/mLの合成ペプチド((2)で合成したペプチド)、及び上記(3)で分離した2×106/mLのCD8+T細胞とAIM-V培地(応用実施例2等と同じ)にて刺激培養する。
(9)活性化CD8+T細胞とペプチド刺激DC細胞を共に回収した後、生理食塩水にて洗浄した後、該がん患者に点滴による静脈注入を行なう。
本実施例により、以下が達成される。
(1)悪性リンパ腫患者の腫瘍細胞由来BCR遺伝子のCDR3領域からHLA結合ペプチドを同定する。
(2)患者末梢血からCD8+T細胞分離用磁気ビーズにより2x106細胞のCD8陽性細胞を回収する。純度は98%以上である。
(3)患者末梢血から単球を分離し、分化培養培地で培養することで、MHC DR+, CD40+,CD80/CD86+であるDCへの分化が確認する。
(4)患者単球由来DC、CD8+細胞、ペプチドを混合培養することで、腫瘍特異的CTL及びDCを増殖することができる。
(5)ペプチド刺激CD8+細胞とDC細胞を共に患者に静脈移入し、腫瘍細胞数の減少、臨床症状の改善を見ることができる。
本実施例から、本発明により以下のような効果が達成されることが理解される。
(1)患者癌細胞由来のBCR配列を使って、治療用個別化がんぺプチドを作製することができ、HLA型や抗原の発現に関係なく幅広い患者に患者自己免疫細胞療法を行なうことができる。
(2)患者のHLAに適合したペプチドを用いるため、より患者に適合し、癌細胞に特異性の高い、効果的な患者自己免疫細胞療法が実現できる。
(3)BCR解析から得られた遺伝子配列から直接抗原ペプチドを化学合成できるため、安全性が高く、抗原同定などの必要がない。
(4)DC細胞とCTL細胞の相乗効果が期待でき、高い治療効果が見込める。
本実施例では、本発明のレパトア解析を用いたオーダーメイドがん特異的T細胞受容体遺伝子の単離、in vitro抗原刺激によるがん特異的TCR遺伝子の単離の実証例を行う。以下にその手順を説明する(図66参照)。
(1)がん患者から常法により腫瘍細胞を摘出する。
(2)該患者由来腫瘍細胞を培養培地(RPMI1640,11875-093, Invitrogen、以下「培養液」と称することもある)中で細断した後、0.70μmのフィルター(Falconセルストレイナー、コーニング)でろ過することにより、単一細胞に分離する。培養液中に10μg/mlのマイトマイシンC(注射用マイトマイシンC、協和発酵キリン)により37℃で2時間不活化処理をする。
(3)該がん患者の全血10mLからFicoll-Paque比重遠心分離により末梢血単核球細胞(PBMC)を分離する。PBMCは培養培地(RPMI1640)で洗浄後2×106/mLの濃度で懸濁する。
(4)PBMCの一部(1×106)を未処理コントロール試料として、細胞よりRNAをTrizol RNA抽出キット(Invitrogen)により抽出する。
(5)不活化腫瘍細胞と末梢血細胞を低濃度IL-2の存在下10%FCS(16000-044, Invitrogen)を含むRPMI1640培地(RPMI1640,11875-093, Invitrogen)で1週間培養することにより腫瘍特異的T細胞を抗原刺激し、細胞増殖させる。
(6)T細胞活性化の後、培養培地から生細胞を回収し、PBS(045-29795, 和光純薬)にて洗浄後、細胞よりRNAを抽出する。
(7)(4)および(6)で抽出したRNA試料を用いて、本発明の次世代レパトア解析を実施する(その条件は、本明細書解析試験例、解析実施例1~5に記載のものを使用することができる。)
(8)本発明の次世代レパトア解析により得られたTCR遺伝子配列データについて、コントロール試料に比し刺激試料で大きく増加したTCR遺伝子を抽出し、それらをランキング処理した後に、上位のTCRαおよびTCRβ遺伝子を選択する。
(9)これらの全長TCRαおよびTCRβ遺伝子をクローニングし、遺伝子発現用レトロウイルスベクター(Retro-X Vectors and Systems, Clonetech)にそれぞれ導入する。
(10)(9)で調製したこれらTCRα及びTCRβ組換えプラスミドベクターを用いてパッケージング細胞GP2-293細胞株(631458, Clonetech)を形質転換し、遺伝子導入用レトロウイルスを作製する。
(11)成分採血装置(テルモアフェレーシス装置AC-555)により分離したリンパ球細胞を用いて、遺伝子組換えTCRαレトロウイルス及び遺伝子組換えTCRβレトロウイルスを単独で、連続して感染させることにより、機能的αβTCRを発現するリンパ球集団を得る。
(12)細胞表面におけるTCRα/TCRβヘテロダイマーの発現とその陽性細胞の割合をFACS(応用実施例5を参照、同じ条件を使用することができる)により確認する。
(13)目的のTCRα/TCRβを発現する腫瘍特異的患者リンパ球を患者に細胞移入する
(結果)
本実施例により、以下が達成される。
(1)患者腫瘍組織で刺激した試料とコントロール試料との対比から、腫瘍組織で増加するTCR遺伝子を選択し、ランキングした結果、末梢血細胞に多く存在するTCRが除かれ、腫瘍特異的TCR遺伝子が多数抽出される。
(2)これら抽出された遺伝子について、同程度のランキングにあるTCRαおよびTCRβ遺伝子を選択し、腫瘍特異的TCR遺伝子導入リンパ球の作製に利用する。
(3)レトロウイルス発現ベクター内に全長のTCRαおよびβ鎖遺伝子をクローニングすることができ、パッケージングにより高力価のTCRαレトロウイルス及びTCRβレトロウイルスが作製される。
(4)該患者リンパ球に混合レトロウイルスを感染させ、組換え型TCRα/TCRβの発現をFACSにて確認する。
(5)一連の工程により製造された腫瘍特異的TCR遺伝子組換えリンパ球を患者に移入することで、腫瘍細胞数の減少、臨床症状の改善を見ることができる。
本実施例から、本発明により以下のような効果が達成されることが理解される。
(1)患者自身の癌細胞及びT細胞を使って、治療用の腫瘍特異的TCR遺伝子導入リンパ球を作製することができ、HLA型や抗原の発現に関係なく幅広い患者にTCR遺伝子治療を行なうことができる。
(2)患者試料に存在するTCR配列を利用するため、患者のHLAにあったTCR遺伝子を利用することができ、癌細胞に特異性の高い、効果的な治療が実現できる。
(3)患者試料に存在するTCR配列を利用するため、患者正常細胞に反応しない、安全性の高いTCR遺伝子治療が実現できる。
(3)TCR解析から得られた遺伝子配列を直接利用するため、抗原の同定が必要なく、特定の抗原を用いたTCR遺伝子の取得が必要ない。
本実施例では、本発明のレパトア解析を用いたin vitro抗原刺激による単離されたがん特異的TCR遺伝子の調製の実証例を行う。以下にその手順を説明する(図67参照)。
(1)HLAが同一のがん患者からそれぞれ腫瘍細胞を摘出し、同時に末梢血を分離する。
(2)腫瘍細胞浸潤T細胞を含む腫瘍組織、あるいはリンパ球細胞からTrizol Reagent(invitrogen)を用いてRNAを抽出する。
(3)RNAから本明細書における調製実施例等で説明されているAdaptor-ligation PCR法でTCR遺伝子(調製実施例等と同じ)を増幅し、GS Juniorベンチトップシステム(Roche)等次世代シーケンスによるレパトア解析を行なう。
(4)それぞれの使用から得られたTCR遺伝子配列について新規に開発したTCR/BCRレパトア解析ソフトウェア(Repertoire Genesis、本明細書の解析実施例1~5を参照)を用いて、V,DおよびCDR3領域の配列を決定し、同一配列の存在頻度に基づいてランキングする。
(5)個々の患者において末梢血細胞に比べ腫瘍細胞で高い存在頻度(ここでは、具体的な例として、存在頻度>10倍、かつ腫瘍組織でのランキング上位のもの)を示したTCR遺伝子を検索し、腫瘍特異的として同定する。
(6)それら腫瘍特異的TCR遺伝子について、複数の同一のHLAを有するがん患者間で共通するTCR遺伝子配列を検索する。
(7)最も多くの癌患者で共通する腫瘍特異的TCR遺伝子を治療用腫瘍特異的TCRとして選択する。
(8)これらの全長TCRαおよびTCRβ遺伝子をクローニングし、遺伝子発現用レトロウイルスベクターに導入する(応用実施例6と同じものを使用することができる)。
(9)これらTCRα及びTCRβ遺伝子発現レトロウイルスベクターから遺伝子導入用ウイルスを上記応用実施例6(10)の方法に従い作製する。
(10)該患者から採取したリンパ球に上記(9)により作製したTCRαレトロウイルスを含有する培養液とTCRβレトロウイルスを含有する培養液を等量混合し、37℃で4時間培養する。その後PBSにて細胞を洗浄し、さらに、37℃で24時間培養する。
(11)細胞表面における遺伝子組換えTCRαβ分子の発現を確認する。IOTest Beta MarkTCR Vβレパトワ解析キット(Multi-analysisTCR Vβ antibodies, IM3497, Beckman Courter)および抗ヒトCD8抗体(CD8α,6602385, Beckman Courter)を用いたFACS解析によって、CD8陽性細胞における遺伝子導入するTCRβ鎖陽性細胞の割合を確認する。
(12)(11)で目的TCRαβの発現が確認された細胞を、RPMI1640培地にて0.5 x 106細胞濃度で37℃の条件下で細胞培養を行なう。腫瘍特異的TCR遺伝子導入リンパ球ををPBSで洗浄した後、該がん患者に点滴による静脈注入(テルフュージョン輸液システム、テルモ)により細胞移入する。
本実施例により、以下が達成される。
(1)患者腫瘍組織間で共通するTCR遺伝子を選択し、ランキングした結果、末梢血細胞に多く存在するTCRが除かれ、腫瘍特異的TCR遺伝子が多数抽出される。
(2)これら抽出された遺伝子について、同程度のランキングにあるTCRαおよびTCRβ遺伝子のペア、かつ同一患者に存在するペアを選択し、腫瘍特異的TCR遺伝子導入リンパ球の作製に利用する。
(3)レトロウイルス発現ベクター内に全長のTCRαおよびβ鎖遺伝子をクローニングすることができ、パッケージングにより高力価のTCRαレトロウイルス及びTCRβレトロウイルスを作製する。
(4)該患者リンパ球に混合レトロウイルスを感染させ、組換え型TCRα/TCRβ発現をFACSにて確認する。
(5)一連の工程により製造された腫瘍特異的TCR遺伝子組換えリンパ球を患者に移入することで、腫瘍細胞数の減少、臨床症状の改善を見ることができる。
本実施例から、本発明により以下のような効果が達成されることが理解される。
(1)患者自身の癌細胞及びT細胞を使って、治療用の腫瘍特異的TCR遺伝子導入リンパ球を作製することができ、HLA型や抗原の発現に関係なく幅広い患者にTCR遺伝子治療を行なうことができる。
(2)患者試料に存在するTCR配列を利用するため、患者のHLAにあったTCR遺伝子を利用することができ、癌細胞に特異性の高い、効果的な治療が実現できる。
(3)患者試料に存在するTCR配列を利用するため、患者正常細胞に反応しない、安全性の高いTCR遺伝子治療が実現できる。
(3)TCR解析から得られた遺伝子配列を直接利用するため、抗原の同定が必要なく、特定の抗原を用いたTCR遺伝子の取得が必要ない。
本実施例では、本発明のレパトア解析を用いた細胞加工療法の実証例を行う。以下にその手順を説明する(図68参照)。
(1) 応用実施例6に従い遺伝子導入用レトロウイルスを作製し、機能的αβTCRを発現するリンパ球集団を作出する。
(2)応用実施例6(1)-(2)の手順に従って分離、不活化した患者由来腫瘍細胞をRPMI1640培地(11875-093, Invitrogen)で希釈する。
(3)ELISPOTキット(IFN-γ, Human,ELISpot Kit, EL285, R&D Systems社)を用いて、(1)で作製した腫瘍特異的TCR遺伝子導入リンパ球と不活化患者腫瘍細胞を、1x106/mLの細胞濃度で、リンパ球と腫瘍細胞の細胞比(E:T比)を2:1、1:1、0.5:1の比で混合したものを、37℃で24時間培養する。
(4)24時間後、細胞を除去し、PVFD膜上のINFγの産生を発色法により検出し、IFNγ産生細胞数をカウントすることで、腫瘍特異的TCR遺伝子導入リンパ球の腫瘍特異性について評価する。
(5)5%以下の細胞でIFNγの産生が見られない場合は、応用実施例6(8)で採用したTCR以外のTCR遺伝子の中で、ランキング上位で、かつTCRαとTCRβが同程度の存在比率を示すペアを選択し、応用実施例6(9)-(11)の工程を経て、新たな腫瘍特異的TCR遺伝子導入リンパ球を作製する。
(6)このTCRαおよびTCRβについて、上記(1)-(4)の工程を実施し、腫瘍特異的TCR遺伝子導入リンパ球の腫瘍特異性について評価する。
本実施例により、以下が達成される。
(1)腫瘍特異的TCR遺伝子導入リンパ球を作製し、不活化腫瘍細胞に対する反応性を調べる。該TCR遺伝子導入リンパ球が、腫瘍に反応して、IFNγを産生することがわかる。
(2)腫瘍特異的TCR遺伝子導入リンパ球を患者に移入し、抗腫瘍効果並びに臨床症状の改善を見る。
本実施例から、本発明により以下のような効果が達成されることが理解される。
(1)患者自身の癌細胞及びT細胞を使って、治療用の腫瘍特異的TCR遺伝子導入リンパ球を作製することができ、HLA型や抗原の発現に関係なく幅広い患者にTCR遺伝子治療を行なうことができる。
(2)患者試料に存在するTCR配列を利用するため、患者のHLAにあったTCR遺伝子を利用することができ、癌細胞に特異性の高い、効果的な治療が実現できる。
(3)患者試料に存在するTCR配列を利用するため、患者正常細胞に反応しない、安全性の高いTCR遺伝子治療が実現できる。
(3)TCR解析から得られた遺伝子配列を直接利用するため、抗原の同定が必要なく、特定の抗原を用いたTCR遺伝子の取得が必要ない
本実施例では、本発明のレパトア解析を用いたインビトロで刺激試験での有効性および/または安全性評価の実証例を提供する。以下にその手順を説明する(図69参照)。
(1) 応用実施例6に従い遺伝子導入用レトロウイルスを作製し、腫瘍特異的αβTCRを発現するリンパ球集団を作出する。
(1)有効性を評価する場合、患者由来の癌細胞を摘出・分離し、培養液(RPMI1640,11875-093, Invitrogen)中で細断した後、0.70μmのフィルター(Falconセルストレイナー、コーニングでろ過することにより、単一細胞に分離する。培養液中に10μg/mlのマイトマイシンC(注射用マイトマイシンC、協和発酵キリン)により37℃で2時間不活化処理をする。不活化処理を行なった後、応用実施例6に記載されるように作製した腫瘍特異的TCR遺伝子導入Tリンパ球と混合培養する。
(2)応用実施例8に示したELISPOT法によって、腫瘍細胞に対する反応性を評価する。すなわち、ELISPOTキット(IFN-γ, Human,ELISpot Kit, EL285, R&D Systems社)を用いて、応用実施例6に従い作製した腫瘍特異的TCR遺伝子導入リンパ球と不活化患者腫瘍細胞を、1x106/mLの細胞濃度で、リンパ球と腫瘍細胞の細胞比(E:T比)を2:1、1:1、0.5:1の比で混合したものを、37℃で24時間培養する。
(3)24時間後、細胞を除去し、PVFD膜上のINFγの産生を発色法により検出し、IFNγ産生細胞数をカウントすることで、腫瘍特異的TCR遺伝子導入リンパ球の腫瘍特異性について評価する。ELISPOT法以外に、MTT試験(CellProliferation Kit I, MTT assay, 11465007001, Roche Diagnostics)あるいはIL-2産生試験(ヒトIL-2 ELISAシステム, GEヘルスケア, RPN5965)などの細胞増殖試験によっても評価することができる。
本実施例により、以下が達成される。
(1)腫瘍特異的TCR遺伝子組換えリンパ球の不活化腫瘍細胞に対する反応性を調べた結果、高頻度なIFNγの産生が認められる。
(2)培養時間に経時的にIFNγ陽性細胞数は増加し、24時間でプラトーに達する。
(考察)
本実施例から、本発明により以下のような効果が達成されることが理解される。
(1)腫瘍特異的TCR遺伝子導入リンパ球を使った遺伝子治療を施す前に、患者自身の細胞を使った有効性の評価ができ、治療前に有効性を予測できる。
(2)有効性を評価することで、TCR遺伝子を選択して利用することができ、より効果的なTCR遺伝子治療ができる。
(1’)安全性を評価する場合、対照となる既存の細胞株、患者癌細胞を含んでいないと考えられる正常組織(腫瘍摘出の過程で採取される正常組織の一部)、あるいは固形腫瘍の場合は患者末梢血細胞を用いて(1)(2)に同様の試験を実施する。
(2’)正常組織に対する腫瘍特異的TCR遺伝子導入Tリンパ球の反応性をELISPOT法で定量し、評価する。
(3’)正常細胞への反応性が低く、腫瘍細胞への反応性が高い腫瘍特異的TCR遺伝子導入Tリンパ球を選択し、患者治療に用いる。
本実施例により、以下が達成される。
(1)腫瘍特異的TCR遺伝子導入リンパ球を作製し、不活化正常細胞に対する反応性を調べた結果、IFNγが産生されず、正常細胞に対してほとんど反応性を示さないことがわかる。
本実施例から、本発明により以下のような効果が達成されることが理解される。
(1)腫瘍特異的TCR遺伝子導入リンパ球を使ったハイリスクの遺伝子治療を施す前に、患者自身の細胞を使った安全性の評価ができ、より安全な治療を実現できる。
(2)安全性を評価することで、リスクの高いTCR遺伝子を除外でき、より安全なTCR遺伝子を使った治療ができる。
配列番号20~31:BCRリードのCDR3アミノ酸配列
配列番号32~38:実施例2で使用されるプライマー配列(表2)
配列番号39:Adaptor-Aの配列
配列番号40~60:シーケンス用プライマー(表6)
配列番号61~1164:BCRリードのCDR3アミノ酸配列(表1H)
配列番号1165~1324:Molt-4細胞段階希釈試料におけるTCRリード
配列番号1325~1374:分子同定(MID Tag)配列の例
配列番号1375:Adaptor-Bの配列
配列番号1376~1379:TCRの各全長配列
配列番号1381~1386:BCRの各全長配列
配列番号1387:CM3-GS(配列番号7)中の特異的配列(CM3)
配列番号1388:CA3-GS(配列番号10)中の特異的配列(CA3)
配列番号1389:CG3-GS(配列番号13)中の特異的配列(CG3)
配列番号1390:CD3-GS(配列番号16)中の特異的配列(CD3)
配列番号1391:CE3-GS(配列番号19)中の特異的配列(CE3)
配列番号1392:標的配列TRBC、名称TRBC2*01、膜結合型
配列番号1393:標的配列TRBC、名称TRBC2*02、膜結合型
配列番号1394:標的配列TRGC、名称TRGC1*02、膜結合型
配列番号1395:標的配列TRGC、名称TRGC2*01、膜結合型
配列番号1396:標的配列TRGC、名称TRGC2*02、膜結合型
配列番号1397:標的配列TRGC、名称TRGC2*03、膜結合型
配列番号1398:標的配列TRGC、名称TRGC2*04、膜結合型
配列番号1399:標的配列TRGC、名称TRGC2*05、膜結合型
配列番号1400:標的配列IGHA、名称IGHA2*01、分泌型
配列番号1401:標的配列IGHA、名称IGHA2*02s、分泌型
配列番号1402:標的配列IGHA、名称IGHA2*02、膜結合型
配列番号1403:標的配列IGHA、名称IGHA2*03、分泌型
配列番号1404:標的配列IGHD、名称IGHD*01、分泌型
配列番号1405:標的配列IGHD、名称IGHD*02、分泌型
配列番号1406:標的配列IGHD、名称IGHD*02、膜結合型
配列番号1407:標的配列IGHE、名称IGHE*01、膜結合型
配列番号1408:標的配列IGHE、名称IGHE*02、分泌型
配列番号1409:標的配列IGHE、名称IGHE*03、膜結合型
配列番号1410:標的配列IGHE、名称IGHE*04、分泌型
配列番号1411:標的配列IGHE、名称IGHE*04、膜結合型
配列番号1412:標的配列IGHG、名称IGHG1*02、分泌型
配列番号1413:標的配列IGHG、名称IGHG1*03、分泌型
配列番号1414:標的配列IGHG、名称IGHG2*0、分泌型
配列番号1415:標的配列IGHG、名称IGHG2*01、膜結合型
配列番号1416:標的配列IGHG、名称IGHG2*02、分泌型
配列番号1417:標的配列IGHG、名称IGHG2*03、分泌型
配列番号1418:標的配列IGHG、名称IGHG2*04、分泌型
配列番号1419:標的配列IGHG、名称IGHG2*05、分泌型
配列番号1420:標的配列IGHG、名称IGHG2*06、分泌型
配列番号1421:標的配列IGHG、名称IGHG2*06、膜結合型
配列番号1422:標的配列IGHG、名称IGHG3*01、分泌型
配列番号1423:標的配列IGHG、名称IGHG3*01、膜結合型
配列番号1424:標的配列IGHG、名称IGHG3*03、分泌型
配列番号1425:標的配列IGHG、名称IGHG3*03、膜結合型
配列番号1426:標的配列IGHG、名称IGHG3*04、分泌型
配列番号1427:標的配列IGHG、名称IGHG3*05、分泌型
配列番号1428:標的配列IGHG、名称IGHG3*06、分泌型
配列番号1429:標的配列IGHG、名称IGHG3*07、分泌型
配列番号1430:標的配列IGHG、名称IGHG3*08、分泌型
配列番号1431:標的配列IGHG、名称IGHG3*09、分泌型
配列番号1432:標的配列IGHG、名称IGHG3*10、分泌型
配列番号1433:標的配列IGHG、名称IGHG3*11、分泌型
配列番号1434:標的配列IGHG、名称IGHG3*12、分泌型
配列番号1435:標的配列IGHG、名称IGHG3*13、分泌型
配列番号1436:標的配列IGHG、名称IGHG3*14、分泌型
配列番号1437:標的配列IGHG、名称IGHG3*15、分泌型
配列番号1438:標的配列IGHG、名称IGHG3*16、分泌型
配列番号1439:標的配列IGHG、名称IGHG3*17、分泌型
配列番号1440:標的配列IGHG、名称IGHG3*18、分泌型
配列番号1441:標的配列IGHG、名称IGHG3*19、分泌型
配列番号1442:標的配列IGHG、名称IGHG4*01、分泌型
配列番号1443:標的配列IGHG、名称IGHG4*02、分泌型
配列番号1444:標的配列IGHG、名称IGHG4*03、分泌型
配列番号1445:標的配列IGHG、名称IGHG4*04、分泌型
配列番号1446:標的配列IGHG、名称IGHG4*04、膜結合型
配列番号1447:標的配列IGHM、名称IGHM*01、膜結合型
配列番号1448:標的配列IGHM、名称IGHM*03、分泌型
配列番号1449:標的配列IGHM、名称IGHM*03、膜結合型
配列番号1450~1499:TRAリード(上位50位)(表3-1)
配列番号1500~1549:TRBリード(上位50位)(表3-2)
配列番号1550~1587:健常人における重複TCRα鎖リード配列(表3-7)
配列番号1588~1626:健常人における重複TCRβ鎖リード配列(表3-8)
配列番号1627~1647:インバリアントTCR候補遺伝子(表3-9)
配列番号1648~1860:がん患者における重複TCRαリード配列とがん特異的TCR
αリード(表3-11)
配列番号1861~1909:がん患者における重複TCRβリード配列とがん特異的TCR
β(表3-12)
配列番号1910~1921:P5-P20EAプライマー
配列番号1922~1929:P7-CA3プライマー
配列番号1930~1937:P7-CB3プライマー
配列番号1938~1992:解析システムの実施例5で同定されたパブリックTCRα配列において観察されるインバリアントTCRの配列
Claims (46)
- データベースを用いて被験体のT細胞レセプター(TCR)またはB細胞レセプター(BCR)の可変領域のレパトア(repertoire)を定量的に解析する方法であって、該方法は、
(1)該被験者から非バイアス的に増幅した、T細胞レセプター(TCR)またはB細胞レセプター(BCR)の核酸配列を含む核酸試料を提供する工程;
(2)該核酸試料に含まれる該核酸配列を決定する工程;および
(3)決定された該核酸配列にもとづいて、各遺伝子の出現頻度またはその組み合わせを算出し、該被験体のTCRもしくはBCRレパトアを導出する工程
を包含する、方法。 - 前記核酸試料は、複数種類のT細胞レセプター(TCR)またはB細胞レセプター(BCR)の核酸配列を含み、前記(2)は単一の配列決定により前記核酸配列が決定される、請求項1に記載の方法。
- 前記単一の配列決定は、前記核酸試料から配列決定用の試料への増幅において、プライマーとして使用する配列は少なくとも一方がC領域をコードする核酸配列またはその相補鎖と同一配列を有することを特徴とする、請求項2に記載の方法。
- 前記単一の配列決定は、共通アダプタープライマーを用いて行われることを特徴とする、請求項3に記載の方法。
- 前記非バイアス的な増幅が、V領域特異的な増幅ではない、請求項1に記載の方法。
- 前記レパトアはBCRの可変領域のレパトアであり、前記核酸配列はBCRの核酸配列である、請求項1に記載の方法。
- データベースを用いて被験体のT細胞レセプター(TCR)またはB細胞レセプター(BCR)の可変領域のレパトア(repertoire)を定量的に解析するためのシステムであって、該システムは、
(1)該被験者から非バイアス的に増幅した、T細胞レセプター(TCR)またはB細胞レセプター(BCR)の核酸配列を含む核酸試料を提供するためのキット;
(2)該核酸試料に含まれる該核酸配列を決定するための装置;および
(3)決定された該核酸配列にもとづいて、各遺伝子の出現頻度またはその組み合わせを算出し、該被験体のTCRもしくはBCRレパトアを導出するための装置を備えるシステム。 - 請求項7に記載のシステムと、該システムに基づいて導出されたTCRもしくはBCRレパトアに基づいて前記被験者の疾患、障害または状態を分析する手段とを備える、被験者の疾患、障害または状態を分析するシステム。
- 請求項8に記載のシステムで決定された被験者の疾患、障害または状態と、前記TCRもしくはBCRレパトアとを定量的に関連付ける手段、および該定量的な関連から、適切な処置または予防のための手段を選択する手段とを備える、該被験者の疾患、障害または状態を処置または予防するためのシステム。
- 前記(1)は、以下の工程
(1-1)標的となる細胞に由来するRNA試料を鋳型として相補的DNAを合成する工程;
(1-2)該相補的DNAを鋳型として二本鎖相補的DNAを合成する工程;
(1-3)該二本鎖相補的DNAに共通アダプタープライマー配列を付加してアダプター付加二本鎖相補的DNAを合成する工程;
(1-4)該アダプター付加二本鎖相補的DNAと、該共通アダプタープライマー配列からなる共通アダプタープライマーと、第1のTCRまたはBCRのC領域特異的プライマーとを用いて第1のPCR増幅反応を行う工程であって、
該第1のTCRまたはBCRのC領域特異的プライマーは、該TCRまたはBCRの目的とするC領域に十分に特異的であり、かつ、他の遺伝子配列に相同性のない配列を含み、かつ、増幅された場合に下流にサブタイプ間に不一致塩基を含むよう設計される、工程
(1-5)(1-4)のPCR増幅産物と、該共通アダプタープライマーと、第2のTCRまたはBCRのC領域特異的プライマーとを用いて第2のPCR増幅反応を行う工程であって、該第2のTCRまたはBCRのC領域特異的プライマーは、該第1のTCRまたはBCRのC領域特異的プライマーの配列より下流の配列において該TCRまたはBCRのC領域に完全マッチの配列を有するが他の遺伝子配列に相同性のない配列を含み、かつ
、増幅された場合に下流にサブタイプ間に不一致塩基を含むよう設計される、工程;および
(1-6)(1-5)のPCR増幅産物と、該共通アダプタープライマーの核酸配列に第1の追加アダプター核酸配列を含む付加共通アダプタープライマーと、第2の追加アダプター核酸配列および分子同定(MID Tag)配列を第3のTCRまたはBCRのC領域特異的配列に付加したアダプター付の第3のTCRまたはBCRのC領域特異的プライマーとを用いて第3のPCR増幅反応を行う工程であって、
該第3のTCRまたはBCRのC領域特異的プライマーは、該第2のTCRまたはBCRのC領域特異的プライマーの配列より下流の配列において該TCRまたはBCRのC領域に完全マッチの配列を有するが他の遺伝子配列に相同性のない配列を含み、かつ、増幅された場合に下流にサブタイプ間に不一致塩基を含むよう設計され、
該第1の追加アダプター核酸配列は、DNA捕捉ビーズへの結合およびemPCR反応に適切な配列であり、
該第2の追加アダプター核酸配列は、emPCR反応に適切な配列であり、
該分子同定(MID Tag)配列は、増幅産物が同定できるように、ユニークさを付与するための配列である、工程;
を包含する、
請求項1に記載の方法。 - 前記(1)キットは、以下:
(1-1)標的となる細胞に由来するRNA試料を鋳型として相補的DNAを合成する手段;
(1-2)該相補的DNAを鋳型として二本鎖相補的DNAを合成する手段;
(1-3)該二本鎖相補的DNAに共通アダプタープライマー配列を付加してアダプター付加二本鎖相補的DNAを合成する手段;
(1-4)該アダプター付加二本鎖相補的DNAと、該共通アダプタープライマー配列からなる共通アダプタープライマーと、第1のTCRまたはBCRのC領域特異的プライマーとを用いて第1のPCR増幅反応を行う手段であって、
該第1のTCRまたはBCRのC領域特異的プライマーは、該TCRまたはBCRの目的とするC領域に十分に特異的であり、かつ、他の遺伝子配列に相同性のない配列を含み、かつ、増幅された場合に下流にサブタイプ間に不一致塩基を含むよう設計される、手段
(1-5)(1-4)のPCR増幅産物と、該共通アダプタープライマーと、第2のTCRまたはBCRのC領域特異的プライマーとを用いて第2のPCR増幅反応を行う手段であって、該第2のTCRまたはBCRのC領域特異的プライマーは、該第1のTCRまたはBCRのC領域特異的プライマーの配列より下流の配列において該TCRまたはBCRのC領域に完全マッチの配列を有するが他の遺伝子配列に相同性のない配列を含み、かつ、増幅された場合に下流にサブタイプ間に不一致塩基を含むよう設計される、手段;および
(1-6)(1-5)のPCR増幅産物と、該共通アダプタープライマーの核酸配列に第1の追加アダプター核酸配列を含む付加共通アダプタープライマーと、第2の追加アダプター核酸配列および分子同定(MID Tag)配列を第3のTCRまたはBCRのC領域特異的配列に付加したアダプター付の第3のTCRまたはBCRのC領域特異的プライマーとを用いて第3のPCR増幅反応を行う手段であって、
該第3のTCRまたはBCRのC領域特異的プライマーは、該第2のTCRまたはBCRのC領域特異的プライマーの配列より下流の配列において該TCRまたはBCRのC領域に完全マッチの配列を有するが他の遺伝子配列に相同性のない配列を含み、かつ、増幅された場合に下流にサブタイプ間に不一致塩基を含むよう設計され、
該第1の追加アダプター核酸配列は、DNA捕捉ビーズへの結合およびemPCR反応に適切な配列であり、
該第2の追加アダプター核酸配列は、emPCR反応に適切な配列であり、
該分子同定(MID Tag)配列は、増幅産物が同定できるように、ユニークさを付与するための配列である、手段;
を包含する、
請求項7に記載のシステム。 - (3)前記TCRもしくはBCRレパトアの導出は以下の工程
(3-1)V領域、D領域、J領域および必要に応じてC領域の少なくとも1つを含む遺伝子領域ごとの参照データベースを提供する工程:
(3-2)必要に応じてトリミングを行い、必要に応じて適切な長さのものを抽出した入力配列セットを提供する工程;
(3-3)該入力配列セットについて、該遺伝子領域ごとに、該参照データベースと相同性検索を行い、近似する参照アリルおよび/または該参照アリルの配列とのアラインメントを記録する工程;
(3-4)該入力配列セットについてV領域およびJ領域をアサインし、アサイン結果に基づいて、D領域の核酸配列を抽出する工程;
(3-5)該D領域の核酸配列をアミノ酸配列に翻訳し、該アミノ酸配列を利用してD領域を分類する工程;
(3-6)(3-5)での分類に基づいて、V領域、D領域、J領域および必要に応じてC領域の各出現頻度、もしくはその組合せの出現頻度を算出することで、TCRもしくはBCRレパトアを導出する工程;
を包含する方法によって達成される、請求項1または10に記載の方法。 - (3)前記TCRもしくはBCRレパトアの導出のための装置は以下
(3-1)V領域、D領域、J領域および必要に応じてC領域の少なくとも1つを含む遺伝子領域ごとの参照データベースを提供する手段:
(3-2)必要に応じてトリミングを行い、必要に応じて適切な長さのものを抽出した入力配列セットを提供する手段;
(3-3)該入力配列セットについて、該遺伝子領域ごとに、該参照データベースと相同性検索を行い、近似する参照アリルおよび/または該参照アリルの配列とのアラインメントを記録する手段;
(3-4)該入力配列セットについてV領域およびJ領域をアサインし、アサイン結果に基づいて、D領域の核酸配列を抽出する手段;
(3-5)該D領域の核酸配列をアミノ酸配列に翻訳し、該アミノ酸配列を利用してD領域を分類する手段;
(3-6)(3-5)での分類に基づいて、V領域、D領域、J領域および必要に応じてC領域の各出現頻度、もしくはその組合せの出現頻度を算出することで、TCRもしくはBCRレパトアを導出する手段;
を備える、請求項7または11に記載のシステム。 - TCRもしくはBCRレパトアを解析する方法であって、以下のステップ:
(1)V領域、D領域、J領域および必要に応じてC領域の少なくとも1つを含む遺伝子領域ごとの参照データベースを提供するステップ:
(2)必要に応じてトリミングを行い、必要に応じて適切な長さのものを抽出した入力配列セットを提供するステップ;
(3)該入力配列セットについて、該遺伝子領域ごとに、該参照データベースと相同性検索を行い、近似する参照アリルおよび/または該参照アリルの配列とのアラインメントを記録するステップ;
(4)該入力配列セットについてV領域およびJ領域をアサインし、アサイン結果に基づいて、D領域の核酸配列を抽出するステップ;
(5)該D領域の核酸配列をアミノ酸配列に翻訳し、該アミノ酸配列を利用してD領域を分類するステップ;
(6)(5)での分類に基づいて、V領域、D領域、J領域および必要に応じてC領域の各出現頻度、もしくはその組合せの出現頻度を算出することで、TCRもしくはBCRレパトアを導出するステップ;
を包含する、方法。 - 前記入力配列セットは、非バイアス配列セットである、請求項14に記載の方法。
- 前記トリミングは、リード両端から低クオリティ領域を削除し;リード両端からアダプタ配列と10bp以上マッチする領域を削除し;および残った長さが200bp以上(TCR)もしくは300bp以上(BCR)なら高クオリティとして解析に使用するステップによって達成される、請求項14に記載の方法。
- 前記低クオリティは、QV値の7bp移動平均が30未満のものである、請求項16に記載の方法。
- TCRもしくはBCRレパトアを解析するシステムであって、該システムは:
(1)V領域、D領域、J領域および必要に応じてC領域の少なくとも1つを含む遺伝子領域ごとの参照データベースを提供する手段:
(2)必要に応じてトリミングを行い、必要に応じて適切な長さのものを抽出した入力配列セットを提供する手段;
(3)該入力配列セットについて、該遺伝子領域ごとに、該参照データベースと相同性検索を行い、近似する参照アリルおよび/または該参照アリルの配列とのアラインメントを記録する手段;
(4)該入力配列セットについてV領域およびJ領域をアサインし、アサイン結果に基づいて、D領域の核酸配列を抽出する手段;
(5)該D領域の核酸配列をアミノ酸配列に翻訳し、該アミノ酸配列を利用してD領域を分類する手段;
(6)入力配列セットにおいて、V領域、D領域、J領域および必要に応じてC領域の各出現頻度、もしくはその組合せの出現頻度を算出することで、TCRもしくはBCRレパトアを導出する手段;
を包含する、システム。 - TCRもしくはBCRレパトアを解析する方法の処理をコンピュータに実行させるコンピュータプログラムであって、該方法は以下のステップ:
(1)V領域、D領域、J領域および必要に応じてC領域の少なくとも1つを含む遺伝子領域ごとの参照データベースを提供するステップ:
(2)必要に応じてトリミングを行い、必要に応じて適切な長さのものを抽出した入力配列セットを提供するステップ;
(3)該入力配列セットについて、該遺伝子領域ごとに、該参照データベースと相同性検索を行い、近似する参照アリルおよび/または該参照アリルの配列とのアラインメントを記録するステップ;
(4)該入力配列セットについてV領域およびJ領域をアサインし、アサイン結果に基づいて、D領域の核酸配列を抽出するステップ;
(5)該D領域の核酸配列をアミノ酸配列に翻訳し、該アミノ酸配列を利用してD領域を分類するステップ;
(6)入力配列セットにおいて、V領域、D領域、J領域および必要に応じてC領域の各出現頻度、もしくはその組合せの出現頻度を算出することで、TCRもしくはBCRレパトアを導出するステップ;
を包含する、プログラム。 - TCRもしくはBCRレパトアを解析する方法の処理をコンピュータに実行させるコンピュータプログラムを格納する記録媒体であって、該方法は以下のステップ:
(1)V領域、D領域、J領域および必要に応じてC領域の少なくとも1つを含む遺伝子領域ごとの参照データベースを提供するステップ:
(2)必要に応じてトリミングを行い、必要に応じて適切な長さのものを抽出した入力配列セットを提供するステップ;
(3)該入力配列セットについて、該遺伝子領域ごとに、該参照データベースと相同性検索を行い、近似する参照アリルおよび/または該参照アリルの配列とのアラインメントを記録するステップ;
(4)該入力配列セットについてV領域およびJ領域をアサインし、アサイン結果に基づいて、D領域の核酸配列を抽出するステップ;
(5)該D領域の核酸配列をアミノ酸配列に翻訳し、該アミノ酸配列を利用してD領域を分類するステップ;
(6)入力配列セットにおいて、V領域、D領域、J領域および必要に応じてC領域の各出現頻度、もしくはその組合せの出現頻度を算出することで、TCRもしくはBCRレパトアを導出するステップ;
を包含する、記録媒体。 - データベースを用いた遺伝子配列解析により、T細胞レセプター(TCR)またはB細胞レセプター(BCR)の可変領域のレパトア(repertoire)の定量解析を行うための試料を調製するための方法であって、
(1)標的となる細胞に由来するRNA試料を鋳型として相補的DNAを合成する工程;
(2)該相補的DNAを鋳型として二本鎖相補的DNAを合成する工程;
(3)該二本鎖相補的DNAに共通アダプタープライマー配列を付加してアダプター付加二本鎖相補的DNAを合成する工程;
(4)該アダプター付加二本鎖相補的DNAと、該共通アダプタープライマー配列からなる共通アダプタープライマーと、第1のTCRまたはBCRのC領域特異的プライマーとを用いて第1のPCR増幅反応を行う工程であって、
該第1のTCRまたはBCRのC領域特異的プライマーは、該TCRまたはBCRの目的とするC領域に十分に特異的であり、かつ、他の遺伝子配列に相同性のない配列を含み、かつ、増幅された場合に下流にサブタイプ間に不一致塩基を含むよう設計される、工程
(5)(4)のPCR増幅産物と、該共通アダプタープライマーと、第2のTCRまたはBCRのC領域特異的プライマーとを用いて第2のPCR増幅反応を行う工程であって、該第2のTCRまたはBCRのC領域特異的プライマーは、該第1のTCRまたはBCRのC領域特異的プライマーの配列より下流の配列において該TCRまたはBCRのC領域に完全マッチの配列を有するが他の遺伝子配列に相同性のない配列を含み、かつ、増幅された場合に下流にサブタイプ間に不一致塩基を含むよう設計される、工程;および
(6)(5)のPCR増幅産物と、該共通アダプタープライマーの核酸配列に第1の追加アダプター核酸配列を含む付加共通アダプタープライマーと、第2の追加アダプター核酸配列および分子同定(MID Tag)配列を第3のTCRまたはBCRのC領域特異的配列に付加したアダプター付の第3のTCRまたはBCRのC領域特異的プライマーとを用いて第3のPCR増幅反応を行う工程であって、
該第3のTCRまたはBCRのC領域特異的プライマーは、該第2のTCRまたはBCRのC領域特異的プライマーの配列より下流の配列において該TCRまたはBCRのC領域に完全マッチの配列を有するが他の遺伝子配列に相同性のない配列を含み、かつ、増幅された場合に下流にサブタイプ間に不一致塩基を含むよう設計され、
該第1の追加アダプター核酸配列は、DNA捕捉ビーズへの結合およびemPCR反応に適切な配列であり、
該第2の追加アダプター核酸配列は、emPCR反応に適切な配列であり、
該分子同定(MID Tag)配列は、増幅産物が同定できるように、ユニークさを付与するための配列である、工程;
を包含する、方法。 - 前記第1、第2および第3のTCRまたはBCRのC領域特異的プライマーは、それぞれ独立して、TCRまたはBCRのレパトア解析のためのものであり、BCRである場合、IgM,IgG,IgA,IgD,またはIgEの各アイソタイプC領域に完全マッチする配列であって、該IgGおよびIgAの場合は、サブタイプについても完全マッチし、該データベースに含まれる他の配列に相同性を持たない配列であり、かつ、該プライマーの下流においてサブタイプ間に
不一致塩基が含まれるように選択され、
該共通アダプタープライマー配列は、増幅に適切な塩基長であり、ホモダイマーおよび分子内ヘアピン構造が取りにくく、安定して2本鎖を形成できるように設計され、該データベース内のすべてのBCR遺伝子配列と相同性が高くなく、かつ、該C領域特異的プライマーと同じ程度のTmになるように設計される、請求項21に記載の方法。 - 第1のTCRまたはBCRのC領域特異的プライマーは、以下の構造:CM1(配列番号5)、CA1(配列番号8)、CG1(配列番号11)、CD1(配列番号14)、CE1(配列番号17)、CA1(配列番号35)、またはCB1(配列番号37)を有する、請求項14に記載の方法。
- 第2のTCRまたはBCRのC領域特異的プライマーは、以下の構造:CM2(配列番号6)、CA2(配列番号9)、CG2(配列番号12)、CD2(配列番号15)、CE2(配列番号18)、CA2(配列番号35)、またはCB2(配列番号37)を有する、請求項14に記載の方法。
- 第3のTCRまたはBCRのC領域特異的プライマーは、以下の構造:CM3-GS(配列番号7)、CA3-GS(配列番号10)、CG3-GS(配列番号13)、CD3-GS(配列番号16)またはCE3-GS(配列番号19)を有する、請求項14に記載の方法。
- 前記TCRまたはBCRのC領域特異的プライマーは、いずれも、TCRまたはBCRのすべてのサブクラスに対応するセットで提供される、請求項14に記載の方法。
- 請求項14~26のいずれか1項に記載の方法で製造された試料を用いて遺伝子解析を行う方法。
- 前記遺伝子解析は、T細胞レセプター(TCR)またはB細胞レセプター(BCR)の可変領域のレパトア(repertoire)の定量解析である、請求項27に記載の方法。
- 被験者にがんイディオタイプペプチド感作免疫細胞療法に使用するための組成物を調製する方法であって、該方法は、
(1)請求項B1に記載の方法によって、該被験者のT細胞レセプター(TCR)またはB細胞レセプター(BCR)レパトアを解析する工程;
(2)該解析の結果に基づいて、該被験者のがん細胞由来のTCRまたはBCRを決定する工程であって、該決定は該被験者のがん細胞由来のTCRまたはBCR遺伝子の存在頻度ランキングにおいて、上位ランクの配列が、該がん細胞由来のTCRまたはBCRとして選択することによってなされる、工程;
(3)決定された該がん由来のTCRまたはBCRに基づいて、HLA検査ペプチドの候補のアミノ酸配列を決定する工程であって、該決定は、HLA結合ペプチド予測アルゴリズムを用いて算出されたスコアに基づきなされる、工程;および
(4)決定されたペプチドを合成する工程;
を包含する、方法。 - 前記(3)工程のHLA検査ペプチドの候補は、BIMAS、SYFPEITHI、RANKPEPまたはNetMHCを用いて決定される、請求項29に記載の方法。
- 前記(4)工程の後に、前記ペプチドと、前記被験者由来の樹状細胞または抗原提示細胞と、前記被験者由来のCD8+T細胞とを混合して培養する工程を包含する、請求項29に記載の方法。
- 前記(4)工程の後に、前記ペプチドと、前記被験者由来の樹状細胞とを混合して培養する工程を包含する、請求項29に記載の方法。
- 前記(4)工程の後に、前記ペプチドと、前記被験者由来の樹状細胞または抗原提示細胞と、前記被験者由来のCD8+T細胞とを混合して培養してCD8+T細胞-樹状細胞/抗原提示細胞-ペプチド混合物を生産する工程、および前記ペプチドと、前記被験者由来の樹状細胞とを混合して培養して樹状細胞-ペプチド混合物を生産する工程を包含する、請求項29に記載の方法。
- (A)被験者に由来する抗原タンパク質または抗原ペプチドまたは該被験者に由来するリンパ球または請求項C1に記載の前記決定されたペプチドと、該被験者に由来する不活化がん細胞と、該被験者に由来するTリンパ球とを混合し培養して腫瘍特異的T細胞を生産する工程;
(B)該腫瘍特異的T細胞のTCRを請求項1に記載の方法によって解析する工程;および
(C)該解析の結果に基づいて、所望の腫瘍特異的T細胞を単離する工程
を包含する、in vitro抗原刺激による単離されたがん特異的TCR遺伝子を調製する方法。 - (A)工程は、被験者に由来する抗原タンパク質または抗原ペプチドと、該被験者に由来する不活化がん細胞と、該被験者に由来するTリンパ球とを混合し培養して腫瘍特異的T細胞を生産する工程である、請求項34に記載の方法。
- (A)工程は、前記被験者に由来するリンパ球と、該被験者に由来する不活化がん細胞と、該被験者に由来するTリンパ球とを混合し培養して腫瘍特異的T細胞を生産する工程である、請求項34に記載の方法。
- (A)工程は、請求項C1に記載の前記決定されたペプチドと、該被験者に由来する不活化がん細胞と、該被験者に由来するTリンパ球とを混合し培養して腫瘍特異的T細胞を生産する工程である、請求項34に記載の方法。
- (A)共通のHLAを有する被験体から単離されたリンパ球または癌組織を提供する工程;
(B)該リンパ球または癌組織について、該腫瘍特異的T細胞のTCRを請求項1に記載の方法によって解析する工程;および
(C)腫瘍特異的T細胞に共通する配列を有するT細胞を単離する工程
を包含する、共通配列検索による単離されたがん特異的TCR遺伝子を調製する方法。 - A)患者から採取されたTリンパ球を提供する工程;
B)該Tリンパ球を抗原刺激した後に、請求項1に記載の方法に基づいてTCRを解析する工程であって、該抗原刺激は、前記被験者に由来する抗原タンパク質または抗原ペプチド、該被験者に由来する不活性化がん細胞、または腫瘍由来イディオタイプペプチドによってなされる、工程;
C)解析された該TCRにおいて最適抗原および最適TCRを選択する工程; および
D)該最適TCRのTCR遺伝子の腫瘍特異的αおよびβTCR発現ウイルスベクターを生産する工程
を包含する細胞加工療法に使用するための該腫瘍特異的TCR遺伝子導入Tリンパ球を調製する方法。 - 前記抗原刺激は、前記被験者に由来する抗原タンパク質または抗原ペプチドによってなされる、請求項39に記載の方法。
- 前記抗原刺激は、前記被験者に由来する不活性化がん細胞によってなされる、請求項39に記載の方法。
- 前記抗原刺激は、前記腫瘍由来イディオタイプペプチドによってなされる、請求項39に記載の方法。
- C)工程は、前記被験体のがん組織に高発現する抗原を選択することを含む、請求項39に記載の方法。
- C)工程は、抗原特異的リンパ球刺激試験において最も強くT細胞を活性させる抗原を選択することを含む、請求項39に記載の方法。
- C)工程は、抗原刺激前後において請求項1に基づいて行ったレパトア解析から特定のTCRの頻度を最も増加させる抗原を選択することを含む、請求項39に記載の方法。
- 請求項38に記載の方法で単離されたがん特異的TCR遺伝子を用いてインビトロで刺激試験を行い有効性および/または安全性評価を行う方法。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP19181888.9A EP3572510B1 (en) | 2013-11-21 | 2014-11-20 | T cell receptor and b cell receptor repertoire analysis system, and use of same in treatment and diagnosis |
| CN201480073846.6A CN106103711A (zh) | 2013-11-21 | 2014-11-20 | T细胞受体和b细胞受体库分析系统及其在治疗和诊断中的应用 |
| JP2015548994A JP6164759B2 (ja) | 2013-11-21 | 2014-11-20 | T細胞受容体およびb細胞受容体レパトアの解析システムならびにその治療および診断への利用 |
| EP14864397.6A EP3091074B1 (en) | 2013-11-21 | 2014-11-20 | T cell receptor and b cell receptor repertoire analysis system, and use of same in treatment and diagnosis |
| US15/038,422 US11203783B2 (en) | 2013-11-21 | 2014-11-20 | T cell receptor and B cell receptor repertoire analysis system, and use of same in treatment and diagnosis |
| US17/460,067 US20220119884A1 (en) | 2013-11-21 | 2021-08-27 | T cell receptor and b cell receptor repertoire analysis system, and use of same in treatment and diagnosis |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013-241403 | 2013-11-21 | ||
| JP2013241405 | 2013-11-21 | ||
| JP2013-241405 | 2013-11-21 | ||
| JP2013241404 | 2013-11-21 | ||
| JP2013241403 | 2013-11-21 | ||
| JP2013-241404 | 2013-11-21 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/038,422 A-371-Of-International US11203783B2 (en) | 2013-11-21 | 2014-11-20 | T cell receptor and B cell receptor repertoire analysis system, and use of same in treatment and diagnosis |
| US17/460,067 Division US20220119884A1 (en) | 2013-11-21 | 2021-08-27 | T cell receptor and b cell receptor repertoire analysis system, and use of same in treatment and diagnosis |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015075939A1 true WO2015075939A1 (ja) | 2015-05-28 |
Family
ID=53179221
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/005849 WO2015075939A1 (ja) | 2013-11-21 | 2014-11-20 | T細胞受容体およびb細胞受容体レパトアの解析システムならびにその治療および診断への利用 |
Country Status (5)
| Country | Link |
|---|---|
| US (2) | US11203783B2 (ja) |
| EP (2) | EP3572510B1 (ja) |
| JP (5) | JP6164759B2 (ja) |
| CN (1) | CN106103711A (ja) |
| WO (1) | WO2015075939A1 (ja) |
Cited By (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105095687A (zh) * | 2015-06-26 | 2015-11-25 | 南方科技大学 | 一种免疫组库数据分析方法及终端 |
| WO2016136716A1 (ja) * | 2015-02-23 | 2016-09-01 | 国立大学法人東北大学 | 遺伝子特異的非バイアス増幅法 |
| EP2954070A4 (en) * | 2013-02-11 | 2016-10-26 | Cb Biotechnologies Inc | METHOD FOR ASSESSING AN IMMUNOREPERTOIR |
| WO2017046335A1 (en) * | 2015-09-18 | 2017-03-23 | INSERM (Institut National de la Santé et de la Recherche Médicale) | T cell receptors (tcr) and uses thereof for the diagnosis and treatment of diabetes |
| WO2017158103A1 (en) * | 2016-03-16 | 2017-09-21 | Immatics Biotechnologies Gmbh | Transfected t-cells and t-cell receptors for use in immunotherapy against cancers |
| CN107345241A (zh) * | 2016-05-12 | 2017-11-14 | 眭维国 | B细胞抗原受体h链cdr3的处理方法 |
| WO2017212072A1 (en) * | 2016-06-10 | 2017-12-14 | Umc Utrecht Holding B.V. | Human leukocyte antigen restricted gamma delta t cell receptors and methods of use thereof |
| WO2017222056A1 (ja) * | 2016-06-23 | 2017-12-28 | 国立研究開発法人理化学研究所 | ワンステップ逆転写テンプレートスイッチpcrを利用したt細胞受容体およびb細胞受容体レパトア解析システム |
| WO2018026018A1 (ja) * | 2016-08-05 | 2018-02-08 | 国立大学法人東北大学 | T細胞受容体の認識機構を用いたがん又は感染症の治療及び診断 |
| WO2018104478A1 (en) * | 2016-12-08 | 2018-06-14 | Immatics Biotechnologies Gmbh | Novel t cell receptors and immune therapy using the same |
| JP2018525034A (ja) * | 2015-08-10 | 2018-09-06 | ハーエス ダイアグノミクス ゲーエムベーハー | 腫瘍特異的t細胞を提供するための方法 |
| WO2018168949A1 (ja) | 2017-03-15 | 2018-09-20 | 学校法人兵庫医科大学 | がん免疫療法の新規バイオマーカ |
| WO2019073965A1 (ja) | 2017-10-10 | 2019-04-18 | 国立大学法人広島大学 | エフェクターT細胞(Teff)抗原受容体を用いた抗原特異的制御性T細胞(Treg)の作出技術 |
| WO2019073964A1 (ja) | 2017-10-10 | 2019-04-18 | 国立大学法人広島大学 | Platinum TALENを用いたT細胞受容体の完全置換技術 |
| WO2020040302A1 (ja) | 2018-08-24 | 2020-02-27 | Repertoire Genesis株式会社 | T細胞受容体およびb細胞受容体の機能的なサブユニットペア遺伝子の解析方法 |
| WO2020040210A1 (ja) | 2018-08-22 | 2020-02-27 | 国立研究開発法人国立精神・神経医療研究センター | 筋痛性脳脊髄炎/慢性疲労症候群(me/cfs)のバイオマーカー |
| EP3458077A4 (en) * | 2016-05-17 | 2020-04-01 | Chimera Bioengineering Inc. | METHODS OF MANUFACTURING NEW AREAS OF ANTIGEN BINDING |
| US10626160B2 (en) | 2016-03-16 | 2020-04-21 | Immatics Biotechnologies Gmbh | Transfected T-cells and T-cell receptors for use in immunotherapy against cancers |
| CN111599411A (zh) * | 2020-06-08 | 2020-08-28 | 谱天(天津)生物科技有限公司 | 一种检测血液bcr重链和轻链的引物及免疫组库方法、应用 |
| CN111787930A (zh) * | 2017-10-06 | 2020-10-16 | 芝加哥大学 | 针对癌症特异性抗原对t淋巴细胞的筛选 |
| JP2021500406A (ja) * | 2017-10-26 | 2021-01-07 | ユニバーシティ カレッジ カーディフ コンサルタンツ エルティーディー | 新規t細胞受容体 |
| EP3786178A1 (en) * | 2019-08-30 | 2021-03-03 | Max-Delbrück-Centrum für Molekulare Medizin in der Helmholtz-Gemeinschaft | Tcr constructs specific for ebv-derived antigens |
| WO2022149549A1 (ja) * | 2021-01-05 | 2022-07-14 | オンコセラピー・サイエンス株式会社 | SARS-CoV-2蛋白由来ペプチドおよびそれを含むワクチン |
| WO2022270631A1 (ja) | 2021-06-25 | 2022-12-29 | Repertoire Genesis株式会社 | T細胞エピトープ配列を同定する方法およびその応用 |
| US11686724B2 (en) | 2012-03-28 | 2023-06-27 | Gadeta B.V. | Compositions comprising gamma 9 delta 2 T-cell receptors and methods of use thereof to treat cancer |
| US11840577B2 (en) | 2019-08-02 | 2023-12-12 | Immatics Biotechnologies Gmbh | Antigen binding proteins specifically binding MAGE-A |
Families Citing this family (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11390921B2 (en) | 2014-04-01 | 2022-07-19 | Adaptive Biotechnologies Corporation | Determining WT-1 specific T cells and WT-1 specific T cell receptors (TCRs) |
| US20160333409A1 (en) * | 2015-03-09 | 2016-11-17 | Cb Biotechnologies, Inc. | Method for identifying disease-associated cdr3 patterns in an immune repertoire |
| EP3067366A1 (en) * | 2015-03-13 | 2016-09-14 | Max-Delbrück-Centrum Für Molekulare Medizin | Combined T cell receptor gene therapy of cancer against MHC I and MHC II-restricted epitopes of the tumor antigen NY-ESO-1 |
| US11026969B2 (en) * | 2015-12-23 | 2021-06-08 | Fred Hutchinson Cancer Research Center | High affinity T cell receptors and uses thereof |
| CN109072227A (zh) * | 2016-03-15 | 2018-12-21 | 组库创世纪株式会社 | 用于免疫治疗的监测和诊断及治疗剂的设计 |
| CN107435065B (zh) * | 2016-05-10 | 2021-06-25 | 江苏荃信生物医药有限公司 | 鉴定灵长类抗体的方法 |
| CN110870018A (zh) * | 2017-05-19 | 2020-03-06 | 10X基因组学有限公司 | 用于分析数据集的系统和方法 |
| WO2018231958A1 (en) * | 2017-06-13 | 2018-12-20 | Adaptive Biotechnologies Corp. | Determining wt-1 specific t cells and wt-1 specific t cell receptors (tcrs) |
| CN109251980A (zh) * | 2017-07-14 | 2019-01-22 | 中国人民解放军第八医院 | 膀胱癌组织t细胞图谱模型及其构建方法和构建系统 |
| JP2021502802A (ja) * | 2017-10-10 | 2021-02-04 | メモリアル スローン ケタリング キャンサー センター | プライマ抽出およびクローン性検出のためのシステムおよび方法 |
| WO2019133874A1 (en) * | 2017-12-31 | 2019-07-04 | Berkeley Lights, Inc. | General functional assay |
| WO2019210144A1 (en) * | 2018-04-27 | 2019-10-31 | Vanderbilt University | Broadly neutralizing antibodies against hepatitis c virus |
| US20190352712A1 (en) * | 2018-05-04 | 2019-11-21 | Shoreline Biome, Llc | Multiple Specific/Nonspecific Primers for PCR of a Complex Gene Pool |
| CN108624667A (zh) * | 2018-05-15 | 2018-10-09 | 佛山市第人民医院(中山大学附属佛山医院) | 一种基于二代测序的t细胞受体库分析方法及装置 |
| KR102543325B1 (ko) * | 2018-08-13 | 2023-06-13 | 루트패스 제노믹스, 인크. | 쌍을 이룬 이분 면역수용체 폴리뉴클레오티드의 고처리량 클로닝 및 이의 적용 |
| CA3129831A1 (en) * | 2019-02-12 | 2020-08-20 | Tempus Labs, Inc. | An integrated machine-learning framework to estimate homologous recombination deficiency |
| CN113906047A (zh) | 2019-04-05 | 2022-01-07 | 根路径基因组学公司 | 用于t细胞受体基因组装的组合物和方法 |
| US20220308061A1 (en) * | 2019-09-03 | 2022-09-29 | The Regents Of The University Of California | Method to sequence mrna in single cells in parallel with quantification of intracellular phenotype |
| CN111073961A (zh) * | 2019-12-20 | 2020-04-28 | 苏州赛美科基因科技有限公司 | 一种基因稀有突变的高通量检测方法 |
| IL294459A (en) * | 2019-12-31 | 2022-09-01 | Singular Genomics Systems Inc | Polynucleotide barcodes for long read sequencing |
| CN111415707B (zh) * | 2020-03-10 | 2023-04-25 | 四川大学 | 临床个体化肿瘤新抗原的预测方法 |
| WO2021216738A2 (en) * | 2020-04-21 | 2021-10-28 | Intima Bioscience, Inc. | Compositions and methods of generating an immune response |
| JP2023548556A (ja) * | 2020-11-05 | 2023-11-17 | ボード オブ リージェンツ,ザ ユニバーシティ オブ テキサス システム | Egfr抗原を標的とする操作されたt細胞受容体および使用方法 |
| CN112322716B (zh) * | 2020-11-25 | 2021-07-30 | 深圳泛因医学有限公司 | 基于tcr/bcr高通量测序的特定淋巴细胞含量分析方法及装置 |
| CN112863601B (zh) * | 2021-01-15 | 2023-03-10 | 广州微远基因科技有限公司 | 病原微生物耐药基因归属模型及其建立方法和应用 |
| WO2022159773A1 (en) * | 2021-01-22 | 2022-07-28 | 10X Genomics, Inc. | Systems and methods for selecting cells of interest based on visualization of immune cell data |
| CN113122617B (zh) * | 2021-03-15 | 2023-07-14 | 成都益安博生物技术有限公司 | 一种筛选特异bcr/tcr的方法及其系统 |
| WO2023120562A1 (ja) * | 2021-12-22 | 2023-06-29 | 合同会社H.U.グループ中央研究所 | 抗体多様性の算出装置、抗体多様性の算出方法、ならびに抗体多様性の算出プログラム |
| CN116716327B (zh) * | 2023-08-04 | 2023-10-20 | 科士华(南京)生物技术有限公司 | 一种构建tcr载体的方法 |
Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4897445A (en) | 1986-06-27 | 1990-01-30 | The Administrators Of The Tulane Educational Fund | Method for synthesizing a peptide containing a non-peptide bond |
| WO1995018145A1 (en) | 1993-12-24 | 1995-07-06 | Ilexus Pty Ltd | Conjugates of human mucin and a carbohydrate polymer and their use in cancer treatment |
| JPH10229897A (ja) | 1997-02-19 | 1998-09-02 | Igaku Seibutsugaku Kenkyusho:Kk | 免疫グロブリン遺伝子発現の検出方法 |
| US5849589A (en) | 1996-03-11 | 1998-12-15 | Duke University | Culturing monocytes with IL-4, TNF-α and GM-CSF TO induce differentiation to dendric cells |
| US6406705B1 (en) | 1997-03-10 | 2002-06-18 | University Of Iowa Research Foundation | Use of nucleic acids containing unmethylated CpG dinucleotide as an adjuvant |
| JP2007515154A (ja) | 2003-05-13 | 2007-06-14 | モノクワント プロプライアタリー リミティド | 例えばt細胞受容体v/d/j遺伝子内の反復配列によるクローン細胞の同定 |
| WO2009137255A2 (en) | 2008-04-16 | 2009-11-12 | Hudsonalpha Institute For Biotechnology | Method for evaluating and comparing immunorepertoires |
| JP2012508011A (ja) | 2008-11-07 | 2012-04-05 | シーケンタ インコーポレイテッド | 配列解析によって病態をモニターする方法 |
| WO2013033721A1 (en) | 2011-09-02 | 2013-03-07 | Atreca, Inc. | Dna barcodes for multiplexed sequencing |
| WO2013043922A1 (en) | 2011-09-22 | 2013-03-28 | ImmuMetrix, LLC | Compositions and methods for analyzing heterogeneous samples |
| WO2013044234A1 (en) | 2011-09-22 | 2013-03-28 | ImmuMetrix, LLC | Detection of isotype profiles as signatures for disease |
| WO2013059725A1 (en) | 2011-10-21 | 2013-04-25 | Adaptive Biotechnologies Corporation | Quantification of adaptive immune cell genomes in a complex mixture of cells |
| JP2013524848A (ja) | 2010-05-06 | 2013-06-20 | シーケンタ インコーポレイテッド | クロノタイププロファイルを用いた健康状態および疾患状態のモニタリング |
Family Cites Families (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9423085D0 (en) | 1994-11-16 | 1995-01-04 | Stringer Bradley M J | Targeted T lymphocytes |
| GB0128153D0 (en) | 2001-11-23 | 2002-01-16 | Bayer Ag | Profiling of the immune gene repertoire |
| DE10211088A1 (de) | 2002-03-13 | 2003-09-25 | Ugur Sahin | Differentiell in Tumoren exprimierte Genprodukte und deren Verwendung |
| CA2501863A1 (en) | 2002-10-11 | 2004-04-22 | Erasmus Universiteit Rotterdam | Nucleic acid amplification primers for pcr-based clonality studies |
| JP3738308B2 (ja) | 2002-11-29 | 2006-01-25 | 篤 村口 | 抗原特異的リンパ球抗原受容体遺伝子のクローニング方法 |
| FR2863274B1 (fr) | 2003-12-05 | 2012-11-16 | Commissariat Energie Atomique | Procede d'evaluation quantitative d'un rearrangement ou d'une recombinaison genetique ciblee d'un individu et ses applications. |
| GB0328363D0 (en) * | 2003-12-06 | 2004-01-14 | Imp College Innovations Ltd | Therapeutically useful molecules |
| JP4480423B2 (ja) | 2004-03-08 | 2010-06-16 | 独立行政法人科学技術振興機構 | 免疫細胞クローンの拡大の有無の判定方法 |
| JP4069133B2 (ja) | 2004-11-19 | 2008-04-02 | 財団法人富山県新世紀産業機構 | 生物試料に含まれる目的遺伝子を増幅する方法 |
| DE102005013846A1 (de) | 2005-03-24 | 2006-10-05 | Ganymed Pharmaceuticals Ag | Identifizierung von Oberflächen-assoziierten Antigenen für die Tumordiagnose und -therapie |
| JP5026047B2 (ja) | 2006-10-18 | 2012-09-12 | 株式会社膠原病研究所 | 自己免疫疾患の発症に関わる自己応答性t細胞またはt細胞受容体の同定方法、およびその利用 |
| JP2009011236A (ja) | 2007-07-04 | 2009-01-22 | Shizuoka Prefecture | 1細胞レベルでのt細胞抗原レセプター遺伝子の解析・同定方法 |
| DK2567707T3 (en) | 2007-07-27 | 2017-07-31 | Immatics Biotechnologies Gmbh | Composition of tumour-associated peptides and related anti-cancer vaccine |
| EP2062982A1 (fr) | 2007-11-26 | 2009-05-27 | ImmunID | Procédé d'étude de la diversité combinatoire V(D)J |
| US20090216638A1 (en) | 2008-02-27 | 2009-08-27 | Total System Services, Inc. | System and method for providing consumer directed payment card |
| JP2010035472A (ja) | 2008-08-04 | 2010-02-18 | Shizuoka Prefecture | Sst−rex法を用いた新規のがん抗原特異的抗体遺伝子スクリーニング方法 |
| US8748103B2 (en) | 2008-11-07 | 2014-06-10 | Sequenta, Inc. | Monitoring health and disease status using clonotype profiles |
| WO2010151416A1 (en) | 2009-06-25 | 2010-12-29 | Fred Hutchinson Cancer Research Center | Method of measuring adaptive immunity |
| JP6192537B2 (ja) | 2010-08-06 | 2017-09-06 | ルードヴィヒ−マクシミリアン−ウニヴェルズィテート ミュンヘン | T細胞の標的抗原の同定 |
| CA3088393C (en) | 2010-09-20 | 2021-08-17 | Biontech Cell & Gene Therapies Gmbh | Antigen-specific t cell receptors and t cell epitopes |
| EP2663864B8 (en) | 2011-01-14 | 2019-06-05 | iRepertoire, Inc. | Immunodiversity assessment method and its use |
| JP2014514927A (ja) | 2011-04-13 | 2014-06-26 | イミュニカム・エイビイ | 抗原特異的t細胞の増殖のための方法 |
| EP3372694A1 (en) | 2012-03-05 | 2018-09-12 | Adaptive Biotechnologies Corporation | Determining paired immune receptor chains from frequency matched subunits |
| SG10201507700VA (en) | 2012-05-08 | 2015-10-29 | Adaptive Biotechnologies Corp | Compositions and method for measuring and calibrating amplification bias in multiplexed pcr reactions |
| CN105189779B (zh) | 2012-10-01 | 2018-05-11 | 适应生物技术公司 | 通过适应性免疫受体多样性和克隆性表征进行的免疫能力评估 |
-
2014
- 2014-11-20 CN CN201480073846.6A patent/CN106103711A/zh active Pending
- 2014-11-20 EP EP19181888.9A patent/EP3572510B1/en active Active
- 2014-11-20 EP EP14864397.6A patent/EP3091074B1/en active Active
- 2014-11-20 US US15/038,422 patent/US11203783B2/en active Active
- 2014-11-20 JP JP2015548994A patent/JP6164759B2/ja active Active
- 2014-11-20 WO PCT/JP2014/005849 patent/WO2015075939A1/ja active Application Filing
-
2017
- 2017-06-16 JP JP2017118613A patent/JP6661106B2/ja active Active
- 2017-11-20 JP JP2017222833A patent/JP6661107B2/ja active Active
-
2020
- 2020-01-15 JP JP2020004617A patent/JP2020074782A/ja active Pending
-
2021
- 2021-08-27 US US17/460,067 patent/US20220119884A1/en active Pending
-
2022
- 2022-01-18 JP JP2022005700A patent/JP2022062068A/ja active Pending
Patent Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4897445A (en) | 1986-06-27 | 1990-01-30 | The Administrators Of The Tulane Educational Fund | Method for synthesizing a peptide containing a non-peptide bond |
| WO1995018145A1 (en) | 1993-12-24 | 1995-07-06 | Ilexus Pty Ltd | Conjugates of human mucin and a carbohydrate polymer and their use in cancer treatment |
| US5849589A (en) | 1996-03-11 | 1998-12-15 | Duke University | Culturing monocytes with IL-4, TNF-α and GM-CSF TO induce differentiation to dendric cells |
| JPH10229897A (ja) | 1997-02-19 | 1998-09-02 | Igaku Seibutsugaku Kenkyusho:Kk | 免疫グロブリン遺伝子発現の検出方法 |
| US6406705B1 (en) | 1997-03-10 | 2002-06-18 | University Of Iowa Research Foundation | Use of nucleic acids containing unmethylated CpG dinucleotide as an adjuvant |
| JP2013116116A (ja) | 2003-05-13 | 2013-06-13 | Monoquant Pty Ltd | T細胞受容体v/d/j遺伝子内の反復配列によるクローン細胞の同定 |
| JP2007515154A (ja) | 2003-05-13 | 2007-06-14 | モノクワント プロプライアタリー リミティド | 例えばt細胞受容体v/d/j遺伝子内の反復配列によるクローン細胞の同定 |
| WO2009137255A2 (en) | 2008-04-16 | 2009-11-12 | Hudsonalpha Institute For Biotechnology | Method for evaluating and comparing immunorepertoires |
| JP2012508011A (ja) | 2008-11-07 | 2012-04-05 | シーケンタ インコーポレイテッド | 配列解析によって病態をモニターする方法 |
| JP2013524848A (ja) | 2010-05-06 | 2013-06-20 | シーケンタ インコーポレイテッド | クロノタイププロファイルを用いた健康状態および疾患状態のモニタリング |
| JP2013524849A (ja) | 2010-05-06 | 2013-06-20 | シーケンタ インコーポレイテッド | 複雑なアンプリコンの配列解析 |
| WO2013033721A1 (en) | 2011-09-02 | 2013-03-07 | Atreca, Inc. | Dna barcodes for multiplexed sequencing |
| WO2013043922A1 (en) | 2011-09-22 | 2013-03-28 | ImmuMetrix, LLC | Compositions and methods for analyzing heterogeneous samples |
| WO2013044234A1 (en) | 2011-09-22 | 2013-03-28 | ImmuMetrix, LLC | Detection of isotype profiles as signatures for disease |
| WO2013059725A1 (en) | 2011-10-21 | 2013-04-25 | Adaptive Biotechnologies Corporation | Quantification of adaptive immune cell genomes in a complex mixture of cells |
Non-Patent Citations (105)
| Title |
|---|
| "Current Protocols in Molecular Biology (1987-1997)", JOHN WILEY & SONS, article "Supplement 1-38" |
| "DNA Cloning 1: Core Techniques, A Practical Approach, Second Edition,", 1995, OXFORD UNIVERSITY PRESS |
| "DNA Microarrays and Latest PCR Methods", CELLULAR ENGINEERING |
| "Experimental Methods for Transgenesis & Expression Analysis", 1997, YODOSHA, article "Experimental Medicine, Supplemental" |
| "MOLECULAR CLONING : A LABORATORY MANUAL 3rd Ed.", 2001, COLD SPRING HARBOR |
| A. KIBBE: "Handbook of Pharmaceutical Excipients, 3rd ed.,", 2000, AMERICAN PHARMACEUTICAL ASSOCIATION AND PHARMACEUTICAL PRESS |
| ADAMS, R. L. ET AL.: "The Biochemistry of the Nucleic Acids", 1992, CHAPMAN & HALL |
| AHSEN ET AL., CLIN CHEM, vol. 47, no. 11, 2001, pages 1956 - 1961 |
| AHSEN ET AL., CLINCHEM, vol. 47, no. 11, 2001, pages 1956 - 1961 |
| ALLISON AC: "The mode of action of immunological adjuvants", DEV BIOL STAND., vol. 92, 1998, pages 3 - 11 |
| ALTSCHUL ET AL., J.MOL.BIOL., vol. 215, 1990, pages 403 - 410 |
| ANNUAL REVIEW IMMUNOLOGY, vol. 18, 2000, pages 495 - 527 |
| ANNUAL REVIEW IMMUNOLOGY, vol. 6, 1993, pages 309 - 326 |
| ARONESTY E., THE OPEN BIOINFORMATICSJOURNAL, vol. 7, 2013, pages 1 - 8 |
| ARTHUR M. KRIEG: "Therapeutic potential of Toll-like receptor 9 activation", NATUREREVIEWS, DRUG DISCOVERY, vol. 5, 2006, pages 471 - 484 |
| ASSAF C; HUMMEL M; DIPPEL E; GOERDT S; MULLER HH; ANAGNOSTOPOULOS I; ORFANOS CE; STEIN H: "High detection rate of T-cell receptor beta chain rearrangements in T-cell lymphoproliferations by family specific polymerase chain reaction in combination with the GeneScan technique and DNA sequencing", BLOOD, vol. 96, no. 2, 15 July 2000 (2000-07-15), pages 640 - 6 |
| AUSUBEL ET AL.: "Current Protocols in Molecular Biology", 1995, WILEY INTERSCIENCE PUBLISHERS |
| AUSUBEL, F. M.: "Current Protocols in Molecular Biology", 1987, GREENE PUB. ASSOCIATES AND WILEY-INTERSCIENCE |
| AUSUBEL, F. M.: "Short Protocols in Molecular Biology: A Compendium of Methods from Current Protocols in Molecular Biology", 1992, GREENE PUB. ASSOCIATES |
| AUSUBEL, F. M.: "Short Protocols in Molecular Biology: A Compendium of Methods from Current Protocols in Molecular Biology", 1995, GREENE PUB. ASSOCIATES |
| AUSUBEL, F. M.: "Short Protocols in Molecular Biology: A Compendium of Methods from Current Protocols in Molecular Biology", 1999, WILEY |
| AUSUBEL, F.M.: "Short Protocols in Molecular Biology: A Compendium of Methods from Current Protocols in Molecular Biology", 1989, GREENE PUB. ASSOCIATES AND WILEY-INTERSCIENCE |
| BATZER ET AL., NUCLEIC ACID RES., vol. 19, 1991, pages 5081 |
| BLACKBURN, G. M. ET AL.: "Nucleic Acids in Chemistry and Biology", 1996, OXFORD UNIVERSITY PRESS |
| BOLOTIN D.A. ET AL.: "Next generation sequencing for TCR repertoire profiling: platform-specific features and correction algorithms.", EUR. J. IMMUNOL., vol. 42, no. 11, 2012, pages 3073 - 3083, XP055235351 * |
| BOMMARITO ET AL., NUCLEIC ACIDS RES, vol. 28, no. 9, 2000, pages 1929 - 1934 |
| BRUNSVIG PF; AAMDAL S; GJERTSEN MK; KVALHEIM G; MARKOWSKI-GRIMSRUD CJ; SVE I; DYRHAUG M; TRACHSEL S; MULLER M; ERIKSEN JA: "Telomerase peptide vaccination: a phase I/II study in patients with non-small cell lung cancer", CANCER IMMUNOLIMMUNOTHER., vol. 55, no. 12, 2006, pages 1553 - 1564 |
| BURCHELL ET AL.: "Breast Cancer, Advances in biology and therapeutics", 1996, JOHN LIBBEY EUROTEXT, pages: 309 - 313 |
| CELL, vol. 76, 1994, pages 287 - 299 |
| CHEE-SENG, KU; EN YUN, LOY; YUDI, PAWITAN; KEE-SENG, CHIA: "Encyclopedia of Life Sciences (ELS", April 2010, JOHN WILEY SONS, LTD, article "Next Generation Sequencing Technologies and Their Applications" |
| CHUSAINOW J; YANG YS; YEO JH; TOH PC; ASVADI P; WONG NS; YAP MG: "A study of monoclonal antibody-producing CHO cell lines: what makes a stable high producer?", BIOTECHNOL BIOENG, vol. 102, no. 4, 1 March 2009 (2009-03-01), pages 1182 - 96 |
| COLIGAN ET AL.: "Current Protocols In Protein Science", 1995, JOHN WILEY& SONS |
| COLWELL, R. K. ET AL., JOURNAL OF PLANT ECOLOGY, vol. 5, pages 3 - 21 |
| CONDON ET AL., NATURE MEDICINE, vol. 2, 1996, pages 1122 - 1127 |
| CONRY ET AL., SEMINARS IN ONCOLOGY, vol. 23, 1996, pages 135 - 147 |
| DUPUIS M; MURPHY TJ; HIGGINS D; UGOZZOLI M; VAN NEST G; OTT G; MCDONALD DM: "Dendritic cells internalize vaccine adjuvant after intramuscular injection", CELL IMMUNOL., vol. 186, no. 1, 1998, pages 18 - 27 |
| ECKSTEIN, F.: "Oligonucleotides and Analogues: A Practical Approach", 1991, IRL PRESS |
| FREEMAN J.D. ET AL.: "Profiling the T- cell receptor beta-chain repertoire by massively parallel sequencing.", GENOME RES., vol. 19, no. 10, 2009, pages 1817 - 1824, XP002636496 * |
| GABRILOVICH DI; CUNNINGHAM HT: "Carbone DP; IL-12 and mutant P53 peptide-pulsed dendritic cells for the specific immunotherapy of cancer", J IMMUNOTHER EMPHASIS TUMOR IMMUNOL., 1996, pages 414 - 418 |
| GAIT, M. J.: "Oligonucleotide Synthesis: A Practical Approach", 1985, IRL PRESS |
| GAIT, M. J.: "Oligonucleotide Synthesis: A Practical Approach", 1990, IRL PRESS |
| GONG ET AL., NATURE MEDICINE, vol. 3, 1997, pages 558 - 561 |
| GRAHAM ET AL., INT J. CANCER, vol. 65, 1996, pages 664 - 670 |
| HARRIS. T.D., SCIENCE, 2008, pages 320 - 160,109 |
| HERMANSON, G. T.: "Bioconjugate Techniques", 1996, ACADEMIC PRESS |
| HUMAN IMMUNOLOGY, vol. 56, 1997, pages 57 - 69 |
| HUTTER D ET AL., NUCLEOSIDES NUCLEOTIDES NUCLEIC ACID, vol. 29, no. 11, 2010, pages 879 - 95 |
| IMMUNOLOGY, vol. 96, 1999, pages 465 - 72 |
| INNIS, M. A. ET AL.: "PCR Strategies", 1995, ACADEMIC PRESS |
| INNIS, M. A.: "PCR Protocols: A Guide to Methods and Applications", 1990, ACADEMIC PRESS |
| ISHIDA I; TOMIZUKA K; YOSHIDA H; TAHARA T; TAKAHASHI N; OHGUMA A; TANAKA S; UMEHASHI M; MAEDA H; NOZAKI C: "Production of human monoclonal and polyclonal antibodies in TransChromo animals", CLONING STEM CELLS, vol. 4, no. 1, 2002, pages 91 - 102 |
| ITO M; HIRAMATSU H; KOBAYASHI K; SUZUE K; KAWAHATA M; HIOKI K; UEYAMA Y; KOYANAGI Y; SUGAMURA K; TSUJI K: "NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells", BLOOD, vol. 100, no. 9, 1 November 2002 (2002-11-01), pages 3175 - 82 |
| J. SAMBROOK ET AL.,: "Molecular Cloning: A Laboratory Manual, 2nd ed.,", 1989, COLD SPRING HARBOR |
| JAYAPAL KP; WLASCHIN KF; HU W-S; YAP MGS: "Recombinant protein therapeutics from CHO cells-20 years and counting", CHEM ENG PROG., vol. 103, 2007, pages 40,47 |
| JOURNAL OF IMMUNOLOGICAL METHODS, vol. 169, 1994, pages 17 - 23 |
| JOURNAL OF IMMUNOLOGICAL METHODS, vol. 201, 1997, pages 145 - 15 |
| KITAURA K. ET AL.: "A new method for quantitative analysis of the T cell receptor V region repertoires in healthy common marmosets by microplate hybridization assay.", J. IMMUNOL. METHODS, vol. 384, no. 1-2, 2012, pages 81 - 91, XP055345569 * |
| LAGISETTY KH; MORGAN RA: "Cancer therapy with genetically-modified T cells for the treatment of melanoma", J GENE MED., vol. 14, no. 6, June 2012 (2012-06-01), pages 400 - 4 |
| LANGERAK AW ET AL., BLOOD, vol. 98, 2001, pages 165 - 173 |
| LEUKEMIA RESEARCH, vol. 27, 2003, pages 305 - 312 |
| LINNEMANN C; SCHUMACHER TN; BENDLE GM: "T-cell receptor gene therapy: critical parameters for clinical success", J INVEST DERMATOL., vol. 131, no. 9, September 2011 (2011-09-01), pages 1806 - 16 |
| LITOSH VA ET AL., NUCLEIC ACIDS RES., vol. 39, no. 6, March 2011 (2011-03-01), pages E39 |
| LONGENECKER ET AL., ANN. NY ACAD. SCI., vol. 690, 1993, pages 276 - 291 |
| LU ET AL., J. ORG. CHEM., vol. 46, 1981, pages 3433 |
| M. STAEHLER; A. STENZL; P. Y. DIETRICH; T.EISEN; A. HAFERKAMP; J. BECK; A. MAYER; S. WALTER; H. SINGH; J. FRISCH: "An open label study to evaluate the safety and immunogenicity of the peptide based cancer vaccine IMA901", ASCO MEETING, 2007 |
| MACISAAC C ET AL., J IMMUNOL METHODS, vol. 283, 2003, pages 9 - 15 |
| MARKET, PROC NATL ACAD SCI U S A., vol. 81, no. 18, September 1984 (1984-09-01), pages 5662 - 5666 |
| MATSUTANI T ET AL., BR J HAEMATOL, vol. 109, 2000, pages 759 - 769 |
| MATSUTANI T ET AL., HUM IMMUNOL, vol. 56, 1997, pages 57 - 69 |
| MATSUTANI T ET AL., MOL IMMUNOL, vol. 44, 2007, pages 2378 - 2387 |
| MATSUTANI T ET AL., MOL IMMUNOL, vol. 48, 2011, pages 623 - 629 |
| METZKER ML: "Sequencing technologies - the next generation", NAT REV GENET., vol. 11, no. 1, January 2010 (2010-01-01), pages 31 - 46 |
| MEZIERE ET AL., J. IMMUNOL., vol. 159, 1997, pages 3230 - 3237 |
| NAT GENET., vol. 32, December 2002 (2002-12-01), pages 526 - 32 |
| NEEDLEMAN; WUNSCH, J.MOL.BIOL., vol. 48, 1970, pages 443 - 453 |
| NUCLEIC ACID RESEARCH, vol. 37, no. LL, 2009, pages D1006 - D1012 |
| OHTSUKA ET AL., J.BIOL.CHEM., vol. 260, 1985, pages 2605 - 2608 |
| PEARSON; LIPMAN, PROC.NATL.ACAD.SCI., USA, vol. 85, 1988, pages 2444 - 2448 |
| PROC NATL ACAD SCI, vol. 90, 1993, pages 2385 - 2388 |
| PUSKAS LG ET AL., BIOTECHNIQUES, vol. 32, no. 6, June 2002 (2002-06-01), pages 1330 - 4,1336,1338,1340 |
| PUSKAS LG; ZVARA A; HACKLER L JR; VAN HUMMELEN P: "RNA amplification results in reproducible microarray data with slight ratio bias", BIOTECHNIQUES, vol. 32, no. 6, June 2002 (2002-06-01), pages 1330 - 4,1336,1338,1340 |
| R. LUNDBLAD: "Chemical Reagents for Protein Modification, 3rd ed.,", 2005, CRC PRESS |
| REBAI N ET AL., PROC NATL ACAD SCI USA, vol. 91, 1994, pages 1529 - 1533 |
| ROSSOLINI ET AL., MOL.CELL.PROBES, vol. 8, 1994, pages 91 - 98 |
| ROTH ET AL., SCAND. J. IMMUNOLOGY, vol. 43, 1996, pages 646 - 651 |
| SAMBROOK ET AL.: "Molecular Cloning, A Laboratory Manual", 1989, COLD SPRING HARBOR LABORATORY |
| SAMBROOK ET AL.: "Molecular Cloning: A Laboratory Manual, Third Ed.,", vol. 1, 2001, COLD SPRING HARBOR LABORATORY PRESS, pages: 7.42 - 7.45 |
| SAMBROOK J. ET AL.: "Molecular Cloning: A Laboratory Manual", 1989, COLD SPRING HARBOR |
| SANTA LUCIA, J. PROC NATL ACAD SCI USA, vol. 95, no. 4, 1998, pages 1460 - 1465 |
| SHABAROVA, Z. ET AL.: "Advanced Organic Chemistry of Nucleic Acids", 1994, WEINHEIM |
| SHUJUNSHA, SAIBO KOGAKU BESSATSU: "DNA Maikuroarei to Saishin PCR ho ("DNA Microarrays and Latest PCR Methods")", CELLULAR ENGINEERING, EXTRA ISSUE, |
| SMITH; WATERMAN, J.MOL.BIOL., vol. 147, 1981, pages 195 - 197 |
| SNINSKY, J. J. ET AL.: "PCR Applications: Protocols for Functional Genomics", 1999, ACADEMIC PRESS |
| TANG DT; PLESSY C; SALIMULLAH M; SUZUKI AM; CALLIGARIS R; GUSTINCICH S; CARNINCI P: "Suppression of artifacts and barcode bias in high-throughput transcriptome analyses utilizing template switching", NUCLEIC ACIDS RES., vol. 41, no. 3, 1 February 2013 (2013-02-01), pages E44 |
| TEMBHARE P ET AL., AM J CLIN PATHOL, vol. 135, 2011, pages 890 - 900 |
| THOMAS AM; SANTARSIERO LM; LUTZ ER; ARMSTRONG TD; CHEN YC; HUANG LQ; LAHERU DA; GOGGINS M; HRUBAN RH; JAFFEE EM: "Mesothelin-specific CD8(+) T cell responses provide evidence of in vivo cross-priming by antigen-presenting cells in vaccinated pancreatic cancer patients", J EXP MED., vol. 200, no. 3, 2 August 2004 (2004-08-02), pages 297 - 306 |
| TUNNACLIFFE A; KEFFORD R; MILSTEIN C; FORSTER A; RABBITTS TH: "Sequence and evolution of the human T-cell antigen receptor beta-chain genes", PROC NATL ACAD SCI USA., vol. 82, no. 15, August 1985 (1985-08-01), pages 5068 - 72 |
| UTTENTHAL BJ; CHUA I; MORRIS EC; STAUSS HJ: "Challenges in T cell receptor gene therapy", J GENE MED., vol. 14, no. 6, June 2012 (2012-06-01), pages 386 - 99 |
| VAN DEN BEEMD R ET AL., CYTOMETRY, vol. 40, 2000, pages 336 - 345 |
| VILLANYI Z; MAI, A; SZABAD J: "Repeated template switching: Obstacles in cDNA libraries and ways to avoid them", THE OPEN GENOMICS JOURNAL, vol. 5, 2012, pages 1 - 6 |
| WANG ET AL., SCIENCE, vol. 224, no. 4656, 29 June 1984 (1984-06-29), pages 1431 - 1433 |
| YUSUKE NAKAMURA,: "Genomu Kaiseki Jikkenho Nakamura Yusuke Labo Manyuaru", 2002, YODOSHA |
| ZHAI ET AL., J.IMMUNOL., vol. 156, 1996, pages 700 - 710 |
| ZHOU ET AL., BLOOD, vol. 86, 1995, pages 3295 - 3301 |
| ZOLLER ET AL., NUCLEIC ACIDS RES., vol. 10, no. 20, 25 October 1982 (1982-10-25), pages 6487 - 6500 |
Cited By (62)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11686724B2 (en) | 2012-03-28 | 2023-06-27 | Gadeta B.V. | Compositions comprising gamma 9 delta 2 T-cell receptors and methods of use thereof to treat cancer |
| EP2954070A4 (en) * | 2013-02-11 | 2016-10-26 | Cb Biotechnologies Inc | METHOD FOR ASSESSING AN IMMUNOREPERTOIR |
| WO2016136716A1 (ja) * | 2015-02-23 | 2016-09-01 | 国立大学法人東北大学 | 遺伝子特異的非バイアス増幅法 |
| JPWO2016136716A1 (ja) * | 2015-02-23 | 2017-11-30 | 国立大学法人東北大学 | 遺伝子特異的非バイアス増幅法 |
| CN105095687A (zh) * | 2015-06-26 | 2015-11-25 | 南方科技大学 | 一种免疫组库数据分析方法及终端 |
| JP2018525034A (ja) * | 2015-08-10 | 2018-09-06 | ハーエス ダイアグノミクス ゲーエムベーハー | 腫瘍特異的t細胞を提供するための方法 |
| WO2017046335A1 (en) * | 2015-09-18 | 2017-03-23 | INSERM (Institut National de la Santé et de la Recherche Médicale) | T cell receptors (tcr) and uses thereof for the diagnosis and treatment of diabetes |
| US11078251B2 (en) | 2015-09-18 | 2021-08-03 | INSERM (Institut National de la Santé et de la Recherche Médicale) | T cell receptors (TCR) and uses thereof for the diagnosis and treatment of diabetes |
| KR102259109B1 (ko) | 2016-03-16 | 2021-06-01 | 이매틱스 바이오테크놀로지스 게엠베하 | 암에 대한 면역요법에서의 사용을 위하여 형질주입된 t 세포 및 t 세포 수용체 |
| US10538573B2 (en) | 2016-03-16 | 2020-01-21 | Immatics Biotechnologies Gmbh | Transfected T-cells and T-cell receptors for use in immunotherapy against cancers |
| TWI826891B (zh) * | 2016-03-16 | 2023-12-21 | 德商英麥提克生物技術股份有限公司 | 轉染 t 細胞和 t 細胞受體用於癌症免疫治療 |
| AU2017235467B2 (en) * | 2016-03-16 | 2021-04-08 | Immatics Biotechnologies Gmbh | Transfected T-cells and T-cell receptors for use in immunotherapy against cancers |
| US10889629B2 (en) | 2016-03-16 | 2021-01-12 | Immatics Biotechnologies Gmbh | Transfected T-cells and T-cell receptors for use in immunotherapy against cancers |
| KR20180118783A (ko) * | 2016-03-16 | 2018-10-31 | 이매틱스 바이오테크놀로지스 게엠베하 | 암에 대한 면역요법에서의 사용을 위하여 형질주입된 t 세포 및 t 세포 수용체 |
| CN108884136A (zh) * | 2016-03-16 | 2018-11-23 | 伊玛提克斯生物技术有限公司 | 用于癌症免疫治疗的转染t细胞和t细胞受体 |
| US11945854B2 (en) | 2016-03-16 | 2024-04-02 | Immatics Biotechnologies Gmbh | Transfected T-cells and T-cell receptors for use in immunotherapy against cancers |
| WO2017158103A1 (en) * | 2016-03-16 | 2017-09-21 | Immatics Biotechnologies Gmbh | Transfected t-cells and t-cell receptors for use in immunotherapy against cancers |
| CN108884136B (zh) * | 2016-03-16 | 2023-03-17 | 伊玛提克斯生物技术有限公司 | 用于癌症免疫治疗的转染t细胞和t细胞受体 |
| US10626160B2 (en) | 2016-03-16 | 2020-04-21 | Immatics Biotechnologies Gmbh | Transfected T-cells and T-cell receptors for use in immunotherapy against cancers |
| TWI764886B (zh) * | 2016-03-16 | 2022-05-21 | 德商英麥提克生物技術股份有限公司 | 轉染 t 細胞和 t 細胞受體用於癌症免疫治療 |
| CN107345241A (zh) * | 2016-05-12 | 2017-11-14 | 眭维国 | B细胞抗原受体h链cdr3的处理方法 |
| EP3458077A4 (en) * | 2016-05-17 | 2020-04-01 | Chimera Bioengineering Inc. | METHODS OF MANUFACTURING NEW AREAS OF ANTIGEN BINDING |
| WO2017212072A1 (en) * | 2016-06-10 | 2017-12-14 | Umc Utrecht Holding B.V. | Human leukocyte antigen restricted gamma delta t cell receptors and methods of use thereof |
| US11596654B2 (en) | 2016-06-10 | 2023-03-07 | Gadeta B.V. | Human leukocyte antigen restricted gamma delta T cell receptors and methods of use thereof |
| US11166984B2 (en) | 2016-06-10 | 2021-11-09 | Umc Utrecht Holding B.V. | Method for identifying δT-cell (or γT-cell) receptor chains or parts thereof that mediate an anti-tumour or an anti-infective response |
| TWI758298B (zh) * | 2016-06-23 | 2022-03-21 | 國立研究開發法人理化學研究所 | 利用單步驟逆轉錄模板置換pcr之t細胞受體及b細胞受體多樣性解析系統 |
| EP3486321A4 (en) * | 2016-06-23 | 2020-01-15 | Riken | SYSTEM FOR T CELL RECEPTOR AND B CELL RECEPTOR ANALYSIS USING ONE-STEP RT-PCR WITH MATRIX SWITCHING |
| JP7066139B2 (ja) | 2016-06-23 | 2022-05-13 | 国立研究開発法人理化学研究所 | ワンステップ逆転写テンプレートスイッチpcrを利用したt細胞受容体およびb細胞受容体レパトア解析システム |
| JPWO2017222056A1 (ja) * | 2016-06-23 | 2019-04-11 | 国立研究開発法人理化学研究所 | ワンステップ逆転写テンプレートスイッチpcrを利用したt細胞受容体およびb細胞受容体レパトア解析システム |
| CN109312327A (zh) * | 2016-06-23 | 2019-02-05 | 国立研究开发法人理化学研究所 | 使用一步逆转录模板转换pcr的t细胞受体和b细胞受体库分析系统 |
| WO2017222056A1 (ja) * | 2016-06-23 | 2017-12-28 | 国立研究開発法人理化学研究所 | ワンステップ逆転写テンプレートスイッチpcrを利用したt細胞受容体およびb細胞受容体レパトア解析システム |
| JP7012364B2 (ja) | 2016-08-05 | 2022-02-14 | 国立大学法人東北大学 | T細胞受容体の認識機構を用いたがん又は感染症の治療及び診断 |
| WO2018026018A1 (ja) * | 2016-08-05 | 2018-02-08 | 国立大学法人東北大学 | T細胞受容体の認識機構を用いたがん又は感染症の治療及び診断 |
| JPWO2018026018A1 (ja) * | 2016-08-05 | 2019-06-27 | 国立大学法人東北大学 | T細胞受容体の認識機構を用いたがん又は感染症の治療及び診断 |
| US11325961B2 (en) | 2016-08-05 | 2022-05-10 | Tohoku University | Natural killer cell function enhancer |
| AU2017373815C1 (en) * | 2016-12-08 | 2021-08-26 | Immatics Biotechnologies Gmbh | Novel T cell receptors and immune therapy using the same |
| WO2018104478A1 (en) * | 2016-12-08 | 2018-06-14 | Immatics Biotechnologies Gmbh | Novel t cell receptors and immune therapy using the same |
| AU2017373815B2 (en) * | 2016-12-08 | 2020-12-10 | Immatics Biotechnologies Gmbh | Novel T cell receptors and immune therapy using the same |
| US10527623B2 (en) | 2016-12-08 | 2020-01-07 | Immatics Biotechnologies Gmbh | T cell receptors and immune therapy using the same |
| US10725044B2 (en) | 2016-12-08 | 2020-07-28 | Immatics Biotechnologies Gmbh | T cell receptors and immune therapy using the same |
| WO2018168949A1 (ja) | 2017-03-15 | 2018-09-20 | 学校法人兵庫医科大学 | がん免疫療法の新規バイオマーカ |
| CN111787930A (zh) * | 2017-10-06 | 2020-10-16 | 芝加哥大学 | 针对癌症特异性抗原对t淋巴细胞的筛选 |
| US11788076B2 (en) | 2017-10-10 | 2023-10-17 | Hiroshima University | Full replacement technique for T cell receptor using platinum TALEN |
| JP7333495B2 (ja) | 2017-10-10 | 2023-08-25 | 国立大学法人広島大学 | エフェクターT細胞(Teff)抗原受容体を用いた抗原特異的制御性T細胞(Treg)の作出技術 |
| JPWO2019073965A1 (ja) * | 2017-10-10 | 2021-01-14 | 国立大学法人広島大学 | エフェクターT細胞(Teff)抗原受容体を用いた抗原特異的制御性T細胞(Treg)の作出技術 |
| WO2019073965A1 (ja) | 2017-10-10 | 2019-04-18 | 国立大学法人広島大学 | エフェクターT細胞(Teff)抗原受容体を用いた抗原特異的制御性T細胞(Treg)の作出技術 |
| WO2019073964A1 (ja) | 2017-10-10 | 2019-04-18 | 国立大学法人広島大学 | Platinum TALENを用いたT細胞受容体の完全置換技術 |
| JP7289311B2 (ja) | 2017-10-26 | 2023-06-09 | ユニバーシティ カレッジ カーディフ コンサルタンツ エルティーディー | 新規t細胞受容体 |
| JP2021500406A (ja) * | 2017-10-26 | 2021-01-07 | ユニバーシティ カレッジ カーディフ コンサルタンツ エルティーディー | 新規t細胞受容体 |
| CN112840033A (zh) * | 2018-08-22 | 2021-05-25 | 国立研究开发法人国立精神·神经医疗研究中心 | 肌痛性脑脊髓炎/慢性疲劳综合症(me/cfs)的生物标志物 |
| JPWO2020040210A1 (ja) * | 2018-08-22 | 2021-08-10 | 国立研究開発法人国立精神・神経医療研究センター | 筋痛性脳脊髄炎/慢性疲労症候群(me/cfs)のバイオマーカー |
| WO2020040210A1 (ja) | 2018-08-22 | 2020-02-27 | 国立研究開発法人国立精神・神経医療研究センター | 筋痛性脳脊髄炎/慢性疲労症候群(me/cfs)のバイオマーカー |
| WO2020040302A1 (ja) | 2018-08-24 | 2020-02-27 | Repertoire Genesis株式会社 | T細胞受容体およびb細胞受容体の機能的なサブユニットペア遺伝子の解析方法 |
| JP7328631B2 (ja) | 2018-08-24 | 2023-08-17 | Repertoire Genesis株式会社 | T細胞受容体およびb細胞受容体の機能的なサブユニットペア遺伝子の解析方法 |
| JPWO2020040302A1 (ja) * | 2018-08-24 | 2021-08-10 | Repertoire Genesis株式会社 | T細胞受容体およびb細胞受容体の機能的なサブユニットペア遺伝子の解析方法 |
| US11840577B2 (en) | 2019-08-02 | 2023-12-12 | Immatics Biotechnologies Gmbh | Antigen binding proteins specifically binding MAGE-A |
| WO2021038031A1 (en) * | 2019-08-30 | 2021-03-04 | Max-Delbrück-Centrum Für Molekulare Medizin In Der Helmholtz-Gemeinschaft | Tcr constructs specific for ebv-derived antigens |
| EP3786178A1 (en) * | 2019-08-30 | 2021-03-03 | Max-Delbrück-Centrum für Molekulare Medizin in der Helmholtz-Gemeinschaft | Tcr constructs specific for ebv-derived antigens |
| CN111599411A (zh) * | 2020-06-08 | 2020-08-28 | 谱天(天津)生物科技有限公司 | 一种检测血液bcr重链和轻链的引物及免疫组库方法、应用 |
| CN111599411B (zh) * | 2020-06-08 | 2023-04-14 | 谱天(天津)生物科技有限公司 | 一种检测血液bcr重链和轻链的引物及免疫组库方法、应用 |
| WO2022149549A1 (ja) * | 2021-01-05 | 2022-07-14 | オンコセラピー・サイエンス株式会社 | SARS-CoV-2蛋白由来ペプチドおよびそれを含むワクチン |
| WO2022270631A1 (ja) | 2021-06-25 | 2022-12-29 | Repertoire Genesis株式会社 | T細胞エピトープ配列を同定する方法およびその応用 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20220119884A1 (en) | 2022-04-21 |
| JP2018085985A (ja) | 2018-06-07 |
| EP3572510B1 (en) | 2022-09-21 |
| US20160289760A1 (en) | 2016-10-06 |
| JP6661107B2 (ja) | 2020-03-11 |
| JPWO2015075939A1 (ja) | 2017-03-16 |
| CN106103711A (zh) | 2016-11-09 |
| JP2017212988A (ja) | 2017-12-07 |
| EP3091074B1 (en) | 2019-08-07 |
| JP6661106B2 (ja) | 2020-03-11 |
| EP3572510A1 (en) | 2019-11-27 |
| US11203783B2 (en) | 2021-12-21 |
| JP2022062068A (ja) | 2022-04-19 |
| EP3091074A4 (en) | 2017-06-21 |
| EP3091074A1 (en) | 2016-11-09 |
| JP6164759B2 (ja) | 2017-07-19 |
| JP2020074782A (ja) | 2020-05-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6661107B2 (ja) | T細胞受容体およびb細胞受容体レパトアの解析のための方法およびそのためのソフトウェア | |
| Au et al. | Determinants of anti-PD-1 response and resistance in clear cell renal cell carcinoma | |
| JP7307048B2 (ja) | 腫瘍におけるhlaアレルの分析及びそれらの使用 | |
| US11083784B2 (en) | Peptides and combination of peptides for use in immunotherapy against CLL and other cancers | |
| US20210104294A1 (en) | Method for predicting hla-binding peptides using protein structural features | |
| US20210382068A1 (en) | Hla single allele lines | |
| AU2015314776A1 (en) | Personalized cancer vaccines and methods therefor | |
| Schaettler et al. | TCR-engineered adoptive cell therapy effectively treats intracranial murine glioblastoma | |
| WO2019008365A1 (en) | METHOD OF TREATING CANCER WITH A NEO-ANTIGEN INDEL PHASE | |
| US20210172961A1 (en) | Methods for identifying rna editing-derived epitopes that elicit immune responses in cancer | |
| US20240024439A1 (en) | Administration of anti-tumor vaccines | |
| TW202126676A (zh) | 用於抗cll及其他癌症之免疫治療的新穎胜肽及胜肽組合物 | |
| US20230248814A1 (en) | Compositions and methods for treating merkel cell carcinoma (mcc) using hla class i specific epitopes | |
| AU2022348080A1 (en) | Novel tumor-specific antigens for colorectal cancer and uses thereof | |
| WO2022226055A1 (en) | Personalized allogeneic immunotherapy |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14864397 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2015548994 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15038422 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2014864397 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014864397 Country of ref document: EP |