WO2013147141A1 - 粒状ポリアリーレンスルフィド及びその製造方法 - Google Patents
粒状ポリアリーレンスルフィド及びその製造方法 Download PDFInfo
- Publication number
- WO2013147141A1 WO2013147141A1 PCT/JP2013/059497 JP2013059497W WO2013147141A1 WO 2013147141 A1 WO2013147141 A1 WO 2013147141A1 JP 2013059497 W JP2013059497 W JP 2013059497W WO 2013147141 A1 WO2013147141 A1 WO 2013147141A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polyarylene sulfide
- polymerization
- phase separation
- sieve
- pas
- Prior art date
Links
Images
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G75/00—Macromolecular compounds obtained by reactions forming a linkage containing sulfur with or without nitrogen, oxygen, or carbon in the main chain of the macromolecule
- C08G75/02—Polythioethers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G75/00—Macromolecular compounds obtained by reactions forming a linkage containing sulfur with or without nitrogen, oxygen, or carbon in the main chain of the macromolecule
- C08G75/14—Polysulfides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G75/00—Macromolecular compounds obtained by reactions forming a linkage containing sulfur with or without nitrogen, oxygen, or carbon in the main chain of the macromolecule
- C08G75/02—Polythioethers
- C08G75/0204—Polyarylenethioethers
- C08G75/0209—Polyarylenethioethers derived from monomers containing one aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G75/00—Macromolecular compounds obtained by reactions forming a linkage containing sulfur with or without nitrogen, oxygen, or carbon in the main chain of the macromolecule
- C08G75/02—Polythioethers
- C08G75/0204—Polyarylenethioethers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G75/00—Macromolecular compounds obtained by reactions forming a linkage containing sulfur with or without nitrogen, oxygen, or carbon in the main chain of the macromolecule
- C08G75/02—Polythioethers
- C08G75/0204—Polyarylenethioethers
- C08G75/0231—Polyarylenethioethers containing chain-terminating or chain-branching agents
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G75/00—Macromolecular compounds obtained by reactions forming a linkage containing sulfur with or without nitrogen, oxygen, or carbon in the main chain of the macromolecule
- C08G75/02—Polythioethers
- C08G75/0204—Polyarylenethioethers
- C08G75/025—Preparatory processes
- C08G75/0268—Preparatory processes using disulfides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/12—Powdering or granulating
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L81/00—Compositions of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing sulfur with or without nitrogen, oxygen or carbon only; Compositions of polysulfones; Compositions of derivatives of such polymers
- C08L81/02—Polythioethers; Polythioether-ethers
Definitions
- the present invention relates to a granular polyarylene sulfide and a method for producing the same. More specifically, the present invention is a granular polyarylene sulfide having good thermal stability, low gas generation during molding, low halogen content and nitrogen content, low melt viscosity, and high performance balance. About. Furthermore, the present invention is a method for producing a granular polyarylene sulfide in which a sulfur source and a dihaloaromatic compound are polymerized in an organic amide solvent, wherein the polymerization reaction is performed in the presence of a disulfide compound in the polymerization step.
- the present invention relates to a method for producing a granular polyarylene sulfide which is screened and recovered with high yield.
- PAS Polyarylene sulfide
- PPS polyphenylene sulfide
- PPS polyphenylene sulfide
- Engineering plastic with excellent stability.
- PAS can be molded into various molded products, films, sheets, fibers, etc. by general melt processing methods such as extrusion molding, injection molding, compression molding, etc., so it can be used in a wide range of fields such as electrical / electronic equipment and automotive equipment. It is widely used.
- PAS As a typical production method of PAS, a method of reacting a sulfur source with a dihaloaromatic compound in an organic amide solvent such as N-methyl-2-pyrrolidone (hereinafter abbreviated as “NMP”) is known. ing.
- NMP N-methyl-2-pyrrolidone
- the PAS obtained by this method usually tends to have a structure in which a halogen is bonded to the end of the polymer. Therefore, even if the separation and recovery after the polymerization reaction are sufficiently performed, the PAS has a high halogen content. When such a PAS having a high halogen content is used, environmental pollution has become a problem as seen in recent halogen regulations.
- PAS has been increasingly used as a compound containing a filler, for example, glass fiber, in many fields such as electric and electronic equipment.
- a filler for example, glass fiber
- Such a compound usually contains about 30 to 50% by mass of glass fiber and is used in the field of electrical and electronic equipment, so that it not only has a problem of reducing the halogen content from the viewpoint of environmental regulations.
- PAS PAS with good thermal stability, low generation of gas during molding, and low melt viscosity. This is because when such a compound is melt-formed, if the melt viscosity of the PAS is high, the PAS is likely to be thermally deteriorated due to local high temperatures due to friction, etc. due to kneading with hard glass fibers.
- problems such as deterioration in thermal stability and increase in generated gas cause problems such as failure to obtain stable and good melt molding conditions.
- Patent Document 1 in order to obtain a PAS having a low halogen content, a mercapto compound, a metal salt of a mercapto compound, a phenol compound, a metal salt of a phenol compound, and a disulfide compound are selected. It has been proposed to add one or more compounds.
- Example 7 uses phenol
- the chlorine content is only improved to about 1,200 to 2,100 ppm.
- Example 8 which is the only example using the disulfide compound (diphenyl disulfide) of Patent Document 1, it was reported that the chlorine content was 1,800 ppm, and the chlorine content was still high. Amount.
- Patent Document 1 as described in Example 1, “A powdered PAS... Was obtained”, the manufactured PAS was considered to be fine, not granular. It is done.
- Patent Document 1 Furthermore, the thiophenol used in Patent Document 1 is easily oxidized during storage and handling (handling). Therefore, when industrial production is carried out, the molecular weight of PAS and halogen (chlorine) depend on the degree of oxidation. Fluctuations occur in the reduction effect, and there are difficulties in industrial production within a certain standard product.
- the organic amide solvent such as NMP and the alkali metal hydroxide react by heating
- a compound containing nitrogen element is generated as an impurity.
- NMP and sodium hydroxide (NaOH) react with each other NMP opens and sodium methylaminobutanoate [(CH 3 ) NH—CH 2 —CH 2 —CH 2 —COONa] is generated.
- This compound reacts with p-dichlorobenzene, a dihaloaromatic compound, to produce sodium chlorophenylmethylaminobutanoate.
- they can be incorporated at the polymer ends during the PAS polymerization reaction. Dirt such as molds and dies caused by such a compound containing nitrogen atoms adversely affects the quality of the molded product, and thus requires frequent cleaning.
- An object of the present invention is to provide a granular PAS having a low halogen content, good thermal stability, less gas generation during molding, and having a low melt viscosity, and efficiently obtaining the granular PAS.
- An object of the present invention is to provide a method for producing granular PAS. That is, both a low halogen content and a low melt viscosity (high fluidity) can be achieved, and these characteristics can be stably obtained with a high yield and a PAS with a low nitrogen content. It is an object of the present invention to obtain the above.
- the present inventors have created a liquid-liquid phase separation state in which a liquid polymer phase and a liquid polymer phase are mixed in the presence of a phase separation agent.
- a low halogen content is obtained by performing a polymerization reaction in the presence of a disulfide compound, and sieving the produced polymer with a sieve having a specific sieve opening to obtain a sieved product. It was found that granular PAS having good thermal stability, less gas generation during molding, and low melt viscosity can be recovered in high yield.
- the inventors of the present invention have developed a PAS filter paper recovery method and a granular method for PAS having a certain range of melt viscosity (3 to 100 Pa ⁇ s) measured under conditions of a temperature of 310 ° C. and a shear rate of 1,200 sec ⁇ 1.
- the chlorine content and the like were examined for the recovery method (the sieve top with a sieve opening of 38 ⁇ m and the product with a sieve opening of 38 ⁇ m), the following surprising findings were obtained.
- the fine powder that passed through the sieve and passed through the sieve by sieving contains a large amount of low molecular weight substances and oligomers having chlorine bonded to the end of the molecule.
- the fine powder under the sieve has a chlorine content exceeding 20,000 ppm.
- the chlorine content of the sieved material remaining on the sieve without passing through the sieve having a sieve opening of 38 ⁇ m was lower than 1,500 ppm.
- the sieve is sieved with a sieve having a sieve opening of 150 ⁇ m, and the chlorine content of the sieved product remaining on the sieve without passing through the sieve is sieved with a sieve having a sieve opening of 38 ⁇ m and passed through the sieve.
- the chlorine content of the sieved product remaining on the sieve was further reduced.
- a polymerization reaction is performed in the presence of a disulfide compound, and the separation and recovery after polymerization have a specific sieve opening.
- a sieve By sieving the produced polymer with a sieve, it is possible to efficiently produce granular PAS with a low halogen (low chlorine) content, good thermal stability, little gas generation during molding, and low melt viscosity. It can be recovered at a rate.
- the present invention basically combines the three elements of occurrence of a liquid-liquid phase separation state in the polymerization process, presence of a disulfide compound in the polymerization process, and sieving in the separation / recovery process. There is a big effect.
- the thermal stability mentioned here can be evaluated by a method for evaluating the thermal stability of conventional PAS or ordinary thermoplastic resin.
- TGA thermogravimetric analysis
- the generated gas at the time of molding can be evaluated substantially by the amount of mold deposit at the time of injection molding, or in the laboratory by generated gas analysis.
- the granular PAS of the present invention has a low halogen content.
- Strict adjustment of the ratio of dihaloaromatic compound / sulfur source reduced the number of polymer terminals having halogen.
- the molecular weight was adjusted with a disulfide compound.
- the molecular weight was adjusted by strict adjustment of the ratio of dihaloaromatic compound / sulfur source.
- the yield was improved by adjusting the ratio of the phase separation agent for causing the liquid-liquid phase separation state and by strictly adjusting the ratio of the water / sulfur source.
- the liquid-liquid phase separation state was changed by the disulfide compound, the separation of the low molecular weight product / oligomer and the polymer component became efficient, and the granular PAS component was efficiently recovered.
- the present invention basically combines three elements: the occurrence of a liquid-liquid phase separation state in the polymerization process, the presence of a disulfide compound in the polymerization process, and sieving in the separation / recovery process.
- the following great effects can be achieved.
- the liquid-liquid phase separation state occurs due to the presence of the phase separation agent in the latter half of the polymerization, at the end of the polymerization or at the beginning of temperature reduction, and granulation is performed by cooling and solidification from the liquid-liquid phase separation state.
- the present invention has been completed based on these findings.
- a granular PAS comprising: (I) the granular PAS contains a substituent of —S— cleaved from a disulfide compound at the end; (Ii) The granular PAS is a sieved product after sieving with a sieve having a sieve opening of 38 ⁇ m or more, (Iii) the granular PAS has a halogen content of 1,500 ppm or less, and (Iv) A granular PAS having a melt viscosity of 3 to 100 Pa ⁇ s measured under conditions of a temperature of 310 ° C. and a shear rate of 1,200 sec ⁇ 1 is provided.
- the polymerization step of polymerizing the sulfur source and the dihaloaromatic compound in the organic amide solvent, the cooling step of cooling the liquid phase containing the produced polymer after the polymerization step, and the produced polymer are separated.
- a liquid-liquid phase separation state in which a phase separation agent is present during the polymerization step and / or before the cooling step, and the product polymer rich phase and the product polymer dilute phase coexist.
- a process for producing a granular PAS comprising the step of causing (I) performing the polymerization reaction in the presence of a disulfide compound in the polymerization step; and (Ii) In the separation / recovery step, the above-mentioned method for producing granular PAS is provided, in which the produced polymer is sieved with a sieve having a sieve opening of 38 ⁇ m or more to obtain a sieved product.
- a granular PAS having a high thermal stability, a low gas generation during molding, a low halogen content and a low melt viscosity, and a high balance between these properties is obtained in a high yield. be able to.
- granular PAS with a reduced nitrogen content can be obtained.
- the granular PAS of the present invention having a low halogen (low chlorine) content, good thermal stability, little gas generation during molding, and low melt viscosity is particularly required in the electric and electronic equipment field in recent years. It is useful as a low halogen (low chlorine) and low melt viscosity PAS.
- FIG. 1 is a graph plotting the chlorine content and melt viscosity of sieve tops of 150 ⁇ m sieve sieves in Examples 1 to 4 and Comparative Examples 1, 6, and 7.
- At least one sulfur source selected from the group consisting of alkali metal sulfides and alkali metal hydrosulfides is used as the sulfur source.
- the alkali metal sulfide include lithium sulfide, sodium sulfide, potassium sulfide, rubidium sulfide, cesium sulfide, and a mixture of two or more thereof.
- the alkali metal hydrosulfide include lithium hydrosulfide, sodium hydrosulfide, potassium hydrosulfide, rubidium hydrosulfide, cesium hydrosulfide, and a mixture of two or more thereof.
- the alkali metal sulfide any of an anhydride, a hydrate, and an aqueous solution may be used. Among these, sodium sulfide and lithium sulfide are preferable because they can be obtained industrially at low cost.
- the alkali metal sulfide is preferably used as an aqueous mixture such as an aqueous solution (that is, a mixture with fluid water) from the viewpoint of processing operation, measurement, and the like.
- the alkali metal hydrosulfide may be any of anhydride, hydrate, and aqueous solution. Among these, sodium hydrosulfide and lithium hydrosulfide are preferable because they can be obtained industrially at low cost.
- the alkali metal hydrosulfide is preferably used as an aqueous mixture such as an aqueous solution (that is, a mixture with fluid water) from the viewpoint of processing operation, measurement, and the like.
- alkali metal hydrosulfide In the production process of alkali metal sulfide, generally, a small amount of alkali metal hydrosulfide is by-produced. A small amount of alkali metal hydrosulfide may be contained in the alkali metal sulfide used in the present invention. In this case, the total molar amount of the alkali metal sulfide and the alkali metal hydrosulfide becomes the charged sulfur source in the charging step after the dehydration step described later.
- a small amount of alkali metal sulfide is generally produced as a by-product in the production process of alkali metal hydrosulfide.
- a small amount of alkali metal sulfide may be contained in the alkali metal hydrosulfide used in the present invention.
- the total molar amount of the alkali metal hydrosulfide and the alkali metal sulfide becomes the charged sulfur source in the charging step after the dehydration step.
- a mixture of both serves as a charged sulfur source.
- an alkali metal hydroxide is used in combination.
- the alkali metal hydroxide include lithium hydroxide, sodium hydroxide, potassium hydroxide, rubidium hydroxide, cesium hydroxide, and a mixture of two or more thereof.
- sodium hydroxide and lithium hydroxide are preferable because they can be obtained industrially at low cost.
- the alkali metal hydroxide is preferably used as an aqueous mixture such as an aqueous solution.
- the dihaloaromatic compound used in the present invention is a dihalogenated aromatic compound having two halogen atoms directly bonded to an aromatic ring.
- Specific examples of the dihaloaromatic compound include, for example, o-dihalobenzene, m-dihalobenzene, p-dihalobenzene, dihalotoluene, dihalonaphthalene, methoxy-dihalobenzene, dihalobiphenyl, dihalobenzoic acid, dihalodiphenyl ether, dihalodiphenyl sulfone. , Dihalodiphenyl sulfoxide, dihalodiphenyl ketone and the like.
- the halogen atom refers to each atom of fluorine, chlorine, bromine and iodine, and in the same dihaloaromatic compound, the two halogen atoms may be the same or different. Of these halogen atoms, a chlorine atom is preferred. These dihaloaromatic compounds can be used alone or in combination of two or more. p-Dichlorobenzene (p-DCB) is usually often used.
- the charged amount of the dihaloaromatic compound is usually 1.005 to 1.040 mol, preferably 1.008 to 1.035 mol, more preferably 1.010 to 1.030 mol, especially 1 mol per mol of the charged sulfur source.
- the amount is preferably 1.012 to 1.028 mol.
- Branching / crosslinking agent and molecular weight control agent In order to introduce a branched or crosslinked structure into PAS, a polyhalo compound having 3 or more halogen atoms bonded thereto (not necessarily an aromatic compound), an active hydrogen-containing halogenated aromatic A compound, a halogenated aromatic nitro compound, or the like can be used in combination.
- the polyhalo compound as the branching / crosslinking agent is preferably trihalobenzene.
- a monohalo organic compound can be added at any stage of the polymerization process.
- monohalo organic compounds include monohalo-substituted saturated or unsaturated aliphatic hydrocarbons such as monohalopropane, monohalobutane, monohaloheptane, monohalohexane, aryl halide and chloroprene; monohalo-substituted saturated cyclic such as monohalocyclohexane and monohalodecalin Hydrocarbon: monohalobenzene, monohalonaphthalene, 4-chlorobenzoic acid, methyl 4-chlorobenzoate, 4-chlorodiphenylsulfone, 4-chlorobenzonitrile, 4-chlorobenzotrifluoride, 4-chloronitrobenzene, 4-chloro Monohalo-substituted aromatic hydrocarbons such as chloroacetophenone, 4-chlorobenzophenone and
- Halogen atoms refer to fluorine, chlorine, bromine, and iodine atoms. Of these halogen atoms, a chlorine atom is preferred. In addition, an organic compound substituted with one chlorine atom and having a substituent such as trifluoromethane, which is extremely less reactive than the chlorine atom, is also incorporated into the monohalo organic compound for convenience. I will do it.
- Organic Amide Solvent in the present invention, an organic amide solvent that is an aprotic polar organic solvent is used as a solvent for the dehydration reaction and the polymerization reaction.
- the organic amide solvent is preferably stable to alkali at high temperatures.
- organic amide solvent examples include amide compounds such as N, N-dimethylformamide and N, N-dimethylacetamide; N-alkylcaprolactam compounds such as N-methyl- ⁇ -caprolactam; N-methyl-2-pyrrolidone, N-alkylpyrrolidone compounds or N-cycloalkylpyrrolidone compounds such as N-cyclohexyl-2-pyrrolidone; N, N-dialkylimidazolidinone compounds such as 1,3-dialkyl-2-imidazolidinone; tetramethylurea, etc. Tetraalkylurea compounds; hexaalkylphosphoric acid triamide compounds such as hexamethylphosphoric acid triamide. These organic amide solvents may be used alone or in combination of two or more.
- N-alkylpyrrolidone compounds, N-cycloalkylpyrrolidone compounds, N-alkylcaprolactam compounds, and N, N-dialkylimidazolidinone compounds are preferable, and in particular, N-methyl-2-pyrrolidone, N-methyl- ⁇ -caprolactam and 1,3-dialkyl-2-imidazolidinone are preferably used.
- the amount of the organic amide solvent used in the polymerization reaction of the present invention is usually in the range of 0.1 to 10 kg, preferably 0.15 to 5 kg, per mole of sulfur source. If the amount of the organic amide solvent used is less than 0.1 kg, it is difficult to carry out the polymerization reaction stably, and if it exceeds 10 kg, the production cost increases.
- phase separation agents can be used in order to generate a liquid-liquid phase separation state and to obtain a PAS having a low halogen content and an adjusted melt viscosity in a short time.
- a phase separation agent is a compound that dissolves in an organic amide solvent by itself or in the presence of a small amount of water and has an action of reducing the solubility of PAS in an organic amide solvent.
- the phase separation agent itself is a compound that is not a solvent for PAS.
- phase separation agent generally known compounds can be used as the phase separation agent for PAS.
- phase separation agents include water, organic carboxylic acid metal salts such as alkali metal carboxylates, organic sulfonic acid metal salts, alkali metal halides such as lithium halides, alkaline earth metal halides, and aromatic carboxylic acids. Alkaline earth metal salts, alkali metal phosphates, alcohols, paraffin hydrocarbons and the like can be mentioned.
- phase separation agents can be used alone or in combination of two or more. Among these, water and organic carboxylic acid metal salts are preferable because they are inexpensive.
- organic carboxylic acid metal salt organic sulfonic acid metal salt, alkali metal halide such as lithium halide, alkaline earth metal halide, alkaline earth metal salt of aromatic carboxylic acid, alkali metal phosphate, etc. preferable.
- phase separation agent water is particularly preferable.
- organic carboxylic acid metal salts sodium acetate is preferable, and when used, it can be used as an aqueous solution of usually 25 to 35% by mass, preferably 30% by mass.
- the amount of phase separation agent used varies depending on the type of compound used, but is generally in the range of 0.01 to 15 moles per mole of the charged sulfur source.
- the amount is preferably 0.01 to 13 mol, more preferably 0.02 to 12 mol, and particularly preferably 0.03 to 10 mol.
- the amount of the phase separation agent used is less than 0.01 mol, it is difficult to cause a liquid-liquid phase separation state, and when it exceeds 15 mol, it is difficult to proceed the polymerization reaction well.
- the phase separation agent can be present either during the polymerization step and / or before the cooling step described later, thereby obtaining a step in which a liquid-liquid phase separation state occurs. Can do.
- the polymerization reaction in the polymerization step is performed in the presence of a disulfide compound.
- the disulfide compound may be added at any stage of the polymerization process. For example, when the polymerization process includes a two-stage process including a pre-stage polymerization process and a post-stage polymerization process, it may be added in the pre-stage polymerization process or in the post-stage polymerization process. Moreover, you may add to the preparation process at the time of a pre-stage polymerization process start.
- the —S— substituent cleaved from the disulfide compound replaces the halogen group (chlorine group) at the terminal of the resulting PAS, thereby reducing the halogen content of the PAS. Presumed to play a role of reduction.
- the PAS end includes —S—C 6 H 5 that has reacted with the end.
- the terminal group component at the PAS end is mostly —Cl, —SC 6 H 5 , —SH in which the disulfide compound has reacted.
- a nitrogen compound derived from an organic amide solvent can be analyzed quantitatively or qualitatively by elemental analysis, high-temperature NMR analysis, or IR analysis.
- disulfide compounds can be obtained by quantitative analysis of -Cl by elemental analysis, quantitative determination of -SH by titration or derivatization reaction or IR method, and nitrogen analysis of organic amide solvent-derived nitrogen compounds. The amount of —S—C 6 H 5 reacted with can be calculated.
- disulfide compounds are insoluble in water.
- the disulfide compound is distributed to the polymer concentrated phase with a small amount of water component, and efficiently replaces the halogen at the PAS terminal, contributing to low halogenation. It is done.
- the disulfide compound exhibits advantageous effects such as good reactivity even in a liquid-liquid phase separation state. That is, it is one of the features of the present invention that the reactivity of the disulfide compound is well expressed in a liquid-liquid phase separation state.
- the addition timing of the disulfide compound may be determined based on the conversion rate of the dihaloaromatic compound.
- the disulfide compound has a dihaloaromatic compound conversion rate of 0 to 100%, usually 45% or more, preferably 45 to 99.5%, more preferably 60 to 99% in the polymerization step. More preferably, it is added at a point of 70 to 98.5%, particularly preferably 80 to 98%, and can be present in the polymerization step.
- disulfide compound examples include diphenyl disulfide (DPDS), p-p'ditolyl disulfide, dibenzyl disulfide, dibenzoyl disulfide, and dithiobenzoyl disulfide, and diphenyl disulfide is preferable.
- DPDS diphenyl disulfide
- p-p'ditolyl disulfide dibenzyl disulfide
- dibenzoyl disulfide dibenzoyl disulfide
- dithiobenzoyl disulfide diphenyl disulfide
- the amount of the disulfide compound added when the polymerization reaction is carried out in the presence of the disulfide compound is 0.0005 to 0.015 mol, preferably 0.0007 to 0.005 mol per mol of the charged sulfur source. 01 mol, more preferably 0.0008 to 0.008 mol, still more preferably 0.0009 to 0.006 mol, and particularly preferably 0.001 to 0.005 mol.
- the amount of disulfide compound added in this range is a granular PAS with good thermal stability, low gas generation during molding, low halogen content, low melt viscosity, and high performance balance. It is important in getting.
- the production method comprises a polymerization step for polymerizing a sulfur source and a dihaloaromatic compound in an organic amide solvent, a cooling step for cooling the liquid phase containing the produced polymer after the polymerization step, and separation and recovery of the produced polymer. Including a separation / recovery step, and further a phase-separating agent is present during the polymerization step and / or before the cooling step to generate a liquid-liquid phase separation state in which the product polymer rich phase and the product polymer dilute phase coexist. It is a manufacturing method including a process.
- phase separation polymerization in which the polymerization process is continued in a liquid-liquid phase separation state in which the liquid phase in the polymerization reaction system is mixed with a polymer rich phase and a polymer thin phase in the presence of a phase separation agent.
- a process may be included.
- a dehydration process As a pre-process of the polymerization process, it is preferable to arrange a dehydration process to adjust the amount of coexisting water (also referred to as water content) in the reaction system.
- the dehydration step is preferably carried out by heating and reacting a mixture containing an organic amide solvent and an alkali metal sulfide in an inert gas atmosphere, and discharging water out of the system by distillation.
- an alkali metal hydrosulfide is used as the sulfur source
- the reaction is carried out by heating and reacting a mixture containing the alkali metal hydrosulfide and the alkali metal hydroxide, and discharging water out of the system by distillation.
- the amount of water composed of hydrated water (crystal water), an aqueous medium, by-product water, etc. is preferably dehydrated until it falls within the range of the coexisting water amount required in the charging step described later. If the amount of coexisting water falls outside the range required in the preparation step, an additional amount of water shortage may be added.
- an organic amide solvent an alkali metal hydrosulfide, and 0.95 to 1.07 mole of alkali metal hydroxide per mole of the alkali metal hydrosulfide It is preferable to heat and react the mixture containing water and to discharge at least a part of the distillate containing water from the system containing the mixture to the outside of the system.
- the preferred mole of alkali metal hydroxide per mole of alkali metal hydrosulfide charged in this step is 0.96 to 1.06 mole, more preferably 0.97 to 1.05 mole.
- Alkali metal hydrosulfides often contain a small amount of alkali metal sulfide, and the amount of sulfur source is the total amount of alkali metal hydrosulfide and alkali metal sulfide. Even if the alkali metal hydrosulfide contains an alkali metal sulfide, there is no problem as a raw material of PAS, but in order to produce the granular PAS of the present invention, the content is preferably as small as possible. In addition, even if a small amount of alkali metal sulfide is mixed, the present invention calculates the mole with the alkali metal hydroxide based on the content (analytical value) of the alkali metal hydrosulfide, adjust.
- the order in which the raw materials are charged in the dehydration process may be in any order, and each raw material may be additionally charged during the dehydration process.
- An organic amide solvent is used as a solvent used in the dehydration step. This solvent is preferably the same as the organic amide solvent used in the polymerization step, and N-methyl-2-pyrrolidone is particularly preferred.
- the amount of the organic amide solvent used is usually 0.1 to 10 kg, preferably 0.15 to 5 kg, per mole of sulfur source charged into the reaction vessel.
- the mixture after the raw materials are charged into the reaction vessel is usually heated at a temperature of 300 ° C. or lower, preferably 100 to 250 ° C., usually for 15 minutes to 24 hours, preferably 30 minutes to 10 hours. Done.
- a heating method there are a method for maintaining a constant temperature, a stepwise or continuous temperature raising method, or a method in which both are combined.
- the dehydration step is performed by a batch method, a continuous method, or a combination method of both methods.
- the apparatus for performing the dehydration step may be the same as or different from the reaction vessel used in the subsequent polymerization step.
- the material of the device is preferably a corrosion resistant material such as titanium.
- part of the organic amide solvent is usually discharged with the water out of the reaction vessel. At that time, hydrogen sulfide is discharged out of the system as a gas.
- the preparation step is performed according to the present invention “a polymerization step for polymerizing a sulfur source and a dihaloaromatic compound in an organic amide solvent, a cooling step for cooling a liquid phase containing the produced polymer after the polymerization step, and a produced polymer.
- a liquid-liquid phase including a separation phase / recovery step for separating / recovering the liquid and a phase separation agent in the polymerization step and / or before the cooling step to coexist the product polymer rich phase and the product polymer dilute phase.
- the ⁇ Production method including the step of causing a separation state '', the amount of phase separation agent with respect to the charged sulfur source, the amount of coexisting water with respect to the charged sulfur source, the amount of dihaloaromatic compound with respect to the charged sulfur source, and the charged sulfur source required in the polymerization step It is a step of adjusting the amount of alkali metal hydroxide with respect to the amount of disulfide compound with respect to the charged sulfur source.
- an alkali metal hydroxide is generated by the equilibrium reaction and remains in the system. Therefore, it is necessary to accurately grasp the amount of volatilized hydrogen sulfide and determine the molar amount of the alkali metal hydroxide with respect to the sulfur source in the preparation step.
- an alkali metal hydroxide and water can be added to the mixture remaining in the system after the dehydration step, if necessary.
- the amount of coexisting water at the start of the polymerization reaction is usually 0.02 to 2 mol, preferably 0.05 to 1.9 mol, more preferably 0.5 to 1. mol, relative to 1 mol of the charged sulfur source in the charging step.
- the range is preferably 8 mol. Within this range, the amount of coexisting water can be increased during the polymerization reaction.
- the charged amount of the dihaloaromatic compound is usually 1.005 to 1.040 mol, preferably 1.008 to 1.035 mol, more preferably 1.010 to 1.030 mol, especially 1 mol per mol of the charged sulfur source.
- the amount is preferably 1.012 to 1.028 mol.
- the amount of alkali metal hydroxide per mole of the charged sulfur source is preferably 1.005 to 1.080 mol, more preferably 1.010 to 1.075 mol, especially The amount is preferably 1.020 to 1.073 mol. It is preferable to carry out the polymerization reaction with a small excess of alkali metal hydroxide in order to stably carry out the polymerization reaction and obtain a high-quality PAS.
- the amount of the disulfide compound added when the polymerization reaction is carried out in the presence of the disulfide compound is 0.0005 to 0.015 mol, preferably 0.0007 to 0.005 mol per mol of the charged sulfur source. 01 mol, more preferably 0.0008 to 0.008 mol, still more preferably 0.0009 to 0.006 mol, and particularly preferably 0.001 to 0.005 mol.
- the disulfide compound may be added alone in the polymerization step, or may be added as a mixture with an organic amide solvent.
- the amount of the organic amide solvent is 0.1 to 10 kg, preferably 0.15 to 5 kg per mole of the sulfur source or the charged sulfur source.
- the polymerization process is carried out by heating a sulfur source and a dihaloaromatic compound in an organic amide solvent.
- a phase separation polymerization step in which the polymerization step is continued in a liquid-liquid phase separation state in which the liquid phase in the polymerization reaction system is a mixture of a polymer rich phase and a polymer thin phase in the presence of a phase separation agent. It is preferable to include.
- phase separation agent may be added before the cooling step after the polymerization reaction.
- the granular PAS can be separated and recovered by cooling the liquid phase containing the produced polymer in the liquid-liquid phase separation state.
- Phase separation agents include water, organic carboxylic acid metal salts, organic sulfonic acid metal salts, alkali metal halides, alkaline earth metal halides, alkaline earth metal salts of aromatic carboxylic acids, alkali metal phosphates, alcohols, and It is at least one phase separation agent selected from the group consisting of paraffinic hydrocarbons.
- the phase separation agent is usually 0.01 to 15 mol, preferably 0.01 to 13 mol, more preferably 0.02 to 12 mol, particularly preferably 0.03 to 10 mol per mol of the charged sulfur source. Use moles.
- the polymerization reaction needs to be performed in the presence of a disulfide compound at some stage in the polymerization process.
- the polymerization reaction in the polymerization step is 0.0005 to 0.015 mol, preferably 0.0007 to 0.01 mol, more preferably 0.0008 to 0.008 mol, and still more preferably, per mol of the charged sulfur source. It must be carried out in the presence of 0.0009 to 0.006 mol, particularly preferably 0.001 to 0.005 mol of disulfide compound.
- a disulfide compound with a dihaloaromatic compound conversion rate of 45% or more it is preferable to add a disulfide compound with a dihaloaromatic compound conversion rate of 45% or more. Moreover, you may mix a polymerization adjuvant and other additives before a polymerization process or during a polymerization process.
- the polymerization reaction is preferably carried out in a two-step process of a pre-stage polymerization process and a post-stage polymerization process, generally in the range of 170 to 290 ° C.
- a heating method a method of maintaining a constant temperature, a stepwise or continuous temperature raising method, or a combination of both methods is used.
- the polymerization reaction time is generally in the range of 10 minutes to 72 hours, preferably 30 minutes to 48 hours.
- the organic amide solvent used in the polymerization step is usually 0.1 to 10 kg, preferably 0.15 to 5 kg, per mole of the charged sulfur source. Within this range, the amount may be changed during the polymerization reaction.
- a method is preferred in which the polymerization reaction is continued by converting the liquid phase in the polymerization reaction system to a phase-separated state at the stage when the conversion of the dihaloaromatic compound reaches 80 to 99 mol% after the start of the polymerization reaction.
- a phase separation agent or increase the amount of the additive acting as a phase separation agent.
- limit especially as a phase-separation agent Water or an organic carboxylic acid metal salt and these combinations, especially water are preferable at the point which is cheap and the control of a polymerization reaction and post-processing are easy.
- the polymerization reaction is carried out in the presence of a disulfide compound, and the polymerization step is performed at least in the following two-stage steps: (I) A sulfur source and a dihaloaromatic compound in an organic amide solvent are present in an amount of 0.02 to 2 mol of coexisting water and 1.005 to 1.040 mol of a dihaloaromatic compound per mol of the charged sulfur source. In the state, a polymerization reaction is carried out at a temperature of 170 to 270 ° C.
- phase separation agent While present in the range of 0.01 to 10 moles per mole and heating to a temperature of 240 to 290 ° C., the liquid phase in the polymerization reaction system is converted to a phase separation state and the polymerization reaction is continued.
- Post-polymerization step It is preferable to carry out by.
- Prepolymerization step The amount of coexisting water in the reaction system in the prepolymerization step is 0.02 to 2 mol, preferably 0.05 to 1.9 mol, more preferably 0.5 to 1.8 mol, per mol of the charged sulfur source. Range. If the amount of coexisting water is too small, an undesirable reaction such as decomposition of PAS is likely to occur. Conversely, if it exceeds 2 moles, the polymerization rate is remarkably reduced, or the organic amide solvent and the produced PAS are likely to be decomposed. Neither is preferred.
- the charged amount of the dihaloaromatic compound is usually 1.005 to 1.040 mol, preferably 1.008 to 1.035 mol, more preferably 1.010 to 1.030 mol, in particular, per mol of the charged sulfur source.
- the amount is preferably 1.012 to 1.028 mol.
- Polymerization is performed within a temperature range of 170 to 270 ° C., preferably 180 to 265 ° C. If the polymerization temperature is too low, the polymerization rate becomes too slow. Conversely, if the polymerization temperature is higher than 270 ° C., the produced PAS and the organic amide solvent are liable to decompose, and the degree of polymerization of the produced PAS becomes extremely low.
- the polymerization temperature in the pre-stage polymerization step is preferably controlled within the range of 200 to 255 ° C.
- the pre-stage polymerization step is a stage in which the conversion of the dihaloaromatic compound reaches 80 to 99%, preferably 85 to 98%, more preferably 90 to 97% after the start of the polymerization reaction, and the liquid phase Is a step before the phase separation state occurs.
- the conversion rate of the dihaloaromatic compound was determined by gas chromatography to determine the amount of the dihaloaromatic compound remaining in the reaction mixture, and based on the residual amount, the charged amount of the dihaloaromatic compound, and the charged amount of the sulfur source, It is a value calculated by an equation.
- conversion rate [[DHA charge (mol) ⁇ DHA remaining amount (mol)] / [DHA charge (mol)]] ⁇ 100 To calculate the conversion.
- a polymer (also referred to as “prepolymer”) having a melt viscosity of usually 0.5 to 30 Pa ⁇ s measured at a temperature of 310 ° C. and a shear rate of 1,200 sec ⁇ 1 is produced. Is desirable.
- the liquid phase in the polymerization reaction system includes a polymer rich phase having a high content of polymer (prepolymer) produced by the previous polymerization and a polymer dilute phase having a low content of the polymer. Phase separate. The phase separation state can be clearly observed visually.
- the phase separation agent is generally used in an amount of 0.01 to 10 mol, preferably 0.03 to 8 mol, more preferably 0.04 to 7 mol, per mol of the charged sulfur source.
- the amount of coexisting water in the reaction system in the latter polymerization step is usually 2 to 5 mol, preferably 2.1 to 4.5 mol, per mol of the charged sulfur source. More preferably, it is desirable to adjust to the range of 2.2 to 4 mol, particularly preferably 2.3 to 3.5 mol.
- the amount of coexisting water in the reaction system is less than 2 mol or more than 5 mol, the degree of polymerization of the produced PAS decreases.
- phase separation agent other than water (organic carboxylic acid metal salt, organic sulfonic acid metal salt, alkali metal halide, alkaline earth metal halide, alkaline earth metal salt of aromatic carboxylic acid, phosphorus
- the phase separation agent is added in an amount of 0.01 to 3 per mole of the charged sulfur source. Mole, preferably 0.02 to 2 mol, more preferably 0.03 to 1 mol, particularly preferably 0.04 to 0.5 mol, is preferably present.
- the phase separation agent water and other phase separation agents other than water can be used in combination.
- the amount of coexisting water in the reaction system is 0.01 to 7 mol, preferably 0.1 to 4 mol, more preferably 1 to 3.5 mol, and other than water, per mol of the charged sulfur source.
- the phase separation agent is preferably present in the range of 0.01 to 3 mol, preferably 0.02 to 1 mol, more preferably 0.03 to 0.5 mol, per mol of the charged sulfur source. Strict adjustment of the ratio of phase separation agent / feeding sulfur source leads to reduction of low molecular weight substances and oligomers.
- the polymerization temperature in the post-stage polymerization step is in the range of 240 to 290 ° C. If the polymerization temperature in the subsequent polymerization step is less than 240 ° C, it is difficult to obtain a PAS having an adjusted melt viscosity, and if it exceeds 290 ° C, the produced PAS and the organic amide solvent may be decomposed. Further, a temperature range of 245 to 280 ° C., particularly 250 to 275 ° C. is preferable because a PAS having an adjusted melt viscosity is easily obtained.
- the latter polymerization step in the present invention is not a simple fractionation / granulation step of the PAS prepolymer produced in the former polymerization step, but is for raising the degree of polymerization of the PAS prepolymer.
- the polymerization reaction is continued in a phase separation state in which the liquid phase in the polymerization reaction system is mixed with the produced polymer rich phase and the produced polymer dilute phase in the presence of the phase separation agent.
- the PAS concentration of the concentrated phase is usually 30 to 70% by mass, preferably 40 to 60% by mass, more preferably 45 to 55% by mass.
- the PAS concentration of the dilute phase is usually 0.1 to 15% by mass, preferably 0.5 to 10% by mass, more preferably 1 to 8% by mass.
- the polymerization reaction method may be a batch method, a continuous method, or a combination of both methods.
- a system using two or more reaction vessels can be used as desired.
- Cooling step In the present invention, after the polymerization step and before the cooling step, the above-described phase separation agent is present to cause a liquid-liquid phase separation state in which the product polymer rich phase and the product polymer dilute phase coexist. be able to. If necessary, the occurrence of a liquid-liquid phase separation state can be adjusted by stirring.
- the liquid phase containing the produced polymer is usually cooled from a high temperature state after the polymerization step.
- the slow cooling it is preferable to cool the liquid phase by controlling the temperature decreasing rate at 2.0 to 0.1 ° C./min.
- the slow cooling can be performed by a method in which the polymerization reaction system is exposed to an ambient temperature (for example, room temperature).
- an ambient temperature for example, room temperature.
- the temperature of the liquid phase in the polymerization reaction system is the polymerization temperature in the liquid-liquid phase separation polymerization process, or the temperature at which PAS is solidified and granulated from the liquid-liquid phase separation state (hereinafter referred to as “solidification / granulation”).
- solidification / granulation the temperature at which PAS is solidified and granulated from the liquid-liquid phase separation state
- granulation temperature for example, until it drops to about 240 to 200 ° C., preferably 2.0 to 0.1 ° C./min, more preferably 1.5 to 0.2 ° C./min, and more It is desirable to gradually cool the liquid phase by controlling the temperature drop rate at 1.3 to 0.3 ° C./min. Such control of the cooling rate can promote the granulation of the polymer.
- the liquid phase can be cooled to a desired temperature without temperature control.
- the polymerization reaction system can be allowed to stand at the ambient temperature, or the rate of temperature drop of the liquid phase can be increased.
- the final cooling temperature is a temperature not lower than room temperature and lower than 220 ° C. at which separation and recovery processes such as sieving are easy.
- it is 40 degreeC or more, More preferably, it is 45 degreeC or more.
- the upper limit is preferably 200 ° C. or lower, and a slurry containing PAS that is sufficiently granulated can be obtained by setting the washing to preferably less than 100 ° C.
- a method of separating / recovering granular PAS from a reaction solution by a method of sieving using a specific sieve opening sieve is provided.
- Sieving may be performed while the product slurry is in a high temperature state (eg, a temperature of room temperature or higher and lower than 220 ° C.).
- the produced PAS is sieved with a sieve having a sieve opening of 38 ⁇ m or more, and recovered as a sieved product after sieving.
- the sieving may be performed after washing, which will be described later, or after drying.
- sieving may be performed at each stage before washing, after washing, and after drying.
- PAS may be washed with hot water or the like.
- the produced PAS can also be treated with a salt such as an organic acid or ammonium chloride.
- the organic acid is preferably acetic acid. After washing, it is dried according to a conventional method.
- Granular PAS is a sieved product after sieving with a sieve having a sieve opening of 38 ⁇ m or more.
- the sieve used for recovering the granular PAS is usually a sieve having a sieve opening selected from a range of 38 ⁇ m to 2,800 ⁇ m, preferably a sieve selected from a range of 38 ⁇ m to 1,500 ⁇ m.
- a sieve most preferably a sieve having a sieve opening selected from the range of 38 ⁇ m to 300 ⁇ m is employed.
- a sieve used for collecting a sieve having an opening specifically, a sieve having a sieve opening of 150 ⁇ m (100 mesh (number of meshes / inch)), a sieve opening of 105 ⁇ m (145 mesh (number of meshes / number) Inch)), a sieve having a sieve opening of 75 ⁇ m (200 mesh (number of meshes / inch)), and a sieve having a sieve opening of 38 ⁇ m (400 mesh (number of meshes / inch)), etc.
- the oligomer can be removed. More preferably, it is desirable to use a sieve having a sieve opening of 150 ⁇ m (100 mesh (number of meshes / inch)) that can efficiently remove fine by-product salt.
- the granular polymer collected as a sieved product by sieving with a sieve having a sieve opening of 38 ⁇ m or more is usually 80% by mass or more, preferably with respect to the total amount before sieving. It can be recovered in a yield of 80 to 99.5% by mass, more preferably 83 to 99% by mass, particularly preferably 85 to 98% by mass.
- the granular PAS collected by a sieve having a sieve opening of 150 ⁇ m or more is usually 80% by mass or more, specifically 80 to 98% by mass, preferably 83 to 97% by mass, and particularly preferably 85 to 97% by mass. It can be recovered in a yield of 96% by weight.
- the yields typified by these are also called sieved products (mass%).
- the granular PAS sieve top (mass%) is obtained by sieving the PAS mass (theoretical amount) when it is assumed that all of the effective sulfur components in the charged sulfur source present in the reaction vessel after the dehydration step have been converted to PAS.
- the total amount of PAS before dividing was used as a standard.
- the sieved product (mass%) was calculated by (sieved product) / (total amount of PAS before sieving: PAS mass (theoretical amount)). If the charged sulfur source is charged in an excess molar ratio than the dihaloaromatic compound, not all of the charged sulfur source can be converted to PAS.
- the sieved material (% by mass) is calculated based on the amount. Also in the case of filter paper collection, the recovery rate is calculated by the filter paper top / (total amount of PAS before sieving: PAS mass (theoretical amount)).
- Polyarylene sulfide is a granular PAS, wherein (i) the granular PAS contains a substituent of -S- cleaved from a disulfide compound at the end, and (ii) the granular PAS is 38 ⁇ m or more. (Iii) The granular PAS has a halogen content of 1,500 ppm or less, and (iv) the granular polyarylene sulfide has a temperature of A granular PAS having a melt viscosity of 3 to 100 Pa ⁇ s measured under conditions of 310 ° C. and a shear rate of 1,200 sec ⁇ 1 is obtained.
- the granular PAS is a polymerization step for polymerizing a sulfur source and a dihaloaromatic compound in an organic amide solvent, a cooling step for cooling a liquid phase containing the produced polymer after the polymerization step, And a recovery step for separating and recovering the produced polymer, and further a liquid-liquid in which a phase separating agent is present during the polymerization step and / or before the cooling step to mix the produced polymer rich phase and the produced polymer dilute phase.
- a method for producing granular PAS comprising a step of causing a phase separation state, wherein (i) a polymerization reaction is performed in the presence of a disulfide compound in the polymerization step, and (ii) in the separation / recovery step
- the produced polymer is sieved with a sieve having a sieve opening of 38 ⁇ m or more to obtain a sieved product, which is produced by a method for producing granular PAS.
- the melt viscosity measured at a temperature of 310 ° C. and a shear rate of 1,200 sec ⁇ 1 is usually 3 to 100 Pa ⁇ s, preferably 7 to 80 Pa ⁇ s, more preferably 10 to 70 Pa.
- a granular PAS of s, particularly preferably 13 to 60 Pa ⁇ s, more preferably 15 to 55 Pa ⁇ s, and most preferably 17 to 50 Pa ⁇ s can be obtained.
- the halogen content (chlorine content) of the obtained granular PAS is 1,500 ppm or less, preferably 1,300 ppm or less, more preferably 1,250 ppm or less. Depending on the application, it may be 1,000 ppm or less, preferably 900 ppm or less, or 850 ppm or less.
- the lower limit of the halogen content is usually about 100 ppm or 200 ppm.
- the nitrogen content of the obtained granular PAS is 1,000 ppm or less, preferably 800 ppm or less, more preferably 700 ppm or less, even more preferably 650 ppm or less, and particularly preferably 600 ppm or less.
- the lower limit of the nitrogen content is about 1 ppm or 2 ppm.
- the average particle size of the obtained granular PAS is 50 to 2,500 ⁇ m, preferably 70 to 1,000 ⁇ m, more preferably 100 to 800 ⁇ m, particularly preferably 280 to 550 ⁇ m, most preferably. 300 to 500 ⁇ m.
- the granular PAS of the present invention is used as it is or after being oxidatively cross-linked, either alone or by blending various synthetic resins, various fillers, various additives, and extrusion molding of various injection molded products, sheets, films, fibers, pipes, etc. Can be formed into a product.
- Granular PAS is also useful as a sealing agent or coating agent for electronic components.
- PPS is particularly preferable.
- Resin Composition When the granular PAS of the present invention is used as a composition, the other components are as follows.
- thermoplastic resins that are stable at high temperatures are preferable. Specific examples thereof include aromatic polyesters such as polyethylene terephthalate and polybutylene terephthalate; polytetrafluoroethylene, tetrafluoroethylene / hexafluoropropylene copolymers, Tetrafluoroethylene / perfluoroalkyl vinyl ether copolymer, polychlorotrifluoroethylene, polyvinylidene fluoride, vinylidene fluoride / hexafluoropropylene copolymer, propylene / tetrafluoroethylene copolymer, vinylidene fluoride / chlorotrifluoroethylene Fluororesin such as copolymer, ethylene / hexafluoropropylene copolymer; polyacetal, polystyrene, poly
- thermoplastic resins can be used alone or in combination of two or more.
- an inorganic filler such as glass fiber is blended
- various synthetic resins are used so that good moldability can be obtained in accordance with the characteristics of the low melt viscosity PAS which is a feature of the present invention. It is important to select a material that has a low melt viscosity and an intrinsic viscosity and that provides a good melting behavior.
- Examples of the various fillers include inorganic fiber materials such as glass fiber, carbon fiber, asbestos fiber, silica fiber, alumina fiber, zirconia fiber, boron nitride fiber, silicon nitride fiber, boron fiber, potassium titanate whisker, stainless steel,
- Examples include fibrous fillers such as metal fibrous materials such as aluminum, titanium, steel, and brass; high-melting organic fibrous materials such as polyamide, fluororesin, polyester resin, and acrylic resin;
- Examples of the filler include mica, silica, talc, alumina, kaolin, calcium sulfate, calcium carbonate, titanium oxide, ferrite, glass powder, zinc oxide, nickel carbonate, iron oxide, quartz powder, magnesium carbonate, and barium sulfate.
- a granular or plate-like filler such as clay.
- These fillers can be used alone or in combination of two or more.
- These fillers may be treated with a sizing agent or a surface treatment agent as necessary.
- the sizing agent or surface treatment agent include functional compounds such as epoxy compounds, isocyanate compounds, silane compounds, and titanate compounds. These compounds may be used after having been subjected to surface treatment or focusing treatment on the filler in advance, or may be added simultaneously when the composition is adjusted.
- the filler is usually blended in an amount of 0 to 800 parts by weight, preferably 0 to 500 parts by weight, more preferably 0 to 300 parts by weight with respect to 100 parts by weight of the granular PAS.
- an inorganic fibrous filler such as glass fiber is blended as a filler, a resin composition and a molded article excellent in mechanical properties such as tensile strength can be obtained.
- a compound containing a filler such as glass fiber it is particularly preferable to use as a compound containing a filler such as glass fiber.
- the regulated value of the halogen content is set to 900 ppm or less.
- the blending amount of glass fiber is about 30 to 50% by mass, so that when the granular PAS of the present invention is used, the chlorine content is sufficiently below the regulation value.
- Various fillers include pigments, dyes, antioxidants, UV absorbers, lubricants, nucleating agents, flame retardants, resin modifiers, coupling agents, antistatic agents, conductive materials, carbon precursors, mold release agents, Examples include plasticizers.
- the measuring method of physical properties and characteristics in the present invention is as follows.
- the sieved product was calculated by (sieved product) / (total amount of PAS before sieving: PAS mass (theoretical amount)). If the charged sulfur source is charged in an excess molar ratio than the dihaloaromatic compound, not all of the charged sulfur source can be converted to PAS. The amount on the sieve (% by mass) was calculated based on the amount. Also in the case of filter paper recovery, the recovery rate was calculated as the filter paper top / (total amount of PAS before sieving: PAS mass (theoretical amount)).
- the average particle diameter of the produced polymer (granular PAS) recovered in the separation / recovery step is 2,800 ⁇ m (7 mesh (number of meshes / inch)) as the sieve used, and 1 as the sieve opening.
- Example 1 In a 20 liter autoclave, 6,001 g of N-methyl-2-pyrrolidone (hereinafter abbreviated as “NMP”), 2,000 g of aqueous sodium hydrosulfide (NaSH; purity 62 mass%), aqueous sodium hydroxide (NaOH; purity) (74.0% by mass) 1,171 g was charged. NaOH / NaSH (sulfur source) has a molar ratio of 0.98.
- the content of the autoclave is cooled to 150 ° C., 3,264 g of p-dichlorobenzene (hereinafter abbreviated as “p-DCB”), 2,707 g of NMP, 19 g of sodium hydroxide, and 167 g of water are added, While stirring, the reaction was carried out at 220 ° C. for 5 hours to carry out prepolymerization.
- the water / feed sulfur source is 1.5 in molar ratio.
- the NaOH / feed sulfur source is 1.05 in molar ratio.
- the NMP / feed sulfur source is 0.37 kg / mol.
- the p-DCB / source sulfur source is 1.020 in molar ratio.
- the conversion rate of p-DCB at the end of the previous polymerization was 93%.
- DPDS diphenyl disulfide
- NMP N-phenyl disulfide
- DPDS / feed sulfur source 0.001 in molar ratio.
- 443 g of water was injected, the temperature was raised to 255 ° C., and the reaction was carried out for 5 hours to carry out post polymerization.
- the water / feed sulfur source is 2.63 in molar ratio.
- the mixture was gradually cooled to 220 ° C. at a temperature drop rate of 1 ° C./min, and cooled to about room temperature from 220 ° C., and the contents were respectively set to 38 ⁇ m (400 mesh) and 150 ⁇ m (100 mesh) mesh opening ), And the granular PAS was washed with acetone three times and then with water three times. The granular PAS was washed once with an acetic acid aqueous solution adjusted to pH 4 and washed three times with water to obtain a washed granular PAS. The granular PAS thus obtained was dried at 100 ° C. overnight.
- the average particle size of the granular PAS thus obtained was 442 ⁇ m.
- a sieve opening of 150 ⁇ m (100 mesh) sieve has a melt viscosity of 39 Pa ⁇ s, a chlorine content of 1,150 ppm, a nitrogen content of 530 ppm, and a sieve opening of 38 ⁇ m (400 mesh) sieve is melted.
- the viscosity was 38 Pa ⁇ s
- the chlorine content was 1,200 ppm
- the nitrogen content was 560 ppm.
- the sieved product (mass%) of granular PAS was 91% with a sieved product of 150 ⁇ m (100 mesh), and was 93% with a sieved product with a sieve opening of 38 ⁇ m (400 mesh).
- Example 2 Similar to Example 1, except that 14.3 g of DPDS was added when the conversion rate of p-DCB at the end of the pre-polymerization was 92%, and the molar ratio of DPDS / feeding sulfur source was 0.003. Went to.
- the average particle size of the granular PAS thus obtained was 476 ⁇ m. Further, the sieved product having a sieve opening of 150 ⁇ m (100 mesh) had a melt viscosity of 19 Pa ⁇ s, a chlorine content of 950 ppm, and a nitrogen content of 550 ppm.
- the sieve top with a sieve opening of 38 ⁇ m (400 mesh) has a melt viscosity of 19 Pa ⁇ s, a chlorine content of 1,100 ppm, a nitrogen content of 575 ppm, and passed through a sieve with a sieve opening of 38 ⁇ m (400 mesh) Recovered from the filter paper had a melt viscosity of less than 1 Pa ⁇ s and a chlorine content of 20,500 ppm.
- the sieved product (mass%) of the granular PAS was 89% with a sieved product having a sieve opening of 150 ⁇ m (100 mesh) and 91% with a sieved product of 38 ⁇ m (400 mesh).
- Example 3 The process up to the dehydration step was performed in the same manner as in Example 1. After the dehydration step, the contents of the autoclave are cooled to 150 ° C., p-DCB 3,280 g, NMP 2,708 g, sodium hydroxide 19 g, water 167 g and DPDS 9.5 g are added and reacted at 220 ° C. for 5 hours with stirring. And pre-stage polymerization was performed. When DPDS is added, that is, when the polymerization reaction starts, the conversion rate of p-DCB is 0%. The water / charged sulfur source is 1.50 in molar ratio. The NaOH / feed sulfur source is 1.05 in molar ratio.
- the NMP / feed sulfur source is 0.38 kg / mol.
- the p-DCB / charged sulfur source is 1.025 in molar ratio.
- the DPDS / feed sulfur source is 0.002 in molar ratio.
- the conversion rate of p-DCB at the end of the previous polymerization was 92%.
- the average particle size of the granular PAS thus obtained was 398 ⁇ m.
- the sieve opening of 150 ⁇ m (100 mesh) sieve has a melt viscosity of 41 Pa ⁇ s, chlorine content of 1,200 ppm, nitrogen content of 530 ppm, and sieve opening of 38 ⁇ m (400 mesh) sieve is melted.
- the viscosity was 40 Pa ⁇ s
- the chlorine content was 1,200 ppm
- the nitrogen content was 560 ppm.
- the sieved product (mass%) of the granular PAS was 91% for the sieved product having a sieve opening of 150 ⁇ m (100 mesh) and 93% for the sieved product having a sieve opening of 38 ⁇ m (400 mesh).
- Example 4 The process up to the dehydration step was performed in the same manner as in Example 1. After the dehydration step, the contents of the autoclave are cooled to 150 ° C., 3,248 g of p-DCB, 2,707 g of NMP, 19 g of sodium hydroxide, and 167 g of water are added and reacted at 220 ° C. for 5 hours with stirring to perform pre-stage polymerization. Went.
- the water / charged sulfur source is 1.50 in molar ratio.
- the NaOH / feed sulfur source molar ratio is 1.05.
- the NMP / feed sulfur source is 0.37 kg / mol.
- the molar ratio of p-DCB / feeding sulfur source is 1.015.
- the conversion rate of p-DCB at the end of the previous polymerization was 94%.
- the DPDS / feed sulfur source has a molar ratio of 0.002.
- 443 g of water was injected, the temperature was raised to 255 ° C., and the reaction was carried out for 5 hours to carry out post polymerization.
- the water / feed sulfur source is 2.63 in molar ratio.
- Example 2 After the post-stage polymerization was completed, the same procedure as in Example 1 was performed to obtain granular PAS.
- the granular PAS thus obtained had an average particle size of 356 ⁇ m.
- the sieve opening of 150 ⁇ m (100 mesh) has a melt viscosity of 23 Pa ⁇ s, a chlorine content of 800 ppm, and a nitrogen content of 580 ppm, and the sieve opening of 38 ⁇ m (400 mesh) has a melt viscosity of 22 Pa. S, chlorine content 800 ppm, nitrogen content 600 ppm.
- the sieved product (mass%) of the granular PAS was 88% for the sieved product having a sieve opening of 150 ⁇ m (100 mesh), and 91% for the sieved product having a sieve opening of 38 ⁇ m (400 mesh).
- Example 1 The same procedure as in Example 3 was performed except that DPDS was not added.
- the obtained granular PAS had an average particle diameter of 651 ⁇ m.
- the sieve opening 150 ⁇ m (100 mesh) sieve top has a melt viscosity of 128 Pa ⁇ s, chlorine content 1,100 ppm, nitrogen content 575 ppm, and sieve opening 38 ⁇ m (400 mesh) sieve top is melted.
- the viscosity was 120 Pa ⁇ s
- the chlorine content was 1,150 ppm
- the nitrogen content was 580 ppm.
- the sieved product (mass%) of the granular PAS was 89% with a sieved product having a sieve opening of 150 ⁇ m (100 mesh) and 91% with a sieved product having a sieve opening of 38 ⁇ m (400 mesh).
- the conversion rate of p-DCB at the end of the previous polymerization was 92%.
- 4.8 g of DPDS and 762 g of NMP were injected and reacted.
- the DPDS / feed sulfur source is 0.001 in molar ratio.
- 443 g of water was injected, the temperature was raised to 255 ° C., and the reaction was carried out for 5 hours to carry out post polymerization.
- the water / feed sulfur source is 2.63 in molar ratio.
- Post-stage polymerization, cooling, and separation / recovery were performed in the same manner as in Example 1 to obtain granular PAS.
- the granular PAS thus obtained had an average particle size of 344 ⁇ m.
- the sieve opening of 150 ⁇ m (100 mesh) sieve has a melt viscosity of 15 Pa ⁇ s, a chlorine content of 3,000 ppm, a nitrogen content of 650 ppm, and a sieve opening of 38 ⁇ m (400 mesh) sieve is melted.
- the viscosity was 14 Pa ⁇ s
- the chlorine content was 3,100 ppm
- the nitrogen content was 700 ppm.
- the sieved product (mass%) of granular PAS was 88% with a sieved product having a sieve opening of 150 ⁇ m (100 mesh) and 90% with a sieved product having a sieve opening of 38 ⁇ m (400 mesh).
- Example 3 The same procedure as in Example 1 was performed until the previous polymerization. When the conversion rate of p-DCB was 93%, 4.8 g of DPDS and 762 g of NMP were injected and reacted. The DPDS / feed sulfur source is 0.001 in molar ratio. Next, while continuing stirring, the amount of water at the time of charging was kept without adding water, and the temperature was raised to 255 ° C. and allowed to react for 5 hours to carry out post polymerization.
- the mixture was cooled to around room temperature, and the contents were sieved using a sieve having a sieve opening of 150 ⁇ m (100 mesh).
- the sieving material containing fine powder (filter paper recovery, recovery rate 99%) had a chlorine content of 2,950 ppm and a nitrogen content of 2,000 ppm.
- the melt viscosity was 5 Pa ⁇ s.
- Example 4 The same procedure as in Example 1 was performed until the previous polymerization. The conversion rate of p-DCB at the end of the previous polymerization is 92%. Next, 95 g of DPDS and 762 g of NMP were injected and reacted. The DPDS / feed sulfur source is 0.020 in molar ratio. Next, while stirring, 443 g of water was injected, the temperature was raised to 255 ° C., and the reaction was carried out for 5 hours to carry out post polymerization. The water / feed sulfur source is 2.63 in molar ratio.
- the same procedure as in Example 1 was performed to obtain granular PAS.
- the granular PAS thus obtained had an average particle size of 268 ⁇ m.
- the sieve opening of 150 ⁇ m (100 mesh) sieve has a melt viscosity of 1.5 Pa ⁇ s, a chlorine content of 1,000 ppm, a nitrogen content of 580 ppm, and a sieve opening of 38 ⁇ m (400 mesh).
- the melt viscosity was 1 Pa ⁇ s
- the chlorine content was 1,100 ppm
- the nitrogen content was 630 ppm.
- the sieved product (mass%) of the granular PAS was 51% for the sieved product having a sieve opening of 150 ⁇ m (100 mesh) and 55% for the sieved product of 38 ⁇ m (400 mesh).
- Example 5 The same procedure as in Example 2 was performed except that the filter paper was used for collection.
- the PAS of filter paper recovery (recovery rate 99%) had a melt viscosity of 8 Pa ⁇ s, a chlorine content of 2,800 ppm, and a nitrogen content of 1,030 ppm.
- a sieve opening of 150 ⁇ m (100 mesh) sieve has a melt viscosity of 30 Pa ⁇ s, a chlorine content of 3,800 ppm, a nitrogen content of 830 ppm, and a sieve opening of 38 ⁇ m (400 mesh) sieve is melted.
- the viscosity was 27 Pa ⁇ s
- the chlorine content was 3,950 ppm
- the nitrogen content was 870 ppm.
- the granular PAS sieve top (mass%) was 88% with a sieve opening of 150 ⁇ m (100 mesh), and 93% with a sieve opening of 38 ⁇ m (400 mesh).
- Example 7 After completion of the dehydration step, the same procedure as in Example 1 was performed except that 3263 g of p-DCB was added, 4750 g of NMP was added, and DPDS was not added. After completion of the pre-polymerization, 650 g of water was injected while stirring, and the temperature was raised to 255 ° C. and reacted for 5 hours to carry out post-polymerization.
- the water / feed sulfur source is 3.16 in molar ratio.
- Example 2 After the post-stage polymerization was completed, the same procedure as in Example 1 was performed to obtain granular PAS.
- the granular PAS thus obtained had an average particle size of 1,200 ⁇ m.
- the sieve opening of 150 ⁇ m (100 mesh) sieve has a melt viscosity of 300 Pa ⁇ s, a chlorine content of 700 ppm, and a nitrogen content of 350 ppm, and the sieve opening of 38 ⁇ m (400 mesh) sieve has a melt viscosity of 290 Pa. S, chlorine content 800 ppm, nitrogen content 380 ppm.
- the sieved product (mass%) of the granular PAS was 84% for the sieved product having a sieve opening of 150 ⁇ m (100 mesh) and 85% for the sieved product having a sieve opening of 38 ⁇ m (400 mesh).
- Comparative Example 1 is a case where DPDS was not added. In this case, the melt viscosity of the obtained granular PAS becomes a value outside the upper limit range of the present invention.
- Comparative Example 2 the value of the dihaloaromatic compound relative to the charged sulfur source is outside the upper limit range. In this case, the chlorine content of the obtained granular PAS becomes a value outside the upper limit range of the present invention.
- Comparative Example 3 is a case where the amount of coexisting water in the subsequent polymerization step is outside the lower limit of the present invention. When sieving with a sieve having a sieve opening of 150 ⁇ m, there is no sieve top as granular PAS.
- Comparative Example 4 is a case where the amount of DPDS added is outside the upper limit range of the present invention. In this case, the melt viscosity of the obtained granular PAS becomes a value outside the lower limit of the present invention, and the yield is not good.
- Comparative Example 5 is a case where sieving is not performed in Example 2.
- the melt viscosity of PAS recovered from filter paper is outside the lower limit of the present invention, and the chlorine content is outside the upper limit of the present invention.
- Comparative Examples 6 and 7 are cases where DPDS was not added.
- Comparative Example 6 is a case where the value of the dihaloaromatic compound relative to the charged sulfur source is outside the upper limit range.
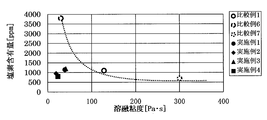
- Comparative Examples 1, 6, and 7 show conventional technical levels that do not use DPDS, which are different from the present invention (hereinafter, indicated by numerical values of a sieve opening of 150 ⁇ m). That is, when the melt viscosity is reduced from 300 Pa ⁇ s (Comparative Example 7) to 128 Pa ⁇ s (Comparative Example 1) and then 30 Pa ⁇ s (Comparative Example 6), the chlorine content is changed from 700 ppm (Comparative Example 7) to 1100 ppm. (Comparative Example 1) and then rise to 3800 ppm (Comparative Example 6). That is, low melt viscosity (high fluidization) and low halogenation are in a trade-off relationship. This is clearly seen in FIG.
- Examples 1 to 4 show a specific effect at a low melt viscosity and a low chlorine content, whereas in the comparative example, at a low melt viscosity, a high chlorine content ( Comparative Example 6) When the melt viscosity is high, the chlorine content is low (Comparative Example 7). Moreover, even if melt viscosity and chlorine content are reduced within the range of the prior art (Comparative Example 1), the low melt viscosity and low chlorine content of the examples are not reached.
- the granular PAS of the present invention is suitable in a wide range of fields such as electrical / electronic equipment and automobile equipment because a granular PAS with a good balance of melt viscosity, halogen content, nitrogen content, thermal stability and yield can be obtained. Can be used.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Polymers With Sulfur, Phosphorus Or Metals In The Main Chain (AREA)
- Processes Of Treating Macromolecular Substances (AREA)
Abstract
Description
(ii)それに比べて、38μm篩篩上物では塩素含有量及び窒素含有量が少なく、38μm篩通過品や濾紙回収品とは大きな差異が存在する。これらの知見は、一般的に認識されている事実、すなわち低分子量になるに従いそれに比例してPAS分子数が多くなる、すなわち末端数が増えるため、塩素含有量が順次大きくなることと様相を異にする。
(iii)また、ジスルフィド化合物の相分離状態における、良好な反応性などの特異的効果により、分子の末端に塩素が結合した低分子量物やオリゴマー等の多くを効率的に排除すること等により、さらに低塩素化され、熱安定性がよく、成形加工時のガス発生の少ない粒状ポリマーを高収率で回収することができる。
(ii)低溶融粘度化(高流動性)の実現。
(iii)低溶融粘度化(高流動性)と低ハロゲン(塩素)化の両立達成。
(iv)重合工程でのジスルフィド化合物による相分離状態の制御。
(v)粒状回収物(篩目開き38μm(400メッシュ)以上の篩篩上物)の顕著な収率向上。
(i)該粒状PASが、末端に、ジスルフィド化合物が開裂した-S-の置換基を含み、
(ii)該粒状PASが、38μm以上の篩目開きを有する篩での篩い分け後の篩上物であり、
(iii)該粒状PASが、ハロゲン含有量1,500ppm以下であり、かつ、
(iv)該粒状PASが、温度310℃及び剪断速度1,200sec-1の条件下で測定した溶融粘度が、3~100Pa・sである粒状PASが提供される。
(i)該重合工程の中で、重合反応をジスルフィド化合物の存在下で行い、かつ、
(ii)該分離・回収工程において、生成ポリマーを、38μm以上の篩目開きを有する篩で篩い分け、篩上物を得る、前記の粒状PASの製造方法が提供される。
1-1.硫黄源
本発明では、硫黄源としてアルカリ金属硫化物及びアルカリ金属水硫化物からなる群より選ばれる少なくとも一種の硫黄源を使用する。アルカリ金属硫化物としては、硫化リチウム、硫化ナトリウム、硫化カリウム、硫化ルビジウム、硫化セシウム、及びこれらの2種以上の混合物などを挙げることができる。アルカリ金属水硫化物としては、水硫化リチウム、水硫化ナトリウム、水硫化カリウム、水硫化ルビジウム、水硫化セシウム、及びこれらの2種以上の混合物などを挙げることができる。
本発明で使用するジハロ芳香族化合物は、芳香環に直接結合した2個のハロゲン原子を有するジハロゲン化芳香族化合物である。ジハロ芳香族化合物の具体例としては、例えば、o-ジハロベンゼン、m-ジハロベンゼン、p-ジハロベンゼン、ジハロトルエン、ジハロナフタレン、メトキシ-ジハロベンゼン、ジハロビフェニル、ジハロ安息香酸、ジハロジフェニルエーテル、ジハロジフェニルスルホン、ジハロジフェニルスルホキシド、ジハロジフェニルケトン等が挙げられる。
PASに分岐または架橋構造を導入するために、3個以上のハロゲン原子が結合したポリハロ化合物(必ずしも芳香族化合物でなくてもよい)、活性水素含有ハロゲン化芳香族化合物、ハロゲン化芳香族ニトロ化合物等を併用することができる。分岐・架橋剤としてのポリハロ化合物として、好ましくはトリハロベンゼンが挙げられる。
本発明では、脱水反応及び重合反応の溶媒として、非プロトン性極性有機溶媒である有機アミド溶媒を用いる。有機アミド溶媒は、高温でアルカリに対して安定なものが好ましい。
本発明では、液-液相分離状態を生起させ、低ハロゲン含有量で、溶融粘度を調整したPASを短時間で得るために、各種相分離剤を用いることができる。相分離剤とは、それ自身でまたは少量の水の共存下に、有機アミド溶媒に溶解し、PASの有機アミド溶媒に対する溶解性を低下させる作用を有する化合物である。相分離剤それ自体は、PASの溶媒ではない化合物である。
本発明では、重合工程での重合反応は、ジスルフィド化合物の存在下で行われる。ジスルフィド化合物の添加は、重合工程のどの段階でも良い。例えば、重合工程が、前段重合工程、後段重合工程の二段階工程を含む場合は、前段重合工程で添加しても良いし、後段重合工程で添加しても良い。また、前段重合工程開始時、すなわち、仕込み工程に添加してもよい。
製造方法は、有機アミド溶媒中で硫黄源とジハロ芳香族化合物とを重合する重合工程、該重合工程後の生成ポリマーを含有する液相を冷却する冷却工程及び生成ポリマーを分離・回収する分離・回収工程を含み、さらに、該重合工程中及び/または冷却工程前に相分離剤を存在させて、生成ポリマー濃厚相と生成ポリマー希薄相とが混在する液-液相分離状態を生起させる工程を含む製造方法である。
重合工程の前工程として、脱水工程を配置して反応系内の共存水量(水分量ともいう)を調節することが好ましい。脱水工程は、望ましくは不活性ガス雰囲気下、有機アミド溶媒とアルカリ金属硫化物とを含む混合物を加熱して反応させ、蒸留により水を系外へ排出する方法により実施する。硫黄源としてアルカリ金属水硫化物を用いる場合には、アルカリ金属水硫化物とアルカリ金属水酸化物とを含む混合物を加熱して反応させ、蒸留により水を系外へ排出する方法により実施する。
仕込み工程は、本発明の「有機アミド溶媒中で硫黄源とジハロ芳香族化合物とを重合する重合工程、該重合工程後の生成ポリマーを含有する液相を冷却する冷却工程、及び生成ポリマーを分離・回収する分離・回収工程を含み、さらに、該重合工程中及び/または冷却工程前に相分離剤を存在させて、生成ポリマー濃厚相と生成ポリマー希薄相とが混在する液-液相分離状態を生起させる工程を含む製造方法」において、重合工程で必要とされる、仕込み硫黄源に対する相分離剤量、仕込み硫黄源に対する共存水量、仕込み硫黄源に対するジハロ芳香族化合物量、仕込み硫黄源に対するアルカリ金属水酸化物量、仕込み硫黄源に対するジスルフィド化合物量等を調整する工程である。
重合工程は、有機アミド溶媒中で硫黄源とジハロ芳香族化合物を加熱することにより行われる。重合工程が、相分離剤の存在下に、重合反応系内の液相に生成ポリマー濃厚相と生成ポリマー希薄相とが混在する液-液相分離状態で重合反応を継続する相分離重合工程を含むことが好ましい。
(I)有機アミド溶媒中で硫黄源とジハロ芳香族化合物とを、仕込み硫黄源1モル当たり0.02~2モルの共存水量、1.005~1.040モルのジハロ芳香族化合物が存在する状態で、170~270℃の温度で重合反応させて、該ジハロ芳香族化合物の転化率が80~99%のポリマーを生成させる前段重合工程;及び
(II)相分離剤を、仕込み硫黄源1モル当たり、0.01~10モルの範囲で存在させるとともに、240~290℃の温度に加熱することにより、重合反応系内の液相を相分離状態に転換して重合反応を継続して行う後段重合工程;
によって行うことが好ましい。
前段重合工程における反応系の共存水量は、仕込み硫黄源1モル当たり、0.02~2モル、好ましくは0.05~1.9モル、より好ましくは0.5~1.8モルの範囲である。共存水量が少なすぎると、PASの分解等の望ましくない反応が起こり易く、逆に、2モルを超過すると、重合速度が著しく小さくなったり、有機アミド溶媒や生成PASの分解が生じ易くなるので、いずれも好ましくない。ジハロ芳香族化合物の仕込量は、仕込み硫黄源1モル当たり、通常1.005~1.040モル、好ましくは1.008~1.035モル、より好ましくは1.010~1.030モル、特に好ましくは1.012~1.028モルである。
転化率=[〔DHA仕込み量(モル)-DHA残存量(モル)〕/〔DHA仕込み量(モル)-DHA過剰量(モル)〕]×100
によって転化率を算出する。
転化率=[〔DHA仕込み量(モル)-DHA残存量(モル)〕/〔DHA仕込み量(モル)〕]×100
によって転化率を算出する。
後段重合工程で、相分離剤は、仕込み硫黄源1モル当たり、通常0.01~10モル、好ましくは0.03~8モル、より好ましくは、0.04~7モル用いる。
本発明では、重合工程後であって、冷却工程前に、前述した相分離剤を存在させて、生成ポリマー濃厚相と生成ポリマー希薄相とが混在する液-液相分離状態を生起させることができる。必要ならば、撹拌を行うことにより、液-液相分離状態の生起を調整することができる。
本発明の製造方法によれば、粒状PASを生成させることができるため、特定の篩目開きの篩を用いて篩い分けする方法により粒状PASを反応液から分離・回収する方法を採用する。生成物スラリーが高温状態(例えば、室温以上220℃未満の温度)にある間に、篩い分けを行っても良い。本発明の製造方法においては、38μm以上の篩目開きを有する篩で生成PASの篩い分けを行い、篩い分け後の篩上物として回収する。篩い分けは、後述する洗浄後、または乾燥後に行ってもよい。また、篩い分けを、洗浄前、洗浄後、乾燥後の各段階で行ってよい。
本発明によれば、粒状PASであって、(i)該粒状PASが、末端に、ジスルフィド化合物が開裂した-S-の置換基を含み、(ii)該粒状PASが、38μm以上の篩目開きを有する篩での篩い分け後の篩上物であり、(iii)該粒状PASが、ハロゲン含有量1,500ppm以下であり、かつ、(iv)該粒状ポリアリーレンスルフィドが、温度310℃及び剪断速度1,200sec-1の条件下で測定した溶融粘度が、3~100Pa・sである粒状PASが得られる。また、本発明によれば、該粒状PASは、有機アミド溶媒中で硫黄源とジハロ芳香族化合物とを重合する重合工程、該重合工程後の生成ポリマーを含有する液相を冷却する冷却工程、及び生成ポリマーを分離・回収する回収工程を含み、さらに、該重合工程中及び/または冷却工程前に相分離剤を存在させて、生成ポリマー濃厚相と生成ポリマー希薄相とが混在する液-液相分離状態を生起させる工程を含む粒状PASの製造方法であって、(i)該重合工程の中で、重合反応をジスルフィド化合物の存在下で行い、かつ、(ii)該分離・回収工程において、生成ポリマーを、38μm以上の篩目開きを有する篩で篩い分け、篩上物を得る、粒状PASの製造方法で製造される。
本発明の粒状PASを組成物として用いる場合、他の成分は次のとおりである。各種合成樹脂としては、高温において安定な熱可塑性樹脂が好ましく、その具体例としては、ポリエチレンテレフタレートやポリブチレンテレフタレート等の芳香族ポリエステル;ポリテトラフルオロエチレン、テトラフルオロエチレン/ヘキサフルオロプロピレン共重合体、テトラフルオロエチレン/パーフルオロアルキルビニルエーテル共重合体、ポリクロロトリフルオロエチレン、ポリフッ化ビニリデン、フッ化ビニリデン/ヘキサフルオロプロピレン共重合体、プロピレン/テトラフルオロエチレン共重合体、フッ化ビニリデン/クロロトリフルオロエチレン共重合体、エチレン/ヘキサフルオロプロピレン共重合体等のフッ素樹脂;ポリアセタール、ポリスチレン、ポリアミド、ポリカーボネート、ポリフェニレンエーテル、ポリアルキルアクリレート、ABS樹脂、ポリ塩化ビニルなどを挙げることができる。これらの熱可塑性樹脂は、それぞれ単独で、あるいは2種以上を組み合わせて使用することができる。ガラス繊維などの無機充填剤を配合したコンパウンドとして使用する場合には、本発明の特徴である低溶融粘度PASの特性に合わせて、良好な成形加工性が得られるように、上記各種の合成樹脂は、溶融粘度や固有粘度が低く良好な溶融挙動が得られるものを選択することが重要である。
反応後、生成ポリマー(PAS)を、篩目開き38μm(400メッシュ(目数/インチ))または篩目開き150μm(100メッシュ(目数/インチ))の篩で篩別して分別し、洗浄した。粒状PASの篩上物(質量%)は、脱水工程後の反応缶中に存在する仕込み硫黄源中の有効硫黄成分の全てがPASに転換したと仮定したときのPAS質量(理論量)を篩い分けする前のPASの全量として基準とした。篩上物(質量%)は、(篩上物)/(篩い分けする前のPASの全量:PAS質量(理論量))で算出した。仕込み硫黄源がジハロ芳香族化合物よりも過剰のモル比で仕込まれた場合は、仕込み硫黄源の全てがPASに転換することはあり得ない場合もあるが、その場合でも、一応仕込み硫黄源の量を基準として篩上物(質量%)を算出することとした。濾紙回収の場合も、濾紙上物/(篩い分けする前のPASの全量:PAS質量(理論量))で、回収率を算出した。
粒状PAS中のハロゲン含有量として、塩素含有量を燃焼イオンクロマト法により測定した。
(測定条件)
イオンクロマトグラフ:DIONEX製 DX320
燃焼用前処理装置:三菱化学製 AQF-100,ABC,WS-100,GA-100
試料:10mg
ヒーター:Inlet Temp/900℃,Outlet Temp/1000℃
吸収液:H2O2 900ppm,内標準PO4 3- 25ppm
粒状PASの溶融粘度は、キャピラリーとして1.0mmφ、長さ10.0mmのノズルを装着した(株)東洋精機製作所製キャピログラフ1C(登録商標)により溶融粘度を測定した。設定温度を310℃とした。ポリマー試料を装置内に導入し、5分間保持した後、剪断速度1,200sec-1で溶融粘度を測定した。
分離・回収工程で回収した生成ポリマー(粒状PAS)の平均粒径は、使用篩として、篩目開き2,800μm(7メッシュ(目数/インチ))、篩目開き1,410μm(12メッシュ(目数/インチ))、篩目開き1,000μm(16メッシュ(目数/インチ))、篩目開き710μm(24メッシュ(目数/インチ))、篩目開き500μm(32メッシュ(目数/インチ))、篩目開き250μm(60メッシュ(目数/インチ))、篩目開き150μm(100メッシュ(目数/インチ))、篩目開き105μm(145メッシュ(目数/インチ))、篩目開き75μm(200メッシュ(目数/インチ))、篩目開き38μm(400メッシュ(目数/インチ))の篩を用いた篩分法により測定し、各篩の篩上物の質量から、累積質量が50%質量となる時の平均粒径を算出した。
粒状PAS10mgを微量窒素硫黄分析計(アステック株式会社製、機種「ANTEK7000」)を用いて窒素の含有量を測定した。(基準物質はピリジン)
20リットルのオートクレーブに、N-メチル-2-ピロリドン(以下、「NMP」と略記)6,001g、水硫化ナトリウム水溶液(NaSH;純度62質量%)2,000g、水酸化ナトリウム水溶液(NaOH;純度74.0質量%)1,171gを仕込んだ。NaOH/NaSH(硫黄源)は、モル比で0.98である。
前段重合終了時のp-DCBの転化率が92%の時に、DPDS14.3gを加えたこと、及びDPDS/仕込み硫黄源のモル比を0.003としたことを除いて、実施例1と同様に行った。
脱水工程までは、実施例1と同様に行った。上記脱水工程後、オートクレーブの内容物を150℃まで冷却し、p-DCB3,280g、NMP2,708g、水酸化ナトリウム19g、及び水167g、DPDS9.5gを加え、撹拌しながら220℃で5時間反応させ、前段重合を行った。DPDS添加時、すなわち、重合反応開始時のp-DCBの転化率は0%である。水/仕込み硫黄源は、モル比で1.50である。NaOH/仕込み硫黄源は、モル比で1.05である。NMP/仕込み硫黄源は、0.38kg/モルである。p-DCB/仕込み硫黄源は、モル比で、1.025である。DPDS/仕込み硫黄源は、モル比で0.002である。前段重合終了時のp-DCBの転化率は92%であった。
脱水工程までは、実施例1と同様に行った。上記脱水工程後、オートクレーブの内容物を150℃まで冷却し、p-DCB3,248g、NMP2,707g、水酸化ナトリウム19g、及び水167gを加え、撹拌しながら220℃で5時間反応させ、前段重合を行った。水/仕込み硫黄源は、モル比で1.50である。NaOH/仕込み硫黄源のモル比は、1.05である。NMP/仕込み硫黄源は、0.37kg/モルである。p-DCB/仕込み硫黄源はモル比で、1.015である。前段重合終了時のp-DCBの転化率は94%であった。
DPDSを添加しないことを除いて、実施例3と同様に行った。得られた粒状PASは、平均粒径が651μmであった。また、篩目開き150μm(100メッシュ)篩上物は、溶融粘度が128Pa・s、塩素含有量1,100ppm、窒素含有量575ppmであり、篩目開き38μm(400メッシュ)篩上物は、溶融粘度120Pa・s、塩素含有量1,150ppm、窒素含有量580ppmであった。粒状PASの篩上物(質量%)は、篩目開き150μm(100メッシュ)の篩上物で89%であり、篩目開き38μm(400メッシュ)の篩上物で91%であった。
脱水工程までは、実施例1と同様に行った。上記脱水工程後、オートクレーブの内容物を150℃まで冷却し、p-DCB3,360g、NMP2,707g、水酸化ナトリウム19g、及び水167gを加え、撹拌しながら220℃で5時間反応させ、前段重合を行った。水/仕込み硫黄源は、モル比で1.50である。NaOH/仕込み硫黄源のモル比は、1.05である。NMP/仕込み硫黄源は、0.38kg/モルである。p-DCB/仕込み硫黄源はモル比で、1.050である。前段重合終了時のp-DCBの転化率は92%であった。次に、DPDS4.8g、NMP762gを圧入し反応させた。DPDS/仕込み硫黄源は、モル比で0.001である。次に撹拌を続けながら水443gを圧入し、255℃に昇温し5時間反応させ、後段重合を行った。水/仕込み硫黄源は、モル比で2.63である。
前段重合までは、実施例1と同様に行った。p-DCBの転化率が93%の時、DPDS4.8g、NMP762gを圧入し反応させた。DPDS/仕込み硫黄源は、モル比で0.001である。次に撹拌を続けながら、仕込み時の水量のまま、水を加えることなく、255℃に昇温し5時間反応させ、後段重合を行った。
前段重合までは、実施例1と同様に行った。前段重合終了時のp-DCBの転化率は92%である。次に、DPDS95g、NMP762gを圧入し反応させた。DPDS/仕込み硫黄源は、モル比で0.020である。次に撹拌を続けながら水443gを圧入し、255℃に昇温し5時間反応させ、後段重合を行った。水/仕込み硫黄源は、モル比で2.63である。
実施例2において濾紙を用いて回収した以外は同様に行った。濾紙回収(回収率99%)のPASは、溶融粘度8Pa・s、塩素含有量2,800ppm、窒素含有量1,030ppmであった。
脱水工程終了後、p-DCBを3392g添加し、DPDSを添加しなかったこと以外は実施例1と同様に行った。
脱水工程終了後、p-DCBを3263g添加し、NMPを4750g添加し、DPDSを添加しなかったこと以外は実施例1と同様に行った。前段重合終了後、撹拌を続けながら水650gを圧入し、255℃に昇温し5時間反応させ、後段重合を行った。水/仕込み硫黄源は、モル比で3.16である。


比較例1は、DPDSを添加しなかった場合である。この場合、得られた粒状PASの溶融粘度が、本発明の上限の範囲外の値となる。比較例2は、仕込み硫黄源に対するジハロ芳香族化合物の値が上限の範囲外の場合である。この場合、得られた粒状PASの塩素含有量が本発明の上限の範囲外の値となる。比較例3は、後段重合工程での共存水量が本発明の下限の範囲外の場合である。篩目開き150μmの篩で篩い分けた場合、粒状PASとしての篩上物はない。濾紙回収のPASの溶融粘度は、本発明の下限の範囲外となり、塩素含有量は、本発明の上限の範囲外となる。比較例4は、DPDSの添加量を本発明の上限の範囲外にした場合である。この場合、得られた粒状PASの溶融粘度は、本発明の下限の範囲外の値となり、また、収率が良くない。比較例5は、実施例2において、篩分けを行わない場合である。濾紙回収のPASの溶融粘度は、本発明の下限の範囲外となり、塩素含有量は、本発明の上限の範囲外となる。比較例6、7は、DPDSを添加しなかった場合である。加えて、比較例6は、仕込み硫黄源に対するジハロ芳香族化合物の値が上限の範囲外の場合である。
Claims (21)
- 粒状ポリアリーレンスルフィドであって、
(i)該粒状ポリアリーレンスルフィドが、末端に、ジスルフィド化合物が開裂した-S-の置換基を含み、
(ii)該粒状ポリアリーレンスルフィドが、38μm以上の篩目開きを有する篩での篩い分け後の篩上物であり、
(iii)該粒状ポリアリーレンスルフィドが、ハロゲン含有量1,500ppm以下であり、かつ、
(iv)該粒状ポリアリーレンスルフィドが、温度310℃及び剪断速度1,200sec-1の条件下で測定した溶融粘度が、3~100Pa・sである粒状ポリアリーレンスルフィド。 - 有機アミド溶媒中で硫黄源とジハロ芳香族化合物とを重合する重合工程及び該重合工程後の生成ポリマーを含有する液相を冷却する冷却工程を含み、さらに、該重合工程中及び/または冷却工程前に相分離剤を存在させて、生成ポリマー濃厚相と生成ポリマー希薄相とが混在する液-液相分離状態を生起させる工程を含み、かつ、該重合工程の中で、重合反応をジスルフィド化合物の存在下で行う製造方法で製造された粒状ポリアリーレンスルフィドである請求項1記載の粒状ポリアリーレンスルフィド。
- 重合工程が、相分離剤の存在下に、重合反応系内の液相に生成ポリマー濃厚相と生成ポリマー希薄相とが混在する液-液相分離状態で重合反応を継続する相分離重合工程を含む請求項1または2記載の粒状ポリアリーレンスルフィド。
- 粒状ポリアリーレンスルフィドが、窒素含有量650ppm以下である請求項1乃至3のいずれか1項に記載の粒状ポリアリーレンスルフィド。
- ハロゲンが、塩素である請求項1乃至4のいずれか1項に記載の粒状ポリアリーレンスルフィド。
- 篩が、150μm以上の篩目開きを有する篩である請求項1乃至5のいずれか1項に記載の粒状ポリアリーレンスルフィド。
- 塩素含有量1,300ppm以下である請求項1乃至6のいずれか1項に記載の粒状ポリアリーレンスルフィド。
- 温度310℃及び剪断速度1,200sec-1の条件下で測定した溶融粘度が、15~55Pa・sである請求項1乃至7のいずれか1項に記載の粒状ポリアリーレンスルフィド。
- 有機アミド溶媒中で硫黄源とジハロ芳香族化合物とを重合する重合工程、該重合工程後の生成ポリマーを含有する液相を冷却する冷却工程、及び生成ポリマーを分離・回収する分離・回収工程を含み、さらに、該重合工程中及び/または冷却工程前に相分離剤を存在させて、生成ポリマー濃厚相と生成ポリマー希薄相とが混在する液-液相分離状態を生起させる工程を含む粒状ポリアリーレンスルフィドの製造方法であって、
(i)該重合工程の中で、重合反応をジスルフィド化合物の存在下で行い、かつ、
(ii)該分離・回収工程において、生成ポリマーを、38μm以上の篩目開きを有する篩で篩い分け、篩上物を得る、請求項1記載の粒状ポリアリーレンスルフィドの製造方法。 - 重合工程が、相分離剤の存在下に、重合反応系内の液相に生成ポリマー濃厚相と生成ポリマー希薄相とが混在する液-液相分離状態で重合反応を継続する相分離重合工程を含む請求項9記載の粒状ポリアリーレンスルフィドの製造方法。
- 相分離剤が、水、有機カルボン酸金属塩、有機スルホン酸金属塩、アルカリ金属ハライド、アルカリ土類金属ハライド、芳香族カルボン酸のアルカリ土類金属塩、リン酸アルカリ金属塩、アルコール類、及びパラフィン系炭化水素からなる群より選ばれる少なくとも一種の相分離剤である請求項9または10記載の粒状ポリアリーレンスルフィドの製造方法。
- 相分離剤を、仕込み硫黄源1モル当たり、0.01~15モル用いる請求項9乃至11のいずれか1項に記載の粒状ポリアリーレンスルフィドの製造方法。
- 重合工程での重合反応が、仕込み硫黄源1モル当たり、0.0005~0.015モルのジスルフィド化合物の存在下で行われる請求項9乃至12のいずれか1項に記載の粒状ポリアリーレンスルフィドの製造方法。
- ジハロ芳香族化合物の転化率が45%以上となった時点で、ジスルフィド化合物を添加し、重合工程に存在させる請求項9乃至13のいずれか1項に記載の粒状ポリアリーレンスルフィドの製造方法。
- ジスルフィド化合物が、ジフェニルジスルフィドである請求項9乃至14のいずれか1項に記載の粒状ポリアリーレンスルフィドの製造方法。
- 篩が、150μm以上の篩目開きを有する篩である請求項9乃至15のいずれか1項に記載の粒状ポリアリーレンスルフィドの製造方法。
- 150μm以上の篩目開きを有する篩での篩い分け後の篩上物が、篩い分け前の全量に対して、80質量%以上である請求項9乃至16のいずれか1項に記載の粒状ポリアリーレンスルフィドの製造方法。
- 重合工程を少なくとも下記の2段階工程:
(I)有機アミド溶媒中で硫黄源とジハロ芳香族化合物とを、仕込み硫黄源1モル当たり0.02~2モルの共存水量、1.005~1.040モルのジハロ芳香族化合物が存在する状態で、170~270℃の温度で重合反応させて、該ジハロ芳香族化合物の転化率が80~99%のポリマーを生成させる前段重合工程;及び
(II)相分離剤を、仕込み硫黄源1モル当たり、0.01~10モルの範囲で存在させるとともに、240~290℃の温度に加熱することにより、重合反応系内の液相を相分離状態に転換して重合反応を継続して行う後段重合工程;
によって行う請求項9~17記載の粒状ポリアリーレンスルフィドの製造方法。 - 相分離剤が水の場合、仕込み硫黄源1モル当たり2~5モルの水が存在する状態となるように重合反応系内の共存水量を調整する請求項12記載の粒状ポリアリーレンスルフィドの製造方法。
- 相分離剤が、有機カルボン酸金属塩、有機スルホン酸金属塩、アルカリ金属ハライド、アルカリ土類金属ハライド、芳香族カルボン酸のアルカリ土類金属塩、リン酸アルカリ金属塩、アルコール類、及びパラフィン系炭化水素からなる群より選ばれる少なくとも一種の相分離剤の場合、仕込み硫黄源1モル当たり、0.01~3モルの範囲で存在させる請求項12記載の粒状ポリアリーレンスルフィドの製造方法。
- 水と水以外の他の相分離剤を併用する場合、共存水量を、仕込み硫黄源1モル当り0.01~7モルに調整するとともに、水以外の他の相分離剤を、仕込み硫黄源1モル当り0.01~3モルの範囲で存在させる請求項12記載の粒状ポリアリーレンスルフィドの製造方法。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020147021810A KR101660614B1 (ko) | 2012-03-30 | 2013-03-29 | 입상 폴리아릴렌 설파이드 및 그 제조방법 |
| EP13768784.4A EP2840105A4 (en) | 2012-03-30 | 2013-03-29 | GRANULAR POLY (ARYLENE SULFIDE) AND METHOD FOR PRODUCING THE SAME |
| CN201380005933.3A CN104144970B (zh) | 2012-03-30 | 2013-03-29 | 粒状聚亚芳基硫醚及其制造方法 |
| US14/378,854 US9422400B2 (en) | 2012-03-30 | 2013-03-29 | Granular polyarylene sulfide and process for manufacturing the same |
| JP2014508086A JP6062924B2 (ja) | 2012-03-30 | 2013-03-29 | 粒状ポリアリーレンスルフィド及びその製造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012083010 | 2012-03-30 | ||
| JP2012-083010 | 2012-03-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013147141A1 true WO2013147141A1 (ja) | 2013-10-03 |
Family
ID=49260383
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/059497 WO2013147141A1 (ja) | 2012-03-30 | 2013-03-29 | 粒状ポリアリーレンスルフィド及びその製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US9422400B2 (ja) |
| EP (1) | EP2840105A4 (ja) |
| JP (1) | JP6062924B2 (ja) |
| KR (1) | KR101660614B1 (ja) |
| CN (1) | CN104144970B (ja) |
| WO (1) | WO2013147141A1 (ja) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015050053A1 (ja) * | 2013-10-01 | 2015-04-09 | 株式会社クレハ | 分岐型ポリアリーレンスルフィド樹脂及びその製造方法、並びにその高分子改質剤としての使用 |
| WO2015147090A1 (ja) * | 2014-03-25 | 2015-10-01 | 株式会社クレハ | 熱処理微粉ポリアリーレンスルフィド、及び該熱処理微粉ポリアリーレンスルフィドを製造する製造方法 |
| US9388283B2 (en) | 2013-09-25 | 2016-07-12 | Ticona Llc | Method of polyarylene sulfide crystallization |
| US9403948B2 (en) | 2013-09-25 | 2016-08-02 | Ticona Llc | Salt byproduct separation during formation of polyarylene sulfide |
| WO2016159234A1 (ja) * | 2015-03-31 | 2016-10-06 | 株式会社クレハ | 微粉ポリアリーレンスルフィドを製造する方法及び微粉ポリアリーレンスルフィド |
| US9562139B2 (en) | 2013-09-25 | 2017-02-07 | Ticona Llc | Process for forming low halogen content polyarylene sulfides |
| US9587074B2 (en) | 2013-09-25 | 2017-03-07 | Ticona Llc | Multi-stage process for forming polyarylene sulfides |
| KR20170027724A (ko) | 2014-06-30 | 2017-03-10 | 도레이 카부시키가이샤 | 폴리아릴렌설파이드 및 그 제조 방법 |
| US9604156B2 (en) | 2013-09-25 | 2017-03-28 | Ticona Llc | Method and system for separation of a polymer from multiple compounds |
| US9617387B2 (en) | 2013-09-25 | 2017-04-11 | Ticona Llc | Scrubbing process for polyarylene sulfide formation |
| JP2019507825A (ja) * | 2016-12-30 | 2019-03-22 | 浙江新和成特種材料有限公司Zhejiang Nhu Special Materials Co., Ltd. | 低塩素含有量のポリフェニレンスルフィド及びその製造方法、樹脂組成物並びに成形体 |
| WO2020026918A1 (ja) * | 2018-07-31 | 2020-02-06 | 東レ株式会社 | ポリアリーレンスルフィドの製造方法、ポリアリーレンスルフィドプレポリマーおよびその製造方法 |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6626444B2 (ja) * | 2013-08-27 | 2019-12-25 | ティコナ・エルエルシー | 射出成形用の耐熱性強化熱可塑性組成物 |
| WO2016133738A1 (en) | 2015-02-19 | 2016-08-25 | Ticona Llc | Method for forming a low viscosity polyarylene sulfide |
| WO2016133739A1 (en) | 2015-02-19 | 2016-08-25 | Ticona Llc | Method for forming a high molecular weight polyarylene sulfide |
| WO2016133740A1 (en) | 2015-02-19 | 2016-08-25 | Ticona Llc | Method of polyarylene sulfide precipitation |
| WO2016153610A1 (en) | 2015-03-25 | 2016-09-29 | Ticona Llc | Technique for forming a high melt viscosity polyarylene sulfide |
| JP7071235B2 (ja) * | 2018-07-03 | 2022-05-18 | ポリプラスチックス株式会社 | 多孔質成形体及びその製造方法 |
| KR102251404B1 (ko) | 2018-07-03 | 2021-05-12 | 주식회사 엘지화학 | 폴리아릴렌 설파이드의 제조 방법 |
| KR102306017B1 (ko) * | 2018-09-28 | 2021-09-27 | 주식회사 엘지화학 | 폴리아릴렌 설파이드의 제조 방법 |
| KR102306016B1 (ko) * | 2018-09-28 | 2021-09-27 | 주식회사 엘지화학 | 폴리아릴렌 설파이드의 제조 방법 |
| US11407861B2 (en) | 2019-06-28 | 2022-08-09 | Ticona Llc | Method for forming a polyarylene sulfide |
| JP2023508316A (ja) | 2019-12-20 | 2023-03-02 | ティコナ・エルエルシー | ポリアリーレンスルフィドを形成するための方法 |
| JP7262664B2 (ja) * | 2020-03-24 | 2023-04-21 | 株式会社クレハ | ポリアリーレンスルフィドの製造方法 |
| US20230022693A1 (en) * | 2021-07-06 | 2023-01-26 | Northwestern University | Polymer compositions for vertical channel organic electrochemical transistors and complementary logic circuits |
| WO2023038889A1 (en) | 2021-09-08 | 2023-03-16 | Ticona Llc | Extraction technique for recovering an organic solvent from a polyarylene sulfide waste sludge |
| JP2024535216A (ja) | 2021-09-08 | 2024-09-30 | ティコナ・エルエルシー | ポリアリーレンスルフィド廃棄汚泥から有機溶媒を回収するための逆溶媒技術 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS59215323A (ja) | 1983-05-17 | 1984-12-05 | バイエル・アクチエンゲゼルシヤフト | ポリアリ−レンサルフアイドの製造法 |
| JPS62106929A (ja) * | 1985-11-02 | 1987-05-18 | Toyo Soda Mfg Co Ltd | ポリフエニレンスルフイドおよびその製造方法 |
| JP2002201274A (ja) * | 2000-12-28 | 2002-07-19 | Dic Ep Inc | ポリアリーレンスルフィドの製造法 |
| JP2010053335A (ja) * | 2008-07-30 | 2010-03-11 | Toray Ind Inc | ポリアリーレンスルフィド樹脂の製造方法 |
| JP2010126621A (ja) | 2008-11-27 | 2010-06-10 | Dic Corp | ポリアリーレンスルフィドの製造方法 |
| JP2011148870A (ja) * | 2010-01-20 | 2011-08-04 | Toray Ind Inc | ポリアリーレンスルフィドの製造方法 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4820801A (en) * | 1985-11-02 | 1989-04-11 | Tosoh Corp. | Polyphenylene sulfide containing covalently bonded chlorine in a reduced amount |
| JPH0816156B2 (ja) * | 1988-12-22 | 1996-02-21 | 財団法人生産開発科学研究所 | ポリアリーレンチオエーテルの製造法 |
| US5280104A (en) * | 1992-06-30 | 1994-01-18 | Phillips Petroleum Company | Process for the preparation of poly(arylene sulfide) with low metal contamination and polymer produced |
| US20020183481A1 (en) * | 1999-12-30 | 2002-12-05 | Vidaurri Fernando C. | Method to decrease corrosiveness of reactants in poly(arylene sulfide) polymer production |
| EP1440996B1 (en) * | 2001-09-27 | 2010-05-26 | Kureha Corporation | Process for production of polyarylene sulfide |
| US8680230B2 (en) * | 2008-07-22 | 2014-03-25 | Kureha Corporation | Production process of poly(arylene sulfide) whose content of terminal halogen group has been reduced |
| KR101549205B1 (ko) * | 2008-12-23 | 2015-09-02 | 에스케이케미칼 주식회사 | 폴리아릴렌 설파이드의 제조 방법 |
| KR101554010B1 (ko) * | 2008-12-31 | 2015-09-18 | 에스케이케미칼 주식회사 | 유리 요오드 저감 폴리아릴렌 설파이드의 제조 방법 |
-
2013
- 2013-03-29 US US14/378,854 patent/US9422400B2/en active Active
- 2013-03-29 JP JP2014508086A patent/JP6062924B2/ja active Active
- 2013-03-29 EP EP13768784.4A patent/EP2840105A4/en not_active Withdrawn
- 2013-03-29 WO PCT/JP2013/059497 patent/WO2013147141A1/ja active Application Filing
- 2013-03-29 KR KR1020147021810A patent/KR101660614B1/ko active IP Right Grant
- 2013-03-29 CN CN201380005933.3A patent/CN104144970B/zh active Active
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS59215323A (ja) | 1983-05-17 | 1984-12-05 | バイエル・アクチエンゲゼルシヤフト | ポリアリ−レンサルフアイドの製造法 |
| JPS62106929A (ja) * | 1985-11-02 | 1987-05-18 | Toyo Soda Mfg Co Ltd | ポリフエニレンスルフイドおよびその製造方法 |
| JP2002201274A (ja) * | 2000-12-28 | 2002-07-19 | Dic Ep Inc | ポリアリーレンスルフィドの製造法 |
| JP2010053335A (ja) * | 2008-07-30 | 2010-03-11 | Toray Ind Inc | ポリアリーレンスルフィド樹脂の製造方法 |
| JP2010126621A (ja) | 2008-11-27 | 2010-06-10 | Dic Corp | ポリアリーレンスルフィドの製造方法 |
| JP2011148870A (ja) * | 2010-01-20 | 2011-08-04 | Toray Ind Inc | ポリアリーレンスルフィドの製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2840105A4 |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9617387B2 (en) | 2013-09-25 | 2017-04-11 | Ticona Llc | Scrubbing process for polyarylene sulfide formation |
| US9388283B2 (en) | 2013-09-25 | 2016-07-12 | Ticona Llc | Method of polyarylene sulfide crystallization |
| US9403948B2 (en) | 2013-09-25 | 2016-08-02 | Ticona Llc | Salt byproduct separation during formation of polyarylene sulfide |
| US9604156B2 (en) | 2013-09-25 | 2017-03-28 | Ticona Llc | Method and system for separation of a polymer from multiple compounds |
| US9562139B2 (en) | 2013-09-25 | 2017-02-07 | Ticona Llc | Process for forming low halogen content polyarylene sulfides |
| US9587074B2 (en) | 2013-09-25 | 2017-03-07 | Ticona Llc | Multi-stage process for forming polyarylene sulfides |
| US9938379B2 (en) | 2013-09-25 | 2018-04-10 | Ticona Llc | Salt byproduct separation during formation of polyarylene sulfide |
| US9868824B2 (en) | 2013-09-25 | 2018-01-16 | Ticona Llc | Method of polyarylene sulfide crystallization |
| WO2015050053A1 (ja) * | 2013-10-01 | 2015-04-09 | 株式会社クレハ | 分岐型ポリアリーレンスルフィド樹脂及びその製造方法、並びにその高分子改質剤としての使用 |
| US10081710B2 (en) | 2013-10-01 | 2018-09-25 | Kureha Corporation | Branched polyarylene sulfide resin, method for manufacturing same and use as polymer modifier |
| JPWO2015050053A1 (ja) * | 2013-10-01 | 2017-03-09 | 株式会社クレハ | 分岐型ポリアリーレンスルフィド樹脂及びその製造方法、並びにその高分子改質剤としての使用 |
| WO2015147090A1 (ja) * | 2014-03-25 | 2015-10-01 | 株式会社クレハ | 熱処理微粉ポリアリーレンスルフィド、及び該熱処理微粉ポリアリーレンスルフィドを製造する製造方法 |
| JPWO2015147090A1 (ja) * | 2014-03-25 | 2017-04-13 | 株式会社クレハ | 熱処理微粉ポリアリーレンスルフィド、及び該熱処理微粉ポリアリーレンスルフィドを製造する製造方法 |
| EP3162839A4 (en) * | 2014-06-30 | 2018-01-10 | Toray Industries, Inc. | Polyarylene sulfide and method for manufacturing same |
| KR20170027724A (ko) | 2014-06-30 | 2017-03-10 | 도레이 카부시키가이샤 | 폴리아릴렌설파이드 및 그 제조 방법 |
| JPWO2016159234A1 (ja) * | 2015-03-31 | 2017-10-19 | 株式会社クレハ | 微粉ポリアリーレンスルフィドを製造する方法及び微粉ポリアリーレンスルフィド |
| WO2016159234A1 (ja) * | 2015-03-31 | 2016-10-06 | 株式会社クレハ | 微粉ポリアリーレンスルフィドを製造する方法及び微粉ポリアリーレンスルフィド |
| US10280264B2 (en) | 2015-03-31 | 2019-05-07 | Kureha Corporation | Method for manufacturing fine polyarylene sulfide powder, and fine polyarylene sulfide powder |
| JP2019507825A (ja) * | 2016-12-30 | 2019-03-22 | 浙江新和成特種材料有限公司Zhejiang Nhu Special Materials Co., Ltd. | 低塩素含有量のポリフェニレンスルフィド及びその製造方法、樹脂組成物並びに成形体 |
| WO2020026918A1 (ja) * | 2018-07-31 | 2020-02-06 | 東レ株式会社 | ポリアリーレンスルフィドの製造方法、ポリアリーレンスルフィドプレポリマーおよびその製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US9422400B2 (en) | 2016-08-23 |
| EP2840105A1 (en) | 2015-02-25 |
| JP6062924B2 (ja) | 2017-01-18 |
| JPWO2013147141A1 (ja) | 2015-12-14 |
| CN104144970B (zh) | 2016-10-12 |
| EP2840105A4 (en) | 2015-12-02 |
| KR101660614B1 (ko) | 2016-09-27 |
| US20150065664A1 (en) | 2015-03-05 |
| KR20140109476A (ko) | 2014-09-15 |
| CN104144970A (zh) | 2014-11-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6062924B2 (ja) | 粒状ポリアリーレンスルフィド及びその製造方法 | |
| JP5221877B2 (ja) | ポリアリーレンスルフィドの製造方法 | |
| JP5623277B2 (ja) | 粒状ポリアリーレンスルフィドの製造方法 | |
| JP5731196B2 (ja) | 末端ハロゲン基含量が低減されたポリアリーレンスルフィドの製造方法 | |
| JP6517337B2 (ja) | 粒状ポリアリーレンスルフィドを製造する方法、及び粒状ポリアリーレンスルフィド | |
| US9587074B2 (en) | Multi-stage process for forming polyarylene sulfides | |
| KR101827231B1 (ko) | 폴리아릴렌 설파이드의 제조 방법 및 폴리아릴렌 설파이드 | |
| JP5189293B2 (ja) | 分岐型ポリアリーレンスルフィド樹脂及びその製造方法、並びにその高分子改質剤としての使用 | |
| CN108602954B (zh) | 粒状聚亚芳基硫醚的制造方法、粒状聚亚芳基硫醚的平均粒径增大方法、粒状聚亚芳基硫醚的粒子强度提高方法以及粒状聚亚芳基硫醚 | |
| EP3524632B1 (en) | Polyarylene sulfide preparation method | |
| JP2016536443A (ja) | ポリアリーレンスルフィド結晶化方法 | |
| JP6306601B2 (ja) | 分岐型ポリアリーレンスルフィド樹脂及びその製造方法、並びにその高分子改質剤としての使用 | |
| JP7357695B2 (ja) | ポリアリーレンスルフィドの製造方法 | |
| JP6889271B2 (ja) | ポリアリーレンスルフィドの製造方法 | |
| JP2024021683A (ja) | ポリアリーレンスルフィド樹脂の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13768784 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2014508086 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20147021810 Country of ref document: KR Kind code of ref document: A |
|
| REEP | Request for entry into the european phase |
Ref document number: 2013768784 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013768784 Country of ref document: EP Ref document number: 14378854 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |