WO2013141105A1 - (メタ)アクリル酸エステル系共重合体の製造方法 - Google Patents
(メタ)アクリル酸エステル系共重合体の製造方法 Download PDFInfo
- Publication number
- WO2013141105A1 WO2013141105A1 PCT/JP2013/056958 JP2013056958W WO2013141105A1 WO 2013141105 A1 WO2013141105 A1 WO 2013141105A1 JP 2013056958 W JP2013056958 W JP 2013056958W WO 2013141105 A1 WO2013141105 A1 WO 2013141105A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- meth
- acrylate
- anionic polymerization
- aluminum
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/52—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides selected from boron, aluminium, gallium, indium, thallium or rare earths
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F220/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride ester, amide, imide or nitrile thereof
- C08F220/02—Monocarboxylic acids having less than ten carbon atoms; Derivatives thereof
- C08F220/10—Esters
- C08F220/12—Esters of monohydric alcohols or phenols
- C08F220/14—Methyl esters, e.g. methyl (meth)acrylate
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/46—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides selected from alkali metals
- C08F4/48—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides selected from alkali metals selected from lithium, rubidium, caesium or francium
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D4/00—Coating compositions, e.g. paints, varnishes or lacquers, based on organic non-macromolecular compounds having at least one polymerisable carbon-to-carbon unsaturated bond ; Coating compositions, based on monomers of macromolecular compounds of groups C09D183/00 - C09D183/16
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J4/00—Adhesives based on organic non-macromolecular compounds having at least one polymerisable carbon-to-carbon unsaturated bond ; adhesives, based on monomers of macromolecular compounds of groups C09J183/00 - C09J183/16
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F222/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by a carboxyl radical and containing at least one other carboxyl radical in the molecule; Salts, anhydrides, esters, amides, imides, or nitriles thereof
- C08F222/10—Esters
- C08F222/1006—Esters of polyhydric alcohols or polyhydric phenols
- C08F222/102—Esters of polyhydric alcohols or polyhydric phenols of dialcohols, e.g. ethylene glycol di(meth)acrylate or 1,4-butanediol dimethacrylate
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/54—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with other compounds thereof
- C08F4/56—Alkali metals being the only metals present, e.g. Alfin catalysts
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/54—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with other compounds thereof
- C08F4/56—Alkali metals being the only metals present, e.g. Alfin catalysts
- C08F4/565—Lithium being present, e.g. butyllithium + sodiumphenoxide
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2312/00—Crosslinking
- C08L2312/06—Crosslinking by radiation
Definitions
- the present invention relates to a method for producing a (meth) acrylic acid ester copolymer.
- the (meth) acrylic acid ester-based copolymer obtained by the production method of the present invention is useful, for example, as a constituent component of the photocurable resin composition.
- a photo-curable resin composition that is cured by irradiation with ultraviolet rays or electron beams is known.
- Photocurable resin compositions have been developed for applications such as adhesives, pressure-sensitive adhesives, paints, and coating materials in the fields of electricity, electronics, optics, and the like.
- the component constituting the photocurable resin composition include monomers, oligomers, and polymers having one or more polymerizable functional groups. For example,
- the asymmetric dimethacrylate represented by is anionic polymerized in toluene in the presence of t-butyllithium and methylaluminum bis (2,6-di-t-butylphenoxide), so that a plurality of methacryloyl groups can be used as polymerizable functional groups. It is known that a polymer having a group in the side chain can be obtained (see Non-Patent Document 1).
- Non-Patent Document 1 requires anionic polymerization at a low temperature of ⁇ 60 ° C. in order to suppress the polymerization of side chain methacryloyl groups during anionic polymerization, and there is room for improvement in the industry. Therefore, an object of the present invention is to provide a (meth) acrylic acid ester copolymer having one or more polymerizable functional groups in the side chain and capable of being cured by irradiation with ultraviolet rays or electron beams. Another object of the present invention is to provide an industrially advantageous production method.
- Di (meth) acrylate (1) (hereinafter referred to as “di (meth) acrylate (1)”) represented by the following general formula (2)
- alkyl (meth) acrylate (2) (wherein R 5 represents a hydrogen atom or a methyl group, and R 6 represents an alkyl group having 1 to 6 carbon atoms)
- alkyl (meth) acrylate (2) represented by the formula: an organic lithium compound (L), the following general formula (3):
- a method for producing a (meth) acrylic acid ester copolymer (X) hereinafter referred to as copolymer (X)
- copolymer (X) which is anionic polymerized in the presence of A method for producing the copolymer (X) of the above [
- the copolymer (X) useful as a constituent component of the photocurable resin composition can be industrially advantageously produced.
- a mixture of di (meth) acrylate (1) and alkyl (meth) acrylate (2) is prepared from an organolithium compound (L), an aluminum compound (A), and a Lewis base (B). Anionic polymerization occurs in the presence.
- di (meth) acrylate (1) a (meth) acryloyl group to which R 2 is bonded is selectively polymerized, and a (meth) acryloyl group bonded to the carbon atom to which R 3 and R 4 are bonded. Is suppressed and remains in the side chain of the resulting copolymer (X), so that the copolymer (X) can be cured by irradiating with ultraviolet rays or electron beams.
- Examples of the linear alkylene group having 1 to 5 carbon atoms represented by R 2 include a methylene group, an ethylene group, a trimethylene group, a tetramethylene group, and a pentamethylene group, and a methylene group or an ethylene group is preferable.
- Examples of the hydrocarbon group having 1 to 6 carbon atoms independently represented by R 3 and R 4 include a methyl group, an ethyl group, an n-propyl group, an isopropyl group, an n-butyl group, an isobutyl group, and a sec-butyl group.
- R 5 represents a hydrogen atom or a methyl group, and is preferably the same atom or functional group as R 1 .
- Examples of the alkyl group having 1 to 6 carbon atoms represented by R 6 include methyl group, ethyl group, n-propyl group, isopropyl group, n-butyl group, isobutyl group, sec-butyl group, t-butyl group, and 2-methylbutyl.
- a methyl group or an ethyl group is preferable.
- the alkyl group having 1 to 6 carbon atoms represented by R 3 and R 4 and the alkyl group having 1 to 6 carbon atoms represented by R 6 may have a substituent.
- the substituent is not particularly limited as long as it does not inhibit the production method of the present invention.
- alkoxy groups such as methoxy group, ethoxy group, isopropoxy group, t-butoxy group; chlorine atom, bromine atom and the like And the like.
- the molar ratio of di (meth) acrylate (1) to alkyl (meth) acrylate (2) in the mixture to be subjected to anionic polymerization depends on the high polymerization rate and the resulting copolymer (X ) Is preferably in the range of 5:95 to 90:10, more preferably in the range of 10:90 to 80:20, and in the range of 20:80 to 70:30. More preferably.
- other monomers other than di (meth) acrylate (1) and alkyl (meth) acrylate (2) are further allowed to coexist within a range not inhibiting the production method of the present invention.
- the other monomer is not particularly limited as long as it is an anion-polymerizable monomer, and these other monomers may be added to the polymerization system separately even if used in a mixture. May be used.
- the other monomer include aromatic vinyl compounds such as styrene and ⁇ -methylstyrene; conjugated dienes such as butadiene and isoprene.
- the amount of such other monomers used is not particularly limited, but is usually 0.05 to 2 masses of the total amount of di (meth) acrylate (1) and alkyl (meth) acrylate (2) in the mixture. From the viewpoint of photocurability of the resulting copolymer (X), a range of 1 mass times or less is preferable.
- the monomer used in the production method of the present invention allows the polymerization to proceed smoothly. From the viewpoint, it is preferable to perform a drying treatment in advance under an inert gas atmosphere or the like. In the drying treatment, a dehydrating agent or a drying agent such as calcium hydride, molecular sieves, activated alumina or the like is preferably used.
- the organolithium compound (L) used in the method of the present invention acts as an anionic polymerization initiator.
- the organic lithium compound (L) include t-butyl lithium, 1,1-dimethylpropyl lithium, 1,1-diphenylhexyl lithium, 1,1-diphenyl-3-methylpentyl lithium, ethyl ⁇ -lithioisobutyrate, Butyl ⁇ -lithioisobutyrate, methyl ⁇ -lithioisobutyrate, isopropyl lithium, sec-butyl lithium, 1-methylbutyl lithium, 2-ethylpropyl lithium, 1-methylpentyl lithium, cyclohexyl lithium, diphenylmethyl lithium, ⁇ -Methylbenzyl lithium, methyl lithium, n-propyl lithium, n-butyl lithium, n-pentyl lithium and the like.
- An organolithium compound having 3 to 40 carbon atoms having a chemical structure having a secondary carbon atom as an anion center is preferred, and sec-butyllithium is particularly preferred.
- an organic lithium compound (L) may be used individually by 1 type, or may use 2 or more types together.
- the amount of the organolithium compound (L) used is the monomer in the mixture used (that is, di (meth) acrylate (1) and alkyl (meth) acrylate (2) and other monomers as optional components). It is preferable to use it in a proportion within the range of 0.0001 to 0.2 mole times the total of the above from the viewpoint that the copolymer (X) can be produced smoothly.
- the aluminum compound (A) used in the production method of the present invention is the monomer used (that is, di (meth) acrylate (1) and alkyl (meth) acrylate (2) and other monomers as optional components).
- the monomer used that is, di (meth) acrylate (1) and alkyl (meth) acrylate (2) and other monomers as optional components.
- the following general formula (A-1) is the monomer used (that is, di (meth) acrylate (1) and alkyl (meth) acrylate (2) and other monomers as optional components).
- R 7 represents a monovalent saturated hydrocarbon group, a monovalent aromatic hydrocarbon group, an alkoxy group, an aryloxy group or an N, N-disubstituted amino group
- R 8 and R 9 are each independently An aryloxy group or an aryleneoxy group bonded to each other.
- R 10 represents an aryloxy group
- R 11 and R 12 each independently represents a monovalent saturated hydrocarbon group, a monovalent aromatic hydrocarbon group, an alkoxy group or an N, N-disubstituted amino group. Represents a group.
- an organoaluminum compound hereinafter referred to as an aluminum compound (A-2)
- A-1 aluminum compound
- Examples of the aryloxy group represented by R 7 , R 8 , R 9 and R 10 include a phenoxy group, a 2-methylphenoxy group, a 4-methylphenoxy group, a 2,6-dimethylphenoxy group, and a 2,4-dioxy group.
- Examples of the aryleneoxy group in which R 8 and R 9 are bonded to each other include 2,2′-biphenol, 2,2′-methylenebisphenol, 2,2′-methylenebis (4-methyl-6-t-butylphenol) ), (R)-(+)-1,1′-bi-2-naphthol, (S)-( ⁇ )-1,1′-bi-2-naphthol and the like, hydrogen in two phenolic hydroxyl groups Examples include groups other than atoms.
- the above aryloxy group and aryleneoxy group may have one or more substituents, and examples of the substituent include a methoxy group, an ethoxy group, an isopropoxy group, and a t-butoxy group.
- Alkoxy groups such as halogen atoms such as chlorine and bromine.
- Examples of the monovalent saturated hydrocarbon group represented by R 7 , R 11 and R 12 include, for example, methyl group, ethyl group, n-propyl group, isopropyl group, n-butyl group, isobutyl group, sec-butyl group, t Examples thereof include alkyl groups such as -butyl group, 2-methylbutyl group, 3-methylbutyl group, n-octyl group and 2-ethylhexyl group; cycloalkyl groups such as cyclohexyl group and the like.
- R ⁇ 7 >, R ⁇ 11 > and R ⁇ 12 > represent, aryl groups, such as a phenyl group; Aralkyl groups, such as a benzyl group, etc. are mentioned, for example.
- alkoxy group represented by R 7 , R 11 , and R 12 include a methoxy group, an ethoxy group, an isopropoxy group, and a t-butoxy group.
- N, N-disubstituted amino group represented by R 7 , R 11 and R 12 include dialkylamino groups such as dimethylamino group, diethylamino group and diisopropylamino group; and bis (trimethylsilyl) amino group.
- Examples of the functional group represented by R 7 , R 11 and R 12 include an alkoxy group such as a methoxy group, an ethoxy group, an isopropoxy group and a t-butoxy group; and a substituent such as a halogen atom such as chlorine and bromine. You may have.
- Examples of the aluminum compound (A-1) include ethylbis (2,6-di-t-butyl-4-methylphenoxy) aluminum, ethylbis (2,6-di-t-butylphenoxy) aluminum, ethyl [2,2 '-Methylenebis (4-methyl-6-t-butylphenoxy)] aluminum, isobutylbis (2,6-di-t-butyl-4-methylphenoxy) aluminum, isobutylbis (2,6-di-t-butyl) Phenoxy) aluminum, isobutyl [2,2′-methylenebis (4-methyl-6-t-butylphenoxy)] aluminum, n-octylbis (2,6-di-t-butyl-4-methylphenoxy) aluminum, n- Octylbis (2,6-di-t-butylphenoxy) aluminum, n-octyl [2,2′-methylenebi (4-Methyl-6-t-butylphenoxy
- isobutyl bis (2,6-di-t-butyl-4-methylphenoxy) aluminum, isobutyl bis (2,6 -Di-t-butylphenoxy) aluminum, isobutyl [2,2'-methylenebis (4-methyl-6-t-butylphenoxy)] aluminum and the like are preferable.
- Examples of the aluminum compound (A-2) include diethyl (2,6-di-t-butyl-4-methylphenoxy) aluminum, diethyl (2,6-di-t-butylphenoxy) aluminum, diisobutyl (2,6 -Di-t-butyl-4-methylphenoxy) aluminum, diisobutyl (2,6-di-t-butylphenoxy) aluminum, di-n-octyl (2,6-di-t-butyl-4-methylphenoxy) aluminum And di-n-octyl (2,6-di-t-butylphenoxy) aluminum.
- These aluminum compounds (A) may be used individually by 1 type, or may use 2 or more types together.
- the aluminum compound (A) can be produced according to a known method.
- isobutyl bis (2,6-di-t-butyl-4-methylphenoxy) aluminum which is an aluminum compound (A-1)
- 25 ml of dry toluene distilled under an argon atmosphere and 11 g of 2,6-di-t-butyl-4-methylphenol were added and dissolved while stirring at room temperature, and 6.8 ml of triisobutylaluminum was added to the resulting solution.
- It can be prepared as a 0.6 mol / l toluene solution by adding and stirring at 80 ° C. for about 18 hours.
- the amount of the aluminum compound (A) used can be appropriately selected according to the type of organic solvent and other various anionic polymerization conditions, but the stability of anionic polymerization and the R of di (meth) acrylate (1) From the viewpoint of selectively polymerizing the (meth) acryloyl group to which 2 is bonded, it is preferably used in an amount of 1.0 to 10.0 moles per mole of the organolithium compound (L). It is more preferably used in the range of 2 to 6.0 mol, more preferably in the range of 2.0 to 3.5 mol, and particularly preferably in the range of 2.2 to 3.0 mol.
- the Lewis base (B) used in the production method of the present invention is selected from the group consisting of ethers and tertiary polyamines.
- the ether can be appropriately selected from compounds having an ether bond in the molecule, but from the viewpoint of high polymerization initiation efficiency and living property of the polymerization terminal anion, a cyclic ether having two or more ether bonds in the molecule or 1 Acyclic ethers having at least one ether bond in the molecule are preferred.
- Examples of the cyclic ether having two or more ether bonds in the molecule include crown ethers such as 12-crown-4, 15-crown-5, and 18-crown-6.
- acyclic ether having one or more ether bonds in the molecule examples include acyclic monoethers such as dimethyl ether, diethyl ether, diisopropyl ether, dibutyl ether, and anisole; 1,2-dimethoxyethane, 1,2-diethyl Ethoxyethane, 1,2-diisopropoxyethane, 1,2-dibutoxyethane, 1,2-diphenoxyethane, 1,2-dimethoxypropane, 1,2-diethoxypropane, 1,2-diisopropoxy Propane, 1,2-dibutoxypropane, 1,2-diphenoxypropane, 1,3-dimethoxypropane, 1,3-diethoxypropane, 1,3-diisopropoxypropane, 1,3-dibutoxypropane, 1,3-diphenoxypropane, 1,4-dimethoxybutane, 1,4-diethoxybutane
- Acyclic diethers such as 1,4-di
- the tertiary polyamine that can be used as the Lewis base (B) means a compound having two or more tertiary amine structures in the molecule.
- the tertiary polyamine include N, N, N ′, N′-tetramethylethylenediamine, N, N, N ′, N′-tetraethylethylenediamine, N, N, N ′, N ′′, N ′′ -pentamethyldiethylenetriamine 1,1,4,7,10,10-hexamethyltriethylenetetraamine, chain polyamines such as tris [2- (dimethylamino) ethyl] amine; 1,3,5-trimethylhexahydro-1,3 , 5-triazine, 1,4,7-trimethyl-1,4,7-triazacyclononane, 1,4,7,10,13,16-hexamethyl-1,4,7,10,13,16- Non-aromatic heterocyclic compounds such as hexaazacyclo
- Lewis base (B) a compound having one or more ether bonds and one or more tertiary amine structures in the molecule may be used as the Lewis base (B).
- examples of such a compound include tris [2- (2-methoxyethoxy) ethyl] amine.
- a Lewis base (B) may be used individually by 1 type, or may use 2 or more types together.
- the use amount of the Lewis base (B) is preferably in the range of 0.3 to 5 mol with respect to 1 mol of the organolithium compound (L) from the viewpoints of polymerization initiation efficiency, living property of the polymerization terminal anion, and the like.
- the range of 0.5 to 3 mol is more preferable, the range of 0.8 to 2.5 mol is more preferable, and the range of 1.0 to 2.0 mol is particularly preferable. If the amount of the Lewis base (B) used exceeds 5 moles relative to 1 mole of the organolithium compound (L), it tends to be disadvantageous in terms of economy, and if it is less than 0.3 moles, the initiation efficiency of anionic polymerization decreases. Tend to.
- the use amount of the Lewis base (B) is preferably in the range of 0.2 to 1.2 mol times, and in the range of 0.3 to 1.0 mol times with respect to the use amount of the aluminum compound (A). It is more preferable that
- the production method of the present invention is preferably carried out in the presence of an organic solvent from the viewpoint of controlling the temperature of anionic polymerization and homogenizing the inside of the system to facilitate the anionic polymerization.
- Organic solvents include hydrocarbons such as toluene, xylene, cyclohexane, and methylcyclohexane from the viewpoints of safety, separability from water in washing the reaction mixture after anionic polymerization, and ease of recovery and reuse; chloroform Halogenated hydrocarbons such as methylene chloride and carbon tetrachloride; esters such as dimethyl phthalate are preferred.
- These organic solvents may be used alone or in combination of two or more.
- the amount of the organic solvent used depends on the monomer used (that is, di (meth) acrylate (1) and alkyl (meth) acrylate (2) and other monomers as optional components), the organic lithium compound used (L ), Aluminum compound (A), Lewis base (B), organic solvent, etc., but can be appropriately adjusted, such as smooth progress of anionic polymerization, separation and acquisition of the produced copolymer (X), waste liquid treatment, etc. From the viewpoint, it is preferably in the range of 200 to 3000 parts by mass with respect to 100 parts by mass of the mixture used.
- a method of adding a mixture of an organolithium compound (L), an aluminum compound (A), a Lewis base (B) and a di (meth) acrylate (1) and an alkyl (meth) acrylate (2) to a reaction system for anionic polymerization Is not particularly limited.
- the Lewis base (B) is preferably added so as to come into contact with the aluminum compound (A) before contact with the organolithium compound (L).
- the aluminum compound (A) may be added to the anionic polymerization reaction system before or simultaneously with the mixture composed of the di (meth) acrylate (1) and the alkyl (meth) acrylate (2). .
- the aluminum compound (A) is added to the reaction system for anionic polymerization simultaneously with the mixture comprising the di (meth) acrylate (1) and the alkyl (meth) acrylate (2), the aluminum compound (A) is separately mixed with the mixture. It may be added afterwards.
- additives may be present in the anionic polymerization reaction system, if necessary.
- the other additives include inorganic salts such as lithium chloride; metal alkoxides such as lithium methoxyethoxy ethoxide and potassium t-butoxide; tetraethylammonium chloride and tetraethylphosphonium bromide.
- the anionic polymerization is preferably performed at ⁇ 30 to 25 ° C.
- the temperature is lower than ⁇ 30 ° C.
- the polymerization rate decreases and the productivity tends to decrease.
- the temperature is higher than 25 ° C., it is difficult to suppress polymerization of the (meth) acryloyl group bonded to the carbon atom to which R 3 and R 4 of di (meth) acrylate (1) are bonded, and the resulting copolymer is obtained.
- the photocurability of (X) tends to decrease.
- the anionic polymerization is preferably performed in an atmosphere of an inert gas such as nitrogen, argon or helium. Furthermore, it is preferable to carry out it under sufficient stirring conditions so that the reaction system of anionic polymerization becomes uniform.
- the production method of the present invention can stop anionic polymerization by adding a polymerization terminator to the reaction mixture in accordance with a known anionic polymerization method.
- a polymerization terminator for example, a protic compound such as methanol; acetic acid or hydrochloric acid in methanol; or the like can be used.
- the amount of the polymerization terminator used is usually preferably in the range of 1 to 100 mol per 1 mol of the organic lithium compound (L).
- the reaction mixture is poured into a poor solvent of the copolymer (X) to precipitate the copolymer (X), and the organic solvent is distilled off from the reaction mixture to obtain the copolymer (X). Methods and the like.
- the metal component derived from the organolithium compound (L) or the aluminum compound (A) remains in the copolymer (X) obtained separately, the physical properties of the copolymer (X) are deteriorated and the transparency is increased. Defects may occur. Therefore, it is preferable to remove the metal component derived from the organolithium compound (L) and the aluminum compound (A) after the anionic polymerization is stopped.
- a method for removing the metal component it is effective to subject the copolymer (X) to a washing treatment using an acidic aqueous solution, an adsorption treatment using an adsorbent such as an ion exchange resin, or the like.
- acidic aqueous solution hydrochloric acid, sulfuric acid aqueous solution, nitric acid aqueous solution, acetic acid aqueous solution, propionic acid aqueous solution, citric acid aqueous solution etc. can be used, for example.
- a copolymer (X) having an arbitrary molecular weight can be produced.
- the number average molecular weight is in the range of 1,000 to 1,000,000. It is preferable from the viewpoints of handleability, fluidity, mechanical properties and the like of the combined (X).
- a copolymer (X) having a narrow molecular weight distribution is usually obtained, and a copolymer (X) having a molecular weight distribution (Mw / Mn) of 1.5 or less is produced. Can do.
- the copolymer (X) obtained by the production method of the present invention is useful as a component of a photocurable resin composition used for, for example, an adhesive, a pressure-sensitive adhesive, a paint, a coating material, and the like.
- a photocurable resin composition used for, for example, an adhesive, a pressure-sensitive adhesive, a paint, a coating material, and the like.
- cured material is obtained by irradiating an ultraviolet-ray, an electron beam, etc.
- the consumption rate C 11 (mol%) of di (meth) acrylate (1) was calculated by the following formula.
- C 11 100 (1-I 11 / I 10 )
- I 10 and I 11 are integrals of proton peaks derived from protons bonded to the carbon atom adjacent to the oxygen atom in R 2 of di (meth) acrylate (1) at the start of anion polymerization and at the end of anion polymerization, respectively.
- the ratio between the value and the integral value of the proton peak derived from the benzene ring of toluene used for anionic polymerization is shown.
- the consumption rate C 21 (mol%) of the alkyl (meth) acrylate (2) was calculated by the following formula.
- I 10 and I 11 represent the above-described ratios, respectively
- I 20 and I 21 represent R in di (meth) acrylate (1) observed to overlap each other at the start of anionic polymerization and at the end of anionic polymerization, respectively.
- the overall consumption rate C (mol%) was calculated by the following formula using the consumption rates C 11 and C 21 obtained by the above-described method and N.
- C ⁇ C 11 ⁇ N + C 21 ⁇ (100 ⁇ N) ⁇ / 100
- the consumption rate C 11 (mol%) of di (meth) acrylate (1) was the consumption rate C (mol%).
- the side chain functional group reaction rate C 31 (mol%) of di (meth) acrylate (1) was calculated.
- I 30 and I 31 are respectively the integral value of the proton peak derived from the olefin of the methacryloyl group bonded to the carbon atom to which R 3 and R 4 are bonded and the anionic polymerization at the start of anion polymerization and at the stop of the anion polymerization.
- the ratio with the integral value of the proton peak derived from the benzene ring of toluene used is shown.
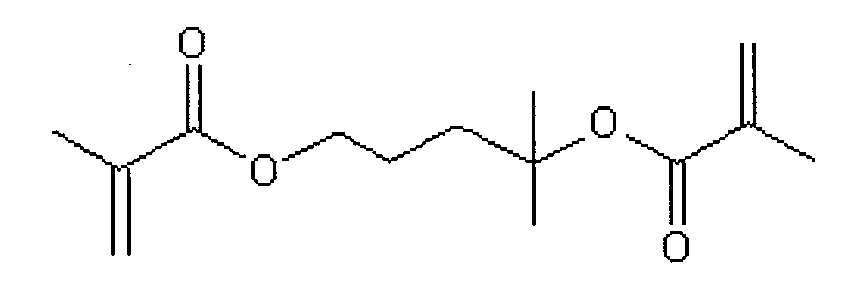
- reaction solution 120 minutes after the end of the addition of the mixture, the reaction solution was sampled to obtain a consumption rate C of 1,1-dimethylpropane-1,3-diol dimethacrylate and methyl methacrylate and 1,1-dimethylpropane-1,3-diol di
- the side chain functional group reaction rate of methacrylate was measured, and 10.0 ml of methanol was added to the reaction solution to stop anionic polymerization.
- the resulting solution was poured into 1 liter of hexane, and the produced copolymer (X) was precipitated and collected.
- Example 2 7.22 ml of 0.450 mol / L toluene solution of isobutylbis (2,6-di-t-butyl-4-methylphenoxy) aluminum and 1,1,4,7,10,10-hexamethyltriethylenetetramine A copolymer (X) was obtained in the same manner as in Example 1 except that the amount was changed to 0.35 ml (1.30 mmol).
- the consumption rate C of 1,1-dimethylpropane-1,3-diol dimethacrylate and methyl methacrylate in the mixture at the end of anionic polymerization was 100%, and the side chain functionality of 1,1-dimethylpropane-1,3-diol dimethacrylate The group reaction rate was 14.0%.
- Mn in the obtained copolymer (X) was 8,000, and Mw / Mn was 1.14.
- Example 4 The temperature in the reactor before addition of the mixture and the temperature for anionic polymerization were 25 ° C., and the mixture was a mixture of 1.24 ml of 1,1-dimethylpropane-1,3-diol dimethacrylate and 2.21 ml of methyl methacrylate.
- a copolymer (X) was obtained in the same manner as in Example 1 except that the total amount was changed to 3.45 ml.
- Example 5 A copolymer (X) was obtained in the same manner as in Example 3, except that the temperature in the reactor before addition of the mixture and the temperature for anionic polymerization were set to -22 ° C.
- the consumption rate C of 1,1-dimethylpropane-1,3-diol dimethacrylate and methyl methacrylate in the mixture at the end of anionic polymerization was 100%, and the side chain functionality of 1,1-dimethylpropane-1,3-diol dimethacrylate The group reaction rate was 9.0%.
- Mn in the obtained copolymer (X) was 3,000, and Mw / Mn was 1.10.
- Example 6 Copolymer (X) in the same manner as in Example 1 except that the 0.450 mol / L toluene solution of isobutylbis (2,6-di-t-butyl-4-methylphenoxy) aluminum was changed to 5.78 ml. Got.
- the consumption rate C of 1,1-dimethylpropane-1,3-diol dimethacrylate and methyl methacrylate in the mixture at the end of anionic polymerization was 100%, and the side chain functionality of 1,1-dimethylpropane-1,3-diol dimethacrylate The group reaction rate was 11.3%.
- Mn in the obtained copolymer (X) was 7,100, and Mw / Mn was 1.14.
- Example 7 Copolymer (X) in the same manner as in Example 1 except that the 0.450 mol / L toluene solution of isobutylbis (2,6-di-t-butyl-4-methylphenoxy) aluminum was changed to 3.18 ml. Got.
- the consumption rate C of 1,1-dimethylpropane-1,3-diol dimethacrylate and methyl methacrylate in the mixture at the end of anionic polymerization was 100%, and the side chain functionality of 1,1-dimethylpropane-1,3-diol dimethacrylate The group reaction rate was 9.6%.
- Mn in the obtained copolymer (X) was 6,900, and Mw / Mn was 1.18.
- Example 8 Anionic polymerization was carried out in the same manner as in Example 1 except that the toluene solution of isobutylbis (2,6-di-t-butyl-4-methylphenoxy) aluminum 0.450 mol / L was changed to 2.17 ml. Sampling was performed 120 minutes later, and the monomer consumption rate was 92%, and the remaining monomer was confirmed. Further, the anionic polymerization was continued for 2 hours, but since the increase in the monomer consumption rate was hardly observed, it was judged that the anionic polymerization was almost stopped, and 5 hours after the start of the anionic polymerization, the same as in Example 1 Then, anionic polymerization was stopped to obtain a methacrylic ester polymer.
- the consumption rate C of 1,1-dimethylpropane-1,3-diol dimethacrylate in the mixture at the end of anionic polymerization was 94%, and the side chain functional group reaction rate of 1,1-dimethylpropane-1,3-diol dimethacrylate was 5.9%.
- Mn in the obtained methacrylate polymer was 6,800, and Mw / Mn was 1.14.
- Example 1 except that instead of the mixture of 1,1-dimethylpropane-1,3-diol dimethacrylate and methyl methacrylate, only 7.73 ml of 1,1-dimethylpropane-1,3-diol dimethacrylate was used. Anionic polymerization was carried out in the same manner as described above. Sampling was conducted after 120 minutes, and the monomer consumption rate was 51%, and the remaining monomer was confirmed.
- anionic polymerization was continued for 2 hours, but since the increase in the monomer consumption rate was hardly observed, it was judged that the anionic polymerization was almost stopped, and 5 hours after the start of the anionic polymerization, the same as in Example 1 Then, anionic polymerization was stopped to obtain a methacrylic ester polymer.
- the consumption rate C of 1,1-dimethylpropane-1,3-diol dimethacrylate was 53%, and the side chain functional group reaction rate was 3.3%.
- Mn in the obtained methacrylate polymer was 7,800, and Mw / Mn was 1.16.
- anionic polymerization was continued for 2 hours, but since the increase in the monomer consumption rate was hardly observed, it was judged that the anionic polymerization was almost stopped, and 5 hours after the start of the anionic polymerization, the same as in Example 1 Then, anionic polymerization was stopped to obtain a methacrylic ester polymer.
- the consumption rate C of 1,1-dimethylpropane-1,3-diol dimethacrylate was 74%, and the side chain functional group reaction rate was 0.9%.
- Mn in the obtained methacrylate polymer was 7,600 and Mw / Mn was 1.08.
- the consumption rate of the monomer is adjusted such that the amount of the aluminum compound (A) used is 2.2 mol or more with respect to 1 mol of the organolithium compound (L). C becomes 100%, and it turns out that the side chain functional group reaction rate of di (meth) acrylate (1) becomes low by setting it as 3.0 mol or less.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Life Sciences & Earth Sciences (AREA)
- Wood Science & Technology (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
- Polymerization Catalysts (AREA)
Abstract
Description
例えば、
[1]下記一般式(1)

で示されるジ(メタ)アクリレート(1)と(以下、「ジ(メタ)アクリレート(1)」と称する)、下記一般式(2)

で示されるアルキル(メタ)アクリレート(2)(以下、「アルキル(メタ)アクリレート(2)」と称する)とからなる混合物を、有機リチウム化合物(L)、下記一般式(3)
で示される化学構造を分子中に含む三級有機アルミニウム化合物(A)(以下、アルミニウム化合物(A)と称する)並びにエーテルおよび三級ポリアミンからなる群から選ばれる少なくとも1種のルイス塩基(B)の存在下で、アニオン重合することを特徴とする(メタ)アクリル酸エステル系共重合体(X)(以下、共重合体(X)と称する)の製造方法;並びに
[2]前記混合物中のジ(メタ)アクリレート(1)とアルキル(メタ)アクリレート(2)とのモル比が、5:95~90:10の範囲である、上記[1]の共重合体(X)の製造方法
を提供することにより達成される。
本発明の製造方法では、ジ(メタ)アクリレート(1)と、アルキル(メタ)アクリレート(2)とからなる混合物を、有機リチウム化合物(L)、アルミニウム化合物(A)、ルイス塩基(B)の存在下でアニオン重合する。ジ(メタ)アクリレート(1)においては、R2が結合している(メタ)アクリロイル基が選択的に重合され、R3およびR4が結合している炭素原子に結合する(メタ)アクリロイル基の重合は抑制されて、得られる共重合体(X)の側鎖に残留するので、かかる共重合体(X)は紫外線や電子線等を照射することで硬化させることができる。
で示される有機アルミニウム化合物(以下、アルミニウム化合物(A-1)と称する)または下記一般式(A-2)
で示される有機アルミニウム化合物(以下、アルミニウム化合物(A-2)と称する)を使用することが好ましく、アルミニウム化合物(A-1) を使用することがより好ましい。
以下の実施例等において、原料は常法により乾燥精製し、窒素にて脱気したものを使用し、移送および供給は窒素雰囲気下にて行った。
アニオン重合後のジ(メタ)アクリレート(1)およびアルキル(メタ)アクリレート(2)の消費率並びにジ(メタ)アクリレート(1)の側鎖官能基反応率は、反応液0.5mlを採取してメタノール0.5ml中に入れて混合後、該混合液から0.1mlを採取して、重クロロホルム0.5mlに溶解させて1H-NMR(ECX400(400MHz)JEOL製、測定温度=25℃、スキャン回数=16、緩和時間=4秒)を測定した結果から算出した。
ジ(メタ)アクリレート(1)の消費率C11(mol%)は、下記式により算出した。
C11=100(1-I11/I10)
ここで、I10、I11はそれぞれアニオン重合開始時およびアニオン重合停止時における、ジ(メタ)アクリレート(1)のR2における酸素原子に隣接する炭素原子に結合するプロトン由来のプロトンピークの積分値と、アニオン重合に用いたトルエンのベンゼン環由来のプロトンピークの積分値との比を示す。
アルキル(メタ)アクリレート(2)の消費率C21(mol%)は、下記式により算出した。
C21=100[1-{I21×(100-N×I11/I10)}/{I20×(100-N)}]
ここで、I10、I11はそれぞれ上記した比を示し、I20、I21はそれぞれアニオン重合開始時およびアニオン重合停止時における、互いに重なって観測されるジ(メタ)アクリレート(1)におけるR2が結合している(メタ)アクリロイル基のオレフィンに由来するプロトンピークとアルキル(メタ)アクリレート(2)のメタクリロイル基におけるオレフィンに由来するプロトンピークとの積分値の合計と、アニオン重合に用いたトルエンのベンゼン環由来のプロトンピークの積分値との比を示し、Nは反応開始時におけるジ(メタ)アクリレート(1)とアルキル(メタ)アクリレート(2)の総量に対するジ(メタ)アクリレート(1)のモル分率(mol%)を示す。
全体の消費率C(mol%)を、上記した方法で求めた消費率C11、C21および上記Nを用いて、下記式により算出した。
C={C11×N+C21×(100-N)}/100
なお、比較例においては、ジ(メタ)アクリレート(1)の消費率C11(mol%)
を消費率C(mol%)とした。
下記式により、ジ(メタ)アクリレート(1)の側鎖官能基反応率C31(mol%)
を算出した。
C31=100-100×I31/I30
ここでI30、I31はそれぞれアニオン重合開始時およびアニオン重合停止時における、R3およびR4が結合している炭素原子に結合するメタクロイル基のオレフィン由来のプロトンピークの積分値とアニオン重合に用いたトルエンのベンゼン環由来のプロトンピークの積分値との比を示す。
得られた共重合体(X)のGPC(ゲルパーミュエーションクロマトグラフィー、HLC-8220GPC(東ソー製)、カラム:TSK-gel SuperMultiporeHZ-M(東ソー製)(カラム径=4.6mm、カラム長=15cm)、測定条件:流速=0.35ml/min、温度=40℃、溶離液=テトラヒドロフラン)を測定し、標準ポリスチレン換算の数平均分子量分布(Mn)および分子量分布(Mw/Mn)を求めた。
内部を乾燥し、窒素置換した300mlのフラスコに、トルエン100ml、ルイス塩基(B)として1,1,4,7,10,10-ヘキサメチルトリエチレンテトラミン0.20ml(0.715mmol)、アルミニウム化合物(A)としてイソブチルビス(2,6-ジ-t-ブチル-4-メチルフェノキシ)アルミニウムの0.450mol/Lトルエン溶液を4.33ml加えて0℃に冷却した。これに有機リチウム化合物(L)としてsec-ブチルリチウムの1.30mol/Lシクロヘキサン溶液0.50ml(0.65mmol)を加えた。フラスコ内の混合液を激しく攪拌しながら、0℃で1,1-ジメチルプロパン-1,3-ジオールジメタクリレート3.09ml(13.0mmol)とメチルメタクリレート1.38ml(13.0mmol)との混合物4.47mlを加え、アニオン重合を開始した。0℃で攪拌を続けると、反応液は当初黄色の溶液となり、さらに窒素雰囲気下で攪拌を続けると30分後にほぼ無色となった。混合物の添加終了から120分後に、反応液をサンプリングして1,1-ジメチルプロパン-1,3-ジオールジメタクリレートおよびメチルメタクリレートの消費率Cおよび1,1-ジメチルプロパン-1,3-ジオールジメタクリレートの側鎖官能基反応率を測定すると共に、反応液中にメタノールを10.0ml加えることにより、アニオン重合を停止させた。得られた溶液を1リットルのヘキサン中に注ぎ、生成した共重合体(X)を沈殿させ、回収した。
イソブチルビス(2,6-ジ-t-ブチル-4-メチルフェノキシ)アルミニウムの0.450mol/Lトルエン溶液を7.22ml、1,1,4,7,10,10-ヘキサメチルトリエチレンテトラミンを0.35ml(1.30mmol)に変更した以外は、実施例1と同様にして共重合体(X)を得た。
イソブチルビス(2,6-ジ-t-ブチル-4-メチルフェノキシ)アルミニウムの0.450mol/Lトルエン溶液を3.18mlに変更し、混合物を1,1-ジメチルプロパン-1,3-ジオールジメタクリレート0.77mlとメチルメタクリレート1.04mlとの混合物(計1.81ml)に変更した以外は、実施例1と同様にして共重合体(X)を得た。
混合物を添加する前の反応器内温度およびアニオン重合する温度を25℃とし、混合物を1,1-ジメチルプロパン-1,3-ジオールジメタクリレート1.24mlと、メチルメタクリレートと2.21mlとの混合物(計3.45ml)に変更した以外は、実施例1と同様にして共重合体(X)を得た。
混合物を添加する前の反応器内温度およびアニオン重合する温度を-22℃とした以外は、実施例3と同様にして共重合体(X)を得た。
イソブチルビス(2,6-ジ-t-ブチル-4-メチルフェノキシ)アルミニウムの0.450mol/Lトルエン溶液を5.78mlに変更した以外は、実施例1と同様にして共重合体(X)を得た。
イソブチルビス(2,6-ジ-t-ブチル-4-メチルフェノキシ)アルミニウムの0.450mol/Lトルエン溶液を3.18mlに変更した以外は、実施例1と同様にして共重合体(X)を得た。
イソブチルビス(2,6-ジ-t-ブチル-4-メチルフェノキシ)アルミニウム0.450mol/Lのトルエン溶液を2.17mlに変更した以外は、実施例1と同様にしてアニオン重合を実施した。120分後にサンプリングしたが、単量体消費率が92%であり、単量体の残存が確認された。さらにアニオン重合を2時間継続したが、単量体消費率の上昇は殆ど見られなかったので、アニオン重合がほぼ停止していると判断して、アニオン重合開始から5時間後に実施例1と同様にアニオン重合を停止し、メタクリル酸エステル系重合体を得た。
1,1-ジメチルプロパン-1,3-ジオールジメタクリレートおよびメチルメタクリレートの混合物に代えて、1,1-ジメチルプロパン-1,3-ジオールジメタクリレート7.73mlのみを用いた以外は、実施例1と同様にしてアニオン重合を実施した。120分後にサンプリングしたが、単量体消費率が51%であり、単量体の残存が確認された。さらにアニオン重合を2時間継続したが、単量体消費率の上昇は殆ど見られなかったので、アニオン重合がほぼ停止していると判断して、アニオン重合開始から5時間後に実施例1と同様にアニオン重合を停止し、メタクリル酸エステル系重合体を得た。
1,1-ジメチルプロパン-1,3-ジオールジメタクリレートの量を6.19mlとし、該1,1-ジメチルプロパン-1,3-ジオールジメタクリレートを添加する前の反応器内温度およびアニオン重合する温度を-22℃とした以外は、比較例1と同様にしてアニオン重合を実施した。120分後にサンプリングしたが、単量体消費率が72%であり、単量体の残存が確認された。さらにアニオン重合を2時間継続したが、単量体消費率の上昇は殆ど見られなかったので、アニオン重合がほぼ停止していると判断して、アニオン重合開始から5時間後に実施例1と同様にアニオン重合を停止し、メタクリル酸エステル系重合体を得た。

Claims (2)
- 下記一般式(1)

(式中、R1は水素原子またはメチル基を表し、R2は炭素数1~5の直鎖アルキレン基を表し、R3及びR4はそれぞれ独立して炭素数1~6の炭化水素基を表す。)
で示されるジ(メタ)アクリレート(1)と、下記一般式(2)

(式中、R5は水素原子またはメチル基を表し、R6は炭素数1~6のアルキル基を表す)
で示されるアルキル(メタ)アクリレート(2)とからなる混合物を、有機リチウム化合物(L)、下記一般式(3)

(式中、Arは芳香族環を表す)
で示される化学構造を分子中に含む三級有機アルミニウム化合物(A)並びにエーテルおよび三級ポリアミンからなる群から選ばれる少なくとも1種のルイス塩基(B)の存在下で、アニオン重合することを特徴とする(メタ)アクリル酸エステル系共重合体(X)の製造方法。 - 前記混合物中のジ(メタ)アクリレート(1)とアルキル(メタ)アクリレート(2)とのモル比が、5:95~90:10の範囲であることを特徴とする請求項1に記載の(メタ)アクリル酸エステル系共重合体(X)の製造方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201380015313.8A CN104204005B (zh) | 2012-03-22 | 2013-03-13 | (甲基)丙烯酸酯类共聚物的制造方法 |
| JP2014506166A JP6132831B2 (ja) | 2012-03-22 | 2013-03-13 | (メタ)アクリル酸エステル系共重合体の製造方法 |
| EP13764845.7A EP2829559B1 (en) | 2012-03-22 | 2013-03-13 | Method for producing (meth)acrylic acid ester-based copolymer |
| US14/385,348 US9051398B2 (en) | 2012-03-22 | 2013-03-13 | Method for producing (meth)acrylate copolymer |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012064826 | 2012-03-22 | ||
| JP2012-064826 | 2012-03-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013141105A1 true WO2013141105A1 (ja) | 2013-09-26 |
Family
ID=49222565
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/056958 WO2013141105A1 (ja) | 2012-03-22 | 2013-03-13 | (メタ)アクリル酸エステル系共重合体の製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US9051398B2 (ja) |
| EP (1) | EP2829559B1 (ja) |
| JP (1) | JP6132831B2 (ja) |
| CN (1) | CN104204005B (ja) |
| WO (1) | WO2013141105A1 (ja) |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20160002378A1 (en) * | 2013-03-18 | 2016-01-07 | Kuraray Co., Ltd. | (meth) acrylic block copolymer and process for producing the same |
| WO2016027602A1 (ja) * | 2014-08-19 | 2016-02-25 | 株式会社クラレ | 活性エネルギー線硬化性組成物 |
| JP2016041794A (ja) * | 2014-08-19 | 2016-03-31 | 株式会社クラレ | 活性エネルギー線硬化性組成物 |
| JP2016050270A (ja) * | 2014-09-01 | 2016-04-11 | 株式会社クラレ | (メタ)アクリル系ブロック共重合体および活性エネルギー線硬化性組成物 |
| JP2016050290A (ja) * | 2014-09-02 | 2016-04-11 | 株式会社クラレ | ブロック共重合体および活性エネルギー線硬化性組成物 |
| JP2016056250A (ja) * | 2014-09-08 | 2016-04-21 | 株式会社クラレ | 活性エネルギー線硬化性ハードコート剤およびハードコートフィルム |
| JP2016056251A (ja) * | 2014-09-08 | 2016-04-21 | 株式会社クラレ | メタクリル樹脂成形体およびその製造方法 |
| JP2016058143A (ja) * | 2014-09-05 | 2016-04-21 | 株式会社クラレ | 導電ペースト |
| JP2016056313A (ja) * | 2014-09-11 | 2016-04-21 | 株式会社クラレ | 活性エネルギー線硬化型アンダーコート剤および表面被覆成形体 |
| JP2016060797A (ja) * | 2014-09-17 | 2016-04-25 | 株式会社クラレ | 活性エネルギー線硬化性組成物 |
| JP2016210926A (ja) * | 2015-05-12 | 2016-12-15 | 株式会社クラレ | ブロック共重合体ならびにその製造方法および用途 |
| WO2017138424A1 (ja) * | 2016-02-08 | 2017-08-17 | 日本曹達株式会社 | (メタ)アクリル酸エステル重合体の製造方法 |
| US20180208700A1 (en) * | 2015-07-22 | 2018-07-26 | Kuraray Co., Ltd. | (meth)acrylic block copolymer |
| WO2019098153A1 (ja) | 2017-11-15 | 2019-05-23 | 株式会社クラレ | (メタ)アクリル系ブロック共重合体およびそれを含有する活性エネルギー線硬化性組成物 |
| US10426712B2 (en) | 2014-09-17 | 2019-10-01 | Kuraray Noritake Dental Inc. | Dental polymerizable composition |
| WO2020189486A1 (ja) | 2019-03-19 | 2020-09-24 | 株式会社クラレ | 活性エネルギー線架橋性熱可塑性重合体およびそれを含有する組成物 |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018178134A (ja) * | 2018-08-27 | 2018-11-15 | 株式会社クラレ | アニオン重合方法および重合体の製造方法 |
| KR102285438B1 (ko) | 2018-11-27 | 2021-08-02 | 주식회사 엘지화학 | 아크릴산 에스테르 화합물 제조 방법 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004035728A (ja) * | 2002-07-03 | 2004-02-05 | Mitsubishi Gas Chem Co Inc | ジ(メタ)アクリレートモノマー、及びその製造法 |
| JP2004091613A (ja) * | 2002-08-30 | 2004-03-25 | Mitsubishi Rayon Co Ltd | 架橋性モノマー、およびそれを用いた熱可塑性架橋ポリマー |
| JP2008522003A (ja) * | 2004-12-03 | 2008-06-26 | ストックハウゼン ゲーエムベーハー | 高吸水性ポリマー用架橋剤 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1552046A (en) * | 1977-02-02 | 1979-09-05 | Ciba Geigy Ag | Film adhesives |
| CA2265310C (en) * | 1998-03-23 | 2007-12-18 | Kuraray Co., Ltd. | Preparation process of acrylic acid ester polymer |
| US6555637B1 (en) * | 1999-08-24 | 2003-04-29 | Kuraray Co., Ltd. | Anionic polymerization process, and process for producing a polymer by the anionic polymerization process |
| CA2318720C (en) * | 1999-09-20 | 2008-10-14 | Kuraray Co., Ltd. | Process for polymerizing a methacrylic ester or an acrylic ester |
| JP4389357B2 (ja) * | 2000-06-16 | 2009-12-24 | 日油株式会社 | 架橋剤、架橋樹脂、脱架橋方法およびその再利用方法 |
| WO2006009083A1 (ja) * | 2004-07-16 | 2006-01-26 | Kuraray Co., Ltd. | アクリル系重合体を含む潤滑油添加剤および潤滑油組成物 |
| JP4713235B2 (ja) * | 2005-06-15 | 2011-06-29 | 東京応化工業株式会社 | ポジ型レジスト組成物およびレジストパターン形成方法 |
| CN101238155B (zh) * | 2005-07-22 | 2011-05-04 | 可乐丽股份有限公司 | 从反应液中获取(甲基)丙烯酸酯系聚合物的方法 |
| JP2012155047A (ja) * | 2011-01-25 | 2012-08-16 | Jsr Corp | 感放射線性組成物及びその製造方法 |
-
2013
- 2013-03-13 WO PCT/JP2013/056958 patent/WO2013141105A1/ja active Application Filing
- 2013-03-13 EP EP13764845.7A patent/EP2829559B1/en active Active
- 2013-03-13 JP JP2014506166A patent/JP6132831B2/ja active Active
- 2013-03-13 CN CN201380015313.8A patent/CN104204005B/zh active Active
- 2013-03-13 US US14/385,348 patent/US9051398B2/en active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004035728A (ja) * | 2002-07-03 | 2004-02-05 | Mitsubishi Gas Chem Co Inc | ジ(メタ)アクリレートモノマー、及びその製造法 |
| JP2004091613A (ja) * | 2002-08-30 | 2004-03-25 | Mitsubishi Rayon Co Ltd | 架橋性モノマー、およびそれを用いた熱可塑性架橋ポリマー |
| JP2008522003A (ja) * | 2004-12-03 | 2008-06-26 | ストックハウゼン ゲーエムベーハー | 高吸水性ポリマー用架橋剤 |
Non-Patent Citations (1)
| Title |
|---|
| POLYMER INTERNATIONAL, vol. 49, 2000, pages 11 - 47 |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20160002378A1 (en) * | 2013-03-18 | 2016-01-07 | Kuraray Co., Ltd. | (meth) acrylic block copolymer and process for producing the same |
| US9988477B2 (en) * | 2013-03-18 | 2018-06-05 | Kuraray Co., Ltd. | (Meth) acrylic block copolymer and process for producing the same |
| WO2016027602A1 (ja) * | 2014-08-19 | 2016-02-25 | 株式会社クラレ | 活性エネルギー線硬化性組成物 |
| JP2016041794A (ja) * | 2014-08-19 | 2016-03-31 | 株式会社クラレ | 活性エネルギー線硬化性組成物 |
| CN106574029A (zh) * | 2014-08-19 | 2017-04-19 | 株式会社可乐丽 | 活性能量射线固化性组合物 |
| JP2016050270A (ja) * | 2014-09-01 | 2016-04-11 | 株式会社クラレ | (メタ)アクリル系ブロック共重合体および活性エネルギー線硬化性組成物 |
| JP2016050290A (ja) * | 2014-09-02 | 2016-04-11 | 株式会社クラレ | ブロック共重合体および活性エネルギー線硬化性組成物 |
| JP2016058143A (ja) * | 2014-09-05 | 2016-04-21 | 株式会社クラレ | 導電ペースト |
| JP2016056251A (ja) * | 2014-09-08 | 2016-04-21 | 株式会社クラレ | メタクリル樹脂成形体およびその製造方法 |
| JP2016056250A (ja) * | 2014-09-08 | 2016-04-21 | 株式会社クラレ | 活性エネルギー線硬化性ハードコート剤およびハードコートフィルム |
| JP2016056313A (ja) * | 2014-09-11 | 2016-04-21 | 株式会社クラレ | 活性エネルギー線硬化型アンダーコート剤および表面被覆成形体 |
| JP2016060797A (ja) * | 2014-09-17 | 2016-04-25 | 株式会社クラレ | 活性エネルギー線硬化性組成物 |
| US10426712B2 (en) | 2014-09-17 | 2019-10-01 | Kuraray Noritake Dental Inc. | Dental polymerizable composition |
| JP2016210926A (ja) * | 2015-05-12 | 2016-12-15 | 株式会社クラレ | ブロック共重合体ならびにその製造方法および用途 |
| US20180208700A1 (en) * | 2015-07-22 | 2018-07-26 | Kuraray Co., Ltd. | (meth)acrylic block copolymer |
| WO2017138424A1 (ja) * | 2016-02-08 | 2017-08-17 | 日本曹達株式会社 | (メタ)アクリル酸エステル重合体の製造方法 |
| WO2019098153A1 (ja) | 2017-11-15 | 2019-05-23 | 株式会社クラレ | (メタ)アクリル系ブロック共重合体およびそれを含有する活性エネルギー線硬化性組成物 |
| US11613602B2 (en) | 2017-11-15 | 2023-03-28 | Kuraray Co., Ltd. | (Meth)acrylic block copolymer and active energy ray curable composition containing the same |
| WO2020189486A1 (ja) | 2019-03-19 | 2020-09-24 | 株式会社クラレ | 活性エネルギー線架橋性熱可塑性重合体およびそれを含有する組成物 |
| US11845818B2 (en) | 2019-03-19 | 2023-12-19 | Kuraray Co., Ltd. | Active energy ray crosslinkable thermoplastic polymer and composition containing the same |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2013141105A1 (ja) | 2015-08-03 |
| EP2829559A4 (en) | 2015-08-12 |
| EP2829559A1 (en) | 2015-01-28 |
| US9051398B2 (en) | 2015-06-09 |
| EP2829559B1 (en) | 2018-12-12 |
| JP6132831B2 (ja) | 2017-05-24 |
| CN104204005B (zh) | 2016-04-20 |
| US20150038658A1 (en) | 2015-02-05 |
| CN104204005A (zh) | 2014-12-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6132831B2 (ja) | (メタ)アクリル酸エステル系共重合体の製造方法 | |
| JP5226731B2 (ja) | アニオン重合方法および該重合方法による重合体の製造方法 | |
| CA2318720C (en) | Process for polymerizing a methacrylic ester or an acrylic ester | |
| CN111356712B (zh) | (甲基)丙烯酸类嵌段共聚物和含有其的活性能量射线固化性组合物 | |
| CN107849203B (zh) | (甲基)丙烯酸系嵌段共聚物 | |
| CN112601765B (zh) | 阴离子聚合方法和聚合物的制造方法 | |
| JP6309402B2 (ja) | (メタ)アクリル系ブロック共重合体および活性エネルギー線硬化性組成物 | |
| JP4549507B2 (ja) | メタクリル酸エステル又はアクリル酸エステルの重合方法 | |
| JP5154179B2 (ja) | ブロック共重合体の製造方法 | |
| JP4549508B2 (ja) | メタクリル酸エステル又はアクリル酸エステルの重合方法 | |
| JP4573967B2 (ja) | アニオン重合方法および該重合方法による重合体の製造方法 | |
| JP2019089952A (ja) | (メタ)アクリル系ブロック共重合体の製造方法 | |
| TW201734067A (zh) | (甲基)丙烯酸系嵌段共聚物及含有該(甲基)丙烯酸系嵌段共聚物之活性能量射線硬化性組成物 | |
| JP2023028492A (ja) | (メタ)アクリル酸エステル重合体の製造方法 | |
| JP2016210926A (ja) | ブロック共重合体ならびにその製造方法および用途 | |
| JP2019035029A (ja) | 活性エネルギー線硬化性組成物 | |
| JP2019052248A (ja) | 活性エネルギー線硬化性組成物 | |
| JP2019108444A (ja) | 活性エネルギー線硬化性組成物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201380015313.8 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13764845 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14385348 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2014506166 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013764845 Country of ref document: EP |