WO2013133367A1 - 新規トリアジン誘導体 - Google Patents
新規トリアジン誘導体 Download PDFInfo
- Publication number
- WO2013133367A1 WO2013133367A1 PCT/JP2013/056266 JP2013056266W WO2013133367A1 WO 2013133367 A1 WO2013133367 A1 WO 2013133367A1 JP 2013056266 W JP2013056266 W JP 2013056266W WO 2013133367 A1 WO2013133367 A1 WO 2013133367A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituent
- group
- mmol
- compound
- added
- Prior art date
Links
- 0 CC(N)=NC(*)=NC(Cl)=C Chemical compound CC(N)=NC(*)=NC(Cl)=C 0.000 description 6
- ZSXGLVDWWRXATF-UHFFFAOYSA-N CN(C)C(OC)OC Chemical compound CN(C)C(OC)OC ZSXGLVDWWRXATF-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D251/00—Heterocyclic compounds containing 1,3,5-triazine rings
- C07D251/02—Heterocyclic compounds containing 1,3,5-triazine rings not condensed with other rings
- C07D251/12—Heterocyclic compounds containing 1,3,5-triazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D251/26—Heterocyclic compounds containing 1,3,5-triazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with only hetero atoms directly attached to ring carbon atoms
- C07D251/40—Nitrogen atoms
- C07D251/42—One nitrogen atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/53—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with three nitrogens as the only ring hetero atoms, e.g. chlorazanil, melamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D251/00—Heterocyclic compounds containing 1,3,5-triazine rings
- C07D251/02—Heterocyclic compounds containing 1,3,5-triazine rings not condensed with other rings
- C07D251/12—Heterocyclic compounds containing 1,3,5-triazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D251/14—Heterocyclic compounds containing 1,3,5-triazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hydrogen or carbon atoms directly attached to at least one ring carbon atom
- C07D251/16—Heterocyclic compounds containing 1,3,5-triazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hydrogen or carbon atoms directly attached to at least one ring carbon atom to only one ring carbon atom
- C07D251/18—Heterocyclic compounds containing 1,3,5-triazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hydrogen or carbon atoms directly attached to at least one ring carbon atom to only one ring carbon atom with nitrogen atoms directly attached to the two other ring carbon atoms, e.g. guanamines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D251/00—Heterocyclic compounds containing 1,3,5-triazine rings
- C07D251/02—Heterocyclic compounds containing 1,3,5-triazine rings not condensed with other rings
- C07D251/12—Heterocyclic compounds containing 1,3,5-triazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D251/14—Heterocyclic compounds containing 1,3,5-triazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hydrogen or carbon atoms directly attached to at least one ring carbon atom
- C07D251/22—Heterocyclic compounds containing 1,3,5-triazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hydrogen or carbon atoms directly attached to at least one ring carbon atom to two ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
Definitions
- the present invention relates to a pharmaceutical, particularly a novel triazine derivative having a BTK inhibitory action or a pharmaceutically acceptable salt thereof.
- Bruton's tyrosine kinase is a member of the Tec family of non-receptor tyrosine kinases and is an important expression expressed in all hematopoietic cell types except T lymphocytes and natural killer cells. It is a signaling enzyme. BTK is an important regulator of B cell survival, differentiation, proliferation, activation, and the like, and plays an important role in B cell signal transduction (Non-patent Documents 1 and 2).
- B-cell receptor (BCR) on the cell surface transmits a signal into the cell via BTK existing downstream of the B-cell receptor; therefore, abnormal activation of the B cell signal transduction pathway is It is thought to promote the growth and survival of cancer cells such as B cell lymphoma and chronic lymphocytic leukemia (Non-patent Document 3).
- BTK is also known to play an important role in the signal pathways of many other cells and is said to be involved in allergic diseases, autoimmune diseases and inflammatory diseases.
- Non-Patent Document 1 For example, BTK plays an important role in high affinity IgE receptor (Fc ⁇ RI) signaling in mast cells. BTK-deficient mast cells have reduced degranulation and production of pro-inflammatory cytokines.
- Fc ⁇ RI IgE receptor
- Non-patent Document 5 systemic lupus erythematosus
- BTK mutant mice are resistant to the development of collagen-induced arthritis (Non-patent Document 6). Therefore, a compound having BTK inhibitory activity is useful for the treatment of diseases involving BTK signals, such as cancer, B cell lymphoma and chronic lymphocytic leukemia, as well as allergic diseases, autoimmune diseases and inflammation. It is also useful for the treatment of sexual diseases.
- diseases involving BTK signals such as cancer, B cell lymphoma and chronic lymphocytic leukemia, as well as allergic diseases, autoimmune diseases and inflammation. It is also useful for the treatment of sexual diseases.
- An object of the present invention is to provide a pharmaceutical, particularly a novel triazine derivative having a BTK inhibitory action or a pharmaceutically acceptable salt thereof.
- the present invention is achieved by the following (1) to (4).
- R 1 has an aryl group which may have a substituent, a heterocyclic ring which may have a substituent, a heterocyclic condensed ring which may have a substituent, and a substituent.
- R 2 represents a hydrogen atom, a halogen atom, a lower alkyl group which may have a substituent, an alkoxy group which may have a substituent, and R 3 has a substituent.
- R 4 represents a hydrogen atom or a lower group which may have a substituent.
- R 1 represents an alkyl group, an alkoxy group which may have a substituent, an amino group which may have a substituent, or a halogen atom
- R 5 represents a hydrogen atom or a lower alkyl group which may have a substituent.
- carded or form a bond with R 1, also is a saturated or unsaturated optionally having a substituent group, 5 to form a 6-membered ring, By, it may form a polycyclic condensed ring.
- R 1 is an aryl group which may have a substituent.
- R 2 is a lower alkyl group which may have a substituent.
- R 5 forms a polycyclic fused ring by forming a bond with R 1 and forming a saturated or unsaturated 5- or 6-membered ring which may have a substituent. Or a pharmaceutically acceptable salt thereof.
- the novel triazine derivative represented by the formula (I) and pharmaceutically acceptable salts thereof have an excellent BTK inhibitory action.
- the present invention was completed.
- the compounds provided by the present invention against diseases known to be associated with abnormal cellular responses mediated by BTK such as autoimmune diseases, inflammatory diseases, bone diseases, cancers such as lymphoma, etc. It is useful as a preventive or therapeutic drug (pharmaceutical composition). Further, it is useful as a BTK inhibitor in reagents for experiments and research.
- novel triazine derivative of the present invention is a compound represented by the following formula (I).
- R 1 has an aryl group which may have a substituent, a heterocyclic ring which may have a substituent, a heterocyclic condensed ring which may have a substituent, and a substituent.
- R 2 represents a hydrogen atom, a halogen atom, a lower alkyl group which may have a substituent, an alkoxy group which may have a substituent, and R 3 has a substituent.
- R 4 represents a hydrogen atom or a lower group which may have a substituent.
- R 5 represents a hydrogen atom or a lower alkyl group which may have a substituent.
- R 1 also is a saturated or unsaturated optionally having a substituent group, 5 to form a 6-membered ring, By, it may form a polycyclic condensed ring.
- the halogen atom include fluorine, chlorine, bromine and the like.
- the aryl group part of the aryl group which may have a substituent may be any aryl group having 6 to 14 carbon atoms, and specific examples include phenyl, naphthyl, indenyl and the like.
- heterocyclic portion of the heterocyclic ring which may have a substituent examples include an alicyclic heterocyclic group and an aromatic heterocyclic group.
- the alicyclic heterocyclic group include a nitrogen atom, a sulfur atom and And a 3- to 8-membered heterocyclic group containing at least one heteroatom selected from oxygen atoms.
- Specific examples include pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, thiomorpholinyl and the like.
- aromatic heterocyclic group examples include a 5- or 6-membered monocyclic aromatic heterocyclic group containing at least one heteroatom selected from a nitrogen atom, a sulfur atom and an oxygen atom.
- Specific examples include imidazolyl, pyrazolyl, thienyl, thiazolyl, pyridyl and the like.
- the heterocyclic condensed ring portion of the heterocyclic condensed ring which may have a substituent is, for example, a bicyclic condensed 3- to 8-membered ring and selected from a nitrogen atom, a sulfur atom and an oxygen atom
- a condensed heterocyclic group containing at least one hetero atom is exemplified. Specific examples include tetrahydroisoquinolyl, benzothiophenyl, benzimidazolyl, benzoxazolyl, benzothiazolyl, indolyl, isoquinolyl, phthalimide and the like.
- the lower alkyl group part of the lower alkyl group which may have a substituent may be any of linear, branched or cyclic alkyl groups having 1 to 3 carbon atoms, specifically, a methyl group, An isopropyl group, a cyclopropyl group, etc. can be mentioned.
- the alkoxy group part of the alkoxy group which may have a substituent may be any alkoxy group having a linear, branched or cyclic alkyl group having 1 to 3 carbon atoms, specifically, methoxy Group, ethoxy group, isopropyloxy group, cyclopropyloxy group and the like.
- the amino group which may have a substituent may be any amino group having a linear, branched or cyclic alkyl group having 1 to 3 carbon atoms, specifically, , Amino group, methylamino group, dimethylamino group and the like.
- alkynyl group part of the alkynyl group which may have a substituent include linear or branched alkynyl groups having 2 to 6 carbon atoms, and specific examples include ethynyl, propargyl, 2-butynyl and the like. Can be mentioned.
- Examples of the substituent of the alkynyl group which may have a substituent include an aryl group which may have a substituent, a heterocyclic ring which may have a substituent, and a heterocyclic condensation which may have a substituent.
- a ring is mentioned, Specifically, an aryl group etc. can be mentioned.
- substituent of the alkoxy group which may have a substituent or the amino group which may have a substituent one or two or more arbitrary types of substituents may be chemically possible unless otherwise specified. When there are two or more substituents, each substituent may be the same or different, for example, a halogen atom, a substituted or unsubstituted alkyl group, a substituted or non-substituted group.
- Examples of the polycyclic fused ring formed by forming a saturated or unsaturated 5- or 6-membered ring in which R 5 forms a bond with R 1 and may have a substituent include, for example, a nitrogen atom, a sulfur atom And a condensed heterocyclic group in which a 3- to 8-membered ring containing a hetero atom such as an oxygen atom is condensed.
- Specific examples include oxoisoquinolyl, oxodihydroisoquinolyl, oxophthalazyl, oxothienopyrrolyl and the like.
- the compound (I) of the present invention may have an isomer depending on, for example, the type of substituent.
- the chemical structure of only one form of those isomers may be described, but the present invention includes all isomers (geometric isomers, optical isomers, tautomers) that can occur structurally. Etc.) and also includes isomers alone or a mixture thereof.
- Examples of the pharmaceutically acceptable salt of the compound (I) of the present invention include inorganic acid salts with hydrochloric acid, sulfuric acid, carbonic acid, phosphoric acid, fumaric acid, maleic acid, methanesulfonic acid, ptoluenesulfonic acid, and the like. And organic acid salts. Also, alkali metal salts with sodium, potassium, etc., alkaline earth metal salts with magnesium, calcium, etc., organic amine salts with lower alkyl amines, lower alcohol amines, etc., basic amino acid salts with lysine, arginine, ornithine, etc. In addition, ammonium salts and the like are also included in the present invention.
- the compound (I) of the present invention and pharmaceutically acceptable salts thereof can be produced, for example, by the following method.
- a method usually used in organic synthetic chemistry for example, a functional group Protection, deprotection [T. W. Greene, Protective Groups in Organic Synthesis 3rd Edition, John Wiley & Sons, Inc. , 1999] can be easily manufactured. Further, the order of reaction steps such as introduction of substituents can be changed as necessary.
- R 1 , R 2 , R 3 , R 4 and R 5 are as defined above, and W represents a boronyl group or a boronic ester group.
- the compound (I) of the present invention can be produced by a cross-coupling reaction such as a Suzuki coupling reaction using the compound (II) and the compound (III) (for example, known conditions for the conditions of the Suzuki coupling reaction) (See N. Miyaura, et al, J. Am. Chem. Soc., 107, 972 (1985), N. Miyaura, A. Suzuki, Chem. Rev. 95, 2457 (1995))). That is, it can be carried out using a base and an additive as necessary in the presence of a metal catalyst such as palladium or nickel.
- a cross-coupling reaction such as a Suzuki coupling reaction using the compound (II) and the compound (III) (for example, known conditions for the conditions of the Suzuki coupling reaction) (See N. Miyaura, et al, J. Am. Chem. Soc., 107, 972 (1985), N. Miyaura, A. Suzuki, Chem. Rev. 95, 2457 (1995)). That is,
- Examples of the solvent used for the reaction include THF, dioxane, toluene, dimethoxyethane, methanol, ethanol, acetonitrile and the like. It is also suitable to use a mixture of two or more of these solvents, or a mixture of these with water. Preferred is a mixed solvent of THF and water, a mixed solvent of toluene, methanol and water, or dioxane.
- Compound (II) is preferably used in an equivalent amount or an excess amount relative to compound (III), more preferably 1 equivalent to 10 equivalents.
- a base may be added to accelerate the reaction, and sodium carbonate, cesium carbonate, potassium carbonate, etc. are usually used as the base.
- the amount of the base to be used is 1 to 10 equivalents, preferably 1 to 5 equivalents, relative to compound (III).
- a commercially available palladium catalyst used for cross coupling for example, PdCl 2 (dppf), Pd 2 (dba) 3 , Pd (PPh 3 ) 4, etc.
- a catalytic amount that is, 0.1 equivalent to 0.5 equivalent relative to compound (III).
- an additive can be added as necessary.
- the additive include rac-BINAP and the like, and 0.01 to 1 equivalent can be used with respect to compound (III).
- the reaction can be synthesized by reacting between 0 ° C. and 200 ° C. for several minutes to several days, preferably between 10 ° C. and 100 ° C. for 1 hour to 36 hours.
- the synthesis can also be performed by using a microwave synthesizer, for example, by reacting for several minutes to several hours under a temperature condition of 60 ° C. to 150 ° C.
- the compound (II) used as a raw material of Scheme 1 can be produced by, for example, the method shown in Scheme 2.
- Compound (II) can be produced by subjecting amine (IV) and carboxylic acid (R 1 COOH) or acid chloride (R 1 COCl) to an amidation reaction often used in ordinary organic chemistry.
- compound (II) is prepared by using 1 to 5 molar equivalents, preferably 1 to 1.5 molar equivalents of carboxylic acid (R 1 COOH) and amine (IV) in a solvent in the presence of a base such as triethylamine.
- a base such as triethylamine.
- the solvent may be any solvent as long as it is inert to the reaction, and is not particularly limited. For example, chloroform, dichloromethane, diethyl ether, or THF alone or a mixed solvent thereof can be used.
- N, N′-dicyclohexylcarbodiimide DCC
- EDC 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride
- CDI 1,1-carbonyldiimidazole
- PPA 2-chloro-1-methylpyridinium iodine, 1-propylphosphonic acid cyclic anhydride
- the reaction can be carried out at a temperature between ⁇ 10 ° C. and the boiling point of the solvent used for 1 hour to 1 week, but is preferably synthesized by reacting at a temperature between 0 ° C. and room temperature for 1 hour to 1 day. be able to. If necessary, it can also be synthesized by adding a reaction reagent such as 1-hydroxybenzotriazole (HOBt).
- a reaction reagent such as 1-hydroxybenzotriazole (HOBt).
- Compound (II) reacts with amine (IV) in an amount of 1 to 5 molar equivalents, preferably 1 to 1.5 molar equivalents of acid chloride (R 1 COCl) in a solvent in the presence of a base such as pyridine or triethylamine.
- a base such as pyridine or triethylamine.
- the solvent is not particularly limited, for example, chloroform, dichloromethane, diethyl ether, pyridine or THF alone or a mixed solvent thereof can be used.
- the reaction can be carried out at a temperature between ⁇ 10 ° C. and the boiling point of the solvent used for 1 hour to 1 week, but is preferably synthesized by reacting at a temperature between 0 ° C. and room temperature for 1 hour to 1 day. be able to.
- compound (II) can be synthesized from amine (IV) and carboxylic acid (R 1 COOH) by a mixed acid anhydride method. These amidation reactions are preferably carried out under anhydrous conditions in an inert gas (argon, nitrogen, etc.) atmosphere.
- Amine (IV) can be obtained as a commercial product or by a known method or a method analogous thereto.
- Compound (II) can also be produced by introducing W into compound (V) as shown in Scheme 3, for example.
- R 1 , R 2 , R 5 and W are as defined above, and X represents halogen.
- Compound (II) can be produced by activating compound (V) with n-butyllithium or the like and then reacting with borate ester. That is, compound (II) is lithiated by treating compound (V) with 1 to 5 molar equivalents, preferably 1 to 1.5 molar equivalents of n-butyllithium, and 1 to 5 molar equivalents, preferably 1 to 5 molar equivalents. It can be obtained by reacting with 1.5 molar equivalent borate ester.
- the solvent may be any solvent as long as it is inert to the reaction, and is not particularly limited, but preferably THF can be used.
- the reaction temperature is usually ⁇ 100 ° C. to ⁇ 30 ° C., preferably ⁇ 80 ° C. to ⁇ 60 ° C.
- reaction time is not specifically limited, Usually, 0.1 to 12 hours are illustrated, and 0.2 to 6 hours are mentioned as a preferable example.
- compound (II) has a boiling point of a solvent in which compound (V) and 1 to 5 molar equivalents, preferably 1 to 1.5 molar equivalents of metal magnesium and a catalytic amount of iodine are used in an ether solvent from ⁇ 10 ° C. It can also be obtained by reacting at a temperature between 1 to 5 molar equivalents, preferably 1 to 1.5 molar equivalents of boric acid ester.
- the reaction temperature is generally ⁇ 30 ° C. to ⁇ 100 ° C., preferably ⁇ 60 ° C. to ⁇ 80 ° C.
- reaction time is not specifically limited, Usually, 0.1 to 12 hours are illustrated, and 0.2 to 6 hours are mentioned as a preferable example.
- compound (II) is coupled with 1 to 5 molar equivalents, preferably 1 to 3 molar equivalents of a diboron ester in an organic solvent in the presence of a metal catalyst such as palladium or nickel and a base. It can be obtained by reacting.
- the metal catalyst a commercially available palladium catalyst (for example, PdCl 2 (dppf), Pd 2 (dba) 3 , Pd (PPh 3 ) 4 etc.) used for cross coupling can be used, and a compound ( It is preferred to add a catalytic amount, ie 0.1 equivalent to 0.5 equivalent, relative to V).
- a catalytic amount ie 0.1 equivalent to 0.5 equivalent, relative to V.
- potassium acetate or the like is usually used. With respect to the dose of the base used, it can be 1 to 10 equivalents, preferably 1 to 5 equivalents, relative to compound (V).
- the solvent is not particularly limited as long as it is inert to the reaction, but dioxane can be preferably used.
- reaction temperature is usually 0 ° C. to 200 ° C., preferably 10 ° C. to 100 ° C.
- reaction time is not specifically limited, Usually, 0.2 to 48 hours are illustrated, and 1 to 36 hours are preferable examples.
- These reactions are desirably carried out under an inert gas (argon, nitrogen, etc.) atmosphere under anhydrous conditions.
- Compound (V) can be obtained as a commercial product, or by a known method or a method analogous thereto.
- Compound (III) requires amine (R 3 NH 2 ) and 1 to 5 molar equivalents, preferably 1 to 1.5 molar equivalents of 2,4-dichloro-1,3,5-triazine in a polar solvent. Accordingly, it is obtained by reacting in the presence of a base catalyst.
- the solvent is not particularly limited as long as it is inert to the reaction, but dimethylformamide can be preferably used.
- the reaction temperature is usually 0 ° C. to 200 ° C., preferably 10 ° C. to 100 ° C. Although reaction time is not specifically limited, Usually, 0.2 to 48 hours are illustrated, and 1 to 36 hours are preferable examples.
- R 1 , R 2 , R 3 and R 5 are as defined above, and R 4 is a hydrogen atom.
- Compound (I) can be obtained by reacting compound (VI) with 1 to 5 molar equivalents, preferably 1 to 1.5 molar equivalents of compound (VII) in an organic solvent in the presence of a base.
- the solvent is not particularly limited as long as it is inert to the reaction, but dioxane can be preferably used.
- Examples of the base used include sodium methoxide, sodium ethoxide, and potassium tert-butoxide, and preferably potassium tert-butoxide can be used.
- reaction temperature is usually 0 ° C. to 200 ° C., preferably 10 ° C. to 100 ° C.
- reaction time is not specifically limited, Usually, 0.2 to 48 hours are illustrated, and 1 to 36 hours are preferable examples.
- Compound (VII) can be obtained as a commercially available product or by a known method or a method analogous thereto.
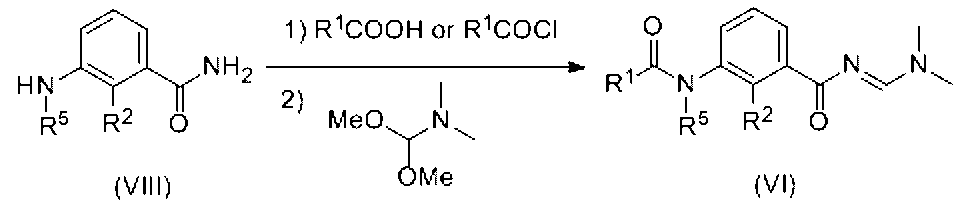
- Compound (VI) is produced by reacting a compound obtained by the reaction of amine (VIII) with carboxylic acid (R 1 COOH) or acid chloride (R 1 COCl) with N, N-dimethylformamide dimethyl acetal. can do.
- the amide obtained above can be obtained by reacting 1 to 10 molar equivalents of N, N-dimethylformamide dimethyl acetal in an organic solvent or without solvent.
- the solvent is not particularly limited as long as it is inert to the reaction, but THF can be preferably used.
- the reaction temperature is usually 0 ° C. to 200 ° C., preferably 10 ° C. to 100 ° C. Although reaction time is not specifically limited, Usually, 0.2 to 48 hours are illustrated, and 1 to 36 hours are preferable examples.
- the reaction is also preferably carried out under microwave irradiation conditions.
- Amine (VIII) can be obtained as a commercial product, or by a known method or a method analogous thereto.
- R 1 , R 2 , R 3 , R 4 and R 5 are as defined above.
- Compound (I) of the present invention can be produced by subjecting amine (IX) and carboxylic acid (R 1 COOH) or acid chloride (R 1 COCl) to an amidation reaction.
- the conditions for the amidation reaction are the same as those in the method for producing compound (II) in Scheme 2 above.
- Compound (IX) used as a starting material of Scheme 7 can be produced, for example, by the method shown in Scheme 8.
- Compound (IX) can be produced by a cross-coupling reaction between compound (IV) and compound (III).
- compound (IX) can also be produced by protecting and deprotecting the amino group of compound (IV) by appropriately combining methods commonly used in organic synthetic chemistry as necessary. .
- protection and deprotection of the functional group of the amino group of compound (IV) [T. W. Greene, Protective Groups in Organic Synthesis 3rd Edition, John Wiley & Sons, Inc. , 1999] or a nitro group derivative which is an amino group precursor of the compound (IV).
- Compound (IV) can be obtained as a commercial product, or by a known method or a method analogous thereto.
- Compound (V) used as a raw material of Scheme 3 can be produced by, for example, the method shown in Scheme 9.
- Compound (V) is obtained by reacting amide (X) with 1 to 5 molar equivalents, preferably 1.5 to 3 molar equivalents of compound (XI) in a polar solvent in the presence of a metal catalyst. Is obtained.
- the solvent is not particularly limited as long as it is inert to the reaction, and DMSO can be preferably used.
- compound (V) can also be produced by protecting and deprotecting R 2 of compound (XI) by appropriately combining methods commonly used in organic synthetic chemistry as necessary. .
- R 2 of compound (XI) for example, protection and deprotection of the functional group of the hydroxyl group and amino group of compound (XI) [T. W. Greene, Protective Groups in Organic Synthesis 3rd Edition, John Wiley & Sons, Inc. , 1999] or an aldehyde derivative which is a hydroxyl precursor of compound (XI).
- the reaction is usually carried out by reacting at 80 to 200 ° C. for 0.5 to 200 hours, preferably 100 to 150 ° C. for 1 to 100 hours.
- the reaction is also preferably carried out under microwave irradiation conditions.
- metal catalyst to be used a commercially available palladium catalyst (for example, PdCl 2 (dppf), Pd 2 (dba) 3 , Pd (PPh 3 ) 4 etc.) or copper iodide (I) can be used. It is preferable to add 0.01 to 2 equivalents with respect to amide (X).
- Examples of the base to be used include potassium carbonate, sodium carbonate, cesium carbonate and sodium hydrogen carbonate.
- cesium carbonate and sodium hydrogen carbonate can be used, and 1 to 10 molar equivalents, preferably 1 to 10 molar equivalents relative to the amide (X). Examples are 2 to 5 molar equivalents.
- it is compoundable even if 0.1 equivalent of 0.5 equivalent of xanthophos is added as needed.
- Amide (X) and compound (XI) can be obtained as commercial products, or by a known method or a method analogous thereto.
- the boronyl group represented by W may be in the form of an alkali metal or alkaline earth metal salt.
- boronic acid ester groups include boronic acid dimethyl ester group, boronic acid diethyl ester group, boron Acid dibutyl ester group, boronic acid dicyclohexyl group, boronic acid ethylene glycol ester group, boronic acid propylene glycol ester group (boronic acid 1,2-propanediol ester group, boronic acid 1,3-propanediol ester group), boronic acid neo Boronic acid ester groups such as pentyl glycol ester group, boronic acid catechol ester group, boronic acid glycerin ester group, boronic acid trimethylolethane ester group, boronic acid diethanolamine ester group, boronic acid triethanolamine ester group; Group, and the like.
- the above methods are appropriately combined, and methods commonly used in organic synthetic chemistry (for example, alkylation reaction of amino group, reaction of oxidizing alkylthio group to sulfoxide group or sulfone group, alkoxy group to hydroxyl group, or vice versa)
- alkylation reaction of amino group for example, alkylation reaction of amino group, reaction of oxidizing alkylthio group to sulfoxide group or sulfone group, alkoxy group to hydroxyl group, or vice versa
- the compound (I) of the present invention having a desired functional group at a desired position can be obtained.
- Compound (I) or a pharmaceutically acceptable salt thereof of the present invention can be prepared in the form of a conventional pharmaceutical preparation (pharmaceutical composition) suitable for oral administration, parenteral administration or topical administration.
- Preparations for oral administration include solid preparations such as tablets, granules, powders and capsules, and liquid preparations such as syrups. These formulations can be prepared by conventional methods. Solid preparations can be prepared by using conventional pharmaceutical carriers such as lactose, starch such as corn starch, crystalline cellulose such as microcrystalline cellulose, hydroxypropylcellulose, calcium carboxymethylcellulose, talc, magnesium stearate, etc. it can. Capsules can be prepared by wrapping the granules or powders thus prepared in capsules. A syrup can be prepared by dissolving or suspending the compound (I) of the present invention or a pharmaceutically acceptable salt thereof in an aqueous solution containing sucrose, carboxymethylcellulose and the like.
- Preparations for parenteral administration include infusions such as instillation.
- Injectable formulations can also be prepared by conventional methods, including isotonic agents (eg, mannitol, sodium chloride, glucose, sorbitol, glycerol, xylitol, fructose, maltose, mannose), stabilizers (eg, sodium sulfite, Albumin) and preservatives (eg, benzyl alcohol, methyl p-oxybenzoate).
- isotonic agents eg, mannitol, sodium chloride, glucose, sorbitol, glycerol, xylitol, fructose, maltose, mannose
- stabilizers eg, sodium sulfite, Albumin
- preservatives eg, benzyl alcohol, methyl p-oxybenzoate
- the dose of the compound (I) of the present invention or a pharmaceutically acceptable salt thereof can be varied according to the severity of the disease, the age and weight of the patient, the dosage form, etc., but is usually 1 mg per day in an adult. It is in the range of ⁇ 1,000 mg, which can be administered once or divided into two or three times by the oral or parenteral route.
- the compound (I) of the present invention or a pharmaceutically acceptable salt thereof can also be used as a BTK inhibitor, as a reagent for experiment or research.
- Reference Example 1 and Examples 1 to 5, 33, 39, 42, 43 and 44 were produced as follows.
- Reference Example 1 (Synthesis of raw material compounds) 4- (tert-Butyl) -N- [2-methyl-3- (4,4,5,5-tetramethyl-1,3,2-dioxaboran-2-yl) phenyl] benzamide
- reaction solution is diluted with DCM (100 mL), washed sequentially with water (2 ⁇ 30 mL), 1N hydrochloric acid (30 mL), saturated aqueous sodium hydrogen carbonate solution (30 mL), saturated aqueous sodium chloride solution (30 mL), and anhydrous sodium sulfate. Dried. The solid obtained by concentrating the solvent under reduced pressure was suspended in hexane and filtered. The solid was washed with hexane and dried to give N- (3-bromo-2-methylphenyl) -4- (tert-butyl) benzamide (9.0 g).
- reaction was carried out at 110 ° C. for 20 minutes using a microwave reactor. Water was added to the reaction solution, and the mixture was extracted twice with ethyl acetate. The combined organic layers were washed with saturated aqueous sodium hydrogen carbonate solution and saturated brine, and dried over anhydrous sodium sulfate.
- the solvent was concentrated under reduced pressure, and the resulting residue was diluted with DCM (10 mL), trifluoroacetic acid (10 mL) was added, and the mixture was stirred at room temperature for 10 min. Triethylsilane (6.4 mL) was added to the reaction solution, and the mixture was stirred at room temperature for 30 minutes.
- the reaction solution was concentrated under reduced pressure, and the resulting residue was diluted with DCM (100 mL), washed sequentially with water (50 mL) and saturated aqueous sodium chloride solution (20 mL), and dried over anhydrous sodium sulfate.
- the reaction solution is diluted with DCM (300 mL), washed successively with water (100 mL), 1N hydrochloric acid (100 mL), saturated aqueous sodium hydrogen carbonate solution (100 mL), saturated aqueous sodium chloride solution (100 mL), and dried over anhydrous sodium sulfate. It was. The residue obtained by concentrating the solvent under reduced pressure was dissolved in dioxane (39 mL) under a nitrogen atmosphere, and bis (pinacolato) diboron (2.98 g, 11.75 mmol), [1,1′-bis (diphenylphosphino)].
- Ferrocene] palladium (II) dichloride dichloromethane adduct (480 mg, 0.588 mmol) and potassium acetate (1.73 g, 17.63 mmol) were added, and the mixture was stirred at 80 ° C. for 16 hours.
- the obtained crude alcohol product (2.61 g) was dissolved in DCM (50 mL), and acetyl chloride (1.39 mL, 19.6 mmol) and pyridine (1.39 mL, 19.6 mmol) were added under ice cooling. In addition, the mixture was stirred for 1 hour while returning to room temperature. The reaction solution was added to ice water and extracted twice with chloroform. The obtained organic layers were combined, washed with saturated brine, and dried over anhydrous sodium sulfate. The solvent was distilled off under reduced pressure, an ethyl acetate-diisopropyl ether mixed solution (1: 5) was added to the obtained residue, and the precipitated solid was collected by filtration.
- Example compounds [Table 1-1] to [Table 1-27] were prepared by using the corresponding starting materials (commercially available products or compounds derivatized from commercially available compounds by known methods or equivalent methods). According to the method described in the examples, the methods usually used in organic synthetic chemistry were appropriately combined as necessary.
- Test example 1 Activity inhibition test against BTK (method for measuring kinase activity)
- the kinase activity was measured by a mobility shift assay (MSA) method using QuickScct Screening Assist TM (trademark) MSA (commercially available kit manufactured by Carna Biosciences).
- MSA mobility shift assay
- Assay buffer [20 mM HEPES, 0.01% Triton X-100 TM, 2 mM dithiothreitol, pH 7.5], adjusting to be substrate (4 ⁇ M), MgCl 2 (20 mM), ATP (120 ⁇ M), A substrate mixture was prepared.
- an enzyme solution was prepared by diluting a kinase (BTK; manufactured by Carna Biosciences, catalog No. 08-080) with an assay buffer so as to have a concentration of 0.2 nM. From 10 mM DMSO solution of test compound to 10 concentrations (0.00003 mM, 0.0001 mM, 0.0003 mM, 0.001 mM, 0.003 mM, 0.01 mM, 0.03 mM, 0.1 mM, 0.3 mM, 1 mM) Further diluted with DMSO, each was diluted 25-fold with assay buffer to give a drug solution (4% DMSO solution).
- BTK kinase
- the height of each peak of the separated substrate and phosphorylated substrate was defined as S and P, respectively, and a blank added with assay buffer instead of the enzyme solution was measured.
- IC 50 value was calculated by regression analysis of inhibition rate and test compound concentration (logarithm).
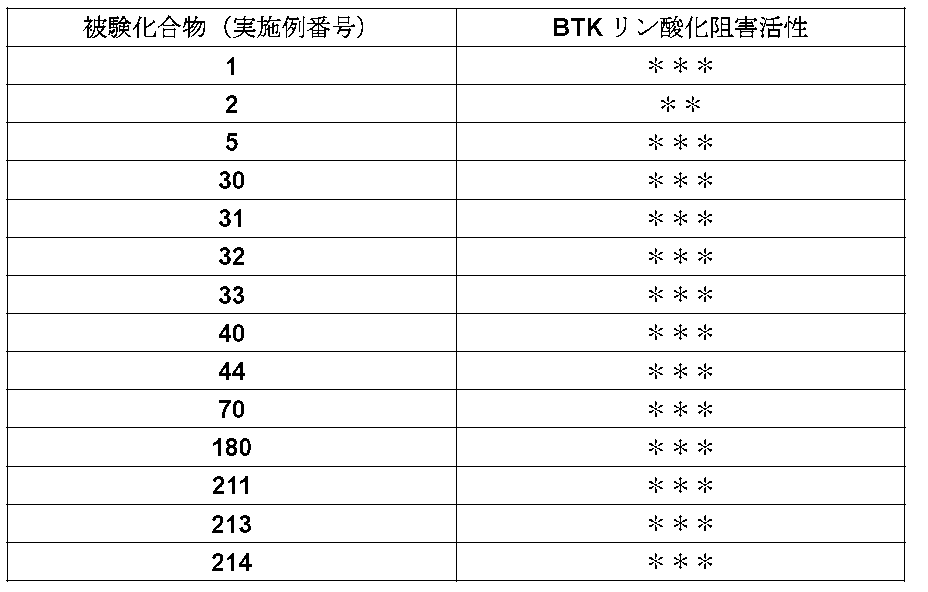
- the IC 50 value for BTK of the compound of the present invention showed a strong inhibitory activity of 1 ⁇ M or less.
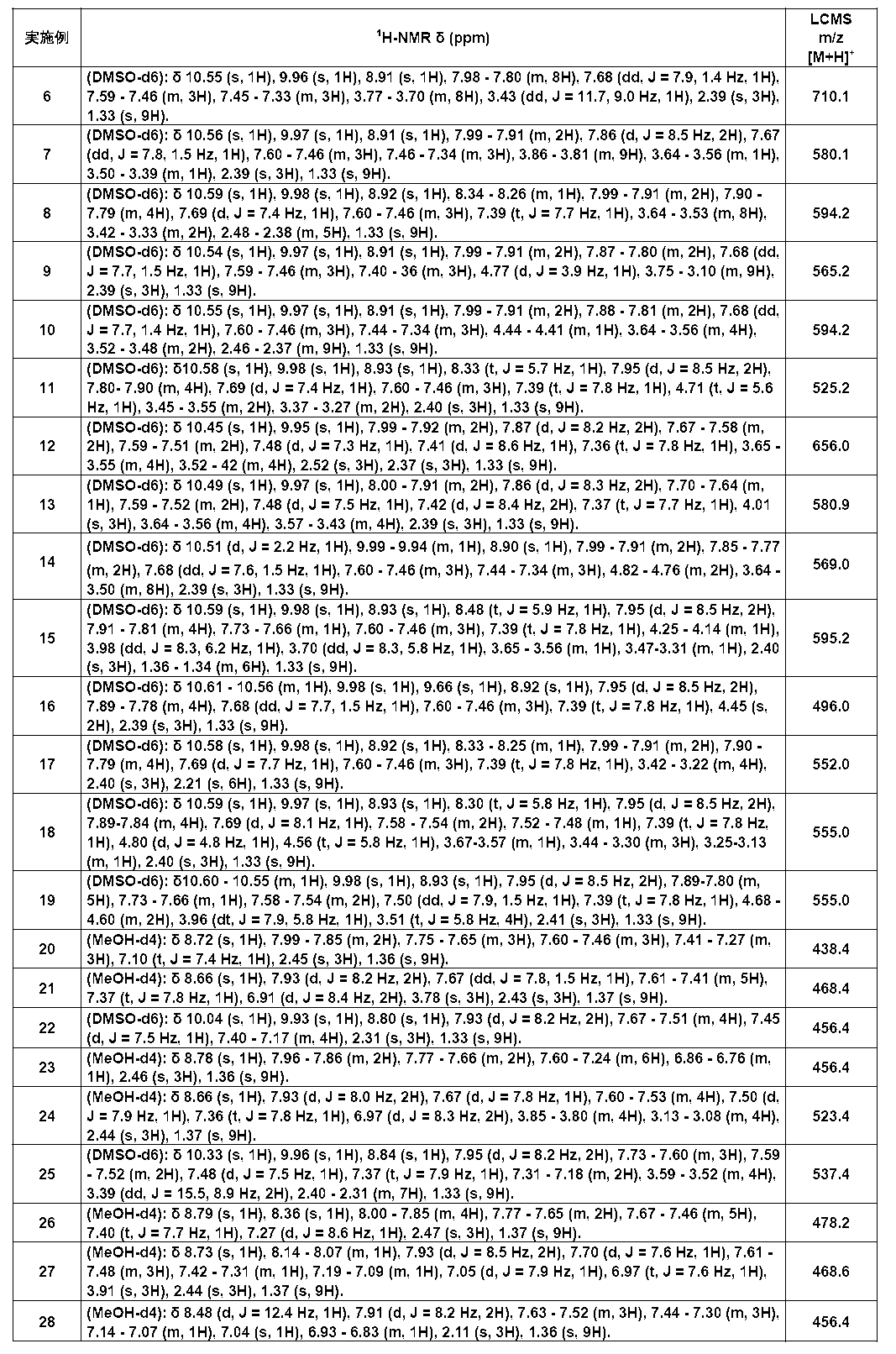
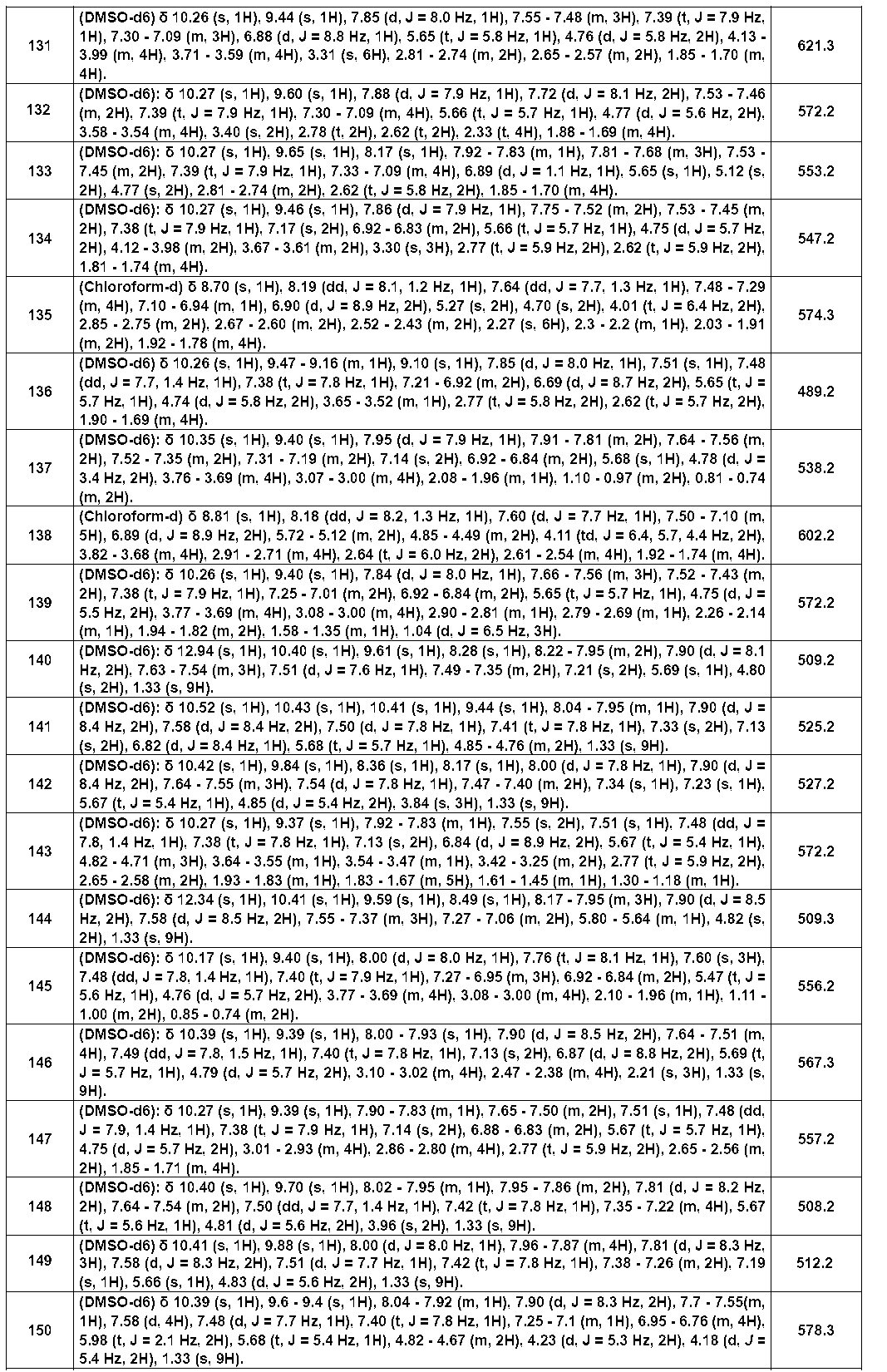
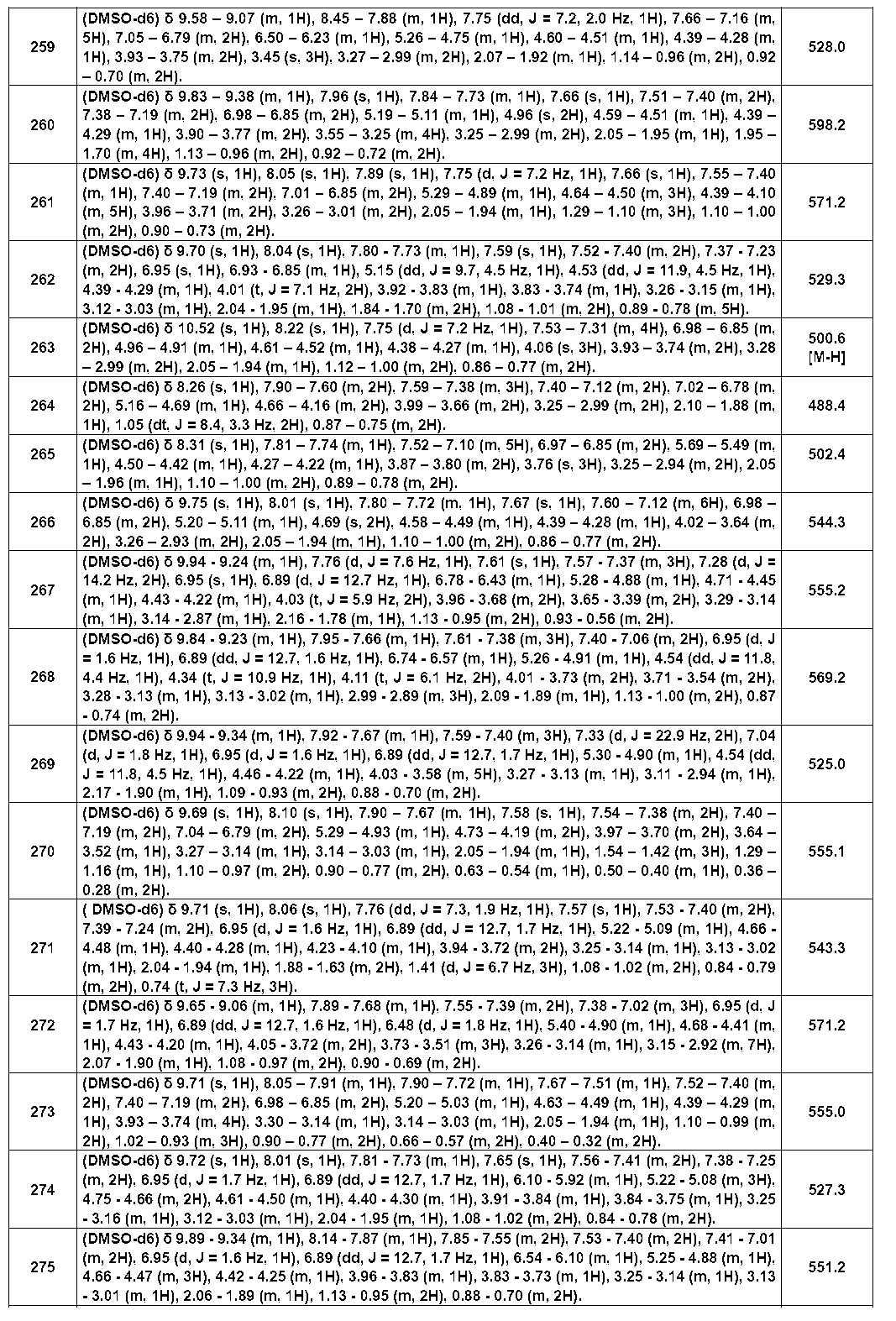
- Table 3 shows the BTK inhibitory activity of representative compounds.
- test compound compound (I) of the present invention
- compound (I) of the present invention has a strong BTK inhibitory activity
- Test example 2 Activity inhibition test for dephosphorylated BTK (adjustment of dephosphorylated BTK)
- Dephosphorylated BTK contains 10 U / ⁇ g and 2 mM of biotinylated BTK protein BTN-BTK (manufactured by Carna Biosciences), ⁇ protein phosphate (manufactured by New England BioLabs, Code No. P0753S) and MnCl 2 respectively.
- the mixture was reacted at 4 ° C. overnight, and after removing ⁇ protein phosphate by anti-DYKDDDDK-tag antibody agarose gel chromatography, the buffer was exchanged using 10DG Desalting Column.
- the method for measuring the kinase activity and the method for evaluating the dephosphorylated BTK inhibitory activity were performed according to Test Example 1. However, in the measurement of dephosphorylated kinase activity, ATP was adjusted to 200 ⁇ M, and dephosphorylated BTK was adjusted to 0.6 nM instead of kinase (BTK; Carna Biosciences, catalog No. 08-080). did.
- the IC 50 value of the compound of the present invention against dephosphorylated BTK is 1 ⁇ M or less, and it was found that the compound of the present invention exhibits a strong inhibitory activity.
- the dephosphorylation BTK inhibitory activity of representative compounds is shown in Tables 4-1 and 4-2.
- Ramos cells (2G6.4C10, ATCC No. CRL-1923) use RPMI-1640 medium (GIBCO) supplemented with 10% FBS (AusGene) and 5% penicillin streptomycin (Nacalai) in a T75 flask. The cells were cultured in a 5% CO 2 incubator.
- the cultured Ramos cells were diluted with RPM-1640 medium (hereinafter referred to as medium) excluding serum so that the cell density was 7.5 ⁇ 10 6 cells / mL, and incubated at 37 ° C. for 45 minutes. After subdividing the cell suspension into 2.0 mL tubes, add 500 ⁇ L of a test compound solution in which a 1 mM DMSO solution of the test compound is diluted to 3 ⁇ M in the medium, and the final concentration of the test compound is 1 ⁇ M. Incubate for 1 hour at 37 ° C. Thereafter, IgM (Invitrogen, H15100) diluted in medium was added to a final concentration of 10 ⁇ g / mL and incubated at 37 ° C. for 10 minutes.
- medium RPM-1640 medium
- serum excluding serum
- the pellets obtained by collecting the cells by centrifugation were added to a Lysis buffer [RIPA Buffer ( ⁇ 1) (Cell Signaling Technology), 1% Phosphase Inhibitor Cocktail 3 (Sigma, No. P0044), 1% Phosphaitase 100 ⁇ L of (Nacalai Co., No. 07575) and 1 mM phenylmethylsulfonyl fluoride (PMSF) added] was added, stirred gently, and allowed to stand for 10 minutes. The supernatant was collected by centrifugation (15,000 rpm, 15 minutes), and the amount of protein was quantified. The sample was mixed with SDS-sample buffer and reacted at 95 ° C.
- RIPA Buffer ( ⁇ 1) Cell Signaling Technology
- Phosphase Inhibitor Cocktail 3 Sigma, No. P0044
- PMSF phenylmethylsulfonyl fluoride
- the detected band was quantified by densitometry (ATTO CS Analyzer ver3.0), and the luminescence of the phosphorylated BTK band with no compound added and IgM-stimulated group was 100%, phosphorylated with no compound added and IgM-unstimulated group
- the inhibition rate was calculated from the intensity of the band in each group, assuming that the emission of the BTK band was 0%.
- Each phosphorylated BTK band was corrected by total BTK.
- the combinations and dilution concentrations of the primary antibody and the secondary antibody used in this test are as follows.
- the compound of the present invention strongly inhibited the autophosphorylation activity of intracellular BTK at a concentration of 1 ⁇ M.
- Test Example 3-2 Intracellular BTK autophosphorylation activity inhibition test 2 Test compounds were prepared and added as follows, and protein extraction and detection of BTK or phosphorylated BTK were performed according to Test Example 3-1, and the inhibition rate of each test compound was calculated.
- the cultured Ramos cells were diluted with RPM-1640 medium (hereinafter referred to as medium) excluding serum so that the cell density was 7.5 ⁇ 10 6 cells / mL, and incubated at 37 ° C. for 45 minutes. After subdividing the cell suspension into 1 mL portions in a 2.0 mL tube, 500 ⁇ L of a test compound solution in which a 0.3 mM DMSO solution of the test compound is diluted to 0.9 ⁇ M in the medium is added to obtain a final concentration of the test compound. Was incubated at 37 ° C. for 1 hour under the condition of 0.3 ⁇ M. Thereafter, IgM (Invitrogen, H15100) diluted in medium was added to a final concentration of 10 ⁇ g / mL and incubated at 37 ° C. for 10 minutes.
- medium RPM-1640 medium
- serum excluding serum
- Intracellular BTK autophosphorylation inhibitory activity is indicated by ** for 70% or more, ** for 50% or more and less than 70%, and * for 30% or more and less than 50%.
- the compound of the present invention strongly inhibited the autophosphorylation activity of intracellular BTK at a concentration of 0.3 ⁇ M.
- Test Examples 3-1 and 3-2 show that the compound of the present invention has a strong inhibitory action on “autophosphorylation activity of intracellular BTK”.
- the compounds provided by the present invention against diseases known to be associated with abnormal cellular responses mediated by BTK such as autoimmune diseases, inflammatory diseases, bone diseases, cancers such as lymphoma, etc. It is useful as a preventive or therapeutic drug (pharmaceutical composition). Further, it is useful as a BTK inhibitor in reagents for experiments and research.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Oncology (AREA)
- Hematology (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Transplantation (AREA)
- Physical Education & Sports Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description
(1)下式(I)で示されるトリアジン誘導体またはその薬学的に許容される塩。
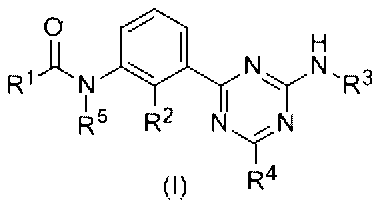
(2)R1が置換基を有してもよいアリール基である(1)に記載のトリアジン誘導体またはその薬学的に許容される塩。
(3)R2が置換基を有してもよい低級アルキル基である(1)に記載のトリアジン誘導体またはその薬学的に許容される塩。
(4)R5は、R1と結合を形成し、置換基を有することもある飽和もしくは不飽和の、5ないし6員環を形成することによって、多環性縮合環を形成する、(1)に記載のトリアジン誘導体またはその薬学的に許容される塩。
本発明の新規なトリアジン誘導体は、下式(I)で示される化合物である。

前記式(I)において、
ハロゲン原子としては、フッ素、塩素、臭素などが挙げられる。
置換基を有してもよいアルキニル基のアルキニル基部分としては、炭素数2から6の直鎖状もしくは分枝状のアルキニル基が挙げられ、具体的には、エチニル、プロパルギル、2-ブチニル等を挙げることができる。置換基を有してもよいアルキニル基の置換基としては、置換基を有してもよいアリール基、置換基を有してもよい複素環、置換基を有してもよい複素環式縮合環が挙げられ、具体的には、アリール基等を挙げることができる。
DCM : ジクロロメタン
DCC : N,N’-ジシクロヘキシルカルボジイミド
EDC : 1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩
HOBt : 1-ヒドロキシベンゾトリアゾール
THF : テトラヒドロフラン
DIEA : N,N-ジイソプロピルエチルアミン
DMF : ジメチルホルムアミド
DMSO : ジメチルスルホキシド
TEA : トリエチルアミン
CDCl3 : 重クロロホルム
式(I)で表される本発明の化合物は、例えばスキーム1によって製造することができる。
[スキーム1]
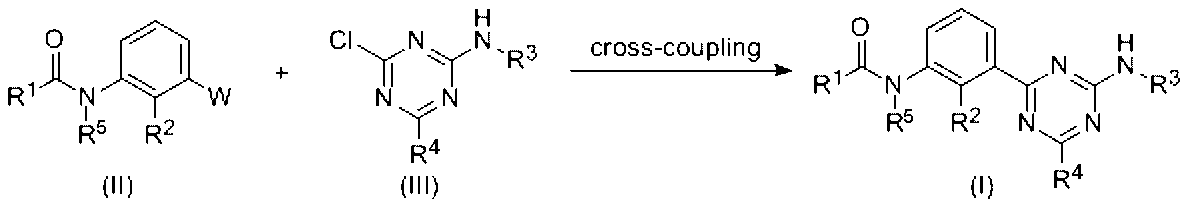
また、スキーム1の原料として用いられる化合物(II)は、例えばスキーム2に表す方法によって製造することができる。
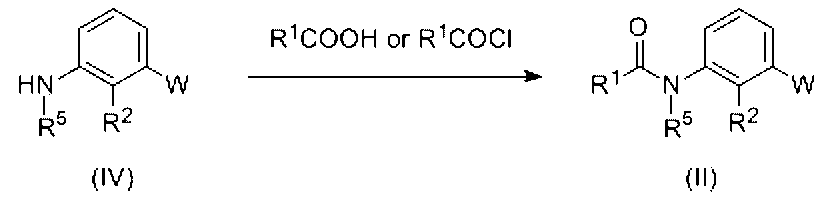
さらに、化合物(II)は、アミン(IV)とカルボン酸(R1COOH)から、混合酸無水物法によっても合成できる。
これらのアミド化反応は、いずれも不活性ガス(アルゴン、窒素等)雰囲気下、無水条件で行うことが望ましい。
アミン(IV)は市販品として、または公知の方法もしくはそれに準じた方法により得ることができる。

溶媒は、反応に不活性なものであればいずれでもよく、特に限定されるものではないが、好ましくはTHFを用いることができる。
溶媒は反応に不活性なものであればいずれでもよく、特に限定されるものではないが、好ましくはジオキサンを用いることができる。
これらの反応は、いずれも不活性ガス(アルゴン、窒素等)雰囲気下、無水条件で行うことが望ましい。
化合物(V)は市販品として、または公知の方法もしくはそれに準じた方法により得ることができる。

溶媒は反応に不活性なものであればいずれでもよく、特に限定されるものではないが、好ましくはジメチルホルムアミドを用いることができる。
R4が水素原子である本発明の化合物(I)は、例えばスキーム5によっても製造することができる。

溶媒は反応に不活性なものであればいずれでもよく、特に限定されるものではないが、好ましくはジオキサンを用いることができる。
化合物(VII)は、市販品として、または公知の方法もしくはそれに準じた方法により得ることができる。
溶媒は反応に不活性なものであればいずれでもよく、特に限定されるものではないが、好ましくはTHFを用いることができる。
アミン(VIII)は、市販品として、または公知の方法もしくはそれに準じた方法により得ることができる。

スキーム7の原料として用いられる化合物(IX)は、例えばスキーム8に表す方法によって製造することができる。

化合物(IX)は、化合物(IV)と化合物(III)とのクロスカップリング反応によって製造することができる。
化合物(IV)は、市販品として、または公知の方法もしくはそれに準じた方法により得ることができる。
スキーム3の原料として用いられる化合物(V)は、例えばスキーム9に表す方法によって製造することができる。

溶媒は反応に不活性なものであればいずれでもよく、特に限定されるものではないが、好ましくはDMSOを用いることができる。
本発明の化合物(I)またはその薬学的に許容される塩は、経口投与、非経口投与または局所的投与に適した従来の薬学製剤(医薬組成物)の形態に調製することができる。
また、本発明の化合物(I)またはその薬学的に許容される塩は、BTK阻害剤として、実験用、研究用の試薬として用いることもできる。
参考例1(原料化合物の合成)
4-(tert-ブチル)-N-[2-メチル-3-(4,4,5,5-テトラメチル-1,3,2-ジオキサボラン-2-イル)フェニル]ベンズアミド
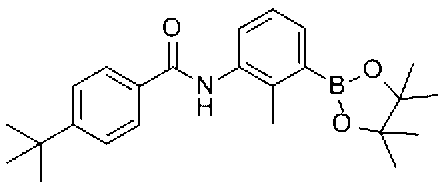
窒素雰囲気下、3-ブロモ-2-メチルアニリン(5.01g,26.9mmol)のDCM溶液(100ml)に、氷冷下、TEA(7.5mL,53.9mmol)および4-tert-ブチルベンゾイルクロリド(5.3g,26.9mmol)を加え0℃で4時間攪拌した。反応溶液をDCM(100mL)で希釈し、水(2×30mL)、1規定塩酸(30mL)、飽和炭酸水素ナトリウム水溶液(30mL)、飽和塩化ナトリウム水溶液(30mL)で順に洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を減圧濃縮して得られた固体をヘキサンに懸濁し、濾過した。固体をヘキサンで洗浄し、乾燥させ、N-(3-ブロモ-2-メチルフェニル)-4-(tert-ブチル)ベンズアミドを得た(9.0g)。
窒素雰囲気下、第1工程で製造したN-(3-ブロモ-2-メチルフェニル)-4-(tert-ブチル)ベンズアミド(500mg,1.44mmol)のジオキサン溶液(10ml)に、ビス(ピナコラト)ジボロン(733mg,2.89mmol)、[1,1’-ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリド ジクロロメタン付加物(118mg,0.14mmol)および酢酸カリウム(424mg,4.33mmol)を加え、16時間加熱還流した。溶媒を減圧留去し、得られた残渣をカラムクロマトグラフィー(シリカゲル、ヘキサン/酢酸エチル)で精製して、4-(tert-ブチル)-N-[2-メチル-3-(4,4,5,5-テトラメチル-1,3,2-ジオキサボラン-2-イル)フェニル]ベンズアミド(410mg)を得た。
4-(tert-ブチル)-N-(2-メチル-3-{4-[ (3,4,5-トリメトキシフェニル)アミノ]-1,3,5-トリアジン-2-イル}フェニル)ベンズアミド

氷冷下、2,4-ジクロロ-1,3,5-トリアジン(300mg,2.0mmol)のDMF溶液(5mL)に、DIEA(0.5mL,3.0mmol)および3,4,5-トリメトキシアニリン(366mg,2.0mmol)を加え、0℃で16時間攪拌した。反応溶液を水(15mL)で希釈し、酢酸エチル(3×50mL)で抽出した。得られた有機層を水(20mL)で洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を減圧留去し、得られた残渣をカラムクロマトグラフィー(シリカゲル、ヘキサン/酢酸エチル)で精製して、4-クロロ-N-(3,4,5-トリメトキシフェニル)-1,3,5-トリアジン-2-アミン(400mg)を得た。
4-クロロ-N-(3,4,5-トリメトキシフェニル)-1,3,5-トリアジン-2-アミン(50.0mg,0.17mmol)および参考例1で製造した4-(tert-ブチル)-N-[2-メチル-3-(4,4,5,5-テトラメチル-1,3,2-ジオキサボラン-2-イル)フェニル]ベンズアミド(66mg,0.17mmol)のジメトキシエタン溶液(3mL)に、テトラキス(トリフェニルホスフィン)パラジウム(0)(19mg,0.017mmol)および炭酸カリウム(47mg,0.34mmol)の水溶液(1mL)を加え、マイクロウェーブ反応装置を用いて110℃で20分間反応させた。反応溶液を酢酸エチル(2×40mL)で抽出し、得られた有機層を水(5mL)で洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を減圧留去し、得られた残渣をカラムクロマトグラフィー(シリカゲル、ヘキサン/酢酸エチル)で精製して、標記化合物(27mg)を得た。
4-(tert-ブチル)-N-[2-メチル-3-(4-{[4-(モルホリン-4-カルボニル)フェニル]アミノ}-1,3,5-トリアジン-2-イル)フェニル]ベンズアミド

4-[(4-{3-[4-(tert-ブチル)ベンズアミド]-2-メチルフェニル}-1,3,5-トリアジン-2-イル)アミノ]安息香酸
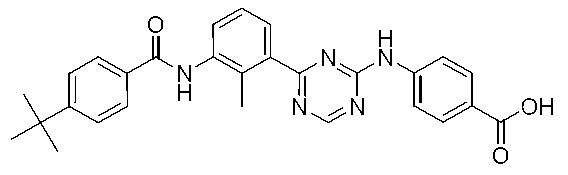
氷冷下、2,4-ジクロロ-1,3,5-トリアジン(2.0g,13.3mmol)のDMF溶液(33mL)に、DIEA(3.5mL,20.0mmol)および4-アミノ安息香酸エチル(2.2g,13.3mmol)を加え、0℃で20分間攪拌した。析出した固体をろ取し、水で洗浄した。得られた固体を減圧下、乾燥させて、4-[(4-クロロ-1,3,5-トリアジン-2-イル)アミノ]安息香酸エチル(2.45g)を得た。
参考例1で製造した4-(tert-ブチル)-N-[2-メチル-3-(4,4,5,5-テトラメチル-1,3,2-ジオキサボラン-2-イル)フェニル]ベンズアミド(353mg,0.9mmol)をジメトキシエタン溶液(15mL)と水(1mL)の混合溶媒に溶解し、第1工程で製造した4-[(4-クロロ-1,3,5-トリアジン-2-イル)アミノ]安息香酸エチル(250mg,0.9mmol)、テトラキス(トリフェニルホスフィン)パラジウム(0)(104mg,0.09mmol)および炭酸カリウム(248mg,1.79mmol)を加え、反応容器内を減圧脱気および窒素置換し、マイクロウェーブ反応装置を用いて110℃で20分間反応させた。反応溶液に水を加え、酢酸エチルで2回抽出し、合わせた有機層を飽和炭酸水素ナトリウム水溶液および飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を減圧留去し、得られた残渣をカラムクロマトグラフィー(シリカゲル、ヘキサン/酢酸エチル)で精製して、4-[(4-{3-[4-(tert-ブチル)ベンズアミド]-2-メチルフェニル}-1,3,5-トリアジン-2-イル)アミノ]安息香酸エチル(183mg)を得た。
4-[(4-{3-[4-(tert-ブチル)ベンズアミド]-2-メチルフェニル}-1,3,5-トリアジン-2-イル)アミノ]安息香酸エチル(730mg)のTHF溶液(16.4mL)にエタノール(8.2mL)および2規定水酸化ナトリウム水溶液(4.1mL)を加え、室温で終夜撹拌した。反応溶液を2規定塩酸で中和して水を加えたのち、酢酸エチルで2回抽出した。得られた有機層を、水および飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を減圧留去し、標記化合物(690mg)を得た。
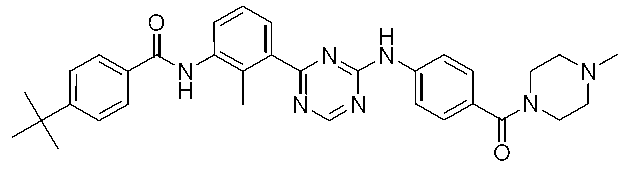
4-(tert-ブチル)-N-[2-メチル-3-(4-{[4-(4-メチルピペラジン-1-カルボニル)フェニル]アミノ}-1,3,5-トリアジン-2-イル)フェニル]ベンズアミド
N-[3-(4-アミノ-6-{[4-(モルホリン-4-カルボニル)フェニル]アミノ}-1,3,5-トリアジン-2-イル)-2-メチルフェニル]-4-(tert-ブチル)ベンズアミド
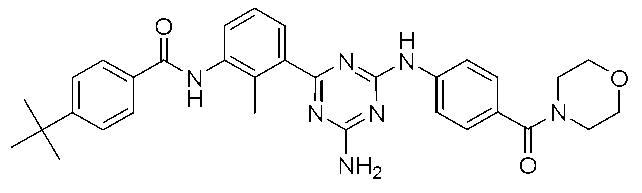
氷冷下、2-アミノ-4,6-ジクロロ-1,3,5-トリアジン(300mg,1.82mmol)のDMF溶液(3mL)に、DIEA(0.23mL,1.33mmol)および(4-アミノフェニル)モルホリン-4-イル-メタノン(250mg,1.21mmol)を加え、室温で16時間攪拌した。反応溶液を酢酸エチル(100mL)で希釈し、水および飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を減圧留去して、{4-[(4-アミノ-6-クロロ-1,3,5-トリアジン-2-イル)アミノ]フェニル}(モルホリノ)メタノン(325mg)を得た。
{4-[(4-アミノ-6-クロロ-1,3,5-トリアジン-2-イル)アミノ]フェニル}(モルホリノ)メタノン(50.0mg,0.15mmol)および参考例1で製造した4-(tert-ブチル)-N-[2-メチル-3-(4,4,5,5-テトラメチル-1,3,2-ジオキサボラン-2-イル)フェニル]ベンズアミド(59mg,0.15mmol)のジメトキシエタン溶液(3mL)に、テトラキス(トリフェニルホスフィン)パラジウム(0)(17mg,0.015mmol)および炭酸カリウム(41mg,0.3mmol)の水溶液(1mL)を加え、マイクロウェーブ反応装置を用いて110℃で20分間反応させた。反応溶液を酢酸エチル(2×40mL)で抽出し、得られた有機層を水(5mL)で洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を減圧留去し、得られた残渣をカラムクロマトグラフィー(シリカゲル、ヘキサン/酢酸エチル)で精製して、標記化合物(27mg)を得た。
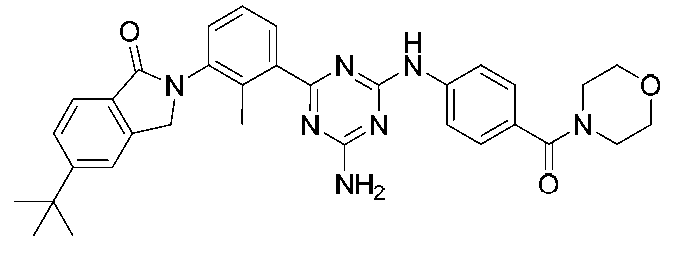
2-[3-(4-アミノ-6-{[4-(モルホリン-4-カルボニル)フェニル]アミノ}-1,3,5-トリアジン-2-イル)-2-メチルフェニル]-5-(tert-ブチル)イソインドリン-1-オン
4-tert-ブチルフタル酸無水物(4.39g,21.5mmol)の酢酸溶液(40mL)に、3-ブロモ-2-メチルアニリン(2.65mL,21.5mmol)を加え、100℃で1時間攪拌した。反応溶液を減圧濃縮したのち、得られた残渣を酢酸エチル(300mL)で希釈し、水(100mL)、飽和炭酸水素ナトリウム水溶液(100mL)、飽和塩化ナトリウム水溶液(100mL)で順に洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を減圧濃縮して得られた固体をヘキサンに懸濁し、ろ取した。固体をヘキサンで洗浄し、乾燥させて、2-(3-ブロモ-2-メチルフェニル)-5-(tert-ブチル)イソインドリン-1,3-ジオン(7.0g)を得た。
2-(3-ブロモ-2-メチルフェニル)-5-(tert-ブチル)イソインドリン-1,3-ジオン(1.0g,2.7mmol)をメタノール(20mL)に懸濁させ、水素化ホウ素ナトリウム(203mg,5.37mmol)を加え、室温にて10分間攪拌した。反応溶液を減圧濃縮後、得られた残渣をDCM(200mL)で希釈し、水(100mL)、飽和塩化ナトリウム水溶液(100mL)で順に洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を減圧濃縮して得られた残渣をDCM(10mL)で希釈し、トリフルオロ酢酸(10mL)を加え、室温にて10分間撹拌した。反応溶液にトリエチルシラン(6.4mL)を加え、室温にて30分間撹拌した。反応溶液を減圧濃縮し、得られた残渣をDCM(100mL)で希釈し、水(50mL)、飽和塩化ナトリウム水溶液(20mL)で順に洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を減圧留去し、得られた残渣をカラムクロマトグラフィー(シリカゲル、ヘキサン/酢酸エチル)で精製して、2-(3-ブロモ-2-メチルフェニル)-5-(tert-ブチル)イソインドリン-1-オン(242mg)を得た。
窒素雰囲気下、2-(3-ブロモ-2-メチルフェニル)-5-(tert-ブチル)イソインドリン-1-オン(200mg,0.56mmol)のジオキサン溶液(3.7mL)に、ビス(ピナコラト)ジボロン(284mg,1.12mmol)、[1,1’-ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリド ジクロロメタン付加物(46mg,0.056mmol)および酢酸カリウム(164mg,1.68mmol)を加え、16時間加熱還流した。反応溶液に水(50mL)を加え、酢酸エチル(3×25mL)で抽出した。得られた有機層を合わせ、飽和炭酸水素ナトリウム水溶液および飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を減圧留去し、得られた残渣をカラムクロマトグラフィー(シリカゲル、ヘキサン/酢酸エチル)で精製して、5-(tert-ブチル)-2-[2-メチル-3-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)フェニル]イソインドリン-1-オン(206mg)を得た。
実施例5の第1工程で製造した{4-[(4-アミノ-6-クロロ-1,3,5-トリアジン-2-イル)アミノ]フェニル}(モルホリノ)メタノン(69.0mg,0.21mmol)および5-(tert-ブチル)-2-[2-メチル-3-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)フェニル]イソインドリン-1-オン(126mg,0.31mmol)のジメトキシエタン溶液(3mL)に、テトラキス(トリフェニルホスフィン)パラジウム(0)(24mg,0.021mmol)および炭酸カリウム(57mg,0.4mmol)の水溶液(1mL)を加え、マイクロウェーブ反応装置を用いて110℃で20分間反応させた。反応溶液に水(50mL)を加え、酢酸エチル(3×25mL)で抽出した。得られた有機層を合わせ、飽和炭酸水素ナトリウム水溶液および飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を減圧留去し、得られた残渣をカラムクロマトグラフィー(シリカゲル、ヘキサン/酢酸エチル)で精製して、標記化合物(30mg)を得た。
N-[3-(4-アミノ-6-{[4-(モルホリン-4-カルボニル)フェニル]アミノ}-1,3,5-トリアジン-2-イル)-2-(ヒドロキシメチル)フェニル]-4-(tert-ブチル)ベンズアミド

(2-ブロモ-6-ニトロベンジルオキシ)(tert-ブチル)ジメチルシラン(14.5g,41.9mmol)のエタノール溶液(291mL)に、鉄粉(23.4g,419mmol)、塩化アンモニウム(44.8g,837mmol)および水(58mL)を加え、80℃で3時間攪拌した。反応溶液をろ過後、ろ液を減圧濃縮した。得られた残渣に水(500mL)を加え、クロロホルム(3×300mL)で抽出し、合わせた有機層を水および飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を減圧留去し、3-ブロモ-2-{[(tert-ブチルジメチルシリル)オキシ]メチル}アニリン(12.1g)を得た。
窒素雰囲気下、3-ブロモ-2-{[(tert-ブチルジメチルシリル)オキシ]メチル}アニリン(2.0g,6.32mmol)のTHF溶液(63mL)に、氷冷下、TEA(1.76mL,12.65mmol)および4-tert-ブチルベンゾイルクロリド(1.36mL,6.96mmol)を加え0℃で4時間攪拌した。反応溶液をDCM(300mL)で希釈し、水(100mL)、1規定塩酸(100mL)、飽和炭酸水素ナトリウム水溶液(100mL)、飽和塩化ナトリウム水溶液(100mL)で順に洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を減圧濃縮して得られた残渣を窒素雰囲気下、ジオキサン(39mL)に溶解し、ビス(ピナコラト)ジボロン(2.98g,11.75mmol)、[1,1’-ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリド ジクロロメタン付加物(480mg,0.588mmol)および酢酸カリウム(1.73g,17.63mmol)を加え、80℃にて16時間撹拌した。反応溶液に水(100mL)を加え、酢酸エチル(3×100mL)で抽出した。得られた有機層を合わせ、飽和炭酸水素ナトリウム水溶液および飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を減圧留去し、得られた残渣をカラムクロマトグラフィー(シリカゲル、ヘキサン/酢酸エチル)で精製して、4-(tert-ブチル)-N-(2-{[(tert-ブチルジメチルシリル)オキシ]メチル}-3-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)フェニル)ベンズアミド(3.0g)を得た。
実施例5の第1工程と同様の方法で製造した{4-[(4-アミノ-6-クロロ-1,3,5-トリアジン-2-イル)アミノ]フェニル}(モルホリノ)メタノン(256mg,0.76mmol)および4-(tert-ブチル)-N-(2-{[(tert-ブチルジメチルシリル)オキシ]メチル}-3-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)フェニル)ベンズアミド(400mg,0.76mmol)のジメトキシエタン溶液(11.5mL)に、テトラキス(トリフェニルホスフィン)パラジウム(0)(88mg,0.076mmol)および炭酸カリウム(211mg,1.53mmol)の水溶液(3.8mL)を加え、マイクロウェーブ反応装置を用いて110℃で20分間反応させた。反応溶液に水(100mL)を加え、酢酸エチル(3×50mL)で抽出した。得られた有機層を合わせ、飽和炭酸水素ナトリウム水溶液および飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を減圧留去し、得られた残渣をカラムクロマトグラフィー(シリカゲル、ヘキサン/酢酸エチル)で精製して、N-[3-(4-アミノ-6-{[4-(モルホリン-4-カルボニル)フェニル]アミノ}-1,3,5-トリアジン-2-イル)-2-{[(tert-ブチルジメチルシリル)オキシ]メチル}フェニル]-4-(tert-ブチル)ベンズアミド(144mg)を得た。
N-[3-(4-アミノ-6-{[4-(モルホリン-4-カルボニル)フェニル]アミノ}-1,3,5-トリアジン-2-イル)-2-{[(tert-ブチルジメチルシリル)オキシ]メチル}フェニル]-4-(tert-ブチル)ベンズアミド(144mg,0.2mmol)のTHF溶液(4.1mL)に、テトラブチルアンモニウムフルオリドテトラヒドロフラン溶液(1mol/L、0.41mL)を加え、室温で16時間撹拌した。溶媒を減圧留去し、得られた残渣をカラムクロマトグラフィー(シリカゲル、ヘキサン/酢酸エチル)で精製して、標記化合物(62mg)を得た。
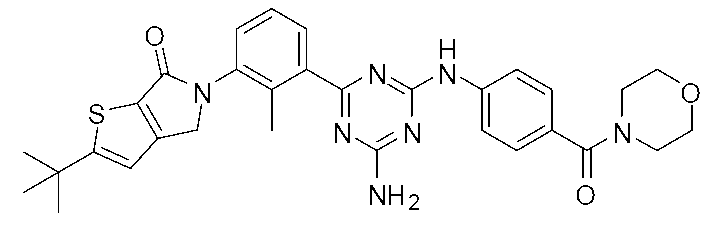
5-[3-(4-アミノ-6-{[4-(モルホリン-4-カルボニル)フェニル]アミノ}-1,3,5-トリアジン-2-イル)-2-メチルフェニル]-2-(tert-ブチル)-4H-チエノ[2,3-c]ピロール-6(5H)-オン
窒素雰囲気下、塩化アルミニウム(6.4g,48.0mmol)のDCM溶液(7.3mL)に、3-メチルチオフェン-2-カルボン酸メチル(5.0g,32.0mmol)のDCM溶液(3.6mL)を-80℃で5分間かけて滴加し、5分間攪拌した。本反応液に、2-クロロ-2-メチルプロパン(3.55g,38.4mmol)のDCM溶液(3.6mL)を5分間かけて滴加し、徐々に昇温させながら室温で14時間攪拌した。反応溶液を氷に加え、DCM(3×300mL)で抽出した。得られた有機層を合わせ、水、飽和炭酸水素ナトリウム水溶液および飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を減圧留去し、得られた残渣をカラムクロマトグラフィー(シリカゲル、ヘキサン/酢酸エチル)で精製して、5-(tert-ブチル)-3-メチルチオフェン-2-カルボン酸メチル(4.74g)を得た。
5-(tert-ブチル)-3-メチルチオフェン-2-カルボン酸メチル(3.15g,14.84mmol)を四塩化炭素(40mL)に懸濁させ、N-ブロモスクシンイミド(3.17g,17.8mmol)及び2,2’-アゾビス(2-メチルプロピオニトリル)(122mg,0,74mmol)を加え、85℃で14時間攪拌した。反応溶液をろ過した後、ろ液を減圧濃縮した。得られた残渣をカラムクロマトグラフィー(シリカゲル、ヘキサン/酢酸エチル)で精製して、3-(ブロモメチル)-5-(tert-ブチル)チオフェン-2-カルボン酸メチル(4.15g)を得た。
(第3工程)
窒素雰囲気下、3-(ブロモメチル)-5-(tert-ブチル)チオフェン-2-カルボン酸メチル(1.5g,5.15mmol)のアセトニトリル溶液(25mL)に、3-ブロモ-2-メチルアニリン(2.88g,15.45mmol)および炭酸セシウム(1.85g,5.67mmol)を加え、室温で16時間攪拌した。溶媒を減圧留去し、得られた残渣をカラムクロマトグラフィー(シリカゲル、ヘキサン/酢酸エチル)で精製して、3-{[(3-ブロモ-2-メチルフェニル)アミノ]メチル}-5-(tert-ブチル)チオフェン-2-カルボン酸メチル(1.43g)を得た。
3-{[(3-ブロモ-2-メチルフェニル)アミノ]メチル}-5-(tert-ブチル)チオフェン-2-カルボン酸メチル(1.39g,3.5mmol)のTHF-メタノールの混合溶液(1:1、10mL)に、水酸化リチウム(838mg,35mmol)の水溶液(5mL)を加え、45℃で14時間攪拌した。反応溶液を減圧濃縮し、得られた残渣に2規定塩酸(20mL)を加えた。析出した固体をろ取し、ヘキサンで洗浄後、乾燥させて、3-{[(3-ブロモ-2-メチルフェニル)アミノ]メチル}-5-(tert-ブチル)チオフェン-2-カルボン酸(1.15g)を得た。
窒素雰囲気下、3-{[(3-ブロモ-2-メチルフェニル)アミノ]メチル}-5-(tert-ブチル)チオフェン-2-カルボン酸(1.04g,2.72mmol)のDCM溶液(25mL)に、塩化チオニル(0.79mL,10.9mmol)を加え、室温で16時間攪拌した。反応溶液を減圧濃縮し、得られた残渣をカラムクロマトグラフィー(シリカゲル、ヘキサン/酢酸エチル)で精製して、5-(3-ブロモ-2-メチルフェニル)-2-(t-ブチル)-4H-チエノ[2,3-c]ピロール-6(5H)-オン(605mg)を得た。
窒素雰囲気下、5-(3-ブロモ-2-メチルフェニル)-2-(t-ブチル)-4H-チエノ[2,3-c]ピロール-6(5H)-オン(460mg,1.26mmol)のジオキサン溶液(8.0mL)に、ビス(ピナコラト)ジボロン(641mg,2.53mmol)、[1,1’-ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリド ジクロロメタン付加物(103mg,0.126mmol)および酢酸カリウム(372mg,3.79mmol)を加え、80℃で16時間加熱した。反応溶液に水(50mL)を加え、酢酸エチル(2×25mL)で抽出した。得られた有機層を合わせ、飽和炭酸水素ナトリウム水溶液および飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を減圧留去し、得られた残渣をカラムクロマトグラフィー(シリカゲル、ヘキサン/酢酸エチル)で精製して、2-(tert-ブチル)-5-[2-メチル-3-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)フェニル]-4H-チエノ[2,3-c]ピロール-6(5H)-オン(210mg)を得た。
実施例5の第1工程で製造した{4-[(4-アミノ-6-クロロ-1,3,5-トリアジン-2-イル)アミノ]フェニル}(モルホリノ)メタノン(81.1mg,0.24mmol)および2-(tert-ブチル)-5-[2-メチル-3-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)フェニル]-4H-チエノ[2,3-c]ピロール-6(5H)-オン(100mg,0.24mmol)のジメトキシエタン溶液(3mL)に、テトラキス(トリフェニルホスフィン)パラジウム(0)(28mg,0.024mmol)および炭酸カリウム(67mg,0.49mmol)の水溶液(1.2mL)を加え、マイクロウェーブ反応装置を用いて110℃で40分間反応させた。反応溶液に水(50mL)を加え、酢酸エチル(2×25mL)で抽出した。得られた有機層を合わせ、飽和炭酸水素ナトリウム水溶液および飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を減圧留去し、得られた残渣をカラムクロマトグラフィー(シリカゲル、ヘキサン/酢酸エチル)で精製して、標記化合物(50mg)を得た。
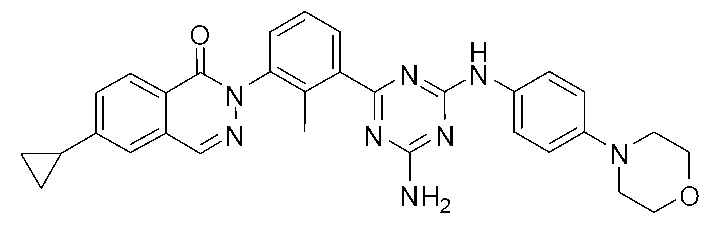
2-(3-{4-アミノ-6-[(4-モルホリノフェニル)アミノ]-1,3,5-トリアジン-2-イル}-2-メチルフェニル)-6-シクロプロピルフタラジン-1(2H)-オン
6-ブロモフタラジン-1(2H)-オン(1.00g,4.44mmol)、
シクロプロピルボロン酸(0.57g,6.67mmol)、トリシクロヘキシルフォスフィン(0.13g,0.44mmol)および無水リン酸カリウム(0.57g,6.67mmol)のトルエン(20mL)-水(1.5mL)混合溶液に、窒素雰囲気下室温にて、酢酸パラジウム(0.20g,0.89mmol)を加え、100℃で3時間反応させた。室温に戻した後、反応溶液を濾過し、不溶物を水ならびに酢酸エチルで洗浄した。ろ液を酢酸エチルで抽出した後、水、ついで飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させ、溶媒を減圧留去した。得られた粗生成物をカラムクロマトグラフィー(シリカゲル、ヘキサン/酢酸エチル)で精製して、6-シクロプロピルフタラジン-1(2H)-オン(0.13g)を得た。
6-シクロプロピルフタラジン-1(2H)-オン(56mg,0.30mmol)、1,3-ジブロモ-2-メチルベンゼン(150mg,0.60mmol)、炭酸セシウム(195mg,0.60mmol)およびヨウ化銅(I)(11mg,0.06mmol)のDMSO溶液(2mL)を5分間窒素で置換した後、150℃で18時間加熱攪拌させた。室温に戻した後、反応溶液を冷水に添加し、酢酸エチルで2回抽出した。得られた有機層を合わせ、飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を減圧留去し、得られた残渣をカラムクロマトグラフィー(シリカゲル、ヘキサン/酢酸エチル)で精製して、2-(3-ブロモ-2-メチルフェニル)-6-シクロプロピルフタラジン-1(2H)-オン(17mg)を得た。
2-(3-ブロモ-2-メチルフェニル)-6-シクロプロピルフタラジン-1(2H)-オン(16mg,0.045mmol)の1,4-ジオキサン溶液(0.45mL)に、ビス(ピナコラト)ジボロン(23mg,0.09mmol)、[1,1’-ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリド ジクロロメタン付加物(3.7mg,0.0045mmol)および酢酸カリウム(13mg,0.13mmol)を加え、80℃にて20時間撹拌した。室温に戻した後、反応溶液に水を加え、酢酸エチルで抽出した。得られた有機層を無水硫酸ナトリウムで乾燥させ、溶媒を減圧留去し、得られた残渣をカラムクロマトグラフィー(シリカゲル、ヘキサン/酢酸エチル)で精製して、6-シクロプロピル-2-[2-メチル-3-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)フェニル]フタラジン-1(2H)-オン(9mg)を得た。
氷冷下、2-アミノ-4,6-ジクロロ-1,3,5-トリアジン(124mg,0.75mmol)のTHF溶液(1.2mL)に、DIEA(0.18mL,1.33mmol)および4-モルホリノアニリン(89mg,0.50mmol)のTHF溶液(0.8mL)をゆっくり加え、そのまま氷温で1時間、さらに室温で16時間攪拌した。析出した固体を濾取し、酢酸エチルで洗浄、乾燥させて、2-アミノ-4-クロロ-6-[(4-モルホリノフェニル)アミノ]-1,3,5-トリアジン(124mg)を得た。
2-アミノ-4-クロロ-6-[(4-モルホリノフェニル)アミノ]-1,3,5-トリアジン(7mg,0.022mmol)および上記第3工程にて得られた6-シクロプロピル-2-[2-メチル-3-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)フェニル]フタラジン-1(2H)-オン(9mg,0.022mmol)のジメトキシエタン溶液(0.6mL)に、窒素雰囲気下、室温にてテトラキス(トリフェニルホスフィン)パラジウム(0)(2.6mg,0.0022mmol)および炭酸カリウム(6mg,0.45mmol)の水溶液(0.2mL)を加え、マイクロウェーブ反応装置を用いて110℃で20分間反応させた。室温に戻した後、不溶物を濾過して除き、分取用HPLC(水/メタノール、ギ酸添加)で精製して、標記化合物(2.7mg)を得た。
2-(3-{4-アミノ-6-[(4-モルホリンノフェニル)アミノ]-1,3,5-トリアジン-2-イル}-2-(ヒドロキシメチル)フェニル)-6-シクロプロピル-8-フルオロ-3,4-ジヒドロイソキノリン-1(2H)-オン

6-シクロプロピル-8-フルオロ-3,4-ジヒドロイソキノリン-1(2H)-オン(1.56g,7.6mmol)、1,6-ジブロモベンズアルデヒド(4.0g,15.2mmol)、ヨウ化銅(I)(1.45g,7.6mmol)、ならびに炭酸水素ナトリウム(1.28g,15.2mmol)のDMSO懸濁液(15mL)を、窒素雰囲気下、110℃で2日間加熱攪拌した。反応溶液を室温まで放冷後、反応混合物に水と酢酸エチルを加えた。セライト濾過で析出物を除去し、濾液を酢酸エチルで3回抽出した。得られた有機層を合わせ、飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を減圧留去して得られた残渣をカラムクロマトグラフィー(シリカゲル、クロロホルム~ヘキサン/酢酸エチル)で精製して、2-ブロモ-6-(6-シクロプロピル-8-フルオロ-1-オキソ-3,4-ジヒドロイソキノリン-1(2H)-イル)ベンズアルデヒド(2.07g)を得た。
2-ブロモ-6-(6-シクロプロピル-8-フルオロ-1-オキソ-3,4-ジヒドロイソキノリン-1(2H)-イル)ベンズアルデヒド(2.53g,6.52mmol)のDCM/イソプロパノール混合溶液(2:1,36mL)に、氷冷下、水素化ホウ素ナトリウム(0.12g,3.26mmol)を加え、室温に戻しながら1時間激しく攪拌した。反応溶液を氷水に加え、クロロホルムで2回抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧留去した。得られたアルコール体の粗生成物(2.61g)をDCM(50mL)に溶解し、氷冷下、塩化アセチル(1.39mL,19.6mmol)、ピリジン(1.39mL,19.6mmol)を加え、室温に戻しながら1時間攪拌した。反応溶液を氷水に加え、クロロホルムで2回抽出した。得られた有機層を合わせ、飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を減圧留去して得られた残渣に酢酸エチル-ジイソプロピルエーテル混合溶液(1:5)を加え、析出した固体を濾取した。固体をジイソプロピルエーテルで洗浄し、乾燥させて、2-ブロモ-6-(6-シクロプロピル-8-フルオロ-1-オキソ-3,4-ジヒドロイソキノリン-1(2H)-イル)ベンジルアセテート(2.20g)を得た。
窒素雰囲気下、2-ブロモ-6-(6-シクロプロピル-8-フルオロ-1-オキソ-3,4-ジヒドロイソキノリン-2(1H)-イル)ベンジルアセテート(2.10g,4.86mmol)のジオキサン溶液(24.0mL)に、ビス(ピナコラト)ジボロン(2.47g,9.72mmol)、[1,1’-ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリド ジクロロメタン付加物(397mg,0.486mmol)および酢酸カリウム(1.43g,14.6mmol)を加え、95℃で24時間加熱撹拌した。反応を完結させるために、ビス(ピナコラト)ジボロン(2.47g,9.72mmol)、[1,1’-ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリド ジクロロメタン付加物(397mg,0.486mmol)および酢酸カリウム(1.43g,14.6mmol)を追加し、95℃でさらに24時間加熱撹拌した。反応溶液を室温まで冷却後、反応混合物に水を加え、酢酸エチルで抽出した。得られた有機層を合わせ、飽和炭酸水素ナトリウム水溶液および飽和食塩水で順に洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を減圧留去して得られた残渣をカラムクロマトグラフィー(シリカゲル、ヘキサン/酢酸エチル)で精製して、2-(6-シクロプロピル-8-フルオロ-1-オキソ-3,4-ジヒドロイソキノリン-2(1H)-イル)-6-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ベンジルアセテート(2.02g)を得た。
実施例43の第4工程と同様の方法で製造した2-アミノ-4-クロロ-6-[(4-モルホリノフェニル)アミノ]-1,3,5-トリアジン(1.31g,4.26mmol)および2-(6-シクロプロピル-8-フルオロ-1-オキソ-3,4-ジヒドロイソキノリン-2(1H)-イル)-6-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ベンジルアセテート(2.0g,4.26mmol)のジメトキシエタン溶液(15mL)に、テトラキス(トリフェニルホスフィン)パラジウム(0)(246mg,0.21mmol)および炭酸カリウム(1.18g,8.51mmol)の水溶液(7.5mL)を加え、100℃で6時間加熱した。反応溶液に水を加え、析出した固体をろ取し、水で洗浄した。得られた残渣をカラムクロマトグラフィー(シリカゲル、ヘキサン/酢酸エチル)で精製して、2-{4-アミノ-6-[(4-モルホリノフェニル)アミノ]-1,3,5-トリアジン-2-イル}-6-(6-シクロプロピル-8-フルオロ-1-オキソ-3,4-ジヒドロイソキノリン-2(1H)-イル)ベンジルアセテートとその脱アセチル体の混合物(2.0g)を得た。得られた混合物(2.0g)のメタノール溶液(15mL)に、炭酸カリウム(828mg,6mmol)を加え、70℃で30分間加熱した。反応溶液に水を加え、析出した固体をろ取し、水及びヘキサンで順に洗浄した。得られた固体を乾燥して、標記化合物(1.59g)を得た。
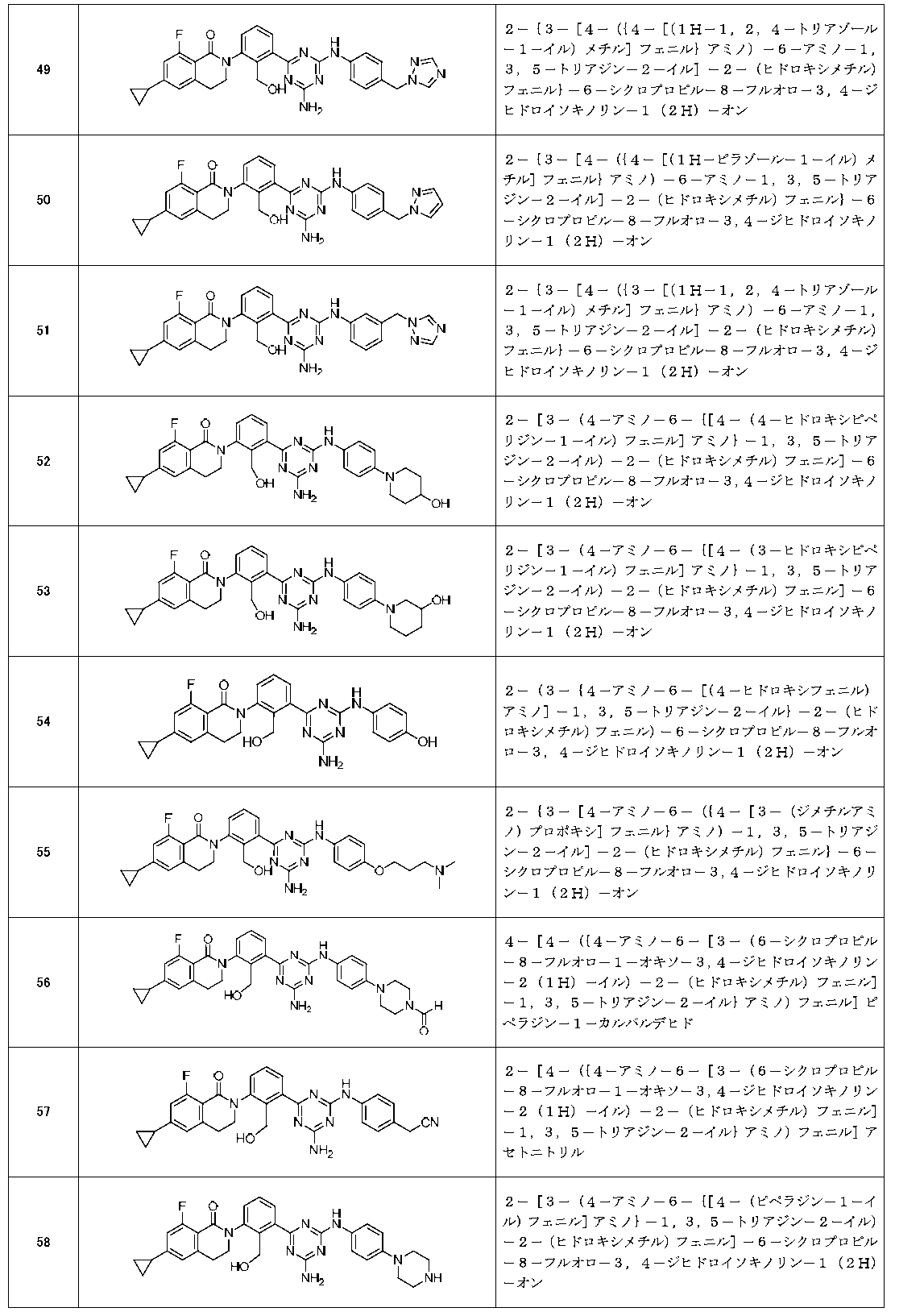
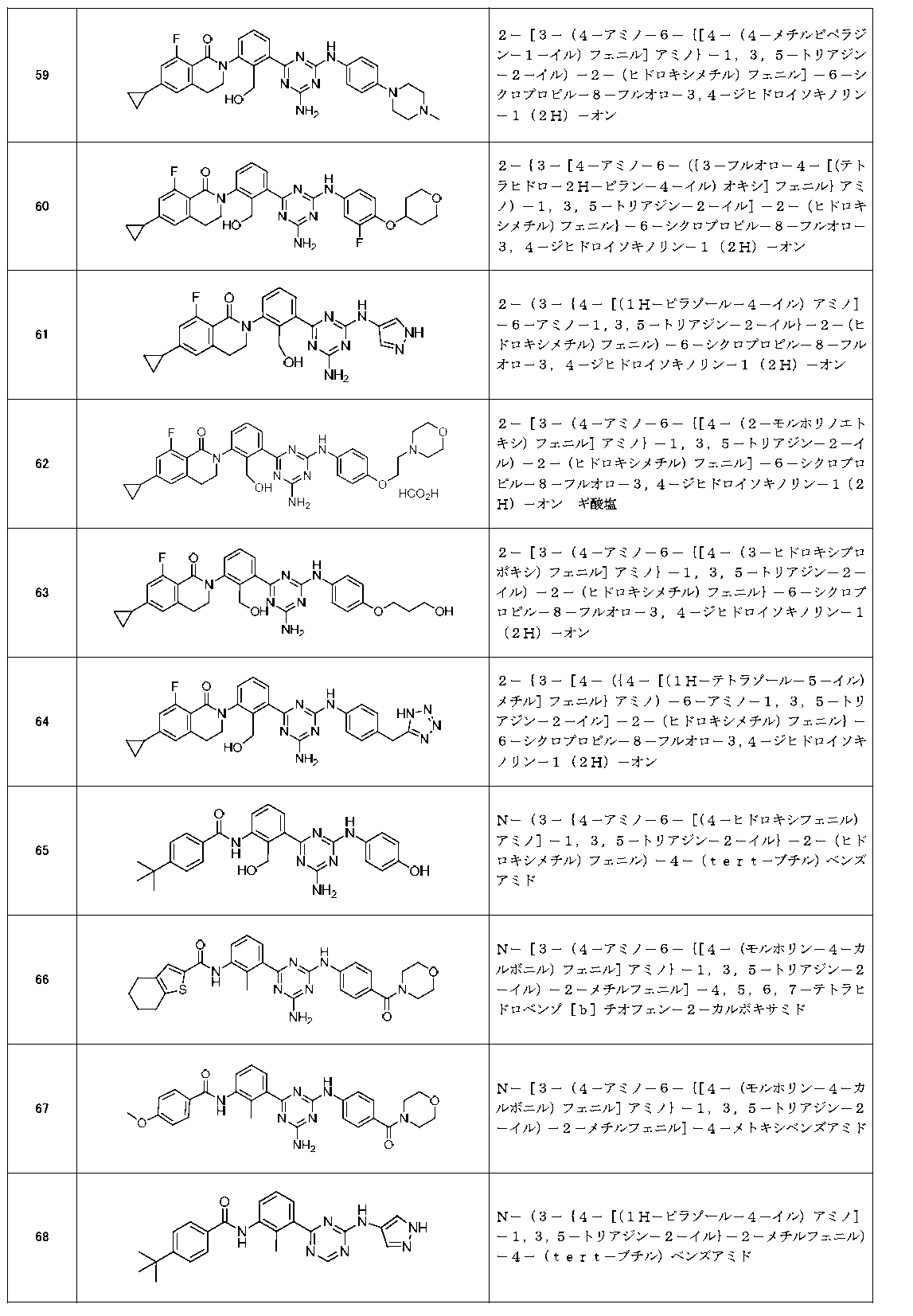
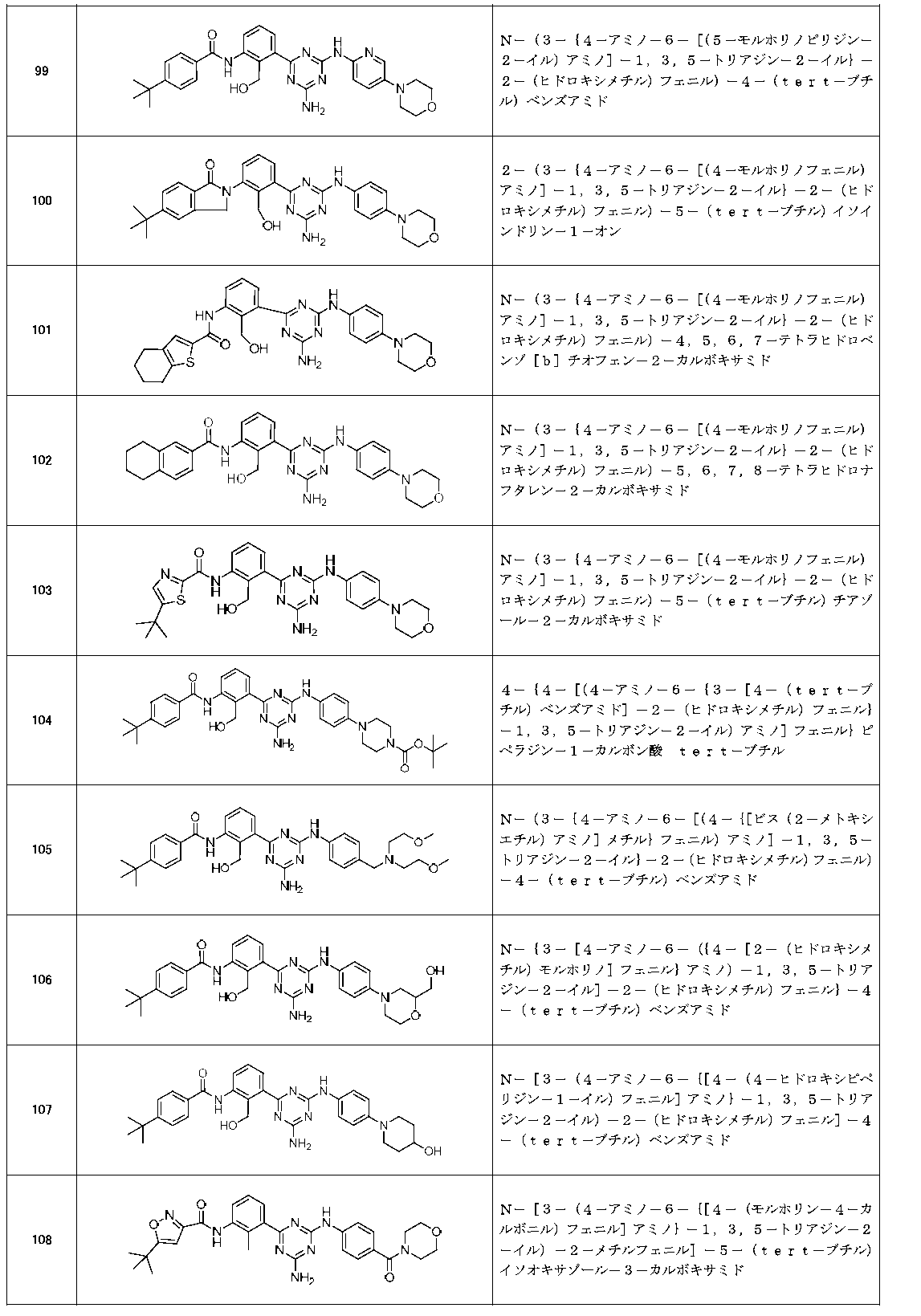
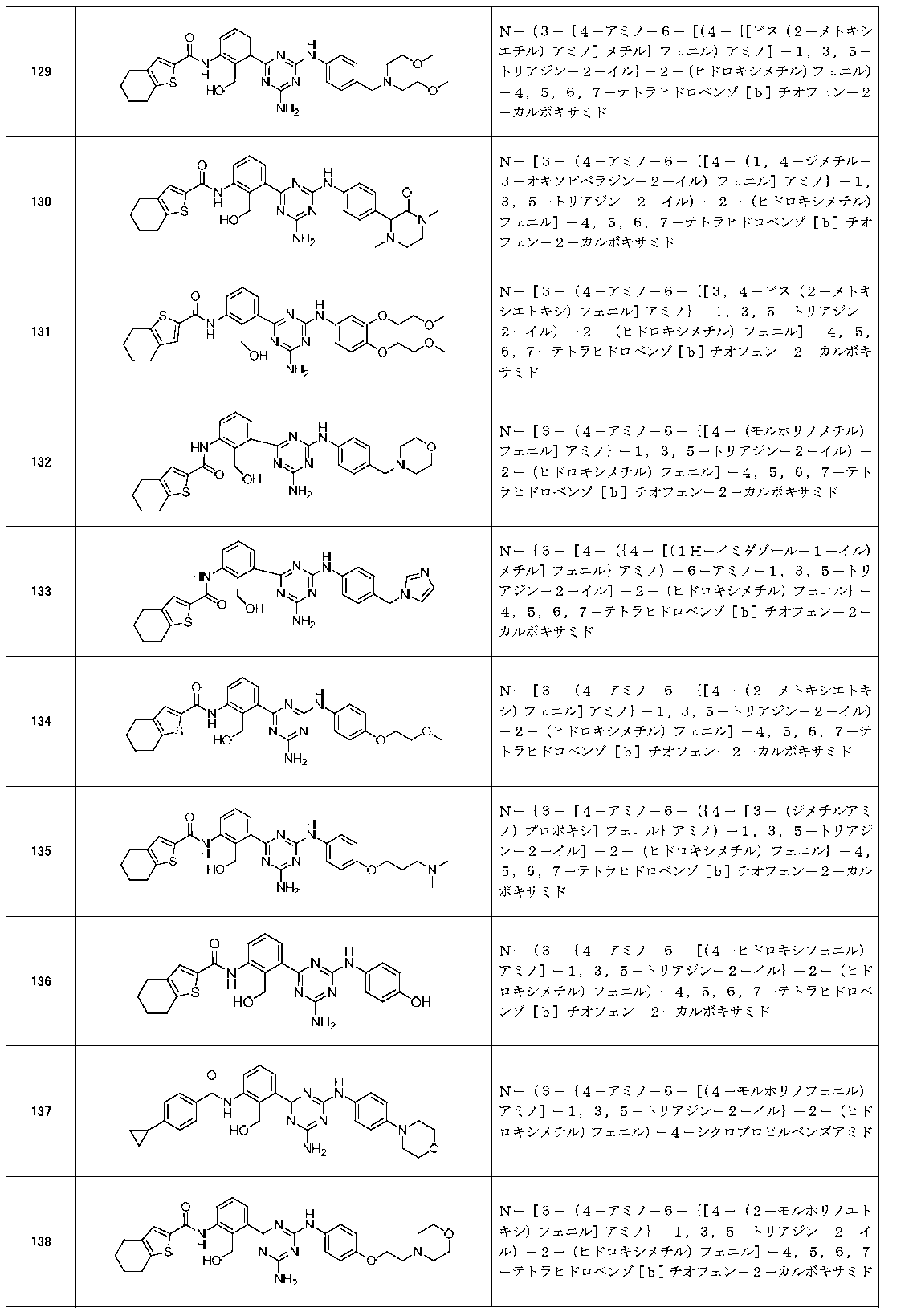
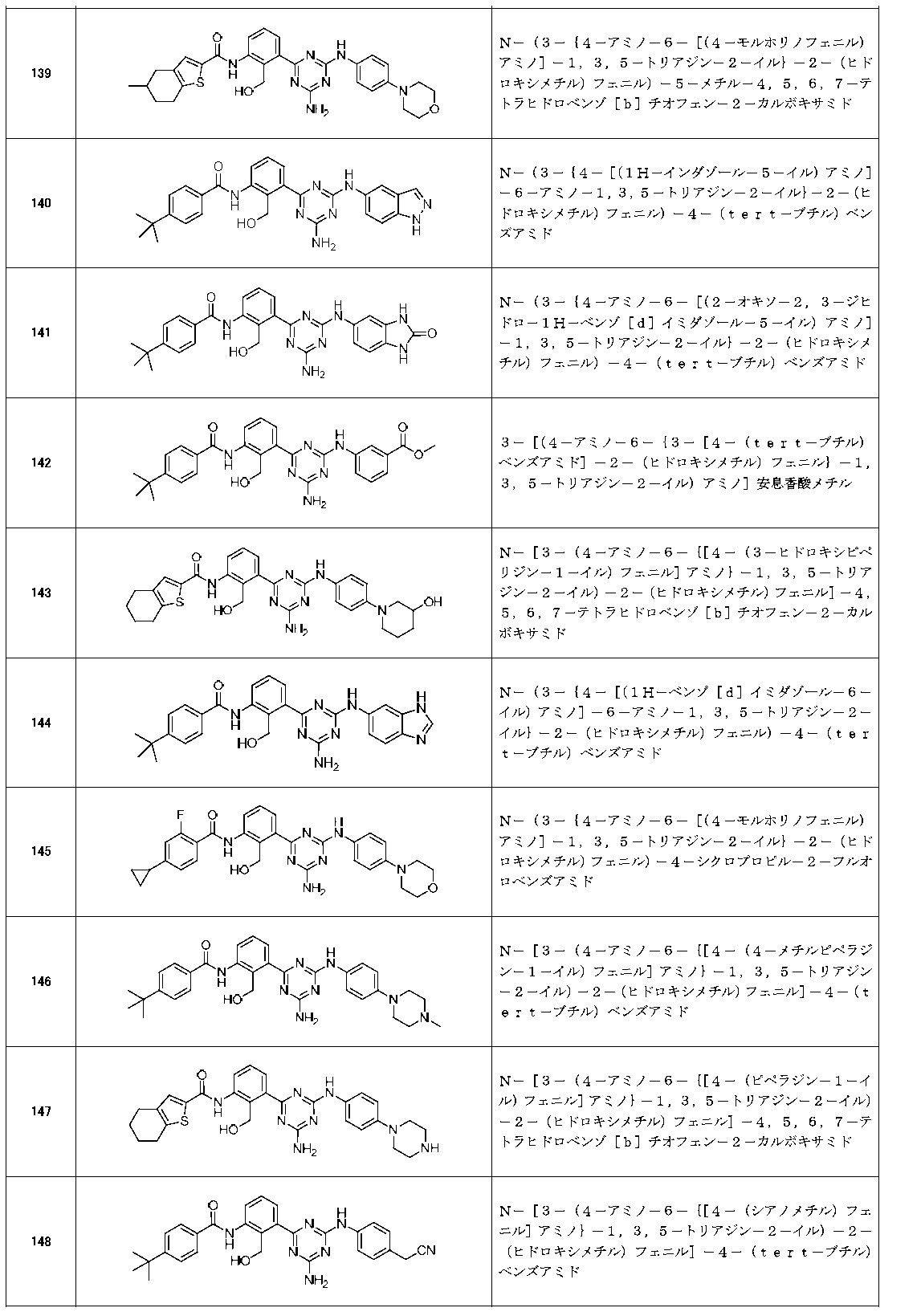
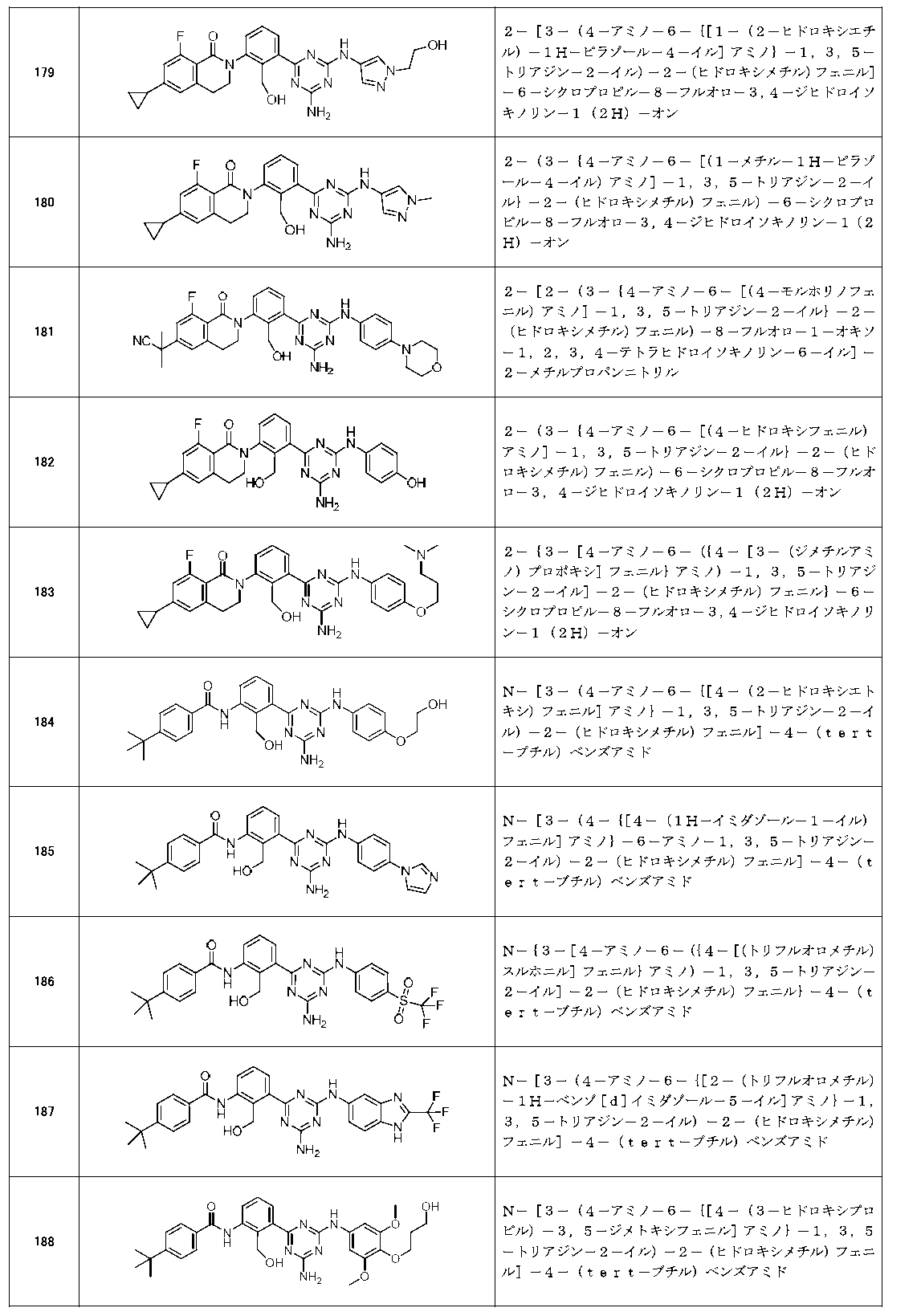
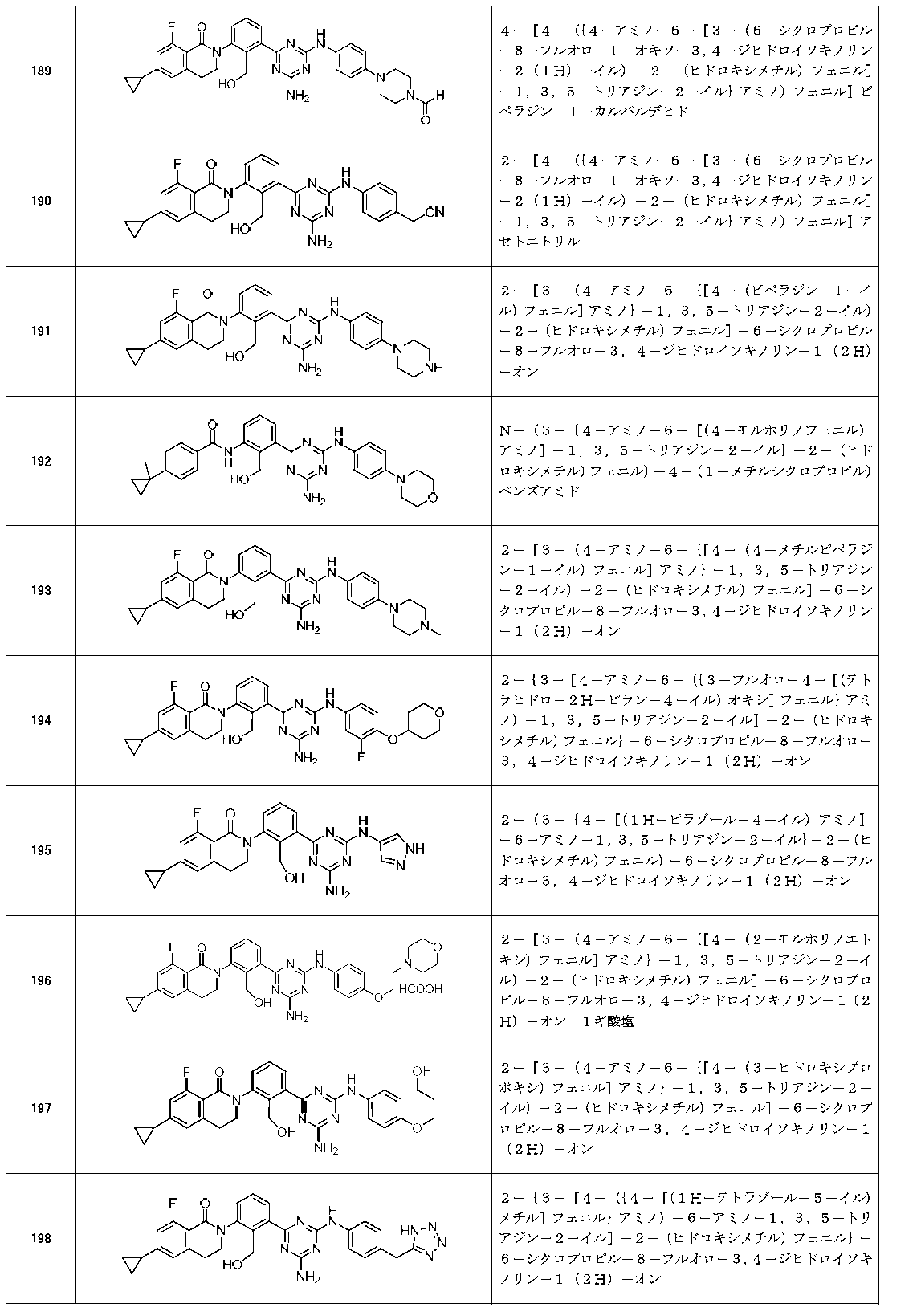
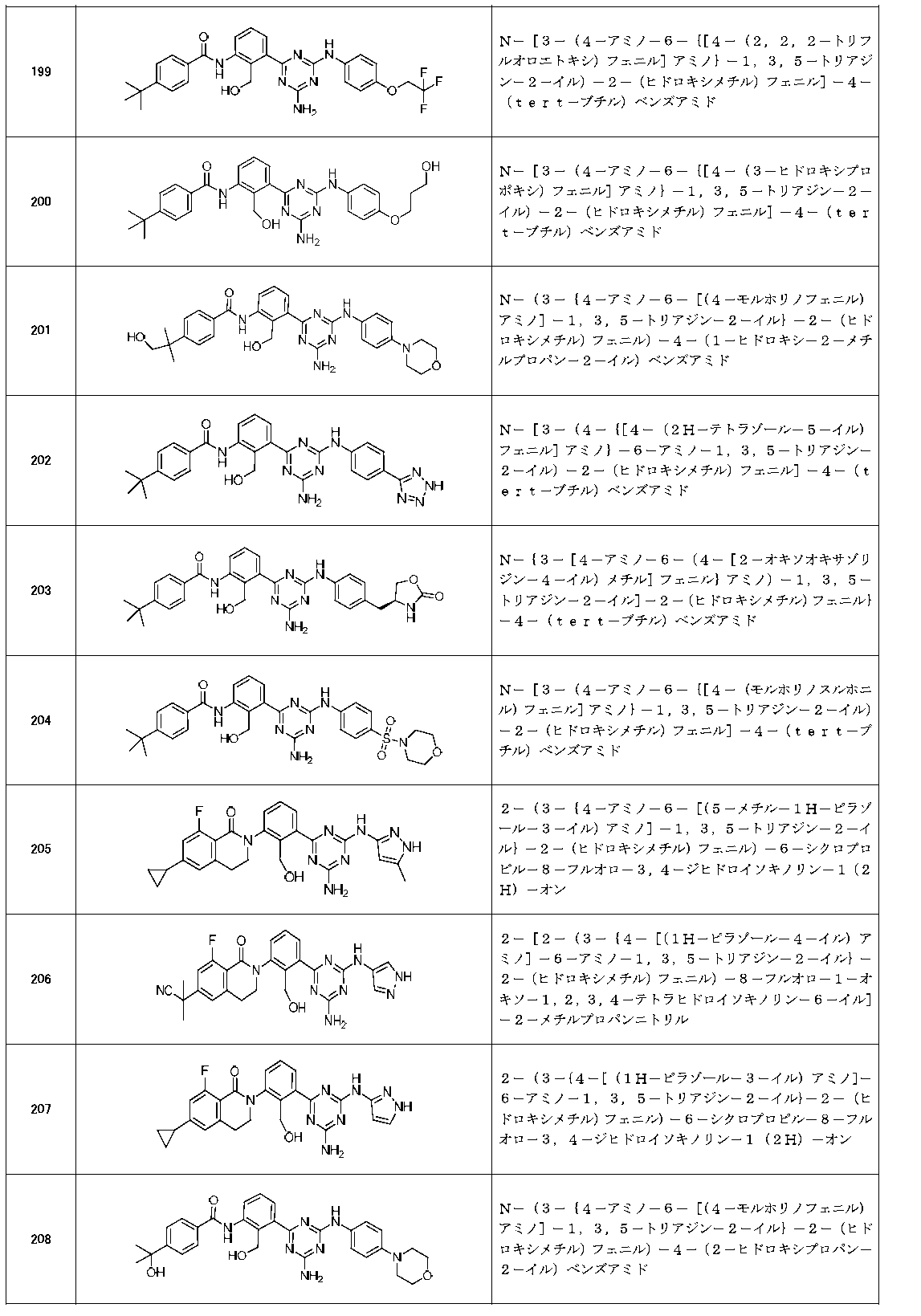
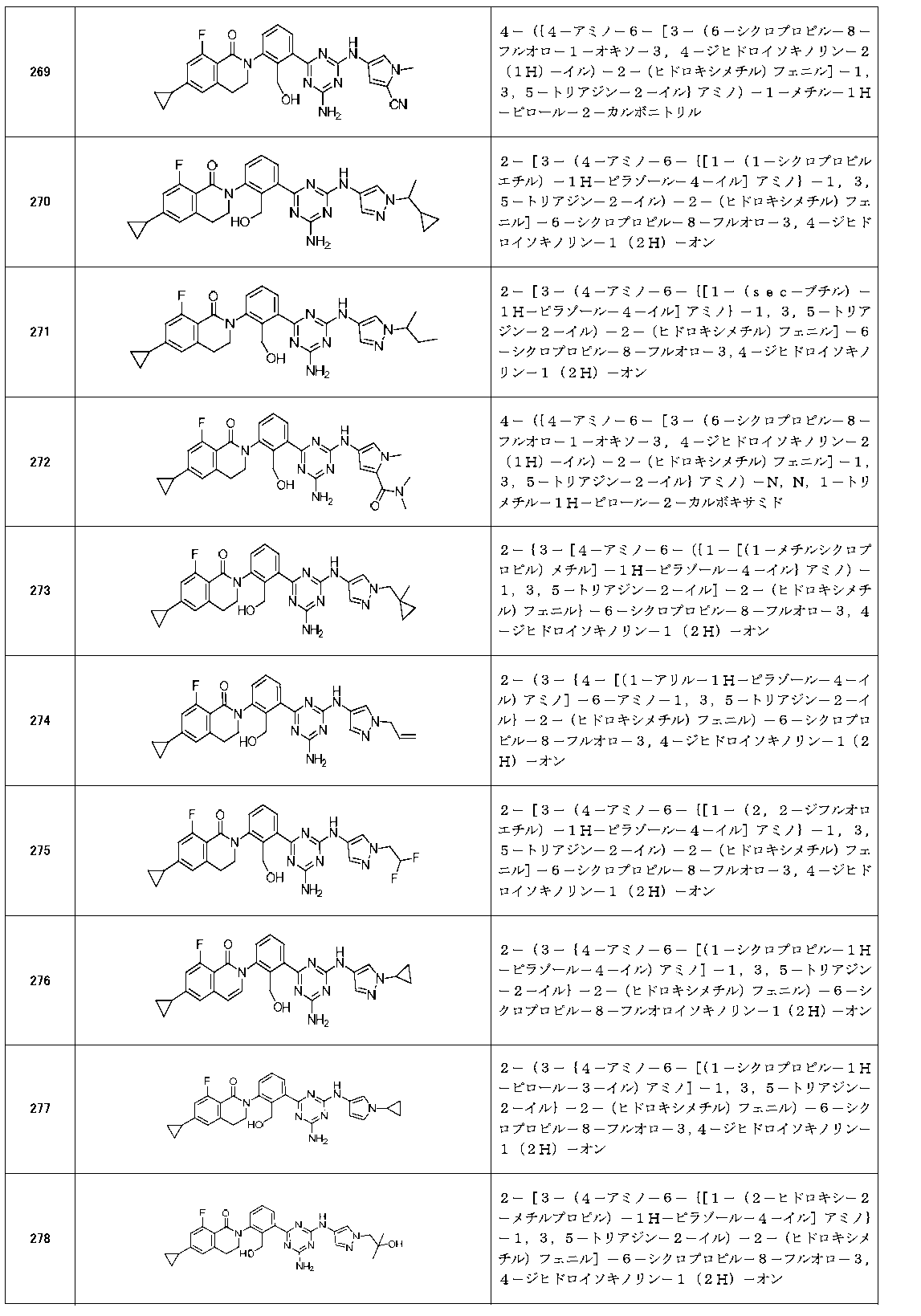
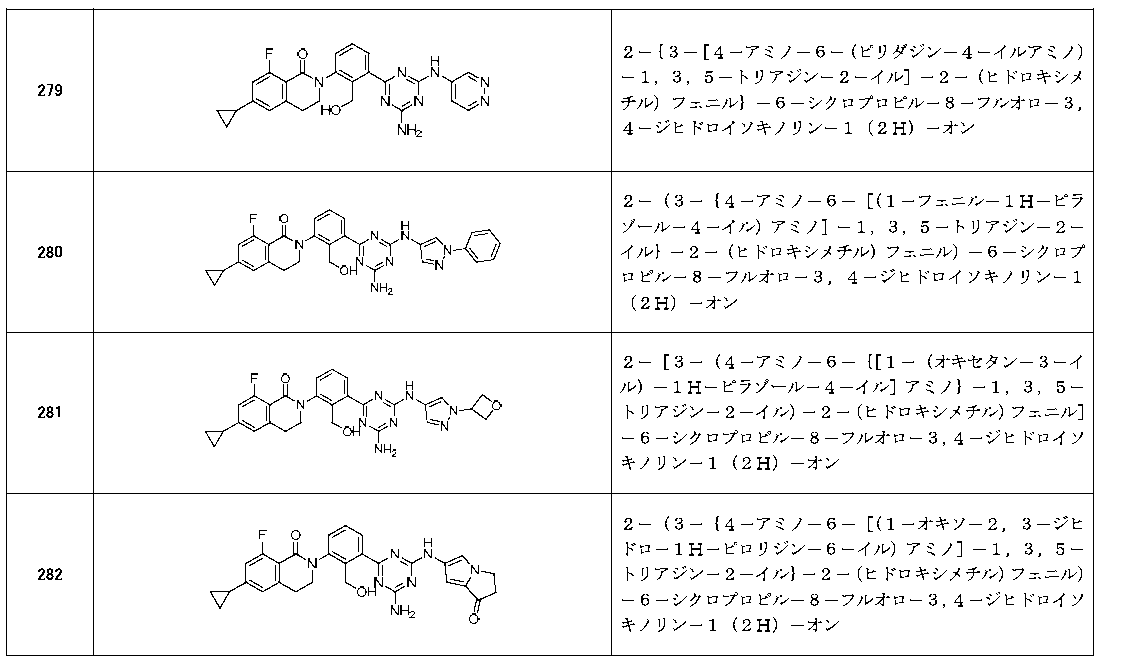
以下の実施例化合物[表1-1]~[表1-27]は、それぞれ対応する原料(市販品、または市販化合物から公知の方法もしくはそれに準じた方法により誘導体化した化合物)を用い、上述の実施例記載の方法に従い、必要に応じて、有機合成化学で通常用いられる方法を適宜組み合わせて製造した。


























BTKに対する活性阻害試験
(キナーゼ活性の測定方法)
分離された基質およびリン酸化された基質の各ピークの高さをそれぞれSおよびPとし、またブランクとして酵素溶液の代わりにアッセイバッファーを添加したものを測定した。
被験化合物の阻害率(%)は、次の式に従って算出した。
阻害率(%)=(1-(C-A)/(B-A))×100
ただし、A、B、Cは、それぞれブランクウェルのP/(P+S)、コントロール溶液ウェルのP/(P+S)、化合物添加ウェルのP/(P+S)を示す。
また、IC50値は、阻害率と被験化合物濃度(対数)の回帰分析により算出した。
本発明化合物のBTKに対するIC50値は、1μM以下の強い阻害活性を示した。代表化合物のBTK阻害活性を表3に示す。

脱リン酸化BTKに対する活性阻害試験
(脱リン酸化BTKの調整)
本発明化合物の脱リン酸化BTKに対するIC50値は、1μM以下であり、本発明化合物は、強い阻害活性を示すことが判明した。代表化合物の脱リン酸化BTK阻害活性を表4-1および4-2に示す。
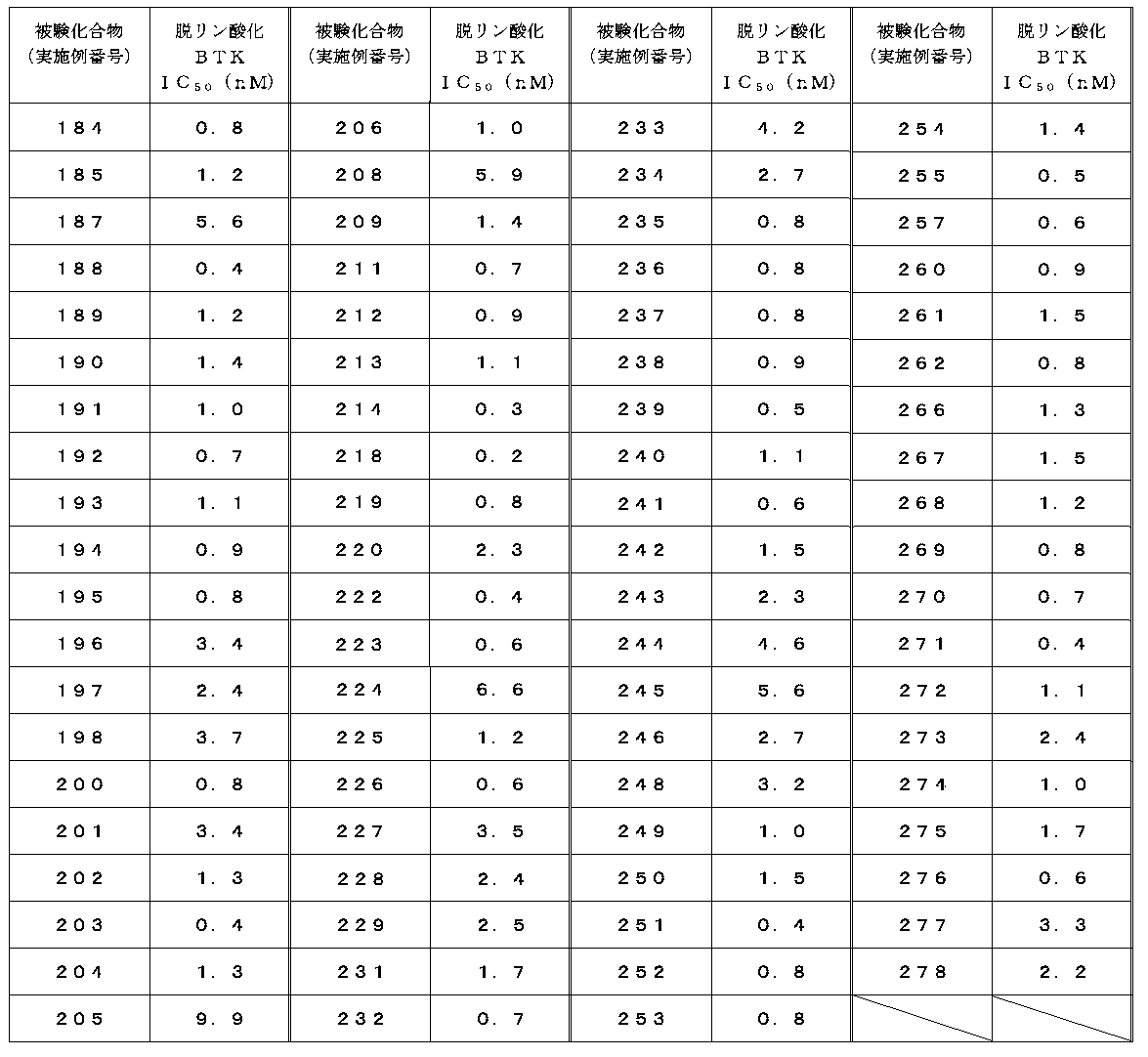
細胞内BTKの自己リン酸化活性阻害試験
Ramos細胞(2G6.4C10、ATCC社No.CRL-1923)は、T75フラスコ中、10%FBS(AusGene社)および5%ペニシリンストレプトマイシン(ナカライ社)を添加したRPMI-1640培地(GIBCO社)を用いて5%CO2インキュベーター内で培養した。
培養したRamos細胞を細胞密度7.5×106cells/mLになるように、血清を除いたRPM-1640培地(以後、培地)で希釈して、45分間37℃で保温した。細胞懸濁液を2.0mLチューブに1mLずつ小分けした後、培地で3μMになるように、被験化合物の1mM DMSO溶液を希釈した被験化合物溶液を500μL添加し、被験化合物の最終濃度が1μMの条件下で1時間37℃インキュベーションした。その後、培地で希釈したIgM(Invitrogen、 H15100)を最終濃度が10μg/mLになるように添加して、10分間37℃でインキュベーションした。
遠心操作により細胞を回収して得られたペレットにLysisバッファー[RIPA Buffer(×1)(Cell Signaling Technology社)に、1% Phosphatase inhibitor Cacktail 3(Sigma社、No.P0044)、1% Phosphatase inhibitor Cacktail (ナカライ社、No.07575)および1mM フッ化フェニルメチルスルホニル(PMSF)を添加したもの]を100μL添加し、軽く攪拌したのち10分間静置した。遠心操作(15,000rpm、15分間)により上清を回収し、タンパク質量を定量した。SDS-サンプルバッファーと混合し、95℃5分間反応させてタンパク質を変性させて、サンプル溶液とした。4-20%のグラジエントアクリルアミドゲル(コスモバイオ社、No.414879)の各ウェルにサンプル溶液を5μLずつアプライし、電気泳動を行った。その後、iBlotゲルトランスファーシステム(ライフテクノロジーズ社)を用いてPVDF膜にゲル中のタンパク質を転写した。
転写したPVDF膜を2%ECL prime blocking Reagent(GEヘルスケア社)でブロッキング処理した後、一次抗体として抗BTKマウス抗体(BDtransduction laboratory社、No.611116)もしくは抗リン酸化BTKウサギ抗体(pY223、EPITOMICS社、No.2207-1)を用い、4℃で1晩反応させた。未反応の一次抗体をTBSTバッファー(10mM Tris-HCl(pH7.5)、150mM NaCl、0.1% Tween20)で洗浄後、二次抗体としてHRPラベルした抗マウスIgGヤギ抗体(ライフテクノロジーズ社、No.62-6520)あるいは抗ウサギIgGヤギ抗体(ライフテクノロジーズ社、No.65-6120)を用い、2%ECL prime blocking Reagentを添加したTBSTバッファー中で、室温で1時間反応させた。未反応の二次抗体をTBSTバッファーで洗浄後、ECL Prime Western Blotting Detection System(GEヘルスケア社)を用いて添付のプロトコールどおりに反応させた後、CCDカメラ(ATTO、Light-CaptureII)を用いて、それぞれのバンドを化学発光で検出した。検出されたバンドをデンシトメトリー(ATTO CS Analyzer ver3.0)により数値化し、化合物非添加かつIgM刺激群のリン酸化BTKのバンドの発光を100%、化合物非添加かつIgM無刺激群のリン酸化BTKのバンドの発光を0%として、各群におけるバンドの強度から阻害率を算出した。なお、それぞれのリン酸化BTKのバンドは、総BTKにより補正を行なった。
本試験で用いた一次抗体と二次抗体の組み合わせおよび希釈濃度は以下の通りである。

細胞内BTKの自己リン酸化活性阻害試験2
以下のように被験化合物の調整および添加を行い、タンパク質の抽出、BTKまたはリン酸化BTKの検出は試験例3-1に従って実施し、各被験化合物の阻害率を算出した。
培養したRamos細胞を細胞密度7.5×106cells/mLになるように、血清を除いたRPM-1640培地(以後、培地)で希釈して、45分間37℃で保温した。細胞懸濁液を2.0mLチューブに1mLずつ小分けした後、培地で0.9μMになるように、被験化合物の0.3mM DMSO溶液を希釈した被験化合物溶液を500μL添加し、被験化合物の最終濃度が0.3μMの条件下で1時間37℃インキュベーションした。その後、培地で希釈したIgM(Invitrogen、 H15100)を最終濃度が10μg/mLになるように添加して、10分間37℃でインキュベーションした。
Claims (4)
- 下式(I)で示されるトリアジン誘導体またはその薬学的に許容される塩。
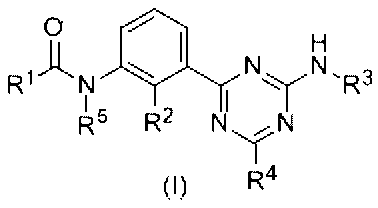
(式中、R1は、置換基を有してもよいアリール基、置換基を有してもよい複素環、置換基を有してもよい複素環式縮合環、置換基を有してもよいアルキニル基を表し、R2は、水素原子、ハロゲン原子、置換基を有してもよい低級アルキル基、置換基を有してもよいアルコキシ基を表し、R3は、置換基を有してもよいアリール基、置換基を有してもよい複素環、置換基を有してもよい複素環式縮合環を表し、R4は、水素原子、置換基を有してもよい低級アルキル基、置換基を有してもよいアルコキシ基、置換基を有してもよいアミノ基、ハロゲン原子を表し、R5は、水素原子、置換基を有してもよい低級アルキル基を表すか、あるいはR1と結合を形成し、置換基を有することもある飽和もしくは不飽和の、5ないし6員環を形成することによって、多環性縮合環を形成してもよい。) - R1が置換基を有してもよいアリール基である請求項1に記載のトリアジン誘導体またはその薬学的に許容される塩。
- R2が置換基を有してもよい低級アルキル基である請求項1に記載のトリアジン誘導体またはその薬学的に許容される塩。
- R5は、R1と結合を形成し、置換基を有することもある飽和もしくは不飽和の、5ないし6員環を形成することによって、多環性縮合環を形成する、請求項1に記載のトリアジン誘導体またはその薬学的に許容される塩。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/383,860 US20150011751A1 (en) | 2012-03-09 | 2013-03-07 | Novel triazine derivative |
| KR1020147025303A KR20140131955A (ko) | 2012-03-09 | 2013-03-07 | 신규 트리아진 유도체 |
| EP13758233.4A EP2824099A4 (en) | 2012-03-09 | 2013-03-07 | NOVEL TRIAZINE DERIVATIVE |
| IN1897MUN2014 IN2014MN01897A (ja) | 2012-03-09 | 2013-03-07 | |
| CN201380012641.2A CN104169260A (zh) | 2012-03-09 | 2013-03-07 | 新三嗪衍生物 |
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012-053543 | 2012-03-09 | ||
| JP2012053543 | 2012-03-09 | ||
| JP2012-100646 | 2012-04-26 | ||
| JP2012100646 | 2012-04-26 | ||
| JP2012147111 | 2012-06-29 | ||
| JP2012-147111 | 2012-06-29 | ||
| JP2012-280811 | 2012-12-25 | ||
| JP2012280811 | 2012-12-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013133367A1 true WO2013133367A1 (ja) | 2013-09-12 |
Family
ID=49116836
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/056266 WO2013133367A1 (ja) | 2012-03-09 | 2013-03-07 | 新規トリアジン誘導体 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20150011751A1 (ja) |
| EP (1) | EP2824099A4 (ja) |
| JP (1) | JPWO2013133367A1 (ja) |
| KR (1) | KR20140131955A (ja) |
| CN (1) | CN104169260A (ja) |
| IN (1) | IN2014MN01897A (ja) |
| WO (1) | WO2013133367A1 (ja) |
Cited By (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015006592A1 (en) * | 2013-07-11 | 2015-01-15 | Agios Pharmaceuticals, Inc. | N,6-bis(aryl or heteroaryl)-1,3,5-triazine-2,4-diamine compounds as idh2 mutants inhibitors for the treatment of cancer |
| WO2015012149A1 (ja) | 2013-07-26 | 2015-01-29 | カルナバイオサイエンス株式会社 | 新規トリアジン誘導体 |
| WO2015033888A1 (ja) * | 2013-09-03 | 2015-03-12 | カルナバイオサイエンス株式会社 | 新規2,6-ジアミノピリミジン誘導体 |
| WO2015041155A1 (ja) | 2013-09-20 | 2015-03-26 | カルナバイオサイエンス株式会社 | 新規トリアジン誘導体 |
| WO2015136398A1 (en) | 2014-03-12 | 2015-09-17 | Novartis Ag | Combination comprising a btk inhibitor and an akt inhibitor |
| US9434979B2 (en) | 2009-10-21 | 2016-09-06 | Shin-San Michael Su | Methods and compositions for cell-proliferation-related disorders |
| US9474779B2 (en) | 2012-01-19 | 2016-10-25 | Agios Pharmaceuticals, Inc. | Therapeutically active compositions and their methods of use |
| US9512107B2 (en) | 2012-01-06 | 2016-12-06 | Agios Pharmaceuticals, Inc. | Therapeutically active compositions and their methods of use |
| US9512084B2 (en) | 2013-11-29 | 2016-12-06 | Novartis Ag | Amino pyrimidine derivatives |
| US9579324B2 (en) | 2013-07-11 | 2017-02-28 | Agios Pharmaceuticals, Inc | Therapeutically active compounds and their methods of use |
| US9662327B2 (en) | 2011-06-17 | 2017-05-30 | Agios Pharmaceuticals, Inc | Phenyl and pyridinyl substituted piperidines and piperazines as inhibitors of IDH1 mutants and their use in treating cancer |
| US9738625B2 (en) | 2013-08-02 | 2017-08-22 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| US9751863B2 (en) | 2015-08-05 | 2017-09-05 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| US9856279B2 (en) | 2011-06-17 | 2018-01-02 | Agios Pharmaceuticals, Inc. | Therapeutically active compositions and their methods of use |
| US9968595B2 (en) | 2014-03-14 | 2018-05-15 | Agios Pharmaceuticals, Inc. | Pharmaceutical compositions of therapeutically active compounds |
| US9980961B2 (en) | 2011-05-03 | 2018-05-29 | Agios Pharmaceuticals, Inc. | Pyruvate kinase activators for use in therapy |
| WO2018097234A1 (ja) | 2016-11-25 | 2018-05-31 | カルナバイオサイエンス株式会社 | 新規オキソイソキノリン誘導体 |
| US10017495B2 (en) | 2013-07-11 | 2018-07-10 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| US10029987B2 (en) | 2009-06-29 | 2018-07-24 | Agios Pharmaceuticals, Inc. | Therapeutic compounds and compositions |
| US10202339B2 (en) | 2012-10-15 | 2019-02-12 | Agios Pharmaceuticals, Inc. | Therapeutic compounds and compositions |
| US10376510B2 (en) | 2013-07-11 | 2019-08-13 | Agios Pharmaceuticals, Inc. | 2,4- or 4,6-diaminopyrimidine compounds as IDH2 mutants inhibitors for the treatment of cancer |
| US10610125B2 (en) | 2009-03-13 | 2020-04-07 | Agios Pharmaceuticals, Inc. | Methods and compositions for cell-proliferation-related disorders |
| US10653710B2 (en) | 2015-10-15 | 2020-05-19 | Agios Pharmaceuticals, Inc. | Combination therapy for treating malignancies |
| US10689414B2 (en) | 2013-07-25 | 2020-06-23 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| US10980788B2 (en) | 2018-06-08 | 2021-04-20 | Agios Pharmaceuticals, Inc. | Therapy for treating malignancies |
| JP2021512158A (ja) * | 2018-01-25 | 2021-05-13 | レデックス・ファーマ・パブリック・リミテッド・カンパニーRedx Pharma PLC | Rho関連プロテインキナーゼのモジュレーター |
| JP2021523206A (ja) * | 2018-05-14 | 2021-09-02 | バイオジェン・エムエイ・インコーポレイテッドBiogen MA Inc. | ブルトン型チロシンキナーゼに対する阻害剤 |
| US11234976B2 (en) | 2015-06-11 | 2022-02-01 | Agios Pharmaceuticals, Inc. | Methods of using pyruvate kinase activators |
| US11419859B2 (en) | 2015-10-15 | 2022-08-23 | Servier Pharmaceuticals Llc | Combination therapy for treating malignancies |
| US11844758B2 (en) | 2013-07-11 | 2023-12-19 | Servier Pharmaceuticals Llc | Therapeutically active compounds and their methods of use |
| US11872192B2 (en) | 2018-04-03 | 2024-01-16 | Blueprint Medicines Corporation | RET inhibitor for use in treating cancer having a RET alteration |
Families Citing this family (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TW201718581A (zh) | 2015-10-19 | 2017-06-01 | 英塞特公司 | 作為免疫調節劑之雜環化合物 |
| LT3377488T (lt) | 2015-11-19 | 2023-01-10 | Incyte Corporation | Heterocikliniai junginiai, kaip imunomoduliatoriai |
| SI3394033T1 (sl) | 2015-12-22 | 2021-03-31 | Incyte Corporation | Heterociklične spojine kot imunomodulatorji |
| MA44860A (fr) | 2016-05-06 | 2019-03-13 | Incyte Holdings Corp | Composés hétérocycliques utilisés comme immunomodulateurs |
| WO2017205464A1 (en) | 2016-05-26 | 2017-11-30 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| EP4137489A1 (en) | 2016-06-20 | 2023-02-22 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| WO2018013789A1 (en) | 2016-07-14 | 2018-01-18 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| EP3504198B1 (en) | 2016-08-29 | 2023-01-25 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| ES2927104T3 (es) | 2016-09-09 | 2022-11-02 | Incyte Corp | Derivados de pirazolopiridina como moduladores de HPK1 y usos de los mismos para el tratamiento del cáncer |
| AR109595A1 (es) | 2016-09-09 | 2018-12-26 | Incyte Corp | Compuestos de pirazolopirimidina y usos de estos como inhibidores de hpk1 |
| WO2018049214A1 (en) | 2016-09-09 | 2018-03-15 | Incyte Corporation | Pyrazolopyridine derivatives as hpk1 modulators and uses thereof for the treatment of cancer |
| WO2018049191A1 (en) | 2016-09-09 | 2018-03-15 | Incyte Corporation | Pyrazolopyridone derivatives as hpk1 modulators and uses thereof for the treatment of cancer |
| TWI798192B (zh) | 2016-12-22 | 2023-04-11 | 美商英塞特公司 | 免疫調節劑化合物及使用方法 |
| ES2874756T3 (es) | 2016-12-22 | 2021-11-05 | Incyte Corp | Derivados de triazolo[1,5-A]piridina como inmunomoduladores |
| MX2019007416A (es) | 2016-12-22 | 2019-12-11 | Incyte Corp | Derivados de benzooxazol como inmunomoduladores. |
| US20180179179A1 (en) | 2016-12-22 | 2018-06-28 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| WO2018152220A1 (en) | 2017-02-15 | 2018-08-23 | Incyte Corporation | Pyrazolopyridine compounds and uses thereof |
| WO2018149284A1 (zh) * | 2017-02-16 | 2018-08-23 | 四川科伦博泰生物医药股份有限公司 | 激酶抑制剂及其制备方法和用途 |
| WO2019051199A1 (en) | 2017-09-08 | 2019-03-14 | Incyte Corporation | 6-CYANO-INDAZOLE COMPOUNDS AS HEMATOPOIETIC PROGENITOR KINASE 1 (HPK1) MODULATORS |
| US10745388B2 (en) | 2018-02-20 | 2020-08-18 | Incyte Corporation | Indazole compounds and uses thereof |
| SI3755703T1 (sl) | 2018-02-20 | 2022-11-30 | Incyte Corporation | N-(fenil)-2-(fenil)pirimidin-4-karboksamidni derivati in sorodne spojine kot zaviralci HPK1 za zravljenje raka |
| US10752635B2 (en) | 2018-02-20 | 2020-08-25 | Incyte Corporation | Indazole compounds and uses thereof |
| ES2940750T3 (es) | 2018-03-30 | 2023-05-11 | Incyte Corp | Compuestos heterocíclicos como inmunomoduladores |
| US11299473B2 (en) | 2018-04-13 | 2022-04-12 | Incyte Corporation | Benzimidazole and indole compounds and uses thereof |
| HRP20230306T1 (hr) | 2018-05-11 | 2023-05-12 | Incyte Corporation | Derivati tetrahidro-imidazo[4,5-c]piridina kao pd-l1 imunomodulatori |
| US10899755B2 (en) | 2018-08-08 | 2021-01-26 | Incyte Corporation | Benzothiazole compounds and uses thereof |
| MA53726A (fr) | 2018-09-25 | 2022-05-11 | Incyte Corp | Composés pyrazolo[4,3-d]pyrimidine en tant que modulateurs des alk2 et/ou fgfr |
| TW202114680A (zh) | 2019-08-06 | 2021-04-16 | 美商英塞特公司 | Hpk1抑制劑之固體形式 |
| CA3150434A1 (en) | 2019-08-09 | 2021-02-18 | Incyte Corporation | Salts of a pd-1/pd-l1 inhibitor |
| TW202126652A (zh) | 2019-09-30 | 2021-07-16 | 美商英塞特公司 | 作為免疫調節劑之吡啶并[3,2—d]嘧啶化合物 |
| KR20220101664A (ko) | 2019-11-11 | 2022-07-19 | 인사이트 코포레이션 | Pd-1/pd-l1 억제제의 염 및 결정질 형태 |
| US11866434B2 (en) | 2020-11-06 | 2024-01-09 | Incyte Corporation | Process for making a PD-1/PD-L1 inhibitor and salts and crystalline forms thereof |
| WO2022099018A1 (en) | 2020-11-06 | 2022-05-12 | Incyte Corporation | Process of preparing a pd-1/pd-l1 inhibitor |
| US11760756B2 (en) | 2020-11-06 | 2023-09-19 | Incyte Corporation | Crystalline form of a PD-1/PD-L1 inhibitor |
| CN113004246B (zh) * | 2021-02-22 | 2022-02-01 | 广西医科大学 | 1,3,5-三嗪-2-胺-4,6取代衍生物或其药学上可接受的盐和用途 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008076883A2 (en) * | 2006-12-15 | 2008-06-26 | Abraxis Bioscience, Inc. | Triazine derivatives and their therapeutical applications |
| WO2008121742A2 (en) * | 2007-03-28 | 2008-10-09 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| WO2009091388A2 (en) * | 2007-12-21 | 2009-07-23 | Progenics Pharmaceuticals, Inc. | Triazines and related compounds having antiviral activity, compositions and methods thereof |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2011529073A (ja) * | 2008-07-24 | 2011-12-01 | ブリストル−マイヤーズ スクイブ カンパニー | キナーゼ調節因子として有用な縮合ヘテロ環化合物 |
-
2013
- 2013-03-07 US US14/383,860 patent/US20150011751A1/en not_active Abandoned
- 2013-03-07 CN CN201380012641.2A patent/CN104169260A/zh active Pending
- 2013-03-07 JP JP2014503539A patent/JPWO2013133367A1/ja active Pending
- 2013-03-07 WO PCT/JP2013/056266 patent/WO2013133367A1/ja active Application Filing
- 2013-03-07 EP EP13758233.4A patent/EP2824099A4/en not_active Withdrawn
- 2013-03-07 KR KR1020147025303A patent/KR20140131955A/ko not_active Application Discontinuation
- 2013-03-07 IN IN1897MUN2014 patent/IN2014MN01897A/en unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008076883A2 (en) * | 2006-12-15 | 2008-06-26 | Abraxis Bioscience, Inc. | Triazine derivatives and their therapeutical applications |
| WO2008121742A2 (en) * | 2007-03-28 | 2008-10-09 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| WO2009091388A2 (en) * | 2007-12-21 | 2009-07-23 | Progenics Pharmaceuticals, Inc. | Triazines and related compounds having antiviral activity, compositions and methods thereof |
Non-Patent Citations (11)
| Title |
|---|
| DAVIS R. E. ET AL., NATURE, vol. 463, 2010, pages 88 - 92 |
| ELLMEIER W. ET AL., FEBS J., vol. 278, 2011, pages 1990 - 2000 |
| HALCOMB K. E., MOL. IMMUNOL., vol. 46, no. 2, 2008, pages 233 - 241 |
| JANSSON L.; HOLMDAHL R., CLIN. EXP. IMMUNOL., vol. 94, 1993, pages 459 - 465 |
| KUROSAKI T., CURR. OPIN. IMMUNOL., vol. 12, 2000, pages 276 - 281 |
| N. MIYAURA ET AL., AM. CHEM. SOC., vol. 107, 1985, pages 972 |
| N. MIYAURA; A. SUZUKI, CHEM. REV., vol. 95, 1995, pages 2457 |
| SATTERTHWAITE, A. B.; WITTE, O. N., IMMUNOL. REV., vol. 175, 2000, pages 120 - 127 |
| See also references of EP2824099A4 * |
| T. W. GREENE: "Protective Groups in Organic Synthesis", 1999, JOHN WILEY & SONS, INC. |
| T. W. GREENE: "Protective Groups in Organic Synthesis", 1999, JOHN WILEY&SONS, INC. |
Cited By (73)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10610125B2 (en) | 2009-03-13 | 2020-04-07 | Agios Pharmaceuticals, Inc. | Methods and compositions for cell-proliferation-related disorders |
| US11866411B2 (en) | 2009-06-29 | 2024-01-09 | Agios Pharmaceutical, Inc. | Therapeutic compounds and compositions |
| USRE49582E1 (en) | 2009-06-29 | 2023-07-18 | Agios Pharmaceuticals, Inc. | Therapeutic compounds and compositions |
| US10029987B2 (en) | 2009-06-29 | 2018-07-24 | Agios Pharmaceuticals, Inc. | Therapeutic compounds and compositions |
| US10988448B2 (en) | 2009-06-29 | 2021-04-27 | Agios Pharmaceuticals, Inc. | Therapeutic compounds and compositions |
| US9434979B2 (en) | 2009-10-21 | 2016-09-06 | Shin-San Michael Su | Methods and compositions for cell-proliferation-related disorders |
| US10711314B2 (en) | 2009-10-21 | 2020-07-14 | Agios Pharmaceuticals, Inc. | Methods for diagnosing IDH-mutant cell proliferation disorders |
| US9982309B2 (en) | 2009-10-21 | 2018-05-29 | Agios Pharmaceuticals, Inc. | Method for treating cell proliferation related disorders |
| US9980961B2 (en) | 2011-05-03 | 2018-05-29 | Agios Pharmaceuticals, Inc. | Pyruvate kinase activators for use in therapy |
| US10632114B2 (en) | 2011-05-03 | 2020-04-28 | Agios Pharmaceuticals, Inc. | Pyruvate kinase activators for use in therapy |
| US11793806B2 (en) | 2011-05-03 | 2023-10-24 | Agios Pharmaceuticals, Inc. | Pyruvate kinase activators for use in therapy |
| US9856279B2 (en) | 2011-06-17 | 2018-01-02 | Agios Pharmaceuticals, Inc. | Therapeutically active compositions and their methods of use |
| US9662327B2 (en) | 2011-06-17 | 2017-05-30 | Agios Pharmaceuticals, Inc | Phenyl and pyridinyl substituted piperidines and piperazines as inhibitors of IDH1 mutants and their use in treating cancer |
| US11505538B1 (en) | 2012-01-06 | 2022-11-22 | Servier Pharmaceuticals Llc | Therapeutically active compounds and their methods of use |
| US9512107B2 (en) | 2012-01-06 | 2016-12-06 | Agios Pharmaceuticals, Inc. | Therapeutically active compositions and their methods of use |
| US9732062B2 (en) | 2012-01-06 | 2017-08-15 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| US10294215B2 (en) | 2012-01-06 | 2019-05-21 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| US9656999B2 (en) | 2012-01-06 | 2017-05-23 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| US10640534B2 (en) | 2012-01-19 | 2020-05-05 | Agios Pharmaceuticals, Inc. | Therapeutically active compositions and their methods of use |
| US9474779B2 (en) | 2012-01-19 | 2016-10-25 | Agios Pharmaceuticals, Inc. | Therapeutically active compositions and their methods of use |
| US10717764B2 (en) | 2012-01-19 | 2020-07-21 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| US9850277B2 (en) | 2012-01-19 | 2017-12-26 | Agios Pharmaceuticals, Inc. | Therapeutically active compositions and their methods of use |
| US11667673B2 (en) | 2012-01-19 | 2023-06-06 | Servier Pharmaceuticals Llc | Therapeutically active compounds and their methods of use |
| US10202339B2 (en) | 2012-10-15 | 2019-02-12 | Agios Pharmaceuticals, Inc. | Therapeutic compounds and compositions |
| EA032070B1 (ru) * | 2013-07-11 | 2019-04-30 | Аджиос Фармасьютикалз, Инк. | Соединения n,6-бис(арил или гетероарил)-1,3,5-триазин-2,4-диамина в качестве ингибиторов мутантов idh2 для лечения ракового заболевания |
| US10172864B2 (en) | 2013-07-11 | 2019-01-08 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| JP2016523977A (ja) * | 2013-07-11 | 2016-08-12 | アギオス ファーマシューティカルス,インコーポレーテッド | 癌の治療のためのidh2変異体阻害剤としてのn,6−ビス(アリール又はヘテロアリール)−1,3,5−トリアジン−2,4−ジアミン化合物 |
| US11844758B2 (en) | 2013-07-11 | 2023-12-19 | Servier Pharmaceuticals Llc | Therapeutically active compounds and their methods of use |
| US10017495B2 (en) | 2013-07-11 | 2018-07-10 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| US10028961B2 (en) | 2013-07-11 | 2018-07-24 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| US10946023B2 (en) | 2013-07-11 | 2021-03-16 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| US10376510B2 (en) | 2013-07-11 | 2019-08-13 | Agios Pharmaceuticals, Inc. | 2,4- or 4,6-diaminopyrimidine compounds as IDH2 mutants inhibitors for the treatment of cancer |
| US10111878B2 (en) | 2013-07-11 | 2018-10-30 | Agios Pharmaceuticals, Inc. | N,6-bis(aryl or heteroaryl)-1,3,5-triazine-2,4-diamine compounds as IDH2 mutants inhibitors for the treatment of cancer |
| US9579324B2 (en) | 2013-07-11 | 2017-02-28 | Agios Pharmaceuticals, Inc | Therapeutically active compounds and their methods of use |
| US9724350B2 (en) | 2013-07-11 | 2017-08-08 | Agios Pharmaceuticals, Inc. | N,6-bis(aryl or heteroaryl)-1,3,5-triazine-2,4-diamine compounds as IDH2 mutants inhibitors for the treatment of cancer |
| WO2015006592A1 (en) * | 2013-07-11 | 2015-01-15 | Agios Pharmaceuticals, Inc. | N,6-bis(aryl or heteroaryl)-1,3,5-triazine-2,4-diamine compounds as idh2 mutants inhibitors for the treatment of cancer |
| US11021515B2 (en) | 2013-07-25 | 2021-06-01 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| US10689414B2 (en) | 2013-07-25 | 2020-06-23 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| CN105683178B (zh) * | 2013-07-26 | 2019-04-12 | 卡尔那生物科学株式会社 | 三嗪衍生物 |
| US9656995B2 (en) | 2013-07-26 | 2017-05-23 | Carna Biosciences, Inc. | Triazine derivative |
| CN105683178A (zh) * | 2013-07-26 | 2016-06-15 | 卡尔那生物科学株式会社 | 新型三嗪衍生物 |
| WO2015012149A1 (ja) | 2013-07-26 | 2015-01-29 | カルナバイオサイエンス株式会社 | 新規トリアジン誘導体 |
| JPWO2015012149A1 (ja) * | 2013-07-26 | 2017-03-02 | カルナバイオサイエンス株式会社 | 新規トリアジン誘導体 |
| US10730854B2 (en) | 2013-08-02 | 2020-08-04 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| US9738625B2 (en) | 2013-08-02 | 2017-08-22 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| US10093654B2 (en) | 2013-08-02 | 2018-10-09 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| WO2015033888A1 (ja) * | 2013-09-03 | 2015-03-12 | カルナバイオサイエンス株式会社 | 新規2,6-ジアミノピリミジン誘導体 |
| CN105745203A (zh) * | 2013-09-20 | 2016-07-06 | 卡尔那生物科学株式会社 | 新三嗪衍生物 |
| WO2015041155A1 (ja) | 2013-09-20 | 2015-03-26 | カルナバイオサイエンス株式会社 | 新規トリアジン誘導体 |
| JPWO2015041155A1 (ja) * | 2013-09-20 | 2017-03-02 | カルナバイオサイエンス株式会社 | 新規トリアジン誘導体 |
| US11180460B2 (en) | 2013-11-29 | 2021-11-23 | Novartis Ag | Amino pyrimidine derivatives |
| US9512084B2 (en) | 2013-11-29 | 2016-12-06 | Novartis Ag | Amino pyrimidine derivatives |
| US10457647B2 (en) | 2013-11-29 | 2019-10-29 | Novartis Ag | Amino pyrimidine derivatives |
| US11673868B2 (en) | 2013-11-29 | 2023-06-13 | Novartis Ag | Amino pyrimidine derivatives |
| WO2015136398A1 (en) | 2014-03-12 | 2015-09-17 | Novartis Ag | Combination comprising a btk inhibitor and an akt inhibitor |
| US11504361B2 (en) | 2014-03-14 | 2022-11-22 | Servier Pharmaceuticals Llc | Pharmaceutical compositions of therapeutically active compounds |
| US10449184B2 (en) | 2014-03-14 | 2019-10-22 | Agios Pharmaceuticals, Inc. | Pharmaceutical compositions of therapeutically active compounds |
| US10799490B2 (en) | 2014-03-14 | 2020-10-13 | Agios Pharmaceuticals, Inc. | Pharmaceutical compositions of therapeutically active compounds |
| US9968595B2 (en) | 2014-03-14 | 2018-05-15 | Agios Pharmaceuticals, Inc. | Pharmaceutical compositions of therapeutically active compounds |
| US11234976B2 (en) | 2015-06-11 | 2022-02-01 | Agios Pharmaceuticals, Inc. | Methods of using pyruvate kinase activators |
| US9751863B2 (en) | 2015-08-05 | 2017-09-05 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| US10653710B2 (en) | 2015-10-15 | 2020-05-19 | Agios Pharmaceuticals, Inc. | Combination therapy for treating malignancies |
| US11419859B2 (en) | 2015-10-15 | 2022-08-23 | Servier Pharmaceuticals Llc | Combination therapy for treating malignancies |
| WO2018097234A1 (ja) | 2016-11-25 | 2018-05-31 | カルナバイオサイエンス株式会社 | 新規オキソイソキノリン誘導体 |
| KR20190104142A (ko) | 2016-11-25 | 2019-09-06 | 카나 바이오사이언스, 인코포레이션 | 신규 옥소이소퀴놀린 유도체 |
| US10793575B2 (en) | 2016-11-25 | 2020-10-06 | Carna Biosciences, Inc. | Oxoisoquinoline derivatives |
| JP2021512158A (ja) * | 2018-01-25 | 2021-05-13 | レデックス・ファーマ・パブリック・リミテッド・カンパニーRedx Pharma PLC | Rho関連プロテインキナーゼのモジュレーター |
| JP7518324B2 (ja) | 2018-01-25 | 2024-07-18 | レデックス・ファーマ・リミテッド | Rho関連プロテインキナーゼのモジュレーター |
| US11872192B2 (en) | 2018-04-03 | 2024-01-16 | Blueprint Medicines Corporation | RET inhibitor for use in treating cancer having a RET alteration |
| US11963958B2 (en) | 2018-04-03 | 2024-04-23 | Rigel Pharmaceuticals, Inc. | RET inhibitor for use in treating cancer having a RET alteration |
| JP7359782B2 (ja) | 2018-05-14 | 2023-10-11 | バイオジェン・エムエイ・インコーポレイテッド | ブルトン型チロシンキナーゼに対する阻害剤 |
| JP2021523206A (ja) * | 2018-05-14 | 2021-09-02 | バイオジェン・エムエイ・インコーポレイテッドBiogen MA Inc. | ブルトン型チロシンキナーゼに対する阻害剤 |
| US10980788B2 (en) | 2018-06-08 | 2021-04-20 | Agios Pharmaceuticals, Inc. | Therapy for treating malignancies |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2824099A4 (en) | 2015-11-11 |
| JPWO2013133367A1 (ja) | 2015-07-30 |
| IN2014MN01897A (ja) | 2015-07-10 |
| KR20140131955A (ko) | 2014-11-14 |
| EP2824099A1 (en) | 2015-01-14 |
| CN104169260A (zh) | 2014-11-26 |
| US20150011751A1 (en) | 2015-01-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2013133367A1 (ja) | 新規トリアジン誘導体 | |
| EP3889134B1 (en) | Compound for inhibiting pge2/ep4 signaling transduction inhibiting, preparation method therefor, and medical uses thereof | |
| JP6986032B2 (ja) | Jak阻害剤としてのピロロピリミジン化合物の結晶 | |
| KR20130062951A (ko) | 키누레닌 생성 억제 작용을 갖는 함질소 복소환 화합물 | |
| JP7015060B2 (ja) | 新規オキソイソキノリン誘導体 | |
| CN110156656B (zh) | 五元杂芳环衍生物、其制备方法、药物组合物及应用 | |
| JP2021525247A (ja) | 複素縮合ピリドン化合物及びidh阻害剤としてのこれらの使用 | |
| KR20140086002A (ko) | Fms 키나아제 저해 활성을 갖는 피리다진 접합고리 유도체 | |
| CN116438174A (zh) | 化合物及其作为mif抑制剂的用途 | |
| JP6156953B2 (ja) | 新規トリアジン誘導体 | |
| WO2015033888A1 (ja) | 新規2,6-ジアミノピリミジン誘導体 | |
| WO2013161848A1 (ja) | 新規1,2,4-トリアジン誘導体 | |
| CN113061098B (zh) | 酰胺化合物及其衍生物,制备方法、药物组合物和应用 | |
| WO2022208382A1 (ko) | 신규한 디알콕시나프토[2,3-c]퓨란-1(3h)-온 유도체 및 이를 포함하는 호흡기 질환 또는 sars-cov-2 감염 질환의 예방 또는 치료용 약제학적 조성물 | |
| CN110467629B (zh) | 苯醌衍生物、其药物组合物及应用 | |
| CN109956929B (zh) | 杂环衍生物、其制备方法、药物组合物及应用 | |
| WO2015041155A1 (ja) | 新規トリアジン誘導体 | |
| WO2011093365A1 (ja) | 含窒素複素環化合物 | |
| KR20090018612A (ko) | 단백질 키나제 억제제로서의 치환된 3-시아노피리딘 | |
| CN110066271B (zh) | 吡咯衍生物、其制备方法、药物组合物及应用 | |
| CN115960117B (zh) | 含硫并环类衍生物抑制剂、其制备方法和应用 | |
| WO2022067063A1 (en) | Mutant selective egfr inhibitors and methods of use thereof | |
| CN112209934A (zh) | 含有氮杂螺庚烷的btk抑制剂 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13758233 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2013758233 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013758233 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2014503539 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20147025303 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14383860 Country of ref document: US |