WO2012036152A1 - 高純度ヘパリンおよびその製造方法 - Google Patents
高純度ヘパリンおよびその製造方法 Download PDFInfo
- Publication number
- WO2012036152A1 WO2012036152A1 PCT/JP2011/070851 JP2011070851W WO2012036152A1 WO 2012036152 A1 WO2012036152 A1 WO 2012036152A1 JP 2011070851 W JP2011070851 W JP 2011070851W WO 2012036152 A1 WO2012036152 A1 WO 2012036152A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- heparin
- ethanol
- nitrite
- mucopolysaccharide
- volume
- Prior art date
Links
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B37/00—Preparation of polysaccharides not provided for in groups C08B1/00 - C08B35/00; Derivatives thereof
- C08B37/006—Heteroglycans, i.e. polysaccharides having more than one sugar residue in the main chain in either alternating or less regular sequence; Gellans; Succinoglycans; Arabinogalactans; Tragacanth or gum tragacanth or traganth from Astragalus; Gum Karaya from Sterculia urens; Gum Ghatti from Anogeissus latifolia; Derivatives thereof
- C08B37/0063—Glycosaminoglycans or mucopolysaccharides, e.g. keratan sulfate; Derivatives thereof, e.g. fucoidan
- C08B37/0075—Heparin; Heparan sulfate; Derivatives thereof, e.g. heparosan; Purification or extraction methods thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B37/00—Preparation of polysaccharides not provided for in groups C08B1/00 - C08B35/00; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/715—Polysaccharides, i.e. having more than five saccharide radicals attached to each other by glycosidic linkages; Derivatives thereof, e.g. ethers, esters
- A61K31/726—Glycosaminoglycans, i.e. mucopolysaccharides
- A61K31/727—Heparin; Heparan
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B37/00—Preparation of polysaccharides not provided for in groups C08B1/00 - C08B35/00; Derivatives thereof
- C08B37/0003—General processes for their isolation or fractionation, e.g. purification or extraction from biomass
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B37/00—Preparation of polysaccharides not provided for in groups C08B1/00 - C08B35/00; Derivatives thereof
- C08B37/006—Heteroglycans, i.e. polysaccharides having more than one sugar residue in the main chain in either alternating or less regular sequence; Gellans; Succinoglycans; Arabinogalactans; Tragacanth or gum tragacanth or traganth from Astragalus; Gum Karaya from Sterculia urens; Gum Ghatti from Anogeissus latifolia; Derivatives thereof
- C08B37/0063—Glycosaminoglycans or mucopolysaccharides, e.g. keratan sulfate; Derivatives thereof, e.g. fucoidan
- C08B37/0075—Heparin; Heparan sulfate; Derivatives thereof, e.g. heparosan; Purification or extraction methods thereof
- C08B37/0078—Degradation products
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N30/00—Investigating or analysing materials by separation into components using adsorption, absorption or similar phenomena or using ion-exchange, e.g. chromatography or field flow fractionation
- G01N30/02—Column chromatography
- G01N30/04—Preparation or injection of sample to be analysed
- G01N30/06—Preparation
- G01N30/14—Preparation by elimination of some components
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N30/00—Investigating or analysing materials by separation into components using adsorption, absorption or similar phenomena or using ion-exchange, e.g. chromatography or field flow fractionation
- G01N30/02—Column chromatography
- G01N30/88—Integrated analysis systems specially adapted therefor, not covered by a single one of the groups G01N30/04 - G01N30/86
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T436/00—Chemistry: analytical and immunological testing
- Y10T436/14—Heterocyclic carbon compound [i.e., O, S, N, Se, Te, as only ring hetero atom]
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T436/00—Chemistry: analytical and immunological testing
- Y10T436/14—Heterocyclic carbon compound [i.e., O, S, N, Se, Te, as only ring hetero atom]
- Y10T436/142222—Hetero-O [e.g., ascorbic acid, etc.]
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T436/00—Chemistry: analytical and immunological testing
- Y10T436/14—Heterocyclic carbon compound [i.e., O, S, N, Se, Te, as only ring hetero atom]
- Y10T436/142222—Hetero-O [e.g., ascorbic acid, etc.]
- Y10T436/143333—Saccharide [e.g., DNA, etc.]
Definitions
- the present invention relates to high-purity heparin that is substantially free of a causative agent of a side effect, is highly safe, and is useful as a pharmaceutical, a cosmetic, a research reagent, and the like, and a method for producing the same.
- Heparin is an acidic mucopolysaccharide present in the liver, small intestine, lungs, skin and the like, including sulfated D-glucosamine, D-glucuronic acid, L-iduronic acid and the like. Heparin has strong blood anticoagulant activity, treatment of generalized intravascular blood coagulation syndrome (DIC), various thromboembolism (venous thrombosis, myocardial infarction, pulmonary embolism, cerebral embolism, In addition to the treatment and prevention of limb arterial thromboembolism, intraoperative and postoperative thromboembolism, etc., blood during use of extracorporeal circulation devices such as hemodialysis and cardiopulmonary bypass, blood vessel catheter insertion, blood transfusion and blood tests, etc.
- DIC generalized intravascular blood coagulation syndrome
- various thromboembolism venous thrombosis, myocardial infarction, pulmonary embolism, cerebral embolism
- extracorporeal circulation devices such as hemodia
- heparin Used to prevent coagulation.
- heparin has lipoprotein lipase activation, antiplatelet aggregation, blood pressure lowering, anticomplement, cancer metastasis, degranulation inhibition from mast cells, local inflammation It is also known to have many physiological activities such as suppression, analgesia, and blood circulation promoting action of muscle tissue.
- Heparin is mainly produced by extraction / fractionation from the tissues of healthy edible animals, but since the BSE (bovine spongiform encephalopathy) problem, the origin of heparin as a pharmaceutical is almost derived from porcine small intestinal mucosa.
- porcine small intestinal mucosa is suspended in an aqueous solvent, digested with protein, and then adsorbent is added (Non-patent Document 1) to combine heparin and other mucopolysaccharides (mainly chondroitin sulfate family, heparan sulfate, etc.) Extract as a body to make a crude material. This is then batch mixed / fractionated to obtain heparin (so-called “unfractionated heparin”).
- Heparin (unfractionated heparin) obtained by the above method contains mucopolysaccharides (mainly heparan sulfate, chondroitin sulfate B and C) other than heparin, and it is known that the content varies depending on the raw material and the production method. It is. However, the approximate side effects of the impurities have been identified and tolerated, so that unfractionated heparin has long been used as a pharmaceutical.
- mucopolysaccharides mainly heparan sulfate, chondroitin sulfate B and C
- Non-Patent Documents 2 to 4 This is a substance that does not exist in nature, and is said to have a high possibility of being mixed during the manufacture of the drug substance.
- Non-Patent Document 5 In Japan, OSCS is identified as a causative substance of an adverse event, and confirmation / purity tests are being conducted by the government and pharmaceutical manufacturers (Non-patent Documents 7 and 8).
- Patent Document 1 As a method for producing or purifying heparin, methods described in 1) Non-Patent Documents 1, 2) Non-Patent Document 9, 3) Non-Patent Document 10, 4) Patent Document 1, etc. are known. However, a method that can easily and effectively remove impurities such as OSCS and chondroitin sulfate from heparin has not been known so far. Furthermore, there is no known method for easily detecting or measuring such impurities in heparin.
- An object of the present invention is to provide high-purity heparin that is substantially free of impurities such as OSCS and chondroitin sulfate and has high safety, a method for producing the same, and a method for confirming the purity of heparin in the production process.
- the present inventors fractionated heparin using an organic solvent such as ethanol under predetermined conditions, thereby easily and effectively removing impurities such as OSCS and chondroitin sulfate. Furthermore, since these impurities are resistant to nitrous acid decomposition under given conditions, the presence and amount of these impurities can be determined by HPLC analysis after the nitrous acid decomposition. The inventors have found that it can be confirmed and measured, and have completed the present invention. That is, the present invention includes the following: [1] Heparin substantially free of nitrite degradation resistant impurities.
- a 0.2 to 1 volume (volume) of an organic solvent selected from ethanol, methanol, isopropanol, acetone and a mixed solvent thereof is mixed with a 5 to 30% by weight heparin aqueous solution.
- the heparin according to [3], wherein the salt is selected from sodium chloride and sodium acetate.
- heparin according to any one of the above [1] to [5], which has a molecular weight in the range of 3000 to 30000 daltons.
- a 5 to 30% by weight heparin aqueous solution is mixed with 0.2 to 1 volume (volume) of an organic solvent selected from ethanol, methanol, isopropanol, acetone, and a mixed solvent thereof.
- a method for producing heparin comprising obtaining a colloidal precipitate.
- An organic solvent selected from 0.2 to 1 volume (volume) of ethanol, methanol, isopropanol, acetone and a mixed solvent thereof is mixed with 5 to 30% by weight of heparin aqueous solution, and heparin is added.
- a 0.2 to 1 volume (volume) of an organic solvent selected from ethanol, methanol, isopropanol, acetone and a mixed solvent thereof is mixed with 5 to 30% by weight of heparin aqueous solution, A method for producing heparin, comprising obtaining a supernatant liquid.
- a medicament comprising the heparin according to any one of [1] to [6] and [8] to [11].
- a pharmaceutical composition comprising the heparin according to any one of [1] to [6] and [8] to [11].
- a method for detecting or measuring a nitrite-resistant mucopolysaccharide or a nitrite-decomposable mucopolysaccharide contained in the mucopolysaccharide which comprises decomposing the mucopolysaccharide into nitrite.
- the high-purity heparin of the present invention does not contain OSCS or the like which is a causative agent of side effects, it has high safety and can be used very suitably as a pharmaceutical product, cosmetics, research reagent or the like.
- the method for producing heparin of the present invention high-purity heparin substantially free of nitrite degradation resistant impurities can be obtained easily.
- the said method can be utilized industrially.
- the mucopolysaccharide detection or measurement method of the present invention the presence or absence of other mucopolysaccharides having different characteristics against nitrite degradation can be easily confirmed in the mucopolysaccharide product. Safety and the like can be ensured.
- the process of producing the target mucopolysaccharide it is possible to easily confirm the contamination of other mucopolysaccharides that exhibit different properties against nitrite degradation, so that the production process of the target mucopolysaccharide can be effectively performed. It can be managed and contamination of other mucopolysaccharides in the intermediate material and the final product can be avoided.
- FIG. 1 shows the ethanol content of unfractionated heparin (Na salt, UFN-SP) containing other mucopolysaccharides (mainly heparan sulfate (HS) / chondroitin sulfate B (CSB) / chondroitin sulfate C (CSC)).
- HS heparan sulfate
- CSB chondroitin sulfate B
- CSC chondroitin sulfate C
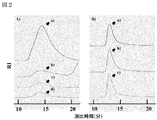
- FIG. 2 shows an OSCS control substance by nitrous acid decomposition, that is, OSCS-containing unfractionated heparin (Na salt, OSHP-SH, OSCS content about 12.5%), OSCS standard product (OSCS-STD), OSCS and chondroitin sulfate
- OSCS-STD OSCS standard product
- OSCS chondroitin sulfate
- OSHP-SH and OSCS-STD are as follows: A) OSHP-SH and OSCS-STD a) OSHP-SH before nitrous acid decomposition (solid line) b) OSHP-SH after nitrous acid decomposition (dashed line) c) OSCS-STD before nitrous acid decomposition (dotted line) d) OSCS-STD after nitrous acid decomposition (one-dot broken line) B) CSMS-CE1 and -CE2 a) CSMS-CE1 after nitrous acid decomposition (solid line) b) Nitrous acid CSMS-CE2 (broken line) after disassembly c) CSB-STD (dotted line) FIG.
- FIG. 3 is a HPLC chart showing the molecular weight changes of mucopolysaccharide control substances, ie, chondroitin sulfate family (CSA, CSB, CSC, CSD and CSE), heparan sulfate (HS) and keratan sulfate (KS) due to nitrite degradation. is there.
- CSA chondroitin sulfate family
- CSC heparan sulfate
- KS keratan sulfate
- FIG. 4 is a HPLC chart showing the molecular weight distribution of each unfractionated heparin Na salt (UFN 1-5) and Ca salt (UFC) before nitrous acid decomposition before ethanol fractionation.
- UFN 1-5 unfractionated heparin Na salt
- UAC Ca salt
- FIG. 5 is a HPLC chart showing the molecular weight distribution after nitrous acid decomposition of Na salt (UFN 1 to 5) and Ca salt (UFC) of each unfractionated heparin before ethanol fractionation. Each symbol in FIG.
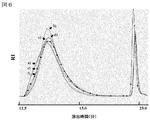
- FIG. 6 is a HPLC chart showing the molecular weight distribution of each unfractionated heparin Na salt (UFN 1-5) and Ca salt (UFC) before ethanol decomposition after ethanol fractionation. Each symbol in FIG.
- FIG. 7 is a HPLC chart showing the molecular weight distribution after nitrous acid decomposition of Na salt (UFN 1 to 5) and Ca salt (UFC) of each unfractionated heparin after ethanol fractionation. Each symbol in FIG.
- UFN7 is as follows: a) UFN1 (solid line), b) UFN2 (dashed line), c) UFN3 (dotted line), d) UFN4 (one-dot chain line), e) UFN5 (two-dot chain line) , F) UFC ( ⁇ solid line)
- the high-purity heparin of the present invention is characterized by being substantially free of nitrite degradation resistance impurities.
- the “heparin” in the present invention is not particularly limited and may be obtained from conventionally known raw materials and production methods. For example, so-called “unfractionated heparin”, “low molecular weight heparin”, and “heparin” Among “heparan sulfate” having a similar constituent sugar composition and binding mode, “heparan sulfate” having a high molecular weight or a high sulfuric acid content is particularly mentioned.
- unfractionated heparin means heparin that has not been depolymerized and usually has a molecular weight in the range of 3000 to 30000 daltons.
- the “low molecular weight heparin” is obtained by depolymerizing unfractionated heparin to reduce the molecular weight, and usually has a molecular weight in the range of 1500 to 12,000 daltons.
- the “heparan sulfate” usually has a molecular weight in the range of 3000 to 30000 daltons.
- the heparin in the present invention generally exhibits a physiological activity or pharmacological activity substantially the same as the free form in vivo, for example, heparin derivatives and pharmaceutically acceptable salts, addition salts, hydration Things etc. are included in the technical scope of the present invention.
- the molecular weight here is a weight average molecular weight determined by an HPLC method by size exclusion gel chromatography using an aqueous solvent.
- Nirite decomposition in the present invention may be any nitrous acid decomposition treatment under conditions that substantially decompose heparin and do not decompose impurities such as OSCS and chondroitin sulfate described later. pH 1.0 to 7.0 (preferably pH 2.0 to 5.0), reaction temperature ⁇ 10 to 40 ° C. (preferably ⁇ 5 to 10 ° C.), reaction time 0.5 to 60 minutes (preferably 5 to 15) Min), the amount of nitrous acid (especially sodium nitrite) used per 1 g of the target substance (heparin, etc.), such as 10 to 1000 mg (preferably 50 to 100 mg), etc. It means nitrous acid decomposition treatment.
- nitrite degradation resistant impurity means an impurity in heparin that is resistant to the above nitrous acid degradation.
- heparin is subjected to HPLC (for example, under conditions described later) after the above nitrous acid degradation.
- HPLC for example, under conditions described later
- it means a substance eluted at a position corresponding to the elution position of heparin before nitrite decomposition.
- chondroitin sulfate examples include persulfated chondroitin sulfate (OSCS), chondroitin sulfate A (chondroitin-4-sulfate: CSA), B (dermatan sulfate: CSB), C (chondroitin-6-sulfate: CSC), Disaccharide unit structure of galactosamine and uronic acid (glucuronic acid or iduronic acid) in mucopolysaccharides such as D (chondroitin-2,6-sulfate: CSD), E (chondroitin-4,6-sulfate: CSE), etc. And keratan sulfate (KC) having a disaccharide unit structure of glucosamine and galactose in the basic skeleton.
- OSCS persulfated chondroitin sulfate
- A chondroitin-4-sulfate: CSA
- B dermatan sulfate: CSB
- C chon
- the target heparin is “molecular weight” of the “parnaparin sodium” standard test method described in the 15th Amendment Manual of the Japanese Pharmacy Law. Detected by the refractive index (RI) appearing at the elution position of heparin obtained when analyzed by high performance liquid chromatography (HPLC) with reference to the conditions (for example, in the following specific examples, the elution time is 10 to 20 minutes)
- RI refractive index
- HPLC high performance liquid chromatography
- the total area value of the peak appearing at the elution position of heparin is 5% or less, preferably It means 1% or less, more preferably 0.5% or less.
- HPLC conditions include the following conditions: Detection system: SHIMADZU management system (LC solution), differential refractometer (RI: RID-10A) Column and guard column: TSK gel G-2000SWXL and TSK guard column SWXL manufactured by TOSO Column temperature: 40 ° C Mobile phase: 0.2 mol / L Na sulfate (pH 5.0) Flow rate: 0.5 mL / min.
- the high-purity heparin of the present invention can be produced, for example, by a method including fractionating raw material heparin containing nitrite degradation-resistant impurities and the like using an organic solvent such as ethanol under predetermined conditions.
- organic solvent such as ethanol
- examples of the organic solvent used for the fractionation include ethanol, methanol, isopropanol, acetone, and mixtures thereof. Among these, ethanol is most preferable in consideration of the residue in the final product.
- the ethanol fraction is 0.2 to 1 times (preferably 0.4 to 0.6 times) of 5 to 30% by weight (preferably 10 to 20% by weight) heparin aqueous solution ( Volumetric ethanol is mixed to obtain a colloidal precipitate of heparin.
- This method is simple and industrially applicable.
- ethanol precipitation is used to obtain heparin as a white precipitate.
- the heparin concentration in the heparin aqueous solution to which the conventional ethanol precipitation method is applied is 1 to 5% by weight, which is significantly lower than the ethanol fraction in the present invention.
- the amount of ethanol mixed in the heparin aqueous solution is 2 to 10 times (volume), which is significantly larger than the ethanol fraction in the present invention. That is, the ethanol fraction in the present invention is clearly distinguished from the conventional ethanol precipitation method.
- the raw material heparin in the ethanol fraction is not particularly limited, and various purification steps such as raw material of unfractionated heparin, unfractionated heparin, low molecular weight heparin, and heparin having an impurity concentration can be used.
- the molecular weight of the raw material heparin is preferably 3000 to 30000 daltons, more preferably 5000 to 15000 daltons.
- the raw material heparin is dissolved in water such as purified water and water for injection so as to be in the above concentration range to prepare a heparin aqueous solution.
- the above heparin aqueous solution has an acidic to neutral pH because the precipitation of the solvent is more sensitive as the pH of the aqueous solution increases due to the properties of heparin, and the precipitation of the solvent is slower as the pH decreases. It is preferably near, for example, pH 2.5 to 7.5, preferably pH 4.0 to 7.0.
- the above heparin aqueous solution is more sensitive to precipitation due to the solvent as the ionic strength increases due to the properties of heparin, and more slowly when the ionic strength decreases, and when the salt concentration is low. Since it is difficult to form a colloidal precipitate and a centrifugal operation or the like is required, it is preferable that the salt is dissolved.
- the salt concentration is, for example, 50 to 500 mM, preferably 100 to 250 mM.
- the salt include pharmaceutically acceptable salts such as sodium chloride and sodium acetate because heparin is mainly used as a medicine. Therefore, the heparin aqueous solution may be one in which heparin is dissolved in physiological saline.
- the treatment temperature and treatment time in the ethanol fraction are not particularly limited as long as the colloidal precipitate of heparin can be obtained, but it is, for example, ⁇ 10 to 40 ° C. (preferably 5 to 25 ° C.).
- the temperature can be, for example, 0.5 to 48 hours (preferably 4 to 24 hours).
- nitrite degradation resistant impurities which are impurities, remain in the supernatant, but heparin is colloidal and precipitates. Therefore, by separating the colloidal precipitate from the supernatant, high-purity heparin that is substantially free of nitrite degradation resistant impurities can be obtained.
- the “colloidal” heparin or “colloidal” precipitate in the present invention is one in which heparin forms a colloidal dispersed phase, and in the mixture of water and organic solvent used in the fractionation in the present invention.
- a filter paper such as an ultrafiltration membrane (for example, a molecular weight cut size of 500 to 50,000 MW) is used instead of a commonly used micro order molecular size sieve.
- the fractionation with an organic solvent such as ethanol is preferably carried out as follows.
- the organic solvent used for the fractionation include ethanol, methanol, isopropanol, acetone, or a mixture thereof. Among these, ethanol is most preferable in consideration of the residue in the final product.
- the above raw material heparin is dissolved in water such as purified water and water for injection so as to be in the above concentration range to prepare a heparin aqueous solution.
- the pH is acidic to neutral, from the viewpoint that the precipitation by the solvent becomes more sensitive as the pH of the aqueous solution increases, and the precipitation by the solvent becomes slower as the pH decreases.
- the pH is 2.5 to 7.5, preferably pH 4.0 to 7.0.
- the precipitation by the solvent becomes more sensitive as the ionic strength increases, and the precipitation by the solvent becomes slower as the ionic strength decreases. Since the operation and the like are necessary, it is not suitable for batch operation, and therefore it is preferable that the salt is dissolved.
- the salt concentration is, for example, 50 to 500 mM, preferably 100 to 250 mM.
- the salt include pharmaceutically acceptable salts such as sodium chloride and sodium acetate.
- the ethanol fraction is 0.2 to 1 volume (preferably 0.25 to 0.6 volume) (volume) of 5 to 30 wt% (preferably 10 to 20 wt%) heparin aqueous solution.
- Ethanol is mixed to obtain a precipitate of nitrite decomposition resistant impurities.
- nitrite degradation resistant impurities which are impurities, precipitate, but heparin remains in the supernatant. Therefore, by separating the supernatant from the precipitate, it is possible to obtain high-purity heparin that is substantially free of nitrite degradation resistant impurities.
- high-purity heparin substantially free of nitrite degradation-resistant impurities is obtained by performing purification treatment (ethanol precipitation method, etc.), drying treatment (vacuum drying, etc.), etc. according to conventional methods. It can be obtained as a white powder.
- the high-purity heparin obtained according to the present invention is highly safe because it does not substantially contain impurities such as OSCS, which is a causative agent of side effects, and exhibits the same physiological activity as conventional heparin. It can be very suitably applied to the same pharmaceutical use.
- the high-purity heparin of the present invention has strong blood anticoagulant activity, treatment of generalized intravascular blood coagulation syndrome (DIC), various thromboembolism (venous thrombosis, myocardial infarction, lung In addition to the treatment and prevention of embolism, cerebral embolism, extremity thromboembolism, intraoperative and postoperative thromboembolism, etc., when using extracorporeal circulators such as hemodialysis and cardiopulmonary bypass, inserting a vascular catheter or transfusion It can be used to prevent blood coagulation during blood tests.
- DIC generalized intravascular blood coagulation syndrome
- various thromboembolism venous thrombosis, myocardial infarction, lung
- extracorporeal circulators such as hemodialysis and cardiopulmonary bypass
- the high-purity heparin of the present invention has a lipoprotein lipase activation action, an antiplatelet aggregation action, a blood pressure lowering action, an anti-complement action, a cancer metastasis inhibiting action, a degranulation inhibiting action from mast cells, a local inflammation inhibition, an analgesic. Since it has many physiological activities such as blood circulation promoting action of muscle tissue, it can also be used as an agent for preventing or treating various diseases based on these actions.
- the high-purity heparin of the present invention can be formulated by an ordinary method and administered as an injection or an oral preparation, like conventional heparin.
- it is administered by the following administration method, and the dose or administration rate is usually determined by measuring the whole blood coagulation time or whole blood activated partial thromboplastin time after administration of this drug, and the age, case, indication area Or it is determined by the purpose.
- an amount corresponding to 5,000 to 50,000 heparin units of heparin is diluted with 1,000 ml of 5% glucose injection solution, physiological saline or Ringer's solution, and 20 to 30 drops per minute.
- Intravenous infusion at the rate of front and back.
- heparin is injected intravenously every 4 to 8 hours in an amount corresponding to 5,000 to 50,000 heparin units.
- heparin corresponding to 5,000 to 10,000 heparin units is injected subcutaneously or intramuscularly every 4 hours.
- the appropriate amount for each patient in the artificial kidney is calculated based on the results of a heparin sensitivity test before dialysis.
- heparin Prior to dialysis, heparin is administered in an amount corresponding to 1,000 to 3,000 heparin units. After starting dialysis, an amount corresponding to 500 to 1,500 heparin units is continuously or every hour. The amount corresponding to 500 to 1,500 heparin units is intermittently added to the above. In the case of the local heparinization method, an amount corresponding to 1,500 to 2,500 heparin units per hour is continuously infused.
- an amount corresponding to 150 to 300 heparin units / kg is administered depending on the surgical method and method, and additional administration is appropriately performed according to the extension of the extracorporeal circulation time.
- heparin equivalent to 500 to 2,000 heparin units / g once or several times a day In the case of an external preparation, it is used as an ointment of heparin in an amount corresponding to 100 to 500 heparin units / g, and an appropriate amount is applied to a rubbing or gauze once to several times a day.
- heparin in an amount equivalent to 1,000 to 4,000 heparin units / g is applied to the anus or vagina once or twice a day.
- the high-purity heparin of the present invention can be suitably used as cosmetics, research reagents and the like, as in the case of conventional heparin.
- the present invention also provides a method for detecting or measuring a nitrite-resistant mucopolysaccharide or a nitrite-decomposable mucopolysaccharide contained in the mucopolysaccharide, which comprises nitrite-degrading the mucopolysaccharide.
- a nitrite-resistant mucopolysaccharide or a nitrite-decomposable mucopolysaccharide contained in the mucopolysaccharide which comprises nitrite-degrading the mucopolysaccharide.
- the “nitrite degradation resistant mucopolysaccharide” in the above method include mucopolysaccharides exemplified as the nitrite degradation resistant impurity.
- “Nitrite-decomposable mucopolysaccharide” means a mucopolysaccharide that is degraded by nitrous acid decomposition, and examples thereof include heparin and he
- the “nitrous acid decomposition” in the above method is the same as the above nitrous acid decomposition.
- the mucopolysaccharide used in the above method is not particularly limited, and examples thereof include heparin, heparan sulfate, chondroitin sulfate, and keratan sulfate.
- the above-mentioned HPLC method can be used to detect or measure nitrite-resistant mucopolysaccharide or nitrite-decomposable mucopolysaccharide contained in the mucopolysaccharide. That is, when the target mucopolysaccharide is analyzed by HPLC after the above nitrous acid decomposition, the target mucopolysaccharide is a nitrite-decomposable mucopolysaccharide (heparin, etc.), and the nitrite-degrading mucopolysaccharide (chondroitin sulfate) is contained in the subject.
- the peak corresponding to the nitrite-decomposable mucopolysaccharide before nitrite decomposition disappears, and the peak corresponding to the nitrite-decomposable mucopolysaccharide is detected.
- the target mucopolysaccharide is a nitrite-resistant mucopolysaccharide (such as chondroitin sulfate) and the target contains nitrite-degradable mucopolysaccharide (such as heparin)
- nitrite-resistant mucopolysaccharide such as chondroitin sulfate
- nitrite-degradable mucopolysaccharide such as heparin
- the presence or absence of other mucopolysaccharides having different properties against nitrous acid degradation can be easily confirmed in the mucopolysaccharide product. Etc. can be secured.
- Nitrous acid decomposition In order to reduce side reactions in a weakly acidic region (around pH 4.0) at high temperatures, this treatment was all performed under ice cooling. Moreover, in order to avoid accumulation
- HPLC method The molecular weight distribution of the substance contained in each sample was confirmed by the HPLC method.
- the conditions of the HPLC method were in accordance with the “Molecular weight” section of the “Parnaparin sodium” standard test method described in the 15th Amendment Manual of the Japanese Pharmacy Law.
- the HPLC conditions used are shown below.
- Detection system SHIMADZU management system (LC solution), differential refractometer (RI: RID-10A) Column and guard column: TSK gel G-2000SWXL and TSK guard column SWXL manufactured by TOSO Column temperature: 40 ° C
- Mobile phase 0.2 mol / L Na sulfate (pH 5.0)
- Flow rate 0.5 mL / min.
- OSCS Persulfated chondroitin sulfate
- OSCS-STD Japan Public Standards Association
- OSCS-containing unfractionated heparin Na salt, OSHP-SH, OSCS content about 12.5%, lot No. 1060 manufactured by C company
- CSMS-CE1 and -CE2 crude OSCS which is a mixture of OSCS and chondroitin sulfate family
- OSCS-containing unfractionated heparin Na salt: Lot No.
- Example 1 As a sample, unfractionated heparin (Na salt, UFN-SP, Lot No. 1035-0792 manufactured by C Co.) (500 g) containing other mucopolysaccharides (mainly heparan sulfate / chondroitin sulfate B / chondroitin sulfate C) was used. Weighed into a 10 L chemical enamel tank and added physiological saline (Japanese Pharmacopoeia compatible product) to 5 L (pH 6.0).
- physiological saline Japanese Pharmacopoeia compatible product
- Ethanol 2.5 L; Wako Pure Chemical Industries, Ltd., special grade reagent
- the supernatant was transferred to a 30 L chemical enamel tank, ethanol (20 L) was added, and the mixture was vigorously stirred.
- the colloidal precipitate was transferred to a 30 L chemical enamel tank and added with physiological saline (3 L) and stirred, and then ethanol (20 L) was added and stirred vigorously. After each treatment, it was allowed to stand for 24 hours.
- the obtained product is subjected to nitrous acid decomposition by the above method, and the amount of substances (heparin and nitrite decomposition resistant impurities) contained in the product before and after nitrous acid decomposition is subjected to HPLC by the above method.
- the total area of peaks appearing during the elution time of 10 to 20 minutes was obtained. The results are shown in Table 1.
- Examples 2 to 7 For the same samples as in Comparative Examples 1 to 6, ethanol fractionation was performed in the same manner as in Example 1. Next, each product obtained was subjected to nitrous acid decomposition by the above method. The distribution of substances contained in the sample before and after nitrous acid decomposition was confirmed by the HPLC method (FIGS. 6 and 7). In addition, the amount of substances (heparin and nitrite decomposition resistant impurities) contained in the product before and after nitrite decomposition was subjected to HPLC by the above method, and the total of peaks that appeared during the elution time of 10 to 20 minutes was measured. Calculated as area. The results are shown in Table 1.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- General Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Polymers & Plastics (AREA)
- Materials Engineering (AREA)
- Immunology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Pathology (AREA)
- Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- General Physics & Mathematics (AREA)
- Epidemiology (AREA)
- Dermatology (AREA)
- Sustainable Development (AREA)
- Diabetes (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Neurology (AREA)
- Pulmonology (AREA)
- Physical Education & Sports Medicine (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
Abstract
Description
本発明は、副作用の原因物質を実質的に含まない、安全性が高く、医薬品、化粧品、研究試薬等として有用な高純度ヘパリンおよびその製造方法に関する。
ヘパリンは、強い血液抗凝固活性を有しており、汎発性血管内血液凝固症候群(DIC)の治療、種々の血栓塞栓症(静脈血栓症、心筋梗塞症、肺塞栓症、脳塞栓症、四肢動脈血栓塞栓症、術中・術後の血栓塞栓症など)の治療および予防のほか、血液透析・人工心肺などの体外循環装置使用時や血管カテーテル挿入時または輸血および血液検査の際などにおける血液凝固の防止に用いられている。
また、ヘパリンは、血液抗凝固活性の他に、リポ蛋白リパーゼ活性化作用、抗血小板凝集作用、血圧低下作用、抗補体作用、癌転移抑制作用、肥満細胞からの脱顆粒阻害作用、局部炎症抑制、鎮痛および筋組織の血行促進作用などの多くの生理活性を有することも知られている。
日本国内においては重度の副作用報告は無かったが、一部の未分画ヘパリン製剤および低分子量ヘパリン(Low molecular weight heparin:LMWH)製剤が回収となり、市場への安定供給問題が深刻化した。
しかしながら、ヘパリンからOSCS、コンドロイチン硫酸等の不純物を簡便かつ効果的に除去することができる方法は、これまでに知られていない。さらに、このようなヘパリン中の不純物を、簡便に検出または測定する方法もこれまで知られていない。
すなわち、本発明には、以下のものが含まれる:
〔1〕 亜硝酸分解抵抗性不純物を実質的に含有しないヘパリン。
〔2〕 5~30重量%のヘパリン水溶液に対して、0.2~1倍量(体積)のエタノール、メタノール、イソプロパノール、アセトンおよびこれらの混合溶媒から選択される有機溶媒を混合し、ヘパリンのコロイド状沈殿物を得ることを含む方法により得られるヘパリン。
〔3〕 ヘパリン水溶液中に、塩が50~500mMの濃度で溶解している、上記〔2〕に記載のヘパリン。
〔4〕 塩が、塩化ナトリウムおよび酢酸ナトリウムから選択される、上記〔3〕に記載のヘパリン。
〔5〕 コロイド状である、上記〔1〕~〔4〕のいずれかに記載のヘパリン。
〔6〕 分子量が、3000~30000ダルトンの範囲内にある、上記〔1〕~〔5〕のいずれかに記載のヘパリン。
〔7〕 5~30重量%のヘパリン水溶液に対して、0.2~1倍量(体積)のエタノール、メタノール、イソプロパノール、アセトンおよびこれらの混合溶媒から選択される有機溶媒を混合し、ヘパリンのコロイド状沈殿物を得ることを含む、ヘパリンの製造方法。
〔8〕 5~30重量%のヘパリン水溶液に対して、0.2~1倍量(体積)のエタノール、メタノール、イソプロパノール、アセトンおよびこれらの混合溶媒から選択される有機溶媒を混合し、ヘパリンを含有する上澄み液を得ることを含む方法により得られるヘパリン。
〔9〕 ヘパリン水溶液中に、塩が50~500mMの濃度で溶解している、上記〔8〕に記載のヘパリン。
〔10〕 塩が、塩化ナトリウムおよび酢酸ナトリウムから選択される、上記〔9〕に記載のヘパリン。
〔11〕 分子量が、1500~12000ダルトンの範囲内にある、上記〔8〕~〔10〕のいずれかに記載のヘパリン。
〔12〕 5~30重量%のヘパリン水溶液に対して、0.2~1倍量(体積)のエタノール、メタノール、イソプロパノール、アセトンおよびこれらの混合溶媒から選択される有機溶媒を混合し、ヘパリンを含有する上澄み液を得ることを含む、ヘパリンの製造方法。
〔13〕 上記〔1〕~〔6〕および〔8〕~〔11〕のいずれかに記載のヘパリンを含んでなる医薬。
〔14〕 医薬として使用するための、上記〔1〕~〔6〕および〔8〕~〔11〕のいずれかに記載のヘパリン。
〔15〕 上記〔1〕~〔6〕および〔8〕~〔11〕のいずれかに記載のヘパリンを含有する医薬組成物。
〔16〕 ムコ多糖を亜硝酸分解することを特徴とする、当該ムコ多糖中に含まれる亜硝酸分解抵抗性ムコ多糖または亜硝酸分解性ムコ多糖の検出または測定方法。
本発明のヘパリンの製造方法によれば、亜硝酸分解抵抗性不純物を実質的に含まない高純度ヘパリンを簡便に得ることができる。また、当該方法は、工業的に利用可能である。
本発明のムコ多糖の検出または測定方法によれば、ムコ多糖製品中に、亜硝酸分解に対して異なる特性を示す他のムコ多糖の混入の有無を簡便に確認することができるので、製品の安全性等を確保することができる。また、目的のムコ多糖を製造する過程においても、亜硝酸分解に対して異なる特性を示す他のムコ多糖の混入を簡便に確認することができるので、目的のムコ多糖の製造工程を効果的に管理でき、中間材料や最終生成物中への他のムコ多糖の混入を回避することができる。
本発明における「ヘパリン」は、特に限定されず、従来既知の原料および製造方法から得られたものでよいが、例えば、いわゆる「未分画ヘパリン」、「低分子量ヘパリン」、また「ヘパリン」と類似した構成糖組成と結合様式を有する「ヘパラン硫酸」の中で、特に高分子量あるいは高硫酸含量の「ヘパラン硫酸」等が挙げられる。
上記「未分画ヘパリン」は、解重合処理されていないヘパリンを意味し、通常、分子量が3000~30000ダルトンの範囲内にあるものである。
上記「低分子量ヘパリン」は、未分画ヘパリンを解重合処理して低分子化したものであり、通常、分子量が1500~12000ダルトンの範囲内にあるものである。
上記「ヘパラン硫酸」は、通常、分子量が3000~30000ダルトンの範囲内にあるものである。
また、本発明におけるヘパリンには、一般に生体内において遊離形と実質的に同様の生理活性または薬理活性を発揮するもの、例えば、ヘパリンの誘導体および医薬的に許容される塩、付加塩、水和物などは本発明の技術的範囲に含まれる。
なお、ここでいう分子量は、水性溶媒によるサイズ排除ゲルクロマトグラフィーによるHPLC法で決定される重量平均分子量である。
上記HPLCの条件の具体例としては、例えば、以下の条件が挙げられる:
検出システム:SHIMADZU製管理システム(LC solution)、示差屈折計(RI:RID-10A)
カラムおよびガードカラム:TOSO製TSK gel G-2000SWXLおよびTSK guard column SWXL
カラム温度:40℃
移動相:0.2mol/L 硫酸Na(pH 5.0)
流量:0.5mL/分。
上記分画に用いる有機溶媒としては、例えば、エタノール、メタノール、イソプロパノールもしくはアセトンまたはこれらの混合物が挙げられる。なかでも、最終生成物への残留を考慮した場合、エタノールが最も好ましい。
従来、ヘパリンを白色沈殿として得るために、エタノール沈殿法が利用されている。しかしながら、従来のエタノール沈殿法が適用されるヘパリン水溶液中のヘパリン濃度は、1~5重量%であり、本発明におけるエタノール分画に比べて大幅に低い。また、ヘパリン水溶液に混合するエタノールの量は、2~10倍量(体積)であり、本発明におけるエタノール分画に比べて顕著に大きい。すなわち、本発明におけるエタノール分画は、従来のエタノール沈殿法と明確に区別される。
上記ヘパリン水溶液は、ヘパリンの特性により水溶液のpHの上昇に伴い溶媒による沈殿生成がより鋭敏に、またpHの低下に伴い溶媒による沈殿生成がより緩慢になる点から、そのpHが酸性~中性付近であることが好ましく、例えば、pH2.5~7.5、好ましくはpH4.0~7.0である。
また、上記ヘパリン水溶液は、ヘパリンの特性によりイオン強度の上昇に伴い溶媒による沈殿生成がより鋭敏に、またイオン強度の低下に伴い溶媒による沈殿生成がより緩慢になること、また塩濃度が低い場合はコロイド沈殿を形成しづらくなり遠心操作等が必要となることから、塩が溶解していることが好ましい。その塩濃度としては、例えば、50~500mM、好ましくは100~250mMである。塩としては、ヘパリンが主に医薬品として使用されることから、薬学的に許容される塩、例えば、塩化ナトリウム、酢酸ナトリウム、等が挙げられる。
したがって、ヘパリン水溶液は、ヘパリンを生理食塩水に溶解させたものであってもよい。
当該分画に用いる有機溶媒としては、例えば、エタノール、メタノール、イソプロパノールもしくはアセトンまたはこれらの混合物が挙げられる。なかでも、最終生成物への残留を考慮した場合、エタノールが最も好ましい。
また、イオン強度の上昇に伴い溶媒による沈殿生成がより鋭敏に、またイオン強度の低下に伴い溶媒による沈殿生成がより緩慢になること、また塩濃度が低い場合はコロイド沈殿を形成しづらくなり遠心操作等が必要となることからバッチ操作に適さないため、塩が溶解していることが好ましい。その塩濃度としては、例えば、50~500mM、好ましくは100~250mMである。塩としては、薬学的に許容される塩、例えば、塩化ナトリウム、酢酸ナトリウム等が挙げられる。
エタノール分画は、5~30重量%(好ましくは10~20重量%)のヘパリン水溶液に対して、0.2~1倍量(好ましくは0.25~0.6倍量)(体積)のエタノールを混合し、亜硝酸分解抵抗性不純物の沈殿物を得る。
この場合、不純物である亜硝酸分解抵抗性不純物は沈殿するが、ヘパリンは上澄み液中に残る。したがって、上澄み液を沈殿物と分離することにより、亜硝酸分解抵抗性不純物を実質的に含まない高純度ヘパリンを得ることができる。
上記方法における「亜硝酸分解抵抗性ムコ多糖」としては、上記亜硝酸分解抵抗性不純物として例示されたムコ多糖等が挙げられる。また、「亜硝酸分解性ムコ多糖」は、上記亜硝酸分解によって分解されるムコ多糖を意味し、例えば、ヘパリン、ヘパラン硫酸等が挙げられる。
また、上記方法における「亜硝酸分解」は、上記亜硝酸分解と同様である。
上記方法に使用されるムコ多糖としては、特に限定されないが、例えば、ヘパリン、ヘパラン硫酸、コンドロイチン硫酸、ケラタン硫酸等が挙げられる。
以下に、実施例を挙げて本発明をさらに詳しく説明するが、本発明は、これらに限定されるものではない。
(1)亜硝酸分解
高温下における弱酸性域(pH4.0付近)での副反応を低減するため、本処理は全て氷冷下で行った。また、反応終了後の過剰の亜硝酸Naの蓄積を避けるため、各試料に対する亜硝酸Naの添加量は、試料1gあたり60mgとした。
予め注射用水(日本薬局方適合品)に溶かした各試料溶液に、所定量の亜硝酸Naを加えて攪拌した後、HClでpHを1.5前後に調整して反応を開始した。30分後、NaOHでpHを5.0に調整して反応を終了させ、エタノールを添加して固化、乾燥して白色粉末を得た。
(2)HPLC法
各試料中に含まれる物質の分子量分布をHPLC法により確認した。HPLC法の条件は、日本薬局法第15改正解説書に記載された「パルナパリンナトリウム」規格試験法の「分子量」の項に準じた。使用したHPLCの条件を以下に示す。
検出システム:SHIMADZU製管理システム(LC solution)、示差屈折計(RI:RID-10A)
カラムおよびガードカラム:TOSO製TSK gel G-2000SWXLおよびTSK guard column SWXL
カラム温度:40℃
移動相:0.2mol/L 硫酸Na(pH 5.0)
流量:0.5mL/分。
過硫酸化コンドロイチン硫酸(OSCS)標準品(OSCS-STD、日本公定書協会)、OSCS含有未分画ヘパリン(Na塩、OSHP-SH、OSCS含量約12.5%、C社製ロットNo.1060-07-0033)、OSCSおよびコンドロイチン硫酸ファミリーの混合物である粗OSCS(CSMS-CE1および-CE2、N社製OSCS含有未分画ヘパリン(Na塩:ロットNo.PH-64107およびpH-64507)からヘパリン/ヘパラン硫酸の含量を約95%以下に低減して調製)を、上記の方法で亜硝酸分解し、上記の方法でHPLCを行い、亜硝酸分解前後の分子量変化を確認した(図2)。その結果、OSCS-STD、CSMS-CE1および-CE2について、2糖単位のピークシフトを伴う分解および低分子化は確認されなかった。OSHP-SH(OSCS含量約12.5%)について、2糖単位のピークシフトを伴う分解および低分子化が確認され、RIで検出された亜硝酸分解未分解物のピークの面積値は、亜硝酸分解前のピークの面積値の約12.1%であった。また、OSHP-SHの未分解物のピークは、OSCS-STDのピークと近い分子量を示した(図2-A)。また、CSMS-CE1および-CE2のピークは、コンドロイチン硫酸B(CSB-STB、純度>95%、豚小腸粘膜抽出物から調製)と近い分子量を示した(図2-B)。
コンドロイチン硫酸ファミリー(試薬特級品)のA、B、C、DおよびE型(CSA、CSB、CSC、CSDおよびCSE)、ヘパラン硫酸(HS)およびケラタン硫酸(KS)を生化学工業(株)から購入し、上記の方法で亜硝酸分解し、上記の方法でHPLCを行い、亜硝酸分解前後の分子量変化を確認した(図3、点線より左側)。その結果、コンドロイチン硫酸ファミリーおよびKSについて、2糖単位のピークシフトを伴う分解および低分子化は確認されなかった。一方、HSは2糖単位のピークシフトを伴う分解および低分子化が確認された(図3、点線より右側)。
試料として、他のムコ多糖(主にヘパラン硫酸/コンドロイチン硫酸B/コンドロイチン硫酸C)を含有する未分画ヘパリン(Na塩、UFN-SP、C社製ロットNo.1035-0792)(500g)を10L化学ホーロータンクに量り取り、生理食塩液(日本薬局方適合品)を加えて5Lとした(pH6.0)。この溶液に、エタノール(2.5L;和光純薬(株)、試薬特級品)を加えて攪拌した後、室温(25℃)で24時間以上静置した(エタノール分画)。コロイド状沈殿物(下層)と上澄み液(上層)の二層に分配されたことを確認した後、上澄み液を30L化学ホーロータンクに移してエタノール(20L)を加えて激しく攪拌した。コロイド状沈殿物を30L化学ホーロータンクに移して生理食塩液(3L)を加えて攪拌した後、エタノール(20L)を加えて激しく攪拌した。各々の処理後、24時間静置した。両方のタンク底に沈殿した白色析出物をそれぞれブフナーロート上で回収した後、エタノールで洗浄し、五酸化リン存在下、室温で24時間減圧乾燥した。最終的にコロイド状沈殿物から418.2gの白色粉末を回収(回収率83.6%)した。
上記UFN-SPについて、エタノール分画前、エタノール分画後の上層(上澄み液)および下層(コロイド状沈殿物)に含まれる物質の分布を上記HPLC法で確認した。その結果を図1に示す。
また、得られた生成物を上記の方法で亜硝酸分解し、亜硝酸分解前後の生成物中に含まれる物質(ヘパリンおよび亜硝酸分解抵抗性不純物)の量を、上記の方法でHPLCを行い、溶出時間10~20分の間に現れたピークの総面積として求めた。その結果を表1に示す。
上記UFN-SPを、上記の方法で亜硝酸分解した。亜硝酸分解の前後の試料中に含まれる物質の分布を上記HPLC法で確認した。また、亜硝酸分解前後の生成物中に含まれる物質(ヘパリンおよび亜硝酸分解抵抗性不純物)の量を、上記の方法でHPLCを行い、溶出時間10~20分の間に現れたピークの総面積として求めた。その結果を表1に示す。
1H-NMR法を用いた試験においてOSCS由来のシグナルが目視上未検出あるいはヘパリンの13C由来のサテライトシグナルでないことが確認済のNa塩5試料(UFN1~5)およびCa塩1試料(UFC)を、上記の方法で亜硝酸分解した。亜硝酸分解の前後の試料中に含まれる物質の分布を上記HPLC法で確認した(図4および5)。また、亜硝酸分解前後の生成物中に含まれる物質(ヘパリンおよび亜硝酸分解抵抗性不純物)の量を、上記の方法でHPLCを行い、溶出時間10~20分の間に現れたピークの総面積として求めた。その結果を表1に示す。
上記比較例1~6と同様の試料について、実施例1と同様に、エタノール分画を行った。次いで、得られた各生成物を、上記の方法で亜硝酸分解した。亜硝酸分解の前後の試料中に含まれる物質の分布を上記HPLC法で確認した(図6および7)。また、亜硝酸分解前後の生成物中に含まれる物質(ヘパリンおよび亜硝酸分解抵抗性不純物)の量を、上記の方法でHPLCを行い、溶出時間10~20分の間に現れたピークの総面積として求めた。その結果を表1に示す。

Claims (16)
- 亜硝酸分解抵抗性不純物を実質的に含有しないヘパリン。
- 5~30重量%のヘパリン水溶液に対して、0.2~1倍量(体積)のエタノール、メタノール、イソプロパノール、アセトンおよびこれらの混合溶媒から選択される有機溶媒を混合し、ヘパリンのコロイド状沈殿物を得ることを含む方法により得られるヘパリン。
- ヘパリン水溶液中に、塩が50~500mMの濃度で溶解している、請求項2に記載のヘパリン。
- 塩が、塩化ナトリウムおよび酢酸ナトリウムから選択される、請求項3に記載のヘパリン。
- コロイド状である、請求項1~4のいずれかに記載のヘパリン。
- 分子量が、3000~30000ダルトンの範囲内にある、請求項1~5のいずれかに記載のヘパリン。
- 5~30重量%のヘパリン水溶液に対して、0.2~1倍量(体積)のエタノール、メタノール、イソプロパノール、アセトンおよびこれらの混合溶媒から選択される有機溶媒を混合し、ヘパリンのコロイド状沈殿物を得ることを含む、ヘパリンの製造方法。
- 5~30重量%のヘパリン水溶液に対して、0.2~1倍量(体積)のエタノール、メタノール、イソプロパノール、アセトンおよびこれらの混合溶媒から選択される有機溶媒を混合し、ヘパリンを含有する上澄み液を得ることを含む方法により得られるヘパリン。
- ヘパリン水溶液中に、塩が50~500mMの濃度で溶解している、請求項8に記載のヘパリン。
- 塩が、塩化ナトリウムおよび酢酸ナトリウムから選択される、請求項9に記載のヘパリン。
- 分子量が、1500~12000ダルトンの範囲内にある、請求項8~10のいずれかに記載のヘパリン。
- 5~30重量%のヘパリン水溶液に対して、0.2~1倍量(体積)のエタノール、メタノール、イソプロパノール、アセトンおよびこれらの混合溶媒から選択される有機溶媒を混合し、ヘパリンを含有する上澄み液を得ることを含む、ヘパリンの製造方法。
- 請求項1~6および8~11のいずれかに記載のヘパリンを含んでなる医薬。
- 医薬として使用するための、請求項1~6および8~11のいずれかに記載のヘパリン。
- 請求項1~6および8~11のいずれかに記載のヘパリンを含有する医薬組成物。
- ムコ多糖を亜硝酸分解することを特徴とする、当該ムコ多糖中に含まれる亜硝酸分解抵抗性ムコ多糖または亜硝酸分解性ムコ多糖の検出または測定方法。
Priority Applications (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP16191158.1A EP3144325B1 (en) | 2010-09-14 | 2011-09-13 | High purity heparin and production method therefor |
| EP11825154.5A EP2617737B1 (en) | 2010-09-14 | 2011-09-13 | High purity heparin and production method therefor |
| DK11825154.5T DK2617737T3 (en) | 2010-09-14 | 2011-09-13 | HIGH PURPOSE HEPARIN AND PREPARATION PROCEDURE |
| US13/822,825 US8932867B2 (en) | 2010-09-14 | 2011-09-13 | High purity heparin and production method therefor |
| AU2011303081A AU2011303081B2 (en) | 2010-09-14 | 2011-09-13 | High purity heparin and production method therefor |
| CA2811186A CA2811186C (en) | 2010-09-14 | 2011-09-13 | High purity heparin and production method therefor |
| KR1020177007476A KR101794877B1 (ko) | 2010-09-14 | 2011-09-13 | 고순도 헤파린 및 그 제조방법 |
| JP2012534011A JPWO2012036152A1 (ja) | 2010-09-14 | 2011-09-13 | 高純度ヘパリンおよびその製造方法 |
| KR1020137007805A KR101847846B1 (ko) | 2010-09-14 | 2011-09-13 | 고순도 헤파린 및 그 제조방법 |
| CN201180054488.0A CN103209997B (zh) | 2010-09-14 | 2011-09-13 | 高纯度肝素及其制备方法 |
| KR1020187009094A KR102082276B1 (ko) | 2010-09-14 | 2011-09-13 | 고순도 헤파린 및 그 제조방법 |
| US14/565,671 US9540454B2 (en) | 2010-09-14 | 2014-12-10 | High purity heparin and production method therefor |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010-205310 | 2010-09-14 | ||
| JP2010205310 | 2010-09-14 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/822,825 A-371-Of-International US8932867B2 (en) | 2010-09-14 | 2011-09-13 | High purity heparin and production method therefor |
| US14/565,671 Division US9540454B2 (en) | 2010-09-14 | 2014-12-10 | High purity heparin and production method therefor |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012036152A1 true WO2012036152A1 (ja) | 2012-03-22 |
Family
ID=45831611
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/070851 WO2012036152A1 (ja) | 2010-09-14 | 2011-09-13 | 高純度ヘパリンおよびその製造方法 |
Country Status (9)
| Country | Link |
|---|---|
| US (2) | US8932867B2 (ja) |
| EP (2) | EP2617737B1 (ja) |
| JP (5) | JPWO2012036152A1 (ja) |
| KR (3) | KR101794877B1 (ja) |
| CN (1) | CN103209997B (ja) |
| AU (1) | AU2011303081B2 (ja) |
| CA (3) | CA2811186C (ja) |
| DK (2) | DK3144325T3 (ja) |
| WO (1) | WO2012036152A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103145877A (zh) * | 2012-12-08 | 2013-06-12 | 青岛九龙生物医药有限公司 | 正丙醇萃取法降低肝素钠中的半乳糖胺含量的方法 |
| CN103923230A (zh) * | 2013-01-11 | 2014-07-16 | 青岛亚博生物科技有限公司 | 一种肝素钠的精制方法 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102298179B1 (ko) | 2017-11-27 | 2021-09-06 | (주)우리비앤비 | 비분획 헤파린의 제조방법 |
| WO2020020152A1 (zh) * | 2018-07-24 | 2020-01-30 | 深圳市海普瑞药业集团股份有限公司 | 一种达肝素钠亚硝酸降解产物的分析方法及其应用 |
| CN109232773A (zh) * | 2018-11-29 | 2019-01-18 | 淮安麦德森制药有限公司 | 肝素锂的制备方法 |
| KR102104367B1 (ko) | 2019-09-02 | 2020-04-24 | 팜앤바이오 주식회사 | 헤파린나트륨의 제조장치 및 제조방법 |
| KR20220147996A (ko) | 2021-04-28 | 2022-11-04 | 주식회사 휴메딕스 | 고순도 헤파린나트륨의 제조방법 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002293804A (ja) | 2001-03-28 | 2002-10-09 | Itoham Foods Inc | 低分子量ヘパリンの製造法 |
| JP2010205310A (ja) | 2009-02-27 | 2010-09-16 | Sony Corp | シャーシ構造及び電子機器 |
Family Cites Families (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5446809A (en) | 1977-08-08 | 1979-04-13 | Choay Sa | Purifying of heparine |
| GB1602439A (en) * | 1977-08-08 | 1981-11-11 | Choay Sa | Purified heparin |
| FR2482611B1 (fr) * | 1980-05-14 | 1986-03-07 | Pharmindustrie | Nouveaux polysaccharides sulfates, procedes pour leur preparation et leur utilisation comme medicaments |
| FR2482603A1 (fr) * | 1980-05-14 | 1981-11-20 | Pharmindustrie | Nouveaux esters d'heparine utilisables pour la preparation de medicaments, et procedes pour leur preparation |
| FR2572080B1 (fr) * | 1984-10-18 | 1987-06-26 | Dropic | Procede de preparation de compositions de mucopolysaccharides dotees d'une activite antithrombotique elevee, les compositions obtenues et leur application en tant que medicaments |
| IT1214609B (it) * | 1985-05-17 | 1990-01-18 | Opocrin Spa | Esosaminoglicani solfati depolimerizzati ad attivita'antitrombotica, fibrinolitica, antinfiammatoria, loro procedimento di preparazione e composizioni farmaceutiche che li contengono. |
| JPS624703A (ja) * | 1985-07-02 | 1987-01-10 | Tooa Eiyoo Kk | 低分子量へパリン画分の製法 |
| IT1234508B (it) * | 1988-06-10 | 1992-05-19 | Alfa Wassermann Spa | Derivati eparinici e procedimento per la loro preparazione |
| FR2634485B1 (fr) * | 1988-07-21 | 1992-02-28 | Sanopi | Glycosaminoglycanes selectivement o-acyles, leur preparation et compositions pharmaceutiques les contenant |
| IT1234826B (it) * | 1989-01-30 | 1992-05-29 | Alfa Wassermann Spa | Derivati eparinici e procedimento per la loro preparazione |
| FR2675806B1 (fr) * | 1991-04-23 | 1994-06-10 | Rhone Poulenc Rorer Sa | Polysaccharides sulfates, procede de preparation, composition pharmaceutique et utilisation. |
| FR2687158B1 (fr) * | 1992-02-07 | 1995-06-30 | Rhone Poulenc Rorer Sa | Polysaccharides sulfates, procede de preparation, composition pharmaceutique et utilisation. |
| IT1260137B (it) | 1992-04-17 | 1996-03-28 | Alfa Wassermann Spa | Glicosaminoglicani semisintetici a struttura eparinica od eparanica modificati nella posizione 2 dell'acido alfa-l-iduronico-2-0-solfato |
| IT1260136B (it) | 1992-04-17 | 1996-03-28 | Alfa Wassermann Spa | Glicosaminoglicani semisintetici contenenti acido alfa-l-galatturonicosostituito con radicali nucleofili in posizione 3 |
| FR2704861B1 (fr) * | 1993-05-07 | 1995-07-28 | Sanofi Elf | Fractions d'héparine purifiées, procédé d'obtention et compositions pharmaceutiques les contenant. |
| JP4897991B2 (ja) * | 1999-07-23 | 2012-03-14 | ラボラトリオス ファルマセウティコス ロビ ソシエダッド アノニマ | 超低分子量ヘパリン組成物 |
| FR2811992B1 (fr) * | 2000-07-21 | 2003-07-04 | Aventis Pharma Sa | Melanges de polysaccharides derives d'heparine, leur preparation et les compositions pharmaceutiques les contenant |
| EP1319183B1 (en) * | 2000-09-12 | 2009-03-25 | Massachusetts Institute Of Technology | Methods and products related to low molecular weight heparin |
| JP2003096104A (ja) * | 2001-09-26 | 2003-04-03 | Chisso Corp | 低分子ヘパリンまたはその塩の製造方法 |
| JP4186460B2 (ja) | 2001-12-06 | 2008-11-26 | チッソ株式会社 | 精製された、ヘパリン、その塩もしくはヘパリン誘導体の製造方法および精製された、低分子ヘパリン、その塩もしくはヘパリン誘導体の製造方法 |
| CA2478700C (en) * | 2002-03-11 | 2012-10-16 | Momenta Pharmaceuticals, Inc. | Analysis of sulfated polysaccharides |
| FR2845686B1 (fr) * | 2002-10-10 | 2013-08-30 | Aventis Pharma Sa | Melanges de polysaccharides derives d'heparine, leur preparation et les compositions pharmaceutiques les contenant |
| FR2857971B1 (fr) * | 2003-07-24 | 2005-08-26 | Aventis Pharma Sa | Melanges d'oligosaccharides derives d'heparine, leur preparation et les compositions pharmaceutiques les contenant |
| AR043110A1 (es) * | 2004-02-04 | 2005-07-20 | Syntex Sa | Sal de heparina de bajo peso molecular con trietanolamina util como agente terapeutico-antitrombotico de administracion topica, procedimientos para preparar dichas sales, proceso para la eliminacion de la higroscopicidad de la sal de heparina, composiciones farmaceuticas para uso topico en terapia a |
| US7468358B2 (en) * | 2004-06-16 | 2008-12-23 | Paringenix, Inc. | Method and medicament for sulfated polysaccharide treatment of heparin-induced thrombocytopenia (HIT) syndrome |
| FR2912408A1 (fr) * | 2007-02-14 | 2008-08-15 | Sanofi Aventis Sa | Heparine comprenant au moins une liaison covalente avec la biotine ou un derive de la biotine,leur procede de preparation,leur utilisation |
| CN101575385B (zh) * | 2008-05-09 | 2011-01-12 | 青岛九龙生物医药有限公司 | 萃取法分离肝素钠中的多硫酸软骨素的方法 |
| US20120183976A1 (en) * | 2009-04-16 | 2012-07-19 | He Zhou | Methods of assessing activity of a polysaccharide composition |
| CN101824098B (zh) * | 2010-02-12 | 2011-06-22 | 淮安麦德森化学有限公司 | 快速沉淀分离肝素钠中多硫酸软骨素的方法 |
-
2011
- 2011-09-13 DK DK16191158.1T patent/DK3144325T3/da active
- 2011-09-13 CA CA2811186A patent/CA2811186C/en not_active Expired - Fee Related
- 2011-09-13 AU AU2011303081A patent/AU2011303081B2/en not_active Ceased
- 2011-09-13 DK DK11825154.5T patent/DK2617737T3/en active
- 2011-09-13 WO PCT/JP2011/070851 patent/WO2012036152A1/ja active Application Filing
- 2011-09-13 KR KR1020177007476A patent/KR101794877B1/ko active IP Right Grant
- 2011-09-13 CA CA3020369A patent/CA3020369C/en not_active Expired - Fee Related
- 2011-09-13 EP EP11825154.5A patent/EP2617737B1/en active Active
- 2011-09-13 US US13/822,825 patent/US8932867B2/en active Active
- 2011-09-13 JP JP2012534011A patent/JPWO2012036152A1/ja active Pending
- 2011-09-13 EP EP16191158.1A patent/EP3144325B1/en active Active
- 2011-09-13 CN CN201180054488.0A patent/CN103209997B/zh active Active
- 2011-09-13 CA CA2974062A patent/CA2974062C/en not_active Expired - Fee Related
- 2011-09-13 KR KR1020137007805A patent/KR101847846B1/ko active IP Right Grant
- 2011-09-13 KR KR1020187009094A patent/KR102082276B1/ko active IP Right Grant
-
2014
- 2014-12-10 US US14/565,671 patent/US9540454B2/en active Active
-
2015
- 2015-03-09 JP JP2015046245A patent/JP6082416B2/ja active Active
-
2016
- 2016-08-05 JP JP2016154813A patent/JP6511418B2/ja active Active
-
2019
- 2019-01-22 JP JP2019008601A patent/JP6755980B2/ja active Active
- 2019-01-22 JP JP2019008602A patent/JP2019065306A/ja active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002293804A (ja) | 2001-03-28 | 2002-10-09 | Itoham Foods Inc | 低分子量ヘパリンの製造法 |
| JP2010205310A (ja) | 2009-02-27 | 2010-09-16 | Sony Corp | シャーシ構造及び電子機器 |
Non-Patent Citations (13)
| Title |
|---|
| BEYER, T. ET AL., JOURNAL OF PHARMACEUTICAL AND BIOMEDICAL ANALYSIS, vol. 48, 2008, pages 13 - 19 |
| GUERRINI, M. ET AL., NATURE BIOTECHNOLOGY, vol. 26, 2008, pages 669 - 675 |
| GUERRINI, M. ET AL.: "Orthogonal analytical approaches to detect potential contaminants in heparin", PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES OF THE UNITED STATES OF AMERICA, vol. 106, no. 40, 6 October 2009 (2009-10-06), pages 16956 - 16961, XP002551175 * |
| HASHII NORITAKA ET AL., IYAKU KENKYU, vol. 39, no. 10, 2008, pages 651 - 659 |
| JIA, H., NATURE BIOTECHNOLOGY, vol. 26, 2008, pages 477 - 478 |
| NATURE BIOTECHNOLOGY, vol. 26, 2008, pages 669 - 675 |
| RODEN, L.; DORFMAN, A., ACTA CHEMI. SCAND., vol. 13, 1959, pages 2121 |
| SCHILLER, S. ET AL., J. BIOL. CHEM., vol. 236, 1961, pages 983 |
| SCHMIDT, M; DMOCHOWSKI, A, BIOCHIM. BIOPHYS. ACTA, vol. 83, 1964, pages 137 |
| See also references of EP2617737A4 * |
| THE NEW ENGLAND JOURNAL OF MEDICINE, vol. 358, 2008, pages 2457 - 2467 |
| THE NEW ENGLAND JOURNAL OF MEDICINE, vol. 359, 2008, pages 2674 - 2684 |
| ZHANG Z. ET AL.: "Analysis of Pharmaceutical Heparins and Potential Contaminants Using 1H-NMR and PAGE", JOURNAL OF PHARMACEUTICAL SCIENCES, vol. 98, no. 11, 2009, pages 4017 - 4026, XP055080932 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103145877A (zh) * | 2012-12-08 | 2013-06-12 | 青岛九龙生物医药有限公司 | 正丙醇萃取法降低肝素钠中的半乳糖胺含量的方法 |
| CN103923230A (zh) * | 2013-01-11 | 2014-07-16 | 青岛亚博生物科技有限公司 | 一种肝素钠的精制方法 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6755980B2 (ja) | 高純度ヘパリンおよびその製造方法 | |
| EP2794666B1 (en) | Use of chemically modified heparin derivates in sickle cell disease | |
| WO2000006608A1 (fr) | Nouveau glycosaminoglycan et compositions medicamenteuses le contenant | |
| JPH0323528B2 (ja) | ||
| JPH03243601A (ja) | 血液凝固の制御能をもつムコ多糖組成物およびその製造方法 | |
| CN103285031B (zh) | 解聚海参糖胺聚糖在制备防治血栓栓塞疾病药物中的应用 | |
| ITCO960031A1 (it) | Fucani a basso peso molecolare aventi attivita' anticoagulante antitrombinica e antitrombotica | |
| AU2015203727B2 (en) | High Purity Heparin and Production Method Therefor | |
| US5547945A (en) | Remitting agent for nephrotic syndrome and hepatopathy symptoms | |
| US20160082051A1 (en) | Use of sea cucumber glycosaminoglycan in preparing medicine for prevention and treatment of thromboembolic disease | |
| JPH0748265A (ja) | ネフローゼ症候群及び肝障害症状の寛解剤 | |
| JPH08165243A (ja) | 抗炎症剤 | |
| JPH0748403A (ja) | 丹参由来多糖類、その製造法および用途 | |
| CN104147039A (zh) | 黑海参糖胺聚糖在制备防治血栓栓塞疾病药物中的应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180054488.0 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11825154 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2811186 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2012534011 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13822825 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2011825154 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011825154 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20137007805 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2011303081 Country of ref document: AU Date of ref document: 20110913 Kind code of ref document: A |