WO2012029819A1 - 新規加水分解酵素タンパク質 - Google Patents
新規加水分解酵素タンパク質 Download PDFInfo
- Publication number
- WO2012029819A1 WO2012029819A1 PCT/JP2011/069680 JP2011069680W WO2012029819A1 WO 2012029819 A1 WO2012029819 A1 WO 2012029819A1 JP 2011069680 W JP2011069680 W JP 2011069680W WO 2012029819 A1 WO2012029819 A1 WO 2012029819A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- amino acid
- seq
- acid sequence
- protein
- hydrolase protein
- Prior art date
Links
- JFZPKTIPFQSYEI-UHFFFAOYSA-N CC1(CCCCC1)C=C Chemical compound CC1(CCCCC1)C=C JFZPKTIPFQSYEI-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P7/00—Preparation of oxygen-containing organic compounds
- C12P7/62—Carboxylic acid esters
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/16—Hydrolases (3) acting on ester bonds (3.1)
- C12N9/18—Carboxylic ester hydrolases (3.1.1)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P41/00—Processes using enzymes or microorganisms to separate optical isomers from a racemic mixture
- C12P41/003—Processes using enzymes or microorganisms to separate optical isomers from a racemic mixture by ester formation, lactone formation or the inverse reactions
- C12P41/005—Processes using enzymes or microorganisms to separate optical isomers from a racemic mixture by ester formation, lactone formation or the inverse reactions by esterification of carboxylic acid groups in the enantiomers or the inverse reaction
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/52—Improvements relating to the production of bulk chemicals using catalysts, e.g. selective catalysts
Definitions
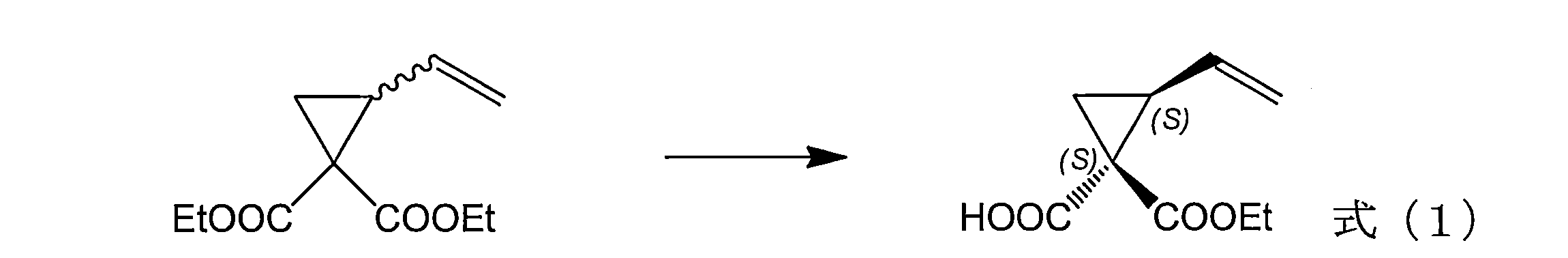
- the present invention relates to a novel hydrolase protein that can be used for the production of (1S, 2S) -1-alkoxycarbonyl-2-vinylcyclopropanecarboxylic acid, and the use thereof.
- Non-Patent Document 1 describes a method for hydrolyzing dimethyl 2-vinylcyclopropane-1,1-dicarboxylic acid with an enzyme to obtain (1S, 2S) -1-methoxycarbonyl-2-vinylcyclopropanecarboxylic acid.
- the optical selectivity of the enzyme is insufficient, and the obtained (1S, 2S) -1-methoxycarbonyl-2-vinylcyclopropanecarboxylic acid has an optical purity of 90% ee, which is an intermediate of a pharmaceutical product. It was unsuitable as an industrial production method.
- Bacillus subtilis is ATCC23857
- the amino acid sequence of paranitrobenzyl esterase derived from the Bacillus ⁇ ⁇ subtilis ATCC23857 strain (hereinafter sometimes referred to as “PNBE23857”). Is registered in GenBank Accession No. ZP_03593235.
- the present invention is useful as an intermediate for the production of a therapeutic agent for hepatitis C by hydrolyzing a dialkyl ⁇ 2-vinylcyclopropane-1,1-dicarboxylic acid with an enzyme.
- paranitrobenzyl hydrolase protein derived from Bacillus subtilis has good selectivity and diethyl 2-vinylcyclopropane-1,1- It was found that (1S, 2S) -1-ethoxycarbonyl-2-vinylcyclopropanecarboxylic acid can be efficiently obtained by hydrolyzing dicarboxylic acid.
- a hydrolase comprising the amino acid sequence shown in any of SEQ ID NOs: 2 to 5 and having an activity of catalyzing the reaction shown in formula (1) with higher selectivity than the protein consisting of the amino acid sequence shown in SEQ ID NO: 1. protein.
- amino acid sequence shown in any one of SEQ ID NOs: 1 to 5 amino acid numbers 70, 106, 107, 108, 219, 270, 271, 272, 273, 273, 274, 275
- the amino acid sequence of Nos. 276 and 313 has an amino acid sequence in which one or more amino acids are substituted with amino acids having a lower side chain than the wild-type amino acid, and the reaction shown in Formula (1) is performed.
- amino acid sequence shown in any one of SEQ ID NOs: 1 to 5 the amino acid numbers 188, 190, 193, 215, 216, 217, 314, 358, 362, and 363
- One or more of the amino acids consists of an amino acid sequence substituted with an amino acid having a bulky side chain compared to the wild-type amino acid, and the reaction shown in Formula (1) is converted from the amino acid sequence shown in SEQ ID NO: 1.
- a hydrolase protein having an activity of catalyzing with higher selectivity than the protein.
- amino acid numbers 70, 106, 107, 108, 219, 270, 271, 272, 273, 273, 274, 275 One or more of amino acids No. 276, No. 313 is substituted with an amino acid whose side chain is less bulky than wild-type amino acids, and amino acid Nos. 188, 190, 193,
- amino acids Nos. 215, 216, 217, 314, 358, 362, and 363 are substituted with amino acids having a higher bulk side chain than wild-type amino acids.
- a hydrolase protein comprising an amino acid sequence and having an activity of catalyzing the reaction represented by the formula (1) with higher selectivity than a protein comprising the amino acid sequence represented by SEQ ID NO: 1.
- the leucine of amino acid number 270 is substituted with any amino acid of serine, glutamine, glutamic acid, or alanine, according to [5] or [6] Hydrolase protein.
- the leucine at amino acid number 70 is aspartic acid
- the leucine at 270 number is any one of glutamine, glutamic acid, or alanine
- the 273th leucine is at arginine
- a hydrolase protein comprising an amino acid sequence in which the 313th leucine is substituted with methionine, and having an activity of catalyzing the reaction represented by formula (1) with higher selectivity than the protein comprising the amino acid sequence represented by SEQ ID NO: 1.
- a hydrolase protein according to any one of [1] to [12] is allowed to act on a dialkyl 2-vinylcyclopropane-1,1-dicarboxylic acid to produce (1S, 2S) -1-alkoxycarbonyl.
- the hydrolase protein of the present invention when hydrolyzing dialkyl 2-vinylcyclopropane-1,1-dicarboxylic acid to produce 1-alkoxycarbonyl-2-vinylcyclopropanecarboxylic acid, (1S, 2S) 1-alkoxycarbonyl-2-vinylcyclopropanecarboxylic acid has high selectivity that it can be produced preferentially. That is, according to the hydrolase protein of the present invention, (1S, 2S) -1-alkoxycarbonyl-2-vinylcyclopropanecarboxylic acid, which is useful as an intermediate for producing a therapeutic agent for hepatitis C, is industrially put into practical use. And can be manufactured more efficiently than conventional methods.
- the method for producing (1S, 2S) -1-alkoxycarbonyl-2-vinylcyclopropanecarboxylic acid using the hydrolase protein of the present invention can be used for the production of a therapeutic agent for hepatitis C and its intermediate. it can.
- the compound produced by the production method of the present invention can be used as a raw material or an intermediate in the production of a therapeutic agent for hepatitis C.
- hydrolase protein of the present invention comprises the amino acid sequence shown in any one of SEQ ID NOs: 2 to 5, or an amino acid sequence obtained by mutating the amino acid sequence, and a reaction represented by the formula (1) Is characterized in that it has an activity of catalyzing with higher selectivity than a protein consisting of the amino acid sequence shown in SEQ ID NO: 1.
- SEQ ID NO: 1 is an amino acid sequence of paranitrobenzyl esterase (PNBE23857, GenBank Accession No. ZP_03593235) derived from Bacillus subtilis ATCC23857 strain.
- Et represents an ethyl group.
- selectivity means optical selectivity and means a property of producing an optically active compound by preferentially producing a certain isomer. Specifically, it means the property of preferentially producing (1S, 2S) -1-alkoxycarbonyl-2-vinylcyclopropanecarboxylic acid by hydrolyzing dialkyl 2-vinylcyclopropane-1,1-dicarboxylic acid. To do. Dialkyl 2-vinylcyclopropane-1,1-dicarboxylic acid is required for hydrolase protein to hydrolyze (1S, 2S) -1-alkoxycarbonyl-2-vinylcyclopropanecarboxylic acid The following points are mentioned as selectivity.
- dialkyl 2-vinylcyclopropane-1,1-dicarboxylic acid has two types of stereoisomers, (2R) and (2S), but there are special conditions that cause steric inversion at the 2-position. Unless otherwise, only the (2S) isomer can be the starting material for the desired (1S, 2S) -1-alkoxycarbonyl-2-vinylcyclopropanecarboxylic acid.
- the hydrolase protein is pro-R among the two alkoxycarbonyl groups bonded to the 1-position prochiral carbon.
- Pro-R is a notation that distinguishes two Xs on CX 2 YZ, and the outline is as follows. Priorities are determined for each substituent on C according to the CIP rule. Then, assume that one of the two Xs has a higher priority than the other. The priority relationship with Y and Z is not changed. Based on provisional priorities, the central notation chirality is determined to be R or S using RS notation.
- the priority X at that time is designated as pro-R
- pro-S the priority X at that time
- This selectivity can be compared using the ratio of (1S, 2S) and (1R, 2S) isomers of 1-alkoxycarbonyl-2-vinylcyclopropanecarboxylic acid produced by hydrolysis as an index, and (1S, 2S )
- the yield of (1S, 2S) -1-alkoxycarbonyl-2-vinylcyclopropanecarboxylic acid is improved as the ratio of the amount of (1R, 2S) produced is lower than the amount produced. Is advantageous.
- the hydrolase protein is a pro-R alkoxycarbonyl group out of two alkoxycarbonyl groups bonded to the 1-position prochiral carbon of dialkyl (2R) -2-vinylcyclopropane-1,1-dicarboxylic acid.
- any selectivity can be compared by the enantiomeric excess (% ee) of the (1S, 2S) form of the 1-alkoxycarbonyl-2-vinylcyclopropanecarboxylic acid produced by hydrolysis to the (1R, 2R) form.
- the hydrolase protein is pro-R among the two alkoxycarbonyl groups bonded to the 1-position prochiral carbon of dialkyl (2R) -2-vinylcyclopropane-1,1-dicarboxylic acid.
- Hydroxyloxycarbonyl group can be hydrolyzed to produce (1S, 2R) -1-alkoxycarbonyl-2-vinylcyclopropanecarboxylic acid corresponding to the desired (1S, 2S) diastereomer.
- This selectivity can be compared by the ratio of the generated amount of (1S, 2S) isomer and (1S, 2R) isomer of 1-alkoxycarbonyl-2-vinylcyclopropanecarboxylic acid to be generated.
- Those having a lower production amount of (1S, 2R) body relative to the production amount of 2S) body are less likely to adversely affect the subsequent production process and the physiological activity of the manufactured pharmaceutical, and are advantageous in industrialization.
- the selectivity in the present invention is an index of “1R2S / 1S2S ratio”, “1S2S optical purity (% ee)”, or “1S2R / 1S2S ratio” in 1-alkoxycarbonyl-2-vinylcyclopropanecarboxylic acid. It is what. That is, “the activity of catalyzing the reaction shown in Formula (1) with higher selectivity than the protein consisting of the amino acid sequence shown in SEQ ID NO: 1” of the present invention is the product 1-ethoxycarbonyl-2-vinylcyclohexane.
- the “1R2S / 1S2S ratio”, “1S2S optical purity (% ee)”, or “1S2R / 1S2S ratio” in propanecarboxylic acid can be determined as an index.
- Preferred embodiments of the hydrolase protein of the present invention include the following.
- Reaction consisting of an amino acid sequence in which one or more of the amino acids of Nos. 276 and 313 is substituted with an amino acid having a lower side chain bulk than the wild-type amino acid, and represented by the above formula (1)
- a hydrolase protein having an activity of catalyzing with higher selectivity than a protein comprising (C) In the amino acid sequence shown in any one of SEQ ID NOs: 1 to 5, amino acid numbers 70, 106, 107, 108, 219, 270, 271, 272, 273, 274, 275 One or more of amino acids No. 276, No. 313 is substituted with an amino acid whose side chain is less bulky than wild-type amino acids, and amino acid Nos. 188, 190, 193, One or more of amino acids Nos. 215, 216, 217, 314, 358, 362, and 363 are substituted with amino acids having a higher bulk side chain than wild-type amino acids.
- a hydrolase protein comprising an amino acid sequence and having an activity of catalyzing the reaction represented by the above formula (1) with higher selectivity than a protein comprising the amino acid sequence represented by SEQ ID NO: 1.
- D The hydrolase protein of (a) or (c) above, wherein at least one amino acid of amino acids Nos. 70, 270, 273, and 313 is substituted.
- a hydrolase protein comprising an amino acid sequence and having an activity of catalyzing the reaction represented by the above formula (1) with higher selectivity than a protein comprising the amino acid sequence represented by SEQ ID NO: 1.
- the hydrolase protein (d) is particularly preferable.
- “Bulk” in “an amino acid having a lower side chain bulk than a wild-type amino acid” or “an amino acid having a higher side chain bulk than a wild-type amino acid” specifically refers to “J. Theoret. Biol. , 1968, 21, 170, the value shown as Bulkiness in Table 3 can be used as an index. Specifically, the Bulkiness value of each amino acid is as follows.
- substitution with an amino acid whose side chain is lower in bulk than the wild-type amino acid means that the amino acid has been substituted with an amino acid having a smaller Bulkiness value than the wild-type amino acid.
- substitution with an amino acid having a bulky side chain compared to the wild-type amino acid means that the amino acid has been substituted with an amino acid having a greater Bulkiness value than the wild-type amino acid.
- the hydrolase protein of the present invention includes one or more of leucine or isoleucine having amino acid numbers 70, 270, 273, or 313 in the amino acid sequence shown in any one of SEQ ID NOs: 1 to 5. However, those substituted with an amino acid having a smaller Bulkiness value than these are preferred.
- hydrolase proteins include the following.
- E The hydrolase protein of (d) above, wherein in the amino acid sequence shown in any one of SEQ ID NOs: 1 to 5, at least leucine of amino acid number 70 is aspartic acid, asparagine, serine, threonine, or glycine A hydrolase protein in which any amino acid is substituted.
- F The hydrolase protein of (d) or (e) above, wherein in the amino acid sequence shown in SEQ ID NO: 3 or 4, the leucine at amino acid number 270 is one of serine, glutamine, glutamic acid, or alanine A hydrolase protein that is substituted with an amino acid.
- (G) The hydrolase protein of (d) or (e) above, wherein in the amino acid sequence shown in SEQ ID NO: 1, 2, or 5, isoleucine of amino acid number 270 is serine, glutamine, glutamic acid, Alternatively, a hydrolase protein substituted with any amino acid of alanine.
- (H) The hydrolase protein of any one of (d) to (g) above, wherein in the amino acid sequence shown in any one of SEQ ID NOs: 1 to 5, leucine of amino acid number 273 is arginine or histidine A hydrolase protein substituted with any amino acid.
- leucine at amino acid number 70 is aspartic acid
- leucine at 270 is any amino acid of glutamine, glutamic acid, or alanine
- leucine at 273 is arginine.
- the hydrolase protein of the present invention further includes an amino acid sequence having 90% or more homology with the amino acid sequence of the hydrolase protein of the present invention described above, or the amino acid sequence of the hydrolase protein of the present invention described above. It consists of an amino acid sequence having 1 to several amino acid substitutions, deletions and / or additions, and catalyzes the reaction shown in formula (1) with higher selectivity than the protein consisting of the amino acid sequence shown in SEQ ID NO: 1. Those having the activity of
- amino acid sequence having 90% or more homology with the above-mentioned amino acid sequence of the hydrolase protein of the present invention means the amino acid of the hydrolase protein of the present invention. It means that the homology or identity with the sequence is 90% or more, preferably 93% or more, more preferably 95% or more, particularly preferably 98% or more.
- substitution, deletion and / or addition of one to several amino acids means, for example, about 1 to 40, preferably about 1 to 20 More preferably, it means about 1 to 10, more preferably about 1 to 5.
- the origin of the hydrolase protein of the present invention is not particularly limited, and may be a natural enzyme derived from a microorganism or a genetically modified protein.
- the natural enzyme can be preferably obtained from Bacillus subtilis. That is, the hydrolase protein of the present invention includes, for example, Bacillus subtilis NBRC3026, Bacillus ⁇ subtilis 3 NBRC3108, Bacillus ⁇ subtilis NBRC3027, and Bacillus ⁇ subtilis 13 NBRC3027, and Bacillus ⁇ subtilis NBRC3027.
- Specific purification methods include, for example, salting out with ammonium sulfate (ammonium sulfate) or sodium sulfate, centrifugation, dialysis, ultrafiltration, adsorption chromatography, ion exchange chromatography, hydrophobic chromatography, reverse phase chromatography. Processing operations such as chromatography, gel filtration, gel permeation chromatography, affinity chromatography, electrophoresis, zymography, and combinations thereof.
- ammonium sulfate ammonium sulfate
- sodium sulfate sodium sulfate
- centrifugation dialysis
- ultrafiltration ultrafiltration
- adsorption chromatography ion exchange chromatography
- hydrophobic chromatography hydrophobic chromatography
- reverse phase chromatography reverse phase chromatography.
- Processing operations such as chromatography, gel filtration, gel permeation chromatography, affinity chromatography, electrophoresis, zymography, and combinations thereof.
- the hydrolase protein of the present invention can be obtained as a recombinant protein by cloning the gene of the hydrolase protein of the present invention according to a known method, introducing the gene into a suitable host and expressing it.
- a probe or primer specific to the gene of the hydrolase protein of the present invention is designed based on the information on the base sequence of the gene encoding the hydrolase protein of the present invention, and the probe or primer is used to DNA libraries (e.g., genomic libraries or cDNA such as Bacillus subtilis NBRC3026, Bacillus subtilis NBRC3108, Bacillus subtilis NBRC3027, and Bacillus subtilis NBRC3013)
- the DNA encoding the hydrolase protein of the present invention is isolated or amplified from the rally, etc., put into a vector by gene recombination technology, introduced into a host cell, and expressed therein, thereby allowing the hydrolysis of the present invention. Enzyme protein can be obtained.
- DNA of the present invention DNA encoding the hydrolase protein of the present invention described above is provided.
- Specific examples of the DNA of the present invention include DNA comprising the following base sequences.
- substitution of one to several bases in the base sequence described in SEQ ID NOs: 7 to 10 lack of A base sequence having a loss and / or an addition, which encodes a hydrolase protein having an activity of catalyzing the reaction shown in Formula (1) with higher selectivity than the protein consisting of the amino acid sequence shown in SEQ ID NO: 1 Base sequence:
- DNA encoding the hydrolase protein of the present invention includes Bacillus subtilis NBRC3026, Bacillus ⁇ subtilis NBRC3108, Bacillus ⁇ subtilis NBRC3027, and Bacillus ⁇ subtilis 13 NBRC3027 and Bacillus ⁇ subtilis (Bacillus RC RC30 It is a cloned DNA or a mutation introduced into the DNA.
- Bacillus RC RC30 It is a cloned DNA or a mutation introduced into the DNA.
- the base sequence is determined according to the present invention, it is possible to synthesize based on this base sequence.
- DNA hybridization of a microorganism such as Bacillus subtilis is performed by hybridization using an oligonucleotide prepared based on this base sequence as a probe, or by PCR using an oligonucleotide prepared based on the sequence as a primer. It can also be isolated from a library (eg, genomic DNA library or cDNA library).
- the DNA of the hydrolase protein of the present invention is not limited to those isolated from natural microorganisms, but also known DNA synthesis methods such as the methods described in US Pat. No. 6,472,184 and US Pat. No. 5,750,380. It may be synthesized using.
- DNA capable of hybridizing under stringent conditions refers to DNA having the nucleotide sequence set forth in SEQ ID NOs: 7 to 10 or a complementary sequence thereof and 80% or more, preferably 90% by BLAST analysis. % Or more, more preferably 95% or more of DNA containing a base sequence having homology.
- hybridization under stringent conditions is a reaction in a normal hybridization buffer at a temperature of 40 to 70 ° C., preferably 60 to 65 ° C., and a salt concentration of 15 mM to 300 mM, preferably The washing can be performed in a washing solution of 15 mM to 60 mM or the like.
- “1 to several” in the “base sequence having substitution, deletion and / or addition of 1 to several bases” means, for example, about 1 to 30, preferably 1 Means about 20, more preferably about 1 to 10, more preferably about 1 to 5.
- substitution, deletion, and / or addition of amino acids that do not substantially harm the enzyme activity can be easily selected by those skilled in the art.
- DNA encoding the hydrolase protein of the present invention having amino acid substitutions, deletions, and / or additions that do not substantially affect enzyme activity should be obtained from, for example, natural mutants or variants. Can do.
- DNA having a base sequence having 1 to several base substitutions, deletions and / or additions can be prepared by using a mutagen or site-specific mutation. It can be obtained by a usual mutation operation such as a method. These can be easily performed with a commercially available kit such as PrimeSTAR Mutagenesis Basal Kit (Takara Bio) or Quick Change Site Directed Muta Genesis Kit (Stratagene).
- the present invention further provides a recombinant vector having the DNA of the present invention.
- the DNA of the present invention is usually added to the vector together with a promoter suitable for the host microorganism so that the 5 ′ end of the coding region of the DNA of the present invention is linked downstream of this promoter. insert.
- the DNA of the present invention may be inserted into an expression vector containing a promoter.
- the expression vector is not particularly limited as long as it can replicate and proliferate in the host microorganism, and examples thereof include plasmid vectors, shuttle vectors, and phage vectors.
- Specific plasmid vectors include pBR322, pUC18, pHSG298, pUC118, pSTV28, pTWV228, pHY300PLK (the above plasmid vectors can be purchased from, for example, Takara Bio Inc.), pET expression vector series (manufactured by Novagen), and the like. Can do. Examples of E.
- coli-coryneform bacterium shuttle vectors include plasmid pCRY30 described in JP-A-3-210184; plasmids pCRY21, pCRY2KE, pCRY2KX, pCRY31, pCRY3KE and pCRY3KX described in JP-A-2-276575; Plasmids pCRY2 and pCRY3 described in Kaihei 1-191686; pAM330 described in JP-A-58-67679; pHM1519 described in JP-A-58-77895; described in JP-A-58-192900 PAJ655, pAJ611 and pAJ1844; pCG1 described in JP-A-57-134500; pCG2 described in JP-A-58-35197; pCG4 and pCG1 described in JP-A-57-183799 Etc., or it may be derivatives thereof.
- the phage vector include ( ⁇ FixI
- a promoter for expressing the DNA encoding the hydrolase protein of the present invention a promoter possessed by the host microorganism can be generally used, but is not limited thereto, and the promoter of the hydrolase protein DNA of the present invention is not limited thereto. Any promoter may be used as long as it is a prokaryotic base sequence for initiating transcription. Specifically, a lactose operon promoter, a tryptophan operon promoter, a lambda phage-derived PL promoter, a tryptophan lactose hybrid (tac) promoter [H. A. Bose et al., Proc. Natl. Acad. Sci. U.
- inducible promoters can be used for the purpose of improving the expression efficiency.
- gene expression can be induced by adding lactose or isopropyl- ⁇ -D-thiogalactoside (IPTG).
- a transformant obtained by introducing the DNA or recombinant vector of the present invention into a host cell.
- the host into which the DNA or recombinant vector of the present invention is introduced is not particularly limited, but dialkyl 2-vinylcyclopropane-1,1-dicarboxylic acid is converted to (1S, 2S) -1-alkoxycarbonyl-2- Those having activity of hydrolyzing other than vinylcyclopropanecarboxylic acid are not suitably used. Further, those having an activity of changing the produced (1S, 2S) -1-alkoxycarbonyl-2-vinylcyclopropanecarboxylic acid into another compound are also not preferable.
- the host that can be used in the present invention is brought into contact with a dialkyl 2-vinylcyclopropane-1,1-dicarboxylic acid or the desired product (1S, 2S) -1-alkoxycarbonyl-2-vinylcyclopropanecarboxylic acid. As a result, it can be selected by analyzing the resulting compound.
- Specific examples of host microorganisms that can be used in the present invention include Escherichia bacteria (for example, Escherichia coli), Actinomycetes bacteria, Bacillus bacteria, Serratia.
- (Serratia) bacteria Pseudomonas bacteria, Corynebacterium bacteria, Brevibacterium bacteria, Rhodococcus bacteria, Lactobacillus bacteria, Streptomyces ( Streptomyces bacteria, Thermus bacteria, Streptococcus bacteria and the like can also be used as hosts for introducing the DNA or recombinant vector of the present invention.
- a competent cell method [Journalourof Molecular Biology, logyVol.53, p.159 (1970)], pulse wave energization method [J. Indust.Microbiol., Vol.5, p.159 (1990)], etc., transduction methods using phages [E. Ohtsubo, Genetics, Vol.64, p.189 (1970)], conjugation transfer method [J. G. C. Ottow , Ann. Rev. Microbiol., Vol. 29, p. 80 (1975)], cell fusion method [MH Gabor, J. Bacteriol., Vol. 137, p. 1346 (1979)] and the like can be used. From these methods, a method suitable for the host microorganism may be appropriately selected.
- homologous recombination technology or transposon or insertion sequence for directly introducing the DNA encoding the hydrolase protein of the present invention linked to a promoter into the chromosome of the host microorganism It can also be expressed by a technique introduced using the above. Therefore, the transformant of the present invention is not limited as long as the enzyme of the present invention is expressed, and the method of gene introduction is not limited.
- the transformant obtained as described above is cultured, and the hydrolase protein of the present invention is obtained from the culture. Can be collected.
- the transformant can be cultured in a normal nutrient medium containing a carbon source, nitrogen source, inorganic salt, various vitamins, etc.
- a carbon source include sugars such as glucose, sucrose, fructose and maltose, ethanol Alcohols such as methanol, citric acid, malic acid, succinic acid, maleic acid, organic acids such as fumaric acid, molasses, etc. are used.
- the nitrogen source for example, ammonia, ammonium sulfate, ammonium chloride, ammonium nitrate, urea or the like is used alone or in combination.
- inorganic salts that can be used include potassium monohydrogen phosphate, potassium dihydrogen phosphate, and magnesium sulfate.
- nutrients such as various vitamins such as peptone, meat extract, yeast extract, corn steep liquor, casamino acid, and biotin can be added to the medium.
- Cultivation is usually carried out under aerobic conditions such as aeration stirring and shaking.
- the culture temperature is not particularly limited as long as the host microorganism can grow, and the pH during the culture is not particularly limited as long as the host microorganism can grow.
- the pH adjustment during the culture can be performed by adding an acid or an alkali.
- the enzyme can be collected from the culture by a known collection method using the enzyme activity as an index.
- the enzyme does not necessarily need to be purified to homogeneity, and may be purified to a degree of purification according to the application.
- the crudely purified fraction or purified enzyme used in the present invention not only the cells isolated from the culture solution in which the transformant was cultured, but also the culture solution and the cells are crushed by means of ultrasonic waves, crushing or the like.
- the resulting crushed product, the extract containing the hydrolase protein of the present invention obtained by extracting the crushed product with water, and the like, and further subjected to treatment such as ammonium sulfate salting out and column chromatography.
- the obtained crude enzyme preparation or purified enzyme preparation of the hydrolase protein of the present invention may be used.
- carrier can also be used.
- Immobilization of these cells, disrupted cells, extract or purified enzyme is carried out by immobilizing the cells on an appropriate carrier such as acrylamide monomer, alginic acid, or carrageenan in accordance with a known and commonly used method. It can be performed by the method of making it. For example, when the cells are immobilized on a carrier, they are recovered from the culture or washed with an appropriate buffer, for example, a phosphate buffer (pH 6 to 10) of about 0.02 M to 0.2 M. Can be used.
- an appropriate buffer for example, a phosphate buffer (pH 6 to 10) of about 0.02 M to 0.2 M.
- Examples of the optionally substituted alkyl group having 1 to 10 carbon atoms represented by R include a substituted or unsubstituted linear, branched or cyclic alkyl group having 1 to 10 carbon atoms, and more preferable. Includes a substituted or unsubstituted linear, branched or cyclic alkyl group having 1 to 6 carbon atoms.
- a normal hexyl group, a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclohexyl group and the like are preferable, and an alkyl group having 1 to 4 carbon atoms is particularly preferable.
- the optionally substituted aralkyl group having 7 to 20 carbon atoms represented by R is preferably a substituted or unsubstituted aralkyl group having 7 to 12 carbon atoms.
- benzyl group 4-methylbenzyl group, 4-methoxybenzyl group, 4-chlorobenzyl group, 4-bromobenzyl group, 2-phenylethyl group, 1-phenylethyl group, 3-phenylpropyl group, etc.
- benzyl group 4-methylbenzyl group, 4-methoxybenzyl group, 4-chlorobenzyl group, 4-bromobenzyl group, 2-phenylethyl group, 1-phenylethyl group, 3-phenylpropyl group, etc.
- benzyl group 4-methylbenzyl group, 4-methoxybenzyl group, 4-chlorobenzyl group, 4-bromobenzyl group, 2-phenylethyl group, 1-phenylethyl group, 3-phenyl
- R examples of the optionally substituted aryl group having 6 to 12 carbon atoms represented by R include a phenyl group, 1-naphthyl group, 2-naphthyl group, o-methylphenyl group, m-methylphenyl group, p -Methylphenyl group, o-methoxyphenyl group, m-methoxyphenyl group, p-methoxyphenyl group, 2,3-dimethylphenyl group, 2,4-dimethylphenyl group, 2,5-dimethylphenyl group, 2,6 -Dimethylphenyl group, 3,4-dimethylphenyl group, 3,5-dimethylphenyl group, 2,3,5-trimethylphenyl group, 2,3,6-trimethylphenyl group, 2,4,6-trimethylphenyl group And o-nitrophenyl group, m-nitrophenyl group, p-nitrophenyl group and the like are preferable.
- R is preferably
- the hydrolase protein of the present invention When the hydrolase protein of the present invention is allowed to act on dialkyl 2-vinylcyclopropane-1,1-dicarboxylic acid, the hydrolase protein of the present invention, which has been purified or roughly purified, is produced.
- (1S) by allowing a microorganism (eg, a transformant having a DNA encoding the hydrolase protein of the present invention) or a treated product thereof to act on a dialkyl 2-vinylcyclopropane-1,1-dicarboxylic acid.
- 2S -1-alkoxycarbonyl-2-vinylcyclopropanecarboxylic acid.
- the reaction method is not particularly limited, and a dialkyl ⁇ 2-vinylcyclopropane-1,1-dicarboxylic acid serving as a substrate is added to a liquid containing the hydrolase protein of the present invention, and an appropriate temperature (for example, 10 ° C. to 40 ° C.). Or about pressure (eg about atmospheric pressure). Thereby, (1S, 2S) -1-alkoxycarbonyl-2-vinylcyclopropanecarboxylic acid can be produced.
- the method for fractionating the desired (1S, 2S) -1-alkoxycarbonyl-2-vinylcyclopropanecarboxylic acid from the reaction mixture is not particularly limited, and any method for separation or purification known to those skilled in the art may be used. it can. For example, it can be carried out by solvent extraction, crystallization, resin adsorption, column chromatography, etc., but is not limited thereto.
- the hydrolase protein of the present invention is a method for producing (1S, 2S) -1-ethoxycarbonyl-2-vinylcyclopropanecarboxylic acid by hydrolyzing diethyl 2-vinylcyclopropane-1,1-dicarboxylic acid In particular, it can be suitably used.
- the dialkyl 2-vinylcyclopropane-1,1-dicarboxylic acid of the present invention can be produced by the reaction of the following formula (3). That is, it can be produced by reacting the raw material compound of the formula (3) with malonic acid esters in the presence of an alkali metal alkoxide or an alkali metal hydride.
- R 3 represents an optionally substituted arylsulfonyl group having 6 to 12 carbon atoms, an alkylsulfonyl group having 1 to 10 carbon atoms, and an aralkylsulfonyl group having 7 to 20 carbon atoms.
- Examples include benzenesulfonyl group, p-toluenesulfonyl group, 1-naphthalenesulfonyl group, 2-naphthalenesulfonyl group, methanesulfonyl group, ethanesulfonyl group, propanesulfonyl group, trifluoromethanesulfonyl group, and benzylsulfonyl group.
- R 3 is preferably a methanesulfonyl group, a benzenesulfonyl group or a p-toluenesulfonyl group, particularly preferably a p-toluenesulfonyl group.
- R is the same as defined above.
- the starting compound of the formula (3) can be produced according to a known method, for example, a method described in Frederic Dolle et al., Bioorg. Med. Chem. 2006, 14, 1115-1125 and the like. Moreover, it can manufacture by reaction of the following formula
- 1,4-butenediol is reacted with a compound represented by R 3 X and then crystallized, or a commercially available raw material compound is hydrolyzed with an acid or a base to produce 1,4-butenediol. It can be produced by obtaining butenediol and further reacting with a compound represented by R 3 X, followed by crystallization.
- X 1 represents a hydrogen atom or R 1
- X 2 represents a hydrogen atom or R 2
- R 1 and R 2 each independently have 2 to 11 carbon atoms that may be substituted
- X 1 is R 1 and X 2 is R 2 , more preferably R 1 and R 2 are an acetyl group, an ethylcarbonyl group, a tert-butylcarbonyl group, and a benzoyl group, and more preferably an acetyl group It is.
- (1S, 2S) -1-ethoxycarbonyl-2-vinylcyclopropanecarboxylic acid obtained in the present invention is useful for the production of various HCV NS3 protease inhibitors and the like that are under development as therapeutic agents for hepatitis C.
- An intermediate (1R, 2S) / (1S, 2R) -1-amino-1-ethoxycarbonyl-2-vinylcyclopropane and a salt thereof can be produced.
- Example 1 Cloning of paranitrobenzyl esterase gene (pnbA; SEQ ID NOs: 6 to 10) (1) Cloning of gene Bacillus subtilis ATCC23857, Bacillus subtilis NBRC3026, Bacillus subtilis (Bacillus subtilis) ) DNeasy Blood & Tissue Kit from cells obtained by culturing NBRC3108, Bacillus subtilis NBRC3027, and Bacillus subtilis NBRC3013 overnight in a liquid medium designated by each strain preservation institution. Chromosomal DNA was prepared using (Qiagen).
- pnbA23857 SEQ ID NO: 6
- PNBE23857 GenBank Accession No. ZP_03593235, SEQ ID NO: 1
- Primers pnbA F and pnbA R for amplifying the full length of the esterase gene were designed and synthesized.
- the respective base sequences are shown in SEQ ID NOs: 11 (pnbAF) and 12 (pnbA R) in the sequence listing.
- PCR polymerase chain reaction
- PCR was performed using KOD-plus-Ver.2 (manufactured by Toyobo Co., Ltd.) according to the conditions of the attached instruction manual. Regarding the temperature condition, after holding at 94 ° C. for 2 minutes, (94 ° C., 15 seconds; 58 ° C., 30 seconds; 68 ° C., 4 minutes) was repeated 30 cycles, and the temperature was held at 68 ° C. for 5 minutes to finish.
- pnbA3026 For the plasmids ppnbA3026, ppnbA3108, ppnbA3027, and ppnbA3013, the inserted DNA sequence was analyzed, and it was confirmed that each gene contained a 1467 bp ORF. These genes were named pnbA3026, pnbA3108, pnbA3027, and pnbA3013, respectively. Each sequence was as shown by SEQ ID NO: 7 (pnbA3026), 8 (pnbA3108), 9 (pnbA3027) and 10 (pnbA3013). The amino acid sequences encoded by the respective DNA sequences were named PNBE3026, PNBE3108, PNBE3027, and PNBE3013.
- Each sequence was as shown in SEQ ID NO: 2 (PNBE3026), 3 (PNBE3108), 4 (PNBE3027) and 5 (PNBE3013).
- Each amino acid sequence showed a sequence identity of 97%, 91%, 90%, and 98% with respect to the known amino acid sequence of PNBE23857 (SEQ ID NO: 1), respectively.
- Example 2 Comparison of Selectivity of Novel Paranitrobenzyl Esterase Using the five plasmids obtained in Example 1, Escherichia coli JM109 (manufactured by Takara Bio Inc.) was transformed according to a conventional method. Each of the obtained recombinant Escherichia coli was cultured by shaking at 30 ° C. in a liquid LB medium containing 20 mg / l kanamycin and 0.2 mM IPTG, and collected at 20 hours of culture.
- VCPDE 100 mM phosphoric acid containing 5 g / l racemic 1,1-diethoxycarbonyl-2-vinylcyclopropane
- VCPDE 5 g / l racemic 1,1-diethoxycarbonyl-2-vinylcyclopropane
- VCPME 1-ethoxycarbonyl-2-vinylcyclopropanecarboxylic acid
- the three enzymes PNBE3026, PNBE3108, and PNBE3027 showed higher values in the “1S2S optical purity” than the enzyme PNBE23857 with a known sequence.
- PNBE3013 showed a lower value in the “1S2R / 1S2S ratio” than the enzyme PNBE23857 with a known sequence.
- Synthesis Example VCPDE used in Example 1 was synthesized as follows. A 2 L 4-neck flask was charged with 93.4 g (583 mmol) of malonic acid diethyl ester and 1000 mL of toluene, and 223 mL (569 mmol) of a 20% sodium ethoxide ethanol solution was added as a base. After stirring at room temperature for 1.5 hours, 59.4 g of trans-1,4-dibromo-2-butene (278 mmol, Aldrich reagent) was added. After stirring at room temperature for 2 hours, 109 mL (278 mmol) of a 20% sodium ethoxide ethanol solution was further added.
- Example 3 Three-dimensional structure modeling of the hydrolase protein of the present invention (SEQ ID NO: 4)
- the amino acid sequence of PNBE3027 was subjected to a BLAST search against Protein Data Bank (PDB), and the following three types of crystal structures were referenced. Selected as.
- the notation A56V indicates that the 56th amino acid residue alanine (A) is substituted with valine (V).
- the S-form and R-form of the substrate VCPDE are generated in the direction of (1S, 2S) -VCPME and the direction of (1R, 2R) -VCPME. It was created by coordinating to the initial structure and minimizing energy. When performing energy minimization, the following distance constraint conditions were set in consideration of the hydrolysis reaction mechanism of esterase.
- Example 4 Estimation of amino acid residues involved in substrate recognition in paranitrobenzyl esterase First, in each of the “1S2S model” and “1R2R model” obtained in Example 3, the carbon at the 2-position of the vinyl group of VCPDE All amino acid residues that were partially or wholly within 8 cm from the atom were extracted. Use Discovery Studio Visualizer v2.5.5.9350 to calculate the distance. Among the extracted amino acid residues, other than the hydrogen atom closest to the carbon atom at the 2-position of the vinyl group of VCPDE of each model The distance from each atom was defined as the distance from each amino acid residue.
- Example 5 Modification of paranitrobenzyl esterase gene by mutagenesis Using the plasmid ppnbA3027 obtained in Example 1 as a template, a primer (L70FW) shown in SEQ ID NO: 13 and a primer (L70RV) shown in SEQ ID NO: 14 were used. Then, a random mutation was introduced into the base encoding leucine with amino acid number 70 by QuikChange Multi Site-Directed Mutagenesis Kit (Stratagene). About 200 colonies obtained were each cultured with shaking at 30 ° C. in a liquid LB medium containing 20 mg / l kanamycin and 0.2 mM IPTG, and collected at 20 hours of culture.
- reaction was performed in 100 mM potassium phosphate buffer containing 5 g / l or 30 g / l racemic VCPDE and 5% dimethyl sulfoxide at 30 ° C and pH 7 for 21 hours. I let you. Further, in the same manner as described above, a primer (L313FW) shown in SEQ ID NO: 15 and a primer (L313RV) shown in SEQ ID NO: 16 are used to randomly generate a base encoding the leucine residue of amino acid number 313. About 200 colonies were reacted in the same manner as described above.
- SEQ ID Nos: 1 to 20 in the present specification are as follows.
- SEQ ID No. 1 Amino acid sequence of Bacillus subtilis ATCC23857-derived paranitrobenzyl esterase (PNBE23857, GenBank Accession No.
- SEQ ID NO: 2 Amino acid sequence PNBE3026 of a hydrolase protein derived from Bacillus subtilis NBRC3026
- SEQ ID NO: 3 Amino acid sequence PNBE3108 of the hydrolase protein derived from Bacillus subtilis NBRC3108
- SEQ ID NO: 4 Amino acid sequence PNBE3027 of the hydrolase protein derived from Bacillus subtilis NBRC3027
- SEQ ID NO: 5 Amino acid sequence PNBE3013 of the hydrolase protein derived from Bacillus subtilis NBRC3013
- SEQ ID NO: 1 SEQ ID NO: 7: Base sequence of a DNA encoding a hydrolase protein derived from Bacillus subtilis NBRC3026 pnbA3026 SEQ ID NO: 8: Base sequence of DNA encoding a hydrolase protein derived from Bacillus subtilis NBRC3108 pnbA3108 SEQ ID NO: 9: Base sequence of DNA pnbA3027 encoding a hydrolase protein derived from Bacillus subtilis NBRC3027 SEQ ID NO: 10: Base sequence of DNA encoding the hydrolase protein derived from Bacillus subtilis NBRC3013 pnbA3013 SEQ ID NO: 11: Primer pnbA F for amplifying the full length of the paranitrobenzyl esterase gene SEQ ID NO: 12: Primer pnbA R for amplifying the full length of paranitrobenzyl esterase gene Sequence number 13: Primer (L70FW) Sequence number 14: Primer (L)
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- General Engineering & Computer Science (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Molecular Biology (AREA)
- Biomedical Technology (AREA)
- Analytical Chemistry (AREA)
- Enzymes And Modification Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
Description
[1] 配列番号2~5のいずれかに示すアミノ酸配列からなり、式(1)に示す反応を、配列番号1に示すアミノ酸配列からなるタンパク質より高い選択性で触媒する活性を有する加水分解酵素タンパク質。


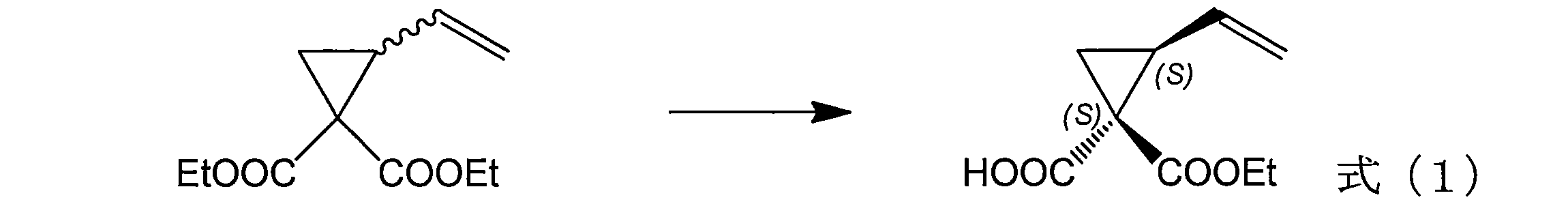


(a)配列番号7から10に記載の塩基配列;
(b)配列番号7から10に記載の塩基配列からなるDNAと、ストリンジェントな条件化でハイブリダイズすることができるDNAであって、式(1)に示す反応を、配列番号1に示すアミノ酸配列からなるタンパク質より高い選択性で触媒する活性を有する加水分解酵素タンパク質をコードする塩基配列:又は
(c)配列番号7から10に記載の塩基配列において、1から数個の塩基の置換、欠失、及び/又は付加を有する塩基配列であって、式(1)に示す反応を、配列番号1に示すアミノ酸配列からなるタンパク質より高い選択性で触媒する活性を有する加水分解酵素タンパク質をコードする塩基配列:

(1)本発明の加水分解酵素タンパク質
本発明の加水分解酵素タンパク質は、配列番号2~5のいずれかに示すアミノ酸配列、又はそれに変異を施したアミノ酸配列からなり、式(1)に示す反応を、配列番号1に示すアミノ酸配列からなるタンパク質より高い選択性で触媒する活性を有することを特徴とする。ここで、配列番号1は、バシラス・サチリス (Bacillus subtilis) ATCC23857株由来のパラニトロベンジルエステラーゼ(PNBE23857、GenBank Accession No. ZP_03593235)のアミノ酸配列である。

ジアルキル 2-ビニルシクロプロパン-1,1-ジカルボン酸を加水分解して(1S,2S)-1-アルコキシカルボニル-2-ビニルシクロプロパンカルボン酸を効率的に得るための加水分解酵素タンパク質に求められる選択性としては以下の点が挙げられる。
まず、ジアルキル 2-ビニルシクロプロパン-1,1-ジカルボン酸には、(2R)体及び(2S)体の2種類の立体異性体が存在するが、2位での立体反転が起こる特殊な条件で無い限り、基本的には(2S)体のみが目的とする(1S,2S)-1-アルコキシカルボニル-2-ビニルシクロプロパンカルボン酸の原料となり得る。
ジアルキル (2S)-2-ビニルシクロプロパン-1,1-ジカルボン酸を加水分解する際、加水分解酵素タンパク質は、1位のプロキラルな炭素に結合した二つのアルコキシカルボニル基のうち、pro-Rなアルコキシカルボニル基のみを優先的に加水分解し、(1S,2S)-1-アルコキシカルボニル-2-ビニルシクロプロパンカルボン酸を優先的に生成する選択性を有する必要がある。なお、pro-Rとは、CX2YZ 上にある 2個の X について、それらを区別する表記法であり、概要は以下の通りである。C上の各置換基についてCIP則により優先順位を決める。そのとき、2個の X のうち一方の優先順位を他方よりも高いものと仮定する。Y、Z との優先順位の関係は変えない。仮の優先順位に基づき、RS表記法により中心炭素のキラリティが R か S かを決める。R体の場合は、そのときに優先させた X を pro-R とし、S体の場合はpro-S とする。
本選択性は、加水分解により生成する1-アルコキシカルボニル-2-ビニルシクロプロパンカルボン酸の(1S,2S)体と(1R,2S)体の比率を指標として比較可能であり、(1S,2S)体の生成量に比して(1R,2S)体の生成量の比率が低いものほど(1S,2S)-1-アルコキシカルボニル-2-ビニルシクロプロパンカルボン酸の収率が向上し、工業化において有利である。
次に、ラセミ体のジアルキル 2-ビニルシクロプロパン-1,1-ジカルボン酸を基質とする際に、(2R)体を加水分解酵素タンパク質により加水分解して、(1S,2R)体及び(1R,2R)体を生成し得るが、目的とする(1S,2S)体のエナンチオマーにあたる(1R,2R)体はその生成量を極力低く抑えることが好ましい。即ち、加水分解酵素タンパク質は、ジアルキル (2R)-2-ビニルシクロプロパン-1,1-ジカルボン酸の1位のプロキラルな炭素に結合した二つのアルコキシカルボニル基のうち、pro-Rなアルコキシカルボニル基のみを優先的に加水分解し、(1S,2R)-1-アルコキシカルボニル-2-ビニルシクロプロパンカルボン酸を優先的に生成する選択性、及び/又はジアルキル (2R)-2-ビニルシクロプロパン-1,1-ジカルボン酸に比してジアルキル (2S)-2-ビニルシクロプロパン-1,1-ジカルボン酸を優先的に加水分解する選択性を有する必要がある。いずれの選択性も、加水分解により生成する1-アルコキシカルボニル-2-ビニルシクロプロパンカルボン酸の(1S,2S)体の(1R,2R)体に対するエナンチオマー過剰率(% e.e.)により比較可能であり、(1S,2S)体のエナンチオマー過剰率が高いものほど後の製造工程や製造された医薬品の生理活性に悪影響が生じる可能性が低く、工業化において有利である。
また、上記したように、加水分解酵素タンパク質はジアルキル (2R)-2-ビニルシクロプロパン-1,1-ジカルボン酸の1位のプロキラルな炭素に結合した二つのアルコキシカルボニル基のうち、pro-Rなアルコキシカルボニル基を加水分解し、目的とする(1S,2S)体のジアステレオマーにあたる(1S,2R)-1-アルコキシカルボニル-2-ビニルシクロプロパンカルボン酸を生成し得るが、後段の製造工程への影響を軽減するため、この(1S,2R)体の混入も極力低減させることが好ましい。
本選択性は、生成する1-アルコキシカルボニル-2-ビニルシクロプロパンカルボン酸の(1S,2S)体の生成量と(1S,2R)体の生成量の比率により比較可能であり、(1S,2S)体の生成量に対する(1S,2R)体の生成量が低いものの方が後の製造工程や製造された医薬品の生理活性に悪影響が生じる可能性が低く、工業化において有利である。
即ち、本発明の「式(1)に示す反応を、配列番号1に示すアミノ酸配列からなるタンパク質より高い選択性で触媒する活性」とは、生成物である1-エトキシカルボニル-2-ビニルシクロプロパンカルボン酸における、「1R2S/1S2S比」、「1S2S体光学純度(% e.e.)」、あるいは「1S2R/1S2S比」を指標として決定することができる。
(a)配列番号1~5のいずれかに示すアミノ酸配列において、アミノ酸番号70番、106番、107番、108番、219番、270番、271番、272番、273番、274番、275番、276番、及び313番のアミノ酸のうち1つ以上が、野生型のアミノ酸に比べて側鎖の嵩高さが低いアミノ酸に置換されているアミノ酸配列からなり、上記式(1)に示す反応を、配列番号1に示すアミノ酸配列からなるタンパク質より高い選択性で触媒する活性を有する加水分解酵素タンパク質。
(b)配列番号1~5のいずれかに示すアミノ酸配列において、アミノ酸番号188番、190番、193番、215番、216番、217番、314番、358番、362番、及び363番のアミノ酸のうち1つ以上が、野生型のアミノ酸に比べて側鎖の嵩高さが高いアミノ酸に置換されているアミノ酸配列からなり、上記式(1)に示す反応を、配列番号1に示すアミノ酸配列からなるタンパク質より高い選択性で触媒する活性を有する加水分解酵素タンパク質。
(c)配列番号1~5のいずれかに示すアミノ酸配列において、アミノ酸番号70番、106番、107番、108番、219番、270番、271番、272番、273番、274番、275番、276番、及び313番のアミノ酸のうち1つ以上が、野生型のアミノ酸に比べて側鎖の嵩高さが低いアミノ酸に置換されていて、かつアミノ酸番号188番、190番、193番、215番、216番、217番、314番、358番、362番、及び363番のアミノ酸のうち1つ以上が、野生型のアミノ酸に比べて側鎖の嵩高さが高いアミノ酸に置換されているアミノ酸配列からなり、上記式(1)に示す反応を、配列番号1に示すアミノ酸配列からなるタンパク質より高い選択性で触媒する活性を有する加水分解酵素タンパク質。
(d)上記(a)又は(c)の加水分解酵素タンパク質であって、少なくとも、アミノ酸番号70番、270番、273番、及び313番のアミノ酸のうち1つ以上のアミノ酸が置換されているアミノ酸配列からなり、上記式(1)に示す反応を、配列番号1に示すアミノ酸配列からなるタンパク質より高い選択性で触媒する活性を有する加水分解酵素タンパク質。
本発明においては、特に(d)の加水分解酵素タンパク質が好ましい。
Arg(アルギニン):14.28
Asp(アスパラギン酸):11.68
Asn(アスパラギン):12.82
Cys(システイン):13.46
Glu(グルタミン酸):13.57
Gln(グルタミン):14.45
Gly(グリシン): 3.40
His(ヒスチジン):13.69
Leu(ロイシン):21.40
Ile(イソロイシン):21.40
Lys(リシン):15.71
Met(メチオニン):16.25
Phe(フェニルアラニン):19.80
Pro(プロリン):17.43
Ser(セリン): 9.47
Thr(スレオニン):15.77
Trp(トリプトファン):21.67
Tyr(チロシン):18.03
Val(バリン):21.57
特に、本発明の加水分解酵素タンパク質としては、配列番号1~5のいずれかに示すアミノ酸配列において、アミノ酸番号70番、270番、273番、又は313番のロイシン又はイソロイシンのうちの1つ以上が、これらよりも上記Bulkinessの値が小さいアミノ酸に置換されているものが好ましい。
(e)上記(d)の加水分解酵素タンパク質であって、配列番号1~5のいずれかに示すアミノ酸配列において、少なくともアミノ酸番号70番のロイシンが、アスパラギン酸、アスパラギン、セリン、スレオニン、又はグリシンのいずれかのアミノ酸に置換されている加水分解酵素タンパク質。
(f)上記(d)又は(e)の加水分解酵素タンパク質であって、配列番号3又は4に示すアミノ酸配列において、アミノ酸番号270番のロイシンが、セリン、グルタミン、グルタミン酸、又はアラニンのいずれかのアミノ酸に置換されている加水分解酵素タンパク質。
(g)上記(d)又は(e)の加水分解酵素タンパク質であって、配列番号1、2又は5のいずれかに示すアミノ酸配列において、アミノ酸番号270番のイソロイシンが、セリン、グルタミン、グルタミン酸、又はアラニンのいずれかのアミノ酸に置換されている加水分解酵素タンパク質。
(h)上記(d)から(g)のいずれかの加水分解酵素タンパク質であって、配列番号1~5のいずれかに示すアミノ酸配列において、アミノ酸番号273番のロイシンが、アルギニン、又はヒスチジンのいずれかのアミノ酸に置換されている加水分解酵素タンパク質。
(i)上記(d)から(h)のいずれかの加水分解酵素タンパク質であって、配列番号1~5のいずれかに示すアミノ酸配列において、アミノ酸番号313番のロイシンが、メチオニン、アラニン、又はプロリンのいずれかのアミノ酸に置換されている加水分解酵素タンパク質。
本発明によれば、上記した本発明の加水分解酵素タンパク質をコードするDNAが提供される。
本発明のDNAの具体例としては、以下の塩基配列からなるDNAが挙げられる。
(a)配列番号7から10に記載の塩基配列;
(b)配列番号7から10に記載の塩基配列からなるDNAと、ストリンジェントな条件化でハイブリダイズすることができるDNAであって、式(1)に示す反応を、配列番号1に示すアミノ酸配列からなるタンパク質より高い選択性で触媒する活性を有する加水分解酵素タンパク質をコードする塩基配列:又は
(c)配列番号7から10に記載の塩基配列において、1から数個の塩基の置換、欠失、及び/又は付加を有する塩基配列であって、式(1)に示す反応を、配列番号1に示すアミノ酸配列からなるタンパク質より高い選択性で触媒する活性を有する加水分解酵素タンパク質をコードする塩基配列:

本発明によればさらに、本発明のDNAを有する組み換えベクターが提供される。組み換えベクターを調製する際には、通常、本発明のDNAを宿主微生物に適したプロモーターとともに、このプロモーターの下流に本発明のDNAのコード領域の5'末端側が連結されるようにして、ベクターに挿入する。あるいはプロモーターを含む発現ベクターを用い、これに本発明のDNAを挿入してもよい。
本発明で用いることができる宿主は、ジアルキル 2-ビニルシクロプロパン-1,1-ジカルボン酸、あるいは目的物である(1S,2S)-1-アルコキシカルボニル-2-ビニルシクロプロパンカルボン酸と接触させ、その結果、得られる化合物を分析することにより選択することができる。
本発明で用いることができる宿主微生物としては、具体的には、エシェリヒア(Escherichia)属細菌(例えば、大腸菌)が挙げられ、また、放線菌(Actinomycetes)属細菌、バチルス(Bacillus)属細菌、セラチア(Serratia)属細菌、シュードモナス(Pseudomonas)属細菌、コリネバクテリウム(Corynebacterium)属細菌、ブレビバクテリウム(Brevibacterium)属細菌、ロドコッカス(Rhodococcus)属細菌、ラクトバチルス(Lactobacillus)属細菌、ストレプトマイセス(Streptomyces)属細菌、サーマス(Thermus)属細菌、ストレプトコッカス(Streptococcus)属細菌等も、上記性質を有するものは、本発明のDNA又は組み換えベクターを導入する宿主として用いることができる。
また、上記微生物において、ジアルキル 2-ビニルシクロプロパン-1,1-ジカルボン酸を(1S,2S)-1-アルコキシカルボニル-2-ビニルシクロプロパンカルボン酸以外に加水分解する活性や生成された(1S,2S)-1-アルコキシカルボニル-2-ビニルシクロプロパンカルボン酸をさらに別の化合物に変化させる活性を有するものは、該活性を有する酵素タンパク質をコードする遺伝子を既存の方法により破壊することにより、本発明のDNA又は組み換えベクターを導入する宿主として用いることができる。
本発明によれば、上記のようにして得られる形質転換体を培養し、その培養物から本発明の加水分解酵素タンパク質を採取することができる。
本発明によれば、式(2)で表される製造、即ち、本発明の加水分解酵素タンパク質をジアルキル 2-ビニルシクロプロパン-1,1-ジカルボン酸に作用させて(1S,2S)-1-アルコキシカルボニル-2-ビニルシクロプロパンカルボン酸を製造することを含む、(1S,2S)-1-アルコキシカルボニル-2-ビニルシクロプロパンカルボン酸の製造方法が提供される。

Rとしては、好ましくはメチル基、エチル基、tert-ブチル基、及びベンジル基であり、さらに好ましくはエチル基である。


式(4)中、X1は水素原子又はR1を示し、X2は水素原子又はR2を示し、R1及びR2はそれぞれ独立して置換されていてもよい炭素数2~11のアルキルカルボニル基、炭素数8~21のアラルキルカルボニル基、又は炭素数7~13のアリールカルボニル基を示す。但し、X1及びX2が同時に水素原子となる場合を除く。好ましくは、X1がR1で且つX2がR2であり、より好ましくはR1及びR2がアセチル基、エチルカルボニル基、tert-ブチルカルボニル基、ベンゾイル基であり、さらに好ましくはアセチル基である。
(1)遺伝子のクローニング
バシラス・サチリス (Bacillus subtilis) ATCC23857、バシラス・サチリス (Bacillus subtilis) NBRC3026、バシラス・サチリス (Bacillus subtilis) NBRC3108、バシラス・サチリス (Bacillus subtilis) NBRC3027、及びバシラス・サチリス (Bacillus subtilis) NBRC3013を各菌株保存機関が指定する液体培地でそれぞれ一晩培養して得られた菌体より、DNeasy Blood & Tissue Kit (キアゲン社製)を用いて、染色体DNAをそれぞれ調製した。
調製した各菌株由来の染色体DNAを鋳型とし、pnbA F、pnbA Rをプライマーとして、ポリメラーゼ連鎖反応(PCR)により、それぞれ約1.5kbpのDNA断片を増幅した。
このときのPCRは、Easy-AハイフィデリティPCRクローニング酵素(アジレント・テクノロジー社製)を使用し、添付の取り扱い説明書の条件に従って反応を実施した。温度条件については、94℃で2分間保持の後、(94℃、30秒;50℃、30秒;72℃、90秒)を35サイクル繰り返し、72℃で5分間保持して終了した。
特開2005-34025号公報記載の方法に準じて調製したプラスミドpKV32をテンプレートとし、配列番号19に示すプライマー(pKVXmaIFW)と配列番号20に示すプライマー(pKVXmaIRV)を用いて、PCRにより約4kbpの断片を増幅した。増幅された断片を制限酵素XmaIにより消化した後、Ligation-Convenience Kit(ニッポンジーン社製)により自己閉環させてプラスミドを得た。得られたプラスミドをpKW32と命名した。
このときのPCRは、KOD -plus- Ver.2(東洋紡社製)を使用し、添付の取り扱い説明書の条件に従って反応を実施した。温度条件については、94℃で2分間保持の後、(94℃、15秒;58℃、30秒;68℃、4分)を30サイクル繰り返し、68℃で5分間保持して終了した。
上記(1)で得られたDNA断片はそれぞれ、Ligation-Convenience Kit(ニッポンジーン社製)を用いて、(2)で調製したクローニングベクターpKW32に導入した。以下、得られた各プラスミドをそれぞれppnbA23857、ppnbA3026、ppnbA3108、ppnbA3027、及びppnbA3013とする。
実施例1で得られた5種のプラスミドを用い、大腸菌(Escherichia coli) JM109(タカラバイオ株式会社製) を常法に従い形質転換した。得られた組換え大腸菌それぞれをカナマイシン 20 mg/l、IPTG 0.2mMを含む液体LB培地を用いて30℃で振盪培養し、培養20時間目に集菌した。
得られた菌体を用いて、5 g/lのラセミ体1,1-ジエトキシカルボニル-2-ビニルシクロプロパン(以下、「VCPDE」と称する。)及び5%のジメチルスルホキシドを含む100mM リン酸カリウム緩衝液中で、30℃、pH 7の条件で21時間反応させた。
カラム: CHIRALPAK AD-3(4.6 x 250 mm、ダイセル化学社製)
溶離液: ヘキサン : エタノール : トリフルオロ酢酸 = 95 : 5 : 0.1
流速: 0.8 ml/min
温度: 30℃
検出: UV 210 nm
また、PNBE3013は「1S2R/1S2S比」において、配列既知の酵素PNBE23857に比べて低い値を示した。

実施例1で使用したVCPDEは以下のようにして合成した。
2L4口フラスコに、マロン酸ジエチルエステル93.4g(583mmol)、トルエン1000mLを仕込み、塩基として20%ナトリウムエトキシドエタノール溶液223mL(569mmol)を加えた。室温下、1.5時間撹拌後、trans-1,4-ジブロモ-2―ブテン59.4g(278mmol、アルドリッチ社製試薬)を加えた。室温下、2時間撹拌後、さらに20%ナトリウムエトキシドエタノール溶液109mL(278mmol)を加えた。室温下、1時間撹拌後、trans-1,4-ジブロモ-2―ブテン29.1g(136mmol、アルドリッチ社製試薬)を加えた。室温下、4時間撹拌後、1M水酸化ナトリウム水溶液347mLを添加して14時間撹拌後、有機層を分離した。次いで有機層を水132mLで2回洗浄し、無水硫酸ナトリウムで乾燥後に濾過し、得られた濾液を40℃にて減圧濃縮し、淡黄色油状物の粗生成物として1,1-ジ-エトキシカルボニル-2-ビニルシクロプロパン99.3gを得た。この粗生成物の中に1,1-ジ-エトキシカルボニル-2-ビニルシクロプロパンは83.2g(収率94.9%)含まれていた。また、マロン酸ジエチルエステルは含まれていなかった。
PNBE3027のアミノ酸配列について、Protein Data Bank (PDB)に対してBLAST検索を行い、以下の3種の結晶構造を参照構造として選択した。なお、以下においてA56Vという表記は、56番目のアミノ酸残基アラニン(A)がバリン(V)に置換していることを示す。
・para-nitrobenzyl esterase Mutation: A56V, I60V, T73K, L144M, L313F, H322Y, A343V, M358V, Y370F, A400T, G412E, I437T, T459S(PDB id: 1C7J) 配列一致度89%
・para-nitrobenzyl esterase Mutation: I60V, L144M, P317S, H322R, L334S, M358V, Y370F (PDB id: 1C7I) 配列一致度90%
・para-nitrobenzyl esterase (PDB id: 1QE3) 配列一致度91%
・活性部位Ser189のアルコール酸素原子と加水分解されるエステル結合のカルボニル炭素の距離を2.0Å以上3.0Å以内とする。
以上の処理により得られた構造を「1S2Sモデル」及び「1R2Rモデル」とした。
まず、実施例3で得られた「1S2Sモデル」及び「1R2Rモデル」それぞれにおいて、VCPDEのビニル基の2位の炭素原子から8Å以内に一部もしくは全部が存在するアミノ酸残基を全て抽出した。距離の計算にはDiscovery Studio Visualizer v2.5.5.9350を使用し、抽出された各アミノ酸残基の中でそれぞれのモデルのVCPDEのビニル基の2位の炭素原子からの距離が最も近い水素原子以外の原子との距離を各アミノ酸残基との距離と規定した。抽出された各アミノ酸残基について、規定した距離を「1S2Sモデル」における距離と「1R2Rモデル」における距離とで比較した結果、「1R2Rモデル」に比べて「1S2Sモデル」における距離が近いアミノ酸残基を表2に、「1S2Sモデル」に比べて「1R2Rモデル」における距離が近いアミノ酸残基を表3に示した。


CA = Carbon, Alpha
CB = Carbon, Beta
CG = Oxygen, Gamma
CD = Carbon, Delta
CE = Carbon, Epsilon
CZ = Carbon, Zeta
OE = Oxygen, Epsilon
実施例1で得たプラスミドppnbA3027を鋳型として、配列表の配列番号13に示すプライマー(L70FW)と配列番号14に示すプライマー(L70RV)を用いて、QuikChange Multi Site-Directed Mutagenesis Kit (ストラタジーン社製)によりアミノ酸番号70番のロイシンをコードする塩基にランダムな変異を導入した。得られた約200個のコロニーをそれぞれカナマイシン 20 mg/l、IPTG 0.2mMを含む液体LB培地を用いて30℃で振盪培養し、培養20時間目に集菌した。
得られた菌体を用いて、5 g/lもしくは30 g/lのラセミ体VCPDE、5%のジメチルスルホキシドを含む100mM リン酸カリウム緩衝液中で、30℃、pH 7の条件で21時間反応させた。
また、上記と同様にして、配列表の配列番号15に示すプライマー(L313FW)と配列番号16に示すプライマー(L313RV)を用いて、アミノ酸番号313番のロイシン残基をコードする塩基に対してランダムな変異を導入し、約200個のコロニーを上記と同様の方法で反応させた。
さらに、上記と同様にして、配列表の配列番号17に示すプライマー(L270L273FW)と配列番号18に示すプライマー(L270L273RV)を用いて、アミノ酸番号270と273番のロイシン残基をコードする塩基に対してランダムな変異を導入し、約400個のコロニーを上記と同様の方法で反応させた。


P:プロリン
G:グリシン
T:スレオニン
S:セリン
N:アスパラギン
A:アラニン
M:メチオニン
D:アスパラギン酸
F:フェニルアラニン
H:ヒスチジン
R:アルギニン
E:グルタミン酸
Q:グルタミン
配列番号1:バシラス・サチリス (Bacillus subtilis) ATCC23857由来パラニトロベンジルエステラーゼ(PNBE23857、GenBank Accession No. ZP_03593235)のアミノ酸配列
配列番号2:バシラス・サチリス (Bacillus subtilis) NBRC3026由来加水分解酵素タンパク質のアミノ酸配列PNBE3026
配列番号3:バシラス・サチリス (Bacillus subtilis) NBRC3108由来加水分解酵素タンパク質のアミノ酸配列PNBE3108
配列番号4:バシラス・サチリス (Bacillus subtilis) NBRC3027由来加水分解酵素タンパク質のアミノ酸配列PNBE3027
配列番号5:バシラス・サチリス (Bacillus subtilis) NBRC3013由来加水分解酵素タンパク質のアミノ酸配列PNBE3013
配列番号6:バシラス・サチリス (Bacillus subtilis) ATCC23857由来パラニトロベンジルエステラーゼ(PNBE23857、GenBank Accession No. ZP_03593235、配列番号1)をコードする遺伝子配列(pnbA23857)
配列番号7:バシラス・サチリス (Bacillus subtilis) NBRC3026由来加水分解酵素タンパク質をコードするDNAの塩基配列pnbA3026
配列番号8:バシラス・サチリス (Bacillus subtilis) NBRC3108由来加水分解酵素タンパク質をコードするDNAの塩基配列pnbA3108
配列番号9:バシラス・サチリス (Bacillus subtilis) NBRC3027由来加水分解酵素タンパク質をコードするDNAの塩基配列pnbA3027
配列番号10:バシラス・サチリス (Bacillus subtilis) NBRC3013由来加水分解酵素タンパク質をコードするDNAの塩基配列pnbA3013
配列番号11:パラニトロベンジルエステラーゼ遺伝子の全長を増幅させるためのプライマーpnbA F
配列番号12:パラニトロベンジルエステラーゼ遺伝子の全長を増幅させるためのプライマーpnbA R
配列番号13:プライマー(L70FW)
配列番号14:プライマー(L70RV)
配列番号15:プライマー(L313FW)
配列番号16:プライマー(L313RV)
配列番号17:プライマー(L270L273FW)
配列番号18:プライマー(L270L273RV)
配列番号19:プライマー(pKVXmaIFW)
配列番号20:プライマー(pKVXmaIRV)
Claims (15)
- 配列番号2~5のいずれかに示すアミノ酸配列からなり、式(1)に示す反応を、配列番号1に示すアミノ酸配列からなるタンパク質より高い選択性で触媒する活性を有する加水分解酵素タンパク質。

- 配列番号1~5のいずれかに示すアミノ酸配列において、アミノ酸番号70番、106番、107番、108番、219番、270番、271番、272番、273番、274番、275番、276番、及び313番のアミノ酸のうち1つ以上が、野生型のアミノ酸に比べて側鎖の嵩高さが低いアミノ酸に置換されているアミノ酸配列からなり、式(1)に示す反応を、配列番号1に示すアミノ酸配列からなるタンパク質より高い選択性で触媒する活性を有する加水分解酵素タンパク質。

- 配列番号1~5のいずれかに示すアミノ酸配列において、アミノ酸番号188番、190番、193番、215番、216番、217番、314番、358番、362番、及び363番のアミノ酸のうち1つ以上が、野生型のアミノ酸に比べて側鎖の嵩高さが高いアミノ酸に置換されているアミノ酸配列からなり、式(1)に示す反応を、配列番号1に示すアミノ酸配列からなるタンパク質より高い選択性で触媒する活性を有する加水分解酵素タンパク質。

- 配列番号1~5のいずれかに示すアミノ酸配列において、アミノ酸番号70番、106番、107番、108番、219番、270番、271番、272番、273番、274番、275番、276番、及び313番のアミノ酸のうち1つ以上が、野生型のアミノ酸に比べて側鎖の嵩高さが低いアミノ酸に置換されていて、かつアミノ酸番号188番、190番、193番、215番、216番、217番、314番、358番、362番、及び363番のアミノ酸のうち1つ以上が、野生型のアミノ酸に比べて側鎖の嵩高さが高いアミノ酸に置換されているアミノ酸配列からなり、式(1)に示す反応を、配列番号1に示すアミノ酸配列からなるタンパク質より高い選択性で触媒する活性を有する加水分解酵素タンパク質。
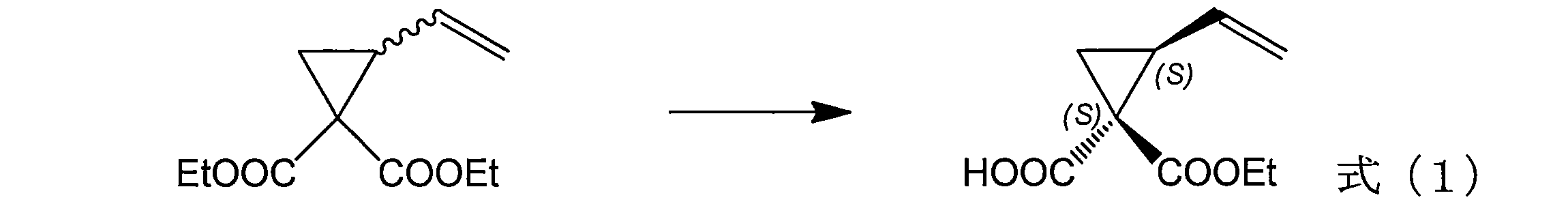
- 配列番号1~5のいずれかに示すアミノ酸配列において、少なくとも、アミノ酸番号70番、270番、273番、及び313番のアミノ酸のうち1つ以上のアミノ酸が置換されている、請求項2又は4に記載の加水分解酵素タンパク質。
- 配列番号1~5のいずれかに示すアミノ酸配列において、少なくともアミノ酸番号70番のロイシンが、アスパラギン酸、アスパラギン、セリン、スレオニン、又はグリシンのいずれかのアミノ酸に置換されている、請求項5に記載の加水分解酵素タンパク質。
- 配列番号3又は4に示すアミノ酸配列において、アミノ酸番号270番のロイシンが、セリン、グルタミン、グルタミン酸、又はアラニンのいずれかのアミノ酸に置換されている、請求項5又は6に記載の加水分解酵素タンパク質。
- 配列番号1、2又は5のいずれかに示すアミノ酸配列において、アミノ酸番号270番のイソロイシンが、セリン、グルタミン、グルタミン酸、又はアラニンのいずれかのアミノ酸に置換されている、請求項5又は6に記載の加水分解酵素タンパク質。
- 配列番号1~5のいずれかに示すアミノ酸配列において、アミノ酸番号273番のロイシンが、アルギニン、又はヒスチジンのいずれかのアミノ酸に置換されている、請求項5から8のいずれか1項に記載の加水分解酵素タンパク質。
- 配列番号1~5のいずれかに示すアミノ酸配列において、アミノ酸番号313番のロイシンが、メチオニン、アラニン、又はプロリンのいずれかのアミノ酸に置換されている、請求項5から9のいずれか1項に記載の加水分解酵素タンパク質。
- 配列番号4に示すアミノ酸配列において、アミノ酸番号70番のロイシンがアスパラギン酸に、270番のロイシンがグルタミン、グルタミン酸、又はアラニンのいずれかのアミノ酸に、273番目のロイシンがアルギニンに、かつ313番目のロイシンがメチオニンに置換されているアミノ酸配列からなり、式(1)に示す反応を、配列番号1に示すアミノ酸配列からなるタンパク質より高い選択性で触媒する活性を有する加水分解酵素タンパク質。

- 請求項1から11の加水分解酵素タンパク質のアミノ酸配列と90%以上の相同性を有するアミノ酸配列、又は請求項1から11の加水分解酵素タンパク質のアミノ酸配列に対して1から数個のアミノ酸の置換、欠失、及び/又は付加を有するアミノ酸配列からなり、式(1)に示す反応を、配列番号1に示すアミノ酸配列からなるタンパク質より高い選択性で触媒する活性を有する加水分解酵素タンパク質。

- 請求項1から12の何れかに記載の加水分解酵素タンパク質をコードするDNA。
- 以下の塩基配列からなる、請求項13に記載のDNA。
(a)配列番号7から10に記載の塩基配列;
(b)配列番号7から10に記載の塩基配列からなるDNAと、ストリンジェントな条件化でハイブリダイズすることができるDNAであって、式(1)に示す反応を、配列番号1に示すアミノ酸配列からなるタンパク質より高い選択性で触媒する活性を有する加水分解酵素タンパク質をコードする塩基配列:又は
(c)配列番号7から10に記載の塩基配列において、1から数個の塩基の置換、欠失、及び/又は付加を有する塩基配列であって、式(1)に示す反応を、配列番号1に示すアミノ酸配列からなるタンパク質より高い選択性で触媒する活性を有する加水分解酵素タンパク質をコードする塩基配列:

- 請求項1から12のいずれか1項に記載の加水分解酵素タンパク質をジアルキル 2-ビニルシクロプロパン-1,1-ジカルボン酸に作用させて(1S,2S)-1-アルコキシカルボニル-2-ビニルシクロプロパンカルボン酸を製造することを含む、(1S,2S)-1-アルコキシカルボニル-2-ビニルシクロプロパンカルボン酸の製造方法。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| SG2013007422A SG187652A1 (en) | 2010-08-31 | 2011-08-31 | Novel hydrolase protein |
| IN757CHN2013 IN2013CN00757A (ja) | 2010-08-31 | 2011-08-31 | |
| EP11821834.6A EP2612913B1 (en) | 2010-08-31 | 2011-08-31 | Novel hydrolase protein |
| JP2011539573A JP5657560B2 (ja) | 2010-08-31 | 2011-08-31 | 新規加水分解酵素タンパク質 |
| US13/813,047 US9029107B2 (en) | 2010-08-31 | 2011-08-31 | Hydrolase protein |
| CN201180009868.2A CN102834515B (zh) | 2010-08-31 | 2011-08-31 | 新的水解酶蛋白 |
| US14/637,429 US9334509B2 (en) | 2010-08-31 | 2015-03-04 | Hydrolase protein |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010194630 | 2010-08-31 | ||
| JP2010-194630 | 2010-08-31 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/813,047 A-371-Of-International US9029107B2 (en) | 2010-08-31 | 2011-08-31 | Hydrolase protein |
| US14/637,429 Division US9334509B2 (en) | 2010-08-31 | 2015-03-04 | Hydrolase protein |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012029819A1 true WO2012029819A1 (ja) | 2012-03-08 |
Family
ID=45772898
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/069680 WO2012029819A1 (ja) | 2010-08-31 | 2011-08-31 | 新規加水分解酵素タンパク質 |
Country Status (7)
| Country | Link |
|---|---|
| US (2) | US9029107B2 (ja) |
| EP (1) | EP2612913B1 (ja) |
| JP (1) | JP5657560B2 (ja) |
| CN (1) | CN102834515B (ja) |
| IN (1) | IN2013CN00757A (ja) |
| SG (1) | SG187652A1 (ja) |
| WO (1) | WO2012029819A1 (ja) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013137330A1 (ja) * | 2012-03-14 | 2013-09-19 | 積水メディカル株式会社 | 光学活性2-ビニルシクロプロパン-1,1-ジカルボン酸エステルの製造法 |
| WO2014098188A1 (ja) | 2012-12-20 | 2014-06-26 | 株式会社エーピーアイ コーポレーション | cis-5-ヒドロキシ-2-ピペリジンカルボン酸誘導体の製造方法およびcis-5-ヒドロキシ-2-ピペリジンカルボン酸の精製方法 |
| WO2014129459A1 (ja) | 2013-02-19 | 2014-08-28 | 株式会社エーピーアイ コーポレーション | L-リジン水酸化酵素およびそれを利用したヒドロキシ-l-リジンの製造法およびヒドロキシ-l-ピペコリン酸の製造法 |
| JP2016503662A (ja) * | 2013-01-18 | 2016-02-08 | コデクシス, インコーポレイテッド | カルバペネム合成に有用な人口生体触媒 |
| WO2019189724A1 (ja) * | 2018-03-30 | 2019-10-03 | 株式会社エーピーアイ コーポレーション | 新規加水分解酵素及びそれを利用した(1s,2s)-1-アルコキシカルボニル-2-ビニルシクロプロパンカルボン酸の製造方法 |
| WO2021107133A1 (ja) * | 2019-11-29 | 2021-06-03 | 日本マイクロバイオファーマ株式会社 | メイタンシノールの酵素的生産方法 |
| CN114480343A (zh) * | 2020-10-27 | 2022-05-13 | 湖北大学 | 具提升活性的pet水解酶 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9029107B2 (en) | 2010-08-31 | 2015-05-12 | Api Corporation | Hydrolase protein |
| TWI758313B (zh) * | 2016-10-12 | 2022-03-21 | 美商陶氏農業科學公司 | 一種用於製備(1r,3r)-及(1s,3s)-2,2-二鹵基-3-(經取代之苯基)環丙烷甲酸的方法 |
| DK3607097T3 (da) * | 2017-04-07 | 2023-09-18 | Dupont Nutrition Biosci Aps | Bacillus-værtsceller, der danner beta-galactosidaser og lactaser i fravær af p-nitrobenzylesterase-sideaktivitet |
| CN110283804A (zh) * | 2019-07-18 | 2019-09-27 | 江苏省农业科学院 | 酯酶基因及编码蛋白与应用 |
| CN115109765B (zh) * | 2022-06-24 | 2023-10-13 | 浙江工业大学 | 酯酶pnbA-BS突变体及其在制备l-薄荷醇中的应用 |
Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS57134500A (en) | 1981-02-12 | 1982-08-19 | Kyowa Hakko Kogyo Co Ltd | Plasmid pcg1 |
| JPS57183799A (en) | 1981-04-17 | 1982-11-12 | Kyowa Hakko Kogyo Co Ltd | Novel plasmid |
| JPS5835197A (ja) | 1981-08-26 | 1983-03-01 | Kyowa Hakko Kogyo Co Ltd | プラスミドpcg2 |
| JPS5867679A (ja) | 1981-09-30 | 1983-04-22 | アメリカン・サイアナミド・カンパニ− | イソシアン酸を三量化してシアヌル酸をつくる方法 |
| JPS5877895A (ja) | 1981-11-02 | 1983-05-11 | Ajinomoto Co Inc | プラスミドphm1519 |
| JPS58192900A (ja) | 1982-05-04 | 1983-11-10 | Ajinomoto Co Inc | 複合プラスミド |
| WO1987006269A1 (en) * | 1986-04-16 | 1987-10-22 | Sumitomo Chemical Company, Limited | Process for preparing optically active cyclopropanecarboxylic acids |
| JPH01191686A (ja) | 1988-01-26 | 1989-08-01 | Mitsubishi Petrochem Co Ltd | 複合プラスミド |
| JPH02276575A (ja) | 1989-01-31 | 1990-11-13 | Kibun Kk | 耐熱性の高いヌクレアーゼ画分の製造方法 |
| JPH03210184A (ja) | 1990-01-11 | 1991-09-13 | Mitsubishi Petrochem Co Ltd | 新規プラスミドベクター |
| US5750380A (en) | 1981-06-30 | 1998-05-12 | City Of Hope Research Institute | DNA polymerase mediated synthesis of double stranded nucleic acids |
| US6472184B1 (en) | 1997-08-22 | 2002-10-29 | Peter Hegemann | Method for producing nucleic acid polymers |
| JP2005034025A (ja) | 2003-07-18 | 2005-02-10 | Mitsubishi Pharma Corp | 大腸菌での異種遺伝子発現を促進させるプラスミドベクター、組換え大腸菌、該組換え大腸菌を用いた化学物質製造法及び組換えタンパク質の製造法 |
| WO2007017181A2 (de) * | 2005-08-05 | 2007-02-15 | Henkel Kommanditgesellschaft Auf Aktien | Verwendung von esterasen zur spaltung von kunststoffen |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5180671A (en) | 1986-04-16 | 1993-01-19 | Sumitomo Chemical Company, Limited | Method for producing optically active cyclopropane carboxylic acid |
| JPH0679557B2 (ja) | 1986-04-16 | 1994-10-12 | 住友化学工業株式会社 | 光学活性なシス−シクロプロパンカルボン酸の製造方法 |
| US5468632A (en) * | 1991-12-20 | 1995-11-21 | Eli Lilly And Company | Recombinant DNA compounds and expression vectors encoding para-nitrobenzyl esterase activity from bacillus |
| US5741691A (en) * | 1996-01-23 | 1998-04-21 | California Institute Of Technology | Para-nitrobenzyl esterases with enhanced activity in aqueous and nonaqueous media |
| US5945325A (en) * | 1998-04-20 | 1999-08-31 | California Institute Of Technology | Thermally stable para-nitrobenzyl esterases |
| EP1712630A4 (en) | 2004-02-04 | 2008-04-16 | Api Corp | PROCESS FOR THE PRODUCTION OF OPTICALLY ACTIVE ALCOHOL AND OPTICALLY ACTIVE CARBOXYLIC ACID |
| EP2537825B1 (en) * | 2010-02-16 | 2016-09-14 | API Corporation | Method for producing 1-amino-1-alkoxycarbonyl-2-vinylcyclopropane |
| US9029107B2 (en) | 2010-08-31 | 2015-05-12 | Api Corporation | Hydrolase protein |
-
2011
- 2011-08-31 US US13/813,047 patent/US9029107B2/en active Active
- 2011-08-31 IN IN757CHN2013 patent/IN2013CN00757A/en unknown
- 2011-08-31 CN CN201180009868.2A patent/CN102834515B/zh active Active
- 2011-08-31 SG SG2013007422A patent/SG187652A1/en unknown
- 2011-08-31 EP EP11821834.6A patent/EP2612913B1/en active Active
- 2011-08-31 WO PCT/JP2011/069680 patent/WO2012029819A1/ja active Application Filing
- 2011-08-31 JP JP2011539573A patent/JP5657560B2/ja active Active
-
2015
- 2015-03-04 US US14/637,429 patent/US9334509B2/en not_active Expired - Fee Related
Patent Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS57134500A (en) | 1981-02-12 | 1982-08-19 | Kyowa Hakko Kogyo Co Ltd | Plasmid pcg1 |
| JPS57183799A (en) | 1981-04-17 | 1982-11-12 | Kyowa Hakko Kogyo Co Ltd | Novel plasmid |
| US5750380A (en) | 1981-06-30 | 1998-05-12 | City Of Hope Research Institute | DNA polymerase mediated synthesis of double stranded nucleic acids |
| JPS5835197A (ja) | 1981-08-26 | 1983-03-01 | Kyowa Hakko Kogyo Co Ltd | プラスミドpcg2 |
| JPS5867679A (ja) | 1981-09-30 | 1983-04-22 | アメリカン・サイアナミド・カンパニ− | イソシアン酸を三量化してシアヌル酸をつくる方法 |
| JPS5877895A (ja) | 1981-11-02 | 1983-05-11 | Ajinomoto Co Inc | プラスミドphm1519 |
| JPS58192900A (ja) | 1982-05-04 | 1983-11-10 | Ajinomoto Co Inc | 複合プラスミド |
| WO1987006269A1 (en) * | 1986-04-16 | 1987-10-22 | Sumitomo Chemical Company, Limited | Process for preparing optically active cyclopropanecarboxylic acids |
| JPH01191686A (ja) | 1988-01-26 | 1989-08-01 | Mitsubishi Petrochem Co Ltd | 複合プラスミド |
| JPH02276575A (ja) | 1989-01-31 | 1990-11-13 | Kibun Kk | 耐熱性の高いヌクレアーゼ画分の製造方法 |
| JPH03210184A (ja) | 1990-01-11 | 1991-09-13 | Mitsubishi Petrochem Co Ltd | 新規プラスミドベクター |
| US6472184B1 (en) | 1997-08-22 | 2002-10-29 | Peter Hegemann | Method for producing nucleic acid polymers |
| JP2005034025A (ja) | 2003-07-18 | 2005-02-10 | Mitsubishi Pharma Corp | 大腸菌での異種遺伝子発現を促進させるプラスミドベクター、組換え大腸菌、該組換え大腸菌を用いた化学物質製造法及び組換えタンパク質の製造法 |
| WO2007017181A2 (de) * | 2005-08-05 | 2007-02-15 | Henkel Kommanditgesellschaft Auf Aktien | Verwendung von esterasen zur spaltung von kunststoffen |
Non-Patent Citations (14)
| Title |
|---|
| ALUPEI V. ET AL.: "Cyclodextrins in polymer synthesis: synthesis and influence of methylated p-cyclodextrin on the radical polymerization behavior of 1,1-disubstituted 2-vinylcyclopropane in aqueous medium.", MACROMOL. RAPID COMMUN., vol. 22, no. 16, 2001, pages 1350 - 1353, XP002695900 * |
| BEAULIEU P.L. ET AL.: "Synthesis of (1R,2S)-1-amino-2-vinylcyclopropanecarboxylic acid (vinyl-ACCA) derivatives: key intermediates for the preparation of inhibitors of the hepatitis C virus NS3 protease.", J. ORG. CHEM., vol. 70, no. 15, 2005, pages 5869 - 5879, XP002686607 * |
| C. FLICHE ET AL., SYNTH. COMMUN., vol. 24, no. 20, 1994, pages 2873 - 2876 |
| E. OHTSUBO, GENETICS, vol. 64, 1970, pages 189 |
| FLICHE C. ET AL.: "Enantioselective synthesis of (1R,2S) and (1S,2S) dehydrocoronamic acids.", SYNTH. COMMUN., vol. 24, no. 20, 1994, pages 2873 - 2876, XP000857016 * |
| FREDERIC DOLLE ET AL., BIOORG. MED. CHEM., vol. 14, 2006, pages 1115 - 1125 |
| H. A. BOSE ET AL., PROC. NATL. ACAD. SCI. U. S. A., vol. 80, 1983, pages 21 |
| J. G. C. OTTOW, ANN. REV. MICROBIOL., vol. 29, 1975, pages 80 |
| J. INDUST. MICROBIOL., vol. 5, 1990, pages 159 |
| J. THEORET. BIOL., vol. 21, 1968, pages 170 |
| JOURNAL OF MOLECULAR BIOLOGY, vol. 53, 1970, pages 159 |
| M. H. GABOR, J. BACTERIOL., vol. 137, 1979, pages 1346 |
| See also references of EP2612913A4 * |
| ZOCK J. ET AL.: "The Bacillus subtilis pnbA gene encoding p-nitrobenzyl esterase: cloning, sequence and high-level expression in Escherichia coli.", GENE, vol. 151, no. 1-2, 1994, pages 37 - 43, XP023541807 * |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013137330A1 (ja) * | 2012-03-14 | 2013-09-19 | 積水メディカル株式会社 | 光学活性2-ビニルシクロプロパン-1,1-ジカルボン酸エステルの製造法 |
| JPWO2013137330A1 (ja) * | 2012-03-14 | 2015-08-03 | 積水メディカル株式会社 | 光学活性2−ビニルシクロプロパン−1,1−ジカルボン酸エステルの製造法 |
| WO2014098188A1 (ja) | 2012-12-20 | 2014-06-26 | 株式会社エーピーアイ コーポレーション | cis-5-ヒドロキシ-2-ピペリジンカルボン酸誘導体の製造方法およびcis-5-ヒドロキシ-2-ピペリジンカルボン酸の精製方法 |
| JP2016503662A (ja) * | 2013-01-18 | 2016-02-08 | コデクシス, インコーポレイテッド | カルバペネム合成に有用な人口生体触媒 |
| EP2946006A4 (en) * | 2013-01-18 | 2016-08-24 | Codexis Inc | MANIPULATED BIO-CATALYSTS FOR CARBAPENE SYNTHESIS |
| US9540622B2 (en) | 2013-01-18 | 2017-01-10 | Codexis, Inc. | Engineered biocatalysts useful for carbapenem synthesis |
| US9663771B2 (en) | 2013-01-18 | 2017-05-30 | Codexis, Inc. | Engineered biocatalysts useful for carbapenem synthesis |
| WO2014129459A1 (ja) | 2013-02-19 | 2014-08-28 | 株式会社エーピーアイ コーポレーション | L-リジン水酸化酵素およびそれを利用したヒドロキシ-l-リジンの製造法およびヒドロキシ-l-ピペコリン酸の製造法 |
| WO2019189724A1 (ja) * | 2018-03-30 | 2019-10-03 | 株式会社エーピーアイ コーポレーション | 新規加水分解酵素及びそれを利用した(1s,2s)-1-アルコキシカルボニル-2-ビニルシクロプロパンカルボン酸の製造方法 |
| JPWO2019189724A1 (ja) * | 2018-03-30 | 2021-04-01 | 株式会社エーピーアイ コーポレーション | 新規加水分解酵素及びそれを利用した(1s,2s)−1−アルコキシカルボニル−2−ビニルシクロプロパンカルボン酸の製造方法 |
| EP3778898A4 (en) * | 2018-03-30 | 2022-02-09 | API Corporation | NOVEL HYDROLASE AND PROCESS FOR THE PREPARATION OF (1S,2S)-1-ALKOXYCARBONYL-2-VINYLCYCLOPROPANECARBONIC ACID THEREFORE |
| US11572551B2 (en) | 2018-03-30 | 2023-02-07 | Api Corporation | Hydrolase and method for producing (1S,2S)-1-alkoxycarbonyl-2-vinylcyclopropane carboxylic acid using same |
| JP7458680B2 (ja) | 2018-03-30 | 2024-04-01 | 株式会社エーピーアイ コーポレーション | 新規加水分解酵素及びそれを利用した(1s,2s)-1-アルコキシカルボニル-2-ビニルシクロプロパンカルボン酸の製造方法 |
| WO2021107133A1 (ja) * | 2019-11-29 | 2021-06-03 | 日本マイクロバイオファーマ株式会社 | メイタンシノールの酵素的生産方法 |
| CN114480343A (zh) * | 2020-10-27 | 2022-05-13 | 湖北大学 | 具提升活性的pet水解酶 |
| CN114480343B (zh) * | 2020-10-27 | 2023-07-04 | 湖北大学 | 具提升活性的pet水解酶 |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2012029819A1 (ja) | 2013-10-31 |
| IN2013CN00757A (ja) | 2015-04-24 |
| EP2612913B1 (en) | 2016-07-27 |
| CN102834515A (zh) | 2012-12-19 |
| US20130130338A1 (en) | 2013-05-23 |
| CN102834515B (zh) | 2016-08-03 |
| JP5657560B2 (ja) | 2015-01-21 |
| US9334509B2 (en) | 2016-05-10 |
| EP2612913A4 (en) | 2014-03-26 |
| SG187652A1 (en) | 2013-03-28 |
| US9029107B2 (en) | 2015-05-12 |
| EP2612913A1 (en) | 2013-07-10 |
| US20150252393A1 (en) | 2015-09-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5657560B2 (ja) | 新規加水分解酵素タンパク質 | |
| CN109609475B (zh) | 草铵膦脱氢酶突变体及其合成l-草铵膦的应用 | |
| CN104099305A (zh) | 一种羰基还原酶突变体及其基因和应用 | |
| JP6476110B2 (ja) | L−リジン水酸化酵素およびそれを利用したヒドロキシ−l−リジンの製造法およびヒドロキシ−l−ピペコリン酸の製造法 | |
| CN110592036A (zh) | 一种草铵膦脱氢酶突变体及在氧化-还原多酶偶联生产l-草铵膦中的应用 | |
| JP4809660B2 (ja) | 3−キヌクリジノン還元酵素およびこれを用いる(r)−3−キヌクリジノールの製造方法 | |
| CN110885803A (zh) | 重组草铵膦脱氢酶、基因工程菌及其在制备l-草铵膦中的应用 | |
| US20230323317A1 (en) | Novel hydrolase and method for producing (1s,2s)-1-alkoxycarbonyl-2-vinylcyclopropane carboxylic acid using same | |
| JP5903298B2 (ja) | N−サクシニル−dl−アミノ酸に対する向上されたd体選択性を有する改変型d−サクシニラーゼ | |
| US9890406B2 (en) | Method for producing cathine | |
| US9783796B2 (en) | Amidase, gene for the same, vector, transformant, and method for production of optically active carboxylic acid amide and optically active carboxylic acid by using any one of those items | |
| US20080003640A1 (en) | Deinococcus N-acylamino acid racemase and use of preparing L-amino acid | |
| CN114774491B (zh) | 制备(2s,3r)-2-(邻苯二甲酰亚胺基甲基)-3-羟基丁酸酯的方法 | |
| KR102703236B1 (ko) | 입체 선택성이 향상된 칸디다 안타르티카 지방분해효소 b 변이체 | |
| EP1130108A1 (en) | Methods for racemizing N-acylamino acids and producing optically active amino acids | |
| US20100144010A1 (en) | Enzymatic Asymmetric Decarboxylation of Disubstituted Malonic Acids | |
| JP4618753B2 (ja) | N−アシル−アミノ酸のラセミ化方法、および光学活性アミノ酸の製造方法 | |
| US20160186217A1 (en) | Method for biocatalytic synthesis of substituted or unsubstituted phenylacetic acids and ketones having enzymes of microbial styrene degradation | |
| JP2024014657A (ja) | L-アルギニンまたはl-シトルリン生産能が向上したコリネバクテリウム属微生物およびこれを用いたl-アルギニンまたはl-シトルリンの生産方法 | |
| JP4270910B2 (ja) | 光学活性2−ヒドロキシ−2−トリフルオロ酢酸類の製造方法 | |
| Nojiri | Studies on enzymatic synthesis of optically active amides for pharmaceutical intermediates | |
| JP2024014656A (ja) | L-アルギニンまたはl-シトルリン生産能が向上したコリネバクテリウム属微生物およびこれを用いたl-アルギニンまたはl-シトルリンの生産方法 | |
| CN112048537A (zh) | (r)-泛酸内酯乙酸酯的合成 | |
| JP2008194037A (ja) | 生体触媒による4−ハロ−3−ヒドロキシ酪酸エステルの光学分割法 | |
| JP2015139426A (ja) | 新規β−アミノ酸の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180009868.2 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011539573 Country of ref document: JP |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11821834 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13813047 Country of ref document: US |
|
| REEP | Request for entry into the european phase |
Ref document number: 2011821834 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011821834 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |