WO2012017872A1 - 金属錯体色素、光電変換素子及び光電気化学電池 - Google Patents
金属錯体色素、光電変換素子及び光電気化学電池 Download PDFInfo
- Publication number
- WO2012017872A1 WO2012017872A1 PCT/JP2011/067009 JP2011067009W WO2012017872A1 WO 2012017872 A1 WO2012017872 A1 WO 2012017872A1 JP 2011067009 W JP2011067009 W JP 2011067009W WO 2012017872 A1 WO2012017872 A1 WO 2012017872A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- general formula
- dye
- integer
- layer
- Prior art date
Links
- VQMPTVQCADWACQ-UHFFFAOYSA-N CCC(C)(CC)N Chemical compound CCC(C)(CC)N VQMPTVQCADWACQ-UHFFFAOYSA-N 0.000 description 1
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F15/00—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic System
- C07F15/0006—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic System compounds of the platinum group
- C07F15/0046—Ruthenium compounds
- C07F15/0053—Ruthenium compounds without a metal-carbon linkage
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09B—ORGANIC DYES OR CLOSELY-RELATED COMPOUNDS FOR PRODUCING DYES, e.g. PIGMENTS; MORDANTS; LAKES
- C09B57/00—Other synthetic dyes of known constitution
- C09B57/10—Metal complexes of organic compounds not being dyes in uncomplexed form
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01G—CAPACITORS; CAPACITORS, RECTIFIERS, DETECTORS, SWITCHING DEVICES OR LIGHT-SENSITIVE DEVICES, OF THE ELECTROLYTIC TYPE
- H01G9/00—Electrolytic capacitors, rectifiers, detectors, switching devices, light-sensitive or temperature-sensitive devices; Processes of their manufacture
- H01G9/20—Light-sensitive devices
- H01G9/2059—Light-sensitive devices comprising an organic dye as the active light absorbing material, e.g. adsorbed on an electrode or dissolved in solution
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/30—Coordination compounds
- H10K85/341—Transition metal complexes, e.g. Ru(II)polypyridine complexes
- H10K85/344—Transition metal complexes, e.g. Ru(II)polypyridine complexes comprising ruthenium
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01G—CAPACITORS; CAPACITORS, RECTIFIERS, DETECTORS, SWITCHING DEVICES OR LIGHT-SENSITIVE DEVICES, OF THE ELECTROLYTIC TYPE
- H01G9/00—Electrolytic capacitors, rectifiers, detectors, switching devices, light-sensitive or temperature-sensitive devices; Processes of their manufacture
- H01G9/20—Light-sensitive devices
- H01G9/2004—Light-sensitive devices characterised by the electrolyte, e.g. comprising an organic electrolyte
- H01G9/2013—Light-sensitive devices characterised by the electrolyte, e.g. comprising an organic electrolyte the electrolyte comprising ionic liquids, e.g. alkyl imidazolium iodide
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01G—CAPACITORS; CAPACITORS, RECTIFIERS, DETECTORS, SWITCHING DEVICES OR LIGHT-SENSITIVE DEVICES, OF THE ELECTROLYTIC TYPE
- H01G9/00—Electrolytic capacitors, rectifiers, detectors, switching devices, light-sensitive or temperature-sensitive devices; Processes of their manufacture
- H01G9/20—Light-sensitive devices
- H01G9/2027—Light-sensitive devices comprising an oxide semiconductor electrode
- H01G9/2031—Light-sensitive devices comprising an oxide semiconductor electrode comprising titanium oxide, e.g. TiO2
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E10/00—Energy generation through renewable energy sources
- Y02E10/50—Photovoltaic [PV] energy
- Y02E10/542—Dye sensitized solar cells
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Power Engineering (AREA)
- Microelectronics & Electronic Packaging (AREA)
- Crystallography & Structural Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Plural Heterocyclic Compounds (AREA)
- Hybrid Cells (AREA)
- Photovoltaic Devices (AREA)
Abstract
【課題】変換効率が高く、さらに耐久性に優れた金属錯体色素、それを用いた光電変換素子及び光電気化学電池を提供する。 【解決手段】 下記一般式(1)で表される金属錯体色素。 M(LL1)m1(LL2)m2(X)m3・CI 一般式(1) [一般式(1)において、Mは金属原子を表し、LL1は特定の2座または3座の配位子、LL2は特定の2座または3座の配位子、Xは特定の1座または2座の配位子を表し、m1は1~3の整数を表し、m2は0~2の整数を表し、m3は0~3の整数を表し、CIは電荷を中和させるのに対イオンが必要な場合の対イオンを表、LL1、LL2、Xのいずれか1つは、少なくとも1つの酸性基を有する。
Description
本発明は、変換効率が高く、耐久性に優れた金属錯体色素、光電変換素子及び光電気化学電池に関する。
光電変換素子は各種の光センサー、複写機、太陽電池等に用いられている。この光電変換素子には金属を用いたもの、半導体を用いたもの、有機顔料や色素を用いたもの、あるいはこれらを組み合わせたものなどの様々な方式が実用化されている。中でも、非枯渇性の太陽エネルギーを利用した太陽電池は、燃料が不要であり、無尽蔵なクリーンエネルギーを利用したものとして、その本格的な実用化が大いに期待されている。この中でも、シリコン系太陽電池は古くから研究開発が進められてきた。各国の政策的な配慮もあって普及が進んでいる。しかし、シリコンは無機材料であり、スループット及び分子修飾には自ずと限界がある。
そこで色素増感型太陽電池の研究が精力的に行われている。とくに、スイスのローザンヌ工科大学のGraetzel等がポーラス酸化チタン薄膜の表面にルテニウム錯体からなる色素を固定した色素増感型太陽電池を開発し、アモルファスシリコン並の変換効率を実現した。これにより、色素増感型太陽電池が一躍世界の研究者から注目を集めるようになった。
特許文献1には、ルテニウム錯体色素の光電変換能を改善する方法が開示されている。
特許文献1では、2,2′-ビピリジン骨格上に特定の置換基を持つ2座または3座の配位子を利用して共役を拡大することにより、高いモル吸光係数を示し、長波長域においても優れた吸収能を有し、光電変換素子に使用した場合に耐久性に優れた金属錯体色素が得られることが報告されている。
しかしながら、長波長化効果、耐久性のいずれにおいても、特許文献1の金属錯体色素および光電変換素子はまだ十分とはいえない。
特許文献1では、2,2′-ビピリジン骨格上に特定の置換基を持つ2座または3座の配位子を利用して共役を拡大することにより、高いモル吸光係数を示し、長波長域においても優れた吸収能を有し、光電変換素子に使用した場合に耐久性に優れた金属錯体色素が得られることが報告されている。
しかしながら、長波長化効果、耐久性のいずれにおいても、特許文献1の金属錯体色素および光電変換素子はまだ十分とはいえない。
本発明の課題は、変換効率が高く、耐久性に優れ、さらに半導体微粒子への吸着安定性性に優れた金属錯体色素、それを用いた光電変換素子及び光電気化学電池を提供することにある。
本発明者等は、鋭意検討を重ねた結果、直線性の高い剛直な置換基で置換された配位子を有する金属錯体色素が、導電性支持体上に形成された多孔質半導体微粒子に配向吸着して、色素を剥離する原因となる水や色素を分解する求核種などの攻撃を受けにくいため、変換効率が高く、耐久性に優れる光電変換素子及び光電気化学電池を提供することができることを見出した。本発明はこの知見に基づきなされたものである。
本発明によれば、以下の手段が提供される。
<1>下記一般式(1)で表される金属錯体色素。
M(LL1)m1(LL2)m2(X)m3・CI 一般式(1)
[一般式(1)において、
Mは金属原子を表し、
LL1は下記一般式(2)で表される2座または3座の配位子を表し、
LL2は下記一般式(7)で表される2座または3座の配位子を表し、
Xはアシルオキシ基、アシルチオ基、チオアシルオキシ基、チオアシルチオ基、アシルアミノオキシ基、チオカルバメート基、ジチオカルバメート基、チオカルボネート基、ジチオカルボネート基、トリチオカルボネート基、アシル基、チオシアネート基、イソチオシアネート基、シアネート基、イソシアネート基、シアノ基、アルキルチオ基、アリールチオ基、アルコキシ基およびアリールオキシ基からなる群から選ばれた基で配位する1座または2座の配位子、あるいはハロゲン原子、カルボニル、ジアルキルケトン、1,3-ジケトン、カルボンアミド、チオカルボンアミドまたはチオ尿素からなる1座または2座の配位子を表し、
m1は1~3の整数を表し、m1が2以上のときLL1は同じでも異なっていてもよく、
m2は0~2の整数を表し、m2が2のときLL2は同じでも異なっていてもよく、
m3は0~3の整数を表し、m3が2以上のときXは同じでも異なっていてもよく、またX同士が連結していてもよく、
CIは電荷を中和させるのに対イオンが必要な場合の対イオンを表す。
LL1、LL2、Xのいずれか1つは、少なくとも1つの酸性基を有する。
M(LL1)m1(LL2)m2(X)m3・CI 一般式(1)
[一般式(1)において、
Mは金属原子を表し、
LL1は下記一般式(2)で表される2座または3座の配位子を表し、
LL2は下記一般式(7)で表される2座または3座の配位子を表し、
Xはアシルオキシ基、アシルチオ基、チオアシルオキシ基、チオアシルチオ基、アシルアミノオキシ基、チオカルバメート基、ジチオカルバメート基、チオカルボネート基、ジチオカルボネート基、トリチオカルボネート基、アシル基、チオシアネート基、イソチオシアネート基、シアネート基、イソシアネート基、シアノ基、アルキルチオ基、アリールチオ基、アルコキシ基およびアリールオキシ基からなる群から選ばれた基で配位する1座または2座の配位子、あるいはハロゲン原子、カルボニル、ジアルキルケトン、1,3-ジケトン、カルボンアミド、チオカルボンアミドまたはチオ尿素からなる1座または2座の配位子を表し、
m1は1~3の整数を表し、m1が2以上のときLL1は同じでも異なっていてもよく、
m2は0~2の整数を表し、m2が2のときLL2は同じでも異なっていてもよく、
m3は0~3の整数を表し、m3が2以上のときXは同じでも異なっていてもよく、またX同士が連結していてもよく、
CIは電荷を中和させるのに対イオンが必要な場合の対イオンを表す。
LL1、LL2、Xのいずれか1つは、少なくとも1つの酸性基を有する。

一般式(2)において、
R1およびR2はそれぞれ独立に酸性基を表し、
R3およびR4はそれぞれ独立に連結基を表し、
Y1およびY2はそれぞれ独立に一般式(3)~(6)のいずれかで表される環より水素原子を2つ脱離して得られる置換基を表し、
Y3およびY4は水素原子または置換基を表す。
L1またはL2はそれぞれ独立にアルキニレン、アリーレンまたはそれらの組み合わせからなる共役鎖を表す。
a1およびa2はそれぞれ独立に0~3の整数を表し、a1が2以上のときR1は同じでも異なっていてもよく、a2が2以上のときR2は同じでも異なっていてもよい。
b1およびb2はそれぞれ独立に0~3の整数を表し、b1が2以上のときR3は同じでも異なっていてもよく、互いに連結して環を形成してもよく、b2が2以上のときR4は同じでも異なっていてもよく、互いに連結して環を形成してもよく、b1およびb2が共に1以上のときR3とR4が連結して環を形成してもよい。
n1およびn2はそれぞれ独立に1以上の整数を表し、n3、n4はそれぞれ独立に1以上の整数を表す。ただし、n3が2以上の場合、複数のY1は同じでも異なってもよく、n4が2以上の場合、複数のY2は同じでも異なっても良い。
zは0または1を表す。
R1およびR2はそれぞれ独立に酸性基を表し、
R3およびR4はそれぞれ独立に連結基を表し、
Y1およびY2はそれぞれ独立に一般式(3)~(6)のいずれかで表される環より水素原子を2つ脱離して得られる置換基を表し、
Y3およびY4は水素原子または置換基を表す。
L1またはL2はそれぞれ独立にアルキニレン、アリーレンまたはそれらの組み合わせからなる共役鎖を表す。
a1およびa2はそれぞれ独立に0~3の整数を表し、a1が2以上のときR1は同じでも異なっていてもよく、a2が2以上のときR2は同じでも異なっていてもよい。
b1およびb2はそれぞれ独立に0~3の整数を表し、b1が2以上のときR3は同じでも異なっていてもよく、互いに連結して環を形成してもよく、b2が2以上のときR4は同じでも異なっていてもよく、互いに連結して環を形成してもよく、b1およびb2が共に1以上のときR3とR4が連結して環を形成してもよい。
n1およびn2はそれぞれ独立に1以上の整数を表し、n3、n4はそれぞれ独立に1以上の整数を表す。ただし、n3が2以上の場合、複数のY1は同じでも異なってもよく、n4が2以上の場合、複数のY2は同じでも異なっても良い。
zは0または1を表す。

一般式(3)~(6)において、R5~R11は置換基を表し、n5~n11はそれぞれ独立に0~2の整数を表し、複数のR5~R11は隣接する置換基と互いに結合して環を形成してもよい。n6とn7の和は2以下である。n8とn9の和は2以下である。n10とn11の和は2以下である。

一般式(7)において、Za、ZbおよびZcはそれぞれ独立に5または6員環を形成しうる非金属原子群を表し、それぞれ独立に置換基を有していても良い。cは0または1を表す。]
<2>前記一般式(1)において、前記LL1が一般式(2A)で表される2座または3座の配位子である<1>記載の金属錯体色素。
<2>前記一般式(1)において、前記LL1が一般式(2A)で表される2座または3座の配位子である<1>記載の金属錯体色素。

[一般式(2A)において、R1、R2、R3、R4、Y1、Y2、Y3、Y4、L1、L2、a1、a2、b1、b2、n1、n2、n3、n4は、一般式(2)におけるものと同義である。]
<3>前記一般式(2)において、L1、L2がアリーレンを表す<1>記載の金属錯体色素。
<4>前記一般式(2A)において、L1、L2がアリーレンを表す<2>記載の金属錯体色素。
<5>前記一般式(1)において、LL1が下記一般式(8)で表される2座または3座の配位子である<1>または<3>に記載の金属錯体色素。
<3>前記一般式(2)において、L1、L2がアリーレンを表す<1>記載の金属錯体色素。
<4>前記一般式(2A)において、L1、L2がアリーレンを表す<2>記載の金属錯体色素。
<5>前記一般式(1)において、LL1が下記一般式(8)で表される2座または3座の配位子である<1>または<3>に記載の金属錯体色素。

[一般式(8)において、R1、R2、R3、R4、Y1、Y2、Y3、Y4、a1、a2、b1、b2、n1、n2、n3、n4、zは、一般式(2)におけるものと同義である。]
<6>前記一般式(1)において、LL1が下記一般式(8A)で表される2座または3座の配位子である<1>~<5>のいずれか1項に記載の金属錯体色素。
<6>前記一般式(1)において、LL1が下記一般式(8A)で表される2座または3座の配位子である<1>~<5>のいずれか1項に記載の金属錯体色素。

[一般式(8A)において、R1、R2、R3、R4、Y1、Y2、Y3、Y4、a1、a2、b1、b2、n1、n2、n3、n4は、一般式(2)におけるものと同義である。]
<7>前記一般式(1)において、Mがルテニウムを表す<1>~<6>のいずれか1項に記載の金属錯体色素。
<8>前記一般式(3)で表される環より水素原子を2つ脱離して得られる連結基が下記一般式(3A)で表される<1>~<7>項のいずれか1項に記載の金属錯体色素。
<7>前記一般式(1)において、Mがルテニウムを表す<1>~<6>のいずれか1項に記載の金属錯体色素。
<8>前記一般式(3)で表される環より水素原子を2つ脱離して得られる連結基が下記一般式(3A)で表される<1>~<7>項のいずれか1項に記載の金属錯体色素。

[一般式(3A)において、n5は一般式(3)のものと同義である。*はL1またはL2への結合部を表し、**はY3またはY4への結合部を表す。]
<9>前記一般式(2)において、Y1、Y2が下記一般式(9)~(12)のいずれかで表される<1>~<8>項のいずれか1項に記載の金属錯体色素。
<9>前記一般式(2)において、Y1、Y2が下記一般式(9)~(12)のいずれかで表される<1>~<8>項のいずれか1項に記載の金属錯体色素。

(式中、R12、R13、R15、R16、R18、R19、R21、R22は水素原子または置換基を表す。*はL1またはL2への結合部を表し、**はY3またはY4への結合部を表す。)
<10>前記一般式(9)~(12)のいずれかで表される置換基が少なくとも一つ以上の炭素原子数5以上の脂肪族基を持つ<9>記載の金属錯体色素。
<11><1>~<10>のいずれか1項に記載の金属錯体色素と半導体微粒子とを有する感光層を具備した、光電気変換素子。
<12>前記一般式(1)記載の金属錯体色素と他の色素を組み合わせて用いる<11>記載の光電変換素子。
<13>前記他の色素が一般式(13)で表される<12>に記載の光電変換素子。
Mz(LL11)m11(LL12)m12(X11)m13・CI11 一般式(13)
[一般式(13)において、
Mzは金属原子を表し、
LL11は下記一般式(14)で表される2座又は3座の配位子を表し、
LL12は下記一般式(15)で表される2座又は3座の配位子を表し、
X11はアシルオキシ基、アシルチオ基、チオアシルオキシ基、チオアシルチオ基、アシルアミノオキシ基、チオカルバメート基、ジチオカルバメート基、チオカルボネート基、ジチオカルボネート基、トリチオカルボネート基、アシル基、チオシアネート基、イソチオシアネート基、シアネート基、イソシアネート基、シアノ基、アルキルチオ基、アリールチオ基、アルコキシ基およびアリールオキシ基からなる群から選ばれた基で配位する1座又は2座の配位子、あるいはハロゲン原子、カルボニル、ジアルキルケトン、1,3-ジケトン、カルボンアミド、チオカルボンアミドまたはチオ尿素からなる1座または2座の配位子を表し、
m11は0~3の整数を表し、m11が2以上のとき、LL11は同じでも異なっていてもよく、
m12は0~2の整数を表し、m12が2のとき、LL12は同じでも異なっていてもよい。ただし、m11とm12のうち少なくとも一方は1以上の整数である。
m13は0~3の整数を表し、m13が2以上のとき、X11は同じでも異なっていてもよく、X11同士が連結していてもよい。
CI11は一般式(13)において、電荷を中和させるのに対イオンが必要な場合の対イオンを表す。
LL11、LL12、X11のいずれか1つは、少なくとも1つの酸性基を有する。]
<10>前記一般式(9)~(12)のいずれかで表される置換基が少なくとも一つ以上の炭素原子数5以上の脂肪族基を持つ<9>記載の金属錯体色素。
<11><1>~<10>のいずれか1項に記載の金属錯体色素と半導体微粒子とを有する感光層を具備した、光電気変換素子。
<12>前記一般式(1)記載の金属錯体色素と他の色素を組み合わせて用いる<11>記載の光電変換素子。
<13>前記他の色素が一般式(13)で表される<12>に記載の光電変換素子。
Mz(LL11)m11(LL12)m12(X11)m13・CI11 一般式(13)
[一般式(13)において、
Mzは金属原子を表し、
LL11は下記一般式(14)で表される2座又は3座の配位子を表し、
LL12は下記一般式(15)で表される2座又は3座の配位子を表し、
X11はアシルオキシ基、アシルチオ基、チオアシルオキシ基、チオアシルチオ基、アシルアミノオキシ基、チオカルバメート基、ジチオカルバメート基、チオカルボネート基、ジチオカルボネート基、トリチオカルボネート基、アシル基、チオシアネート基、イソチオシアネート基、シアネート基、イソシアネート基、シアノ基、アルキルチオ基、アリールチオ基、アルコキシ基およびアリールオキシ基からなる群から選ばれた基で配位する1座又は2座の配位子、あるいはハロゲン原子、カルボニル、ジアルキルケトン、1,3-ジケトン、カルボンアミド、チオカルボンアミドまたはチオ尿素からなる1座または2座の配位子を表し、
m11は0~3の整数を表し、m11が2以上のとき、LL11は同じでも異なっていてもよく、
m12は0~2の整数を表し、m12が2のとき、LL12は同じでも異なっていてもよい。ただし、m11とm12のうち少なくとも一方は1以上の整数である。
m13は0~3の整数を表し、m13が2以上のとき、X11は同じでも異なっていてもよく、X11同士が連結していてもよい。
CI11は一般式(13)において、電荷を中和させるのに対イオンが必要な場合の対イオンを表す。
LL11、LL12、X11のいずれか1つは、少なくとも1つの酸性基を有する。]

[一般式(14)において、
R101及びR102はそれぞれ独立に酸性基を表し、
R103及びR104はそれぞれ独立に置換基を表し、
R105及びR106はそれぞれ独立にアルキル基、アリール基、又はヘテロ環基を表す。
L11及びL12はそれぞれ独立に、アリーレン基、ヘテロアリーレン基、エテニレン基及びエチニレン基から選ばれた少なくとも1種からなる共役鎖を表す。
a11及びa12はそれぞれ独立に0~3の整数を表し、a11が2以上のときR101は同じでも異なっていてもよく、a12が2以上のときR102は同じでも異なっていてもよい。
b11及びb12はそれぞれ独立に0~3の整数を表し、b11が2以上のときR103は同じでも異なっていてもよく、R103は互いに連結して環を形成してもよく、b12が2以上のときR104は同じでも異なっていてもよく、R104は互いに連結して環を形成してもよい。b11及びb12が共に1以上のとき、R103とR104が連結して環を形成してもよい。
d11及びd12はそれぞれ独立に0~5の整数を表す。
d13は0または1を表す。
R101及びR102はそれぞれ独立に酸性基を表し、
R103及びR104はそれぞれ独立に置換基を表し、
R105及びR106はそれぞれ独立にアルキル基、アリール基、又はヘテロ環基を表す。
L11及びL12はそれぞれ独立に、アリーレン基、ヘテロアリーレン基、エテニレン基及びエチニレン基から選ばれた少なくとも1種からなる共役鎖を表す。
a11及びa12はそれぞれ独立に0~3の整数を表し、a11が2以上のときR101は同じでも異なっていてもよく、a12が2以上のときR102は同じでも異なっていてもよい。
b11及びb12はそれぞれ独立に0~3の整数を表し、b11が2以上のときR103は同じでも異なっていてもよく、R103は互いに連結して環を形成してもよく、b12が2以上のときR104は同じでも異なっていてもよく、R104は互いに連結して環を形成してもよい。b11及びb12が共に1以上のとき、R103とR104が連結して環を形成してもよい。
d11及びd12はそれぞれ独立に0~5の整数を表す。
d13は0または1を表す。

一般式(15)において、Zd、Ze及びZfはそれぞれ独立に、5又は6員環を形成しうる非金属原子群を表し、それぞれ独立に置換基を有していてもよい。fは0又は1を表す。]
<14>導電性支持体上に、前記感光体層、電荷移動体及び対極をこの順序で積層した構造を有する、前記<11>~<13>のいずれか1項に記載の光電変換素子。
<15><11>~<14>のいずれか1項記載の光電変換素子を備える光電気化学電池。
<14>導電性支持体上に、前記感光体層、電荷移動体及び対極をこの順序で積層した構造を有する、前記<11>~<13>のいずれか1項に記載の光電変換素子。
<15><11>~<14>のいずれか1項記載の光電変換素子を備える光電気化学電池。
本発明の金属錯体色素を用いると、長波長領域に吸収を示し、半導体上に安定に吸着し、変換効率が高く、耐久性に優れた光電変換素子および光電気化学電池を提供することができる。
本発明の上記及び他の特徴及び利点は、適宜添付の図面を参照して、下記の記載からより明らかになるであろう。
本発明の上記及び他の特徴及び利点は、適宜添付の図面を参照して、下記の記載からより明らかになるであろう。
本発明の色素を光電変換素子に組み込んで用いたときに、増感色素として機能する一般式(1)で表される金属錯体色素において、その配位子と吸着基(結合基)とが特有の相互作用を示し、光電変換効率と耐久性との両立を実現する。その詳細な原理は以下のように推定される。まず、配位子LL1はジピリジンリガンドがさらに直線性の高い剛直な構造を置換基として有し、ジピリジンリガンドと共に共役系を形成している。これにより長波長側のピークの吸収領域が長波長側へさらに拡大し、高い長波長吸収効率(分子吸光係数ε)を示すものと考えられる。一方、吸着基(結合基)は、半導体微粒子との強固な配向吸着状態を実現している。配位子LL1は上記直線性の高い剛直な置換基の効果により吸着基の吸着状態を保護すると考えられる。つまり、直線性の高い剛直な置換基が密に配向することとなり、吸着基が剥離する原因となる水や色素を分解する求核種などの攻撃を受けにくくし、耐久性を改善する効果があると考えられる。また、色素分子同士の非効率的な会合による変換効率低下も抑制されると考えられる。さらに、直線性の高い剛直な構造の末端側にチエニル基等の含硫黄複素環を有することにより、一電子酸化状態が安定となり、上記長波長吸収効果や耐久性がさらに高まることにつながったと推定される。
以下、本発明の好ましい実施形態について詳細に説明する。
以下、本発明の好ましい実施形態について詳細に説明する。
本発明の光電変換素子の好ましい実施態様を、図面を参照して説明する。図1に示すように、光電変換素子10は、導電性支持体1、導電性支持体1上にその順序で配された、感光体層2、電荷移動体層3、及び対極4からなる。前記導電性支持体1と感光体2とにより受光電極5を構成している。その感光体2は導電性微粒子22と増感色素21とを有しており、色素21はその少なくとも一部において導電性微粒子22に吸着している(色素は吸着平衡状態になっており、一部電荷移動体層に存在していてもよい。)。感光体2が形成された導電性支持体1は光電変換素子10において作用電極として機能する。この光電変換素子10を外部回路6で仕事をさせるようにして、光電気化学電池100として作動させることができる。
なお、光電変換素子の上下は特に定めなくてもよいが、本明細書において、図示したものに基づいて言えば、受光側となる対極4の側を上部(天部)の方向とし、支持体1の側を下部(底部)の方向とする。
なお、光電変換素子の上下は特に定めなくてもよいが、本明細書において、図示したものに基づいて言えば、受光側となる対極4の側を上部(天部)の方向とし、支持体1の側を下部(底部)の方向とする。
受光電極5は、導電性支持体1および導電性支持体上に塗設される色素21の吸着した半導体微粒子22の感光層(半導体膜)2よりなる電極である。感光体(半導体膜)2に入射した光は色素を励起する。励起色素はエネルギーの高い電子を有している。そこでこの電子が色素21から半導体微粒子22の伝導帯に渡され、さらに拡散によって導電性支持体1に到達する。このとき色素21の分子は酸化体となっている。電極上の電子が外部回路で仕事をしながら色素酸化体に戻ることにより、光電気化学電池として作用する。この際、受光電極5はこの電池の負極として働く。
本実施形態の光電変換素子は、導電性支持体上に後述の金属錯体増感色素が吸着された多孔質半導体微粒子の層を有する感光体を有する。このとき色素において一部電解質中に解離したもの等があってもよいことは上述のとおりである。感光体は目的に応じて設計され、単層構成でも多層構成でもよい。本実施形態の光電変換素子の感光体には、特定の金属錯体増感色素が吸着した半導体微粒子を含み、感度が高く、光電気化学電池として使用する場合に、高い変換効率を得ることができ、さらに高い耐久性を有する。
以下、本発明について説明する。
(A)色素
(A1)一般式(1)の構造を有する金属錯体色素
M(LL1)m1(LL2)m2(X)m3・CI 一般式(1)
一般式(1)の構造を有する色素は、金属原子に、配位子LL1、場合により配位子LL2および特定の官能基Xが配位しており、必要な場合はCIにより電気的に中性に保たれている。
(A1-1)金属原子M
Mは金属原子を表す。Mは好ましくは4配位または6配位が可能な金属であり、より好ましくはRu、Fe、Os、Cu、W、Cr、Mo、Ni、Pd、Pt、Co、Ir、Rh、Re、Mn又はZnである。特に好ましくは、Ru、Os、Zn又はCuであり、最も好ましくはRuである。
(A1)一般式(1)の構造を有する金属錯体色素
M(LL1)m1(LL2)m2(X)m3・CI 一般式(1)
一般式(1)の構造を有する色素は、金属原子に、配位子LL1、場合により配位子LL2および特定の官能基Xが配位しており、必要な場合はCIにより電気的に中性に保たれている。
(A1-1)金属原子M
Mは金属原子を表す。Mは好ましくは4配位または6配位が可能な金属であり、より好ましくはRu、Fe、Os、Cu、W、Cr、Mo、Ni、Pd、Pt、Co、Ir、Rh、Re、Mn又はZnである。特に好ましくは、Ru、Os、Zn又はCuであり、最も好ましくはRuである。
(A1-2)配位子LL1
配位子LL1は、下記一般式(2)により表される2座または3座の配位子であり、好ましくは2座配位子である。
配位子LL1の数を表すm1は1~3の整数であり、1~2であるのが好ましく、1であるのがより好ましい。m1が2以上のとき、LL1は同じでも異なっていてもよい。
配位子LL1は、下記一般式(2)により表される2座または3座の配位子であり、好ましくは2座配位子である。
配位子LL1の数を表すm1は1~3の整数であり、1~2であるのが好ましく、1であるのがより好ましい。m1が2以上のとき、LL1は同じでも異なっていてもよい。

一般式(2)において、R1およびR2はそれぞれ独立に酸性基(解離性のプロトンを有する置換基)を表す。酸性基としては、カルボキシル基、スルホン酸基、ヒドロキシル基、ヒドロキサム酸基、ホスホリル基またはホスホニル基が挙げられる。R1またはR2として、好ましくはホスホニル基、カルボキシル基であり、さらに好ましくはカルボキシル基である。なお、これらの酸性基は連結基を介してピリジン環に導入されているものであってもよい。
a1およびa2はそれぞれ独立に0~3の整数を表す。a1が2以上のときR1は同じでも異なっていてもよい。a2が2以上のときR2は同じでも異なっていてもよい。a1は0又は1であるのが好ましく、さらに好ましくは0である。a2は0又は1であるのが好ましく、さらに好ましくは0である。
a1およびa2はそれぞれ独立に0~3の整数を表す。a1が2以上のときR1は同じでも異なっていてもよい。a2が2以上のときR2は同じでも異なっていてもよい。a1は0又は1であるのが好ましく、さらに好ましくは0である。a2は0又は1であるのが好ましく、さらに好ましくは0である。
R3およびR4はそれぞれ独立に置換基を表す。置換基として、好ましくはアルキル基(好ましくは炭素原子数1~20のアルキル基、例えばメチル、エチル、イソプロピル、t-ブチル、ペンチル、ヘプチル、1-エチルペンチル、ベンジル、2-エトキシエチル、1-カルボキシメチル等であって、アラルキル基などの置換アルキルを含む概念である)、アルケニル基(好ましくは炭素原子数2~20のアルケニル基、例えば、ビニル、アリル、オレイル等)、アルキニル基(好ましくは炭素原子数2~20のアルキニル基、例えば、エチニル、ブタジイニル、フェニルエチニル等)、シクロアルキル基(好ましくは炭素原子数3~20のシクロアルキル基、例えば、シクロプロピル、シクロペンチル、シクロヘキシル、4-メチルシクロヘキシル等)、アリール基(好ましくは炭素原子数6~26のアリール基、例えば、フェニル、1-ナフチル、4-メトキシフェニル、2-クロロフェニル、3-メチルフェニル等)、ヘテロ環基(好ましくは炭素原子数2~20のヘテロ環基、例えば、2-ピリジル、4-ピリジル、2-イミダゾリル、2-ベンゾイミダゾリル、2-チアゾリル、2-オキサゾリル等)、アルコキシ基(好ましくは炭素原子数1~20のアルコキシ基、例えば、メトキシ、エトキシ、イソプロピルオキシ、ベンジルオキシ等)、アリールオキシ基(好ましくは炭素原子数6~26のアリールオキシ基、例えば、フェノキシ、1-ナフチルオキシ、3-メチルフェノキシ、4-メトキシフェノキシ等)、アルコキシカルボニル基(好ましくは炭素原子数2~20のアルコキシカルボニル基、例えば、エトキシカルボニル、2-エチルヘキシルオキシカルボニル等)、アミノ基(好ましくは炭素原子数0~20のアミノ基、例えば、アミノ、N,N-ジメチルアミノ、N,N-ジエチルアミノ、N-エチルアミノ、アニリノ等)、スルホンアミド基(好ましくは炭素原子数0~20のスルホンアミド基、例えば、N,N-ジメチルスルホンアミド、N-フェニルスルホンアミド等)、アシルオキシ基(好ましくは炭素原子数1~20のアシルオキシ基、例えば、アセチルオキシ、ベンゾイルオキシ等)、カルバモイル基(好ましくは炭素原子数1~20のカルバモイル基、例えば、N,N-ジメチルカルバモイル、N-フェニルカルバモイル等)、アシルアミノ基(好ましくは炭素原子数1~20のアシルアミノ基、例えば、アセチルアミノ、ベンゾイルアミノ等)、シアノ基、又はハロゲン原子(例えばフッ素原子、塩素原子、臭素原子、ヨウ素原子等)であり、より好ましくはアルキル基、アルケニル基、アリール基、ヘテロ環基、アルコキシ基、アリールオキシ基、アルコキシカルボニル基、アミノ基、アシルアミノ基、シアノ基又はハロゲン原子であり、特に好ましくはアルキル基、アルケニル基、ヘテロ環基、アルコキシ基、アルコキシカルボニル基、アミノ基、アシルアミノ基又はシアノ基である。これらの基はさらに置換されていてもよく、置換基の例としてはR3およびR4の置換基が挙げられる。
b1およびb2はそれぞれ独立に0~3の整数を表し、0~2の整数であるのが好ましい。b1が2以上のときR3は同じでも異なっていてもよく、互いに連結して環を形成してもよい。b1は好ましくは0~1であり、特に好ましくは0である。b2が2以上のときR4は同じでも異なっていてもよく、互いに連結して環を形成してもよい。b2は好ましくは0~1であり、特に好ましくは0である。b1およびb2が共に1以上のときR3とR4が連結して環を形成してもよい。R3またはR4が連結して形成する環の好ましい例としては、ベンゼン環、ピリジン環、チオフェン環、ピロール環、シクロヘキサン環、シクロペンタン環等が挙げられる。
L1またはL2はそれぞれ独立にアルキニレン、アリーレンまたはこれらの組み合わせからなる共役鎖を表す。アルキニレンとしては、炭素原子数2~6のアルキニレンが好ましく、炭素原子数2~4のアルキニレンがより好ましい。さらに好ましくはエチニレン、1,3-ブタジイニレン、ヘキシニレンであり、特に好ましくはエチニレンである。アリーレンとしては炭素原子数6~30のアリーレンが好ましく、炭素原子数6~24のアリーレンがより好ましい。さらに好ましくはフェニレン、ナフチレンであり、特に好ましくはフェニレンである。アルキニレン及びアリーレンの組み合わせからなる共役鎖としては、エチニレンとフェニレンを1つずつ組み合わせた共役鎖が好ましい。L1またはL2はアリーレンであることが好ましい。
n1、n2はそれぞれ独立に1以上の整数を表す。好ましくはn1は1~3であり、より好ましくは1~2である。また、好ましくはn2は1~3であり、より好ましくは1~2である。
zは0または1を表す。zは好ましくは1である。
zが1の場合、LL1は下記一般式(2A)で表される2座または3座の配位子である。
n1、n2はそれぞれ独立に1以上の整数を表す。好ましくはn1は1~3であり、より好ましくは1~2である。また、好ましくはn2は1~3であり、より好ましくは1~2である。
zは0または1を表す。zは好ましくは1である。
zが1の場合、LL1は下記一般式(2A)で表される2座または3座の配位子である。

一般式(2A)において、R1、R2、R3、R4、Y1、Y2、Y3、Y4、L1、L2、a1、a2、b1、b2、n1、n2、n3、n4は、一般式(2)におけるものと同義である。
一般式(2)において、Y1およびY2はそれぞれ独立に一般式(3)~(6)のいずれかで表される環より水素原子を2つ脱離して得られる連結基を表す。

一般式(2)において、n3、n4はそれぞれ独立に1以上の整数を表す。n3は、好ましくは1~5であり、より好ましくは1~3であり、さらに好ましくは1~2である。また、n4は、好ましくは1~5であり、より好ましくは1~3であり、さらに好ましくは1~2である。n3が2以上の場合、複数のY1は同じでも異なってもよく、n4が2以上の場合、複数のY2は同じでも異なっても良い。
一般式(3)~(6)において、R5~R11は置換基を表す。置換基の例としては、R3およびR4の例として挙げた置換基が挙げられる。
好ましくはR5~R11は、アルキル基、アルケニル基、アルキニル基、アリール基、アシル基であり、より好ましくは、アルキル基、アルケニル基、アルキニル基であり、さらに好ましくはアルキル基、アルキニル基である。アルキル基としては、炭素原子数2~18のものが好ましく、より好ましくは炭素原子数5~18、さらに好ましくは炭素原子数6~18、特に好ましくは炭素原子数6~12である。アルキル基の例としてはn-ブチル、t-ブチル、ペンチル、ヘキシル、シクロヘキシル、デシルが挙げられる。アルケニル基としては、炭素原子数2~18のものが好ましく、より好ましくは炭素原子数5~18、さらに好ましくは炭素原子数6~18、特に好ましくは炭素原子数6~12である。アルケニル基の例としては、ビニル、アリル、オレイルが挙げられる。アルキニル基としては、炭素原子数2~18のものが好ましく、より好ましくは炭素原子数5~18、さらに好ましくは炭素原子数6~18、特に好ましくは炭素原子数6~12である。アルキニル基の例としてはエチニル、ブタジイニル、フェニルエチニルが挙げられる。アリール基の例としては、フェニル基、トリフェニルアミン基が挙げられる。これらの基はさらに置換されていてもよく、置換基の例としてはR5~R11の置換基が挙げられる。
一般式(3)~(6)において、R5~R11は置換基を表す。置換基の例としては、R3およびR4の例として挙げた置換基が挙げられる。
好ましくはR5~R11は、アルキル基、アルケニル基、アルキニル基、アリール基、アシル基であり、より好ましくは、アルキル基、アルケニル基、アルキニル基であり、さらに好ましくはアルキル基、アルキニル基である。アルキル基としては、炭素原子数2~18のものが好ましく、より好ましくは炭素原子数5~18、さらに好ましくは炭素原子数6~18、特に好ましくは炭素原子数6~12である。アルキル基の例としてはn-ブチル、t-ブチル、ペンチル、ヘキシル、シクロヘキシル、デシルが挙げられる。アルケニル基としては、炭素原子数2~18のものが好ましく、より好ましくは炭素原子数5~18、さらに好ましくは炭素原子数6~18、特に好ましくは炭素原子数6~12である。アルケニル基の例としては、ビニル、アリル、オレイルが挙げられる。アルキニル基としては、炭素原子数2~18のものが好ましく、より好ましくは炭素原子数5~18、さらに好ましくは炭素原子数6~18、特に好ましくは炭素原子数6~12である。アルキニル基の例としてはエチニル、ブタジイニル、フェニルエチニルが挙げられる。アリール基の例としては、フェニル基、トリフェニルアミン基が挙げられる。これらの基はさらに置換されていてもよく、置換基の例としてはR5~R11の置換基が挙げられる。
Y3およびY4は水素原子または置換基を表す。Y3またはY4が表す置換基の例としては、R5~R11の置換基の例として挙げたものが挙げられる。
一般式(3)~(6)で表される置換基は、R5~R11、Y3、Y4として、炭素原子数5以上(より好ましくは炭素原子数6~18、さらに好ましくは炭素原子数6~12)の脂肪族基を少なくとも一つ(好ましくは1~2つ、より好ましくは1つ)持つことが好ましい。炭素原子数5以上の脂肪族基としては、上述のアルキル基、アルケニル基、アルキニル基が挙げられ、好ましくは、アルキル基、アルキニル基であり、より好ましくはアルキニル基である。アルキニル基はさらにアルキル基により置換されていることが好ましく、炭素原子数5以上のアルキル基により置換されていることが好ましい。
一般式(3)~(6)で表される置換基は、R5~R11、Y3、Y4として、炭素原子数5以上(より好ましくは炭素原子数6~18、さらに好ましくは炭素原子数6~12)の脂肪族基を少なくとも一つ(好ましくは1~2つ、より好ましくは1つ)持つことが好ましい。炭素原子数5以上の脂肪族基としては、上述のアルキル基、アルケニル基、アルキニル基が挙げられ、好ましくは、アルキル基、アルキニル基であり、より好ましくはアルキニル基である。アルキニル基はさらにアルキル基により置換されていることが好ましく、炭素原子数5以上のアルキル基により置換されていることが好ましい。
n5~n11はそれぞれ独立に0~2の整数を表す。n5は、好ましくは1~2、特に好ましくは1である。n6は、好ましくは1~2、特に好ましくは1である。n7は、好ましくは1~2、特に好ましくは1である。n8は、好ましくは1~2、特に好ましくは1である。n9は、好ましくは1~2、特に好ましくは1である。n10は、好ましくは1~2、特に好ましくは1である。n11は、好ましくは1~2、特に好ましくは1である。n6とn7の和は2以下である。n8とn9の和は2以下である。n10とn11の和は2以下である。
R5~R11が複数ある場合、複数のR5~R11は同じでも異なっていてもよい。また、複数のR5~R11はそれぞれ隣接する置換基と互いに結合して環を形成してもよい。R5~R11の形成する環の好ましい例としては、ベンゼン環、ピリジン環、チオフェン環、ピロール環、シクロヘキサン環、シクロペンタン環等が挙げられる。
R5~R11が複数ある場合、複数のR5~R11は同じでも異なっていてもよい。また、複数のR5~R11はそれぞれ隣接する置換基と互いに結合して環を形成してもよい。R5~R11の形成する環の好ましい例としては、ベンゼン環、ピリジン環、チオフェン環、ピロール環、シクロヘキサン環、シクロペンタン環等が挙げられる。
配位子LL1がアルキル基、アルケニル基等を含むとき、これらは直鎖状でも分岐状でもよく、置換されていても無置換でもよい。また配位子LL1がアリール基、ヘテロ環基等を含むとき、それらは単環でも縮環でもよく、置換されていても無置換でもよい。
一般式(3)で表される環より水素原子を2つ脱離して得られる連結基は、R5がチエニレン基と結合して縮合環を形成していることが好ましい。R5とチエニレン基とが結合して形成する環の好ましい例としては、ベンゼン環、インドール環、ジオキサン環が挙げられる。
一般式(3)で表される環より水素原子を2つ脱離して得られる連結基は、下記一般式(3A)で表される連結基であることが好ましい。

前記一般式(2)において、Y1、Y2は、一般式(3)または一般式(5)で表される連結基であることが好ましく、一般式(3)で表される連結基であることがさらに好ましい。
前記一般式(2)において、Y1、Y2は下記一般式(9)~(12)のいずれかで表される置換基であることが好ましい。

一般式(9)~(12)で表される連結基は、R12、R13、R15、R16、R18、R19、R21、R22、Y3、Y4として、炭素原子数5以上(より好ましくは炭素原子数6~18、さらに好ましくは6~12)の脂肪族基を少なくとも一つ(好ましくは1~2、より好ましくは1つ)持つことが好ましい。炭素原子数5以上の脂肪族基の例としては、アルキル基、アルケニル基、アルキニル基が挙げられ、好ましくは、アルキル基、アルキニル基であり、より好ましくはアルキニル基である。
前記一般式(2)において、Y1、Y2は、一般式(9)または一般式(11)で表される連結基であることが好ましく、一般式(9)で表される連結基であることがさらに好ましい。
一般式(1)における配位子LL1は、下記一般式(8)で表される配位子であることが好ましい。

一般式(8)において、R1、R2、R3、R4、Y1、Y2、Y3、Y4、a1、a2、b1、b2、n1、n2、n3、n4、zは、一般式(2)におけるものと同義であり、好ましい範囲も同様である。
一般式(1)における配位子LL1は、下記一般式(8A)で表される配位子であることがさらに好ましい。
一般式(1)における配位子LL1は、下記一般式(8A)で表される配位子であることがさらに好ましい。

一般式(8A)において、R1、R2、R3、R4、Y1、Y2、Y3、Y4、a1、a2、b1、b2、n1、n2、n3、n4は、一般式(2)におけるものと同義であり、好ましい範囲も同様である。
(A1-3)配位子LL2
一般式(1)中、LL2は下記一般式(7)により表される2座又は3座の配位子を表す。配位子LL2の数を表すm2は0~2の整数であり、0又は1であるのが好ましい。m2が2のときLL2は同じでも異なっていてもよい。
一般式(1)中、LL2は下記一般式(7)により表される2座又は3座の配位子を表す。配位子LL2の数を表すm2は0~2の整数であり、0又は1であるのが好ましい。m2が2のときLL2は同じでも異なっていてもよい。

一般式(7)中、Za、ZbおよびZcはそれぞれ独立に5員環又は6員環を形成しうる非金属原子群を表す。形成される5員環又は6員環は置換されていても無置換でもよく、単環でも縮環していてもよい。Za、Zb及びZcは炭素原子、水素原子、窒素原子、酸素原子、硫黄原子、リン原子及び/又はハロゲン原子で構成されることが好ましく、芳香族環を形成するのが好ましい。5員環の場合はイミダゾール環、オキサゾール環、チアゾール環又はトリアゾール環を形成するのが好ましく、6員環の場合はピリジン環、ピリミジン環、ピリダジン環又はピラジン環を形成するのが好ましい。なかでもイミダゾール環又はピリジン環がより好ましい。
Za、ZbおよびZcで表される非金属原子群は、置換基を有してもよく、酸性基を有していることが好ましい。置換基としては、一般式(1)のR3およびR4の例としてあげた基が挙げられる。酸性基としては、例えばカルボキシル基、スルホン酸基、ヒドロキシル基、ヒドロキサム酸基、ホスホリル基及びホスホニル基等が挙げられる。
Za、ZbおよびZcで表される非金属原子群は、置換基を有してもよく、酸性基を有していることが好ましい。置換基としては、一般式(1)のR3およびR4の例としてあげた基が挙げられる。酸性基としては、例えばカルボキシル基、スルホン酸基、ヒドロキシル基、ヒドロキサム酸基、ホスホリル基及びホスホニル基等が挙げられる。
一般式(7)中、cは0または1を表す。cは0であるのが好ましく、LL2は2座配位子であるのが好ましい。
配位子LL2は好ましくは、下記一般式C-1~C-10のいずれかで表される有機配位子を表す。

一般式C-1~C-10において、R11~R22は置換基を表す。
置換基としては、ハロゲン原子、炭素原子数1~12の置換又は無置換のアルキル基、炭素原子数2~12の置換又は無置換のアルケニル基、炭素原子数7~12の置換又は無置換のアラルキル基、炭素原子数6~12の置換又は無置換のアリール基、あるいは前述の酸性基(これらの酸性基は塩を形成していてもよい)が挙げられる。アルキル基及びアラルキル基のアルキル部分は直鎖状でも分岐状でもよい。また、アリール基およびアラルキル基のアリール部分は単環でも多環(縮合環、環集合)でもよい。R11~R22で表される置換基はさらに置換されていてもよく、さらに置換する置換基としてはR11~R22として挙げた置換基であり、酸性基で置換されていることが好ましい。R11~R22で表される置換基は、それ自身が酸性基であるか、または酸性基を置換基として有する基であることが好ましい。
e1~e12は0以上の整数を表す。具体的には、e1およびe2はそれぞれ独立に0~4の整数を表し、e3は0~3の整数を表し、e4およびe5はそれぞれ独立に0~4の整数を表し、e6は0~2の整数を表し、e7およびe8はそれぞれ独立に0~3の整数を表し、e9は0~4の整数を表し、e10~e12はそれぞれ独立に0~6の整数を表す。e1~e12が2以上のとき、R11~R22はそれぞれ同じでも異なっていてもよい。e1~e12はそれぞれ独立に1である場合が好ましい。
R11とR12は同じでも異なっていてもよいが、同じであるのが好ましい。R13~R15は同じでも異なっていても良いが、同じであるのが好ましい。R16~R18は同じでも異なっていても良いが、同じであるのが好ましい。R19とR20は同じでも異なっていても良いが、同じであるのが好ましい。R21とR22は同じでも異なっていても良いが、同じであるのが好ましい。
置換基としては、ハロゲン原子、炭素原子数1~12の置換又は無置換のアルキル基、炭素原子数2~12の置換又は無置換のアルケニル基、炭素原子数7~12の置換又は無置換のアラルキル基、炭素原子数6~12の置換又は無置換のアリール基、あるいは前述の酸性基(これらの酸性基は塩を形成していてもよい)が挙げられる。アルキル基及びアラルキル基のアルキル部分は直鎖状でも分岐状でもよい。また、アリール基およびアラルキル基のアリール部分は単環でも多環(縮合環、環集合)でもよい。R11~R22で表される置換基はさらに置換されていてもよく、さらに置換する置換基としてはR11~R22として挙げた置換基であり、酸性基で置換されていることが好ましい。R11~R22で表される置換基は、それ自身が酸性基であるか、または酸性基を置換基として有する基であることが好ましい。
e1~e12は0以上の整数を表す。具体的には、e1およびe2はそれぞれ独立に0~4の整数を表し、e3は0~3の整数を表し、e4およびe5はそれぞれ独立に0~4の整数を表し、e6は0~2の整数を表し、e7およびe8はそれぞれ独立に0~3の整数を表し、e9は0~4の整数を表し、e10~e12はそれぞれ独立に0~6の整数を表す。e1~e12が2以上のとき、R11~R22はそれぞれ同じでも異なっていてもよい。e1~e12はそれぞれ独立に1である場合が好ましい。
R11とR12は同じでも異なっていてもよいが、同じであるのが好ましい。R13~R15は同じでも異なっていても良いが、同じであるのが好ましい。R16~R18は同じでも異なっていても良いが、同じであるのが好ましい。R19とR20は同じでも異なっていても良いが、同じであるのが好ましい。R21とR22は同じでも異なっていても良いが、同じであるのが好ましい。
(A1-4)配位子X
一般式(1)中、Xは1座又は2座の配位子を表す。配位子Xの数を表すm3は0~3の整数を表し、m3は好ましくは1又は2である。Xが1座配位子のとき、m3は2であるのが好ましく、Xが2座配位子のとき、m3は1であるのが好ましい。m3が2以上のとき、Xは同じでも異なっていてもよく、X同士が連結していてもよい。
一般式(1)中、Xは1座又は2座の配位子を表す。配位子Xの数を表すm3は0~3の整数を表し、m3は好ましくは1又は2である。Xが1座配位子のとき、m3は2であるのが好ましく、Xが2座配位子のとき、m3は1であるのが好ましい。m3が2以上のとき、Xは同じでも異なっていてもよく、X同士が連結していてもよい。
配位子Xは、アシルオキシ基(好ましくは炭素原子数1~20のアシルオキシ基、例えば、アセチルオキシ、ベンゾイルオキシ、サリチル酸、グリシルオキシ、N,N-ジメチルグリシルオキシ、オキザリレン(―OC(O)C(O)O―)等)、アシルチオ基(好ましくは炭素原子数1~20のアシルチオ基、例えば、アセチルチオ、ベンゾイルチオ等)、チオアシルオキシ基(好ましくは炭素原子数1~20のチオアシルオキシ基、例えば、チオアセチルオキシ基(CH3C(S)O―)等))、チオアシルチオ基(好ましくは炭素原子数1~20のチオアシルチオ基、例えば、チオアセチルチオ(CH3C(S)S―)、チオベンゾイルチオ(PhC(S)S―)等))、アシルアミノオキシ基(好ましくは炭素原子数1~20のアシルアミノオキシ基、例えば、N-メチルベンゾイルアミノオキシ(PhC(O)N(CH3)O―)、アセチルアミノオキシ(CH3C(O)NHO―)等))、チオカルバメート基(好ましくは炭素原子数1~20のチオカルバメート基、例えば、N,N-ジエチルチオカルバメート等)、ジチオカルバメート基(好ましくは炭素原子数1~20のジチオカルバメート基、例えば、N-フェニルジチオカルバメート、N,N-ジメチルジチオカルバメート、N,N-ジエチルジチオカルバメート、N,N-ジベンジルジチオカルバメート等)、チオカルボネート基(好ましくは炭素原子数1~20のチオカルボネート基、例えば、エチルチオカルボネート等)、ジチオカルボネート(好ましくは炭素原子数1~20のジチオカルボネート、例えば、エチルジチオカルボネート(C2H5OC(S)S―)等)、トリチオカルボネート基(好ましくは炭素原子数1~20のトリチオカルボネート基、例えば、エチルトリチオカルボネート(C2H5SC(S)S-)等)、アシル基(好ましくは炭素原子数1~20のアシル基、例えば、アセチル、ベンゾイル等)、チオシアネート基、イソチオシアネート基、シアネート基、イソシアネート基、シアノ基、アルキルチオ基(好ましくは炭素原子数1~20のアルキルチオ基、例えばメタンチオ、エチレンジチオ等)、アリールチオ基(好ましくは炭素原子数6~20のアリールチオ基、例えば、ベンゼンチオ、1,2-フェニレンジチオ等)、アルコキシ基(好ましくは炭素原子数1~20のアルコキシ基、例えばメトキシ等)及びアリールオキシ基(好ましくは炭素原子数6~20のアリールオキシ基、例えばフェノキシ、キノリン-8-ヒドロキシル等)からなる群から選ばれた基で配位された1座又は2座の配位子、若しくはハロゲン原子(好ましくは塩素原子、臭素原子、ヨウ素原子等)、カルボニル(…CO)、ジアルキルケトン(好ましくは炭素原子数3~20のジアルキルケトン、例えばアセトン((CH3)2CO…)等)、1,3-ジケトン(好ましくは炭素原子数3~20の1,3-ジケトン、例えば、アセチルアセトン(CH3C(O…)CH=C(O―)CH3)、トリフルオロアセチルアセトン(CF3C(O…)CH=C(O―)CH3)、ジピバロイルメタン(tC4H9C(O…)CH=C(O―)t-C4H9)、ジベンゾイルメタン(PhC(O…)CH=C(O―)Ph)、3-クロロアセチルアセトン(CH3C(O…)CCl=C(O―)CH3)等)、カルボンアミド(好ましくは炭素原子数1~20のカルボンアミド、例えば、CH3N=C(CH3)O―、―OC(=NH)―C(=NH)O―等)、チオカルボンアミド(好ましくは炭素原子数1~20のチオカルボンアミド、例えば、CH3N=C(CH3)S―等)、またはチオ尿素(好ましくは炭素原子数1~20のチオ尿素、例えば、NH(…)=C(S―)NH2、CH3N(…)=C(S―)NHCH3、(CH3)2N―C(S…)N(CH3)2等)からなる配位子を表す。なお、「…」は配位結合を示す。
配位子Xは、好ましくはアシルオキシ基、チオアシルチオ基、アシルアミノオキシ基、ジチオカルバメート基、ジチオカルボネート基、トリチオカルボネート基、チオシアネート基、イソチオシアネート基、シアネート基、イソシアネート基、シアノ基、アルキルチオ基、アリールチオ基、アルコキシ基およびアリールオキシ基からなる群から選ばれた基で配位する配位子、あるいはハロゲン原子、カルボニル、1,3-ジケトンまたはチオ尿素からなる配位子であり、より好ましくはアシルオキシ基、アシルアミノオキシ基、ジチオカルバメート基、チオシアネート基、イソチオシアネート基、シアネート基、イソシアネート基、シアノ基またはアリールチオ基からなる群から選ばれた基で配位する配位子、あるいはハロゲン原子、1,3-ジケトンまたはチオ尿素からなる配位子であり、特に好ましくはジチオカルバメート基、チオシアネート基、イソチオシアネート基、シアネート基およびイソシアネート基からなる群から選ばれた基で配位する配位子、あるいはハロゲン原子または1,3-ジケトンからなる配位子であり、最も好ましくは、ジチオカルバメート基、チオシアネート基およびイソチオシアネート基からなる群から選ばれた基で配位する配位子、あるいは1,3-ジケトンからなる配位子である。なお配位子Xがアルキル基、アルケニル基、アルキニル基、アルキレン基等を含む場合、それらは直鎖状でも分岐状でもよく、置換されていても無置換でもよい。またアリール基、ヘテロ環基、シクロアルキル基等を含む場合、それらは置換されていても無置換でもよく、単環でも縮環していてもよい。
Xが2座配位子のとき、Xはアシルオキシ基、アシルチオ基、チオアシルオキシ基、チオアシルチオ基、アシルアミノオキシ基、チオカルバメート基、ジチオカルバメート基、チオカルボネート基、ジチオカルボネート基、トリチオカルボネート基、アシル基、アルキルチオ基、アリールチオ基、アルコキシ基およびアリールオキシ基からなる群から選ばれた基で配位する配位子、あるいは1,3-ジケトン、カルボンアミド、チオカルボンアミド、またはチオ尿素からなる配位子であるのが好ましい。
Xが1座配位子のとき、Xはチオシアネート基、イソチオシアネート基、シアネート基、イソシアネート基、シアノ基、アルキルチオ基、アリールチオ基からなる群から選ばれた基で配位する配位子、あるいはハロゲン原子、カルボニル、ジアルキルケトン、チオ尿素からなる配位子であるのが好ましい。
本発明の金属錯体色素においては、Xがイソチオシアネート、イソシアネートまたはイソセレノシアネートであることが特に好ましい。
Xが1座配位子のとき、Xはチオシアネート基、イソチオシアネート基、シアネート基、イソシアネート基、シアノ基、アルキルチオ基、アリールチオ基からなる群から選ばれた基で配位する配位子、あるいはハロゲン原子、カルボニル、ジアルキルケトン、チオ尿素からなる配位子であるのが好ましい。
本発明の金属錯体色素においては、Xがイソチオシアネート、イソシアネートまたはイソセレノシアネートであることが特に好ましい。
(A1-5)対イオンCI
一般式(1)において、CIは電荷を中和させるのに対イオンが必要な場合の対イオンを表す。一般に、色素が陽イオン又は陰イオンであるか、あるいは正味のイオン電荷を有するかどうかは、色素中の金属、配位子および置換基に依存する。
一般式(1)において、CIは電荷を中和させるのに対イオンが必要な場合の対イオンを表す。一般に、色素が陽イオン又は陰イオンであるか、あるいは正味のイオン電荷を有するかどうかは、色素中の金属、配位子および置換基に依存する。
置換基が解離性基を有することなどにより、一般式(1)の色素は解離して負電荷を持ってもよい。この場合、一般式(1)の色素全体の電荷はCIにより電気的に中性とされる。
対イオンCIが正の対イオンの場合、例えば、対イオンCIは、無機又は有機のアンモニウムイオン(例えばテトラアルキルアンモニウムイオン、ピリジニウムイオン等)、アルカリ金属イオン又はプロトンである。
対イオンCIが負の対イオンの場合、例えば、対イオンCIは、無機陰イオンでも有機陰イオンでもよい。例えば、ハロゲン陰イオン(例えば、フッ化物イオン、塩化物イオン、臭化物イオン、ヨウ化物イオン等)、置換アリールスルホン酸イオン(例えばp-トルエンスルホン酸イオン、p-クロロベンゼンスルホン酸イオン等)、アリールジスルホン酸イオン(例えば1,3-ベンゼンジスルホン酸イオン、1,5-ナフタレンジスルホン酸イオン、2,6-ナフタレンジスルホン酸イオン等)、アルキル硫酸イオン(例えばメチル硫酸イオン等)、硫酸イオン、チオシアン酸イオン、過塩素酸イオン、テトラフルオロホウ酸イオン、ヘキサフルオロホスフェートイオン、ピクリン酸イオン、酢酸イオン、トリフルオロメタンスルホン酸イオン等が挙げられる。さらに電荷均衡対イオンとして、イオン性ポリマーあるいは色素と逆電荷を有する他の色素を用いてもよく、金属錯イオン(例えばビスベンゼン-1,2-ジチオラトニッケル(III)等)も使用可能である。
(A1-6)結合基
一般式(1)で表される構造を有する色素は、半導体微粒子の表面に対する適当な酸性基(結合基、interlocking group)を1つ以上有する。すなわち、LL1、LL2、およびXの少なくとも1つに、少なくとも1つの酸性基を有する。この結合基を色素中に1~6個有するのがより好ましく、1~4個有するのが特に好ましい。上記結合基として、カルボキシル基、スルホン酸基、ヒドロキシル基、ヒドロキサム酸基(例えば―CONHOH等)、ホスホリル基(例えば―OP(O)(OH)2等)、ホスホニル基(例えば―P(O)(OH)2等)等の酸性基(解離性のプロトンを有する置換基)を色素中に有することが好ましい。配位子LL2上に上記酸性基を有するのが好ましい。なかでも、ホスホニル基、カルボキシル基を配位子LL2上に有することが好ましい。
一般式(1)で表される構造を有する色素は、半導体微粒子の表面に対する適当な酸性基(結合基、interlocking group)を1つ以上有する。すなわち、LL1、LL2、およびXの少なくとも1つに、少なくとも1つの酸性基を有する。この結合基を色素中に1~6個有するのがより好ましく、1~4個有するのが特に好ましい。上記結合基として、カルボキシル基、スルホン酸基、ヒドロキシル基、ヒドロキサム酸基(例えば―CONHOH等)、ホスホリル基(例えば―OP(O)(OH)2等)、ホスホニル基(例えば―P(O)(OH)2等)等の酸性基(解離性のプロトンを有する置換基)を色素中に有することが好ましい。配位子LL2上に上記酸性基を有するのが好ましい。なかでも、ホスホニル基、カルボキシル基を配位子LL2上に有することが好ましい。
本発明で用いる一般式(1)で表される金属錯体色素の具体例を以下に示すが、本発明はこれらに限定されるものではない。なお、下記具体例における色素がプロトン解離性基を有する配位子を含む場合、該配位子は必要に応じて解離しプロトンを放出してもよい。
*は複素環への結合部を表す。**はアルキル基への結合部を表す。
*は複素環への結合部を表す。**はアルキル基への結合部を表す。






本発明の一般式(1)により表される色素は、種々カップリング反応で調製した配位子を用いて、特開2001-291534号公報や当該公報に引用された方法を参考にして合成することができる。
また、一般式(1)の構造を有する色素は、溶液中における長波長側のピークの極大吸収波長が、500~1000nmの範囲であり、より好ましくは550~800nmの範囲である。
さらに、一般式(1)で表される金属錯体色素の光吸収波長域は、350~1200nmの範囲にあることが好ましく、400~1200nmの範囲にあることがより好ましい。
なお、本明細書において化合物(錯体、色素を含む)の表示については、当該化合物そのもののほか、その塩、錯体(錯体以外のとき)、そのイオンを含む意味に用いる。また、所望の効果を奏する範囲で、所定の形態で修飾された化合物を含む意味である。また、本明細書において置換・無置換を明記していない置換基については、その基に任意の置換基を有していてもよい意味である。これは置換・無置換を明記していない化合物についても同義である。好ましい置換基の例としては、R3およびR4の例として挙げた置換基が挙げられる。
さらに、一般式(1)で表される金属錯体色素の光吸収波長域は、350~1200nmの範囲にあることが好ましく、400~1200nmの範囲にあることがより好ましい。
なお、本明細書において化合物(錯体、色素を含む)の表示については、当該化合物そのもののほか、その塩、錯体(錯体以外のとき)、そのイオンを含む意味に用いる。また、所望の効果を奏する範囲で、所定の形態で修飾された化合物を含む意味である。また、本明細書において置換・無置換を明記していない置換基については、その基に任意の置換基を有していてもよい意味である。これは置換・無置換を明記していない化合物についても同義である。好ましい置換基の例としては、R3およびR4の例として挙げた置換基が挙げられる。
(A2)一般式(13)で表される構造を有する色素
本発明の光電変換素子及び光電気化学電池においては、上記一般式(1)の構造を有する色素とともに下記一般式(13)で表される構造を有する色素を用いることが好ましい。
Mz(LL11)m11(LL12)m12(X11)m13・CI11 一般式(13)
一般式(13)の構造を有する色素は、金属原子に、配位子LL11及び/又は配位子LL12と、場合により特定の官能基X11が配位しており、必要な場合はCI11により電気的に中性に保たれている。
(A2-1)金属原子Mz
Mzは金属原子を表す。Mzは好ましくは4配位または6配位が可能な金属であり、より好ましくはRu、Fe、Os、Cu、W、Cr、Mo、Ni、Pd、Pt、Co、Ir、Rh、Re、Mn又はZnである。特に好ましくは、Ru、Os、Zn又はCuであり、最も好ましくはRuである。
本発明の光電変換素子及び光電気化学電池においては、上記一般式(1)の構造を有する色素とともに下記一般式(13)で表される構造を有する色素を用いることが好ましい。
Mz(LL11)m11(LL12)m12(X11)m13・CI11 一般式(13)
一般式(13)の構造を有する色素は、金属原子に、配位子LL11及び/又は配位子LL12と、場合により特定の官能基X11が配位しており、必要な場合はCI11により電気的に中性に保たれている。
(A2-1)金属原子Mz
Mzは金属原子を表す。Mzは好ましくは4配位または6配位が可能な金属であり、より好ましくはRu、Fe、Os、Cu、W、Cr、Mo、Ni、Pd、Pt、Co、Ir、Rh、Re、Mn又はZnである。特に好ましくは、Ru、Os、Zn又はCuであり、最も好ましくはRuである。
(A2-2)配位子LL11
配位子LL11は、下記一般式(14)により表される2座または3座の配位子であり、好ましくは2座配位子である。配位子LL11の数を表すm11は0~3の整数であり、1~3であるのが好ましく、1であるのがより好ましい。m11が2以上のとき、LL11は同じでも異なっていてもよい。ただし、m11と、後述の配位子LL12の数を表すm12のうち少なくとも一方は1以上の整数である。したがって金属原子に、配位子LL11及び/又は配位子LL12が配位している。
なお、一般式(14)で表される配位子には、一般式(2)で表される配位子と同じ構造となるものを含まない。
配位子LL11は、下記一般式(14)により表される2座または3座の配位子であり、好ましくは2座配位子である。配位子LL11の数を表すm11は0~3の整数であり、1~3であるのが好ましく、1であるのがより好ましい。m11が2以上のとき、LL11は同じでも異なっていてもよい。ただし、m11と、後述の配位子LL12の数を表すm12のうち少なくとも一方は1以上の整数である。したがって金属原子に、配位子LL11及び/又は配位子LL12が配位している。
なお、一般式(14)で表される配位子には、一般式(2)で表される配位子と同じ構造となるものを含まない。
一般式(14)中のR101及びR102はそれぞれ独立に酸性基を表す。酸性基としては、カルボキシル基、スルホン酸基、ヒドロキシル基、ヒドロキサム酸基(好ましくは炭素原子数1~20のヒドロキサム酸基、例えば、―CONHOH、―CONCH3OH等)、ホスホリル基(例えば―OP(O)(OH)2等)またはホスホニル基(例えば―P(O)(OH)2等)が挙げられる。好ましくはカルボキシル基、ホスホニル基であり、より好ましくはカルボキシル基が挙げられる。R101およびR102はピリジン環上のどの炭素原子に置換してもよい。また、これらの酸性基は連結基を介してピリジン環に導入されているものであってもよい。

式中、R103、R104はそれぞれ独立に置換基を表す。前記置換基の具体例としては、好ましくはアルキル基(好ましくは炭素原子数1~20のアルキル基、例えばメチル、エチル、イソプロピル、t-ブチル、ペンチル、ヘプチル、1-エチルペンチル、ベンジル、2-エトキシエチル、1-カルボキシメチル等)、アルケニル基(好ましくは炭素原子数2~20のアルケニル基、例えば、ビニル、アリル、オレイル等)、アルキニル基(好ましくは炭素原子数2~20のアルキニル基、例えば、エチニル、ブタジイニル、フェニルエチニル等)、シクロアルキル基(好ましくは炭素原子数3~20のシクロアルキル基、例えば、シクロプロピル、シクロペンチル、シクロヘキシル、4-メチルシクロヘキシル等)、アリール基(好ましくは炭素原子数6~26のアリール基、例えば、フェニル、1-ナフチル、4-メトキシフェニル、2-クロロフェニル、3-メチルフェニル等)、ヘテロ環基(好ましくは炭素原子数2~20のヘテロ環基、例えば、2-ピリジル、4-ピリジル、2-イミダゾリル、2-ベンゾイミダゾリル、2-チアゾリル、2-オキサゾリル等)、アルコキシ基(好ましくは炭素原子数1~20のアルコキシ基、例えば、メトキシ、エトキシ、イソプロピルオキシ、ベンジルオキシ等)、アリールオキシ基(好ましくは炭素原子数6~26のアリールオキシ基、例えば、フェノキシ、1-ナフチルオキシ、3-メチルフェノキシ、4-メトキシフェノキシ等)、アルコキシカルボニル基(好ましくは炭素原子数2~20のアルコキシカルボニル基、例えば、エトキシカルボニル、2-エチルヘキシルオキシカルボニル等)、アミノ基(好ましくは炭素原子数0~20のアミノ基、例えば、アミノ、N,N-ジメチルアミノ、N,N-ジエチルアミノ、N-エチルアミノ、アニリノ等)、スルホンアミド基(好ましくは炭素原子数0~20のスルホンアミド基、例えば、N,N-ジメチルスルホンアミド、N-フェニルスルホンアミド等)、アシルオキシ基(好ましくは炭素原子数1~20のアシルオキシ基、例えば、アセチルオキシ、ベンゾイルオキシ等)、カルバモイル基(好ましくは炭素原子数1~20のカルバモイル基、例えば、N,N-ジメチルカルバモイル、N-フェニルカルバモイル等)、アシルアミノ基(好ましくは炭素原子数1~20のアシルアミノ基、例えば、アセチルアミノ、ベンゾイルアミノ等)、シアノ基、又はハロゲン原子(例えばフッ素原子、塩素原子、臭素原子、ヨウ素原子等)であり、より好ましくはアルキル基、アルケニル基、アリール基、ヘテロ環基、アルコキシ基、アリールオキシ基、アルコキシカルボニル基、アミノ基、アシルアミノ基、シアノ基又はハロゲン原子であり、特に好ましくはアルキル基、アルケニル基、ヘテロ環基、アルコキシ基、アルコキシカルボニル基、アミノ基、アシルアミノ基又はシアノ基である。
配位子LL11がアルキル基、アルケニル基等を含むとき、これらは直鎖状でも分岐状でもよく、置換されていても無置換でもよい。また配位子LL11がアリール基、ヘテロ環基等を含むとき、それらは単環でも縮環でもよく、置換されていても無置換でもよい。
一般式(14)中、R105及びR106はそれぞれ独立にアルキル基、アリール基、又はヘテロ環基を表す。一般式(14)において、アルキル基、アリール基、およびヘテロ環基は、それぞれ独立に、さらに置換基を有しても良い。アルキル基としては、炭素原子数5~18(好ましくは6~18、より好ましくは6~12)のアルキル基が好ましく、例えばへキシル、シクロヘキシル、デカン等が挙げられる。アリール基としては、炭素原子数6~30(好ましくは6~24、より好ましくは6~18)のアリール基が好ましく、例えば、フェニル、置換フェニル、ナフチル、置換ナフチル等が挙げられる。ヘテロ環基としては、炭素原子数3~30(好ましくは5~25、より好ましくは5~20)の5又は6員環のヘテロ環基が好ましく、例えば、2-チエニル、2-ピロリル、2-イミダゾリル、1-イミダゾリル、4-ピリジル、3-インドリルが挙げられる。また、R105およびR106は、上述のアルキル基、アリール基、およびヘテロ環基の2つ以上の組合せからなる基であっても良い。
R105及びR106は、置換基を有するアリール基またはヘテロ環基であることが好ましい。アリール基またはヘテロ環基上の置換基としては、アルキル基、アルケニル基、アルキニル基、シクロアルキル基、アルコキシ基、アリールオキシ基、アミノ基、アシルアミノ基(以上好ましい例はR103及びR104の場合と同様)またはヒドロキシル基であるのが好ましく、アルキル基、アルコキシ基、アミノ基またはヒドロキシル基であるのがより好ましく、アルキル基であるのが特に好ましい。R105とR106は同じであっても異なっていてもよいが、同じであるのが好ましい。
R105及びR106は、置換基を有するアリール基またはヘテロ環基であることが好ましい。アリール基またはヘテロ環基上の置換基としては、アルキル基、アルケニル基、アルキニル基、シクロアルキル基、アルコキシ基、アリールオキシ基、アミノ基、アシルアミノ基(以上好ましい例はR103及びR104の場合と同様)またはヒドロキシル基であるのが好ましく、アルキル基、アルコキシ基、アミノ基またはヒドロキシル基であるのがより好ましく、アルキル基であるのが特に好ましい。R105とR106は同じであっても異なっていてもよいが、同じであるのが好ましい。
R105とR106は、直接ピリジン環に結合していてもよい。R105とR106は、L11及び/又はL12を介してピリジン環に結合していてもよい。
ここでL11及びL12はそれぞれ独立に、アリーレン基、ヘテロアリーレン基、エテニレン基及びエチニレン基から選ばれた少なくとも1種からなる共役鎖を表す。エテニレン基は置換基を有していてもよい。エテニレン基が置換基を有する場合、該置換基はアルキル基であるのが好ましく、メチルであるのがより好ましい。アリーレン基の例としてはL1またはL2の例として挙げたものが好ましい。ヘテロアリーレン基の例としては、チエニレン基が挙げられる。
L11及びL12はそれぞれ独立に、炭素原子数2~6個の共役鎖であるのが好ましく、エテニレン、ブタジエニレン、エチニレン、ブタジイニレン、メチルエテニレン又はジメチルエテニレンがより好ましく、エテニレン又はブタジエニレンが特に好ましく、エテニレンが最も好ましい。L11とL12は同じであっても異なっていてもよいが、同じであるのが好ましい。なお、共役鎖が炭素―炭素二重結合を含む場合、各二重結合はトランス体であってもシス体であってもよく、これらの混合物であってもよい。
d11及びd12はそれぞれ独立に0~5の整数を表す。d11及びd12は好ましくは1~3であり、さらに好ましくは1~2である。d11が2以上のときL11は同じでも異なっていてもよい。d12が2以上のときL12は同じでも異なっていてもよい。
L11及びL12はそれぞれ独立に、炭素原子数2~6個の共役鎖であるのが好ましく、エテニレン、ブタジエニレン、エチニレン、ブタジイニレン、メチルエテニレン又はジメチルエテニレンがより好ましく、エテニレン又はブタジエニレンが特に好ましく、エテニレンが最も好ましい。L11とL12は同じであっても異なっていてもよいが、同じであるのが好ましい。なお、共役鎖が炭素―炭素二重結合を含む場合、各二重結合はトランス体であってもシス体であってもよく、これらの混合物であってもよい。
d11及びd12はそれぞれ独立に0~5の整数を表す。d11及びd12は好ましくは1~3であり、さらに好ましくは1~2である。d11が2以上のときL11は同じでも異なっていてもよい。d12が2以上のときL12は同じでも異なっていてもよい。
一般式(14)において、d13は0又は1を表し、好ましくは1である。
一般式(14)において、a11及びa12はそれぞれ独立に0~3の整数を表す。a11が2以上のときR101は同じでも異なっていてもよく、a12が2以上のときR102は同じでも異なっていてもよい。a11は0又は1であるのが好ましく、a12は0~2の整数であるのが好ましい。特に、d13が0のときa12は1又は2であるのが好ましく、d13が1のときa12は0又は1であるのが好ましい。a11とa12の和は0~2の整数であるのが好ましい。
一般式(14)において、a11及びa12はそれぞれ独立に0~3の整数を表す。a11が2以上のときR101は同じでも異なっていてもよく、a12が2以上のときR102は同じでも異なっていてもよい。a11は0又は1であるのが好ましく、a12は0~2の整数であるのが好ましい。特に、d13が0のときa12は1又は2であるのが好ましく、d13が1のときa12は0又は1であるのが好ましい。a11とa12の和は0~2の整数であるのが好ましい。
b11及びb12はそれぞれ独立に0~3の整数を表し、0~2の整数であるのが好ましい。b11が2以上のとき、R103は同じでも異なっていてもよく、互いに連結して環を形成していてもよい。b12が2以上のとき、R104は同じでも異なっていてもよく、互いに連結して環を形成していてもよい。またb11及びb12がともに1以上のとき、R103とR104が連結して環を形成していてもよい。形成する環の好ましい例としては、ベンゼン環、ピリジン環、チオフェン環、ピロール環、シクロヘキサン環、シクロペンタン環等が挙げられる。
a11とa12の和が1以上であって、配位子LL11が酸性基を少なくとも1個有するときは、一般式(13)中のm11は2または3であるのが好ましく、2であるのがより好ましい。
(A2-3)配位子LL12
一般式(13)中、LL12は2座又は3座の配位子を表す。配位子LL12の数を表すm12は0~2の整数であり、0又は1であるのが好ましい。m12が2のときLL12は同じでも異なっていてもよい。ただし、m12と、前述の配位子LL11の数を表すm11のうち少なくとも一方は1以上の整数である。
一般式(13)中、LL12は2座又は3座の配位子を表す。配位子LL12の数を表すm12は0~2の整数であり、0又は1であるのが好ましい。m12が2のときLL12は同じでも異なっていてもよい。ただし、m12と、前述の配位子LL11の数を表すm11のうち少なくとも一方は1以上の整数である。
配位子LL12は、下記一般式(15)で表される2座又は3座の配位子である。

一般式(15)中、Zd、Ze及びZfはそれぞれ独立に、5員環又は6員環を形成しうる非金属原子群を表す。形成される5員環又は6員環は置換されていても無置換でもよく、単環でも縮環していてもよい。Zd、Ze及びZfは炭素原子、水素原子、窒素原子、酸素原子、硫黄原子、リン原子及び/又はハロゲン原子で構成されることが好ましく、芳香族環を形成するのが好ましい。5員環の場合はイミダゾール環、オキサゾール環、チアゾール環又はトリアゾール環を形成するのが好ましく、6員環の場合はピリジン環、ピリミジン環、ピリダジン環又はピラジン環を形成するのが好ましい。なかでもイミダゾール環又はピリジン環がより好ましい。
Zd、Ze及びZfで表される非金属原子群は、置換基を有してもよく、酸性基を有していることが好ましい。置換基としては、一般式(1)のR3およびR4の例としてあげた基が挙げられる。酸性基としては、例えばカルボキシル基、スルホン酸基、ヒドロキシル基、ヒドロキサム酸基、ホスホリル基及びホスホニル基等が挙げられる。また、これらの酸性基は連結基を介して導入されているものであってもよい。
Zd、Ze及びZfで表される非金属原子群は、置換基を有してもよく、酸性基を有していることが好ましい。置換基としては、一般式(1)のR3およびR4の例としてあげた基が挙げられる。酸性基としては、例えばカルボキシル基、スルホン酸基、ヒドロキシル基、ヒドロキサム酸基、ホスホリル基及びホスホニル基等が挙げられる。また、これらの酸性基は連結基を介して導入されているものであってもよい。
一般式(15)中、fは0または1を表す。fは0であるのが好ましく、LL12は2座配位子であるのが好ましい。
一般式(15)の好ましい態様は、一般式(7)のものと同様である。
(A2-4)配位子X11
一般式(13)中、X11は1座又は2座の配位子を表す。配位子X11の数を表すm13は0~2の整数を表し、m13は好ましくは1又は2である。X11が1座配位子のとき、m13は2であるのが好ましく、X11が2座配位子のとき、m13は1であるのが好ましい。m13が2のとき、X11は同じでも異なっていてもよく、X11同士が連結していてもよい。
一般式(13)中、X11は1座又は2座の配位子を表す。配位子X11の数を表すm13は0~2の整数を表し、m13は好ましくは1又は2である。X11が1座配位子のとき、m13は2であるのが好ましく、X11が2座配位子のとき、m13は1であるのが好ましい。m13が2のとき、X11は同じでも異なっていてもよく、X11同士が連結していてもよい。
配位子X11としては、一般式(1)のXの例として挙げた置換基が挙げられ、その好ましい範囲も同様である。
(A2-5)対イオンCI11
一般式(14)中のCI11は電荷を中和させるのに対イオンが必要な場合の対イオンを表す。一般に、色素が陽イオン又は陰イオンであるか、あるいは正味のイオン電荷を有するかどうかは、色素中の金属、配位子および置換基に依存する。
一般式(14)中のCI11は電荷を中和させるのに対イオンが必要な場合の対イオンを表す。一般に、色素が陽イオン又は陰イオンであるか、あるいは正味のイオン電荷を有するかどうかは、色素中の金属、配位子および置換基に依存する。
置換基が解離性基を有することなどにより、一般式(14)の色素は解離して負電荷を持ってもよい。この場合、一般式(14)の色素全体の電荷はCI11により電気的に中性とされる。
対イオンCI11としては、一般式(1)のCIの例として挙げたものが挙げられ、その好ましい範囲も同様である。
(A2-6)結合基
一般式(13)で表される構造を有する色素は、半導体微粒子の表面に対する適当な酸性基(結合基、interlocking group)を1つ以上有する。すなわち、LL11、LL12、およびX11の少なくとも1つに、少なくとも1つの酸性基を有する。この結合基を色素中に1~6個有するのがより好ましく、1~4個有するのが特に好ましい。上記結合基として、カルボキシル基、スルホン酸基、ヒドロキシル基、ヒドロキサム酸基(例えば―CONHOH等)、ホスホリル基(例えば―OP(O)(OH)2等)、ホスホニル基(例えば―P(O)(OH)2等)等の酸性基(解離性のプロトンを有する置換基)を色素中に有することが好ましい。配位子LL12上に上記酸性基を有するのが好ましい。なかでも、ホスホニル基、カルボキシル基を配位子LL12上に有することが好ましい。また、これらの酸性基は連結基を介して導入されているものであってもよい。
一般式(13)で表される構造を有する色素は、半導体微粒子の表面に対する適当な酸性基(結合基、interlocking group)を1つ以上有する。すなわち、LL11、LL12、およびX11の少なくとも1つに、少なくとも1つの酸性基を有する。この結合基を色素中に1~6個有するのがより好ましく、1~4個有するのが特に好ましい。上記結合基として、カルボキシル基、スルホン酸基、ヒドロキシル基、ヒドロキサム酸基(例えば―CONHOH等)、ホスホリル基(例えば―OP(O)(OH)2等)、ホスホニル基(例えば―P(O)(OH)2等)等の酸性基(解離性のプロトンを有する置換基)を色素中に有することが好ましい。配位子LL12上に上記酸性基を有するのが好ましい。なかでも、ホスホニル基、カルボキシル基を配位子LL12上に有することが好ましい。また、これらの酸性基は連結基を介して導入されているものであってもよい。
本発明で用いる一般式(13)で表される構造を有する色素の具体例を以下に示すが、本発明はこれらに限定されるものではない。なお、下記具体例における色素がプロトン解離性基を有する配位子を含む場合、該配位子は必要に応じて解離しプロトンを放出してもよい。

本発明の一般式(13)により表される色素は、特開2001-291534号公報や当該公報に引用された方法を参考にして合成することができる。
一般式(13)の構造を有する色素は、溶液における極大吸収波長が、好ましくは300~1000nmの範囲であり、より好ましくは350~950nmの範囲であり、特に好ましくは370~900nmの範囲である。
さらに、一般式(13)で表される金属錯体色素の光吸収波長域は、350~1200nmの範囲にあることが好ましく、400~900nmの範囲にあることがより好ましい。
さらに、一般式(13)で表される金属錯体色素の光吸収波長域は、350~1200nmの範囲にあることが好ましく、400~900nmの範囲にあることがより好ましい。
(A3)その他の色素
本発明の光電変換素子及び光電気化学電池においては、上記一般式(1)の構造を有する色素や一般式(13)の構造を有する色素とともに、通常の色素を併用することもできる。
本発明の光電変換素子及び光電気化学電池においては、上記一般式(1)の構造を有する色素や一般式(13)の構造を有する色素とともに、通常の色素を併用することもできる。
本発明の光電変換素子及び光電気化学電池においては、一般式(1)の構造を有する金属錯体色素を必須成分として用いる。さらに好ましくは、一般式(13)の構造を有する色素を併用することにより、広範囲の波長の光を利用でき、高い変換効率を確保するとともに、変換効率の低下率を低減することできる。
一般式(1)の構造を有する金属錯体色素と、一般式(13)の構造を有する色素の配合割合は、前者をR、後者をSとすると、モル%の比で、R/S=90/10~10/90、好ましくはR/S=80/20~20/80、さらに好ましくはR/S=70/30~30/70、より一層好ましくはR/S=60/40~40/60、最も好ましくはR/S=55/45~45/55であり、通常は両者を等モル使用する。
(B)電荷移動体
本発明の光電変換素子に用いられる電解質組成物には、酸化還元対として、例えばヨウ素とヨウ化物(例えばヨウ化リチウム、ヨウ化テトラブチルアンモニウム、ヨウ化テトラプロピルアンモニウム等)との組み合わせ、アルキルビオローゲン(例えばメチルビオローゲンクロリド、ヘキシルビオローゲンブロミド、ベンジルビオローゲンテトラフルオロボレート)とその還元体との組み合わせ、ポリヒドロキシベンゼン類(例えばハイドロキノン、ナフトハイドロキノン等)とその酸化体との組み合わせ、2価と3価の鉄錯体(例えば赤血塩と黄血塩)の組み合わせ等が挙げられる。これらのうちヨウ素とヨウ化物との組み合わせが好ましい。
本発明の光電変換素子に用いられる電解質組成物には、酸化還元対として、例えばヨウ素とヨウ化物(例えばヨウ化リチウム、ヨウ化テトラブチルアンモニウム、ヨウ化テトラプロピルアンモニウム等)との組み合わせ、アルキルビオローゲン(例えばメチルビオローゲンクロリド、ヘキシルビオローゲンブロミド、ベンジルビオローゲンテトラフルオロボレート)とその還元体との組み合わせ、ポリヒドロキシベンゼン類(例えばハイドロキノン、ナフトハイドロキノン等)とその酸化体との組み合わせ、2価と3価の鉄錯体(例えば赤血塩と黄血塩)の組み合わせ等が挙げられる。これらのうちヨウ素とヨウ化物との組み合わせが好ましい。
ヨウ素塩のカチオンは5員環又は6員環の含窒素芳香族カチオンであるのが好ましい。特に、一般式(1)により表される化合物がヨウ素塩でない場合は、WO95/18456号、特開平8-259543号、電気化学,第65巻,11号,923頁(1997年)等に記載されているピリジニウム塩、イミダゾリウム塩、トリアゾリウム塩等のヨウ素塩を併用するのが好ましい。
本発明の光電変換素子に使用される電解質組成物中には、ヘテロ環4級塩化合物と共にヨウ素を含有するのが好ましい。ヨウ素の含有量は電解質組成物全体に対して0.1~20質量%であるのが好ましく、0.5~5質量%であるのがより好ましい。
本発明の光電変換素子に用いられる電解質組成物は溶媒を含んでいてもよい。電解質組成物中の溶媒含有量は組成物全体の50質量%以下であるのが好ましく、30質量%以下であるのがより好ましく、10質量%以下であるのが特に好ましい。
溶媒としては低粘度でイオン移動度が高いか、高誘電率で有効キャリアー濃度を高めることができるか、あるいはその両方であるために優れたイオン伝導性を発現できるものが好ましい。このような溶媒としてカーボネート化合物(エチレンカーボネート、プロピレンカーボネート等)、複素環化合物(3-メチル-2-オキサゾリジノン等)、エーテル化合物(ジオキサン、ジエチルエーテル等)、鎖状エーテル類(エチレングリコールジアルキルエーテル、プロピレングリコールジアルキルエーテル、ポリエチレングリコールジアルキルエーテル、ポリプロピレングリコールジアルキルエーテル等)、アルコール類(メタノール、エタノール、エチレングリコールモノアルキルエーテル、プロピレングリコールモノアルキルエーテル、ポリエチレングリコールモノアルキルエーテル、ポリプロピレングリコールモノアルキルエーテル等)、多価アルコール類(エチレングリコール、プロピレングリコール、ポリエチレングリコール、ポリプロピレングリコール、グリセリン等)、ニトリル化合物(アセトニトリル、グルタロジニトリル、メトキシアセトニトリル、プロピオニトリル、ベンゾニトリル、ビスシアノエチルエーテル等)、エステル類(カルボン酸エステル、リン酸エステル、ホスホン酸エステル等)、非プロトン性極性溶媒(ジメチルスルホキシド(DMSO)、スルフォラン等)、水、特開2002-110262記載の含水電解液、特開2000-36332号公報、特開2000-243134号公報、及び再公表WO/00-54361号公報記載の電解質溶媒などが挙げられる。これらの溶媒は二種以上を混合して用いてもよい。
また、電解質溶媒として、室温において液体状態であり、及び/又は室温よりも低い融点を有する電気化学的に不活性な塩を用いても良い。例えば、1-エチルー3-メチルイミダゾリウムトリフルオロメタンスルホネート、1-ブチルー3-メチルイミダゾリウムトリフルオロメタンスルホネート等にイミダゾリウム塩、ピリジニウム塩などの含窒素ヘテロ環四級塩化合物、又はテトラアルキルアンモニウム塩などが挙げられる。
本発明の光電変換素子に用いられる電解質組成物には、ポリマーやオイルゲル化剤を添加したり、多官能モノマー類の重合やポリマーの架橋反応等の手法によりゲル化(固体化)してもよい。
ポリマーを添加することにより電解質組成物をゲル化させる場合、Polymer Electrolyte Reviews-1及び2(J. R. MacCallumとC. A. Vincentの共編、ELSEVIER APPLIED SCIENCE)に記載された化合物等を添加することができる。この場合、ポリアクリロニトリル又はポリフッ化ビニリデンを用いるのが好ましい。
オイルゲル化剤を添加することにより電解質組成物をゲル化させる場合は、オイルゲル化剤としてJ. Chem. Soc. Japan, Ind. Chem. Soc., 46779 (1943)、J. Am. Chem. Soc., 111, 5542 (1989)、J. Chem. Soc., Chem. Commun., 390 (1993)、Angew. Chem. Int.Ed. Engl., 35, 1949 (1996)、Chem. Lett., 885, (1996)、J. Chem. Soc., Chem. Commun., 545, (1997)等に記載された化合物を使用することができ、アミド構造を有する化合物を用いるのが好ましい。
多官能モノマー類の重合によって電解質組成物をゲル化する場合は、多官能モノマー類、重合開始剤、電解質及び溶媒から溶液を調製し、キャスト法、塗布法、浸漬法、含浸法等の方法により色素を担持した電極上にゾル状の電解質層を形成し、その後多官能モノマーのラジカル重合によってゲル化させる方法が好ましい。多官能モノマー類はエチレン性不飽和基を2個以上有する化合物であることが好ましく、ジビニルベンゼン、エチレングリコールジアクリレート、エチレングリコールジメタクリレート、ジエチレングリコールジアクリレート、ジエチレングリコールジメタクリレート、トリエチレングリコールジアクリレート、トリエチレングリコールジメタクリレート、ペンタエリスリトールトリアクリレート、トリメチロールプロパントリアクリレート等が好ましい。
ゲル電解質は上記多官能モノマー類の他に単官能モノマーを含む混合物の重合によって形成してもよい。単官能モノマーとしては、アクリル酸又はα-アルキルアクリル酸(アクリル酸、メタクリル酸、イタコン酸等)或いはそれらのエステル又はアミド(メチルアクリレート、エチルアクリレート、n-プロピルアクリレート、i-プロピルアクリレート、n-ブチルアクリレート、i-ブチルアクリレート、t-ブチルアクリレート、n-ペンチルアクリレート、3-ペンチルアクリレート、t-ペンチルアクリレート、n-ヘキシルアクリレート、2,2-ジメチルブチルアクリレート、n-オクチルアクリレート、2-エチルヘキシルアクリレート、4-メチル-2-プロピルペンチルアクリレート、セチルアクリレート、n-オクタデシルアクリレート、シクロヘキシルアクリレート、シクロペンチルアクリレート、ベンジルアクリレート、ヒドロキシエチルアクリレート、2-ヒドロキシプロピルアクリレート、2-メトキシエチルアクリレート、2-エトキシエチルアクリレート、2-メトキシエトキシエチルアクリレート、フェノキシエチルアクリレート、3-メトキシブチルアクリレート、エチルカルビトールアクリレート、2-メチル-2-ニトロプロピルアクリレート、2,2,2-トリフルオロエチルアクリレート、オクタフルオロペンチルアクリレート、ヘプタデカフルオロデシルアクリレート、メチルメタクリレート、n-ブチルメタクリレート、i-ブチルメタクリレート、t-ブチルメタクリレート、t-ペンチルメタクリレート、n-オクタデシルメタクリレート、ベンジルメタクリレート、ヒドロキシエチルメタクリレート、2-ヒドロキシプロピルメタクリレート、2-メトキシエチルメタクリレート、2-エトキシエチルメタクリレート、2-メトキシエトキシエチルメタクリレート、ジメチルアミノエチルメタクリレート、2,2,2-トリフルオロエチルメタクリレート、テトラフルオロプロピルメタクリレート、ヘキサフルオロプロピルメタクリレート、ヘプタデカフルオロデシルメタクリレート、エチレングリコールエチルカーボネートメタクリレート、2-イソボルニルメタクリレート、2-ノルボルニルメチルメタクリレート、5-ノルボルネン-2-イルメチルメタクリレート、3-メチル-2-ノルボルニルメチルメタクリレート、アクリルアミド、N-i-プロピルアクリルアミド、N-n-ブチルアクリルアミド、N-t-ブチルアクリルアミド、N,N-ジメチルアクリルアミド、N-メチロールアクリルアミド、ジアセトンアクリルアミド、2-アクリルアミド-2-メチルプロパンスルホン酸、アクリルアミドプロピルトリメチルアンモニウムクロライド、メタクリルアミド、N-メチルメタクリルアミド、N-メチロールメタクリルアミド等)、ビニルエステル類(酢酸ビニル等)、マレイン酸又はフマル酸或いはそれらから誘導されるエステル類(マレイン酸ジメチル、マレイン酸ジブチル、フマル酸ジエチル等)、p-スチレンスルホン酸のナトリウム塩、アクリロニトリル、メタクリロニトリル、ジエン類(ブタジエン、シクロペンタジエン、イソプレン等)、芳香族ビニル化合物(スチレン、p-クロロスチレン、t-ブチルスチレン、α-メチルスチレン、スチレンスルホン酸ナトリウム等)、N-ビニルホルムアミド、N-ビニル-N-メチルホルムアミド、N-ビニルアセトアミド、N-ビニル-N-メチルアセトアミド、ビニルスルホン酸、ビニルスルホン酸ナトリウム、アリルスルホン酸ナトリウム、メタクリルスルホン酸ナトリウム、ビニリデンフルオライド、ビニリデンクロライド、ビニルアルキルエーテル類(メチルビニルエーテル等)、エチレン、プロピレン、ブテン、イソブテン、N-フェニルマレイミド等が使用可能である。
多官能モノマーの配合量は、モノマー全体に対して0.5~70質量%とすることが好ましく、1.0~50質量%であるのがより好ましい。上述のモノマーは、大津隆行・木下雅悦共著「高分子合成の実験法」(化学同人)や大津隆行「講座重合反応論1ラジカル重合(I)」(化学同人)に記載された一般的な高分子合成法であるラジカル重合によって重合することができる。本発明で使用するゲル電解質用モノマーは加熱、光又は電子線によって、或いは電気化学的にラジカル重合させることができるが、特に加熱によってラジカル重合させるのが好ましい。この場合、好ましく使用できる重合開始剤は2,2’-アゾビスイソブチロニトリル、2,2’-アゾビス(2,4-ジメチルバレロニトリル)、ジメチル2,2’-アゾビス(2-メチルプロピオネート)、ジメチル2,2’-アゾビスイソブチレート等のアゾ系開始剤、ラウリルパーオキシド、ベンゾイルパーオキシド、t-ブチルパーオクトエート等の過酸化物系開始剤等である。重合開始剤の好ましい添加量はモノマー総量に対し0.01~20質量%であり、より好ましくは0.1~10質量%である。
ゲル電解質に占めるモノマーの重量組成範囲は0.5~70質量%であるのが好ましい。より好ましくは1.0~50質量%である。ポリマーの架橋反応により電解質組成物をゲル化させる場合は、組成物に架橋可能な反応性基を有するポリマー及び架橋剤を添加するのが好ましい。好ましい反応性基はピリジン環、イミダゾール環、チアゾール環、オキサゾール環、トリアゾール環、モルホリン環、ピペリジン環、ピペラジン環等の含窒素複素環であり、好ましい架橋剤は窒素原子が求核攻撃できる官能基を2つ以上有する化合物(求電子剤)であり、例えば2官能以上のハロゲン化アルキル、ハロゲン化アラルキル、スルホン酸エステル、酸無水物、酸クロライド、イソシアネート等である。
本発明の電解質組成物には、金属ヨウ化物(LiI、NaI、KI、CsI、CaI2等)、金属臭化物(LiBr、NaBr、KBr、CsBr、CaBr2等)、4級アンモニウム臭素塩(テトラアルキルアンモニウムブロマイド、ピリジニウムブロマイド等)、金属錯体(フェロシアン酸塩-フェリシアン酸塩、フェロセン-フェリシニウムイオン等)、イオウ化合物(ポリ硫化ナトリウム、アルキルチオール-アルキルジスルフィド等)、ビオロゲン色素、ヒドロキノン-キノン等を添加してよい。これらは混合して用いてもよい。
また、本発明ではJ. Am. Ceram. Soc., 80, (12), 3157-3171 (1997)に記載のt-ブチルピリジンや、2-ピコリン、2,6-ルチジン等の塩基性化合物を添加してもよい。塩基性化合物を添加する場合の好ましい濃度範囲は0.05~2Mである。
また、本発明において電解質としては、正孔導体物質を含む電荷輸送層を用いても良い。正孔導体物質として、9,9’-スピロビフルオレン誘導体などを用いることができる。
また、電極層、光電変換層、ホール輸送層、伝導層、対極層を順次に積層することができる。p型半導体として機能するホール輸送材料をホール輸送層としてもちいることができる。好ましいホール輸送層としては、例えば無機系又は有機系のホール輸送材料を用いることができる。無機系ホール輸送材料としては、CuI、CuO,NiO等が挙げられる。また、有機系ホール輸送材料としては、高分子系と低分子系のものが挙げられ、高分子系のものとしては、例えばポリビニルカルバゾール、ポリアミン、有機ポリシラン等が挙げられる。また、低分子系のものとしては、例えばトリフェニルアミン誘導体、スチルベン誘導体、ヒドラゾン誘導体、フェナミン誘導体等が挙げられる。この中でも有機ポリシランは、従来の炭素系高分子と異なり、主鎖のSiに沿って非局化されたσ電子が光伝導に寄与し、高いホール移動度を有するため、好ましい(Phys. Rev. B, 35, 2818(1987))。
本発明における伝導層は、導電性のよいものであれば特に限定されないが、例えば無機導電性材料、有機導電性材料、導電性ポリマー、分子間電荷移動錯体等が挙げられる。中でもドナー材料とアクセプター材料とから形成された分子間電荷移動錯体が好ましい。この中でも、有機ドナーと有機アクセプターとから形成されたものを好ましく用いることができる。
ドナー材料は、分子構造内で電子がリッチなものが好ましい。例えば、有機ドナー材料としては、分子のπ電子系に、置換若しくは無置換アミン基、水酸基、エーテル基、セレン又は硫黄原子を有するものが挙げられ、具体的には、フェニルアミン系、トリフェニルメタン系、カルバゾール系、フェノール系、テトラチアフルバレン系材料が挙げられる。アクセプター材料としては、分子構造内で電子不足なものが好ましい。例えば、有機アクセプター材料としては、フラーレン、分子のπ電子系にニトロ基、シアノ基、カルボキシル基又はハロゲン基等の置換基を有するものが挙げられ、具体的にはPCBM、ベンゾキノン系、ナフトキノン系等のキノン系、フロオレノン系、クロラニル系、ブロマニル系、テトラシアノキノジメタン系、テトラシアノンエチレン系等が挙げられる。
伝導層の厚みは、特に限定されないが、多孔質を完全に埋めることができる程度が好ましい。
(C)導電性支持体
図1に示すように、本発明の光電変換素子には、導電性支持体1上には多孔質の半導体微粒子22に色素21が吸着された感光体2が形成されている。後述する通り、例えば、半導体微粒子の分散液を導電性支持体に塗布・乾燥後、本発明の色素溶液に浸漬することにより、感光層を製造することができる。
図1に示すように、本発明の光電変換素子には、導電性支持体1上には多孔質の半導体微粒子22に色素21が吸着された感光体2が形成されている。後述する通り、例えば、半導体微粒子の分散液を導電性支持体に塗布・乾燥後、本発明の色素溶液に浸漬することにより、感光層を製造することができる。
導電性支持体としては、金属のように支持体そのものに導電性があるものか、または表面に導電膜層を有するガラスや高分子材料を使用することができる。導電性支持体は実質的に透明であることが好ましい。実質的に透明であるとは光の透過率が10%以上であることを意味し、50%以上であることが好ましく、80%以上が特に好ましい。導電性支持体としては、ガラスや高分子材料に導電性の金属酸化物を塗設したものを使用することができる。このときの導電性の金属酸化物の塗布量は、ガラスや高分子材料の支持体1m2当たり、0.1~100gが好ましい。透明導電性支持体を用いる場合、光は支持体側から入射させることが好ましい。好ましく使用される高分子材料の一例として、テトラアセチルセルロース(TAC)、ポリエチレンテレフタレート(PET)、ポリエチレンナフタレート(PEN)、シンジオタクチックポリスチレン(SPS)、ポリフェニレンスルフィド(PPS)、ポリカーボネート(PC)、ポリアリレート(PAR)、ポリスルフォン(PSF)、ポリエステルスルフォン(PES)、ポリエーテルイミド(PEI)、環状ポリオレフィン、ブロム化フェノキシ等を挙げることができる。導電性支持体上には、表面に光マネージメント機能を施してもよく、例えば、特開2003-123859記載の高屈折膜及び低屈折率の酸化物膜を交互に積層した反射防止膜、特開2002-260746記載のライトガイド機能が挙げられる。
この他にも、金属支持体も好ましく使用することができる。その一例としては、チタン、アルミニウム、銅、ニッケル、鉄、ステンレスを挙げることができる。これらの金属は合金であってもよい。さらに好ましくは、チタン、アルミニウム、銅が好ましく、特に好ましくは、チタンやアルミニウムである。
導電性支持体上には、紫外光を遮断する機能を持たせることが好ましい。例えば、紫外光を可視光に変えることが出来る蛍光材料を透明支持体中または、透明支持体表面に存在させる方法や紫外線吸収剤を用いる方法も挙げられる。
導電性支持体上には、さらに特開平11-250944号公報等に記載の機能を付与してもよい。
好ましい導電膜としては金属(例えば白金、金、銀、銅、アルミニウム、ロジウム、インジウム等)、炭素、もしくは導電性の金属酸化物(インジウム-スズ複合酸化物、酸化スズにフッ素をドープしたもの等)が挙げられる。
導電膜層の厚さは0.01~30μmであることが好ましく、0.03~25μmであることが更に好ましく、特に好ましくは0.05~20μmである。
導電性支持体は表面抵抗が低い程よい。好ましい表面抵抗の範囲としては50Ω/cm2以下であり、さらに好ましくは10Ω/cm2以下である。この下限に特に制限はないが、通常0.1Ω/cm2程度である。
導電膜の抵抗値はセル面積が大きくなると大きくなる為、集電電極を配置してもよい。支持体と透明導電膜の間にガスバリア膜及び/又はイオン拡散防止膜を配置しても良い。ガスバリア層としては、樹脂膜や無機膜を使用することができる。
また、透明電極と多孔質半導体電極光触媒含有層を設けてもよい。透明導電層は積層構造でも良く、好ましい方法としてたとえば、ITO上にFTOを積層することができる。
(D)半導体微粒子
図1に示すように、本発明の光電変換素子には、導電性支持体1上には多孔質の半導体微粒子22に色素21が吸着された感光層2が形成されている。後述する通り、例えば、半導体微粒子の分散液を前記の導電性支持体に塗布・乾燥後、本発明の色素溶液に浸漬することにより、感光体を製造することができる。
図1に示すように、本発明の光電変換素子には、導電性支持体1上には多孔質の半導体微粒子22に色素21が吸着された感光層2が形成されている。後述する通り、例えば、半導体微粒子の分散液を前記の導電性支持体に塗布・乾燥後、本発明の色素溶液に浸漬することにより、感光体を製造することができる。
半導体微粒子としては、好ましくは金属のカルコゲニド(例えば酸化物、硫化物、セレン化物等)またはペロブスカイトの微粒子が用いられる。金属のカルコゲニドとしては、好ましくはチタン、スズ、亜鉛、タングステン、ジルコニウム、ハフニウム、ストロンチウム、インジウム、セリウム、イットリウム、ランタン、バナジウム、ニオブ、もしくはタンタルの酸化物、硫化カドミウム、セレン化カドミウム等が挙げられる。ペロブスカイトとしては、好ましくはチタン酸ストロンチウム、チタン酸カルシウム等が挙げられる。これらのうち酸化チタン、酸化亜鉛、酸化スズ、酸化タングステンが特に好ましい。
半導体には伝導に関わるキャリアーが電子であるn型とキャリアーが正孔であるp型が存在するが、本発明の素子ではn型を用いることが変換効率の点で好ましい。n型半導体には、不純物準位をもたず伝導帯電子と価電子帯正孔によるキャリアーの濃度が等しい固有半導体(あるいは真性半導体)の他に、不純物に由来する構造欠陥により電子キャリアー濃度の高いn型半導体が存在する。本発明で好ましく用いられるn型の無機半導体は、TiO2、TiSrO3、ZnO、Nb2O3、SnO2、WO3、Si、CdS、CdSe、V2O5、ZnS、ZnSe、SnSe、KTaO3、FeS2、PbS、InP、GaAs、CuInS2、CuInSe2などである。これらのうち最も好ましいn型半導体はTiO2、ZnO、SnO2、WO3、ならびにNb2O3である。また、これらの半導体の複数を複合させた半導体材料も好ましく用いられる。
半導体微粒子の粒径は、半導体微粒子分散液の粘度を高く保つ目的で、一次粒子の平均粒径が2nm以上50nm以下であることが好ましく、また一次粒子の平均粒径が2nm以上30nm以下の超微粒子であることがより好ましい。粒径分布の異なる2種類以上の微粒子を混合してもよく、この場合小さい粒子の平均サイズは5nm以下であるのが好ましい。また、入射光を散乱させて光捕獲率を向上させる目的で、上記の超微粒子に対して平均粒径が50nmを越える大きな粒子を、低含率で添加、又は別層塗布することもできる。この場合、大粒子の含率は、平均粒径が50nm以下の粒子の質量の50%以下であることが好ましく、20%以下であることがより好ましい。上記の目的で添加混合する大粒子の平均粒径は、100nm以上が好ましく、250nm以上がより好ましい。
光散乱用の大粒子を用いることで、ヘイズ率60%以上となることが好ましい。ヘイズ率とは(拡散透過率)÷(全光透過率)で表される。
半導体微粒子の作製法としては、作花済夫の「ゾル・ゲル法の科学」アグネ承風社(1998年)等に記載のゲル・ゾル法が好ましい。またDegussa社が開発した塩化物を酸水素塩中で高温加水分解により酸化物を作製する方法も好ましい。半導体微粒子が酸化チタンの場合、上記ゾル・ゲル法、ゲル・ゾル法、塩化物の酸水素塩中での高温加水分解法はいずれも好ましいが、さらに清野学の「酸化チタン 物性と応用技術」技報堂出版(1997年)に記載の硫酸法および塩素法を用いることもできる。さらにゾル・ゲル法として、バルべ等のジャーナル・オブ・アメリカン・セラミック・ソサエティー,第80巻,第12号,3157~3171頁(1997年)に記載の方法や、バーンサイドらのケミストリー・オブ・マテリアルズ,第10巻,第9号,2419~2425頁に記載の方法も好ましい。
この他に、半導体微粒子の製造方法として、例えば、チタニアナノ粒子の製造方法として好ましくは、四塩化チタンの火炎加水分解による方法、四塩化チタンの燃焼法、安定なカルコゲナイド錯体の加水分解、オルトチタン酸の加水分解、可溶部と不溶部から半導体微粒子を形成後可溶部を溶解除去する方法、過酸化物水溶液の水熱合成、またはゾル・ゲル法によるコア/シェル構造の酸化チタン微粒子の製造方法が挙げられる。
チタニアの結晶構造としては、アナターゼ型、ブルッカイト型、または、ルチル型があげられ、アナターゼ型、ブルッカイト型が好ましい。
チタニアナノチューブ・ナノワイヤー・ナノロッドをチタニア微粒子に混合してもよい。
チタニアは、非金属元素などによりドーピングされていても良い。チタニアへの添加剤としてドーパント以外に、ネッキングを改善する為のバインダーや逆電子移動防止の為に表面へ添加剤を用いても良い。好ましい添加剤の例としては、ITO、SnO粒子、ウイスカー、繊維状グラファイト・カーボンナノチューブ、酸化亜鉛ネッキング結合子、セルロース等の繊維状物質、金属、有機シリコン、ドデシルベンゼンスルホン酸、シラン化合物等の電荷移動結合分子、及び電位傾斜型デンドリマーなどが挙げられる。
チタニア上の表面欠陥を除去するなどの目的で、色素吸着前にチタニアを酸塩基又は酸化還元処理しても良い。エッチング、酸化処理、過酸化水素処理、脱水素処理、UV-オゾン、酸素プラズマなどで処理してもよい。
(E)半導体微粒子分散液
本発明においては、半導体微粒子以外の固形分の含量が、半導体微粒子分散液全体の10質量%以下よりなる半導体微粒子分散液を前記の導電性支持体に塗布し、適度に加熱することにより、多孔質半導体微粒子塗布層を得ることができる。
本発明においては、半導体微粒子以外の固形分の含量が、半導体微粒子分散液全体の10質量%以下よりなる半導体微粒子分散液を前記の導電性支持体に塗布し、適度に加熱することにより、多孔質半導体微粒子塗布層を得ることができる。
半導体微粒子分散液を作製する方法としては、前述のゾル・ゲル法の他に、半導体を合成する際に溶媒中で微粒子として析出させそのまま使用する方法、微粒子に超音波などを照射して超微粒子に粉砕する方法、あるいはミルや乳鉢などを使って機械的に粉砕しすり潰す方法、等が挙げられる。分散溶媒としては、水および/または各種の有機溶媒を用いることができる。有機溶媒としては、メタノール,エタノール,イソプロピルアルコール,シトロネロール,ターピネオールなどのアルコール類、アセトンなどのケトン類、酢酸エチルなどのエステル類、ジクロロメタン、アセトニトリル等が挙げられる。
分散の際、必要に応じて例えばポリエチレングリコール、ヒドロキシエチルセルロース、カルボキシメチルセルロースのようなポリマー、界面活性剤、酸、またはキレート剤等を分散助剤として少量用いてもよい。しかし、これらの分散助剤は、導電性支持体上へ製膜する工程の前に、ろ過法や分離膜を用いる方法、あるいは遠心分離法などによって大部分を除去しておくことが好ましい。半導体微粒子分散液は、半導体微粒子以外の固形分の含量が分散液全体の10質量%以下とすることができる。この濃度は好ましくは5%以下であり、さらに好ましくは3%以下であり、特に好ましくは1%以下である。さらに好ましくは0.5%以下であり、特に好ましくは0.2%である。すなわち、半導体微粒子分散液中に、溶媒と半導体微粒子以外の固形分を半導体微粒子分散液全体の10質量%以下とすることができる。実質的に半導体微粒子と分散溶媒のみからなることが好ましい。
半導体微粒子分散液の粘度が高すぎると分散液が凝集してしまい製膜することができず、逆に半導体微粒子分散液の粘度が低すぎると液が流れてしまい製膜することができないことがある。したがって分散液の粘度は、25℃で10~300N・s/m2が好ましい。さらに好ましくは、25℃で50~200N・s/m2である。
半導体微粒子分散液の塗布方法としては、アプリケーション系の方法としてローラ法、ディップ法等を使用することができる。またメータリング系の方法としてエアーナイフ法、ブレード法等を使用することができる。またアプリケーション系の方法とメータリング系の方法を同一部分にできるものとして、特公昭58-4589号に開示されているワイヤーバー法、米国特許2681294号明細書等に記載のスライドホッパー法、エクストルージョン法、カーテン法等が好ましい。また汎用機を使用してスピン法やスプレー法で塗布するのも好ましい。湿式印刷方法としては、凸版、オフセットおよびグラビアの3大印刷法をはじめ、凹版、ゴム版、スクリーン印刷等が好ましい。これらの中から、液粘度やウェット厚さに応じて、好ましい製膜方法を選択する。また本発明の半導体微粒子分散液は粘度が高く、粘稠性を有するため、凝集力が強いことがあり、塗布時に支持体とうまく馴染まない場合がある。このような場合に、UVオゾン処理で表面のクリーニングと親水化を行うことにより、塗布した半導体微粒子分散液と導電性支持体表面の結着力が増し、半導体微粒子分散液の塗布が行い易くなる。
半導体微粒子層全体の好ましい厚さは0.1~100μmである。半導体微粒子層の厚さはさらに1~30μmが好ましく、2~25μmがより好ましい。半導体微粒子の支持体1m2当りの担持量は0.5g~400gが好ましく、5~100gがより好ましい。
塗布した半導体微粒子の層に対し、半導体微粒子同士の電子的接触の強化と、支持体との密着性の向上のため、また塗布した半導体微粒子分散液を乾燥させるために、加熱処理が施される。この加熱処理により多孔質半導体微粒子層を形成することができる。その他、部材の特性や用途に応じて適宜公知の方法により半導体微粒子層を形成してもよい。例えば、特開2001-291534号公報に記載の材料や調製方法、作製方法を参照することができ、本明細書に引用する。
また、加熱処理に加えて光のエネルギーを用いることもできる。例えば、半導体微粒子として酸化チタンを用いた場合に、紫外光のような半導体微粒子が吸収する光を与えることで表面を活性化してもよいし、レーザー光などで半導体微粒子表面のみを活性化することができる。半導体微粒子に対して該微粒子が吸収する光を照射することで、粒子表面に吸着した不純物が粒子表面の活性化によって分解され、上記の目的のために好ましい状態とすることができる。加熱処理と紫外光を組み合わせる場合は、半導体微粒子に対して該微粒子が吸収する光を照射しながら、加熱が100℃以上250℃以下あるいは好ましくは100℃以上150℃以下で行われることが好ましい。このように、半導体微粒子を光励起することによって、微粒子層内に混入した不純物を光分解により洗浄するとともに、微粒子の間の物理的接合を強めることができる。
また、半導体微粒子分散液を前記の導電性支持体に塗布し、加熱や光を照射する以外に他の処理を行ってもよい。好ましい方法として例えば、通電、化学的処理などが挙げられる。
塗布後に圧力をかけても良く、圧力をかける方法としては、特表2003-500857号公報等が挙げられる。光照射の例としては、特開2001-357896号公報等が挙げられる。プラズマ・マイクロ波・通電の例としては、特開2002-353453号公報等が挙げられる。化学的処理としては、例えば特開2001-357896号公報が挙げられる。
上述の半導体微粒子を導電性支持体上に塗設する方法は、上述の半導体微粒子分散液を導電性支持体上に塗布する方法のほか、特許第2664194号公報に記載の半導体微粒子の前駆体を導電性支持体上に塗布し空気中の水分によって加水分解して半導体微粒子膜を得る方法などの方法を使用することができる。
前駆体として例えば、(NH4)2TiF6、過酸化チタン、金属アルコキシド・金属錯体・金属有機酸塩等が挙げられる。
また、金属有機酸化物(アルコキシドなど)を共存させたスラリーを塗布し加熱処理、光処理などで半導体膜を形成する方法、無機系前駆体を共存させたスラリー、スラリーのpHと分散させたチタニア粒子の性状を特定した方法が挙げられる。これらスラリーには、少量であればバインダーを添加しても良く、バインダーとしては、セルロース、フッ素ポリマー、架橋ゴム、ポリブチルチタネート、カルボキシメチルセルロースなどが挙げられる。
半導体微粒子又はその前駆体層の形成に関する技術としては、コロナ放電、プラズマ、UVなどの物理的な方法で親水化する方法、アルカリやポリエチレンジオキシチオフェンとポリスチレンスルホン酸などによる化学処理、ポリアニリンなどの接合用中間膜の形成などが挙げられる。
半導体微粒子を導電性支持体上に塗設する方法として、上述の(1)湿式法とともに、(2)乾式法、(3)その他の方法を併用しても良い。(2)乾式法として好ましくは、特開2000-231943号公報等が挙げられる。(3)その他の方法として、好ましくは、特開2002-134435号公報等が挙げられる。
乾式法としては、蒸着やスパッタリング、エアロゾルデポジション法などが挙げられる。また、電気泳動法・電析法を用いても良い。
また、耐熱基板上でいったん塗膜を作製した後、プラスチック等のフィルムに転写する方法を用いても良い。好ましくは、特開2002-184475号公報記載のEVAを介して転写する方法、特開2003-98977号公報記載の紫外線、水系溶媒で除去可能な無機塩を含む犠牲基板上に半導体層・導電層を形成後、有機基板に転写後、犠牲基板を除去する方法などが挙げられる。
半導体微粒子は多くの色素を吸着することができるように表面積の大きいものが好ましい。例えば半導体微粒子を支持体上に塗設した状態で、その表面積が投影面積に対して10倍以上であることが好ましく、100倍以上であることがより好ましい。この上限には特に制限はないが、通常5000倍程度である。好ましい半導体微粒子の構造としては、特開2001-93591号公報等が挙げられる。
一般に、半導体微粒子の層の厚みが大きいほど単位面積当たりに担持できる色素の量が増えるため光の吸収効率が高くなるが、発生した電子の拡散距離が増すため電荷再結合によるロスも大きくなる。半導体微粒子層の好ましい厚みは素子の用途によって異なるが、典型的には0.1~100μmである。光電気化学電池として用いる場合は1~50μmであることが好ましく、3~30μmであることがより好ましい。半導体微粒子は、支持体に塗布した後に粒子同士を密着させるために、100~800℃の温度で10分~10時間加熱してもよい。支持体としてガラスを用いる場合、製膜温度は400~600℃が好ましい。
支持体として高分子材料を用いる場合、250℃以下で製膜後加熱することが好ましい。その場合の製膜方法としては、(1)湿式法、(2)乾式法、(3)電気泳動法(電析法を含む)の何れでも良く、好ましくは、(1)湿式法、又は(2)乾式法であり、更に好ましくは、(1)湿式法である。
なお、半導体微粒子の支持体1m2当たりの塗布量は0.5~500g、さらには5~100gが好ましい。
半導体微粒子に色素を吸着させるには、溶液と本発明の色素よりなる色素吸着用色素溶液の中に、よく乾燥した半導体微粒子を長時間浸漬するのが好ましい。色素吸着用色素溶液に使用される溶液は、本発明の色素が溶解できる溶液なら特に制限なく使用することができる。例えば、エタノール、メタノール、イソプロパノール、トルエン、t-ブタノール、アセトニトリル、アセトン、n-ブタノールなどを使用することができる。その中でも、エタノール、トルエンを好ましく使用することができる。
溶液と本発明の色素よりなる色素吸着用色素溶液は必要に応じて50℃ないし100℃に加熱してもよい。色素の吸着は半導体微粒子の塗布前に行っても塗布後に行ってもよい。また、半導体微粒子と色素を同時に塗布して吸着させてもよい。未吸着の色素は洗浄によって除去する。塗布膜の焼成を行う場合は色素の吸着は焼成後に行うことが好ましい。焼成後、塗布膜表面に水が吸着する前にすばやく色素を吸着させるのが特に好ましい。吸着する色素は上記の色素A1の1種類でもよいし、さらに色素A2を混合しても、さらにほかの色素を混合してもよい。光電変換の波長域をできるだけ広くするように、混合する色素が選ばれる。色素を混合する場合は、すべての色素が溶解するようにして、色素吸着用色素溶液とすることが好ましい。
色素の使用量は、全体で、支持体1m2当たり0.01~100ミリモルが好ましく、より好ましくは0.1~50ミリモル、特に好ましくは0.1~10ミリモルである。この場合、本発明の色素の使用量は5モル%以上とすることが好ましい。
また、色素の半導体微粒子に対する吸着量は半導体微粒子1gに対して0.001~1ミリモルが好ましく、より好ましくは0.1~0.5ミリモルである。
このような色素量とすることによって、半導体における増感効果が十分に得られる。これに対し、色素量が少ないと増感効果が不十分となり、色素量が多すぎると、半導体に付着していない色素が浮遊し増感効果を低減させる原因となる。
また、会合など色素同士の相互作用を低減する目的で無色の化合物を共吸着させてもよい。共吸着させる疎水性化合物としてはカルボキシル基を有するステロイド化合物(例えばコール酸、ピバル酸(pivalic acid))等が挙げられる。
色素を吸着した後に、アミン類を用いて半導体微粒子の表面を処理してもよい。好ましいアミン類としては4-tert-ブチルピリジン、ポリビニルピリジン等が挙げられる。これらは液体の場合はそのまま用いてもよいし有機溶媒に溶解して用いてもよい。
対向電極は、光電気化学電池の正極として働くものである。対向電極は、通常前述の導電性支持体と同義であるが、強度が十分に保たれるような構成では支持体は必ずしも必要でない。ただし、支持体を有する方が密閉性の点で有利である。対向電極の材料としては、白金、カーボン、導電性ポリマー、などがあげられる。好ましい例としては、白金、カーボン、導電性ポリマーが挙げられる。
対極の構造としては、集電効果が高い構造が好ましい。好ましい例としては、特開平10-505192号公報などが挙げられる。
受光電極は酸化チタンと酸化スズ(TiO2/SnO2)などの複合電極を用いても良く、チタニアの混合電極として例えば、特開2000-113913号公報等が挙げられる。チタニア以外の混合電極として例えば、特開2001-185243号公報、特開2003-282164号公報等が挙げられる。
また、素子の構成としては、第1電極層、第1光電変換層、導電層、第2光電変換層、第2電極層を順次積層した構造を有していても良い。この場合、第1光電変換層と第2光電変換層に用いる色素は同一または異なっていてもよく、異なっている場合には、吸収スペクトルが異なっていることが好ましい。その他、適宜この種の電気化学素子に適用される構造や部材を適用することができる。
受光電極は、入射光の利用率を高めるなどのためにタンデム型にしても良い。好ましいタンデム型の構成例としては、特開2000-90989、特開2002-90989号公報等に記載の例が挙げられる。
受光電極層内部で光散乱、反射を効率的に行う光マネージメント機能を設けてもよい。好ましくは、特開2002-93476号公報に記載のものが挙げられる。
導電性支持体と多孔質半導体微粒子層の間には、電解液と電極が直接接触することによる逆電流を防止する為、短絡防止層を形成することが好ましい。好ましい例としては、特開平06-507999号公報等が挙げられる。
受光電極と対極の接触を防ぐ為に、スペーサーやセパレータを用いることが好ましい。好ましい例としては、特開2001-283941号公報が挙げられる。
セル、モジュールの封止法としては、ポリイソブチレン系熱硬化樹脂、ノボラック樹脂、光硬化性(メタ)アクリレート樹脂、エポキシ樹脂、アイオノマー樹脂、ガラスフリット、アルミナにアルミニウムアルコキシドを用いる方法、低融点ガラスペーストをレーザー溶融する方法などが好ましい。ガラスフリットを用いる場合、粉末ガラスをバインダーとなるアクリル樹脂に混合したものでもよい。
以下、本発明を実施例に基づきさらに詳細に説明するが、本発明はこれらに限定されるものではない。
<例示色素の調製>
本発明の金属錯体色素として、以下の色素を調製した。
以下の説明において、NBSはN-ブロモスクシンイミド、DMFはN,N-ジメチルホルムアミド、PPhはトリフェニルフォスフィン、DMEはジメチルエーテル、THFはテトラヒドロフラン、dpppは1,3-ビス(ジフェニルホスフィノ)プロパン、bpyはビピリジン、TMSはトリメチルシリル、TEAはトリエチルアミンを表す。
(中間体A-15aの調製)
下記の方法に従って中間体A-15aを調製した。
本発明の金属錯体色素として、以下の色素を調製した。
以下の説明において、NBSはN-ブロモスクシンイミド、DMFはN,N-ジメチルホルムアミド、PPhはトリフェニルフォスフィン、DMEはジメチルエーテル、THFはテトラヒドロフラン、dpppは1,3-ビス(ジフェニルホスフィノ)プロパン、bpyはビピリジン、TMSはトリメチルシリル、TEAはトリエチルアミンを表す。
(中間体A-15aの調製)
下記の方法に従って中間体A-15aを調製した。

A-15eの調製
A-15d 28gをDMF300mLに溶解させ、NBSを18g添加し、室温で5時間攪拌した。その後、水と酢酸エチルを加え、分液を行い有機層を濃縮した。これをカラムクロマトグラフィーで精製し、A-15e 30gを得た。
A-15gの調製
A-15e 30g、A-15f 25gを1,2-ジメトキシエタン(300mL)に溶解させた後にPd(PPh3)4を触媒量、10%Na2CO3溶液を60mL加え、75℃で20時間撹拌した。酢酸エチルと水を加え、分液抽出後に濃縮した有機相をカラムクロマトグラフィーで精製しA-15g 34gを得た。
A-15hの調製
A-15g 34gをDMF350mLに溶解させ、NBSを15g添加し、室温で5時間攪拌した。その後、水と酢酸エチルを加え、分液抽出した有機層を濃縮した。これをカラムクロマトグラフィーで精製し、A-15h 35gを得た。
A-15iの調製
A-15h 35gをTHF1000mLに溶解させ-78℃に冷却した。ここに窒素雰囲気下1.6M n-ブチルリチウムヘキサン溶液を250mL滴下した。室温にした後に1時間攪拌した。再度-78℃に冷却後ClSn(n-Bu)3を25g加えた。これを室温にし12時間攪拌後に水とジエチルエーテルを加え、分液抽出後に濃縮したものをカラムクロマトグラフィーで精製し、A-15i 45gを得た。
A-15kの調製
A-15i 36g、A-15j 13gをジオキサン500mLに溶解させた。ここにPd(PPh3)4を触媒量加え、窒素雰囲気化加熱還流させ五時間攪拌した。室温まで冷却後、酢酸エチル、フッ化カリウム水溶液を加え30分間攪拌した後に分液を行い、さらに有機相を水で洗った。これをカラムクロマトグラフィーで精製しA-15k 15gを得た。
A-15aの調製
A-15k 15g、Mg 0.8gをTHF300mL中50℃で1時間攪拌した。これをA-15l 4g、触媒量のNiCl2(dppp)のTHF溶液を50℃に滴下した。これを5時間攪拌後に酢酸エチルと水を加え分液抽出した有機相を濃縮した。これをカラムクロマトグラフィーで精製し、A-15a 20gを得た。
A-15d 28gをDMF300mLに溶解させ、NBSを18g添加し、室温で5時間攪拌した。その後、水と酢酸エチルを加え、分液を行い有機層を濃縮した。これをカラムクロマトグラフィーで精製し、A-15e 30gを得た。
A-15gの調製
A-15e 30g、A-15f 25gを1,2-ジメトキシエタン(300mL)に溶解させた後にPd(PPh3)4を触媒量、10%Na2CO3溶液を60mL加え、75℃で20時間撹拌した。酢酸エチルと水を加え、分液抽出後に濃縮した有機相をカラムクロマトグラフィーで精製しA-15g 34gを得た。
A-15hの調製
A-15g 34gをDMF350mLに溶解させ、NBSを15g添加し、室温で5時間攪拌した。その後、水と酢酸エチルを加え、分液抽出した有機層を濃縮した。これをカラムクロマトグラフィーで精製し、A-15h 35gを得た。
A-15iの調製
A-15h 35gをTHF1000mLに溶解させ-78℃に冷却した。ここに窒素雰囲気下1.6M n-ブチルリチウムヘキサン溶液を250mL滴下した。室温にした後に1時間攪拌した。再度-78℃に冷却後ClSn(n-Bu)3を25g加えた。これを室温にし12時間攪拌後に水とジエチルエーテルを加え、分液抽出後に濃縮したものをカラムクロマトグラフィーで精製し、A-15i 45gを得た。
A-15kの調製
A-15i 36g、A-15j 13gをジオキサン500mLに溶解させた。ここにPd(PPh3)4を触媒量加え、窒素雰囲気化加熱還流させ五時間攪拌した。室温まで冷却後、酢酸エチル、フッ化カリウム水溶液を加え30分間攪拌した後に分液を行い、さらに有機相を水で洗った。これをカラムクロマトグラフィーで精製しA-15k 15gを得た。
A-15aの調製
A-15k 15g、Mg 0.8gをTHF300mL中50℃で1時間攪拌した。これをA-15l 4g、触媒量のNiCl2(dppp)のTHF溶液を50℃に滴下した。これを5時間攪拌後に酢酸エチルと水を加え分液抽出した有機相を濃縮した。これをカラムクロマトグラフィーで精製し、A-15a 20gを得た。
例示色素A-15の調製
上記で得られた化合物A-15a 11.7g、Z-1 3.2g、をDMF120mlに加え、窒素雰囲気下、70℃で4時間攪拌した。その後4-4’-(COOH)2-2,2’-ビピリジン 2.4g、DMF50mlを加え、窒素雰囲気下、160℃で3.5時間加熱攪拌した。その後チオシアン酸アンモニウム 53.2gを加え、窒素雰囲気下、130℃で5時間攪拌した。濃縮後、水を5ml加えろ過し、ジエチルエーテルで洗った。粗精製物をTBAOH(水酸化テトラブチルアンモニウム)と共にメタノール溶液に溶解し、SephadexLH-20カラムで精製した。主層の分画を回収し濃縮後硝酸0.2Mを添加して、沈殿物をろ過後、水及びジエチルエーテルで洗い、A-15のテトラブチルアンモニウム塩を 1.5gを得た。精製物をメタノール溶液に溶解し、硝酸1Mを添加して沈殿物をろ過後、水及びジエチルエーテルで洗い、A-15を1.3g得た。MS-ESI m/z = 1644.61(M-H)+
上記で得られた化合物A-15a 11.7g、Z-1 3.2g、をDMF120mlに加え、窒素雰囲気下、70℃で4時間攪拌した。その後4-4’-(COOH)2-2,2’-ビピリジン 2.4g、DMF50mlを加え、窒素雰囲気下、160℃で3.5時間加熱攪拌した。その後チオシアン酸アンモニウム 53.2gを加え、窒素雰囲気下、130℃で5時間攪拌した。濃縮後、水を5ml加えろ過し、ジエチルエーテルで洗った。粗精製物をTBAOH(水酸化テトラブチルアンモニウム)と共にメタノール溶液に溶解し、SephadexLH-20カラムで精製した。主層の分画を回収し濃縮後硝酸0.2Mを添加して、沈殿物をろ過後、水及びジエチルエーテルで洗い、A-15のテトラブチルアンモニウム塩を 1.5gを得た。精製物をメタノール溶液に溶解し、硝酸1Mを添加して沈殿物をろ過後、水及びジエチルエーテルで洗い、A-15を1.3g得た。MS-ESI m/z = 1644.61(M-H)+

(例示色素A-1の調製)
下記のスキームの方法に従ってA-1aを調製し、以下例示色素A-15と同様にしてA-1を調製した。MS-ESI m/z = 843.94(M-H)+
下記のスキームの方法に従ってA-1aを調製し、以下例示色素A-15と同様にしてA-1を調製した。MS-ESI m/z = 843.94(M-H)+

A-1dの調製
A-1b 16gをTHF500mLに溶解させ、触媒量のPd(PPh3)2Cl2、触媒量のCuI、TEA 80mLを添加した後にA-1c 9.8gを加え、80℃で24時間攪拌した後に水とジクロロメタンを加え分液抽出した有機層を濃縮後にカラムクロマトグラフィーで精製しA-1d 14gを得た。
A-1fの調製
A-1d 13gをMeOH 200mLに溶解させ、炭酸カリウム 25gを加え、40℃で5時間攪拌した。水と酢酸エチルを加え、分液抽出した有機相を濃縮後にカラムクロマトグラフィーで精製し,A-1f 6.0gを得た。
A-1aの調製
A-15l 9gをTHF 100mLに溶解させ、触媒量のPd(PPh3)2Cl2、触媒量のCuI、TEA20mLを添加した後にA-1f 6.0gを加え、80℃で24時間攪拌した後に水とジクロロメタンを加え分液抽出した有機層を濃縮後にカラムクロマトグラフィーで精製しA-1a 12gを得た。
A-1b 16gをTHF500mLに溶解させ、触媒量のPd(PPh3)2Cl2、触媒量のCuI、TEA 80mLを添加した後にA-1c 9.8gを加え、80℃で24時間攪拌した後に水とジクロロメタンを加え分液抽出した有機層を濃縮後にカラムクロマトグラフィーで精製しA-1d 14gを得た。
A-1fの調製
A-1d 13gをMeOH 200mLに溶解させ、炭酸カリウム 25gを加え、40℃で5時間攪拌した。水と酢酸エチルを加え、分液抽出した有機相を濃縮後にカラムクロマトグラフィーで精製し,A-1f 6.0gを得た。
A-1aの調製
A-15l 9gをTHF 100mLに溶解させ、触媒量のPd(PPh3)2Cl2、触媒量のCuI、TEA20mLを添加した後にA-1f 6.0gを加え、80℃で24時間攪拌した後に水とジクロロメタンを加え分液抽出した有機層を濃縮後にカラムクロマトグラフィーで精製しA-1a 12gを得た。
(例示色素A-23の調製)
下記のスキームの方法に従ってA-23aを調製し、以下例示色素A-15と同様にしてA-23を調製した。MS-ESI m/z = 1081.02(M-H)+
下記のスキームの方法に従ってA-23aを調製し、以下例示色素A-15と同様にしてA-23を調製した。MS-ESI m/z = 1081.02(M-H)+

A-23cの調製
A-23b 13.4gをDMF200mLに溶解させ、NBSを 18g添加し、室温で5時間攪拌した。その後、水と酢酸エチルを加え、分液を行い有機層を濃縮した。これをカラムクロマトグラフィーで精製し、A-23c 19gを得た。
A-23dの調製
A-23c 19gをTHF200mLに溶解させ-78℃に冷却した。ここに窒素雰囲気下1.6M n-ブチルリチウムヘキサン溶液を60mL滴下した。室温にした後に1時間攪拌した。再度-78℃に冷却後ClSn(n-Bu)3を 28g加えた。これを室温にし12時間攪拌後に水とジエチルエーテルを加え、分液抽出後に濃縮したものをカラムクロマトグラフィーで精製し、A-23d 26gを得た。
A-23eの調製
A-23d 21g、A-15j 13gをジオキサン300mlに溶解させた。ここにPd(PPh3)4を触媒量加え、窒素雰囲気化加熱還流させ五時間攪拌した。室温まで冷却後、酢酸エチル、フッ化カリウム水溶液を加え30分間攪拌した後に分液を行い、さらに有機相を水で洗った。これをカラムクロマトグラフィーで精製しA-23e 10gを得た。
A-23fの調製
A-23e 7gをTHF100mLに溶解させ、触媒量のPd(PPh3)2Cl2、触媒量のCuI、TEA 20mLを添加した後にA-1c 2.5gを加え、80℃で24時間攪拌した後に水とジクロロメタンを加え分液抽出した有機層を濃縮後にカラムクロマトグラフィーで精製しA-23f 6.0gを得た。
A-23gの調製
A-23f 6.0gをMeOH100mLに溶解させ、炭酸カリウム 3.0gを加え、40℃で5時間攪拌した。水と酢酸エチルを加え、分液抽出した有機相を濃縮後にカラムクロマトグラフィーで精製し,A-23g 4.0gを得た。
A-23aの調製
A-23h 2.3gをTHF50mLに溶解させ、触媒量のPd(PPh3)2Cl2、触媒量のCuI、TEA 10mLを添加した後にA-15l 1.5gを加え、80℃で24時間攪拌した後に、水とジクロロメタンを加え分液抽出した有機層を濃縮後にカラムクロマトグラフィーで精製しA-23a 2.5gを得た。
A-23b 13.4gをDMF200mLに溶解させ、NBSを 18g添加し、室温で5時間攪拌した。その後、水と酢酸エチルを加え、分液を行い有機層を濃縮した。これをカラムクロマトグラフィーで精製し、A-23c 19gを得た。
A-23dの調製
A-23c 19gをTHF200mLに溶解させ-78℃に冷却した。ここに窒素雰囲気下1.6M n-ブチルリチウムヘキサン溶液を60mL滴下した。室温にした後に1時間攪拌した。再度-78℃に冷却後ClSn(n-Bu)3を 28g加えた。これを室温にし12時間攪拌後に水とジエチルエーテルを加え、分液抽出後に濃縮したものをカラムクロマトグラフィーで精製し、A-23d 26gを得た。
A-23eの調製
A-23d 21g、A-15j 13gをジオキサン300mlに溶解させた。ここにPd(PPh3)4を触媒量加え、窒素雰囲気化加熱還流させ五時間攪拌した。室温まで冷却後、酢酸エチル、フッ化カリウム水溶液を加え30分間攪拌した後に分液を行い、さらに有機相を水で洗った。これをカラムクロマトグラフィーで精製しA-23e 10gを得た。
A-23fの調製
A-23e 7gをTHF100mLに溶解させ、触媒量のPd(PPh3)2Cl2、触媒量のCuI、TEA 20mLを添加した後にA-1c 2.5gを加え、80℃で24時間攪拌した後に水とジクロロメタンを加え分液抽出した有機層を濃縮後にカラムクロマトグラフィーで精製しA-23f 6.0gを得た。
A-23gの調製
A-23f 6.0gをMeOH100mLに溶解させ、炭酸カリウム 3.0gを加え、40℃で5時間攪拌した。水と酢酸エチルを加え、分液抽出した有機相を濃縮後にカラムクロマトグラフィーで精製し,A-23g 4.0gを得た。
A-23aの調製
A-23h 2.3gをTHF50mLに溶解させ、触媒量のPd(PPh3)2Cl2、触媒量のCuI、TEA 10mLを添加した後にA-15l 1.5gを加え、80℃で24時間攪拌した後に、水とジクロロメタンを加え分液抽出した有機層を濃縮後にカラムクロマトグラフィーで精製しA-23a 2.5gを得た。
(色素の極大吸収波長の測定)
用いた色素の極大吸収波長を測定した。その結果を表1に示す。測定は、分光光度計(U-4100(商品名)、日立ハイテク社製)によって行い、溶液はTHF:エタノール=1:1を用い、濃度が2μMになるように調整した。
用いた色素の極大吸収波長を測定した。その結果を表1に示す。測定は、分光光度計(U-4100(商品名)、日立ハイテク社製)によって行い、溶液はTHF:エタノール=1:1を用い、濃度が2μMになるように調整した。

[実験1]
図1に示す光電変換素子10を以下のようにして作製した。
ガラス基板上に、透明導電膜としてフッ素をドープした酸化スズをスパッタリングにより形成し、これをレーザーでスクライブして、透明導電膜を2つの部分に分割した。
次に、水とアセトニトリルの容量比4:1からなる混合溶媒100mlにアナターゼ型酸化チタン(日本アエロジル社製のP-25(商品名))32gを配合し、自転/公転併用式のミキシングコンディショナーを使用して均一に分散、混合し、半導体微粒子分散液を得た。この分散液を透明導電膜に塗布し、500℃で加熱して受光電極を作製した。
その後、同様にシリカ粒子とルチル型酸化チタンとを40:60(質量比)で含有する分散液を作製し、この分散液を前記の受光電極に塗布し、500℃で加熱して絶縁性多孔体を形成した。次いで対極として炭素電極を形成した。
次に、下記表2に記載された増感色素のエタノール溶液(3×10-4mol/L)に、上記の絶縁性多孔体が形成されたガラス基板を48時間浸漬した。増感色素の染着したガラスを4-tert-ブチルピリジンの10%エタノール溶液に30分間浸漬した後、エタノールで洗浄し自然乾燥させた。このようにして得られる感光体層の厚さは10μmであり、半導体微粒子の塗布量は20g/m2であった。電解液は、ヨウ化ジメチルプロピルイミダゾリウム(0.5mol/L)、ヨウ素(0.1mol/L)のメトキシプロピオニトリル溶液を用いた。
図1に示す光電変換素子10を以下のようにして作製した。
ガラス基板上に、透明導電膜としてフッ素をドープした酸化スズをスパッタリングにより形成し、これをレーザーでスクライブして、透明導電膜を2つの部分に分割した。
次に、水とアセトニトリルの容量比4:1からなる混合溶媒100mlにアナターゼ型酸化チタン(日本アエロジル社製のP-25(商品名))32gを配合し、自転/公転併用式のミキシングコンディショナーを使用して均一に分散、混合し、半導体微粒子分散液を得た。この分散液を透明導電膜に塗布し、500℃で加熱して受光電極を作製した。
その後、同様にシリカ粒子とルチル型酸化チタンとを40:60(質量比)で含有する分散液を作製し、この分散液を前記の受光電極に塗布し、500℃で加熱して絶縁性多孔体を形成した。次いで対極として炭素電極を形成した。
次に、下記表2に記載された増感色素のエタノール溶液(3×10-4mol/L)に、上記の絶縁性多孔体が形成されたガラス基板を48時間浸漬した。増感色素の染着したガラスを4-tert-ブチルピリジンの10%エタノール溶液に30分間浸漬した後、エタノールで洗浄し自然乾燥させた。このようにして得られる感光体層の厚さは10μmであり、半導体微粒子の塗布量は20g/m2であった。電解液は、ヨウ化ジメチルプロピルイミダゾリウム(0.5mol/L)、ヨウ素(0.1mol/L)のメトキシプロピオニトリル溶液を用いた。
(変換効率の測定)
500Wのキセノンランプ(ウシオ製)の光をAM1.5Gフィルター(商品名、Oriel社製)及びシャープカットフィルター(KenkoL-42、商品名)を通すことにより紫外線を含まない模擬太陽光を発生させた。この光の強度は89mW/cm2であった。作製した光電変換素子にこの光を照射し、電流電圧測定装置(ケースレー238型、商品名、ケースレー社製)で、光電変換特性を測定した。
光電気化学電池の変換効率を測定した結果を下記の表2に示した。
変換効率が6.0%以上のものをA、5%以上6%未満のものをB、4%以上5%未満のものをC、3%以上4%未満のものをD、1.5%以上3%未満のものをE、1.5%未満のものをFとして表示し、変換効率D以上のものを合格とし、D未満のものを不合格とした。
また、耐久性として変換効率の初期値に対する45%RH下で400時間連続照射後の変換効率を評価した。初期値に対する400時間後の変換効率が90%以上のものをA、85%以上90%未満のものをB、80%以上85%未満のものをC、70%以上80%未満のものをD、70%未満のものをEとして評価した。
また、吸着安定性は、電解液の含水率を1.0%に調整したサンプルについて、変換効率の初期値に対する200時間連続照射後の変換効率により評価した。初期値に対する低下率が10%以下のものを◎、20~10%のものを○、30~20%のものを△、30%以上のものを×とした。
500Wのキセノンランプ(ウシオ製)の光をAM1.5Gフィルター(商品名、Oriel社製)及びシャープカットフィルター(KenkoL-42、商品名)を通すことにより紫外線を含まない模擬太陽光を発生させた。この光の強度は89mW/cm2であった。作製した光電変換素子にこの光を照射し、電流電圧測定装置(ケースレー238型、商品名、ケースレー社製)で、光電変換特性を測定した。
光電気化学電池の変換効率を測定した結果を下記の表2に示した。
変換効率が6.0%以上のものをA、5%以上6%未満のものをB、4%以上5%未満のものをC、3%以上4%未満のものをD、1.5%以上3%未満のものをE、1.5%未満のものをFとして表示し、変換効率D以上のものを合格とし、D未満のものを不合格とした。
また、耐久性として変換効率の初期値に対する45%RH下で400時間連続照射後の変換効率を評価した。初期値に対する400時間後の変換効率が90%以上のものをA、85%以上90%未満のものをB、80%以上85%未満のものをC、70%以上80%未満のものをD、70%未満のものをEとして評価した。
また、吸着安定性は、電解液の含水率を1.0%に調整したサンプルについて、変換効率の初期値に対する200時間連続照射後の変換効率により評価した。初期値に対する低下率が10%以下のものを◎、20~10%のものを○、30~20%のものを△、30%以上のものを×とした。


本発明の色素は、変換効率、耐久性に優れ、水による脱着等の影響を受けにくく吸着安定性が優れていることがわかった。
[実験2]
色素を二種類用いて、表3記載の濃度でエタノール中に溶解させた色素溶液にガラス基盤を浸透させ染着した以外は実験1と同様の方法で光電気変換素子の作成及び測定を行った。結果を表3に示す。変換効率が7.0%以上のものをA、6.0%以上7.0%未満のものをB、5.0%以上6.0%未満のものをC、6.5%未満のものをDとして評価した。
色素を二種類用いて、表3記載の濃度でエタノール中に溶解させた色素溶液にガラス基盤を浸透させ染着した以外は実験1と同様の方法で光電気変換素子の作成及び測定を行った。結果を表3に示す。変換効率が7.0%以上のものをA、6.0%以上7.0%未満のものをB、5.0%以上6.0%未満のものをC、6.5%未満のものをDとして評価した。

[実験3]
ガラス基板上にITO膜を作製し、その上にFTO膜を積層することにより、透明導電膜を作製した。その後透明導電膜上に酸化物半導体多孔質膜を形成することにより、透明電極板を得た。そしてその透明電極板を使用して光電気化学電池を作製し、変換効率を測定した。その方法は以下の(1)~(5)の通りである。
(1)ITO(インジウム・スズ・オキサイド)膜用原料化合物溶液の調製
塩化インジウム(III)四水和物5.58gと塩化スズ(II)二水和物0.23gとをエタノール100mlに溶解して、ITO膜用原料化合物溶液とした。
(2)FTO(フッ素ドープ酸化スズ)膜用原料化合物溶液の調製
塩化スズ(IV)五水和物0.701gをエタノール10mlに溶解し、これにフッ化アンモニウム0.592gの飽和水溶液を加え、この混合物を超音波洗浄機に約20分間かけ、完全に溶解して、FTO膜用原料化合物溶液とした。
(3)ITO/FTO透明導電膜の作製
厚さ2mmの耐熱ガラス板の表面を化学洗浄し、乾燥した後、このガラス板を反応器内に置き、ヒータで加熱した。ヒータの加熱温度が450℃になったところで、(1)で得られたITO膜用原料化合物溶液を、口径0.3mmのノズルから圧力0.06MPaで、ガラス板までの距離を400mmとして、25分間噴霧した。
このITO膜用原料化合物溶液の噴霧後、2分間(この間ガラス基板表面にエタノールを噴霧し続け、基板表面温度の上昇を抑えるようにした。)経過し、ヒータの加熱温度が530℃になった時に、(2)で得られたFTO膜用原料化合物溶液を同様の条件で2分30秒間噴霧した。これにより、耐熱ガラス板上に厚さ530nmのITO膜、厚さ170nmのFTO膜が順次形成された透明電極板が得られた。
比較のために、厚さ2mmの耐熱ガラス板上に同様に、厚さ530nmのITO膜のみを成膜した透明電極板と、同じく厚さ180nmのFTO膜のみを成膜した透明電極板とをそれぞれ作製した。
これら3種の透明電極板を加熱炉にて、450℃で2時間加熱した。
(4)光電気化学電池の作製
次に、上記3種の透明電極板を用いて、特許第4260494号明細書中の図2に示した構造の光電気化学電池を作製した。酸化物半導体多孔質膜の形成は、平均粒径約230nmの酸化チタン微粒子をアセトニトリルに分散してペーストとし、これを透明電極11上にバーコート法により厚さ15μmに塗布し、乾燥後450℃で1時間焼成して行った。その後、この酸化物半導体多孔質膜に表4記載の色素を担持した。色素溶液への浸漬条件は前記実験1と同じとした。
さらに、対極には、ガラス板上にITO膜とFTO膜とを積層した導電性基板を使用し、電解質層には、ヨウ素/ヨウ化物の非水溶液からなる電解液を用いた。光電気化学電池の平面寸法は25mm×25mmとした。
(5)光電気化学電池の評価
(4)で得られた光電気化学電池について、擬似太陽光(AM1.5)を照射し、実験1と同様の方法で光電変換特性を測定し、変換効率を求めた。その結果を表4に示す。変換効率が6.0%以上のものをA、5%以上6%未満のものをB、4%以上5%未満のものをC、3%以上4%未満のものをD、1.5%以上3%未満のものをE、1.5%未満のものをFとして表示し、変換効率D以上のものを合格とし、D未満のものを不合格とした。また、耐久性として変換効率の初期値に対し400時間後の変換効率が90%以上のものをA、85%以上90%未満のものをB、80%以上85%未満のものをC、70%以上80%未満のものをD、70%未満のものをEとして評価した。
ガラス基板上にITO膜を作製し、その上にFTO膜を積層することにより、透明導電膜を作製した。その後透明導電膜上に酸化物半導体多孔質膜を形成することにより、透明電極板を得た。そしてその透明電極板を使用して光電気化学電池を作製し、変換効率を測定した。その方法は以下の(1)~(5)の通りである。
(1)ITO(インジウム・スズ・オキサイド)膜用原料化合物溶液の調製
塩化インジウム(III)四水和物5.58gと塩化スズ(II)二水和物0.23gとをエタノール100mlに溶解して、ITO膜用原料化合物溶液とした。
(2)FTO(フッ素ドープ酸化スズ)膜用原料化合物溶液の調製
塩化スズ(IV)五水和物0.701gをエタノール10mlに溶解し、これにフッ化アンモニウム0.592gの飽和水溶液を加え、この混合物を超音波洗浄機に約20分間かけ、完全に溶解して、FTO膜用原料化合物溶液とした。
(3)ITO/FTO透明導電膜の作製
厚さ2mmの耐熱ガラス板の表面を化学洗浄し、乾燥した後、このガラス板を反応器内に置き、ヒータで加熱した。ヒータの加熱温度が450℃になったところで、(1)で得られたITO膜用原料化合物溶液を、口径0.3mmのノズルから圧力0.06MPaで、ガラス板までの距離を400mmとして、25分間噴霧した。
このITO膜用原料化合物溶液の噴霧後、2分間(この間ガラス基板表面にエタノールを噴霧し続け、基板表面温度の上昇を抑えるようにした。)経過し、ヒータの加熱温度が530℃になった時に、(2)で得られたFTO膜用原料化合物溶液を同様の条件で2分30秒間噴霧した。これにより、耐熱ガラス板上に厚さ530nmのITO膜、厚さ170nmのFTO膜が順次形成された透明電極板が得られた。
比較のために、厚さ2mmの耐熱ガラス板上に同様に、厚さ530nmのITO膜のみを成膜した透明電極板と、同じく厚さ180nmのFTO膜のみを成膜した透明電極板とをそれぞれ作製した。
これら3種の透明電極板を加熱炉にて、450℃で2時間加熱した。
(4)光電気化学電池の作製
次に、上記3種の透明電極板を用いて、特許第4260494号明細書中の図2に示した構造の光電気化学電池を作製した。酸化物半導体多孔質膜の形成は、平均粒径約230nmの酸化チタン微粒子をアセトニトリルに分散してペーストとし、これを透明電極11上にバーコート法により厚さ15μmに塗布し、乾燥後450℃で1時間焼成して行った。その後、この酸化物半導体多孔質膜に表4記載の色素を担持した。色素溶液への浸漬条件は前記実験1と同じとした。
さらに、対極には、ガラス板上にITO膜とFTO膜とを積層した導電性基板を使用し、電解質層には、ヨウ素/ヨウ化物の非水溶液からなる電解液を用いた。光電気化学電池の平面寸法は25mm×25mmとした。
(5)光電気化学電池の評価
(4)で得られた光電気化学電池について、擬似太陽光(AM1.5)を照射し、実験1と同様の方法で光電変換特性を測定し、変換効率を求めた。その結果を表4に示す。変換効率が6.0%以上のものをA、5%以上6%未満のものをB、4%以上5%未満のものをC、3%以上4%未満のものをD、1.5%以上3%未満のものをE、1.5%未満のものをFとして表示し、変換効率D以上のものを合格とし、D未満のものを不合格とした。また、耐久性として変換効率の初期値に対し400時間後の変換効率が90%以上のものをA、85%以上90%未満のものをB、80%以上85%未満のものをC、70%以上80%未満のものをD、70%未満のものをEとして評価した。
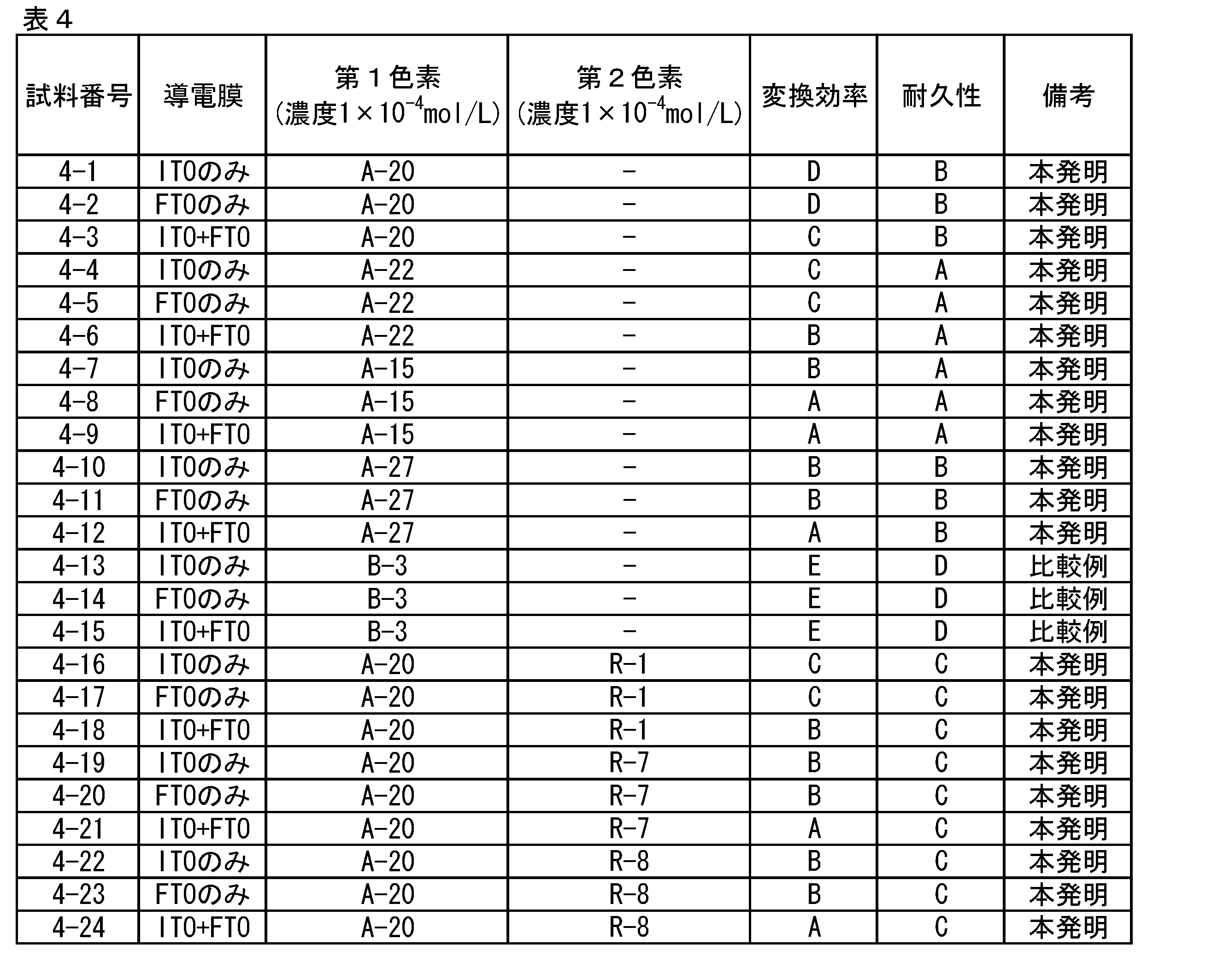
比較色素では変換効率が低いのに対し、本発明の例示色素を使用したものでは良好な結果を示すことがわかった。透明電極板として、ITO膜とFTO膜とを積層したものを用いた光電気化学電池では、ITO膜のみもしくはFTO膜のみを成膜したものを用いた場合に比べ特に変換効率が高く、本発明の色素でその効果が高いことがわかった。
[実験4]
FTO膜上に集電電極を配し、光電気化学電池を作製し、変換効率を評価した。評価は以下の通り、試験セル(i)と試験セル(iv)の2種類を用いた。
FTO膜上に集電電極を配し、光電気化学電池を作製し、変換効率を評価した。評価は以下の通り、試験セル(i)と試験セル(iv)の2種類を用いた。
(試験セル(i))
長さ100mm×幅100mm×厚さ2mmの耐熱ガラス板の表面を化学洗浄し、乾燥した後、このガラス板を反応器内に置き、ヒータで加熱した後、上記の実験2で調製したFTO(フッ素ドープ酸化スズ)膜用原料化合物溶液を、口径0.3mmのノズルから圧力0.06MPaで、ガラス板までの距離を400mmとして、25分間噴霧し、FTO膜付きガラス基板を用意した。
その表面に、エッチング法により深さ5μmの溝を格子回路パターン状に形成した。フォトリソグラフでパターン形成した後に、フッ酸を用いてエッチングを行った。これに、めっき形成を可能とするためにスパッタ法により金属導電層(シード層)を形成し、更にアディティブめっきにより金属配線層を形成した。金属配線層は、透明基板表面から凸レンズ状に3μm高さまで形成した。回路幅は60μmとした。この上から、遮蔽層としてFTO膜を400nmの厚さでSPD法により形成して、電極基板(i)とした。なお、電極基板(i)の断面形状は、特開2004-146425中の図2に示すものとなっていた。
長さ100mm×幅100mm×厚さ2mmの耐熱ガラス板の表面を化学洗浄し、乾燥した後、このガラス板を反応器内に置き、ヒータで加熱した後、上記の実験2で調製したFTO(フッ素ドープ酸化スズ)膜用原料化合物溶液を、口径0.3mmのノズルから圧力0.06MPaで、ガラス板までの距離を400mmとして、25分間噴霧し、FTO膜付きガラス基板を用意した。
その表面に、エッチング法により深さ5μmの溝を格子回路パターン状に形成した。フォトリソグラフでパターン形成した後に、フッ酸を用いてエッチングを行った。これに、めっき形成を可能とするためにスパッタ法により金属導電層(シード層)を形成し、更にアディティブめっきにより金属配線層を形成した。金属配線層は、透明基板表面から凸レンズ状に3μm高さまで形成した。回路幅は60μmとした。この上から、遮蔽層としてFTO膜を400nmの厚さでSPD法により形成して、電極基板(i)とした。なお、電極基板(i)の断面形状は、特開2004-146425中の図2に示すものとなっていた。
電極基板(i)上に、平均粒径25nmの酸化チタン分散液を塗布・乾燥し、450℃で1時間加熱・焼結した。これを表5に示す色素のエタノール溶液中に40分間浸漬して色素を担持させた。
50μm厚の熱可塑性ポリオレフィン樹脂シートを介して、白金スパッタFTO基板と上記基板を対向して配置し、樹脂シート部を熱溶融させて両極板を固定した。
あらかじめ白金スパッタ極側に開けておいた電解液の注液口から、0.5Mのヨウ化塩と0.05Mのヨウ素とを主成分に含むメトキシアセトニトリル溶液を注液し、電極間に満たした。さらに周辺部及び電解液注液口をエポキシ系封止樹脂で封止し、集電端子部に銀ペーストを塗布して、試験セル(i)とした。実験1と同様の方法で、AM1.5の疑似太陽光を試験セル(i)に照射し、変換効率を測定した。その結果を表5に示す。
50μm厚の熱可塑性ポリオレフィン樹脂シートを介して、白金スパッタFTO基板と上記基板を対向して配置し、樹脂シート部を熱溶融させて両極板を固定した。
あらかじめ白金スパッタ極側に開けておいた電解液の注液口から、0.5Mのヨウ化塩と0.05Mのヨウ素とを主成分に含むメトキシアセトニトリル溶液を注液し、電極間に満たした。さらに周辺部及び電解液注液口をエポキシ系封止樹脂で封止し、集電端子部に銀ペーストを塗布して、試験セル(i)とした。実験1と同様の方法で、AM1.5の疑似太陽光を試験セル(i)に照射し、変換効率を測定した。その結果を表5に示す。
(試験セル(iv))
試験セル(i)と同様の方法で、長さ100×幅100mmのFTO膜付きガラス基板を用意した。そのFTOガラス基板上に、アディティブめっき法により金属配線層(金回路)を形成した。この金属配線層(金回路)は基板表面に格子状に形成し、回路幅50μm、回路厚5μmとした。この表面に、厚さ300nmのFTO膜を遮蔽層として、SPD法により形成して電極基板(iv)とした。電極基板(iv)の断面をSEM-EDXを用いて確認したところ、配線底部でめっきレジストの裾引きに起因すると思われる潜り込みがあり、影部分にはFTOが被覆されていなかった。
電極基板(iv)を用い、試験セル(i)と同様に、試験セル(iv)を作製した。実験1と同様の方法でAM1.5の疑似太陽光を照射し、変換効率を測定した。その結果を表5に示す。
光電気化学電池の変換効率を測定した結果を下記の表5に示した。変換効率が6.0%以上のものをA、5%以上6%未満のものをB、4%以上5%未満のものをC、3%以上4%未満のものをD、1.5%以上3%未満のものをE、1.5%未満のものをFとして表示し、変換効率D以上のものを合格とし、D未満のものを不合格とした。また、耐久性として、変換効率の初期値に対し45%RH下で400時間連続照射後の変換効率が90%以上のものをA、85%以上90%未満のものをB、80%以上85%未満のものをC、70%以上80%未満のものをD、70%未満のものをEとして評価した。
試験セル(i)と同様の方法で、長さ100×幅100mmのFTO膜付きガラス基板を用意した。そのFTOガラス基板上に、アディティブめっき法により金属配線層(金回路)を形成した。この金属配線層(金回路)は基板表面に格子状に形成し、回路幅50μm、回路厚5μmとした。この表面に、厚さ300nmのFTO膜を遮蔽層として、SPD法により形成して電極基板(iv)とした。電極基板(iv)の断面をSEM-EDXを用いて確認したところ、配線底部でめっきレジストの裾引きに起因すると思われる潜り込みがあり、影部分にはFTOが被覆されていなかった。
電極基板(iv)を用い、試験セル(i)と同様に、試験セル(iv)を作製した。実験1と同様の方法でAM1.5の疑似太陽光を照射し、変換効率を測定した。その結果を表5に示す。
光電気化学電池の変換効率を測定した結果を下記の表5に示した。変換効率が6.0%以上のものをA、5%以上6%未満のものをB、4%以上5%未満のものをC、3%以上4%未満のものをD、1.5%以上3%未満のものをE、1.5%未満のものをFとして表示し、変換効率D以上のものを合格とし、D未満のものを不合格とした。また、耐久性として、変換効率の初期値に対し45%RH下で400時間連続照射後の変換効率が90%以上のものをA、85%以上90%未満のものをB、80%以上85%未満のものをC、70%以上80%未満のものをD、70%未満のものをEとして評価した。
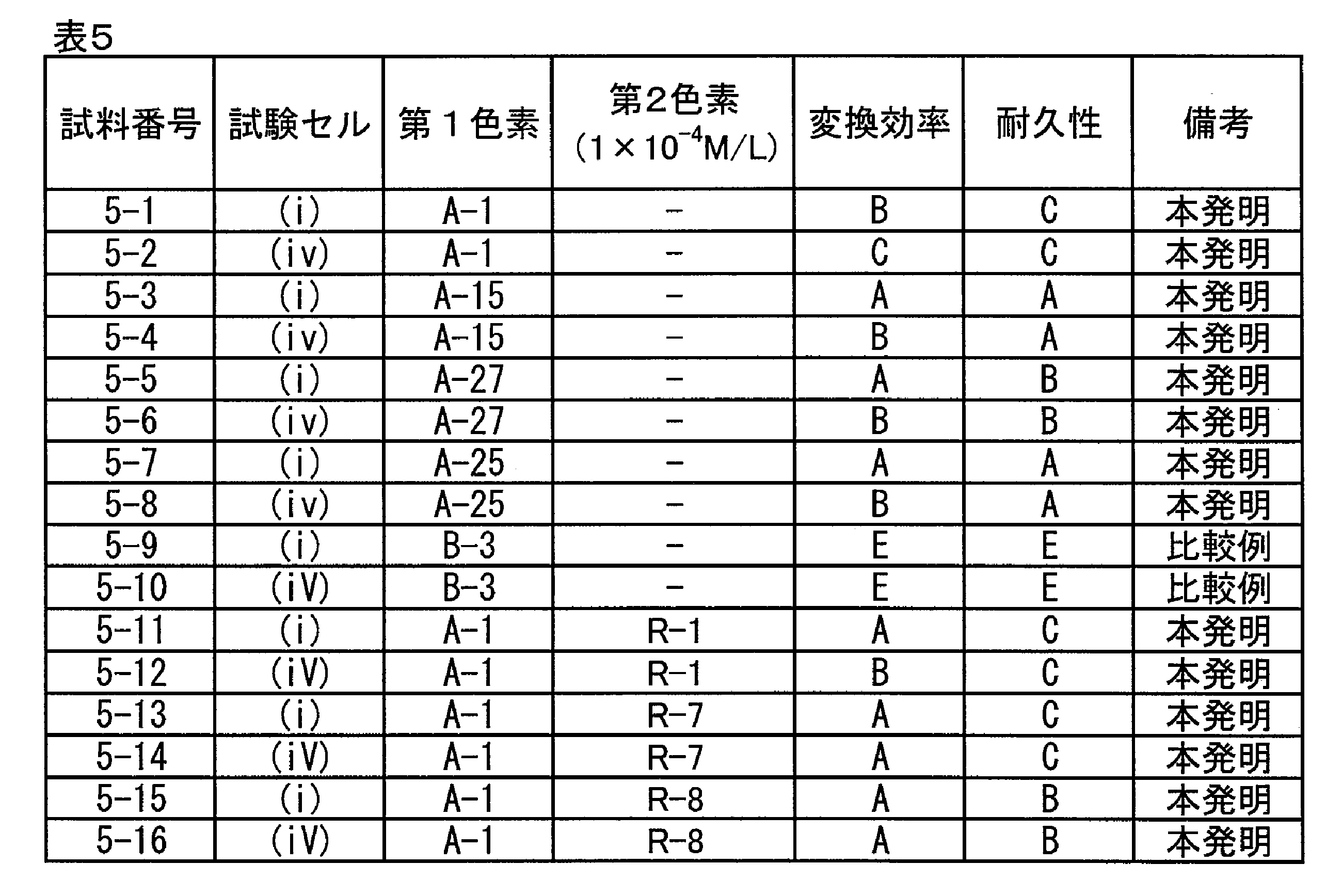
本発明の色素は、セルが変わっても比較色素に対し高い性能を示した。
[実験5]
ペルオキソチタン酸及び酸化チタン微粒子を作製し、これを用いて酸化物半導体膜を作製した。これを用いて光電気化学電池(A)~(D)を作製し、評価した。
(光電気化学電池(A))
(1)酸化物半導体膜形成用塗布液(A)の調製
5gの水素化チタンを1リットルの純水に懸濁し、5質量%の過酸化水素液400gを30分かけて添加し、ついで80℃に加熱して溶解してペルオキソチタン酸の溶液を調製した。この溶液の全量から90容積%を分取し、濃アンモニア水を添加してpH9に調整し、オートクレーブに入れ、250℃で5時間、飽和蒸気圧下で水熱処理を行ってチタニアコロイド粒子(A)を調製した。得られたチタニアコロイド粒子は、X線回折により結晶性の高いアナターゼ型酸化チタンであった。
ペルオキソチタン酸及び酸化チタン微粒子を作製し、これを用いて酸化物半導体膜を作製した。これを用いて光電気化学電池(A)~(D)を作製し、評価した。
(光電気化学電池(A))
(1)酸化物半導体膜形成用塗布液(A)の調製
5gの水素化チタンを1リットルの純水に懸濁し、5質量%の過酸化水素液400gを30分かけて添加し、ついで80℃に加熱して溶解してペルオキソチタン酸の溶液を調製した。この溶液の全量から90容積%を分取し、濃アンモニア水を添加してpH9に調整し、オートクレーブに入れ、250℃で5時間、飽和蒸気圧下で水熱処理を行ってチタニアコロイド粒子(A)を調製した。得られたチタニアコロイド粒子は、X線回折により結晶性の高いアナターゼ型酸化チタンであった。
次に、上記で得られたチタニアコロイド粒子(A)を10質量%まで濃縮し、前記ペルオキソチタン酸溶液を混合し、この混合液中のチタンをTiO2換算し、TiO2質量の30質量%となるように膜形成助剤としてヒドロキシプロピルセルロースを添加して半導体膜形成用塗布液(A)を調製した。
(2)酸化物半導体膜(A)の作製
次いで、フッ素ドープした酸化スズが電極層として形成された透明ガラス基板上に前記塗布液(A1)を塗布し、自然乾燥し、引き続き低圧水銀ランプを用いて6000mJ/cm2の紫外線を照射してペルオキソ酸を分解させ、塗膜を硬化させた。塗膜を300℃で30分間加熱してヒドロキシプロピルセルロースの分解およびアニーリングを行って酸化物半導体膜(A)をガラス基板に形成した。
次いで、フッ素ドープした酸化スズが電極層として形成された透明ガラス基板上に前記塗布液(A1)を塗布し、自然乾燥し、引き続き低圧水銀ランプを用いて6000mJ/cm2の紫外線を照射してペルオキソ酸を分解させ、塗膜を硬化させた。塗膜を300℃で30分間加熱してヒドロキシプロピルセルロースの分解およびアニーリングを行って酸化物半導体膜(A)をガラス基板に形成した。
(3)酸化物半導体膜(A)への色素の吸着
次に、分光増感色素として本発明の色素の濃度3×10-4モル/リットルのエタノール溶液を調製した。この色素溶液を100rpmスピナーで、金属酸化物半導体膜(A3)上へ塗布して乾燥した。この塗布および乾燥工程を5回行った。
次に、分光増感色素として本発明の色素の濃度3×10-4モル/リットルのエタノール溶液を調製した。この色素溶液を100rpmスピナーで、金属酸化物半導体膜(A3)上へ塗布して乾燥した。この塗布および乾燥工程を5回行った。
(4)電解質溶液の調製
アセトニトリルと炭酸エチレンとの体積比が1:5の混合溶媒に、テトラプロピルアンモニウムアイオダイドを0.46モル/リットル、ヨウ素を0.07モル/リットルの濃度となるように溶解して電解質溶液を調製した。
アセトニトリルと炭酸エチレンとの体積比が1:5の混合溶媒に、テトラプロピルアンモニウムアイオダイドを0.46モル/リットル、ヨウ素を0.07モル/リットルの濃度となるように溶解して電解質溶液を調製した。
(5)光電気化学電池(A)の作製
(3)で作製した、色素を吸着させた酸化物半導体膜(A)が形成されたガラス基板を一方の電極とし、他方の電極として、フッ素ドープした酸化スズを電極として形成しその上に白金を担持した透明ガラス基板を対向して配置し、側面を樹脂にてシールし、電極間に(4)の電解質溶液を封入し、さらに電極間をリード線で接続して光電気化学電池(A)を作製した。
(3)で作製した、色素を吸着させた酸化物半導体膜(A)が形成されたガラス基板を一方の電極とし、他方の電極として、フッ素ドープした酸化スズを電極として形成しその上に白金を担持した透明ガラス基板を対向して配置し、側面を樹脂にてシールし、電極間に(4)の電解質溶液を封入し、さらに電極間をリード線で接続して光電気化学電池(A)を作製した。
(6)光電気化学電池(A)の評価
光電気化学電池(A)は、ソーラーシュミレーターで100W/m2の強度の光を照射して、η(変換効率)を測定し、その結果を表6に示した。
(光電気化学電池(B))
紫外線を照射してペルオキソ酸を分解させ、膜を硬化させた後、Arガスのイオン照射(日新電気製:イオン注入装置、200eVで10時間照射)を行った以外は酸化物半導体膜(A)と同様にして酸化物半導体膜(B)を形成した。
酸化物半導体膜(A)と同様に、酸化物半導体膜(B)に色素の吸着を行った。
その後光電気化学電池(A)と同様の方法で光電気化学電池(B)を作成し、ηを測定した。その結果を表6に示した。
光電気化学電池(A)は、ソーラーシュミレーターで100W/m2の強度の光を照射して、η(変換効率)を測定し、その結果を表6に示した。
(光電気化学電池(B))
紫外線を照射してペルオキソ酸を分解させ、膜を硬化させた後、Arガスのイオン照射(日新電気製:イオン注入装置、200eVで10時間照射)を行った以外は酸化物半導体膜(A)と同様にして酸化物半導体膜(B)を形成した。
酸化物半導体膜(A)と同様に、酸化物半導体膜(B)に色素の吸着を行った。
その後光電気化学電池(A)と同様の方法で光電気化学電池(B)を作成し、ηを測定した。その結果を表6に示した。
(光電気化学電池(C))
18.3gの4塩化チタンを純水で希釈して、TiO2換算で1.0質量%含有する水溶液を得た。この水溶液を撹拌しながら、15質量%のアンモニア水を添加し、pH9.5の白色スラリーを得た。このスラリーを濾過洗浄し、TiO2換算で、10.2質量%の水和酸化チタンゲルのケーキを得た。このケーキと5質量%過酸化水素液400gを混合し、ついで80℃に加熱して溶解してペルオキソチタン酸の溶液を調製した。この溶液全量から90体積%を分取し、これに濃アンモニア水を添加してpH9に調整し、オートクレーブに入れ、250℃で5時間、飽和蒸気圧下で水熱処理を行ってチタニアコロイド粒子(C)を調製した。
次に、上記で得られたペルオキソチタン酸溶液とチタニアコロイド粒子(C)を使用して酸化物半導体膜(A)と同様にして酸化物半導体膜(C)を形成し、金属酸化物半導体膜(A)と同様にして、分光増感色素として本発明の色素の吸着を行った。
その後光電気化学電池(A)と同様の方法で光電気化学電池(C)を作製し、ηを測定した。その結果を表6に示した。
18.3gの4塩化チタンを純水で希釈して、TiO2換算で1.0質量%含有する水溶液を得た。この水溶液を撹拌しながら、15質量%のアンモニア水を添加し、pH9.5の白色スラリーを得た。このスラリーを濾過洗浄し、TiO2換算で、10.2質量%の水和酸化チタンゲルのケーキを得た。このケーキと5質量%過酸化水素液400gを混合し、ついで80℃に加熱して溶解してペルオキソチタン酸の溶液を調製した。この溶液全量から90体積%を分取し、これに濃アンモニア水を添加してpH9に調整し、オートクレーブに入れ、250℃で5時間、飽和蒸気圧下で水熱処理を行ってチタニアコロイド粒子(C)を調製した。
次に、上記で得られたペルオキソチタン酸溶液とチタニアコロイド粒子(C)を使用して酸化物半導体膜(A)と同様にして酸化物半導体膜(C)を形成し、金属酸化物半導体膜(A)と同様にして、分光増感色素として本発明の色素の吸着を行った。
その後光電気化学電池(A)と同様の方法で光電気化学電池(C)を作製し、ηを測定した。その結果を表6に示した。
(光電気化学電池(D))
18.3gの4塩化チタンを純水で希釈してTiO2換算で1.0質量%含有する水溶液を得た。これを撹拌しながら、15質量%のアンモニア水を添加し、pH9.5の白色スラリーを得た。このスラリーを濾過洗浄した後、純水に懸濁してTiO2として0.6質量%の水和酸化チタンゲルのスラリーとし、これに塩酸を加えてpH2とした後、オートクレーブに入れ、180℃で5時間、飽和蒸気圧下で水熱処理を行ってチタニアコロイド粒子(D)を調製した。
次に、チタニアコロイド粒子(D)を10質量%まで濃縮し、これに、TiO2に換算して、30質量%となるように膜形成助剤としてヒドロキシプロピルセルロースを添加して、半導体膜形成用塗布液を調製した。次いで、フッ素ドープした酸化スズが電極層として形成された透明ガラス基板上に、前記塗布液を塗布し、自然乾燥し、引き続き低圧水銀ランプを用いて6000mJ/cm2の紫外線を照射し、膜を硬化させた。さらに、300℃で30分間加熱してヒドロキシプロピルセルロースの分解およびアニーリングを行い、酸化物半導体膜(D)を形成した。
18.3gの4塩化チタンを純水で希釈してTiO2換算で1.0質量%含有する水溶液を得た。これを撹拌しながら、15質量%のアンモニア水を添加し、pH9.5の白色スラリーを得た。このスラリーを濾過洗浄した後、純水に懸濁してTiO2として0.6質量%の水和酸化チタンゲルのスラリーとし、これに塩酸を加えてpH2とした後、オートクレーブに入れ、180℃で5時間、飽和蒸気圧下で水熱処理を行ってチタニアコロイド粒子(D)を調製した。
次に、チタニアコロイド粒子(D)を10質量%まで濃縮し、これに、TiO2に換算して、30質量%となるように膜形成助剤としてヒドロキシプロピルセルロースを添加して、半導体膜形成用塗布液を調製した。次いで、フッ素ドープした酸化スズが電極層として形成された透明ガラス基板上に、前記塗布液を塗布し、自然乾燥し、引き続き低圧水銀ランプを用いて6000mJ/cm2の紫外線を照射し、膜を硬化させた。さらに、300℃で30分間加熱してヒドロキシプロピルセルロースの分解およびアニーリングを行い、酸化物半導体膜(D)を形成した。
次に、酸化物半導体膜(A)と同様にして分光増感色素として、本発明の色素の吸着を行った。その後、光電気化学電池(A)と同様の方法で、光電気化学電池(D)を作製した。
光電気化学電池(A)~(D)について、擬似太陽光(AM1.5)を照射し、実験1と同様の方法で光電変換特性を測定し、変換効率を求めた。変換効率が6.0%以上のものをA、5%以上6%未満のものをB、4%以上5%未満のものをC、3%以上4%未満のものをD、1.5%以上3%未満のものをE、1.5%未満のものをFとして表示し、変換効率D以上のものを合格とし、D未満のものを不合格とした。
また、耐久性として、変換効率の初期値に対し45%RH下で400時間後の変換効率が90%以上のものをA、85%以上90%未満のものをB、80%以上85%未満のものをC、70%以上80%未満のものをD、70%未満のものをEとして評価した。
また、耐久性として、変換効率の初期値に対し45%RH下で400時間後の変換効率が90%以上のものをA、85%以上90%未満のものをB、80%以上85%未満のものをC、70%以上80%未満のものをD、70%未満のものをEとして評価した。

表6より、本発明の色素の場合には、光電気化学電池(A)~(C)を使用した場合により変換効率が高いことがわかった。
[実験6]
方法を変えて酸化チタンの調製を行い、得られた酸化チタンから酸化物半導体膜を作製し、光電気化学電池とし、その評価を行った。
(1)熱処理法による酸化チタンの調製
(酸化チタン1(ブルーカイト型)等)
市販のアナターゼ型酸化チタン(石原産業社製、商品名ST-01)を用い、これを約900℃に加熱してブルーカイト型の酸化チタンに変換し、さらに約1,200℃に加熱してルチル型の酸化チタンとした。それぞれ順に、比較酸化チタン1(アナターゼ型)、酸化チタン1(ブルーカイト型)、比較酸化チタン2(ルチル型)とする。
(2)湿式法による酸化チタンの合成
(酸化チタン2(ブルーカイト型))
蒸留水954mlを還流冷却器付きの反応槽に装入し、95℃に加温する。撹拌速度を約200rpmに保ちながら、この蒸留水に四塩化チタン(Ti含有量:16.3質量%、比重1.59、純度99.9%)水溶液46mlを約5.0ml/minの速度で反応槽に滴下した。このとき、反応液の温度が下がらないように注意した。その結果、四塩化チタン濃度が0.25mol/リットル(酸化チタン換算2質量%)であった。反応槽中では反応液が滴下直後から、白濁し始めたがそのままの温度で保持を続け、滴下終了後さらに昇温し沸点付近(104℃)まで加熱し、この状態で60分間保持して完全に反応を終了した。
方法を変えて酸化チタンの調製を行い、得られた酸化チタンから酸化物半導体膜を作製し、光電気化学電池とし、その評価を行った。
(1)熱処理法による酸化チタンの調製
(酸化チタン1(ブルーカイト型)等)
市販のアナターゼ型酸化チタン(石原産業社製、商品名ST-01)を用い、これを約900℃に加熱してブルーカイト型の酸化チタンに変換し、さらに約1,200℃に加熱してルチル型の酸化チタンとした。それぞれ順に、比較酸化チタン1(アナターゼ型)、酸化チタン1(ブルーカイト型)、比較酸化チタン2(ルチル型)とする。
(2)湿式法による酸化チタンの合成
(酸化チタン2(ブルーカイト型))
蒸留水954mlを還流冷却器付きの反応槽に装入し、95℃に加温する。撹拌速度を約200rpmに保ちながら、この蒸留水に四塩化チタン(Ti含有量:16.3質量%、比重1.59、純度99.9%)水溶液46mlを約5.0ml/minの速度で反応槽に滴下した。このとき、反応液の温度が下がらないように注意した。その結果、四塩化チタン濃度が0.25mol/リットル(酸化チタン換算2質量%)であった。反応槽中では反応液が滴下直後から、白濁し始めたがそのままの温度で保持を続け、滴下終了後さらに昇温し沸点付近(104℃)まで加熱し、この状態で60分間保持して完全に反応を終了した。
反応により、得られたゾルを濾過し、次いで60℃の真空乾燥器を用いて粉末とした。この粉末をX線回折法により定量分析した結果、(ブルーカイト型121面のピーク強度)/(三本が重なる位置でのピーク強度)比は0.38、(ルチル型のメインピーク強度)/(三本が重なる位置でのピーク強度)比は0.05であった。これらから求めると酸化チタンは、ブルーカイト型が約70.0質量%、ルチル型が約1.2質量%、アナターゼ型が約28.8質量%の結晶性であった。また、透過型電子顕微鏡でこの微粒子を観察したところ、1次粒子の平均粒径は0.015μmであった。
(酸化チタン3(ブルーカイト型))
三塩化チタン水溶液(Ti含有量:28質量%、比重1.5、純度99.9%)を蒸留水で希釈し、チタン濃度換算で0.25モル/Lの溶液とした。このとき、液温が上昇しないよう氷冷して、50℃以下に保った。次に、この溶液を還流冷却器付きの反応槽に500ml投入し、85℃に加温しながらオゾンガス発生装置から純度80%のオゾンガスを1L/minでバブリングし、酸化反応を行なった。この状態で2時間保持し、完全に反応を終了した。得られたゾルをろ過、真空乾燥し、粉末とした。この粉末をX線回折法により定量分析した結果、(ブルーカイト型121面のピーク強度)/(三本が重なる位置でのピーク強度)比は0.85、(ルチル型のメインピーク強度)/(三本が重なる位置でのピーク強度)比は0であった。これらから求めると二酸化チタンは、ブルーカイト型が約98質量%、ルチル型が0質量%、アナターゼ型が0質量%であり、約2%は無定形であった。また、透過型電子顕微鏡でこの微粒子を観察したところ、1次粒子の平均粒径は0.05μmであった。
三塩化チタン水溶液(Ti含有量:28質量%、比重1.5、純度99.9%)を蒸留水で希釈し、チタン濃度換算で0.25モル/Lの溶液とした。このとき、液温が上昇しないよう氷冷して、50℃以下に保った。次に、この溶液を還流冷却器付きの反応槽に500ml投入し、85℃に加温しながらオゾンガス発生装置から純度80%のオゾンガスを1L/minでバブリングし、酸化反応を行なった。この状態で2時間保持し、完全に反応を終了した。得られたゾルをろ過、真空乾燥し、粉末とした。この粉末をX線回折法により定量分析した結果、(ブルーカイト型121面のピーク強度)/(三本が重なる位置でのピーク強度)比は0.85、(ルチル型のメインピーク強度)/(三本が重なる位置でのピーク強度)比は0であった。これらから求めると二酸化チタンは、ブルーカイト型が約98質量%、ルチル型が0質量%、アナターゼ型が0質量%であり、約2%は無定形であった。また、透過型電子顕微鏡でこの微粒子を観察したところ、1次粒子の平均粒径は0.05μmであった。
(色素増感型光電変換素子の作製および評価)
上記の方法で調製した酸化チタン1~3を半導体として特開2000-340269号公報記載の図1に示す構成の光電変換素子を用いた光電気化学電池を以下の方法で作製した。
ガラス基板上にフッ素ドープの酸化スズをコートし、導電性透明電極とした。電極面上にそれぞれの酸化チタン粒子を原料としたペーストを作成し、バーコート法で厚さ50μmに塗布した後、500℃で焼成して膜厚約20μmの薄層を形成した。
上記の方法で調製した酸化チタン1~3を半導体として特開2000-340269号公報記載の図1に示す構成の光電変換素子を用いた光電気化学電池を以下の方法で作製した。
ガラス基板上にフッ素ドープの酸化スズをコートし、導電性透明電極とした。電極面上にそれぞれの酸化チタン粒子を原料としたペーストを作成し、バーコート法で厚さ50μmに塗布した後、500℃で焼成して膜厚約20μmの薄層を形成した。
表7に示す色素のエタノール溶液(色素濃度3×10-4mol/L、色素を2種使用している場合には、第1色素濃度は1×10-1mol/Lとした。)を調製し、これに上記の酸化チタンの薄層を形成したガラス基板を浸漬し、12時間室温で保持した。その結果、酸化チタンの薄層上にこれらの色素を吸着させた。
電解液としてテトラプロピルアンモニウムのヨウ素塩とヨウ化リチウムのアセトニトリル溶液を用い、白金を対極として特開2000-340269号公報の図1に示す構成を有する光電変換素子を作製した。光電変換は160Wの高圧水銀ランプの光(フィルターで赤外線部をカット)を上記の素子に照射し、実験1と同様の方法で変換効率を測定した。その結果を表7に示す。
変換効率が6.0%以上のものをA、5%以上6%未満のものをB、4%以上5%未満のものをC、3%以上4%未満のものをD、1.5%以上3%未満のものをE、1.5%未満のものをFとして表示し、変換効率D以上のものを合格とし、D未満のものを不合格とした。
また、耐久性として、変換効率の初期値に対し45%RH下、連続照射400時間後の変換効率が90%以上のものをA、85%以上90%未満のものをB、80%以上85%未満のものをC、70%以上80%未満のものをD、70%未満のものをEとして評価した。
また、耐久性として、変換効率の初期値に対し45%RH下、連続照射400時間後の変換効率が90%以上のものをA、85%以上90%未満のものをB、80%以上85%未満のものをC、70%以上80%未満のものをD、70%未満のものをEとして評価した。

[実験7]
下記の粒径の異なる酸化チタンを用いて、半導体微粒子が分散したペーストを作製した。これを用いて光電気化学電池を作製し、その特性を評価した。
下記の粒径の異なる酸化チタンを用いて、半導体微粒子が分散したペーストを作製した。これを用いて光電気化学電池を作製し、その特性を評価した。
[ペーストの調製]
(ペースト1)
球形のTiO2粒子(アナターゼ型、平均粒径;25nm、以下、球形TiO2粒子1という)を硝酸溶液に入れて撹拌することによりチタニアスラリーを調製した。次に、チタニアスラリーに増粘剤としてセルロース系バインダーを加え、混練してペースト1を調製した。
(ペースト1)
球形のTiO2粒子(アナターゼ型、平均粒径;25nm、以下、球形TiO2粒子1という)を硝酸溶液に入れて撹拌することによりチタニアスラリーを調製した。次に、チタニアスラリーに増粘剤としてセルロース系バインダーを加え、混練してペースト1を調製した。
(ペースト2)
球形TiO2粒子1と、別の球形のTiO2粒子(アナターゼ型、平均粒径;200nm、以下、球形TiO2粒子2という)とを硝酸溶液に入れて撹拌することによりチタニアスラリーを調製した。次に、チタニアスラリーに増粘剤としてセルロース系バインダーを加え、混練してペースト2(TiO2粒子1の質量:TiO2粒子2の質量=30:70)を調製した。
球形TiO2粒子1と、別の球形のTiO2粒子(アナターゼ型、平均粒径;200nm、以下、球形TiO2粒子2という)とを硝酸溶液に入れて撹拌することによりチタニアスラリーを調製した。次に、チタニアスラリーに増粘剤としてセルロース系バインダーを加え、混練してペースト2(TiO2粒子1の質量:TiO2粒子2の質量=30:70)を調製した。
(ペースト3)
ペースト1と棒状のTiO2粒子(アナターゼ型、直径;100nm、アスペクト比;5、以下、棒状TiO2粒子1という)とを混合し、棒状TiO2粒子1の質量:ペースト1の質量=10:90のペースト3を調製した。
ペースト1と棒状のTiO2粒子(アナターゼ型、直径;100nm、アスペクト比;5、以下、棒状TiO2粒子1という)とを混合し、棒状TiO2粒子1の質量:ペースト1の質量=10:90のペースト3を調製した。
(ペースト4)
ペースト1と棒状TiO2粒子1とを混合し、棒状TiO2粒子1の質量:ペースト1の質量=30:70のペースト4を調製した。
ペースト1と棒状TiO2粒子1とを混合し、棒状TiO2粒子1の質量:ペースト1の質量=30:70のペースト4を調製した。
(ペースト5)
ペースト1と棒状TiO2粒子1を混合し、棒状TiO2粒子1の質量:ペースト1の質量=50:50のペースト5を調製した。
ペースト1と棒状TiO2粒子1を混合し、棒状TiO2粒子1の質量:ペースト1の質量=50:50のペースト5を調製した。
(ペースト6)
ペースト1と板状のマイカ粒子(直径;100nm、アスペクト比;6、以下、板状マイカ粒子1という)とを混合し、板状マイカ粒子1の質量:ペースト1の質量=20:80のペースト6を調製した。
ペースト1と板状のマイカ粒子(直径;100nm、アスペクト比;6、以下、板状マイカ粒子1という)とを混合し、板状マイカ粒子1の質量:ペースト1の質量=20:80のペースト6を調製した。
(ペースト7)
ペースト1と棒状のTiO2粒子(アナターゼ型、直径;30nm、アスペクト比;6.3、以下、棒状TiO2粒子2という)とを混合し、棒状TiO2粒子2の質量:ペースト1の質量=30:70のペースト7を調製した。
ペースト1と棒状のTiO2粒子(アナターゼ型、直径;30nm、アスペクト比;6.3、以下、棒状TiO2粒子2という)とを混合し、棒状TiO2粒子2の質量:ペースト1の質量=30:70のペースト7を調製した。
(ペースト8)
ペースト1と棒状のTiO2粒子(アナターゼ型、直径;50nm、アスペクト比;6.1、以下、棒状TiO2粒子3という)を混合し、棒状TiO2粒子3の質量:ペースト1の質量=30:70のペースト8を調製した。
ペースト1と棒状のTiO2粒子(アナターゼ型、直径;50nm、アスペクト比;6.1、以下、棒状TiO2粒子3という)を混合し、棒状TiO2粒子3の質量:ペースト1の質量=30:70のペースト8を調製した。
(ペースト9)
ペースト1と棒状のTiO2粒子(アナターゼ型、直径;75nm、アスペクト比;5.8、以下、棒状TiO2粒子4という)とを混合し、棒状TiO2粒子4の質量:ペースト1の質量=30:70のペースト9を調製した。
ペースト1と棒状のTiO2粒子(アナターゼ型、直径;75nm、アスペクト比;5.8、以下、棒状TiO2粒子4という)とを混合し、棒状TiO2粒子4の質量:ペースト1の質量=30:70のペースト9を調製した。
(ペースト10)
ペースト1と棒状のTiO2粒子(アナターゼ型、直径;130nm、アスペクト比;5.2、以下、棒状TiO2粒子5という)とを混合し、棒状TiO2粒子5の質量:ペースト1の質量=30:70のペースト10を調製した。
ペースト1と棒状のTiO2粒子(アナターゼ型、直径;130nm、アスペクト比;5.2、以下、棒状TiO2粒子5という)とを混合し、棒状TiO2粒子5の質量:ペースト1の質量=30:70のペースト10を調製した。
(ペースト11)
ペースト1と棒状のTiO2粒子(アナターゼ型、直径;180nm、アスペクト比;5、以下、棒状TiO2粒子6という)とを混合し、棒状TiO2粒子6の質量:ペースト1の質量=30:70のペースト11を調製した。
ペースト1と棒状のTiO2粒子(アナターゼ型、直径;180nm、アスペクト比;5、以下、棒状TiO2粒子6という)とを混合し、棒状TiO2粒子6の質量:ペースト1の質量=30:70のペースト11を調製した。
(ペースト12)
ペースト1と棒状のTiO2粒子(アナターゼ型、直径;240nm、アスペクト比;5、以下、棒状TiO2粒子7という)とを混合し、棒状TiO2粒子7の質量:ペースト1の質量=30:70のペースト12を調製した。
ペースト1と棒状のTiO2粒子(アナターゼ型、直径;240nm、アスペクト比;5、以下、棒状TiO2粒子7という)とを混合し、棒状TiO2粒子7の質量:ペースト1の質量=30:70のペースト12を調製した。
(ペースト13)
ペースト1と棒状のTiO2粒子(アナターゼ型、直径;110nm、アスペクト比;4.1、以下、棒状TiO2粒子8という)とを混合し、棒状TiO2粒子8の質量:ペースト1の質量=30:70のペースト13を調製した。
ペースト1と棒状のTiO2粒子(アナターゼ型、直径;110nm、アスペクト比;4.1、以下、棒状TiO2粒子8という)とを混合し、棒状TiO2粒子8の質量:ペースト1の質量=30:70のペースト13を調製した。
(ペースト14)
ペースト1と棒状のTiO2粒子(アナターゼ型、直径;105nm、アスペクト比;3.4、以下、棒状TiO2粒子9という)とを混合し、棒状TiO2粒子9の質量:ペースト1の質量=30:70のペースト14を調製した。
ペースト1と棒状のTiO2粒子(アナターゼ型、直径;105nm、アスペクト比;3.4、以下、棒状TiO2粒子9という)とを混合し、棒状TiO2粒子9の質量:ペースト1の質量=30:70のペースト14を調製した。
(光電気化学電池1)
以下に示す手順により、特開2002-289274号公報の図5に記載の光電極12と同様の構成を有する光電極を作製し、更に、光電極を用いて、当該光電極以外は特開2002-289274号公報の図3に記載の色素増感型太陽電池20と同様の構成を有する10×10mmのスケールの光電気化学電池1を作製した。
以下に示す手順により、特開2002-289274号公報の図5に記載の光電極12と同様の構成を有する光電極を作製し、更に、光電極を用いて、当該光電極以外は特開2002-289274号公報の図3に記載の色素増感型太陽電池20と同様の構成を有する10×10mmのスケールの光電気化学電池1を作製した。
ガラス基板上にフッ素ドープされたSnO2導電膜(膜厚;500nm)を形成した透明電極を準備した。
このSnO2導電膜上に、上記のペースト2をスクリーン印刷し、次いで乾燥させた。その後、空気中、450℃の条件のもとで焼成した。更に、ペースト4を用いてこのスクリーン印刷と焼成とを繰り返すことにより、SnO2導電膜上に上記特許文献の図5に示す半導体電極2と同様の構成の半導体電極(受光面の面積;10mm×10mm、層厚;10μm、半導体層の層厚;6μm、光散乱層の層厚;4μm、光散乱層に含有される棒状TiO2粒子1の含有率;30質量%)を形成し、増感色素を含有していない光電極を作製した。
このSnO2導電膜上に、上記のペースト2をスクリーン印刷し、次いで乾燥させた。その後、空気中、450℃の条件のもとで焼成した。更に、ペースト4を用いてこのスクリーン印刷と焼成とを繰り返すことにより、SnO2導電膜上に上記特許文献の図5に示す半導体電極2と同様の構成の半導体電極(受光面の面積;10mm×10mm、層厚;10μm、半導体層の層厚;6μm、光散乱層の層厚;4μm、光散乱層に含有される棒状TiO2粒子1の含有率;30質量%)を形成し、増感色素を含有していない光電極を作製した。
次に、半導体電極に色素を以下のようにして吸着させた。まずマグネシウムエトキシドで脱水した無水エタノールを溶媒として、これに表8記載の色素のそれぞれの濃度が3×10-4mol/L(第2色素を併用する場合は、第1色素の濃度1×10-4mol/L、第2色素の濃度1×10-4mol/L)となるように溶解し、色素溶液を調製した。次に、この溶液に半導体電極を浸漬し、これにより、半導体電極に色素が全量で約1.5×10-7mol/cm2吸着し、光電極10を完成させた。
次に、対極として上記の光電極と同様の形状と大きさを有する白金電極(Pt薄膜の厚さ;100nm)、電解質Eとして、ヨウ素及びヨウ化リチウムを含むヨウ素系レドックス溶液を調製した。更に、半導体電極の大きさに合わせた形状を有するデュポン社製のスペーサS(商品名:「サーリン」)を準備し、特開2002-289274号公報の図3に示すように、光電極10と対極CEとスペーサSを介して対向させ、内部に上記の電解質を充填して光電気化学電池1を完成させた。
(光電気化学電池2)
半導体電極の製造を以下のようにして行ったこと以外は、光電気化学電池1と同様の手順により特開2002-289274号公報記載の図1に示した光電極10を作製し、特開2002-289274号公報記載の図3に示した色素増感型太陽電池20と同様の構成を有する光電気化学電池2を作製した。
半導体電極の製造を以下のようにして行ったこと以外は、光電気化学電池1と同様の手順により特開2002-289274号公報記載の図1に示した光電極10を作製し、特開2002-289274号公報記載の図3に示した色素増感型太陽電池20と同様の構成を有する光電気化学電池2を作製した。
ペースト2を半導体層形成用ペーストとして使用した。そして、SnO2導電膜上に、ペースト2をスクリーン印刷し、次いで乾燥させた。その後、空気中、450℃の条件のもとで焼成し、半導体層を形成した。
ペースト3を光散乱層の最内部の層形成用ペーストとして使用した。また、ペースト5を光散乱層の最外部の層形成用ペーストとして使用した。そして、光電気化学電池1と同様にして半導体層上に光散乱層を形成した。
そして、SnO2導電膜上に、特開2002-289274号公報記載の図1に示す半導体電極2と同様の構成の半導体電極(受光面の面積;10mm×10mm、層厚;10μm、半導体層の層厚;3μm、最内部の層の層厚;4μm、最内部の層に含有される棒状TiO2粒子1の含有率;10質量%、最外部の層の層厚;3μm、最内部の層に含有される棒状TiO2粒子1の含有率;50質量%)を形成し、増感色素を含有していない光電極を作製した。光電気化学電池1と同様に、光電極と対極CEとスペーサSを介して対向させ、内部に上記の電解質を充填して光電気化学電池2を完成させた。
(光電気化学電池3)
半導体電極の製造に際して、ペースト1を半導体層形成用ペーストとして使用し、ペースト4を光散乱層形成用ペーストとして使用したこと以外は、光電気化学電池1と同様の手順により、特開2002-289274号公報の図5に示した光電極10を作製し、特開2002-289274号公報記載の図3に示した光電気化学電池20と同様の構成を有する光電気化学電池3を作製した。
なお、半導体電極は、受光面の面積;10mm×10mm、層厚;10μm、半導体層の層厚;5μm、光散乱層の層厚;5μm、光散乱層に含有される棒状TiO2粒子1の含有率;30質量%であった。
半導体電極の製造に際して、ペースト1を半導体層形成用ペーストとして使用し、ペースト4を光散乱層形成用ペーストとして使用したこと以外は、光電気化学電池1と同様の手順により、特開2002-289274号公報の図5に示した光電極10を作製し、特開2002-289274号公報記載の図3に示した光電気化学電池20と同様の構成を有する光電気化学電池3を作製した。
なお、半導体電極は、受光面の面積;10mm×10mm、層厚;10μm、半導体層の層厚;5μm、光散乱層の層厚;5μm、光散乱層に含有される棒状TiO2粒子1の含有率;30質量%であった。
(光電気化学電池4)
半導体電極の製造に際して、ペースト2を半導体層形成用ペーストとして使用し、ペースト6を光散乱層形成用ペーストとして使用したこと以外は、光電気化学電池1と同様の手順により図5に示した光電極10及び特開2002-289274記載の図3に示した光電気化学電池20と同様の構成を有する光電極及び光電気化学電池4を作製した。なお、半導体電極は、受光面の面積;10mm×10mm、層厚;10μm、半導体層の層厚;6.5μm、光散乱層の層厚;3.5μm、光散乱層に含有される板状マイカ粒子1の含有率;20質量%であった。
半導体電極の製造に際して、ペースト2を半導体層形成用ペーストとして使用し、ペースト6を光散乱層形成用ペーストとして使用したこと以外は、光電気化学電池1と同様の手順により図5に示した光電極10及び特開2002-289274記載の図3に示した光電気化学電池20と同様の構成を有する光電極及び光電気化学電池4を作製した。なお、半導体電極は、受光面の面積;10mm×10mm、層厚;10μm、半導体層の層厚;6.5μm、光散乱層の層厚;3.5μm、光散乱層に含有される板状マイカ粒子1の含有率;20質量%であった。
(光電気化学電池5)
半導体電極の製造に際して、ペースト2を半導体層形成用ペーストとして使用し、ペースト8を光散乱層形成用ペーストとして使用したこと以外は、光電気化学電池1と同様の手順により光電気化学電池5を作製した。なお、半導体電極の光散乱層に含有される棒状TiO2粒子3の含有率;30質量%であった。
半導体電極の製造に際して、ペースト2を半導体層形成用ペーストとして使用し、ペースト8を光散乱層形成用ペーストとして使用したこと以外は、光電気化学電池1と同様の手順により光電気化学電池5を作製した。なお、半導体電極の光散乱層に含有される棒状TiO2粒子3の含有率;30質量%であった。
(光電気化学電池6)
半導体電極の製造に際して、ペースト2を半導体層形成用ペーストとして使用し、ペースト9を光散乱層形成用ペーストとして使用したこと以外は、光電気化学電池1と同様の手順により光電気化学電池6を作製した。なお、半導体電極の光散乱層に含有される棒状TiO2粒子4の含有率;30質量%であった。
半導体電極の製造に際して、ペースト2を半導体層形成用ペーストとして使用し、ペースト9を光散乱層形成用ペーストとして使用したこと以外は、光電気化学電池1と同様の手順により光電気化学電池6を作製した。なお、半導体電極の光散乱層に含有される棒状TiO2粒子4の含有率;30質量%であった。
(光電気化学電池7)
半導体電極の製造に際して、ペースト2を半導体層形成用ペーストとして使用し、ペースト10を光散乱層形成用ペーストとして使用したこと以外は、光電気化学電池1と同様の手順により光電気化学電池7を作製した。なお、半導体電極の光散乱層に含有される棒状TiO2粒子5の含有率;30質量%であった。
半導体電極の製造に際して、ペースト2を半導体層形成用ペーストとして使用し、ペースト10を光散乱層形成用ペーストとして使用したこと以外は、光電気化学電池1と同様の手順により光電気化学電池7を作製した。なお、半導体電極の光散乱層に含有される棒状TiO2粒子5の含有率;30質量%であった。
(光電気化学電池8)
半導体電極の製造に際して、ペースト2を半導体層形成用ペーストとして使用し、ペースト11を光散乱層形成用ペーストとして使用したこと以外は、光電気化学電池1と同様の手順により光電気化学電池8を作製した。なお、半導体電極の光散乱層に含有される棒状TiO2粒子6の含有率;30質量%であった。
半導体電極の製造に際して、ペースト2を半導体層形成用ペーストとして使用し、ペースト11を光散乱層形成用ペーストとして使用したこと以外は、光電気化学電池1と同様の手順により光電気化学電池8を作製した。なお、半導体電極の光散乱層に含有される棒状TiO2粒子6の含有率;30質量%であった。
(光電気化学電池9)
半導体電極の製造に際して、ペースト2を半導体層形成用ペーストとして使用し、ペースト13を光散乱層形成用ペーストとして使用したこと以外は、光電気化学電池1と同様の手順により光電気化学電池9を作製した。なお、半導体電極の光散乱層に含有される棒状TiO2粒子8の含有率;30質量%であった。
半導体電極の製造に際して、ペースト2を半導体層形成用ペーストとして使用し、ペースト13を光散乱層形成用ペーストとして使用したこと以外は、光電気化学電池1と同様の手順により光電気化学電池9を作製した。なお、半導体電極の光散乱層に含有される棒状TiO2粒子8の含有率;30質量%であった。
(光電気化学電池10)
半導体電極の製造に際して、ペースト2を半導体層形成用ペーストとして使用し、ペースト14を光散乱層形成用ペーストとして使用したこと以外は、光電気化学電池1と同様の手順により光電気化学電池10を作製した。なお、半導体電極の光散乱層に含有される棒状TiO2粒子9の含有率;30質量%であった。
半導体電極の製造に際して、ペースト2を半導体層形成用ペーストとして使用し、ペースト14を光散乱層形成用ペーストとして使用したこと以外は、光電気化学電池1と同様の手順により光電気化学電池10を作製した。なお、半導体電極の光散乱層に含有される棒状TiO2粒子9の含有率;30質量%であった。
(光電気化学電池11)
半導体電極の製造に際して、ペースト2のみを用いて半導体層のみからなる半導体電極(受光面の面積;10mm×10mm、層厚;10μm)を作製したこと以外は、光電気化学電池1と同様の手順により光電気化学電池11を作製した。
半導体電極の製造に際して、ペースト2のみを用いて半導体層のみからなる半導体電極(受光面の面積;10mm×10mm、層厚;10μm)を作製したこと以外は、光電気化学電池1と同様の手順により光電気化学電池11を作製した。
(電気化学電池12)
半導体電極の製造に際して、ペースト2を半導体層形成用ペーストとして使用し、ペースト7を光散乱層形成用ペーストとして使用したこと以外は、光電気化学電池1と同様の手順により光電極及び比較光電気化学電池12を作製した。なお、半導体電極の光散乱層に含有される棒状TiO2粒子2の含有率;30質量%であった。
半導体電極の製造に際して、ペースト2を半導体層形成用ペーストとして使用し、ペースト7を光散乱層形成用ペーストとして使用したこと以外は、光電気化学電池1と同様の手順により光電極及び比較光電気化学電池12を作製した。なお、半導体電極の光散乱層に含有される棒状TiO2粒子2の含有率;30質量%であった。
[特性の試験及び評価]
光電気化学電池1~12について、ソーラーシミュレータ(WACOM製、WXS-85H(商品名))を用いて、AM1.5フィルターを通したキセノンランプから1000W/m2の疑似太陽光を照射した。I-Vテスターを用いて電流-電圧特性を測定し、変換効率を求めた。その結果を表8に示す。
光電気化学電池1~12について、ソーラーシミュレータ(WACOM製、WXS-85H(商品名))を用いて、AM1.5フィルターを通したキセノンランプから1000W/m2の疑似太陽光を照射した。I-Vテスターを用いて電流-電圧特性を測定し、変換効率を求めた。その結果を表8に示す。
変換効率が6.0%以上のものをA、5%以上6%未満のものをB、4%以上5%未満のものをC、3%以上4%未満のものをD、1.5%以上3%未満のものをE、1.5%未満のものをFとして表示し、変換効率D以上のものを合格とし、D未満のものを不合格とした。
耐久性として、変換効率の初期値に対しRH45%下連続照射400時間後の変換効率が90%以上のものをA、85%以上90%未満のものをB、80%以上85%未満のものをC、70%以上80%未満のものをD、70%未満のものをEとして評価した。
耐久性として、変換効率の初期値に対しRH45%下連続照射400時間後の変換効率が90%以上のものをA、85%以上90%未満のものをB、80%以上85%未満のものをC、70%以上80%未満のものをD、70%未満のものをEとして評価した。

[実験8]
金属酸化物微粒子に金属アルコキシドを加えスラリー状としたものを導電性基板に塗布し、その後、UVオゾン照射、UV照射又は乾燥を行い、電極を作製した。その後、光電気化学電池を作製し、変換効率を測定した。
金属酸化物微粒子に金属アルコキシドを加えスラリー状としたものを導電性基板に塗布し、その後、UVオゾン照射、UV照射又は乾燥を行い、電極を作製した。その後、光電気化学電池を作製し、変換効率を測定した。
(金属酸化物微粒子)
金属酸化物微粒子としては、酸化チタンを用いた。酸化チタンは、質量比で、30%ルチル型及び70%アナターゼ型、平均粒径25nmのP25粉末(Degussa社製、商品名)を用いた。
金属酸化物微粒子としては、酸化チタンを用いた。酸化チタンは、質量比で、30%ルチル型及び70%アナターゼ型、平均粒径25nmのP25粉末(Degussa社製、商品名)を用いた。
(金属酸化物微粒子粉末の前処理)
金属酸化物微粒子をあらかじめ熱処理することで表面の有機物と水分を除去した。酸化チタン微粒子の場合は450℃のオーブンで大気下、30分間加熱した。
金属酸化物微粒子をあらかじめ熱処理することで表面の有機物と水分を除去した。酸化チタン微粒子の場合は450℃のオーブンで大気下、30分間加熱した。
(金属酸化物微粒子に含まれる水分量の測定)
温度26℃、湿度72%の環境に保存しておいた酸化チタン、P25粉末(Degussa社製、商品名)に含まれる水分量を、熱重量測定における重量減少、及び300℃に加熱したときに脱着した水分量のカールフィッシャー滴定により定量した。
温度26℃、湿度72%の環境に保存しておいた酸化チタン、P25粉末(Degussa社製、商品名)に含まれる水分量を、熱重量測定における重量減少、及び300℃に加熱したときに脱着した水分量のカールフィッシャー滴定により定量した。
酸化チタン、P25粉末(Degussa社製、商品名)を300℃で加熱したときに脱着する水分量をカールフィッシャー滴定によって定量したところ、0.1033gの酸化チタン微粉末中に0.253mgの水が含まれていた。すなわち、酸化チタン微粉末は約2.5質量%の水分を含んでいた。30分間熱処理し、冷却後デシケーター中に保存して用いた。
(金属アルコキシドペーストの調製)
金属酸化物微粒子を結合する役割をする金属アルコキシドとしては、チタン原料としてはチタン(IV)テトライソプロポキシド(TTIP)、ジルコニウム原料としてはジルコニウム(IV)テトラn-プロポキシド、ニオブ原料としてはニオブ(V)ペンタエトキシド(全てAldrich社製)をそれぞれ用いた。
金属酸化物微粒子を結合する役割をする金属アルコキシドとしては、チタン原料としてはチタン(IV)テトライソプロポキシド(TTIP)、ジルコニウム原料としてはジルコニウム(IV)テトラn-プロポキシド、ニオブ原料としてはニオブ(V)ペンタエトキシド(全てAldrich社製)をそれぞれ用いた。
金属酸化物微粒子と金属アルコキシドのモル濃度比は、金属アルコキシドの加水分解によって生じるアモルファス層が過度に厚くならず、かつ粒子同士の結合が十分行えるように、金属酸化物微粒子径に応じて適宜調節した。なお、金属アルコキシドはすべて、0.1Mのエタノール溶液とした。酸化チタン微粒子とチタン(IV)テトライソプロポキシド(TTIP)とを混合する場合には、酸化チタン微粒子1gに対し、3.55gの0.1M TTIP溶液を混合した。このとき、得られたペースト中の酸化チタン濃度は約22質量%となり、塗布に適当な粘度となった。また、このときの酸化チタンとTTIPとエタノールは、質量比で1:0.127:3.42、モル比で1:0.036:5.92であった。
同様に、酸化チタン微粒子とTTIP以外のアルコキシドの混合ペーストについても微粒子濃度が22質量%となるように調製した。酸化亜鉛及び酸化スズ微粒子を用いたペーストでは16質量%とした。酸化亜鉛及び酸化スズの場合は、金属酸化物微粒子1gに対して、金属アルコキシド溶液5.25gの比で混合した。
金属酸化物微粒子と金属アルコキシド溶液は、密閉容器中においてマグネチックスターラーによって2時間攪拌して均一なペーストを得た。導電性基板へのペーストの塗布方法は、ドクターブレード法、スクリーン印刷法、スプレー塗布法などを用いることが可能であり、適当なペースト粘度は塗布方法によって適宜選択した。ここでは簡便にガラス棒で塗布する方法(ドクターブレード法に類似)を用いた。この場合、適当なペースト粘度を与える金属酸化物微粒子の濃度は概ね5~30質量%の範囲となった。
金属アルコキシドの分解によって生成するアモルファス金属酸化物の厚さは本実験では0.1~0.6nm程度の範囲にあり、適切な範囲の厚さとすることができた。
(導電性基板上へのペーストの塗布と風乾処理)
スズドープ酸化インジウム(ITO)導電膜付きポリエチレンテレフタレート(PET)フィルム基板(20Ω/cm2)又はフッ素ドープ酸化スズ(FTO)導電膜付きガラス基板(10Ω/cm2)に、スペーサとして粘着テープ2枚を一定間隔で平行に貼り付け、上記の方法に従って調製した各ペーストを、ガラス棒を用いて均一に塗布した。
ペーストを塗布後、色素吸着前に、UVオゾン処理、UV照射処理、又は乾燥処理の有無について条件を変えて多孔質膜を作製した。
スズドープ酸化インジウム(ITO)導電膜付きポリエチレンテレフタレート(PET)フィルム基板(20Ω/cm2)又はフッ素ドープ酸化スズ(FTO)導電膜付きガラス基板(10Ω/cm2)に、スペーサとして粘着テープ2枚を一定間隔で平行に貼り付け、上記の方法に従って調製した各ペーストを、ガラス棒を用いて均一に塗布した。
ペーストを塗布後、色素吸着前に、UVオゾン処理、UV照射処理、又は乾燥処理の有無について条件を変えて多孔質膜を作製した。
(乾燥処理)
導電性基板へ塗布した後の膜を大気中室温において2分程度で風乾した。この過程でペースト中の金属アルコキシドが大気中の水分によって加水分解を受け、Tiアルコキシド、Zrアルコキシド、Nbアルコキシドからそれぞれアモルファスの酸化チタン、酸化ジルコニウム、酸化ニオブが形成された。
生成したアモルファス金属酸化物が、金属酸化物微粒子同士及び膜と導電性基板を接着する役割を果たすため、風乾するのみで機械的強度と付着性に優れた多孔質膜が得られた。
導電性基板へ塗布した後の膜を大気中室温において2分程度で風乾した。この過程でペースト中の金属アルコキシドが大気中の水分によって加水分解を受け、Tiアルコキシド、Zrアルコキシド、Nbアルコキシドからそれぞれアモルファスの酸化チタン、酸化ジルコニウム、酸化ニオブが形成された。
生成したアモルファス金属酸化物が、金属酸化物微粒子同士及び膜と導電性基板を接着する役割を果たすため、風乾するのみで機械的強度と付着性に優れた多孔質膜が得られた。
(UVオゾン処理)
UVオゾン処理には日本レーザー電子社製のNL-UV253 UVオゾンクリーナーを用いた。UV光源には185nmと254nmに輝線を持つ4.5W水銀ランプ3個を備えており、試料を光源から約6.5センチの距離に水平に配置した。チャンバー中に酸素気流を導入することでオゾンが発生する。本実施例においてはこのUVオゾン処理を2時間行なった。なお、このUVオゾン処理によるITO膜及びFTO膜の導電性の低下は全く見られなかった。
UVオゾン処理には日本レーザー電子社製のNL-UV253 UVオゾンクリーナーを用いた。UV光源には185nmと254nmに輝線を持つ4.5W水銀ランプ3個を備えており、試料を光源から約6.5センチの距離に水平に配置した。チャンバー中に酸素気流を導入することでオゾンが発生する。本実施例においてはこのUVオゾン処理を2時間行なった。なお、このUVオゾン処理によるITO膜及びFTO膜の導電性の低下は全く見られなかった。
(UV処理)
チャンバー中を窒素置換して処理を行う以外は同様に、前記UVオゾン処理と同様に、2時間処理を行った。このUV処理によるITO膜及びFTO膜の導電性の低下はまったく見られなかった。
チャンバー中を窒素置換して処理を行う以外は同様に、前記UVオゾン処理と同様に、2時間処理を行った。このUV処理によるITO膜及びFTO膜の導電性の低下はまったく見られなかった。
(色素吸着)
増感色素として表9に記載の増感色素を用いて、各増感色素の0.3mMのエタノール溶液を調製した(第2色素を併用する場合には第1色素の濃度を0.1mM、第2色素の濃度を0.1mMとした)。本実験では上記のプロセスで作製した多孔質膜を100℃のオーブンで1時間乾燥した後に増感色素の溶液に浸漬し、そのまま室温で50分間放置して酸化チタン表面に色素を吸着させた。色素吸着後の試料はエタノールで洗浄し、風乾した。
増感色素として表9に記載の増感色素を用いて、各増感色素の0.3mMのエタノール溶液を調製した(第2色素を併用する場合には第1色素の濃度を0.1mM、第2色素の濃度を0.1mMとした)。本実験では上記のプロセスで作製した多孔質膜を100℃のオーブンで1時間乾燥した後に増感色素の溶液に浸漬し、そのまま室温で50分間放置して酸化チタン表面に色素を吸着させた。色素吸着後の試料はエタノールで洗浄し、風乾した。
(光電気化学電池の作製と電池特性評価)
色素吸着後の多孔質膜が形成された導電性基板を光電極とし、これと白金微粒子をスパッタリングにより修飾したITO/PETフィルム又はFTO/ガラス対極を対向させて、光電気化学電池を試作した。上記光電極の実効面積は約0.2cm2とした。電解質溶液には0.5MのLiI、0.05MのI2、0.5Mのt-ブチルピリジンを含む3-メトキシプロピオニトリルを用い、毛管現象によって両電極間のギャップに導入した。
色素吸着後の多孔質膜が形成された導電性基板を光電極とし、これと白金微粒子をスパッタリングにより修飾したITO/PETフィルム又はFTO/ガラス対極を対向させて、光電気化学電池を試作した。上記光電極の実効面積は約0.2cm2とした。電解質溶液には0.5MのLiI、0.05MのI2、0.5Mのt-ブチルピリジンを含む3-メトキシプロピオニトリルを用い、毛管現象によって両電極間のギャップに導入した。
電池性能の評価は、一定フォトン数(1016cm-2)照射下での光電流作用スペクトル測定及びAM1.5擬似太陽光(100mW/cm2)照射下でのI-V測定により行なった。これらの測定には分光計器社製のCEP-2000型分光感度測定装置を用いた。得られた変換効率を表9に示す。
変換効率が3.5%以上のものをA、3.0%以上3.5%未満のものをB、2.5%以上3.0%未満のものをC、2.0%以上2.5%未満のものをD、1.5%以上2.0%未満のものをE、1.5%未満のものをFとして表示した。
また、耐久性として、変換効率の初期値に対する400時間連続照射後の変換効率が90%以上のものをA、85%以上90%未満のものをB、80%以上85%未満のものをC、70%以上80%未満のものをD、70%未満のものをEとして評価した。
また、耐久性として、変換効率の初期値に対する400時間連続照射後の変換効率が90%以上のものをA、85%以上90%未満のものをB、80%以上85%未満のものをC、70%以上80%未満のものをD、70%未満のものをEとして評価した。

表9において、「UVオゾン」、「UV」、「乾燥」の欄はそれぞれ、多孔質膜の形成後、増感色素吸着前における、UVオゾン処理、UV照射処理、乾燥処理の有無を表す。処理したものが「○」であり、処理なしのものが「×」である。
表9の「酸化チタン前処理」の欄は、酸化チタン微粒子の前処理(450℃のオーブンで30分間熱処理)の有無を示す。試料9-6、9-14、9-22、9-30、9-38は、高TTIP濃度(酸化チタン:TTIPのモル比が1:0.356)のペーストを用いた試料を表す。他の試料(試料9-1~9-5、9-7~9-13、9-15~9-21、9-23~9-29、9-31~9-37、9-39、9-40)は全て酸化チタン:TTIP=1:0.0356のペーストを用いた。
表9からわかるように、本発明の色素を用いた光電気化学電池は、多孔質膜の形成後、増感色素吸着前における、UVオゾン処理、UV照射処理、乾燥処理の有無にかかわらず、常に光電気化学電池の変換効率が高く、合格レベルの変換効率が得られることがわかった。さらに400時間経過後の変換効率が初期値の70%以上と、優れた耐久性を示した。
[実験9]
溶媒としてアセトニトリルを用い、ヨウ化リチウム0.1mol/l、ヨウ素0.05mol/l、ヨウ化ジメチルプロピルイミダゾリウム0.62mol/lを溶解した電解質溶液を調製した。ここに下記に示すNo.1~No.8のベンズイミダゾール系化合物をそれぞれ濃度0.5mol/lになるように別々に添加し、溶解した。
溶媒としてアセトニトリルを用い、ヨウ化リチウム0.1mol/l、ヨウ素0.05mol/l、ヨウ化ジメチルプロピルイミダゾリウム0.62mol/lを溶解した電解質溶液を調製した。ここに下記に示すNo.1~No.8のベンズイミダゾール系化合物をそれぞれ濃度0.5mol/lになるように別々に添加し、溶解した。

ガラス基板上に、透明導電膜としてフッ素をドープした酸化スズをスパッタリングにより、導電膜を形成した。この導電膜上にアナターゼ型酸化チタン粒子を含有する分散液(水とアセトニトリルの容量比4:1からなる混合溶媒100mlにアナターゼ型酸化チタン(日本アエロジル社製のP-25(商品名))を32g配合し、自転/公転併用式のミキシングコンディショナーを使用して均一に分散、混合して得た、半導体微粒子分散液)を塗布し、その後500℃で焼結して厚さ15μmの感光層を形成した。次に、下記表に記載された増感色素のエタノール溶液(濃度:1×10-4mol/L;2種併用の場合の第1色素、第2色素の濃度はそれぞれ0.1mMとした。)に、感光層を24時間浸した。この感光層に、No.1~No.8のベンズイミダゾール系化合物電解液を、滴下した。
ここにポリエチレンフィルム製のフレーム型スペーサー(厚さ25μm)をのせ、白金対電極でこれを覆い、光電変換素子を作製した。
ここにポリエチレンフィルム製のフレーム型スペーサー(厚さ25μm)をのせ、白金対電極でこれを覆い、光電変換素子を作製した。
得られた光電変換素子に、Xeランプを光源として強度100mW/cm2の光を照射し、開放電圧と光電変換効率を測定した。その結果を表10に示した。
(結果の評価)
開放電圧は、7.0V以上のものを◎、6.5V以上7.0V未満のものを○、6.0V以上6.5V未満のものを△、6.0V未満のものを×として評価し、6.5V以上を合格とした。
変換効率が6.0%以上のものをA、5%以上6%未満のものをB、4%以上5%未満のものをC、3%以上4%未満のものをD、1.5%以上3%未満のものをE、1.5%未満のものをFとして表示し、変換効率D以上のものを合格とし、D未満のものを不合格とした。
また、耐久性は、変換効率の初期値に対するRH45%下400時間連続照射後の変換効率が90%以上のものをA、85%以上90%未満のものをB、80%以上85%未満のものをC、70%以上80%未満のものをD、70%未満のものをEとして評価した。
なお、表10には、ベンズイミダゾール系化合物を加えていない電解液を用いた光電変換素子の結果も示した。
開放電圧は、7.0V以上のものを◎、6.5V以上7.0V未満のものを○、6.0V以上6.5V未満のものを△、6.0V未満のものを×として評価し、6.5V以上を合格とした。
変換効率が6.0%以上のものをA、5%以上6%未満のものをB、4%以上5%未満のものをC、3%以上4%未満のものをD、1.5%以上3%未満のものをE、1.5%未満のものをFとして表示し、変換効率D以上のものを合格とし、D未満のものを不合格とした。
また、耐久性は、変換効率の初期値に対するRH45%下400時間連続照射後の変換効率が90%以上のものをA、85%以上90%未満のものをB、80%以上85%未満のものをC、70%以上80%未満のものをD、70%未満のものをEとして評価した。
なお、表10には、ベンズイミダゾール系化合物を加えていない電解液を用いた光電変換素子の結果も示した。

表10からわかるように、本発明の色素を用いた光電気化学電池は、開放電圧及び変換効率の初期値がともに合格レベルであり、さらに400時間経過後の変換効率が初期値の80%以上と、優れた耐久性を示した。
これに対して、比較色素を用いた場合には、開放電圧は合格レベルであるが、耐久性に問題があることがわかった。本発明の色素はチオフェンによる吸収域拡大、ε増加効果と、一電子酸化状態の安定化に加え、アルキニレンまたはアリーレンの直線性の高い平面構造により、密な吸着状態を作ることで、水の接近や求核種による変換効率の低下を防ぎ、高変換効率・高耐久性を示していると考えられる。
これに対して、比較色素を用いた場合には、開放電圧は合格レベルであるが、耐久性に問題があることがわかった。本発明の色素はチオフェンによる吸収域拡大、ε増加効果と、一電子酸化状態の安定化に加え、アルキニレンまたはアリーレンの直線性の高い平面構造により、密な吸着状態を作ることで、水の接近や求核種による変換効率の低下を防ぎ、高変換効率・高耐久性を示していると考えられる。
[実験10]
(光電気化学電池21)
以下に示す手順により、特開2004-152613号公報の図1に示した光電極10と同様の構成を有する光電極(ただし、半導体電極2を2層構造とした。)を作製し、更に、この光電極を用いた以外は特開2004-152613号公報の図1に示した色素増感型太陽電池20と同様の構成を有する光電気化学電池(半導体電極2の受光面F2の面積:1cm2)を作製した。なお、当該2層構造を有する半導体電極2の各層について、透明電極1に近い側に配置される層を「第1の層」、対極CEに近い側に配置される層を「第2の層」という。
(光電気化学電池21)
以下に示す手順により、特開2004-152613号公報の図1に示した光電極10と同様の構成を有する光電極(ただし、半導体電極2を2層構造とした。)を作製し、更に、この光電極を用いた以外は特開2004-152613号公報の図1に示した色素増感型太陽電池20と同様の構成を有する光電気化学電池(半導体電極2の受光面F2の面積:1cm2)を作製した。なお、当該2層構造を有する半導体電極2の各層について、透明電極1に近い側に配置される層を「第1の層」、対極CEに近い側に配置される層を「第2の層」という。
まず、平均粒子径25nmのP25粉末(Degussa社製、商品名)と、これと粒子径の異なる酸化チタン粒子、P200粉末(平均粒子径:200nm、Degussa社製、商品名)とを用い、P25とP200の合計の含有量が15質量%で、P25とP200との質量比が、P25:P200=30:70となるように、これらにアセチルアセトン、イオン交換水、界面活性剤(東京化成社製、商品名;「Triton-X」)を加え、混練して第2の層形成用のスラリー、以下、「スラリー1」とする)を調製した。
次に、P200を使用せず、P25のみを使用したこと以外は、前述のスラリー1と同様の調製手順により第1の層形成用のスラリー(P1の含有量;15質量%、以下、「スラリー2」とする)を調製した。
次に、P200を使用せず、P25のみを使用したこと以外は、前述のスラリー1と同様の調製手順により第1の層形成用のスラリー(P1の含有量;15質量%、以下、「スラリー2」とする)を調製した。
一方、ガラス基板(透明導電性ガラス)上に、フッ素ドープされたSnO2導電膜(膜厚:700nm)を形成した透明電極(厚さ:1.1mm)を準備した。そして、このSnO2導電膜上に、上述のスラリー2をバーコータで塗布し、次いで乾燥させた。その後、大気中、450℃で30分間焼成した。このようにして、透明電極上に、半導体電極2の第1の層を形成した。
更に、スラリー1を用いて、上述と同様の塗布と焼成とを繰り返すことにより、第1の層上に、第2の層を形成した。このようにして、SnO2導電膜上に半導体電極2(受光面の面積;1.0cm2、第1層と第2層の合計厚さ:10μm(第1の層の厚さ:3μm、第2の層の厚さ:7μm))を形成し、増感色素を含有していない状態の光電極10を作製した。
次に、色素として表11記載の色素のエタノール溶液(各増感色素の濃度;3×10-4mol/L。第2色素を併用する場合には第1色素の濃度1×10-4mol/L、第2色素の濃度1×10-4mol/L)を調製した。この溶液に前記光電極10を浸漬し、80℃の温度条件のもとで20時間放置した。これにより、半導体電極の内部に増感色素を合計で約1.0×10-7mol/cm2吸着させた。
次に、上記の光電極と同様の形状と大きさを有する対極CEを作製した。先ず、透明導電性ガラス上に、塩化白金酸六水和物のイソプロパノール溶液を滴下し、大気中で乾燥した後に450℃で30分焼成処理することにより、白金焼結対極CEを得た。なお、この対極CEには予め電解質Eの注入用の孔(直径1mm)を設けておいた。
次に、溶媒となるメトキシアセトニトリルに、ヨウ化亜鉛と、ヨウ化-1,2-ジメチル-3-プロピルイミダゾリウムと、ヨウ素と、4-tert-ブチルピリジンとを溶解させて液状電解質(ヨウ化亜鉛の濃度:10mmol/L、ヨウ化ジメチルプロピルイミダゾリウムの濃度:0.6mol/L、ヨウ素の濃度:0.05mol/L、4-tert-ブチルピリジン濃度:1mol/L)を調製した。
次に、半導体電極の大きさに合わせた形状を有する三井デュポンポリケミカル社製のスペーサS(商品名:「ハイミラン」,エチレン/メタクリル酸ランダム共重合体アイオノマーフィルム)を準備し、特開2004-152613号公報の図1に示すように、光電極と対極とをスペーサを介して対向させ、それぞれを熱溶着により張り合わせて電池の筐体(電解質未充填)を得た。
次に、液状電解質を対極の孔から筐体内に注入した後、孔をスペーサと同素材の部材で塞ぎ、更に対極の孔にこの部材を熱溶着させて孔を封止し、光電気化学電池21を完成させた。
次に、液状電解質を対極の孔から筐体内に注入した後、孔をスペーサと同素材の部材で塞ぎ、更に対極の孔にこの部材を熱溶着させて孔を封止し、光電気化学電池21を完成させた。
(光電気化学電池22)
液状電解質におけるヨウ化亜鉛の濃度を50mmol/Lとしたこと以外は、光電気化学電池21と同様の手順及び条件で光電気化学電池22を作製した。
液状電解質におけるヨウ化亜鉛の濃度を50mmol/Lとしたこと以外は、光電気化学電池21と同様の手順及び条件で光電気化学電池22を作製した。
(光電気化学電池23)
液状電解質におけるヨウ化亜鉛の代わりにヨウ化リチウムを添加し、液状電解質におけるヨウ化リチウムの濃度を20mmol/Lとしたこと以外は、光電気化学電池21と同様の手順及び条件で光電気化学電池23を作製した。
液状電解質におけるヨウ化亜鉛の代わりにヨウ化リチウムを添加し、液状電解質におけるヨウ化リチウムの濃度を20mmol/Lとしたこと以外は、光電気化学電池21と同様の手順及び条件で光電気化学電池23を作製した。
(電気化学電池24)
液状電解質におけるヨウ化亜鉛の代わりにヨウ化リチウムを添加し、液状電解質におけるヨウ化リチウムの濃度を100mmol/Lとしたこと以外は、光電気化学電池21と同様の手順及び条件で光電気化学電池24を作製した。
液状電解質におけるヨウ化亜鉛の代わりにヨウ化リチウムを添加し、液状電解質におけるヨウ化リチウムの濃度を100mmol/Lとしたこと以外は、光電気化学電池21と同様の手順及び条件で光電気化学電池24を作製した。
(試験と評価)
以下の手順により、光電気化学電池21~24を用いた試料について、変換効率を測定した。
電池特性評価試験は、ソーラーシミュレータ(ワコム製、商品名;「WXS-85-H型」)を用い、AMフィルター(AM1.5)を通したキセノンランプ光源からの疑似太陽光の照射条件を、100mW/cm2とする(いわゆる「1Sun」の照射条件)測定条件の下で行った。
以下の手順により、光電気化学電池21~24を用いた試料について、変換効率を測定した。
電池特性評価試験は、ソーラーシミュレータ(ワコム製、商品名;「WXS-85-H型」)を用い、AMフィルター(AM1.5)を通したキセノンランプ光源からの疑似太陽光の照射条件を、100mW/cm2とする(いわゆる「1Sun」の照射条件)測定条件の下で行った。
各光電気化学電池について、I-Vテスターを用いて室温にて電流-電圧特性を測定し、これらから変換効率を求めた。得られた結果を表11(1Sunの照射条件)の「初期値」として示す。
また、60℃、1Sun照射で、10Ω負荷での作動条件で500時間経過後の変換効率の結果も表11に示す。変換効率が6.0%以上のものをA、5%以上6%未満のものをB、4%以上5%未満のものをC、3%以上4%未満のものをD、1.5%以上3%未満のものをE、1.5%未満のものをFとして表示し、変換効率D以上のものを合格とし、D未満のものを不合格とした。なお、500時間経過後の変換効率が初期値に対し70%以上維持しているものを合格とした。
また、60℃、1Sun照射で、10Ω負荷での作動条件で500時間経過後の変換効率の結果も表11に示す。変換効率が6.0%以上のものをA、5%以上6%未満のものをB、4%以上5%未満のものをC、3%以上4%未満のものをD、1.5%以上3%未満のものをE、1.5%未満のものをFとして表示し、変換効率D以上のものを合格とし、D未満のものを不合格とした。なお、500時間経過後の変換効率が初期値に対し70%以上維持しているものを合格とした。

表11からわかるように、本発明の色素を用いた光電気化学電池は、変換効率の初期値がともに合格レベルであり、さらに500時間経過後の変換効率も初期値の70%以上と、優れた耐久性を示した。
これに対して、比較色素を用いた場合には、変換効率・耐久性に問題があることがわかった。
また、チエニレン基上に炭素原子数5以上の置換基を持つものがさらに高い耐久性・変換効率を示す事が明らかとなった。配向吸着による安定化・吸着量増大と、適度な非効率会合抑制効果があると考えられる。
これに対して、比較色素を用いた場合には、変換効率・耐久性に問題があることがわかった。
また、チエニレン基上に炭素原子数5以上の置換基を持つものがさらに高い耐久性・変換効率を示す事が明らかとなった。配向吸着による安定化・吸着量増大と、適度な非効率会合抑制効果があると考えられる。
表11に示した結果から明らかなように、本発明の色素は電解質にヨウ化亜鉛を添加した場合でも優れたものであることがわかった。
[実験11]
1.二酸化チタン分散液の調製
内側をフッ素樹脂コーティングした内容積200mlのステンレス製容器に二酸化チタン微粒子(日本アエロジル(株)製,Degussa P-25)15g、水45g、分散剤(アルドリッチ社製、Triron X-100)1g、直径0.5mmのジルコニアビーズ(ニッカトー社製)30gを入れ、サンドグラインダーミル(アイメックス社製)を用いて1500rpmで2時間分散処理した。得られた分散液からジルコニアビーズを濾別した。得られた分散液中の二酸化チタン微粒子の平均粒径は2.5μmであった。なお粒径はMALVERN社製のマスターサイザーにより測定した。
1.二酸化チタン分散液の調製
内側をフッ素樹脂コーティングした内容積200mlのステンレス製容器に二酸化チタン微粒子(日本アエロジル(株)製,Degussa P-25)15g、水45g、分散剤(アルドリッチ社製、Triron X-100)1g、直径0.5mmのジルコニアビーズ(ニッカトー社製)30gを入れ、サンドグラインダーミル(アイメックス社製)を用いて1500rpmで2時間分散処理した。得られた分散液からジルコニアビーズを濾別した。得られた分散液中の二酸化チタン微粒子の平均粒径は2.5μmであった。なお粒径はMALVERN社製のマスターサイザーにより測定した。
2.色素を吸着した酸化チタン微粒子層(電極A)の作製
フッ素をドープした酸化スズを被覆した20mm×20mmの導電性ガラス板(旭ガラス(株)製,商品名:TCOガラス-U,表面抵抗:約30Ω/m2)を準備し、その導電層側の両端(端から3mmの幅の部分)にスペーサー用粘着テープを張った後で、導電層上にガラス棒を用いて上記分散液を塗布した。分散液の塗布後、粘着テープを剥離し、室温で1日間風乾した。次にこの半導体塗布ガラス板を電気炉(ヤマト科学(株)製マッフル炉FP-32型)に入れ、450℃で30分間焼成した。半導体塗布ガラス板を取り出し冷却した後、表12に示す増感色素のエタノール溶液(濃度:3×10-4mol/L。第2色素を併用する場合には、第1色素の濃度1×10-4mol/L、第2色素の濃度1×10-4mol/L)に3時間浸漬した。色素が吸着した半導体塗布ガラス板を4-tert-ブチルピリジンに15分間浸漬した後、エタノールで洗浄し、自然乾燥させた。このようにして得られた色素増感酸化チタン微粒子層の厚さは10μmであり、酸化チタン微粒子の塗布量は20g/m2であった。また色素の吸着量は、その種類に応じて0.1~10mmol/m2の範囲内であった。
フッ素をドープした酸化スズを被覆した20mm×20mmの導電性ガラス板(旭ガラス(株)製,商品名:TCOガラス-U,表面抵抗:約30Ω/m2)を準備し、その導電層側の両端(端から3mmの幅の部分)にスペーサー用粘着テープを張った後で、導電層上にガラス棒を用いて上記分散液を塗布した。分散液の塗布後、粘着テープを剥離し、室温で1日間風乾した。次にこの半導体塗布ガラス板を電気炉(ヤマト科学(株)製マッフル炉FP-32型)に入れ、450℃で30分間焼成した。半導体塗布ガラス板を取り出し冷却した後、表12に示す増感色素のエタノール溶液(濃度:3×10-4mol/L。第2色素を併用する場合には、第1色素の濃度1×10-4mol/L、第2色素の濃度1×10-4mol/L)に3時間浸漬した。色素が吸着した半導体塗布ガラス板を4-tert-ブチルピリジンに15分間浸漬した後、エタノールで洗浄し、自然乾燥させた。このようにして得られた色素増感酸化チタン微粒子層の厚さは10μmであり、酸化チタン微粒子の塗布量は20g/m2であった。また色素の吸着量は、その種類に応じて0.1~10mmol/m2の範囲内であった。
3.光電気化学電池aの作製
溶媒としては、アセトニトリルと3-メチル-2-オキサゾリジノンとの体積比90/10の混合物を用いた。この溶媒に、ヨウ素と電解質塩として、1-メチル-3-ヘキシルイミダゾリウムのヨウ素塩を加えて、0.5mol/Lの電解質塩および0.05mol/Lのヨウ素を含んだ溶液を調製した。この溶液に、(溶媒+窒素含有高分子化合物+塩)100質量部に対し、窒素含有高分子化合物(α)を10質量部加えた。さらに窒素含有高分子化合物の反応性窒素原子に対する求電子剤(β)を0.1モル混合し、均一な反応溶液とした。
溶媒としては、アセトニトリルと3-メチル-2-オキサゾリジノンとの体積比90/10の混合物を用いた。この溶媒に、ヨウ素と電解質塩として、1-メチル-3-ヘキシルイミダゾリウムのヨウ素塩を加えて、0.5mol/Lの電解質塩および0.05mol/Lのヨウ素を含んだ溶液を調製した。この溶液に、(溶媒+窒素含有高分子化合物+塩)100質量部に対し、窒素含有高分子化合物(α)を10質量部加えた。さらに窒素含有高分子化合物の反応性窒素原子に対する求電子剤(β)を0.1モル混合し、均一な反応溶液とした。
一方、導電性ガラス板上に形成された色素増感酸化チタン微粒子層の上にスペーサーを介して白金を蒸着したガラス板からなる対極の白金薄膜側を載置し、導電性ガラス板と白金蒸着ガラス板とを固定した。得られた組立体の開放端を上記電解質溶液に浸漬し、毛細管現象により色素増感酸化チタン微粒子層中に反応溶液を浸透させた。
次いで80℃で30分間加熱して、架橋反応を行った。このようにして、特開2000-323190号公報の図2に示すような、導電性ガラス板10の導電層12上に、色素増感酸化チタン微粒子層20、電解質層30、白金薄膜42及びガラス板41からなる対極40が順に積層された本発明の光電気化学電池a(試料番号12-1)を得た。
また色素と電解質組成物の組成の組み合わせを表12に示すように変更した以外は上記工程を繰り返すことにより、異なる感光体および/または電荷移動体を有する光電気化学電池a(12-4、12-7、12-10、12-13、12-16~12-18)を得た。
次いで80℃で30分間加熱して、架橋反応を行った。このようにして、特開2000-323190号公報の図2に示すような、導電性ガラス板10の導電層12上に、色素増感酸化チタン微粒子層20、電解質層30、白金薄膜42及びガラス板41からなる対極40が順に積層された本発明の光電気化学電池a(試料番号12-1)を得た。
また色素と電解質組成物の組成の組み合わせを表12に示すように変更した以外は上記工程を繰り返すことにより、異なる感光体および/または電荷移動体を有する光電気化学電池a(12-4、12-7、12-10、12-13、12-16~12-18)を得た。
4.光電気化学電池b、cの作製
(1)光電気化学電池b
前述のようにして本発明の色素により色素増感された酸化チタン微粒子層からなる電極A(20mm×20mm)を同じ大きさの白金蒸着ガラス板にスペーサーを介して重ねあわせた。次に両ガラス板の隙間に毛細管現象を利用して電解液(アセトニトリルと3-メチル-2-オキサゾリジノンとの体積比90/10の混合物を溶媒としたヨウ素0.05mol/L、ヨウ化リチウム0.5mol/Lの溶液)を浸透させて、光電気化学電池b(試料番号12-2)を作製した。また色素を表12に示すように変更した以外は上記工程を繰り返すことにより、光電気化学電池b(試料番号12-5、12-8、12-11、12-14)を得た。
(1)光電気化学電池b
前述のようにして本発明の色素により色素増感された酸化チタン微粒子層からなる電極A(20mm×20mm)を同じ大きさの白金蒸着ガラス板にスペーサーを介して重ねあわせた。次に両ガラス板の隙間に毛細管現象を利用して電解液(アセトニトリルと3-メチル-2-オキサゾリジノンとの体積比90/10の混合物を溶媒としたヨウ素0.05mol/L、ヨウ化リチウム0.5mol/Lの溶液)を浸透させて、光電気化学電池b(試料番号12-2)を作製した。また色素を表12に示すように変更した以外は上記工程を繰り返すことにより、光電気化学電池b(試料番号12-5、12-8、12-11、12-14)を得た。
(2)光電気化学電池c(特開平9-27352号に記載の電解質)
前述のようにして本発明の色素により色素増感された酸化チタン微粒子層からなる電極A(20mm×20mm)上に、電解液を塗布し、含浸させた。なお電解液は、ヘキサエチレングリコールメタクリル酸エステル(日本油脂化学(株)製,ブレンマーPE-350)1gと、エチレングリコール1gと、重合開始剤として2-ヒドロキシ-2-メチル-1-フェニル-プロパン-1-オン(日本チバガイギー(株)製,ダロキュア1173)20mgを含有した混合液に、ヨウ化リチウム500mgを溶解し10分間真空脱気することにより得た。次に前記混合溶液を含浸させた多孔性酸化チタン層を減圧下に置くことにより、多孔性酸化チタン層中の気泡を除き、モノマーの浸透を促した後、紫外光照射により重合して高分子化合物の均一なゲルを多孔性酸化チタン層の微細空孔内に充填した。このようにして得られたものをヨウ素雰囲気に30分間曝して、高分子化合物中にヨウ素を拡散させた後、白金蒸着ガラス板を重ね合わせ、光電気化学電池cを得た(試料番号12-3)。また色素を表12に示すように変更した以外は上記工程を繰り返すことにより、光電気化学電池c(試料番号12-6、12-9、12-12、12-15)を得た。
前述のようにして本発明の色素により色素増感された酸化チタン微粒子層からなる電極A(20mm×20mm)上に、電解液を塗布し、含浸させた。なお電解液は、ヘキサエチレングリコールメタクリル酸エステル(日本油脂化学(株)製,ブレンマーPE-350)1gと、エチレングリコール1gと、重合開始剤として2-ヒドロキシ-2-メチル-1-フェニル-プロパン-1-オン(日本チバガイギー(株)製,ダロキュア1173)20mgを含有した混合液に、ヨウ化リチウム500mgを溶解し10分間真空脱気することにより得た。次に前記混合溶液を含浸させた多孔性酸化チタン層を減圧下に置くことにより、多孔性酸化チタン層中の気泡を除き、モノマーの浸透を促した後、紫外光照射により重合して高分子化合物の均一なゲルを多孔性酸化チタン層の微細空孔内に充填した。このようにして得られたものをヨウ素雰囲気に30分間曝して、高分子化合物中にヨウ素を拡散させた後、白金蒸着ガラス板を重ね合わせ、光電気化学電池cを得た(試料番号12-3)。また色素を表12に示すように変更した以外は上記工程を繰り返すことにより、光電気化学電池c(試料番号12-6、12-9、12-12、12-15)を得た。
5.光電変換効率の測定
500Wのキセノンランプ(ウシオ電機(株)製)の光をAM1.5フィルター(Oriel社製)およびシャープカットフィルター(Kenko L-42)を通すことにより、紫外線を含まない模擬太陽光とした。光強度は89mW/cm2とした。
500Wのキセノンランプ(ウシオ電機(株)製)の光をAM1.5フィルター(Oriel社製)およびシャープカットフィルター(Kenko L-42)を通すことにより、紫外線を含まない模擬太陽光とした。光強度は89mW/cm2とした。
前述の光電気化学電池の導電性ガラス板10と白金蒸着ガラス板40にそれぞれワニ口クリップを接続し、各ワニ口クリップを電流電圧測定装置(ケースレーSMU238型(商品名))に接続した。これに導電性ガラス板10側から模擬太陽光を照射し、発生した電気を電流電圧測定装置により測定した。これにより求められた光電気化学電池の変換効率の初期値と、500時間連続照射時の変換効率の低下率を表12に示す。
変換効率が6.0%以上のものをA、5%以上6%未満のものをB、4%以上5%未満のものをC、3%以上4%未満のものをD、1.5%以上3%未満のものをE、1.5%未満のものをFとして表示し、変換効率D以上のものを合格とし、D未満のものを不合格とした。
また、500時間経過後の変換効率の低下率を表12に示した。低下率が30%以下の場合を合格、30%を越える場合を不合格とした。
変換効率が6.0%以上のものをA、5%以上6%未満のものをB、4%以上5%未満のものをC、3%以上4%未満のものをD、1.5%以上3%未満のものをE、1.5%未満のものをFとして表示し、変換効率D以上のものを合格とし、D未満のものを不合格とした。
また、500時間経過後の変換効率の低下率を表12に示した。低下率が30%以下の場合を合格、30%を越える場合を不合格とした。

窒素含有高分子化合物α、求電子剤βは以下の化合物を表す。


[実験12]
ゾル-ゲル法によって調製した懸濁液を用いてスクリーン印刷によりTiO2の多孔質層をFTOガラス上に塗布し450℃で焼成した。これに本発明の色素化合物A-9、及び増感色素B-1の10-4mol/Lエタノール溶液中に浸漬することで、色素を吸着させた。
100mgの2,2′,7,7′-テトラキス(ジフェニルアミノ)-9,9′-スピロビフルオレンを5mlのクロロホルムに溶解した。溶液を、色素を吸着させた多孔質層の表面に軽く塗ることによって、この溶液を層の細孔内にしみこませた。さらに溶液の一滴を直接表面に置いて室温で乾燥した。ついで被覆支持体を蒸着装置に装着して約10-5ミリバールの真空下の熱蒸着によってさらに厚さ100nmの2,2′,7,7′-テトラキス(ジフェニルアミノ)-9,9′-スピロビフルオレンの層を適用した。さらに蒸着装置内でこの被覆支持体に対極として厚さ200nmの金の層を被覆した。
ゾル-ゲル法によって調製した懸濁液を用いてスクリーン印刷によりTiO2の多孔質層をFTOガラス上に塗布し450℃で焼成した。これに本発明の色素化合物A-9、及び増感色素B-1の10-4mol/Lエタノール溶液中に浸漬することで、色素を吸着させた。
100mgの2,2′,7,7′-テトラキス(ジフェニルアミノ)-9,9′-スピロビフルオレンを5mlのクロロホルムに溶解した。溶液を、色素を吸着させた多孔質層の表面に軽く塗ることによって、この溶液を層の細孔内にしみこませた。さらに溶液の一滴を直接表面に置いて室温で乾燥した。ついで被覆支持体を蒸着装置に装着して約10-5ミリバールの真空下の熱蒸着によってさらに厚さ100nmの2,2′,7,7′-テトラキス(ジフェニルアミノ)-9,9′-スピロビフルオレンの層を適用した。さらに蒸着装置内でこの被覆支持体に対極として厚さ200nmの金の層を被覆した。
このように調製した試料を高圧ランプ、光学フィルター、レンズ及びマウンティングを含む光学装置に取り付けた。フィルターの使用及びレンズの移動によって強度を変えることができた。金の層とSnO2層とに接点を付け、試料を照射している間電流測定装置に示した装置に取り付けた。測定のために、適当な光学フィルターを用い波長が430nm未満の光を遮断した。さらに放射線の強度を約1000W/m2にほぼ一致するように装置を調整した。
金の層及びSnO2層に接点を付け、また試料を照射している間は両接点をポテンシオスタットに接続した。外部電圧をかけずに増感色素B-1を用いた試料では約90nAの電流を生じたが、本発明の色素化合物A-9を用いた試料では約190nAの電流を生じた。どちらの試料の場合も照射しないと電流は消失した。
金の層及びSnO2層に接点を付け、また試料を照射している間は両接点をポテンシオスタットに接続した。外部電圧をかけずに増感色素B-1を用いた試料では約90nAの電流を生じたが、本発明の色素化合物A-9を用いた試料では約190nAの電流を生じた。どちらの試料の場合も照射しないと電流は消失した。
[実験13]
特開2000-90989の実施例1と同様に作成したタンデムセルにおいても、比較色素B-1に比べ本発明の色素A-9では変換効率が高いことが確認できた。
特開2000-90989の実施例1と同様に作成したタンデムセルにおいても、比較色素B-1に比べ本発明の色素A-9では変換効率が高いことが確認できた。
[実験14]
チタンイソプロポキシド125mlを0.1M-硝酸水溶液(キシダ化学株式会社製)750mlに滴下し、80℃で8時間加熱して、加水分解反応をさせることにより、ゾル液を調製した。得られたゾル液をチタン製オートクレーブにて250℃で15時間保持し、粒子成長させ、その後、超音波分散を30分間行うことにより、平均一次粒径20nmの酸化チタン粒子を含むコロイド溶液を得た。
チタンイソプロポキシド125mlを0.1M-硝酸水溶液(キシダ化学株式会社製)750mlに滴下し、80℃で8時間加熱して、加水分解反応をさせることにより、ゾル液を調製した。得られたゾル液をチタン製オートクレーブにて250℃で15時間保持し、粒子成長させ、その後、超音波分散を30分間行うことにより、平均一次粒径20nmの酸化チタン粒子を含むコロイド溶液を得た。
得られた酸化チタン粒子を含むコロイド溶液を、エバポレーターにて、酸化チタンが10wt%の濃度になるまでゆっくりと濃縮した後、ポリエチレングレコール(キシダ化学株式会社製、重量平均分子量:200,000)を酸化チタンに対する重量比で40%添加し、攪拌することにより、酸化チタン粒子が分散した懸濁液を得た。
透明導電膜としてSnO2膜を形成したガラス基板1の透明導電膜側に、調製した酸化チタン懸濁液をドクターブレード法で塗布し、面積10mm×10mm程度の塗膜を得た。この塗膜を120℃で30分間予備乾燥し、さらに酸素雰囲気下、500℃で30分間焼成し、第1層多孔質光電変換層の第1層多孔質半導体層となる、膜厚が10μm程度の酸化チタン膜を形成した。
次に、市販の酸化チタン微粒子(テイカ社製、製品名:TITANIX JA-1、粒径約180nm)4.0gと酸化マグネシウム粉末(キシダ化学株式会社製)0.4gを蒸留水20mlに入れ、塩酸でpH=1に調整した。さらに、ジルコニアビーズを加え、この混合溶液をペイントシェイカーで8時間分散処理した。得られた分散液からジルコニアビーズを濾別した。その後、ポリエチレングレコール(キシダ化学株式会社製、重量平均分子量:200,000)を酸化チタンに対する重量比で40%添加し、攪拌することにより、酸化チタン粒子が分散した懸濁液を得た。
第1層多孔質半導体層の酸化チタン膜を形成したガラス基板1の第1層多孔質半導体層上に、調製した酸化チタン懸濁液をドクターブレード法で塗布し、塗膜を得た。この塗膜を80℃で20分間予備乾燥し、さらに酸素雰囲気下、約500℃で60分間焼成し、第2層多孔質光電変換層の第2層多孔質半導体層となる、膜厚が22μm程度の酸化チタン膜1を形成した。多孔質半導体層のへイズ率を測定したところ、84%であった。
吸収スペクトルにおける最大感度吸収波長領域を短波長側に有する色素(第1色素)として、下記式で表されるメロシアニン系色素S-2をエタノールに溶解して、濃度3×10-4モル/リットルの第1色素の吸着用色素溶液を調製した。

透明導電膜と多孔質半導体層を具備したガラス基板1を、約50℃に加温した第1色素の吸着用色素溶液に25℃で10分間浸漬させて、多孔質半導体層に第1色素を吸着させた。その後、ガラス基板1を無水エタノールで数回洗浄し、約60℃で約20分間乾燥させた。次いで、ガラス基板1を0.5N-塩酸に約10分間浸漬させ、その後エタノールで洗浄して、第2層多孔質半導体層に吸着された第1色素を脱着した。さらに、ガラス基板1を約60℃で約20分間乾燥させた。
次に、吸収スペクトルにおける最大感度吸収波長領域を長波長側に有する色素(第2色素)として、比較色素B-1、及び本発明の色素(A-9、A-15、A-25またはA-27)をエタノールに溶解して、濃度3×10-4モル/リットルの第2色素の吸着用色素溶液を調製した。
透明導電膜と第1色素を吸着した多孔質半導体層を具備したガラス基板1を、室温、常圧で第2色素の吸着用色素溶液に15分間浸漬させて、多孔質半導体層に第2色素を吸着させた。その後、ガラス基板1を無水エタノールで数回洗浄し、約60℃で約20分間乾燥させた。ここで多孔質半導体層のへイズ率を測定したところ、84%(B-1を使用した場合)、85%(本発明の色素を使用した場合)であった。
次に、3-メトキシプロピオニトリル溶媒に、ジメチルプロピルイミダゾリウムヨージドが濃度0.5モル/リットル、ヨウ化リチウムが濃度0.1モル/リットル、ヨウ素が濃度0.05モル/リットルになるように溶解させて、酸化還元性電解液を調製した。第1色素と第2色素を吸着させた多孔質半導体層を具備したガラス基板1の多孔質半導体層側と、対向電極層8として白金を具備したITOガラスからなる対極側支持体の白金側とが対向するように設置し、その間に調製した酸化還元性電解液を注入し、周囲をエポキシ系樹脂の封止材により封止して、色素増感型光電気化学電池を完成した。
また、第2層多孔質半導体層を第1多孔質半導体層と同じ層とする、すなわち第1多孔質半導体層を形成する酸化チタン懸濁液を用いて第2層多孔質半導体層を形成すること以外は、酸化チタン膜1と同様に酸化チタン膜2を作成し、これを用いて同様に光電気化学電池を作製し、評価した。多孔質光電変換層のヘイズ率は15%(B-1を使用した場合)、16%(本発明の色素を使用した場合)であった。
得られた光電気化学電池を測定条件:AM-1.5(100mW/cm2)で評価した結果を表13に示した。変換効率は、3.5%以上のものを◎、2.5%以上3.5%未満のものを○、2.0%以上2.5%未満のものを△、2.0%未満のものを×として表示した。

本発明の色素は光電変換効率に優れ、この系でも有効であることがわかる。
[実験15]
市販の酸化チタン粒子(テイカ株式会社製、平均粒径20nm)4.0gとジエチレングリコールモノメチルエーテル20mlとを、硬質ガラスビーズを使用してペイントシェイカーにより6時間分散させて酸化チタン懸濁液を作成した。次いで、得られた懸濁液から硬質ガラスビーズを濾別し、この酸化チタン懸濁液を、ドクターブレードを用いて、予め酸化スズ導電層を付着させたガラス板(電極層)に塗布し、100℃で30分予備乾燥した後、電気炉で500℃で40分間焼成し、ガラス板上に酸化チタン膜(半導体材料)を形成した。これとは別に、表14に示す本発明の増感色素及び比較色素をエタノールに溶解して光増感色素溶液を得た。
[実験15]
市販の酸化チタン粒子(テイカ株式会社製、平均粒径20nm)4.0gとジエチレングリコールモノメチルエーテル20mlとを、硬質ガラスビーズを使用してペイントシェイカーにより6時間分散させて酸化チタン懸濁液を作成した。次いで、得られた懸濁液から硬質ガラスビーズを濾別し、この酸化チタン懸濁液を、ドクターブレードを用いて、予め酸化スズ導電層を付着させたガラス板(電極層)に塗布し、100℃で30分予備乾燥した後、電気炉で500℃で40分間焼成し、ガラス板上に酸化チタン膜(半導体材料)を形成した。これとは別に、表14に示す本発明の増感色素及び比較色素をエタノールに溶解して光増感色素溶液を得た。
この光増感色素溶液の濃度はそれぞれ1×10-4モル/リットルであった(第2色素を併用する場合は、第1色素の濃度1×10-4モル/リットル、第2色素の濃度1×10-4モル/リットルとした)。次に、この溶液中に、膜状の酸化チタンが形成された前記のガラス板を入れ、60℃で60分間色素吸着を行った後、乾燥することにより、ガラス板上に半導体材料及び光増感色素からなる光電変換層を形成した(試料A)。前記試料Aの光電変換層上に、ホール輸送材料としてのポリビニルカルバゾール(重量平均分子量3,000)のトルエン溶液(1%)を塗布して、減圧乾燥してホール輸送層を形成した(試料B)。分子間電荷移動錯体としてのエチルカルバゾール1.95g及び5-ニトロナフトキノン2.03gを100mlアセトンに溶解して、得られた溶液を試料Bのホール輸送層上に繰り返し塗布して伝導層を形成した。次いで、伝導層上に金電極(対電極)を蒸着して光電変換素子を得た(試料C)。得られた光電変換素子(試料C)にソーラーシミュレーターで100W/m2の強度の光を照射して、変換率を測定した。結果を表14に示した。変換効率は、1.5%以上のものを◎、1.0%以上1.5%未満のものを○、0.5%以上1.0%未満のものを△、0.5%未満のものを×として表示した。

本発明の色素は光電変換効率に優れ、この系でも有効であることがわかる。
[実験16]
(1)第1光電変換層の形成
市販の酸化チタン粒子(テイカ株式会社製、平均粒径30nm)4.0gとジエチレングリコールモノメチルエーテル20mlを、硬質ガラスビーズを使用しペイントシェイカーにより6時間分散させ、酸化チタン懸濁液を作成した。次いで、この酸化チタン懸濁液をから硬質ガラスビーズを濾別し、ドクターブレードを用いて、予め酸化スズ導電層が付着されたガラス板に塗布し、100℃で30分予備乾燥した後、電気炉で500℃で40分間焼成し、酸化チタン膜を得た。
[実験16]
(1)第1光電変換層の形成
市販の酸化チタン粒子(テイカ株式会社製、平均粒径30nm)4.0gとジエチレングリコールモノメチルエーテル20mlを、硬質ガラスビーズを使用しペイントシェイカーにより6時間分散させ、酸化チタン懸濁液を作成した。次いで、この酸化チタン懸濁液をから硬質ガラスビーズを濾別し、ドクターブレードを用いて、予め酸化スズ導電層が付着されたガラス板に塗布し、100℃で30分予備乾燥した後、電気炉で500℃で40分間焼成し、酸化チタン膜を得た。
これとは別に、R-1をエタノールに溶解し、濃度を3×10-4mol/Lとした。次に、この溶液中に膜状の酸化チタンを形成した前記のガラス板を入れ、60℃で720分間色素吸着を行ってから乾燥し、第1光電変換層(試料A)を得た。
(2)第2光電変換層の形成
市販の酸化ニッケル粒子(キシダ化学社製、平均粒径100nm)4.0gとジエチレングリコールモノメチルエーテル20mlとを、ガラスビーズを使用してペイントシェイカーで8時間分散させ、酸化ニッケル懸濁液を作成した。次いで、この酸化ニッケル懸濁液からガラスビーズを濾別し、ドクターブレードを用いて、予め酸化スズ導電層が付着されたガラス板に塗布し、100℃で30分予備乾燥した後、電気炉で300℃で30分間焼成し、酸化ニッケル膜を得た。
市販の酸化ニッケル粒子(キシダ化学社製、平均粒径100nm)4.0gとジエチレングリコールモノメチルエーテル20mlとを、ガラスビーズを使用してペイントシェイカーで8時間分散させ、酸化ニッケル懸濁液を作成した。次いで、この酸化ニッケル懸濁液からガラスビーズを濾別し、ドクターブレードを用いて、予め酸化スズ導電層が付着されたガラス板に塗布し、100℃で30分予備乾燥した後、電気炉で300℃で30分間焼成し、酸化ニッケル膜を得た。
上記R-1とは別に、表15に示す本発明の色素及び比較色素B-3をジメチルスルホキシドに溶解した。
これらの色素の濃度をそれぞれ1×10-4mol/Lとした。次に、この溶液中に膜状の酸化ニッケルを形成した前記のガラス板を入れ、70℃で60分間色素吸着を行ってから乾燥し、本発明の第2光電変換層を形成し、試料Bを得た。
(3)光電変換素子の作成
前記の試料A上に試料Bを位置させた。これら2つの電極の間に液体電解質を入れ、この側面を樹脂で封止した後、リード線を取付けて、光電変換素子(素子構成C)を作成した。なお、液体電解質は、アセトニトリル/炭酸エチレンの混合溶媒(体積比が1:4)に、テトラプロピルアンモニウムアイオダイドとヨウ素とを、それぞれの濃度が0.46mol/L、0.06mol/Lとなるように溶解したものを用いた。
前記の試料A上に試料Bを位置させた。これら2つの電極の間に液体電解質を入れ、この側面を樹脂で封止した後、リード線を取付けて、光電変換素子(素子構成C)を作成した。なお、液体電解質は、アセトニトリル/炭酸エチレンの混合溶媒(体積比が1:4)に、テトラプロピルアンモニウムアイオダイドとヨウ素とを、それぞれの濃度が0.46mol/L、0.06mol/Lとなるように溶解したものを用いた。
また、前記の試料Aを一方の電極として備え、対極として白金を担持した透明導電性ガラス板を用いた。2つの電極の間に液体電解質を入れ、この側面を樹脂で封止した後、リード線を取付けて、光電変換素子(素子構成D)を作成した。
得られた光電変換素子にソーラーシミュレーターで1000W/m2の強度の光を照射し、変換効率を測定した。その結果を表15に示す。なお、変換効率は、6.5%以上のものを◎、6.0%以上6.5%未満のものを○、5.0%以上6.0%未満のものを△、5.0%未満のものを×として表示した。

本発明の色素を用いた光電気化学電池は光電変換効率に優れ、この系でも有効であることがわかる。
[実験17]
高分子電解質を用いた色素増感型光電気化学電池の作製した例について説明する。
[実験17]
高分子電解質を用いた色素増感型光電気化学電池の作製した例について説明する。
酸化チタン膜を作製する塗液は、市販の酸化チタン粒子(テイカ社製、商品名AMT-600、アナターゼ型結晶、平均粒径30nm、比表面積50m2/g)4.0gとジエチレングリコールモノメチルエーテル20mlとをガラスビーズを使用し、ペイントシェイカーで7時間分散させ、酸化チタン懸濁液を調製した。この酸化チタン懸濁液から、ガラスビーズを濾別し、ドクターブレードを用いて、11μm程度の膜厚、10mm×10mm程度の面積で、SnO2を透明導電膜としてガラス基板1上に作製された基板上に、透明導電膜側に塗布し、100℃で30分間予備乾燥した後、460℃で40分間酸素下で焼成し、その結果、膜厚が8μm程度の酸化チタン膜Aを作製した。
次に本発明の色素及び比較の色素B-3を無水エタノールに濃度3×10-4モル/リットルでそれぞれ溶解させ吸着用色素溶液を作製した。この吸着用色素溶液と、上述で得られた酸化チタン膜と透明導電膜を具備した透明基板とを容器にそれぞれ入れ、約4時間浸透させることにより色素を吸着させた。その後、無水エタノールで数回洗浄し約60℃で約20分間乾燥させた。
次に、下記一般式(105)で表されるメタクリレート系モノマー単位において、Rがメチル基であり、Aが8個のポリエチレンオキサイド基と2個のポリプロピレンオキサイド基と中心核としてブタンテトライル基とにより構成されるモノマー単位からなる高分子化合物を用意した。

(式中、Rはメチル基であり、Aはエステル基と炭素原子で結合している残基であり、nは2~4である。)
上記モノマー単位をプロピレンカーボネート(以下、PCと記載する)に20wt%の濃度で溶解させ、また、熱重合開始剤としてアゾビスイソブチロニトリル(AIBN)をモノマー単位に対して1wt%の濃度で溶解させモノマー溶液を作製した。このモノマー溶液を以下の方法で、前記の酸化チタン膜に含浸させた。
真空容器内にビーカー等の容器を設置し、その中に透明導電膜を具備した透明基板上の酸化チタン膜Aを入れ、ロータリーポンプで約10分間真空引きする。真空容器内を真空状態に保ちながらモノマー溶液をビーカー内に注入し、約15分間含浸させ酸化チタン中に単量体溶液を十分に染み込ませた。ポリエチレン製セパレーター、PETフィルムと押さえ板を設置し冶具にて固定した。その後、約85℃で30分間加熱することにより、熱重合させ、酸化チタン膜上に高分子化合物層を形成した。
次に、高分子化合物に含浸させる酸化還元性電解液を作製した。酸化還元性電解液は、PCを溶媒として濃度0.5モル/リットルのヨウ化リチウムと濃度0.05モル/リットルのヨウ素を溶解させて作製した。この溶液中に上述の酸化チタン膜Aに作製した高分子化合物を約2時間浸すことにより、高分子化合物中に酸化還元性電解液を染み込ませて高分子電解質を作製した。
その後、白金膜を具備した導電性基板を設置し、エポキシ系の封止剤にて周囲を封止し素子Aを作成した。
また、酸化チタン膜Aを色素吸着後、単量体処理を行わずに、PCを溶媒として濃度0.5モル/リットルのヨウ化リチウムと濃度0.05モル/リットルのヨウ素を溶解させて作製した酸化還元電解液をそのまま対極との間に注入して封止して素子Bを作成した。素子A、Bを用いて、ソーラーシミュレーターで1000W/m2の強度の光を照射して、変換効率を測定した。結果を表16に示す。
変換効率は、3.5%以上のものを◎、2.5%以上3.5%未満のものを○、2.0%以上2.5%未満のものを△、2.0%未満のものを×として表示した。
また、酸化チタン膜Aを色素吸着後、単量体処理を行わずに、PCを溶媒として濃度0.5モル/リットルのヨウ化リチウムと濃度0.05モル/リットルのヨウ素を溶解させて作製した酸化還元電解液をそのまま対極との間に注入して封止して素子Bを作成した。素子A、Bを用いて、ソーラーシミュレーターで1000W/m2の強度の光を照射して、変換効率を測定した。結果を表16に示す。
変換効率は、3.5%以上のものを◎、2.5%以上3.5%未満のものを○、2.0%以上2.5%未満のものを△、2.0%未満のものを×として表示した。

本発明の色素を用いた素子は光電変換効率に優れ、この系でも有効であることがわかる。
[実験18]
(光電変換素子の作製)
図1に示す光電変換素子10を以下のようにして作製した。
ガラス基板上に、透明導電膜としてフッ素をドープした酸化スズをスパッタリングにより形成し、これをレーザーでスクライブして、透明導電膜を2つの部分に分割した。
次に、水とアセトニトリルの容量比4:1からなる混合溶媒100mlにアナターゼ型酸化チタン(日本アエロジル社製のP-25(商品名))32gを配合し、自転/公転併用式のミキシングコンディショナーを使用して均一に分散、混合し、半導体微粒子分散液を得た。この分散液を透明導電膜に塗布し、500℃で加熱して受光電極を作製した。
その後、同様にシリカ粒子とルチル型酸化チタンとを40:60(質量比)で含有する分散液を作製し、この分散液を前記の受光電極に塗布し、500℃で加熱して絶縁性多孔体を形成した。次いで対極として炭素電極を形成した。
次に、下記表17に記載された増感色素(複数混合または単独)のエタノール溶液に、上記の絶縁性多孔体が形成されたガラス基板を5時間浸漬した。増感色素の染着したガラスを4-tert-ブチルピリジンの10%エタノール溶液に30分間浸漬した後、エタノールで洗浄し自然乾燥させた。このようにして得られる感光体層の厚さは10μmであり、半導体微粒子の塗布量は20g/m2であった。電解液は、ヨウ化ジメチルプロピルイミダゾリウム(0.5mol/L)、ヨウ素(0.1mol/L)のメトキシプロピオニトリル溶液を用いた。
[実験18]
(光電変換素子の作製)
図1に示す光電変換素子10を以下のようにして作製した。
ガラス基板上に、透明導電膜としてフッ素をドープした酸化スズをスパッタリングにより形成し、これをレーザーでスクライブして、透明導電膜を2つの部分に分割した。
次に、水とアセトニトリルの容量比4:1からなる混合溶媒100mlにアナターゼ型酸化チタン(日本アエロジル社製のP-25(商品名))32gを配合し、自転/公転併用式のミキシングコンディショナーを使用して均一に分散、混合し、半導体微粒子分散液を得た。この分散液を透明導電膜に塗布し、500℃で加熱して受光電極を作製した。
その後、同様にシリカ粒子とルチル型酸化チタンとを40:60(質量比)で含有する分散液を作製し、この分散液を前記の受光電極に塗布し、500℃で加熱して絶縁性多孔体を形成した。次いで対極として炭素電極を形成した。
次に、下記表17に記載された増感色素(複数混合または単独)のエタノール溶液に、上記の絶縁性多孔体が形成されたガラス基板を5時間浸漬した。増感色素の染着したガラスを4-tert-ブチルピリジンの10%エタノール溶液に30分間浸漬した後、エタノールで洗浄し自然乾燥させた。このようにして得られる感光体層の厚さは10μmであり、半導体微粒子の塗布量は20g/m2であった。電解液は、ヨウ化ジメチルプロピルイミダゾリウム(0.5mol/L)、ヨウ素(0.1mol/L)のメトキシプロピオニトリル溶液を用いた。
(変換効率の測定)
500Wのキセノンランプ(ウシオ製)の光をAM1.5Gフィルター(商品名、Oriel社製)及びシャープカットフィルター(KenkoL-42、商品名)を通すことにより紫外線を含まない模擬太陽光を発生させた。この光の強度は89mW/cm2であった。作製した光電変換素子にこの光を照射し、発生した電気を電流電圧測定装置(ケースレー238型、商品名)にて測定した。これにより求められた光電気化学電池の変換効率を測定した結果を下記表17に示した。結果は、変換効率が7.5%以上のものを◎、7.3%以上7.5%未満のものを○、7.1%以上7.3%未満のものを△、7.1%未満のものを×として評価した。
500Wのキセノンランプ(ウシオ製)の光をAM1.5Gフィルター(商品名、Oriel社製)及びシャープカットフィルター(KenkoL-42、商品名)を通すことにより紫外線を含まない模擬太陽光を発生させた。この光の強度は89mW/cm2であった。作製した光電変換素子にこの光を照射し、発生した電気を電流電圧測定装置(ケースレー238型、商品名)にて測定した。これにより求められた光電気化学電池の変換効率を測定した結果を下記表17に示した。結果は、変換効率が7.5%以上のものを◎、7.3%以上7.5%未満のものを○、7.1%以上7.3%未満のものを△、7.1%未満のものを×として評価した。

本発明の色素を用いて作製された電気化学電池は、表17に示されているように、本件の一般式(1)で表される色素と一般式(13)で表される色素の組み合わせを使用した場合は、変換効率は7.5%以上と高い値を示した。その他のそれに対して、比較例は、変換効率は7.1%未満と不十分であった。
[実験19]
1.二酸化チタン分散液の調製
内側をフッ素樹脂コーティングした内容積200mlのステンレス製容器に二酸化チタン微粒子(日本アエロジル(株)製,Degussa P-25)15g、水45g、分散剤(アルドリッチ社製、Triron X-100)1g、直径0.5mmのジルコニアビーズ(ニッカトー社製)30gを入れ、サンドグラインダーミル(アイメックス社製)を用いて1500rpmで2時間分散処理した。得られた分散液からジルコニアビーズを濾別した。得られた分散液中の二酸化チタン微粒子の平均粒径は2.5μmであった。なお粒径はMALVERN社製のマスターサイザー(商品名)により測定した。
1.二酸化チタン分散液の調製
内側をフッ素樹脂コーティングした内容積200mlのステンレス製容器に二酸化チタン微粒子(日本アエロジル(株)製,Degussa P-25)15g、水45g、分散剤(アルドリッチ社製、Triron X-100)1g、直径0.5mmのジルコニアビーズ(ニッカトー社製)30gを入れ、サンドグラインダーミル(アイメックス社製)を用いて1500rpmで2時間分散処理した。得られた分散液からジルコニアビーズを濾別した。得られた分散液中の二酸化チタン微粒子の平均粒径は2.5μmであった。なお粒径はMALVERN社製のマスターサイザー(商品名)により測定した。
2.色素を吸着した酸化チタン微粒子層(電極A)の作製
フッ素をドープした酸化スズを被覆した20mm×20mmの導電性ガラス板(旭ガラス(株)製,商品名:TCOガラス-U,表面抵抗:約30Ω/m2)を準備し、その導電層側の両端(端から3mmの幅の部分)にスペーサー用粘着テープを張った後で、導電層上にガラス棒を用いて上記分散液を塗布した。分散液の塗布後、粘着テープを剥離し、室温で1日間風乾した。次にこの半導体塗布ガラス板を電気炉(ヤマト科学(株)製マッフル炉FP-32型)に入れ、450℃で30分間焼成した。半導体塗布ガラス板を取り出し冷却した後、表18に示す色素のエタノール溶液(濃度:3×10-4mol/L)に3時間浸漬した。色素が吸着した半導体塗布ガラス板を4-tert-ブチルピリジンに15分間浸漬した後、エタノールで洗浄し、自然乾燥させて、色素を吸着した酸化チタン微粒子層(電極A)を得た。電極Aの色素増感酸化チタン微粒子層の厚さは10μmであり、酸化チタン微粒子の塗布量は20g/m2であった。また色素の吸着量は、その種類に応じて0.1~10mmol/m2の範囲内であった。
フッ素をドープした酸化スズを被覆した20mm×20mmの導電性ガラス板(旭ガラス(株)製,商品名:TCOガラス-U,表面抵抗:約30Ω/m2)を準備し、その導電層側の両端(端から3mmの幅の部分)にスペーサー用粘着テープを張った後で、導電層上にガラス棒を用いて上記分散液を塗布した。分散液の塗布後、粘着テープを剥離し、室温で1日間風乾した。次にこの半導体塗布ガラス板を電気炉(ヤマト科学(株)製マッフル炉FP-32型)に入れ、450℃で30分間焼成した。半導体塗布ガラス板を取り出し冷却した後、表18に示す色素のエタノール溶液(濃度:3×10-4mol/L)に3時間浸漬した。色素が吸着した半導体塗布ガラス板を4-tert-ブチルピリジンに15分間浸漬した後、エタノールで洗浄し、自然乾燥させて、色素を吸着した酸化チタン微粒子層(電極A)を得た。電極Aの色素増感酸化チタン微粒子層の厚さは10μmであり、酸化チタン微粒子の塗布量は20g/m2であった。また色素の吸着量は、その種類に応じて0.1~10mmol/m2の範囲内であった。
3.色素増感光電気化学電池の作製
上述のように作製した色素増感電極A(20mm×20mm)をこれと同じ大きさの白金蒸着ガラスと重ね合わせた。次に、両ガラスの隙間に毛細管現象を利用して電解質組成物を染み込ませ、電解質を酸化チタン電極中に導入した。これにより、図1に示すように、導電性ガラスからなる導電性支持体1(ガラスの透明基板上に導電層が設層されたもの)、感光体2、電荷移動体3、白金からなる対極4及びガラスの透明基板(図示せず)を順に積層しエポキシ系封止剤で封止した色素増感光電気化学電池を作製した。ただし、電解質組成物の粘度が高く毛細管現象を利用して電解質組成物を染み込ませることが困難な場合は、電解質組成物を50℃に加温し、これを酸化チタン電極に塗布した後、この電極を減圧下に置き電解質組成物が十分浸透し電極中の空気が抜けた後、白金蒸着ガラス(対極)を重ね合わせて同様に色素増感光電気化学電池を作製した。
上述のように作製した色素増感電極A(20mm×20mm)をこれと同じ大きさの白金蒸着ガラスと重ね合わせた。次に、両ガラスの隙間に毛細管現象を利用して電解質組成物を染み込ませ、電解質を酸化チタン電極中に導入した。これにより、図1に示すように、導電性ガラスからなる導電性支持体1(ガラスの透明基板上に導電層が設層されたもの)、感光体2、電荷移動体3、白金からなる対極4及びガラスの透明基板(図示せず)を順に積層しエポキシ系封止剤で封止した色素増感光電気化学電池を作製した。ただし、電解質組成物の粘度が高く毛細管現象を利用して電解質組成物を染み込ませることが困難な場合は、電解質組成物を50℃に加温し、これを酸化チタン電極に塗布した後、この電極を減圧下に置き電解質組成物が十分浸透し電極中の空気が抜けた後、白金蒸着ガラス(対極)を重ね合わせて同様に色素増感光電気化学電池を作製した。
色素を変更して上述の工程を行い、試料番号18-1~18-9の色素増感光電気化学電池を作製した。各色素増感光電気化学電池に用いた電解質組成物としては、下記のヘテロ環4級塩化合物を98質量%及びヨウ素を2質量%含有するものを用いた。

4.光電変換効率の測定
500Wのキセノンランプ(ウシオ電機社(株)製)の光をAM1.5フィルター(Oriel社製)及びシャープカットフィルター(Kenko L-37)ことにより紫外線を含まない模擬太陽光を発生させた。この光の強度は70mW/cm2であった。この模擬太陽光を、50℃で色素増感光電気化学電池に照射し、発生した電気を電流電圧測定装置(ケースレーSMU238型)で測定した。
また、85℃で1000時間暗所保存後の変換効率の低下率、及び500時間連続光照射後の変換効率の低下率も測定した。
これらの結果を表18に示す。
500Wのキセノンランプ(ウシオ電機社(株)製)の光をAM1.5フィルター(Oriel社製)及びシャープカットフィルター(Kenko L-37)ことにより紫外線を含まない模擬太陽光を発生させた。この光の強度は70mW/cm2であった。この模擬太陽光を、50℃で色素増感光電気化学電池に照射し、発生した電気を電流電圧測定装置(ケースレーSMU238型)で測定した。
また、85℃で1000時間暗所保存後の変換効率の低下率、及び500時間連続光照射後の変換効率の低下率も測定した。
これらの結果を表18に示す。

表18より、本発明の色素増感光電気化学電池は、比較例に比べて耐久性が向上していることがわかった。
[実験20]
下記の方法に従って、色素増感光電気化学電池を作製し、評価した。その結果を表19に示す。
(1)透明導電性支持体の作製
感光性電極用支持体として、表面がフッ素コートされた厚さ0.4mmのシートの片面に、導電性の酸化スズの薄膜を厚さ200nmで均一にコーティングして可撓性のある透明導電性支持体を使用した。
下記の方法に従って、色素増感光電気化学電池を作製し、評価した。その結果を表19に示す。
(1)透明導電性支持体の作製
感光性電極用支持体として、表面がフッ素コートされた厚さ0.4mmのシートの片面に、導電性の酸化スズの薄膜を厚さ200nmで均一にコーティングして可撓性のある透明導電性支持体を使用した。
(2)対極用の導電性シートの作製
厚さ0.4mmのポリイミド製カプトン(登録商標)フィルムの片面に、真空スパッタリング法によって厚さ300nmの白金膜で均一に被覆した。面抵抗は5Ω/cm2であった。
厚さ0.4mmのポリイミド製カプトン(登録商標)フィルムの片面に、真空スパッタリング法によって厚さ300nmの白金膜で均一に被覆した。面抵抗は5Ω/cm2であった。
(3)半導体微粒子分散液の調製
C.J.BarbeらのJ.Am.Ceramic Soc.80巻、p.3157の論文に記載の製造方法に従い、チタン原料にチタニウムテトライソプロポキシドを用い、オートクレーブ中での重合反応の温度を230℃に設定して、二酸化チタン濃度11質量%のアナターゼ型二酸化チタンの分散液を合成した。得られた二酸化チタン粒子の一次粒子のサイズは10~30nmであった。得られた分散液を、超遠心分離機にかけて、粒子を分離し、凝集物を乾燥した後、メノウ乳鉢上で粉砕して白色粉末の半導体微粒子aを得た。水とアセトニトリルの容量比4:1からなる混合溶媒100mlに、半導体微粒子aを溶媒100mlあたり32gの濃度で添加し、自転/公転併用式のミキシングコンディショナーを使って均一に分散、混合した。この結果、得られた白色の半導体微粒子分散液は、50~150N・s/m2の高粘度のペースト状となり、このまま塗布に用いるのに適した液物性をもっていることがわかった。試料番号19-3、19-10では、平均分子量が50万のポリエチレングリコール(PEG)の粉末を、溶媒100ml当たり7.7g配合した。その他の半導体微粒子分散液には、半導体微粒子以外の固形分は加えなかった。
C.J.BarbeらのJ.Am.Ceramic Soc.80巻、p.3157の論文に記載の製造方法に従い、チタン原料にチタニウムテトライソプロポキシドを用い、オートクレーブ中での重合反応の温度を230℃に設定して、二酸化チタン濃度11質量%のアナターゼ型二酸化チタンの分散液を合成した。得られた二酸化チタン粒子の一次粒子のサイズは10~30nmであった。得られた分散液を、超遠心分離機にかけて、粒子を分離し、凝集物を乾燥した後、メノウ乳鉢上で粉砕して白色粉末の半導体微粒子aを得た。水とアセトニトリルの容量比4:1からなる混合溶媒100mlに、半導体微粒子aを溶媒100mlあたり32gの濃度で添加し、自転/公転併用式のミキシングコンディショナーを使って均一に分散、混合した。この結果、得られた白色の半導体微粒子分散液は、50~150N・s/m2の高粘度のペースト状となり、このまま塗布に用いるのに適した液物性をもっていることがわかった。試料番号19-3、19-10では、平均分子量が50万のポリエチレングリコール(PEG)の粉末を、溶媒100ml当たり7.7g配合した。その他の半導体微粒子分散液には、半導体微粒子以外の固形分は加えなかった。
(4)半導体微粒子分散液中の固形分の測定
厚さ1.9mmの無アルカリガラスの基板に分散液をアプリケーターで塗布し、40~70μmの厚さで塗布し、室温で1時間乾燥させた。その後、空気中、350℃で0.5時間加熱し、加熱前後の重量変化を測定したところ、前記試料番号19-3、19-10の半導体微粒子以外の固形分含量は1%であった。それ以外試料の半導体微粒子以外の固形分含量は、0.3%であった。
厚さ1.9mmの無アルカリガラスの基板に分散液をアプリケーターで塗布し、40~70μmの厚さで塗布し、室温で1時間乾燥させた。その後、空気中、350℃で0.5時間加熱し、加熱前後の重量変化を測定したところ、前記試料番号19-3、19-10の半導体微粒子以外の固形分含量は1%であった。それ以外試料の半導体微粒子以外の固形分含量は、0.3%であった。
(5)半導体微粒子層の作製
(1)で用意した透明導電性支持体に、(3)で調製した分散液をアプリケータで塗布し、室温下で1時間乾燥させることにより、40~70μmの均一な厚さの塗布層を形成した。さらに、この塗布層を表19記載の条件で処理して、色素増感のための多孔質半導体微粒子層を作製した。多孔質半導体微粒子層の最終的な平均膜厚は、いずれも6~7μmであった。
(1)で用意した透明導電性支持体に、(3)で調製した分散液をアプリケータで塗布し、室温下で1時間乾燥させることにより、40~70μmの均一な厚さの塗布層を形成した。さらに、この塗布層を表19記載の条件で処理して、色素増感のための多孔質半導体微粒子層を作製した。多孔質半導体微粒子層の最終的な平均膜厚は、いずれも6~7μmであった。
(6)色素吸着溶液の調製
表19に示した色素を乾燥したアセトニトリル:t-ブタノール:エタノールを体積比で2:1:1の混合溶媒に、色素濃度が3×10-4モル/リットルとなるように溶解した。この色素溶液に添加剤として、p-C9H19-C6H4-O-(CH2CH2-O)3-(CH2)4-SO3Naの構造の有機スルホン酸誘導体を0.025モル/リットルの濃度となるように溶解して、色素吸着用溶液を調製した。
表19に示した色素を乾燥したアセトニトリル:t-ブタノール:エタノールを体積比で2:1:1の混合溶媒に、色素濃度が3×10-4モル/リットルとなるように溶解した。この色素溶液に添加剤として、p-C9H19-C6H4-O-(CH2CH2-O)3-(CH2)4-SO3Naの構造の有機スルホン酸誘導体を0.025モル/リットルの濃度となるように溶解して、色素吸着用溶液を調製した。
(7)色素の吸着
上記の多孔質半導体微粒子層を塗設した基板を、上記の吸着用色素溶液に浸漬して、攪拌下40℃で3時間放置した。
このようにして半導体微粒子層に色素を吸着させ、感光層に用いる色素増感電極(感光性電極)を作製した。
上記の多孔質半導体微粒子層を塗設した基板を、上記の吸着用色素溶液に浸漬して、攪拌下40℃で3時間放置した。
このようにして半導体微粒子層に色素を吸着させ、感光層に用いる色素増感電極(感光性電極)を作製した。
(8)色素増感光電気化学電池の作製
色素吸着した多孔質半導体微粒子層をかき落として、受光面積1.0cm2(直径約1.1cm)の円型の感光性電極を形成した。この電極に対して、対極の白金蒸着ガラス基板を、熱圧着性のポリエチレンフイルム製のフレーム型スペーサー(厚さ20μm)を挿入して重ね合わせ、スペーサー部分を120℃に加熱し両基板を圧着した。さらにセルのエッジ部をエポキシ樹脂接着剤でシールした。対極の基板のコーナー部にあらかじめ設けた電解液注液用の小孔を通して、電解液として、後述するいずれかのイミダゾリウムイオンE1~E4/ヨウ素=50:1(質量比)の組成から成る室温溶融塩を基板の小孔から毛細管現象を利用して電極間の空間にしみこませた。
E1:1,2-ジメチル-3-プロピルイミダゾリウムヨージド
E2:1-ブチル-3-メチルイミダゾリウムヨージド
E3:1-メチル-3-プロピルイミダゾリウムヨージド
E4:1,3-ジ(2-(2-(2-メトキシエトキシ)エトキシ)エチル)イミダゾリウムヨージド
以上のセル組立工程と、電解液注入の工程をすべて上記の露点-60℃の乾燥空気中で実施した。溶融塩の注入後、真空下でセルを数時間吸引し感光性電極および溶融塩を含めたセル内部の脱気を行い、最終的に小孔を低融点ガラスで封じた。これにより、導電性支持体、色素が吸着された多孔質半導体微粒子電極(感光性電極)、電解液、対極および支持体が順に積層された色素増感光電気化学電池を作製した。
色素吸着した多孔質半導体微粒子層をかき落として、受光面積1.0cm2(直径約1.1cm)の円型の感光性電極を形成した。この電極に対して、対極の白金蒸着ガラス基板を、熱圧着性のポリエチレンフイルム製のフレーム型スペーサー(厚さ20μm)を挿入して重ね合わせ、スペーサー部分を120℃に加熱し両基板を圧着した。さらにセルのエッジ部をエポキシ樹脂接着剤でシールした。対極の基板のコーナー部にあらかじめ設けた電解液注液用の小孔を通して、電解液として、後述するいずれかのイミダゾリウムイオンE1~E4/ヨウ素=50:1(質量比)の組成から成る室温溶融塩を基板の小孔から毛細管現象を利用して電極間の空間にしみこませた。
E1:1,2-ジメチル-3-プロピルイミダゾリウムヨージド
E2:1-ブチル-3-メチルイミダゾリウムヨージド
E3:1-メチル-3-プロピルイミダゾリウムヨージド
E4:1,3-ジ(2-(2-(2-メトキシエトキシ)エトキシ)エチル)イミダゾリウムヨージド
以上のセル組立工程と、電解液注入の工程をすべて上記の露点-60℃の乾燥空気中で実施した。溶融塩の注入後、真空下でセルを数時間吸引し感光性電極および溶融塩を含めたセル内部の脱気を行い、最終的に小孔を低融点ガラスで封じた。これにより、導電性支持体、色素が吸着された多孔質半導体微粒子電極(感光性電極)、電解液、対極および支持体が順に積層された色素増感光電気化学電池を作製した。
(9)色素増感光電気化学電池の評価
500Wのキセノンランプ(ウシオ電機社製)に太陽光シミュレーション用補正フィルター(Oriel社製AM1.5direct(商品名))を装着し、上記色素増感光電気化学電池に対し、入射光強度が100mW/cm2の模擬太陽光を、多孔質半導体微粒子電極(感光性電極)の側から照射した。素子は恒温装置のステージ上に密着して固定し、照射中の素子の温度を50℃に制御した。電流電圧測定装置(ケースレー社製ソースメジャーユニット238型(商品名))を用いて、素子に印加するDC電圧を10mV/秒の定速でスキャンし、素子の出力する光電流を計測することにより、光電流-電圧特性を測定した。これにより求められた上記の各種素子のエネルギー変換効率(η)を、セルの構成要素(半導体微粒子、増感色素)の内容とともに表19に記載した。変換効率が6.0%以上のものをA、5%以上6%未満のものをB、4%以上5%未満のものをC、3%以上4%未満のものをD、1.5%以上3%未満のものをE、1.5%未満のものをFとして表示し、変換効率D以上のものを合格とし、D未満のものを不合格とした。120時間連続光照射後の変換効率の低下率も測定した。これらの結果を表19に示す。
500Wのキセノンランプ(ウシオ電機社製)に太陽光シミュレーション用補正フィルター(Oriel社製AM1.5direct(商品名))を装着し、上記色素増感光電気化学電池に対し、入射光強度が100mW/cm2の模擬太陽光を、多孔質半導体微粒子電極(感光性電極)の側から照射した。素子は恒温装置のステージ上に密着して固定し、照射中の素子の温度を50℃に制御した。電流電圧測定装置(ケースレー社製ソースメジャーユニット238型(商品名))を用いて、素子に印加するDC電圧を10mV/秒の定速でスキャンし、素子の出力する光電流を計測することにより、光電流-電圧特性を測定した。これにより求められた上記の各種素子のエネルギー変換効率(η)を、セルの構成要素(半導体微粒子、増感色素)の内容とともに表19に記載した。変換効率が6.0%以上のものをA、5%以上6%未満のものをB、4%以上5%未満のものをC、3%以上4%未満のものをD、1.5%以上3%未満のものをE、1.5%未満のものをFとして表示し、変換効率D以上のものを合格とし、D未満のものを不合格とした。120時間連続光照射後の変換効率の低下率も測定した。これらの結果を表19に示す。

表19に示すように、導電性高分子製の導電性支持体に本発明の色素を吸着させた多孔質半導体微粒子層を形成した場合に、実用レベルの光電変換効率を有する色素増感光電気化学電池が得られた。特に半導体微粒子以外の固形分含量が0.3%の分散液を支持体に塗布し、熱処理を120~150℃で行いその後紫外線照射し、その後本発明の色素を吸着させて多孔質半導体微粒子層を作製した場合は、光電変換効率が高くなった。
また、固形分の含量が1.0質量%の分散液を導電性高分子製の支持体に塗布し加熱することにより多孔質半導体微粒子層を作製し、本発明の色素を吸着させた場合も、比較色素を吸着させた場合と比較して、高い変換効率の色素増感光電気化学電池が得られることがわかった。さらに比較色素を用いた色素増感光電気化学電池の場合は、連続光照射後の変換効率の低下率が35%以上と高くなったのに対し、本発明の色素を用いた色素増感光電気化学電池の場合は、連続光照射後の変換効率の低下率が20%以下で、耐久性に優れることがわかった。
また、固形分の含量が1.0質量%の分散液を導電性高分子製の支持体に塗布し加熱することにより多孔質半導体微粒子層を作製し、本発明の色素を吸着させた場合も、比較色素を吸着させた場合と比較して、高い変換効率の色素増感光電気化学電池が得られることがわかった。さらに比較色素を用いた色素増感光電気化学電池の場合は、連続光照射後の変換効率の低下率が35%以上と高くなったのに対し、本発明の色素を用いた色素増感光電気化学電池の場合は、連続光照射後の変換効率の低下率が20%以下で、耐久性に優れることがわかった。
[実験21]
[実験19]のエポキシ系封止剤として、エピコート828((商品名)、ジャパンエポキシレジン社製)、硬化剤及びプラスチックペーストからなる樹脂組成物中に直径25μmのガラス球体がほぼ均一に分散された封止剤ペーストを用いたこと以外は同様にして、色素増感光電気化学電池を作製し、光電変換効率の測定を行った。変換効率が6.0%以上のものをA、5%以上6%未満のものをB、4%以上5%未満のものをC、3%以上4%未満のものをD、1.5%以上3%未満のものをE、1.5%未満のものをFとして表示し、変換効率D以上のものを合格とし、D未満のものを不合格とした。
これにより求めた各色素増感太陽学電池の変換効率(η)、初期値に対する85℃で1000時間暗所保存後の変換効率の低下率、及び初期値に対する500時間連続光照射後の変換効率の低下率を表20に示す。
[実験19]のエポキシ系封止剤として、エピコート828((商品名)、ジャパンエポキシレジン社製)、硬化剤及びプラスチックペーストからなる樹脂組成物中に直径25μmのガラス球体がほぼ均一に分散された封止剤ペーストを用いたこと以外は同様にして、色素増感光電気化学電池を作製し、光電変換効率の測定を行った。変換効率が6.0%以上のものをA、5%以上6%未満のものをB、4%以上5%未満のものをC、3%以上4%未満のものをD、1.5%以上3%未満のものをE、1.5%未満のものをFとして表示し、変換効率D以上のものを合格とし、D未満のものを不合格とした。
これにより求めた各色素増感太陽学電池の変換効率(η)、初期値に対する85℃で1000時間暗所保存後の変換効率の低下率、及び初期値に対する500時間連続光照射後の変換効率の低下率を表20に示す。

表20より、本発明の色素増感光電気化学電池は、変換効率の初期値はいずれも7.0%以上と高い値を示した。また、暗所保存後及び連続光照射後において、低下率は15%以下および20%以下であり、比較例に比べて耐久性が優れていることがわかった。
本発明をその実施態様とともに説明したが、我々は特に指定しない限り我々の発明を説明のどの細部においても限定しようとするものでなく、添付の請求項の範囲に示した発明の精神と範囲に反することなく幅広く解釈されるべきであると考える。
本願は、2010年8月3日に日本で特許出願された特願2010-174833に基づく優先権を主張するものであり、これはここに参照してその内容を本明細書の記載の一部として取り込む。
1 導電性支持体
2 感光体層
21 色素
22 半導体微粒子
3 電荷移動体層
4 対極
5 受光電極
6 回路
10 光電変換素子
100 光電気化学電池
2 感光体層
21 色素
22 半導体微粒子
3 電荷移動体層
4 対極
5 受光電極
6 回路
10 光電変換素子
100 光電気化学電池
Claims (15)
- 下記一般式(1)で表される金属錯体色素。
M(LL1)m1(LL2)m2(X)m3・CI 一般式(1)
[一般式(1)において、
Mは金属原子を表し、
LL1は下記一般式(2)で表される2座または3座の配位子を表し、
LL2は下記一般式(7)で表される2座または3座の配位子を表し、
Xはアシルオキシ基、アシルチオ基、チオアシルオキシ基、チオアシルチオ基、アシルアミノオキシ基、チオカルバメート基、ジチオカルバメート基、チオカルボネート基、ジチオカルボネート基、トリチオカルボネート基、アシル基、チオシアネート基、イソチオシアネート基、シアネート基、イソシアネート基、シアノ基、アルキルチオ基、アリールチオ基、アルコキシ基およびアリールオキシ基からなる群から選ばれた基で配位する1座または2座の配位子、あるいはハロゲン原子、カルボニル、ジアルキルケトン、1,3-ジケトン、カルボンアミド、チオカルボンアミドまたはチオ尿素からなる1座または2座の配位子を表し、
m1は1~3の整数を表し、m1が2以上のときLL1は同じでも異なっていてもよく、
m2は0~2の整数を表し、m2が2のときLL2は同じでも異なっていてもよく、
m3は0~3の整数を表し、m3が2以上のときXは同じでも異なっていてもよく、またX同士が連結していてもよく、
CIは電荷を中和させるのに対イオンが必要な場合の対イオンを表す。
LL1、LL2、Xのいずれか1つは、少なくとも1つの酸性基を有する。

R1およびR2はそれぞれ独立に酸性基を表し、
R3およびR4はそれぞれ独立に置換基を表し、
Y1およびY2はそれぞれ独立に一般式(3)~(6)のいずれかで表される環より水素原子を2つ脱離して得られる置換基を表し、
Y3およびY4は水素原子または置換基を表す。
L1またはL2はそれぞれ独立にアルキニレン、アリーレンまたはそれらの組み合わせからなる共役鎖を表す。
a1およびa2はそれぞれ独立に0~3の整数を表し、a1が2以上のときR1は同じでも異なっていてもよく、a2が2以上のときR2は同じでも異なっていてもよい。
b1およびb2はそれぞれ独立に0~3の整数を表し、b1が2以上のときR3は同じでも異なっていてもよく、互いに連結して環を形成してもよく、b2が2以上のときR4は同じでも異なっていてもよく、互いに連結して環を形成してもよく、b1およびb2が共に1以上のときR3とR4が連結して環を形成してもよい。
n1およびn2はそれぞれ独立に1以上の整数を表し、n3、n4はそれぞれ独立に1以上の整数を表す。ただし、n3が2以上の場合、複数のY1は同じでも異なってもよく、n4が2以上の場合、複数のY2は同じでも異なっても良い。
zは0または1を表す。


- 前記一般式(1)において、前記LL1が一般式(2A)で表される2座または3座の配位子である請求項1記載の金属錯体色素。

- 前記一般式(2)において、L1、L2がアリーレンを表す請求項1記載の金属錯体色素。
- 前記一般式(2A)において、L1、L2がアリーレンを表す請求項2記載の金属錯体色素。
- 前記一般式(1)において、LL1が下記一般式(8)で表される2座または3座の配位子である請求項1または3に記載の金属錯体色素。

- 前記一般式(1)において、LL1が下記一般式(8A)で表される2座または3座の配位子である請求項1~5のいずれか1項に記載の金属錯体色素。

- 前記一般式(1)において、Mがルテニウムを表す請求項1~6のいずれか1項に記載の金属錯体色素。
- 前記一般式(3)で表される環より水素原子を2つ脱離して得られる置換基が下記一般式(3A)で表される請求項1~7項のいずれか1項に記載の金属錯体色素。

- 前記一般式(2)において、Y1、Y2が下記一般式(9)~(12)のいずれかで表される請求項1~8項のいずれか1項に記載の金属錯体色素。

- 前記一般式(9)~(12)のいずれかで表される置換基が少なくとも一つ以上の炭素原子数5以上の脂肪族基を持つ請求項9記載の金属錯体色素。
- 請求項1~10のいずれか1項に記載の金属錯体色素と半導体微粒子とを有する感光層を具備した、光電気変換素子。
- 前記一般式(1)記載の金属錯体色素と他の色素を組み合わせて用いる請求項11記載の光電変換素子。
- 前記他の色素が一般式(13)で表される請求項12に記載の光電変換素子。
Mz(LL11)m11(LL12)m12(X11)m13・CI11 一般式(13)
[一般式(13)において、
Mzは金属原子を表し、
LL11は下記一般式(14)で表される2座又は3座の配位子を表し、
LL12は下記一般式(15)で表される2座又は3座の配位子を表し、
X11はアシルオキシ基、アシルチオ基、チオアシルオキシ基、チオアシルチオ基、アシルアミノオキシ基、チオカルバメート基、ジチオカルバメート基、チオカルボネート基、ジチオカルボネート基、トリチオカルボネート基、アシル基、チオシアネート基、イソチオシアネート基、シアネート基、イソシアネート基、シアノ基、アルキルチオ基、アリールチオ基、アルコキシ基およびアリールオキシ基からなる群から選ばれた基で配位する1座又は2座の配位子、あるいはハロゲン原子、カルボニル、ジアルキルケトン、1,3-ジケトン、カルボンアミド、チオカルボンアミドまたはチオ尿素からなる1座または2座の配位子を表し、
m11は0~3の整数を表し、m11が2以上のとき、LL11は同じでも異なっていてもよく、
m12は0~2の整数を表し、m12が2のとき、LL12は同じでも異なっていてもよい。ただし、m11とm12のうち少なくとも一方は1以上の整数である。
m13は0~3の整数を表し、m13が2以上のとき、X11は同じでも異なっていてもよく、X11同士が連結していてもよい。
CI11は一般式(13)において、電荷を中和させるのに対イオンが必要な場合の対イオンを表す。
LL11、LL12、X11のいずれか1つは、少なくとも1つの酸性基を有する。]

R101及びR102はそれぞれ独立に酸性基を表し、
R103及びR104はそれぞれ独立に置換基を表し、
R105及びR106はそれぞれ独立にアルキル基、アリール基、又はヘテロ環基を表す。
L11及びL12はそれぞれ独立に、アリーレン基、ヘテロアリーレン基、エテニレン基及びエチニレン基から選ばれた少なくとも1種からなる共役鎖を表す。
a11及びa12はそれぞれ独立に0~3の整数を表し、a11が2以上のときR101は同じでも異なっていてもよく、a12が2以上のときR102は同じでも異なっていてもよい。
b11及びb12はそれぞれ独立に0~3の整数を表し、b11が2以上のときR103は同じでも異なっていてもよく、R103は互いに連結して環を形成してもよく、b12が2以上のときR104は同じでも異なっていてもよく、R104は互いに連結して環を形成してもよい。b11及びb12が共に1以上のとき、R103とR104が連結して環を形成してもよい。
d11及びd12はそれぞれ独立に0~5の整数を表す。
d13は0または1を表す。

- 導電性支持体上に、前記感光体層、電荷移動体及び対極をこの順序で積層した構造を有する、請求項11~13のいずれか1項に記載の光電変換素子。
- 請求項11~14のいずれか1項記載の光電変換素子を備える光電気化学電池。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012527679A JP5869481B2 (ja) | 2010-08-03 | 2011-07-26 | 金属錯体色素、光電変換素子及び光電気化学電池 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010174833 | 2010-08-03 | ||
| JP2010-174833 | 2010-08-03 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012017872A1 true WO2012017872A1 (ja) | 2012-02-09 |
Family
ID=45559374
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/067009 WO2012017872A1 (ja) | 2010-08-03 | 2011-07-26 | 金属錯体色素、光電変換素子及び光電気化学電池 |
Country Status (3)
| Country | Link |
|---|---|
| JP (1) | JP5869481B2 (ja) |
| TW (1) | TW201211165A (ja) |
| WO (1) | WO2012017872A1 (ja) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102660132A (zh) * | 2012-04-25 | 2012-09-12 | 天津大学 | 单边不对称结构的三苯胺桥接联吡啶钌配合物光敏染料及制备方法 |
| CN102660135A (zh) * | 2012-04-25 | 2012-09-12 | 天津大学 | 三苯胺桥接联吡啶钌配合物光敏染料及制备方法 |
| GB2518837A (en) * | 2013-10-01 | 2015-04-08 | G24 Power Ltd | Electrolyte and photovoltaic cell comprising the same |
| JP2015093937A (ja) * | 2013-11-12 | 2015-05-18 | 株式会社豊田中央研究所 | 金属錯体、色素増感型太陽電池、色素増感型太陽電池モジュール、金属錯体の製造方法及び配位子 |
| WO2016006511A1 (ja) * | 2014-07-07 | 2016-01-14 | 富士フイルム株式会社 | 光電変換素子、色素増感太陽電池、金属錯体色素、色素溶液、およびターピリジン化合物またはそのエステル化物 |
| WO2016006512A1 (ja) * | 2014-07-07 | 2016-01-14 | 富士フイルム株式会社 | 光電変換素子、色素増感太陽電池、金属錯体色素、色素溶液、およびターピリジン化合物またはそのエステル化物 |
| WO2016148100A1 (ja) * | 2015-03-17 | 2016-09-22 | 富士フイルム株式会社 | ルテニウム錯体色素、色素溶液、光電変換素子および色素増感太陽電池 |
| CN106463270A (zh) * | 2014-06-11 | 2017-02-22 | 富士胶片株式会社 | 光电转换元件、色素增感太阳能电池、金属络合物色素和色素溶液 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008174734A (ja) * | 2006-12-18 | 2008-07-31 | Sumitomo Chemical Co Ltd | 化合物、光電変換素子及び光電気化学電池 |
| JP2009215539A (ja) * | 2008-02-19 | 2009-09-24 | National Central Univ | 感光剤染料 |
| WO2010055471A1 (en) * | 2008-11-11 | 2010-05-20 | Ecole Polytechnique Federale De Lausanne (Epfl) | Novel anchoring ligands for sensitizers of dye-sensitized photovoltaic devices |
-
2011
- 2011-07-26 JP JP2012527679A patent/JP5869481B2/ja not_active Expired - Fee Related
- 2011-07-26 WO PCT/JP2011/067009 patent/WO2012017872A1/ja active Application Filing
- 2011-08-02 TW TW100127418A patent/TW201211165A/zh unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008174734A (ja) * | 2006-12-18 | 2008-07-31 | Sumitomo Chemical Co Ltd | 化合物、光電変換素子及び光電気化学電池 |
| JP2009215539A (ja) * | 2008-02-19 | 2009-09-24 | National Central Univ | 感光剤染料 |
| WO2010055471A1 (en) * | 2008-11-11 | 2010-05-20 | Ecole Polytechnique Federale De Lausanne (Epfl) | Novel anchoring ligands for sensitizers of dye-sensitized photovoltaic devices |
Non-Patent Citations (4)
| Title |
|---|
| ANDREA BARBIERI ET AL.: "Mononuclear and Binuclear Wirelike Ruthenium(II) Complexes with Oligo-diethynyl-thiophene Bridged Back- to-Back Terpyridine Ligands: Synthesis and Electrochemical and Photophysical Properties", INORGANIC CHEMISTRY, vol. 43, no. 23, 2004, pages 7359 - 7368, XP055234638, DOI: doi:10.1021/ic0493043 * |
| ANTHONY HARRIMAN ET AL.: "Photophysical Properties of Ruthenium(II) Tris (2,2'- bipyridine) Complexes Bearing Conjugated Thiophene Appendages", INORGANIC CHEMISTRY, vol. 45, no. 24, 2006, pages 9729 - 9741 * |
| SEBASTIEN GOEB ET AL.: "Energy Transfer in Hybrids Based on a Thiophene-Substituted Ethynylbipyridine Dimer Decorated with Re(I), Ru(II), and Os(II) Units", INORGANIC CHEMISTRY, vol. 45, no. 3, 2006, pages 1173 - 1183 * |
| SIMON J. DUNNE ET AL.: "Stille cross-coupling of metal complexes", INORGANIC CHEMISTRY COMMUNICATIONS, vol. 1, no. 5, 1998, pages 167 - 169 * |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102660132A (zh) * | 2012-04-25 | 2012-09-12 | 天津大学 | 单边不对称结构的三苯胺桥接联吡啶钌配合物光敏染料及制备方法 |
| CN102660135A (zh) * | 2012-04-25 | 2012-09-12 | 天津大学 | 三苯胺桥接联吡啶钌配合物光敏染料及制备方法 |
| GB2518837A (en) * | 2013-10-01 | 2015-04-08 | G24 Power Ltd | Electrolyte and photovoltaic cell comprising the same |
| JP2015093937A (ja) * | 2013-11-12 | 2015-05-18 | 株式会社豊田中央研究所 | 金属錯体、色素増感型太陽電池、色素増感型太陽電池モジュール、金属錯体の製造方法及び配位子 |
| TWI661010B (zh) * | 2014-06-11 | 2019-06-01 | 日商富士軟片股份有限公司 | 光電轉換組件、色素增感太陽能電池、金屬絡合物色素和色素溶液 |
| EP3157028A4 (en) * | 2014-06-11 | 2017-05-31 | Fujifilm Corporation | Photoelectric conversion element, dye-sensitized solar cell, metal-complex pigment, and pigment solution |
| CN106463270A (zh) * | 2014-06-11 | 2017-02-22 | 富士胶片株式会社 | 光电转换元件、色素增感太阳能电池、金属络合物色素和色素溶液 |
| KR20170018000A (ko) * | 2014-07-07 | 2017-02-15 | 후지필름 가부시키가이샤 | 광전 변환 소자, 색소 증감 태양 전지, 금속 착체 색소, 색소 용액, 및 터피리딘 화합물 또는 그 에스터화물 |
| KR20170016441A (ko) * | 2014-07-07 | 2017-02-13 | 후지필름 가부시키가이샤 | 광전 변환 소자, 색소 증감 태양 전지, 금속 착체 색소, 색소 용액, 및 터피리딘 화합물 또는 그 에스터화물 |
| JPWO2016006511A1 (ja) * | 2014-07-07 | 2017-05-25 | 富士フイルム株式会社 | 光電変換素子、色素増感太陽電池、金属錯体色素、色素溶液、およびターピリジン化合物またはそのエステル化物 |
| WO2016006512A1 (ja) * | 2014-07-07 | 2016-01-14 | 富士フイルム株式会社 | 光電変換素子、色素増感太陽電池、金属錯体色素、色素溶液、およびターピリジン化合物またはそのエステル化物 |
| KR101982944B1 (ko) | 2014-07-07 | 2019-05-27 | 후지필름 가부시키가이샤 | 광전 변환 소자, 색소 증감 태양 전지, 금속 착체 색소, 색소 용액, 및 터피리딘 화합물 또는 그 에스터화물 |
| KR101982945B1 (ko) | 2014-07-07 | 2019-05-27 | 후지필름 가부시키가이샤 | 광전 변환 소자, 색소 증감 태양 전지, 금속 착체 색소, 색소 용액, 및 터피리딘 화합물 또는 그 에스터화물 |
| WO2016006511A1 (ja) * | 2014-07-07 | 2016-01-14 | 富士フイルム株式会社 | 光電変換素子、色素増感太陽電池、金属錯体色素、色素溶液、およびターピリジン化合物またはそのエステル化物 |
| WO2016148100A1 (ja) * | 2015-03-17 | 2016-09-22 | 富士フイルム株式会社 | ルテニウム錯体色素、色素溶液、光電変換素子および色素増感太陽電池 |
| JPWO2016148100A1 (ja) * | 2015-03-17 | 2018-01-11 | 富士フイルム株式会社 | ルテニウム錯体色素、色素溶液、光電変換素子および色素増感太陽電池 |
Also Published As
| Publication number | Publication date |
|---|---|
| TW201211165A (en) | 2012-03-16 |
| JPWO2012017872A1 (ja) | 2013-10-03 |
| JP5869481B2 (ja) | 2016-02-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5572029B2 (ja) | 金属錯体色素、光電変換素子及び光電気化学電池 | |
| JP5620316B2 (ja) | 光電変換素子、光電気化学電池及び色素 | |
| JP5620314B2 (ja) | 光電変換素子、光電気化学電池、光電変換素子用色素及び光電変換素子用色素溶液 | |
| JP5869481B2 (ja) | 金属錯体色素、光電変換素子及び光電気化学電池 | |
| JP5681716B2 (ja) | 金属錯体色素、光電変換素子及び光電気化学電池 | |
| JP5620469B2 (ja) | 光電変換素子の製造方法、光電変換素子及び光電気化学電池 | |
| JP5721717B2 (ja) | 金属錯体色素、光電変換素子及び光電気化学電池 | |
| JP5689351B2 (ja) | 光電変換素子及び光電気化学電池 | |
| JP5620315B2 (ja) | 光電変換素子及び光電気化学電池 | |
| JP5620496B2 (ja) | 金属錯体色素、光電変換素子及び光電気化学電池 | |
| JP5649368B2 (ja) | 光電変換素子及び光電気化学電池 | |
| WO2012017874A1 (ja) | 金属錯体色素、光電変換素子及び光電気化学電池 | |
| JP5816111B2 (ja) | 金属錯体色素組成物、光電変換素子及び光電気化学電池 | |
| JP2012051952A (ja) | 色素、光電変換素子及び光電気化学電池 | |
| WO2011118580A1 (ja) | 光電変換素子及び光電気化学電池 | |
| WO2012017873A1 (ja) | 金属錯体色素、光電変換素子及び光電気化学電池 | |
| JP5662728B2 (ja) | 色素、これを用いた光電変換素子及び光電気化学電池 | |
| JP5572028B2 (ja) | 光電変換素子及びこれを用いた光電気化学電池、光電変換素子用組成物 | |
| JP2012038436A (ja) | 光電変換素子及びこれを用いた光電気化学電池、光電変換素子用組成物 | |
| JP2012038435A (ja) | 光電変換素子、光電気化学電池及び光電変換素子用色素溶液 | |
| JP2012036239A (ja) | 金属錯体色素、光電変換素子及び光電気化学電池 | |
| JP5756766B2 (ja) | 光電変換素子、光電気化学電池及び色素 | |
| JP5572027B2 (ja) | 光電変換素子及びこれに用いられる光電変換素子用組成物 | |
| WO2012017870A1 (ja) | 色素、光電変換素子及び光電気化学電池 | |
| JP2012036238A (ja) | 金属錯体色素、光電変換素子及び光電気化学電池 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11814500 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012527679 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11814500 Country of ref document: EP Kind code of ref document: A1 |