WO2009134086A2 - 심혈관계 질환 치료용 약제학적 제제 - Google Patents
심혈관계 질환 치료용 약제학적 제제 Download PDFInfo
- Publication number
- WO2009134086A2 WO2009134086A2 PCT/KR2009/002278 KR2009002278W WO2009134086A2 WO 2009134086 A2 WO2009134086 A2 WO 2009134086A2 KR 2009002278 W KR2009002278 W KR 2009002278W WO 2009134086 A2 WO2009134086 A2 WO 2009134086A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cellulose
- pharmaceutical formulation
- release
- copolymer
- enteric
- Prior art date
Links
Images



























Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
- A61K47/38—Cellulose; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
Definitions
- the present invention provides a pharmaceutical preparation comprising (1) a prerelease compartment containing simvastatin and a delayed release compartment containing rozatan, and (2) a delayed release compartment containing a prior release compartment containing simvastatin and olmesartan.
- compositions comprising a compartment, (3) a pharmaceutical formulation comprising a prior release compartment containing simvastatin and a delayed release compartment containing valsartan, (4) a prior release compartment containing simvastatin and a candesartan
- a pharmaceutical preparation comprising a delayed-release compartment comprising: (5) a pharmaceutical preparation comprising a prior-release compartment containing roschvastatin and a delayed-release compartment containing rozatan, and (6) a rochevastatin
- Pharmaceutical formulations comprising a prior release compartment and a delayed release compartment containing ibesartan, (7) a prior release compartment containing rochevastatin and a candesartan It is directed to a pharmaceutical preparation comprising a delayed-release compartment.
- Hypertension is a condition caused by blood pressure being maintained above a normal range, and generally means when systolic blood pressure is 140 mmHg or more or diastolic blood pressure is 90 mmHg or more.
- systolic blood pressure 140 mmHg or more or diastolic blood pressure is 90 mmHg or more.
- One out of five adults in Korea is a chronic circulatory disease with high incidence, and the frequency of its occurrence is increasing worldwide.
- hypertension is a disease that requires more active management and treatment because it can cause fatal complications such as stroke, heart failure, and coronary artery disease, even though there are no symptoms.
- Hypertension is a condition that is caused by multiple causes. Therefore, it is difficult to determine in advance what will be the consequences of using a single anticompressant [Journal of hypertension 1995: 9: S33-S36]. More than two-thirds of patients with hypertension have been reported to require two or more hypertension medications that are not controlled as a single agent and are classified differently. It is difficult to lower blood pressure to the desired level as a single drug hypertension drug, and in order to obtain a significant therapeutic effect, two or more drugs with different classifications must be combined.
- blood pressure tends to rise with age. In people over 60, about 63% develop high blood pressure. In particular, the systolic blood pressure increases around 60 years old, the diastolic blood pressure is rather low isolated systolic hypertension. This is called senile hypertension. Geriatric hypertension can help you to maintain your blood pressure 24 hours a day and at night to prevent sudden heart attacks that may occur during sleep and prevent strokes caused by hypertension caused by intense stress during the day.
- non-dipper type patients with hypertension that do not lower blood pressure during sleep have a high risk of complications such as ischemic heart disease and stroke, and should be treated in consideration of biorhythm [Adv. Drug Deliv. . Rev., 2007: 904-922.
- hypertension treatment should understand the multifactoriality and polymorphism of the disease and formulate it appropriately for the condition and administer it at the optimal time, thereby maintaining blood pressure evenly for 24 hours and thereby preventing fatal complications.
- hypertension treatment is not the only purpose to lower blood pressure.
- the purpose of the treatment of hypertension is to prevent cardiovascular diseases such as myocardial infarction, heart failure, stroke, and premature death, which are prone to hypertension, and to prevent the worsening of the condition.
- a combination prescription is essential.
- the use of a single agent is effective for only 26% of patients, but a combination regimen can help prevent complications by maintaining the target blood pressure in as many as 74% of patients. [Hypertension Optimal Treatment, United Kingdom Prospective Diabetes Study , Large clinical].
- the US FDA has recognized the need for a combination formulation for 30 years, based on the so-called Fixed-dose Combination Therapy.
- the fixed-rate compound principle is that when combining drugs with different pharmacological actions, each compound should be combined in the same amount as when prescribed alone. This is called a fixed ratio combination formulation, and as long as the efficacy and safety of a single formulation are already recognized and the combination prescription is carried out by the prescribing physicians, such combination formulations are approved without separate experiments.
- Combination formulations can reduce the risk of developing circulatory complications, thereby reducing long-term prevention costs.
- calcium channel blocker As the above-mentioned pharmacologically active ingredient effective in hypertension of multifactorial and polymorphism, calcium channel blocker, angiotensin II receptor blocker renin blocker, beta adrenergic blocker, angiotensin converting enzyme inhibitor, diuretic agent, etc., depending on the similarity or mechanism of the chemical structure Can be mentioned.
- Combinations of combinations recommended for the treatment of hypertension also include statin-based lipid inhibitors and angiotensin II receptor blockers, HMG-CoA reductase inhibitors.
- HMG-CoA reductase strongly inhibit the catalysis of the conversion of mevalonate from the HMG-CoA reductase 3-hydroxy-3-methylglutaryl-coenzyme (HMG-CoA), resulting in cholesterol in the liver. It has the effect of inhibiting the production and lowering the low density lipoprotein cholesterol (LDL-C).
- Statin-based lipid inhibitors include simvastatin, roschvastatin, atorvastatin, pitavastatin, fluvastatin, lovastatan and pravastatin. Specifically, simvastatin and roschvastatin are as follows.
- Simvastatin strongly inhibits the conversion of HMG-CoA reductase [3-hydroxy-3-methylglutaryl-coenzyme reductase] to mevalonate, which inhibits liver cholesterol production and lower-density lipoprotein cholesterol (LDL- C) is one of statin-based lipid lowering agents that has an effect of lowering [Lancet 1995; 346: 750-753, Am J Cardiol 1998; 82: 57T-59T, Am J Cardiol 1995; 76: 107C-112C, Hypertens Res 2003; 26: 699-704, Hypertens Res 2003; 26: 273-280.] Br Med Bull 2001; 59: 3-16, Am J Med 1998; 104 (Suppl 1): 6S-8S, Clin Pharmacokinet 2002; 41: 343-370], which are known to be very effective in the treatment and prevention of complex hyperlipidemia, atherosclerosis and coronary heart disease ("Scandinavian Simvastatin Survival Study" published in the Lancet, vol. 344
- simvastatin by itself, is inactive, but in the form of ⁇ -hydroxy acid, which is active in the liver, is metabolized by the enzyme cytochrome P450 3A4 in the liver and excreted from the liver, acting in the liver.
- cytochrome P450 3A4 the enzyme that catalyzes the oxidation of simvastatin.
- the presence of high concentrations of simvastatin in the blood, which are not metabolized by cytochrome P450 3A4 increases the risk of inducing muscle disorders such as myolysis, which is a side effect of simvastatin.
- Rochevastatin is (3R, 5S, 6E) -7- [4- (4-fluorophenyl) -2- (N-methylmethanesulfoamido) -6- (propan-2-yl) pyrimidine -5-yl] -3,5-dihydrooxyhept-6-enoic acid, which is an HMG-CoA reductase inhibitor.
- Rochevastatin is a representative drug that inhibits HMG-CoA reductase, which regulates the cholesterol synthesis pathway, and inhibits cholesterol production in the liver.
- Roschvastatin has the effect of reducing total cholesterol, low density lipoprotein-cholesterol (LDL-C), and triglycerides and increasing high density lipoprotein-cholesterol (HDL-C). Judy et al., Clinical Therapeutics. vol. 26 (9) (2004), p1368-1387. Due to this lipid control effect, Rochevastatin is used for the treatment of primary hyperlipidemia, mixed dyslipidemia and familial hypercholesterolemia.
- LDL-C low density lipoprotein-cholesterol
- HDL-C high density lipoprotein-cholesterol
- rosuvastatin When used in combination with drugs metabolized by the cytochrome P450 2C9 enzyme, rosuvastatin inhibits the metabolism of rosuvastatin in the liver, leading to increased blood levels, which can cause side effects in combinations that were unpredictable in a single agent.
- repeated doses result in the accumulation of unmetabolized drugs, which leads to an increase in steady-state blood concentrations compared with the use of single agents, thereby avoiding the side effects and adverse effects of high doses.
- the drug metabolism is not normal, so careful administration of the dose is considered. Since this is rarely known, the patients who take it are taking it with little awareness.
- angiotensin-II-receptor blockers and roschvastatin can be used. Because of its high clinical synergy, the combination has been administered despite the risk of adverse effects of elevated blood levels.
- Angiotensin-II Receptor Blockers ARBs block the action of vasoconstrictor factors Blocks the action of aldosterone, which increases angiotensin-II, a blood pressure booster, to relax blood vessels. Since the angiotensin-II receptor blocker inhibits RAAS (Renin and Angiotensin System) excited state during sleep after midnight, it is suitable for patients with non-dipper type hypertension due to its strong anti-pressure effect after midnight.
- RAAS Renin and Angiotensin System
- Angiotensin-II-receptor blocker (ARB) medications lower blood pressure while preventing and treating heart failure, arrhythmias after myocardial infarction, diabetic complications and prevention, renal failure prevention and treatment, stroke prevention and treatment, antiplatelet action, atherosclerosis prevention Action, suppresses harmful effects of aldosterone, reduces the effects of metabolic syndrome, prevents circulatory disease chain deterioration, etc.
- ARB Angiotensin-II-receptor blocker
- Lozatan [2-butyl-4chloro-1- [2- (1H-tetrazol-5-yl) biphenyl-4-ylmethyl] -1H-imidazol-5-methanol] is an angiotensin divalent vascular wall receptor. It is an antihypertensive agent that antagonizes binding. This angiotensin-II is a factor causing blood pressure increase, left ventricular hypertrophy, vascular hypertrophy, atherosclerosis, renal failure, stroke and the like (US Patent No. 5,138,069).
- Rozatan prevents and treats heart failure, arrhythmias after myocardial infarction, prevents and treats diabetic complications, prevents and treats renal failure, prevents and treats stroke, antiplatelet action, prevents atherosclerosis, inhibits aldosterone harmful effects, alleviates the effects of metabolic syndrome, circulation It is known as a drug that exhibits a wide range of actions such as preventing mechanical disease cascading [Clin, Exp. Hypertens. 1998, 20, p. 205-221. Circulation, 2000; 101, p. 1653-1659.
- lojatan When lojatan is absorbed, it first enters the liver. Some of them are the active lozatan molecules, which flow out into the blood and reach their highest concentration in one hour. However, some remain metabolized by two enzymes, the liver enzymes cytochromes P450 2C9 and 3A4, to reach the highest blood levels 3-4 hours after being converted to the more active rojatan carboxylic acid. That is, the pharmacological action of rojatan is the pharmacological action of rojatan and rojatan carboxylic acid mixture which is a rojatan active metabolite.
- Rozatan has an antihypertensive effect on myocardial systolic and diastolic at moderate doses, additional heart failure prevention and treatment related to all symptoms of hypertension, prevention of arrhythmia and heart failure after myocardial infarction, prevention of diabetic complications, and prevention of renal failure. , Prevents stroke, prevents antiplatelet action, prevents atherosclerosis, inhibits aldosterone harmful effects, alleviates metabolic syndrome, prevents circulatory aggression, and sleep disorders caused by urination. : Clin, Exp. Hypertens., Vol. 20 (1998), [p. 205-221]; J. Hypertens., Vol. 13 (8) (1995), [p.891-899]; Kidney lnt., Vol.
- Ibesartan is a representative non-peptide angiotensin-II-receptor blocker, which relaxes blood vessels by selectively inhibiting angiotensin-II binding to receptors in tissues such as vascular smooth muscle cells and adrenal glands. [M burnier et al., The Lancet. vol.355 (2000), p637-645] Because of this vasorelaxation, Ibesatan is used to treat hypertension and nephropathy in Type 2 Diabetic Patients.
- Valsartan in angiotensin-II receptor blocker [Formula: N- (1-oxopentyl) -N-[[2 '-(1H-tetrazol-5-yl) [biphenyl-4-yl] methyl] -L- Valine] is an anti-pressure agent that relaxes blood vessels by blocking the action of vasoconstrictors and blocking the action of aldosterone, which increases angiotensin II, a blood pressure raising substance.
- Angiotensin II is a factor causing blood pressure increase, left ventricular hypertrophy, vascular hypertrophy, atherosclerosis, renal failure, stroke and the like (US Patent No. 5,399,578).
- Valsartan is a drug belonging to angiotensin-II receptor antagonists, first released in Germany in 1996 and approved by the US FDA in 1996.
- Valsartan is used to treat a wider range of cardiovascular diseases, including heart failure and myocardial infarction, with excellent blood pressure-enhancing effects, and a clinical study published in the 2003 American Academy of Cardiology showed that valsartan reduced mortality in patients after myocardial infarction by 25 percent.
- Valsartan with this feature is known to have a strong blood pressure lowering effect from midnight to dawn [Hypertension, 2003; 42: 283-290, Chronobiol. Int., 2005; 22: 755-776.
- Valsartan one of the ARBs, has a strong blood pressure lowering effect from midnight to early morning when RAAS (Renin and angiotensin system) works strongly [J. Hypertens, 2005; 23: 1913-1922, Hypertension, 2003; 42: 283-290, Chronobiol. Int. 2005; 22: 755-776.
- RAAS Renin and angiotensin system
- Candesartan [2-ethyloxy-1-(# 4- [2- (2H-1,2,3,4-tetrazol-5-yl) phenyl] -phenyl ⁇ -1H-1,3-benzodia Sol-6-carboxylic acid] is a representative drug of non-peptide angiotensin-II-receptor blocker, which relaxes blood vessels by selectively inhibiting angiotensin-II binding to receptors in tissues such as vascular smooth muscle cells and adrenal glands. [M burnier et al., The Lancet. Vol. 355 (2000), p637-645] With this vasorelaxation, candesartan is a nephropathy in Type 2 Diabetic Patients. Used for treatment.
- Candesartan is commercially available in the form of a prodrug of candesartan due to its low bioavailability (15% candesartan cilexetin tablets, 40% solution). It is absorbed as candesartan from the small intestine wall and the absorption rate is as fast as Tmax 3-4 hours. Therefore, in order to prevent hypertension, stroke treatment and other complications with candesartan administration, blood pressure drop should be continued from midnight until morning when angiotensin and aldosterone are secreted. Therefore, candesartan requires administration after evening (Easthope SE et al .: Candesartan Cilexetil: An Update of its Use in Essential Hypertension, Drugs Volume 62 (8) 2002 pp 1253-1287).
- Olmesartan is a selective angiotensin II receptor (type AT1) antagonist among angiotensin-II receptor blockers. In particular, it is a very good drug to co-administer with drugs that are not metabolized by the Cytochrome P450 system.
- statin-based anti-lipid drug an angiotensin-II receptor blocker-based drug is preferred for the treatment of hypertension, there are various problems when the formulation is simply mixed.
- additives such as acidic substances and glidants should be additionally used to improve this.
- the angiotensin-II receptor blocker which is effective in the evening session
- the optimal time period of dihydropyridine statin-based lipid inhibitor which is effective in the morning regimen
- the present inventors completed the present invention to solve the problem of the simple combination preparation and at the same time to develop a more effective combination formulation for the treatment of cardiovascular diseases such as hypertension.
- the present invention relates to a technology for formulating a functional combination that can suppress the decrease in drug efficacy due to drug interactions and prevent side effects from occurring when two drugs are simultaneously administered.
- the drug passes through the barrier in the first stage, enters the liver in the second stage, metabolizes and activates in the liver cells in the tertiary stage, and the biliary tract in the fourth stage Efflux transporters, influx transporters, and metabolic enzymes that absorb, metabolize, and excrete drugs everywhere, such as when exiting cells, exist everywhere.
- one component may interfere with the absorption, distribution, and metabolism of the other, thereby reducing the efficacy or increasing the side effects. Therefore, one component must be passed first, and the other component must be passed at a time difference to eliminate drug interaction.
- the purpose of the present invention is to determine the dissolution order and maintain the time difference between the two components with the aim of realizing the ideal combination method when all the drugs are heterogeneously administered. It is absorbed to enable functional combinations that maximize the efficacy and minimize side effects.
- transporters and drug metabolizing enzymes that have been tested or reviewed for the preparation of the functional combination of the present invention are as follows.
- P-gp P-glycoprotein
- MDR Multidrug resistance
- MRP Multidrug resistance associated protein
- Influx Transporter Organic anion transport protein (OATP), Sodium taurocholate cotransporting polypeptide (NTCP), Organic cation transporter (OCT)
- OATP Organic anion transport protein
- NTCP Sodium taurocholate cotransporting polypeptide
- OCT Organic cation transporter
- Uridine-5-phophate-glucuronosyltransferase UDP-gt
- Sulfatase Sulfotransferase (1a1, 2a1, 1e1)
- the technical problem to be solved by the present invention is to minimize the side effects of co-administration of each drug, to induce an optimal pharmacological effect, to obtain a clinical synergistic effect by administering the drug at the time of expression of the pharmacological effect of each drug It is possible to provide a pharmaceutical formulation capable of increasing drug compliance.
- the present invention relates to a controlled release pharmaceutical preparation comprising a prior release compartment containing an HMG CoA reductase inhibitor as a pharmacologically active ingredient, and a delayed release compartment containing angiotensin-2 receptor blocker as a pharmacologically active ingredient. It is about.
- the present invention provides a prior-release compartment containing simvastatin or a pharmaceutically acceptable salt thereof as the pharmacologically active ingredient, and rozatan, an isomer thereof or a pharmaceutically acceptable salt thereof as the pharmacologically active ingredient.
- a pharmaceutical formulation A is provided comprising a delayed-release compartment.
- the present invention also provides a prior-release compartment containing simvastatin or a pharmaceutically acceptable salt thereof as the pharmacologically active ingredient, and olmesartan, an isomer thereof, a pharmaceutically acceptable salt thereof or a prodrug thereof as the pharmacologically active ingredient.
- a pharmaceutical formulation B comprising a delayed-release compartment containing is provided.
- the present invention also provides a prior-release compartment containing simvastatin or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient, and a delayed-release compartment containing valsartan, an isomer thereof or a pharmaceutically acceptable salt thereof as the pharmacologically active ingredient. It provides a pharmaceutical formulation C comprising a.
- the present invention also provides a prior-release compartment containing simvastatin or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient, and candesartan, an isomer thereof, a pharmaceutically acceptable salt thereof or a prodrug thereof as a pharmacologically active ingredient.
- a pharmaceutical formulation D comprising a delayed-release compartment containing is provided.
- the present invention provides a prior-release compartment containing Rochevastatin or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient, and a delay containing Rozatan, an isomer thereof or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient.
- a pharmaceutical formulation E comprising a release compartment is provided.
- the present invention also provides a prior-release compartment containing Roschvastatin or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient, and Ivesartan, an isomer thereof or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient.
- a pharmaceutical formulation F comprising a delayed-release compartment is provided.
- the present invention also provides a prior-release compartment containing Rochevastatin or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient, and candesartan, an isomer thereof, or a pharmaceutically acceptable salt thereof or a pro pharmaceutically active ingredient thereof.
- a pharmaceutical formulation G comprising a delayed-release compartment containing a drug.
- salts referred to herein refer to salts commonly used in the pharmaceutical industry, for example, inorganic ionic salts made of calcium, potassium, sodium and magnesium, hydrochloric acid, nitric acid, phosphoric acid, bromic acid.
- OO prodrug means to be broken down into its active ingredient “OO” due to enzymes and chemicals in the body.
- candersartan prodrug means that it is broken down in the body to become the active ingredient candersartan.
- a pharmacologically active ingredient name is construed to include all of its isomers, pharmaceutically acceptable salts and optionally prodrugs thereof.
- hereinafter even if described as "rojatan", if present, it is interpreted to include both isomers of rozatan and pharmaceutically acceptable salts of rozatan.
- the pharmaceutical formulation of the present invention releases the angiotensin-II-receptor blocker of the delayed-release compartment after a period of time after the release of the statin-based lipid inhibitors of the prior-release compartment, thereby having a release suitable for the characteristics of each drug.
- the pharmaceutical formulation of the present invention provides a physical compartment that controls the release between two active ingredients, thereby improving the problem of co-administration or co-administration of existing single agents, resulting in an excellent therapeutic or prophylactic effect. That is, while using the two drugs in combination, by varying their release rate to prevent the antagonism and side effects between the drugs at the same time can obtain a synergistic effect, it is easy to take the patient.
- the present invention provides pharmaceutical formulations A, B, C, D, E, F, G.
- the present invention provides a prior-release compartment comprising simvastatin or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient, and a delayed-release compartment comprising rozatan, an isomer thereof or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient. It provides a pharmaceutical formulation A comprising.
- the present invention provides a pharmaceutical formulation A wherein at least 80% and preferably at least 85% of the total amount of simvastatin is released within 30 minutes after the start of simvastatin dissolution.
- the present invention provides a pharmaceutical preparation, wherein the rozatan is eluted within 20% of the total amount of lozatan in the unit preparation up to 2 hours after the start of simvastatin elution orally, preferably within 10% upon oral administration.
- the present invention provides pharmaceutical formulation A, wherein lozatan is absorbed in the liver 2 to 4 hours later than simvastatin.
- the active ingredient simvastatin in the prior release compartment comprises 1 to 160 mg of the formulation (100 mg to 1,000 mg total) on an adult (65-75 kg adult male) daily basis, preferably 5 To 80 mg.
- the active ingredient in the delayed-release compartment may comprise from about 12.5 to 200 mg of lojatan in the unit formulation, with 25 to 100 mg being preferred.
- the present invention includes a prior-release compartment comprising simvastatin or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient, and olmesartan, an isomer thereof, a pharmaceutically acceptable salt thereof or a prodrug thereof as a pharmacologically active ingredient.
- a pharmaceutical formulation B comprising a delayed-release compartment is provided.
- the olmesartan prodrug is decomposed into olmesartan, which is the active ingredient in the body, and is preferably olmesartan medoxomil.
- the present invention provides a pharmaceutical formulation B wherein at least 80% and preferably at least 90% of the total amount of simvastatin is released within 30 minutes after initiation of simvastatin dissolution.
- the present invention provides a pharmaceutical formulation B wherein olmesartan is released within 5% of the total amount of olmesartan in the unit formulation up to 2 hours after the start of simvastatin elution upon oral administration, preferably olmesart after 2 hours after initiation of simvastatin elution.
- Pharmaceutical Formulation B in which carbon is released.
- the present invention provides a pharmaceutical formulation B wherein olmesartan is absorbed in the liver 2 to 4 hours later than simvastatin.
- simvastatin the active ingredient in the prior release compartment, comprises 1 to 160 mg of the formulation (200 mg to 1,200 mg total) on an adult (65-75 kg adult male) daily basis, preferably 5 To 80 mg.
- the active ingredient in the delayed-release compartment may comprise about 2.5-80 mg of olmesartan medoxomil in the unit formulation, with 5-40 mg being preferred.
- the present invention provides a prior release compartment comprising simvastatin or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient, and a delayed release compartment comprising valsartan, an isomer thereof or a pharmaceutically acceptable salt thereof as the pharmacologically active ingredient. It provides a pharmaceutical formulation C comprising.
- the present invention provides a pharmaceutical formulation C wherein at least 80% and preferably at least 90% of the total amount of simvastatin is released within 30 minutes after initiation of simvastatin dissolution.
- the present invention also provides a pharmaceutical formulation C wherein valsartan is eluted 2 hours after the start of simvastatin elution upon oral administration.
- the present invention provides a pharmaceutical formulation C wherein valsartan is absorbed in the liver 2 to 4 hours later than simvastatin. do.
- the active ingredient simvastatin in the prior-release compartment comprises 1 to 160 mg of the formulation (200 mg to 1,200 mg total) on a daily basis for adults (65-75 kg adult male), preferably 5 To 80 mg.
- the active ingredient in the delayed-release compartment may comprise 1 to 800 mg of valsartan in the unit formulation, with 20 to 640 mg being preferred.
- the present invention includes a prior-release compartment comprising simvastatin or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient, and candesartan, an isomer thereof, a pharmaceutically acceptable salt thereof or a prodrug thereof as a pharmacologically active ingredient.
- a pharmaceutical formulation D comprising a delayed-release compartment is provided.
- the candesartan prodrug is decomposed into the active ingredient candesartan in the body, and is preferably candesartan cilexetil.
- the present invention provides a pharmaceutical formulation D wherein at least 80% and preferably at least 85% of the total amount of simvastatin is released within 30 minutes after initiation of simvastatin dissolution.
- the present invention provides a pharmaceutical formulation D wherein candesartan is released within 35% of the total amount of candesartan in a unit formulation up to 2 hours after the start of simvastatin dissolution upon oral administration. to provide.
- the present invention provides a pharmaceutical formulation D wherein candesartan is absorbed in the liver 2 to 4 hours later than simvastatin.
- simvastatin the active ingredient in the prior release compartment, comprises 1 to 160 mg of the formulation (100 mg to 800 mg total) on a daily basis for adults (65-75 kg adult male), preferably 5 to 80 mg.
- the active ingredient in the delayed-release compartment may comprise from about 2 to 64 mg of candesartan in the unit formulation, with 4 to 32 mg being preferred.
- the present invention relates to a sustained release compartment comprising roschvastatin or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient, and a delayed release composition comprising rozatan, an isomer thereof or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient.
- a pharmaceutical formulation E comprising a compartment is provided.
- the present invention provides a pharmaceutical formulation E wherein at least 80% and preferably at least 85% of the total amount of simvastatin is released within 30 minutes after the start of simvastatin dissolution.
- the present invention also provides a pharmaceutical formulation E wherein the release of rozatan, the active ingredient of the delayed-release compartment, is initiated about 2 hours after the onset of release of roschvastatin, the active ingredient of the prior-release compartment, preferably about 3 hours. to provide.
- the present invention also relates to a process in which rozatan, the active ingredient of the delayed-release compartment, is about 2 hours after the start of the release of roschvastatin, the active ingredient of the prior-release compartment, preferably about 3 hours, About 0 to 40% of the total amount, preferably about 0 to 20%, is provided to provide pharmaceutical preparation E.
- the pharmacologically active ingredient of the prior release compartment contains about 2.5 to 80 mg, preferably about 5 to 40 mg, Rochevastatin in the unit dosage form, and the active ingredient in the delayed release compartment comprises the unit dosage form.
- Potassium salt of losaltan about 10 to 200 mg, preferably about 25 to 100 mg.
- the present invention provides a prior-release compartment comprising Rochevastatin or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient, and a delay comprising Ivesartan, an isomer thereof or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient.
- Pharmaceutical formulations F comprising a release compartment are provided.
- the release of ibesartan, the active ingredient of the delayed-release compartment is about 2 to 10 hours after the start of release of roschvastatin, the active ingredient of the prior-release compartment, preferably between about 2 to 6 hours. , More preferably, between about 2 and 5 hours.
- the active ingredient of the delayed-release compartment is until about 2 hours after the start of release of roschvastatin, which is the active ingredient of the prior-release compartment, preferably about 4 hours, Pharmaceutical Formulation F is provided, which releases about 0-20% of the total amount of carbon.
- the active ingredient in the pre-release compartment may comprise about 0.1 to 200 mg of Roschvastatin in the unit formulation, preferably about 0.2 to 100 mg, more preferably about 5 to 40 mg
- the active ingredient in the delayed-release compartment may comprise about 1 to 1000 mg of ibesartan, or a pharmaceutically acceptable salt thereof, in the unit formulation, preferably about 2 to 500 mg, more preferably about 75 to 300 mg. can do.
- the present invention provides a prior-release compartment comprising Rochevastatin or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient, and candesartan, an isomer thereof, or a pharmaceutically acceptable salt thereof or a prodrug thereof as a pharmacologically active ingredient. It provides a pharmaceutical formulation G comprising a delayed-release compartment comprising a.
- the pharmaceutical formulation of the present invention provides that the release of candesartan, the active ingredient of the delayed-release compartment, is between about 1 hour and 8 hours after initiation of the release of roschvastatin, the active ingredient of the prior-release compartment, preferably from about 2 hours to Pharmaceutical Formulation G, which is initiated between 4 hours, is provided.
- the present invention also provides about 0 to about the total amount of candesartan in the unit formulation until the candesartan of the delayed-release compartment is about 2 hours after initiation of the release of roschvastatin in the prior release compartment, preferably about 3 hours.
- a pharmaceutical formulation G is provided in which 40% is released.
- the pharmacologically active ingredient of the prior release compartment comprises about 0.1 to 500 mg of Roschvastatin in the unit formulation, preferably about 0.2 to 100 mg, more preferably about 5 to 40 mg
- the active ingredient in the delayed-release compartment comprises about 1-1000 mg, preferably about 2-500 mg, more preferably about 4-32 mg, as candesartan cilexetil in the unit formulation.
- Pre-release compartment refers to the compartment that is released before the delayed-release compartment in the pharmaceutical formulation of the present invention.
- Pre-release compartments include pharmacologically active ingredients and, if necessary, pharmaceutically acceptable additives and other excipients.
- the pharmacologically active ingredient contained in the prior release compartment is first released with sufficient time difference to exhibit rapid efficacy prior to the pharmacologically active ingredient contained in the delayed release compartment.
- the prior release compartment is in the form of a mixture, granules, pellets, or tablets through conventional procedures for preparing oral administration agents such as mixing, coalescing, drying and granulation together with pharmaceutically acceptable additives in addition to the pharmacologically active ingredient. It can be prepared as. In addition, in the case where the fluidity is not good and tableting is not possible directly, it may be compressed, granulated, and granulated to granulate.
- Simvastatin may be used as a prior release compartment in Pharmaceutical Formulation A, Pharmaceutical Formulation B, Pharmaceutical Formulation C, and Pharmaceutical Formulation D.
- Pre-release compartments include simvastatin, isomers thereof or pharmaceutically acceptable salts as pharmacologically active ingredients.
- the active ingredient simvastatin in the prior release compartment in the formulation of the present invention comprises 1 to 160 mg in an adult (65-75 kg adult male) unit dosage unit, preferably 5 to 80 mg.
- Rochevastatin may be used as a prior release compartment in Pharmaceutical Formulation E, Pharmaceutical Formulation F, Pharmaceutical Formulation G.
- the pharmacologically active ingredient of the prior release compartment comprises roschvastatin, isomers thereof, or a pharmaceutically acceptable salt thereof, and preferably roschvastatin or a pharmaceutically acceptable salt thereof, more preferably Roche It may contain a vastatin calcium salt.
- the pharmacologically active ingredient of the prior release compartment comprises about 0.1 to 500 mg of Roschvastatin in the unit formulation, preferably about 0.2 to 100 mg, more preferably about 2.5 to 80 mg, even more preferably about 5 to 40 mg.
- the pre-release compartment of the present invention may use pharmaceutically acceptable diluents, binders, disintegrants, and additives of lubricants within the scope of not impairing the effects of the present invention.
- a pH adjusting agent an antioxidant, a dissolution aid, etc.
- a stabilizer may be additionally used in addition to the additive.
- the additive comprises 0.01-100 parts by weight with respect to 1 part by weight of the active ingredient.
- the diluent in the pre-release compartment of the present invention is a sugar, starch, microcrystalline cellulose (microcrystalline cellulose), lactose, lactose monohydrate, glucose, di-mannitol, alginate, alkaline earth metal salt, clay, polyethylene glycol, dicalcium phosphate, anhydrous Calcium hydrogen phosphate, mixtures thereof, and the like.
- the binder is starch, microcrystalline cellulose, highly dispersible silica, mannitol, di-mannitol, sucrose, lactose hydrate, polyethylene glycol, polyvinylpyrrolidone (povidone), polyvinylpyrrolidone copolymer (co) Povidone), hypromellose, hydroxypropyl cellulose, natural gum, synthetic gum, copovidone, gelatin, mixtures thereof, and the like.
- the disintegrating agent in the prior-release compartment of the present invention may be a starch or modified starch, such as sodium starch glycolate, corn starch, potato starch or pregelatinized starch; Clay such as bentonite, montmorillonite, or veegum; Celluloses such as microcrystalline cellulose, hydroxypropyl cellulose or carboxymethyl cellulose; Algins such as sodium alginate or alginic acid; Crosslinked celluloses such as croscarmellose sodium; Gums such as guar gum and xanthan gum; Crosslinked polymers such as crosslinked polyvinylpyrrolidone (crospovidone); Effervescent agents such as sodium bicarbonate, citric acid, or mixtures thereof can be used.
- a starch or modified starch such as sodium starch glycolate, corn starch, potato starch or pregelatinized starch
- Clay such as bentonite, montmorillonite, or veegum
- Celluloses such as microcrystalline cellulose,
- the lubricant is talc, stearic acid (stearic acid), magnesium stearate (magnesium stearate), calcium stearate (calcium stearate), sodium lauryl sulfate, hydrogenated vegetable oil, sodium benzoate, sodium stearyl fuma Latex, glyceryl behenate, glyceryl monorate, glyceryl monostearate, glyceryl palmitostearate, colloidal silicon dioxide, polyethylene glycol or mixtures thereof and the like can be used.
- the pH adjusting agent is selected from acetic acid, adipic acid, ascorbic acid (ascorbic acid), sodium ascorbate (sodium ascorbate), sodium ether, malic acid, succinic acid, tartaric acid, fumaric acid, citric acid (citric acid) And acidifying agents such as precipitated calcium carbonate, ammonia water, meglumine, sodium carbonate, magnesium oxide, magnesium carbonate, sodium citrate, calcium tribasic phosphate, and the like.
- the antioxidant may be dibutyl hydroxy toluene (butylate hydroxy toluene), butylated hydroxyanisole, tocopherol acetate, tocopherol, propyl gallate, sodium hydrogen sulfite, sodium pyrosulfite, or the like.
- the dissolution aid may be polyoxyethylene sorbitan fatty acid esters such as sodium lauryl sulfate, polysorbate, docusate sodium, poloxamer and the like.
- pharmaceutically acceptable stabilizers may be used ascorbic acid, citric acid, butylhydroxy anisole, butylhydroxy toluene, tocopherol derivatives and the like.
- formulation of the present invention may be formulated by selecting and using a pharmaceutically acceptable additive as various additives selected from colorants and fragrances.
- the range of additives usable in the pre-release compartments of the present invention is not limited to the use of such additives, and the additives described above may be formulated containing a range of doses in a usual range by selection.
- Delayed-release compartment refers to a compartment in which the active ingredient is released from a predetermined time after the start of release of the active ingredient in the prior-release compartment in the pharmaceutical formulation according to the present invention. Delayed-release compartments include (1) pharmacologically active ingredients; (2-a) a release controlling substance or (2-b) an osmotic pressure regulator and a semipermeable membrane coating base; (3) If necessary, it may further include a pharmaceutically acceptable additive. The pharmacologically active ingredient contained in the delayed-release compartment is released after sufficient time has elapsed after the start of release of the pharmacologically active ingredient contained in the prior-release compartment.
- Rozatan may be used as a delayed-release compartment in Pharmaceutical Formulation A, Pharmaceutical Formulation E.
- the pharmacologically active ingredient of the delayed-release compartment includes rozatan or a pharmaceutically acceptable salt, wherein the active ingredient in the delayed-release compartment may contain about 10 to 200 mg of locotan in the unit formulation and preferably about 25 to 100 mg.
- Representative salts of pharmaceutically acceptable examples include rozatan potassium.
- Olmesartan can be used as a delayed-release compartment in Pharmaceutical Formulation B.
- the pharmacologically active ingredient of the delayed-release compartment comprises olmesartan, a prodrug thereof or a pharmaceutically acceptable salt thereof, wherein the active ingredient in the delayed-release compartment comprises about 2.5-80 mg of olmesartan medoxomil in the unit formulation. 5 to 40 mg is preferred.
- Valsartan may be used as a delayed-release compartment in Pharmaceutical Formulation C.
- the pharmacologically active ingredient of the delayed-release compartment comprises the isomer of valsartan and a pharmaceutically acceptable salt, wherein the active ingredient in the delayed-release compartment may comprise 1 to 800 mg of valsartan in the unit formulation, preferably 20 to 640 mg.
- Candesartan can be used as a delayed-release compartment in Pharmaceutical Formulation D, Pharmaceutical Formulation G.
- the pharmacologically active ingredient of the delayed-release compartment includes candesartan, a prodrug thereof or a pharmaceutically acceptable salt thereof, wherein the active ingredient in the delayed-release compartment may contain about 2 to 64 mg of candesartan in the unit dosage form. 4 to 32 mg is preferred.
- Ivesartan can be used as a delayed-release compartment in Pharmaceutical Formulation F.
- the pharmacologically active ingredient of the delayed-release compartment comprises ibesartan, or a pharmaceutically acceptable salt thereof.
- Imaxartan Tmax is about 2 hours.
- the active ingredient in the delayed-release compartment may comprise about 1 to 1000 mg of ibesartan, or a pharmaceutically acceptable salt thereof, in the unit formulation, preferably about 2 to 500 mg, more preferably about 75 to 300 mg. can do.
- the delayed-release compartment in the pharmaceutical formulation of the present invention comprises at least one release controlling substance selected from the group consisting of enteric polymers, water insoluble polymers, hydrophobic compounds, and hydrophilic polymers. And at least one release controlling material selected from water insoluble polymers and enteric polymers.
- the release controlling substance may be used in an amount of 0.05 to 100 parts by weight based on 1 part by weight of the active ingredient. If the amount is less than the above range, sufficient delayed release property cannot be obtained. Release is delayed and no significant clinical effect is obtained.
- the release controlling substance is 0.01 to 20 parts by weight based on 1 part by weight of rozatan, and the release controlling substance is 0.1 to 50 parts by weight based on 1 part by weight of Ibesartan in the case of Ibesartan.
- the emission control material includes 0.1 to 50 parts by weight based on 1 part by weight of candesartan.
- hydroxypropylmethylcellulose and “hypromellose” are synonymous.
- hydroxypropylmethylcellulose 0000 which also includes hydroxypropylmethylcellulose, is synonymous with “hypromelloseOOOO”.
- hydroxypropylmethylcellulose phthalate is synonymous with “hypromellose phthalate.”
- the enteric polymer is insoluble or stable under acidic conditions of less than pH 5, and refers to a polymer that is dissolved or decomposed under specific pH conditions of pH 5 or higher.
- the enteric polymer that can be used in the present invention is at least one selected from the group consisting of an enteric cellulose derivative, an enteric acrylic acid copolymer, an enteric polymethacrylate copolymer, an enteric maleic acid copolymer and an enteric polyvinyl derivative, and the enteric cellulose derivative Hypromellose acetate succinate, hypromellose phthalate (hydroxypropyl methyl cellulose phthalate), hydroxymethyl ethyl cellulose phthalate, cellulose acetate phthalate, cellulose acetate succinate, cellulose acetate maleate, cellulose benzoate phthalate, cellulose Among propionate phthalate, methyl cellulose phthalate, carboxymethylethyl cellulose and ethyl hydroxyethyl cellulose phthalate and
- the enteric maleic acid-based copolymer is a vinyl acetate-maleic anhydride copolymer, styrene-maleic anhydride copolymer, styrene-maleic acid monoester copolymer, 1 selected from vinyl methyl ether maleic anhydride copolymer, ethylene maleic anhydride copolymer, vinyl butyl ether maleic anhydride copolymer, acrylonitrile-methyl acrylate maleic anhydride copolymer, and butyl styrene-maleic anhydride copolymer Species or more;
- the enteric polyvinyl derivative is at least one selected from polyvinyl alcohol phthalate, polyvinylacetate phthalate, polyvinyl butyrate phthalate and
- the water insoluble polymer refers to a polymer that does not dissolve in pharmaceutically acceptable water (eg purified water) that controls the release of the drug.
- the water-insoluble polymers usable in the present invention include polyvinylacetate (eg, colicoat SR30D), water-insoluble polymethacrylate copolymers [eg, poly (ethylacrylate-methyl methacrylate) copolymers (eg, Eudragit) NE30D), poly (ethylacrylate-methyl methacrylate-trimethylaminoethylmethacrylate chloride) copolymer (e.g.
- Eudragit RSPO, RL, RS), etc. ethyl cellulose, cellulose ester, cellulose ether, cellulose acyl At least one member selected from the group consisting of latex, cellulose dicylate, cellulose triacylate, cellulose acetate, cellulose diacetate and cellulose triacetate.
- the hydrophobic compound refers to a substance that does not dissolve in pharmaceutically acceptable water that controls the release of the drug.
- the hydrophobic compounds usable in the present invention are selected from the group consisting of fatty acids and fatty acid esters, fatty alcohols, waxes, inorganic substances, and mixtures thereof, and the fatty acids and fatty acid esters are glyceryl palmitostearate, glycerol.
- the fatty acid alcohols are at least one selected from cetostearyl alcohol, cetyl alcohol and stearyl alcohol;
- the wax is at least one selected from carnauba wax, beeswax, and microcrystalline wax;
- the inorganic substance is at least one selected from talc, precipitated calcium carbonate, calcium dihydrogen phosphate, zinc oxide, titanium oxide, kaolin, bentonite, montmorillonite and non-gum.
- the hydrophilic polymer refers to a polymeric material that is dissolved in pharmaceutically acceptable water that controls the release of the drug.
- the hydrophilic polymer usable in the present invention is selected from the group consisting of sugars, cellulose derivatives, gums, proteins, polyvinyl derivatives, hydrophilic polymethacrylate copolymers, polyethylene derivatives, carboxyvinyl polymers, and mixtures thereof, At least one selected from dextrin, polydextrin, dextran, pectin and pectin derivatives, alginate, polygalacturonic acid, xylan, arabinoxylan, arabinogalactan, starch, hydroxypropylstarch, amylose, and amylopectin ;
- the cellulose derivative is at least one selected from hydroxypropyl methyl cellulose (hypromellose), hydroxypropyl cellulose , hydroxymethyl cellulose, hydroxyethyl cellulose, methyl cellulose and sodium carboxymethyl cellulose;
- Preferred emission control materials in the embodiments of the present invention are as follows.
- the release controlling substance preferably comprises one or more selected from water insoluble polymers and enteric polymers.
- the enteric polymer is preferably hypromellose phthalate or methyl methacrylate acrylic acid copolymer.
- the enteric polymer according to the present invention may be included in an amount of 0.1 to 20 parts by weight, preferably 0.5 to 10 parts by weight, and less than 0.1 parts by weight, when the enteric polymer according to the present invention is easily dissolved at a pH of less than 5 parts by weight, 20 parts by weight. In the case of excessive excess, the total weight of the preparation is unnecessarily large or there is a problem that excessive dissolution is delayed.
- the water-insoluble polymer is preferably a poly (ethylacrylate-methyl methacrylate-trimethylaminoethylmethacrylate chloride) copolymer, ethylcellulose or cellulose acetate.
- the water-insoluble polymer according to the present invention may be included in an amount of 0.1 to 30 parts by weight, preferably 0.5 to 20 parts by weight, and less than 0.1 parts by weight relative to 1 part by weight of rozatan. If the amount is more than the weight part, there is a problem that excessive dissolution is delayed.
- the hydrophobic compound is preferably carnauba wax.
- the hydrophobic compound according to the present invention may be included in an amount of 0.1 to 20 parts by weight, preferably 0.5 to 10 parts by weight, and less than 0.1 parts by weight relative to 1 part by weight of rozatan. In the case of negative excess, there is a problem in that elution is excessively delayed.
- the hydrophilic polymer is preferably one or more selected from carbomer and hypromellose.
- Hydrophilic polymer according to the present invention may be included in 0.05 to 30 parts by weight, preferably 0.5 to 20 parts by weight with respect to 1 part by weight of rozatan, when less than 0.05 parts by weight has a problem that the release rate is not controlled, 30 weight In the case of excessive excess, there is a problem in that the release rate is not controlled, and in the case of more than 30 parts by weight, dissolution is excessively delayed.
- the release controlling substance preferably comprises one or more selected from water insoluble polymers and enteric polymers.
- the enteric polymer is preferably polyvinylacetate phthalate, hypromellose phthalate or methyl methacrylate acrylic acid copolymer.
- the enteric polymer according to the present invention may be included in an amount of 0.1 to 20 parts by weight, preferably 0.5 to 10 parts by weight relative to 1 part of olmesartan, and when it is less than 0.1 parts by weight, there is a problem in that it is easily dissolved at a pH of less than 5 and 20 parts by weight. In the case of excessive excess, the total weight of the preparation is unnecessarily large or there is a problem that excessive dissolution is delayed.
- the water insoluble polymer is preferably polyvinylacetate, ethylcellulose or cellulose acetate.
- the water-insoluble polymer according to the present invention may be included in an amount of 0.1 to 30 parts by weight, preferably 0.5 to 20 parts by weight relative to 1 part of olmesartan, and when less than 0.1 part by weight, there is a problem in that the release of the drug is not controlled. If the amount is more than the weight part, there is a problem that excessive dissolution is delayed.
- the hydrophobic compound may be included in an amount of 0.1 to 20 parts by weight, preferably 0.5 to 10 parts by weight relative to 1 part of olmesartan, and when less than 0.1 part by weight, the release of the drug is not controlled. If it exceeds 20 parts by weight, there is a problem in that elution is excessively delayed.
- the hydrophilic polymer is preferably one or more selected from carbomer, hydroxypropylcellulose, hypromellose.
- the hydrophilic polymer according to the present invention may be included in an amount of 0.05 to 30 parts by weight, preferably 0.5 to 20 parts by weight with respect to 1 part by weight of olmesartan, and when less than 0.05 parts by weight, there is a problem that the release rate is not controlled. In the case of more than part by weight, there is a problem in that the release rate is not controlled, in the case of more than 30 parts by weight, dissolution is excessively delayed.
- the release controlling substance comprises at least one selected from water insoluble polymers and enteric polymers.
- the enteric polymer is preferably hypromellose phthalate.
- the enteric polymer according to the present invention may be included in an amount of 0.1 to 20 parts by weight, preferably 0.5 to 10 parts by weight, compared to 1 part by weight of valsartan, and when it is less than 0.1 parts by weight, it is easily dissolved at a pH of less than 5 parts, and 20 parts by weight. If exceeded, there is a problem in that the total weight of the formulation is unnecessarily large or excessively delayed dissolution.
- the water-insoluble polymer is preferably polyvinylacetate or poly (ethylacrylate-methyl methacrylate-trimethylaminoethylmethacrylate chloride) copolymer.
- the water-insoluble polymer according to the present invention may be included in an amount of 0.1 to 30 parts by weight, preferably 0.5 to 20 parts by weight with respect to 1 part by weight of valsartan, and when less than 0.1 part by weight, there is a problem in that the release of the drug is not controlled and 30 parts by weight. In the case of negative excess, there is a problem in that elution is excessively delayed.
- the hydrophobic compound is preferably carnauba wax.
- Hydrophobic compound according to the present invention may be included 0.1 to 20 parts by weight, preferably 0.5 to 10 parts by weight relative to 1 part by weight of valsartan, when less than 0.1 parts by weight has a problem that the release of the drug is not controlled, 20 parts by weight If it exceeds, there is a problem that excessive dissolution is delayed.
- the hydrophilic polymer is preferably one or more selected from hydroxypropyl cellulose.
- the hydrophilic polymer according to the present invention may be included in an amount of 0.05 to 30 parts by weight, preferably 0.5 to 20 parts by weight with respect to 1 part by weight of valsartan, and when it is less than 0.05 parts by weight, the release rate is not controlled, and 30 parts by weight. If it exceeds, there is a problem that the release rate is not controlled, if more than 30 parts by weight excessive dissolution is delayed.
- the release controlling substance preferably comprises one or more selected from water insoluble polymers and enteric polymers.
- the enteric polymer is preferably hypromellose acetate succinate or methyl methacrylate acrylic acid copolymer.
- the enteric polymer according to the present invention may be included in an amount of 0.1 to 20 parts by weight, preferably 0.5 to 10 parts by weight, compared to 1 part by weight of candesartan, and when it is less than 0.1 parts by weight, it is easily dissolved at a pH of less than 5, 20 If the amount is more than the weight part, there is a problem in that the total weight of the preparation is unnecessarily large or excessively delayed dissolution.
- the water-insoluble polymer is preferably a poly (ethyl acrylate-methyl methacrylate-triethylaminoethyl- methacrylate chloride) copolymer, polyvinylacetate or ethylcellulose.
- the water-insoluble polymer according to the present invention may be included in an amount of 0.1 to 30 parts by weight, preferably 0.5 to 20 parts by weight, compared to 1 part by weight of candesartan, and when less than 0.1 parts by weight, there is a problem in that the release of the drug is not controlled. In the case of more than 30 parts by weight, there is a problem that excessive dissolution is delayed.
- the preferred hydrophobic compound is carnaubawax.
- Hydrophobic compound according to the present invention may be included 0.1 to 20 parts by weight, preferably 0.5 to 10 parts by weight compared to candesartan, when less than 0.1 parts by weight there is a problem that the release of the drug is not controlled, more than 20 parts by weight In this case, there is a problem in that elution is excessively delayed.
- the preferred hydrophilic polymer is hypromellose.
- the hydrophilic polymer according to the present invention may be included in an amount of 0.05 to 30 parts by weight, preferably 0.5 to 20 parts by weight with respect to 1 part by weight of candesartan, and when it is less than 0.05 parts by weight, there is a problem in that the release rate is not controlled. In the case of more than part by weight, there is a problem in that the release rate is not controlled, in the case of more than 30 parts by weight, dissolution is excessively delayed.
- enteric polymers are hypromellose phthalate, hypromellose acetate succinate, carboxymethylethyl cellulose, methyl methacrylate acrylic acid copolymer, methacrylic acid-methyl methacrylate copolymer, More preferably, it is a hypromellose phthalate and a methyl methacrylate acrylic acid copolymer.
- Enteric polymer according to the present invention may be included in 1 to 50% by weight, preferably 3 to 30% by weight based on the total weight of the formulation, when less than 1% by weight has a problem that is not dissolved or stable under acidic conditions, 50 weight If more than%, there is a problem that does not dissolve even under basic conditions.
- the preferred water insoluble polymer may be at least one selected from polyvinyl acetate, ethyl cellulose, and cellulose acetate, and more preferably at least one selected from polyvinyl acetate, ethyl cellulose, cellulose acetate. .
- Water-insoluble polymer according to the present invention may be included in about 1 to 50% by weight, preferably about 3 to 30% by weight relative to the total weight of the formulation, when less than 1% by weight is difficult to have a sufficient delay time, 50 If it is more than the weight%, there is a problem that the release of the drug does not occur or becomes too long to be 9 hours or more of the delay time.
- preferred hydrophobic compounds are at least one selected from glyceryl palmitostearate, glyceryl behenate, carnauba wax, beeswax, more preferably selected from glyceryl palmitostearate, carnauba wax More than one.
- Hydrophobic compound according to the present invention may be included in about 1 to 50% by weight, preferably about 3 to 30% by weight relative to the total weight of the formulation, if less than 1% by weight has a problem that does not affect the release of the drug at all In case of more than 50% by weight, there is a problem that the release of the drug does not occur or is difficult to formulate.
- preferred hydrophilic compounds are dextrin, dextran, pectin hypromellose, hydroxypropylcellulose, guar gum, gum arabic, xanthan gum, gelatin, poly (butyl methacrylate- (2-dimethylaminoethyl Methacrylate-methyl methacrylate copolymer, carbomer may be one or more selected from dextrin, hypromellose, xanthan gum, carbomer.
- hydrophilic polymer may be included in about 1 to 70% by weight, preferably about 3 to 50% by weight relative to the total weight of the formulation, when less than 1% by weight does not affect the disintegration of the tablet at all, 70 If it is more than% by weight, it is difficult to control disintegration and release.
- preferred release controlling substances are carbomer, hypromellose acetate succinate, polyvinylacetate, carnauba wax, hypromellose, hydroxypropyl cellulose, ethyl cellulose, hypromellose phthalate , Methyl methacrylate acrylic acid copolymer, and mixtures thereof, and more preferred release control materials are selected from the group consisting of polyvinylacetate, hypromellose acetate succinate, carbomer and mixtures thereof. .
- enteric polymers include hypromellose phthalate, hypromellose acetate succinate, cellulose acetate phthalate, methacrylic acid acrylic acid copolymer, methacrylic acid and ethyl acrylate copolymer, styrene-maleic acid monoester. It may be at least one selected from a copolymer, polyvinylacetate phthalate, more preferably selected from hypromellose phthalate, hypromellose acetate succinate, methacrylic acid acrylate copolymer, methacrylic acid and ethyl acrylate copolymer. It may be one or more.
- Enteric polymer according to the present invention may be included in about 5 to 80% by weight, preferably about 10 to 30% by weight based on the total weight of the formulation, when less than 5% by weight has a problem that is not dissolved or stable under acidic conditions, If it is more than 80% by weight there is a problem that does not dissolve even under basic conditions.
- preferred water-insoluble polymers are preferably polyvinyl acetate, poly (ethylacrylate-methyl methacrylate) copolymer, poly (ethylacrylate-methyl methacrylate-trimethylaminoethylmethacrylate ) Copolymer, poly (ethyl acrylate-methyl methacrylate-trimethylaminoethyl methacrylate) copolymer, ethyl cellulose, cellulose acetate may be one or more selected from, and more preferably polyvinyl acetate, poly (ethylacryl Latex-methyl methacrylate-trimethylaminoethyl methacrylate chloride) copolymer, ethyl cellulose, cellulose acetate may be at least one selected from the group.
- Water-insoluble polymer according to the present invention may be included in about 5 to 80% by weight, preferably about 10 to 30% by weight based on the total weight of the formulation, when less than 5% by weight is difficult to have a sufficient delay time, 80 If it is more than the weight%, there is a problem that the release of the drug does not occur or becomes too long to be 9 hours or more of the delay time.
- the preferred hydrophobic compound may be at least one selected from glyceryl palmitostearate, glyceryl behenate, stearic acid, cetyl alcohol, carnauba wax, gum, more preferably glyceryl behenate It may be at least one selected from stearic acid, cetyl alcohol, carnauba wax.
- Hydrophobic compound according to the present invention may be included in about 5 to 80% by weight, preferably about 10 to 30% by weight relative to the total weight of the formulation, if less than 5% by weight has a problem that does not affect the release of the drug at all In the case of more than 80% by weight, there is a problem that the release of the drug does not occur or is difficult to formulate.
- preferred hydrophilic polymers are preferably hypromellose, hydroxypropylcellulose, guar gum, xanthan gum, gelatin, polyvinyl pyrrolidone, poly (butyl methacrylate- (2-dimethylamino At least one selected from ethyl) methacrylate-methylmethacrylate) copolymer, and more preferably at least one selected from hypromellose, hydroxypropyl cellulose, xanthan gum, and polyvinyl pyrrolidone. have.
- Hydrophilic polymer according to the present invention may be included in about 5 to 80% by weight, preferably about 10 to 30% by weight relative to the total weight of the formulation, when less than 5% by weight does not affect the disintegration of the tablet at all In the case of more than 80% by weight, disintegration and release are difficult to control.
- preferred release control substances include hypromellose acetate succinate, hypromellose phthalate, methyl methacrylate copolymer, polyvinylacetate, ethyl cellulose, cellulose acetate, carnauba wax, hips. Romeose (hydroxypropylmethylcellulose), hydroxypropyl cellulose, polyvinyl pyrrolidone, and mixtures thereof, and more preferably hypromellose acetate succinate, hypromelo Osphthalate, methyl methacrylate acrylate, polyvinylacetate, ethyl cellulose, carnauba wax, hypromellose, hydroxypropyl cellulose, and mixtures thereof.
- the release controlling substance is preferably an enteric polymer, a water insoluble polymer, a hydrophilic polymer, and a mixture of hydrophobic compounds and hydrophilic polymers.
- preferred enteric polymers are preferably hydroxypropylmethylcellulose phthalate (hypromellose phthalate), hydroxypropylmethylcellulose acetate succinate (hypromellose acetate succinate), cellulose acetate phthalate, acrylic acid It may be at least one selected from methacrylic acid copolymer, methacrylic acid and ethyl acrylate copolymer, styrene-maleic acid monoester copolymer, polyvinylacetate phthalate, more preferably hydroxypropyl methyl cellulose phthalate, hydroxypropyl methyl It may be at least one selected from cellulose acetate succinate, methacrylic acid acrylate copolymer, methacrylic acid and ethyl acrylate copolymer.
- Enteric polymer according to the present invention may be included in about 5 to 80% by weight, preferably about 10 to 30% by weight based on the total weight of the formulation, when less than 5% by weight has a problem that is not dissolved or stable under acidic conditions, If it is more than 80% by weight there is a problem that does not dissolve even under basic conditions.
- preferred water-insoluble polymers are preferably polyvinyl acetate, poly (ethylacrylate-methyl methacrylate) copolymer, poly (ethylacrylate-methyl methacrylate-trimethylaminoethylmethacrylatechloride 1) at least one selected from copolymers, ethyl cellulose and cellulose acetate, and more preferably at least one selected from polyvinyl acetate, poly (ethyl acrylate-methyl methacrylate) copolymer, ethyl cellulose and cellulose acetate.
- polyvinyl acetate poly (ethylacrylate-methyl methacrylate) copolymer
- poly (ethyl acrylate-methyl methacrylate) copolymer ethyl cellulose and cellulose acetate.
- Water-insoluble polymer according to the present invention may be included in about 5 to 80% by weight, preferably about 10 to 30% by weight based on the total weight of the formulation, when less than 5% by weight is difficult to have a sufficient delay time, 80 If it is more than the weight%, there is a problem that the release of the drug does not occur or becomes too long to be 9 hours or more of the delay time.
- the preferred hydrophobic compound may preferably be at least one selected from glyceryl palmitostearate, glyceryl behenate, stearic acid, cetyl alcohol, carnauba wax, more preferably glyceryl behenate It may be at least one selected from stearic acid, carnauba wax.
- Hydrophobic compound according to the present invention may be included in about 5 to 80% by weight, preferably about 10 to 30% by weight relative to the total weight of the formulation, if less than 5% by weight has a problem that does not affect the release of the drug at all In the case of more than 80% by weight, there is a problem that the release of the drug does not occur or is difficult to formulate.
- preferred hydrophilic polymers are hydroxypropylmethylcellulose, hydroxypropylcellulose, guar gum, xanthan gum, gelatin, polyvinyl pyrrolidone, poly (butyl methacrylate- (2-dimethylaminoethyl) meta Acrylate-methyl methacrylate) copolymer may be one or more selected from hydroxypropyl methyl cellulose, hydroxypropyl cellulose, xanthan gum, polyvinyl pyrrolidone.
- Hydrophilic polymer according to the present invention may be included in about 5 to 80% by weight, preferably about 10 to 30% by weight based on the total weight of the formulation, when less than 5% by weight does not affect the disintegration of the tablet at all In case of more than 80% by weight, disintegration and release are difficult to control.
- preferred release controlling substances are hydroxypropylmethylcellulose acetate succinate, hydroxypropylmethylcellulose phthalate, methyl methacrylate copolymer, polyvinylacetate, ethylcellulose, cellulose acetate, carnauba wax, Hydroxypropylmethylcellulose, hydroxypropylcellulose, polyvinyl pyrrolidone, and mixtures thereof; More preferred release control materials are hydroxypropylmethylcellulose acetate succinate, hydroxypropylmethylcellulose phthalate, methyl methacrylate acrylic acid, polyvinylacetate, ethylcellulose, carnauba wax, hydroxypropylmethylcellulose, hydroxy Propylcellulose, and mixtures thereof; More preferred release controlling substances are selected from the group consisting of hydroxypropylmethylcellulose acetate succinate, hydroxypropyl cellulose, polyvinylacetate, and mixtures thereof.
- the delayed-release compartment of the present invention includes an osmotic pressure control agent and may be a compartment coated with a semipermeable membrane coating base.
- the difference in the osmotic pressure in the digestive tract and the tablet by the osmotic pressure regulator causes water to pass through the semipermeable membrane on the surface of the tablet, increasing the pressure in the tablet.
- the drug may be released through the osmotic transport hole or the pores of the coating film or the coating layer may collapse when the pressure exceeds the elasticity of the coating base.
- the osmotic pressure regulating agent refers to a component used to control the release rate of the drug using the principle of osmotic pressure, for example magnesium sulfate, magnesium chloride, sodium chloride, lithium chloride, potassium sulfate, sodium sulfate, lithium sulfate At least one selected from the group consisting of sodium sulfate and mixtures thereof.
- the semi-permeable membrane coating base is a pharmaceutically usable coating base, which is formulated into the coating layer of the pharmaceutical formulation to be used to form a film which passes some components but does not pass other components.
- the semipermeable membrane coating base in the present invention is, for example, polyvinyl acetate, water-insoluble polymethacrylate copolymer, ethyl cellulose, cellulose ester, cellulose ether, cellulose acylate, cellulose dicylate, cellulose triacylate, cellulose acetate, cellulose di And at least one selected from the group consisting of acetate, cellulose triacetate, and mixtures thereof.
- the content of a preferred osmotic pressure control agent and a semipermeable membrane coating base is as follows.
- preferred osmotic agents are sodium chloride, sodium sulfate.
- the osmotic pressure control agent may be included in 0.05 parts by weight to 30 parts by weight, preferably 0.1 to 20 parts by weight, and less than 0.1 parts by weight of rojatan. In this case, there is a problem in that it is impossible to unnecessarily increase the total weight of the formulation or to realize a suitable drug release rate.
- the preferred semipermeable membrane coating base is ethylcellulose or cellulose acetate.
- the semi-permeable membrane coating base may be included in an amount of 0.05 parts by weight to 30 parts by weight, preferably 0.1 parts by weight to 20 parts by weight, and less than 0.05 parts by weight, with respect to 1 part by weight of rozatan. And, if it is more than 30 parts by weight, there is a problem that the release of the drug does not occur or the delay time is over 9 hours or longer.
- the preferred osmotic agent is sodium chloride or sodium sulfate.
- Osmotic pressure regulator may be included in 0.05 parts by weight to 30 parts by weight, preferably 0.1 to 20 parts by weight in 1 part by weight of olmesartan, when less than 0.1 parts by weight has a weak osmotic effect is weak, more than 30 parts by weight There is a problem that unnecessarily increase the total weight of the formulation or implement a suitable drug release rate.
- the preferred semipermeable membrane coating base is ethylcellulose or cellulose acetate.
- the semi-permeable membrane coating base may be included in an amount of 0.05 parts by weight to 30 parts by weight, preferably 0.1 parts by weight to 20 parts by weight with respect to 1 part by weight of olmesartan, and less than 0.05 parts by weight, it is difficult to have a sufficient delay time. If there is more than 30 parts by weight, there is a problem that the release of the drug does not occur or the delay time becomes over 9 hours or longer.
- preferred osmotic agents are sodium chloride or sodium sulfate.
- Osmotic pressure control agent may be included in 0.05 to 30 parts by weight, preferably 0.1 to 20 parts by weight to 1 part of valsartan, if less than 0.1 parts by weight has a problem that the effect of generating osmotic pressure is weak, if it is more than 30 parts by weight There is a problem that it is not possible to increase the formulation gross weight or to implement a suitable drug release rate.
- the preferred semipermeable membrane coating base is ethylcellulose.
- the semi-permeable coating agent may be included in an amount of 0.05 parts by weight to 30 parts by weight, preferably 0.1 parts by weight to 20 parts by weight, and less than 0.05 parts by weight with respect to 1 part by weight of valsartan. In case of more than 30 parts by weight, there is a problem in that the release of the drug does not occur or the delay time becomes over 9 hours or longer.
- the preferred osmotic agent is sodium chloride or sodium sulfate.
- the osmotic pressure control agent may be included in 0.05 parts by weight to 30 parts by weight, preferably 0.1 to 20 parts by weight in 1 part by weight of candesartan, and if less than 0.1 parts by weight, the osmotic pressure generating effect is weak, and more than 30 parts by weight In case of, there is a problem in that it is impossible to unnecessarily increase the total weight of the formulation or to realize a suitable drug release rate.
- the preferred semipermeable membrane coating base is ethylcellulose or cellulose acetate.
- the semi-permeable membrane coating base may be included in 0.05 parts by weight to 30 parts by weight, preferably 0.1 parts by weight to 20 parts by weight with respect to 1 part by weight of candesartan, and less than 0.05 parts by weight is difficult to have a sufficient delay time If there is more than 30 parts by weight, there is a problem that the release of the drug does not occur or the delay time becomes over 9 hours or longer.
- the preferred osmotic pressure regulator is sodium chloride.
- Osmotic pressure control agent according to the present invention may be included in about 0.1 to 50% by weight, preferably about 1 to 30% by weight relative to the total weight of the formulation, when less than 0.1% by weight there is a problem that the osmotic pressure is not formed, 50% by weight If it is exceeded, the semipermeable membrane is damaged due to the formation of a large osmotic pressure, so there is a problem in that the controlled release is not controlled.
- the preferred semipermeable membrane coating base may be at least one selected from polyvinyl acetate, ethyl cellulose and cellulose triacetate, and more preferably at least one selected from polyvinyl acetate and ethyl cellulose.
- the semi-permeable membrane coating base according to the present invention may be included in about 1 to 50% by weight, preferably about 3 to 30% by weight based on the total weight of the formulation, and when less than 1% by weight, it is difficult to form a desired semi-permeable membrane. If it is more than 50% by weight, there is a problem that all components may not pass.
- the preferred osmotic pressure regulator is sodium chloride.
- Osmotic pressure control agent according to the present invention may be included in about 1 to 80% by weight, preferably about 2 to 50% by weight based on the total weight of the formulation, there is a problem that the osmotic pressure is not formed when less than 1% by weight.
- the preferred semipermeable membrane coating base may be at least one selected from polyvinylacetate, water-insoluble polymethacrylate copolymer, ethylcellulose, cellulose triacetate, more preferably polyvinyl acetate, water-insoluble poly It may be at least one selected from methacrylate copolymer and ethyl cellulose.
- Semi-permeable membrane coating base according to the present invention may be included in about 5 to 80% by weight, preferably about 10 to 30% by weight based on the total weight of the formulation, when less than 5% by weight has a problem that it is difficult to form the desired semi-permeable membrane If it is more than 80% by weight, there is a problem that all components may not pass.
- the preferred osmotic pressure regulator is sodium chloride.
- Osmotic pressure control agent according to the present invention may be included in about 1 to 80% by weight, preferably about 2 to 50% by weight based on the total weight of the formulation, there is a problem that the osmotic pressure is not formed when less than 1% by weight.
- the preferred semipermeable membrane coating base may be at least one selected from polyvinyl acetate, polymethacrylate copolymer, ethylcellulose, cellulose triacetate, more preferably polyvinyl acetate, polymethacrylate air It may be at least one selected from coalesced and ethyl cellulose.
- Semi-permeable membrane coating base according to the present invention may be included in about 5 to 80% by weight, preferably about 10 to 30% by weight based on the total weight of the formulation, when less than 5% by weight has a problem that it is difficult to form the desired semi-permeable membrane If it is more than 80% by weight, there is a problem that all components may not pass.
- the formulations of the present invention are diluents, binders, disintegrating agents other than those mentioned as pharmaceutically acceptable (2-a) release controlling substances and (2-b) osmotic pressure regulators and semipermeable membrane coating agents within the scope of not impairing the effects of the present invention.
- Commonly used additives such as releases, lubricants, pH adjusters, antifoams, dissolution aids and the like can be formulated further using within a range not departing from the nature of delayed release.
- sugar starch, microcrystalline cellulose, lactose, lactose monohydrate, glucose, mannitol, di-mannitol, alginate, alkaline earth metal salt, clay, polyethylene glycol, dicalcium phosphate, anhydrous calcium hydrogen phosphate, or these Mixtures thereof and the like can be used.
- binders starch, microcrystalline cellulose, highly dispersible silica, mannitol, sucrose, lactose monohydrate, polyethylene glycol, polyvinylpyrrolidone, polyvinylpyrrolidone copolymer, hypromellose, hydroxypropyl cellulose, natural gum, synthetic Gum, copovidone, povidone, gelatin, mixtures thereof, and the like.
- starch or modified starches such as sodium starch glycolate, corn starch, potato starch, or pregelatinized starch (starch gelatinized starch); Clay such as bentonite, montmorillonite, or veegum; Celluloses such as microcrystalline cellulose, hydroxypropyl cellulose or carboxymethyl cellulose; Algins such as sodium alginate or alginic acid; Crosslinked celluloses such as croscarmellose sodium; Gums such as guar gum and xanthan gum; Crosslinked polymers such as crosslinked polyvinylpyrrolidone (crospovidone); Effervescent agents such as sodium bicarbonate, citric acid, or mixtures thereof can be used.
- Clay such as bentonite, montmorillonite, or veegum
- Celluloses such as microcrystalline cellulose, hydroxypropyl cellulose or carboxymethyl cellulose
- Algins such as sodium alginate or alginic acid
- Crosslinked celluloses such as croscar
- Talc stearic acid, magnesium stearate, calcium stearate, sodium lauryl sulfate, hydrogenated vegetable oil, sodium benzoate, colloidal silicon dioxide, sodium stearyl fumarate, glyceryl behenate, glyceryl monolate, glyceryl monostearate , Glyceryl palmitostearate, polyethylene glycol, magnesium aluminate silicate and the like can be used.
- the pH adjusting agent may be selected from an acidifying agent such as acetic acid, adipic acid, ascorbic acid, ascorbic acid nacrium, sodium ether, malic acid, succinic acid, tartaric acid, fumaric acid, citric acid (citric acid) and Basic agents such as precipitated calcium carbonate, ammonia water, meglumine, sodium carbonate, magnesium oxide, magnesium carbonate, sodium citrate, calcium tribasic phosphate, and the like can be used.
- an acidifying agent such as acetic acid, adipic acid, ascorbic acid, ascorbic acid nacrium, sodium ether, malic acid, succinic acid, tartaric acid, fumaric acid, citric acid (citric acid)
- Basic agents such as precipitated calcium carbonate, ammonia water, meglumine, sodium carbonate, magnesium oxide, magnesium carbonate, sodium citrate, calcium tribasic phosphate, and the like can be used.
- the antifoaming agent may use dimethicone, oleyl alcohol, propylene glycol alginate, simethicone such as simethicone emulsion and the like.
- the dissolution aid may be used polyoxyethylene sorbitan fatty acid esters such as sodium lauryl sulfate, polysorbate, docusate sodium, poloxamer and the like. It is also possible to add a plasticizer such as triethyl citrate and polyethylene glycol.
- the formulation of the present invention may be formulated by selecting and using a pharmaceutically acceptable additive as various additives selected from colorants and fragrances.
- a pharmaceutically acceptable additive as various additives selected from colorants and fragrances.
- the range of additives usable in the present invention is not limited to the use of such additives, and the above additives may be formulated to contain a range of dosages, usually by selection.
- purified water, ethanol, methanol, methylene chloride, and the like may be used as a solvent of the binding solvent and the delayed-release additive, and more preferably, purified water and ethanol are preferable.
- the range of usable additives is not limited to the use of such additives, and the above-mentioned additives may be formulated to contain a range of dosages by selection.
- formulations of the present invention can be prepared in a variety of formulations, for example, can be formulated in tablets, powders, granules, capsules and the like, such as uncoated tablets, coated tablets, multi-layered tablets, or nucleated tablets.
- the preparation of the present invention is a tabletting by selectively mixing additives such as granules constituting the pre-release compartment and granules constituting the delayed-release compartment and the like to have a pre-release compartment and a delayed-release compartment in a single tablet, and thus the active ingredient of each compartment.
- additives such as granules constituting the pre-release compartment and granules constituting the delayed-release compartment and the like to have a pre-release compartment and a delayed-release compartment in a single tablet, and thus the active ingredient of each compartment.
- This may be in the form of uncoated tablets will be eluted separately to show the respective effects.
- the formulation of the present invention may be in the form of a biphasic matrix tablet consisting of the delayed-release compartment and the pre-release compartment surrounding it.
- the formulation of the present invention may be in the form of a film coated tablet consisting of a film consisting of a tablet consisting of a delayed-release compartment and a pre-release compartment surrounding the outside of the tablet, simbastatin of the film coating layer as the film coating layer is dissolved It will elute first.
- the formulation of the present invention is obtained by mixing the pharmaceutical additives in the granules constituting the delayed-release compartment and the prior-release compartment, and tableting into double or triple wells using a multiple tableting machine, delayed-release compartment and pre-release
- the compartments may be in the form of multi-layered tablets forming a multilayer structure. Each layer constituting the multilayer tablet may be in a parallel state.
- This formulation is a tablet for oral administration which is formulated to enable pre-release and delayed release in layers.
- the formulation of the present invention may be in the form of a nucleated tablet consisting of an inner core consisting of a delayed-release compartment and an outer layer composed of a prior-release compartment surrounding the outer surface of the inner core.
- the nucleated tablet may be an osmotic nucleated tablet, and the osmotic nucleated tablet contains an osmotic pressure control agent inside the tablet for delayed release, followed by tableting, followed by coating the surface of the tablet with an osmotic semipermeable membrane to make it an inner core.
- the granules constituting the pre-release compartment are mixed with pharmaceutical additives and compressed into an outer layer to have a delayed-release inner core, and the surface of the inner core is surrounded by a pre-release layer.
- the formulations of the present invention may be in the form of particles, granules, pellets, or capsules comprising tablets and particles, granules, pellets, or tablets, which consist of delayed-release compartments.
- the tablet consisting of the delayed-release compartment of the capsule may include an osmotic pressure-controlling agent within the tablet and an osmotic coated tablet having a semipermeable membrane coating base on the surface of the tablet.
- the material of the capsule may be one selected from gelatin, succinate gelatin, or hypromellose, or a mixture thereof.
- the formulations of the present invention may further form a coating layer on the outside of the delayed release compartment and / or the prior release compartment. That is, the surface of particles, granules, pellets, or tablets, etc., which are composed of delayed-release compartments and / or pre-release compartments, may be coated for the purpose of delayed release or stabilization of the formulation.
- the formulation according to the present invention may be provided in a state such as uncoated tablet without additional coating, but may be in the form of a coated tablet further comprising a coating layer by forming a coating layer on the outside of the formulation, if necessary.
- a coating layer By forming the coating layer, it is possible to provide a formulation that can further ensure the stability of the active ingredient.
- the method of forming the coating layer may be appropriately selected by a person skilled in the art from the method of forming a film-like coating layer on the surface of the tablet layer, a method such as a fluidized bed coating method, a fan coating method may be applied, and preferably Fan coating can be applied.
- the coating layer may be formed using a coating agent, a coating aid, or a mixture thereof.
- the coating agent may be a cellulose derivative such as hypromellose, hydroxypropyl cellulose, sugar derivatives, polyvinyl derivatives, waxes, fats, Gelatin, mixtures thereof, and the like;
- Coating aids may be polyethylene glycol, ethyl cellulose, glycerides, titanium oxide, talc, diethyl phthalate, or mixtures thereof.
- the coating layer may include 0.5 to 15% by weight based on the total weight of the tablet.
- the pharmaceutical formulation of the present invention may be formulated using a time-dose dosing principle as disclosed in Chrontherpeutics (2003, Peter Redfern, PhP) by any suitable method in the art, and specifically in a method comprising the following steps Can be prepared by
- the angiotensin-II-receptor blocker is mixed, combined and dried by administering one or two release controlling substances selected from an enteric polymer, a water insoluble polymer, a hydrophobic compound, and a hydrophilic polymer and a conventional additive used in pharmaceuticals. Delayed-release granules or tablets through granulation, coating, or tableting, or angiotensin-II-receptor blockers may be mixed, combined, dried, formulated or tableted by administration of osmotic agents and conventional additives used pharmaceutically. After the semi-permeable membrane coating base coating step to obtain a delayed-release granules or tablets.
- the second step is a pre-release granule obtained through a conventional procedure for producing oral solids by mixing, coalescing, drying, granulating or coating by administering a statin-based lipid inhibitor and a pharmaceutically acceptable conventional additive. Or obtaining a tablet.
- the granules or tablets obtained in the first step and the second step are mixed with pharmaceutical excipients, tableted or filled to obtain a preparation for oral administration.
- the first step and the second step may be reversed or executed simultaneously.
- the composite formulation of the present invention may be prepared by the above process, and the formulation method is described in more detail as follows, but is not limited thereto.
- the particles or granules obtained in the first step are further coated as they are or with a release controlling material, and then mixed with the granules prepared in the second step and compressed into a certain amount of weight to prepare a tablet.
- the obtained tablet can be film coated as necessary for the purpose of improving stability or property.
- the coated tablets or granules obtained in the first step are further coated as they are or with a release control material, dried, and then compressed into a predetermined amount to prepare tablets as they are or additionally coated, and then separately dissolved simvastatin in an aqueous film coating solution and dispersed
- a release control material dried, and then compressed into a predetermined amount to prepare tablets as they are or additionally coated, and then separately dissolved simvastatin in an aqueous film coating solution and dispersed
- the granules obtained in the first step as they are or are additionally coated and dried with a release controlling substance and the granules obtained in the second step can be prepared in a double tablet using a tablet press.
- Coated multi-layered tablets can be prepared by formulating or coating triple or more multi-layered tablets by adding a release aid layer as required by the formulation design or needs.
- the coated tablet or granules obtained in the first step are additionally coated as it is or with a release control material, dried, and then compressed into a predetermined amount to be coated as it is or additionally to the inner core, followed by a nucleated tableting machine together with the granules obtained in the second step.
- the coated nucleated tablet may be prepared by preparing or coating a nucleated tablet in a form in which a pre-release layer surrounds the surface of the first-stage tablet.
- the granules obtained in the first step are additionally coated as is or with a release controlling substance, and the dried granules or tablets and the granules or tablets obtained in the second step are placed in a capsule charger and filled into capsules of a predetermined size by an effective amount of each active ingredient in an appropriate amount.
- the angiotensin-II-receptor blocker-containing preparation obtained in the first step and the statin-lipid inhibitor-containing preparation obtained in the second step can be prepared together with a foil, a blister, a bottle, and the like to be taken at the same time.
- the human dosage of the preparation of the present invention is appropriately selected according to the absorbency, inactivation rate and excretion rate of the active ingredient in the body, the age, sex and condition of the patient, for example, as follows.
- Pharmaceutical Formulation A is generally administered to adults in the total amount of rozatan and simvastatin, 17.5 to 360 mg per day, preferably 35 to 180 mg per day, so that it can exert anti-compressive action and prevent complications.
- Pharmaceutical preparation B is generally administered to adults in the total amount of olmesartan and simvastatin, 7.5 to 240 mg per day, preferably 15 to 120 mg per day to provide anti-pressure and prevent complications have.
- Pharmaceutical Formulation C is generally administered to adults in the total amount of valsartan and simvastatin, 2.0 to 960 mg per day, preferably 22 to 700 mg per day to provide anti-pressure and anti-complications.
- Pharmaceutical formulation D is generally administered to adults in the total amount of candesartan and simvastatin, 7 to 224 mg per day, preferably 14 to 112 mg per day to provide anti-pressure and prevent complications have.
- Pharmaceutical Formulation E is generally administered to adults about 5 to 40 mg per day with Rochevastatin and about 25 to 100 mg per day with Rozatan for anti-pressure, hypolipidemic and complication prevention effects. You can do it.
- Pharmaceutical Formulation F is generally administered to adults about 5 to 40 mg per day with roschvastatin and about 75 to 300 mg per day with ibesartan for anti-pressure, hypolipidemic and complication-preventing effects. I can do it.
- Pharmaceutical Formulation G is generally administered to adults about 5 to 40 mg per day with roschvastatin and about 4 to 32 mg per day with candesartan for anti-pressure, hypolipidemic and complication prevention. I can do it.
- the combined drug system of the present invention includes two different drugs as the active ingredient, and is formulated into a single compound so that only one dose is administered. Due to the difference in the release time of the drug does not occur between the antagonism between the side effects due to the antagonism can be reduced, the effect of each drug is shown to be improved than the effect of their own alone.
- the formulation of the present invention is a combination formulation of components having different pharmacology, it may not only counteract side effects, but also reduce the risk factors of the development of circulatory complications, thereby reducing the long-term prevention cost, and the single formulation. It is very economically efficient by reducing the packaging cost and maintaining the time required for the administration of high-quality personnel.
- the present invention also provides a pharmaceutical formulation for administration in the evening hours, that is, from 5 pm to 11 pm (17 to 23 pm).
- the present invention also provides a method for treating a cardiovascular disease comprising administering a pharmaceutical agent of the present invention to a mammal.
- the present invention provides a method for treating hypertension and hyperlipidemia or consequent cardiovascular disease or metabolic syndrome, comprising administering a pharmaceutical preparation of the present invention to a mammal at 5 pm to 11 pm once a day.
- the cardiovascular disease is a very broad disease that refers to both cardiovascular and other vascular diseases including cerebrovascular disease.
- Types of cardiac diseases include hypertension, heart failure, arrhythmia, cardiomyopathy, and endocarditis, including ischemic heart disease (myocardial infarction, angina pectoris, etc.) due to the progression of atherosclerosis. .
- ischemic heart disease myocardial infarction, angina pectoris, etc.
- the pharmaceutical formulations of the present invention are very useful in the prevention or treatment of diseases, pharmacologically, clinically, scientifically and economically, than single and simple combination formulations of each drug.
- the pharmaceutical formulations of the present invention prevent antagonism and side effects between the two drugs and exhibit optimal efficacy.
- the pharmaceutical formulation of the present invention can be taken at a time, so that medication guidance and medication for the patient are easy.
- Example 1 is a graph showing the dissolution rate of zoco (simvastatin mono), koza (rozatan mono), pharmaceutical preparations prepared in Example I-1.
- FIG. 3 is a graph showing the dissolution rate of the commercially available zoco (simvastatin mono), koza (rozatan mono), pharmaceutical preparations prepared in Example I-7.
- FIG. 4 is a graph showing the dissolution rate of the commercially available zoco (simvastatin mono), koza (rozatan mono), pharmaceutical preparations prepared in Example I-10.
- FIG. 5 is a graph showing the dissolution rate of the pharmaceutical preparations prepared in Zoco (simvastatin mono), Koza (Rozatan mono), Examples I-12 and I-14.
- FIG. 6 is a graph comparing systolic blood pressure between administration routes as an animal test result according to Experimental Example I-6.
- Example 7 is a graph comparing diastolic blood pressure between administration routes as an animal test result according to Experimental Example I-6.
- Figure 9 shows the dissolution rate of the pharmaceutical preparation (tablet) of the present invention prepared according to Example II-1 and the control agent simvastatin monotherapy (Merck: Zocor), olmesartan medoxomil monotherapy (Daiichi Sankyo: Benicar, Olmetec) The graph shown.
- a pharmaceutical preparation (capsule) of the present invention prepared according to Examples II-5 or II-6 and a control agent simvastatin monotherapy (Merck: Zocor), olmesartan medoxomil monotherapy (Daiichi Sankyo: Benicar, Olmetec) is a graph showing the dissolution rate.
- FIG. 11 shows simvastatin mono (Merck: Zocor), olmesartan medoxomil mono (Daiichi Sankyo: Benicar, which is a pharmaceutical preparation (capsule) of the present invention prepared according to Examples II-7 or II-8) Olmetec) is a graph showing the dissolution rate.
- 13 is a pharmaceutical preparation (film-coated tablets, osmo-core tablets) of the present invention prepared according to Examples II-10 and II-11 and simvastatin monotherapy (Merck: Zocor), olmesartan medoxomil monotherapy ( Daiichi Sankyo: Benicar, Olmetec).
- FIG. 14 is a graph showing dissolution rates of simvastatin / valsartan nucleated tablets prepared according to Example III-1 and valsartan of simvastatin and diovan of each single agent Koza.
- FIG. 15 is a graph showing the dissolution rates of simvastatin / valsartan multi-layered tablets prepared according to Example III-3 and valsartan of simvastatin and dioban of each single agent Koza.
- FIG. 16 is a graph showing the dissolution rate of simvastatin / valsartan matrix tablets prepared according to Example III-5 and valsartan of simovastatin and diovan of each single agent Koza.
- Figure 17 is a graph showing the dissolution rate of the capsule containing simvastatin valsartan tablets prepared according to Example III-8 and the valsartan of simovastatin and dioban of each single agent Koza.
- Fig. 18 is a graph showing the dissolution patterns of the preparations of Zoco (simvastatin monotherapy), Atacane (candesartan monotherapy), and Example IV-1.
- FIG. 20 is a graph showing the dissolution profiles of commercially available Zoco (simvastatin mono), Atacan (candesartan mono), Example IV-7.
- Fig. 21 is a graph showing the dissolution patterns of the preparations of the commercially available zoco (simvastatin mono), atacane (candesartan mono), and Example IV-10.
- FIG. 22 is a graph showing the elution profile of the preparations of Zoco (simvastatin mono), Atacane (candesartan mono), Examples IV-12 and IV-14.
- FIG. 23 shows the Rozatan component and Rocheba of Koza and Cresto, each single agent, as a combined controlled release formulation of Rozatan-Roschvastatin and a control prepared according to Examples V-2, V-4, and V-8. It is a graph which shows the curve of the comparative elution of a statin component.
- FIG. 24 shows the Rozatan component and Roshuba of Koza and Cresto, each single agent, as a combined controlled release formulation of Rozatan-Roschvastatin and a control prepared according to Examples V-6, V-15, and V-17. It is a graph which shows the curve of the comparative elution of a statin component.
- FIG. 25 is a graph showing dissolution rates of roschvastatin and ibesartan in Crest tablets and Aprobel tablets as pharmaceutical preparations and control agents of Examples VI-2, VI-4, VI-8, and VI-9.
- FIG. 26 is a graph showing the dissolution rate of Rochevastatin and Ivesartan in the pharmaceutical preparations and the control agents Crest tablets and Aprobel tablets of Examples VI-6, VI-17, VI-19, and VI-20.
- FIG. 27 is a graph showing dissolution rates of roschvastatin and candesartan in the pharmaceutical formulations and the control formulations of Examples VII-2, VII-4, and VII-8, Cresto tablet and Atacan tablet.
- FIG. 28 is a graph showing dissolution rates of roschvastatin and candesartan in the pharmaceutical and control formulations of Examples VII-6, VII-15, and VII-17, Cresto tablet and Atacan tablet.
- simvastatin and the excipient microcrystalline cellulose, lactose and corn starch were sieved through a No. 35 sieve and mixed with a high speed mixer. Separately, hydroxypropyl cellulose and citric acid were dissolved in purified water (60 mg per tablet) to prepare a binding solution, which was added to a high speed mixer together with the main ingredient mixture and combined. After association, granulation was carried out using an oscillator in No. 18 and dried at 30 ° C. using a hot water dryer. After the drying was finished again using a F-type sizer equipped with No. 20 sieve was prepared simvastatin pre-release granules.
- potassium rozatan, sodium starch glycolate and microcrystalline cellulose were sieved through a No. 20 sieve and mixed for 15 minutes in a double cone mixer to prepare a mixture.
- hydroxypropyl cellulose was dissolved in purified water (140 mg per tablet) to prepare a binding solution.
- the mixture was put into a fluidized bed granulator and granulated by the addition of a binder solution. High speed mixers are optionally used in the assembly process.
- the fluidized bed granulator was a top-spray system using GPCG-1 (Glatt, Germany). After the granules were added, they were preheated under the following conditions.
- the air flow was 80 m 3 / hour, the inlet air temperature was 40 ° C and the filter shaking (delta P filter ⁇ 500pa) was carried out for 5 seconds in 30 seconds in asynchronous mode.
- the bonding liquid was assembled while spraying at 1.0 to 10 g / min.
- the atomizing air was controlled at 1.0 to 2.0 bar and the coating liquid spray angle was adjusted.
- Air flow increases from 80 m 3 / h to 120 m 3 / h as the process proceeds, and the filter shaking (delta P filter ⁇ 4000 pa) is kept in concurrency mode in 1 minute to prevent loss. It was assembled while performing for 5 seconds.
- the fluid bed dryer assembly was dried after assembly was complete.
- GPCG-1 (Glatt, Germany) was used for the fluid bed granule dryer, and the granulation was carried out under the following conditions.
- the air flow was 120 m 3 / hour
- the inlet air temperature was 65 °C
- the filter shaking (delta P filter ⁇ 4000 pa) was performed in asynchronous mode for 5 seconds in 30 seconds.
- the product temperature reaches 40 °C
- the sample was taken and completed if it meets the criteria of 2.5% or less of drying loss.
- a coating solution is prepared by dissolving and dispersing polyethylene glycol 6000 and hypromellose phthalate in ethanol and purified water (8: 2 (v / v)) mixed solvent (300 mg per tablet).
- the granules were subjected to a fluid bed granulation coater (GPCG-1: Glatt, Germany).
- Fluidized bed granulation coater was using a bottom-spray system using GPCG-1 (Glatt, Germany).
- the plate to be adjusted according to the size of granule is B or C type
- the partition gap is 25 mm
- the spray nozzle is 1 mm.
- the granules were added and then preheated under the following preheating conditions. Air flow was 100 m 3 / hour, inlet air temperature was 45 ⁇ 60 °C, product temperature was 40 ⁇ 50 °C, filter shaking (delta P filter ⁇ 500 pa) was performed in asynchronous mode for 5 seconds in 30 seconds. . When the product temperature reached 35 ° C. in the preheating process, the film was coated while spraying the coating liquid at 1 to 5 g per minute.
- the product temperature was maintained at 34 ⁇ 38 °C, when the coating was completed, the product temperature was maintained at 40 °C about 1 hour drying and surface work.
- the coating was completed to prepare rojatan potassium delayed-release granules.
- Two granules of 1) and 2) prepared by the above method and butylate hydroxyanisole were placed in a double cone mixer and mixed. Magnesium stearate was added to the mixture for final mixing. The final mixture was compressed using a rotary tablet press (MRC-33: Sejong).
- a coating solution was prepared by dissolving and dispersing hypromellose 2910, hydroxypropyl cellulose, titanium oxide, and talc in purified water (430 mg per tablet) .
- the above tablet was filmed as a high coater (SFC-30N: Sejong Machinery, Korea).
- a coating layer was formed to prepare a biphasic matrix tablet.
- Potassium rozatan, sodium starch glycolate, microcrystalline cellulose were sieved through a No. 20 sieve and mixed for 15 minutes in a double cone mixer as in the ingredients and contents shown in Table I-1. Separately, hydroxypropyl cellulose was dissolved in purified water (280 mg per tablet) to prepare a binding solution. The mixture was put into a fluidized bed granulator and granulated by the addition of a binder solution. High speed mixers are optionally used in the assembly process. The fluidized bed granulator was a top-spray system using GPCG-1 (Glatt, Germany). After the granules were added, they were preheated under the following conditions.
- the air flow was 80 m 3 / hour, the inlet air temperature was 40 ° C and the filter shaking (delta P filter ⁇ 500pa) was carried out for 5 seconds in 30 seconds in asynchronous mode.
- the bonding liquid was assembled while spraying at 1.0 to 10 g / min.
- the atomizing air was controlled at 1.0 to 2.0 bar and the coating liquid spray angle was adjusted.
- Air flow increases from 80 m 3 / h to 120 m 3 / h as the process proceeds, and the filter shaking (delta P filter ⁇ 4000 pa) is kept in concurrency mode in 1 minute to prevent loss. It was assembled while performing for 5 seconds.
- the fluid bed dryer assembly was dried after assembly was complete.
- GPCG-1 (Glatt, Germany) was used for the fluid bed granule dryer, and the granulation was carried out under the following conditions.
- the air flow was 120 m 3 / hour
- the inlet air temperature was 65 °C
- the filter shaking (delta P filter ⁇ 4000 pa) was performed in asynchronous mode for 5 seconds in 30 seconds.
- the product temperature reaches 40 °C
- the sample was taken and completed if it meets the criteria of 2.5% or less of drying loss.
- a coating solution is prepared by dissolving and dispersing cellulose acetate (acetyl group 32%) and cellulose acetate (acetyl group 39.8%) in a mixed solvent of ethanol and methylene chloride (400 mg per tablet).
- the granules were subjected to a fluid bed granulation coater (GPCG-1: Glatt, Germany).
- Fluidized bed granulation coater was using a bottom-spray system using GPCG-1 (Glatt, Germany).
- the plate to be adjusted according to the size of granule is B or C type
- the partition gap is 25 mm
- the spray nozzle is 1 mm.
- the granules were added and then preheated under the following preheating conditions. Air flow was 100 m 3 / hour, inlet air temperature was 45 ⁇ 60 °C, product temperature was 40 ⁇ 50 °C, filter shaking (delta P filter ⁇ 500 pa) was performed in asynchronous mode for 5 seconds in 30 seconds. . When the product temperature reached 35 ° C. in the preheating process, the film was coated while spraying the coating liquid at 1 to 5 g per minute.
- the product temperature was maintained at 34 ⁇ 38 °C, when the coating was completed, the product temperature was maintained at 40 °C about 1 hour drying and surface work.
- the coating was completed to prepare rojatan potassium delayed-release granules.
- Example I-1 The components and contents described in Table I-1 were carried out in the same manner as in the method of 3) of Example I-1 to prepare a biphasic matrix tablet of the title.
- excipients microcrystalline cellulose, lactose and corn starch were sieved through a No. 35 sieve and mixed with a high speed mixer.
- hydroxypropyl cellulose and citric acid were dissolved in purified water (60 mg per tablet) to prepare a binding solution, which was added to a high speed mixer together with the main ingredient mixture and combined.
- granulation was carried out using an oscillator in No. 18 and dried at 30 ° C. using a hot water dryer. After drying, it was established using an F-type sizer equipped with No. 20 body again. This formulation was placed in a double cone mixer, mixed with butylate hydroxyanisole, and then mixed with magnesium stearate and finally mixed to prepare a simvastatin pre-release compartment.
- Potassium rozatan, sodium starch glycolate, microcrystalline cellulose, and lactose were mixed for 15 minutes in a double cone mixer as in the components and contents shown in Table I-1.
- polyvinylpyrrolidone was dissolved in purified water (40 mg per tablet) to obtain a binding solution.
- the mixture was put into a fluidized bed granulator, granulated by adding a binder solution, and dried.
- the dried product was placed in a fluidized bed coater, and a solution (160 mg per tablet) of ethyl cellulose and Eudragit RL in ethanol and methylene chloride mixture (1: 1 (w / w)) was prepared to prepare the granulated material as a fluidized bed coating. Coating.
- Example I-1 delayed-release granules The conditions of fluid bed granulation, drying, coating and the like are the same as those of Example I-1 delayed-release granules.
- the colloidal silicon dioxide was mixed with the coating by a double cone mixer for 15 minutes.
- Magnesium stearate was added to the mixture, followed by final mixing in a double cone mixer.
- Example I-1 The components and contents described in Table I-1 were carried out in the same manner as in 1) of Example I-3 to prepare the titled simvastatin pre-release compartment.
- Example I-3 The components and contents described in Table I-1 were carried out in the same manner as in 3) of Example I-3, to prepare the title multilayer tablet.
- Example I-1 The components and contents described in Table I-1 were carried out in the same manner as in 1) of Example I-3 to prepare the titled simvastatin pre-release compartment.
- Example I-3 The components and contents described in Table I-1 were carried out in the same manner as in 3) of Example I-3, to prepare the title multilayer tablet.
- Example I-1 The components and contents described in Table I-1 were carried out in the same manner as in 1) of Example I-3 to prepare the titled simvastatin pre-release compartment.
- Example I-3 The components and contents described in Table I-1 were carried out in the same manner as in 3) of Example I-3, to prepare the title multilayer tablet.
- simvastatin and the excipient microcrystalline cellulose, sodium starch glycolate, lactose and corn starch were sieved through a No. 35 sieve and mixed with a high speed mixer.
- hydroxypropyl cellulose and citric acid were dissolved in purified water (100 mg per tablet) to prepare a binding solution, which was added to a high speed mixer with the main ingredient mixture and fed together.
- granulation was carried out using an oscillator in No. 18 and dried at 30 ° C. using a hot water dryer. After drying, it was established using an F-type sizer equipped with No. 20 body again. This formulation was placed in a double cone mixer, mixed with butylate hydroxyanisole, and then mixed with magnesium stearate and finally mixed to prepare a simvastatin pre-release compartment.
- lozatan potassium, polyvinylpyrrolidone copolymer, microcrystalline cellulose, pregelatinized starch and colloidal silicon dioxide were mixed for 15 minutes in a double cone mixer.
- the mixture and magnesium stearate are put into a double cone mixer, and after final mixing, it is compressed into tablets using a rotary tableting machine (MRC-33: Sejong) to make an inner core tablet.
- MRC-33: Sejong rotary tableting machine
- a solution of hypromellose dissolved in ethanol and purified water (8: 2 (v / v)) mixed solvent (20 mg per tablet) and acrylic acid in purified water (90 mg per tablet) were prepared, respectively.
- a high coater SFC-30N: Sejong
- a coating solution for 2 times to prepare a delayed-release coating inner core tablet of the inner core tablet form.
- nucleus tablet tableting machine (RUD-1: Kilian) as the inner layer of the rozatan potassium-coated core tablet in step 2) and the composition containing simvastatin of step 1) as an outer layer
- Hypromellose 2910, hydroxypropyl cellulose, titanium oxide, and talc were dissolved and dispersed in purified water (420 mg per tablet) to prepare a coating solution.
- the nucleated tablets above were added to a high coater (SFC-30N: Sejong) and coated with a coating solution to complete the film-coated nucleated tablets.
- Example I-7 The components and the contents shown in Table I-1 are compressed in the same manner as in 2) of Example I-7 to obtain the inner core tablets.
- a solution of Eudragit RL in ethanol and purified water (8: 2 (v / v)) mixed solvent (20 mg per tablet) and Eudragit RS in purified water (200 mg per tablet) were prepared.
- the inner core tablet was introduced into a high coater (SFC-30N: Sejong) and then coated with a coating solution for 2 times to prepare a delayed-release coated inner core tablet in the form of a core tablet.
- Example I-7 The ingredients and contents described in Table I-1 were carried out in the same manner as in 3) of Example I-7, to thereby prepare the nucleated tablet of the title.
- Example I-7 The components and the contents shown in Table I-1 are compressed in the same manner as in 2) of Example I-7 to obtain the inner core tablets. Separately, a solution of carbomer dissolved in purified water (80 mg per tablet) and an acrylic is dissolved in purified water (160 mg per tablet), respectively.
- the above inner core tablets are added to a high coater (SFC-30N: Sejong) and then coated with a coating solution. Coating over two times to prepare a delayed-release coating inner core tablet in the form of inner core tablets.
- Example I-7 The ingredients and contents described in Table I-1 were carried out in the same manner as in 3) of Example I-7, to thereby prepare the nucleated tablet of the title.
- lozatan potassium, polyvinylpyrrolidone copolymer, microcrystalline cellulose, pregelatinized starch, colloidal silicon dioxide, and sodium chloride were mixed with a double cone mixer for 15 minutes.
- the mixture and magnesium stearate are put into a double cone mixer, and after final mixing, it is compressed into tablets using a rotary tableting machine (MRC-33: Sejong) to make an inner core tablet.
- MRC-33 Sejong
- ethyl cellulose was dispersed in ethanol and methylene chloride (1: 1 (w / w)) mixed solvent (500mg per tablet) and then coated with inner core using a high coater (SFC-30N, Sejong Machinery, Korea). Was coated to prepare an osmotic coated inner core tablet.
- the title osmotic film-coated nucleated tablet was prepared by the method of Example I-7 3) using the rozatan potassium osmotic coated inner core tablet of step 2) as the inner core and the composition containing simvastatin of step 1) as the outer layer. It was.
- the sugar seeds were sieved through a No. 35 sieve according to the ingredients and contents of Table I-2, and charged into a fluidized bed granulator (GPCG 1: Glatt), and then separately mixed with ethanol and purified water (8: 2 (v / v)) (550 mg per tablet).
- GPCG 1 Glatt
- ethanol and purified water 8: 2 (v / v)
- the combined solution in which hypromellose and rozatan potassium were dissolved was sprayed to form rozatan potassium-containing pellets and dried.
- the granules of lozatan potassium retardant pellets were prepared by spraying a solution of hypromellose phthalate in ethanol and purified water (8: 2) (v / v) mixed solvent (300 mg per tablet).
- steps 1) and 2) were mixed in a double cone mixer by the ingredients and contents of Table I-2. Magnesium stearate was added to the mixture and finally mixed with a double cone mixer. The final mixed mixture was placed in a powder feeder and filled using a capsule charger to complete the preparation of the controlled release formulation in capsule form.
- Table I-2 The seeds and components of Table I-2 were sieved through a No. 35 sieve and placed in a fluid bed granulator (GPCG 1: Glatt), followed by dissolving citric acid in ethanol (100 mg per tablet). Simbastatin-dissolved binder solution was sprayed and dried to prepare simvastatin-containing pre-release pellets.
- GPCG 1 Glatt
- the seeds and ingredients of Table I-2 were sieved through a No. 35 sieve and placed in a fluidized bed granulator (GPCG 1: Glatt), followed by separately mixing ethanol and purified water (8: 2 (v / v)) solvent (1400 mg per tablet).
- GPCG 1 Glatt
- the combined solution in which hypromellose and rozatan potassium were dissolved was sprayed to form rozatan potassium-containing pellets and dried.
- the granules were sprayed with acrylide in a mixture of ethanol and purified water (8: 2 (v / v)) in a mixed solvent (300 mg per tablet) to prepare a lozatan potassium delayed pellet.
- steps 1) and 2) were mixed in a double cone mixer by the ingredients and contents of Table I-2. Butyrate hydroxyanisole and magnesium stearate were added to the mixture, followed by final mixing in a double cone mixer. The final mixed mixture was placed in a powder feeder and filled using a capsule charger to complete the preparation of the controlled release formulation in capsule form.
- simvastatin pre-release granules prepared according to the method of Example 1-1 and magnesium stearate were added to a double cone mixer, and finally mixed with a rotary tableting machine (MRC-33: Sejong Machinery , Korea) was compressed into tablets to prepare a prior-release tablet.
- MRC-33 Sejong Machinery , Korea
- the sugar seeds were sieved through a No. 35 sieve according to the ingredients and contents of Table I-2, and charged into a fluidized bed granulator (GPCG 1: Glatt), and then separately mixed with ethanol and purified water (8: 2 (v / v)) (550 mg per tablet).
- GPCG 1 Glatt
- ethanol and purified water 8: 2 (v / v)
- the combined solution in which hypromellose and rozatan potassium were dissolved was sprayed to form rozatan potassium-containing pellets and dried.
- the granules of carnauba wax were dissolved in ethanol and purified water (8: 2 (v / v)) mixed solvent (420 mg per tablet), and then sprayed lozatan potassium delayed pellets were prepared.
- Example 1-2 2 a dried product was prepared according to the method of Example 1-2 2).
- the dried product was put in a fluidized bed coater, and separately cellulose acetate (acetyl group 32%), cellulose acetate (acetyl group 39.8%), and hypromellose were mixed with ethanol and methylene chloride (8: 2 (v / v)) (per tablet).
- the coating solution dissolved and dispersed in 900mg) was prepared and the coating was completed according to the method of 2) of Example I-2 to prepare rozatan potassium delayed-release granules.
- Ethyl cellulose, Eudragit RL and hypromellose phthalate were dissolved and dispersed in ethanol and methylene chloride (1: 1 (w / w)) mixed solvent (400 mg per tablet) according to the components and contents of Table I-2.
- the coating solution was prepared to prepare lozatan potassium delayed-release granules according to the method of Example 1-1 2).
- Carbomer and hypromellose phthalate were dissolved and dispersed in a mixed solvent of ethanol and methylene chloride (1: 1 (w / w)) (800 mg per tablet) according to the ingredients and contents shown in Table I-2.
- Lozatan potassium delayed-release granules were prepared according to the method of Example 1-1 2).
- Eudragit RS was mixed with ethanol and purified water (8: 2 (v / v)) separately. Prepare liquids dissolved in solvent (50mg per tablet) and acrylic acid in purified water (100mg per tablet), respectively.
- the above uncoated tablets are added to a high coater (SFC-30N: Sejong) and then coated with a coating solution. Lozatan potassium delayed-release tablets were prepared.
- ethylcellulose was separately mixed with ethanol and methylene chloride (1: 1 (w / w)) solvent. (50mg per tablet) and acrylic acid dissolved in purified water (200mg per tablet) are prepared respectively, and the above uncoated tablet is added to a high coater (SFC-30N: Sejong) and then coated with a coating solution twice. Rosaza potassium delayed-release tablets were prepared.
- steps 1) and 2) were mixed with a double cone mixer.
- the mixture was placed in a powder feeder and filled using a capsule charger to complete the preparation of the controlled release formulation in capsule form.
- simvastatin pre-release granules of Example I-3 and 2) rozatan potassium delayed-release granule final composition tabletting was carried out using a rotary tablet press (MRC-33: Sejong), respectively, and placed in a blister packaging container. Each tablet was included and packaged for simultaneous use.
- Test method Paddle method, 50 revolutions / minute
- Dissolution test basis Dissolution test method of General Test Method
- Test Method Paddle Method, 50 Turns / Min
- Test drug 0.01M hydrochloric acid solution, 750mL (artificial gas solution)
- the dissolution test showed that the two-phase matrix tablet simvastatin component of the present invention showed almost the same elution characteristics compared to the crude crude tablets, but the lozatan component is very delayed dissolution rate compared to the control formulation Koza You can check As a result of dissolution test of rozatan component, the dissolution rate of rozatan component up to 120 minutes, which is the artificial gastric juice interval, is within 10% in the simvastatin / rozatan biphasic matrix tablet of the present invention, but the control agent is about 60%.
- the dissolution rate of rozatan component in the artificial serous section was 100% in 150 minutes in the control formulation, but it was found to be much slower in the simvastatin / rozatan biphasic matrix tablet of the present invention at about 20% in 240 minutes in total. .
- matrix tablets of simvastatin / rozatan of the present invention can delay the initial release of rozatan by the intended time. Therefore, since the initial release of rozatan is much slower than simvastatin, simvastatin is first metabolized in the liver and is a pharmaceutical agent that can secure sufficient time for regeneration of the metabolic enzyme cytochrome P450.
- lojatan exhibited a more delayed dissolution rate as the amount of ethyl cellulose increased, and specifically, a lozatan dissolution rate within 20% for a total of 240 minutes.
- the pharmaceutical preparation of the present invention is a pharmaceutical preparation capable of sufficiently securing time for regeneration of metabolic enzyme cytochrome P450 after simvastatin is first metabolized in the liver since the initial release of rozatan is much slower than simvastatin.
- simvastatin component of the nucleated tablet of the present invention was found to exhibit almost the same elution characteristics as that of the crude crude tablet, but the rozatan component was compared to Koza, a control formulation, Experimental Example I-1 We can see the very delayed dissolution rate.
- simvastatin / rozatan nucleated tablets of the present invention unlike the dissolution of the combination of rozatan monotherapy and simvastatin monotherapy, simultaneously simbastatin is first metabolized in the liver because the initial release of rozatan is much slower than simvastatin After receiving, it is a pharmaceutical agent that can sufficiently secure time for regeneration of metabolic enzyme cytochrome P450.
- the simvastatin component of the osmotic nucleated tablet of the present invention was found to exhibit almost the same dissolution characteristics as compared to the crude crude tablet, but the rozatan component was a control formulation. Compared to koza, you can see the very delayed dissolution rate.
- simvastatin / rozatan osmotic nucleus tablets of the present invention unlike the dissolution of the combination of rozatan monotherapy and simvastatin monotherapy simultaneously, simvastatin metabolizes the liver first because the initial release of rozatan is much slower than simvastatin. It is a pharmaceutical preparation that can ensure sufficient time to regenerate the cytochrome P450 metabolism-related enzyme after receiving.
- Simvastatin / Rozatan capsules pellet-pellet or capsules (granules-granules) prepared according to Examples I-12 or I-14 above, and each component monoclonal control drug (Zokoco, Korea MSD: Simvastatin mono / Comparative dissolution test of Koza-jung, Korea MSD: Rozatan single agent) was performed. Dissolution test method for each component is the same as Experimental Example I-1, and the results are shown in FIG.
- the simvastatin component of the capsule (pellet-pellet) and (granule-granule) of the present invention was found to exhibit almost the same dissolution properties as compared to the crude crude tablet.
- the lojatan component can be found to have a very delayed dissolution rate when compared to the control agent Koza.
- simvastatin / rozatan capsule pellet-pellet
- granule-granule of the present invention, unlike the dissolution of when the combination of rozatan single drug and simvastatin single drug, the initial release of rozatan than simvastatin Because it is very slow, simvastatin is first metabolized in the liver, and then it is a pharmaceutical agent that has sufficient time to regenerate the metabolic enzyme cytochrome P450.
- This experiment is an embodiment of the present invention as a simultaneous administration group of simvastatin and rozatan and an experimental group to be similar to the release time of each of the commercially available control ingredients (zoco-tablet, Korean MSD: simvastatin monolithic / koza-crystal, Korean MSD: rojatan monolith).
- Korean MSD simvastatin monolithic / koza-crystal
- Korean MSD rojatan monolith
- This test is a rat model animal test, which is designed by dividing light condition and dark condition. The biorhythms of rats and humans are opposite, so when applied to humans, the time zone is reversed.
- the evening time difference administration group (light condition) was the most excellent blood pressure lowering effect among the four groups.
- the pharmaceutical preparations according to the present invention have an optimal blood pressure lowering effect during the time from the morning of the morning until the middle of the day after the administration of the average blood pressure, unlike the conventional co-administration group.
- Example I-7 This experiment was carried out according to Example I-7 of the present invention as a commercially administered group of 'zoco-tablet' (simvastatin 20mg; MSD) alone and 'co-co-tablet' and 'co-crystal' (rozatan potassium 50mg; MSD).
- the provided compositions were administered to conduct clinical trials as shown in Table I-5.
- Lipids measured on day 42 (fasting) at the time of dosing identified by this comparative clinical study are shown in Table I-6.
- the clinical results of the simultaneous administration of simvastatin and rozatan in the pharmaceutical formulation of Example I-7 and the single-component single-administration group showed that the pharmaceutical formulations of the present invention were related to blood pressure drop, lipid reduction, and side effects related biomarkers (Biomakers). In all respects, there were no significant side effects other than the mild side effects that simvastatin and rozatan were administered in each of the eight clinical trials.
- the pharmaceutical formulation of the present invention at the same dosage shows an elevated anti-hyperlipidemic effect of simvastatin and an elevated blood pressure lowering effect of simvastatin than simultaneous administration of simvastatin and rozatan single agent.
- simvastatin Simvastatin, Ranbaxy Laboratories LTD, India
- microcrystalline cellulose AvicelPH, FMC Biopolymer, USA
- mannitol Pearlitol, Roquette America INC, USA
- apples with No. 35 sieve and fluidized bed
- GPCG-1 granulator
- HPC-L hydroxypropyl cellulose
- citric acid Citric acid, Merck, Germany
- High speed mixers (Lab. Pharma Mixer P, Diosna, Germany) can optionally be used in the assembly process.
- a fluid-bed granulator was used for the bottom-spray system. After the granules were added, they were preheated under the following conditions. The air flow was 80 m 3 / hour, the inlet air temperature was 40 ° C and the filter shaking (delta P filter ⁇ 500pa) was carried out for 5 seconds in 30 seconds in asynchronous mode. When the product temperature reached 35 ° C. in the preheating process, the bonding liquid was assembled while spraying at 1.0 to 10 g / min. The atomizing air was controlled at 1.0 to 2.0 bar and the coating liquid spray angle was adjusted.
- Air flow increases from 80 m 3 / h to 120 m 3 / h as the process proceeds, and the filter shaking (delta P filter ⁇ 4000 pa) is kept in concurrency mode in 1 minute to prevent loss. It was assembled while performing for 5 seconds.
- the fluid bed dryer (GPCG-1, Glatt, Germany) was put in the assembly and proceeded under the following conditions.
- the air flow was 120 m 3 / hour
- the inlet air temperature was 65 °C
- the filter shaking (delta P filter ⁇ 4000 pa) was performed in asynchronous mode for 5 seconds in 30 seconds.
- the product temperature reaches 40 °C
- the sample was taken and completed if it meets the criteria of 2.5% or less of drying loss.
- the dried product was established using a No. 20 sieve F-type sizer (KYK-60, Korea Medi, Korea) to prepare simvastatin pre-release granules.
- the finished granules are placed in a double cone mixer (Dasan Pharmatech, Korea), butylhydroxyanisole (Merck, Germany), sodium starch glycolate (JRS, German colloidal silicon oxide (Aerosil 200VV) , Degussa, Germany) was added and mixed, and then mixed with magnesium stearate (Magnesium Stearate, Hwawon Pharm., Korea) for 4 minutes to prepare a simvastatin pre-release final mixture.
- JRS German colloidal silicon oxide
- Hwawon Pharm., Korea Magnerosil 200VV
- Olmesartan Medoxomil (MSL Laboratiories LTD, India), Lactose (Parmatose, DMV Pharma, Netherlands), Sodium croscarmellose (Vivasol, JRS PHARMA, Germany), crosslinking with the ingredients and contents shown in Table II-1 Polyvinylpyrrolidone (Crospovidone, BASF, Germany) and low-substituted hydroxypropyl cellulose (L-HPC, Shin-etsu, Japan) were apologized to No. 35 and placed on a fluid bed granulator (GPCG-1, Glatt, Germany).
- hydroxypropyl cellulose (HPC-L, Nippon soda, Japan, not shown in Table II-1) was separately dissolved in purified water to obtain a binding solution.
- the conditions of fluid bed granulator and fluid bed drying are the same as those of simvastatin immediate-release granules.Separately cellulose acetate 320S (acetal group 32%) (Eastman Chemical Company, USA), cellulose acetate 398NF10 (acetal group 39.8%) (Eastman Chemical) Company, USA), prepared by dissolving hypromellose in a mixture of ethanol and methylene chloride (1: 1 (w / w)) to granulate in the fluidized bed granulation coater (GPCG-1, Platt, Germany) Coating was carried out under the same conditions.
- GPCG-1 fluidized bed granulation coater
- the spray method used a top-spray system.
- the plate to be adjusted according to the size of granule is B or C type, the partition gap is 25 mm and the spray nozzle is 1 mm.
- the granules were added and then preheated under the following preheating conditions. Air flow was 100 m 3 / hour, inlet air temperature was 45 ⁇ 60 °C, product temperature was 40 ⁇ 50 °C, filter shaking (delta P filter ⁇ 500 pa) was performed in asynchronous mode for 5 seconds in 30 seconds. . When the product temperature reached 35 ° C. in the preheating process, the film was coated while spraying the coating liquid at 1 to 5 g per minute.
- the product temperature was maintained at 34 ⁇ 38 °C, when the coating was completed, the product temperature was maintained at 40 °C about 1 hour drying and surface work.
- magnesium stearate was added to the prepared olmesartan medoxomill delayed-release granules, followed by mixing for 4 minutes, thereby preparing an olmesartan medoxomill delayed-release final mixture.
- simvastatin pre-release final mixture was prepared according to the preparation method of Example II-1, according to the preparation method of Example II-1. The mixtures were prepared respectively.
- olmesartan medoxomil delayed-release granules and simvastatin pre-release granules into the granule inlet of the rotary multi- tablet tableting machine [MRC-37T, Sejong Pharmatech, Korea], respectively, and then tableting the tablets with hypromelo OS 2910 and polyethylene glycol 6,000 were dissolved in ethanol and purified water mixture (8: 2 (v / v)) and coated using a coating solution prepared by dispersing titanium oxide.
- Example II-2 Prepared according to the method (2) of Example II-2 with the ingredients and contents shown in Table II-1 to prepare an olmesartan medoxo mill delayed-release final mixture.
- the finished olmesartan medoxomil delayed-release final mixture was stacked in the middle layer (second layer), and the simvastatin rapid-release final mixture prepared according to the method of Example II-1 (1) was divided into one and three layers.
- the tablet into the granule inlet of the triple tablet press (MRC-37T: Sejong Pharmatech, Korea), respectively, and then, after tableting, the tablets with hypromellose 2910 and polyethylene glycol 6,000 were mixed with ethanol and purified water (8: 2 (v / v)) and then coated with a coating solution prepared by dispersing titanium oxide to prepare a tablet.
- a simvastatin pre-release final mixture was prepared using a high speed mixer.
- Olmesartan medoxomil, lactose, croscarmellose sodium, cross-linked polyvinylpyrrolidone, low-substituted hydroxypropyl cellulose as the ingredients and the contents shown in Table II-1 were identified as No. 35 and a high-speed mixer (Lab. Pharma Mixer P) , Diosna, Germany), mixed at high speed for 5 minutes, and then separately added to a binder solution prepared by dissolving hydroxypropyl cellulose in purified water. After the completed granules were dried in a fluidized bed dryer, olmesartan delayed-release granules were prepared using an F-type sizer (KYK-60, Korea Medi, Korea) equipped with a No. 20 sieve.
- F-type sizer KYK-60, Korea Medi, Korea
- Magnesium stearate was added to the finished granules, followed by mixing for 4 minutes to prepare the olmesartan delayed-release final mixture, and tableting with a rotary tablet press (MRC-30, Sejong Pharmatech, Korea) to prepare an inner core.
- a rotary tablet press MRC-30, Sejong Pharmatech, Korea
- the inner core was administered to a coating machine (SFC-30F, Sejong Pharmatech, Korea), and separately cellulose acetate 320S (acetal group 32%), cellulose acetate 398NF10 (acetal group 39.8%), and hypromellose were dissolved in ethanol and methylene chloride. Coated with a coating solution to prepare a coated tablet.
- a coating machine SFC-30F, Sejong Pharmatech, Korea
- cellulose acetate 320S acetal group 32%)
- cellulose acetate 398NF10 acetal group 39.8%
- hypromellose hypromellose
- the above-described simvastatin pre-release final mixture and olmesartan medoxomill delayed-release inner core were compressed together, and then the tablets were tableted and hypromellose 2910, polyethylene glycol 6,000 Was dissolved in ethanol and purified water mixture (8: 2 (v / v)) and then coated with a coating liquid prepared by dispersing titanium oxide to prepare a tablet.
- simvastatin, microcrystalline cellulose, and mannitol to the ingredients and contents shown in Table II-1, apologize with No. 35, put into a high speed mixer (Lab. Pharma Mixer P, Diosna, Germany), mix for 5 minutes at high speed, and then hydroxy Propyl cellulose and citric acid were dissolved in purified water, and then prepared by adding a binding solution.
- Simvastatin pre-release granules were prepared by using an F-type sizer equipped with No. 20 sieve, and granules, butyrate hydroxyanisole, and starch were mixed in a double cone mixer (Dasan Pharmatech, Korea). Sodium lyconate and colloidal silicon oxide were added and mixed for 60 minutes, and magnesium stearate was added and mixed for 4 minutes to prepare a simvastatin pre-release final mixture.
- simvastatin pre-release granules were compressed in a rotary tablet press (MRC-30: Sejong Machinery, Korea).
- the completed granules were dried in a fluidized bed dryer, and then sized using an F-type sizer equipped with No. 20 sieve to prepare olmesartan medoxomil delayed-release granules.
- the granules were put again in a double cone mixer, and Carbomer 71G (Carboxy Vinyl Polymer, Lubrizol, USA) was added in a powder state, mixed for 10 minutes, magnesium stearate was added, mixed for 4 minutes, and olmesartan medoc.
- the low-density delayed-release final mixture was prepared and compressed using a rotary tablet press (MRC-30: Sejong Pharmatech, Korea).
- the tablets are administered to a coating machine (SFC-30F, Sejong Pharmatech, Korea), and olmesar by coating a solution of hypromellose phthalate (HPMCP, Shin-etsu, Japan) in 80% (v / v) ethanol with a coating solution. Burnt Medoxomil delayed-release tablets were prepared.
- Simvastatin pre-release granules were prepared by the same method as the granule preparation method of Example II-5 (method up to olmesartan medoxomill delayed-release granule manufacturing step) using the ingredients and contents shown in Table II-1, and again.
- carbomer 71G was added in a powder state and mixed for 10 minutes to prepare an olmesartan medoxo mill delayed-release final mixture.
- Olmesartan Medoxomil delayed-release tablets were prepared according to the method (2) of Example II-5 with the ingredients and contents shown in Table II-1.
- Simulstatin-olmesartan is filled with the No. 1 hard gelatine capsule by using the capsule filling machine (SFN-8N, Sejong Pharmatech, Korea) using the capsule-filling machine (SFN-8N, Sejong Pharmatech, Korea).
- Medoxomil capsules tablettes + granules were prepared.
- Simvastatin pre-release mixtures were prepared in accordance with the method and the method shown in Table II-1 according to the method of Example II-6 (1) simbastatin pre-release final mixture.
- Olmesartan Medoxomil Delayed-Release Granules contains olmesartan Medoxomil, Lactose, Croscarmellose Sodium, Crosslinked Polyvinylpyrrolidone, and Low Substituted-Hydroxypropylcellulose as the ingredients and contents shown in Table II-1. Apples were placed in a fluidized bed granulator and mixed for 5 minutes to prepare a mixture. Separately, hydroxypropyl cellulose was dissolved in purified water to obtain a binding solution. Conditions such as fluidized bed granulator and fluidized bed drying are the same as those of olmesartan medoxomill delayed-release bed granules.
- the finished product was placed in a fluidized bed coater, and separately prepared by dissolving cellulose acetate 320S, cellulose acetate 398NF10 (acetal group 39.8%), and hypromellose in ethanol and methylene chloride mixture (1: 1 (w / w)).
- the granulated material was placed in a fluidized bed granulator coater and coated under the conditions of Example II-1 (2).
- Simvastatin pre-release granules were prepared with simvastatin pre-release final mixture according to the method (1) of Example II-7 with the ingredients and contents shown in Table II-1.
- olmesartan medoxomill delayed release pellets were sieved through a Sugar Seed (Sugar sphere, NP Pharmaceutical, France) with a No. 35 sieve and placed in a fluid bed coater (GPCG-1, Glatt, Germany), followed by dipping in purified water and ethanol separately.
- the combined solution obtained by dissolving or suspending romeose and olmesartan medoxomil was sprayed to form an olmesartan medoxomil containing pellet and dried.
- the granules were sprayed with a solution of hypromellose phthalate in a mixture of ethanol and methylene chloride (1: 1 (w / w)) to prepare an olmesartan medoxomill delayed-release pellet.
- simvastatin pre-release final mixture prepared by the above method and olmesartan medoxomil delayed release pellets were mixed and filled into capsule No. 1 capsules with a capsule filling machine for simvastatin-olmesartan medoxomil capsules.
- a tablet (granule + pellet) was prepared.
- Simvastatin microcrystalline cellulose, mannitol, lactose and ascorbic acid (Ascorbic acid, Merck, Germany) were weighed into apples with No. 35 sieve and mixed in a double cone mixer for 60 minutes, followed by butylate. Hydroxyanisole, sodium starch glycolate and colloidal silicon oxide were added and mixed again for 60 minutes. After adding magnesium stearate, the mixture was mixed for 4 minutes to prepare a simvastatin pre-release final mixture.
- Olmesartan medoxomil, lactose, croscarmellose sodium, cross-linked polyvinylpyrrolidone, low-substituted hydroxypropyl cellulose as apples No. 35 were mixed with the ingredients and contents shown in Table II-1, and mixed with a double cone mixer for 30 minutes. To prepare a mixture. Separately, hydroxypropyl cellulose was dissolved in purified water to obtain a binding solution. After the above mixture was administered to a fluidized bed granulator or a high speed mixer, the binder was sprayed to prepare granules.
- Carbomer 71G was added to the dried product in a powder state, mixed for 10 minutes, and then sieved to a constant size.
- hypromellose was dissolved in a mixture of ethanol and purified water (8: 2 (v / v)) to prepare a primary coating solution, and acrylic acid (Acryl-eze, Colorcon, USA) was dissolved in 80% (v / v) ethanol.
- a secondary coating solution was prepared. After the preparation of the coating solution was completed, the granules were administered to the fluidized bed coater and coated with the primary coating solution, and then the secondary coating solution was subjected to the secondary coating. After completion of the coating, after adding magnesium stearate and mixing for 4 minutes to prepare an olmesartan medoxo mill delayed-release final mixture.
- simvastatin mixture was mixed with olmesartan medoxomil delayed-release final mixture and then compressed in a rotary tablet press (MRC-30: Sejong Pharmatech, Korea). After tableting was completed, hypromellose 2910 and polyethylene glycol 6,000 were dissolved in a purified water mixture (8: 2 (v / v)) and coated with a coating solution in which titanium oxide was dispersed.
- the dried product is prepared by simulstatin pre-release granules using an F-type granulator equipped with a No. 20 sieve, and the grains, butyrate hydroxyanisole, sodium starch glycolate, and colloidal silicon oxide are mixed in a double cone mixer. After mixing and mixing for 60 minutes, magnesium stearate was added and mixed for 4 minutes to prepare a simvastatin pre-release final mixture.
- simvastatin pre-release granules were compressed in a rotary tablet press (MRC-30: Sejong Pharmatech, Korea).
- Olmesartan medoxomil, croscarmellose sodium, lactose, cross-linked polyvinylpyrrolidone, low-substituted-hydroxypropyl cellulose as apples No. 35 and the ingredients and contents shown in Table II-1 were administered to a fluidized bed granulator, Separately, hydroxypropyl cellulose was dissolved in purified water to prepare a granule by spraying the binding solution. After the granules were dried in a fluidized bed dryer, the dried product was separately prepared by dissolving a polyvinylacetate phthalate (Phthalavin, Colorcon, USA) in ethanol and coated under the same conditions of (2) of Example II-1.
- a polyvinylacetate phthalate Phthalavin, Colorcon, USA
- Carbomer 71G was added to the granules in a powder state and mixed for 10 minutes, and then sieved to a constant size.
- magnesium stearate was added thereto, and mixed for 4 minutes to prepare olmesartan medoxo-million delayed-release granules.
- the finished olmesartan medoxomill delayed-release granules were compressed using a rotary tablet press (MRC-30: Sejong Pharmatech, Korea).
- Each coated tablet was packaged in one PTP (Press Through Pack) packaging container to prepare a packaging kit that can be used at the same time.
- PTP Pressure Through Pack
- the prior-release granules were prepared according to the granulation process of the method (1) of Example II-10 with the ingredients and contents shown in Table II-1.
- Olmesartan medoxomil, croscarmellose sodium, lactose, cross-linked polyvinylpyrrolidone, sodium chloride, low-substituted-hydroxypropyl cellulose as the ingredients and contents of Table II-1 were mixed with a No. 35 sieve and mixed with a double cone mixer. Magnesium stearate was added thereto and finally mixed with a double cone mixer, and the final mixture was compressed using a rotary tablet press (MRC-30, Sejong Pharmatech, Korea).
- ethyl cellulose (Aquacoat ECD, FMC, USA) was dissolved in a mixture of ethanol and methylene chloride (1: 1 (w / w)) as an osmotic coating base, and then a coating machine (SFC-30F, Sejong Pharmatech, Korea) was used. Coated on the inner core to produce an osmotic core tablet.
- Nucleated tablets were prepared by using a nucleated tablet tableting machine (RUD-1: Kilian) as an inner core of an olmesartan medoxomilable osmotic nucleus, tableting with simvastatin prior-release granules as an outer layer, and then forming a film coating layer as a coating machine. After tableting was completed, hypromellose 2910 and polyethylene glycol 6,000 were dissolved in ethanol and purified water mixture (8: 2 (v / v)) and coated using a coating solution prepared by dispersing titanium oxide.
- ROD-1 nucleated tablet tableting machine
- Comparative dissolution test was performed using the simvastatin single agent (Merck: Zocor) and Olmesartan Medoxomil (Sankyo: Benicar, Olmetec) as the pharmaceutical preparation prepared in Example II.
- simvastatin dissolution test the dissolution test was conducted based on the USP30, and in the case of the olmesartan dissolution test, the elution solution was dissolved in 0.1 N hydrochloric acid solution (acid environment) at pH 6.8 (phosphate solution).
- the dissolution test was performed by changing the buffer to simvastatin and olmesartan medoxomil. The results are shown in Figs. 9, 10, 11, 12, and 13 (the test population is 12 each).
- Test method Paddle method, 50 revolutions / minute
- Dissolution test basis The complex preparation obtained in Example II and olmesartan medoxoid tablets are subjected to the dissolution test method of the nine general test methods of the Korean Pharmacopoeia.
- the detailed test method was performed in artificial gastric solution (1st solution of 9 tablets disintegration test method) and warmed to 37 ⁇ 0.5 °C for 2 hours, and then in artificial intestine solution (2nd solution of test method). Dissolution test was conducted.
- Test method Paddle method, 50 revolutions / minute
- Test solution Test solution: 0.1 N hydrochloric acid solution, 500 mL (0 ⁇ 2 hours), 6.8 artificial intestine solution, 900 mL (after 2 hours)
- simvastatin in the pharmaceutical formulation prepared in Example II was eluted 90% of the total amount of simvastatin in the formulation within 30 minutes after the start of elution in the same manner as the control.
- olmesartan is olmesartan up to 2 hours after initiation of simvastatin, unlike olmetec, which is a control agent, releases 90% or more of the total amount of olmesartan medoxomill within 30 minutes after initiation of simvastatin elution. Less than 5% of the total amount of medoc forests was eluted.
- the pharmaceutical formulation of the present invention unlike the dissolution of the simultaneous treatment of the olmesartan medoxomil mono- and simvastatin mono-control drug, simvastatin first in the liver because the initial release of olmesartan medoxomil is much slower than simvastatin After receiving metabolism, it is a pharmaceutical agent capable of sufficiently securing time for regeneration of the metabolic enzyme cytochrome P450.
- simvastatin Simvastatin, Biocon, India
- microcrystalline cellulose Vivapur 12, JRS, Germany
- di-mannitol Pearlitol, Roquette, France
- a high speed mixer Hydroxypropylcellulose Kermel LF, Hercules, USA
- citric acid Citric acid monohydrate, Merck, USA
- Butylated hydroxyanisole (BHA, Merck, USA), sodium starch glycolate (Explotab, JRS, Germany) and colloidal silicon dioxide (Aerosil pharma 200, Degussa, USA) were mixed and magnesium stearate The final mixture was put into a double cone mixer.
- valsartan Dr. Reddy's, India
- Table III-1 valsartan binding solution in which hydroxypropyl cellulose was dissolved in purified water was added to a high-speed mixer, coalesced, granulated using an oscillator, and dried in a fluid bed dryer.
- GPCG-1 (Glatt, Germany) was used for the fluid bed granule dryer, and the granulation was carried out under the following conditions.
- the air flow was 120 m 3 / hour
- the inlet air temperature was 65 °C
- the filter shaking (delta P filter ⁇ 4000 pa) was performed in asynchronous mode for 5 seconds in 30 seconds.
- the product temperature reaches 40 °C
- the sample was taken and completed if it meets the criteria of 2.5% or less of drying loss.
- the dried product is sized using an F-type sizer equipped with a No. 20 body, and put into a double cone mixer, microcrystalline cellulose, crosslinked polyvinylpyrrolidone (Crospovidone, BASF, Germany), colloidal silicon dioxide , Sodium lauryl sulfate (Jeelate, Jeen, USA) was added and mixed with a double cone mixer, and then magnesium stearate was added to the mixture and finally mixed.
- the final mixture was compressed into tablets using a rotary tablet press (MRC-33: Sejong) and used as a nuclear tablet.
- nucleated tablet tablet machine (RUD-1: Kilian) as an inner core of valsartan nuclear tablets and a composition containing simvastatin as an outer layer, followed by hypromellose 2910 (Pharmacoat 603, Shin-Etsu, Japan), hydroxypropyl A coating solution made by dissolving cellulose, titanium oxide (Fanglian, JHP, China), and talc (Talc, Nippon soda, Japan) in a mixture of ethanol and purified water (8: 2 (v / v)). Machine, Korea) to form a film coating layer to produce a nucleated tablet.
- ROD-1 Kilian
- the dried product was sized using an F type sizer equipped with a No. 20 sieve, and put into a fluidized bed granulator (GPCG 1: Glatt).
- the granules were sprayed onto the granules by spraying a hypromellose phthalate solution dissolved in a 1: 1 (w / w) mixture of ethanol and methylene chloride.
- the granules were placed in a double cone mixer, microcrystalline cellulose, cross-linked polyvinylpyrrolidone, colloidal silicon dioxide, and lauryl sulfate sodium were added and mixed.
- Magnesium stearate was added thereto, mixed with a final double cone mixer, and the final mixture was compressed into tablets using a rotary tablet press (MRC-33: Sejong), which was used as an inner core tablet.
- Simvastatin-valsartan nucleated tablets were prepared in the same manner as in Example III-1, 3) Tableting and Coating Method.
- valsartan, microcrystalline cellulose and poloxamer 188 (Lutrol, BASF, USA) were apples in No. 35, mixed with a double cone mixer, and hypromellose was dissolved in purified water.
- the binding solution was sprayed to form granules and dried.
- the granules were sprayed onto the granules by spraying a solution of hypromellose phthalate dissolved in a 1: 1 (w / w) mixture of ethanol and methylene chloride.
- Magnesium stearate was added and mixed in a final double cone mixer.
- the tablets were compressed using a multi-layer tablet press (MRC-37T: Sejong).
- the composition containing simvastatin was placed in a primary powder feeder, and the composition containing valsartan was placed in a secondary powder feeder to minimize mixing between layers.
- It is a coating solution prepared by dissolving hypromellose 2910, hydroxypropyl cellulose, titanium oxide, and talc in ethanol and purified water mixture (8: 2 (v / v)) under the condition that it can be used. , Sejong Machine, Korea) to form a film coating layer to prepare a delayed-release tablet in the form of a multi-layered tablet.
- valsartan, microcrystalline cellulose, and poloxamer 188 were appled in a No. 35 sieve and mixed in a double cone mixer as shown in Table III-1, and the granules were sprayed separately by spraying a binding solution prepared by dissolving hypromellose in water. Formed and dried. The granules were coated by spraying Eudragit RS PO (Evonik, USA) solution dissolved in a 1: 1 (w / w) mixture of ethanol and methylene chloride. Magnesium stearate was added thereto and mixed in a final double cone mixer.
- Simvastatin-valsartan multi-layered tablets were prepared in the same manner as in Example III-3, 3) Tableting and Coating Method.
- simvastatin, microcrystalline cellulose, and di-mannitol were apples mixed with a No. 35 sieve and mixed with a high-speed mixer, as shown in Table III-1, to prepare a binder solution by dissolving hydroxypropyl cellulose and citric acid in purified water.
- the mixture was added to a high-speed mixer and fed together, granulated using No. 20 sieve using an oscillator, dried at 60 ° C. using a hot water dryer, and then re-established into No. 20 sieve, and butylated hydroxyanisole was added thereto. Mixed.
- valsartan, microcrystalline cellulose and poloxamer 188 were appled in a No. 35 sieve and mixed in a double cone mixer as shown in Table III-1.
- the mixture was poured into a fluidized bed granulator (GPCG 1: Glatt) and sprayed with a bonding liquid prepared by dissolving hypromellose in purified water, to form granules and dried. Again, the granules were sprayed onto the granules by spraying a Eudragit RS PO solution dissolved in a 1: 1 (w / w) mixture of ethanol and methylene chloride.
- GPCG 1 Glatt
- Each final composition prepared above was mixed with a double cone mixer, sodium starch glyconate and colloidal silicon dioxide were mixed, and magnesium stearate was added thereto, followed by final mixing with a double cone mixer.
- the final mixture was compressed into tablets using a rotary tablet press (MRC-33: Sejong), and then a high coater (SFC-30N) was prepared by dissolving hypromellose 2910, hydroxypropyl cellulose, titanium oxide, and talc in ethanol and purified water. , Sejong Machine, Korea) to form a film coating layer to prepare a two-phase matrix tablet.
- MRC-33 Sejong
- SFC-30N high coater
- Valsartan, microcrystalline cellulose and poloxamer 188 were appleted in a No. 35 sieve and mixed in a double cone mixer as shown in Table III-1 below.
- the mixture was added to a fluidized bed granulator (GPCG 1: Glatt), and separately Granules were formed by spraying a combined solution prepared by dissolving melose in purified water.
- the granules were dried by spraying a solution of hypromellose phthalate dissolved in a (1: 1 (w / w)) mixture of ethanol and methylene chloride. Granules were coated.
- Simvastatin-valsartan biphasic matrix tablets were prepared by following the 3) post-mixing, tableting and coating methods of Example III-5.
- the final compositions of steps 1) and 2) were mixed with a double cone mixer, sodium starch glycolate was added thereto, mixed with a double cone mixer, colloidal silicon dioxide was mixed, and magnesium stearate was added to the final mixture. .
- the final mixed mixture was put into a powder feeder and filled into No. 1 gelatin hard capsules using a capsule charger.
- simvastatin, microcrystalline cellulose, and di-mannitol were appled into No. 35 sieve and mixed with a high speed mixer.
- hydroxypropyl cellulose and citric acid are dissolved in water to prepare a binding solution, which is combined with a main ingredient mixture in a high-speed mixer, fed together, granulated using No. 20 sieve using an oscillator, and dried at 60 ° C using a hot water dryer, and then again.
- Butylated hydroxyanisole, sodium starch glycolate, and colloidal silicon dioxide were mixed, and magnesium stearate was added and finally mixed with a double cone mixer, and the final mixture was subjected to a rotary tablet press (MRC-33).
- Tablet was prepared by dissolving and dispersing hypromellose 2910, polyethylene glycol 6000 (PEG6000, Duksan, Korea), titanium oxide and talc in 80 (w / w)% ethanol. It was coated with a high coater (SFC-30N, Sejong Machinery, Korea).
- the simvastatin tablet of step 1) and the valsartan tablet of step 2) were filled into No. 1 hard gelatin capsules using a capsule charger.
- the apples were mixed with simvastatin, microcrystalline cellulose and di-mannitol No. 35 as shown in Table III-1, and mixed in a high speed mixer. Separately, hydroxypropyl cellulose and citric acid were dissolved in purified water to prepare a binder solution, and the main component mixture was prepared. The mixture was put into a high-speed mixer and combined, granulated using No. 20 sieve using an oscillator, dried at 60 ° C. using a hot water dryer, and then re-established into No. 20 sieve. Here, butylated hydroxyanisole and starch glycone were added. Sodium acid and colloidal silicon dioxide were mixed, and magnesium stearate was added thereto, followed by final mixing in a double cone mixer.
- Simvastatin tablets of step 1) and valsartan tablets of step 2) were filled into 1st hypromellose hard capsules using a capsule charger.
- simvastatin As shown in Table III-1, simvastatin, butylated hydroxyanisole, colloidal silicon oxide, hypromellose 2910, polyethylene glycol 6000, titanium oxide, talc, ethanol and methylene chloride (1: 1 (w / w)) dissolved and dispersed in a mixed solution to prepare a pre-release simvastatin coating solution.
- Valsartan tablets prepared above were administered to a high coater (SFC-30N, Sejong Machinery, Korea) and then first coated with simvastatin coating solution.
- Hypomellose 2910, hydroxypropyl cellulose, titanium oxide, and talc described in the coating layer of Table III-1 were first coated with a coating solution prepared by dissolving ethanol and purified water mixture ((8: 2 (v / v)). The secondary coating on to prepare a film coated tablets.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Epidemiology (AREA)
- Diabetes (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Emergency Medicine (AREA)
- Cardiology (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Heart & Thoracic Surgery (AREA)
- Inorganic Chemistry (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
본 발명은 (1) 심바스타틴을 함유하는 선방출성 구획과 로자탄을 함유하는 지연방출성 구획을 포함하는 약제학적 제제, (2) 심바스타틴을 함유하는 선방출성 구획과 올메사르탄을 함유하는 지연방출성 구획을 포함하는 약제학적 제제, (3) 심바스타틴을 함유하는 선방출성 구획과 발사르탄을 함유하는 지연방출성 구획을 포함하는 약제학적 제제, (4) 심바스타틴을 함유하는 선방출성 구획과 칸데사르탄을 함유하는 지연방출성 구획을 포함하는 약제학적 제제, (5) 로슈바스타틴을 함유하는 선방출성 구획과 로자탄을 함유하는 지연방출성 구획을 포함하는 약제학적 제제, (6) 로슈바스타틴을 함유하는 선방출성 구획과 이베사르탄을 함유하는 지연방출성 구획을 포함하는 약제학적 제제, (7) 로슈바스타틴을 함유하는 선방출성 구획과 칸데사르탄을 함유하는 지연방출성 구획을 포함하는 약제학적 제제를 제공한다. 본 발명의 약제학적 제제는 제제 내 다른 구획내 포함된 상이한 약물이 시간차를 두고 방출됨으로써 약물 상호간 길항작용 및 부작용을 최소화하고, 약물 본래의 효과를 최대화한다. 본 발명의 두가지 약물의 특정 조합에 의한 약제학적 제제는, 각각의 단독 투여 및 단순 복합 투여에 비하여 현저히 우수한 질환의 치료 내지 예방 효과를 나타낸다.
Description
본 발명은 (1) 심바스타틴을 함유하는 선방출성 구획과 로자탄을 함유하는 지연방출성 구획을 포함하는 약제학적 제제, (2) 심바스타틴을 함유하는 선방출성 구획과 올메사르탄을 함유하는 지연방출성 구획을 포함하는 약제학적 제제, (3) 심바스타틴을 함유하는 선방출성 구획과 발사르탄을 함유하는 지연방출성 구획을 포함하는 약제학적 제제, (4) 심바스타틴을 함유하는 선방출성 구획과 칸데사르탄을 함유하는 지연방출성 구획을 포함하는 약제학적 제제, (5) 로슈바스타틴을 함유하는 선방출성 구획과 로자탄을 함유하는 지연방출성 구획을 포함하는 약제학적 제제, (6) 로슈바스타틴을 함유하는 선방출성 구획과 이베사르탄을 함유하는 지연방출성 구획을 포함하는 약제학적 제제, (7) 로슈바스타틴을 함유하는 선방출성 구획과 칸데사르탄을 함유하는 지연방출성 구획을 포함하는 약제학적 제제에 관한 것이다.
고혈압이란 혈압이 정상 범위를 넘어 높게 유지됨에 따라 초래되는 병증으로, 일반적으로 수축기 혈압이 140mmHg 이상이거나 확장기 혈압이 90mmHg 이상인 경우를 의미한다. 우리나라 성인의 5명중 1명이 그 대상일 정도로 발생빈도가 높은 만성 순환기계 질환이며, 그 발생 빈도는 전세계적으로 증가하고 있는 추세이다. 또한 고혈압은 중증도 이하의 환자에서도 외견상의 증상이 없음에도 불구하고 뇌졸증, 심부전, 관상동맥질환등 치명적인 합병증을 유발할 수 있기 때문에 보다 적극적인 환자의 관리와 치료가 요구되는 질환이다.
고혈압은 매우 다양한 원인에 의해 중복적으로 유발되는 증상이다. 따라서 단일 항압제를 사용하는 경우 어떤 결과가 나오는지 미리 판단하기 어렵다[Journal of hypertension 1995: 9: S33-S36]. 고혈압 환자의 2/3이상은 상기와 같은 하나의 단일제제의 투약으로서 조절되지 않으며 서로 분류가 다른 2가지 이상의 고혈압 약이 필요하다고 보고되었다. 단일제 고혈압 약으로서는 혈압을 목적하는 수준으로까지 내리기 어려우며, 유의적인 치료효과를 얻기 위해서는 서로 분류가 다른 2가지 이상의 약물의 복합투여가 이루어져야 한다.
일반적으로 혈압은 나이와 함께 상승하는 경향이 있다. 60세 이상에서는 약 63%가 고혈압의 증상이 나타난다. 특히, 60세 전후에 수축기 혈압은 높아지고, 확장기 혈압은 오히려 낮은 고립성 수축기 고혈압이 나타나게 된다. 이를 노인성 고혈압이라고 한다. 노인성 고혈압은 낮과 밤동안 24시간을 균형있게 혈압을 유지시켜야 수면 중에 나타날 수 있는 급발성 심장마비도 예방해 주고, 낮 활동시의 과격한 스트레스로 발생한 고혈압으로 인한 뇌졸중도 예방해 줄 수 있다.
한편, 고혈압 환자들이 노화할수록 신기능이 저하된다. 이로 인해 혈압 상승인자인 안지오텐신Ⅱ의 분비가 증가되어 혈압을 더욱 올라가게 한다. 이러한 경우 단일제 고혈압 약으로서는 수축기 혈압을 목적하는 수준으로까지 내리기 어려우며, 고혈압 약의 복합 처방으로 원하는 항압 효과를 기대할 수 있다.
고혈압 환자의 치료에서 또 다른 중요한 요소는 혈압의 생체리듬(Circadian variation of blood pressure)을 고려해야 함이다. 환자를 포함한 일반인의 하루 중 혈압은 수면-기상 주기(sleep-wake cycle)에 맞춰 밤과 새벽 사이에 혈압이 하강하고, 기상 후 오전 중의 혈압 상승을 시작으로 하여 일과시간 중(활동 중)에는 혈압이 상승해 최고조에 이르게 된다. 이러한 이유로 고혈압 합병병증의 관리의 치료는 혈압이 최고조에 이르는 발생 위험시간은 이른 아침시간부터 오전일과 시간대를 중심으로 이루어 져야 하는 것으로 알려져 있다 [Adv. Drug Deliv. Rev., 2007: 923-939].
또한, 일반 고혈압 환자와 달리 수면 중 혈압이 저하되지 않는 유형의 고혈압인 Non-dipper형 환자의 경우도 허혈성 심질환, 뇌졸증등 합병증 발현의 위험이 높아 생체리듬을 감안한 치료가 이루어져야 한다[Adv.Drug Deliv. Rev., 2007: 904-922].
따라서, 고혈압 치료는 질환의 다인성, 다형성을 이해하고 병증에 적합한 제제화를 통하여, 이를 최적의 시간대에 투여함으로써, 24시간 균등하게 혈압을 유지시켜 주고 나아가 이를 통하여 치명적인 합병증을 예방하여야 한다. 또한, 고혈압 치료는 혈압만 내리는 것이 목적이 아니다. 고혈압 환자에게 합병되기 쉬운 심근 경색, 심부전, 뇌졸중, 조기 사망 등의 심혈관계 질환을 예방해 주고 그 병태의 악화를 막아 주어 건강하게 오래 살 수 있도록 하는 것이 고혈압 치료의 목적인 것이다.
고혈압 치료를 위한 지침으로서 복합 처방의 필요성을 요약하면 다음과 같다[J. Hum. Hypertens 1995: 33-36].
1) 동일 환자라 할지라도 다양한 원인이 중복되어 고혈압을 일으키고 있다.
2) 단일제제가 다양한 병태를 모두 다스릴 수는 없다.
3) 단일제제의 효과는 처방 환자의 50% 이하에게 유효하다.
4) 복합제제의 효과는 처방 환자의 80% 이상에게 유효하다.
5) 특히, 당뇨병 등 합병증을 지닌 자의 고혈압에 대해 단일제제는 목적하는 바의 항압 효과를 얻을 수 없을 뿐 아니라 합병증을 예방하기는 더욱 어렵다.
6) 저용량의 단일제제의 사용으로 효과를 보지 못한 경우 용량을 증가시키면 부작용만 증가시키는 경우가 많지만 복합제 사용으로 이같은 부작용을 줄일 수 있다.
7) 복합제제는 약리 작용을 달리하는 약효군을 복합함으로써 다양한 원인을 제거함과 동시에 합병증을 예방하고 부작용을 상쇄시킬 수가 있다. 따라서, 고혈압을 처음부터 치료하는 경우에도 단일제로 시작하는 것보다 복합제로 시작하는 것이 최선의 치료법이라고 미국 심장학회(American Heart Association)는 강조하고 있다.
8) 특히, 합병증을 지닌 고혈압 환자는 합병증이 없는 고혈압 환자보다 혈압을 더 내려야 한다. 이러한 경우는 반드시 복합 처방이 필수적이다. 단일 제제를 사용하는 경우 26%의 환자에게만 효과를 볼 수 있지만 복합 처방은 무려 74%의 환자에게 목표로 하는 혈압을 유지시켜 합병증의 악화를 예방 할 수 있다[ Hypertension Optimal Treatment, United Kingdom Prospective Diabetes Study, 대단위 임상].
9) 미국 FDA는 30년 전부터 이른바 고정 비율 복합 원리(Fixed-dose Combination Therapy)에 의거 복합 제제의 필요성을 인정해 왔다. 고정 비율 복합 원리란 약리 작용이 서로 다른 약물을 복합시킬 때는 단일제제 각각을 단독으로 처방 할 때와 같은 양으로 복합시켜야 한다는 원리다. 이를 고정 비율 복합제제라 하며, 단일제제의 약효와 안전성이 이미 인정되어 있고 처방의들에 의해 복합 처방이 실시되고 있는 한 이러한 복합제제는 별도의 실험 없이 허가되고 있다.
10) 고정 비율 복합 항압제는 단일제제보다 혈압 강하 작용이 우수하다는 것은 주지되어 있다.
11) 개별 성분의 용량을 증가시키지 아니하므로 개별 성분의 부작용 출현을 현저히 예방할 수 있다.
12) 항압제의 부작용은 상당수가 순환기계에 대한 부작용이다. 따라서, 서로 다른 약리를 지닌 성분을 복합함으로써 서로의 부작용을 상쇄시키는 경우가 많다.
13) 복합제제는 환자의 복약 준수를 쉽게 해준다. 노년 인구 증가에 따라 복약 지도에 소요되는 처방의의 시간 낭비를 줄일 수 있다.
14) 복합제제는 순환기계 합병증의 발병위험 인자를 감소시켜 줄 수 있으므로 장기간의 예방 경비를 절감시켜 줄 수 있다.
15) 단일제제를 각각 유지하는 포장비용 절감과 고급 인력의 투약 조제 시간 절감이 가능하다.
상기에서 언급한 다인성, 다형성의 고혈압에 효과적인 약리활성 성분으로서, 그 화학구조의 유사성 또는 치료 기전에 따라 칼슘채널차단제, 안지오텐신 Ⅱ 수용체 차단제 레닌 차단제, 베타 아드레날린 차단제, 안지오텐신 전환효소 억제제, 이뇨제 등을 들 수 있다.
또한, 고혈압 치료를 위해 권장되는 복합 처방의 조합으로 HMG-CoA 환원효소 억제제인 스타틴계 지질저해제와 안지오텐신 II 수용체 차단제가 있다.
1. HMG-CoA 환원효소 억제제인 스타틴계 지질 저해제
HMG-CoA 환원효소의 억제제는 HMG-CoA 환원효소인 3-하이드록시-3-메틸글루타릴-코엔자임에이(HMG-CoA)에서 메발로네이트의 전환을 촉매하는 것을 강력히 억제하여 간에서의 콜레스테롤 생성을 억제하고 저밀도 지단백 콜레스테롤(LDL-C)을 저하시키는 효과를 나타내며, 이에 속하는 스타틴계 지질저해제에는 심바스타틴, 로슈바스타틴, 아토르바스타틴, 피타바스타틴, 플루바스타틴, 로바스타탄, 프라바스타틴 등이 있으며, 구체적으로 심바스타틴과 로슈바스타틴에 대해 살펴보면 다음과 같다.
1) 심바스타틴(Simvastatin)
심바스타틴은 HMG-CoA 환원효소[3-하이드록시-3-메틸글루타릴-코엔자임에이 환원효소]가 메발로네이트로 전환시키는 것을 강력히 억제하여 간에서의 콜레스테롤 생성을 억제하고 저밀도 지단백질 콜레스테롤(LDL-C)을 저하시키는 효과를 나타내는 스타틴계 지질 저하제 중 하나이며[참조: Lancet 1995; 346: 750-753, Am J Cardiol 1998; 82: 57T-59T, Am J Cardiol 1995; 76: 107C-112C, Hypertens Res 2003; 26: 699-704, Hypertens Res 2003; 26: 273-280.]Br Med Bull 2001; 59: 3-16, Am J Med 1998; 104 (Suppl 1): 6S-8S, Clin Pharmacokinet 2002; 41: 343-370], 복합 고지혈증, 동맥경화장애 및 관상 심장 질환의 치료 및 예방에 매우 효과적인 약물로 알려져 있다[참조: "Scandinavian Simvastatin Survival Study" published in the Lancet, vol.344,(1994), p.1383-89; Lancet, 1994; 344: 1383-1389].
또한, 심바스타틴은 그 자체로는 불활성이나, 간에서 활성형인 β-히드록시산 형태로 변하며, 간 내의 효소인 사이토크롬 P450 3A4에 의해 대사되고 간 내에서 작용하면서 간으로부터 배설된다. 한편, 사이토크롬 P450 3A4에 의해 대사되지 않은 심바스타틴이 혈중에 고농도로 존재하면 심바스타틴의 부작용인 근육 융해증과 같은 근육 장애의 유발 위험이 더욱 증가된다[참조: Drug Metab Dispos 1990; 18: 138-145, Drug Metab Dispos 1990; 18: 476-483, Drug Metab Dispos 1997; 25: 1191-1199, Clin Pharmacol Ther 1998; 63: 332-341 ; Clin Pharmacol Ther 1998; 64: 177-182 ; Physicians Desk Reference 2006 (Zocor) ; J Pharmacol Exp Ther 1997; 282: 294-300 ; Pharmacol Exp Ther 1999; 290: 1116-1125 ; Life Sci 2004; 76: 281-292].
2) 로슈바스타틴(Rosuvastatin)
로슈바스타틴의 화학명은 (3R,5S,6E)-7-[4-(4-플루오로페닐)-2-(N-메틸메탄술포아미도)-6-(프로판-2-일)피리미딘-5-일]-3,5-디히드로옥시헵트-6-에노산으로, HMG-CoA 환원효소 억제제이다. 로슈바스타틴은 콜레스테롤 합성경로를 조절하는 HMG-CoA 환원효소를 억제하는 약물 중 대표적인 약물로 간에서 콜레스테롤 생성을 억제한다. 이러한 작용으로 로슈바스타틴은 총콜레스테롤(Total-C), 저밀도지질단백질-콜레스테롤(LDL-C), 및 트리글리세라이드를 감소시키고 고밀도지질단백질-콜레스테롤(HDL-C)를 증가시키는 작용을 지니고 있다[Judy et al., Clinical Therapeutics. vol.26(9)(2004), p1368-1387]. 이러한 지질조절효과로 로슈바스타틴은 원발성 고지혈증(primary hyperlipidemia), 복합형 이상지혈증(mixed dyslipidemia) 및 가족성 고콜레스테롤혈증(familial hypercholesterolemia)의 치료에 사용되고 있다.
로슈바스타틴을 사이토크롬 P450 2C9 효소에 의해 대사되는 약물과 병용하는 경우 로슈바스타틴의 간 내 대사가 억제되어 혈중 농도가 증가하게 되어 단일제에서는 예측할 수 없었던 복합제에서의 부작용을 유발할 수 있다. 또한 반복 병용 투여시에는 대사되지 않고 남은 약물이 축적되어 정상상태의 혈중농도가 단일제를 복용할 때보다 증가하는 현상을 보이게 되며 이로 인하여 고용량을 복용 했을 시에 나타나는 부작용 및 이상작용을 피할 수가 없게 된다. 특히 신부전환자나 간부전환자의 경우에는 약물대사능이 정상적이지 않아 복용량에 대한 신중한 투여가 고려되는데 이러한 점은 거의 알려져 있지 않기 때문에 복용하는 환자는 거의 인지하지 못한 채 복용하고 있다. 병용 투여의 유익성이 병용 투여로 인하여 증가되는 위험성을 상회하지 않는다면 다른 약물과의 병용 투여는 피해야 하는 것이 병용 투여의 원칙이나, 안지오텐신―Ⅱ―수용체 차단제와 로슈바스타틴의 병용 투여시 얻을 수 있는 임상적인 상승효과가 크기 때문에 혈중농도 상승에 따른 부작용의 위험에도 불구하고 병용 투여를 해왔다.안지오텐신―Ⅱ―수용체 차단제(Angiotensin-Ⅱ Receptor blocker, 이하 'ARB')는 혈관수축 인자의 작용을 차단시키고 혈압상승물질인 안지오텐신-II를 증가시키는 알도스테론 작용을 차단시켜 혈관을 이완시키는 작용을 한다. 이러한 안지오텐신-II 수용체 차단제는 자정 이후 수면 중의 RAAS(Renin and Angiotensin System)계 흥분상태를 억제하므로 자정 이후 항압효과가 강하여 Non-dipper형 고혈압 환자에 적합하다.
2. 안지오텐신-II 수용체 차단제
안지오텐신-II-수용체 차단제(ARB) 약물은 혈압 강하 작용을 하면서 심부전 예방 및 치료, 심근 경색 후 부정맥, 당뇨병성 합병증 예방 및 치료, 신부전 예방 및 치료, 뇌졸중 예방 및 치료, 항 혈소판 작용, 동맥 경화 예방 작용, 알도스테론 유해 작용 억제, 대사 증후군 영향력 완화, 순환기계 질환 연쇄적 악화 예방 등 광범위한 작용을 나타내는 약물이다[Clin,Exp.Hypertens.,vol.20(1998), p.205-221, J.Hypertens., vol. 13 (8) (1995), p.891-899, Kidney Int., vol.57(2)(2000), p.601-606, Am.J. Hypertens., vol.10 (12PT2) Suppl. (1997), p.325-331, Circulation, vol. 101(14)(2000), p.1653-1659, J.Hypertension., vol 17 (7) (1999), p.907-716, Circulation, vol.101(2000), p.2349].
1) 로자탄(Losartan)
로자탄[2-부틸-4클로로-1-[2-(1H-테트라졸-5-일)비페닐-4-일메틸]-1H-이미다졸-5-메탄올]은 안지오텐신2가 혈관벽 수용체에 결합하는 것을 길항하는 항고혈압제이다. 이 안지오텐신-II는 혈압 상승, 좌심실 비대, 혈관 비대, 죽상 경화, 신부전, 뇌졸중 등을 유발하는 인자이다[미국특허 제 5,138,069호 공보]. 로자탄은 심부전 예방 및 치료, 심근 경색 후 부정맥, 당뇨병성 합병증 예방 및 치료, 신부전 예방 및 치료, 뇌졸중 예방 및 치료, 항혈소판 작용, 동맥 경화 예방 작용, 알도스테론 유해 작용 억제, 대사 증후군 영향력 완화, 순환기계 질환 연쇄적 악화 예방 등 광범위한 작용을 나타내는 약물로 알려져 있다[Clin,Exp.Hypertens. 1998, 20, p.205-221. Circulation, 2000;101,p.1653-1659].
로자탄은 흡수되면 일차로 간으로 들어간다. 그 중 일부는 활성형 자체인 로자탄 분자 그대로 혈중으로 유출되어 1시간 내에 혈중 최고 농도에 이르게 된다. 그러나 나머지 일부는 간 내 효소 사이토크롬 P450 2C9 와 3A4 라는 두 가지 효소에 의해 대사를 받아 더욱 활성이 높은 로자탄 카르복실산으로 변화된 후 3-4 시간 후에 최고 혈중 농도에 이르게 된다. 즉, 로자탄의 약리 작용은 로자탄과 로자탄 활성대사체인 로자탄 카르복실산 혼합체의 약리 작용이다. 경구 투여 용량의 약 14%가 간내 효소에 의해 활성형 대사체인 로자탄 카르복실산의 형태로 전환되며,활성형 대사체는 로자탄의 40배에 해당하는 약리 작용을 나타낸다. 혈중 소실 속도도 로자탄이 600mL/min이고, 활성형 대사체가 50mL/min 으로서 활성형 대사체가 더욱 느린 소실속도를 나타내어 지속적인 작용 시간의 유지에 중요한 역할을 한다.
로자탄은 적정 투여량에서 심근 수축기와 확장기에 대한 혈압강하 효과를 보이며, 고혈압의 제반증상에 관련된 부가적인 심부전 예방 및 치료, 심근 경색 후 부정맥과 심부전 예방 치료, 당뇨병성 합병증 예방 치료, 신부전 예방 치료, 뇌졸중 예방 치료, 항 혈소판 작용, 동맥 경화 예방 작용, 알도스테론 유해 작용억제, 대사 증후군 작용 완화, 순환기계 질환 연쇄적 악화예방 효과, 야(夜)뇨로 인한 수면장애 등 광범위한 작용을 나타내는 약물이다[참조: Clin,Exp.Hypertens., vol.20(1998), [p.205-221] ; J.Hypertens .,vol. 13 (8) (1995), [p.891-899] ; Kidney lnt., vol.57(2)(2000),[p.601-606] ; Am.J.Hypertens.,vol.10 (12PT2) Suppl. (1997), [p.325-331] ; Circulation, vol. 101(14) (2000), [p.1653-1659] ; J.Hypertension., vol 17 (7) (1999),[p.907-716] ; Circulation, vol.101(2000),p.2349].
2) 이베사탄(Irbesartan)
이베사탄은 비펩타이드성 안지오텐신-II-수용체 차단제중 대표적인 약물로서, 혈관 평활근 세포와 부신과 같은 조직에서 안지오텐신-II가 수용체에 결합하는 것을 선택적으로 억제하여 혈관을 이완시키는 작용을 한다.[M burnier et al., The Lancet. vol.355(2000), p637-645] 이러한 혈관이완작용으로 이베사탄은 고혈압 및 제2형 당뇨병 환자의 신질환(nephropathy in Type 2 Diabetic Patients) 치료에 사용되고 있다.
3) 발사르탄(Valsartan)
안지오텐신-II 수용체 차단제 중 발사르탄 [화학식명 : N-(1-옥소펜틸)-N-[[2'-(1H-테트라졸-5-일)[비페닐-4-일]메틸]-L-발린]은 혈관수축인자의 작용을 차단시키고 혈압 상승 물질인 안지오텐신 Ⅱ를 증가시키는 알도스테론 작용을 차단시켜 혈관을 이완시키는 항압제이다. 안지오텐신Ⅱ는 혈압 상승, 좌심실 비대, 혈관 비대, 죽상 경화, 신부전, 뇌졸중 등을 유발하는 인자이다[미국특허 제 5,399,578 호 공보].
발사르탄은 안지오텐신-II 수용체 길항제에 속하는 약물로 1996년 독일에서 처음 발매됐고 96년에 미 FDA 승인을 받은 매우 뛰어난 혈압 강하 효과를 보이는 항고혈압제이다. 경증에서 중등도의 고혈압이 있는 61~80세 노인들을 대상으로 16주간 진행한 임상 연구에서 수축기 혈압을 평균 18.6㎜Hg, 이완기 혈압은 13.7㎜Hg 낮추는 것으로 확인됐다. 발사르탄은 뛰어난 혈압 강화 효과와 함께 심부전, 심근경색증 등 더욱 광범위한 심혈관 질환의 치료제로 사용되고 있고 2003년 미 심장학회에서 발표된 임상 연구에서 발사르탄은 심근경색 후 환자의 사망률을 25% 낮추는 것으로 나타났다. 31개국의 환자 1만5000여명을 대상으로 한 임상 연구에서 발사르탄이 장기적인 심장 보호 효과도 있는 것으로 밝혀졌다. 이러한 특징을 가진 발사르탄은 한밤중 이후 새벽까지의 혈압 강하 작용이 강한 것으로 알려져 있다[Hypertension, 2003; 42: 283-290], [Chronobiol. Int., 2005; 22: 755-776].
ARB 중 하나인 발사르탄은 RAAS(Renin and angiotensin system)가 강하게 작동하는 한밤중 이후 새벽까지 강한 혈압 강하 효과를 발휘한다[J. Hypertens, 2005; 23: 1913-1922], [Hypertension, 2003; 42: 283-290], [Chronobiol. Int. 2005; 22: 755-776].
4) 칸데사르탄(Candesartan)
칸데사르탄[2-에틸옥시-1-({4-[2-(2H-1,2,3,4-테트라졸-5-일)페닐]-페닐}-1H-1,3-벤조디아졸-6-카르복실산]은 비펩타이드성 안지오텐신-II-수용체 차단제 중 대표적인 약물로서, 혈관 평활근 세포와 부신과 같은 조직에서 안지오텐신-II가 수용체에 결합하는 것을 선택적으로 억제하여 혈관을 이완시키는 작용을 한다.[M burnier et al., The Lancet. vol.355(2000), p637-645] 이러한 혈관이완작용으로 칸데사르탄은 고혈압 및 제2형 당뇨병 환자의 신질환(nephropathy in Type 2 Diabetic Patients) 치료에 사용된다.
칸데사르탄은 생체이용률이 낮기 때문에 칸데사르탄의 프로드럭 형태로 시판되고 있다(칸데사르탄 실렉세틴 정제 15%, 용액 40%). 이는 소장 벽에서 칸데사르탄으로 분리 되어 흡수되며 흡수 속도는 Tmax 3~4시간으로 빠르다. 따라서 칸데사르탄 투여로 고혈압, 뇌졸중 치료 및 기타 합병증을 예방하기 위해서는 안지오텐신과 알도스테론이 분비되는 자정 이후부터 아침까지 혈압 강하를 지속시켜야 한다. 따라서, 칸데사르탄은 저녁 이후 투여가 필요하다[참조: Easthope SE et al.: Candesartan Cilexetil: An Update of its Use in Essential Hypertension, Drugs Volume 62(8)2002 pp 1253-1287 ].
5) 올메사르탄(Olmesartan)
올메사르탄은 안지오텐신-II 수용체 차단제 중 선택적인 안지오텐신 II 수용체(type AT1)길항제이다. 특히 Cytochrome P450 시스템에 의해 대사되지 않는 약물로 병용투여하기에 매우 우수한 약물이다.
고혈압 치료를 위해 스타틴계 지질저해제 계열의 약물과 안지오텐신-II 수용체 차단제 계열의 약물의 조합이 바람직함에도 불구하고, 양자를 단순히 혼합하여 제제화할 경우에는 다음과 같은 여러가지 문제점이 있다.
첫째, 두 약물의 단순 혼합시 약물의 용해도가 감소되므로, 이를 개선하기 위해서 추가로 산성물질과 유동화제 등 첨가제들이 추가로 사용되어야 한다.
둘째, 두 약물이 동시에 방출됨으로써, 저녁시간대 복용이 효과적인 안지오텐신-II 수용체 차단제와 아침시간대 복용이 효과적인 디히드로피리딘계 스타틴계 지질저해제의 최적 복용시간대를 동시에 만족시키지 못하므로 각 약물의 효과를 제대로 발휘하게 할 수 없다.
셋째, 두 약물이 동시에 방출되기 때문에 약물 상호간의 대사적인 간섭에 의해 최고 혈중농도 또는 생체 이용률과 같은 약물동태에 영향을 주게 되어 서로의 약효에 직접적인 영향을 미칠 수 있으며, 부작용을 초래할 수 있다.
이에, 본 발명자들은 단순 복합제제의 문제점을 해결함과 동시에 고혈압 등의 심혈관질환 치료에 보다 효과적인 복합제제를 개발하기 위한 연구결과 본 발명을 완성하였다.
본 발명은 두 약물을 동시에 투여할 때 약물상호작용으로 인해 약효가 감소하는 것을 억제하고 부작용이 발생하는 것을 억제할 수 있는 기능성 복합제의 제제화 기술에 대한 발명이다.
모든 약물은 두 성분 이상을 복용할 때 개개 약물의 흡수, 대사, 분포, 약효발현 및 배설에 관여하는 약물수송체(Transporter), 약물대사효소(Metabolic Enzyme), 유전자 등의 작용 특성을 면밀히 검토하여 배합 성분의 용출 순서와 시간차를 결정하고, 흡수 시간차를 유지할 수 있도록 해주어야 약물간의 체내에서의 상호 길항 작용을 최소해 줄 수 있다 그 결과 약물배합으로 인한 부작용을 감소시키고 약효를 증강시켜줄 수 있음은 주지의 사실이다.
더욱 상세하게 서명한다면 약물이 1차 단계로 장벽을 통과할, 2차단계로 간으로 유입될 때, 3차 단계로 간 세포 내에서 대사되어 활성화될 때, 그리고 4차 단계로 담도 등을 통해 간 세포 밖으로 빠져나갈 때 등 각 단계마다 약물을 곳곳에서 흡수, 대사, 배설시키는 배출수송체(Efflux transporter), 흡수수송체(Influx transporter), 대사효소 등이 곳곳에 존재한다.
그러나 약물의 종류에 따라서는 이러한 수송체와 효소와 유전자의 작용을 억제하기도 한다.
따라서 두 종류의 약물이 동시에 각 단계에 통과하는 경우, 한 성분이 다른 성분의 흡수, 분포, 대사를 방해하여 약효를 감소하거나 부작용을 증가시킬 수가 있다. 따라서 한 성분을 먼저 통과시키고 다른 성분은 시간차를 두고 통과시켜 약물간 상호길항작용을 없애주어야 한다.
이러한 수송체는 현재 300여종 발견되어 있고, 대사효소는 500여종 발견되어 있으며, 유전자도 57종이 발견되어 있다. 머지않아 유전자 게놈 해석기술을 통해 모든 약물에 대한 수송체와 효소가 발견될 것이다.
따라서 현재 대사 효소나 수송체가 알려져 있지 않은 어떤 약물이라 할지라도 곧 발견 될 것임은 사필귀정이므로 두 종 이상의 약물의 상호작용이 없다라고 주장할 수는 없다. 따라서 대사 효소나 수송체가 알려져 있지 아니한 두 성분의 약물이라 할지라도 두 성분간 용출 시간차이를 두고 흡수시키는 것이 합리적이다.
본 발명의 배경은 이상 언급한 바와같이 모든 약물을 이종 이상 복합 투여할 때 이상적인 복합 방식을 실현시키려는 목적으로 두 성분간 상호 길항 작용이 확실한 것 들을 중심으로 성분간 용출순서를 결정하고 시간차를 유지시켜 흡수되어 약효를 극대화하고 부작용을 극소화한 기능성 복합제를 가능하게 하였다.
본 발명의 기능성 복합제를 제제화하기 위한 실험을 하였거나 그 자료를 검토한 수송체와 약물 대사 효소를 예시하면 아래와 같다.
1) 배출수송체(Efflux Transporter): P-glycoprotein(P-gp), Multidrug resistance(MDR), Multidrug resistance associated protein(MRP)
2) 흡수수송체(Influx Transporter): Organic anion transport protein(OATP), Sodium taurocholate cotransporting polypeptide(NTCP), Organic cation transporter(OCT)
3) 약물대사효소: Cytochrome P450 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2E1, 3A4/5
4) 기타 대사효소: Uridine-5-phophate-glucuronosyltransferase(UDP-gt), Sulfatase, Sulfotransferase(1a1, 2a1, 1e1)
5) 핵수용체(Nucleic Receptor): Pregnane-X-Receptor(PXR), Constitutive Androstane Receptor(CAR)
본 발명이 해결하고자 하는 기술적 과제는 각각의 약물을 병용 투여시의 부작용은 최소화하고, 최적의 약리효과를 유도하며, 각 약물의 약리효과를 발현하는 시간대에 약물을 투여하여 임상적인 상승효과를 얻을 수 있고, 복약순응도를 높일 수 있는 약제학적 제제를 제공하는 것이다.
본 발명은 약리학적 활성성분으로서 HMG CoA 환원효소 억제제를 함유하는 선방출성 구획, 및 약리학적 활성성분으로서 안지오텐신-2 수용체 차단제를 함유하는 지연 방출성 구획을 포함하는 방출성이 조절된 약제학적 제제에 관한 것이다.
보다 상세하게, 본 발명은 약리학적 활성성분으로 심바스타틴 또는 그의 약학적으로 허용되는 염을 함유하는 선방출성 구획, 및 약리학적 활성성분으로 로자탄, 이의 이성질체 또는 이의 약학적으로 허용되는 염을 함유하는 지연방출성 구획을 포함하는 약제학적 제제A를 제공한다.
또한 본 발명은 약리학적 활성성분으로 심바스타틴 또는 이의 약학적으로 허용되는 염을 함유하는 선방출성 구획, 및 약리학적 활성성분으로 올메사르탄, 이의 이성질체, 이의 약학적으로 허용되는 염 또는 이의 프로드럭을 함유하는 지연방출성 구획을 포함하는 약제학적 제제B를 제공한다.
또한 본 발명은 약리학적 활성성분으로 심바스타틴 또는 이의 약학적으로 허용되는 염을 함유하는 선방출성 구획, 및 약리학적 활성성분으로 발사르탄, 이의 이성질체 또는 이의 약학적으로 허용되는 염을 함유하는 지연방출성 구획을 포함하는 약제학적 제제C를 제공한다.
또한 본 발명은 약리학적 활성성분으로 심바스타틴 또는 이의 약학적으로 허용되는 염을 함유하는 선방출성 구획, 및 약리학적 활성성분으로 칸데사르탄, 이의 이성질체, 이의 약학적으로 허용되는 염 또는 이의 프로드럭을 함유하는 지연방출성 구획을 포함하는 약제학적 제제D를 제공한다.
또한 본 발명은 약리학적 활성성분으로 로슈바스타틴 또는 이의 약학적으로 허용되는 염을 함유하는 선방출성 구획, 및 약리학적 활성성분으로 로자탄, 이의 이성질체 또는 이의 약학적으로 허용되는 염 을 함유하는 지연방출성 구획을 포함하는 약제학적 제제E를 제공한다.
또한 본 발명은 약리학적 활성성분으로 로슈바스타틴 또는 이의 약학적으로 허용되는 염을 함유하는 선방출성 구획, 및 약리학적 활성성분으로 이베사르탄, 이의 이성질체 또는 이의 약학적으로 허용되는 염을 함유하는 지연방출성 구획을 포함하는 약제학적 제제F를 제공한다.
또한 본 발명은 약리학적 활성성분으로 로슈바스타틴 또는 이의 약학적으로 허용되는 염을 함유하는 선방출성 구획, 및 약리학적 활성성분으로 칸데사르탄, 이의 이성질체, 이의 약학적으로 허용되는 염 또는 이의 프로드럭을 함유하는 지연방출성 구획을 포함하는 약제학적 제제G를 제공한다.
본 명세서에서 언급되는 약학적으로 허용되는 염은 의약업계에서 통상적으로 사용되는 염을 의미하며, 예를 들어 칼슘, 칼륨, 나트륨 및 마그네슘 등으로 제조된 무기이온염, 염산, 질산, 인산, 브롬산, 요오드산, 과염소산, 주석산 및 황산 등으로 제조된 무기산염, 메탄설폰산, 에탄설폰산, 벤젠설폰산, p-톨루엔술폰산, 나프탈렌설폰산, 트리플루오로아세트산, 시트르산, 말레인산, 숙신산, 옥살산, 벤조산, 타르타르산, 푸마르산, 만데르산, 프로피온산, 구연산, 젖산, 글리콜산 , 글루콘산, 갈락투론산, 글루탐산, 글루타르산, 글루쿠론산, 아스파르트산, 아스코르브산, 카본산, 바닐릭산, 하이드로 아이오딕산 등으로 제조된 유기산염, 메탄설폰산, 에탄설폰산, 벤젠설폰산, p-톨루엔설폰산 및 나트탈렌설폰산 등으로 제조된 설폰산염, 글리신, 아르기닌, 라이신 등으로 제조된 아미노산염 및 트리메틸아민, 트리에틸아민, 암모니아, 피리딘, 피콜린 등으로 제조된 아민염 등이 있으나, 이들의 예로 본 발명의 약학적으로 허용되는 염이 제한되는 것은 아니다.
본 발명에서, "OO프로드럭"은 신체 내에서 효소·화학 물질로 인해 그 활성성분인 "OO"으로 분해되는 것을 의미한다. 예를 들어 설명하면, 칸데르사르탄 프로드럭은 체내에서 분해되어 그의 활성성분인 칸데르사르탄이 되는 것을 의미한다.
이하에서, 특별한 언급이 없는 한 약리학적 활성성분명의 언급은 이의 이성질체, 이의 약학적으로 허용되는 염 및 경우에 따라서는 이의 프로드럭을 모두 포함하는 의미로 해석된다. 예를 들어, 이하에서,‘로자탄’이라고 기재되어 있더라도, 만약 존재한다면 로자탄의 이성질체 및 로자탄의 약학적으로 허용되는 염까지 모두 포함하는 것으로 해석된다.
본 발명의 약제학적 제제는 선방출성 구획의 스타틴계 지질저해제가 방출되고 일정시간이 지난 후 지연방출성 구획의 안지오텐신-II-수용체 차단제가 방출되어 각각의 약물의 특성에 적합한 방출을 갖게된다.
본 발명의 약제학적 제제는 두 활성성분 간의 방출성을 제어하는 물리적인 구획을 제공함으로써, 기존 단일제제의 병용투여 또는 동시투여의 문제점을 개선하여 우수한 치료 또는 예방 효과를 나타낸다. 즉, 두 약물을 복합하여 사용하면서도, 이들의 방출속도를 달리함으로써 약물 상호간의 길항 작용 및 부작용을 방지함과 동시에 약효의 상승 작용을 얻을 수 있으며, 환자의 복약이 용이하다.
또한, 구체적으로 본 발명은 약제학적 제제A, B, C, D, E, F, G를 제공한다.
1. 약제학적 제제A
본 발명은 약리학적 활성성분으로 심바스타틴 또는 이의 약학적으로 허용되는 염을 포함하는 선방출성 구획, 및 약리학적 활성성분으로 로자탄, 이의 이성질체 또는 이의 약학적으로 허용되는 염을 포함하는 지연방출성 구획을 포함하는 약제학적 제제A를 제공한다.
본 발명의 심바스타틴 및 로자탄을 함유하는 약제학적 제제A에 사용된 약리활성성분의 약리 작용은 다음 표1과 같다.
[규칙 제26조에 의한 보정 18.05.2009]

본 발명은 심바스타틴 용출 개시 후 30분 이내에 심바스타틴 총량의 80% 이상 바람직하게는 85% 이상이 방출되는 약제학적 제제A를 제공한다. 또한, 본 발명은 경구투여시 로자탄이 심바스타틴 용출개시 후 2시간까지 단위제제 내 로자탄의 총량의 20% 이내로 용출되는 약제학적 제제, 바람직하게는 10% 이내로 용출되는 약제학적 제제A를 제공한다. 본 발명은 로자탄이 심바스타틴보다 2 내지 4시간 늦게 간에서 흡수되는 약제학적 제제A를 제공한다.
약제학적 제제A에서, 선방출성 구획 중 활성성분인 심바스타틴은 성인(체중 65~75 kg의 성인 남자) 1일 기준으로 제제(전체 100mg~1,000mg) 중 1 내지 160mg을 포함하고, 바람직하기로는 5 내지 80 mg 을 포함한다.
약제학적 제제A에서, 지연방출성 구획 중 활성성분은 단위제제 중 로자탄은 약12.5 내지 200mg 포함될 수 있으며 25 내지 100mg이 바람직하다.
2. 약제학적 제제B
본 발명은 약리학적 활성성분으로 심바스타틴 또는 이의 약학적으로 허용되는 염을 포함하는 선방출성 구획, 및 약리학적 활성성분으로 올메사르탄, 이의 이성질체, 이의 약학적으로 허용되는 염 또는 이의 프로드럭을 포함하는 지연방출성 구획을 포함하는 약제학적 제제B를 제공한다.
여기서, 올메사르탄 프로드럭은 체내에서 활성성분인 올메사르탄으로 분해되는 것으로, 올메사르탄 메독소밀인 것이 바람직하다.
본 발명은 심바스타틴 용출 개시 후 30분 이내에 심바스타틴 총량의 80% 이상 바람직하게는 90% 이상이 방출되는 약제학적 제제B를 제공한다. 또한, 본 발명은 경구투여시 올메사르탄이 심바스타틴 용출개시 후 2시간까지 단위 제제 내 올메사르탄 총량의 5% 이내로 방출되는 약제학적 제제B, 바람직하게는 심바스타틴 용출 개시후 2시간 이후에 올메사르탄이 방출되는 약제학적 제제B를 제공한다. 본 발명은 올메사르탄이 심바스타틴보다 2 내지 4시간 늦게 간에서 흡수되는 약제학적 제제B를 제공한다.
약제학적 제제B에서, 선방출성 구획 중 활성성분인 심바스타틴은 성인(체중 65~75 kg의 성인 남자) 1일 기준으로 제제(전체 200mg~1,200mg) 중 1 내지 160mg을 포함하고, 바람직하기로는 5 내지 80 mg 을 포함한다.
약제학적 제제B에서, 지연방출성 구획 중 활성성분은 단위제제 중 올메사르탄 메독소밀로서 약 2.5 내지 80mg 포함될 수 있으며 5 내지 40mg이 바람직하다.
3. 약제학적 제제C
본 발명은 약리학적 활성성분으로 심바스타틴 또는 이의 약학적으로 허용되는 염을 포함하는 선방출성 구획, 및 약리학적 활성성분으로 발사르탄, 이의 이성질체 또는 이의 약학적으로 허용되는 염을 포함하는 지연방출성 구획을 포함하는 약제학적 제제C를 제공한다.
본 발명의 약제학적 제제C에 사용된 약리활성성분의 약리 작용은 다음 표2와 같다.
[규칙 제26조에 의한 보정 18.05.2009]

본 발명은 심바스타틴 용출 개시 후 30분 이내에 심바스타틴 총량의 80% 이상 바람직하게는 90% 이상이 방출되는 약제학적 제제C를 제공한다. 또한, 본 발명은 경구투여시 발사르탄이 심바스타틴 용출개시 후 2시간 이후에 용출되는 약제학적 제제C를 제공한다.본 발명은 발사르탄이 심바스타틴보다 2 내지 4시간 늦게 간에서 흡수되는 약제학적 제제C를 제공한다.
약제학적 제제C에서, 선방출성 구획 중 활성성분인 심바스타틴은 성인(체중 65~75 kg의 성인 남자) 1일 기준으로 제제(전체 200mg~1,200mg) 중 1 내지 160mg을 포함하고, 바람직하기로는 5 내지 80 mg 을 포함한다.
약제학적 제제C에서, 지연방출성 구획 중 활성성분은 단위제제 중 발사르탄은 1 내지 800mg 포함될 수 있으며 20 내지 640mg이 바람직하다.
4. 약제학적 제제D
본 발명은 약리학적 활성성분으로 심바스타틴 또는 이의 약학적으로 허용되는 염을 포함하는 선방출성 구획, 및 약리학적 활성성분으로 칸데사르탄, 이의 이성질체, 이의 약학적으로 허용되는 염 또는 이의 프로드럭을 포함하는 지연방출성 구획을 포함하는 약제학적 제제D를 제공한다.
여기서, 칸데사르탄 프로드럭은 체내에서 활성성분인 칸데사르탄으로 분해되는 것으로, 칸데사르탄 실렉세틸인 것이 바람직하다.
본 발명은 심바스타틴 용출 개시 후 30분 이내에 심바스타틴 총량의 80% 이상 바람직하게는 85% 이상이 방출되는 약제학적 제제D를 제공한다. 또한, 본 발명은 경구투여시 칸데사르탄이 심바스타틴 용출개시 후 2시간까지 단위 제제내 칸데사르탄 총량의 35% 이내로 방출되는 약제학적 제제D, 바람직하게는 20% 이내로 용출되는 약제학적 제제D를 제공한다. 본 발명은 칸데사르탄이 심바스타틴보다 2 내지 4시간 늦게 간에서 흡수되는 약제학적 제제D를 제공한다.
약제학적 제제D에서, 선방출성 구획 중 활성성분인 심바스타틴은 성인(체중 65~75 kg의 성인 남자) 1일 기준으로 제제(전체 100mg~800mg) 중 1 내지 160mg을 포함하고, 바람직하기로는 5 내지 80 mg 을 포함한다.
약제학적 제제D에서, 지연방출성 구획 중 활성성분은 단위제제 중 칸데사르탄은 약 2 내지 64mg 포함될 수 있으며 4 내지 32mg이 바람직하다.
5. 약제학적 제제E
본 발명은 약리학적 활성성분으로 로슈바스타틴 또는 이의 약학적으로 허용되는 염을 포함하는 선방출성 구획, 및 약리학적 활성성분으로 로자탄, 이의 이성질체 또는 이의 약학적으로 허용되는 염 포함하는 지연방출성 구획을 포함하는 약제학적 제제E를 제공한다.
본 발명은 심바스타틴 용출 개시 후 30분 이내에 심바스타틴 총량의 80% 이상 바람직하게는 85% 이상이 방출되는 약제학적 제제E를 제공한다. 또한 본 발명은 지연방출성 구획의 활성성분인 로자탄의 방출이 선방출성 구획의 활성성분인 로슈바스타틴 방출 개시 약 2시간 이후에, 바람직하게는 약 3시간 이후에 개시되는 약제학적 제제E를 제공한다.
본 발명은 또한 지연방출성 구획의 활성성분인 로자탄이 선방출성 구획의 활성성분인 로슈바스타틴 방출 개시 후 약 2시간 경과시까지, 바람직하게는 약 3시간 경과시까지, 단위 제형 중 로자탄 총량의 약 0 내지 40%가, 바람직하게는 약 0 내지 20%가 방출되는 약제학적 제제E를 제공한다.
약제학적 제제E에서, 선방출성 구획의 약리학적 활성성분은 단위 제제 중 로슈바스타틴으로 약 2.5 ~ 80 mg, 바람직하게는 약 5 ~ 40 mg을 포하고, 지연방출성 구획 중 활성성분은 단위제제 중 로살탄 칼륨으로 약 10 ~ 200 mg, 바람직하게는 약 25~ 100 mg으로 포함한다.
6. 약제학적 제제F
본 발명은 약리학적 활성성분으로 로슈바스타틴 또는 이의 약학적으로 허용되는 염을 포함하는 선방출성 구획, 및 약리학적 활성성분으로 이베사르탄, 이의 이성질체 또는 이의 약학적으로 허용되는 염을 포함하는 지연방출성 구획을 포함하는 약제학적 제제F를 제공한다.
본 발명은 지연방출성 구획의 활성성분인 이베사르탄의 방출이 선방출성 구획의 활성성분인 로슈바스타틴의 방출 개시 후 약 2시간 10시간 사이에, 바람직하게는 약 2시간 내지 6시간 사이에, 보다 바람직하게는 약 2시간 내지 5시간 사이에 개시되는 약제학적 제제F를 제공한다.
본 발명은 지연방출성 구획의 활성성분인 이베사르탄이 선방출성 구획의 활성성분인 로슈바스타틴 방출개시 후 약 2시간 경과시까지, 바람직하게는 약 4시간 경과시까지, 단위제제 중 이베사르탄 총량의 약 0 내지 20% 방출되는 약제학적 제제F를 제공한다.
약제학적 제제F에서, 선방출성 구획 중 활성성분은 단위제제 중 로슈바스타틴으로 약 0.1 ~ 200mg 포함할 수 있으며, 바람직하게는 약 0.2~100mg, 보다 바람직하게는 약 5~40mg 포함할 수 있으며, 지연방출성 구획 중 활성성분은 단위제제 중 이베사르탄, 또는 이의 약제학적으로 허용가능한 염으로 약 1 ~ 1000mg 포함할 수 있으며, 바람직하게는 약 2~500mg, 보다 바람직하게는 약 75~300mg 포함할 수 있다.
7. 약제학적 제제G
본 발명은 약리학적 활성성분으로 로슈바스타틴 또는 이의 약학적으로 허용되는 염을 포함하는 선방출성 구획, 및 약리학적 활성성분으로 칸데사르탄, 이의 이성질체, 이의 약학적으로 허용되는 염 또는 이의 프로드럭을 포함하는 지연방출성 구획을 포함하는 약제학적 제제G를 제공한다.
본 발명의 약제학적 제제는 지연방출성 구획의 활성성분인 칸데사르탄의 방출이 선방출성 구획의 활성성분인 로슈바스타틴 방출 개시 후 약 1시간 내지 8시간 사이에, 바람직하게는 약 2시간 내지 4시간 사이에 개시되는 약제학적 제제G를 제공한다.
본 발명은 또한 지연방출성 구획의 칸데사르탄이 선방출성구획의 로슈바스타틴 방출 개시 후 약 2시간 경과시까지, 바람직하게는 약 3시간 경과시까지 단위제제 중 칸데사르탄 총량의 약 0 내지 40%가 방출되는 약제학적 제제G를 제공한다.
약제학적 제제G에서, 선방출성 구획의 약리학적 활성성분은 단위제제 중 로슈바스타틴으로 약 0.1 ~ 500 mg 포함하고, 바람직하게는 약 0.2 ~ 100 mg을, 보다 바람직하게는 약 5 ~ 40 mg을 포함하고, 지연방출성 구획 중 활성성분은 단위제제 중 칸데사르탄실렉세틸으로 약 1~ 1000 mg, 바람직하게는 약 2 ~ 500 mg을, 보다 바람직하게는 약4 ~ 32 mg을 포함한다.
이하에서는 본 발명의 약제학적 제제의 선방출성 구획 및 지연방출성 구획에 대해 보다 상세히 설명한다.
I. 선(先)방출성 구획
선방출성 구획은 본 발명의 약제학적 제제에 있어서 지연방출성 구획보다 먼저 방출되는 구획을 의미한다. 선방출성 구획은 약리학적 활성성분 및 필요에 따라 약학적으로 허용가능한 첨가제, 기타 부형제를 포함한다. 선방출성 구획 내 포함된 약리학적 활성성분은 충분한 시간의 차이를 두고 먼저 방출되어, 지연방출성 구획 내 포함된 약리학적 활성성분에 앞서 신속한 약효를 나타낸다.
본 발명에서 선방출성 구획은 약리학적 활성성분 외에 약제학적을 허용되는 첨가제와 함께 혼합, 연합, 건조 및 제립 등의 경구투여제를 제조하기 위한 통상의 과정을 통하여 혼합물, 과립, 펠렛, 또는 정제 형태로 제조할 수 있다. 또한, 유동성이 좋지 않아 직접 타정이 가능하지 않은 경우는 압착, 제립, 및 정립하여 과립화할 수 있다.
1. 약리학적 활성성분
(1) 심바스타틴
약제학적 제제A, 약제학적 제제B, 약제학적 제제C,약제학적 제제D에서 선방출성 구획으로 심바스타틴을 사용할 수 있다.
선방출성 구획은 약리학적 활성성분으로 심바스타틴, 그의 이성질체 또는 약제학적으로 허용 가능한 염을 포함한다. 본 발명의 제제에서 선방출성 구획 중 활성성분인 심바스타틴은 성인(체중 65~75 kg의 성인 남자) 단위제제 중 1 내지 160mg을 포함하고, 바람직하기로는 5 내지 80 mg을 포함한다.
(2) 로슈바스타틴
약제학적 제제E, 약제학적 제제F, 약제학적 제제G에서 선방출성 구획으로 로슈바스타틴을 사용할 수 있다.
선방출성 구획의 약리학적 활성성분은 로슈바스타틴, 그의 이성질체, 또는 그의 약제학적으로 허용되는 염을 포함하며, 바람직하게는 로슈바스타틴 또는 그의 약학적으로 허용가능한 그의 염을, 보다 바람직하게는 로슈바스타틴 칼슘염을 포함 할 수 있다. 선방출성 구획의 약리학적 활성성분은 단위제제 중 로슈바스타틴으로 약 0.1 ~ 500 mg 포함하고, 바람직하게는 약 0.2 ~ 100 mg을, 보다 바람직하게는 약 2.5 내지 80mg을, 보다 더 바람직하게는 약 5 ~ 40 mg을 포함한다.
2. 약제학적으로 허용가능한 첨가제
본 발명의 선방출성 구획은 본 발명의 효과를 해치지 않는 범위 안에서 약학적으로 허용 가능한 희석제, 결합제, 붕해제, 윤활제의 첨가제를 사용할 수 있다.
활성성분이 심바스타틴인 경우, 상기 첨가제 이외에 pH 조절제, 산화방지제, 용해보조제 등을 추가할 수 있으며, 활성성분이 로슈바스타틴인 경우, 상기 첨가제 이외에 안정화제를 추가로 사용할 수 있다.
본 발명의 선방출성 구획에서, 첨가제는 활성성분 1중량부에 대하여 0.01-100 중량부를 포함한다.
본 발명의 선방출성 구획에서 희석제는 슈가, 전분, 미결정셀룰로오스(미세결정셀룰로오스), 유당,유당수화물, 포도당, 디-만니톨, 알기네이트, 알칼리토금속류염, 클레이, 폴리에틸렌글리콜, 디칼슘포스페이트, 무수인산수소칼슘, 또는 이들의 혼합물 등을 사용할 수 있다.
본 발명의 선방출성 구획에서 결합제는 전분, 미결정셀룰로오스, 고분산성 실리카, 만니톨, 디-만니톨, 자당, 유당수화물, 폴리에틸렌글리콜, 폴리비닐피롤리돈(포비돈), 폴리비닐피롤리돈 공중합체(코포비돈), 히프로멜로오스, 히드록시프로필셀룰로오스, 천연검, 합성검, 코포비돈, 젤라틴, 또는 이들의 혼합물 등을 사용할 수 있다.
본 발명의 선방출성 구획에서 붕해제는 전분글리콘산나트륨, 옥수수전분, 감자전분 또는 전호화전분 등의 전분 또는 변성전분; 벤토나이트, 몬모릴로나이트, 또는 비검(veegum) 등의 클레이; 미결정셀룰로오스, 히드록시프로필셀룰로오스 또는 카르복시메틸셀룰로오스 등의 셀룰로오스류; 알긴산나트륨 또는 알긴산 등의 알긴류; 크로스카멜로스(croscarmellose)나트륨 등의 가교 셀룰로오스류; 구아검, 잔탄검 등의 검류; 가교 폴리비닐피롤리돈(crospovidone) 등의 가교 중합체; 중탄산나트륨, 시트르산 등의 비등성 제제, 또는 이들의 혼합물을 사용할 수 있다.
본 발명의 선방출성 구획에서 윤활제는 탈크, 스테아린산(스테아르산), 스테아린산 마그네슘(스테아르산 마그네슘), 스테아린산 칼슘(스테아르산 칼슘), 라우릴설페이트나트륨, 수소화식물성오일, 나트륨벤조에이트, 나트륨스테아릴푸마레이트, 글리세릴 베헤네이트, 글리세릴 모노레이트, 글리세릴모노스테아레이트, 글리세릴 팔미토스테아레이트, 콜로이드성 이산화규소, 폴리에틸렌글리콜 또는 이들의 혼합물 등을 사용할 수 있다.
본 발명의 선방출성 구획에서, pH 조절제는 초산, 아디프산, 아스코르빈산(아스코르브산), 아스코르빈산 나트륨(아스코르브산 나트륨), 에테르산 나트륨, 사과산, 숙신산, 주석산, 푸마르산, 구연산(시트르산)과 같은 산성화제와 침강 탄산 칼슘, 암모니아수, 메글루민, 탄산 나트륨, 산화 마그네슘, 탄산 마그네슘, 구연산 나트륨, 삼염기칼슘인산염과 같은 염기성화제 등을 사용할 수 있다.
본 발명의 선방출성 구획에서 산화방지제는 디부틸 히드록시 톨루엔(부틸레이트 히드록시 톨루엔), 부틸레이티드 히드록시아니솔, 초산 토코페롤, 토코페롤, 프로필 갈레이트, 아황산수소나트륨, 피로아황산나트륨 등을 사용할 수 있다.본 발명의 선방출성 구획에서, 용해보조제는 라우릴황산나트륨, 폴리소르베이트 등의 폴리옥시에틸렌 소르비탄 지방산 에스테류, 도큐세이트 나트륨, 폴록사머(poloxamer) 등을 사용할 수 있다.
본 발명의 선방출성 구획에서, 약학적으로 허용가능한 안정화제는 아스코르빈산, 구연산, 부틸히드록시 아니솔, 부틸히드록시 톨루엔, 토코페롤 유도체 등을 사용할 수 있다.
이외에도 착색제, 향료 중에서 선택된 다양한 첨가제로서 약학적으로 허용 가능한 첨가제를 선택 사용하여 본 발명의 제제를 제제화할 수 있다.
본 발명의 선방출성 구획에서 사용가능한 첨가제의 범위가 상기 첨가제를 사용하는 것으로 한정되는 것은 아니며, 상기한 첨가제를 선택에 의하여 통상 범위의 용량을 함유하여 제제화할 수 있다.
II. 지연방출성 구획
지연방출성 구획은 본 발명에 의한 약제학적 제제에 있어서 선방출성 구획 내 활성성분의 방출 개시 후 일정 시간부터 그 활성성분이 방출되는 구획을 의미한다. 지연방출성 구획은 (1) 약리학적 활성성분; (2-a) 방출제어물질, 또는 (2-b) 삼투압 조절제 및 반투과성막 코팅기제를 포함하며; (3) 필요에 따라 약제학적으로 허용 가능한 첨가제를 추가로 포함할 수 있다. 지연방출성 구획 중 내 포함된 약리학적 활성성분은 선방출성 구획내 포함된 약리학적 활성성분의 방출 개시하고 충분한 시간이 경과된 이후에 방출된다.
1. 약리학적 활성성분
(1) 로자탄
약제학적 제제A, 약제학적 제제E에서 지연방출성 구획으로 로자탄을 사용할 수 있다.
지연방출성 구획의 약리학적 활성성분은 로자탄 또는 약학적으로 허용 가능한 염을 포함하며, 지연방출성 구획 중 활성성분은 단위제제 중 로자탄은 약 10 내지 200mg 포함될 수 있으며 약 25 내지 100mg이 바람직하다. 약제학적으로 허용가능한 예의 대표적인 염에는 로자탄 칼륨이 있다.
(2) 올메사르탄
약제학적 제제B에서 지연방출성 구획으로 올메사르탄을 사용할 수 있다.
지연방출성 구획의 약리학적 활성성분은 올메사르탄, 그의 프로드럭 또는 약제학적으로 허용 가능한 염을 포함하며, 지연방출성 구획 중 활성성분은 단위제제 중 올메사르탄 메독소밀로서 약 2.5 내지 80mg 포함될 수 있으며 5 내지 40mg이 바람직하다.
(3) 발사르탄
약제학적 제제C에서 지연방출성 구획으로 발사르탄을 사용할 수 있다.
지연방출성 구획의 약리학적 활성성분은 발사르탄 그의 이성질체 및 약학적으로 허용 가능한 염을 포함하며, 지연방출성 구획 중 활성성분은 단위제제 중 발사르탄은 1 내지 800mg 포함될 수 있으며 20 내지 640mg이 바람직하다.
(4) 칸데사르탄
약제학적 제제D, 약제학적 제제G에서 지연방출성 구획으로 칸데사르탄을 사용할 수 있다.
지연방출성 구획의 약리학적 활성성분은 칸데사르탄, 그의 프로드럭 또는 약제학적으로 허용 가능한 염을 포함하며, 지연방출성 구획 중 활성성분은 단위제제 중 칸데사르탄은 약 2 내지 64mg 포함될 수 있으며 4 내지 32mg이 바람직하다.
(5) 이베사탄
약제학적 제제F에서 지연방출성 구획으로 이베사르탄을 사용할 수 있다.
지연방출성 구획의 약리학적 활성성분은 이베사르탄, 또는 그의 약학적으로 허용 가능한 염을 포함한다. 이베사르탄의 Tmax는 약 2시간이다.
지연방출성 구획 중 활성성분은 단위제제 중 이베사르탄, 또는 그의 약제학적으로 허용가능한 염으로 약 1 ~ 1000mg 포함할 수 있으며, 바람직하게는 약 2~500mg, 보다 바람직하게는 약 75~300mg 포함할 수 있다.
2-a. 방출제어물질
본 발명의 약제학적 제제 중 지연방출성 구획은 장용성 고분자, 수불용성 중합체, 소수성 화합물, 및 친수성 고분자로 이루어진 군에서 선택된 1종 이상의 방출제어물질을 포함한다. 바람직하게는 수불용성 중합체 및 장용성 고분자 중에서 선택된 1종 이상의 방출제어물질을 포함한다.
본 발명의 지연방출성 구획에서 방출제어물질은 활성성분 1 중량부에 대하여 0.05~ 100 중량부 사용가능한데, 사용량이 상기 범위 미만이면 충분한 지연방출성을 얻을 수 없고, 사용량이 상기 범위를 초과하면 약물방출이 지연되어 유의성 있는 임상적 효과를 얻을 수 없다. 바람직하게는 활성성분이 로자탄인 경우, 방출제어물질은 로자탄 1중량부에 대하여 0.01 내지 20 중량부이며, 이베사르탄인 경우 방출제어물질은 이베사르탄 1중량부에 대하여 0.1 내지 50 중량부, 칸데사르탄인 경우 방출제어물질은 칸데사르탄 1 중량부에 대하여 0.1 내지 50 중량부를 포함한다.
이하에서, "히드록시프로필메틸셀룰로오스"와 "히프로멜로오스"는 동의어이다. 또한 히드록시프로필메틸셀룰로오스를 포함하는 용어 "히드록시프로필메틸셀룰로오스0000"은 "히프로멜로오스OOOO" 와 동의어이다. 예를 들어, "히드록시프로필메틸셀룰로오스 프탈레이트"는 "히프로멜로오스 프탈레이트"와 동의어이다.
상기 장용성 고분자는 pH5 미만의 산성 조건하에서 불용성이거나 또는 안정한 것으로, pH5 이상인 특정 pH 조건하에서 용해되거나 또는 분해되는 고분자를 말한다. 본 발명에서 사용가능한 장용성 고분자는 장용성 셀룰로오스 유도체, 장용성 아크릴산계 공중합체, 장용성 폴리메타크릴레이트 공중합체, 장용성 말레인산계 공중합체 및 장용성 폴리비닐 유도체로 이루어진 군에서 선택된 1종 이상이며, 상기 장용성 셀룰로오스 유도체는 히프로멜로오스아세테이트숙시네이트, 히프로멜로오스프탈레이트(히드록시프로필메틸셀룰로오스 프탈레이트), 히드록시메틸에틸셀룰로오스프탈레이트, 셀룰로오스아세테이트프탈레이트, 셀룰로오스아세테이트숙시네이트, 셀룰로오스아세테이트말레이트, 셀룰로오스벤조에이트프탈레이트, 셀룰로오스프로피오네이트프탈레이트, 메틸셀룰로오스프탈레이트, 카르복시메틸에틸셀룰로오스 및 에틸히드록시에틸셀룰로오스프탈레이트 및 메틸히드록시에틸셀룰로오스 중에서 선택된 1종 이상이고; 상기 장용성 아크릴산계 공중합체는 스티렌-아크릴산 공중합체, 아크릴산메틸-아크릴산 공중합체, 아크릴산메틸메타크릴산 공중합체(예컨대, 아크릴-이즈), 아크릴산부틸-스티렌-아크릴산 공중합체, 및 아크릴산메틸-메타크릴산-아크릴산옥틸공중합체 중에서 선택된 1종 이상이며; 상기 장용성 폴리메타크릴레이트 공중합체는 폴리(메타크릴산-메틸 메타크릴레이트) 공중합체(예컨대, 유드라짓 L100, 유드라짓 S, 에보닉, 독일), 폴리 (메타크릴산- 에틸아크릴레이트) 공중합체 (예컨대, 유드라짓 L100-55) 중에서 선택된 1종 이상이며, 상기 장용성 말레인산계 공중합체는 아세트산비닐-말레인산 무수물 공중합체, 스티렌-말레인산 무수물 공중합체, 스티렌-말레인산모노에스테르 공중합체, 비닐메틸에테르-말레인산 무수물 공중합체, 에틸렌-말레인산 무수물 공중합체, 비닐부틸에테르-말레인산 무수물 공중합체, 아크릴로니트릴-크릴산메틸ㆍ말레인산 무수물 공중합체 및 아크릴산부틸-스티렌-말레인산 무수물 공중합체 중에서 선택된 1종 이상이고; 상기 장용성 폴리비닐 유도체는 폴리비닐알콜프탈레이트, 폴리비닐아세테이트프탈레이트, 폴리비닐부틸레이트프탈레이트 및 폴리비닐아세트아세탈프탈레이트 중에서 선택된 1종 이상이다.
상기 수불용성 중합체는 약물의 방출을 제어하는 약제학적으로 허용가능한 물(예를 들어, 정제수)에 용해되지 않는 고분자를 말한다. 본 발명에서 사용가능한 수불용성 중합체는 폴리비닐아세테이트(예컨대, 콜리코트 SR30D), 수불용성 폴리메타크릴레이트 공중합체[예컨대, 폴리(에틸아크릴레이트-메틸 메타크릴레이트) 공중합체(예컨대, 유드라짓 NE30D), 폴리(에틸아크릴레이트-메틸 메타크릴레이트-트리메틸아미노에틸메타크릴레이트 클로라이드)공중합체(예컨대, 유드라짓RSPO, RL, RS)등], 에틸셀룰로오스, 셀룰로오스 에스테르, 셀룰로오스 에테르, 셀룰로오스 아실레이트, 셀룰로오스 디아실레이트, 셀룰로오스 트리아실레이트, 셀룰로오스 아세테이트, 셀룰로오스 디아세테이트 및 셀룰로오스 트리아세테이트로 이루어진 군에서 선택된 1 종 이상인 것이다.
상기 소수성 화합물은 약물의 방출을 제어하는 약제학적으로 허용가능한 물에 용해되지 않는 물질을 말한다. 본 발명에서 사용가능한 소수성 화합물은 지방산 및 지방산 에스테르류, 지방산 알코올류, 왁스류, 무기물질, 및 이들의 혼합물로 이루어진 군에서 선택된 것이며, 상기 지방산 및 지방산 에스테르류는 글리세릴 팔미토스테아레이트, 글리세릴 스테아레이트, 글리세릴 비헤네이트, 세틸 팔미테이트, 글리세릴 모노올레이트 및 스테아린산 중에서 선택된 1종 이상이고; 상기 지방산 알코올류는 세토스테아릴 알코올, 세틸알코올 및 스테아릴알코올 중에서 선택된 1종 이상이며; 상기 왁스류는 카르나우바왁스, 밀납, 및 미결정왁스 중에서 선택된 1종 이상이고; 상기 무기물질은 탈크, 침강탄산칼슘, 인산일수소칼슘, 산화아연, 산화티탄, 카올린, 벤토나이트, 몬모릴로나이트 및 비검 중에서 선택된 1종 이상이다.
상기 친수성 고분자는 약물의 방출을 제어하는 약제학적으로 허용가능한 물에 용해되는 고분자 물질을 말한다. 본 발명에서 사용가능한 친수성 고분자는 당류, 셀룰로오스 유도체, 검류, 단백질류, 폴리비닐 유도체, 친수성 폴리메타크릴레이트 공중합체, 폴리에틸렌 유도체, 카르복시비닐폴리머 및 이들의 혼합물로 이루어진 군에서 선택된 것이며, 상기 당류는 덱스트린, 폴리덱스트린, 덱스트란, 펙틴 및 펙틴 유도체, 알긴산염, 폴리갈락투론산, 자일란, 아라비노자일란, 아라비노갈락탄, 전분, 히드록시프로필스타치, 아밀로오스, 및 아밀로펙틴 중에서 선택된 1종 이상이고 ; 상기 셀룰로오스 유도체는 히드록시프로필메틸셀룰로오스(히프로멜로오스), 히드록시프로필셀룰로오스, 히드록시메틸셀룰로오스, 히드록시에틸셀룰로오스, 메틸셀룰로오스 및 카르복시메틸셀룰로오스 나트륨 중에서 선택된 1종 이상이며; 상기 검류는 구아검, 로커스트 콩 검, 트라가칸타, 카라기난, 아카시아검, 아라비아검, 젤란검, 및 잔탄검 중에서 선택된 1종 이상이고; 상기 단백질류는 젤라틴, 카제인, 및 제인 중에서 선택된 1종 이상이며; 상기 폴리비닐 유도체는 폴리비닐 알코올, 폴리비닐 피롤리돈, 및 폴리비닐아세탈디에틸아미노아세테이트 중에서 선택된 1종 이상이고; 상기 친수성 폴리메타크릴레이트 공중합체는 폴리(부틸 메타크릴레이트-(2-디메틸아미노에틸)메타크릴레이트-메틸메타크릴레이트) 공중합체(예컨대, 유드라짓E100, 에보닉, 독일)이고; 상기 폴리에틸렌 유도체는 폴리에틸렌 글리콜, 및 폴리에틸렌 옥사이드 중에서 선택된 1종 이상이며; 및 상기 카르복시비닐폴리머는 카보머를 사용한다.
본 발명 구현에서 바람직한 방출제어물질은 다음과 같다.
(1) 약제학적 제제 A(심바스타틴 및 로자탄 함유 약제학적 제제)
약제학적 제제A에서, 바람직하게는 방출제어물질은 수불용성 중합체 및 장용성 고분자 중에서 선택된 1종 이상을 포함한다.
약제학적 제제A에서, 장용성 고분자는 히프로멜로오스프탈레이트 또는 아크릴산메틸메타크릴산 공중합체가 바람직하다. 본 발명에 의한 장용성 고분자는 로자탄 1중량부 대비 0.1~20 중량부, 바람직하게는 0.5~10 중량부로 포함될 수 있으며, 0.1 중량부 미만인 경우에는 pH 5 미만에서 쉽게 용해되는 문제점이 있고, 20중량부 초과인 경우에는 불필요하게 제제 총중량이 커지거나 과도하게 용출이 지연되는 문제점이 있다.
약제학적 제제A에서, 수불용성 중합체는 폴리(에틸아크릴레이트-메틸 메타크릴레이트-트리메틸아미노에틸메타크릴레이트 클로라이드)공중합체, 에틸셀룰로오스 또는 셀룰로오스 아세테이트가 바람직하다. 본 발명에 의한 수불용성 중합체는 로자탄 1중량부 대비 0.1~30 중량부, 바람직하게는 0.5~20 중량부로 포함될 수 있으며, 0.1중량부 미만인 경우에는 약물의 방출이 제어되지 않는 문제점이 있고, 30 중량부 초과인 경우에는 과도하게 용출이 지연되는 문제점이 있다.
약제학적 제제A에서, 소수성 화합물은 카르나우바왁스가 바람직하다. 본 발명에 의한 소수성 화합물은 로자탄 1중량부 대비 0.1~20 중량부, 바람직하게는 0.5~10 중량부 포함될 수 있으며, 0.1 중량부 미만인 경우에는 약물의 방출이 제어되지 않는 문제점이 있고, 20중량부 초과인 경우에는 과도하게 용출이 지연되는 문제점이 있다.
약제학적 제제A에서, 친수성 고분자는 카보머 및 히프로멜로오스 중에서 선택된 1종 이상이 바람직하다. 본 발명에 의한 친수성 고분자는 로자탄 1중량부에 대하여 0.05~30 중량부, 바람직하게는 0.5~20중량부로 포함될 수 있으며, 0.05중량부 미만인 경우에는 방출속도가 조절되지 않는 문제점이 있고, 30중량부 초과인 경우에는 방출속도가 조절되지 않는 문제점이 있고, 30중량부 초과인 경우에는 과도하게 용출이 지연되는 문제점이 있다.
(2) 약제학적 제제 B(심바스타틴 및 올메사르탄 함유 약제학적 제제)
약제학적 제제B에서, 바람직하게는 방출제어물질은 수불용성 중합체 및 장용성 고분자 중에서 선택된 1종 이상을 포함한다.
약제학적 제제B에서, 장용성 고분자는 폴리비닐아세테이트프탈레이트, 히프로멜로오스 프탈레이트 또는 아크릴산메틸메타크릴산 공중합체가 바람직하다. 본 발명에 의한 장용성 고분자는 올메사르탄 1중량 대비 0.1~20 중량부, 바람직하게는 0.5~10 중량부로 포함될 수 있으며, 0.1 중량부 미만인 경우에는 pH 5 미만에서 쉽게 용해되는 문제점이 있고, 20중량부 초과인 경우에는 불필요하게 제제 총중량이 커지거나 과도하게 용출이 지연되는 문제점이 있다.
약제학적 제제B에서, 수불용성 중합체는 폴리비닐아세테이트, 에틸셀룰로오스 또는 셀룰로오스 아세테이트가 바람직하다. 본 발명에 의한 수불용성 중합체는 올메사르탄 1중량 대비 0.1~30 중량부, 바람직하게는 0.5~20 중량부로 포함될 수 있으며, 0.1중량부 미만인 경우에는 약물의 방출이 제어되지 않는 문제점이 있고, 30 중량부 초과인 경우에는 과도하게 용출이 지연되는 문제점이 있다.
약제학적 제제B에서, 소수성 화합물은 올메사르탄 1중량 대비 0.1~20 중량부, 바람직하게는 0.5~10 중량부 포함될 수 있으며, 0.1 중량부 미만인 경우에는 약물의 방출이 제어되지 않는 문제점이 있고, 20중량부 초과인 경우에는 과도하게 용출이 지연되는 문제점이 있다.
약제학적 제제B에서, 친수성 고분자는 카보머, 히드록시프로필셀룰로오스, 히프로멜로오스 중에서 선택된 1종 이상이 바람직하다. 본 발명에 의한 친수성 고분자는 올메사르탄 1중량부에 대하여 0.05~30 중량부, 바람직하게는 0.5~20중량부로 포함될 수 있으며, 0.05중량부 미만인 경우에는 방출속도가 조절되지 않는 문제점이 있고, 30중량부 초과인 경우에는 방출속도가 조절되지 않는 문제점이 있고, 30중량부 초과인 경우에는 과도하게 용출이 지연되는 문제점이 있다.
(3) 약제학적 제제 C(심바스타틴 및 발사르탄 함유 약제학적 제제)
약제학적 제제C에서, 바람직하게는 방출제어물질은 수불용성 중합체 및 장용성 고분자 중에서 선택된 1종 이상을 포함한다.
약제학적 제제C에서, 장용성 고분자는 히프로멜로오스프탈레이트가 바람직하다. 본 발명에 의한 장용성 고분자는 발사르탄 1중량부 대비 0.1~20 중량부, 바람직하게는 0.5~10 중량부로 포함될 수 있으며, 0.1 중량부 미만인 경우에는 pH 5 미만에서 쉽게 용해되는 문제점이 있고, 20중량부 초과인 경우에는 불필요하게 제제 총중량이 커지거나 과도하게 용출이 지연되는 문제점이 있다.
약제학적 제제C에서, 수불용성 중합체는 폴리비닐아세테이트 또는 폴리(에틸아크릴레이트-메틸 메타크릴레이트-트리메틸아미노에틸메타크릴레이트 클로라이드)공중합체 가 바람직하다. 본 발명에 의한 수불용성 중합체는 발사르탄 1중량부 대비 0.1~30 중량부, 바람직하게는 0.5~20 중량부로 포함될 수 있으며, 0.1중량부 미만인 경우에는 약물의 방출이 제어되지 않는 문제점이 있고, 30 중량부 초과인 경우에는 과도하게 용출이 지연되는 문제점이 있다.
약제학적 제제C에서, 소수성 화합물은 카르나우바왁스가 바람직하다. 본 발명에 의한 소수성 화합물은 발사르탄 1중량부 대비 0.1~20 중량부, 바람직하게는 0.5~10 중량부 포함될 수 있으며, 0.1 중량부 미만인 경우에는 약물의 방출이 제어되지 않는 문제점이 있고, 20중량부 초과인 경우에는 과도하게 용출이 지연되는 문제점이 있다.
약제학적 제제C에서, 친수성 고분자는 히드록시프로필셀룰로오스 중에서 선택된 1종 이상이 바람직하다. 본 발명에 의한 친수성 고분자는 발사르탄 1중량부에 대하여 0.05~30 중량부, 바람직하게는 0.5~20중량부로 포함될 수 있으며, 0.05중량부 미만인 경우에는 방출속도가 조절되지 않는 문제점이 있고, 30중량부 초과인 경우에는 방출속도가 조절되지 않는 문제점이 있고, 30중량부 초과인 경우에는 과도하게 용출이 지연되는 문제점이 있다.
(4) 약제학적 제제 D(심바스타틴 및 칸데사르탄 함유 약제학적 제제)
약제학적 제제D에서, 바람직하게는 방출제어물질은 수불용성 중합체 및 장용성 고분자 중에서 선택된 1종 이상을 포함한다.
약제학적 제제D에서, 장용성 고분자는 히프로멜로오스 아세테이트 숙시네이트 또는 아크릴산메틸메타크릴산 공중합체가 바람직하다. 본 발명에 의한 장용성 고분자는 칸데사르탄 1중량부 대비 0.1~20 중량부, 바람직하게는 0.5~10 중량부로 포함될 수 있으며, 0.1 중량부 미만인 경우에는 pH 5 미만에서 쉽게 용해되는 문제점이 있고, 20중량부 초과인 경우에는 불필요하게 제제 총중량이 커지거나 과도하게 용출이 지연되는 문제점이 있다.
약제학적 제제D에서, 수불용성 중합체는 폴리(에틸 아크릴레이트-메틸 메타크릴레이드-트리에틸아미노에틸- 메타크릴레이트 클로라이드) 공중합체, 폴리비닐아세테이트 또는 에틸셀룰로오스가 바람직하다. 본 발명에 의한 수불용성 중합체는 칸데사르탄 1중량부 대비 0.1~30 중량부, 바람직하게는 0.5~20 중량부로 포함될 수 있으며, 0.1중량부 미만인 경우에는 약물의 방출이 제어되지 않는 문제점이 있고, 30 중량부 초과인 경우에는 과도하게 용출이 지연되는 문제점이 있다.
약제학적 제제D에서, 바람직한 소수성 화합물은 카르나우바왁스이다. 본 발명에 의한 소수성 화합물은 칸데사르탄 대비 0.1~20 중량부, 바람직하게는 0.5~10 중량부 포함될 수 있으며, 0.1 중량부 미만인 경우에는 약물의 방출이 제어되지 않는 문제점이 있고, 20중량부 초과인 경우에는 과도하게 용출이 지연되는 문제점이 있다.
약제학적 제제D에서, 바람직한 친수성 고분자는 히프로멜로오스이다. 본 발명에 의한 친수성 고분자는 칸데사르탄 1중량부에 대하여 0.05~30 중량부, 바람직하게는 0.5~20중량부로 포함될 수 있으며, 0.05중량부 미만인 경우에는 방출속도가 조절되지 않는 문제점이 있고, 30중량부 초과인 경우에는 방출속도가 조절되지 않는 문제점이 있고, 30중량부 초과인 경우에는 과도하게 용출이 지연되는 문제점이 있다.
(5) 약제학적 제제 E(로슈바스타틴 및 로자탄 함유 약제학적 제제)
약제학적 제제E에 있어서, 바람직한 장용성 고분자는 히프로멜로오스프탈레이트, 히프로멜로오스아세테이트숙시네이트, 카르복시메틸에틸셀룰로오스, 아크릴산메틸메타크릴산 공중합체, 메타크릴산-메타크릴산메틸 공중합체이고, 더 바람직하게는 히프로멜로오스프탈레이트, 아크릴산메틸메타크릴산 공중합체이다. 본 발명에 의한 장용성 고분자는 제제 총중량에 대하여 1~50중량%, 바람직하게는 3~30중량%로 포함될 수 있으며, 1중량% 미만인 경우에는 산성조건에서 용해되거나 안정하지 못한 문제점이 있고, 50중량% 초과인 경우에는 염기성조건 하에서도 용해되지 않는 문제점이 있다.
약제학적 제제E에서, 바람직한 수불용성 중합체는 폴리비닐 아세테이트, 에틸셀룰로오스, 및 셀룰로오스아세테이트 중에서 선택된 1종 이상일 수 있고, 보다 바람직하게는 폴리비닐 아세테이트, 에틸셀룰로오스, 셀룰로오스아세테이트 중에서 선택된 1종 이상 일 수 있다.
본 발명에 의한 수불용성 중합체는 제제 총중량에 대하여 약 1~50중량%, 바람직하게는 약 3~30중량%로 포함될 수 있으며, 1중량% 미만인 경우에는 충분한 지연 시간을 갖기 어려운 문제점이 있고, 50중량% 초과인 경우에는 약물의 방출이 일어나지 않거나 지연시간의 9시간 이상이 되어 지나치게 길어지는 문제점이 있다.
약제학적 제제E에서, 바람직한 소수성 화합물은 글리세릴 팔미토스테아레이트, 글리세릴 베헤네이트, 카르나우바왁스, 밀납으로부터 선택된 하나 이상, 보다 바람직하게는 글리세릴 팔미토스테아레이트, 카르나우바왁스로부터 선택된 하나 이상이다. 본 발명에 의한 소수성 화합물은 제제 총중량에 대하여 약 1~50중량%, 바람직하게는 약 3~30중량%로 포함될 수 있으며, 1중량% 미만인 경우에는 약물의 방출에 전혀 영향을 주지 못하는 문제점이 있고, 50중량% 초과인 경우에는 약물의 방출이 일어나지 않거나 제형화 하기 어려운 문제점이 있다.
약제학적 제제E에서, 바람직한 친수성 화합물은 덱스트린, 덱스트란, 펙틴 히프로멜로오스, 히드록시프로필셀룰로오스, 구아검, 아라비아검, 잔탄검, 젤라틴, 폴리(부틸 메타크릴레이트-(2-디메틸아미노에틸)메타크릴레이트-메틸메타크릴레이트 공중합체, 카보머 중에서 선택된 1종이상일 수 있고, 보다 바람직하게는 덱스트린, 히프로멜로오스, 잔탄검, 카보머 중에서 선택된 1종 이상 일 수 있다. 본 발명에 의한 친수성 고분자는 제제 총중량에 대하여 약 1~70중량%, 바람직하게는 약 3~50중량%로 포함될 수 있으며, 1중량% 미만인 경우에는 정제의 붕해에 전혀 영향을 주지 못 하는 문제점이 있고, 70중량% 초과인 경우에는 붕해 및 방출을 제어하기 힘든 문제점이 있다.
약제학적 제제E에 있어서, 바람직한 방출제어물질은 카보머, 히프로멜로오스아세테이트숙시네이트, 폴리비닐아세테이트, 카라나우바왁스, 히프로멜로오스, 히드록시프로필셀룰로오스, 에틸셀룰로오스, 히프로멜로오스프탈레이트, 아크릴산메틸메타크릴산 공중합체, 및 이들의 혼합물로 구성된 군에서 선택된 것이며, 보다 바람직한 방출제어물질은 폴리비닐아세테이트, 히프로멜로오스아세테이트숙시네이트, 카보머 및 이들의 혼합물로 구성된 군으로부터 선택된 것이다.
(6) 약제학적 제제 F(로슈바스타틴 및 이베사르탄 함유 약제학적 제제)
약제학적 제제F에 있어서, 바람직한 장용성 고분자는 히프로멜로오스프탈레이트, 히프로멜로오스아세테이트숙시네이트, 셀룰로오스아세테이트프탈레이트, 아크릴산메타크릴산 공중합체, 메타크릴산ㆍ아크릴산에틸공중합체, 스티렌-말레인산모노에스테르 공중합체, 폴리비닐아세테이트프탈레이트 중에서 선택된 1종 이상일 수 있고, 보다 바람직하게는 히프로멜로오스프탈레이트, 히프로멜로오스아세테이트숙시네이트, 아크릴산메타크릴산 공중합체, 메타크릴산ㆍ아크릴산에틸공중합체 중에서 선택된 1종 이상 일 수 있다. 본 발명에 의한 장용성 고분자는 제제 총중량에 대하여 약 5~80중량%, 바람직하게는 약 10~30중량%로 포함될 수 있으며, 5중량% 미만인 경우에는 산성조건에서 용해되거나 안정하지 못한 문제점이 있고, 80중량% 초과인 경우에는 염기성조건 하에서도 용해되지 않는 문제점이 있다.
약제학적 제제F에 있어서, 바람직한 수불용성 중합체는 바람직하게는 폴리비닐 아세테이트, 폴리(에틸아크릴레이트-메틸 메타크릴레이트) 공중합체, 폴리(에틸아크릴레이트-메틸 메타크릴레이트-트리메틸아미노에틸메타크릴레이트)공중합체, 폴리(에틸아크릴레이트-메틸 메타크릴레이트-트리메틸아미노에틸메타크릴레이트)공중합체, 에틸셀룰로오스, 셀룰로오스아세테이트 중에서 선택된 1종 이상일 수 있고, 보다 바람직하게는 폴리비닐 아세테이트, 폴리(에틸아크릴레이트-메틸 메타크릴레이트-트리메틸아미노에틸메타크릴레이트클로라이드)공중합체, 에틸셀룰로오스, 셀룰로오스아세테이트 중에서 선택된 1종 이상 일 수 있다. 본 발명에 의한 수불용성 중합체는 제제 총중량에 대하여 약 5~80중량%, 바람직하게는 약 10~30중량%로 포함될 수 있으며, 5중량% 미만인 경우에는 충분한 지연 시간을 갖기 어려운 문제점이 있고, 80중량% 초과인 경우에는 약물의 방출이 일어나지 않거나 지연시간의 9시간 이상이 되어 지나치게 길어지는 문제점이 있다.
약제학적 제제F에 있어서, 바람직한 소수성 화합물은 글리세릴 팔미토스테아레이트, 글리세릴 베헤네이트, 스테아린산, 세틸알코올, 카르나우바왁스, 비검 중에서 선택된 1종 이상일 수 있고, 보다 바람직하게는 글리세릴 베헤네이트, 스테아린산, 세틸알코올, 카르나우바왁스 중에서 선택된 1종 이상 일 수 있다. 본 발명에 의한 소수성 화합물은 제제 총중량에 대하여 약 5~80중량%, 바람직하게는 약 10~30중량%로 포함될 수 있으며, 5중량% 미만인 경우에는 약물의 방출에 전혀 영향을 주지 못하는 문제점이 있고, 80중량% 초과인 경우에는 약물의 방출이 일어나지 않거나 제형화 하기 어려운 문제점이 있다.
약제학적 제제F에 있어서, 바람직한 친수성 고분자는 바람직하게는 히프로멜로오스, 히드록시프로필셀룰로오스, 구아검, 잔탄검, 젤라틴, 폴리비닐 피롤리돈, 폴리(부틸 메타크릴레이트-(2-디메틸아미노에틸)메타크릴레이트-메틸메타크릴레이트) 공중합체 중에서 선택된 1종 이상일 수 있고, 보다 바람직하게는 히프로멜로오스, 히드록시프로필셀룰로오스, 잔탄검, 폴리비닐 피롤리돈 중에서 선택된 1종 이상 일 수 있다. 본 발명에 의한 친수성 고분자는 제제 총중량에 대하여 약 5~80중량%, 바람직하게는 약 10~30중량%로 포함될 수 있으며, 5중량% 미만인 경우에는 정제의 붕해에 전혀 영향을 주지 못하는 문제점이 있고, 80중량% 초과인 경우에는 붕해 및 방출을 제어 하기 힘든 문제점이 있다.
약제학적 제제 F에 있어서, 바람직한 방출제어물질은 히프로멜로오스아세테이트숙시네이트,히프로멜로오스프탈레이트, 아크릴산메틸메타크릴산 공중합체, 폴리비닐아세테이트, 에틸셀룰로오스, 셀룰로오스 아세테이트, 카르나우바왁스, 히프로멜로오스(히드록시프로필메틸셀룰로오스), 히드록시프로필셀룰로오스, 폴리비닐 피롤리돈, 및 이들의 혼합물로 이루어진 군으로부터 선택된 것일 수 있고, 보다 바람직하게는 히프로멜로오스아세테이트숙시네이트, 히프로멜로오스프탈레이트, 아크릴산메틸메타크릴산 공중합체, 폴리비닐아세테이트, 에틸셀룰로오스, 카르나우바왁스, 히프로멜로오스, 히드록시프로필셀룰로오스, 및 이들의 혼합물로 이루어진 군으로부터 선택된 것일 수 있다.
(7) 약제학적 제제 G(로슈바스타틴 및 칸데르사르탄 함유 약제학적 제제)
약제학적 제제G에서, 방출제어물질은 장용성 고분자, 수불용성 중합체, 친수성 고분자, 및 소수성 화합물과 친수성고분자의 혼합물이 바람직하다.
약제학적 제제G에서, 바람직한 장용성 고분자는 바람직하게는 히드록시프로필메틸셀룰로오스프탈레이트(히프로멜로오스 프탈레이트), 히드록시프로필메틸셀룰로오스아세테이트숙시네이트(히프로멜로오스 아세테이트숙시네이트), 셀룰로오스아세테이트프탈레이트, 아크릴산메타크릴산 공중합체, 메타크릴산ㆍ아크릴산에틸공중합체, 스티렌-말레인산모노에스테르 공중합체, 폴리비닐아세테이트프탈레이트 중에서 선택된 1종 이상일 수 있고, 보다 바람직하게는 히드록시프로필메틸셀룰로오스프탈레이트, 히드록시프로필메틸셀룰로오스아세테이트숙시네이트, 아크릴산메타크릴산 공중합체, 메타크릴산ㆍ아크릴산에틸공중합체 중에서 선택된 1종 이상 일 수 있다. 본 발명에 의한 장용성 고분자는 제제 총중량에 대하여 약 5~80중량%, 바람직하게는 약 10~30중량%로 포함될 수 있으며, 5중량% 미만인 경우에는 산성조건에서 용해되거나 안정하지 못한 문제점이 있고, 80중량% 초과인 경우에는 염기성조건 하에서도 용해되지 않는 문제점이 있다.
약제학적 제제G에서, 바람직한 수불용성 중합체는 바람직하게는 폴리비닐 아세테이트, 폴리(에틸아크릴레이트-메틸 메타크릴레이트) 공중합체, 폴리(에틸아크릴레이트-메틸 메타크릴레이트-트리메틸아미노에틸메타크릴레이트클로라이드)공중합체, 에틸셀룰로오스, 셀룰로오스아세테이트 중에서 선택된 1종 이상일 수 있고, 보다 바람직하게는 폴리비닐 아세테이트, 폴리(에틸아크릴레이트-메틸 메타크릴레이트) 공중합체, 에틸셀룰로오스, 셀룰로오스아세테이트 중에서 선택된 1종 이상 일 수 있다. 본 발명에 의한 수불용성 중합체는 제제 총중량에 대하여 약 5~80중량%, 바람직하게는 약 10~30중량%로 포함될 수 있으며, 5중량% 미만인 경우에는 충분한 지연 시간을 갖기 어려운 문제점이 있고, 80중량% 초과인 경우에는 약물의 방출이 일어나지 않거나 지연시간의 9시간 이상이 되어 지나치게 길어지는 문제점이 있다.
약제학적 제제G에서, 바람직한 소수성 화합물은 바람직하게는 글리세릴 팔미토스테아레이트, 글리세릴 베헤네이트, 스테아린산, 세틸알코올, 카르나우바왁스 중에서 선택된 1종 이상일 수 있고, 보다 바람직하게는 글리세릴 베헤네이트, 스테아린산, 카르나우바왁스 중에서 선택된 1종 이상 일 수 있다. 본 발명에 의한 소수성 화합물은 제제 총중량에 대하여 약 5~80중량%, 바람직하게는 약 10~30중량%로 포함될 수 있으며, 5중량% 미만인 경우에는 약물의 방출에 전혀 영향을 주지 못하는 문제점이 있고, 80중량% 초과인 경우에는 약물의 방출이 일어나지 않거나 제형화 하기 어려운 문제점이 있다.
약제학적 제제G에서, 바람직한 친수성 고분자는 히드록시프로필메틸셀룰로오스, 히드록시프로필셀룰로오스, 구아검, 잔탄검, 젤라틴, 폴리비닐 피롤리돈, 폴리(부틸 메타크릴레이트-(2-디메틸아미노에틸)메타크릴레이트-메틸메타크릴레이트) 공중합체 중에서 선택된 1종 이상일 수 있고, 보다 바람직하게는 히드록시프로필메틸셀룰로오스, 히드록시프로필셀룰로오스, 잔탄검, 폴리비닐 피롤리돈 중에서 선택된 1종 이상 일 수 있다. 본 발명에 의한 친수성 고분자는 제제 총중량에 대하여 약 5~80중량%, 바람직하게는 약 10~30중량%로 포함될 수 있으며, 5중량% 미만인 경우에는 정제의 붕해에 전혀 영향을 주지 못 하는 문제점이 있고, 80중량% 초과인 경우에는 붕해 및 방출을 제어하기 힘든 문제점이 있다.
약제학적 제제G에서, 바람직한 방출제어물질은 히드록시프로필메틸셀룰로오스아세테이트숙시네이트, 히드록시프로필메틸셀룰로오스프탈레이트, 아크릴산메틸메타크릴산 공중합체, 폴리비닐아세테이트, 에틸셀룰로오스, 셀룰로오스 아세테이트, 카르나우바왁스, 히드록시프로필메틸셀룰로오스, 히드록시프로필셀룰로오스, 폴리비닐 피롤리돈, 및 이들의 혼합물로 이루어진 군으로부터 선택된 것이며; 보다 바람직한 방출제어물질은 히드록시프로필메틸셀룰로오스아세테이트숙시네이트, 히드록시프로필메틸셀룰로오스프탈레이트, 아크릴산메틸메타크릴산 공중합체, 폴리비닐아세테이트, 에틸셀룰로오스, 카르나우바왁스, 히드록시프로필메틸셀룰로오스, 히드록시프로필셀룰로오스, 및 이들의 혼합물로 이루어진 군으로부터 선택된 것이고; 보다 바람직한 방출제어물질은 히드록시프로필메틸셀룰로오스아세테이트숙시네이트, 히드록시프로필셀룰로오스, 폴리비닐아세테이트, 및 이들의 혼합물로 이루어진 군으로부터 선택된 것이다.
2-b. 삼투압 조절제 및 반투과성막 코팅기제
본 발명의 지연방출성 구획은 삼투압조절제를 포함하며, 반투과성막 코팅기제로 코팅된 구획일 수 있다.
삼투압 조절제에 의한 정제와 소화관 내의 삼투압 차이로 인해 물이 정제 표면의 반투과성막을 통과하여 정제 내의 압력이 증가한다. 이로 인해 약물은 삼투 수송 구멍이나 코팅막의 세공을 통하여 방출되거나 코팅 기제의 탄성을 넘는 압력이 발생하면 코팅층이 붕괴되어 방출될 수 있다.
본 발명의 지연방출성 구획에서, 삼투압조절제는 삼투압의 원리를 이용하여 약물의 방출속도를 조절하는데 사용되는 성분을 말하며, 예컨대 황산마그네슘, 염화마그네슘, 염화나트륨, 염화리튬, 황산칼륨, 황산나트륨, 황산리튬, 황산나트륨 및 이들의 혼합물로 이루어진 군에서 선택된 1종 이상의 것이다.
본 발명의 지연방출성 구획에서, 반투과성막 코팅기제는 약학적으로 사용가능한 코팅기제로서, 약학적 제제의 코팅층에 배합하여 일부 성분은 통과시키지만, 다른 성분은 통과시키지 않는 막을 형성하는데 사용하는 물질을 말한다. 본 발명에서 반투과성막 코팅기제는 예컨대 폴리비닐 아세테이트, 수불용성 폴리메타크릴레이트 공중합체, 에틸셀룰로오스, 셀룰로오스 에스테르, 셀룰로오스 에테르, 셀룰로오스 아실레이트, 셀룰로오스 디아실레이트, 셀룰로오스 트리아실레이트, 셀룰로오스 아세테이트, 셀룰로오스 디아세테이트, 셀룰로오스 트리아세테이트 및 이들의 혼합물로 이루어진 군에서 선택된 1종 이상을 들 수 있다.
본 발명의 구체적 구현예에서 바람직한 삼투압 조절제 및 반투과성막 코팅기제의 함량은 다음과 같다.
(1) 약제학적 제제 A(심바스타틴 및 로자탄 함유 약제학적 제제)
약제학적 제제A에서, 바람직한 삼투압조절제는 염화나트륨, 황산나트륨이다. 여기서, 삼투압조절제는 로자탄 1중량부에 0.05 내지 30 중량부, 바람직하게는 0.1 내지 20중량부로 포함될 수 있으며, 0.1중량부 미만인 경우에는 삼투압 발생 효과가 미약한 문제점이 있고, 30중량부 초과인 경우에는 불필요하게 제제 총중량을 증가시키거나 적합한 약물 방출속도를 구현할 수 없는 문제점이 있다.
약제학적 제제A에서, 바람직한 반투과성막 코팅기제는 에틸셀룰로오스 또는 셀룰로오스 아세테이트이다. 여기서, 반투과성막 코팅기제는 로자탄1 중량부에 대해서 0.05 중량부 ~ 30 중량부, 바람직하게는 0.1 중량부 ~ 20 중량부로 포함될 수 있으며, 0.05 중량부 미만인 경우에는 충분한 지연 시간을 갖기 어려운 문제점이 있고, 30 중량부 초과인 경우에는 약물의 방출이 일어나지 않거나 지연시간이 9시간 이상이 되어 지나치게 길어지는 문제점이 있다.
(2) 약제학적 제제 B(심바스타틴 및 올메사르탄 함유 약제학적 제제)
약제학적 제제B에서, 바람직한 삼투압조절제는 염화나트륨 또는 황산나트륨이다. 삼투압조절제는 올메사르탄 1중량부에 0.05 내지 30 중량부, 바람직하게는 0.1 내지 20중량부로 포함될 수 있으며, 0.1중량부 미만인 경우에는 삼투압 발생 효과가 미약한 문제점이 있고, 30중량부 초과인 경우에는 불필요하게 제제 총중량을 증가시키거나 적합한 약물 방출속도를 구현할 수 없는 문제점이 있다.
약제학적 제제B에서, 바람직한 반투과성막 코팅기제는 에틸셀룰로오스 또는 셀룰로오스 아세테이트이다. 여기서, 반투과성막 코팅기제는 올메사르탄 1 중량부에 대해서 0.05 중량부 ~ 30 중량부, 바람직하게는 0.1 중량부 ~ 20 중량부로 포함될 수 있으며, 0.05 중량부 미만인 경우에는 충분한 지연 시간을 갖기 어려운 문제점이 있고, 30 중량부 초과인 경우에는 약물의 방출이 일어나지 않거나 지연시간이 9시간 이상이 되어 지나치게 길어지는 문제점이 있다.
(3) 약제학적 제제 C(심바스타틴 및 발사르탄 함유 약제학적 제제)
약제학적 제제C에서, 바람직한 삼투압조절제는 염화나트륨 또는 황산나트륨이다. 삼투압조절제는 발사르탄1 중량부에 0.05 내지 30 중량부, 바람직하게는 0.1 내지 20중량부로 포함될 수 있으며, 0.1중량부 미만인 경우에는 삼투압 발생 효과가 미약한 문제점이 있고, 30중량부 초과인 경우에는 불필요하게 제제 총중량을 증가시키거나 적합한 약물 방출속도를 구현할 수 없는 문제점이 있다.
약제학적 제제C에서, 바람직한 반투과성막 코팅기제는 에틸셀룰로오스이다. 여기서, 반투과성막 코팅기제는 발사르탄1 중량부에 대해서 0.05 중량부 ~ 30 중량부, 바람직하게는 0.1 중량부 ~ 20 중량부로 포함될 수 있으며, 0.05 중량부 미만인 경우에는 충분한 지연 시간을 갖기 어려운 문제점이 있고, 30 중량부 초과인 경우에는 약물의 방출이 일어나지 않거나 지연시간이 9시간 이상이 되어 지나치게 길어지는 문제점이 있다.
(4) 약제학적 제제 D(심바스타틴 및 칸데르사르탄 함유 약제학적 제제)
약제학적 제제D에서, 바람직한 삼투압조절제는 염화나트륨 또는 황산나트륨이다. 여기서, 삼투압조절제는 칸데사르탄 1중량부에 0.05 내지 30 중량부, 바람직하게는 0.1 내지 20중량부로 포함될 수 있으며, 0.1중량부 미만인 경우에는 삼투압 발생 효과가 미약한 문제점이 있고, 30중량부 초과인 경우에는 불필요하게 제제 총중량을 증가시키거나 적합한 약물 방출속도를 구현할 수 없는 문제점이 있다.
약제학적 제제D에서, 바람직한 반투과성막 코팅기제는 에틸셀룰로오스 또는 셀룰로오스 아세테이트이다. 여기서, 반투과성막 코팅기제는 칸데사르탄 1 중량부에 대해서 0.05 중량부 ~ 30 중량부, 바람직하게는 0.1 중량부 ~ 20 중량부로 포함될 수 있으며, 0.05 중량부 미만인 경우에는 충분한 지연 시간을 갖기 어려운 문제점이 있고, 30 중량부 초과인 경우에는 약물의 방출이 일어나지 않거나 지연시간이 9시간 이상이 되어 지나치게 길어지는 문제점이 있다.
(5) 약제학적 제제 E(로슈바스타틴 및 로자탄 함유 약제학적 제제)
약제학적 제제E에서, 바람직한 삼투압 조절제는 염화나트륨이다. 본 발명에 의한 삼투압조절제는 제제 총중량에 대하여 약 0.1~50중량%, 바람직하게는 약 1~30중량%로 포함될 수 있으며, 0.1중량% 미만인 경우에는 삼투압이 형성되지 않는 문제점이 있고, 50중량% 초과인 경우에는 큰 삼투압의 형성으로 반투막이 손상되어 제어방출조절이 되지 않는 문제점이 있다.
약제학적 제제E에서, 바람직한 반투과성막 코팅기제는 폴리비닐 아세테이트, 에틸셀룰로오스, 셀룰로오스 트리아세테이트 중에서 선택된 1종 이상일 수 있고, 보다 바람직하게는 폴리비닐 아세테이트, 에틸셀룰로오스 중에서 선택된 1종 이상 일 수 있다. 본 발명에 의한 반투과성막 코팅기제는 제제 총중량에 대하여 약 1~50중량%, 바람직하게는 약 3~30중량%로 포함될 수 있으며, 1중량% 미만인 경우에는 목적하는 반투과성막을 형성하기 어려운 문제점이 있고, 50중량% 초과인 경우에는 모든 성분이 통과 되지 않을 수 있는 문제점이 있다.
(6) 약제학적 제제 F(로슈바스타틴 및 이베사르탄 함유 약제학적 제제)
약제학적 제제F에서, 바람직한 삼투압 조절제는 염화나트륨이다. 본 발명에 의한 삼투압조절제는 제제 총중량에 대하여 약 1~80중량%, 바람직하게는 약 2~50중량%로 포함될 수 있으며, 1중량% 미만인 경우에는 삼투압이 형성되지 않는 문제점이 있다.
약제학적 제제F에서, 바람직한 반투과성막 코팅기제는 폴리비닐아세테이트, 수불용성 폴리메타크릴레이트 공중합체, 에틸셀룰로오스, 셀룰로오스 트리아세테이트 중에서 선택된 1종 이상일 수 있고, 보다 바람직하게는 폴리비닐 아세테이트, 수불용성 폴리메타크릴레이트 공중합체, 에틸셀룰로오스 중에서 선택된 1종 이상 일 수 있다. 본 발명에 의한 반투과성막 코팅기제는 제제 총중량에 대하여 약 5~80중량%, 바람직하게는 약 10~30중량%로 포함될 수 있으며, 5중량% 미만인 경우에는 목적하는 반투과성막을 형성하기 어려운 문제점이 있고, 80중량% 초과인 경우에는 모든 성분이 통과 되지 않을 수 있는 문제점이 있다.
(7) 약제학적 제제 G(로슈바스타틴 및 칸데르사르탄 함유 약제학적 제제)
약제학적 제제G에서, 바람직한 삼투압 조절제는 염화나트륨이다. 본 발명에 의한 삼투압조절제는 제제 총중량에 대하여 약 1~80중량%, 바람직하게는 약 2~50중량%로 포함될 수 있으며, 1중량% 미만인 경우에는 삼투압이 형성되지 않는 문제점이 있다.
약제학적 제제G에서, 바람직한 반투과성막 코팅기제는 폴리비닐 아세테이트, 폴리메타크릴레이트 공중합체, 에틸셀룰로오스, 셀룰로오스 트리아세테이트 중에서 선택된 1종 이상일 수 있고, 보다 바람직하게는 폴리비닐 아세테이트, 폴리메타크릴레이트 공중합체, 에틸셀룰로오스 중에서 선택된 1종 이상 일 수 있다. 본 발명에 의한 반투과성막 코팅기제는 제제 총중량에 대하여 약 5~80중량%, 바람직하게는 약 10~30중량%로 포함될 수 있으며, 5중량% 미만인 경우에는 목적하는 반투과성막을 형성하기 어려운 문제점이 있고, 80중량% 초과인 경우에는 모든 성분이 통과 되지 않을 수 있는 문제점이 있다.
3. 약제학적으로 허용가능한 첨가제
본 발명의 제제는 본 발명의 효과를 해치지 않는 범위 안에서 약학적으로 허용 가능한 (2-a) 방출제어물질 및 (2-b) 삼투압 조절제 및 반투과성막 코팅기제로 언급한 것 이외의 희석제, 결합제, 붕해제, 윤활제, pH 조절제, 소포제, 용해보조제 등의 통상적으로 사용되는 첨가제를 지연방출성의 성격을 벗어나지 않는 범위내에서 추가로 사용하여 제제화할 수 있다.
예를 들어, 희석제로 슈가, 전분, 미결정셀룰로오스, 유당, 유당수화물, 포도당, 만니톨, 디-만니톨, 알기네이트, 알칼리토금속류염, 클레이, 폴리에틸렌글리콜, 디칼슘포스페이트, 무수인산수소칼슘, 또는 이들의 혼합물 등을 사용할 수 있다.
결합제로는 전분, 미결정셀룰로오스, 고분산성 실리카, 만니톨, 자당, 유당수화물, 폴리에틸렌글리콜, 폴리비닐피롤리돈, 폴리비닐피롤리돈 공중합체, 히프로멜로오스, 히드록시프로필셀룰로오스, 천연검, 합성검, 코포비돈, 포비돈, 젤라틴, 또는 이들의 혼합물 등을 사용할 수 있다.
붕해제로는 전분글리콘산나트륨, 옥수수전분, 감자전분 또는 전호화전분(전젤라틴화전분) 등의 전분 또는 변성전분; 벤토나이트, 몬모릴로나이트, 또는 비검(veegum) 등의 클레이; 미결정셀룰로오스, 히드록시프로필셀룰로오스 또는 카르복시메틸셀룰로오스 등의 셀룰로오스류; 알긴산나트륨 또는 알긴산 등의 알긴류; 크로스카멜로스(croscarmellose)나트륨 등의 가교 셀룰로오스류; 구아검, 잔탄검 등의 검류; 가교 폴리비닐피롤리돈(crospovidone) 등의 가교 중합체; 중탄산나트륨, 시트르산 등의 비등성 제제, 또는 이들의 혼합물을 사용할 수 있다.
윤활제로 탈크, 스테아린산, 스테아린산 마그네슘, 스테아린산 칼슘, 라우릴황산나트륨, 수소화식물성오일, 나트륨벤조에이트, 콜로이드성 이산화규소, 나트륨스테아릴푸마레이트, 글리세릴 베헤네이트, 글리세릴 모노레이트, 글리세릴모노스테아레이트, 글리세릴 팔미토스테아레이트, 폴리에틸렌글리콜, 메타규산알루민산마그네슘 등을 사용할 수 있다.
본 발명의 약제학적으로 허용 가능한 첨가제로서 pH 조절제는 초산, 아디프산, 아스코르빈산, 아스코르빈산 나크륨, 에테르산 나트륨, 사과산, 숙신산, 주석산, 푸마르산, 구연산(시트르산)과 같은 산성화제와 침강 탄산 칼슘, 암모니아수, 메글루민, 탄산 나트륨, 산화 마그네슘, 탄산 마그네슘, 구연산 나트륨, 삼염기칼슘인산염과 같은 염기성화제 등을 사용할 수 있다.
본 발명의 약제학적 허용 가능한 첨가제로서 소포제는 디메시콘, 올레일 알코올, 프로필렌글리콜 알지네이트, 시메티콘 에멀젼과 같은 시메티콘류 등을 사용할 수 있다.
본 발명의 약제학적으로 허용가능한 첨가제에서, 용해보조제는 라우릴황산나트륨, 폴리소르베이트 등의 폴리옥시에틸렌 소르비탄 지방산 에스테르류, 도큐세이트 나트륨, 폴록사머 등을 사용할 수 있다. 또한 트리에틸시트레이트, 폴리에틸렌글리콜과 같은 가소제를 첨가할 수도 있다.
이외에도 착색제, 향료 중에서 선택된 다양한 첨가제로서 약학적으로 허용 가능한 첨가제를 선택 사용하여 본 발명의 제제를 제제화할 수 있다. 본 발명에서 사용가능한 첨가제의 범위가 상기 첨가제를 사용하는 것으로 한정되는 것은 아니며, 상기한 첨가제는 선택에 의하여 통상 범위의 용량을 함유하여 제제화할 수 있다.
또한 본 발명의 지연방출제제에서, 결합용매와 지연 방출성 첨가제의 용매로 정제수, 에탄올, 메탄올, 염화메틸렌 등을 사용할 수 있으며, 더욱 바람직하게는 정제수, 에탄올이 바람직하다.
본 발명의 약제학적으로 허용가능한 첨가제에서, 사용가능한 첨가제의 범위가 상기 첨가제를 사용하는 것으로 한정되는 것은 아니며, 상기한 첨가제는 선택에 의하여 통상 범위의 용량을 함유하여 제제화할 수 있다.
본 발명의 제제는 다양한 제형으로 제조할 수 있으며, 예를 들어 나정, 코팅정, 다층정, 또는 유핵정 등의 정제, 분말제, 과립제, 또는 캡슐제 등으로 제형화할 수 있다.
본 발명의 제제는 선방출성 구획을 이루는 과립 등과 지연방출성 구획을 이루는 과립 등에 선택적으로 첨가제를 후혼합하여 타정한 것으로 단일 정제 내에 선방출성 구획과 지연방출성 구획을 갖게 되어 각각의 구획의 활성성분이 별도로 용출하게 되어 각각의 약효를 나타내게 되는 나정 형태일 수 있다.
본 발명의 제제는 상기 지연방출성 구획과, 이를 둘러싸는 선방출성 구획으로 이루어진 2상의 매트릭스 정제 형태일 수 있다.
또한, 본 발명의 제제는 지연방출성 구획으로 이루어진 정제와 상기 정제의 외부를 둘러싸는 선방출성 구획으로 이루어진 필름코팅층으로 구성된 필름코팅정 형태일 수 있으며, 필름코팅층이 용해함에 따라 필름코팅층의 심바스타틴이 먼저 용출되게 된다.
또한, 본 발명의 제제는 지연방출성 구획과 선방출성 구획을 구성하는 과립물에 약제학적인 첨가제를 혼합하고, 다중 타정기를 사용하여 2중정 혹은 3중정으로 타정하여 얻어진, 지연방출성 구획과 선방출성 구획이 다층구조를 이루는 다층정 형태일 수 있다. 상기 다층정을 이루는 각각의 층은 평행한 상태일 수 있다. 이 제제는 층별로 선방출과 지연방출이 가능하도록 제제화된 경구 투여용 정제이다.
또한, 본 발명의 제제는 지연방출성 구획으로 이루어진 내핵과 상기 내핵의 외면을 둘러싸고 있는 선방출성 구획으로 이루어진 외층으로 구성된 유핵정 형태일 수 있다. 상기 유핵정은 삼투성 유핵정일 수 있으며, 상기 삼투성 유핵정은 지연방출을 위해 삼투압 조절제를 정제의 내부에 함유하게 하여 타정한 후, 삼투성 반투막 코팅기제로 정제의 표면을 코팅하여 이를 내핵으로 하고, 선방출성 구획을 구성하는 과립물을 약제학적인 첨가제와 혼합한 뒤 외층으로 하여 타정함으로써 지연방출성의 내핵을 갖고 상기 내핵의 표면을 선방출층이 둘러싼 형태의 제형이다.
본 발명의 제제는 지연방출성 구획으로 이루어진 입자, 과립, 펠렛, 또는 정제와 선방출성 구획으로 이루어진 입자, 과립, 펠렛, 또는 정제를 포함하는 캡슐제 형태일 수 있다.
상기 캡슐제의 지연방출성 구획으로 이루어진 정제는 삼투압 조절제를 정제 내부에 포함하고, 정제의 표면에 반투과성막 코팅기제를 갖는 삼투성 코팅정일 수 있다.
상기 캡슐제의 재질은 젤라틴, 숙시네이트 젤라틴, 또는 히프로멜로오스, 그 혼합물 중에서 선택된 하나일 수 있다.
본 발명의 제제는 지연방출성 구획, 또는/및 선방출성 구획의 외부에 코팅층을 추가로 형성할 수 있다. 즉 지연방출성 구획, 또는/및 선방출성 구획으로 이루어진 입자, 과립, 펠렛, 또는 정제 등의 표면에 방출지연 또는 제제 안정을 위한 목적으로 코팅을 할 수 있다.
본 발명에 따른 제제는, 추가의 코팅이 없는 나정 등의 상태로도 제공되지만, 필요에 따라 상기 제제의 외부에 코팅층을 형성시켜, 코팅층을 추가로 포함하는 코팅정 형태의 제제일 수 있다. 코팅층을 형성함으로써, 활성성분의 안정성을 더욱 확보할 수 있는 제제를 제공할 수 있다.
코팅층을 형성하는 방법은 정제층의 표면에 필름상의 코팅층을 형성할 수 있는 방법 중에서 당업자의 선택에 의하여 적절히 선택할 수 있으며, 유동층 코팅법, 팬 코팅법 등의 방법을 적용할 수 있으며, 바람직하게는 팬 코팅법을 적용할 수 있다.
코팅층은 피막제, 피막 보조제 또는 이들의 혼합물을 사용하여 형성할 수 있으며, 예를 들어, 피막제는 히프로멜로오스, 히드록시프로필셀룰로오스 등과 같은 셀룰로오스 유도체, 당 유도체, 폴리비닐 유도체, 왁스류, 지방류, 젤라틴, 또는 이들의 혼합물 등을 사용할 수 있고; 피막 보조제는 폴리에틸렌글리콜, 에틸셀룰로오스, 글리세라이드류, 산화티탄, 탈크, 디에틸프탈레이트, 또는 이들의 혼합물 등을 사용할 수 있다.
코팅층은 정제 총 중량에 대하여 0.5 ~ 15 중량% 범위로 포함할 수 있다.
본 발명의 약제학적 제제는 당 분야의 적절한 방법으로, 일예로 Chrontherpeutics(2003, Peter Redfern, PhP) 에 개시되어 있는 시간차 투약 원리를 이용하여 제제화 할 수 있으며, 구체적으로 이하의 단계를 포함하는 방법에 의해 제조될 수 있다.
제 1 단계는 안지오텐신-II-수용체 차단제를 장용성 고분자, 수불용성 중합체, 소수성 화합물, 친수성 고분자 중에서 선택된 방출제어물질 1종 또는 2종과 약제학적으로 사용되는 통상의 첨가제를 투여하여 혼합, 연합, 건조, 제립 또는 코팅, 및 타정을 통해 지연방출성 과립 또는 정제를 얻거나, 안지오텐신-II-수용체 차단제를 삼투압조절제와 약제학적으로 사용되는 통상의 첨가제를 투여하여 혼합, 연합, 건조, 정립 또는 타정한 후 반투과성막 코팅기제로 코팅하여 지연방출성 과립 또는 정제를 얻는 단계이다.
제 2 단계는 스타틴계 지질저해제와 약제학적으로 허용되는 통상의 첨가제를 투여하여 혼합, 연합, 건조, 제립 혹은 코팅, 및 타정을 통해 경구 고형제를 생산하기 위한 통상의 과정을 통하여 얻어진 선방출성 과립 또는 정제를 얻는 단계이다.
제 3 단계는 상기 제 1 단계 및 제 2 단계에서 얻어진 각각의 과립 혹은 정제를 약제학적인 부형제와 혼합하여 타정 또는 충전하여 경구 투여용 제제를 얻는 단계이다.
상기 제 1 단계와 상기 제 2 단계는 순서를 바꾸거나, 동시에 실시할 수 있다.
상기 과정에 의하여 본 발명의 복합제제가 제조될 수 있으며, 제제화 방법을 보다 상세하게 설명하면 다음과 같으나, 이에 한정되는 것은 아니다.
[가] 2상의 매트릭스 정제의 제조
제 1 단계에서 얻어진 입자 또는 과립을 그대로 또는 방출제어물질로 추가 코팅한 후, 제 2 단계에서 제조한 과립과 혼합하여 일정량의 무게로 타정하여 정제를 제조한다. 얻어진 정제를 안정성 또는 성상 개선의 목적으로 필요에 따라 필름 코팅을 할 수 있다.
[나] 활성성분을 함유한 필름코팅정의 제조
제 1 단계에서 얻어진 코팅정 또는 과립을 그대로 또는 방출제어물질로 추가 코팅하고 건조한 후 일정량으로 타정하여 그대로 혹은 추가로 코팅하여 정제를 제조한 후, 따로 심바스타틴을 수용성의 필름코팅용액에 용해 후 분산시켜 제 1 단계에서 얻은 정제 외층에 코팅함으로써 필름코팅에 활성성분을 함유한 경구투여형 필름코팅정제를 제조할 수 있다.
[다] 다층정의 제조
제 1 단계에서 얻어진 과립을 그대로 또는 방출제어물질로 추가 코팅하고 건조하여 얻은 과립과 제 2 단계에서 얻어진 과립을 타정기를 이용하여 2중정으로 제조할 수 있다. 제형설계 또는 필요에 따라 방출 보조층을 추가하여 3중 또는 그 이상의 다층정을 제조하거나 코팅하여 코팅 다층정을 제조할 수 있다.
[라] 유핵정의 제조
제 1 단계에서 얻어진 코팅정 또는 과립을 그대로 또는 방출제어물질로 추가 코팅하고 건조한 후 일정량으로 타정하여 그대로 혹은 추가로 코팅을 하여 내핵으로 한 후, 제 2 단계에서 얻은 과립과 함께 유핵정타정기로 타정하여 1단계 정제의 표면을 선방출층이 둘러싼 형태의 유핵정을 제조하거나 코팅하여 코팅 유핵정을 제조할 수 있다.
[마] 캡슐제(과립 또는 정제 함유)의 제조
제 1 단계에서 얻어진 과립을 그대로 또는 방출제어물질로 추가 코팅하고 건조한 과립 또는 정제와, 제 2 단계에서 얻은 과립 또는 정제를 캡슐충전기에 넣고 일정 크기의 캡슐에 각 주성분 유효량 해당 량만큼 충전하여 캡슐제를 제조할 수 있다.
[바] 캡슐제(펠렛)의 제조
(1) 안지오텐신-II-수용체 차단제와 방출제어물질, 필요에 따라 약제학적으로 허용 가능한 첨가제를 물, 유기용매, 또는 혼합용매에 용해시키거나 현탁시켜 슈가 시드에 코팅, 건조 후 필요에 따라 방출제어물질 단독 또는 2종 이상을 사용하여 물, 유기용매, 또는 혼합용매에 용해시킨 후 코팅, 건조한 후 제 2 단계에서 얻은 과립 또는 제 3 단계에서 얻은 정제와 혼합 후 캡슐충진기로 캡슐에 충전하여 캡슐제를 제조할 수 있다.
(2) 스타틴계 지질저해제와 약제학적으로 허용 가능한 첨가제를 물, 유기용매, 또는 혼합용매에 용해시키거나 현탁시켜 슈가 시드에 코팅, 건조 후 상기(1)의 안지오텐신-II-수용체 차단제를 함유한 방출제어 펠렛과 혼합 후 캡슐충진기로 캡슐에 충전하여 캡슐제를 제조할 수 있다.
[사] 키트의 제조
제 1 단계에서 얻어진 안지오텐신-II-수용체 차단제 함유 제제와, 제 2 단계에서 얻은 스타틴지질저해제 함유 제제를 호일, 블리스터, 병 등에 같이 충전하여 동시에 복용이 가능한 키트로 제조할 수 있다.
본 발명 제제의 인체 투여량은 체내에서 활성성분의 흡수도, 불활성화율 및 배설속도, 환자의 연령, 성별 및 상태 등에 따라 적절히 선택되며, 예를 들어 설명하면 다음과 같다.
약제학적 제제A는, 일반적으로 성인에게 로자탄과 심바스타틴의 합계량으로, 1일 17.5 ~ 360 mg 투여하며, 바람직하게는 1일 35 ~ 180mg을 투여하여 항압작용과 합병증 예방 작용을 발휘토록 할 수 있다. 약제학적 제제B는, 일반적으로 성인에게 올메사르탄과 심바스타틴의 합계량으로, 1일 7.5 ~ 240 mg 투여하며, 바람직하게는 1일 15 ~ 120mg을 투여하여 항압작용과 합병증 예방 작용을 발휘토록 할 수 있다. 약제학적 제제C는, 일반적으로 성인에게 발사르탄과 심바스타틴의 합계량으로, 1일 2.0 ~ 960 mg 투여하며, 바람직하게는 1일 22 ~ 700mg을 투여하여 항압작용과 합병증 예방 작용을 발휘토록 할 수 있다. 약제학적 제제D는, 일반적으로 성인에게 칸데사르탄 과 심바스타틴의 합계량으로, 1일 7 내지 224 mg 투여하며, 바람직하게는 1일 14 내지 112mg을 투여하여 항압작용과 합병증 예방 작용을 발휘토록 할 수 있다.
약제학적 제제E는,일반적으로 성인에게 로슈바스타틴으로 1일 약 5 ~ 40 mg을 투여하며, 로자탄으로 1일 약 25 ~ 100 mg을 투여하여 항압작용, 지질저하작용 및 합병증 예방 작용을 발휘토록 할 수 있다.
약제학적 제제F는,일반적으로 성인에게 로슈바스타틴으로 1일 약 5 ~ 40 mg을 투여하며, 이베사르탄으로 1일 약 75 ~ 300 mg을 투여하여 항압작용, 지질저하작용 및 합병증 예방 작용을 발휘토록 할 수 있다.
약제학적 제제G는, 일반적으로 성인에게 로슈바스타틴으로 1일 약 5 ~ 40 mg을 투여하며, 칸데사르탄으로 1일 약 4 ~ 32 mg을 투여하여 항압작용, 지질저하작용 및 합병증 예방 작용을 발휘토록 할 수 있다.
상기와 같은 본 발명의 복합 약물 시스템은 서로 다른 두 약물을 활성성분으로 포함하여 복합 제제화되어 단 1회씩 투여케 함으로써 각 활성성분을 별도로 제제화하여 이를 동시에 복용케 하는 경우보다 복약지도가 쉽고, 또한 약물의 방출시간의 차이로 인해 약물 상호간의 길항작용이 일어나지 않아 길항작용에 따른 부작용을 감소시킬 수 있으며, 각 약물이 가지는 효과가 이들의 자체가 단독으로 가지는 효과보다 향상되어 나타난다.
또한, 본 발명의 제제는 서로 다른 약리를 지닌 성분의 복합제제이므로 부작용을 상쇄시켜 줄 뿐 아니라 순환기계 합병증의 발병 위험 인자를 감소시켜 줄 수 있으므로 장기간의 예방 경비를 절감시켜 줄 수 있으며, 단일 제제를 각각 유지하는 포장비용 절감과 고급 인력의 투약 조제 시간을 절감시켜 경제적인 면에서 매우 효율적이다.
또한, 본 발명은 저녁시간대, 즉, 오후 5시 내지 11시(17 내지 23시) 투여용 약제학적 제제를 제공한다.
또한, 본 발명은 본 발명의 약제학적 제제를 포유류에게 투여하는 단계를 포함하는 심혈관계 질환의 치료방법을 제공한다. 바람직하게는 본 발명의 약제학적 제제를 포유류에게 1일 1회 오후 5시 내지 11시에 투여하는 단계를 포함하는 고혈압 및 고지혈증 또는 그로 인한 심혈관계 질환 또는 대사증후군의 치료방법을 제공한다.
상기 심혈관계 질환은 심장혈관과 뇌혈관을 포함한 기타 혈관질환을 모두 일컫는 매우 광범위한 질환이다. 심장질환의 종류에는 동맥경화 진행에 의한 허혈성 심장질환(심근경색, 협심증 등)을 대표로 하여 고혈압, 심부전, 부정맥, 심근증, 심내막염 등이 있으며 혈관질환에는 뇌졸중(중풍)과 말초혈관질환 등이 있다. 또한, 고혈압, 당뇨병, 비만증, 고지혈증, 관상 동맥 질환 등이 복합적으로 나타나는 이른바 대사성 증후군을 지닌 자들의 고혈압과 합병증 등을 포함한다.
상술한 바와 같이, 본 발명의 약제학적 제제는 질환의 예방 또는 치료에서 약리학적, 임상학적, 과학적 및 경제적으로 각각의 약물의 단일 제제 및 단순 복합 제제보다 매우 유용하다. 본 발명의 약제학적 제제는 두 약물 상호간의 길항 작용 및 부작용을 방지하고, 최적의 약효를 나타낸다. 또한, 본 발명의 약제학적 제제는 한 번에 복용할 수 있으므로 환자에 대한 복약지도 및 복약이 용이하다.
도 1은 조코(심바스타틴 단일제), 코자(로자탄 단일제), 실시예 I-1에서 제조된 약제학적 제제의 용출률을 나타낸 그래프이다.
도 2는 실시예 I-3 내지 I-6의 약제학적 제제에서의 로자탄의 용출률을 나타낸 그래프이다.
도 3은 시판중인 조코(심바스타틴 단일제), 코자(로자탄 단일제), 실시예 I-7에서 제조된 약제학적 제제의 용출률을 나타낸 그래프이다.
도 4는 시판중인 조코(심바스타틴 단일제), 코자(로자탄 단일제), 실시예 I-10에서 제조된 약제학적 제제의 용출률을 나타낸 그래프이다.
도 5는 조코(심바스타틴 단일제), 코자(로자탄 단일제), 실시예 I-12 및 I-14에서 제조된 약제학적 제제의 용출률을 나타낸 그래프이다.
도 6은 실험예 I-6에 의한 동물시험결과로서 투여경로간 수축기 혈압을 비교한 그래프이다.
도 7은 실험예 I-6에 의한 동물시험결과로서 투여경로간 이완기 혈압을 비교한 그래프이다.
도 8은 실험예 I-6에 의한 동물시험결과로서 투여경로간 평균 혈압을 비교한 그래프이다.
도 9는 실시예 II-1에 따라 제조된 본발명의 약제학적 제제(정제)와 대조약인 심바스타틴 단일제(Merck: Zocor), 올메사르탄 메독소밀 단일제(Daiichi Sankyo: Benicar, 올메텍)의 용출률을 나타낸 그래프이다.
도 10은 실시예 II-5 또는 II-6에 따라 제조된 본발명의 약제학적 제제(캡슐제)와 대조약인 심바스타틴 단일제(Merck: Zocor), 올메사르탄 메독소밀 단일제( Daiichi Sankyo: Benicar, 올메텍)의 용출률을 나타낸 그래프이다.
도 11은 실시예 II-7 또는 II-8에따라 제조된 본 발명의 약제학적 제제(캡슐제)와 대조약인 심바스타틴 단일제(Merck: Zocor), 올메사르탄 메독소밀 단일제( Daiichi Sankyo: Benicar, 올메텍)의 용출률을 나타낸 그래프이다.
도 12는 실시예 II-2, II-3, II-9에 따라 제조된 본발명의 약제학적 제제(이중정, 다층정, 단일정)와 대조약인 심바스타틴 단일제(Merck: Zocor), 올메사르탄 메독소밀 단일제( Daiichi Sankyo: Benicar, 올메텍)의 용출률을 나타낸 그래프이다.
도 13은 실시예 II-10, II-11에 따라 제조된 본 발명의 약제학적 제제(필름코팅정, 삼투내핵정)와 대조약인 심바스타틴 단일제(Merck: Zocor), 올메사르탄 메독소밀 단일제( Daiichi Sankyo: Benicar, 올메텍)의 용출률을 나타낸 그래프이다.
도 14는 실시예 III-1따라 제조된 심바스타틴/발사르탄 유핵정과 각각의 단일제인 코자의 심바스타틴과 디오반의 발사르탄의 용출률을 나타낸 그래프이다.
도 15는 실시예 III-3에 따라 제조된 심바스타틴/발사르탄 다층정과 각각의 단일제인 코자의 심바스타틴과 디오반의 발사르탄의 용출률을 나타낸 그래프이다.
도 16은 실시예 III-5에따라 제조된 심바스타틴/발사르탄 매트릭스 정제와 각각의 단일제인 코자의 심바스타틴과 디오반의 발사르탄의 용출률을 나타낸 그래프이다.
도 17은 실시예 III-8에 따라 제조된 심바스타틴 발사르탄 정제를 함유한 캡슐제와 각각의 단일제인 코자의 심바스타틴과 디오반의 발사르탄의 용출률을 나타낸 그래프이다.
도 18은 조코(심바스타틴 단일제), 아타칸(칸데사르탄 단일제), 실시예 IV-1의 제제의 용출양상을 나타낸 그래프이다.
도 19는 실시예 IV-3 내지 IV-6의 제제의 용출양상을 나타낸 그래프이다.
도 20은 시판중인 조코(심바스타틴 단일제), 아타칸(칸데사르탄 단일제), 실시예 IV-7의 제제 용출양상을 나타낸 그래프이다.
도 21은 시판중인 조코(심바스타틴 단일제), 아타칸(칸데사르탄 단일제), 실시예 IV-10의 제제의 용출양상을 나타낸 그래프이다.
도 22는 조코(심바스타틴 단일제), 아타칸(칸데사르탄 단일제), 실시예 IV-12 및 IV-14의 제제의 용출양상을 나타낸 그래프이다.
도 23은 실시예 V-2, V-4, 및 V-8에 따라 제조된 로자탄-로슈바스타틴의 복합 제어 방출 제제와 대조약으로서 각각의 단일제인 코자와 크레스토의 로자탄 성분과 로슈바스타틴 성분의 비교 용출의 곡선을 나타낸 그래프이다.
도 24는 실시예 V-6, V-15, 및 V-17에 따라 제조된 로자탄-로슈바스타틴의 복합 제어 방출 제제와 대조약으로서 각각의 단일제인 코자와 크레스토의 로자탄 성분과 로슈바스타틴 성분의 비교 용출의 곡선을 나타낸 그래프이다.
도 25은 실시예 VI-2, VI-4, VI-8, VI-9의 약제학적 제제 및 대조제제인 크레스토정, 아프로벨정에서 로슈바스타틴, 이베사르탄의 용출률을 나타낸 그래프이다.
도 26는 실시예 VI-6, VI-17, VI-19, VI-20의 약제학적 제제 및 대조제제인 크레스토정, 아프로벨정에서 로슈바스타틴, 이베사르탄의 용출률을 나타낸 그래프이다.
도 27은 실시예 VII-2, VII-4, VII-8의 약제학적 제제 및 대조제제인 크레스토정, 아타칸정에서 로슈바스타틴, 칸데사르탄의 용출률을 나타낸 그래프이다.
도 28은 실시예 VII-6, VII-15, VII-17의 약제학적 제제 및 대조제제인 크레스토정, 아타칸정에서 로슈바스타틴, 칸데사르탄의 용출률을 나타낸 그래프이다.
본 발명의 이점 및 특징, 그리고 그것들을 달성하는 방법은 상세하게 후술되어 있는 실시예들을 참조하면 명확해질 것이다. 그러나 본 발명은 이하에서 개시되는 실시예 들에 한정되는 것이 아니라 서로 다른 다양한 형태로 구현될 것이며, 단지 본 실시예들은 본 발명의 개시가 완전하도록 하고, 본 발명이 속하는 기술 분야에서 통상의 지식을 가진 자에게 발명의 범주를 완전하게 알려주기 위해 제공되는 것이며, 본 발명은 청구항의 범주에 의해 정의될 뿐이다.
[실시예 및 실험예 I] 심바스타틴 및 로자탄 함유 약제학적 제제
<실시예 I- 1> 2상 매트릭스 정제의 제조
1) 심바스타틴 선방출성 과립의 제조
표 I-1에 기재된 성분 및 함량과 같이, 심바스타틴과 부형제인 미결정셀룰로오스, 유당, 옥수수전분을 35호체로 체과하고 고속혼합기로 혼합하였다. 따로 히드록시프로필셀룰로오스, 시트르산을 정제수 (1정당 60mg)에 녹여 결합액을 제조하고 이를 주성분 혼합물과 함께 고속혼합기에 투입하고 연합하였다. 연합이 끝나면 18호체로 오실레이터를 이용하여 제립하고 이를 온수 건조기를 이용하여 30 ℃에서 건조하였다. 건조가 끝나면 다시 20호체가 장착된 F형 정립기를 사용하여 정립하여 심바스타틴 선방출성 과립을 제조하였다.
2) 로자탄 칼륨의 지연방출성 과립의 제조
표 I-1에 기재된 성분 및 함량과 같이, 로자탄 칼륨, 전분글리콘산 나트륨, 미결정셀룰로오스를 20호체로 체과하고 더블콘믹서로 15분간 혼합하여 혼합물을 제조하였다. 따로 히드록시프로필셀룰로오스를 정제수 (1정당 140mg)에 녹여 결합액을 제조하였다. 상기 혼합물을 유동층과립기에 넣고 결합액을 가하여 조립하였다. 조립 공정에서 고속혼합기를 선택적으로 사용한다. 유동층 과립기는 GPCG-1(Glatt, Germany)을 사용하여 탑-스프레이 시스템(Top-spray system)을 사용하였다. 과립을 넣은 후, 다음과 같은 조건에서 예열하였다. Air flow는 80 m3/시간, Inlet air 온도는 40℃, 필터 shaking(delta P 필터 < 500pa 로 유지)은 비동시성 모드로 30초에 5초간 진행하였다. 예열 공정에서 제품온도가 35℃에 도달하면 결합액을 분당 1.0 ~ 10 g으로 분사하면서 조립하고 분무된 공기(atomizing air)는 1.0 ~ 2.0 bar 범위에서 조절하며 코팅액 분사각을 조절하였다. 공정이 진행되면서 입자가 생성되기 때문에 Air flow는 80 m3/h에서 120 m3/h로 증가시키고, 손실을 막기 위해 필터 shaking(delta P 필터 <4000 pa로 유지)을 동시성 모드로 1분에 5초간 실시하면서 조립하였다.
조립이 완료된 후 유동층 건조기 조립물을 건조시켰다.
유동층 과립 건조기는 GPCG-1(Glatt, Germany)을 사용하였고 조립물을 넣은 후 다음과 같은 조건에서 진행하였다. Air flow는 120 m3/시간, Inlet air 온도는 65 ℃, 필터 shaking(delta P 필터 < 4000 pa로 유지)은 비동시성 모드로 30초에 5초간 진행하였다. 제품온도가 40 ℃에 이르면 샘플을 채취하여 건조 감량 2.5% 이하 기준에 적합하면 완료하고, 초과시에는 더 진행한 후 재측정하여 건조를 완료하였다. 건조가 완료되면 건조물을 유동층 코팅기에 넣고, 따로 폴리에틸렌글리콜6000, 히프로멜로오스 프탈레이트를 에탄올과 정제수(8:2(v/v))혼합용매(1정당 300mg)에 용해 및 분산시킨 코팅액을 조제하여 위의 과립물을 유동층 과립 코팅기(GPCG-1: Glatt, Germany)하였다.
유동층 과립코팅기는 GPCG-1(Glatt, Germany)을 사용하여 바틈-스프레이 시스템(bottom-spray system)을 이용하였다. 과립의 크기에 따라 조절하여야 하는 plate는 B 또는 C 타입, Partition gap은 25 mm 위치, 분사노즐은 1 mm 크기를 장착하여 사용하였다. 과립을 넣은 후 다음과 같은 예열 조건에서 예열하였다. Air flow는 100 m3/시간, Inlet air 온도는 45 ~ 60 ℃, 제품온도는 40 ~ 50 ℃, 필터 shaking(delta P 필터 < 500 pa로 유지)은 비동시성 모드로 30초에 5초간 진행하였다. 예열 공정에서 제품온도가 35℃에 도달하면 필름 코팅액을 분당 1 ~ 5 g으로 분사하면서 코팅하고 분무된 공기(atomizing air)는 1.0 ~ 1.5 bar 범위에서 조절하며 코팅액 분사각을 조절하였다. 공정이 진행되는 동안에는 제품온도를 34 ~ 38 ℃로 유지시키고, 코팅이 완료되면 제품온도를 40 ℃로 유지하면서 약 1시간 정도 건조 및 표면 작업을 하였다. 코팅 완료하여 로자탄 칼륨 지연방출 과립을 제조하였다.
3) 타정 및 코팅
위의 방법으로 제조된 1), 2)의 두 과립물과 부틸레이트히드록시아니솔을 더블콘믹서에 넣고 혼합하였다. 이 혼합물에 스테아린산 마그네슘을 넣어 최종 혼합하였다. 최종 혼합물을 로타리 타정기(MRC-33: 세종)를 사용하여 타정하였다. 따로 히프로멜로오스 2910, 히드록시프로필셀룰로오스, 산화티탄, 탈크를 정제수 (1정당 430mg)에 용해 및 분산시킨 코팅액을 조제하여 위의 정제를 하이코터(SFC-30N: 세종 기계, 한국)로서 필름 코팅 층을 형성하여 2상 매트릭스 정제를 제조하였다.
<실시예 I- 2> 2상 매트릭스 정제의 제조
1) 심바스타틴 선방출성 과립의 제조
표 I-1에 기재된 성분 및 함량을 실시예 I- 1의 1)의 방법과 동일하게 수행하여 표제의 선방출성과립을 제조하였다.
2) 로자탄 칼륨의 지연방출성 과립의 제조
표 I-1에 기재된 성분 및 함량과 같이 로자탄 칼륨, 전분글리콘산 나트륨, 미결정셀룰로오스를 20호체로 체과하고 더블콘믹서로 15분간 혼합하여 혼합물을 제조하였다. 따로 히드록시프로필 셀룰로오스를 정제수 (1정당 280mg)에 녹여 결합액을 제조하였다. 상기 혼합물을 유동층과립기에 넣고 결합액을 가하여 조립하였다. 조립 공정에서 고속혼합기를 선택적으로 사용한다. 유동층 과립기는 GPCG-1(Glatt, Germany)을 사용하여 탑-스프레이 시스템(Top-spray system)을 사용하였다. 과립을 넣은 후, 다음과 같은 조건에서 예열하였다. Air flow는 80 m3/시간, Inlet air 온도는 40℃, 필터 shaking(delta P 필터 < 500pa 로 유지)은 비동시성 모드로 30초에 5초간 진행하였다. 예열 공정에서 제품온도가 35℃에 도달하면 결합액을 분당 1.0 ~ 10 g으로 분사하면서 조립하고 분무된 공기(atomizing air)는 1.0 ~ 2.0 bar 범위에서 조절하며 코팅액 분사각을 조절하였다. 공정이 진행되면서 입자가 생성되기 때문에 Air flow는 80 m3/h에서 120 m3/h로 증가시키고, 손실을 막기 위해 필터 shaking(delta P 필터 <4000 pa로 유지)을 동시성 모드로 1분에 5초간 실시하면서 조립하였다.
조립이 완료된 후 유동층 건조기 조립물을 건조시켰다.
유동층 과립 건조기는 GPCG-1(Glatt, Germany)을 사용하였고 조립물을 넣은 후 다음과 같은 조건에서 진행하였다. Air flow는 120 m3/시간, Inlet air 온도는 65 ℃, 필터 shaking(delta P 필터 < 4000 pa로 유지)은 비동시성 모드로 30초에 5초간 진행하였다. 제품온도가 40 ℃에 이르면 샘플을 채취하여 건조 감량 2.5% 이하 기준에 적합하면 완료하고, 초과시에는 더 진행한 후 재측정하여 건조를 완료하였다. 건조가 완료되면 건조물을 유동층 코팅기에 넣고, 따로 셀룰로오스 아세테이트 (아세틸기 32%), 셀룰로오스 아세테이트 (아세틸기 39.8%)를 에탄올과 염화메틸렌 혼합용매 (1정당 400mg)에 용해 및 분산 시킨 코팅액을 조제하여 위의 과립물을 유동층 과립 코팅기(GPCG-1: Glatt, Germany)하였다.
유동층 과립코팅기는 GPCG-1(Glatt, Germany)을 사용하여 바틈-스프레이 시스템(bottom-spray system)을 이용하였다. 과립의 크기에 따라 조절하여야 하는 plate는 B 또는 C 타입, Partition gap은 25 mm 위치, 분사노즐은 1 mm 크기를 장착하여 사용하였다. 과립을 넣은 후 다음과 같은 예열 조건에서 예열하였다. Air flow는 100 m3/시간, Inlet air 온도는 45 ~ 60 ℃, 제품온도는 40 ~ 50 ℃, 필터 shaking(delta P 필터 < 500 pa로 유지)은 비동시성 모드로 30초에 5초간 진행하였다. 예열 공정에서 제품온도가 35℃에 도달하면 필름 코팅액을 분당 1 ~ 5 g으로 분사하면서 코팅하고 분무된 공기(atomizing air)는 1.0 ~ 1.5 bar 범위에서 조절하며 코팅액 분사각을 조절하였다. 공정이 진행되는 동안에는 제품온도를 34 ~ 38 ℃로 유지시키고, 코팅이 완료되면 제품온도를 40 ℃로 유지하면서 약 1시간 정도 건조 및 표면 작업을 하였다. 코팅 완료하여 로자탄 칼륨 지연방출 과립을 제조하였다.
3) 타정 및 코팅
표 I-1에 기재된 성분 및 함량을 실시예 I- 1의 3)의 방법과 동일하게 수행하여 표제의 2상메트릭스 정제를 제조하였다.
<실시예 I- 3> 다층정 제조
1) 심바스타틴 선방출성 구획의 제조
표 I-1에 기재된 성분 및 함량과 같이 부형제인 미결정셀룰로오스, 유당, 옥수수전분을 35호체로 체과하고 고속혼합기로 혼합하였다. 따로 히드록시프로필셀룰로오스, 시트르산을 정제수 (1정당 60mg)에 녹여 결합액을 제조하고 이를 주성분 혼합물과 함께 고속혼합기에 투입하고 연합하였다. 연합이 끝나면 18호체로 오실레이터를 이용하여 제립하고 이를 온수 건조기를 이용하여 30 ℃에서 건조하였다. 건조가 끝나면 다시 20호체가 장착된 F형 정립기를 사용하여 정립하였다. 이 정립물을 더블콘믹서에 넣고 부틸레이트 히드록시아니솔을 넣고 혼합 한 뒤 이 혼합물에 스테아린산 마그네슘을 넣고 최종 혼합하여 심바스타틴 선방출성 구획을 제조하였다.
2) 로자탄 칼륨 지연방출성 구획의 제조
표 I-1에 기재된 성분 및 함량과 같이 로자탄 칼륨, 전분글리콘산 나트륨, 미결정셀룰로오스, 유당을 더블콘믹서로 15분간 혼합하였다. 따로 폴리비닐피롤리돈을 정제수 (1정당 40mg)에 녹여 결합액으로 하였다. 상기 혼합물을 유동층 과립기에 넣고 결합액을 가하여 조립하고 건조 하였다. 상기 건조물을 유동층 코팅기에 넣고, 별도로 에틸셀룰로오스, 유드라짓 RL을 에탄올과 염화메틸렌 혼액(1: 1(w/w))에 녹인 액 (1정당 160mg)을 조제하여 상기 조립물을 유동층 코팅으로 코팅한다. 유동층 과립, 건조, 코팅 등의 조건은 실시예 I- 1 지연방출성 과립의 공정과 동일하다. 코팅물에 콜로이드성 이산화규소를 더블콘믹서로 15분간 혼합하였다. 혼합물에 스테아린산 마그네슘을 넣어 더블콘믹서로 최종 혼합하였다.
3) 타정 및 코팅
표 I-1에 기재된 성분 및 함량과 같이 반제품을 1차 분말공급기에 넣고,2) 반제품을 2차 분말 공급기에 넣어 층간의 혼입을 최소화할 수 있는 조건으로 다층정 타정기(MRC-37T: 세종)를 사용하여 타정하였다. 따로 히프로멜로오스 2910, 히드록시프로필셀룰로오스, 산화티탄, 탈크를 정제수에 용해 및 분산시킨 코팅액을 조제하여 위의 정제를 하이코터(SFC-30N: 세종 기계, 한국)로서 필름 코팅 층을 형성하여 다층정제를 제조하였다.
<실시예 I- 4> 다층정 제조
1) 심바스타틴 선방출성 구획의 제조
표 I-1에 기재된 성분 및 함량을 실시예 I-3의 1)과 동일한 방법으로 수행하여, 표제의 심바스타틴 선방출성 구획을 제조하였다.
2) 로자탄 칼륨 지연방출성 구획의 제조
표 I-1에 기재된 성분 및 함량을 실시예 I-3의 2)과 동일한 방법으로 수행하여, 표제의 로자탄칼슘 지연방출성 구획을 제조하였다.
3) 타정 및 코팅
표 I-1에 기재된 성분 및 함량을 실시예 I-3의 3)과 동일한 방법으로 수행하여, 표제의 다층정을 제조하였다.
<실시예 I- 5> 다층정 제조
1) 심바스타틴 선방출성 구획의 제조
표 I-1에 기재된 성분 및 함량을 실시예 I-3의 1)과 동일한 방법으로 수행하여, 표제의 심바스타틴 선방출성 구획을 제조하였다.
2) 로자탄 칼륨 지연방출성 구획의 제조
표 I-1에 기재된 성분 및 함량을 실시예 I-3의 2)과 동일한 방법으로 수행하여, 표제의 로자탄칼슘 지연방출성 구획을 제조하였다.
3) 타정 및 코팅
표 I-1에 기재된 성분 및 함량을 실시예 I-3의 3)과 동일한 방법으로 수행하여, 표제의 다층정을 제조하였다.
<실시예 I- 6> 다층정 제조
1) 심바스타틴 선방출성 구획의 제조
표 I-1에 기재된 성분 및 함량을 실시예 I-3의 1)과 동일한 방법으로 수행하여, 표제의 심바스타틴 선방출성 구획을 제조하였다.
2) 로자탄 칼륨 지연방출성 구획의 제조
표 I-1에 기재된 성분 및 함량을 실시예 I-3의 2)과 동일한 방법으로 수행하여, 표제의 로자탄칼슘 지연방출성 구획을 제조하였다.
3) 타정 및 코팅
표 I-1에 기재된 성분 및 함량을 실시예 I-3의 3)과 동일한 방법으로 수행하여, 표제의 다층정을 제조하였다.
<실시예 I- 7> 유핵정 제조
1) 심바스타틴 선방출성 과립의 제조
표 I-1에 기재된 성분 및 함량과 같이, 심바스타틴과 부형제인 미결정셀룰로오스, 전분글리콘산 나트륨, 유당, 옥수수전분을 35호체로 체과하고 고속혼합기로 혼합하였다. 따로 히드록시프로필셀룰로오스, 시트르산을 정제수 (1정당 100mg)에 녹여 결합액을 제조하고 이를 주성분 혼합물과 함께 고속혼합기에 투입하고 연합하였다. 연합이 끝나면 18호체로 오실레이터를 이용하여 제립하고 이를 온수 건조기를 이용하여 30 ℃에서 건조하였다. 건조가 끝나면 다시 20호체가 장착된 F형 정립기를 사용하여 정립하였다. 이 정립물을 더블콘믹서에 넣고 부틸레이트 히드록시아니솔을 넣고 혼합 한 뒤 이 혼합물에 스테아린산 마그네슘을 넣고 최종 혼합하여 심바스타틴 선방출성 구획을 제조하였다.
2) 로자탄 칼륨 지연방출성 코팅 내핵정의 제조
표 I-1에 기재된 성분 및 함량과 같이, 로자탄 칼륨, 폴리비닐피롤리돈 공중합체, 미결정셀룰로오스, 전호화전분, 콜로이드성 이산화규소를 더블콘믹서로 15분간 혼합하였다. 상기 혼합물과 스테아린산 마그네슘을 더블콘믹서에 넣고 최종혼합 후 로타리 타정기(MRC-33: 세종)를 사용하여 타정하여 이를 내핵정으로 한다. 따로 히프로멜로오스를 에탄올과 정제수(8:2(v/v)) 혼합용매(1정당 20mg)에 녹인 액과 아크릴이즈를 정제수 (1정당 90mg)에 녹인 액을 각각 조제하여 위의 내핵정을 하이코터(SFC-30N: 세종)에 투입한 후 코팅액으로 2차에 걸쳐 코팅하여 내핵정 형태의 지연방출성 코팅 내핵정을 제조하였다.
3) 타정 및 코팅
유핵정 타정기(RUD-1: Kilian)를 사용하여 공정2)의 로자탄 칼륨 코팅 핵정을 내핵정으로 하고 공정1)의 심바스타틴을 포함하는 조성물을 외층으로 하여 유핵정제의 제조를 완료한 후, 따로 히프로멜로오스 2910, 히드록시프로필셀룰로오스, 산화티탄, 탈크를 정제수 (1정당 420mg)에 용해 및 분산시켜 코팅액을 조제하였다. 위의 유핵정을 하이코터(SFC-30N: 세종)에 투입한 후 코팅액으로 코팅하여 필름 코팅 유핵정 제조를 완료하였다.
<실시예 I- 8> 유핵정 제조
1) 심바스타틴 선방출성 구획의 제조
표 I-1에 기재된 성분 및 함량을 실시예 I-7의 1)과 동일한 방법으로 수행하여, 표제의 심바스타틴 선방출성 구획을 제조하였다.
2) 로자탄 칼륨 지연방출성 구획의 제조
표 I-1에 기재된 성분 및 함량을 실시예 I-7의 2)과 동일한 방법으로 타정하여 이를 내핵정으로 한다. 따로 유드라짓 RL를 에탄올과 정제수(8:2(v/v)) 혼합용매(1정당 20mg)에 녹인 액과 유드라짓 RS를 정제수 (1정당 200mg)에 녹인 액을 각각 조제하여 위의 내핵정을 하이코터(SFC-30N: 세종)에 투입한 후 코팅액으로 2차에 걸쳐 코팅하여 내핵정 형태의 지연방출성 코팅 내핵정을 제조하였다.
3) 타정 및 코팅
표 I-1에 기재된 성분 및 함량을 실시예 I-7의 3)과 동일한 방법으로 수행하여, 표제의 유핵정을 제조하였다.
<실시예 I- 9> 유핵정 제조
1) 심바스타틴 선방출성 구획의 제조
표 I-1에 기재된 성분 및 함량을 실시예 I-7의 1)과 동일한 방법으로 수행하여, 표제의 심바스타틴 선방출성 구획을 제조하였다.
2) 로자탄 칼륨 지연방출성 구획의 제조
표 I-1에 기재된 성분 및 함량을 실시예 I-7의 2)과 동일한 방법으로 타정하여 이를 내핵정으로 한다. 따로 카보머를 정제수(1정당 80mg)에 녹인 액과 아크릴이즈를 정제수 (1정당 160mg)에 녹인 액을 각각 조제하여 위의 내핵정을 하이코터(SFC-30N: 세종)에 투입한 후 코팅액으로 2차에 걸쳐 코팅하여 내핵정 형태의 지연방출성 코팅 내핵정을 제조하였다.
3) 타정 및 코팅
표 I-1에 기재된 성분 및 함량을 실시예 I-7의 3)과 동일한 방법으로 수행하여, 표제의 유핵정을 제조하였다.
<실시예 I- 10> 삼투성 유핵정 제조
1) 심바스타틴 선방출성 구획의 제조
표 I-1의 성분 및 함량으로, 실시예 I- 7의 1)의 동일한 방법에 따라 선방출성 구획을 제조하였다.
2) 로자탄 칼륨 지연방출성 삼투성 코팅 내핵정의 제조
표 I-1의 성분 및 함량으로, 로자탄 칼륨, 폴리비닐피롤리돈 공중합체, 미결정셀룰로오스, 전호화전분, 콜로이드성 이산화규소, 염화나트륨을 더블콘믹서로 15분간 혼합하였다. 상기 혼합물과 스테아린산 마그네슘을 더블콘믹서에 넣고 최종혼합 후 로타리 타정기(MRC-33: 세종)를 사용하여 타정하여 이를 내핵정으로 한다. 삼투성 코팅기제로서 에틸셀룰로오스를 에탄올과 염화메틸렌 (1:1(w/w)) 혼합용매 (1정당 500mg)에 분산시킨 후 하이코터(SFC-30N, 세종 기계, 한국)를 이용하여 내핵 나정을 코팅하여 삼투성 코팅 내핵정을 제조하였다.
3) 타정 및 코팅
공정2)의 로자탄 칼륨 삼투성 코팅 내핵정을 내핵으로 하고 공정1)의 심바스타틴을 포함하는 조성물을 외층으로 하여 실시예 I- 7의 3)의 방법으로 표제의 삼투성 필름 코팅 유핵정을 제조하였다.
<실시예 I- 11> 캡슐제 제조 (과립-펠렛)
1) 심바스타틴 선방출성 과립의 제조
표 I-2의 성분 및 함량으로, 실시예 I- 1의 1)의 방법에 따라 과립을 제조 한 후 상기 정립물에 부틸레이트 히드록시아니솔을 더블콘믹서에 넣고 최종혼합 하여 심바스타틴 선방출성 과립을 제조하였다.
2) 로자탄 칼륨 지연방출성 펠렛의 제조
표 I-2의 성분 및 함량으로 슈가 시드를 35호체로 체과하고 유동층 과립기(GPCG 1: Glatt)에 투입한 뒤, 따로 에탄올와 정제수 (8:2(v/v)) 혼합용매 (1정당 550mg)에 히프로멜로오스와 로자탄 칼륨을 용해시킨 결합액을 분무하여 로자탄 칼륨 함유 펠렛을 형성, 건조하였다. 다시 상기의 과립에 히프로멜로오스 프탈레이트를 에탄올과 정제수 (8:2)(v/v) 혼합용매 (1정당 300mg)에 녹인 액을 분무하여 로자탄 칼륨 지연성 펠렛을 제조하였다.
3) 혼합 및 캡슐 충전
표 I-2의 성분 및 함량으로 공정 1)과 2)의 최종 조성물을 더블콘믹서로 혼합하였다. 혼합물에 스테아린산 마그네슘을 투입하고 더블콘믹서로 최종 혼합하였다. 최종 혼합된 혼합물을 분말 공급기에 투입하고 캡슐충전기를 이용하여 충전하여 캡슐형태의 제어 방출 제제의 제조를 완료하였다.
<실시예 I- 12> 캡슐제 제조 (팰렛-펠렛)
1) 심바스타틴 선방출성 팰렛의 제조
표 I-2의 성분 및 함량으로시드를 35호체로 체과하고 유동층 과립기(GPCG 1: Glatt)에 투입한 뒤, 따로 에탄올 (1정당 100mg)에 시트르산을 용해시킨 후 이 용액에 히드록시프로필셀룰로오스과 심바스타틴을 용해시킨 결합액을 분무, 건조하여 심바스타틴 함유 선방출성 펠렛을 제조하였다.
2) 로자탄 칼륨 지연방출성 펠렛의 제조
표 I-2의 성분 및 함량으로시드를 35호체로 체과하고 유동층 과립기(GPCG 1: Glatt)에 투입한 뒤, 따로 에탄올과 정제수 (8:2(v/v)) 혼합용매 (1정당 1400mg)에 히프로멜로오스와 로자탄 칼륨을 용해시킨 결합액을 분무하여 로자탄 칼륨 함유 펠렛을 형성, 건조하였다. 다시 상기의 과립에 아크릴이즈를 에탄올과 정제수 (8:2(v/v)) 혼합용매 (1정당 300mg)에 녹인 액을 분무하여 로자탄 칼륨 지연성 펠렛을 제조하였다.
3) 혼합 및 캡슐 충전
표 I-2의 성분 및 함량으로 공정 1)과 2)의 최종 조성물을 더블콘믹서로 혼합하였다. 혼합물에 부틸레이트 히드록시아니솔, 스테아린산 마그네슘을 투입하고 더블콘믹서로 최종 혼합하였다. 최종 혼합된 혼합물을 분말 공급기에 투입하고 캡슐충전기를 이용하여 충전하여 캡슐형태의 제어 방출 제제의 제조를 완료하였다.
<실시예 I- 13> 캡슐제 제조 (정제-펠렛)
1) 심바스타틴 선방출성 정제의 제조
표 I-2의 성분 및 함량으로, 실시예 I- 1의 1)의 방법에 따라 제조한 심바스타틴 선방출성 과립과 스테아린산 마그네슘을 더블콘믹서에 넣고 최종 혼합한 후 로타리 타정기 (MRC-33:세종기계, 한국)를 사용하여 타정하여 선방출성 정제를 제조하였다.
2) 로자탄 칼륨 지연방출성 펠렛의 제조
표 I-2의 성분 및 함량으로 슈가 시드를 35호체로 체과하고 유동층 과립기(GPCG 1: Glatt)에 투입한 뒤, 따로 에탄올와 정제수 (8:2(v/v)) 혼합용매 (1정당 550mg)에 히프로멜로오스와 로자탄 칼륨을 용해시킨 결합액을 분무하여 로자탄 칼륨 함유 펠렛을 형성, 건조하였다. 다시 상기의 과립에 카르나우바왁스를 에탄올과 정제수 (8:2(v/v)) 혼합용매 (1정당 420mg)에 녹인 액을 분무하여 로자탄 칼륨 지연성 펠렛을 제조하였다.
3) 혼합 및 캡슐 충전
실시예 I- 11의 3)의 방법에 따라 제조하였다.
<실시예 I- 14> 캡슐제 제조 (과립-과립)
1) 심바스타틴 선방출성 과립의 제조
표 I-2의 성분 및 함량으로 실시예 I- 11의 1)의 방법에 따라 제조하였다.
2) 로자탄 칼륨 지연방출성 과립의 제조
표 I-2의 성분 및 함량으로, 실시예 I- 2의 2)의 방법에 따라 건조물을 제조하였다. 건조물을 유동층 코팅기에 넣고, 따로 셀룰로오스 아세테이트 (아세틸기 32%), 셀룰로오스 아세테이트 (아세틸기 39.8%), 히프로멜로오스를 에탄올과 염화메틸렌 (8:2(v/v)) 혼합용매 (1정당 900mg)에 용해 및 분산 시킨 코팅액을 조제하여 실시예 I- 2의 2)의 방법에 따라 코팅 완료하여 로자탄 칼륨 지연방출성 과립을 제조하였다.
3) 혼합 및 캡슐 충전
실시예 I- 11의 3)의 방법에 따라 제조하였다.
<실시예 I- 15> 캡슐제 제조 (팰렛-과립)
1) 심바스타틴 선방출성 팰렛의 제조
표 I-2의 성분 및 함량으로 실시예 I- 12의 1)의 방법에 따라 제조하였다.
2) 로자탄 칼륨 지연방출성 과립의 제조
표 I-2의 성분 및 함량으로, 에틸셀룰로오스, 유드라짓 RL, 히프로멜로오스 프탈레이트를 에탄올과 염화메틸렌(1:1 (w/w)) 혼합용매 (1정당 400mg)에 용해 및 분산시킨 코팅액을 조제하여 실시예 I- 1의 2)의 방법에 따라 로자탄 칼륨 지연방출성 과립을 제조하였다.
3) 혼합 및 캡슐 충전
실시예 I- 12의 3)의 방법에 따라 제조하였다.
<실시예 I- 16> 캡슐제 제조 (정제-과립)
1) 심바스타틴 선방출성 정제의 제조
표 I-2의 성분 및 함량으로 실시예 I- 13의 1)의 방법에 따라 제조하였다.
2) 로자탄 칼륨 지연방출성 과립의 제조
표 I-2의 성분 및 함량으로, 카보머, 히프로멜로오스 프탈레이트를 에탄올과 염화메틸렌 (1:1(w/w))혼합용매 (1정당 800mg)에 용해 및 분산시킨 코팅액을 조제하여 실시예 I- 1의 2)의 방법에 따라 로자탄 칼륨 지연방출성 과립을 제조하였다.
3) 혼합 및 캡슐 충전
실시예 I- 11의 3)의 방법에 따라 제조하였다.
<실시예 I- 17> 캡슐제 제조 (과립-정제)
1) 심바스타틴 선방출성 과립의 제조
표 I-2의 성분 및 함량으로 실시예 I- 14의 1)의 방법에 따라 제조하였다.
2) 로자탄 칼륨 지연방출성 정제의 제조
표 I-2의 성분 및 함량으로, 실시예 I- 7의1)의 방법에 따라 로자탄 칼륨 나정을 타정한 후 따로 유드라짓 RS를 에탄올과 정제수(8:2(v/v)) 혼합용매(1정당 50mg)에 녹인 액과 아크릴이즈를 정제수 (1정당 100mg)에 녹인 액을 각각 조제하여 위의 나정을 하이코터(SFC-30N: 세종)에 투입한 후 코팅액으로 2차에 걸쳐 코팅하여 로자탄 칼륨 지연방출성 정제를 제조하였다.
3) 혼합 및 캡슐 충전
실시예 I- 11의 3)의 방법에 따라 제조하였다.
<실시예 I- 18> 캡슐제 제조 (팰렛-정제)
1) 심바스타틴 선방출성 팰렛의 제조
표 I-2의 성분 및 함량으로 실시예 I- 12의 1)의 방법에 따라 제조 하였다.
2) 로자탄 칼륨 지연방출성 정제의 제조
표 I-2의 성분 및 함량으로, 실시예 I- 7의 1)의 방법에 따라 로자탄 칼륨 나정을 타정한 후 따로 카보머를 에탄올과 정제수(8:2) 혼합용매(1정당 50mg)에 녹인 액과 히프로멜로오스 프탈레이트를 에탄올과 정제수(8:2(v/v)) 혼합용매 (1정당 100mg)에 녹인 액을 각각 조제하여 위의 나정을 하이코터(SFC-30N: 세종)에 투입한 후 코팅액으로 2차에 걸쳐 코팅하여 로자탄 칼륨 지연방출성 정제를 제조하였다.
3) 혼합 및 캡슐 충전
실시예 I- 12의 3)의 방법에 따라 제조하였다.
<실시예 I- 19> 캡슐제 제조 (정제-정제)
1) 심바스타틴 선방출성 정제의 제조
표 I-2의 성분 및 함량으로 실시예 I- 13의 1)의 방법에 따라 제조하였다.
2) 로자탄 칼륨 지연방출성 정제의 제조
표 I-2의 성분 및 함량으로, 실시예 I- 7의 1)의 방법에 따라 로자탄 칼륨 나정을 타정한 후 따로 에틸셀룰로오스를 에탄올과 염화메틸렌(1:1(w/w)) 혼합용매 (1정당 50mg)에 녹인 액과 아크릴이즈를 정제수 (1정당 200mg)에 녹인 액을 각각 조제하여 위의 나정을 하이코터(SFC-30N: 세종)에 투입한 후 코팅액으로 2차에 걸쳐 코팅하여 로자탄 칼륨 지연방출성 정제를 제조하였다.
3) 캡슐 충전
공정 1)과 2)의 최종 조성물을 더블콘믹서로 혼합하였다. 혼합물을 분말 공급기에 투입하고 캡슐충전기를 이용하여 충전하여 캡슐형태의 제어 방출 제제의 제조를 완료하였다.
<실시예 I- 20> 로자탄 칼륨-심바스타틴 블러스터 포장 키트
실시예 I- 3의 1) 심바스타틴 선방출 과립과 2) 로자탄 칼륨 지연방출 과립 최종 조성물을 혼합하여 타정하는 대신에 각각 로타리 타정기(MRC-33: 세종)를 사용하여 타정하고 블리스터 포장용기에 각각의 정제가 포함되어 동시복용 가능하도록 포장하였다.
[규칙 제26조에 의한 보정 18.05.2009]
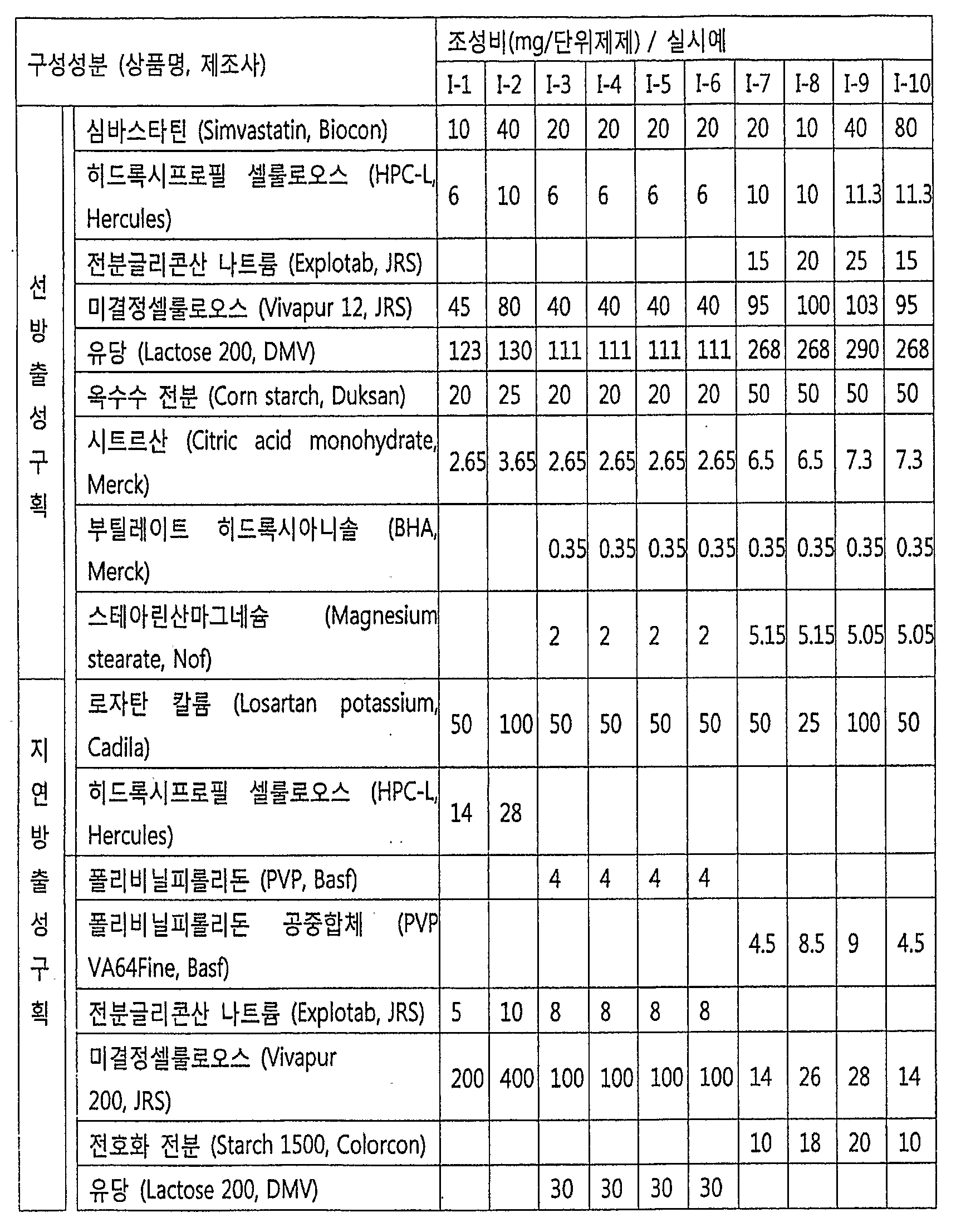
[규칙 제26조에 의한 보정 18.05.2009]
[규칙 제26조에 의한 보정 18.05.2009]

[규칙 제26조에 의한 보정 18.05.2009]

<실험예 I- 1> 비교 용출시험(comparative dissolution profile test)
상기 실시예 I- 1에 따라 제조된 심바스타틴/로자탄 2상 매트릭스 정제와 각 성분 단일제 대조약(조코정, 한국 MSD: 심바스타틴 단일제 / 코자정, 한국 MSD: 로자탄 단일제)의 비교 용출시험을 실시하였다. 심바스타틴 성분 용출시험의 경우 미국약전 (USP31)에 근거하여 용출시험을 진행하였고, 로자탄 성분 용출 시험의 경우 120분을 기점으로 용출액을 인공 위액에서 인공 장액으로 변경하여 총 480분간 용출시험을 진행하였다. 각 성분의 용출시험 방법은 아래와 같으며, 그 결과를 도 1에 나타내었다.
[심바스타틴 선방출 구획 시험방법]
용출시험 근거: 미국약전(USP30)중의 '심바스타틴 정제'항
시험 방법: 패들법(Paddle method), 50회전/분
시험약: pH=7.0 완충액 (조성 = 계면활성제로서 라우릴설페이트 나트륨 0.5% 중량/중량을 함유하는 0.01M 인산이수소나트륨 용액), 900mL
분석 방법: 자외가시부흡광광도법 (검출 파장 = 최대 247, 최소 257nm)
[로자탄 지연방출 구획 시험방법]
용출시험 근거: 대한약전 제 8개정 중 일반시험법의 용출시험법
시험 방법: 패들법, 50회전/분
시험약: 0.01M 염산용액, 750mL (인공위액)
pH 6.8 인산완충액, 총 1000mL (인공장액)
분석 방법: 자외가시부흡광광도법
도 1에 의하면 상기 용출 시험 시 본 발명의 2상 매트릭스 정제 심바스타틴 성분은 대조 제제인 조코정과 비교하여 거의 동등한 용출특성을 나타내는 것으로 확인되었으나, 로자탄 성분은 대조 제제인 코자와 비교할 때 매우 지연된 용출 속도를 확인 할 수 있다. 로자탄 성분의 용출 시험 결과를 보면, 인공 위액 구간인 120분까지의 로자탄 성분의 용출률은 본 발명의 심바스타틴/로자탄 2상 매트릭스 정제에서 10% 이내이나, 대조 제제는 약 60%임을 확인할 수 있었고, 이후 인공 장액 구간에서 로자탄 성분의 용출률은 대조제제에서 총 150분에 100%이나, 본 발명의 심바스타틴/로자탄 2상 매트릭스 정제에서는 총 240분에 약 20%로 훨씬 느림을 확인할 수 있었다.
따라서 본 발명의 심바스타틴/로자탄의 매트릭스 정제는 로자탄의 초기 방출을 의도한 시간만큼 지연 방출시킬 수 있다. 따라서 로자탄의 초기 방출이 심바스타틴보다 매우 느리기 때문에 심바스타틴이 먼저 간에서 대사를 받은 후 대사 관련 효소인 사이토크롬 P450이 재생될 시간을 충분히 확보할 수 있는 약제학적 제제이다.
<실험예 I- 2> 비교 용출시험(comparative dissolution profile test)
상기 실시예 I- 3 내지 I-6에서 제조된 약제학적 제제 내의 로자탄의 비교 용출시험을 실시하였다. 로자탄의 용출시험 방법은 실험예 I- 1과 같으며, 그 결과를 도 2에 나타내었다.
도 2에 의하면 본 발명의 다층정에서 로자탄은 에틸셀룰로오스의 사용량이 증가함에 따라 더욱 지연된 용출 속도를 나타내었으며, 구체적으로 총 240분까지 20% 이내의 로자탄 용출률을 나타내었다.
따라서 본 발명의 약제학적 제제는 로자탄의 초기 방출이 심바스타틴보다 매우 느리기 때문에 심바스타틴이 먼저 간에서 대사를 받은 후 대사 관련 효소인 사이토크롬 P450이 재생될 시간을 충분히 확보할 수 있는 약제학적 제제이다.
<실험예 I- 3> 비교 용출시험(comparative dissolution profile test)
상기 실시예 I- 7에 따라 제조된 심바스타틴/로자탄 유핵정과 각 성분 단일제 대조약(조코정, 한국 MSD: 심바스타틴 단일제 / 코자정, 한국 MSD: 로자탄 단일제)의 비교 용출시험을 실시하였다. 각 성분별 용출시험 방법은 실험예 I- 1과 같으며, 그 결과를 도 3에 나타내었다.
도 3에 의하면 상기 용출 시험 시 본 발명의 유핵정 중 심바스타틴 성분은 대조 제제인 조코정과 비교하여 거의 동등한 용출특성을 나타내는 것으로 확인되었으나, 로자탄 성분은 대조 제제인 코자와 비교할 때 실험예 I- 1과 같이 매우 지연된 용출 속도를 확인할 수 있다.
따라서, 본 발명의 심바스타틴/로자탄 유핵정은 대조약인 로자탄 단일제와 심바스타틴 단일제를 동시 복용하였을 경우의 용출 양상과는 달리 로자탄의 초기 방출이 심바스타틴보다 매우 느리기 때문에 심바스타틴이 먼저 간에서 대사를 받은 후 대사 관련 효소인 사이토크롬 P450이 재생될 시간을 충분히 확보할 수 있는 약제학적 제제이다.
<실험예 I- 4> 비교 용출시험(comparative dissolution profile test)
상기 실시예 I- 10에 따라 제조된 심바스타틴/로자탄 삼투성 유핵정과 각 성분 단일제 대조약(조코정, 한국 MSD: 심바스타틴 단일제 / 코자정, 한국 MSD: 로자탄 단일제)의 비교 용출시험을 실시하였다. 각 성분별 용출시험 방법은 실험예 I- 1과 같으며, 그 결과를 도 4에 나타내었다.
도 4에 의하면 실험예 I- 1의 조건에서 용출 시험 시 본 발명의 삼투성 유핵정 중 심바스타틴 성분은 대조 제제인 조코정과 비교하여 거의 동등한 용출특성을 나타내는 것으로 확인되었으나, 로자탄 성분은 대조 제제인 코자정과 비교할 때 매우 지연된 용출 속도를 확인할 수 있다.
따라서 본 발명의 심바스타틴/로자탄 삼투성 유핵정은 대조약인 로자탄 단일제와 심바스타틴 단일제를 동시 복용하였을 경우의 용출 양상과는 달리 로자탄의 초기 방출이 심바스타틴보다 매우 느리기 때문에 심바스타틴이 먼저 간에서 대사를 받은 후 대사 관련 효소인 사이토크롬 P450이 재생될 시간을 충분히 확보할 수 있는 약제학적 제제이다.
<실험예 I- 5> 비교 용출시험(comparative dissolution profile test)
상기 실시예 I- 12 또는 I-14에 따라 제조된 심바스타틴/로자탄 캡슐제(펠렛-펠렛) 또는 캡슐제(과립-과립)이며 이와 각 성분 단일제 대조약(조코정, 한국 MSD: 심바스타틴 단일제 / 코자정, 한국 MSD: 로자탄 단일제)의 비교 용출시험을 실시하였다. 각 성분별 용출시험 방법은 실험예 I- 1과 같으며, 그 결과를 도 5에 나타내었다.
도 5에 의하면 실험예 I- 1의 조건에서 용출 시험시 본 발명의 캡슐제(펠렛-펠렛),(과립-과립)중 심바스타틴 성분은 대조 제제인 조코정과 비교하여 거의 동등한 용출특성을 나타내는 것으로 확인되었으나, 로자탄 성분은 대조 제제인 코자와 비교할 때 매우 지연된 용출 속도를 확인 할 수 있다.
따라서 본 발명의 심바스타틴/로자탄 캡슐제(펠렛-펠렛),(과립-과립)는 대조약인 로자탄 단일제와 심바스타틴 단일제를 동시 복용하였을 경우의 용출 양상과는 달리 로자탄의 초기 방출이 심바스타틴보다 매우 느리기 때문에 심바스타틴이 먼저 간에서 대사를 받은 후 대사 관련 효소인 사이토크롬 P450이 재생될 시간을 충분히 확보할 수 있는 약제학적 제제이다.
<실험예 I-6> 동물 시험(Animal Study)
본 실험은 시판중인 각 성분 대조약(조코정, 한국 MSD: 심바스타틴 단일제 / 코자정, 한국 MSD: 로자탄 단일제)의 방출시간과 유사하도록 심바스타틴과 로자탄의 동시 투여군과 실험군으로서 본 발명의 실시예 I에 의하여 제공되는 조성물의 방출 시간과 동일하도록 심바스타틴과 로자탄을 시간차 투여함으로써 본 발명에 의한 조성물로서의 효과를 확인하기 위해, 동물시험을 표 I-3와 같이 실시하였다.
[규칙 제26조에 의한 보정 18.05.2009]

본 비교 동물 임상 결과 밝혀진 동물 임상결과의 약동학/약력학은 표 I-4 및 도 6 내지 8과 같다.
[규칙 제26조에 의한 보정 18.05.2009]

<본 시험은 쥐를 모델로 한 동물시험으로 명조건과 암조건으로 나누어 설계하였으며, 쥐와 사람의 생체리듬은 정반대이므로 사람에게 적용시 시간대를 반대로 적용함.>
상기 표I-4에 나타난바와 같이, 저녁 시간차 투여군(명조건)이 4그룹 중에서 가장 혈압강하 효과가 뛰어났다.
따라서, 본 발명에 의한 약제학적 제제가 통상적인 동시투여군과는 달리, 평균혈압이 최고조에 이르는 투여 익일 아침부터 한낮에 이르는 시간 동안의 최적의 혈압강하효과를 가짐을 알 수 있다.
<실험예 I- 7> 임상시험(Preliminary Clinical Test)
본 실험은 시판중인 `조코정'(심바스타틴 20mg; MSD) 단독 투여군과 '조코정' 및 '코자정'(로자탄 칼륨 50mg; MSD) 동시투여군 및 실험군으로서 본 발명의 실시예 I- 7에 의하여 제공되는 조성물을 투여하여 표 I-5과 같이 임상시험을 실시하였다.

본 실험은 발명의 효과를 뒷받침하는 실험으로서 시판을 목적으로 의약품의 허가를 위해 진행되는 국내외 임상시험기준과 실험군수를 줄여 실시되었으나, 시험기간 중 피험자 관리 및 기타 임상시험으로써의 기준은 엄수하여 실시되었다.
본 비교 임상 결과 밝혀진 투약 42일째(공복시) 측정한 지질은 표 I-6에 나타냈다.
[표I-6]

본 비교 임상 결과 밝혀진 투약 42일째 측정한 혈압, 맥박 및 맥압을 표 I-7에 나타냈다.
[표I-7]

본 비교 임상 결과 밝혀진 투약 42일째 측정한 바이오마커는 표 I-8에 나타냈다.
[표I-8]

이상과 같이 본 발명의 실시예I-7의 심바스타틴과 로자탄의 약제학적 제제와 각 성분 단일제 동시 투여군의 비교 임상 결과, 본 발명의 약제학적 제제가 혈압 강하, 지질 감소, 부작용 관련 바이오마커(Biomakers) 모든 면에서 각 성분의 단일제 동시 투여군에 비해 우수하였으며, 시험에 참여한 모든 임상군 중 8명에게서 심바스타틴과 로자탄을 각각 투여했을 경우 일반적으로 나타나는 경증의 부작용 이외 중대한 부작용은 없었다.
따라서, 동일한 투여용량에서 본 발명의 약제학적 제제는 심바스타틴과 로자탄 단일 제제의 동시 투여보다 상승된 심바스타틴의 항고지혈증 효과 및 상승된 로자탄의 혈압강하 효과를 나타낸다.
실시예 및 실험예 II] 심바스타틴 및 올메사르탄 함유 약제학적 제제
<실시예 II- 1> 심바스타틴 - 올메사르탄 메독소밀매트릭스 정제 제조
(1) 심바스타틴 선방출성 혼합물 제조
표 II-1에 나타난 성분 및 함량으로 심바스타틴(Simvastatin, Ranbaxy Laboratories LTD, 인도), 미결정셀룰로오스(AvicelPH, FMC Biopolymer, 미국), 만니톨(Pearlitol, Roquette America INC, 미국)을 달아 35 호체로 사과하고 유동층 과립기(GPCG-1,Glatt, 독일)에 넣고 5분간 혼합하여 혼합물을 제조한 후, 별도로 히드록시프로필셀룰로오스(HPC-L, Nippon soda, 일본)와 구연산(Citric acid, Merck, 독일)을 정제수에 녹여 결합액을 제조하였다. 상기 혼합물을 유동층과립기에 넣고 결합액을 가하여 조립하였다. 조립 공정에서 고속혼합기(Lab. Pharma Mixer P, 디오스나, 독일) 를 선택적으로 사용할 수 있다. 유동층 과립기를 사용하여 바틈-스프레이 시스템(bottom-spray system)을 사용하였다. 과립을 넣은 후, 다음과 같은 조건에서 예열하였다. Air flow는 80 m3/시간, Inlet air 온도는 40℃, 필터 shaking(delta P 필터 < 500pa 로 유지)은 비동시성 모드로 30초에 5초간 진행하였다. 예열 공정에서 제품온도가 35℃에 도달하면 결합액을 분당 1.0 ~ 10 g으로 분사하면서 조립하고 분무된 공기(atomizing air)는 1.0 ~ 2.0 bar 범위에서 조절하며 코팅액 분사각을 조절하였다. 공정이 진행되면서 입자가 생성되기 때문에 Air flow는 80 m3/h에서 120 m3/h로 증가시키고, 손실을 막기 위해 필터 shaking(delta P 필터 <4000 pa로 유지)을 동시성 모드로 1분에 5초간 실시하면서 조립하였다.
조립이 완료된 후 유동층 건조기(GPCG-1, Glatt, 독일) 조립물을 넣은 후 다음과 같은 조건에서 진행하였다. Air flow는 120 m3/시간, Inlet air 온도는 65 ℃, 필터 shaking(delta P 필터 < 4000 pa로 유지)은 비동시성 모드로 30초에 5초간 진행하였다. 제품온도가 40 ℃에 이르면 샘플을 채취하여 건조 감량 2.5% 이하 기준에 적합하면 완료하고, 초과시에는 더 진행한 후 재측정하여 건조를 완료하였다.
건조가 완료되면 건조물을 20호체가 장착된 F형 정립기(KYK-60, 코리아메디, 한국)를 사용하여 정립하여 심바스타틴 선방출성 과립을 제조하였다.
완성된 과립은 더블콘믹서(다산파마텍, 한국)에 넣고, 부틸레이트히드록시아니솔( Butylhydroxyanisole, Merck, 독일), 전분글리콘산나트륨(sodium starch glycolate, JRS, 독일콜로이드성이산화규소(Aerosil 200VV, Degussa, 독일 )를 넣고 혼합한 후, 스테아린산 마그네슘( Magnesium Stearate, 화원약품, 한국)을 투입 후 4분간 혼합하여 심바스타틴 선방출성 최종혼합물을 제조하였다.
(2) 올메사르탄 메독소밀 지연방출성 혼합물 제조
표 II- 1에 나타난 성분 및 함량으로 올메사르탄 메독소밀(Olmesartan Medoxomil, MSN Laboratiories LTD, 인도), 유당(Parmatose, DMV Pharma, Netherlands), 크로스카멜로스나트륨(Vivasol, JRS PHARMA, 독일), 가교 폴리비닐피롤리돈(Crospovidone, BASF, 독일), 저치환-히드록시프로필셀룰로스(L-HPC, Shin-etsu, 일본)을 35호체로 사과하고 유동층 과립기(GPCG-1,Glatt, 독일)에 넣고 5분간 혼합하여 혼합물을 제조한 후, 별도로 히드록시프로필셀룰로오스(HPC-L, Nippon soda, 일본, 표 II- 1에 없음)를 정제수에 녹여 결합액으로 하였다. 유동층 과립기 및 유동층 건조 등의 조건은 심바스타틴 속방층 과립의 공정과 동일하다.별도로 셀룰로오스아세테이트320S(아세탈기 32%)(Eastman Chemical Company, 미국), 셀룰로오스아세테이트398NF10(아세탈기 39.8%)(Eastman Chemical Company, 미국), 히프로멜로오스를 에탄올과 염화메틸렌 혼액(1:1(w/w))에 녹인 액을 조제하여 상기 유동층 과립 코팅기(GPCG-1,Glatt, 독일) 안의 조립물을다음과 같은 조건으로 코팅하였다.
스프레이 방식은 탑-스프레이 시스템(Top-spray system)을 이용하였다. 과립의 크기에 따라 조절하여야 하는 plate는 B 또는 C 타입, Partition gap은 25 mm 위치, 분사노즐은 1 mm 크기를 장착하여 사용하였다. 과립을 넣은 후 다음과 같은 예열 조건에서 예열하였다. Air flow는 100 m3/시간, Inlet air 온도는 45 ~ 60 ℃, 제품온도는 40 ~ 50 ℃, 필터 shaking(delta P 필터 < 500 pa로 유지)은 비동시성 모드로 30초에 5초간 진행하였다. 예열 공정에서 제품온도가 35℃에 도달하면 필름 코팅액을 분당 1 ~ 5 g으로 분사하면서 코팅하고 분무된 공기(atomizing air)는 1.0 ~ 1.5 bar 범위에서 조절하며 코팅액 분사각을 조절하였다. 공정이 진행되는 동안에는 제품온도를 34 ~ 38 ℃로 유지시키고, 코팅이 완료되면 제품온도를 40 ℃로 유지하면서 약 1시간 정도 건조 및 표면 작업을 하였다. 코팅 완료 후 제조된 올메사르탄 메독소밀 지연방출성 과립에 스테아린산 마그네슘을 투입 후 4분간 혼합하여 올메사르탄 메독소밀 지연방출성 최종혼합물을 제조하였다.
(3) 타정 및 코팅
상기 (1), (2)의 두 과립물을 혼합한 후 로타리 타정기(MRC-30: 세종파마텍, 한국)에서 타정하였다. 타정이 완료된 정제를 히프로멜로오스 2910(Pharmacoat, Shin-etsu, 일본), 폴리에틸렌글리콜 6,000(Lutrol 6000, BASF, 독일)을 에탄올과 정제수 혼액(8:2(v/v))에 용해 후 산화티탄(Tioside Americas, 미국)을 분산 시켜 제조한 코팅액을 사용하여 코팅(SFC-30F, 세종파마텍, 한국)하였다.
<실시예 II- 2> 심바스타틴 - 올메사르탄 메독소밀 이중정제 제조
표 II- 1에 나타난 성분과 함량으로 상기 실시예 II- 1의 (1) 제조 방법에 따라 심바스타틴 선방출성 최종혼합물을, 실시예 II- 1의 (2) 제조 방법에 따라 올메사르탄 메독소밀최종혼합물을 각각 제조 하였다.
완성된 올메사르탄 메독소밀 지연방출성 과립과 심바스타틴 선방출성 과립을 로타리 다중정 타정기[MRC-37T, 세종파마텍, 한국]의 다른 과립 주입구에 각각 넣고 타정한 후, 타정이 완료된 정제를 히프로멜로오스 2910, 폴리에틸렌글리콜 6,000을 에탄올과 정제수혼액(8:2(v/v))에 용해 후 산화티탄을 분산 시켜 제조한 코팅액을 사용하여 코팅하였다.
<실시예 II- 3> 심바스타틴 - 올메사르탄 메독소밀제조
표 II- 1에 나타난 성분과 함량으로 상기 실시예 II- 2의 (2) 방법에 따라 제조하여 올메사르탄 메독소밀 지연방출성 최종혼합물를 제조하였다.
완성된 올메사르탄 메독소밀 지연방출 최종혼합물 중간층(2번째층)으로 쌓고, 실시예 II- 1의 (1)의 방법에 따라 제조된 심바스타틴 속방출 최종혼합물을 1층 및 3층으로 분할하여 로타리 삼중정 타정기(MRC-37T: 세종파마텍, 한국)의 다른 과립 주입구에 각각 넣고 타정한 후, 타정이 완료된 정제를 히프로멜로오스 2910, 폴리에틸렌글리콜 6,000을 에탄올 및 정제수 혼액(8:2(v/v))에 용해 후 산화티탄을 분산시켜 제조한 코팅액을 사용하여 코팅한 정제를 제조하였다.
<실시예 II- 4> 심바스타틴 - 올메사르탄 메독소밀 유핵정제 제조
(1) 심바스타틴 선방출성 혼합물 제조
II- 1의 (1) 방법으로 조립 공정은 고속혼합기를 사용하여, 심바스타틴 선방출성 최종혼합물을 제조하였다.
(2) 올메사르탄 메독소밀 지연방출성 내핵정 제조
표 II- 1에 나타난 성분 및 함량으로 올메사르탄 메독소밀, 유당, 크로스카멜로스나트륨, 가교 폴리비닐피롤리돈, 저치환-히드록시프로필셀룰로스을35호체로 사과하고 고속혼합기(Lab. Pharma Mixer P, 디오스나, 독일)에 넣고 5분간 고속으로 혼합한 뒤, 별도로 히드록시프로필셀룰로오스를 정제수에 녹여 제조한 결합액을 가하여 조립하였다. 완료된 조립물을 유동층 건조기에서 건조한 후, 20호체가 장착된 F형 정립기(KYK-60, 코리아메디, 한국)를 사용하여 올메사르탄 지연방출성 과립을 제조하였다.
완료된 과립에 스테아린산 마그네슘을 투입 후 4분간 혼합하여 올메사르탄 지연방출성 최종혼합물을 제조하고, 로타리 타정기(MRC-30, 세종파마텍, 한국)로 타정을 하여 내핵을 제조하였다.
(3) 내핵정 코팅
내핵을 코팅기(SFC-30F, 세종파마텍, 한국)에 투여하고, 별도로 셀룰로오스아세테이트 320S(아세탈기 32%), 셀룰로오스아세테이트 398NF10(아세탈기 39.8%), 히프로멜로오스를 에탄올과 염화메틸렌에 녹인 액을 코팅액으로 코팅하여 코팅정을 제조하였다.
(4) 유핵정 제조
유핵정타정기(RUD-1, Killian, 독일)에서 상기의 심바스타틴 선방출성 최종혼합물과 올메사르탄 메독소밀 지연방출성 내핵을 함께 타정한 후, 타정이 완료된 정제를 히프로멜로오스 2910, 폴리에틸렌글리콜 6,000을 에탄올 및 정제수 혼액(8:2(v/v))에 용해 후 산화티탄을 분산시켜 제조한 코팅액을 사용하여 코팅한 정제를 제조하였다.
<실시예 II- 5> 심바스타틴 - 올메사르탄 메독소밀 캡슐제(정제 + 정제)의 제조
(1) 심바스타틴 선방출성 정제 제조
표 II- 1에 나타난 성분 및 함량으로 심바스타틴, 미결정셀룰로오스, 만니톨을 달아 35 호체로 사과하고 고속혼합기(Lab. Pharma Mixer P, 디오스나, 독일)에 넣고 5분간 고속으로 혼합한 뒤, 별도로 히드록시프로필셀룰로오스와 구연산을 정제수에 녹여 제조한 결합액을 가하여 조립하였다. 완료된 조립물을 유동층 건조기에서 건조한 후, 20호체가 장착된 F형 정립기를 사용하여 심바스타틴 선방출성 과립을 제조하고, 더블콘믹서(다산파마텍, 한국)에 과립과 부틸레이트히드록시아니솔, 전분글리콘산나트륨, 콜로이드성이산화규소를 넣고 60분간 혼합한 후, 스테아린산 마그네슘을 투입 후 4분간 혼합하여 심바스타틴 선방출성 최종혼합물을 제조하였다.
상기의 심바스타틴 선방출성 과립을 로타리 타정기(MRC-30:세종기계, 한국)에서 타정하였다.
(2) 올메사르탄 메독소밀 지연방출성 정제 제조
표 II- 1에 나타난 성분 및 함량으로 올메사르탄 메독소밀, 유당, 크로스카멜로스나트륨, 히프로멜로오스, 가교 폴리비닐피롤리돈, 저치환-히드록시프로필셀룰로오스를 35호체로 사과하고 고속혼합기(Lab. Pharma Mixer P, 디오스나, 독일)에 넣고 5분간 고속으로 혼합한 뒤, 별도로 히드록시프로필셀룰로오스를 정제수에 녹여 제조한 결합액을 가하여 조립하였다.
완료된 조립물을 유동층 건조기에서 건조한 후, 20호체가 장착된 F형 정립기를 사용하여 정립하여 올메사르탄 메독소밀 지연방출성 과립을 제조하였다.
다시 상기의 과립을 더블콘믹서에 넣고, 카보머 71G(카복시비닐폴리머, 루브리졸, 미국)를 분말상태로 투입하여 10분간 혼합한 후, 스테아린산 마그네슘을 투입 하여 4분간 혼합하여 올메사르탄 메독소밀 지연방출성 최종혼합물을 제조한 후 로타리 타정기(MRC-30: 세종파마텍, 한국)로 타정하였다.
정제를 코팅기(SFC-30F, 세종파마텍, 한국)에 투여하고, 히프로멜로오스프탈레이트(HPMCP, Shin-etsu, 일본)를 80%(v/v)에탄올에 녹인 액을 코팅액으로 코팅하여 올메사르탄 메독소밀 지연방출형 정제을 제조하였다.
(3) 캡슐제 제조
타정이 완료된 두 정제를 캡슐충진기(SFN-8N, 세종파마텍, 한국)를 이용하여 1호의 경질 젤라틴캡슐에 충전하여 심바스타틴 - 올메사르탄 메독소밀 캡슐제(정제 + 정제)를 제조하였다.
<실시예 II- 6> 심바스타틴 - 올메사르탄 메독소밀캡슐제(과립 + 정제)의 제조
(1) 심바스타틴 선방출성 혼합물 제조
표 II- 1에 나타난 성분 및 함량으로 실시예 II- 5의 (1) 과립제조방법(올메사르탄 메독소밀 지연방출성 과립제조 단계까지의 방법)과 동일한 방법으로심바스타틴 선방출성 과립를 제조하고, 다시 더블콘믹서에 넣고, 카보머 71G를 분말상태로 투입하여 10분간 혼합하여 올메사르탄 메독소밀 지연방출성 최종혼합물을 제조하였다.
(2) 올메사르탄 메독소밀 지연방출성 정제 제조
표 II- 1에 나타난 성분 및 함량으로 실시예 II- 5의 (2)방법에 따라 올메사르탄 메독소밀 지연방출성 정제를 제조하였다.
(3) 캡슐제 제조
완성된 올메사르탄 메독소밀 지연방출성 정제와 상기의 심바스타틴 선방출성 최종혼합물을 캡슐충진기(SFN-8N, 세종파마텍, 한국)를 이용하여 1호의 경질젤라킨 캡슐로 충전하여 심바스타틴 - 올메사르탄 메독소밀 캡슐제(정제 + 과립)를 제조하였다.
<실시예 II- 7> 심바스타틴 - 올메사르탄 메독소밀 캡슐제(과립 + 과립)의 제조
(1) 심바스타틴 선방출성 혼합물 제조
심바스타틴 선방출성 혼합물은 표 II- 1에 나타난 성분 및 함량으로 상기 실시예 II- 6의 (1) 방법에 따라 심바스타틴 선방출성 최종혼합물을 제조하였다.
(2) 올메사르탄 메독소밀 지연방출성 혼합물 제조
올메사르탄 메독소밀 지연방출성 과립은 표 II- 1에 나타난 성분 및 함량으로 올메사르탄 메독소밀, 유당, 크로스카멜로스나트륨, 가교 폴리비닐피롤리돈, 저치환-히드록시프로필셀룰로스을 35호체로 사과하고 유동층 과립기에 넣고 5분간 혼합하여 혼합물을 제조한 후, 별도로 히드록시프로필셀룰로오스를 정제수에 녹여 결합액으로 하였다. 유동층 과립기 및 유동층 건조 등의 조건은 올메사르탄 메독소밀 지연방출층 과립의 공정과 동일하다. 다시 완료된 건조물을 유동층 코팅기에 넣고, 별도로 셀룰로오스아세테이트320S, 셀룰로오스아세테이트398NF10(아세탈기 39.8%), 히프로멜로오스를 에탄올과 염화메틸렌 혼액(1:1(w/w))에 녹인 액을 조제하여 상기 조립물을 유동층 과립 코팅기에 넣고실시예 II- 1의 (2) 조건으로 코팅하였다.
(3) 캡슐제 제조
완성된 두 혼합물을 1호 캡슐에 캡슐충진기로 충전하여 심바스타틴 - 올메사르탄 메독소밀 캡슐제(과립 + 과립)를 제조하였다.
<실시예 II- 8> 심바스타틴 - 올메사르탄 메독소밀 캡슐제(과립+펠렛)의 제조
(1) 심바스타틴 선방출성 혼합물의 제조
심바스타틴 선방출성 과립은 표 II- 1에 나타난 성분 및 함량으로 상기 실시예 II- 7의 (1) 방법에 따라 심바스타틴 선방출성 최종혼합물을 제조하였다.
(2) 올메사르탄 메독소밀 지연 방출 펠렛의 제조
올메사르탄 메독소밀 지연 방출 펠렛의 제조는 슈가 시드(Sugar sphere, NP Pharmaceutical, 프랑스)를 35호체로 체과하고 유동층 코팅기(GPCG-1, Glatt, 독일)에 투입한 뒤, 따로 정제수와 에탄올에 히프로멜로오스와 올메사르탄 메독소밀을 용해 또는 현탁시킨 결합액을 분무하여 올메사르탄 메독소밀 함유 펠렛을 형성하고, 건조하였다. 다시 상기의 과립에 히프로멜로오스프탈레이트를 에탄올과 염화메틸렌 혼액(1:1(w/w))에 녹인 액을 분무하여 올메사르탄 메독소밀 지연 방출 펠렛을 제조하였다.
(3) 캡슐제 제조
표 II- 1의 성분과 함량으로, 상기 제조 방법으로 제조된 심바스타틴 선방출성 최종혼합물과 올메사르탄 메독소밀 지연 방출 펠렛을 혼합하여 1호 캡슐에 캡슐충진기로 충전하여 심바스타틴 - 올메사르탄 메독소밀 캡슐제(과립+펠렛)를 제조하였다.
<실시예 II- 9> 심바스타틴 - 올메사르탄 메독소밀 2상 매트릭스 정제의 제조
(1) 심바스타틴 선방출성 혼합물의 제조
표 II- 1에 나타난 성분 및 함량으로 심바스타틴, 미결정셀룰로오스, 만니톨, 유당, 아스코르빈산(Ascorbic acid, Merck, 독일)을 칭량하여 35 호체로 사과하고 더블콘믹서에 60분간 혼합한 뒤, 부틸레이트히드록시아니솔, 전분글리콘산나트륨, 콜로이드성이산화규소를 넣고 60 분간 다시 혼합하였다. 스테아린산 마그네슘을 투입 후 4분간 혼합하여 심바스타틴 선방출성 최종혼합물을 제조하였다.
(2) 올메사르탄 메독소밀 지연방출성 과립 제조
표 II- 1에 나타난 성분 및 함량으로 올메사르탄 메독소밀, 유당, 크로스카멜로스나트륨, 가교 폴리비닐피롤리돈, 저치환-히드록시프로필셀룰로스를 35호체로 사과하고 더블콘믹서로 30분간 혼합하여 혼합물을 제조하였다. 별도로 히드록시프로필셀룰로오스를 정제수에 녹여 결합액으로 하였다. 위의 혼합물을 유동층 과립기 또는 고속혼합기에 투여한 후, 결합액을 분무하며 과립을 제조하였다. 완료된 과립을 유동층 건조기에서 건조한 후, 다시 상기의 건조물에 카보머 71G를 분말상태로 투입하여 10분간 혼합한 후, 일정한 크기로 체과하였다. 별도로 히프로멜로오스를 에탄올 및 정제수 혼액(8:2(v/v))에 녹여 1차 코팅액을 제조하고 아크릴이즈(Acryl-eze, Colorcon, 미국)를 80%(v/v)에탄올에 녹여 2차 코팅액을 제조하였다. 코팅액 제조가 완료된 후, 상기 과립을 유동층 코팅기에 투여하고 1차 코팅액으로 코팅한 후, 2차 코팅액으로 2차 코팅을 하였다. 코팅 완료 후 스테아린산 마그네슘 투입 후 4분간 혼합하여 올메사르탄 메독소밀 지연방출성 최종혼합물을 제조하였다.
(3) 타정 및 코팅
상기의 심바스타틴 혼합물과 올메사르탄 메독소밀 지연방출성 최종혼합물을 혼합한 후 로타리 타정기(MRC-30: 세종파마텍, 한국)에서 타정하였다. 타정이 완료된 정제를 히프로멜로오스 2910, 폴리에틸렌글리콜 6,000을정제수 혼액(8:2(v/v))에 용해시킨 후 산화티탄을 분산시킨 코팅액으로 코팅하였다.
<실시예 II- 10> 심바스타틴 - 올메사르탄 메독소밀블리스터 포장 키트의 제조
(1) 심바스타틴 선방출성 정제 제조
표 II- 1에 나타난 성분 및 함량으로 심바스타틴, 미결정셀룰로오스, 유당, 전젤라틴화전분(Starch 1500, Colorcon,미국)을 칭량하여 35호체로 사과하고, 고속혼합기(Lab. Pharma Mixer P, 디오스나, 독일)에 넣고, 5분 간 혼합한 뒤, 별도로 히드록시프로필셀룰로오스와 구연산을 정제수에 녹여 제조한 결합액을 이용하여 조립하였다. 조립이 완료된 후 유동층 건조기에 조립물을 넣은 후 실시예 II- 1과 같은 조건에서 건조하였다.
건조가 완료되면 건조물을 20호체가 장착된 F형 정립기를 사용하여 심바스타틴 선방출성 과립을 제조하고, 더블콘믹서에 정립물과 부틸레이트히드록시아니솔, 전분글리콘산나트륨, 콜로이드성이산화규소를 넣고 60분간 혼합한 후, 스테아린산 마그네슘을 투입 후 4분간 혼합하여 심바스타틴 선방출성 최종혼합물을 제조하였다.
상기의 심바스타틴 선방출성 과립을 로타리 타정기(MRC-30: 세종파마텍, 한국)에서 타정하였다.
(2) 올메사르탄 메독소밀 지연방출성 정제 제조
표 II- 1에 나타난 성분 및 함량으로 올메사르탄 메독소밀, 크로스카멜로스나트륨, 유당, 가교 폴리비닐피롤리돈, 저치환-히드록시프로필셀룰로스을 35호체로 사과하고, 유동층 과립기에 투여한 후, 별도로 히드록시프로필셀룰로오스를 정제수에 녹여 결합액을 분무하여 과립을 제조하였다. 완료된 과립을 유동층 건조기에서 건조한 후, 건조물을 별도로 폴리비닐아세테이트프탈레이트(Phthalavin, Colorcon, 미국)를 에탄올에 녹인 액을 조제하여 실시예 II- 1의 (2)의 동일한 조건으로 코팅하였다.
다시 상기의 과립에 카보머 71G를 분말상태로 투입하여 10분간 혼합한 후, 일정한 크기로 체과하였다.
체과 후 스테아린산 마그네슘 투입하고 4분간 혼합하여 올메사르탄 메독소밀지연방출 과립을 제조하였다.
완성된 올메사르탄 메독소밀 지연방출성 과립은 로타리 타정기(MRC-30: 세종파마텍, 한국)로 타정하였다.
(3) 타정 및 코팅 후 포장 키트 제조
코팅이 완료된 각각의 정제는 하나의 PTP(Press Through Pack)포장용기에 포장하여 동시복용이 가능한 포장키트를 제조하였다.
<실시예 II- 11> 심바스타틴 - 올메사르탄 메독소밀 삼투성 유핵정의 제조
(1) 심바스타틴 선방출성 과립의 제조
표 II- 1에 나타난 성분 및 함량으로 실시예 II- 10의 (1)방법의 과립과정에 따라 선방출성 과립을 제조하였다.
(2) 올메사르탄 메독소밀 지연방출성 정제의 제조(내핵)
표 II- 1의 성분 및 함량으로 올메사르탄 메독소밀, 크로스카멜로스나트륨, 유당, 가교 폴리비닐피롤리돈, 염화나트륨, 저치환-히드록시프로필셀룰로스을 35호체로 사과하고 더블콘믹서로 혼합한 후 여기에 스테아린산 마그네슘을 넣어 최종적으로 더블콘믹서로 혼합하고, 상기 최종 혼합물을 로타리 타정기(MRC-30, 세종파마텍, 한국)를 사용하여 타정하였다. 타정 후 삼투성 코팅기제로서 에틸셀룰로오스(Aquacoat ECD, FMC, 미국)를 에탄올과 염화메틸렌 혼액(1:1(w/w))에 용해시킨 후 코팅기(SFC-30F, 세종파마텍, 한국)를 이용하여 내핵에 코팅하여 삼투성 핵정을 제조하였다.
(3) 타정 및 코팅
유핵정 타정기(RUD-1: Kilian)를 사용하여 올메사르탄 메독소밀삼투성 핵정을 내핵으로 하고 심바스타틴 선방출성 과립을 외층으로 하여 타정한 다음 코팅기로서 필름 코팅층을 형성하여 유핵정을 제조하였다. 타정이 완료된 정제를 히프로멜로오스 2910, 폴리에틸렌글리콜 6,000을 에탄올과 정제수 혼액(8:2(v/v))에 용해 후 산화티탄을 분산 시켜 제조한 코팅액을 사용하여 코팅하였다.

실험예 II- 1: 비교 용출시험(comparative dissolution profile test)
상기 실시예 II에서 제조한 약제학적 제제와 대조약으로 심바스타틴 단일제(Merck: Zocor) 및 올메사르탄 메독소밀Sankyo: Benicar, Olmetec)을 사용하여 비교 용출시험을 실시하였다. 심바스타틴 성분 용출 시험의 경우 미국약전 (USP30)의 근거하여 용출시험을 진행하였고, 올메살탄 성분 용출시험의 경우 2시간을 기점으로 용출액을 0.1 N-염산용액(산성환경)에서 pH 6.8(인공장액)완충액으로 변경하여 심바스타틴과 올메사르탄 메독소밀을 용출시험을 진행하였다. 그 결과를 도 9, 10, 11, 12, 13에 나타내었다(시험 개체수는 각각 12개)
[심바스타틴 시험방법]
용출시험 근거: 미국약전(USP30)중의 '심바스타틴 정제'항
시험 방법: 패들법(Paddle method), 50회전/분
시험약: pH=7.0 완충액 (조성 = 계면활성제로서 라우릴 황산 나트륨 0.5% 중량/중량을 함유하는 0.01M 인산이수소나트륨 용액), 900mL
분석 방법: 자외가시부흡광광도법 (검출 파장 = 238nm)
[올메사르탄 메독소밀 용출시험방법]
용출시험 근거: 상기 실시예 II 에서 얻은 복합제제와 올메사르탄 메독소밀정제를 대한약전 9개정 일반시험법 중 용출시험법에 따라 진행한다.
상세한 시험방법은 37± 0.5 ℃로 가온한 인공위액(대한약전 9개정 붕해시험법 제 1액)에서 2시간 동안 용출시험을 진행한 후 인공장액(대한약전 9개정 붕해시험법 제 2액)에서 용출시험을 진행하였다.
시험 방법: 패들법(Paddle method), 50회전/분
시험액: 시험액 : 0.1 N 염산용액, 500 mL (0~2시간), 6.8 인공장액, 900 mL (2시간 이후)
분석방법: 고성능액체크로마토그래피분석
컬럼: C-18, 4.6mm, 25cm, 5μm, 검출파장(올메사르탄 메독소밀) =237 nm
도 9,10,11,12,13,에 의하면 실시예 II 에서 제조된 약제학적 제제에서 심바스타틴은 대조제인 조코와 동일하게 용출시작후 30분이내에 제제중 심바스타틴 총량의 90%가 용출되었다. 한편, 본 발명의 약제학적 제제에서 올메사르탄은 대조제인 올메텍이 심바스타틴 용출 개시후 30분 이내에 올메사르탄 메독소밀 총량의 90% 이상 방출된 것과 달리, 심바스타틴 용출 개시후 2시간 이내까지 올메사르탄 메독소림 총량의 5%미만이 용출되었다.
따라서 본 발명의 약제학적 제제는 대조약인 올메사르탄 메독소밀 단일제와 심바스타틴 단일제를 동시 복용하였을 경우의 용출 양상과는 달리 올메사르탄 메독소밀의 초기 방출이 심바스타틴보다 매우 느리기 때문에 심바스타틴이 먼저 간에서 대사를 받은 후 대사 관련 효소인 사이토크롬 P450이 재생될 시간을 충분히 확보할 수 있는 약제학적 제제이다.
[실시예 및 실험예 III] 심바스타틴 및 발사르탄 함유 약제학적 제제
<실시예 III- 1> 심바스타틴 - 발사르탄 유핵정의 제조
1)선방출성 구획의 제조
다음 표 III- 1에 나타난 성분 및 함량과 같이 심바스타틴(Simvastatin, Biocon, India), 미결정셀룰로오스(Vivapur 12, JRS, Germany), 디-만니톨(Pearlitol, Roquette, France)을 35호체로 사과하고 고속혼합기로 혼합하였다.히드록시프로필셀룰로오스(Klucel LF, Hercules, USA)와 구연산(Citric acid monohydrate, Merck, USA)을 정제수에 녹여 결합액을 제조하고 이를 주성분 혼합물과 함께 고속혼합기에 투입하고 연합한 후 20호체로 오실레이터를 이용하여 제립하고 이를 온수 건조기를 이용하여 60℃에서 건조한 다음 다시 20호체로 정립하였다. 여기에 부틸레이티드히드록시아니솔(BHA, Merck, USA), 전분글리콘산나트륨(Explotab, JRS, Germany), 콜로이드성 이산화규소(Aerosil pharma 200, Degussa, USA) 를 혼합하고, 스테아린산 마그네슘을 넣어 더블콘믹서로 최종 혼합하였다.
2)지연방출성 구획의 제조
다음 표 III- 1에 나타난 성분 및 함량과 같이 발사르탄(Dr.Reddy's, India)을 35호체로 사과하고 고속 혼합기에 투입하였다. 따로 히드록시프로필셀룰로오스를 정제수에 녹인 결합액을 고속혼합기에 투입하고 연합한 후 20호체로 오실레이터를 이용하여 제립하고 이를 유동층 건조기로 건조시켰다
유동층 과립 건조기는 GPCG-1(Glatt, Germany)을 사용하였고 조립물을 넣은 후 다음과 같은 조건에서 진행하였다. Air flow는 120 m3/시간, Inlet air 온도는 65 ℃, 필터 shaking(delta P 필터 < 4000 pa로 유지)은 비동시성 모드로 30초에 5초간 진행하였다. 제품온도가 40 ℃에 이르면 샘플을 채취하여 건조 감량 2.5% 이하 기준에 적합하면 완료하고, 초과시에는 더 진행한 후 재측정하여 건조를 완료하였다.
건조가 완료되면 건조물을 20호체가 장착된 F형 정립기를 사용하여 정립하고, 더블콘믹서에 정립물을 넣고, 미결정셀룰로오스, 가교 폴리비닐피롤리돈(Crospovidone, BASF, Germany), 콜로이드성 이산화규소, 라우릴설페이트나트륨(Jeelate, Jeen, USA)을 투입 후 더블콘믹서로 혼합한 후 상기 혼합물에 스테아린산 마그네슘을 넣고 최종 혼합하였다. 상기의 최종 혼합물은 로타리 타정기(MRC-33: 세종)를 사용하여 타정하고 이를 핵정으로 하였다.
다시 상기의 정제에 에탄올과 염화메틸렌의 1:1(w/w)혼액에 용해시킨 히프로멜로오스프탈레이트(HPMC-P, Shin-Etsu, Japan) 용액을 하이코터(SFC-30N, 세종 기계, 한국)를 사용하여 필름 코팅 층을 형성하여 핵정을 제조하였다.
3)타정 및 코팅
유핵정 타정기(RUD-1: Kilian)를 사용하여 발사르탄 핵정을 내핵으로 하고 심바스타틴을 포함하는 조성물을 외층으로 하여 타정한 다음 히프로멜로오스2910(Pharmacoat 603, Shin-Etsu, Japan), 히드록시프로필셀룰로오스, 산화티탄(Fanglian, JHP, China), 탈크(Talc, Nippon soda, Japan)를 에탄올 및 정제수 혼액(8:2(v/v))에 녹여 제조한 코팅액으로 하이코터(SFC-30N, 세종 기계, 한국)를 사용하여 필름 코팅 층을 형성하여 유핵정을 제조하였다.
<실시예 III- 2> 심바스타틴 - 발사르탄 유핵정의 제조
1)선방출성 구획의 제조
다음 표 III- 1에 나타난 성분 및 함량과 실시예 III- 1의 1) 심바스타틴 선방출성 구획의 제조 방법에 따라 제조하였다.
2)지연방출성 구획의 제조 (내핵)
다음 표 III- 1에 나타난 성분 및 함량과 같이 발사르탄을 35호체로 사과하고 고속 혼합기에 투입하였다. 따로 히드록시프로필셀룰로오스 정제수에 녹인 결합액을 고속혼합기에 투입하고 연합한 후 20호체로 오실레이터를 이용하여 제립하고 이를 유동층 건조기로 건조시켰다.
건조가 완료되면 건조물을 20호체가 장착된 F형 정립기를 사용하여 정립하고, 유동층과립기(GPCG 1: Glatt)에 투입하였다. 상기의 과립에 에탄올과 염화메틸렌의 1:1(w/w)혼액에 용해시킨 히프로멜로오스프탈레이트 용액을 분무하여 과립을 코팅하였다. 상기의 과립은 더블콘믹서에 넣고, 미결정셀룰로오스, 가교 폴리비닐피롤리돈, 콜로이드성 이산화규소, 라우릴설페이트나트륨을 투입하고 혼합하였다.
여기에 스테아린산 마그네슘을 넣어 최종 더블콘믹서로 혼합한 후 상기 최종 혼합물을 로타리 타정기(MRC-33: 세종)를 사용하여 타정하고 이를 내핵정으로 하였다.
3)타정 및 코팅
실시예 III- 1의 3) 타정 및 코팅 방법에 따라 조작하여 심바스타틴-발사르탄 유핵정을 제조하였다.
<실시예 III- 3> 심바스타틴 - 발사르탄 다층정의 제조
1)선방출성 구획의 제조
다음 표 III- 1에 나타난 성분 및 함량과 실시예 III- 1의 1) 심바스타틴 선방출성 구획의 제조 방법에 따라 제조하였다.
2)지연방출성 구획의 제조
다음 표 III- 1에 나타난 성분 및 함량과 같이 발사르탄, 미결정셀룰로오스와 폴록사머 188(Lutrol, BASF, USA)를 35호체로 사과하고 더블콘믹서로 혼합하고, 따로 히프로멜로오스를 정제수에 녹여 만든 결합액을 분무하여 과립을 형성, 건조하였다. 상기의 과립에 에탄올과 염화메틸렌의 1:1(w/w)혼액에 용해시킨 히프로멜로오스프탈레이트 용액을 분무하여 과립을 코팅하였다.스테아린산 마그네슘을 넣어 최종 더블 콘믹서로 혼합하였다
3)타정 및 코팅
본 공정에서는 다층정 타정기(MRC-37T: 세종)를 사용하여 타정하였다.상기 심바스타틴을 포함하는 조성물을 1차 분말공급기에 넣고, 발사르탄을 포함하는 조성물을 2차 분말 공급기에 넣어 층간의 혼입을 최소화할 수 있는 조건으로 타정하고, 히프로멜로오스2910, 히드록시프로필셀룰로오스, 산화티탄, 탈크를 에탄올 및 정제수 혼액(8:2(v/v))에 녹여 제조한 코팅액으로 하이코터(SFC-30N, 세종 기계, 한국)로서 필름 코팅층을 형성하여 다층정 형태의 지연방출성 정제를 제조하였다.
<실시예 III- 4> 심바스타틴 - 발사르탄 다층정의 제조
1)선방출성 구획의 제조
다음 표 III- 1에 나타난 성분 및 함량과 실시예 III- 1의 1) 심바스타틴 선방출성 구획의 제조 방법에 따라 제조하였다.
2)지연방출성 구획의 제조
다음 표 III- 1에 나타난 성분 및 함량과 같이 발사르탄, 미결정 셀룰로오스와 폴록사머 188을 35호체로 사과하고 더블콘믹서로 혼합하고, 따로 히프로멜로오스를 물에 녹여 만든 결합액을 분무하여 과립을 형성, 건조하였다. 상기의 과립에 에탄올과 염화메틸렌의 1:1(w/w)혼액에 용해시킨 유드라짓 RS PO(Eudragit RS PO, Evonik, USA) 용액을 분무하여 과립을 코팅하였다. 여기에 스테아린산 마그네슘을 넣어 최종 더블 콘믹서로 혼합하였다.
3)타정 및 코팅
실시예 III- 3의 3) 타정 및 코팅 방법에 따라 조작하여 심바스타틴-발사르탄 다층정을 제조하였다.
<실시예 III- 5> 심바스타틴 - 발사르탄 2상 매트릭스 정제의 제조
1)선방출성 구획의 제조
다음 표 III- 1에 나타낸 성분 및 함량과 같이 심바스타틴, 미결정셀룰로오스, 디-만니톨을 35호체로 사과하고 고속혼합기로 혼합하였다.히드록시프로필셀룰로오스와 구연산을 정제수에 녹여 결합액을 제조하고 이를 상기 주성분의 혼합물과 함께 고속혼합기에 투입하고 연합한 후 20호체로 오실레이터를 이용하여 제립하고 이를 온수 건조기를 이용하여 60℃에서 건조한 다음 다시 20호체로 정립하고, 여기에 부틸레이티드히드록시아니솔을 넣고 혼합하였다.
2)지연방출성 구획의 제조
다음 표 III- 1에 나타난 성분 및 함량과 같이 발사르탄, 미결정셀룰로오스와 폴록사머 188을 35호체로 사과하고 더블콘믹서로 혼합하였다.
상기의 혼합물을 유동층과립기(GPCG 1: Glatt) 에 투입하고 따로 히프로멜로오스를 정제수에 녹여 만든 결합액을 분무하여 과립을 형성, 건조하였다. 다시 상기의 과립에 에탄올과 염화메틸렌의 1:1(w/w)혼액에 용해시킨 유드라짓 RS PO 용액을 분무하여 과립을 코팅하였다.
3)후혼합, 타정 및 코팅
상기 제조된 각각의 최종 조성물을 더블콘믹서로 혼합하고, 전분 글리콘산 나트륨, 콜로이드성 이산화규소를 혼합한 후 스테아린산 마그네슘을 넣어 더블콘믹서로 최종 혼합하였다.
상기 최종 혼합물을 로타리 타정기(MRC-33: 세종)를 사용하여 타정한 다음 히프로멜로오스2910, 히드록시프로필셀룰로오스, 산화티탄, 탈크를 에탄올 및 정제수에 녹여 제조한 코팅액으로 하이코터(SFC-30N, 세종 기계, 한국)로서 필름 코팅 층을 형성하여 2상 매트릭스 정제를 제조하였다.
<실시예 III- 6> 심바스타틴 - 발사르탄 2 상 매트릭스 정제의 제조
1)선방출성 구획의 제조
다음 표 III- 1에 나타난 성분 및 함량과 실시예 III- 5의 1) 심바스타틴 선방출성 구획의 제조 방법에 따라 제조하였다
2)지연방출성 구획의 제조
다음 표 III- 1에 나타난 성분 및 함량과 같이 발사르탄, 미결정 셀룰로오스와 폴록사머 188을 35호체로 사과하고 더블콘믹서로 혼합하였다.혼합물을 유동층과립기(GPCG 1: Glatt) 에 투입하고 따로 히프로멜로오스를 정제수에 녹여 만든 결합액을 분무하여 과립을 형성, 건조하였다.상기의 과립에 에탄올과 염화메틸렌의 (1:1(w/w))혼액에 용해시킨 히프로멜로오스프탈레이트 용액을 분무하여 과립을 코팅하였다.
3) 타정 및 코팅
실시예 III- 5의 3) 후혼합, 타정 및 코팅 방법에 따라 조작하여 심바스타틴-발사르탄 2상 매트릭스 정제를 제조하였다.
<실시예 III- 7> 심바스타틴 - 발사르탄 2상 캡슐 제제
1)선방출성 구획의 제조 (과립)
다음 표 III- 1에 나타난 성분 및 함량과 실시예 III- 5의 1) 심바스타틴 선방출성 구획의 제조 방법에 따라 제조하였다.
2) 발사르탄 지연방출성 구획의 제조 (과립)
다음 표 III- 1에 나타난 성분 및 함량과 실시예 III- 6의 2) 발사르탄 지연방출성 구획의 제조 방법에 따라 제조하였다.
3)혼합 및 캡슐충전
공정 1)과 2)의 최종 조성물을 더블콘믹서로 혼합하고, 여기에 전분글리콘산나트륨을 투입한 후 더블콘믹서로 혼합하고, 콜로이드성 이산화규소를 혼합하고, 스테아린산 마그네슘을 넣어 최종 혼합하였다. 최종 혼합된 혼합물을 분말 공급기에 투입하고 캡슐충전기를 이용하여 1호 젤라틴 경질캡슐에 충전하였다.
<실시예 III- 8> 심바스타틴 - 발사르탄 정제를 함유한 캡슐제
1)선방출성 구획의 제조 (정제)
다음 표 III- 1에 나타난 성분 및 함량과 같이 심바스타틴, 미결정셀룰로오스, 디-만니톨을 35호체로 사과하고 고속혼합기로 혼합하였다. 따로 히드록시프로필셀룰로오스와 구연산을 물에 녹여 결합액을 제조하고 이를 주성분 혼합물과 함께 고속혼합기에 투입하고 연합한 후 20호체로 오실레이터를 이용하여 제립하고 이를 온수 건조기를 이용하여 60℃에서 건조한 다음 다시 20호체로 정립하였다.부틸레이티드히드록시아니솔, 전분글리콘산나트륨, 콜로이드성 이산화규소를 혼합하고, 스테아린산 마그네슘을 넣어 더블콘믹서로 최종 혼합한 후 상기 최종 혼합물을 로타리 타정기(MRC-33: 세종)를 사용하여 타정하였다.정제는 히프로멜로오스2910, 폴리에틸렌글리콜 6000(PEG6000, Duksan, Korea), 산화티탄 및 탈크를 80(w/w)% 에탄올에 용해 및 분산시켜 제조한 코팅액을 이용하여 하이코터(SFC-30N, 세종 기계, 한국)로 코팅하였다.
2)지연방출성 구획의 제조 (정제)
다음 표 III- 1에 나타난 성분 및 함량과 실시예 III- 1의 2) 발사르탄 지연방출성 구획의 제조 방법에 따라 제조하였다.
3) 캡슐충전
공정 1)의 심바스타틴 정제와 공정 2)의 발사르탄 정제를 캡슐충전기를 이용하여 1호의 경질 젤라틴 캡슐에 충전하였다.
<실시예 III- 9> 심바스타틴-발사르탄 캡슐제
1)선방출성 구획의 제조 (과립)
다음 표 III- 1에 나타난 성분 및 함량과 같이 심바스타틴, 미결정셀룰로오스, 디-만니톨 35호체로 사과하고 고속혼합기로 혼합하였다.따로 히드록시프로필셀룰로오스와 구연산을 정제수에 녹여 결합액을 제조하고 이를 주성분 혼합물과 함께 고속혼합기에 투입하고 연합한 다음 20호체로 오실레이터를 이용하여 제립하고 이를 온수 건조기를 이용하여 60℃에서 건조한 후 다시 20호체로 정립하였다.여기에 부틸레이티드히드록시아니솔, 전분 글리콘산 나트륨, 콜로이드성 이산화규소를 혼합하고, 스테아린산 마그네슘을 넣어 더블콘믹서로 최종 혼합하였다.
2)지연방출성 구획의 제조 (정제)
다음 표 III- 1에 나타난 성분 및 함량과 실시예 III- 1의 2) 발사르탄 지연방출성 구획의 제조 방법에 따라 제조하였다.
3) 캡슐충전
공정 1)의 심바스타틴 정제와 공정 2)의 발사르탄 정제를 캡슐충전기를 이용하여 1호의 히프로멜로오스 경질캡슐에 충전하였다.
<실시예 III- 10> 심바스타틴-발사르탄 코팅정
1)심바스타틴 선방출성 코팅액의 제조
다음 표 III- 1에 나타난 성분 및 함량과 같이 심바스타틴, 부틸레이티드히드록시아니솔, 콜로이드성이산화규소, 히프로멜로오스2910, 폴리에틸렌클리콜 6000, 산화티탄, 탈크를 에탄올과 염화메틸렌(1:1 (w/w)) 혼액에 용해 및 분산시켜 선방출성 심바스타틴 코팅액을 제조하였다.
2)지연방출성 구획의 제조 (정제)
다음 표 III- 1에 나타난 성분 및 함량과 실시예 III- 1의 2) 발사르탄 지연방출성 구획의 제조 방법에 따라 제조하였다.
3) 1차코팅
위에서 제조한 발사르탄 정을 하이코터(SFC-30N, 세종 기계, 한국)에 투여한 후 심바스타틴 코팅액으로 1차 코팅하였다.
4)2차 코팅
표 III- 1의 코팅층에 기재된 히프로멜로오스2910, 히드록시프로필셀룰로오스, 산화티탄, 탈크를 에탄올 및 정제수 혼액((8:2(v/v))에 녹여 제조한 코팅액으로 1차 코팅완료된 정제에 2차 코팅을 하여 필름코팅정을 제조하였다.
<실시예 III- 11> 심바스타틴 선방출-발사르탄 삼투성 유핵정의 제조
1)선방출성 구획의 제조
다음 표 III- 1에 나타난 성분 및 함량과 실시예 III- 1의 1) 심바스타틴 선방출성 구획의 제조 방법에 따라 제조하였다.
2) 발사르탄 지연방출성 구획의 제조 (정제)
다음 표 III- 1에 나타난 성분 및 함량과 같이 발사르탄을 35호체로 사과하고 고속 혼합기에 투입하였다. 따로 히드록시프로필셀룰로오스를 정제수에 녹인 결합액을 고속혼합기에 투입하고 연합한 후 20호체로 오실레이터를 이용하여 제립하고 이를 유동층 건조기로 건조시켰다.
건조가 완료되면 건조물을 20호체가 장착된 F형 정립기를 사용하였다. 상기의 과립은 더블콘믹서에 넣고, 미결정셀룰로오스, 가교 폴리비닐피롤리돈, 콜로이드성 이산화규소, 라우릴설페이트나트륨, 염화나트륨을 투입하고 혼합하였다.
여기에 스테아린산 마그네슘을 넣어 최종 더블콘믹서로 혼합한 후 상기 최종 혼합물을 로타리 타정기(MRC-33: 세종)를 사용하여 타정하였다.
타정 후 삼투성 기제로서 콜리코드 SR 30D(폴리비닐 아세테이트 30% 현탁액,바스프)와 트리에틸시트레이트(Citroflex 2, Morimura BROS, USA)를 정제수에 분산시킨 후 하이코터(SFC-30N, 세종 기계, 한국)를 이용하여 내핵에 코팅하여 삼투성 핵정을 제조하였다.
3) 타정 및 코팅
실시예 III- 1의 3) 타정 및 코팅 방법에 따라 조작하여 심바스타틴 선방출-발사르탄 삼투성 유핵정제를 제조하였다.
<실시예 III- 12> 심바스타틴-발사르탄 블리스터 포장 키트
다음 표 III- 1에 나타난 성분 및 함량과 같이 제조하되, 실시예 III- 8의 발사르탄 정과 심바스타틴 정제를 캡슐에 동시 충전하는 것 대신 블리스터 포장용기에 동시복용 가능하도록 포장하는 것을 제외하고는 실시예 III- 8와 같이 제조하였다.


<실험예 III- 1> 용출 양상 시험 (dissolution profile test)
상기 실시예 III-1, III-3, III-5, III-8의 여러 제형의 정제와 대조약(조코: 심바스타틴 단일제, 디오반: 발사르탄 단일제) 을 사용하여 비교 용출시험을 실시하였다. 심바스타틴 성분 용출시험의 경우 미국약전(USP 30)을 근거하여 용출 시험을 진행하였고 발사르탄 성분 용출시험의 경우 2시간을 기점으로 용출액을 0.1 N-염산용액(산성환경)에서 pH 6.8(인공장액)완충액으로 변경하여 용출시험을 진행하였다(시험 개체수는 각각 12개).
[심바스타틴 시험방법]
용출시험 근거: 미국약전(USP30)중의 '심바스타틴 정제'항
시험 방법: 패들법(Paddle method), 50회전/분
시험약: pH=7.0 완충액 (조성 = 계면활성제로서 라우릴 황산 나트륨 0.5% 중량/중량을 함유하는 0.01M 인산이수소나트륨 용액), 900mL
분석 방법: 자외가시부흡광광도법 (검출 파장 = 최대 247, 최소 257nm)
[발사르탄 시험방법]
용출시험 근거: 미국약전(USP 31)중의 'Valsartan and Hydrochlorothiazide tablet'항
시험 방법: 패들법(Paddle method), 50회전/분
시험액: 0.01 N 염산용액, 1000ml (0~2시간)
pH=6.8 완충액(인산염 용액), 1000 mL (2시간 이후)
분석방법: 자외가시부흡광광도법 (검출파장 = 최대 270, 최소 250 nm)
도 14, 15, 16, 17에 의하면 실시예 III-1, III-3, III-5, III-8의 정제는 하기 조건에서 용출 시험시 심바스타틴 성분은 대조 제제인 조코와 비교하여 거의 동등한 용출특성을 나타내는 것으로 확인 되었다. 한편, 대조제인 디오반의 120분까지의 용출률이 약 30%인 것에 비하여, 본 발명의 심바스타틴/발사르탄 제제에서 발사르탄 성분의 용출률은 인공위액 구간인 120분까지 약 5% 미만으로 약2시간 이상의 지연방출효과가 있는 것으로 확인되었다.
[실시예 및 실험예 IV] 심바스타틴 및 칸데사르탄 함유 약제학적 제제
<실시예 IV- 1> 2상 매트릭스 정제의 제조
1) 심바스타틴 선방출성 과립의 제조
표 IV-1에 나타낸 성분 및 함량으로 심바스타틴과 미결정셀룰로오스, 유당, 옥수수전분을 35호체로 체과하고 고속혼합기로 혼합하였다. 따로 히드록시프로필셀룰로오스, 시트르산을 정제수 (1정당 60mg)에 녹여 결합액을 제조하고 이를 주성분 혼합물과 함께 고속혼합기에 투입하고 연합하였다. 연합이 끝나면 18호체로 오실레이터를 이용하여 제립하고 이를 온수 건조기를 이용하여 30 ℃에서 건조하였다. 건조가 끝나면 다시 20호체가 장착된 F형 정립기를 사용하여 정립하여 심바스타틴 선방출성 과립을 제조하였다.
2) 칸데사르탄 실렉세틸의 지연방출성 과립의 제조
표 IV-1에 나타낸 성분 및 함량으로 유당, 옥수수전분, 및 카르복시메틸셀룰로오스 칼슘을 35호체로 체과하고 더블콘믹서로 혼합하였다. 따로 폴리에틸렌글리콜6000을 정제수 (1정당 310mg)에 녹힌 후 칸데사르탄 실렉세틸을 분산시킨 다음 유동층 과립기에 혼합물을 넣고 이 액을 분무하였다. 얻어진 물질에 따로 히드록시프로필셀룰로오스를 정제수 (1정당 20mg)에 녹인 액을 분무하여 과립을 형성하였다. 따로 폴리비닐아세테이트, 가교 폴리비닐피롤리돈, 및 라우릴설페이트 나트륨을 정제수 (1정당 600mg)에 분산시킨 후 위에서 형성된 과립에 분무하여 과립을 코팅한 다음 건조 하였다. 건조가 끝나면 다시 20호체가 장착된 F형 정립기를 사용하여 정립하여 칸데사르탄 실렉세틸 지연방출성 과립을 제조하였다.
3) 타정 및 코팅
표 IV-1에 나타낸 성분 및 함량으로 위의 1), 2)의 방법으로 제조된 두 과립물과 항산화제 부틸레이트 히드록시아니솔을 더블콘믹서에 넣고 혼합하였다. 이 혼합물에 스테아린산 마그네슘을 넣어 최종 혼합하였다. 최종 혼합물을 로타리 타정기(MRC-33: 세종)를 사용하여 타정하였다. 따로 히프로멜로오스 2910, 히드록시프로필셀룰로오스, 산화티탄, 탈크를 정제수 (1정당 350mg)에 용해 및 분산시킨 코팅액을 조제하여 위의 정제를 하이코터(SFC-30N: 세종 기계, 한국)로서 필름 코팅 층을 형성하여 2상 매트릭스 정제를 제조하였다.
<실시예 IV- 2> 2상 매트릭스 정제의 제조
1) 심바스타틴 선방출성 과립의 제조
표 IV-1에 나타낸 성분 및 함량을 이용하여 실시예 IV-1의 1) 방법으로 표제의 심바스타틴 선방출성 과립을 제조하였다.
2) 칸데사르탄 실렉세틸의 지연방출성 과립의 제조
표 IV-1에 나타낸 성분 및 함량으로 유당, 옥수수전분, 및 카르복시메틸셀룰로오스 칼슘을 35호체로 체과하고 더블콘믹서로 혼합하였다. 따로 폴리에틸렌글리콜6000을 정제수 (1정당 310mg)에 녹힌 후 칸데사르탄 실렉세틸을 분산시킨 다음 유동층 과립기에 혼합물을 넣고 이 액을 분무하였다. 얻어진 물질에 따로 히드록시프로필셀룰로오스를 정제수 (1정당 20mg)에 녹인 액을 분무하여 과립을 형성하였다. 따로 카르바우나 왁스, 가교 폴리비닐피롤리돈, 및 라우릴설페이트 나트륨을 정제수 (1정당 265mg)에 분산시킨 후 위에서 형성된 과립에 분무하여 과립을 코팅한 다음 건조 하였다. 따로 히프로멜로오스를 정제수 (1정당 300mg)에 녹인 액을 분무하여 2차 코팅한다. 건조가 끝나면 다시 20호체가 장착된 F형 정립기를 사용하여 정립하여 칸데사르탄 실렉세틸 지연방출성 과립을 제조하였다.
3) 타정 및 코팅
표 IV-1에 나타낸 성분 및 함량을 이용하여, 위의 1) 및 2)의 방법으로 제조된 두과립물을 실시예 IV-1의 3)의 방법으로 표제의 2상 매트릭스 정제를 제조하였다.
<실시예 IV- 3> 다층정 제조
1) 심바스타틴 선 방출성 층의 제조
표 IV-1에 나타낸 성분 및 함량으로,부형제인 미결정셀룰로오스, 유당, 옥수수전분을 35호체로 체과하고 고속혼합기로 혼합하였다. 따로 히드록시프로필셀룰로오스, 시트르산을 정제수 (1정당 60mg)에 녹여 결합액을 제조하고 이를 주성분 혼합물과 함께 고속혼합기에 투입하고 연합하였다. 연합이 끝나면 18호체로 오실레이터를 이용하여 제립하고 이를 온수 건조기를 이용하여 30 ℃에서 건조하였다. 건조가 끝나면 다시 20호체가 장착된 F형 정립기를 사용하여 정립하였다. 이 정립물을 더블콘믹서에 넣고 부틸레이트 히드록시아니솔을 넣고 혼합 한 뒤 이 혼합물에 스테아린산 마그네슘을 넣고 최종 혼합하여 심바스타틴 선방출성 층을 제조하였다.
2) 칸데사르탄 실렉세틸 지연방출성 층의 제조
표 IV-1에 나타낸 성분 및 함량으로, 유당, 옥수수전분, 및 카르복시메틸셀룰로오스 칼슘, 염화나트륨을 35호체로 체과하고 더블콘믹서로 혼합하였다. 따로 폴리에틸렌글리콜6000을 정제수 (1정당 310mg)에 녹힌 후 칸데사르탄 실렉세틸을 분산시킨 다음 유동층 과립기에 혼합물을 넣고 이 액을 분무하였다. 얻어진 물질에 따로 히드록시프로필셀룰로오스를 정제수 (1정당 20mg)에 녹인 액을 분무하여 과립을 형성하였다. 따로 에틸셀룰로오스와 유드라짓 RL을 메틸렌클로라이드와 에탄올의 1:1 혼합액 (1정당 270mg)에 용해시킨 후 위에서 형성된 과립에 분무하여 과립을 코팅한 다음 건조 하였다. 건조가 끝나면 다시 20호체가 장착된 F형 정립기를 사용하여 정립한다. 상기 정립물을 더블콘 믹서에 넣고 스테아르산마그네슘 투입한 뒤 혼합하여 칸데사르탄 실렉세틸 지연방출성 층을 제조하였다.
3) 타정 및 코팅
상기 1)의 반제품을 1차 분말공급기에 넣고,반제품을 2차 분말 공급기에 넣어 층간의 혼입을 최소화할 수 있는 조건으로 다층정 타정기(MRC-37T: 세종)를 사용하여 타정하였다. 따로 히프로멜로오스 2910, 히드록시프로필셀룰로오스, 산화티탄, 탈크를 정제수 (1정당 270mg)에 용해 및 분산시킨 코팅액을 조제하여 위의 정제를 하이코터(SFC-30N: 세종 기계, 한국)로서 필름 코팅 층을 형성하여 다층정제를 제조하였다.
<실시예 IV-4 내지 IV-6> 다층정의 제조
표 IV-1에 기재된 성분 및 함량으로 실시예 IV-3과 동일한 방법으로 제조하여 표제의 다층정을 제조하였다.
<실시예 IV-7> 유핵정 제조
1) 심바스타틴 선방출성 과립의 제조
표 IV-1에 기재된 성분 및 함량으로, 심바스타틴과 부형제인 미결정셀룰로오스, 전분글리콘산 나트륨, 유당, 옥수수전분을 35호체로 체과하고 고속혼합기로 혼합하였다. 따로 히드록시프로필셀룰로오스, 시트르산을 정제수 (1정당 100mg)에 녹여 결합액을 제조하고 이를 주성분 혼합물과 함께 고속혼합기에 투입하고 연합하였다.
연합이 끝나면 18호체로 오실레이터를 이용하여 제립하고 이를 온수 건조기를 이용하여 30 ℃에서 건조하였다. 건조가 끝나면 다시 20호체가 장착된 F형 정립기를 사용하여 정립하였다. 이 정립물을 더블콘믹서에 넣고 부틸레이트 히드록시아니솔을 넣고 혼합 한 뒤 이 혼합물에 스테아린산 마그네슘을 넣고 최종 혼합하여 심바스타틴 선방출성 과립을 제조하였다.
2) 칸데사르탄 실렉세틸 지연방출성 코팅 내핵정의 제조
표 IV-1에 기재된 성분 및 함량으로, 칸데사르탄 실렉세틸, 유당, 옥수수전분, 및 카르복시메틸셀룰로오스 칼슘을 35호체로 체과하고 고속혼합기로 혼합하였다.
따로 폴리에틸렌글리콜6000, 히드록시프로필셀룰로오스를 정제수 (1정당 300mg)에 녹여 결합액을 제조하고 이를 주성분 혼합물과 함께 고속혼합기에 투입하고 연합하였다. 연합이 끝나면 18호체로 오실레이터를 이용하여 제립하고 이를 온수 건조기를 이용하여 30 ℃에서 건조하였다. 건조가 끝나면 다시 20호체가 장착된 F형 정립기를 사용하여 정립하였다. 이 정립물을 더블콘믹서에 넣고 스테아린산 마그네슘을 넣고 최종혼합 후 로타리 타정기(MRC-33: 세종)를 사용하여 타정하였다.
타정이 완료된 정제에 히프로멜로오스 아세테이트 숙시네이트를 에탄올과 정제수 (8:2(v/v)) 혼합용매 (1정당 200mg)에 용해시켜 제조한 코팅액으로 코팅하여 칸데사르탄 실렉세틸 지연방출성 코팅 내핵정을 제조하였다.
3) 타정 및 코팅
유핵정 타정기(RUD-1: Kilian)를 사용하여 공정2)의 칸데사르탄 실렉세틸 코팅 핵정을 내핵정으로 하고 공정1)의 심바스타틴을 포함하는 조성물을 외층으로 하여 유핵정제의 제조를 완료한 후, 따로 히프로멜로오스 2910, 히드록시프로필셀룰로오스, 산화티탄, 탈크를 정제수 (1정당 470mg)에 용해 및 분산시켜 코팅액을 조제하였다. 위의 유핵정을 하이코터(SFC-30N: 세종)에 투입한 후 코팅액으로 코팅하여 필름 코팅 유핵정 제조를 완료하였다.
<실시예 IV-8> 유핵정의 제조
1) 심바스타틴 선방출성 과립의 제조
표 IV-1에 나타낸 성분 및 함량을 이용하여 실시예 IV-7의 1) 방법으로 표제의 심바스타틴 선방출성 과립을 제조하였다.
2) 칸데사르탄 실렉세틸의 지연방출성 코팅 내핵정의 제조
표 IV-1에 나타낸 성분 및 함량으로 유당, 옥수수전분, 및 카르복시메틸셀룰로오스 칼슘을 35호체로 체과하고 더블콘믹서로 혼합하였다. 따로 폴리에틸렌글리콜6000을 정제수 (1정당 310mg)에 녹힌 후 칸데사르탄 실렉세틸을 분산시킨 다음 유동층 과립기에 혼합물을 넣고 이 액을 분무하였다. 얻어진 물질에 따로 히드록시프로필셀룰로오스를 정제수 (1정당 20mg)에 녹인 액을 분무하여 과립을 형성한 다음 건조 하였다. 건조가 끝나면 더블콘믹서에 가교 폴리비닐피롤리돈, 라우릴설페이트 나트륨을 투입한 후 혼합한다. 혼합이 끝나면 스테아린산 마그네슘을 다시 투입하고 최종혼합을 한 후 로타리 타정기(MRC-33: 세종)를 사용하여 타정하였다. 타정이 완료된 정제를 하이코터(SFC-30N: 세종)에 투입한 후 따로 폴리비닐아세테이트를 정제수 (1정당 360mg)에 용해시켜 제조한 코팅액으로 코팅하여 칸데사르탄 실렉세틸 지연방출성 코팅 내핵정을 제조하였다.
3) 타정 및 코팅
표 IV-1에 나타낸 성분 및 함량을 실시예 IV-7의 3)과 동일한 방법으로 수행하여, 표제의 필름 코팅 유핵정을 제조하였다.
<실시예 IV-9> 유핵정의 제조
1) 심바스타틴 선방출성 과립의 제조
표 IV-1에 나타낸 성분 및 함량을 이용하여 실시예 IV-7의 1) 방법으로 표제의 심바스타틴 선방출성 과립을 제조하였다.
2) 칸데사르탄 실렉세틸의 지연방출성 코팅 내핵정의 제조
표 IV-1에 나타낸 성분 및 함량으로 유당, 옥수수전분, 및 카르복시메틸셀룰로오스 칼슘을 35호체로 체과하고 더블콘믹서로 혼합하였다. 따로 폴리에틸렌글리콜6000을 정제수 (1정당 310mg)에 녹힌 후 칸데사르탄 실렉세틸을 분산시킨 다음 유동층 과립기에 혼합물을 넣고 이 액을 분무하였다. 얻어진 물질에 따로 히드록시프로필셀룰로오스를 정제수 (1정당 20mg)에 녹인 액을 분무하여 과립을 형성한 다음 건조 하였다. 건조가 끝나면 더블콘믹서에 스테아린산 마그네슘을 투입하여 최종혼합을 한 후 로타리 타정기(MRC-33: 세종)를 사용하여 타정하였다. 타정이 완료된 정제를 하이코터(SFC-30N: 세종)에 투입한 후 따로 카르바우나 왁스를 정제수 (1정당 80mg)에 용해시켜 제조한 코팅액으로 코팅한 다음 건조 하였다. 추가적으로 히프로멜로오스를 정제수 (1정당 80mg)에 녹인 액을 코팅액으로 하여 2차 코팅하여 칸데사르탄 실렉세틸 지연방출성 코팅 내핵정을 제조하였다.
3) 타정 및 코팅
표 IV-1에 나타낸 성분 및 함량을 실시예 IV-7의 3)과 동일한 방법으로 수행하여, 표제의 필름 코팅 유핵정을 제조하였다.
<실시예 IV-10> 반투과성막 유핵정 제조
1) 심바스타틴 선 방출성 과립의 제조
표 IV- 1에 기재된 성분 및 함량으로, 실시예 IV- 7의 1)의 동일한 방법에 따라 선방출성 과립을 제조하였다.
2) 칸데사르탄 실렉세틸 지연방출성 반투과성막 코팅 내핵정의 제조
표 IV-1에 기재된 성분 및 함량으로, 칸데사르탄 실렉세틸, 유당, 옥수수전분, 및 카르복시메틸셀룰로오스 칼슘, 염화나트륨을 35호체로 체과하고 고속혼합기로 혼합하였다. 따로 폴리에틸렌글리콜6000, 히드록시프로필셀룰로오스를 정제수 (1정당 300mg)에 녹여 결합액을 제조하고 이를 주성분 혼합물과 함께 고속혼합기에 투입하고 연합하였다. 연합이 끝나면 18호체로 오실레이터를 이용하여 제립하고 이를 온수 건조기를 이용하여 30 ℃에서 건조하였다. 건조가 끝나면 다시 20호체가 장착된 F형 정립기를 사용하여 정립하였다. 이 정립물을 더블콘믹서에 넣고 스테아린산 마그네슘을 넣고 최종혼합 후 로타리 타정기(MRC-33: 세종)를 사용하여 타정하였다. 타정이 완료된 정제에 에틸셀룰로오스를 메틸렌클로라이드와 에탄올의 1:1 (w/w)혼합액 (1정당 200mg)에 용해시켜 제조한 코팅액으로 코팅하여 칸데사르탄 실렉세틸 지연방출성 반투과성막 코팅 내핵정을 제조하였다.
3) 타정 및 코팅
공정2)의 칸데사르탄 실렉세틸 반투과성막 코팅 내핵정을 내핵으로 하고 공정1)의 심바스타틴을 포함하는 조성물을 외층으로 하여 실시예 IV- 7의 3) 공정으로 반투과성막 필름 코팅 유핵정을 제조하였다.
<실시예 IV-11> 캡슐제 제조 (과립-펠렛)
1) 심바스타틴 선방출성 과립의 제조
표 IV- 2의 성분 및 함량으로, 실시예 IV- 1의 1)의 방법에 따라 과립을 제조 한 후 상기 정립물에 부틸레이트 히드록시아니솔을 더블콘믹서에 넣고 최종혼합 하여 심바스타틴 선방출성 과립을 제조하였다.
2) 칸데사르탄 실렉세틸 지연방출성 펠렛의 제조
표 IV-2에 기재된 성분 및 함량으로,시드를 35호체로 체과하고 유동층 과립기(GPCG 1: Glatt)에 투입한 뒤, 따로 에탄올과 정제수 (8:2(v/v)) 혼합용매 (1정당 530mg)에 칸데사르탄 실렉세틸, 히드록시프로필셀룰로오스, 카르복시메틸셀룰로오스 칼슘, 폴리에틸렌글리콜6000을 용해시킨 결합액을 분무하여 칸데사르탄 실렉세틸 함유 펠렛을 형성, 건조하였다. 다시 상기의 과립에 히프로멜로오스 아세테이트 숙시네이트를 에탄올과 염화메틸렌 (1:1(w/w)) 혼합용매 (1정당 760mg)에 녹인 액을 분무하여 칸데사르탄 실렉세틸 지연방출성 펠렛을 제조하였다.
3) 혼합 및 캡슐 충전
표 IV- 2에 기재된 성분 및 함량으로 공정 1)과 2)의 최종 조성물을 더블콘믹서로 혼합하였다. 혼합물에 스테아린산 마그네슘을 투입하고 더블콘믹서로 최종 혼합하였다. 최종 혼합된 혼합물을 분말 공급기에 투입하고 캡슐충전기를 이용하여 제1호의 경질 젤라틴 캡슐에 충전하여 캡슐형태의 제어 방출 제제의 제조를 완료하였다.
<실시예 IV-12> 캡슐제 제조 (팰렛-펠렛)
1) 심바스타틴 선방출성 팰렛의 제조
표 IV- 2에 기재된 성분 및 함량으로,시드를 35호체로 체과하고 유동층 과립기(GPCG 1: Glatt)에 투입한 뒤, 따로 에탄올 (1정당 100mg)에 시트르산, 용해시킨 후 이 용액에 히드록시프로필셀룰로오스과 심바스타틴을 용해시킨 결합액을 분무, 건조하여 심바스타틴 함유 선방출성 펠렛을 제조하였다.
2) 칸데사르탄 실렉세틸 지연방출성 펠렛의 제조
표 IV- 2에 기재된 성분 및 함량으로시드를 35호체로 체과하고 유동층 과립기(GPCG 1: Glatt)에 투입한 뒤, 따로 에탄올과 정제수 (8:2(v/v)) 혼합용매 (1정당 530mg)에 칸데사르탄 실렉세틸, 히드록시프로필셀룰로오스, 카르복시메틸셀룰로오스 칼슘, 폴리에틸렌글리콜6000을 용해시킨 결합액을 분무하여 칸데사르탄 실렉세틸 함유 펠렛을 형성, 건조하였다. 다시 상기의 과립에 아크릴이즈를 정제수 (1정당 500mg)에 녹인 액을 분무하여 칸데사르탄 실렉세틸 지연방출성 펠렛을 제조하였다.
3) 혼합 및 캡슐 충전
표 IV- 2에 기재된 성분 및 함량으로 공정 1)과 2)의 최종 조성물을 더블콘믹서로 혼합하였다. 혼합물에 부틸레이트 히드록시아니솔, 스테아린산 마그네슘을 투입하고 더블콘믹서로 최종 혼합하였다. 최종 혼합된 혼합물을 분말 공급기에 투입하고 캡슐충전기를 이용하여 제1호의 경질 젤라틴 캡슐에 충전하여 캡슐형태의 제어 방출 제제의 제조를 완료하였다.
<실시예 IV-13> 캡슐제 제조 (정제-펠렛)
1) 심바스타틴 선방출성 정제의 제조
표 IV- 2에 기재된 성분 및 함량으로, 실시예 IV- 11의 1)의 방법에 따라 제조한 심바스타틴 선방출성 과립과 스테아린산 마그네슘을 더블콘믹서에 넣고 최종 혼합한 후 로타리 타정기 (MRC-33:세종기계, 한국)를 사용하여 타정하여 선방출성 정제를 제조하였다.
2) 칸데사르탄 실렉세틸 지연방출성 펠렛의 제조
표 IV- 2에 기재된 성분 및 함량으로시드를 35호체로 체과하고 유동층 과립기(GPCG 1: Glatt)에 투입한 뒤, 따로 에탄올과 정제수 (8:2(v/v)) 혼합용매 (1정당 530mg)에 칸데사르탄 실렉세틸, 히드록시프로필셀룰로오스, 카르복시메틸셀룰로오스 칼슘, 폴리에틸렌글리콜6000을 용해시킨 결합액을 분무하여 칸데사르탄 실렉세틸 함유 펠렛을 형성, 건조하였다. 다시 상기의 과립에 폴리비닐아세테이트, 히프로멜로오스를 에탄올과 염화메틸렌 (1:1)(w/w)혼합용매 (1정당 550mg)에 녹인 액을 분무하여 칸데사르탄 실렉세틸 지연방출성 펠렛을 제조하였다.
3) 혼합 및 캡슐 충전
실시예 IV- 11의 3)의 방법에 따라 제조하였다.
<실시예 IV-14> 캡슐제 제조 (과립-과립)
1) 심바스타틴 선방출성 과립의 제조
표 IV- 2에 기재된 성분 및 함량으로 실시예 IV- 11의 1)의 방법에 따라 제조하였다.
2) 칸데사르탄 실렉세틸 지연방출성 과립의 제조
표 IV- 2에 기재된 성분 및 함량으로, 유당, 옥수수전분, 및 카르복시메틸셀룰로오스 칼슘을 35호체로 체과하고 더블콘믹서로 혼합하였다. 따로 폴리에틸렌글리콜6000을 정제수 (1정당 310mg)에 녹인 후 칸데사르탄 실렉세틸을 분산시킨 다음 유동층 과립기에 혼합물을 넣고 이 액을 분무하였다. 얻어진 물질에 따로 히드록시프로필셀룰로오스를 정제수 (1정당 20mg)에 녹인 액을 분무하여 과립을 형성하였다. 따로 히프로멜로오스를 정제수(1정당 500mg)에 분산시킨 후 위에서 형성된 과립에 분무하여 과립을 코팅한 다음 건조 하였다. 건조가 끝나면 다시 20호체가 장착된 F형 정립기를 사용하여 정립하여 칸데사르탄 실렉세틸 지연방출성 과립을 제조하였다.
3) 혼합 및 캡슐 충전
실시예 IV- 11의 3)의 방법에 따라 제조하였다.
<실시예 IV-15> 캡슐제 제조 (팰렛-과립)
1) 심바스타틴 선방출성 팰렛의 제조
표 IV- 2에 기재된 성분 및 함량으로 실시예 IV- 12의 1)의 방법에 따라 제조하였다.
2) 칸데사르탄 실렉세틸 지연방출성 과립의 제조
표 IV- 2에 기재된 성분 및 함량으로,옥수수전분, 및 카르복시메틸셀룰로오스 칼슘, 염화나트륨을 35호체로 체과하고 더블콘믹서로 혼합하였다. 따로 폴리에틸렌글리콜6000을 정제수 (1정당 350mg)에 녹인 후 칸데사르탄 실렉세틸을 분산시킨 다음 유동층 과립기에 혼합물을 넣고 이액을 분무하였다. 얻어진 물질에 따로 히드록시프로필셀룰로오스를 정제수 (1정당 20mg)에 녹인 액을 분무하여 과립을 형성하였다. 따로 에틸셀룰로오스를 메틸렌클로라이드와 에탄올의 1:1(w/w) 혼합액 (1정당 300mg)에 용해시킨 후 위에서 형성된 과립에 분무하여 과립을 코팅한 다음 건조 하였다. 건조가 끝나면 다시 20호체가 장착된 F형 정립기를 사용하여 정립하여 칸데사르탄 실렉세틸 지연방출성 과립을 제조하였다.
3) 혼합 및 캡슐 충전
실시예 IV- 12의 3)의 방법에 따라 제조하였다.
<실시예 IV-16> 캡슐제 제조 (정제-과립)
1) 심바스타틴 선방출성 정제의 제조
표 IV- 2에 기재된 성분 및 함량으로 실시예 IV- 13의 1)의 방법에 따라 제조하였다.
2) 칸데사르탄 실렉세틸 지연방출성 과립의 제조
표 IV- 2에 기재된 성분 및 함량으로 실시예 IV-14의 2)방법 중 히프로멜로오스 이용한 코팅액 대신에, 히프로멜로오스 아세테이트 숙시네이트를 에탄올과 정제수 (8:2(v/v)) 혼합용매 (1정당 500mg)에 용해시킨 코팅액을 조제하여 사용한 것을 제외하고는 실시예 IV- 14의 2)의 방법과 동일하게 수행하여 칸데사르탄 실렉세틸 지연방출성 과립을 제조하였다.
3) 혼합 및 캡슐 충전
실시예 IV- 11의3)의 방법에 따라 제조하였다.
<실시예 IV-17> 캡슐제 제조 (과립-정제)
1) 심바스타틴 선방출성 과립의 제조
표 IV- 2에 기재된 성분 및 함량으로 실시예 IV- 11의 1)의 방법에 따라 제조하였다.
2) 칸데사르탄 실렉세틸 지연방출성 정제의 제조
표 IV- 2에 기재된성분 및 함량으로, 실시예 IV- 7의 2)에 기재된 방법에 따라 칸데사르탄실렉세틸 나정을 타정한다. 타정이 완료된 정제를 하이코터(SFC-30N: 세종)에 투입한 후 따로 카르바우나 왁스를 정제수 (1정당 65mg)에 용해시켜 제조한 코팅액으로 코팅한 다음 건조 하였다. 추가적으로 히프로멜로오스를 정제수 (1정당 65mg)에 녹인 액을 코팅액으로 하여 2차 코팅하여 칸데사르탄 실렉세틸 지연방출성 정제를 제조하였다.
3) 혼합 및 캡슐 충전
실시예 IV- 11의 3)의 방법에 따라 제조하였다.
<실시예 IV-18> 캡슐제 제조 (팰렛-정제)
1) 심바스타틴 선방출성 팰렛의 제조
표 IV- 2에 기재된 성분 및 함량으로 실시예 IV- 12의 1)의 방법에 따라 제조하였다.
2) 칸데사르탄 실렉세틸 지연방출성 정제의 제조
표 IV- 2에 기재된성분 및 함량으로, 실시예 IV- 7의 2)에 기재된 방법에 따라 칸데사르탄실렉세틸 나정을 타정한다. 타정이 완료된 정제를 하이코터(SFC-30N: 세종)에 투입한 후 따로 유드라짓 RL을 정제수 (1정당 40mg)에 용해시켜 제조한 코팅액으로 코팅한 다음 건조 하였다. 추가적으로 아크릴이즈를 정제수 (1정당 150mg)에 녹인 액을 코팅액으로 하여 2차 코팅하여 칸데사르탄 실렉세틸 지연방출성 정제를 제조하였다.
3) 혼합 및 캡슐 충전
실시예 IV- 12의 3)의 방법에 따라 제조하였다.
<실시예 IV-19> 캡슐제 제조 (정제-정제)
1) 심바스타틴 선방출성 정제의 제조
표 IV- 2에 기재된 성분 및 함량으로 실시예 IV- 13의 1)의 방법에 따라 제조하였다.
2) 칸데사르탄 실렉세틸 지연방출성 정제의 제조
표 IV- 2에 기재된 성분 및 함량으로, 실시예 IV- 7의 2)의 방법에 따라 칸데사르탄 실렉세틸 나정을 타정한다. 타정이 완료된 정제를 하이코터(SFC-30N: 세종)에 투입한 후 따로 폴리비닐아세테이트를 에탄올과 염화메틸렌 (1:1(w/w))혼합용매 (1정당 250mg)에 녹인 액을 조제하여 위의 나정을 하이코터(SFC-30N, 세종)에 투입한 후 코팅액으로 코팅하여 칸데사르탄 실렉세틸 지연방출성 정제를 제조하였다.
3) 캡슐 충전
공정 1)과 2)의 최종 조성물을 더블콘믹서로 혼합하였다. 혼합물을 분말 공급기에 투입하고 캡슐충전기를 이용하여 충전하여 캡슐형태의 제어 방출 제제의 제조를 완료하였다.
<실시예 IV- 20> 칸데사르탄 실렉세틸-심바스타틴 블러스터 포장 키트
실시예 IV- 3의 1) 심바스타틴 선방출 과립과 실시예 IV-3의 2) 칸데사르탄 실렉세틸 지연방출 과립 최종 조성물을 혼합하여 타정하는 대신에 각각 로타리 타정기(MRC-33: 세종)를 사용하여 타정하고 블리스터 포장용기에 각각의 정제가 포함되어 동시복용 가능하도록 포장하였다.




<실험예 IV- 1> 비교 용출시험(comparative dissolution profile test)
상기 실시예 IV- 1에 따라 제조된 심바스타틴/칸데사르탄 2상 매트릭스 정제이며 이와 각 성분 단일제 대조약(조코정, 한국 MSD: 심바스타틴 단일제 / 아타칸정, 한국아스트라제네카: 칸데사르탄 단일제)의 비교 용출시험을 실시하였다. 심바스타틴 성분 용출시험의 경우 미국약전 (USP31)에 근거하여 용출시험을 진행하였고, 칸데사르탄 성분 용출 시험의 경우 칸데사르탄이 난용성 물질이므로 계면 활성제 폴리소르베이트 80을 1% 농도로 용출액에 첨가하여 120분을 기점으로 용출액을 인공 위액에서 인공 장액으로 변경하여 총 360분간 용출시험을 진행하였다. 각 성분의 용출시험 방법은 아래와 같으며, 그 결과를 도 18에 나타내었다.
[심바스타틴 선방출 구획 시험방법]
용출시험 근거: 미국약전(USP30)중의 '심바스타틴 정제'항
시험 방법: 패들법(Paddle method), 50회전/분
시험약: pH=7.0 완충액 (조성 = 계면활성제로서 라우릴설페이트 나트륨 0.5% 중량/중량을 함유하는 0.01M 인산이수소나트륨 용액), 900mL
분석 방법: 자외가시부흡광광도법 (검출 파장 = 최대 247, 최소 257nm)
[칸데사르탄 지연방출 구획 시험방법]
용출시험 근거: 대한약전 제 8개정 중 일반시험법의 용출시험법
시험 방법: 패들법, 50회전/분
시험약: 1% 폴리소르베이트80을 함유한 0.01M 염산용액, 750mL (인공위액) / 1% 폴리소르베이트80을 함유한 pH 6.8 인산완충액, 총 1000mL (인공장액)
분석 방법: 자외가시부흡광광도법
도 18에 의하면 상기 용출 시험 시 본 발명의 2상 매트릭스 정제 심바스타틴 성분은 대조 제제인 조코정과 비교하여 거의 동등한 용출특성을 나타내는 것으로 확인되었으나, 칸데사르탄 성분은 대조 제제인 아타칸정과 비교할 때 매우 지연된 용출 속도를 확인 할 수 있다. 칸데사르탄 성분의 용출 시험 결과를 보면, 인공 위액 구간인 120분까지의 칸데사르탄 성분의 용출률은 본 발명의 심바스타틴/칸데사르탄 2상 매트릭스 정제에서 칸데사르탄 총량의 30% 이내이나, 대조 제제는 약 70%임을 확인할 수 있었고, 이후 인공 장액 구간에서 칸데사르탄 성분의 용출률은 대조제제에서 총 150분에 100%이나, 본 발명의 심바스타틴/칸데사르탄 2상 매트릭스 정제에서는 총 150분에 약 40%로 훨씬 느림을 확인할 수 있었다.
<실험예 IV- 2> 비교 용출시험(comparative dissolution profile test)
상기 실시예 IV- 3 내지 IV-6에서 제조된 약제학적 제제(다층정)에서의 칸데사르탄의 비교 용출시험을 실시하였다. 칸데사르탄의 용출시험 방법은 실험예 IV- 1과 같으며, 그 결과를 도 19에 나타내었다.
도 19에 의하면 실험예 IV- 1의 조건에서 용출 시험 시 본 발명의 다층정은 에틸셀룰로오스의 사용량이 증가함에 따라 칸데사르탄 성분이 다소 지연된 용출 속도를 보임을 확인할 수 있었다. 즉, 에틸셀룰로오스를 사용하여 코팅한 본발명의 약제학적 제제에서 칸데사르탄의 용출률은 용출시험 시작후 150분까지 40% 이내였다.
<실험예 IV- 3> 비교 용출시험(comparative dissolution profile test)
상기 실시예 IV- 7에 따라 제조된 심바스타틴/칸데사르탄 유핵정과 각 성분 단일제 대조약(조코정, 한국 MSD: 심바스타틴 단일제 / 아타칸정, 한국 아스트라제네카: 칸데사르탄 단일제)의 비교 용출시험을 실시하였다. 각 성분별 용출시험 방법은 실험예 IV- 1과 같으며, 그 결과를 도 20에 나타내었다.
도 20에 의하면 상기 용출 시험 시 본 발명의 유핵정 중 심바스타틴 성분은 대조 제제인 조코정과 비교하여 거의 동등한 용출특성을 나타내는 것으로 확인되었으나, 칸데사르탄 성분은 대조 제제인 아타칸와 비교할 때 실험예 IV- 1과 같이 매우 지연된 용출 속도를 확인 할 수 있다.
<실험예 IV-4> 비교 용출시험(comparative dissolution profile test)
상기 실시예 IV- 10에 따라 제조된 심바스타틴/칸데사르탄 반투과성막 유핵정과 각 성분 단일제 대조약(조코정, 한국 MSD: 심바스타틴 단일제 / 아타칸정, 한국 아스트라제네카: 칸데사르탄 단일제)의 비교 용출시험을 실시하였다. 각 성분별 용출시험 방법은 실험예 IV- 1과 같으며, 그 결과를 도 21에 나타내었다.
도 21에 의하면 실험예 IV- 1의 조건에서 용출 시험 시 본 발명의 반투과성막 유핵정 중 심바스타틴 성분은 대조 제제인 조코정과 비교하여 거의 동등한 용출특성을 나타내는 것으로 확인되었으나, 칸데사르탄 성분은 대조 제제인 아타칸정과 비교할 때 매우 지연된 용출 속도를 확인 할 수 있다.
<실험예 IV-5> 비교 용출시험(comparative dissolution profile test)
상기 실시예 IV-12, IV-14에 따라 제조된 심바스타틴/칸데사르탄 캡슐제(펠렛-펠렛) 또는 캡슐제(과립-과립)이며 이와 각 성분 단일제 대조약(조코정, 한국 MSD: 심바스타틴 단일제 / 아타칸정, 한국 아스트라제네카: 칸데사르탄 단일제)의 비교 용출시험을 실시하였다. 각 성분별 용출시험 방법은 실험예 IV- 1과 같으며, 그 결과를 도 22와 같이 나타내었다.
도 22에 의하면 실험예 IV- 1의 조건에서 용출 시험 시 본 발명의 캡슐제(펠렛-펠렛),(과립-과립)중 심바스타틴 성분은 대조 제제인 조코정과 비교하여 거의 동등한 용출특성을 나타내는 것으로 확인되었다. 그에 반해, 본 발명의 약제학적 제제에서 칸데사르탄은 대조 제제인 아타칸와 비교할 때 매우 지연된 용출 속도를 확인할 수 있다.
따라서 본 발명의 약제학적 제제는 대조약인 칸데사르탄 단일제와 심바스타틴 단일제를 동시 복용하였을 경우의 용출 양상과는 달리 칸데사르탄의 초기 방출이 심바스타틴보다 매우 느리기 때문에 심바스타틴이 먼저 간에서 대사를 받은 후 대사 관련 효소인 사이토크롬 P450이 재생될 시간을 충분히 확보할 수 있는 약제학적 제제이다.
[실시예 및 실험예 V] 로슈바스타틴 및 로자탄 함유 약제학적 제제
<제조예 V- 1> 로슈바스타틴칼슘 선방출성 과립 제조
표 V- 1에 나타난 성분 및 함량으로 로슈바스타틴칼슘(Rosuvastatin calcium, Ranbaxy), 미결정셀룰로오스 (Vivapur 101, JRS), 유당수화물(Lactose 200, DMV), 삼염기칼슘인산염(TCP, Rhodia), 및 크로스포비돈(Kollidon CL)을 혼합하여 혼합물을 제조하였다. 별도로 히드록시프로필셀룰로오스(HPC-L, Hercules)를 정제수(단위제형 당 약 20mg 상당)에 녹인 후 위 혼합물과 연합한 다음 건조하였다. 건조된 과립물을 정립한 후 스테아르산마그네슘(Magnesium stearate, Nof)을 투입한 뒤 혼합하여 로슈바스타틴칼슘 선방출성 과립을 제조하였다.
<제조예 V- 2> 로슈바스타틴칼슘 선방출성 과립 제조
표 V- 1에 나타난 성분 및 함량으로 제조예 V-1과 동일한 방법으로 정제수(단위제형 당 약 70mg 상당)를 사용하여 제조하였다.
<제조예 V- 3> 로슈바스타틴칼슘 선방출성 과립 제조
표 V- 1에 나타난 성분 및 함량으로 제조예 V-1과 동일한 방법으로 정제수(단위제형 당 약 100mg 상당)를 사용하여 제조하였다.
[표V-1]

<제조예 V- 4> 로슈바스타틴칼슘의 선방출성 펠렛 제조
표 V- 2에 나타난 성분 및 함량으로 슈가비드(Non-pareil-101, Freund)에 로슈바스타틴칼슘, 삼염기칼슘인산염, 히드록시프로필셀룰로오스, 및 크로스포비돈을 정제수(단위제형 당 약 800mg 상당)에 분산 및 용해시킨 액을 유동층 과립기(GPCG1, Glatt, 이하 동일기종 사용)를 이용하여 분무하여 코팅한 다음 건조하여 로슈바스타틴칼슘 선방출성 펠렛을 제조하였다.
[표V-2]

<제조예 V- 5> 친수성 고분자 및 장용성 고분자를 포함하는 로자탄칼륨 지연방출성 과립 제조
표 V- 3에 나타난 성분 및 함량으로 로자탄칼륨(Losartan potassium, Cipla), 미결정셀룰로오스, 유당수화물, 전호화전분, 및 전분글리콜산나트륨, 카보머(Carbopol 71G, Lubrizol)을 혼합하여 혼합물을 제조하였다. 별도로 히드록시프로필셀룰로오스를 정제수(단위제형 당 약 20mg 상당)에 녹인 후 이 액을 위 로자탄칼륨, 미결정셀룰로오스, 유당수화물, 전호화전분, 및 전분글리콜산나트륨 혼합물에 유동층 과립기를 이용하여 분무하여 과립을 형성하였다. 따로 히프로멜로오스아세테이트숙시네이트(HPMC-AS LF, Shinetsu)를 80%에탄올(단위제형 당 약 1200mg 상당)에 용해시킨 후 위에서 형성된 과립에 분무하여 과립을 코팅한 다음 건조 하였다. 상기 과립물에 스테아르산마그네슘 투입한 뒤 혼합하여 로자탄칼륨 지연방출성 과립을 제조하였다.
<제조예 V- 6> 친수성 고분자 및 장용성 고분자를 포함하는 로자탄칼륨 지연방출성 정제 제조
표 V- 3에 나타난 성분 및 함량으로 로자탄칼륨, 미결정셀룰로오스, 유당수화물, 전호화전분, 및 전분글리콜산나트륨을 혼합하여 혼합물을 제조하였다. 별도로 히드록시프로필셀룰로오스를 정제수(단위제형 당 약 20mg 상당)에 녹인 후 위 혼합물과 연합한 다음 건조하였다. 건조된 과립물을 정립한 후 카보머, 스테아르산마그네슘을 투입한 뒤 혼합하여 직경 6.0 mm 펀치가 장착된 로타리 타정기(MRC-33, 세종기계, 이하 동일기종 사용)에서 타정하였다. 타정이 완료된 정제에 히프로멜로오스아세테이트숙시네이트를 80%에탄올(단위제형 당 약 300mg 상당)에 용해시켜 제조한 코팅액으로 코팅기(SFC-30F, 세종기계, 이하 동일기종 사용)를 사용하여 로자탄칼륨 지연방출성 정제를 제조하였다.
<제조예 V- 7> 친수성 고분자 및 장용성 고분자 드라이코팅을 포함하는 로자탄 지연방출성 정제 제조
표 V- 3에 나타난 성분 및 함량으로 로자탄칼륨, 미결정셀룰로오스, 유당수화물, 전호화전분, 및 전분글리콜산나트륨을 혼합하여 혼합물을 제조하였다. 별도로 히드록시프로필셀룰로오스, 카보머를 정제수(단위제형 당 약 20mg 상당)에 녹인 후 위 혼합물과 연합한 다음 건조하였다. 건조된 과립물을 정립한 후 스테아르산마그네슘을 투입한 뒤 혼합하여 직경 6.0 mm 펀치가 장착된 로타리 타정기에서 타정하였다. 타정이 완료된 정제를 내핵으로 하고 히프로멜로오스아세테이트숙시네이트, 미결정셀룰로오스(Vivapur102, JRS), 및 스테아르산마그네슘의 혼합물과 함께 11 mm 펀치가 장착된 유핵정타정기(RUD-1, Kilian, 이하 동일기종 사용)에서 타정하여 드라이코팅 된 로자탄 지연방출성 유핵정을 제조하였다.
[표V-3]

<제조예 V- 8> 수불용성 중합체를 포함하는 로자탄칼륨 지연방출성 과립 제조
표 V- 4에 나타난 성분 및 함량으로 로자탄칼륨, 미결정셀룰로오스, 유당수화물, 전호화전분, 및 전분글리콜산나트륨을 혼합하여 혼합물을 제조하였다. 별도로 히드록시프로필셀룰로오스를 정제수(단위제형 당 약 20mg 상당)에 녹인 후 이 액을 위 로자탄칼륨, 미결정셀룰로오스, 유당수화물, 전호화전분, 및 전분글리콜산나트륨 혼합물에 유동층 과립기를 이용하여 분무하여 과립을 형성하였다. 따로 콜리코트 SR30D (Kollicoat SR 30D, 폴리비닐아세테이트 27%, 포비돈 2.7%, 소디움라우릴설페이트 0.3%, 및 정제수 70%의 조성, Basf)를 위에서 형성된 과립에 분무하여 과립을 코팅한 다음 건조 하였다. 상기 과립물에 스테아르산마그네슘 투입한 뒤 혼합하여 로자탄칼륨 지연방출성 과립을 제조하였다.
<제조예 V- 9> 수불용중합체를 포함하는 로자탄칼륨 지연방출성 정제 제조
표 V- 4에 나타난 성분 및 함량으로 로자탄칼륨, 미결정셀룰로오스, 유당수화물, 전호화전분, 및 전분글리콜산나트륨을 혼합하여 혼합물을 제조하였다. 별도로 히드록시프로필셀룰로오스를 정제수(단위제형 당 약 18mg 상당)에 녹인 후 위 혼합물과 연합한 다음 건조하였다. 건조된 과립물을 정립한 후 스테아르산마그네슘을 투입한 뒤 혼합하여 직경 6.0 mm 펀치가 장착된 로타리 타정기에서 타정하였다. 타정이 완료된 정제에 콜리코트 SR30D로 코팅하여 로자탄칼륨 지연방출성 정제를 제조하였다.
[표V-4]

<제조예 V- 10> 소수성화합물 및 친수성고분자를 포함하는 로자탄칼륨 지연방출성 과립 제조
표 V- 5에 나타난 성분 및 함량으로 로자탄칼륨, 미결정셀룰로오스, 유당수화물, 전호화전분, 및 전분글리콜산나트륨을 혼합하여 혼합물을 제조하였다. 별도로 히드록시프로필셀룰로오스를 정제수(단위제형 당 약 20mg 상당)에 녹인 후 이 액을 위 로자탄칼륨, 미결정셀룰로오스, 유당수화물, 전호화전분, 및 전분글리콜산나트륨 혼합물에 유동층 과립기를 이용하여 분무하여 과립을 형성하였다. 따로 카르나우바왁스(Cavawax W6, ISP), 히프로멜로오스(Pharmacoat 603, Shinetsu) 및 폴리에틸렌글리콜6000(PEG6000, Duksan)을 정제수(단위제형 당 약 1000mg 상당)에 분산시킨 후 위에서 형성된 과립에 분무하여 과립을 코팅한 다음 건조 하였다. 상기 과립물에 스테아르산마그네슘 투입한 뒤 혼합하여 로자탄칼륨 지연방출성 과립을 제조하였다.
<제조예 V- 11> 소수성화합물 및 친수성고분자를 포함하는 로자탄칼륨 지연방출성 정제 제조
표 V- 5에 나타난 성분 및 함량으로 로자탄칼륨, 미결정셀룰로오스, 유당수화물, 전호화전분, 및 전분글리콜산나트륨을 혼합하여 혼합물을 제조하였다. 별도로 히드록시프로필셀룰로오스를 정제수(단위제형 당 약 24mg 상당)에 녹인 후 위 혼합물과 연합한 다음 건조하였다. 건조된 과립물을 정립한 후 스테아르산마그네슘을 투입한 뒤 혼합하여 직경 6.0 mm 펀치가 장착된 로타리 타정기에서 타정하였다. 타정이 완료된 정제에 카르나우바왁스, 히프로멜로오스, 및 폴리에틸렌글리콜6000을 정제수(단위제형 당 약 300mg 상당)에 분산시켜 제조한 코팅액으로 코팅하여 로자탄칼륨 지연방출성 정제를 제조하였다.
[표V-5]

<제조예 V- 12> 친수성고분자를 포함하는 로자탄칼륨 지연방출성 과립 제조
표 V- 6에 나타난 성분 및 함량으로 로자탄칼륨, 미결정셀룰로오스, 유당수화물, 전호화전분, 및 전분글리콜산나트륨을 혼합하여 혼합물을 제조하였다. 별도로 히드록시프로필셀룰로오스를 정제수(단위제형 당 약 20mg 상당)에 녹인 후 이 액을 위 로자탄칼륨, 미결정셀룰로오스, 유당수화물, 전호화전분, 및 전분글리콜산나트륨 혼합물에 유동층 과립기를 이용하여 분무하여 과립을 형성하였다. 따로 히드록시프로필셀룰로오스, 및 폴리에틸렌글리콜6000을 정제수(단위제형 당 약 1000mg 상당)에 녹인 후 위에서 형성된 과립에 분무하여 과립을 코팅한 다음 건조 하였다. 상기 과립물에 스테아르산마그네슘 투입한 뒤 혼합하여 로자탄칼륨 지연방출성 과립을 제조하였다.
<제조예 V- 13> 친수성고분자를 포함하는 로자탄칼륨 지연방출성 정제 제조
표 V- 6에 나타난 성분 및 함량으로 로자탄칼륨, 미결정셀룰로오스, 유당수화물, 전호화전분, 및 전분글리콜산나트륨을 혼합하여 혼합물을 제조하였다. 별도로 히드록시프로필셀룰로오스를 정제수(단위제형 당 약 20mg 상당)에 녹인 후 위 혼합물과 연합한 다음 건조하였다. 건조된 과립물을 정립한 후 스테아르산마그네슘을 투입한 뒤 혼합하여 직경 6.0 mm 펀치가 장착된 로타리 타정기에서 타정하였다. 타정이 완료된 정제에 히드록시프로필셀룰로오스 및 폴리에틸렌글리콜6000(단위제형 당 약 360mg 상당)을 정제수에 녹여 제조한 코팅액으로 코팅하여 로자탄칼륨 지연방출성 정제를 제조하였다.
[표V-6]

<제조예 V- 14> 반투과성막 코팅기제 및 삼투압 조절제를 포함하는 로자탄칼륨 지연방출성 과립 제조
표 V- 7에 나타난 성분 및 함량으로 로자탄칼륨, 미결정셀룰로오스, 유당수화물, 전호화전분, 전분글리콜산나트륨 및 염화나트륨을 혼합하여 혼합물을 제조하였다. 별도로 히드록시프로필셀룰로오스(단위제형 당 약 20mg 상당)를 정제수에 녹인 후 이 액을 위 로자탄칼륨, 미결정셀룰로오스, 유당수화물, 전호화전분, 및 전분글리콜산나트륨 혼합물에 유동층 과립기를 이용하여 분무하여 과립을 형성하였다. 따로 에틸셀룰로오스(Ethocel, Colorcon)를 메틸렌클로라이드와 에탄올의 1:1 혼합액(단위제형 당 약 600mg 상당)에 용해시킨 후 위에서 형성된 과립에 분무하여 과립을 코팅한 다음 건조 하였다. 상기 과립물에 스테아르산마그네슘 투입한 뒤 혼합하여 로자탄칼륨 지연방출성 과립을 제조하였다.
<제조예 V- 15> 반투과성막 코팅기제 및 삼투압 조절제를 포함하는 로자탄칼륨 지연방출성 정제 제조
표 V- 7에 나타난 성분 및 함량으로 로자탄칼륨, 미결정셀룰로오스, 유당수화물, 전호화전분, 전분글리콜산나트륨 및 염화나트륨을 혼합하여 혼합물을 제조하였다. 별도로 히드록시프로필셀룰로오스를 정제수(단위제형 당 약 18mg 상당)에 녹인 후 위 혼합물과 연합한 다음 건조하였다. 건조된 과립물을 정립한 후 스테아르산마그네슘을 투입한 뒤 혼합하여 직경 6.0 mm 펀치가 장착된 로타리 타정기에서 타정하였다. 타정이 완료된 정제에 에틸셀룰로오스를 메틸렌클로라이드와 에탄올의 1:1 혼합액(단위제형 당 약 240mg 상당)에 용해시켜 제조한 코팅액으로 코팅하여 로자탄칼륨 지연방출성 정제를 제조하였다.
[표V-7]

<제조예 V- 16> 장용성고분자를 포함하는 로자탄칼륨의 지연방출성 펠렛 제조
표 V- 8에 나타난 성분 및 함량으로 슈가비드에 로자탄칼륨, 히드록시프로필셀룰로오스, 및 전분글리콜산나트륨을 정제수에 분산 및 용해시킨 액을 유동층 과립기를 이용하여 분무하여 코팅한다. 별도로 히프로멜로오스아세테이트숙시네이트를 80%에탄올(단위제형 당 약 750mg 상당)에 용해시켜 제조한 액을 위 비드에 다시 분무하여 코팅한 후 건조하여 로자탄칼륨의 지연방출성 펠렛을 제조하였다.
[표V-8]

<실시예 V-1> 로슈바스타틴칼슘 - 로자탄칼륨 2상의 매트릭스 정제 제조
상기 제조예 V- 2의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로 10mg 해당량)과 제조예 V- 5의 장용성고분자를 포함하는 로자탄칼륨 지연방출성 과립(로자탄칼륨으로 50mg 해당량)을 혼합한 다음 직경 10 mm 펀치가 장착된 로타리 타정기로 타정하였다. 타정이 완료된 2상의 메트릭스 정제를 표 V-9와 같이 히프로멜로오스, 폴리에틸렌글리콜6000, 산화티탄(Fanglian, JHP)으로 정제수(단위제형 당 약 300mg 상당)를 이용하여 코팅하여 2상 매트릭스 정제를 제조하였다.
[표V-9]

<실시예 V-2> 로슈바스타틴칼슘을 코팅층에 함유한 로자탄칼륨 필름코팅정의 제조
상기 제조예 V- 9의 수불용중합체를 포함하는 로자탄칼륨 지연방출성 정제에 표 V-10에 나타난 성분 및 함량으로 정제수(단위제형 당 약 800mg 상당)를 이용하여 코팅하여 로슈바스타틴칼슘을 코팅층에 함유한 필름코팅정 정제를 제조하였다.
[표V-10]

<실시예 V-3> 로슈바스타틴칼슘을 코팅층에 함유한 로자탄 유핵필름코팅정의 제조
상기 제조예 V- 7의 장용성고분자를 드라이코팅으로 포함하는 로자탄 지연방출성 유핵정에 표 V-10에 나타난 성분 및 함량으로 정제수(단위제형 당 약 800mg 상당)를 이용하여 코팅하여 로슈바스타틴칼슘을 코팅층에 함유한 유핵필름코팅정을 제조하였다.
<실시예 V-4> 로슈바스타틴칼슘 - 로자탄칼륨 2층정 제조
상기 제조예 V- 2의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로 10mg 해당량)과 제조예 V- 8의 수불용중합체를 포함하는 로자탄칼륨 지연방출성 과립(로자탄칼륨으로 50mg 해당량)을 직경 10 mm 펀치가 장착된 로타리 삼중정 타정기의 다른 과립 주입구에 각각 넣고 타정하여 2층정 제조하였다. 타정이 완료된 정제를 표 V-9에 나타난 성분 및 함량으로 정제수(단위제형 당 약 300mg 상당)를 이용하여 코팅하였다.
<실시예 V-5> 로슈바스타틴칼슘 - 로자탄칼륨 다층정 제조
상기 제조예 V- 2의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로 10mg 해당량)을 1층 및 3층으로 1/2씩 분할투입하고 제조예 V- 10의 소수성화합물과 친수성고분자를 포함하는 로자탄칼륨 지연방출성 과립(로자탄칼륨으로 50mg 해당량)을 중간층(2번째층)으로 하여 직경 10 mm 펀치가 장착된 로타리 삼중정 타정기의 다른 과립 주입구에 각각 넣고 타정하여 다층정을 제조하였다. 타정이 완료된 정제를 표 V-9에 나타난 성분 및 함량으로 정제수(단위제형 당 약 300mg 상당)를 이용하여 코팅하였다.
<실시예 V-6> 로슈바스타틴칼슘 - 로자탄칼륨 유핵정 제조
상기 제조예 V- 6의 장용성고분자를 포함하는 로자탄칼륨 지연방출성 정제를 내핵으로 하여 제조예 V- 3의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로 10mg 해당량)과 함께 11 mm 펀치가 장착된 유핵정타정기에서 타정하여 유핵정을 제조하였다. 타정이 완료된 정제를 표 V-9에 나타난 성분 및 함량으로 정제수(단위제형 당 약 300mg 상당)를 이용하여 코팅하였다.
<실시예 V-7> 로슈바스타틴칼슘 - 로자탄 유핵2층정 제조
상기 제조예 V- 7의 장용성고분자를 드라이코팅으로 포함하는 로자탄 지연방출성 유핵정의 타정시 제조예 V-1의 로슈바스타틴 선방출성 과립(로슈바스타틴으로 10mg 해당량)을 또 다른 주입구로 가하여 2층정 중 한 층이 유핵정이 되도록 타정하여 유핵2층정을 제조하였다. 타정이 완료된 정제를 표 V-9에 나타난 성분 및 함량으로 정제수(단위제형 당 약 300mg 상당)를 이용하여 코팅하였다.
<실시예 V-8> 로슈바스타틴칼슘 - 로자탄칼륨 캡슐제(정제+정제) 제조
상기 제조예 V- 1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로 10mg 해당량)을 6 mm 펀치가 장착된 로타리 타정기에서 타정한 후 그 정제를, 제조예 V- 13의 친수성고분자를 포함하는 로자탄칼륨 지연방출성 정제와 함께 0호 캡슐에 충전하여 캡슐제를 제조하였다.
<실시예 V-9> 로슈바스타틴칼슘 - 로자탄칼륨 캡슐제(과립+정제) 제조
상기 제조예 V- 1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로 10mg 해당량)을 제조예 V- 11의 소수성화합물과 친수성고분자를 포함하는 로자탄칼륨 지연방출성 정제와 함께 0호 캡슐에 충전하여 캡슐제를 제조하였다.
<실시예 V-10> 로슈바스타틴칼슘 - 로자탄칼륨 캡슐제(정제+과립) 제조
상기 제조예 V- 1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로 10mg 해당량)을 6 mm 펀치가 장착된 로타리 타정기에서 타정한 후 그 정제를, 제조예 V- 12의 친수성고분자를 포함하는 로자탄칼륨 지연방출성 과립(로자탄칼륨으로 50mg 해당량)과 함께 0호 캡슐에 충전하여 캡슐제를 제조하였다.
<실시예 V-11> 로슈바스타틴칼슘 - 로자탄칼륨 캡슐제(과립+과립) 제조
상기 제조예 V- 1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로 10mg 해당량)과 제조예 V- 14의 반투과성막 코팅기제와 삼투압 조절제를 포함하는 로자탄칼륨 지연방출성 과립(로자탄칼륨으로 50mg 해당량)과 함께 0호 캡슐에 충전하여 캡슐제를 제조하였다.
<실시예 V-12> 로슈바스타틴칼슘 - 로자탄칼륨 캡슐제(펠렛+정제) 제조
상기 제조예 V- 4의 로슈바스타틴칼슘 선방출성 펠렛(로슈바스타틴으로 10mg 해당량)과 제조예 V- 15의 반투과성막 코팅기제와 삼투압 조절제를 포함하는 로자탄칼륨 지연방출성 정제와 함께 0호 캡슐에 충전하여 캡슐제를 제조하였다.
<실시예 V-13> 로슈바스타틴칼슘 - 로자탄칼륨 캡슐제(펠렛+과립) 제조
상기 제조예 V- 4의 로슈바스타틴칼슘 선방출성 펠렛(로슈바스타틴으로 10mg 해당량)과 제조예 V- 5의 장용성고분자를 포함하는 로자탄칼륨 지연방출성 과립(로자탄칼륨으로 50mg 해당량)과 함께 0호 캡슐에 충전하여 캡슐제를 제조하였다.
<실시예 V-14> 로슈바스타틴칼슘 - 로자탄칼륨 캡슐제(정제+펠렛) 제조
상기 제조예 V- 1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로 10mg 해당량)을 6 mm 펀치가 장착된 로타리 타정기에서 타정한 후 그 정제를, 제조예 V- 16의 장용성고분자를 포함하는 로자탄칼륨 지연방출성 펠렛(로자탄칼륨으로 50mg 해당량)과 함께 0호 캡슐에 충전하여 캡슐제를 제조하였다.
<실시예 V-15> 로슈바스타틴칼슘 - 로자탄칼륨 캡슐제(과립+펠렛) 제조
상기 제조예 V- 1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로 10mg 해당량)과 제조예 V- 16의 장용성고분자를 포함하는 로자탄칼륨 지연방출성 펠렛(로자탄칼륨으로 50mg 해당량)과 함께 0호 캡슐에 충전하여 캡슐제를 제조하였다.
<실시예 V-16> 로슈바스타틴칼슘 - 로자탄칼륨 캡슐제(과립+캡슐) 제조
상기 제조예 V- 5의 장용성고분자를 포함하는 로자탄칼륨 지연방출성 과립(로자탄칼륨으로 50mg 해당량)을 2호 캡슐에 충전하고 제조예 V- 1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로 10mg 해당량)과 함께 0호 캡슐에 충전하여 캡슐제를 제조하였다.
<실시예 V-17> 로슈바스타틴칼슘 - 로자탄칼륨 캡슐제(과립+지연방출성 캡슐) 제조
표 V- 11에 나타난 성분 및 함량으로 로자탄칼륨, 유당수화물, 전분글리콜산나트륨, 및 이산화규소를 2호 캡슐에 충전한 다음 히프로멜로오스아세테이트숙시네이트를 80%에탄올(단위제형 당 약 450mg 상당)에 용해시켜 제조한 코팅액을 이용하여 코팅한다. 장용성고분자로 코팅된 캡슐을 제조예 V- 1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로 10mg 해당량)과 함께 0호 캡슐에 충전하여 캡슐제를 제조하였다.
[표V-11]

<실시예 V-18> 로슈바스타틴칼슘 - 로자탄칼륨 블리스터 포장 키트의 제조
상기 제조예 V- 1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로 10mg 해당량)을 6 mm 펀치가 장착된 로타리 타정기에서 타정하고 제조예 V- 6의 장용성고분자를 포함하는 로자탄칼륨 지연방출성 정제(로자탄칼륨으로 50mg 해당량)와 함께 하나의 PTP(Press Through Pack)포장용기에 포장하여 동시복용이 가능한 포장키트를 제조하였다.
<실험예 V- 1> 용출 양상 시험1 (dissolution profile test)
상기 실시예 V-2, 및 실시예 V-4에서 얻은 정제와 실시예 V-8에서 얻은 캡슐제를 대조제제로 코자정(MSD : 로자탄칼륨 단일제)와 크레스토정(Crestor : 로슈바스타틴칼슘 단일제)을 사용하여 아래 조건에 따라 비교 용출시험을 실시하였다.
생체의 약물의 흡수경로와 유사하게 위의 산성조건은 0.1N 염산용액 및 장관 조건은 pH 6.8(대한약전)액으로 설정하여 시험을 진행하였으며 위 체류시간을 고려하여 0.1N HCl액에서 2시간동안 용출을 진행하고 이후 인산나트륨용액 및 수산화나트륨 또는 염산용액을 추가하여 pH 6.8이 되도록 조절한 후 이후 시험을 진행하는 방법으로 용출시험을 진행하였다.
[로자탄 시험방법]
용출시험 근거: 대한약전 제 8 개정 중 일반시험법의 용출시험법
시험 방법: 패들법(Paddle method), 50 회전/분
시험액: 0.1N 염산용액 750 mL (0~2시간), 0.1N 염산용액 750 mL과 0.2 mol/L 인산나트륨용액 250mL 및 필요에 따라 1 mol/L 수산화나트륨용액 또는 2 mol/L 염산용액으로 pH6.8을 맞춘 용액 (2시간 이후)
분석방법: HPLC-UV (검출파장 = 최대 254 nm)
[로슈바스타틴 시험방법]
용출시험 근거: 대한약전 제 8 개정 중 일반시험법의 용출시험법
시험 방법: 패들법(Paddle method), 50회전/분
시험액: 0.1 N-염산용액(산성환경)), 1000 mL
분석방법: HPLC-UV (검출파장 = 244 nm)
용출시험결과 다음 도 23과 같은 결과를 얻을 수 있었으며 실시예 V-2, V-4, V-8의 로슈바스타틴 성분은 대조 제제인 크레스토정과 비교하여 동등한 용출특성을 나타냈다. 하지만 로자탄 성분은 대조 제제인 코자정과 비교할 때 120분~180분의 방출지연시간을 가짐을 확인할 수 있었다.
<실험예 V- 2> 용출 양상 시험2 (dissolution profile test)
상기 실시예 V-6에서 얻은 정제와 실시예 V-15, 및 실시예 V-17에서 얻은 캡슐제의 용출시험을 실시하였으며 용출시험 조건 및 분석조건은 실험예 V-1과 동일하게 진행하였다.
용출시험결과 다음 도24와 같은 결과를 얻을 수 있었으며 실시예 V-6, V-15, 및 V-17의 로슈바스타틴 성분은 대조 제제인 크레스토정과 비교하여 동등한 용출특성을 나타냈다. 하지만 로자탄 성분은 대조 제제인 코자정과 비교할 때 120분~180분의 방출지연시간을 가짐을 확인할 수 있었다.
[실시예 및 실험예 VI] 로슈바스타틴 및 이사베르탄 함유 약제학적 제제
<제조예 VI- 1> 로슈바스타틴칼슘 선방출성 과립 제조
표 VI- 1에 나타난 성분 및 함량으로 로슈바스타틴칼슘, 미결정셀룰로오스, 유당수화물, 삼염기칼슘인산염, 및 크로스포비돈을 혼합하여 혼합물을 제조하였다. 별도로 히드록시프로필셀룰로오스를 정제수에 녹인 후 위 혼합물과 스피드믹서(Lab. Pharma Mixer, Diosna, 이하 동일기종 사용)를 사용하여 연합한 다음 건조하였다. 건조된 과립물을 정립한 후 스테아르산마그네슘을 투입한 뒤 혼합하여 로슈바스타틴칼슘 선방출성 과립을 제조하였다.
<제조예 VI- 2> 로슈바스타틴칼슘 선방출성 과립 제조
표 VI- 1에 나타난 성분 및 함량으로 제조예 VI-1과 동일한 방법으로 제조하였다.
<제조예 VI- 3> 로슈바스타틴칼슘 선방출성 과립 제조
표 VI- 1에 나타난 성분 및 함량으로 제조예 VI-1과 동일한 방법으로 제조하였다.
[표VI-1]

<제조예 VI- 4> 로슈바스타틴칼슘의 선방출성펠렛 제조
표 VI- 2에 나타난 성분 및 함량으로 슈가비드에 로슈바스타틴칼슘, 삼염기칼슘인산염, 히드록시프로필셀룰로오스, 및 크로스포비돈을 정제수에 10%(v/v)가 되도록 분산 및 용해시킨 액을 유동층 과립기(GPCG 1, Glatt, 이하 동일기종 이용)를 이용하여 분무하여 코팅한 다음 건조하여 로슈바스타틴칼슘 선방출성 펠렛을 제조하였다.
[표VI-2]

<제조예 VI- 5> 장용성 고분자를 이용한 이베사르탄 지연방출성 과립 제조
표 VI- 3에 나타난 성분 및 함량으로 이베사르탄, 유당수화물, 미결정셀룰로오스, 전호화전분, 및 크로스카멜로스나트륨을 혼합하여 혼합물을 제조하였다. 별도로 폴록사머188을 정제수에 녹인 후 이 액을 위 이베사르탄, 유당수화물, 미결정셀룰로오스, 전호화전분, 및 크로스카멜로스나트륨 혼합물에 유동층 과립기를 이용하여 분무하여 과립을 형성하였다. 따로 히프로멜로오스아세테이트숙시네이트를 80%(v/v)에탄올에 8%(w/w)가 되도록 용해시킨 후 위에서 형성된 과립에 분무하여 과립을 코팅한 다음 건조하였다. 상기 과립물에 스테아르산마그네슘 투입한 뒤 혼합하여 이베사르탄 지연방출성 과립을 제조하였다.
<제조예 VI- 6> 장용성 고분자를 이용한 이베사르탄 지연방출성 정제 제조
표 VI- 3에 나타난 성분 및 함량으로 이베사르탄, 유당수화물, 미결정셀룰로오스, 전호화전분, 및 크로스카멜로스나트륨을 혼합하여 혼합물을 제조하였다. 별도로 폴록사머188을 정제수에 녹인 후 위 혼합물과 연합 한 다음 건조하였다. 건조된 과립물을 정립한 후 이산화규소, 및 스테아르산마그네슘을 투입한 뒤 혼합하여 직경 6.0 mm 펀치가 장착된 로타리 타정기(MRC-33, Sejong 이하 동일기종 사용)에서 타정하였다. 타정이 완료된 정제에 히프로멜로오스아세테이트숙시네이트를 80%에탄올에 8%(w/w)가 되도록 용해시켜 제조한 액을 가지고 코팅기(SFC-30F, Sejong)를 이용하여 코팅하여 이베사르탄 지연방출성 정제를 제조하였다.
<제조예 VI- 7> 장용성 고분자를 드라이코팅으로 이용한 이베사르탄 지연방출성 정제 제조
표 VI- 3에 나타난 성분 및 함량으로 이베사르탄, 유당수화물, 미결정셀룰로오스, 전호화전분, 및 크로스카멜로스나트륨을 혼합하여 혼합물을 제조하였다. 별도로 폴록사머188을 정제수에 녹인 후 위 혼합물과 연합한 다음 건조하였다. 건조된 과립물을 정립한 후 이산화규소, 및 스테아르산마그네슘을 투입한 뒤 혼합하여 직경 6.0 mm 펀치가 장착된 로타리 타정기에서 타정하였다. 타정이 완료된 정제를 내핵으로 하고 히프로멜로오스아세테이트숙시네이트, 미결정셀룰로오스, 및 스테아르산마그네슘의 혼합물과 함께 11 mm 펀치가 장착된 유핵정타정기에서 타정하여 드라이코팅된 이베사르탄 지연방출성 유핵정을 제조하였다.
[표VI-3]

<제조예 VI- 8> 수불용성 중합체를 이용한 이베사르탄 지연방출성 과립 제조
표 VI- 4에 나타난 성분 및 함량으로 이베사르탄, 유당수화물, 미결정셀룰로오스, 전호화전분, 및 크로스카멜로스나트륨을 혼합하여 혼합물을 제조하였다. 별도로 폴록사머188을 정제수에 녹인 후 이 액을 위 이베사르탄, 유당수화물, 미결정셀룰로오스, 전호화전분, 및 크로스카멜로스나트륨 혼합물에 유동층 과립기를 이용하여 분무하여 과립을 형성하였다. 따로 콜리코트 SR30D(Kollicoat SR 30D, 폴리비닐아세테이트 27%, 포비돈 2.7%, 소디움라우릴설페이트 0.3%, 및 정제수 70%, Basf)를 위에서 형성된 과립에 분무하여 과립을 코팅한 다음 건조 하였다. 상기 과립물에 스테아르산마그네슘 투입한 뒤 혼합하여 이베사르탄 지연방출성 과립을 제조하였다.
<제조예 VI- 9> 수불용성 중합체를 이용한 이베사르탄 지연방출성 정제 제조
표 VI- 4에 나타난 성분 및 함량으로 이베사르탄, 유당수화물, 미결정셀룰로오스, 전호화전분, 및 크로스카멜로스나트륨을 혼합하여 혼합물을 제조하였다. 별도로 폴록사머188을 정제수에 녹인 후 위 혼합물과 연합 한 다음 건조하였다. 건조된 과립물을 정립한 후 이산화규소, 및 스테아르산마그네슘을 투입한 뒤 혼합하여 직경 6.0 mm 펀치가 장착된 로타리 타정기에서 타정하였다. 타정이 완료된 정제에 콜리코트 SR30D로 코팅하여 이베사르탄 지연방출성 정제를 제조하였다.
[표VI-4]

*정제수량 제외한 함량 (이하 제조예 VI- 동일)
<제조예 VI- 10> 소수성화합물 및 친수성고분자를 이용한 이베사르탄 지연방출성 과립 제조
표 VI- 5에 나타난 성분 및 함량으로 이베사르탄, 유당수화물, 미결정셀룰로오스, 전호화전분, 및 크로스카멜로스나트륨을 혼합하여 혼합물을 제조하였다. 별도로 폴록사머188을 정제수에 녹인 후 이 액을 위 이베사르탄, 유당수화물, 미결정셀룰로오스, 전호화전분, 및 크로스카멜로스나트륨 혼합물에 유동층 과립기를 이용하여 분무하여 과립을 형성하였다. 따로 카르나우바왁스, 히프로멜로오스, 및 폴리에틸렌글리콜6000을 정제수에 10%(v/v)가 되도록 분산시킨 후 위에서 형성된 과립에 분무하여 과립을 코팅한 다음 건조 하였다. 상기 과립물에 스테아르산마그네슘을 투입한 뒤 혼합하여 이베사르탄 지연방출성 과립을 제조하였다.
<제조예 VI- 11> 소수성화합물 및 친수성고분자를 이용한 이베사르탄 지연방출성 정제 제조
표 VI- 5에 나타난 성분 및 함량으로 이베사르탄, 유당수화물, 미결정셀룰로오스, 전호화전분, 및 크로스카멜로스나트륨을 혼합하여 혼합물을 제조하였다. 별도로 폴록사머188을 정제수에 녹인 후 위 혼합물과 연합 한 다음 건조하였다. 건조된 과립물을 정립한 후 이산화규소, 및 스테아르산마그네슘을 투입한 뒤 혼합하여 직경 6.0 mm 펀치가 장착된 로타리 타정기에서 타정하였다. 타정이 완료된 정제에 카르나우바왁스, 히프로멜로오스, 및 폴리에틸렌글리콜6000을 정제수에 10%(v/v)가 되도록 분산시켜 제조한 코팅액으로 코팅하여 이베사르탄 지연방출성 정제를 제조하였다.
[표VI-5]

<제조예 VI- 12> 친수성고분자를 이용한 이베사르탄 지연방출성 과립 제조
표 VI- 6에 나타난 성분 및 함량으로 이베사르탄, 유당수화물, 미결정셀룰로오스, 전호화전분, 및 크로스카멜로스나트륨을 혼합하여 혼합물을 제조하였다. 별도로 폴록사머188을 정제수에 녹인 후 이 액을 위 이베사르탄, 유당수화물, 미결정셀룰로오스, 전호화전분, 및 크로스카멜로스나트륨 혼합물에 유동층 과립기를 이용하여 분무하여 과립을 형성하였다. 따로 히드록시프로필셀룰로오스, 및 폴리에틸렌글리콜6000을 정제수에 10%(v/v)가 되도록 녹인 후 위에서 형성된 과립에 분무하여 과립을 코팅한 다음 건조 하였다. 상기 과립물에 스테아르산마그네슘 투입한 뒤 혼합하여 이베사르탄 지연방출성 과립을 제조하였다.
<제조예 VI- 13> 친수성고분자를 이용한 이베사르탄 지연방출성 정제 제조
표 VI- 6에 나타난 성분 및 함량으로 이베사르탄, 유당수화물, 미결정셀룰로오스, 전호화전분, 및 크로스카멜로스나트륨을 혼합하여 혼합물을 제조하였다. 별도로 폴록사머188을 정제수에 녹인 후 위 혼합물과 연합 한 다음 건조하였다. 건조된 과립물을 정립한 후 이산화규소, 및 스테아르산마그네슘을 투입한 뒤 혼합하여 직경 6.0 mm 펀치가 장착된 로타리 타정기에서 타정하였다. 타정이 완료된 정제에 히드록시프로필셀룰로오스 및 폴리에틸렌글리콜6000을 정제수에 10%(v/v)가 되도록 녹여 제조한 코팅액으로 코팅하여 이베사르탄 지연방출성 정제를 제조하였다.
[표VI-6]

<제조예 VI- 14> 반투과성막 코팅기제 및 삼투압 조절제를 이용한 이베사르탄 지연방출성 과립 제조
표 VI- 7에 나타난 성분 및 함량으로 이베사르탄, 유당수화물, 미결정셀룰로오스, 전호화전분, 염화나트륨, 및 크로스카멜로스나트륨을 혼합하여 혼합물을 제조하였다. 별도로 폴록사머188을 정제수에 녹인 후 이 액을 위 이베사르탄, 유당수화물, 미결정셀룰로오스, 전호화전분, 및 크로스카멜로스나트륨 혼합물에 유동층 과립기를 이용하여 분무하여 과립을 형성하였다. 따로 에틸셀룰로오스를 메틸렌클로라이드와 에탄올의 1:1혼합액에 8%(w/w)가 되도록 용해시킨 후 위에서 형성된 과립에 분무하여 과립을 코팅한 다음 건조하였다. 상기 과립물에 스테아르산마그네슘 투입한 뒤 혼합하여 이베사르탄 지연방출성 과립을 제조하였다.
<제조예 VI- 15> 반투과성막 코팅기제 및 삼투압 조절제를 이용한 이베사르탄 지연방출성 정제 제조
표 VI- 7에 나타난 성분 및 함량으로 이베사르탄, 유당수화물, 미결정셀룰로오스, 전호화전분, 염화나트륨, 및 크로스카멜로스나트륨을 혼합하여 혼합물을 제조하였다. 별도로 폴록사머188을 정제수에 녹인 후 위 혼합물과 연합 한 다음 건조하였다. 건조된 과립물을 정립한 후 이산화규소, 및 스테아르산마그네슘을 투입한 뒤 혼합하여 직경 6.0 mm 펀치가 장착된 로타리 타정기에서 타정하였다. 타정이 완료된 정제에 에틸셀룰로오스를 메틸렌클로라이드와 에탄올의 1:1혼합액에 8%(w/w)가 되도록 용해시켜 제조한 코팅액으로 코팅하여 이베사르탄 지연방출성 정제를 제조하였다.
[표VI-7]

<제조예 VI- 16> 장용성고분자를 이용한 이베사르탄의 지연방출성 펠렛 제조
표 VI- 8에 나타난 성분 및 함량으로 슈가비드에 이베사르탄, 폴록사머188, 히드록시프로필셀룰로오스, 및 크로스카멜로스나트륨을 정제수에 분산 및 용해시킨 액을 유동층 과립기를 이용하여 분무하여 코팅한다. 별도로 히프로멜로오스아세테이트숙시네이트를 80%에탄올에 8%(w/w)가 되도록 용해시켜 제조한 액을 위 비드에 다시 분무하여 코팅한 후 건조하여 이베사르탄의 지연방출성 펠렛을 제조하였다.
[표VI-8]

<실시예 VI- 1> 로슈바스타틴칼슘 - 이베사르탄 2상의 메트릭스 정제 제조
상기 제조예 VI- 2의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴칼슘으로서 10.4 mg 해당량)과 제조예 VI- 5의 장용성고분자를 이용한 이베사르탄 지연방출성 과립(이베사르탄으로서 75 mg 해당량)을 혼합한 다음 직경 10 mm 펀치가 장착된 로타리 타정기로 타정하였다. 타정이 완료된 2상의 메트릭스 정제를 표 VI-9에 나타난 성분 및 함량으로 정제수에 10%(v/v)가 되도록 녹인 코팅액으로 코팅하였다.
[표VI-9]

<실시예 VI- 2> 로슈바스타틴칼슘을 코팅층에 함유한 이베사르탄 필름코팅정의 제조
상기 제조예 VI- 9의 수불용성 중합체를 이용한 이베사르탄 지연방출성 정제에 표 VI-10에 나타난 성분 및 함량으로 정제수에 10%(v/v)가 되도록 분산 용해시킨 코팅액으로 코팅하여 로슈바스타틴칼슘을 코팅층에 함유한 필름코팅정 정제를 제조하였다.
<실시예 VI- 3> 로슈바스타틴칼슘을 코팅층에 함유한 이베사르탄 유핵필름코팅정의 제조
상기 제조예 VI- 7의 장용성고분자를 드라이코팅으로 이용한 이베사르탄 지연방출성 유핵정에 표 VI-10에 나타난 성분 및 함량으로 정제수에 10%(v/v)가 되도록 분산 용해시킨 코팅액으로 코팅하여 로슈바스타틴칼슘을 코팅층에 함유한 유핵필름코팅정을 제조하였다.
[표VI-10]

<실시예 VI- 4> 로슈바스타틴칼슘 - 이베사르탄 2층정 제조
상기 제조예 VI- 2의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴칼슘으로서 10.4 mg 해당량)과 제조예 VI- 8의 수불용성 중합체를 이용한 이베사르탄 지연방출성 과립(이베사르탄으로서 75 mg 해당량)을 직경 10 mm 펀치가 장착된 로타리 삼중정 타정기(MRC-37T, Sejong, 이하 동일 기종 사용)의 다른 과립 주입구에 각각 넣고 타정하여 2층정 제조하였다. 타정이 완료된 정제를 표 VI-9에 나타난 성분 및 함량으로 정제수에 10%(v/v)가 되도록 용해시킨 코팅액으로 코팅하였다.
<실시예 VI- 5> 로슈바스타틴칼슘 - 이베사르탄 다층정 제조
상기 제조예 VI- 2의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴칼슘으로서 10.4 mg 해당량)을 1층 및 3층으로 1/2씩 분할투입하고 제조예 VI- 10의 소수성화합물과 친수성고분자를 이용한 이베사르탄 지연방출성 과립(이베사르탄으로서 75 mg 해당량)을 중간층(2번째층)으로 하여 직경 10 mm 펀치가 장착된 로타리 삼중정 타정기의 다른 과립 주입구에 각각 넣고 타정하여 다층정을 제조하였다. 타정이 완료된 정제를 표 VI-9에 나타난 성분 및 함량으로 정제수에 10%(v/v)가 되도록 용해시킨 코팅액으로 코팅하였다.
<실시예 VI- 6> 로슈바스타틴칼슘 - 이베사르탄 유핵정 제조
상기 제조예 VI- 6의 장용성고분자를 이용한 이베사르탄 지연방출성 정제(이베사르탄으로서 75 mg 해당량)를 내핵으로 하여 제조예 VI- 3의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴칼슘으로서 10.4 mg 해당량)과 함께 11 mm 펀치가 장착된 유핵정타정기에서 타정하여 유핵정을 제조하였다. 타정이 완료된 정제를 표 VI-9에 나타난 성분 및 함량으로 정제수에 10%(v/v)가 되도록 용해시킨 코팅액으로 코팅하였다.
<실시예 VI- 7> 로슈바스타틴칼슘 - 이베사르탄 유핵2층정 제조
상기 제조예 VI- 7의 장용성고분자를 드라이코팅으로 이용한 이베사르탄 지연방출성 유핵정(이베사르탄으로서 75 mg 해당량)의 타정 시 제조예 VI-1의 로슈바스타틴 선방출성 과립(로슈바스타틴칼슘으로서 10.4 mg 해당량)을 또 다른 주입구로 가하여 2층정 중 한 층이 유핵정이 되도록 타정하여 유핵2층정을 제조하였다. 타정이 완료된 정제를 표 VI-9에 나타난 성분 및 함량으로 정제수에 10%(v/v)가 되도록 용해시킨 코팅액으로 코팅하였다.
<실시예 VI- 8> 로슈바스타틴칼슘 - 이베사르탄 캡슐제(정제+정제) 제조
상기 제조예 VI- 1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴칼슘으로서 10.4 mg 해당량)을 6 mm 펀치가 장착된 로타리 타정기에서 타정하여 제조예 VI- 13의 친수성고분자를 이용한 이베사르탄 지연방출성 정제(이베사르탄으로서 75 mg 해당량)와 함께 0호 캡슐에 충전하여 정제2개가 함유되어 있는 캡슐제를 제조하였다.
<실시예 VI- 9> 로슈바스타틴칼슘 - 이베사르탄 캡슐제(과립+정제) 제조
상기 제조예 VI- 1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴칼슘으로서 10.4 mg 해당량)을 제조예 VI- 11의 소수성화합물과 친수성고분자를 이용한 이베사르탄 지연방출성 정제(이베사르탄으로서 75 mg 해당량)와 함께 0호 캡슐에 충전하여 과립과 정제가 함유되어 있는 캡슐제를 제조하였다.
<실시예 VI- 10> 로슈바스타틴칼슘 - 이베사르탄 캡슐제(정제+과립) 제조
상기 제조예 VI- 1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴칼슘으로서 10.4 mg 해당량)을 6 mm 펀치가 장착된 로타리 타정기에서 타정하여 제조예 VI- 12의 친수성고분자를 이용한 이베사르탄 지연방출성 과립(이베사르탄으로서 75 mg 해당량)과 함께 0호 캡슐에 충전하여 정제와 과립이 함유되어 있는 캡슐제를 제조하였다.
<실시예 VI- 11> 로슈바스타틴칼슘 - 이베사르탄 캡슐제(과립+과립) 제조
상기 제조예 VI- 1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴칼슘으로서 10.4 mg 해당량)과 제조예 VI- 14의 반투과성막 코팅기제와 삼투압 조절제를 이용한 이베사르탄 지연방출성 과립(이베사르탄으로서 75 mg 해당량)과 함께 0호 캡슐에 충전하여 과립과 과립이 함유되어 있는 캡슐제를 제조하였다.
<실시예 VI- 12> 로슈바스타틴칼슘 - 이베사르탄 캡슐제(펠렛+정제) 제조
상기 제조예 VI- 4의 로슈바스타틴칼슘 선방출성 펠렛(로슈바스타틴칼슘으로서 10.4 mg 해당량)과 제조예 VI- 15의 반투과성막 코팅기제와 삼투압 조절제를 이용한 이베사르탄 지연방출성 정제(이베사르탄으로서 75 mg 해당량)와 함께 0호 캡슐에 충전하여 펠렛과 정제가 함유되어 있는 캡슐제를 제조하였다.
<실시예 VI- 13> 로슈바스타틴칼슘 - 이베사르탄 캡슐제(펠렛+과립) 제조
상기 제조예 VI- 4의 로슈바스타틴칼슘 선방출성 펠렛(로슈바스타틴칼슘으로서 10.4 mg 해당량)과 제조예 VI- 5의 장용성고분자를 이용한 이베사르탄 지연방출성 과립(이베사르탄으로서 75 mg 해당량)과 함께 0호 캡슐에 충전하여 펠렛과 과립이 함유되어 있는 캡슐제를 제조하였다.
<실시예 VI- 14> 로슈바스타틴칼슘 - 이베사르탄 캡슐제(정제+펠렛) 제조
상기 제조예 VI- 1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴칼슘으로서 10.4 mg 해당량)을 6 mm 펀치가 장착된 로타리 타정기에서 타정하여 제조예 VI- 16의 장용성고분자를 이용한 이베사르탄 지연방출성 펠렛(이베사르탄으로서 75 mg 해당량)과 함께 0호 캡슐에 충전하여 정제와 펠렛이 함유되어 있는 캡슐제를 제조하였다.
<실시예 VI- 15> 로슈바스타틴칼슘 - 이베사르탄 캡슐제(과립+펠렛) 제조
상기 제조예 VI- 1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴칼슘으로서 10.4 mg 해당량)과 제조예 VI- 16의 장용성고분자를 이용한 이베사르탄 지연방출성 펠렛(이베사르탄으로서 75 mg 해당량)과 함께 0호 캡슐에 충전하여 과립과 펠렛이 함유되어 있는 캡슐제를 제조하였다.
<실시예 VI- 16> 로슈바스타틴칼슘 - 이베사르탄 캡슐제(과립+캡슐) 제조
상기 제조예 VI- 5의 장용성고분자를 이용한 이베사르탄 지연방출성 과립(이베사르탄으로서 75 mg 해당량)을 2호 캡슐에 충전하고 제조예 VI- 1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴칼슘으로서 10.4 mg 해당량)과 함께 0호 캡슐에 충전하여 과립과 캡슐이 함유되어 있는 캡슐제를 제조하였다.
<실시예 VI- 17> 로슈바스타틴칼슘 - 이베사르탄 캡슐제(과립+지연방출성 캡슐) 제조
표 VI- 11에 나타난 성분 및 함량으로 이베사르탄, 유당수화물, 폴록사며188, 크로스카멜로스나트륨, 이산화규소, 및 스테아르산마그네슘을 2호 캡슐에 충전한 다음 히프로멜로오스아세테이트숙시네이트를 80% 에탄올에 8%(w/w)가 되도록 용해시켜 제조한 코팅액을 이용하여 코팅하였다다. 장용성고분자로 코팅된 캡슐을 제조예 VI- 1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴칼슘으로서 10.4 mg 해당량)과 함께 0호 캡슐에 충전하여 과립과 지연방출성 캡슐이 함유되어 있는 캡슐제를 제조하였다.
[표VI-11]

<실시예 VI- 18> 로슈바스타틴칼슘 - 이베사르탄 블리스터 포장 키트의 제조
상기 제조예 VI- 1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴칼슘으로서 10.4 mg 해당량)을 6 mm 펀치가 장착된 로타리 타정기에서 타정하고 제조예 VI- 6의 장용성고분자를 이용한 이베사르탄 지연방출성 정제(이베사르탄으로서 75 mg 해당량)와 함께 블리스터 포장용기(Minister A, 흥아엔지니어링)를 사용하여 동시복용이 가능한 포장키트로 제조하였다.
<실시예 VI- 19> 로슈바스타틴칼슘 이베사르탄 유핵정 제조
표 VI- 12에 나타난 성분 및 함량으로 이베사르탄, 유당수화물, 미결정셀룰로오스, 전호화전분, 및 크로스카멜로스나트륨을 혼합하여 혼합물을 제조하였다. 별도로 폴록사머188을 정제수에 녹인 후 위 혼합물과 연합 한 다음 건조하였다. 건조된 과립물을 정립한 후 이산화규소, 및 스테아르산마그네슘을 투입한 뒤 혼합하여 직경 6.0 mm 펀치가 장착된 로타리 타정기에서 타정하였다. 타정이 완료된 정제에 히프로멜로오스아세테이트숙시네이트, 및 콜리코트 SR30D를 80%에탄올에 8%(w/w)가 되도록 용해시켜 제조한 액을 가지고 코팅기를 이용하여 코팅하여 이베사르탄 지연방출성 정제를 제조하였다. 장용성고분자와수불용중합체가포함되도록제조된정제를제조예 VI- 3의 로슈바스타틴칼슘 선방출성 과립과 함께 11 mm 펀치가 장착된 유핵정 타정기에서 타정하여 유핵정을 제조하였다. 타정이 완료된 정제를 표 VI-9에 나타난 성분 및 함량으로 정제수에 10%(v/v)가 되도록 녹인 코팅액으로 코팅하였다.
<실시예 VI- 20> 로슈바스타틴칼슘 이베사르탄 유핵정 제조
표 VI- 12에 나타난 성분 및 함량으로 이베사르탄, 유당수화물, 미결정셀룰로오스, 전호화전분, 및 크로스카멜로스나트륨을 혼합하여 혼합물을 제조하였다. 별도로 폴록사머188을 정제수에 녹인 후 위 혼합물과 연합 한 다음 건조하였다. 건조된 과립물을 정립한 후 이산화규소, 및 스테아르산마그네슘을 투입한 뒤 혼합하여 직경 6.0 mm 펀치가 장착된 로타리 타정기에서 타정하였다. 타정이 완료된 정제에 히프로멜로오스아세테이트숙시네이트, 및 카르나우바왁스를 80%에탄올에 8%(w/w)가 되도록 용해시켜 제조한 액을 가지고 코팅기를 이용하여 코팅하여 이베사르탄 지연방출성 정제를 제조하였다. 장용성고분자와수불용중합체가포함되도록제조된정제를제조예 VI- 3의 로슈바스타틴칼슘 선방출성 과립과 함께 11 mm 펀치가 장착된 유핵정 타정기에서 타정하여 유핵정을 제조하였다. 타정이 완료된 정제를 표 VI-9에 나타난 성분 및 함량으로 정제수에 10%(v/v)가 되도록 녹인 코팅액으로 코팅하였다.
[표VI-12]

<실험예 VI- 1> 용출 양상 시험1 (dissolution profile test)
상기 실시예 VI-2, VI-4에서 얻은 정제와 실시예 VI-8, VI-9에서 얻은 캡슐제를 대조제제로 크레스토정(Astrazeneca: 로슈바스타틴 단일제)와 아프로벨정(sanofi-aventis : 이베사르탄 단일제)을 사용하여 아래 조건에 따라 비교 용출시험을 실시하였다.
생체의 약물의 흡수경로와 유사하게 위의 산성조건은 pH 1.2액(대한약전 9개정 용출시험법의 제1액) 및 장관 조건은 pH 6.8(대한약전 9개정 용출시험법의 제2액)액으로 설정하여 시험을 진행하였으며 위 체류시간을 고려하여 pH 1.2액에서 2시간동안 용출을 진행하고 이후 pH 6.8에서 시험을 진행하는 방법으로 용출시험을 진행하였다.
용출시험 조건
시험법: 패들법
검 체: 실시예 VI-2, VI-4의 정제, 실시예 VI-8, VI-9의 캡슐제, 및 대조제제
검체수: 각각 12개 회전수 : 50회전/분
시험액: pH 1.2액, 및 pH 6.8액
액 량: 900 mL
용출시험에서 얻어진 검액을 다음 조건에 따라 액체크로마토그래프법으로 정량하여 각각 제형의 용출율을 구하였다.
분석조건
분석법: 액체크로마토그래프
유 속: 1 mL/분
컬 럼: C18, 150 mm x 4.5 mm, 5 μm
주입량: 10 μL
검출기: 자외부흡광광도계(측정파장 220 nm)
이동상: 인산완충액과 아세토니트릴의 60 : 40 혼합액
인산완충액: 5.5 mL g의 인산을 정제수 950 mL 에 가하고 트리에틸아민으로 pH를 3.0으로 조절하고 정제수를 가하여 총량을 1,000 mL로 한다.
용출시험결과 다음 도 25과 같은 결과를 얻을 수 있었으며 실시예 VI-2, VI-4, VI-8, VI-9의 로슈바스타틴 성분은 대조 제제인 크레스토정과 비교하여 동등한 용출특성을 나타냈다. 하지만 이베사르탄 성분은 대조 제제인 아프로벨정과 비교할 때 120분~300분의 방출지연시간을 가짐을 확인할 수 있었다.
또한 지연방출성 물질의 종류와 제형에 따라 방출지연시간을 제어할 수 있음을 알 수 있었으며 이를 통하여 실시예 VI 의 모든 제형으로 본 발명의 약제학적 제제를 개발할 수 있음을 확인할 수 있다.
<실험예 VI- 2> 용출 양상 시험2 (dissolution profile test)
상기 실시예 VI-6, VI-19, VI-20에서 얻은 정제, 및 실시예 VI-17에서 얻은 캡슐제의 용출시험을 실시하였으며 용출시험 조건 및 분석조건은 실험예 VI-1과 동일하게 진행하였다.
용출시험결과 다음 도 26와 같은 결과를 얻을 수 있었으며 실시예 VI-6, VI-17, VI-19, VI-20의 로슈바스타틴 성분은 대조 제제인 크레스토정과 비교하여 동등한 용출특성을 나타냈다. 하지만 이베사르탄 성분은 대조 제제인 아프로벨정과 비교할 때 120분~300분의 방출지연시간을 가짐을 확인할 수 있었다.
또한 제형에 따라 방출지연시간을 제어할 수 있음을 알 수 있었으며 이를 통하여 실시예 VI의 모든 제형으로 본 발명의 약제학적 제제를 개발할 수 있음을 확인할 수 있다.
[실시예 및 실험예 VII] 로슈바스타틴 및 칸데사르탄 함유 약제학적 제제
<제조예 VII-1> 로슈바스타틴칼슘 선방출성 과립 제조
표 VII- 1에 나타난 성분 및 함량으로 로슈바스타틴칼슘, 미결정셀룰로오스, 유당수화물, 삼염기칼슘인산염, 및 크로스포비돈을 혼합하여 혼합물을 제조하였다. 별도로 히드록시프로필셀룰로오스를 정제수에 녹인 후 위 혼합물과 스피드믹서(Lab. Pharma Mixer, Diosna, 이하 동일기종 사용)를 사용하여 연합 한 다음 건조하였다. 건조된 과립물을 정립한 후 스테아르산마그네슘을 투입한 뒤 혼합하여 로슈바스타틴칼슘 선방출성 과립을 제조하였다.
<제조예 VII-2> 로슈바스타틴칼슘 선방출성 과립 제조
표 VII- 1에 나타난 성분 및 함량으로 제조예 VII-1과 동일한 방법으로 제조하였다.
<제조예 VII-3> 로슈바스타틴칼슘 선방출성 과립 제조
표 VII- 1에 나타난 성분 및 함량으로 제조예VII-1과 동일한 방법으로 제조하였다.
[표VII-1]

<제조예 VII-4> 로슈바스타틴칼슘의 선방출성 펠렛 제조
표 VII- 2에 나타난 성분 및 함량으로 슈가비드에 로슈바스타틴칼슘, 삼염기칼슘인산염, 히드록시프로필셀룰로오스, 및 크로스포비돈을 정제수에 10%(v/v)가 되도록 분산 및 용해시킨 액을 유동층 과립기(GPCG 1, Glatt, 이하 동일기종 사용)를 이용하여 분무하여 코팅한 다음 건조하여 로슈바스타틴칼슘 선방출성 펠렛을 제조하였다.
[표VII-2]

<제조예 VII-5> 장용성 고분자를 포함하는 칸데사르탄실렉세틸 지연방출성 과립 제조
표 VII- 3에 나타난 성분 및 함량으로 유당수화물, 옥수수전분, 및 카르복시메틸셀룰로오스 칼슘을 혼합하여 혼합물을 제조하였다. 별도로 폴리에틸렌글리콜6000을 정제수에 녹이고 이 액에 칸데사르탄실렉세틸을 분산시킨 다음 이 분산액을 위 유당일수화물, 옥수수전분, 및 카르복시메틸셀룰로오스 칼슘 혼합물에 유동층 과립기를 이용하여 분무하였다. 얻어진 물질에 추가로 히드록시프로필셀룰로오스 수용액(5%(v/v))으로 분무하여 과립을 형성하였다. 따로 히프로멜로오스아세테이트숙시네이트를 80%에탄올에 8%(w/w)가 되도록 용해시킨 후 위에서 형성된 과립에 분무하여 과립을 코팅한 다음 건조 하였다. 상기 과립물에 스테아르산마그네슘 투입한 뒤 혼합하여 칸데사르탄실렉세틸 지연방출성 과립을 제조하였다.
<제조예 VII-6> 장용성 고분자를 필름코팅으로 포함하는 칸데사르탄실렉세틸 지연방출성 정제 제조
표 VII- 3에 나타난 성분 및 함량으로 칸데사르탄실렉세틸, 유당수화물, 옥수수전분, 및 카르복시메틸셀룰로오스 칼슘을 혼합하여 혼합물을 제조하였다. 별도로 폴리에틸렌글리콜6000 및 히드록시프로필셀룰로오스를 정제수에 녹인 후 위 혼합물과 연합 한 다음 건조하였다. 건조된 과립물을 정립한 후 스테아르산마그네슘을 투입한 뒤 혼합하여 직경 6.0 mm 펀치가 장착된 로타리 타정기(MRC-33, Sejong, 이하 동일기종 사용)에서 타정하였다. 타정이 완료된 정제에 히프로멜로오스아세테이트숙시네이트를 80%에탄올에 8%(w/w)가 되도록 용해시켜 제조한 액을 가지고 코팅기(SFC-30F, Sejong, 이하 동일기종 사용)를 이용하여 코팅하여 칸데사르탄실렉세틸 지연방출성 정제를 제조하였다.
<제조예 VII-7> 장용성 고분자를 드라이코팅으로 포함하는 칸데사르탄실렉세틸 지연방출성 유핵정 제조
표 VII- 3에 나타난 성분 및 함량으로 칸데사르탄실렉세틸, 유당수화물, 옥수수전분, 및 카르복시메틸셀룰로오스 칼슘을 혼합하여 혼합물을 제조하였다. 별도로 폴리에틸렌글리콜6000 및 히드록시프로필셀룰로오스를 정제수에 녹인 후 위 혼합물과 연합 한 다음 건조하였다. 건조된 과립물을 정립한 후 스테아르산마그네슘을 투입한 뒤 혼합하여 직경 6.0 mm 펀치가 장착된 로타리 타정기에서 타정하였다. 타정이 완료된 정제를 내핵으로 하고 히프로멜로오스아세테이트숙시네이트, 미결정셀룰로오스, 및 스테아르산마그네슘의 혼합물과 함께 11 mm 펀치가 장착된 유핵정타정기(RUD-1, Killian, 이하 동일기종 사용)에서 타정하여 드라이코팅 된 칸데사르탄실렉세틸 지연방출성 유핵정을 제조하였다.
[표VII-3]

<제조예 VII-8> 수불용중합체를 포함하는 칸데사르탄실렉세틸 지연방출성 과립 제조
표 VII- 4에 나타난 성분 및 함량으로 유당수화물, 옥수수전분, 및 카르복시메틸셀룰로오스 칼슘을 혼합하여 혼합물을 제조하였다. 별도로 폴리에틸렌글리콜6000을 정제수에 녹이고 이 액에 칸데사르탄실렉세틸을 분산시킨 다음 이 분산액을 위 유당일수화물, 옥수수전분, 및 카르복시메틸셀룰로오스 칼슘 혼합물에 유동층 과립기를 이용하여 분무하였다. 얻어진 물질에 추가로 히드록시프로필셀룰로오스 수용액(5%(v/v))으로 분무하여 과립을 형성하였다. 따로 콜리코트 SR30D (Kollicoat SR 30D, 폴리비닐에세테이트 27%, 포비돈 2.7%, 소디움라우릴설페이트 0.3%, 및 정제수 70%의 조성, Basf)를 위에서 형성된 과립에 분무하여 과립을 코팅한 다음 건조 하였다. 상기 과립물에 스테아르산마그네슘 투입한 뒤 혼합하여 칸데사르탄실렉세틸 지연방출성 과립을 제조하였다.
<제조예 VII-9> 수불용중합체를 포함하는 칸데사르탄실렉세틸 지연방출성 정제 제조
표 VII- 4에 나타난 성분 및 함량으로 칸데사르탄실렉세틸, 유당수화물, 옥수수전분, 및 카르복시메틸셀룰로오스 칼슘을 혼합하여 혼합물을 제조하였다. 별도로 폴리에틸렌글리콜6000 및 히드록시프로필셀룰로오스를 정제수에 녹인 후 위 혼합물과 연합 한 다음 건조하였다. 건조된 과립물을 정립한 후 스테아르산마그네슘을 투입한 뒤 혼합하여 직경 6.0 mm 펀치가 장착된 로타리 타정기에서 타정하였다. 타정이 완료된 정제에 콜리코트 SR30D로 코팅하여 칸데사르탄실렉세틸 지연방출성 정제를 제조하였다.
[표VII-4]

<제조예 VII-10> 소수성화합물 및 친수성고분자를 포함하는 칸데사르탄실렉세틸 지연방출성 과립 제조
표 VII- 5에 나타난 성분 및 함량으로 유당수화물, 옥수수전분, 및 카르복시메틸셀룰로오스 칼슘을 혼합하여 혼합물을 제조하였다. 별도로 폴리에틸렌글리콜6000을 정제수에 녹이고 이 액에 칸데사르탄실렉세틸을 분산시킨 다음 이 분산액을 위 유당일수화물, 옥수수전분, 및 카르복시메틸셀룰로오스 칼슘 혼합물에 유동층 과립기를 이용하여 분무하였다. 얻어진 물질에 추가로 히드록시프로필셀룰로오스 수용액(5%(v/v))으로 분무하여 과립을 형성하였다. 따로 카르나우바왁스, 히프로멜로오스, 및 폴리에틸렌글리콜6000을 정제수에 10%(v/v)가 되도록 분산시킨 후 위에서 형성된 과립에 분무하여 과립을 코팅한 다음 건조 하였다. 상기 과립물에 스테아르산마그네슘 투입한 뒤 혼합하여 칸데사르탄실렉세틸 지연방출성 과립을 제조하였다.
<제조예 VII-11> 소수성화합물 및 친수성고분자를 포함하는 칸데사르탄실렉세틸 지연방출성 정제 제조
표 VII- 5에 나타난 성분 및 함량으로 칸데사르탄실렉세틸, 유당수화물, 옥수수전분, 및 카르복시메틸셀룰로오스 칼슘을 혼합하여 혼합물을 제조하였다. 별도로 폴리에틸렌글리콜6000 및 히드록시프로필셀룰로오스를 정제수에 녹인 후 위 혼합물과 연합 한 다음 건조하였다. 건조된 과립물을 정립한 후 스테아르산마그네슘을 투입한 뒤 혼합하여 직경 6.0 mm 펀치가 장착된 로타리 타정기에서 타정하였다. 타정이 완료된 정제에 카르나우바왁스, 히프로멜로오스, 및 폴리에틸렌글리콜6000을 정제수에 10%(v/v)가 되도록 분산시켜 제조한 코팅액으로 코팅하여 칸데사르탄실렉세틸 지연방출성 정제를 제조하였다.
[표VII-5]

<제조예 VII-12> 친수성고분자를 포함하는 칸데사르탄실렉세틸 지연방출성 과립 제조
표 VII- 6에 나타난 성분 및 함량으로 유당수화물, 옥수수전분, 및 카르복시메틸셀룰로오스 칼슘을 혼합하여 혼합물을 제조하였다. 별도로 폴리에틸렌글리콜6000을 정제수에 녹이고 이 액에 칸데사르탄실렉세틸을 분산시킨 다음 이 분산액을 위 유당일수화물, 옥수수전분, 및 카르복시메틸셀룰로오스 칼슘 혼합물에 유동층 과립기를 이용하여 분무하였다. 얻어진 물질에 추가로 히드록시프로필셀룰로오스 수용액(5%(v/v))으로 분무하여 과립을 형성하였다. 따로 히드록시프로필셀룰로오스 및 폴리에틸렌글리콜6000을 정제수에 10%(v/v)가 되도록 녹인 후 위에서 형성된 과립에 분무하여 과립을 코팅한 다음 건조 하였다. 상기 과립물에 스테아르산마그네슘 투입한 뒤 혼합하여 칸데사르탄실렉세틸 지연방출성 과립을 제조하였다.
<제조예 VII-13> 친수성고분자를 포함하는 칸데사르탄실렉세틸 지연방출성 정제 제조
표 VII- 6에 나타난 성분 및 함량으로 칸데사르탄실렉세틸, 유당수화물, 옥수수전분, 및 카르복시메틸셀룰로오스 칼슘을 혼합하여 혼합물을 제조하였다. 별도로 폴리에틸렌글리콜6000 및 히드록시프로필셀룰로오스를 정제수에 녹인 후 위 혼합물과 연합 한 다음 건조하였다. 건조된 과립물을 정립한 후 스테아르산마그네슘을 투입한 뒤 혼합하여 직경 6.0 mm 펀치가 장착된 로타리 타정기에서 타정하였다. 타정이 완료된 정제에 히드록시프로필셀룰로오스 및 폴리에틸렌글리콜6000을 정제수에 10%(v/v)가 되도록 녹여 제조한 코팅액으로 코팅하여 칸데사르탄실렉세틸 지연방출성 정제를 제조하였다.
[표VII-6]

<제조예 VII-14> 반투과성막 코팅기제 및 삼투압 조절제를 포함하는 칸데사르탄실렉세틸 지연방출성 과립 제조
표 VII- 7에 나타난 성분 및 함량으로 유당수화물, 옥수수전분, 카르복시메틸셀룰로오스 칼슘, 및 염화나트륨을 혼합하여 혼합물을 제조하였다. 별도로 폴리에틸렌글리콜6000을 정제수에 녹이고 이 액에 칸데사르탄실렉세틸을 분산시킨 다음 이 분산액을 위 유당일수화물, 옥수수전분, 및 카르복시메틸셀룰로오스 칼슘 혼합물에 유동층 과립기를 이용하여 분무하였다. 얻어진 물질에 추가로 히드록시프로필셀룰로오스 수용액(5%(v/v))으로 분무하여 과립을 형성하였다. 따로 에틸셀룰로오스를 메틸렌클로라이드와 에탄올의 1:1 혼합액에 8%(w/w)가 되도록 용해시킨 후 위에서 형성된 과립에 분무하여 과립을 코팅한 다음 건조 하였다. 상기 과립물에 스테아르산마그네슘 투입한 뒤 혼합하여 칸데사르탄실렉세틸 지연방출성 과립을 제조하였다.
<제조예 VII-15> 반투과성막 코팅기제 및 삼투압 조절제를 포함하는 칸데사르탄실렉세틸 지연방출성 정제 제조
표 VII- 7에 나타난 성분 및 함량으로 칸데사르탄실렉세틸, 유당수화물, 옥수수전분, 카르복시메틸셀룰로오스 칼슘, 및 염화나트륨을 혼합하여 혼합물을 제조하였다. 별도로 폴리에틸렌글리콜6000, 및 히드록시프로필셀룰로오스를 정제수에 녹인 후 위 혼합물과 연합 한 다음 건조하였다. 건조된 과립물을 정립한 후 스테아르산마그네슘을 투입한 뒤 혼합하여 직경 6.0 mm 펀치가 장착된 로타리 타정기에서 타정하였다. 타정이 완료된 정제에 에틸셀룰로오스를 메틸렌클로라이드와 에탄올의 1:1 혼합액8%(w/w)가 되도록 용해시켜 제조한 코팅액으로 코팅하여 칸데사르탄실렉세틸 지연방출성 정제를 제조하였다.
[표VII-7]

<제조예 VII-16> 장용성 고분자를 포함하는 칸데사르탄실렉세틸의 지연방출성 펠렛 제조
표 VII- 8에 나타난 성분 및 함량으로 슈가비드에 칸데사르탄실렉세틸, 폴리에틸렌글리콜6000, 히드록시프로필셀룰로오스, 및 카르복시메틸셀룰로오스칼슘을 정제수에 분산 및 용해시킨 액을 유동층 과립기를 이용하여 분무하여 코팅한다. 별도로 히프로멜로오스아세테이트숙시네이트를 80%에탄올에 8%(w/w)가 되도록 용해시켜 제조한 액을 위 비드에 다시 분무하여 코팅한 후 건조하여 칸데사르탄실렉세틸의 지연방출성 펠렛을 제조하였다.
[표VII-8]

<실시예 VII-1> 로슈바스타틴칼슘 - 칸데사르탄실렉세틸 2상의 매트릭스 정제 제조
상기 제조예 VII-2의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로서 10.4 mg 해당량)과 제조예 VII-5의 장용성 고분자를 포함하는 칸데사르탄실렉세틸 지연방출성 과립(칸데사르탄실렉세틸로서 16 mg 해당량)을 혼합한 다음 직경 10 mm 펀치가 장착된 로타리 타정기로 타정하였다. 타정이 완료된 2상의 메트릭스정제를 표 VII-9에 나타난 성분 및 함량으로 정제수에 10%(v/v)가 되도록 녹인 코팅액으로 코팅하였다.
[표VII-9]

<실시예 VII-2> 로슈바스타틴칼슘을 코팅층에 함유한 칸데사르탄실렉세틸 필름코팅정의 제조
상기 제조예VII-9의 수불용중합체를 포함하는 칸데사르탄실렉세틸 지연방출성 정제에 표 VII-10에 나타난 성분 및 함량으로 정제수에 10%(v/v)가 되도록 분산 용해시킨 코팅액으로 코팅하여 로슈바스타틴칼슘을 코팅층에 함유한 필름코팅정 정제를 제조하였다.
[표VII-10]

<실시예 VII-3> 로슈바스타틴칼슘을 코팅층에 함유한 칸데사르탄실렉세틸 유핵필름코팅정의 제조
상기 제조예 VII-7의 장용성 고분자를 드라이코팅으로 포함하는 칸데사르탄실렉세틸 지연방출성 유핵정에 표 VII-10에 나타난 성분 및 함량으로 정제수에 10%(v/v)가 되도록 녹인 코팅액으로 코팅하여 로슈바스타틴칼슘을 코팅층에 함유한 유핵필름코팅정을 제조하였다.
<실시예 VII-4> 로슈바스타틴칼슘 - 칸데사르탄실렉세틸 2층정 제조
상기 제조예 VII-2의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로서 10.4 mg 해당량)과 제조예 VII-8의 수불용중합체를 포함하는 칸데사르탄실렉세틸 지연방출성 과립(칸데사르탄실렉세틸로서 16 mg 해당량)을 직경 10 mm 펀치가 장착된 로타리 삼중정 타정기(모델명, 제조사명, 이하 동일기종 사용)의 다른 과립 주입구에 각각 넣고 타정하여 2층정 제조하였다. 타정이 완료된 정제를 표 VII-9에 나타난 성분 및 함량으로 정제수에 10%(v/v)가 되도록 녹인 코팅액으로 코팅하였다.
<실시예 VII-5> 로슈바스타틴칼슘 - 칸데사르탄실렉세틸 다층정 제조
상기 제조예 VII-2의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로서 10.4 mg 해당량)을 1층 및 3층으로 1/2씩 분할투입하고 제조예 VII-10의 소수성화합물과 친수성고분자를 포함하는 칸데사르탄실렉세틸 지연방출성 과립(칸데사르탄실렉세틸로서 16 mg 해당량)을 중간층(2번째층)으로 하여 직경 10 mm 펀치가 장착된 로타리 삼중정 타정기의 다른 과립 주입구에 각각 넣고 타정하여 다층정을 제조하였다. 타정이 완료된 정제를 표 VII-9에 나타난 성분 및 함량으로 정제수에 10%(v/v)가 되도록 녹인 코팅액으로 코팅하였다.
<실시예 VII-6> 로슈바스타틴칼슘 - 칸데사르탄실렉세틸 유핵정 제조
상기 제조예 VII-6의 장용성 고분자를 포함하는 칸데사르탄실렉세틸 지연방출성 정제를 내핵으로 하여 제조예 VII-3의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로서 10.4 mg 해당량)과 함께 11 mm 펀치가 장착된 유핵정타정기에서 타정하여 유핵정을 제조하였다. 타정이 완료된 정제를 표 VII-9에 나타난 성분 및 함량으로 정제수에 10%(v/v)가 되도록 녹인 코팅액으로 코팅하였다.
<실시예 VII-7> 로슈바스타틴칼슘 - 칸데사르탄실렉세틸 유핵2중정 제조
상기 제조예 VII-7의 장용성 고분자를 드라이코팅으로 포함하는 칸데사르탄실렉세틸 지연방출성 유핵정의 타정시 제조예VII-1의 로슈바스타틴 선방출성 과립(로슈바스타틴으로서 10.4 mg 해당량)을 또 다른 주입구로 가하여 2중정 중 한 층이 유핵정이 되도록 타정하여 유핵2중정을 제조하였다. 타정이 완료된 정제를 표 VII-9에 나타난 성분 및 함량으로 정제수에 10%(v/v)가 되도록 녹인 코팅액으로 코팅하였다.
<실시예 VII-8> 로슈바스타틴칼슘 - 칸데사르탄실렉세틸 캡슐제(정제+정제) 제조
상기 제조예 VII-1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로서 10.4 mg 해당량)을 6 mm 펀치가 장착된 로타리 타정기에서 타정한 후 그 정제를, 제조예 VII-13의 친수성고분자를 포함하는 칸데사르탄실렉세틸 지연방출성 정제와 함께 0호 캡슐에 충전하여 캡슐제를 제조하였다.
<실시예 VII-9> 로슈바스타틴칼슘 - 칸데사르탄실렉세틸 캡슐제(과립+정제) 제조
상기 제조예 VII-1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로서 10.4 mg 해당량)을 제조예 VII-11의 소수성화합물과 친수성고분자를 포함하는 칸데사르탄실렉세틸 지연방출성 정제와 함께 0호 캡슐에 충전하여 캡슐제를 제조하였다.
<실시예 VII-10> 로슈바스타틴칼슘 - 칸데사르탄실렉세틸 캡슐제(정제+과립) 제조
상기 제조예 VII-1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로서 10.4 mg 해당량)을 6 mm 펀치가 장착된 로타리 타정기에서 타정한 후 그 정제를, 제조예 VII-12의 친수성고분자를 포함하는 칸데사르탄실렉세틸 지연방출성 과립(칸데사르탄실렉세틸로서 16 mg 해당량)과 함께 0호 캡슐에 충전하여캡슐제를 제조하였다.
<실시예 VII-11> 로슈바스타틴칼슘 - 칸데사르탄실렉세틸 캡슐제(과립+과립) 제조
상기 제조예 VII-1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로서 10.4 mg 해당량)과 제조예 VII-14의 반투과성막 코팅기제와 삼투압 조절제를 포함하는 칸데사르탄실렉세틸 지연방출성 과립(칸데사르탄실렉세틸로서 16 mg 해당량)과 함께 0호 캡슐에 충전하여 캡슐제를 제조하였다.
<실시예 VII-12> 로슈바스타틴칼슘 - 칸데사르탄실렉세틸 캡슐제(펠렛+정제) 제조
상기 제조예 VII-4의 로슈바스타틴칼슘 선방출성 펠렛(로슈바스타틴으로서 10.4 mg 해당량)과 제조예 VII-15의 반투과성막 코팅기제와 삼투압 조절제를 포함하는 칸데사르탄실렉세틸 지연방출성 정제와 함께 0호 캡슐에 충전하여 캡슐제를 제조하였다.
<실시예 VII-13> 로슈바스타틴칼슘 - 칸데사르탄실렉세틸 캡슐제(펠렛+과립) 제조
상기 제조예 VII-4의 로슈바스타틴칼슘 선방출성 펠렛(로슈바스타틴으로서 10.4 mg 해당량)과 제조예 VII-5의 장용성 고분자를 포함하는 칸데사르탄실렉세틸 지연방출성 과립(칸데사르탄실렉세틸로서 16 mg 해당량)과 함께 0호 캡슐에 충전하여 캡슐제를 제조하였다.
<실시예 VII-14> 로슈바스타틴칼슘 - 칸데사르탄실렉세틸 캡슐제(정제+펠렛) 제조
상기 제조예 VII-1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로서 10.4 mg 해당량)을 6 mm 펀치가 장착된 로타리 타정기에서 타정한 후 그 정제를, 제조예 VII-16의 장용성 고분자를 포함하는 칸데사르탄실렉세틸 지연방출성 펠렛(칸데사르탄실렉세틸로서 16 mg 해당량)과 함께 0호 캡슐에 충전하여 캡슐제를 제조하였다.
<실시예 VII-15> 로슈바스타틴칼슘 - 칸데사르탄실렉세틸 캡슐제(과립+펠렛) 제조
상기 제조예 VII-1의 로슈바스타틴칼슘 선방출성과립(로슈바스타틴으로서 10.4 mg 해당량)과 제조예 VII-16의 장용성 고분자를 포함하는 칸데사르탄실렉세틸 지연방출성 펠렛(칸데사르탄실렉세틸로서 16 mg 해당량)과 함께 0호 캡슐에 충전하여 캡슐제를 제조하였다.
<실시예 VII-16> 로슈바스타틴칼슘 - 칸데사르탄실렉세틸 캡슐제(과립+캡슐) 제조
상기 제조예 VII-5의 장용성 고분자를 포함하는 칸데사르탄실렉세틸 지연방출성 과립(칸데사르탄실렉세틸로서 16 mg 해당량)을 2호 캡슐에 충전하고 그 캡슐을, 제조예 VII-1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로서 10.4 mg 해당량)과 함께 0호 캡슐에 충전하여 캡슐제를 제조하였다.
<실시예 VII-17> 로슈바스타틴칼슘 - 칸데사르탄실렉세틸 캡슐제(과립+지연방출성 캡슐) 제조
표 VII- 11에 나타난 성분 및 함량으로 칸데사르탄실렉세틸, 유당수화물, 폴리에틸렌글리콜6000, 카르복시메틸셀룰로오스칼슘, 및 스테아르산마그네슘을 2호 캡슐에 충전한 다음 그 캡슐을 히프로멜로오스아세테이트숙시네이트를 80%에탄올에 8%(w/w)가 되도록 용해시켜 제조한 코팅액을 이용하여 코팅하였다. 장용성 고분자로 코팅된 캡슐을 제조예 VII-1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로서 10.4 mg 해당량)과 함께 0호 캡슐에 충전하여 캡슐제를 제조하였다.
[표VII-11]

<실시예 VII-18> 로슈바스타틴칼슘 - 칸데사르탄실렉세틸 블리스터 포장 키트의 제조
상기 제조예 VII-1의 로슈바스타틴칼슘 선방출성 과립(로슈바스타틴으로서 10.4 mg 해당량)을 6 mm 펀치가 장착된 로타리 타정기에서 타정한 후 그 정제를, 제조예 VII-6의 장용성 고분자를 포함하는 칸데사르탄실렉세틸 지연방출성 정제와 함께 하나의 PTP(Press Through Pack)포장용기(Minister A, 흥아엔지니어링)에 포장하여 동시복용이 가능한 포장키트를 제조하였다.
<실험예 VII-1> 용출 양상 시험1 (dissolution profile test)
상기 실시예 VII-2과 실시예 VII-4에서 얻은 정제와 실시예 VII-8에서 얻은 캡슐제를 대조제제로 크레스토정(Astrazeneca : 로슈바스타틴 단일제)와 아타칸정(Astrazeneca : 칸데사르탄 단일제)을 사용하여 아래 조건에 따라 비교 용출시험을 실시하였다.
생체의 약물의 흡수경로와 유사하게 위의 산성조건은 pH 1.2액(대한약전 9개정 용출시험법의 제1액) 및 장관 조건은 pH 6.8(대한약전 9개정 용출시험법의 제2액)액으로 설정하여 시험을 진행하였으며 위 체류시간을 고려하여 pH 1.2액에서 2시간동안 용출을 진행하고 이후 pH 6.8에서 시험을 진행하는 방법으로 용출시험을 진행하였다.
용출시험 조건은 아래와 같다. 칸데사르탄이 난용성 물질이므로 계면활성제인 폴리소르베이트80을 1%농도로 용출액에 첨가하여 시험하였다.
용출시험 조건
시험법: 패들법
검체수: 각각 12개
회전수: 50회전/분
시험액: 1% 폴리소르베이트80을 함유한 pH 1.2액, 및 pH 6.8액
액 량: 900 mL
용출시험에서 얻어진 검액을 다음 조건에 따라 액체크로마토그래프법으로 정량하여 각각 제형의 용출률을 구하였다.
분석조건
분석법: 액체크로마토그래프
유 속: 1 mL/분
컬 럼: C18, 150 mm × 4.5 mm (5 μm)
주입량: 10 μL
검출기: 자외부흡광광도계(측정파장 257 nm)
이동상: 초산완충액과 아세토니트릴의 45 : 55 혼합액
초산완충액: 1.54 g의 초산암모늄을 정제수에 녹여 1,000 mL로 하고 초산을 이용하여 pH를 4.5로 조절한다.
용출시험결과 다음 도 27과 같은 결과를 얻을 수 있었으며 실시예 VII-2, VII-4, VII-8의 로슈바스타틴 성분은 대조 제제인 크레스토정과 비교하여 동등한 용출특성을 나타냈다. 하지만 칸데사르탄 성분은 대조 제제인 아타칸정과 비교할 때 120분~180분의 방출지연시간을 가짐을 확인할 수 있었다.
또한 지연방출성 물질의 종류와 제형에 따라 방출지연시간을 제어할 수 있음을 알 수 있었으며 이를 통하여 제조예의 모든 제형으로 본 발명의 약제학적 제제를 개발할 수 있음을 확인할 수 있다.
<실험예 VII- 2> 용출 양상 시험2 (dissolution profile test)
상기 실시예 VII-6에서 얻은 정제와 실시예 VII-15 및 실시예 VII-17에서 얻은 캡슐제의 용출시험을 실시하였으며 용출시험 조건 및 분석조건은 실험예 VII-1과 동일하게 진행하였다.
용출시험결과 다음 도 28과 같은 결과를 얻을 수 있었으며 실시예 VII-6, VII-15, VII-17의 로슈바스타틴 성분은 대조 제제인 크레스토정과 비교하여 동등한 용출특성을 나타냈다. 하지만 칸데사르탄 성분은 대조 제제인 아타칸정과 비교할 때 120분~150분의 방출지연시간을 가짐을 확인할 수 있었다.
또한 제형에 따라 방출지연시간을 제어할 수 있음을 알 수 있었으며 이를 통하여 제조예의 모든 제형으로 본 발명의 약제학적 제제를 개발할 수 있음을 확인할 수 있다.
Claims (183)
- 약리학적 활성성분으로 심바스타틴(Simvastatin), 그의 이성질체 또는 그의 약학적으로 허용하는 염 을 함유하는 선방출성 구획, 및 약리학적 활성성분으로 로자탄(Losartan), 그의 이성질체 또는 그의 약학적으로 허용하는 염을 함유하는 지연방출성 구획을 포함하는 방출성이 제어된 약제학적 제제.
- 제 1 항에 있어서, 상기 심바스타틴이 용출 개시 후 30분 이내에 심바스타틴 총량의 80% 이상이 방출되는 약제학적 제제.
- 제 1 항에 있어서, 상기 로자탄이 경구투여시 심바스타틴 용출개시 후 2시간까지 단위제제 내 로자탄의 총량의20% 이내로 방출되는 약제학적 제제.
- 제 1 항에 있어서, 상기 심바스타틴은 단위제제 중 1 내지 160mg 인 약제학적 제제.
- 제 1 항에 있어서, 상기 로자탄은 단위제제 중 12.5 내지 200mg인 약제학적 제제.
- 제 1 항에 있어서, 상기 지연방출성 구획은 장용성 고분자, 수불용성 중합체, 소수성 화합물 및 친수성 고분자로 이루어진 군에서 선택된 1종 이상의 방출제어물질을 포함하는 약제학적 제제.
- 제 6 항에 있어서, 상기 방출제어물질이 장용성 고분자 및 수불용성 중합체 중에서 선택된 1종 이상인 약제학적 제제.
- 제 6 항에 있어서, 상기 방출제어물질이 로자탄 1중량부에 대하여 0.05중량부 내지 100중량부로 포함되는 것인 약제학적 제제.
- 제 6 항에 있어서, 상기 장용성 고분자는 장용성 셀룰로오스 유도체, 장용성 아크릴산계 공중합체, 장용성 폴리메타크릴레이트 공중합체, 장용성 말레인산계 공중합체 및 장용성 폴리비닐 유도체 중에서 선택된 1종 이상인 것인 약제학적 제제.
- 제 9 항에 있어서, 상기 장용성 셀룰로오스 유도체는 히프로멜로오스아세테이트숙시네이트, 히프멜로오스프탈레이트, 히드록시메틸에틸셀룰로오스프탈레이트, 셀룰로오스아세테이트프탈레이트, 셀룰로오스아세테이트숙시네이트, 셀룰로오스아세테이트말레이트, 셀룰로오스벤조에이트프탈레이트, 셀룰로오스프로피오네이트프탈레이트, 메틸셀룰로오스프탈레이트, 카르복시메틸에틸셀룰로오스, 에틸히드록시에틸셀룰로오스프탈레이트 및 메틸히드록시에틸셀룰로오스 중에서 선택된 1종 이상이고; 상기 장용성 아크릴산계 공중합체는 스티렌-아크릴산 공중합체, 아크릴산메틸-아크릴산 공중합체, 아크릴산메틸메타크릴산 공중합체, 아크릴산부틸-스티렌-아크릴산 공중합체, 및 아크릴산메틸-메타크릴산-아크릴산옥틸공중합체 중에서 선택된 1종 이상이며; 상기 장용성 폴리메타크릴레이트 공중합체는 폴리(메타크릴산 메틸 메타크릴레이트) 공중합체, 폴리 (메타크릴산 에틸아크릴레이트) 공중합체 중에서 선택된 1종 이상이며, 상기 장용성 말레인산계 공중합체는 아세트산비닐-말레인산 무수물 공중합체, 스티렌-말레인산 무수물 공중합체, 스티렌-말레인산모노에스테르 공중합체, 비닐메틸에테르-말레인산 무수물 공중합체, 에틸렌-말레인산 무수물 공중합체, 비닐부틸에테르-말레인산 무수물 공중합체, 아크릴로니트릴-크릴산메틸ㆍ말레인산 무수물 공중합체 및 아크릴산부틸-스티렌-말레인산 무수물 공중합체중에서 선택된 1종 이상이고; 또는 상기 장용성 폴리비닐 유도체는 폴리비닐알콜프탈레이트, 폴리비닐아세테이트프탈레이트, 폴리비닐부틸레이트프탈레이트, 및 폴리비닐아세트아세탈프탈레이트 중에서 선택된 1종 이상인 약제학적 제제.
- 제 10 항에 있어서, 장용성 고분자가 히프로멜로오스프탈레이트 또는 아크릴산메틸메타크릴산 공중합체인 약제학적 제제.
- 제 9 항에 있어서, 상기 장용성 고분자가 로자탄1 중량부에 대해서 0.1 중량부 내지 20 중량부인 약제학적 제제.
- 제 6 항에 있어서, 상기 수불용성 중합체는 폴리비닐아세테이트, 수불용성 폴리메타크릴레이트 공중합체, 에틸셀룰로오스, 셀룰로오스 에스테르, 셀룰로오스 에테르, 셀룰로오스 아실레이트, 셀룰로오스 디아실레이트, 셀룰로오스 트리아실레이트, 셀룰로오스아세테이트, 셀룰로오스 디아세테이트, 및 셀룰로오스 트리아세테이트 중에서 선택된 1 종 이상인 약제학적 제제.
- 제 13 항에 있어서, 상기 수불용성 중합체는 수불용성 폴리메타크릴레이트 공중합체인 폴리(에틸아크릴레이트-메틸 메타크릴레이트-트리메틸아미노에틸메타크릴레이트 클로라이드)공중합체, 에틸셀룰로오스 또는 셀룰로오스 아세테이트인 약제학적 제제.
- 제 13 항에 있어서, 상기 수불용성 중합체가 로자탄1 중량부에 대해서 0.1 중량부 내지 30 중량부인 약제학적 제제.
- 제 14 항에 있어서, 상기 수불용성 중합체가 에틸셀룰로오스인 약제학적 제제.
- 제 6 항에 있어서, 상기 소수성 화합물은 지방산 및 지방산 에스테르류, 지방산 알코올류, 왁스류 및 무기물질 중에서 선택된 1종 이상인 약제학적 제제.
- 제 17 항에 있어서, 상기 지방산 및 지방산 에스테르류로는 글리세릴 팔미토스테아레이트, 글리세릴 스테아레이트, 글리세릴 비헤네이트, 세틸 팔미테이트, 글리세릴 모노 올레이트 및 스테아린산 중에서 선택된 1종 이상 이고, 상기 지방산 알코올류는 세토스테아릴 알코올, 세틸알코올 및 스테아릴알코올 중에서 선택된 1종 이상 이며;, 상기 왁스류는 카르나우바왁스, 밀납 및 미결정왁스 중에서 선택된 1종 이상 이고; 상기 무기물질은 탈크, 침강탄산칼슘, 인산일수소칼슘, 산화아연, 산화티탄, 카올린, 벤토나이트, 몬모릴로나이트 및 비검중에서 선택된 1종 이상인 약제학적 제제.
- 제 17 항에 있어서, 소수성 화합물이 로자탄1 중량부에 대해서 0.1 중량부 내지 20 중량부인 약제학적 제제.
- 제 18 항에 있어서, 상기 소수성 화합물이 카르나우바왁스인 약제학적 제제.
- 제 6 항에 있어서, 상기 친수성 고분자는 당류, 셀룰로오스 유도체, 검류, 단백질류, 폴리비닐 유도체, 친수성 폴리메타크릴레이트 공중합체, 폴리에틸렌 유도체 및 카르복시비닐폴리머 중에서 선택된 1종 이상인 약제학적 제제.
- 제 21 항에 있어서, 상기 당류는 덱스트린, 폴리덱스트린, 덱스트란, 펙틴 및 펙틴 유도체, 알긴산염, 폴리갈락투론산, 자일란, 아라비노자일란, 아라비노갈락탄, 전분, 히드록시프로필스타치, 아밀로오스 및 아밀로펙틴 중에서 선택된 1종 이상이고; 상기 셀룰로오스 유도체는 히프로멜로오스, 히드록시프로필셀룰로오스, 히드록시메틸셀룰로오스, 히드록시에틸셀룰로오스, 메틸셀룰로오스, 카르복시메틸셀룰로오스 나트륨, 히드록시프로필 메틸셀룰로오스 아세테이트 숙시네이트 및 히드록시에틸메틸셀룰로오스에서 선택된 1종 이상이며; 상기 검류는 구아검, 로커스트 콩검, 트라가칸타, 카라기난, 아카시아검, 아라비아검, 젤란검 및 잔탄검 중에서 선택된 1종 이상이고; 상기 단백질류는 젤라틴, 카제인 및 제인 중에서 선택 1종 이상이며; 상기 폴리비닐 유도체는 폴리비닐 알코올, 폴리비닐 피롤리돈 및 폴리비닐아세탈디에틸아미노아세테이트 중에서 선택된 1종 이상이며; 상기 친수성 폴리메타크릴레이트 공중합체는 폴리(부틸 메타크릴레이트-(2-디메틸아미노에틸)메타크릴레이트-메틸메타크릴레이트) 공중합체이고; 상기 폴리에틸렌 유도체는 폴리에틸렌글리콜 및 폴리에틸렌 옥사이드 중에서 선택된 1종 이상이며; 상기 카르복시비닐폴리머는 카보머인 것인 약제학적 제제.
- 제 22 항에 있어서, 상기 친수성 고분자는 카보머 또는 히프로멜로오스인 약제학적 제제.
- 제 21 항에 있어서, 친수성 고분자가 로자탄1 중량부에 대해서 0.05 중량부 내지 30 중량부인 약제학적 제제.
- 제 1 항에 있어서, 상기 지연방출성 구획은 삼투압 조절제를 포함하며 반투과성막 코팅기제로 코팅된 것인 약제학적 제제.
- 제 25 항에 있어서, 상기 삼투압 조절제는 황산마그네슘, 염화마그네슘, 염화나트륨, 염화리튬, 황산칼륨, 황산나트륨, 황산리튬, 황산나트륨, 및 이들의 혼합물로 이루어진 군에서 선택된 1종 이상인 것인 약제학적 제제.
- 제 26 항에 있어서, 상기 삼투압 조절제는 염화나트륨, 황산나트륨인 약제학적 제제.
- 제 25 항에 있어서, 상기 반투과성막 코팅기제는 폴리비닐 아세테이트, 수불용성 폴리메타크릴레이트 공중합체, 에틸셀룰로오스, 셀룰로오스 에스테르, 셀룰로오스 에테르, 셀룰로오스 아실레이트, 셀룰로오스 디아실레이트, 셀룰로오스 트리아실레이트, 셀룰로오스 아세테이트, 셀룰로오스 디아세테이트, 셀룰로오스 트리아세테이트, 및 이들의 혼합물로 이루어진 군에서 선택된 1종 이상인 것인 약제학적 제제.
- 제 28 항에 있어서, 상기 반투과성막 코팅기제는 에틸셀룰로오스, 셀룰로오스 아세테이트인 약제학적 제제.
- 약리학적 활성성분으로 심바스타틴(Simvastatin), 그의 이성질체 또는 그의 약학적으로 허용하는 염 을 함유하는 선방출성 구획, 및 약리학적 활성성분으로 올메사르탄 (Olmesartan), 그의 프로드럭 또는 그의 약제학적으로 허용하는 염을 함유하는 지연방출성 구획을 포함하는 방출성이 제어된 약제학적 제제.
- 제 30 항에 있어서, 상기 심바스타틴이 용출 개시 후 30분 이내에 심바스타틴 총량의 80% 이상이 방출되는 약제학적 제제.
- 제 30 항에 있어서, 상기 올메사르탄이 경구투여시 심바스타틴 용출개시 후 2시간까지 단위제제내 올메사르탄의 총량의 5% 이내로 방출되는 약제학적 제제.
- 제 30 항에 있어서, 상기 올메사르탄 프로드럭이 올메사르탄 메독소밀인 약제학적 제제.
- 제 30 항에 있어서, 상기 심바스타틴은 제제 중 1 내지 160mg 인 약제학적 제제.
- 제 30 항에 있어서, 상기 올메사르탄은 제제 중 2.5 내지 80mg인 약제학적 제제.
- 제 30 항에 있어서, 상기 지연방출성 구획은 장용성 고분자, 수불용성 중합체, 소수성 화합물 및 친수성 고분자로 이루어진 군에서 선택된 1종 이상의 방출제어물질을 포함하는 약제학적 제제.
- 제 36 항에 있어서, 상기 방출제어물질이 장용성 고분자 및 수불용성 중합체 중에서 선택된 1종 이상인 약제학적 제제.
- 제 36 항에 있어서, 상기 방출제어물질이 올메사르탄 1중량부에 대하여 0.05중량부 내지 100중량부로 포함되는 것인 약제학적 제제.
- 제 36 항에 있어서, 상기 장용성 고분자는 장용성 셀룰로오스 유도체, 장용성 아크릴산계 공중합체, 장용성 폴리메타크릴레이트 공중합체, 장용성 말레인산계 공중합체 및 장용성 폴리비닐 유도체 중에서 선택된 1종 이상인 것인 약제학적 제제.
- 제 39 항에 있어서, 상기 장용성 셀룰로오스 유도체는 히프로멜로오스아세테이트숙시네이트, 히프멜로오스프탈레이트, 히드록시메틸에틸셀룰로오스프탈레이트, 셀룰로오스아세테이트프탈레이트, 셀룰로오스아세테이트숙시네이트, 셀룰로오스아세테이트말레이트, 셀룰로오스벤조에이트프탈레이트, 셀룰로오스프로피오네이트프탈레이트, 메틸셀룰로오스프탈레이트, 카르복시메틸에틸셀룰로오스, 에틸히드록시에틸셀룰로오스프탈레이트 및 메틸히드록시에틸셀룰로오스 중에서 선택된 1종 이상이고; 상기 장용성 아크릴산계 공중합체는 스티렌-아크릴산 공중합체, 아크릴산메틸-아크릴산 공중합체, 아크릴산메틸메타크릴산 공중합체, 아크릴산부틸-스티렌-아크릴산 공중합체 및 및 아크릴산-메타크릴산-아크릴산옥틸공중합체중에서 선택된 1종이상이고; 상기 장용성 메타크릴레이트 공중합체는 폴리메타크릴산 메틸 메타크릴레이트)공중합체 및 폴리 (메타크릴산ㆍ에틸아크레이트 )공중합체, 중에서 선택된 1종 이상이며; 상기 장용성 말레인산계 공중합체는 아세트산비닐-말레인산 무수물 공중합체, 스티렌-말레인산 무수물 공중합체, 스티렌-말레인산모노에스테르 공중합체, 비닐메틸에테르-말레인산 무수물 공중합체, 에틸렌-말레인산 무수물 공중합체, 비닐부틸에테르-말레인산 무수물 공중합체, 아크릴로니트릴-크릴산메틸ㆍ말레인산 무수물 공중합체 및 아크릴산부틸-스티렌-말레인산 무수물 공중합체중에서 선택된 1종 이상이고; 또는 상기 장용성 폴리비닐 유도체는 폴리비닐알콜프탈레이트, 폴리비닐아세테이트프탈레이트, 폴리비닐부틸레이트프탈레이트, 및 폴리비닐아세트아세탈프탈레이트 중에서 선택된 1종 이상인 약제학적 제제.
- 제 40 항에 있어서, 장용성 고분자가 폴리비닐아세테이트프탈레이트, 히프로멜로오스프탈레이트 또는 아크릴산메틸메타크릴산 공중합체인 약제학적 제제.
- 제 39 항에 있어서, 상기 장용성 고분자가 올메사르탄 1 중량부에 대해서 0.1 중량부 내지 20 중량부인 약제학적 제제.
- 제 36 항에 있어서, 상기 수불용성 중합체는 폴리비닐아세테이트, 수불용성 폴리메타크릴레이트 공중합체, 에틸셀룰로오스, 셀룰로오스 에스테르, 셀룰로오스 에테르, 셀룰로오스 아실레이트, 셀룰로오스 디아실레이트, 셀룰로오스 트리아실레이트, 셀룰로오스아세테이트, 셀룰로오스 디아세테이트, 및 셀룰로오스 트리아세테이트 중에서 선택된 1 종 이상인 약제학적 제제.
- 제 43 항에 있어서, 상기 수불용성 중합체는 폴리비닐아세테이트, 에틸셀룰로오스 또는 셀룰로오스 아세테이트인 약제학적 제제.
- 제 43 항에 있어서, 상기 수불용성 중합체가 올메사르탄 1 중량부에 대해서 0.1 중량부 내지 30 중량부인 약제학적 제제.
- 제 36 항에 있어서, 상기 소수성 화합물은 지방산 및 지방산 에스테르류, 지방산 알코올류, 왁스류 및 무기물질 중에서 선택된 1종 이상인 약제학적 제제.
- 제 46 항에 있어서, 상기 지방산 및 지방산 에스테르류로는 글리세릴 팔미토스테아레이트, 글리세릴 스테아레이트, 글리세릴 비헤네이트, 세틸 팔미테이트, 글리세릴 모노 올레이트 및 스테아린산 중에서 선택된 1종 이상 이고;, 상기 지방산 알코올류는 세토스테아릴 알코올, 세틸알코올 및 스테아릴알코올 중에서 선택된 1종 이상 이며;, 상기 왁스류는 카르나우바왁스, 밀납 및 미결정왁스 중에서 선택된 1종 이상 이고; 상기 무기물질은 탈크, 침강탄산칼슘, 인산일수소칼슘, 산화아연, 산화티탄, 카올린, 벤토나이트, 몬모릴로나이트 및 비검중에서 선택된 1종 이상인 약제학적 제제.
- 제 46 항에 있어서, 소수성 화합물이 올메사르탄 1 중량부에 대해서 0.1 중량부 내지 20 중량부인 약제학적 제제.
- 제 36 항에 있어서, 상기 친수성 고분자는 당류, 셀룰로오스 유도체, 검류, 단백질류, 폴리비닐 유도체, 친수성 폴리메타크릴레이트 공중합체, 폴리에틸렌 유도체 및 카르복시비닐폴리머 중에서 선택된 1종 이상인 약제학적 제제.
- 제 49 항에 있어서, 상기 당류는 덱스트린, 폴리덱스트린, 덱스트란, 펙틴 및 펙틴 유도체, 알긴산염, 폴리갈락투론산, 자일란, 아라비노자일란, 아라비노갈락탄, 전분, 히드록시프로필스타치, 아밀로오스 및 아밀로펙틴 중에서 선택된 1종 이상이고; 상기 셀룰로오스 유도체는 히프로멜로오스, 히드록시프로필셀룰로오스, 히드록시메틸셀룰로오스, 히드록시에틸셀룰로오스, 메틸셀룰로오스 및 카르복시메틸셀룰로오스 나트륨 중에서 선택된 1종 이상이며; 상기 검류는 구아검, 로커스트 콩검, 트라가칸타, 카라기난, 아카시아검, 아라비아검, 젤란검 및 잔탄검 중에서 선택된 1종 이상이고; 상기 단백질류는 젤라틴, 카제인 및 제인 중에서 선택 1종 이상이며; 상기 폴리비닐 유도체는 폴리비닐 알코올, 폴리비닐 피롤리돈 및 폴리비닐아세탈디에틸아미노아세테이트 중에서 선택된 1종 이상이며; 상기 친수성 폴리메타크릴레이트 공중합체는 폴리(부틸 메타크릴레이트-(2-디메틸아미노에틸)메타크릴레이트-메틸메타크릴레이트 ) 공중합체이고; 상기 폴리에틸렌 유도체는 폴리에틸렌글리콜 및 폴리에틸렌 옥사이드 중에서 선택된 1종 이상이며; 상기 카르복시비닐폴리머는 카보머인 것인 약제학적 제제.
- 제 49 항에 있어서, 친수성 고분자가 올메사르탄 1 중량부에 대해서 0.05 중량부 내지 30 중량부인 약제학적 제제.
- 제 30 항에 있어서, 상기 지연방출성 구획은 삼투압 조절제를 포함하며 반투과성막 코팅기제로 코팅된 것인 약제학적 제제.
- 제 52 항에 있어서, 상기 삼투압 조절제는 황산마그네슘, 염화마그네슘, 염화나트륨, 염화리튬, 황산칼륨, 황산나트륨, 황산리튬, 황산나트륨, 및 이들의 혼합물로 이루어진 군에서 선택된 1종 이상인 것인 약제학적 제제.
- 제 52 항에 있어서, 상기 반투과성막 코팅기제는 폴리비닐 아세테이트, 수불용성 폴리메타크릴레이트 공중합체, 에틸셀룰로오스, 셀룰로오스 에스테르, 셀룰로오스 에테르, 셀룰로오스 아실레이트, 셀룰로오스 디아실레이트, 셀룰로오스 트리아실레이트, 셀룰로오스 아세테이트, 셀룰로오스 디아세테이트, 셀룰로오스 트리아세테이트, 및 이들의 혼합물로 이루어진 군에서 선택된 1종 이상인 것인 약제학적 제제.
- 약리학적 활성성분으로 심바스타틴(Simvastatin), 그의 이성질체 또는 그의 약학적으로 허용하는 염 을 함유하는 선방출성 구획, 및 약리학적 활성성분으로 발사르탄(Valsartan), 그의 이성질체 또는 그의 약학적으로 허용하는 염을 함유하는 지연방출성 구획을 포함하는 방출성이 제어된 약제학적 제제.
- 제 55 항에 있어서, 상기 심바스타틴이 용출 개시 후 30분 이내에 심바스타틴 총량의 80% 이상이 방출되는 약제학적 제제.
- 제 55 항에 있어서, 상기 발사르탄이 경구투여시 심바스타틴 용출개시 후 2시간 이후에 방출되는 약제학적 제제.
- 제 55 항에 있어서, 상기 심바스타틴은 제제 중 1 내지 160mg 인 약제학적 제제.
- 제 55 항에 있어서, 상기 발사르탄은 제제 중 1 내지 800mg인 약제학적 제제.
- 제 55 항에 있어서, 상기 지연방출성 구획은 장용성 고분자, 수불용성 중합체, 소수성 화합물 및 친수성 고분자로 이루어진 군에서 선택된 1종 이상의 방출제어물질을 포함하는 약제학적 제제.
- 제 60 항에 있어서, 상기 방출제어물질이 장용성 고분자 및 수불용성 중합체 중에서 선택된 1종 이상인 약제학적 제제.
- 제 60 항에 있어서, 상기 방출제어물질이 발사르탄 1중량부에 대하여 0.05중량부 내지 100중량부로 포함되는 것인 약제학적 제제.
- 제 60 항에 있어서, 상기 장용성 고분자는 장용성 셀룰로오스 유도체, 장용성 아크릴산계 공중합체, 장용성 폴리메타크릴레이트 공중합체, 장용성 말레인산계 공중합체 및 장용성 폴리비닐 유도체 중에서 선택된 1종 이상인 것인 약제학적 제제.
- 제 63 항에 있어서, 상기 장용성 셀룰로오스 유도체는 히프로멜로오스아세테이트숙시네이트, 히프멜로오스프탈레이트, 히드록시메틸에틸셀룰로오스프탈레이트, 셀룰로오스아세테이트프탈레이트, 셀룰로오스아세테이트숙시네이트, 셀룰로오스아세테이트말레이트, 셀룰로오스벤조에이트프탈레이트, 셀룰로오스프로피오네이트프탈레이트, 메틸셀룰로오스프탈레이트, 카르복시메틸에틸셀룰로오스, 에틸히드록시에틸셀룰로오스프탈레이트 및 메틸히드록시에틸셀룰로오스 중에서 선택된 1종 이상이고; 상기 장용성 아크릴산계 공중합체는 스티렌-아크릴산 공중합체, 아크릴산메틸-아크릴산 공중합체, 아크릴산메틸메타크릴산 공중합체, 아크릴산부틸-스티렌-아크릴산 공중합체, 및 아크릴산메틸-메타크릴산-아크릴산옥틸공중합체 중에서 선택된 1종 이상이며; 상기 장용성 폴리메타크릴레이트 공중합체는 폴리(메타크릴산-메틸 메타크릴산메틸) 공중합체, 폴리(메타크릴산ㆍ에틸아크릴레이트)공중합체 중에서 선택된 1종 이상이며; 상기 장용성 말레인산계 공중합체는 아세트산비닐-말레인산 무수물 공중합체, 스티렌-말레인산 무수물 공중합체, 스티렌-말레인산모노에스테르 공중합체, 비닐메틸에테르-말레인산 무수물 공중합체, 에틸렌-말레인산 무수물 공중합체, 비닐부틸에테르-말레인산 무수물 공중합체, 아크릴로니트릴-크릴산메틸ㆍ말레인산 무수물 공중합체 및 아크릴산부틸-스티렌-말레인산 무수물 공중합체중에서 선택된 1종 이상이고; 또는 상기 장용성 폴리비닐 유도체는 폴리비닐알콜프탈레이트, 폴리비닐아세탈프탈레이트, 폴리비닐부틸레이트프탈레이트, 및 폴리비닐아세트아세탈프탈레이트 중에서 선택된 1종 이상인 약제학적 제제.
- 제 64 항에 있어서, 장용성 고분자가 히프로멜로오스프탈레이트인 약제학적 제제.
- 제 63 항에 있어서, 상기 장용성 고분자가 발사르탄1 중량부에 대해서 0.1 중량부 내지 20 중량부인 약제학적 제제.
- 제 60 항에 있어서, 상기 수불용성 중합체는 폴리비닐아세테이트, 수불용성 폴리메타크릴레이트 공중합체, 에틸셀룰로오스, 셀룰로오스 에스테르, 셀룰로오스 에테르, 셀룰로오스 아실레이트, 셀룰로오스 디아실레이트, 셀룰로오스 트리아실레이트, 셀룰로오스아세테이트, 셀룰로오스 디아세테이트, 및 셀룰로오스 트리아세테이트 중에서 선택된 1 종 이상인 약제학적 제제.
- 제 67 항에 있어서, 상기 수불용성 중합체는 폴리비닐아세테이트 또는 수불용성 폴리메타크릴레이트인 폴리(에틸아크릴레이트-메틸 메타크릴레이트-트리메틸아미노에틸메타크릴레이트 클로라이드)공중합체 인 약제학적 제제.
- 제 67 항에 있어서, 상기 수불용성 중합체가 발사르탄1 중량부에 대해서 0.1 중량부 내지 30 중량부인 약제학적 제제.
- 제 60 항에 있어서, 상기 소수성 화합물은 지방산 및 지방산 에스테르류, 지방산 알코올류, 왁스류 및 무기물질 중에서 선택된 1종 이상인 약제학적 제제.
- 제 70 항에 있어서, 상기 지방산 및 지방산 에스테르류로는 글리세릴 팔미토스테아레이트, 글리세릴 스테아레이트, 글리세릴 비헤네이트, 세틸 팔미테이트, 글리세릴 모노 올레이트 및 스테아린산 중에서 선택된 1종 이상 이고; 상기 지방산 알코올류는 세토스테아릴 알코올, 세틸알코올 및 스테아릴알코올 중에서 선택된 1종 이상 이며;, 상기 왁스류는 카르나우바왁스, 밀납 및 미결정왁스 중에서 선택된 1종 이상 이고; 상기 무기물질은 탈크, 침강탄산칼슘, 인산일수소칼슘, 산화아연, 산화티탄, 카올린, 벤토나이트, 몬모릴로나이트 및 비검중에서 선택된 1종 이상인 약제학적 제제.
- 제 70 항에 있어서, 소수성 화합물이 발사르탄1 중량부에 대해서 0.1 중량부 내지 20 중량부인 약제학적 제제.
- 제 60 항에 있어서, 상기 친수성 고분자는 당류, 셀룰로오스 유도체, 검류, 단백질류, 폴리비닐 유도체, 친수성 폴리메타크릴레이트 공중합체, 폴리에틸렌 유도체 및 카르복시비닐폴리머 중에서 선택된 1종 이상인 약제학적 제제.
- 제 73 항에 있어서, 상기 당류는 덱스트린, 폴리덱스트린, 덱스트란, 펙틴 및 펙틴 유도체, 알긴산염, 폴리갈락투론산, 자일란, 아라비노자일란, 아라비노갈락탄, 전분, 히드록시프로필스타치, 아밀로오스 및 아밀로펙틴 중에서 선택된 1종 이상이고; 상기 셀룰로오스 유도체는 히프로멜로오스, 히드록시프로필셀룰로오스, 히드록시메틸셀룰로오스, 히드록시에틸셀룰로오스, 메틸셀룰로오스 및 카르복시메틸셀룰로오스 나트륨 중에서 선택된 1종 이상이며; 상기 검류는 구아검, 로커스트 콩검, 트라가칸타, 카라기난, 아카시아검, 아라비아검, 젤란검 및 잔탄검 중에서 선택된 1종 이상이고; 상기 단백질류는 젤라틴, 카제인 및 제인 중에서 선택 1종 이상이며; 상기 폴리비닐 유도체는 폴리비닐 알코올, 폴리비닐 피롤리돈 및 폴리비닐아세탈디에틸아미노아세테이트 중에서 선택된 1종 이상이며; 상기 친수성 폴리메타크릴레이트 공중합체는 폴리(부틸 메타크릴레이트-(2-디메틸아미노에틸)메타크릴레이트-메틸메타크릴레이트) 공중합체이고; 상기 폴리에틸렌 유도체는 폴리에틸렌글리콜 및 폴리에틸렌 옥사이드 중에서 선택된 1종 이상이며; 상기 카르복시비닐폴리머는 카보머인 것인 약제학적 제제.
- 제 73 항에 있어서, 친수성 고분자가 발사르탄1 중량부에 대해서 0.05 중량부 내지 30 중량부인 약제학적 제제.
- 제 55 항에 있어서, 상기 지연방출성 구획은 삼투압 조절제를 포함하며 반투과성막 코팅기제로 코팅된 것인 약제학적 제제.
- 제 76 항에 있어서, 상기 삼투압 조절제는 황산마그네슘, 염화마그네슘, 염화나트륨, 염화리튬, 황산칼륨, 황산나트륨, 황산리튬, 황산나트륨, 및 이들의 혼합물로 이루어진 군에서 선택된 1종 이상인 것인 약제학적 제제.
- 제 76 항에 있어서, 상기 반투과성막 코팅기제는 폴리비닐 아세테이트, 수불용성 폴리메타크릴레이트 공중합체, 에틸셀룰로오스, 셀룰로오스 에스테르, 셀룰로오스 에테르, 셀룰로오스 아실레이트, 셀룰로오스 디아실레이트, 셀룰로오스 트리아실레이트, 셀룰로오스 아세테이트, 셀룰로오스 디아세테이트, 셀룰로오스 트리아세테이트, 및 이들의 혼합물로 이루어진 군에서 선택된 1종 이상인 것인 약제학적 제제.
- 약리학적 활성성분으로 심바스타틴(Simvastatin), 그의 이성질체 또는 그의 약학적으로 허용하는 염 을 함유하는 선방출성 구획, 및 약리학적 활성성분으로 칸데사르탄 (Candesartan), 그의 프로드럭 및 그의 약제학적으로 허용하는 염.
- 제 79 항에 있어서, 상기 심바스타틴이 용출 개시 후 30분 이내에 심바스타틴 총량의 80% 이상이 방출되는 약제학적 제제.
- 제 79 항에 있어서, 상기 칸데사르탄이 경구투여시 심바스타틴 용출개시 후 2시간까지 단위제제 내 칸데사르탄 총량의 35% 이내로 방출되는 약제학적 제제.
- 제 79 항에 있어서, 상기 칸데사르탄 프로드럭이 칸데사르탄 실렉세틸인 약제학적 제제.
- 제 79 항에 있어서, 상기 심바스타틴은 단위제제 중 1 내지 160mg 인 약제학적 제제.
- 제 79 항에 있어서, 상기 칸데사르탄은 단위제제 중 2 내지 64mg인 약제학적 제제.
- 제 79 항에 있어서, 상기 지연방출성 구획은 장용성 고분자, 수불용성 중합체, 소수성 화합물 및 친수성 고분자로 이루어진 군에서 선택된 1종 이상의 방출제어물질을 포함하는 약제학적 제제.
- 제 85 항에 있어서, 상기 방출제어물질이 장용성 고분자 및 수불용성 중합체 중에서 선택된 1종 이상인 약제학적 제제.
- 제 85 항에 있어서, 상기 방출제어물질이 칸데사르탄 1중량부에 대하여 0.05중량부 내지 100중량부로 포함되는 것인 약제학적 제제.
- 제 85 항에 있어서, 상기 장용성 고분자는 장용성 셀룰로오스 유도체, 장용성 아크릴산계 공중합체, 장용성 폴리메타크릴레이트 공중합체, 장용성 말레인산계 공중합체 및 장용성 폴리비닐 유도체 중에서 선택된 1종 이상인 것인 약제학적 제제.
- 제 88 항에 있어서, 상기 장용성 셀룰로오스 유도체는 히프로멜로오스아세테이트숙시네이트, 히프멜로오스프탈레이트, 히드록시메틸에틸셀룰로오스프탈레이트, 셀룰로오스아세테이트프탈레이트, 셀룰로오스아세테이트숙시네이트, 셀룰로오스아세테이트말레이트, 셀룰로오스벤조에이트프탈레이트, 셀룰로오스프로피오네이트프탈레이트, 메틸셀룰로오스프탈레이트, 카르복시메틸에틸셀룰로오스, 에틸히드록시에틸셀룰로오스프탈레이트 및 메틸히드록시에틸셀룰로오스 중에서 선택된 1종 이상이고; 상기 장용성 아크릴산계 공중합체는 스티렌-아크릴산 공중합체, 아크릴산메틸-아크릴산 공중합체, 아크릴산메틸메타크릴산 공중합체, 아크릴산부틸-스티렌-아크릴산 공중합체, 및 아크릴산메틸-메타크릴산-아크릴산옥틸공중합체 중에서 선택된 1종 이상이며; 상기 장용성 폴리메타크릴레이트 공중합체는 폴리(메타크릴산 메틸 메타크릴레이트) 공중합체, 폴리 (메타크릴산 에틸아크릴레이트) 공중합체 중에서 선택된 1종 이상이며, 상기 장용성 말레인산계 공중합체는 아세트산비닐-말레인산 무수물 공중합체, 스티렌-말레인산 무수물 공중합체, 스티렌-말레인산모노에스테르 공중합체, 비닐메틸에테르-말레인산 무수물 공중합체, 에틸렌-말레인산 무수물 공중합체, 비닐부틸에테르-말레인산 무수물 공중합체, 아크릴로니트릴-크릴산메틸ㆍ말레인산 무수물 공중합체 및 아크릴산부틸-스티렌-말레인산 무수물 공중합체중에서 선택된 1종 이상이고; 또는 상기 장용성 폴리비닐 유도체는 폴리비닐알콜프탈레이트, 폴리비닐아세탈프탈레이트, 폴리비닐부틸레이트프탈레이트, 및 폴리비닐아세트아세탈프탈레이트 중에서 선택된 1종 이상인 약제학적 제제.
- 제 89 항에 있어서, 장용성 고분자가 히프로멜로오스 아세테이트 숙시네이트 또는 아크릴산메틸메타크릴산 공중합체인 약제학적 제제.
- 제 88 항에 있어서, 상기 장용성 고분자가 칸데사르탄 1 중량부에 대해서 0.1 중량부 내지 20 중량부인 약제학적 제제.
- 제 85 항에 있어서, 상기 수불용성 중합체는 폴리비닐아세테이트, 수불용성 폴리메타크릴레이트 공중합체, 에틸셀룰로오스, 셀룰로오스 에스테르, 셀룰로오스 에테르, 셀룰로오스 아실레이트, 셀룰로오스 디아실레이트, 셀룰로오스 트리아실레이트, 셀룰로오스아세테이트, 셀룰로오스 디아세테이트, 및 셀룰로오스 트리아세테이트 중에서 선택된 1 종 이상인 약제학적 제제.
- 제 92 항에 있어서, 상기 수불용성 중합체는 수불용성 폴리메타크릴레이트인 폴리(에틸아크릴레이트-메틸 메타크릴레이트-트리메틸아미노에틸메타크릴레이트 클로라이드), 폴리비닐아세테이트 또는 에틸셀룰로오스 인 약제학적 제제.
- 제 92 항에 있어서, 상기 수불용성 중합체가 칸데사르탄 1 중량부에 대해서 0.1 중량부 내지 30 중량부인 약제학적 제제.
- 제 85 항에 있어서, 상기 소수성 화합물은 지방산 및 지방산 에스테르류, 지방산 알코올류, 왁스류 및 무기물질 중에서 선택된 1종 이상인 약제학적 제제.
- 제 95 항에 있어서, 상기 지방산 및 지방산 에스테르류로는 글리세릴 팔미토스테아레이트, 글리세릴 스테아레이트, 글리세릴 비헤네이트, 세틸 팔미테이트, 글리세릴 모노 올레이트 및 스테아린산 중에서 선택된 1종 이상 이고; 상기 지방산 알코올류는 세토스테아릴 알코올, 세틸알코올 및 스테아릴알코올 중에서 선택된 1종 이상 이며; 상기 왁스류는 카르나우바왁스, 밀납 및 미결정왁스 중에서 선택된 1종 이상 이고; 상기 무기물질은 탈크, 침강탄산칼슘, 인산일수소칼슘, 산화아연, 산화티탄, 카올린, 벤토나이트, 몬모릴로나이트 및 비검중에서 선택된 1종 이상인 약제학적 제제.
- 제 95 항에 있어서, 소수성 화합물이 칸데사르탄 1 중량부에 대해서 0.1 중량부 내지 20 중량부인 약제학적 제제.
- 제 96 항에 있어서, 상기 소수성 화합물이 카르나우바왁스인 약제학적 제제.
- 제 85 항에 있어서, 상기 친수성 고분자는 당류, 셀룰로오스 유도체, 검류, 단백질류, 폴리비닐 유도체, 친수성 폴리메타크릴레이트 공중합체, 폴리에틸렌 유도체 및 카르복시비닐폴리머 중에서 선택된 1종 이상인 약제학적 제제.
- 제 99 항에 있어서, 상기 당류는 덱스트린, 폴리덱스트린, 덱스트란, 펙틴 및 펙틴 유도체, 알긴산염, 폴리갈락투론산, 자일란, 아라비노자일란, 아라비노갈락탄, 전분, 히드록시프로필스타치, 아밀로오스 및 아밀로펙틴 중에서 선택된 1종 이상이고; 상기 셀룰로오스 유도체는 히프로멜로오스, 히드록시프로필셀룰로오스, 히드록시메틸셀룰로오스, 히드록시에틸셀룰로오스, 메틸셀룰로오스, 카르복시메틸셀룰로오스 나트륨, 히드록시프로필 메틸셀룰로오스 아세테이트 숙시네이트 및 히드록시에틸메틸셀룰로오스에서 선택된 1종 이상이며; 상기 검류는 구아검, 로커스트 콩검, 트라가칸타, 카라기난, 아카시아검, 아라비아검, 젤란검 및 잔탄검 중에서 선택된 1종 이상이고; 상기 단백질류는 젤라틴, 카제인 및 제인 중에서 선택 1종 이상이며; 상기 폴리비닐 유도체는 폴리비닐 알코올, 폴리비닐 피롤리돈 및 폴리비닐아세탈디에틸아미노아세테이트 중에서 선택된 1종 이상이며; 상기 친수성 폴리메타크릴레이트 공중합체는 폴리(부틸 메타크릴레이트-(2-디메틸아미노에틸)메타크릴레이트-메틸메타크릴레이트) 공중합체 이고; 상기 폴리에틸렌 유도체는 폴리에틸렌글리콜 및 폴리에틸렌 옥사이드 중에서 선택된 1종 이상이며; 상기 카르복시비닐폴리머는 카보머인 것인 약제학적 제제.
- 제 100 항에 있어서, 친수성 고분자는 히프로멜로스 인 약제학적 제제.
- 제 99 항에 있어서, 친수성 고분자가 칸데사르탄 1 중량부에 대해서 0.05 중량부 내지 30 중량부인 약제학적 제제.
- 제 79 항에 있어서, 상기 지연방출성 구획은 삼투압 조절제를 포함하며 반투과성막 코팅기제로 코팅된 것인 약제학적 제제.
- 제 103 항에 있어서, 상기 삼투압 조절제는 황산마그네슘, 염화마그네슘, 염화나트륨, 염화리튬, 황산칼륨, 황산나트륨, 황산리튬, 황산나트륨, 및 이들의 혼합물로 이루어진 군에서 선택된 1종 이상인 것인 약제학적 제제.
- 제 104 항에 있어서, 상기 삼투압 조절제는 염화나트륨 또는 황산나트륨인 약제학적 제제.
- 제 103 항에 있어서, 상기 반투과성막 코팅기제는 폴리비닐 아세테이트, 수불용성 폴리메타크릴레이트 공중합체, 에틸셀룰로오스, 셀룰로오스 에스테르, 셀룰로오스 에테르, 셀룰로오스 아실레이트, 셀룰로오스 디아실레이트, 셀룰로오스 트리아실레이트, 셀룰로오스 아세테이트, 셀룰로오스 디아세테이트, 셀룰로오스 트리아세테이트, 및 이들의 혼합물로 이루어진 군에서 선택된 1종 이상인 것인 약제학적 제제.
- 제 106 항에 있어서, 상기 반투과성막 코팅기제는 에틸셀룰로오스 또는 셀룰로오스 아세테이트인 약제학적 제제.
- 약리학적 활성성분으로 로슈바스타틴(Rosuvastatin), 그의 이성질체 또는 그의 약학적으로 허용하는 염을 함유하는 선방출성 구획, 및 약리학적 활성성분으로 로자탄(losartan) 또는 그의 약학적으로 을 함유하는 지연방출성 구획을 포함하는 약제학적 제제.
- 제108항에 있어서, 상기 로자탄의 방출이 상기 로슈바스타틴 방출 개시 2시간 이후에 개시되는 약제학적 제제.
- 제108항에 있어서, 상기 로자탄의 방출이 상기 로슈바스타틴 방출 개시 3시간 이후에 개시되는 약제학적 제제.
- 제108항에 있어서, 상기 로자탄이 상기 로슈바스타틴 방출 개시 후 3시간 경과시 까지 단위 제형 중 로자탄 총량의 0 내지 40%가 방출되는 약제학적 제제.
- 제108항에 있어서, 상기 로자탄이 상기 로슈바스타틴 방출 개시 후 3시간 경과시 까지 단위 제형 중 로자탄 총량의 0 내지 20%가 방출되는 약제학적 제제.
- 제 108 항에 있어서, 상기 지연방출성 구획은 장용성 고분자, 수불용성 중합체, 소수성 화합물, 친수성 고분자, 및 이들의 혼합물로 이루어진 군에서 선택된 방출제어물질을 포함하는 것인 약제학적 제제.
- 제 113 항에 있어서, 상기 방출제어물질은 로자탄 1 중량부에 대하여, 0.05 내지 100 중량부로 포함되는 것인 약제학적 제제.
- 제 113 항에 있어서, 상기 장용성 고분자는 장용성 셀룰로오스 유도체, 장용성 아크릴산계 공중합체, 장용성 폴리메타크릴레이트 공중합체, 장용성 말레인산계 공중합체, 장용성 폴리비닐 유도체, 및 이들의 혼합물로 이루어진 군에서 선택된 것인 약제학적 제제.
- 제 115 항에 있어서, 상기 장용성 셀룰로오스 유도체는 히프로멜로오스아세테이트숙시네이트, 히프멜로오스프탈레이트, 히드록시메틸에틸셀룰로오스프탈레이트, 셀룰로오스아세테이트프탈레이트, 셀룰로오스아세테이트숙시네이트, 셀룰로오스아세테이트말레이트, 셀룰로오스벤조에이트프탈레이트, 셀룰로오스프로피오네이트프탈레이트, 메틸셀룰로오스프탈레이트, 카르복시메틸에틸셀룰로오스, 에틸히드록시에틸셀룰로오스프탈레이트 및 메틸히드록시에틸셀룰로오스 중에서 선택된 1종 이상이고; 상기 장용성 아크릴산계 공중합체는 스티렌-아크릴산 공중합체, 아크릴산메틸-아크릴산 공중합체, 아크릴산메틸메타크릴산 공중합체, 아크릴산부틸-스티렌-아크릴산 공중합체, 및 아크릴산메틸-메타크릴산-아크릴산옥틸공중합체 중에서 선택된 1종 이상이며; 상기 장용성 폴리메타크릴레이트 공중합체는 폴리(메타크릴산-메틸 메타크릴산메틸) 공중합체, 폴리(메타크릴산ㆍ에틸아크릴레이트)공중합체 중에서 선택된 1종 이상이며; 상기 장용성 말레인산계 공중합체는 아세트산비닐-말레인산 무수물 공중합체, 스티렌-말레인산 무수물 공중합체, 스티렌-말레인산모노에스테르 공중합체, 비닐메틸에테르-말레인산 무수물 공중합체, 에틸렌-말레인산 무수물 공중합체, 비닐부틸에테르-말레인산 무수물 공중합체, 아크릴로니트릴-크릴산메틸ㆍ말레인산 무수물 공중합체 및 아크릴산부틸-스티렌-말레인산 무수물 공중합체중에서 선택된 1종 이상이고; 또는 상기 장용성 폴리비닐 유도체는 폴리비닐알콜프탈레이트, 폴리비닐아세탈프탈레이트, 폴리비닐부틸레이트프탈레이트, 및 폴리비닐아세트아세탈프탈레이트 중에서 선택된 1종 이상인 약제학적 제제.
- 제 113 항에 있어서, 상기 수불용성 중합체는 폴리비닐아세테이트, 수불용성 폴리메타크릴레이트 공중합체, 에틸셀룰로오스, 셀룰로오스 에스테르, 셀룰로오스 에테르, 셀룰로오스 아실레이트, 셀룰로오스 디아실레이트, 셀룰로오스 트리아실레이트, 셀룰로오스아세테이트, 셀룰로오스 디아세테이트, 및 셀룰로오스 트리아세테이트 중에서 선택된 1 종 이상인 약제학적 제제.
- 제 113 항에 있어서, 상기 소수성 화합물은 지방산 및 지방산 에스테르류, 지방산 알코올류, 왁스류, 무기물질, 및 이들의 혼합물로 이루어진 군에서 선택된 것인 약제학적 제제.
- 제 118 항에 있어서, 상기 지방산 및 지방산 에스테르류는 글리세릴 팔미토스테아레이트, 글리세릴 스테아레이트, 글리세릴 비헤네이트, 세틸 팔미테이트, 글리세릴 모노 올레이트 및 스테아린산 중에서 선택된 하나 이상; 상기 지방산 알코올류는 세토스테아릴 알코올, 세틸알코올 및 스테아릴알코올 중에서 선택된 하나 이상; 상기 왁스류는 카르나우바왁스, 밀납, 및 미결정왁스 중에서 선택된 하나 이상; 또는 상기 무기물질은 탈크, 침강탄산칼슘, 인산일수소칼슘, 산화아연, 산화티탄, 카올린, 벤토나이트, 몬모릴로나이트 및 비검 중에서 선택된 하나 이상인 약제학적 제제.
- 제 113 항에 있어서, 상기 친수성 고분자는 당류, 셀룰로오스 유도체, 검류, 단백질류, 폴리비닐 유도체, 친수성 폴리메타크릴레이트 공중합체, 폴리에틸렌 유도체, 카르복시비닐폴리머, 및 이들의 혼합물로 이루어진 군에서 선택된 것인 약제학적 제제.
- 제 120 항에 있어서, 상기 당류는 덱스트린, 폴리덱스트린, 덱스트란, 펙틴 및 펙틴 유도체, 알긴산염, 폴리갈락투론산, 자일란, 아라비노자일란, 아라비노갈락탄, 전분, 히드록시프로필스타치, 아밀로오스, 및 아밀로펙틴 중에서 선택된 하나 이상; 상기 셀룰로오스 유도체는 히프로멜로오스, 히드록시프로필셀룰로오스, 히드록시메틸셀룰로오스, 히드록시에틸셀룰로오스, 메틸셀룰로오스, 카르복시메틸셀룰로오스 나트륨, 히드록시프로필 메틸셀룰로오스 아세테이트 숙시네이트, 및 히드록시에틸메틸셀룰로오스 중에서 선택된 하나 이상이고; 상기 검류는 구아검, 로커스트 콩 검, 트라가칸타, 카라기난, 아카시아검, 아라비아검, 젤란검, 및 잔탄검 중에서 선택된 하나 이상이며; 상기 단백질류는 젤라틴, 카제인, 및 제인 중에서 선택된 하나 이상이고; 상기 폴리비닐 유도체는 폴리비닐 알코올, 폴리비닐 피롤리돈, 및 폴리비닐아세탈디에틸아미노아세테이트 중에서 선택된 하나 이상이며; 상기 친수성 폴리메타크릴레이트 공중합체는 폴리(부틸 메타크릴레이트-(2-디메틸아미노에틸)메타크릴레이트-메틸메타크릴레이트) 공중합체이고; 상기 폴리에틸렌 유도체는 폴리에틸렌 글리콜, 및 폴리에틸렌 옥사이드 중에서 선택된 하나 이상이며; 또는 상기 카르복시비닐폴리머는 카보머인 약제학적 제제.
- 제 108 항에 있어서, 상기 지연방출성 구획은 삼투압 조절제를 포함하며 반투과성막 코팅기제로 코팅된 것인 약제학적 제제.
- 제 122 항에 있어서, 상기 반투과성막 코팅기제는 폴리비닐 아세테이트, 수불용성 폴리메타크릴레이트 공중합체, 에틸셀룰로오스, 셀룰로오스 에스테르, 셀룰로오스 에테르, 셀룰로오스 아실레이트, 셀룰로오스 디아실레이트, 셀룰로오스 트리아실레이트, 셀룰로오스 아세테이트, 셀룰로오스 디아세레이티드, 셀룰로오스 트리아세테이트, 및 이들의 혼합물로 이루어진 군에서 선택된 것인 약제학적 제제.
- 제 122 항에 있어서, 상기 삼투압 조절제는 황산마그네슘, 염화마그네슘, 염화나트륨, 염화리튬, 황산칼륨, 황산나트륨, 황산리튬, 황산나트륨, 및 이들의 혼합물로 이루어진 군에서 선택된 것인 약제학적 제제.
- 제 108 항에 있어서, 상기 로슈바스타틴은 제제 중 2.5 내지 80 mg 포함되는 것인 약제학적 제제.
- 제 108 항에 있어서, 상기 로자탄은 로자탄 칼륨으로 제제 중 10 내지 200 mg 포함되는 것인 약제학적 제제.
- 제 113 항에 있어서, 상기 방출제어물질은 카보머, 히프로멜로오스아세테이트숙시네이트, 폴리비닐아세테이트, 카라나우바왁스, 히프로멜로오스, 히드록시프로필셀룰로오스, 에틸셀룰로오스, 히프로멜로오스프탈레이트, 아크릴산메틸메타크릴산 공중합체 및 이들의 혼합물로 구성된 군에서 선택된 것인 약제학적 제제.
- 제 127 항에 있어서, 상기 방출제어물질은 히프로멜로오스아세테이트숙시네이트, 폴리비닐아세테이트, 카보머 및 이들의 혼합물로 구성된 군에서 선택된 것인 약제학적 제제.
- 제128항에 있어서, 상기 지연방출성 구획은 폴리비닐아세테이트를 방출제어물질로 포함하는 것인 약제학적 제제.
- 약리학적 활성성분으로 로슈바스타틴(Rosuvastatin), 그의 이성질체 또는 그의 약학적으로 허용하는 염 을 함유하는 선방출성 구획, 및 약리학적 활성성분으로 이베사르탄(Irbesartan) 또는 그의 약학적으로 허용 가능한 염을 함유하는 지연방출성 구획을 포함하는 방출성이 제어된 약제학적 제제.
- 제130항에 있어서, 상기 이베사르탄의 방출이 상기 로슈바스타틴 방출 개시 후 2시간 내지 6시간 사이에 개시되는 약제학적 제제.
- 제130항에 있어서, 상기 이베사르탄의 방출이 상기 로슈바스타틴 방출개시 후 2시간 이상 내지 5시간 사이에 개시되는 약제학적 제제.
- 제130항에 있어서, 상기 이베사르탄이 상기 로슈바스타틴 방출 개시 후 2시간 경과시까지 단위제제 중 이베사르탄총량의 0 내지 20%방출되는 약제학적 제제.
- 제130항에 있어서, 상기 이베사르탄이 상기 로슈바스타틴 방출개시 후 4시간 경과시까지 단위제제 중 이베사르탄 총량의 0 내지 20% 방출되는 약제학적 제제.
- 제 130 항에 있어서, 상기 지연방출성 구획은 장용성고분자, 수불용성 중합체, 소수성 화합물, 친수성 고분자, 및 이들의 혼합물로 이루어진 군에서 선택된 방출제어물질을 포함하는 것인 약제학적 제제.
- 제 135 항에 있어서, 상기 방출제어물질은 이베사르탄 1 중량부에 대하여, 0.05 내지 100 중량부로 포함되는 것인 약제학적 제제.
- 제 135 항에 있어서, 상기 장용성고분자는 장용성셀룰로오스유도체, 장용성아크릴산계공중합체, 장용성 폴레메타크릴레이트 공중합체, 장용성말레인산계공중합체, 장용성폴리비닐유도체, 및 이들의 혼합물로 이루어진 군에서 선택된 것인 약제학적 제제.
- 제 137 항에 있어서, 상기 장용성 셀룰로오스 유도체는 히프로멜로오스아세테이트숙시네이트, 히프멜로오스프탈레이트, 히드록시메틸에틸셀룰로오스프탈레이트, 셀룰로오스아세테이트프탈레이트, 셀룰로오스아세테이트숙시네이트, 셀룰로오스아세테이트말레이트, 셀룰로오스벤조에이트프탈레이트, 셀룰로오스프로피오네이트프탈레이트, 메틸셀룰로오스프탈레이트, 카르복시메틸에틸셀룰로오스, 에틸히드록시에틸셀룰로오스프탈레이트 및 메틸히드록시에틸셀룰로오스 중에서 선택된 1종 이상이고; 상기 장용성 아크릴산계 공중합체는 스티렌-아크릴산 공중합체, 아크릴산메틸-아크릴산 공중합체, 아크릴산메틸메타크릴산 공중합체, 아크릴산부틸-스티렌-아크릴산 공중합체, 및 아크릴산메틸-메타크릴산-아크릴산옥틸공중합체 중에서 선택된 1종 이상이며; 상기 장용성 폴리메타크릴레이트 공중합체는 폴리(메타크릴산-메틸 메타크릴산메틸) 공중합체, 폴리(메타크릴산ㆍ에틸아크릴레이트)공중합체 중에서 선택된 1종 이상이며; 상기 장용성 말레인산계 공중합체는 아세트산비닐-말레인산 무수물 공중합체, 스티렌-말레인산 무수물 공중합체, 스티렌-말레인산모노에스테르 공중합체, 비닐메틸에테르-말레인산 무수물 공중합체, 에틸렌-말레인산 무수물 공중합체, 비닐부틸에테르-말레인산 무수물 공중합체, 아크릴로니트릴-크릴산메틸ㆍ말레인산 무수물 공중합체 및 아크릴산부틸-스티렌-말레인산 무수물 공중합체중에서 선택된 1종 이상이고; 또는 상기 장용성 폴리비닐 유도체는 폴리비닐알콜프탈레이트, 폴리비닐아세탈프탈레이트, 폴리비닐부틸레이트프탈레이트, 및 폴리비닐아세트아세탈프탈레이트 중에서 선택된 1종 이상인 약제학적 제제.
- 제 135 항에 있어서, 상기 수불용성 중합체는 폴리비닐아세테이트, 수불용성 폴리메타크릴레이트 공중합체, 에틸셀룰로오스, 셀룰로오스 에스테르, 셀룰로오스 에테르, 셀룰로오스 아실레이트, 셀룰로오스 디아실레이트, 셀룰로오스 트리아실레이트, 셀룰로오스아세테이트, 셀룰로오스 디아세테이트, 및 셀룰로오스 트리아세테이트 중에서 선택된 1 종 이상인 약제학적 제제.
- 제 135 항에 있어서, 상기 소수성 화합물은 지방산 및 지방산 에스테르류, 지방산 알코올류, 왁스류, 무기물질, 및 이들의 혼합물로 이루어진 군에서 선택된 것인 약제학적 제제.
- 제 140 항에 있어서, 상기 지방산 및 지방산 에스테르류는 글리세릴 팔미토스테아레이트, 글리세릴 스테아레이트, 글리세릴 비헤네이트, 세틸 팔미테이트, 글리세릴 모노 올레이트 및 스테아린산 중에서 선택된 하나 이상; 상기 지방산 알코올류는 세토스테아릴 알코올, 세틸알코올 및 스테아릴알코올 중에서 선택된 하나 이상; 상기 왁스류는 카르나우바왁스, 밀납, 및 미결정왁스 중에서 선택된 하나 이상; 또는 상기 무기물질은 탈크, 침강탄산칼슘, 인산일수소칼슘, 산화아연, 산화티탄, 카올린, 벤토나이트, 몬모릴로나이트 및 비검 중에서 선택된 하나 이상인 약제학적 제제.
- 제 135 항에 있어서, 상기 친수성 고분자는 당류, 셀룰로오스 유도체, 검류, 단백질류, 폴리비닐 유도체, 친수성 폴리메타크릴레이트 공중합체, 폴리에틸렌 유도체, 카르복시비닐폴리머, 및 이들의 혼합물로 이루어진 군에서 선택된 것인 약제학적 제제.
- 제 142 항에 있어서, 상기 당류는 덱스트린, 폴리덱스트린, 덱스트란, 펙틴 및 펙틴 유도체, 알긴산염, 폴리갈락투론산, 자일란, 아라비노자일란, 아라비노갈락탄, 전분, 히드록시프로필스타치, 아밀로오스, 및 아밀로펙틴 중에서 선택된 하나 이상; 상기 셀룰로오스 유도체는 히프로멜로오스, 히드록시프로필셀룰로오스, 히드록시메틸셀룰로오스, 히드록시에틸셀룰로오스, 메틸셀룰로오스, 카르복시메틸셀룰로오스 나트륨, 히드록시프로필 메틸셀룰로오스 아세테이트 숙시네이트, 및 히드록시에틸메틸셀룰로오스 중에서 선택된 하나 이상; 상기 검류는 구아검, 로커스트 콩 검, 트라가칸타, 카라기난, 아카시아검, 아라비아검, 젤란검, 및 잔탄검 중에서 선택된 하나 이상; 상기 단백질류는 젤라틴, 카제인, 및 제인 중에서 선택된 하나 이상; 상기 폴리비닐 유도체는 폴리비닐 알코올, 폴리비닐 피롤리돈, 및 폴리비닐아세탈디에틸아미노아세테이트 중에서 선택된 하나 이상; 상기 친수성 폴리메타크릴레이트 공중합체는 폴리(부틸 메타크릴레이트-(2-디메틸아미노에틸)메타크릴레이트-메틸메타크릴레이트) 공중합체; 상기 폴리에틸렌 유도체는 폴리에틸렌 글리콜, 및 폴리에틸렌 옥사이드 중에서 선택된 하나 이상이며; 상기 카르복시비닐폴리머는 카보머인 약제학적 제제.
- 제 130 항에 있어서, 상기 지연방출성 구획은 삼투압 조절제를 포함하며 반투과성막 코팅기제로 코팅된 것인 약제학적 제제.
- 제 144 항에 있어서, 상기 반투과성막 코팅기제는 폴리비닐 아세테이트, 수불용성폴리메타크릴레이트 공중합체, 에틸셀룰로오스, 셀룰로오스 에스테르, 셀룰로오스 에테르, 셀룰로오스 아실레이트, 셀룰로오스 디아실레이트, 셀룰로오스 트리아실레이트, 셀룰로오스 아세테이트, 셀룰로오스 디아세레이티드, 셀룰로오스 트리아세테이트, 및 이들의 혼합물로 이루어진 군에서 선택된 것인 약제학적 제제.
- 제 144 항에 있어서, 상기 삼투압 조절제는 황산마그네슘, 염화마그네슘, 염화나트륨, 염화리튬, 황산칼륨, 황산나트륨, 황산리튬, 황산나트륨, 및 이들의 혼합물로 이루어진 군에서 선택된 것인 약제학적 제제.
- 제 130 항에 있어서, 상기 로슈바스타틴은 제제 중 0.1 내지 200 mg 포함되는 것인 약제학적 제제.
- 제 130 항에 있어서, 상기 이베사르탄은 제제 중 1 내지 1000 mg 포함되는 것인 약제학적 제제.
- 제 130 항에 있어서, 상기 약제학적 제제는 이층정 또는 유핵정인 약제학적 제제.
- 제135항에 있어서, 상기 방출제어물질은 히프로멜로오스아세테이트숙시네이트, 히프로멜로오스프탈레이트, 아크릴산메틸메타크릴산 공중합체, 폴리비닐아세테이트, 에틸셀룰로오스, 셀룰로오스 아세테이트, 카르나우바왁스, 히프로멜로오스, 히드록시프로필셀룰로오스, 폴리비닐 피롤리돈, 및 이들의 혼합물로 이루어진 군으로부터 선택된 것인 약제학적 제제.
- 제150항에 있어서, 상기 방출제어물질은 폴리비닐아세테이트, 히프로멜로오스아세테이트숙시네이트, 카르나우바왁스, 히프로멜로오스, 및 이들의 혼합물로 이루어진 군으로부터 선택된 것인 약제학적 제제.
- 제130항에 있어서, 상기 지연방출성 구획은 방출제어물질로 코팅된 것인 약제학적 제제.
- 제152항에 있어서, 상기 방출제어물질은 히프로멜로오스아세테이트숙시네이트, 히프로멜로오스프탈레이트, 아크릴산메틸메타크릴산 공중합체, 폴리비닐아세테이트, 에틸셀룰로오스, 셀룰로오스 아세테이트, 카르나우바왁스, 히프로멜로오스, 히드록시프로필셀룰로오스, 폴리비닐 피롤리돈, 및 이들의 혼합물로 이루어진 군으로부터 선택된 것인 약제학적 제제.
- 약리학적 활성성분으로 로슈바스타틴(Rosuvastatin), 그의 이성질체 또는 그의 약학적으로 허용하는 염 을 함유하는 선방출성 구획, 및 약리학적 활성성분으로 칸데사르탄(Candesartan), 그의 약학적으로 허용 가능한 염 또는 그의 프로드럭을 함유하는 지연방출성 구획을 포함하는 방출성이 제어된 약제학적 제제.
- 제154항에 있어서, 상기 칸데사르탄의 방출이 상기 로슈바스타틴 방출 개시 후 1시간 내지 8시간 사이에 개시되는 약제학적 제제.
- 제154항에 있어서, 상기 칸데사르탄의 방출이 상기 로슈바스타틴 방출 개시 후 2시간 내지 4시간 사이에 개시되는 약제학적 제제.
- 제154항에 있어서, 상기 칸데사르탄이 상기 로슈바스타틴 방출 개시 후 2시간 경과시까지 단위제제 중 칸데사르탄 총량의 0 내지 40% 이내로 방출되는 약제학적 제제.
- 제154항에 있어서, 상기 칸데사르탄이 상기 로슈바스타틴 방출개시 후 3시간 경과시까지 단위제제 중 칸데사르탄 총량의 0 내지 40% 이내로 방출되는 약제학적 제제.
- 제 154 항에 있어서, 상기 지연방출성 구획은 장용성 고분자, 수불용성 중합체, 소수성 화합물, 친수성 고분자, 및 이들의 혼합물로 이루어진 군에서 선택된 방출제어물질을 포함하는 것인 약제학적 제제.
- 제 159 항에 있어서, 상기 방출제어물질은 칸데사르탄 1 중량부에 대하여, 0.05 ~ 100 중량부로 포함되는 것인 약제학적 제제.
- 제 159 항에 있어서, 상기 장용성 고분자는 장용성 셀룰로오스 유도체, 장용성 아크릴산계 공중합체, 장용성 폴레메타크릴레이트 공중합체, 장용성 말레인산계 공중합체, 장용성 폴리비닐 유도체, 및 이들의 혼합물로 이루어진 군에서 선택된 것인 약제학적 제제.
- 제 161 항에 있어서, 상기 장용성 셀룰로오스 유도체는 히프로멜로오스아세테이트숙시네이트, 히프멜로오스프탈레이트, 히드록시메틸에틸셀룰로오스프탈레이트, 셀룰로오스아세테이트프탈레이트, 셀룰로오스아세테이트숙시네이트, 셀룰로오스아세테이트말레이트, 셀룰로오스벤조에이트프탈레이트, 셀룰로오스프로피오네이트프탈레이트, 메틸셀룰로오스프탈레이트, 카르복시메틸에틸셀룰로오스, 에틸히드록시에틸셀룰로오스프탈레이트 및 메틸히드록시에틸셀룰로오스 중에서 선택된 1종 이상이고; 상기 장용성 아크릴산계 공중합체는 스티렌-아크릴산 공중합체, 아크릴산메틸-아크릴산 공중합체, 아크릴산메틸메타크릴산 공중합체, 아크릴산부틸-스티렌-아크릴산 공중합체, 및 아크릴산메틸-메타크릴산-아크릴산옥틸공중합체 중에서 선택된 1종 이상이며; 상기 장용성 폴리메타크릴레이트 공중합체는 폴리(메타크릴산-메틸 메타크릴산메틸) 공중합체, 폴리(메타크릴산ㆍ에틸아크릴레이트)공중합체 중에서 선택된 1종 이상이며; 상기 장용성 말레인산계 공중합체는 아세트산비닐-말레인산 무수물 공중합체, 스티렌-말레인산 무수물 공중합체, 스티렌-말레인산모노에스테르 공중합체, 비닐메틸에테르-말레인산 무수물 공중합체, 에틸렌-말레인산 무수물 공중합체, 비닐부틸에테르-말레인산 무수물 공중합체, 아크릴로니트릴-크릴산메틸ㆍ말레인산 무수물 공중합체 및 아크릴산부틸-스티렌-말레인산 무수물 공중합체중에서 선택된 1종 이상이고; 또는 상기 장용성 폴리비닐 유도체는 폴리비닐알콜프탈레이트, 폴리비닐아세탈프탈레이트, 폴리비닐부틸레이트프탈레이트, 및 폴리비닐아세트아세탈프탈레이트 중에서 선택된 1종 이상인 약제학적 제제.
- 제 159 항에 있어서, 상기 수불용성 중합체는 폴리비닐아세테이트, 수불용성 폴리메타크릴레이트 공중합체, 에틸셀룰로오스, 셀룰로오스 에스테르, 셀룰로오스 에테르, 셀룰로오스 아실레이트, 셀룰로오스 디아실레이트, 셀룰로오스 트리아실레이트, 셀룰로오스아세테이트, 셀룰로오스 디아세테이트, 및 셀룰로오스 트리아세테이트 중에서 선택된 1 종 이상인 약제학적 제제.
- 제 159 항에 있어서, 상기 소수성 화합물은 지방산 및 지방산 에스테르류, 지방산 알코올류, 왁스류, 무기물질, 및 이들의 혼합물로 이루어진 군에서 선택된 것인 약제학적 제제.
- 제 164 항에 있어서, 상기 지방산 및 지방산 에스테르류는 글리세릴 팔미토스테아레이트, 글리세릴 스테아레이트, 글리세릴 비헤네이트, 세틸 팔미테이트, 글리세릴 모노 올레이트 및 스테아린산 중에서 선택된 하나 이상; 상기 지방산 알코올류는 세토스테아릴 알코올, 세틸알코올 및 스테아릴알코올 중에서 선택된 하나 이상; 상기 왁스류는 카르나우바왁스, 밀납, 및 미결정왁스 중에서 선택된 하나 이상; 또는 상기 무기물질은 탈크, 침강탄산칼슘, 인산일수소칼슘, 산화아연, 산화티탄, 카올린, 벤토나이트, 몬모릴로나이트 및 비검 중에서 선택된 하나 이상인 약제학적 제제.
- 제 159 항에 있어서, 상기 친수성 고분자는 당류, 셀룰로오스 유도체, 검류, 단백질류, 폴리비닐 유도체, 친수성 폴리메타크릴레이트 공중합체, 폴리에틸렌 유도체, 카르복시비닐폴리머, 및 이들의 혼합물로 이루어진 군에서 선택된 것인 약제학적 제제.
- 제 166 항에 있어서, 상기 당류는 덱스트린, 폴리덱스트린, 덱스트란, 펙틴 및 펙틴 유도체, 알긴산염, 폴리갈락투론산, 자일란, 아라비노자일란, 아라비노갈락탄, 전분, 히드록시프로필스타치, 아밀로오스, 및 아밀로펙틴 중에서 선택된 하나 이상; 상기 셀룰로오스 유도체는 히드록시프로필메틸셀룰로오스, 히드록시프로필셀룰로오스, 히드록시메틸셀룰로오스, 히드록시에틸셀룰로오스, 메틸셀룰로오스, 카르복시메틸셀룰로오스 나트륨, 히드록시프로필 메틸셀룰로오스 아세테이트 숙시네이트, 및 히드록시에틸메틸셀룰로오스 중에서 선택된 하나 이상; 상기 검류는 구아검, 로커스트 콩 검, 트라가칸타, 카라기난, 아카시아검, 아라비아검, 젤란검, 및 잔탄검 중에서 선택된 하나 이상; 상기 단백질류는 젤라틴, 카제인, 및 제인 중에서 선택된 하나 이상; 상기 폴리비닐 유도체는 폴리비닐 알코올, 폴리비닐 피롤리돈, 및 폴리비닐아세탈디에틸아미노아세테이트 중에서 선택된 하나 이상; 상기 친수성 폴리메타크릴레이트 공중합체는 폴리(부틸 메타크릴레이트-(2-디메틸아미노에틸)메타크릴레이트-메틸메타크릴레이트) 공중합체; 상기 폴리에틸렌 유도체는 폴리에틸렌 글리콜, 및 폴리에틸렌 옥사이드 중에서 선택된 하나 이상; 또는 상기 카르복시비닐폴리머는 카보머인 약제학적 제제.
- 제 154 항에 있어서, 상기 지연방출성 구획은 삼투압 조절제를 포함하며 반투과성막 코팅기제로 코팅된 것인 약제학적 제제.
- 제 168 항에 있어서, 상기 반투과성막 코팅기제는 폴리비닐 아세테이트, 수불용성 폴리메타크릴레이트 공중합체, 에틸셀룰로오스, 셀룰로오스 에스테르, 셀룰로오스 에테르, 셀룰로오스 아실레이트, 셀룰로오스 디아실레이트, 셀룰로오스 트리아실레이트, 셀룰로오스 아세테이트, 셀룰로오스 디아세레이티드, 셀룰로오스 트리아세테이트, 및 이들의 혼합물로 이루어진 군에서 선택된 것인 약제학적 제제.
- 제 168 항에 있어서, 상기 삼투압 조절제는 황산마그네슘, 염화마그네슘, 염화나트륨, 염화리튬, 황산칼륨, 황산나트륨, 황산리튬, 황산나트륨, 및 이들의 혼합물로 이루어진 군에서 선택된 것인 약제학적 제제.
- 제 154 항에 있어서, 상기 로슈바스타틴은 제제 중 5 ~ 40 mg 포함되는 것인 약제학적 제제.
- 제 154 항에 있어서, 상기 칸데사르탄은 칸데사르탄실렉세틸로 제제 중 4 ~ 32 mg 포함되는 것인 약제학적 제제.
- 제159항에 있어서, 상기 방출제어물질은 히드록시프로필메틸셀룰로오스아세테이트숙시네이트, 히드록시프로필메틸셀룰로오스프탈레이트, 아크릴산메틸메타크릴산 공중합체, 폴리비닐아세테이트, 에틸셀룰로오스, 셀룰로오스 아세테이트, 카르나우바왁스, 히드록시프로필메틸셀룰로오스, 히드록시프로필셀룰로오스, 폴리비닐 피롤리돈, 및 이들의 혼합물로 이루어진 군으로부터 선택된 것인 약제학적 제제.
- 제 1 항, 제30항, 제55항, 제79항, 제108항, 제130항 및 154항 중 어느 한 항에 있어서, 상기 약제학적 제제는 상기 지연방출성 구획과, 이를 둘러싸는 선방출성 구획으로 이루어진 2상의 매트릭스 정제 형태인 약제학적 제제.
- 제 1 항, 제30항, 제55항, 제79항, 제108항, 제130항 및 154항 중 어느 한 항에 있어서, 상기 약제학적 제제는 지연방출성 구획으로 이루어진 정제와 상기 정제의 외부를 둘러싸는 선방출성 구획으로 이루어진 필름코팅층으로 구성된 필름코팅정 형태인 약제학적 제제.
- 제 1 항, 제30항, 제55항, 제79항, 제108항, 제130항 및 154항 중 어느 한 항에 있어서, 상기 약제학적 제제는 상기 지연방출성 구획과 상기 선방출성 구획이 층을 이루는 다층정 형태인 약제학적 제제.
- 제 1 항, 제30항, 제55항, 제79항, 제108항, 제130항 및 154항 중 어느 한 항에 있어서, 상기 제제는 지연방출성 구획으로 이루어진 내핵과, 상기 내핵의 외면을 둘러싸고 있는 선방출성 구획으로 이루어진 외층으로 구성된 유핵정 형태인 약제학적 제제.
- 제 177 항에 있어서, 상기 유핵정은 반투과성막 유핵정인 약제학적 제제.
- 제 1 항, 제30항, 제55항, 제79항, 제108항, 제130항 및 154항 중 어느 한 항에 있어서, 상기 약제학적 제제는 지연방출성 구획으로 이루어진 입자, 과립, 펠렛, 또는 정제와, 선방출성 구획으로 이루어진 입자, 과립, 펠렛, 또는 정제를 포함하는 캡슐제 형태인 약제학적 제제.
- 제 1 항, 제30항, 제55항, 제79항, 제108항, 제130항 및 154항 중 어느 한 항에 있어서, 상기 지연방출성 구획, 또는 상기 선방출성 구획 중 하나 이상의 외부에 코팅층을 추가로 포함하는 약제학적 제제.
- 제 1 항, 제30항, 제55항, 제79항, 제108항, 제130항 및 154항 중 어느 한 항에 있어서, 외부에 코팅층을 추가로 포함하는 코팅정 형태인 약제학적 제제.
- 제 1 항, 제30항, 제55항, 제79항, 제108항, 제130항 및 154항 중 어느 한 항에 있어서, 상기 제제는 오후 5시 내지 11시 투여용인 약제학적 제제.
- 제 1 항, 제30항, 제55항, 제79항, 제108항, 제130항 및 154항 중 어느 한 항의 약제학적 제제를 포유류에게 투여하는 단계를 포함하는 심혈관계 질환 치료방법.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR10-2008-0039864 | 2008-04-29 | ||
| KR20080039864 | 2008-04-29 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| WO2009134086A2 true WO2009134086A2 (ko) | 2009-11-05 |
| WO2009134086A3 WO2009134086A3 (ko) | 2010-01-21 |
| WO2009134086A9 WO2009134086A9 (ko) | 2010-03-11 |
Family
ID=41255555
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/KR2009/002256 WO2009134076A2 (ko) | 2008-04-29 | 2009-04-29 | 방출성이 제어된 안지오텐신 - II - 수용체 차단제와 H M G - C o A 환원 효소 억제제의 복합제 조성물 |
| PCT/KR2009/002261 WO2009134079A2 (ko) | 2008-04-29 | 2009-04-29 | 약제학적 제제 |
| PCT/KR2009/002278 WO2009134086A2 (ko) | 2008-04-29 | 2009-04-29 | 심혈관계 질환 치료용 약제학적 제제 |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/KR2009/002256 WO2009134076A2 (ko) | 2008-04-29 | 2009-04-29 | 방출성이 제어된 안지오텐신 - II - 수용체 차단제와 H M G - C o A 환원 효소 억제제의 복합제 조성물 |
| PCT/KR2009/002261 WO2009134079A2 (ko) | 2008-04-29 | 2009-04-29 | 약제학적 제제 |
Country Status (2)
| Country | Link |
|---|---|
| KR (15) | KR101230731B1 (ko) |
| WO (3) | WO2009134076A2 (ko) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103002883A (zh) * | 2010-05-14 | 2013-03-27 | 韩美科学株式会社 | 包含HMG-CoA还原酶抑制剂和厄贝沙坦的双层片剂形式的药物制剂 |
| KR102304910B1 (ko) * | 2021-01-11 | 2021-09-27 | 알리코제약(주) | 칸데사르탄을 함유하는 안정한 약학적 조성물 |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9498464B2 (en) * | 2011-12-09 | 2016-11-22 | Artskin D.O.O. | Treatment of arterial wall by combination of RAAS inhibitor and HMG-CoA reductase inhibitor |
| KR20130076015A (ko) * | 2011-12-28 | 2013-07-08 | 주식회사 삼양바이오팜 | 높은 경도를 갖는 속붕정 및 이의 제조 방법 |
| KR20140028971A (ko) * | 2012-08-31 | 2014-03-10 | 한미약품 주식회사 | 아토바스타틴, 이르베살탄 및 탄산마그네슘을 포함하는 이층정 복합제제 |
| KR20140030505A (ko) * | 2012-08-31 | 2014-03-12 | 한미약품 주식회사 | 이베살탄 및 HMG-CoA 환원효소 억제제를 포함하는 약제학적 캡슐 복합제제 |
| KR20160105662A (ko) * | 2015-02-27 | 2016-09-07 | 대원제약주식회사 | 에페리손과 펠루비프로펜을 함유하는 약제학적 조성물 |
| KR102145853B1 (ko) * | 2018-06-19 | 2020-08-19 | 한국유나이티드제약 주식회사 | 실로스타졸과 스타틴계약물을 함유하는 약학 조성물 |
| KR102042626B1 (ko) | 2019-06-26 | 2019-11-11 | 한미약품 주식회사 | 이베살탄 및 HMG-CoA 환원효소 억제제를 포함하는 약제학적 캡슐 복합제제 |
| KR102104507B1 (ko) * | 2019-08-23 | 2020-04-24 | 브릿지바이오테라퓨틱스(주) | 팔미토일-l-프롤릴-l-프롤릴-글리실-l-타이로신 나트륨을 포함하는 약제학적 제제 및 이의 제조방법 |
| EP4255495A1 (en) | 2020-12-03 | 2023-10-11 | Battelle Memorial Institute | Polymer nanoparticle and dna nanostructure compositions and methods for non-viral delivery |
| JP2024516108A (ja) | 2021-04-07 | 2024-04-12 | バテル・メモリアル・インスティテュート | 非ウイルス性担体を同定および使用するための迅速な設計、構築、試験、および学習技術 |
| CN119564625B (zh) * | 2025-02-08 | 2025-06-13 | 山东祥维斯医药科技有限公司 | 一种具有降血压作用的甜菜碱组合物及其制备方法 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20010022508A (ko) * | 1997-07-31 | 2001-03-15 | 추후보정 | 3-히드록시-3-메틸글루타릴 조효소 a 환원 효소 억제제와니코틴산 화합물의 조성물 및 야간에 1일 1회 투여하여고지질혈증을 치료하는 방법 |
| US6682759B2 (en) * | 2002-02-01 | 2004-01-27 | Depomed, Inc. | Manufacture of oral dosage forms delivering both immediate-release and sustained-release drugs |
| US20050013863A1 (en) * | 2003-07-18 | 2005-01-20 | Depomed, Inc., A Corporation Of The State Of California | Dual drug dosage forms with improved separation of drugs |
| KR100582347B1 (ko) * | 2004-12-30 | 2006-05-22 | 한미약품 주식회사 | 3-하이드록시-3-메틸글루타릴 조효소 a 환원효소 억제제및 고혈압 치료제의 복합제제 및 그의 제조방법 |
| KR100648825B1 (ko) * | 2005-02-22 | 2006-11-24 | 한국유나이티드제약 주식회사 | 당뇨병 환자의 동맥경화증 예방을 위한 혈당강하제, 에이치엠지이-씨오에이 환원효소 저해제 및 장용코팅된 아스피린을 함유하는 복합 펠렛 |
| KR100762847B1 (ko) * | 2006-01-27 | 2007-10-04 | 씨제이 주식회사 | 멀티플 유닛 타입 서방성 경구 제제 및 그 제조방법 |
| WO2008023869A1 (en) * | 2006-08-24 | 2008-02-28 | Hanall Pharmaceutical Co., Ltd. | Combined pharmeceutical formulation with controlled-release comprising dihydropyridine calcium channel blockers and hmg-coa reductase inhibitors |
-
2009
- 2009-04-29 KR KR1020090037379A patent/KR101230731B1/ko not_active Expired - Fee Related
- 2009-04-29 KR KR1020090037422A patent/KR20090114333A/ko not_active Withdrawn
- 2009-04-29 WO PCT/KR2009/002256 patent/WO2009134076A2/ko active Application Filing
- 2009-04-29 KR KR1020090037380A patent/KR101238156B1/ko not_active Expired - Fee Related
- 2009-04-29 KR KR1020090037376A patent/KR20090114324A/ko not_active Withdrawn
- 2009-04-29 KR KR1020090037381A patent/KR20090114329A/ko not_active Withdrawn
- 2009-04-29 KR KR1020090037375A patent/KR20090114323A/ko not_active Withdrawn
- 2009-04-29 WO PCT/KR2009/002261 patent/WO2009134079A2/ko active Application Filing
- 2009-04-29 KR KR1020090037382A patent/KR20090114330A/ko not_active Withdrawn
- 2009-04-29 WO PCT/KR2009/002278 patent/WO2009134086A2/ko active Application Filing
- 2009-04-29 KR KR1020090037378A patent/KR20090114326A/ko not_active Withdrawn
- 2009-04-29 KR KR1020090037421A patent/KR20090114332A/ko not_active Withdrawn
- 2009-04-29 KR KR1020090037372A patent/KR20090114320A/ko not_active Withdrawn
- 2009-04-29 KR KR1020090037373A patent/KR20090114321A/ko not_active Withdrawn
- 2009-04-29 KR KR1020090037374A patent/KR20090114322A/ko not_active Withdrawn
- 2009-04-29 KR KR1020090037371A patent/KR101205633B1/ko not_active Expired - Fee Related
- 2009-04-29 KR KR1020090037420A patent/KR20090114331A/ko not_active Withdrawn
- 2009-04-29 KR KR1020090037377A patent/KR20090114325A/ko not_active Withdrawn
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103002883A (zh) * | 2010-05-14 | 2013-03-27 | 韩美科学株式会社 | 包含HMG-CoA还原酶抑制剂和厄贝沙坦的双层片剂形式的药物制剂 |
| KR102304910B1 (ko) * | 2021-01-11 | 2021-09-27 | 알리코제약(주) | 칸데사르탄을 함유하는 안정한 약학적 조성물 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20090114322A (ko) | 2009-11-03 |
| KR20090114331A (ko) | 2009-11-03 |
| KR20090114330A (ko) | 2009-11-03 |
| KR20090114328A (ko) | 2009-11-03 |
| KR20090114323A (ko) | 2009-11-03 |
| WO2009134079A9 (ko) | 2010-03-11 |
| WO2009134086A9 (ko) | 2010-03-11 |
| KR20090114333A (ko) | 2009-11-03 |
| WO2009134076A2 (ko) | 2009-11-05 |
| KR101205633B1 (ko) | 2012-11-27 |
| KR101230731B1 (ko) | 2013-02-07 |
| KR101238156B1 (ko) | 2013-02-27 |
| KR20090114319A (ko) | 2009-11-03 |
| KR20090114320A (ko) | 2009-11-03 |
| WO2009134076A3 (ko) | 2010-02-18 |
| KR20090114329A (ko) | 2009-11-03 |
| WO2009134079A2 (ko) | 2009-11-05 |
| WO2009134079A3 (ko) | 2010-01-21 |
| KR20090114332A (ko) | 2009-11-03 |
| KR20090114327A (ko) | 2009-11-03 |
| KR20090114321A (ko) | 2009-11-03 |
| KR20090114324A (ko) | 2009-11-03 |
| KR20090114325A (ko) | 2009-11-03 |
| WO2009134086A3 (ko) | 2010-01-21 |
| KR20090114326A (ko) | 2009-11-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2009134086A9 (ko) | 심혈관계 질환 치료용 약제학적 제제 | |
| WO2009134057A2 (ko) | 안지오텐신-ⅱ-수용체 차단제를 포함하는 약제학적 제제 | |
| WO2009125981A2 (ko) | 약제학적 제제 | |
| KR102695126B1 (ko) | 글루코키나제 활성화제 및 sglt-2 억제제를 포함하는 약제학적 병용물, 조성물, 및 병용 제제, 및 이의 제조방법 및 용도 | |
| WO2012124973A2 (en) | Combined formulation with improved stability | |
| WO2009104932A2 (ko) | 복합제제 | |
| WO2010008203A2 (ko) | 칼슘채널길항제를 포함하는 약제학적 제제 | |
| WO2009104939A2 (ko) | 약제학적 제제 | |
| CN101005829A (zh) | 喹硫平制剂 | |
| WO2009104916A9 (ko) | 심혈관계 질환 치료용 약제학적 제제 | |
| WO2009125987A9 (ko) | 약제학적 제제 | |
| WO2015012633A1 (ko) | 서방형 메트포르민과 속방형 HMG-CoA 환원효소 억제제를 포함하는 복합제제 | |
| WO2018151580A1 (ko) | 이토프리드 염산염을 포함하는 속효성과 지속성을 갖는 약학적 제제 | |
| WO2009125944A9 (ko) | 비디히드로피리딘계 칼슘 채널 차단제 및 안지오텐신-2 수용체 차단제를 포함하는 약제학적 제제 | |
| WO2009142421A2 (ko) | 약제학적 제제 | |
| WO2009134056A9 (ko) | 약제학적 제제 | |
| WO2013169082A1 (ko) | 보센탄 제어방출성 경구제제 | |
| WO2010021473A2 (ko) | 약제학적 제제 | |
| WO2013187700A1 (ko) | 메트포르민 및 HMG-CoA 환원효소 억제제를 포함하는 약제학적 복합제제 | |
| WO2009151295A2 (ko) | 아젤니디핀과 HMG-CoA 환원효소 억제제 또는 안지오텐신 Ⅱ 수용체 차단제를 포함하는 약제학적 제제 | |
| WO2021230664A1 (ko) | 사쿠비트릴, 발사르탄 및 네비보롤을 포함하는 심부전 및 허혈성 심질환의 예방 또는 치료용 약제학적 조성물 및 이를 포함하는 약제학적 복합제제 | |
| RU2770043C9 (ru) | Фармацевтическая комбинация, композиция и комбинированная композиция, содержащая активатор глюкокиназы и ингибитор sglt-2, и способы их приготовления и их применения | |
| RU2781638C2 (ru) | Фармацевтическая комбинация, композиция и комбинированный состав, содержащие активатор глюкокиназы и активатор рецептора ppar, и способ их приготовления и их применение | |
| RU2775603C2 (ru) | Фармацевтическая комбинация, композиция и состав, содержащие активатор глюкокиназы и ингибитор альфа-глюкозидазы, способы приготовления и их применение | |
| WO2020213868A1 (ko) | 에제티미브 및 로사르탄을 포함하는 약제학적 복합제제 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09738992 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 09738992 Country of ref document: EP Kind code of ref document: A2 |