WO2002059094A1 - Nouveau procede de preparation d'un acide quinolone-carboxylique - Google Patents
Nouveau procede de preparation d'un acide quinolone-carboxylique Download PDFInfo
- Publication number
- WO2002059094A1 WO2002059094A1 PCT/CN2001/001156 CN0101156W WO02059094A1 WO 2002059094 A1 WO2002059094 A1 WO 2002059094A1 CN 0101156 W CN0101156 W CN 0101156W WO 02059094 A1 WO02059094 A1 WO 02059094A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mol
- formula
- compound
- dichloro
- group
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/48—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
- C07D215/54—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen attached in position 3
- C07D215/56—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen attached in position 3 with oxygen atoms in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/76—Ketones containing a keto group bound to a six-membered aromatic ring
- C07C49/80—Ketones containing a keto group bound to a six-membered aromatic ring containing halogen
- C07C49/807—Ketones containing a keto group bound to a six-membered aromatic ring containing halogen all halogen atoms bound to the ring
Definitions
- the present invention relates to a process for the preparation of quinolonecarboxylic acid, and more particularly to a process for preparing a quinolonecarboxylic acid using polyhaloacetophenone as a starting material.
- Quinolone anti-infective drugs have received widespread attention in recent years due to their excellent properties.
- Quinolone carboxylic acid is an important intermediate for such anti-infective drugs.
- Various quinolones are not derived from these intermediates and are synthesized by several steps. Therefore, the synthesis of these quinolonecarboxylic acids is obviously very important in the preparation of quinolones.
- One of the objects of the present invention is to provide a novel process for the preparation of quinolonecarboxylic acid from the synthesis of quinolonecarboxylic acid from the starting polyhaloacetophenone.
- the method steps of the present invention are relatively small, and a relatively desirable yield can be achieved.
- a process for preparing a quinolonecarboxylic acid of the formula (I) of the present invention is relatively small, and a relatively desirable yield can be achieved.
- ⁇ is a halogen atom, an amino group
- R 2 is a halogen atom
- R 3 is hydrogen, a halogen atom, ( ⁇ - ⁇ oxy, CN,
- 1 4 is 3 - (3 6 cyclodecyl, fluorenyl, (C 4 methoxy CC 4 fluorenyl, cc 4 cc 4
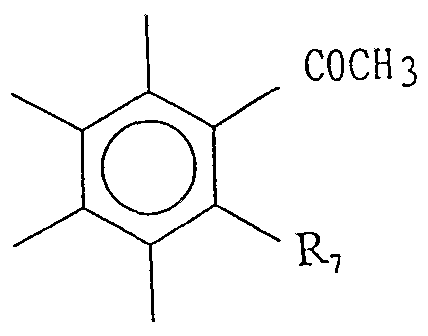
- RR 2 and R 3 have the same meanings as defined above, and R 7 is a halogen atom.
- R 8 represents a methyl group or an ethyl group
- Rj is H, C1;
- R 2 is C1
- R 3 is F, ( ⁇ -( 4 alkoxy, CN;
- the compound of the formula (I) is preferably hydrogen, fluorine or amino, preferably fluorine or chlorine; R 3 is preferably fluorine, CN or OCH 3 ; preferably cyclopropane Base, cyclohexyl.
- the reaction of the compound of the formula (II) with a carbonate can be carried out with methyl carbonate or ethyl ester, and the reaction is usually carried out in an organic solvent in the presence of a base. It is preferred to use the carbonate itself as a solvent, and the base used may be selected from sodium hydride or sodium alkoxide.
- the reaction of the compound of formula (III) with the orthoformate is usually carried out in the presence of acetic anhydride, and after the reaction, it is directly reacted with R 4 NH 2 of the formula (IV) to give a compound of the formula (V).
- the cyclization of the reactant of the formula (V) is carried out in an organic solvent under basic conditions, preferably DMF is used as the solvent, and anhydrous potassium carbonate is optionally used as the base to obtain a compound of the formula (VI).
- anhydrous potassium carbonate is optionally used as the base to obtain a compound of the formula (VI).
- the corresponding quinolone carboxylate is hydrolyzed to the corresponding carboxylic acid of the formula 00 by conventional methods in the art.
- R 1 when R 1 is NH 2 , it can be converted from a compound of the formula (VI) wherein the halogen is corresponding, and the reaction is as follows:
- R 3 is preferably F, OCH 3 , CN.
- the formula ( ⁇ ) is carried out by a plurality of methods in the preparation of the compound.
- the present invention is carried out from a readily available polyhalogenated benzene.
- the invention is further illustrated by the following specific examples. Preparation of quinacridone
- reaction mixture was poured into 800 ml of ice water, and the solid was filtered, washed with water (100 ml), and then dried, and then evaporated to 15 5,7,8-tetrafluoro-1,4-dihydro-4-oxoquinoline-3-carboxylic acid ethyl ester 53.0 g (0.161 mol), mp 169-171 ° C, yield 93%.
- the solvent was evaporated, and the residue was crystalljjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjj
- the material was poured into 170 g of crushed ice containing 75 ml of 25% sulfuric acid and stirred for 5 minutes.
- the organic phase was separated and the aqueous layer was extracted with toluene.
- the organic layer was combined, washed sequentially with aqueous sodium chloride solution, aqueous sodium hydrogen carbonate and water and dried over anhydrous sodium sulfate.
- the material was poured into a 1,000 ml aqueous solution containing 12.4 g of NaHSO 3 , allowed to stand overnight, filtered, washed 3-4 times with warm water (about 30 Torr), and dried to obtain 2,4,6-trichloro-3,5- 34.1 g (0.115 mol) of difluorobromobenzene, yield 92.9%.
- reaction temperature was raised to room temperature and stirred for 3 hours.
- the reaction mass was poured into 150 g of crushed ice containing 75 ml of dilute sulfuric acid (25%) and stirred for 5 minutes.
- the organic layer was separated and the aqueous layer was extracted with toluene 30 ml.
- the organic layers were combined, washed successively with 45ml, 45ml ,, saturated aqueous NaHC0 3 and saturated aqueous NaCl 45ml water 25ml with saturated aqueous NaCl.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Quinoline Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Description






Claims






Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN01814188.9A CN1219769C (zh) | 2000-08-16 | 2001-07-06 | 新的喹诺酮羧酸的制备方法 |
| EP01980134A EP1319656B1 (en) | 2000-08-16 | 2001-07-06 | A new process for preparing a quinolone-carboxylic acid |
| DE60122539T DE60122539T2 (de) | 2000-08-16 | 2001-07-06 | Verfahren zur Herstellung von Chinolincarbonsäuren |
| JP2002559396A JP2004517149A (ja) | 2000-08-16 | 2001-07-06 | キノリンカルボン酸の製造方法 |
| US10/344,643 US6699992B2 (en) | 2000-08-16 | 2001-07-06 | Process for preparing quinolonecarboxylic acids |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN00123446.3 | 2000-08-16 | ||
| CN00123446.3A CN1338455A (zh) | 2000-08-16 | 2000-08-16 | 新的喹诺酮羧酸的制备方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2002059094A1 true WO2002059094A1 (fr) | 2002-08-01 |
Family
ID=4589879
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2001/001156 WO2002059094A1 (fr) | 2000-08-16 | 2001-07-06 | Nouveau procede de preparation d'un acide quinolone-carboxylique |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US6699992B2 (zh) |
| EP (1) | EP1319656B1 (zh) |
| JP (1) | JP2004517149A (zh) |
| CN (2) | CN1338455A (zh) |
| AT (1) | ATE337304T1 (zh) |
| DE (1) | DE60122539T2 (zh) |
| WO (1) | WO2002059094A1 (zh) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR100519158B1 (ko) * | 2002-12-21 | 2005-10-06 | 주식회사유한양행 | 퀴놀론 카르복실레이트 유도체의 제조방법 |
| US8187483B2 (en) * | 2006-08-11 | 2012-05-29 | Jason Plumhoff | Method to minimize CD etch bias |
| US8063066B2 (en) | 2007-03-19 | 2011-11-22 | Takeda Pharmaceutical Company Limited | MAPK/ERK kinase inhibitors |
| US20090043646A1 (en) * | 2007-08-06 | 2009-02-12 | International Business Machines Corporation | System and Method for the Automated Capture and Clustering of User Activities |
| CN102260172B (zh) * | 2011-03-30 | 2013-11-06 | 山东先达农化股份有限公司 | 一种2-氯-5-烷基羰基乙酰基-4氟苯氧乙酰化合物的制备方法 |
| CN103450068B (zh) * | 2012-05-27 | 2015-09-16 | 重庆常捷医药化工有限公司 | 一种齐拉西酮中间体的合成方法 |
| CN107987074B (zh) * | 2017-10-27 | 2020-12-29 | 浙江美诺华药物化学有限公司 | 一种普拉沙星的合成方法 |
| CN111989314B (zh) | 2018-04-25 | 2024-04-30 | 拜耳动物保健有限责任公司 | 喹诺酮甲酸酯水解的方法 |
| CN114933543B (zh) * | 2022-07-21 | 2022-10-28 | 山东国邦药业有限公司 | 一种3-环丙胺基-2-(2,4-二氯-5-氟苯甲酰基)丙烯酸甲酯的合成方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4730000A (en) * | 1984-04-09 | 1988-03-08 | Abbott Laboratories | Quinoline antibacterial compounds |
| ES2049640A1 (es) * | 1992-06-18 | 1994-04-16 | Genesis Para La Investigacion | Procedimiento paras la preparacion de acidos quinolin carboxilicos. |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NZ225926A (en) * | 1987-09-08 | 1990-04-26 | Sterling Drug Inc | 7-pyridinyl-4-oxo-3-quinolinecarboxylic acid derivatives, and pharmaceutical compositions |
| US5075319A (en) * | 1987-09-08 | 1991-12-24 | Sterling Drug Inc. | Pyridinyl-quinolone compounds, their preparation and use |
| ATE190972T1 (de) * | 1994-08-02 | 2000-04-15 | Procter & Gamble | Verfahren zur herstellung von antimikrobiellen verbindungen |
| JP2000119221A (ja) * | 1998-10-14 | 2000-04-25 | Kyorin Pharmaceut Co Ltd | (2,4,5−トリフルオロ−3−メトキシベンゾイル)酢酸エステル誘導体の製造方法及びその製造中間体 |
-
2000
- 2000-08-16 CN CN00123446.3A patent/CN1338455A/zh active Pending
-
2001
- 2001-07-06 CN CN01814188.9A patent/CN1219769C/zh not_active Expired - Fee Related
- 2001-07-06 WO PCT/CN2001/001156 patent/WO2002059094A1/zh active IP Right Grant
- 2001-07-06 US US10/344,643 patent/US6699992B2/en not_active Expired - Fee Related
- 2001-07-06 EP EP01980134A patent/EP1319656B1/en not_active Expired - Lifetime
- 2001-07-06 AT AT01980134T patent/ATE337304T1/de not_active IP Right Cessation
- 2001-07-06 JP JP2002559396A patent/JP2004517149A/ja active Pending
- 2001-07-06 DE DE60122539T patent/DE60122539T2/de not_active Expired - Lifetime
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4730000A (en) * | 1984-04-09 | 1988-03-08 | Abbott Laboratories | Quinoline antibacterial compounds |
| ES2049640A1 (es) * | 1992-06-18 | 1994-04-16 | Genesis Para La Investigacion | Procedimiento paras la preparacion de acidos quinolin carboxilicos. |
Also Published As
| Publication number | Publication date |
|---|---|
| DE60122539D1 (de) | 2006-10-05 |
| EP1319656A1 (en) | 2003-06-18 |
| CN1219769C (zh) | 2005-09-21 |
| DE60122539T2 (de) | 2007-05-03 |
| EP1319656B1 (en) | 2006-08-23 |
| JP2004517149A (ja) | 2004-06-10 |
| CN1338455A (zh) | 2002-03-06 |
| EP1319656A4 (en) | 2004-09-29 |
| ATE337304T1 (de) | 2006-09-15 |
| US6699992B2 (en) | 2004-03-02 |
| CN1447797A (zh) | 2003-10-08 |
| US20030166936A1 (en) | 2003-09-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI411602B (zh) | 製造整合酶抑制劑的方法及彼所用的中間物 | |
| KR101023635B1 (ko) | 4-옥소퀴놀린 화합물 제조 방법 | |
| KR940007305B1 (ko) | 8-위치에서 치환된 퀴놀론카복실산 유도체, 이의 제조방법 및 이를 함유하는 약제학적 조성물 | |
| KR930004652B1 (ko) | 퀴놀론 카복실산의 제조방법 | |
| EP0191185B1 (en) | Quinoline-carboxylic acid derivatives | |
| WO2002059094A1 (fr) | Nouveau procede de preparation d'un acide quinolone-carboxylique | |
| KR910003616B1 (ko) | 할로겐화 퀴놀론 카복실산의 제조방법 | |
| HU204512B (en) | New process for producing quinoline-carboxylic acid derivatives | |
| JP4305597B2 (ja) | 弗素置換安息香酸の製造法 | |
| EP0184035B1 (en) | Quinolonecarboxylic acid derivatives and process for their preparation | |
| JPS6261948A (ja) | テトラフルオロ安息香酸の改良された製法 | |
| HU195643B (en) | Process for producing quinolon-carboxylic acid derivatives | |
| US4885386A (en) | Process for the synthesis of 3-chloro-2,4,5-trifluorobenzoic acid | |
| JPS63264439A (ja) | 3,5,6−トリフルオロ−4−ヒドロキシフタル酸の製造法 | |
| HU196165B (en) | Process for preparing new benzoyl acetic acid ester derivatives | |
| JP3573249B2 (ja) | 2,3,4−トリフルオロ−5−ヨ−ド安息香酸、そのエステル類及びその製造法 | |
| JP2556330B2 (ja) | アニソ−ル誘導体並びにその製造方法 | |
| JP2594017B2 (ja) | 8−メトキシキノロンカルボン酸誘導体の製造中間体 | |
| JP2716952B2 (ja) | 8−メトキシキノロンカルボン酸誘導体の製造中間体 | |
| KR930000439B1 (ko) | 퀴놀론 유도체의 제조방법 | |
| JP2589007B2 (ja) | キノロンカルボン酸誘導体の製造中間体 | |
| JPH09100255A (ja) | 8−メトキシキノロンカルボン酸誘導体の製造中間体 | |
| KR950014221B1 (ko) | 8-메톡시퀴놀론 카르복실산 유도체의 제조 중간체의 제조방법 | |
| JP2021521238A (ja) | キノロンカルボン酸エステルの加水分解プロセス | |
| CN116178265A (zh) | 一种3-(二氟甲基)-1-甲基-1h-吡唑-4-羧酸的合成方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2001980134 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10344643 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 2002 559396 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002559396 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 018141889 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2001980134 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2001980134 Country of ref document: EP |