KR930011489B1 - 치환된 벤즈아제핀, 이의 제조방법 및 이를 함유하는 약제학적 조성물 - Google Patents
치환된 벤즈아제핀, 이의 제조방법 및 이를 함유하는 약제학적 조성물 Download PDFInfo
- Publication number
- KR930011489B1 KR930011489B1 KR1019880701534A KR880701534A KR930011489B1 KR 930011489 B1 KR930011489 B1 KR 930011489B1 KR 1019880701534 A KR1019880701534 A KR 1019880701534A KR 880701534 A KR880701534 A KR 880701534A KR 930011489 B1 KR930011489 B1 KR 930011489B1
- Authority
- KR
- South Korea
- Prior art keywords
- compound
- formula
- alkyl
- alkoxy
- pharmaceutically acceptable
- Prior art date
Links
- 0 C*(C)C1=CC(C)C(*)C(*)=C1 Chemical compound C*(C)C1=CC(C)C(*)C(*)=C1 0.000 description 8
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D223/00—Heterocyclic compounds containing seven-membered rings having one nitrogen atom as the only ring hetero atom
- C07D223/14—Heterocyclic compounds containing seven-membered rings having one nitrogen atom as the only ring hetero atom condensed with carbocyclic rings or ring systems
- C07D223/16—Benzazepines; Hydrogenated benzazepines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/26—Psychostimulants, e.g. nicotine, cocaine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D223/00—Heterocyclic compounds containing seven-membered rings having one nitrogen atom as the only ring hetero atom
- C07D223/14—Heterocyclic compounds containing seven-membered rings having one nitrogen atom as the only ring hetero atom condensed with carbocyclic rings or ring systems
- C07D223/32—Heterocyclic compounds containing seven-membered rings having one nitrogen atom as the only ring hetero atom condensed with carbocyclic rings or ring systems containing carbocyclic rings other than six-membered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/56—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/08—1,2,4-Triazoles; Hydrogenated 1,2,4-triazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Neurosurgery (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biomedical Technology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Neurology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
Abstract
내용 없음.
Description
[발명의 명칭]
치환된 벤즈아제핀, 이의 제조방법 및 이를 함유하는 약제학적 조성물
[발명의 상세한 설명]
[발명의 배경]
본 발명은 신규의 1- 또는 5- 치환된 -2,3,4,5,-테트라하이드로-1H-3-벤즈아제핀, 이의 제조방법 및 이를 함유하는 약제학적 조성물에 관한 것이다. 본 화합물은 정신병, 우울증, 통증 및 고혈압의 치료에 있어 귀중한 약제학적 특성을 갖는다.
치환된 1-페닐-2,3,4,5-테트라하이드로-1H-3-벤즈아제핀은 당해 기술분야에 밝혀져 있다[참조 : 미합중국 특허 제3,393,192호, 제3,609,138호, 제4,011,319호, 제4,284,555호, 제4,477,378호 및 영국 특허 제1,118,688호]. 이들 특허에 공지된 본 화합물에 대해 논의된 활성은 항균성, 중앙 신경계 효과 및 혈압강하 효과를 포함한다.
문헌[참조 : Wenistock et al., in Drugs of the Future, Vo1. 10, No. 8, pp 645-697(1985)]에서는 1-페닐치환체가 특정 형태의 벤즈아제핀의 도파민적 활성에 대해 가지는 중요한 효과를 논하고 있다(P686의 표 2 참조).
유럽 특허원 제83105610.6(공개번호 제0 096 838호)는 제7- 및 제-8 위치에 H 및/ 또는 알콕시 치환체를 갖는 특정한 1-아릴옥시 치환된 2,3,4,5-테트라하이드로-3-벤즈아제핀을 기술하고 있다. 본 화합물들은 우울증 치료에 효과적이라고 공지되어 있다.
[본 발명의 요약]
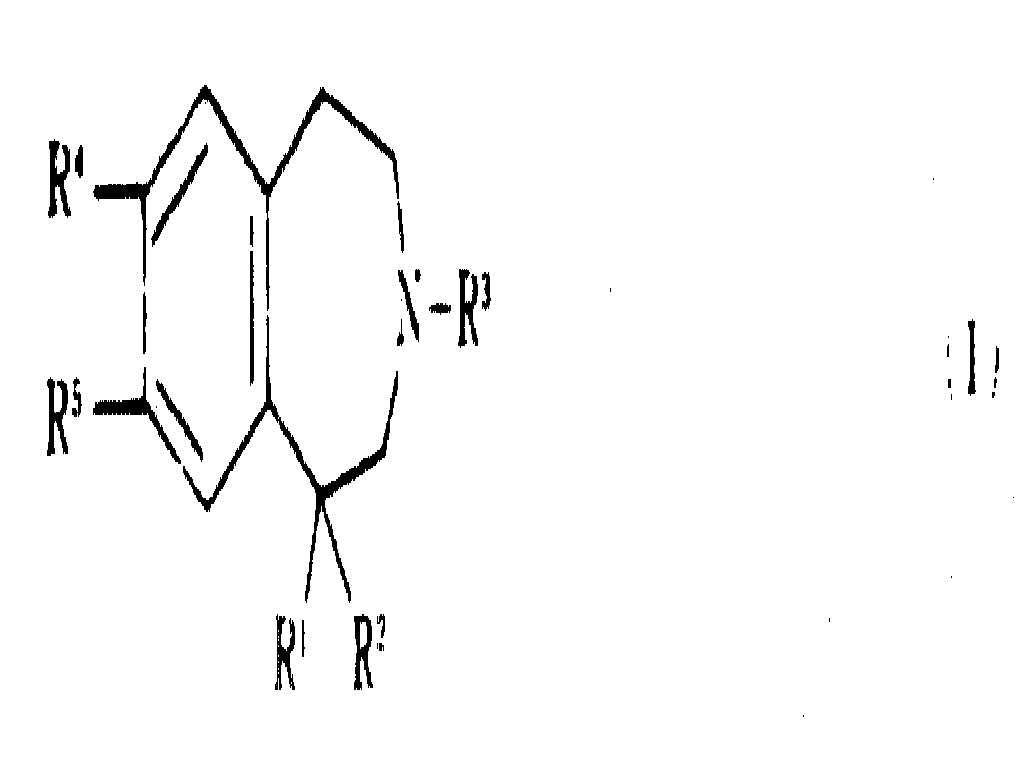
놀랍게도 1-페닐 치환체가 결핍된 특정한 신규의 벤즈아제핀은 우수한 항-도파민적 활성을 갖으며, 특히, 도파민 수용체의 D-1 아종에 대한 놀라운 선택성을 나타냄이 밝혀졌다. 따라서, 본 발명의 한 태양으로서 본 발명은 신규한 하기 일반식(Ⅰ)의 벤즈아제핀 및 약제학적으로 허용되는 이의 염을 제공한다 :

상기식에서, R1은 -XR6, -CHR7R8, 사이클로알킬, 사이클로알케닐, -H, -CH, -(CO)OR9, -O(CO)R9, -O(CO)N(R9)2,


본 명세서 및 특허청구범위에서 사용된 하기 용어들은 다른 언급이 없는 한 다음과 같은 의미를 갖는다 : 할로(할로알킬의 할로를 포함)는 플루오로, 클로로, 브로모 또는 요오드이고; 알킬(사이클로알킬알킬, 사이클로아케닐알키, 헤테로아릴알킬, 알콕시, 알콕시알킬 등의 알킬잔기를 포함)은 탄소수 1 내지 6의 직쇄 또는 측쇄 탄소쇄이며; 사이클로알킬(사이클로알킬알킬의 사이클로알킬 잔기를 포함)은 탄소수 3 내지 8의 포화 카보사이클환이고; 사이클로알케닐(사이크로알키넬알킬의 사이클로알케닐 잔기를 포함)은 탄소수가 5 내지 8이며 탄소-탄소 이중결합을 포함하는 카보사이클환이며; 알케닐(아르알케닐의 알케닐 잔기를 포함)은 하나 이상이 탄소-탄소 이중결합을 갖으며 탄소수가 1 내지 6인 직쇄 또는 측쇄 탄소쇄이고; 알키닐(아르알키닐의 알키닐 잔기 포함)은 하나 이상의 탄소-탄소 삼중결합을 갖으며 탄소수가 2 내지 6인 직쇄 또는 측쇄 탄소쇄이며; 아릴은(아르알킬, 아르알케닐 및 아르알키닐의 아릴 잔기를 포함)은 비치환된 페닐 또는 치환된 페닐이고; 치환된 페닐은 알킬, 하이드록시, 알콕시, 알킬티오, 할로, 트리플루오로메틸 또느 이의 조합체에 의해 일 또는 이-치환된 페닐이며; 카보닐 산소는 =O 그룹이고; 할로알킬은 가능한 할로겐화 부위에 따라 상기 정의한 알킬 그룹의 수소중 일부 또는 전부를 치환하는 1 내지 5개의 할로 그룹(바람직하게는 클로로 또는 플루오로)을 함유하는 상기 정의된 알킬그룹(예 : CF2CH2Cl 등)이며; 알칸디일은 2가의, 탄소수 1 내지 6의 직쇄 또는 측쇄 탄화수소쇄이고, 이의 동일 또는 상이한 탄소원자로부터 2개의 결합이 가능하고(예 : 메틸렌, 에틸렌, 에틸린덴, 등); 헤테로아릴(헤테로아릴알킬의 헤테로아릴 잔기 포함)은 카보사이클릭 그룹에 삽입되는 하나 이상의 O,S 및/ 또는 N을 갖고 방향족 특성을 제공하는 충분한 수의 비편재된 π-전자를 갖는 방향족 헤테로사이클릭 그룹, 바람직하게는 탄소수 2 내지 14의 방향족 헤테로사이클릭 그룹(예 : 2-, 3- 또는 4- 피리딜, 2- 또는 3-푸릴, 2- 또는 3-티에닐, 2-, 4- 또는 5-티아졸일, 1-, 2- 또는 4-이미다졸일, 2-, 4-, 5- 또는 6-피리미디닐, 2- 또는 3-피라지닐, 3- 또는 4-피리다지닐, 3-, 5- 또는 6-[1-, 2-, 4-트리아지닐], 2-, 3-, 4-, 5-, 6- 또는 7-벤조푸라닐, 2-, 3-, 4-, 5-, 6- 또는 7-인돌일, 1-, 3-, 4- 또는 5-피라졸일, 2-, 4- 또는 5-옥사졸일 등)이며, 이의 모든 유용한 치환가능 탄소 또는 질소는 벤즈아제핀환 시스템에 부착 가능한 점이다.
등); 헤테로아릴(헤테로아릴알킬의 헤테로아릴 잔기 포함)은 카보사이클릭 그룹에 삽입되는 하나 이상의 O,S 및/ 또는 N을 갖고 방향족 특성을 제공하는 충분한 수의 비편재된 π-전자를 갖는 방향족 헤테로사이클릭 그룹, 바람직하게는 탄소수 2 내지 14의 방향족 헤테로사이클릭 그룹(예 : 2-, 3- 또는 4- 피리딜, 2- 또는 3-푸릴, 2- 또는 3-티에닐, 2-, 4- 또는 5-티아졸일, 1-, 2- 또는 4-이미다졸일, 2-, 4-, 5- 또는 6-피리미디닐, 2- 또는 3-피라지닐, 3- 또는 4-피리다지닐, 3-, 5- 또는 6-[1-, 2-, 4-트리아지닐], 2-, 3-, 4-, 5-, 6- 또는 7-벤조푸라닐, 2-, 3-, 4-, 5-, 6- 또는 7-인돌일, 1-, 3-, 4- 또는 5-피라졸일, 2-, 4- 또는 5-옥사졸일 등)이며, 이의 모든 유용한 치환가능 탄소 또는 질소는 벤즈아제핀환 시스템에 부착 가능한 점이다.
바람직한 본 발명의 태양에서, R1은 -XR6, -CHR7RS, 사이클로알킬 또는 사이클로알케닐(여기에서, R6은 -H, 페닐, 치환된 페닐, 아르알킬, 알킬, 할로알킬 또는 알콕시 알킬을 나타내고, X는 -O- 또는 -S-을 나타내며, R7은 H 또는 알킬을 나타내고, R8은 사이클로알킬, 사이클로알케닐, 할로아킬 또는 알콕시알킬을 나타낸다)을 나타낸다. 특히 바람직한 R1은 사이클로알킬 및 사이클로알케닐, 특히 사이클로헥실 및 사이클로헥세닐이다. R1이 XR6일때, 바람직한 R8은 알킬, 특히 메틸 및 에틸 및 사이클로알킬, 특히 사이클로헥실이고, 바람직한 X는 -O- 및 -S-이다.
추가의 바람직한 본 발명의 태양에서, R1은 (여기에서, m은 1이고 R9는 수소 또는 알킬이다)을 나타낸다. 또다른 추가의 바람직한 본 발명의 태양에서 R은 1-피롤일이다.
(여기에서, m은 1이고 R9는 수소 또는 알킬이다)을 나타낸다. 또다른 추가의 바람직한 본 발명의 태양에서 R은 1-피롤일이다.
R2는 바람직하게는 -H이고, R3는 바람직하게는 -CH3이다. R4는 바람직하게는 할로겐, 특히 클로로이고, R5는 바람직하게는 -OH, -O.CO.R9또는 -OC(R7)2OCOR13(여기에서, R9는 알킬, 알콕시 또는 알콕시알킬을 나타내고, R7은 수소를 나타내며 R13은 알킬을 나타낸다)이다.
바람직한 일반식(Ⅰ)의 화합물은 다음과 같다 :
8-클로로-5-메톡시-3-메틸-2,3,4,5-테트라하이드로- -3-벤즈아제핀-7-올,
-3-벤즈아제핀-7-올,
8-클로로-5-에톡시-3-메틸-2,3,4,5-테트라하이드로- -3-벤즈아제핀-7-올,
-3-벤즈아제핀-7-올,
8-클로로-5-에틸티오-3-메틸-2,3,4,5-테트라하이드로- -3-벤즈아제핀-7-올,
-3-벤즈아제핀-7-올,
8-클로로-3-메틸-5-페녹시-2,3,4,5-테트라하이드로- -3-벤즈아제핀-7-올,
-3-벤즈아제핀-7-올,
8-클로로-3-메틸-5-페닐티오-2,3,4,5-테트라하이드로- -3-벤즈아제핀-7-올,
-3-벤즈아제핀-7-올,
7-클로로-8-디메틸카바모일-1-에톡시-메틸-2,3,4,5-테트라하이드로- -3-벤즈아제핀
-3-벤즈아제핀
8-클로로-3-메틸-5-(1-피페리디닐)-2,3,4,5-테트라하이드로- -3-벤즈아제핀-7-올,
-3-벤즈아제핀-7-올,
8-클로로-5-사이클로헥실-3-메틸-2,3,4,5-테트라하이드로- -3-벤즈아제핀-7-올,
-3-벤즈아제핀-7-올,
8-클로로-5-사이클로헥실옥시-3-메틸-2,3,4,5-테트라하이드로- -3-벤즈아제핀-7-올,
-3-벤즈아제핀-7-올,
8-클로로-5-(N,N-디메틸아미노프로필)-3-메틸-2,3,4,5-테트라하이드로- -3-벤즈아제핀-7-올,
-3-벤즈아제핀-7-올,
8-클로로-5-(2-사이클로헥세닐)-3-메틸-2,3,4,5-테트라하이드로- -3-벤즈아제핀-7-올,
-3-벤즈아제핀-7-올,
8-클로로-5-알릴-3-메틸-2,3,4,5-테트라하이드로- -3-벤즈아제핀-7-올,
-3-벤즈아제핀-7-올,
8-클로로-5-(2,2,2-트리플루오로에톡시)-3-메틸-2,3,4,5-테트라하이드로- -3-벤즈아제핀-7-올,
-3-벤즈아제핀-7-올,
8-클로로-5-벤질옥시-3-메틸-2,3,4,5-테트라하이드로- -3-벤즈아제핀-7-올,
-3-벤즈아제핀-7-올,
8-클로로-5-(펜에틸옥시)-3-메틸-2,3,4,5-테트라하이드로- -3-벤즈아제핀-7-올,
-3-벤즈아제핀-7-올,
8-클로로-5-(1-피롤일)-3-메틸-2,3,4,5-테트라하이드로- -3-벤즈아제핀-7-올,
-3-벤즈아제핀-7-올,
8-클로로-7-하이드록시-3-메틸-2,3,4,5-테트라하이드로-스피로[ -3-벤즈아제핀-5,5'-사이클로펜탄],
-3-벤즈아제핀-5,5'-사이클로펜탄],
8-클로로-7-(에톡시-포밀옥시)-5-사이클로헥실-3-메틸-2,3,4,5-테트라하이드로- -3-벤즈아제핀,
-3-벤즈아제핀,
8-클로로-7-(이소프로필-포밀옥시)-5-알릴-3-메틸-2,3,4,5-테트라하이드로- -3-벤즈아제핀,
-3-벤즈아제핀,
8-클로로-7-(메톡시-아세톡시)-5-알릴-3-메틸-2,3,4,5-테트라하이드로- -3-벤즈아제핀,
-3-벤즈아제핀,
8-클로로-7-아세톡시-5-(3-메틸-2-부테닐)-3-메틸-2,3,4,5-테트라하이드로- -3-벤즈아제핀,
-3-벤즈아제핀,
8-클로로-7-(3급-부티릴옥시-메톡시)-5-알릴-3-메틸-2,3,4,5-테트라하이드로- -3-벤즈아제핀 및 약제학적으로 허용되는 이들의 염.
-3-벤즈아제핀 및 약제학적으로 허용되는 이들의 염.
본 발명의 또다른 태양에서, 본 발명은
A) 하기 일반식(a)의 카보닐 화합물의 환원반응,
B) 하기 일반식(b)의 에스테르의 환원반응,
C) 하기 일반식(c)의 염의 이중결합에서의 환원반응,
D) 하기 일반식(d)의 화합물에 있어서 HD의 제거 및 아제핀 환 형성을 수반하는 분자내 축합반응, 및
E) 하기 일반식(e)의 화합물의 올레핀 이중결합에서의 환원 반응중에서 선택되는 반응을 포함하며, 이 반응후에, 필요에 따라
(i) 질소원자에 존재하는 특정 보호 그룹의 제거,
(ii) R3가 수송인 경우 R3가 알킬, 알릴 또는 사이클로프로필이 되도록 하는 질소원자에서의 알킬화,
(iii) R1의 -OH이고 R2가 -H일때 상응하는 에테르 또는 티올이 되도록하는 R1이 에테르화 또는 티오에테르화,
(iv) R5가 -OH 일때 R5의 에스테르화,
(v) R4가 -H 일때 R4의 할로겐화,
(vi) R4가 -H 일때 R4를 하이드록시메틸화한후 그렇게 도입된 하이드록시메닐 그룹을 환원시키는 것중 하나이상의 임의의 반응을 수행하며, R5a가 알콕시인 경우 상기 임의의 단계 또는 단계들 전 또는 후에 R5a를 탈알킬화하여 일반식(Ⅰ)의 화합물을 제조하고 그렇게 수득된 일반식(Ⅰ)의 화합물이 유리형태 또는 약제학적으로 허용되는 염의 형태로 분리됨을 특징으로 하는 일반식(Ⅰ)에 따른 화합물의 제조방법을 제공하는 것이다.





상기식에서, 아제핀환중의 점선은 임의의 이중결합을 나타내고, R1,R2,R3,R4및 R13은 일반식(Ⅰ)에 대해 정의된 바와 같으며, R30는 R3또는 COOR13이고, R5a는 일반식(Ⅰ)에 대해 정의된 바와 같이 R5이거나 또는 알콕시이며, L3는 음이온, 바람직하게는 할로산 또는 설폰산중에서 유도된 음이온이고, D는 아제핀 환이 형성될때 DH로서 제거될 수 있는 반응성 그룹이며, Z는 R1또는 R2이다.
본 발명은 또한 일반식(Ⅰ)의 화합물의 제조에 유용한 중간체, 즉 하기 일반식(Ⅱ)의 중간체를 포함한다 :

상기식에서, R3a는 상기에 정의한 R3또는 -COOR14(여기에서, R14는 알킬, 아릴, 아르알킬 또는 할로알킬이다)을 나타내고; R4는 상기에 정의한 바와 같고; R5a는 상기에 정의한 R5또는 알콕시를 나타내고; Q는 H, 할로 또는 -SO2R"(여기에서, R"는 CH3, CF3, 페닐 또는 톨일이다)을 나타낸다. Q는 바람직하게는 클로로 또는 브로모를 나타낸다. 바람직한 중간체는 구조식(Ⅱa)의 화합물이다.

일반식(Ⅰ)의 화합물은 진통제, 콜린억제제, 항공격소제 및 종합적 진통제 특성을 지닌다. 따라서 본 발명은 약제학적으로 허용되는 담체와 배합하여 일반식(Ⅰ)의 화합물을 함유하는 약제학적 조성물, 및 유효량의 일반식(Ⅰ)의 화합물을 병에 걸린 포유동물에 투여하여 포유동물에 있어서 정신이상, 정신분열증 또는 우울증을 포함한 정신장애를 치료하는 방법, 또는 포유동물에 있어서 통증 또는 불안을 억제시키는 방법을 포함한다.
[상세한 설명]
특정한 본 발명의 화합물(예 : R1및 R2가 상이한 경우)이 이성체 형태로 존재할 수 있다. 본 발명은 순수한 형태 및 라세믹 혼합물을 포함한 형태의 모든 이성체를 포함한다.
일반식(Ⅰ)의 화합물은 수화형태(예 : 헤미하이드레이트)를 포함하여 용매화물 형태는 물론 비용매화물형태로 존재할 수 있다. 일반적으로, 물, 에탄올등과 같은 약제학적으로 허용되는 용매와의 용매화물 형태는 본 발명의 목적을 위해 비용매화물형태에 상응한다.
일반식(Ⅰ)의 화합물은 유기 및 무기산과 함께 약제학적으로 허용되는 염을 형성할 수 있다. 염 형성에 적합한 산의 예는 당해 분야에 잘 공지되어 있는 염화수소산, 황산, 인산, 아세트산, 시트르산, 말론산, 살리실산, 말산, 푸마르산, 석신산, 아스코르브산, 말레산, 메탄설폰산 및 기타 무기 및 카복실산이다. 염은 유리염기 형태와 충분한 양의 바람직한 산을 접촉시켜 통상적인 방법으로 염을 생성하여 제조한다. 유리 염기 또는 수산화 나트륨, 탄산 칼륨, 암모니아 및 중탄산나트륨과 같은 적합한 염기 수용액으로 염을 처리하여 재생할 수 있다. 유리 염기 형태는 극성 용매중에서의 용해도와 같은 특정 물리적 특성에 있어 그들 각각의 염 형태와 차이가 있으나, 그 염은 다른 면에서는 본 발명의 목적을 위한 각각의 유리 염기와 동등하다.
상기 일반식(Ⅰ)의 화합물은 다음 A 내지 E의 방법으로 제조할 수 있다 :
A. 하기 일반식(Ⅲ)의 화합물을 적합한 환원제와 반응시켜 카보닐 산소를 환원시킨다.

적합한 환원제는 BH3/THF, LiAlH4, NaBH4/피리딘, NaAlH2(OCH2CH2OCH2H5)2등을 포함한다. 반응은 약 0℃ 내지 약 120℃의 어떠한 적합한 온도에서도 수행할 수 있고, THF, 에테르등과 같은 불활성용매중에서 수행할 수 있다.
일반식(Ⅲ) 화합물은 하기한 방법으로 제조할 수 있다 :
예를 들어, 하기한 일반식(Ⅳ)의 화합물을 일반식(Ⅴ)의 화합물과 반응시켜 일반식(Ⅵ)의 화합물을 형성시킨다 :

상기식에서, R12는 메틸 또는 에틸과 같은 알킬 그룹이다. 이 반응은 약 0℃ 내지 약 50℃의 어떠한 적합한 온도에서도 수행할 수 있다. 일반적으로 DMF, CH2Cl2등과 같은 불활성 용매를 사용하나, 반응을 또한 순수하게 수행시킬 수도 있다. 반응은 결합제 또는 탈수화제(예 : 디사이클로헥실카보디이미드, N-에틸-N'-(디메틸아미노) 에틸카보디이미드 등)의 존재하에 수행한다.
또한, 일반식(Ⅵ)의 화합물은 일반식(Ⅳ)의 화합물을 예를 들어 SOCl2또는 (COCl)2와 반응시켜 하기 일반식(Ⅳa)의 산 클로라이드를 수득한 후 이를 일반식(Ⅴ)의 화합물과 반응시킴으로써 제조될 수 있다.
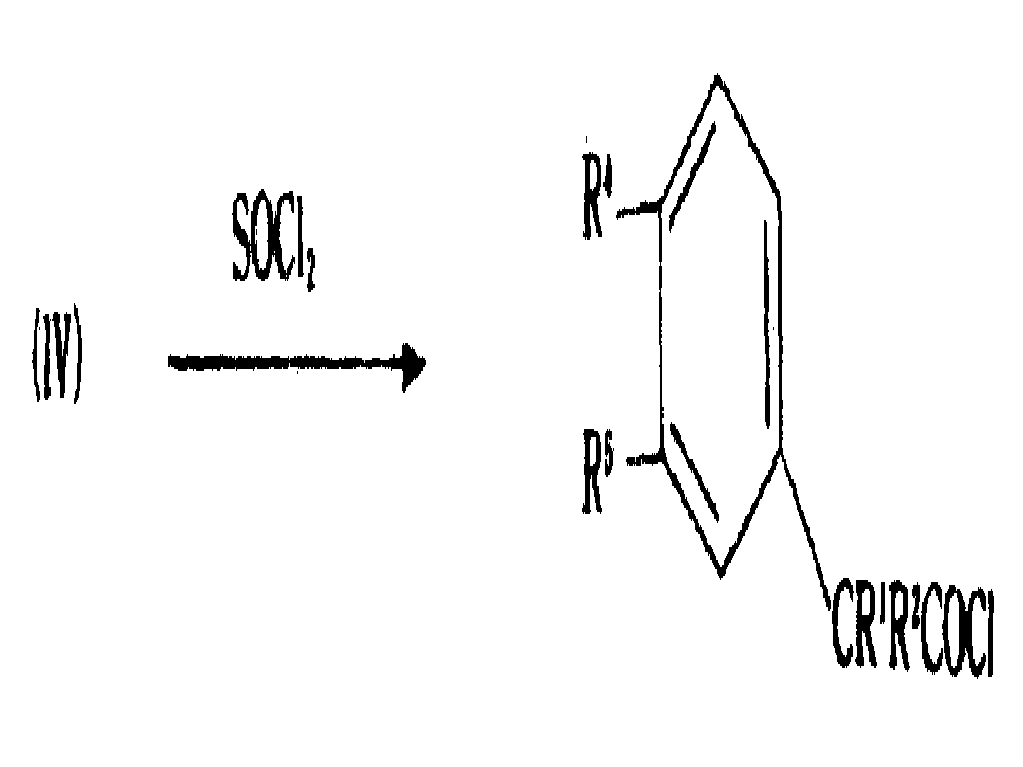
이 반응에서, 결합제는 필요치 않다.
일반식(Ⅳ)의 화합물은 공지되어 있거나 당해분야의 통상적 기술로 제조할 수 있다. 일반식(Ⅴ)의 아세탈은 또한 공지되어 있거나 통상적 기술로 제조할 수 있다[참조 : 미합중국 특허 제4,490,369호].
일반식(Ⅵ)의 아세탈은 CF3SO3H, HCl등과 같은 강산과 반응하여 하기 일반식(Ⅶ)의 화합물을 생성한다 :

본 반응은 순수하게 즉, 용매로서 산과 함께, 또는 아세트산과 같은 용매의 존재하게 수행된다. 약 0℃내지 약 50℃의 어떠한 적합한 온도에서도 수행할 수 있다.
일반식(Ⅶ)의 화합물을 이어서, 일반식(Ⅶ)에서 카보닐은 환원시키지 않고 올레핀 결합을 환원시킬 적합한 수소화제(예 H2/PtO2, H2/Pd-C 등)를 사용하여 하기 일반식(Ⅲ)의 화합물로 환원시킨다 :

또는 , 일반식(Ⅲ)의 화합물은 하기에 나타내 바와 같이, 일반식(Ⅳb)의 화합물을 일반식(Ⅴ)의 화합물과 반응시켜 하기 일반식(Ⅵa)의 화합물을 제조한 후, 강산과 반응시키고 나서 환원제와 반응시켜 하기 일반식(Ⅶa) 및 (Ⅷ)의 화합물을 형성시키는 순차적 단계에 의해 제조될 수 있다 :

상기식에서, R12는 상기한 바와 같다. 이 반응은 각각의 반응에 대해 전술한 조건하에서 수행할 수 있다.
일반식(Ⅷ)의 화합물은 SO2Y2(예 : SO2Cl2SO2Br 등)과 같은 할로겐화제와 반응하여 하기 일반식(Ⅸ)의 화합물을 생성한다 :

이 반응은 어떠한 적합한 온도에서도 수행될 수 있으며 통상적으로 CH2Cl2, CHCl3등과 같은 불활성 용매중에서 수행한다.
일반식(Ⅸ)의 화합물에서 Y그룹을 OH그룹으로 가수분해 시킨 후 적합한 설포닐 할라이드 또는 무수물(예 : 톨일설포닐 클로라이드 또는 메탄설포닐 클로라이드)과 반응시켜 기타 상기한 일반식(Ⅱ)의 중간체를 수득한다.
상기 문장에서 기술한 일반식(Ⅸ)의 화합물 또는 이의 설포닐 유도체를 적합한 친핵체(nu)와 반응시켜 Y가 치환된 하기 일반식(X)의 화합물을 제조할 수 있다 :
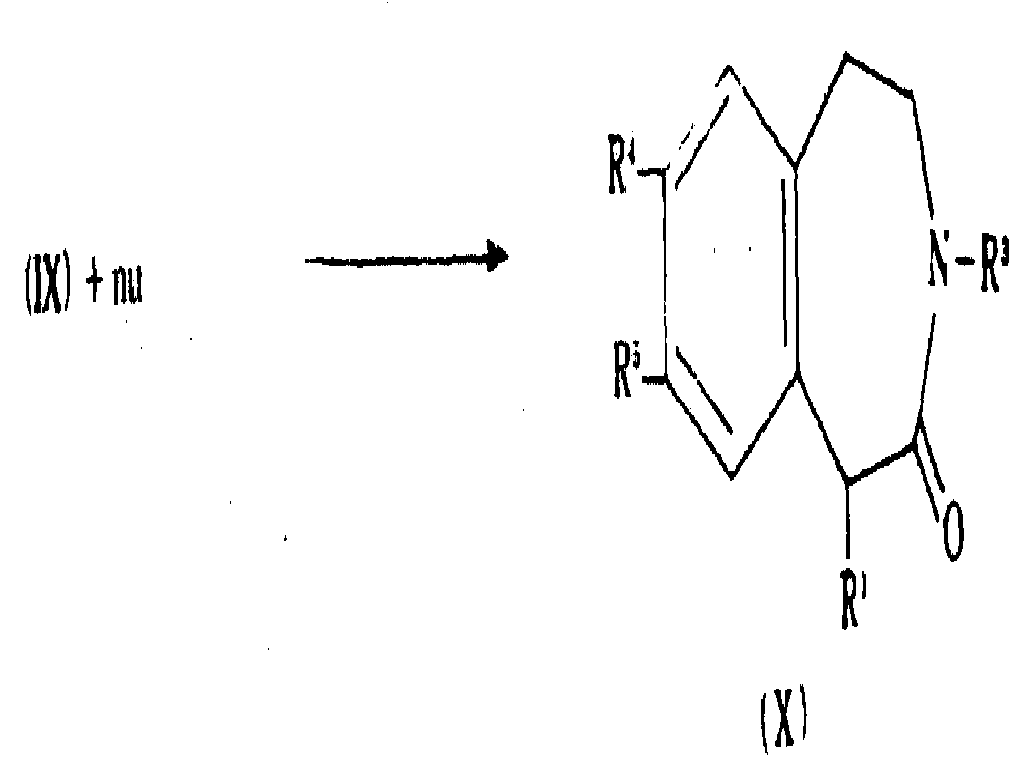
친핵체는 그룹 R1의 전구체이고, 예를 들어 알카놀, 일급 또는 이급 아민, 티올, 소디오에틸말로네이트, 시아나이드 등일 수 있다.
R2가 수소 이외인 것이 요구될 경우, 일반식(X)의 화합물을 전술한 바의 적합한 할로겐화제와 반응시켜 하기 일반식(XI)의 화합물을 형성시킨다 :

일반식(XI)의 화합물을 이어서 그룹 R2의 전구체인 친핵체(nu)와 반응시켜 Y그룹을 치환하고 하기 일반식(Ⅲ)의 화합물을 제조한다 :

이 두 단계에 대한 반응조건은 상기에 기술한 바와 같다. 또한, 친핵체의 반응순서는 R2그룹이 먼저 첨가되고 R1그룹이 후에 첨가되도록 역으로 할 수도 있다.
또다른 경로로서는, 일반식(Ⅷ)의 화합물은 L1이 이탈그룹(예 : Cl,Br 또는 I와 같은 할로겐, 또는 토실옥시, 메탄설포닐옥시 등과 같은 설포노닐옥시 그룹)인 일반식 R1L1의 화합물과 친전자 치환 반응으로 반응시켜 하기 일반식(X)의 화합물을 생성한다.

본 발명은 NaH,KH, 칼륨 3급 부톡사이드 등과 같은 강염기, M+L-의 존재하에 수행한다. 반응은 약 0℃ 내지 약 100℃의 온도에서 수행될 수 있으며 THF,DMF 등과 같은 불활성 용매 중에서 또는 순수하게 수행될 수 있다.
R2가 수소가 아닌 다른 것이 요구될 때, 일반식(X)의 화합물을 화합물 R2L1(여기서, L1은 상기한 바와 같다)과 또 다른 친전자 치환반응으로 반응시켜 하기 일반식(Ⅲ)의 화합물을 제조한다 :

이 두번째 친전자적 치환반응은 전술한 바와 기본적으로 동일한 조건하에서 수행된다. 또한, R2L1및 R2L1반응물의 반응순서를 R2그룹을 먼저 가하고 R1그룹은 후에 가하는 식으로 역으로 할 수도 있다.
상기 일반식(Ⅶ)의 화합물은 하기 일반식(Ⅶ)화합물의 올레핀 결합 및 카보닐 그룹 모두를 환원시킬 보다 강한 환원제를 사용하여 일반식(Ⅰ)의 화합물로 직접 전환될 수도 있다 :

적합한 보다 강한 환원조건은, 예를 들어 승온 및 승압하에서 예를 들어 약 25˚내지 약 100℃ 및 20 내지 100기압에서 라니니켈에 의한 촉매적 수소화를 포함한다. 본 환원 반응은 에탄올과 같은 불활성 용매중에서 수행할 수 있다.
B. R3가 CH3인 일반식(Ⅰ)의 화합물을 제조하기 위해, 하기 일반식(Ⅶ)의 화합물을 하기 일반식(Ⅷ)의 화합물로 환원시킨다 :

상기식에서, R은 알킬 또는 아릴 그룹(예 : 메틸, 에틸, 페닐 등)이다. 예를 들어 LiAlH4등과 같은 어떠한 적합한 환원제도 적합한 용매(예 : 에테르, THF등)중에서 0℃ 내지 반응 혼합물의 환류 온도 미만의 온도에서 사용할 수 있다.
일반식(XII)의 화합물은 다수의 상이한 기술로 제조할 수 있다. 예를 들어, 하기 화합물(XIV)을 화합물(XV)과 반응시켜 화합물(XⅥ)을 수득한다 :

일반식(XV)에서 L2는 Cl,Br,l, 토실옥시, 메탄설포닐옥시 등과 같은 적절한 이탈 그룹을 나타내며, R12는 알킬을 나타낸다. 에테르, CH2Cl2CHCl 등과 같은 어떠한 불활성 용매를 사용할 수 있다.
일반식(XVI)의 화합물은 HCl,CF3SO3H등과 같은 강산으로 고리화 반응시켜 하기 일반식(XⅦ) 및 (XⅧ)의 화합물(화합물(XⅦ)는 또한 이하에 더욱 상세히 기술될 본 발명의 공정 D에 따라 제조된 일반식(I)의 최종화합물이다)을 수득한다 :

일반식(XⅦ)의 화합물을 분리하여 일반식 의 화합물과 이어서 세르 암모늄 니트레이트 및 나트륨 브로메이트와 같은 산화제와 반응시켜 하기 일반식(XIX)의 화합물을 생성한다 :
의 화합물과 이어서 세르 암모늄 니트레이트 및 나트륨 브로메이트와 같은 산화제와 반응시켜 하기 일반식(XIX)의 화합물을 생성한다 :

일반식(XIX)의 화합물의 카보닐 그룹을 적합한 환원제(예 : NaBH4)에 의해 하이드록실 그룹으로 환원시켜 하기 일반식(XX)의 화합물을 생성시킨다.

일반식(XX)의 화합물을 R1이 OR6a이고 R6a가 페닐, 치환된 페닐 또는 나프틸이고, R2가 H인 하기 일반식(XIIa)의 화합물로 전환시키기 위해, 일반식(XX)의 화합물을 디에틸 아조디카복실레이트(DEAD)의 존재하에 일반식 R6aOH 화합물과 반응시킨다 :

일반식(XX)의 화합물을 R2가 H이고, R1이 R1a이며 R1a가 OR6a가 아닌 R1이 일반식(XIIb)의 화합물로 전환시키기 위해, 일반식(XX)의 화합물을 토실 클로라이드(TsCl)과 같은 설포닐 할라이드와 반응시켜 일반식(XXI)의 화합물을 형성시킨다. 일반식(XXI)의 화합물을 이어서 R1a그룹의 전구체인 적합한 친핵체(nu)(예 : 메틸아민과 같은 HNR6R10, 메탄올, 에탄올 또는 벤질알콜과 같은 알칸올, 메탄티올과 같은 티올 NaCN과 같은 시아나이드 등)와 반응시켜 하기 일반식(XIIb)의 화합물을 생성한다 :

일반식(XIIa) 및 (XIIb)의 화합물은 R2가 H가 아닌 알빈식(XII)의 화합물로 전환시키기 위하여, 상기 두문단에서 기술한 반응을 적합한 친핵체로 반복하여 수행하여 목적하는 R2그룹을 생성시킨다. 역시 R1및 R2그룹의 첨가는 역으로 할 수 있다.
C. 일반식(XXIIa) 또는 (XXIIb)의 화합물을 화합물(R2L3) 또는 (R1L3)와 각기 반응시킨 후, 0℃ 내지 반응 혼합물의 환류온도에서 저급 알코올과 같은 불활성 용매 중에서 적합한 수소화제(예 : NaBH4)와 반응시켜 일반식(Ⅰ)의 화합물을 생성한다 :

상기식에서, L3는 할로산 또는 설폰산(예 : Br, 토실옥시, Cl등)으로부터 유도된 음이온과 같은 적합한 이탈 그룹을 나타낸다.
일반식(XXIIa) 또는 (XXIIb)의 화합물은 일반식(XVIII)의 화합물을 적합한 친전자체 R1L3또는 R2L3(L3는 상술한 바와 같다)와 각기 반응시켜 제조할 수 있다. 적합한 친전자체는, 예를 들어 벤질 브로마이드를 포함한다. 본 반응은 탄산칼륨과 같은 염기 및 아세토니트릴과 같은 불활성 용매의 존재하에 수행할 수 있다.
또 다른 방법은, 즉 하기 일반식(XXIIIa) 또는 (XXIIb)의 화합물을 생성하기에 적합한 공정 E에 의하여, 일반식(XXIIa) 및 (XXIIb) 화합물의 올레핀 이중-결합을 0℃ 내지 반응혼합물의 환류 온도하에서 불활성 매체중의 아세트산과 같은 카르복실산의 존재하에서 나트륨 보로하이드라이드로 처리함 같이, 당해 분야의 통상적 기술로 포화시킬 수 있다 :

D. 일반식(Ⅰ)의 화합물 또한 일반식(XVI)의 화합물의 분자내 축합에 의해 제조할 수 있다 :

상기식에서, D는 아제핀의 환의 형성시에 DH로서 제거될 수 있는 반응성 그룹이다. 전형적으로 D는 하이드록시, 치환된 하이드록시 그룹, 특히 알콕시, 염소 또는 브롬과 같은 할로겐 또는 -O-토실 또는 -O-메실 그룹과 같은 설폰산 에스테르일 수 있다. 축합은 적합하게는 일반식(XVI)의 화합물을 0℃ 내지 반응혼합물의 환류온도에서 불활성 매체중에서 HCl, CF3SO3H와 같은 강산으로 처리한 후, 일반식(I)의 목적 화합물을 분리하여 실시한다.
끝처리 단계로서, 일반식(XXIV)의 화합물을 일반식(XXV)의 화합물과 반응시켜 일반식(I)의 화합물을 생성시킨다 :
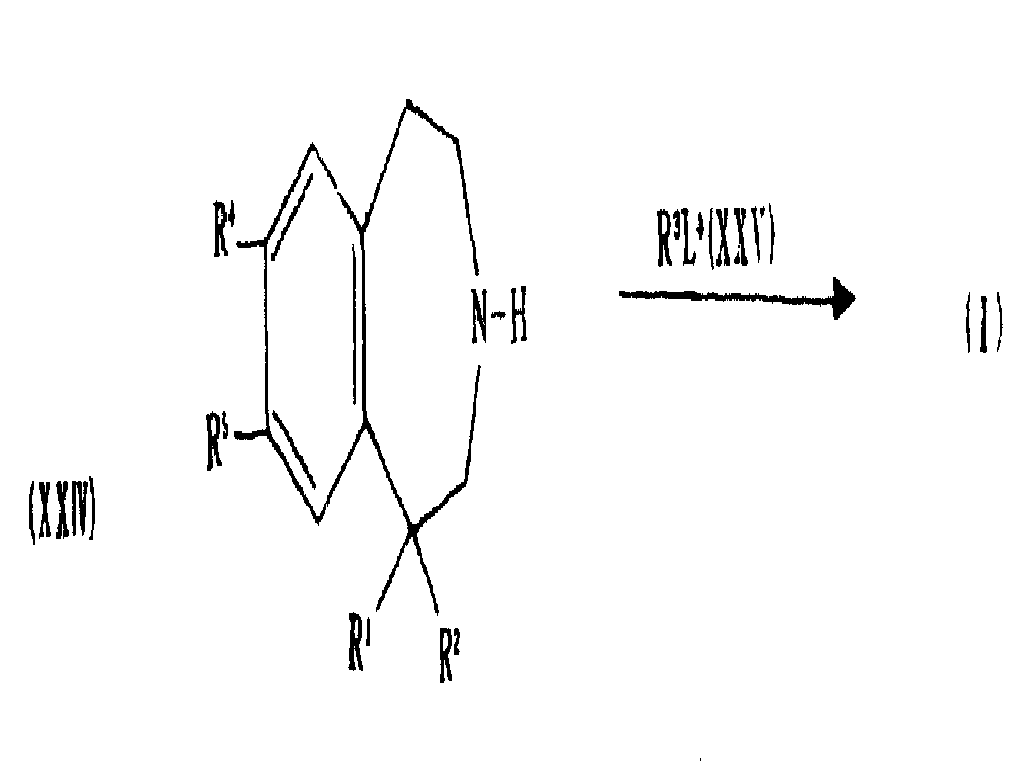
상기식에서, L4는 Br, 토실옥시, Cl 등과 같은 이탈 그룹이다. 상기 일반식(XXIV)의 화합물은, 예를 들어 일반식(XII)의 화합물을 염기(예 : 수성 또는 알코올성 KOH 또는 NaOH)와 같은 가수분해제로 처리하여 제조될 수 있다.
상기 공정 A 내지 E에서, 반응도중 특정 R1,R2,R3,R4및 R5를 보호하는 것이 때때로 바람직하고/하거나 필요하다. 통상적인 보호그룹을 적용한다. 예를 들어, 다음 표의 첫째열에 제시한 그룹을 표의 둘째열에 나타낸 바와 같이 보호한다.

물론, 당해분야에서 잘 알려져 있는 기타 보호그룹을 이용할 수 있다. 반응 또는 반응들 후에, 보호그룹은 표준 방법으로 제거될 수 있다.
또한, 일반식(I)의 R1,R2,R3,R4및 R5그룹들은 본 화합물을 출발물질을 적절하게 선택하여 변화될 수 있거나, 일반식(I)와 화합물을 치환체의 또다른 R1,R2,R3,R4및 R5그룹으로의 목적한 전환을 수행할 적합한 시약과 반응시켜 변화될 수 있다.
일반식(I)의 화합물의 용도는 항-정신병 및 항-우울증 활성을 암시하도록 선정된 시험 방법에 따라 기술될 수 있다.
[래트(rat)에 있어 조건 회피 억제]
임상적으로 활성인 정신병 치료제는 탈출반응을 지연시키지 않은 용량으로 뚜렷한 시도 회피 행동을 억제한다고 알려져 있다[참조 : Ann. N. Y. Acad. Sci.66, 740(1957)]. 일련의 실험을 수행하여 본 발명 화합물의 래트에 있어서의 조건 회피 반응(CAR) 억제 능력을 평가한다.
[재료 및 방법]
래트를 10- 초간의 발에 대한 쇼크(0.6ma)를 피하기 위해 5-초간의 긴장상태에 대한 반응으로 실험실의 바닥위 6.75인치(17.5cm)에 놓인 플랫폼상으로 점프시킨다. 20회 시도로 이루어진 각 실험 기간은 30초 간격으로 한다. 정확한 CAR을 래트가 긴장 상태중(발에 대한 쇼크에 선행) 플랫폼상으로 점프할때 마다 기록한다. 탈출 반응을 래트가 쇼크중에 플랫폼으로 점프할때 기록한다. 반응 실패는 10- 초간의 쇼크 기간중 탈출반응의 결핍으로 정의된다.
6 내지 8마리의 래트 그룹을 2일간 연속 훈련시킨다(총 40회 시도). 제2일에 표준에 도달한 래트(+20회 시도중 16회 이상의 정확한 CAR)를 제3일에 시험 약제 또는 담체로 처리한다. CAR의 억제는 약제-처리대 담체-처리 래트의 수행력을 비교하는 스튜던트 t-테스트를 사용하여 통계학적으로 분석한다. 각 약제에 대한 최소유효랑(MED)은 회피 반응을 유의하게(P<0.05) 감소시키는 것으로 시험된 최소 용량으로서 정의된다.
[결과]
상기 방법으로 시험했을때 본 발명의 대표적인 화합물은 하기 표 1에 기술한 바와 같이 조건회피 반응의 용량-관련 특이적 억제율을 나타낸다.
[표 1]

*Ph=페닐
**POM=t-BuㆍCOOㆍCH2O-
[경쟁적 억제 분석]
신경 조직상에 재현가능한 생리학적 변화에 영향을 미칠 수 있는 많은 화합물들은 하나이상의 수용체 부위에 결합함으로써 작용한다고 생각된다. 타겟(target)기관 또는 구조의 동종물을 사용하여 시험관에서 시험수행시 이들 수용체 부위와 강하게 상호작용하는 화합물은 생체내 투여시 유사한 특성을 나타낼 것으로 기대되며, 따라서 강력안 치료 및/ 또는 진단제로서 계속 연구대상이 되고 있다.
시험관내에서 화합물이 수용체 부위에 결합함은 유효한 부위의 결합 특이성 및 포화성으로 표현된다. 결합의 특성화에 대한 방법론 및 데이타의 해설이 문헌[참조 : Billard et all., Life Sciences 35, 1885(1984)]에 기술되어 있고, 여기서, 벤즈아제핀(R)-(+)-8-클로로-2,3,4,5-테트라하이드로-3-메틸-5-페닐- -3-벤즈아제핀-7-을 헤미말리에이트(SCH 23390)의 도파핀 D-1 수용체에 대한 결합이 특성화된다.
-3-벤즈아제핀-7-을 헤미말리에이트(SCH 23390)의 도파핀 D-1 수용체에 대한 결합이 특성화된다.
[재료 및 방법]
삼중수소화된 SCH 23390 및 삼중수소화된 스피페론(강력한 D-2 리간드)을 빌랴드 등의 상기 참조 문헌에 기술된 바와 같이 수득하여 0.05M 트리스 완충액(PH 7.4)중에서 연속적으로 희석시킨다. 본 발명의 화합물을 0.05M 트리스 완충액(요구되는 PH 7.4)중에서 희석시킨다.
[조직 제조]
챨스 리버 브리딩 래보러터리즈(Charles River Breeding Laboratories, Mass)로부터 스프라그-돌리 래트 숫놈(200 내지 250g)의 뇌조직을 얻어 이를 사용한다. 래트를 인력으로 치사하여 그의 뇌를 분리하고 얼음위에 둔다. 줄문 조직을 절개하여 PH 7.4(25℃에서)의 냉각된 50mM 트리스 완충액 100용량(W/V)중에서 모우고 균질화시킨다(Brinkman Polytron, 10초). 균질물을 20,000Xg에서 10분동안 원심분리시킨다. 생성된 펠릿을 트리스 완충액 중에서 재균질화시키고 다시 원심분리시킨다. 최종 펠릿을 120mM NaCl, 5mM KCl, 2mM CaCl2, 및 1mM MgCl2를 함유한 PH 7.4의 50mM 트리스 완충액 중에서 재현탁시킨다.
[분석]
폴리프로필렌 항온 튜브에 4mg/ml 메틸셀룰로오즈, 트리스 완충액내의3H-SCH 23390 용액 100μl(최종 반응 혼합물의 농도=0.3nM) 또는 트리스 완충액 내의3H-스피페론 용액 100μl(최종 농도=0.2nM) 및 조직 현탁액 800μl(약 3mg/분석)를 함유한 PH 7.4이 0.05M 트리스 중에서 현탁 또는 용해된 다양한 농도의 개개의 시험 화합물 100μl을 얻는다. 튜브를 15분간 37℃에서 항온시키고 와트만 GF/B 여과지를 통과시켜 신속히 진공여과시키고 PH 7.4의 냉각된 50mM 트리스 완충액 4ml로 4회 세척한다. 여과지를 신틸레이션 바이얼에 이동시키고, 신틸런트(Scintosol, Isolab, Inc.) 10ml로 25℃에서 16시간 평형시키고 액체 신틸레이션 계수기에서 방사활성을 측정한다. Ki치는 Ki=IC50/(1+([L]/KD)) (여기에서, IC50=특이적으로 결합된3H-Sch 23390의 50%를 치환하는데 필요한 시험 약제의 농도, [L]=분석에서 사용된 방사성 리간드의 농도, 및 KD=해리상수)의 관계를 이용하여 빌랴드 등에 의해 기술된 바와 같이 구한다.
[결과]
본 발명의 화합물에 대한 분석으로부터 구한 억제상수(Ki)는 하기 표 2에 나타낸 바와 같다.
[표 2]

*Ph=페닐
**POM=t-BuㆍCOOㆍCH2O-
SCH 23390과의 경쟁적 결합 분서에서 본 화합물의 비교적 작은 Ki치는 일반식(I)의 화합물이 D-1 수용체 부위에 강력히 결합됨을 의미한다. 스피페론이 높은 선택성을 갖는 D-2 부위에 대한 비교적 높은 Ki치는 본 화합물이 그 수용체 부위에 특이적으로 결합되지 않음을 의미한다.
본 발명의 우울증 치료 방법은 하기에 설명하는 바와 같이, 예를들어 마우스에 있어서 테트라벤아진(TBZ)-유도된 하수증에 대한 화합물의 효과를 측정하거나 래트에 있어서 설치류방제(muricide) 활성에 대한 화합물의 효과를 측정하는 시험방법으로 확인한다.
[우울증 치료 능력]
마우스에 있어서 테트라벤아진(TAZ)-유도된 하수증에 대한 효력
임상적으로 활성인 우울증 치료제는 마우스에 있어 TBZ-유도된 하수증을 저지한다고 알려져 있다(참조 : Psychosomatic Medicine, Nodine and Moyer, Eds., Lea and Febiger, Philadelphia, 1962, pp 683-90). 본 시험의 활성을 사람에 있어서 우울증 치료제 활성을 예견하는 데에 이용한다.
[방법 및 재료]
5마리의 마우스 그룹에 시험약제를 투여한지 30분 후에 테트라벤아진을 30mg/kg으로 복강내 주사한다. 30분 후에, 하수증의 정도를 검사한다. 처리된 각각의 그룹에 대한 억제율(%)을 이용하여 ED50값 (마우스의 하수증을 50% 억제시키는 용량)을 결정한다. 프로빗 분석으로 ED50값 및 95% 신뢰한계를 계산한다.
[래트에 있어서 설치류방제 작용에 대한 효력]
래트에 있어서 설치류방제(마우스-치사)작용의 방해정도를 약제의 우울증 치료 활성을 평가하는 측정치로서 사용한다[참조 : Int. J. Neuro-pharmacol.,5, 405-11(1966)].
[방법 및 재료]
5마리의 래트 그룹에 시험약제를 복강내로 투여하고 30분 및 60분후에 설치류방제 작용 여부를 시험한다. 이들 시각에서 수득한 데이타를 사용하여 처리된 각각의 그룹에 대한 억제율(%)을 계산하고 용량-반응 데이타를 이용하여 각각의 ED50을 결정한다. ED50은 처리된 래트의 설치류방제 작용을 50% 저지시키는 용량이고 프로빗 분석을 이용하여 계산된다.
일반식(I)의 화합물의 진통효과 및 진통효과를 제공하는 방법은 하기 기술된 마우스에 있어서 아세트산 비틀림 시험(Acetic Acid Writhing Test)으로 예시될 수 있다.
[마우스에 있어서 아세트산 비틀림 시험]
아세트산 복강내 주사에 의해 유도된 비틀림의 억제는 항유해수용기 약제(고통 감각의 인식 또는 전도를 억제시키는 약제)의 차폐에 대한 정립된 실험적 동물 모델이다[참조 : Hendershot et al.,J.Pharmacol.Exp.Therap.125: 237, (1959) 및 Koster et al.,Fed.Proc.18: 412, (1959)].
[방법 및 재료]
시험대상 화합물을 수성 0.4% 메틸셀룰로오즈 담체중에 용해시키거나 현탁시킨다. 경구투여용 용량은 체중 1kg당 20mg의 총용적으로 선택된 화합물 중량이 투여되게끔 제조한다. 피하 또는 복강내 투여용 용량은 체중 1kg당 10ml의 용적으로 선택된 화합물 중량이 투여되게끔 제조한다.
시험 방법은 문헌[참조 : 상기의 Hendershot et al.]에 기술되어 있고, 단 페닐퀴논을 아세트산으로 대체하였다. 5마리의 그룹의 숫컷 CFl 마우스(20 내지 26g)에 시험약제를 경구투여하고 15분후에 0.6% 수성 아세트산을 주사한다(10mg/kg). 대량 관찰 비이커내에 마우스를 놓아두고, 아세트산을 주사한지 3분후 부터 시작하여 10분 간격으로 각각의 동물에 대한 비틀림 수를 계수한다. 비틀림은 연속적인 등구부림, 골반 회전 및 후지(hindlimb)확대로서 정의된다. 30mg/kg의 용량을 사용하여 초기 차폐를 수행한다. 이 용량으로 대조군과 비교한 비틀림의 수가 50% 이상 감소할 경우, 동물은 보호되어지는 것을 간주되므로, 보다 낮은 용량의 로그순서를 이용하여 용량 반응곡선을 도시하며, 보간법으로 ED50값을 결정한다.
독성에 관하여는, 본 발명의 화합물은 치료용량에서 무독성이다.
일반식(I)의 화합물로부터 약제학적 조성물을 제조할 경우, 불활성인 약제학으로 허용되는 담체를 활성화합물과 혼합시킬 수 있다. 약제학적으로 허용되는 담체는 고체 또는 액체일 수 있다. 고형 제제로는 산제, 정제, 분산과립제, 캡슐제, 카세 및 좌제가 있다. 고체담체는 또한 희석제, 향미제, 용해제, 윤활제, 현탁화제, 결합제 또는 정제 붕해제로서 작용할 수 있는 하나 이상의 물질일 수 있고, 캡슐화 물질일 수도 있다. 산제에서, 담체는 미분된 활성화합물과 혼합되는 미분된 고체이다. 정제에서, 활성화합물은 필요한 결합특성을 갖는 담체와 적합한 비율로 혼합되어 목적하는 형태 및 크기로 만들어진다. 산제 및 정제는 활성화합물의 효능, 투여대상의 체격 및 연령, 및 특정한 치료에 요구되는 용량범위에 따라서, 통상적으로 활성성분 5 내지 약 70%를 함유한다. 적합한 고체담체는 탄산마그네슘, 마그네슘 스테아레이트, 탈크, 당, 락토오즈, 펙틴, 덱스트린, 전분, 젤라틴, 트라가칸드 고무, 메틸셀룰로오즈, 나트륨 카복시메틸 셀룰로오즈, 저 용융 왁스, 코코아 버터 및 약제학적 분야에 통상적으로 사용된 기타의 물질이다. "제제"라는 용어는 캡슐에서 활성성분이 담체로 둘러싸져서(다른 담체와 함께 또는 다른 담체 없이), 담체와 연합될 경우, 활성물질을 캡슐화 물질인 담체와 함께 제형화한 것을 포함한다. 유사하게, 카세도 포함된다. 경구투여용으로 적합한 고체용량형으로 정제, 산제, 카세 및 캡슐제를 사용할 수 있다.
좌제를 제조하기 위해, 지방산 글리세라이드 또는 코코아 버터의 혼합물과 같은 저 용융 왁스를 먼저 용융시키고 이에 활성성분을 교반시켜 균질하게 분산시킨다. 그다음 용융된 균질 혼합물을 편리한 크기의 주형내에 붓고 냉각시킴으로써 고체화한다.
액체형 제제로는 용제, 현탁제 및 유화제가 있다. 예로서, 비경구 주사용 물 또는 물-프로필렌 글리콜 용액을 언급할 수 있다. 액제는 또한 수성 폴리에틸렌 글리콜 용액중의 용액으로 제형화될 수 있다. 경구투여용으로 적합한 수성 제제는 물에 활성성분을 가하고 바람직한 바의 적합한 착색제, 향미제, 안정화제, 감미제, 용해제 및 증진제를 가함으로써 제조될 수 있다. 경구투여용으로 적합한 수성 현탁제는 미분된 활성성분을 물중에 점성물질(예 : 천연 또는 합성고무, 수지, 메틸셀룰로오즈, 나트륨 카복시메틸셀룰로오즈 및 기타 공지된 현탁화제)과 함께 분산시켜 제조될 수 있다.
또한, 투여하기 전에 즉시, 경우 또는 비경구 투여용 액체형 제제로 전환될 수 있는 고형제제가 포함된다. 이러한 액체형으로 용제, 현탁제 및 유화제가 있다. 이들 특정한 고형제제는 단위용량형으로 가장 편리하게 제공되어지며 그 자체가 사용되어 단일 액체 용량 단위를 제공한다. 한편, 액체형으로 전환된 후, 다수의 개별적 액체용량을, 예비 측정된 용적의 액체형 제제를 시린지(syringe), 티스푼 또는 다른 부피측정용기로 측정하여 수득할 수 있도록 충분한 고체가 제공될 수 있다. 액체형으로 전환될 수 있는 고형제제는 활성물질에 추가로, 향미제, 착색제, 안정화제, 완충제, 인공 및 천연감미제, 분산화제, 중진제, 용해제 등을 포함한다. 액체형제제를 제조하는데 사용되는 용매는 물, 등장성 수성염 용액, 에탄올, 글리세린, 프로필렌글리콜 등 및 이의 혼합물이다. 사용되는 용매는 투여경로를 고려하여 선택할 수 있다. 예를들어, 다량의 에탄올을 함유한 액체제제는 일반적으로 비경구용으로 적합하지 않다.
본 발명은 또한 경피투여를 포함하여 다른 투여계를 고려할 수 있으나, 이에만 제한되는 것은 아니다. 경피 조성물은 크림, 로션 및/ 또는 유제의 형태를 취할 수 있으며 본 목적을 위해 당해 기술분야에서 통상적인 메트릭스 또는 저장형의 경피 고약에 포함시킬 수 있다.
바람직하게는, 약제학적 제제는 단위 용량형이다. 그러한 형태에서, 제제는 적합한 양의 활성 성분을 함유하는 단위용량으로 세분된다. 단위용량형은 바이얼 또는 앰플중에서 패킷된 정제, 캡슐 및 분말과 같은 분별 정량의 제제를 함유하는 패키지 제제일 수 있다. 단위 용량형은 또한 캡슐, 카세 또는 정제자체일 수 있으며, 또한 이들중 적합한 얼마간은 패키지 형태일 수 있다.
단위투여 제제중의 활성 성분량은 특정 적용 및 활성 성분의 강도 및 의도하는 치료에 따라 1mg 내지 100mg으로 변화 또는 조정할 수 있다. 약 0.02 내지 약 2.0mg/kg 바람직하게는 약 0.02 내지 약 0.2mg/kg의 용량을 사용하여 1일당 1 내지 3회 투여로 나누어 사용할 수 있다. 조성물은, 필요에 따라, 기타 치료제를 또한 함유할 수 있다.
용량은 환자의 요구상태, 치료될 질환의 심각성 및 사용될 특정 화합물에 따라 변화시킬 수 있다. 특정상황에 대한 적합한 용량의 결정은 의학분야의 전문가들의 영역이다. 편리하게는 1일 총 투여량은 하루에 수회로 나누어 또는 연속적 주입에 의해 분배하여 투여할 수 있다.
본 명세서에 기술된 본 발명은 하기 실시예에 의해 예시되며, 이는 본 발명의 범주를 제한하는 것으로 간주되어서는 안된다. 본 출원 발명자의 목적하는 한도내에서 대치적 기전 경로 및 유사 구조물도 당해분야의 숙련인들에게 명백할 것이다.
[제조실시예 1]

상기 구조식 A의 화합물 30.0g, 브로모아세트알데히드 디에틸아세탈 32.8g, 무수 K2CO350g 및 무수 디메틸포름아미드(DMF) 150ml의 혼합물을 교반시키고 질소하게 120℃까지 가열한다. 2시간후, 용액을 여과하고, 그 여과물을 물속에 붓는다. 그 혼합물을 에테르 200ml로 2회 추출하고, 혼합된 에테르층을 염수로 세척하고, 건조시켜 오일로 농축시킨다(38.4g). 얇은 막 크로마토그라피는 상기 구조식(B)의 화합물이 유일한 주생성물임을 보여준다(CHCl3/CH3OH/NH4OH 1000 : 4 : 3으로 전개된 Rf=0.73).
[실시예 1]

제조실시예 1에서와 같이 제조된 구조식 B의 화합물(3.4g)을 빙욕 온도에서 메탄설폰산 10ml와 혼합한후, 생성 용액을 70℃까지 가열한다. 2시간후, 생성된 혼합물을 과잉의 냉각된 포화 NaCO3용액에 주입한다. 혼합물을 에테르로 추출한다. 추출물을 염수로 세척하고, 건조시켜 오일로 농축시킨다(2.7g). 생성물을 에테르에 용해시키고 약간 과잉의 에테르성 HCl로 처리한다. 황색 고무를 분리 및 결정화시킨다. 고체를 여과하고 2-부타논으로부터 재결정하여 융점이 195 내지 197℃인 하이드로클로라이드 염으로써 상기 구조식(C)의 화합물을 수득한다.

디메틸포름아미드 20ml중의 실시예 1A에서 제조된 구조식 C의 화합물(750mg)을 광유중의 60% 나트륨 하이드라이드(490mg) 및 디메틸포름아미드 20ml중의 에탄 티올(0.9ml)로부터 제조된 용액에 적가한다. 생성된 혼합물을 130℃에서 10시간동안 가열하고 물에 부은후, 에테르로 추출한다. 이어서 수성층을 HCl로 pH 1로 산성으로 만들고 에테르로 다시 추출한다. 수성층을 고체 NaHCO3로 재염기성화하고 침전된 오일을 에틸 아세테이트로 추출한다. 추출물을 염수로 세척하고, 무수 MgSO4상에서 건조시키고, 오일로 농축시켜, 이를 에테르에 용해시키고 에테르성 HCl로 처리한다. 침전된 염을 웃물을 가만히 따라내어 분리하고, 아세톤으로부터 재결정화하여 융점이 235 내지 236℃인 염화수소산염으로 상기 구조식 D의 화합물을 수득한다.
기본적으로 실시예 1에서 기술한 바와 동일한 방법을 이용하고, 적합한 브로모아세트알데하이드의 디메틸아세탈을 사용하여, 하기 5-알콕시벤즈아제핀-7-올을 제조한다 :

[제조실시예 2]

벤젠 250ml중의 상기 구조식 C의 화합물(19.5g)의 용액을 환류하에 에틸 클로로포메이트 20.7ml로 처리한다. 생성된 용액을 3시간동안 환류하에 가열한후, 용매를 진공중에서 제거하고, 그 잔사를 에테르 및 5% HCl 사이에 분배한다. 에테르층을 분리하고, 염수로 세척하고, 건조시켜 진한 고무상태로 농축시키고, 이를 석유 에테르중에 용해시키고 다코(Darco) 및 플로리실(Florisil)로 처리하고 여과시킨다. 그 여과물을 황색 오일로 농축시킨다(16.9g). 얇은 막 크로마토그라피는 Rf=0.4(헥산/에틸아세테이트-2 : 1)인 단일 지점으로서 상기 구조식 E의 화합물을 나타낸다.

상기 구조식 E의 화합물(16.9g)을 아세토니트릴 175ml중에 용해시키고 물 75ml중의 나트륨 브로메이트 8.45g 및 세릭 암모늄 니트레이트 548mg의 용액으로 처리한다. 2- 상 혼합물을 24시간동안 환류하에 교반시킨다.
냉각된 혼합물을 물 250ml로 희석시키고 에테르 250ml로 2회 추출한다. 에테르상을 염수로 세척하고, 건조시켜 고무로 될때까지 농축시킨다. 에테르/석유 에테프로 연마하여 각기둥 형태로서 구조식 F의 화합물(6.0g)의 수득한다(웅점 : 134 내지 135℃).
[실시예 2]

상기 구조식 F의 화합물(1.0g)(제조실시예 2B에서 기술한 바와 같이 제조하며 R1은 R2와 함께 카보닐을 나타낸다)을 순수한 에탄올 20ml중에서 현탁시키고 교반시키며 나트륨 보로하이드라이드 140mg으로 일부분씩 처리한다. 혼합물을 40℃까지 가온하고 20분동안 교반시킨후, 5% HCl 10ml 및 얼음 약 10g을 첨가한다. 추가의 30분간 교반시킨후, 고형 생성물을 여과하고, 건조시켜 융점이 143 내지 144℃인 상기 구조식 G의 화합물 930mg을 수득한다.

무수 디메틸포름아미드 10ml중의 상기 구조식 G의 화합물 0.45g의 용액을 디메틸포름아미드 10ml중에서 60% NaH/광유 287mg 및 에탄디올 0.56ml로부터 제조한 나트륨 티오에톡사이드 용액에 가한다. 생성된 용액을 125℃에서 밤새 가열한다. 이어서 물속에 붓고 에테르로 추출한다. 수성상을 pH 1로 산성화시키고 고형 NaHCO3로 재염기화시킨다. 오일상 생성물을 에틸 아세테이트로 추출하고 용액을 증발시켜 조생성물로 상기 구조식 H의 화합물을 수득한다(0.4g). 이 화합물을 약간 과잉의 에테르성 HCl로 에테르성 용액처리를 하여 그의 하이드로클로라이드로 전환시킨다. 생성된 염을 여과하고 진공중에서 건조시켜 백색의 무정형 고체를 생성한다.
[실시예 3]

실시예 2에서 기술한 바와 같이 제조한 구조식 H의 화합물(0.4g)을 디메틸포름아미드 5ml중에 용해시키고, 트리에틸아민 0.2ml를 첨가한후 메틸 요오다이드 0.093ml를 가한다. 생성된 혼합물을 실온에서 밤새 방치시킨후 이를 물속에 붓는다. 혼합물을 에틸 아세테이트로 추출하고, 건조시켜 오일로 농축시킨다. 이 물질(250mg)을 25g의 머크실리카겔 60-G상에서 CHCl3/CH3OH/NH4CH-1000 : 50 : 3으로 용출시켜 크로마토그라피한다. TLC 균일질(Rf=0.69, 동일 용매계)인 성분을 수득하여(105mg) 에테르에 용해시키고 45mg 말레산의 에테르성 용액으로 처리한다. 침전된 고체를 여과하고 진공에서 건조시켜 말리에이트 염으로서 상기 구조식(J)의 화합물 88mg을 수득한다.
C13H18NOCl. C4H4O4에 대한 분석
계산치 : C; 52.64, H; 5.72, N; 3.61.
실측치 : C; 52.14, H; 5.60, N; 3.46.
FAB질량 스펙트럼 m/e+1=272.
[제조실시예 4]

트리페닐포스핀(0.57g) 및 페놀(0.21g)을 벤젠 30ml중의 실시예 2A에 기술한 바와 같이 제조된 구조식 G의 화합물(0.6g)의 용액에 첨가한다. 이어서 이 용액에 5분에 거쳐 또다른 벤젠 10ml중의 디이소프로필 아조디카복실레이트(0.433ml)의 용액을 첨가한다. 생성된 혼합물을 실온에서 밤새 방치한 후 고무 상태로 농축시킨다. 본 생성물을 에틸 아세테이트/헥산-1 : 4로 용출시키며 머크실라카겔 60G 100g상에서 크로마토그라피하여 황색 오일로서 상기 구조식 K의 화합물 0.5g을 수득한다.
[실시예 4]

에테르 20ml중의 상기 구조식 K의 화합물(430mg)을 에테르 20ml중의 리튬 알루미늄 하이드라이드(53mg)의 현탁액에 첨가한다. 그 혼합물을 3시간동안 실온에서 교반시킨후 모든 고체가 용해되어 상기 분리될때까지 냉각된 10% NaOH 용액으로 처리하여 분해시킨다. 수성상을 에틸 아세테이트로 추출하고 혼합된 유기층을 염수로 세척하고, 건조하여 오일로 농축시키고(350mg) 진공중에서 밤새 건조하여 고체화시켜 상기 구조식 L의 화합물을 수득한다.

디메틸포름아미드 10ml중의 상기 구조식 L의 화합물(350mg) 용액을 디메틸포름아미드 5ml중의 에탄티올 0.408ml 및 132mg의 50% NaH/광유 분산액으로부터 제조한 나트륨 티오에톡사이드 용액에 적가한다. 생성된 혼합물을 질소 기압하에 100℃에서 3시간동안 교반시킨다. 용매를 진공중에서 제거하고, 그 잔사를 물과 에테르 사이에 분배하여 상기 구조식 M의 화합물을 수득한다(융점 : 166 내지 168℃).
제조실시예 4에서 페놀 대신 하기 표 3의 첫째열에 나타낸 치환된 페놀을 사용하고 기본적으로 제조실시예 4 및 실시예 4에 기술된 바와 동일한 공정을 이용하여, 표 3의 두번째 열에 나타낸 화합물을 또한 제조한다 :
[표 3]

[제조실시예 5]

벤젠(10ml)중의 트리-n-부틸포스핀(0.485g, 2.4mmole)의 교반된 용액에 고체 N-(페닐티오)석신이미드(475mg, 2.4mmole)를 한번에 가한다. 생성된 용액을 주변온도에서 5분간 교반시킨 후 실시예 2A에서 기술한 바와 같이 제조한 구조식 G(554mg, 1.8mmole)의 화합물로 모두 한번에 가한다. 혼합물을 주위온도에서 약 12시간 동안 교반한다. 추가의 트리-n-부틸포스핀 0.2ml을 가하고 추가로 2시간동안 교반시킨다.
생성된 혼합물을 농축시켜 건조시키고 물 및 에테르 헥산 1 : 1을 가한다. 유기상을 염수로 세척하고, 건조시켜 고무(0.8g)로 농축시킨다. 고무를 헥산, 이어서 헥산 에틸 아세테이트(1 : 4)로 용출시키며 약 8.0g의 머크 실리카겔 G상에서 크로마토그라피하여 상기 구조식 N의 화합물 0.6g을 수득하고, NMR 및 TLC로 특성화한다(Rf=0.3, 에틸 아세테이트 헥산 1 : 3 용액 중에서).
[실시예 5]

에테르 20ml 중의 제조실시예 5의 상기 구조식 N의 크로마토그라피된 화합물(0.55g, 1.4mmole)을 에테르 20ml중의 리튬 알루미늄 하이드라이드 70mg(1.8mmole)의 냉각된 슬러리에 가한다. 탁한 용액을 주변 온도에서 약 50시간동안 교반시킨다. 추가의 에테르중의 LiAlH440mg을 가한다. 30분후 TLC는 완료된 반응임을 나타낸다. 냉각된 10% NaOH를 모든 고체가 용해될 때까지 가한다. 수성상을 분리하고 에틸 아세테이트로 추출한다. 혼합된 유기층을 염수로 세척하고, 건조 및 농축시켜 고무로써 구조식 P의 화합물을 수득한다(0.427g).

상기 구조식 P의 화합물(0.42g, 1.25mmole)을 DMF 5ml 중에서 나트륨 티오에톡사이드의 용액(오일 분산액 중의 60% NaH 100mg(2.5mmole) 및 DMF 10ml중의 에탄티올 0.185ml(2.5mmole)으로부터 제조)에 가하고 맑은 용액을 약 32시간동안 100 내지 110℃에서 교반시킨다. 추가의 나트륨 티오에톡사이드(상기한 바와 같이 제조) 2.5mmole을 가하고 반응 혼합물을 추가의 3시간동안 가열하는데, 이때 TLC로부터 실질적으로 완전 반응임을 알 수 있다.
그 혼합물을 물에 붓고 헥산으로 추출한다. 염기성 수용액을 5% HCl로 pH 1로 산성화시키고 헥산으로 재-추출한다. 산성 상을 고체 NaHCO3으로 염기성으로 만들고 에틸 아세테이트로 추출하여 오일상 생성물 400mg을 수득한다. 방치중, 물질이 결정화한다. 고체를 에테르/석유 에테르로부터 재결정화시켜 상기 구조식 Q의 화합물 170mg을 수득한다(융점 : 158 내지 160℃).
[제조실시예 6]

티오닐 클로라이드 85ml을 무수 디클로로메탄 100ml 중의 상기 구조식 R의 산 64.0g의 용액에 교반시키며 적가한다. 혼합물을 실온에서 3시간 더 교반시킨 후 증기욕에서 2시간동안 가볍게 환류시켜 가열한다. 저비등 물질(용매 및 과잉 SO2Cl2)을 약 50℃의 진공하에 증류제거한다. 그 잔사를 실온 및 진공하에서 2시간 더 건조시킨다.
생성된 농축 산 클로라이드를 CH2Cl2120ml에 용해시킨 후 메틸렌 클로라이드 350ml 중의 N-메틸아미노아세트 알데하이드 디메틸아세탈 50ml 및 트리에틸아민 80ml(50% 과잉)의 교반 용액에 20 내지 25℃에서 때때로 냉각시키며 1.5시간에 걸쳐 적가한다. 혼합물을 실온에서 한시간 더 교반시킨다. 반응 혼합물을 물 500ml로 2회 추출하고 MgSO4상에서 건조시키고, 여과시킨후 건조할 때까지 교반 증발시켜 점성 시럽으로서 상기 구조식 S의 화합물 약 100g을 수득한다.

점성의 시럽을 진한 HCl(빙욕내에서 미리 냉각시킴) 500ml에 냉각 및 교반(빙욕중에서(시키며 조금씩 가한다. 이를 아세트산 500ml로 추가로 희석시킨다. 그 혼합물을 실온에서 밤새 교반시킨다. 반응 혼합물을 얼음 물 8ℓ에 30분가 교반시키며 주입한다. 고무상 고체를 여과분리하고 물로 세척한다. 여과물을 CH2Cl21ℓ로 추출하고 건조할 때까지 회전증발시킨다. 회전증발된 잔사 및 습윤 고무상 고체를 혼합하고 에테르 700ml 중에 재용해시킨다. 에테르를 물 300ml로 2회 추출하고, 에테르 용액을 K2CO3상에서 건조시키고, 활성탄화 및 여과시킨후 건조할 때까지 회전증발시켜 추출시 결정화되어 상기 구조식 T의 화합물을 제공할 점성 시럽 68.0g을 수득한다.

상기 제조실시예 6B으로부터의 구조식 T의 화합물 68.0g을 에탄올 600ml 중에 용해시킨 뒤 동등한 두부분으로 나누고 각 부분을 PtO22.5g상에서 H2로 환원시킨다.
촉매를 제거한후, 두가지 여과물을 혼합하여 TLC로 체크하고 회전증발시켜 건조시킨다. 그 잔사를 걸러내며 냉각된 에틸 아세테이트 150ml로 교반시킨다. 그 용액을 빙욕중에서 냉각시키고, 여과하고 고체를 냉각된 에틸 아세테이트로 세척하여 구조식 U의 화합물 약 28.0g을 수득한다. 본 물질 24.0g 및 다른 배취로부터의 12.0g을 혼합하고 비등 에틸 아세테이트 100ml 중에 용해시킨다. 혼합물을 냉동기중에서 냉각시켜 여과하여, 그 고체를 냉각 에틸 아세테이트로 세척한다. 고체를 실온에서 1시간동안 건조시키고 구조식 U의 화합물 22.50g을 수득한다(융점; 104 내지 105℃).

CH2Cl2300ml 중의 상기 구조식 U의 화합물 용액에 약 25분에 걸쳐 CH2Cl235ml 중의 SO2Cl215ml의 용액을 가한다. 반응 혼합물을 실온에서 2 1/2시간 더욱 교반시킨후 교반시키면서 빙수 500ml에 주입한다. 유기층을 MgSO4상에서 건조시키고, 여과한 후 회전증발시켜 건조시킨다. 잔사를 분별 결정화시킨다. 혼합물을 이어서 냉각된 에틸 아세테이트 40ml로 연마하고 여과하여, 분리된 고체를 냉각 에틸 아세테이트 10ml로 세척하여 융점이 162 내지 164℃인 구조식 W의 화합물 13.90g을 수득한다.
여과물을 냉동기에서 밤새 유지시킨 후 여과하여 순도가 떨어지는 구조식 W의 화합물 추가의 1.2g을 수득한다.

DMF 10ml중의 상기 구조식 W의 화합물 1.2g 및 K2CO32.0g의 교반된 현탁액에 피페리딘 430mg을 한번에 가한다. 혼합물을 실온에서 교반시키고 물 700ml중에 교반하면서 가한다. 고무상 고체를 여과 분리한다. 본 습윤 고체를 CHCl250ml 중에 용해시키고 물 50ml로 추출한다. 유기층을 분리하여 K2CO3상에서 추출하고, 여과시킨 후 회전증발시켜 건조시킨다. 그 잔사를 아세토니트릴(10ml)로부터 재결정화시켜 융점이 139 내지 141℃인 구조식 Z의 화합물 700mg을 수득한다.
[실시예 6]

THF 20ml 중의 구조식 Z의 화합물(제조실시예 6E에서 기술한 바와 같이 제조딤) 2.95g 및 THF(1.06M)중의 디보란 40ml의 용액을 환류하에 18시간동안 증기욕 내에서 가열한다. 혼합물을 증류하여 건조시킨다. 그 잔사를 4N HCl 25ml로 주의하여 처리한 후 30분간 교반하면서 증기욕 내에서 가열한다. 혼합물을 냉각시키고 물 30ml로 희석시키며 NaOH로 염기성이 되게 한후 에테르 50ml로 2회 추출한다. 에테르층을 혼합하고, K2CO3상에서 건조시키고, 여과시킨 후 건조될 때까지 회전증발시켜 오일상 시럽 약 2.0g을 수득하며, 이를 CH2Cl2/C2H5OH/NH4OH(100/5/2)로 용출시키며 TLC 그레이드 실리카겔 100g의 컬럼을 통과시켜 정제한다. 목적 성분을 포함하는 분획을 혼합한 후 건조할 때까지 회전증발시켜 상기 구조식 AA의 목적물질 540mg을 수득한다.

수성 48% HBr 10ml 중의 상기 구조식 AA의 화합물 480mg을 130℃에서 6 1/2시간동안 교반시키며 가열한다. 혼합물을 빙수 100ml에 가하고 pH를 NaHCO3로 약 8로 조절한다. 혼합물을 CH2Cl240ml로 2회 추출한다. 건조된 혼합 추출물을 회전증발시켜 오렌지색의 고무상 물질 290mg를 남겨서 이를 CH2Cl2/C2H5OH/NH4OH(50/3/1)로 용출시키며 TLC 그레이드 실리카겔 30mg을 통과시켜 정제한다. 목적 성분을 함유한 분획을 회전증발시킨 잔사를 에테르 30ml 중에 용해시키고 서서히 5ml 까지 증발시킨다. 수득된 고체를 여과 분리하고, 90℃에서 5시간동안 건조시켜 융점이 155 내지 157℃인 상기 구조식 AB의 화합물 75mg을 제공한다.
제조실시예 6E의 피페리딘 대신 하기 표 4의 첫째열에 나타낸 반응물을 사용하고 제조실시예 6E 및 상기 실시예 6에 기술된 동일 방법으로, 표 4의 두번째 열에 나타낸 화합물을 합성할 수 있다.
[표 4]

[제조실시예 7]

DMF 및 H2O 각각의 20ml 중의 구조식 W의 화합물(제조실시예 6D에서 기술된 바와 같이 제조됨) 1.40g, NaHCO34.0g 및 나트륨 디티오나이트 1.75g을 실온에서 1 1/2시간 교반시킨다. 물 200ml를 교반하며 첨가한다. 혼합물을 여과하고 분리된 고체를 물로 세척하여 약 1.09g의 고체를 수득하고, 이를 아세토니트릴로부터 재결정하여 소량의 구조식 AC의 목적 화합물을 수득한다(융점; 117 내지 118℃). 아세토니트릴중에서의 재결정화 여과물로부터 덜 순수한 구조식 AC의 화합물 950mg을 수득한다.

N2하에, NaH(876mg, 60% 오일 분산액)을 실온에서 THF/DMF(10 : 1) 150ml 중의 구조식 AC(2.5g)의 화합물 용액에 가한다. THF/DMF(10cc)중의 사이클로헥실 브로마이드(1.5cc) 용액을 상기 혼합물에 낙하 판넬을 통해 상기 혼합물에서 첨가한다. 그 혼합물을 80℃에서 오일욕 중에서 가열한다. 2시간후 반응이 종결된다. 용매를 40℃에서 회전증발기 상에서 제거하고(펌프 설치됨) 그 잔사를 빙수 200CC로 신속히 희석시킨다. 생성된 혼합물을 CH2Cl2200ml로 추출하고 CH2Cl2층을 분리하고 MgSO4상에서 건조시킨다. CH2Cl2층을 회전증발시켜 무정형 고체 3g을 얻고 이를 용출액으로서 에틸 아세테이트/헥산(40 : 60)을 사용하며 키젤겔 60G 상에서 크로마토그라피하여 구조식 AD 의 생성물 약 1.54g을 수득한다.
[실시예 7]

구조식 AD의 화합물(1.53g)(제조실시예 7B에서 기술한 바와 같에 제조), 무수 THF(50cc) 및 디보란 (THF 중의 1M 용액 16cc)을 2시간동안 환류시킨다. 반응 혼합물을 실온으로 냉각시키고 H2O 5cc를 신중히 첨가한다. 용매를 약 30℃의 회전증발기상에서 제거한다. 에탄올(100cc) 및 4N HCl 25cc를 잔사에 첨가하고, 그 혼합물을 증기욕에서 1 1/2시간동안 환류시킨다. 에탄올을 50 내지 60℃의 회전증발기에서 제거하고, 잔류 수성 부분을 빙수 100ml로 희석시킨다. 혼합물을 10% NaOH 용액으로 약 pH 8로 염기화하여 CH2Cl2100cc로 2회 추출한다. 혼합된 추출물을 MgSO4상에서 건조시키고, 증발시켜 오일로서 상기 구조식 AE의 화합물 1.26g을 수득한다.

디메틸포름아미드(DMF) 6ml 중의 상기 구조식 AE의 사이클로헥실 화합물(1.2g)의 용액을 광유 및 에탄디올 1.4ml중의 60% 나트륨 하이드라이드 757mg으로부터 제조한 DMF 6ml 중의 나트륨 티오에톡사이드 용액에 첨가한다. 생성된 혼합물을 오일욕중에서 4시간동안 120℃에서 가열하고, 냉각시키고, 빙-수100ml로 희석시키고 헥산 50ml 세척한다. 5% HCl을 분리된 수성층에 첨가하여 pH를 7.5 내지 8로 조정한다. 혼합물을 CH2Cl2200ml씩으로 2회 추출하고, 혼합된 추출물을 MgSO4상에서 건조시키고, 여과하고 증발시켜 오일을 생성하여, 이를 고진공중에서 건조시킨다. 오일을 부분적으로 결정화하고 에테르-석유 에테르로부터 재결정하여 융점이 144 내지 147℃인 상기 구조식 AE의 생성물 454mg을 수득한다.
하기 표 5의 첫째열에 기재된 반응물을 상기 제조 실시예 7B중의 사이클로헥실 브로마이드 대신 사용하고 제조실시예 7B 및 실시예 7에 기술된 바와 기본적으로 동일한 방법으로, 표 5의 두번째 열에 나타내 화합물을 또한 합성한다.
[표 5]

제조실시예 7B의 방법중 사이클로헥실 브로마이드 대신 CH2=CH-CO2CH3을 사용하고 제조실시예 7B 및 실시예 7에 기술된 바와 기본적으로 동일한 반응을 이용하여, 하기 나타낸 화합물 AG를 또한 제조할 수 있다 :

[제조실시예 8]

순수한 에탄올 30ml중의 나트륨 에톡사이드의 용액을 나트륨 253mg을 사용하여 제조하고, 구조식 W의 화합물(2.75g)(제조실시예 6D에 기술된 바와 같이 제조)을 이 용액에 가한 후, 반응 혼합물을 3시간동안 환류하에 가열한다. 혼합물을 회전증발시켜 건조시킨다. 그 잔사를 H2O 및 CH2Cl2각기 50ml로 처리한다.
CH2Cl2부분을 MgSO4로 건조시키고 농축시켜 고체 잔사 2.50g을 제조하여 이를 아세토니트릴 15ml로 부터 재결정화하여 융점이 106 내지 108℃인 상기 구조식 AH의 화합물 약 780mg을 수득한다.
[실시예 8]

THF 20ml중의 구조식 AH의 화합물(제조 실시예 78에서 기술한 바와 같이 제조)775mg의 용액을 디보란/THF(1.06M) 15ml에 교반하며 가한다. 혼합물을 5 1/2시간 동안 더 환류하에 가열시킨 후 증류하여 건조시킨다. 잔사를 4N HCl로 처리한 후 30분간 교반시키며 증기욕 내에서 가열한다. HCl 혼합물을 H2O 20ml로 희석시키고, 냉각시킨 후 NaOH로 염기성으로 만든다. 이 혼합물을 에테르 50ml로 추출한다. 에테르를 회전증발시켜 제거하여 NMR에 의해 확인되는 구조식 AI의 조생성물은 오일상 시럽 450mg을 얻는다.

NaSO2H5를 NaH 1.50g(오일 60%) 및 DMF 30ml중의 에탄티올 3.0ml를 사용하여, DMF 중에서 제조한다. 이 용액 4g에 DMF 2ml중의 상기 일반식 AI의 화합물 1.20g의 용액을 가한다. 혼합물을 130 내지 140℃의 오일욕 중에서 4시간 동안 가열한 후 실온에서 냉각시킨 후 물 150ml에 가한다. 아세트산을 적하하여 pH를 약 8로 조정한다. 이 혼합물을 CH2Cl230ml로 2회 추출한다. CH2Cl2층을 MgSO4상에서 건조시키고, 여과한 후 약 4 내지 5ml가 될때까지 회전증발시킨 후 90℃의 10mm에서 증류시켜 건조시킨다. 잔사를 CH2Cl2/C2H5OH/NH4OH(50/2.5/1)로 용출시키며 TLC 그레이드 실리카겔 50g의 컬럼을 통해 정제한다. 목적 화합물을 함유한 분획을 혼합한 후 회전 증발로 건조시켜 상기 구조식 AJ의 조생성물은 점성잔사 약 800mg을 수득한다. 화합물을 무수 HCl 약간의 과잉량으로 에테르성 용액중에서 하이드로클로라이드 염으로 전환시킨다. 생성염을 여과하고 진공중에서 건조시켜 융점이 235 내지 236℃(분해)인 HCl 염을 수득한다.
[실시예 9]

무수 디메톡시에탄 20ml중의 구조식 AJ 화합물(실시예 8에서 기술한 바와 같이 제조)의 하이드로클로라이드 0.8g(2.7mmole)의 현탁액에 NaH(오일 분산액 60%) 210mg(5.5mmole)을 교반시키며 나누어 가한다. 기체의 증발이 멈춘 후(약 10분) 10ml 디메톡시에탄중의 디메틸카바모일 클로라이드(0.290g, 0.248ml, 2.7mmole)의 용액을 가하고 혼합물을 주변온도에서 밤새 교반시킨 후 3시간 동안 50℃까지 가열한다.
반응 혼합물을 여과시키고(고체인 이론적 NaCl 0.32g 수득) 거의 건조될때까지 증발시킨다. 에테르 및 희석된 NaOH를 첨가한다. 상을 분리한다. 에테르상을 염수로 세척하고, MgSO4상에서 건조시키고, 탈색시키고(Darco 및 Florisil) NMR에 의해 확인되는 구조식 AK의 화합물인 0.9g의 고무상이 될때까지 농축시킨다.
상기 실시예 9에 따른 구조식 AK의 본 고무질 물질(0.9g)을 에테르중에 용해시키고 약간 과잉의 에테르성 HCl로 처리한 후 여과시킨다. 분리된 흡습성 고체를 아세토니트릴 약 20ml중에 즉각 재용해시키고, 약 100ml 에테르로 희석시키고, 냉각시키며 여과하여 상기 구조식 AK의 목적화합물 520mg을 하이드로클로라이드로서 수득한다(융점 : 199 내지 202℃).
[제조실시예 10]

나트륨 하이드라이드(1.16g, 60% 오일 분산액)를 질소하에 실온에서 THF/DMF(10 : 1)의 35ml중에 구조식 AC의 화합물(3.5g, 제조실시예 7A에 기술한 바와 같이 제조)의 용액에 첨가한다. THF/DMF(10 : 1)10ml중의 알릴 브로마이드(1.4cc)의 용액을 이어서 시린지를 통해 첨가하고 그 혼합물을 50℃에서 0.5시간 가열시킨 후 65℃에서 1시간 동안 가열시킨다. 용매를 40℃에서 회전 증발기상에서 제거하고 얼음 200cc를 잔사에 빠르게 가한다. 생성된 혼합물을 CH2Cl2150cc 씩 2회 추출한 후 혼합된 추출물을 물 50cc로 세척한후 MgSO4상에서 건조시킨다. CH2Cl2추출액을 회전 증발시켜 오일 3.6g을 수득한다. 에틸 아세테이트/헥산 혼합물(40 : 60)로 부터 재결정하여 일반식 AL의 생성물 2.8g을 수득한다.
[실시예 10]

THF(30cc)중의 화합물 AL(2.8)(제조실시예 10에 기술된 바와 같이 제조)의 용액을 실온에서 THF(50cc)중의 LiAlH4(1.1g)의 현탁액에 첨가한다. 1시간 후 얇은 막 크로마토그라피에 의해 지시되는 바와 같이 반응이 종결된다. 생성된 반응 혼합물에 물 1.1cc, 15% NaOH 용액 1.1cc, 이어서 물 3.3cc를 가한다. 침전을 여과 분리시키고, THF를 회전 증발기상에서 제거시킨 후 물 200cc를 잔사에 가한다. 이어서 혼합물을 두개의 150cc CH2Cl2분획으로 추출하고, 혼합한 추출액을 물 75cc로 세척한 후 MgSO4상에서 건조시킨다. 건조된 CH2Cl2층을 회전증발시켜 오일을 생성하고, 이를 용출액으로서 에틸 아세테이트 및 헥산의 1 : 1 혼합물을 사용하여 실리카 컬럼상에서 크로마토그라피하여 오일로서 목적 생성물 AM을 수득한다(1.52g).

NaSC2H5는 0.89g의 NaH(60% 오일 분산액)을 DMF 20cc중의 에탄티올 1.6cc의 냉각된 용액에 배취식으로 첨가하여 제조한다. 반응 혼합물을 15분동안 방치시킨 후 DMF 30cc중의 구조식 AM의 화합물 1.45g의 용액을 혼합물에 주입한다. 생성된 반응 혼합물을 120℃의 오일욕에서 2시간 동안 가열하고 실온으로 냉각시킨 후 물 400cc를 가한다. 생성 혼합물의 pH를 H2SO4로 1로 조정하고, 그 혼합물을 디에틸 에테르 150cc로 1회 추출한 후 고체 NaHCO3를 사용하여 염기성화시켜 pH 8로 만든다. 이 혼합물을 2개의 150cc 에틸 아세테이트 분획으로 추출한 후 혼합한 추출물을 MgSO4상에서 건조시키고 증발시켜 오일(840mg)을 수득한다. 오일을 용출액으로서 에틸 아세테이트; 헥산(1 : 1)을 사용하여 실리카 컬럼상에서 크로마토그라피하여 조생성물 440mg을 생성하고 이를 에틸 아세테이트/헥산 혼합물로부터 재결정화하여 목적 생성물 AN(융점 : 141 내지 143℃) 310mg을 수득한다.
상기 실시예에 기술된 방법들을 이용하여, 하기 표 6에 기술된 일반식(I)의 화합물을 제조할 수 있다.
[표 6]

본 발명을 상기한 바와 같이 특정 태양에 관하여 기술하였으나, 이의 많은 개량, 변경 및 변화를 가할 수 있음은 본 기술 분야의 일반 숙련인들에게 명백한 것이다. 그러한 모든 개량, 변경 및 변화는 본 발명의 정신 및 범주내에 포함되어야 한다.
Claims (15)
- 하기 일반식(I)의 화합물 및 약제학적으로 허용되는 이의 염.
 상기식에서, R1은 -XR6, -CHR7R8, 사이클로알킬, 사이클로알케닐, -H, -CN, -(CO)OR9, -(CO)R9, -O(CO)N(R9)2, -C=CR9, -(CO)N(R9)2,이미다졸일 또는 피롤일을 나타내고, R2는 -H(R1이 아닌 경우), -OH(R1이 -OH 또는 -SH가 아닌 경우) 또는 알콕시를 나타내며; 또한, R1및 R2는 함께 카보닐 산소, =CH-아릴 그룹, 또는 일반식
상기식에서, R1은 -XR6, -CHR7R8, 사이클로알킬, 사이클로알케닐, -H, -CN, -(CO)OR9, -(CO)R9, -O(CO)N(R9)2, -C=CR9, -(CO)N(R9)2,이미다졸일 또는 피롤일을 나타내고, R2는 -H(R1이 아닌 경우), -OH(R1이 -OH 또는 -SH가 아닌 경우) 또는 알콕시를 나타내며; 또한, R1및 R2는 함께 카보닐 산소, =CH-아릴 그룹, 또는 일반식 의 그룹(여기에서, B는 알칸디일이고 W는 -O-, -S-, 또는 -CH2-이다)을 나타내고; R3는 H, 알킬, 알릴 또는 사이클로프로필메틸을 나타내며; R4는 H, 할로, 알킬, 할로알킬 또는 알콕시를 나타내고; R5는 -OR10, -N(R9)2또는 -OㆍC(R7)2ㆍOCOR13을 나타내며; R6는 -H, 아릴, 헤테로아릴, 나프틸, 아르알킬, 헤테로아릴알킬, 알킬, 사이클로알킬, 사이클로알케닐, 할로알킬, 알케닐, 알키닐, 사이클로알킬알킬, 사이클로알케닐알킬, 알콕시 알킬 또는 -(CH2)nR11을 나타내고; R7는 -H 또는 알킬을 나타내며; R8는 사이클로알킬, 사이클로알케닐, 사이클로알킬알킬, 사이클로알케닐알킬, 아르알케닐, 아르알키닐, 헤테로아릴, 헤테토아릴알킬, 알케닐, 알키닐, 할로알킬, 알콕시 알킬 또는 -(CH2)nR12을 나타내고; R9은 각기 독립적으로 H, 알킬, 알콕시, 알콕시알킬, 아르알킬 또는 아릴을 나타내며; R10은 H, COR9또는 -CON(R9)2를 나타내고; R11은 -(CO)OR9, -COR9, -(CO)N(R9)2, -CN, O(CO)N(R9)2, -O(CO)R9, -N(R9)2, -OR9또는 -SR9을 나타내며(단, n이 1일때 R11은 -N(R9)2, -OR9또는 -SR9는 아니다); R12는 -(CO)OR9, -COR9, -(CO)N(R9)2, -CN, -O(CO)N(R9)2, -O(CO)R9, -N(R9)2, -OR9또는 -SR9을 나타내고; R13은 알킬, 아르알킬 또는 아릴을 나타내며; X는 - O-, -S-, 또는 -N(R9)-을 나타내며; m은 0 또는 1이며; n은 1 내지 4의 정수이고; Y는 N 또는 CH를 나타내며; Z는 CH2(Y가 CH를 나타내지 않는 경우) 또는 NR9를 나타내고; P 및 q는 Y가 N이고 Z가 NR9일때 p와 q의 합은 1 내지 5이고 p및 q는 모두 1을 나타내지는 않도록 하며 각기 독립적으로 1 내지 3의 정수를 나타낸다.
의 그룹(여기에서, B는 알칸디일이고 W는 -O-, -S-, 또는 -CH2-이다)을 나타내고; R3는 H, 알킬, 알릴 또는 사이클로프로필메틸을 나타내며; R4는 H, 할로, 알킬, 할로알킬 또는 알콕시를 나타내고; R5는 -OR10, -N(R9)2또는 -OㆍC(R7)2ㆍOCOR13을 나타내며; R6는 -H, 아릴, 헤테로아릴, 나프틸, 아르알킬, 헤테로아릴알킬, 알킬, 사이클로알킬, 사이클로알케닐, 할로알킬, 알케닐, 알키닐, 사이클로알킬알킬, 사이클로알케닐알킬, 알콕시 알킬 또는 -(CH2)nR11을 나타내고; R7는 -H 또는 알킬을 나타내며; R8는 사이클로알킬, 사이클로알케닐, 사이클로알킬알킬, 사이클로알케닐알킬, 아르알케닐, 아르알키닐, 헤테로아릴, 헤테토아릴알킬, 알케닐, 알키닐, 할로알킬, 알콕시 알킬 또는 -(CH2)nR12을 나타내고; R9은 각기 독립적으로 H, 알킬, 알콕시, 알콕시알킬, 아르알킬 또는 아릴을 나타내며; R10은 H, COR9또는 -CON(R9)2를 나타내고; R11은 -(CO)OR9, -COR9, -(CO)N(R9)2, -CN, O(CO)N(R9)2, -O(CO)R9, -N(R9)2, -OR9또는 -SR9을 나타내며(단, n이 1일때 R11은 -N(R9)2, -OR9또는 -SR9는 아니다); R12는 -(CO)OR9, -COR9, -(CO)N(R9)2, -CN, -O(CO)N(R9)2, -O(CO)R9, -N(R9)2, -OR9또는 -SR9을 나타내고; R13은 알킬, 아르알킬 또는 아릴을 나타내며; X는 - O-, -S-, 또는 -N(R9)-을 나타내며; m은 0 또는 1이며; n은 1 내지 4의 정수이고; Y는 N 또는 CH를 나타내며; Z는 CH2(Y가 CH를 나타내지 않는 경우) 또는 NR9를 나타내고; P 및 q는 Y가 N이고 Z가 NR9일때 p와 q의 합은 1 내지 5이고 p및 q는 모두 1을 나타내지는 않도록 하며 각기 독립적으로 1 내지 3의 정수를 나타낸다.
- 제1항에 있어서, R1은 -XR6, -CHR7R8, 사이클로알킬 또는 사이클로알케닐(여기에서, X는 -O-, 또는 -S-를 나타내고, R6는 -H, 페닐, 치환된 페닐, 아르알킬, 알킬, 사이클로알킬, 할로알킬 또는 알콕시알킬을 나타내고, R7은 H 또는 알킬, 바람직하게는 H를 나타내며, R8은 사이클로알킬, 사이클로알케닐, 할로알킬, 알콕시알킬, 알케닐 또는 알키닐을 나타낸다)를 나타내는 화합물.
- 제2항에 있어서, R1이 사이클로헥실 또는 사이클로헥세닐을 나타내는 화합물.
- 제1항에 있어서, R1이또는 피롤일(여기서, m은 1이고 R9는 수소 또는 알킬을 나타낸다)을 나타내는 화합물.

- 제1항 내지 제4항중 어느 한 항에 있어서, R2가 -H를 나타내고 R3가 -CH3를 나타내는 화합물.
- 제1항 내지 제4항중 어느 한 항에 있어서, R4가 할로겐, 바람직하게는 클로로를 나타내고, R5는 -OH-, -OCRㆍR9또는 -OㆍC(R7)2ㆍOCOR13(여기에서, R9는 알킬, 알콕시 또는 알콕시알킬을 나타내고, R7은 수소를 나타내며, R13는 알킬을 나타낸다)를 나타내는 화합물.
- 제1항에 있어서, 상기 화합물이 하기 화합물 및 약제학적으로 허용되는 그의 염중에서 선택되는 화합물.

- 하기 일반식(a)의 카보닐 화합물을 환원반응시킴을 특징으로 하여, 제1항에 따른 일반식(Ⅰ)의 화합물 또는 약제학적으로 허용되는 이의 염을 제조하는 방법.
 상기식에서, 아제핀환 중의 점선은 임의의 이중결합을 나타내고, R1,R2,R3,R4및 R5는 제1항에서 정의한 바와 같으며 R3a는 R3또는 COOR13이고, R13은 제1항에서 정의한 바와 같으며, R5a는 R5이거나 알콕시이다.
상기식에서, 아제핀환 중의 점선은 임의의 이중결합을 나타내고, R1,R2,R3,R4및 R5는 제1항에서 정의한 바와 같으며 R3a는 R3또는 COOR13이고, R13은 제1항에서 정의한 바와 같으며, R5a는 R5이거나 알콕시이다. - 일반식(II)의 화합물 및 약제학적으로 허용되는 임의 염.
 상기식에서, Q는 H, 할로 또는 -OSO2R"(여기에서, R"는 CH3,CF3, 페닐 또는 톨일이다)를 나타내고; R3a는 H, 알킬, 알릴, 사이클로프로필메틸 또는 COOR14(여기에서, R10는 알킬, 아릴, 아르알킬 또는 할로알킬이다)을 나타내며; R4는 H, 할로, 알킬, 할로알킬 또는 알콕시를 나타내고; R5a는 -OR10, -N(R9)2ㆍOC(R7)2ㆍOCOR13또는 알콕시[여기서, R7은 제1항에서 정의한 바왁 같으며, R9는 각기 독립적으로 H, 알킬, 알콕시, 알콕시알킬, 아르알킬 또는 아릴을 나타내고; R14은 H, -COR9또는 -CON(R9)2를 나타내며; R13은 알킬, 아르알킬 또는 아릴을 나타낸다]를 나타낸다.
상기식에서, Q는 H, 할로 또는 -OSO2R"(여기에서, R"는 CH3,CF3, 페닐 또는 톨일이다)를 나타내고; R3a는 H, 알킬, 알릴, 사이클로프로필메틸 또는 COOR14(여기에서, R10는 알킬, 아릴, 아르알킬 또는 할로알킬이다)을 나타내며; R4는 H, 할로, 알킬, 할로알킬 또는 알콕시를 나타내고; R5a는 -OR10, -N(R9)2ㆍOC(R7)2ㆍOCOR13또는 알콕시[여기서, R7은 제1항에서 정의한 바왁 같으며, R9는 각기 독립적으로 H, 알킬, 알콕시, 알콕시알킬, 아르알킬 또는 아릴을 나타내고; R14은 H, -COR9또는 -CON(R9)2를 나타내며; R13은 알킬, 아르알킬 또는 아릴을 나타낸다]를 나타낸다. - 활성 성분으로서 제1항내지 제7항중 어느 한 항의 화합물과 함께 약제학적으로허용되는 담체를 함유하는 약제학적 조성물.
- 정신병 또는 우울증의 치료, 또는 진통효과의 제공, 특히 정신병 치료에 사용되는 약제학적 조성물을 제조하기 위한 제1항 내지 제7항중 어느 한 항에 따른 화합물의 용도.
- 하기 일반식(b)의 에스테르를 환원반응시킴을 특징으로 하여, 제1항에 따른 일반식(I)의 화합물 또는 약제학적으로 허용되는 이의 염을 제조하는 방법.

 상기식에서, R1,R2,R3,R4,R5및 R13은 제1항에서의 정의한 바와 같고, R5a는 R5이거나 알콕시이다.
상기식에서, R1,R2,R3,R4,R5및 R13은 제1항에서의 정의한 바와 같고, R5a는 R5이거나 알콕시이다. - 하기 일반식(c)의 염을 이중결합에서 환원반응시킴을 특징으로 하여, 제1항에 따른 일반식(I)의 화합물 또는 약제학적으로 허용되는 이의 염을 제조하는 방법.

 상기식에서, R1,R2,R3,R4및 R5는 제1항에서 정의한 바와 같고, R5a는 R5이거나 알콕시이며, L3은 음이온, 바람직하게는 할로산 또는 설폰산으로부터 유도된 음이온이다.
상기식에서, R1,R2,R3,R4및 R5는 제1항에서 정의한 바와 같고, R5a는 R5이거나 알콕시이며, L3은 음이온, 바람직하게는 할로산 또는 설폰산으로부터 유도된 음이온이다. - 하기 일반식(d)의 화합물을 HD의 제거 및 아제핀환의 형성이 수반되는 분자내 축합반응시킴을 특징으로 하여, 제1항에 따른 일반식(I)의 화합물 또는 약제학적으로 허용되는 이의 염을 제조하는 방법.

 상기식에서, R1,R2,R3,R4및 R5는 제1항에서 정의한 바와 같고, R3a는 R3또는 COOR13이며, R13은 제1항에서 정의한 바와 같고, R5a는 R5이거나 알콕시이며, D는 아제핀환의 형성과 함께 DH로서 제걸될 수 있는 반응성 그룹이다.
상기식에서, R1,R2,R3,R4및 R5는 제1항에서 정의한 바와 같고, R3a는 R3또는 COOR13이며, R13은 제1항에서 정의한 바와 같고, R5a는 R5이거나 알콕시이며, D는 아제핀환의 형성과 함께 DH로서 제걸될 수 있는 반응성 그룹이다. - 하기 일반식(e)의 화합물을 올레핀 이중결합에서 환원반응시킴을 특징으로 하여, 제1항에 따른 일반식(I)의 화합물 또는 약제학적으로 허용되는 이의 염을 제조하는 방법.

 상기식에서, R1,R2,R3,R4및 R5는 제1항에서 정의한 바와 같고, R5a는 R5이거나 알콕시이며, Z는 R1또는 R2이다.
상기식에서, R1,R2,R3,R4및 R5는 제1항에서 정의한 바와 같고, R5a는 R5이거나 알콕시이며, Z는 R1또는 R2이다.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US3213587A | 1987-03-27 | 1987-03-27 | |
| PCT/US1988/000899 WO1988007526A1 (en) | 1987-03-27 | 1988-03-24 | Substituted benzazepines, their preparation and pharmaceutical compositions containing them |
| US032135 | 1993-03-17 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| KR890700577A KR890700577A (ko) | 1989-04-25 |
| KR930011489B1 true KR930011489B1 (ko) | 1993-12-08 |
Family
ID=21863286
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1019880701534A KR930011489B1 (ko) | 1987-03-27 | 1988-03-24 | 치환된 벤즈아제핀, 이의 제조방법 및 이를 함유하는 약제학적 조성물 |
Country Status (18)
| Country | Link |
|---|---|
| EP (2) | EP0285919B1 (ko) |
| JP (1) | JPH0662574B2 (ko) |
| KR (1) | KR930011489B1 (ko) |
| AT (1) | ATE112766T1 (ko) |
| AU (1) | AU619744B2 (ko) |
| CA (1) | CA1321195C (ko) |
| DE (1) | DE3851769T2 (ko) |
| DK (1) | DK165688C (ko) |
| FI (1) | FI894566A0 (ko) |
| HU (1) | HU205744B (ko) |
| IL (1) | IL85855A (ko) |
| MY (1) | MY103357A (ko) |
| NO (1) | NO174507B (ko) |
| NZ (1) | NZ224038A (ko) |
| PH (1) | PH27337A (ko) |
| PT (1) | PT87068B (ko) |
| WO (1) | WO1988007526A1 (ko) |
| ZA (1) | ZA882080B (ko) |
Families Citing this family (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK107688D0 (da) * | 1988-03-01 | 1988-03-01 | Novo Industri As | Carbaminsyreestere af substituerede 7-hydroxy-2,3,4,5-tetrahydro-1h-3-benzazepiner |
| DK67489D0 (da) * | 1989-02-14 | 1989-02-14 | Novo Industri As | Nye benzazepinderivater |
| US5068326A (en) * | 1989-04-03 | 1991-11-26 | Kung Hank F | Dopamine receptor ligands and imaging agents |
| AU8201591A (en) * | 1990-06-15 | 1992-01-07 | Schering Corporation | 8-lower alkyl-5-cycloalkyl or 5-cycloalkenyl substitued benzazepines and pharmaceutical compositions containing them |
| JPH0615530B2 (ja) * | 1990-09-25 | 1994-03-02 | シェリング・コーポレーション | 多環式ベンズアゼピン |
| US5241065A (en) * | 1992-02-25 | 1993-08-31 | Schering Corporation | 2,3,4,5-tetrahydro-1h-3-benzazepines having anti-psychotic activity |
| ATE305454T1 (de) * | 2000-07-05 | 2005-10-15 | Ortho Mcneil Pharm Inc | Nichtpeptidische substituierte spirobenzoazepine als vasopressin antagonisten |
| US6953787B2 (en) * | 2002-04-12 | 2005-10-11 | Arena Pharmaceuticals, Inc. | 5HT2C receptor modulators |
| PL220722B1 (pl) * | 2002-12-20 | 2015-12-31 | Glaxo Group Ltd | Benzoazepinowe pochodne, zawierająca je kompozycja farmaceutyczna i ich zastosowania do wytwarzania leku i do leczenia chorób neurologicznych |
| CN1823059A (zh) | 2003-05-22 | 2006-08-23 | 先灵公司 | 作为治疗肥胖和CNS疾病的选择性D<sub>1</sub>/D<sub>5</sub>受体拮抗剂的5-H-苯并[D]萘并[2,1-B]氮杂䓬衍生物 |
| EA016485B1 (ru) | 2003-06-17 | 2012-05-30 | Арена Фармасьютикалз, Инк. | Способы получения 3-бензазепинов |
| AU2012200805B2 (en) * | 2003-06-17 | 2015-01-22 | Arena Pharmaceuticals, Inc. | Benzazepine derivatives useful for the treatment of 5HT2C receptor associated diseases |
| EP2332920A3 (en) * | 2003-06-17 | 2011-12-21 | Arena Pharmaceuticals, Inc. | Processes for preparing 3-benzazepines |
| WO2005042491A1 (en) * | 2003-10-22 | 2005-05-12 | Arena Pharmaceuticals, Inc. | Benzazepine derivatives and methods of prophylaxis or treatment of 5ht2c receptor associated diseases |
| WO2005042490A1 (en) * | 2003-10-22 | 2005-05-12 | Arena Pharmaceuticals, Inc. | Benzazepine derivatives and methods of prophylaxis or treatment of 5ht2c receptor associated diseases |
| CN102321023A (zh) | 2004-12-21 | 2012-01-18 | 艾尼纳制药公司 | (r)-8-氯-1-甲基-2,3,4,5-四氢-1h-3-苯并氮杂卓盐酸盐的晶型 |
| EA201100129A1 (ru) | 2004-12-23 | 2011-10-31 | Арена Фармасьютикалз, Инк. | Композиции и способы применения модулятора рецептора 5ht-2c |
| EP2518053A1 (en) | 2006-04-03 | 2012-10-31 | Arena Pharmaceuticals, Inc. | Process for the preparation of 8-chloro-1-methyl-2,3,4,5-tetrahydro-1h-3-benzazepine |
| JP5404414B2 (ja) | 2006-12-05 | 2014-01-29 | アリーナ ファーマシューティカルズ, インコーポレイテッド | (r)−8−クロロ−1−メチル−2,3,4,5−テトラヒドロ−1h−3−ベンゾアゼピンおよびその中間体を調製するための方法 |
| JP5491421B2 (ja) | 2008-03-04 | 2014-05-14 | アリーナ ファーマシューティカルズ, インコーポレイテッド | 5−ht2cアゴニストである(r)−8−クロロ−1−メチル−2,3,4,5−テトラヒドロ−1h−3−ベンゾアゼピンに関連する中間体の調製のためのプロセス |
| WO2010148207A2 (en) | 2009-06-18 | 2010-12-23 | Arena Pharmaceuticals, Inc. | Processes for the preparation of 5-ht2c receptor agonists |
| KR20130112848A (ko) | 2010-06-02 | 2013-10-14 | 아레나 파마슈티칼스, 인크. | 5-ht2c 수용체 아고니스트의 제조 방법 |
| KR20130101524A (ko) | 2010-09-01 | 2013-09-13 | 아레나 파마슈티칼스, 인크. | 5-ht2c 아고니스트의 비-흡습성 염 |
| WO2012030938A1 (en) | 2010-09-01 | 2012-03-08 | Arena Pharmaceuticals, Inc. | Salts of lorcaserin with optically active acids |
| AU2011296003B2 (en) | 2010-09-01 | 2015-11-12 | Arena Pharmaceuticals, Inc. | Modified-release dosage forms of 5-HT2C agonist useful for weight management |
| EP2939677A1 (en) | 2010-09-01 | 2015-11-04 | Arena Pharmaceuticals, Inc. | Administration of lorcaserin to indviduals with renal impairment |
| KR20190128001A (ko) | 2012-10-09 | 2019-11-13 | 에자이 알앤드디 매니지먼트 가부시키가이샤 | 체중 관리 방법 |
| HU230826B1 (hu) | 2014-11-19 | 2018-07-30 | Richter Gedeon Nyrt. | Eljárás benzazepin származékok előállítására |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1268243A (en) * | 1968-03-11 | 1972-03-22 | Wallace & Tiernan Inc | 0,2,4,5,-tetrahydro-3h,3-benzazepines |
| DE1934150A1 (de) * | 1968-07-10 | 1970-01-15 | Pennwalt Corp | Neue 1-Alkanoyloxy-1,2,4,5-tetrahydro-3H,3-benzazepine |
| US4988690A (en) * | 1982-06-14 | 1991-01-29 | Hoechst-Roussel Pharmaceuticals Inc. | 1-aryloxy-2,3,4,5-tetrahydro-3-benzazepines and anti-depressant use thereof |
| DK180485D0 (da) * | 1985-04-22 | 1985-04-23 | Novo Industri As | Nitrogenholdige forbindelser |
| US4707483A (en) * | 1985-12-20 | 1987-11-17 | Smithkline Beckman Corporation | 1-phenyl-3-benzazepines and their use for treating gastrointestinal motility disorders |
| GB8807922D0 (en) * | 1988-04-05 | 1988-05-05 | Fujisawa Pharmaceutical Co | Isoquinoline compound & process for preparation thereof |
-
1988
- 1988-03-23 ZA ZA882080A patent/ZA882080B/xx unknown
- 1988-03-23 PH PH36675A patent/PH27337A/en unknown
- 1988-03-24 WO PCT/US1988/000899 patent/WO1988007526A1/en not_active Application Discontinuation
- 1988-03-24 PT PT87068A patent/PT87068B/pt not_active IP Right Cessation
- 1988-03-24 MY MYPI88000305A patent/MY103357A/en unknown
- 1988-03-24 EP EP88104758A patent/EP0285919B1/en not_active Expired - Lifetime
- 1988-03-24 CA CA000562352A patent/CA1321195C/en not_active Expired - Fee Related
- 1988-03-24 DE DE3851769T patent/DE3851769T2/de not_active Expired - Fee Related
- 1988-03-24 AU AU15964/88A patent/AU619744B2/en not_active Ceased
- 1988-03-24 AT AT88104758T patent/ATE112766T1/de not_active IP Right Cessation
- 1988-03-24 KR KR1019880701534A patent/KR930011489B1/ko not_active IP Right Cessation
- 1988-03-24 EP EP88903596A patent/EP0357641A1/en active Pending
- 1988-03-24 JP JP63503399A patent/JPH0662574B2/ja not_active Expired - Lifetime
- 1988-03-24 HU HU882812A patent/HU205744B/hu not_active IP Right Cessation
- 1988-03-24 IL IL85855A patent/IL85855A/xx not_active IP Right Cessation
- 1988-03-25 NZ NZ224038A patent/NZ224038A/xx unknown
- 1988-11-15 NO NO885096A patent/NO174507B/no unknown
- 1988-11-23 DK DK652688A patent/DK165688C/da active
-
1989
- 1989-09-27 FI FI894566A patent/FI894566A0/fi not_active Application Discontinuation
Also Published As
| Publication number | Publication date |
|---|---|
| FI894566A (fi) | 1989-09-27 |
| EP0285919A1 (en) | 1988-10-12 |
| PH27337A (en) | 1993-06-08 |
| NO885096L (no) | 1988-11-15 |
| DK652688A (da) | 1988-11-23 |
| NO885096D0 (no) | 1988-11-15 |
| CA1321195C (en) | 1993-08-10 |
| DK165688B (da) | 1993-01-04 |
| ATE112766T1 (de) | 1994-10-15 |
| EP0357641A1 (en) | 1990-03-14 |
| HUT53882A (en) | 1990-12-28 |
| PT87068A (pt) | 1988-04-01 |
| DE3851769D1 (de) | 1994-11-17 |
| EP0285919B1 (en) | 1994-10-12 |
| NO174507C (ko) | 1994-05-18 |
| NZ224038A (en) | 1990-10-26 |
| DK652688D0 (da) | 1988-11-23 |
| DE3851769T2 (de) | 1995-03-09 |
| ZA882080B (en) | 1989-04-26 |
| DK165688C (da) | 1993-05-24 |
| JPH02502723A (ja) | 1990-08-30 |
| PT87068B (pt) | 1992-07-31 |
| AU1596488A (en) | 1988-11-02 |
| FI894566A0 (fi) | 1989-09-27 |
| AU619744B2 (en) | 1992-02-06 |
| KR890700577A (ko) | 1989-04-25 |
| HU205744B (en) | 1992-06-29 |
| JPH0662574B2 (ja) | 1994-08-17 |
| NO174507B (no) | 1994-02-07 |
| IL85855A (en) | 1993-02-21 |
| MY103357A (en) | 1993-06-30 |
| WO1988007526A1 (en) | 1988-10-06 |
| IL85855A0 (en) | 1988-09-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR930011489B1 (ko) | 치환된 벤즈아제핀, 이의 제조방법 및 이를 함유하는 약제학적 조성물 | |
| US5015639A (en) | Substituted benzazepines, their preparation and pharmaceutical compositions containing them | |
| US5247080A (en) | Substituted benzazepines useful as intermediates for producing pharmaceutically active compounds | |
| JPH08507049A (ja) | 低脂血症性化合物1,4−チアゼピン | |
| CA2190865A1 (fr) | Derives d'imidazole (ant)agonistes du recepteur h3 de l'histamine | |
| JPH05501566A (ja) | 3―置換―1―(アリールまたはアリールアルキル)―2(1h)―キノリノン | |
| KR900007781B1 (ko) | 융합된 벤즈아제핀 | |
| KR850000626B1 (ko) | 시클로헥센 유도체의 제조방법 | |
| EP1960387A1 (fr) | Derives de isoquinoline et benzo[h]isoquinoline, leur preparation et leur utilisation en therapeutique en tant qu antagonistes du recepteur de l histamine h3 | |
| JPH0613474B2 (ja) | 4,5―シクロアルカノ―3―ベンズアゼピン―7―オール誘導体およびその使用 | |
| JPH09501954A (ja) | イミダゾ[5,1−c][1,4]ベンゾキサジン−1−オンのイミダゾリルアルキル誘導体およびそれらの製造法 | |
| WO1991016323A1 (fr) | Derives du naphtosultame antagonistes de la serotonine, leur preparation et les medicaments les contenant | |
| US5374722A (en) | Bridged benzazepines | |
| HU180467B (en) | Process for producing new ergol-8-ene- and ergoline-sceleted compounds | |
| JPH11515030A (ja) | シス−ベンズ[e]インドール化合物のエナンチオマー、これらの製造、及びドーパミン−D3受容体選択的医薬としての利用 | |
| US5789432A (en) | Aromatase-inhibiting composition containing azole derivative | |
| US4318909A (en) | Benzoxazocines | |
| US4626522A (en) | Benzoxazocines intermediates | |
| JPH072776A (ja) | アンジオテンシンii拮抗性ピリジン誘導体 | |
| HU209753B (en) | Process for producing of 3-(4-nitrophenoxy)-propylamine derivatives and pharmaceutical compositions comprising them | |
| JPH0143746B2 (ko) | ||
| JPH0812650A (ja) | 縮合複素環化合物、その製造法および剤 | |
| FR2655652A1 (fr) | Antagonistes de la serotonine, leur preparation et les medicaments les contenant. | |
| FR2633928A1 (fr) | Derives de (aza)naphtalenesultame, leurs procedes de preparation et les medicaments les contenant | |
| KR20010072407A (ko) | N-치환 아자비시클로헵탄 유도체, 그의 제조 및 용도 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A201 | Request for examination | ||
| E902 | Notification of reason for refusal | ||
| G160 | Decision to publish patent application | ||
| E701 | Decision to grant or registration of patent right | ||
| GRNT | Written decision to grant | ||
| LAPS | Lapse due to unpaid annual fee |