JP3676817B2 - 新規因子ix精製法 - Google Patents
新規因子ix精製法 Download PDFInfo
- Publication number
- JP3676817B2 JP3676817B2 JP50062497A JP50062497A JP3676817B2 JP 3676817 B2 JP3676817 B2 JP 3676817B2 JP 50062497 A JP50062497 A JP 50062497A JP 50062497 A JP50062497 A JP 50062497A JP 3676817 B2 JP3676817 B2 JP 3676817B2
- Authority
- JP
- Japan
- Prior art keywords
- eluent
- resin
- factor
- group
- solution
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/48—Hydrolases (3) acting on peptide bonds (3.4)
- C12N9/50—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25)
- C12N9/64—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from animal tissue
- C12N9/6421—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from animal tissue from mammals
- C12N9/6424—Serine endopeptidases (3.4.21)
- C12N9/644—Coagulation factor IXa (3.4.21.22)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y304/00—Hydrolases acting on peptide bonds, i.e. peptidases (3.4)
- C12Y304/21—Serine endopeptidases (3.4.21)
- C12Y304/21022—Coagulation factor IXa (3.4.21.22)
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Genetics & Genomics (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Biomedical Technology (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- General Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- Molecular Biology (AREA)
- Peptides Or Proteins (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Compounds Of Unknown Constitution (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Description
一般的には、本発明は、新規蛋白回収および精製方法に関し、より詳細には、因子IXの回収および精製のための新規方法に関する。
発明の背景
現在、組み換え法の出現は、適当に形質転換された宿主細胞内での高レベルの蛋白の生産を可能にする。分泌蛋白については、目的蛋白の精製は、宿主細胞培地からの単離および精製を必要とする。典型的には、培地は選択栄養素(例えば、ビタミン類、アミノ酸、コファクター、無機質)およびさらなる増殖因子/補足物(インスリンおよび可能なさらなる外来性蛋白を包含)を含有する。ならし培地は目的分泌生成物のみならず、さらに分泌された有意な量の宿主細胞蛋白および他の物質(例えば、核酸、膜小胞)を含有する。高レベルで発現されても、目的生成物はならし培地中に存在するすべての蛋白のうちの少量のものであるかもしれない。形質転換宿主細胞により分泌される蛋白が目的生成物の性質とはまったく異なった性質(例えば、電荷、分子サイズ、アミノ酸組成)を有することは意外なことではない。同様に、選択された分泌宿主細胞蛋白は目的生成物の特性と非常に類似した特性を示すかもしれず、そのことにより重要な課題が精製に用いるプロセスに課せられる。ならし培地からの組み換え蛋白の精製のためのプロセスの開発が行われているが、使用条件(分離のための大きな利益を得るために、分泌蛋白間のわずかな相違を利用するするための使用できる条件)が目的生成物の変性により限定されるということは重要であり、そのことにより、存在する他のすべての宿主細胞蛋白からの目的生成物の分離が困難となっている。
上記分泌宿主蛋白のほかに、ならし培地は、目的生成物をコードしているが種々雑多に発現された遺伝子に由来する生成物を含んでいるかもしれない。これらは最終薬剤物質として望ましいものではなく、例えば、糖鎖付加、硫酸化、ガンマカルボキシレーション、または生物学的活性に潜在的に必要な他の修飾のごときある種の翻訳後修飾を欠く生成物形態を包含する。さらに、目的生成物の蛋白分解的に分解された形態もならし培地に存在するかもしれず、それらを精製途中で除去する必要があるが、それらは目的生成物に非常に近似している。不幸なことに、イオン交換クロマトグラフィー、疎水性相互作用クロマトグラフィー、およびサイズ排除クロマトグラフィーのごときたいていのアプローチは、これらの望ましくない形態から、ヒトの治療という状況に使用されるに必要な目的生成物の分離の程度を提供しないかもしれない。所望生成物と汚染混入物質との間のわずかな相違(例えば、わずかな電荷の相違、分子サイズのわずかな相違)をフルに利用するためには、強力な変性剤の使用がしばしば必要である。しかしながら、かかる変性剤は、生物学的活性の損失、新たな抗原部位の発現、および選択された翻訳後修飾物の化学的分解の潜在的促進をもたらす可能性がある。
類似の特性を有する分子(例えば、発現遺伝子の修飾形態)からの目的生成物の分離のほかに、所望生成物と特異的に反応するならし培地中に存在する成分から所望生成物を分離する必要があることを認識することは重要である。目的蛋白が正に帯電している場合、それは負に帯電している分子に結合する傾向があり、そのことは伝統的方法による生成物の精製を困難にする。
本発明に関する一般的背景は以下のごとし。Yan,USPN4981952(1991年1月1日)およびYan,et al.Bio/Technology 8:655(1990年6月)には、ビタミンK依存性蛋白の精製のための偽アフィニティーアイオン交換クロマトグラフィーの使用が開示されている。Josic,et al.J.Chrom.632:1(1993)には、他のビタミンK依存性蛋白からの因子IXの分離のためのヘパリンアフィニティークロマトグラフィーの使用が開示されている。Suomela,Thromb.Res.7:101(1975);Suomela,Eur.J.Bio.Chem.71:145(1976);およびSuomela,Thrombos.Haemostasis.35:211(1976)には、種々のクロッティング因子および因子IX血漿変種の分離におけるヒドロキシアパタイトの使用が記載されている(炭水化物部分、例えばシアル酸およびガラクトースの含量の相違による電荷の相違に基づく)。しかしながら、Reekers,et al.Haemostasis 1:2(1972)には、ヒドロキシアパタイトが因子II、VIIおよびIXを互いに分離できず、またこれらを他の血漿蛋白からも分離できないことが示されている。Schwinn,et al.USPN 4411794には、濃度50〜200mMのカルシウム存在下においてヒドロキシアパタイトを用いる血液クロッティング因子の部分精製が開示されている。Feldman,et al.Biotech.Blood Proteins 227:63(1993)およびRoberts,et al.Vox Sang 67(suppl.1):69(1994)には、酸性化および銅荷電キレートセファロース(因子IXがヒト・血漿から低収率で得られた)を用いるウイルス感染性の低減が開示されている。
典型的には、研究者は伝統的クロマトグラフィー法を用いて所望生成物を精製してきた。しばしば、かかる方法はヒトの治療用製品に望まれる純度および均一性のレベルまでの生成物の精製が不十分である。研究者は、目的蛋白が特異的相互作用する固定化リガンドに結合するアフィニティークロマトグラフィーの使用によりこの困難を克服しようとした。適当な洗浄後、リガンド−蛋白相互作用を破壊することにより所望生成物を溶離することができ、しばしば、有意に純粋な溶離物が得られる。しかしながら、ならし培地中に存在する修飾形態からの所望生成物の分離の場合に、1工程のアフィニティークロマトグラフィー法は不十分であるかもしれず、他のアフィニティー樹脂および/または伝統的分離法と組み合わせて使用しなければならない。高分解能アフィニティークロマトグラフィー工程(例えば、固定化モノクローアル抗体を用いる免疫アフィニティー精製)でさえも、相互作用部位が共通しているために他の成分から所望生成物の十分に分離できないかもしれない(例えば、目的生成物に存在するエピトープが蛋白分解された形態の生成物にも存在する場合)。
したがって、かかる困難を効果的に克服する蛋白精製法に対する必要性が当該分野において存在し続けている。
発明の簡単な概要
因子IXを含有する溶液をアイオン交換樹脂に適用し、樹脂から因子IXを溶離するのに必要な導電率よりも低い導電率を有する溶液で該アニオン交換樹脂を洗浄し、該アニオン交換樹脂を第1の溶離液で溶離して第1の溶離物を得て、該溶離物をヘパリンまたはヘパリン様(例えば、負に帯電したマトリックス)樹脂に適用し、該ヘパリンまたはヘパリン様樹脂を第2の溶離液で溶離して第2の溶離物を得て、該第2の溶離物をヒドロキシアパタイト樹脂に適用し、次いで、該ヒドロキシアパタイト樹脂を第3の溶離液で溶離して精製因子IXを含有する第3の溶離物を得ることを特徴とする、溶液中の因子IXの精製方法が本発明により提供される。第1の溶離物をヒドロキシアパタイト樹脂に適用してもよい。また、該方法は、第3の溶離物を固定化金属アフィニティー樹脂に適用し、次いで、固定化金属アフィニティー樹脂を第4の溶離液で溶離して精製因子IXを含有する第4の溶離物を得ることをさらに特徴としてもよい。本発明方法によれば、因子IXは血漿由来のもの、培養細胞により発現されたもの、あるいは当業者に知られたように組み換え的に製造されたものであってよい。好ましくは、第1の洗浄液は、因子IXをカラムから溶離するのに必要な導電率よりも低く、一般的には、負荷溶液および第1の溶離液バッファーの導電率よりも高いかまたは同等の導電率を有する溶液を含む。この導電率は、第1の溶離物中に存在することになる混入蛋白の大部分を除去するのに十分なものである。適当な第1の洗浄液は、塩化ナトリウム、塩化カリウム、硫酸ナトリウム、リン酸ナトリウムまたはリン酸カリウムのごとき塩溶液を含み、適当なバッファー剤を含有していもよい。適当な濃度範囲は、因子IXを溶離することなく汚染混入物質を除去するのに有効なものであり、25mMないし200mMの塩、好ましくは200mMの塩化ナトリウムを包含する。第1の溶離液は、カルシウム、マグネシウム、マンガン、ストロンチウム、亜鉛、コバルトおよびニッケルのごとき2価カチオンを含む。適当な濃度範囲は、因子IXの溶離に効果的なものであり、例えば、5ないし100mMの範囲、好ましくは50mMのpH約8.0のTrisのごときバッファー剤、50ないし250mMの範囲、好ましくは100mMのNaCl、および5ないし20mMの範囲、好ましくは約10mMの塩化カルシウムを含有する溶液を包含する。
適当なアニオン交換樹脂は、ジエチルアミノエステル(DEAE)、ポリエチレンイミン(PEI)、および4級アミノエタン(QAE)のごとき正に帯電した基を有する樹脂を包含し、Q-Sepharose Fast Flow、DEAE-Sepharose Fast Flow、Poros-Q、Fractogel-TMAE、Fractogel-DMAE、およびQAE-Toyopearlを包含するが、好ましい樹脂はQ-Sepharose Fast Flow(Pharmacia)である。
第2の溶離液は、塩化ナトリウムおよび塩化カリウムを伴ったTrisのごときバッファー中の適当な塩であってよく、50mM Tris,0.50M NaCl,pH8.0が好ましい。適当なヘパリンまたはヘパリン様樹脂は、ヘパリン、セルロースの硫酸エステル、スルフィルプロピル(SP)、カルボキシル、およびカルボキシメチルのごとき負に帯電した基を有する樹脂を包含し、Matrex Cellufine Sulfate、Heparin Sepharose、Heparin Toyopearl、Carboxy Sulfon、Fractogel EMD-SO3、およびFractogel-EMD COOを包含するか、好ましいのはMatrex Cellfine Sulfateである。
第3の溶離液はリン酸塩および硫酸塩のごとき塩であってよく、0.5Mリン酸カリウム、0.2M NaCl,pH7.2が好ましい。適当なヒドロキシアパタイト樹脂は、セラミック−ヒドロキシアパタイト、Biogel HT等のごときリン酸カルシウムを含有するものを包含するが、セッラミック−HAが好ましい。固定化金属アフィニティー樹脂は、Fractogel-EMD-Chelate、Chelating-Sepharose、Matrex Cellufine Chelate、およびPOROS 20MCを包含するが、本発明にはFractogel-EMD-Chelateが好ましい。第4の溶離液は、イミダゾール、EDTA、EGTA、グリシン、ヒスチジンおよびTrisのごときキレーターを含有するバッファー溶液であるが、好ましいのは20mMリン酸カリウム,15mMイミダゾール、0.1M NaCl,pH7.1である。
本発明方法により製造される因子IX組成物も本発明により提供される。そのようにして製造された因子IXは240〜400U/mgの範囲の比活性を有し、約240U/mgであってもよい。
発明の詳細な説明
本明細書の用語「因子IX」は、血漿、形質転換細胞系から単離された因子IX、ならびに宿主細胞培養培地から単離された組み換え的に生産された因子IXを包含するがこれらに限らない。
本明細書の用語「アニオン交換樹脂」は、ジエチルアミノエタン(DEAE)、イエチレンイミン(PEI)、および4級アミノエタン(QAE)のごとき正に帯電した部分(中性pHにおいて)を有する樹脂を包含し、例えば、Q-Sepharose Fast Flow(Pharmacia)、DEAE-Sepharose Fast Flow、DEAE-Toyopearl、QAE-Toyopearl,POROS-Q、Fractogel-DMAE、Fractogel-TMAE、Matrex Cellufine DEAE等を包含するがこれらに限らない。
本明細書の用語「第1の洗浄液」は、アニオン交換カラムから因子IXを溶離させるのに必要な導電率よりも低い導電率を有する溶液を包含するがこれに限るものではなく、一般的には、その導電率は、負荷溶液の導電率および第1の溶離液の導電率よりも高いかまたはそれらと同等である。この導電率は、第1の溶離物中に存在することになる混入蛋白の大部分を除去するのに十分なものである。適当な第1の洗浄液は塩溶液であってよく、例えば、塩化ナトリウム、塩化カリウム、硫酸ナトリウム、リン酸ナトリウムまたはリン酸カリウムを包含し、適当にバッファー化することができる。典型的には、低濃度(25mM塩)から高濃度(200mM塩)にいたる濃度範囲であり、200mM塩化ナトリウムが好ましい。
本明細書の用語「第1の溶離液」は、約0.05Mの濃度のバッファー剤(例えば、Tris)、2価カチオン不存在下では樹脂から溶離するには十分でない濃度(例えば、約0.10M〜0.20M)の塩(例えば、NaCl)、および低濃度(約0.01M)の2価カチオン(例えば、CaCl2)を含むpH8の溶液を包含するが、これに限らない。好ましくは、「第1の溶離液」は「第1の洗浄液」よりも低い導電率を有する。
本明細書の用語「ヘパリン」樹脂および「ヘパリン様」樹脂を互いに交えて使用し、それらはヘパリン、セルロースの硫酸エステル、スルフィルプロピル(SP)、カルボキシル、およびカルボキシメチルのごとき負に帯電した基を有する樹脂を包含し、Fractogel EMD-SO3、Carboxy Sulfon、Fractogel-EMD COO、Heparin Sepharose、Matrex Cellufine Sulfate、およびを包含するがこれらに限らない。
本明細書の用語「第2の溶離液」は、約0.05Mの濃度のバッファー剤(例えばTris)、因子IXと負に帯電した樹脂支持体との相互作用を破壊するに十分な濃度(例えば0.05M)の塩(例えば、NaCl、KCl、Na2SO4)を含むpH8.0の溶液を包含するが、これに限らない。本発明プロセスに用いる場合には、第2の溶離液はその後のプロセス工程、すなわちヒドロキシアパタイトに適合すべきである。
本明細書の用語「ヒドロキシアパタイトカラム」は、例えばBioGel-HTおよびセラミック−ヒドロキシアパタイトのごときリン酸カルシウムゲル支持体を包含するが、これらに限らない。
本明細書の用語「第3の溶離液」(および「第2のリン酸塩バッファー」)は、因子IXと樹脂との相互作用を破壊するに十分な濃度(例えば、約0.20Mなたはそれ以上)のバッファー剤(例えば、リン酸塩または硫酸塩)および因子IXとヒドロキシアパタイト樹脂との電荷相互作用を最小化するに十分な濃度で存在する塩(例えば、NaCl、KCl)を含む中性pH(pH7.2)の溶液を包含するが、これに限らない。用語「第1のリン酸塩バッファー」は、不活性形態の因子IXをヒドロキシアパタイト樹脂から除去するに十分な濃度のバッファー剤(例えば、リン酸塩または硫酸塩)を含む溶液を包含するが、これに限らない。
本明細書の用語「固定化金属アフィニティー樹脂」(IMAC)は、多価カチオンに結合し、調和して機能しうる固定化官能基(例えば、イミノジ酢酸)を含む樹脂を包含し、Chelating-Sepharose、Fractogel-EMD-Chelate、POROS 20MC、およびMatrex Cellufine Chelateがあるが、これらに限らない。結合金属イオンをいくつかの可能な選択物から選択することができ、銅、ニッケル、カドミウム、コバルト、鉄、亜鉛またはストロンチウムがあるが、これらに限らない。
用語「第4の溶離液」(「置換物質」ともいう)は、IMAC樹脂支持体に結合因している子IXと置換するが、樹脂支持体の固定化されている金属イオンとの置換が最小である化合物を包含し(これに限らない)、グリシン、ヒスチジン、tris、イミダゾール、EDTA、EGTA等があるがこれらに限らない。置換物質の適当濃度は結合アフィニティーにより変化し、実験的に条件を評価することにより確認できることを、当業者は容易に理解する。典型的には、低濃度(例えば、5〜15mMの置換物質)ないし高濃度(例えば、100〜200mMの置換物質)の範囲の濃度である。
因子IXについていう比活性「U/mg」は、プールされた血漿または単離血漿を用い、精製IXを標準物質として用いるインビトロ(APTT)クロッティングアッセイにおいて決定される生物学的活性を包含するが、これに限らない。SEC、RP−HPLC、色素によるアッセイ(例えば、Bradford法、Lowry法)または280nmにおける吸光度を包含するいくつかの適当に確認された方法により蛋白濃度を決定することができる。因子IX欠損血漿を用いるPittman,D.,et al.,Blood 79:389-397(1992)の方法により因子IXの活性を決定する。
図1はプロセスの概要を示す。示された工程の順序は目下好ましい順序であるが、所望ならば順序を入れ替えることができ、工程を省力することができることが当業者により理解されるであろう。
本発明によれば、まず、約0.6μmの孔サイズの十字フロー濾過膜用いる精密濾過(MF)により細胞をならし培地から除去する。深さ0.45μmのフィルターで濾過することにより、精製用の無細胞ならし培地を調製してもよい。次いで、無細胞ならし培地を限外濾過により濃縮することができ、次いで、所望ならば、第1のクロマトグラフィー工程に負荷するための適当なバッファー中に透析濾過する。別法として、適当なバッファーで平衡化した第1のクロマトグラフィーカラムに無細胞ならし培地を直接負荷してもよい。
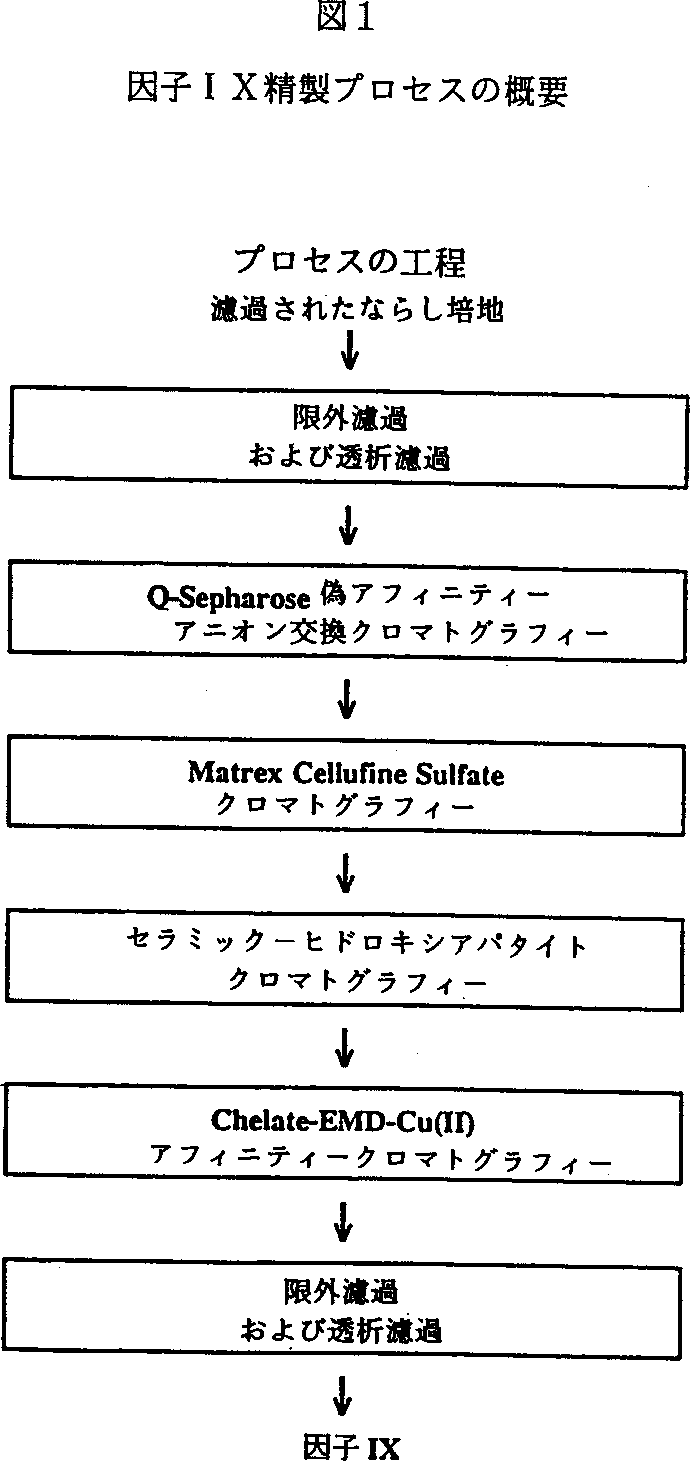
第1のクロマトグラフィー工程、すなわちQ-Sepharose Fast Flow(FF)(Pharmacia)によるアニオン交換において、因子IXが捕捉され、UF/DF#1濃縮物プール中に存在する宿主細胞成分から精製される。Q-SepharoseFFカラムは因子IXを吸着し、操作pHよりも高い等電点を有する混入している宿主細胞蛋白は、カラムを素通りすることによりプロセスの流れから除去される。次いで、緩やかに結合している汚染混入物質を除去する前に、因子IXが吸着しているカラムを洗浄し、溶離用に調製するバッファーの導電率を調節する。
典型的には、バッファーのイオン強度を増加させることにより結合蛋白をQ-Sepharose FFから溶離させる。しかしながら、因子IXの精製プロセスは、例えば塩化カルシウムをバッファーに添加することにより活性因子IXを溶離する偽アフィニティーアニオン交換モードにおいてこの樹脂を使用する。この2価カチオンは、活性形態の因子IXを樹脂から溶離させる。この溶離バッファーとともにいくつかのあまり活性でない因子IXもQ-Sepharose FFカラムから溶離されるかもしれない。選択された不活性形態の因子IXおよび他の混入宿主細胞蛋白はカラムに結合したままである。Q-Sepharose FF工程により因子IXの純度が有意に向上する。
第2のクロマトグラフィー工程において、Q-Sepharose FF溶離物プールを希釈せずに直接Matrex Cellulose Sulfateカラム上に負荷する。因子IXはカラムに吸着するが、他の混入蛋白(例えば、Q-Sepharose FF溶離物中に存在する可溶性PACEおよび他の宿主細胞蛋白)はカラムを素通りすることによりプロセスの流れから除去される。低イオン強度バッファーでカラムを洗浄してすべての未結合蛋白を除去する。塩(例えば、塩化ナトリウム)を用いてバッファーのイオン強度を増加させることにより因子IXを溶離する。
第3のクロマトグラフィー工程、すなわちCeramic-HAカラムクロマトグラフィーを行っている間に不活性な因子IX形態のさらなる除去を行う。Matrex Cellufine Sulfate溶離物プールのpHを約7.5に合わせ、次いで、溶離物プールをCeramic-HAカラム上に直接負荷する。因子IXはカラムに吸着する。Ceramic-HAカラムをバッファーで洗浄して緩やかに結合している汚染混入物質を除去し、次いで、50mMリン酸カリウム,0.185M NaCl(pH7.2)で洗浄して固く結合している汚染混入物質(不活性形態の因子IXを包含する)を除去する。高濃度のリン酸カリウム(例えば、200mMまたはそれ以上,pH7.2)を溶離液として用いるステップ−ワイズ様式で、結合している活性因子IXを溶離させる。
第4のクロマトグラフィー工程、すなわちFractogel EMD-Chelate-Cu(II)クロマトグラフィーにより、まだ生成物の流れに存在している低レベルの混入宿主細胞蛋白を除去する。Ceramic-HA溶離物プールをFractogel EMD-Chelate-Cu(II)カラムに直接負荷する。因子IXおよび多くの汚染混入蛋白がカラムに吸着する。バッファー中の低濃度のイミダゾール(例えば、約15mM)により精製活性因子IXがカラムから溶離され、残存する混入宿主細胞蛋白はカラムに結合したままとなることにより生成物の流れから除去される。
最後に、限外濾過、次いで、ポリソルベート80不含であること以外は)処方バッファーと同じバッファー中への透析濾過(UF/DF#2)によりFractogel EMD-Chelate-Cu(II)溶離物プールを濃縮する。適当な処方バッファーは、ヒスチジン、グリシン、蔗糖、およびポリソルベート80(それぞれ10mM、260mM、1%、および0.005%であってもよい)を含む。透析濾過が完了したら、因子IXを目的濃度にまで濃縮する。生成物プールをUF/DF#2装置から取り、ポリソルベート80を目的濃度0.005%となるまで添加することにより処方する。次いで、因子IX薬剤物質をサンプリングし、ラベルを貼り、次いで、約−80℃で保存する。最終プロセス工程であるUF/DF#2は、有意な蛋白変性または損失なしに精製因子IX薬剤物質を濃縮し透析濾過することにおいて有効である。SDS−PAGE分析(還元および非還元)は、プロセス全体の質を評価するための1の方法である。各工程は80%ないし100%の回収率であり、因子IXの平均の正味の収率は約51%である。精製プロセスに供する前のクロッティング活性と因子IX薬剤物質(プロセス途中の試料および保持液として取ったものを除く)の全クロッティング活性からプロセス全体の収率を決定する。
下記実施例は本発明の実施を説明する。これらの実施例は説明のみを目的とし、特許請求されている本発明の範囲を何ら限定するものではない。
実施例1は限外濾過/透析濾過による蛋白の濃縮を説明する。実施例2はQ-Sepharose Fast Flow偽アフィニティーイオン交換クロマトグラフィーによる因子IXの精製に関する。実施例3はMatrex Cellulose Sulfateクロマトグラフィーによる因子IXの精製を説明する。実施例4はヒドロキシアパタイトを用いる蛋白精製に関する。実施例5は固定化金属アフィニティークロマトグラフィーによる蛋白精製を説明する。実施例6は限外濾過/透析濾過による蛋白の濃縮および処方に関する。
実施例1−限外濾過/透析濾過による蛋白濃縮
十字フロー膜濾過を用いる限外濾過/透析濾過(US/DF#1)を行って無細胞ならし培地を濃縮しバッファー交換することができる。十字フロー装置に使用する膜は、分子量に基づいて物質を分離する選択的透過性フィルターとして役立つ。因子IXのごとき高分子の溶液成分は膜により保持され、無機塩類およびバッファー成分のごとき低分子成分は多孔性膜構造中を自由に通過し、透過液中に除去される。
置換バッファーを保持液に添加するよりも早い速度で十字フロー装置からバッファーを取る場合、蛋白溶液は濃縮される。バッファーが膜を通って出て行くのとほぼ同じ速度で置換バッファーを十字フロー保持液に添加する場合、最初のバッファーは連続的に希釈される(蛋白の透析濾過)。これらの条件下で、低分子量の成分は容易に交換され、蛋白濃度は一定のままである。保持液の5倍の体積のバッファーの添加により、理論上最初のバッファーの99%またはそれ以上が置換される。
使用前に、UF/DF#1システムを50mM Tris,150mM NaCl,pH7.5で平衡化する。無細胞ならし培地を、最初の無細胞ならし培地の体積を基準として約20倍に濃縮する。次いで、濃縮無細胞ならし培地をバッファー中に透析濾過する。保持液の少なくとも5倍の体積のバッファーが膜を通過して時点で透析濾過を完了し、理論上、無細胞ならし培地に存在する塩類および他の低分子量成分の99%またはそれ以上が除去される。
透析濾過が完了したならば、必要な場合、保持液を濃縮する。次いで、残留している因子IX生成物をリザーバーおよび配管から回収するのに十分なバッファーで装置を洗う。次いで、プールをUF/DF容器からポンプで出し、オートクレーブ済み0.2μmフィルターで濾過してきれいな容器中に入れる。さらに処理するまでUF/DF#1プールを2ないし8℃で保存する。
実施例2−Q-Sepharose Fast Flow偽アフィニティーアニオン交換クロマトグラフィーによる因子IXの精製
Q-Sepharose Fast Flow(FF)(Pharmacia)は、短いリンカーを介して4級アミン基を共有結合して誘導体化された架橋アガロースマトリックスからなる強アニオン交換樹脂である。操作pHにおいて正味の負電荷を有する酸性蛋白(因子IXのごとき)および他の多イオン性物質は電荷相互作用によりQ-Sepharose FFに結合する。典型的には、導電率の大きい溶液によりこれらの電荷の相互作用を破壊することにより結合成分はQ-Sepharose FFから個々に溶離される。しかしながら、因子IX精製プロセスには偽アフィニティーモードのQ-Sepharose FF樹脂を用いる。低濃度(10mM)の塩化カルシウムを含有する溶液を用いて因子IXをカラムから溶離する。溶離バッファー中のカルシウムイオンを含めることは因子IXのコンホーメーション変化を引き起こし、樹脂からの溶離を引き起こす。
Q-Sepharose FFを用いてUF/DF#1保持液から因子IXを捕捉し、非荷電汚染混入物質および塩基性汚染混入物質を除去し(カラムへの負荷中に未結合フラクション中に出る)、因子IX(不活性形態の因子IXを包含)を酸性蛋白から分離し(それらは樹脂に結合するが、バッファーへの塩化カルシウム添加によっては溶離されない)、濃縮された因子IXのプロセスの流れを次の精製プロセスであるクロマトグラフィー工程、すなわちMatrex Cellufine Sulfateへと送る。
この工程に関するすべてのクロマトグラフィー操作を2ないし8℃で行ってもよい。まず、Q-Sepharose FFカラムに50mM Tris,2M NaCl,pH8.0をチャージし、次いで、50mM Tris,150mM NaCl,pH8.0で平衡化する。UF/DF#1保持液をQ-Sepharose FFカラムに負荷し、次いで、50mM Tris,200mM NaCl,pH8.0でカラムを洗浄する。この最初の洗浄により、全員荷物がカラムを通り、緩やかに樹脂に結合している負荷物中の非吸着性不純物ならびに汚染混入物質が系から洗い出されることが確実となる。次いで、50mM Tris,100mM NaCl,pH8.0でカラムを洗浄して導電率を下げて溶離に備える。
50mM Tris,100mM NaCl,10mM CaCl2,pH8.0でカラムから因子IXを溶離し、溶離生成物をを1本のピークとして集める。Q-Sepharose FF溶離物をサンプリングし、さらなる処理を行うまで2ないし8℃で保存する。カラムを再生して再使用してもよい。
実施例3−Matrex Cellufine Sulfateクロマトグラフィーによる因子IXの精製
Matrex Cellufine Sulfateは、硫酸エステルで誘導体化された球形セルロースビーズからなる。ヘパリン結合ドメインを含む蛋白のアフィニティー精製のための固定化ヘパリンアナログとしてそれを用いることができる。それは負に帯電した硫酸官能基を有するので、それをカチオン交換クロマトグラフィーに用いてもよい。操作pHにおいて正味の正電荷を有する塩基性蛋白、他の多イオン性物質、ならびにヘパリン結合蛋白は樹脂に結合し、溶液のイオン強度を増加させると溶離する。Q-Sepharose FF溶離物プール中の因子IX以外の宿主細胞蛋白を除去するための因子IX精製プロセスにおいてMatrex Cellufine Sulfate樹脂を用い、さらにヒドロキシアパタイトカラム負荷に適するバッファー条件を提供してもよい。
この工程のすべてのクロマトグラフィー操作を2ないし8℃で行ってもよい。負荷工程の準備のために、Matrex Cellufine Sulfateカラムを50mM Tris,pH8.0で平衡化する。Q-Sepharose FF溶離物プールを平衡化されたMatrex Cellufine Sulfateカラムに直接負荷し、カラムを50mM Tris,150mM NaCl,10mM CaCl2,pH8.0で洗浄して、すべての負荷物がカラムを通り、弱く結合した不純物が系から除去されることを確実にする。次に、溶離の前にカラムを洗浄してカルシウムイオンを除去する。
洗浄工程完了後、Matrex Cellufine Sulfateカラムを50mM Tris,500mM NaCl,pH8.0で溶離し、溶離物を1つのUV吸収溶離物プールとして集める。Matrex Cellufine Sulfate溶離物プールをサンプリングし、さらに処理するまで2ないし8℃で保存する。カラムを再生し、再使用してもよい。
実施例4−ヒドロキシアパタイトクロマトグラフィーを用いる蛋白精製
セラミック−ヒドロキシアパタイト(Ceramic-HA)は、大きな機械的強度を有する球形多孔質粒子からなる合成形態のリン酸カルシウムである。Ceramic-HAは、主に電荷相互作用に基づいて、広範囲の電荷および等電点の蛋白を分離する。因子IXは、ほぼ中性pHにおいてCeramic-HAに結合する酸性蛋白である。典型的には、バッファー溶液にリン酸塩を添加することにより、酸性蛋白はCeramic-HAから溶離される。目的分子の特性により溶離に必要なリン酸塩濃度は変動し、そのことにより結合蛋白が個々に溶離される。Ceramic-HAを因子IX精製プロセスに用いて、Matrex Cellufine Sulfate溶離物プール中の不活性因子IXおよび他の汚染混入物質を除去し、溶離物バッファーを最終クロマトグラフィー工程に適合したものにする。最終クロマトグラフィー工程が固定化金属アフィニティークロマトグラフィーであるので、Ceramic-HAの溶離に使用するバッファーをIMACに適合するように選択する。このことによりプロセスに差し挟む透析濾過または他のバッファー交換法を省略する。金属イオンと固定化リガンドとの相互作用を破壊するので、Tris、グリシン、ヒスチジンのごときバッファーはIMACに適合しない。
負荷のための準備において、Ceramic-HAカラムを50mM Tris,500mM NaCl,pH7.5で平衡化する。Matrex Cellufine Sulfate溶離物プールを希HClで滴定してpH7.5とし、Ceramic-HAカラムに直接負荷する。負荷完了後、カラムをバッファー(すべての負荷物がカラムを通り、)で洗浄して緩く結合した汚染混入物質がカラムから除去されることを確実にする。次に、カラムを50mM KH2PO4,185mM NaCl,pH7.2で洗浄して不活性形態の因子IXをプロセスの流れから除去する。
洗浄工程完了後、結合した因子IXを500mM K2HPO4,200mM NaCl,pH7.2で溶離し、因子IX溶離物を1つのUV吸収溶離物プールとして集める。溶離物プールをサンリングし、さらなる処理を行うまで2ないし8℃に保存する。カラムを再生し、再使用してもよい。
実施例5−固定化金属アフィニティークロマトグラフィーによる蛋白精製
Fractogel-EMD-Chelateは、遷移状態の金属イオンが結合しうるイミノジ酢酸官能基で誘導体化されたメタクリレートポリマーからなる。精製プロセスでの使用準備において、硫酸銅溶液を用いて樹脂に銅をチャージする。固定化銅イオンと相互作用しうる蛋白はカラムに保持され、相互作用しない汚染混入物質は未結合フラクション中に素通りする。イミダゾール含有溶液を用いて結合蛋白を溶離させる。Fractogel-EMD-Chelate-Cu(II)工程を蛋白精製プロセスに用いて、固定化金属イオンに結合しないかまたは因子IXの溶離に必要なイミダゾールよりも高濃度のイミダゾールを必要とする汚染混入物質をプロセスの流れから除去する。このクロマトグラフィー工程を表すために、用語IMAC(固定化金属アフィニティークロマトグラフィー)を使用する。
負荷のための準備において、未チャージの(固定化金属イオンのない)Fractogel-EMD-Chelateカラムを、100mM酢酸、500mM NaCl,pH4.0で洗浄し、次いで、200mM CuSO4,500mM NaClをチャージする。100mM酢酸、500mM NaCl,pH4.0、次いで、200mMイミダゾール,500mM NaCl,pH7.1で洗浄することにより緩く結合した銅イオンを除去する。次いで、Fractogel-EMD-Chelate-Cu(II)樹脂を200mM K2HPO4,200mM NaCl,pH7.1で平衡化する(平衡化V)。Ceramic-HA溶離物プールを、平衡化したFractogel-EMD-Chelate-Cu(II)カラムに直接負荷する。
負荷完了後、平衡化バッファーでカラムを洗浄して、すべての負荷物がカラムを通ることを確実にする。20mM K2HPO4,15mMイミダゾール,100mM NaCl,pH7.1を用いて樹脂に結合した因子IXを溶離する。Fractogel-EMD-Chelate-Cu(II)溶離物を1つのUV吸収プールとして集める。集めた後、溶離物プールを、カラム溶離物1リットルあたり20mLの500mM EDTA,pH8.0で希釈する。さらに処理するまで溶離物プールを室温に保存する。
実施例6−限外濾過/透析濾過#2による蛋白の濃縮および処方
因子IXを選択バッファーに移すために、限外濾過/透析濾過工程を用いる。十字フローUF/DFは非クロマトグラフィー分離法であり、これを用いて溶液中の物質の濃縮およびバッファー交換を行うことができる。供給液を選択的透過性膜表面に平行に流し、膜の保持液側に圧をかけて、膜表面における水および溶質の相対的透過性に基づく輸送を行わせる。これらの環境において、低分子量の供給液中の成分は自由に膜の孔を通過して透過フラクション中に至り、高分子量物質(例えば、因子IX)は膜により保持され、保持フラクションを形成する。この方法において、水およびバッファー塩をFractogel-EMD-Chelate-Cu(II)溶離物プールから除去することができ、因子IXを目的濃度にまで濃縮することができる。
十字フローシステムの螺旋溝カートリッジコンポーネントを、まず10mMヒスチジン,260mMグリシン,1%蔗糖,pH6.8で平衡化する。次いで、Fractogel-EMD-Chelate-Cu(II)溶離物プール(前以て500mM TETAで希釈)を、蛋白濃縮に備えて十字フロー装置のステンレス製保持液加圧容器に移す。
移すことが完了したら、正味の正の経膜圧力をかけながら保持溶液をポンプで連続的に加圧容器から螺旋溝カートリッジを通して加圧容器に戻す。この操作を行っている間、目盛り付き収集容器を用いて透過フラクション体積を測定することにより保持液体積を連続的にモニターする。
目的保持液体積に達したならば、保持液プールを選択バッファー中に透析濾過する。この操作の間、透過液がシステムから流出するのと同じ速度で透析濾過バッファーを加圧容器中にポンプで送り、そのことにより保持液体積を一定に維持する。
透析濾過工程完了後、限外濾過を用いて保持液フラクションを目的体積にまで濃縮する。保持液加圧容器からの流出フラクションを止め、目的体積の選択バッファーで螺旋溝カートリッジ中の保持液フラクションを保持液加圧容器中に洗い込む。濃縮され、透析濾過された因子IX生成物をポンプで加圧容器から風袋を計っておいたプール用ビンに取る。
因子IX生成物プールを10mMヒスチジン,260mMグリシン,1%蔗糖,1%ポリソルベート80,pH6.8で希釈してポリソルベート最終濃度0.005%とする。次いで、生成物を完全に混合し、0.2μmフィルター(前以て10mMヒスチジン,260mMグリシン,1%蔗糖,0.005%ポリソルベート80,pH6.8にて平衡化)で濾過して脱パイロジェン化したテフロン製ビンに入れる。次いで、蛋白をサンプリングし、ラベルを貼り、液体窒素で素早く凍結して−80℃で保存する。
組み換え的に製造された因子IXの形質転換宿主細胞からの精製によって本発明方法を説明したが、該方法は、天然の細胞中に存在する因子IXの精製にも使用でき、溶液または血漿、細胞ホモジネート物、細胞培養上清、あるいは単離細胞サブフラクションからの蛋白の精製に使用できる。本発明を特定の方法および組成物について説明したが、本発明を考慮して当業者が変更および修正を行うことが理解される。
上記の説明的実施例に記載された本発明における多くの修飾および変更は当業者により行われると予想され、したがって、添付した請求の範囲にあるような限定のみが本発明に課されるはずである。したがって、特許請求されている本発明の範囲内にあるすべてのかかる均等な変更を添付した請求の範囲がカバーすると考えられる。
Claims (20)
- 活性型因子IXおよび不活性型因子IXを含有する溶液中の因子IXの精製方法であって、下記工程を特徴とする方法:
該溶液をアニオン交換樹脂に適用すること、
該アニオン交換樹脂を第1の洗浄液で洗浄すること、
該アニオン交換樹脂を第1の溶離液で溶離して第1の溶離物を得ること、
ここに該第1の溶離液は該第1の洗浄液の導電率および該溶液の導電率のうち高い方よりも低い導電率を有するものであり、
該溶離物をヘパリン様樹脂に適用すること、
該ヘパリン様樹脂を第2の溶離液で溶離して第2の溶離物を得ること、
該第2の溶離物をヒドロキシアパタイト樹脂に適用すること、ついで
該ヒドロキシアパタイト樹脂を第3の溶離液で溶離して第3の溶離液を得ること。 - 該第1の洗浄液の導電率が該溶液の導電率よりも高いか等しいものである請求項1の方法。
- 該第1の洗浄液が、塩化ナトリウム、塩化カリウム、硫酸ナトリウム、リン酸ナトリウムまたはリン酸カリウムからなる群より選択される溶液を含むものである請求項2の方法。
- 該第1の洗浄液が200mM塩化ナトリウムである請求項3の方法。
- 下記工程:
該第3の溶離物を固定化金属アフィニティー樹脂に適用し、次いで、
該固定化金属アフィニティー樹脂を第4の溶離液で溶離して第4の溶離物を得ること
をさらに特徴とする請求項1ないし4のいずれか1項の方法。 - 該第1の溶離液が、カルシウム、マグネシウム、マンガン、ストロンチウム、亜鉛、コバルトおよびニッケルからなる群より選択される2価カチオンを含むものである請求項1ないし5のいずれか1項の方法。
- 該第1の溶離液が10mMカルシウムである請求項6の方法。
- 該アニオン交換樹脂が、ジエチルアミノエタン(DEAE)、ポリエチレンイミン(PEI)および4級アミノエタン(QAE)からなる群より選択されるメンバーである正に帯電した基を有するものである請求項1ないし7のいずれか1項の方法。
- 該アニオン交換樹脂がQ-Sepharose Fast Flow(商標)である請求項8の方法。
- 該ヘパリン様樹脂が、ヘパリン、セルロースの硫酸エステル、スルフィルプロピル(SP)、カルボキシルおよびカルボキシメチルからなる群より選択されるメンバーである負に帯電した基を有するものである請求項1ないし9のいずれか1項の方法。
- 該ヘパリン様樹脂がMatrex Cellufine Sulfate(商標)である請求項10の方法。
- 該第2の溶離液が、塩化ナトリウムおよび塩化カリウムからなる群より選択されるメンバーである請求項1ないし11のいずれか1項の方法。
- 該第2の溶離液が50mM Tris,0.50M NaCl,pH8.0である請求項12の方法。
- 該第3の溶離液が、リン酸塩および硫酸塩からなる群から選択されるメンバーである請求項1ないし13のいずれか1項の方法。
- 該第3の溶離液が0.5Mリン酸カリウム,0.2M NaCl,pH7.2である請求項14記載の方法。
- 該ヒドロキシアパタイト樹脂が、セラミック−ヒドロキシアパタイトおよびBioGel-HTからなる群より選択されるメンバーである請求項1ないし15のいずれか1項の方法。
- 該ヒドロキシアパタイト樹脂がセラミック−ヒドロキシアパタイトである請求項16の方法。
- 該固定化金属アフィニティー樹脂が、Fractogel-EMD-Chelate(商標)、Chelating-Sepharose(商標)、Matrex Cellufine Chelate(商標)およびPOROS 20MC(商標)からなる群より選択されるメンバーである請求項5ないし17のいずれか1項の方法。
- 該第4の溶離液が置換物質である請求項5ないし18のいずれか1項の方法。
- 該置換物質が、イミダゾール、EDTA、グリシン、ヒスチジンおよびTrisからなる群より選択されるメンバーである請求項19の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/472,823 US5714583A (en) | 1995-06-07 | 1995-06-07 | Factor IX purification methods |
| US08/472,823 | 1995-06-07 | ||
| PCT/US1996/007305 WO1996040883A1 (en) | 1995-06-07 | 1996-05-21 | Novel factor ix purification methods |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPH11506603A JPH11506603A (ja) | 1999-06-15 |
| JP3676817B2 true JP3676817B2 (ja) | 2005-07-27 |
Family
ID=23877079
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP50062497A Expired - Lifetime JP3676817B2 (ja) | 1995-06-07 | 1996-05-21 | 新規因子ix精製法 |
Country Status (11)
| Country | Link |
|---|---|
| US (2) | US5714583A (ja) |
| EP (2) | EP1571208B1 (ja) |
| JP (1) | JP3676817B2 (ja) |
| AT (2) | ATE393217T1 (ja) |
| AU (1) | AU5754396A (ja) |
| CA (1) | CA2220501C (ja) |
| DE (2) | DE69637509T2 (ja) |
| DK (2) | DK1571208T3 (ja) |
| ES (2) | ES2353798T3 (ja) |
| PT (2) | PT1571208E (ja) |
| WO (1) | WO1996040883A1 (ja) |
Families Citing this family (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5714583A (en) * | 1995-06-07 | 1998-02-03 | Genetics Institute, Inc. | Factor IX purification methods |
| US20030036629A1 (en) * | 1997-12-12 | 2003-02-20 | Barry Foster | Novel tgf-beta protein purification methods |
| AU760048B2 (en) | 1998-05-06 | 2003-05-08 | Genentech Inc. | Protein purification by ion exchange chromatography |
| CN1331602A (zh) | 1998-09-21 | 2002-01-16 | 遗传研究所有限公司 | 负调节针对医用蛋白的免疫反应的方法 |
| US7083787B2 (en) * | 2000-11-15 | 2006-08-01 | Globeimmune, Inc. | Yeast-dendritic cell vaccines and uses thereof |
| KR100451308B1 (ko) * | 2001-12-28 | 2004-10-06 | 선바이오(주) | 단백질 의약품 정제 방법에서의 바이러스 제거 방법 |
| RU2337968C2 (ru) | 2003-01-09 | 2008-11-10 | Дженентек, Инк. | Способ очистки гетерологичного полипептида |
| US20050008580A1 (en) * | 2003-04-09 | 2005-01-13 | Wyeth | Hemophilia treatment by inhalation of coagulation factors |
| EP1765411B2 (en) * | 2004-06-30 | 2017-10-11 | Nektar Therapeutics | Polymer-factor ix moiety conjugates |
| US20080045453A1 (en) | 2005-12-21 | 2008-02-21 | Drohan William N | Method of producing biologically active vitamin K dependent proteins by recombinant methods |
| EP2650305B1 (en) | 2006-03-24 | 2024-05-08 | Bioverativ Therapeutics Inc. | PC5 as a factor IX propeptide processing enzyme |
| WO2008104372A1 (en) * | 2007-02-28 | 2008-09-04 | Baxter International Inc. | Method for the purification of recombinant blood coagulation factor ix enriched in sulfated and/or phosphorylated molecules |
| CA2683423C (en) * | 2007-04-26 | 2020-10-27 | Inspiration Biopharmaceuticals, Inc. | Recombinant vitamin k dependent proteins with high sialic acid content and methods of preparing same |
| DK2215117T4 (en) | 2007-10-30 | 2018-04-09 | Genentech Inc | ANTISTO PURIFICATION BY CATION CHANGE CHROMATOGRAPHY |
| EP2326658A4 (en) * | 2008-09-12 | 2013-04-10 | Ge Healthcare Bio Sciences Ab | IMPROVED PROTEIN-AGGREGATE SEPARATION WITH MULTIMODAL ANION EXCHANGERS IN THE PRESENCE OF ZWITTERIONS EXCLUDED FROM THE PROTEIN |
| FR2946348B1 (fr) | 2009-06-05 | 2011-08-05 | Lab Francais Du Fractionnement | Procede de preparation d'une composition de complexe prothrombique a haut degre de purete |
| CN102471794B (zh) | 2009-07-10 | 2014-10-29 | Csl有限公司 | 提高维生素k依赖性蛋白质的表达产量的方法 |
| WO2011053738A1 (en) | 2009-10-30 | 2011-05-05 | Inspiration Biopharmaceuticals, Inc. | Method of producing recombinant vitamin k dependent proteins |
| US8697844B2 (en) | 2009-11-24 | 2014-04-15 | Novo Nordisk A/S | Method of purifying pegylated proteins |
| RU2731720C2 (ru) * | 2010-03-30 | 2020-09-08 | Октафарма Аг | Способ очистки витамин к-зависимых белков, таких как коагуляционный фактор vii |
| MX2012011065A (es) | 2010-03-30 | 2012-10-10 | Octapharma Ag | Proceso para purificacion de proteina de factor de crecimiento. |
| AU2011247567B2 (en) * | 2010-04-29 | 2014-06-19 | Takeda Pharmaceutical Company Limited | Purification method for divalent cation binding proteins on anion exchange resin |
| WO2012082933A1 (en) | 2010-12-15 | 2012-06-21 | Baxter International, Inc. | Eluate collection using conductivity gradient |
| BRPI1105317A2 (pt) | 2011-01-24 | 2013-04-30 | Fundacco Hemoct De Ribeirco Preto | produÇço estÁvel e em larga escala de fviii humano em linhagem celular humana sk-hep-1 |
| AU2012304763A1 (en) | 2011-09-06 | 2014-03-06 | Medimmune Llc | Methods for processing coagulation factors |
| AU2012322948B2 (en) * | 2011-10-14 | 2014-11-06 | Takeda Pharmaceutical Company Limited | Protein purification by anion exchange chromatography |
| WO2013053888A1 (en) * | 2011-10-14 | 2013-04-18 | Baxter International Inc. | Protein purification by anion exchange chromatography |
| JP6571011B2 (ja) | 2013-03-15 | 2019-09-04 | バクスアルタ インコーポレイテッド | 陰イオン交換クロマトグラフィーによるビタミンk依存性タンパク質の精製方法 |
| US9663553B2 (en) | 2014-01-29 | 2017-05-30 | Hemarus Therapeutics Limited | Integrated process for the production of therapeutics (human albumin, immunoglobulins, clotting factor VIII and clotting factor IX) from human plasma |
| CN114134109B (zh) * | 2021-12-10 | 2023-01-24 | 广州远想生物科技股份有限公司 | Egf间充质干细胞外泌体的纯化方法 |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3101752A1 (de) * | 1981-01-21 | 1982-08-26 | Behringwerke Ag, 3550 Marburg | "verfahren zur reinigung der blutgerinnungsfaktoren ii, vii, ix und/oder x und danach hergestellte praeparationen" |
| US4447416A (en) * | 1982-04-28 | 1984-05-08 | American National Red Cross | Plasma protein concentrates of reduced thrombogenicity and their use in clinical replacement therapy |
| US5055557A (en) * | 1983-03-04 | 1991-10-08 | Scripps Clinic & Research Foundation | Ultrapurification of factor IX and other vitamin K-dependent proteins |
| JPS61500226A (ja) * | 1983-10-28 | 1986-02-06 | ニュ−・イングランド・メディカル・センタ−・ホスピタルズ・インコ−ポレ−テッド | コンホメ−ション特異性抗体を用いる血液凝固タンパク質の精製および単離法 |
| US4721572A (en) * | 1985-07-12 | 1988-01-26 | Miles Laboratories, Inc. | Purfication of blood clotting factors and other blood proteins on non-carbohydrate sulfated matrices |
| US4786726A (en) * | 1986-01-06 | 1988-11-22 | Blood Systems, Inc. | Factor IX therapeutic blood product, means and methods of preparing same |
| US4725673A (en) * | 1986-08-29 | 1988-02-16 | Alpha Therapeutic Corporation | Plasma fraction purification using silica resin bound to a ligand |
| US4831118A (en) * | 1987-08-07 | 1989-05-16 | Scripps Clinic And Research Foundation | Flouroplastic immunoaffinity columns for purification of blood proteins |
| DE3877529T2 (de) * | 1987-10-23 | 1996-11-07 | Centre Regional De Transfusion | Herstellung von hoch-gereingtem menschlichen Faktor IX sowie anderem Plasmaproteinkonzentrat und dessen therapeutische Verwendung. |
| WO1989005652A1 (en) * | 1987-12-21 | 1989-06-29 | American National Red Cross | Pure factor ix product |
| GB8729822D0 (en) * | 1987-12-22 | 1988-02-03 | Central Blood Lab Authority | Chemical process |
| US5118614A (en) * | 1988-01-18 | 1992-06-02 | Tessek Sdruzeni Praha | Concentrates of coagulation factors ii, vii, ix and x, method of their preparation and use |
| DE3826792C1 (ja) * | 1988-08-06 | 1989-07-06 | Biotest Pharma Gmbh, 6072 Dreieich, De | |
| US4981952A (en) * | 1988-10-04 | 1991-01-01 | Eli Lilly And Company | Method for the purification of vitamin K-dependent proteins |
| DE3914869C1 (ja) * | 1989-05-05 | 1990-08-09 | Biotest Pharma Gmbh, 6072 Dreieich, De | |
| AT402261B (de) * | 1991-01-25 | 1997-03-25 | Immuno Ag | Komplex enthaltend den gerinnungsfaktor ix |
| IT1262899B (it) * | 1992-03-27 | 1996-07-22 | Sclavo Spa | Processo per l'isolamento di fattore ix, fattore x e fattore ii altamente purificati dal complesso protrombinico o dal plasma umano |
| US5714583A (en) * | 1995-06-07 | 1998-02-03 | Genetics Institute, Inc. | Factor IX purification methods |
-
1995
- 1995-06-07 US US08/472,823 patent/US5714583A/en not_active Expired - Lifetime
-
1996
- 1996-05-21 PT PT05012614T patent/PT1571208E/pt unknown
- 1996-05-21 ES ES05012614T patent/ES2353798T3/es not_active Expired - Lifetime
- 1996-05-21 AT AT96915892T patent/ATE393217T1/de active
- 1996-05-21 JP JP50062497A patent/JP3676817B2/ja not_active Expired - Lifetime
- 1996-05-21 ES ES96915892T patent/ES2306450T3/es not_active Expired - Lifetime
- 1996-05-21 CA CA2220501A patent/CA2220501C/en not_active Expired - Lifetime
- 1996-05-21 DE DE69637509T patent/DE69637509T2/de not_active Expired - Lifetime
- 1996-05-21 DK DK05012614.3T patent/DK1571208T3/da active
- 1996-05-21 EP EP05012614A patent/EP1571208B1/en not_active Expired - Lifetime
- 1996-05-21 DK DK96915892T patent/DK0832200T3/da active
- 1996-05-21 EP EP96915892A patent/EP0832200B1/en not_active Expired - Lifetime
- 1996-05-21 WO PCT/US1996/007305 patent/WO1996040883A1/en active Application Filing
- 1996-05-21 DE DE69638291T patent/DE69638291D1/de not_active Expired - Lifetime
- 1996-05-21 PT PT96915892T patent/PT832200E/pt unknown
- 1996-05-21 AU AU57543/96A patent/AU5754396A/en not_active Abandoned
- 1996-05-21 AT AT05012614T patent/ATE488582T1/de active
-
1997
- 1997-10-17 US US08/953,143 patent/US6627737B1/en not_active Expired - Lifetime
Also Published As
| Publication number | Publication date |
|---|---|
| WO1996040883A1 (en) | 1996-12-19 |
| DE69637509D1 (de) | 2008-06-05 |
| CA2220501C (en) | 2014-01-07 |
| JPH11506603A (ja) | 1999-06-15 |
| EP1571208A1 (en) | 2005-09-07 |
| EP1571208B1 (en) | 2010-11-17 |
| ES2353798T3 (es) | 2011-03-07 |
| DE69637509T2 (de) | 2009-05-28 |
| ATE488582T1 (de) | 2010-12-15 |
| DK1571208T3 (da) | 2011-01-24 |
| ATE393217T1 (de) | 2008-05-15 |
| EP0832200A1 (en) | 1998-04-01 |
| EP0832200B1 (en) | 2008-04-23 |
| AU5754396A (en) | 1996-12-30 |
| PT832200E (pt) | 2008-07-17 |
| DE69638291D1 (de) | 2010-12-30 |
| US6627737B1 (en) | 2003-09-30 |
| DK0832200T3 (da) | 2008-07-21 |
| PT1571208E (pt) | 2011-01-04 |
| CA2220501A1 (en) | 1996-12-19 |
| ES2306450T3 (es) | 2008-11-01 |
| US5714583A (en) | 1998-02-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3676817B2 (ja) | 新規因子ix精製法 | |
| JP4519646B2 (ja) | プレプロインスリンの精製方法 | |
| JP4559216B2 (ja) | 液体中の夾雑物からアルブミンを分離する方法、使用及びキット | |
| KR101829860B1 (ko) | 응고인자 ⅸ와 같은 비타민 k 의존성 단백질을 정제하는 방법 | |
| CN102234332B (zh) | 一种重组人血白蛋白及其融合蛋白的分离纯化工艺 | |
| JP3768485B2 (ja) | 血清アルブミンの精製方法 | |
| JP2573467B2 (ja) | 第ix因子タンパク質を含有する治療用途に適した薬剤的組成物 | |
| JP7445332B2 (ja) | ベバシズマブ精製の最適化された方法 | |
| US5525500A (en) | Chromatographic process for the copurification of chondroitinase I and II proteins from Proteus vulgaris | |
| AU759379B2 (en) | Novel factor IX purification methods | |
| JP2001139600A (ja) | Il−6r・il−6融合蛋白質の精製方法 | |
| AU685035B2 (en) | A purification process of a human growth hormone | |
| JP4154743B2 (ja) | インターロイキン6レセプターの精製方法 | |
| KR100341297B1 (ko) | 재조합활성형프로인슐린의분리.정제방법 | |
| JPS5810522A (ja) | 不活化トロンビンゲルによるアンチトロンビン3の精製法 | |
| JPS6176419A (ja) | 精製された組織性プラスミノ−ゲンアクチベ−タの製造法 | |
| Nandakumar et al. | Purification of horse radish peroxidase by metal affinity chromatography in an expanded bed system | |
| JPH10265499A (ja) | ヒト成長ホルモンの精製方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20040309 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20040426 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20040607 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20050405 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20050502 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 3676817 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20090513 Year of fee payment: 4 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20100513 Year of fee payment: 5 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20100513 Year of fee payment: 5 |
|
| S202 | Request for registration of non-exclusive licence |
Free format text: JAPANESE INTERMEDIATE CODE: R315201 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20100513 Year of fee payment: 5 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20100513 Year of fee payment: 5 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110513 Year of fee payment: 6 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110513 Year of fee payment: 6 |
|
| R153 | Grant of patent term extension |
Free format text: JAPANESE INTERMEDIATE CODE: R153 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120513 Year of fee payment: 7 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120513 Year of fee payment: 7 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20200513 Year of fee payment: 15 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| EXPY | Cancellation because of completion of term |