JP2017536654A - 好ましい形態を有する不純物含有カソード材料及び不純物含有金属炭酸塩から調製するための方法 - Google Patents
好ましい形態を有する不純物含有カソード材料及び不純物含有金属炭酸塩から調製するための方法 Download PDFInfo
- Publication number
- JP2017536654A JP2017536654A JP2017518496A JP2017518496A JP2017536654A JP 2017536654 A JP2017536654 A JP 2017536654A JP 2017518496 A JP2017518496 A JP 2017518496A JP 2017518496 A JP2017518496 A JP 2017518496A JP 2017536654 A JP2017536654 A JP 2017536654A
- Authority
- JP
- Japan
- Prior art keywords
- sulfur
- sodium
- carbonate
- ratio
- impurities
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 239000012535 impurity Substances 0.000 title claims abstract description 143
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 title claims abstract description 135
- 229910052751 metal Inorganic materials 0.000 title claims description 88
- 239000002184 metal Substances 0.000 title claims description 88
- 238000000034 method Methods 0.000 title claims description 65
- 239000010406 cathode material Substances 0.000 title description 30
- 239000011734 sodium Substances 0.000 claims abstract description 274
- 229910052717 sulfur Inorganic materials 0.000 claims abstract description 136
- 239000002243 precursor Substances 0.000 claims abstract description 126
- 229910052708 sodium Inorganic materials 0.000 claims abstract description 124
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims abstract description 123
- 239000011593 sulfur Substances 0.000 claims abstract description 123
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 claims abstract description 109
- 239000002245 particle Substances 0.000 claims abstract description 72
- 239000000843 powder Substances 0.000 claims abstract description 58
- 150000001875 compounds Chemical class 0.000 claims abstract description 26
- 229910052744 lithium Inorganic materials 0.000 claims abstract description 20
- 229910052748 manganese Inorganic materials 0.000 claims abstract description 13
- 229910052759 nickel Inorganic materials 0.000 claims abstract description 12
- 239000007774 positive electrode material Substances 0.000 claims abstract description 7
- 238000009826 distribution Methods 0.000 claims abstract description 5
- HBBGRARXTFLTSG-UHFFFAOYSA-N Lithium ion Chemical compound [Li+] HBBGRARXTFLTSG-UHFFFAOYSA-N 0.000 claims abstract description 4
- 229910001416 lithium ion Inorganic materials 0.000 claims abstract description 4
- 239000000203 mixture Substances 0.000 claims description 63
- 239000000243 solution Substances 0.000 claims description 32
- 238000000151 deposition Methods 0.000 claims description 18
- 229910021450 lithium metal oxide Inorganic materials 0.000 claims description 16
- 239000007788 liquid Substances 0.000 claims description 13
- 239000002019 doping agent Substances 0.000 claims description 12
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 claims description 12
- 239000002002 slurry Substances 0.000 claims description 12
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 claims description 11
- 150000002500 ions Chemical class 0.000 claims description 11
- 229910052782 aluminium Inorganic materials 0.000 claims description 10
- -1 sulfate compound Chemical class 0.000 claims description 9
- 229910052749 magnesium Inorganic materials 0.000 claims description 7
- 229910052684 Cerium Inorganic materials 0.000 claims description 6
- 229910052796 boron Inorganic materials 0.000 claims description 6
- 229910052791 calcium Inorganic materials 0.000 claims description 6
- 229910052804 chromium Inorganic materials 0.000 claims description 6
- 239000012527 feed solution Substances 0.000 claims description 6
- 238000002156 mixing Methods 0.000 claims description 6
- 229910052758 niobium Inorganic materials 0.000 claims description 6
- 229910052718 tin Inorganic materials 0.000 claims description 6
- 229910052719 titanium Inorganic materials 0.000 claims description 6
- 229910052725 zinc Inorganic materials 0.000 claims description 6
- 229910052726 zirconium Inorganic materials 0.000 claims description 6
- 238000002441 X-ray diffraction Methods 0.000 claims description 5
- 238000003991 Rietveld refinement Methods 0.000 claims description 2
- 229910001415 sodium ion Inorganic materials 0.000 claims description 2
- JDZCKJOXGCMJGS-UHFFFAOYSA-N [Li].[S] Chemical compound [Li].[S] JDZCKJOXGCMJGS-UHFFFAOYSA-N 0.000 claims 2
- 238000001556 precipitation Methods 0.000 description 97
- 239000000523 sample Substances 0.000 description 63
- 210000004027 cell Anatomy 0.000 description 46
- 230000008569 process Effects 0.000 description 44
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 36
- 238000010899 nucleation Methods 0.000 description 27
- 238000012360 testing method Methods 0.000 description 27
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 24
- 239000002253 acid Substances 0.000 description 24
- 239000011572 manganese Substances 0.000 description 22
- PXHVJJICTQNCMI-UHFFFAOYSA-N nickel Substances [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 21
- 239000002244 precipitate Substances 0.000 description 21
- 239000000047 product Substances 0.000 description 20
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 17
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 17
- 230000008021 deposition Effects 0.000 description 16
- 230000002441 reversible effect Effects 0.000 description 16
- 239000013078 crystal Substances 0.000 description 15
- 230000002427 irreversible effect Effects 0.000 description 15
- 238000006243 chemical reaction Methods 0.000 description 14
- 238000004519 manufacturing process Methods 0.000 description 14
- 238000002360 preparation method Methods 0.000 description 14
- 238000001878 scanning electron micrograph Methods 0.000 description 14
- 229910052723 transition metal Inorganic materials 0.000 description 14
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 14
- 239000012071 phase Substances 0.000 description 13
- 229910021529 ammonia Inorganic materials 0.000 description 12
- 235000002639 sodium chloride Nutrition 0.000 description 11
- 238000002474 experimental method Methods 0.000 description 10
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 9
- 230000007423 decrease Effects 0.000 description 9
- 150000003839 salts Chemical group 0.000 description 9
- 150000003624 transition metals Chemical class 0.000 description 9
- 238000005406 washing Methods 0.000 description 9
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 8
- 230000002829 reductive effect Effects 0.000 description 8
- 239000007787 solid Substances 0.000 description 8
- 238000001816 cooling Methods 0.000 description 7
- 238000009792 diffusion process Methods 0.000 description 7
- 238000001035 drying Methods 0.000 description 7
- 238000005516 engineering process Methods 0.000 description 7
- 238000010304 firing Methods 0.000 description 7
- 229910000000 metal hydroxide Inorganic materials 0.000 description 7
- 150000004692 metal hydroxides Chemical class 0.000 description 7
- 238000003860 storage Methods 0.000 description 7
- 239000011149 active material Substances 0.000 description 6
- 238000007792 addition Methods 0.000 description 6
- 238000004458 analytical method Methods 0.000 description 6
- 238000013459 approach Methods 0.000 description 6
- 239000003518 caustics Substances 0.000 description 6
- 239000003792 electrolyte Substances 0.000 description 6
- 238000001914 filtration Methods 0.000 description 6
- 235000015110 jellies Nutrition 0.000 description 6
- 239000008274 jelly Substances 0.000 description 6
- 238000005245 sintering Methods 0.000 description 6
- 229910015118 LiMO Inorganic materials 0.000 description 5
- 150000001768 cations Chemical class 0.000 description 5
- 238000004140 cleaning Methods 0.000 description 5
- 238000005342 ion exchange Methods 0.000 description 5
- 230000006911 nucleation Effects 0.000 description 5
- 239000011148 porous material Substances 0.000 description 5
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- 238000001354 calcination Methods 0.000 description 4
- 230000015556 catabolic process Effects 0.000 description 4
- 238000000576 coating method Methods 0.000 description 4
- 238000006731 degradation reaction Methods 0.000 description 4
- 238000005137 deposition process Methods 0.000 description 4
- 238000000349 field-emission scanning electron micrograph Methods 0.000 description 4
- 239000010410 layer Substances 0.000 description 4
- 229910021437 lithium-transition metal oxide Inorganic materials 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 238000003303 reheating Methods 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- 229910000299 transition metal carbonate Inorganic materials 0.000 description 4
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 3
- 238000010923 batch production Methods 0.000 description 3
- 238000000975 co-precipitation Methods 0.000 description 3
- 239000011248 coating agent Substances 0.000 description 3
- 238000010924 continuous production Methods 0.000 description 3
- 238000013461 design Methods 0.000 description 3
- 239000012467 final product Substances 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 239000002351 wastewater Substances 0.000 description 3
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- PWHULOQIROXLJO-UHFFFAOYSA-N Manganese Chemical compound [Mn] PWHULOQIROXLJO-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 238000000498 ball milling Methods 0.000 description 2
- 238000005452 bending Methods 0.000 description 2
- 238000001739 density measurement Methods 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 238000000840 electrochemical analysis Methods 0.000 description 2
- 239000007772 electrode material Substances 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 238000000445 field-emission scanning electron microscopy Methods 0.000 description 2
- 239000011888 foil Substances 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 238000003780 insertion Methods 0.000 description 2
- 230000037431 insertion Effects 0.000 description 2
- 239000010808 liquid waste Substances 0.000 description 2
- SGDQMWNLOSTDSU-UHFFFAOYSA-L lithium;sodium;sulfate Chemical compound [Li+].[Na+].[O-]S([O-])(=O)=O SGDQMWNLOSTDSU-UHFFFAOYSA-L 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 238000004806 packaging method and process Methods 0.000 description 2
- 238000000634 powder X-ray diffraction Methods 0.000 description 2
- 238000004886 process control Methods 0.000 description 2
- 238000011084 recovery Methods 0.000 description 2
- 239000013074 reference sample Substances 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 2
- 230000007704 transition Effects 0.000 description 2
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- KMTRUDSVKNLOMY-UHFFFAOYSA-N Ethylene carbonate Chemical compound O=C1OCCO1 KMTRUDSVKNLOMY-UHFFFAOYSA-N 0.000 description 1
- 229910008039 Li-M-O Inorganic materials 0.000 description 1
- 229910001290 LiPF6 Inorganic materials 0.000 description 1
- 239000007832 Na2SO4 Substances 0.000 description 1
- 229910016438 Ni0.42Mn0.42Co0.16 Inorganic materials 0.000 description 1
- 229910016774 Ni0.5Mn0.3Co0.2 Inorganic materials 0.000 description 1
- 229910017095 Ni0.6Mn0.2Co0.2 Inorganic materials 0.000 description 1
- 229910015228 Ni1/3Mn1/3CO1/3 Inorganic materials 0.000 description 1
- 229910018539 Ni—Mn—Co Inorganic materials 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 238000000333 X-ray scattering Methods 0.000 description 1
- KFDQGLPGKXUTMZ-UHFFFAOYSA-N [Mn].[Co].[Ni] Chemical compound [Mn].[Co].[Ni] KFDQGLPGKXUTMZ-UHFFFAOYSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 150000008051 alkyl sulfates Chemical class 0.000 description 1
- 239000001099 ammonium carbonate Substances 0.000 description 1
- 235000012501 ammonium carbonate Nutrition 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- GKPXMGUNTQSFGA-UHFFFAOYSA-N but-2-ynyl 1-methyl-3,6-dihydro-2h-pyridine-5-carboxylate;4-methylbenzenesulfonic acid Chemical group CC1=CC=C(S(O)(=O)=O)C=C1.CC#CCOC(=O)C1=CCCN(C)C1 GKPXMGUNTQSFGA-UHFFFAOYSA-N 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 239000006182 cathode active material Substances 0.000 description 1
- 210000005056 cell body Anatomy 0.000 description 1
- 239000000919 ceramic Substances 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 229910017052 cobalt Inorganic materials 0.000 description 1
- 239000010941 cobalt Substances 0.000 description 1
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 1
- MEYVLGVRTYSQHI-UHFFFAOYSA-L cobalt(2+) sulfate heptahydrate Chemical compound O.O.O.O.O.O.O.[Co+2].[O-]S([O-])(=O)=O MEYVLGVRTYSQHI-UHFFFAOYSA-L 0.000 description 1
- 238000002485 combustion reaction Methods 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- IEJIGPNLZYLLBP-UHFFFAOYSA-N dimethyl carbonate Chemical compound COC(=O)OC IEJIGPNLZYLLBP-UHFFFAOYSA-N 0.000 description 1
- 238000007599 discharging Methods 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 239000003517 fume Substances 0.000 description 1
- 229910002804 graphite Inorganic materials 0.000 description 1
- 239000010439 graphite Substances 0.000 description 1
- 230000035876 healing Effects 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000007689 inspection Methods 0.000 description 1
- 238000009533 lab test Methods 0.000 description 1
- 238000006138 lithiation reaction Methods 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- ISPYRSDWRDQNSW-UHFFFAOYSA-L manganese(II) sulfate monohydrate Chemical compound O.[Mn+2].[O-]S([O-])(=O)=O ISPYRSDWRDQNSW-UHFFFAOYSA-L 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 229910001510 metal chloride Inorganic materials 0.000 description 1
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 1
- 238000003801 milling Methods 0.000 description 1
- RRIWRJBSCGCBID-UHFFFAOYSA-L nickel sulfate hexahydrate Chemical compound O.O.O.O.O.O.[Ni+2].[O-]S([O-])(=O)=O RRIWRJBSCGCBID-UHFFFAOYSA-L 0.000 description 1
- 229940116202 nickel sulfate hexahydrate Drugs 0.000 description 1
- 231100000989 no adverse effect Toxicity 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 238000007086 side reaction Methods 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 238000003746 solid phase reaction Methods 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000013112 stability test Methods 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- OMLHKGLZLXMRON-UHFFFAOYSA-N sulfane;dihydrate Chemical compound O.O.S OMLHKGLZLXMRON-UHFFFAOYSA-N 0.000 description 1
- 229910021653 sulphate ion Inorganic materials 0.000 description 1
- 239000002344 surface layer Substances 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 238000010345 tape casting Methods 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- 229910000314 transition metal oxide Inorganic materials 0.000 description 1
- 229910000385 transition metal sulfate Inorganic materials 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
- 238000004065 wastewater treatment Methods 0.000 description 1
- 238000004260 weight control Methods 0.000 description 1
- 238000004804 winding Methods 0.000 description 1
Images




























Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/50—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of manganese
- H01M4/505—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of manganese of mixed oxides or hydroxides containing manganese for inserting or intercalating light metals, e.g. LiMn2O4 or LiMn2OxFy
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G53/00—Compounds of nickel
- C01G53/006—Compounds containing, besides nickel, two or more other elements, with the exception of oxygen or hydrogen
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G53/00—Compounds of nickel
- C01G53/06—Carbonates
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G53/00—Compounds of nickel
- C01G53/40—Nickelates
- C01G53/42—Nickelates containing alkali metals, e.g. LiNiO2
- C01G53/44—Nickelates containing alkali metals, e.g. LiNiO2 containing manganese
- C01G53/50—Nickelates containing alkali metals, e.g. LiNiO2 containing manganese of the type [MnO2]n-, e.g. Li(NixMn1-x)O2, Li(MyNixMn1-x-y)O2
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
- H01M10/0525—Rocking-chair batteries, i.e. batteries with lithium insertion or intercalation in both electrodes; Lithium-ion batteries
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/52—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of nickel, cobalt or iron
- H01M4/525—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of nickel, cobalt or iron of mixed oxides or hydroxides containing iron, cobalt or nickel for inserting or intercalating light metals, e.g. LiNiO2, LiCoO2 or LiCoOxFy
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/50—Solid solutions
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/70—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/70—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data
- C01P2002/72—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data by d-values or two theta-values, e.g. as X-ray diagram
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/01—Particle morphology depicted by an image
- C01P2004/03—Particle morphology depicted by an image obtained by SEM
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/51—Particles with a specific particle size distribution
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/60—Particles characterised by their size
- C01P2004/61—Micrometer sized, i.e. from 1-100 micrometer
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/80—Particles consisting of a mixture of two or more inorganic phases
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/11—Powder tap density
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/12—Surface area
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/40—Electric properties
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/80—Compositional purity
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M2004/026—Electrodes composed of, or comprising, active material characterised by the polarity
- H01M2004/028—Positive electrodes
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
Abstract
Description
2NaHCO3+MSO4→Na2SO4+MCO3+H2CO3 (1)
このプロセスの欠点は、低い効率である。1kgのMCO3を析出させるために、典型的には約1.5kgの重炭酸ナトリウムが必要とされ、一方、炭酸塩プロセス
Na2CO3+MSO4→Na2SO4+MCO3 (2)
は、はるかに少ない量、わずか約900gの炭酸塩を必要とする。更に、重炭酸塩の溶解度は、炭酸ナトリウムの溶解度(約400g/L)よりも非常に低い(90℃で約200g/L)。このことは、炭酸塩析出と比べて、溶液1リットル当たりの重炭酸塩プロセスの最大収率が、3倍低く、これは、濾過及び廃水処理のコストを大幅に増大させ、重炭酸プロセスを完全には競合できないものとなることを意味する。
並びにx+y+z+v=1である。別の実施形態において、炭酸塩前駆体化合物は、一般式MCO3を有することができ、式中、M=NixMnyCOzAvであり、Aはドーパントであり、0.30≦x≦0.60、0.20≦y≦0.50、及び0.1≦z≦0.40であり、v≦0.05、並びにx+y+z+v=1である。1つのサブ実施形態において、v=0である。更に別の実施形態において、炭酸塩前駆体化合物は、一般式MCO3を有することができ、式中、M=NixMnyCOzAvであり、Aはドーパントであり、0.10≦x≦0.30、0.55≦y≦0.80、及び0.1≦z≦0.30であり、v≦0.05、並びにx+y+z+v=1である。1つのサブ実施形態において、v=0である。いくつかの実施形態において、ドーパントAは、Mg、Al、Ti、Zr、Ca、Ce、Cr、Nb、Sn、Zn及びBのうちのいずれか1つ以上であってもよい。
0.10≦a≦0.25、0.10≦x≦0.30、0.55≦y≦0.80、及び0≦z≦0.30である。
1<Na/S<2であり、かつ粉末が、Li2SO4を更に含むかのいずれかであってもよい。
Ni、Mn及びCoイオン並びにA源を含む供給溶液を用意するステップと、
炭酸塩溶液及びNaイオンを含むイオン溶液を用意するステップと、
M’イオンを含むシードを含むスラリーを用意するステップであって、M’=Nix’Mny’COz’A’n’であり、A’がドーパントであり、0≦x’≦1、0≦y’≦1、0≦z’≦1、0≦n’≦1及びx’+y’+z’+n’=1である、ステップと、
反応器内で供給溶液、イオン溶液及びスラリーを混合し、それによって反応性液体混合物を得るステップと、
反応性液体混合物中のシードに炭酸塩を析出させ、それによって反応済み液体混合物及び炭酸塩前駆体を得るステップと、
反応済み液体混合物から炭酸塩前駆体を分離するステップと、を含む方法を提供することができる。一実施形態において、このシードは、0.1〜3μmの中央粒径D50を有する。別の実施形態では、M’イオンは、M’CO3、M’(OH)2、M’酸化物及びM’OOHのうちのいずれか1つである水不溶性化合物中に存在する。Ni、Mn、Co、及びAイオンは、水溶性硫酸塩化合物に存在する可能性がある。更に別の実施形態において、シードスラリー中の金属含有量の供給溶液中の金属含有量に対するモル比(M’seeds/Mfeed)は0.001〜0.1であり、炭酸塩前駆体の中央粒径は、比M’seeds/Mfeedによって決定される。特定の実施形態において、M=M’である。
本発明の第1の態様による炭酸塩前駆体を用意するステップと、
Li前駆体化合物を用意するステップと、
M−炭酸塩とLi前駆体とを混合するステップと、
この混合物を最大で500℃まで加熱するステップであって、300〜500℃の温度上昇が少なくとも1時間行われるステップと、
この混合物を600〜1100℃の温度で少なくとも1時間焼成するステップと、を含む方法を提供することができる。
A Na2CO3+B MSO4→Na2SO4+{M1−2xNa2x}{(CO3)1−y(SO4)y} (3)である。
[実施例1]Na及び硫黄含有炭酸塩の析出
酸対塩基の流量比を変化させて、第1のシリーズの析出(7つのサンプル)を以下の通りに行った:
1)M=Ni0.42Mn0.42Co0.16(552)を有する混合金属硫酸塩の溶液を調製する。金属濃度は2モル/リットルである。
2)1リットル当たり2モルのCO3の濃度で、Na2CO3溶液を調製する。
3)金属硫酸塩及び炭酸塩溶液の連続流を、厳密な撹拌(1000rpm)の下で水を含有する反応器に供給する。反応器を90℃に保つ。総流量を、反応器の容積を、2.8時間以内に置き換えるように選択する。塩基対酸(CO3/SO4)のモル流量比を、0.92〜1.35の値に設定する。流量を重量測定により制御しかつ一定に定める。析出を6時間行う。
4)各動作のそれぞれの時間の後に、少量の試験サンプルを採取する(サンプル1、2、...6を得る)。スラリー内の炭酸塩析出物の粒径を、レーザー回折によってチェックする。
5)最終サンプルを、4時間から6時間まで採取する。最終サンプルを水中で繰り返し洗浄して、あらゆる残存する塩を除去し、濾過し、120℃の空気で乾燥させる。
6)濾過溶液を採取し、非析出金属の含有量を決定するために検査する。過剰の塩基(Na2CO3)もpH滴定によりチェックする。
この実施例においては、析出条件を変更して、Na及びS不純物についての一般的な傾向から派生する可能性をCO3/M流量比の関数として検討する。一部の場合では、Na2CO3の10%が2NaHCO3によって置き換えられる(この場合、Na濃度は、4モル/Lに固定され、流量比は、0.5*Na/SO4と定義される)。一部の場合では、Na2CO3の10%がNaOH3によって置き換えられる(1モルのNa2CO3当たり2モルのNaOH)。一部の場合では、析出温度が変更され(25℃に)、一部の場合では、反応物の濃度が変更され、一部の場合では、シーディング技術が適用され、一部の場合では、反応器の形状が変更され、一部の場合では、滞留時間が変更される。ほとんどの実験について、金属組成NMC=552が使用される。結論は、一般的に、MCO3は不純物を含有し、どんな場合にも不純物を含まないMCO3が得られないということである。図5は、これらの結果をまとめている。
この実施例は、金属炭酸塩沈殿物のPSDを制御することの困難さを示す。好ましい沈殿プロセスは、連続プロセスである(連続流動式反応器としても知られる)。図6は、以下の基準を備えた、連続撹拌槽型反応器(CSTR)の設計を示す。
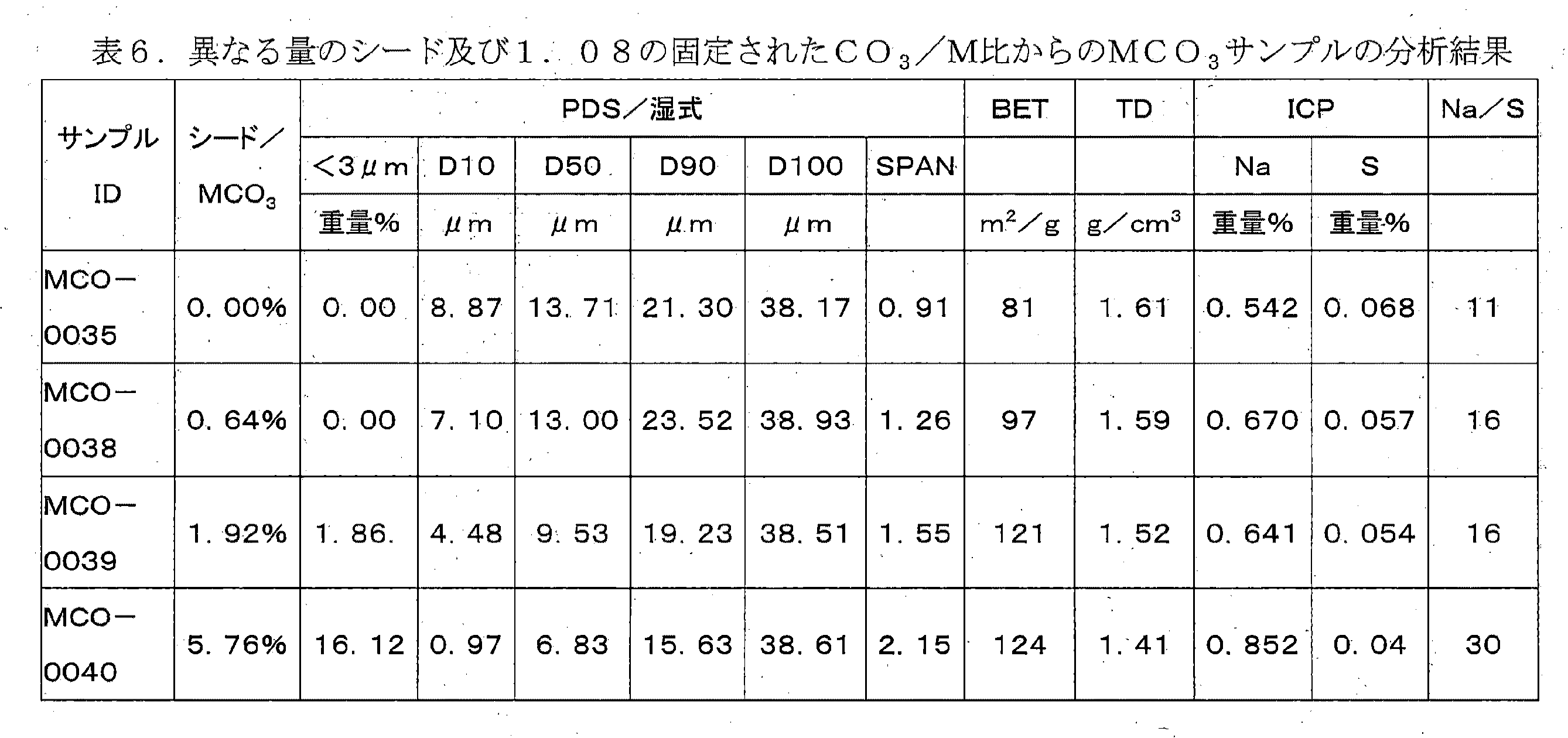
混合水酸化物のための典型的な析出プロセスは、流量(酸対塩基)比を注意深く制御することによって粒径が調整される連続析出である。このアプローチは、ある特定の流量比に関して、明確な定常状態の粒径が得られるという事実に基づく。したがって、塩基/酸比が増加する場合、典型的には、定常状態中の析出物のPSDは減少し、そのため、酸対塩基の流量比の小さな変動が、狭い所望の範囲において粒径を制御するために利用される。基礎となる科学的根拠は、核生成速度のpHへの依存性である。pHが上昇すると、核生成速度は増加し、粒径は減少する。この実施例は、このようなプロセスが、ナトリウム及び硫黄含有混合炭酸塩に対しては実際に不可能であることを示すであろう。本発明の場合、流量比は、所望のナトリウム対硫黄比を達成する必要性によって決定され、流量比はこの意味で調整される。したがって、PSDは、不純物の制御と独立して制御され得ない。実施例4に示すように、これは、連続析出中に得られる粒径が、Na/S比が析出中の決定的な要因である場合には非常に大きいためである。表4は、析出の6時間後の最終的PSDパラメータを示し、Mは、実施例1の552組成である。ほとんどの場合、定常状態には達していなかったため、D50は、析出が継続するならばさらに成長することになる。所望のPSDが10μmであるならば、これは、0.97未満のCO3/Mを選択するだけで達成することができる。しかしながら、これらの条件においては、沈殿した水酸化物の硫黄不純物が非常に高く、Na対S値は所望の0.4比未満である。図8は、D50値の増加を流量CO3/Mの関数として示す。明らかに、Na/S不純物範囲(実施例1、2を参照)並びにPSD範囲(実施例4、5を参照)の両方が、同じ流量比に強く依存する。したがって、所望の不純物レベルを有するMCO3前駆体を析出させ、それと同時に所望の粒径を達成することは不可能である。
前の実施例で示したように、金属炭酸塩析出プロセス(Na2CO3+MSO4→Na2SO4+MCO3)の問題のうちの1つは、PSD制御である。粒径が流量制御((OH)2/M)によって制御される水酸化物析出の場合とは対照的に、炭酸塩析出の場合には、この析出プロセスが、金属水酸化物析出プロセスよりも流量制御に対してはるかに敏感であるために、金属炭酸塩前駆体の異なるサイズを容易に生成することができない。同時係属中の欧州特許第14188028.6号に開示されているように、金属炭酸塩析出中のシーディング技術が、粒径を正確に制御し、定常状態のプロセスに容易に達することを可能にすることが見出されている。
シード調製プロセス:ステップ(1)及び(2)
ステップ(1):金属炭酸塩シードのボールミルプロセス:以前に調製された金属炭酸塩粉末を、ボトル中でセラミックボールで3日間ボールミルする。
ステップ(2):ボールミルした金属炭酸塩スラリーをボトルから採取し、その後ふるい分けする。シーディング技術を用いる金属炭酸塩析出プロセス:ステップ(3)〜(5)
ステップ(3):金属硫酸塩の溶解プロセス:硫酸ニッケル六水和物、硫酸マンガン一水和物及び硫酸コバルト七水和物を、H2O中に溶解する。この溶液の典型的な濃度は2モル/Lである。
ステップ(4):Na2CO3を用いる金属炭酸塩前駆体の析出プロセス:金属炭酸塩析出の典型的な温度は90℃である。CSTR反応器においては、撹拌速度は1000RPMである。滞留時間は2時間である。金属炭酸塩シードスラリーを、1時間に1回反応器に添加する。
ステップ(5):金属炭酸塩前駆体の洗浄及び乾燥プロセス:洗浄用に脱イオン水を使用する。得られたウェットケーキを150℃で16時間超にわたって乾燥させる。
PSDの制御のほかに、金属炭酸塩の析出プロセスの別の問題は、不純物の制御である。硫黄含有量を低下させるために、苛性洗浄が適用され、洗浄された金属炭酸塩前駆体は、表7の苛性洗浄の結果から分かるように、金属水酸化物前駆体と比べて比較的低い硫黄含有量を有する。しかし、金属炭酸塩前駆体のナトリウム含有量は、予想されたものよりも高い。化学作用がナトリウム含有量を低下させることができるかどうか、イオン交換実験で検討するべきである。したがって、この実施例は、不純物を除去する試みで、高いNa対S不純物比を有する前駆体に焦点を当てる。このような前駆体は、流量比CO3/M>1.00に対して得られる。これは大量生産のために興味深く、なぜなら、このような状況では、全ての遷移金属は沈殿し、廃水中の少量の残存するNa2CO3は問題ではないからである。イオン交換実験における洗浄時間、温度、及び添加剤の種類を制御することによって、ナトリウム含有量を低下させようと努力された。しかしながら、これがあまりに困難であることが証明され、ナトリウム不純物を効果的に低減させるためには、あまりに多くの時間、又はあまりに高価な化学物質が必要とされる。
金属炭酸塩前駆体を、4Lの撹拌(1000rpm)反応器を使用して調製する。温度は90℃である。水中に溶解したNa2CO3及びMSO4の正確に制御された2つの流れを、反応器に連続して注入する。塩基対酸の流量CO3/Mは1.03である。MSO4の流れの金属組成は、M=NMC552である。Na2CO3及びMSO4の流れの濃度は、2モル/Lである。滞留時間、すなわち、1つの反応器内容物を置き換えるのに要する時間は、2.75時間である。シード処理技術を使用する。シードは、初期の沈殿から得られたMCO3をボールミリングすることによって得る。シードのD50は、0.5μmである。シードを含有するスラリーを反応器に頻繁に注入し、注入されたシードと析出生成物との間の重量比は0.63%である。反応器の半分が水で満たされた後に、析出を開始する。析出は12時間にわたって行う。オーバーフローする生成物の採取を、4時間目から開始する。12時間後に、反応器の内容物並びに採取されたオーバーフローを繰り返し焼成し、水中で洗浄する。析出を、正確に同様な様式で数回繰り返し、十分な量の生成物を得る。析出中に、PSDを毎時間チェックする。析出プロセスが、2μm未満まで変化するD50で、非常に安定であることが見出され、得られたD50は13±1.5μmであった。濾過及び洗浄後に、生成物を、120℃の空気中で一晩乾燥させる。
1)ナトリウム及び硫黄含有炭酸塩(LX0142、LX0143)前駆体から得られたNMCの表面積が、高密度M(OH)2前駆体から作製された基準NMCのものよりも著しく大きい。SEM顕微鏡写真は、大きなBET表面積は、開放細孔に由来し、これは断面SEMによって確認されることを強く示唆する。
2)開放細孔性及び高BETが、電気化学的性能を著しく改善する。LX0142&143の不可逆容量は、参照LX0031のもよりも著しく小さい。所定の組成については、充電容量が、程度の差はあるが固定値である。故に、不可逆容量が減少する場合、可逆容量は増加する。したがって、ほぼ2.5%低い(=9.94〜12.44)不可逆容量を有するLX0143は、参照と比較して対応する増加した(3.2%)可逆容量をもたらす。
3)不純物含有サンプルLX0143が、不純物を含まないサンプルLX0142よりも1.4%低い容量を有する。より低い容量は、不活性アルキル硫酸塩の存在と一致する。ナトリウム及び硫黄不純物は、硫酸塩Li2NaSO4、LiNaSO4又はNa2SO4として存在し、これらは可逆容量に寄与しない。1.3重量%の塩が存在すると推定される。このことは、LX0142と比べてのサンプルLX0143の観測された、より低い容量(−1.4%)を完全に説明する。
4)LX0142及びLX0143のコインセル試験が、不純物を含まないLX0142と比較して、不純物含有LX0143の良好なバッテリーサイクル安定性を示す。
フルセルを作製する。フルセルは、巻かれたパウチセル型のものであり、約650mAhの容量を有する。3種の異なるカソード材料を試験する:フルセルロット番号AL705は、水酸化物から得られた基準NMCであるLX0031を含有する。フルセルロット番号AL885は、不純物を含まないLX0142を含有し、AL886は、不純物含有カソードLX0143を含有する。全体的に、好ましい領域内にナトリウム及び硫黄不純物を有する所望の形態を備えたNMCであるLX0143を含有するAL886は、優れた結果を示す。セルの以下の詳細において、作製及び試験が列挙され説明される。
フルセル試験目的のために、調製された正極(カソード)が、典型的にはグラファイトタイプのカーボンである負極(アノード)、及び多孔性電気的絶縁膜(セパレータ)と共に組み立てられる。フルセルは、以下の主要ステップにより作製される。(a)電極をスリッティングする、(b)電極を乾燥させる、(c)ジェリーロールに曲げる、(d)パッケージングする。
(a)電極をスリッティングする:NMP又は水系コーティング後、電極活物質は、スリッティングの機械によりスリッティングされ得る。電極の幅及び長さは、バッテリー用途により決定される。
(b)タップを取り付ける:2種類のタップが存在する。アルミニウムタップは正極(カソード)に取り付けられ、銅タップは負極(アノード)に取り付けられる。
(c)電極を乾燥させる:準備された正極(カソード)及び負極(アノード)は、85℃〜120℃で8時間、真空オーブンにおいて乾燥される。
(d)ジェリーロールに曲げる:電極を乾燥後、ジェリーロールが、巻き線機を使用して作製される。
ジェリーロールは少なくとも負極(アノ−ド)、多孔性電気的絶縁膜(セパレータ)、及び正極(カソード)からなる。
(e)パッケージングする:作製されたジェリーロールが、アルミニウム積層フィルムパッケージと共に800mAhのセルに組み込まれ、パウチセルになる。更に、ジェリーロールは、電解質によって含浸される。電解質の量は、正極及び負極、並びに多孔性セパレータの多孔性及び寸法によって算出される。最終的に、パッケージ化されたフルセルは、封止機械によって封止される。
多くの異なるフルセルの評価試験が可能である。本発明は、(a)サイクル安定性、(b)容量及びレート特性、(c)膨張、(d)保管試験、及び(e)DCR抵抗試験の結果を示す。
(a)サイクル安定性:セルは、数百サイクルにわたって完全に充放電される。サイクル試験は、望まない副反応を速めるために、25℃又は高温(例えば45℃)で実行され、これにより、より速い容量損失を強いる。
(b)容量及びレート特性:容量は、0.2Cレートのレートで、4.3V〜2.7Vで測定された放電容量である。効率は、第1の充電と第1の放電容量との間の%で表される比である。レート特性は、0.2Cでのレートの百分率として表される、0.5;1.0;2.0;3.0及び4.0Cのレートにおける放電容量である。0.2Cは、5時間以内に充電されたセルを放電する電流に相当する。例えば、1Cは、0.2Cの電流の5倍大きい電流である。
(c)膨張:パウチセルは、完全に充電され、90℃に加熱されたオーブン内に挿入され、数時間の間その温度に保たれる。充電されたカソードは、90℃で電解質と反応しガスを発生させる。発生ガスが膨張を生じさせる。実施例において、高温への曝露の4時間後に測定された厚さ増加(=膨張)の値を報告する。膨張は多くの用途に関連する問題であり、更に、本執筆者らは、膨張がコーティング中の水への曝露に起因する最終的な表面損傷を検知するための非常に高感度な方法であると予想している。
(d)保管試験、すなわち、残存及び回復容量:セルは完全に充電され、1ヶ月間、60℃で保管される。1ヶ月後、セルは60℃のチャンバから取り除かれ、25℃で試験される。セルは、放電され、放電中、残存容量が測定される。再充電された後、セルは放電され、回復された容量が得られる。この容量チェック後、60℃での保管は更に1ヶ月続けられ、残存及び容量回復率が再び測定され、次いでセルは、3回目保管され、再び測定される。多くの用途との関連性に加えて、保管実験はまた、水系コーティング中のカソードの損傷を評価するためのとても高感度なツールである。
(e)保管試験と連動したDCR抵抗試験:60℃での、1、2、及び3ヶ月保管後の容量測定に加えて、セルのDCR抵抗及びDCRの経時的な発生(初期DCRに対する%として表される)が測定される。DCR抵抗は、電流パルスに対する電圧応答から得られ、使用される手順は、USABC標準(United States Advanced Battery Consortium LLC)によるものである。データは、バッテリー寿命を予知するために、消失する割合を未来に当てはめるのに使用され得ることから、DCR抵抗は、実用的用途にとても関連しており、更には、電解質とアノード又はカソードとの反応の反応生成物が、低導電性表面層として沈殿するので、DCR抵抗は、電極に及ぶ損傷を検知するのにとても感度がよい。
ナトリウム及び硫黄含有MCO3がNMCカソードを作製するために使用されるとき、ナトリウム及び硫黄の大部分が、最終サンプル中に不純物として残存する。ナトリウム対硫黄不純物の比が1未満である場合、ナトリウム不純物がLi2SO4及びLiNaSO4として存在すると予想される。ナトリウム対硫黄不純物の比が1〜2である場合、Na2SO4及びLiNaSCvが共存すると予想される。LiNaSCvは、ICSD番号3814で、結晶学データベースにおいて見出すことができる。主ピークは20=23°周辺にある。これを確認するために、Na2S2O8をLiCO3と400℃において反応させることによってLiNaSO4を調製した。反応式は、Li2CO3+Na2S2O8→2LiNaSO4+CO2+1/2O2である。基本的に、単相のLiNaSO4を得た。図18は、XRDパターンを、ICDSデータベースから番号3814についての格子定数及び原子位置を用いて算出されたパターンと共に示す。LiNaSO4相の主ピークは、2θの23.44°にある。その他の強いピークは、32.74°及び22.66°にある。特に、ナトリウム対硫黄比が、ほぼ1である場合(この場合には、不純物相の大部分がLiNaSO4である)、不純物相のX線散乱は、粉末XRDによって明らかに検出されるのに十分に強い可能性がある。他方では23.44、32.74及び22.66度において異なるピークを検出することは、LiNaSO4相が存在していることの有力な証拠となる。
NMC=261を有するパイロットプラント前駆体を、実施例1&6で説明したシーディング技術を使用して得た。図19は、この前駆体のSEM形態を示す。この前駆体は、3216ppmのナトリウムと5280ppmの硫黄とを含有した(ICPによって測定)。この前駆体をLi2CO3とブレンドした。Li:Mブレンド比は、1.468である。1kgを調製した。このブレンドを、800℃までゆっくりと加熱し、次いで10時間焼成する。冷却後、生成物をふるい分けし、サンプルHLM330を得る。最終生成物の形態を図20に示す。所望の形態が達成されていた。粒子は球状であり、比較的高密度であるが、同時に顕著な細孔性を呈する。BET表面積は比較的大きく、4.8m2/gであり、これは同じ形状の高密度粉末のものよりも非常に大きく、開放メソ細孔構造の存在を示唆している。焼成後に、全体で2重量%の不純物が予想され、不純物の86%(17625ppm)はLiNaSO4として存在し、14%がLi2SO4として存在する。一般に、ナトリウム対硫黄比がほぼ1である場合に、我々はLiNaSO4による最高の寄与を予想している。この材料は、コインセル(実施例8に記載されるように作製された)において優れた性能を有した。80mA/gの電流を使用して、室温(25℃)において3.0〜4.6Vで試験を行ったとき、291mAh/gの可逆容量及び5.4%の不可逆容量が達成される。M(OH)2から調製された同様な材料からこれまでに達成されたいかなるデータよりも著しく高い優れた結果が得られる。サイクル安定性も満足し得るものである:100サイクル当たり16%の消失。
MCO3前駆体を、NMC=552の代わりに、NMC=532の組成を有する炭酸塩が析出される以外は、実施例1に記載されたものと同様な方法によって作製する。シードは、前の析出から得られたボールミリングされたMCO3から得る。塩基/酸(CO3/M)流量比を、1.03に設定する。析出された炭酸塩(サンプルMCO−0099ak)は、D50=16μmを有する。BET=ABET=144m2/gである、著しいナノ細孔性を伴う良好な球状形態が観測された。図23は、析出された炭酸塩のSEMを示す。しかしながら、ICPは、ナトリウム対硫黄不純物比が所望の領域内0.4<Na/S<2にはないことを示した。不純物含有量は、5370ppmのNa及び2400ppmの硫黄である。これは、ナトリウム対硫黄不純物比が3超をもたらす。
サンプルの作製を繰り返した。MCO3前駆体MCO−0099akをLi2CO3とブレンドする。Li:Mブレンド比は1.02である。しかし、ここで、1遷移金属当たり2モル%のLi2SO4をブレンドに添加する。硫酸塩の添加後に、ブレンド中のナトリウム対硫黄不純物比は、所望の範囲0.4〜2.0の範囲内である。このブレンドを875℃で10時間にわたって焼成し、サンプルEX1534を得る。図24は、このサンプルのSEMを示す。図25は、サンプルEX1534の粉末X線回折パターンを示す。スキャン条件は、2時間のスキャン、0.02°のステップ、15〜85°であった。ピークが22.58、23.22、29.53、30.39及び32.53°において観測されたために、明らかに、LiNaSO4二次相が存在する。y軸は、小ピークを強調するために対数目盛で示されている。
実施例12においてサンプルEX1518及びEX1519について不良な結果を得た。この性能が、あまりに高いナトリウム対硫黄不純物比によって引き起こされるかどうかを更に検討するために、所望の領域内よりも低い比を有する前駆体を選択し、実験を繰り返す。
1)塩基対酸流量比(CO3/M)を、1.00に調整して、所望の0.4<Na/S<2領域内のナトリウム及び硫黄不純物の良好なバランスを達成する。
2)シーディングは適用しない。
3)析出は、6時間行い、サンプルを4時間目から採取することを始める。
NMC=111組成を有するナトリウム及び硫黄含有炭酸塩前駆体を、実施例1に記載される通りに作製する。塩基/酸流量比(CO3/M)を1.0として選択して、所望の領域0.4<Na/S<2内のナトリウム対硫黄不純物比を達成する。得られたサンプルMCO−0114gは、2890ppmのナトリウムと3660ppmの硫黄とを含有し、すなわち、ナトリウム対硫黄不純物比は、1.1である。この前駆体をLi2CO3と混合する。Li:Mのブレンド比は、1.1である。このブレンドを、空気流中で850℃で10時間にわたって焼成し、サンプルMX0809を得る。
(a)MCO3をベースとした前駆体の使用に由来する開放メソ細孔性と、
(b)好ましい不純物比の範囲内のナトリウム及び硫黄不純物を有するMCO3前駆体とによるものとする。
前の実施例は、優れた電気化学的性能を有するNMCが、硫黄及びナトリウム含有炭酸塩前駆体から達成され得ることを示した。しかしながら、大量生産における典型的なM(OH)2の析出と比べて、MCO3の析出は、その低い体積効率において主な欠点を有する。主な理由は、NaOHと比べてのNa2CO3の非常に低い溶解度にある。典型的なM(OH)2析出において、供給材料は(1)10MのNaOH、(2)2MのMSO4、及び(3)10MのNH4OHとすることができる。析出は、以下の(簡略化)式に従う:
2NaOH+MSO4+NH4OH→Na2SO4+M(OH)2+NH4OH
Claims (20)
- リチウムイオンバッテリーにおいて活性正電極材料として使用可能なリチウム金属(M)酸化物粉末を製造するための炭酸塩前駆体化合物であって、Mが、20〜90モル%のNi、10〜70モル%のMn、及び10〜40モル%のCoを含み、前記前駆体が、ナトリウム及び硫黄不純物を更に含み、ナトリウム対硫黄のモル比(Na/S)が、0.4<Na/S<2である、炭酸塩前駆体化合物。
- 一般式MCO3を有し、式中、M=NixMnyCozAvであり、Aがドーパントであり、0.20≦x≦0.90、0.10≦y≦0.67、及び0.1≦z≦0.40であり、v≦0.05、並びにx+y+z+v=1である、請求項1に記載の炭酸塩前駆体化合物。
- Aが、Mg、Al、Ti、Zr、Ca、Ce、Cr、Nb、Sn、Zn、及びBのうちのいずれか1つ以上である、請求項2に記載の炭酸塩前駆体化合物。
- リチウムイオンバッテリーにおいて活性正電極材料として使用可能なリチウム金属(M)酸化物粉末を製造するための炭酸塩前駆体化合物であって、一般式MCO3を有し、式中、M=NixMnyCOzAvであり、Aが、ドーパントであり、0.10≦x≦0.30、0.55≦y≦0.80、及び0≦z≦0.30であり、v≦0.05、並びにx+y+z+v=1であり、前記前駆体が、ナトリウム及び硫黄不純物を更に含み、ナトリウム対硫黄のモル比(Na/S)が、0.4<Na/S<2である、炭酸塩前駆体化合物。
- 重量%で表されるナトリウム(Nawt)含有量と硫黄(Swt)含有量との和(2*Nawt)+Swtが、0.4重量%超かつ1.6重量%未満である、請求項1に記載の炭酸塩前駆体化合物。
- ナトリウム含有量が、0.1〜0.7重量%であり、硫黄含有量が、0.2〜0.9重量%である、請求項5に記載の炭酸塩前駆体化合物。
- 再充電可能なバッテリーにおける正電極材料のためのリチウム金属酸化物粉末であって、一般式Li1+aMi1−aO2を有し、式中、M=NixMnyCOzAvであり、Aがドーパントであり、−0.05≦a≦0.25、0.20≦x≦0.90、0.10≦y≦0.67、及び0.10≦z≦0.40であり、v≦0.05、並びにx+y+z+v=1であり、前記粉末が、10μm≦D50≦20μmの粒径分布、0.9≦BET≦5の比表面を有し、前記BETが、m2/gで表され、前記粉末が、ナトリウム及び硫黄不純物を更に含み、重量%で表されるナトリウム(Nawt)含有量と硫黄(Swt)含有量との和(2*Nawt)+Swtが、0.4重量%超かつ1.6重量%未満であり、ナトリウム対硫黄のモル比(Na/S)が、0.4<Na/S<2である、リチウム金属酸化物粉末。
- 二次LiNaSO4相を含む、請求項7に記載のリチウム金属酸化物粉末。
- 前記二次LiNaSO4相の相対重量が、前記粉末のXRDパターンのリートベルト解析によって決定して、少なくとも0.5重量%である、請求項7に記載のリチウム金属酸化物粉末。
- 0.4<Na/S<1であり、かつ前記粉末が、Na2SO4を更に含むか、又は
1<Na/S<2であり、かつ前記粉末が、Li2SO4を更に含むかのいずれかである、請求項7に記載のリチウム金属酸化物粉末。 - Aが、Mg、Al、Ti、Zr、Ca、Ce、Cr、Nb、Sn、Zn、及びBのうちのいずれか1つ以上である、請求項7に記載のリチウム金属酸化物粉末。
- 再充電可能なバッテリーにおける正電極材料のためのリチウム金属酸化物粉末であって、一般式Li1+aMi1−aO2を有し、式中、M=NixMnyCOzAvであり、Aがドーパントであり、0.10≦a≦0.25、0.10≦x≦0.30、0.55≦y≦0.80、及び0≦z≦0.30であり、v<0.05、並びにx+y+z+v=1であり、前記粉末が、10μm≦D50≦20μmの粒径分布、0.9≦BET≦5の比表面を有し、前記BETが、m2/gで表され、前記粉末が、ナトリウム及び硫黄不純物を更に含み、重量%で表されるナトリウム(Nawt)含有量と硫黄(Swt)含有量との和(2*Nawt)+Swtが、0.4重量%超かつ1.6重量%未満であり、ナトリウム対硫黄のモル比(Na/S)が、0.4<Na/S<2である、リチウム金属酸化物粉末。
- 請求項2に記載の炭酸塩前駆体化合物を調製するための方法であって、
Ni、Mn及びCoイオン並びにA源を含む供給溶液を用意するステップであって、前記Ni、Mn、Co及びAイオンが、水溶性硫酸塩化合物中に存在する、ステップと、
炭酸塩溶液及びNaイオンを含むイオン溶液を用意するステップと、
M’イオンを含むシードを含むスラリーを用意するステップであって、M’=Nix’Mny’Coz’A’n’であり、
A’がドーパントであり、0≦x’≦1、0≦y’≦1、0≦z’≦1、0≦n’≦1及びx’+y’+z’+n’=1である、ステップと、
反応器内で前記供給溶液、前記イオン溶液及び前記スラリーを混合し、それによって反応性液体混合物を得るステップと、
前記反応性液体混合物中のシードに炭酸塩を析出させ、それによって反応済み液体混合物及び前記炭酸塩前駆体を得るステップと、
前記反応済み液体混合物から前記炭酸塩前駆体を分離するステップと、
を含む方法。 - 前記M’イオンが、M’CO3、M’(OH)2、M’酸化物及びM’OOHのうちのいずれか1つである水不溶性化合物中に存在する、請求項13に記載の方法。
- 前記シードスラリー中の金属含有量の前記供給溶液中の金属含有量に対するモル比(M’seeds/Mfeed)が0.001〜0.1であり、前記炭酸塩前駆体の中央粒径が、比M’seeds/Mfeedによって決定される、請求項13に記載の方法。
- A及びA’が、Mg、Al、Ti、Zr、Ca、Ce、Cr、Nb、Sn、Zn、及びBのうちのいずれか1つ以上である、請求項13に記載の炭酸塩前駆体化合物。
- 前記反応器中のNH3の濃度が、5.0g/L未満である、請求項13に記載の方法。
- M=M’である、請求項13に記載の方法。
- 前記イオン溶液が、水酸化物及び重炭酸塩溶液のうちのいずれか一方又は両方を更に含み、比OH/CO3、又はOH/HCO3、あるいはこれらの比の両方が、1/10未満である、請求項13に記載の方法。
- 前記シードが、0.1〜3μmの中央粒径D50を有する、請求項13に記載の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14188045 | 2014-10-08 | ||
| EP14188045.0 | 2014-10-08 | ||
| PCT/IB2015/057492 WO2016055911A1 (en) | 2014-10-08 | 2015-09-30 | Impurity containing cathode material with preferred morphology and method to prepare from impurity containing metal carbonate |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2017536654A true JP2017536654A (ja) | 2017-12-07 |
| JP6482659B2 JP6482659B2 (ja) | 2019-03-13 |
Family
ID=51687864
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2017518496A Active JP6482659B2 (ja) | 2014-10-08 | 2015-09-30 | 好ましい形態を有する不純物含有カソード材料及び不純物含有金属炭酸塩から調製するための方法 |
Country Status (9)
| Country | Link |
|---|---|
| US (2) | US10411258B2 (ja) |
| EP (1) | EP3204973B1 (ja) |
| JP (1) | JP6482659B2 (ja) |
| KR (1) | KR102004625B1 (ja) |
| CN (1) | CN106795008B (ja) |
| HU (1) | HUE042152T2 (ja) |
| PL (1) | PL3204973T3 (ja) |
| TW (1) | TWI591883B (ja) |
| WO (1) | WO2016055911A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017530086A (ja) * | 2014-10-08 | 2017-10-12 | ユミコア | リチウム・ニッケル・マンガン・コバルト酸化物カソード材料用のカーボネート前駆体及びその製造方法 |
| JP2021515966A (ja) * | 2018-03-02 | 2021-06-24 | ユミコア | 充電式リチウムイオン電池用の正極材料 |
| US11271203B2 (en) | 2017-12-22 | 2022-03-08 | Umicore | Positive electrode material for rechargeable lithium ion batteries and methods of making thereof |
| JP2023510117A (ja) * | 2019-12-18 | 2023-03-13 | ユミコア | 充電式リチウムイオン電池用の粉末状リチウムコバルト系酸化物カソード活物質粉末及びその製造方法 |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109311696B (zh) * | 2016-07-20 | 2023-06-02 | 托普索公司 | 具有可调粒径分布的电池材料的可升级沉淀合成方法 |
| US11367872B2 (en) | 2017-03-03 | 2022-06-21 | Umicore | Precursor and method for preparing Ni based cathode material for rechargeable lithium ion batteries |
| JP6856763B2 (ja) * | 2017-03-03 | 2021-04-14 | ユミコア | 充電式リチウムイオン電池用のNi系カソード材料を調製するための前駆体及び方法 |
| EP3592706B1 (en) | 2017-03-08 | 2023-06-14 | Umicore | Precursors of cathode materials for a rechargeable lithium ion battery |
| WO2018167533A1 (en) * | 2017-03-14 | 2018-09-20 | Umicore | Precursors for cathode material with improved secondary battery performance and method to prepare the precursors |
| US20190123347A1 (en) * | 2017-07-14 | 2019-04-25 | Umicore | Ni based cathode material for rechargeable lithium-ion batteries |
| HUE051555T2 (hu) * | 2017-07-14 | 2021-03-01 | Umicore Nv | NI alapú katódanyag újratölthetõ lítium-ion akkumulátorokhoz |
| CN109987655B (zh) * | 2017-12-29 | 2021-10-01 | 荆门市格林美新材料有限公司 | 一种碱式碳酸镍的制备工艺 |
| KR20190106638A (ko) | 2018-03-09 | 2019-09-18 | 주식회사 엘지화학 | 리튬 이차전지 |
| WO2019172637A2 (ko) | 2018-03-09 | 2019-09-12 | 주식회사 엘지화학 | 리튬 이차전지 |
| EP3776695B1 (en) * | 2018-03-28 | 2023-08-02 | Umicore | Lithium transition metal composite oxide as a positive electrode active material for rechargeable lithium secondary batteries |
| KR20210006422A (ko) * | 2018-05-04 | 2021-01-18 | 유미코아 | 불소화 전해질을 포함하는 Ni계 리튬 이온 2차 배터리 |
| CN110137489B (zh) * | 2019-06-19 | 2021-06-29 | 厦门厦钨新能源材料股份有限公司 | 具有痕量金属杂质的镍钴锰酸锂材料、制备方法及应用 |
| EP3994747A1 (en) * | 2019-07-03 | 2022-05-11 | Umicore | Lithium nickel manganese cobalt composite oxide as a positive electrode active material for rechargeable lithium ion batteries |
| CN112713269B (zh) * | 2020-12-31 | 2021-10-29 | 浙江帕瓦新能源股份有限公司 | 一种降低正极材料前驱体中钠离子和硫酸根离子含量的生产系统和生产方法 |
| JP2024516811A (ja) * | 2021-08-25 | 2024-04-17 | エスエム ラブ コーポレーション リミテッド | 正極活物質、その製造方法、及びそれを含む正極を含むリチウム二次電池 |
| CN114751465B (zh) * | 2022-05-24 | 2023-06-27 | 荆门市格林美新材料有限公司 | 一种分阶段元素替代实现制备高Al均匀四氧化三钴方法 |
| WO2024066445A1 (zh) * | 2022-09-28 | 2024-04-04 | 中伟新材料股份有限公司 | 钠离子电池正极材料前驱体及其制备方法、钠离子电池正极材料、钠离子电池和涉电设备 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH09245787A (ja) * | 1996-03-07 | 1997-09-19 | Kansai Shokubai Kagaku Kk | リチウム二次電池用正極活物質 |
| JP2001273898A (ja) * | 2000-01-20 | 2001-10-05 | Japan Storage Battery Co Ltd | 非水電解質二次電池用正極活物質およびその製造方法並びにそれを使用した非水電解質二次電池 |
| WO2004064180A1 (ja) * | 2003-01-08 | 2004-07-29 | Nikko Materials Co., Ltd. | リチウム二次電池正極用の材料並びにその製造方法 |
| JP2006503789A (ja) * | 2002-10-31 | 2006-02-02 | エルジー・ケム・リミテッド | 金属成分の組成に勾配を有するリチウム遷移金属酸化物 |
| JP2011096650A (ja) * | 2009-09-30 | 2011-05-12 | Toda Kogyo Corp | 正極活物質粒子粉末及びその製造方法、並びに非水電解質二次電池 |
| JP2011198759A (ja) * | 2010-02-23 | 2011-10-06 | Toda Kogyo Corp | 正極活物質前駆体粒子粉末及び正極活物質粒子粉末、並びに非水電解質二次電池 |
| WO2013069454A1 (ja) * | 2011-11-09 | 2013-05-16 | 株式会社Gsユアサ | 非水電解質二次電池用活物質、その活物質の製造方法、非水電解質二次電池用電極及び非水電解質二次電池 |
| JP2014503451A (ja) * | 2010-11-25 | 2014-02-13 | ビーエーエスエフ ソシエタス・ヨーロピア | 遷移金属複合酸化物の前駆体の製造方法 |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3066915A (en) | 1957-11-18 | 1962-12-04 | Sr Linferd Linabery | Post puller device |
| CN100417595C (zh) | 2002-11-19 | 2008-09-10 | 比亚迪股份有限公司 | 由碳酸盐前躯体制备锂过渡金属复合氧化物的方法 |
| JP4336941B2 (ja) | 2003-01-06 | 2009-09-30 | 株式会社ジェイテクト | 負荷駆動回路 |
| JP4305629B2 (ja) * | 2003-03-27 | 2009-07-29 | 戸田工業株式会社 | 四酸化三マンガン粒子粉末及びその製造法、非水電解質二次電池用正極活物質及びその製造法、並びに非水電解質二次電池 |
| US7709149B2 (en) * | 2004-09-24 | 2010-05-04 | Lg Chem, Ltd. | Composite precursor for aluminum-containing lithium transition metal oxide and process for preparation of the same |
| CN101229928B (zh) | 2007-01-25 | 2010-04-07 | 湖南科力远新能源股份有限公司 | 一种球形镍钴锰酸锂材料的制备方法 |
| JP4968945B2 (ja) | 2008-02-01 | 2012-07-04 | 日本化学工業株式会社 | 複合炭酸塩およびその製造方法 |
| JP4968944B2 (ja) | 2008-02-01 | 2012-07-04 | 日本化学工業株式会社 | 複合炭酸塩およびその製造方法 |
| WO2010015368A1 (en) * | 2008-08-04 | 2010-02-11 | Umicore | Highly crystalline lithium transition metal oxides |
| EP2445041B1 (en) * | 2009-06-17 | 2016-04-13 | Sony Corporation | Nonaqueous electrolyte battery, positive electrode for nonaqueous electrolyte battery, negative electrode for nonaqueous electrolyte battery, separator for nonaqueous electrolyte battery, electrolyte for nonaqueous electrolyte battery, and method for producing separator for nonaqueous electrolyte battery |
| AU2011290195B2 (en) * | 2010-08-10 | 2014-04-10 | Agc Seimi Chemical Co., Ltd. | Production method for a composite compound comprising nickel and cobalt |
| WO2012132155A1 (ja) * | 2011-03-31 | 2012-10-04 | 戸田工業株式会社 | マンガンニッケル複合酸化物粒子粉末およびその製造方法、非水電解質二次電池用正極活物質粒子粉末およびその製造方法、ならびに非水電解質二次電池 |
| JP5921705B2 (ja) * | 2012-02-01 | 2016-05-24 | エルジー・ケム・リミテッド | リチウム複合遷移金属酸化物の前駆体製造用反応器及び前駆体の製造方法 |
-
2015
- 2015-09-30 KR KR1020177012208A patent/KR102004625B1/ko active IP Right Grant
- 2015-09-30 US US15/517,273 patent/US10411258B2/en active Active
- 2015-09-30 JP JP2017518496A patent/JP6482659B2/ja active Active
- 2015-09-30 CN CN201580055057.4A patent/CN106795008B/zh active Active
- 2015-09-30 EP EP15849645.5A patent/EP3204973B1/en active Active
- 2015-09-30 WO PCT/IB2015/057492 patent/WO2016055911A1/en active Application Filing
- 2015-09-30 PL PL15849645T patent/PL3204973T3/pl unknown
- 2015-09-30 HU HUE15849645A patent/HUE042152T2/hu unknown
- 2015-10-05 TW TW104132709A patent/TWI591883B/zh active
-
2019
- 2019-07-25 US US16/521,846 patent/US11462735B2/en active Active
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH09245787A (ja) * | 1996-03-07 | 1997-09-19 | Kansai Shokubai Kagaku Kk | リチウム二次電池用正極活物質 |
| JP2001273898A (ja) * | 2000-01-20 | 2001-10-05 | Japan Storage Battery Co Ltd | 非水電解質二次電池用正極活物質およびその製造方法並びにそれを使用した非水電解質二次電池 |
| JP2006503789A (ja) * | 2002-10-31 | 2006-02-02 | エルジー・ケム・リミテッド | 金属成分の組成に勾配を有するリチウム遷移金属酸化物 |
| WO2004064180A1 (ja) * | 2003-01-08 | 2004-07-29 | Nikko Materials Co., Ltd. | リチウム二次電池正極用の材料並びにその製造方法 |
| JP2011096650A (ja) * | 2009-09-30 | 2011-05-12 | Toda Kogyo Corp | 正極活物質粒子粉末及びその製造方法、並びに非水電解質二次電池 |
| JP2011198759A (ja) * | 2010-02-23 | 2011-10-06 | Toda Kogyo Corp | 正極活物質前駆体粒子粉末及び正極活物質粒子粉末、並びに非水電解質二次電池 |
| JP2014503451A (ja) * | 2010-11-25 | 2014-02-13 | ビーエーエスエフ ソシエタス・ヨーロピア | 遷移金属複合酸化物の前駆体の製造方法 |
| WO2013069454A1 (ja) * | 2011-11-09 | 2013-05-16 | 株式会社Gsユアサ | 非水電解質二次電池用活物質、その活物質の製造方法、非水電解質二次電池用電極及び非水電解質二次電池 |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017530086A (ja) * | 2014-10-08 | 2017-10-12 | ユミコア | リチウム・ニッケル・マンガン・コバルト酸化物カソード材料用のカーボネート前駆体及びその製造方法 |
| US11380882B2 (en) | 2014-10-08 | 2022-07-05 | Umicore | Carbonate precursors for lithium nickel manganese cobalt oxide cathode material and the method of making same |
| US11271203B2 (en) | 2017-12-22 | 2022-03-08 | Umicore | Positive electrode material for rechargeable lithium ion batteries and methods of making thereof |
| JP2021515966A (ja) * | 2018-03-02 | 2021-06-24 | ユミコア | 充電式リチウムイオン電池用の正極材料 |
| JP2021516422A (ja) * | 2018-03-02 | 2021-07-01 | ユミコア | 充電式リチウムイオン電池用の正極材料 |
| JP7055891B2 (ja) | 2018-03-02 | 2022-04-18 | ユミコア | 充電式リチウムイオン電池用の正極材料 |
| JP7091461B2 (ja) | 2018-03-02 | 2022-06-27 | ユミコア | 充電式リチウムイオン電池用の正極材料 |
| US11916224B2 (en) | 2018-03-02 | 2024-02-27 | Umicore | Positive electrode material for rechargeable lithium ion batteries |
| US11949096B2 (en) | 2018-03-02 | 2024-04-02 | Umicore | Positive electrode material for rechargeable lithium ion batteries |
| JP2023510117A (ja) * | 2019-12-18 | 2023-03-13 | ユミコア | 充電式リチウムイオン電池用の粉末状リチウムコバルト系酸化物カソード活物質粉末及びその製造方法 |
| JP7442646B2 (ja) | 2019-12-18 | 2024-03-04 | ユミコア | 充電式リチウムイオン電池用の粉末状リチウムコバルト系酸化物カソード活物質粉末及びその製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US10411258B2 (en) | 2019-09-10 |
| TWI591883B (zh) | 2017-07-11 |
| US20170309909A1 (en) | 2017-10-26 |
| KR102004625B1 (ko) | 2019-07-26 |
| EP3204973A4 (en) | 2018-05-09 |
| HUE042152T2 (hu) | 2019-06-28 |
| EP3204973A1 (en) | 2017-08-16 |
| TW201620183A (zh) | 2016-06-01 |
| EP3204973B1 (en) | 2018-12-05 |
| KR20170065635A (ko) | 2017-06-13 |
| US20190386303A1 (en) | 2019-12-19 |
| US11462735B2 (en) | 2022-10-04 |
| CN106795008B (zh) | 2018-11-27 |
| PL3204973T3 (pl) | 2019-09-30 |
| JP6482659B2 (ja) | 2019-03-13 |
| CN106795008A (zh) | 2017-05-31 |
| WO2016055911A1 (en) | 2016-04-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6482659B2 (ja) | 好ましい形態を有する不純物含有カソード材料及び不純物含有金属炭酸塩から調製するための方法 | |
| JP5938614B2 (ja) | リチウムコバルト酸化物材料 | |
| JP6323117B2 (ja) | 非水電解質二次電池用正極活物質の前駆体の製造方法、及び非水電解質二次電池用正極活物質の製造方法 | |
| JP6287970B2 (ja) | ニッケル複合水酸化物とその製造方法 | |
| US10297825B2 (en) | Process for producing nickel cobalt aluminum composite hydroxide and process for producing positive electrode active material for non-aqueous electrolyte secondary batteries | |
| JP7215169B2 (ja) | ニッケルマンガン複合水酸化物とその製造方法、非水系電解質二次電池用正極活物質とその製造方法、および非水系電解質二次電池 | |
| JP6985406B2 (ja) | 改善された二次電池性能を有するカソード材料の前駆体及び前駆体を調製する方法 | |
| US20090302267A1 (en) | Inorganic Compounds | |
| KR20130097779A (ko) | 혼합 금속 산화 수산화물 및 그 제조 방법 | |
| US10840511B2 (en) | Maganese composite hydroxide and process for producing same, positive electrode active material and process for producing same, and non-aqueous electrolyte secondary battery | |
| US11919783B2 (en) | Beta-nickel hydroxide doped with aluminum | |
| WO2023013494A1 (ja) | リチウム二次電池用正極活物質、リチウム二次電池用正極及びリチウム二次電池 | |
| JP2022504835A (ja) | リチウム遷移金属複合酸化物およびその製造方法 | |
| JP6988370B2 (ja) | ニッケル複合水酸化物、ニッケル複合水酸化物の製造方法 | |
| JP2015173122A (ja) | 非水系電解質二次電池の正極活物質の製造方法 | |
| US11973223B2 (en) | Manganese composite hydroxide and process for producing same, positive electrode active material and process for producing same, and non-aqueous electrolyte secondary battery | |
| WO2016067960A1 (ja) | ニッケル複合水酸化物とその製造方法 | |
| WO2017056021A1 (en) | Hydrated manganese oxide and a method for the production thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20180410 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20180507 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20180703 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20180903 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20181109 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20181126 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20181221 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20190108 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20190128 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20190212 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6482659 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |