JP2012506360A - 無機化合物の製造方法 - Google Patents
無機化合物の製造方法 Download PDFInfo
- Publication number
- JP2012506360A JP2012506360A JP2011532694A JP2011532694A JP2012506360A JP 2012506360 A JP2012506360 A JP 2012506360A JP 2011532694 A JP2011532694 A JP 2011532694A JP 2011532694 A JP2011532694 A JP 2011532694A JP 2012506360 A JP2012506360 A JP 2012506360A
- Authority
- JP
- Japan
- Prior art keywords
- precursor
- group
- ionic liquid
- formula
- sio
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Images


































Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/58—Selection of substances as active materials, active masses, active liquids of inorganic compounds other than oxides or hydroxides, e.g. sulfides, selenides, tellurides, halogenides or LiCoFy; of polyanionic structures, e.g. phosphates, silicates or borates
- H01M4/5825—Oxygenated metallic salts or polyanionic structures, e.g. borates, phosphates, silicates, olivines
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B25/00—Phosphorus; Compounds thereof
- C01B25/16—Oxyacids of phosphorus; Salts thereof
- C01B25/26—Phosphates
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B25/00—Phosphorus; Compounds thereof
- C01B25/16—Oxyacids of phosphorus; Salts thereof
- C01B25/26—Phosphates
- C01B25/37—Phosphates of heavy metals
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B25/00—Phosphorus; Compounds thereof
- C01B25/16—Oxyacids of phosphorus; Salts thereof
- C01B25/26—Phosphates
- C01B25/45—Phosphates containing plural metal, or metal and ammonium
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B25/00—Phosphorus; Compounds thereof
- C01B25/16—Oxyacids of phosphorus; Salts thereof
- C01B25/26—Phosphates
- C01B25/455—Phosphates containing halogen
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B33/00—Silicon; Compounds thereof
- C01B33/20—Silicates
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G1/00—Methods of preparing compounds of metals not covered by subclasses C01B, C01C, C01D, or C01F, in general
- C01G1/10—Sulfates
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G49/00—Compounds of iron
- C01G49/009—Compounds containing, besides iron, two or more other elements, with the exception of oxygen or hydrogen
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G51/00—Compounds of cobalt
- C01G51/006—Compounds containing, besides cobalt, two or more other elements, with the exception of oxygen or hydrogen
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G53/00—Compounds of nickel
- C01G53/006—Compounds containing, besides nickel, two or more other elements, with the exception of oxygen or hydrogen
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/58—Selection of substances as active materials, active masses, active liquids of inorganic compounds other than oxides or hydroxides, e.g. sulfides, selenides, tellurides, halogenides or LiCoFy; of polyanionic structures, e.g. phosphates, silicates or borates
- H01M4/582—Halogenides
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/50—Solid solutions
- C01P2002/52—Solid solutions containing elements as dopants
- C01P2002/54—Solid solutions containing elements as dopants one element only
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/70—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data
- C01P2002/72—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data by d-values or two theta-values, e.g. as X-ray diagram
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/70—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data
- C01P2002/77—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data by unit-cell parameters, atom positions or structure diagrams
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/80—Crystal-structural characteristics defined by measured data other than those specified in group C01P2002/70
- C01P2002/88—Crystal-structural characteristics defined by measured data other than those specified in group C01P2002/70 by thermal analysis data, e.g. TGA, DTA, DSC
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/01—Particle morphology depicted by an image
- C01P2004/04—Particle morphology depicted by an image obtained by TEM, STEM, STM or AFM
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/40—Electric properties
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/29—Coated or structually defined flake, particle, cell, strand, strand portion, rod, filament, macroscopic fiber or mass thereof
- Y10T428/2982—Particulate matter [e.g., sphere, flake, etc.]
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Electrochemistry (AREA)
- Engineering & Computer Science (AREA)
- Crystallography & Structural Chemistry (AREA)
- Materials Engineering (AREA)
- Battery Electrode And Active Subsutance (AREA)
- Secondary Cells (AREA)
- Inorganic Compounds Of Heavy Metals (AREA)
- Compounds Of Iron (AREA)
Abstract
Description
粉末状の材料は、それが無機であるか、有機であるか又は有機金属であるかを問わず、特にそのままで使用されるセラミックとして若しくは焼結を対象としたセラミックとして、又はデータ保存用の磁性材料、ディスプレイ装置の顔料及び蛍光材料、又は電極部品、特にリチウム電池としての用途に極めて有益である。
≡M−OH+HO−M≡→≡M−O−M≡+H2O(M=金属、Si、Ge)。
H3PO3+FeSO4+3LiOH→LiFePO4+Li2SO4+3H2O。
本発明の目的は、従来の粉末の製造方法の欠点を、エネルギー及び出発原料の点で経済的であり、かつ、均質な粒子を得るのを可能にすると同時に空気と反応しやすい試剤の酸化という現象を回避することを可能にする、複合無機酸化物の製造方法を提案することによって克服することである。
(式中:
・Aは、アルカリ金属、アルカリ土類金属、ドーパント元素及び欠落(lacune)から選択される少なくとも1種の成分を表し;
・Mは(T1-tT’t)を表し、Tは1種以上の遷移金属を表し、T’はMg、Ca、Al及び希土類元素から選択される少なくとも1種の元素を表し、0≦t<1であり;
・Yは、S、Se、P、As、Si、Ge及びAlから選択される少なくとも1種の元素を表し;
・Zは、F、O及びOHから選択される少なくとも1種の元素を表し;
・a、m、y及びzは化学両論係数であり、かつ、実数、ゼロ又は正数であるが、ただし次の条件を満たすものとする:
* a、m、t、y及びzは、式(I)の無機酸化物の電気的中性に関連するものであり、
* a≧0;m>0;y>0
* z≧0。)
を、式(I)の無機酸化物の構成成分の先駆物質から出発して製造するための方法であって、
(i)該先駆物質を、陽イオン及び陰イオンから形成された、それらの電荷が平衡した1種以上のイオン液体を含む支持液体に分散させて、該先駆物質の該液体への分散液を得、
(ii)該懸濁液を25〜380℃の温度にまで加熱し、
(iii)該イオン液体と、該先駆物質間の反応により得られた式(I)の無機酸化物とを分離すること
を含むことを特徴とする方法である。
・TiCl4、(NH4)2TiO(C3H4O3)2、(NH4)2TiO(C2O4)2、(NH4)2TiF6、Ti(OR1)4及びTi(NR2)4(ここで、R1基のそれぞれ又はR2基のそれぞれは、互いに独立して、好ましくは1〜10個の炭素原子を有するアルキル基を表す。);
・FeCl3、Fe2(SO4)3、Fe(NO3)3及びNH4Fe(SO4)2並びにそれらの水和物;
・FeCl2、FeSO4、Fe(C2O4)、Fe(CH3CO2)2(特にLiFePO4及びその固溶体の製造について)及びそれらの水和物;
・MnCl2、MnSO4、Mn(C2O4)、Mn(CH3CO2)2、Mn(NO3)2及びそれらの水和物(特にLiMnPO4、LiMnBO3及びそれらの固溶体の製造について);
・CoCl2、CoSO4、Co(C2O4)、Co(CH3CO2)2、Co(NO3)2及びそれらの水和物;
・NiCl2、NiSO4、Ni(C2O4)、Ni(CH3CO2)2、Ni(NO3)2及びそれらの水和物;
・CrCl3、Cr2(SO4)3、Cr(NO3)3及びそれらの水和物;
・VOCl2、VOSO4及びそれらの水和物。
・燐酸塩AaMmPO4、特に化合物AaM1 mPO4(ここで、a=1及びA=Li;mは1〜0.85の範囲であり、M1は、Feそのもの又はMg、Co、Ni、Mn、Al、Cr及びTiから選択される少なくとも1種の他の金属元素との組合せとして表す;
・フルオロ燐酸塩AaMmPO4F、特に化合物LiMmPO4F、例えばLiFePO4F;
・化合物AaMmSO4F、AがLi又はNaであり、MがFe、Mn、Co及びNiから選択される少なくとも1種の元素を表す特に化合物、例えばLiFeSO4F、LiCoSO4F、LiNiSO4F、Li(Fe1-tMnt)SO4F、NaFeSO4F及びNaCoSO4F。
・珪酸塩、例えばファヤライト及びその固溶体、特にかんらん石構造の珪酸塩Fe2-x-zMnxMgwSiO4、(0≦x、w≦2)、及びリチウムとの混合珪酸塩Li2Fe1-x'-w'Mnx'Mgw'SiO4(0≦x’、w’≦1);
・硫酸塩、例えばLi2Fe2(SO4)3及びNa2Fe2(SO4)3;
・珪燐酸塩、例えば化合物Na3+xZr2(P1-xSix)3O12、Li1-xFe1+xP1-xSixO4、Li1+xFeP1-xSixO4、Li2-xFeSi1-xPxO4及びLi2-xMn1-wMgwSi1-xPxO4(ここで、0≦x≦1、0≦w≦1);
・ホスホ硫酸塩、例えば(LiFePO4)2(SO4);
・珪硫酸塩、例えばLi2-2xFeSi1-xSxO4、0≦x≦1;
・ホスホ珪硫酸塩、例えばLiM2P1-x-wSixSwO4、0≦x、w≦1
・混合フルオロ燐酸塩、例えばNa2Fe1-x-wMnxMgwPO4F(0≦x≦1及び0≦w≦0.15)又はLiVPO4(O1-xFx)及びNaVPO4(O1-xF-x)(ここで0≦x≦1)、フルオロ燐酸塩、例えばMPO4F(M=Fe、Mn又はAl)。

・基R4〜R17、R27、R24、R28、R29、R37、R34、R39、R43及びR46〜R57、互いに独立に、C1−C24アルキル、C1−C24アリールアルキル又は(C1−C24)アルキルアリール基を表し;
・基R18〜R22、R23、R25、R26、R30〜R33、R35、R36、R38、R40〜R42、R44及びR45は、水素原子、C1−C24アルキル基、アリール基、C1−C24オキサアルキル基又は基[(CH)2]mQ(ここで、Qは、OH、CN、C(=O)OR58、C(=O)NR59R60、NR61R62又は1−イミダゾイル、3−イミダゾイル若しくは4−イミダゾイル基を表し、mは、0〜12の正の整数である。)を表す;
・基R8〜R16は(C1−C20)アルキルアリール基又は基NR63R64を表すこともでき、
・R58〜R64は、互いに独立に、水素原子又はC1−C20アルキル、アリール又はC1−C20オキサアルキル基を表す。
式中:
・R及びR’は、同一のもの又は異なるものであってよく、C1−C24アルキル、アリール又は(C1−C24)アルキルアリール基を表し、
・Rfは、CnF2n+1(ここで0≦n≦8)、CF3OCF2、HCF2CF2及びC6F5から選択されるフルオロ基である。
イオン液体1−エチル−3−メチルイミダゾリウムビス(トリフルオロメタンスルホニル)イミド中でのLiFePO4の合成
この例では、LiFePO4の合成を50mL丸底フラスコ内での沈殿により行った。
2mLの1,2−プロパンジオール及び0.5gの尿素を含有する1mLの1−エチル−3−メチルイミダゾリウムビス(トリフルオロメタンスルホニル)イミド(又はEMI−TFSI)(Solvionic社が供給)に0.524gの99%LiH2PO4(オールドリッチ)及び1gのFeCl2・4H2Oを添加した。10分間撹拌後に、この混合物(懸濁液)を180℃の温度にまで1℃/分の温度上昇速度で到達させた。温度を180℃で10分間維持し、続いて反応媒体を室温にまで冷却した。ろ過により回収した後、LiFePO4の粉末を5mLのアセトンで洗浄し、続いて50mLの蒸留水で2回洗浄し、最後に5mLのアセトンで洗浄し、そして60℃のオーブン内で乾燥させた。1gのLiFePO4が95%の収率で得られる。
続いて、このようにして得られた化合物を銅陰極を用いてX線回折(XR)により分析した。対応する回折図形を添付図1に示している。無機酸化物LiFePO4が斜方晶系構造の単一相であることを示している。このようにして得られたLiFePO4の形態は次のとおりである:
SG:Pnma(62)
a=10.33235(5)Å;b=6.00502(6)Å;c=4.69804(3)Å。
イオン液体EMI−TFSI中でのLiFePO4の合成
LiFePO4の合成を50mL丸底フラスコ内での沈殿により行った。0.524gの99%LiH2PO4(オールドリッチ)及び0.908gのFe(C2O4)・2H2Oを15mLのEMI−TFSIに添加した。10分間撹拌後に、この懸濁液を1℃/分の温度上昇速度で250℃にした。反応媒体の温度を250℃に24時間にわたって維持し、続いて、この媒体を室温にまで冷却した。ろ過により回収した後、LiFePO4粉末を50mLのアセトン、続いて50mLの熱水で2回、最後に50mLのアセトンで洗浄し、そして60℃のオーブン内で乾燥させた。1.53gのLiFePO4を97%の収率で得た。
このようにして得られた化合物を、銅陰極を用いてX線回折により分析した。対応する回折図形を添付図2に示している。これは、無機酸化物LiFePO4が例1に従って得られた試料と同じ斜方晶系構造を有する単一相であることを示している。
イオン液体を例1と同じ方法で回収した。
EMI−TFSI中における、微量の1−テトラデシル−3−メチルイミダゾリウムビス(トリフルオロメタンスルホニル)イミドの存在下でのLiFePO4の合成
LiFePO4の合成ボンベ内で行った。5×10-3molのLiH2PO4及び5×10-3molのFe(C2O4)・2H2Oを、微量の1−テトラデシル−3−メチルイミダゾリウムビス(トリフルオロメタンスルホニル)イミド(粒子の形成を変化させるための界面活性剤として使用)を含有する10mLのEMI−TFSIに添加した。撹拌後、反応混合物を1℃/分の温度上昇速度で250℃の温度にした。反応媒体の温度を250℃に24時間にわたって維持し、続いて、この媒体を室温にまで冷却した。上記例2で示したとおりに回収、洗浄及び乾燥させた後に、所期の生成物を得た。X線回折による分析からLiFePO4の単一相が示された。
微量の1−テトラデシル−3−メチルイミダゾリウムビス(トリフルオロメタンスルホニル)イミドを含有する1−エチル−3−メチルイミダゾリウムトリフルオロメタンスルホネート(EMI−トリフレート)中でのLiFePO4の合成
LiFePO4の合成をボンベ内で行った。5×10-3molのLiH2PO4及び5×10-3molのFe(C2O4)・2H2Oを微量の1−テトラデシル−3−メチルイミダゾリウムビス(トリフルオロメタンスルホニル)イミド(粒子の形成を変化させるための界面活性剤として使用)を含有する10mLのEMI−トリフレートに添加した。撹拌後、反応混合物を1℃/分の温度上昇速度で250℃の温度にした。反応媒体の温度を250℃に24時間にわたって維持し、続いて、この媒体を室温にまで冷却した。上記例2で示したとおりに回収、洗浄及び乾燥させた後に、所期の生成物を得た。X線回折による分析からLiFePO4の単一相が示された。
FeF2及びNa3PO4からのNa2FePO4Fの合成
1gのFeF2/Na3PO4等モル混合物(10分間の粉砕により得られる)を5mLの1−ブチル−2,3−ジメチルイミダゾリウムビス(トリフルオロメタンスルホニル)イミドに導入する。この混合物を270℃で48時間加熱し、続いて室温にまで冷却させた。ろ過後に回収された粉末を20mLのアセトンで洗浄して微量のイオン液体を除去し、冷却熱水で迅速にすすいで合成中に形成された微量NaFを除去し、20mLのアセトンで洗浄し、続いて60℃のオーブン内で乾燥させた。
図3は、次の反応スキームに従って得られた化合物のX線回折図形を示す:FeF2+Na3PO4→Na2FePO4F+NaF。これは、該化合物が単一の斜方晶系相であることを示しており、そのパラメーターは次のとおりである:SG:P b c n(60);a=5.20681(4)Å;b=13.58217(2)Å;c=11.69389(2)Å。
化合物Na2FePO4Fが20〜50nmの平均サイズを有する粒子の形態で得られる。
FeF2、FeCl2及びNa3PO4からのNa2FePO4Fの製造
例5の手順を、先駆物質の混合物として1/2FeF2、1/2FeCl2及びNa3PO4の等モル混合物1gを使用して繰り返した。
図4は、次の反応スキームに従って得られた化合物のX線回折図形を示している:1/2FeF2+1/2FeCl2+Na3PO4 → Na2FePO4F+NaCl。これは、該化合物が単一の斜方晶系相であることを示しており、そのパラメーターは次のとおりである:SG:P b c n(60);a=5.22576(4)Å;b=13.86986(2)Å;c=11.79141(2)Å.
化合物Na2FePO4Fが1〜3μmの平均サイズを有する粒子の状態で得られる。
MnF2及びNa3PO4からのNa2MnPO4Fの合成
先駆物質の混合物としてMnF2/Na3PO4の等モル混合物1gを使用して例1の手順を繰り返した。
図5は、次の反応スキームに従って得られた化合物のX線回折図形を示している:MnF2+Na3PO4 → Na2MnPO4F+NaCl。これは、該化合物が単一の単斜晶系相であることを示しており、そのパラメーターは次のとおりである:SG:P 121/N1(14);a=13.69172(4)Å;b=5.30686(2)Å;c=13.70873(4)Å;β=119.67074°。
FeF2、FeCl2、MnF2及びNa3PO4からのNa2Fe0.95Mn0.05PO4Fの合成
先駆物質の混合物として0.5FeF2、0.45FeCl2、0.05MnF2及びNa3PO4の等モル混合物1gを使用し、洗浄を変更することによって例6の手順を繰り返した。
形成され、ろ過によって回収された粉末をアセトンで洗浄して微量のイオン液体を除去し、続いて20mLのメタノールで2回洗浄して合成中に形成されたNaClを除去し、続いて20mLのアセトンで洗浄し、最後に60℃のオーブン内で乾燥させた。
図6は、次の反応スキームに従って得られた化合物のX線回折図形を示している:
0.5FeF2+0.45FeCl2+0.05MnCl2+Na3PO4→Na2Fe0.95Mn0.05PO4F+NaCl。
図6は、該化合物が単一の斜方晶系相であることを示しており、そのパラメーターは次のとおりである:SG:P b c n(60);a=5.24863(4)Å;b=13.85132(3)Å;c=11.79877(4)Å。
FeF3及びLi3PO4からのLiFePO4Fの合成
1gのFeF3/Li3PO4等モル混合物(30分間の粉砕により得られる)を5mLの1−ブチル−3−メチルイミダゾリウムトリフルオロメタンスルホネートに導入した。この混合物を260℃で48時間加熱し、続いて室温にまで冷却させた。ろ過後に回収された粉末を20mLのアセトンで洗浄して微量のイオン液体を除去し、冷却熱水で迅速にすすいで合成中に形成された微量のLiFを除去し、20mLのアセトンで洗浄し、続いて60℃のオーブン内で乾燥させた。
図7に示されたX線回折図形は、次の反応スキームに従って得られた化合物のものである:FeF3+Li3PO4→LiFePO4F+2LiF。これは、該化合物が空間群P−1(2)の単一の三斜晶系相であることを示しており、そのパラメーターは次のとおりである:a=5.15616Å、b=5.31041Å、c=7.48189Å、α=67.22507、β=67.33746、γ=81.74728°、V=174.303Å3。
FeSO4・7H2O及びNaFからのNaFeSO4Fの合成
5mLのEMI−TFSI及び2.808gのFeSO4・7H2Oの混合物を開放Parr(商標)ボンベに設置し、そして230℃に加熱した。加熱5時間後、この混合物を室温にまで冷却し、0.42gのNaFを添加し、続いてParr(商標)ボンベを密閉する。10分の磁気撹拌後、混合物を250℃で24時間加熱する。室温にまで冷却後、回収した粉末を20mLのアセトンで2回洗浄し、続いて60℃のオーブン内で乾燥させた。図8に示すX線回折図は、次の格子定数を有する空間群P21/cの単斜晶系格子に新たな結晶相が形成したことを示している:a=6.6798(2)Å、b=8.7061(2)Å、c=7.19124(18)Å、β=113.517(2)°及びV=383.473(18)Å3。
LiTiPO4Fの合成
合成を260℃のParr(商標)ボンベ内で実施する。LiTiPO4Fの合成の制限因子は反応温度である。標準的なイオン液体との反応を完全にするためには、300℃よりも高い温度が必要である。しかし、弗素化材料は280℃以上の温度で分解する。OH(ヒドロキシル)基の存在下で2位においてCH3基で保護されたイオン液体を使用すると、先駆物質の溶解度を増加させることによって反応温度を低下させることが可能になる。
30分間の粉砕によって得られたTiF3及びLi3PO4の1gの等モル混合物を5mLの1,2−ジメチル(3−ヒドロキシプロピル)イミダゾリウムビス(トリフルオロメタンスルホニル)イミドに添加する。20分間撹拌後に、混合物を260℃で48時間加熱し、続いて室温にまで冷却する。ろ過により回収した粉末を20mLのアセトンで洗浄して微量のイオン液体を除去し、冷却熱水ですすいで合成中に形成された微量のLiFを除去し、20mLのアセトンで洗浄し、次いでオーブン内において60℃で乾燥させた。
図9は、次の反応スキームに従って得られた化合物LiTiPO4FのX線回折図形を示している:TiF3+Li3PO4→LiTiPO4F+2LiF。これは、該化合物が空間群P−1(2)の単一の三斜晶系相であることを示しており、そのパラメーターは次のとおりである:a=5.24979Å、b=5.31177Å、c=7.43029Å;α=68.07435°、β=68.01394°、γ=83.37559° V=178.161Å3。
この化合物は、ナノメートル粒子の形態である。
例5及び6に記載した方法により得られた化合物の性能品質を評価した。
物質のそれぞれを、一方では「リチウム」電気化学セルにおけるカソード材料として、他方では「ナトリウム」電気化学セルにおけるカソード材料として使用した。サイクリングは、電子を15時間で交換するC/15レジームで行った。
「リチウム」電池は次のものを備える:
・リチウム金属のシートから形成されたアノード;
・LiPF6の炭酸エチル及び炭酸ジメチルの1/1(質量)混合物への1M溶液から形成された電解質。
「ナトリウム」電池は次のものを備える:
・スチールディスクに適用されたナトリウム金属によって形成されたアノード;
・NaClO4の炭酸プロピレンへの1M溶液によって形成された電解質。
図のそれぞれにおいて、電位Pの変化(Vで表す)は、最初の2サイクルについてのアルカリ金属の含有量xに応じて与えられている(図10では化合物(Li,Na)xFePO4Fについて、図11では化合物NaxFePO4Fについて)。差込は、サイクリングレジームRに応じたキャパシタンスCの変化(mAh/gで表す)を示す。
LiFeSO4Fの製造
合成
予備工程において、FeSO4・7H2OをEMI−TFSI中において250℃で10時間にわたり熱処理し、続いて280℃で24時間にわたり熱処理した。形成された一水和物FeSO4・H2Oを遠心分離により回収し、酢酸エチルで洗浄し、次いで真空下において100℃で乾燥させた。
このようにして得られた0.85gのFeSO4・H2O及び0.148gのLiF(1/1.14モル比)を乳鉢において互いに混合し、この混合物をParr(商標)ボンベに導入し、そして5mLのエチルメチルイミダゾリウムビス(トリフルオロメタンスルホニル)イミド(EMI−TFSI)を添加した。混合物を20分間室温で撹拌し、これらの相を2時間にわたって沈降させ、続いて、この混合物を300℃で2時間にわたり開放ボンベ内で撹拌することなく加熱した。
この反応混合物を室温にまで冷却した後に、得られた粉末を遠心分離で分離し、20mLのジクロロメタンで3回洗浄し、次いでオーブン内において60℃で乾燥させた。
得られた生成物は淡緑色の粉末の状態である。これを様々な分析に付した。
図12は、SEMによって得られた画像を示しており、これは、粉末がマイクロメートル粒子から形成された凝集体の状態にあることを示している。
図13aは、TEM画像、特に対応するSAED図を示しており、またこれは、粒子が多数の結晶から形成されていることを示している。図13bはEDSスペクトルを示しており、これは、Fが存在することを示している。強度は、x軸のエネルギーE(keVで表す)の関数としてy軸(任意単位)に与えている。
図14は、X線回折図、及び、差込の形で、得られた化合物の構造を示している。この構造は、Li+イオンが位置したトンネルを有する独立のFeO4F2八面体、SO4四面体を有する。
図15は、質量分析と一緒になったTGAによる化合物のキャラクタリゼーション中に得られた線図を示している。上部の曲線(−1.14%、0.07%などの値を有する)はTGA分析に相当し、中程の曲線(458.5℃及び507.4℃の値を有する)は示差走査熱量測定(DSC)に相当し、下の曲線(m48及びm64の表示を有する)は質量分析に相当する。これらの曲線は、SO2の損失(これは、質量分析計の電子衝撃下では、SOに部分的に断片化された状態になる)に相当する23.41%の重量損失が400℃〜700℃で起こることを示している。350℃よりも高い温度についてのTGA及びDSC曲線のうねりは、化合物の熱不安定化の開始を示す。
*Fe2O3(79−1741)
|Fe2O3(25−1402)
↓Li2SO4(32−064)+FeF3・3H2O(32−0464)
・LiHSO4(31−0721)。
無水FeSO4及びLiFの等モル混合物を製造し、そしてこれを空気中において450℃で15分間加熱した。
図17は、出発反応体の混合物についてのX線回折図(図17a)及び熱処理後に得られた生成物についてのX線回折図(図17b)を示す。それぞれFeSO4及びLiFに相当するピークが図17aで見られるのに対し、図17bは、それぞれLiF、Li2SO4、Fe2O3及びLi2S2O7に相当するピークを示している。
この例は、Fe及びSの先駆物質と、Fの先駆物質との混合物をセラミック手段により処理により処理しても、米国特許出願公開第2005/0163699号で主張されたこととは対称的に、化合物LiFeSO4Fが得られないことを確認するものである。
EMI−TFSI中でのFeSO4・7H2O及びLiFからのLiFeSO4Fの合成
乳鉢において調製された1.404gのFeSO4・7H2O及び0.149gのLiFの混合物を3mLの1−エチル−3−メチルイミダゾリウムビス(トリフルオロメタンスルホニル)イミド(EMI−TFSI)を含有するPTFEフラスコに置き、この混合物を20分間にわたって室温で磁気撹拌し、撹拌を停止し、続いて2mLのイオン液体(EMI−TFSI)を添加し、そしてこの混合物を撹拌することなく室温で30分間保持した。続いて、全体を200℃のオーブン内に置き、オーブンの温度を20分ごとに10℃ずつ275℃まで上昇させ、この値を12時間保持し、続いてゆっくりと冷却させた。
熱処理の間に形成された粉末を遠心分離によりイオン液体から分離し、10mLのジクロロメタンで3回洗浄し、続いて60℃のオーブン内で乾燥させた。
銅陰極で実施されたX線回折スペクトルの精密化(図18に示す)から、2つの相LiFeSO4F及びFeSO4・H2Oが等しい割合で存在していることが示された。
三斜晶系、空間群:P−1(2)
A=5.1819(5)Å、b=5.4853(4)Å、c=7.2297(4)Å、
α=106.4564(3)°、β=107.134(6)°、γ=97.922(5)°
V=182.761(4)Å3。
三斜晶系、空間群:P−1(2)
A=5.178(7)Å、b=5.176(7)Å、c=7.599(7)Å;
α=107.58(6)°、β=107.58(8)°、γ=93.34(6)°
V=182.56(4)Å3。
EMI−TFSI中においてFeSO4・H2O及びLiFから出発するLiFeSO4Fの合成
乳鉢において調製された0.85gのFeSO4・H2O及び0.149gのLiF(1/1.14モル比)の混合物を、3mLの1−エチル−3−メチルイミダゾリウムビス(トリフルオロメタンスルホニル)イミド(EMI−TFSI)を含有するPTFEフラスコに導入し、この混合物を20分間にわたって室温で磁気撹拌し、撹拌を停止し、続いて2mLのイオン液体(EMI−TFSI)を添加し、そしてこの混合物を撹拌することなく室温で30分間保持した。続いて、全体を200℃のオーブンに導入し、そしてオーブンの温度を20分ごとに10℃ずつ275℃まで上昇させ、この値を12時間保持し、続いてゆっくりと冷却させた。
熱処理の間に形成された粉末を遠心分離によりイオン液体から分離し、10mLのジクロロメタンで3回洗浄し、続いて60℃のオーブン内で乾燥させた。
銅陰極で実施されたX線回折スペクトルの精密化(図19に示す)から、単一相のLiFeSO4F相の存在が示された。その格子定数は次の通りである:
三斜晶系、空間群:P−1(2)
a=5.1827(7)Å、b=5.4946(6)Å、c=7.2285(7)Å,
α=106.535(7)°、β=107.187(6)°、γ=97.876(5)°
V=182.95(4)Å3。
FeSO4・H2O及びLiFから出発するLiFeSO4Fの合成
乳鉢において調製された0.85gのFeSO4・H2O及び0.149gのLiF(1/1.14モル比)の混合物を3mLの1−エチル−3−メチルイミダゾリウムビス(トリフルオロメタンスルホニル)イミド(EMI−TFSI)を含有するオートクレーブに置き、この混合物を30分間にわたって室温で磁気撹拌し、撹拌を停止し、続いて2mLのイオン液体(EMI−TFSI)を添加し、そしてこの混合物を撹拌することなく室温で30分間保持した。このオートクレーブをアルゴン下で閉じた後に、全体を200℃のオーブン内に置き、そしてこのオーブンの温度を20分毎に10℃ずつ280℃にまで上昇させ、この値で48時間保持し、続いてゆっくりと冷却させた。
熱処理の間に形成された粉末を遠心分離によりイオン液体から分離し、10mLのジクロロメタンで3回洗浄し、続いて60℃のオーブン内で乾燥させた。
得られた生成物は、白みがかった粉末の状態である。この例1の試料との僅かな色の相違は、操作条件によっては相が非化学量論的になる傾向があることを示している。
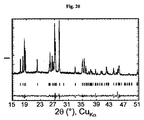
銅陰極で実施されたX線回折スペクトルの精密化(図20に示す)から、単一のLiFeSO4F相の存在が示される。その格子定数は次の通りである:
三斜晶系、空間群:P−1(2)
a=5.1782(4)Å、b=5.4972(4)Å、c=7.2252(4)Å、
α=106.537(4)°、β=107.221(4)°、γ=97.788(3)°
V=182.82(4)Å3。
1−ブチル−3−メチルイミダゾリウムトリフルオロメタンスルホネート(トリフレート)中におけるFeSO4・H2O及びLiFから出発するLiFeSO4Fの合成
乳鉢において調製された0.85gのFeSO4・H2O及び0.149gのLiF(1/1.14モル比)の混合物を3mLの1−ブチル−3−メチルイミダゾリウムトリフルオロメタンスルホネート(トリフレート)を含有するオートクレーブに導入し、この混合物を30分間にわたって室温で磁気撹拌し、撹拌を停止し、続いて2mLのイオン液体EMI−Tfを添加し、そしてこの混合物を撹拌することなく室温で30分間保持した。このオートクレーブをアルゴン下で閉じた後に、全体を200℃のオーブン内に置き、そしてこのオーブンの温度を20分毎に10℃ずつ270℃にまで上昇させ、この値で48時間保持し、続いてゆっくりと冷却させた。
熱処理の間に形成された粉末を遠心分離によりイオン液体から分離し、10mLのジクロロメタンで3回洗浄し、続いて60℃のオーブン内で乾燥させた。
コバルトカソードで実施されたX線回折スペクトルの精密化(図21に示す)から、LiFeSO4F相(約50質量%を占める)及び2つの「無水FeSO4」相の存在が示される。
相2:斜方晶系、空間群Cmcm(63)
相3:斜方晶系、空間群Pbnm(62)
EMI−TFSI中におけるCoSO4・H2O及びLiFから出発するLiCoSO4Fの合成
使用した先駆物質CoSO4・H2OをCoSO4・7H2Oから真空下において160℃で2時間にわたり加熱することによって製造した。
乳鉢において調製された0.86gのCoSO4・H2O及び0.149gのLiF(1/1.13モル比)の混合物を5mLの1−エチル−3−メチルイミダゾリウムビス(トリフルオロメタンスルホニル)イミド(EMI−TFSI)を含有するPTFEフラスコに置き、この混合物を20分間にわたって室温で磁気撹拌し、そして撹拌を停止させた。続いてこのフラスコをアルゴン下で閉じ、そして反応混合物を撹拌することなく室温で30分間保持した。続いて、全体を250℃のオーブンに導入し、このオーブンの温度を10分毎に5℃ずつ275℃にまで上昇させ、この値で36時間保持し、続いてゆっくりと冷却させた。
熱処理の間に形成された粉末を遠心分離によりイオン液体から分離し、10mLの酢酸エチルで3回洗浄し、次いでオーブン内において60℃で乾燥させた。
コバルトカソードで実施されたX線回折スペクトルの精密化(図22に示す)から、三斜格子(P−1)LiCoSO4Fの単一相の存在が示される。その格子定数は次のとおりである:
a=5.1719(6)Å、b=5.4192(6)Å、c=7.1818(7)Å、
α=106.811(7)°、β=107.771(7)°、γ=97.975(5)°
V=177.71(3)Å3。
EMI−TFSI中におけるNiSO4・H2O及びLiFから出発するLiNiSO4Fの合成
先駆物質として使用する一水和物NiSO4・H2OをNiSO4・7H2Oから真空下において240℃で2時間にわたり加熱することによって製造する。
乳鉢において調製された0.86gのNiSO4・H2O及び0.149gのLiF(1/1.13モル比)の混合物を5mLの1−エチル−3−メチルイミダゾリウムビス(トリフルオロメタンスルホニル)イミド(EMI−TFSI)を含有するPTFEフラスコに置き、この混合物を20分間にわたって室温で磁気撹拌し、そして撹拌を停止させた。続いてフラスコをアルゴン下で閉じ、そして反応混合物を撹拌することなく室温で30分間保持した。続いて、全体を250℃のオーブン内に置き、そしてオーブンの温度を285℃にまで2時間にわたって上昇させ、この値で36時間保持し、続いてゆっくりと冷却させた。
熱処理の間に形成された粉末を遠心分離によりイオン液体から分離し、10mLの酢酸エチルで3回洗浄し、続いて60℃のオーブン内で乾燥させた。
コバルトカソードで作製されたX線回折図(図25に示す)は、得られた化合物がLiFeSO4F又はLiCoSO4Fと同様の相を90.95%よりも多く含有することを示している。この相の格子定数は次の通りである:
三斜晶系、空間群:P−1(2)
a=5.173(1)Å、b=5.4209(5)Å、c=7.183(1)Å、
α=106.828(9)°、β=107.776(8)°、γ=97.923(8)°
V=177。85(5)Å3。
LiFe1-yMnySO4Fの固溶体
化合物LiFe1-yMnySO4FをLiF及び先駆物質としての固溶体Fe1-yMnySO4・H2Oから製造した。
1−yモルのFeSO4・7H2O及びyモルのMnSO4・H2Oを、Fe(II)の酸化を防止するためにアルゴンで予め脱気された2mLの熱水に溶解させ、続いて20mLのエタノールを添加した。エタノールの添加中に沈殿により形成された粉末を遠心分離によって回収し、20mLのエタノールで2回洗浄し、続いて真空下において200℃で1時間加熱した。
yの値を変化させることによって、様々な試料を調製した。
これらの試料をX線回折で分析した。得られた試料「y=0.5」の回折図形を図28に示す。これは格子定数が次のとおりの固溶体Fe0.5Mn0.5SO4・H2Oであることを示している:
三斜晶系;空間群:P−1(2)
a=5.2069Å、b=5.2056Å、c=7.6725Å、
α=107.7196°、β=107.4498°、γ=93.08°
V=186.56Å3。
合成は、先駆物質の様々な試料について、イオノサーマル手段により270℃のオートクレーブ内で行った。
乳鉢において調製された0.85gのFe0.5Mn0.5SO4・H2O及び0.149gのLiF(1/1.14モル比)の混合物を、3mLの1−エチル−3−メチルイミダゾリウムビス(トリフルオロメタンスルホニル)イミド(EMI−TFSI)を含有するオートクレーブに置き、この混合物を20分間にわたって室温で磁気撹拌し、撹拌を停止し、続いて2mLのイオン液体(EMI−TFSI)を添加し、そしてこの混合物を撹拌することなく室温で30分間保持した。このオートクレーブをアルゴン下で閉じた後に、全体を200℃のオーブン内に置き、そしてこのオーブンの温度を20分毎に10℃ずつ270℃にまで上昇させ、この値で48時間保持し、続いてゆっくりと冷却させた。
熱処理の間に形成された粉末を遠心分離によりイオン液体から分離し、10mLのジクロロメタンで3回洗浄し、続いて60℃のオーブン内で乾燥させた。
X線回折から、固溶体LiFe1-yMnySO4Fが低いy値(特にy<0.1)で形成すること、及び混合相がそれよりも高いy値で形成すること(特にy>0.25)が示される。
FeSO4Fの製造
この化合物をアセトニトリル中において室温でNO2OF4により化学的脱リチオ化することによって製造する。図29に示すX線回折スペクトルから、この化合物は、パラメーターが次のとおりの格子で結晶化することが示される:
三斜晶系、空間群:P−1(2)
A=5.0682Å、b=5.0649Å、c=7.255Å
α=69.36°、β=68.80°、γ=88.16°
V=161.52Å3。
電気化学的試験
例16に従って製造された化合物LiFeSO4Fの試料を、電極がリチウムホイルであり、2個の電極が1/1炭酸エチレン/炭酸ジメチルEC−DMC混合物へのLiPF6の1M溶液が浸漬されたポリプロピレンセパレータによって隔離されたスウェージロックセルにおける正極材料として試験した。正極を製造するために、80mgのLiFeSO4F(1μmの平均直径を有する粒子の状態)及び20mgの炭素をSPEX 1800ミル内での15分間の機械的粉砕により互いに混合し、1cm2当たり8mgのLiFeSO4Fに相当する量の混合物をアルミニウム電流コレクタに適用した。
Claims (15)
- 式(I)AaMm(YO4)yZz(I)の無機酸化物
(式中:
・Aは、アルカリ金属、アルカリ土類金属、ドーパント元素及び欠落(lacune)から選択される少なくとも1種の元素を表し;
・Mは(T1-tT’t)を表し、Tは1種以上の遷移金属を表し、T’はMg、Ca、Al及び希土類元素から選択される少なくとも1種の元素を表し、0≦t<1であり;
・Yは、S、Se、P、As、Si、Ge及びAlから選択される少なくとも1種の元素を表し;
・Zは、F、O及びOHから選択される少なくとも1種の成分を表し;
・a、m、y及びzは化学両論係数であり、かつ、実数、ゼロ又は正数であるが、ただし次の条件を満たすものとする:
*a、m、t、y及びzは、式(I)の無機酸化物の電気的中性に関連するものであり、
*a≧0;m>0;y>0
*z≧0
である。)
を、式(I)の無機酸化物の構成成分の先駆物質から出発して製造するための方法であって、次の工程:
(i)該先駆物質を、陽イオン及び陰イオンから形成された、それらの電荷が平衡した1種以上のイオン液体を含む支持液体に分散させて、該先駆物質の該液体への懸濁液を得、
(ii)該懸濁液を25〜380℃の温度にまで加熱し、
(iii)該イオン液体と、該先駆物質間の反応により得られた式(I)の無機酸化物とを分離すること
を含むことを特徴とする方法。 - 前記アルカリ金属又はアルカリ土類金属Aの先駆物質が、炭酸塩、炭酸水素塩、水酸化物、過酸化物及び硝酸塩などの熱不安定性陰イオンの塩;酢酸塩及び蟻酸塩などの揮発性有機酸の塩;蓚酸塩、マロン酸塩及びクエン酸塩などの、加熱されると分解し得る酸の塩から選択されることを特徴とする、請求項1に記載の方法。
- 前記先駆物質がLi2CO3、LiHCO3、LiOH、Li2O2、LiNO3、LiCH3CO2、LiCHO2、Li2C2O4、Li3C6H5O7、Na2CO3、NaOH、Na2O2、NaNO3、NaCH3CO2、NaCHO2、Na2C2O4、Na3C6H5O7、K2CO3、KOH、K2O2、KO2KNO3、KCH3CO2、KCHO2、K2C2O4、K3C6H5O7及びそれらの水和物から選択されることを特徴とする、請求項2に記載の方法。
- 前記遷移金属M及び希土類元素の先駆物質が揮発性無機酸の塩、すなわち硝酸塩及び炭酸塩、揮発性有機酸の塩、すなわち酢酸塩及び蟻酸塩、加熱されると分解し得る酸の塩、すなわち蓚酸塩塩、マロン酸塩及びクエン酸塩、並びに無機酸の塩、すなわち硫酸塩、塩化物及び臭化物から選択されることを特徴とする、請求項1〜3のいずれかに記載の方法。
- 前記オキシアニオンYO4の先駆物質が、H2SO4、H3PO4などの対応する酸;熱不安定性アンモニウム、アミン、イミダゾール又はピリミジン塩、例えばNH4HSO4、(NH4)2SO4、NH4HSeO4、(NH4)2SeO4、NH4H2PO4、(NH4)2HPO4、NH4H2AsO4又は(NH4)2HAsO4;ナノメートルSiO2又はGeO2の形態の珪素又はゲルマニウム誘導体;(R3O)4Si及び(R3O)4Ge又は重合体−Si[OR3)2−I]p(ここで0≦p≦104であり、R3は好ましくは1〜10個の炭素原子を含有するアルキル又はアルキルオキシアルキル基を表す。)などのテトラアルコキシシラン又はゲルマン誘導体から選択されることを特徴とする、請求項1〜4のいずれかに記載の方法。
- 前記オキシアニオン先駆物質がAHSO4、A2SO4、AHSeO4、A2SeO4、AH2PO4、A2HPO4、A3PO4、AH2AsO4、A2HAsO4、A3AsO4、A4SiO4、A4GeO4、A2SiO3、A2GeO3及びM2Si5O13(ここでAはアルカリ金属又はアルカリ土類金属を表す。)から選択されることを特徴とする、請求項1〜5のいずれかに記載の方法。
- 前記オキシアニオン先駆物質がLiHSO4、Li2SO4、LiH2PO4、Li3PO4、Li4SiO4、Li2SiO3、Li2Si5O13から選択されることを特徴とする、請求項6に記載の方法。
- 前記工程(i)の間に前記イオン液体中に存在する先駆物質の量が0.01質量%〜85質量%であることを特徴とする、請求項1〜7のいずれかに記載の方法。
- 前記式(I)の酸化物が、燐酸塩AaMmPO4、フルオロ燐酸塩AaMmPO4F又はMPO4F、フルオロ硫酸塩AaMmSO4F、かんらん石構造の珪酸塩Fe2-x-zMnxMgwSiO4(0≦x、w≦2)、リチウムとの混合珪酸塩Li2Fe1-x'-w'Mnx'Mgw'SiO4(0≦x’、w’≦1)、硫酸塩、珪燐酸塩、ホスホ硫酸塩、珪硫酸塩及びホスホ珪硫酸塩から選択されることを特徴とする、請求項1〜8のいずれかに記載の方法。
- 前記イオン液体の陽イオンが次式の陽イオン:
・R4〜R17、R27、R24、R28、R29、R37、R34、R39、R43及びR46〜R57は、互いに独立に、C1−C24アルキル、C1−C24アリールアルキル又は(C1−C24)アルキルアリール基を表し;
・基R18〜R22、R23、R25、R26、R30〜R33、R35、R36、R38、R40〜R42、R44及びR45は、水素原子、C1−C24アルキル基、アリール基、C1−C24オキサアルキル基又は基[(CH)2]mQ(ここで、Qは、OH、CN、C(=O)OR58、C(=O)NR59R60、NR61R62又は1−イミダゾイル、3−イミダゾイル若しくは4−イミダゾイル基を表し、mは、0〜12の正の整数である。)を表し;
・また、基R8〜R16は(C1−C20)アルキルアリール基又は基NR63R64を表すことができ、
・R58〜R64は、互いに独立に、水素原子又はC1−C20アルキル、アリール又はC1−C20オキサアルキル基を表す。)
から選択されることを特徴とする、請求項1〜9のいずれかに記載の方法。 - 前記イオン液体の陰イオンが、Cl、Br、I、RSO3 -、ROSO3 -、[RPO2]-、[R(R’O)PO2]-、[(RO)2PO2]-、BF4 -、RfBF3 -、PF6 -、RfPF5 -、(Rf)2PF4 -、(Rf)3PF3 -、RfCO2 -、RfSO3 -、[(RfSO2)2N]-、[(RfSO2)2CH]-、[(RfSO2)2C(CN)]-、[RfSO2C(CN)2]-、[(RfSO2)3C]-、N(CN)2 -、C(CN)3 -、[(C2O4)2B]-
(式中:
・R及びR'は、同一のもの又は異なるものであってよく、C1−C24アルキル、アリール又は(C1−C24)アルキルアリール基を表し、
・Rfは、CnF2n+1(ここで0≦n≦8)、CF3OCF2、HCF2CF2及びC6F5から選択されるフルオロ基である。)
から選択されることを特徴とする、請求項1〜10のいずれかに記載の方法。 - 前記イオン液体が糖類などの単炭水化物及びデンプン及びセルロースなどの重合炭水化物から選択される1種以上の炭素先駆物質を含有することを特徴とする、請求項1〜11のいずれかに記載の方法。
- 前記加熱工程(ii)を、380℃を超えて続行することを特徴とする、請求項16に記載の方法。
- 前記加熱工程(ii)を不活性雰囲気下において大気圧で実施することを特徴とする、請求項1〜13のいずれかに記載の方法。
- 前記加熱工程(ii)の所要時間が10分〜200時間の範囲にあることを特徴とする、請求項1〜14のいずれかに記載の方法。
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR0805875A FR2937631B1 (fr) | 2008-10-23 | 2008-10-23 | Procede d'elaboration d'oxydes inorganiques en milieu liquide ionique |
| FR0805875 | 2008-10-23 | ||
| FR0953529 | 2009-05-28 | ||
| FR0953529A FR2946038B1 (fr) | 2009-05-28 | 2009-05-28 | Procede de preparation d'un materiau fluore utile comme matiere active d'electrode. |
| FR0955233 | 2009-07-27 | ||
| FR0955233A FR2948353B1 (fr) | 2009-07-27 | 2009-07-27 | Fluorosulfates, leur preparation, leur utilisation comme materiau d'electrode |
| PCT/FR2009/052038 WO2010046608A1 (fr) | 2008-10-23 | 2009-10-23 | Procede d'elaboration de composes inorganiques |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2012506360A true JP2012506360A (ja) | 2012-03-15 |
| JP5706327B2 JP5706327B2 (ja) | 2015-04-22 |
Family
ID=41540498
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2011532694A Expired - Fee Related JP5706327B2 (ja) | 2008-10-23 | 2009-10-23 | 無機化合物の製造方法 |
| JP2011532696A Expired - Fee Related JP5607058B2 (ja) | 2008-10-23 | 2009-10-23 | 電極材料として有用なフルオロ硫酸塩 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2011532696A Expired - Fee Related JP5607058B2 (ja) | 2008-10-23 | 2009-10-23 | 電極材料として有用なフルオロ硫酸塩 |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US9590245B2 (ja) |
| EP (2) | EP2349924B1 (ja) |
| JP (2) | JP5706327B2 (ja) |
| CN (2) | CN102282097B (ja) |
| ES (1) | ES2450134T3 (ja) |
| WO (2) | WO2010046610A1 (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010260761A (ja) * | 2009-05-01 | 2010-11-18 | Kyushu Univ | 非水電解質二次電池用正極の製造方法及びそれを用いた非水電解質二次電池 |
| JP2013163602A (ja) * | 2012-02-09 | 2013-08-22 | National Institute Of Advanced Industrial Science & Technology | 鉄含有複合リン酸フッ化物、その製造方法、及びそれを正極活物質として用いた二次電池 |
| WO2015037489A1 (ja) * | 2013-09-11 | 2015-03-19 | 国立大学法人 東京大学 | ナトリウムイオン二次電池用正極材料 |
| WO2018030425A1 (ja) * | 2016-08-09 | 2018-02-15 | 花王株式会社 | 薄膜状無機酸化物の製造方法 |
| US11066302B2 (en) | 2016-08-09 | 2021-07-20 | Kao Corporation | Method for producing inorganic oxide in form of thin film |
Families Citing this family (51)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9308511B2 (en) | 2009-10-14 | 2016-04-12 | Arizona Board Of Regents, A Body Corporate Of The State Of Arizona Acting For And On Behalf Of Arizona State University | Fabricating porous materials using thixotropic gels |
| US9242900B2 (en) | 2009-12-01 | 2016-01-26 | Arizona Board Of Regents, A Body Corporate Of The State Of Arizona Acting For And On Behalf Of Arizona State University | Porous geopolymer materials |
| FR2959991B1 (fr) * | 2010-05-17 | 2012-06-08 | Centre Nat Rech Scient | Procede de preparation de fluorosulfates de metal alcalin et de metal de transition |
| US9365691B2 (en) | 2010-08-06 | 2016-06-14 | Arizona Board Of Regents, A Body Corporate Of The State Of Arizona Acting For And On Behalf Of Arizona State University | Fabricating porous materials using intrepenetrating inorganic-organic composite gels |
| FR2972441B1 (fr) * | 2011-03-08 | 2013-04-05 | Centre Nat Rech Scient | Nouveau materiau fluore utilisable comme matiere active d'electrode |
| CN102332580B (zh) * | 2011-03-15 | 2013-11-27 | 中国科学院物理研究所 | 氟化硫酸铁盐化合物、制备方法及用途 |
| US10096821B2 (en) * | 2011-05-02 | 2018-10-09 | Toyota Jidosha Kabushiki Kaisha | Lithium secondary battery |
| CN102280616B (zh) * | 2011-07-01 | 2013-09-18 | 合肥工业大学 | 一种镍钴锰酸锂正极材料的制备方法 |
| US8580886B2 (en) | 2011-09-20 | 2013-11-12 | Dow Corning Corporation | Method for the preparation and use of bis (alkoxysilylorgano)-dicarboxylates |
| KR102060844B1 (ko) | 2011-09-21 | 2019-12-30 | 아리조나 보드 오브 리전트스, 아리조나주의 아리조나 주립대 대행법인 | 지오폴리머 수지 재료, 지오폴리머 재료, 및 그에 의해 제조된 재료 |
| KR101350168B1 (ko) * | 2011-09-21 | 2014-01-17 | 전자부품연구원 | 리튬이차전지용 양극재료 및 이의 제조방법 |
| KR101316066B1 (ko) * | 2011-09-27 | 2013-10-11 | 전자부품연구원 | 이차전지용 양극재료 및 이의 제조방법 |
| JP5839227B2 (ja) | 2011-11-10 | 2016-01-06 | トヨタ自動車株式会社 | リチウム二次電池とその製造方法 |
| KR101375701B1 (ko) * | 2011-11-11 | 2014-03-20 | 에스케이씨 주식회사 | 플루오르화 포스페이트 함유 리튬이차전지용 양극활물질 및 이의 제조방법 |
| CN104024265B (zh) | 2011-12-02 | 2017-03-01 | 道康宁公司 | 酯官能化硅烷及其制备和用途;以及亚胺化合物作为相转移催化剂的用途 |
| TWI442616B (zh) * | 2011-12-23 | 2014-06-21 | Ind Tech Res Inst | 混成型儲能元件 |
| GB201201717D0 (en) * | 2012-02-01 | 2012-03-14 | Faradion Ltd | Sulfate electrodes |
| FR2987498B1 (fr) * | 2012-02-29 | 2017-08-11 | Univ Picardie | Sulfates utiles comme materiaux d'electrode |
| JP5710535B2 (ja) * | 2012-03-28 | 2015-04-30 | 株式会社東芝 | 非水電解質二次電池及び電池パック |
| FR2990064B1 (fr) * | 2012-04-25 | 2014-05-23 | Commissariat Energie Atomique | Accumulateur electrochimique au lithium du type lithium-air |
| EP2704245A1 (en) | 2012-09-03 | 2014-03-05 | Agencia Estalal Consejo Superior de Investigaciones Cientificas | Method for preparing carbon coated electrode active material particles |
| CN103682293B (zh) * | 2012-09-24 | 2016-01-13 | 华为技术有限公司 | 一种富锂固溶体正极材料及其制备方法、锂离子电池正极材料和锂离子电池 |
| CN103199229B (zh) * | 2013-03-19 | 2015-04-15 | 南开大学 | 聚阴离子掺杂的富锂层状氧化物正极材料及其制备和应用 |
| JP6076928B2 (ja) | 2013-03-26 | 2017-02-08 | 株式会社東芝 | 電池用活物質材料、非水電解質電池、電池パック及び自動車 |
| EP2813468B1 (en) * | 2013-06-14 | 2016-03-30 | CIC Energigune | New high capacity materials based on transition metals of oxinitrides |
| US10170759B2 (en) | 2013-06-21 | 2019-01-01 | Arizona Board Of Regents On Behalf Of Arizona State University | Metal oxides from acidic solutions |
| WO2015040679A1 (ja) * | 2013-09-17 | 2015-03-26 | 株式会社 東芝 | 非水電解質電池及び電池パック |
| JP6054540B2 (ja) | 2013-09-20 | 2016-12-27 | 株式会社東芝 | 正極活物質、非水電解質電池及び電池パック |
| CN103887493A (zh) * | 2014-02-24 | 2014-06-25 | 杭州电子科技大学 | 一种锂离子电池正极材料及其制备方法 |
| CN103996847B (zh) * | 2014-04-25 | 2017-01-11 | 中南大学 | 水系锂离子电池LiyTi2-xMx(PO4)3/C负极材料及其制备方法 |
| US10926241B2 (en) | 2014-06-12 | 2021-02-23 | Arizona Board Of Regents On Behalf Of Arizona State University | Carbon dioxide adsorbents |
| CN104795564B (zh) * | 2014-09-23 | 2018-02-13 | 中国科学院物理研究所 | 一种水溶液二次电池的正极材料、极片、二次电池和用途 |
| CN107546372B (zh) * | 2016-06-29 | 2020-11-24 | 中国科学院大连化学物理研究所 | 一种阴离子掺杂的磷酸钛锂负极材料及其制备和应用 |
| CN107611427A (zh) * | 2016-07-12 | 2018-01-19 | 南通亨利锂电新材料有限公司 | 一种微晶结构金属磷酸盐及其制备方法 |
| CN108091760B (zh) | 2016-11-23 | 2019-11-22 | 清华大学 | 调控含氢过渡金属氧化物相变的方法 |
| CN108091870B (zh) | 2016-11-23 | 2021-02-26 | 清华大学 | 含氢过渡金属氧化物、制备方法及原电池 |
| CN108091759B (zh) | 2016-11-23 | 2019-07-09 | 清华大学 | 相变电子器件 |
| CN108091913B (zh) | 2016-11-23 | 2020-01-21 | 清华大学 | 固态燃料电池及固态电解质的制备方法 |
| WO2018136695A1 (en) | 2017-01-20 | 2018-07-26 | Seo Dong Kyun | Aluminosilicate nanorods |
| CN106673074B (zh) * | 2017-03-03 | 2021-02-09 | 广东佳纳能源科技有限公司 | 一种一水硫酸钴的生产方法 |
| CN107230779B (zh) * | 2017-05-03 | 2021-02-19 | 武汉理工大学 | 一种高温稳定的相变型氟硫酸铁锂电池材料的制备方法及电极片与锂离子电池的使用方法 |
| CN108063230B (zh) * | 2017-12-14 | 2020-02-14 | 成都新柯力化工科技有限公司 | 一种低温锂电池改性氟硫酸铁锂正极材料及其制备方法 |
| CN109004189A (zh) * | 2018-07-10 | 2018-12-14 | 河北师范大学 | 一种煅烧含离子液体的镍化合物制备锂电池负极材料的方法 |
| CN108913138B (zh) * | 2018-08-10 | 2021-06-04 | 河南科技学院 | 一种Eu3+激活的硅酸盐基红色荧光粉及其制备方法 |
| CN109638275B (zh) * | 2018-12-17 | 2021-10-15 | 中科廊坊过程工程研究院 | 一种硒、硅酸根共掺杂高镍正极材料及其制备方法和应用 |
| US11094998B2 (en) * | 2019-06-19 | 2021-08-17 | GM Global Technology Operations LLC | Ceramic-coated separators for lithium-containing electrochemical cells and methods of making the same |
| CN110342486B (zh) * | 2019-07-22 | 2022-09-30 | 湖北百杰瑞新材料股份有限公司 | 一种二氟磷酸锂的制备方法 |
| CN113373443B (zh) * | 2021-05-20 | 2022-09-06 | 浙江锋锂新能源科技有限公司 | 一种气液混合处理锂金属表面的方法及锂金属电池 |
| CN113943977B (zh) * | 2021-10-09 | 2022-12-06 | 中国科学院福建物质结构研究所 | KMgSO4F化合物、KMgSO4F非线性光学晶体及其制法和用途 |
| CN116417579A (zh) | 2021-12-29 | 2023-07-11 | 宁德时代新能源科技股份有限公司 | 正极活性材料、制备正极材料的方法、正极极片、二次电池、电池模块、电池包和用电装置 |
| CN114628623B (zh) * | 2022-02-16 | 2023-05-23 | 南京师范大学 | 一种碳纳米管穿插的KFeSO4F材料的制法及应用 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004035303A (ja) * | 2002-07-02 | 2004-02-05 | Sangaku Renkei Kiko Kyushu:Kk | 無機酸化物中空粒子とその製造方法 |
| JP2004509447A (ja) * | 2000-09-26 | 2004-03-25 | ハイドロ−ケベック | 制御されたサイズを持つ炭素によって被覆された、酸化還元物質の合成方法 |
| JP2006511421A (ja) * | 2002-12-16 | 2006-04-06 | コミッサリア ア レネルジー アトミーク | アルカリ金属の挿入化合物の調製方法、これを含む活物質およびこの活物質を含む素子 |
| JP2008130526A (ja) * | 2006-11-27 | 2008-06-05 | Hitachi Maxell Ltd | 電気化学素子用活物質、その製造方法、および電気化学素子 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6514640B1 (en) * | 1996-04-23 | 2003-02-04 | Board Of Regents, The University Of Texas System | Cathode materials for secondary (rechargeable) lithium batteries |
| US6777132B2 (en) * | 2000-04-27 | 2004-08-17 | Valence Technology, Inc. | Alkali/transition metal halo—and hydroxy-phosphates and related electrode active materials |
| US20050163699A1 (en) | 2004-01-23 | 2005-07-28 | Jeremy Barker | Fluorosulfate-based electrode active materials and method of making the same |
| DE102006011754B4 (de) * | 2006-03-13 | 2013-12-05 | Evonik Degussa Gmbh | Mikrowellen-Synthesen kristalliner Metalloxidpartikel in lonischen Flüssigkeiten (ILs) |
| CN101037509A (zh) * | 2006-03-15 | 2007-09-19 | 华东理工大学 | 新型有机-无机杂化透明导电薄膜及其制备方法 |
| EP2128092A1 (en) * | 2008-05-22 | 2009-12-02 | Universität Potsdam | Zinc silicate microcrystals and method of preparation |
-
2009
- 2009-10-23 ES ES09760203.1T patent/ES2450134T3/es active Active
- 2009-10-23 CN CN200980152321.0A patent/CN102282097B/zh not_active Expired - Fee Related
- 2009-10-23 WO PCT/FR2009/052040 patent/WO2010046610A1/fr active Application Filing
- 2009-10-23 EP EP09760205.6A patent/EP2349924B1/fr not_active Not-in-force
- 2009-10-23 WO PCT/FR2009/052038 patent/WO2010046608A1/fr active Application Filing
- 2009-10-23 US US13/124,725 patent/US9590245B2/en not_active Expired - Fee Related
- 2009-10-23 JP JP2011532694A patent/JP5706327B2/ja not_active Expired - Fee Related
- 2009-10-23 CN CN200980152322.5A patent/CN102282098B/zh not_active Expired - Fee Related
- 2009-10-23 US US13/124,723 patent/US9293767B2/en not_active Expired - Fee Related
- 2009-10-23 JP JP2011532696A patent/JP5607058B2/ja not_active Expired - Fee Related
- 2009-10-23 EP EP09760203.1A patent/EP2349923B1/fr not_active Not-in-force
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004509447A (ja) * | 2000-09-26 | 2004-03-25 | ハイドロ−ケベック | 制御されたサイズを持つ炭素によって被覆された、酸化還元物質の合成方法 |
| JP2004509058A (ja) * | 2000-09-26 | 2004-03-25 | ハイドロ−ケベック | Lixm1−ym’y(xo4)nを主成分とする物質の合成法 |
| JP2004035303A (ja) * | 2002-07-02 | 2004-02-05 | Sangaku Renkei Kiko Kyushu:Kk | 無機酸化物中空粒子とその製造方法 |
| JP2006511421A (ja) * | 2002-12-16 | 2006-04-06 | コミッサリア ア レネルジー アトミーク | アルカリ金属の挿入化合物の調製方法、これを含む活物質およびこの活物質を含む素子 |
| JP2008130526A (ja) * | 2006-11-27 | 2008-06-05 | Hitachi Maxell Ltd | 電気化学素子用活物質、その製造方法、および電気化学素子 |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010260761A (ja) * | 2009-05-01 | 2010-11-18 | Kyushu Univ | 非水電解質二次電池用正極の製造方法及びそれを用いた非水電解質二次電池 |
| JP2013163602A (ja) * | 2012-02-09 | 2013-08-22 | National Institute Of Advanced Industrial Science & Technology | 鉄含有複合リン酸フッ化物、その製造方法、及びそれを正極活物質として用いた二次電池 |
| WO2015037489A1 (ja) * | 2013-09-11 | 2015-03-19 | 国立大学法人 東京大学 | ナトリウムイオン二次電池用正極材料 |
| JPWO2015037489A1 (ja) * | 2013-09-11 | 2017-03-02 | 国立大学法人 東京大学 | ナトリウムイオン二次電池用正極材料 |
| TWI633697B (zh) * | 2013-09-11 | 2018-08-21 | 國立大學法人東京大學 | 鈉離子二次電池用正極材料 |
| WO2018030425A1 (ja) * | 2016-08-09 | 2018-02-15 | 花王株式会社 | 薄膜状無機酸化物の製造方法 |
| JP6424298B2 (ja) * | 2016-08-09 | 2018-11-14 | 花王株式会社 | 薄膜状無機酸化物の製造方法 |
| JPWO2018030425A1 (ja) * | 2016-08-09 | 2019-01-10 | 花王株式会社 | 薄膜状無機酸化物の製造方法 |
| US11066302B2 (en) | 2016-08-09 | 2021-07-20 | Kao Corporation | Method for producing inorganic oxide in form of thin film |
| US11084733B2 (en) | 2016-08-09 | 2021-08-10 | Kao Corporation | Method for producing inorganic oxide in form of thin film |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2010046610A1 (fr) | 2010-04-29 |
| US9590245B2 (en) | 2017-03-07 |
| JP5607058B2 (ja) | 2014-10-15 |
| EP2349923B1 (fr) | 2013-12-04 |
| EP2349924A1 (fr) | 2011-08-03 |
| CN102282097B (zh) | 2015-04-29 |
| EP2349924B1 (fr) | 2017-02-08 |
| WO2010046608A1 (fr) | 2010-04-29 |
| ES2450134T3 (es) | 2014-03-24 |
| JP2012506361A (ja) | 2012-03-15 |
| US20120129050A1 (en) | 2012-05-24 |
| WO2010046608A9 (fr) | 2010-06-17 |
| CN102282097A (zh) | 2011-12-14 |
| WO2010046610A9 (fr) | 2010-06-17 |
| EP2349923A1 (fr) | 2011-08-03 |
| US20120007020A1 (en) | 2012-01-12 |
| JP5706327B2 (ja) | 2015-04-22 |
| CN102282098A (zh) | 2011-12-14 |
| US9293767B2 (en) | 2016-03-22 |
| CN102282098B (zh) | 2014-12-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5706327B2 (ja) | 無機化合物の製造方法 | |
| KR101708561B1 (ko) | 고속 충/방전 Li- 및 Na-이온 배터리 애노드용의 Sb 나노결정 또는 Sb-합금 나노결정 | |
| JP6097306B2 (ja) | マンガン含有金属リン酸塩及びその製造方法 | |
| US9444102B2 (en) | Fluoro material which may be used as an electrode active material | |
| EP2882683B1 (en) | Process for the colloidal synthesis of lithium iron phosphate | |
| TWI485919B (zh) | 用於鋰二次電池之正極活性物質其製造方法 | |
| JP2008105912A (ja) | ナノ複酸化物AxMyOzの製造方法 | |
| KR101786806B1 (ko) | 알칼리 금속 및 전이 금속 플루오로술페이트의 제조 방법 | |
| JP2013119492A (ja) | リチウムシリケート系化合物及びその製造方法 | |
| Piffet et al. | High temperature X-ray diffraction study of the formation of Na2Ti3O7 from a mixture of sodium carbonate and titanium oxide | |
| FR2937631A1 (fr) | Procede d'elaboration d'oxydes inorganiques en milieu liquide ionique | |
| Zharov et al. | New Approaches to Synthesizing Nanostructured Electrode Materials Based on Double Lithium and Cobalt Phosphates in Salt Melts | |
| JP2023183400A (ja) | アルカリ金属イオン電池用オキソ酸系正極活物質の製造方法 | |
| FR2946038A1 (fr) | Procede de preparation d'un materiau fluore utile comme matiere active d'electrode. | |
| FR2948353A1 (fr) | Fluorosulfates, leur preparation, leur utilisation comme materiau d'electrode |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20121012 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20131217 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20140415 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140714 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20150127 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20150226 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5706327 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |