ES2740325T3 - Derivados de 1-(5-terc-butil-2-aril-pirazol-3-il)-3-[2-fluoro-4-[(3-oxo-4h-pirido[2,3 b]piracin-8-il)oxi]fenil]urea como inhibidores raf para el tratamiento del cáncer - Google Patents
Derivados de 1-(5-terc-butil-2-aril-pirazol-3-il)-3-[2-fluoro-4-[(3-oxo-4h-pirido[2,3 b]piracin-8-il)oxi]fenil]urea como inhibidores raf para el tratamiento del cáncer Download PDFInfo
- Publication number
- ES2740325T3 ES2740325T3 ES14806358T ES14806358T ES2740325T3 ES 2740325 T3 ES2740325 T3 ES 2740325T3 ES 14806358 T ES14806358 T ES 14806358T ES 14806358 T ES14806358 T ES 14806358T ES 2740325 T3 ES2740325 T3 ES 2740325T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- treatment
- cancer
- braf
- vol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000011282 treatment Methods 0.000 title claims description 227
- 206010028980 Neoplasm Diseases 0.000 title claims description 142
- 201000011510 cancer Diseases 0.000 title claims description 74
- 239000003112 inhibitor Substances 0.000 title claims description 56
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 title description 42
- 239000004202 carbamide Substances 0.000 title description 22
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 title description 20
- 150000001875 compounds Chemical class 0.000 claims abstract description 266
- 150000003839 salts Chemical class 0.000 claims abstract description 18
- 239000012453 solvate Substances 0.000 claims abstract description 15
- 150000001204 N-oxides Chemical class 0.000 claims abstract description 14
- 150000004677 hydrates Chemical class 0.000 claims abstract description 11
- 125000004765 (C1-C4) haloalkyl group Chemical group 0.000 claims abstract description 5
- 125000004767 (C1-C4) haloalkoxy group Chemical group 0.000 claims abstract description 4
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 174
- 238000000034 method Methods 0.000 claims description 145
- 102100027103 Serine/threonine-protein kinase B-raf Human genes 0.000 claims description 141
- 101000984753 Homo sapiens Serine/threonine-protein kinase B-raf Proteins 0.000 claims description 137
- 208000035475 disorder Diseases 0.000 claims description 116
- 102100033479 RAF proto-oncogene serine/threonine-protein kinase Human genes 0.000 claims description 101
- 230000005764 inhibitory process Effects 0.000 claims description 74
- 230000035772 mutation Effects 0.000 claims description 67
- 239000000203 mixture Substances 0.000 claims description 66
- 201000001441 melanoma Diseases 0.000 claims description 63
- 101000744436 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) Trans-acting factor D Proteins 0.000 claims description 62
- 201000010099 disease Diseases 0.000 claims description 58
- 230000004913 activation Effects 0.000 claims description 48
- 101100523539 Mus musculus Raf1 gene Proteins 0.000 claims description 40
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 claims description 37
- 230000037361 pathway Effects 0.000 claims description 33
- 230000002062 proliferating effect Effects 0.000 claims description 33
- 108010053099 Vascular Endothelial Growth Factor Receptor-2 Proteins 0.000 claims description 32
- 238000002560 therapeutic procedure Methods 0.000 claims description 31
- 206010061218 Inflammation Diseases 0.000 claims description 19
- 208000027866 inflammatory disease Diseases 0.000 claims description 19
- 230000004054 inflammatory process Effects 0.000 claims description 18
- 239000002829 mitogen activated protein kinase inhibitor Substances 0.000 claims description 16
- BJHCYTJNPVGSBZ-YXSASFKJSA-N 1-[4-[6-amino-5-[(Z)-methoxyiminomethyl]pyrimidin-4-yl]oxy-2-chlorophenyl]-3-ethylurea Chemical compound CCNC(=O)Nc1ccc(Oc2ncnc(N)c2\C=N/OC)cc1Cl BJHCYTJNPVGSBZ-YXSASFKJSA-N 0.000 claims description 15
- 208000006673 asthma Diseases 0.000 claims description 15
- 230000000306 recurrent effect Effects 0.000 claims description 15
- 208000011231 Crohn disease Diseases 0.000 claims description 13
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 claims description 12
- 206010009944 Colon cancer Diseases 0.000 claims description 12
- 201000004681 Psoriasis Diseases 0.000 claims description 12
- 208000001333 Colorectal Neoplasms Diseases 0.000 claims description 11
- 208000034578 Multiple myelomas Diseases 0.000 claims description 11
- 239000003085 diluting agent Substances 0.000 claims description 11
- 102000043136 MAP kinase family Human genes 0.000 claims description 10
- 108091054455 MAP kinase family Proteins 0.000 claims description 10
- 241001465754 Metazoa Species 0.000 claims description 10
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 10
- 208000036142 Viral infection Diseases 0.000 claims description 10
- 229920006395 saturated elastomer Polymers 0.000 claims description 10
- 230000009385 viral infection Effects 0.000 claims description 10
- 229940124647 MEK inhibitor Drugs 0.000 claims description 9
- 206010046851 Uveitis Diseases 0.000 claims description 9
- 206010041823 squamous cell carcinoma Diseases 0.000 claims description 9
- 201000009030 Carcinoma Diseases 0.000 claims description 8
- 206010033128 Ovarian cancer Diseases 0.000 claims description 8
- 208000002780 macular degeneration Diseases 0.000 claims description 8
- 206010009900 Colitis ulcerative Diseases 0.000 claims description 7
- 201000003883 Cystic fibrosis Diseases 0.000 claims description 7
- 208000032612 Glial tumor Diseases 0.000 claims description 7
- 206010018338 Glioma Diseases 0.000 claims description 7
- 208000003019 Neurofibromatosis 1 Diseases 0.000 claims description 7
- 208000024834 Neurofibromatosis type 1 Diseases 0.000 claims description 7
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 7
- 206010060862 Prostate cancer Diseases 0.000 claims description 7
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 7
- 201000006704 Ulcerative Colitis Diseases 0.000 claims description 7
- 206010064930 age-related macular degeneration Diseases 0.000 claims description 7
- 201000010989 colorectal carcinoma Diseases 0.000 claims description 7
- 208000005017 glioblastoma Diseases 0.000 claims description 7
- 208000023356 medullary thyroid gland carcinoma Diseases 0.000 claims description 7
- 230000001394 metastastic effect Effects 0.000 claims description 7
- 206010061289 metastatic neoplasm Diseases 0.000 claims description 7
- 206010006187 Breast cancer Diseases 0.000 claims description 6
- 208000026310 Breast neoplasm Diseases 0.000 claims description 6
- 208000015943 Coeliac disease Diseases 0.000 claims description 6
- 206010012688 Diabetic retinal oedema Diseases 0.000 claims description 6
- 206010012689 Diabetic retinopathy Diseases 0.000 claims description 6
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 6
- 206010033701 Papillary thyroid cancer Diseases 0.000 claims description 6
- 206010039491 Sarcoma Diseases 0.000 claims description 6
- 201000008937 atopic dermatitis Diseases 0.000 claims description 6
- 201000005667 central retinal vein occlusion Diseases 0.000 claims description 6
- 208000035250 cutaneous malignant susceptibility to 1 melanoma Diseases 0.000 claims description 6
- 201000011190 diabetic macular edema Diseases 0.000 claims description 6
- 239000003937 drug carrier Substances 0.000 claims description 6
- 230000003176 fibrotic effect Effects 0.000 claims description 6
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 6
- 208000002154 non-small cell lung carcinoma Diseases 0.000 claims description 6
- 201000002528 pancreatic cancer Diseases 0.000 claims description 6
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 6
- 208000004644 retinal vein occlusion Diseases 0.000 claims description 6
- 208000030045 thyroid gland papillary carcinoma Diseases 0.000 claims description 6
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 claims description 6
- 206010006458 Bronchitis chronic Diseases 0.000 claims description 5
- 206010016935 Follicular thyroid cancer Diseases 0.000 claims description 5
- 201000009794 Idiopathic Pulmonary Fibrosis Diseases 0.000 claims description 5
- 201000003793 Myelodysplastic syndrome Diseases 0.000 claims description 5
- 208000024770 Thyroid neoplasm Diseases 0.000 claims description 5
- 206010006451 bronchitis Diseases 0.000 claims description 5
- 208000007451 chronic bronchitis Diseases 0.000 claims description 5
- 206010073071 hepatocellular carcinoma Diseases 0.000 claims description 5
- 231100000844 hepatocellular carcinoma Toxicity 0.000 claims description 5
- 208000026278 immune system disease Diseases 0.000 claims description 5
- 208000036971 interstitial lung disease 2 Diseases 0.000 claims description 5
- 201000008968 osteosarcoma Diseases 0.000 claims description 5
- 210000000496 pancreas Anatomy 0.000 claims description 5
- 208000030901 thyroid gland follicular carcinoma Diseases 0.000 claims description 5
- 208000031261 Acute myeloid leukaemia Diseases 0.000 claims description 4
- 206010003571 Astrocytoma Diseases 0.000 claims description 4
- 206010008342 Cervix carcinoma Diseases 0.000 claims description 4
- 206010010744 Conjunctivitis allergic Diseases 0.000 claims description 4
- 206010012442 Dermatitis contact Diseases 0.000 claims description 4
- 208000003556 Dry Eye Syndromes Diseases 0.000 claims description 4
- 206010013774 Dry eye Diseases 0.000 claims description 4
- 208000007766 Kaposi sarcoma Diseases 0.000 claims description 4
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 4
- 208000001344 Macular Edema Diseases 0.000 claims description 4
- 208000009018 Medullary thyroid cancer Diseases 0.000 claims description 4
- 208000002454 Nasopharyngeal Carcinoma Diseases 0.000 claims description 4
- 206010029260 Neuroblastoma Diseases 0.000 claims description 4
- 208000006265 Renal cell carcinoma Diseases 0.000 claims description 4
- 206010039085 Rhinitis allergic Diseases 0.000 claims description 4
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 claims description 4
- 125000000217 alkyl group Chemical group 0.000 claims description 4
- 208000002205 allergic conjunctivitis Diseases 0.000 claims description 4
- 201000010105 allergic rhinitis Diseases 0.000 claims description 4
- 208000024998 atopic conjunctivitis Diseases 0.000 claims description 4
- 208000010668 atopic eczema Diseases 0.000 claims description 4
- 201000010881 cervical cancer Diseases 0.000 claims description 4
- 201000005202 lung cancer Diseases 0.000 claims description 4
- 208000020816 lung neoplasm Diseases 0.000 claims description 4
- 208000004748 plexiform neurofibroma Diseases 0.000 claims description 4
- 201000009410 rhabdomyosarcoma Diseases 0.000 claims description 4
- 201000002510 thyroid cancer Diseases 0.000 claims description 4
- 238000002054 transplantation Methods 0.000 claims description 4
- 206010052747 Adenocarcinoma pancreas Diseases 0.000 claims description 3
- 206010005003 Bladder cancer Diseases 0.000 claims description 3
- 208000002177 Cataract Diseases 0.000 claims description 3
- 206010010741 Conjunctivitis Diseases 0.000 claims description 3
- 206010011017 Corneal graft rejection Diseases 0.000 claims description 3
- 206010012434 Dermatitis allergic Diseases 0.000 claims description 3
- 206010012438 Dermatitis atopic Diseases 0.000 claims description 3
- 206010014561 Emphysema Diseases 0.000 claims description 3
- 206010064212 Eosinophilic oesophagitis Diseases 0.000 claims description 3
- 201000004066 Ganglioglioma Diseases 0.000 claims description 3
- 208000010412 Glaucoma Diseases 0.000 claims description 3
- 206010022941 Iridocyclitis Diseases 0.000 claims description 3
- 206010025415 Macular oedema Diseases 0.000 claims description 3
- 208000007054 Medullary Carcinoma Diseases 0.000 claims description 3
- 201000007288 Pleomorphic xanthoastrocytoma Diseases 0.000 claims description 3
- 206010038389 Renal cancer Diseases 0.000 claims description 3
- 208000000208 Wet Macular Degeneration Diseases 0.000 claims description 3
- 125000003545 alkoxy group Chemical group 0.000 claims description 3
- 201000004612 anterior uveitis Diseases 0.000 claims description 3
- 208000029771 childhood onset asthma Diseases 0.000 claims description 3
- 208000029742 colonic neoplasm Diseases 0.000 claims description 3
- 208000011325 dry age related macular degeneration Diseases 0.000 claims description 3
- 201000000708 eosinophilic esophagitis Diseases 0.000 claims description 3
- 208000014829 head and neck neoplasm Diseases 0.000 claims description 3
- 230000000968 intestinal effect Effects 0.000 claims description 3
- 206010023332 keratitis Diseases 0.000 claims description 3
- 201000010666 keratoconjunctivitis Diseases 0.000 claims description 3
- 230000002197 limbic effect Effects 0.000 claims description 3
- 206010024627 liposarcoma Diseases 0.000 claims description 3
- 201000010230 macular retinal edema Diseases 0.000 claims description 3
- 230000036210 malignancy Effects 0.000 claims description 3
- 201000002094 pancreatic adenocarcinoma Diseases 0.000 claims description 3
- 201000007407 panuveitis Diseases 0.000 claims description 3
- 208000002815 pulmonary hypertension Diseases 0.000 claims description 3
- 201000003651 pulmonary sarcoidosis Diseases 0.000 claims description 3
- 206010039083 rhinitis Diseases 0.000 claims description 3
- 201000009890 sinusitis Diseases 0.000 claims description 3
- 208000017572 squamous cell neoplasm Diseases 0.000 claims description 3
- 208000010570 urinary bladder carcinoma Diseases 0.000 claims description 3
- 206010001150 Adenocarcinoma gastric Diseases 0.000 claims description 2
- 208000008334 Dermatofibrosarcoma Diseases 0.000 claims description 2
- 206010057070 Dermatofibrosarcoma protuberans Diseases 0.000 claims description 2
- 208000013452 Fallopian tube neoplasm Diseases 0.000 claims description 2
- 201000010915 Glioblastoma multiforme Diseases 0.000 claims description 2
- 206010061306 Nasopharyngeal cancer Diseases 0.000 claims description 2
- 201000004404 Neurofibroma Diseases 0.000 claims description 2
- 201000007286 Pilocytic astrocytoma Diseases 0.000 claims description 2
- 208000002151 Pleural effusion Diseases 0.000 claims description 2
- 208000020790 biliary tract neoplasm Diseases 0.000 claims description 2
- 208000022033 carcinoma of urethra Diseases 0.000 claims description 2
- 208000006990 cholangiocarcinoma Diseases 0.000 claims description 2
- 208000010247 contact dermatitis Diseases 0.000 claims description 2
- 201000011610 giant cell glioblastoma Diseases 0.000 claims description 2
- 208000002409 gliosarcoma Diseases 0.000 claims description 2
- 208000026037 malignant tumor of neck Diseases 0.000 claims description 2
- 201000011216 nasopharynx carcinoma Diseases 0.000 claims description 2
- 201000004662 neurofibroma of spinal cord Diseases 0.000 claims description 2
- 201000008129 pancreatic ductal adenocarcinoma Diseases 0.000 claims description 2
- 208000010918 peritoneal neoplasm Diseases 0.000 claims description 2
- 201000010174 renal carcinoma Diseases 0.000 claims description 2
- 201000007433 ureter carcinoma Diseases 0.000 claims description 2
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 abstract description 3
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 abstract 1
- 210000004027 cell Anatomy 0.000 description 136
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 92
- -1 TBAP compounds Chemical class 0.000 description 92
- 102000016914 ras Proteins Human genes 0.000 description 66
- 101150040459 RAS gene Proteins 0.000 description 46
- 101150076031 RAS1 gene Proteins 0.000 description 46
- 239000003814 drug Substances 0.000 description 44
- 230000000694 effects Effects 0.000 description 39
- 230000033115 angiogenesis Effects 0.000 description 38
- 108090000744 Mitogen-Activated Protein Kinase Kinases Proteins 0.000 description 36
- 102000004232 Mitogen-Activated Protein Kinase Kinases Human genes 0.000 description 36
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 36
- 230000011664 signaling Effects 0.000 description 33
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 31
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 description 31
- 230000015572 biosynthetic process Effects 0.000 description 31
- 238000009472 formulation Methods 0.000 description 31
- 239000000243 solution Substances 0.000 description 31
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 30
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 30
- GPXBXXGIAQBQNI-UHFFFAOYSA-N vemurafenib Chemical compound CCCS(=O)(=O)NC1=CC=C(F)C(C(=O)C=2C3=CC(=CN=C3NC=2)C=2C=CC(Cl)=CC=2)=C1F GPXBXXGIAQBQNI-UHFFFAOYSA-N 0.000 description 30
- 108090000623 proteins and genes Proteins 0.000 description 28
- 238000012360 testing method Methods 0.000 description 28
- 230000002018 overexpression Effects 0.000 description 26
- 238000006243 chemical reaction Methods 0.000 description 25
- 238000003786 synthesis reaction Methods 0.000 description 25
- 229960003862 vemurafenib Drugs 0.000 description 25
- 235000013877 carbamide Nutrition 0.000 description 24
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 24
- 239000003921 oil Substances 0.000 description 23
- 235000019198 oils Nutrition 0.000 description 23
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 23
- 210000001519 tissue Anatomy 0.000 description 23
- 102000007665 Extracellular Signal-Regulated MAP Kinases Human genes 0.000 description 22
- 108010007457 Extracellular Signal-Regulated MAP Kinases Proteins 0.000 description 22
- 229940079593 drug Drugs 0.000 description 21
- 230000007246 mechanism Effects 0.000 description 21
- 230000001404 mediated effect Effects 0.000 description 21
- 239000007787 solid Substances 0.000 description 21
- 230000006870 function Effects 0.000 description 20
- 238000004519 manufacturing process Methods 0.000 description 20
- 230000004663 cell proliferation Effects 0.000 description 19
- 102000002574 p38 Mitogen-Activated Protein Kinases Human genes 0.000 description 19
- 108010068338 p38 Mitogen-Activated Protein Kinases Proteins 0.000 description 19
- 108010014186 ras Proteins Proteins 0.000 description 19
- 102000005962 receptors Human genes 0.000 description 19
- 108020003175 receptors Proteins 0.000 description 19
- 239000003795 chemical substances by application Substances 0.000 description 18
- 230000019491 signal transduction Effects 0.000 description 18
- 230000014509 gene expression Effects 0.000 description 17
- 230000001225 therapeutic effect Effects 0.000 description 17
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 16
- 239000003102 growth factor Substances 0.000 description 16
- 239000003446 ligand Substances 0.000 description 16
- 208000002193 Pain Diseases 0.000 description 15
- 238000001727 in vivo Methods 0.000 description 15
- 235000018102 proteins Nutrition 0.000 description 15
- 102000004169 proteins and genes Human genes 0.000 description 15
- 238000011160 research Methods 0.000 description 15
- 102000001301 EGF receptor Human genes 0.000 description 14
- 108060006698 EGF receptor Proteins 0.000 description 14
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 14
- 108091000080 Phosphotransferase Proteins 0.000 description 14
- 230000027455 binding Effects 0.000 description 14
- 230000012010 growth Effects 0.000 description 14
- 238000000338 in vitro Methods 0.000 description 14
- 239000007788 liquid Substances 0.000 description 14
- 102000020233 phosphotransferase Human genes 0.000 description 14
- 230000004083 survival effect Effects 0.000 description 14
- 201000001320 Atherosclerosis Diseases 0.000 description 13
- 102100023593 Fibroblast growth factor receptor 1 Human genes 0.000 description 13
- 101710182386 Fibroblast growth factor receptor 1 Proteins 0.000 description 13
- 241000699670 Mus sp. Species 0.000 description 13
- 108010091528 Proto-Oncogene Proteins B-raf Proteins 0.000 description 13
- 102000001332 SRC Human genes 0.000 description 13
- BFSMGDJOXZAERB-UHFFFAOYSA-N dabrafenib Chemical compound S1C(C(C)(C)C)=NC(C=2C(=C(NS(=O)(=O)C=3C(=CC=CC=3F)F)C=CC=2)F)=C1C1=CC=NC(N)=N1 BFSMGDJOXZAERB-UHFFFAOYSA-N 0.000 description 13
- 239000000543 intermediate Substances 0.000 description 13
- 239000008194 pharmaceutical composition Substances 0.000 description 13
- 230000004044 response Effects 0.000 description 13
- 206010039073 rheumatoid arthritis Diseases 0.000 description 13
- 102000018233 Fibroblast Growth Factor Human genes 0.000 description 12
- 108050007372 Fibroblast Growth Factor Proteins 0.000 description 12
- 101710182389 Fibroblast growth factor receptor 2 Proteins 0.000 description 12
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 12
- 238000005481 NMR spectroscopy Methods 0.000 description 12
- 108060006706 SRC Proteins 0.000 description 12
- 102100040247 Tumor necrosis factor Human genes 0.000 description 12
- 206010003246 arthritis Diseases 0.000 description 12
- 230000033228 biological regulation Effects 0.000 description 12
- 239000000872 buffer Substances 0.000 description 12
- 235000019439 ethyl acetate Nutrition 0.000 description 12
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 12
- 208000004296 neuralgia Diseases 0.000 description 12
- 230000035755 proliferation Effects 0.000 description 12
- 102000009929 raf Kinases Human genes 0.000 description 12
- 108010077182 raf Kinases Proteins 0.000 description 12
- 108010087686 src-Family Kinases Proteins 0.000 description 12
- 239000000725 suspension Substances 0.000 description 12
- 102100023600 Fibroblast growth factor receptor 2 Human genes 0.000 description 11
- 229960002465 dabrafenib Drugs 0.000 description 11
- 239000002158 endotoxin Substances 0.000 description 11
- 125000000524 functional group Chemical group 0.000 description 11
- 230000002757 inflammatory effect Effects 0.000 description 11
- 230000002401 inhibitory effect Effects 0.000 description 11
- 230000003211 malignant effect Effects 0.000 description 11
- 208000021722 neuropathic pain Diseases 0.000 description 11
- 230000002246 oncogenic effect Effects 0.000 description 11
- 102000009076 src-Family Kinases Human genes 0.000 description 11
- 239000003826 tablet Substances 0.000 description 11
- 229940125431 BRAF inhibitor Drugs 0.000 description 10
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 10
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 10
- 241000700605 Viruses Species 0.000 description 10
- 238000003556 assay Methods 0.000 description 10
- 230000001419 dependent effect Effects 0.000 description 10
- 150000002148 esters Chemical class 0.000 description 10
- 229940126864 fibroblast growth factor Drugs 0.000 description 10
- ARRNBPCNZJXHRJ-UHFFFAOYSA-M hydron;tetrabutylazanium;phosphate Chemical class OP(O)([O-])=O.CCCC[N+](CCCC)(CCCC)CCCC ARRNBPCNZJXHRJ-UHFFFAOYSA-M 0.000 description 10
- 229920006008 lipopolysaccharide Polymers 0.000 description 10
- 231100000590 oncogenic Toxicity 0.000 description 10
- 230000036407 pain Effects 0.000 description 10
- 230000003389 potentiating effect Effects 0.000 description 10
- 102200055464 rs113488022 Human genes 0.000 description 10
- 239000002904 solvent Substances 0.000 description 10
- 102000004127 Cytokines Human genes 0.000 description 9
- 108090000695 Cytokines Proteins 0.000 description 9
- 101100291385 Drosophila melanogaster p38a gene Proteins 0.000 description 9
- 102100039788 GTPase NRas Human genes 0.000 description 9
- 102000009465 Growth Factor Receptors Human genes 0.000 description 9
- 108010009202 Growth Factor Receptors Proteins 0.000 description 9
- 101000744505 Homo sapiens GTPase NRas Proteins 0.000 description 9
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 9
- 206010040070 Septic Shock Diseases 0.000 description 9
- 108091008605 VEGF receptors Proteins 0.000 description 9
- 230000006907 apoptotic process Effects 0.000 description 9
- 239000000306 component Substances 0.000 description 9
- 210000002889 endothelial cell Anatomy 0.000 description 9
- 239000004615 ingredient Substances 0.000 description 9
- 239000000463 material Substances 0.000 description 9
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 9
- 239000000651 prodrug Substances 0.000 description 9
- 229940002612 prodrug Drugs 0.000 description 9
- 230000001105 regulatory effect Effects 0.000 description 9
- 238000012552 review Methods 0.000 description 9
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 8
- 238000005160 1H NMR spectroscopy Methods 0.000 description 8
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 8
- ZEOWTGPWHLSLOG-UHFFFAOYSA-N Cc1ccc(cc1-c1ccc2c(n[nH]c2c1)-c1cnn(c1)C1CC1)C(=O)Nc1cccc(c1)C(F)(F)F Chemical compound Cc1ccc(cc1-c1ccc2c(n[nH]c2c1)-c1cnn(c1)C1CC1)C(=O)Nc1cccc(c1)C(F)(F)F ZEOWTGPWHLSLOG-UHFFFAOYSA-N 0.000 description 8
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 8
- 102000018471 Proto-Oncogene Proteins B-raf Human genes 0.000 description 8
- 230000003213 activating effect Effects 0.000 description 8
- 239000006071 cream Substances 0.000 description 8
- 239000003995 emulsifying agent Substances 0.000 description 8
- 230000004927 fusion Effects 0.000 description 8
- 230000001976 improved effect Effects 0.000 description 8
- 239000000546 pharmaceutical excipient Substances 0.000 description 8
- 230000008569 process Effects 0.000 description 8
- 238000000746 purification Methods 0.000 description 8
- 239000011541 reaction mixture Substances 0.000 description 8
- 239000000523 sample Substances 0.000 description 8
- 239000003981 vehicle Substances 0.000 description 8
- 101001047681 Homo sapiens Tyrosine-protein kinase Lck Proteins 0.000 description 7
- 241001502974 Human gammaherpesvirus 8 Species 0.000 description 7
- 241000725303 Human immunodeficiency virus Species 0.000 description 7
- 102100021001 Kinase suppressor of Ras 1 Human genes 0.000 description 7
- 206010027476 Metastases Diseases 0.000 description 7
- 108700020796 Oncogene Proteins 0.000 description 7
- 108091007960 PI3Ks Proteins 0.000 description 7
- 102000038030 PI3Ks Human genes 0.000 description 7
- 108090000430 Phosphatidylinositol 3-kinases Proteins 0.000 description 7
- 102100024036 Tyrosine-protein kinase Lck Human genes 0.000 description 7
- 201000005969 Uveal melanoma Diseases 0.000 description 7
- 210000004369 blood Anatomy 0.000 description 7
- 239000008280 blood Substances 0.000 description 7
- 210000004204 blood vessel Anatomy 0.000 description 7
- 239000000969 carrier Substances 0.000 description 7
- 230000001684 chronic effect Effects 0.000 description 7
- 238000011161 development Methods 0.000 description 7
- 230000018109 developmental process Effects 0.000 description 7
- 239000012636 effector Substances 0.000 description 7
- 229910052739 hydrogen Inorganic materials 0.000 description 7
- 230000007062 hydrolysis Effects 0.000 description 7
- 238000006460 hydrolysis reaction Methods 0.000 description 7
- 230000001965 increasing effect Effects 0.000 description 7
- 208000015181 infectious disease Diseases 0.000 description 7
- 239000002609 medium Substances 0.000 description 7
- 210000000056 organ Anatomy 0.000 description 7
- 239000012044 organic layer Substances 0.000 description 7
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 7
- 230000026731 phosphorylation Effects 0.000 description 7
- 238000006366 phosphorylation reaction Methods 0.000 description 7
- 239000006187 pill Substances 0.000 description 7
- 239000000843 powder Substances 0.000 description 7
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 7
- 201000010028 Acrocephalosyndactylia Diseases 0.000 description 6
- 108091008794 FGF receptors Proteins 0.000 description 6
- 102100030708 GTPase KRas Human genes 0.000 description 6
- 101000584612 Homo sapiens GTPase KRas Proteins 0.000 description 6
- 101001137642 Homo sapiens Kinase suppressor of Ras 1 Proteins 0.000 description 6
- 241000699666 Mus <mouse, genus> Species 0.000 description 6
- 102000001253 Protein Kinase Human genes 0.000 description 6
- 208000000102 Squamous Cell Carcinoma of Head and Neck Diseases 0.000 description 6
- 210000001744 T-lymphocyte Anatomy 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 230000001154 acute effect Effects 0.000 description 6
- 238000013459 approach Methods 0.000 description 6
- 239000012131 assay buffer Substances 0.000 description 6
- 239000002585 base Substances 0.000 description 6
- 239000012267 brine Substances 0.000 description 6
- 210000002808 connective tissue Anatomy 0.000 description 6
- 239000000839 emulsion Substances 0.000 description 6
- RCXMILPYEKVQLB-UHFFFAOYSA-N ethyl 5-tert-butyl-1h-pyrazole-3-carboxylate Chemical compound CCOC(=O)C=1C=C(C(C)(C)C)NN=1 RCXMILPYEKVQLB-UHFFFAOYSA-N 0.000 description 6
- 238000002474 experimental method Methods 0.000 description 6
- 239000003925 fat Substances 0.000 description 6
- 238000004128 high performance liquid chromatography Methods 0.000 description 6
- 208000037903 inflammatory enteropathy Diseases 0.000 description 6
- 239000007924 injection Substances 0.000 description 6
- 238000002347 injection Methods 0.000 description 6
- 230000003834 intracellular effect Effects 0.000 description 6
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 6
- 210000004185 liver Anatomy 0.000 description 6
- 210000002540 macrophage Anatomy 0.000 description 6
- 201000008482 osteoarthritis Diseases 0.000 description 6
- 125000006239 protecting group Chemical group 0.000 description 6
- 108060006633 protein kinase Proteins 0.000 description 6
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 6
- 239000003381 stabilizer Substances 0.000 description 6
- 125000001493 tyrosinyl group Chemical group [H]OC1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 6
- 208000000094 Chronic Pain Diseases 0.000 description 5
- 229910052693 Europium Inorganic materials 0.000 description 5
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 5
- 102100029974 GTPase HRas Human genes 0.000 description 5
- 101000584633 Homo sapiens GTPase HRas Proteins 0.000 description 5
- 241000701044 Human gammaherpesvirus 4 Species 0.000 description 5
- 208000019693 Lung disease Diseases 0.000 description 5
- 241000283973 Oryctolagus cuniculus Species 0.000 description 5
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical group CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 5
- 108010029485 Protein Isoforms Proteins 0.000 description 5
- 102000001708 Protein Isoforms Human genes 0.000 description 5
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 5
- 108010053100 Vascular Endothelial Growth Factor Receptor-3 Proteins 0.000 description 5
- 230000005856 abnormality Effects 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 239000000853 adhesive Substances 0.000 description 5
- 230000001070 adhesive effect Effects 0.000 description 5
- 238000004458 analytical method Methods 0.000 description 5
- 230000009286 beneficial effect Effects 0.000 description 5
- 230000008901 benefit Effects 0.000 description 5
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 5
- 231100000504 carcinogenesis Toxicity 0.000 description 5
- 230000024245 cell differentiation Effects 0.000 description 5
- 210000000170 cell membrane Anatomy 0.000 description 5
- 235000009508 confectionery Nutrition 0.000 description 5
- 239000000356 contaminant Substances 0.000 description 5
- 230000006378 damage Effects 0.000 description 5
- 230000004069 differentiation Effects 0.000 description 5
- OGPBJKLSAFTDLK-UHFFFAOYSA-N europium atom Chemical compound [Eu] OGPBJKLSAFTDLK-UHFFFAOYSA-N 0.000 description 5
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical compound O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 5
- 230000006872 improvement Effects 0.000 description 5
- 208000014674 injury Diseases 0.000 description 5
- 102000006495 integrins Human genes 0.000 description 5
- 108010044426 integrins Proteins 0.000 description 5
- 125000000468 ketone group Chemical group 0.000 description 5
- 210000004072 lung Anatomy 0.000 description 5
- 230000009401 metastasis Effects 0.000 description 5
- 239000002674 ointment Substances 0.000 description 5
- 229920000136 polysorbate Polymers 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- 210000002307 prostate Anatomy 0.000 description 5
- 230000002685 pulmonary effect Effects 0.000 description 5
- 125000004076 pyridyl group Chemical group 0.000 description 5
- 108700042226 ras Genes Proteins 0.000 description 5
- 230000009467 reduction Effects 0.000 description 5
- 238000007634 remodeling Methods 0.000 description 5
- 230000036303 septic shock Effects 0.000 description 5
- 210000003491 skin Anatomy 0.000 description 5
- 239000011550 stock solution Substances 0.000 description 5
- 208000011580 syndromic disease Diseases 0.000 description 5
- 230000008685 targeting Effects 0.000 description 5
- 210000004881 tumor cell Anatomy 0.000 description 5
- 230000004614 tumor growth Effects 0.000 description 5
- 238000011144 upstream manufacturing Methods 0.000 description 5
- 150000003672 ureas Chemical class 0.000 description 5
- 230000002792 vascular Effects 0.000 description 5
- 210000005166 vasculature Anatomy 0.000 description 5
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 4
- HUTNOYOBQPAKIA-UHFFFAOYSA-N 1h-pyrazin-2-one Chemical compound OC1=CN=CC=N1 HUTNOYOBQPAKIA-UHFFFAOYSA-N 0.000 description 4
- 208000024827 Alzheimer disease Diseases 0.000 description 4
- 208000023275 Autoimmune disease Diseases 0.000 description 4
- 208000005623 Carcinogenesis Diseases 0.000 description 4
- ZBNZXTGUTAYRHI-UHFFFAOYSA-N Dasatinib Chemical compound C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1Cl ZBNZXTGUTAYRHI-UHFFFAOYSA-N 0.000 description 4
- 208000009386 Experimental Arthritis Diseases 0.000 description 4
- 102100028138 F-box/WD repeat-containing protein 7 Human genes 0.000 description 4
- 206010016654 Fibrosis Diseases 0.000 description 4
- 102000013446 GTP Phosphohydrolases Human genes 0.000 description 4
- 108091006109 GTPases Proteins 0.000 description 4
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 4
- 102100021866 Hepatocyte growth factor Human genes 0.000 description 4
- 101000771237 Homo sapiens Serine/threonine-protein kinase A-Raf Proteins 0.000 description 4
- 101000851007 Homo sapiens Vascular endothelial growth factor receptor 2 Proteins 0.000 description 4
- 206010065390 Inflammatory pain Diseases 0.000 description 4
- 102100039688 Insulin-like growth factor 1 receptor Human genes 0.000 description 4
- 239000002067 L01XE06 - Dasatinib Substances 0.000 description 4
- 201000005099 Langerhans cell histiocytosis Diseases 0.000 description 4
- 229920001213 Polysorbate 20 Polymers 0.000 description 4
- 102000001788 Proto-Oncogene Proteins c-raf Human genes 0.000 description 4
- 108010029869 Proto-Oncogene Proteins c-raf Proteins 0.000 description 4
- 101710141955 RAF proto-oncogene serine/threonine-protein kinase Proteins 0.000 description 4
- 241000700159 Rattus Species 0.000 description 4
- 102100029437 Serine/threonine-protein kinase A-Raf Human genes 0.000 description 4
- 108010090091 TIE-2 Receptor Proteins 0.000 description 4
- 108091023040 Transcription factor Proteins 0.000 description 4
- 102000040945 Transcription factor Human genes 0.000 description 4
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 4
- 102000009484 Vascular Endothelial Growth Factor Receptors Human genes 0.000 description 4
- 102100033178 Vascular endothelial growth factor receptor 1 Human genes 0.000 description 4
- 208000027418 Wounds and injury Diseases 0.000 description 4
- 230000002159 abnormal effect Effects 0.000 description 4
- 238000002835 absorbance Methods 0.000 description 4
- 230000009471 action Effects 0.000 description 4
- 206010069351 acute lung injury Diseases 0.000 description 4
- 150000001412 amines Chemical class 0.000 description 4
- 239000004037 angiogenesis inhibitor Substances 0.000 description 4
- 229940121369 angiogenesis inhibitor Drugs 0.000 description 4
- 230000003143 atherosclerotic effect Effects 0.000 description 4
- 210000000988 bone and bone Anatomy 0.000 description 4
- 210000004556 brain Anatomy 0.000 description 4
- 230000036952 cancer formation Effects 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 208000019425 cirrhosis of liver Diseases 0.000 description 4
- 230000006552 constitutive activation Effects 0.000 description 4
- 231100000433 cytotoxic Toxicity 0.000 description 4
- 230000001472 cytotoxic effect Effects 0.000 description 4
- 229960002448 dasatinib Drugs 0.000 description 4
- 230000002074 deregulated effect Effects 0.000 description 4
- 206010012601 diabetes mellitus Diseases 0.000 description 4
- 238000006471 dimerization reaction Methods 0.000 description 4
- 239000000499 gel Substances 0.000 description 4
- HHLFWLYXYJOTON-UHFFFAOYSA-N glyoxylic acid Chemical compound OC(=O)C=O HHLFWLYXYJOTON-UHFFFAOYSA-N 0.000 description 4
- 239000008187 granular material Substances 0.000 description 4
- 239000001963 growth medium Substances 0.000 description 4
- JXXWJMUPNZDILL-UHFFFAOYSA-N imidazo[4,5-b]pyridin-2-one Chemical compound C1=CC=NC2=NC(=O)N=C21 JXXWJMUPNZDILL-UHFFFAOYSA-N 0.000 description 4
- 239000012535 impurity Substances 0.000 description 4
- 238000011534 incubation Methods 0.000 description 4
- 230000003993 interaction Effects 0.000 description 4
- 239000013067 intermediate product Substances 0.000 description 4
- 230000000155 isotopic effect Effects 0.000 description 4
- 238000000021 kinase assay Methods 0.000 description 4
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 4
- 210000000214 mouth Anatomy 0.000 description 4
- 239000002324 mouth wash Substances 0.000 description 4
- 210000002569 neuron Anatomy 0.000 description 4
- OVLXOTUWFLHWQT-UHFFFAOYSA-N oxazolo[4,5-b]pyridin-2(3H)-one Chemical compound C1=CC=C2OC(=O)NC2=N1 OVLXOTUWFLHWQT-UHFFFAOYSA-N 0.000 description 4
- 239000006072 paste Substances 0.000 description 4
- 230000001575 pathological effect Effects 0.000 description 4
- AHWALFGBDFAJAI-UHFFFAOYSA-N phenyl carbonochloridate Chemical compound ClC(=O)OC1=CC=CC=C1 AHWALFGBDFAJAI-UHFFFAOYSA-N 0.000 description 4
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 4
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 4
- 208000005069 pulmonary fibrosis Diseases 0.000 description 4
- 230000008261 resistance mechanism Effects 0.000 description 4
- 208000037803 restenosis Diseases 0.000 description 4
- 230000035945 sensitivity Effects 0.000 description 4
- 239000007921 spray Substances 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- 239000000829 suppository Substances 0.000 description 4
- 238000001308 synthesis method Methods 0.000 description 4
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 4
- LIRYPHYGHXZJBZ-UHFFFAOYSA-N trametinib Chemical compound CC(=O)NC1=CC=CC(N2C(N(C3CC3)C(=O)C3=C(NC=4C(=CC(I)=CC=4)F)N(C)C(=O)C(C)=C32)=O)=C1 LIRYPHYGHXZJBZ-UHFFFAOYSA-N 0.000 description 4
- 210000003932 urinary bladder Anatomy 0.000 description 4
- SKVGLOFWEJFQKU-UHFFFAOYSA-N (3-fluorophenyl)hydrazine;hydron;chloride Chemical compound Cl.NNC1=CC=CC(F)=C1 SKVGLOFWEJFQKU-UHFFFAOYSA-N 0.000 description 3
- 244000215068 Acacia senegal Species 0.000 description 3
- 206010001052 Acute respiratory distress syndrome Diseases 0.000 description 3
- 229920000936 Agarose Polymers 0.000 description 3
- 108010048154 Angiopoietin-1 Proteins 0.000 description 3
- 102000009088 Angiopoietin-1 Human genes 0.000 description 3
- 102100022014 Angiopoietin-1 receptor Human genes 0.000 description 3
- 208000025490 Apert syndrome Diseases 0.000 description 3
- 201000006474 Brain Ischemia Diseases 0.000 description 3
- 206010008120 Cerebral ischaemia Diseases 0.000 description 3
- 102100031480 Dual specificity mitogen-activated protein kinase kinase 1 Human genes 0.000 description 3
- 206010014824 Endotoxic shock Diseases 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 102000044168 Fibroblast Growth Factor Receptor Human genes 0.000 description 3
- 108090000379 Fibroblast growth factor 2 Proteins 0.000 description 3
- 102100027842 Fibroblast growth factor receptor 3 Human genes 0.000 description 3
- 101710182396 Fibroblast growth factor receptor 3 Proteins 0.000 description 3
- 102000016621 Focal Adhesion Protein-Tyrosine Kinases Human genes 0.000 description 3
- 108010067715 Focal Adhesion Protein-Tyrosine Kinases Proteins 0.000 description 3
- 102000018898 GTPase-Activating Proteins Human genes 0.000 description 3
- 108091006094 GTPase-accelerating proteins Proteins 0.000 description 3
- 229920000084 Gum arabic Polymers 0.000 description 3
- 241000711549 Hepacivirus C Species 0.000 description 3
- 102000003745 Hepatocyte Growth Factor Human genes 0.000 description 3
- 108090000100 Hepatocyte Growth Factor Proteins 0.000 description 3
- 101000753291 Homo sapiens Angiopoietin-1 receptor Proteins 0.000 description 3
- 101001060231 Homo sapiens F-box/WD repeat-containing protein 7 Proteins 0.000 description 3
- 101000753253 Homo sapiens Tyrosine-protein kinase receptor Tie-1 Proteins 0.000 description 3
- 101000851018 Homo sapiens Vascular endothelial growth factor receptor 1 Proteins 0.000 description 3
- 241000701806 Human papillomavirus Species 0.000 description 3
- 206010020751 Hypersensitivity Diseases 0.000 description 3
- 101710184277 Insulin-like growth factor 1 receptor Proteins 0.000 description 3
- 108090001007 Interleukin-8 Proteins 0.000 description 3
- 102000004890 Interleukin-8 Human genes 0.000 description 3
- 229930194542 Keto Natural products 0.000 description 3
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 3
- 230000005723 MEK inhibition Effects 0.000 description 3
- 102000002274 Matrix Metalloproteinases Human genes 0.000 description 3
- 108010000684 Matrix Metalloproteinases Proteins 0.000 description 3
- 206010027480 Metastatic malignant melanoma Diseases 0.000 description 3
- 241001529936 Murinae Species 0.000 description 3
- 206010029748 Noonan syndrome Diseases 0.000 description 3
- 208000008589 Obesity Diseases 0.000 description 3
- 201000004014 Pfeiffer syndrome Diseases 0.000 description 3
- 102100033237 Pro-epidermal growth factor Human genes 0.000 description 3
- 102000042888 RAF family Human genes 0.000 description 3
- 108091082327 RAF family Proteins 0.000 description 3
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 208000005718 Stomach Neoplasms Diseases 0.000 description 3
- 230000006044 T cell activation Effects 0.000 description 3
- 108091008874 T cell receptors Proteins 0.000 description 3
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 3
- 102000012753 TIE-2 Receptor Human genes 0.000 description 3
- 102100022007 Tyrosine-protein kinase receptor Tie-1 Human genes 0.000 description 3
- 102000016549 Vascular Endothelial Growth Factor Receptor-2 Human genes 0.000 description 3
- 102100033179 Vascular endothelial growth factor receptor 3 Human genes 0.000 description 3
- 235000010489 acacia gum Nutrition 0.000 description 3
- 239000000205 acacia gum Substances 0.000 description 3
- 210000000577 adipose tissue Anatomy 0.000 description 3
- 208000007128 adrenocortical carcinoma Diseases 0.000 description 3
- 239000000443 aerosol Substances 0.000 description 3
- 208000026935 allergic disease Diseases 0.000 description 3
- 239000012491 analyte Substances 0.000 description 3
- 230000001093 anti-cancer Effects 0.000 description 3
- 230000000840 anti-viral effect Effects 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 230000005540 biological transmission Effects 0.000 description 3
- 239000000090 biomarker Substances 0.000 description 3
- 230000000903 blocking effect Effects 0.000 description 3
- 210000000481 breast Anatomy 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 150000001735 carboxylic acids Chemical class 0.000 description 3
- 230000000747 cardiac effect Effects 0.000 description 3
- 150000001768 cations Chemical class 0.000 description 3
- 230000006369 cell cycle progression Effects 0.000 description 3
- 230000001413 cellular effect Effects 0.000 description 3
- 206010008118 cerebral infarction Diseases 0.000 description 3
- 125000003636 chemical group Chemical group 0.000 description 3
- 229960002271 cobimetinib Drugs 0.000 description 3
- 238000007398 colorimetric assay Methods 0.000 description 3
- 239000012230 colorless oil Substances 0.000 description 3
- 238000004440 column chromatography Methods 0.000 description 3
- 238000002648 combination therapy Methods 0.000 description 3
- 230000001086 cytosolic effect Effects 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 239000003623 enhancer Substances 0.000 description 3
- 150000002085 enols Chemical class 0.000 description 3
- 208000030533 eye disease Diseases 0.000 description 3
- 239000012091 fetal bovine serum Substances 0.000 description 3
- 102000052178 fibroblast growth factor receptor activity proteins Human genes 0.000 description 3
- 230000004761 fibrosis Effects 0.000 description 3
- 206010017758 gastric cancer Diseases 0.000 description 3
- 238000010914 gene-directed enzyme pro-drug therapy Methods 0.000 description 3
- 235000011187 glycerol Nutrition 0.000 description 3
- 201000000459 head and neck squamous cell carcinoma Diseases 0.000 description 3
- 239000005556 hormone Substances 0.000 description 3
- 238000003119 immunoblot Methods 0.000 description 3
- XKTZWUACRZHVAN-VADRZIEHSA-N interleukin-8 Chemical compound C([C@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@@H](NC(C)=O)CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N[C@@H](CCSC)C(=O)N1[C@H](CCC1)C(=O)N1[C@H](CCC1)C(=O)N[C@@H](C)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC=1C=CC(O)=CC=1)C(=O)N[C@H](CO)C(=O)N1[C@H](CCC1)C(N)=O)C1=CC=CC=C1 XKTZWUACRZHVAN-VADRZIEHSA-N 0.000 description 3
- 229940096397 interleukin-8 Drugs 0.000 description 3
- 210000000936 intestine Anatomy 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- 230000000302 ischemic effect Effects 0.000 description 3
- 210000003734 kidney Anatomy 0.000 description 3
- 229940043355 kinase inhibitor Drugs 0.000 description 3
- 239000006210 lotion Substances 0.000 description 3
- 210000004698 lymphocyte Anatomy 0.000 description 3
- 238000012423 maintenance Methods 0.000 description 3
- 210000004962 mammalian cell Anatomy 0.000 description 3
- 239000003550 marker Substances 0.000 description 3
- 230000000873 masking effect Effects 0.000 description 3
- 102000006240 membrane receptors Human genes 0.000 description 3
- 208000021039 metastatic melanoma Diseases 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 201000006417 multiple sclerosis Diseases 0.000 description 3
- 208000010125 myocardial infarction Diseases 0.000 description 3
- 235000020824 obesity Nutrition 0.000 description 3
- 230000011164 ossification Effects 0.000 description 3
- 230000008506 pathogenesis Effects 0.000 description 3
- 230000002085 persistent effect Effects 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 3
- 239000011505 plaster Substances 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 102000004196 processed proteins & peptides Human genes 0.000 description 3
- 108090000765 processed proteins & peptides Proteins 0.000 description 3
- 230000004224 protection Effects 0.000 description 3
- 230000002829 reductive effect Effects 0.000 description 3
- 230000026267 regulation of growth Effects 0.000 description 3
- 239000000741 silica gel Substances 0.000 description 3
- 229910002027 silica gel Inorganic materials 0.000 description 3
- 210000000130 stem cell Anatomy 0.000 description 3
- 230000000638 stimulation Effects 0.000 description 3
- 201000011549 stomach cancer Diseases 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 238000001356 surgical procedure Methods 0.000 description 3
- 238000002626 targeted therapy Methods 0.000 description 3
- 229940124597 therapeutic agent Drugs 0.000 description 3
- 229960004066 trametinib Drugs 0.000 description 3
- 230000005747 tumor angiogenesis Effects 0.000 description 3
- 230000003827 upregulation Effects 0.000 description 3
- 230000006459 vascular development Effects 0.000 description 3
- VEEGZPWAAPPXRB-BJMVGYQFSA-N (3e)-3-(1h-imidazol-5-ylmethylidene)-1h-indol-2-one Chemical compound O=C1NC2=CC=CC=C2\C1=C/C1=CN=CN1 VEEGZPWAAPPXRB-BJMVGYQFSA-N 0.000 description 2
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- KKVYYGGCHJGEFJ-UHFFFAOYSA-N 1-n-(4-chlorophenyl)-6-methyl-5-n-[3-(7h-purin-6-yl)pyridin-2-yl]isoquinoline-1,5-diamine Chemical compound N=1C=CC2=C(NC=3C(=CC=CN=3)C=3C=4N=CNC=4N=CN=3)C(C)=CC=C2C=1NC1=CC=C(Cl)C=C1 KKVYYGGCHJGEFJ-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- TYEYBOSBBBHJIV-UHFFFAOYSA-M 2-oxobutanoate Chemical compound CCC(=O)C([O-])=O TYEYBOSBBBHJIV-UHFFFAOYSA-M 0.000 description 2
- MXZMACXOMZKYHJ-UHFFFAOYSA-N 4,4-dimethyl-3-oxopentanenitrile Chemical compound CC(C)(C)C(=O)CC#N MXZMACXOMZKYHJ-UHFFFAOYSA-N 0.000 description 2
- MNPLTKHJEAFOCA-UHFFFAOYSA-N 4-amino-3-fluorophenol Chemical compound NC1=CC=C(O)C=C1F MNPLTKHJEAFOCA-UHFFFAOYSA-N 0.000 description 2
- DIRINUVNYFAWQF-UHFFFAOYSA-N 4-chloro-3-nitropyridin-2-amine Chemical compound NC1=NC=CC(Cl)=C1[N+]([O-])=O DIRINUVNYFAWQF-UHFFFAOYSA-N 0.000 description 2
- LWDWTDHJKYVOFT-UHFFFAOYSA-N 5-tert-butyl-2-(2-oxo-1H-pyridin-4-yl)pyrazole-3-carboxylic acid hydrochloride Chemical compound Cl.CC(C)(C)c1cc(C(O)=O)n(n1)-c1cc[nH]c(=O)c1 LWDWTDHJKYVOFT-UHFFFAOYSA-N 0.000 description 2
- IAVMYZXRASMAQF-UHFFFAOYSA-N 5-tert-butyl-2-(3-fluorophenyl)pyrazol-3-amine Chemical compound N1=C(C(C)(C)C)C=C(N)N1C1=CC=CC(F)=C1 IAVMYZXRASMAQF-UHFFFAOYSA-N 0.000 description 2
- CGOZYYXGKRKZRR-UHFFFAOYSA-N 5-tert-butyl-2-(3-fluorophenyl)pyrazole-3-carboxylic acid Chemical compound N1=C(C(C)(C)C)C=C(C(O)=O)N1C1=CC=CC(F)=C1 CGOZYYXGKRKZRR-UHFFFAOYSA-N 0.000 description 2
- QOVGZKZWSXKFGS-UHFFFAOYSA-N 5-tert-butyl-2-(3-methoxyphenyl)pyrazole-3-carboxylic acid Chemical compound C(C)(C)(C)C1=NN(C(=C1)C(=O)O)C1=CC(=CC=C1)OC QOVGZKZWSXKFGS-UHFFFAOYSA-N 0.000 description 2
- BTGMFQHKNSUENT-UHFFFAOYSA-N 5-tert-butyl-2-[3-(trifluoromethyl)phenyl]pyrazole-3-carboxylic acid Chemical compound C(C)(C)(C)C1=NN(C(=C1)C(=O)O)C1=CC(=CC=C1)C(F)(F)F BTGMFQHKNSUENT-UHFFFAOYSA-N 0.000 description 2
- 208000030507 AIDS Diseases 0.000 description 2
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 2
- 102100034594 Angiopoietin-1 Human genes 0.000 description 2
- 201000003076 Angiosarcoma Diseases 0.000 description 2
- 241000416162 Astragalus gummifer Species 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 241001115070 Bornavirus Species 0.000 description 2
- 102100032912 CD44 antigen Human genes 0.000 description 2
- 102000008203 CTLA-4 Antigen Human genes 0.000 description 2
- 108010021064 CTLA-4 Antigen Proteins 0.000 description 2
- 101100381481 Caenorhabditis elegans baz-2 gene Proteins 0.000 description 2
- 201000002927 Cardiofaciocutaneous syndrome Diseases 0.000 description 2
- 208000006029 Cardiomegaly Diseases 0.000 description 2
- 108010001857 Cell Surface Receptors Proteins 0.000 description 2
- 206010052358 Colorectal cancer metastatic Diseases 0.000 description 2
- 201000003874 Common Variable Immunodeficiency Diseases 0.000 description 2
- 208000009283 Craniosynostoses Diseases 0.000 description 2
- 206010049889 Craniosynostosis Diseases 0.000 description 2
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 2
- 241000701022 Cytomegalovirus Species 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- 101100481408 Danio rerio tie2 gene Proteins 0.000 description 2
- 208000032131 Diabetic Neuropathies Diseases 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 2
- 238000002965 ELISA Methods 0.000 description 2
- 241000710188 Encephalomyocarditis virus Species 0.000 description 2
- 206010014733 Endometrial cancer Diseases 0.000 description 2
- 206010014759 Endometrial neoplasm Diseases 0.000 description 2
- 241000792859 Enema Species 0.000 description 2
- 241000283073 Equus caballus Species 0.000 description 2
- 208000025127 Erdheim-Chester disease Diseases 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- 108090000386 Fibroblast Growth Factor 1 Proteins 0.000 description 2
- 102000003974 Fibroblast growth factor 2 Human genes 0.000 description 2
- 108090000385 Fibroblast growth factor 7 Proteins 0.000 description 2
- 101710113436 GTPase KRas Proteins 0.000 description 2
- 208000018522 Gastrointestinal disease Diseases 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- 102000006395 Globulins Human genes 0.000 description 2
- 108010044091 Globulins Proteins 0.000 description 2
- 206010018364 Glomerulonephritis Diseases 0.000 description 2
- 239000004471 Glycine Substances 0.000 description 2
- 201000005569 Gout Diseases 0.000 description 2
- 208000009329 Graft vs Host Disease Diseases 0.000 description 2
- 108010067218 Guanine Nucleotide Exchange Factors Proteins 0.000 description 2
- 102000016285 Guanine Nucleotide Exchange Factors Human genes 0.000 description 2
- WZUVPPKBWHMQCE-UHFFFAOYSA-N Haematoxylin Chemical compound C12=CC(O)=C(O)C=C2CC2(O)C1C1=CC=C(O)C(O)=C1OC2 WZUVPPKBWHMQCE-UHFFFAOYSA-N 0.000 description 2
- 206010019280 Heart failures Diseases 0.000 description 2
- 208000001258 Hemangiosarcoma Diseases 0.000 description 2
- 102100026122 High affinity immunoglobulin gamma Fc receptor I Human genes 0.000 description 2
- 208000017604 Hodgkin disease Diseases 0.000 description 2
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 2
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 2
- 101000924552 Homo sapiens Angiopoietin-1 Proteins 0.000 description 2
- 101000868273 Homo sapiens CD44 antigen Proteins 0.000 description 2
- 101100066427 Homo sapiens FCGR1A gene Proteins 0.000 description 2
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 2
- 101001050288 Homo sapiens Transcription factor Jun Proteins 0.000 description 2
- 206010021143 Hypoxia Diseases 0.000 description 2
- 208000029462 Immunodeficiency disease Diseases 0.000 description 2
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 2
- 208000009289 Jackson-Weiss syndrome Diseases 0.000 description 2
- 206010023856 Laryngeal squamous cell carcinoma Diseases 0.000 description 2
- 208000018142 Leiomyosarcoma Diseases 0.000 description 2
- 206010025323 Lymphomas Diseases 0.000 description 2
- 108010068342 MAP Kinase Kinase 1 Proteins 0.000 description 2
- 102000019149 MAP kinase activity proteins Human genes 0.000 description 2
- 108040008097 MAP kinase activity proteins Proteins 0.000 description 2
- 206010025654 Malignant melanoma of sites other than skin Diseases 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 208000001145 Metabolic Syndrome Diseases 0.000 description 2
- 101100381978 Mus musculus Braf gene Proteins 0.000 description 2
- 101100481410 Mus musculus Tek gene Proteins 0.000 description 2
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 2
- 208000037538 Myelomonocytic Juvenile Leukemia Diseases 0.000 description 2
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 2
- 108010057466 NF-kappa B Proteins 0.000 description 2
- 102000003945 NF-kappa B Human genes 0.000 description 2
- 238000011789 NOD SCID mouse Methods 0.000 description 2
- 206010028851 Necrosis Diseases 0.000 description 2
- 206010029113 Neovascularisation Diseases 0.000 description 2
- 208000012902 Nervous system disease Diseases 0.000 description 2
- 208000030880 Nose disease Diseases 0.000 description 2
- 229910003850 O-nPr Inorganic materials 0.000 description 2
- 102000043276 Oncogene Human genes 0.000 description 2
- 102100040479 P2X purinoceptor 2 Human genes 0.000 description 2
- 101710189968 P2X purinoceptor 2 Proteins 0.000 description 2
- 208000037581 Persistent Infection Diseases 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- 208000004550 Postoperative Pain Diseases 0.000 description 2
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 2
- 108091008611 Protein Kinase B Proteins 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 101100372762 Rattus norvegicus Flt1 gene Proteins 0.000 description 2
- 206010063837 Reperfusion injury Diseases 0.000 description 2
- 239000006146 Roswell Park Memorial Institute medium Substances 0.000 description 2
- 102000038012 SFKs Human genes 0.000 description 2
- 108091008118 SFKs Proteins 0.000 description 2
- 108010017324 STAT3 Transcription Factor Proteins 0.000 description 2
- 206010040047 Sepsis Diseases 0.000 description 2
- 102100024040 Signal transducer and activator of transcription 3 Human genes 0.000 description 2
- 208000000453 Skin Neoplasms Diseases 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 2
- 206010041849 Squamous cell carcinoma of the hypopharynx Diseases 0.000 description 2
- 208000036844 Squamous cell carcinoma of the larynx Diseases 0.000 description 2
- 206010041857 Squamous cell carcinoma of the oral cavity Diseases 0.000 description 2
- 208000006011 Stroke Diseases 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- 208000007536 Thrombosis Diseases 0.000 description 2
- 229920001615 Tragacanth Polymers 0.000 description 2
- 102100023132 Transcription factor Jun Human genes 0.000 description 2
- 206010052779 Transplant rejections Diseases 0.000 description 2
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 2
- 102220531375 Uncharacterized protein KIAA2012_Q16K_mutation Human genes 0.000 description 2
- 102000016663 Vascular Endothelial Growth Factor Receptor-3 Human genes 0.000 description 2
- 206010047115 Vasculitis Diseases 0.000 description 2
- 201000000690 abdominal obesity-metabolic syndrome Diseases 0.000 description 2
- 230000001594 aberrant effect Effects 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 239000012190 activator Substances 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- 208000009956 adenocarcinoma Diseases 0.000 description 2
- 208000020990 adrenal cortex carcinoma Diseases 0.000 description 2
- 150000001299 aldehydes Chemical class 0.000 description 2
- 208000002029 allergic contact dermatitis Diseases 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 150000001409 amidines Chemical class 0.000 description 2
- 235000001014 amino acid Nutrition 0.000 description 2
- 150000001413 amino acids Chemical group 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 239000002870 angiogenesis inducing agent Substances 0.000 description 2
- 230000002491 angiogenic effect Effects 0.000 description 2
- 125000000129 anionic group Chemical group 0.000 description 2
- 239000005557 antagonist Substances 0.000 description 2
- 230000001772 anti-angiogenic effect Effects 0.000 description 2
- 229940121363 anti-inflammatory agent Drugs 0.000 description 2
- 239000002260 anti-inflammatory agent Substances 0.000 description 2
- 230000000259 anti-tumor effect Effects 0.000 description 2
- 239000000427 antigen Substances 0.000 description 2
- 238000010913 antigen-directed enzyme pro-drug therapy Methods 0.000 description 2
- 108091007433 antigens Proteins 0.000 description 2
- 102000036639 antigens Human genes 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 238000003782 apoptosis assay Methods 0.000 description 2
- 239000012300 argon atmosphere Substances 0.000 description 2
- 210000001367 artery Anatomy 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- 230000003305 autocrine Effects 0.000 description 2
- 239000011324 bead Substances 0.000 description 2
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 2
- 101150048834 braF gene Proteins 0.000 description 2
- 208000029028 brain injury Diseases 0.000 description 2
- HQABUPZFAYXKJW-UHFFFAOYSA-N butan-1-amine Chemical compound CCCCN HQABUPZFAYXKJW-UHFFFAOYSA-N 0.000 description 2
- 208000035269 cancer or benign tumor Diseases 0.000 description 2
- 210000000845 cartilage Anatomy 0.000 description 2
- 125000002091 cationic group Chemical group 0.000 description 2
- 230000021164 cell adhesion Effects 0.000 description 2
- 238000000423 cell based assay Methods 0.000 description 2
- 230000010261 cell growth Effects 0.000 description 2
- 239000006285 cell suspension Substances 0.000 description 2
- 230000036755 cellular response Effects 0.000 description 2
- 230000005754 cellular signaling Effects 0.000 description 2
- 230000002490 cerebral effect Effects 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- 208000024063 childhood astrocytic tumor Diseases 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- BSMCAPRUBJMWDF-KRWDZBQOSA-N cobimetinib Chemical compound C1C(O)([C@H]2NCCCC2)CN1C(=O)C1=CC=C(F)C(F)=C1NC1=CC=C(I)C=C1F BSMCAPRUBJMWDF-KRWDZBQOSA-N 0.000 description 2
- 210000001072 colon Anatomy 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- OPQARKPSCNTWTJ-UHFFFAOYSA-L copper(ii) acetate Chemical compound [Cu+2].CC([O-])=O.CC([O-])=O OPQARKPSCNTWTJ-UHFFFAOYSA-L 0.000 description 2
- 230000008878 coupling Effects 0.000 description 2
- 238000010168 coupling process Methods 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- 210000004714 cranial suture Anatomy 0.000 description 2
- 230000034994 death Effects 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 238000010511 deprotection reaction Methods 0.000 description 2
- 230000003831 deregulation Effects 0.000 description 2
- 238000000502 dialysis Methods 0.000 description 2
- 235000005911 diet Nutrition 0.000 description 2
- 230000037213 diet Effects 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 239000000539 dimer Substances 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- 230000003511 endothelial effect Effects 0.000 description 2
- 210000003038 endothelium Anatomy 0.000 description 2
- 239000007920 enema Substances 0.000 description 2
- 229940095399 enema Drugs 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 210000003743 erythrocyte Anatomy 0.000 description 2
- GOBBDDYNEWCTIT-UHFFFAOYSA-N ethyl 5-tert-butyl-2-(3-methoxyphenyl)pyrazole-3-carboxylate Chemical compound CCOC(=O)C1=CC(=NN1C1=CC=CC(OC)=C1)C(C)(C)C GOBBDDYNEWCTIT-UHFFFAOYSA-N 0.000 description 2
- LROVBOCWVSRJNA-UHFFFAOYSA-N ethyl 5-tert-butyl-2-(3-methylphenyl)pyrazole-3-carboxylate Chemical compound CCOC(=O)C1=CC(=NN1C1=CC=CC(C)=C1)C(C)(C)C LROVBOCWVSRJNA-UHFFFAOYSA-N 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 2
- 230000007717 exclusion Effects 0.000 description 2
- 239000003889 eye drop Substances 0.000 description 2
- 229940012356 eye drops Drugs 0.000 description 2
- 210000002950 fibroblast Anatomy 0.000 description 2
- 230000003352 fibrogenic effect Effects 0.000 description 2
- 239000000945 filler Substances 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 125000003709 fluoroalkyl group Chemical group 0.000 description 2
- 239000006260 foam Substances 0.000 description 2
- 230000037406 food intake Effects 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 201000006585 gastric adenocarcinoma Diseases 0.000 description 2
- 230000002496 gastric effect Effects 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 2
- 208000024908 graft versus host disease Diseases 0.000 description 2
- 201000011066 hemangioma Diseases 0.000 description 2
- 239000000833 heterodimer Substances 0.000 description 2
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 2
- 125000000487 histidyl group Chemical group [H]N([H])C(C(=O)O*)C([H])([H])C1=C([H])N([H])C([H])=N1 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 2
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 2
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 2
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 2
- 230000037417 hyperactivation Effects 0.000 description 2
- 208000013403 hyperactivity Diseases 0.000 description 2
- 206010020718 hyperplasia Diseases 0.000 description 2
- 230000003463 hyperproliferative effect Effects 0.000 description 2
- 230000007954 hypoxia Effects 0.000 description 2
- 230000028993 immune response Effects 0.000 description 2
- 210000000987 immune system Anatomy 0.000 description 2
- 238000003364 immunohistochemistry Methods 0.000 description 2
- 230000001771 impaired effect Effects 0.000 description 2
- 230000006698 induction Effects 0.000 description 2
- 230000028709 inflammatory response Effects 0.000 description 2
- 230000009545 invasion Effects 0.000 description 2
- 238000011835 investigation Methods 0.000 description 2
- 229960005386 ipilimumab Drugs 0.000 description 2
- 208000002551 irritable bowel syndrome Diseases 0.000 description 2
- 208000028867 ischemia Diseases 0.000 description 2
- 239000012948 isocyanate Substances 0.000 description 2
- 150000002513 isocyanates Chemical class 0.000 description 2
- 201000011628 juvenile astrocytoma Diseases 0.000 description 2
- 201000005992 juvenile myelomonocytic leukemia Diseases 0.000 description 2
- 210000000867 larynx Anatomy 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 230000003902 lesion Effects 0.000 description 2
- 208000032839 leukemia Diseases 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 230000000527 lymphocytic effect Effects 0.000 description 2
- 208000003747 lymphoid leukemia Diseases 0.000 description 2
- 239000006166 lysate Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 230000035800 maturation Effects 0.000 description 2
- 210000002752 melanocyte Anatomy 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- 230000004060 metabolic process Effects 0.000 description 2
- 208000037970 metastatic squamous neck cancer Diseases 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 239000011859 microparticle Substances 0.000 description 2
- 235000013336 milk Nutrition 0.000 description 2
- 239000008267 milk Substances 0.000 description 2
- 210000004080 milk Anatomy 0.000 description 2
- 230000009456 molecular mechanism Effects 0.000 description 2
- 150000004682 monohydrates Chemical class 0.000 description 2
- 230000000877 morphologic effect Effects 0.000 description 2
- 238000000465 moulding Methods 0.000 description 2
- 208000025113 myeloid leukemia Diseases 0.000 description 2
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 210000003928 nasal cavity Anatomy 0.000 description 2
- 229940097496 nasal spray Drugs 0.000 description 2
- 239000007922 nasal spray Substances 0.000 description 2
- 230000017074 necrotic cell death Effects 0.000 description 2
- 239000013642 negative control Substances 0.000 description 2
- 230000014399 negative regulation of angiogenesis Effects 0.000 description 2
- 230000009826 neoplastic cell growth Effects 0.000 description 2
- 230000001537 neural effect Effects 0.000 description 2
- 230000004770 neurodegeneration Effects 0.000 description 2
- 208000015122 neurodegenerative disease Diseases 0.000 description 2
- 230000004112 neuroprotection Effects 0.000 description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 2
- 210000001331 nose Anatomy 0.000 description 2
- 239000002773 nucleotide Substances 0.000 description 2
- 125000003729 nucleotide group Chemical group 0.000 description 2
- 238000011275 oncology therapy Methods 0.000 description 2
- 150000002891 organic anions Chemical class 0.000 description 2
- 210000000963 osteoblast Anatomy 0.000 description 2
- 230000002611 ovarian Effects 0.000 description 2
- 210000001672 ovary Anatomy 0.000 description 2
- 238000012261 overproduction Methods 0.000 description 2
- 230000000803 paradoxical effect Effects 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 230000007170 pathology Effects 0.000 description 2
- 230000007310 pathophysiology Effects 0.000 description 2
- 230000035515 penetration Effects 0.000 description 2
- 239000002953 phosphate buffered saline Substances 0.000 description 2
- 238000002428 photodynamic therapy Methods 0.000 description 2
- 230000035790 physiological processes and functions Effects 0.000 description 2
- 229920000371 poly(diallyldimethylammonium chloride) polymer Polymers 0.000 description 2
- 229920005862 polyol Polymers 0.000 description 2
- 150000003077 polyols Chemical class 0.000 description 2
- 229920001184 polypeptide Polymers 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 239000013641 positive control Substances 0.000 description 2
- 229940069328 povidone Drugs 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 230000005522 programmed cell death Effects 0.000 description 2
- 230000000750 progressive effect Effects 0.000 description 2
- 230000000770 proinflammatory effect Effects 0.000 description 2
- 230000001737 promoting effect Effects 0.000 description 2
- AMMGCGVWJMRTQI-UHFFFAOYSA-N prop-1-en-2-yl carbonochloridate Chemical compound CC(=C)OC(Cl)=O AMMGCGVWJMRTQI-UHFFFAOYSA-N 0.000 description 2
- 230000000069 prophylactic effect Effects 0.000 description 2
- 238000011321 prophylaxis Methods 0.000 description 2
- LOUPRKONTZGTKE-LHHVKLHASA-N quinidine Chemical compound C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@H]2[C@@H](O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-LHHVKLHASA-N 0.000 description 2
- 239000002994 raw material Substances 0.000 description 2
- 230000007420 reactivation Effects 0.000 description 2
- 229940124617 receptor tyrosine kinase inhibitor Drugs 0.000 description 2
- 210000003289 regulatory T cell Anatomy 0.000 description 2
- 230000003938 response to stress Effects 0.000 description 2
- 230000037390 scarring Effects 0.000 description 2
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- CYOHGALHFOKKQC-UHFFFAOYSA-N selumetinib Chemical compound OCCONC(=O)C=1C=C2N(C)C=NC2=C(F)C=1NC1=CC=C(Br)C=C1Cl CYOHGALHFOKKQC-UHFFFAOYSA-N 0.000 description 2
- 230000009758 senescence Effects 0.000 description 2
- 208000013223 septicemia Diseases 0.000 description 2
- 108091006024 signal transducing proteins Proteins 0.000 description 2
- 102000034285 signal transducing proteins Human genes 0.000 description 2
- 201000000849 skin cancer Diseases 0.000 description 2
- 208000000649 small cell carcinoma Diseases 0.000 description 2
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 2
- 239000000600 sorbitol Substances 0.000 description 2
- 235000010356 sorbitol Nutrition 0.000 description 2
- 210000000278 spinal cord Anatomy 0.000 description 2
- 230000000087 stabilizing effect Effects 0.000 description 2
- 238000010186 staining Methods 0.000 description 2
- 230000004936 stimulating effect Effects 0.000 description 2
- 210000002536 stromal cell Anatomy 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 230000001629 suppression Effects 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 238000010189 synthetic method Methods 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 238000003419 tautomerization reaction Methods 0.000 description 2
- HLZKNKRTKFSKGZ-UHFFFAOYSA-N tetradecan-1-ol Chemical compound CCCCCCCCCCCCCCO HLZKNKRTKFSKGZ-UHFFFAOYSA-N 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- 235000010487 tragacanth Nutrition 0.000 description 2
- 239000000196 tragacanth Substances 0.000 description 2
- 229940116362 tragacanth Drugs 0.000 description 2
- 206010044652 trigeminal neuralgia Diseases 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- 241000712461 unidentified influenza virus Species 0.000 description 2
- 230000004862 vasculogenesis Effects 0.000 description 2
- LLDWLPRYLVPDTG-UHFFFAOYSA-N vatalanib succinate Chemical compound OC(=O)CCC(O)=O.C1=CC(Cl)=CC=C1NC(C1=CC=CC=C11)=NN=C1CC1=CC=NC=C1 LLDWLPRYLVPDTG-UHFFFAOYSA-N 0.000 description 2
- 208000008662 verrucous carcinoma Diseases 0.000 description 2
- 208000017802 verrucous carcinoma of oral cavity Diseases 0.000 description 2
- 230000029812 viral genome replication Effects 0.000 description 2
- 230000003612 virological effect Effects 0.000 description 2
- 239000003039 volatile agent Substances 0.000 description 2
- 235000012431 wafers Nutrition 0.000 description 2
- 239000001993 wax Substances 0.000 description 2
- 230000029663 wound healing Effects 0.000 description 2
- 229940034727 zelboraf Drugs 0.000 description 2
- DHQMUJSACXTPEA-UHFFFAOYSA-N (2-methoxypyridin-4-yl)boronic acid Chemical compound COC1=CC(B(O)O)=CC=N1 DHQMUJSACXTPEA-UHFFFAOYSA-N 0.000 description 1
- YUXKOWPNKJSTPQ-AXWWPMSFSA-N (2s,3r)-2-amino-3-hydroxybutanoic acid;(2s)-2-amino-3-hydroxypropanoic acid Chemical compound OC[C@H](N)C(O)=O.C[C@@H](O)[C@H](N)C(O)=O YUXKOWPNKJSTPQ-AXWWPMSFSA-N 0.000 description 1
- KNXQDJCZSVHEIW-UHFFFAOYSA-N (3-fluorophenyl)boronic acid Chemical compound OB(O)C1=CC=CC(F)=C1 KNXQDJCZSVHEIW-UHFFFAOYSA-N 0.000 description 1
- NLLGFYPSWCMUIV-UHFFFAOYSA-N (3-methoxyphenyl)boronic acid Chemical compound COC1=CC=CC(B(O)O)=C1 NLLGFYPSWCMUIV-UHFFFAOYSA-N 0.000 description 1
- BJQCPCFFYBKRLM-UHFFFAOYSA-N (3-methylphenyl)boronic acid Chemical compound CC1=CC=CC(B(O)O)=C1 BJQCPCFFYBKRLM-UHFFFAOYSA-N 0.000 description 1
- QHFKWIKCUHNXAU-UHFFFAOYSA-N (4-nitrophenyl) carbamate Chemical class NC(=O)OC1=CC=C([N+]([O-])=O)C=C1 QHFKWIKCUHNXAU-UHFFFAOYSA-N 0.000 description 1
- QMMJWQMCMRUYTG-UHFFFAOYSA-N 1,2,4,5-tetrachloro-3-(trifluoromethyl)benzene Chemical compound FC(F)(F)C1=C(Cl)C(Cl)=CC(Cl)=C1Cl QMMJWQMCMRUYTG-UHFFFAOYSA-N 0.000 description 1
- DDMOUSALMHHKOS-UHFFFAOYSA-N 1,2-dichloro-1,1,2,2-tetrafluoroethane Chemical compound FC(F)(Cl)C(F)(F)Cl DDMOUSALMHHKOS-UHFFFAOYSA-N 0.000 description 1
- FSSPGSAQUIYDCN-UHFFFAOYSA-N 1,3-Propane sultone Chemical compound O=S1(=O)CCCO1 FSSPGSAQUIYDCN-UHFFFAOYSA-N 0.000 description 1
- IBCZSSWQZVYRBN-UHFFFAOYSA-N 1-(5-tert-butyl-2-phenylpyrazol-3-yl)-3-[2-fluoro-4-[(1-methyl-2-oxo-3h-imidazo[4,5-b]pyridin-7-yl)oxy]phenyl]urea Chemical compound C1=CN=C2NC(=O)N(C)C2=C1OC(C=C1F)=CC=C1NC(=O)NC1=CC(C(C)(C)C)=NN1C1=CC=CC=C1 IBCZSSWQZVYRBN-UHFFFAOYSA-N 0.000 description 1
- TUSDEZXZIZRFGC-UHFFFAOYSA-N 1-O-galloyl-3,6-(R)-HHDP-beta-D-glucose Natural products OC1C(O2)COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC1C(O)C2OC(=O)C1=CC(O)=C(O)C(O)=C1 TUSDEZXZIZRFGC-UHFFFAOYSA-N 0.000 description 1
- LGEZTMRIZWCDLW-UHFFFAOYSA-N 14-methylpentadecyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCCCCCCCCCCCCCC(C)C LGEZTMRIZWCDLW-UHFFFAOYSA-N 0.000 description 1
- JVVRJMXHNUAPHW-UHFFFAOYSA-N 1h-pyrazol-5-amine Chemical class NC=1C=CNN=1 JVVRJMXHNUAPHW-UHFFFAOYSA-N 0.000 description 1
- KOPFEFZSAMLEHK-UHFFFAOYSA-N 1h-pyrazole-5-carboxylic acid Chemical class OC(=O)C=1C=CNN=1 KOPFEFZSAMLEHK-UHFFFAOYSA-N 0.000 description 1
- NHBKXEKEPDILRR-UHFFFAOYSA-N 2,3-bis(butanoylsulfanyl)propyl butanoate Chemical compound CCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCC NHBKXEKEPDILRR-UHFFFAOYSA-N 0.000 description 1
- GFMMXOIFOQCCGU-UHFFFAOYSA-N 2-(2-chloro-4-iodoanilino)-N-(cyclopropylmethoxy)-3,4-difluorobenzamide Chemical compound C=1C=C(I)C=C(Cl)C=1NC1=C(F)C(F)=CC=C1C(=O)NOCC1CC1 GFMMXOIFOQCCGU-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- 125000005273 2-acetoxybenzoic acid group Chemical group 0.000 description 1
- CDAWCLOXVUBKRW-UHFFFAOYSA-N 2-aminophenol Chemical class NC1=CC=CC=C1O CDAWCLOXVUBKRW-UHFFFAOYSA-N 0.000 description 1
- 125000004182 2-chlorophenyl group Chemical group [H]C1=C([H])C(Cl)=C(*)C([H])=C1[H] 0.000 description 1
- RRAKUTCVRYDSFE-UHFFFAOYSA-N 2-fluoro-4-pyridin-4-ylaniline Chemical compound C1=C(F)C(N)=CC=C1C1=CC=NC=C1 RRAKUTCVRYDSFE-UHFFFAOYSA-N 0.000 description 1
- UPHOPMSGKZNELG-UHFFFAOYSA-N 2-hydroxynaphthalene-1-carboxylic acid Chemical compound C1=CC=C2C(C(=O)O)=C(O)C=CC2=C1 UPHOPMSGKZNELG-UHFFFAOYSA-N 0.000 description 1
- MSYGAHOHLUJIKV-UHFFFAOYSA-N 3,5-dimethyl-1-(3-nitrophenyl)-1h-pyrazole-4-carboxylic acid ethyl ester Chemical compound CC1=C(C(=O)OCC)C(C)=NN1C1=CC=CC([N+]([O-])=O)=C1 MSYGAHOHLUJIKV-UHFFFAOYSA-N 0.000 description 1
- WEVYNIUIFUYDGI-UHFFFAOYSA-N 3-[6-[4-(trifluoromethoxy)anilino]-4-pyrimidinyl]benzamide Chemical compound NC(=O)C1=CC=CC(C=2N=CN=C(NC=3C=CC(OC(F)(F)F)=CC=3)C=2)=C1 WEVYNIUIFUYDGI-UHFFFAOYSA-N 0.000 description 1
- 125000004179 3-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(Cl)=C1[H] 0.000 description 1
- 125000004180 3-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(F)=C1[H] 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- ROKOFZNQCIIJMI-UHFFFAOYSA-N 4-(4-fluorophenyl)-1-cycloropropylmethyl-5-(4-pyridyl)-imidazole Chemical compound C1=CC(F)=CC=C1C1=C(C=2C=CN=CC=2)N(CC2CC2)C=N1 ROKOFZNQCIIJMI-UHFFFAOYSA-N 0.000 description 1
- HIQIXEFWDLTDED-UHFFFAOYSA-N 4-hydroxy-1-piperidin-4-ylpyrrolidin-2-one Chemical compound O=C1CC(O)CN1C1CCNCC1 HIQIXEFWDLTDED-UHFFFAOYSA-N 0.000 description 1
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 1
- JKTORXLUQLQJCM-UHFFFAOYSA-N 4-phosphonobutylphosphonic acid Chemical compound OP(O)(=O)CCCCP(O)(O)=O JKTORXLUQLQJCM-UHFFFAOYSA-N 0.000 description 1
- WQUXJXDHYBIPCI-UHFFFAOYSA-N 5-tert-butyl-2-(3-methylphenyl)pyrazole-3-carboxylic acid Chemical compound C(C)(C)(C)C1=NN(C(=C1)C(=O)O)C1=CC(=CC=C1)C WQUXJXDHYBIPCI-UHFFFAOYSA-N 0.000 description 1
- 102100040084 A-kinase anchor protein 9 Human genes 0.000 description 1
- 208000004476 Acute Coronary Syndrome Diseases 0.000 description 1
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 1
- 230000007730 Akt signaling Effects 0.000 description 1
- 208000007848 Alcoholism Diseases 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 229930183010 Amphotericin Natural products 0.000 description 1
- QGGFZZLFKABGNL-UHFFFAOYSA-N Amphotericin A Natural products OC1C(N)C(O)C(C)OC1OC1C=CC=CC=CC=CCCC=CC=CC(C)C(O)C(C)C(C)OC(=O)CC(O)CC(O)CCC(O)C(O)CC(O)CC(O)(CC(O)C2C(O)=O)OC2C1 QGGFZZLFKABGNL-UHFFFAOYSA-N 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 108010009906 Angiopoietins Proteins 0.000 description 1
- 102000009840 Angiopoietins Human genes 0.000 description 1
- 102400000068 Angiostatin Human genes 0.000 description 1
- 108010079709 Angiostatins Proteins 0.000 description 1
- 201000002862 Angle-Closure Glaucoma Diseases 0.000 description 1
- 102000006306 Antigen Receptors Human genes 0.000 description 1
- 108010083359 Antigen Receptors Proteins 0.000 description 1
- 108091023037 Aptamer Proteins 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 206010003210 Arteriosclerosis Diseases 0.000 description 1
- 206010058029 Arthrofibrosis Diseases 0.000 description 1
- 241000132092 Aster Species 0.000 description 1
- 238000012935 Averaging Methods 0.000 description 1
- 208000000412 Avitaminosis Diseases 0.000 description 1
- 208000037157 Azotemia Diseases 0.000 description 1
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 1
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 208000035143 Bacterial infection Diseases 0.000 description 1
- 201000004569 Blindness Diseases 0.000 description 1
- 208000008720 Bone Marrow Neoplasms Diseases 0.000 description 1
- 208000020084 Bone disease Diseases 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 241000282817 Bovidae Species 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- 208000011691 Burkitt lymphomas Diseases 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- 0 CC(C)C(C)c1n[n](C2C=C(*)*=CC2C)c(N)c1 Chemical compound CC(C)C(C)c1n[n](C2C=C(*)*=CC2C)c(N)c1 0.000 description 1
- 206010006895 Cachexia Diseases 0.000 description 1
- 102000000905 Cadherin Human genes 0.000 description 1
- 108050007957 Cadherin Proteins 0.000 description 1
- 101100356682 Caenorhabditis elegans rho-1 gene Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 206010058019 Cancer Pain Diseases 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 201000000274 Carcinosarcoma Diseases 0.000 description 1
- 208000020446 Cardiac disease Diseases 0.000 description 1
- 206010007558 Cardiac failure chronic Diseases 0.000 description 1
- 206010007572 Cardiac hypertrophy Diseases 0.000 description 1
- 208000031229 Cardiomyopathies Diseases 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- 241001049165 Caria Species 0.000 description 1
- 108010067316 Catenins Proteins 0.000 description 1
- 102000016362 Catenins Human genes 0.000 description 1
- 208000001387 Causalgia Diseases 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- 102100025064 Cellular tumor antigen p53 Human genes 0.000 description 1
- 206010008190 Cerebrovascular accident Diseases 0.000 description 1
- 108091006146 Channels Proteins 0.000 description 1
- 208000005243 Chondrosarcoma Diseases 0.000 description 1
- 208000002691 Choroiditis Diseases 0.000 description 1
- 108010077544 Chromatin Proteins 0.000 description 1
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 1
- 244000060011 Cocos nucifera Species 0.000 description 1
- 235000013162 Cocos nucifera Nutrition 0.000 description 1
- 108020004705 Codon Proteins 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 206010010099 Combined immunodeficiency Diseases 0.000 description 1
- 208000023890 Complex Regional Pain Syndromes Diseases 0.000 description 1
- 241000709675 Coxsackievirus B3 Species 0.000 description 1
- 206010066946 Craniofacial dysostosis Diseases 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- 241000699802 Cricetulus griseus Species 0.000 description 1
- 229920002785 Croscarmellose sodium Polymers 0.000 description 1
- 201000006526 Crouzon syndrome Diseases 0.000 description 1
- 208000025962 Crush injury Diseases 0.000 description 1
- 102000005636 Cyclic AMP Response Element-Binding Protein Human genes 0.000 description 1
- 108010045171 Cyclic AMP Response Element-Binding Protein Proteins 0.000 description 1
- 206010058202 Cystoid macular oedema Diseases 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- 108020004414 DNA Proteins 0.000 description 1
- 230000004544 DNA amplification Effects 0.000 description 1
- 238000011238 DNA vaccination Methods 0.000 description 1
- 230000004568 DNA-binding Effects 0.000 description 1
- 241000252212 Danio rerio Species 0.000 description 1
- 201000004624 Dermatitis Diseases 0.000 description 1
- 208000012239 Developmental disease Diseases 0.000 description 1
- 208000002249 Diabetes Complications Diseases 0.000 description 1
- 208000007342 Diabetic Nephropathies Diseases 0.000 description 1
- 206010012655 Diabetic complications Diseases 0.000 description 1
- 239000004338 Dichlorodifluoromethane Substances 0.000 description 1
- 101000876610 Dictyostelium discoideum Extracellular signal-regulated kinase 2 Proteins 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- 206010059866 Drug resistance Diseases 0.000 description 1
- 101710146526 Dual specificity mitogen-activated protein kinase kinase 1 Proteins 0.000 description 1
- 102100023266 Dual specificity mitogen-activated protein kinase kinase 2 Human genes 0.000 description 1
- 239000012591 Dulbecco’s Phosphate Buffered Saline Substances 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- 102000012545 EGF-like domains Human genes 0.000 description 1
- 108050002150 EGF-like domains Proteins 0.000 description 1
- 108010041308 Endothelial Growth Factors Proteins 0.000 description 1
- 208000037487 Endotoxemia Diseases 0.000 description 1
- 101800003838 Epidermal growth factor Proteins 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 101150031329 Ets1 gene Proteins 0.000 description 1
- 241000289695 Eutheria Species 0.000 description 1
- 108700024394 Exon Proteins 0.000 description 1
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 1
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 1
- 101710105178 F-box/WD repeat-containing protein 7 Proteins 0.000 description 1
- 239000001263 FEMA 3042 Substances 0.000 description 1
- 101150021185 FGF gene Proteins 0.000 description 1
- 241000282324 Felis Species 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 102000003971 Fibroblast Growth Factor 1 Human genes 0.000 description 1
- 102100031706 Fibroblast growth factor 1 Human genes 0.000 description 1
- 102100024785 Fibroblast growth factor 2 Human genes 0.000 description 1
- 102000003972 Fibroblast growth factor 7 Human genes 0.000 description 1
- 102100027844 Fibroblast growth factor receptor 4 Human genes 0.000 description 1
- 101710182387 Fibroblast growth factor receptor 4 Proteins 0.000 description 1
- 201000008808 Fibrosarcoma Diseases 0.000 description 1
- 102000039539 Fos family Human genes 0.000 description 1
- 108091067362 Fos family Proteins 0.000 description 1
- 208000034951 Genetic Translocation Diseases 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 108010024636 Glutathione Proteins 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 241000282575 Gorilla Species 0.000 description 1
- 206010018634 Gouty Arthritis Diseases 0.000 description 1
- 208000003807 Graves Disease Diseases 0.000 description 1
- 208000015023 Graves' disease Diseases 0.000 description 1
- 102100025334 Guanine nucleotide-binding protein G(q) subunit alpha Human genes 0.000 description 1
- 208000035895 Guillain-Barré syndrome Diseases 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 206010066476 Haematological malignancy Diseases 0.000 description 1
- 208000030836 Hashimoto thyroiditis Diseases 0.000 description 1
- 206010019233 Headaches Diseases 0.000 description 1
- 208000002125 Hemangioendothelioma Diseases 0.000 description 1
- 208000002250 Hematologic Neoplasms Diseases 0.000 description 1
- 241000700721 Hepatitis B virus Species 0.000 description 1
- 208000005176 Hepatitis C Diseases 0.000 description 1
- 206010019842 Hepatomegaly Diseases 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000890598 Homo sapiens A-kinase anchor protein 9 Proteins 0.000 description 1
- 101000721661 Homo sapiens Cellular tumor antigen p53 Proteins 0.000 description 1
- 101001014196 Homo sapiens Dual specificity mitogen-activated protein kinase kinase 1 Proteins 0.000 description 1
- 101000857888 Homo sapiens Guanine nucleotide-binding protein G(q) subunit alpha Proteins 0.000 description 1
- 101001034652 Homo sapiens Insulin-like growth factor 1 receptor Proteins 0.000 description 1
- 101001052493 Homo sapiens Mitogen-activated protein kinase 1 Proteins 0.000 description 1
- 101000692455 Homo sapiens Platelet-derived growth factor receptor beta Proteins 0.000 description 1
- 101001047090 Homo sapiens Potassium voltage-gated channel subfamily H member 2 Proteins 0.000 description 1
- 101000820294 Homo sapiens Tyrosine-protein kinase Yes Proteins 0.000 description 1
- 241000700588 Human alphaherpesvirus 1 Species 0.000 description 1
- 241000701024 Human betaherpesvirus 5 Species 0.000 description 1
- 241000713772 Human immunodeficiency virus 1 Species 0.000 description 1
- 241000701589 Human papillomavirus type 6b Species 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 241000282620 Hylobates sp. Species 0.000 description 1
- 208000004454 Hyperalgesia Diseases 0.000 description 1
- 208000035150 Hypercholesterolemia Diseases 0.000 description 1
- 206010020772 Hypertension Diseases 0.000 description 1
- 206010020880 Hypertrophy Diseases 0.000 description 1
- 206010021135 Hypovitaminosis Diseases 0.000 description 1
- 208000024934 IgG4-related mediastinitis Diseases 0.000 description 1
- 208000014919 IgG4-related retroperitoneal fibrosis Diseases 0.000 description 1
- 206010061598 Immunodeficiency Diseases 0.000 description 1
- 241000712431 Influenza A virus Species 0.000 description 1
- 102000000589 Interleukin-1 Human genes 0.000 description 1
- 108010002352 Interleukin-1 Proteins 0.000 description 1
- 206010061252 Intraocular melanoma Diseases 0.000 description 1
- 206010073086 Iris melanoma Diseases 0.000 description 1
- 208000032382 Ischaemic stroke Diseases 0.000 description 1
- PIWKPBJCKXDKJR-UHFFFAOYSA-N Isoflurane Chemical compound FC(F)OC(Cl)C(F)(F)F PIWKPBJCKXDKJR-UHFFFAOYSA-N 0.000 description 1
- 206010069755 K-ras gene mutation Diseases 0.000 description 1
- 108010003046 KSR-1 protein kinase Proteins 0.000 description 1
- 208000002260 Keloid Diseases 0.000 description 1
- 206010023330 Keloid scar Diseases 0.000 description 1
- 208000008839 Kidney Neoplasms Diseases 0.000 description 1
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 1
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 241000283953 Lagomorpha Species 0.000 description 1
- 208000006552 Lewis Lung Carcinoma Diseases 0.000 description 1
- 208000028018 Lymphocytic leukaemia Diseases 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 102000002576 MAP Kinase Kinase 1 Human genes 0.000 description 1
- 108010068353 MAP Kinase Kinase 2 Proteins 0.000 description 1
- 239000007993 MOPS buffer Substances 0.000 description 1
- 238000000134 MTT assay Methods 0.000 description 1
- 231100000002 MTT assay Toxicity 0.000 description 1
- 241000289581 Macropus sp. Species 0.000 description 1
- 206010025421 Macule Diseases 0.000 description 1
- 208000035719 Maculopathy Diseases 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 208000006644 Malignant Fibrous Histiocytoma Diseases 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 101150100676 Map2k1 gene Proteins 0.000 description 1
- 101150024075 Mapk1 gene Proteins 0.000 description 1
- 208000002805 Mediastinal fibrosis Diseases 0.000 description 1
- 101001052524 Medicago sativa Mitogen-activated protein kinase homolog MMK1 Proteins 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- 206010027406 Mesothelioma Diseases 0.000 description 1
- 206010027452 Metastases to bone Diseases 0.000 description 1
- 206010059282 Metastases to central nervous system Diseases 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 208000019695 Migraine disease Diseases 0.000 description 1
- 206010049567 Miller Fisher syndrome Diseases 0.000 description 1
- 102100024193 Mitogen-activated protein kinase 1 Human genes 0.000 description 1
- 102100026888 Mitogen-activated protein kinase kinase kinase 7 Human genes 0.000 description 1
- 101100467524 Murine sarcoma virus 3611 V-RAF gene Proteins 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- 101100444898 Mus musculus Egr1 gene Proteins 0.000 description 1
- 102100038895 Myc proto-oncogene protein Human genes 0.000 description 1
- 101710135898 Myc proto-oncogene protein Proteins 0.000 description 1
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 1
- 108050000637 N-cadherin Proteins 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 125000000815 N-oxide group Chemical group 0.000 description 1
- 101150111783 NTRK1 gene Proteins 0.000 description 1
- 206010028729 Nasal cavity cancer Diseases 0.000 description 1
- 208000026344 Nasal disease Diseases 0.000 description 1
- 206010028767 Nasal sinus cancer Diseases 0.000 description 1
- CIZQBFOSWFQZTN-UHFFFAOYSA-N Nc(ccc(Oc(ccnc1N)c1[N+]([O-])=O)c1)c1F Chemical compound Nc(ccc(Oc(ccnc1N)c1[N+]([O-])=O)c1)c1F CIZQBFOSWFQZTN-UHFFFAOYSA-N 0.000 description 1
- 206010061309 Neoplasm progression Diseases 0.000 description 1
- 208000003510 Nephrogenic Fibrosing Dermopathy Diseases 0.000 description 1
- 206010067467 Nephrogenic systemic fibrosis Diseases 0.000 description 1
- 208000028389 Nerve injury Diseases 0.000 description 1
- 208000009905 Neurofibromatoses Diseases 0.000 description 1
- 208000036110 Neuroinflammatory disease Diseases 0.000 description 1
- 208000025966 Neurological disease Diseases 0.000 description 1
- 208000005890 Neuroma Diseases 0.000 description 1
- 206010029488 Nodular melanoma Diseases 0.000 description 1
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 1
- 102000007399 Nuclear hormone receptor Human genes 0.000 description 1
- 108020005497 Nuclear hormone receptor Proteins 0.000 description 1
- 206010030348 Open-Angle Glaucoma Diseases 0.000 description 1
- 208000001132 Osteoporosis Diseases 0.000 description 1
- 208000007571 Ovarian Epithelial Carcinoma Diseases 0.000 description 1
- 239000012661 PARP inhibitor Substances 0.000 description 1
- SUDAHWBOROXANE-SECBINFHSA-N PD 0325901 Chemical compound OC[C@@H](O)CONC(=O)C1=CC=C(F)C(F)=C1NC1=CC=C(I)C=C1F SUDAHWBOROXANE-SECBINFHSA-N 0.000 description 1
- 239000012823 PI3K/mTOR inhibitor Substances 0.000 description 1
- YZDJQTHVDDOVHR-UHFFFAOYSA-N PLX-4720 Chemical compound CCCS(=O)(=O)NC1=CC=C(F)C(C(=O)C=2C3=CC(Cl)=CN=C3NC=2)=C1F YZDJQTHVDDOVHR-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 241000282577 Pan troglodytes Species 0.000 description 1
- 241000845082 Panama Species 0.000 description 1
- 241001504519 Papio ursinus Species 0.000 description 1
- 208000030852 Parasitic disease Diseases 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- 208000031481 Pathologic Constriction Diseases 0.000 description 1
- 208000034038 Pathologic Neovascularization Diseases 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- LRBQNJMCXXYXIU-PPKXGCFTSA-N Penta-digallate-beta-D-glucose Natural products OC1=C(O)C(O)=CC(C(=O)OC=2C(=C(O)C=C(C=2)C(=O)OC[C@@H]2[C@H]([C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)O2)OC(=O)C=2C=C(OC(=O)C=3C=C(O)C(O)=C(O)C=3)C(O)=C(O)C=2)O)=C1 LRBQNJMCXXYXIU-PPKXGCFTSA-N 0.000 description 1
- 206010034464 Periarthritis Diseases 0.000 description 1
- 208000031135 Pfeiffer syndrome type 2 Diseases 0.000 description 1
- 208000004983 Phantom Limb Diseases 0.000 description 1
- 206010056238 Phantom pain Diseases 0.000 description 1
- YGYAWVDWMABLBF-UHFFFAOYSA-N Phosgene Chemical compound ClC(Cl)=O YGYAWVDWMABLBF-UHFFFAOYSA-N 0.000 description 1
- 229940123932 Phosphodiesterase 4 inhibitor Drugs 0.000 description 1
- 241000709664 Picornaviridae Species 0.000 description 1
- 102100026547 Platelet-derived growth factor receptor beta Human genes 0.000 description 1
- 229940121906 Poly ADP ribose polymerase inhibitor Drugs 0.000 description 1
- 208000008601 Polycythemia Diseases 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 229920001214 Polysorbate 60 Polymers 0.000 description 1
- 241000282405 Pongo abelii Species 0.000 description 1
- 208000003971 Posterior uveitis Diseases 0.000 description 1
- 102100022807 Potassium voltage-gated channel subfamily H member 2 Human genes 0.000 description 1
- 102100025067 Potassium voltage-gated channel subfamily H member 4 Human genes 0.000 description 1
- 101710163352 Potassium voltage-gated channel subfamily H member 4 Proteins 0.000 description 1
- 208000026149 Primary peritoneal carcinoma Diseases 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 102100024622 Proenkephalin-B Human genes 0.000 description 1
- 108091007744 Programmed cell death receptors Proteins 0.000 description 1
- 108090000315 Protein Kinase C Proteins 0.000 description 1
- 102000003923 Protein Kinase C Human genes 0.000 description 1
- 102000052575 Proto-Oncogene Human genes 0.000 description 1
- 108700020978 Proto-Oncogene Proteins 0.000 description 1
- 241000125945 Protoparvovirus Species 0.000 description 1
- 241000589517 Pseudomonas aeruginosa Species 0.000 description 1
- 101150111584 RHOA gene Proteins 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 206010037779 Radiculopathy Diseases 0.000 description 1
- 108091005682 Receptor kinases Proteins 0.000 description 1
- 101710100968 Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 1
- 208000033464 Reiter syndrome Diseases 0.000 description 1
- 206010061481 Renal injury Diseases 0.000 description 1
- 241000702263 Reovirus sp. Species 0.000 description 1
- 208000013616 Respiratory Distress Syndrome Diseases 0.000 description 1
- 206010038748 Restrictive cardiomyopathy Diseases 0.000 description 1
- 208000017442 Retinal disease Diseases 0.000 description 1
- 206010038923 Retinopathy Diseases 0.000 description 1
- 206010038933 Retinopathy of prematurity Diseases 0.000 description 1
- 206010038979 Retroperitoneal fibrosis Diseases 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 102000014400 SH2 domains Human genes 0.000 description 1
- 108050003452 SH2 domains Proteins 0.000 description 1
- 102000000395 SH3 domains Human genes 0.000 description 1
- 108050008861 SH3 domains Proteins 0.000 description 1
- RYMZZMVNJRMUDD-UHFFFAOYSA-N SJ000286063 Natural products C12C(OC(=O)C(C)(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 RYMZZMVNJRMUDD-UHFFFAOYSA-N 0.000 description 1
- 206010039710 Scleroderma Diseases 0.000 description 1
- 201000010208 Seminoma Diseases 0.000 description 1
- 208000021712 Soft tissue sarcoma Diseases 0.000 description 1
- NWGKJDSIEKMTRX-AAZCQSIUSA-N Sorbitan monooleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O NWGKJDSIEKMTRX-AAZCQSIUSA-N 0.000 description 1
- 201000002661 Spondylitis Diseases 0.000 description 1
- 208000035518 Squamous cell carcinoma of the oropharynx Diseases 0.000 description 1
- 201000009594 Systemic Scleroderma Diseases 0.000 description 1
- 206010042953 Systemic sclerosis Diseases 0.000 description 1
- 102000005450 TIE receptors Human genes 0.000 description 1
- 108010006830 TIE receptors Proteins 0.000 description 1
- 108010090089 TIE-1 Receptor Proteins 0.000 description 1
- 108700012920 TNF Proteins 0.000 description 1
- 239000004098 Tetracycline Substances 0.000 description 1
- 244000299461 Theobroma cacao Species 0.000 description 1
- 235000009470 Theobroma cacao Nutrition 0.000 description 1
- 238000012338 Therapeutic targeting Methods 0.000 description 1
- 206010062129 Tongue neoplasm Diseases 0.000 description 1
- 206010044248 Toxic shock syndrome Diseases 0.000 description 1
- 231100000650 Toxic shock syndrome Toxicity 0.000 description 1
- 101710150448 Transcriptional regulator Myc Proteins 0.000 description 1
- 206010048873 Traumatic arthritis Diseases 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 239000013504 Triton X-100 Substances 0.000 description 1
- 229920004890 Triton X-100 Polymers 0.000 description 1
- 206010044696 Tropical spastic paresis Diseases 0.000 description 1
- 102000044209 Tumor Suppressor Genes Human genes 0.000 description 1
- 108700025716 Tumor Suppressor Genes Proteins 0.000 description 1
- 206010054094 Tumour necrosis Diseases 0.000 description 1
- 102100030398 Twist-related protein 1 Human genes 0.000 description 1
- 102000011017 Type 4 Cyclic Nucleotide Phosphodiesterases Human genes 0.000 description 1
- 108010037584 Type 4 Cyclic Nucleotide Phosphodiesterases Proteins 0.000 description 1
- 102100021788 Tyrosine-protein kinase Yes Human genes 0.000 description 1
- 208000015778 Undifferentiated pleomorphic sarcoma Diseases 0.000 description 1
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 1
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 1
- 108010053096 Vascular Endothelial Growth Factor Receptor-1 Proteins 0.000 description 1
- 208000032594 Vascular Remodeling Diseases 0.000 description 1
- 102100039037 Vascular endothelial growth factor A Human genes 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- WOAORAPRPVIATR-UHFFFAOYSA-N [3-(trifluoromethyl)phenyl]boronic acid Chemical compound OB(O)C1=CC=CC(C(F)(F)F)=C1 WOAORAPRPVIATR-UHFFFAOYSA-N 0.000 description 1
- SORGEQQSQGNZFI-UHFFFAOYSA-N [azido(phenoxy)phosphoryl]oxybenzene Chemical compound C=1C=CC=CC=1OP(=O)(N=[N+]=[N-])OC1=CC=CC=C1 SORGEQQSQGNZFI-UHFFFAOYSA-N 0.000 description 1
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 208000038016 acute inflammation Diseases 0.000 description 1
- 230000006022 acute inflammation Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000001464 adherent effect Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 208000011341 adult acute respiratory distress syndrome Diseases 0.000 description 1
- 201000000028 adult respiratory distress syndrome Diseases 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 238000001042 affinity chromatography Methods 0.000 description 1
- 108010081667 aflibercept Proteins 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 229910001413 alkali metal ion Inorganic materials 0.000 description 1
- 125000005907 alkyl ester group Chemical group 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 201000009961 allergic asthma Diseases 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 1
- 229940009444 amphotericin Drugs 0.000 description 1
- APKFDSVGJQXUKY-INPOYWNPSA-N amphotericin B Chemical compound O[C@H]1[C@@H](N)[C@H](O)[C@@H](C)O[C@H]1O[C@H]1/C=C/C=C/C=C/C=C/C=C/C=C/C=C/[C@H](C)[C@@H](O)[C@@H](C)[C@H](C)OC(=O)C[C@H](O)C[C@H](O)CC[C@@H](O)[C@H](O)C[C@H](O)C[C@](O)(C[C@H](O)[C@H]2C(O)=O)O[C@H]2C1 APKFDSVGJQXUKY-INPOYWNPSA-N 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 230000003698 anagen phase Effects 0.000 description 1
- 239000003098 androgen Substances 0.000 description 1
- 230000006481 angiogenic pathway Effects 0.000 description 1
- 230000006427 angiogenic response Effects 0.000 description 1
- 238000002399 angioplasty Methods 0.000 description 1
- 150000001449 anionic compounds Chemical class 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 230000002456 anti-arthritic effect Effects 0.000 description 1
- 229940124650 anti-cancer therapies Drugs 0.000 description 1
- 230000003510 anti-fibrotic effect Effects 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 230000002555 anti-neurodegenerative effect Effects 0.000 description 1
- 230000001028 anti-proliverative effect Effects 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 238000011319 anticancer therapy Methods 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- IMOZEMNVLZVGJZ-QGZVFWFLSA-N apremilast Chemical compound C1=C(OC)C(OCC)=CC([C@@H](CS(C)(=O)=O)N2C(C3=C(NC(C)=O)C=CC=C3C2=O)=O)=C1 IMOZEMNVLZVGJZ-QGZVFWFLSA-N 0.000 description 1
- 229960001164 apremilast Drugs 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 235000019568 aromas Nutrition 0.000 description 1
- 150000004982 aromatic amines Chemical class 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 208000011775 arteriosclerosis disease Diseases 0.000 description 1
- 238000006254 arylation reaction Methods 0.000 description 1
- FZCSTZYAHCUGEM-UHFFFAOYSA-N aspergillomarasmine B Natural products OC(=O)CNC(C(O)=O)CNC(C(O)=O)CC(O)=O FZCSTZYAHCUGEM-UHFFFAOYSA-N 0.000 description 1
- 230000001746 atrial effect Effects 0.000 description 1
- 230000001363 autoimmune Effects 0.000 description 1
- 230000004900 autophagic degradation Effects 0.000 description 1
- 230000035578 autophosphorylation Effects 0.000 description 1
- AMEDKBHURXXSQO-UHFFFAOYSA-N azonous acid Chemical compound ONO AMEDKBHURXXSQO-UHFFFAOYSA-N 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 208000022362 bacterial infectious disease Diseases 0.000 description 1
- 239000000022 bacteriostatic agent Substances 0.000 description 1
- 230000003385 bacteriostatic effect Effects 0.000 description 1
- XFILPEOLDIKJHX-QYZOEREBSA-N batimastat Chemical compound C([C@@H](C(=O)NC)NC(=O)[C@H](CC(C)C)[C@H](CSC=1SC=CC=1)C(=O)NO)C1=CC=CC=C1 XFILPEOLDIKJHX-QYZOEREBSA-N 0.000 description 1
- 229950001858 batimastat Drugs 0.000 description 1
- 238000010009 beating Methods 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- 210000003445 biliary tract Anatomy 0.000 description 1
- 238000010256 biochemical assay Methods 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000008033 biological extinction Effects 0.000 description 1
- 238000010170 biological method Methods 0.000 description 1
- 210000000601 blood cell Anatomy 0.000 description 1
- 239000012503 blood component Substances 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 238000006664 bond formation reaction Methods 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 208000019664 bone resorption disease Diseases 0.000 description 1
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- FUFJGUQYACFECW-UHFFFAOYSA-L calcium hydrogenphosphate Chemical compound [Ca+2].OP([O-])([O-])=O FUFJGUQYACFECW-UHFFFAOYSA-L 0.000 description 1
- 230000005907 cancer growth Effects 0.000 description 1
- 239000012830 cancer therapeutic Substances 0.000 description 1
- 208000014729 capillary malformation Diseases 0.000 description 1
- OXZUGZIPIDPXGC-UHFFFAOYSA-N carbamic acid;1-hydroxypyrrolidine-2,5-dione Chemical class NC(O)=O.ON1C(=O)CCC1=O OXZUGZIPIDPXGC-UHFFFAOYSA-N 0.000 description 1
- ZCIHBTYOSGLSMF-UHFFFAOYSA-N carbamic acid;1h-pyrazol-5-amine Chemical class NC(O)=O.NC1=CC=NN1 ZCIHBTYOSGLSMF-UHFFFAOYSA-N 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- SKOLWUPSYHWYAM-UHFFFAOYSA-N carbonodithioic O,S-acid Chemical compound SC(S)=O SKOLWUPSYHWYAM-UHFFFAOYSA-N 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- JJWKPURADFRFRB-UHFFFAOYSA-N carbonyl sulfide Chemical compound O=C=S JJWKPURADFRFRB-UHFFFAOYSA-N 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 238000012754 cardiac puncture Methods 0.000 description 1
- 150000001767 cationic compounds Chemical class 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 239000006143 cell culture medium Substances 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 230000004709 cell invasion Effects 0.000 description 1
- 230000012292 cell migration Effects 0.000 description 1
- 238000001516 cell proliferation assay Methods 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 210000002421 cell wall Anatomy 0.000 description 1
- 230000033077 cellular process Effects 0.000 description 1
- 230000004637 cellular stress Effects 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 208000025669 cerebellar pilocytic astrocytoma Diseases 0.000 description 1
- 208000019065 cervical carcinoma Diseases 0.000 description 1
- 210000003679 cervix uteri Anatomy 0.000 description 1
- AOGYCOYQMAVAFD-UHFFFAOYSA-N chlorocarbonic acid Chemical class OC(Cl)=O AOGYCOYQMAVAFD-UHFFFAOYSA-N 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 210000003483 chromatin Anatomy 0.000 description 1
- 208000037976 chronic inflammation Diseases 0.000 description 1
- 230000006020 chronic inflammation Effects 0.000 description 1
- 201000010902 chronic myelomonocytic leukemia Diseases 0.000 description 1
- LOUPRKONTZGTKE-UHFFFAOYSA-N cinchonine Natural products C1C(C(C2)C=C)CCN2C1C(O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-UHFFFAOYSA-N 0.000 description 1
- 230000007882 cirrhosis Effects 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- 230000008045 co-localization Effects 0.000 description 1
- 239000003245 coal Substances 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- RESIMIUSNACMNW-BXRWSSRYSA-N cobimetinib fumarate Chemical compound OC(=O)\C=C\C(O)=O.C1C(O)([C@H]2NCCCC2)CN1C(=O)C1=CC=C(F)C(F)=C1NC1=CC=C(I)C=C1F.C1C(O)([C@H]2NCCCC2)CN1C(=O)C1=CC=C(F)C(F)=C1NC1=CC=C(I)C=C1F RESIMIUSNACMNW-BXRWSSRYSA-N 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 230000009918 complex formation Effects 0.000 description 1
- 208000014439 complex regional pain syndrome type 2 Diseases 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 239000004020 conductor Substances 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 210000004351 coronary vessel Anatomy 0.000 description 1
- 239000002537 cosmetic Substances 0.000 description 1
- 239000001767 crosslinked sodium carboxy methyl cellulose Substances 0.000 description 1
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 1
- 239000010779 crude oil Substances 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- 208000030381 cutaneous melanoma Diseases 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 201000010206 cystoid macular edema Diseases 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 230000009849 deactivation Effects 0.000 description 1
- SASYSVUEVMOWPL-NXVVXOECSA-N decyl oleate Chemical compound CCCCCCCCCCOC(=O)CCCCCCC\C=C/CCCCCCCC SASYSVUEVMOWPL-NXVVXOECSA-N 0.000 description 1
- 230000000593 degrading effect Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 238000000432 density-gradient centrifugation Methods 0.000 description 1
- 231100000223 dermal penetration Toxicity 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- AQEFLFZSWDEAIP-UHFFFAOYSA-N di-tert-butyl ether Chemical compound CC(C)(C)OC(C)(C)C AQEFLFZSWDEAIP-UHFFFAOYSA-N 0.000 description 1
- 208000033679 diabetic kidney disease Diseases 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 150000004985 diamines Chemical class 0.000 description 1
- 235000019700 dicalcium phosphate Nutrition 0.000 description 1
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 1
- 235000019404 dichlorodifluoromethane Nutrition 0.000 description 1
- 229940042935 dichlorodifluoromethane Drugs 0.000 description 1
- 229940087091 dichlorotetrafluoroethane Drugs 0.000 description 1
- 150000005690 diesters Chemical class 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 210000002249 digestive system Anatomy 0.000 description 1
- 150000004683 dihydrates Chemical class 0.000 description 1
- IJKVHSBPTUYDLN-UHFFFAOYSA-N dihydroxy(oxo)silane Chemical compound O[Si](O)=O IJKVHSBPTUYDLN-UHFFFAOYSA-N 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- MKRTXPORKIRPDG-UHFFFAOYSA-N diphenylphosphoryl azide Chemical compound C=1C=CC=CC=1P(=O)(N=[N+]=[N-])C1=CC=CC=C1 MKRTXPORKIRPDG-UHFFFAOYSA-N 0.000 description 1
- 208000016097 disease of metabolism Diseases 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 239000003534 dna topoisomerase inhibitor Substances 0.000 description 1
- MVCOAUNKQVWQHZ-UHFFFAOYSA-N doramapimod Chemical compound C1=CC(C)=CC=C1N1C(NC(=O)NC=2C3=CC=CC=C3C(OCCN3CCOCC3)=CC=2)=CC(C(C)(C)C)=N1 MVCOAUNKQVWQHZ-UHFFFAOYSA-N 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 230000007783 downstream signaling Effects 0.000 description 1
- 239000006196 drop Substances 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 238000007905 drug manufacturing Methods 0.000 description 1
- 241001492478 dsDNA viruses, no RNA stage Species 0.000 description 1
- 241001493065 dsRNA viruses Species 0.000 description 1
- 229950009791 durvalumab Drugs 0.000 description 1
- 239000000428 dust Substances 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 239000012149 elution buffer Substances 0.000 description 1
- 239000002895 emetic Substances 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 239000008387 emulsifying waxe Substances 0.000 description 1
- 150000002081 enamines Chemical class 0.000 description 1
- 201000010048 endomyocardial fibrosis Diseases 0.000 description 1
- 230000010595 endothelial cell migration Effects 0.000 description 1
- 108091007231 endothelial receptors Proteins 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 230000006353 environmental stress Effects 0.000 description 1
- YQGOJNYOYNNSMM-UHFFFAOYSA-N eosin Chemical compound [Na+].OC(=O)C1=CC=CC=C1C1=C2C=C(Br)C(=O)C(Br)=C2OC2=C(Br)C(O)=C(Br)C=C21 YQGOJNYOYNNSMM-UHFFFAOYSA-N 0.000 description 1
- 108060002566 ephrin Proteins 0.000 description 1
- 102000012803 ephrin Human genes 0.000 description 1
- 229940116977 epidermal growth factor Drugs 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 108700021358 erbB-1 Genes Proteins 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- JIOGOANRZBZWDU-UHFFFAOYSA-N ethyl 5-tert-butyl-2-(2-methoxypyridin-4-yl)pyrazole-3-carboxylate Chemical compound CCOC(=O)C1=CC(=NN1C1=CC=NC(OC)=C1)C(C)(C)C JIOGOANRZBZWDU-UHFFFAOYSA-N 0.000 description 1
- BCDBJUDBGVARTC-UHFFFAOYSA-N ethyl 5-tert-butyl-2-(3-fluorophenyl)pyrazole-3-carboxylate Chemical compound CCOC(=O)C1=CC(C(C)(C)C)=NN1C1=CC=CC(F)=C1 BCDBJUDBGVARTC-UHFFFAOYSA-N 0.000 description 1
- BVGQQDNVKLMVAB-UHFFFAOYSA-N ethyl 5-tert-butyl-2-[3-(trifluoromethyl)phenyl]pyrazole-3-carboxylate Chemical compound CCOC(=O)C1=CC(=NN1C1=CC=CC(=C1)C(F)(F)F)C(C)(C)C BVGQQDNVKLMVAB-UHFFFAOYSA-N 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 238000001400 expression cloning Methods 0.000 description 1
- 210000002744 extracellular matrix Anatomy 0.000 description 1
- 230000008622 extracellular signaling Effects 0.000 description 1
- 210000000887 face Anatomy 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 210000005002 female reproductive tract Anatomy 0.000 description 1
- 210000003754 fetus Anatomy 0.000 description 1
- 230000000893 fibroproliferative effect Effects 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 238000013100 final test Methods 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 210000001650 focal adhesion Anatomy 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 239000012737 fresh medium Substances 0.000 description 1
- NGGMYCMLYOUNGM-CSDLUJIJSA-M fumagillin(1-) Chemical class C([C@H]([C@H]([C@@H]1[C@]2(C)[C@H](O2)CC=C(C)C)OC)OC(=O)\C=C\C=C\C=C\C=C\C([O-])=O)C[C@@]21CO2 NGGMYCMLYOUNGM-CSDLUJIJSA-M 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 238000012268 genome sequencing Methods 0.000 description 1
- 210000004907 gland Anatomy 0.000 description 1
- 229950000918 glembatumumab Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 229960003180 glutathione Drugs 0.000 description 1
- 229940075507 glyceryl monostearate Drugs 0.000 description 1
- 239000004519 grease Substances 0.000 description 1
- 208000035474 group of disease Diseases 0.000 description 1
- 201000009277 hairy cell leukemia Diseases 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- BCQZXOMGPXTTIC-UHFFFAOYSA-N halothane Chemical compound FC(F)(F)C(Cl)Br BCQZXOMGPXTTIC-UHFFFAOYSA-N 0.000 description 1
- 229960003132 halothane Drugs 0.000 description 1
- 210000003128 head Anatomy 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 231100000869 headache Toxicity 0.000 description 1
- 230000035876 healing Effects 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 208000019622 heart disease Diseases 0.000 description 1
- 230000002489 hematologic effect Effects 0.000 description 1
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 1
- 208000034737 hemoglobinopathy Diseases 0.000 description 1
- 230000011132 hemopoiesis Effects 0.000 description 1
- 208000002672 hepatitis B Diseases 0.000 description 1
- 108700025184 hepatitis B virus X Proteins 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- UQEAIHBTYFGYIE-UHFFFAOYSA-N hexamethyldisiloxane Chemical compound C[Si](C)(C)O[Si](C)(C)C UQEAIHBTYFGYIE-UHFFFAOYSA-N 0.000 description 1
- 230000013632 homeostatic process Effects 0.000 description 1
- 239000000710 homodimer Substances 0.000 description 1
- 208000018821 hormone-resistant prostate carcinoma Diseases 0.000 description 1
- 102000050152 human BRAF Human genes 0.000 description 1
- 208000024697 human cytomegalovirus infection Diseases 0.000 description 1
- 239000003906 humectant Substances 0.000 description 1
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical compound [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 239000008309 hydrophilic cream Substances 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 230000002390 hyperplastic effect Effects 0.000 description 1
- 230000009610 hypersensitivity Effects 0.000 description 1
- 206010020871 hypertrophic cardiomyopathy Diseases 0.000 description 1
- 208000003532 hypothyroidism Diseases 0.000 description 1
- 230000002989 hypothyroidism Effects 0.000 description 1
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 1
- 229960002411 imatinib Drugs 0.000 description 1
- 150000002466 imines Chemical class 0.000 description 1
- 229940127121 immunoconjugate Drugs 0.000 description 1
- 230000007813 immunodeficiency Effects 0.000 description 1
- 230000002055 immunohistochemical effect Effects 0.000 description 1
- 229960001438 immunostimulant agent Drugs 0.000 description 1
- 238000009169 immunotherapy Methods 0.000 description 1
- 238000005470 impregnation Methods 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 206010022000 influenza Diseases 0.000 description 1
- 208000018337 inherited hemoglobinopathy Diseases 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 239000002054 inoculum Substances 0.000 description 1
- 229910001412 inorganic anion Inorganic materials 0.000 description 1
- 229910001411 inorganic cation Inorganic materials 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 238000007689 inspection Methods 0.000 description 1
- 229950001014 intetumumab Drugs 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 229940078545 isocetyl stearate Drugs 0.000 description 1
- 229960002725 isoflurane Drugs 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- XUGNVMKQXJXZCD-UHFFFAOYSA-N isopropyl palmitate Chemical compound CCCCCCCCCCCCCCCC(=O)OC(C)C XUGNVMKQXJXZCD-UHFFFAOYSA-N 0.000 description 1
- 210000001117 keloid Anatomy 0.000 description 1
- 201000010982 kidney cancer Diseases 0.000 description 1
- 210000003127 knee Anatomy 0.000 description 1
- 238000011005 laboratory method Methods 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 201000010901 lateral sclerosis Diseases 0.000 description 1
- 201000010260 leiomyoma Diseases 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 108020001756 ligand binding domains Proteins 0.000 description 1
- 238000011542 limb amputation Methods 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 229940057995 liquid paraffin Drugs 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 201000005243 lung squamous cell carcinoma Diseases 0.000 description 1
- 206010025135 lupus erythematosus Diseases 0.000 description 1
- 230000001926 lymphatic effect Effects 0.000 description 1
- 210000004324 lymphatic system Anatomy 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- 125000000040 m-tolyl group Chemical group [H]C1=C([H])C(*)=C([H])C(=C1[H])C([H])([H])[H] 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 230000036244 malformation Effects 0.000 description 1
- 201000002350 malignant ciliary body melanoma Diseases 0.000 description 1
- 208000006178 malignant mesothelioma Diseases 0.000 description 1
- 201000005282 malignant pleural mesothelioma Diseases 0.000 description 1
- 210000001161 mammalian embryo Anatomy 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 241001515942 marmosets Species 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 201000000638 mature B-cell neoplasm Diseases 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 210000001370 mediastinum Anatomy 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000028161 membrane depolarization Effects 0.000 description 1
- 108020004084 membrane receptors Proteins 0.000 description 1
- 230000005906 menstruation Effects 0.000 description 1
- 208000030159 metabolic disease Diseases 0.000 description 1
- 230000037353 metabolic pathway Effects 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 206010027599 migraine Diseases 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 230000002297 mitogenic effect Effects 0.000 description 1
- 239000007932 molded tablet Substances 0.000 description 1
- 230000004784 molecular pathogenesis Effects 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 1
- 125000000896 monocarboxylic acid group Chemical group 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- 208000005264 motor neuron disease Diseases 0.000 description 1
- 229940124303 multikinase inhibitor Drugs 0.000 description 1
- 239000003471 mutagenic agent Substances 0.000 description 1
- 231100000707 mutagenic chemical Toxicity 0.000 description 1
- 230000003505 mutagenic effect Effects 0.000 description 1
- 230000036438 mutation frequency Effects 0.000 description 1
- 230000000869 mutational effect Effects 0.000 description 1
- 206010028417 myasthenia gravis Diseases 0.000 description 1
- 206010028537 myelofibrosis Diseases 0.000 description 1
- 201000000050 myeloid neoplasm Diseases 0.000 description 1
- 230000002071 myeloproliferative effect Effects 0.000 description 1
- 208000031225 myocardial ischemia Diseases 0.000 description 1
- 229940043348 myristyl alcohol Drugs 0.000 description 1
- 208000009091 myxoma Diseases 0.000 description 1
- 208000001611 myxosarcoma Diseases 0.000 description 1
- PWDYHMBTPGXCSN-VCBMUGGBSA-N n,n'-bis[3,5-bis[(e)-n-(diaminomethylideneamino)-c-methylcarbonimidoyl]phenyl]decanediamide Chemical compound NC(N)=N/N=C(\C)C1=CC(C(=N/N=C(N)N)/C)=CC(NC(=O)CCCCCCCCC(=O)NC=2C=C(C=C(C=2)C(\C)=N\N=C(N)N)C(\C)=N\N=C(N)N)=C1 PWDYHMBTPGXCSN-VCBMUGGBSA-N 0.000 description 1
- GTWJETSWSUWSEJ-UHFFFAOYSA-N n-benzylaniline Chemical compound C=1C=CC=CC=1CNC1=CC=CC=C1 GTWJETSWSUWSEJ-UHFFFAOYSA-N 0.000 description 1
- 201000000494 nasal cavity squamous cell carcinoma Diseases 0.000 description 1
- 239000007923 nasal drop Substances 0.000 description 1
- 229940100662 nasal drops Drugs 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- 230000001338 necrotic effect Effects 0.000 description 1
- 230000017095 negative regulation of cell growth Effects 0.000 description 1
- 230000013152 negative regulation of cell migration Effects 0.000 description 1
- 230000035407 negative regulation of cell proliferation Effects 0.000 description 1
- 230000019079 negative regulation of cytokine secretion Effects 0.000 description 1
- 230000031990 negative regulation of inflammatory response Effects 0.000 description 1
- 208000010915 neoplasm of mature B-cells Diseases 0.000 description 1
- 210000005170 neoplastic cell Anatomy 0.000 description 1
- 230000001613 neoplastic effect Effects 0.000 description 1
- 201000008383 nephritis Diseases 0.000 description 1
- 210000005036 nerve Anatomy 0.000 description 1
- 230000008764 nerve damage Effects 0.000 description 1
- 206010061311 nervous system neoplasm Diseases 0.000 description 1
- 230000000626 neurodegenerative effect Effects 0.000 description 1
- 201000004931 neurofibromatosis Diseases 0.000 description 1
- 230000001272 neurogenic effect Effects 0.000 description 1
- 230000003959 neuroinflammation Effects 0.000 description 1
- 230000002232 neuromuscular Effects 0.000 description 1
- 230000002981 neuropathic effect Effects 0.000 description 1
- 201000001119 neuropathy Diseases 0.000 description 1
- 230000007823 neuropathy Effects 0.000 description 1
- 230000003472 neutralizing effect Effects 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 125000000018 nitroso group Chemical group N(=O)* 0.000 description 1
- GQPLMRYTRLFLPF-UHFFFAOYSA-N nitrous oxide Inorganic materials [O-][N+]#N GQPLMRYTRLFLPF-UHFFFAOYSA-N 0.000 description 1
- 229960003301 nivolumab Drugs 0.000 description 1
- 201000000032 nodular malignant melanoma Diseases 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- 230000000414 obstructive effect Effects 0.000 description 1
- 201000002575 ocular melanoma Diseases 0.000 description 1
- 239000003883 ointment base Substances 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid group Chemical group C(CCCCCCC\C=C/CCCCCCCC)(=O)O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 230000006548 oncogenic transformation Effects 0.000 description 1
- 229950002104 ontuxizumab Drugs 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000002892 organic cations Chemical class 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 210000003300 oropharynx Anatomy 0.000 description 1
- 208000008798 osteoma Diseases 0.000 description 1
- 230000016087 ovulation Effects 0.000 description 1
- 150000002923 oximes Chemical class 0.000 description 1
- 230000008050 pain signaling Effects 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-N palmitic acid group Chemical group C(CCCCCCCCCCCCCCC)(=O)O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 1
- RUVINXPYWBROJD-UHFFFAOYSA-N para-methoxyphenyl Natural products COC1=CC=C(C=CC)C=C1 RUVINXPYWBROJD-UHFFFAOYSA-N 0.000 description 1
- 230000003076 paracrine Effects 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 210000003695 paranasal sinus Anatomy 0.000 description 1
- 208000022697 paranasal sinus squamous cell carcinoma Diseases 0.000 description 1
- 230000003071 parasitic effect Effects 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 230000001991 pathophysiological effect Effects 0.000 description 1
- ACNHBCIZLNNLRS-UBGQALKQSA-N paxilline Chemical compound N1C2=CC=CC=C2C2=C1[C@]1(C)[C@@]3(C)CC[C@@H]4O[C@H](C(C)(O)C)C(=O)C=C4[C@]3(O)CC[C@H]1C2 ACNHBCIZLNNLRS-UBGQALKQSA-N 0.000 description 1
- 229960002621 pembrolizumab Drugs 0.000 description 1
- 210000000578 peripheral nerve Anatomy 0.000 description 1
- 210000001428 peripheral nervous system Anatomy 0.000 description 1
- 208000033808 peripheral neuropathy Diseases 0.000 description 1
- 230000002572 peristaltic effect Effects 0.000 description 1
- 201000002628 peritoneum cancer Diseases 0.000 description 1
- 239000008251 pharmaceutical emulsion Substances 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 230000006611 pharmacological activation Effects 0.000 description 1
- BSCCSDNZEIHXOK-UHFFFAOYSA-N phenyl carbamate Chemical compound NC(=O)OC1=CC=CC=C1 BSCCSDNZEIHXOK-UHFFFAOYSA-N 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 239000002587 phosphodiesterase IV inhibitor Substances 0.000 description 1
- 201000007291 pilocytic astrocytoma of cerebellum Diseases 0.000 description 1
- 239000002574 poison Substances 0.000 description 1
- 231100000614 poison Toxicity 0.000 description 1
- 208000037244 polycythemia vera Diseases 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 230000026341 positive regulation of angiogenesis Effects 0.000 description 1
- 230000029279 positive regulation of transcription, DNA-dependent Effects 0.000 description 1
- 230000002980 postoperative effect Effects 0.000 description 1
- 235000008476 powdered milk Nutrition 0.000 description 1
- 230000002028 premature Effects 0.000 description 1
- 108010074732 preproenkephalin Proteins 0.000 description 1
- 150000003138 primary alcohols Chemical class 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 210000002248 primary sensory neuron Anatomy 0.000 description 1
- 208000029340 primitive neuroectodermal tumor Diseases 0.000 description 1
- 238000004393 prognosis Methods 0.000 description 1
- KWTBUMWSEOCASE-UHFFFAOYSA-N prop-1-en-2-yl carbamate Chemical compound CC(=C)OC(N)=O KWTBUMWSEOCASE-UHFFFAOYSA-N 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 1
- 238000002731 protein assay Methods 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- 230000006337 proteolytic cleavage Effects 0.000 description 1
- 125000004289 pyrazol-3-yl group Chemical group [H]N1N=C(*)C([H])=C1[H] 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 150000003222 pyridines Chemical class 0.000 description 1
- YEYHFKBVNARCNE-UHFFFAOYSA-N pyrido[2,3-b]pyrazine Chemical compound N1=CC=NC2=CC=CN=C21 YEYHFKBVNARCNE-UHFFFAOYSA-N 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 229960001404 quinidine Drugs 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 208000002574 reactive arthritis Diseases 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 230000025053 regulation of cell proliferation Effects 0.000 description 1
- 230000011663 regulation of signaling Effects 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 210000002345 respiratory system Anatomy 0.000 description 1
- 208000023504 respiratory system disease Diseases 0.000 description 1
- 210000001525 retina Anatomy 0.000 description 1
- 210000001210 retinal vessel Anatomy 0.000 description 1
- 210000000574 retroperitoneal space Anatomy 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 102200006532 rs112445441 Human genes 0.000 description 1
- 102220197833 rs112445441 Human genes 0.000 description 1
- 102200006531 rs121913529 Human genes 0.000 description 1
- 102200006537 rs121913529 Human genes 0.000 description 1
- 102200006539 rs121913529 Human genes 0.000 description 1
- 102200006538 rs121913530 Human genes 0.000 description 1
- 102200006541 rs121913530 Human genes 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 210000004761 scalp Anatomy 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 238000010956 selective crystallization Methods 0.000 description 1
- 229950010746 selumetinib Drugs 0.000 description 1
- 229950011005 semapimod Drugs 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 208000002491 severe combined immunodeficiency Diseases 0.000 description 1
- 238000007493 shaping process Methods 0.000 description 1
- 208000007056 sickle cell anemia Diseases 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 1
- 229960002855 simvastatin Drugs 0.000 description 1
- 238000009097 single-agent therapy Methods 0.000 description 1
- 210000002027 skeletal muscle Anatomy 0.000 description 1
- 230000012488 skeletal system development Effects 0.000 description 1
- 208000017520 skin disease Diseases 0.000 description 1
- 201000003708 skin melanoma Diseases 0.000 description 1
- 201000010106 skin squamous cell carcinoma Diseases 0.000 description 1
- 102000030938 small GTPase Human genes 0.000 description 1
- 108060007624 small GTPase Proteins 0.000 description 1
- 150000003384 small molecules Chemical group 0.000 description 1
- 210000002460 smooth muscle Anatomy 0.000 description 1
- 210000000329 smooth muscle myocyte Anatomy 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- 239000012265 solid product Substances 0.000 description 1
- 230000000392 somatic effect Effects 0.000 description 1
- 229960003787 sorafenib Drugs 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 238000005507 spraying Methods 0.000 description 1
- 241001147420 ssDNA viruses Species 0.000 description 1
- 238000011255 standard chemotherapy Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 210000004500 stellate cell Anatomy 0.000 description 1
- 230000036262 stenosis Effects 0.000 description 1
- 208000037804 stenosis Diseases 0.000 description 1
- 239000003270 steroid hormone Substances 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 150000005846 sugar alcohols Polymers 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 230000017960 syncytium formation Effects 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 210000002437 synoviocyte Anatomy 0.000 description 1
- 201000004595 synovitis Diseases 0.000 description 1
- 239000012622 synthetic inhibitor Substances 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000015523 tannic acid Nutrition 0.000 description 1
- LRBQNJMCXXYXIU-NRMVVENXSA-N tannic acid Chemical compound OC1=C(O)C(O)=CC(C(=O)OC=2C(=C(O)C=C(C=2)C(=O)OC[C@@H]2[C@H]([C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)O2)OC(=O)C=2C=C(OC(=O)C=3C=C(O)C(O)=C(O)C=3)C(O)=C(O)C=2)O)=C1 LRBQNJMCXXYXIU-NRMVVENXSA-N 0.000 description 1
- 229920002258 tannic acid Polymers 0.000 description 1
- 229940033123 tannic acid Drugs 0.000 description 1
- 239000008399 tap water Substances 0.000 description 1
- 235000020679 tap water Nutrition 0.000 description 1
- 208000001608 teratocarcinoma Diseases 0.000 description 1
- UIYOVVYZPVVUMJ-UHFFFAOYSA-N tert-butyl carbamoyl carbonate Chemical compound CC(C)(C)OC(=O)OC(N)=O UIYOVVYZPVVUMJ-UHFFFAOYSA-N 0.000 description 1
- FGTJJHCZWOVVNH-UHFFFAOYSA-N tert-butyl-[tert-butyl(dimethyl)silyl]oxy-dimethylsilane Chemical compound CC(C)(C)[Si](C)(C)O[Si](C)(C)C(C)(C)C FGTJJHCZWOVVNH-UHFFFAOYSA-N 0.000 description 1
- 108091008743 testicular receptors 4 Proteins 0.000 description 1
- 229960002180 tetracycline Drugs 0.000 description 1
- 229930101283 tetracycline Natural products 0.000 description 1
- 235000019364 tetracycline Nutrition 0.000 description 1
- 150000003522 tetracyclines Chemical class 0.000 description 1
- 230000004797 therapeutic response Effects 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 208000037816 tissue injury Diseases 0.000 description 1
- 201000006134 tongue cancer Diseases 0.000 description 1
- 239000012049 topical pharmaceutical composition Substances 0.000 description 1
- 229940044693 topoisomerase inhibitor Drugs 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000011830 transgenic mouse model Methods 0.000 description 1
- 206010044412 transitional cell carcinoma Diseases 0.000 description 1
- 102000027257 transmembrane receptors Human genes 0.000 description 1
- 108091008578 transmembrane receptors Proteins 0.000 description 1
- 230000017105 transposition Effects 0.000 description 1
- 230000008733 trauma Effects 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical compound OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 description 1
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 description 1
- 229940029284 trichlorofluoromethane Drugs 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- 150000004684 trihydrates Chemical class 0.000 description 1
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 208000006961 tropical spastic paraparesis Diseases 0.000 description 1
- 210000005239 tubule Anatomy 0.000 description 1
- 230000005748 tumor development Effects 0.000 description 1
- 230000005751 tumor progression Effects 0.000 description 1
- 238000002604 ultrasonography Methods 0.000 description 1
- 238000000825 ultraviolet detection Methods 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 241001529453 unidentified herpesvirus Species 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 230000009107 upstream regulation Effects 0.000 description 1
- 208000009852 uremia Diseases 0.000 description 1
- 201000005112 urinary bladder cancer Diseases 0.000 description 1
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 1
- 208000023747 urothelial carcinoma Diseases 0.000 description 1
- 238000002255 vaccination Methods 0.000 description 1
- 239000004474 valine Substances 0.000 description 1
- 150000003679 valine derivatives Chemical class 0.000 description 1
- 210000003556 vascular endothelial cell Anatomy 0.000 description 1
- 230000006444 vascular growth Effects 0.000 description 1
- 210000004509 vascular smooth muscle cell Anatomy 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 210000003501 vero cell Anatomy 0.000 description 1
- 208000030401 vitamin deficiency disease Diseases 0.000 description 1
- 239000011534 wash buffer Substances 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 238000007482 whole exome sequencing Methods 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4985—Pyrazines or piperazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/04—Artificial tears; Irrigation solutions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/16—Otologicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Pulmonology (AREA)
- Ophthalmology & Optometry (AREA)
- Dermatology (AREA)
- Oncology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Virology (AREA)
- Communicable Diseases (AREA)
- Rheumatology (AREA)
- Pain & Pain Management (AREA)
- Otolaryngology (AREA)
- Hematology (AREA)
- Immunology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Un compuesto seleccionado de compuestos de la siguiente fórmula, y sales farmacéuticamente aceptables, N-óxidos, hidratos y solvatos de los mismos:**Fórmula** donde: =X- es independientemente =CH- o =N-; -Y es independientemente -Y1, -Y2, -Y3, -Y4, -Y5 o -Y6; -Y1 es independientemente -F, -Cl, -Br o -I; -Y2 es alquilo C1-4 saturado lineal o ramificado; -Y3 es haloalquilo C1-4 saturado lineal o ramificado; -Y4 es -OH; -Y5 es alcoxi C1-4 saturado lineal o ramificado; e -Y6 es haloalcoxi C1-4 saturado lineal o ramificado.
Description
DESCRIPCIÓN
Derivados de 1 -(5-terc-butil-2-aril-pirazol-3-il)-3-[2-fluoro-4-[(3-oxo-4h-pirido[2,3-b]piracin-8-il)oxi]fenil]urea como inhibidores raf para el tratamiento del cáncer
CAMPO TÉCNICO
La presente invención trata generalmente del campo de los compuestos terapéuticos. Más específicamente, la presente invención trata de ciertos compuestos de 1-(5-terc-butil-2-aril-pirazol-3-il)-3-[2-fluoro-4-[(3-oxo-4H-pirido[2,3-b]piracin-8-il)oxi]fenil]urea (denominados en la presente “compuestos de TBAP”) que, entre otras cosas, inhiben RAF (p. ej., BRAF, CRAF, etc.). La presente invención también trata de composiciones farmacéuticas que comprenden tales compuestos, y tales compuestos y composiciones para el uso, tanto in vitro como in vivo, para inhibir RAF (p. ej., BRAF, CRAf , etc.) y para tratar trastornos que incluyen: trastornos proliferativos; cáncer (incluyendo, p. ej., melanoma maligno, carcinoma colorrectal, adenocarcinoma pancreático); inflamación; trastornos inmunológicos; infecciones virales; trastornos fibróticos; trastornos asociados con una forma mutada de RAF (p. ej., BRAF, CRAF, etc.); trastornos mejorados por la inhibición de RAF (p. ej., BRAF, CRAF, etc.); trastornos mejorados por la inhibición de BRAF mutante; trastornos mejorados por la inhibición de BRAF y CRAF; trastornos asociados con mutaciones de RAS y/o activación de la ruta de MAPK; trastornos mejorados por la inhibición de SRC, p38, FGFRA, VEGFR-2 (KDR) y/o LCK; etc.
ANTECEDENTES
Se cita en la presente un número de publicaciones a fin de describir y divulgar más a fondo la invención y el estado de la técnica de la que trata la invención.
A lo largo de esta memoria descriptiva, incluyendo las reivindicaciones que siguen, a menos que el contexto requiera otra cosa, se entenderá que la palabra “comprenden” y variaciones tales como “comprende” y “que comprende” implican la inclusión de un número entero o etapa o grupo de números enteros o etapas indicados pero no la exclusión de ningún otro número entero o etapa o grupo de números enteros o etapas.
Se debe apuntar que, según se usa en la memoria descriptiva y las reivindicaciones adjuntas, las formas singulares “un”, “uno(a)” y “el(la)” incluyen las referencias plurales a menos que el contexto dicte claramente otra cosa. Así, por ejemplo, la referencia a “un portado farmacéutico” incluye mezclas de dos o más de tales portadores, y similares.
Los intervalos a menudo se expresan en la presente como de “aproximadamente” un valor particular y/o hasta “aproximadamente” otro valor particular. Cuando se expresa tal intervalo, otra realización incluye desde un valor particular y/o hasta el otro valor particular. De forma similar, cuando los valores se expresan como aproximaciones, mediante el uso de “aproximadamente” precedente, se entenderá que el valor particular forma otra realización.
Esta divulgación incluye información que puede ser útil para comprender la presente invención. No es una admisión de que cualquier información proporcionada en la presente sea técnica anterior o pertinente para la invención actualmente reivindicada, o que cualquier publicación mencionada específicamente o implícitamente sea técnica anterior.
RAF, Trastornos Proliferativos y Cáncer
Se considera generalmente que las mutaciones en los genes que controlan directamente o indirectamente el crecimiento y la diferenciación celular son la principal causa del cáncer. Los tumores malignos se desarrollan a través de una serie de cambios escalonados progresivos que conducen a la pérdida del control del crecimiento característica de las células cancerosas, es decir, proliferación desregulada continua, la capacidad para invadir tejidos circundantes, la capacidad para metastatizarse en diferentes zonas orgánicas, la estimulación de la angiogénesis, la resistencia a la apoptosis, la capacidad para evadir el sistema inmunitario, rutas metabólicas anormales e inflamación local. Estudios in vitro cuidadosamente controlados han ayudado a definir los factores que caracterizan el crecimiento de células normales y neoplásticas y han conducido a la identificación de proteínas específicas que controlan el crecimiento y la diferenciación celular.
RAF es una diana aguas abajo clave para las proteínas RAS de unión a nucleótidos de guanina / GTPasa y media en la activación de la cascada de MAP cinasa que consiste en RAF-MEK-ERK. La ERK activada es una cinasa que posteriormente tiene como diana un número de proteínas responsables de mediar en, entre otras cosas, el crecimiento, la supervivencia y las funciones de transcripción de la ruta. Estas incluyen los factores de transcripción ELK1, C-JUN, la familia Ets (incluyendo Ets 1, 2 y 7) y la familia FOS. La ruta de transducción de señales RAS-RAF-MEK-ERK se activa en respuesta a muchos estímulos celulares incluyendo factores de crecimiento tales como EGF, PDGF, KGF, etc. Debido a que la ruta es una diana principal para la acción de factores de crecimiento, se ha encontrado que la actividad de RAF-MEK-ERK se regula al alza en muchos tumores dependientes de factores. La observación de que aproximadamente 20% de todos los tumores han sufrido una mutación activadora en una de las proteínas RAS indica que la ruta es más ampliamente importante en la tumorigénesis.
La familia de oncogenes RAF incluye tres genes muy conservados denominados ARAF., BRAF y CRAF (también denominado Raf-1). Los genes RAF codifican proteína cinasas que se cree que representan papeles reguladores importantes en procesos de transducción de señales que regulan la proliferación celular. Los genes RAF codifican proteína cinasas específicas para serina-treonina muy conservadas, que son incorporadas en la membrana plasmática después de la unión directa a RAS, que es el episodio iniciador en la activación de RAF. Las proteínas RAF son parte de una ruta de transducción de señales que se cree que consiste en tirosina cinasas receptoras, cinasas p21 RAS, RAF, Mek1 (activador de ERK, o MAPk K) y cinasas ERK (MAPK), que finalmente fosforilan varios sustratos celulares, incluyendo factores de transcripción. La señalización a través de esta ruta puede mediar en la diferenciación, la proliferación o la transformación oncogénica en diferentes contextos celulares. Así, se cree que las cinasas RAF representan un papel fundamental en la ruta de transducción de señales celular normal, acoplando una multitud de factores de crecimiento a su efecto neto, la proliferación celular. Debido a que las proteína RAF son efectores aguas abajo directos de proteína RAS, se cree que las terapias dirigidas contra cinasas RAF son útiles en el tratamiento de tumores dependientes de RAS.
Las cinasas RAF se regulan y expresan diferencialmente. CRAF se expresa en todos los órganos y en todas las líneas celulares que se han examinado. ARAF y BRAF también parecen ser ubicuas, pero se expresan lo más intensamente en tejidos urogenitales y cerebrales, respectivamente. Debido a que BRAF se expresa mucho en tejidos neurales, se creyó una vez que se limitaba a estos tejidos, peso se ha encontrado desde entonces que se expresa más ampliamente. Aunque todas las proteínas RAF pueden unirse a RAS activa, BRAF es más fuertemente activada por RAS oncogénica.
BRAF es importante en el cáncer, debido a que se muta en aproximadamente la mitad de los melanomas malignos y carcinomas papilares tiroideos, 30% del cáncer de ovario de baja malignidad, 15% de cánceres de colon, y con frecuencia muy alta en tricoleucemia, así como ocurre a frecuencias menores en un número de otros cánceres, totalizando 7% de los cánceres humanos. Véase, p. ej., http://www.sanger.ac.uk/genetics/CGP/cosmic/. Un subtipo específico de cánceres pancreáticos, el carcinoma medular natural del páncreas KRAS2, presenta mutaciones de BRAF en 30% de las muestras (véase, p. ej., Calhoun y cols., 2003). En contraste, las mutaciones de ARAF y CRAF son muy raras en el cáncer humano.


Se han descrito más de 100 mutaciones diferentes en BRAF en el cáncer, pero una sola mutación (una sustitución de la valina (V) por ácido glutámico (E) en la posición 600) explica aproximadamente 80% de las mutaciones de BRAF totales en el cáncer. Este mutante activa BRAF 500 veces, y permite que estimule la señalización constitutiva de ERK y NFkB, estimulando la supervivencia y la proliferación. Por consiguiente, V600EBRAF puede transformar células tales como fibroblastos y melanocitos. La inhibición de V600EBRAF en células cancerosas inhibe la proliferación celular e induce apoptosis in vitro; in vivo, suprime el crecimiento de células tumorales, validando 600EBRAF como una diana terapéutica.
Otras mutaciones V600 BRAF identificada en melanoma son V600K, V600D y V600R (véase, p. ej., Davies y cols., 2002; Wan y cols., 2004; Long y cols., 2011; Rubinstein y cols., 2010). Un subgrupo secundario de melanomas también se identificó con mutaciones de BRAF en posiciones distintas a 600. Estos mutantes de BRAF de posición distinta a V600 no activan necesariamente la actividad de BRAF cinasa directamente, sino que requieren la presencia de CRAF para transactivar su señalización de MAPK (véase, p. ej., Smalley y cols., 2009). En tal caso, la inhibición de la actividad de RAF seguiría siendo un objetivo beneficioso en el tratamiento del cáncer.
De forma importante, se ha mostrado que los fármacos que inhiben BRAF mutante tales como vemurafenib (PLX4032, RG7204, Zelboraf) (véase, p. ej., Flaherty y cols., 2010) y dabrafenib (GSK-2118436) (véase, p. ej., Falchook y cols., 2012) pueden mediar en respuestas impresionantes en pacientes cuyos tumores expresan BRAF oncogénica (revisado en Salama y cols., 2013). En particular, el vemurafenib ha mostrado resultados prometedores en melanoma accionado por BRAF mutante (véanse, p. ej., Chapman y cols., 2011; Sosman y cols., 2012). Fue aprobado en 2011 por the USA Food and Drug Administration (FDA) para el tratamiento de melanoma metastásico o inextirpable en fase tardía por mutante de BRAF V600E, y en 2012 por la Agencia Europea del Medicamento (EMA, por sus siglas en inglés) como monoterapia para el tratamiento de pacientes adultos con cualquier melanoma inextirpable o metastásico positivo a mutación BRAF V600. El dabrafenib fue aprobado por la FDA y la EMA en 2013 para la misma indicación.
Estos datos validan BRAF mutante como una diana terapéutica en el melanoma y una diana potencial para otros cánceres y enfermedades proliferativas en los que la BRAF se muta. Esta y otra evidencia sugieren que la inhibición de la actividad de RAF (p. ej., BRAF) sería beneficiosa en el tratamiento del cáncer, y que la inhibición de la actividad de RAF (p. ej., BRAF) podría ser particularmente beneficiosa en los cánceres que contienen una mutación de BRAF activada constitutivamente.
Resistencia a Inhibidores de BRAF
A pesar de ser capaz de mediar en respuestas clínicas significativas, la mayoría de los pacientes tratados con vermurafenib y dabrafenib finalmente empeoran durante el tratamiento (véanse p. ej., Flaherty y cols., 2010; Sosman y cols., 2012) debido a la adquisición de resistencia que puede estar mediada por varios mecanismos (véase, p. ej., Sullivan y cols., 2011). Por otra parte, aproximadamente 30% de los pacientes presentan resistencia primaria y no responden a pesar de la presencia de una mutación de BRAF (véase, p. ej., Chapman y cols., 2011). Las mutaciones en KRAS (G12S, G12V, G12D, G12A, G12C, G13A, G13D) se han sugerido como marcadores predictivos para identificar tumores que no son sensibles al tratamiento con inhibidores de BRAF mutante (véase, p. ej., Hatzivassiliou y cols., 2011). La mutación Q16K de NRAS confiere resistencia al inhibidor de BRAF vemurafenib (véase, p. ej., Nazarian y cols., 2010). De forma similar, la resistencia al tratamiento con el inhibidor de BRAF dabrafenib es predicha por las mutaciones Q16K y A146T de la proteína NRAS (véase, p. ej., Greger y cols., 2012). La activación de RAS a través de mutaciones condujo a un incremento en la dimerización de RAF (formación del heterodímero de CRAF con la proteína BRAF y/o el homodímero CRAF/BRAF) con un incremento de la señalización a través de la cascada de MAPK y un incremento de la proliferación celular (véase, p. ej., Poulikakos y cols. 2010). La regulación al alza de (y las mutaciones en) CRAF es otro mecanismo de resistencia observado en melanomas resistentes tratados con inhibidores de BRAF (véanse, p. ej., Heidorn y cols., 2010; Montagut y cols., 2008; Antony y cols. 2013). Por lo tanto, es probable que los inhibidores de panRAF de múltiples isoformas de RAF (BRAF y CRAF especialmente) tengan un efecto mejorado en melanomas mutados en RAS y otros cánceres mutados en RAS, y se dirijan a un mecanismo de resistencia clave para inhibidores de BRAF selectivos.
El aumento del número de copias del gen BRAFV600E está asociado con la resistencia a inhibidores de BRAF en melanoma con mutante de BRAF (véase, p. ej., Shi y cols., 2012) y carcinoma colorrectal (véase, p. ej., Corcoran y cols., 2010). Ambos modelos son sensibles a la inhibición concomitante de BRAF y MEK, pero solo el melanoma con
muíante de BRAF amplificado es sensible al inhibidor de MEK solo. Es probable que este mecanismo de resistencia sea sensible a la inhibición de panRAF que solamente a la inhibición de BRAF.
En algunos melanomas, la resistencia de vemurafenib se adquiría a través de la expresión de isoformas variantes de empalme de BRAFV600E. Una variante de empalme de 61kDa de BRAFV600E (p61BRAFV600E) carecía de los exones 4-8 que codifican el dominio de unión a RAS, y era resistente a vemurafenib. p61BRAFV600E se dimeriza constitutivamente en ausencia de RAS activada. Se observó que la dimerización de p61BRAFV600E era crítica para mediar en la resistencia a inhibidores de BRAF (véase, p. ej., Poulikakos y cols., 2011).
La fusión génica de BRAF y CRAF es un mecanismo alternativo de la activación de la ruta de MAPK. Estos productos de fusión génica activadores se han identificado en el cáncer de próstata, el cáncer gástrico y el melanoma (SLC45A3-BRAF y ESRP1-RAF1) (véase, p. ej., Palanisamy y cols., 2010), cánceres tiroideos (AKAP9-BRAF) (véase, p. ej., Ciampi y cols., 2005) y astrocitomas pediátricos (KIAA1549-BRAF) (véase, p. ej., Sievert y cols., 2013). Algunos de los modelos que expresan fusiones de RAF (por ejemplo SLC45A3-BRAF) son sensibles a la inhibición de BRAF y MEK; en contraste, el modelo KIAA1549-BRAF es resistente a PLX4720, pero es sensible a un inhibidor de BRAF de segunda generación.
El supresor de cinasa de Ras (KSR) es un modulador positivo conservado de la ruta RAS-RAF-MEK-ERK. KSR1 interactúa constitutivamente con MEK y se sabe que representa un papel importante en la colocalización de MEK con RAF en la membrana plasmática. La KSR1 está implicada en la activación de la ruta de MAPK por inhibidores de BRAF en mutante de RAS o células RAS activadas (véase, p. ej., McKay y cols., 2011). Se han propuesto dos mecanismos de activación farmacológica de la ruta. Un mecanismo implica la formación del dímero CRAF-KSR1, la formación de complejos con MEK y la fosforilación de MEK mediante el dímero KSR1 -CRAF (véase, p. ej., Hu y cols., 2011). En el otro mecanismo, KSR1 se dimeriza con BRAF, y compiten con el heterodímero BRAF-CRAf que es el conductor de la fosforilación de MEK. Se ha sugerido que las células activadas por RAS con menor expresión de KSR1 mostrarán más activación de la ruta paradójica (véase, p. ej., McKay y cols., 2011). En ambos mecanismos, es probable que los inhibidores de panRAF reduzcan la activación de la ruta independientemente del nivel de expresión de KSR1.
La sobreexpresión de tirosina cinasas receptoras (RTK) es otro mecanismo de resistencia a inhibidores de BRAF. La sobreexpresión de EGFR (receptor del factor de crecimiento epidérmico) conduce a la reactivación de la ruta de MAPK mediada por EGFR y la resistencia a vemurafenib en cánceres colorrectales con mutantes de BRAF (véase, p. ej., Corcoran y cols., 2012). En líneas celulares de melanoma con mutantes de BRAF resistentes a fármacos, la señalización de EGFR-SFK-STAT3 puede mediar en la resistencia a inhibidores de BRAF in vitro e in vivo, en el melanoma (véase, p. ej., Girotti y cols., 2013). Las cinasas de la familia Src SFK representan un papel clave para mediar en la resistencia a inhibidores de BRAF en células de melanoma (véase, p. ej., Girotti y cols., 2013; Vergani y cols., 2011). Se observa una fosforilación elevada de las SFK LYN, YES y fYn en las líneas resistentes a vemurafenib. El crecimiento de células resistentes es sensible a la inhibición de SFK: El dasatinib y el agotamiento de SRC y LYN suprimen ambos la invasión de las células resistentes in vitro. De importancia crítica, la señalización de SFK se incrementaba en un tumor de un paciente con resistencia intrínseca a vemurafenib, y el dasatinib inhibía el crecimiento y la metástasis de este tumor en ratones.
Corcoran y cols., 2012, “EGFR-mediated reactivation of MAPK signaling contributes to insensitivity of BRAF-mutant cáncer colorrectals to RAF inhibition with vemurafenib”, Cancer Discoverv. Vol. 2, pp. 227-235.
Se encontró que PDGFR-p se sobreexpresaba y se hiperfosforilaba en líneas celulares de BRAF mutante resistentes a vemurafenib, y se regulan al alza en varios casos de tumores resistentes a vemurafenib procedentes de pacientes, sugiriendo que este mecanismo puede ser clínicamente pertinente (véase, p. ej., Nazarian y cols., 2010). La regulación al alza de PDGFR-p puede conducir la resistencia al activar otras rutas aguas abajo independientes de ERK1/2 (PI3K, PLCy).
Los estudios mecanicistas mostraron que la señalización de IGFR1 mediaba en la señalización de PI3K/AKT incrementada en células que adquirían resistencia adquirida a inhibidores de BRAF y que la resistencia se podría invertir al tratar las células con la combinación de una PI3K y un inhibidor de MEK o un lGF1 R y un inhibidor de MEK (véase, p. ej., Villanueva y cols., 2011). La importancia aplicada de este hallazgo se confirmó por la observación de que 1 de 5 especímenes con melanoma de pacientes que fracasaban con vemurafenib expresaban niveles incrementados de IGFR1 (véase, p. ej., Villanueva y cols., 2011).
La regulación al alza de factores de crecimiento por el estroma es un mecanismo de resistencia. Se ha observado una correlación significativa entre la expresión de células estromáticas de HGF en pacientes con melanoma con mutante de BRAF y resistencia innata al tratamiento con inhibidores de RAF (véase, p. ej., Wilson y cols., 2012). cMET y/o sus ligandos se reivindican como marcadores predictivos para identificar tumores que no son vulnerables a un inhibidor de BRAF (véanse, p. ej., Hatzivassiliou y cols., 2011; Straussman y cols., 2012). La inhibición doble de RAF y bien HGF o bien MET dio como resultado la inversión de la resistencia al fármaco, sugiriendo la terapia de combinación inhibidora de RAF más HGF o MET como una estrategia terapéutica potencial (véanse, p. ej., Hatzivassiliou y cols., 2011; Straussman y cols., 2012).
FGFR1 está implicado en el empeoramiento de melanomas, y la desactivación de FGFR1 da como resultado la inhibición del crecimiento de melanomas in vivo (véase, p. ej., Wang y cols., 1997). El factor de crecimiento de fibroblastos (FGF, por sus siglas en inglés) rescata algunas células mutantes en BRAF del tratamiento con PLX4032 (véase, p. ej., Wilson y cols., 2012) y la inhibición de FGFR1 es sinérgica con el inhibidor de múltiples cinasas/BRAF sorafenib y el inhibidor de BRAF específico RG7204 (véase, p. ej., Metzner y cols., 2012). Estos hallazgos sugieren qua la inhibición de RTK tales como EGFR, PDGFR-p, HGFR, IGF1R y FGFR, y de SFKs debe tener como diana un número de mecanismos de resistencia para inhibidores de BRAF selectivos y por consiguiente ser de utilidad en tumores con mutantes de BRAF que se han vuelto resistentes a inhibidores selectivos de BRAF.
Cáncer y RAS
Las proteínas RAS son proteínas pequeñas que se unen a nucleótidos de guanina, que están aguas abajo de receptores de factores de crecimiento, citocinas y hormonas. Estos receptores de la superficie celular activan proteínas llamadas factores de intercambio de nucleótidos de guanina (GNEF, por sus siglas en inglés), que reemplazan GDP por GTP en proteínas RAS, estimulando la activación de RAS. Otras proteínas llamadas proteínas activadoras de GTPasa (GAP, por sus siglas en inglés) estimulan la actividad intrínseca para GTPasa de RAS, promoviendo de ese modo la hidrólisis de GTP y devolviendo RAS a su estado inactivo unido a GDP. La RAS activada se une a varias proteínas efectoras, incluyendo fosfoinosítido 3-cinasa (PI3K, por sus siglas en inglés), la familia RAF de proteína cinasas y el factor de intercambio de nucleótidos de guanina Ral. A su vez, estos efectores regulan la actividad de las rutas de señalización que controlan la proliferación, la senescencia, la supervivencia y la diferenciación celulares. Hay tres genes RAS en mamíferos denominados HRAS, KRAS y NRAS y cumplen funciones solapadas pero no conservadas.
Las proteínas RAS también son importantes en el cáncer. 20-30% de los tumores humanos albergan mutaciones somáticas de ganancia de función en uno de los genes RAS. Lo más comúnmente, estas implican a los codones para glicina 12 (G12), glicina 13 (G13) y glutamina 61 (Q61) y estas mutaciones dificultan, a través de diferentes mecanismos, la actividad intrínseca de GTPasa estimulada por GAP de RAS, reteniéndola en el estado activo unido a GTP y permitiendo que promueva la tumorigénesis. Véanse, p. ej., Downward y cols., 2003; Young y cols., 2009; Bos y cols., 1989.


(*) La RAS más frecuentemente mutada citada (KRAS o NRAS o HRAS).
Otros cánceres tienen mutaciones menos frecuentes de los genes de la familia RAS, pero su mutación predice un pronóstico, por ejemplo neuroblastoma (8% de mutación de NRAS), adenocarcinoma de estómago (6% de mutante de KRAS).
RAS y RAF
Las proteínas RAS activas activan varios efectores aguas abajo, incluyendo las proteínas de la familia RAF. Hay tres proteínas RAF, ARAF, BRAF y CRAF. RAF activada fosforila y activa una segunda proteína cinasa llamada MEK, que a continuación fosforila y activa una tercera proteína cinasa llamada ERK. ERK fosforila una multitud de sustratos citosólicos y nucleares, regulando de ese modo procesos celulares tales como proliferación, supervivencia, diferenciación y senescencia.
Notablemente, sin embargo, en las células cancerosas, la RAS oncogénica no señala a través de BRAF, sino que en cambio señala exclusivamente a través de CRAF para activar MEK.
En la gran mayoría de los cánceres, las mutaciones en BRAF y RAS son mutuamente exclusivas. Esto proporciona una evidencia genética para sugerir que estas proteínas están en la misma ruta y que conducen los mismos procesos en células cancerosas. Sin embargo, hay diferencias claras entre las funciones de BRAF oncogénica y RAS oncogénica en células cancerosas. En primer lugar, RAS activa carias rutas, mientras que solo se sabe que BRAF active la ruta MEK/ERK. Como consecuencia, las células con mutantes en BRAF son más dependientes de la señalización de MEK/ERK y así son considerablemente más sensibles a inhibidores de BRAF o MEK que la célula en la que se muta RAS. Véanse, p. ej., Garnett y cols., 2004; Wellbrock y cols., 2004; Gray-Schopfer y cols., 2007; Solit y cols., 2006.
Aparte de las mutaciones, las proteínas de señalización en la cascada de MAPK se sobreexpresan en un número de enfermedades malignas. Por ejemplo, HRAS y NRAS se sobreexpresan en cánceres de cuello uterino. Las mutaciones en RAS son raras en el carcinoma adrenocortical, pero la población colectiva de tumores con mutaciones en RAS, BRAF y EGFR muestran un incremento de la señalización a través de la ruta y pueden ser una diana para inhibidores de la ruta de MAPK (véase, p. ej., Kotoula y cols., 2009). Los cánceres ováricos de baja malignidad y el cáncer peritoneal responden al bloqueo de la ruta de MAPK con el inhibidor de MEK selumetinib independientemente del estado de mutación de RAS/RAF. En el melanoma uveal, la ruta de MAPK es activada a través de la mutación de GNAQ que explica 50% de los melanomas uveales (véase, p. ej., Gaudi y cols., 2011). cRAF se sobreexpresa en una variedad de cánceres humanos primarios, tales como de pulmón, hígado, próstata, tumores neurodérmicos primitivos, carcinoma de células escamosas de cabeza y cuello (véanse, p. ej., Damodar
Reddy y cois., 2001; Hwang y cois., 2004; Mukterjee y cois., 2005; Schreck y cois., 2006; Riva y cois., 1995). La ruta de MAPK se activa en 74% de ias muestras de pacientes con ieucemia mieioide aguda (véase, p. ej., Miieiia y cois., 2001). En ia neurofibromatosis tipo 1, ia pérdida dei gen supresor de tumores NF1 conduce a una hiperactivación de ia señaiización de RAS, y una desreguiarización de ia señaiización de Ras/ERK que es crítica para ei crecimiento de tumores de ios nervios periféricos NF1 (véase, p. ej., Jessen y cois., 2013). Un inhibidor de panRAF provoca un bioqueo efectivo de ia ruta de MAPK para tumores con mutantes en BRAF y para tumores con mutantes en RAS y tiene una ampiia apiicación para cánceres con desreguiación de ia ruta de señaiización de MAPK.
Por io tanto, ios cánceres con mutaciones activadoras de RAS, RAF y EGFR o sobreexpresión de RAS, RAF y EGFR, inciuyendo cuaiquiera de ias isoformas de ias mismas, pueden ser particuiarmente sensibies a ia inhibición de panRAF (p. ej., CRAF y BRAF). ios cánceres con otras anormaiidades que conducen a una ruta de RAF-MEK-ERK desreguiada también pueden ser particuiarmente sensibies ai tratamiento con inhibidores de ia actividad de panRAF (p. ej., CRAF y BRAF). Ejempios de taies anormaiidades inciuyen ia activación constitutiva de un receptor de factor de crecimiento; ia sobreexpresión de uno o más receptores de factores de crecimiento; ia sobreexpresión de uno o más factores de crecimiento; ia activación de rutas mediadas por KSR; y ias fusiones génicas de BRAF o CRAF.
Ruta de MAPK en Otras Enfermedades
La ruta RAF-MEK-ERK funciona aguas abajo de muchos receptores y estímuios indicando un ampiio papei en ia reguiación de ia función ceiuiar. Por esta razón, ios inhibidores de RAF pueden encontrar utiiidad en otras afecciones que están asociadas con ia reguiación ai aiza de ia señaiización a través de esta ruta. La ruta RAF-MEK-ERK también es un componente importante de ia respuesta normai de céiuias no transformadas a ia acción de factores de crecimiento. Por io tanto, ios inhibidores de RAF se pueden usar en enfermedades en ias que hay una proiiferación inapropiada o excesiva de tejido normai. Estas inciuyen, por ejempio, giomeruionefritis y psoriasis. La función de ias céiuias infiamatorias está controiada por muchos factores, cuyos efectos están mediados por diferentes rutas de transducción de señaies. Aunque aigunas funciones proinfiamatorias ciave están mediadas por p38 Map cinasa (p. ej., iiberación de TNF), otras están mediadas por otras rutas. La ruta RAF-MEK-ERK, en particuiar, es una importante señai activadora y proiiferativa en muchas céiuias infiamatorias. Los iinfocitos B y T, en particuiar, requieren ia activación de ia ruta RAF-MEK-ERK para ia expansión cionai y ia generación de pobiaciones efectoras (véanse, p. ej., Cantreii, 2003; Genot y cois., 2000). La ruta de señaiización ceiuiar de ia que forma parte RAF se ha reiacionado con trastornos infiamatorios caracterizados por ia proiiferación de céiuias T (activación y crecimiento de céiuias T), taies como rechazo de injertos tisuiares, choque endotóxico y nefritis giomeruiar.
La activación de ia señaiización de MAPK/ERK se ha demostrado en muchos modeios de modeios de enfermedad, y se ha mostrado que ia inhibición de ia ruta, usando, por ejempio, inhibidores de MEK, es potenciaimente beneficiosa en estas enfermedades diversas taies como:
• Doior: Evidencia de Eficacia en Modeios de Doior: La ruta de MEK es reguiada ai aiza en neuronas dei asta posterior en ei doior persistente (véanse, p. ej., Ji y cois., 2002; Song y cois., 2005; Ma y cois., 2005; Karim y cois., 2006); Inhibidores de Mek en ei doior neuropático (véase, p. ej., Dixon y cois., 2001).
• Apopiejía: Evidencia de Eficacia en Modeios de Apopiejía Neuroprotección Significativa contra Lesión Cerebrai Isquémica por Inhibición de ia MEK (véanse, p. ej., Wang y cois., 2003; Wang y cois., 2004; Maddahi y cois., 2010).
• Diabetes: Evidencia en Compiicaciones Diabéticas (véase, p. ej., Fujita y cois., 2004).
• Infiamación: Evidencia de Eficacia en Modeios de Infiamación (véanse, p. ej., Jaffee y cois., 2000; Thaihamer y cois., 2008; Geppert y cois., 1994). •
• Artritis: Evidencia de eficacia en osteoartritis experimentai (véase, p. ej., Peiietier y cois., 2003); modeio de artritis reumatoide (véanse, p. ej., Chun y cois., 2002; Dudiey y cois., 2000); revisado en Thaihamer y cois., 2008.
• Remodeiación cardíaca, por ejempio, en ei síndrome metabóiico (véase, p. ej., Asrih y cois., 2013).
• Lesión orgánica, por ejempio, en una iesión renai inducida por cispiatino (véase, p. ej., Jo y cois., 2005).
• Hemogiobinopatías: drepanocitosis, p-taiasemia, hemogiobinopatía H (véase, p. ej., Zennadi y cois., 2012).
• Asma (véase, p. ej., Bridges y cois., 2000).
• Rechazo de traspiantes (véase, p. ej., Giibertsen y cois., 2000).
• Choque séptico (véase, p. ej., Geppert y cois., 1994).
• Infección virai, por ejempio hepatitis B (véase, p. ej., Benn y cois., 1994), hepatitis C (véase, p. ej., Zhang y cois., 2012), virus de inmunodeficiencia humana (HIV, por sus sigias en ingiés) (véase, p. ej., Yang y cois., 1999), virus de Epstein-Barr (EBV, por sus sigias en ingiés) (véase, p. ej., Fukuda y cois., 2007), HPV (véase, p. ej., Payne y cois.,
2001), herpesvirus humano-8 (HHV, por sus siglas en inglés) asociado con sarcoma de Kaposi (véase, p. ej., Akula y cois., 2004), citomegalovirus humano (véase, p. ej., Johnson y cols., 2001), virus de Coxsackie B3 (véase, p. ej., Luo y cols., 2002), virus de Borna (véase, p. ej., Planz y cols., 2001), virus de la gripe (véase, p. ej., Pleschka y cols., 2001).
• infecciones crónicas y enfermedades autoinmunitarias, por ejemplo, al inhibir la actividad de células T reguladoras (véase, p. ej., Kjetil y cols., 2013).
• Aterosclerosis (véase, p. ej., Miura y cols., 2004).
• Reestenosis (véase, p. ej., Graf y cols., 1997).
• Cardiomiopatía (véase, p. ej., Lorenz y cols., 2009).
• Lesión por reperfusión isquémica cardíaca (véase, p. ej., Zouki y cols., 2000).
• Psoriasis (véase, p. ej., Haase y cols., 2001).
• Enfermedad de Alzheimer (véase, p. ej., Mei y cols., 2006) y otros trastornos neurológicos inducidos tales como mielopatía asociada a HTLV-I/enfermedades parasitarias espáticas tropicales o neurodegenerativas tales como enfermedad de Parkinson o esclerosis lateral amiloide a través de modulación de variantes de empalme de CD44 (véase, p. ej., Pinner y cols., 2009).
• Enfermedad pulmonar obstructiva crónica (véase, p. ej., Mercer y cols., 2006).
• Enteropatía inflamatoria (véase, p. ej., Lowenberg y cols., 2005).
• Enfermedades fibrogenéticas, tales como fibrosis quística (véase, p. ej., Li y cols., 1998), fibrosis hepática, por ejemplo, cirrosis hepática (véase, p. ej., Davies y cols., 1996).
• Las mutaciones en RAS hereditarias conducen a un grupo de enfermedades llamadas colectivamente rasopatía. Elegir como diana la ruta de MAPK en estas enfermedades se ha propuesto como un enfoque terapéutico en este tipo de enfermedades tales como el síndrome de Noonan (véase, p. ej., Gu y cols., 2013), el síndrome cardiofaciocutáneo (véase, p. ej., Anastasaki y cols., 2012) y malformaciones capilares (véase, p. ej., Vikkula y cols., 2004).
RTKs
Las tirosina cinasas receptoras (RTK) son importantes en la transmisión de señales bioquímica a través de la membrana plasmática de las células. Estas moléculas transmembranarias consisten característicamente en un dominio de unión a ligando extracelular conectado a través de un segmento en la membrana plasmática a un dominio de tirosina cinasa intracelular. La unión del ligando al receptor da como resultado la estimulación de la actividad de tirosina cinasa asociada al receptor que conduce a la fosforilación de residuos de tirosina tanto en el receptor como en otras proteínas intracelulares, conduciendo a una variedad de respuestas celulares. Hasta la fecha, se han identificado al menos diecinueve subfamilias de RTK distintas, definidas por la homología de la secuencia de aminoácidos.
FGFR
La familia de factores de crecimiento de fibroblastos (FGF) de polipéptidos de señalización regula una serie diversa de funciones fisiológicas incluyendo mitogénesis, curación de heridas, diferenciación celular y angiogénesis, y desarrollo. El crecimiento así como la proliferación de células tanto normales como malignas están afectados por cambios en la concentración local de estas moléculas de señalización extracelular, que actúan como factores autocrinos así como paracrinos. La señalización de FGF autocrina puede ser particularmente importante en el empeoramiento de cánceres dependientes de hormonas esteroideas y para un estado independiente de hormonas (véase, p. ej., Powers y cols., 2000).
Los FGF y sus receptores se expresan en niveles incrementados en varios tejidos y líneas celulares y se cree que la sobreexpresión contribuye al fenotipo maligno. Por otra parte, un número de oncogenes son homólogos de genes que codifican receptores de factores de crecimiento, y hay un potencial para la activación aberrante de la señalización dependiente de FGF en cáncer pancreático humano (véase, p. ej., Ozawa y cols., 2001).
Los dos miembros prototípicos son factor de crecimiento de fibroblastos ácido (aFGF o FGF1) y factor de crecimiento de fibroblastos básico (bFGF o FGF2) y, hasta la fecha, se han identificado al menos veinte miembros distintos de la familia de FGF. La respuesta celular a FGF se transmite a través de cuatro tipos de receptores de factor de crecimiento de fibroblastos de tirosina cinasa transmembranaria de alta afinidad numerados de 1 a 4 (FGFR-1 a FGFR-4). Al unirse al ligando, los receptores se dimerizan y auto- o trans-fosforilan residuos de tirosina citoplásmicos específicos para transmitir una señal intracelular que finalmente alcanza efectores de factores de transcripción nucleares.
La interrupción de la ruta de FGFR-1 (FGFRA) afecta a la proliferación de células tumorales debido a que esta cinasa se activa en muchos tipos de tumores además de células endoteliales proliferativas. La sobreexpresión y la activación de FGFR-1 en la vasculatura asociada a tumores ha sugerido un papel de estas moléculas en la angiogénesis tumoral.
FGFR-2 tiene gran afinidad por los factores de crecimientos de fibroblastos ácidos y/o básicos, así como los ligandos de factor de crecimiento de queratinocitos. FGFR-2 también propaga los potentes efectos oncogénicos de los FGF durante el crecimiento y la diferenciación de osteoblastos. Se observó que las mutaciones en FGFR-2, que conducen a alteraciones funcionales complejas, inducen osificación anormal de suturas craneales (craneosinostosis), que implican un papel principal de la señalización de FGFR en la formación ósea intramembranosa. Por ejemplo, en el síndrome de Apert (AP, por sus siglas en inglés), caracterizado por una osificación prematura de la sutura craneal, la mayoría de los casos se asocian con mutación puntuales que engendran ganancia de función en FGFR-2 (véase, p. ej., Lemonnier y cols., 2001).
Lemonnier y cols., 2001, “Role of N-cadherin and protein kinase C in osteoblast gene activation induced by the S252W fibroblast growth factor receptor 2 mutation in Apert craniosynostosis”, J. Bone Miner. Res., Vol. 16, pp. 832-845.
Varias anormalidades graves en el desarrollo esquelético humano, incluyendo síndromes de Apert, Crouzon, Jackson-Weiss, piel corrugada del cuero cabelludo de Beare-Stevenson y Pfeiffer se asocian a la presencia de mutaciones en FGFR-2. La mayoría, si no todos, los casos de síndrome de Pfeiffer (PS, por sus siglas en inglés) también están provocados por la mutación de novo del gen de FGFR-2 (véanse, p. ej., Meyers y cols., 1996; Plomp y cols., 1998), y se observó recientemente que las mutaciones en FGFR-2 rompen una de las reglas cardinales que gobiernan la especificidad de los ligandos. A saber, dos formas de empalme mutantes de receptor de factor de crecimiento de fibroblastos, FGFR2c y FGFR2b, han adquirido la capacidad de unirse a y ser activadas por ligandos de FGF atípicos. Esta pérdida de especificidad conduce a una señalización aberrante y sugiere que los fenotipos graves de estos síndromes patológicos resultan de la activación dependiente de ligandos ectópicos de FGFR-2 (véase, p. ej., Yu y cols., 2000).
Mutaciones activadoras del receptor la tirosina cinasa receptora FGFR-3 tales como translocaciones cromosómicas o mutaciones puntuales producen receptores FGFR-3 constitutivamente activos desregulados que se han relacionado con el mieloma múltiple y con carcinomas de vejiga urinaria y cuello uterino (véase, p. ej., Powers y cols., 2000). Según esto, la inhibición de FGFR-3 sería útil en el tratamiento del mieloma múltiple y carcinomas de vejiga urinaria y cuello uterino.
Angiogénesis
Las enfermedades proliferativas crónicas a menudo están acompañadas por una angiogénesis profunda, que puede contribuir a o mantener un estado inflamatorio y/o proliferativo, OK conduce a la destrucción de tejidos a través de la proliferación invasiva de vasos sanguíneos. Véanse, p. ej., Folkman, 1995; Folkman, 1997; Folkman y cols., 1992. Angiogénesis se usa generalmente para describir el desarrollo de vasos sanguíneos nuevos o de sustitución, o neovascularización. Es un proceso necesario y fisiológicamente normal por el que se establece la vasculatura en el embrión. En general, la angiogénesis no se produce en la mayoría de los tejidos adultos normales, siendo excepciones las zonas de ovulación, menstruación y curación de heridas. Sin embargo, muchas enfermedades se caracterizan por una angiogénesis persistente y desregulada. Por ejemplo, en la artritis, nuevos vasos sanguíneos capilares invaden la articulación y destruyen el cartílago (véase, p. ej., Colville-Nash and Scott, 1992). En la diabetes (y en muchas enfermedades oculares diferentes), nuevos vasos sanguíneos invaden la mácula o la retina u otras estructuras oculares, y pueden provocar ceguera (véase, p. ej., Alon y cols., 1995). El proceso de la aterosclerosis se ha ligado a la angiogénesis (véase, p. ej., Kahlon y cols., 1992). Se ha encontrado que el crecimiento y la metástasis de tumores son dependientes de la angiogénesis (véanse, p. ej., Folkman, 1992; Denekamp, 1993; Fidler and Ellis, 1994).
El reconocimiento de la implicación de la angiogénesis en enfermedades importantes se ha acompañado por la investigación para identificar y desarrollar inhibidores de la angiogénesis. Estos inhibidores se clasifican generalmente en respuesta a dianas discretas en la cascada de angiogénesis, tales como activación de células endoteliales por una señal angiogénica; síntesis y liberación de enzimas degradativas; migración de células endoteliales; proliferación de células endoteliales; y formación de túbulos capilares. Por lo tanto, la angiogénesis se produce en muchos estadios y se están haciendo intentos de descubrir y desarrollar compuestos que trabajen para bloquear la angiogénesis en estos diversos estadios.
Hay muchas publicaciones que muestran que los inhibidores de la angiogénesis, que trabajan mediante diversos mecanismos, son beneficiosos en enfermedades tales como cáncer y metástasis (véanse, p. ej., O’Reilly y cols., 1994; Ingber y cols., 1990), enfermedades oculares (véase, p. ej., Friedlander y cols., 1995), artritis (véanse, p. ej., Peacock y cols., 1992; Peacock y cols., 1995) y hemangioma (véase, p. ej., Taraboletti y cols., 1995).
VEGFR
El factor de crecimiento endotelial vascular (VEGF, por sus siglas en inglés), un polipéptido, es mitogénico para células endoteliales in vitro y estimula respuestas angiogénicas in vivo. El VEGF también se ha relacionado con una angiogénesis inapropiada (véase, p. ej., Pinedo y cols., 2000). Los VEGFR son tirosina cinasas receptoras (RTK). Las RTK catalizan la fosforilación de residuos de tirosilo específicos en proteínas implicadas en la regulación del crecimiento y la diferenciación celular (véanse, p. ej., Wilks y cols., 1990; Courtneidge y cols., 1993; Cooper y cols., 1994; Paulson y cols., 1995; Chan y cols., 1996).
Se han identificados tres receptores RTK para VEGF: VEGFR-1 (Flt-1), VEGFR-2 (Flk-1 o KDR) y VEGFR-3 (Flt-4). Estos receptores están implicados en la angiogénesis y participan en la transducción de señales (véase, p. ej., Mustonen y cols., 1995).
De particular interés es VEGFR-2 (KDR), que es una RTK receptora transmembranaria expresada principalmente en células endoteliales. La activación de VEGFR-2 por VEGF es una etapa crítica en la ruta de transducción de señales que inicia la angiogénesis tumoral. La expresión de VEGF puede ser constitutiva a las células tumorales y también puede ser regulada al alza en respuesta a ciertos estímulos. Uno de tales estímulos es la hipoxia, donde la expresión de VEGF es regulada al alza en tejidos hospedadores tanto tumorales como asociados. El ligando VEGF activa VEGFR-2 al unirse a su centro de unión a VEGF extracelular. Esto conduce a la dimerización del receptor de los VEGFR y autofosforilación de residuos de tirosina en el dominio de cinasa intracelular de VEGFR-2. El dominio de cinasa funciona para transferir un fosfato desde ATP hasta los residuos de tirosina, proporcionando así centros de unión para proteínas de señalización aguas abajo de VEGFR-2 conduciendo finalmente al inicio de la angiogénesis (véase, p. ej., McMahon y cols., 2000).
La inhibición en el centro de unión al dominio de cinasa de VEGFR-2 bloquearía la fosforilación de residuos de tirosina y serviría para interrumpir el inicio de la angiogénesis.
VEGFR-2 (y VEGFR-3) se localizan principalmente en la vasculatura tumoral (sanguínea y/o linfática) que soporta la mayoría de los cánceres sólidos, y se regula al alza significativamente. Es probable que el mecanismo de acción clínico primario de los inhibidores de la señalización de VEGF sea a través de la elección como diana de vasos tumorales en lugar de células tumorales (véase, p. ej., Smith y cols., 2010), aunque se han descrito otros mecanismos. Se ha observado que los agentes que tienen como diana el factor de crecimiento endotelial vascular growth factor (VEGF), administrados bien como agentes únicos o bien en combinación con quimioterapia, benefician a los pacientes con enfermedades malignas en estadio avanzado (véase, p. ej., Ellis y cols., 2008).
KDR representa un papel crucial en otras enfermedades, y los inhibidores de KDR pueden encontrar utilidad en estas afecciones.
Aterosclerosis: KDR se expresa intensamente tanto en células endoteliales durante la angiogénesis como en el endotelio luminal de vasos ateroscleróticos humanos, pero no en arterias o venas normales (véase, p. ej., Belgore y cols., 2004). La interacción entre VEGF y el receptor de VEGF 2 (KDR, humano; Flk-1, ratón) es clave para la angiogénesis patológica y se ha relacionado con el desarrollo de lesiones ateroscleróticas (véase, p. ej., Inoue y cols., 1998). La vacunación contra KDR dio como resultado la activación de células T, la supresión de la neoangiogénesis y una notable reducción en la aterosclerosis que era independiente de la hipercolesterolemia en ratones tanto macho como hembra (véase, p. ej., Petrovan y cols., 2007).
Obesidad: La formación de nuevos vasos en tejidos grasos durante la obesidad inducida por la dieta se debe en gran parte a la angiogénesis en lugar de a la vasculogénesis de novo. El tratamiento antiangiogénico mediante el bloqueo de VEGFR2 pero no VEGFR1 puede limitar la expansión del tejido adiposo (véase, p. ej., Tam y cols., 2009).
Retinopatía y Maculopatía: La activación anormal del sistema VEGF-VEGFR está íntimamente implicada en la progresión de la degeneración macular asociada a la edad (AMD, por sus siglas en inglés). Por lo tanto, un aptámero contra VEGF-A^s, un anticuerpo neutralizador de VEGF (tipo Fab) y VEGF-Trap están ahora aprobados para el tratamiento de la AMD (véase, p. ej., Masabumi y cols., 2013). El bevazucimab, un anticuerpo anti-VEGF, se usa extraoficialmente en afecciones tales como la AMD, la retinopatía diabética y el edema macular diabético (DME, por sus siglas en inglés) (véase, p. ej., Rotsos y cols., 2008).
Síndrome de dolor neuropático: VEGF y VEGFR2 están implicados en la patogénesis del dolor neuropático. El tratamiento anti-rVEGF en ratas CCI puede aliviar el dolor neuropático crónico al disminuir las expresiones de VEGFR2 y receptores de P2X2/3 en neuronas del DRG para inhibir la transmisión de la señalización del dolor neuropático (véase, p. ej., Lin y cols., 2010).
Artritis reumatoide: PTK787/ZK222584, un inhibidor de tirosina cinasa receptora con actividad específica contra los VEGFR, y que exhibe una fuerte inhibición de VEGF-R2 (KDR) y una inhibición ligeramente más débil de VEGFR1 (Flt-1), Flk-1 (el homólogo de ratón de KDR) y Flt-4 (el receptor encontrado en el sistema linfático), inhibió la hinchazón de rodilla en 40%, puntuaciones de gravedad (en 51%) y puntuaciones histológicas globales en ratones con artritis inducida por colágeno (véase, p. ej., Grosios y cols., 2004)
TIE
La angiopoyetina 1 (Ang1), un ligando para la tirosina cinasa receptora específica del endotelio TIE-2, en un factor angiogénico (véanse, p. ej., Davis y cols., 1996; Partanen y cols., 1992; Davis y cols., 1994; Davis y cols., 1996; Alitalo y cols., 1996; Godowski y cols., 1997). El acrónimo TIE representa “dominios de homología de Ig y EGF que contienen tirosina cinasa”. TIE se usa para identificar una clase de tirosina cinasas receptoras, que se expresan exclusivamente en células endoteliales vasculares y células hematopoyéticas tempranas. Típicamente, las tirosina cinasas receptoras TIE se caracterizan por la presencia de un dominio similar a EGF y un dominio similar a inmunoglobulina (IG), que consiste en unidades de plegamiento extracelulares, estabilizados por enlaces disulfuro intracatenarios (véase, p. ej., Partanen y cols., 1999). A diferencia de VEGF, que funciona durante los estadios iniciales del desarrollo vascular, Ang1 y su receptor TIE-2 funcionan en los estadios tardíos del desarrollo vascular, es decir, durante la remodelación vascular (remodelación se refiere a la formación de una luz vascular) y la maduración (véanse, p. ej., Yancopoulos y cols., 1998; Peters y cols., 1998; Suri y cols., 1996).
Por consiguiente, se esperaría que la inhibición de TIE-2 sirviera para interrumpir la remodelación y la maduración de nueva vasculatura iniciada por angiogénesis interrumpiendo de ese modo el proceso angiogénico.
p38
p38 es un miembro de la familia MAPK de 38 kDa que se activa en respuesta al estrés y representa un papel importante en la respuesta inmunitaria y la supervivencia y la diferenciación celular. Se han descrito cuatro p38 MAPK cinasas; estas proteínas compartes un alto grado de homología (p38a, p, y y 5). Las p38 MAPK se pueden activar mediante diferentes estímulos tales como factores de crecimiento, citocinas inflamatorias o una variedad de estreses ambientales. Las p38 MAPK pueden activar a su vez un número de dianas aguas abajo, incluyendo proteína cinasas, sustratos citosólicos, factores de transcripción y factores de remodelación de cromatina. La fuerte activación de las p38 MAPKs por citocinas y estreses celulares promueve generalmente la inhibición del crecimiento celular e induce la apoptosis (véase, p. ej., la revisión en Cuadrado y cols., 2010). Más recientemente, se ha encontrado que p38a representa papeles importantes en el mantenimiento de la homeostasis y patologías relacionadas. El papel más conocido y más ampliamente presentado de p38a en la enfermedad está relacionado con su función en la señalización de citocinas y la promoción de la inflamación patológica. Varios estudios han mostrado cómo p38a puede mediar en una serie de modelos de enfermedad, incluyendo artritis reumatoide, psoriasis, enfermedad de Alzheimer, enteropatía inflamatoria, enfermedad de Crohn, tumorigénesis, enfermedad cardiovascular y apoplejía. Por otra parte, hay una evidencia de un papel de la p38 MAPK en el desarrollo y el mantenimiento de un número de enfermedades pulmonares, tales como asma, fibrosis quística, fibrosis pulmonar idiopática y enfermedad pulmonar obstructiva crónica. Así, p38a es una diana farmacéutica interesante especialmente debido a su importante papel en enfermedades inflamatorias (véase, p. ej., la revisión en Oeztuerk-Winder y cols., 2012). Fármacos de piridinilimidazol tales como SB203580 fueron los primeros inhibidores de p38 MAPK en ser identificados que se unen competitivamente en el bolsillo de unión a ATP, y se han usado ampliamente para estudiar las funciones de p38 MAPK (véase, p. ej., Coulthard y cols., 2009).
SRC
c-SRC pertenece a las cinasas de la familia SRC no receptoras (SFK). Estas proteínas están implicadas en muchos episodios celulares tales como proliferación, supervivencia y movilidad celular. Así, la hiperactivación de la señalización de SRC contribuye a diversos aspectos del desarrollo de tumores. La función más destacada de c-SRC es su interacción intensiva con tirosina cinasas receptoras (RTK) transmembranarias en la membrana celular a través de sus dominios SH2 y SH3. c-SRC interactúa con muchas RTK incluyendo el receptor del factor de crecimiento epidérmico (EGFR, por sus siglas en inglés), el receptor del factor de crecimiento epidérmico humano 2 (HER2, por sus siglas en inglés), el receptor del factor de crecimiento derivado de plaquetas (PDGFR, por sus siglas en inglés), el receptor del factor de crecimiento insulinoide 1 (IGF-1R, por sus siglas en inglés) y el receptor de c-Met/factor de crecimiento de hepatocitos (HGFR, por sus siglas en inglés). A través de estas interacciones, c-SRC integra y regula la señalización de RTK y transduce directamente señales de supervivencia a efectores aguas abajo tales como fosfoinosítido 3-cinasas (PI3Ks), Akt, y transductor de señales y activador de la transcripción 3 (STAT3, por sus siglas en inglés) (véase, p. ej., Zhang y cols., 2012). Otros receptores membranarios tales como integrinas también pueden activar c-SRC desencadenando así una cascada de señales que regula la migración, la adherencia y la invasión celular. La activación de c-Src a través de la interacción con catenina p120 promueve la disociación de uniones adherentes célula-célula mejorando asó la movilidad celular. c-SRC fosforila directamente la cinasa de adherencia focal (FAK, por sus siglas en inglés) estabilizando complejos de adherencia focales, que consisten en FAK, paxilina, RhoA y otros componentes, y mejora la adherencia celular a la matriz extracelular. Por otra parte, c-SRC representa un importante papel el la regulación del microambiente tumoral. La activación de c-SRC en la hipoxia promueve la angiogénesis a través de la estimulación de la expresión del factor de crecimiento endotelial vascular (VEGF), metaloproteinasa de matriz (MMP, por sus siglas en inglés) e interleucina-8 (IL-8) (véase, p. ej., Yeatman y cols., 2004).
La elección como diana de SFK es un enfoque terapéutico bien establecido para muchos tipos de cáncer. El dasatinib es un inhibidor de molécula pequeña de múltiples cinasas que inhibe potentemente cinasas de la familia SRC (SRC, LCK, YES, FYN), pero también BCR-ABL, c-KIT, PDGFR-a y p, y cinasa receptora de efrina (véase, p. ej., Lindauer y cols., 2010). Estudios más recientes han presentado que Src también está implicada en la ruta de señalización relacionada con la inflamación. Muchos estudios han mostrado que c-SRC representa un papel crítico
en respuestas inflamatorias mediadas por macrófagos. De forma importante, una variedad de enfermedades inflamatorias está estrechamente relacionada con la activación de macrófagos; por lo tanto, la inhibición de c-SRC puede representar una estrategia útil para enfermedades mediadas por macrófagos (véase, p. ej., Byeon y cols., 2012)
Lck
La Lck (cinasa específica de linfocitos, por sus siglas en inglés) es una cinasa de las SFK que es crítica para la activación de células T, y su actividad está inducida por el receptor de células T (TCR, por sus siglas en inglés). Las señales de TCR iniciadas por Lck conducen a episodios de regulación génica que dan como resultado liberación de citocinas, proliferación y supervivencia de células T específicas para antígenos, amplificando de ese modo respuestas inmunitarias específicas. Se espera que la inhibición de Lck ofrezca un nuevo enfoque terapéutico para el tratamiento de trastornos autoinmunitarios e inflamatorios mediados por células T y/o rechazo de trasplantes de órganos (véase, p. ej., Martin y cols., 2010).
Compuestos conocidos
Niculescu-Duvaz y cols., 2006, describe ciertos compuestos de imidazo[4,5-b]piridin-2-ona y oxazolo[4,5-b]piridin-2-ona que, entre otras cosas, inhiben la actividad de RAF (p. ej., BRAF), y que son útiles en el tratamiento de trastornos proliferativos tales como cáncer. Un número de compuestos mostrados allí tienen un grupo 5-(terc-butil)-2-(fenil)-pirazol-3-ilo o un grupo 5-(terc-butil)-2-(piridil)-pirazol-3-ilo. Sin embargo, en todos los casos, el grupo fenilo y piridilo no está sustituido, está sustituido en para o está disustituido en orto,para; en ninguno de los compuestos, está sustituido en meta. Se muestran los siguientes compuestos:



Niculescu-Duvaz y cois., 2007, describe ciertos compuestos de imidazo[4,5-b]piridin-2-ona y oxazolo[4,5-b]piridin-2-ona que, entre otras cosas, inhiben la actividad de RAF (p. ej., BRAF), y que son útiles en el tratamiento de trastornos proliferativos tales como cáncer. Un número de compuestos mostrados allí tienen un grupo 5-(terc-butil)-2-(fenil)-pirazol-3-ilo. Sin embargo, en todos los casos, el grupo fenilo no está sustituido o está sustituido en para; en ninguno de los compuestos, está sustituido en meta. Se muestran los siguientes compuestos:

Springer y cols., 2009, describe ciertos compuestos de sustituidos en 8 con pirido[2,3-b]piracina que, entre otras cosa, inhiben la actividad de RAF (p. ej., BRAF), y que son útiles en el tratamiento de trastornos proliferativos tales como cáncer. Un número de compuestos mostrados allí tienen un grupo 5-(terc-butil)-2-(fenil)-pirazol-3-ilo o un grupo 5-(terc-butil)-2-(piridil)-pirazol-3-ilo. Sin embargo, en todos los casos, el grupo fenilo y el grupo piridilo no está
sustituido o está sustituido en para; en ninguno de los compuestos, está sustituido en meta. Se muestran los siguientes compuestos:

actividad de RAF (p. ej., BRAF), y que son útiles en el tratamiento de trastornos proliferativos tales como cáncer. Un número de compuestos mostrados allí tienen un grupo 5-(terc-butil)-2-(fenil)-pirazol-3-ilo. Sin embargo, en todos los
casos, el grupo fenilo no está sustituido o está sustituido en para; en ninguno de los compuestos está sustituido en meta. Se muestran los siguientes compuestos:

Springer y cois., 2011, describe ciertos compuestos de 1-(5-terc-butil-2-fenil-2H-pirazol-3-il)-3-[2-fluoro-4-(1-metil-2-oxo-2,3-dihidro-1H-imidazo[4,5-b]piridin-7-iloxi)-fenil]urea que, entre otras cosas, inhiben la actividad de RAF (p. ej., BRAF), y que son útiles en el tratamiento de trastornos proliferativos tales como cáncer. Se muestra el siguiente compuesto:

Murray y cols., 2011, describe ciertos compuestos de para el uso en el tratamiento de una enfermedad inflamatoria o un trastorno respiratorio. Unos pocos de los compuestos mostrados allí tienen un grupo 5-(terc-butil)-2-(fenil)-pirazol-3-ilo o un grupo 5-(terc-butil)-2-(piridil)-pirazol-3-ilo. Sin embargo, en todos los casos, el grupo fenilo y piridilo no está sustituido, está sustituido en para o está sustituido en meta, para; en ninguno de los compuestos, está sustituido en meta, no sustituido en para. Se muestran los siguientes compuestos:

Se conoce un número de compuestos que tienen un grupo 5-(terc-butil)-2-(3-fluoro-fenil)-pirazol-3-ilo, incluyendo los siguientes:

SUMARIO DE LA INVENCI N
Un aspecto de la invención trata de ciertos compuestos de 1-(5-terc-butil-2-aril-pirazol-3-il)-3-[2-fluoro-4-[(3-oxo-4H-pirido[2,3-b]piracin-8-il)oxi]fenil]urea (denominados en la presente “compuestos de TBAP”), según se describe en la presente.
Otro aspecto de la invención trata de una composición (p. ej., una composición farmacéutica) que comprende un compuesto de TBAP, según se describe en la presente, y un portador o diluyente farmacéuticamente aceptable. También se describe en la presente un método para preparar una composición (p. ej., una composición farmacéutica) que comprende la etapa de mezclar un compuesto de TBAP, según se describe en la presente, y un portador o diluyente farmacéuticamente aceptable.
También se describe en la presente un método para inhibir la función de RAF (p. ej., BRAF, CRAF, etc.) (p. ej., en una célula), in vitro o in vivo, que comprende poner el contacto la célula con una cantidad eficaz de un compuesto de TBAP, según se describe en la presente.
Otro aspecto de la presente invención trata de un compuesto de TBAP según se describe en la presente para el uso en un método de tratamiento del cuerpo de un ser humano o un animal mediante terapia, por ejemplo, para el uso en un método de tratamiento de un trastorno (p. ej., una enfermedad) según se describe en la presente.
También se describe en la presente el uso de un compuesto de TBAP, según se describe en la presente, en la fabricación de un medicamento, por ejemplo, para el uso en un método de tratamiento, por ejemplo, para el uso en un método de tratamiento de un trastorno (p. ej., una enfermedad) según se describe en la presente.
También se describe en la presente un método de tratamiento, por ejemplo, un método de tratamiento de un trastorno (p. ej., una enfermedad) según se describe en la presente, que comprende administrar a un sujeto que necesite tratamiento una cantidad terapéuticamente eficaz de un compuesto de TBAP, según se describe en la presente, preferiblemente en la forma de una composición farmacéutica.
También se describe en la presente un estuche que comprende (a) un compuesto de TBAP, según se describe en la presente, preferiblemente proporcionado como una composición farmacéutica y en un recipiente adecuado y/o con un envase adecuado; y (b) instrucciones de uso, por ejemplo, instrucciones escritas sobre cómo administrar el compuesto.
También se describe en la presente un compuesto de TBAP obtenible mediante un método de síntesis según se describe en la presente, o un método que comprende un método de síntesis según se describe en la presente. También se describe en la presente un compuesto de TBAP obtenido mediante un método de síntesis según se describe en la presente, o un método que comprende un método de síntesis según se describe en la presente. También se describen en la presente nuevos productos intermedios, según se describe en la presente, que son adecuados para el uso en los métodos de síntesis descritos en la presente.
También se describe en la presente el uso de tales nuevos productos intermedios, según se describe en la presente, en los métodos de síntesis descritos en la presente.
Como será apreciado por un experto en la técnica, características y realizaciones preferidas de un aspecto de la invención también tratarán de otros aspectos de la invención.
DESCRIPCIÓN DETALLADA DE LA INVENCIÓN
Compuestos
Un aspecto de la presente invención se refiere a ciertos compuestos de 1-(5-terc-butil-2-aril-pirazol-3-il)-3-[2-fluoro-4-[(3-oxo-4H-pirido[2,3-b]piracin-8-il)oxi]fenil]urea que están estructuralmente relacionados con los siguientes compuestos:

1-(5-terc-butil-2-fen¡l-pirazol-3-il)- 1-(5-terc-butil-2-(4-p¡r¡d¡l)pirazol-3-il)-3-[2-fluoro-4-[(3-oxo-4H-pirido[2,3- 3-[2-fluoro-4-[(3-oxo-4H-pirido[2,3-b]piracin-8-il)oxi]fenil]urea b]piracin-8-il)oxi]fenil]urea
Más particularmente, la presente invención se refiere a ciertos compuestos relacionados que tienen adicionalmente un solo sustituyente en meta (indicado en la presente como -Y).
Así, un aspecto de la presente invención trata de compuestos seleccionados de compuestos de la siguiente fórmula, y sales farmacéuticamente aceptables, N-óxidos, hidratos y solvatos de los mismos, en donde =X- e -Y son como se definen en la presente (por comodidad, denominados colectivamente “compuestos de TBAP”):

Algunas realizaciones de la invención incluyen las siguientes:
(1) Un compuesto seleccionado de compuestos de la siguiente fórmula, y sales farmacéuticamente aceptables, N-óxidos, hidratos y solvatos de los mismos:

=X- es independientemente =CH- o =N-;
-Y es independientemente -Y1, -Y2, -Y3, -Y4, -Y5 o -Y6;
-Y1 es independientemente -F, -Cl, -Br o -I;
-Y2 es alquilo C1-4 saturado lineal o ramificado;
-Y3 es haloalquilo C1-4 saturado lineal o ramificado;
-Y4 es -OH;
-Y5 es alcoxi C1-4 saturado lineal o ramificado; y
-Y6 es haloalcoxi C1-4 saturado lineal o ramificado.
Nótese que es posible la tautomerización en el grupo 3-oxo-3,4-dihidropirido[3,2-¿>]piracin-8-ilo, como se muestra posteriormente. A menos que se indique otra cosa, una referencia a un tautómero pretende ser una referencia a ambos tautómeros.

Nótese que cuando -X= es -N= e -Y es -Y4 (es decir, -OH), es posible la tautomerización en el grupo 2-hidroxi-pirid-4-ilo resultante, como se muestra posteriormente. A menos que se indique otra cosa, una referencia a un tautómero pretende ser una referencia a ambos tautómeros.

Nótese que cuando =X- es =N-, el grupo resultante es un grupo piridil-4-ilo grupo, y se puede formar un N-óxido, según se muestra posteriormente.

Para evitar dudas, el término “haloalquilo C1-4 saturado lineal o ramificado” se refiere a un grupo alquilo C1-4 saturado lineal o ramificado que tiene 1 o más (p. ej., 1,2, 3, etc.) sustituyentes halógeno (p. ej., -F, -Cl, -Br, -I). Un ejemplo de tal grupo es -CF3.
Para evitar dudas, el término “alcoxi C1-4 saturado lineal o ramificado” se refiere a un grupo -OR, donde R es un grupo alquilo C1-4 saturado lineal o ramificado. Un ejemplo de tal grupo es -OMe.
De forma similar, el término “haloalcoxi C1-4 saturado lineal o ramificado” se refiere a un grupo -OR, donde R es un grupo haloalquilo C1-4 saturado lineal o ramificado. Un ejemplo de tal grupo es -OCF3.
Para evitar dudas: metilo se abrevia como -Me; etilo se abrevia como -Et; n-propilo se abrevia como -nPr; /'sopropilo se abrevia como -iPr; n-butilo se abrevia como -nBu; /so-butilo se abrevia como -iBu; sec-butilo se abrevia como -sBu; ferc-butilo se abrevia como -tBu; y fenilo se abrevia como -Ph.
El Grupo =X-(2) Un compuesto según (1), en el que =X- es =CH-.
(3) Un compuesto según (1), en el que =X- es =N-.
El Grupo -Y
(4) Un compuesto según uno cualquiera de (1) a (3), en el que -Y es -Y1.
(5) Un compuesto según uno cualquiera de (1) a (3), en el que -Y es -Y2.
(6) Un compuesto según uno cualquiera de (1) a (3), en el que -Y es -Y3.
(7) Un compuesto según uno cualquiera de (1) a (3), en el que -Y es -Y4.
(8) Un compuesto según uno cualquiera de (1) a (3), en el que -Y es -Y5.
(9) Un compuesto según uno cualquiera de (1) a (3), en el que -Y es -Y6.
El Grupo -Y1
(10) Un compuesto según uno cualquiera de (1) a (9), en el que -Y1, si está presente, es independientemente -F, -Cl, -Br.
(11) Un compuesto según uno cualquiera de (1) a (9), en el que -Y1, si está presente, es independientemente -F o -Cl.
(12) Un compuesto según uno cualquiera de (1) a (9), en el que -Y1, si está presente, es -F.
(13) Un compuesto según uno cualquiera de (1) a (9), en el que -Y1, si está presente, es -Cl.
(14) Un compuesto según uno cualquiera de (1) a (9), en el que -Y1, si está presente, es -Br.
(15) Un compuesto según uno cualquiera de (1) a (9), en el que -Y1, si está presente, es -I.
El Grupo -Y2
(16) Un compuesto según uno cualquiera de (1) a (15), en el que -Y2, si está presente, es independientemente -Me, -Et, -nPr, -iPr, -nBu, -iBu, -sBu o -tBu.
(17) Un compuesto según uno cualquiera de (1) a (15), en el que -Y2, si está presente, es independientemente -Me, -Et, -nPr o -iPr.
(18) Un compuesto según uno cualquiera de (1) a (15), en el que -Y2, si está presente, es independientemente -Me o -Et.
(19) Un compuesto según uno cualquiera de (1) a (15), en el que -Y2, si está presente, es -Me.
El Grupo -Y3
(20) Un compuesto según uno cualquiera de (1) a (19), en el que -Y3, si está presente, es fluoroalquilo C1-4 saturado lineal o ramificado.
(21) Un compuesto según uno cualquiera de (1) a (19), en el que -Y3, si está presente, es independientemente -CH2F, -CHF2, -CF3, -CH2CH2F, -CH2CHF2 o -CH2CF3.
(22) Un compuesto según uno cualquiera de (1) a (19), en el que -Y3, si está presente, es independientemente -CH2F, -CHF2 o -CF3.
(23) Un compuesto según uno cualquiera de (1) a (19), en el que -Y3, si está presente, es -CF3.
El Grupo -Y5
(24) Un compuesto según uno cualquiera de (1) a (23), en el que -Y5, si está presente, es independientemente -O-Me, -O-Et, -O-nPr, -O-iPr, -O-nBu, -O-iBu, -O-sBu o -O-tBu.
(25) Un compuesto según uno cualquiera de (1) a (23), en el que -Y5, si está presente, es independientemente -O-Me, -O-Et, -O-nPr o -O-iPr.
(26) Un compuesto según uno cualquiera de (1) a (23), en el que -Y5, si está presente, es independientemente -O-Me o -O-Et.
(27) Un compuesto según uno cualquiera de (1) a (23), en el que -Y5, si está presente, es -O-Me.
El Grupo -Y6
(28) Un compuesto según uno cualquiera de (1) a (27), en el que -Y6, si está presente, es fluoroalquilo C1-4 saturado lineal o ramificado.
(29) Un compuesto según uno cualquiera de (1) a (27), en el que -Y6, si está presente, es independientemente -O-CH2F, -O-CHF2, -O-CF3, -O-CH2CH2F, -O-CH2CHF2 o -O-CH2CF3.
(30) Un compuesto según uno cualquiera de (1) a (27), en el que -Y6, si está presente, es independientemente -O-CH2F, -O-CHF2 o -O-CF3.
(31) Un compuesto según uno cualquiera de (1) a (27), en el que -Y6, si está presente, es -O-CF3.
Algunos Compuestos Preferidos
(32) Un compuesto según (1), seleccionado de compuestos de las siguientes fórmulas y sales farmacéuticamente aceptables, N-óxidos, hidratos y solvatos de los mismos:


Combinaciones
Se aprecia que ciertas características de la invención, que, por claridad, se describen en el contexto de realizaciones separadas, también se pueden proporcionar en combinación en una sola realización. A la inversa, diversas características de la invención, que, por brevedad, se describen en el contexto de una sola realización, también se pueden proporcionar separadamente o en cualquier subcombinación adecuada. Todas las combinaciones de las realizaciones que tratan de los grupos químicos representados por las variables (p. ej., =X-, -Y, -Y1, -Y2, -Y3, -Y4, -Y5, -Y6, etc.) son abarcadas específicamente por la presente invención y se divulgan en la presente justamente como si todas y cada una de las combinaciones se divulgaran individualmente y explícitamente, hasta el punto de que tales combinaciones abarcan compuestos que son compuestos estables (es decir, compuestos que se pueden aislar, caracterizar y probar con respecto a la actividad biológica). Además, todas las subcombinaciones de los grupos químicos listados en las realizaciones que describen tales variables también son abarcadas específicamente por la presente invención y se divulgan en la presente justamente como si todas y cada una de las subcombinaciones de grupos químicos se divulgaran individualmente y explícitamente en la presente.
Formas Sustancialmente Purificadas
Un aspecto de la presente invención trata de compuestos de TBAP, según se describe en la presente, en forma sustancialmente purificada y/o en una forma sustancialmente libre de contaminantes.
En una realización, el compuesto está en forma sustancialmente purificada y/en una forma sustancialmente libre de contaminantes.
En una realización, el compuesto está en una forma sustancialmente purificada con una pureza de al menos 50% en peso, p. ej., al menos 60% en peso, p. ej., al menos 70% en peso, p. ej., al menos 80% en peso, p. ej., al menos 90% en peso, p. ej., al menos 95% en peso, p. ej., al menos 97% en peso, p. ej., al menos 98% en peso, p. ej., al menos 99% en peso.
En una realización, el compuesto está en una forma sustancialmente libre de contaminantes en la que los contaminantes representan no más de 50% en peso, p. ej., no más de 40% en peso, p. ej., no más de 30% en peso, p. ej., no más de 20% en peso, p. ej., no más de 10% en peso, p. ej., no más de 5% en peso, p. ej., no más de 3% en peso, p. ej., no más de 2% en peso, p. ej., no más de 1% en peso. A menos que se especifique, los contaminantes se refieren a otros compuestos.
Isómeros
Ciertos compuestos pueden existir en una o más formas geométricas, ópticas, enantiómeras, diastereoisómeras, epímeras, atrópicas, estereoisómeras, tautómeras, conformacionales o anómeras particulares, incluyendo, pero no limitadas a, formas cis y trans; formas E y Z; formas c, t y r; formas endo y exo; formas R, S y meso; formas D y L; formas d y l; formas (+) y (-); formas ceto, enol y enolato; formas sin y anti; formas sinclinales y anticlinales; formas a y p; formas axiales y ecuatoriales; formas de bote, silla, trenza, sobre y media silla; y combinaciones de las mismas, posteriormente denominadas en la presente “isómeros” (o “formas isómeras”).
Nótese que, excepto cuando se analiza posteriormente para las formas tautómeras, se excluyen específicamente del término “isómeros”, según se usa en la presente, los isómeros estructurales (o generales) (es decir, isómeros que difieren en las conexiones entre átomos en vez de meramente por la posición de los átomos en el espacio). Por ejemplo, una referencia a un grupo metoxi, -OCH3, no se debe considerar como una referencia a su isómero estructural, un grupo hidroximetilo, -CH2OH. De forma similar, una referencia a orto-clorofenilo no se debe considerar como una referencia a su isómero estructural, meta-clorofenilo. Sin embargo, una referencia a una clase de estructuras puede incluir perfectamente formas estructuralmente isómeras que estén dentro de esa clase (p. ej., alquilo C1-7 incluye n-propilo e isopropilo; butilo incluye n-, iso-, sec- y terc-butilo; metoxifenilo incluye orto-, meta- y para-metoxifenilo).
La exclusión anterior no trata de formas tautómeras, por ejemplo, formas ceto, enol y enolato, como, por ejemplo, en los siguientes pares tautómeros: ceto/enol (ilustrado posteriormente), imina/enamina, amida/iminoalcohol, amidina/amidina, nitroso/oxima, tiocetona/enotiol, N-nitroso/hidroxiazo y nitro/acinitro.

Nótese que en el término "isómero" se incluyen específicamente compuestos con una o más sustituciones isotópicas. Por ejemplo, H puede estar en cualquier forma isotópica, incluyendo 1H, 2H (D) y 3H (T); C puede estar en cualquier forma isotópica, incluyendo 12C, 13C y 14C; O puede estar en cualquier forma isotópica, incluyendo 16O y 18O; y similares.
A menos que se especifique otra cosa, una referencia a un compuesto particular incluye todas estas formas isómeras, incluyendo mezclas de las mismas. Métodos para la preparación y separación de tales formas isómeras bien son conocidos en la técnica o bien se pueden obtener fácilmente adaptando los métodos mostrados en la presente o métodos conocidos, de un modo conocido.
Sales
Puede ser conveniente o deseable preparar, purificar y/o manejar una sal correspondiente del compuesto, por ejemplo, una sal farmacéuticamente aceptable. Ejemplos de sales farmacéuticamente aceptables se analizan en Berge y cols., 1977, “Pharmaceutically Acceptable Salts”, J. Pharm. Sci., Vol. 66, pp. 1 -19.
Por ejemplo, si el compuesto es aniónico o tiene un grupo funcional que puede ser aniónico (p. ej., -COOH puede ser -COO-), entonces se puede formar una sal con un catión adecuado. Ejemplos de cationes inorgánicos adecuados incluyen, pero no se limitan a, iones de metales alcalinos tales como Na+ y K+, cationes alcalinotérreos tales como Ca2+ y Mg2+, y otros cationes tales como Al3+. Ejemplos de cationes orgánicos adecuados incluyen, pero no se limitan a, ion amonio (es decir, NH4+) e iones amonio sustituidos (p. ej., NH3R+, NH2R2+, NHR3+, NR4+). Ejemplos algunos iones amonio sustituido adecuados son los derivados de: etilamina, dietilamina, diciclohexilamina, trietilamina, butilamina, etilendiamina, etanolamina, dietanolamina, piperacina, bencilamina, fenilbencilamina, colina, meglumina y trometamina, así como aminoácidos, tales como lisina y arginina. Un ejemplo de un ion amonio cuaternario común es N(CH3)4+.
Si el compuesto es catiónico o tiene un grupo funcional que puede ser catiónico (p. ej., -NH2 puede ser -NH3+), entonces se puede formar una sal con un anión adecuado. Ejemplos de aniones inorgánicos adecuados incluyen, pero no se limitan a, los derivados de los siguientes ácidos inorgánicos: clorhídrico, bromhídrico, yodhídrico, sulfúrico, sulfuroso, nítrico, nitroso, fosfórico y fosforoso.
Ejemplos de aniones orgánicos adecuados incluyen, pero no se limitan a, los derivados de los siguientes ácidos orgánicos: 2-acetoxibenzoico, acético, ascórbico, aspártico, benzoico, canforsulfónico, cinámico, cítrico, edético, etanodisulfónico, etanosulfónico, fórmico, fumárico, glucoheptónico, glucónico, glutámico, glicólico, hidroximaleico, hidroxinaftalenocarboxílico, isetiónico, láctico, lactobiónico, láurico, maleico, málico, metanosulfónico, múcico, oleico, oxálico, palmítico, pamoico, pantoténico, fenilacético, fenilsulfónico, propiónico, pirúvico, salicílico, esteárico, succínico, sulfanílico, tartárico, toluenosulfónico y valérico. Ejemplos de aniones orgánicos poliméricos adecuados incluyen, pero no se limitan a, los derivados de los siguientes ácidos poliméricos: ácido tánico, carboximetilcelulosa. A menos que se especifique otra cosa, una referencia a un compuesto particular también incluye formas salinas del mismo.
N-Óxidos
Puede ser conveniente o deseable preparar, purificar y/o manejar un N-óxido correspondiente del compuesto. Por ejemplo, un compuesto que tiene un grupo piridilo se puede preparar, purificar y/o manejar como el N-óxido correspondiente.

A menos que se especifique otra cosa, una referencia a un compuesto particular también incluye formas de N-óxido del mismo.
Hidratos y Solvatos
Puede ser conveniente o deseable preparar, purificar y/o manejar un solvato correspondiente del compuesto. El término “solvato” se usa en la presente en el sentido convencional para referirse a un complejo de soluto (p. ej., compuesto, sal de compuesto) y disolvente. Si el disolvente es agua, el solvato se puede denominar convenientemente un hidrato, por ejemplo, un monohidrato, un dihidrato, un trihidrato, etc.
A menos que se especifique otra cosa, una referencia a un compuesto particular también incluye las formas de solvato e hidrato del mismo.
Formas Químicamente Protegidas
Puede ser conveniente o deseable preparar, purificar y/o manejar el compuesto en una forma químicamente protegida. El término “forma químicamente protegida” se usa en la presente en el sentido químico convencional y trata de un compuesto en el que uno o más grupos funcionales reactivos se protegen de reacciones químicas no deseables bajo condiciones especificadas (p. ej., pH, temperatura, radiación, disolvente y similares). En la práctica, se emplean métodos químicos muy conocidos para hacer reversiblemente no reactivo a un grupo funcional, que de otro modo sería reactivo, bajo condiciones especificadas. En una forma químicamente protegida, uno o más grupos funcionales reactivos están en la forma de un grupo protegido o protector (también conocido como un grupo enmascarado o enmascarante o un grupo bloqueado o bloqueante). Al proteger un grupo funcional reactivo, se
pueden realizar reacciones que implican otros grupos funcionales reactivos desprotegidos, sin afectar al grupo protegido; el grupo protector se puede retirar, habitualmente en una etapa posterior, sin afectar sustancialmente al resto de la molécula. Véase, por ejemplo, Protective Groups in Organic Synthesis (T. Greene y P. Wuts; 4a Edición; John Wiley and Sons, 2006).
Una amplia variedad de tales métodos de “protección”, “bloqueo” o “enmascaramiento” son ampliamente usados y muy conocidos en la síntesis orgánica. Por ejemplo, un compuesto que tiene dos grupos funcionales reactivos no equivalentes, ambos de los cuales serían reactivos bajo condiciones especificadas, se puede derivar para hacer que uno de los grupos funcionales esté “protegido”, y de ese modo no sea reactivo, bajo las condiciones especificadas; así protegido, el compuesto se puede usar como un reaccionantes que tiene efectivamente sólo un grupo funcional reactivo. Después de que se termine la reacción deseada (que implica al otro grupo funcional), el grupo protegido se puede “desproteger” para volver a su funcionalidad original.
Por ejemplo, un grupo hidroxi se puede proteger como un éter (-OR) o un éster (-OC(=O)R), por ejemplo, como: un éter t-butílico; un éter bencílico, benzhidrílico (difenilmetílico) o tritílico (trifenilmetílico); un éter trimetilsilílico o t-butildimetilsilílico; o un éster acetílico (-OC(=O)CH3, -OAc).
Por ejemplo, un grupo aldehído o cetona se puede proteger como un acetal (R-CH(OR)2) o cetal (R2C(OR)2), respectivamente, en los que el grupo carbonilo (>C=O) se convierte en un diéter (>C(OR)2), mediante una reacción con, por ejemplo, un alcohol primario. El grupo aldehído o cetona se regenera fácilmente mediante hidrólisis usando un gran exceso de agua en presencia de ácido.
Profármacos
Puede ser conveniente o deseable preparar, purificar y/o manejar el compuesto en la forma de un profármaco. El término “profármaco”, según se usa en la presente, trata de un compuesto que, cuando se metaboliza (p. ej., in vivo), da el compuesto activo deseado. Típicamente, el profármaco es inactivo o menos activo que el compuesto activo deseado, pero puede proporcionar propiedades de manejo, administración o metabólicas ventajosas.
Por ejemplo, algunos profármacos son ésteres del compuesto activo (p. ej., un éster metabólicamente lábil fisiológicamente aceptable). Durante el metabolismo, el grupo éster (-C(=O)OR) se escinde para dar el fármaco activo. Tales ésteres se pueden formar mediante esterificación, por ejemplo, de cualquiera de los grupos ácido carboxílico (-C(=O)OH) en el compuesto originario, con, cuando sea apropiado, antes de la protección de cualesquiera otros grupos reactivos presentes en el compuesto originario, seguido por desprotección si se requiere. Además, algunos profármacos se activan enzimáticamente para dar el compuesto activo o un compuesto que, durante una reacción química adicional, da el compuesto activo (por ejemplo, como en ADEPT, GDEPT, LIDEPT, etc.). Por ejemplo, el profármaco puede ser un derivado de azúcar u otro conjugado glicosídico o puede ser un derivado de éster de aminoácido.
Síntesis Química
Se describen en la presente métodos para la síntesis química de compuestos de la presente invención. Estos y/u otros métodos muy conocidos se pueden modificar y/o adaptar de maneras conocidas a fin de facilitar la síntesis de compuestos adicionales dentro del alcance de la presente invención.
Descripciones de métodos y procedimientos de laboratorio generales, útiles para la preparación de los compuestos descritos en la presente, se proporcionan en Vogel's Textbook of Practical Organic Chemistry, 5a Edición, 1989, (Editores: Furniss, Hannaford, Smith y Tatchell) (publicado por Longmann, Reino Unido).
Métodos para la síntesis de compuestos de piridina en particular se describen en Heterocyclic Chemistry, 3a Edición, 1998, (Editores: Joule, Mills y Smith) (publicado por Chapman & Hall, Reino Unido).
Los compuestos de TBAP descritos en la presente se pueden preparar a través del producto intermedio (2) clave. Este producto intermedio se puede preparar a partir de una materia prima disponible comercialmente, 2-amino-3-nitro-4-cloropiridina (1) y 3-fluoro-4-aminofenol. El producto intermedio (2) se puede proteger selectivamente en el grupo amino, por ejemplo como un carbamato BOC, para proporcionar el producto intermedio (3).
Un ejemplo de tal método se ilustra en el siguiente esquema.
Esquema 1

El producto intermedio (3) también se puede obtener directamente a partir de 2-amino-3-nitro-4-cloropiridina (1) y 3-fluoro-4-aminofenol protegido con BOC en N.
Un ejemplo de tal método se ilustra en el siguiente esquema.
Esquema 2

El grupo nitro del producto intermedio protegido (3) se puede reducir para dar un grupo amino, por ejemplo, con Pd/C y formiato amónico o hidrógeno o con NiCl2 y NaBH4, para dar el producto intermedio diaminado (4).
Un ejemplo de tal método se ilustra en el siguiente esquema.
Esquema 3

Se pueden obtener piridopiracinonas a partir del producto intermedio (4) mediante la reacción con glioxilato de etilo o ácido glioxílico. Ambos isómeros (5) y (6) se pueden obtener a partir de la reacción de (4) con glioxalato de etilo o ácido glioxílico. La relación de los dos isómeros puede estar influenciada por la elección de los reactivos y los disolventes, de modo que se obtenga uno preferentemente. El isómero (5) deseado se puede separar de la mezcla mediante cromatografía en columna o cristalización selectiva de la mezcla.
Un ejemplo de tal método se ilustra en el siguiente esquema.
Esquema 4

La desprotección del grupo protector (PG), por ejemplo, con fluoruro de tetrabutilamonio (TBAF) para un grupo protector Boc, produce el producto intermedio (7) común.
Un ejemplo de tal método se ilustra en el siguiente esquema.
Esquema 5

5 7
El producto intermedio (7) clave se hace reaccionar con 3-terc-butil-5-isocyanato-1-aril-1H-pirazols (10) para proporcionar las ureas (11) correspondientes.
Un ejemplo de tal método se ilustra en el siguiente esquema.
Esquema 6

7 10 11
Los isocianatos (10) respectivos se pueden obtener, por ejemplo, mediante la reacción de aminas (9) con fosgeno, trifosgeno o sus derivados o mediante la conversión de los ácidos carboxílicos (8) correspondientes en azidas de acilo con, por ejemplo, difenilfosforilazida, seguido por transposición de Curtius. Estas reacciones se identifican solamente para ilustración, y se debe apuntar que se conocen en la técnica otros reactivos adecuados que también se pueden usar para convertir aminas o ácidos carboxílicos en isocianatos.
Ejemplos de tales métodos se ilustran en el siguiente esquema.
Esquema 7

Los ácidos carboxílicos (8) deseados se pueden obtener, por ejemplo, mediante la reacción de los ácidos fenil- o piridil-borónicos sustituidos en meta (R es H) o ésteres borónicos (R es alquilo) (12) correspondientes con éster de -íerc-butil-1 H-pirazol-5-carboxilato seguido por la hidrólisis del éster en ácido carboxílico. Los ésteres borónicos, B(OR)2 , incluyen ésteres cíclicos, tales como 4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-ilo.
Un ejemplo de tal método se ilustra en el siguiente esquema.
Esquema 8

Las aminas (9) deseadas se pueden obtener, por ejemplo, mediante la reacción de las fenil- o piridil-hidracinas (14) sustituidas en meta correspondientes con 4,4-dimetil-3-oxopentanonitrilo.
Un ejemplo de tal método se ilustra en el siguiente esquema.
Esquema 9

14 9
En un enfoque alternativo, el producto intermedio (7) se hace reaccionar con carbamatos activados de 3-terc-butil-5-amino-1 -aril-1 H-pirazols para proporcionar las ureas correspondientes.
Un ejemplo de tal método se ilustra en el siguiente esquema.
Esquema 10

Los carbamatos activados respectivos se pueden obtener, por ejemplo, mediante la reacción de aminas (9) con cloroformiatos, por ejemplo, con cloroformiato de fenilo para formar (3-(terc-butil)-1-aril-1H-pirazol-5-il)carbamato de fenilo (15) o con cloroformiato de 1 -metilvinilo para formar (3-(terc-butil)-1-aril-1H-pirazol-5-il)carbamato de 1 metilvinilo.
Alternativamente, la posición amino del producto intermedio (7) común se puede activar mediante reacción, por ejemplo, con cloroformiato de fenilo o cloroformiato de 1 -metilvinilo.
Un ejemplo de tal método se ilustra en el siguiente esquema.
Esquema 11

El carbamato activado así formado se puede hacer reaccionar a continuación con una amina aromática para proporcionar la urea correspondiente.
Un ejemplo de tal método se ilustra en el siguiente esquema.
Esquema 12

Los carbamatos activados mostrados en los Esquemas 10-12 anteriores son meramente ejemplos. También se pueden usar otros carbamatos activados conocidos en la técnica, incluyendo, por ejemplo, carbamatos de 4-nitrofenilo y carbamatos de N-hidroxisuccinimida.
En un enfoque alternativo, la urea se forma en primer lugar, antes de la ciclación.
Un ejemplo de tal método se ilustra en el siguiente esquema.
Esquema 13

En un enfoque alternativo, los aminofenoles se pueden convertir en ureas para formar el producto intermedios (20). Un ejemplo de tal método se ilustra en el siguiente esquema.
Esquema 14

The producto intermedios (20) se puede acoplar a continuación con (1) para proporcionar (17). Una conversión adicional, por ejemplo, según se describe anteriormente en el Esquema 13, conduce al producto (11).
Un ejemplo de tal método se ilustra en el siguiente esquema.
Esquema 15

Composiciones
Un aspecto de la presente invención trata de una composición (p. ej., una composición farmacéutica) que comprende un compuesto de TBAP, según se describe en la presente, y un portador, diluyente o excipiente farmacéuticamente aceptable.
También se describe en la presente un método para preparar una composición (p. ej., una composición farmacéutica) que comprende mezclar un compuesto de TBAP, según se describe en la presente, y un portador, diluyente o excipiente farmacéuticamente aceptable.
Usos
Los compuestos de TBAP descritos en la presente son útiles en el tratamiento de, por ejemplo, trastornos proliferativos (como “agentes antiproliferativos”), cáncer (como “agentes anticancerosos”), enfermedades inflamatorias (como “agentes antiinflamatorios”), infecciones virales (como “agentes antivirales”), enfermedades neurodegenerativas (como “agentes antineurodegenerativos”), enfermedades fibróticas (como “agentes antifibróticos”), etc.
Uso en Métodos para Inhibir RAF (p. ej., BRAF, CRAF, etc.)
También se describe en la presente un método para inhibir la función de RAF (p. ej., BRAF, CRAF, etc.) (p. ej., en una célula), in vitro o in vivo, que comprende poner en contacto la célula con una cantidad eficaz de un compuesto de TBAP, según se describe en la presente.
Un experto normal en la técnica es capaz de determinar fácilmente si un compuesto candidato inhibe o no RAF (p. ej., BRAF, CRAF, etc.). Por ejemplo, ensayos adecuados se describen en la presente o se conocen en la técnica. En una realización, el método se realiza in vitro.
En una realización, el método se realiza in vivo.
En una realización, el compuesto de TBAP se proporciona en la forma de una composición farmacéuticamente aceptable.
Se puede tratar cualquier tipo de célula, incluyendo adiposa, pulmonar, gastrointestinal (incluyendo, p. ej., intestino, colon), de mama (mamarias), ováricas, prostáticas, de hígado (hepáticas), de riñón (renales), de vejiga urinaria, pancráticas, cerebrales y cutáneas.
Por ejemplo, una muestra de células se puede hacer crecer in vitro y un compuesto se puede poner en contacto con dichas células, y se puede observar el efecto del compuesto sobre esas células. Como un ejemplo de “efecto”, se puede determinar el estado morfológico de las células (p. ej., vivas o muertas, etc.). Cuando se encuentra que el compuesto ejerce una influencia sobre las células, esto se puede usar como un marcador de pronóstico o diagnóstico de la eficacia del compuesto en métodos para tratar a un paciente que tiene células del mismo tipo celular.
Uso en Métodos para Inhibir la Proliferación Celular, etc.
Los compuestos de TBAP descritos en la presente, p. ej., (a) regulan (p. ej., inhiben) la proliferación celular; (b) inhiben la progresión del ciclo celular; (c) promueven la apoptosis; o (d) una combinación de uno o más de estos. También se describe en la presente un método para regular (p. ej., inhibir) la proliferación celular (p. ej., la proliferación de una célula), inhibir la progresión del ciclo celular, promover la apoptosis o una combinación de uno o más de estos, in vitro o in vivo, que comprende poner en contacto una célula con una cantidad eficaz de un compuesto de TBAP, según se describe en la presente.
En una realización, el método es un métodos para regular (p. ej., inhibir) la proliferación celular (p. ej., la proliferación de una célula), in vitro o in vivo, que comprende poner en contacto una célula con una cantidad eficaz de un compuesto de TBAP, según se describe en la presente.
En una realización, el método se realiza in vitro.
En una realización, el método se realiza in vivo.
En una realización, el compuesto de TBAP se proporciona en la forma de una composición farmacéuticamente aceptable.
Se puede tratar cualquier tipo de célula, incluyendo pulmonar, gastrointestinal (incluyendo, p. ej., intestino, colon), de mama (mamaria), ovárica, prostática, de hígado (hepática), de riñón (renal), de vejiga urinaria, pancreática, cerebral y cutánea.
Un experto normal en la técnica es capaz de determinar fácilmente si un compuesto candidato regula (p. ej., inhibe) o no la proliferación celular, etc. Por ejemplo, se describen en la presente ensayos que se pueden usar convenientemente para evaluar la actividad ofrecida por un compuesto particular.
Por ejemplo, una muestra de células (p. ej., procedente de un tumor) se puede hacer crecer in vitro y un compuesto se puede poner en contacto con dichas células, y se puede observar el efecto del compuesto sobre esas células. Como un ejemplo de “efecto”, se puede determinar el estado morfológico de las células (p. ej., vivas o muertas, etc.). Cuando se encuentra que el compuesto ejerce una influencia sobre las células, esto se puede usar como un marcador de pronóstico o diagnóstico de la eficacia del compuesto en métodos para tratar a un paciente que tiene células del mismo tipo celular.
Uso en Métodos de Terapia
Otro aspecto de la presente invención trata de un compuesto de TBAP, según se describe en la presente, para el uso en un método de tratamiento del cuerpo de un ser humano o un animal mediante terapia, por ejemplo, para el uso en un método de tratamiento de un trastorno (p. ej., una enfermedad) según se describe en la presente.
Uso en la Fabricación de Medicamentos
También se describe en la presente el uso de un compuesto de TBAP, según se describe en la presente, en la fabricación de un medicamento, por ejemplo, para el uso en un método de tratamiento, por ejemplo, para el uso en un método de tratamiento de un trastorno (p. ej., una enfermedad) según se describe en la presente.
En una realización, el medicamento comprende el compuesto de TBAP.
Métodos de Tratamiento
También se describe en la presente un método de tratamiento, por ejemplo, un método de tratamiento de un trastorno (p. ej., una enfermedad) según se describe en la presente, que comprende administrar a un sujeto que necesite tratamiento una cantidad terapéuticamente eficaz de un compuesto de TBAP, según se describe en la presente, preferiblemente en la forma de una composición farmacéutica.
Trastornos Tratados - Trastornos Proliferativos
En una realización (p. ej., para el uso en métodos de terapia, de uso en la fabricación de medicamentos, de métodos de tratamiento), el tratamiento es el tratamiento de un trastorno proliferativo.
El término “trastorno proliferativo”, según se usa en la presente, trata de una proliferación celular no querida o descontrolada de células excesivas o anormales que no se desea, tal como crecimiento neoplástico o hiperplástico. En una realización, el tratamiento es el tratamiento de: un trastorno proliferativo caracterizado por proliferación celular benigna, premaligna o maligna.
En una realización, el tratamiento es el tratamiento de: hiperplasia; un neoplasma; un tumor (p. ej., un histocitoma, un glioma, un astrocitoma, un osteoma); cáncer; psoriasis; una enfermedad ósea; un trastorno fibroproliferativo (p. ej., de tejidos conectivos); fibrosis pulmonar; aterosclerosis; o proliferación celular del músculo liso en los vasos sanguíneos (p. ej., estenosis o reestenosis después de angioplastia).
Trastornos Tratados - Cáncer
En una realización (p. ej., de uso en métodos de terapia, de uso en la fabricación de medicamentos, de métodos de tratamiento), el tratamiento es el tratamiento de cáncer.
En una realización, el tratamiento es el tratamiento de metástasis cancerosa.
Entre los cánceres se incluyen:
(1) Carcinomas, incluyendo tumores derivados de epitelios escamosos estratificados (carcinomas de células escamosas) y tumores que surgen dentro de órganos o glándulas (adenocarcinomas). Ejemplos incluyen mama, colorrectal, pulmón, páncreas, próstata, ovario.
(2) Sarcomas, incluyendo: osteosarcoma y sarcoma osteogénico (hueso); condrosarcoma (cartílago); leiomiosarcoma (músculo liso); rabdomiosarcoma (músculo esquelético); sarcoma mesotelial y mesotelioma (revestimiento membranoso de cavidades corporales); fibrosarcoma (tejido fibroso); angiosarcoma y hemangioendotelioma (vasos sanguíneos); liposarcoma (tejido adiposo); glioma y astrocitoma (tejido conectivo neurogénico encontrado en el cerebro); mixosarcoma (tejido conectivo embrionario primitivo); tumor mesenquimal y mesodérmico mixto (tipos de tejido conectivo mixtos).
(3) Mieloma.
(4) Melanomas incluyendo, p. ej., melanoma de propagación superficial, melanoma nodular, melanoma lentiginoso maligno, melanoma acral y melanoma uveal.
(5) Tumores hemtopoyéticos, incluyendo: leucemia mielógena y granulocítica (enfermedad maligna de la serie mieloide y de glóbulos blancos granulocíticos); leucemia linfática, linfocítica y linfoblástica (enfermedad maligna de la serie linfoide y glóbulos rojos linfocíticos); policitemia verdadera (también conocida como eritremia) (enfermedad maligna de diversos productos celulares sanguíneos, pero predominando los glóbulos rojos).
(6) Linfomas, incluyendo: linfomas hodgkiniano y no hodgkiniano.
(7) Tipos Mixtos, incluyendo, p. ej., carcinoma adenoescamoso; tumor mesodérmico mixto; carcinosarcoma; teratocarcinoma.
En una realización, el cáncer se caracteriza por o se caracteriza además por células madre cancerosas.
En una realización, el tratamiento es el tratamiento de cáncer que es resistente al tratamiento con un anticuerpo, p. ej., un anticuerpo conocido, p. ej., un anticuerpo aprobado legislativamente. En una realización, el tratamiento es el tratamiento de melanoma que es resistente al tratamiento con un anticuerpo, p. ej., un anticuerpo conocidos, p. ej., un anticuerpo aprobado legislativamente. Ejemplos de tales anticuerpos que son conocidos para tratar el melanoma incluyen: anticuerpos que se unen a CTLA-4 (antígeno asociado a linfocitos T citotóxicos 4, por sus siglas en inglés), tales como ipilimumab (aprobado); anticuerpos que se unen a PD-1 (receptor de muerte celular programada 1), tales como pembrolizumab (aprobado) y nivolumab (aprobado); anticuerpos que se unen a PD-L1 (ligando de muerte celular programada 1, por sus siglas en inglés), tales como MEDI4736 (en estudios clínicos) y MPDL3280A (en estudios clínicos); anticuerpos y conjugados de anticuerpo que se unen a glicoproteína de antígeno de melanoma NMB, tales como glembatumumab vedotina (en estudios clínicos); anticuerpos que se unen a marcador endotelial antitumoral 1, tales como ontuxizumab (en estudios clínicos); anticuerpos que se unen a VEGF, tales como bevacizumab, solos o en combinación con quimioterapia estándar o IFN-a2b en bajas dosis (en estudios clínicos); anticuerpos que se unen a gangliósido GD3, tales como KW-2871 (en estudios clínicos); anticuerpos que se unen a isoformas de integrina, tales como avp1, avp3, avp5 y avp6, tales como intetumumab (en estudios clínicos).
El efecto anticancerosos puede surgir a través de uno o más mecanismos, incluyendo, pero no limitados a, la regulación de la proliferación celular, la inhibición de la progresión del ciclo celular, la inhibición de la angiogénesis (la formación de nuevos vasos sanguíneos), la inhibición de metástasis (la propagación de un tumor desde su origen), la inhibición de la migración celular (la propagación de células cancerosas a otras partes del cuerpo), la inhibición de la invasión (la propagación de células tumorales en estructuras normales próximas), la promoción de la apoptosis (muerte celular programada), la muerte por necrosis o la inducción de muerte por autofagia. Los compuestos descritos en la presente se pueden usar en el tratamiento de los cánceres descritos en la presente, independientemente de los mecanismos analizados en la presente.
Trastornos Tratados - Inflamación
En una realización (p. ej., para el uso en métodos de terapia, de uso en la fabricación de medicamentos, de métodos de tratamiento), el tratamiento es el tratamiento de la inflamación (p. ej., un trastorno o reacción inflamatorio).
En una realización, el tratamiento es el tratamiento de la inflamación aguda (p. ej., mediada por infección aguda). En una realización, el tratamiento es el tratamiento de la inflamación crónica (p. ej., mediada por infección crónica). En una realización, la enfermedad inflamatoria se selecciona de enfermedades inflamatorias del pulmón (p. ej., asma; enfermedad pulmonar obstructiva crónica (COPD, por sus siglas en inglés)); el ojo (p. ej., uveítis); y el tracto gastrointestinal (p. ej., enfermedad de Crohn; colitis ulcerativa).
En una realización, la enfermedad inflamatoria se selecciona de:
(i) enfermedades o trastornos pulmonares que tienen un componente inflamatorio, tales como fibrosis quística, hipertensión pulmonar, sarcoidosis pulmonar, fibrosis pulmonar idiopática y, particularmente, COPD (incluyendo bronquitis crónica y enfisema), asma y asma pediátrica;
(ii) enfermedades o trastornos cutáneos que tienen un componente inflamatorio, tales como dermatitis atópica, dermatitis alérgica, dermatitis de contacto y psoriasis;
(iii) enfermedades o trastornos nasales que tienen un componente inflamatorio, tales como rinitis alérgica, rinitis y sinusitis;
(iv) enfermedades o trastornos oculares que tienen un componente inflamatorio, tales como conjuntivitis, conjuntivitis alérgica, queratoconjuntivitis seca (ojo seco), glaucoma, retinopatía diabética, edema macular (incluyendo edema macular diabético), oclusión de la vena retinal central (CRVO, por sus siglas en inglés), degeneración macular seca y/o húmeda asociada a la edad (AM O), inflamación después de una operación de cataratas y, particularmente, uveítis (incluyendo uveítis posterior, anterior y panuveítis), rechazo de injertos corneal y trasplante de células límbicas; y
(v) enfermedades o trastornos gastrointestinales que tienen un componente inflamatorio, tales como enteropatía sensible al gluten (enfermedad celíaca), esofagitis eosinofílica, enfermedad intestinal del injertos contra el hospedador y, particularmente, enfermedad de Crohn y colitis ulcerativa.
En una realización, la enfermedad inflamatoria se selecciona de: fibrosis quística; hipertensión pulmonar; sarcoidosis pulmonar; fibrosis pulmonar idiopática; enfermedad pulmonar obstructiva crónica (COPD) (incluyendo bronquitis crónica y enfisema); asma; asma pediátrica; dermatitis atópica; dermatitis alérgica; dermatitis de contacto; psoriasis; rinitis alérgica; rinitis; sinusitis; conjuntivitis; conjuntivitis alérgica; queratoconjuntivitis seca (ojo seco); glaucoma; retinopatía diabética; edema macular (incluyendo edema macular diabético); oclusión de la vena retinal central (CRVO); degeneración macular seca y/o húmeda asociada a la edad (AMD); inflamación después de una operación de cataratas; uveítis (incluyendo uveítis posterior, anterior y panuveítis); rechazo de injerto corneal y trasplante de células límbicas; enteropatía sensible al gluten (enfermedad celíaca); esofagitis eosinofílica; enfermedad intestinal del injerto contra el hospedador; enfermedad de Crohn; y colitis ulcerativa.
En una realización, la enfermedad inflamatoria es asma o COPD.
En una realización, la enfermedad inflamatoria es uveítis, enfermedad de Crohn o colitis ulcerativa.
Trastornos Tratados - Trastornos Inmunológicos
En una realización (p. ej., para el uso en métodos de terapia, de uso en la fabricación de medicamentos, de métodos de tratamiento), el tratamiento es el tratamiento de un trastorno inmunológico.
En una realización, el tratamiento es el tratamiento de una alergia.
En una realización, el tratamiento es el tratamiento de una enfermedad inflamatoria de las vías respiratorias, tal como asma.
En una realización, el tratamiento es el tratamiento de dermatitis de contacto alérgica.
En una realización, el tratamiento es el tratamiento de una enfermedad del sistema inmunitario.
En una realización, el tratamiento es el tratamiento de una enfermedad autoinmunitaria, p. ej., artritis reumatoide; lupus eritematoso sistémico (lupus); enteropatía inflamatoria (IBD, por sus siglas en inglés); esclerosis múltiple (MS, por sus siglas en inglés); diabetes mellitus tipo 1; síndrome de Guillain-Barre; psoriasis; enfermedad de Graves, tiroiditis de Hashimoto; miastenia grave; vasculitis; una enfermedad de inmunodeficiencia; inmunodeficiencia combinada grave (SCID, por sus siglas en inglés); inmunodeficiencia variable común (CVID, por sus siglas en inglés); virus de inmunodeficiencia humana (HIV); síndrome de inmunodeficiencia adquirida (sida); inmunodeficiencia inducida por fármacos; o síndrome del injerto contra el hospedador.
Trastornos Tratados - Infección Viral
En una realización (p. ej., para el uso en métodos de terapia, de uso en la fabricación de medicamentos, de métodos de tratamiento), el tratamiento es el tratamiento de una infección viral.
En una realización, el tratamiento es el tratamiento de una infección viral por:
(Grupo I:) un virus de dsDNA, p. ej., un adenovirus, un herpesvirus, un poxvirus;
(Grupo II:) un virus de ssDNA, p. ej., un parvovirus;
(Grupo III:) un virus de dsRNA, p. ej., un reovirus;
(Grupo IV:) un virus de (+)ssRNA, p. ej., un picornavirus, un togavirus;
(Grupo V:) un virus de (-)ssRNA, p. ej., un ortomixovirus, un rabdovirus;
(Grupo VI:) un virus de ssRNA-RT, p. ej., un retrovirus; o
(Grupo VII:) un virus de dsDNA-RT, p. ej., a hepadnavirus.
Según se usa anteriormente: ds: doble hebra; ss: hebra ; (+)ssRNA: RNA de hebra ; (-)ssRNA: RNA de hebra -; ssRNA-RT: RNA (hebra ) con producto intermedio de DNA en el ciclo vital.
En una realización, el tratamiento es el tratamiento de: virus de inmunodeficiencia humana (HIV); virus de hepatitis B (HBV, por sus siglas en inglés); virus de hepatitis C (HCV, por sus siglas en inglés); virus del papiloma humano (HPV, por sus siglas en inglés); citomegalovirus (CMV, por sus siglas en inglés); o virus de Epstein-Barr (EBV); herpesvirus humano 8 (HHV) asociado con sarcoma de Kaposi; virus de Coxsackie B3; virus de Borna; virus de la gripe.
Trastornos Tratados - Trastornos Fibróticos
En una realización (p. ej., para el uso en métodos de terapia, de uso en la fabricación de medicamentos, de métodos de tratamiento), el tratamiento es el tratamiento de un trastorno fibrótico (p. ej., un trastorno caracterizado por fibrosis excesiva, p. ej., un exceso de tejido conectivo fibroso en un tejido u órgano, p. ej., desencadenado por un proceso reparador o reactivo, p. ej., en respuesta a una lesión (p. ej., cicatrización, curación) o tejido fibrótico en exceso que surge de una sola línea celular (p. ej., fibroma)).
En una realización, el tratamiento es el tratamiento de:
(para los pulmones:) fibrosis pulmonar; fibrosis pulmonar secundaria a fibrosis quística; fibrosis pulmonar idiopática; fibrosis masiva progresiva del minero del carbón;
(para el hígado:) cirrosis;
(para el corazón:) fibrosis endomiocárdica; antiguo infarto de miocardio; fibrosis auricular;
(para el mediastino:) fibrosis mediastinal;
(para el hueso:) mielofibrosis;
(para el retroperitoneo:) fibrosis retroperitoneal;
(para la piel:) fibrosis sistémica nefrogénica; cicatrización queloidea; esclerosis sistémica; escleroderma;
(para el intestino:) enfermedad de Crohn;
(para el tejido conectivo:) artrofibrosis; o capsulitis.
Trastornos Tratados - Trastornos Mejorados por la Inhibición de BRAF Mutante
En una realización (p. ej., para el uso en métodos de terapia, de uso en la fabricación de medicamentos, de métodos de tratamiento), el tratamiento es el tratamiento de un trastorno (p. ej., una enfermedad) (p. ej., un trastorno proliferativo, p. ej., cáncer) que está asociado con una forma mutada de RAF (p. ej., BRAF), tal como, por ejemplo, las mutaciones descritas en Davies y cols., 2002; Wan y cols., 2004; y Stratton y cols., 2003.
En una realización (p. ej., para el uso en métodos de terapia, de uso en la fabricación de medicamentos, de métodos de tratamiento), el tratamiento es el tratamiento de un trastorno (p. ej., una enfermedad) (p. ej., un trastorno proliferativo, p. ej., cáncer) que se mejora mediante la inhibición de RAF (p. ej., BRAF).
En una realización, el tratamiento es el tratamiento de: un trastorno (p. ej., una enfermedad) que se caracteriza por células que sobreexpresan RAF mutante (p. ej., BRAF) (p. ej., en comparación con células normales correspondientes; p. ej., en el que la sobreexpresión en un factor de 1.5, 2, 3, 5, 10, 20 o 50).
Trastornos proliferativos:
En una realización, el tratamiento es el tratamiento de: melanoma maligno; carcinoma colorrectal; carcinoma colorrectal metastásico; cáncer tiroideo folicular; cáncer tiroideo insular; cáncer tiroideo papilar; carcinoma ovárico; carcinoma ovárico de baja malignidad; cáncer pulmonar no microcítico; tricoleucemia; colangiocarcinoma; glioma pediátrico de baja malignidad (p. ej., astrocitoma pilocítico; ganglioglioma; xantoastrocitoma pleomórfico); mieloma múltiple; o carcinoma medular del páncreas. En una realización, el tratamiento es el tratamiento de: adenocarcinoma ductal pancreático.
En una realización, el tratamiento es el tratamiento de un trastorno (p. ej., una enfermedad) que está asociado con una forma mutada de RAF (p. ej., BRAF, CRAF, etc.) pero que es resistente al tratamiento con un inhibidor de RAF conocido (p. ej., aprobado) (p. ej., BRAF, CRAF, etc.). Ejemplos inhibidores de BRAF conocidos (p. ej., aprobados) incluyen vemurafenib (PLX4032, RG7204, Zelboraf) (aprobado) y dabrafenib (GSK-2118436) (aprobado).
En una realización, el tratamiento es el tratamiento de un trastorno (p. ej., una enfermedad) que está asociado con una forma mutada de RAF (p. ej., BRAF, CRAF, etc.) pero que es resistente al tratamiento con una combinación de un inhibidor de RAF conocidos (p. ej., aprobado) (p. ej., BRAF, CRAF, etc.) y un inhibidor de MEK conocido (p. ej., aprobado). Ejemplos de inhibidores de MEK incluyen: trametinib (GSK 1120212) (aprobado); selumetinib (AZD6244) (en estudios clínicos); PD 325901 (en estudios clínicos); cobimetinib (GDC 0973, XL 518) (en estudios clínicos); y CI 1040 (PD184352) (en estudios clínicos).
En una realización, el tratamiento es el tratamiento de: melanoma por mutante de BRAF intrínsecamente resistente a vemurafenib; melanoma por mutante de BRAF que adquiere resistencia al tratamiento con vemurafenib; melanoma por mutante de BRAF intrínsecamente resistente a dabrafenib; melanoma por mutante de BRAF que adquiere resistencia al tratamiento con dabrafenib; o melanoma por mutante de BRAF que adquiere resistencia a una combinación de un inhibidor de BRAF y un inhibidor de MEK (p. ej., dabrafenib y trametinib).
Otros Trastornos:
En una realización, el tratamiento es el tratamiento de: histiocitosis de células de Langerhans (LCH, por sus siglas en inglés) o enfermedad de Erdheim-Chester.
Trastornos Tratados - Trastornos Mejorados por la Inhibición tanto de BRAF como de CRAF
Los cánceres con, por ejemplo, mutaciones activadoras de RAS, RAF y EGFR o la sobreexpresión de RAS, RAF y EGFR, incluyendo cualquiera de las isoformas de los mismos, pueden ser particularmente sensibles a la inhibición de panRAF (p. ej., CRAF y BRAF). Los cánceres con otras anormalidades que conducen a una señal de la ruta RAF-MEK-ERK regulada al alza también pueden ser particularmente sensibles al tratamiento con inhibidores de la actividad de panRAF (p. ej., CRAF y BRAF). Ejemplos de tales anormalidades incluyen la activación constitutiva de un receptor de factor de crecimiento; la sobreexpresión de uno o más receptores de factores de crecimiento; la sobreexpresión de uno o más factores de crecimiento; activación de la ruta mediada por KSR; y las fusiones génicas de BRAF o CRAF.
En una realización (p. ej., para el uso en métodos de terapia, de uso en la fabricación de medicamentos, de métodos de tratamiento), el tratamiento es el tratamiento de un trastorno (p. ej., una enfermedad) (p. ej., un trastorno proliferativo, p. ej., cáncer) que se mejora mediante la inhibición tanto de BRAF como de CRAF.
En una realización, el tratamiento es el tratamiento de: un trastorno (p. ej., una enfermedad) (p. ej., un trastorno proliferativo, p. ej., cáncer) que se caracteriza por la activación constitutivo de un receptor de factor de crecimiento; la sobreexpresión de uno o más receptores de factores de crecimiento (p. ej., en comparación con células normales correspondientes; p. ej., en el que la sobreexpresión es en un factor de 1.5, 2, 3, 5, 10, 20 o 50); la sobreexpresión de uno o más factores de crecimiento (p. ej., en comparación con células normales correspondientes; p. ej., en el que la sobreexpresión es en un factor de 1.5, 2, 3, 5, 10, 20 o 50); y/o fusiones génicas activadoras de BRAF y/o CRAF.
En una realización, el tratamiento es el tratamiento de: un trastorno (p. ej., una enfermedad) (p. ej., un trastorno proliferativo, p. ej., cáncer) que se caracteriza por uno o más o la totalidad de:
(a) mutantes activadores de RAS y/o RAF;
(b) regulación al alza de RAS y/o RAF;
(c) regulación al alza de señales de la ruta RAF-MEK-ERK; y
(d) regulación al alza de receptores de factores de crecimiento (p. ej., ERBB2 y EGFR).
En una realización, el tratamiento es el tratamiento de: una enfermedad inflamatoria; una infección; un trastorno autoinmunitario; apoplejía; isquemia; un trastorno cardíaco; un trastorno neurológico; un trastorno fibrogenético, un trastorno proliferativo; un trastorno hiperproliferativo; un trastorno hiperproliferativo no canceroso; un tumor; leucemia; un neoplasma; cáncer; carcinoma; una enfermedad metabólica; una enfermedad maligna; reestenosis vascular; psoriasis; aterosclerosis; artritis reumatoide; osteoartritis; insuficiencia cardíaca; dolor crónico; dolor neuropático; ojo seco; glaucoma de ángulo cerrado; o glaucoma de ángulo amplio.
Trastornos Tratados - Trastornos Asociados con Mutaciones de RAS y/o Activación de la Ruta de MAPK
En una realización (p. ej., para el uso en métodos de terapia, de uso en la fabricación de medicamentos, de métodos de tratamiento), el tratamiento es el tratamiento de un trastorno (p. ej., una enfermedad) que está asociado con una
mutación de RAS (p. ej., KRAS, NRAS, HRAS) y/o activación de la ruta de MAPK (p. ej., hiperactividad de la ruta de MAPK).
Trastornos proliferativos:
En una realización, el tratamiento es el tratamiento de un trastorno proliferativo (p. ej., cáncer) que está asociado con una mutación de RAS (p. ej., KRAS, NRAS, HRAS) y/o la activación de la ruta de MAPK (p. ej., hiperactividad de la ruta de MAPK).
En una realización, el tratamiento es el tratamiento de: carcinoma no microcítico; cáncer colorrectal; cáncer colorrectal metastásico; carcinoma hepatocelular; adenocarcinoma pancreático; melanoma maligno; una enfermedad maligna hematológica (p. ej., leucemia mielomonocítica juvenil (JMML); leucemia mielomonocítica crónica (CMML); síndrome mielodisplástico (MDS, por sus siglas en inglés); leucemia linfoblástica aguda (ALL, por sus siglas en inglés); mieloma múltiple (MM); linfoma de Burkitt; linfoma de Hodgkin); cáncer ovárico epitelial tipo I; cáncer peritoneal primario; adenocarcinoma del tracto biliar; cáncer tiroideo folicular; cáncer tiroideo papilar indiferenciado; sarcoma de tejidos blandos (p. ej., angiosarcoma; leiomiosarcoma; rabdomiosarcoma; mixoma; histiocitoma fibroso maligno); neurofibromatosis tipo 1 (NF1); neurofibromas plexiformes (PN, por sus siglas en inglés) inoperables; melanoma uveal; melanoma de los cuerpos ciliares; melanoma coroidal; melanoma del iris; melanoma intraocular metastásico; carcinoma adrenocortical; cáncer renal; seminoma; cáncer de la vejiga urinaria; cáncer endometrial; cáncer del cuello uterino; neuroblastoma; adenocarcinoma de estómago; carcinoma de células escamosos de cabeza y cuello; o cáncer de próstata.
Otros Trastornos:
En una realización, el tratamiento es el tratamiento de: rechazo de trasplantes (p. ej., xenoinjerto; piel; un miembro; un órgano; médula ósea); osteoartritis; artritis reumatoide; fibrosis quística; una complicación de la diabetes (p. ej., retinopatía diabética; nefropatía diabética); hepatomegalia; cardiomegalia; síndrome de Noonan; síndrome cardiofaciocutáneo; cardiomiopatía hipertrófica; apoplejía (p. ej., apoplejía isquémica focal aguda; isquemia cerebral global); insuficiencia cardíaca; choque séptico; asma; trastorno pulmonar obstructivo crónico; enfermedad de Alzheimer; dolor crónico (p. ej., dolor idiopático; dolor asociado con alcoholismo crónico, deficiencia de vitaminas, uremia o hipotiroidismo; dolor crónico asociado con inflamación; dolor posoperatorio crónico); o dolor neuropático (p. ej., asociado con inflamación; dolor posoperatorio; dolor del miembro fantasma; dolor por quemaduras; gota; neuralgia trigeminal; dolor herpético agudo; dolor posherpético; causalgia; neuropatía diabética; avulsión del plexo; neuroma; vasculitis; infección viral; lesión por aplastamiento; lesión por constricción; lesión tisular; amputación de miembros; lesión nerviosa entre el sistema nervioso periférico y el sistema nervioso central).
Trastornos Tratados - Trastornos Mejorados por la Inhibición de SRC
En una realización (p. ej., para el uso en métodos de terapia, de uso en la fabricación de medicamentos, de métodos de tratamiento), el tratamiento es el tratamiento de un trastorno (p. ej., una enfermedad) que es mejorado por la inhibición de SRC.
En una realización, el tratamiento es el tratamiento de: un trastorno (p. ej., una enfermedad) (p. ej., un trastorno proliferativo) que está asociado con una mutación de SRC; la sobreexpresión de SRC (p. ej., en comparación con células normales correspondientes; p. ej., en el que la sobreexpresión es en un factor de 1.5, 2, 3, 5, 10, 20 o 50); o la activación de la ruta aguas arriba de SRC (p. ej., por señalización elevada de RTK).
En una realización, el tratamiento es el tratamiento de: cáncer endometrial; carcinoma no microcítico; mesotelioma pleural maligno; melanoma maligno; leucemia mieloide crónica (p. ej., resistente a imatinib); metástasis óseas; cáncer de próstata resistente a hormonas; cáncer de próstata recurrente; osteosarcoma recurrente; leucemia linfoblástica aguda; cáncer colorrectal; cáncer colorrectal metastásico; cáncer de mama; cáncer ovárico; cáncer de cabeza y cuello recurrente o metastásico (p. ej., cáncer de cuello escamoso metastásico con carcinoma celular escamoso primario oculto; cáncer de cuello escamoso metastásico recurrente con primario oculto; carcinoma de células escamosas recurrente de la hipofaringe; carcinoma de células escamosas recurrente de la laringe; carcinoma de células escamosas recurrente de los labios y la cavidad oral; carcinoma de células escamosas recurrente de la nasofaringe; carcinoma de células escamosas recurrente de la orofaringe; carcinoma de células escamosas recurrente del seno paranasal y la cavidad nasal; carcinoma verrugoso recurrente de la laringe; carcinoma verrugoso recurrente de la cavidad oral; carcinoma de células escamosas de la hipofarínge; carcinoma de células escamosas de la laringe; carcinoma de células escamosas de los labios y la cavidad oral; carcinoma de células escamosas de la nasofaringe; carcinoma de células escamosas de la orofaringe; carcinoma de células escamosas del seno paranasal y la cavidad nasal; carcinoma verrugoso de la laringe; carcinoma verrugoso de la cavidad oral; cáncer de lengua); cáncer de piel recurrente; carcinoma de células escamosas de la piel; leucemia mielógena aguda; glioblastoma; o glioma pontino intrínseco difuso.
Trastornos Tratados - Trastornos Mejorados por la Inhibición de p38
En una realización (p. ej., para el uso en métodos de terapia, de uso en la fabricación de medicamentos, de métodos de tratamiento), el tratamiento es el tratamiento de un trastorno (p. ej., una enfermedad) que es mejorado por la inhibición de p38 (p. ej., p38a, p38y).
En una realización, el tratamiento es el tratamiento de: un trastorno (p. ej., una enfermedad) (p. ej., un trastorno proliferativo) que está asociado con una mutación en p38 (p. ej., p38a, p38y); la sobreexpresión de p38 (p. ej., p38a, p38Y) (p. ej., en comparación con células normales correspondientes; p. ej., en el que la sobreexpresión es en un factor de 1.5, 2, 3, 5, 10, 20 o 50); o la activación de la ruta aguas arriba de p38 (p. ej., p38a, p38y).
Trastornos proliferativos:
En una realización, el tratamiento es el tratamiento de: cáncer ovárico; cáncer de células escamosas de la cavidad oral; mieloma múltiple; neoplasmas de la médula ósea; o síndrome mielodisplástico.
Otros Trastornos:
En una realización, el tratamiento es el tratamiento de: un trastorno inflamatorio caracterizado por proliferación de células T (p. ej., activación y crecimiento de células T).
En una realización, el tratamiento es el tratamiento de: artritis reumatoide; osteoartritis; artritis psoriásica; síndrome de Reiter; artritis traumática; artritis rubeólica; sinovitis aguda; artritis gotosa; o espondilitis.
En una realización, el tratamiento es el tratamiento de: psoriasis; eccema; rinitis alérgica; conjuntivitis alérgica; asma; síndrome de dificulta respiratoria del adulto; lesión pulmonar aguda (ALI, por sus siglas en inglés); síndrome de dificultad respiratoria aguda (ARDS, por sus siglas en inglés); enfermedad pulmonar crónica; enfermedad pulmonar obstructiva crónica; caquexia sistémica; glomerulonefritis; insuficiencia cardíaca crónica; aterosclerosis; síndrome coronario agudo; isquemia cardíaca; o infarto de miocardio.
En una realización, el tratamiento es el tratamiento de: endotoxemia; síndrome de choque tóxico; enteropatía inflamatoria; aterosclerosis; síndrome del intestino irritable; enfermedad de Crohn; colitis ulcerativa; una enfermedad de resorción ósea; osteoporosis; diabetes; lesión por reperfusión; reacción del injerto contra el hospedador; rechazo de aloinjertos; septicemia; choque séptico; choque endotóxico; septicemia gramnegativa; glomerulonefritis; reestenosis; o trombosis.
En una realización, el tratamiento es el tratamiento de dolor.
En una realización, el tratamiento es el tratamiento de: dolor crónico; dolor neuromuscular; cefalea; dolor por cáncer; dolor inflamatorio agudo o crónico asociado con osteoartritis o artritis reumatoide; dolor inflamatorio posoperatorio; dolor neuropático; neuropatía diabética; neuralgia trigeminal; neuralgia poshepática; neuropatía inflamatoria; dolor migrañoso; radiculopatía lumbosacra; dolor dental; traumatismo nervioso; o isquemia neural.
Trastornos Tratados - Trastornos Mejorados por la Inhibición de FGFR1
En una realización (p. ej., para el uso en métodos de terapia, de uso en la fabricación de medicamentos, de métodos de tratamiento), el tratamiento es el tratamiento de un trastorno (p. ej., una enfermedad) que es mejorado por la inhibición de FGFR1.
En una realización, el tratamiento es el tratamiento de: un trastorno (p. ej., una enfermedad) (p. ej., un trastorno proliferativo) que está asociado con una mutación en FGFR1; la sobreexpresión de FGFR1 (p. ej., en comparación con células normales correspondientes; p. ej., en el que la sobreexpresión es en un factor de 1.5, 2, 3, 5, 10, 20 o 50); o la activación de la ruta aguas arriba de FGFR1.
En una realización, el tratamiento es el tratamiento de: cáncer de mama; cáncer pulmonar escamoso; cáncer de estómago; carcinoma urotelial; mieloma múltiple; síndrome mieloproliferativo 8p11; o carcinoma hepatocelular. Trastornos Tratados - Trastornos Mejorados por la Inhibición de VEGFR-2 (KDR)
En una realización (p. ej., para el uso en métodos de terapia, de uso en la fabricación de medicamentos, de métodos de tratamiento), el tratamiento es el tratamiento de un trastorno (p. ej., una enfermedad) que es mejorado por la inhibición de VEGFR-2 (KDR).
En una realización, el tratamiento es el tratamiento de: un trastorno (p. ej., una enfermedad) (p. ej., un trastorno proliferativo) que está asociado con una mutación en VEGFR-2; la sobreexpresión de VEGFR-2 (p. ej., en comparación con células normales correspondientes; p. ej., en el que la sobreexpresión es en un factor de 1.5, 2, 3, 5, 10, 20 o 50); o la activación de la ruta aguas arriba de VEGFR-2.
En una realización, el tratamiento es el tratamiento de: un trastorno (p. ej., una enfermedad) que se caracteriza por un incremento en la producción de VEGF (p. ej., bien por células cancerosas o bien por células estromáticas).
Trastornos proliferativos:
En una realización, el tratamiento es el tratamiento de: cáncer pancreático; cáncer pulmonar no microcítico (NSCLC, por sus siglas en inglés); neoplasmas ováricos; neoplasma peritoneales; neoplasmas de las trompas de Falopio; cáncer pulmonar y efusión pleural asociada; cáncer de células escamosas recurrente o metastásico de la cabeza y el cuello; carcinoma nasofaríngeo localmente avanzado; glioblastoma (p. ej., glioblastoma multiforme; glioblastoma de células gigantes); gliosarcoma; glioma pontino intrínseco difuso; sarcoma de Kaposi relacionado con HIV; mieloma múltiple; carcinoma de células renales; adenocarcinoma gástrico metastásico; leucemia mieloide aguda (AML); carcinoma hepatocelular; dermatofibrosarcoma; cáncer tiroideo medular (MTC, por sus siglas en inglés); cáncer tiroideo papilar; cáncer tiroideo folicular; síndrome mielodisplástico; neurofibromatosis tipo 1; neurofibroma plexiforme; neurofibroma de la médula espinal; cáncer de mama; neoplasmas del tracto biliar; cáncer de cuello uterino; cáncer de próstata; melanoma; carcinoma de vejiga urinaria; carcinoma de uretra; carcinoma de uréter; carcinoma renal; carcinoma pélvico; sarcoma; liposarcoma; cáncer de colon; osteosarcoma; carcinoma sinovial; neuroblastoma; o rabdomiosarcoma.
Otros Trastornos:
En una realización, el tratamiento es el tratamiento de: aterosclerosis; obesidad; síndrome de dolor neuropático; degeneración macular asociada a la edad; retinopatía diabética; edema macular diabético; o artritis reumatoide. Trastornos Tratados - Trastornos Mejorados por la Inhibición de LCK
En una realización (p. ej., para el uso en métodos de terapia, de uso en la fabricación de medicamentos, de métodos de tratamiento), el tratamiento es el tratamiento de un trastorno (p. ej., una enfermedad) que es mejorado por la inhibición de LCK.
En una realización, el tratamiento es el tratamiento de: un trastorno (p. ej., una enfermedad) que está asociado con una mutación en LCK; la sobreexpresión de LCK (p. ej., en comparación con células normales correspondientes; p. ej., en el que la sobreexpresión es en un factor de 1.5, 2, 3, 5, 10, 20 o 50); o la activación de la ruta aguas arriba de LCK.
En una realización, el tratamiento es el tratamiento de: una enfermedad afección patológica inmunológica que implica un componente inmunológico.
En una realización, el tratamiento es el tratamiento de: artritis reumatoide; enteropatía inflamatoria (p. ej., colitis ulcerativa; enfermedad de Crohn); psoriasis; artritis psoriásica; rechazo de trasplantes de tejidos u órganos (incluyendo, p. ej., prevención de); enfermedad del injerto contra el hospedador aguda o crónica; rechazo de aloinjertos; rechazo de xenoinjertos; asma alérgica; esclerosis múltiple; diabetes tipo 1; fibrosis pulmonar; o una reacción de hipersensibilidad de la piel.
Tratamiento
El término “tratamiento”, según se usa en la presente en el contexto de tratar un trastorno, se relaciona generalmente con el tratamiento de un ser humano o un animal (p. ej., en aplicaciones veterinarias), en el que se alcanza algún efecto terapéutico deseado, por ejemplo, la inhibición de progreso del trastorno, e incluye una reducción en la velocidad del progreso, una interrupción en la velocidad de progreso, un alivio de los síntomas del trastorno, una mejora del trastorno y una cura del trastorno. También se incluye el tratamiento como una medida profiláctica (es decir, profilaxis). Por ejemplo, el uso con pacientes que todavía no han desarrollado el trastorno, pero que tienen riesgo de desarrollar el trastorno, abarcado por el término “tratamiento”.
Por ejemplo, el tratamiento incluye la profilaxis de cáncer, reduciendo la incidencia del cáncer, aliviando los síntomas del cáncer, etc.
El término “cantidad terapéuticamente eficaz”, según se usa en la presente, trata de la cantidad de un compuesto o un material, una composición o una forma de dosificación que comprende un compuesto, que es eficaz para producir algún efecto terapéutico deseado, de acuerdo con una relación beneficio/riesgo razonable, cuando se administra según un régimen de tratamiento deseado.
Terapias Combinadas
El término “tratamiento” incluye tratamientos y terapias combinados, en los que se combinan dos o más tratamientos o terapias, por ejemplo, secuencialmente o simultáneamente. Por ejemplo, los compuestos descritos en la presente también se pueden usar en terapias combinadas, p. ej., junto con otros agentes. Ejemplos de tratamientos y terapias incluyen quimioterapia (la administración de agentes activos, incluyendo, p. ej., fármacos, anticuerpos (p. ej., como en la inmunoterapia), profármacos (p. ej., como en la terapia fotodinámica, GDEPT, ADEPT, etc.); cirugía; radioterapia; terapia fotodinámica; terapia génica; y dietas controladas.
También se describe en la presente un compuesto según se describe en la presente, en combinación con uno o más (p. ej., 1,2, 3, 4, etc.) agentes terapéuticos adicionales, según se describe posteriormente.
La combinación particular será a discreción del médico, que seleccionará las dosificaciones usando su conocimiento general común y regímenes de dosificación conocidos por un profesional experto.
Los agentes (es decir, el compuesto descrito en la presente más uno o más de otros agentes) se pueden administrar simultáneamente o secuencialmente, y se pueden administrar en esquemas de dosificación que varían individualmente y a través de diferentes vías. Por ejemplo, cuando se administran secuencialmente, los agentes se pueden administrar a intervalos poco separados (p. ej., a lo largo de un período de 5-10 minutos) o a intervalos mayores (p. ej., 1, 2, 3, 4 o más horas separados o incluso separados períodos aún mayores cuando se requiera), estando el régimen de dosificación preciso de acuerdo con las propiedades del agente o los agentes terapéuticos. Los agentes (es decir, el compuesto descrito en la presente más uno o más de otros agentes) se pueden formular conjuntamente en una sola forma de dosificación o alternativamente, los agentes individuales se pueden formular separadamente y se pueden presentar conjuntamente en la forma de un estuche, opcionalmente con instrucciones para su uso.
Ejemplos de agentes/terapias adicionales que se pueden coadministrar/combinar con el tratamiento con los compuestos de TBAP descritos en la presente memoria incluyen los siguientes: antimetabolitos; agentes alquilantes; venenos para el huso; inhibidores topoisomerasas; agentes de unión a DNA; inhibidores de cinasas; anticuerpos terapéuticos; inhibidores de PARP; inhibidores del metabolismo de NAD; inhibidores metabólicos; agentes elegidos como diana; agentes endocrinos; etc.
Otros Usos
Los compuestos de TBAP descritos en la presente también se pueden usar como aditivos de cultivos celulares para inhibir rAf (p. ej., BRAF, CRAF, etc.).
Los compuestos de TBAP descritos en la presente también se pueden usar como parte de un ensayo in vitro, por ejemplo, a fin de determinar si un hospedante candidato es propenso a beneficiarse del tratamiento con el compuesto en cuestión.
Los compuestos de TBAP descritos en la presente también se pueden usar como un patrón, por ejemplo, en un ensayo, a fin de identificar otros compuestos activos, distintos a inhibidores de RAF (p. ej., BRAF, CRAF, etc.), etc. Estuches
También se describe en la presente un estuche que comprende (a) un compuesto de TBAP según se describe en la presente o una composición que comprende un compuesto de TBAP según se describe en la presente, p. ej., proporcionado preferiblemente en un recipiente adecuado y/o con un envase adecuado; y (b) instrucciones de uso, p. ej., instrucciones escritas sobre cómo administrar el compuesto la composición.
Las instrucciones escritas también pueden incluir una lista de indicaciones para las que el ingrediente activo es un tratamiento adecuado.
Vías de Administración
El compuesto de TBAP o la composición farmacéutica que comprende el compuesto de TBAP se puede administrar a un sujeto mediante cualquier vía de administración conveniente, ya sea sistémicamente/periféricamente o tópicamente (es decir, en la zona de acción deseada).
Ejemplos de vías de administración incluyen oral (p. ej., mediante ingestión); yugal; sublingual; transdérmica (incluyendo, p. ej., mediante un parche, un emplasto, etc.); transmucosa (incluyendo, p. ej., mediante un parche, un emplasto, etc.); intranasal (p. ej., mediante pulverización nasal); ocular (p. ej., mediante gotas oculares); pulmonar (p. ej., mediante terapia de inhalación o insuflación usando, p. ej., un aerosol, p. ej., a través de la boca o la nariz); rectal (p. ej., mediante un supositorio o enema); vaginal (p. ej., mediante un pesario); parenteral, por ejemplo, mediante inyección, incluyendo subcutánea, intradérmica, intramuscular, intravenosa, intraarterial, intracardíaca, intratecal, intraespinal, intracapsular, subcapsular, intraorbital, intraperitoneal, intratraqueal, subcuticular, intraarticular, subaracnoidea e intraesternal; mediante el implante de un depósito o una reserva, por ejemplo, subcutáneamente o intramuscularmente.
El Sujeto/Paciente
El sujeto/paciente puede ser un cordado, un vertebrado, un mamífero, un mamífero placentario, un marsupial (p. ej., canguro, wómbat), un roedor (p. ej., una cobaya, un hámster, una rata, un ratón), un múrido (p. ej., un ratón), un lagomorfo (p. ej., un conejo), un ave (p. ej., un pájaro), un cánido (p. ej., un perro), un felino (p. ej., un gato), un equino (p. ej., un caballo), un porcino (p. ej., un cerdo), un óvido (p. ej., una oveja), un bóvido (p. ej., una vaca), un
primate, simio (p. ej., un simio inferior o un simio superior), un simio inferior (p. ej., tití, babuino), un simio superior (p. ej., gorila, chimpancé, orangután, gibón) o un ser humano.
Por otra parte, el sujeto/paciente puede estar en cualquiera de sus formas de desarrollo, por ejemplo, un feto.
En una realización preferida, el sujeto/paciente es un ser humano.
Formulaciones
Aunque es posible que un compuesto de TBAP se administre solo, es preferible presentarlo como una formulación farmacéutica (p. ej., composición, preparación, medicamento) que comprenda al menos un compuesto de TBAP, según se describe en la presente, junto con uno o más de otros ingredientes farmacéuticamente aceptable muy conocidos para los expertos en la técnica, incluyendo portadores, diluyentes, excipientes, adyuvantes, cargas, tampones, conservantes, antioxidantes, lubricantes, estabilizantes, solubilizantes, tensioactivos (p. ej., agentes humectantes), agentes enmascarantes, agentes colorantes, agentes aromatizantes y agentes edulcorantes farmacéuticamente aceptables. La formulación puede comprender además otros agentes activos, por ejemplo, otros agentes terapéuticos o profilácticos.
Así, la presente invención proporciona además composiciones farmacéuticas, según se definen anteriormente, y métodos para elaborar una composición farmacéutica que comprenden mezclar al menos un compuesto de TBAP, según se describe en la presente, junto con uno o más de otros ingredientes farmacéuticamente aceptables muy conocidos por los expertos en la técnica, p. ej., portadores, diluyentes, excipientes, etc. Si se formulan como unidades discretas (p. ej., comprimidos, etc.), cada unidad contiene una cantidad predeterminada (dosificación) del compuesto.
El término “farmacéuticamente aceptable”, según se usa en la presente, trata de compuestos, ingredientes, materiales, composiciones, formas de dosificación, etc., que son, dentro del alcance del juicio médico razonable, adecuados para el uso en contacto con los tejidos del sujeto en cuestión (p. ej., el ser humano) sin toxicidad, irritación, respuesta alérgica u otro problema o complicación excesivos, acorde con una relación beneficio/riesgo razonable. Cada portador, diluyente, excipiente, etc. también debe ser “aceptable” en el sentido de ser compatible con los otros ingredientes de la formulación.
Portadores, diluyentes, excipientes, etc. adecuados se pueden encontrar en textos farmacéuticos estándar, por ejemplo, Remington's Pharmaceutical Sciences, 18a edición, Mack Publishing Company, Easton, Pa., 1990; y Handbook of Pharmaceutical Excipients, 5a edición, 2005.
Las formulaciones se pueden preparar mediante cualesquiera métodos muy conocidos en la técnica de la farmacia. Tales métodos incluyen la etapa de asociar el compuesto con un portador que constituye uno o más ingredientes accesorios. En general, las formulaciones se preparan al asociar uniformemente e íntimamente el compuesto con los portadores (p. ej., portadores líquidos, un portador sólido finamente dividido, etc.), y a continuación conformar el producto, si es necesario.
La formulación se puede preparar para proporcionar una liberación rápida o lenta; liberación inmediata, retardada, temporizada o sostenida; o una combinación de las mismas.
Las formulaciones pueden estar adecuadamente en forma de líquidos, soluciones (p. ej., acuosas, no acuosas), suspensiones (p. ej., acuosas, no acuosas), emulsiones (p. ej., aceite en agua, agua en aceite), elixires, jarabes, electuarios, colutorios, gotas, comprimidos (incluyendo, p. ej., comprimidos revestidos), gránulos, polvos, caramelos para chupar, pastillas, cápsulas (incluyendo, p. ej., cápsulas de gelatina duras y blandas), obleas, píldoras, ampollas, bolos, supositorios, pesarios, tinturas, geles, pastas, pomadas, cremas, lociones, aceites, espumas, pulverizaciones, nebulizaciones o aerosoles.
Las formulaciones se pueden proporcionar adecuadamente como un parche, un emplasto adhesivo, un vendaje, un apósito o similar que está impregnado con uno o más compuestos y opcionalmente uno o más de otros ingredientes farmacéuticamente aceptables, incluyendo, por ejemplo, mejoradores de la penetración, la impregnación y la absorción. Las formulaciones también se pueden proporcionar adecuadamente en la forma de un depósito o una reserva.
El compuesto puede estar disuelto en, suspendido en o mezclado con uno o más de otros ingredientes farmacéuticamente aceptables. El compuesto se puede presentar en un liposoma u otro material en forma de micropartículas que está diseñado para dirigir el compuesto, por ejemplo, a componentes de la sangre o uno o más órganos.
Formulaciones adecuadas para la administración oral (p. ej., mediante ingestión) incluyen líquidos, soluciones (p. ej., acuosas, no acuosas), suspensiones (p. ej., acuosas, no acuosas), emulsiones (p. ej., aceite en agua, agua en aceite), elixires, jarabes, electuarios, comprimidos, gránulos, polvos, cápsulas, obleas, píldoras, ampollas, bolos.
Formulaciones adecuadas para la administración yugal incluyen colutorios, caramelos para chupar, pastillas, así como parches, emplastos adhesivos, depósitos y reservas. Los caramelos para chupar comprenden típicamente el compuesto en una base aromatizada, habitualmente sacarosa y goma arábiga o tragacanto. Las pastillas comprende típicamente el compuesto en una matriz inerte, tal como gelatina y glicerina o sacarosa y goma arábiga. Los colutorios comprenden típicamente el compuesto en un portador líquido adecuado.
Formulaciones adecuadas para la administración sublingual incluyen comprimidos, caramelos para chupar, pastillas, cápsulas y píldoras.
Formulaciones adecuadas para la administración transmucosa oral incluyen líquidos, soluciones (p. ej., acuosas, no acuosas), suspensiones (p. ej., acuosas, no acuosas), emulsiones (p. ej., aceite en agua, agua en aceite), colutorios, caramelos para chupar, pastillas, así como parches, emplastos adhesivos, depósitos y reservas.
Formulaciones adecuadas para la administración transmucosa no oral incluyen líquidos, soluciones (p. ej., acuosas, no acuosas), suspensiones (p. ej., acuosas, no acuosas), emulsiones (p. ej., aceite en agua, agua en aceite), supositorios, pesarios, geles, pastas, pomadas, cremas, lociones, aceites, así como parches, emplastos adhesivos, depósitos y reservas.
Formulaciones adecuadas para la administración transdérmica incluyen geles, pastas, pomadas, cremas, lociones y aceites, así como parches, emplastos adhesivos, vendajes, apósitos, depósitos y reservas.
Los comprimidos se pueden elaborar por medios convencionales, p. ej., compresión o moldeo, opcionalmente con uno o más ingredientes accesorios. Los comprimidos en forma comprimida se pueden preparar al comprimir en una máquina adecuada el compuesto en una forma que fluye libremente tal como un polvo o gránulos, opcionalmente mezclados con uno o más aglutinantes (p. ej., povidona, gelatina, goma arábiga, sorbitol, tragacanto, hidroxipropilmetilcelulosa); cargas o diluyentes (p. ej., lactosa, celulosa microcristalina, hidrogenofosfato cálcico); lubricantes (p. ej., estearato magnésico, talco, sílice); desintegrantes (p. ej., almidón glicolato sódico, povidona reticulada, carboximetilcelulosa sódica reticulada); agentes tensioactivos o dispersantes o humectantes (p. ej., laurilsulfato sódico); conservantes (p. ej., p-hidroxibenzoato de metilo, p-hidroxibenzoato de propilo, ácido sórbico); aromas, agentes mejoradores del aroma y edulcorantes. Los comprimidos moldeados se pueden elaborar al moldear en una máquina adecuada una mezcla del compuesto en polvo humedecido con un diluyente líquido inerte. Opcionalmente, los comprimidos pueden estar revestidos o ranurados y se pueden formular a fin de proporcionar una liberación lenta o controlada del compuesto de los mismos usando, por ejemplo, hidroxipropilmetilcelulosa en proporciones variables para proporcionar el perfil de liberación deseado. Los comprimidos se pueden proveer opcionalmente de un revestimiento, por ejemplo, para afectar a la liberación, por ejemplo un revestimiento entérico, para proporcionar liberación en partes del sistema digestivo diferentes al estómago.
Las pomadas se preparan típicamente a partir del compuesto y una base para pomadas parafínica o miscible con agua.
Las cremas se preparan típicamente a partir del compuesto y una base para cremas de aceite en agua. Si se desea, la fase acuosa de la base para cremas puede incluir, por ejemplo, al menos aproximadamente 30% p/p de un alcohol polihidroxilado, es decir, un alcohol que tiene dos o más grupos hidroxilo tal como propilenglicol, butano-1,3-diol, manitol, sorbitol, glicerol y polietilenglicol y mezclas de los mismos. Las formulaciones tópicas pueden incluir deseablemente un compuesto que mejore la absorción o la penetración del compuesto a través de la piel u otras áreas afectadas. Ejemplos de tales mejoradores de la penetración dérmica incluyen dimetilsulfóxido y análogos relacionados.
Las emulsiones se preparan típicamente a partir del compuesto y una fase oleosa, que opcionalmente puede comprender simplemente un emulsionante (conocido de otro modo como un emulgente) o puede comprender una mezcla de al menos un emulsionante con una grasa o un aceite o tanto con una grasa como con un aceite. Preferiblemente, se incluye un emulsionante hidrófilo junto con un emulsionante lipófilo que actúa como un estabilizante. También se prefiere incluir tanto un aceite como una grasa. Conjuntamente, el emulsionante o los emulsionantes con o sin estabilizante o estabilizantes constituyen la llamada cera emulsionante, y la cera junto con el aceite y/o la grasa constituyen la llamada base para pomadas emulsionante que forma la fase dispersada oleosa de las formulaciones en crema.
Emulgentes y estabilizantes de la emulsión adecuados incluyen Tween 60, Span 80, alcohol cetoestearílico, alcohol miristílico, monoestearato de glicerilo y laurilsulfato sódico. La elección de aceites o grasas adecuados para la formulación se basa en alcanzar las propiedades cosméticas deseadas, puesto que la solubilidad del compuesto en la mayoría de los aceites con probabilidades de ser usados en formulaciones farmacéuticas en emulsión puede ser muy baja. Así, la crema debe ser preferiblemente un producto no graso, que no manche y lavable con consistencia adecuada para evitar la pérdida de los tubos u otros recipientes. Se pueden usar ésteres alquílicos mono- o dibásicos de cadena lineal o ramificada tales como diisoadipato, estearato de isocetilo, diéster propilenglicólico de ácidos grasos de coco, miristato de isopropilo, oleato de decilo, palmitato de isopropilo, estearato de butilo, palmitato de 2-etilhexilo o una combinación de ésteres de cadena ramificada conocida como Crodamol CAP, siendo los tres
últimos ásteres preferidos. Estos se pueden usar solos o en combinación dependiendo de las propiedades requeridas. Alternativamente, se pueden usar lípidos de alto punto de fusión tales como parafina blanda blanca y/o parafina líquida u otros aceites minerales.
Formulaciones adecuadas para la administración intranasal, en los que el portador es un líquido, incluyen, por ejemplo, una pulverización nasal, gotas nasales o mediante administración en aerosol mediante un nebulizador, incluyen soluciones acuosas u oleosas del compuesto.
Formulaciones adecuadas para la administración intranasal, en las que el portador es un sólido, incluyen, por ejemplo, las presentadas como un polvo grueso que tiene un tamaño de partícula, por ejemplo, en el intervalo de aproximadamente 20 a aproximadamente 500 micras que se administra del modo en el que se toma el rapé, es decir, mediante inhalación rápida a través de la fosa nasal desde un recipiente del polvo mantenido cerca de la nariz. Formulaciones adecuadas para la administración pulmonar (p. ej., mediante terapia de inhalación o insuflación) incluyen las presentadas como una pulverización en aerosol desde un envase presurizado, con el uso de un propelente adecuado, tal como diclorodifluorometano, triclorofluorometano, diclorotetrafluoroetano, dióxido de carbono u otros gases adecuados.
Formulaciones adecuadas para la administración ocular incluyen gotas oculares en las que el compuesto está disuelto o suspendido en un portador adecuado, especialmente un disolvente acuoso para el compuesto.
Formulaciones adecuadas para la administración rectal se pueden presentar como un supositorio con una base adecuada que comprende, por ejemplo, aceites naturales o endurecidos, ceras, grasas, polioles semilíquidos o líquidos, por ejemplo, mantaca de cacao o un salicilato; o como una solución o suspensión para el tratamiento mediante un enema.
Formulaciones adecuadas para la administración vaginal se pueden presentar como pesarios, tampones, cremas, geles, pastas, espumas o formulaciones para pulverización que contienen además del compuesto portadores tales como los que se conocen en la técnica como apropiados.
Formulaciones adecuadas para la administración parenteral (p. ej., mediante inyección) incluyen líquidos (p. ej., soluciones, suspensiones) acuosos o no acuosos, isotónicos, libres de pirógenos, estériles, en los que el compuesto está disuelto, suspendido o proporcionado de otro modo (p. ej., en un liposoma u otro material en forma de micropartículas). Tales líquidos pueden contener adicionalmente otros ingredientes farmacéuticamente aceptables, tales como antioxidantes, tampones, conservantes, estabilizantes, bacteriostáticos, agentes de suspensión, agentes espesantes y solutos que hacen la formulación isotónica con la sangre (u otro fluido corporal pertinente) del receptor pretendido. Ejemplos de excipientes incluyen, por ejemplo, agua, alcoholes, polioles, glicerol, aceites vegetales y similares. Ejemplos de portadores isotónicos adecuados para el uso en tales formulaciones incluyen inyección de cloruro sódico, inyección de solución de Ringer o inyección de solución de Ringer con lactato. Típicamente, la concentración del compuesto en el líquido es de aproximadamente 1 ng/ml a aproximadamente 10 pg/ml, por ejemplo de aproximadamente 10 ng/ml to aproximadamente 1 pg/ml. Las formulaciones se pueden presentar en recipientes sellados de monodosis o de múltiples dosis, por ejemplo, ampollas y viales, y se pueden almacenar en un estado secado por congelación (liofilizado) que sólo requiere la adición del portador líquido estéril, por ejemplo agua para inyecciones, inmediatamente antes del uso. Las soluciones y suspensiones para inyección extemporánea se pueden preparar a partir de polvos estériles, gránulos y comprimidos.
Dosificación
Será apreciado por el experto en la técnica que las dosificaciones apropiadas de los compuestos de TBAP y las composiciones que comprenden los compuestos de TBAP pueden variar de paciente a paciente. Determinar la dosificación óptima implicará generalmente el equilibrio del nivel de beneficio terapéutico frente a cualquier riesgo o efectos secundarios perjudiciales. El nivel de dosificación seleccionado dependerá de una variedad de factores incluyendo la actividad del compuesto de TBAP particular, la vía de administración, el momento de la administración, la velocidad de excreción del compuesto de TBAP, la duración del tratamiento, otros fármacos, compuestos y/o materiales usados en combinación, la gravedad del trastorno, y la especie, el sexo, la edad, el peso, el estado, la salud general y los antecedentes médicos del paciente. La cantidad de compuesto de TBAP y la vía de administración estarán finalmente a discreción del médico, veterinario o profesional clínico, aunque generalmente la dosificación se seleccionará para alcanzar concentraciones locales en la zona de acción que alcanzan el efecto deseado sin provocar efectos secundarios dañinos o perjudiciales sustanciales.
La administración se puede efectuar en una dosis, continuamente o intermitentemente (p. ej., en dosis divididas a intervalos apropiados) a lo largo del curso del tratamiento. Métodos para determinar los medios más eficaces y la dosificación de administración son muy conocidos por los expertos en la técnica y variarán con la formulación usada para la terapia, el propósito de la terapia, la célula o células diana que se traten y el sujeto que se trate. Se pueden llevar a cabo administraciones únicas o múltiples siendo seleccionados el nivel y el patrón de la dosis por el médico, veterinario o profesional clínico responsable.
En general, una dosis adecuada del compuesto de TBAP es en intervalo de aproximadamente 10 pg a aproximadamente 250 mg (más típicamente de aproximadamente 100 pg a aproximadamente 25 mg) por kilogramo de peso corporal del sujeto al día. Cuando el compuesto es una sal, un éster, una amida, un profármaco o similares, la cantidad administrada se calcula basándose en el compuesto originario y así el peso real que se va a usar se incrementa proporcionalmente.
Síntesis Química
Todas las materias primas, los reactivos y los disolventes para las reacciones eran de calidad para reactivos y se usaron según se adquirieron. Los disolventes de cromatografía eran de calidad para HPLC y se usaron sin purificación adicional. Las reacciones se verificaron mediante análisis por cromatografía en capa fina (TLC, por sus siglas en inglés) usando placas de capa fina F-254 de gel de sílice 60 de Merck. La cromatografía en columna de desarrollo rápido se llevó a cabo en gel de sílice 60 de Merck (0.015-0.040 mm) o en columna de gel de sílice desechables Isolute Flash Si y Si II. Los análisis por LCMS se realizaron en un sistema de HPLC Micromass LCT / Alliance 2795 de Water con una columna de 5 pm, C18, 50 mm x 4,6 mm d. i. Discovery de Supelco a una temperatura de 22°C usando el siguiente sistema de disolventes: Disolvente A: Metanol; Disolvente B: ácido fórmico al 0,1% en agua a un caudal de 1 ml/min. Gradiente comenzando con 10% de A / 90% de B (en volumen) de 0 - 0,5 minutos y a continuación 10% de A / 90% de B a 90% de A / 10% de B de 0,5 minutos a 6,5 minutos y continuando a 90% de A / 10% de B hasta 10 minutos. De 10-10.5 minutos el gradiente se volvió hasta 10% de A / 90% de donde las concentraciones permanecieron hasta 12 minutos. La detección UV era a 254 nm y la ionización era electropulverización iónica positiva o negativa. El intervalo de exploración del peso molecular es 50-1.000. Las muestras se suministraron como 1 mg/ml en DMSO o metanol con 3 pl inyectados en un relleno parcial del circuito. Los espectros de NMR se registraron en DMSO-d6 en un espectrómetro de 500 MHz de Bruker Advance.
Parte(I): N-arilación de ásteres de pirazolcarboxilato
Método A: 3-terc-butil-1 H-pirazol-5-carboxilato de etilo (1 equiv.), el ácido borónico deseado (2 equiv.), acetato de cobre (II) (1,5 equiv) y DMF seca se añadieron bajo remoción para dar una solución azul. Se añadió pindina seca (2 equiv.), con lo que el color se volvía verde, seguido por una cucharada de tamices moleculares de 4 Á (0.4 nm) en polvo secados al horno. La mezcla se agitó a temperatura ambiente bajo atmósfera de argón hasta la terminación de la reacción, según se verificó por LC-MS. Después de la terminación de la reacción, la mezcla se diluyó con solución de AcOEt y NH4CL La fase orgánica se aisló, se lavó con solución de NH4 G , NaHCO3 ac. sat., se secó (MgSO4 con resina atrapadora de Cu), se filtró y se evaporó para dar una sustancia oleosa, que en algunos casos se purificó adicionalmente mediante cromatografía en columna.
Síntesis 1
3-terc-Butil-1-(3-metoxifenil)-1 H-pirazol-5-carboxilato de etilo

Se usó el Método A con 3-terc-butil-1 H-pirazol-5-carboxilato de etilo (320 mg, 1.631 mmol) y ácido 3 metoxifenilborónico (307 mg, 2.020 mmol). La reacción se terminó después de remover a temperatura ambiente durante 22 horas. Después del tratamiento, el aceite amarillo resultante se disolvió en DCM / hexano y se cargó en una columna SNAP de 50 g, que se eluyó con 2 ® 20% (en volumen) de EtOAc en hexano. El compuesto del epígrafe se obtuvo como un aceite incoloro.
Rendimiento: 462 mg (94%, 92% puro). 1H-NMR (DMSO-d6), 5 (ppm), J (Hz): 1.17 (t, 3H, 3Jh h=7.1, CH3), 1.30 (s, 9H, terc-Bu), 3.79 (s, 3H, OCH3), 4.17 (q, 2H, 3Jh h=7.1, OCH2CH3), 6.98 (m, 4H, HAr), 7.36 (t, 1H, 3Jh h=8.1, HAr), LC-MS (2.79 min): m/z calc. para C17H22N2O3 [M+H]+ : 303.1; encontrado: 303.2.
Síntesis 2
3-terc-Butil-1-(3-trifluorometilfenil)-1 H-pirazol-5-carboxilato de etilo

El Método A se usó con 3-terc-butilpirazol-5-carboxilato de etilo (320 mg, 1.60 mmol), and 3-trifluorometilfenilborónico (307 mg, 1.60 mmol). Después de 20 horas, la mezcla de reacción se diluyó con AcOEt (20 ml), se lavó con 2 x 20 ml de agua, NaHCO3 (20 ml, conc.) y finalmente con 20 ml de salmuera. La capa orgánica se secó (MgSO4) y se evaporó hasta sequedad para dar un aceite (417 mg). El compuesto se usó para la etapa de hidrólisis posterior sin purificación adicional.
Síntesis 3
3-terc-Butil-1-(3-metilfenil)-1H-pirazol-5-carboxilato de etilo
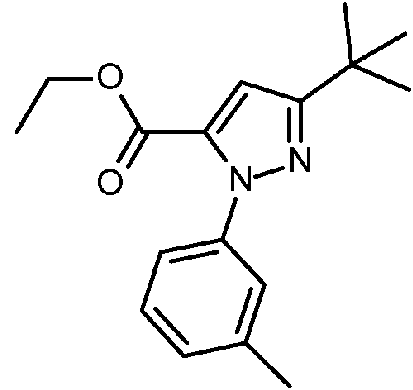
Se usó el Método A con 3-terc-butilpirazol-5-carboxilato de etilo (320 mg, 1.60 mmol) y ácido 3-metilfenilborónico (218 mg, 1.60 mmol). Después de 20 horas, la mezcla de reacción se diluyó con AcOEt (20 ml), se lavó con 2 x 20 ml de agua, NaHCO3 (20 ml; conc.) y finalmente con 20 ml de salmuera. La capa orgánica se secó (MgSO4) y se evaporó hasta sequedad para dar un aceite (466 mg). El compuesto se usó en la etapa de hidrólisis posterior sin purificación adicional.
Síntesis 4
3-terc-Butil-1-(3-fluorofenil)-1 H-pirazol-5-carboxilato de etilo

Se usó el Método A con 3-terc-butilpirazol-5-carboxilato de etilo (320 mg, 1.60 mmol) y ácido 3-fluorofenilborónico (224 mg, 1.60 mmol). Después de 20 horas, la mezcla de reacción se diluyó con AcOEt (20 ml), se lavó con 2 x 20 ml de agua, NaHCO3 (20 ml; conc.) y finalmente con 20 ml de salmuera. La capa orgánica se secó (MgSO4) y se evaporó hasta sequedad para dar un aceite (463 mg). El compuesto se usó en la etapa de hidrólisis posterior sin purificación adicional.
Síntesis 5
3-terc-Butil-1-(2-metoxipiridin-4-yl)-1 H-pirazol-5-carboxilato de etilo

Se usó el Método A con 3-terc-butilpirazol-5-carboxilato de etilo (202 mg, 1.03 mmol) y ácido 2-metoxipiridin-4-ilborónico (208 mg, 1.360 mmol). La purificación con 2 ® 50% (en volumen) EtOAc en hexano dio el compuesto del epígrafe como un aceite incoloro.
Rendimiento: 243 mg (59%). 1H-NMR (DMSO-d6), 5 (ppm), J (Hz): 1.22 (t, 3H, 3Jh h=7.1, CH3), 1.29 (s, 9H, terc-Bu), 3.90 (s, 3H, OCH3), 4.23 (q, 2H, 3Jh h=7.1, OCH2CH3), 6.94 (d, 1H, 3Jh h=1.7, HPir), 7.07 (s, 1H, HAr), 7.13 (dd, 1H, 3Jh h=5.6, 1.7, HPir), 8.23 (d, 1H, 3Jh h=5.6, HPir). LC-MS (2.83 min): m/z calc. para C16H21N3O3 [M+H]+: 304.1; encontrado: 303.1.
Parte(II): Hidrólisis del éster etílico
Método B. El 3-terc-butil-1H-pirazol-5-carboxilato de etilo sustituido en 1 apropiado (1 equiv.) se disolvió en una mezcla 4 : 1 : 1 de THF / MeOH / H2O, se añadió monohidrato de hidróxido de litio (1.1 equiv.) y la mezcla incolora se agitó durante 16 horas a temperatura ambiente. Las materias volátiles se evaporaron posteriormente, y el sólido resultante se redisolvió en H2O y el pH de la solución se ajustó hasta 1 con HCl acuoso al 10%. La mezcla lechosa resultante se extrajo con EtOAc dos veces y la fracción orgánica combinada se lavó con salmuera, se secó y se concentró hasta sequedad para dar un sólido cristalino blanco.
Método C. El 3-terc-butilpirazol-5-carboxilato de etilo sustituido en 1 apropiado (1 equiv.) se sometió a reflujo durante 30 minutos en 10 ml de EtOH y 3 ml de solución de NaOH (2 M). Después de enfriar a temperatura ambiente, la mezcla de reacción se neutralizó hasta pH 4.0 (AcOH), se diluyó con 20 ml de agua y se extrajo con AcOEt. La capa orgánica se lavó con 2 x 20 ml de agua, se secó (MgSO4) y se evaporó hasta sequedad y el residuo así obtenido se purificó usando un sistema Biotage Isolera.
Síntesis 6
Ácido 3-terc-butil-1-(3-metoxifenil)-1 H-pirazol-5-carboxílico

Usar el Método B, 3-terc-butil-1-(4-metoxifenil)-1 H-pirazol-5-carboxilato de etilo (442 mg, 1.345 mmol) dio el compuesto del epígrafe como cristales blancos.
Rendimiento: 300 mg (81%). 1H-NMR (DMSO-d6), 5 (ppm), J (Hz): 1.29 (s, 9H, terc-Bu), 3.78 (s, 3H, OCH3), 6.90 (s, 1H, HAr), 6.97 (m, 4H, HAr), 7.35 (t, 1H, 3Jh h=8.1, HAr), 13.14 (s, 1H, COOH). LC-MS (2.51 min): m/z calc. para C15H18N2O3 [M+H]+: 275.1; encontrado: 275.0.
Síntesis 7
Ácido 3-terc-butil-1 -(3-trifluorometilfenil)-1 H-pirazol-5-carboxílico

Usando el Método C con 3-terc-butil-1-(3-trifluorometilfenil)-1 H-pirazol-5-carboxilato de etilo en bruto (471 mg), se obtuvo un producto sólido que se sometió a purificación adicional usando un sistema Biotage Isolera y una mezcla de ciclohexano:EtOAc 1:1 como eluyente (modo isocrático) y dio el compuesto del epígrafe.
Rendimiento: 133 mg (26.6% a lo largo de 2 etapas). 1H NMR (DMSO), 5h (ppm), J (Hz): 1.31 (s, 9H, (CH^C), 6.99 (s, 1H, H Pir), 7.70 (t, 1H, H5 Arom., J=7.7Hz), 7.76-7.82 (m, 3H, H2+4+6 Arom.), 13.32 (s, 1H, CO2H Pir.). Masa ex.: (C15H16F3N2O2 ) calc. 313.1157, encontrada 313.1155.
Síntesis 8
Ácido 3-terc-butil-1-(3-metilfenil)-1 H-pirazol-5-carboxílico

Usando el Método C con 3-terc-butil-1-(3-metilfenil)-1 H-pirazol-5-carboxilato de etilo en bruto (460 mg), se obtuvo el compuesto del epígrafe después de la purificación usando un sistema Biotage Isolera y una mezcla 1:1 de ciclohexano:EtOAc como eluyente (modo isocrático).
Rendimiento: 101 mg (24% a lo largo de 2 etapas). 1H NMR (DMSO), 5h (ppm), J (Hz): 1.29 (s, 9H, (CH3)3C), 2.35 (s, 3H, 3 -CH3), 6.89 (s, 1H, Pir-H), 7.18 (d, 1H, H2 Arom., J=7.2Hz), 7.20-7.24 (m, 2H, H4+6 Arom.), 7.32 (t, 1H, H5 Arom. J=7.3Hz), 13.09 (s, 1H, CO2H Pir.). Masa ex.: (C15H18N2O2) calc. 258.1368, encontrada 258.1373.
Síntesis 9
Ácido 3-terc-butil-1 -(3-fluorofenil)-1 H-pirazol-5-carboxílico

Usando el Método C con 3-terc-butil-1-(3-fluorofenil)-1H-pirazol-5-carboxilato de etilo en bruto (463 mg), se obtuvo el compuesto del epígrafe. Véase, p. ej., Springer y cols., 2011.
Rendimiento 166 mg (39.6% a lo largo de 2 etapas). 1H NMR (DMSO), 5h (ppm), J (Hz): 1.29 (s, 9H, (CH^C), 6.95 (s, 1H, Pir-H), 7.23-7.30 (m, 2H, Arom-^+s), 7.35 (d, 1H, H2 Arom., J=9.8 Hz), 7.44-7.53 (m, 1H, H6 Arom.), 13.24 (s, 1H, CO2H Pir.). Masa ex: (C14H15FN2O2) calc. 262.1118, encontrada 262.1117.
Síntesis 10
Hidrocloruro de ácido 3-terc-butil-1-(2-oxo-1,2-dihyd ropiridin-4-yl)-1 H-pirazol-5-carboxílico

Método D: Se disolvió 3-terc-butil-1-(2-metoxipiridin-4-yl)-1H-pirazol-5-carboxilato de etilo (110 mg, 0.363 mmol) en HCl 6 M en H2O (4.5 ml, 27.00 mmol) y la solución incolora se calentó hasta 90°C durante 48 h. Todas las materias volátiles se evaporaron posteriormente y el aceite incoloro resultante se coevaporó con DCM (10 ml) y a continuación con Et2O (10 ml), lo que dio el compuesto del epígrafe como un sólido blanco.
Rendimiento: 93 mg (98%). 1H-NMR (DMSO-d6), 5 (ppm), J (Hz): 1.28 (s, 9H, terc-Bu), 6.32 (d, 1H, J=1.9, HAr), 6.37 (dd, 1H, J=7.1, 1.9, HAr), 6.99 (s, 1H, HAr), 7.46 (d, 1H, J=7.1, HAr). LC-MS (2.14 min): m/z calc. para C13H16N3O3 [M-Cl]+: 262.1; encontrado: 262.0;
Parte(III): Formación de 5-aminopirazoles
Síntesis 11
3-(terc-Butil)-1 -(3-fluorofenil)-1 H-pirazol-5-amina

Método E: Una mezcla de 4,4-dimetil-3-oxopentanonitrilo (77 g, 0.62 mol) e hidrocloruro de 3-fluorofenilhidracina (100 g, 0.62 mol) se añadió a tolueno (1 l) y se calentó hasta 100oC durante 24 horas, punto después del cual se dejó que la reacción se enfriara hasta 20°C. La mezcla se filtró, se lavó con tolueno (2 x 250 ml) se secó. La sal de HCl en bruto se combinó con una partida previa (realizada usando 180 g de hidrocloruro de 3-fluorofenilhidracina y 234 g de hidrocloruro de 3-fluorofenilhidracina) y se repartió entre DCM (4 l) y NaHCO3 ac. sat. (4 l). La mezcla se agitó hasta que no quedaba sólido. La capa de DCM se separó, se secó (MgSO4), se filtró y se concentró a vacío para proporcionar el compuesto del epígrafe como un sólido naranja (210g) con 52% de rendimiento. Pureza >95% (sobre una base molar) mediante NMR y 94.4% (sobre una base molar) mediante LCMS.
Parte(IV): Formación de carbamatos de 5-aminopirazol
Síntesis 12
N-[3-terc-Butil-1 -(3-fluorofenil)-1 H-pirazol-5-il]carbamato de fenilo
Método F: Se disolvió 3-(terc-butil)-1-(3-fluorofenil)-1 H-pirazol-5-amina (210 g, 0.90 mol) en THF (5 l) a 0°C antes de la adición de piridina (146 ml, 1.80 mol). Se cargó cloroformiato de fenilo (113 ml, 0.90 mol) en THF (300 ml) gota a gota a 0-5°C a lo largo de 30 minutos. La mezcla de reacción se agitó a 0°C durante 30 minutos y a continuación se dejó calentar hasta temperatura ambiente. Después de 4 horas, la HPLC mostró que quedaba 8% del estadio 1. Se añadió una carga adicional de cloroformiato de fenilo (11 ml, 0.088 mol) y, después de 30 minutos, el análisis de HPLC indicó que la reacción había terminado. Se cargó EtOAc (5 l) y la capa orgánica se lavó con HCl 1 M (2 x 1.2 l), agua (1.2 l), NaHCO3 ac. sat. (1.2 l) y salmuera sat. (1.2 l). La capa orgánica se secó (MgSO4), se filtró y se concentró a vacío. El aceite en bruto se recogió en una mezcla 1 : 3 de EtOAc / heptano y se concentró a vacío para dar un sólido. El sólido se suspendió en heptano (2.5 l) durante 1 hora, se filtró y se lavó con heptano (200 ml). El material se secó a 40°C durante la noche para dar el compuesto del epígrafe (286 g) con 90% de rendimiento. Pureza >95% por NMR.
Parte(V): Acoplamiento de fragmentos de arilpirazol y 4-aminofenoxi-piridopiracinona con formación de un conectar de urea
Síntesis 13
1 -[3-terc-butil-1 -[(3-fluoro-fenil)-1 H-pirazol-5-il]3-[2-fluoro-4(3-oxo-3,4-dihidropirido[2,3-b]piracin-8-iloxi)fenil]urea (TBAP-001)
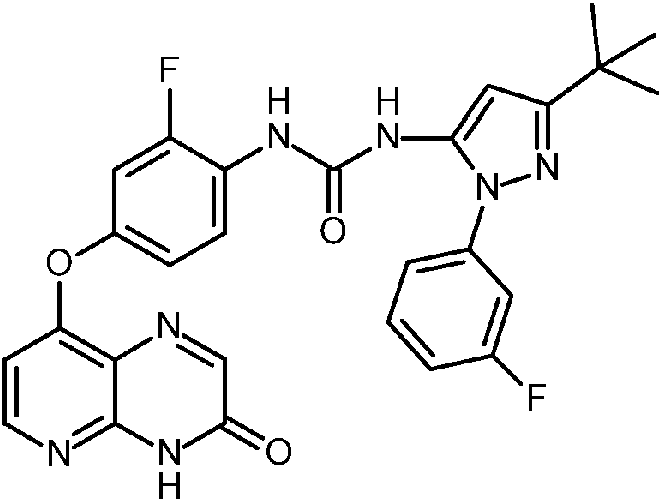
Método G: Se disolvieron 81 mg (0.31 mmol) de ácido 3-terc-butil-1-(3-fluorofenil)-pirazol-5-carboxílico en 2 ml de DMF en un tubo Carousel bajo remoción y atmósfera inerte. A continuación, se añadieron 0.044 ml (0.32 mmol) de trietilamina y 0.067 ml (0.032 mmol) de DPPA y la remoción continuó durante 30 minutos a 0°C y durante 1 hora adicional a temperatura ambiente. Se añadió de una vez a esta mezcla de reacción la 4-(3-fluoro-4-aminofenil)-piridino-[2.3-b]-piracin-2-ona (40 mg, 0.15 mmol) (véase, p. ej., Zambon y cols., 2010) y el tubo con la mezcla de reacción se calentó a 100°C durante 30 minutos, bajo remoción y una atmósfera de argón. Después de enfriar a temperatura ambiente, la solución se diluyó con 10 ml de AcOEt. La capa orgánica se lavó con 2 x 10 ml de salmuera, se secó (MgSO4) y se evaporó hasta sequedad. El residuo así obtenido se trituró con Et2O y se filtró para dar el compuesto del epígrafe como un sólido amorfo pardo claro.
Rendimiento: 66 mg (83.0%). 1H NMR (500 MHz, DMSO-d6) 5: 1.29 (s, 9H, t-Bu), 6.41 (s, 1H, H ^ zol), 6.64 (d, 1H, HPin J=5.6Hz), 7.°2-7.07 (m "1^ HArom central^ 7.22-7.30 (m 2H HArom pirazol^ 7.40-7.44 (m 2H HArom central+ HArom pirazol^ 7.53-7.60 (m "1^ HArom pirazol^ 8.14 (t 1 H HArom centrad J=9.1Hz), 8.17 (s, 1 H, Hpiracinona), 8.36 (d, 1H, Hpir, J=5.6 Hz), 8.87 (s, 1 H, NHurea), 8.98 (s, 1 H, NHurea), 12.90 (s, 1 H, NH). LC-MS, tR = 2.61 min, m/z: 531.2 (M)+, calc. para C27H23F2N7O3. HRMS: (M)+ calc. para C27H23F2N7O3 , 531.1830, encontrado: 531.1832.
Método H: Se cargaron N-[3-terc-butil-1-(3-fluorofenil)-1H-pirazol-5-il]carbamato de fenilo (220 g, 0.623 mol) y DMSO (1.7 l) a 4-(3-fluoro-4-aminofenil)-piridino-[2.3-b]-piracin-2-ona (169.5 g, 0.623 mol). La mezcla de reacción se agitó a 20-22°C durante la noche. La 1H NMR indicó que la reacción había terminado. La reacción se desactivó en agua (8.6 l) y se agitó durante 1 hora antes de filtrarse y lavarse con agua (2 x 2 l). El material se secó a 60°C a lo largo del fin de semana. El sólido se suspendió en EtOAc (3.39 l) durante 1 hora, se filtró y se lavó con EtOAc (750 ml) para dar 320 g del compuesto del epígrafe. La NMR indicó que todavía había fenol presente. El material se resuspendió en EtOAc (3.2 l) durante 1 hora, se filtró y se lavó con EtOAc (500 ml) y se secó para proporcionar 293 g del compuesto del epígrafe (9% en peso de EtOAc) por NMR, una sola impureza 0.8% en peso). El sólido se recristalizó en THF (5.7 l) y heptano (2.85 l), dejando que la partida se enfriara hasta temperatura ambiente antes de separar los sólidos por filtración. La torta filtrante se lavó con heptano (2.85 l) y se secó a 45°C durante la noche para dar 221 g del compuesto del epígrafe. El análisis por HPLC mostró que la impureza previa en 0.8% (en peso) se reducía hasta 0.23% (en peso); sin embargo, la impureza de urea se enriquecía hasta 0.58% (en peso). La 1H NMR mostró 5% de heptano (en peso). El material se secó a 110°C durante 12 horas para llevar el nivel de heptano hasta < 0.5% (en peso) por NMR, dando un total de 211 g del compuesto del epígrafe como un sólido cristalino blanco con un rendimiento de 64%. Pureza por HPLC 98.8% (en peso), una sola impureza 0.58% (en peso).
Síntesis 14
1-[3-íerc-Butil-1-[(3-metil-fenil)-1 H-pirazol-5-il]3-[2-fluoro-4(3-oxo-3,4-dihidropirido[2,3-b]piracin-8-iloxi)fenil]urea (TBAP-002)
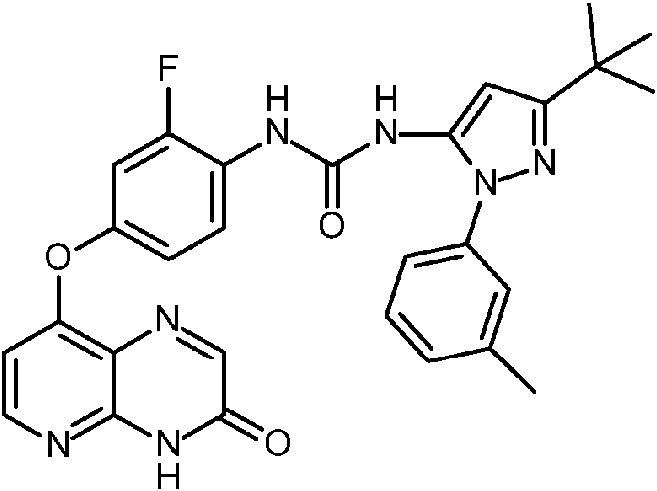
Usando el Método G, con ácido 3-íerc-butil-1-(3-metilfenil)-1H-pirazol-5-carboxílico (80 mg, 0.31 mmol) y 4-(3-fluoro-4-aminofenil)-piridino-[2,3-b]-piracin-2-ona (40 mg, 0.15 mmol), se obtuvo el compuesto del epígrafe como un sólido blancuzco.
Rendimiento: 69 mg (87.4%). 1H NMR (500 MHz, DMSO-d6) 5 : 1.28 (s, 9H, t-Bu), 2.40 (s, 3H, 3 -CH3), 6.39 (s, 1H, HPirazol), 6.64 (d, 1 H, HPir, J=5.6Hz), 7.02-7.06 (m, 1H, HArom central), 7.22-7.36 (m, 4H, 3 HArom pirazol +1 HArom central), 7.43 (t 1H, HArom pirazol, J=7.7Hz), 8.16 (t, 1H, HArom central, J=9.1Hz), 8.17 (s, 1H, Hpiracinona), 8.36 (d, 1H, Hpir, J=5.6 Hz), 8.81 (s, 1H, NHurea), 8.98 (s, 1H, NHurea), 12.90 (s, 1H, NH), LC-MS, ír = 2.65 min, m/z: 527.2 (M)+, calc. para C28H26FN7O3, HRMS: (M+H)+ calc. para C28H26FN7O3, 527.2081, encontrado: 527.2088.
Síntesis 15
1-[3-íerc-Butil-1-[(3-trifluorometil-fenil)-1 H-pirazol-5-il]3-[2-fluoro-4(3-oxo-3,4-dihidropirido[2,3-b]piracin-8-iloxi)fenil]urea (TBAP-003)
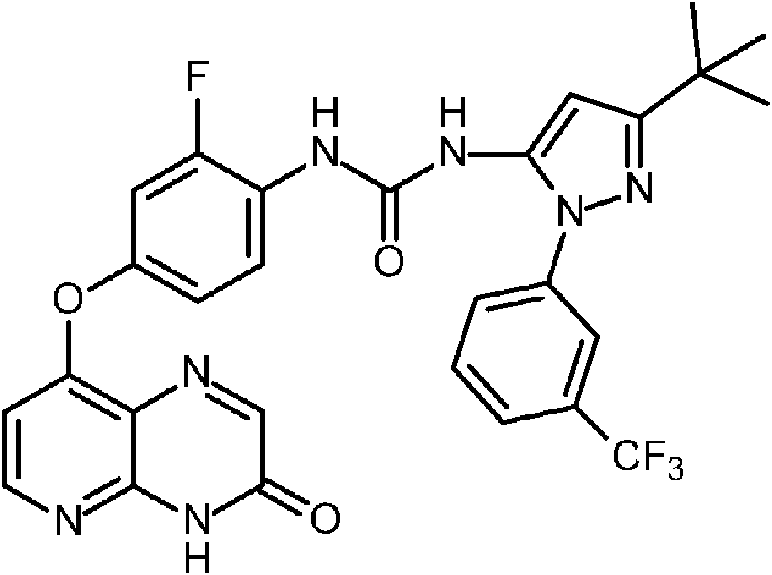
Usando el Método G, con ácido 3-íerc-butil-1-(3-trifluorometilfenil)-1 H-pirazol-5-carboxílico (97 mg, 0.31 mmol)y 4-(3-fluoro-4-aminofenil)-piridino-[2,3-b]-piracin-2-ona (40 mg, 0.15 mmol), se obtuvo el compuesto del epígrafe como un sólido blancuzco.
Rendimiento: 70 mg (80.4%). 1H NMR (500 MHz, DMSO-d6) 5 : 1.30 (s, 9H, t-Bu), 6.42 (s, 1H, Hpirazol), 6.64 (d, 1H, HPih J=5.6Hz) 7.°2-7.°6 (m 1^ HArom central), 7.27-7.36 (m, 1H, HArom central), 7.74-7.80 (m, 2H, HArom pirazol), 7.86-7.90 (m, 2H, HArom pirazol), 8.05 (t, 1H, HArom central, J=9.1Hz), 8.17 (s, 1H, Hpiracinona), 8.36 (d, 1H, Hpir, J=5.6 Hz), 8.87 (s, 1H, NHurea), 8.93 (s, 1H, NHurea), 12.90 (s, 1H, NH), LC-MS, ÍR = 2.71 min, m/z: 581.2 (M)+, calc. para C28H23F4N7O3. HRMS: (M+H)+ calc. para C28H23F4N7O3, 581.1798, encontrado: 581.1796.
Síntesis 16
1-(3-íerc-butil-1-(3-metoxifenil)-1 H-pirazol-5-il)-3-(2-fluoro-4-(3-oxo-3,4-dihidropirido[3,2-£>]piracin-8-iloxi)fenil)urea (TBAP-004)

Usando el Método G con ácido 3-terc-butil-1-(3-metoxifenil)-1 H-pirazol-5-carboxílico (60 mg, 0.22 mmol) y 8-(4-amino-3-fluorofenoxi)-pirido[2,3-£>]piracin-3(4H)-ona (31.6 mg, 0.12 mmol), el compuesto del epígrafe se obtuvo como un sólido amarillo claro.
Rendimiento: 50 mg (84%). 1H-NMR (DMSO-d6), 5 (ppm), J (Hz): 1.29 (s, 9H, terc-Bu), 3.83 (s, 3H, OCH3), 6.41 (s, 1H, HPir), 6 .66 (d, '3Jhh=5.7, 1H, HPir), 7.01-7.12 (m, 4H, HAr), 7.31 (m, 1H, HAr), 7.46 (t, 3Jhh=8.1, 1H, HAr), 8.18 (m, 2H, HAr), 8.38 (d, 3Jhh=5.7, 1H, HPir), 8.85 (s an, 1H, NH), 9.03 (s an, 1H, NH), 12.92 (s an, 1H, NH);_19F-NMR (DMSO-d6), 5 (ppm): -124.8. LC-MS (2.59 min): m/z calc. para C28H27FN7O4 [M+H]+ : 544.1; encontrado: 544.1. HRMS (3.19 min): m/z calc. para C28H27FN7O4 [M+H]+: 544.21031; encontrado: 544.21029.
Síntesis 17
1-(3-terc-Butil-1-(2-oxo-1,2-dihidropiridin-4-il)-1 H-pirazol-5-yl)-3-(2-fluoro-4-(3-oxo-3,4-dihidropirido[3,2-£>]piracin-8-iloxi)fenil)urea (TBAP-005)

Usando el Método G con hidrocloruro de ácido 3-terc-butil-1-(2-oxo-1,2-dihidropiridin-4-il)-1 H-pirazol-5-carboxílico (70.8 mg, 0.238 mmol) y 8-(4-amino-3-fluorofenoxi)-pirido[2,3-£>]piracin-3(4H)-ona (32.4 mg, 0.119 mmol), se obtuvo un sólido que se purificó mediante cromatografía en columna sobre gel de sílice, eluyendo con 5 ® 30% (en volumen) de MeOH en DCM, para dar el compuesto del epígrafe como un sólido blanco.
Rendimiento: 15 mg (24%), 1H-NMR (DMSO-de), 5 (ppm), J (Hz): 1.28 (s, 9H, terc-Bu), 6.43 (s, 1H, HPir), 6.50 (d, 3Jhh=2.2, 1H, HAr), 6.58 (dd, 3Jhh=7.2, 2.2, 1H, HAr), 6 .66 (d, 3Jhh=5.6, 1H, HPir), 7.06 (m, 1H, HAr), 7.32 (m, 1H, HAr), 7.50 (d, 3Jhh=7.2, 1H, HAr), 8.13 (m, 1H, HAr), 8.18 (m, 1H, HAr), 8.37 (d, 3Jhh=5.6, 1H, HPir), 9.05 (s an, 1H, NH), 9.13 (s, 1H, NH), 11.66 (s an, 1H, NH), 12.90 (s an, 1H, NH). LC-MS (2.36 min): m/z calc. para C26H24FN8O4 [M+H]+: 531.1; encontrado: 531.2. HRMS (2.97 min): m/z calc. para C26H24FN8O4 [M+H]+: 531.18991; encontrado: 531.18952.
Métodos y Datos Biológicos
Ensayo de cinasa DELFIA
Los compuestos se evaluaron en un ensayo de cinasa realizado según el siguiente protocolo.
Preparación de V600EBRAF:
Se generó ^ ^B R A F mediante la infección de células de insecto SF9 cultivadas en medio SF-900 II (Invitrogen, Paisley, Escocia) con un baculovirus que contenía BRAF humana de longitud completa con una cola de histidina N-terminal y se purificó mediante cromatografía de afinidad de níquel-agarosa.
Preparación de GST-MEK:
La proteína MEK1 de conejo de longitud completa se expresó con una cola de GST en el extremo N y una cola de histidina C-terminal en bacterias Escherichia coli JM109 y se purificó mediante cromatografía de afinidad de níquelagarosa.
Purificación de V600EBRAFy GST-MEK:
Procedimiento:
1. Sométanse las células a lisis en tampón de resuspensión (1 ml por 10 ml de cultivo de SF9 o cultivo de JM109 para BRAF o MEK, respectivamente), sométanse a ultrasonidos durante 1-2 minutos y centrifúguense a 14.000 rpm (en tubos de 2 ml) durante 10 minutos.
2. Tómense 1.5 ml de ‘cuentas’ de níquel-agarosa por 10 ml de lisado y añádanse a la columna (Bio-rad).
3. Lávese la columna con tampón de resuspensión 3 veces.
4. Añádase el lisado a la columna.
5. Lávese 3 veces con 10 ml de tampón de lavado.
6. Añádanse 10 ml de tampón de elución a las cuentas y recójanse en tubos de 2 ml.
7. Compruébese la concentración de proteína de las eluciones y dialícese durante la noche a 4°C en tampón de diálisis.
Tampones:

Tampón de Diálisis (mezclado y almacenado en una habitación fría):

Tampón de Cinasa DELFIA (DKB):

MOPS = cido 3-[N-morfolino]propanosulfónico (Sigma M3183).
EGTA = Ácido etilenglicol-bis(2-aminoetileter)-N,N,N',N'-tetraacético (Sigma E3889).
DKB1 (DKB con proteína BRAFy MEK):
Combínense 4950 gl de DKB y 50 gl de solución de reserva de 2.5 mg/ml de GST-MEK obtenida como se describe anteriormente (para dar 1 mg de MEK por 40 gl). A continuación, añádanse 22.5 gl de solución de reserva de BRAF obtenida como se describe anteriormente para dar ~0.2 gl de BRAF por 40 gl.
DKB2 (DKB con proteína MEK):
Combínense 4950 gl de DKB y 50 gl de solución de reserva de 2.5 mg/ml de GST-MEK (para dar 1 mg de MEK por 40 gl). Úsense 500 gl de esta para la extinción ("blow out" (BO)) y el control del vector vacío (EV, por sus siglas en inglés).
ATP:
Dilúyase solución de reserva de ATP 100 mM en agua destilada hasta 500 gM para dar una concentración final de 100 gM en el ensayo.
Inhibidores (Compuestos de Prueba):
Dilúyase solución de reserva 100 mM hasta 10, 3, 1, 0.3, 0.1, 0.03, 0.01, 0.003, 0.001, 0.0003 y 0.0001 mM en DMSO en una placa para fármacos, dando como resultado concentraciones de 100, 30, 10, 3, 1,0.3, 0.1,0.03, 0.01, 0.003 y 0.001 gM en el ensayo.
Anticuerpo primario:
Fosfo-MEK1/2 CST N° 9121S diluido 1:1000 en tampón de ensayo (AB, por sus siglas en inglés) DELFIA. Preincúbese el anticuerpo en el AB durante 30 minutos a temperatura ambiente antes de usar.
Anticuerpo secundario:
Anticuerpo secundario marcado con europio contra globulinas de conejo Perkin Elmer N° AD0105 diluido 1:1000 en tampón de ensayo (AB) DELFIA. Preincúbese el anticuerpo en el AB durante 30 minutos a temperatura ambiente antes de usar. (Los anticuerpos primario y secundario se incubaron conjuntamente.)
Tween:
0. 1 . de Tween 20 en agua.
Tampón de Ensayo:
Tampón de ensayo DELFIA Perkin Elmer N° 4002-0010.
Solución de Mejora:
Solución de mejora DELFIA Perkin Elmer N° 4001-0010.
Placas de Ensayo:
Placa negra revestida con glutationa de 96 pocillos Perbio N° 15340.
Procedimiento:
1. Prebloquéense los pocillos con 5% de leche en TBS durante 1 hora.
2. Lávense los pocillos 3 veces con 200 pl de TBS.
3. Siémbrense 40 pl de DKB1 para todos los inhibidores (compuestos de prueba), control de DMSO y opcionalmente otros compuestos de control.
4. Siémbrense 40 pl de DKB2 para pocillos de BO y EV.
5. Añádanse los inhibidores (compuestos de prueba) en 0,5 pl por pocillo según la disposición deseada de la placa.
6. Añádanse 0.5 pl de DMSO a los pocillos de control con vehículo.
7. Añádanse 2 pl de BRAF a pocillos de BO y EV.
8. Preincúbese con los compuestos de prueba durante 10 minutos a temperatura ambiente con agitación.
9. Añádanse 10 pl de solución de reserva de ATP 500 pM, en DKB, para dar una concentración de ensayo de 100 pM.
10. Séllense las placas con TopSeal e incúbese a temperatura ambiente con agitación durante 45 minutos.
11. Lávense las placas 3 veces con 200 pl de Tween20 al 0.1%/agua para terminar la reacción.
12. Añádanse 50 pl por pocillo de mezcla de anticuerpos e incúbese durante 1 hora a temperatura ambiente con agitación.
13. Lávense las placas 3 veces con 200 pl de Tween20 al 0.1%/agua.
14. Añádanse 100 pl de solución de mejora DELFIA por pocillo, cúbrase con papel de aluminio e incúbese a temperatura ambiente durante 30 minutos con agitación.
15. Léase en un lector de placas Victor (Perkin-Elmer, Turku, Finlandia) usando el protocolo del europio.
Los valores para muestra sin analito (Vector vacío) se sustraen de todos los valores. Los controles de DMSO se fijan como 100% de actividad y los puntos de ensayo (la respuesta) se calculan como un porcentaje del control de DMSO. Los datos se representan usando el programa Graphpad Prism y se calcula una línea de regresión no lineal usando una ecuación de respuesta a la dosis sigmoidea de pendiente variable:
Y = Inferior [Superior - Inferior] / [ 1 10A((LogEC50 - X) * Ladera)]
donde X es el logaritmo de la concentración e Y es la respuesta. La IC50 generada por este procedimiento es la concentración del fármaco que produce un valor del porcentaje de control de fluorescencia a medio camino entre la saturación y la meseta de efecto cero. Habitualmente se realizan tres ensayos independientes y se presenta la IC50 media.
Los datos se resumen en la siguiente tabla.
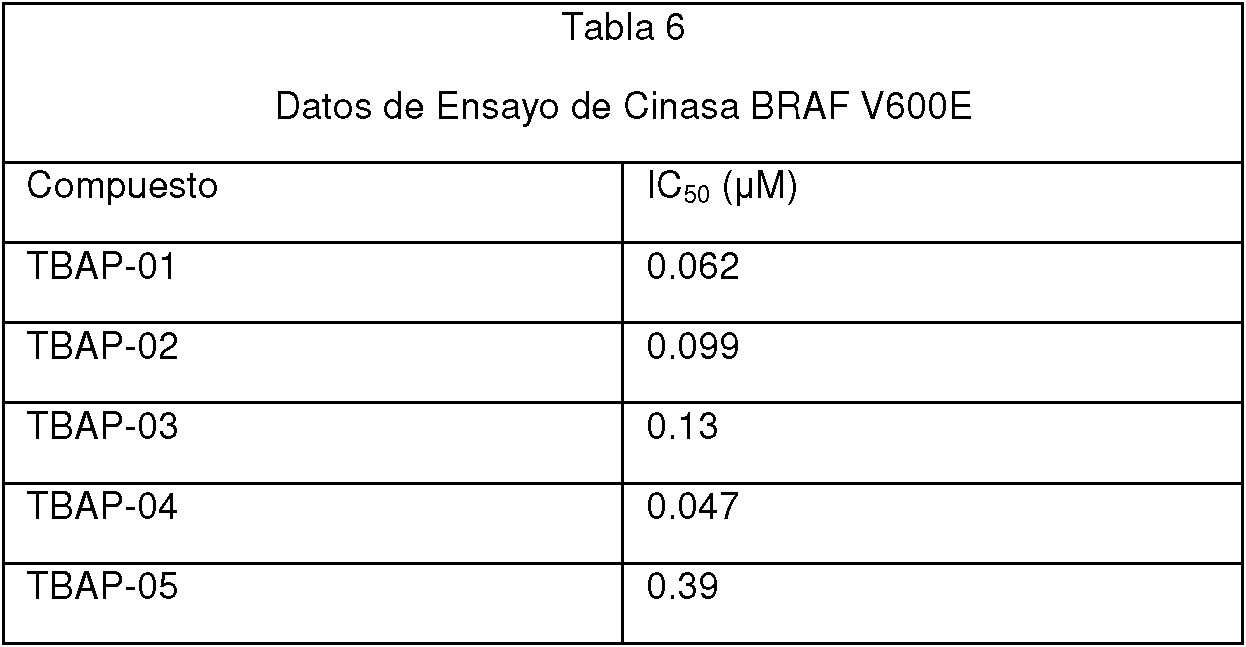
Ensayo de Fosfo-ERK Basado en Células (Células BRAF WM266.4 mutantes)
Los compuestos se ensayaron usando un ensayo basado en células que se realizó según el siguiente protocolo. Día 0:
Siémbrense 16.000 células BRAF WM266.4 mutantes/pocillo en 99 pl de medio en una placa de 96 pocillos.
Día 1:
1. Añádase 1 pl de compuesto de prueba a las células (1 pl de solución total).
2. Incúbense las células con el compuesto de prueba durante 6 horas a 37°C.
3. Aspírese la solución de todos los pocillos.
4. Fíjense las células con 100 pl de formaldehído al 4%/Triton X-100 al 0.25% en PBS por pocillo.
5. Incúbese la placa durante 1 hora a 4°C.
6. Aspírese la solución de fijación y añádanse 300 pl de TBS por pocillo.
7. Déjese la placa durante la noche a 4°C.
Día 2:
1. Lávese la placa 2 veces con 200 pl de PBS por pocillo.
2. Bloquéese con 100 pl de leche secada al 5% en TBS.
3. Incúbese la placa durante 20 minutos a 37°C.
4. Lávese la placa 2 veces con Tween al 0.1%/H2O.
5. Añádanse 50 pl de 3 pg/ml de anticuerpo primario pERK (Sigma M8159), diluido en leche en polvo al 5%/TBS, a cada pocillo.
6. Incúbese la placa durante 2 horas a 37°C.
7. Lávese la placa 3 veces con Tween al 0.1%/H2O.
8. Añádanse a cada pocillo 50 pl de 0.45 pg/ml de anticuerpo secundario contra globulinas de ratón marcado con europio (Perkin Elmer).
9. Incúbese la placa durante 1 hora a 37°C.
10. Lávese la placa 3 veces con Tween al 0.1%/H2O.
11. Añádanse a cada pocillo 100 pl de solución de mejora (Perkin Elmer).
12. Déjese la placa durante aproximadamente 10 minutos a temperatura ambiente antes de batir suavemente la placa.
13. Léase la fluorescencia resuelta con el tiempo con europio en un lector de placas Victor2 (Perkin-Elmer, Turku, Finlandia).
14. Lávese la placa 2 veces con Tween al 0.1%/H2Ü.
15. Mídase la concentración de proteína con el ensayo de ácido bicinconínico (BCA, Sigma) al añadir 200 gl de solución por pocillo.
16. Incúbese la placa durante 30 minutos a 37°C.
17. Léanse los niveles de absorbancia a 570 nm en un lector de placas.
Nótese que los recuentos de europio se normalizan para los niveles de proteína al dividir los recuentos por la absorbancia.
Los valores para la muestra sin analito (sin células) se sustraen de todos los valores. Los controles de DMSO se fijan como 100% de actividad y los puntos de ensayo (las respuestas) se calculan como un porcentaje del control de DMSO. Los datos se representan usando el programa Graphpad Prism y se calcula una línea de regresión no lineal usando una ecuación de respuesta a la dosis sigmoidea de pendiente variable:
Y = Inferior [Superior - Inferior] / [1 10A((LogEC50 - X) * Ladera)]
donde X es el logaritmo de la concentración e Y es la respuesta). La IC50 generada por este procedimiento es la concentración del fármaco que produce un valor del porcentaje de control de fluorescencia a medio camino entre la saturación y la meseta de efecto cero. Habitualmente se realizan tres ensayos independientes y se presenta la IC50 media.
Los datos se resumen en la siguiente tabla.

Ensayo de Proliferación Celular de SRB (GI50 de SRB)
Las líneas celulares se cultivan normalmente en DMEM o RPMI1640 complementados con 10% de suero bovino fetal a 37°C en una atmósfera de CO2 al 10% saturada con agua. Los cultivos se mantienen en fase de crecimiento exponencial al subcultivar antes de haberse vuelto confluentes (intervalos de 3-5 días). Se preparan suspensiones de células simples al recoger un matraz de cultivo tisular de 80 cm2 con 5 ml de tripsina-EDTA comercial. Después de 5 minutos, las células separadas se mezclan con 5 ml de medio de cultivo totalmente complementado y se nodulizan centrífugamente (1000 rpm durante 7 minutos). Después de aspirar el sobrenadante, el nódulo celular se resuspende en 10 ml de medio reciente y las células se desagregan completamente al hacer correr hacia arriba/hacia abajo 5 veces todo el volumen a través de una aguja de calibre 19. La concentración de las células se determina usando un hemocitómetro (dilución 1/10). Un volumen adecuado para dar un exceso de al menos 2 veces para el número de pruebas que se efectúan, típicamente 100-200 ml, se prepara al diluir la suspensión celular hasta 10.000-40.000/ml, y se aportan 100 gl/pocillo a placas de 96 pocillos usando una bomba peristáltica programable de 8 canales, dando 1.000-4.000 células/pocillo, dejando la columna 12 sin tratamiento. Las placas se devuelven a la incubadora durante 24 horas para permitir que las células se unan de nuevo.
Los compuestos que se prueban se preparan a 10 mM en DMSO. Se diluyen partes alícuotas (24 gl) en 1.2 ml de medio de cultivo dando 200 gM, y se realizan 10 diluciones en serie de 3 veces al transferir 80 gl a 160 gl. Se añaden partes alícuotas (100 gl) de cada dilución a los pocillos, usando un pipeteador de 8 canales, realizando así una solución adicional de 2 veces, y dando dosis que varían de 100 gM a 0.005 gM. La columna 11 recibe
solamente medio de cultivo simple. Cada compuesto se prueba por cuadruplicado, siendo cada repetición el promedio de cuatro pocillos.
Después de un crecimiento adicional de 5 días, las placas se vacían, y las células se fijan en ácido tricloroacético al 10% durante 30 minutos a 4°C. Después de un enjuague a fondo en agua corriente del grifo, las placas se secan, y se tiñen al añadir 50 pl de una solución de sulforrodamina-B al 0.1% en ácido acético al 1%, durante 10 minutos a temperatura ambiente. La mancha se vierte y las placas se enjuagan a fondo bajo una corriente de ácido acético al 1% (retirando así la mancha no unida) y se secan. La mancha unida se recoge en solución mediante la adición de 100 pl de tampón de Tris pH 8, seguido por 10 minutos en un agitador de placas (aproximadamente 500 rpm). La absorbancia a 540 nm en cada pocillo (que es proporcionar al número de células presente) se determina usando un lector de placas.
Después de promediar los valores sin analito en la columna 12, esto se sustrajo de todos los valores, y los resultados se expresaron como un porcentaje del valor no tratado (columna 11). Los 10 valores así derivados (por cuadruplicado) se representan frente al logaritmo de la concentración de fármaco, y se analizan mediante regresión no lineal para una ecuación logística de cuatro parámetros, fijando restricciones si es sugerido por una inspección. La GI 50 generada por este procedimiento es la concentración del fármaco que produce un porcentaje de control A540 a medio camino entre la saturación y la meseta de efecto cero.
Los resultados para una gama de líneas celulares se resumen posteriormente.






Estudios de Xenoinjertos
Para líneas celulares estándar, las células se inocularon subcutáneamente en suspensión (0.2 ml) en el costado de ratones hembra atímicos o con inmunodeficiencia combinada grave. Grupos de 7-8 ratones se asignaron al tratamiento siguiendo una asignación estratificada de volúmenes tumorales. El tratamiento con TBAP-01 comenzó entre los días 11-14 después de la administración de las células. Se administraron por sonda 200 pl de un suspensión (DMSO : agua, 1 : 19, v/v en 10 ml/kg). Los animales de control recibieron una dosificación similar de vehículo (DMSO : agua, 1 : 19, v/v). El tratamiento con TBAP-01 se continuó una vez al día durante 24 dosis.
Para xenoinjertos derivados de pacientes (PDX, por sus siglas en inglés), se recogió tejido reciente inmediatamente después de la cirugía en RPMI complementado con FBS al 10%. El tejido se transfirió a una placa de Petri estéril. Las partes necróticas del tumor se retiraron y un trozo de 5 x 5 x 5 mm se implantó subcutáneamente en el costado de un ratón Cb 17 NOD SCID. Cuando el tumor alcanzaba los límites de tamaño autorizados por el ministerio del interior, se extirpaba, y el tejido viable se disecó en cubos de 5 x 5 x 5 mm y se trasplantó en ratones Cb 17 NOD SCID adicionales usando el mismo procedimiento. Los análisis genómicos e histológicos conformaron que los tumores en cada punto se derivaban del material de partida. Después del trasplante, se dejó que el tumor RM-2 (LÍNEA 2) (mutante de BRAF, xenoinjerto derivado de paciente con resistencia intrínseca a vemurafenib), el tumor RM-17 (LÍNEA 3) (mutante de BRAF, xenoinjerto derivado de paciente resistente a una combinación de dabrafenib trametinib) y RM33S (tipo BRAF natural Ras natural de un paciente que es refractario a ipilimumab) crecieran hasta aproximadamente 50-60mm3 antes del inicio del tratamiento mediante sonda orogástrica diaria de TBAP-01 en 20 mg/kg/día o vehículo durante 24 o 17 días, respectivamente. La célula derivada de paciente LP2-CL2 (LÍNEA 1) (mutante de BRAF, derivado de un paciente que adquirió resistencia a vemurafenib en la clínica después de 3 meses de tratamiento) se estableció a partir de tejido reciente recogido después de la cirugía. Las células se hicieron crecer en RPMI complementado con FBS al 10%.
Los resultados se resumen en la siguiente tabla.


Estudios de Biomarcadores
Las células se inocularon subcutáneamente en suspensión (0.2 ml) en el costado de ratones atímicos hembra. Grupos de 3-6 ratones se asignaron al tratamiento con una sola dosis de compuesto de prueba (para los estudios de inmunotransferencia presentados en la Tabla 13) o 4 dosis diarias (para los estudios inmunohistoquímicos presentados en la Tabla 13) 14-21 días después de la administración de las células. Se administraron por sonda 200 pl de 40-50 mg/kg de suspensión de TBAP-01 en DMSO : agua. Los animales de control recibieron una dosificación similar de vehículo (DMSO : agua, 1 :19, v/v). Los tumores se recogieron 2-8 horas después de la dosificación y se sometieron a lisis en tampón de lisis NP40 al 1% (100 pl de tampón / 15 mg de tejido) usando un homogeneizador de tejidos (Precellys 24). El contenido de proteína total se midió usando el 660 nm Protein Assay (Pierce) y 40 pg de proteína total se cargaron en una SDS-PAGE para inmunotransferencia adicional. Se usaron anticuerpos para ERK2 (Santa Cruz Technologies), fosfo-MEK (Cell Signaling) y fosfo-ERK (Sigma) para la inmunotransferencia; la señal se reveló usando anticuerpos secundarios fluorescentes (Invitrogen y Li-cor) en el sistema Odissey (Li-cor). Alternativamente, los tumores se recogieron 1 hora después de la dosificación final al acabar la terapia (24 dosis diarias) y se procesaron de un modo similar al descrito anteriormente.
Inmunohistoquímica (IHC): Los tumores se fijaron con formalina y se prepararon como se describe en otras partes (véase p. ej., Dhomen y cols., 2009) para tinción con hematoxilina y eosina, pSRC (Invitrogen 44660G) y pERK (Cell Signaling 20G11) de conejo. Se incluyeron controles positivos y negativos en cada experimento. La puntuación del patrón y la intensidad de la tinción se realizó de un modo enmascarado.
Los datos se resumen en la siguiente tabla, que muestra el porcentaje de reducción de fosfo-MEK (ppMEK) y fosfo-ERK (ppERK), en comparación con los controles tratados con vehículo, para TBAP-01. pMEK y pERK se normalizan a ERK total en las muestras tratadas así como en las muestras de control.

Estudios Farmacocinéticos
Se usaron ratones BALB/cAnNCrl hembra de al menos 6 semanas de edad para los análisis farmacocinéticos. Los ratones fueron dosificados intravenosamente (2 mg/kg, en DMSO:Tween 20:agua 10:1:89 v/v) u oralmente por sonda. Se recogieron muestras en 7 u 8 momentos entre los 5 minutos y las 18 o 24 horas para la vía intravenosa y en 6 u 8 momentos entre los 15 minutos y las 18 o 24 horas para la vía oral. Se usaron tres ratones por momento por vía. Se sometieron a anestesia con halotano o isoflurano y se recogió sangre para la preparación de plasma mediante punción cardíaca terminal en jeringas heparinizadas. Las muestras se plasma se congelaron instantáneamente en nitrógeno líquido y a continuación se almacenaron a -70°C antes del análisis. Todos los procedimientos que involucraban a animales se realizaron según las regulaciones nacionales del Ministerio del interior bajo the Animals (Scientific Procedures) Act 1986 y dentro de las directrices indicadas por the Institute’s Animal Ethics Committee y the United Kingdom Coordinating Committee for Cancer Research’s ad hoc Committee on the Welfare of Animals in Experimental Neoplasia.
Los datos se resumen en la siguiente tabla.

Inhibición de hERG
Se efectuaron estudios en Cyprotex Discovery en Cheshire, Reino Unido, según el protocolo del contratista. Los estudios se realizaron en un instrumento IonWorks™ HT (Molecular Devices Corporation), que realiza automáticamente medidas electrofisiológicas en 48 células individuales simultáneamente en una placa de 384
pocilios especializada (PatchPlate™). La células usadas eran células de ovario de hámster chino (CHO, por sus siglas en inglés) transfectadas establemente con hERG (línea celular obtenida de Cytomyx, Reino Unido). Se preparó una suspensión de células individuales en solución extracelular (solución salina tamponada con fosfato de Dulbecco con calcio y magnesio pH 7-7.2) y se añadieron automáticamente partes alícuotas a cada pocillo de una PatchPlate™. A continuación, las células se colocaron sobre un pequeño hueco en el fondo de cada pocillo al aplicar un vacío debajo de la placa para formar una junta eléctrica. El vacío se aplicó a través de un solo compartimento común a todos los pocillos que se rellenó con solución intracelular (tamponada hasta pH 7.2 con HEPES). La resistencia de cada junta se midió a través de un electrodo con toma de tierra común en el compartimento intracelular y se pusieron electrodos individuales en cada uno de los pocillos superiores.
El acceso eléctrico a la célula se alcanzó a continuación al hacer circular un agente de perforación, anfotericina, por debajo de la PatchPlate™ y a continuación medir la corriente de hERG antes del compuesto. Se colocó un electrodo en el compartimento extracelular y se aplicó un potencial de mantenimiento de -80 mV aplicado durante 15 segundos. Los canales de hERG se activaron a continuación al aplicar una etapa de despolarización hasta 40 mV durante 5 segundos y a continuación se pinzaron a -50 mV durante 4 segundos para provocar la corriente de cola de hERG, antes de volver a -80 mV durante 0.3 segundos. A continuación, el compuesto de prueba se añadió automáticamente a los pocillos superiores de la PatchPlate™ desde una placa de microvaloración de 96 pocillos que contenía un intervalo de concentraciones de compuesto TBAP-01. Se incluyó quinidina, un inhibidor de hERG establecido, como un control experimental. Se disolvió TBAP-01 en DMSO y se ensayó en concentraciones finales que variaban de 100 pM a 32 nM en DMSO al 0.25%. Se incluyó tampón que contenía DMSO al 0.25% como un control negativo. El compuesto de prueba se dejó en contacto con las células durante 300 segundos antes de registrar las corrientes usando el mismo protocolo de etapas de voltaje que en el barrido anterior al compuesto. Cada concentración se probó en 4 pocillos repetidos.
Las corrientes posteriores al compuesto se expresaron como un porcentaje de las corrientes anteriores al compuesto y se representaron frente a la concentración de cada compuesto. Cuando se observaba inhibición dependiente de la concentración, los datos se ajustaron a la siguiente ecuación:
y = ( ymáx - ymín ) / ( 1 ( x / x50 ) ) ymín.
en la que:
y = (corriente posterior al compuesto / corriente anterior al compuesto) x 100;
x = concentración;
x50 = concentración requerida para inhibir la corriente en 50% (IC50); y
s = pendiente de la gráfica.
Los datos se resumen en la siguiente tabla.


Actividad Contra Otras Dianas
Se efectuaron estudios en Life Technologies en Paisley según el protocolo del contratista. Se disolvió TBAP-01 en DMSO y se ensayó en concentraciones finales que variaban de 10 pM a 0.5 nM en DMSO al 1%, en presencia de una concentración de ATP de 100 pM. Los valores de IC50 para los compuestos de prueba se determinaron usando el ensayo bioquímico Z'-LYTE® empleando un formato asociado a enzimas basado en fluorescencia, basado en la sensibilidad diferencial de péptidos fosforilados y no fosforilados a la escisión proteolítica.
Se efectuaron estudios adicionales en The International Centre for Kinase Profiling en Dundee según el protocolo del contratista. Se disolvió TBAP-01 en DMSO y se ensayó a una concentración final de 1 pM en DMSO al 2% frente a 131 cinasas. Los ensayos se llevaron a cabo usando un ensayo radiactivo de unión al filtro (33P-ATP).
Los datos se resumen en las siguientes tablas.

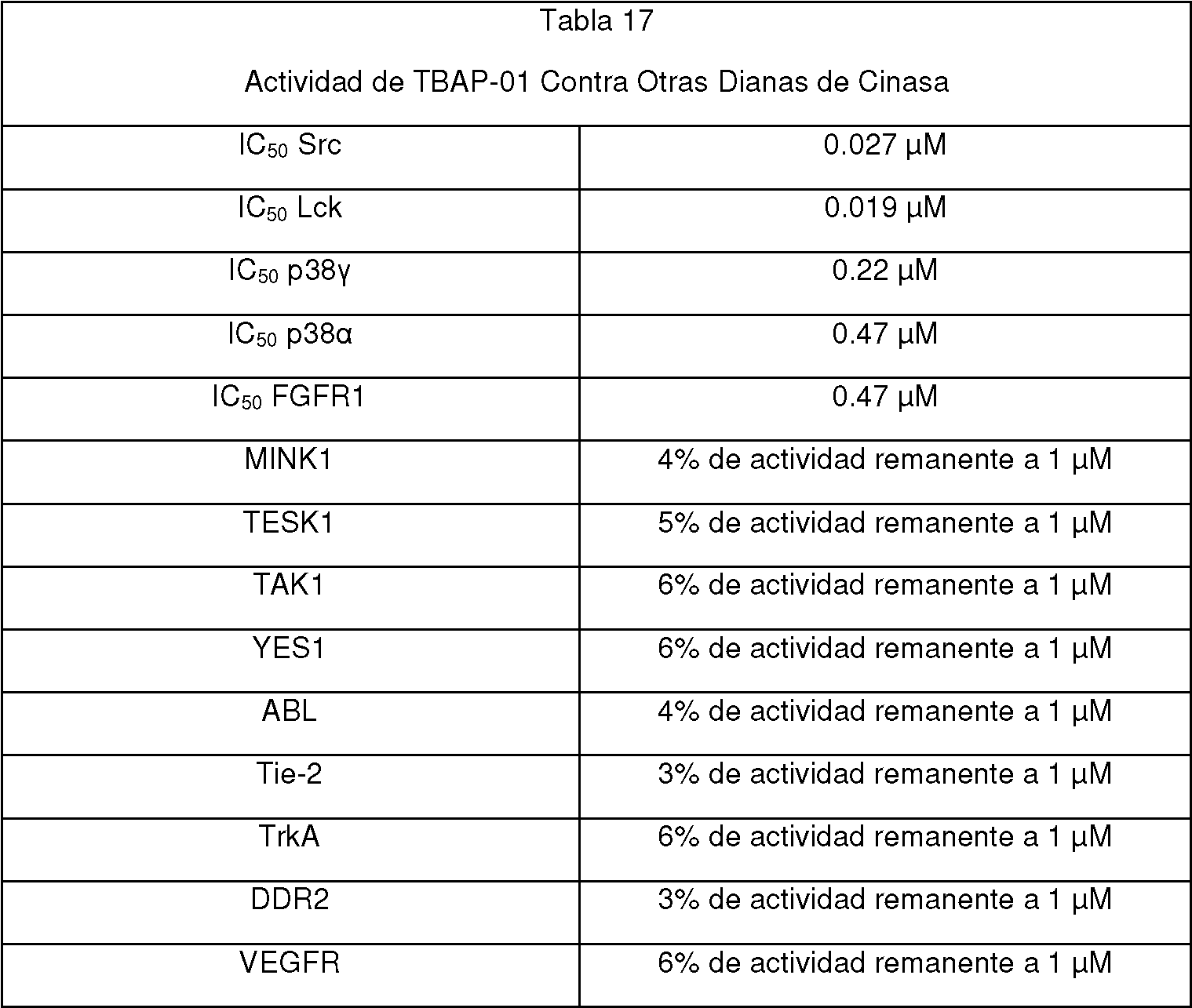
Nótese, p. ej., que se sabe bien que: TAK1 es una diana en cánceres tales como linfoma y cáncer colorrectal y pancreático; TrkA es una diana en cáncer de pulmón y de mama; DDR2 es una diana en cánceres tales como cáncer pulmonar de células escamosas; VEGFR y Tie-2 son dianas antiangiogénicas; ABL es una diana en la leucemia; y YES1 es una diana en cánceres tales como melanoma y cáncer de mama.
Actividad Antiviral
La actividad antiviral de los compuestos contra la infección por el virus de la encefalomiocarditis (ECMV, por sus siglas en inglés) ATCC® VR-129B de células HepG2; la infección por el virus de herpes simples HSV-1, SC16 de células Vero; y virus de la gripe A, A/Panama/2007/99 (H3N2) en células MDCK; se evaluó en KWSBiotest (Bristol, Reino Unido), según el protocolo del contratista.
Las células que la replicación viral se hicieron crecer hasta números suficientes en medio de crecimiento con complementos. Una vez que las células eran confluentes, se sembraron en placas de fondo plano de 96 pocillos. Para la determinación de la EC50 (Concentración Eficaz, 50%), el medio se retiró y los compuestos se añadieron a
10 veces la concentración final en DMSO al 0.4% 10 minutos antes de la infección viral. Una hora después de la infección, se añadió a los pocillos medio superpuesto para dar 1 vez la concentración de los compuestos a lo largo de la duración del estudio. Los pocillos con vehículo y control positivo se establecieron para controlar cualquier influencia sobre la viabilidad celular. Para la determinación de CC50 (Concentración Citotóxica, 50%), se siguió el mismos procedimiento, excepto que sólo se añadía medio en lugar de inóculo viral.
A continuación, los pocillos individuales se evaluaron usando el ensayo de MTT, que es un ensayo colorimétrico cuantitativo para la supervivencia de células de mamífero. Las células se incubaron durante 3 horas con solución de 1 mg/ml de MTT. A continuación, se determinó la intensidad cromático al cuantificar la absorbancia a la longitud de onda apropiada. El resultado proporciona una indicación de la eficacia antiviral de cada compuesto como una EC50, así como una CC50 para mostrar cualquier efecto citotóxico de los compuestos sobre las células en ausencia de infección viral. Los pocillos infectados viralmente también se inspeccionaron visualmente para cualquier CPE o formación de sincitios.
Concentración Eficaz (EC50): La capacidad de los compuestos para reducir la muerte celular inducida por virus se evaluó usando el ensayo colorimétrico de MTT para la supervivencia de células de mamífero. El resultado del ensayo se cuantificó usando un lector de placas de ELISA, y se determinó la EC50 para cada uno de los compuestos que se evalúan con cada virus. Los resultados se presentaron gráficamente junto con el error estándar de la media (SEM, por sus siglas en inglés) para cada. Se calculó la significación estadística de la eficacia de cada compuesto. Concentración Citotóxica (CC50): El efecto citotóxico de los compuestos se evaluó usando el ensayo colorimétrico de MTT para la supervivencia de células de mamífero. El resultado del ensayo se cuantificó usando un lector de placas de ELISA, y se determinó la CC50 para cada uno de los compuestos que se evalúan. Los resultados se presentaron gráficamente junto con el error estándar de la media (SEM) para cada grupo. Se calculó la significación estadística de la eficacia de cada compuesto.
Liberación de TNF-a Estimulada por LPS de Células Mononucleares de Sangre Periférica (PBMC, por sus siglas en inglés)Humanas
El factor de necrosis tumoral-a (TNF-a), una citocina secretada de 17 kDa, representa un papel importante en enfermedades inflamatorias y trastornos inmunitarios. El TNF-a es secretado principalmente por macrófagos activados (véase, p. ej., Shakhov y cols., 1990) y monocitos (véase, p. ej., Yao y cols., 1997) en respuesta a varios estímulos inflamatorios e inmunológicos. Por ejemplo, durante la infección bacteriana, el lipopolisacárido (LPS), un componente de la pared celular bacteriana gramnegativa, induce la liberación de TNF-a (véase, p. ej., Martich y cols., 1991).
La sobreproducción de citocinas inflamatorias, tales como TNF-a, se ha relacionado con trastornos inflamatorios tales como la enfermedad de Crohn (CD, por sus siglas en inglés) y la enteropatía inflamatoria (véase, p. ej., Kam y cols., 2000; Nakamura y cols., 2006), la artritis reumatoide (véase, p. ej., Keffer y cols., 1991; McCann y cols., 2010), el choque séptico (véase, p. ej., Link y cols., 2008; Shapira y cols., 1996), el asma (véase, p. ej., Berry y cols., 2007), la bronquitis crónica (CB, por sus siglas en inglés), la enfermedad pulmonar obstructiva crónica (COPD), la lesión pulmonar aguda (ALI) y el síndrome de dificultad respiratoria agudo (ARDS) (véase, p. ej., Mukhopadhyay y cols., 2006). La reducción de los niveles de TNF-a se ha asociado con una mejora en estas afecciones.
La actividad del compuesto TBAP-01 en la liberación de TNFa estimulada por LPS de células mononucleares de sangre periférica (PBMC) humanas se determinó en Argenta/Charles River, Cowley, Oxford, según el protocolo del contratista. Las PBMC se aislaron de sangre de voluntarios humanos sanos usando una técnica de centrifugación con gradiente de densidad estándar. Las PBMC se suspendieron en medio y se aportaron a una placa de 96 pocillos y se incubaron a 37°C durante 3 horas en una incubadora humidificada. Después de la incubación, el medio se reemplazó y el compuesto de prueba, el compuesto de referencia (BIRB796) o el vehículo apropiado se añadieron a las células y la placa se incubó a 37°C durante 1 hora. Después de la incubación, se añadieron LPS (E coli 0111:84, 10 ng/ml) o un control de vehículo apropiado a las células a continuación y la placa se devolvió a la incubadora para la incubación durante la noche. Después de la incubación, la placa se centrifugó a 300 x g durante 4 minutos a temperatura ambiente. Los sobrenadantes libres de células se retiraron y se almacenaron (congeladas) hasta que se ensayaban con respecto a los niveles de TNF-a usando un estuche EUSA disponible comercialmente (R&D Systems).
El compuesto de prueba se disolvió en DMSO y se almacenaron congeladas partes alícuotas. Una parte alícuota separada se usó para cada experimento. Para cada experimento, el compuesto de prueba se diluyó en DMSO (hasta 1.000 veces la concentración de ensayo final), y a continuación se diluyó en medio de cultivo celular para dar las concentraciones requeridas mientras se mantenía una concentración constante de DMSO (concentración final de DMSO al 0.1% en el ensayo).
Se realizó una curva de respuesta a la dosis de 8 puntos, con tres experimentos separados (n=3). El efecto del compuesto de prueba en cada experimento se expresó como un porcentaje de inhibición de la respuesta estimulada por LPS. Los datos del porcentaje de inhibición para cada compuesto de prueba en cada experimento se reunieron para determinar un solo valor de IC50 para cada compuesto de prueba.
Se encontró que el compuesto TBAP-01 exhibía una potente inhibición usando este ensayo, con una IC50 de 3.4 nM y un intervalo de confianza al 95% de 2.0-5.7 nM.
Datos de Comparación - 1
Datos para TBAP-01 y compuestos conocidos estructuralmente relacionados (AA-04 en Springer y cols., 2011; y AA-018 , AA-019, AA-062, AA-084 en Springer y cols., 2009) se resumen posteriormente.





Datos de Comparación - 2
En comparación con el Compuesto AA-04 en Springer y cois., 2011, TBAP-01 es: (a) 10 veces más potente en el ensayo de cinasa BRAF;
(b) 8 veces más potente en el ensayo celular de pERK; y
(c) 5 veces más potente en el ensayo de inhibición de la proliferación celular.

Datos de Comparación - 3
En comparación con el Compuesto AA-018 en Springer y cois., 2009, TBAP-01 es:
(a) 7 veces más eficaz en xenoinjerto de melanoma por BRAF mutante A375M a la dosis eficaz máxima; y
(b) 2 veces más biodisponibilidad oral.

Datos de Comparación - 4
En comparación con el Compuesto AA-019 en Springer y cols., 2009, TBAP-01 es:
(a) tolerado in vivo en dosis 2 veces superiores (es decir, 40-50 mg/kg), a pesar de tener una Cmáx y un AUC superiores que AA-019 a la misma dosis;
(b) hasta 16 veces más potente en la inhibición de la proliferación celular en líneas celulares vírgenes o resistentes a fármacos aprobados mutantes en BRAF y líneas celulares de RAS mutante;
(c) 2 veces más soluble (es decir, tiene una solubilidad termodinámica 2 veces superior);
(d) 2 veces más eficaz en xenoinjerto de melanoma con BRAF mutante A375M y xenoinjerto de melanoma con BRAF mutante WM266.4 a la dosis eficaz máxima;
(e) 1.2 veces más eficaz sobre xenoinjerto colorrectal con RAS mutante SW620 a la dosis eficaz máxima;
(f) 1.2-1.5 veces más eficaz sobre xenoinjerto de melanoma con BRAF mutante derivado de un paciente resistente a vemurafenib RM-2 (LÍNEA 2) y xenoinjerto de melanoma con BRAF mutante A375R a la dosis eficaz máxima; y (g) 2.4 veces más eficaz sobre xenoinjerto de melanoma con BRAF mutante derivado de un paciente resistente a dabrafenib+trametinib RM-17 (LÍNEA 3) a la dosis eficaz máxima.
(h) >6.5 veces más eficaz en la inhibición del biomarcador pERK en el aloinjerto de PDAC R172H pancreático a la dosis eficaz máxima.
(i) 2.5 veces más eficaz en la inhibición del biomarcador pSRC en el aloinjerto PDAC R172H pancreático a la dosis eficaz máxima.

Datos de Comparación - 5
En comparación con el Compuesto AA-062 en Springer y cols., 2009, TBAP-01 es 9 veces más eficaz sobre xenoinjerto de melanoma con BRAF mutante A375M a la dosis eficaz máxima. Esto es sorprendente e inesperado debido a que AA-062 tiene una Cmáx 5 veces superior y una AUC 12 veces superior en comparación con TBAP-01.

Datos de Comparación - 6
En comparación con el Compuesto AA-084 en Springer y cols., 2009, TBAP-01 es:
(a) 11 veces más potente sobre el ensayo de cinasa BRAF;
(b) 8 veces más potente sobre el ensayo celular de pERK; y
(c) 5 veces más potente sobre el ensayo de inhibición de la proliferación celular.

REFERENCIAS
Se cita en la presente un número de publicaciones a fin de describir y divulgar más a fondo la invención y el estado de la técnica de la que trata la invención. Las citas completas de estas referencias se proporcionan posteriormente. Akula y cols., 2004, “Raf promotes human herpesvirus-8 (HHV-8/KSHV) infection”, Oncogene, Vol. 23, pp. 5227-5241.
Alitalo y cols., 1996, “Promoter for the receptor tyrosine kinase, Tie”, Patente de EE. UU. N° 5.877.020 concedida el 2 de marzo de 1999.
Alon y cols., 1995, “Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity”, Nature Med., Vol. 1, pp. 1024-1028.
Anastasaki y cols., 2012, “Continual low-level MEK inhibition ameliorates cardio-facio-cutaneous phenotypes in zebrafish”, Disease Models & Mechanisms, Vol. 5, pp. 546-552.
Antony y cols., 2013, “C-RAF Mutations Confer Resistance to RAF Inhibitors”, Cancer Research, Vol. 73, pp. 4840-4851.
Arcaini y cols., 2012, “The BRAF V600E mutation in hairy cell leukemia and other mature B-cell neoplasms”, Blood, Vol. 119, pp. 188-191.
Asrih y cols., 2013, “Role of Mitogen-Activated Protein Kinase Pathways in multifactorial adverse cardiac remodeling associated with Metabolic Syndrome”, Mediators of Inflammation, Vol. 2013, Article ID No. 367245.
Badalian-Very y cols., 2011, “Recent advances in the understanding of Langerhans cell histiocytosis”, British Journal of Haematology. Vol. 156, pp. 163-172.
Belgore y cols., 2004, “Localisation of members of the vascular endothelial growth factor (VEGF) family and their receptors in human atherosclerotic arteries”, J. Clin. Pathol., Vol. 57, pp. 266-272.
Benn y cols., 1994, “Hepatitis B virus HBx protein activates Ras-GTP complex formation and establishes a Ras, Raf, MAP kinase signaling cascade”, PNAS, Vol. 91, pp. 10350-10354.
Berry y cols., 2007, “TNF-alpha in asthma”, Curr. Opin. Pharmacol., Vol. 7, No. 3, pp. 279-282.
Bos y cols., 1989, “Ras oncogenes in human cancer: a review”, Cancer Research, Vol. 49, pp. 4682-4689.
Bridges y cols., 2000, “Treatment of asthma with MEK inhibitors ”, publicación de solicitud de patente internacional (PCT) número WO 00/40235, publicada el 13 de julio de 2000.
Byeon y cols., 2012, “The role of Src kinase in macrophage-mediated inflammatory responses”, Mediators of Inflammation, Vol. 2012, Article ID No. 512926.
Calhoun y cols., 2003, “BRAF and FBXW7 (CDC4, FBW7, AGO, SEL10) Mutations in Distinct Subsets of Pancreatic Cancer”, American Journal of Pathologv, Vol. 163, pp. 1255-1260.
Cantin y cols., 2007, “Pirazolyl urea derivatives useful in the treatment of cancer”, publicación de solicitud de patente internacional (PCT) número WO 2007/059202 A2, publicada el 24 de mayo de 2007.
Cantrell, 2003, “GTPases and T cell activation”, Immunol. Rev., Vol. 192, pp. 122-130.
Chan y cols., 1996, “Regulation of antigen receptor signal transduction by protein tyrosine kinases”, Curr.
Opin.ImmunoL Vol. 8, No. 3, pp. 394-401.
Chapman y cols., 2011, “Improved survival with vemurafenib in melanoma with BRAF V600E mutation”, New England Journal of Medicine, Vol. 364, pp. 2507-2516.
Chapman y cols., 2011, “Initial genome sequencing and analysis of multiple myeloma”, Nature, Vol. 471, pp. 467-472.
Chun y cols., 2002, “Pharmaceutical composition for prevention and treatment of joint arthritis and a screening method thereof”, publicación de patente internacional número WO 02/102367, publicada el 27 de febrero de 2002.
Ciampi y cols., 2005, “Oncogenic AKAP9-BRAF fusion is a novel mechanism of MAPK pathway activation in thyroid cancer”, J. Clin. Invest., Vol. 115, pp. 94-101.
Colville-Nash and Scott, 1992, “Angiogenesis and rheumatoid arthritis: pathogenic and therapeutic implications”, Ann. Rhum. Dis., Vol. 51, p. 919.
Cooper y cols., 1994, “Membrane-associated tyrosine kinases as molecular switches”, Semin. Cell Biol.. Vol. 5, No. 6, pp. 377-387.
Corcoran y cols., 2010, “BRAF Gene Amplification Can Promote Acquired Resistance to MEK Inhibitors in Cancer Cells Harboring the BRAF V600E Mutation”, Sci. Signal., Vol. 3, ra84.
Coulthard y cols., 2009, “p38MAPK: stress responses from molecular mechanisms to therapeutics”, Trends Mol.
Med., Vol. 15, pp. 369-379.
Courtneidge y cols., 1993, “The Src family of protein tyrosine kinases: regulation and functions”, Dev. Suppl., pp. 57 64.
Cuadrado y cols., 2010, “Mechanisms and functions of p38 MAPK signalling”, Biochem. J., Vol. 429, pp. 403-417. Damodar Reddy y cols., 2001, “Role of MAP kinase pathways in primitive neuroectodermal tumours”, Anticancer Research, Vol. 21, pp. 2733-2738.
Davies y cols., 1996, “Raf and Mitogen-activated Protein Kinase Regulate Stellate Cell Collagen Gene Expression”, J. Biol. Chem., Vol. 271, pp. 11039-11042.
Davies y cols., 2002, “Mutations of the BRAF gene in human cancer”, Nature, Vol. 417, pp. 949-954.
Davis y cols., 1994, “Tie-2 ligand 1 ”, Patente de EE. UU. N° 5.879.672 concedida el 09 de marzo de 1999.
Davis y cols., 1994, “TIE-2 ligand, and method of making”, Patente de EE. UU. N° 5,521,073 concedida el 28 de mayo de 1996.
Davis y cols., 1996, “Isolation of Angiopoietin-1, a Ligand for the TIE2 Receptor, by Secretion-Trap Expression Cloning”, CeJJ, Vol. 87, pp. 1161-1169.
Denekamp, 1993, “Angiogenesis, neovascular proliferation and vascular pathophysiology as targets for cancer therapy”, Br. J. Rad., Vol. 66, pp. 181 -196.
Dhomen y cols., 2009, “Oncogenic Braf induces melanocyte senescence and melanoma in mice”, Cancer Cell, Vol. 15, pp. 294-303.
Dixon y cols., 2001, “Method for treating chronic pain using MEK inhibitors”, publicación de solicitud de patente internacional (PCT) número WO 01/05392, publicada el 25 de enero de 2001.
Downward y cols., 2003, “Targeting RAS signalling pathways in cancer therapy”, Nature Reviews Cancer, Vol. 3, pp. 11-22.
Dudley y cols., 2000, “Treatment of arthritis with MEK inhibitors”, publicación de solicitud de patente internacional (PCT) número WO 00/35436, publicada el 22 de junio de 2000.
Ellis y cols., 2008, “VEGF-targeted therapy: mechanisms of anti-tumour activity”, Nature Reviews Cancer, Vol. 8, pp. 579-591.
Falchook y cols., 2012, “Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours:
a phase 1 dose-escalation trial”, The Lancet, Vol. 379, pp. 1893-1901.
Fernandes-Medarde, 2011, “Ras in Cancer and Developmental Diseases”, Genes & Cancer, Vol. 2, pp. 344-358. Fidler y Ellis, 1994, “The implications of angiogenesis for the biology and therapy of cancer metastasis”, Cell, Vol. 79, pp. 185-188.
Flaherty y cols., 2010, “Inhibition of mutated, activated BRAF in metastatic melanoma”, New England Journal of Medicine, Vol. 363, pp. 809-819.
Flynn y cols., 2008, “Enzyme modulators and treatments”, publicación se solicitud de patente de EE. UU. número US 2008/0113967 A1, publicada el 15 de mayo de 2008.
Folkman y cols., 1992, “Angiogenesis”, J. Biol. Chem., Vol. 267, pp. 10931-10934.
Folkman, 1992, “The role of angiogenesis in tumour growth”, Semin. Cancer Biol., Vol. 3, pp. 65-71.
Folkman, 1995, “Angiogenesis in cancer, vascular, rheumatoid and other disease”, Nature Medicine, Vol. 1, pp. 27-31.
Folkman, 1997, “Angiogenesis and angiogenesis inhibition: an overview”, EXS, Vol. 79, pp. 1-81.
Friedlander y cols., 1995, “Definition of two angiogenic pathways by distinct alpha v integrins”, Science, Vol. 270, pp. 1500-1502.
Fujita y cols., 2004, “ERK and p38 mediate high-glucose-induced hypertrophy and TGF-a expression in renal tubular cells”, Am. J. Physiol. Renal. Physiol., Vol. 286, p. F120.
Fukuda y cols., 2007, “Epstein-Barr Virus Latent Membrane Protein 2A Mediates Transformation through Constitutive Activation of the Ras/PI3-K/Akt Pathway”, J. Virol., Vol. 81, pp. 9299-9306.
Furuta y cols., 2012, “Nitrogenated aromatic heterocyclic ring derivative”, publicación de solicitud de patente internacional (PCT) número WO 2012/008564 A1 , publicada el 19 de enero de 2012.
Garnett y cols., 2004, “Guilty as charged: B-RAF es a human oncogene”, Cancer Cell, Vol. 6, pp. 313-319.
Gaudi y cols., 2011, “Molecular Bases of Cutaneous and Uveal Melanomas”, Pathology Research International, Vol. 2011, ID Artículo N2159421.
Genot y cols., 2000, “Ras regulation and function in lymphocytes”, Curr. Opin. Immunol., Vol. 12, pp. 289-294.
Geppert y cols., 1994, “Lipopolysaccharide signals activation of Tumour Necrosis Factor biosíntesis through the Ras/Raf-1/MEK/MAPK pathway”, Mol. Med., Vol. 1, pp. 93-103.
Gilbertsen y cols., 2000, “Use of a MEK inhibitor for preventing transplant rejection”, publicación de solicitud de patente internacional (PCT) número WO 00/35435, publicada el 22 de junio de 2000.
Girotti y cols., 2013, “Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma”, Cancer Discoverv. Vol. 3, pp. 158-167.
Godowski y cols., 1997, “Tie ligand homologues”, Patente de EE. UU. N° 6.030.831 concedida el 29 de febrero de 2000.
Graf y cols., 1997, “Mitogen-Activated Protein Kinase Activation es Involved in Platelet-Derived Growth Factor-Directed Migration by Vascular Smooth Muscle Cells”, Hypertension, Vol. 29, pp. 334-339.
Gray-Schopfer y cols., 2007, “Melanoma biology and new targeted therapy”, Nature, Vol. 445, pp. 851 -857.
Greger y cols., 2012, “Combinations of BRAF, MEK, and PI3K/mTOR Inhibitors Overcome Acquired Resistance to the BRAF Inhibitor GSK2118436 Dabrafenib, Mediated by NRAS or MEK Mutations”, Molecular Cancer Therapeutics, Vol. 11, pp. 909-920.
Grosios y cols., 2004, “Angiogenesis inhibition by the novel VEGF receptor tyrosine kinase inhibitor, PTK787/ZK222584, causes significant anti-arthritic effects in models of rheumatoid arthritis”, Inflamm. Res., Vol. 53, pp. 133-142.
Gu y cols., 2013, “Use of organic compound for the treatment of Noonan syndrome”, publicación de solicitud de patente internacional (PCT) número WO 2013/033133, publicada el 07 de marzo de 2013.
Haase y cols., 2001, “A role for mitogen-activated protein kinase activation by integrins in the pathogenesis of psoriasis”, J. Clin. Invest., Vol. 108, pp. 527-536.
Haroche y cols., 2012, “High prevalence of BRAF V600E mutations in Erdheim-Chester disease but not in other non-Langerhans cell histiocytoses”, Blood, Vol. 120, pp. 2700-2703.
Hatzivassiliou y cols., 2011, “Determining sensitivity of cells to B-Raf inhibitor treatment by detecting K-ras mutation and RTK expression levels”, publicación de solicitud de patente internacional (PCT) número WO 2011/028540 publicada el 10 de marzo de 2011.
Hatzivassiliou y cols., 2011, “Determining sensitivity of cells to B-Raf inhibitor treatment by detecting K-ras mutation and RTK expression levels”, publicación de solicitud de patente internacional (PCT) número WO 2011/028540 publicada el 10 de marzo de 2011.
Heidorn y cols., 2010, “Kinase-dead BRAF and oncogenic RAS cooperate to drive tumour progression through CRAF”, CeJ], Vol. 140, pp. 209-221.
Hu y cols., 2011, “Mutation that blocks ATP binding creates a pseudokinase stabilizing the scaffolding function of kinase suppressor of Ras, CRAF and BRAF”, PNAS, Vol. 18, pp. 6067-6072.
Hwang y cois., 2004, “Over-expression of c-raf-1 proto-oncogene in liver cirrhosis and hepatocellular carcinoma”, Hepatoloav Research, Vol. 29, pp. 113-121.
Ingber y cols., 1990, “Synthetic analogues of fumagillin that inhibit angiogenesis and suppress tumour growth”, Nature, Vol. 348, pp. 555-557.
Inoue y cols., 1998, “Vascular endothelial growth factor (VEGF) expression in human coronary atherosclerotic lesions:
possible pathophysiological significance of VEGF in progression of atherosclerosis”, Circulation, Vol. 98, pp. 2108-2116.
Jaffee y cols., 2000, “Inhibition of MAP Kinase Kinase (MEK) Results in an Anti-inflammatory Response in Vivo”, Biochem. Biophys. Res. Com., Vol. 268, p. 647.
Jessen y cols., 2013, “MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumours”, Journal of Clinical Investigation, Vol. 123, pp. 340-347.
Ji y cols., 2002, “ERK MAP Kinase Activation in Superficial Spinal Cord Neurons Induces Prodynorphin and NK-1 Upregulation and Contributes to Persistent Inflammatory Pain Hypersensitivity”, J. Neurosci., Vol. 22, p. 478. Jo y cols., 2005, “MEK inhibitor, U0126, attenuates cisplatin-induced renal injury by decreasing inflammation and apoptosis, Kidney Intl., Vol. 67, pp. 458-466.
Johnson y cols., 2001, “The role of MKK1/2 kinase activity in human cytomegalovirus infection”, J. Gen. Virol., Vol. 82, pp. 493-497.
Kahlon y cols., 1992, “Angiogenesis in atherosclerosis”, Can. J. Cardiol., Vol. 8, p. 60.
Kam y cols., 2000, “TNF-alpha antagonists for the treatment of Crohn's disease”, Expert Opin. Pharmacother., Vol. 1, pp. 615-622.
Karim y cols., 2006, “Impaired inflammatory pain and thermal hyperalgesia in mice expressing neuron-specific dominant negative mitogen activated protein kinase kinase (MEK)”, Mol. Pain., Vol. 2, p. 2.
Keffer y cols., 1991, “Transgenic mice expressing human necrosis factor: a predictive genetic model of arthritis”, EMBO J., Vol. 10, No. 13, pp. 4025-4031.
Kjetil y cols., 2013, “Métodos and compositions for inhibition of activation of regulatory T cells”, publicación de solicitud de patente internacional (PCT) número WO 2013/001372, publicada el 03 de enero de 2013.
Kotoula y cols., 2009, “Mutational analysis of the BRAF, RAS and EGFR genes in human adrenocortical carcinomas”, Endocrine-Related Cancer, Vol. 16, pp. 565-572.
Li y cols., 1998, “Activation of NF-kB via a Src-dependent Ras-MAPK-pp90rsk pathway is required for Pseudomonas aeruginosa-induced mucin overproduction in epithelial cells”, PNAS, Vol. 95, pp. 5718-5723.
Lin y cols., 2010, “VEGF and its receptor-2 involved in neuropathic pain transmission mediated by P2X2/3 receptor of primary sensory neurons”, Brain Research Bulletin, Vol. 83, pp. 284-291.
Lindauer y cols., 2012, “Dasatinib”, Recent Results in Cancer Research, Vol. 184, pp. 83-102.
Link y cols., 2008, “Phosphodiesterase 4 inhibition but not beta-adrenergic stimulation suppresses tumor necrosis factor-alpha release in peripheral blood mononuclear cells in septic shock”, Crit. Care, Vol. 12, No. 6, R159. Long y cols., 2011, “Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma”, J.
Clin. Oncol., Vol. 29, N210, pp. 1239-1246.
Lorenz y cols., 2009, “Cardiac hypertrophy: Targeting Raf/MEK/ERK1/2-signaling”, The International Journal of Biochemistry& Cell Biology. Vol. 41, pp. 2351-2355.
Lowenberg y cols., 2005, “Specific Inhibition of c-Raf Activity by Semapimod Induces Clinical Remission in Severe Crohn’s Disease”, J. Immunol., Vol. 175, pp. 2293-2300.
Luo y cols., 2002, “Coxsackievirus B3 Replication es Reduced by Inhibition of the Extracellular Signal-Regulated Kinase (ERK) Signaling Pathway”, J. Virol., Vol. 76, pp. 3365-3373.
Ma y cols., 2005, “The ERK/MAPK pathway, as a target for the treatment of neuropathic pain”, Expert Opin Ther Targets, Vol. 9, p. 699.
Maddahi y cols., 2010, “Cerebral ischemia induces microvascular pro-inflammatory cytokine expression via the MEK/ERK pathway, J. Neuroinflammation, Vol. 7, pp. 1-14.
Mammas y cois., 2005, “Involvement of the ras genes in female genital tract cáncer (Review)”, International Journal of Oncoloav. Vol. 26, pp. 1241-1255.
Martich y cols., 1991, “Detection of interleukin 8 and tumor necrosis factor in normal humans after intravenous endotoxin: the effect of antiinflammatory agents”, J. Exp. Med., Vol. 173, pp 1021-1024.
Martin y cols., 2010, “Update on lymphocyte specific kinase inhibitors: a patent survey”, Expert Opin. Ther. Pat., Vol. 20, pp. 1573-1593.
Masabumi y cols., 2013, “Vascular endothelial growth factor and its receptor system: physiological functions in angiogenesis and pathological roles in various diseases”, J. Biochem., Vol. 153, pp. 13-19.
McCann y cols., 2010, “Apremilast, a novel PDE4 inhibitor, inhibits spontaneous production of tumour necrosis factoralpha from human rheumatoid synovial cells and ameliorates experimental arthritis”, Arthritis Res. Ther., Vol.
12, No. 3, R107.
McKay y cols., 2011, “Complexity in KSR function revealed by Raf inhibitor and KSR structure studies”, Small GTPases, Vol. 2, pp. 276-281.
McMahon y cols., 2000, “VEGF receptor signaling in tumour angiogenesis”, The Oncoloaist. Vol. 5, pp. 3-10.
Mei y cols., 2006, “Distribution, levels and phosphorylation of Raf-1 in Alzheimer’s disease”, J. Neurochem., Vol. 99, pp. 1377-1388.
Mercer y cols., 2006, “Emerging role of MAP kinase pathways as therapeutic targets in COPD”, Int. J. of COPD, Vol. 1, pp. 137-150.
Metzner y cols., 2012, “Fibroblast Growth Factor Receptors as Therapeutic Targets in Human Melanoma: Synergism with BRAF Inhibition”, Journal of Investigative Dermatoloav. Vol. 131, pp. 2087-2095.
Meyers y cols., 1996, “FGFR2 exon IIIa and IIIc mutations in Crouzon, Jackson-Weiss, and Pfeiffer syndromes:
evidence for missense changes, insertions, and a deletion due to alternative RNA splicing”, Am. J. Hum. Genet., Vol. 58, pp. 491-498.
Milella y cols., 2001, “Therapeutic targeting of the MEK/MAPK signal transduction module in acute myeloid leukemia”, Journal of Clinical Investigation, Vol. 108, pp. 851-859.
Miura y cols., 2004, “Simvastatin suppresses coronary artery endothelial tube formation by disrupting Ras/Raf/ERK signaling”, Atherosclerosis, Vol. 175, pp. 235-243.
Montagut y cols., 2008, “Elevated CRAF as a Potential Mechanism of Acquired Resistance to BRAF Inhibition in Melanoma”, Cancer Research, Vol. 68, pp. 4853-4861.
Mukherjee y cols., 2005, “Raf-1 expression may influence progression to androgen insensitive prostate cancer”, Prostate, Vol. 64, pp. 101-107.
Mukhopadhyay y cols., 2006, “Role of TNFalpha in pulmonary pathophysiology”, Respir. Res., Vol. 7, p. 125.
Murray y cols., 2011, “Respiratory formulations and compuestos for use therein”, publicación de solicitud de patente internacional (PcT) número WO 2011/158044 A2, publicada el 22 de diciembre de 2011.
Mustonen y cols., 1995, “Endothelial receptor tyrosine kinases involved in angiogenesis”, J. Cell Biol., Vol. 129, pp. 895-898.
Nakamura y cols., 2006, “Novel strategies for the treatment of inflammatory bowel disease: Selective inhibition of cytokines and adhesion molecules”, World J. Gastroenterol., Vol. 12, pp. 4628-4235.
Nazarian y cols., 2010, “Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation”, Nature, Vol. 468, pp. 973-979.
Niculescu-Duvaz y cols., 2006, “Imidazo[4,5-b]pyridin-2-one and oxazolo[4,5-b]pyridin-2-one compounds and analogs thereof as therapeutic compounds”, publicación de solicitud de patente internacional (PCT) número WO 2006/043090 A1, publicada el 27 de abril de 2006.
Niculescu-Duvaz y cols., 2007, “Imidazo[4,5-b]pyridin-2-one and oxazolo[4,5-b]pyridin-2-one compounds and analogs thereof as therapeutic compounds”, publicación de solicitud de patente internacional (PCT) número WO 2007/125330 A1, publicada el 08 de noviembre de 2007.
Niculescu-Duvaz y cols., 2009, “Aryl-quinolyl compounds and their use”, publicación de solicitud de patente internacional (PCT) número w O 2009/130487 A1, publicada el 29 de octubre de 2009.
O’Reilly y cois., 1994, “Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma.”, Cell, Vol. 79, pp. 315-328.
Oeztuerk-Winder y cols., 2012, “The many faces of p38 mitogen-activated protein kinase in progenitor/stem cell differentiation”, Biochem. J., Vol. 445, pp. 1-10.
Ozawa y cols., 2001, “Growth factors and their receptors in pancreatic cancer”, Teratog. Carcinog. Mutagen., Vol. 21, pp. 27-44.
Palanisamy y cols., 2010, “Rearrangements of the RAF Kinase Pathway in Prostate Cancer, Gastric Cancer and Melanoma”, Nat. Med., Vol. 16, pp. 793-798.
Partanen y cols., 1992, “A novel endothelial cell surface receptor tyrosine kinase with extracellular epidermal growth factor homology domains”, Mol. Cell Biol., Vol. 12, pp. 1698-1707.
Partanen y cols., 1999, “Functions of Tie1 and Tie2 Receptor Tyrosine Kinases in Vascular Development”, Curr. Topics Microbiol. Immunol., Vol. 237, pp. 159-172.
Paulson y cols., 1995, “Receptor tyrosine kinases and the regulation of hematopoiesis”, Semin. Immunol., Vol. 7, No. 4, pp. 267-277.
Payne y cols., 2001, “Human Papillomavirus Type 6b Virus-Like Particles Are Able To Activate the Ras-MAP Kinase Pathway and Induce Cell Proliferation”, J. Virol., Vol. 75, pp. 4150-4157.
Peacock y cols., 1992, “Angiogenesis inhibition suppresses collagen arthritis”, J. Exp. Med., Vol. 175, pp. 1135-1138. Peacock y cols., 1995, “A novel angiogenesis inhibitor suppresses rat adjuvant arthritis”, Cell. Immunol., Vol. 160, pp. 178-184.
Pelletier y cols., 2003, “In vivo selective inhibition of mitogen-activated protein kinase kinase 1/2 in rabbit experimental osteoarthritis is associated with a reduction in the development of structural changes”, Arthritis & Rheumatism, Vol. 48, p. 1582-1593.
Peters, 1998, “Vascular Endothelial Growth Factor and the Angiopoietins: Working Together to Build a Better Blood Vessel”, Circ. Res., Vol. 83, No. 3, pp. 342-343.
Petrovan y cols., 2007, “DNA Vaccination Against VEGF Receptor 2 Reduces Atherosclerosis in LDL Receptor-Deficient Mice”, Arteriosclerosis, Thrombosis, and Vascular Biology, Vol. 27, pp. 1095-1100.
Pinedo y cols., 2000, “The Role of VEGF in Tumour Angiogenesis”, The Oncologist, Vol. 5, pp. 1 -2.
Pinner y cols., 2009, “CD44 splice variants in neurodegenerative diseases”, publicación de solicitud de patente internacional (PCT) número WO 2009/007934, publicada el 15 de enero de anuary 2009.
Planz y cols., 2001, “MEK-specific inhibitor U0126 blocks spread of Bornuna disease virus in cultured cells”, J. Virol., Vol. 75, pp. 4871-4877.
Pleschka y cols., 2001, “Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade”, Nature Cell Biology. Vol. 3, pp. 301-305.
Plomp y cols., 1998, “Pfeiffer syndrome type 2: further delineation and review of the literature”, Am. J. Med. Genet., Vol. 75, pp. 245-251.
Poulikakos y cols., 2010, “RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF”, Nature, Vol. 464, pp. 427-430.
Poulikakos y cols., 2011, “RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E)”, Nature, Vol. 480, pp. 387-390.
Powers y cols., 2000, “Fibroblast growth factors, their receptors and signaling”, Endocr. Relat. Cancer, Vol. 7, pp. 165-197.
Riva y cols., 1995, “Differential c-myc, c-jun, c-raf and p53 expression in squamous cell carcinoma of the head and neck: Implication in drug and radioresistance”, European Journal of Cancer ParteB: Oral Oncology, Vol. 31, pp. 384-391.
Rotsos y cols., 2008, “Cystoid macular edema”, Clin. OphthalmoL Vol. 2, pp. 919-930.
Rubinstein y cols., 2010, “Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032”, J. Transl. Med., Vol. 8, p. 67.
Salama y cois., 2013, “BRAF in Melanoma: Current strategies and future directions”, Clinical Cáncer Research, Vol. 19, No. 16, pp. 4326-4334.
Schindler y cols., 2011, “Analysis of BRAF V600E mutation in 1,320 nervous system tumours reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma”, Acta Neuropathologica, Vol. 121, pp. 397-405.
Schreck y cols., 2006, “Raf kinases: Oncogenesis and drug discovery”, International Journal of Cancer, Vol. 119, pp. 2261-2271.
Shakhov y cols., 1990, “Kappa B-type enhancers are involved in lipopolysaccharide-mediated transcriptional activation of the tumor necrosis factor alpha gene in primary macrophages”, J. Exp. Med., Vol. 171, pp. 35 47.
Shapira y cols., 1996, “Protection against endotoxic shock and lipopolysaccharide-induced local inflammation by tetracycline: correlation with inhibition of cytokine secretion”, Infect. Immun., Vol. 64, No. 3, pp. 825-828. Shi y cols., 2012, “Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance”, Nature Commun., Vol. 3, p. 724.
Sievert y cols., 2013 “Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas”, PNAS, Vol. 110, pp. 5957-5962.
Smalley y cols., 2009, “CRAF inhibition induces apoptosis in melanoma cells with non-V600E BRAF mutations”, Oncogene, Vol. 28, pp. 85-94.
Smith y cols., 2007, “Urea compounds useful in the treatment of cancer”, publicación de solicitud de patente internacional (PCT) número WO 2007/064872 A2, publicada el 07 de junio de 2007.
Smith y cols., 2010, “Vascular Endothelial Growth Factor Receptors VEGFR-2 and VEGFR-3 Are Localized Primarily to the Vasculature in Human Primary Solid Cancers”, Clin. Cancer Res., Vol. 16, pp. 3548-3561.
Solit y cols., 2006, “BRAF mutation predicts sensitivity to MEK inhibition”, Nature, Vol. 439, pp. 358-362.
Song y cols., 2005, “Activation of ERK/CREB pathway in spinal cord contributes to chronic constrictive injury-induced neuropathic pain in rats”, Acta Pharmacol Sin., Vol. 26, p. 789.
Sosman y cols., 2012, “Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib”, New England Journal of Medicine, Vol. 366, pp. 707-714.
Springer y cols., 2009, “Pyrido[2,3-b]pirazine-8-substituted compounds and their use”, publicación de solicitud de patente internacional (PCT) número WO 2009/077766 A1, publicada el 25 de junio de 2009.
Springer y cols., 2011, “1-(5-tert-Butyl-2-phenyl-2H-pyrazol-3-yl)-3-[2-fluoro-4-(1-methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-7-yloxy)-phenyl]urea and related compuestos and their use in therapy”, publicación de solicitud de patente internacional (PCT) número WO 2011/092469 A1, publicada el 04 de agosto de 2011. Stratton y cols., 2003, “Genes”, publicación de solicitud de patente internacional (PCT) número WO 03/056036 A2 publicada el 10 de julio de 2003.
Straussman y cols., 2012, “Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion”, Nature, Vol. 487, pp. 500-506.
Suijkerbuijk y cols., 2010, “Development of novel, highly potent inhibitors of V-RAF murine sarcoma viral oncogene homologue B1 (BRAF): increasing cellular potency through optimization of a distal heteroaromatic group”, J. Med. Chem., Vol. 53, pp. 2741-2756.
Sullivan y cols., 2011, “BRAF in melanoma: pathogenesis, diagnosis, inhibition, and resistance”, J. Skin Cancer, Vol. 2011, Article ID No. 423239.
Suri y cols., 1996, “Requisite Role of Angiopoietin-1, a Ligand for the TIE2 Receptor, during Embryonic Angiogenesis”, Cell, Vol. 87, pp. 1171-1180.
Tam y cols., 2009, “Blockade of VEGFR2 and Not VEGFR1 Can Limit Diet-Induced Fat Tissue Expansion: Role of Local versus Bone Marrow-Derived Endothelial Cells”, PLoS One, Vol. 4, e4974.
Taraboletti y cols., 1995, “Inhibition of angiogenesis and murine hemangioma growth by batimastat, a synthetic inhibitor of matrix metalloproteinases”, J. Natl. Cancer Inst., Vol. 87, p. 293.
Thalhamer y cois., 2008, “MAPKs and their relevance to arthritis and inflammation”, Rheumatoloav. Vol. 47, pp. 409-414.
Vergani y cols., 2011, “ Identification of MET and SRC activation in melanoma cell lines showing primary resistance to PLX4032”, Neoplasia, Vol. 13, pp. 1132-1142.
Vikkula y cols., 2004, “Medical use of ras antagonists for the treatment of capillary malformation”, publicación de solicitud de patente internacional (PCT) número WO 2004/083458, publicada el 30 de septiembre de 2004. Villanueva y cols., 2011, “Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by co-targeting MEK and IGF-1R/PI3K”, Cancer Cell, Vol. 18, pp. 683-695.
Wan y cols., 2004, “Mechanism of activation of RAF-ERK signalling pathway by oncogenic mutations in B-RAF”, Cell, Vol. 116, pp. 855-867.
Wang y cols., 1997, “Antisense targeting of basic fibroblast growth factor and fibroblast growth factor receptor-1 in human melanomas blocks intratumoural angiogenesis and tumour growth”, Nature Medicine, Vol. 3, pp. 887-893.
Wang y cols., 2003, “Significant Neuroprotection against Ischemic Brain Injury by Inhibition of the MEK1 Protein Kinase in Mice: Exploration of Potential Mechanism Associated with Apoptosis”, J. Pharmacol. Exp. Ther., Vol. 304, p. 172.
Wang y cols., 2004, “Inhibition of MEK/ERK 1/2 pathway reduces pro-inflammatory cytokine interleukin-1 expression in focal cerebral ischemia”, Brain Res., Vol. 996, p. 55.
Ward y cols., 2012, “Targeting oncogenic Ras signaling in hematologic malignantes”, Blood, Vol. 120, pp. 3397-3406.
Wellbrock y cols., 2004, “V599EB-RAF is an oncogene in melanocytes”, Cancer Research, Vol. 64, pp. 2338-2342. Whittaker y cols., 2010, “A novel, selective and efficacious nanomolar piridopiracinone inhibitor of V600EBRAF”, Cancer Research, Vol. 70, No. 20, pp. 80368044.
Wilks y cols., 1990, “Structure and function of the protein tyrosine kinases”, Progress in Growth Factor Research, Vol. 2, pp. 97-111.
Wilson y cols., 2012, “Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors”, Nature, Vol. 487, pp. 505-509.
Xing, 2013, “Molecular pathogenesis and mechanisms of thyroid cancer”, Nature Reviews Cancer, Vol. 13, pp. 184-199.
Yancopoulos y cols., 1998, “Vasculogenesis, Angiogenesis, and Growth Factors: Ephrins Enter the Fray at the Border ”, Cell, Vol. 93, pp. 661-664.
Yang y cols., 1999, “Regulation of Human Immunodeficiency Virus Type 1 Infectivity by the ERK Mitogen-Activated Protein Kinase Signaling Pathway”, J. Virol., Vol. 73, pp. 3460-3466.
Yao y cols., 1997, “Lipopolysaccharide induction of the tumor necrosis factor-alpha promoter in human monocytic cells. Regulation by Egr-1, c-Jun and NF-kappaB transcription factors”, J. Biol. Chem., Vol. 272, pp. 17795 17801.
Yeatman y cols., 2004, “A renaissance for SRC”, Nature Reviews Cancer, Vol. 4, pp. 470-480.
Young y cols., 2009, “Ras signaling and therapies”, Advances in Cancer Research, Vol. 102, pp. 1 -17.
Yu y cols., 2000, “Loss of fibroblast growth factor receptor 2 ligand binding specificity in Apert syndrome”, Proc. Natl.
Acad. Sci. U.S.A., Vol. 97, pp. 14536-14541.
Zambon y cols., 2010, “Novel hinge binder improves activity and pharmacokinetic properties of BRAF inhibitors”, J.
Med. Chem., Vol. 53, No. 15, pp. 5639-5655.
Zennadi y cols., 2012, “Methods of treating hemoglobinopathies”, publicación de solicitud de patente internacional (PCT) número WO 2012/149547, publicada el 01 de noviembre de 2012.
Zhang y cols., 2012, “Activation of the Ras/Raf/MEK Pathway Facilitates Hepatitis C Virus Replication via Attenuation of the Interferon-JAK-STAT Pathway”, J. Virol., Vol. 86, pp. 1544-1554.
Zhang y cois., 2012, “Targeting Src family kinases in anti-cancer therapies: turning promise into triumph”, Trends Pharmacol. Sci. Med., Vol. 33, pp. 122-128.
Zouki y cols., 2000, “Peroxinitrite induces integrin-dependent adhesion of human neutrophils to endothelial cells via activation of the Raf-1/MEK/Erk pathway”, fAs EB J., Vol. 15, pp. 25-27.
Claims (25)
1. Un compuesto seleccionado de compuestos de la siguiente fórmula, y sales farmacéuticamente aceptables, N-óxidos, hidratos y solvatos de los mismos:

donde: =X- es independientemente =CH- o =N-;
-Y es independientemente -Y1, -Y2, -Y3, -Y4, -Y5 o -Y6;
-Y1 es independientemente -F, -Cl, -Br o -I;
-Y2 es alquilo C1-4 saturado lineal o ramificado;
-Y es haloalquilo C1-4 saturado lineal o ramificado;
-Y4 es -OH;
-Y5 es alcoxi C1-4 saturado lineal o ramificado; e
-Y6 es haloalcoxi C1-4 saturado lineal o ramificado.
2. Un compuesto según la reivindicación 1, en el que =X- es =CH-.
3. Un compuesto según la reivindicación 1, en el que =X- es =N-.
4. Un compuesto según una cualquiera de las reivindicaciones 1 a 3, en el que:
-Y1, si está presente, es -F;
-Y , si está presente, es Me;
-Y3, si está presente, es -CF3 ;
-Y5, si está presente, es -O-Me;
-Y6, si está presente, es -O-CF3.
5. Un compuesto según la reivindicación 1, seleccionado del siguiente compuesto y sales farmacéuticamente aceptables, N-óxidos, hidratos y solvatos del mismo:

6. Un compuesto según la reivindicación 1, seleccionado del siguiente compuesto y sales farmacéuticamente aceptables, N-óxidos, hidratos y solvatos del mismo:

7. Un compuesto según la reivindicación 1, seleccionado del siguiente compuesto y sales farmacéuticamente aceptables, N-óxidos, hidratos y solvatos del mismo:

8. Un compuesto según la reivindicación 1, seleccionado del siguiente compuesto y sales farmacéuticamente aceptables, N-óxidos, hidratos y solvatos del mismo:
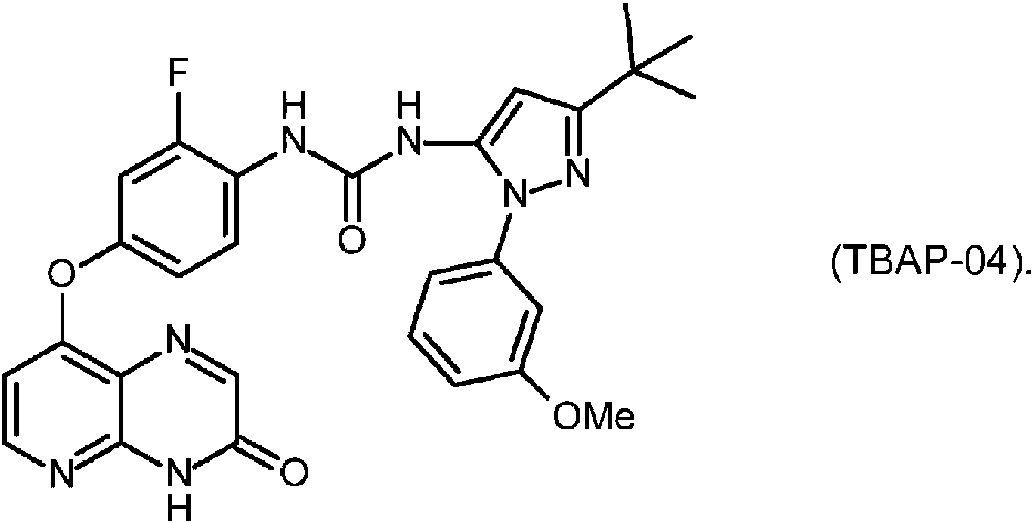
9. Un compuesto según la reivindicación 1, seleccionado del siguiente compuesto y sales farmacéuticamente aceptables, N-óxidos, hidratos y solvatos del mismo:

10. Una composición que comprende un compuesto según una cualquiera de las reivindicaciones 1 a 9, y un portador o diluyente farmacéuticamente aceptable.1
11. Un compuesto según una cualquiera de las reivindicaciones 1 a 9, para el uso en un método de tratamiento del cuerpo de un ser humano o un animal mediante terapia.
12. Un compuesto para el uso según la reivindicación 11, en un método de tratamiento de: un trastorno proliferativo; cáncer; inflamación; un trastorno inmunológico; una infección viral; un trastorno fibrótico; un trastorno asociado con una forma mutada de RAF; un trastorno mejorado por la inhibición de RAF; un trastorno mejorado por la inhibición de BRAF mutante; un trastorno mejorado por la inhibición de BRAF y CRAF; un trastorno asociado con mutaciones en RAS y/o la activación de la ruta de MAPK; un trastorno mejorado por la inhibición de SRC, p38, FGFRA, VEGFR-2 (KDR) y/o LCK.
13. Un compuesto para el uso según la reivindicación 11, en un método de tratamiento de: un trastorno proliferativo.
14. Un compuesto para el uso según la reivindicación 11, en un método de tratamiento de: cáncer.
15. Un compuesto para el uso según la reivindicación 11, en un método de tratamiento de: cáncer pancreático; cáncer pulmonar no microcítico (NSCLC, por sus siglas en inglés); neoplasmas ováricos; neoplasma peritoneales; neoplasmas de las trompas de Falopio; cáncer pulmonar y efusión pleural asociada; cáncer de células escamosas recurrente o metastásico de la cabeza y el cuello; carcinoma nasofaríngeo localmente avanzado; glioblastoma (p. ej., glioblastoma multiforme; glioblastoma de células gigantes); gliosarcoma; glioma pontino intrínseco difuso; sarcoma de Kaposi relacionado con HIV; mieloma múltiple; carcinoma de células renales; adenocarcinoma gástrico metastásico; leucemia mieloide aguda (AML); carcinoma hepatocelular; dermatofibrosarcoma; cáncer tiroideo medular (MTC, por sus siglas en inglés); cáncer tiroideo papilar; cáncer tiroideo folicular; síndrome mielodisplástico; neurofibromatosis tipo 1; neurofibroma plexiforme; neurofibroma de la médula espinal; cáncer de mama; neoplasmas del tracto biliar; cáncer de cuello uterino; cáncer de próstata; melanoma; carcinoma de vejiga urinaria; carcinoma de uretra; carcinoma de uréter; carcinoma renal; carcinoma pélvico; sarcoma; liposarcoma; cáncer de colon; osteosarcoma; carcinoma sinovial; neuroblastoma; o rabdomiosarcoma.
16. Un compuesto para el uso según la reivindicación 11, en un método de tratamiento de: melanoma maligno; carcinoma colorrectal; carcinoma colorrectal metastásico; cáncer tiroideo folicular; cáncer tiroideo insular; cáncer tiroideo papilar; carcinoma ovárico; carcinoma ovárico de baja malignidad; cáncer pulmonar no microcítico; tricoleucemia; colangiocarcinoma; glioma pediátrico de baja malignidad (p. ej., astrocitoma pilocítico; ganglioglioma; xantoastrocitoma pleomórfico); mieloma múltiple; carcinoma medular del páncreas; o adenocarcinoma ductal pancreático.
17. Un compuesto para el uso según la reivindicación 11, en un método de tratamiento de: melanoma maligno.
18. Un compuesto para el uso según la reivindicación 11, en un método de tratamiento de: carcinoma colorrectal.
19. Un compuesto para el uso según la reivindicación 11, en un método de tratamiento de: adenocarcinoma pancreático.
20. Un compuesto para el uso según la reivindicación 11, en un método de tratamiento de: un trastorno asociado con una forma mutada de RAF.
21. Un compuesto para el uso según la reivindicación 11, en un método de tratamiento de: un trastorno resistente al tratamiento con un inhibidor de RAF conocido.
22. Un compuesto para el uso según la reivindicación 11, en un método de tratamiento de: un trastorno resistente al tratamiento con una combinación de un inhibidor de RAF conocido y un inhibidor de MEK conocido.
23. Un compuesto para el uso según la reivindicación 11, en un método de tratamiento de: un trastorno resistente al tratamiento con un anticuerpo conocido.
24. Un compuesto para el uso según la reivindicación 11, en un método de tratamiento de: una enfermedad inflamatoria seleccionada de: fibrosis quística; hipertensión pulmonar; sarcoidosis pulmonar; fibrosis pulmonar idiopática; enfermedad pulmonar obstructiva crónica (COPD) (incluyendo bronquitis crónica y enfisema); asma; asma pediátrica; dermatitis atópica; dermatitis alérgica; dermatitis de contacto; psoriasis; rinitis alérgica; rinitis; sinusitis; conjuntivitis; conjuntivitis alérgica; queratoconjuntivitis seca (ojo seco); glaucoma; retinopatía diabética; edema macular (incluyendo edema macular diabético); oclusión de la vena retinal central (CRVO); degeneración macular seca y/o húmeda asociada a la edad (AMD); inflamación después de una operación de cataratas; uveitis (incluyendo uveitis posterior, anterior y panuveitis); rechazo de injerto corneal y trasplante de células límbicas; enteropatía sensible al gluten (enfermedad celíaca); esofaguitis eosinofílica; enfermedad intestinal de injerto contra el hospedador; enfermedad de Crohn; y colitis ulcerativa.
25. Un compuesto para el uso según la reivindicación 11, en un método de tratamiento de: asma o COPD.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB1320729.5A GB201320729D0 (en) | 2013-11-25 | 2013-11-25 | Therapeutic compounds and their use |
| PCT/GB2014/053490 WO2015075483A1 (en) | 2013-11-25 | 2014-11-25 | 1 -(5-tert-butyl-2-aryl-pyrazol-3-yl)-3-[2-fluoro-4-[(3-oxo-4h-pyrido[2, 3-b]pyrazin-8-yl)oxy]phenyl]urea derivatives as raf inhibitors for the treatment of cancer |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2740325T3 true ES2740325T3 (es) | 2020-02-05 |
Family
ID=49918123
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14806358T Active ES2740325T3 (es) | 2013-11-25 | 2014-11-25 | Derivados de 1-(5-terc-butil-2-aril-pirazol-3-il)-3-[2-fluoro-4-[(3-oxo-4h-pirido[2,3 b]piracin-8-il)oxi]fenil]urea como inhibidores raf para el tratamiento del cáncer |
Country Status (19)
| Country | Link |
|---|---|
| US (2) | US9725447B2 (es) |
| EP (1) | EP3074396B1 (es) |
| JP (1) | JP6389529B2 (es) |
| KR (1) | KR102327096B1 (es) |
| CN (1) | CN105793260B (es) |
| AU (1) | AU2014351571B2 (es) |
| BR (1) | BR112016011078B1 (es) |
| CA (1) | CA2928009C (es) |
| DK (1) | DK3074396T3 (es) |
| EA (1) | EA034216B1 (es) |
| ES (1) | ES2740325T3 (es) |
| GB (1) | GB201320729D0 (es) |
| HU (1) | HUE045596T2 (es) |
| IL (1) | IL245062B (es) |
| MX (1) | MX372942B (es) |
| PL (1) | PL3074396T3 (es) |
| PT (1) | PT3074396T (es) |
| SA (1) | SA516371189B1 (es) |
| WO (1) | WO2015075483A1 (es) |
Families Citing this family (58)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2520940T3 (es) | 2007-12-19 | 2014-11-12 | Cancer Research Technology Limited | Compuestos de pirido[2,3-b]pirazina 8-sustituida y su uso |
| NZ601085A (en) | 2010-02-01 | 2015-04-24 | Cancer Rec Tech Ltd | 1-(5-tert-butyl-2-phenyl-2h-pyrazol-3-yl)-3-[2-fluoro-4-(1-methyl-2-oxo-2,3-dihydro-1h-imidazo[4,5-b]pyridin-7-yloxy)-phenyl]-urea and related compounds and their use in therapy |
| GB201320732D0 (en) | 2013-11-25 | 2014-01-08 | Cancer Rec Tech Ltd | Methods of chemical synthesis |
| GB201320729D0 (en) * | 2013-11-25 | 2014-01-08 | Cancer Rec Tech Ltd | Therapeutic compounds and their use |
| AU2016334041B2 (en) * | 2015-10-08 | 2023-02-09 | Macrogenics, Inc. | Combination therapy for the treatment of cancer |
| JP2019508491A (ja) * | 2016-02-05 | 2019-03-28 | エヴォール・サイエンス・エルエルシー | がん治療用組み合わせ |
| WO2017144877A1 (en) | 2016-02-23 | 2017-08-31 | Cancer Research Technology Limited | Dietary product devoid of at least two non essential amino acids |
| EP3443114A1 (en) | 2016-04-15 | 2019-02-20 | H. Hoffnabb-La Roche Ag | Diagnostic and therapeutic methods for cancer |
| KR101796684B1 (ko) * | 2016-05-19 | 2017-11-10 | 건국대학교 산학협력단 | 케라틴 8 인산화 억제제를 포함하는 황반변성 예방 또는 치료용 약학 조성물 및 황반변성 치료제의 스크리닝 방법 |
| MX387210B (es) | 2016-12-22 | 2025-03-18 | Amgen Inc | Benzoisotiazol, isotiazolo[3,4-b]piridina, quinazolina, ftalazina, pirido[2,3-d]piridazina y derivados de pirido[2,3-d]pirimidina como inhibidores de kras g12c para tratar el cáncer de pulmón, pancreático o colorrectal. |
| CA3049926A1 (en) | 2017-01-17 | 2018-07-26 | Heparegenix Gmbh | Protein kinase inhibitors for promoting liver regeneration or reducing or preventing hepatocyte death |
| AU2018217963B2 (en) * | 2017-02-10 | 2020-11-19 | Novartis Ag | 1-(4-amino-5-bromo-6-(1 H-pyrazol-1-yl)pyrimidin-2-yl)-1 H-pyrazol-4-ol and use thereof in the treatment of cancer |
| CN106986860A (zh) * | 2017-05-19 | 2017-07-28 | 南京大学 | 一类含酰胺键的吡唑类抗肿瘤化合物的设计、合成 |
| JOP20190272A1 (ar) | 2017-05-22 | 2019-11-21 | Amgen Inc | مثبطات kras g12c وطرق لاستخدامها |
| JP2020530014A (ja) * | 2017-08-07 | 2020-10-15 | エヴォール・サイエンス・エルエルシー | がんを治療するための組み合わせ |
| WO2019051296A1 (en) | 2017-09-08 | 2019-03-14 | Genentech, Inc. | DIAGNOSTIC AND THERAPEUTIC METHODS OF CANCER |
| EP4141005B1 (en) * | 2017-09-08 | 2024-04-03 | Amgen Inc. | Inhibitors of kras g12c and methods of using the same |
| EP3700901A1 (en) | 2017-10-24 | 2020-09-02 | Genentech, Inc. | (4-hydroxypyrrolidin-2-yl)-heterocyclic compounds and methods of use thereof |
| WO2019084030A1 (en) | 2017-10-24 | 2019-05-02 | Genentech, Inc. | (4-HYDROXYPYRROLIDIN-2-YL) -HYDROXAMATE COMPOUNDS AND METHODS OF USE |
| WO2019092455A1 (en) | 2017-11-13 | 2019-05-16 | Cancer Research Technology Ltd | Dietary product |
| WO2019158579A1 (en) | 2018-02-13 | 2019-08-22 | Vib Vzw | Targeting minimal residual disease in cancer with rxr antagonists |
| WO2019183523A1 (en) | 2018-03-23 | 2019-09-26 | Genentech, Inc. | Hetero-bifunctional degrader compounds and their use as modulators of targeted ubiquination (vhl) |
| MA52501A (fr) | 2018-05-04 | 2021-03-10 | Amgen Inc | Inhibiteurs de kras g12c et leurs procédés d'utilisation |
| PT3788116T (pt) * | 2018-05-04 | 2022-09-22 | Univ Do Porto | Fotossensibilizadores quimioluminescentes à base de imidazopirazinona com estados excitados de singleto e tripleto disponíveis |
| WO2019213526A1 (en) | 2018-05-04 | 2019-11-07 | Amgen Inc. | Kras g12c inhibitors and methods of using the same |
| US10988485B2 (en) | 2018-05-10 | 2021-04-27 | Amgen Inc. | KRAS G12C inhibitors and methods of using the same |
| JP7360396B2 (ja) | 2018-06-01 | 2023-10-12 | アムジエン・インコーポレーテツド | Kras g12c阻害剤及び同一物の使用方法 |
| GB201809295D0 (en) | 2018-06-06 | 2018-07-25 | Institute Of Cancer Res Royal Cancer Hospital | Lox inhibitors |
| CA3100390A1 (en) | 2018-06-12 | 2020-03-12 | Amgen Inc. | Kras g12c inhibitors encompassing piperazine ring and use thereof in the treatment of cancer |
| GB201818651D0 (en) | 2018-11-15 | 2019-01-02 | Univ Sheffield | Compounds |
| GB201818649D0 (en) | 2018-11-15 | 2019-01-02 | Univ Sheffield | Compounds |
| GB201818750D0 (en) | 2018-11-16 | 2019-01-02 | Institute Of Cancer Res Royal Cancer Hospital | Lox inhibitors |
| JP7516029B2 (ja) | 2018-11-16 | 2024-07-16 | アムジエン・インコーポレーテツド | Kras g12c阻害剤化合物の重要な中間体の改良合成法 |
| JP7377679B2 (ja) | 2018-11-19 | 2023-11-10 | アムジエン・インコーポレーテツド | がん治療のためのkrasg12c阻害剤及び1種以上の薬学的に活性な追加の薬剤を含む併用療法 |
| MX2021005700A (es) | 2018-11-19 | 2021-07-07 | Amgen Inc | Inhibidores de kras g12c y metodos de uso de los mismos. |
| JP2022523100A (ja) * | 2019-02-01 | 2022-04-21 | グラクソスミスクライン、インテレクチュアル、プロパティー、ディベロップメント、リミテッド | ベランタマブマフォドチンおよび抗ox40抗体を含むがんの併用治療ならびにその使用および方法 |
| KR102192576B1 (ko) * | 2019-03-21 | 2020-12-17 | 경북대학교 산학협력단 | 다브라페닙을 유효성분으로 포함하는 알레르기 질환 예방, 치료 또는 개선용 조성물 |
| EP3738593A1 (en) | 2019-05-14 | 2020-11-18 | Amgen, Inc | Dosing of kras inhibitor for treatment of cancers |
| JOP20210310A1 (ar) | 2019-05-21 | 2023-01-30 | Amgen Inc | أشكال الحالة الصلبة |
| US20230089368A1 (en) | 2019-07-19 | 2023-03-23 | Anagenesis Biotechnologies S.A.S. | Polyaromatic urea derivatives and their use in the treatment of muscle diseases |
| CA3166636A1 (en) | 2020-01-10 | 2021-07-15 | Immuneering Corporation | Mek inhibitors and therapeutic uses thereof |
| JP2023530235A (ja) | 2020-06-03 | 2023-07-14 | フェス・セラピューティクス,インコーポレーテッド | 個別化された処置方法のための製剤 |
| EP4161495A4 (en) | 2020-06-04 | 2024-07-10 | Faeth Therapeutics, Inc. | PERSONALIZED CANCER TREATMENT METHODS |
| EP4029501A1 (en) | 2021-01-19 | 2022-07-20 | Anagenesis Biotechnologies | Combination of polyaromatic urea derivatives and glucocorticoid or hdac inhibitor for the treatment of diseases or conditions associated with muscle cells and/or satellite cells |
| KR20240041917A (ko) | 2021-07-27 | 2024-04-01 | 도레이 카부시키가이샤 | 암의 치료 및/또는 예방을 위한 의약품 |
| CN119156372A (zh) | 2022-03-17 | 2024-12-17 | 斯普林渥克斯治疗有限公司 | 使用米达美替尼的儿科患者的1型神经纤维瘤病(nf1)相关的丛状神经纤维瘤(pn)的治疗 |
| CN115400122B (zh) * | 2022-04-29 | 2023-04-18 | 佛山病原微生物研究院 | 一种tak-632在制备用于抗腺病毒感染的药物中的用途 |
| EP4532470A1 (en) | 2022-05-25 | 2025-04-09 | Ikena Oncology, Inc. | Mek inhibitors and uses thereof |
| GB202208347D0 (en) | 2022-06-07 | 2022-07-20 | Univ Court Univ Of Glasgow | Targets for cancer therapy |
| GB202209622D0 (en) | 2022-06-30 | 2022-08-17 | Institute Of Cancer Res Royal Cancer Hospital | Compounds |
| GB202209624D0 (en) | 2022-06-30 | 2022-08-17 | Institute Of Cancer Res Royal Cancer Hospital | Prodrugs |
| WO2024033381A1 (en) | 2022-08-10 | 2024-02-15 | Vib Vzw | Inhibition of tcf4/itf2 in the treatment of cancer |
| WO2024186693A1 (en) * | 2023-03-03 | 2024-09-12 | Immuneering Corporation | Methods of treating cancer with a ras mutation |
| WO2024220470A1 (en) | 2023-04-17 | 2024-10-24 | Ikena Oncology, Inc. | Tead inhibitors and uses thereof in the treatment of proliferative disorders |
| WO2024229406A1 (en) | 2023-05-04 | 2024-11-07 | Revolution Medicines, Inc. | Combination therapy for a ras related disease or disorder |
| US20250049810A1 (en) | 2023-08-07 | 2025-02-13 | Revolution Medicines, Inc. | Methods of treating a ras protein-related disease or disorder |
| US20250154171A1 (en) | 2023-10-12 | 2025-05-15 | Revolution Medicines, Inc. | Ras inhibitors |
| CN119899194B (zh) * | 2025-03-31 | 2025-06-24 | 浙江省人民医院 | 双环杂芳基类braf负变构调节剂及其制备方法与应用 |
Family Cites Families (88)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4082845A (en) | 1977-04-25 | 1978-04-04 | Merck & Co., Inc. | 3-(1-Piperazinyl)-pyrido[2,3-b]pyrazines |
| JPS5665863A (en) | 1979-10-31 | 1981-06-03 | Tokyo Organ Chem Ind Ltd | Novel aniline derivative, its preparation and pesticide containing the same |
| JPS5738777A (en) | 1980-08-19 | 1982-03-03 | Sogo Yatsukou Kk | 2-sufanilamidopyrathyn derivative |
| US4666828A (en) | 1984-08-15 | 1987-05-19 | The General Hospital Corporation | Test for Huntington's disease |
| US4683202A (en) | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| US4801531A (en) | 1985-04-17 | 1989-01-31 | Biotechnology Research Partners, Ltd. | Apo AI/CIII genomic polymorphisms predictive of atherosclerosis |
| US5272057A (en) | 1988-10-14 | 1993-12-21 | Georgetown University | Method of detecting a predisposition to cancer by the use of restriction fragment length polymorphism of the gene for human poly (ADP-ribose) polymerase |
| US5192659A (en) | 1989-08-25 | 1993-03-09 | Genetype Ag | Intron sequence analysis method for detection of adjacent and remote locus alleles as haplotypes |
| US5879672A (en) | 1994-10-07 | 1999-03-09 | Regeneron Pharmaceuticals, Inc. | Tie-2 ligand 1 |
| US5643755A (en) | 1994-10-07 | 1997-07-01 | Regeneron Pharmaceuticals Inc. | Nucleic acid encoding tie-2 ligand |
| ATE294236T1 (de) | 1994-09-22 | 2005-05-15 | Licentia Ltd | Promotor der tie rezeptor protein kinase |
| US6218529B1 (en) | 1995-07-31 | 2001-04-17 | Urocor, Inc. | Biomarkers and targets for diagnosis, prognosis and management of prostate, breast and bladder cancer |
| CA2262403C (en) | 1995-07-31 | 2011-09-20 | Urocor, Inc. | Biomarkers and targets for diagnosis, prognosis and management of prostate disease |
| ZA971896B (en) | 1996-03-26 | 1998-09-07 | Du Pont Merck Pharma | Aryloxy-and arythio-fused pyridines and pyrimidines and derivatives |
| US6809097B1 (en) | 1996-09-25 | 2004-10-26 | Zeneca Limited | Quinoline derivatives inhibiting the effect of growth factors such as VEGF |
| US6030831A (en) | 1997-09-19 | 2000-02-29 | Genetech, Inc. | Tie ligand homologues |
| AU748849B2 (en) | 1997-09-26 | 2002-06-13 | Zentaris Gmbh | Azabenzimidazole-based compounds for modulating serine/threonine protein kinase function |
| GB9721437D0 (en) | 1997-10-10 | 1997-12-10 | Glaxo Group Ltd | Heteroaromatic compounds and their use in medicine |
| WO2000035436A2 (en) | 1998-12-16 | 2000-06-22 | Warner-Lambert Company | Treatment of arthritis with mek inhibitors |
| US6696440B1 (en) | 1999-01-07 | 2004-02-24 | Warner-Lambert Company | Treatment of asthma with MEK inhibitors |
| AU3475900A (en) | 1999-01-29 | 2000-08-18 | University Of Akron, The | Polyimides used as microelectronic coatings |
| HUP0202623A3 (en) | 1999-07-16 | 2003-03-28 | Warner Lambert Co | Method for treating chronic pain using mek inhibitors |
| GB2356398A (en) | 1999-11-18 | 2001-05-23 | Lilly Dev Ct S A | Preparation of arylsulfamides |
| EP1244672B1 (en) | 1999-12-21 | 2005-07-20 | Sugen, Inc. | 4-substituted 7-aza-indolin-2-ones and their use as protein kinase inhibitors |
| US6492529B1 (en) | 2000-01-18 | 2002-12-10 | Boehringer Ingelheim Pharmaceuticals, Inc. | Bis pyrazole-1H-pyrazole intermediates and their synthesis |
| US7238813B2 (en) | 2000-11-29 | 2007-07-03 | Smithkline Beecham Corporation | Chemical compounds |
| KR20020096367A (ko) | 2001-06-19 | 2002-12-31 | 주식회사 티지 바이오텍 | 관절염 예방 또는 치료제 및 그것의 스크리닝 방법 |
| EP1456364B1 (en) | 2001-12-21 | 2011-03-23 | The Wellcome Trust | Mutations in the b-raf gene |
| AU2003257170B2 (en) | 2002-08-09 | 2008-12-11 | Merck Sharp & Dohme Corp. | Tyrosine kinase inhibitors |
| WO2004083458A1 (en) | 2003-03-20 | 2004-09-30 | Universite Catholique De Louvain | Medical use of ras antagonists for the treatment of capillary malformation |
| MXPA05013349A (es) | 2003-06-13 | 2006-03-09 | Novartis Ag | Derivados de 2-amino-pirimidina como inhibidores de quinasa raf. |
| JPWO2005080377A1 (ja) | 2004-02-20 | 2007-10-25 | キリンホールディングス株式会社 | TGFβ阻害活性を有する化合物およびそれを含んでなる医薬組成物 |
| TW200616974A (en) | 2004-07-01 | 2006-06-01 | Astrazeneca Ab | Chemical compounds |
| MX2007002434A (es) | 2004-08-31 | 2007-05-04 | Astrazeneca Ab | Derivados de quinazolinona y su uso como inhibidores de b-raf. |
| CA2583096A1 (en) | 2004-10-15 | 2006-04-20 | Astrazeneca Ab | Quinoxalines as b raf inhibitors |
| GB0423554D0 (en) * | 2004-10-22 | 2004-11-24 | Cancer Rec Tech Ltd | Therapeutic compounds |
| GB0428082D0 (en) | 2004-12-22 | 2005-01-26 | Welcome Trust The Ltd | Therapeutic compounds |
| CA2592118C (en) | 2004-12-23 | 2015-11-17 | Deciphera Pharmaceuticals, Llc | Urea derivatives as enzyme modulators |
| WO2007059202A2 (en) | 2005-11-15 | 2007-05-24 | Bayer Healthcare Ag | Pyrazolyl urea derivatives useful in the treatment of cancer |
| JP2009518298A (ja) | 2005-12-01 | 2009-05-07 | バイエル ヘルスケア リミティド ライアビリティ カンパニー | 癌治療に有用な尿素化合物 |
| BRPI0619514A2 (pt) | 2005-12-08 | 2011-10-04 | Millennium Pharm Inc | compostos bicìclicos com atividade inibidora cinase, composição farmacêutica contendo os mesmos e uso de ditos compostos |
| US7989461B2 (en) | 2005-12-23 | 2011-08-02 | Amgen Inc. | Substituted quinazolinamine compounds for the treatment of cancer |
| US7951819B2 (en) | 2006-04-26 | 2011-05-31 | Cancer Research Technology Limited | Imidazo[4, 5-B]pyridin-2-one and oxazolo[4, 5-B] pyridin-2-one compounds and analogs thereof as cancer therapeutic compounds |
| EP1992628A1 (en) | 2007-05-18 | 2008-11-19 | Glaxo Group Limited | Derivatives and analogs of N-ethylquinolones and N-ethylazaquinolones |
| US8188113B2 (en) * | 2006-09-14 | 2012-05-29 | Deciphera Pharmaceuticals, Inc. | Dihydropyridopyrimidinyl, dihydronaphthyidinyl and related compounds useful as kinase inhibitors for the treatment of proliferative diseases |
| WO2008044688A1 (fr) | 2006-10-11 | 2008-04-17 | Daiichi Sankyo Company, Limited | Dérivé de l'urée |
| CA2692861A1 (en) | 2007-07-10 | 2009-01-15 | Neurim Pharmaceuticals (1991) Ltd. | Cd44 splice variants in neurodegenerative diseases |
| ES2520940T3 (es) | 2007-12-19 | 2014-11-12 | Cancer Research Technology Limited | Compuestos de pirido[2,3-b]pirazina 8-sustituida y su uso |
| GB0807609D0 (en) | 2008-04-25 | 2008-06-04 | Cancer Rec Tech Ltd | Therapeutic compounds and their use |
| CA2738828A1 (en) | 2008-10-02 | 2010-04-08 | Respivert Limited | P38 map kinase inhibitors |
| GB0818033D0 (en) | 2008-10-02 | 2008-11-05 | Respivert Ltd | Novel compound |
| KR20110094130A (ko) | 2008-12-11 | 2011-08-19 | 레스피버트 리미티드 | P38 map 키나제 억제제 |
| GB0905955D0 (en) | 2009-04-06 | 2009-05-20 | Respivert Ltd | Novel compounds |
| WO2011004276A1 (en) | 2009-07-06 | 2011-01-13 | Pfizer Limited | Hepatitis c virus inhibitors |
| SG10201402917XA (en) | 2009-08-24 | 2014-08-28 | Genentech Inc | Determining sensitivity of cells to b-raf inhibitor treatment by detecting kras mutation and rtk expression levels |
| GB0918249D0 (en) | 2009-10-19 | 2009-12-02 | Respivert Ltd | Compounds |
| GB0921731D0 (en) | 2009-12-11 | 2010-01-27 | Respivert Ltd | Theraputic uses |
| GB0921730D0 (en) | 2009-12-11 | 2010-01-27 | Respivert Ltd | Method of treatment |
| NZ601085A (en) | 2010-02-01 | 2015-04-24 | Cancer Rec Tech Ltd | 1-(5-tert-butyl-2-phenyl-2h-pyrazol-3-yl)-3-[2-fluoro-4-(1-methyl-2-oxo-2,3-dihydro-1h-imidazo[4,5-b]pyridin-7-yloxy)-phenyl]-urea and related compounds and their use in therapy |
| GB201005589D0 (en) | 2010-04-01 | 2010-05-19 | Respivert Ltd | Novel compounds |
| WO2011124923A2 (en) | 2010-04-08 | 2011-10-13 | Respivert Limited | Novel compounds |
| US9024041B2 (en) | 2010-04-08 | 2015-05-05 | Respivert Ltd. | P38 MAP kinase inhibitors |
| WO2011158044A2 (en) * | 2010-06-17 | 2011-12-22 | Respivert Limited | Respiratory formulations and compounds for use therein |
| GB201010193D0 (en) | 2010-06-17 | 2010-07-21 | Respivert Ltd | Medicinal use |
| GB201010196D0 (en) | 2010-06-17 | 2010-07-21 | Respivert Ltd | Methods |
| WO2012008564A1 (ja) | 2010-07-16 | 2012-01-19 | 協和発酵キリン株式会社 | 含窒素芳香族複素環誘導体 |
| UY33337A (es) | 2010-10-18 | 2011-10-31 | Respivert Ltd | DERIVADOS SUSTITUIDOS DE 1H-PIRAZOL[ 3,4-d]PIRIMIDINA COMO INHIBIDORES DE LAS FOSFOINOSITIDA 3-QUINASAS |
| US9592236B2 (en) | 2011-04-28 | 2017-03-14 | Duke University | Methods of treating hemoglobinopathies |
| EA025266B1 (ru) | 2011-06-23 | 2016-12-30 | Ваймет Фармасьютикалс, Инк. | Соединения, ингибирующие металлоферменты |
| WO2013001372A2 (en) | 2011-06-30 | 2013-01-03 | University Of Oslo | Methods and compositions for inhibition of activation of regulatory t cells |
| WO2013033133A1 (en) | 2011-09-01 | 2013-03-07 | Novartis Ag | Use of organic compound for the treatment of noonan syndrome |
| PL2763984T3 (pl) | 2011-10-03 | 2016-10-31 | 1-pirazolilo-3-(4-((2-anilinopirymidyno-4-ilo)oksy)naftaleno-1-ilo)moczniki jako inhibitory kinazy p38 MAP | |
| EP2578582A1 (en) | 2011-10-03 | 2013-04-10 | Respivert Limited | 1-Pyrazolyl-3-(4-((2-anilinopyrimidin-4-yl)oxy)napththalen-1-yl)ureas as p38 MAP kinase inhibitors |
| GB201214750D0 (en) | 2012-08-17 | 2012-10-03 | Respivert Ltd | Compounds |
| GB201215357D0 (en) | 2012-08-29 | 2012-10-10 | Respivert Ltd | Compounds |
| EP2890701B1 (en) | 2012-08-29 | 2017-03-29 | Respivert Limited | Kinase inhibitors |
| EP2890460B1 (en) | 2012-08-29 | 2017-02-22 | Respivert Limited | Kinase inhibitors |
| WO2014033447A2 (en) | 2012-08-29 | 2014-03-06 | Respivert Limited | Kinase inhibitors |
| WO2014076484A1 (en) | 2012-11-16 | 2014-05-22 | Respivert Limited | Kinase inhibitors |
| WO2014140582A1 (en) | 2013-03-14 | 2014-09-18 | Respivert Limited | Kinase inhibitors |
| TW201522341A (zh) | 2013-03-15 | 2015-06-16 | Respivert Ltd | 化合物 |
| TWI641592B (zh) | 2013-04-02 | 2018-11-21 | 英商瑞斯比維特有限公司 | 激酶抑制劑 |
| WO2014162121A1 (en) | 2013-04-02 | 2014-10-09 | Topivert Pharma Limited | Kinase inhibitors based upon n-alkyl pyrazoles |
| GB201320729D0 (en) | 2013-11-25 | 2014-01-08 | Cancer Rec Tech Ltd | Therapeutic compounds and their use |
| CA2934199A1 (en) | 2013-12-20 | 2015-06-25 | Respivert Limited | Urea derivatives useful as kinase inhibitors |
| CA2939355A1 (en) | 2014-02-14 | 2015-08-20 | Topivert Pharma Limited | Substituted 1-pyrazolyl-3-naphthyl-urea compound with anti-inflammatory properties |
| WO2016051186A1 (en) | 2014-10-01 | 2016-04-07 | Respivert Limited | N-phenyl-3-quinazolin-6-yl-benzamide derivatives as p38 kinase inhibitors |
| MA40774A (fr) | 2014-10-01 | 2017-08-08 | Respivert Ltd | Dérivés de diaryle-urée en tant qu'inhibiteurs de kinase p38 |
-
2013
- 2013-11-25 GB GBGB1320729.5A patent/GB201320729D0/en not_active Ceased
-
2014
- 2014-11-25 EA EA201690531A patent/EA034216B1/ru not_active IP Right Cessation
- 2014-11-25 WO PCT/GB2014/053490 patent/WO2015075483A1/en active Application Filing
- 2014-11-25 BR BR112016011078-1A patent/BR112016011078B1/pt active IP Right Grant
- 2014-11-25 MX MX2016006119A patent/MX372942B/es active IP Right Grant
- 2014-11-25 PT PT14806358T patent/PT3074396T/pt unknown
- 2014-11-25 PL PL14806358T patent/PL3074396T3/pl unknown
- 2014-11-25 CN CN201480062652.6A patent/CN105793260B/zh active Active
- 2014-11-25 HU HUE14806358A patent/HUE045596T2/hu unknown
- 2014-11-25 KR KR1020167011667A patent/KR102327096B1/ko active Active
- 2014-11-25 DK DK14806358.9T patent/DK3074396T3/da active
- 2014-11-25 EP EP14806358.9A patent/EP3074396B1/en active Active
- 2014-11-25 CA CA2928009A patent/CA2928009C/en active Active
- 2014-11-25 AU AU2014351571A patent/AU2014351571B2/en active Active
- 2014-11-25 JP JP2016554928A patent/JP6389529B2/ja active Active
- 2014-11-25 ES ES14806358T patent/ES2740325T3/es active Active
- 2014-11-25 US US15/038,872 patent/US9725447B2/en active Active
-
2016
- 2016-04-12 IL IL245062A patent/IL245062B/en active IP Right Grant
- 2016-05-23 SA SA516371189A patent/SA516371189B1/ar unknown
-
2017
- 2017-06-29 US US15/637,365 patent/US10167282B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| JP6389529B2 (ja) | 2018-09-12 |
| US10167282B2 (en) | 2019-01-01 |
| PL3074396T3 (pl) | 2019-11-29 |
| JP2016538345A (ja) | 2016-12-08 |
| IL245062A0 (en) | 2016-06-30 |
| EP3074396A1 (en) | 2016-10-05 |
| EP3074396B1 (en) | 2019-05-01 |
| CN105793260A (zh) | 2016-07-20 |
| AU2014351571A1 (en) | 2016-05-19 |
| HUE045596T2 (hu) | 2020-01-28 |
| DK3074396T3 (da) | 2019-07-29 |
| CN105793260B (zh) | 2018-04-10 |
| CA2928009A1 (en) | 2015-05-28 |
| KR20160079792A (ko) | 2016-07-06 |
| AU2014351571B2 (en) | 2019-02-14 |
| BR112016011078A8 (pt) | 2020-04-22 |
| EA034216B1 (ru) | 2020-01-17 |
| US20160355510A1 (en) | 2016-12-08 |
| KR102327096B1 (ko) | 2021-11-16 |
| US20170298066A1 (en) | 2017-10-19 |
| IL245062B (en) | 2019-09-26 |
| CA2928009C (en) | 2022-08-30 |
| US9725447B2 (en) | 2017-08-08 |
| SA516371189B1 (ar) | 2018-06-30 |
| PT3074396T (pt) | 2019-08-06 |
| MX2016006119A (es) | 2016-12-14 |
| EA201690531A1 (ru) | 2016-08-31 |
| GB201320729D0 (en) | 2014-01-08 |
| MX372942B (es) | 2020-04-03 |
| BR112016011078B1 (pt) | 2022-12-06 |
| WO2015075483A1 (en) | 2015-05-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2740325T3 (es) | Derivados de 1-(5-terc-butil-2-aril-pirazol-3-il)-3-[2-fluoro-4-[(3-oxo-4h-pirido[2,3 b]piracin-8-il)oxi]fenil]urea como inhibidores raf para el tratamiento del cáncer | |
| ES2520940T3 (es) | Compuestos de pirido[2,3-b]pirazina 8-sustituida y su uso | |
| KR101473662B1 (ko) | 피리도피라진 유도체 및 신호 전달 경로의 조절제로서의 이의 용도 | |
| JP6151919B2 (ja) | ヘタリールアミノナフチリジン | |
| JP5256047B2 (ja) | ピロロ[3,2−c]ピリジン−4−オン2−インドリノン(indolinone)プロテインキナーゼ阻害剤 | |
| JP5827627B2 (ja) | TGF−β受容体キナーゼ阻害剤としてのヘテロアリールアミノキノリン | |
| JP6054406B2 (ja) | Fgfrキナーゼ阻害を介した抗癌ベンゾピラジン | |
| US20110130406A1 (en) | Pyrazolo-pyridines as tyrosine kinase inhibitors | |
| CN112142744A (zh) | 取代的稠合杂芳双环化合物作为激酶抑制剂及其应用 | |
| JP2011525535A (ja) | PI3K/mTOR阻害剤 | |
| US20100226881A1 (en) | PYRAZOLO[1,5-a]PYRIDINES AND THEIR USE IN CANCER THERAPY | |
| KR20240009929A (ko) | Parp 억제제로서 치환된 융합 이환 화합물 및 이의 용도 | |
| CN112912142B (zh) | 作为腺苷受体拮抗剂的5-氮杂吲唑衍生物 | |
| TWI565698B (zh) | 喹啉化合物,其製造方法及用途 | |
| AU2013207082A1 (en) | Therapeutic use of imidazopyridine derivatives | |
| HK1229330A1 (en) | 1 -(5-tert-butyl-2-aryl-pyrazol-3-yl)-3-[2-fluoro-4-[(3-oxo-4h-pyrido[2, 3-b]pyrazin-8-yl)oxy]phenyl]urea derivatives as raf inhibitors for the treatment of cancer | |
| HK1229330B (en) | 1 -(5-tert-butyl-2-aryl-pyrazol-3-yl)-3-[2-fluoro-4-[(3-oxo-4h-pyrido[2, 3-b]pyrazin-8-yl)oxy]phenyl]urea derivatives as raf inhibitors for the treatment of cancer | |
| CN120035595A (zh) | 噻唑并[5,4-d]嘧啶化合物、包含其的组合物及其用途 | |
| TW201524962A (zh) | 喹唑啉化合物,其製備方法及用途 |