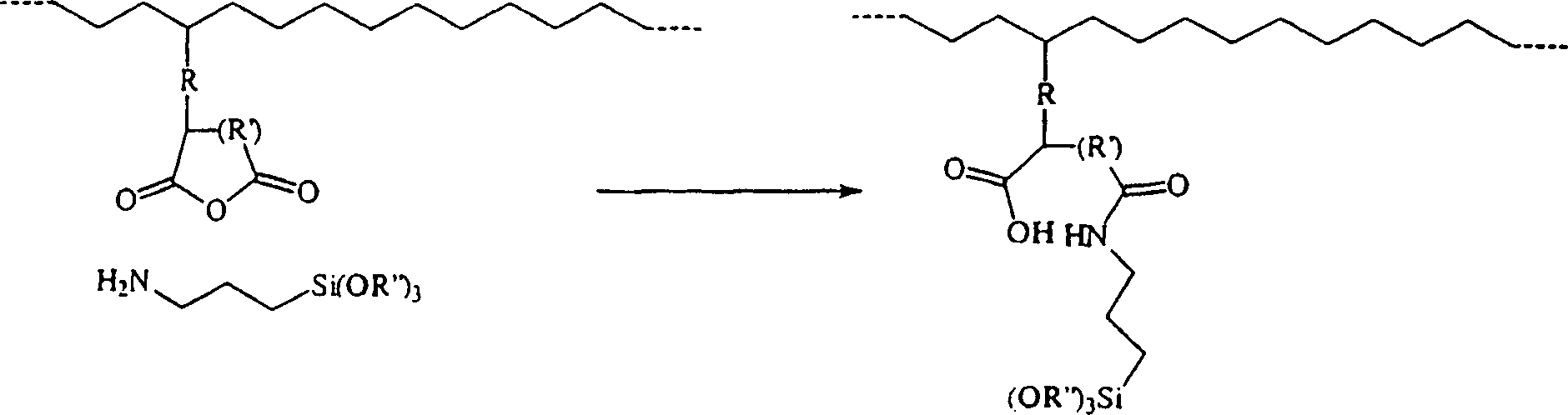
-
Hintergrund
der Erfindung
-
Thermoplastische
Elastomere (TPE) zeigen die funktionellen Eigenschaften von herkömmlichen
wärmehärtenden
Kautschuken, sie können
jedoch rasch geschmolzen werden und sind deshalb zur Verarbeitung in
einer herkömmlichen
thermoplastischen Fabrikationsausrüstung geeignet. Der größte Teil
der TPE besteht aus zwei Phasen, einer, die aus einem Kautschukmaterial
(Elastomer) besteht, die in der anderen unlöslich ist, und einem fließfähigen thermoplastischen
Material. Das Kautschukmaterial liegt als dispergierte Phase vor, und
der Thermoplast ist die kontinuierliche Phase.
-
Obwohl
es im Prinzip nicht erforderlich ist, den Kautschuk in einem TPE
zu vernetzen, hat es sich als wirksam erwiesen, Vernetzungstechniken
zu verwenden, um eine bessere chemische Beständigkeit, bessere mechanische
Eigenschaften und eine bessere Steuerung der Phasentrennung zu erhalten.
Derartige TPE-Zusammensetzungen werden, wenn eine Vernetzungsreaktion
und ein Verfahren verwendet werden, um eine Phasentrennung in aufgeteilte
Bereiche zu erzielen, als thermoplastische Vulkanisate (TPV) bezeichnet.
Um deren thermoplastischen Charakter zu erhalten, ist es wesentlich,
dass nur die Kautschukphase vernetzt wird. Bezüglich einer umfangreichen und
detaillierten Beschreibung und Übersichtsbesprechung
der TPV-Technologie siehe beispielsweise 5. Abdou-Sabet, R. C. Puydak
und C. P. Rader in Rubber Chemistry and Technology, Bd. 69, S. 476–493, 1996.
-
Weiter
wurde demonstriert, dass sich das mechanische Verhalten von TPVs
mit dem Vernetzungsgrad der Gummiphase und mit dem Inversen der
Teilchengröße der Gummibereiche
verbessert. Eine dynamische Vernetzung (die aus einem innigen Mischen
einer Mischung von kompatiblen Polymeren, dann dem Einführen eines
Vernetzungssystems in die Mischung, während das Mischverfahren fortgesetzt
wird, besteht) wird verwendet, um die fein dispergierte, hoch vernetzte
Gummiphase aus einer homogenen Mischung von Polymeren zu erzeugen.
-
Aus
thermodynamischen und hydrodynamischen Gründen wird es bevorzugt, dass
die Polymerviskosität
erhöht
wird, während
die Vernetzung stattfindet, da die Teilchen dazu tendieren zu agglomerieren,
während
die Phasen getrennt werden. Wenn darüber hinaus bei der Vernetzung
ein Phasenumkehrprozess stattfinden kann, ist dies für die Bildung
von faserigen Gummibereichen günstig,
die für
spezielle mechanische Eigenschaften sorgen können. Jedoch wurde gefunden,
dass es vorzuziehen ist, einen Vernetzungsmechanismus zu wählen, an
dem die thermoplastische Phase teilweise beteiligt ist, nicht bis
zu dem Punkt, an dem der thermoplastische Charakter des TPV entfernt
wird, sondern lediglich, um eine bessere Haftung und Kompatibilität der Polymere
zu erzielen.
-
Die
Wahl eines Vernetzungsverfahrens und von Chemikalien wird von Verarbeitungserfordernissen
regiert, z. B. der Reaktionsgeschwindigkeit bei der Verarbeitungstemperatur;
der Kompatibilität
mit dem Elastomer; den Nebenreaktionen mit dem Thermoplasten; der
Effizienz (Zahl der Vernetzungen, die von jedem Molekülvernetzer
erzeugt werden); der Abwesenheit von unerwünschten Reaktionen; der Toxizität und dem
Gefahrenpotential; der Farbe und dem Geruch.
-
Ein
Beispiel für
derartige TPVs ist EPDM/PP, das im U.S.-Patent Nr. 3,130,535 beschrieben
ist.
-
EPDM
und PP werden innig in einem Innenkneter gemischt, und ein Peroxid
wird dazugegeben, um das EPDM zu vernetzen. Überschüssiges Peroxid und/oder eine übermäßig hohe
Verarbeitungstemperatur und/oder übermäßig reaktive Polymere verursachen
einen Abbau der PP-Phase und/oder Scorch. Im Gegensatz dazu verursachen
eine unzureichende Menge an Peroxid und/oder eine zu niedrige Verarbeitungstemperatur
und/oder ein wenig reaktives EPDM eine unzureichende Vernetzung.
-
Ein
Nachteil der TPVs auf Polyolefin-Basis ist, dass sie nicht ohne
eine vorangehende Oberflächenbehandlung
aufgestrichen werden können.
Es wurde im U.S.-Patent Nr. 4,311,628 offenbart, dass andere Vernetzungsmittel
verwendet werden können,
z. B. Dimethyloloctylphenol-Harz und Schwefel. Es konnten überlegene
mechanische Eigenschaften erzielt werden, aber leider leiden beide
Systeme an einem übermäßigen Geruch
und/oder einer Gelbfärbung
der resultierenden Materialien sowie an der schwierigen Steuerung
der Schwefel-Vulkanisationsreaktionen.
-
Im
europäischen
Patent 0 324 434 wurde offenbart, Silan-gepfropfte Polymere in der
thermoplastischen Phase zu verwenden. Nach dem Mischen wird das
Material geformt, und man lässt
es mit atmosphärischer
Feuchtigkeit reagieren. So war es möglich, nach einer Wasserhärtung ein
stärker
elastomeres Material zu erhalten. Jedoch enthält der erhaltene wassergehärtete Gegenstand
kein thermoplastisches Elastomer mehr und kann nicht wiederverarbeitet
werden. Um diese Beschränkung
zu überwinden,
offenbarte das europäische
Patent 0 409 542 das Mischen eines EPR (Ethylen-Propylen-Kautschuks) oder
EPDM mit einem kristallinen Ethylen-Propylen-Thermoplasten, einem
organofunktionellen Silan und einem Radikalerzeuger. Das Silan wird
durch den Radikalerzeuger auf das Harz gepfropft, und die Vernetzung
findet durch die Reaktion des Silans mit Wasser statt.
-
Eine
Verfeinerung der obigen Verfahren ist im europäischen Patent 0 510 559 offenbart,
in dem zuerst EPR oder EPDM gepfropft wird, dann dem thermoplastischen
PP und einem vernetzenden Additiv, das Wasser umfasst, beigemischt
wird. Das gleiche Verfahren ist unter Verwendung von Polyethylen
sehr niedriger oder ultraniedriger Dichte (VLDPE oder ULDPE) offenbart,
um die Ausgangsmaterialkosten zu verringern und die Mischtemperaturen
zu erniedrigen. Siehe die
DE
44 02 943 . Es wird auch vorgeschlagen, die PP-Komponente und
die PE-Komponente zusammen mit Silan und dem Radikalerzeuger als
trockene Mischung zuzusetzen, wobei die Zugabe von Wasser und des
Kondensationskatalysators in einer anschließenden Stufe vorgenommen wird.
Jedoch ist die Zugabe von Wasser in einen Extruder bei Temperaturen
gut über
dessen Siedepunkt ein schwieriges Verfahren. Darüber hinaus ist die benötigte Wassermenge
so gering, dass ihre Zudosierung eine anspruchsvolle Ausrüstung erfordert,
im Widerspruch zum Ziel des Patents.
-
Das
U.S.-Patent Nr. 4,146,529 an Yamamoto et al. offenbart die Umsetzung
eines Säure-modifizierten Polypropylens
mit einem Amino- oder Epoxysilan, aber der Zweck einer derartigen
Reaktion besteht darin, die Alkoxygruppen zu verwenden, um die Füllstoffe
zu binden und das nicht-gepfropfte Carbonsäureanhydrid umzusetzen, um
nicht-flüchtige
Produkte mit geringem Geruch zu bilden, nicht um die Alkoxy-Funktionalitäten unter
sich zu vernetzen. Der Zweck dieser Zusammensetzungen besteht darin,
Mineralstoffe anzubinden und nicht, thermoplastische Vulkanisate
zu bilden; oder in Abwesenheit von Füllstoff, die Reaktion des Amino
oder Epoxy des Silans mit freier, flüchtiger, nicht-gepfropfter
Säure oder
freiem, flüchtigem,
nicht-gepfropftem Anhydrid zu begünstigen.
-
Das
deutsche Patent
DE 196 29 429 lehrt
(unter anderen Gegenständen)
die Verwendung von Vormischungen von Vinylsilanen, Aminosilan und
ungesättigten
Carbonsäureanhydriden,
die jeweils für
die Vernetzung von Polyolefinen verwendet werden.
-
Kurze Beschreibung
der Zeichnungen
-
Die 1–5 veranschaulichen
physikalische Eigenschaften von TPVs, die in den Beispielen hergestellt
werden.
-
Zusammenfassung
der Erfindung
-
Die
vorliegende Erfindung lehrt die Herstellung von TPVs unter Verwendung
von Polymeren, Carbonsäureanhydrid
und einem Aminosilan.
-
Detaillierte
Beschreibung der Erfindung
-
Die
Ziele der vorliegenden Erfindung bestehen darin, neue TPVs mit einem
großen
Bereich von Eigenschaften, preiswerte TPVs, anstreichbare TPVs herzustellen,
die Herstellung von TPVs in herkömmlichen Mischern
ohne das Erfordernis für
eine teure Zusatzausrüstung
zu ermöglichen,
die Verwendung von hohen Mengen an Vernetzungsmitteln (z. B. metallorganischen
Verbindungen oder Peroxiden) bei der Herstellung von TPVs zu vermeiden
und stabile TPV-Zusammensetzungen zu erhalten. ZUSAMMENSETZUNG
-
Die
TPVs sind eine Mischung aus
- (a) einem ersten
Polymer (Kautschukphase);
- (b) einem kristallinen oder teilkristallinen thermoplastischen
Polymer (thermoplastische Phase);
- (c) einem Carbonsäureanhydrid,
das der Komponente (a) als Comonomer einverleibt wird oder auf die Komponente
(a) gepfropft wird; und
- (d) einem Aminosilan, das kein tertiäres Aminosilan ist;
welche
man vernetzen lässt.
-
A. Polymere
-
Geeignete
Polyolefin-Kautschukphasen-Komponenten (a) sind alle Polymere, die
so umgesetzt werden können,
dass ein Carbonsäureanhydrid-haltiges
Polymer geliefert wird, wie z. B. Ethylen-Propylen-Copolymer (EPR);
Ethylen-Propylen-Dien-Terpolymer (EPDM); Butylkautschuk (BR); Naturkautschuk
(NR); chlorierte Polyethylene (CPE); Siliconkautschuk; Isoprenkautschuk
(IR); Butadienkautschuk (BR); Styrol-Butadien-Kautschuk (SBR); Ethylen-Vinylacetat
(EVA); Ethylen-Butylacrylat (EBA); Ethylen-Methacrylat (EMA); Ethylen-Ethylacrylat
(EEA); Ethylen-alpha-Olefin-Copolymere (z. B. EXACT und ENGAGE,
LLDPE (lineares Polyethylen niedriger Dichte)), Polyethylen hoher
Dichte (HDPE) und Nitrilkautschuk (NBR). Polypropylen ist als diese
Phase nicht geeignet, da es eine Tendenz aufweist, sich bei der
Vernetzung abzubauen; wenn jedoch das Polypropylen ein Copolymer
oder Pfropfcopolymer von Propylen mit einem Säureanhydrid ist, kann es verwendet
werden. Vorzugsweise ist das Polymer ein Ethylen-Polymer oder -Copolymer
mit mindestens 50% Ethylen-Gehalt (bezüglich Monomer), bevorzugter
sind mindestens 70% der Monomere Ethylen.
-
Geeignete
thermoplastische Polymere (b) sind Polypropylen (PP); Polyethylen,
insbesondere hoher Dichte (PE); Polystyrol (PS); Acrylnitril-Butadien-Styrol
(ABS); Styrol-Acrylnitril (SAN); Polymethylmethacrylat (PMMA); thermoplastische
Polyester (PET, PBT); Polycarbonat (PC); und Polyamid (PA).
-
Derartige
Polymere können
durch jedes in der Technik bekannte Verfahren hergestellt werden,
einschließlich
einer Festphasen-, Aufschlämmungsphasen-,
Gasphasen-, Lösungsmittelphasen-,
Grenzphasenpolymerisation (radikalisch, ionisch, Metall-initiiert
(z. B. Metallocen, Zielger-Natta)), Polykondensation, Polyaddition
oder Kombinationen dieser Methoden.
-
Es
ist möglich,
dass die Polymere für
die zwei Phasen die gleichen sind, wobei das Säureanhydrid zuvor zu einem
Teil des Polymers gegeben wird, wobei das so umgesetzte Polymer
als die Kautschukphase innerhalb des TPV wirken wird. Eine solche
vorherige Zugabe schließt
die Möglichkeiten
ein, dass das Säureanhydrid
als Comonomer in dem Polymer anwesend ist oder dass das Säureanhydrid
zuvor mit dem Polymer umgesetzt wird. In beiden Fällen wäre die Zugabe
des getrennten Säureanhydrids
nicht erforderlich, da es in dem Polymer vorliegt. Angesichts dieses
Grades von Komplexität
wird es bevorzugt, dass die zwei Polymere voneinander verschieden
sind.
-
Eine
dritte Alternative besteht darin, dass das Polymer der Kautschukphase
und der thermoplastischen Phase das gleiche Polymer sein können, dass
aber das Säureanhydrid
dem Polymer als Ganzem zugesetzt wird. In einem derartigen Fall
würde,
wenn das Silan zugesetzt wird, ein Teil des Polymers die Kautschukphase
bilden, während
ein anderer Teil nicht reagieren würde (im Hinblick auf die relativ
geringe vorhandene Menge an Anhydrid und Silan). Es ist wichtig,
dass während
des Verfahrens ein geeigneter Grad der Phasentrennung zwischen der
Kautschuk- und der thermoplastischen Phase geschaffen wird. Dieses
Verfahren würde
nicht notwendigerweise zu einem TPV führen, da man nicht notwendigerweise
die erforderliche Phase erzielen würde, ohne die Verfahrenskomplexität bedeutend
zu erhöhen,
und deshalb ist es nicht bevorzugt.
-
Im
Fall von zwei verschiedenen Polymeren wird das Polymer, das mit
dem Säureanhydrid
reaktiver ist, durch das Säureanhydrid
gepfropft und wirkt als die Kautschukphase in dem TPV.
-
Das
Polymer, welches die Kautschukphase bilden soll, muss extrudierbar
sein und sollte in der Lage sein, mit dem Säureanhydrid gepfropft zu werden.
-
Der
Schmelzpunkt der thermoplastischen Phase sollte geringer sein als
die Zersetzungstemperatur des Aminosilans sowie die Zersetzungstemperatur
des Säureanhydrids
(falls nicht das Säureanhydrid
ein Comonomer in dem Polymer ist).
-
Die
Polymere können
unimodale, bimodale oder multimodale Molekulargewichtsverteilungen
aufweisen. Der Schmelzfluss der Polymere kann irgendeiner von denjenigen
sein, die in der Technik zur Verwendung bei der Bildung von Thermoplasten
und Kautschuken bekannt sind.
-
B. Carbonsäureanhydride
-
Alle
Carbonsäureanhydride,
die durch irgendeinen möglichen
Mechanismus auf das Polymer gepfropft werden können, das die Kautschukphase
bilden soll, können
verwendet werden. Es wird bevorzugt, dass entweder in dem Polymer
oder bevorzugter in dem Säureanhydrid
eine Unsättigung
vorliegt, um dieses Pfropfen zu bewerkstelligen. Die Unsättigung
des Carbonsäureanhydrids
kann innerhalb oder außerhalb
einer Ringstruktur, falls vorhanden, vorliegen, solange sie die
Reaktion mit dem Polymer ermöglicht.
Das Säureanhydrid
kann Halogenide einschließen.
Mischungen von verschiedenen Carbonsäureanhydriden können verwendet
werden. Beispielhafte ungesättigte
Carbonsäureanhydride
zur Verwendung sind Isobutenylbernstein-, (+/–)-2-Octen-1-ylbernstein-,
Itacon-, 2-Dodecen-1-ylbernstein-, cis-1,2,3,6-Tetrahydrophthal-,
cis-5-Norbornen-endo-2,3-dicarbon-, endo-Bicyclo[2.2.2]oct-5-en-2,3-dicarbon-, Methyl-5-norbornen-2,3-carbon-, exo-3,6-Epoxy-1,2,3,6-tetrahydronaphthalin-,
Malein-, Citracon-, 2,3-Dimethylmalein-, 1-Cyclopenten-1,2-dicarbon-,
3,4,5,6-Tetrahydrophthal-, Brommalein- und Dichlormaleinsäureanhydrid.
-
Diese
Säureanhydride
können
als Comonomer in dem Polymer der Kautschukphase vorliegen oder auf
das Polymer, das die Kautschukphase bilden wird, gepfropft werden.
-
Die
zu verwendende Menge an Säureanhydrid
beträgt
0,01 bis 1,0 Gew.-%, bezogen auf die Gesamtmenge an vorhandenem
Polymer.
-
C. Aminosilane
-
Die
Aminosilane zur Verwendung hierin weisen mindestens eine hydrolysierbare
Gruppe, z. B. Alkoxy, Acetoxy oder Halogen, bevorzugt Alkoxy, auf.
Vorzugsweise liegen mindestens zwei derartige hydrolysierbare Gruppen
vor, die eine Vernetzungskondensationsreaktion eingehen können, so
dass die resultierende Verbindung eine solche Vernetzung eingehen
kann. Eine Mischung von verschiedenen Aminosilanen kann verwendet
werden.
-
Das
Amin muss eine ausreichende Reaktionsgeschwindigkeit mit dem Säureanhydrid
aufweisen. Tertiäre
Amine reagieren nicht geeignet mit dem Säureanhydrid und sind nicht
Teil der Erfindung. Die Aminogruppe kann durch eine verzweigte Gruppe
mit dem Siliciumatom verbrückt
sein, um die Gelbfärbung
der resultierenden Zusammensetzung zu verringern.
-
Das
Silan kann durch die Formel YNHBSi(OR)a(X)3_a dargestellt werden,
worin a = 1 bis 3, vorzugsweise 3, Y Wasserstoff, ein Alkyl, Alkenyl,
Hydroxyalkyl, Alkaryl, Alkylsilyl, Alkylamin, C(=O)OR oder C(=O)NR ist,
R ein Acyl, Alkyl, Aryl oder Alkaryl ist, X für R oder Halogen stehen kann,
B ein zweiwertige Verbrückungsgruppe
ist, die vorzugsweise Alkylen ist, die verzweigt (z. B. Neohexylen)
oder cyclisch sein kann. B kann Heteroatombrücken, z. B. eine Etherbindung,
enthalten. Bevorzugt ist B Propylen. Bevorzugt ist R Methyl oder Ethyl.
Methoxy-haltige Silane können
ein besseres Vernetzungsverhalten als Ethoxygruppen sicherstellen.
Bevorzugt ist Y ein Aminoalkyl, Wasserstoff oder Alkyl. Bevorzugter
ist Y Wasserstoff oder ein primäres
Aminoalkyl (z. B. Aminoethyl). Vorzuziehende X sind Cl und Methyl,
bevorzugter Methyl. Beispielhafte Silane sind γ-Aminopropyltrimethoxysilan
(SILQUEST® A-1110-Silan von Witco
Corp., Greenwich, CT, USA); γ-Aminopropyltriethoxysilan
(SILQUEST A-1100); γ-Aminopropylmethyldiethoxysilan;
4-Amino-3,3-dimethylbutyltriethoxysilan; 4-Amino-3,3-dimethylbutylmethyldiethoxysilan;
N-β-(Aminoethyl)-γ-aminopropyltrimethoxysilan
(SILQUEST A-1120); H2NCH2CH2NHCH2CH2NH(CH2)3Si(OCH3)3 (SILQUEST A-1130)
und N-β-(Aminoethyl)-γ-aminopropylmethyldimethoxysilan
(SILQUEST A-2120). Andere geeignete Aminosilane sind wie folgt: 3-(N-Allylamino)propyltrimethoxysilan,
4-Aminobutyltriethoxysilan, 4-Aminobutyltrimethoxysilan, (Aminoethylaminomethyl)phenethyltrimethoxysilan,
Aminophenyltrimethoxysilan, 3-(1-Aminopropoxy)-3,3-dimethyl-1-propenyltrimethoxysilan,
Bis[(3-trimethoxysilyl)propyl]ethylendiamin, N-Methylaminopropyltrimethoxysilan, Bis(γ-triethoxysilylpropyl)amin
(SILQUEST A-1170) und N-Phenyl-γ-aminopropyltrimethoxysilan
(SILQUEST Y-9669).
-
Wenn
das Aminosilan ein latentes Aminosilan, d. h. ein Ureidosilan oder
ein Carbamatosilan, ist, muss die Mischungstemperatur ausreichend
sein, damit die jeweilige Blockierungsgruppe das Amin verlässt und
ermöglicht,
dass das Amin mit der Säureanhydrid-Funktionalität reagiert,
etwa 150 bis 230°C.
Beispiele für
derartige latente Aminosilane sind tert-Butyl-N-(3-trimethoxysilylpropyl)carbamat,
Ureidopropyltriethoxysilan und Ureidopropyltrimethoxysilan. Andere
Carbamatosilane, die verwendet werden können, sind im U.S. Patent Nr. 5,220,047
offenbart. Vorzugsweise ist, um die zusätzliche Komplexität der Deblockierung
zu vermeiden, das Aminosilan kein derartiges latentes Aminosilan.
-
Das
Aminosilan sollte zu 250 bis 25000 ppm, bezogen auf das Gewicht
beider Polymere, vorliegen. Es sollte auch in einem Moläquivalent-Verhältnis zu
dem Säureanhydrid
von etwa 0,1 bis 10, bevorzugter 0,9 bis 1,1, am bevorzugtesten
in einem Verhältnis
von etwa 1 : 1 vorliegen.
-
Das
Silan kann auf einem Träger,
wie einem porösen
Polymer, Siliciumdioxid, Titandioxid oder Ruß, vorliegen, so dass es leicht
bei dem Mischverfahren zu dem Polymer zu geben ist. Beispiele für ein derartiges Material
sind ACCUREL-Polyolefin (Akzo Nobel), STAMYPOR-Polyolefin (DSM)
und VALTEC-Polyolefin (Montell),
SPHERILENE-Polyolefin (Montell), AEROSIL-Siliciumdioxid (Degussa),
MICRO-CEL E (Manville) und ENSACO 350G-Ruß (MMM Carbon).
-
E. Fakultative Additive
-
Ein
Radikalerzeuger wäre
erforderlich, wenn das Carbonsäureanhydrid
durch einen Radikalmechanismus auf das Polymer gepfropft wird, ist
aber nicht erforderlich, wenn das Säureanhydrid entweder über einen anderen
Mechanismus gepfropft wird oder ein Comonomer des Polymers ist.
Geeignete Radikalkatalysatoren können
aus der Gruppe von wasserlöslichen
oder öllöslichen
Peroxiden ausgewählt
sein, wie Wasserstoffperoxid, Ammoniumpersulfat, Kaliumpersulfat,
verschiedenen organischen Peroxy-Katalysatoren, wie Dialkylperoxide,
z. B. Diisopropylperoxid, Dilaurylperoxid, Di-t-butylperoxid, Di-(2-t-butylperoxyisopropyl)benzol; 3,3,5-Trimethyl-1,1-di(tert-butylperoxy)cyclohexan;
2,5-Dimethyl-2,5-di(t-butylperoxy)hexan;
2,5-Dimethyl-2,5-di(t-butylperoxy)hexin-3; Dicumylperoxid, Alkylhydrogenperoxiden,
wie t-Butylhydrogenperoxid, t-Amylhydrogenperoxid, Cumylhydrogenperoxid,
Diacylperoxide, z. B. Acetylperoxid, Lauroylperoxid, Benzoylperoxid,
Peroxyestern, wie Ethylperoxybenzoat, und den Azo-Verbindungen,
wie 2-Azobis(isobutyronitril).
-
Der
Radikalerzeuger kann zu 1/100 bis 1/1, bezogen auf die Molmenge
des Säureanhydrids,
vorliegen.
-
Standard-Additive,
wie Stabilisatoren (UV, Licht oder Alterung), Antioxidantien, Metalldesaktivatoren, Verarbeitungshilfsmittel,
Wachse, Füllstoffe
(Siliciumdioxid, TiO2, CaCO3,
Mg(OH)2, Ruß usw.) und Färbemittel können den
TPVs zugesetzt werden. Zusätzlich
können
den Polymeren Treibmittel zugesetzt werden, so dass, wenn sie extrudiert
werden, das Polymer einen Schaumstoff bildet. Beispiele für derartige
Treibmittel sind flüchtige
Kohlenwasserstoffe, Fluorkohlenwasserstoffe und Chlorfluorkohlenstoffe.
Herkömmlich
bekannte Schäumungsmittel,
wie Azocarbonamid oder Natriumbicarbonat (auch als Natriumhydrogencarbonat
bekannt), zersetzen sich bei erhöhten
Temperaturen unter Lieferung gasförmiger Produkte. Dieses sind
alle chemische Schäumungsverfahren.
Schaumstoffe können
auch durch Injektion von flüssigem
oder gasförmigem Schäumungsmittel
in die Polymerschmelze erzeugt werden. Beispiele sind z. B. Butan,
CO2, Stickstoff, Wasser, Helium usw. Die
Menge eines derartigen Treibmittels sollte bei 0,1 bis 50 Gewichtsprozent
der Polymere liegen.
-
Verfahren
-
In
einer ersten Reaktion wird das Carbonsäureanhydrid auf das Kautschukphasen-Polymer
gepfropft (am bevorzugtesten durch einen Radikalmechanismus). Diese
Reaktion kann vorgenommen werden, während beide Polymere anwesend
sind oder während
die zwei Polymer getrennt sind, obwohl es bevorzugt wird, dies zu
bewerkstelligen, wenn beide Polymere anwesend sind. Wie oben angeführt, kann
dieser Schritt alternativ wirksam durch Einschluss des Carbonsäureanhydrids
als Comonomer in dem Kautschukphasen-Polymer bewerkstelligt werden
(in welchem Fall kein Radikalerzeuger erforderlich ist). Das Polymer
sollte vor der Reaktion mit Aminosilan mit Carbonsäureanhydrid
gepfropft/copolymerisiert werden, da das Reaktionsprodukt zwischen Säureanhydrid
und Aminosilan nur einen schlechten Pfropfwirkungsgrad aufweist.
Eine vorherige Reaktion zwischen Aminosilan und Säureanhydrid
hätte die
Bildung eines Semiamids zur Folge, das unterlegene Pfropfeigenschaften
aufweisen könnte.
In diesem Fall würde
keine Vernetzung stattfinden. Im Gegenteil, ein teilweiser Abbau
des Polymers und/oder die weichmachende Wirkung des Semiamids könnte zu
einer Erhöhung
des Schmelzflussindex (MFI) führen.
-
Es
ist vorzuziehen, während
des Pfropfschrittes mit dem Anhydrid Radikalerzeuger zuzusetzen,
um das Pfropfen des Säureanhydrids
auf das Kautschukphasen-Polymer zu induzieren.
-
Wenn
das zweite thermoplastische Polymer bei der Pfropfung nicht anwesend
ist, sollte es mit dem gepfropften Kautschukphasen-Polymer vor der
Zugabe des Aminosilans gemischt werden; jedoch leidet ein derartiges
Verfahren an Nachteilen im Hinblick auf die mechanischen Eigenschaften
des resultierenden TPV.
-
Der
zweite Schritt ist die Zugabe des Aminosilans zu der Mischung aus
gepfropftem Kautschukphasen-Polymer/thermoplastischem Polymer. Es
müssen
kein Wasser und/oder Katalysator zugesetzt werden. Dies ist eine
sehr schnelle Reaktion zwischen Aminosilan und dem gepfropften Säureanhydrid.
Die Reaktion einer gepfropften Säureanhydrid-Einheit
mit γ-Aminopropylsilan
kann veranschaulicht werden als:
-
-
Die
Reaktion zwischen dem Silan und der gepfropften Säureanhydrid-Einheit
muss schnell, um ein vernetzbares Material zu erhalten, und schneller
als die Reaktion zwischen den Alkoxygruppen und dem Säureanhydrid
sein. Eine verzögerte
Reaktion könnte
gleichzeitig mit der Vernetzung der Alkoxy-Einheiten stattfinden,
was eine Vernetzung verhindern würde.
Die Reaktion zwischen Aminogruppe und Anhydrid ist sehr schnell,
wenn z. B. primäre
Aminogruppen verwendet werden. Die Reaktion kann durch Verwendung
von sekundären
Aminogruppen verlangsamt werden. Darüber hinaus sollte dieser Schritt
bei einer erhöhten
Temperatur, z. B. von 50 bis 200°C,
abhängig
von der Polymermischung, stattfinden.
-
Vorzugsweise
liegt die Polymermischung in einem Schmelzstadium vor, wenn das
Aminosilan zu dem Polymer gegeben wird.
-
Nachdem
das Aminosilan auf das eine Polymer gepfropft worden ist, sollte
man es vernetzen lassen, um die Gelphase des vernetzten Polymers
zu bilden. Es muss keine getrennte Feuchtigkeitshärtung stattfinden.
Ein Kondensationskatalysator kann verwendet werden, um den Vernetzungsprozess
zu beschleunigen, obwohl das Semiamid ein ausreichender Katalysator
sein sollte. Eine bis zehn Minuten bei einer erhöhten Temperatur von 60 bis
200°C sollten
sicherstellen, dass eine solche Vernetzung stattfindet.
-
Es
sollte bemerkt werden, dass die Gesamtmenge an Additiven nur 0,4%
der Gesamt-Zusammensetzung
beträgt,
etwa fünfmal
weniger als die Menge, die für
eine Peroxid- oder Vinylsilan-Härtung erforderlich ist.
Dies führt
auf zweierlei Weise zu Vorteilen, zu einer Verringerung der Gesamtkosten
und zu einer Verringerung von flüchtigen
Peroxiden, die Sicherheitsprobleme darstellen können.
-
Während und
nach der Vernetzung sollten alle Bestandteile in einem Innenmischer
gemischt werden. Der Mischer kann ein Extruder (Einzelschnecken,
Doppelschnecken usw.), ein BUSS KOKNEADER-Mischer oder ein einfacher
Innenmischer sein. Die Bedingungen für das Mischen hängen von
den Polymeren und dem Vernetzungsgrad ab.
-
Eigenschaften
-
Das
resultierende Produkt ist ein thermoplastisches Vulkanisat mit ausgezeichneten
mechanischen Eigenschaften. Die vernetzten Materialien weisen einen
signifikanten Gelgehalt und einen viel niedrigeren MFI als die Ausgangspolymere
auf, was die Kriechfestigkeit verbessern, für eine höhere Reißfestigkeit sorgen und Materialien
bereitstellen sollte, die härter
als nicht-vernetzte Polymermischungen sind. Das Endprodukt weist elastische
Eigenschaften (d. h. eine Bruchdehnung von mehr als 400%) auf, kann
aber mit Verfahren, die normalerweise in der Technik für Thermoplasten
bekannt sind, schmelzverarbeitet werden. Der bevorzugte Gelgehalt
des Endprodukts (d. h. Gummigehalt) liegt zwischen 10 und 50 Gew.-%,
am bevorzugtesten bei 25–35 Gew.-%.
Der Zug- und Biegmodul in der Maschinen- und Querrichtung sind verbessert,
ebenso wie die Fallbolzen-Schlagzähigkeit des Materials.
-
Die
TPVs sind verstreichbar und weisen eine bessere Ölbeständigkeit auf. Die TPVs können z.
B. in Klebstoffen und Dichtungsmitteln, Kabelisolierungen, Rohren,
Profilen, Formteilen, geschäumten
Teilen, Folien usw. verwendet werden.
-
Das
modifizierte Gummiphasen-Aminosilanpolymer tendiert dazu, mit dem
thermoplastischen Polymer kompatibler zu sein, was ein festeres
TPV bereitstellt.
-
Beispiele
-
Alle
verwendeten Silane sind oben angeführt, außer SILQUEST A-186 (γ-(3,4-Epoxycyclohexyl)ethyltrimethoxysilan),
SILQUEST A-187 (γ-Glycidoxypropyltrimethoxysilan)
und SILQUEST A-189 (γ-Mercaptopropyltrimethoxysilan).
Compoundierungsausrüstung:
Brabender-Kopf 50 cm3 mit Banbury-Messern. Compoundierungsparameter:
Brabender-Kopf bei 190°C/120
U/min. Alle Prozentsätze
sind Gewichtsprozentsätze,
falls nicht anders angegeben.
-
Verfahren
I: Gesamtmengen der Bestandteile der Formulierung: 55 g. Alle Bestandteile
wurden in den Brabender-Kopf eingespeist, mit Ausnahme des Polypropylen-Homopolymers
VALTEC HL003 (+ 5% Silan). Nach 5 min (Brabender-Drehmomentwert
wurde vermerkt) wurde das VALTEC HL003 (+5% Silan) in den Brabender-Kopf
gegeben. Die Zeit und der Wert des maximalen Brabender-Drehmoments
wurden als Indikator für
die Vernetzung vermerkt. Nachdem das Brabender-Drehmoment auf das
anfängliche
Niveau zurückgefallen
war und die Homogenisierung des Compounds erzielt worden war (ca.
10 min), wurde das Compound aus dem Brabender entfernt. Ein Prüfling, 1,5
mm Dicke, wurde in einer Presse bei 210°C/20 Bar geformt.
-
Verfahren
II: Gleiches Verfahren wie Verfahren I, aber alle Bestandteile wurden
gleichzeitig in den Brabender-Kopf eingespeist. Compoundierungszeit
15 min. Ein Prüfling,
1,5 mm Dicke, wurde in einer Presse bei 210°C/100 Bar über 200 s geformt.
-
Die
Beispiele 1–3
werden angegeben, um zu zeigen, dass ein großer Bereich von Eigenschaften
erhalten werden kann, abhängig
von den Polymergüten.
Gewichtsprozente, die für
das Material angegeben sind, das auf einem anderen Material getragen
ist, sind auf das Gewicht des Trägers
und des getragenen Materials bezogen.
-
Beispiel
1: Zusammensetzung: 75% ENGAGE 8452 Polyethylen ultraniedriger Dichte
(von Dupont Dow Elastomers, Schmelzindex (190°C/2,16 kg): 3 g/10 min; Dichte:
0,875 g/cm3), 17% VALTEC HL003 Polypropylen-Homopolymer
(SPHERIPOL® poröse Granalien
von Montell: Schmelzindex (230°C/2,16
kg): 0,7 g/10 min; Dichte: 0,900 g/cm3),
1% VALTEC HL003 (+ 5% aufgesaugtes INTEROX DHBP (2,5-Dimethyl-2,5-di(tert-butylperoxy)hexan
von Peroxid-Chemie, München)),
2% VALTEC HL003 (+ 5% aufgesaugtes Maleinsäureanhydrid), 5% VALTEC HL003
(+ 5% aufgesaugtes A-1100), Verfahren I. Das Maleinsäureanhydrid
wurde durch Schmelzen des Anhydrids (F. p. 53°C), Mischen desselben mit dem
Polymer und anschließendes
Abkühlen
in das Polymer absorbiert.
-
Beispiel
2: Zusammensetzung: 75% NORDEL 2722 Ethylen-Propylen-Dien-Monomer-Kautschuk
(Dupont Dow Elastomers: Dichte: 0,88 g/cm3,
Mooney-Viskosität
ML 1 + 4 bei 121°C:
28), 17% VALTEC HL003, 1% VALTEC HL003 (+ 5% aufgesaugtes INTEROX
DHBP), 2% VALTEC HL003 (+5% aufgesaugtes Maleinsäureanhydrid), 5% VALTEC HL003
(+ 5% aufgesaugtes A-1100), Verfahren I.
-
Beispiel
3: Zusammensetzung 75% ENGAGE D8842.00 Polyethylen ultraniedriger
Dichte (von Dupont Dow Elastomers: Schmelzindex (190°C/2,16 kg):
1,0 g/10 min; Dichte: 0,857 g/cm3), 17%
VALTEC HL003, 1% VALTEC HL003 (+ 5% aufgesaugtes INTEROX DHBP),
2% VALTEC HL003 (+ 5% aufgesaugtes Maleinsäureanhydrid), 5% VALTEC HL003
(+ 5% aufgesaugtes A-1100), Verfahren I.
-
Die
Beispiele 4–7
erläutern
den Einfluss des Verfahrens und der Bestandteile.
-
Beispiel
4 (Vergl.): Zusammensetzung: 75% ENGAGE 8452, 25% VALTEC HL003,
Verfahren II.
-
Beispiel
5 (Vergl.): Zusammensetzung: 75% ENGAGE 8452, 22% VALTEC HL003,
1% VALTEC HL003 (+ 5% aufgesaugtes INTEROX DHBP), 2% VALTEC HL003
(+ 5% aufgesaugtes Maleinsäureanhydrid). Verfahren
I.
-
Beispiel
6 (Vergl.): Zusammensetzung: 75% ENGAGE 8452, 17% VALTEC HL003,
1% VALTEC HL003 (+ 5% aufgesaugtes INTEROX DHBP), 2% VALTEC HL003
(+ 5% aufgesaugtes Maleinsäureanhydrid), 5%
VALTEC HL003 (+ 5% aufgesaugtes A-1100), Verfahren II.
-
Beispiel
7 (Vergl.): Zusammensetzung: 75% ENGAGE 8452, 17% VALTEC HL003,
1% VALTEC HL003 (+ 5% aufgesaugtes INTEROX DHBP), 2% VALTEC HL003
(+ 5% aufgesaugtes Maleinsäureanhydrid), 5%
VALTEC HL003 (+ 5% aufgesaugtes Hexadecylamin), Verfahren I.
-
Die
Beispiele 8–17
erläutern
den Einfluss einer Abwandlung des Silan-Typs. Die Beispiele 14–17 sind vergleichend.
Alle Silane wurden als 5gewichtsprozentige Vormischung auf der Basis
von VALTEC HL003 zugesetzt. Die Herstellung der Verbindungen war
gemäß Verfahren
I.
-
PHYSIKALISCHE
EIGENSCHAFTEN
-
Die
physikalischen Eigenschaften wurden gemäß den folgenden Normen gemessen:
| Dehnung
und Reißfestigkeit | ISO
37 (50 mm/min) |
| MFI | ISO
1827-1, Nr. 18 T |
| SHORE
A | ISO
868 |
| GELGEHALT | ISO
6427 |
| Drehmoment | Drehmomentmesser
auf Brabender |
-
ERGEBNISSE
ABWANDLUNG DER SILANE
-
Tabelle
1 zeigt die Eigenschaften der Materialien, die gemäß der im
experimentellen Teil erwähnten Verfahren
erhalten wurden. Die Vergleichsbeispiele (Vergl.-Bsp.) bestanden
aus:
Bsp. 4) der reinen Mischung von ENGAGE 8452 (PE) und VALTEC
HL003 (PP);
Bsp. 5) Mischung von ENGAGE, VALTEC, Peroxid und
Maleinsäureanhydrid;
Bsp.
6) Mischung von ENGAGE, VALTEC, Peroxid, Maleinsäureanhydrid und A-1100, aber
alles auf einmal gemischt, und
Bsp. 7) Mischung von ENGAGE,
VALTEC, Peroxid, Maleinsäureanhydrid
und Hexadecylamin.
-
Bsp.
4 dient dazu, die Eigenschaften der reinen Polymermischung anzugeben,
Bsp. 5, den Einfluss des Silans allein zu bewerten, Bsp. 6, den
Einfluss der Verarbeitung zu zeigen und Vergl.-Bsp. 7, die Notwendigkeit
zu belegen, dass ein Silan in der Mischung vorliegt.
-
TABELLE
1: Eigenschaften der Verbindungen
-
-
1 zeigt die erhaltenen Gelgehalte
für die
verschiedenen Silane, 2 zeigt
das maximale Drehmoment, das während
der Herstellungen der Compounds beobachtet wurde. Die in 1 und 2 gezeigten Daten zeigen, dass die Erhöhung des
maximalen Drehmoments, die während
der Compoundierung beobachtet wird, mit einer Vernetzungsreaktion
in Beziehung steht. Es ist offensichtlich, dass alle primären Amine
(SILQUEST A-1100-, A-1110-, A-1120-, A-1130- und A-2120-Silan) eine
unmittelbare Vernetzung induzieren, wenn sie nach Pfropfen des Maleinsäureanhydrids
in die Polymermischung eingemischt werden.
-
Im
Gegensatz dazu führte
das Mischen aller Materialien zur gleichen Zeit (Vergl.-Bsp. 6)
nicht zu irgendeiner signifikanten Vernetzung. Die leichten Unterschiede
des Vernetzungsgrades im Vergleich zu A-1100-Silan können entweder auf verschiedenen
Molmengen, einer verschiedenen Zahl und/oder Art von Alkoxygruppe
und Zahl von Amino-Funktionen pro Molekül beruhen. Das Material, das
unter Verwendung des sekundären
Aminosilans A-1170 erhalten wurde, zeigt ebenfalls einen relativ
hohen Vernetzungsgrad, wohingegen Y-9669 (sekundäres Aminosilan) keine signifikante
Vernetzung bewirkt. Es wird angemerkt, dass, da das Molekulargewicht
von A-1170 beträchtlich
höher ist
als das der anderen verwendeten Silane und die Experimente mit gleichen
Silangewichten durchgeführt
wurden, die Molmenge von A-1170 geringer ist als die der anderen
Silane und bei äquivalenten
Molmengen eine erhöhte
Gelbildung produziert. Das Letztere gilt auch für die anderen getesteten Silane
sowie für
den Fall, in dem Hexadecylamin anstelle eines Silans verwendet wurde.
-
Die
in den 3–5 dargestellten Daten zeigen
bei allen Fällen,
bei denen eine Vernetzung beobachtet wurde, eine signifikante Änderung
der Materialeigenschaften gegenüber
der reinen Mischung der beiden Polymere. Der Schmelzflussindex fällt auf
etwa 1/10, im Fall von A-1110-Silan sogar auf 1/30. Die Materialien
sind auch signifikant härter,
weisen eine höhere
Reißfestigkeit
und eine niedrigere Bruchdehnung auf. Die Änderungen der Eigenschaften
stehen mit dem relativ hohen Vernetzungsgrad in diesen Compounds
in Übereinstimmung.
-
Mit
Bezug auf die anderen verwendeten Silane können die Eigenschaften der
resultierenden Materialien nicht einem hohen Vernetzungsgrad zugeschrieben
werden. Jedoch bewirken alle Silane eine Abnahme des MFI, was anzeigt,
dass etwas Kettenverlängerung
stattgefunden hat. Dies wird aus dem Vergleich mit allen Vergleichsbeispielen
und insbesondere mit Vergl.-Bsp. 5 ersichtlich (nur Peroxid und
Maleinsäureanhydrid),
in dem sogar ein gewisses Maß an
Abbau stattgefunden hatte. Man bemerke, dass ein hoher MFI auch
gemessen wurde, als A-1100-Silan zur gleichen Zeit wie die anderen
Bestandteile eingemischt wurde.
-
Der
Austausch des Aminosilans durch Hexadecylamin (Vergl.-Bsp. 7) führt ebenfalls
zu einem Material mit überlegenen
mechanischen Eigenschaften und hoher Härte. Die Verbesserung der Eigenschaften
steht vermutlich mit der Anbringung langer Seitenketten an der Polymer-Hauptkette
("Kamm-Polymere") in Beziehung, da
keine Vernetzung möglich
ist. Demgemäß wurde
kein TPV hergestellt (kein Gelgehalt).
-
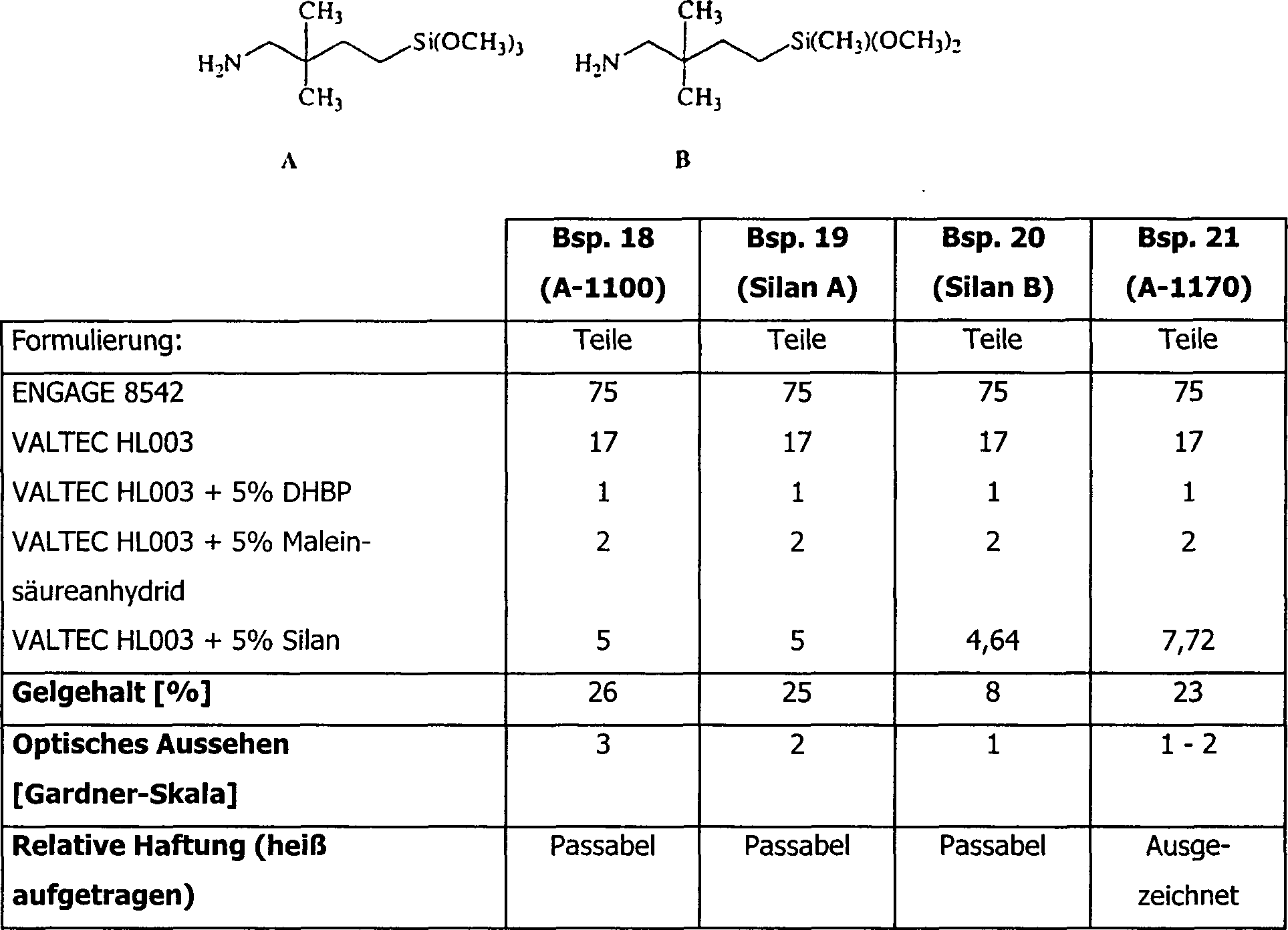
Beispiele 18–21 – ABWANDLUNG
VON AMINOSILANEN
-
Die
TPVs wurden gemäß dem oben
beschriebenen Verfahren hergestellt. Bei allen Ansätzen wurde die
Menge an Aminosilanen gleichgehalten (auf Mol-Basis). Die Silane
A und B sind wie folgt:
-
-
Die
Silane A, B und A-1170 ergaben Produkte, die etwas weniger gelb
sind als dasjenige, das unter Verwendung von A-1100 erhalten wurde.
A-1170 als Vernetzer führt
zu Materialien mit sehr verbesserten Klebeeigenschaften, wenn sie
mit den anderen Proben verglichen werden.
-
SCHAUMSTOFFANWENDUNG – Beispiele
22–25
-
Die
Verwendung von TPV-Produkten für
Schaumstoffanwendungen wurde bewertet (Beispiele 22 und 23) und
mit Formulierungen verglichen, die nur mit Peroxid und Maleinsäureanhydrid
allein (Beispiel 24) bzw. Peroxid (Beispiel 25) umgesetzt wurden.
Die Materialien wurden, mit Ausnahme des Treibmittels Azodicarbonamid,
bei 180°C
in dem Brabender umgesetzt (Einzelheiten der Herstellung der TPVs
wie oben). Nach 10 min wurde die Formulierung auf 160°C abgekühlt, und
das Treibmittel wurde zugesetzt. In Beispiel 23 wurde das Aminosilan
nach der Zugabe des Treibmittels zugesetzt. Nach weiterem 3-minütigen Mischen
wurden Testplatten bei 170°C/100
Bar/100 s hergestellt. Die Platten wurden in einem Ofen 3 bis 5
Minuten bei 200°C
geschäumt.
-
Formulierung
wie nachstehend angegeben
-
Die
Schmelzfestigkeit ist hoch genug, um die Zersetzungstemperatur von
Azodicarbonamid (200°C über mehrere
Minuten) zu tragen. Die Form verblieb innerhalb ihrer ursprünglichen
Abmessungen und klebt nicht an dem Träger. Im Gegensatz dazu änderten
die nicht-vernetzten Proben ihre Abmessungen auf Grund von Schäumen, was
zu einer schlechteren Zellenstruktur und zum Kleben an der Trägeroberfläche führte. Die Reihenfolge
der Zugabe (Silan vor oder nach Azodicarbonamid) scheint das Aussehen
des resultierenden Schaumstoffes oder die Menge an Gel nicht signifikant
zu beeinflussen.
-
Beispiele 26–29 – TPVs mit
einem einzigen Polymer
-
Das
allgemeine Verfahren ist das gleiche wie oben angegeben, jedoch
wurde in den Beispielen 27–29 nur
ein Polymer verwendet, in Beispiel 27 ein Polyethylen, in Beispiel
28 HDPE LUPOLEN 5031L von Elenac: MFI (190°C, 2,16 kg) = 6,5; Dichte: 0,952,
und in Beispiel 29 Polypropylen.
-
-
Das
System gepfropftes Maleinsäureanhydrid/Aminosilan
kann verwendet werden, um reines ENGAGE-Harz sowie reines HDPE zu
vernetzen, führte
aber nicht zu irgendeinem Gel, wenn es in Polypropylen verwendet
wurde.