-
Gebiet der Erfindung
-
Die
vorliegende Erfindung bezieht sich auf sauerstoffhaltige heterocyclische
Verbindungen, welche Hemmaktivität
für Phosphodiesterase
(PDE) IV haben, und welche brauchbar sind als ein therapeutisches
Mittel für
allergische Entzündungszustände wie
Bronchialasthma, allergische Rhinitis, atopische Dermatitis und Nephritis;
Autoimmunkrankheiten wie chronische obstruktive Lungenkreislaufkrankheit,
Rheumatismus, Multiple Sclerose, Crohnsche Krankheit, Psoriasis
und systemische Lupus Erythematosus; Krankheiten des zentralen Nervensystems
wie Depression, Amnesie und Demens; Organopathie, verbunden mit
ischämischem Reflux,
hervorgerufen durch Herzversagen, Schock und cerebrovaskuläre Krankheit
und dergleichen; insulinresistente Diabetes; Wunden, AIDS, Osteoporose,
Harnstein, Harninkontinenz und dergleichen; sowie als ein Wiederherstellungsmittel
bei Ermüdungszuständen, Unwohlsein
und dergleichen.
-
Stand der
Technik
-
Bislang
ist es bekannt, daß die
Funktionen von zahlreichen Hormonen und Neurotransmittern durch eine
Zunahme der Konzentration von Adenosin-3',5'-cyclischem
Monophosphat (cAMP) oder Guanosin-3',5'-cyclischem
Monophosphat (cGMP) ausgedrückt
werden, welche beide die sekundären
Messenger in Zellen sind. Die Zellkonzentrationen von cAMP und cGMP
werden durch die Erzeugung und Zersetzung hiervon gesteuert, und
ihre Zersetzung wird durch Phosphodiesterase (PDE) bewirkt. Wenn
daher PDE gehemmt wird, steigen die Konzentrationen dieser sekundären Zellmessenger
an. Bislang wurden acht Arten von PDE-Isozymen gefunden, und es
wird erwartet, daß die
Isozym-selektiven PDE-Inhibitoren pharmazeutischen Effekt, basierend
auf ihrer physiologischen Signifikanz und Zerteilung in vivo zeigen
[TiPS, 11,150 (1990), ibid. 12, 19 (1991) und Biochemical & Biophysical Research
Communications, 250, 751 (1998)].
-
Es
ist bekannt, daß die
Aktivierung von inflammatorischen Leukocyten durch Erhöhung der
Konzentration des Zell-cAMP
unterdrückt
werden kann. Die außerordentliche
Aktivierung von Leukocyten bewirkt die Sekretion von inflammatorischen
Cytokinen wie dem Tumornecrosefaktor (TNF) und die Expression von
Zellhaftmolekülen
wie interzellulären
Haftmolekülen
(ICAM), gefolgt von zellulärer
Infiltration [J. Mol. Cell. Cariol., 12 (Suppl. II), S61 (1989)].
-
Es
ist bekannt, daß die
Kontraktion der glatten Atmungsmuskeln durch Erhöhung der Konzentration des
Zell-cAMP unterdrückt
werden kann (T. J. Torphy in Directions for New Anti-Asthma Drugs, Herausgeber S.R.
O'Donell und C.
G. A. Persson, 1988, 37, Birkhauser-Verlag). Die außergewöhnliche
Kontraktion eines glatten Atmungsmuskels ist ein Hauptsymptom von
Bronchialasthma. Infiltration von inflammatorischen Leukocyten wie
Neutrophilen wird bei Schädigungen
von Organopathie, verbunden mit ischämischem Reflux wie myocardialer
Ischämie
beobachtet. Es wurde gefunden, daß der Typ IV PDE (PDE IV) hauptsächlich bei
der Zersetzung von cAMP in diesen inflammatorischen Zellen und in
trachealen glatten Muskelzellen teilnimmt. Daher wird erwartet,
daß die
für PDE
IV selektiven Inhibitoren thermapeutischen und/oder präventiven
Effekt auf Entzündungskrankheiten,
obstruktive Atmungskrankheiten und ischämische Krankheiten haben.
-
Es
wird erwartet, daß die
PDE IV-Inhibitoren den Fortschritt und die Ausbreitung der inflammatorischen
Reaktion, welche durch inflammatorische Cytokine wie TNFα und Interleukin
(IL)-8 übertragen
werden, hemmen, weil die PDE IV-Inhibitoren
die Sekretion dieser Cytokine durch Erhöhung der Konzentration von cAMP
unterdrücken.
Beispielsweise ist angegeben, daß TNFα ein Faktor von insulinresistenter
Diabetes ist, da es den phosphorylierenden Mechanismus von Insulinrezeptoren
in Muskeln und Fettzellen herabsetzt [J. Clin. Invest., 94, 1543
(1994)]. In gleicher Weise wird angenommen, daß die PDE IV-Inhibitoren für Autoimmunkrankheiten
nützlich
sein könnten,
wie für
rheumatoide Arthritis, Multiple Sclerose und Chrohnsche Krankheiten,
da TNFα bei
dem Start und dem Fortschreiten dieser Krankheiten teilnimmt [Nature
Medicine, 1, 211 (1995) und ibid. 1, 244 (1995)].
-
Ebenfalls
wurde die Teilnahme von TNFα bei
dem Ermüdungsgefühl nach
Dialyse und demjenigen von an Krebs leidenden Patienten ebenfalls
berichtet [International Journal of Artificial Organs, 21, 83 (1998)
und Oncology Nursing Forum, 19, 419 (1992)]. Daher ist zu erwarten,
daß ein
PDE IV-Inhibitor
effektiv bei der Verbesserung von Ermüdungszuständen, Unwohlsein und dergleichen
ist.
-
Es
wurde angegeben, daß ein
Wirkstoff, welcher cAMP erhöht,
die Heilung von Wunden fördert
[The 68th Annual Meeting of Japan Pharmacological
Society (in Nagoya), Presentation P3-116 (1995)].
-
PDE-IV-Inhibitoren
zeigen einen therapeutischen Effekt gegenüber karcinomatösen Osteopenia-Modell,
Ischiasnerv-Excisionsmodell
oder Ovarialmodell, welche Tiermodelle für Osteoporose sind, und ihre
Möglichkeit
als ein therapeutisches Mittel für
Osteoporose wird vorgeschlagen [Jpn. J. Pharmacol., 79, 477 (1999)].
-
Es
ist bekannt, daß Relaxation
des Harnleiters die Exkretion von Blasensteinen fördert, während ein PDE
IV-Inhibitor die vermikuläre
Bewegung des Harnleiters unterdrückt,
und daher gibt es eine Annahme für die
Wahrscheinlichkeit, daß er
für die
Therapie und/oder Verhütung
von Harnsteinen wirksam ist [J. Urol., 160, 920 (1998)].
-
Die
japanischen veröffentlichten
nicht-geprüften
Patentanmeldungen Nr. 95/242543 und 95/242655 beschreiben 1,4-Benzodioxanderivate
als ein therapeutisches Mittel für
Hepatitiskrankheiten. Die WO 92/10494 beschreibt, daß 1,4-Benzodioxanderivate
eine antagonistische Wirkung gegen Serotonin-(5HAT)3-rezeptoren
haben.
-
In
der
US 5 166 367 sind
1,4-Benzodioxanderivate beschrieben, welche eine Anti-Halluzinationswirkung
haben.
-
In
der japanischen veröffentlichten
nicht-geprüften
Patentanmeldung Nr. 88/179868 sind 1,4-Benzodioxanderivate beschrieben,
welche vasodilatierende Wirkung haben.
-
Die
AU 521 225 beschreibt 1,4-Benzodioxanderivate als Zwischenprodukte
für die
Synthese von Cinnamoylpiperazin.
-
Die
WO 98/22455 beschreibt 1,4-Benzodioxanderivate, welche Hemmaktivität für PDE IV
haben.
-
Beschreibung der Erfindung
-
Es
wird erwartet, daß neue
und nützliche
PDE IV-Inhibitoren einen präventiven
oder therapeutischen Effekt für
Krankheiten eines breiten Bereiches haben. Eine Aufgabe der vorliegenden
Erfindung ist die Bereitstellung von neuen sauerstoffhaltigen heterocyclischen
Verbindungen, welche eine bronchodilatierende oder anti-inflammatorische
Wirkung als Folge des Vorhandenseins einer selektiven Hemmwirkung
für PDE
IV haben, so daß cAMP-Konzentrationen
in Zellen erhöht
werden.
-
Die
vorliegende Erfindung bezieht sich auf sauerstoffhaltige heterocyclische
Verbindungen, wiedergegeben durch die folgende Formel (I):
worin:
m eine ganze
Zahl von 0 bis 4 darstellt;
R
1, R
2, R
3 und R
4 unabhängig
darstellen: ein Wasserstoffatom, substituiertes oder unsubstituiertes
Niederalkyl, substituiertes oder unsubstituiertes Cycloalkyl, Polycycloalkyl,
substituiertes oder unsubstituiertes Niederalkoxycarbonyl, substituiertes
oder unsubstituiertes Niederalkanoyl, substituiertes oder unsubstituiertes
Niederalkanoyloxy, Cyano, Hydroxy, substituiertes oder unsubstituiertes
Niederalkoxy, substituiertes oder unsubstituiertes Niederalkenyl,
substituiertes oder unsubstituiertes Cycloalkenyl, substituiertes
oder unsubstituiertes Aryl, eine substituierte oder unsubstituierte
aromatische heterocyclische Gruppe oder substituiertes oder unsubstituiertes
Aralkyl; zwei Gruppen, die an demselben Kohlenstoffatom von R
1, R
2, R
3 und
R
4 vorhanden sind, kombiniert sind, um einen
gesättigten
Spiro-Kohlenstoffring zusammen mit diesem Kohlenstoffatom zu bilden; zwei
Gruppen, die an den benachbarten Kohlenstoffatomen von R
1, R
2, R
3 und
R
4 vorhanden sind, kombiniert sind, um einen
gesättigten
Kohlenstoffring zusammen mit diesen benachbarten zwei Kohlenstoffatomen
zu bilden; zwei Gruppen, die an den benachbarten Kohlenstoffatomen
unter R
1, R
2, R
3 und R
4 vorhanden
sind, kombiniert sind, um eine Einzelbindung (bildend eine Doppelbindung
zusammen mit der bereits vorhandenen Bindung) zu bilden; oder R
1, R
2, R
3 und
R
4 unabhängig
darstellen: -CONR
7R
8 (worin
R
7 und R
8 unabhängig darstellen:
ein Wasserstoffatom, substituiertes oder unsubstituiertes Niederalkyl,
substituiertes oder unsubstituiertes Niederalkanoyl, substituiertes
oder unsubstituiertes Cycloalkyl, substituiertes oder unsubstituiertes
Aryl, eine substituierte oder unsubstituierte aromatische heterocyclische
Gruppe oder substituiertes oder unsubstituiertes Aralkyl, oder R
7 und R
8 miteinander
kombiniert sind, um eine substituierte oder unsubstituierte heterocyclische
Gruppe zusammen mit dem benachbarten Stickstoffatom darzustellen);
R
5 darstellt: Hydroxy oder substituiertes
oder unsubstituiertes Niederalkoxy;
R
6 darstellt:
ein Wasserstoffatom oder Halogen;
Y darstellt: die folgende
Formel (II):
worin R
9 darstellt:
Cyano, Ethinyl oder Carbamoyl, und R
10 darstellt:
ein Wasserstoffatom, oder R
9 und R
10 kombiniert sind, um eine Einzelbindung
(bildend eine Doppelbindung zusammen mit der bereits existierenden
Bindung) darzustellen, R
11 darstellt: Hydroxy,
Formyl, substituiertes oder unsubstituiertes Niederalkoxy, substituiertes
oder unsubstituiertes Tetrazolyl, -NR
13R
14 (worin R
13 und
R
14 unabhängig darstellen: ein Wasserstoffatom,
substituiertes oder unsubstituiertes Niederalkyl, substituiertes
oder unsubstituiertes Niederalkanoyl, substituiertes oder unsubstituiertes
Cycloalkyl, substituiertes oder unsubstituiertes Aryl, eine substituierte
oder unsubstituierte aromatische heterocyclische Gruppe oder substituiertes
oder unsubstituiertes Aralkyl, oder R
13 und
R
14 kombiniert sind, um eine substituierte
oder unsubstituierte heterocyclische Gruppe zusammen mit dem benachbarten
Stickstoffatom darzustellen), -COOR
15 (worin
R
5 darstellt: ein Wasserstoffatom oder substituiertes
oder unsubstituiertes Niederalkyl), -CONR
16R
17 (worin R
16 und
R
17 unabhängig darstellen: ein Wasserstoffatom,
substituiertes oder unsubstituiertes Niederalkyl, substituiertes
oder unsubstituiertes Niederalkanoyl, substituiertes oder unsubstituiertes
Cycloalkyl, substituiertes oder unsubstituiertes Aryl, eine substituierte
oder unsubstituierte aromatische heterocyclische Gruppe oder substituiertes
oder unsubstituiertes Aralkyl, oder R
16 und
R
17 kombiniert sind, um eine substituierte
oder unsubstituierte heterocyclische Gruppe zusammen mit dem benachbarten
Stickstoffatom darzustellen), oder -CH
2COOR
18 (worin R
18 darstellt:
ein Wasserstoffatom oder substituiertes oder unsubstituiertes Niederalkyl),
R
12 darstellt: ein Wasserstoffatom, oder
substituiertes oder unsubstituiertes Niederalkoxy, oder R
11 und R
12 miteinander
kombiniert sind, um darzustellen: -OCH
2(CH
2)
PO- (worin p eine
ganze Zahl von 1 bis 3 darstellt), -CR
19R
20O- (worin R
19 und
R
20 unabhängig darstellen: ein Wasserstoffatom
oder Cyano), =CHOR
21 (worin R
21 darstellt:
substituiertes oder unsubstituiertes Niederalkyl, substituiertes
oder unsubstituiertes Niederalkenyl oder substituiertes oder unsubstituiertes
Aralkyl), =CHCOOR
22 (worin R
22 darstellt:
ein Wasserstoffatom oder substituiertes oder unsubstituiertes Niederalkyl)
oder =O; oder
pharmazeutisch annehmbare Salze hiervon.
-
Die
vorliegende Erfindung bezieht sich auf sauerstoffhaltige heterocyclische
Verbindungen, in denen Y in der Formel (I) die Formel (II) ist,
oder auf pharmazeutisch annehmbare Salze hiervon. Unter den oben genannten
sind sauerstoffhaltige heterocyclische Verbindungen, worin R9 Cyano ist, oder pharmazeutisch annehmbare
Salze hiervon bevorzugt.
-
In
der vorliegenden Erfindung sind ebenfalls bevorzugte Beispiele sauerstoffhaltige
heterocyclische Verbindungen, worin m = 0 oder 1 in der Formel (I)
ist, oder pharmazeutisch annehmbare Salze hiervon, sauerstoffhaltige
heterocyclische Verbindungen, in denen alle Gruppen R1,
R2, R3 und R4 Wasserstoffatome sind, oder pharmazeutisch
annehmbare Salze hiervon, sowie sauerstoffhaltige heterocyclische
Verbindungen, in denen eine Gruppe von R1,
R2, R3 und R4 substituiertes oder unsubstituiertes Niederalkyl
ist, während
die anderen drei Gruppen Wasserstoffatome sind, oder pharmazeutisch
annehmbare Salze hiervon.
-
Weiterhin
sind bei der Gruppe der oben genannten Verbindungen sauerstoffhaltige
heterocyclische Verbindungen, in de nen R11 Carboxy
oder Hydroxy darstellt, oder R11 und R12 miteinander kombiniert sind, um =O darzustellen,
oder pharmazeutisch annehmbare Salze hiervon ebenfalls bevorzugt.
-
Die
vorliegende Erfindung bezieht sich weiterhin auf eine pharmazeutische
Zusammensetzung, welche wenigstens eine sauerstoffhaltige heterocyclische
Verbindung, wiedergegeben durch die Formel (I), oder pharmazeutisch
annehmbare Salze hiervon enthält.
-
Die
vorliegende Erfindung bezieht sich weiterhin auf einen Phosphodiesterase-(PDE)-IV-inhibitor,
welcher wenigstens eine sauerstoffhaltige heterocyclische Verbindung,
wiedergegeben durch die Formel (I), oder pharmazeutisch annehmbare
Salze hiervon enthält.
-
Im
folgenden werden Verbindungen, welche durch die Formel (I) wiedergegeben
werden, als eine Verbindung (I) bezeichnet. Dasselbe gilt für Verbindungen
der anderen Formelzahlen.
-
In
den Definitionen der Gruppen in der Formel (I) schließen die
Niederalkyl- und Niederalkyleinheit von Niederalkoxy, das Niederalkanoyl,
das Niederalkanoyloxy- und das Niederakoxycarbonyl geradkettige
oder verzweigtkettige Alkylgruppen mit 1 bis 8 Kohlenstoffatomen
ein, wie Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, sec-Butyl,
tert-Butyl, Pentyl, Hexyl, Heptyl und Octyl; das Cycloalkyl schließt Cycloalkylgruppen
mit 3 bis 10 Kohlenstoffatomen ein wie Cyclopropyl, Cyclobutyl,
Cyclopentyl, Cyclohexyl, Cycloheptyl, Cyclooctyl, Cyclononyl und
Cyclodecyl; und das Polycycloalkyl schließt Polycycloalkylgruppen mit
5 bis 12 Kohlenstoffatomen ein wie Bicyclo[3.2.1]octyl, Bicyclo[4.3.2]undecyl,
Adamantyl und Noradamantyl. Das Niederalkenyl schließt geradkettige
oder verzweigtkettige Alkenylgruppen mit 2 bis 8 Kohlenstoffatomen
ein wie Vinyl, 1-Propenyl, Allyl, Methacryl, 1-Butenyl, Crotyl, Pentenyl, Isoprepyl,
Hexenyl, Heptenyl und Octenyl; und das Cycloalkenyl schließt Cycloalkenylgruppen
mit 4 bis 10 Kohlenstoffatomen ein wie Cyclobutenyl, Cyclo pentenyl,
Cyclohexenyl, Cycloheptenyl, Cyclooctenyl, Cyclononenyl und Cyclodecenyl.
Das Aryl schließt
beispielsweise Phenyl und Naphthyl ein; und das Aralkyl schließt Aralkylgruppen
mit 7 bis 15 Kohlenstoffatomen ein wie Benzyl, Phenethyl, Benzhydryl
und Naphthylmethyl. Die aromatische heterocyclische Gruppe schließt ein:
5- oder 6-gliedrige monocyclische aromatische heterocyclische Gruppen
mit 1 bis 2 Sauerstoffatom/en, 5- oder 6-gliedrige monocyclische
aromatische heterocyclische Gruppen mit 1 bis 2 Schwefelatom/en,
5- oder 6-gliedrige
monocyclische aromatische heterocyclische Gruppen mit 1 bis 4 Stickstoffatom/en,
kondensierte bicyclische aromatische heterocyclische Gruppen, bestehend
aus 5- und 6-gliedrigen
Ringen, und kondensierte bicyclische aromatische heterocyclische
Gruppen, bestehend aus 6- und 6-gliedrigen
Ringen ein, worin Sauerstoff, Schwefel und Stickstoff gemischt hierin
vorhanden sein können.
Spezifische Beispiele hiervon schließen ein: Furyl, Thienyl, Pyridyl,
Pyrazinyl, Pyrimidinyl, Pyridazinyl, Chinolyl, Isochinolyl, Phthalazinyl,
Chinazolinyl, Chinoxalinyl, Naphthylidinyl, Pyrrolyl, Pyrazolyl,
Imidazolyl, Triazolyl, Tetrazolyl, Thiazolyl, Oxazolyl, Indolyl,
Indazolyl, Benzimidazolyl, Benzotriazolyl und Purinyl.
-
Die
heterocyclische Gruppe, welche zusammen mit dem benachbarten Stickstoffatom
gebildet wird, schließt
ein: 5-, 6- oder
7-gliedrige monocyclische heterocyclische Gruppen und kondensierte
heterocyclische Gruppen, bestehend aus 6- und 6-gliedrigen Ringen, wie Pyrrolidinyl,
Piperidino, Piperazinyl, Morpholino, Thiomorpholino, Homopiperidino,
Homopiperazinyl, Tetrahydropyridyl, Tetrahydrochinolyl und Tetrahydroisochinolyl.
-
Der
gesättigte
Spirokohlenstoffring, welcher durch zwei Gruppen, die an demselben
Kohlenstoffatom vorhanden sind, zusammen mit diesem Kohlenstoffatom
gebildet wird, und der gesättigte
Kohlenstoffring, welcher durch zwei Gruppen, die an benachbarten
Kohlenstoffatomen vorliegen, zusammen mit diesen zwei Kohlenstoffatomen
gebildet wird, schließen
solche mit 3 bis 10 Kohlenstoffatomen ein wie Cyclopropan, Cyclobutan,
Cyclopentan, Cyclohexan, Cycloheptan, Cyclooctan, Cyclononan und
Cyclodecan. Halogen schließt
Fluor-, Chlor-, Brom- und Jodatome ein.
-
Die
Substituenten in dem substituierten Niederalkyl, dem substituierten
Niederalkoxy, dem substituierten Niederalkoxycarbonyl, dem substituierten
Niederalkanoyl, dem substituierten Niederalkanoyloxy, dem substituierten
Niederalkenyl, dem substituierten Cycloalkyl und dem substituierten
Cycloalkenyl sind dieselben oder unterschiedliche 1 bis 3 Substituent/en,
wie Niederalkyl, Niederalkenyl, Cyano, Cycloalkyl, Cycloalkenyl, Hydroxy,
Niederalkoxy, Carboxy und Halogen, worin das Niederalkyl, das Niederalkenyl,
das Cycloalkyl, das Cycloalkenyl, das Niederalkoxy und das Halogen
dieselben Bedeutungen, wie oben definiert, haben.
-
Die
Substituenten in dem substituierten Aryl, dem substituierten Tetrazolyl,
der substituierten aromatischen heterocyclischen Gruppe, der substituierten
heterocyclischen Gruppe, welche zusammen mit dem benachbarten Stickstoffatom
gebildet wird, und dem substituierten Aralkyl sind dieselben oder
unterschiedlichen 1 bis 3 Substituent/en, wie substituiertes oder
unsubstituiertes Niederalkyl, Hydroxy, Niederalkoxy, Niederalkanoyl,
Niederalkoxycarbonyl, Carboxy, Carbamoyl, Trifluormethyl, Amino,
mono- oder di-niederalkyl-substituiertes Amino, Cyano, Nitro und
Halogen. Das Niederalkyl, die Niederalkyleinheit des Niederalkoxy,
das Niederalkanoyl, das Niederalkoxycarbonyl und das mono- oder
di-niederalkylsubstituierte Amino und das Halogen haben dieselben
Bedeutungen, wie oben definiert, wo der/die Substituent/en in dem
substituierten Niederalkyl dieselbe Bedeutung/en, wie oben definiert,
hat/haben.
-
Die
pharmazeutisch annehmbaren Salze der Verbindung (I) schließen pharmazeutisch
annehmbare Säureadditionssalze,
Me tallsalze, Ammoniumsalze und Additionssalze mit organischem Amin
ein.
-
Die
pharmazeutisch annehmbaren Säureadditionssalze
der Verbindung (I) schließen
anorganische Säureadditionssalze
wie ein Hydrochlorid, ein Sulfat, ein Nitrat und ein Phosphat, sowie
organische Säureadditionssalze
wie ein Acetat, ein Maleat, ein Fumarat und ein Citrat ein; die
pharmazeutisch annehmbaren Metallsalze schließen Alkalimetallsalze wie ein
Natriumsalz und ein Kaliumsalz, Erdalkalimetallsalze wie ein Magnesiumsalz
und ein Calciumsalz, ein Aluminiumsalz und ein Zinksalz ein; die
pharmazeutisch annehmbaren Ammoniumsalze schließen Ammonium und Tetramethylammonium
ein; und die pharmazeutisch annehmbaren Säureadditionssalze mit organischem
Amin schließen
ein Additionssalz mit Morpholin oder Piperidin ein.
-
Verfahren
zur Herstellung der Verbindung (I) werden im folgenden beschrieben.
-
Herstellungsmethode:
Die Verbindung (I) kann nach dem folgenden Verfahren erhalten werden.
-
-
(In
den Formeln haben m, R1, R2,
R3, R4, R5 und R6 dieselben
Bedeutungen wie oben definiert.)
-
Die
Ausgangsverbindung (V) kann nach einer bekannten Methode [Khimiya
Geterotsiklicheskikh Soedinenii, 12, 1614 (1982), etc.] oder nach
einer hierzu vergleichbaren Methode erhalten werden.
-
Nachdem
die Formylgruppe der Verbindung (V) direkt in das entsprechende
halogenierte Methylderivat umgewandelt wor den ist, oder nachdem
die Formylgruppe der Verbindung (V) reduziert wurde und das resultierende
Hydroxymethylderivat zu dem entsprechenden Halogenid- oder Sulfonatderivat
umgewandelt worden ist, wird es mit Metallcyanid umgesetzt, woraufhin
die Verbindung (VI) erhalten werden kann.
-
Die
Verbindung (V) wird mit einem Äquivalent
bis zu einem großen Überschuß von Trialkylsilanhalogenid
oder Triarylsilanhalogenid oder mit einem Äquivalent bis zu einem großen Überschuß eines
halogenierten Salzes und einem Äquivalent
bis zu einem großen Überschuß von Trimethylsilylchlorid
in einem inerten Lösungsmittel
bei der Temperatur zwischen –50°C und dem
Siedepunkt des verwendeten Lösungsmittels
für 5 Minuten
bis 5 Stunden hergestellt, gefolgt von der Behandlung mit einem Äquivalent
bis zu einem großen Überschuß von einem
reduzierenden Mittel bei der Temperatur zwischen –50°C und dem
Siedepunkt des verwendeten Lösungsmittels
für 5 Minuten
bis 48 Stunden, woraufhin das entsprechende Halogenid erhalten werden
kann.
-
Alternativ
wird die Verbindung (V) mit einem Äquivalent bis zu einem großen Überschuß eines
reduzierenden Mittels in einem inerten Lösungsmittel bei der Temperatur
zwischen –50°C und dem
Siedepunkt des verwendeten Lösungsmittels
für 5 Minuten
bis 48 Stunden behandelt, woraufhin das entsprechende Hydroxylmethylderivat
erhalten wird. Das resultierende Hydroxylmethylderivat wird mit
einem Äquivalent
bis zu einem großen Überschuß von einem
halogenierenden Mittel in einem inerten Lösungsmittel bei der Temperatur
zwischen –30°C und dem
Siedepunkt des verwendeten Lösungsmittels
für 5 Minuten
bis 120 Stunden umgesetzt, um das entsprechende Halogenid zu erhalten.
-
Alternativ
wird das resultierende Hydroxymethylderivat zur Reaktion mit einem Äquivalent
bis zu einem großen Überschuß eines
Alkylsulfonylchlorids oder eines Arylsulfonylchlorids in Anwesenheit
von einem Äquivalent
bis zu einem großen Überschuß einer
Base in einem inerten Lösungsmittel bei
der Temperatur zwischen –30°C und dem
Siedepunkt des verwendeten Lösungsmittels
für 5 Minuten
bis 120 Stunden gebracht, woraufhin das entsprechende Sulfonatderivat
erhalten wird.
-
Das
resultierende Halogenid- oder Sulfonatderivat wird zur Reaktion
mit einem Äquivalent
bis zu einem großen Überschuß eines
Metallcyanids in einem inerten Lösungsmittel
bei der Temperatur zwischen –30°C und dem
Siedepunkt des verwendeten Lösungsmittels
für 5 Minuten
bis 120 Stunden gebracht, woraufhin die Verbindung (VI) erhalten
werden kann.
-
Beispiele
des Trialkylsilanhalogenids oder des Triarylsilanhalogenids sind:
Trimethylsilylchlorid, Trimethylsilylbromid, Trimethylsilyljodid,
Triethylsilylchlorid, Dimethylethylsilylchlorid und Triphenylsilylchlorid.
-
Beispiele
des halogenierten Salzes sind Lithiumbromid, Natriumbromid, Kaliumbromid,
Lithiumchlorid, Natriumchlorid, Kaliumchlorid, Lithiumjodid, Natriumjodid
und Kaliumjodid.
-
Beispiele
des reduzierenden Mittels sind 1,1,3,3-Tetramethyldis loxan, Triethylsilan,
Natriumborhydrid, Natriumcyanoborhydrid, Triacetoxyborhydrid und
Lithiumaluminiumhydrid.
-
Beispiele
des halogenierenden Mittels sind Chlorwasserstoffsäure, Bromwasserstoff,
Jodwasserstoff, Thionylchlorid, Phosphoroxychlorid und Phosphortribromid.
-
Beispiele
der Base sind Triethylamin, Diisopropylethylamin, 1,8-Diazabicyclo[5.4.0]-7-undecen
(im folgenden als DBU abgekürzt),
Kaliumcarbonat und Natriumhydrid.
-
Beispiele
des Alkylsulfonylchlorids oder Arylsulfonylchlorids sind Methansulfonylchlorid,
p-Toluolsulfonylchlorid und Benzolsulfonylchlorid.
-
Beispiele
des Metallcyanids sind Natriumcyanid, Kaliumcyanid und Kupfercyanid.
-
Beispiele
des inerten Lösungsmittels
sind Tetrahydrofuran (im folgenden abgekürzt als THF), Dioxan, 1,2-Dimethoxyethan,
Diethylether, Acetonitril, Dimethylformamid (im folgenden abgekürzt als
DMF), Dimethylsulfoxid (im folgenden ab gekürzt als DMSO), Methanol, Ethanol,
Propanol, Dichlormethan, Chloroform, Benzol, Toluol, Pyridin und
Ethylacetat.
-
-
(In
den Formeln haben m, R1, R2,
R3, R4, R5 und R6 dieselben
Bedeutungen wie oben definiert, und R24 steht
für das
Niederalkyl, welches dieselbe Bedeutung wie oben definiert hat.)
-
Die
Verbindung (VIII) kann nach der folgenden Methode erhalten werden.
-
Die
Verbindung (VI) wird zur Reaktion mit der Verbindung (VII) in einem
inerten Lösungsmittel
in Anwesenheit einer katalytischen Menge bis zu einer großen Überschußmenge einer
Base bei der Temperatur zwischen 0°C und dem Siedepunkt des verwendeten
Lösungsmittels
für 5 Minuten
bis 48 Stunden gebracht, woraufhin die Verbindung (VIII) erhalten
werden kann.
-
Beispiele
der Base sind Benzyltrimethylammoniumhydroxid (Triton B), Natriumhydroxid,
Kaliumhydroxid, Natriumhydrid, Kaliumhydrid, Natriummethoxid, Lithiumdiisopropylamid
(im folgenden abgekürzt
als LDA), Pyridin, Kalium-tert-butoxid, DBU, Triethylamin und Diisopropylethylamin.
-
Beispiele
des inerten Lösungsmittels
sind THF, Dioxan, Diethylether, Methanol, Ethanol, 1-Propanol, 2-Propanol,
n-Butanol, tert-Butylalkohol,
Pyridin, Acetonitril, DMF, DMSO, 1,2-Dimethoxyethan, Diethylenglycolmethylether,
Dichlormethan, Chloroform, Benzol und Toluol.
-
-
(In
den Formeln haben m, R1, R2,
R3, R4, R5, R6 und R24 dieselben Bedeutungen wie oben definiert.)
-
Die
Verbindung (IX) kann nach der folgenden Methode aus der Verbindung
(VIII) erhalten werden.
-
Die
Verbindung (VIII) wird zur Reaktion in einem inerten Lösungsmittel
in Anwesenheit von einem Äquivalent
bis zu einem großen Überschuß einer
Base bei der Temperatur zwischen 0°C und dem Siedepunkt des verwendeten
Lösungsmittels
für 5 Minuten
bis 48 Stunden gebracht, woraufhin die Verbindung (IX) erhalten
werden kann.
-
Beispiele
der Base sind Natriumhydrid, Kaliumhydrid, Natriumhydroxid, Kaliumhydroxid,
Natriummethoxid, Natriumethoxid, LDA, Pyridin, Kalium-tert-butoxid,
DBU, Triethylamin und Diisopropylethylamin.
-
Beispiele
des inerten Lösungsmittels
sind THF, Dioxan, Pyridin, Diethylether, Methanol, Ethanol, 1-Propanol,
2-Propanol, 1-Butanol,
tert-Butylalkohol, Acetonitril, DMF, DMSO, 1,2-Dimethoxyethan, Diethylenglycolmethylether,
Dichlormethan, Chloroform, Benzol und Toluol.
-
-
(In
den Formeln haben m, R1, R2,
R3, R4, R5, R6 und R24 dieselben Bedeutungen wie oben definiert.)
-
Die
Verbindung (Ia) kann entsprechend der folgenden Reaktionsstufe erhalten
werden.
-
Die
Verbindung (IX) wird in einem inerten Lösungsmittel in Anwesenheit
von einem Äquivalent
bis zu einem großen Überschuß von Wasser
bei der Temperatur zwischen 60°C
und dem Siedepunkt des verwendeten Lösungsmittels für 5 Minuten
bis 120 Stunden behandelt, woraufhin die Verbindung (Ia) erhalten
werden kann. Erforderlichenfalls kann eine katalytische Menge bis
zu einer Überschußmenge eines
Salzes wie Natriumchlorid, Lithiumchlorid, Natriumjodid, Lithiumjodid
oder Natriumcyanid hinzugesetzt werden.
-
Beispiele
des inerten Lösungsmittels
sind Dioxan, Toluol, DMF, DMSO, tert-Butylalkohol, Acetonitril, 1,2-Dimethoxyethan, Diethylenglycolmethylether,
Ethylenglycol, Triethylenglycol und Wasser.
-
-
(In
den Formeln haben m, R1, R2,
R3, R4, R5 und R6 dieselben
Bedeutungen wie oben definiert.)
-
Die
Verbindung (X) kann entsprechend der folgenden Reaktionsstufe erhalten
werden.
-
2-Trimethylsilyl-1,3-dithian
wird mit einer Base in einem inerten Lösungsmittel bei der Temperatur
zwischen –100°C und 0°C behandelt,
gefolgt von der Reaktion mit der Verbindung (Ia) bei der Temperatur
zwischen –100°C und 30°C für 1 Minute
bis 12 Stunden, woraufhin die Verbindung (X) erhalten werden kann.
-
Beispiele
der Base sind Natriumhydrid, Kaliumhydrid, Natriumhydroxid, Kaliumhydroxid,
Natriummethoxid, Butyllithium, LDA, Lithium-bistrimethylsilylamid,
Natrium-bistrimethylsilylamid, Kalium-bistrimethylsilylamid, Kalium-tert-butoxid,
DBU, Triethylamin, Diisopropylethylamin und Ethylmagnesiumbromid.
-
Beispiele
des inerten Lösungsmittels
sind THF, Dioxan, Diethylether, 1,2-Dimethoxyethan und Diisopropylether.
-
-
(In
den Formeln haben m, R1, R2,
R3, R4, R5 und R6 dieselben
Bedeutungen wie oben definiert, und R25 stellt
dasselbe Niederalkyl wie oben definiert dar.)
-
Die
Verbindung (Ib) kann entsprechend der folgenden Reaktionsstufe erhalten
werden.
-
Die
Verbindung (X) wird in einem Lösungsmittel
[mit Bezug auf dieses Lösungsmittel
wird ein niederer Alkohol später
erwähnt,
der alleine oder als Mischlösungsmittel,
welches den niederen Alkohol (Dioxan/niederen Alkohol, THF/niederen
Alkohol und dergleichen) enthält,
eingesetzt werden, und dieser niedere Alkohol wirkt ebenfalls als
ein Reagens für
die Veresterung der Carboxylgruppe, welche durch die Reaktion erhalten wird]
in Anwesenheit von einem Äquivalent
bis zu einer Überschußmenge eines
zweiwertigen Quecksilbersalzes und einer Säure bei der Temperatur zwischen
0°C und
dem Siedepunkt des verwendeten Lösungsmittels für 5 Minuten
bis 48 Stunden behandelt, woraufhin die Verbindung (Ib) erhalten
werden kann.
-
Beispiele
des zweiwertigen Quecksilbersalzes sind Quecksilberchlorid (HgCl2) und Quecksilberacetat [Hg(OCOCH3)2]. Beispiele der
Säure sind
Perchlorsäure,
Schwefelsäure,
Salzsäure,
Trifluoressigsäure,
p-Toluolsulfonsäure,
Methansulfonsäure
und Bortrifluorid.
-
Beispiele
des Lösungsmittels
sind niedere Alkohole (Methanol, Ethanol, 1-Propanol, 2-Propanol,
1-Butanol, 2-Methyl-1-propanol,
2-Butanol, tert-Butylalkohol, 1-Pentanol, 1-Hexanol, 1-Heptanol,
1-Octanol und dergleichen), ein Mischlösungsmittel von Dioxan/einem
niederen Alkohol (wobei der niedere Alkohol dieselbe Bedeutung wie
oben definiert besitzt) und ein Mischlösungsmittel von THF/einem niederen
Alkohol (worin der niedere Alkohol dieselbe Bedeutung wie oben definiert
besitzt).
-
-
(In
den Formeln haben m, R1, R2,
R3, R4, R5, R6 und R25 dieselben Bedeutungen wie oben definiert.)
-
Die
Verbindung (Ic) kann entsprechend der folgenden Reaktionsstufe erhalten
werden.
-
Die
Verbindung (Ib) wird zur Reaktion mit einer wässrigen Lösung eines Alkali in einem
inerten Lösungsmittel
bei der Temperatur zwischen 0°C
und dem Siedepunkt des verwendeten Lösungsmittels für 5 Minuten
bis 48 Stunden gebracht, woraufhin die Verbindung (Ic) erhalten
werden kann.
-
Beispiele
der wässrigen
Lösung
eines Alkalis sind wässrige
Lösungen
von Natriumhydroxid, Kaliumhydroxid und Lithiumhydroxid, während Beispiele
des inerten Lösungsmittels
Ethanol, Dioxan, Methanol, THF, ein Mischlösungsmittel von Et hanol/THF,
ein Mischlösungsmittel
von Methanol/THF und DMSO sind.
-
-
(In
den Formeln haben m, R1, R2,
R3, R4, R5 und R6 dieselben
Bedeutungen wie oben definiert.)
-
Die
Verbindung (Id) kann entsprechend der folgenden Reaktionsstufe erhalten
werden.
-
Ein Äquivalent
bis zu einer Überschußmenge von
Methoxymethyltriphenylphosphoniumchlorid wird mit einem Äquivalent
bis zu einer Überschußmenge einer
Base in einem inerten Lösungsmittel
bei der Temperatur zwischen –100°C und dem
Siedepunkt des verwendeten Lösungsmittels
behandelt, gefolgt von der Reaktion mit der Verbindung (Ia) bei
der Temperatur zwischen –100°C und dem
Siedepunkt des verwendeten Lösungsmittels
für 5 Minuten
bis 12 Stunden, woraufhin die Verbindung (Id) erhalten werden kann.
-
Beispiele
der Base sind Natriumhydrid, Kaliumhydrid, Natriumhydroxid, Kaliumhydroxid,
Natriummethoxid, Butyllithium, LDA, Lithium-bistrimethylsilylamid,
Natrium-bistrimethylsilylamid, Kalium-bistrimethylsilylamid, Kalium-tert-butoxid,
DBU, Natriumamid und Natriumethoxid.
-
Beispiele
des inerten Lösungsmittels
sind THF, Dioxan, Diethylether, 1,2-Dimethoxyethan, DMF und Diisopropylether.
-
-
(In
den Formeln haben m, R1, R2,
R3, R4, R5 und R6 dieselben
Bedeutungen wie oben definiert.)
-
Die
Verbindung (Ie) kann entsprechend der folgenden Reaktionsstufe erhalten
werden.
-
Die
Verbindung (Id) wird zur Reaktion mit einer katalytischen Menge
bis zu einer Überschußmenge einer
Säure bei
Abwesenheit eines Lösungsmittels
oder in einem inerten Lösungsmittel
bei der Temperatur zwischen 0°C
und dem Siedepunkt des verwendeten Lösungsmittels für 5 Minuten
bis 48 Stunden gebracht, woraufhin die Verbindung (Ie) erhalten
werden kann.
-
Beispiele
der Säure
sind Salzsäure,
Schwefelsäure,
Essigsäure,
Trifluoressigsäure,
p-Toluolsulfonsäure,
Methansulfonsäure,
10-Kampfersulfonsäure,
Bortrifluorid und Aluminiumchlorid.
-
Beispiele
des inerten Lösungsmittels
sind THF, Aceton, Acetonitril, Methanol, Ethanol, Dioxan und ein Mischlösungsmittel
wie ein inertes Lösungsmittel
mit Wasser.
-
-
(In
den Formeln haben m, R1, R2,
R3, R4, R5 und R6 dieselben
Bedeutungen wie oben definiert.)
-
Die
Verbindung (If) kann entsprechend der folgenden Reaktionsstufe erhalten
werden.
-
Die
Verbindung (Ia) wird zur Reaktion mit einem Äquivalent bis zu einem großen Überschuß von Trimethylsulfoxoniumjodid
oder Trimethylsulfoniumjodid in Anwesenheit von einem Äquivalent
bis zu einem großen Überschuß einer
Base in einem inerten Lösungsmittel
bei der Temperatur zwischen –30°C und dem
Siedepunkt des verwendeten Lösungsmittels
für 5 Minuten
bis 48 Stunden gebracht, woraufhin die Verbindung (If) erhalten
werden kann.
-
Beispiele
der Base sind Natriumhydrid, Kaliumhydrid, Natriumhydroxid, Kaliumhydroxid,
Natriummethoxid, Butyllithium, LDA, Lithium-bistrimethylsilylamid,
Natrium-bistrimethylsilylamid, Kalium-bistrimethylsilylamid, Kalium-tert-butoxid,
DBU, Natriumamid und Natriumethoxid.
-
Beispiele
des inerten Lösungsmittels
sind THF, Dioxan, Diethylether, 1,2-Dimethoxyethan, DMF und Diisopropylether.
-
-
(In
den Formeln haben m, R1, R2,
R3, R4, R5 und R6 dieselben
Bedeutungen wie oben definiert.)
-
Die
Verbindung (Ie) kann entsprechend der folgenden Reaktionsstufe erhalten
werden.
-
Die
Verbindung (If) wird zur Reaktion mit einem Äquivalent bis zu einer Überschußmenge einer
Säure bei
Fehlen eines Lösungsmittels
oder in einem inerten Lösungsmittel
bei der Temperatur zwischen 0°C
und dem Siedepunkt des verwendeten Lösungsmittels für 5 Minuten
bis 48 Stunden gebracht, woraufhin die Verbindung (Ie) erhalten
werden kann.
-
Beispiele
der Säure
sind Salzsäure,
Schwefelsäure,
Bromidwasserstoff, Magnesiumchlorid, Magnesiumbromid, Lithiumbromid,
Trifluoressigsäure,
Lithiumperchlorat, p-Toluolsulfonsäure, Methansulfonsäure, 10-Kampfersulfonsäure, Bortrifluorid,
Aluminiumchlorid und Silikagel.
-
Beispiele
des inerten Lösungsmittels
sind THF, Aceton, Acetonitril, Methanol, Ethanol und Dioxan.
-
-
(In
den Formeln haben m, R1, R2,
R3, R4, R5 und R6 dieselben
Bedeutungen wie oben definiert.)
-
Die
Verbindung (Ic) kann entsprechend der folgenden Reaktionsstufe erhalten
werden.
-
Die
Verbindung (Ie) wird zur Reaktion mit einem Äquivalent bis zu einer Überschußmenge eines
Oxidationsmittels in einem inerten Lösungsmittel bei der Temperatur
zwischen 0°C
und dem Siedepunkt des verwendeten Lösungsmittels für 5 Minuten
bis 48 Stunden gebracht, woraufhin die Verbindung (Ic) erhalten
werden kann.
-
Beispiele
des Oxidationsmittels sind chlorige Säure, Kaliumpermanganat und
Wasserstoffperoxid.
-
Wenn
die chlorige Säure
als ein Oxidationsmittel verwendet wird, können ein Äquivalent bis zu einer Überschußmenge von
2-Methyl-2-buten, Sulfaminsäure,
DMSO, einer wässrigen
Lösung
von Wasserstoffperoxid oder dergleichen zugesetzt werden, falls
erforderlich, oder weiterhin kann ein Äquivalent bis zu einer Überschußmenge von
Natriumdihydrogenphosphat hinzugesetzt werden.
-
Beispiele
des inerten Lösungsmittels
sind tert-Butylalkohol, Essigsäure,
DMSO, Aceton und Acetonitril.
-
-
(In
den Formeln haben m, R1, R2,
R3, R4, R5 und R6 dieselben
Bedeutungen wie oben definiert.)
-
Die
Verbindung (Ig) kann entsprechend der folgenden Reaktionsstufe erhalten
werden.
-
Die
Verbindung (Ia) wird zur Reaktion mit einem Äquivalent bis zu einem großen Überschuß von Chloracetonitril
in Anwesenheit von einem Äquivalent
bis zu einem großen Überschuß einer
Base in einem inerten Lösungsmittel
bei der Temperatur zwischen –10°C und dem
Siedepunkt des verwendeten Lösungsmittels
für 5 Minuten
bis 48 Stunden gebracht, woraufhin die Verbindung (Ig) erhalten
werden kann. Erforderlichenfalls kann eine katalytische Menge bis
zu einer Überschußmenge eines
Salzes, wie Benzyltriethylammoniumchlorid, Benzyltriethylammoniumbromid,
Benzyltrimethylammoniumchlorid, Benzyltrimethylammoniumbromid; Tetrabutylammoniumchlorid,
Tetrabutylammoniumbromid, Tetraethylammoniumchlorid oder Triethylmethylammoniumbromid
hinzugesetzt werden.
-
Beispiele
der Base sind Kaliumcarbonat, Natriumcarbonat, Natriumhydrid, Kaliumhydrid,
Lithiumhydroxid, Natriumhydroxid, Kaliumhydroxid, Calciumhydroxid,
Natriummethoxid, Butyllithium, Kalium-tert-butoxid, DBU und Natriumethoxid.
-
Beispiele
des inerten Lösungsmittels
sind Methanol, Ethanol, 1-Propanol, 2-Propanol, tert-Butylalkohol,
Ethylace tat, Toluol, THF, 1,2-Dimethoxyethan, DMF, DMSO und Diisopropylether.
-
-
(In
den Formeln haben m, R1, R2,
R3, R4, R5 und R6 dieselben
Bedeutungen wie oben definiert.)
-
Die
Verbindung (Ic) kann entsprechend der folgenden Reaktionsstufe erhalten
werden.
-
Die
Verbindung (Ig) wird zur Reaktion mit einem Äquivalent bis zu einer Überschußmenge von
Magnesiumbromid oder Lithiumbromid bei Fehlen eines Lösungsmittels
oder in einem inerten Lösungsmittel
in Anwesenheit von einem Äquivalent
bis zu einer Überschußmenge von
Wasser bei der Temperatur zwischen 0°C und dem Siedepunkt des verwendeten
Lösungsmittels
für 5 Minuten
bis 48 Stunden gebracht, woraufhin die Verbindung (Ic) erhalten
werden kann.
-
Beispiele
des inerten Lösungsmittels
sind THF, DMF, Aceton, Acetonitril, Methanol, Ethanol, Dioxan und
ein Mischlösungsmittel
von DMF/Acetonitril.
-
-
(In
den Formeln haben m, R1, R2,
R3, R4, R5, R6, R11 und
R12 jeweils dieselben Bedeutungen wie oben definiert,
und L1 stellt Chlor, Brom oder Jod dar.)
-
Die
Verbindung (XIII) kann entsprechend der folgenden Reaktionsstufe
erhalten werden.
-
Die
Ausgangsverbindung (XI) kann entsprechend einem bekannten Verfahren
(WO 98/22455) oder einem hiermit vergleichbaren Verfahren erhalten
werden. Eine kommerziell erhältliche
Verbindung kann als Verbindung (XII) verwendet werden.
-
Die
Verbindung (XI) wird mit einem Äquivalent
bis zu einer Überschußmenge einer
Base in einem inerten Lösungsmittel
bei der Temperatur zwischen –100°C und Zimmertemperatur
für 5 Minuten
bis 10 Stunden behandelt, gefolgt von der Reaktion mit einem Äquivalent
bis zu einer Überschußmenge der
Verbindung (XII) bei der Temperatur zwischen –100°C und Zimmertemperatur für 5 Minuten
bis 30 Stunden, woraufhin die Verbindung (XIII) erhalten werden
kann. Erforderlichenfalls können
Tetramethylethylendiamin, Cerchlorid oder dergleichen hinzugesetzt
werden.
-
Beispiele
der Base sind Lithium, Magnesium, Methyllithium, Methylmagnesiumbromid,
Ethylmagnesiumbromid und Butyllithium.
-
Beispiele
des inerten Lösungsmittels
sind THF, Dioxan, Diethylether, 1,2-Dimethoxyethan, Diethylenglycoldimethylether,
Benzol, Toluol und Hexan.
-
-
(In
den Formeln haben m, R1, R2,
R3, R4, R5, R6, R11 und
R12 dieselben Bedeutungen wie oben definiert.)
-
Die
Verbindung (Ih) kann entsprechend der folgenden Reaktionsstufe erhalten
werden.
-
Die
Verbindung (XIII) wird zur Reaktion mit einem Äquivalent bis zu einer Überschußmenge einer
Säure bei
Fehlen eines Lösungsmittels
oder in einem inerten Lösungsmittel
bei der Temperatur zwischen 0°C
und dem Siedepunkt des verwendeten Lösungsmittels für 5 Minuten
bis 48 Stunden gebracht, woraufhin die Verbindung (Ih) erhalten
werden kann. Erforderlichenfalls kann Wasser hinzugegeben werden.
-
Beispiele
der Säure
sind Salzsäure,
Schwefelsäure,
10-Kampfersulfonsäure, Essigsäure, Ameisensäure, Trifluoressigsäure, p-Toluolsulfonsäure, Methansulfonsäure, Bortrifluorid
und Aluminiumchlorid.
-
Beispiele
des inerten Lösungsmittels
sind THF, Aceton, Acetonitril, Toluol, Xylol, Methanol, Ethanol, 1-Propanol,
2-Propanol, 1-Butanol,
2-Butanol, tert-Butylalkohol, 1-Pentanol,
1-Hexanol, 1-Heptanol, 1-Octanol und Dioxan.
-
-
(In
den Formeln haben m, R1, R2,
R3, R4, R5, R6, R11 und
R12 dieselben Bedeutungen wie oben definiert.)
-
Die
Verbindung (Ii) kann entsprechend der folgenden Reaktionsstufe erhalten
werden.
-
Die
Verbindung (XIII) wird zur Reaktion mit einem Äquivalent bis zu einer Überschußmenge eines
Cyanids in Anwesenheit von einem Äquivalent bis zu einer Überschußmenge einer
Säure in
einem inerten Lösungsmittel
bei der Temperatur zwischen –100°C und dem
Siedepunkt des verwendeten Lösungsmittels
für 5 Minuten
bis 48 Stunden gebracht, woraufhin die Verbindung (Ii) erhalten
werden kann.
-
Beispiele
der Säure
sind Salzsäure,
Schwefelsäure,
10-Kampfersulfonsäure, Essigsäure, Ameisensäure, Trifluoressigsäure, p-Toluolsulfonsäure, Methansulfonsäure, Titantetrachlorid,
Bortrifluorid und Aluminiumchlorid.
-
Beispiele
des Cyanids sind Trimerthylsilylcyanid, Natriumcyanid und Kaliumcyanid.
-
Beispiele
des inerten Lösungsmittels
sind THF, Dioxan, Diethylether, 1,2-Dimethoxyethan, Methanol, Ethanol,
Acetonitril, Dichlormethan, 1,2-Dichlorethan, Chloroform und Toluol.
-
-
(In
den Formeln haben m, p, R1, R2,
R3, R4, R5 und R6 dieselben
Bedeutungen wie oben definiert.)
-
Die
Verbindung (Ia) kann entsprechend der folgenden Reaktionsstufe erhalten
werden.
-
Die
Ausgangsverbindung (Iia) kann in einer solchen Weise synthetisiert
werden, daß eine
Verbindung (XIII), worin R11 und R12 eine Ketalstruktur haben, unter Verwendung
einer Verbindung (XII), worin R11 und R12 eine Ketalstruktur haben, als ein Ausgangsmaterial
im Verfahren 15 erhalten wird, und eine in Verfahren 17 erwähnte Methode
wird angewandt, wobei die Verbindung (XIII), worin R11 und
R12 eine Ketalstruktur haben, als ein Ausgangsmaterial
verwendet wird.
-
Die
Verbindung (Iia) wird zur Reaktion mit einem Äquivalent bis zu einer Überschußmenge einer
Säure bei
Abwesenheit eines Lösungsmittels
oder in einem inerten Lösungsmittel
bei der Temperatur zwischen 0°C und
dem Siedepunkt des verwendeten Lösungsmittels
für 5 Minuten
bis 48 Stunden gebracht, woraufhin die Verbindung (Ia) erhalten
werden kann.
-
Beispiele
der Säure
sind Salzsäure,
Schwefelsäure,
10-Kampfersulfonsäure, Essigsäure, Ameisensäure, Trifluoressigsäure, p-Toluolsulfonsäure, Methansulfonsäure, Bortrifluorid
und Aluminiumchlorid.
-
Beispiele
des inerten Lösungsmittels
sind THF, Aceton, Acetonitril, Toluol, Methanol, Ethanol, 1-Propanol,
2- Propanol, 1-Butanol,
2-Butanol, tert-Butylalkohol, 1-Pentanol,
1-Hexanol, 1-Heptanol, 1-Octanol, Dioxan und ein Mischlösungsmittel
eines solchen inerten Lösungsmittels
mit Wasser.
-
-
(In
den Formeln haben m, R1, R2,
R3, R4, R5 und R6 dieselben
Bedeutungen wie oben definiert.)
-
Die
Verbindung (Ij) kann entsprechend der folgenden Reaktionsstufe erhalten
werden.
-
Die
Verbindung (Ia) wird zur Reaktion mit einem Äquivalent bis zu einer Überschußmenge eines
Reduktionsmittels in einem inerten Lösungsmittel bei der Temperatur
zwischen –100°C und dem
Siedepunkt des verwendeten Lösungsmittels
für 5 Minuten
bis 48 Stunden gebracht, woraufhin die Verbindung (Ij) erhalten werden
kann.
-
Beispiele
des Reduktionsmittels sind 1,1,3,3-Tetramethyldisiloxan, Triethylsilan,
Natriumborhydrid, Natriumcyanoborhydrid, Triacetoxyborhydrid und
Lithiumaluminiumhydrid.
-
Beispiele
des inerten Lösungsmittels
sind Methanol, Ethanol, 1-Propanol, 2-Propanol, 1-Butanol, 2-Butanol,
tert-Butylalkohol,
1-Pentanol, 1-Hexanol, 1-Heptanol, 1-Octanol und Dioxan.
-
-
(In
den Formeln haben m, n, R1, R2,
R3, R4, R5, R6, L1 und
X jeweils dieselben Bedeutungen wie oben definiert, und L2 stellt Chlor, Brom, Jod oder eine Trifluormethansulfonatgruppe
dar.)
-
Die
Verbindung (Ik) kann entsprechend der folgenden Reaktionsstufe erhalten
werden.
-
Mit
Bezug auf die Verbindung (XIV) kann die kommerziell erhältliche
Verbindung verwendet werden, oder sie kann entsprechend einer bekannten
Methode [Tetrahedron Lett., 30, 5499 (1992)] erhalten werden.
-
Nachdem
die Verbindung (XI) mit einer Base in einem inerten Lösungsmittel
bei der Temperatur zwischen –100°C und Zimmertemperatur
für 5 Minuten
bis 10 Stunden behandelt worden ist, wird die resultierende Verbindung
zur Reaktion mit einem Metallhalogenid oder einer Borverbindung
bei der Temperatur zwischen –100°C und dem
Siedepunkt des verwendeten Lösungsmittels
für 5 Minuten
bis 30 Stunden gebracht, gefolgt von weiterer Reaktion mit der Verbindung
(XIV) in einem inerten Lösungsmittel
bei Anwesenheit einer katalytischen Menge bis zu einer Überschußmenge eines
Palladiumkomplexes bei der Temperatur zwischen Zimmertemperatur
und dem Siedepunkt des verwendeten Lösungsmittels für 5 Minuten
bis 30 Stunden, woraufhin die Verbindung (Ik) erhalten werden kann.
Gegebenenfalls kann bei der oben genannten Reaktion, welche bei Anwesenheit
einer katalytischen Menge bis zu einer Überschußmenge eines Palladiumkomplexes
durchgeführt
wird, ein Salz wie Lithiumchlorid oder Silberoxid erforderlichenfalls
zugegeben werden.
-
Beispiele
der Base sind Lithium, Magnesium, Methyllithium, Methylmagnesiumbromid,
Ethylmagnesiumbromid und Butyllithium.
-
Beispiele
des Metallhalogenids sind halogenierte Alkylzinnderivate wie Chlortributylzinn
und Chlortrimethylzinn und halogenierte Zinkderivate wie Zinkchlorid,
Zinkbromid und Zinkjodid, während
Beispiele der Borverbindung Trimethoxybor, Phenylborsäure und
Borsäure
sind.
-
Beispiele
des Palladiumkomplexes sind:
Tetrakis(triphenylphosphin)palladium,
Dichlor-bis(triphenylphosphin)palladium,
Dichlor-bis(acetonitril)palladium,
[1,1'-Bis(diphenylphosphino)ferrocen]dichlorpalladium
und
Palladiumacetat.
-
Beispiele
des bei der Reaktion verwendeten Lösungsmittels mit einem Metallhalogenid
oder einer Borverbindung sind THF, Dioxan, Diethylether, 1,2-Dimethoxyethan,
Diethylenglycoldimethylether, Benzol, Toluol und Hexan.
-
Beispiele
des bei der Reaktion in Anwesenheit eines Palladiumkomplexes verwendeten
inerten Lösungsmittels
sind THF, Dioxan, Diethylether, Ethylenglycol, Triethylenglycol,
1,2-Dimethoxyethan,
Diethylenglycoldimethylether, Methanol, Ethanol, 1-Butanol, 2-Propanol,
Dichlormethan, Chloroform, Acetonitril, Benzol, Toluol, Dimethylacetamid,
DMF und DMSO.
-
-
(In
den Formeln haben m, R1, R2,
R3, R4, R5, R6, L1 und
Z1-Z2-Z3 jeweils
dieselben Bedeutungen wie oben definiert, Z4 stellt
ein Sauerstoffatom oder ein Schwefelatom dar, und L3 und
L4 stellen Chlor, Brom, Jod oder eine Trifluormethansulfonatgruppe
dar.)
-
Die
Verbindung (Im) und die Verbindung (In) können entsprechend der folgenden
Reaktionsstufe erhalten werden.
-
Im
Hinblick auf die Verbindung (XV) und die Verbindung (XVI) können die
kommerziell erhältlichen
Verbindungen verwendet werden, oder sie können entsprechend bekannten
Methoden erhalten werden [J.Chem.Soc., Perkin Trans. 1, 1954 (1973);
J. Org. Chem., 60(7), 1936 (1995; Tetrahedron Lett., 30(42), 7719 (1994);
Chem. Pharm. Bull., 40(10), 2597 (1992); J. Heterocyclic Chem, 7,
815 (1970): J. Chem. Soc. Chem. Comm., 1183 (1985); etc.].
-
Die
Verbindung (XI) wird mit einer Base in einem inerten Lösungsmittel
bei der Temperatur zwischen –100°C und Zimmertemperatur
für 5 Minuten
bis 10 Stunden behandelt, gefolgt von Reaktion mit einem Metallhalogenid
oder einer Borverbindung bei der Temperatur zwischen –100°C und dem
Siedepunkt des verwendeten Lösungsmittels
für 5 Minuten
bis 30 Stunden. Die resultierende Verbindung wird zur Reaktion mit
der Verbindung (XV) in einem inerten Lösungsmittels bei Anwesenheit
einer katalytischen Menge bis zu einer Überschußmenge eines Palladiumkomplexes
oder eines Nickelkomplexes bei der Temperatur zwischen Zimmertemperatur
und dem Siedepunkt des verwendeten Lösungsmittels für 5 Minuten
bis 30 Stunden gebracht, woraufhin die Verbindung (Im) erhalten
werden kann. Gegebenenfalls, wenn die Verbindung (XVI) anstelle
der Verbindung (XV) verwendet wird, und dieselbe Reaktion wie im
Fall der Verbindung (XV) durchgeführt wird, wird die Verbindung
(In) erhalten.
-
Bei
der oben genannten Reaktion, welche in Anwesenheit einer katalytischen
Menge bis zu einer Überschußmenge eines
Palladiumkomplexes oder eines Nickelkomplexes durchgeführt wird,
kann ein Salz wie Lithiumchlorid oder Silberoxid erforderlichenfalls
hinzugesetzt werden.
-
Beispiele
der Base sind Lithium, Magnesium, Methyllithium, Methylmagnesiumbromid,
Ethylmagnesiumbromid und Butyllithium.
-
Beispiele
des Metallhalogenids sind halogenierte Alkylzinnderivate wie Chlortributylzinn
und Chlortrimethylzinn und halogenierte Zinkderivate wie Zinkchlorid,
Zinkbromid und Zinkjodid, während
Beispiele der Borverbindung Trimethoxybor, Phenylborsäure und
Borsäure
sind.
-
Beispiele
des Palladiumkomplexes sind:
Tetrakis(triphenylphosphin)palladium,
Dichlor-bis(triphenylphosphin)palladium,
Dichlor-bis(acetonitril)palladium,
[1,1'-Bis(diphenylphosphino)ferocen]dichlorpalladium
und
Palladiumacetat.
-
Beispiele
des Nickelkomplexes sind [1,1'-Bis(diphenylphosphino)ferrocen]dichlornickel
und Dichlor-bis(triphenylphosphin)nickel.
-
Beispiele
des bei der Reaktion mit einem Metallhalogenid oder einer Borverbindung
verwendeten inerten Lösungsmittels
sind THF, Dioxan, Diethylether, 1,2-Dimethoxyethan, Diethylenglycoldimethylether,
Benzol, Toluol und Hexan.
-
Beispiele
des bei der Reaktion in Anwesenheit eines Palladiumkomplexes oder
eines Nickelkatalysators verwendeten inerten Lösungsmittels sind THF, Dioxan,
Diethylether, Ethylenglycol, Triethylenglycol, 1,2-Dimethoxyethan,
Diethylenglycoldimethylether, Methanol, Ethanol, 1-Butanol, 2-Propanol, Dichlormethan, Chloroform,
Acetonitril, Benzol, Toluol, Dimethylacetamid, DMF und DMSO.
-
Die
Verbindung (Ima), welche die Verbindung (Im) ist, worin Z1-Z2-Z3 =
C(=O)-NH-C(=O) ist, kann ebenfalls in einer solchen Weise erhalten
werden, daß eine
Reaktion ähnlich
zu der im Verfahren 21 erwähnten durchgeführt wird,
wobei die Verbindung (XI) und ein halogeniertes Diethylphthalat
wie Diethylbromphthalat verwendet wird, gefolgt von Hydrolyse, und
das hierdurch erhaltene Produkt zur Reaktion mit Harnstoff gebracht
wird.
-
Die
Verbindung (I), worin R9 Carbamoyl ist,
kann unter Verwendung der Verbindung (I), worin R9 =
Cyano ist, entsprechend einer bekannten Methode ["Jikken Kagaku Koza
(Handbook of Experimental Chemistry)", 4. Aufl., herausgegeben von der Chemischen
Gesellschaft von Japan, 22, 151-154 (1992)] oder einer hierzu vergleichbaren
Methode erhalten werden.
-
Die
Verbindung (I), worin R9 = Cyano ist, wird
zu der Verbindung umgewandelt, worin die R9 entsprechende
Einheit ein Aldehyd ist, entsprechend einer bekannten Methode ["Jikken Kagaku Koza
(Handbook of Experimental Chemistry)", 4.Aufl., herausgegeben von der Chemischen
Gesellschaft von Japan, 21, 89-94 (1992)] oder einer hierzu ähnlichen
Methode umgewandelt, und dann kann die Verbindung (I), worin R9 = Ethinyl ist, entsprechend einer bekannten
Methode erhalten werden ["Jikken
Kagaku Koza (Handbook of Experimental Chemistry)", 4.Aufl., herausgegeben von der Chemischen
Gesellschaft von Japan, 19, 306-307 (1992)] oder einer hiermit ähnlichen
Methode erhalten werden.
-
Die
Zwischenprodukte und die gewünschten
Verbindungen in jedem der oben genannten Verfahren können mittels
Trennungs- und Reinigungsmethoden
isoliert und gereinigt werden, wie sie konventionellerweise in der
synthetischen organischen Chemie angewandt werden, wie durch Filtration,
Extraktion, Waschen, Trocknen, Konzentrieren, Umkristallisation
und verschiedene Arten der Chromatographie. Die Zwischenprodukte
können
der nachfolgenden Reaktion ohne besondere Reinigung unterworfen
werden.
-
Wenn
es gewünscht
wird, ein Salz der Verbindung (I) zu erhalten, wird die Verbindung
(I) in einem geeigneten Lösungsmittel
aufgelöst
oder suspendiert, dann wird eine Säure oder eine Base hinzugegeben,
und das resultierende Salz kann isoliert und gereinigt werden.
-
Weiterhin
können
die Verbindung (I) und pharmazeutisch annehmbare Salze hiervon ebenfalls
in Form von Addukten mit Wasser und verschiedenen Lösungsmitteln
existieren, welche ebenfalls im Umfang der vorliegenden Erfindung
liegen.
-
Spezifische
Beispiele der Verbindung (I), erhalten entsprechend der vorliegenden
Erfindung, sind in Tabelle 1 gezeigt.
-
-
-
-
-
-
Die
pharmakologischen Aktivitäten
einer der repräsentativen
Verbindungen (I) sind mehr im einzelnen anhand eines Testbeispiels
beschrieben.
-
Testbeispiel 1: Hemmtest
auf rekombinantes menschliches PDE-IV-Enzym
-
Menschliche
Phosphodiesterase cDNA (HSPDE4A) wurde aus Hoden isoliert. Ihre
vorhergesagte Aminosäuresequenz
ist identisch mit der Sequenz (HSPDE4A5), die von Bolger, G. et
al. (Mol. Cell. Biol., 6558 (1993)) berichtet wurde, ausgenommen,
daß 223
Aminosäuren
vom N-Ende hiervon entfernt worden waren. Dieses rekombinante Protein
wurde durch ein E. coli Expressionsplasmid exprimiert und dann gereinigt.
Die PDE-Aktivität wurde
in dem folgenden 2-Stufenprozeß entsprechend
der Methode von Kincaid, R. und Manganiello, V. [Method. Enzymol.,
159, 457 (1988)] gemessen. Das verwendete Substrat war [3H]cAMP (Endkonzentration: 1 μMol/Liter)
und die Reaktion wurde in einer Standardmischung durchgeführt, welche N,N-Bis(2-hydroxyethyl)-2-aminoethansulfonsäure (50
mMol/Liter, pH 7,2), MgCl2 (1 mMol/Liter)
und Sojabohnentrypsininhibitor (0,1 mg/ml) enthielt. Die Reaktion
wurde durch Zugabe des Enzyms hierzu initiiert, und das Gemisch
wurde bei 30°C
für 10
bis 30 Minuten inkubiert. Die Reaktion wurde mit Salzsäure abgebrochen,
und das gebildete 5'-AMP
wurde vollständig
mit 5'-Nucleosidase
zersetzt. Diese Probe wurde der Chromatographie auf DEAE-Sephadex
A-25 unterzogen und das eluierte [3H]Adenosin
wurde mit einem Szintillationszähler
ausgezählt.
Die Testverbindung wurde zugesetzt, nachdem sie in DMSO (Konzentration:
1,7%) aufgelöst
worden war.
-
Bei
dieser Untersuchung zeigte Verbindung 5 Einzymhemmaktivität von mehr
als 87% bei einer Wirkstoffkonzentration von 1 μMol/Liter.
-
Obwohl
die Verbindung (I) oder pharmazeutisch annehmbare Salze hiervon
ebenfalls als solche appliziert werden können, ist es üblicherweise
erwünscht,
sie in Form von verschiedenen pharmazeutischen Präparationen
bereitzustellen. Solche pharmazeutischen Präparationen können für Tiere
und Menschen verwendet werden.
-
Die
pharmazeutischen Präparationen
gemäß der vorliegenden
Erfindung können
die Verbindung (I) oder ein pharmazeutisch annehmbares Salz hiervon
als einen aktiven Inhaltsstoff, alleine oder als Mischung mit anderen
therapeutisch wirksamen Komponenten, enthalten. Weiterhin werden
solche pharmazeutischen Präparationen
mittels beliebiger Einrichtungen, welche auf dem technischen Gebiet
der Arzneimittel wohlbekannt sind, nach dem Mischen des aktiven
Inhaltsstoffes mit einem oder mehreren pharmazeutisch annehmbaren
Trägern
erhalten.
-
Beispiele
des effektiven Inhaltsstoffes, welcher hiermit gemischt werden soll,
sind ein Serotonin-(5HAT)3-Rezeptorantagonist,
ein Serotonin-(5HAT)4-Rezeptoragonist, ein
Serotonin-(5HAT)1A-Rezeptoragonist, ein
Dopamin-(D)2-Rezeptorantagonist, ein Histamin-(H)1-Rezeptorantagonist, ein Muscarin-Rezeptorantagonist,
ein Neurokinin-(NK)1-Rezeptorantagonist
und ein Endothelin-(ET)A-Rezeptorantagonist.
-
Es
ist erwünscht,
den Applikationsweg anzuwenden, welcher am effektivsten in der Therapie
ist, wie orale Applikation und parenterale Applikation, wobei diese
intrabukale, intratracheale, intrarektale, subkutane, intramuskuläre und intravenöse Applikation
einschließen.
-
Die
Applikationsform schließt
Sprays, Kapseln, Tabletten, Granulen, Sirupprodukte, Emulsionen,
Suppositorien, Injektionen, Salben und Pflaster ein.
-
Flüssige Präparationen
wie Emulsionen und Sirupprodukte, welche für die orale Applikation geeignet sind,
können
unter Verwendung von Wasser, Zuckern wie Saccharose, Sorbit und
Fructose, Glycolen wie Polyethylenglycol und Propylenglycol, Ölen wie
Sesamöl,
Olivenöl
und Sojabohnenöl,
Konservierungsstoffen wie p-Hydroxybenzoat und Aromastoffen wie
Erdbeer und Pfefferminze erhalten werden. Kapseln, Tabletten, Pulver
und Granulen können
unter Verwendung von Verdünnungsstoffen
wie Lactose, Glucose, Saccharose und Mannit, Verteilungshilfsmitteln
wie Stärke
und Natriumalginat, Gleitmitteln wie Magnesiumstearat und Talk, Bindemitteln
wie Polyvinylalkohol, Hydroxypropylcellulose und Gelatine, Tensiden,
wie Fettsäureestern
und Weichmachern wie Glycerin erhalten werden.
-
Für die parenterale
Applikation geeignete Präparationen
umfassen ein sterilisiertes wässriges
Mittel, welches die aktive Verbindung enthält, wobei dies bevorzugt für das Blut
eines Patienten isotonisch ist. Beispielsweise wird eine Lösung für die Injektion
unter Verwendung eines Trägers
wie einer Salzlösung,
einer Glucoselösung
oder einer Mischung einer Salzlösung
und einer Glucoselösung
hergestellt. Präparationen
für die intrarektale
Applikation werden unter Verwendung eines Trägers wie Kakaofett, hydriertem
Fett und hydrierter Carbonsäure
hergestellt und als Suppositorien geliefert. Sprays werden unter
Verwendung einer aktiven Verbindung selbst oder einer aktiven Verbindung
mit einem Träger,
welcher die aktive Verbindung als feine Teilchen zur Erleichterung
der Absorption ohne Stimulierung von oraler oder den Atemweg betreffenden
Schleimbildung dispergiert, hergestellt. Beispiele solcher Träger sind
Lactose und Glycerin. Präparationen
wie Aerosol und trockenes Pulver können in Abhängigkeit von den Eigenschaften
der verwendeten aktiven Verbindung und der Träger angewandt werden.
-
Diese
parenteralen Präparationen
können
ebenfalls einen oder mehrere Hilfskomponenten enthalten, ausgewählt aus
Verdünnungsmitteln,
Aromastoffen, Konservierungsstoffen, Verdünnungsmitteln, Desintegratoren,
Gleitmitteln, Bindemitteln, Tensiden und Weichmachern, die alle
oben bei den oralen Präparationen
erwähnt
wurden.
-
Die
effektive Dosis und das Applikationsprogramm der Verbindung (I)
oder eines pharmazeutisch annehmbaren Salzes hiervon kann in Abhängigkeit
von der Form der Applikation, dem Alter und dem Körpergewicht
eines Patienten und dem Typ oder dem Ausmaß des zu behandelnden Leidens
variieren, jedoch wird im Fall von oraler Applikation die Verbindung
(I) oder ein pharmazeutisch annehmbares Salz hiervon in einer Dosis
von 0,01 mg bis 1 g/Erwachsenem/Tag, bevorzugt 0,05 bis 50 mg/Erwachsenem/Tag
appliziert, entweder auf einmal oder in mehreren Teilmengen. Im
Fall von parenteraler Applikation wie intravenöser Applikation wird die Verbindung
(I) oder ein pharmazeutisch annehmbares Salz hiervon in einer Dosis
von 0,001 bis 100 mg/Erwachsenem/Tag, bevorzugt 0,01 bis 10 mg/Erwachsenem/Tag
appliziert, entweder auf einmal oder in mehreren Teilmengen. Diese
Dosiswerte variieren in Abhängigkeit
von den verschiedenen oben beschriebenen Bedingungen.
-
Im
folgenden wird die Ausführung
der vorliegenden Erfindung anhand von Beispielen beschrieben.
-
Beispiel 1. 4-Cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)-cyclohexanon
(Verbindung 1)
-
(Stufe A) Synthese von
2-(8-Methoxy-1,4-benzodioxan-5-yl)-acetonitril (Verbindung 1a)
-
Zu
einer Lösung
von 12 g (62 mMol) von 8-Methoxy-1,4-benzodioxan-5-carbaldehyd in 140 ml
Acetonitril wurden 12 g (110 mMol) Lithiumbromid zugesetzt, und
dann wurden 12 ml (95 mMol) Trimethylsilylchlorid tropfenweise hinzugegeben.
Nach 15 Minuten wurde das Gemisch in Eis gekühlt und 19 ml (110 mMol) von 1,1,3,3-Tetramethyldisiloxan
wurden tropfenweise hinzugegeben, gefolgt von Rühren bei Zimmertemperatur für 2 Stunden.
Das Gemisch wurde mit Methylenchlorid verdünnt, und dann wurde es durch
Celite filtriert. Das Lösungsmittel
wurde im Vakuum von dem Filtrat abgedampft, um eine blaßgelbe ölige Substanz
zu ergeben. Zu einer Lösung
des erhaltenen rohen 5-Brommethyl-8-methoxy-1,4-benzodioxan
in 180 ml DMF wurden 9,2 g (190 mMol) Natriumcyanid zugesetzt, gefolgt
von Rühren
bei Zimmertemperatur für
60 Stunden. Zu dem Gemisch wurde Wasser unter Eiskühlung zugegeben,
und ein hieraus abgeschiedener Feststoff wurde durch Filtration
gesammelt, um 6,8 g (53%) von Verbindung 1a als einen Feststoff
mit Aschefarbe zu erhalten.
Schmelzpunkt: 121–125°C
1H-NMR (CDCl3, δ, ppm) 3,60
(s, 2H), 3,88 (s, 3H), 4,33 (s, 4H), 6,50 (d, J = 8 Hz, 1H), 6,86
(d, J = 8 Hz, 1H).
MASS (m/z) 205 (M+).
-
(Stufe B) Synthese von
Dimethyl-4-cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)-pimelat (Verbindung
1b)
-
Zu
einer Lösung
von 6,2 g (30 mMol) von Verbindung 1a, erhalten in Stufe A, in 94
ml Acetonitril wurden 1,4 ml (3,0 mMol) einer 40%igen methanolischen
Lösung
von Triton B und 27 ml (300 mMol) von Methylacrylat zugegeben, gefolgt
von Erhitzen unter Rückfluß für 5 Stunden.
Das Gemisch wurde zum Abkühlen
stehengelassen, und dann in Wasser eingegossen, gefolgt von Extraktion
mit Ethylacetat. Die organische Schicht wurde mit Salzlösung gewaschen
und über
Natriumsulfat getrocknet, und das Lösungsmittel wurde im Vakuum abgedampft.
Der Rückstand
wurde mittels Kieselgel-Säulenchromatographie
(eluiert mit Hexan/Ethylacetat = 2/1) gereinigt, um 6,4 g (56%)
von Verbindung 1b als eine blaßgelbe ölige Substanz
zu erhalten.
1H-NMR (CDCl3, δ, ppm) 2,05-2,37
(m, 4H), 2,39-2,59 (m, 2H), 2,62-2,82 (m, 2H), 3,60 (s, 6H), 3,87
(s, 3H), 4,20-4,40 (m, 4H), 6,48 (d, J = 9 Hz, 1H), 7,01 (d, J =
9 Hz, 1H).
MASS (m/z) 377 (M+).
-
(Stufe C) Synthese von
4-Cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)-2-methoxycarbonylcyclohexanon
(Verbindung 1c)
-
Zu
einer Lösung
von 6,4 g (17 mMol) von Verbindung 1b, erhalten in Stufe B, in 96
ml 1,2-Dimethoxyethan wurden 2,0 g (50 mMol) von 60%igen Natriumhydrid
zugesetzt. Nach Erhitzen unter Rückfluß für 3 Stunden
wurde das Gemisch zum Abkühlen
stehengelassen, in Eiswasser eingegossen, mit einer 6 Mol/Liter
wässrigen
Salzsäure
angesäuert
und mit Ethylacetat extrahiert. Die organische Schicht wurde mit
Salzlösung
gewaschen und über
Natriumsulfat getrocknet, und das Lösungsmittel wurde abgedampft.
Der Rückstand
wurde durch Kieselgel-Säulenchromatographie
(eluiert mit Hexan/Ethylacetat = 2/1) gereinigt, um 5,0 g (86%)
von Verbindung 1c als weißen
Feststoff zu erhalten.
Schmelzpunkt: 129–132°C
1H-NMR
(CDCl3, δ,
ppm) 2,21-2,50 (m, 3H), 2,61-2,89 (m, 2H), 3,11 (d, J = 15 Hz, 1H,
3,79 (s, 3H), 3,89 (s, 3H), 4,37 (s, 4H), 6,49 (d, J = 9 Hz, 1H),
6,84 (d, J = 9 Hz, 1H), 12,2 (s, 1H).
MASS (m/z) 345 (M+).
-
(Stufe D) Synthese von
Verbindung 1
-
Eine
Mischung von 5,0 g (15 mMol) von Verbindung 1c, erhalten in Stufe
C, 50 ml DMSO, 5 ml Wasser und 5,0 g Natriumchlorid wurde bei 150°C für 5 Stunden
gerührt.
Das Gemisch wurde zum Abkühlen
stehengelassen, und Wasser wurde hinzugesetzt, gefolgt von Extraktion
mit Ethylacetat. Die organische Schicht wurde mit Salzlösung gewaschen
und über
Natriumsulfat getrocknet, und das Lösungsmittel wurde im Vakuum abgedampft.
Der Rückstand
wurde durch Kieselgel-Säulenchromatographie
(eluiert mit Hexan/Ethylacetat = 3/1) gereinigt, um 3,6 g (86%)
von Verbindung 1 als weißen
Feststoff zu erhalten.
Schmelzpunkt: 157–161°C
1H-NMR
(CDCl3, δ,
ppm) 2,21-2,41 (m, 2H), 2,45-2,72 (m, 4H), 2, 81-3,00 (m, 2H), 3,89
(s, 3H), 4,37 (s, 4H), 6,51 (d, J = 9 Hz, 1H), 6,88 (d, J = 9 Hz,
1H).
MASS (m/z) 287 (M+).
-
Beispiel 2. 4-Cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)-cyclohexanonethylenketal
(Verbindung 2)
-
(Stufe A) Synthese von
4-Hydroxy-4-(8-methoxy-1,4-benzodioxan-5-yl)cyclohexanonethylenketal
(Verbindung 2a)
-
In
65 ml THF wurden 10 g (41 mMol) 5-Brom-8-methoxy-1,4-benzodioxan aufgelöst, und
28 ml (45 mMol) einer 1,59 Mol/Liter Lösung von n-Butyllithium in
Hexan wurde tropfenweise bei –78°C hinzugesetzt. Nach
15 Minuten wurde eine Lösung
von 9,6 g (61 mMol) 1,4-Cyclohexadionmonoethylenketal in 50 ml THF tropfenweise
hinzugegeben. Das Gemisch wurde für 1 Stunde gerührt, gefolgt
von Rühren
bei Zimmertemperatur für
20 Minuten. Es wurde Wasser hinzugegeben, das Gemisch wurde mit
Ethylacetat extrahiert, und der Extrakt wurde mit Salzlösung gewaschen
und über
Natriumsulfat getrocknet. Das Lösungsmittel
wurde hieraus abgedampft, und der Rückstand wurde durch Kieselgel-Säulenchromatographie
(eluiert mit Hexan/Ethylacetat = 1/1) gereinigt, um 9,0 g (68%)
von Verbindung 2a als weißen
Feststoff zu erhalten.
Schmelzpunkt: 94–96°C
1H-NMR
(CDCl3, δ,
ppm) 1,58-1,72 (m, 2H), 1,88-2,28 (m, 6H), 3,57 (s, 1H), 3,86 (s,
3H), 3,90-4,07 (m, 4H), 4,35 (s, 4H), 6,46 (d, J = 9 Hz, 1H), 6,82
(d, J = 9 Hz, 1H).
MASS (m/z) 322 (M+).
-
(Stufe B) Synthese von
Verbindung 2
-
In
4,9 ml Methylenchlorid wurden 0,49 g (1,5 mMol) von Verbindung 2a,
erhalten in Stufe A, aufgelöst, 0,26
ml (1,9 mMol) Trimethylsilylcyanid wurden bei –78°C hinzugegeben, dann wurden
0,20 ml (1,6 mMol) eines Bortrifluorid-Ethyletherkomplexes tropfenweise
hinzugefügt,
und das Gemisch wurde für
10 Minuten gerührt,
gefolgt von Rühren
bei Zimmertemperatur für
10 Minuten. Eine gesättigte
wässrige
Lösung
von Natriumbicarbonat wurde hinzugegeben, und das Gemisch wurde
mit Ethylacetat extrahiert. Der Extrakt wurde mit Salzlösung gewaschen
und über
Natriumsulfat getrocknet, und das Lösungsmittel wurde abgedampft.
Der Rückstand
wurde durch Kieselgel-Säulenchromatographie
(eluiert mit Hexan/Ethylacetat = 2/1) gereinigt, um 0,30 g (61%)
von Verbindung 2 als farblose ölige
Substanz zu erhalten.
1H-NMR (CDCl3, δ,
ppm) 1,79-1,95 (m, 2H), 2,06-2,20 (m, 4H), 2,30-2,46 (m, 2H), 3,87
(s, 3H), 3,90-4,07 (m, 4H), 4,36 (s, 4H), 6,48 (d, J = 9 Hz, 1H),
6,82 (d, J = 9 Hz, 1H).
MASS (m/z) 331 (M+).
-
Beispiel 3. Verbindung
1
-
In
2,9 ml Aceton wurden 0,29 g (0,87 mMol) von Verbindung 2, erhalten
in Beispiel 2, aufgelöst,
1,2 ml (7,2 mMol) einer 6 Mol/Liter wässrigen Salzsäure wurden
hinzugegeben, und das Gemisch wurde unter Rückfluß für 3 Stunden erhitzt. Das Gemisch
wurde zum Abkühlen
stehengelassen und in eine gesättigte
wässrige Lösung von
Natriumbicarbonat gegossen, das Gemisch wurde mit Ethylacetat extrahiert,
und der Extrakt wurde mit Salzlösung
gewaschen. Das Gemisch wurde über
Natriumsulfat getrocknet, und das Lösungsmittel wurde verdampft,
um 0,23 g (92%) von Verbindung 1 als einen weißen Feststoff zu erhalten.
-
Beispiel 4. Methyl-cis-4-cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)-cyclohexancarboxylat
(Verbindung 3) und Methyl-trans-4-cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)-cyclohexancarboxylat
(Verbindung 4)
-
(Stufe A) Synthese von
2-[4-Cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)-cyclohexyliden]-1,3-dithian
(Verbindung 3a)
-
Zu
einer Lösung
von 5,0 ml (25 mMol) von 2-Trimethylsilyl-1,3-dithian in 50 ml THF
wurden tropfenweise 17 ml (26 mMol) einer 1,54 Mol/Liter Lösung von
n-Butyllithium in Hexan unter Eiskühlung hinzugegeben. Nach 10
Minuten wurde das Gemisch auf –78°C abgekühlt, und
eine Lösung
von 3,6 g (13 mMol) von Verbindung 1, erhalten in Beispiel 1, in
40 ml THF wurde tropfenweise hinzugegeben. Nach 10 Minuten wurde zu
dem Gemisch Salzlösung
zugesetzt, gefolgt von Zugabe von Wasser bei Zimmertemperatur. Das
Gemisch wurde mit Ethylacetat extrahiert, der Extrakt wurde über Natriumsulfat
getrocknet, und das Lösungsmittel
wurde abgedampft. Der Rückstand
wurde mittels Kieselgel-Säulenchromatographie
(eluiert mit Hexan/Ethylacetat = 4/1) gereinigt, um 3,9 g (79%)
von Verbindung 3a als weißen
Feststoff zu erhalten.
Schmelzpunkt: 164–166°C
1H-NMR
(CDCl3, δ,
ppm) 1,70-1,92 (m, 2H), 2,05-2,24 (m, 2H), 2,28-2,53 (m, 4H), 2,89
(t, J = 6 Hz, 4H), 3,18-3,38 (m, 2H), 3,87 (s, 3H), 4,36 (s, 4H),
6,47 (d, J = 9 Hz, 1H), 6,79 (d, J = 9 Hz, 1H).
MASS (m/z)
389 (M+).
-
(Stufe B) Synthese von
Verbindung 3 und von Verbindung 4
-
In
120 ml Methanol wurden 3,9 g (10 mMol) von Verbindung 3a, erhalten
in Stufe A, suspendiert, 1,7 ml (20 mMol) von 70%iger Perchlorsäure und
4,3 g (16 mMol) von Quecksilberchlorid (HgCl2)
wurden hinzugegeben, und das Gemisch wurde für 4 Stunden gerührt. Das
Gemisch wurde mit Methylenchlorid verdünnt und durch Celite filtriert,
das Filtrat wurde in ei ne gesättigte
wässrige
Lösung
von Natriumbicarbonat eingegossen, und das Gemisch wurde mit Methylenchlorid
extrahiert. Die organische Schicht wurde mit Salzlösung gewaschen
und über
Natriumsulfat getrocknet, und das Lösungsmittel wurde abgedampft.
Der Rückstand
wurde mittels Kieselgel-Säulenchromatographie
(eluiert mit Hexan/Ethylacetat = 1/1) gereinigt, um die rohe Verbindung
3 als weißen
Feststoff zu erhalten und weiterhin 0,18 g (5,5%) von Verbindung
4 als eine farblose transparente ölige Substanz zu erhalten.
Verbindung 3 wurde weiter aus Ethylacetat umkristallisiert, um 0,57
g (17%) von weißen
Kristallen zu ergeben.
-
Verbindung 3
-
- Schmelzpunkt: 123–124°C
- 1H-NMR (CDCl3, δ, ppm) 1,75-2,22
(m, 6H), 2,27-2,51 (m, 3H), 3,71 (s, 3H), 3,88 (s, 3H), 4,36 (s,
4H), 6,48 (d, J = 9 Hz, 1H), 6,84 (d, J = 9 Hz, 1H).
- MASS (m/z) 331 (M+).
-
Verbindung 4
-
- 1H-NMR (CDCl3, δ, ppm) 1,92-2,38
(m, 8H), 2,70-2,88 (m, 1H), 3,69 (s, 3H), 3,87 (s, 3H), 4,36 (s,
4H), 6,48 (d, J = 9 Hz, 1H), 6,81 (d, J = 9 Hz, 1H).
- MASS (m/z) 331 (M+).
-
Beispiel 5. Cis-4-Cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)-cyclohexancarbonsäure (Verbindung
5)
-
Zu
einer Mischung von 0,55 g (1,7 mMol) von Verbindung 3, erhalten
in Beispiel 4, und 3,3 ml Methanol wurden 3,3 ml THF zum Auflösen hiervon
zugesetzt. Zu dem Gemisch wurden tropfenweise 2,6 ml einer 1,3 Mol/Liter
wässrige
Lösung
von Kaliumhydroxid zugegeben, gefolgt von Rühren bei Zimmertemperatur für 1 Stunde.
Das Gemisch wurde in Wasser eingegossen, Ethylacetat wurde hinzugefügt, und
eine wässrige Schicht
wurde extrahiert. Die wässrige
Schicht wurde mit einer 1 Mol/ Liter wässrigen Salzsäure angesäuert, und
der ausgefällte
Feststoff wurde durch Filtration gesammelt und mit Ethanol erneut
aufgeschlämmt,
um 0,45 g (86%) von Verbindung 5 als weißen Feststoff zu erhalten.
Schmelzpunkt:
228–230°C
1H-NMR (DMSO-d6, δ, ppm) 1,59-1,90
(m, 4H), 1,94-2,10 (m, 2H), 2,20-2,45 (m, 3H), 3,75 (s, 3H), 4,27
(dd, J = 5, 12 Hz, 4H), 6,60 (d, J = 9 Hz, 1H), 6,79 (d, J = 9 Hz,
1H), 12,2 (br s, 1H).
MASS (m/z) 317 (M+).
Elementaranalyse : C17J19NO5
Gefunden (%) C : 64,09, H : 6,01,
N : 4,51
Berechnet (%) C : 64,34, H : 6,03, N : 4,41
-
Beispiel 6. 1-(8-Methoxy-1,4-benzodioxan-5-yl)-4-methoxy-methylencyclohexancarbonitril
(Verbindung 6)
-
In
320 ml THF wurden 34 g (99 mMol) von Methoxymethyltriphenylphosphoniumchlorid
suspendiert, und 11 g (99 mMol) Kalium-tert-butoxid wurden hinzugegeben.
Das Gemisch wurde bei Zimmertemperatur für 15 Minuten gerührt, und
eine Lösung
von 15 g (52 mMol) von Verbindung 1, erhalten in Beispiel 1, in
150 ml THF wurde tropfenweise hinzugegeben, gefolgt von Rühren bei
Zimmertemperatur für
45 Minuten. Das Reaktionsgemisch wurde zu Wasser zugesetzt, und
das Gemisch wurde mit Ethylacetat extrahiert. Die organische Schicht
wurde mit Salzlösung
gewaschen und über
Natriumsulfat getrocknet. Das Lösungsmittel
wurde im Vakuum abgedampft, und der Rückstand wurde mittels Kieselgel-Säulenchromatographie
(eluiert mit Hexan/Ethylacetat = 3/1) gereinigt, um 14 g (82%) von
Verbindung 6 als einen weißen
Feststoff zu erhalten.
Schmelzpunkt: 112–113°C
1H-NMR
(CDCl3, δ,
ppm) 1,67-1,82 (m, 2H), 2,08-2,60 (m, 5H), 2,82-2,98 (m, 1H), 3,57
(s, 3H), 3,87 (s, 3H), 4,36 (s, 4H), 5,84 (s, 1H), 6,47 (d, J =
9 Hz, 1H), 6,80 (d, J = 9 Hz, 1H).
MASS (m/z) 315 (M+).
-
Beispiel 7. Cis-4-Cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)-cyclohexancarbaldehyd
(Verbindung 7) und trans-4-Cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)-cyclohexancarbaldehyd
(Verbindung 8)
-
In
100 ml Aceton wurden 10 g (32 mMol) von Verbindung 6, erhalten in
Beispiel 6, aufgelöst,
210 ml einer 6 Mol/Liter wässrigen
Salzsäure
wurden tropfenweise hinzugegeben, und das Gemisch wurde bei Zimmertemperatur
für 2 Stunden
gerührt,
durch Zugabe einer 5 Mol/Liter wässrigen
Lösung
von Natriumhydroxid neutralisiert und mit Ethylacetat extrahiert.
Die organische Schicht wurde mit einer gesättigten wässrigen Lösung von Natriumbicarbonat
und dann mit Salzlösung
gewaschen. Das Gemisch wurde über
Magnesiumsulfat getrocknet, das Lösungsmittel wurde im Vakuum
abgedampft, und der Rückstand
wurde mittels Kieselgel-Säulenchromatographie
(eluiert mit Hexan/Ethylacetat = 2/1) gereinigt, um 7,7 g (0%) von
Verbindung 7 und 1,7 g (18%) von Verbindung 8 zu erhalten, wovon
jede eine farblose ölige
Substanz war.
-
Verbindung 7
-
- 1H-NMR (CDCl3, δ, ppm) 1,80-2,35
(m, 7H), 2,38-2,57 (m, 2H), 3,88 (s, 3H), 4,36 (s, 4H), 6,49 (d,
J = 9 Hz, 1H), 6,85 (d, J = 9 Hz, 1H), 9,68 (s, 1H).
- MASS (m/z) 301 (M+).
-
Verbindung 8
-
- 1H-NMR (CDCl3, δ, ppm) 1,75-1,96
(m, 2H), 2,06-2,37 (m, 6H), 2,56-2,65 (m, 1H), 3,87 (s, 3H), 4,33
(s, 4H), 6,46 (d, J = 9 Hz, 1H), 6,78 (d, J = 9 Hz, 1H), 9,73 (s,
1H).
- MASS (m/z) 301 (M+).
-
Beispiel 8. Cis-4-Cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)-cyclohexancarbonsäure (Verbindung
5)
-
In
155 ml tert-Butylalkohol wurden 7,7 g (26 mMol) von Verbindung 7,
erhalten in Beispiel 7, aufgelöst, 3,1
g (26 mMol) von Natriumdihydrogenphosphat und 12 ml (120 mMol) von
2-Methyl-2-buten, 46 ml einer 80%igen wässrigen Lösung von 3,2 g (28 mMol) chlorige
Säure wurden
tropfenweise unter Eiskühlung
hinzugesetzt, und das Gemisch wurde bei Zimmertemperatur für 1 Stunde
gerührt.
Zu dem Gemisch wurden 5,3 g (51 mMol) Natriumhydrogensulfit zugesetzt,
gefolgt von Rühren
für 15
Minuten, eine 2 Mol/ml wässrige
Lösung von
Natriumhydroxid wurde hinzugesetzt, und das resultierende Gemisch
wurde mit Ethylacetat gewaschen. Das Gemisch wurde auf pH 3,5 mit
einer 6 Mol/Liter wässrigen
Salzsäure
eingestellt, und der hieraus ausgeschiedene Feststoff wurde durch
Filtration gesammelt und aus Ethanol umkristallisiert, um 5,5 g
(67%) von Verbindung 5 als weiße
Kristalle zu ergeben.
-
Beispiel 9. Cis-4-Cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)-cyclohexanol (Verbindung
9)
-
In
8,4 ml Methanol wurden 0,42 g (1,5 mMol) von Verbindung 1 (erhalten
in Beispiel 1) aufgelöst,
und 0,11 g (3,0 mMol) Natriumborhydrid wurden unter Eiskühlung hinzugesetzt.
Das Gemisch wurde bei Zimmertemperatur für 1 Stunde gerührt, 0,057
g (1,5 mMol) Natriumborhydrid wurden erneut unter Eiskühlung zugesetzt,
gefolgt von Rühren
bei Zimmertemperatur für
30 Minuten, eine 1 Mol/Liter wässrige
Salzsäure
wurde unter Eiskühlung
hinzugegeben, und das Gemisch wurde mit Ethylacetat extrahiert.
Die organische Schicht wurde mit Salzlösung gewaschen und über Natriumsulfat
getrocknet, und das Lösungsmittel
wurde im Vakuum abgedampft. Der Rückstand wurde mittels Kieselgel-Säulenchromatographie
(eluiert mit Hexan/ Ethylacetat = 1/2) gereinigt, gefolgt von Umkristallisation
aus Ethanol, um 0,20 g (48%) von Verbindung 9 als weiße Kristalle zu
ergeben.
Schmelzpunkt: 137–138°C
1H-NMR (CDCl3, δ, ppm) 1,75-1,99
(m, 4H), 2,01-2,22 (m, 2H), 2,30-2,54 (m, 2H), 3,56-3,79 (m, 1H),
3,87 (s, 3H), 4,38 (s, 4H), 6,48 (d, J = 8 Hz, 1H), 6,83 (d, J =
8 Hz, 1H).
MASS (m/z) 289 (M+).
Elementaranalyse
: C16H19NO4·0,1
H2O
Gefunden (%) C : 66,21, H : 6,94,
N : 4,82
Berechnet (%) C : 66,01, H : 6,65, N : 4,81
-
Beispiel 10. Ethyl-4-cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)-cyclohexanylidenacetat
(Verbindung 10)
-
In
9,4 ml THF wurden 0,72 ml (3,6 mMol) von Triethylphosphonoacetat
aufgelöst,
und 0,15 g (3,6 mMol) von 60%igem Natriumhydrid wurde hierzu unter
Eiskühlung
zugegeben. Das Gemisch wurde bei Zimmertemperatur für 15 Minuten
gerührt,
0,94 g (3,3 mMol) von Verbindung 1, erhalten in Beispiel 1, wurden
unter Eiskühlung
hinzugegeben, und das Gemisch wurde bei Zimmertemperatur für 30 Minuten
gerührt.
Dann wurde Wasser unter Eiskühlung
hinzugegeben, das Gemisch wurde mit Ethylacetat extrahiert, und
die organische Schicht wurde mit Salzlösung gewaschen und über Natriumsulfat
getrocknet. Das Lösungsmittel
wurde im Vakuum abgedampft, und der Rückstand wurde mittels Kieselgel-Säulenchromatographie
(eluiert mit Hexan/Ethylacetat = 2/1) gereinigt, um 1,4 g (97%)
von Verbindung 10 als weißen
Feststoff zu erhalten.
Schmelzpunkt: 110–112°C
1H-NMR
(CDCl3 δ,
ppm) 1,28 (t, J = 7 Hz, 3H), 1,85-2,10 (m, 2H), 2,31-2,60 (m, 4H),
2,67-2,89 (m, 1H), 3,87 (s, 3H), 3,92-4,12 (m, 1H), 4,16 (q, J =
7 Hz, 2H), 4,36 (s, 4H), 5,71 (s, 1H), 6,48 (d, J = 9 Hz, 1H), 6,81
(d, J = 9 Hz, 1H).
MASS (m/z) 357 (M+).
-
Beispiel 11. 4-Cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)-cyclohexanylidenessigsäure (Verbindung
11)
-
In
3,1 ml Ethanol und 3,1 ml THF wurden 0,31 g (0,87 mMol) von Verbindung
10, erhalten in Beispiel 10, suspendiert, dann wurden 0,65 ml (1,3
mMol) einer 2 Mol/Liter wässrigen
Lösung
von Natriumhydroxid hinzugegeben, das Gemisch wurde bei 70°C für 1 Stunde
gerührt,
und dann wurden 0,65 ml (1,3 mMol) einer 2 Mol/Liter wässrigen
Lösung
von Natriumhydroxid weiter hinzugegeben, gefolgt von Rühren bei
70°C für 1 Stunde.
Eine 1 Mol/Liter wässrige
Salzsäurelösung wurde
tropfenweise unter Eiskühlung
hinzugegeben, und das Gemisch wurde mit Ethylacetat extrahiert.
Die organische Schicht wurde mit Salzlösung gewaschen und über Natriumsulfat
getrocknet, das Lösungsmittel
wurde im Vakuum abgedampft, und der Rückstand wurde mit Ethanol verrieben,
um 0,23 g (82%) von Verbindung 11 als weißen Feststoff zu ergeben.
Schmelzpunkt:
203–204°C
1H-NMR (DMSO-d6, δ, ppm) 1,75-2,00
(m, 2H), 2,05-2,67 (m, 5H), 3,74 (s, 3H), 3,80-4,00 (m, 1H), 4,15-4,43 (m,
4H), 5,69 (s, 1H), 6,59 (d, J = 9 Hz, 1H), 6,81 (d, J = 9 Hz, 1H),
12,1 (br s, 1H).
MASS (m/z) 329 (M+).
Elementaranalyse
: C18H19NO5·0,1
H2O
Gefunden (%) C : 65,30, H : 6,09,
N : 4,19
Berechnet (%) C : 65,29, H : 5,84, N : 4,23
-
Beispiel 12. 4-Cyano-4-(2,2-dimethyl-7-methoxy-1,3-benzodioxol-4-yl)-cyclohexanon
(Verbindung 12)
-
(Stufe A) Synthese von
2-(2,2-Dimethyl-7-methoxy-1,3-benzodioxol-4-yl)-acetonitril
(Verbindung 12a)
-
Zu
einer Lösung
von 9,3 g (45 mMol) von 2,2-Dimethyl-7-methoxy-1,3-benzodioxol-4-carbaldehyd,
erhalten nach der in der japanischen veröffentlichten nicht-geprüften Patentanmeldung
Nr. 98/147585 erwähnten Methode
und nach einer hiermit ähnlichen
Methode, in 47 ml Acetonitril wurden 8,9 g (85 mMol) Lithiumbromid zugesetzt,
und dann wurden 8,5 ml (67 mMol) Trimethylsilylchlorid tropfenweise
hinzugegeben. Nach 15 Minuten wurde das Gemisch eisgekühlt, und
13 ml (76 mMol) von 1,1,3,3-Tetramethyldisiloxan wurden tropfenweise
hinzugegeben, gefolgt von Rühren
bei Zimmertemperatur für
2 Stunden. Das resultierende Gemisch wurde mit Toluol verdünnt und
durch Celite filtriert. Das Lösungsmittel
wurde im Vakuum von dem Filtrat abgedampft, um eine blaßgelbe ölige Substanz
zu ergeben. Zu einer Lösung
dieses rohen 7-Brommethyl-2,2-dimethyl-4-methoxy-1,3-benzodioxols in 73 ml
DMF wurden 5,0 g (102 mMol) Natriumcyanid hinzugesetzt, und das Gemisch
wurde bei Zimmertemperatur für
18 Stunden gerührt.
Unter Eiskühlung
wurde Wasser hinzugegeben, und das Gemisch wurde mit Ethylacetat
extrahiert. Die organische Schicht wurde mit Salzlösung gewaschen und über Natriumsulfat
getrocknet, und das Lösungsmittel
wurde im Vakuum abgedampft. Der Rückstand wurde mittels Kieselgel-Säulenchromatographie
(eluiert mit Hexan/Ethylacetat = 5/1) gereinigt, um 9,1 g (92%) von
Verbindung 12a als einen Feststoff mit Aschefärbung zu erhalten.
Schmelzpunkt:
58–59°C
1H-NMR (CDCl3, δ, ppm) 1,71
(s, 6H), 3,60 (s, 2H), 3,88 (s, 3H), 6,50 (d, J = 9 Hz, 1H), 6,76
(d, J = 9 Hz, 1H).
MASS (m/z) 219 (M+).
-
(Stufe B) Synthese von
Dimethyl-4-cyano-4-(2,2-dimethyl-7-methoxy-1,3-benzodioxol-4-yl)-pimelat
(Verbindung 12b)
-
Zu
einer Lösung
von 9,0 g (41 mMol) von Verbindung 12a, erhalten in Stufe A, in
135 ml Acetonitril wurden 1,9 ml (4,1 mMol) einer 40%igen methanolischen
Lösung
von Triton B und 37 ml (410 mMol) Methylacrylat zugesetzt, und das
Gemisch wurde bei Zimmertemperatur für 30 Minuten gerührt. Wasser
und eine 1 Mol/Liter wässrige
Salzsäure
wurden hinzugegeben, gefolgt von Extraktion mit Ethylacetat. Die
organische Schicht wurde mit Salzlösung gewaschen und über Natriumsulfat
getrocknet, und das Lösungsmittel
wurde im Vakuum abgedampft. Der Rückstand wurde mittels Kieselgel-Säulenchromatographie
(eluiert mit Hexan/Ethylacetat = 5/2) gereinigt, um 11 g (68%) von
Verbindung 12b als blaßgelbe ölige Substanz
zu erhalten.
1H-NMR (CDCl3, δ, ppm) 1,69
(s, 6H), 2,05-2,31 (m, 4H), 2,40-2,64
(m, 4H), 3,60 (s, 6H), 3,89 (s, 3H), 6,49 (d, J = 9 Hz, 1H), 6,88
(d, J = 9 Hz, 1H).
MASS (m/z) 391 (M+).
-
(Stufe C) Synthese von
4-Cyano-4-(2,2-dimethyl-7-methoxy-1,3-benzodioxol-4-yl)-2-methoxycarbonylcyclohexanon
(Verbindung 12c)
-
Zu
einer Lösung
von 11 g (27 mMol) von Verbindung 12b, erhalten in Stufe B, in 161
ml 1,2-Dimethoxyethan wurden 3,3 g (83 mMol) 60%iges Natriumhydrid
zugegeben. Nach Erhitzen unter Rückfluß für 3 Stunden
wurde das Gemisch zum Abkühlen
stehengelassen und in Eiswasser eingegossen, und das Gemisch wurde
mit einer 6 Mol/Liter wässrigen
Salzsäure
angesäuert
und mit Ethylacetat extrahiert. Die organische Schicht wurde mit
Salzlösung
gewaschen und über
Natriumsulfat getrocknet, und das Lösungsmittel wurde abgedampft.
Der Rückstand
wurde mittels Kieselgel-Säulenchromatographie
(eluiert mit Hexan/Ethylacetat = 3/1) gereinigt, um 8,4 g (86%)
von Verbindung 12c als weißen
Feststoff zu erhalten.
Schmelzpunkt: 146–147°C
1H-NMR
(CDCl3, δ,
ppm) 1,71 (s, 3H), 1,73 (s, 3H), 2,12-2,27 (m, 1H), 2,32-2,55 (m,
2H), 2,67-3,00 (m, 3H), 3,78 (s, 3H), 3,89 (s, 3H), 6,51 (d, J =
9 Hz, 1H), 6,92 (d, J = 9 Hz, 1H), 12,2 (s, 1H).
MASS (m/z)
359 (M+).
-
(Stufe D) Synthese von
Verbindung 12
-
Ein
Gemisch von 8,3 g (23 mMol) von Verbindung 12c, erhalten in der
Stufe C, 83 ml DMSO, 8,3 ml Wasser und 8,3 g Natriumchlorid wurde
bei 150°C
für 12
Stunden gerührt.
Das Gemisch wurde zum Abkühlen stehengelassen,
Wasser wurde hinzugefügt,
gefolgt von Extraktion mit Ethylacetat. Die organische Schicht wurde
mit Salzlösung
gewaschen und über
Natriumsulfat getrocknet, und das Lösungsmittel wurde abgedampft.
Der Rückstand
wurde mittels Kieselgel-Säulenchromatographie
(eluiert mit Hexan/Ethylacetat = 3/1) gereinigt, um 5,6 g (81%)
von Verbindung 12 als einen weißen
Feststoff zu erhalten.
Schmelzpunkt: 153–154°C
1H-NMR
(CDCl3, δ,
ppm) 1,72 (s, 6H), 2,35-2,61 (m, 6H), 2,77-2,97 (m, 2H), 3,90 (s, 3H), 6,53 (d,
J = 9 Hz, 1H), 6,97 (d, J = 9 Hz, 1H).
MASS (m/z) 301 (M+).
-
Beispiel 13. Methyl-cis-4-cyano-4-(2,2-dimethyl-7-methoxy-1,3-benzodioxol-4-yl)-cyclohexancarboxylat
(Verbindung 13) und trans-4-Cyano-4-(2,2-dimethyl-7-methoxy-1,3-benzodioxol-4-yl)-cyclohexancarboxylat
(Verbindung 14)
-
(Stufe A) Synthese von
2-[4-Cyano-4-(2,2-dimethyl-7-methoxy-1,3-benzodioxol-4-yl)-cyclohexyliden]-1,3-dithian (Verbindung
13a)
-
Zu
einer Lösung
von 0,56 ml (3,0 mMol) von 2-Trimethylsilyl-1,3-dithian in 5,6 ml
THF wurden tropfenweise 1,9 ml (3,0 mMol) einer Lösung von
1,54 Mol/Liter von Butyllithium in Hexan unter Eiskühlung hinzugegeben.
Nach 15 Minuten wurde das Gemisch auf –78°C abgekühlt, und eine Lösung von
0,42 g (1,4 mMol) von Verbindung 12, erhalten in Beispiel 12, in
0,6 ml THF wurde tropfenweise hinzugefügt. Nach 20 Minuten wurde Salzlösung hinzugegeben
und dann wurde bei Zimmertemperatur Wasser hinzugegeben. Das Gemisch
wurde mit Ethylacetat extrahiert, der Extrakt wurde über Natriumsulfat
getrocknet, und das Lösungsmittel
wurde abgedampft. Der Rückstand
wurde mittels Kieselgel-Säulenchromatographie
(eluiert mit Hexan/Ethylacetat = 6/1) gereinigt, um 0,53 g (93%)
von Verbindung 13a als weißen
Feststoff zu erhalten.
Schmelzpunkt: 194–197°C
1H-NMR
(CDCl3, δ,
ppm) 1,72 (s, 6H), 1,86-2,53 (m, 8H), 2,90 (t, J = 6 Hz, 4H), 3,17-3,33
(m, 2H), 3,88 (s, 3H), 6,48 (d, J = 9 Hz, 1H), 6,86 (d, J = 9 Hz,
1H).
MASS (m/z) 403 (M+).
-
(Stufe B) Synthese von
Verbindung 13 und Verbindung 14
-
In
105 ml Methanol wurden 3,0 g (7,4 mMol) von Verbindung 13a, erhalten
in Stufe A, suspendiert, dann wurden 1,3 ml (15 mMol) von 70%iger
Perchlorsäure
und 3,2 g (12 mMol) von Quecksilberchlorid (HgCl2) hinzugegeben,
und das Gemisch wurde bei 60°C
für 1 Stunde
gerührt.
Das Gemisch wurde mit Methylenchlorid verdünnt und durch Celite filtriert,
und das Filtrat wurde in gesättigtes
wässriges
Natriumbicarbonat eingegossen, gefolgt von Extraktion mit Methylenchlorid.
Die organische Schicht wurde mit Salzlösung gewaschen und über Natriumsulfat
getrocknet, und das Lösungsmittel
wurde abgedampft. Der Rückstand
wurde mittels Kieselgel-Säulenchromatographie
(eluiert mit Hexan/Ethylacetat = 3/1) gereinigt, um 1,4 g (53%)
von Verbindung 13 als einen weißen
Feststoff und 0,31 g (12%) von Verbindung 14 als eine farblose transparente ölige Substanz
zu ergeben.
-
Verbindung 13
-
- Schmelzpunkt: 79–80°C
- 1H-NMR (CDCl3, δ, ppm) 1,72
(s, 6H), 1,87-2,27 (m, 8H), 2,30-2,43
(m, 1H), 3,71 (s, 3H), 3,88 (s, 3H), 6,49 (d, J = 9 Hz, 1H), 6,92
(d, J = 9 Hz, 1H).
- MASS (m/z) 345 (M+).
-
Verbindung 14
-
- 1H-NMR (CDCl3, δ, ppm) 1,71
(s, 6H), 1,90-2,26 (m, 8H), 2,72-2,80
(m, 1H), 3,70 (s, 3H), 3,88 (s, 3H), 6,48 (d, J = 9 Hz, 1H), 6,83
(d, J = 9 Hz, 1H).
- MASS (m/z) 345 (M+).
-
Beispiel 14. Cis-4-Cyano-4-(2,2-dimethyl-7-methoxy-1,3-benzodioxol-4-yl)-cyclohexancarbonsäure (Verbindung
15)
-
Zu
einem Gemisch von 1,7 g (5,0 mMol) von Verbindung 13, erhalten in
Beispiel 13, und 10 ml Methanol wurden 5,2 ml THF zum Auflösen hiervon
zugesetzt. Zu dem Gemisch wurden tropfenweise 7,7 ml (10 mMol) einer
1,3 Mol/Liter wässrigen
Lösung
von Kaliumhydroxid zugegeben, gefolgt von Rühren bei Zimmertemperatur für 2 Stunden.
Es wurde Wasser hinzugegeben, das Gemisch wurde mit Ethylacetat
gewaschen, und die wässrige
Schicht wurde mit einer 1 Mol/Liter wässrigen Salzsäure angesäuert. Das
Gemisch wurde mit Ethylacetat extrahiert, und der Extrakt wurde
mit Salzlösung
gewaschen und über
Natriumsulfat getrocknet. Das Lösungsmittel
wurde abgedampft, und der Rückstand
wurde aus Ethanol umkristallisiert, um 1,1 g (64%) von Verbindung
15 als weiße
Kristalle zu ergeben.
Schmelzpunkt: 195–198°C
1H-NMR
(DMSO-d6, δ, ppm) 1,50-1,78 (m, 8H), 1,79-2,13
(m, 4H), 2,15-2,37 (m, 3H), 3,79 (s, 3H), 6,64 (d, J = 9 Hz, 1H),
6,81 (d, J = 9 Hz, 1H), 12,3 (br s, 1H).
MASS (m/z) 331 (M+).
Elementaranalyse : C18H21NO5
Gefunden
(%) C : 65,33, H : 6,40, N : 4,27
Berechnet (%) C : 65,24,
H : 6,39, N : 4,23
-
Beispiel 15. 4-(8-Methoxy-1,4-benzodioxan-5-yl)-3-cyclohexenon
(Verbindung 16)
-
Zu
1,2 g (3,6 mMol) von Verbindung 2a, erhalten in Stufe A von Beispiel
2, und 1,2 mg (0,0063 mMol) p-Toluolsulfonsäuremonohydrat wurden 70 ml
Wasser und 140 ml Toluol zugegeben, gefolgt von Erhitzen unter Rückfluß für 4 Stunden.
Das Gemisch wurde zum Abkühlen
stehengelassen, das Gemisch wurde mit Toluol extrahiert, und der
Extrakt wurde mit gesättigtem
wässrigem
Natriumbicarbonat und dann mit Salzlösung gewaschen. Das Gemisch
wurde über
Natriumsulfat getrocknet, das Lösungsmittel
wurde mittels Kieselgel-Säulenchromatographie
(eluiert mit Hexan/Ethylacetat = 2/1) gereinigt, um 0,88 g (94%)
von Verbindung 16 als blaßgelben
Feststoff zu ergeben.
Schmelzpunkt: 56–59°C
1H-NMR
(CDCl3, δ,
ppm) 2,59 (t, J = 7 Hz, 2H), 2,75-2,85 (m, 2H), 2,97-3,10 (m, 2H),
3,88 (s, 3H), 4,25-4,37 (m, 4H), 5,83 (t, J = 4 Hz, 1H), 6,48 (d,
J = 8 Hz, 1H), 6,81 (d, J = 8 Hz, 1H).
MASS (m/z) 260 (M+).
-
Beispiel 16. 5-(8-Methoxy-1,4-benzodioxan-5-yl)-1-indanon
(Verbindung 17) (keine Verbindung gemäß der Erfindung)
-
Zu
einem Gemisch von 0,30 g (1,8 mMol) von Verbindung A, erhalten in
Vergleichsbeispiel 1, und 0,39 g (1,8 mMol) von kommerziell erhältlichem
5-Brom-1-indanon wurden 3,0 ml DMF unter einer Stickstoffatmosphäre zu ihrer
Auflösung
hinzugesetzt, und dann wurden 0,39 g (3,7 mMol) Natriumcarbonat
und 0,020 g (0,090 mMol) Palladiumacetat hinzugegeben. Das Ge misch
wurde bei 100°C
für 2 Stunden
gerührt,
gefolgt von Rühren
bei 110°C
für 0,9
Stunden. Ethylacetat wurde zu dem Reaktionsgemisch zugegeben, der
Feststoff wurde abfiltriert, und die resultierende Lösung wurde
der Verteilung unterzogen. Die organische Schicht wurde mit Salzlösung gewaschen
und über
Natriumsulfat getrocknet. Nachdem der Feststoff abfiltriert worden
war, wurde das Filtrat im Vakuum konzentriert, und das Konzentrat
wurde mittels Kieselgel-Säulenchromatographie (eluiert
mit Hexan/Ethylacetat = 3/1) gereinigt und aus Aceton umkristallisiert,
um 0,15 g (28%) von Verbindung 17 als weißen Feststoff zu ergeben.
Schmelzpunkt:
168°C
1H-NMR (CDCl3, δ, ppm) 2,70-2,74
(m, 2H), 3,18 (br t, 2H), 3,93 (s, 3H), 4,28-4,32 (m, 2H), 4,36-4,39
(m, 2H), 6,61 (d, J = 9 Hz, 1H), 6,90 (d, J = 9 Hz, 1H), 7,53 (dd,
J = 2, 8 Hz, 1H), 7,61 (d, J = 2 Hz, 1H), 7,78 (d, J = 8 Hz, 1H).
MASS
(m/z) 296 (M+).
-
Beispiel 17. 6-(8-Methoxy-1,4-benzodioxan-5-yl)-1-tetralon
(Verbindung 18) (keine Verbindung gemäß der Erfindung)
-
Zu
einem Gemisch von 0,37 g (1,7 mMol) von Verbindung A, erhalten in
Vergleichsbeispiel 1, und 0,51 g (1,7 mMol) von 6-Trifluormethansulfonyl-1-tetralon
[Synthetic Communication, 23(21), 2965 (1993) etc.] wurden 3,7 ml
DMF zu ihrer Auflösung
unter einer Stickstoffatmosphäre
zugesetzt, und dann wurden 0,37 g (3,5 mMol) Natriumcarbonat und
0,020 g (0,090 mMol) Palladiumacetat hinzugegeben. Das Gemisch wurde
bei 100°C
für 1,5
Stunden gerührt,
gefolgt von Rühren
bei 120°C
für 2,3
Stunden. Ethylacetat wurde zu dem Reaktionsgemisch zugesetzt, der
Feststoff wurde abfiltriert, und die resultierende Lösung wurde
mit Wasser und mit Salzlösung
gewaschen und über
Natriumsulfat getrocknet. Der Feststoff wurde abfiltriert, das Filtrat
wurde im Vakuum eingeengt, und das Kon zentrat wurde mittels Kieselgel-Säulenchromatographie
(eluiert mit Hexan/Ethylacetat = 3/1) gereinigt und aus Aceton und
dann aus Ethanol umkristallisiert, um 0,070 g (8,0%) von Verbindung
18 als einen weißen
Feststoff zu ergeben. Schmelzpunkt: 156–158°C
1H-NMR
(CDCl3, δ,
ppm) 2,12-2,21 (m, 2H), 2,67 (t, J = 7 Hz, 2H), 3,01 (t, J = 6 Hz,
2H), 3,93 (s, 3H), 4,28-4,31 (m, 2H), 4,36-4,38 (m, 2H), 6,59 (d,
J = 9 Hz, 1H), 6,88 (d, J = 9 Hz, 1H), 7,39 (d, J = 1 Hz, 1H), 7,46
(dd, J = 2, 8 Hz, 1H), 8,06 (d, J = 8 Hz, 1H).
MASS (m/z) 310
(M+).
-
Beispiel 18. 7-(8-Methoxy-1,4-benzodioxan-5-yl)-1-benzosuberon
(Verbindung 19) (keine Verbindung gemäß der Erfindung)
-
Zu
einem Gemisch von 0,37 g (1,6 mMol) von Verbindung A, erhalten in
Vergleichsbeispiel 1, und 0,38 g (1,6 mMol) von 7-Brom-1-benzosuberon
wurden 3,3 ml DMF unter einer Stickstoffatmosphäre zur Auflösung hiervon zugegeben, und
0,33 g (3,1 mMol) Natriumcarbonat und 0,020 g (0,080 mMol) Palladiumacetat
wurden hinzugefügt.
Das Gemisch wurde bei 100°C
für 6,5
Stunden gerührt,
Ethylacetat wurde zu dem Reaktionsgemisch zugegeben, und der Feststoff
wurde abfiltriert. Die resultierende Lösung wurde mit Wasser und mit
Salzlösung
gewaschen und über
Natriumsulfat getrocknet. Der Feststoff wurde abfiltriert, das Filtrat
wurde im Vakuum konzentriert, und das Konzentrat wurde mittels Kieselgel-Säulenchromatographie
(eluiert mit Hexan/Ethylacetat = 3/1) gereinigt und aus Ethanol
und dann aus Aceton umkristallisiert, um 0,16 g (32%) von Verbindung
19 als einen weißen
Feststoff zu ergeben.
Schmelzpunkt: 124–126°C
1H-NMR
(CDCl3, δ,
ppm) 1,83-1,94 (m, 4H), 2,74-2,78 (m, 2H), 2,96-3,01 (m, 2H), 3,93
(s, 3H), 4,28-4,31 (m, 2H), 4,36-4,39 (m, 2H), 6,59 (d, J = 9 Hz,
1H), 6,89 (d, J = 9 Hz, 1H), 7,36 (d, J = 2 Hz, 1H), 7,47 (dd, J
= 2,8 Hz, 1H), 7,79 (d, J = 8 Hz, 1H).
MASS (m/z) 324 (M+).
-
Beispiel 19. Ethyl-2-[4-cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)-cyclohexan-1-yl]-acetat
(Verbindung 20)
-
In
5,5 ml Ethanol und 8,0 ml Aceton wurden 0,55 g (1,5 mMol) von Verbindung
10, erhalten in Beispiel 10, aufgelöst, 0,11 g von 10% Palladium-Kohle
(enthaltend 50% Wasser) wurde hinzugefügt und das Gemisch wurde einer
Hydrierungsreaktion bei Zimmertemperatur und üblichem Druck für 3 Stunden
unterworfen. Nach Entfernung des Katalysators wurde das Lösungsmittel
im Vakuum aus dem Filtrat abgedampft, um 0,54 g (100) von Verbindung
20 als ein Gemisch von Isomeren in Form einer blaßgelben öligen Substanz
zu ergeben.
NMR (CDCl3, δ, ppm) 1,18-1,33
(m, 3H), 1,45-1,73 (m, 3H), 1,77-1,97 (m, 4H), 2,25-2,42 (m, 4H),
3,87 und 3,88 (jeweils s, insgesamt 3H), 4,04-4,27 (m, 2H), 4,35
(s, 4H), 6,48 und 6,49 (jeweils d, J = 9 Hz, insgesamt 1H), 6,84
und 6,88 (jeweils d, J = 9 Hz, insgesamt 1H).
MASS (m/z) 359
(M+).
-
Beispiel 20. 2-[4-Cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)-cyclohexan-1-yl]-essigsäure (Verbindung
21)
-
In
4,1 ml Ethanol wurden 0,41 g (1,1 mMol) von Verbindung 20, erhalten
in Beispiel 19, aufgelöst,
1,1 ml einer 2 Mol/Liter wässrigen
Lösung
von Natriumhydroxid wurde hinzugegeben, und das Gemisch wurde bei Zimmertemperatur
für 1 Stunde
gerührt
und dann bei 60°C
für 20
Minuten gerührt.
Das Gemisch wurde zum Abkühlen
stehengelassen, und Wasser und Ethylacetat wurden hinzugegeben,
gefolgt von Trennen in eine organische Schicht und eine wässrige Schicht.
Die wässrige Schicht
wurde mit einer 6 Mol/Liter wässrigen
Salzsäure
angesäuert
und mit Ethylacetat extrahiert, und der Extrakt wurde mit Salzlösung gewaschen
und über Natriumsulfat
getrocknet. Das Lösungsmittel
wurde im Vakuum abgedampft, Toluol wurde zu dem Rückstand hinzugesetzt,
und das Lösungsmittel
wurde im Vakuum abgedampft. Zu dem Rückstand wurde Diisopropylether
zugesetzt, und dann wurde der ausgefällte Feststoff durch Filtration
gesammelt und aus Ethanol umkristallisiert, um 0,13 g (36%) von
Verbindung 21 als eine Mischung von Isomeren in Form von weißen Kristallen zu
ergeben.
Schmelzpunkt: 149–150°C
NMR
(DMSO-d6, δ, ppm) 1,25-1,57 (m, 2H), 1,63-1,95
(m, 5H), 2,08-2,35 (m, 4H), 3,74 (s, 3H), 4,15-4,42 (m, 4H), 6,59
(d, J = 9 Hz, 1H), 6,81 und 6,86 (jeweils d, J = 9 Hz, insgesamt
1H), 12,1 (s, 1H).
MASS (m/z) 359 (M+).
-
Beispiel 21. 4-(8-Methoxy-1,4-benzodioxan-5-yl)-1(3H)-isobenzofuranon
(Verbindung 22) (keine Verbindung gemäß der Erfindung)
-
Zu
0,61 g Ethyl-4-(8-methoxy-1,4-benzodioxan-5-yl)-2-acetoxymethylbenzoat,
welches durch Behandlung von 0,40 g (1,9 mMol) von in Vergleichsbeispiel
1 erhaltener Verbindung A erhalten worden war, und 0,57 g (1,9 mMol)
von in Vergleichsbeispiel 3 erhaltener Verbindung C, in derselben
Weise wie in Beispiel 17, wurden 6,0 ml Ethanol und 1,2 ml einer
5 Mol/Liter Lösung
von Kaliumhydroxid zugesetzt, das Gemisch wurde bei Zimmertemperatur
für 35
Minuten gerührt,
und das Lösungsmittel
wurde im Vakuum abgedampft, und Wasser wurde zu dem Rückstand
zugesetzt. Zu der erhaltenen Lösung
wurden 6 Mol/Liter Salzsäure
zur Einstellung hiervon auf pH 2 zugegeben, und der ausgefallene
Feststoff wurde durch Filtration gesammelt und im Vakuum getrocknet,
um 0,46 g eines weißen
Feststoffes zu ergeben.
-
Zu
diesem Feststoff wurden 4,2 ml Essigsäure zugesetzt, gefolgt von
Rühren
bei 90°C.
Eine gesättigte wässrige Lösung von
Natriumbicarbonat wurde zu dem Reaktionsgemisch zugesetzt, so daß Essigsäure neutralisiert
wurde, und Ethylacetat wurde hierzu zur Verteilung zugegeben. Die
organische Schicht wurde mit einer gesättigten wässrigen Lösung von Natriumbicarbonat
und Salzlösung
gewaschen und über
Magnesiumsulfat getrocknet. Der Feststoff wurde abfiltriert, das
Filtrat wurde im Vakuum konzentriert, und das Konzentrat wurde mittels
Kieselgel-Säulenchromatographie
(eluiert mit Hexan/Ethylacetat = 3/2) gereinigt und aus Ethanol
umkristallisiert, um 0,33 g (70%) von Verbindung 22 als einen weißen Feststoff
zu ergeben.
Schmelzpunkt: 158–160°C
NMR (CDCl3 δ, ppm) 3,94
(s, 3H), 4,28-4,32 (m, 2H), 4,37-4,40
(m, 2H), 5,35 (s, 2H), 6,62 (d, J = 8,6 Hz, 2H), 6,89 (d, J = 8,6
Hz, 2H), 7,62 (s, 1H), 7,67 (d, J = 8,3 Hz, 1H), 7,93 (d, J = 8,3
Hz, 1H).
MASS (m/z) 298 (M+).
Elementaranalyse
: C17H14O5
Gefunden (%) C : 68,55, H : 4,61
Berechnet
(%) C : 68,45, H : 4,73
-
Beispiel 22. 4-(8-Methoxy-1,4-benzodioxan-5-yl)-phthalimid
(Verbindung 23) (keine Verbindung gemäß der Erfindung)
-
Zu
0,12 g (0,4 mMol) von Verbindung B, erhalten in Vergleichsbeispiel
2, wurden 0,6 ml Ethylenglycol und 0,02 g (0,4 mMol) Harnstoff in
einer Argonströmung
zugegeben, gefolgt von Rühren
bei 150°C
für 5,3 Stunden.
Ethanol wurde zu dem Reaktionsgemisch bei Zimmertemperatur zugesetzt,
und der hieraus abgetrennte Feststoff wurde durch Filtration gesammelt
und im Vakuum getrocknet, um 0,08 g (73%) von Verbindung 23 als
einen gelben Feststoff zu ergeben.
Schmelzpunkt: 285–287°C
NMR
(CDCl3, δ,
ppm) 3,94 (s, 3H), 4,29-4,32 (m, 2H), 4,37-4,40 (m, 2H), 6,62 (d, J = 8,6 Hz, 1H),
6,91 (d, J = 8,6 Hz, 1H), 7,27 (br s, 1H), 7,84-7,88 (br t, 2H),
8,04 (s, 1H).
MASS (m/z) 311 (M+).
Elementaranalyse
: C17H13NO5
Gefunden (%) C: 65,49, H: 4,25, N:
4,22
Berechnet (%) C: 65,59, H: 4,21, N: 4,50
-
Vergleichsbeispiel 1.
(8-Methoxy-1,4-benzodioxan-5-yl)-borsäure (Verbindung
A)
-
Zu
7,4 g (30 mMol) von 5-Brom-8-methoxy-1,4-benzodioxan wurden 74 ml
THF zu seiner Auflösung zugesetzt,
und nach Abkühlen
des Gemisches auf –78°C wurden
23 ml (35 mMol) einer 1,54 Mol/Liter Lösung von n-Butyllithium in
Hexan tropfenweise zugegeben, gefolgt von Rühren bei –78°C für 0,5 Stunden.
-
Zu
diesem Reaktionsgemisch wurde eine Lösung von 4,8 ml (42 mMol) Trimethylborat
in 15 ml THF bei –78°C zugegeben,
und das Gemisch wurde für
1,7 Stunden gerührt,
gefolgt von Rühren
für weitere
1,8 Stunden bei Zimmertemperatur. Salzsäure wurde zur Einstellung des
Gemisches auf pH 1 hinzugegeben, das Gemisch wurde mit Ethylacetat
extrahiert, und der Extrakt wurde mit Wasser und Salzlösung gewaschen
und über
Magnesiumsulfat getrocknet. Der Feststoff wurde abfiltriert, und
das Lösungsmittel
wurde im Vakuum aus der resultierenden Lösung abgedampft, gefolgt von
Umkristallisation aus Toluol, um 4,35 g (68%) von Verbindung A als
einen milchig weißen
Feststoff zu ergeben.
Schmelzpunkt: > 300°C
1H-NMR (CDCl3, δ, ppm) 3,91
(s, 3H), 4,34-4,41 (m, 4H), 5,73 (s, 2H), 6,59 (d, J = 8 Hz, 1H),
7,37 (d, J = 8 Hz, 1H).
MASS (m/z) 210 (M+).
-
Vergleichsbeispiel 2.
4-(8-Methoxy-1,4-benzodioxan-5-yl)-phthalsäure (Verbindung B)
-
Zu
0,69 g (1,8 mMol) von Diethyl-4-(8-methoxy-1,4-benzodioxan-5-yl)-phthalat,
erhalten durch Behandlung von 0,70 g (3,3 mMol) von in Vergleichsbeispiel
1 erhaltener Verbindung A und 1,00 g (3,3 mMol) von bekanntem Diethyl-4-bromphthalat
nach derselben Reaktion wie in Beispiel 17, wurden 7,0 ml Ethanol
und 1,4 ml einer 6 Mol/Liter wässrigen
Lösung
von Kaliumhydroxid zugegeben, das Gemisch wurde bei Zimmertemperatur
für 2 Stunden
gerührt,
und Ethanol wurde im Vakuum abgedampft. Wasser wurde zu der resultierenden
Lösung
zugegeben, das Gemisch wurde auf pH 2 mit einer 6 Mol/Liter Salzsäure eingestellt,
und die hieraus abgetrennten Kristalle wurden durch Filtration gesammelt
und im Vakuum getrocknet, um 0,55 g (94%) von Verbindung B als einen
weißen
Feststoff zu erhalten.
Schmelzpunkt: 192–196°C
NMR (DMSO-d6, δ, ppm) 3,79
(s, 3H), 4,26 (s, 4H), 4,37-4,40 (m, 2H), 6,69 (d, J = 8,6 Hz, 1H),
6,89 (d, J = 8,6 Hz, 1H), 7,63-7,72 (m, 3H), 13,07 (br s, 1H).
MASS
(m/e) 312 (M+-18).
-
Vergleichsbeispiel 3.
Ethyl-2-acetoxymethyl-4-brombenzoat (Verbindung C)
-
(Stufe A) Ethyl-4-brom-2-brommethylbenzoat
(Verbindung Ca)
-
Zu
10,0 g (41 mMol) von Ethyl-4-brom-2-methylbenzoat wurden 300 ml
von α,α,α-Trifluortoluol,
7,54 g (42 mMol) N-Bromsuccinimid
und 2,0 g (12 mMol) Azobisisobutyronitril zugegeben, gefolgt von
Rühren
bei 80°C
für 5,5
Stunden. Wasser wurde zu dem Reaktionsgemisch für die Verteilung zugegeben,
und die organische Schicht wurde mit Wasser und Salzlösung gewaschen
und über
Magnesiumsulfat getrocknet. Der Feststoff wurde abfiltriert, das
Filtrat wurde im Vakuum konzentriert, und das Konzentrat wurde mittels
Kieselgel-Säulenchromatographie
(eluiert mit Hexan/Ethylacetat = 20/1) gereinigt, gefolgt von Umkristallisation
aus Hexan, um 4,07 g (31%) von Verbindung Ca als weißen Feststoff
zu ergeben.
Schmelzpunkt: 56,0–57,0°C
NMR (CDCl3, δ, ppm) 1,
42 (t, J = 7, 1 Hz, 3H), 4, 40 (q, J = 7,1 Hz, 2H), 4,89 (s, 2H),
7,50 (dd, J = 2,0, 8,4 Hz, 1H), 7,62 (d, J = 2, 0 Hz, 1H), 7,84
(d, J = 8, 4 Hz, 1H).
MASS (m/e) 300 (M+).
-
(Stufe B) Verbindung C
-
Zu
1,0 g (3,2 mMol) von Verbindung Ca wurden 10 ml Essigsäure und
4,0 g (49 mMol) Natriumacetat zugegeben, gefolgt von Rühren bei
120°C für 0,5 Stunden.
Eine gesättigte
wässrige
Lösung
von Natriumbicarbonat wurde zu dem Reaktionsgemisch zugegeben, so
daß die
Essigsäure
neutralisiert wurde, und dann wurde Ethylacetat hierzu für die Verteilung
zugegeben. Die organische Schicht wurde mit einer gesättigten
wässrigen
Lösung
von Natriumbicarbonat und dann mit Salzlösung gewaschen und über Magnesiumsulfat
getrocknet. Der Feststoff wurde abfiltriert, das Filtrat wurde im
Vakuum konzentriert, und das Konzentrat wurde mittels Kieselgel-Säulenchromatographie
(eluiert mit Hexan/Ethylacetat = 25/1) gereinigt, um 0,69 g (71%)
von Verbindung C als einen weißen
Feststoff zu ergeben.
Schmelzpunkt: 51,5–52,5°C
NMR (CDCl3, δ, ppm) 1,39
(t, J = 7,1 Hz, 3H), 2,17 (s, 3H), 4,36 (q, J = 7,1 Hz, 2H), 5,50
(s, 2H), 7,51 (dd, J = 2,0, 8,4 Hz, 1H), 7,65 (d, J = 2,0 Hz, 1H),
7,87 (d, J = 8, 4 Hz, 1H).
MASS (m/e) 300 (M+).
-
Industrielle Anwendbarkeit
-
Gemäß der vorliegenden
Erfindung können
sauerstoffhaltige heterocyclische Verbindungen geliefert werden,
welche Hemmaktivität
für Phosphodiesterase-(PDE)-IV
besitzen, und welche brauchbar sind als ein therapeutisches Mittel
für: inflammatorische
allergische Krankheiten wie Bronchialasthma, allergische Rhinitis, atopische
Dermatitis und Nephritis; Autoimmunkrankheiten wie chronisches obstruktives
Lungenleiden, Rheumatismus, Multiple Sclerose, Crohnsche Krankheit,
Psoriasis und systematische Lupuserythematose; Krankheiten des zentralen
Nervensystems wie Depression, Amnesie und Demens; Organopathie,
verbunden mit ischämischem
Reflux, hervorgerufen durch Herzversagen, Schock und cerebrovaskuläres Leiden
und dergleichen; insulinresistente Diabetes; Wunden, AIDS, Osteoporose;
Harnstein, Harninkontinenz und dergleichen; oder als ein Erholungsmittel
bei Ermüdung,
Unwohlsein und dergleichen.

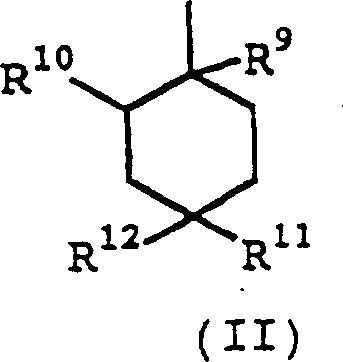
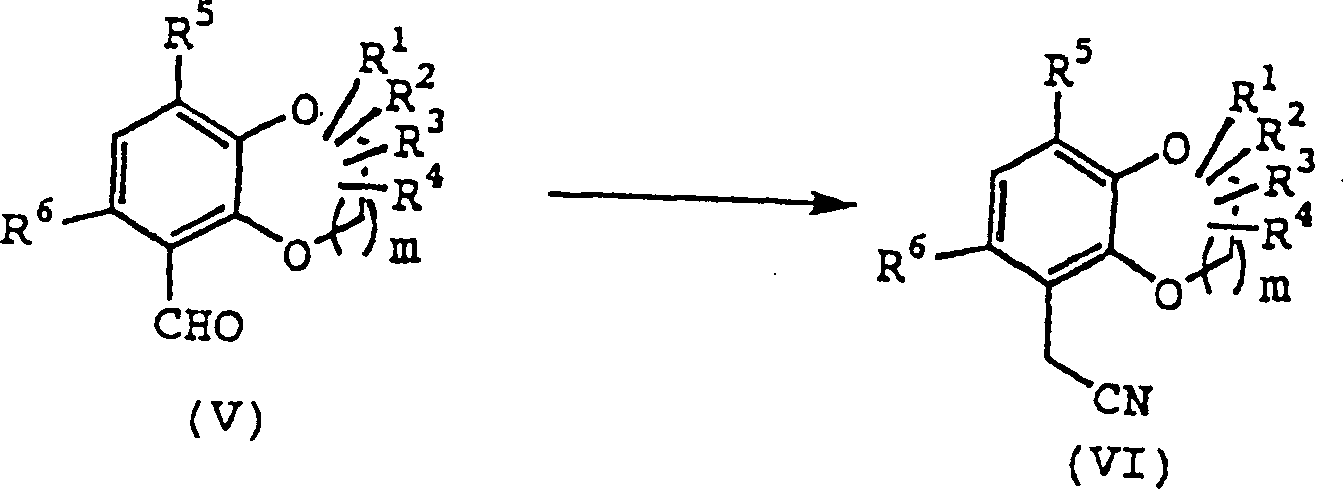
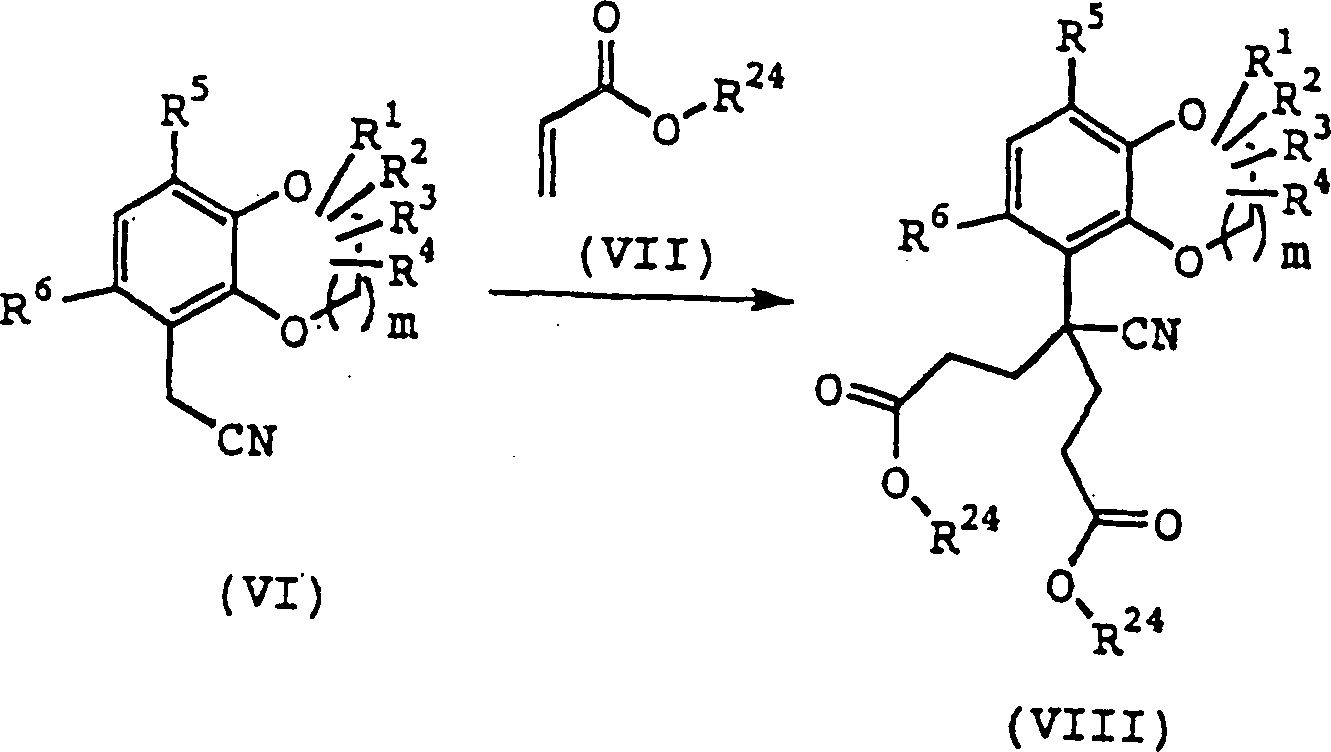
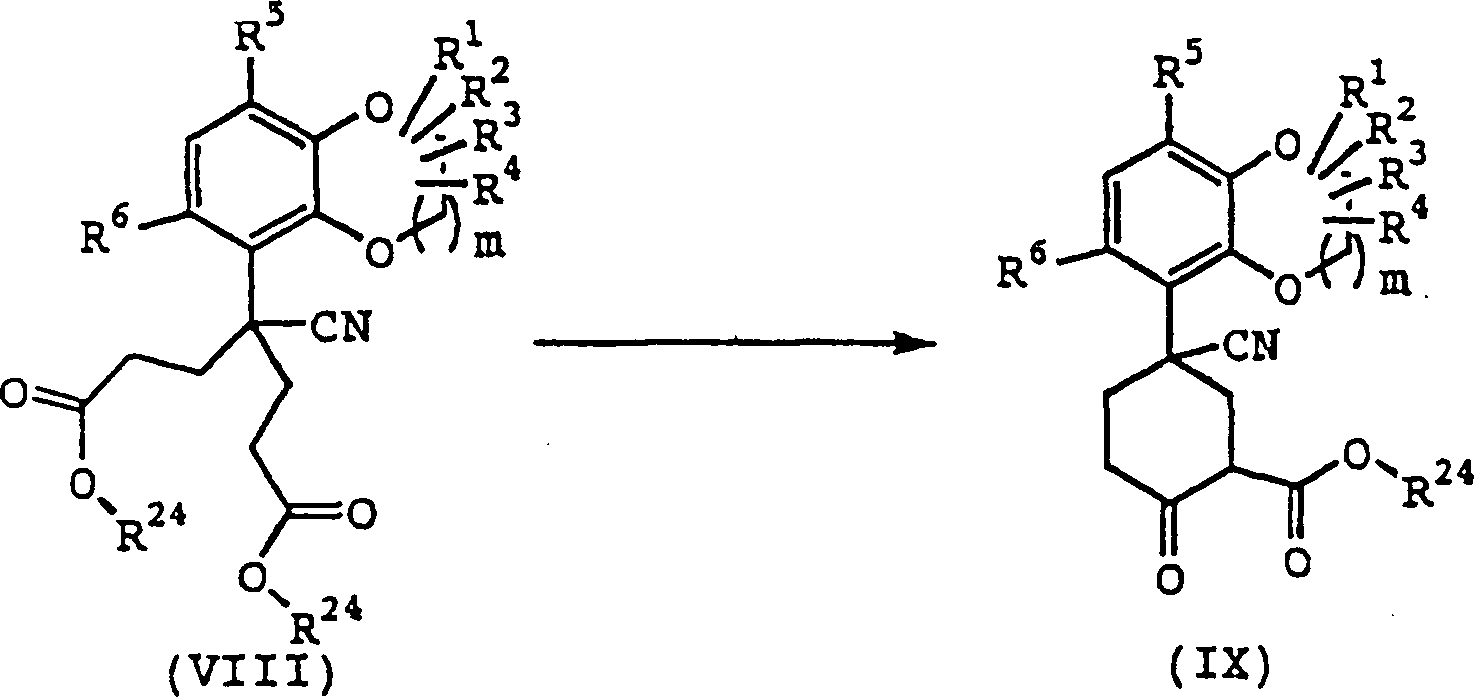
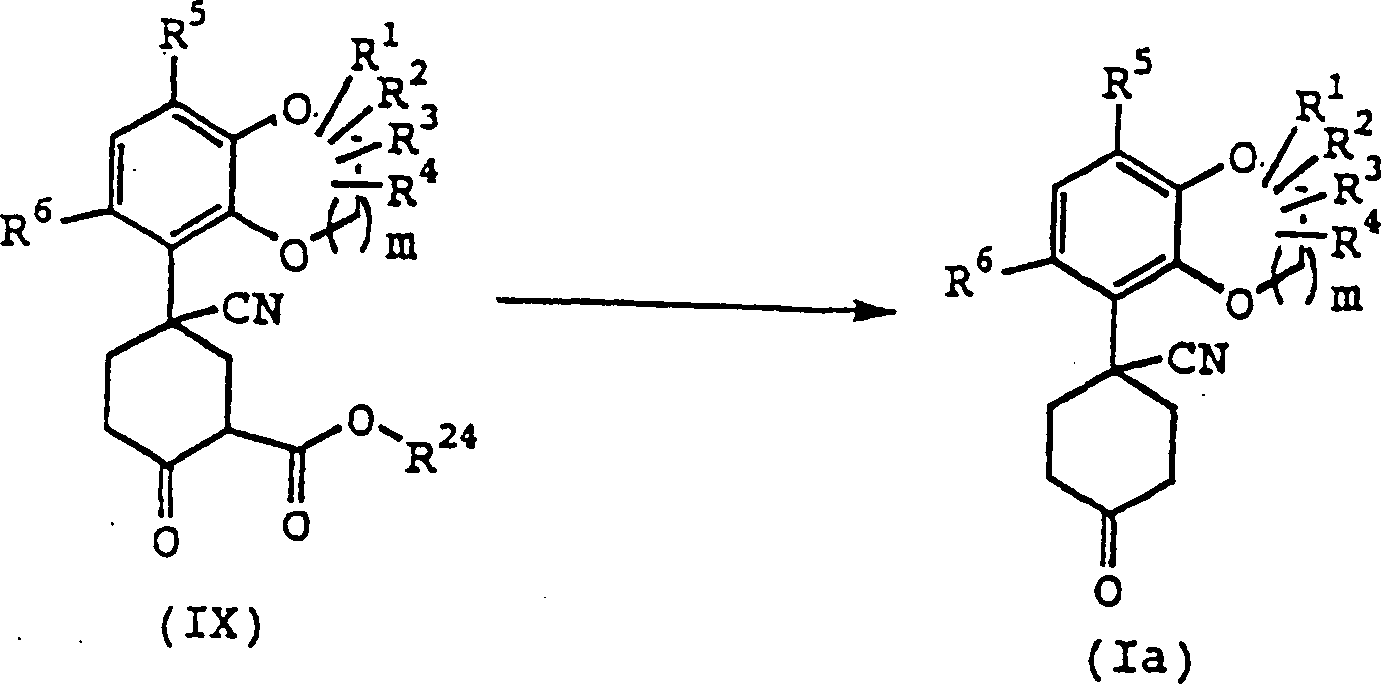
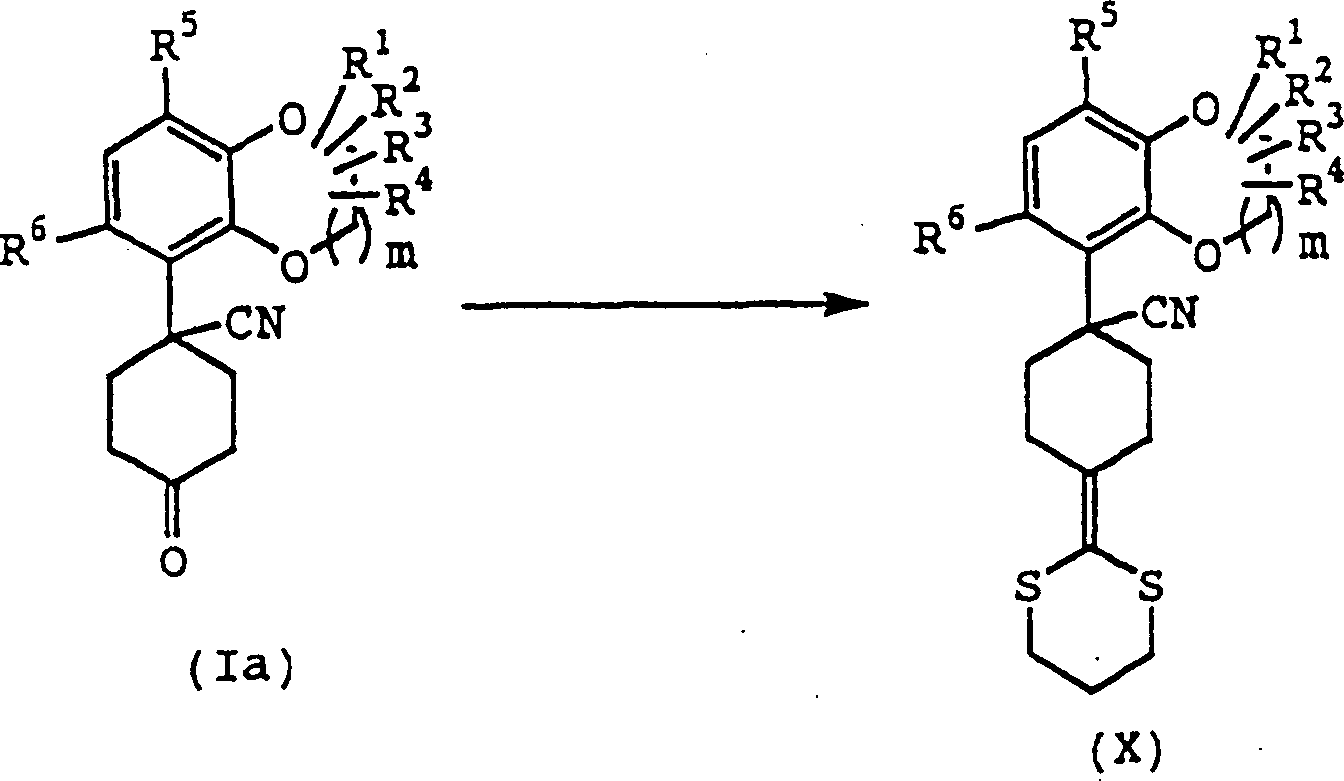
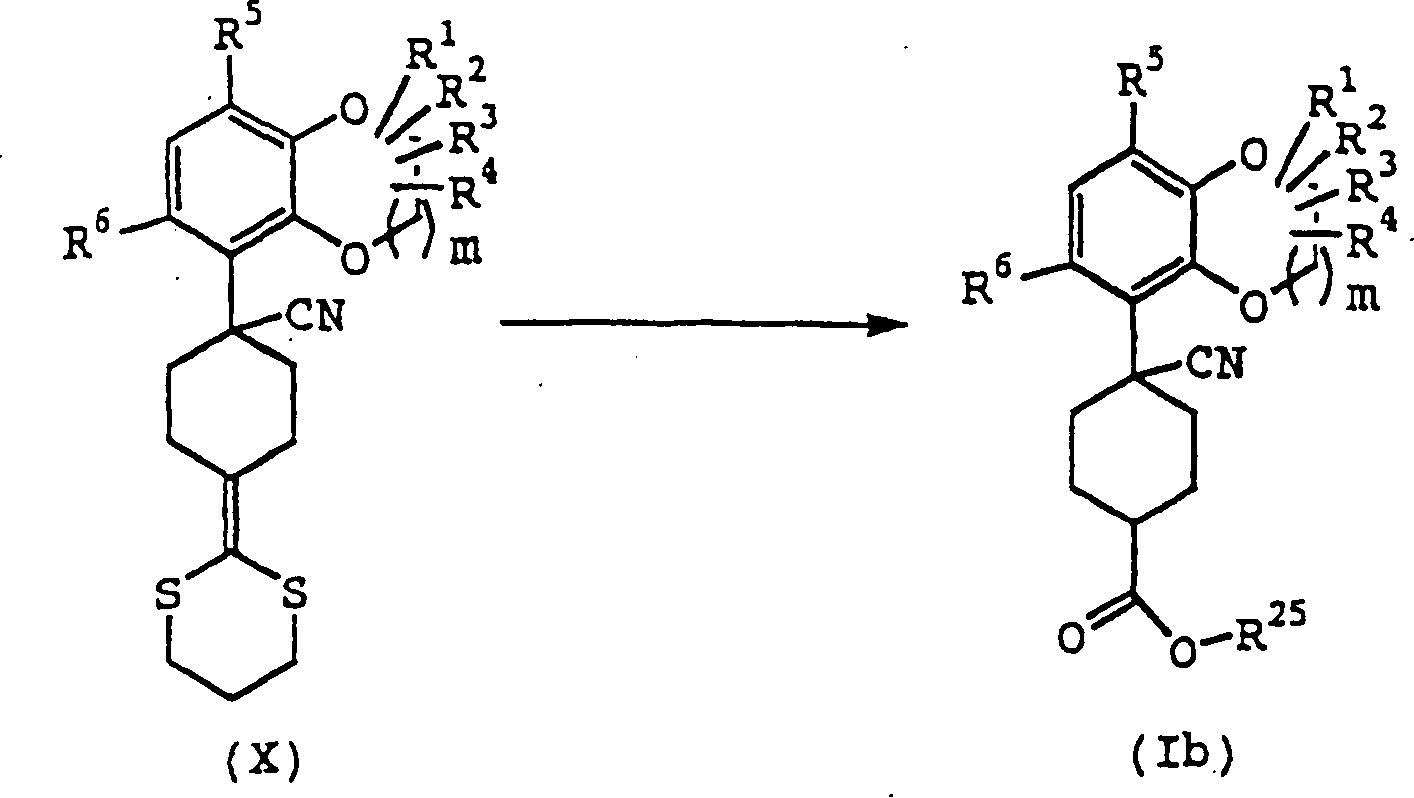

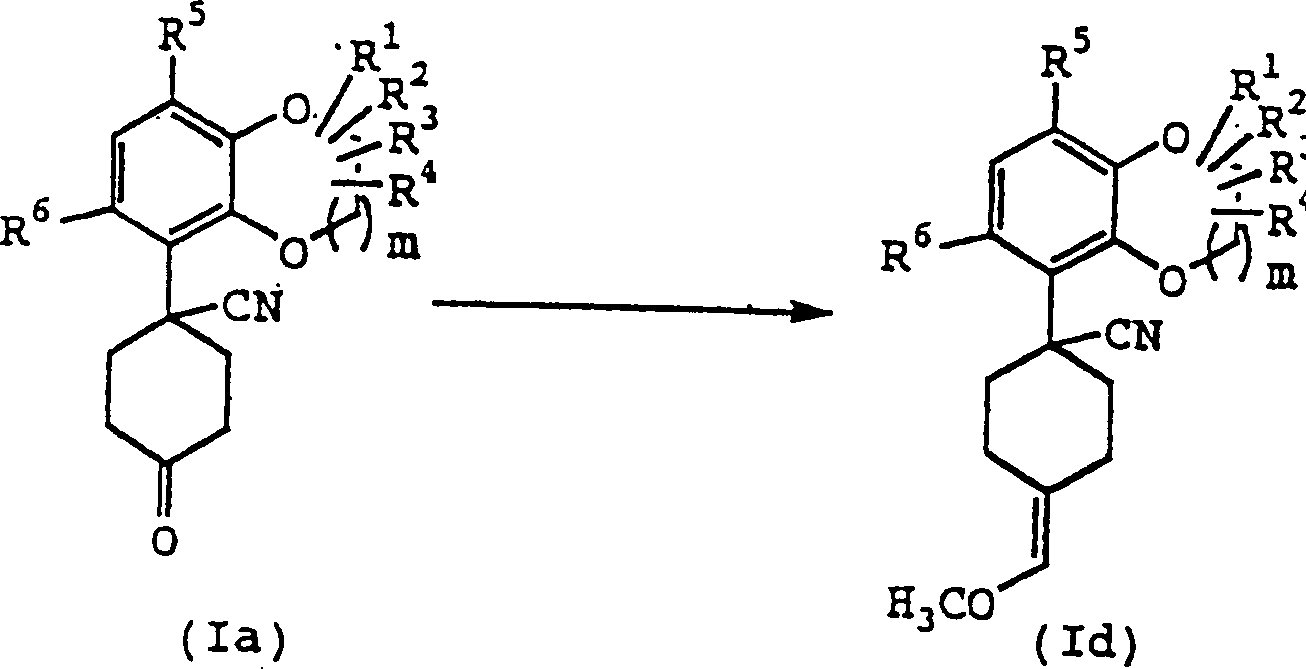
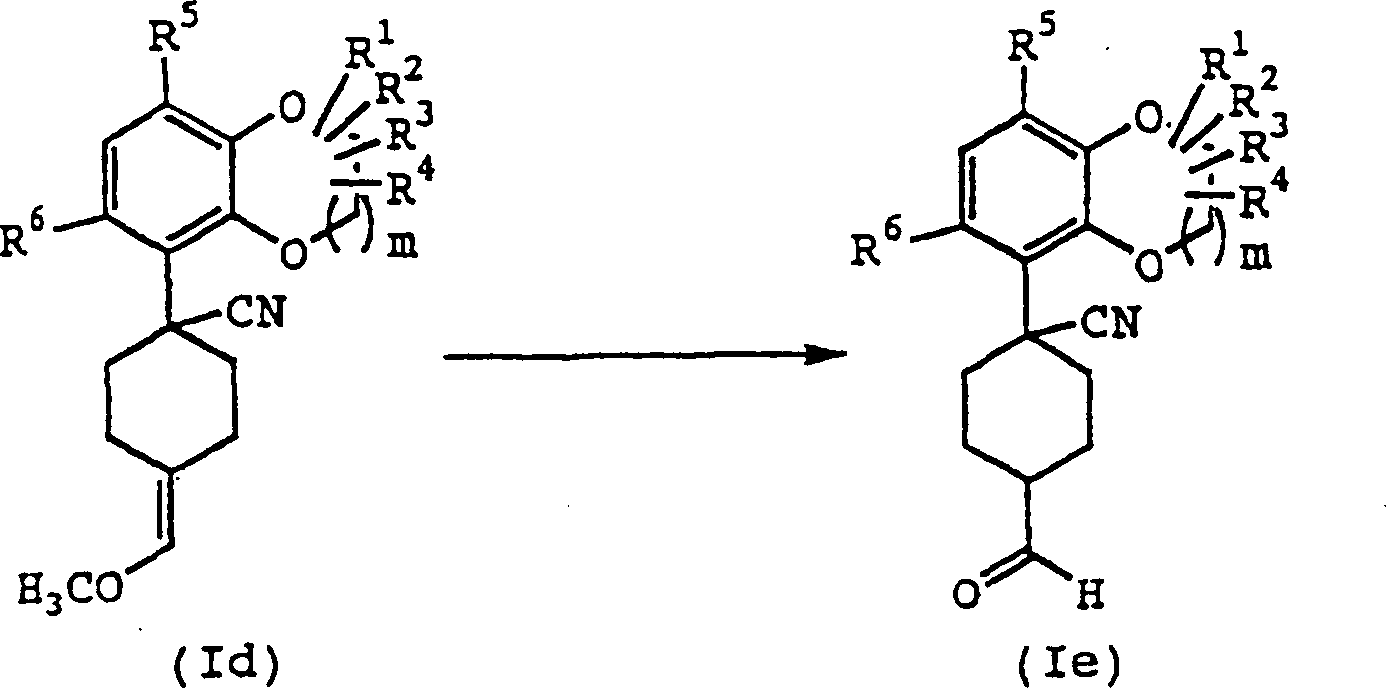
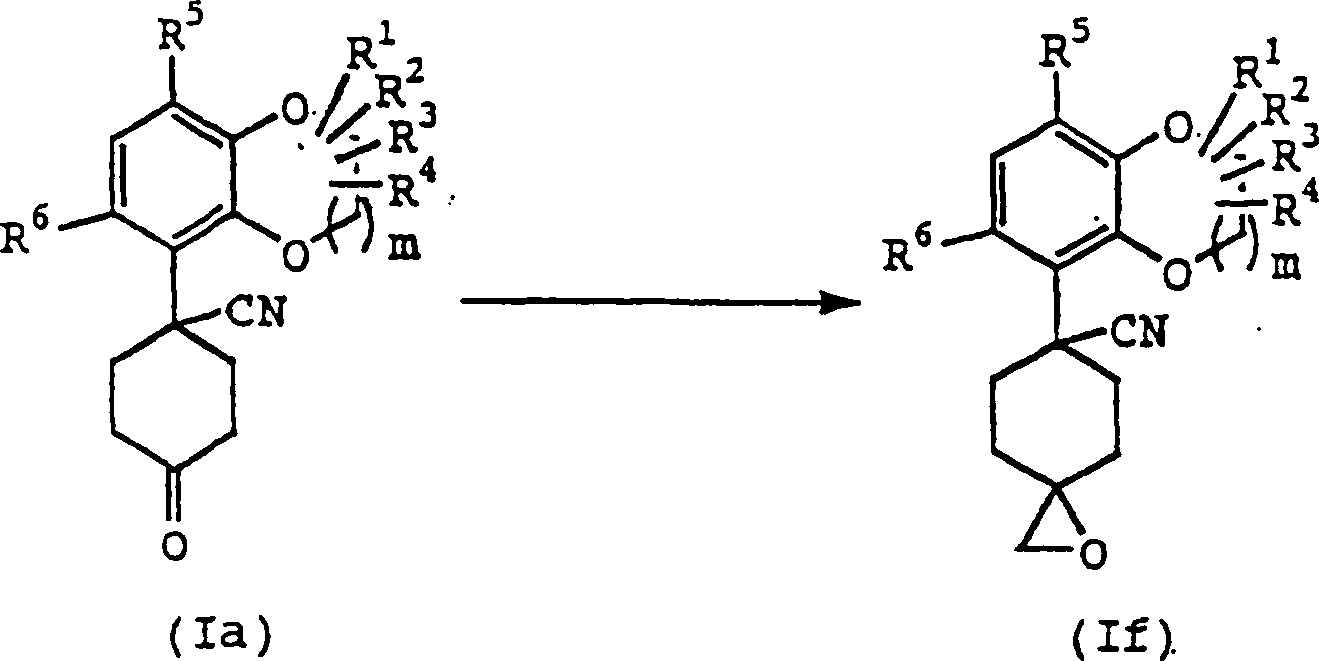
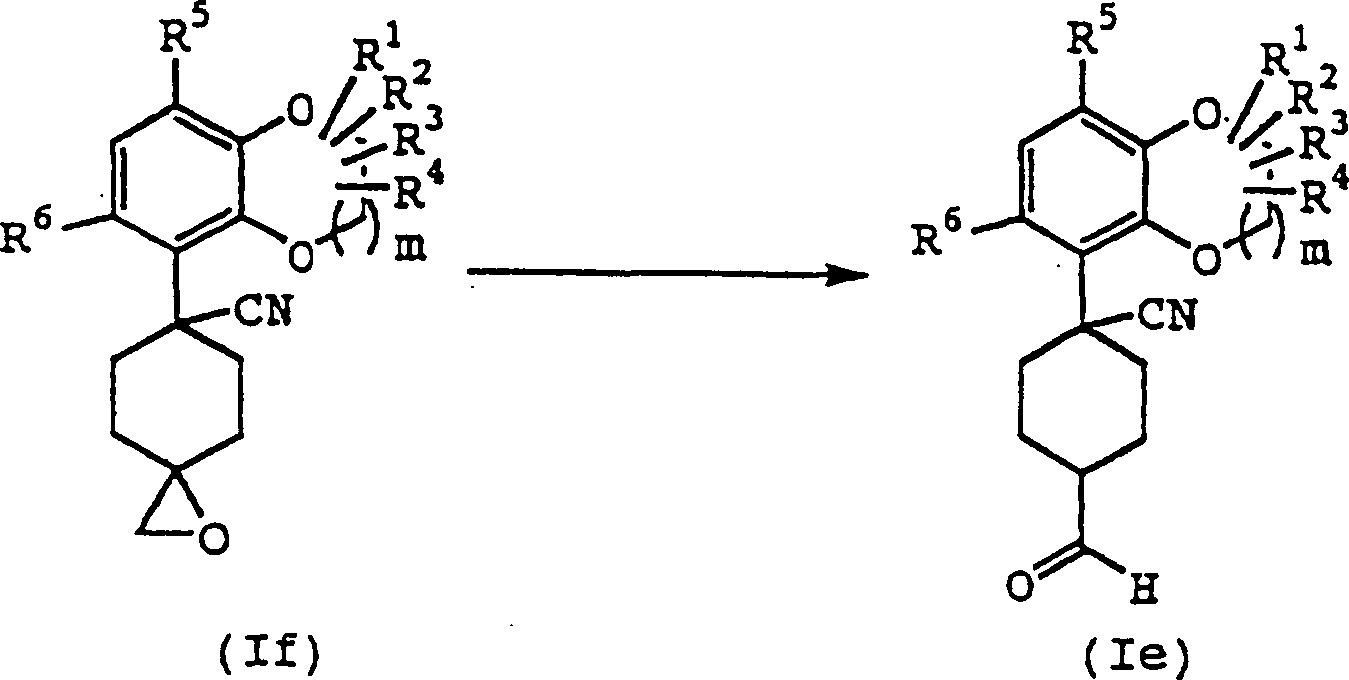

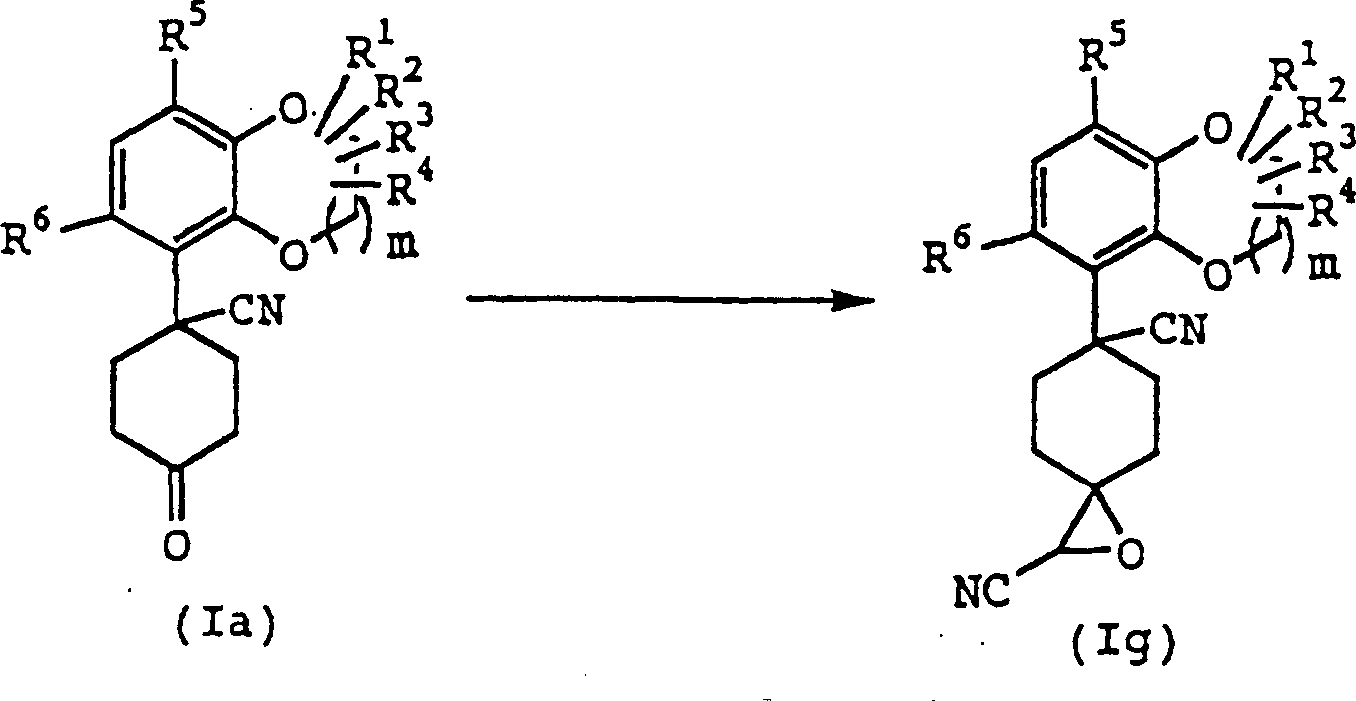


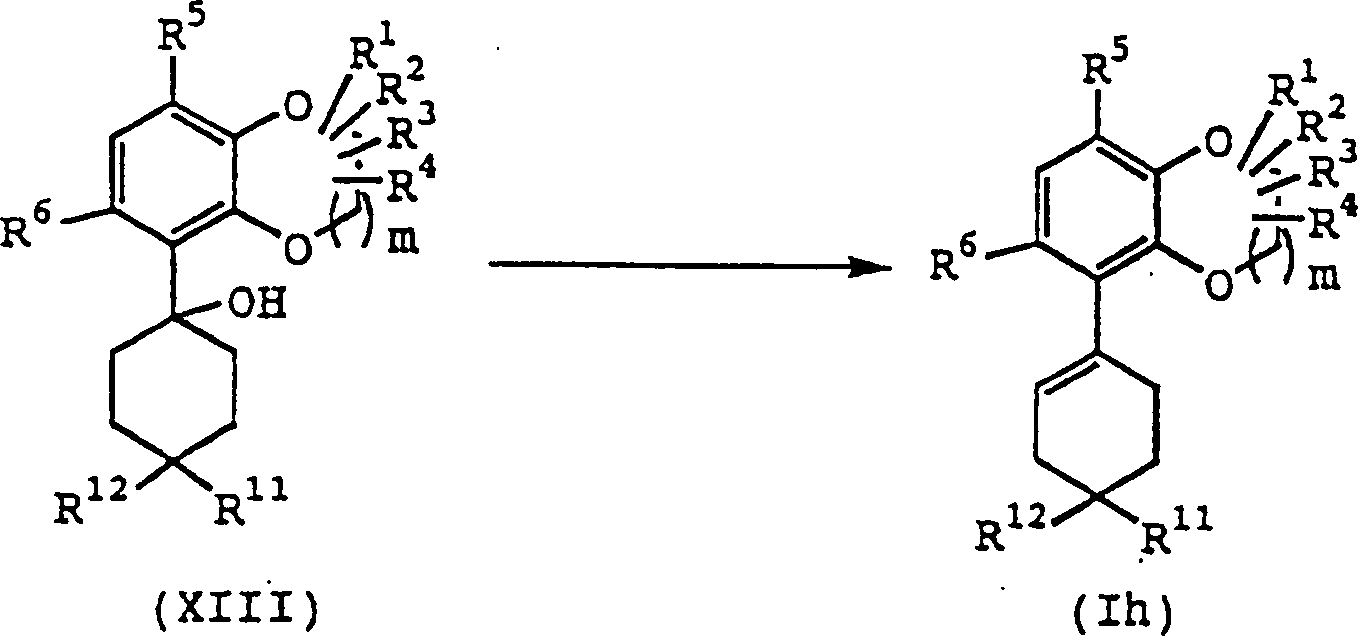

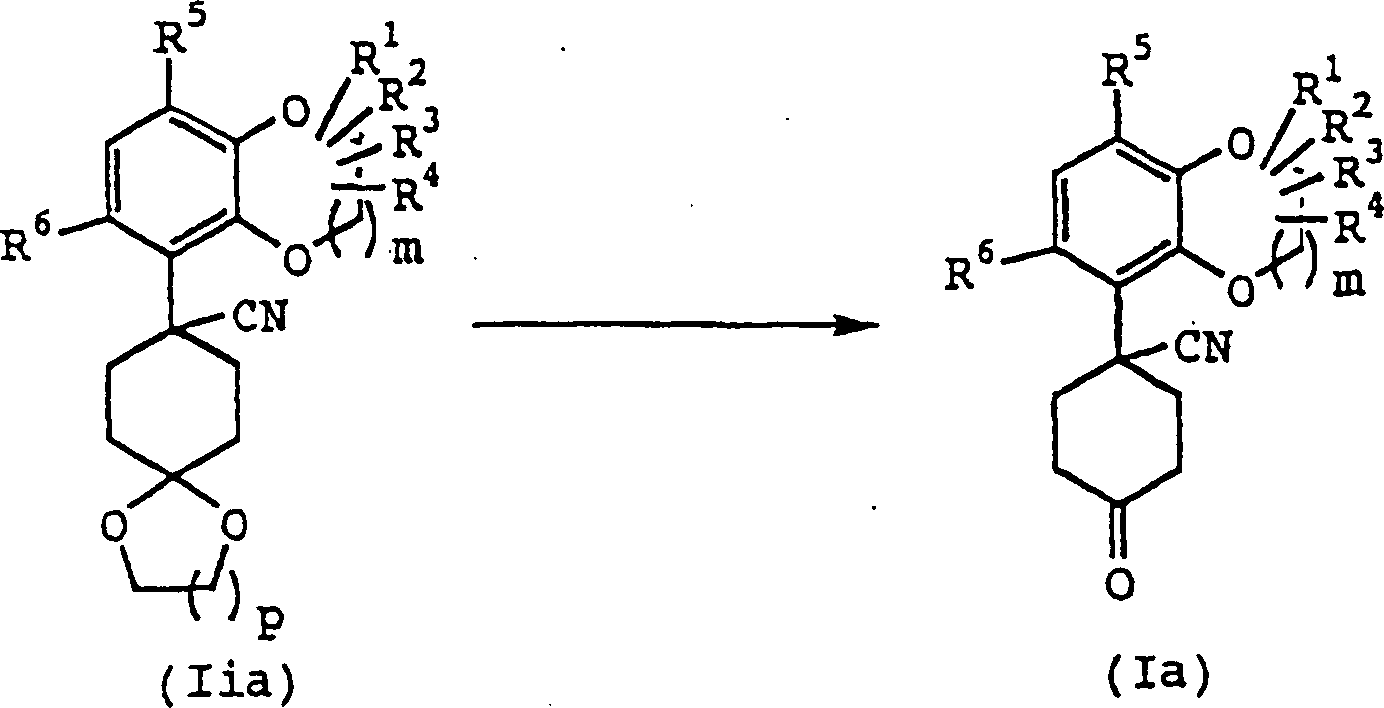

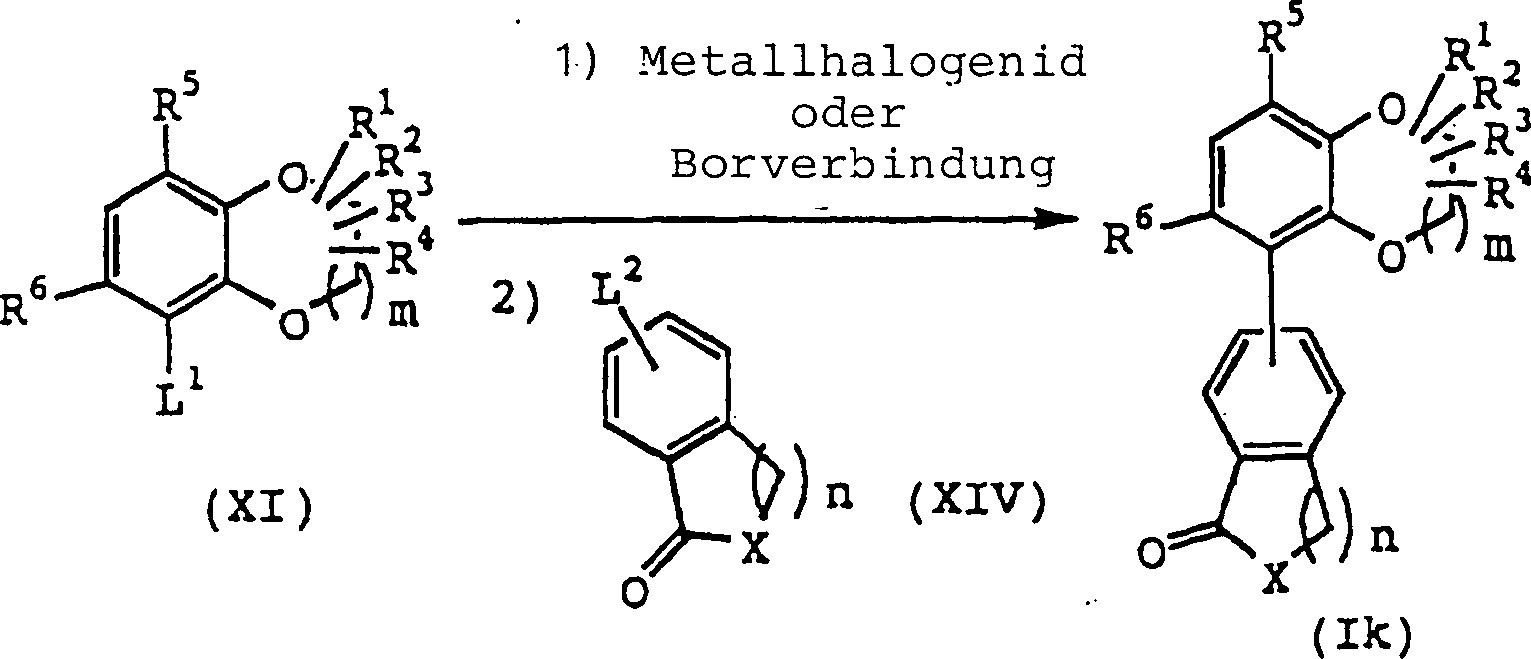
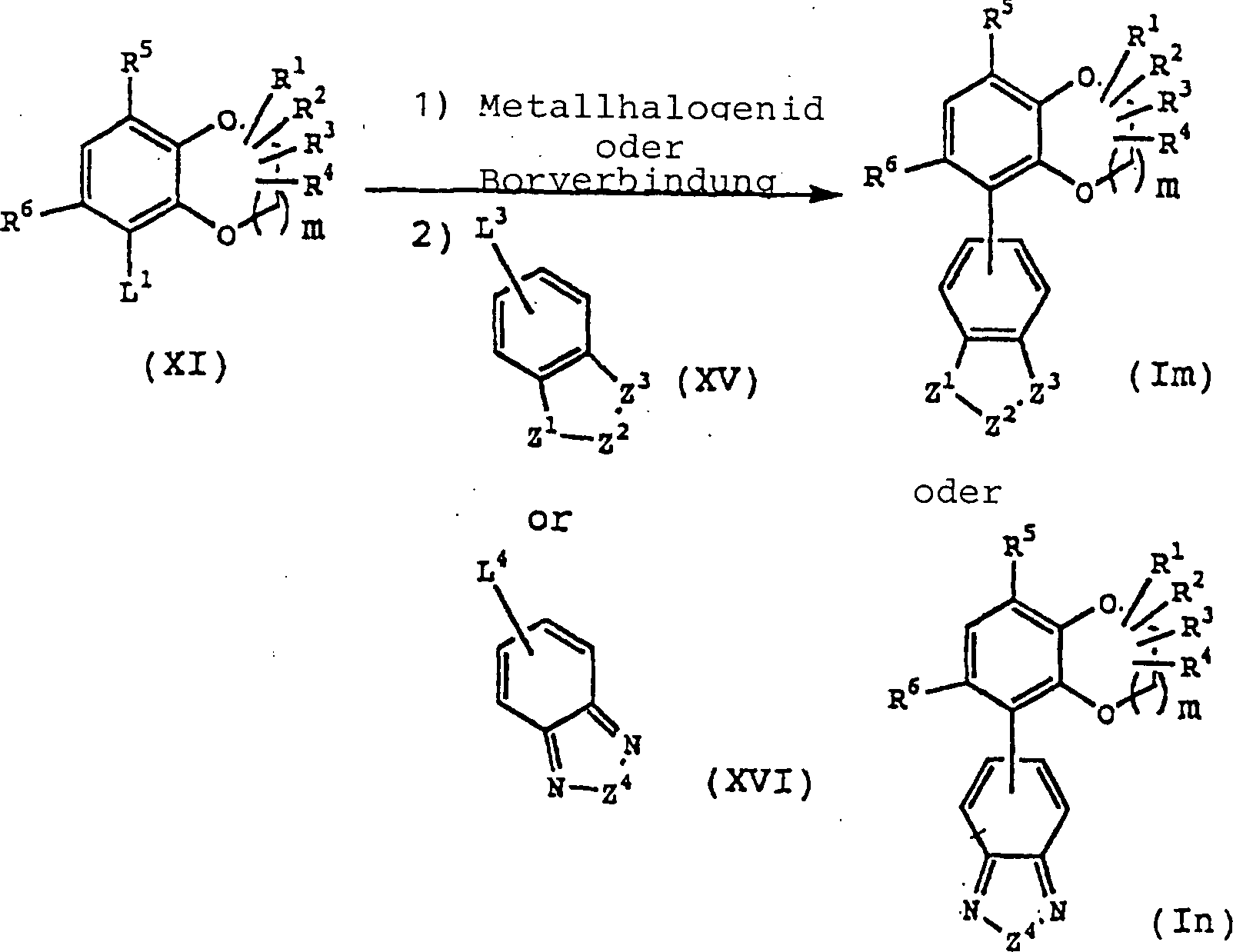


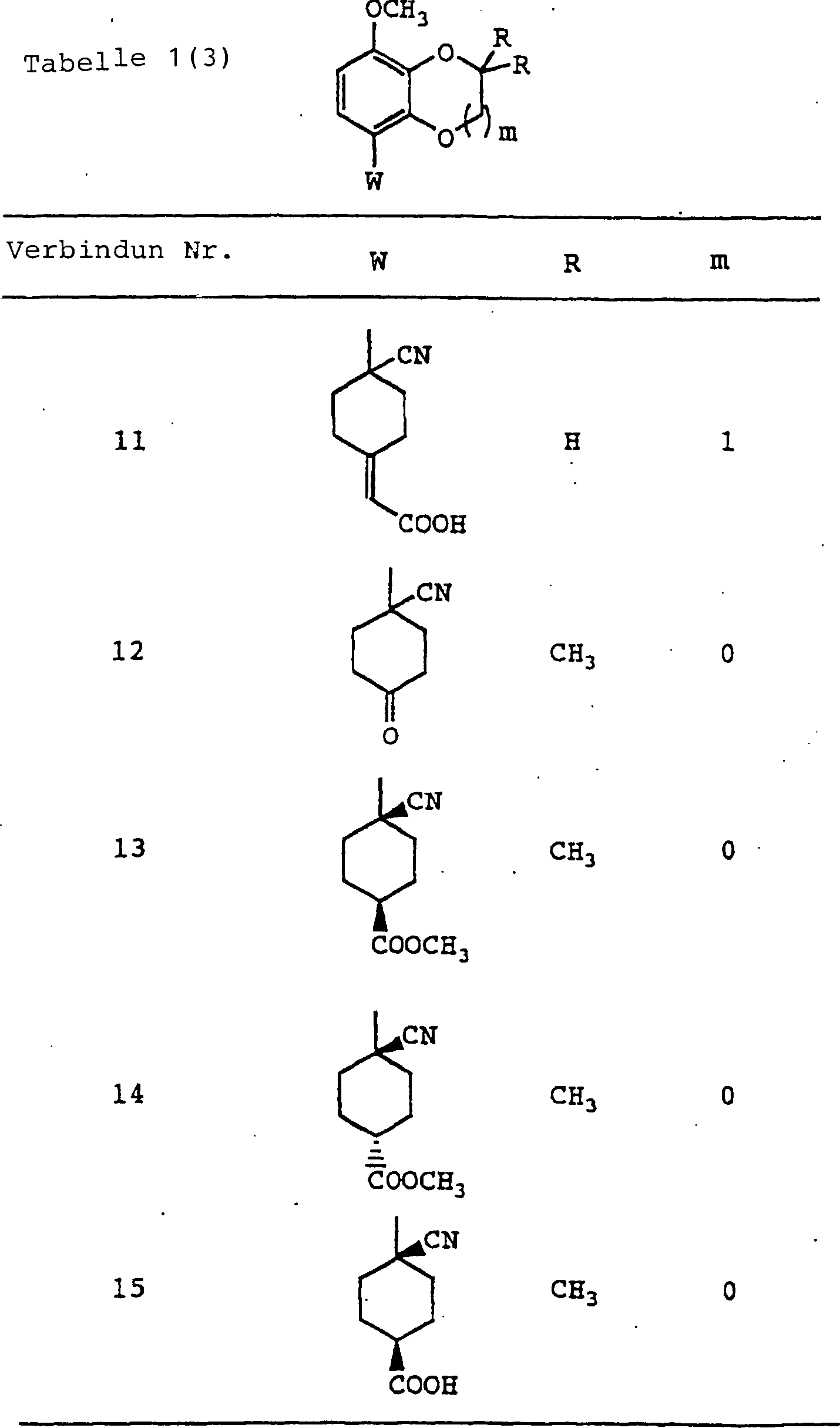
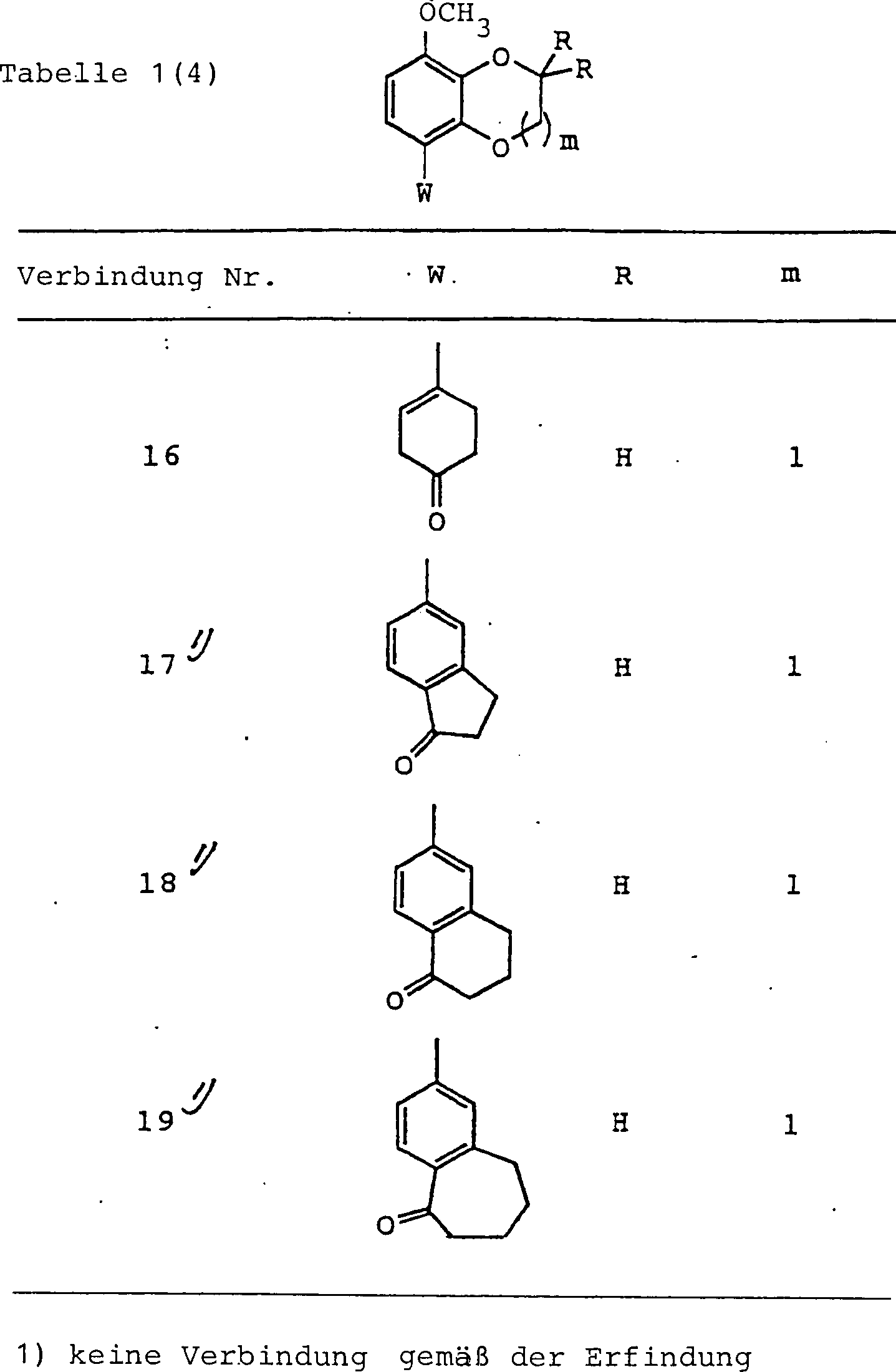
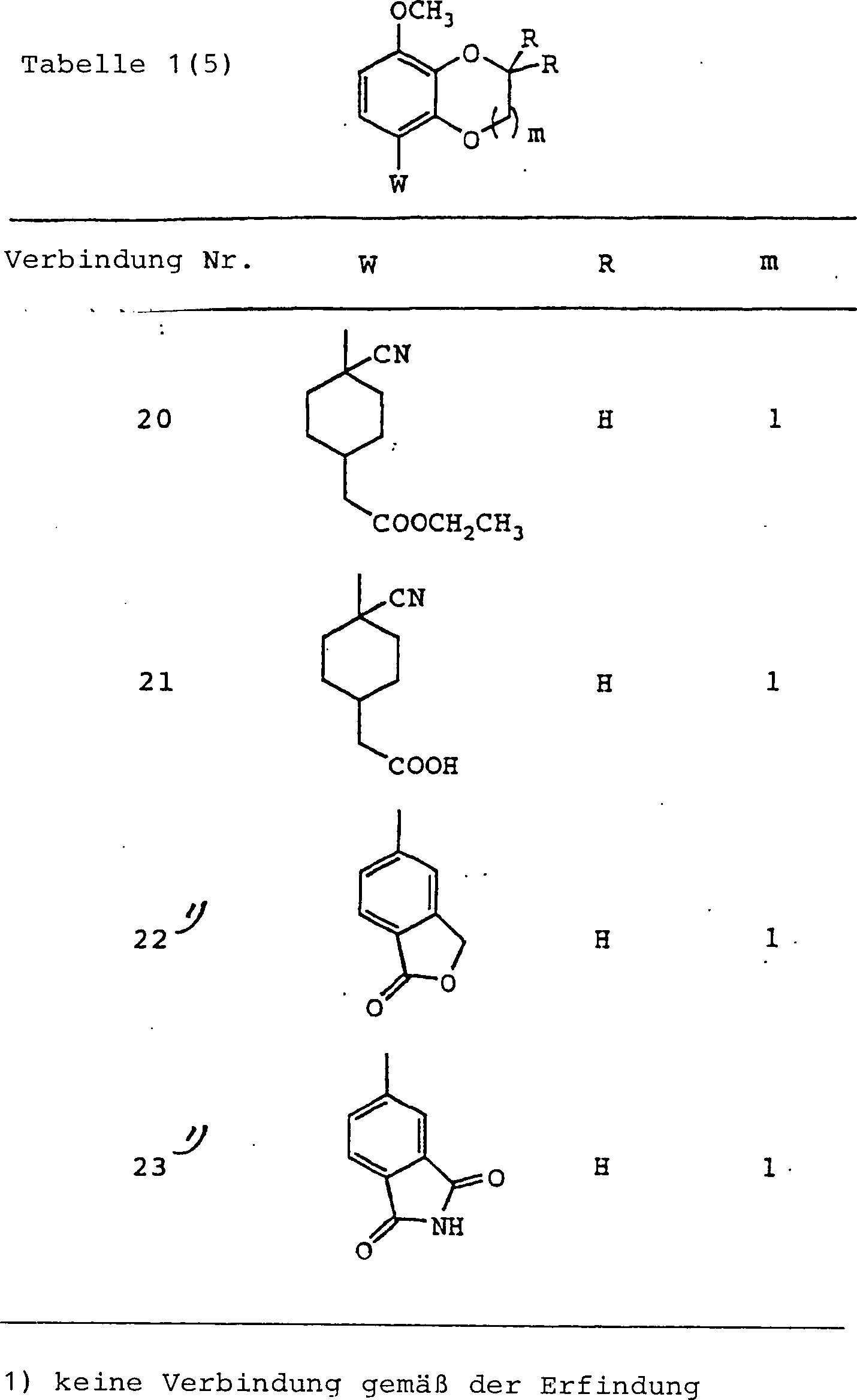
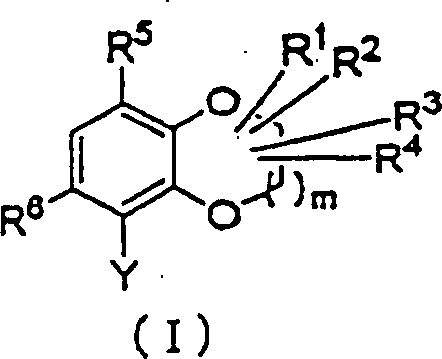 worin R9 darstellt: Cyano, Ethinyl oder Carbamoyl, und R10 darstellt: ein Wasserstoffatom, R11 darstellt: Hydroxy, Formyl, substituiertes oder unsubstituiertes C1-8-Alkoxy, substituiertes oder unsubstituiertes Tetrazolyl, -R13R14 (worin R13 und R14 unabhängig darstellen: ein Wasserstoffatom, substituiertes oder unsubstituiertes C1-8-Alkyl substituiertes oder unsubstituiertes C2-9-Alkanoyl, substituiertes oder unsubstituiertes Cycloalkyl, substituiertes oder unsubstituiertes Aryl, eine substituierte oder unsubstituierte aromatische heterocyclische Gruppe oder substituiertes oder unsubstituiertes Aralkyl, oder R13 und R14 kombiniert sind, um eine substituierte oder unsubstituierte heterocyclische Gruppe zusammen mit dem benachbarten Stickstoffatom darzustellen), -COOR15 (worin R15 darstellt: ein Wasserstoffatom oder substituiertes oder unsubstituiertes C1-8-Alkyl), -CONR16R17 (worin R16 und R17 unabhängig darstellen: ein Wasserstoffatom, substituiertes oder unsubstituiertes C1-8-Alkyl, substituier tes oder unsubstituiertes C2-9-Alkanoyl, substituiertes oder unsubstituiertes Cycloalkyl, substituiertes oder unsubstituiertes Aryl, eine substituierte oder unsubstituierte aromatische heterocyclische Gruppe oder substituiertes oder unsubstituiertes Aralkyl, oder R16 und R17 kombiniert sind, um eine substituierte oder unsubstituierte heterocyclische Gruppe zusammen mit dem benachbarten Stickstoffatom darzustellen) oder -CH2COOR18 (worin R18 darstellt: ein Wasserstoffatom oder substituiertes oder unsubstituiertes C1-8-Alkyl), R12 darstellt: ein Wasserstoffatom oder substituiertes oder unsubstituiertes C1-8-Alkoxy oder R11 und R12 zusammen kombiniert sind, um darzustellen: -OCH2(CH2)pO- (worin p eine ganze Zahl von 1 bis 3 darstellt), -CR19R20O- (worin R19 und R20 unabhängig darstellen: ein Wasserstoffatom oder Cyano), =CHOR21 (worin R21 darstellt: substituiertes oder unsubstituiertes C1-8-Alkyl, substituiertes oder unsubstituiertes C2-8-Alkenyl oder substituiertes oder unsubstituiertes Aralkyl), =CHCOOR22 (worin R22 darstellt: ein Wasserstoffatom oder substituiertes oder unsubstituiertes C1-8-Alkyl) oder =O; oder ein pharmazeutisch annehmbares Salz hiervon.
worin R9 darstellt: Cyano, Ethinyl oder Carbamoyl, und R10 darstellt: ein Wasserstoffatom, R11 darstellt: Hydroxy, Formyl, substituiertes oder unsubstituiertes C1-8-Alkoxy, substituiertes oder unsubstituiertes Tetrazolyl, -R13R14 (worin R13 und R14 unabhängig darstellen: ein Wasserstoffatom, substituiertes oder unsubstituiertes C1-8-Alkyl substituiertes oder unsubstituiertes C2-9-Alkanoyl, substituiertes oder unsubstituiertes Cycloalkyl, substituiertes oder unsubstituiertes Aryl, eine substituierte oder unsubstituierte aromatische heterocyclische Gruppe oder substituiertes oder unsubstituiertes Aralkyl, oder R13 und R14 kombiniert sind, um eine substituierte oder unsubstituierte heterocyclische Gruppe zusammen mit dem benachbarten Stickstoffatom darzustellen), -COOR15 (worin R15 darstellt: ein Wasserstoffatom oder substituiertes oder unsubstituiertes C1-8-Alkyl), -CONR16R17 (worin R16 und R17 unabhängig darstellen: ein Wasserstoffatom, substituiertes oder unsubstituiertes C1-8-Alkyl, substituier tes oder unsubstituiertes C2-9-Alkanoyl, substituiertes oder unsubstituiertes Cycloalkyl, substituiertes oder unsubstituiertes Aryl, eine substituierte oder unsubstituierte aromatische heterocyclische Gruppe oder substituiertes oder unsubstituiertes Aralkyl, oder R16 und R17 kombiniert sind, um eine substituierte oder unsubstituierte heterocyclische Gruppe zusammen mit dem benachbarten Stickstoffatom darzustellen) oder -CH2COOR18 (worin R18 darstellt: ein Wasserstoffatom oder substituiertes oder unsubstituiertes C1-8-Alkyl), R12 darstellt: ein Wasserstoffatom oder substituiertes oder unsubstituiertes C1-8-Alkoxy oder R11 und R12 zusammen kombiniert sind, um darzustellen: -OCH2(CH2)pO- (worin p eine ganze Zahl von 1 bis 3 darstellt), -CR19R20O- (worin R19 und R20 unabhängig darstellen: ein Wasserstoffatom oder Cyano), =CHOR21 (worin R21 darstellt: substituiertes oder unsubstituiertes C1-8-Alkyl, substituiertes oder unsubstituiertes C2-8-Alkenyl oder substituiertes oder unsubstituiertes Aralkyl), =CHCOOR22 (worin R22 darstellt: ein Wasserstoffatom oder substituiertes oder unsubstituiertes C1-8-Alkyl) oder =O; oder ein pharmazeutisch annehmbares Salz hiervon.