-
Die
Erfindung betrifft ein Verfahren zur Gewinnung von L-Dihydroorotsäure (im
Nachstehenden „L-DHO" bezeichnet) durch
Chromatographie an einem anionischen Austauschmaterial in einem
Basen-Wasser-Gemisch unter einem Druck von 1,1 MPa bis 40 MPa. Das
Verfahren kann verwendet werden, um die in-vitro- und in-vivo-Aktivität von N-(4-Trifluormethylphenyl)-5-methylisoxazol-4-carboxamid,
N-(4-Trifluormethylphenyl)-2-cyano-3-hydroxycrotonamid
und ähnliche
Verbindungen zu untersuchen.
-
L-DHO
kann durch ein Kieselgel-Chromatographieverfahren mit anschließender chemischer
Derivatisierung und colorimetrischer Bestimmung (Kesner, L., Aronson,
F.L., Silverman, M., Chan, P.C., Clin. Chem 21/3 (1975) 353) bestimmt
werden. Ein weiteres Verfahren wandelt L-DHO enzymatisch zu Orotsäure durch L-Dihydroorotsäuredehydrogenase
(im Nachstehenden „DHODH"), hergestellt aus
Rattenleber, um und weist nach chemischer Derivatisierung Orotat
durch colorimetrische Veränderungen
nach (Rogers, L.E., Nicolaisen, K., Experientia 28/10 (1972) 1259).
Die Nachteile von diesen Verfahren sind die Störung von anderen Materialien
in komplexen physiologischen Lösungen.
Außerdem
sind die erwähnten
Verfahren auf Grund von arbeitsaufwändiger Probenherstellung sehr
zeitraubend und deshalb für
Routineanalysen in großen
klinischen Studien nicht anwendbar.
-
Bei
der Bemühung
zur Bereitstellung von verbesserten Trennungs- und Isolierungsverfahren
zur Gewinnung von L-Dihydroorotsäure
wurde nun gefunden, dass dieselben durch Chromatographie von L-DHO
in einem Basen-Wasser-Gemisch an einem anionischen Austauschmaterial
und bei einem Druck von 1,1 MPa bis 40 MPa erzielt werden können. Das
Verfahren kann für
die quantitative Bestimmung von L-DHO in Zelllysaten, Säugerserum
und Humanserum verwendet werden. Das Verfahren ist sehr reproduzierbar,
empfindlich und verlässlich.
-
Die
Erfindung, wie in den Ansprüchen
erläutert,
löst die
Aufgabe durch ein chromatographisches Verfahren, umfassend die Schritte
von:
- a) Bereitstellen einer Säule, umfassend
druckstabiles anionisches Austauschmaterial;
- b) Beladen der Säule
mit einer Probenlösung,
die L-Dihydroorotsäure
einschließt;
- c) Ausführen
von Chromatographie;
- d) Eluieren der L-Dihydroorotsäure mit einer eluierenden Lösung, enthaltend
ein Basen-Wasser-Gemisch;
wobei das Verfahren unter einem
Druck von 1,1 MPa bis 40 MPa ausgeführt wird.
-
Der
Begriff druckstabiles anionisches Austauschmaterial bedeutet beispielsweise
Materialien, wie makroporöses
(2 000 Å)
Divinylbenzol/Ethylvinylbenzolpolymer oder ein mikroporöses Polyvinylbenzylammoniumpolymer,
vernetzt mit Divinylbenzol oder Gemischen davon, die mit Alkanol-quaternärem Ammonium
modifiziert sind; oder Vinylbenzylchlorid/Divinylbenzol-makroporöses Polymer;
oder vernetztes Polyethyliminopolymer; oder Siliziumdioxid, modifiziert
mit Propyltrimethylammonium; oder Poly(styrol-divinylbenzol)trimethylammonium.
-
Die
nachstehenden Produkte sind besonders bevorzugt:
Ion Pac AS
11, CarboPac PA 1 oder CarboPac MA 1 Anionenaustauschsäulen, bezogen
von Dionex Corporation, Idstein, Deutschland,
GROM – SIL, starkes
Anion, oder GROM – SIL,
schwaches Anion; bezogen von Grom P 1000 SAX, Ionospher SA oder
Chrompack PA; bezogen von Chrompack PRP-X100 oder RCX-10, bezogen
von Hamilton.
-
Die
Elutionslösung
enthält
ein Basen-Wasser-Gemisch. Geeignete Basen sind abgeleitet von Alkalimetallen
oder Erdalkalimetallen, wie Natriumhydroxid, Kaliumhydroxid, Magnesiumhydroxid
oder Calciumhydroxid. Die Konzentration der Base ist 1 mMol/l bis
etwa 200 mMol/l, bezogen auf Wasser als Lösungsmittel, vorzugsweise 2
mMol/l bis etwa 120 mMol/l, besonders bevorzugt ist 100 mMol/l.
-
Die
Temperatur während
des Chromatographieverfahrens ist etwa 0°C bis etwa 50°C, vorzugsweise etwa
15°C bis
etwa 30°C,
besonders bevorzugt etwa 19°C
bis etwa 25°C.
Der Arbeitsdruck während
der Chromatographie ist im Wesentlichen konstant. Die Chromatographie
kann unter Verwendung von verschiedenen Drücken ausgeführt werden; beispielsweise
kann die Chromatographie unter einem Druck von 1,1 × 108 Pa (1,1 MPa) bis 40 × 106 Pa
(40 MPa), insbesondere von 4,1 MPa bis 5,5 MPa, ausgeführt werden.
Die Elutionsmittelfließgeschwindigkeiten
sind von etwa 0,2 ml/min bis etwa 3 ml/min, vorzugsweise 1 ml/min.
-
Die
Beladung der Säulen,
Chromatographie und Elution von dem L-DHO findet durch bekannte,
herkömmliche
technische Verfahren statt.
-
Eine
geeignete Elution ist jene, worin die Elution einen Zeitgradienten
der Basenkonzentration, vorzugsweise mit einem linearen Verlauf,
zeigt. Dieser Konzentrationsgradient kann beispielsweise durch eine niedere
Basenkonzentration (null im Grenzfall), die bei der Elution am Beginn
der Elution vorliegt, und durch Erhöhen der Basenkonzentration
während
des Elutionsverfahrens angewendet werden. Es ist auf diese Weise möglich, eine
besonders wirksame Trennung von der L-DHO in Proben, abgeleitet
von Serum oder Zelllysaten, zu erreichen. Ein bevorzugter Basengradient
variiert von nahe 1% NaOH (100 mMol/l) und 99% Wasser (am Beginn
der Elution) bis etwa 60% NaOH und 40% Wasser (am Ende der Elution),
wobei der besonders bevorzugte Bereich etwa 1% NaOH und 99% Wasser
(am Beginn der Elution) bis etwa 15% NaOH und 75% Wasser (am Ende
der Elution) ist. Der Basen-Wasser-Gradient wird in einer li nearen
Weise von 2,5 Minuten bis etwa 14 Minuten und von 14 Minuten bis
etwa 25 Minuten verändert,
wobei das Abfallen des Gradienten während dieser 2 Zeiträume verschieden
ist.
-
Eine
besonders geeignete Elution kann durch Anwenden einer niedrigen
Basenkonzentration am Beginn des Trennverfahrens von etwa 1% für einen
Zeitraum von etwa 2,5 Minuten erreicht werden. Das Ergebnis ist
Eluieren von dem Meisten des störenden
Materials von der biologischen Matrix von der Säule. Die Abtrennung des Analyten
wird durch langsames Erhöhen
des Gradienten bis etwa 23% der Base innerhalb eines Zeitraums von
14 Minuten Gesamtanalysenzeit erreicht. Dann wird die Basenkonzentration
auf etwa 60% innerhalb 4 Minuten erhöht, um die Elution von stark
gebundenem Material zu erlauben. 60% Base sollten für nicht länger als
6 Minuten angewendet werden, bis erneute Gleichgewichtseinstellung
durch 1% Basen-Wasser-Gemisch erfolgt. Die nächste Analyse beginnt nach
45 Minuten Gesamtanalysenzeit.
-
Das
Wasser in dem Basen-Wasser-Gradienten wurde des-ionisiert und entgast.
-
Das
Abtrennungsverfahren gemäß der Erfindung
findet in einem Säulenverfahren
statt. Die Temperatur, die vorzugsweise während der anionischen Austauschchromatographie
konstant gehalten wird, kann innerhalb eines breiten Bereichs variiert
werden. Ein bevorzugter Temperaturbereich ist etwa –10°C bis etwa 50°C, insbesondere
etwa 15°C
bis etwa 25°C.
-
Die
Elution von L-DHO findet von 10 Minuten bis 12 Minuten nach dem
Beginn des Gradienten statt. Die Versuchszeit des Elutionsverfahrens
ist 13 min bis 25 min. Die L-DHO wird durch einen Leitfähigkeitsdetektor,
wie ein Modell CD20 von Dionex Cooperation, nachgewiesen. Um die
Grundlinienverschiebung zu minimieren und die Hintergrundleitfähigkeit
zu senken, kann ein Anionenselbstregenerierungssupressor, wie Modell
ASRS-I, 4 mm, von Dionex Cooperation, angewendet werden.
-
Das
erfindungsgemäße Verfahren
ist besonders für
analytische Chromatographie geeignet, kann jedoch auch für prä parative
Chromatographie, insbesondere, wenn das erfindungsgemäße Verfahren
mit einem präparativen
Hochdruck-Flüssigchromatographiesäule-(HPLC)-System
ausgeführt
wird, verwendet werden. Der Begriff „präparative Chromatographie" bedeutet ein Reinigungsverfahren
mit dem Ziel der Gewinnung und nicht nur Analyse von reinen Produkten.
Die Menge der reinen Produkte kann innerhalb breiter Grenzen, beispielsweise
1 mg bis 1000 g, vorzugsweise zwischen 50 mg und 500 mg, variieren.
-
Das
erfindungsgemäße Verfahren
kann verwendet werden, um Veränderungen
in intrazellulären
oder extrazellulären
L-DHO-Konzentrationen
auf Grund der Inhibierung von Dihydroorotsäuredehydrogenase (DHO-DH) nachzuweisen.
Das Enzym DHO-DH
ist für
die Umwandlung von L-Dihydroorotsäure zu Orotsäure während der
De-novo-Pyrimidinsynthese verantwortlich. Die Inhibierung von DHO-DH
führt zur
Akkumulation von L-DHO. Das erfindungsgemäße Verfahren kann für die Herstellung
eines diagnostischen Assays verwendet werden. Das erfindungsgemäße Verfahren
kann zum Bestimmen der Aktivität
von DHO-DH-Inhibitoren verwendet werden. DHO-DH-Inhibitoren sind
beispielsweise N-4-(Trifluormethylphenyl)-5-methylisoxazol-4-carboxylamid,
Brequinar, N-(4-Trifluormethylphenyl)-2-cyano-3-hydroxyhept-2-en-6-in-carboxamid, 2-Cyano-3-cyclopropyl-3-hydroxyacrylsäure-(4-cyanophenyl)-amid
oder N-(4-Trifluormethylphenyl)-2-cyano-3-hydroxycrotonamid. Das
erfindungsgemäße Verfahren
kann zum Bestimmen von L-DHO-Konzentrationen in Pflanzen, Zelllinien,
Tieren und Menschen verwendet werden. L-DHO-Bestimmung kann verwendet werden, um
Aktivität
von DHO-DH-Inhibitoren
in Pflanzen, Säugern
und Menschen zu verfolgen.
-
Das
erfindungsgemäße Verfahren
wird genauer in den folgenden Beispielen beschrieben. Sofern nicht anders
ausgewiesen, betreffen Prozentsatzdaten das Gewicht.
-
Beispiel 1
-
1.1. Chemikalien und Reagenzien
-
Chemikalien
und Reagenzien wurden wie nachstehend ausgewiesen bezogen:
| Carbonat-freies | Bader,
Holland |
| NaOH
und KOH | |
| L-Dihydroorotsäure | Sigma,
München |
| (L-DHO) | |
| HClO4 | Riedel
de Haen, Seelze |
| Eosin,
Chloroform | Riedel
de Haen, Seelze |
| RPMI
1640 Medium | Gibco,
Eggenstein |
| Fötales Kalbsserum
(FCS) | Bio
Whitaker, Verviers, |
| | Belgien |
-
Desionisiertes
Wasser musste durch Helium vor der Verwendung entgast werden.
-
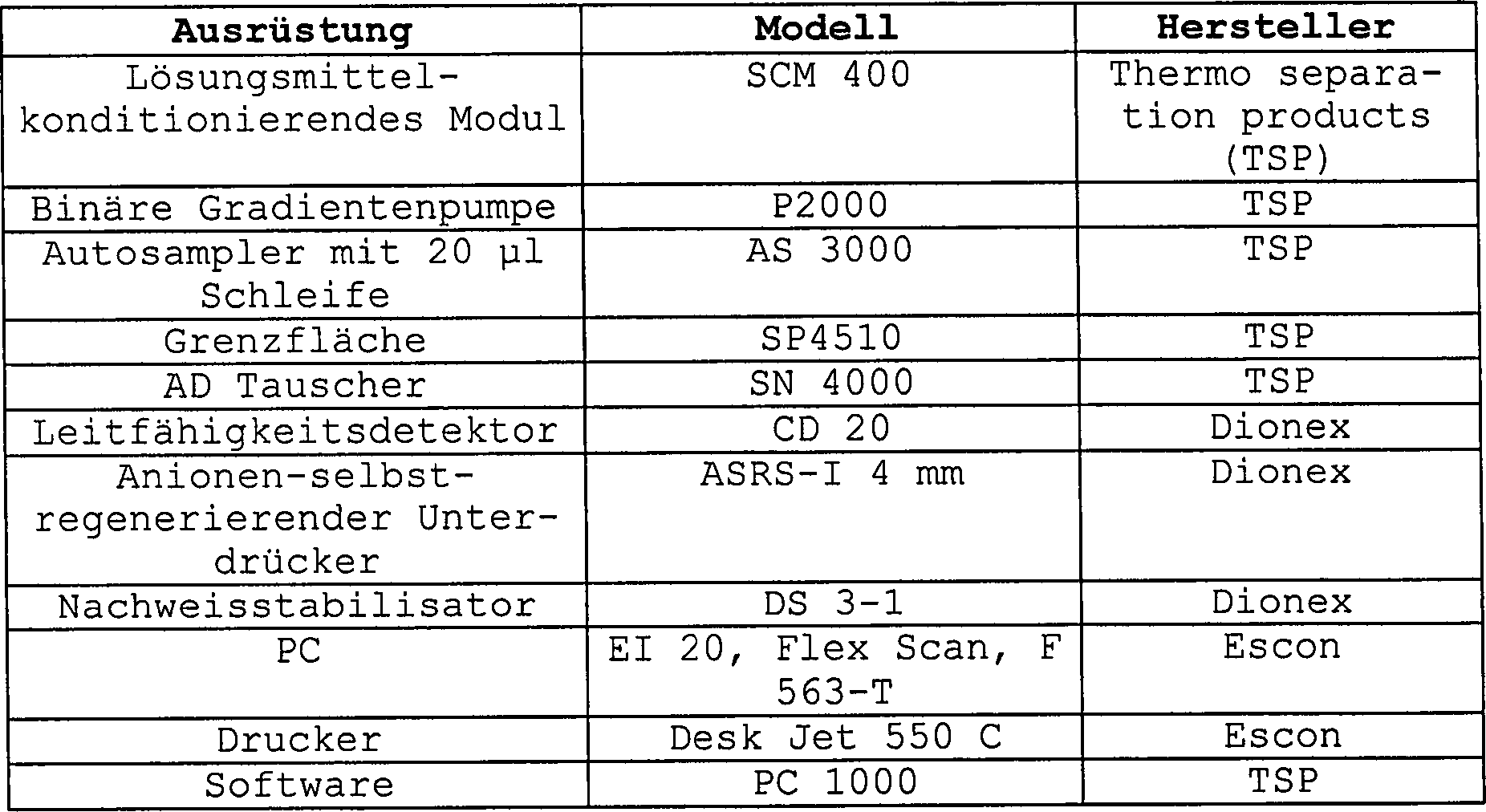
1.2. Chromatographische
Ausrüstung
-
Das
HPLC-System bestand aus den nachstehenden Instrumenten:
-
Das
Peakmaterial wurde innerhalb der gesamten Versuche verwendet.
-
1.3. HPLC-Bedingungen
-
Die
chromatographische Abtrennung wurde mit einer 250 × 4 mm I.D.
IonPac AS 11 Anionenaustauschsäule
(Teilchengröße 13 μm; P/N 044076,
Dionex), ausgerüstet
mit einer IonPac AG 11, 50 × 4
mm I.D. Vorsäule
(Teilchengröße 13 μm; P/N 044078,
Dionex), ausgeführt.
Zusätzlich
wurde eine Anionenfallensäule ATC-1
(P/N 037151, Dionex) zwischen der Gradientenpumpe und dem Einspritzventil
installiert. Um die Grundlinienverschiebung zu minimieren und die
Hintergrundleitfähigkeit
zu senken, wurde der ASRS-I-Suppressor installiert, der bei 300
mA betrieben wurde. Der Bereich des Detektors ist mit 10 μS ausgewiesen.
Der Autosampler wurde auf 14°C
gekühlt,
jedoch die Analyse selbst wurde bei Raumtemperatur ausgeführt. Die
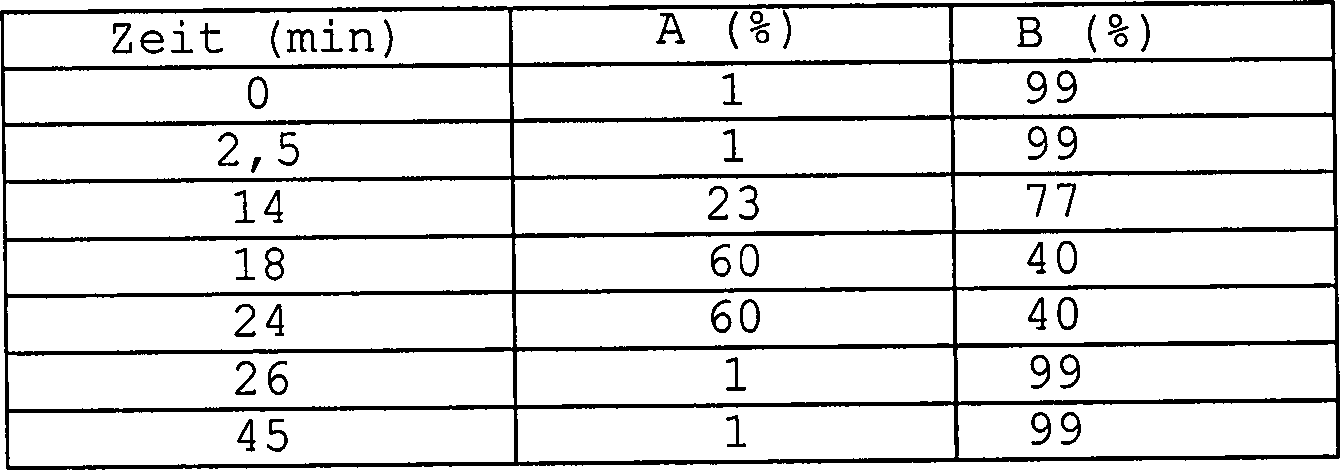
mobile Phase war aus 100 mM NaOH (A) und desionisiertem und entgastem
Wasser (B) zusammengesetzt. Mit dem System wurde der nachstehende
Gradient erzeugt:
-
Die
Fließgeschwindigkeit
war 1 ml/min; Versuchszeit war 45 min.
-
1.4. Standards und Qualitätskontrollproben
-
Eine
Standardstammlösung
wurde durch Auflösen
von 1 mg L-DHO in 1 ml Wasser hergestellt. Aliquote Mengen von 400 μl wurden
auf –20°C gefroren.
Die Stabilität
von diesen Lösungen
wurde für
mindestens 4 Wochen garantiert. Definierte Mengen der Stammlösung wurden
zu den Zelllysaten und zu Human- oder Rattenserum gegeben und zum
Bewerten der Linearität
zwischen dem Anstieg des Signals und der festgesetzten L-DHO-Konzentration bewertet.
Die Präzision
und Genauigkeit des Verfahrens wurden durch Anwenden von Qualitätskontroll-(QC)-Proben mit einem
L-DHO-Gehalt in dem unteren, dem mittleren und dem hohen Konzentrationsbereich
der Signal/Konzentrations-Linearitätskurve untersucht.
-
1.5. Probenzubereitungen
-
1.5.1. Zellen
-
Jurkat-Zellen
wurden von ATCC (TIB 182) erhalten und wie in 1.5.1.1. beschrieben
gezüchtet.
-
1.5.1.1 Gewebskulturbedingungen
-
Jurkat-Zellen
wurden bei 5 × 105/ml angesetzt und 24 Stunden in RPMI 1640
Medium und 10%igem fötalem
Kalbsserum (FCS) gezüchtet.
Die Zellen wurden in frischem Medium zur Zelllyse vereinigt und
die Menge der Zellen und der Prozentsatz an toten Zellen durch Vital-Mikroskopie
mit Eosin berechnet. Nach 24 Stunden hatten sich Zellzahlen um 1,6–1,8-fach innerhalb weniger
als 7% toter Zellen erhöht.
Jurkat-Zellen, die
zur L-DHO-Bestimmung genommen wurden, hatten ein Proliferationsverhältnis von
1,6–1,8-fach
in 24 Stunden und weniger als 7% tote Zellen.
-
1.5.1.2 Herstellung von
Zelllysaten
-
Zellen
wurden in einem definierten Mediumvolumen suspendiert und die Zelldichte
der Proben wurde über
Vitalmikroskopie unter Verwendung von Eosin bestimmt. Etwa 10 × 106 Zellen wurden entfernt, mit 5 Minuten-Zentrifugation
bei 350 × G
pelletisiert und der Überstand
wurde verworfen. Die Lyse der Zellen wurde durch Resuspendieren
des Zellpellets in 500 μl
1,2M HClO4 ausgeführt. Dieses Gemisch wurde in
2,0 ml Eppendorf-Safe-Lock-Becher (Sicherheitsverschlussbecher) überführt und
das Protein wurde nach 2 Minuten Hochgeschwindigkeitszentrifugierung
ausgefällt.
Der Überstand
wurde vollständig
entfernt, in Glasfläschchen überführt und
nach Zusatz von 500 μl
Chloroform sorgfältig
vermischt durch 2 Minuten Vortexbehandlung. Zelllipide wurden durch
Chloroform, gefolgt von 10 Minuten Zentrifugierung (1502 G) bei
10°C extrahiert.
Der gereinigte Überstand
wurde in 2 ml Eppendorf-Bechern gesammelt und bei –20°C bis zur
weiteren Verwendung gelagert. Für
die HPLC-Analyse wurden 100 μl
dieses Überstands
mit 30 μl
6M KOH neutralisiert. Nach Schütteln
für etwa
5 s wurden die Proben auf Eis für
etwa 30 Minuten gelagert. Anschließend wurden sie 5 min bei 15
000 U/min zentrifugiert. Von dem klaren Überstand wurden 20 μl für die HPLC-Analyse
verwendet.
-
1.5.2. Serum
-
Um
den Proteingehalt zu vermindern, wurden 200 μl Serum zu einem Microconfilter
(10 000 D, Modell 10, Code 42407, Amicon) gegeben und 30 min bei
13 000 Umdrehungen pro Minute (U/min) zentrifugiert. Der Durchfluss
bestand aus etwa 150 μl
und enthält
den Analyten L-DHO. Von dieser Flüssigkeit wurden 20 μl für die HPLC-Analyse
verwendet.
-
1.6. Quanitifizierung
-
Der
Integrator bestimmte die Peakhöhe
des Analyten. Eichkurven wurden erhalten durch Auftragen der gemessenen
Peakhöhen
(y) gegen die Analytenkonzentration in verschiedenen biologischen
Matrizes. Gewichtete lineare Regression (1/y) wurde verwendet, um
die L-DHO-Konzentration in Standardproben sowie in Qualitätskontrollen
zurückzurechnen.
Der übliche
Korrelationskoeffizient R wurde durch PROC GLM, bezogen auf die
Analyse des Kovarianzmodells unter Verwendung des Wägefaktors,
bereitgestellt.
-
1.7. Stabilität
-
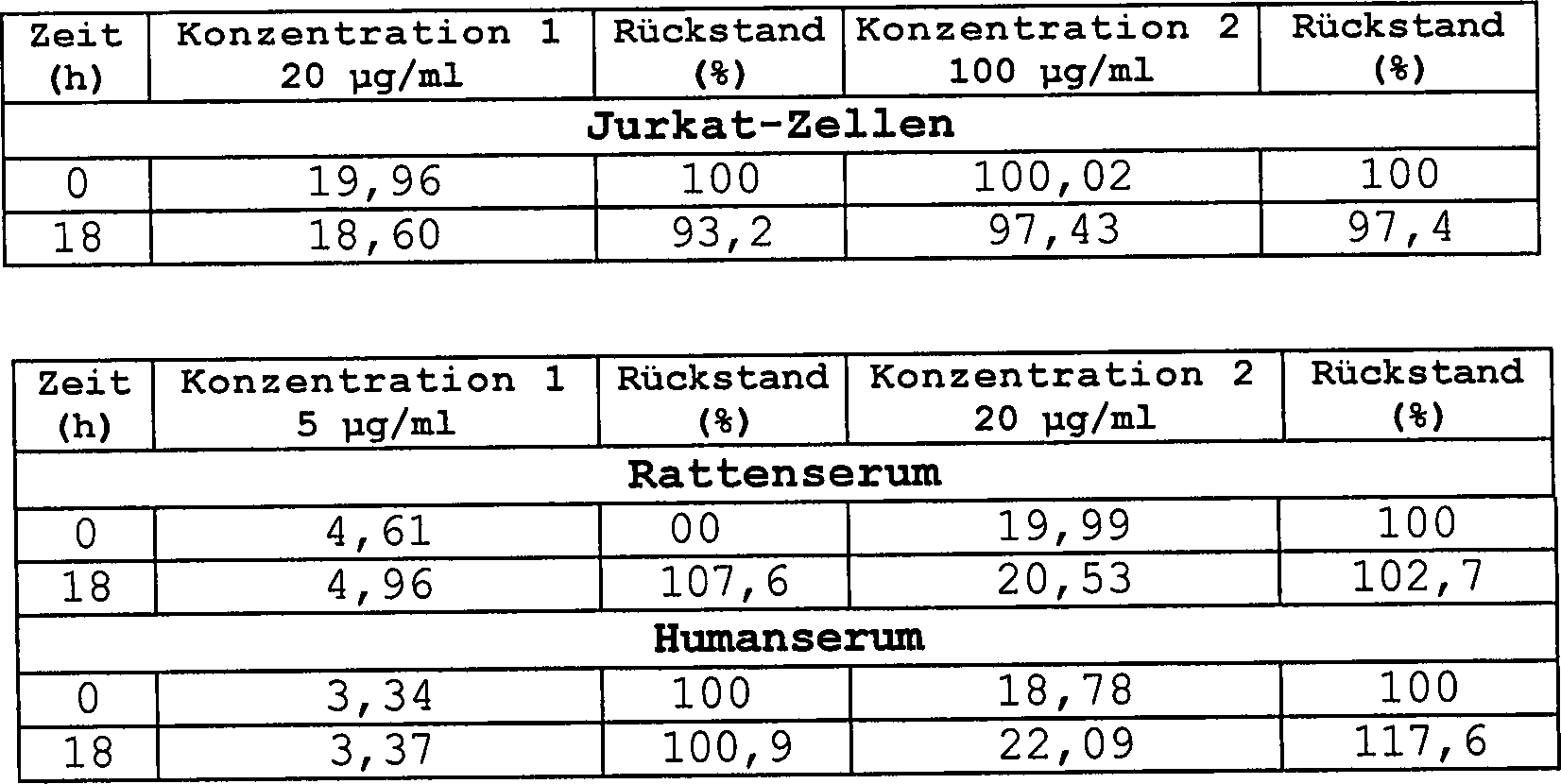
Tabelle
1 zeigt die Stabilitätsdaten
des Analyten bei –20°C in Zelllysaten
und Serumproben nach 2–3 Gefrier/Auftauzyklen
von einer Probe. L-DHO war unter den vorstehend erwähnten Bedingungen
in Jurkat-Zellen über
einen Zeitraum von mindestens 4 Wochen stabil. Die nachgewiesene
Erhöhung
in der Konzentration von einigen Rattenserumproben und einer Humanserumprobe
nach einigen Auftauzyklen von mehr als 10% kann nicht erklärt werden
und zeigt, dass die Genauigkeit in solchen Fällen bis zu 15% im Allgemeinen
und bis zu 29% im schlechtesten Fall vermindert sein kann. Aus diesen
Gründen
kann nur mitgeteilt werden, dass bei Ratten- und Hu manserumproben
L-DHO letztendlich für
nur mindestens 1 Woche stabil ist. Tabelle
1: Stabilitätsdaten
in Zelllysaten und Serum bei –20°C (n = 1)
-
Der
Rückstand
(%) ist ein Prozentsatz der Konzentration, verglichen mit der Anfangsanalyse.
n.d.
= nicht bestimmt
-
Um
die Bedingungen der Proben zu simulieren, wenn sie in dem Autosampler
auf die tatsächliche
Analyse warten, wurde die Stabilität während 18 h bei 14°C bestimmt.
Aus dem Grund wurden die Zelllysate versetzt und dann mit 30 μl 6M KOH/100 μl Lysat vor
dem Beginn der Analyse behandelt. Entsprechend wurden die Serumproben
versetzt und dann, wie in 1.5.2. beschrieben, deproteinisiert.
-
Wie
in Tabelle 2 ersichtlich werden kann, ist der L-DHO-Gehalt leicht in Zellen unter diesen
Bedingungen bis zu einem Maximum von etwa 7% vermindert. In Serumproben
ist der Analyt unter diesen Bedingungen stabil. Aus diesen Gründen nur
wurden so viele HPLC-Proben hergestellt, dass die maximale Länge des
Aufenthalts in dem Autosampler unter 18 h war. Tabelle
2: Stabilität
für 18
h bei 14°C
(n = 1)
-
1.8. Selektivität
-
Der
Vergleich von nicht versetzten Jurkat-Zelllysaten mit entsprechenden
Chromatogrammen, unter Verwendung von Zelllysaten, die mit 50 μg L-DHO/ml
versetzt wurden, zeigte, dass es einen kleinen Peak bei 11,983 min
gab, was die gleiche Retentionszeit wie L-DHO ist. Es ist sehr nahe
liegend, dass sich dieser Peak aus dem natürlichen Gehalt des Analyten
in diesen Zellen ergibt. Zusätzlich
kann ersichtlich werden, dass sich auf Grund der Behandlung der
Zelllysate mit KOH der L-DHO-Peak in zwei Peaks spaltet. Die KOH-Behandlung
jedoch ist wesentlich, um das saure Zelllysat vor der HPLC-Analyse
zu neutralisieren. Es konnte gezeigt werden, dass unter den beschriebenen
Bedingungen die Entwicklung der zweiten Peakhöhe (Retentionszeit (RT) = 11,954
min) angewendet werden kann, um verbesserte Ergebnisse bezüglich Linearität und Reproduzierbarkeit
zu erhalten.
-
Auch
im Fall von Ratten- und Humanserum wurde ein Blindwert mit der gleichen
Retentionszeit wie L-DHO gefunden. Es wird vermutet, dass sich dies
auf den natürlichen
L-DHO-Gehalt des
Organismus bezieht. Die Bestimmung von mindestens 10 verschiedenen
Proben von beiden Arten zeigte, dass der natürliche L-DHO-Gehalt unter der
Nachweisgrenze von 1 μg/ml
war.
-
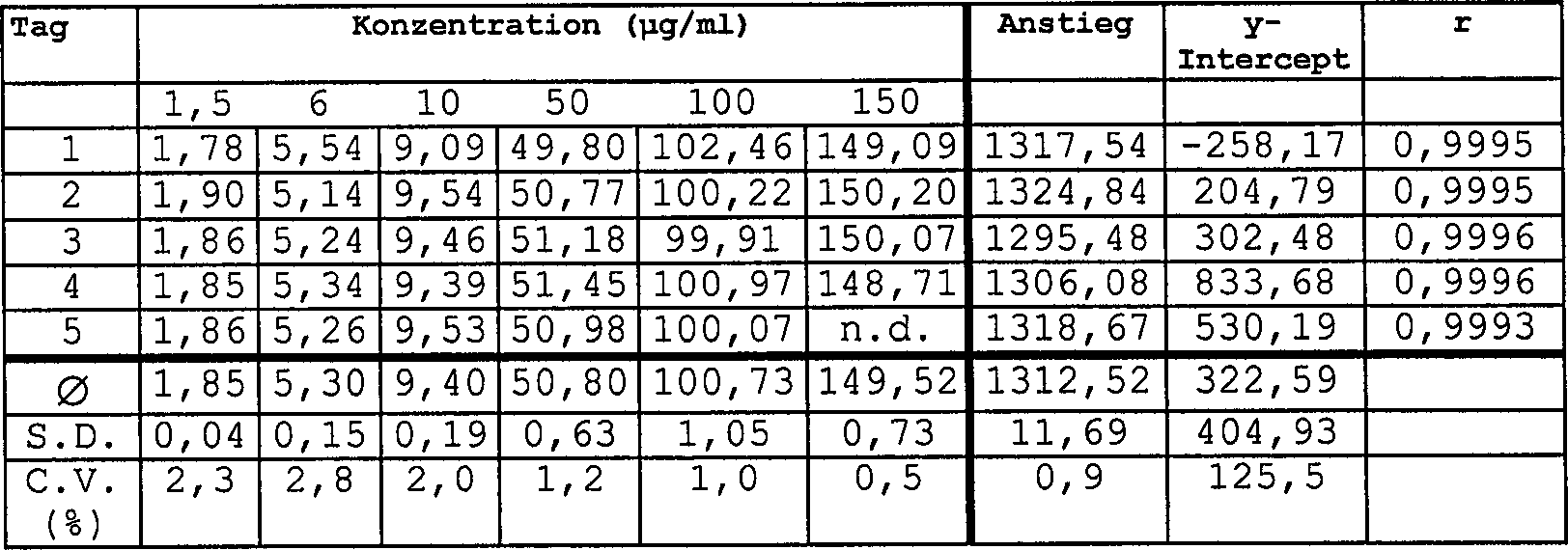
1.9. Linearität
-
Die
Linearität
der Bestimmung wurde an fünf
Eichkurven für
die Zelllinie und verschiedenen Serumproben bewertet. Die Proben
wurden hergestellt und an fünf
verschiedenen Tagen mit L-DHO-Konzentrationen im Bereich von 1,5–150 μg/ml (Zelllysate)
und 1–30 μg/ml (Serumproben)
laufen lassen. Die Ergebnisse werden in Tabellen 3–5 gezeigt.
Für die
Bestimmung der Regressionslinie wurde die Peakhöhe verwendet. Basierend auf
den entsprechenden Konzentrationen der verschiedenen Standards wurde
es, wie in verschiedenen Tabellen ausgewiesen, zurückberechnet.
-
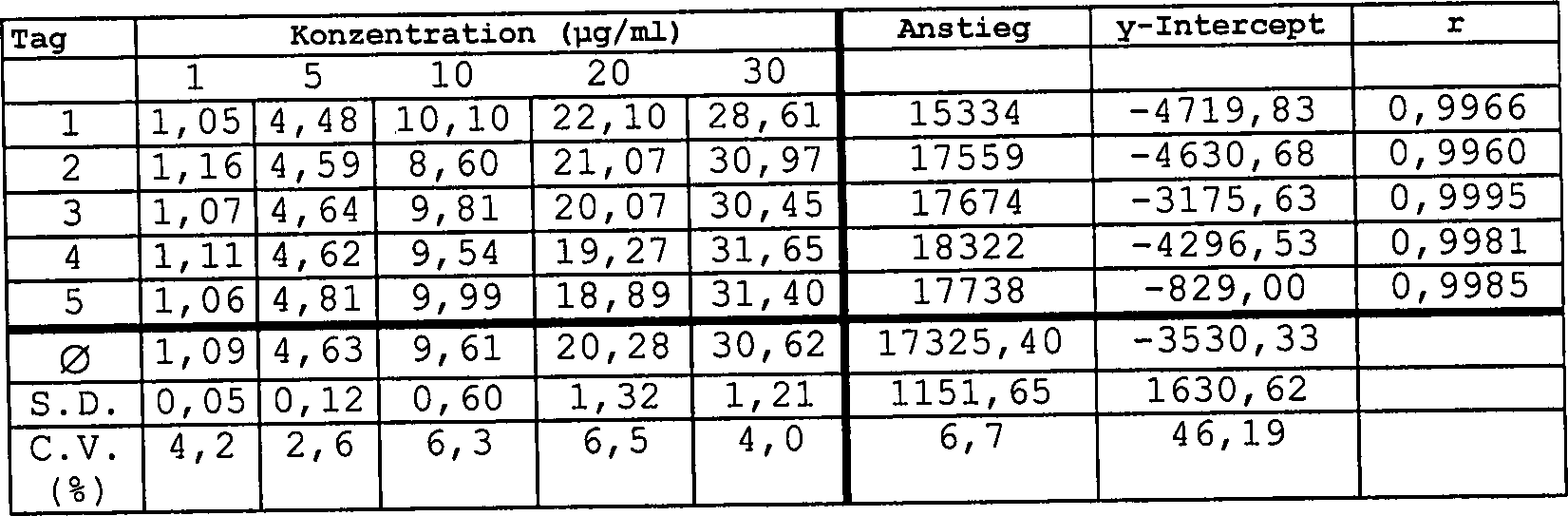
Tabelle 3 Linearität von L-DHO-Bestimmungen
in Jurkat-Zelllysaten
-
Nach
Versetzen wurden die Proben mit 30 μl 6M KOH behandelt und dann
einmal, wie in 1.3. beschrieben, analysiert.
-
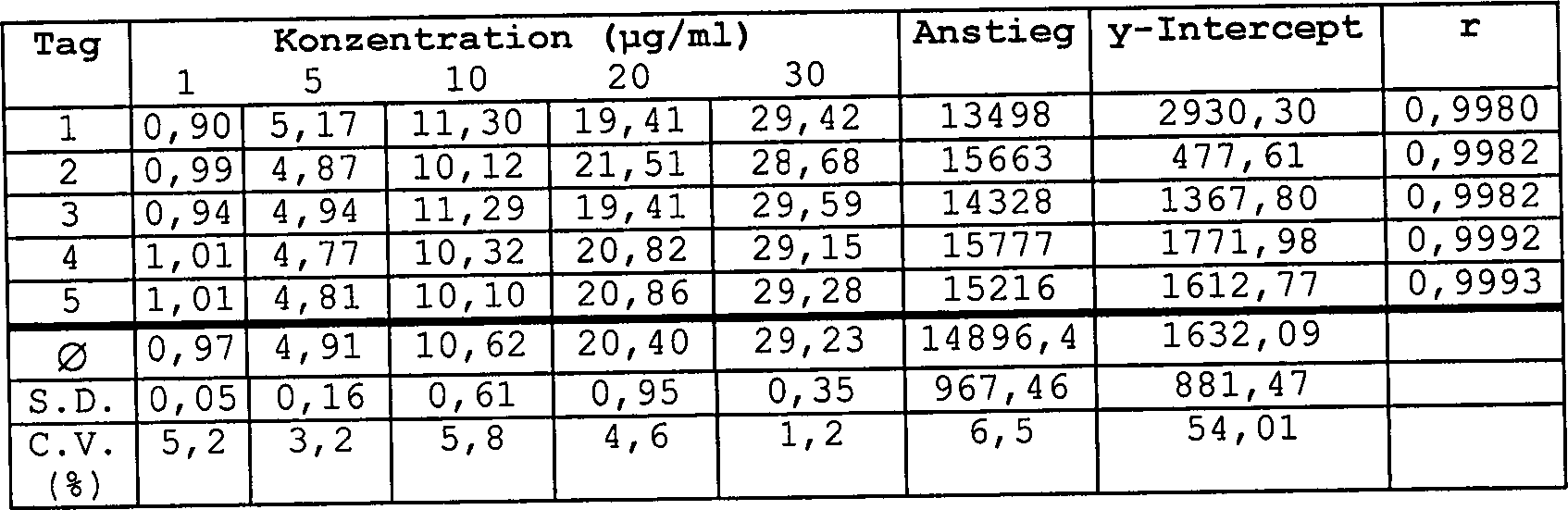
Tabelle 4 Linearität von L-DHO-Bestimmungen
in Rattenserum
-
Nach
Versetzen wurde das Serum von den Proteinen, wie in 1.5.2. beschrieben,
gereinigt.
-
Tabelle 5 Linearität von L-DHO-Bestimmungen
in Humanserum
-
Nach
Versetzen wurde das Serum von Proteinen, wie in 1.5.2. beschrieben,
gereinigt.
- r
= 0,9986·Standardkonzentration
(μg/ml) – nicht
bestimmt
-
Wie
in diesen Tabellen ersichtlich werden kann, war die Linearität in jedem
Fall erwiesen. Dies wird auf die jeweiligen Korrelationskoeffizienten „r" zurückgeführt, die
in allen Fällen > 0,99 sind. Die mittlere
lineare Regressionslinie für
die an 5 verschiedenen Tagen erhaltenen Konzentrationskurven wird
in jeder Tabelle beschrieben und zeigt, dass der Anstieg mit einer
maximalen Variation von 6,7% sehr gut reproduzierbar war.
-
Die
zurückgerechneten
Standardkonzentrationen zeigten im Mittelwert ein C.V. von weniger
als 6,5%, was die hohe Genauigkeit der Werte zeigt. Der übliche Korrelationswert
r von mehr als 0,99 drückt
die sehr hohe Präzision
und Reproduzierbarkeit des Verfahrens aus.
-
1.10. Quantifizierungsgrenze
-
Bezogen
auf diese Ergebnisse, ist die Quantifizierungsgrenze in Jurkat-Zellen
1,5 μg/ml.
In Ratten- und Humanserumproben kann 1 μg/ml L-DHO nachgewiesen werden.
Versetzte Proben bei dieser Konzentration zeigten ein Signal zu
Geräusch-Verhältnis von
mindestens 1:3.
-
1.12. Genauigkeit und
Präzision
-
Genauigkeit
und Präzision
von wiederholten Bestimmungen von L-DHO bei drei verschiedenen Konzentrationen
an fünf
verschiedenen Tagen wird in Tabelle 6–8 zusammengefasst. Die Genauigkeit
wird als der Prozent Unterschied von gefundener zu zugegebener Menge
an L-DHO (Wiedergewinnung) ausgedrückt. Die Intra-Tag-Präzision,
ausgedrückt
als C.V. (%) wurde unter Verwendung der zwei Werte, die erhalten
werden, wenn eine Probe zweimal am Tag gemessen wurde, berechnet.
Die Inter-Tag-Präzision
wurde auch wie C.V. (%) ausgedrückt
und wurde unter Verwendung des Durchschnitts für Werte von jeder Kontrollprobe
an 5 verschiedenen Tagen berechnet.
-
Tabelle 6:
-
Genauigkeit
und Präzision
in Jurkat-Zelllysaten nach Versetzen und anschließender Neutralisation
mit 30 μl
6M KOH. n = 2 bedeutet, dass eine Zelllysatprobe mit der entsprechenden
Konzentration versetzt und zweimal gemessen wurde.
-
-
Tabelle 7:
-
Genauigkeit
und Präzision
in Rattenserum nach Versetzen und anschließender Deproteinisierung; n =
2 bedeutet, dass eine Serumprobe mit der entsprechenden Konzentration
versetzt und zweimal gemessen wurde.
-
-
Tabelle 8:
-
Genauigkeit
und Präzision
in Humanserum nach Versetzen und anschließender Deproteinisierung; n =
2 bedeutet, dass eine Serumprobe mit der entsprechenden Konzentration
versetzt und zweimal gemessen wurde.
-
-
Die
hier dargestellten Ergebnisse zeigen, dass in den meisten Fällen die
Kontrollwerte ± 10%
Variation bei einem Maximum aufwiesen und dass das Verfahren deshalb
sehr genau ist. Nur in einem Fall unterschied sich die Wiedergewinnung
in Jurkat-Zellenbestimmungen etwas von jenem Wert (–11,7%).
Dieser Effekt kann durch die hohe Inter-Tag-Variation von 12% erklärt werden.
Die Inter-Tag-Präzision
in Jurkat-Zellen, Ratte- oder Humanserum war niedriger als 5%, 7%
bzw. 10%. Die Inter-Tag-Präzision
in allen untersuchten Matrizes war sehr niedrig mit einem C.V. von
weniger als 6,0%. Dies zeigt, dass die erhaltenen Ergebnisse sehr
stark reproduzierbar und präzise
sind.
-
Beispiel 2
-
2.1. Gewebskulturbedingungen:
-
- – Herstellung
von serumfreiem Medium: pulverisiertes Iscove-Medium (Biochrom)
wurde in 10 Litern bidestilliertem Wasser, ergänzt mit 18,95 g NaCl, 11,43
g NaHCO3, 700 mg KCl, 10 ml 35%ige NaOH-Lösung und
0,5 ml 1M Mercaptoethanollösung
(Riedel de Haen), gelöst
und steril filtriert. Zu einem Liter hergestelltem Iscove-Medium
wurden vor der Anwendung 32 mg Human-Holotransferrin, 1 g Rinderalbumin
und 1,5 ml Lipide (Sigma) gegeben.
- – Zellkultur:
A20.2.J-Zellen wurden in serumfreiem Medium (37°C, 5% CO2)
in einer Expansionskultur bei logarithmischem Zellwachstum gezüchtet. Die
Zellen, die für
das Assay genommen werden, hatten eine 2,2-fache Proliferationsrate
in 24 h. Der Prozentsatz an toten Zellen war <8% (3).
- – Behandlung
von Zellen mit N-(4-Trifluormethyl)-2-cyano-3-hydroxy-crotonamid, hergestellt
wie in EP-0 529 500, anschließend
A77 1726, beschrieben. A77 1726 wurde in bidestilliertem Wasser
(10 mM) gelöst und
in serumfreiem Medium weiter verdünnt. Den Zellen wurde dann
die geeignete Menge an A77 1726 zugegeben und bei 37°C und 5%igem
CO2 inkubiert.
-
2.2. Herstellung von Zelllysaten
für DHO-Bestimmung:
-
Hergestellte
Zellen wurden in einem definierten Mediumvolumen und Zelldichte
resuspendiert. In Abhängigkeit
von dem erwarteten DHO-Gehalt, wurden zwischen 1–50 Millionen Zellen entfernt,
pelletisiert (5 min, 350 × G)
und der Überstand
verworfen. Die Zellen wurden lysiert durch Zugeben von 500 μl 1,2M HClO4. Die Lysate wurden in 2 ml Eppendorf-Safe-Lock-Becher (Sicherheitsverschlussbecher) überführt und
das Protein mit 2 min Hochgeschwindigkeitszentrifugierung ausgefällt. Die
angesäuerten
Lysate wurden vollständig entfernt,
in Glasfläschchen überführt und
nach Zusatz von 500 μl
Chloroform sorgfältig
vermischt durch 2 Minuten Vortexbehandlung. Zelllipide wurden nach
10 Minuten Kaltzentrifugierung (1502 G; 10°C) extrahiert. Die gereinigten Überstände wurden
in 2 ml Eppendorf-Bechern zur Lagerung bei –20°C gesammelt bis zur Hochdruck-Flüssig-Chromatographie-(HPLC)-Bestimmung.
-
2.3. HPLC-Bestimmung von
DHO:
-
Die
chromatographische Trennung wurde wie in Beispiel 1 beschrieben
ausgeführt.
Der Bereich des Leitfähigkeitsdetektors
wurde auf 10 μS
eingestellt. Die Analyse wurde bei Raumtemperatur ausgeführt. Die mobile
Phase war aus 100 mM NaOH (A) und Wasser (B) zusammengesetzt. Mit
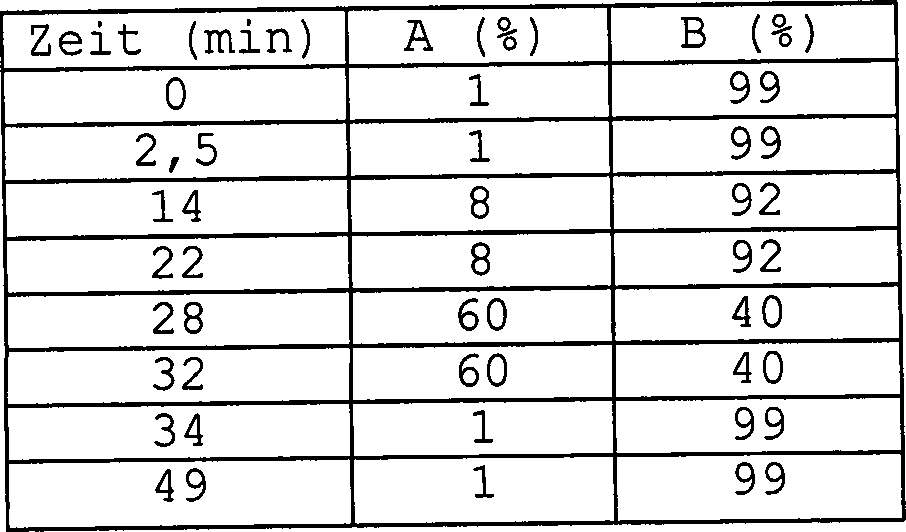
dem System wurde der nachstehende Gradient hergestellt:
-
Die
Fließgeschwindigkeit
war 1 ml/min; Versuchszeit war 49 min.
-
2.4. Ergebnisse
-
A20.2.J-Zellen,
inkubiert mit A77 1726, hatten erhöhte Mengen an intrazellulärer DHO
(Tabellen 9–11). Die
in Tabelle 9 angeführten
Ergebnisse zeigen, dass DHO-Spiegel direkt mit der Zahl der extrahierten
Zellen korrelierten. Tabelle
9: Korrelation von intrazellulären
DHO-Konzentrationen und Anzahl der Zellen, die mit A77 1726 gezüchtet wurden
-
A20.2.J-Zellen
wurden mit 5 μM
A77 1726 behandelt und 24 Stunden (37°C, 5% CO2)
gezüchtet
und dann für
die DHO-Extraktion
(n = 3) zubereitet.
-
Um
die Zellkulturverfahren zu optimieren und das beste Zelle/A77 1726-Molaritätsverhältnis zu
bestimmen, wurden variierende Dichten von A20.2.J-Zellen, zusammen
mit A77 1726 (5 μM),
inkubiert. Die Zellen wurden bei verschiedenen Zeitpunkten abgezogen
und DHO-Konzentrationen bestimmt (Tabelle 10). Auf Grund der Tatsache,
dass DHO-Konzentrationen direkt mit der Menge an extrahierten Zellen
korrelierten (siehe Tabelle 9), wurden für die nachstehenden Versuche
DHO-Konzentrationen auf 10 × 10
6 Zellen in μg/ml extrapoliert. Der beste
lineare Anstieg des DHO-Spiegels wurde mit einer Dichte von 1 × 10
6 Zellen/ml gefunden. Unter Anwendung dieser
Zelldichte wurde die Zeit, in Abhängigkeit von der Erhöhung der
intrazellulären DHO-Spiegel
in Zellen, die mit A77 1726 inkubiert wurden, untersucht. Nachweisbare
Mengen an DHO konnten nach 1 Stunde Inkubation, unabhängig von
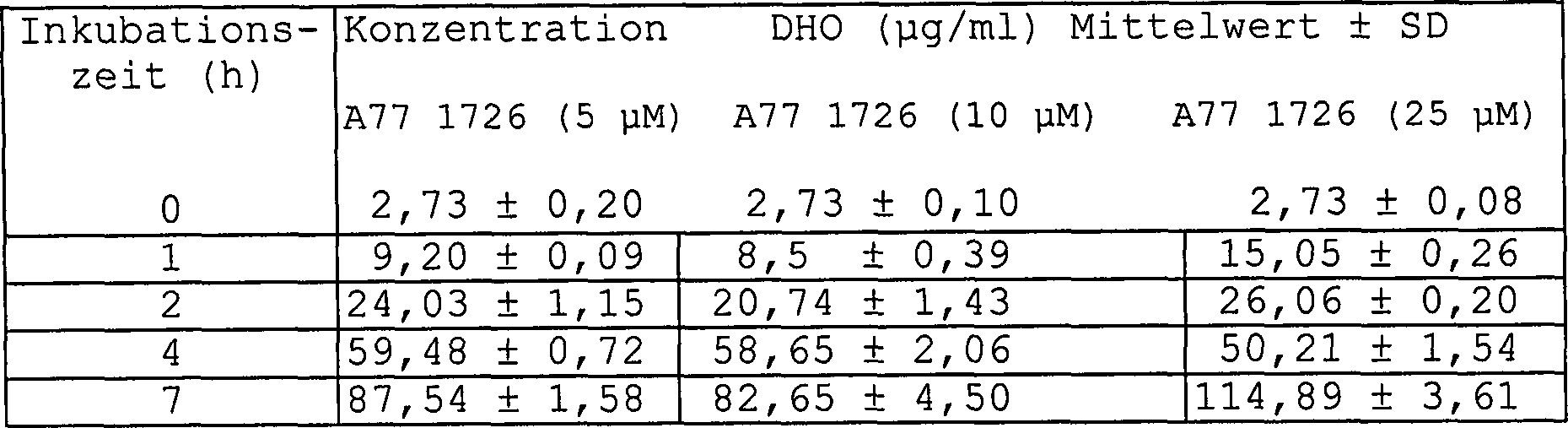
der Arzneistoffkonzentration (Tabelle 11) bestimmt werden. Eine
lineare Erhöhung
wurde mit der größten Menge
an DHO, bestimmt nach 6 h (Tabelle 11), registriert. Nach diesem
Zeitraum wurde eine Sättigung
ohne weitere Erhöhung
von DHO beobachtet. Tabelle
10: Zelldichte und zeitabhängige
Erhöhung
von intrazellulären
DHO-Konzentrationen:
-
Verschiedene
Mengen an A20.2.J-Zellen wurden zusammen mit A77 1726 (5 μM) für die vorstehend angegebenen
Zeiträume
inkubiert und deren intrazelluläre
DHO-Konzentrationen für
jeden Probenpunkt bestimmt. (* Alle Werte extrapoliert auf 10 × 10
6 Zellen) (n = 2). Tabelle
11: Die zeitabhängige
Erhöhung
von intrazellulären
L-DHO-Konzentrationen:
-
Eine
Million A20.2.J-Zellen/ml wurden, zusammen mit variierenden Konzentrationen
von A77 1726 und deren intrazellulären DHO-Konzentrationen, die
bei verschiedenen Zeitpunkten bestimmt wurden, inkubiert. Die Daten
werden als μg/ml
DHO ± SD,
extrapoliert auf zehn Millionen Zellen (0 – 4 h = n = 2, 7 h = n = 4),
angegeben.
-
Inkubation
von A20.2.J-Tumorzellen mit A77 1726 ergab schnelle Akkumulation
von L-DHO auf Grund von DHO-DH-Inhibierung. Die intrazellulären L-DHO-Konzentrationen
korrelierten mit der Zellzahl und waren zeitabhängig. L-DHO-Verfolgen ist ein
Surrogatmarker für
A77 1726 Immunomodulierungsaktivität bei Patienten.
-
Beispiel 3
-
Tiere:
Männliche
Wistar-Lewis-Ratten (Mollegaard Breading Center Ltd. Ejby, DK) mit
einem Körpergewicht
von 160–200
g.
-
Adjuvans
Arthritis: Die Erkrankung wurde durch Injizieren von 0,1 ml Freund'schem Adjuvans (6
ml Mycobacterium smegmatis, suspendiert in 1 ml schwerem, weißem Paraffinöl (Merck,
Darmstadt)), in die Schwanzwurzel von Wistar-Lewis-Ratten induziert.
Pathologische Symptome treten im Allgemeinen zwischen 10 und 14
Tagen nach Krankheitsauslösung
auf.
-
Arzneistoffbehandlung:
Arzneistoffe wurden in 1%iger Carboxymethylcellulose (COMC) suspendiert. Gesunde
Tiere (n = 18) und Adjuvans-erkrankte (n = 18) Ratten (Tag 9 der
Störung)
wurden 10 mg/kg p.o. N-4-(Trifluormethylphenyl)-5-methylisoxazol-4-carboxamid,
anschließend
Leflunomid zweimal täglich
(7:30 und 13:30) für
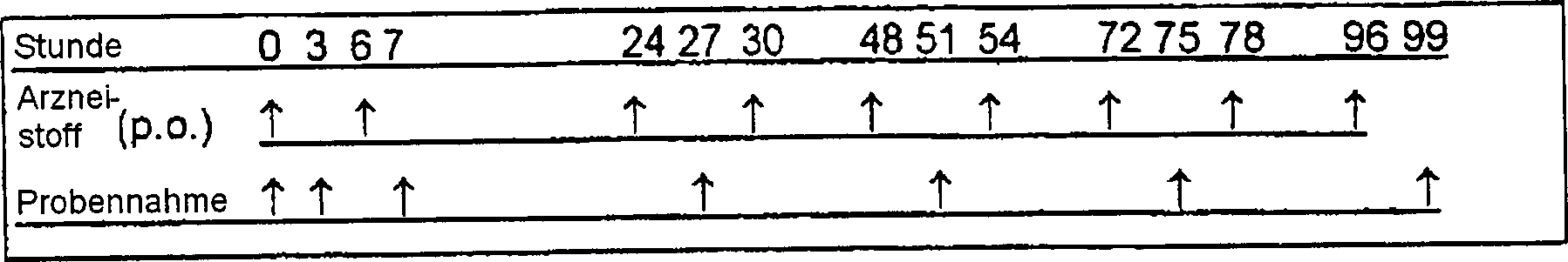
5 Tage; d.h. bei Zeitpunkten: 0 h, 6 h, 24 h, 30 h, 48 h, 54 h,
72 h, 78 h, 96 h (siehe Tabelle 12), gegeben. Zum Zeitpunkt 0 Stunden
erkrankten 3 von zwei Gruppen und nicht erkrankte Gruppen wurden
geopfert, um die Grundlinienspiegel zu bestimmen. Weitere 3 gesunde
und 3 erkrankte Tiere wurden mit Placebo (nur COMC) für 5 Tage
behandelt.
-
Probennehmen:
Drei Tiere pro Gruppe wurden bei jedem Probennahmezeitpunkt geopfert.
Serum und Splenozyten wurden bei 3 h, 7 h, 27 h, 51 h, 75 h und
99 h (siehe Tabelle 12) genommen. Mit Ausnahme des Werts von 7 h,
wurden die Proben drei Stunden nach der letzten Arzneistoffapplikation
genommen. Der Wert 7 h wurde eine Stunde nach dem zweiten Dosieren
genommen. Proben der Placebo behandelten Tiere wurden bei den Zeitpunkten
0 h (n = 3) und 99 h (n = 3) genommen. Tabelle
12: Verabreichung von Leflunomid und Gewebsprobenahmezeitpunkte
-
Herstellung der Proben:
-
- – Blut,
gesammelt durch Herzpunktur, wurde 30 min bei 4°C gelagert und dann 10 min bei
3000 U/min zentrifugiert. Serum wurde abgetrennt und in Eppendorf-Bechern
bei –20°C (3) gelagert.
Vor HPLC-Analyse wurde das gefrorene Serum aufgetaut, und, um die
Proteine zu entfernen, wurden 200 μl Serum zu einem Microconfilter
(Modell 10, Code 42407, Amicon) gegeben und 30 min bei 13 000 U/min
zentrifugiert.
- – Die
Milzen wurden entnommen (n = 3) und zur L-DHO-Analyse vereinigt. Die Zellen, die durch
Zerlegen (Durchleiten durch ein Edelstahlsieb) getrennt wurden,
wurden mit 0,17 M NH4Cl zur Lyse der Erythrozyten behandelt.
Aliquote Mengen von 50 Millionen Milzzellen pro Gruppe wurden hergestellt,
in Becher zur Zentrifugierung gegeben und der Überstand verworfen. Das Zellpellet
wurde unter permanentem Mixen zu 500 μl einer 1,2 M HClO4-Lösung zur
Lyse der Zellen gegeben und 2 min zentrifugiert. Das saure Zelllysat
wurde vollständig
zu Glasfläschchen überführt, 500 μl Chloroform
zugesetzt und 2 min mit einem Vortexmischer vermischt. Zelluläre Lipide
wurden durch Zentrifugierung (10 min 1502 × G und 10°C) ausgefällt. Der Überstand wurde in 2 ml-Becher
gegeben und bei –20°C gelagert.
-
Die
Bestimmung von A77 1726 Serumkonzentrationen wurde wie nachstehend
durchgeführt:
Serumproben
wurden auf Raumtemperatur gebracht und sorgfältig unter Anwendung eines
Vortexmischers vermischt. Das Serum wurde in Eppendorf-Becher pipettiert
(200 μl)
und der innere Standard (A77 1726, 2 μg in 400 μl Acetonitril) zugegeben. Die
Röhrchen
wurden dann in einem Vortexmischer vermischt und bei 2500 U/min
(Raumtemperatur) für
10 Minuten zentrifugiert. Zur HPLC-Analyse wurde der Überstand
(400 μl)
in ein Fläschchen überführt und
Wasser (400 μl)
zugegeben und vermischt. Die HPLC-Bedingungen waren wie nachstehend:
Die Hardware bestand aus einer TSP P2000 Pumpe, einem TSP AS1000
Autosampler, einem TSP SP4270 Integrator und einem TSP UV100 UV-Detektor.
Die Detektion war bei 292 nm Wellenlänge. Die mobile Phase bestand
aus 650 ml Methanol (CHROMASOLV), 2,42 g Tetrabutylammoniumbromid
und 350 ml 0,05 M Ammoniumacetat. Die Fließgeschwindigkeit war 0,5 ml/min.
Eine CHROMPACK Spherisorb ODS-2 Säule von 10 cm mit einer 1 cm
Umkehrphasen(R2)-Guardsäule
wurde verwendet. 100 μl
wurden in die Säule gespritzt
und die Laufzeit war 7 Minuten.
-
HPLC-Bestimmung
von L-DHO-Konzentrationen: Die chromatographische Trennung wurde
wie in Beispiel 1 beschrieben ausgeführt. Der Bereich des Leitfähigkeitsdetektors
wurde auf 10 μS
eingestellt. Die Analyse wurde bei Raumtemperatur ausgeführt. Die
mobile Phase war aus 100 mM NaOH (A) und Wasser (B) zusammengesetzt.
Mit dem System wurden die nachstehenden Gradienten erzeugt:
Serum Splenozyten
-
Die
Fließgeschwindigkeit
war 1 ml/min.
-
Sowohl
gesunde als auch erkrankte Ratten hatten erhöhte zelluläre (Tabelle 13) und Serum-(Tabelle 14)
L-DHO-Konzentrationen
nach oraler Verabreichung von Leflunomid. Diese Erhöhung korrelierte
mit den A77 1727-Serumkonzentrationen, die in diesen Tieren (Tabelle
15) bestimmt wurden. Anfänglich
erreichte 3 Stunden nach oraler Verabreichung A77 1726 eine Konzentration
von etwa 26 μg/ml
in Adjuvanserkrankten Ratten und 31 μg/ml in nicht erkrankten Ratten.
Diese Werte zeigten einen Peak eine Stunde nach dem zweiten Dosieren
(7 h), fielen jedoch auf Werte zwischen 7 und 12 μg/ml für die Dauer
des Versuchs in sowohl erkrankten als auch nicht erkrankten Ratten
ab. Erkrankte Tiere nahmen 51 h in Anspruch zum Erreichen dieser Konzentration;
wohingegen nicht erkrankte Tiere immer dieselbe nach 27 h erreichten
(Tabelle 15). Die A77 1726-Serumkonzentrationen korrelierten mit
den L-DHO-Konzentrationen in dem Serum von Adjuvanserkrankten und
nicht erkrankten Nagern. Im Gegensatz zu den L-DHO-Serumkonzentrationen,
die zu etwa 5 μg/ml
für die
Dauer des Versuchs äquilibrierten,
fiel die Menge an in den Splenozyten gefundener L-DHO unter die
Nachweisgrenze (1,5 μg/ml)
nach 99 h. Tabelle
13: L-DHO-Konzentrationen in Splenozyten [50 × 10
6 Zellen]
von Ratten, behandelt mit Leflunomid
-
Tiere
wurden mit Leflunomid oder Placebo behandelt, die Milzen entfernt
(n = 3) und wie beschrieben vereinigt. Die vereinigten Splenozyten
wurden in zweifacher Ausführung
bestimmt. DL = Nachweisgrenze (1,5 μg/ml); Tabelle
14: L-DHO-Konzentrationen in Serum von Ratten, behandelt mit Leflunomid
-
Tiere
wurden mit Leflunomid oder Placebo behandelt und Blut genommen,
präpariert
und wie beschrieben bestimmt. L-DHO-Serumkonzentrationen wurden
für jedes
Tier einzeln bestimmt. DL = Nachweisgrenze (0,5 μg/ml); Tabelle
15: A77 1726-Konzentrationen in Serum von mit Leflunomid behandelten
Ratten
-
Die
Tiere wurden mit Leflunomid oder Placebo behandelt und Blut genommen,
präpariert
und wie beschrieben bestimmt. A77 1726-Serumkonzentrationen wurden
für jedes
Tier einzeln bestimmt.
-
Oral
verabreichtes Leflunomid wird sehr schnell in vivo zu A77 1726 umgewandelt.
A77 1726 ist der aktive Metabolit von Leflunomid (
US 5 679 709 ). Inkubation von entweder
Adjuvans erkrankten oder nicht erkrankten Ratten mit Leflunomid
ergab sehr schnelle Akkumulation von L-DHO in deren Serum und Splenozyten.
L-DHO-Konzentration korrelierte mit Blutserumkonzentrationen von
A77 1726, was somit eine aktive Absenkung von DHO-DH durch dieses
Molekül
in vivo zeigt. In klinischen Humanstudien kann L-DHO-Verfolgen ein
Surrogatmarker für
Leflunomid-immunomodulierende Aktivität bei Patienten sein.