DE69729007T2 - Hydroxamsäure- und carbonsäure-derivate mit mmp und tnf hemmender wirkung - Google Patents
Hydroxamsäure- und carbonsäure-derivate mit mmp und tnf hemmender wirkung Download PDFInfo
- Publication number
- DE69729007T2 DE69729007T2 DE69729007T DE69729007T DE69729007T2 DE 69729007 T2 DE69729007 T2 DE 69729007T2 DE 69729007 T DE69729007 T DE 69729007T DE 69729007 T DE69729007 T DE 69729007T DE 69729007 T2 DE69729007 T2 DE 69729007T2
- Authority
- DE
- Germany
- Prior art keywords
- alkyl
- optionally substituted
- heterocycloalkyl
- heteroaryl
- cycloalkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- NEAQRZUHTPSBBM-UHFFFAOYSA-N 2-hydroxy-3,3-dimethyl-7-nitro-4h-isoquinolin-1-one Chemical compound C1=C([N+]([O-])=O)C=C2C(=O)N(O)C(C)(C)CC2=C1 NEAQRZUHTPSBBM-UHFFFAOYSA-N 0.000 title abstract description 5
- 230000000694 effects Effects 0.000 title description 26
- 239000002253 acid Substances 0.000 title description 5
- 201000010099 disease Diseases 0.000 claims abstract description 13
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 13
- 102000002274 Matrix Metalloproteinases Human genes 0.000 claims abstract description 12
- 108010000684 Matrix Metalloproteinases Proteins 0.000 claims abstract description 12
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 10
- 230000001404 mediated effect Effects 0.000 claims abstract description 6
- 150000001875 compounds Chemical class 0.000 claims description 101
- 125000000217 alkyl group Chemical group 0.000 claims description 76
- -1 R 5 -C 2 -6 alkenyl Chemical group 0.000 claims description 34
- 125000003118 aryl group Chemical group 0.000 claims description 28
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 25
- 125000001072 heteroaryl group Chemical group 0.000 claims description 25
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 25
- 102100040247 Tumor necrosis factor Human genes 0.000 claims description 13
- 229910052736 halogen Inorganic materials 0.000 claims description 11
- 150000002367 halogens Chemical class 0.000 claims description 11
- 238000011282 treatment Methods 0.000 claims description 11
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 10
- 150000003839 salts Chemical class 0.000 claims description 10
- 125000001424 substituent group Chemical group 0.000 claims description 10
- 108060008683 Tumor Necrosis Factor Receptor Proteins 0.000 claims description 9
- 238000006243 chemical reaction Methods 0.000 claims description 9
- 102000003298 tumor necrosis factor receptor Human genes 0.000 claims description 9
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 8
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 8
- 108060008682 Tumor Necrosis Factor Proteins 0.000 claims description 7
- 239000003814 drug Substances 0.000 claims description 7
- 125000005213 alkyl heteroaryl group Chemical group 0.000 claims description 6
- 125000000392 cycloalkenyl group Chemical group 0.000 claims description 6
- 125000004366 heterocycloalkenyl group Chemical group 0.000 claims description 6
- 238000004519 manufacturing process Methods 0.000 claims description 6
- 229910052760 oxygen Inorganic materials 0.000 claims description 6
- 125000005544 phthalimido group Chemical group 0.000 claims description 6
- 229910052717 sulfur Inorganic materials 0.000 claims description 6
- HCXJFMDOHDNDCC-UHFFFAOYSA-N 5-$l^{1}-oxidanyl-3,4-dihydropyrrol-2-one Chemical group O=C1CCC(=O)[N]1 HCXJFMDOHDNDCC-UHFFFAOYSA-N 0.000 claims description 5
- 102000004190 Enzymes Human genes 0.000 claims description 5
- 108090000790 Enzymes Proteins 0.000 claims description 5
- 108010092694 L-Selectin Proteins 0.000 claims description 5
- 102000016551 L-selectin Human genes 0.000 claims description 5
- 125000005024 alkenyl aryl group Chemical group 0.000 claims description 4
- 201000011510 cancer Diseases 0.000 claims description 4
- 201000008482 osteoarthritis Diseases 0.000 claims description 4
- 239000008194 pharmaceutical composition Substances 0.000 claims description 4
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 4
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 4
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 claims description 3
- 201000001320 Atherosclerosis Diseases 0.000 claims description 3
- 125000003342 alkenyl group Chemical group 0.000 claims description 3
- 229910052739 hydrogen Inorganic materials 0.000 claims description 3
- 201000006417 multiple sclerosis Diseases 0.000 claims description 3
- 201000001245 periodontitis Diseases 0.000 claims description 3
- DBSJXSQIQXSWQQ-UHFFFAOYSA-N 2-[(1-benzoylpiperidin-4-yl)sulfonylmethyl]-n-hydroxy-5-phenylpentanamide Chemical compound C1CN(C(=O)C=2C=CC=CC=2)CCC1S(=O)(=O)CC(C(=O)NO)CCCC1=CC=CC=C1 DBSJXSQIQXSWQQ-UHFFFAOYSA-N 0.000 claims description 2
- GLODGHWUWHRQAJ-UHFFFAOYSA-N 2-[[1-[(4-cyanophenyl)methyl]piperidin-4-yl]sulfonylmethyl]-n-hydroxy-5-phenylpentanamide Chemical compound C1CN(CC=2C=CC(=CC=2)C#N)CCC1S(=O)(=O)CC(C(=O)NO)CCCC1=CC=CC=C1 GLODGHWUWHRQAJ-UHFFFAOYSA-N 0.000 claims description 2
- KPEWKOVGNOMRFJ-UHFFFAOYSA-N 5-phenyl-2-(piperidin-4-ylsulfonylmethyl)pentanoic acid Chemical compound C1CNCCC1S(=O)(=O)CC(C(=O)O)CCCC1=CC=CC=C1 KPEWKOVGNOMRFJ-UHFFFAOYSA-N 0.000 claims description 2
- 206010048998 Acute phase reaction Diseases 0.000 claims description 2
- 208000024827 Alzheimer disease Diseases 0.000 claims description 2
- 206010002198 Anaphylactic reaction Diseases 0.000 claims description 2
- 208000023275 Autoimmune disease Diseases 0.000 claims description 2
- 206010006895 Cachexia Diseases 0.000 claims description 2
- 206010007559 Cardiac failure congestive Diseases 0.000 claims description 2
- 206010009900 Colitis ulcerative Diseases 0.000 claims description 2
- 206010010744 Conjunctivitis allergic Diseases 0.000 claims description 2
- 208000011231 Crohn disease Diseases 0.000 claims description 2
- 206010012438 Dermatitis atopic Diseases 0.000 claims description 2
- 201000009273 Endometriosis Diseases 0.000 claims description 2
- 206010014989 Epidermolysis bullosa Diseases 0.000 claims description 2
- 208000031886 HIV Infections Diseases 0.000 claims description 2
- 206010019280 Heart failures Diseases 0.000 claims description 2
- 208000032843 Hemorrhage Diseases 0.000 claims description 2
- 206010061218 Inflammation Diseases 0.000 claims description 2
- 102000010781 Interleukin-6 Receptors Human genes 0.000 claims description 2
- 108010038501 Interleukin-6 Receptors Proteins 0.000 claims description 2
- 201000009906 Meningitis Diseases 0.000 claims description 2
- 206010027476 Metastases Diseases 0.000 claims description 2
- 208000019695 Migraine disease Diseases 0.000 claims description 2
- 208000001132 Osteoporosis Diseases 0.000 claims description 2
- 201000004681 Psoriasis Diseases 0.000 claims description 2
- 206010037660 Pyrexia Diseases 0.000 claims description 2
- 206010063837 Reperfusion injury Diseases 0.000 claims description 2
- 208000017442 Retinal disease Diseases 0.000 claims description 2
- 206010038923 Retinopathy Diseases 0.000 claims description 2
- 208000006011 Stroke Diseases 0.000 claims description 2
- 208000002847 Surgical Wound Diseases 0.000 claims description 2
- 201000006704 Ulcerative Colitis Diseases 0.000 claims description 2
- 206010064996 Ulcerative keratitis Diseases 0.000 claims description 2
- 206010047115 Vasculitis Diseases 0.000 claims description 2
- 230000001154 acute effect Effects 0.000 claims description 2
- 208000002205 allergic conjunctivitis Diseases 0.000 claims description 2
- 230000036783 anaphylactic response Effects 0.000 claims description 2
- 208000003455 anaphylaxis Diseases 0.000 claims description 2
- 208000022531 anorexia Diseases 0.000 claims description 2
- 230000002785 anti-thrombosis Effects 0.000 claims description 2
- 208000006673 asthma Diseases 0.000 claims description 2
- 208000024998 atopic conjunctivitis Diseases 0.000 claims description 2
- 201000008937 atopic dermatitis Diseases 0.000 claims description 2
- 230000009400 cancer invasion Effects 0.000 claims description 2
- 229910052799 carbon Inorganic materials 0.000 claims description 2
- 238000005345 coagulation Methods 0.000 claims description 2
- 230000015271 coagulation Effects 0.000 claims description 2
- 206010061428 decreased appetite Diseases 0.000 claims description 2
- 239000003085 diluting agent Substances 0.000 claims description 2
- ZZVUWRFHKOJYTH-UHFFFAOYSA-N diphenhydramine Chemical group C=1C=CC=CC=1C(OCCN(C)C)C1=CC=CC=C1 ZZVUWRFHKOJYTH-UHFFFAOYSA-N 0.000 claims description 2
- 239000003937 drug carrier Substances 0.000 claims description 2
- 208000007565 gingivitis Diseases 0.000 claims description 2
- 208000015181 infectious disease Diseases 0.000 claims description 2
- 208000027866 inflammatory disease Diseases 0.000 claims description 2
- 230000004054 inflammatory process Effects 0.000 claims description 2
- 206010027599 migraine Diseases 0.000 claims description 2
- JVLQTFWIBKTBDE-UHFFFAOYSA-N n-hydroxy-2-(oxan-4-ylsulfonylmethyl)-5-phenylpentanamide Chemical group C1COCCC1S(=O)(=O)CC(C(=O)NO)CCCC1=CC=CC=C1 JVLQTFWIBKTBDE-UHFFFAOYSA-N 0.000 claims description 2
- 230000004770 neurodegeneration Effects 0.000 claims description 2
- 208000037803 restenosis Diseases 0.000 claims description 2
- 206010039083 rhinitis Diseases 0.000 claims description 2
- 230000035939 shock Effects 0.000 claims description 2
- 239000012453 solvate Substances 0.000 claims description 2
- GBMIKQJFAKXAKB-UHFFFAOYSA-N tert-butyl 4-[2-(hydroxycarbamoyl)-5-phenylpentyl]sulfonylpiperidine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCC1S(=O)(=O)CC(C(=O)NO)CCCC1=CC=CC=C1 GBMIKQJFAKXAKB-UHFFFAOYSA-N 0.000 claims description 2
- 238000002560 therapeutic procedure Methods 0.000 claims description 2
- 230000004614 tumor growth Effects 0.000 claims description 2
- 230000029663 wound healing Effects 0.000 claims description 2
- 125000002877 alkyl aryl group Chemical group 0.000 claims 3
- 125000006704 (C5-C6) cycloalkyl group Chemical group 0.000 claims 1
- JMYBQGJMVLYIGH-UHFFFAOYSA-N 2-[(1-benzylpiperidin-4-yl)sulfonylmethyl]-n-hydroxy-5-phenylpentanamide Chemical compound C1CN(CC=2C=CC=CC=2)CCC1S(=O)(=O)CC(C(=O)NO)CCCC1=CC=CC=C1 JMYBQGJMVLYIGH-UHFFFAOYSA-N 0.000 claims 1
- VNHDFBIMLUHZNY-UHFFFAOYSA-N 2-[2,6-dimethyl-4-(3-methyl-4-oxophthalazin-1-yl)phenoxy]-n-hydroxyacetamide Chemical compound CC1=C(OCC(=O)NO)C(C)=CC(C=2C3=CC=CC=C3C(=O)N(C)N=2)=C1 VNHDFBIMLUHZNY-UHFFFAOYSA-N 0.000 claims 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 claims 1
- 201000006474 Brain Ischemia Diseases 0.000 claims 1
- 206010008120 Cerebral ischaemia Diseases 0.000 claims 1
- 206010012434 Dermatitis allergic Diseases 0.000 claims 1
- 208000037357 HIV infectious disease Diseases 0.000 claims 1
- 206010026673 Malignant Pleural Effusion Diseases 0.000 claims 1
- 206010025538 Malignant ascites Diseases 0.000 claims 1
- 206010064390 Tumour invasion Diseases 0.000 claims 1
- 229960001138 acetylsalicylic acid Drugs 0.000 claims 1
- 230000033115 angiogenesis Effects 0.000 claims 1
- 206010008118 cerebral infarction Diseases 0.000 claims 1
- 230000001684 chronic effect Effects 0.000 claims 1
- 230000007850 degeneration Effects 0.000 claims 1
- 230000002526 effect on cardiovascular system Effects 0.000 claims 1
- 208000030533 eye disease Diseases 0.000 claims 1
- 125000004446 heteroarylalkyl group Chemical group 0.000 claims 1
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 claims 1
- 208000031225 myocardial ischemia Diseases 0.000 claims 1
- 230000002265 prevention Effects 0.000 claims 1
- 230000028327 secretion Effects 0.000 claims 1
- 125000000547 substituted alkyl group Chemical group 0.000 claims 1
- 239000003112 inhibitor Substances 0.000 abstract description 49
- 150000001732 carboxylic acid derivatives Chemical class 0.000 abstract description 3
- 208000015122 neurodegenerative disease Diseases 0.000 abstract description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 70
- 239000000543 intermediate Substances 0.000 description 69
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 66
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 58
- 239000000243 solution Substances 0.000 description 55
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 51
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 46
- 239000000203 mixture Substances 0.000 description 41
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 39
- 238000000034 method Methods 0.000 description 27
- 235000019439 ethyl acetate Nutrition 0.000 description 20
- 230000005764 inhibitory process Effects 0.000 description 18
- 239000007787 solid Substances 0.000 description 18
- 210000004027 cell Anatomy 0.000 description 17
- 239000012895 dilution Substances 0.000 description 17
- 238000010790 dilution Methods 0.000 description 17
- 239000000725 suspension Substances 0.000 description 17
- 239000002904 solvent Substances 0.000 description 16
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 15
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 14
- 239000012267 brine Substances 0.000 description 14
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 14
- 239000012230 colorless oil Substances 0.000 description 13
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 12
- 239000000706 filtrate Substances 0.000 description 11
- 150000002148 esters Chemical class 0.000 description 10
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical compound CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 10
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 9
- 108060005980 Collagenase Proteins 0.000 description 9
- 102000029816 Collagenase Human genes 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 9
- 108091007196 stromelysin Proteins 0.000 description 9
- 108010026132 Gelatinases Proteins 0.000 description 8
- 102000013382 Gelatinases Human genes 0.000 description 8
- 102000000422 Matrix Metalloproteinase 3 Human genes 0.000 description 8
- 239000004480 active ingredient Substances 0.000 description 8
- 125000004432 carbon atom Chemical group C* 0.000 description 8
- 229960002424 collagenase Drugs 0.000 description 8
- 239000003921 oil Substances 0.000 description 8
- 235000019198 oils Nutrition 0.000 description 8
- 102000008186 Collagen Human genes 0.000 description 7
- 108010035532 Collagen Proteins 0.000 description 7
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 7
- 229920001436 collagen Polymers 0.000 description 7
- 238000004440 column chromatography Methods 0.000 description 7
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 7
- 235000019341 magnesium sulphate Nutrition 0.000 description 7
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 6
- 102000005741 Metalloproteases Human genes 0.000 description 6
- 108010006035 Metalloproteases Proteins 0.000 description 6
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 6
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 6
- 239000000284 extract Substances 0.000 description 6
- 230000002401 inhibitory effect Effects 0.000 description 6
- 239000010410 layer Substances 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- 125000006239 protecting group Chemical group 0.000 description 6
- 239000000741 silica gel Substances 0.000 description 6
- 229910002027 silica gel Inorganic materials 0.000 description 6
- 239000006228 supernatant Substances 0.000 description 6
- 238000008157 ELISA kit Methods 0.000 description 5
- 241000700159 Rattus Species 0.000 description 5
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 5
- 239000012298 atmosphere Substances 0.000 description 5
- 229910002091 carbon monoxide Inorganic materials 0.000 description 5
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 description 5
- 235000014113 dietary fatty acids Nutrition 0.000 description 5
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 5
- 238000002474 experimental method Methods 0.000 description 5
- 239000000194 fatty acid Substances 0.000 description 5
- 229930195729 fatty acid Natural products 0.000 description 5
- 150000004665 fatty acids Chemical class 0.000 description 5
- 229920000609 methyl cellulose Polymers 0.000 description 5
- 235000010981 methylcellulose Nutrition 0.000 description 5
- 239000001923 methylcellulose Substances 0.000 description 5
- 239000000546 pharmaceutical excipient Substances 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- IEQAICDLOKRSRL-UHFFFAOYSA-N 2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-(2-dodecoxyethoxy)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethanol Chemical compound CCCCCCCCCCCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCO IEQAICDLOKRSRL-UHFFFAOYSA-N 0.000 description 4
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 4
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 4
- 101000611023 Homo sapiens Tumor necrosis factor receptor superfamily member 6 Proteins 0.000 description 4
- 241000699670 Mus sp. Species 0.000 description 4
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- 239000007983 Tris buffer Substances 0.000 description 4
- 102100040403 Tumor necrosis factor receptor superfamily member 6 Human genes 0.000 description 4
- 125000003545 alkoxy group Chemical group 0.000 description 4
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 4
- 239000003086 colorant Substances 0.000 description 4
- 239000007859 condensation product Substances 0.000 description 4
- 239000002270 dispersing agent Substances 0.000 description 4
- 239000002158 endotoxin Substances 0.000 description 4
- 229940088598 enzyme Drugs 0.000 description 4
- 235000003599 food sweetener Nutrition 0.000 description 4
- 238000009472 formulation Methods 0.000 description 4
- 125000004475 heteroaralkyl group Chemical group 0.000 description 4
- 238000000338 in vitro Methods 0.000 description 4
- 229920006008 lipopolysaccharide Polymers 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 239000002480 mineral oil Substances 0.000 description 4
- 235000010446 mineral oil Nutrition 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 230000036961 partial effect Effects 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 239000003755 preservative agent Substances 0.000 description 4
- 239000012279 sodium borohydride Substances 0.000 description 4
- 229910000033 sodium borohydride Inorganic materials 0.000 description 4
- 239000000375 suspending agent Substances 0.000 description 4
- 239000003765 sweetening agent Substances 0.000 description 4
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 4
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 4
- 210000004881 tumor cell Anatomy 0.000 description 4
- 230000006433 tumor necrosis factor production Effects 0.000 description 4
- 239000000080 wetting agent Substances 0.000 description 4
- JYWKEVKEKOTYEX-UHFFFAOYSA-N 2,6-dibromo-4-chloroiminocyclohexa-2,5-dien-1-one Chemical compound ClN=C1C=C(Br)C(=O)C(Br)=C1 JYWKEVKEKOTYEX-UHFFFAOYSA-N 0.000 description 3
- 0 C/C(/C=C1)=C\CCN(*)C1=O Chemical compound C/C(/C=C1)=C\CCN(*)C1=O 0.000 description 3
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 3
- 229920001917 Ficoll Polymers 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 229920000084 Gum arabic Polymers 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 3
- 229940124761 MMP inhibitor Drugs 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- 235000019483 Peanut oil Nutrition 0.000 description 3
- 229920001213 Polysorbate 20 Polymers 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical class [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 3
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical class OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 3
- 102100033733 Tumor necrosis factor receptor superfamily member 1B Human genes 0.000 description 3
- 101710187830 Tumor necrosis factor receptor superfamily member 1B Proteins 0.000 description 3
- 235000010489 acacia gum Nutrition 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 239000002671 adjuvant Substances 0.000 description 3
- 125000002723 alicyclic group Chemical group 0.000 description 3
- 239000002168 alkylating agent Substances 0.000 description 3
- 229940100198 alkylating agent Drugs 0.000 description 3
- 239000007900 aqueous suspension Substances 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- RYFCSKVXWRJEOB-UHFFFAOYSA-N dibenzyl propanedioate Chemical compound C=1C=CC=CC=1COC(=O)CC(=O)OCC1=CC=CC=C1 RYFCSKVXWRJEOB-UHFFFAOYSA-N 0.000 description 3
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- 210000003743 erythrocyte Anatomy 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 229920000159 gelatin Polymers 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 235000019322 gelatine Nutrition 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 229940057995 liquid paraffin Drugs 0.000 description 3
- 210000004072 lung Anatomy 0.000 description 3
- 239000003475 metalloproteinase inhibitor Substances 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 239000004006 olive oil Substances 0.000 description 3
- 235000008390 olive oil Nutrition 0.000 description 3
- 239000012044 organic layer Substances 0.000 description 3
- 239000003960 organic solvent Substances 0.000 description 3
- 239000000312 peanut oil Substances 0.000 description 3
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 3
- 239000002953 phosphate buffered saline Substances 0.000 description 3
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 3
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- 238000011321 prophylaxis Methods 0.000 description 3
- 102000005962 receptors Human genes 0.000 description 3
- 108020003175 receptors Proteins 0.000 description 3
- 238000010561 standard procedure Methods 0.000 description 3
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 2
- IZHVBANLECCAGF-UHFFFAOYSA-N 2-hydroxy-3-(octadecanoyloxy)propyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)COC(=O)CCCCCCCCCCCCCCCCC IZHVBANLECCAGF-UHFFFAOYSA-N 0.000 description 2
- LVTIZXGNLIKUQZ-UHFFFAOYSA-N 4-bromopiperidine;hydrobromide Chemical compound Br.BrC1CCNCC1 LVTIZXGNLIKUQZ-UHFFFAOYSA-N 0.000 description 2
- MPANSNMWQPWRIZ-UHFFFAOYSA-N 5-phenyl-2-(pyrrolidin-1-ylsulfonylmethyl)pentanoic acid Chemical compound C1CCCN1S(=O)(=O)CC(C(=O)O)CCCC1=CC=CC=C1 MPANSNMWQPWRIZ-UHFFFAOYSA-N 0.000 description 2
- 102100026802 72 kDa type IV collagenase Human genes 0.000 description 2
- 244000215068 Acacia senegal Species 0.000 description 2
- 235000006491 Acacia senegal Nutrition 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 241000416162 Astragalus gummifer Species 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Chemical compound CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- 102100027995 Collagenase 3 Human genes 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 2
- 208000001382 Experimental Melanoma Diseases 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical class Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- 108010015302 Matrix metalloproteinase-9 Proteins 0.000 description 2
- 102000003843 Metalloendopeptidases Human genes 0.000 description 2
- 108090000131 Metalloendopeptidases Proteins 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 206010033128 Ovarian cancer Diseases 0.000 description 2
- 102000035195 Peptidases Human genes 0.000 description 2
- 108091005804 Peptidases Proteins 0.000 description 2
- 239000004365 Protease Substances 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- 102100030416 Stromelysin-1 Human genes 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- 210000001744 T-lymphocyte Anatomy 0.000 description 2
- 229920001615 Tragacanth Polymers 0.000 description 2
- 230000002152 alkylating effect Effects 0.000 description 2
- 206010003246 arthritis Diseases 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 229910000389 calcium phosphate Inorganic materials 0.000 description 2
- 235000011010 calcium phosphates Nutrition 0.000 description 2
- JYYOBHFYCIDXHH-UHFFFAOYSA-N carbonic acid;hydrate Chemical compound O.OC(O)=O JYYOBHFYCIDXHH-UHFFFAOYSA-N 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 239000007795 chemical reaction product Substances 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- BFPSDSIWYFKGBC-UHFFFAOYSA-N chlorotrianisene Chemical compound C1=CC(OC)=CC=C1C(Cl)=C(C=1C=CC(OC)=CC=1)C1=CC=C(OC)C=C1 BFPSDSIWYFKGBC-UHFFFAOYSA-N 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 238000003818 flash chromatography Methods 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- 239000012634 fragment Substances 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 125000005843 halogen group Chemical group 0.000 description 2
- 125000004404 heteroalkyl group Chemical group 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 2
- 210000000548 hind-foot Anatomy 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- UEXQBEVWFZKHNB-UHFFFAOYSA-N intermediate 29 Natural products C1=CC(N)=CC=C1NC1=NC=CC=N1 UEXQBEVWFZKHNB-UHFFFAOYSA-N 0.000 description 2
- 235000010445 lecithin Nutrition 0.000 description 2
- 239000000787 lecithin Substances 0.000 description 2
- 229940067606 lecithin Drugs 0.000 description 2
- 210000000265 leukocyte Anatomy 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 239000007937 lozenge Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- 230000023881 membrane protein ectodomain proteolysis Effects 0.000 description 2
- 206010061289 metastatic neoplasm Diseases 0.000 description 2
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 2
- 210000001616 monocyte Anatomy 0.000 description 2
- 239000012053 oil suspension Substances 0.000 description 2
- 230000002018 overexpression Effects 0.000 description 2
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 2
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 2
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 2
- 229920000053 polysorbate 80 Polymers 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- 235000019419 proteases Nutrition 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 239000012312 sodium hydride Substances 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- 230000008354 tissue degradation Effects 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- 235000010487 tragacanth Nutrition 0.000 description 2
- 229940116362 tragacanth Drugs 0.000 description 2
- 239000012581 transferrin Substances 0.000 description 2
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 235000015112 vegetable and seed oil Nutrition 0.000 description 2
- 239000008158 vegetable oil Substances 0.000 description 2
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- IBWOBPCKANNSAF-UHFFFAOYSA-N (4-bromopiperidin-1-yl)-phenylmethanone Chemical compound C1CC(Br)CCN1C(=O)C1=CC=CC=C1 IBWOBPCKANNSAF-UHFFFAOYSA-N 0.000 description 1
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- APQIUTYORBAGEZ-UHFFFAOYSA-N 1,1-dibromoethane Chemical compound CC(Br)Br APQIUTYORBAGEZ-UHFFFAOYSA-N 0.000 description 1
- ZORQXIQZAOLNGE-UHFFFAOYSA-N 1,1-difluorocyclohexane Chemical compound FC1(F)CCCCC1 ZORQXIQZAOLNGE-UHFFFAOYSA-N 0.000 description 1
- 125000004973 1-butenyl group Chemical group C(=CCC)* 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- 125000006017 1-propenyl group Chemical group 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- LTMRRSWNXVJMBA-UHFFFAOYSA-L 2,2-diethylpropanedioate Chemical compound CCC(CC)(C([O-])=O)C([O-])=O LTMRRSWNXVJMBA-UHFFFAOYSA-L 0.000 description 1
- CCBVXBHZCNQCBC-UHFFFAOYSA-N 2-(1-benzoylpiperidin-4-yl)-5-phenyl-2-(sulfanylmethyl)pentanoic acid Chemical compound C(C1=CC=CC=C1)(=O)N1CCC(CC1)C(C(=O)O)(CCCC1=CC=CC=C1)CS CCBVXBHZCNQCBC-UHFFFAOYSA-N 0.000 description 1
- VKJCJJYNVIYVQR-UHFFFAOYSA-N 2-(3-bromopropyl)isoindole-1,3-dione Chemical compound C1=CC=C2C(=O)N(CCCBr)C(=O)C2=C1 VKJCJJYNVIYVQR-UHFFFAOYSA-N 0.000 description 1
- RNAMYOYQYRYFQY-UHFFFAOYSA-N 2-(4,4-difluoropiperidin-1-yl)-6-methoxy-n-(1-propan-2-ylpiperidin-4-yl)-7-(3-pyrrolidin-1-ylpropoxy)quinazolin-4-amine Chemical compound N1=C(N2CCC(F)(F)CC2)N=C2C=C(OCCCN3CCCC3)C(OC)=CC2=C1NC1CCN(C(C)C)CC1 RNAMYOYQYRYFQY-UHFFFAOYSA-N 0.000 description 1
- OPPRJXSIOPFPAD-UHFFFAOYSA-N 2-(bromomethyl)-5-(1,3-dioxoisoindol-2-yl)pentanoic acid Chemical compound C1=CC=C2C(=O)N(CCCC(CBr)C(=O)O)C(=O)C2=C1 OPPRJXSIOPFPAD-UHFFFAOYSA-N 0.000 description 1
- YSFDBCZCGQKQAZ-UHFFFAOYSA-N 2-(bromomethyl)-5-phenylpentanoic acid Chemical compound OC(=O)C(CBr)CCCC1=CC=CC=C1 YSFDBCZCGQKQAZ-UHFFFAOYSA-N 0.000 description 1
- PNBPWJACAVHUPD-UHFFFAOYSA-N 2-(furan-2-ylmethylsulfanyl)-2-methyl-5-phenylpentanoic acid Chemical compound O1C(=CC=C1)CSC(C(=O)O)(CCCC1=CC=CC=C1)C PNBPWJACAVHUPD-UHFFFAOYSA-N 0.000 description 1
- SOYGTWKKLYPHKJ-UHFFFAOYSA-N 2-(oxan-4-ylsulfanylmethyl)-5-phenylpentanoic acid Chemical compound C1COCCC1SCC(C(=O)O)CCCC1=CC=CC=C1 SOYGTWKKLYPHKJ-UHFFFAOYSA-N 0.000 description 1
- CMZZDRGYBCAEFS-UHFFFAOYSA-N 2-(oxan-4-ylsulfonylmethyl)-5-phenylpentanoic acid Chemical compound C1COCCC1S(=O)(=O)CC(C(=O)O)CCCC1=CC=CC=C1 CMZZDRGYBCAEFS-UHFFFAOYSA-N 0.000 description 1
- ZFFTZDQKIXPDAF-UHFFFAOYSA-N 2-Furanmethanethiol Chemical compound SCC1=CC=CO1 ZFFTZDQKIXPDAF-UHFFFAOYSA-N 0.000 description 1
- DTZYBBWXZAYJAQ-UHFFFAOYSA-N 2-[(1-benzoylpiperidin-4-yl)sulfonylmethyl]-5-phenylpentanoic acid Chemical compound C1CN(C(=O)C=2C=CC=CC=2)CCC1S(=O)(=O)CC(C(=O)O)CCCC1=CC=CC=C1 DTZYBBWXZAYJAQ-UHFFFAOYSA-N 0.000 description 1
- KUQWMVDFSMVDAQ-UHFFFAOYSA-N 2-[(1-benzylpiperidin-4-yl)sulfonylmethyl]-5-phenylpentanoic acid Chemical compound C1CN(CC=2C=CC=CC=2)CCC1S(=O)(=O)CC(C(=O)O)CCCC1=CC=CC=C1 KUQWMVDFSMVDAQ-UHFFFAOYSA-N 0.000 description 1
- BYUIDTUPOLLPLU-UHFFFAOYSA-N 2-[(4-benzoylphenyl)sulfanylmethyl]-5-(1,3-dioxoisoindol-2-yl)pentanoic acid Chemical compound O=C1C2=CC=CC=C2C(=O)N1CCCC(C(=O)O)CSC(C=C1)=CC=C1C(=O)C1=CC=CC=C1 BYUIDTUPOLLPLU-UHFFFAOYSA-N 0.000 description 1
- ZOZVXTYPDINXPB-UHFFFAOYSA-N 2-[(4-benzoylphenyl)sulfonylmethyl]-5-(1,3-dioxoisoindol-2-yl)-n-hydroxypentanamide Chemical compound O=C1C2=CC=CC=C2C(=O)N1CCCC(C(=O)NO)CS(=O)(=O)C(C=C1)=CC=C1C(=O)C1=CC=CC=C1 ZOZVXTYPDINXPB-UHFFFAOYSA-N 0.000 description 1
- OKNZAHMIAUSTEH-UHFFFAOYSA-N 2-[(4-benzoylphenyl)sulfonylmethyl]-5-(1,3-dioxoisoindol-2-yl)pentanoic acid Chemical compound O=C1C2=CC=CC=C2C(=O)N1CCCC(C(=O)O)CS(=O)(=O)C(C=C1)=CC=C1C(=O)C1=CC=CC=C1 OKNZAHMIAUSTEH-UHFFFAOYSA-N 0.000 description 1
- TZYVWBZMYLFEDQ-UHFFFAOYSA-N 2-[[1-[(2-methylpropan-2-yl)oxycarbonyl]piperidin-4-yl]sulfonylmethyl]-5-phenylpentanoic acid Chemical compound C1CN(C(=O)OC(C)(C)C)CCC1S(=O)(=O)CC(C(O)=O)CCCC1=CC=CC=C1 TZYVWBZMYLFEDQ-UHFFFAOYSA-N 0.000 description 1
- VKUYLANQOAKALN-UHFFFAOYSA-N 2-[benzyl-(4-methoxyphenyl)sulfonylamino]-n-hydroxy-4-methylpentanamide Chemical compound C1=CC(OC)=CC=C1S(=O)(=O)N(C(CC(C)C)C(=O)NO)CC1=CC=CC=C1 VKUYLANQOAKALN-UHFFFAOYSA-N 0.000 description 1
- 125000005999 2-bromoethyl group Chemical group 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- 125000006022 2-methyl-2-propenyl group Chemical group 0.000 description 1
- BLXPDRMJKIQIKT-UHFFFAOYSA-N 2-methylidene-5-phenylpentanoic acid Chemical compound OC(=O)C(=C)CCCC1=CC=CC=C1 BLXPDRMJKIQIKT-UHFFFAOYSA-N 0.000 description 1
- ZMRFRBHYXOQLDK-UHFFFAOYSA-N 2-phenylethanethiol Chemical compound SCCC1=CC=CC=C1 ZMRFRBHYXOQLDK-UHFFFAOYSA-N 0.000 description 1
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- NUSZCZOBSDRJEK-UHFFFAOYSA-N 3-(benzenesulfonyl)-n-hydroxypropanamide Chemical compound ONC(=O)CCS(=O)(=O)C1=CC=CC=C1 NUSZCZOBSDRJEK-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- ZXDFVBJYBXOOJK-UHFFFAOYSA-N 3-(iminomethylideneamino)-n,n-dimethylpentan-2-amine Chemical compound N=C=NC(CC)C(C)N(C)C ZXDFVBJYBXOOJK-UHFFFAOYSA-N 0.000 description 1
- XMZQWZJMTBCUFT-UHFFFAOYSA-N 3-bromopropylbenzene Chemical compound BrCCCC1=CC=CC=C1 XMZQWZJMTBCUFT-UHFFFAOYSA-N 0.000 description 1
- WZWIQYMTQZCSKI-UHFFFAOYSA-N 4-cyanobenzaldehyde Chemical compound O=CC1=CC=C(C#N)C=C1 WZWIQYMTQZCSKI-UHFFFAOYSA-N 0.000 description 1
- IZKKJNOOMWATBZ-UHFFFAOYSA-N 5-(1,3-dioxoisoindol-2-yl)-2-methylidenepentanoic acid Chemical compound C1=CC=C2C(=O)N(CCCC(=C)C(=O)O)C(=O)C2=C1 IZKKJNOOMWATBZ-UHFFFAOYSA-N 0.000 description 1
- WLNKDIGNAIRLGT-UHFFFAOYSA-N 5-phenyl-2-(2-phenylethylsulfanylmethyl)pentanoic acid Chemical compound C=1C=CC=CC=1CCSCC(C(=O)O)CCCC1=CC=CC=C1 WLNKDIGNAIRLGT-UHFFFAOYSA-N 0.000 description 1
- HEPZYEZEUMVYDV-UHFFFAOYSA-N 5-phenyl-2-(sulfanylmethyl)pentanoic acid Chemical compound OC(=O)C(CS)CCCC1=CC=CC=C1 HEPZYEZEUMVYDV-UHFFFAOYSA-N 0.000 description 1
- 101710151806 72 kDa type IV collagenase Proteins 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 108091022885 ADAM Proteins 0.000 description 1
- 102000029791 ADAM Human genes 0.000 description 1
- 102000036664 ADAM10 Human genes 0.000 description 1
- 108091007504 ADAM10 Proteins 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 1
- 102000034473 Adamalysin Human genes 0.000 description 1
- 108030001653 Adamalysin Proteins 0.000 description 1
- LIWMQSWFLXEGMA-WDSKDSINSA-N Ala-Val Chemical compound CC(C)[C@@H](C(O)=O)NC(=O)[C@H](C)N LIWMQSWFLXEGMA-WDSKDSINSA-N 0.000 description 1
- 241001263092 Alchornea latifolia Species 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 208000006386 Bone Resorption Diseases 0.000 description 1
- 239000011547 Bouin solution Substances 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 108050005238 Collagenase 3 Proteins 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 102000005927 Cysteine Proteases Human genes 0.000 description 1
- 108010005843 Cysteine Proteases Proteins 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 201000004624 Dermatitis Diseases 0.000 description 1
- YXHKONLOYHBTNS-UHFFFAOYSA-N Diazomethane Chemical compound C=[N+]=[N-] YXHKONLOYHBTNS-UHFFFAOYSA-N 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 238000005863 Friedel-Crafts acylation reaction Methods 0.000 description 1
- 244000068988 Glycine max Species 0.000 description 1
- 235000010469 Glycine max Nutrition 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- LFVLUOAHQIVABZ-UHFFFAOYSA-N Iodofenphos Chemical compound COP(=S)(OC)OC1=CC(Cl)=C(I)C=C1Cl LFVLUOAHQIVABZ-UHFFFAOYSA-N 0.000 description 1
- RRHGJUQNOFWUDK-UHFFFAOYSA-N Isoprene Chemical compound CC(=C)C=C RRHGJUQNOFWUDK-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 101150014058 MMP1 gene Proteins 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 238000006683 Mannich reaction Methods 0.000 description 1
- 102000000380 Matrix Metalloproteinase 1 Human genes 0.000 description 1
- 108010016113 Matrix Metalloproteinase 1 Proteins 0.000 description 1
- 108010076503 Matrix Metalloproteinase 13 Proteins 0.000 description 1
- 108010016165 Matrix Metalloproteinase 2 Proteins 0.000 description 1
- 108010016160 Matrix Metalloproteinase 3 Proteins 0.000 description 1
- 102000001776 Matrix metalloproteinase-9 Human genes 0.000 description 1
- 102100030412 Matrix metalloproteinase-9 Human genes 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-M Methanesulfonate Chemical compound CS([O-])(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-M 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 108010083674 Myelin Proteins Proteins 0.000 description 1
- 102000006386 Myelin Proteins Human genes 0.000 description 1
- 108010032605 Nerve Growth Factor Receptors Proteins 0.000 description 1
- 102100030411 Neutrophil collagenase Human genes 0.000 description 1
- 101710118230 Neutrophil collagenase Proteins 0.000 description 1
- 108030001564 Neutrophil collagenases Proteins 0.000 description 1
- 102000056189 Neutrophil collagenases Human genes 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical class CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 1
- 101150085390 RPM1 gene Proteins 0.000 description 1
- 241000700157 Rattus norvegicus Species 0.000 description 1
- 241000219061 Rheum Species 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- 102000012479 Serine Proteases Human genes 0.000 description 1
- 108010022999 Serine Proteases Proteins 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 101710108790 Stromelysin-1 Proteins 0.000 description 1
- 230000006044 T cell activation Effects 0.000 description 1
- 102000004338 Transferrin Human genes 0.000 description 1
- 108090000901 Transferrin Proteins 0.000 description 1
- 102100023935 Transmembrane glycoprotein NMB Human genes 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- 102100033725 Tumor necrosis factor receptor superfamily member 16 Human genes 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 150000001252 acrylic acid derivatives Chemical class 0.000 description 1
- 239000011149 active material Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 230000002001 anti-metastasis Effects 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 230000002917 arthritic effect Effects 0.000 description 1
- 210000001188 articular cartilage Anatomy 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 239000012131 assay buffer Substances 0.000 description 1
- 239000000305 astragalus gummifer gum Substances 0.000 description 1
- 208000010668 atopic eczema Diseases 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 235000013871 bee wax Nutrition 0.000 description 1
- 239000012166 beeswax Substances 0.000 description 1
- 150000001558 benzoic acid derivatives Chemical class 0.000 description 1
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- 230000027455 binding Effects 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000002051 biphasic effect Effects 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 230000024279 bone resorption Effects 0.000 description 1
- 201000008275 breast carcinoma Diseases 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 235000010216 calcium carbonate Nutrition 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 239000012876 carrier material Substances 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 150000001860 citric acid derivatives Chemical class 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 230000007012 clinical effect Effects 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 108700004333 collagenase 1 Proteins 0.000 description 1
- 235000009508 confectionery Nutrition 0.000 description 1
- 210000002808 connective tissue Anatomy 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 235000013365 dairy product Nutrition 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 238000006114 decarboxylation reaction Methods 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- MCEIBRVLVAPEGM-UHFFFAOYSA-N dibenzyl 2-[3-(1,3-dioxoisoindol-2-yl)propyl]propanedioate Chemical compound O=C1C2=CC=CC=C2C(=O)N1CCCC(C(=O)OCC=1C=CC=CC=1)C(=O)OCC1=CC=CC=C1 MCEIBRVLVAPEGM-UHFFFAOYSA-N 0.000 description 1
- 150000001991 dicarboxylic acids Chemical class 0.000 description 1
- 239000002027 dichloromethane extract Substances 0.000 description 1
- WGLUMOCWFMKWIL-UHFFFAOYSA-N dichloromethane;methanol Chemical compound OC.ClCCl WGLUMOCWFMKWIL-UHFFFAOYSA-N 0.000 description 1
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 1
- 150000005332 diethylamines Chemical class 0.000 description 1
- 125000005043 dihydropyranyl group Chemical group O1C(CCC=C1)* 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 239000000890 drug combination Substances 0.000 description 1
- 235000006694 eating habits Nutrition 0.000 description 1
- 239000003974 emollient agent Substances 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 210000002683 foot Anatomy 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 230000000855 fungicidal effect Effects 0.000 description 1
- 239000000417 fungicide Substances 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 229940074045 glyceryl distearate Drugs 0.000 description 1
- 229940075507 glyceryl monostearate Drugs 0.000 description 1
- 229920000591 gum Polymers 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 239000002035 hexane extract Substances 0.000 description 1
- FBPFZTCFMRRESA-UHFFFAOYSA-N hexane-1,2,3,4,5,6-hexol Chemical compound OCC(O)C(O)C(O)C(O)CO FBPFZTCFMRRESA-UHFFFAOYSA-N 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 229940102213 injectable suspension Drugs 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 210000004731 jugular vein Anatomy 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 150000003893 lactate salts Chemical class 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 244000144972 livestock Species 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 150000002690 malonic acid derivatives Chemical class 0.000 description 1
- 239000003771 matrix metalloproteinase inhibitor Substances 0.000 description 1
- 229940121386 matrix metalloproteinase inhibitor Drugs 0.000 description 1
- 235000013372 meat Nutrition 0.000 description 1
- SQBDDCLTIMQETN-UHFFFAOYSA-N methyl 2-(acetylsulfanylmethyl)-5-phenylpentanoate Chemical compound CC(=O)SCC(C(=O)OC)CCCC1=CC=CC=C1 SQBDDCLTIMQETN-UHFFFAOYSA-N 0.000 description 1
- METCVQAYKKZJLQ-UHFFFAOYSA-N methyl 2-(bromomethyl)-5-phenylpentanoate Chemical compound COC(=O)C(CBr)CCCC1=CC=CC=C1 METCVQAYKKZJLQ-UHFFFAOYSA-N 0.000 description 1
- QDBLPEJNKICFEF-UHFFFAOYSA-N methyl 2-(chlorosulfonylmethyl)-5-phenylpentanoate Chemical compound COC(=O)C(CS(Cl)(=O)=O)CCCC1=CC=CC=C1 QDBLPEJNKICFEF-UHFFFAOYSA-N 0.000 description 1
- XHUSHCLFYZFIHO-UHFFFAOYSA-N methyl 5-phenyl-2-(pyrrolidin-1-ylsulfonylmethyl)pentanoate Chemical compound C1CCCN1S(=O)(=O)CC(C(=O)OC)CCCC1=CC=CC=C1 XHUSHCLFYZFIHO-UHFFFAOYSA-N 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 239000002324 mouth wash Substances 0.000 description 1
- 210000005012 myelin Anatomy 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 229940053128 nerve growth factor Drugs 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 238000010899 nucleation Methods 0.000 description 1
- SSUCKKNRCOFUPT-UHFFFAOYSA-N o-[tert-butyl(dimethyl)silyl]hydroxylamine Chemical compound CC(C)(C)[Si](C)(C)ON SSUCKKNRCOFUPT-UHFFFAOYSA-N 0.000 description 1
- GYCKQBWUSACYIF-UHFFFAOYSA-N o-hydroxybenzoic acid ethyl ester Natural products CCOC(=O)C1=CC=CC=C1O GYCKQBWUSACYIF-UHFFFAOYSA-N 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 150000003891 oxalate salts Chemical class 0.000 description 1
- LMYJGUNNJIDROI-UHFFFAOYSA-N oxan-4-ol Chemical compound OC1CCOCC1 LMYJGUNNJIDROI-UHFFFAOYSA-N 0.000 description 1
- HDEOKHIQTDMWTQ-UHFFFAOYSA-N oxane-4-thiol Chemical compound SC1CCOCC1 HDEOKHIQTDMWTQ-UHFFFAOYSA-N 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 229920002866 paraformaldehyde Polymers 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical class OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 1
- 210000003200 peritoneal cavity Anatomy 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- YPMBDBMGNAVBAX-UHFFFAOYSA-N phenyl-(4-sulfanylpiperidin-1-yl)methanone Chemical compound C1CC(S)CCN1C(=O)C1=CC=CC=C1 YPMBDBMGNAVBAX-UHFFFAOYSA-N 0.000 description 1
- 125000003884 phenylalkyl group Chemical group 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- 125000004344 phenylpropyl group Chemical group 0.000 description 1
- 235000021317 phosphate Nutrition 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- IUBQJLUDMLPAGT-UHFFFAOYSA-N potassium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([K])[Si](C)(C)C IUBQJLUDMLPAGT-UHFFFAOYSA-N 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 235000018102 proteins Nutrition 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- KOUKXHPPRFNWPP-UHFFFAOYSA-N pyrazine-2,5-dicarboxylic acid;hydrate Chemical compound O.OC(=O)C1=CN=C(C(O)=O)C=N1 KOUKXHPPRFNWPP-UHFFFAOYSA-N 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 238000011552 rat model Methods 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- SDWFMPSNHFWVOV-UHFFFAOYSA-N s-(1-benzoylpiperidin-4-yl) ethanethioate Chemical compound C1CC(SC(=O)C)CCN1C(=O)C1=CC=CC=C1 SDWFMPSNHFWVOV-UHFFFAOYSA-N 0.000 description 1
- BYUNVFSCFPTRBA-UHFFFAOYSA-N s-(oxan-4-yl) ethanethioate Chemical compound CC(=O)SC1CCOCC1 BYUNVFSCFPTRBA-UHFFFAOYSA-N 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 238000004062 sedimentation Methods 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 229910001467 sodium calcium phosphate Inorganic materials 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- HRZFUMHJMZEROT-UHFFFAOYSA-L sodium disulfite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])(=O)=O HRZFUMHJMZEROT-UHFFFAOYSA-L 0.000 description 1
- 229940001584 sodium metabisulfite Drugs 0.000 description 1
- 235000010262 sodium metabisulphite Nutrition 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 235000011069 sorbitan monooleate Nutrition 0.000 description 1
- 239000001593 sorbitan monooleate Substances 0.000 description 1
- 229940035049 sorbitan monooleate Drugs 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 150000003890 succinate salts Chemical class 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 239000012622 synthetic inhibitor Substances 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 150000003892 tartrate salts Chemical class 0.000 description 1
- SJAKIGKVIJINMY-UHFFFAOYSA-N tert-butyl 4-acetylsulfanylpiperidine-1-carboxylate Chemical compound CC(=O)SC1CCN(C(=O)OC(C)(C)C)CC1 SJAKIGKVIJINMY-UHFFFAOYSA-N 0.000 description 1
- KZBWIYHDNQHMET-UHFFFAOYSA-N tert-butyl 4-bromopiperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(Br)CC1 KZBWIYHDNQHMET-UHFFFAOYSA-N 0.000 description 1
- USYCVFFWCMHPPG-UHFFFAOYSA-N tert-butyl 4-sulfanylpiperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(S)CC1 USYCVFFWCMHPPG-UHFFFAOYSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- DUYAAUVXQSMXQP-UHFFFAOYSA-M thioacetate Chemical compound CC([S-])=O DUYAAUVXQSMXQP-UHFFFAOYSA-M 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-M toluene-4-sulfonate Chemical compound CC1=CC=C(S([O-])(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-M 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 108091007466 transmembrane glycoproteins Proteins 0.000 description 1
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical compound OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 230000005747 tumor angiogenesis Effects 0.000 description 1
- 230000005748 tumor development Effects 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
- 238000012447 xenograft mouse model Methods 0.000 description 1
- 230000004572 zinc-binding Effects 0.000 description 1
- 150000007934 α,β-unsaturated carboxylic acids Chemical class 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C317/00—Sulfones; Sulfoxides
- C07C317/44—Sulfones; Sulfoxides having sulfone or sulfoxide groups and carboxyl groups bound to the same carbon skeleton
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/04—Drugs for skeletal disorders for non-specific disorders of the connective tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/14—Decongestants or antiallergics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/46—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with hetero atoms directly attached to the ring nitrogen atom
- C07D207/48—Sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/06—Systems containing only non-condensed rings with a five-membered ring
- C07C2601/08—Systems containing only non-condensed rings with a five-membered ring the ring being saturated
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Physical Education & Sports Medicine (AREA)
- Biomedical Technology (AREA)
- Ophthalmology & Optometry (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Immunology (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Quinoline Compounds (AREA)
- Pyridine Compounds (AREA)
- Pyrane Compounds (AREA)
- Indole Compounds (AREA)
- Furan Compounds (AREA)
- Heterocyclic Compounds Containing Sulfur Atoms (AREA)
- Thiazole And Isothizaole Compounds (AREA)
- Hydrogenated Pyridines (AREA)
Description
- Gebiet der Erfindung
- Die Erfindung betrifft Hydroxamsäure- und Carbonsäurederivate und ihre Verwendung in der Medizin.
- Hintergrund der Erfindung
- Metalloproteinasen, einschließlich Matrix-Metalloproteinase (MMP), (menschliche Fibroblasten)-Collagenase, Gelatinase und TNF-Convertase (TACE), und ihre Wirkungsweisen sowie die Inhibitoren davon und ihre klinischen Wirkungen sind in WO-A-9611209, WO-A-9712902 und WO-A-9719075 beschrieben, deren Inhalt hier als Referenz mitumfasst ist. MMP-Inhibitoren können auch zur Inhibition von anderen Säuger-Metalloproteinasen geeignet sein, wie die Adamalysinfamilie (oder ADAMs), deren Mitglieder TNF-Convertase (TACE) und ADAM-10, die die Freisetzung von TNF-α aus Zellen herbeiführen können, und andere umfassen, von denen gezeigt wurde, dass sie durch menschliche artikuläre Knorpelzellen exprimiert werden und ferner an der Zerstörung von auf Myelin basierendem Protein, ein Phänomen, das mit Multipler Sklerose einhergeht, beteiligt sind.
- Von Verbindungen, die die Eigenschaft der Inhibition der Wirkung der am Bindegewebsabbau beteiligten Metalloproteinasen, wie Collagenase, Stromelysin und Gelatinase, aufweisen, wurde gezeigt, dass sie die Freisetzung von TNF sowohl in vitro als auch in vivo hemmen. Siehe Gearing et al. (1994), Nature 370: 555–557; McGeehan et al (1994), Nature 370: 558–561; GB-A-2268934; und WO-A-9320047. Alle diese beschriebenen Inhibitoren enthalten ebenso wie die in WO-A-9523790 offenbarten Imidazol-subsituierten Verbindungen eine Hydroxamsäure-Zink-Bindungsgruppe. Weitere Verbindungen, die MMP und/oder TNF hemmen, sind in WO-A-9513289, WO-A-9611209, WO-A-96035687, WO-A-96035711, WO-A-96035712 und WO-A-96035714 beschrieben.
- WO-A-9718188 offenbart MMP-Inhibitoren der Formel
- EP-A-0780386 offenbart Verbindungen mit MMP- und TNF-inhibitorischer Aktivität der Formel
- Die Verbindungen von EP-A-0780386, die zuerst in der am 20. Dezember 1995 eingereichten US-Anmeldung Nr. 8939 offenbart wurden, besitzen die gleiche Formel, worin R1 H ist; R2 H, Niedrigalkyl, Aralkyl, Cycloalkyl, Cycloalkylalkyl, Heterocyclo oder NR6R7 ist; R3 H, Niedrigalkyl, Cycloalkyl, Cycloalkylalkyl oder Aralkyl ist; R4 H oder Niedrigalkyl ist; und R5 Niedrigalkyl, Aryl, Aralkyl, Heteroaryl oder Heteroaralkyl ist.
- US-A-4325964 offenbart, dass bestimmte Benzhydrylsulphinylhydroxamate bei neuropsychischen Leiden von Nutzen sind.
- Zayed et al., Zeitschrift für Naturforschung (1966) 180–182, offenbaren 3-Phenylsulphonylpropansäure-N-hydroxyamid als Fungizid.
- Zusammenfassung der Erfindung
- Die Erfindung betrifft neue Verbindungen der Formel (I), die geeignete Inhibitoren von Matrix-Metalloproteinasen und/oder TNF-α-vermittelten Krankheiten, einschließlich von degenerativen Krankheiten und bestimmten Krebsarten, sind.
- Die neuen erfindungsgemäßen Verbindungen sind vom allgemeinen, durch die Formel (I) dargestellten Typ:
X O oder S(O)0-2 ist;
Y NHOH ist;
R1 H oder eine Gruppe (gegebenenfalls substituiert mit R9) ist, die ausgewählt ist aus C1-6-Alkyl, C2-6-Alkenyl, Aryl, Aryl-C1-6-alkyl, Heteroaryl, Heteroaryl-C1-6-alkyl, Heterocycloalkyl, Heterocycloalkyl-C1-6-alkyl, Cycloalkyl und Cycloalkyl-C1-6-alkyl ist;
R2 H oder C1-6-Alkyl ist;
B Cycloalkenyl, Heterocycloalkenyl, Heterocycloalkyl oder Heterocycloalkyl-C1-6-alkyl ist, wovon die Gruppen jeweils gegebenenfalls mit einem Substituenten substituiert sind, der ausgewählt ist aus R5, R5-C1-6-Alkyl, R5-C2-6-Alkenyl, Aryl (gegebenenfalls substituiert mit R5), R5-C1-6-Alkyl, Aryl, Aryl-C1-6-alkyl (gegebenenfalls substituiert mit R5), Heteroaryl-C1-6-alkyl (gegebenenfalls substituiert mit R5), R7-C2-6-Alkenylaryl, Heteroaryl (gegebenenfalls substituiert mit R5), R5-C1-6-Alkylheteroaryl, Cycloalkyl (gegebenenfalls substituiert mit R5), Benzo-kondensiertem Cycloalkyl (gegebenenfalls substituiert mit R5), Heterocycloalkyl (gegebenenfalls substituiert mit R5), Benzo-kondensiertem Heterocycloalkyl (gegebenenfalls substituiert mit R5) und den Gruppen:mit der Maßgabe, dass B nicht Benzhydryl ist, wenn X SO ist, und R1 H ist;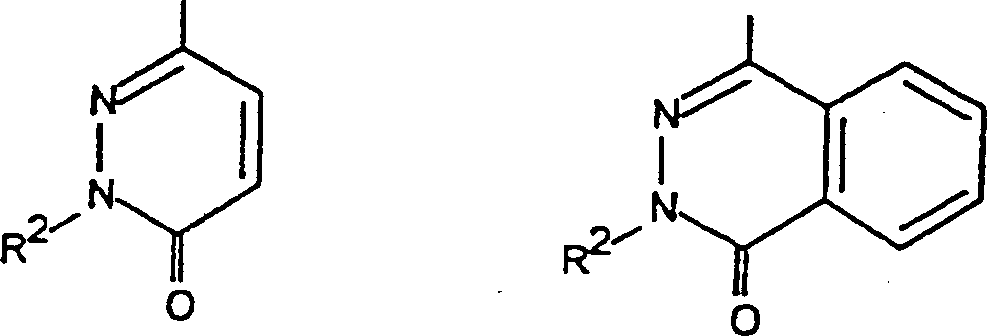
R5 C1-6-Alkyl, R7-C2-6-Alkenyl, Halogen, CN, NO2, N(R6)2, OR6, COR6, CO2R2, CON(R6)2, NR6R7, S(O)0-2R8 oder SO2N(R6)2 ist;
R6 H oder eine Gruppe ist, die ausgewählt ist aus C1-6-Alkyl, Aryl, Aryl-C1-6-alkyl, Heteroaryl, Heteroaryl-C1-6-alkyl, Cycloalkyl, Cycloalkyl-C1-6-alkyl, Heterocycloalkyl und Heterocycloalkyl-C1-6-alkyl, worin die Gruppe gegebenenfalls substituiert ist mit R8, COR8, SO0-2R8, CO2R8, OR8, CONR2R8, NR2R8, Halogen, CN, SO2NR2R8 oder NO2, und für jeden Fall von N(R6)2 die R6-Gruppen gleich oder verschieden sind oder N(R6)2 Heterocycloalkyl ist, das gegebenenfalls substituiert ist mit R8, COR8, SO0-2R8, CO2R8, OR8, CONR2R8, NR2R8, Halogen, CN, SO2NR2R8 oder NO2;
R7 COR6, CON(R6)2, CO2R8 oder SO2R8 ist;
R8 C1-6-Alkyl, Aryl, Aryl-C1-6-alkyl, Heteroaryl oder Heteroaryl-C1-6-alkyl ist; und
R9 OR6, COR6, CO2R2, NR6R7, S(O)0-2R8, SO2N(R6)2, Phthalimido, Succinimido oder die Gruppeist;
und die Salze, Solvate, Hydrate, geschützten Amino- und geschützten Carboxyderivate davon. - Kombinationen von Substituenten und/oder Variablen sind nur zulässig, wenn solche Kombinationen stabile Verbindungen ergeben.
- Beschreibung der Erfindung
- Die bevorzugten erfindungsgemäßen Verbindungen sind diejenigen, wobei eines oder mehreres von Folgendem zutrifft:
X ist S, SO oder SO2;
R1 ist H oder eine Gruppe (gegebenenfalls substituiert mit R9), die ausgewählt ist aus C1-6-Alkyl, C2-6-Alkenyl, Aryl-C1-6-Alkyl, Heteroaryl-C1-6-alkyl, Heterocycloalkyl-C1-6-alkyl und Cycloalkyl-C1-6-alkyl;
B ist Cycloalkenyl oder Heterocycloalkenyl, wovon jede Gruppe gegebenenfalls mit einem Substituenten substituiert ist, der ausgewählt ist aus R5, R5-C1-6-Alkyl, Aryl (gegebenenfalls substituiert mit R5), R5-C1-6-Alkylaryl, Aryl-C1-6-alkyl (gegebenenfalls substituiert mit R5), Heteroaryl-C1-6-alkyl (gegebenenfalls substituiert mit R5), Heteroaryl (gegebenenfalls substituiert mit R5), R5-C1-6-Alkylheteroaryl, R5-C1-6-Alkylheteroaryl, Cycloalkyl (gegebenenfalls substituiert mit R5), Benzo-kondensiertem Cycloalkyl (gegebenenfalls substituiert mit R5), Heterocycloalkyl (gegebenenfalls substituiert mit R5), Benzo-kondensiertem Heterocycloalkyl (gegebenenfalls substituiert mit R5) und den Gruppen:R5 ist Halogen, CN, NO2, N(R6)2, OR6, COR6, CON(R6)2, NR6R7 oder S(O)0-2R8;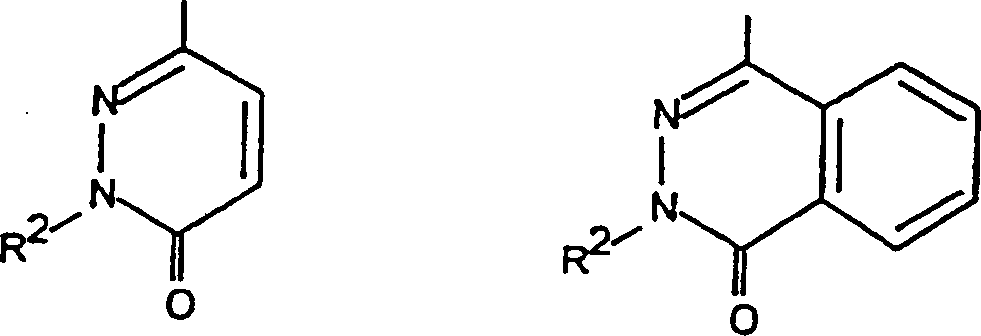
R7 ist COR6; und
R9 ist OR6, CO2R2, Phthalimido, Succinimido oder die Gruppe -
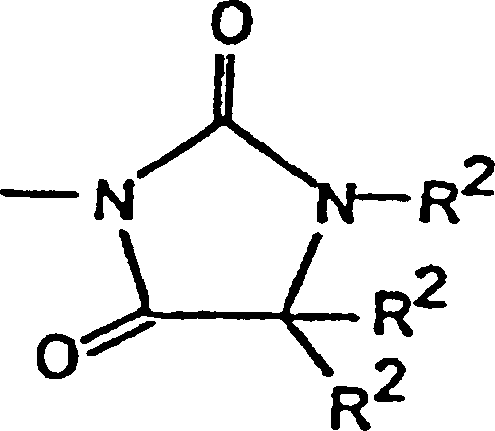
- Die Verbindungen der Beispiele sind besonders bevorzugt.
- Es wird davon ausgegangen, dass die erfindungsgemäßen Verbindungen ein oder mehrere asymmetrische, substituierte Kohlenstoffatome enthalten können. Die Gegenwart von einem oder mehreren dieser asymmetrischen Zentren in einer Verbindung der Formel (I) kann zu Stereoisomeren führen, und in jedem Fall ist die Erfindung so zu verstehen, dass sie alle derartigen Stereoisomere, einschließlich von Enantiomeren und Diastereomeren, und Gemischen, einschließlich von racemischen Gemischen davon, betrifft.
- Wie in dieser Spezifikation verwendet, bedeutet der Begriff "C1-6-Alkyl" allein oder in Kombination eine geradkettige oder verzweigte Alkylgruppierung mit 1 bis 6 Kohlenstoffatomen, einschließlich beispielsweise Methyl, Ethyl, Propyl, Isopropyl, Butyl, tert-Butyl, Pentyl, Hexyl und dergleichen.
- Der Begriff "C2-6-Alkenyl" bezieht sich auf eine geradkettige oder verzweigte Alkylgruppierung mit 2 bis 6 Kohlenstoffatomen und mit zusätzlich einer Doppelbindung von, je nachdem, entweder E- oder Z-Stereochemie. Dieser Begriff würde beispielsweise Vinyl, 1-Propenyl, 1- und 2-Butenyl, 2-Methyl-2-propenyl, etc., einschließen.
- Der Begriff "Cycloalkyl" bezieht sich auf eine gesättigte alicyclische Gruppierung mit 3 bis 6 Kohlenstoffatomen und umfasst beispielsweise Cyclopropyl, Cyclobutyl, Cyclopentyl, Cyclohexyl und dergleichen. "Benzo-kondensiertes Cycloalkyl" umfasst Indanyl und Tetrahydronaphthyl.
- Der Begriff "Heterocycloalkyl" bezieht sich auf eine gesättigte heterocyclische Gruppierung mit 3 bis 6 Kohlenstoffatomen und einem oder mehreren Heteroatomen aus der Gruppe N, O, S und umfasst beispielsweise Azetidinyl, Pyrrolidinyl, Tetrahydrofuranyl, Piperidinyl und dergleichen. "Benzo-kondensiertes Heterocycloalkyl" umfasst Indolinyl und Tetrahydrochinolinyl.
- Der Begriff "Cycloalkenyl" bezieht sich auf eine alicyclische Gruppierung mit 3 bis 6 Kohlenstoffatomen und zusätzlich mit einer Doppelbindung. Dieser Begriff würde beispielsweise Cyclopentenyl oder Cyclohexenyl einschließen.
- Der Begriff "Heterocycloalkenyl" bezieht sich auf eine alicyclische Gruppierung mit 3 bis 6 Kohlenstoffatomen und mit einem oder mehreren Heteroatomen aus der Gruppe N, O, S und zusätzlich mit einer Doppelbindung. Dieser Begriff umfasst beispielsweise Dihydropyranyl.
- Der Begriff "Aryl" bedeutet eine gegebenenfalls substituierte Phenyl- oder Naphthylgruppe, wobei der (die) Substituent(en) beispielsweise aus Halogen, Trifluormethyl, C1-6-Alkyl, Alkoxy, Phenyl und dergleichen ausgewählt sind.
- Der Begriff "Heteroaryl" bezieht sich auf aromatische Ringsysteme mit 5 bis 10 Atomen, wovon mindestens ein Atom aus O, N und S ausgewählt ist, und umfasst beispielsweise Furanyl, Thiophenyl, Pyridyl, Indolyl, Chinolyl und dergleichen.
- Der Begriff "Alkoxy" bezieht sich auf eine geradkettige oder verzweigte Alkoxygruppe, die maximal 6 Kohlenstoffatome enthält, wie Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, tert-Butoxy und dergleichen.
- Der Begriff "Halogen" bedeutet Fluor, Chlor, Brom oder Iod.
- Die Begriffe "geschütztes Amino" und "geschütztes Carboxy" bedeuten Amino- und Carboxygruppen, die auf eine Weise geschützt sein können, die derjenigen, die der Fachwelt bekannt ist, entspricht. Beispielsweise kann eine Aminogruppe durch eine Benzyloxycarbonyl-, tert-Butoxycarbonyl-, Acetyl- oder eine gleichartige Gruppe geschützt sein oder kann in Form einer Phthalimido- oder einer gleichartigen Gruppe vorliegen. Eine Carboxylgruppe kann in Form eines leicht abspaltbaren Esters, wie der Methyl-, Ethyl-, Benzyl- oder tert-Butylester, geschützt sein.
- Die Salze der Verbindungen der Formel (I) umfassen pharmazeutisch verträgliche Salze, beispielsweise Säure-Additionssalze, die sich von anorganischen oder organischen Säuren ableiten, wie Hydrochloride, Hydrobromide, p-Toluol-sulphonate, Phosphate, Sulphate, Perchlorate, Acetate, Trifluoracetate, Propionate, Citrate, Malonate, Succinate, Lactate, Oxalate, Tartrate und Benzoate.
- Die Salze können auch mit Basen gebildet werden. Solche Salze umfassen Salze, die sich von anorganischen oder organischen Basen ableiten, beispielsweise Alkalimetallsalze, wie Magnesium oder Calciumsalze und organische Aminsalze, wie Morpholin-, Piperidin-, Dimethylamin- oder Diethylaminsalze.
- Wenn die "geschützte Carboxy"-Gruppe in den erfindungsgemäßen Verbindungen eine veresterte Carboxygruppe ist, kann es ein metabolisch labiler Ester der Formel CO2R10 sein, worin R10 eine Ethyl-, Benzyl-, Phenethyl-, Phenylpropyl-, α- oder β-Naphthyl-, 2,4-Dimethylphenyl-, 4-tert-Butylphenyl-, 2,2,2-Trifluorethyl-, 1-(Benzyloxy)benzyl-, 1-(Benzyloxy)ethyl-, 2-Methyl-1-propionyloxypropyl-, 2,4,6-Trimethylbenzyloxymethyl- oder eine Pivaloylmethylgruppe sein kann.
- Die Verbindungen der allgemeinen Formel (I) können durch jedes beliebige, geeignete, aus der Technik bekannte Verfahren und/oder durch die folgenden Verfahren hergestellt werden.
- Es wird angenommen, dass dort, wo ein bestimmtes Stereoisomer der Formel (I) erforderlich ist, die hier beschriebenen Syntheseverfahren mit dem entsprechenden homochiralen Ausgangsmaterial verwendet und/oder die Isomere unter Anwendung herkömmlicher Trenntechniken (z. B. HPLC) aus dem Gemisch aufgelöst werden können.
- Die erfindungsgemäßen Verbindungen können durch das folgende Verfahren hergestellt werden. In der Beschreibung und in den Formeln, nachstehend, sind die Gruppen R1, R2, R5, R6, R7, R8, R9, R10, B, X und Y, wie vorstehend definiert, mit der Ausnahme, wo anderweitig angegeben. Es wird angenommen, dass die funktionellen Gruppen, wie Amino-, Hydroxyl- oder Carboxylgruppen, die in den verschiedenen, nachstehend beschriebenen Verbindungen vorhanden sind und deren Erhaltung erwünscht ist, möglicherweise in der geschützten Form vorliegen müssen, bevor eine Reaktion gestartet wird. Unter solchen Umständen kann bei einer bestimmten Reaktion die Entfernung der Schutzgruppe der letzte Schritt sein. Der Fachwelt sind geeignete Schutzgruppen für eine solche Funktionalität bekannt. Spezielle Einzelheiten siehe Greene et al., "Protective Groups in Organic Synthesis", Wiley Interscience.
- Ein Verfahren zur Herstellung von Verbindungen der allgemeinen Formel (I) umfasst die Alkylierung einer Verbindung der Formel B-XH (II), worin B und X wie vorstehend definiert sind, mit einem Alkylierungsmittel der Formel Z-(CH2)n-CHR1-(CH2)m-COY (III) oder (wenn m = 0) einem Acrylat der Formel CH2=CR1-COY (IV), worin R1 wie vorstehend definiert ist, Y eine OR2 oder NHOH-Gruppe ist und Z eine geeignete Abgangsgruppe darstellt (z. B. ein Halogenatom, wie ein Bromatom oder einen Alkylsulphonatester, wie Methansulphonat).
- Die Alkylierungsmittel (III) können in chiraler oder racemischer Form erhalten werden. Viele dieser Derivate können leicht aus im Handel erhältlichen Ausgangsmaterialien unter Anwendung von Verfahren, die der Fachwelt bekannt sind (siehe WO-A-9005719), erhalten werden.
- Acrylate der Formel (IV) können durch die Mannich-Reaktion (d. h. mit Paraformaldehyd und Piperidin in einem geeigneten Lösungsmittel, wie 1,4-Dioxan) mit einer Dicarbonsäure der allgemeinen Formel HO2C-CNR1-CO2H (V) hergestellt werden. Diese Reaktion umfasst einen eliminativen Decarboxylierungsschritt, der direkt zur Bildung einer α,β-ungesättigten Carbonsäure (d. h. wobei Y=OH) führt. Sodann kann die Carbonsäure unter Anwendung der Standardchemie, die der Fachwelt bekannt ist, unter Bereitstellung von Estern (Y=OR2) oder Hydroxamiden (NHOR11), worin R11 eine geeignete Schutzgruppe, wie Benzyl, tert-Butyl- oder tert-Butyldimethylsilyl (TBDMS) ist, aufgearbeitet werden.
- Die Dicarbonsäuren der Formel (V) können durch Alkylierung beispielsweise von Diethylmalonat mit einem Alkylierungsmittel der Formel R1-Z (VI), worin Z wie vorstehend definiert ist, und anschließende Hydrolyse unter basischen Bedingungen hergestellt werden.
- Die Verbindungen der Formel (II), worin B eine Aryl-, Heteroarylfunktion oder eine andere Gruppe als Substituent an einem Kernteil davon (B') umfasst, können durch Paladium-katalysierte Kupplung einer Aryl-, Heteroarylfunktionalisierung oder einer anderen Verbindung mit einer Verbindung der allgemeinen Formel A-B'-XR12 (VII), worin R12 eine geeignete Schutzgruppe, wie Methyl, tert-Butyl, Benzyl oder Trityl und A ein Halogenid, wie Iodid, Bromid oder in einigen Fällen Chlorid, ist, hergestellt werden. Daran anschließend folgt die Entfernung sämtlicher Schutzgruppen.
- Viele solcher Palladium-katalysierten Kupplungsreaktionen sind der Fachwelt bekannt und können Verbindungen der Formel (II) bereitstellen, die Substituenten tragen, die durch R5 beschrieben sind, wie COR6, CO2R2 oder CON(R6)2, sowie Aryl-, Heteroaryl-, Alkenyl- oder Alkylgruppen, die gegebenenfalls mit R5 substituiert sind. Siehe beispielsweise Syn. Comm. (1981) 11: 513; Tet. Lett (1987) 28: 5093; und J. Org. Chem. (1994) 59: 6502. Weitere, durch B und/oder R5 beschriebene Gruppen können durch chemische Standardumwandlungen, die der Fachwelt bekannt sind, eingeführt werden.
- Viele Verbindungen der allgemeinen Formel (II), (VI) und (VII) sind im Handel erhältlich oder können durch aromatische, heteroaromatische oder eine andere, der Fachwelt bekannte Standardchemie aus im Handel erhältlichen Materialien hergestellt werden.
- Falls gewünscht, können die Zwischenstufen der allgemeinen Formel (VIII) und (IX)durch Friedel-Crafts-Acylierung eines einfachen aromatischen Systems Ph-XH (X) mit Phthalsäure- oder Maleinsäureanhydrid und anschließende Behandlung mit einem Hydrazin der allgemeinen Formel (H2N-NHR2 (XI) hergestellt werden.

- Die Verbindungen der Formel (I) können auch durch gegenseitige Umwandlung von anderen Verbindungen der Formel (I) hergestellt werden. So kann beispielsweise eine Verbindung der Formel (I), worin R1 eine C1-6-Alkylgruppe ist, durch Hydrierung (unter Verwendung von Palladium-auf-Kohle in einem geeigneten Lösungsmittel, wie einem Alkohol, z. B. Ethanol) einer Verbindung der Formel (I), worin R1 eine C2-6-Alkenylgruppe ist, hergestellt werden. Außerdem kann eine Verbindung der Formel (I), worin X S(O)1-2 ist, durch Oxidation einer Verbindung der Formel (I), worin X S ist, hergestellt werden.
- Die Carbonsäuren (Y=OH) können unter Anwendung von der Fachwelt bekannten Verfahren in Ester (Y=OR2) oder Hydroxamsäuren (Y=NHOH) übergeführt werden.
- Beliebige Gemische von Endprodukten oder erhaltenen Zwischenstufen können auf der Grundlage der physikalisch-chemischen Unterschiede der Bestandteile auf bekannte Weise, beispielsweise durch Chromatographie, Destillation, fraktionierte Kristallisation oder durch Bildung eines Salzes, falls unter Umständen angemessen oder möglich ist, in die reinen Endprodukte oder Zwischenstufen, aufgetrennt werden.
- Die erfindungsgemäßen Verbindungen zeigen bezüglich der Stromelysine, Collagenasen und Gelatinasen in vitro Inhibitoraktivitäten. Die erfindungsgemäßen Verbindungen zeigen auch eine in vitro Inhibition der TNF-Freisetzung, des TNF-Rezeptor-Shedding, des IL-6-Rezeptor-Shedding und des L-Selectin-Shedding.
- Der 80 kD TNF-Rezeptor (TNFR80) wird an der Zelloberfläche proteolytisch abgespalten (Shedding) und setzt ein lösliches, Liganden-bindendes Rezeptorfragment frei. Interessanterweise wurde gezeigt, dass die Bearbeitung von TNFα und das Shedding von TNFR80 in aktivierten T-Zellen gleichzeitig auftreten, was vermuten lässt, dass möglicherweise eine gemeinsame Protease beteiligt ist. Von Crowe et al., J. Exp. Med., (1995) 181: 1205 wurde gezeigt, dass ein synthetischer Inhibitor der TNF-Bearbeitung ebenfalls das Shedding von TNFR80 blockiert, was nahe legt, dass diese Prozesse während der T-Zellenaktivierung koordiniert reguliert werden können. Die Protease-Spaltstelle in pro-TNF(Ala-Val) ist nämlich auch in der extrazellulären Domäne von TNFR80 (Ala213-Val214) an einer Stelle vorhanden, die mit dem festgestellten Molekulargewicht des abgespaltenen Rezeptorfragments im Einklang steht. Somit können die Metalloproteinase-Inhibitoren auf zwei Ebenen gleichzeitig einen Schutz vor den nachteiligen systemischen Auswirkungen von TNFα bieten, erstens durch Verhinderung der Freisetzung von löslichem TNFα und zweitens durch Blockierung der Akkumulation von abgespaltenem TNFR80.
- In Synergie mit TNF hemmen die Metalloproteinaseinhibitoren auch die Freisetzung des APO-1/Fas(CD95)-Liganden (APO-1L), der die Apoptose in empfindlichen Zielzellen auslöst. Das Shedding von APO-1/Fas (CD95), ein Typ-I-Transmembranglycoprotein (das der Nervenwachstumsfaktor/TNF-Rezeptor-Unterfamilie angehört), wird ebenfalls durch die bekannten Metalloproteinaseninhibitoren, allerdings nicht durch die gemeinsamen Inhibitoren von Serin/Cysteinproteasen, blockiert; siehe Mariani et al., Eur. J. Immunol., (1995) 25: 2303. Von mehreren anderen wichtigen Rezeptoren, die durch aktivierte T- und B-Zellen exprimiert werden, wurde ebenfalls gezeigt, dass sie durch Einwirkung von Metalloproteinasen von der Zelloberfläche abgespalten werden. Diese Enzyme, die kollektiv als Sheddasen bekannt sind, sind die neuen Ziele für die Inhibitoren der Metalloproteinasen, einschließlich für die erfindungsgemäßen Verbindungen.
- Die Aktivität und Selektivität der Verbindungen kann durch die Verwendung des geeigneten Enzym-Inhibitionstests, beispielsweise wie in den Beispielen A–M nachstehend beschrieben, bestimmt werden. Bestimmte erfindungsgemäße Verbindungen besitzen eine selektive inhibitorische Aktivität, insbesondere eine Inhibition von MMP im wesentlichen ohne Inhibition der TNF-Freisetzung und verwandter Aktivitäten, wie vorstehend definiert. Dies kann von besonderem Wert sein, wo solche Aktivitäten mit verminderten Nebenwirkungen einhergehen.
- Die Erfindung betrifft auch ein Verfahren zur Behandlung von Patienten (einschließlich Mensch und/oder tierischen Säugern, die in der Molkerei-, Fleisch- oder Pelzindustrie oder als Haustiere gehalten werden), die an Beschwerden oder Krankheiten leiden, die dem Stromelysin zugeschrieben werden können, wie vorstehend beschrieben, und insbesondere ein Behandlungsverfahren, das die Verabreichung der Matrix-Metalloproteinase-Inhibitoren der Formel (I) als wirksame Bestandteile umfasst.
- Demnach können die Verbindungen der Formel (I) unter anderem bei der Behandlung von Osteoarthritis und rheumatoider Arthritis und bei Krankheiten und Indikationen, die aus der Überexpression dieser Matrix-Metalloproteinasen, wie sie in bestimmten metastatischen Tumor-Zelllinien gefunden werden, resultieren, eingesetzt werden.
- Wie vorstehend erwähnt, sind die Verbindungen der Formel (I) in der Human- oder Veterinärmedizin geeignet, da sie als Inhibitoren von TNF und MMPs wirksam sind. Demnach betrifft die Erfindung nach einem weiteren Aspekt:
- Ein Verfahren zur Handhabung (womit die Behandlung oder Prophylaxe gemeint ist) von Krankheiten oder Zuständen, die durch TNF und/oder MMPs in Säugern, insbesondere in Menschen, vermittelt werden, wobei das Verfahren die Verabreichung einer wirksamen Menge einer Verbindung der Formel (I), vorstehend, oder eines pharmazeutisch verträglichen Salzes davon an den Säuger umfasst; und
eine Verbindung der Formel (I) zur Verwendung in der Human- oder Veterinärmedizin, insbesondere zur Handhabung (womit die Behandlung oder Prophylaxe gemeint ist) von Krankheiten oder Zuständen, die durch TNF und/oder MMPs vermittelt werden; und
die Verwendung einer Verbindung der Formel (I) zur Herstellung eines Mittels zur Handhabung (womit die Behandlung oder Prophylaxe gemeint ist) von Krankheiten oder Zuständen, die durch TNF und/oder MMPs vermittelt werden. - Die Krankheit oder die Zustände, auf die oben Bezug genommen wurde, umfassen entzündliche Erkrankungen, Autoimmunerkrankungen, Krebs, Herz-Kreislauf-Erkrankungen, Erkrankungen, die einen Gewebeabbau einschließen, wie rheumatoide Arthritis, Osteoarthritis, Osteoporose, Neurodegeneration, Alzheimer-Krankheit, Schlaganfall, Vasculitis, Morbus Chrohn, ulcerative Colitis, Multiple Sklerose, Periodontitis, Gingivitis und diejenigen, die einen Gewebeabbau einschließen, wie Knochenresorption, Haemorrhagie, Koagulation, Akutphasen-Reaktion, Kachexie, Anorexie, akute Infektionen, HIV-Infektionen, Fieber, Schockzustände, Transplantat-gegen-Empfänger-Reaktionen, dermatologische Zustände, operative Wundheilung, Psoriasis, atopische Dermatitis, Epidermolysis bullosa, Tumorwachstum, Angiogenese und Invasion durch Sekundärmetastasen, ophthalmologische Erkrankung, Retinopathie, korneale Ulzeration, Reperfusionsverletzung, Migräne, Meningitis, Asthma, Rhinitis, allergische Konjunktivitis, Ekzem, Anaphylaxie, Restenose, kongestives Herzversagen, Endometriose, Artherosklerose, Endosklerose und Asapirin-unabhängige Antithrombose.
- Zur Behandlung der rheumatoiden Arthritis, Osteoarthritis und bei Krankheiten und Indikationen, die aus der Überexpression von Matrix-Metalloendoproteinasen, wie sie in bestimmten metastatischen Tumorzelllinien oder bei anderen Krankheiten vorkommen, die durch Matrix-Metalloendoproteinasen oder erhöhte TNF- Produktion vermittelt werden, resultieren, können die Verbindungen der Formel (I) oral, topisch, parenteral, durch Inhalationsspray oder rektal in Dosierungseinheitsformulierungen verabreicht werden, die nicht toxische pharmazeutisch verträgliche Träger, Hilfsstoffe und Vehikel enthalten. Der Begriff parenteral, wie hier verwendet, umfasst subkutane Injektionen, intravenöse, intramuskuläre, intrasternale Injektions- oder Infusionstechniken. Zusätzlich zu der Behandlung warmblütiger Tiere, wie Mäuse, Ratten, Pferde, Vieh, Schafe, Hunde, Katzen, etc., sind die erfindungsgemäßen Verbindungen bei der Behandlung von Menschen wirksam.
- Die pharmazeutische Zusammensetzung, die den Wirkstoff enthält, kann in einer zur oralen Verwendung geeigneten Form vorliegen, beispielsweise als Tabletten, Lutschpastillen, Pastillen, wässrige oder ölige Suspensionen, dispergierbare Pulver oder Granulatkörner, Emulsionen, harte oder weiche Kapseln oder Sirupe oder Elixiere. Die zur oralen Verwendung beabsichtigten Zusammensetzungen können durch jedes aus der Technik zur Herstellung pharmazeutischer Zusammensetzungen bekannte Verfahren hergestellt werden, und solche Zusammensetzungen können ein oder mehrere Mittel enthalten, die aus der Gruppe ausgewählt sind, bestehend aus Süßstoffen, Aromastoffen, Farbmitteln und Konservierungsmitteln, um pharmazeutisch elegante und wohlschmeckende Präparationen bereitzustellen. Tabletten enthalten den Wirkstoff in einem Gemisch mit nicht toxischen, pharmazeutisch verträglichen Hilfsstoffen, die zur Herstellung von Tabletten geeignet sind. Diese Hilfsstoffe können beispielsweise inerte Verdünnungsmittel, wie Calciumcarbonat, Natriumcarbonat, Lactose, Calciumphosphat oder Natriumphosphat; Granulierungs- und Zerfallshilfen, beispielsweise Maisstärke oder Alginsäure; Bindemittel, beispielsweise Stärke, Gelatine oder Akaziengummi und Gleitmittel, beispielsweise Magnesiumstearat, Stearinsäure oder Talk, sein. Die Tabletten können unbeschichtet sein, oder sie können durch bekannte Techniken zur Verzögerung von Zerfall und Absorption im Gastrointestinaltrakt beschichtet sein und dadurch über einen langen Zeitraum eine verzögerte Wirkung bereitstellen. Beispielsweise kann ein Zeitverzögerungsmaterial, wie Glycerylmonostearat oder Glyceryldistearat, eingesetzt werden. Sie können auch durch die in den US-Patentschriften 4 256 108; 4 166 452; und 4 265 874 beschriebenen Verfahren unter Bildung osmotischer therapeutischer Tabletten zur kontrollierten Freisetzung beschichtet werden.
- Die Formulierungen zur oralen Anwendung können auch als Hartgelatinekapseln, wo der Wirkstoff mit einem inerten festen Verdünnungsmittel, beispielsweise Calciumcarbonat, Calciumphosphat oder Kaolin, vermischt ist, oder als Weichgelatinekapseln, wo der Wirkstoff mit Wasser oder einem öligen Medium, beispielsweise Erdnussöl, Flüssigparaffin oder Olivenöl, vermischt ist, dargereicht werden.
- Wässrige Suspensionen enthalten die wirksamen Materialien in einem Gemisch mit zur Herstellung der wässrigen Suspensionen geeigneten Hilfsstoffen. Solche Hilfsstoffe sind Suspensionsmittel, beispielsweise Natriumcarboxymethylcellulose, Methylcellulose, Hydroxypropylmethylcellulose, Natriumalginatpolyvinylpyrrolidon, Tragacanth und Akaziengummi; die Dispersions- oder Netzmittel können ein natürlich vorkommendes Phosphatid sein, beispielsweise Lecithin oder Kondensationsprodukte eines Alkylenoxids mit Fettsäuren, beispielsweise Polyoxyethylenstearat, oder Kondensationsprodukte von Ethylenoxid mit langkettigen aliphatischen Alkoholen, beispielsweise Heptadecaethylenoxycetanol, oder Kondensationsprodukte von Ethylenoxid mit Teilestern, die sich von Fettsäuren, und einem Hexitol, wie Polyoxyethylen, ableiten, mit Teilestern, die sich von Fettsäuren und Hexitolanhydriden ableiten, beispielsweise Polyoxyethylensorbitanmonooleat. Die wässrigen Suspensionen können auch einen oder mehrere Konservierungsstoffe, beispielsweise Ethyl- oder n-Propyl-p-hydroxybenzoat, ein oder mehrere Farbmittel, ein oder mehrere Aromastoffe und ein oder mehrere Süßstoffe, wie Sacharose oder Sacharin, enthalten.
- Ölsuspensionen können durch Suspendieren des Wirkstoffs in einem Pflanzenöl, beispielsweise Erdnussöl, Olivenöl, Sesamöl oder Kokosöl, oder in einem Mineralöl, wie Flüssigparaffin, formuliert werden. Die Ölsuspensionen können ein Verdickungsmittel, beispielsweise Bienenwachs, Hartparaffin oder Cetylalkohol, enthalten. Süßstoffe, wie diejenigen, die vorstehend aufgeführt sind, und Aromastoffe können unter Bereitstellung einer wohlschmeckenden oralen Präparation zugesetzt werden. Diese Zusammensetzungen können durch die Zugabe eines Antioxidans, wie Ascorbinsäure, konserviert werden.
- Die dispergierbaren Pulver und Granulatkörner, die zur Herstellung einer wässrigen Suspension durch Zugabe von Wasser geeignet sind, stellen den Wirkstoff in einem Gemisch mit einem Dispersions- oder Netzmittel, Suspensionsmittel und einem oder mehreren Konservierungsstoffen bereit. Geeignete Dispersions- oder Netzmittel und Suspensionsmittel sind beispielhaft aufgeführt, beispielsweise können auch Füllstoffe, Aromastoffe und Farbmittel vorhanden sein.
- Die erfindungsgemäßen pharmazeutischen Zusammensetzungen können auch in Form von Öl-in-Wasser-Emulsionen vorliegen. Die ölige Phase kann ein Pflanzenöl, beispielsweise Olivenöl oder Erdnussöl oder ein Mineralöl, beispielsweise Flüssigparaffin, oder Gemische von diesen sein. Geeignete Emulgiermittel können natürlich vorkommende Gummen, beispielsweise Akaziengummi oder Tragacanth, natürlich vorkommende Phosphatide, beispielsweise Sojabohnen, Lecithin und Ester oder Teilester, die sich von Fettsäuren und Hexitolanhydriden ableiten, beispielsweise Sorbitanmonooleat, und die Kondensationsprodukte der Teilester mit Ethylenoxid, beispielsweise Polyoxyethylensorbitanmonooleat, sein. Die Emulsionen können auch Süß- und Aromastoffe enthalten.
- Die Sirupe und Elixiere können mit Süßstoffen formuliert sein, beispielsweise Glycerin, Propylenglycol, Sorbit oder Sacharose. Solche Formulierungen können auch ein Demulcens, einen Konservierungs- und Aromastoff und Farbmittel enthalten. Die pharmazeutischen Zusammensetzungen können auch in Form einer sterilen injizierbaren, wässrigen oder öligen Suspension vorliegen. Diese Suspension kann nach der bekannten Technik unter Verwendung der vorstehend erwähnten geeigneten Dispersions- oder Netzmittel und Suspensionsmittel formuliert werden. Die sterile injizierbare Präparation kann auch in einer sterilen injizierbaren Lösung oder Suspension in einem nicht toxischen, parenteral verträglichen Verdünnungs- oder Lösungsmittel, beispielsweise als Lösung in 1,3-Butandiol, vorliegen. Unter den verträglichen Hilfsstoffen und Lösungsmitteln, die eingesetzt werden können, sind Wasser, Ringer's Lösung und isotone Natriumchloridlösung. Zusätzlich werden üblicherweise sterile, nicht flüssige Öle als Lösungsmittel oder Suspensionsmedium eingesetzt. Für diesen Zweck kann jedes milde, nicht flüssige Öl, einschließlich von synthetischen Mono- oder Diglyceriden eingesetzt werden. Zusätzlich finden Fettsäuren, wie Ölsäure, bei der Herstellung von Injektionslösungen Anwendung.
- Die Verbindungen der Formel (I) können auch in Form von Suppositorien zur rektalen Verabreichung des Arzneimittels verabreicht werden. Die Zusammensetzungen können durch Mischen des Arzneimittels mit einem geeigneten, nicht reizenden Hilfsstoff, der bei gewöhnlichen Temperaturen fest, allerdings bei Rektaltemperatur flüssig ist, hergestellt werden, und sie schmelzen darum im Rektum unter Freisetzung des Arzneimittels. Solche Materialien sind Kakaobutter und Polyethylenglycole.
- Zur topischen Anwendung werden Cremes, Salben, Gele, Lösungen oder Suspensionen etc. eingesetzt, die die Verbindungen der Formel (I) enthalten. Für die Zwecke dieser Beschreibung umfasst die topische Anwendung Mundspülungen und Gurgellösungen.
- Bei der Behandlung der oben angegebenen Zustände sind Dosierungsniveaus in der Größenordnung von etwa 0,05 mg bis etwa 140 mg/kg Körpergewicht/Tag (etwa 2,5 mg bis etwa 7 g/Patient/Tag) geeignet. Beispielsweise kann eine Entzündung durch Verabreichung von etwa 0,01 bis 50 mg der Verbindung/kg/Körpergewicht/Tag (etwa 0,5 mg bis etwa 3,5 g/Patient/Tag) wirksam behandelt werden.
- Die Menge des Wirkstoffs, der mit den Trägermaterialien unter Herstellung einer Einzeldosierungsform kombiniert werden kann, variiert in Abhängigkeit von dem behandelten Wirt und der bestimmten Verabreichungsweise. Beispielsweise kann eine Formulierung, die zur oralen Verabreichung an Menschen beabsichtigt ist, von etwa 5 bis etwa 95% der Gesamtzusammensetzung variieren.
- Dosierungseinheitsformen enthalten in der Regel etwa 1 mg bis etwa 500 mg Wirkstoff.
- Allerdings hängt das spezifische Dosisniveau für einen bestimmten Patienten selbstverständlich von einer Vielzahl von Faktoren ab, einschließlich Aktivität der bestimmten eingesetzten Verbindung, Alter, Körpergewicht, allgemeiner Gesundheitszustand, Geschlecht, Ernährungsgewohnheit, Zeit der Verabreichung, Verabreichungsweg, Ausscheidungsrate, Arzneimittelkombination und Schwere der bestimmten Krankheit, die der Therapie unterzogen wird.
- Die folgenden Beispiele erläutern die Erfindung.
- In den Beispielen werden die folgenden Abkürzungen verwendet:
TNFα Tumornekrosefaktor α LPS Lipopolysaccharid ELISA Enzymimmunosorbens-Assay EDC 1-Ethyl-2-dimethylaminopropylcarbodiimid RT Raumtemperatur - Zwischenstufe 1: 1-(2-Bromethyl)-3,4,4-trimethylhydantoin
- Natriumhydrid (60% Dispersion in Mineralöl, 1,7 g) wurde einer Lösung von 3,4,4-Trimethylhydantoin (5,5 g) in DMF (20 ml) bei Raumtemperatur zugesetzt. Das Gemisch wurde 30 min gerührt, anschließend wurde Dibromethan (3,5 ml) zugetropft, und die Lösung wurde über Nacht gerührt. Das Gemisch wurde zu Wasser (200 ml) gegeben und mit Diethylether extrahiert. Die etherische Schicht wurde mit Wasser gewaschen, getrocknet (MgSO4) und eingedampft, und der Rückstand wurde durch Säulenchromatographie gereinigt, wobei mit Diethylether unter Erhalt der Titelverbindung (4,2 g, 50%) als farbloser Feststoff eluiert wurde.
TLC-Rf 0,5 (Diethylether). - Zwischenstufe 2: 4-Acetylsulfanyltetrahydropyran
- Diethylazodicarboxylat (7,24 ml) wurde einer gerührten Lösung von Triphenylphosphin (12,1 g) in THF (80 ml) zugesetzt und bei Raumtemperatur 10 min gerührt. Thioessigsäure (3,29 ml) und Tetrahydro-4H-pyran-4-ol (2,3 g) wurden zugesetzt (VORSICHT: exotherm), und das Gemisch wurde bei Raumtemperatur weitere 18 h gerührt. Nach Entfernung des THF unter vermindertem Druck ergab Rühren mit Hexan (50 ml) und Wasser (50 ml) einen Niederschlag, der durch Filtration entfernt wurde. Das zweiphasige Filtrat wurde getrennt und die wässrige Phase einmal mit Hexan (50 ml) extrahiert. Die vereinigten Hexanextrakte wurden mit Wasser (30 ml), Salzlösung (10 ml) gewaschen, getrocknet (MgSO4) und eingedampft, wobei eine gelbe Flüssigkeit (2,73 g) zurückblieb. Die Reinigung durch Chromatographie über Kieselgel, wobei mit Hexan/Diethylether (10 : 1) eluiert wurde, lieferte die Titelverbindung als blassgelbe Flüssigkeit (0,98 g, 27%).
TLC-Rf 0,20 (Hexan/Diethylether (10 : 1)). - Zwischenstufe 3: Tetrahydropyran-4-thiol
- Natriumborhydrid (0,278 g) wurde einer gerührten Lösung von Zwischenstufe 2 in Methanol (15 ml) bei 0°C unter Stickstoff zugesetzt. Nach 2 h wurde eine weitere Portion Natriumborhydrid (0,370 g) zugesetzt, und das Gemisch wurde auf Raumtemperatur aufwärmen gelassen und weitere 18 h gerührt. Die Reaktion wurde anschließend mit 1 M Chlorwasserstoffsäure (50 ml) gestoppt und mit Diethylether (2 × 30 ml) extrahiert. Die vereinigten organischen Extrakte wurden mit Salzlösung (10 ml) gewaschen, getrocknet (MgSO4) und unter Bereitstellung der Titelverbindung als farblose Flüssigkeit (0,472 g, 60%) in vacuo konzentriert.
TLC-Rf 0,70 (Hexan/Ethylacetat (3 : 1)). - Zwischenstufe 4: 1-Benzoyl-4-brompiperidin
- Benzoylchlorid (1,41 g), gefolgt von Triethylamin (2,2 g, 2,2 Äq.) wurde einer Lösung von 4-Brompiperidinhydrobromid (2,45 g) in THF (30 ml) bei 0°C zugesetzt. Die Lösung wurde 1 h bei Raumtemperatur gerührt und eingedampft. Der Rückstand wurde in Dichlormethan (100 ml) gelöst, mit Wasser, 1 M HCl und gesättigtem Natriumbicarbonat gewaschen, getrocknet und unter Erhalt der Titelverbindung als farbloses Öl (2,76 g, 100%) eingedampft.
TLC-Rf 0,55 (Ether). - Zwischenstufe 5: 1-Benzoyl-4-(acetylsulfanyl)piperidin
- Kaliumthioacetat (2,3 g) wurde einer Lösung von Zwischenstufe 4 in DMF (50 ml) bei Raumtemperatur zugesetzt. Das Gemisch wurde 3 Tage bei Raumtemperatur gerührt, anschließend zu Wasser gegeben und mit Ether extrahiert. Das Lösungsmittel wurde mit Wasser und Natriumbicarbonatlösung gewaschen, getrocknet und unter Erhalt der Titelverbindung als blass bernsteinfarbenes Öl (2,6 g, 100%) eingedampft.
TLC-Rf 0,45 (Ether). - Zwischenstufe 6: 1-Benzoylpiperidin-4-thiol
- Eine Lösung der Thioacetat-Zwischenstufe 5 (2,6 g) in Methanol (100 ml) wurde mit Natriumborhydrid (1,2 g) behandelt, und das Gemisch wurde bei Raumtemperatur 4 h gerührt, anschließend eingedampft und der Rückstand in Wasser aufgelöst. Die Lösung wurde mit fester Citronensäure angesäuert und mit Dichlormethan (2 × 100 ml) extrahiert. Das Lösungsmittel wurde mit Salzlösung gewaschen, getrocknet und unter Erhalt der Titelverbindung als braunes Öl (2,0 g) eindampft.
TLC-Rf 0,30 (Ether). - Zwischenstufe 7: 1-tert-Butyloxycarbonyl-4-brompiperidin
- Eine Lösung von Di-tert-butyldicarbonat (4,4 g) in Dichlormethan, gefolgt von Triethylamin (5,1 g), wurde einer Suspension von 4-Brompiperidinhydrobromid (5 g) in Dichlormethan (100 ml) bei 0°C zugesetzt. Die Lösung wurde 3 h bei Raumtemperatur gerührt, anschließend mit Wasser, 0,5 M HCl und gesättigtem Natriumbicarbonat gewaschen, getrocknet und unter Erhalt der Titelverbindung als farbloses Öl (5,3 g, 99%) eingedampft.
TLC-Rf 0,70 (Ether). - Zwischenstufe 8: 1-tert-Butyloxycarbonyl-4-(acetylsulfanyl)piperidin
- Kaliumthioacetat (4,4 g) wurde einer Lösung von Zwischenstufe 7 (5,3 g) in DMF (100 ml) zugesetzt, und das Gemisch wurde 4 h auf 100°C erhitzt, anschließend abgekühlt und zu Wasser gegeben. Das Gemisch wurde mit Ether extrahiert und das Lösungsmittel anschließend mit Wasser und Natriumbicarbonat gewaschen, getrocknet und unter Erhalt der Titelverbindung (5,10 g, 96%) eingedampft.
TLC-Rf 0,60 (Ether). - Zwischenstufe 9: Dibenzyl-2-(3-phthalimidopropyl)malonat
- Eine Lösung von Dibenzylmalonat (20 g) in wasserfreiem THF (200 ml) wurde bei 0°C mit Natriumhydrid (60% Dispersion in Mineralöl, 3,1 g) behandelt. Das Gemisch wurde auf Raumtemperatur aufwärmen gelassen und 30 min unter Stickstoff gerührt. Sodann wurde das Gemisch mit einer Lösung von N-(3-Brompropyl)phthalimid (20,5 g) in THF (100 ml) behandelt und 12 h unter Rückfluss erhitzt. Anschließend wurde die Reaktion gekühlt, filtriert und das Filtrat in vacuo eingedampft. Der Rückstand wurde zwischen Ethylacetat (200 ml) und gesättigtem wässrigen Ammoniumchlorid (150 ml) verteilt. Die organische Schicht wurde mit Wasser (100 ml), Salzlösung (100 ml) gewaschen, getrocknet (MgSO4), filtriert und das Filtrat in vacuo eingedampft. Der Rückstand wurde durch Säulenchromatographie über Kieselgel gereinigt, wobei mit 20% Ethylacetat in Hexan unter Erhalt der Titelverbindung (17,5 g, 50%) als weißer Feststoff eluiert wurde.
TLCRf 0,16 (20% Ethylacetat-Hexan). - Gleichermaßen hergestellt wurden:
- Zwischenstufe 10: Dibenzyl-(2-(3,4,4-trimethylhydantoin-1-yl)ethyl)malonat
- Aus Dibenzylmalonat (4,8 g) und Zwischenstufe 1 (4,2 g) als farbloses Öl (7,2 g, 100%).
TLC-Rf 0,4 (Diethylether). - Zwischenstufe 11: Dibenzyl-(3-phenylpropyl)malonat
- Aus Dibenzylmalonat (30 g) und 1-Brom-3-phenylpropan (21 g) als farbloses Öl (34 g, 80%).
TLC-Rf 0,48 (20% Ethylacetat-Hexan). - Zwischenstufe 12: 2-Methylen-5-phthalimidopentansäure
- Eine Lösung von Zwischenstufe 9 (16,75 g) in Dioxan (200 ml) wurde mit 10% Palladium-auf-Kohle (1,7 g) behandelt und bis zum Aufhören der Wasserstoffaufnahme bei Atmosphärendruck hydriert. Der Katalysator wurde durch Filtration über Celite® entfernt und das Filtrat bei Raumtemperatur mit Piperidin (3,2 g) behandelt. Nach 30 min wurde die Reaktion mit Formaldehyd (37% Lösung in Wasser, 15 ml) behandelt, 2 h bei Raumtemperatur gerührt und anschließend 2 h auf 80°C erwärmt. Das Gemisch wurde gekühlt, das Lösungsmittel in vacuo entfernt und der Rückstand zwischen Ethylacetat (200 ml) und 10% wässriger Citronensäure (100 ml) verteilt. Die organische Schicht wurde mit Wasser (100 ml), Salzlösung (100 ml) gewaschen, über Magnesiumsulfat getrocknet, filtriert und das Filtrat in vacuo eingedampft. Der Rückstand wurde durch Säulenchromatographie über Kieselgel gereinigt, wobei mit 40% Ethylacetat in Hexan unter Erhalt der Titelverbindung (12,2 g, 70%) als weißer Feststoff eluiert wurde.
TLC-Rf 0,44 (50% Ethylacetat-Hexan). - Gleichermaßen wurden hergestellt:
- Zwischenstufe 13: 2-Methylen-4-(3,4,4-trimethylhydantoin-1-yl)butansäure
- Aus Zwischenstufe 10 (7,2 g) als farbloser Feststoff (1,5 g, 37%).
TLC-Rf 0,35 (Ethylacetat). - Zwischenstufe 14: 2-Methylen-5-phenylpentansäure
- Aus Zwischenstufe 11 (17,6 g) als farbloses Öl (5,0 g, 60%).
TLC-Rf 0,31 (20% Ethylacetat-Hexan). - Zwischenstufe 15: 2-Brommethyl-5-phthalimidopentansäure
- Zwischenstufe 12 (1,0 g) wurde mit 45% Bromwasserstoff in Essigsäure (30 ml) bei Raumtemperatur behandelt. Nach 3 h wurde die Lösung in Wasser (300 ml) gegossen und das Produkt mit Ethylacetat (3 × 100 ml) extrahiert. Die Extrakte wurden vereinigt, mit Wasser (100 ml), Salzlösung (100 ml) gewaschen, über Magnesiumsulfat getrocknet, filtriert und das Filtrat in vacuo eingedampft. Der Rückstand wurde mit Toluol (2 × 10 ml) unter Erhalt der Titelverbindung als weißer Feststoff (1,3 g, 100%) azeotrop getrocknet.
TLC-Rf 0,34 (50% Ethylacetat-Hexan). - Gleichermaßen wurde hergestellt:
- Zwischenstufe 16: 2-Brommethyl-5-phenylpentansäure
- Aus Zwischenstufe 14 (3,4 g) als farbloses Öl (4,2 g, 87%).
TLC-Rf 0,32 (20% Ethylacetat-Hexan). - Zwischenstufe 17: Methyl-2-(Brommethyl)-5-phenylpentanoat
- Zwischenstufe 16 (5,0 g) wurde mit einer Lösung von Diazomethan in Diethylether behandelt. Die Entfernung des Lösungsmittels in vacuo ergab die Titelverbindung als farbloses Öl (5,2 g, 100%).
TLC-Rf 0,31 (5% Ethylacetat-Hexan). - Zwischenstufe 18: Methyl-2-Acetylsulfanylmethyl-5-phenylpentanoat
- Eine Lösung von Zwischenstufe 17 in DMF wurde mit Kaliumthioacetat (6,0 g) 18 h bei 60°C behandelt. Das Gemisch wurde zu Wasser gegeben und mit Ether extrahiert, anschließend wurde das Lösungsmittel mit Wasser und Salzlösung gewaschen, getrocknet und unter Erhalt der Titelverbindung als braunes Öl (8,5 g, 90%) eingedampft.
TLC-Rf 0,80 (Ether). - Zwischenstufe 19: Methyl-2-Chlorsulfonylmethyl-5-phenylpentanoat
- Chlor wurde 30 min durch eine Suspension von Zwischenstufe 18 (0,7 g) in Eiswasser (20 ml) geleitet. Sodann wurde die gelbe Suspension mit Dichlormethan extrahiert, und das Lösungsmittel wurde mit Wasser, Natriummetabisulfit und Salzlösung gewaschen, anschließend getrocknet und unter Erhalt der Titelverbindung (0,7 g, 100%) als farbloses Öl eingedampft.
TLC-Rf 0,45 (Ether). - Zwischenstufe 20: 1-tert-Butoxycarbonylpiperidin-4-thiol
- Natriumborhydrid (5,0 g) wurde einer Lösung von Zwischenstufe 8 (5,1 g) in Methanol (200 ml) zugesetzt. Die Lösung wurde 2 h bei Raumtemperatur gerührt und sodann in vacuo eingedampft. Der Rückstand wurde sorgfältig in Wasser gelöst und Citronensäure zugesetzt (5,0 g). Das Produkt wurde mit Dichlormethan (2 × 100 ml) extrahiert, die Extrakte wurden vereinigt, über Magnesiumsulfat getrocknet, filtriert, und das Filtrat wurde unter Erhalt der Titelverbindung als blassgelbes Öl (4,70 g, 98%) in vacuo eingedampft.
TLC-Rf 0,40 (Ether). - Zwischenstufe 21: 2-(4-Benzoylphenylsulfanylmethyl)-5-phthalimidopentansäure
- Eine Lösung von Zwischenstufe 15 (1,0 g) in THF (20 ml) wurde 5 min mit Stickstoff gespült und mit Zwischenstufe 15 (0,7 g) und Triethylamin (1,0 ml) behandelt. Das Gemisch wurde bei Raumtemperatur 24 h gerührt und sodann das Lösungsmittel in vacuo entfernt. Der Rückstand wurde zwischen Ethylacetat (100 ml) und Wasser (100 ml) verteilt. Die wässrige Schicht wurde mit Ethylacetat (50 ml) extrahiert, die Extrakte vereinigt, mit Salzlösung gewaschen, über Magnesiumsulfat getrocknet und filtriert. Das Filtrat wurde in vacuo eingedampft, und der Rückstand wurde durch Säulenchromatographie über Kieselgel gereinigt, wobei unter Erhalt der Titelverbindung (0,5 g, 35%) als klarer Gummi mit 50% Ethylacetat in Hexan eluiert wurde.
TLC-Rf 0,33 (50% Ethylacetat-Hexan). - Zwischenstufe 22: 2-(Furan-2-ylmethylsulfanyl)methyl-5-phenylpentansäure
- Kalium-tert-butoxid (415 mg) wurde einer gerührten Lösung von Furfurylthiol (0,186 ml) in Dichlormethan (20 ml) bei 0°C unter einer Stickstoffatmosphäre zugesetzt. Das Gemisch wurde 3 min gerührt, bevor eine Lösung von Zwischenstufe 37 (500 mg) in Dichlormethan (5 ml) zugesetzt wurde. Das Gemisch wurde auf Raumtemperatur aufwärmen gelassen und 6 h gerührt, bevor es in HCl (0,5 M, 100 ml) gegossen wurde. Das Gemisch wurde mit Ethylacetat (2 × 100 ml) extrahiert. Die vereinigten organischen Phasen wurden mit Salzlösung gewaschen und getrocknet (MgSO4). Das Lösungsmittel wurde unter Erhalt der Titelverbindung als gelbes Öl (560 mg, 99%) in vacuo entfernt.
TLC-Rf 0,3 (4 : 1 Hexan-Ethylacetat). - Gleichermaßen wurde hergestellt:
- Zwischenstufe 23: 5-Phenyl-2-[(tetrahydropyran-4-ylsulfanyl)methyl]-pentansäure
- Aus Zwischenstufe 3 (0,40 g) und Zwischenstufe 16 (0,700 g) als blassgelbes Öl (0,99 g, 94%).
TLC-Rf 0,50 (Hexan/Diethylether (1 : 1)).
MS 373 (M + NH4 +) - Zwischenstufe 24: 2-(2-Phenylethylsulfanylmethyl)-5-phenylpentansäure
- Kaliumhexamethyldisilazid (0,5 M in Toluol, 20 ml) wurde einer Lösung von Phenethylmercaptan (0,7 g) in THF (50 ml) bei –78°C zugesetzt, und die Lösung wurde 10 min gerührt, anschließend wurde eine Lösung der Zwischenstufe 16 (1,4 g) in THF (10 ml) zugetropft, und das Gemisch wurde unter Aufwärmen auf Raumtemperatur 3 h gerührt. Das Gemisch wurde zu Wasser gegeben, und die Lösung wurde mit Diethylether gewaschen, mit Essigsäure angesäuert und mit Ether extrahiert. Die etherische Schicht wurde mit Wasser und Salzlösung gewaschen, getrocknet und eingedampft und der Rückstand durch Säulenchromatographie gereinigt, wobei unter Erhalt der Titelverbindung 1,30 g, 78%) als farbloses Öl mit 20% Ethylacetat in Hexan eluiert wurde.
TLC-Rf 0,3 (20% Ethylacetat-Hexan). - Gleichermaßen hergestellt wurden:
- Zwischenstufe 25: 2-(1-Benzoylpiperidin-4-yl)sulfanylmethyl-5-phenylpentansäure
- Aus Zwischenstufe 6 (2,0 g) und Zwischenstufe 16 (2,0 g) als farbloses Öl (1,6 g).
TLC-Rf 0,34 (Ether). - Zwischenstufe 26: 2-((1-tert-Butyloxycarbonyl)piperidin-4-yl)sulfanylmethyl-5-phenylpentansäure
- Aus Zwischenstufe 20 (4,2 g) und Zwischenstufe 16 (5,2 g) als farbloses Öl (4,5 g, 53%).
TLC-Rf 0,3 (Ethylether/Hexane). - Zwischenstufe 27: 2-(4-Benzoylphenylsulfonylmethyl)-5-phthalimidopentansäure
- Eine Lösung von Zwischenstufe 21 (0,4 g) in Methanol (50 ml) wurde bei Raumtemperatur mit einer Lösung von Oxone® (0,52 g) in Wasser (10 ml) behandelt. Das Gemisch wurde 24 h bei Raumtemperatur gerührt und das organische Lösungsmittel in vacuo entfernt. Der Rückstand wurde mit Wasser (100 ml) verdünnt und mit Ethylacetat (3 × 100 ml) extrahiert. Die organischen Extrakte wurden vereinigt, mit Salzlösung (40 ml) gewaschen, über Magnesiumsulfat getrocknet und filtriert. Das Filtrat wurde in vacuo eingedampft, und der Rückstand wurde durch Säulenchromatographie über Kieselgel gereinigt, wobei unter Erhalt der Titelverbindung als weißer Feststoff (0,37 g, 87%) mit 2% Methanol in Dichlormethan eluiert wurde.
TLC-Rf 0,24 (2% Methanol-Dichlormethan).
MS 506 MH+ - Gleichermaßen wurden hergestellt:
- Zwischenstufe 28: 5-Phenyl-2-((tetrahydropyran-4-ylsulfonyl)methyl)-pentansäure
- Aus Zwischenstufe 23 (0,99 g) als wachsartiger, weißer Feststoff (0,62 g, 57%).
TLC-Rf 0,10 (Ethylacetat/Hexan/Essigsäure (49 : 49 : 2)). - Zwischenstufe 29: 2-((1-Benzoylpiperidin-4-yl)sulfonylmethyl)-5-phenylpentansäure
- Aus Zwischenstufe 25 (1,6 g) als weißer Feststoff (1,05 g, 26%).
TLC-Rf 0,57 (EtOAc).
MS 443 (M+) - Zwischenstufe 30: 2-((1-tert-Butyloxycarbonylpiperidin-4-yl)sulfonylmethyl)-5-phenylpentansäure
- Oxon® (13 g) wurde einer Lösung von Zwischenstufe 26 (4,5 g) und Natriumacetat (5 g) in Methanol (200 ml) und Wasser (50 ml) bei Raumtemperatur zugesetzt, und das Gemisch wurde 18 h gerührt, anschließend eingedampft, mit Wasser verdünnt und das Produkt unter Erhalt der Titelverbindung als weißer Feststoff (3,50 g, 75%) durch Filtration gesammelt.
TLC-Rf 0,35 (EtOAc).
MS 439 (M+) - Zwischenstufe 31: Methyl-2-((pyrrolidin-1-yl)sulfonylmethyl)-5-phenyl-pentanoat
- Pyrrolidin (0,2 ml) wurde einer Lösung von Zwischenstufe 19 (0,7 g) und Triethylamin (0,5 ml) in Dichlormethan (20 ml) bei –10°C zugesetzt, und die Lösung wurde 18 h gerührt, anschließend mit Wasser, Natriumbicarbonat und 0,5 M HCl gewaschen. Das Lösungsmittel wurde getrocknet und eingedampft, und der Rückstand wurde durch Flashchromatographie gereinigt, wobei unter Erhalt der Titelverbindung als farbloses Öl (0,16 g, 25%) mit 40% Ether/Hexanen eluiert wurde.
TLC-Rf 0,28 (40% Ether/Hexane). - Zwischenstufe 32: 2-((Pyrrolidin-1-yl)sufonylmethyl)-5-phenylpentansäure
- Lithiumhydroxid (100 mg) wurde einer Lösung der Ester-Zwischenstufe 31 (0,16 g) in Methanol (5 ml) THF (10 ml) und Wasser (5 ml) zugesetzt, und die Lösung wurde 6 h gerührt, anschließend in vacuo eingedampft und der Rückstand in Wasser gelöst. Die wässrige Lösung wurde mit Ether gewaschen, anschließend mit Citronensäure angesäuert und mit Dichlormethan extrahiert. Die Dichlormethanextrakte wurden vereinigt und mit Wasser gewaschen, getrocknet und unter Erhalt des Rohprodukts eingedampft. Das Material wurde durch Flashchromatographie gereinigt, wobei unter Erhalt der Titelverbindung als farbloses Öl (60 mg, 40%) mit 60% Ether/Hexanen eluiert wurde.
TLC-Rf 0,40 (60% Ether/Hexane). - Zwischenstufe 33: 2-(4-Benzoylphenylsulfonylmethyl)-5-phthalimidopentansäure-N-hydroxyamid
- Eine Lösung von Zwischenstufe 27 (0,1 g) in wasserfreiem THF (10 ml) wurde bei Raumtemperatur mit O-(tert-Butyldimethylsilyl)hydroxylamin (0,032 g) und 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimidhydrochlorid (0,042 g) behandelt. Das Gemisch wurde 24 h gerührt und das organische Lösungsmittel in vacuo entfernt. Der Rückstand wurde zwischen Wasser (30 ml) und Ethylacetat (50 ml) verteilt. Die organischen Extrakt wurden mit Salzlösung (40 ml) gewaschen, über Magnesiumsulfat getrocknet und filtriert. Das Filtrat wurde in vacuo eingedampft und der Rückstand in THF (10 ml) gelöst. Die Lösung wurde bei 0°C mit einer 1,0 M Lösung von Tetrabutylammoniumfluorid in THF (1,0 ml) behandelt und 1 h gerührt. Das Lösungsmittel wurde in vacuo entfernt und der Rückstand zwischen Ethylacetat (50 ml) und Wasser (40 ml) verteilt. Die organische Schicht wurde mit Salzlösung (20 ml) gewaschen, über Magnesiumsulfat getrocknet, filtriert und das Filtrat in vacuo eingedampft. Der Rückstand wurde durch Säulenchromatographie über Kieselgel gereinigt, wobei unter Erhalt der Titelverbindung als weißer Feststoff (0,06 g, 58%) mit 2% Methanol in Dichlormethan eluiert wurde.
TLC-Rf 0,74 (Ethylacetat).
MS 521 MH+ - Gleichermaßen wurden hergestellt:
- Beispiel 1: 5-Phenyl-2-((tetrahydropyran-4-yl-sulfonyl)methyl)-pentansäure-N-hydroxyamid
- Aus Zwischenstufe 28 (0,60 g) als weißer Feststoff (0,40 g, 65%).
TLC-Rf 0,20 (Ethylacetat/Hexan (5 : 1) mit Spuren von Essigsäure).
MS 373 (M + NH4 +). - Beispiel 2: 2-((1-Benzoylpiperidin-4-yl)sulfonylmethyl)-5-phenylpentansäure-N-hydroxyamid
- Aus Zwischenstufe 29 (1,05 g) als weißer Feststoff (0,85 g, 80%).
TLC-Rf 0,43 (7% MeOH/Dichlormethan).
MS 458 (M+). - Beispiel 3: 2-((1-tert-Butyloxycarbonylpiperidin-4-yl)sulfonylmethyl)-5-phenylpentansäure-N-hydroxyamid
- Aus Zwischenstufe 30 (3,5 g) als weißer Schaum (2,40 g, 66%).
TLC-Rf 0,7 (7% MeOH/Dichlormethan).
MS 454 (M+). - Beispiel 4: 2-((Pyrrolidin-1-yl)sulfonylmethyl)-5-phenylpentansäure-N-hydroxamid
- Aus Zwischenstufe 32 (60 mg) als farbloser Feststoff (30 mg, 50%).
TLC-Rf 0,40 (7% MeOH/Dichlormethan).
MS 340 (M+). - Beispiel 5: 2-((Piperidin-4-yl)sulfonylmethyl)-5-phenylpentansäure-N-hydroxamidtrifluoracetat
- Trifluoressigsäure (20 ml) wurde einer Lösung der Säure von Beispiel 3 (2,4 g) in Dichlormethan (20 ml) bei Raumtemperatur zugesetzt, und die Lösung wurde 2 h gerührt, anschließend in vacuo eingedampft und der Rückstand mit Toluol (2 × 50 ml) azeotrop gemacht. Anschließend wurde der resultierende Feststoff durch Zugabe von Ether unter Erhalt der Titelverbindung (1,54 g, 65%) aus Methanol ausgefällt.
TLC-Rf 0,25 (10% MeOH/Dichlormethan 2% NH4OH).
MS 323 (MH+). - Beispiel 6: 2-((1-Benzylpiperidin-4-yl)sulfonylmethyl)-5-phenylpentansäure-N-hydroxamid
- Natriumtriacetoxyborhydrid (0,22 g) wurde einer Lösung von Beispiel 5 (0,23 g), Triethylamin (60 mg) und Benzaldehyd (106 mg) in einem Gemisch von Dichlormethan (10 ml) und Methanol (2 ml) zugesetzt. Das Gemisch wurde bei Raumtemperatur 6 h gerührt, anschließend in vacuo eingedampft und der Rückstand zwischen Wasser und Dichlormethan verteilt. Das organische Lösungsmittel wurde mit Wasser gewaschen, getrocknet und eingedampft und der Rückstand durch Chromatographie gereinigt, wobei unter Erhalt der Titelverbindung als weißer Feststoff (60 mg, 25%) mit 6% MeOH in Dichlormethan und 1% NH4OH eluiert wurde.
TLC-Rf 0,35 (6% MeOH/Dichlormethan 1% NH4OH).
MS 445 (M+). - Gleichermaßen wurde hergestellt:
- Beispiel 7: 2-((1-(4-Cyanobenzyl)piperidin-4-yl)sulfonylmethyl)-5-phenylpentansäure-N-hydroxyamid
- Aus Beispiel 5 (0,23 g) und 4-Cyanobenzaldehyd (65 mg) als weißer Feststoff (35 mg, 14%).
TLC-Rf 0,40 (6% MeOH/Dichlormethan).
MS 470 (MH+). - Beispiel A
- Collagenase-inhibitorische Aktivität
- Die Eignung der Verbindungen der allgemeinen Formel (I) als Inhibitoren von Collagenase zu wirken wurde durch die Verfahrensweise von Cawston und Barrett, (Anal. Biochem., 99: 340–345, 1979) bestimmt, wobei eine 1 mM Lösung des zu testenden Inhibitors oder Verdünnungen davon bei 37°C 16 h mit Collagen und Collagenase (gepuffert mit 50 mM Tris, pH 7,6, enthaltend 5 mM CaCl2, 0,05% Brij 35, 60 mM NaCl und 0,02% NaN3) inkubiert wurden. Das Collagen war acetyliertes 3H- oder 14C-Collagen, das durch das Verfahren von Cawston und Murphy (Methods in Enzymolgy, 80: 711, 1981) hergestellt wurde. Die Wahl der radioaktiven Markierung veränderte die Fähigkeit der Collagenase zum Abbau des Collagensubstrats nicht. Die Proben wurden unter Sedimentation des unverdauten Collagens zentrifugiert, und ein Aliquot des radioaktiven Überstands wurde zum Test auf einem Scintillationszähler als Maß für die Hydrolyse entnommen. Die Collagenaseaktivität in Gegenwart von 1 mM Inhibitor oder einer Verdünnung davon wurde mit der Aktivität in einer Kontrolle, in der der Inhibitor fehlte, verglichen, und die Ergebnisse als die Inhibitorkonzentration angegeben, die 50% Inhibition der Collagenase (IC50) bewirkte.
- Beispiel B
- Stromelysin-inhibitorische Aktivität
- Die Eignung der Verbindungen der allgemeinen Formel (I) als Stromelysin-Inhibitoren zu wirken wurde unter Anwendung der Verfahrensweise von Nagase et al (Methods in Enzymology Bd. 254, 1994) bestimmt, wobei eine 0,1 mM Lösung des zu testenden Inhibitors oder Verdünnungen davon bei 37°C 16 h mit Stromelysin und 3H-Transferrin (gepuffert mit 50 mM Tris, pH 7,6, enthaltend 10 mM CaCl2, 150 M NaCl, 0,05% Brij 35 und 0,02% NaN3) inkubiert wurden. Das Transferrin war mit 3H-Iodessigsäure carboxymethyliert. Die Stromelysinaktivität in Gegenwart von 1 mM oder einer Verdünnung davon wurde mit der Aktivität in einer Kontrolle, der Inhibitor fehlte, verglichen, und die Ergebnisse wurden als die Inhibitorkonzentration angegeben, die 50% Inhibition des Stromelysins (IC50) bewirkte.
- Beispiel C
- Gelatinase-inhibitorische Aktivität
- Die Eignung der Verbindungen der allgemeinen Formel (I) als Inhibitoren der Gelatinase zu wirken wurde unter Anwendung der Verfahrensweise von Harris & Krane (Biochem Biophys. Acta, 258: 566–576, 1972) bestimmt, wobei eine 1 mM Lösung des zu testenden Inhibitors oder Verdünnungen davon bei 37°C 16 h mit Gelatinase und Wärme-denaturiertem 3H- oder 14C-acetyliertem Collagen (gepuffert mit 50 mM Tris, pH 7,6, enthaltend 5 mM CaCl2, 0,05% Brij 35 und 0,02% NaN3) inkubiert wurden. Das 3H- oder 14C-Gelatine wurde durch Denaturieren von 3H- oder 14C-Collagen, das nach dem Verfahren von Cawston und Murphy (Methods in Enzymology, 80: 711, 1981) erzeugt wurde, durch Inkubation bei 60°C für 30 min hergestellt. Unverdaute Gelatine wurde durch Zugabe von Trichloressigsäure und Zentrifugation ausgefällt. Die Gelatinaseaktivität in Gegenwart von 1 mM oder einer Verdünnung davon wurde mit der Aktivität in einer Kontrolle, der Inhibitor fehlte, verglichen, und die Ergebnisse wurden als die Inhibitorkonzentration angegeben, die 50% Inhibition der Gelatinase (IC50) bewirkte.
- Beispiel D
- MMP-Inhibitoraktivität-Fluorimetrietest
- Die Eignung der Verbindungen der allgemeinen Formel (I) als Inhibitoren von Collagenase-1(MMP-1), Collagenase-2(MMP-8), Collagenase-3(MMP-13), Gelatinase-A(MMP-2), Gelatinase-B(MMP-9) und Stromelysin-1(MMP-3) zu wirken wurde unter Anwendung der folgenden Verfahrensweise bestimmt:
- Die Inhibitoren werden in Dimethylsulfoxid gelöst, das 0,02% β-Mercaptoethanol enthält, und serielle Verdünnungen werden hergestellt. Aktiviertes Enzym wird in Testpuffer, der 50 mM Tris, pH 7,4, 5 mM CaCl2, 0,002% NaN3 und Brij 35 enthält, in Gegenwart und Abwesenheit von Inhibitor inkubiert. Die Proben werden vor Zugabe des Fluorimetriesubstrats (Mca-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2) bis auf eine Endkonzentration von 10 μM 15 min bei 37°C vorinkubiert. Der Test wird 20–30 min bei 37°C inkubiert und anschließend bei λcx (340 nm) und λcm (405 nm) in einem Fluoroscan II gelesen.
- Die Enzymaktivität wurde mit der Aktivität in einer Kontrolle, der Inhibitor fehlte, verglichen, und die Ergebnisse werden als die Inhibitorkonzentration angegeben, die 50% Inhibition des Stromelysins (IC50) bewirkte.
- Beispiel E
- Inhibition der TNFα-Produktion
- Die Eignung der Verbindungen der allgemeinen Formel (I) als Inhibitoren der TNFα-Produktion zu wirken wird unter Anwendung der folgenden Verfahrensweise bestimmt. 100 μM Lösung des zu testenden Inhibitors oder Verdünnungen davon werden bei 37°C in einer Atmosphäre von 5% CO2 mit THP-1-Zellen (menschliche Monocyten), suspendiert in RPM1-1640-Medium und 20 μM β-Mercaptoethanol bei einer Zelldichte von 1 × 106/ml, inkubiert und mit LPS stimuliert. Nach 18 h wird der Überstand unter Verwendung eines handelsüblichen ELISA-Kit (R & D-Systems) auf die TNFα-Spiegel getestet.
- Die Aktivität in Gegenwart von 0,01 mM Inhibitor oder von Verdünnungen davon wird mit der Aktivität in einer Kontrolle, der Inhibitor fehlt, verglichen und die Ergebnisse werden als die Inhibitorkonzentration angegeben, die 50% Inhibition der TNFα-Produktion bewirkt.
- Beispiel F
- Inhibition des L-Selectin-Shedding
- Die Verbindungen der allgemeinen Formel (I) werden in einem Test des L-Selectin-Shedding durch periphere, mononukleäre Blutzellen (PBMC) bewertet. Die PBMC werden durch Standardverfahrensweisen unter Verwendung von Ficoll aus der Leukozyten- und Erythrozyten-Schicht isoliert. Eine 100 μM Lösung des zu testenden Inhibitors oder Verdünnungen davon werden 20 min bei 37°C in einer Atmosphäre von 5% CO2, mit 4 × 106/ml PBMC, stimuliert mit PMA, inkubiert. Die Zellen werden abzentrifugiert und die Überstände unter Verwendung eines handelsüblichen ELISA-Kit (R & D-Systems) auf L-Selectin getestet.
- Die Aktivität in Gegenwart von 100 μM Inhibitor oder Verdünnungen davon wurde mit der Aktivität in einer Kontrolle, in der Inhibitor fehlte, verglichen, und die Ergebnisse werden als die Inhibitorkonzentration angegeben, die 50% Inhibition des Shedding von sL-Selectin bewirkt.
- Beispiel G
- Inhibition des sII-1RII-Shedding
- Verbindungen der allgemeinen Formel (I) werden in einem Test des sII-1RII-Shedding durch periphere mononukleäre Blutzellen (PBMC) bewertet. Die PBMC werden aus den Leukozyten- und Erythrozyten-Schichten durch Standardverfahrensweisen unter Verwendung von Ficoll isoliert. Eine 100 μM Lösung des zu testenden Inhibitors oder Verdünnungen davon werden 18 h bei 37°C in einer Atmosphäre von 5% CO2 mit 2 × 106/ml PBMC, stimuliert mit II-13, inkubiert. Die Zellen werden abzentrifugiert und die Überstände unter Verwendung eines handelsüblichen ELISA-Kit (R & D-Systems) auf sII-1RII getestet.
- Die Aktivität in Gegenwart von 100 μm Inhibitor oder Verdünnungen davon wird mit der Aktivität in einer Kontrolle, der Inhibitor fehlte, verglichen, und die Ergebnisse werden als die Inhibitorkonzentration angegeben, die 50% Inhibition des Shedding von sII-1RII bewirkt.
- Beispiel H
- Inhibition des II-6R-Shedding
- Die Verbindungen der allgemeinen Formel (I) werden in einem Test des sII-6R-Shedding durch HL-60-Zellen bewertet. Die PBMC werden aus den Leukozyten- und Erythrozyten-Schichten durch Standardverfahrensweisen unter Verwendung von Ficoll isoliert. Eine 100 μM Lösung des zu testenden Inhibitors oder Verdünnungen davon werden 24 h bei 37°C in einer Atmosphäre von 5% CO2 mit 2 × 106/ml HL-60-Zellen, stimuliert mit PMA, inkubiert. Die Zellen werden abzentrifugiert und die Überstände unter Verwendung eines handelsüblichen ELISA-Kit (R & D-Systems) auf sII-6R getestet.
- Die Aktivität in Gegenwart von 100 μM Inhibitor oder von Verdünnungen davon wird mit der Aktivität in einer Kontrolle, der Inhibitor fehlte, verglichen, und die Ergebnisse werden als Inhibitorkonzentration angegeben, die 50% Inhibition des Shedding von II-6R bewirkt.
- Beispiel I
- Inhibition des TNF-RII-Shedding
- Die Eignung der Verbindungen der allgemeinen Formel (I) als Inhibitoren des Shedding von TNF RII zu wirken wird unter Anwendung des folgenden Verfahrens bestimmt. Eine 100 μM Lösung des testenden Inhibitors oder Verdünnungen davon werden bei 37°C in einer Atmosphäre von 5% CO2 mit THP-1-Zellen (menschliche Monocyten), suspendiert in RPM1 1640-Medium und 20 μm β-Mercaptoethanol bei einer Zelldichte von 1 × 106/ml, suspendiert und mit LPS stimuliert. Nach 18 h wird der Überstand unter Verwendung eines im Handel erhältlichen ELISA-Kit (R & D-Systems) auf die sTNF-RII-Spiegel getestet.
- Die Aktivität in Gegenwart von 0,1 mM Inhibitor oder von Verdünnungen davon wird mit der Aktivität in einer Kontrolle, der Inhibitor fehlt, verglichen, und die Ergebnisse werden als die Inhibitorkonzentration angegeben, die 50% Inhibition des TNF-RII-Shedding bewirkt.
- Beispiel J
- Arthritisches Ratten-Hilfsmodell
- Die Verbindungen der allgemeinen Formel (I) wurden in einem Arthritis-Hilfsmodell in Ratten auf der Grundlage der Verfahren bewertet, die von B. B. Newbould (1963), Br. J. Pharmacol, 21, 127–136 und C. M. Pearson und F. D. Wood (1959), Arthritis Rheum, 2, 440–459 eingesetzt wurden. Kurz gesagt, wurde männlichen Wistar-Ratten (180–200 g) Freund's Adjuvans in die Schwanzwurzel injiziert. 12 Tage später wurden die reagierenden Tiere statistisch in Testgruppen aufgeteilt. Die Verbindungen der allgemeinen Formel (I) wurden entweder oral als Suspension in 1% Methylcellulose oder intraperitoneal in 0,2% Carboxymethylcellulose vom Tag 12 bis zum Ende des Experiments am Tag 22 dosiert. Die Hinterpfotenvolumina wurden alle 2 Tage von Tag 12 an gemessen, und vom Hinterfuß wurden nach Abschluss des Experiments Röntgenaufnahmen angefertigt. Die Ergebnisse wurden in Werten der prozentualen Zunahme des Fußvolumens über 12 Tage ausgedrückt.
- Beispiel K
- Ovarialkarzinom-Xenograft-Mausmodell
- Die Verbindungen der allgemeinen Formel (I) wurden in einem Ovarialkarzinom-Xenograftmodell für Krebs auf der Grundlage des Modells bewertet, das von B. Davies et al (1993), Cancer Research, 53, 2087–2091 beschrieben wurde. Dieses Modell besteht kurz gesagt darin, männlichen nu/nu-Mäusen 1 × 109 OVCAR3-icr-Zellen in die Peritonealhöhle zu impfen. Die Verbindungen der allgemeinen Formel (I) werden oral als Suspension in 1% Methylcellulose oder intraperitoneal als Suspension in phosphatgepufferter Salzlösung in 0,01% Tween-20 verabreicht. Am Schluss des Experiments (4–5 Wochen) wird die Anzahl der Peritonealzellen gezählt, und alle festen Tumorablagerungen werden gewogen. In einigen Experimenten wird die Tumorentwicklung durch Messen von tumorspezifischen Antigenen registriert.
- Beispiel L
- Mammakarzinom-Rattenmodell
- Die Verbindungen der allgemeinen Formel (I) wurden in einem HOSP.1-Ratten-Mammakarzinom-Modell für Krebs (S. Eccles et al. (1995), Cancer Research, im Druck) bewertet. Dieses Modell besteht darin, weiblichen CBH/cbi-Ratten 2 × 104 Tumorzellen intravenös in die Jugularvene zu impfen. Die Verbindungen der allgemeinen Formel (I) werden oral als Suspension in 1% Methylcellulose oder intraperitoneal als Suspension in phosphatgepufferter Salzlösung + 0,01% Tween 20 verabreicht. Am Schluss des Experiments (4–5 Wochen) werden die Tiere getötet, die Lungen werden entnommen und die einzelnen Tumore nach 20 h Fixierung in Methacarn gezählt.
- Beispiel M
- B16-Melanom-Mausmodell
- Das antimetastatische Potential der Verbindungen der allgemeinen Formel (I) wird in einem B16-Melanom-Modell in C57BL/6 bewertet. Den Mäusen werden intravenös 2 × 105 B16/F10 Maus-Tumorzellen, geerntet aus in vitro Kulturen, injiziert. Die Inhibitoren werden oral als Suspension in 1% Methylcellulose oder intraperitoneal als Suspension in phosphatgepufferter Salzlösung, pH 7,2 + 0,01 Tween 20, verabreicht. Die Mäuse werden 14 Tage nach dem Zellen-Impfen getötet und die Lungen entfernt und vor dem Fixieren in Bouin's Lösung gewogen. Sodann wird die Anzahl von Kolonien, die auf der Oberfläche von jeder Serie von Lungen vorhanden waren, visuell gezählt.
Claims (24)
- Verbindung der Formel (I)mit der Maßgabe, dass B nicht Benzhydryl ist, wenn X SO ist, und R1 H ist; R5 C1-6-Alkyl, R7-C2-6-Alkenyl, Halogen, CN, NO2, N(R6)2, OR6, COR6, CO2R2, CON(R6)2, NR6R7, S(O)0-2R8 oder SO2N(R6)2 ist; R6 H oder eine Gruppe ist, die ausgewählt ist aus C1-6-Alkyl, Aryl, Aryl-C1-6-alkyl, Heteroaryl, Heteroaryl-C1-6-alkyl, Cycloalkyl, Cycloalkyl-C1-6-alkyl, Heterocycloalkyl und Heterocycloalkyl-C1-6-alkyl, worin die Gruppe gegebenenfalls substituiert ist mit R8, COR8, SO0-2R8, CO2R8, OR8, CONR2R8, NR2R8, Halogen, CN, SO2NR2R8 oder NO2, und für jeden Fall von N(R6)2 die R6-Gruppen gleich oder verschieden sind oder N(R6)2 Heterocycloalkyl ist, das gegebenenfalls substituiert ist mit R8, COR8, SO0-2R8, CO2R8, OR8, CONR2R8, NR2R8, Halogen, CN, SO2NR2R8 oder NO2; R7 COR6, CON(R6)2, CO2R8 oder SO2R8 ist; R8 C1-6-Alkyl, Aryl, Aryl-C1-6-alkyl, Heteroaryl oder Heteroaryl-C1-6-alkyl ist; und R9 OR6, COR6, CO2R2, NR6R7, S(O)0-2R8, SO2N(R6)2, Phthalimido, Succinimido oder die Gruppe
 ist; oder ein Salz, Solvat, Hydrat oder ein geschütztes Amino- oder geschütztes Carboxyderivat davon.
ist; oder ein Salz, Solvat, Hydrat oder ein geschütztes Amino- oder geschütztes Carboxyderivat davon.
- Verbindung nach Anspruch 1, worin R1 H oder gegebenenfalls substituiertes Alkyl, Alkenyl, Aryl, Arylalkyl, Heteroaryl oder Alkylheteroaryl ist; B Heterocycloalkyl ist, das gegebenenfalls substituiert ist mit einem Substituenten, der ausgewählt ist aus R5, R5-C1-6-Alkyl, R5-C2-6-Alkenyl, Aryl (gegebenenfalls substituiert mit R5), R5-C1-6-Alkylaryl, R7-C2-6-Alkenylaryl, Heteroaryl (gegebenenfalls substituiert mit R5), Cycloalkyl (gegebenenfalls substituiert mit R5), Benzo-kondensiertem C5-6-Cycloalkyl (gegebenenfalls substituiert mit R5), Heterocycloalkyl (gegebenenfalls substituiert mit R5), Benzo-kondensiertem C5-6-Heterocycloalkyl (gegebenenfalls substituiert mit R5) und den GruppenR5 C1-6-Alkyl, R5-C2-6-Alkenyl, Halogen, CN, NO2, N(R6)2, OR6, R7OC1-4-Alkyl, COR6, CO2R2, CON(R6)2, NR6R7, S(O)0-2R8 oder SO2N(R6)2 ist; und R6 H, Alkyl, Aryl, Arylalkyl, Heteroaryl oder Heteroarylalkyl ist.
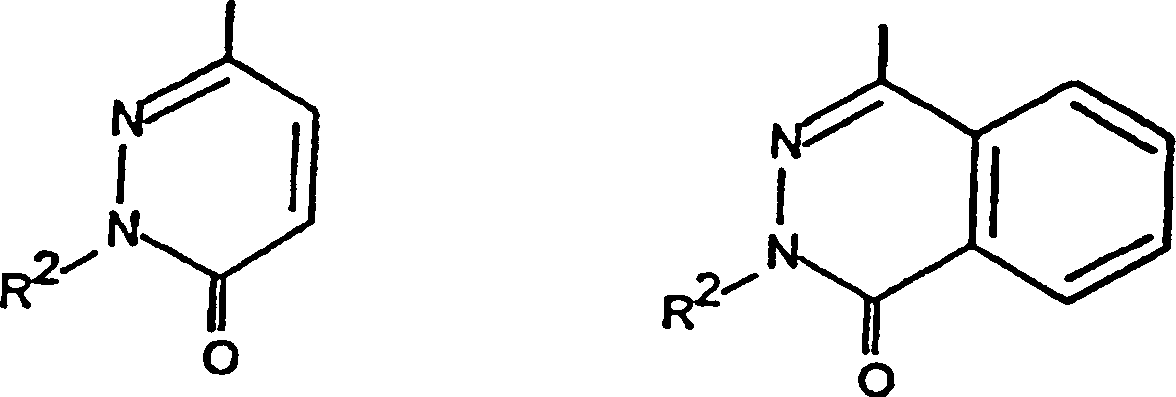
- Verbindung nach Anspruch 1 oder Anspruch 2, worin X S(O)0-2 ist.
- Verbindung nach einem vorhergehenden Anspruch, worin R1 H oder eine Gruppe (gegebenenfalls substituiert mit R9) ist, die ausgewählt ist aus C1-6-Alkyl, C2-6-Alkenyl, Aryl-C1-6-alkyl, Heteroaryl-C1-6-alkyl, Heterocycloalkyl-C1-6-alkyl, Cycloalkyl-C1-6-alkyl.
- Verbindung nach einem vorhergehenden Anspruch worin B Cycloalkenyl oder Heterocycloalkenyl ist, wovon jede der Gruppen gegebenenfalls mit einem Substituenten substituiert ist, der ausgewählt ist aus R5, R5-C1-6-Alkyl, Aryl (gegebenenfalls substituiert mit R5), R5-C1-6-Alkylaryl, Aryl-C1-6-alkyl (gegebenenfalls substituiert mit R5), Heteroaryl-C1-6-alkyl (gegebenenfalls substituiert mit R5), Heteroaryl (gegebenenfalls substituiert mit R5), R5-C1-6-Alkylheteroaryl, R5-C1-6-Alkylheteroaryl, Cycloalkyl (gegebenenfalls substituiert mit R5), Benzo-kondensiertem Cycloalkyl (gegebenenfalls substituiert mit R5), Heterocycloalkyl (gegebenenfalls substituiert mit R5), Benzo-kondensiertem Heterocycloalkyl (gegebenenfalls substituiert mit R5), und den Gruppen:
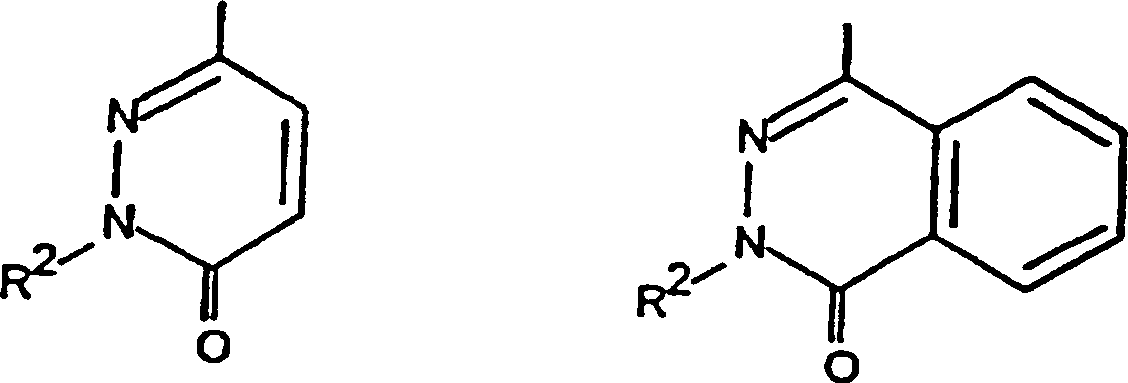
- Verbindung nach einem vorhergehenden Anspruch, worin R5 Halogen, CN, NO2, N(R6)2, OR6, COR6, CON(R6)2, NR6R7, oder S(O)0-2R8 ist.
- Verbindung nach einem vorhergehenden Anspruch, worin R7 COR6 ist.
- Verbindung nach einem vorhergehenden Anspruch, worin R9 OR6, CO2R2, Phthalimido, Succinimido oder die genannte Gruppe ist.
- Verbindung nach einem vorhergehenden Anspruch, worin m 0 ist; R5 C1-6-Alkyl, C2-6-Alkenyl, Halogen, CN, NO2, N(R6)2, OR6, COR6, CO2R2, CON(R6)2, NR6R7, S(O)0-2R8 oder SO2N(R6)2 ist; und R9 OR6, COR6, CO2R2, CON(R6)2, NR6R7, S(O)0-2R8, SO2N(R6)2, Phthalimido oder Succinimido ist.
- Verbindung nach einem vorhergehenden Anspruch, worin X SO2 ist; und R5 COR6 ist.
- Verbindung nach einem vorhergehenden Anspruch, worin X SO2 ist; und B Cycloalkenyl, Heteroycloalkenyl, Heterocycloalkyl oder Heterocycloalkyl-C1-6-alkyl ist, wovon jede Gruppe gegebenenfalls mit einem Substituenten substituiert ist, der ausgewählt ist aus R5, R5-C1-6-Alkyl, R5-C2-6-Alkenyl, Aryl (gegebenenfalls substituiert mit R5), R5-C1-6-Alkylaryl, Aryl-C1-6-alkyl (gegebenenfalls substituiert mit R5), Heteroaryl-C1-6-alkyl (gegebenenfalls substituiert mit R5), R7-C2-6-Alkenylaryl, Heteroaryl (gegebenenfalls substituiert mit R5), R5-C2-6-Alkylheteroaryl, Cycloalkyl (gegebenenfalls substituiert mit R5), Benzo-kondensiertem Cycloalkyl (gegebenenfalls substituiert mit R5), Heterocycloalkyl (gegebenenfalls substituiert mit R5) und Benzo-kondensiertem Heterocycloalkyl (gegebenenfalls substituiert mit R5), mit der Maßgabe, dass B kein unsubstituiertes Cycloalkyl ist.
- Verbindung, die 4-[4-(Hydroxyaminocarbonyl)methoxy-3,5-dimethylphenyl]-2-methyl-1(2H)-phthalazinon ist.
- Verbindung, die ausgewählt ist aus 5-Phenyl-2-((tetrahydropyran-4-ylsulfonyl)methyl)-pentansäure-N-hydroxyamid; 2-((1-Benzoylpiperidin-4-yl)sulfonylmethyl)-5-phenylpentansäure-N-hydroxyamid; 2-((1-tert-Butyloxycarbonylpiperidin-4-yl)sulfonylmethyl)-5-phenylpentansäure-N-hydroxyamid; 2-((Piperidin-4-yl)sulfonylmethyl)-5-phenylpentansäure-N-hydroxyamidtrifluoracetat; 2-((1-Benzylpiperidin-4-yl)sulfonylmethyl)-5-phenylpentansäure-N-hydroxyamid; 2-((1-(4-Cyanobenzyl)piperidin-4-yl)sulfonylmethyl)-5-phenylpentansäure-N-hydroxyamid.
- Verbindung nach einem vorhergehenden Anspruch in Form eines einzelnen Enantiomers oder Diastereomers.
- Pharmazeutische Zusammensetzung zur Verwendung in der Therapie, umfassend eine Verbindung nach einem vorhergehenden Anspruch und ein pharmazeutisch verträgliches Verdünnungsmittel oder einen pharmazeutisch verträglichen Träger.
- Verwendung einer Verbindung nach einem der Ansprüche 1–13 zur Herstellung eines Medikaments zur Behandlung oder Prävention eines Zustands, der mit Matrix-Metalloproteinasen im Zusammenhang steht oder der durch TNF-α oder Enzyme, die an der Ausschüttung von L-Selectin beteiligt sind, die TNF-Rezeptoren oder IL-6-Rezeptoren vermittelt wird.
- Verwendung nach Anspruch 16, wobei der Zustand ausgewählt ist aus Krebs, Entzündung und entzündlichen Krankheiten, Gewebedegeneration, Periodontitis, Augenkrankheit, dermatologischen Erkrankungen, Fieber, cardiovaskulären Wirkungen, Hämorrhagie, Koagulation und Akute-Phasen-Reaktion, Kachexie, Anorexie, akuter Infektion, HIV-Infektion, Schockzuständen, Transplantat-gegen-Empfänger-Reaktionen, Autoimmunkrankheit, Reperfusionsverletzung, Meningitis, Migräne und Aspirinunabhängiger Antithrombose.
- Verwendung nach Anspruch 16, wobei der Zustand ausgewählt ist aus Tumorwachstum, Angiogenese, Tumorinvasion und -ausbreitung, Metastasen, maligner Ascites und malignem Pleuraerguss.
- Verwendung nach Anspruch 16, wobei der Zustand ausgewählt ist aus zerebraler Ischämie, ischämischer Herzkrankheit, rheumatoider Arthritis, Osteoarthritis, Osteoporose, Asthma, Multipler Sklerose, Neurodegeneration, Alzheimer Krankheit, Atherosklerose, Schlaganfall, Vaskulitis, Morbus Crohn und ulzerativer Kolitis.
- Verwendung nach Anspruch 16, wobei der Zustand ausgewählt ist aus Kornea-Ulceration, Retinopathie und operativer Wundheilung.
- Verwendung nach Anspruch 16, wobei der Zustand ausgewählt ist aus Psoriasis, atopischer Dermatitis, chronischen Krebserkrankungen und Epidermolysis bullosa.
- Verwendung nach Anspruch 16, wobei der Zustand ausgewählt ist aus Periodontitis und Gingivitis.
- Verwendung nach Anspruch 16, wobei der Zustand ausgewählt ist aus Rhinitis, allergischer Konjunktivitis, Ekzem und Anaphylaxie.
- Verwendung nach Anspruch 16, wobei der Zustand ausgewählt ist aus Restenose, kongestivem Herzversagen, Endometriose, Atherosklerose und Endosklerose.
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB9616599 | 1996-08-07 | ||
| GBGB9616599.8A GB9616599D0 (en) | 1996-08-07 | 1996-08-07 | Compounds |
| GBGB9707427.2A GB9707427D0 (en) | 1997-04-11 | 1997-04-11 | Compounds having mmp and tnf inhibitory activity |
| GB9707427 | 1997-04-11 | ||
| PCT/GB1997/002129 WO1998005635A1 (en) | 1996-08-07 | 1997-08-07 | Hydroxamic and carboxylic acid derivatives having mmp and tnf inhibitory activity |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE69729007D1 DE69729007D1 (de) | 2004-06-09 |
| DE69729007T2 true DE69729007T2 (de) | 2005-04-07 |
Family
ID=26309836
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE69729007T Expired - Fee Related DE69729007T2 (de) | 1996-08-07 | 1997-08-07 | Hydroxamsäure- und carbonsäure-derivate mit mmp und tnf hemmender wirkung |
Country Status (17)
| Country | Link |
|---|---|
| US (1) | US6118001A (de) |
| EP (1) | EP0968182B1 (de) |
| JP (1) | JP2000517297A (de) |
| KR (1) | KR20000029858A (de) |
| CN (1) | CN1227540A (de) |
| AT (1) | ATE266000T1 (de) |
| AU (1) | AU730464B2 (de) |
| BR (1) | BR9711027A (de) |
| CA (1) | CA2263154A1 (de) |
| CZ (1) | CZ297278B6 (de) |
| DE (1) | DE69729007T2 (de) |
| DK (1) | DK0968182T3 (de) |
| ES (1) | ES2217425T3 (de) |
| NO (1) | NO314452B1 (de) |
| PL (1) | PL193829B1 (de) |
| PT (1) | PT968182E (de) |
| WO (1) | WO1998005635A1 (de) |
Families Citing this family (96)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0960096B1 (de) | 1997-01-22 | 2005-04-06 | Aventis Pharmaceuticals Inc. | Substituierte beta-thiocarbonsäuren |
| WO1998039316A1 (en) | 1997-03-04 | 1998-09-11 | Monsanto Company | N-hydroxy 4-sulfonyl butanamide compounds |
| ES2209122T3 (es) * | 1997-03-04 | 2004-06-16 | Pharmacia Corporation | Compuestos de acido sulfonil alfa-hidroxi hidroxamico aromatico. |
| US7115632B1 (en) | 1999-05-12 | 2006-10-03 | G. D. Searle & Co. | Sulfonyl aryl or heteroaryl hydroxamic acid compounds |
| US6696449B2 (en) | 1997-03-04 | 2004-02-24 | Pharmacia Corporation | Sulfonyl aryl hydroxamates and their use as matrix metalloprotease inhibitors |
| US6794511B2 (en) * | 1997-03-04 | 2004-09-21 | G. D. Searle | Sulfonyl aryl or heteroaryl hydroxamic acid compounds |
| AUPO721997A0 (en) * | 1997-06-06 | 1997-07-03 | Queensland Institute Of Medical Research, The | Anticancer compounds |
| US20030166687A1 (en) | 1997-11-21 | 2003-09-04 | Warpehoski Martha A. | Alpha-hydroxy,-amino and -fluoro derivatives of beta-sulphonyl hydroxamic acids as matrix metalloproteinases inhibitors |
| DE69939190D1 (de) | 1998-06-18 | 2008-09-04 | Hoffmann La Roche | Verfahren für Arylalkylsulfid |
| FR2780402B1 (fr) * | 1998-06-30 | 2001-04-27 | Adir | Nouveaux composes acides carboxyliques et hydroxamiques inhibiteurs de metalloproteases, leur procede de preparation et les compositions pharmaceutiques qui les contiennent |
| WO2000012477A1 (en) * | 1998-08-29 | 2000-03-09 | British Biotech Pharmaceuticals Limited | Hydroxamic acid derivatives as proteinase inhibitors |
| GB9919776D0 (en) * | 1998-08-31 | 1999-10-27 | Zeneca Ltd | Compoujnds |
| US6858598B1 (en) | 1998-12-23 | 2005-02-22 | G. D. Searle & Co. | Method of using a matrix metalloproteinase inhibitor and one or more antineoplastic agents as a combination therapy in the treatment of neoplasia |
| GB9911073D0 (en) * | 1999-05-12 | 1999-07-14 | Darwin Discovery Ltd | Hydroxamic and carboxylic acid derivatives |
| GB9911075D0 (en) | 1999-05-12 | 1999-07-14 | Darwin Discovery Ltd | HYdroxamic and carboxylic acid derivatives |
| GB9916562D0 (en) | 1999-07-14 | 1999-09-15 | Pharmacia & Upjohn Spa | 3-Arylsulfonyl-2-(substituted-methyl) propanoic acid derivatives as matrix metalloproteinase inhibitora |
| GB9922825D0 (en) * | 1999-09-25 | 1999-11-24 | Smithkline Beecham Biolog | Medical use |
| US6667316B1 (en) * | 1999-11-12 | 2003-12-23 | Celgene Corporation | Pharmaceutically active isoindoline derivatives |
| AU3385401A (en) * | 2000-02-21 | 2001-09-03 | Astrazeneca Ab | Arylpiperazines and arylpiperidines and their use as metalloproteinase inhibiting agents |
| GB0009760D0 (en) | 2000-04-19 | 2000-06-07 | Oxford Biomedica Ltd | Method |
| JP2003533525A (ja) | 2000-05-15 | 2003-11-11 | ダーウィン・ディスカバリー・リミテッド | Mmpおよびtnf阻害活性を有するヒドロキサム酸およびカルボン酸誘導体 |
| ES2208595T3 (es) | 2000-05-15 | 2004-06-16 | Darwin Discovery Limited | Derivados del acido hidroxamico. |
| WO2001087844A1 (en) * | 2000-05-15 | 2001-11-22 | Darwin Discovery Limited | Hydroxamic and carboxylic acid derivatives having mmp and tnf inhibitory activity |
| GB0020498D0 (en) | 2000-08-18 | 2000-10-11 | Sterix Ltd | Compound |
| US7842727B2 (en) | 2001-03-27 | 2010-11-30 | Errant Gene Therapeutics, Llc | Histone deacetylase inhibitors |
| CA2344208A1 (en) | 2001-04-30 | 2002-10-30 | Oxford Biomedica (Uk) Limited | Method |
| GB0119474D0 (en) | 2001-08-09 | 2001-10-03 | Astrazeneca Ab | Compounds |
| AU2003247390A1 (en) * | 2002-05-22 | 2003-12-12 | Errant Gene Therapeutics, Llc. | Histone deacetylase inhibitors based on alphachalcogenmethylcarbonyl compounds |
| AU2003243272A1 (en) | 2002-05-22 | 2003-12-12 | Errant Gene Therapeutics, Llc | Histone deacetylase inhibitors based on alpha-ketoepoxide compounds |
| DE10226370B4 (de) | 2002-06-13 | 2008-12-11 | Polyic Gmbh & Co. Kg | Substrat für ein elektronisches Bauteil, Verwendung des Substrates, Verfahren zur Erhöhung der Ladungsträgermobilität und Organischer Feld-Effekt Transistor (OFET) |
| DE10233085B4 (de) | 2002-07-19 | 2014-02-20 | Dendron Gmbh | Stent mit Führungsdraht |
| US8425549B2 (en) | 2002-07-23 | 2013-04-23 | Reverse Medical Corporation | Systems and methods for removing obstructive matter from body lumens and treating vascular defects |
| AU2003261319A1 (en) | 2002-08-01 | 2004-02-23 | Bristol-Myers Squibb Company | Hydantoin derivatives as inhibitors of matrix metalloproteinases and/or tnf-alpha converting enzyme |
| JP2006516548A (ja) | 2002-12-30 | 2006-07-06 | アンジオテック インターナショナル アクツィエン ゲゼルシャフト | 迅速ゲル化ポリマー組成物からの薬物送達法 |
| GB0411562D0 (en) | 2004-05-24 | 2004-06-23 | Sterix Ltd | Compound |
| GB0412492D0 (en) | 2004-06-04 | 2004-07-07 | Sterix Ltd | Compound |
| US20050277897A1 (en) * | 2004-06-14 | 2005-12-15 | Ghannoum Ziad R | Handpiece tip |
| DE102005019181A1 (de) * | 2005-04-25 | 2006-10-26 | Novartis Ag | Peptid-Deformylase (PDF) Inhibitoren 1 |
| RU2409575C2 (ru) * | 2005-04-25 | 2011-01-20 | Новартис Аг | ИМИДАЗО[1,2-a]ПИРИДИНОВЫЕ ПРОИЗВОДНЫЕ, КОТОРЫЕ МОЖНО ИСПОЛЬЗОВАТЬ В КАЧЕСТВЕ ИНГИБИТОРОВ ПЕПТИДДЕФОРМИЛАЗЫ (ПДФ) |
| HUE027013T2 (en) | 2005-05-27 | 2016-10-28 | Ospedale San Raffaele Srl | A gene vector containing MI-RNA |
| GB0513702D0 (en) | 2005-07-04 | 2005-08-10 | Sterix Ltd | Compound |
| GB0526210D0 (en) | 2005-12-22 | 2006-02-01 | Oxford Biomedica Ltd | Vectors |
| GB0604142D0 (en) | 2006-03-01 | 2006-04-12 | Sterix Ltd | Compound |
| WO2007141029A1 (en) * | 2006-06-08 | 2007-12-13 | Helmholtz Zentrum München Deutsches Forschungszentrum Für Gesundheit Und Umwelt (Gmbh) | Specific protease inhibitors and their use in cancer therapy |
| US9873667B2 (en) | 2006-07-27 | 2018-01-23 | Emisphere Technologies Inc. | Arylsulfanyl compounds and compositions for delivering active agents |
| GB0706072D0 (en) | 2007-03-28 | 2007-05-09 | Sterix Ltd | Compound |
| US8088140B2 (en) | 2008-05-19 | 2012-01-03 | Mindframe, Inc. | Blood flow restorative and embolus removal methods |
| US11337714B2 (en) | 2007-10-17 | 2022-05-24 | Covidien Lp | Restoring blood flow and clot removal during acute ischemic stroke |
| GB0722779D0 (en) | 2007-11-20 | 2008-01-02 | Sterix Ltd | Compound |
| EP3266391B1 (de) | 2008-02-22 | 2019-05-01 | Covidien LP | Vorrichtung zur flusswiederherstellung |
| EP3192874B1 (de) | 2008-06-18 | 2019-10-16 | Oxford BioMedica (UK) Limited | Virusreinigung |
| EP2364362B1 (de) | 2008-11-12 | 2015-10-21 | Ospedale San Raffaele S.r.l. | Genvektor zur induktion transgenspezifischer immuntoleranz |
| AU2010243276B2 (en) | 2009-04-30 | 2016-09-15 | Fondazione Telethon Ets | Gene vector |
| GB0914767D0 (en) | 2009-08-24 | 2009-09-30 | Sterix Ltd | Compound |
| US9039749B2 (en) | 2010-10-01 | 2015-05-26 | Covidien Lp | Methods and apparatuses for flow restoration and implanting members in the human body |
| WO2012156839A2 (en) | 2011-05-19 | 2012-11-22 | Ospedale San Raffaele S.R.L. | New generation of splice-less lentiviral vectors for safer gene therapy applications |
| JP6360500B2 (ja) | 2013-02-06 | 2018-07-18 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツングMerck Patent Gesellschaft mit beschraenkter Haftung | 骨関節炎の処置のためのアグリカナーゼ阻害剤としての置換カルボン酸誘導体 |
| AU2014229506B2 (en) | 2013-03-11 | 2017-04-13 | Nobesita As | Natural lipids containing non-oxidizable fatty acids |
| US10076399B2 (en) | 2013-09-13 | 2018-09-18 | Covidien Lp | Endovascular device engagement |
| WO2015059674A1 (en) | 2013-10-24 | 2015-04-30 | Ospedale San Raffaele S.R.L. | Method |
| GB201322798D0 (en) | 2013-12-20 | 2014-02-05 | Oxford Biomedica Ltd | Production system |
| GB201407322D0 (en) | 2014-04-25 | 2014-06-11 | Ospedale San Raffaele | Gene therapy |
| GB201412494D0 (en) | 2014-07-14 | 2014-08-27 | Ospedale San Raffaele And Fond Telethon | Vector production |
| GB201418965D0 (de) | 2014-10-24 | 2014-12-10 | Ospedale San Raffaele And Fond Telethon | |
| GB201608944D0 (en) | 2016-05-20 | 2016-07-06 | Ospedale San Raffaele And Fond Telethon | Gene Tharapy |
| GB201706394D0 (en) | 2017-04-21 | 2017-06-07 | Ospedale San Raffaele Srl | Gene Therapy |
| DE102017211110A1 (de) * | 2017-06-30 | 2019-01-03 | Continental Reifen Deutschland Gmbh | Verfahren zur Herstellung eines Silans, Verfahren zur Modifizierung einer Kieselsäure mit dem Silan und modifizierte Kieselsäure |
| GB201807944D0 (en) | 2018-05-16 | 2018-06-27 | Ospedale San Raffaele Srl | Compositions and methods for haematopoietic stem cell transplantation |
| GB201807945D0 (en) | 2018-05-16 | 2018-06-27 | Ospedale San Raffaele Srl | Vector production |
| CA3104948A1 (en) | 2018-06-25 | 2020-01-02 | Ospedale San Raffaele S.R.L | Gene therapy |
| AU2019358519A1 (en) | 2018-10-11 | 2021-05-27 | Fondazione Telethon Ets | Selection by means of artificial transactivators |
| US20220241240A1 (en) | 2019-01-18 | 2022-08-04 | Societe Des Produits Nestle S.A. | Agents and methods for increasing stem cell function |
| KR20220097492A (ko) | 2019-11-12 | 2022-07-07 | 옥스포드 바이오메디카(유케이) 리미티드 | 생산 시스템 |
| KR20220139911A (ko) | 2020-02-13 | 2022-10-17 | 옥스포드 바이오메디카(유케이) 리미티드 | 렌티바이러스 벡터의 생산 |
| US20230118587A1 (en) | 2020-03-13 | 2023-04-20 | Oxford Biomedica (Uk) Limited | Lentiviral Vectors |
| GB202007106D0 (en) | 2020-05-14 | 2020-07-01 | Ucl Business Plc | Cyclosporine analogues |
| GB202007199D0 (en) | 2020-05-15 | 2020-07-01 | Oxford Biomedica Ltd | Viral vector production |
| CN116391031A (zh) | 2020-09-28 | 2023-07-04 | 雀巢产品有限公司 | 用于增加干细胞功能的组合物和方法 |
| BR112023005370A2 (pt) | 2020-09-28 | 2023-05-09 | Nestle Sa | Composições e métodos para aumentar função das células-tronco |
| GB202017725D0 (en) | 2020-11-10 | 2020-12-23 | Oxford Biomedica Ltd | Method |
| EP4284932A1 (de) | 2021-02-01 | 2023-12-06 | Epsilen Bio S.r.l. | Gen-silencing |
| AU2021202658A1 (en) | 2021-04-28 | 2022-11-17 | Fondazione Telethon | Gene therapy |
| WO2022233767A1 (en) | 2021-05-05 | 2022-11-10 | Société des Produits Nestlé S.A. | Urolithin for increasing stem cell function |
| GB202114530D0 (en) | 2021-10-12 | 2021-11-24 | Oxford Biomedica Ltd | Retroviral vectors |
| GB202114532D0 (en) | 2021-10-12 | 2021-11-24 | Oxford Biomedica Ltd | Lentiviral Vectors |
| GB202114529D0 (en) | 2021-10-12 | 2021-11-24 | Oxford Biomedica Ltd | Lentiviral vectors |
| GB202114534D0 (en) | 2021-10-12 | 2021-11-24 | Oxford Biomedica Ltd | Novel viral regulatory elements |
| GB202114528D0 (en) | 2021-10-12 | 2021-11-24 | Oxford Biomedica Ltd | Lentiviral vectors |
| GB202114972D0 (en) | 2021-10-19 | 2021-12-01 | Ospedale San Raffaele Srl | Gene therapy |
| GB202117844D0 (en) | 2021-12-09 | 2022-01-26 | Oxford Biomedica Ltd | Purification method |
| WO2023209223A1 (en) | 2022-04-28 | 2023-11-02 | Ospedale San Raffaele S.R.L. | Methods for haematopoietic stem cell transplantation |
| GB202206346D0 (en) | 2022-04-29 | 2022-06-15 | Ospedale San Raffaele Srl | Gene therapy |
| GB202209098D0 (en) | 2022-06-21 | 2022-08-10 | Ucl Business Ltd | Cyclosporine analogues |
| GB202211935D0 (en) | 2022-08-16 | 2022-09-28 | Oxford Biomedica Ltd | envelope proteins |
| WO2024079644A1 (en) | 2022-10-11 | 2024-04-18 | Fondazione Telethon Ets | 3d cell culture methods |
| IT202300011790A1 (it) | 2023-06-08 | 2024-12-08 | Fond Telethon Ets | Protocolli di manipolazione genica in cellule immunitarie |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4127722A (en) * | 1975-10-02 | 1978-11-28 | Laboratoire L. Lafon | Benzhydrylsulphinyl derivatives |
| DE2804576A1 (de) * | 1977-02-15 | 1978-08-17 | Lafon Labor | Phenylamidin-derivate, verfahren zu deren herstellung und diese enthaltende pharmazeutische zusammensetzungen |
| WO1993020047A1 (en) * | 1992-04-07 | 1993-10-14 | British Bio-Technology Limited | Hydroxamic acid based collagenase and cytokine inhibitors |
| GB9404046D0 (en) * | 1994-03-03 | 1994-04-20 | Smithkline Beecham Corp | Novel compounds |
| AU692102B2 (en) * | 1995-05-10 | 1998-05-28 | Darwin Discovery Limited | Peptidyl compounds and their therapeutic use |
| US5665777A (en) * | 1995-11-14 | 1997-09-09 | Abbott Laboratories | Biphenyl hydroxamate inhibitors of matrix metalloproteinases |
| ES2183905T3 (es) * | 1995-12-20 | 2003-04-01 | Hoffmann La Roche | Inhibidores de metaloproteasa de matriz. |
| DK0871439T3 (da) * | 1996-01-02 | 2004-08-02 | Aventis Pharma Inc | Substituerede (aryl, heteroaryl, arylmethyl eller heteroarylmethyl) hydroxamsyreforbindelser |
| IL128900A0 (en) * | 1996-09-27 | 2000-01-31 | Upjohn Co | Beta-sulfonyl hydroxamic acids as matrix metalloproteinases inhibitor |
| WO1998039316A1 (en) * | 1997-03-04 | 1998-09-11 | Monsanto Company | N-hydroxy 4-sulfonyl butanamide compounds |
-
1997
- 1997-08-07 DK DK97935666T patent/DK0968182T3/da active
- 1997-08-07 EP EP97935666A patent/EP0968182B1/de not_active Expired - Lifetime
- 1997-08-07 DE DE69729007T patent/DE69729007T2/de not_active Expired - Fee Related
- 1997-08-07 KR KR1019997001020A patent/KR20000029858A/ko not_active Ceased
- 1997-08-07 BR BR9711027A patent/BR9711027A/pt unknown
- 1997-08-07 CZ CZ0036899A patent/CZ297278B6/cs not_active IP Right Cessation
- 1997-08-07 JP JP10507741A patent/JP2000517297A/ja not_active Ceased
- 1997-08-07 US US08/908,397 patent/US6118001A/en not_active Expired - Fee Related
- 1997-08-07 AT AT97935666T patent/ATE266000T1/de not_active IP Right Cessation
- 1997-08-07 PT PT97935666T patent/PT968182E/pt unknown
- 1997-08-07 PL PL97331598A patent/PL193829B1/pl not_active IP Right Cessation
- 1997-08-07 WO PCT/GB1997/002129 patent/WO1998005635A1/en not_active Ceased
- 1997-08-07 CA CA002263154A patent/CA2263154A1/en not_active Abandoned
- 1997-08-07 AU AU38564/97A patent/AU730464B2/en not_active Ceased
- 1997-08-07 ES ES97935666T patent/ES2217425T3/es not_active Expired - Lifetime
- 1997-08-07 CN CN97197048A patent/CN1227540A/zh active Pending
-
1999
- 1999-02-05 NO NO19990543A patent/NO314452B1/no unknown
Also Published As
| Publication number | Publication date |
|---|---|
| AU730464B2 (en) | 2001-03-08 |
| NO990543L (no) | 1999-04-06 |
| JP2000517297A (ja) | 2000-12-26 |
| EP0968182B1 (de) | 2004-05-06 |
| ES2217425T3 (es) | 2004-11-01 |
| US6118001A (en) | 2000-09-12 |
| BR9711027A (pt) | 1999-08-17 |
| CZ297278B6 (cs) | 2006-10-11 |
| EP0968182A1 (de) | 2000-01-05 |
| DK0968182T3 (da) | 2004-08-16 |
| DE69729007D1 (de) | 2004-06-09 |
| KR20000029858A (ko) | 2000-05-25 |
| AU3856497A (en) | 1998-02-25 |
| ATE266000T1 (de) | 2004-05-15 |
| NO990543D0 (no) | 1999-02-05 |
| PT968182E (pt) | 2004-08-31 |
| PL331598A1 (en) | 1999-08-02 |
| CN1227540A (zh) | 1999-09-01 |
| WO1998005635A1 (en) | 1998-02-12 |
| PL193829B1 (pl) | 2007-03-30 |
| CZ36899A3 (cs) | 1999-07-14 |
| CA2263154A1 (en) | 1998-02-12 |
| NO314452B1 (no) | 2003-03-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE69729007T2 (de) | Hydroxamsäure- und carbonsäure-derivate mit mmp und tnf hemmender wirkung | |
| DE69509401T2 (de) | Peptidische verbindungen und deren therapeutische verwendung als metalloproteinase-inhibitoren | |
| DE69422726T2 (de) | Peptidverbindungen und ihre therapeutische verwendung als inhibitoren von metalloproteinasen | |
| DE69620639T2 (de) | Mercaptoalkylpeptidylverbindungen mit einem imidazolsubstituenten und ihre verwendung als inhibitoren der matrix metalloproteinasen (mmp) und/oder des tumor necrosis faktors (tnf) | |
| DE3887307T2 (de) | Sulfhydryl enthaltende tricylische Lactame und ihre pharmakologische Verwendung. | |
| DE69811111T2 (de) | Hydroxamsäure- und carbonsäurederivate mit mmp und tnf hemmender wirkung | |
| DE69625506T2 (de) | Schwefelsubstituierte peptide als inhibitoren für metalloproteinasen und der tnf-freisetzung | |
| DE60030768T2 (de) | Nitrilderivate als cathepsin k hemmer | |
| DE69412469T2 (de) | Hydroxamsaeurederivate als metalloproteinase inhibitoren | |
| DE69624477T2 (de) | Peptide, die metallproteinasen und die tnf-freisetzung hemmen, und ihre therapeutische verwendung | |
| DE69827236T2 (de) | Matrix-metalloproteinase-hemmer | |
| DE60013302T2 (de) | Cystein protease inhibitore | |
| DE69934094T2 (de) | N-hydroxyformamid-derivate | |
| DE9321495U1 (de) | Hydroxaminsäure-Derivat als Metalloproteinase-Inhibitor | |
| DE69832268T2 (de) | Peptidyl2-amino-1hydroxyalkansulfonsäuren als zysteinprotease-inhibitoren | |
| US6566384B1 (en) | Hydroxamic and carboxylic acid derivatives having MMP and TNF inhibitory activity | |
| EP0527458A1 (de) | Neue 3,5-Di-tert.butyl-4-hydroxyphenyl-Derivate, Verfahren zu ihrer Herstellung und Arzneimittel | |
| DE60008548T2 (de) | Hydroxamsäurederivate als matrix-metalloproteinase-inhibitoren | |
| DE60318317T2 (de) | Acylaminothiazolderivate und ihre vewrendung als beta-amyloid inhibitoren | |
| DE69925914T2 (de) | Succinamid hemmstoffe des interleukin-1 beta konvertierenden enzyms | |
| DE69627483T2 (de) | Peptidartige stoffe, die metalloproteinasen und die tnf-freisetzung hemmen, und ihre therapeutische verwendung | |
| DE60101224T2 (de) | Hydroxamsäure-Derivate | |
| DE60002558T2 (de) | Derivate der Phosphonsäure zur Inhibierung von Carboxypeptidase B | |
| DE69615680T2 (de) | Trizyklische verbindungen als matrix-metalloproteinase inhibitoren | |
| DE69724944T2 (de) | Peptidverbindungen mit mmp- und tnf-hemmender wirkung |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition | ||
| 8339 | Ceased/non-payment of the annual fee |