DE60008548T2 - Hydroxamsäurederivate als matrix-metalloproteinase-inhibitoren - Google Patents
Hydroxamsäurederivate als matrix-metalloproteinase-inhibitoren Download PDFInfo
- Publication number
- DE60008548T2 DE60008548T2 DE60008548T DE60008548T DE60008548T2 DE 60008548 T2 DE60008548 T2 DE 60008548T2 DE 60008548 T DE60008548 T DE 60008548T DE 60008548 T DE60008548 T DE 60008548T DE 60008548 T2 DE60008548 T2 DE 60008548T2
- Authority
- DE
- Germany
- Prior art keywords
- sulfonylamino
- alkyl
- group
- compound according
- dibenzofuran
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 239000002253 acid Substances 0.000 title description 16
- NEAQRZUHTPSBBM-UHFFFAOYSA-N 2-hydroxy-3,3-dimethyl-7-nitro-4h-isoquinolin-1-one Chemical compound C1=C([N+]([O-])=O)C=C2C(=O)N(O)C(C)(C)CC2=C1 NEAQRZUHTPSBBM-UHFFFAOYSA-N 0.000 title description 10
- 239000003771 matrix metalloproteinase inhibitor Substances 0.000 title description 3
- 229940121386 matrix metalloproteinase inhibitor Drugs 0.000 title description 3
- 150000001875 compounds Chemical class 0.000 claims description 149
- -1 CF 3 Chemical group 0.000 claims description 73
- 102000002274 Matrix Metalloproteinases Human genes 0.000 claims description 40
- 108010000684 Matrix Metalloproteinases Proteins 0.000 claims description 40
- 229910052799 carbon Inorganic materials 0.000 claims description 37
- 229910052739 hydrogen Inorganic materials 0.000 claims description 27
- 206010019280 Heart failures Diseases 0.000 claims description 24
- 238000011282 treatment Methods 0.000 claims description 22
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 19
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 19
- 125000000217 alkyl group Chemical group 0.000 claims description 18
- 238000002360 preparation method Methods 0.000 claims description 18
- 229910052717 sulfur Inorganic materials 0.000 claims description 18
- 229910052736 halogen Inorganic materials 0.000 claims description 16
- 150000002367 halogens Chemical class 0.000 claims description 16
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 15
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 15
- 201000010099 disease Diseases 0.000 claims description 13
- 229910052760 oxygen Inorganic materials 0.000 claims description 13
- 150000003839 salts Chemical class 0.000 claims description 13
- 206010028980 Neoplasm Diseases 0.000 claims description 12
- 125000001072 heteroaryl group Chemical group 0.000 claims description 12
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 11
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 claims description 10
- 125000005842 heteroatom Chemical group 0.000 claims description 10
- NHLIFYLZAWLMCY-UHFFFAOYSA-N 1-(dibenzofuran-3-ylsulfonylamino)cyclohexane-1-carboxylic acid Chemical compound C=1C=C(C2=CC=CC=C2O2)C2=CC=1S(=O)(=O)NC1(C(=O)O)CCCCC1 NHLIFYLZAWLMCY-UHFFFAOYSA-N 0.000 claims description 9
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 8
- 201000011510 cancer Diseases 0.000 claims description 8
- 239000001257 hydrogen Substances 0.000 claims description 8
- 208000017169 kidney disease Diseases 0.000 claims description 8
- 206010061218 Inflammation Diseases 0.000 claims description 7
- 229910052794 bromium Inorganic materials 0.000 claims description 7
- 125000004432 carbon atom Chemical group C* 0.000 claims description 7
- 229910052801 chlorine Inorganic materials 0.000 claims description 7
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 7
- 239000003814 drug Substances 0.000 claims description 7
- 230000004054 inflammatory process Effects 0.000 claims description 7
- 230000005764 inhibitory process Effects 0.000 claims description 7
- 239000008194 pharmaceutical composition Substances 0.000 claims description 7
- 125000001424 substituent group Chemical group 0.000 claims description 7
- 208000027418 Wounds and injury Diseases 0.000 claims description 6
- 206010003246 arthritis Diseases 0.000 claims description 6
- 229910052731 fluorine Inorganic materials 0.000 claims description 6
- 125000000623 heterocyclic group Chemical group 0.000 claims description 6
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 5
- 230000001684 chronic effect Effects 0.000 claims description 5
- 229910052740 iodine Inorganic materials 0.000 claims description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 5
- 201000006417 multiple sclerosis Diseases 0.000 claims description 5
- LOXWBEWZQJIFOP-UHFFFAOYSA-N 2-methyl-2-(6,7,8,9-tetrahydrodibenzofuran-3-ylsulfonylamino)propanoic acid Chemical compound C1CCCC2=C1C1=CC=C(S(=O)(=O)NC(C)(C)C(O)=O)C=C1O2 LOXWBEWZQJIFOP-UHFFFAOYSA-N 0.000 claims description 4
- 239000004606 Fillers/Extenders Substances 0.000 claims description 4
- 208000001132 Osteoporosis Diseases 0.000 claims description 4
- 125000003545 alkoxy group Chemical group 0.000 claims description 4
- 239000003085 diluting agent Substances 0.000 claims description 4
- 230000002401 inhibitory effect Effects 0.000 claims description 4
- OCBUTXRJOMDGIW-UHFFFAOYSA-N 2-(5a,6,7,8,9,9a-hexahydrodibenzofuran-3-ylsulfonylamino)-2-methylpropanoic acid Chemical compound C1CCCC2OC3=CC(S(=O)(=O)NC(C)(C)C(O)=O)=CC=C3C21 OCBUTXRJOMDGIW-UHFFFAOYSA-N 0.000 claims description 3
- WHNJQGDDFQEEJX-UHFFFAOYSA-N 2-(5a,6,7,8,9,9a-hexahydrodibenzofuran-3-ylsulfonylamino)-n-hydroxy-2-methylpropanamide Chemical compound C1CCCC2OC3=CC(S(=O)(=O)NC(C)(C)C(=O)NO)=CC=C3C21 WHNJQGDDFQEEJX-UHFFFAOYSA-N 0.000 claims description 3
- JLJZTQLCMZBISV-UHFFFAOYSA-N 2-(dibenzofuran-3-ylsulfonylamino)-2-methylpropanoic acid Chemical compound C1=CC=C2C3=CC=C(S(=O)(=O)NC(C)(C)C(O)=O)C=C3OC2=C1 JLJZTQLCMZBISV-UHFFFAOYSA-N 0.000 claims description 3
- INQMWONNAQXTHM-UHFFFAOYSA-N 2-(dibenzofuran-3-ylsulfonylamino)-n-hydroxy-2-methylpropanamide Chemical compound C1=CC=C2C3=CC=C(S(=O)(=O)NC(C)(C)C(=O)NO)C=C3OC2=C1 INQMWONNAQXTHM-UHFFFAOYSA-N 0.000 claims description 3
- 201000001320 Atherosclerosis Diseases 0.000 claims description 3
- 206010064996 Ulcerative keratitis Diseases 0.000 claims description 3
- 206010052428 Wound Diseases 0.000 claims description 3
- 208000007474 aortic aneurysm Diseases 0.000 claims description 3
- 210000004556 brain Anatomy 0.000 claims description 3
- 150000001721 carbon Chemical group 0.000 claims description 3
- 210000000265 leukocyte Anatomy 0.000 claims description 3
- ZJSAKAIGXUWOIN-UHFFFAOYSA-N n-hydroxy-2-methyl-2-(6,7,8,9-tetrahydrodibenzofuran-3-ylsulfonylamino)propanamide Chemical compound C1CCCC2=C1C1=CC=C(S(=O)(=O)NC(C)(C)C(=O)NO)C=C1O2 ZJSAKAIGXUWOIN-UHFFFAOYSA-N 0.000 claims description 3
- 239000001301 oxygen Substances 0.000 claims description 3
- UQWDEKBXMDXMJL-UHFFFAOYSA-N 1-(5a,6,7,8,9,9a-hexahydrodibenzofuran-3-ylsulfonylamino)cyclopentane-1-carboxylic acid Chemical compound C=1C=C2C3CCCCC3OC2=CC=1S(=O)(=O)NC1(C(=O)O)CCCC1 UQWDEKBXMDXMJL-UHFFFAOYSA-N 0.000 claims description 2
- FJGCFIXTQRGUFA-UHFFFAOYSA-N 1-(6,7,8,9,10,11-hexahydrocycloocta[b][1]benzofuran-3-ylsulfonylamino)cyclopentane-1-carboxylic acid Chemical compound C=1C=C2C=3CCCCCCC=3OC2=CC=1S(=O)(=O)NC1(C(=O)O)CCCC1 FJGCFIXTQRGUFA-UHFFFAOYSA-N 0.000 claims description 2
- OMAOAPOTTIUPNS-UHFFFAOYSA-N 2-(2,3-dihydro-1h-cyclopenta[b][1]benzofuran-6-ylsulfonylamino)-2-methylpropanoic acid Chemical compound O1C2=CC(S(=O)(=O)NC(C)(C)C(O)=O)=CC=C2C2=C1CCC2 OMAOAPOTTIUPNS-UHFFFAOYSA-N 0.000 claims description 2
- XTBIJWGIZKJHPR-UHFFFAOYSA-N 2-(2,3-dihydro-1h-cyclopenta[b][1]benzofuran-6-ylsulfonylamino)-n-hydroxy-2-methylpropanamide Chemical compound O1C2=CC(S(=O)(=O)NC(C)(C)C(=O)NO)=CC=C2C2=C1CCC2 XTBIJWGIZKJHPR-UHFFFAOYSA-N 0.000 claims description 2
- UUXJNOIPYDTJDW-UHFFFAOYSA-N 2-(6,7,8,9,10,11-hexahydrocycloocta[b][1]benzofuran-3-ylsulfonylamino)-2-methylpropanoic acid Chemical compound C1CCCCCC2=C1C1=CC=C(S(=O)(=O)NC(C)(C)C(O)=O)C=C1O2 UUXJNOIPYDTJDW-UHFFFAOYSA-N 0.000 claims description 2
- DMCQTSJZJQNSOM-UHFFFAOYSA-N 2-(6,7,8,9,10,11-hexahydrocycloocta[b][1]benzofuran-3-ylsulfonylamino)-n-hydroxy-2-methylpropanamide Chemical compound C1CCCCCC2=C1C1=CC=C(S(=O)(=O)NC(C)(C)C(=O)NO)C=C1O2 DMCQTSJZJQNSOM-UHFFFAOYSA-N 0.000 claims description 2
- USXONGDHWANVIX-UHFFFAOYSA-N 2-[(7-chlorodibenzofuran-2-yl)sulfonylamino]-n-hydroxy-2-methylpropanamide Chemical compound ClC1=CC=C2C3=CC(S(=O)(=O)NC(C)(C)C(=O)NO)=CC=C3OC2=C1 USXONGDHWANVIX-UHFFFAOYSA-N 0.000 claims description 2
- GBVKMNDMKDQYFC-UHFFFAOYSA-N 4-(dibenzofuran-3-ylsulfonylamino)-n-hydroxyoxane-4-carboxamide Chemical compound C=1C=C(C2=CC=CC=C2O2)C2=CC=1S(=O)(=O)NC1(C(=O)NO)CCOCC1 GBVKMNDMKDQYFC-UHFFFAOYSA-N 0.000 claims description 2
- 208000030507 AIDS Diseases 0.000 claims description 2
- 208000024827 Alzheimer disease Diseases 0.000 claims description 2
- 206010059245 Angiopathy Diseases 0.000 claims description 2
- 208000023275 Autoimmune disease Diseases 0.000 claims description 2
- 206010019196 Head injury Diseases 0.000 claims description 2
- 208000023105 Huntington disease Diseases 0.000 claims description 2
- 208000018737 Parkinson disease Diseases 0.000 claims description 2
- 208000004210 Pressure Ulcer Diseases 0.000 claims description 2
- 208000024777 Prion disease Diseases 0.000 claims description 2
- 208000006011 Stroke Diseases 0.000 claims description 2
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 claims description 2
- 208000037979 autoimmune inflammatory disease Diseases 0.000 claims description 2
- 230000010339 dilation Effects 0.000 claims description 2
- 230000035876 healing Effects 0.000 claims description 2
- 230000009545 invasion Effects 0.000 claims description 2
- 208000015122 neurodegenerative disease Diseases 0.000 claims description 2
- 208000028169 periodontal disease Diseases 0.000 claims description 2
- 208000020431 spinal cord injury Diseases 0.000 claims description 2
- CCMDKRCXFVVPIJ-UHFFFAOYSA-N 1-(6,7,8,9-tetrahydrodibenzofuran-3-ylsulfonylamino)cyclopentane-1-carboxylic acid Chemical compound C=1C=C2C=3CCCCC=3OC2=CC=1S(=O)(=O)NC1(C(=O)O)CCCC1 CCMDKRCXFVVPIJ-UHFFFAOYSA-N 0.000 claims 1
- DFNPIMGSMYISHO-UHFFFAOYSA-N 2-(6,7,8,9,10,10a-hexahydro-5ah-cyclohepta[b][1]benzofuran-3-ylsulfonylamino)-2-methylpropanoic acid Chemical compound C1CCCCC2OC3=CC(S(=O)(=O)NC(C)(C)C(O)=O)=CC=C3C21 DFNPIMGSMYISHO-UHFFFAOYSA-N 0.000 claims 1
- 208000002193 Pain Diseases 0.000 claims 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims 1
- 230000001154 acute effect Effects 0.000 claims 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims 1
- 150000002431 hydrogen Chemical class 0.000 claims 1
- 206010028417 myasthenia gravis Diseases 0.000 claims 1
- ANEHMKMFOQDTAP-UHFFFAOYSA-N n-hydroxy-2-methyl-2-(7,8,9,10-tetrahydro-6h-cyclohepta[b][1]benzofuran-3-ylsulfonylamino)propanamide Chemical compound C1CCCCC2=C1C1=CC=C(S(=O)(=O)NC(C)(C)C(=O)NO)C=C1O2 ANEHMKMFOQDTAP-UHFFFAOYSA-N 0.000 claims 1
- 239000011593 sulfur Substances 0.000 claims 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 45
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 42
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 38
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 33
- 230000015572 biosynthetic process Effects 0.000 description 29
- 239000000203 mixture Substances 0.000 description 28
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 25
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 24
- 238000000034 method Methods 0.000 description 24
- 239000011541 reaction mixture Substances 0.000 description 24
- 239000000243 solution Substances 0.000 description 22
- 238000003786 synthesis reaction Methods 0.000 description 22
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 22
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 21
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 20
- 238000005160 1H NMR spectroscopy Methods 0.000 description 19
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 18
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 16
- 102000000424 Matrix Metalloproteinase 2 Human genes 0.000 description 16
- 108010016165 Matrix Metalloproteinase 2 Proteins 0.000 description 16
- 102000000422 Matrix Metalloproteinase 3 Human genes 0.000 description 16
- 238000002844 melting Methods 0.000 description 16
- 230000008018 melting Effects 0.000 description 15
- 239000007787 solid Substances 0.000 description 15
- 230000000694 effects Effects 0.000 description 14
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 13
- 230000015556 catabolic process Effects 0.000 description 13
- 239000003112 inhibitor Substances 0.000 description 13
- 230000002861 ventricular Effects 0.000 description 13
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 12
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 12
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 12
- 230000014509 gene expression Effects 0.000 description 12
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 12
- 210000002744 extracellular matrix Anatomy 0.000 description 10
- 238000001914 filtration Methods 0.000 description 10
- 102000029816 Collagenase Human genes 0.000 description 9
- 108060005980 Collagenase Proteins 0.000 description 9
- 102000005741 Metalloproteases Human genes 0.000 description 9
- 108010006035 Metalloproteases Proteins 0.000 description 9
- 230000003197 catalytic effect Effects 0.000 description 9
- 210000004027 cell Anatomy 0.000 description 9
- 239000003795 chemical substances by application Substances 0.000 description 9
- 229960002424 collagenase Drugs 0.000 description 9
- 210000002808 connective tissue Anatomy 0.000 description 9
- 239000013078 crystal Substances 0.000 description 9
- 238000009472 formulation Methods 0.000 description 9
- 239000007788 liquid Substances 0.000 description 9
- 108090000623 proteins and genes Proteins 0.000 description 9
- 108091007196 stromelysin Proteins 0.000 description 9
- 210000001519 tissue Anatomy 0.000 description 9
- 102000004190 Enzymes Human genes 0.000 description 8
- 108090000790 Enzymes Proteins 0.000 description 8
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 8
- 238000006243 chemical reaction Methods 0.000 description 8
- 238000006731 degradation reaction Methods 0.000 description 8
- 229940088598 enzyme Drugs 0.000 description 8
- 150000002148 esters Chemical class 0.000 description 8
- 210000003584 mesangial cell Anatomy 0.000 description 8
- 239000012074 organic phase Substances 0.000 description 8
- 108090000765 processed proteins & peptides Proteins 0.000 description 8
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 8
- 102000000380 Matrix Metalloproteinase 1 Human genes 0.000 description 7
- 108010016113 Matrix Metalloproteinase 1 Proteins 0.000 description 7
- 108010016160 Matrix Metalloproteinase 3 Proteins 0.000 description 7
- 239000008346 aqueous phase Substances 0.000 description 7
- 239000002775 capsule Substances 0.000 description 7
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 7
- DNIAPMSPPWPWGF-UHFFFAOYSA-N propylene glycol Substances CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 7
- 239000003826 tablet Substances 0.000 description 7
- 208000037260 Atherosclerotic Plaque Diseases 0.000 description 6
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 6
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 150000001408 amides Chemical class 0.000 description 6
- 239000000460 chlorine Substances 0.000 description 6
- 239000011159 matrix material Substances 0.000 description 6
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 5
- 239000005541 ACE inhibitor Substances 0.000 description 5
- 102000008186 Collagen Human genes 0.000 description 5
- 108010035532 Collagen Proteins 0.000 description 5
- 102100027995 Collagenase 3 Human genes 0.000 description 5
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 5
- 102100030417 Matrilysin Human genes 0.000 description 5
- 108090000855 Matrilysin Proteins 0.000 description 5
- 108010015302 Matrix metalloproteinase-9 Proteins 0.000 description 5
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 5
- 238000004458 analytical method Methods 0.000 description 5
- 229940044094 angiotensin-converting-enzyme inhibitor Drugs 0.000 description 5
- 230000037396 body weight Effects 0.000 description 5
- 210000004413 cardiac myocyte Anatomy 0.000 description 5
- 239000012320 chlorinating reagent Substances 0.000 description 5
- 229920001436 collagen Polymers 0.000 description 5
- 239000002552 dosage form Substances 0.000 description 5
- 235000019441 ethanol Nutrition 0.000 description 5
- 230000007062 hydrolysis Effects 0.000 description 5
- 238000006460 hydrolysis reaction Methods 0.000 description 5
- 239000012442 inert solvent Substances 0.000 description 5
- 239000010410 layer Substances 0.000 description 5
- 230000005012 migration Effects 0.000 description 5
- 238000013508 migration Methods 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 239000000651 prodrug Substances 0.000 description 5
- 229940002612 prodrug Drugs 0.000 description 5
- 102000004169 proteins and genes Human genes 0.000 description 5
- 230000002829 reductive effect Effects 0.000 description 5
- 238000010992 reflux Methods 0.000 description 5
- 239000002904 solvent Substances 0.000 description 5
- 0 *C(*)(C(*)=O)N* Chemical compound *C(*)(C(*)=O)N* 0.000 description 4
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 4
- 102000004127 Cytokines Human genes 0.000 description 4
- 108090000695 Cytokines Proteins 0.000 description 4
- 241000588724 Escherichia coli Species 0.000 description 4
- 108010010803 Gelatin Proteins 0.000 description 4
- 102000001776 Matrix metalloproteinase-9 Human genes 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- 229920002472 Starch Polymers 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 125000001931 aliphatic group Chemical group 0.000 description 4
- 125000003118 aryl group Chemical group 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- 239000001768 carboxy methyl cellulose Substances 0.000 description 4
- 230000010343 cardiac dilation Effects 0.000 description 4
- 239000000969 carrier Substances 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 230000018109 developmental process Effects 0.000 description 4
- 239000000796 flavoring agent Substances 0.000 description 4
- 239000008273 gelatin Substances 0.000 description 4
- 229920000159 gelatin Polymers 0.000 description 4
- 229940014259 gelatin Drugs 0.000 description 4
- 235000019322 gelatine Nutrition 0.000 description 4
- 235000011852 gelatine desserts Nutrition 0.000 description 4
- 230000001434 glomerular Effects 0.000 description 4
- 210000000585 glomerular basement membrane Anatomy 0.000 description 4
- 239000000543 intermediate Substances 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 231100000252 nontoxic Toxicity 0.000 description 4
- 230000003000 nontoxic effect Effects 0.000 description 4
- 239000006187 pill Substances 0.000 description 4
- 229920001223 polyethylene glycol Polymers 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 230000000750 progressive effect Effects 0.000 description 4
- 230000002062 proliferating effect Effects 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- 235000017557 sodium bicarbonate Nutrition 0.000 description 4
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 4
- 235000010356 sorbitol Nutrition 0.000 description 4
- 239000000600 sorbitol Substances 0.000 description 4
- 239000008107 starch Substances 0.000 description 4
- 235000019698 starch Nutrition 0.000 description 4
- 229940032147 starch Drugs 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 239000000758 substrate Substances 0.000 description 4
- 210000004509 vascular smooth muscle cell Anatomy 0.000 description 4
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- 241000416162 Astragalus gummifer Species 0.000 description 3
- 206010007559 Cardiac failure congestive Diseases 0.000 description 3
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 3
- 206010016654 Fibrosis Diseases 0.000 description 3
- 102000013382 Gelatinases Human genes 0.000 description 3
- 108010026132 Gelatinases Proteins 0.000 description 3
- 206010018364 Glomerulonephritis Diseases 0.000 description 3
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 3
- 108010076503 Matrix Metalloproteinase 13 Proteins 0.000 description 3
- 206010027476 Metastases Diseases 0.000 description 3
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 3
- 102100028848 Stromelysin-2 Human genes 0.000 description 3
- 210000001744 T-lymphocyte Anatomy 0.000 description 3
- 102000005354 Tissue Inhibitor of Metalloproteinase-2 Human genes 0.000 description 3
- 108010031372 Tissue Inhibitor of Metalloproteinase-2 Proteins 0.000 description 3
- 229920001615 Tragacanth Polymers 0.000 description 3
- 208000025865 Ulcer Diseases 0.000 description 3
- 239000007900 aqueous suspension Substances 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 239000003054 catalyst Substances 0.000 description 3
- 230000004663 cell proliferation Effects 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- 230000006378 damage Effects 0.000 description 3
- 230000034994 death Effects 0.000 description 3
- 231100000517 death Toxicity 0.000 description 3
- 230000007547 defect Effects 0.000 description 3
- 210000002615 epidermis Anatomy 0.000 description 3
- KUAFMPWKUNNUEC-UHFFFAOYSA-N ethyl 1-aminocyclohexane-1-carboxylate Chemical compound CCOC(=O)C1(N)CCCCC1 KUAFMPWKUNNUEC-UHFFFAOYSA-N 0.000 description 3
- 230000004761 fibrosis Effects 0.000 description 3
- 239000012634 fragment Substances 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- 210000003878 glomerular mesangial cell Anatomy 0.000 description 3
- 230000004217 heart function Effects 0.000 description 3
- 239000003906 humectant Substances 0.000 description 3
- 230000006698 induction Effects 0.000 description 3
- 208000014674 injury Diseases 0.000 description 3
- 210000002510 keratinocyte Anatomy 0.000 description 3
- 210000003734 kidney Anatomy 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 3
- 235000019341 magnesium sulphate Nutrition 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- 229920000609 methyl cellulose Polymers 0.000 description 3
- 150000004702 methyl esters Chemical class 0.000 description 3
- 235000010981 methylcellulose Nutrition 0.000 description 3
- 239000001923 methylcellulose Substances 0.000 description 3
- 201000008482 osteoarthritis Diseases 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- 230000035515 penetration Effects 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 239000002516 radical scavenger Substances 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 208000037803 restenosis Diseases 0.000 description 3
- 206010039073 rheumatoid arthritis Diseases 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 229940124530 sulfonamide Drugs 0.000 description 3
- 238000001356 surgical procedure Methods 0.000 description 3
- 239000000375 suspending agent Substances 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 3
- 235000010487 tragacanth Nutrition 0.000 description 3
- 239000000196 tragacanth Substances 0.000 description 3
- 229940116362 tragacanth Drugs 0.000 description 3
- 210000004881 tumor cell Anatomy 0.000 description 3
- 239000001993 wax Substances 0.000 description 3
- LREHXNMBUBVFHA-UHFFFAOYSA-N 1,2,3,4-tetrahydrodibenzofuran Chemical compound O1C2=CC=CC=C2C2=C1CCCC2 LREHXNMBUBVFHA-UHFFFAOYSA-N 0.000 description 2
- FEXVVCVXHKDRRX-UHFFFAOYSA-N 1-(2,3-dihydro-1h-cyclopenta[b][1]benzofuran-6-ylsulfonylamino)cyclopentane-1-carboxylic acid Chemical compound C=1C=C2C=3CCCC=3OC2=CC=1S(=O)(=O)NC1(C(=O)O)CCCC1 FEXVVCVXHKDRRX-UHFFFAOYSA-N 0.000 description 2
- CMCBDXRRFKYBDG-UHFFFAOYSA-N 1-dodecoxydodecane Chemical compound CCCCCCCCCCCCOCCCCCCCCCCCC CMCBDXRRFKYBDG-UHFFFAOYSA-N 0.000 description 2
- FUOOLUPWFVMBKG-UHFFFAOYSA-N 2-Aminoisobutyric acid Chemical compound CC(C)(N)C(O)=O FUOOLUPWFVMBKG-UHFFFAOYSA-N 0.000 description 2
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 2
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 2
- MIIIXQJBDGSIKL-UHFFFAOYSA-N 2-morpholin-4-ylethanesulfonic acid;hydrate Chemical compound O.OS(=O)(=O)CCN1CCOCC1 MIIIXQJBDGSIKL-UHFFFAOYSA-N 0.000 description 2
- YQVLZECCVJWFMV-UHFFFAOYSA-N 2-phenoxycyclohexan-1-ol Chemical compound OC1CCCCC1OC1=CC=CC=C1 YQVLZECCVJWFMV-UHFFFAOYSA-N 0.000 description 2
- BKZGSYYAKPEVPE-UHFFFAOYSA-N 2-phenoxycyclohexan-1-one Chemical compound O=C1CCCCC1OC1=CC=CC=C1 BKZGSYYAKPEVPE-UHFFFAOYSA-N 0.000 description 2
- 125000000389 2-pyrrolyl group Chemical group [H]N1C([*])=C([H])C([H])=C1[H] 0.000 description 2
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 2
- KIUMMUBSPKGMOY-UHFFFAOYSA-N 3,3'-Dithiobis(6-nitrobenzoic acid) Chemical compound C1=C([N+]([O-])=O)C(C(=O)O)=CC(SSC=2C=C(C(=CC=2)[N+]([O-])=O)C(O)=O)=C1 KIUMMUBSPKGMOY-UHFFFAOYSA-N 0.000 description 2
- 125000003682 3-furyl group Chemical group O1C([H])=C([*])C([H])=C1[H] 0.000 description 2
- 125000001397 3-pyrrolyl group Chemical group [H]N1C([H])=C([*])C([H])=C1[H] 0.000 description 2
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 description 2
- FOCDJDFWZHYCOR-UHFFFAOYSA-N 4-(dibenzofuran-3-ylsulfonylamino)oxane-4-carboxylic acid Chemical compound C=1C=C(C2=CC=CC=C2O2)C2=CC=1S(=O)(=O)NC1(C(=O)O)CCOCC1 FOCDJDFWZHYCOR-UHFFFAOYSA-N 0.000 description 2
- KDDQRKBRJSGMQE-UHFFFAOYSA-N 4-thiazolyl Chemical compound [C]1=CSC=N1 KDDQRKBRJSGMQE-UHFFFAOYSA-N 0.000 description 2
- CWDWFSXUQODZGW-UHFFFAOYSA-N 5-thiazolyl Chemical group [C]1=CN=CS1 CWDWFSXUQODZGW-UHFFFAOYSA-N 0.000 description 2
- 244000215068 Acacia senegal Species 0.000 description 2
- 235000006491 Acacia senegal Nutrition 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- 208000025494 Aortic disease Diseases 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 2
- 208000005189 Embolism Diseases 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 2
- 241000206672 Gelidium Species 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 229920000084 Gum arabic Polymers 0.000 description 2
- 102000000589 Interleukin-1 Human genes 0.000 description 2
- 108010002352 Interleukin-1 Proteins 0.000 description 2
- 239000003810 Jones reagent Substances 0.000 description 2
- 229940124761 MMP inhibitor Drugs 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 108010076497 Matrix Metalloproteinase 10 Proteins 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- 102000035195 Peptidases Human genes 0.000 description 2
- 108091005804 Peptidases Proteins 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 102000016611 Proteoglycans Human genes 0.000 description 2
- 108010067787 Proteoglycans Proteins 0.000 description 2
- 101000627863 Rattus norvegicus 72 kDa type IV collagenase Proteins 0.000 description 2
- 101001013135 Rattus norvegicus Interstitial collagenase Proteins 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 2
- 102100028847 Stromelysin-3 Human genes 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- 235000010489 acacia gum Nutrition 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 235000010419 agar Nutrition 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 239000003513 alkali Substances 0.000 description 2
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 2
- 150000001342 alkaline earth metals Chemical class 0.000 description 2
- 150000003973 alkyl amines Chemical class 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 230000004888 barrier function Effects 0.000 description 2
- 239000000440 bentonite Substances 0.000 description 2
- 235000012216 bentonite Nutrition 0.000 description 2
- 229910000278 bentonite Inorganic materials 0.000 description 2
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 239000012267 brine Substances 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 2
- 150000001735 carboxylic acids Chemical class 0.000 description 2
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 2
- 230000000747 cardiac effect Effects 0.000 description 2
- 230000024245 cell differentiation Effects 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 238000010367 cloning Methods 0.000 description 2
- 229940110456 cocoa butter Drugs 0.000 description 2
- 235000019868 cocoa butter Nutrition 0.000 description 2
- 230000008602 contraction Effects 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 230000000875 corresponding effect Effects 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- 230000003412 degenerative effect Effects 0.000 description 2
- 208000037765 diseases and disorders Diseases 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 239000012153 distilled water Substances 0.000 description 2
- 239000002934 diuretic Substances 0.000 description 2
- 229940030606 diuretics Drugs 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 201000002491 encephalomyelitis Diseases 0.000 description 2
- 210000002889 endothelial cell Anatomy 0.000 description 2
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 2
- 239000000945 filler Substances 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 239000012467 final product Substances 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 210000001035 gastrointestinal tract Anatomy 0.000 description 2
- 210000004195 gingiva Anatomy 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 235000001727 glucose Nutrition 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 230000002757 inflammatory effect Effects 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 238000011835 investigation Methods 0.000 description 2
- 230000000302 ischemic effect Effects 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 230000003907 kidney function Effects 0.000 description 2
- 239000007937 lozenge Substances 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 230000008384 membrane barrier Effects 0.000 description 2
- 108020004999 messenger RNA Proteins 0.000 description 2
- NVWZNEDLYYLQJC-UHFFFAOYSA-N methyl 2-amino-2-methylpropanoate;hydrochloride Chemical compound Cl.COC(=O)C(C)(C)N NVWZNEDLYYLQJC-UHFFFAOYSA-N 0.000 description 2
- GMDCKOYTUVYEGA-UHFFFAOYSA-N methyl 2-methyl-2-(6,7,8,9-tetrahydrodibenzofuran-3-ylsulfonylamino)propanoate Chemical compound C1CCCC2=C1C1=CC=C(S(=O)(=O)NC(C)(C)C(=O)OC)C=C1O2 GMDCKOYTUVYEGA-UHFFFAOYSA-N 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 125000002950 monocyclic group Chemical group 0.000 description 2
- 210000000885 nephron Anatomy 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- 229940100688 oral solution Drugs 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 230000003239 periodontal effect Effects 0.000 description 2
- 239000000825 pharmaceutical preparation Substances 0.000 description 2
- FAIAAWCVCHQXDN-UHFFFAOYSA-N phosphorus trichloride Chemical compound ClP(Cl)Cl FAIAAWCVCHQXDN-UHFFFAOYSA-N 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- 230000035755 proliferation Effects 0.000 description 2
- 238000011321 prophylaxis Methods 0.000 description 2
- 235000019833 protease Nutrition 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- 238000005086 pumping Methods 0.000 description 2
- 125000004307 pyrazin-2-yl group Chemical group [H]C1=C([H])N=C(*)C([H])=N1 0.000 description 2
- 125000002206 pyridazin-3-yl group Chemical group [H]C1=C([H])C([H])=C(*)N=N1 0.000 description 2
- 125000004940 pyridazin-4-yl group Chemical group N1=NC=C(C=C1)* 0.000 description 2
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 2
- 125000004527 pyrimidin-4-yl group Chemical group N1=CN=C(C=C1)* 0.000 description 2
- 125000004528 pyrimidin-5-yl group Chemical group N1=CN=CC(=C1)* 0.000 description 2
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 2
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 2
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 2
- 159000000000 sodium salts Chemical class 0.000 description 2
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 2
- 239000004334 sorbic acid Substances 0.000 description 2
- 235000010199 sorbic acid Nutrition 0.000 description 2
- 229940075582 sorbic acid Drugs 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 150000003456 sulfonamides Chemical class 0.000 description 2
- 125000004434 sulfur atom Chemical group 0.000 description 2
- AKEJUJNQAAGONA-UHFFFAOYSA-N sulfur trioxide Chemical compound O=S(=O)=O AKEJUJNQAAGONA-UHFFFAOYSA-N 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 210000001179 synovial fluid Anatomy 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 150000003573 thiols Chemical class 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- 231100000397 ulcer Toxicity 0.000 description 2
- 239000008215 water for injection Substances 0.000 description 2
- 229910052725 zinc Inorganic materials 0.000 description 2
- 239000011701 zinc Substances 0.000 description 2
- MGAXYKDBRBNWKT-UHFFFAOYSA-N (5-oxooxolan-2-yl)methyl 4-methylbenzenesulfonate Chemical compound C1=CC(C)=CC=C1S(=O)(=O)OCC1OC(=O)CC1 MGAXYKDBRBNWKT-UHFFFAOYSA-N 0.000 description 1
- 125000006824 (C1-C6) dialkyl amine group Chemical group 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- ZORQXIQZAOLNGE-UHFFFAOYSA-N 1,1-difluorocyclohexane Chemical compound FC1(F)CCCCC1 ZORQXIQZAOLNGE-UHFFFAOYSA-N 0.000 description 1
- NOGNFTZRPJNCRR-UHFFFAOYSA-N 1-(5a,6,7,8,9,9a-hexahydrodibenzofuran-3-ylsulfonylamino)-n-hydroxycyclopentane-1-carboxamide Chemical compound C=1C=C2C3CCCCC3OC2=CC=1S(=O)(=O)NC1(C(=O)NO)CCCC1 NOGNFTZRPJNCRR-UHFFFAOYSA-N 0.000 description 1
- VJXWPOWOAQSAAV-UHFFFAOYSA-N 1-(6,7,8,9,10,11-hexahydrocycloocta[b][1]benzofuran-3-ylsulfonylamino)-n-hydroxycyclopentane-1-carboxamide Chemical compound C=1C=C2C=3CCCCCCC=3OC2=CC=1S(=O)(=O)NC1(C(=O)NO)CCCC1 VJXWPOWOAQSAAV-UHFFFAOYSA-N 0.000 description 1
- JMTFVHSEOCXLAC-UHFFFAOYSA-N 1-(7,8,9,10-tetrahydro-6h-cyclohepta[b][1]benzofuran-3-ylsulfonylamino)cyclopentane-1-carboxylic acid Chemical compound C=1C=C2C=3CCCCCC=3OC2=CC=1S(=O)(=O)NC1(C(=O)O)CCCC1 JMTFVHSEOCXLAC-UHFFFAOYSA-N 0.000 description 1
- RVISLJHIOJXZTK-UHFFFAOYSA-N 1-[(7-bromodibenzofuran-2-yl)sulfonylamino]-n-hydroxycyclopentane-1-carboxamide Chemical compound C=1C=C2OC3=CC(Br)=CC=C3C2=CC=1S(=O)(=O)NC1(C(=O)NO)CCCC1 RVISLJHIOJXZTK-UHFFFAOYSA-N 0.000 description 1
- HUZNVGNWNVUALF-UHFFFAOYSA-N 1-[(7-bromodibenzofuran-2-yl)sulfonylamino]cyclopentane-1-carboxylic acid Chemical compound C=1C=C2OC3=CC(Br)=CC=C3C2=CC=1S(=O)(=O)NC1(C(=O)O)CCCC1 HUZNVGNWNVUALF-UHFFFAOYSA-N 0.000 description 1
- LDMOEFOXLIZJOW-UHFFFAOYSA-N 1-dodecanesulfonic acid Chemical compound CCCCCCCCCCCCS(O)(=O)=O LDMOEFOXLIZJOW-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- 125000001462 1-pyrrolyl group Chemical group [*]N1C([H])=C([H])C([H])=C1[H] 0.000 description 1
- UOQBPJSYUVKXEU-UHFFFAOYSA-N 2-[(7-chlorodibenzofuran-2-yl)sulfonylamino]-2-methylpropanoic acid Chemical compound ClC1=CC=C2C3=CC(S(=O)(=O)NC(C)(C)C(O)=O)=CC=C3OC2=C1 UOQBPJSYUVKXEU-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- KZMAWJRXKGLWGS-UHFFFAOYSA-N 2-chloro-n-[4-(4-methoxyphenyl)-1,3-thiazol-2-yl]-n-(3-methoxypropyl)acetamide Chemical compound S1C(N(C(=O)CCl)CCCOC)=NC(C=2C=CC(OC)=CC=2)=C1 KZMAWJRXKGLWGS-UHFFFAOYSA-N 0.000 description 1
- ZNQVEEAIQZEUHB-UHFFFAOYSA-N 2-ethoxyethanol Chemical compound CCOCCO ZNQVEEAIQZEUHB-UHFFFAOYSA-N 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 102000017304 72kDa type IV collagenases Human genes 0.000 description 1
- 108050005269 72kDa type IV collagenases Proteins 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 235000019489 Almond oil Nutrition 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 108020000948 Antisense Oligonucleotides Proteins 0.000 description 1
- 206010003162 Arterial injury Diseases 0.000 description 1
- 208000032116 Autoimmune Experimental Encephalomyelitis Diseases 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 206010053567 Coagulopathies Diseases 0.000 description 1
- 206010011091 Coronary artery thrombosis Diseases 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- 240000001879 Digitalis lutea Species 0.000 description 1
- 208000018672 Dilatation Diseases 0.000 description 1
- 201000010046 Dilated cardiomyopathy Diseases 0.000 description 1
- 102000010911 Enzyme Precursors Human genes 0.000 description 1
- 108010062466 Enzyme Precursors Proteins 0.000 description 1
- 102000016359 Fibronectins Human genes 0.000 description 1
- 108010067306 Fibronectins Proteins 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 239000004705 High-molecular-weight polyethylene Substances 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 206010060820 Joint injury Diseases 0.000 description 1
- 208000016593 Knee injury Diseases 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 101150014058 MMP1 gene Proteins 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 240000003183 Manihot esculenta Species 0.000 description 1
- 235000016735 Manihot esculenta subsp esculenta Nutrition 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 108010076502 Matrix Metalloproteinase 11 Proteins 0.000 description 1
- 102100030412 Matrix metalloproteinase-9 Human genes 0.000 description 1
- 108010088571 Membrane-Associated Matrix Metalloproteinases Proteins 0.000 description 1
- 102000008887 Membrane-Associated Matrix Metalloproteinases Human genes 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 208000034827 Neointima Diseases 0.000 description 1
- 102100030411 Neutrophil collagenase Human genes 0.000 description 1
- 101710118230 Neutrophil collagenase Proteins 0.000 description 1
- 102000056189 Neutrophil collagenases Human genes 0.000 description 1
- 108030001564 Neutrophil collagenases Proteins 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 238000000636 Northern blotting Methods 0.000 description 1
- 206010030113 Oedema Diseases 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 208000037273 Pathologic Processes Diseases 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 101000925883 Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Elastase Proteins 0.000 description 1
- 241000219061 Rheum Species 0.000 description 1
- 241000220317 Rosa Species 0.000 description 1
- 241001474728 Satyrodes eurydice Species 0.000 description 1
- 235000002595 Solanum tuberosum Nutrition 0.000 description 1
- 244000061456 Solanum tuberosum Species 0.000 description 1
- 101000693530 Staphylococcus aureus Staphylokinase Proteins 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 101000584292 Streptomyces cacaoi Mycolysin Proteins 0.000 description 1
- 101710108792 Stromelysin-2 Proteins 0.000 description 1
- 108050005271 Stromelysin-3 Proteins 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 241000282898 Sus scrofa Species 0.000 description 1
- 108700005078 Synthetic Genes Proteins 0.000 description 1
- 208000007536 Thrombosis Diseases 0.000 description 1
- 108010031374 Tissue Inhibitor of Metalloproteinase-1 Proteins 0.000 description 1
- 102000005353 Tissue Inhibitor of Metalloproteinase-1 Human genes 0.000 description 1
- 102000005876 Tissue Inhibitor of Metalloproteinases Human genes 0.000 description 1
- 108010005246 Tissue Inhibitor of Metalloproteinases Proteins 0.000 description 1
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 1
- 230000001594 aberrant effect Effects 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 239000003655 absorption accelerator Substances 0.000 description 1
- 239000003070 absorption delaying agent Substances 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 229940022663 acetate Drugs 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 125000004442 acylamino group Chemical group 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- 208000019269 advanced heart failure Diseases 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 125000004183 alkoxy alkyl group Chemical group 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000004644 alkyl sulfinyl group Chemical group 0.000 description 1
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 208000030961 allergic reaction Diseases 0.000 description 1
- 239000008168 almond oil Substances 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- CEGOLXSVJUTHNZ-UHFFFAOYSA-K aluminium tristearate Chemical compound [Al+3].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CEGOLXSVJUTHNZ-UHFFFAOYSA-K 0.000 description 1
- 229940063655 aluminum stearate Drugs 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 230000006229 amino acid addition Effects 0.000 description 1
- 238000002399 angioplasty Methods 0.000 description 1
- 239000002333 angiotensin II receptor antagonist Substances 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000000074 antisense oligonucleotide Substances 0.000 description 1
- 238000012230 antisense oligonucleotides Methods 0.000 description 1
- 210000000709 aorta Anatomy 0.000 description 1
- 206010002906 aortic stenosis Diseases 0.000 description 1
- 125000002029 aromatic hydrocarbon group Chemical group 0.000 description 1
- 206010003119 arrhythmia Diseases 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 230000001363 autoimmune Effects 0.000 description 1
- 210000002469 basement membrane Anatomy 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 239000002876 beta blocker Substances 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M bisulphate group Chemical group S([O-])(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 230000008499 blood brain barrier function Effects 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 210000001218 blood-brain barrier Anatomy 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 235000010216 calcium carbonate Nutrition 0.000 description 1
- FATUQANACHZLRT-KMRXSBRUSA-L calcium glucoheptonate Chemical compound [Ca+2].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O FATUQANACHZLRT-KMRXSBRUSA-L 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 210000000845 cartilage Anatomy 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 108020001778 catalytic domains Proteins 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 239000013522 chelant Substances 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- 230000035602 clotting Effects 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 230000003366 colagenolytic effect Effects 0.000 description 1
- 108700004333 collagenase 1 Proteins 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000004891 communication Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 210000004087 cornea Anatomy 0.000 description 1
- 238000007887 coronary angioplasty Methods 0.000 description 1
- 208000002528 coronary thrombosis Diseases 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 150000001925 cycloalkenes Chemical class 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- ZWAJLVLEBYIOTI-UHFFFAOYSA-N cyclohexene oxide Chemical compound C1CCCC2OC21 ZWAJLVLEBYIOTI-UHFFFAOYSA-N 0.000 description 1
- FWFSEYBSWVRWGL-UHFFFAOYSA-N cyclohexene oxide Natural products O=C1CCCC=C1 FWFSEYBSWVRWGL-UHFFFAOYSA-N 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 230000006735 deficit Effects 0.000 description 1
- 230000000593 degrading effect Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 230000001687 destabilization Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 239000003599 detergent Substances 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 125000005265 dialkylamine group Chemical group 0.000 description 1
- CGJNXSWIIBSXII-UHFFFAOYSA-N dibenzofuran-3-sulfonyl chloride Chemical compound C1=CC=C2C3=CC=C(S(=O)(=O)Cl)C=C3OC2=C1 CGJNXSWIIBSXII-UHFFFAOYSA-N 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- UGMCXQCYOVCMTB-UHFFFAOYSA-K dihydroxy(stearato)aluminium Chemical compound CCCCCCCCCCCCCCCCCC(=O)O[Al](O)O UGMCXQCYOVCMTB-UHFFFAOYSA-K 0.000 description 1
- 201000011304 dilated cardiomyopathy 1A Diseases 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- POULHZVOKOAJMA-UHFFFAOYSA-M dodecanoate Chemical compound CCCCCCCCCCCC([O-])=O POULHZVOKOAJMA-UHFFFAOYSA-M 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 239000008157 edible vegetable oil Substances 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 238000005530 etching Methods 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 210000001723 extracellular space Anatomy 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 239000003885 eye ointment Substances 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 102000013373 fibrillar collagen Human genes 0.000 description 1
- 108060002894 fibrillar collagen Proteins 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 210000000497 foam cell Anatomy 0.000 description 1
- 239000003205 fragrance Substances 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 208000024693 gingival disease Diseases 0.000 description 1
- 208000007565 gingivitis Diseases 0.000 description 1
- 230000024924 glomerular filtration Effects 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- IXCSERBJSXMMFS-UHFFFAOYSA-N hcl hcl Chemical compound Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 230000005802 health problem Effects 0.000 description 1
- 208000019622 heart disease Diseases 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- 230000036732 histological change Effects 0.000 description 1
- 239000008240 homogeneous mixture Substances 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 229960003444 immunosuppressant agent Drugs 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 238000007901 in situ hybridization Methods 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 238000010874 in vitro model Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 230000008595 infiltration Effects 0.000 description 1
- 238000001764 infiltration Methods 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 125000002510 isobutoxy group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])O* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 210000003292 kidney cell Anatomy 0.000 description 1
- 201000006370 kidney failure Diseases 0.000 description 1
- 229940070765 laurate Drugs 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 210000002414 leg Anatomy 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 230000033001 locomotion Effects 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000006609 metabolic stress Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- HBXPTBDRXALAQN-UHFFFAOYSA-N methyl 2-(5a,6,7,8,9,9a-hexahydrodibenzofuran-3-ylsulfonylamino)-2-methylpropanoate Chemical compound C1CCCC2OC3=CC(S(=O)(=O)NC(C)(C)C(=O)OC)=CC=C3C21 HBXPTBDRXALAQN-UHFFFAOYSA-N 0.000 description 1
- AZFBPYNPITWILD-UHFFFAOYSA-N methyl 4-aminooxane-4-carboxylate Chemical compound COC(=O)C1(N)CCOCC1 AZFBPYNPITWILD-UHFFFAOYSA-N 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229960002900 methylcellulose Drugs 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- 208000037891 myocardial injury Diseases 0.000 description 1
- 208000031225 myocardial ischemia Diseases 0.000 description 1
- 210000004165 myocardium Anatomy 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- IJQPRCCBJSWHQA-UHFFFAOYSA-N n-hydroxy-1-(6,7,8,9-tetrahydrodibenzofuran-3-ylsulfonylamino)cyclopentane-1-carboxamide Chemical compound C=1C=C2C=3CCCCC=3OC2=CC=1S(=O)(=O)NC1(C(=O)NO)CCCC1 IJQPRCCBJSWHQA-UHFFFAOYSA-N 0.000 description 1
- ZQYUZMNSJLFCSN-UHFFFAOYSA-N n-hydroxy-1-(7,8,9,10-tetrahydro-6h-cyclohepta[b][1]benzofuran-3-ylsulfonylamino)cyclopentane-1-carboxamide Chemical compound C=1C=C2C=3CCCCCC=3OC2=CC=1S(=O)(=O)NC1(C(=O)NO)CCCC1 ZQYUZMNSJLFCSN-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000005487 naphthalate group Chemical group 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 201000008383 nephritis Diseases 0.000 description 1
- 230000004770 neurodegeneration Effects 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 239000002687 nonaqueous vehicle Substances 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000012053 oil suspension Substances 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- 230000003349 osteoarthritic effect Effects 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- 229940039748 oxalate Drugs 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 239000006201 parenteral dosage form Substances 0.000 description 1
- 239000003182 parenteral nutrition solution Substances 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 230000009054 pathological process Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- 201000001245 periodontitis Diseases 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 229960003742 phenol Drugs 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 230000007505 plaque formation Effects 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000002250 progressing effect Effects 0.000 description 1
- 230000007425 progressive decline Effects 0.000 description 1
- 201000008171 proliferative glomerulonephritis Diseases 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- ALDITMKAAPLVJK-UHFFFAOYSA-N prop-1-ene;hydrate Chemical group O.CC=C ALDITMKAAPLVJK-UHFFFAOYSA-N 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 229940080818 propionamide Drugs 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 1
- 230000017854 proteolysis Effects 0.000 description 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 238000011552 rat model Methods 0.000 description 1
- 239000011535 reaction buffer Substances 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 238000007634 remodeling Methods 0.000 description 1
- 206010038464 renal hypertension Diseases 0.000 description 1
- 230000036454 renin-angiotensin system Effects 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- 150000004760 silicates Chemical class 0.000 description 1
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 235000020374 simple syrup Nutrition 0.000 description 1
- 210000002460 smooth muscle Anatomy 0.000 description 1
- 230000015590 smooth muscle cell migration Effects 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- MWQQWXSAOPTRAQ-UHFFFAOYSA-M sodium;1,2,3,4-tetrahydrodibenzofuran-3-sulfonate Chemical compound [Na+].O1C2=CC=CC=C2C2=C1CC(S(=O)(=O)[O-])CC2 MWQQWXSAOPTRAQ-UHFFFAOYSA-M 0.000 description 1
- QAJGUMORHHJFNJ-UHFFFAOYSA-M sodium;phenoxide;trihydrate Chemical compound O.O.O.[Na+].[O-]C1=CC=CC=C1 QAJGUMORHHJFNJ-UHFFFAOYSA-M 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 235000011069 sorbitan monooleate Nutrition 0.000 description 1
- 239000001593 sorbitan monooleate Substances 0.000 description 1
- 229940035049 sorbitan monooleate Drugs 0.000 description 1
- 229960002920 sorbitol Drugs 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 238000011272 standard treatment Methods 0.000 description 1
- 230000003068 static effect Effects 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- 125000006296 sulfonyl amino group Chemical group [H]N(*)S(*)(=O)=O 0.000 description 1
- 229910021653 sulphate ion Inorganic materials 0.000 description 1
- 230000002889 sympathetic effect Effects 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- CBXCPBUEXACCNR-UHFFFAOYSA-N tetraethylammonium Chemical compound CC[N+](CC)(CC)CC CBXCPBUEXACCNR-UHFFFAOYSA-N 0.000 description 1
- QEMXHQIAXOOASZ-UHFFFAOYSA-N tetramethylammonium Chemical compound C[N+](C)(C)C QEMXHQIAXOOASZ-UHFFFAOYSA-N 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 230000007838 tissue remodeling Effects 0.000 description 1
- 239000002407 tissue scaffold Substances 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000001131 transforming effect Effects 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 230000005747 tumor angiogenesis Effects 0.000 description 1
- 102000003390 tumor necrosis factor Human genes 0.000 description 1
- 231100000588 tumorigenic Toxicity 0.000 description 1
- 230000000381 tumorigenic effect Effects 0.000 description 1
- 210000004231 tunica media Anatomy 0.000 description 1
- 230000007306 turnover Effects 0.000 description 1
- 230000036269 ulceration Effects 0.000 description 1
- 229940070710 valerate Drugs 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- 230000002792 vascular Effects 0.000 description 1
- 210000005167 vascular cell Anatomy 0.000 description 1
- 208000019553 vascular disease Diseases 0.000 description 1
- 230000024883 vasodilation Effects 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 239000011345 viscous material Substances 0.000 description 1
- 230000037314 wound repair Effects 0.000 description 1
- 230000004572 zinc-binding Effects 0.000 description 1
- 238000007805 zymography Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D307/91—Dibenzofurans; Hydrogenated dibenzofurans
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/02—Stomatological preparations, e.g. drugs for caries, aphtae, periodontitis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P23/00—Anaesthetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/08—Vasodilators for multiple indications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/14—Vasoprotectives; Antihaemorrhoidals; Drugs for varicose therapy; Capillary stabilisers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/15—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings
- C07C311/16—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the sulfonamide groups bound to hydrogen atoms or to an acyclic carbon atom
- C07C311/19—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the sulfonamide groups bound to hydrogen atoms or to an acyclic carbon atom to an acyclic carbon atom of a hydrocarbon radical substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/15—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings
- C07C311/20—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the sulfonamide groups bound to a carbon atom of a ring other than a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D307/93—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems condensed with a ring other than six-membered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D407/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00
- C07D407/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings
- C07D407/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/06—Systems containing only non-condensed rings with a five-membered ring
- C07C2601/08—Systems containing only non-condensed rings with a five-membered ring the ring being saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
Description
- Gebiet der Erfindung
- Die vorliegende Erfindung betrifft eine Gruppe von Hydroxamsäureverbindungen und -derivaten, die Matrixmetalloproteinaseenzyme hemmen und daher zur Behandlung von Erkrankungen, die von einem Gewebeabbau herrühren, wie eine Herzerkrankung, Multiple Sklerose, Arthritis, Atherosklerose und Osteoporose, verwendbar sind.
- Hintergrund der Erfindung
- Matrixmetalloproteinasen (die manchmal als MMPs bezeichnet werden) sind natürlich vorkommende Enzyme, die sich bei den meisten Säugern finden. Eine Überexpression und Aktivierung von MMPs oder ein Ungleichgewicht zwischen MMPs und Inhibitoren von MMPs wurden als Faktoren bei der Pathogenese von Erkrankungen, die durch den Abbau der extrazellulären Matrix oder von Bindegeweben gekennzeichnet sind, vorgeschlagen.
- Stromelysin-1 und Gelatinase A sind Mitglieder der Familie der Matrixmetalloproteinasen (MMP). Andere Mitglieder umfassen Fibroblastenkollagenase (MMP-1), Neutrophilenkollagenase (MMP-8), Gelatinase B (92-kDa-Gelatinase) (MMP-9), Stromelysin-2 (MMP-10), Stromelysin-3 (MMP-11), Matrilysin (MMP-7), Kollagenase 3 (MMP-13), TNF-Alpha-Converting-Enzyme (TACE) und andere neu entdeckte membranassoziierte Matrixmetalloproteinasen (H. Sato, T. Takino, Y. Okada, J. Cao, A. Shinagawa, E. Yamamota und M. Seiki, Nature, 1994, 370: 61–65). Diese Enzyme wurden mit einer Zahl von Erkrankungen, die von einem Abbau von Bindegewebe herrühren, die Erkrankungen wie rheumatoide Arthritis, Osteoarthritis, Osteoporose, Periodontitis, Multiple Sklerose, Gingivitis, Corneaepidermis- und Magenulzeration, Atherosklerose, Neointimaproliferation, die zu Restenose und ischämischer Herzinsuffizienz führt, und Tumormetastasen umfassen, impliziert. Es wurde nun erkannt, dass ein Verfahren zur Prophylaxe und Behandlung dieser und anderer Erkrankungen darin besteht, Matrixmetalloproteinaseenzyme zu hemmen, wodurch der Abbau von Bindegeweben, der zu den Erkrankungszuständen führt, herabgesetzt und/oder aufgehoben wird.
- Das katalytische Zink in Matrixmetalloproteinasen ist typischerweise der Brennpunkt für die Inhibitorgestaltung. Die Modifizierung von Substraten durch Einführen von mit Zink ein Chelat bildenden Gruppen erzeugte wirksame Inhibitoren, wie Peptidhydroxamate und Thiol enthaltende Peptide. Peptidhydroxamate und natürliche endogene Inhibitoren von MMPs (TIMPs) wurden erfolgreich zur Behandlung von Tiermodellen von Krebs und Entzündungen verwendet.
- Die Fähigkeit der Matrixmetalloproteinasen zum Abbauen verschiedener Komponenten von Bindegewebe macht sie zu potentiellen Targets für die Kontrolle pathologischer Prozesse. Beispielsweise ist das Losbrechen atherosklerotischer Plaques das häufigste Ereignis, das eine Koronarthrombose auslöst. Die Destabilisierung und der Abbau der extrazellulären Matrix, die diese Plaques umgibt, durch MMPs wurde als eine Ursache für Plaquerissbildung vorgeschlagen. Die Schultern und Regionen einer Schaumzellansammlung in humanen atherosklerotischen Plaques zeigen eine lokal erhöhte Expression von Gelatinase B, Stromelysin-1 und interstiteller Kollagenase. Die In-situ-Zymographie dieses Gewebes ergab eine erhöhte Gelatinolyse- und Caseinolyseaktivität (Z. S. Galla, G. K. Sukhova, M. W. Lark und P. Libby, "Incraesed expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human athero sclerotic plaques", J. Clin. Invest., 1994; 94: 2494–2503). Außerdem wurde ermittelt, dass große Mengen der Stromelysin-RNA-Botschaft in individuellen Zellen in atherosklerotischen Plaques, die Herztransplantationspatienten zum Zeitpunkt der Operation entnommen wurden, lokalisiert sind (A. M. Henney, P. R. Wakeley, M. J. Davies, K. Foster, R. Hembry, G. Murphy und S. Humphries, "Localization of stromelysin gene expression in atherosclerotic plaques by in situ hybridization", Proc. Nat'l. Acad. Sci., 1991; 88: 8154–8158).
- Inhibitoren von Matrixmetalloproteinasen sind bei der Behandlung einer degenerativen Aortaerkrankung, die mit dem Dünnwerden der Media-Aortawand verbunden sind, von Nutzen. Erhöhte Mengen der Pxoteolyseaktitiväten von MMPs wurden bei Patienten mit Aortaaneurysmen und Aortastenose identifiziert (N. Vine und J. T. Powell, "Metalloproteinases in degenerative aortic diseases", Clin. Sci., 1991; 81: 233– 239).
- Herzinsuffizienz ergibt sich aufgrund einer Vielzahl verschiedener Ätiologien, doch ist eine häufige kennzeichnende Eigenschaft eine Herzdilatation, die als ein unabhängiger Risikofaktor für Mortalität identifiziert wurde (T. H. Lee, M. A. Hamilton, L. W. Stevenson, J. D. Moriguchi, G. C. Fonarow, J. S. Child, H. Laks und J. A. Walden, "Impact of left ventricular size on the survival in advanced heart failure", Am. J. Cardiol., 1993; 72: 672–676). Diese Neubildung des versagenden Herzens scheint den Abbau extrazellulärer Matrix zu umfassen. Matrixmetalloproteinasen sind bei Patienten mit sowohl idiopathischer als auch ischämischer Herzinsuffizienz erhöht (A. K. Reddy, S. C. Tyagi, I. E. Tjaha, D. J. Voelker, S. E. Campbell und K. T. Weber, "Activated myocardial collagenase in idiopathic dilated cardiomyopathy", Clin. Res., 1993; 41: 660A; S. C. Tyagi, H. K. Reddy, D. Voelker, I. E. Tjara und K. T. Weber, "Myocardial collagenase in failing human heart", Clin. Res., 1993; 41: 681A). Tiermodelle von Herzinsuffizienz zeigten, dass die Induktion von Gelatinase bei Herzdilatation wichtig ist (P. W. Armstrong, G. W. Moe, R. J. Howard, E. A. Grima und T. F. Cruz, "Structural remodeling in heart failure: gelatinase induction", Can. J. Cardiol., 1994; 10: 214–220), und eine Herzdilatation tiefgreifenden Defiziten der Herzfunktion vorausgeht (H. N. Sabbah, T. Kono, P. D. Stein, G. B. Mancini und S. Goldstein, "Left ventricular shape changes during the course of evolving heart failure", Am. J. Physiol., 1992; 263: H266–H270).
- Stauungsherzinsuffizienz (CHF) ist ein bedeutendes Gesundheitsproblem, das derzeit 7% der gesamten Gesundheitsausgaben in den USA ausmacht. Etwa 400000 neue Fälle von Herzinsuffizienz werden jährlich festgestellt. Die primäre Ursache für die Entwicklung von Herzinsuffizienz ist eine ischämische Herzerkrankung und die meisten neuen Fälle treten nach einem Myokardinfarkt auf. Die Zahl der Krankenhauseinweisungen wegen Herzinsuffizienz stieg von 377000 im Jahre 1979 auf 875000 im Jahre 1993, und die Zahl der Todesfälle während des gleichen Zeitraums stieg um 82,5%. Die durchschnittliche Mortalitätsrate acht Jahre nach der ersten Diagnose beträgt 85% für Männer und 65% für Frauen.
- Die Entwicklung von CHF beginnt als ein Verletzungsprozess des Myokard, der die Herzfunktion (insbesondere die Kontraktions- oder Pumpfunktion) entweder in einer spezifischen Region bzw. spezifischen Regionen oder im gesamten Ausmaß (d. h. global) verringert. Herzinsuffizienz ist vorhanden, wenn die Myokardverletzung von einer derart ausreichenden Schwere ist, dass die Fähigkeit des Herzens zum Pumpen eines adäquaten Blutausstoßes, der die Bedürfnisse der Körpergewebe entweder in Ruhe oder während Bewegung erfüllt, verringert ist. Der Krankheitszustand von Herzinsuffizienz ist keine statische Situation, sondern er wird statt dessen fortschreitend schlechter, bis der Tod entweder plötzlich (beispielsweise durch Herzarrhythmien oder Embolie des Hirns oder der Lunge) oder allmählich aufgrund eines Pumpversagens als solchem auftritt. Die fortschreitende Abnahme der Herzfunktion bei Patienten mit CHF ist durch eine fortschreitende Vergrößerung der Ventrikelkammern (d. h. Ventrikeldilatation) und ein Dünnwerden und Fibrose der Ventrikelmuskulatur gekennzeichnet. Die fortschreitende Ventrikelvergrößerung und die begleitenden histologischen Veränderungen der Ventrikelmuskulatur werden als "Neubildung bzw. Umbildung" bezeichnet, ein Prozess, der Veränderungen der Myokardiocytenstruktur sowie Änderungen der Menge und der Zusammensetzung des umgebenden interstitiellen Bindegewebes umfasst. Ein wichtiger Bestandteil des interstitiellen Bindegewebes ist eine Matrix aus fibrillärem Kollagen, dem "Gewebegerüst", das zum Aufrechterhalten einer passenden Ventrikelgeometrie und der strukturellen Anordnung der anschließenden Kardiamyocyten beiträgt. Die interstitielle Kollagenmatrix unterliegt einem erhöhten Auflösen und Reparieren während der "Neubildung bzw. Umbildung", die zu einer Ventrikelvergrößerung und fortschreitender Herzinsuffizienz führt. Die Verschlechterung der Kollagenmatrix wird durch eine erhöhte Aktivität von Matrixmetalloproteinasen bewirkt, wobei deren Hemmung eine neue Behandlung bei Herzinsuffizienz und Ventrikeldilatation ist. Die Ventrikeldilatation, deren Schwere durch die enddiastolischen und endsystolischen Volumina ermittelt wird, ist ein Prognosemarker der Wahrscheinlichkeit einer folgenden Morbidität und Mortalität. Je größer die Ventrikelkammerabmessungen sind, desto größer ist die Wahrscheinlichkeit anschließender Fälle von Morbidität. Es wird nicht nur die Pumpfunktion durch eine Neubildung bzw. Umbildung und Ventrikeldilatation beeinträchtigt, sondern die vergrößerten Kammern sind für die Bildung von Gerinnungspfropfen, die zu Schlag oder Embolie bei anderen Hauptorganen (beispielsweise Niere, Beine, Intestinaltrakt) führen können, anfällig.
- Eine Standardbehandlung bei Herzinsuffizienz nutzt Diuretika zur Verringerung der Flüssigkeitsretention, Angiotensin-Converting-Enzyme-Inhibitoren (ACE-Is) zur Verringerung der Herzbelastung des versagenden Herzens durch eine Vasodilatation, und in den Endstadien eines Versagens das positive Inotrop Digitalis zum Aufrechterhalten der Herzleistung. Obwohl ACE-Is im Gegensatz zu Diuretika oder positiven Inotropen den Vorteil der Lebensverlängerung aufweisen, ist die vorteilhafte Wirkung von ACE-Is darauf beschränkt, den Tod um nur etwa 18 Monate zu verzögern. Klinische Versuche mit β-adrenergen Blockern wurden vor kurzem auf der Grundlage der Hypothese, dass eine Verringerung des Sympathikusantriebs die metabolische Belastung der Herzmuskelzellen verringert, durchgeführt. Unglücklicherweise wurde auch ermittelt, dass diese Klasse von Verbindungen keine wesentliche Wirkung auf das Fortschreiten von Herzinsuffizienz hat. Der Nichterfolg oder beschränkte Erfolg von früheren Therapien von Herzinsuffizienz zeigt klar, dass der Kontrollmechanismus bzw. die Kontrollmechanismen, die Herzinsuffizienz vermitteln, nicht gezielt erreicht wurden.
- Die Arzneimittelentwicklung für die Behandlung von Herzinsuffizienz konzentrierte sich seit den 60er Jahren auf Herzmuskelzellen. Das Ziel war die Verringerung der Arbeitsbelastung der Zellen, die Verbesserung der Durchblutung der Zellen, die Erhöhung der Kontraktion der Muskulatur, die Verminderung der Stoffwechselbelastung der Kardiomyocyten oder eine Kombination von diesen durch verschiedene Mittel. Die Konzentration auf Kardiomyocyten kann dazu gedient haben, die Aufmerksamkeit auf einen zu weit stromabwärts gelegenen Teil zu konzentrieren. Eine offenkundige Herzinsuffizienz kann durch den Abbau von Herzbindegewebe verursacht sein. Der Abbau bei Herzbindegewebeproteinen vermittelt daher eine Herzdilatation, eines der frühesten Kennzeichen einer Herzinsuffizienz.
- Wir entdeckten nun, dass Verbindungen, die MMPs, die den Abbau von Bindegewebe vermitteln, hemmen, zur Behandlung von Herzinsuffizienz und einer damit verbundenen Ventrikeldilatation verwendbar sind.
- Eine Neointimaproliferation, die zur Restenose führt, entwickelt sich häufig nach einer Koronarangioplastik. Die Wanderung von Gefäßzellen glatter Muskulatur (VSMCs) von der Tunica media zur Neointima ist ein Schlüsselereignis bei der Entwicklung und dem Fortschreiten von vielen Gefäßerkrankungen und eine stark vorhersagbare Konsequenz einer mechanischen Verletzung eines Blutgefäßes (M. P. Bendeck, N. Zempo, A. W. Clowes, R. E. Galardy und M. Reidy, "Smooth muscle cell migration and matrix metalloproteinase expression after arterial injury in the rat", Circulation Research, 1994; 75: 539–545). Northern Blotting und zymographische Analysen zeigten, dass Gelatinase A die Haupt-MMP war, die von diesen Zellen exprimiert und ausgeschüttet wurde. Ferner hemmten Antiseren mit der Fähigkeit zur selektiven Neutralisation von Gelatinase-A-Aktivität auch die VSMC-Migration über die Basalmembranschranke. Nach einer Verletzung des Gefäßes erhöhte sich die Gelatinase-A-Aktivität auf das mehr als 20-fache, wenn VSMCs den Übergang vom ruhenden Zustand zum proliferierenden beweglichen Phänotyp vollzogen (R. R. Pauly, A. Passaniti, C. Bilato, R. Monticone, L. Cheng, N. Papadopoulos, Y. A. Gluzband, L. Smith, C. Weinstein, E. Lakatta und M. T. Crow, "Migration of cultured vascular smooth muscle cells through a basement membrane barrier requires type IV collagenase activity and is inhibited by cellular differentiation", Circulation Research, 1994; 75: 41–54).
- Die normale Nierenfunktion hängt vom Aufrechterhalten von Geweben, die aus differenzierten und hochspezialisierten Nierenzellen, die in dynamischem Gleichgewicht mit den Komponenten der umgebenden extrazellulären Matrix (ECM) sind, aufgebaut sind, ab (M. Davies et al., "Proteinales and glomerular matrix turnover", Kidney Int., 1992; 41: 671– 678). Eine wirksame glomeruläre Filtration erfordert, dass eine semipermeable glomeruläre Basalmembran (GBM), die aus Kollagenen, Fibronectin, Enactin, Laminin und Proteoglykanen besteht, aufrechterhalten wird. Ein strukturelles Gleichgewicht wird durch Ausbalancieren der fortgesetzten Abscheidung von ECM-Proteinen mit deren Abbau durch spezifische Metalloproteinasen (MMP) erreicht. Diese Proteine werden zunächst als Proenzyme sezerniert und anschließend im extrazellulären Raum aktiviert. Diese Proteinasen unterliegen wiederum einer entgegengesetzt ausgleichenden Regulierung ihrer Aktivität durch natürlich vorkommende Inhibitoren, die als TIMPs (Gewebeinhibitoren von Metalloproteinasen) bezeichnet werden.
- Ein Mangel oder Defekte einer Komponente der Filtrationsschranke können katastrophale Folgen für die längerfristige Nierenfunktion haben. Beispielsweise führen bei ererbter Nephritis des Alport-Typs, die mit Mutationen in Genen mit Codierung für ECM-Proteine verbunden ist, Defekte im Kollagenzusammenbau zu fortschreitendem Nierenversagen, das mit einer Aufspaltung der GBM und schließlich glomerulärer und interstitieller Fibrose verbunden ist. Im Gegensatz dazu geht bei entzündlichen Nierenerkrankungen, wie Glomerulonephritis, eine Zellproliferation von Komponenten des Glomerulus häufig einer offensichtlichen ultrastrukturellen Ver änderung der ECM-Matrix voraus. Cytokine und Wachstumsfaktoren, die bei proliferativer Glomerulonephritis impliziert sind, wie Interleukin-1, Tumornekrosefaktor und Umwandlungswachstumsfaktor β, können die Metalloproteinaseexpression in Nierenmesangiumzellen hochregulieren (J. Martin et al., "Enhancement of glomerular mesangial cell neutral proteinase secretion by macrophages: role of Interleukin 1", J. Immunol., 1986; 137: 525–529; H. P. Marti et al., "Homology cloning of rat 72 kDa type IV collagenase: Cytokine and second-messenger inducibility in mesangial cells", Biochem. J., 1993; 291: 441–446; H. P. Marti et al., "Transforming growth factor-b stimulates glomerular mesangial cell synthesis of the 72 kDa type IV collagenase", Am. J. Pathol., 1994; 144: 82–94). Von diesen Metalloproteinasen wird angenommen, dass sie an der fehlerhaften Gewebeumbildung bzw. -neubildung und Zellproliferation, die für Nierenerkrankungen, wie IgA-Nephtopathie, charakteristisch sind, die über den Prozess einer allmählichen glomerulären Fibrose und die Abnahme der funktionellen GBM bis zum Endstadium einer Nierenerkrankung fortschreiten können, beteiligt sind. Die Metalloproteinaseexpression wurde bei einer experimentellen Immunkomplexvermittelten Glomerulonephritis, beispielsweise dem Anti-Thy-1.1-Rattenmodell, gut charakterisiert (W. M. Bagchus, P. J. Hoedemaeker, J. Rozing, W. W. Bakker, "Glumerulonephritis induced by monoclonal anti-Thy 1.1 antibodies: A sequential histological and ultrastructural study in the rat", Lab. Invest., 1986; 55: 680–687; D. H. Lovett, R. J. Johnson, H. P. Marti, J. Martin, M. Davies, W. G. Couser, "Structural characterization of the mesangial cell type IV collagenase and enhanced expression in a model of immune complex mediated glomerulonephritis", Am. J. Pathol., 1992; 141: 85–98).
- Unglücklicherweise bestehen derzeit sehr beschränkte thera peutische Strategien zur Modifizierung des Verlaufs einer fortschreitenden Nierenerkrankung. Obwohl viele Nierenerkrankungen eine entzündliche Komponente aufweisen, sind deren Reaktionen auf Therapien mit Standardimmunsuppressiva nicht vorhersagbar und für einzelne Patienten möglicherweise gefährlich. Die Sekundärfolgen eines allmählichen Nephronversagens, wie eine Aktivierung des Renin-Angiotensin-Systems, begleitet von einer individuellen glomerulären Nephron-Hyperfiltration und Nierenhypertonie, kann mit ACE-Inhibitoren oder Angiotension-II-Rezeptorantagonisten wirksam behandelt werden; doch können diese Verbindungen bestenfalls nur die Rate der GFR-Abnahme verringern.
- Eine neue Strategie zur Behandlung von mindestens einigen Nierenerkrankungen wurde durch vor kurzem erfolgte Beobachtungen des MMP-Verhaltens nahegelegt. Eine Ratten-Mesangialzellen-MMP wurde kloniert (MMP-2), die in gewebespezifischer Weise reguliert wird und im Gegensatz zu anderen zellulären Quellen, wie Tumorzelllinien, durch Cytokine induziert wird (P. D. Brown, A. T. Levy, I. Margulies, L. A. Liotta, W. G. Stetler-Stevenson, "Independent expression and cellular processing of Mr 72,000 type IV collagenase and interstitial collagenase in human tumorigenic cell lines", Cancer Res., 1990; 50: 6148–6191; H. P. Marti et al., "Homology cloning of rat 72 kDa type IV collagenase: Cytokine and second-messenger inducibility in mesangial cells", Biochem. J., 1993; 291: 441–446). Während MMP-2 spezifisch umgebende ECM abbauen kann, beeinflusst sie auch den Phänotyp von angrenzenden Mesangialzellen. Die Hemmung von MMP-2 durch Antisense-Oligonucleotide oder Transfektionsverfahren kann eine Umwandlung des proliferativen Phänotyps von gezüchteten Mesangialzellen in einen ruhenden oder nicht-proliferativen Phänotyp, der das natürliche In-vitro-Verhalten dieser Zellen nachahmt, induzieren (M. Kitamura et al., "Gene transfer of metalloproteinase transin induces aberrant behaviour of cultured mesangial cells", Kidney Int., 1994; 45: 1580–1586; J. Turck et al., "Matrix metalloproteinase 2 (gelatinase A) regulates glomerular mesangial cell proliferation and differentiation", J. Biol. Chem., 1996; 271: 15074–15083).
- Kollagenase- und Stromelysinaktivität wurde in Fibroblasten, die von entzündetem Zahnfleisch isoliert wurden, belegt (V. J. Uitto, R. Applegren und P. J. Robinson, "Collagenase and neutral metalloproteinase activity in extracts from inflamed human gingiva", J. Periodontal Res., 1981; 16: 417–424), und die Enzymkonzentrationen wurden mit der Schwere der Zahnfleischerkrankung korreliert (C. M. Overall, O. W. Wiebkin und J. C. Thonard, "Demonstrations of tissue collagenase activity in vivo and its relationship to inflammation severety in human gingiva", J. Periodontal Res., 1987; 22: 81–88). Ein proteolytischer Abbau der extrazellulären Matrix wurde bei einer Corneaulzeration nach alkalischer Verätzung beobachtet (S. E. Brown, C. A. Weller und H. E. Wasserman, "Collagenolytic activity of alkali burned coreas", Arch. Ophthalmol., 1969; 81: 370–373). Thiol enthaltende Peptide hemmen die Kollagenase, die aus alkaliverätzten Kaninchencorneas isoliert wurde (F. R. Burns, M. S. Stack, R. D. Gray und C. A. Patgerson, Invest. Ophthalmol., 1989; 30: 1569–1575).
- Stromelysin wird durch Basalkeratinocyten bei einer Vielzahl chronischer Ulzera produziert (U. K. Saarialho-Kere, K. Ulpu, A. P. Pentland, H. Birkedal-Hansen, W. C. Parks, H. G. Welgus, "Distinct populations of basal keratinocytes express stromelysin-1 and stromelysin-2 in chronic wounds", J. Clin. Invest., 1994; 94: 79–88).
- Stromelysin-1-mRNA und -Protein wurden in Basalkeratinocyten in der Nähe des Wundrands, jedoch entfernt von die sem, was wahrscheinlich die Stellen der proliferierenden Epidermis darstellt, nachgewiesen. Stromelysin kann daher ein Heilen der Epidermis verhindern.
- Davies et al. (Cancer Res., 1993; 53: 2087–2091) berichteten, dass ein Peptidhydroxamat, BB-94, die Tumorlast verringerte und das Überleben von Mäusen, die humane Eierstockkarzinomxenotransplantate trugen, verlängerte. Ein Peptid der konservierten MMP-Propeptidsequenz war ein schwacher Inhibitor von Gelatinase A und es hemmte das Eindringen von humanen Tumorzellen durch eine Schicht einer wiederhergestellten Basalmembran (A. Melchiori, A. Albili, J. M. Ray und W. G. Stetler-Stevenson, Cancer Res., 1992; 52: 2353–2356), und der natürliche Gewebeinhibitor von Metalloproteinase-2 (TIMP-2) zeigte ebenfalls eine Blockierung des Eindringens von Tumorzellen bei In-vitro-Modellen (Y. A. DeClerck, N. Perez, H. Shimada, Z. C. Boone, K. E. Langley und S. M. Taylor, Cancer Res., 1992; 52: 701–708). Untersuchungen von humanen Krebsarten zeigten, dass Gelatinase A auf der Zelloberfläche eines invasiven Tumors aktiviert ist (A. Y. Strongin, B. L. Marmer, G. A. Grant und G. E. Goldberg, J. Biol. Chem., 1993, 268: 14033–14039) und dort durch Wechselwirkung mit einem rezeptorähnlichen Molekül gehalten wird (W. L. Monsky, T. Kelly, C. Y. Lin, Y. Yeh, W. G. Stetler-Stevenson, S. C. Mueller und W. T. Chen, Cancer Res., 1993; 53: 3159–3164).
- Inhibitoren von MMPs zeigten Aktivität in Modellen von Tumorangiogenese (G. Taraboletti, A. Garofalo, D. Belotti, T. Drudis, P. Borsotti, E. Scanziani, P. D. Brown und R. Giavazzi, Journal of the National Cancer Institute, 1995; 87: 293; und R. Benelli, R. Adatia, B. Ensoli, W. G. Stetler-Stevenson, L. Santi und A. Albini, Oncology Research, 1994; 6; 251–257).
- Mehrere Forscher belegten eine konsistente Erhöhung von Stromelysin und Kollagenase in Synovialflüssigkeiten von Patienten mit rheumatoider und Osteoarthritis im Vergleich zu Kontrollen (L. A. Walakovits, V. L. Moore, N. Bhardwaj, G. S. Gallick und M. W. Lark, "Detection of stromelysin and collagenase in synovial fluid from patients with rheumatoid arthritis and post-traumatic knee injury", Arthritis Rheum., 1992; 35: 35–42; M. Zafarullah, J. P. Pelletier, J. M. Cloutier und J. Marcel-Pelletier, "Elevated metalloproteinases and tissue inhibitor of metalloproteinase mRNA in human osteoarthritic synovia", J. Rheumatol., 1993; 20: 693–697). TIMP-1 und TIMP-2 verhinderten die Bildung von Kollagenfragmenten, jedoch nicht von Proteoglykanfragmenten beim Abbau von sowohl Rindernasen- als auch Schweinegelenkknorpelmodellen für Arthritis, während ein synthetisches Peptidhydroxamat die Bildung beider Fragmente verhindern konnte (H. J. Andrews, T. A. Plumpton, G. P. Harper und T. E. Cawston, Agents Actions, 1992; 37: 147–154; A. J. Ellis, V. A. Curry, E. K. Powell und T. E. Cawston, Biochem. Biophys. Res. Commun., 1994; 201: 94–101).
- Gijbels et al. (J. Clin. Invest., 1994; 94: 2177–2182) beschrieben vor kurzem ein Peptidhydroxamat, GM6001, das die Entwicklung einer experimentellen allergischen Enzephalomyelitis (EAE) in dosisabhängiger Weise unterdrückte oder deren klinische Expression in dosisabhängiger Weise hemmte, was die Verwendung von MMP-Inhibitoren bei der Behandlung von Autoimmunentzündungserkrankungen, wie Multiple Sklerose, nahelegt.
- Eine vor kurzem durchgeführte Untersuchung von Madri erhellte die Rolle von Gelatinase A beim Austreten von T-Zellen aus dem Blutstrom während einer Entzündung (A. M. Ramanic und J. A. Madri, "The Inductionof 72-kD Gelatinase in T-Cells upon Adhesion to Endothelial Cells is VCAM-1 De pendent", J. Cell Biology, 1994; 125: 1165–1178). Diese Transmigration aus der Endothelzellschicht ist mit der Induktion von Gelatinase A koordiniert und sie wird durch Binden an das Gefäßzellen-Adhäsionsmolekül-1 (VCAM-1) vermittelt. Sobald die Schranke überwunden ist, werden Ödem und Entzündung im ZNS produziert. Es ist bekannt, dass die Leukocytenmigration über die Blut-Hirn-Schranke mit der Entzündungsreaktion bei EAE verbunden ist. Eine Hemmung der Metalloproteinase Gelatinase A blockiert den Abbau der extrazellulären Matrix durch aktivierte T-Zellen, der zum Eindringen in das ZNS notwendig ist.
- Diese Untersuchungen lieferten die Basis für die Annahme, dass ein Inhibitor von Stromelysin-1 und/oder Gelatinase A Erkrankungen, die ein Aufbrechen der extrazellulären Matrix, was zu einer Entzündung aufgrund einer Lymphocyteninfiltration, einer ungünstigen Migration von Metastasen- oder aktivierten Zellen oder dem Abbau der für eine Organfunktion notwendigen Strukturintegrität führt, umfassen, behandelt.
- Es besteht ein fortgesetzter Bedarf an Molekülen mit niedrigem Molekulargewicht, die wirtschaftlich hergestellt werden können und dennoch wirksame Inhibitoren von Metalloproteinasen sind. Aufgabe der Erfindung ist die Bereitstellung derartiger Verbindungen, von deren pharmazeutischen Formulierungen und ein Verfahren zur Verwendung derselben zur Behandlung von Erkrankungen, die durch Metalloproteinasen vermittelt werden.
- Zusammenfassung der Erfindung
- Durch die vorliegende Erfindung erfolgt die Bereitstellung von Matrixmetalloproteinaseinhibitoren, die zur Behandlung von Erkrankungen und Störungen, bei denen eine Hemmung von Matrixmetalloproteinasen als vorteilhaft angesehen wird, verwendbar sind.
- Die Verbindungen der Erfindung sind Hydroxamsäureverbindungen mit der Formel I:oder ein pharmazeutisch akzeptables Salz hiervon, worin X ausgewählt ist aus OH und NHOH und d für 1 oder 2 steht.
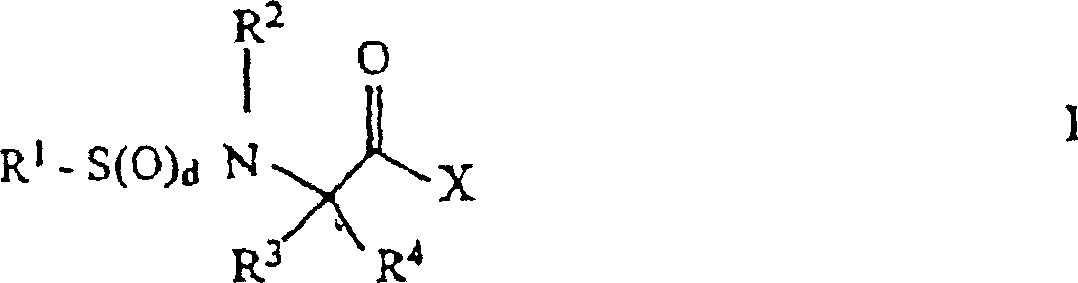
- R1 ist ausgewählt aus der Gruppe:worin Y ausgewählt ist aus O, S, S(O)d (worin d für 1 oder 2 steht), CH2, C(=O) und NRq (worin Rq für H, C1-C6-Alkyl oder C1-C6-Alkylphenyl steht),

R5 ausgewählt ist aus der Gruppe aus H, C1-C10-Alkyl, CF3, CONH2, Halogen, CN, COOH, C1-C4-Alkoxy, CHO, NO2, OH, (CH2)pOH, (CH2)pNH2, Ar und NH2, p für 0 bis 3 steht und Ar ausgewählt ist aus der Gruppe aus (a) Phenyl, (b) Phenyl, das mit C1-C4-Alkyl, C1-C4-Alkoxy, Halogen, NH2, NO2, CN, COOH, CONH2, CF3 oder COOR6 (worin R6 für C1-C10-Alkyl steht) substituiert ist, und (c) Heteroaryl. - R2 ist ausgewählt aus der Gruppe aus (a) Wasserstoff, (b) C1-C4-Alkyl, (c) Benzyl und (d) Benzyl, das mit einem oder mehreren Substituenten aus C1-C4-Alkyl, C1-C4-Alkoxy, F, Cl, Br, I, NH2, NO2, CN, Carboxy und CO2R7 (worin R7 für H oder C1-C4-Alkyl steht) substituiert ist.
- R3 und R4 sind unabhängig voneinander ausgewählt aus der Gruppe aus C1-C20-Alkyl (geradkettig oder verzweigt), C3-C10-Cycloalkyl, Phenyl, Phenyl, das mit C1-C4-Alkyl, C1-C4-Alkoxy, Halogen, NH2, NO2, CN, COOH, CO2R7 (worin R7 wie im vorhergehenden definiert ist) oder CF3 substituiert ist, C3-C10-Heterocyclus und Heteroaryl. R3 und R4 können zusammen eine Ringstruktur (an der das alpha-Kohlenstoffatom angrenzend an die Carbonylgruppe in Formel I beteiligt ist), als -(CH2)s-, das an das Kohlenstoffatom angrenzend an die Carbonylgruppe in Formel I gebunden ist, wobei s eine ganze Zahl von 2 bis 10 ist, bilden. Der Ring kann optional ferner ein oder mehrere Heteroatome umfassen.
- Eine weitere Ausführungsform der Erfindung ist eine pharmazeutische Formulierung, die eine Verbindung der Formel I mit einem Verdünnungsmittel, Träger oder Streckmittel hierfür gemischt umfasst.
- Die Erfindung stellt ferner Verfahren zur Hemmung der Wirkung eines Matrixmetalloproteinaseenzyms bei einem Säuger, die das Verabreichen einer eine Matrixmetalloproteinase hemmenden Menge einer Verbindung der Formel I umfasst, bereit.
- Detaillierte Beschreibung der Erfindung
- Die vorliegende Erfindung ist gerichtet auf Verbindungen mit der Formel I:oder ein pharmazeutisch akzeptables Salz hiervon, worin X ausgewählt ist aus OH und NHOH und d für 1 oder 2 steht.

- R1 ist ausgewählt aus der Gruppe:worin Y ausgewählt ist aus der Gruppe aus O, S, S(O)d (worin d für 1 oder 2 steht), CH2, C(=O) und NRq (worin Rq für H, C1-C6-Alkyl oder C1-C6-Alkylphenyl steht),

R5 ausgewählt ist aus der Gruppe aus H, C1-C10-Alkyl, CF3, CONH2, Halogen, CN, COOH, C1-C4-Alkoxy, CHO, NO2, OH, (CH2)pOH, (CH2)pNH2, Ar und NH2, p für 0 bis 3 steht und Ar ausgewählt ist aus der Gruppe aus (a) Phenyl, (b) Phenyl, das mit C1-C4-Alkyl, C1-C4-Alkoxy, Halogen, NH2, NO2, CN, COOH, CONH2, CF3 oder COOR6 (worin R6 für C1-C10-Alkyl steht) substituiert ist, und (c) Heteroaryl, das, ohne hierauf beschränkt zu sein, 2-, 3- oder 4-Pyridyl, 2-, 4- oder 5-Pyrimidinyl, 3- oder 4-Pyridazinyl, 2-Pyrazinyl, 2- oder 3-Thienyl, 2- oder 3-Furanyl, 1-, 2-, 4- oder 5-Imidazolyl, 1-, 3-, 4- oder 5-Pyrazolyl, 1-, 2- oder 3-Pyrrolyl, 2-, 4- oder 5-Oxazolyl, 2-, 4- oder 5-Thiazolyl, 3-, 4- oder 5-Isoxazolyl, 2-, 3-, 4-, 5-, 6-, 7-, oder 8-Chinolinyl oder 3-, 4- oder 5-Isothiazolyl umfasst. - Beispiele für R1-Gruppen umfassen die folgenden:
-

- R2 ist ausgewählt aus der Gruppe aus (a) Wasserstoff, (b) C1-C4-Alkyl, (c) Benzyl und (d) Benzyl, das mit einem oder mehreren Substituenten aus C1-C4-Alkyl, C1-C4-Alkoxy, F, Cl, Br, I, NH2, NO2, CN, Carboxy und CO2R7 (worin R7 für H oder C1-C4-Alkyl steht) substituiert ist.
- R3 und R4 sind unabhängig voneinander ausgewählt aus der Gruppe aus C1-C20-Alkyl (geradkettig oder verzweigt) , C3-C10-Cycloalkyl, Phenyl, Phenyl, das mit C1-C4-Alkyl, C1-C4-Alkoxy, Halogen, NH2, NO2, CN, COOH, CO2R7 (wobei R7 wie im vorhergehenden definiert ist) oder CF3 substituiert ist, C3-C10-Heterocyclus und Heteroaryl. Ferner können R3 und R4 aneinander gebunden sein oder zusammen einen spirocyclischen Ring (an dem das alpha-Kohlenstoffatom angrenzend an die Carbonylgruppe in Formel I beteiligt ist), wie -(CH2)s-, das an das Kohlenstoffatom angrenzend an die Carbonylgruppe in Formel I gebunden ist, wobei s eine ganze Zahl von 2 bis 10 (vorzugsweise 3 bis 9) bedeutet, bilden. Die Ringsysteme können optional ferner ein oder mehrere Heteroatome umfassen.
- Beispielsweise können R3 und R4 zusammengenommen als -(CH2)s-, wobei s 2 bis 10 bedeutet, einen monocyclischen C3-11-Ring bilden, beispielsweise die folgenden Gruppen (wobei die terminalen Kohlenstoffe an das alpha-Kohlenstoffatom angrenzend an die Carbonylgruppe in Formel I gebunden sind): -CH2CH2-, -CH2CH2CH2-, -CH2CH2CH2CH2-, -CH2CH2CH2CH2CH2- und -CH2CH2CH2CH2CH2CH2- (wobei 3-, 4-, 5-, 6- bzw. 7-gliedrige Ringe gebildet werden).
- R3 und R4 können zusammen einen der im vorhergehenden beschriebenen spirocyclischen Ringe, der des weiteren ein oder mehrere Heteroatome (vorzugsweise 1 bis 6 Heteroatome) an einer beliebigen Stelle in den Ringen umfasst, bilden. Das Heteroatom ist aus der aus O, S und NR8 (wobei R8 aus H und C1-C3-Alkyl ausgewählt ist) bestehenden Gruppe ausge wählt.
- R3 und R4 können auch zusammengenommen die empirische Formel -CH2)sZg-, wobei terminale Kohlenstoffe an das alpha-Kohlenstoffatom gebunden sind, s eine ganze Zahl von 2 bis 10 bedeutet und g eine ganze Zahl von 0 bis 3 bedeutet, bilden. Jedes Z ist ein Heteroatom, das sich an einer beliebigen Position der aus R3 und R4 gebildeten Gruppe befindet, und jedes Z ist unabhängig voneinander aus der aus O, S und NR8 (wobei R8 aus H und C1-C3-Alkyl ausgewählt ist) bestehenden Gruppe ausgewählt.
- Beispielsweise kann ein spirocyclischer Heteroring -(CH2)aZ(CH2)b-, wobei Z das Heteroatom (das aus der aus O, S und R8 bestehenden Gruppe ausgewählt ist) bedeutet, a 1 bis 10 bedeutet und b 1 bis 10 bedeutet und die Summe von a und b nicht mehr als 11 beträgt, umfassen, beispielsweise die folgenden (wobei die terminalen Kohlenstoffe beide an das alpha-Kohlenstaffatom angrenzend an die Carbonylgruppe in Formel I gebunden sind):
-CH2OCH2CH2-, -CH2CH2OCH2CH2-, -CH2CH2OCH2CH2CH2-, -CH2N(H)CH2CH2-, -CH2CH2N(H)CH2CH2-, -CH2CH2N(H)CH2CH2CH2-, -CH2SCH2CH2-, -CH2CH2SCH2CH2-, -CH2CH2SCH2CH2CH2-, -CH2OCH2CH2OCH2-, -CH2N(H)CH2CH2N(H)CH2- und -CH2SCH2CH2SCH2-. Daher können Beispiele für Verbindungen der Erfindung durch die folgende Formel (worin Z das Heteroatom ist und h 0 oder 1 ist) dargestellt werden. -
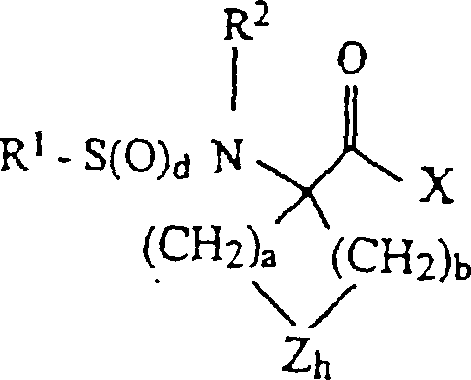
- In bevorzugten Ausführungsformen von Verbindungen der Formel I sind R1-Gruppen aus den folgenden ausgewählt:
-
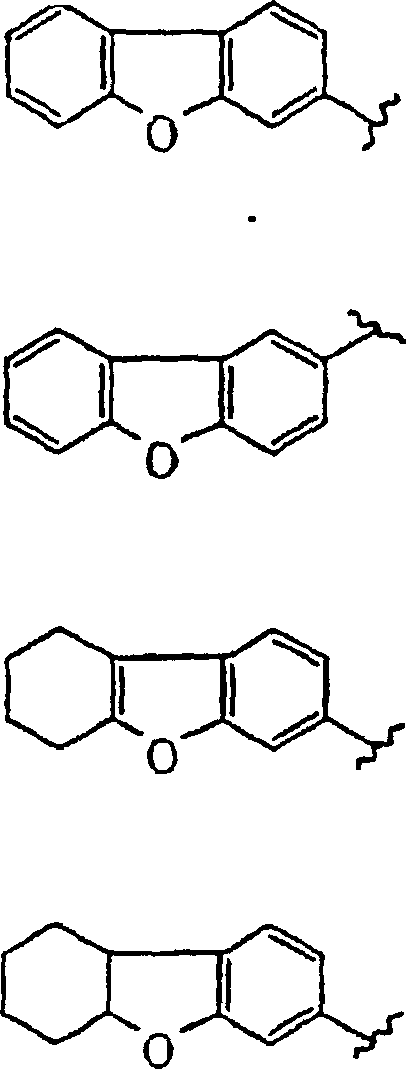
- Vorzugsweise ist d gleich 2, R2 Wasserstoff und X NHOH oder OH. R3 und R4 bilden vorzugsweise zusammengenommen eine Ringstruktur, die vorzugsweise ausgewählt ist aus: -CH2CH2CH2CH2- (wobei die terminalen Kohlenstoffatome an den alpha-Kohlenstoff angrenzend an die Carbonylgruppe in Formel I unter Bildung einer Cyclopentylgruppe gebunden sind); -CH2CH2CH2CH2CH2- (wobei die terminalen Kohlenstoffatome an den alpha-Kohlenstoff angrenzend an die Carbonylgruppe in Formel I unter Bildung einer Cyclohexylgruppe gebunden sind); und -CH2CH2OCH2CH2CH2- (wobei die terminalen Kohlenstoffatome an den alpha-Kohlenstoff angrenzend an die Carbonylgruppe in Formel I unter Bildung eines 6-gliedrigen Heterocyclus gebunden sind). In anderen bevorzugten Ausführungsformen bedeuten R3 und R4 jeweils eine Methylgruppe.
- In den Formeln, die die Verbindungen der Erfindung definieren, bezeichnet Halogen Fluor, Chlor, Brom und Iod, wobei Chlor und Brom bevorzugt sind.
- Der Ausdruck "C1-4-Alkyl" oder "Alkyl-C1-4" bedeutet gerade und verzweigte aliphatische Gruppen mit 1 bis 4 Kohlenstoffatomen, wobei Beispiele derselben Methyl, Ethyl, n-Propyl, Isopropyl, n-Butyl, sek-Butyl und tert-Butyl umfassen. Daher bedeutet der Ausdruck "C1-20-Alkyl" gerade und verzweigte aliphatische Gruppen mit 1 bis 20 Kohlenstoffatomen und der Ausdruck "C1-10-Alkyl" bezeichnet gerade und verzweigte aliphatische Gruppen mit 1 bis 10 Kohlenstoffatomen.
- Der Ausdruck "Alkoxy" bedeutet eine Alkylgruppe, die an ein Sauerstoffatom gebunden ist. Repräsentative Beispiele für Alkoxygruppen umfassen Methoxy, Ethoxy, tert-Butoxy, Propoxy und Isobutoxy.
- Der Ausdruck "Aryl" bedeutet eine aromatische Kohlenwasserstoffgruppe. Repräsentative Beispiele für Arylgruppen umfassen Phenyl und Naphthyl.
- Der Ausdruck "Cycloalkyl" bedeutet eine cyclische aliphatische Gruppe. Beispiele für Cycloalkylgruppen umfassen Cyclopropyl, Cyclobutyl, Cyclopentyl, Cyclohexyl und Cyclooctyl.
- Das Symbol "-" bedeutet eine Bindung.
- Das Symbol "R-" bedeutet eine gebundene R-Gruppe.
- Ein "Heteroatom" ist ein Stickstoff-, Sauerstoff- oder Schwefelatom.
- Der Ausdruck "Heteroaryl" bedeutet ein mono- oder bicyclisches Ringsystem, das 1 oder 2 aromatische Ringe enthält und mindestens 1 Stickstoff-, Sauerstoff- oder Schwefelatom in einem aromatischen Ring enthält. Beispiele für Heteroarylgruppen umfassen, ohne hierauf beschränkt zu sein, 2- oder 3-Thienyl, 2- oder 3-Furanyl, 2- oder 3-Pyrrolyl, 2-, 3- oder 4-Pyridinyl, 2-Pyrazinyl, 2-, 4- oder 5-Pyrimidinyl, 3- oder 4-Pyridazinyl, 1-, 2-, 4- oder 5-Imidazolyl, 1-, 3-, 4- oder 5-Pyrazolyl, 2-, 4- oder 5-Oxazolyl, 2-, 4- oder 5-Thiazolyl, 3-, 4- oder 5-Isoxazolyl, 3-, 4- oder 5-Isothiazolyl oder 2-, 3-, 4-, 5-, 6- oder 7-Indolyl. Ein Heteroaryl kann mit beispielsweise einem oder mehreren, und insbesondere 1 bis 3 Substituenten, wie Halogen, Alkyl, Hydroxy, Hydroxyalkyl, Alkoxy, Alkoxyalkyl, Halogenalkyl, Nitro, Amino, Alkylamino, Acylamino, Alkylthio, Alkylsulfinyl und Alkylsulfonyl, substituiert oder unsubstituiert sein.
- Die MMP hemmenden Verbindungen der Erfindung sind von α,α'-disubstituierten Aminosäuren abgeleitet. Die Verbindungen, worin X gleich NHOH ist, werden ferner als Hydroxamsäureverbindungen bezeichnet. Die Verwendung der Zink bindenden Hydroxamsäureeinheit ergibt vorzugsweise Verbindungen, die alle identifizierten MMPs mit einer gegenüber den entsprechenden Carbonsäuren höheren Wirksamkeit (beispielsweise im nanomolaren Bereich) hemmen.
- Die Verbindungen der vorliegenden Erfindung können als pure Chemikalie therapeutisch verabreicht werden, doch werden Verbindungen der Formel I vorzugsweise als pharmazeutische Zusammensetzung oder Formulierung, wie im folgenden genauer beschrieben wird, verabreicht. Daher erfolgt durch die vorliegenden Erfindung ferner die Bereitstellung pharmazeutischer Formulierungen, die eine Verbindung der Formel I zusammen mit einem oder mehreren pharmazeutisch akzeptablen Trägern und optional anderen therapeutischen und/oder prophylaktischen Bestandteilen umfasst. Die Träger und anderen Zusatzstoffe sind "akzeptabel" in dem Sinne, dass sie mit den anderen Bestandteilen der Formulierungen kompatibel und für den Empfänger derselben nicht schädlich sind.
- Der Ausdruck "pharmazeutisch akzeptable Salze und Prodrugs" bezeichnet die Salze (einschließlich von Carboxylatsalzen und Aminosäureadditionssalzen) und Prodrugs (einschließlich von Estern und Amiden) der Verbindungen der vorliegenden Erfindung, die innerhalb des Umfangs einer tiefgreifenden medizinischen Beurteilung zur Verwendung bei einem Kontakt mit den Geweben eines Patienten ohne eine ungünstige Toxizität, Irritation, allergische Reaktion und dgl. geeignet, mit einem vernünftigen Nutzen/Risiko-Verhältnis behaftet und für die geplante Verwendung wirksam sind, sowie die zwitterionischen Formen, wenn diese möglich sind, der Verbindungen der Erfindung.
- Der Ausdruck "Salze" bezeichnet die relativ nichttoxischen, anorganischen und organischen Säureadditionssalze von Verbindungen der vorliegenden Erfindung. Diese Salze können in sitze während der endgültigen Isolierung und Reinigung der Verbindungen oder durch getrennte Umsetzung der gereinigten Verbindung in der Form der freien Base mit einer geeigneten organischen oder anorganischen Säure und Isolieren des auf diese Weise gebildeten Salzes hergestellt werden. Repräsentative Antionen umfassen Bromid, Chlorid, Sulfat, Bisulfat, Nitrat, Acetat, Oxalat, Valerat, Oleat, Palmitat, Stearat, Laurat, Borat, Benzoat, Lactat, Phosphat, Tosylat, Citrat, Maleat, Fumarat, Succinat, Tartrat, Naphthylat, Mesylat, Glucoheptonat, Lactiobionat und Laurylsulfonat und dgl. Kationen umfassen die Alkali- und Erdalkalimetalle, wie Natrium, Lithium, Kalium, Calcium, Magnesium und dgl., sowie nicht-toxische Ammonium-, quaternäre Ammonium- und Aminkationen einschließlich von, ohne hierauf beschränkt zu sein, Ammonium, Tetramethylammonium, Tetraethylammonium, Methylamin, Dimethylamin, Trimethylamin, Triethylamin, Ethylamin und dgl. Siehe beispielsweise S. M. Berge et al., "Pharmaceutical Salts", J. Pharm. Sci., 1977; 66: 1–19, dessen Offenbarung hierdurch als Bezug aufgenommen ist.
- Der Ausdruck "Prodrugs" bezeichnet Verbindungen, die in vitro, vorzugsweise schnell, umgewandelt werden, wobei die Stammverbindung der obigen Formel, beispielsweise durch Hydrolyse in Blut oder an einer anderen Stelle, erhalten wird. Eine sorgfältige Diskussion ist bei T. Higuchi und V. Stella, "Pro-drugs as Novel Delivery Systems", Band 14 der A. C. S. Symposium-Reihe und bei Bioreversible Carriers in Drug Design, Hrsg. Edward B. Roche, American Pharmaceutical Association und Pergamon Press, 1987, deren Offenbarungen beide hier als Bezug aufgenommen sind, angegeben.
- Ester und Amide können als Prodrugs für die biologisch aktiven Carbonsäuren der vorliegenden Erfindung verwendet werden. Beispiele für pharmazeutisch akzeptable nicht-toxische Ester der Verbindungen der vorliegenden Erfindung umfassen C1-C6-Alkylester, wobei die Alkylgruppe eine gerade oder verzweigte Kette ist. Akzeptable Ester umfassen auch C5-C7-Cycloalkylester sowie Arylalkylester, beispielsweise, ohne hierauf beschränkt zu sein, Benzyl. C1-C4-Alkylester sind bevorzugt. Ester der Verbindungen der vorliegenden Erfindung können nach herkömmlichen Verfahren hergestellt werden.
- Beispiele für pharmazeutisch akzeptable nicht-toxische Amide der Verbindungen der vorliegenden Erfindung umfassen Amide, die von Ammoniak, primären C1-C6-Alkylaminen und sekundären C1-C6-Dialkylaminen, wobei die Alkylgruppen gerad- oder verzweigtkettig sind, abgeleitet sind. Im Falle von sekundären Aminen kann das Amin auch in der Form eines 5- oder 6-gliedrigen Heterocyclus, der ein Stickstoffatom ent hält, sein. Amide, die von Ammoniak, primären C1-C3-Alkylaminen und sekundären C1-C2-Dialkylaminen abgeleitet sind, sind bevorzugt. Amide der Verbindungen der Erfindung können gemäß herkömmlichen Verfahren hergestellt werden.
- Ferner können die Verbindungen der vorliegenden Erfindung in nicht-solvatisierten sowie solvatisierten Formen mit pharmazeutisch akzeptablen Lösemitteln, wie Wasser, Ethanol und dgl., existieren. Im allgemeinen werden die solvatisierten Formen für die Zwecke der vorliegenden Erfindung als den nicht-solvatisierten Formen äquivalent angesehen.
- Wie einem Fachmann klar ist, erstreckt sich ein hier erfolgter Verweis auf eine Behandlung auf die Prophylaxe sowie die Behandlung von etablierten Erkrankungen oder Symptomen. Es ist ferner klar, dass die Menge einer Verbindung gemäß der Erfindung, die zur Verwendung bei einer Behandlung erforderlich ist, mit der Natur der behandelten Erkrankung und mit dem Alter und dem Zustand des Patienten variiert und letztendlich durch den behandelnden Arzt oder Tierarzt bestimmt wird.
- Die pharmazeutische Zubereitung ist vorzugsweise in Einheitsdosisform. Die gewünschte Dosis kann in bequemer Weise in einer Einzeldosis oder als Mehrfachdosen, die in entsprechenden Intervallen, beispielsweise als zwei, drei, vier oder mehr Teildosen pro Tag, verabreicht werden, verabreicht werden. In einer derartigen Form ist die Zubereitung in Einheitsdosen, die entsprechende Mengen der aktiven Komponente enthalten, unterteilt. Die Einheitsdosisform kann eine abgepackte Zubereitung, wobei die Packung diskrete Mengen der Zubereitung enthält, wie abgepackte Tabletten, Kapseln und Pulver in Phiolen oder Ampullen, sein. Auch kann die Einheitsdosisform eine Kapsel, Tablette, ein Cachet oder eine Pastille selbst sein oder es kann die entsprechende Zahl von einer derselben in abgepackter Form sein.
- Bei der therapeutischen Verwendung als Mittel zur Hemmung eines Matrixmetalloproteinaseenzyms zur Behandlung der hier beschriebenen Erkrankungen und Störungen werden die im pharmazeutischen Verfahren der Erfindung verwendeten Verbindungen mit einer Dosis verabreicht, die zur Hemmung der hydrolytischen Aktivität von einem oder mehreren Matrixmetalloproteinaseenzymen wirksam ist. Die Menge einer aktiven Komponente in einer Einheitsdosiszubereitung kann von etwa 0,1 bis etwa 1000 mg, vorzugsweise etwa 10 bis etwa 100 mg, gemäß der speziellen Anwendung und der Wirksamkeit der aktiven Komponente variiert oder eingestellt werden.
- Eine Anfangsdosis von etwa 0,1 bis etwa 500 mg pro kg Körpergewicht pro Tag, beispielsweise etwa 1 bis etwa 100 mg pro kg Körpergewicht pro Tag ist im allgemeinen wirksam. Ein Tagesdosisbereich von etwa 25 bis etwa 75 mg pro kg Körpergewicht kann günstig sein. Die Dosierungen können jedoch in Abhängigkeit von den Bedürfnissen des Patienten, der Schwere der behandelten Störung und der verwendeten Verbindung variieren. Die Bestimmung der geeigneten Dosierung für eine spezielle Situation ist einem Fachmann geläufig. Die Zusammensetzung kann, falls gewünscht, auch andere kompatible therapeutische Mittel enthalten.
- Im allgemeinen wird eine Behandlung mit kleineren Dosierungen, die geringer als die optimale Dosis der Verbindung sind, begonnen. Danach kann die Dosierung in kleinen Inkrementen erhöht werden, bis die unter den Umständen optimale Wirkung erreicht ist. Zur bequemeren Verwendung kann die Gesamttagesdosierung geteilt und in Portionen während des Tages, falls gewünscht, verabreicht werden. Typische Dosierungen betragen von etwa 0,1 bis etwa 1000 mg pro Tag und vorzugsweise etwa 25 bis etwa 250 mg pro Tag, so dass eine Menge bereitgestellt wird, die zur Behandlung der speziellen Erkrankung, die verhindert oder bekämpft werden soll, wirksam ist, bereitgestellt wird. Für einen normalen Erwachsenen mit einem Körpergewicht von etwa 70 kg ist eine Dosierung im Bereich von etwa 0,01 bis etwa 100 mg pro kg Körpergewicht pro Tag bevorzugt. Die speziell verwendete Dosierung kann jedoch variieren. Beispielsweise kann die Dosierung von einer Zahl von Faktoren, die die Bedürfnisse des Patienten, die Schwere der behandelten Erkrankung und die pharmakologische Aktivität der verwendeten Verbindung umfassen, abhängen. Die Bestimmung der optimalen Dosierungen für einen speziellen Patienten ist einem Fachmann bekannt. Der Ausdruck "Patient" umfasst Menschen und Tiere.
- Die Verbindungen und Formulierungen der vorliegenden Erfindung können in einer breiten Vielzahl oraler und parenteraler Dosierungsformen verabreicht werden. Daher können die Verbindungen der vorliegenden Erfindung beispielsweise durch Injektion, d. h. intravenös, intramuskulär, intrakutan, subkutan, intraduodenal, intraarteriell, intramuskulär, intraartikulär und intraperitoneal verabreicht werden. Auch können die Verbindungen der vorliegenden Erfindung durch Inhalation, beispielsweise intranasal oder tiefe Lungeninhalation, verabreicht werden. Ferner können die Verbindungen der vorliegenden Erfindung transdermal verabreicht werden. Die Verbindungen der Erfindung können transmukosal (beispielsweise sublingual oder durch bukkale Verabreichung) oder rektal verabreicht werden. Ophthalmologische Formulierungen, Augensalben, Pulver und Lösungen werden ebenfalls als innerhalb des Schutzumfangs dieser Erfindung liegend betrachtet. Formulierungen der Erfindung werden ebenfalls im folgenden im Detail beschrieben.
- Einem Fachmann ist klar, dass die im folgenden angegebenen Dosierungsformen als aktive Komponente entweder eine Verbindung der Formel I (einschließlich der hier offenbarten bevorzugten Ausführungsformen) oder ein entsprechendes pharmazeutisch akzeptables Salz einer Verbindung der Formel I umfassen können.
- Die Synthese von Beispielen für Sulfonamide der Formel I kann gemäß den folgenden bevorzugten allgemeinen Reaktionsschemata erreicht werden. Falls nicht anders angegeben, wurden alle Ausgangsmaterialien von Handelslieferanten erhalten und ohne weitere Reinigung verwendet.
- Reaktionsschema 1
- Ein Gemisch aus Natriumphenoxid und einem Cycloalkenoxid (wobei n vorzugsweise 1 bis 3 ist), das im folgenden angegeben ist, wird in Wasser unter Rückflusskühlung erhitzt, wobei die Hydroxyetherverbindung der Formel II gebildet wird. Die Hydroxyetherverbindung wird zu einem Keton der Formel III unter Verwendung von Jones-Reagens oder einem geeigneten Oxidationsmittel oxidiert.
-

- Das Keton (III) wird zu einem Gemisch von Schwefelsäure und Phosphorsäure bei 0°C gegeben und sich auf Raumtemperatur erwärmen gelassen, wobei die Verbindung der Formel IV gebildet wird.
-

- Die Verbindung der Formel IV wird in einem inerten Lösemittel, wie 1,2-Dichlorethan oder Dichlormethan, mit Schwefeltrioxid in Dimethylformamid (SO3·DMF) unter Rückflusskühlung erhitzt, wobei ein Sulfonsäuresalz der Formel V gebildet wird.
-

- Ein Sulfonylchlorid kann durch Erhitzen des Natriumsalzes der Formel V mit einem geeigneten Chlorierungsmittel, wie Thionylchlorid (SOCl2), entweder pur oder in einem inerten Lösemittel, wie Toluol (C6H5CH3), unter Rückflusskühlung, wobei das Sulfonychlorid der Formel VI gebildet wird, gebildet werden.
-

- Das Sulfonylchlorid der Formel VI und ein gewählter Aminosäureester werden in einem geeigneten Lösemittel, wie wässriges Tetrahydrofuran oder wasserfreies Tetrahydrofuran ("THF"), mit einem Säurefänger, wie Triethylamin ("NEt3"), gekoppelt, wobei beispielsweise der Methylester der Formel VII gebildet wird.
-

- Aus dem Methylester der Formel VII kann eine Carbonsäure der Formel VIII durch Basenhydrolyse in wässrigem Tetrahydrofuran gebildet werden. Die Hydroxamsäure der Formel IX kann durch zunächst Rühren der Carbonsäure mit Oxalylchlorid ((COCl)2) oder einem anderen geeigneten Chlorierungsmittel in einem inerten Lösemittel, wie 1,2-Dichlorethan oder Dichlormethan, wobei ein Carbonsäurechlorid gebildet wird, das dann mit einer Lösung von Hydroxylamin unter Bildung der gewünschten Hydroxamsäure der Formel IX umgesetzt wird, gebildet werden. Die Verbindungen der Formeln VIII und IX sind bevorzugte Verbindungen der Erfindung, beispielsweise der Formel I.
-
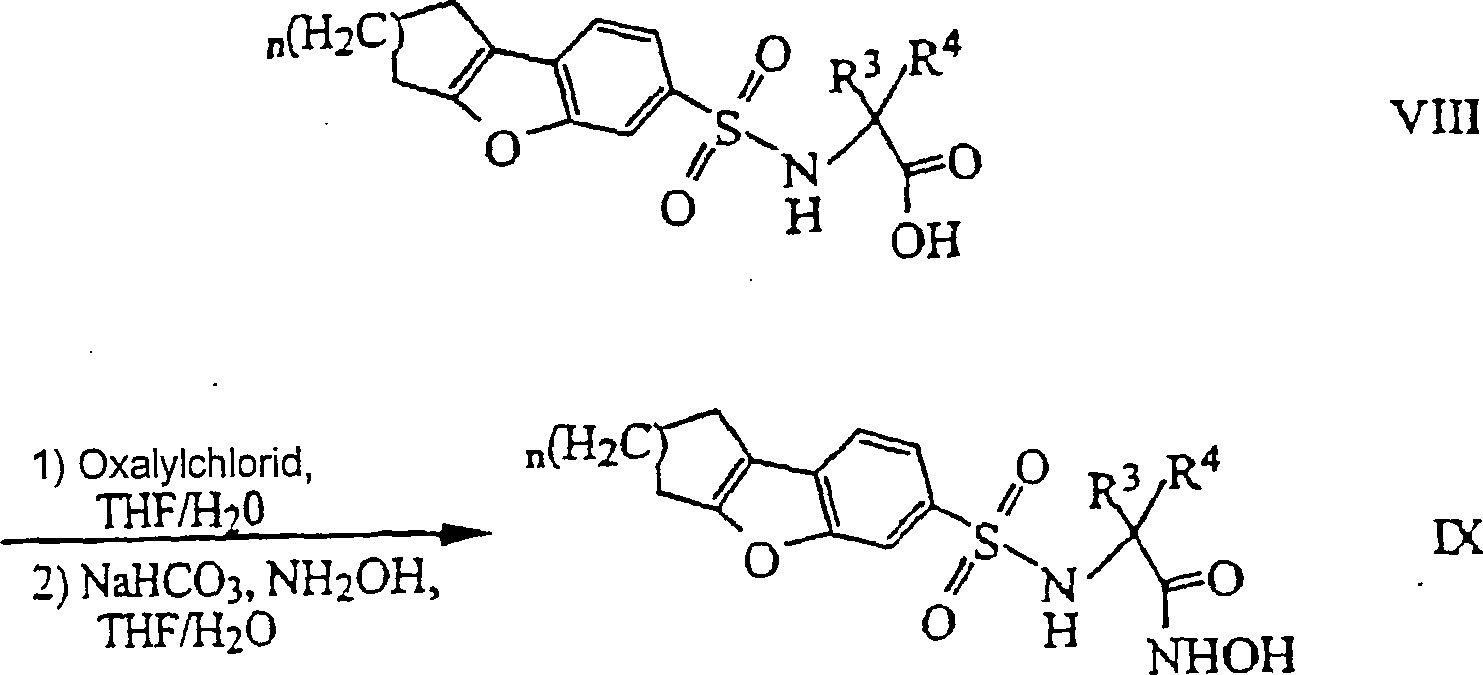
- Reaktionsschema 2
- Ein weiteres bevorzugtes Reaktionsschema zur Herstellung bevorzugter Verbindungen der Erfindung wird im folgenden beschrieben. Eine Verbindung der Formel X kann durch Reduktion des Sulfonsäurenatriumsalzes der Formel V (wobei n vorzugsweise 1 bis 3 ist) ausgehend von Reaktionsschema 1 in einem Lösemittel, wie Ethanol (EtOH) und wässriges Te trahydrofuran, mit Palladium (20% Pd/C) oder anderen geeigneten Metallkatalysatoren unter Wasserstoffgas bei erhöhtem Druck hergestellt werden. Das Sulfonylchlorid der Formel XI kann dann aus der Verbindung der Formel X durch Erhitzen unter Rückflusskühlung mit einem geeigneten Chlorierungsmittel, wie Phosphortrichlorid (PCl3), hergestellt werden.
-

- Das Sulfonylchlorid der Formel XI wird dann mit einem gewählten Aminosäureester in einem Lösemittel, wie wässriges Tetrahydrofuran oder wasserfreies Tetrahydrofuran, mit einem geeigneten Säurefänger, wie Triethylamin (NEt3), vereinigt, wobei ein N-Sulfonamidaminosäureester, beispielsweise der Ester der Formel XII, gebildet wird.
-
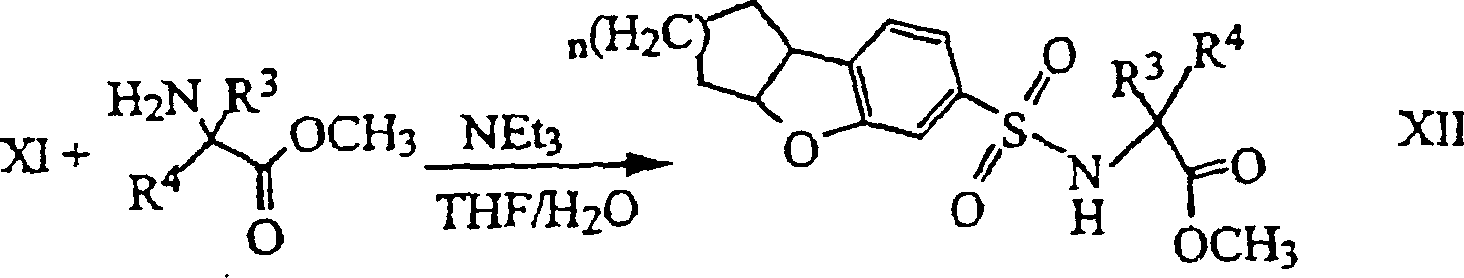
- Die Basenhydrolyse des Esters der Formel XII ergibt die Bildung der Carbonsäure der Formel XIII. Eine Hydroxamsäure, beispielsweise die Säure der Formel XIV, kann dann durch zunächst Rühren der Carbonsäure der Formel XIII mit Oxalylchlorid oder einem anderen geeigneten Chlorierungsmittel in einem inerten Lösemittel, wie 1,2-Dichlorethan oder Dichlormethan, und dann Umsetzen des gebildeten Carbonsäurechlorids mit Hydroxylamin, gebildet werden.
-
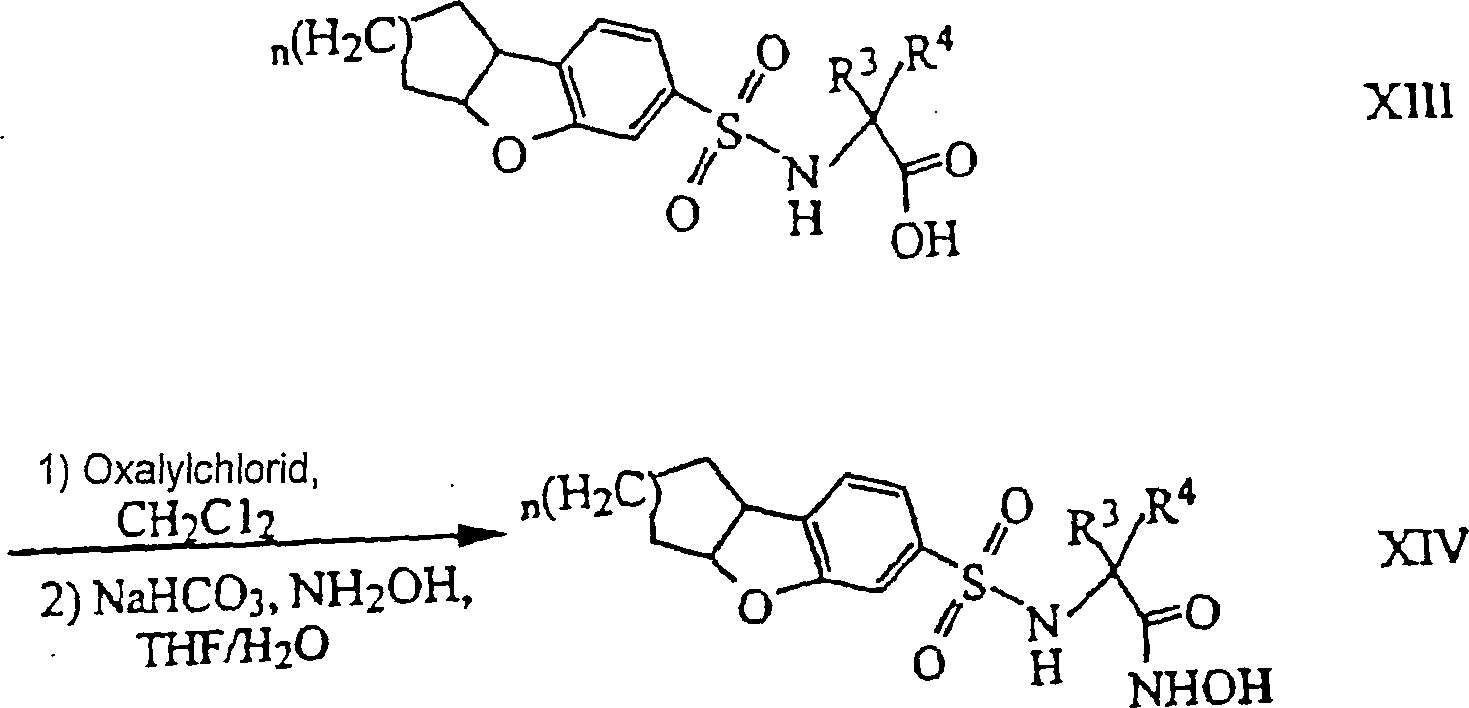
- Bevorzugte Verbindungen der Erfindung können im allgemeinen gemäß den hier beschriebenen Syntheseschemata hergestellt werden. Einem Fachmann ist klar, dass bei jedem Reaktionsschema, wenn dies notwendig ist, Schutzgruppen entsprechend den allgemeinen Prinzipien von Synthesechemie verwendet werden können. Diese Schutzgruppen können in den Endstufen der Synthese unter basischen, sauren oder hydrogenolytischen Bedingungen, die einem Fachmann ohne weiteres klar sind, entfernt werden. Durch Anwendung einer geeigneten Manipulation und eines geeigneten Schutzes von beliebigen chemischen Funktionalitäten kann die Synthese von Verbindungen der Erfindung, die hier nicht speziell angegeben sind, durch zu den hier angegebenen Reaktionsschemata analogen Verfahren erreicht werden.
- Die vorliegende Erfindung umfasst alle möglichen Stereoisomere und geometrischen Isomere von Verbindungen der Formel I und sie umfasst nicht nur racemische Verbindungen, sondern ebenso die optisch aktiven Isomere, wenn beispielsweise die Substituenten R3 und R4 verschieden sind. Wenn eine Verbindung der Formel I als einziges Enantiomer gewünscht wird, kann sie entweder durch Trennen des Endprodukts oder durch stereospezifische Synthese ausgehend von entweder isomerenreinem Ausgangsmaterial oder einem isomerenreinen beliebigen herkömmlichen Zwischenprodukt erhalten werden. Die Auftrennung des Endprodukts, eines Zwischenprodukts oder eines Ausgangsmaterials können nach einem einschlägig bekannten geeigneten Verfahren erreicht werden. Ferner soll in Situationen, in denen Tautomere der Verbindungen in Formel I möglich sind, die vorliegende Erfindung alle tautomeren Formen der Verbindung umfassen.
- Die Synthese von typischen Sulfonamiden der Formel I wird durch die folgenden Beispiele erläutert. Die Beispiele sind nur repräsentativ und sollen in keinster Hinsicht beschränkend sein. Die hier offenbarten Verhältnisse sind Volumenverhältnisse, falls nicht anders angegeben.
- Beispiele 7 und 8
- Synthese von 2-Methyl-2-(6,7,8,9-tetrahydro-dibenzofuran-3-sulfonylamino)-propionsäure
- Stufe (a)
- Cyclohexenoxid (10 g, 0,102 mol) wurde tropfenweise zu einer wässrigen Lösung von Natriumphenoxidtrihydrat (31,6 g, 0,186 mol, 250 ml) gegeben. Das Gemisch wurde über Nacht unter Rückflusskühlung erhitzt, auf Raumtemperatur gekühlt, und ein Niederschlag wurde durch Filtration gewonnen. Der Niederschlag wurde in Ethylacetat gelöst. Die organische Phase wurde mit 1 M NaOH und Kochsalzlösung gewaschen, getrocknet (über MgSO4) und eingeengt, wobei festes 2-Phenoxy-cyclohexanol (eine Verbindung der allgemeinen For mel II) (13,84 g, 70%) erhalten wurde. Der Schmelzpunkt dieser Verbindung wurde als 79–80°C bestimmt. 1H-NMR (CDCl3): δ 7,3 (m, 2H), 6,9 (m, 3H), 4,0 (m, 1H), 3,7 (m, 1H), 2,1 (m, 2H), 1,8 (m, 4H), 1,4 (m, 4H) ppm.
- Stufe (b)
- 2-Phenoxy-cyclohexanol (13,64 g, 0,071 mol) wurde in Aceton (260 ml) gelöst, auf zwischen 0°C bis 5°C gekühlt, und Jones-Reagens (2 M, 78 ml) wurde tropfenweise zugegeben. Das Reaktionsgemisch wurde 4,5 h gerührt. Das Reaktionsgemisch wurde in Wasser (250 ml) gegossen, und Diethylether (500 ml) wurde zugegeben. Die organische Phase wurde von der wässrigen Phase abgetrennt, dann mit Wasser gewaschen, getrocknet (über MgSO4) und zu gelben Kristallen eingeengt. Die Kristalle wurden aus Pentan umkristallisiert, wobei 2-Phenoxy-cyclohexanon (eine Verbindung der allgemeinen Formel III) (10,22 g, 76%) erhalten wurde. 1H-NMR (CDCl3): δ 7,2 (m, 2H), 7,0 (m, 2H), 6,8 (d, 1H), 4,6 (m, 1H), 2,6 (m, 1H), 2,4 (m, 2H), 2,0–1,8 (m, 5H) ppm.
- Stufe (c)
- 2-Phenoxy-cyclohexanon (10,2 g, 0,054 mol) wurde langsam zu einem gekühlten Gemisch von Phosphorsäure und Schwefelsäure (3 : 1, 80 ml) bei 0°C geben. Das Reaktionsgemisch wurde 4 h gerührt und dann über Eis gegossen. Diethylether (300 ml) wurde zugegeben. Die organische Phase wurde von der wässrigen Phase abgetrennt, dann mit Wasser, Natriumbicarbonat und Wasser gewaschen, getrocknet (über MgSO4) und zu einer viskosen Flüssigkeit eingeengt. Das rohe Produkt wurde in Hexan gelöst und durch Silicagelsäulenchromatographie gereinigt, wobei 1,2,3,4-Tetrahydro-dibenzofuran (eine Verbindung der allgemeinen Formel IV) (5,72 g, 61%) erhalten wurde. 1H-NMR (CDCl3): δ 7,4 (m, 2H), 7,2 (m, 2H), 2,7 (m, 4H), 1,9 (m, 4H) ppm.
- Stufe (d)
- 1,2,3,4-Tetrahydro-dibenzofuran (5,7 g, 0,33 mol) wurde tropfenweise zu SO3·DMF-Komplex (16,3 g), der in 1,2-Dichlorethan (100 ml) suspendiert war, gegeben. Das Reaktionsgemisch wurde über Nacht unter Rückflusskühlung erhitzt. Das Reaktionsgemisch wurde dann gekühlt, zu einer dicken Flüssigkeit eingeengt, in Wasser (300 ml) aufgenommen und mit 1 M NaOH (260 ml, pH-Wert 12) basisch gemacht. Eine Kristallisation erfolgte auf einem Eisbad, und Kristalle wurden durch Filtration gewonnen. Die Kristalle wurden in Tetrahydrofuran aufgeschlämmt und durch Filtration gewonnen. Das Produkt wurde getrocknet, wobei 1,2,3,4-Tetrahydro-dibenzofuran-3-sulfonsäurenatriumsalz (eine Verbindung der allgemeinen Formel V) (4,30 g, 47%) erhalten wurde. 1H-NMR (CDCl3): δ 7,6 (s, 1H), 7,4 (m, 2H), 2,6 (m, 4H), 1,9 (m, 4H) ppm.
- Stufe (e)
- Die in der obigen Stufe (d) hergestellte Verbindung (2 g, 7,24 mmol) wurde in Toluol (25 ml) suspendiert und dann tropfenweise mit Thionylchlorid (3,45 g, 28,96 mmol) versetzt. N,N-Dimethylformamid (2 Tropfen) wurde zugegeben, und das Reaktionsgemisch wurde 3 h unter Rückflusskühlung erhitzt, auf Raumtemperatur gekühlt und dann 3 Tage gerührt. Das Reaktionsgemisch wurde eingeengt und zweimal mit Hexan verrieben und reduziert. Die rohen Kristalle wurden mit einem Gemisch von 10% Ethylacetat : Hexan verrieben und dann durch Filtration gewonnen. Das Filtrat wurde eingeengt und mit Hexan gewaschen, wobei eine zweite Charge erhalten wurde. Die Kristalle wurden getrocknet, wobei ein Sulfonylchlorid (eine Verbindung der allgemeinen Formel VI) (1,2 g, 61%) erhalten wurde. 1H-NMR (CDCl3): δ 8,1 (s, 1H), 7,9 (d, 1H), 7,6 (d, 1H), 2,8 (m, 4H), 1,9 (m, 4H) ppm.
- Stufe (f)
- Ein Gemisch aus der in Stufe (e) hergestellten Sulfonylchloridverbindung (1 g, 3,69 mmol) und α-Amino-isobuttersäuremethylesterhydrochlorid (624 mg, 4,06 mmol) wurde mit wässrigem Tetrahydrofuran (4 : 1, 40 ml) verdünnt und tropfenweise mit Triethylamin (785 mg, 7,75 mmol) behandelt. Das Reaktionsgemisch wurde 20 h bei Raumtemperatur gerührt und anschließend mit wässriger Salzsäure (1 M, 20 ml) und Ethylacetat (50 ml) versetzt. Die organische Phase wurde von der wässrigen Phase abgetrennt, getrocknet (über MgSO4) und eingeengt. Die gebildeten Kristalle wurden mit 4 : 1 Hexan : Ethylacetat verrieben und durch Filtration gewonnen, wobei 2-Methyl-2-(6,7,8,9-tetrahydro-dibenzofuran-3-sulfonylamino)-propionsäuremethylester (eine Verbindung der allgemeinen Formel VII) (586 mg, 25%) als weißer Feststoff erhalten wurde. 1H-NMR (CDCl3): δ 7,9 (s, 1H), 7,7 (d, 1H), 7,4 (d, 1H), 3,6 (s, 3H), 2,8 (m, 2H), 2,6 (m, 1H), 1,9 (m, 4H), 1,4 (s, 6H) ppm.
- Stufe (g)
- 2-Methyl-2-(6,7,8,9-tetrahydro-dibenzofuran-3-sulfonylamino)-propionsäuremethylester (586 mg, 1,67 mmol) wurde in wässrigem Tetrahydrofuran (1 : 1, 22 ml) suspendiert und mit wässrigem Natriumhydroxid (1 M, 7 ml) behandelt. Das Reaktionsgemisch wurde 3 Tage bei Raumtemperatur gerührt. Das Tetrahydrofuran wurde unter Vakuum eingeengt. Die wässrige Suspension wurde mit Wasser (40 ml) verdünnt und mit Diethylether gewaschen. Die wässrige Phase wurde von der organischen Phase abgetrennt und angesäuert (auf einen pH-Wert von 1). Der gebildete Niederschlag wurde durch Filtration gewonnen und im Ofen getrocknet, wobei 2-Methyl-2-(6,7,8,9-tetrahydro-dibenzofuran-3-sulfonylamino)-propionsäure (eine Verbindung der allgemeinen Formel VIII) (457 mg, 81%), deren Struktur im folgenden angegeben ist, erhalten wurde. Der Schmelzpunkt dieser Verbindung wurde als 237–240°C bestimmt. 1H-NMR (CDCl3): δ 8,0 (s, 1H), 7,8 (s, 1H), 7,6 (m, 2H), 2,7 (m, 2H), 2,6 (m, 2H), 1,8 (m, 4H), 1,1 (s, 6H) ppm.
-
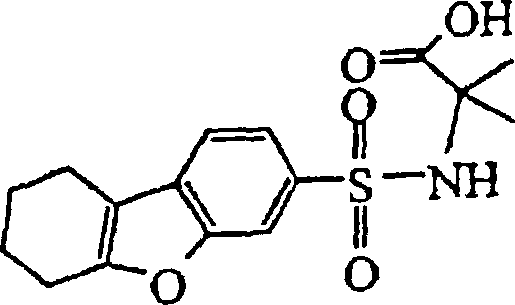
- Synthese von N-Hydroxy-2-methyl-2-(6,7,8,9-tetrahydrodibenzofuran-3-sulfonylamino)-propionamid
- Stufe (h)
- 2-Methyl-2-(6,7,8,9-tetrahydro-dibenzofuran-3-sulfonylamino)-propionsäure (387 mg, 1,15 mmol) wurde in 1,2-Dichlorethan (22 ml) suspendiert und dann tropfenweise mit Oxalylchlorid (438 mg, 3,45 mmol) versetzt. N,N-Dimethylformamid (2 Tropfen) wurde zugegeben und von der Lösung wurde ein Gas entwickelt. Das Reaktionsgemisch wurde gerührt, bis die Feststoffe gelöst waren. Das Reaktionsgemisch wurde durch Eindampfen eingeengt, mit Hexan gewaschen, erneut eingeengt und in Tetrahydrofuran (20 ml) gelöst, und anschließend zu einer wässrigen Tetrahydrofuranlösung (1 : 1, 34 ml, 5°C) von Hydroxylaminhydrochlorid (800 mg, 11,5 mmol) und Natriumbicarbonat (1,8 g, 17,25 mmol) gegeben. Das Reaktionsgemisch wurde über Nacht gerührt, wobei es sich auf Raumtemperatur erwärmen gelassen wurde. Das Tetrahydrofuran wurde unter Vakuum von dem Reaktionsgemisch entfernt. Zu dem Rückstand wurden 1 M HCl und Dichlormethan gegeben. Die organische Phase wurde von der wässrigen Phase abgetrennt, getrocknet (über MgSO4) und eingeengt. Die Kristalle wurden mit Ethylacetat gespült und anschließend durch Filtration gewonnen, wobei N-Hydroxy-2-methyl-2-(6,7,8,9-tetrahydro-dibenzofuran-3-sulfonylamino)-propion amid (eine Verbindung der allgemeinen Formel IX) (235 mg, 58%), dessen Struktur im folgenden angegeben ist, erhalten wurde. Der Schmelzpunkt dieser Verbindung wurde als 175–176°C bestimmt. 1H-NMR (CDCl3): δ 10,0 (s, 1H), 7,6 (s, 1H), 7,4 (d, 1H), 7,2 (m, 2H), 2,6 (m, 2H), 2,4 (m, 2H), 1,6 (m, 4H), 1,0 (s, 6H) ppm.
-
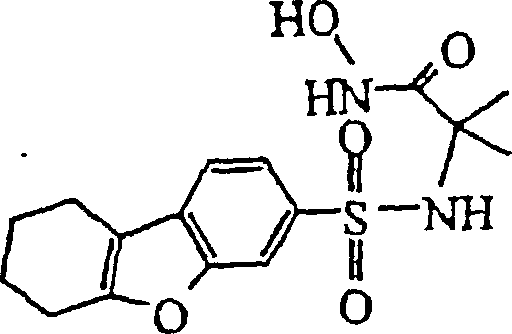
- Unter Verwendung des für Beispiel 7 beschriebenen Verfahrens war die Synthese der Beispiele 9 und 10 möglich.
- Beispiel 9
- 1-(6,7,8,9-Tetrahydro-dibenzofuran-3-sulfonylamino)-cyclopentancarbonsäure
- MS-APCI (negativer Ionenmodus) m/z = 362,1 (M-H).
-

- Beispiel 10
- 2-(6,7,8,9,9a-Hexahydro-4bH-10-oxa-benzo[a]azulen-2-sulfonylamino)-2-methyl-propionsäure
- Der Schmelzpunkt dieser Verbindung wurde als > 200°C bestimmt. 1H-NMR (CDCl3): δ 7,9 (s, 1H), 7,8 (s, 1H), 7,6 (s, 2H), 2,9 (m, 2H), 2,7 (m, 2H), 1,8 (m, 6H) ppm.
-
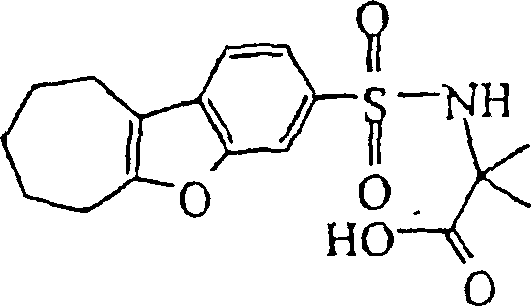
- Beispiel 11
- Unter Verwendung des für Beispiel 8 beschriebenen Verfahrens war die Synthese von Beispiel 11, N-Hydroxy-2-methyl-2-(6,7,8,9-tetrahydro-5H-10-oxa-benzo[a]azulen-2-sulfonylamino)-propionamid, möglich. Der Schmelzpunkt dieser Verbindung wurde als 167–171°C bestimmt. 1H-NMR (CDCl3): δ 7,6 (s, 1H), 7,3 (m, 1H), 7,1 (m, 1H), 6,9 (s, 1H), 2,6 (m, 2H), 2,4 (m, 2H), 1,5 (m, 6H) ppm.
-

- Weitere Verbindungen, die mit dem Verfahren für die Beispiele 7 und 8 hergestellt werden können, sind die folgenden:
- Beispiel 12 2-(2,3-Dihydro-1H-8-oxa-cyclopenta[a]inden-6-sulfonylamino)-2-methyl-propionsäure

- Beispiel 13 2-(2,3-Dihydro-1H-8-oxa-cyclopenta[a]inden-6-sulfonylamino)-N-hydroxy-2-methyl-propionamid

- Beispiel 14 1-(2,3-Dihydro-1H-8-oxa-cyclopenta[a]inden-6-sulfonylamino)-cyclopentancarbonsäure

- Beispiel 15 1-(2,3-Dihydro-1H-8-oxa-cyclopenta[a]inden-6-sulfonylamino)-cyclopentancarbonsäurehydroyxamid

- Beispiel 16 1-(6,7,8,9-Tetrahydro-dibenzofuran-3-sulfonylamino)-cyclopentancarbonsäurehydroxyamid

- Beispiel 17 1-(6,7,8,9-Tetrahydro-5H-10-oxa-benzo[a]azulen-2-sulfonylamino)-cyclopentancarbonsäure

- Beispiel 18 1-(6,7,8,9-Tetrahydro-5H-10-oxa-benzo[a]azulen-2-sulfonylamino)-cyclopentancarbonsäurehydroxyamid

- Beispiel 19 2-(5,6,7,8,9,10-Hexahydro-11-oxa-cycloocta[a]inden-2-sulfonylamino)-2-methyl-propionsäure

- Beispiel 20 2-(5,6,7,8,9,10-Hexahydro-11-oxa-cycloocta[a]inden-2-sulfonylamino)-N-hydroxy-2-methyl-propionamid

- Beispiel 21 1-(5,6,7,8,9,10-Hexahydro-11-oxa-cycloocta[a]inden-2-sulfonylamino)-cyclopentancarbonsäure

- Beispiel 22 1-(5,6,7,8,9,10-Hexahydro-11-oxa-cycloocta[a]inden-2-sulfonylamino)-cyclopentancarbonsäurehydroxyamid

- Beispiel 23
- Synthese von 2-(5a,6,7,8,9,9a-Hexahydro-dibenzofuran-3-sulfonylamino)-2-methyl-propionsäure
- Stufe (a)
- 1,2,3,4-Tetrahydro-dibenzofuran-3-sulfonsäure-natriumsalz (eine Verbindung der allgemeinen Formel V) (2,24 g, 8,17 mmol) wurde in Ethanol (50 ml) und Wasser (50 ml) gelöst. Zu dieser Lösung wurde 20% Pd/C (0,2 g) als Katalysator gegeben, und das gebildete Gemisch wurde mit Wasserstoff unter Druck gesetzt (49,6 psi). Das Reaktionsgemisch wurde 11 h geschüttelt. Der Pd/C-Katalysator wurde abfiltriert, und das Filtrat wurde eingeengt, wobei ein weißer Feststoff (eine Verbindung der allgemeinen Formel X) erhalten wurde (1,95 g, 86%). 1H-NMR (CDCl3): δ 7,1 (s, 2H), 6,9 (s, 1H), 4,6 (m, 1H), 3,2 (m, 1H), 1,8 (m, 4H), 1,3 (m, 4H) ppm.
- Stufe (b)
- Das in Stufe (a) hergestellte Sulfonsäurenatriumsalz (894 mg, 3,24 mmol) wurde langsam zu Phosphortrichlorid (12 ml) gegeben, anschließend 5 Tage unter Rückflusskühlung erhitzt. Das Reaktionsgemisch wurde über Eis (300 ml), zu dem Diethylether (300 ml) gegeben wurde, gegossen. Die organische Phase wurde von der wässrigen Phase abgetrennt, anschließend mit Kochsalzlösung gewaschen, getrocknet (über MgSO4) und eingeengt, wobei das Sulfonylchlorid (eine Verbindung der allgemeinen Formel XI), eine viskose Flüssigkeit, (591 mg, 67%) erhalten wurde. 1H-NMR (CDCl3): δ 7,6 (m, 1H), 7,3 (m, 2H), 4,8 (m, 1H), 3,3 (m, 1H), 1,9 (m, 4H), 1,5 (m, 4H) ppm.
- Stufe (c)
- Triethylamin (769 mg, 7,60 mmol) wurde tropfenweise zu einer Suspension von α-Amino-isobuttersäure (500 mg, 3,26 mmol) in Dichlormethan (20 ml) gegeben. Das in der vorherigen Stufe hergestellte Sulfonylchlorid (591 mg, 2,17 mmol) wurde in Dichlormethan (5 ml) aufgenommen und zu der Aminosäurelösung gegeben. Das Reaktionsgemisch wurde 20 h bei Raumtemperatur gerührt. Das Reaktionsgemisch wurde einge engt, und der Rückstand wurde in Ethylacetat gelöst, mit Salzsäure (1 M) gewaschen, getrocknet (über MgSO4) und eingeengt, wobei 2-(5a,6,7,8,9,9a-Hexahydro-dibenzofuran-3-sulfonylamino)-2-methyl-propionsäuremethylester (eine Verbindung der allgemeinen Formel XII) als viskose orangefarbene Flüssigkeit (420 mg, 55%) erhalten wurde. 1H-NMR (CDCl3): δ 7,4 (m, 1H), 7,2 (m, 3H), 4,7 (m, 1H), 3,6 (s, 3H), 3,2 (m, 1H), 1,9 (m, 4H), 1,5 (m, 4H), 1,4 (s, 6H) ppm.
- Stufe (d)
- Natriumhydroxid (1 M, 6 ml) wurde zu einer Lösung des in Stufe (c) hergestellten Methylesters (420 mg, 1,19 mmol) in wässrigem Tetrahydrofuran (1 : 1, 10 ml) gegeben. Das Reaktionsgemisch wurde 20 h bei Raumtemperatur gerührt. Das Tetrahydrofuran wurde unter Vakuum entfernt, und die eingeengte Lösung wurde mit konzentrierter Salzsäure auf einen pH-Wert von 1 angesäuert. Die Kristalle, die sich bildeten, wurden durch Filtration gewonnen und in einem Vakuumofen getrocknet, wobei 2-(5a,6,7,8,9,9a-Hexahydro-dibenzofuran-3-sulfonylamino)-2-methyl-propionsäure (eine Verbindung der allgemeinen Formel XIII) (297 mg, 73%), deren Struktur im folgenden angegeben ist, erhalten wurde. Der Schmelzpunkt dieser Verbindung wurde als 154–165°C bestimmt. 1H-NMR (CDCl3): δ 7,9 (s, 1H), 7,3 (m, 3H), 7,1 (s, 1H), 4,7 (m, 1H), 2,4 (s, 6H), 1,8 (m, 4H), 1,3 (m, 4H) ppm.
-
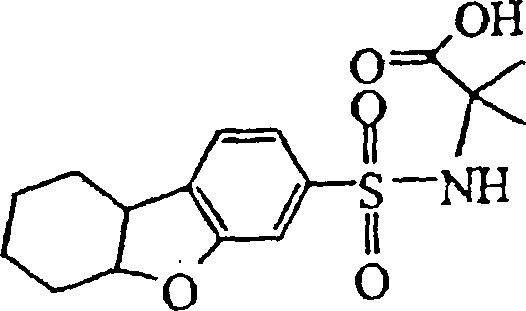
- Beispiel 24
- Synthese von 2-(5a,6,7,8,9,9a-Hexahydro-dibenzofuran-3-sulfonylamino)-N-hydroxy-2-methyl-propionamid
- Die Verbindung von Beispiel 23 (267 mg, 0,79 mmol) wurde in Dichlormethan (13 ml) suspendiert und anschließend mit Oxalylchlorid (301 mg, 2,37 mmol) versetzt. Nach der Zugabe von einem Tropfen DMF wurde von dem Reaktionsgemisch ein Gas entwickelt. Das Reaktionsgemisch wurde bei Raumtemperatur gerührt, bis ein Auflösen erfolgte. Das Gemisch wurde eingeengt, mit Hexan gewaschen und zur Trockne eingeengt. Diese rohe Verbindung wurde in Tetrahydrofuran (6 ml) gelöst und dann zu Hydroxylaminhydrochlorid (550 mg, 7,90 mmol) und Natriumbicarbonat, die in kaltem wässrigem Tetrahydrofuran (1 : 1, 10 ml) gelöst waren, gegeben. Das gebildete Gemisch wurde 15 h gerührt. Das Tetrahydrofuran wurde unter Vakuum von der Lösung entfernt, und dann wurde das Reaktionsgemisch mit Salzsäure (1 M) und Dichlormethan gewaschen. Die organische Phase wurde von der wässrigen Phase abgetrennt, eingeengt, und der Rückstand wurde mit Hexan gewaschen, erneut eingeengt und mit Ethylacetat gewaschen, Das Produkt wurde über Nacht aus Ethylacetat umkristallisiert. Der Feststoff wurde durch Filtration gewonnen und in einem Vakuumofen bei 40°C getrocknet, wobei 2-(5a,6,7,8,9,9a-Hexahydro-dibenzofuran-3-sulfonylamino)-N-hydroxy-2-methyl-propionamid (eine Verbindung der allgemeinen Formel XIV), deren Struktur im folgenden angegeben ist, erhalten wurde. Der Schmelzpunkt dieser Verbindung wurde als 153–159°C bestimmt. 1H-NMR (CDCl3): δ 9,9 (s, 1H), 8,4 (s, 1H), 7,2 (s, 1H), 7,1 (s, 1H), 7,0 (s, 1H), 4,4 (m, 1H), 3,0 (m, 1H), 1,6 (m, 4H), 1,2 (m, 4H), 1,0 (m, 6H) ppm.
-

- Weitere Verbindungen, die mit dem Verfahren für die Beispiele 23 und 24 hergestellt werden können, sind die folgenden:
- Beispiel 25 1-(5a,6,7,8,9,9a-Hexahydro-dibenzofuran-3-sulfonylamino)cyclopentancarbonsäure
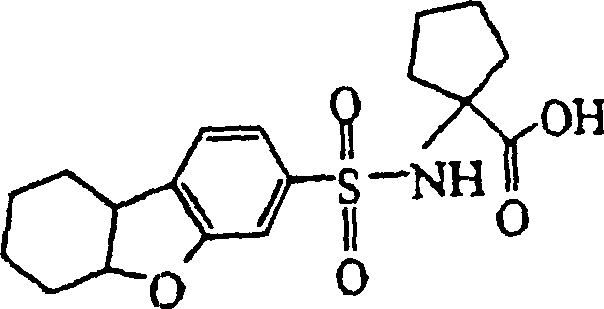
- Beispiel 26 1-(5a,6,7,8,9,9a-Hexahydro-dibenzofuran-3-sulfonylamino)cyclopentancarbonsäurehydroxyamid

- Die nächsten mehreren Beispiele wurden gemäß dem im folgenden angegebenen und beschriebenen allgemeinen Verfahren durchgeführt, um die angegebenen Verbindungen herzustellen. Zunächst wird ein Arylsulfonylchlorid (ArSO2Cl) mit einem α,α'-disubstituierten Aminosäureester unter Verwendung eines inerten Lösemittels, wie Dichlormethan (CH2Cl2), und eines Säurefängers, wie Triethylamin (NEt3), umgesetzt. Das Zwischenprodukt eines Sulfonamidcarbonsäureesters wird mit einer wässrigen Base, wie Natriumhydroxid (NaOH), hydroly siert und anschließend mit einer geeigneten Säure, wie Salzsäure (HCl), angesäuert, wobei das im folgenden angegebene Aminosäuresulfonamid hergestellt wird. Diese Carbonsäure wird dann in die Hydroxamsäure durch zunächst Erzeugen des Säurechlorids unter Verwendung von Oxalylchlorid ((COCl)2) oder eines ähnlichen Chlorierungsmittels und anschließendes Umsetzen des intermediären Säurechlorids mit einer Lösung von freiem Hydroxylamin umgewandelt.
-

- Beispiel 27
- Synthese von 1-(Dibenzofuran-3-sulfonylamino)-cyclohexancarbonsäure
- Ethyl-1-aminocyclohexancarboxylat (0,32 g, 1,9 mmol) und 0,5 g (1,9 mmol) von 3-Dibenzofuransulfonylchlorid wurden in 50 ml Dichlormethan bei Raumtemperatur gemischt. Triethylamin im Überschuss (0,78 ml, 5,7 mmol) wurde zugegeben, und das gebildete Gemisch wurde über Nacht gerührt. Das Reaktionsgemisch wurde zwischen 1 M HCl und Dichlormethan verteilt. Die Dichlormethanschicht wurde abgetrennt und über Magnesiumsulfat getrocknet und anschließend eingeengt. Der Rückstand wurde chromatographiert, wobei 0,28 g des Ethylesters der 1-(Dibenzofuran-3-sulfonylamino)cyclohexancarbonsäureverbindung erhalten wurden. Diese wurde in 20 ml 1 M NaOH suspendiert und 6 h unter Rückflusskühlung erhitzt. Das Produkt wurde mit konzentrierter HCl angesäuert und mit Ethylacetat extrahiert. Die Ethylacetatschicht wurde über Magnesiumsulfat getrocknet und eingeengt, wobei ein weißlicher Feststoff erhalten wurde. Der Feststoff wurde mit 5% Ethylacetat/Hexan verieben, wobei 0,20 g der 1-(Dibenzofuran-3-sulfonylamino)-cyclohexancarbonsäureverbindung, deren Struktur im folgenden angegeben ist, als weißer Feststoff erhalten wurden. Die Verbindung wies einen Schmelzpunkt von etwa 239–243°C auf. 1H-NMR (CDCl3): δ 7,97 (d, 1H), 7,90 (t, 2H), 7,77 (d, 1H), 7,49 (d, 1H), 7,41 (t, 1H), 7,27 (t, 1H), 6,31 (s, 1H), 1,75 (m, 4H), 1,23 (m, 6H) ppm. Eine Analyse zeigte, dass die Verbindung die empirische Formel C19H19N1O5S aufwies.
-
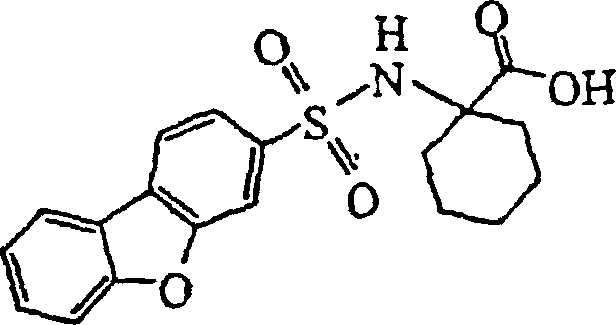
- Beispiel 28
- Synthese von 1-(Dibenzofuran-3-sulfonylamino)-cyclohexancarbonsäurehydroxamid
- 1-(Dibenzofuran-3-sulfonylamino)-cyclohexancarbonsäure (0,18 g, 0,5 mmol) wurde in 50 ml Dichlormethan mit 2 Tropfen Dimethylformamid gelöst. Oxalylchlorid (0,08 ml, 1,0 mmol) wurde zugegeben, und die gebildete Lösung wurde 2 h gerührt und dann unter Vakuum eingeengt. Der gebildete Rückstand wurde in 10 ml Tetrahydrofuran gelöst, und diese Lösung wurde tropfenweise zu einem Gemisch von Natriumbicarbonat (0,61 g, 7,2 mmol) und Hydroxylaminhydrochlorid (0,33 g, 4,8 mmol) in 50 ml eines Tetrahydrofuran/Wasser-Gemischs (Verhältnis 1 : 1) gegeben, das 10 min bei 0°C ge rührt wurde. Das gebildete Gemisch wurde sich auf Raumtemperatur erwärmen gelassen und 64 h gerührt. Das Reaktionsgemisch wurde unter Vakuum eingeengt und zwischen 1 M HCl und Dichlormethan verteilt. Die wässrige Schicht wurde mit weiterem Dichlormethan extrahiert und anschließend wurden die vereinigten organischen Schichten über Magnesiumsulfat getrocknet, filtriert und eingeengt, wobei 0,07 g eines weißlichen Feststoffs, dessen Struktur im folgenden angegeben ist, erhalten wurden. Die Verbindung wies einen Schmelzpunkt von etwa 184–186°C auf. 1H-NMR (CDCl3): δ 7,97 (d, 1H), 7,97 (s, 1H), 7,90 (dd, 2H), 7,75 (d, 1H), 7,48 (d, 1H), 7,41 (t, 1H), 7,27 (t, 1H), 6,65 (s, 1H), 1,79 (m, 4H), 1,14 (m, 6H) ppm. Eine Analyse zeigte, dass die Verbindung die empirische Formel C19H20N2O5S·0,25H2O aufwies.
-

- Beispiel 29
- Synthese von 2-(Dibenzofuran-3-sulfonylamino)-2-methylpropionsäure
- In dem Verfahren von Beispiel 27 wurde Ethyl-1-aminocyclohexancarboxylat durch α-Aminoisobuttersäuremethylesterhydrochlorid ersetzt und 2-(Dibenzofuran-3-sulfonylamino)-N-hydroxy-2-methyl-propionamid erhalten. Die Struktur dieser Verbindung ist im folgenden angegeben. Die Verbindung wies einen Schmelzpunkt von etwa 220–224°C auf. 1H-NMR (CDCl3): δ 8,02 (s, 1H), 7,93 (dd, 2H), 7,83 (d, 1H), 7,53 (d, 1H), 7,46 (t, 1H), 7,31 (t, 1H), 6,26 (s, 1H), 1,36 (s, 6H) ppm. Eine Analyse zeigte, dass die Verbindung die empirische Formel C16H15N1O5S·0,33H2O aufwies.
-
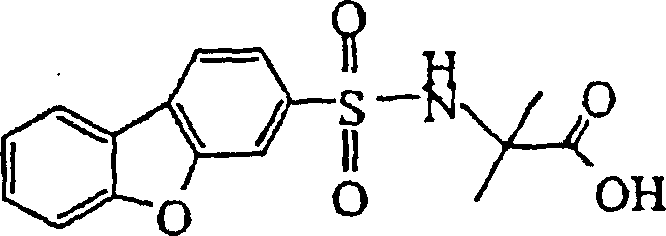
- Beispiel 30
- Synthese von 2-(Dibenzofuran-3-sulfonylamino)-N-hydroxy-2-methyl-propionamid
- In dem Verfahren von Beispiel 28 wurde 1-(Dibenzofuran-3-sulfonylamino)-cyclohexancarbonsäure durch 2-(Dibenzofuran-3-sulfonylamino)-2-methyl-propionsäure ersetzt und 2-(Dibenzofuran-3-sulfonylamino)-2-methyl-propionsäure erhalten. Die Verbindung wies einen Schmelzpunkt von etwa 168– 169°C auf. 1H-NMR (CDCl3): δ 7,96 (s, 1H), 7,87 (dd, 2H), 7,73 (d, 1H), 7,46 (d, 1H), 7,39 (t, 1H), 7,25 (t, 1H), 7,14 (s, 1H), 1,22 (s, 6H) ppm. Eine Analyse zeigte, dass die Verbindung die empirische Formel C16H16N2O5S·1,25H2O aufwies. Die Struktur dieser Verbindung ist im folgenden angegeben.
-

- Beispiel 31
- Synthese von 4-(Dibenzofuran-3-sulfonylamino)-tetrahydropyran-4-carbonsäurehydroxyamid
- In dem Verfahren von Beispiel 27 wurde Ethyl-1-aminocyclohexancarboxylat durch Methyl-4-aminotetrahydro-2H-pyran-4- carboxylat ersetzt und 4-(Dibenzofuran-3-sulfonylamino)-tetrahydro-pyran-4-carbonsäure erhalten und ohne weitere Reinigung verwendet. In dem Verfahren von Beispiel 28 wurde 1-(Dibenzofuran-3-sulfonylamino)-cyclohexancarbonsäure durch 4-(Dibenzofuran-3-sulfonylamino)-tetrahydro-pyran-4-carbonsäure ersetzt und die Titelverbindung erhalten. Die Verbindung wies einen Schmelzpunkt von etwa 160–162°C auf. Eine Analyse zeigte, dass die Verbindung die empirische Formel C18H18N2O6S·2,5H2O aufwies. Die Struktur dieser Verbindung ist im folgenden angegeben.
-

- Beispiel 32
- Synthese von 1-(7-Brom-dibenzofuran-2-sulfonylamino)cyclopentancarbonsäurehydroxyamid
- Wenn in dem Verfahren von Beispiel 28 1-(Dibenzofuran-3-sulfonylamino)-cyclohexancarbonsäure durch 1-(7-Bromdibenzofuran-2-sulfonylamino)-cyclopentancarbonsäure ersetzt wurde, wurde die im folgenden angegebene Titelverbindung erhalten. Die Verbindung wies einen Schmelzpunkt von 177–178°C auf.
-

- Beispiel 33
- Synthese von 2-(7-Chlor-dibenzofuran-2-sulfonylamino)-N-hydroxy-2-methyl-propionamid
- Wenn in dem Verfahren von Beispiel 28 1-(Dibenzofuran-3-sulfonylamino)-cyclohexancarbonsäure durch 2-(7-Chlordibenzofuran-2-sulfonylamino)-2-methyl-propionsäure ersetzt wurde, wurde die im folgenden angegebene Titelverbindung erhalten. Die Verbindung wies einen Schmelzpunkt von 195– 196°C auf.
-

- Biologische Tests
- Die Verbindungen der Erfindung wurden in In-vitro-Standardtests bewertet und es wurde gezeigt, dass sie wirksame Inhibitoren einer Vielzahl von Matrixmetalloproteinaseenzymen sind. Die Verbindungen wurden bewertet, um ihre jeweiligen IC50-Werte, d. h. die mikromolare Konzentration einer Verbindung, die erforderlich ist, um eine Hemmung von 50% der Hydrolyseaktivität der jeweiligen Enzyme zu erreichen, zu bestimmen.
- Experimente wurden mit der vollen Länge ("FL") und/oder katalytischen Domänen ("CD") der Proteinasen durchgeführt. Tabelle 1 zeigt die Aktivität der Verbindungen der Beispiele 27–31 gegenüber MMP-1FL (Kollagenase-1-Enzym voller Länge), MMP-2CD (katalytische Domäne von Gelatinase A), MMP-2FL (Gelatinase-A-Enzym voller Länge), MMP-3CD (katalytische Domäne von Stromelysin-1), MMP-7FL (Matrilysin-Enzym voller Länge), MMP-9FL (Gelatinase-B-Enzym voller Länge), MMP-13CD (katalytische Domäne von Kollagenase-3) und MMP-14CD (katalytische Domäne von MMP-1 des Membrantyps). IC50-Werte wurden unter Verwendung eines Thiopeptolidsubstrats, Ac-Pro-Leu-Gly-Thioester-Leu-Leu-GIy-OEt bestimmt (Q.-Z. Ye, L. L. Johnson, D. J. Hupe und V. Baragi, "Purification and Characterization of the Human Stromelysin Catalytic Domain Expressed in Escherichia coli", Biochemistry, 1992; 31: 11231–11235; Q.-Z. Ye, L. L. Johnson, A. E. Yu und D. J. Hupe, "Reconstructed 19 kDa Catalytic Domain of Gelatinase A IAAP", Biochemistry, 1995; 34: 4702–4708).
- Die getesteten MMPs sind einem Fachmann zugänglich. Beispielsweise kann MMP-1 von Washington University School of Medicine, St. Louis, Missouri erhalten werden. MMP-7 kann gemäß dem bekannten Verfahren, das bei Q.-Z. Ye, L. L. Johnson und V. Baragi, "Gene Syntheses and Expression in E. coli for PUMP, a Human Matrix Metalloproteinase", Bioch. and Biophys. Res. Comm., 1992; 186: 143–149 angegeben ist, erhalten werden. MMP-13 kann gemäß dem bekannten Verfahren, das bei J. M. P. Freije et al., "Molecular Cloning and Expression of Collagenase-3, a Novel Human Matrix Metalloproteinase Produced by Breast Carcinomas", J. Bio. Chem., 1994; 269: 16766-16773 angegeben ist, erhalten werden. MMP-13CD kann auch aus einem synthetischen Gen exprimiert und aus einer Zellkultur von Escherichia coli gereinigt werden, gemäß einem bereits beschriebenen Verfahren (Q.-Z. Ye, L. L. Johnson und V. Baragi, "Gene Synthesis and Expression in E. coli for PUMP, a Human Matrix Metalloproteinase", Biochemical and Biophysical Research Communications, 1992; 186: 143–149). Die Hydrolyse des Thiopeptolidsubstrats Ac-Pro-Leu-Gly-Thioester-Leu-Leu-Gly-OEt (Bachem) wurde als das primäre Screening zum Bestimmen der IC50-Werte für MMP-Inhibitoren verwendet. Ein Reaktionsgemisch von 100 μl enthält 1 mM 5,5'-Dithiobis(2-nitrobenzoesäure) (DTNB), 100 μM Substrat, 0,1% BRIJ® 35, PROTEIN GRADE®, Detergens, 10%- ige Lösung, Sigma, St. Louis, Missouri [Polyoxyethylenglykoldodecylether, Polyoxyethylen(23)laurylether], Enzym und Inhibitor in dem geeigneten Reaktionspuffer. Aktivierte Enzyme voller Länge werden mit 5 nM, die katalytische Domäne von Stromelysin (SCD) mit 10 nM und die katalytische Domäne von Gelatinase A (GaCD) mit 1 nM getestet. Inhibitoren werden von 100 μM bis 1 nM durchmustert. Enzyme voller Länge (beispielsweise MMP-1 und MMP-7) werden in 50 mM 4-(2-Hydroxymethyl)-piperazin-1-ethansulfonsäure (HEPES), 10 mM CaCl2, pH-Wert 7,0; SCD in 50 mM 2-Morpholinoethansulfonsäuremonohydrat (MES), 10 mM CaCl2, pH-Wert 6,0; und GaCD in 50 mM 3-Morpholtriopropansulfonsäure (MOPS), 10 mM CaCl2, 10 μM ZnCl2, pH-Wert 7,0 getestet. Die Änderung der Extinktion bei 405 nm wird auf einer ThermoMax-Mikroplattenlesevorrichtung bei Raumtemperatur kontinuierlich während 20 min überwacht.
- Die Zahl in Klammern gibt die Zahl der getesteten Proben an. Wenn mehrfache Proben getestet wurden, ist das in Tabelle 1 angegebene Ergebnis ein Durchschnittswert.
- Tabelle 1. Matrixmetalloproteinaseenzymhemmung IC50 μM
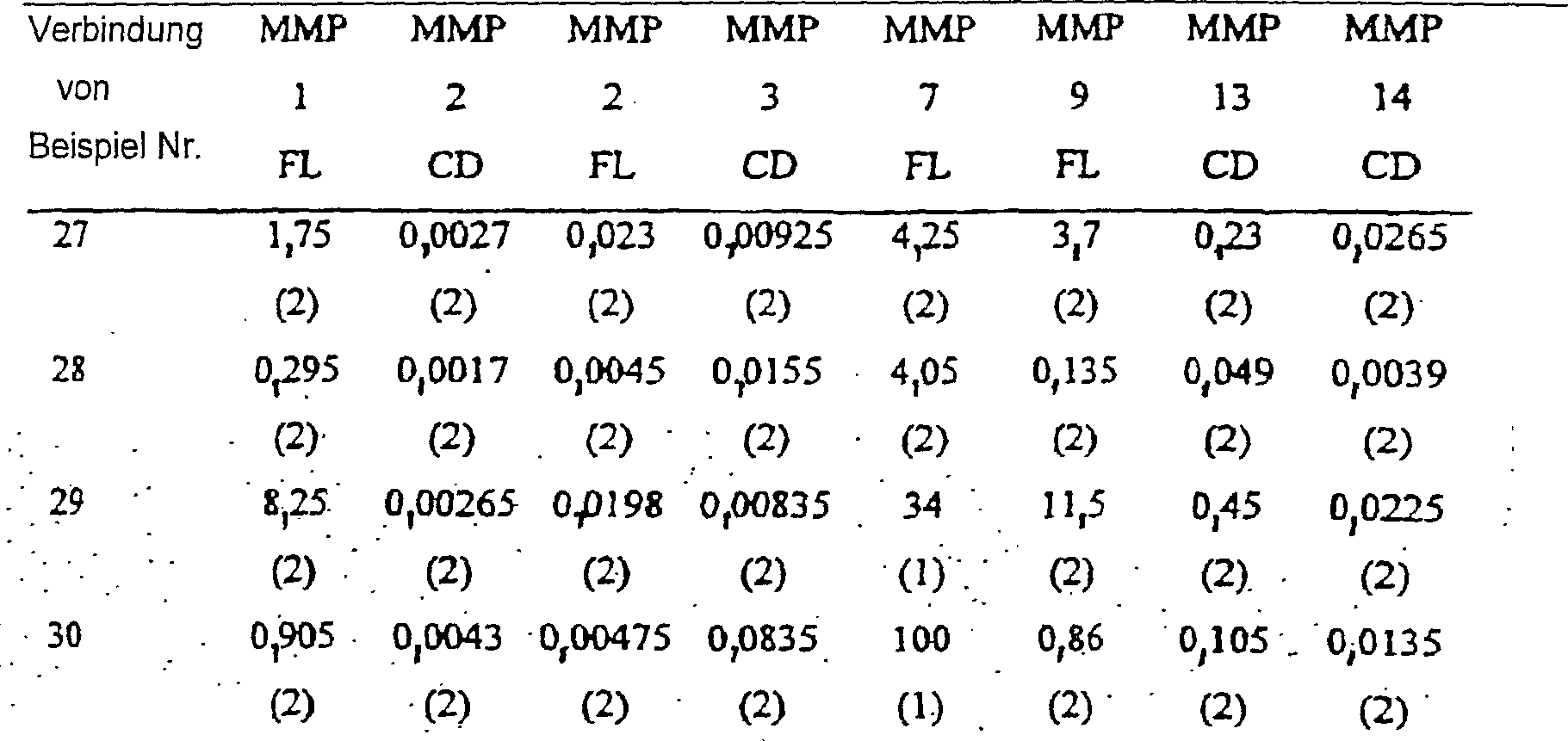
-

- Wenn pharmazeutische Zusammensetzungen und Zubereitungen aus den Verbindungen der vorliegenden Erfindung hergestellt werden, können pharmazeutisch akzeptable Träger entweder fest oder flüssig sein. Zubereitungen in fester Form umfassen Pulver, Tabletten, Pillen, Kapseln, Cachets, Suppositorien, Pastillen und dispergierbare Granulate. Ein fester Träger können eine oder mehrere Substanzen, die auch als Verdünnungsmittel, Aromatisierungsmittel, Solubilisierungsmittel, Gleitmittel, Suspendiermittel, Bindemittel, Konservierungsmittel, den Tablettenzerfall fördernde Mittel fungieren können, oder ein Einkapselungsmaterial sein. Der Ausdruck "Zubereitung" soll die Formulierung der aktiven Verbindung mit Einkapselungsmaterial als Träger, was eine Kapsel ergibt, in der die aktive Komponente mit oder ohne andere Träger von einem Träger umgeben ist, der dadurch mit dieser in Verbindung ist, umfassen.
- In Tabletten und Kapseln wird die aktive Verbindung mit mindestens einem (a) inerten Streckmittel (oder Träger), wie Natriumcitrat oder Dicalciumphosphat; (b) Füllstoff oder Streckmittel, beispielsweise Stärke, Lactose, Saccharose, Glucose, Mannit und Kieselsäure; (c) einem Bindemittel, beispielsweise Sorbit, Tragant, Stärkeschleimlösung, Carboxymethylcellulose, Alginaten, Gelatine, Polyvinylpyrrolidon, Saccharose und Akaziengummi; (d) einem Feuchthaltemittel, beispielsweise Glycerin; (e) einem den Zerfall fördernden Mittel, beispielsweise Agar-Agar, Calciumcarbonat, Kartoffel- oder Tapiokastärke, Alginsäure, bestimmten komplexen Silicaten und Natriumcarbonat; (f) einem Lösungsverzögerungsmittel, beispielsweise Paraffin; (g) einem Absorptionsbeschleuniger, beispielsweise quaternären Ammoniumverbindungen; (h) einem Befeuchtungsmittel, beispielsweise Cetylalkohol, Natriumlaurylsulfat und Glycerinmomostearat; (i) einem Adsorptionsmittel, beispielsweise Kaolin und Bentonit; (j) einem Gleitmittel, beispielsweise Talkum, Calciumstearat, Stearinsäure, Magnesiumstearat, festen Polyethylenglykolen, Natriumlaurylsulfat, Natriumstärkeglykolat; und (k) Gemischen derselben gemischt. Im Falle von Kapseln, Tabletten und Pillen können die Dosierungsformen auch Puffermittel umfassen.
- Feste Zusammensetzungen eines ähnlichen Typs können ebenfalls als Füllstoffe in weichen und harten gefüllten Gelatinekapseln unter Verwendung von Streckmitteln, wie Lactose oder Milchzucker, sowie Polyethylenglykolen mit hohem Molekulargewicht und dgl. verwendet werden.
- Feste Dosierungsformen, wie Tabletten, Dragees, Kapseln, Pillen und Granulate, können mit Überzügen und Hüllen, wie enterischen Überzügen und anderen einschlägig bekannten, hergestellt werden. Die Dosierungsform kann Opakifizierungsmittel enthalten und kann auch derart gestaltet sein, dass die aktive Verbindung oder die aktiven Verbindungen in einen vorbestimmten Teil des Intestinaltrakts in verzögerter Weise freigesetzt werden. Beispiele für Einbettungszusammensetzungen, die verwendet werden können, sind polymere Substanzen und Wachse. Die aktiven Verbindungen können auch in mikroverkapselter Form, wenn dies günstig ist, mit einem oder mehreren der im vorhergehenden genannten Streckmittel sein.
- In Pulvern ist der Träger ein fein zerteilter Feststoff, der im Gemisch mit der fein zerteilten aktiven Komponente vorliegt. Die Pulver und Tabletten enthalten vorzugsweise etwa 5, vorzugsweise etwa 10 bis etwa 70% der aktiven Verbindung. Geeignete Träger sind Magnesiumcarbonat, Magnesiumstearat, Talkum, Zucker, Lactose, Pektin, Dextrin, Stärke, Gelatine, Tragant, Methylcellulose, Natriumcarboxymethylcellulose, ein niedrigschmelzendes Wachs, Kakaobutter und dgl.
- Zur Herstellung von Suppositorien wird ein niedrigschmelzendes Wachs, wie ein Gemisch der Fettsäureglyceride, von Polyethylenglykol oder Kakaobutter, zunächst geschmolzen und die aktive Komponente darin beispielsweise durch Rühren homogen verteilt. Das geschmolzene homogene Gemisch wird dann in Formen geeigneter Größe gegossen, abkühlen gelassen und dadurch verfestigt.
- Zubereitungen in flüssiger Form umfassen Lösungen, Suspensionen, Emulsionen, beispielsweise Wasser- oder Wasser-Propylenglykol-Lösungen. Zur parenteralen Injektion können flüssige Zubereitungen in Lösung in wässriger Polyethylenglykollösung formuliert werden.
- Zur oralen Verwendung geeignete wässrige Lösungen können durch Auflösen der aktiven Komponente in Wasser und die Zugabe geeigneter Farbmittel, Geschmacksstoffe, Stabilisierungsmittel und Dickungsmittel nach Wunsch hergestellt werden. Die Verbindungen der Erfindung können in orale flüssige Zubereitungen, wie beispielsweise wässrige oder Ölsuspensionen, Lösungen, Emulsionen, Sirupe oder Elixiere, eingearbeitet werden. Ferner können diese Verbindungen enthaltende Formulierungen als trockenes Produkt zur Wiederherstellung einer Lösung mit Wasser oder einem anderen geeigneten Vehikel vor der Verwendung präsentiert werden. Diese flüssigen Zubereitungen können herkömmliche Zusatzstoffe, wie (a) Suspendiermittel (wie Sorbitsirup, Methyl cellulose, Glucose/Zuckersirup, Gelatine, Hydroxyethylcellulose, Hydroxypropylmethylcellulose, Carboxymethylcellulose, Aluminiumstearatgel, ethoxylierte Isostearylalkohole, Polyoxyethylensorbit und Sorbitanester, mikrokristalline Cellulose, Aluminiummetahydroxid, Bentonit, Agar-Agar, Tragant und gehärtete essbare Fette), (b) Emulgatoren (wie Lecithin, Sorbitanmonooleat oder Akaziengummi), (c) nichtwässrige Vehikel (die essbare Öle, wie Mandelöl, fraktioniertes Kokosöl, Erdnussöl, Maiskeimöl, Olivenöl, Rizinusöl, Sesamöl, Ölester, Propylenglykol und Ethylalkohol umfassen können) und (d) Konservierungsmittel (wie Methyl- oder Propyl-p-hydroxybenzoat und Sorbinsäure) enthalten. Zur oralen Verwendung geeignete wässrige Suspensionen können durch Dispergieren der fein zerteilten aktiven Komponente in Wasser mit viskosem Material, wie natürlichen oder synthetischen Gummiarten, Harzen, Methylcellulose, Natriumcarboxymethylcellulose und anderen bekannten Suspendiermitteln, hergestellt werden.
- Diese Zusammensetzungen können auch Hilfsstoffe, wie Konservierungsmittel, Befeuchtungsmittel, Emulgatoren, Suspendiermittel und Dispensiermittel sowie Süßungsmittel, Aromatisierungsmittel und Duftmittel enthalten. Eine Vorbeugung vor der Einwirkung von Mikroorganismen kann durch verschiedene antibakterielle und antimykotische Mittel, beispielsweise Parabene, Chlorbutanol, Phenol, Sorbinsäure und dgl., bereitgestellt werden. Es kann auch erwünscht sein, isotonische Mittel, beispielsweise Zucker, Natriumchlorid und dgl., einzuarbeiten. Eine verlängerte Absorption der injizierbaren pharmazeutischen Form kann durch die Verwendung von absorptionsverzögernden Mitteln, beispielsweise Aluminiummonostearat und Gelatine, beigebracht werden.
- Die folgenden Beispiele erläutern typische Formulierungen, die durch die Erfindung bereitgestellt werden.
- Beispiel 35 Zubereitung für eine oralen Lösung

- Zur Herstellung einer oralen Lösung gemäß der Erfindung wird die Sorbitlösung zu 40 ml destilliertem Wasser gegeben und die aktive Verbindung gemäß der Erfindung darin gelöst. Saccharin, das Natriumbenzoat, der Geschmacksstoff und der Farbstoff werden zugegeben und gelöst. Das Volumen wird mit destilliertem Wasser auf 100 ml eingestellt. Jeder Milliliter des Sirups enthält etwa 4 mg der erfindungsgemäßen Verbindung.
- Beispiel 36

- Parenterale Lösung
- In einer Lösung von 700 ml Propylenglykol und 200 ml Wasser zu Injektionszwecken werden 20 g einer aktiven Hydroxamsäu reverbindung gemäß der Erfindung suspendiert. Nachdem die Suspension beendet ist, wird der pH-Wert auf etwa 6,5 mit 1 N Natriumhydroxid eingestellt und das Volumen mit Wasser zu Injektionszwecken auf 1000 ml eingestellt. Die Formulierung wird sterilsiert, in 5,0-ml-Ampullen, die jeweils 2,0 ml enthalten, gefüllt und unter Stickstoff verschlossen.
- Als Matrixmetalloproteinaseinhibitoren sind die Verbindungen der Formel I als Mittel zur Behandlung von Multiple Sklerose verwendbar. Sie sind ebenfalls als Mittel zur Behandlung von Osteoporose, Nierenerkrankungen, atherosklerotischer Plaqueruptur, Herzinsuffizienz, Aortaaneurysma, Linksherzdilatation, Thrombose, Restenose, Periodontiumerkrankung, Corneaulzeration, zur Behandlung von Verbrennungen, Dekubitusulzera, chronischen Ulzera, Wundreparatur, Schlaganfällen, Krebsmetastasen, Tumorangiogenese, Arthritis, die rheumatoide Arthritis und Osteoarthritis umfasst, und Autoimmun- oder entzündlichen Erkrankungen, die die von einem Eindringen von Leukocyzten im Gewebe abhängigen umfassen, verwendbar. Diese Verbindungen sind auch als Mittel zur Behandlung chronischer neurodegenerativer Erkrankungen, die aus der Gruppe von Schlaganfall, Kopftrauma, Rückenmarkverletzungen, Alzheimer-Erkrankung, Amyotropher Lateralsklerose, Gehirnamyloidangiopathie, AIDS, Parkinson-Erkrankung, Chorea Huntington, Prionerkrankungen, Erb-Goldflam-Syndrom und Duchenne-Krankheit ausgewählt sind, verwendbar. Diese Verbindungen können auch während und nach chirurgischen Verfahren, beispielsweise Arterien-Bypass- und Hüftoperationen, sowie einer Angioplastik verwendet werden.
- Die vorhergehende detaillierte Beschreibung ist nur für die Klarheit des Verstehens angegeben, und es sollten daraus keine unnötigen Beschränkungen herausgelesen werden, wobei Modifikationen innerhalb des Umfangs der Erfindung einem Fachmann klar sind.
Claims (25)
- Verbindung der Formel Ioder ein pharmazeutisch akzeptables Salz hiervon, worin X ausgewählt ist aus OH und NHOH; d für 1 oder 2 steht; R1 ausgewählt ist aus der Gruppe:
 worin Y ausgewählt ist aus O, S, S(O)d (worin d für 1 oder 2 steht), CH2, C(=O) und NRq (worin Rq für H, C1-C6-Alkyl oder C1-C6-Alkylphenyl steht); R5 ausgewählt ist aus der Gruppe aus H, C1-C10-Alkyl, CF3, CONH2, Halogen, CN, COOH, C1-C4-Alkoxy, CHO, NO2, OH, (CH2)pOH, (CH2)pNH2, Ar und NH2, p für 0 bis 3 steht und Ar ausgewählt ist aus der Gruppe aus (a) Phenyl, (b) Phenyl, das mit C1-C4-Alkyl, C1-C4-Alkoxy, Halogen, NH2, NO2, CN, COOH, CONH2, CF3 oder COOR6 (worin R6 für C1-C10-Alkyl steht) substituiert ist, und (c) Heteroaryl; R2 ausgewählt ist aus der Gruppe aus (a) Wasserstoff, (b) C1-C4-Alkyl, (c) Benzyl und (d) Benzyl, das mit einem oder mehreren Substituenten aus C1-C4-Alkyl, C1-C4-Alkoxy, F, Cl, Br, I, NH2, NO2, CN, Carboxy und CO2R7 (worin R7 für H oder C1-C4-Alkyl steht) substituiert ist; und R3 und R4 entweder (1) jeweils unabhängig voneinander ausgewählt sind aus der Gruppe aus C1-C20-Alkyl, C3-C10-Cycloalkyl, Phenyl, Phenyl, das mit C1-C4-Alkyl, C1-C4-Alkoxy, Halogen, NH2, NO2, CN, COOH, CO2R7 oder CF3 substituiert ist, C3-C10-Heterocyclus und Heteroaryl oder (2) zusammen eine Gruppe der empirischen Formel -(CH2)sZg- bilden, worin die Substituenten R3 und R4 einen Ring einschließlich dem Kohlenstoffatom, das zu der Carbonylgruppe in Formel I benachbart ist, bilden und wobei s für eine ganze Zahl von 2 bis 10 steht, g 0 bis 6 bedeutet und jeder Rest Z sich an einer beliebigen Position der Substituenten befindet, wobei jeder Rest Z unabhängig voneinander aus O, S und NR8 (worin R8 ausgewählt ist aus H und C1-C3-Alkyl) ausgewählt ist.
worin Y ausgewählt ist aus O, S, S(O)d (worin d für 1 oder 2 steht), CH2, C(=O) und NRq (worin Rq für H, C1-C6-Alkyl oder C1-C6-Alkylphenyl steht); R5 ausgewählt ist aus der Gruppe aus H, C1-C10-Alkyl, CF3, CONH2, Halogen, CN, COOH, C1-C4-Alkoxy, CHO, NO2, OH, (CH2)pOH, (CH2)pNH2, Ar und NH2, p für 0 bis 3 steht und Ar ausgewählt ist aus der Gruppe aus (a) Phenyl, (b) Phenyl, das mit C1-C4-Alkyl, C1-C4-Alkoxy, Halogen, NH2, NO2, CN, COOH, CONH2, CF3 oder COOR6 (worin R6 für C1-C10-Alkyl steht) substituiert ist, und (c) Heteroaryl; R2 ausgewählt ist aus der Gruppe aus (a) Wasserstoff, (b) C1-C4-Alkyl, (c) Benzyl und (d) Benzyl, das mit einem oder mehreren Substituenten aus C1-C4-Alkyl, C1-C4-Alkoxy, F, Cl, Br, I, NH2, NO2, CN, Carboxy und CO2R7 (worin R7 für H oder C1-C4-Alkyl steht) substituiert ist; und R3 und R4 entweder (1) jeweils unabhängig voneinander ausgewählt sind aus der Gruppe aus C1-C20-Alkyl, C3-C10-Cycloalkyl, Phenyl, Phenyl, das mit C1-C4-Alkyl, C1-C4-Alkoxy, Halogen, NH2, NO2, CN, COOH, CO2R7 oder CF3 substituiert ist, C3-C10-Heterocyclus und Heteroaryl oder (2) zusammen eine Gruppe der empirischen Formel -(CH2)sZg- bilden, worin die Substituenten R3 und R4 einen Ring einschließlich dem Kohlenstoffatom, das zu der Carbonylgruppe in Formel I benachbart ist, bilden und wobei s für eine ganze Zahl von 2 bis 10 steht, g 0 bis 6 bedeutet und jeder Rest Z sich an einer beliebigen Position der Substituenten befindet, wobei jeder Rest Z unabhängig voneinander aus O, S und NR8 (worin R8 ausgewählt ist aus H und C1-C3-Alkyl) ausgewählt ist.
- Verbindung nach Anspruch 1, worin R1 die folgende Bedeutung besitzt:

- Verbindung nach Anspruch 1, worin R1 die folgende Bedeutung besitzt:
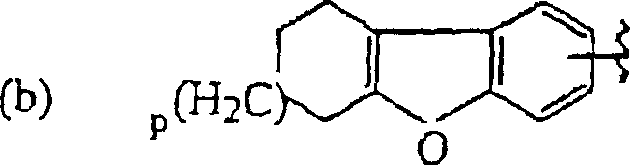
- Verbindung nach Anspruch 1, worin R1 die folgende Bedeutung besitzt:

- Verbindung nach Anspruch 1, worin R1 ausgewählt ist aus der Gruppe:und


- Verbindung nach Anspruch 1, worin R2 für Wasserstoff steht, R3 eine Methylgruppe bedeutet und R4 für eine Methylgruppe steht.
- Verbindung nach Anspruch 1, worin R3 und R4 zusammengenommen als -(CH2)s- (worin s, die Gesamtzahl der Kohlenstoffatome von sowohl R3 als auch R4, 2 bis 10 bedeutet) einen Ring einschließlich des α-Kahlenstoffatoms, das der Carbonylgruppe in Formel I benachbart ist, bilden.
- Verbindung nach Anspruch 7, worin s 4 oder 5 bedeutet.
- Verbindung nach Anspruch 7, worin der Ring ein Heteroatom umfasst, das aus NR8, Sauerstoff und Schwefel ausgewählt ist.
- Verbindung nach Anspruch 1, worin R3 und R4 zusammen einen Ring einschließlich des Kohlenstoffatoms, das der Carbonylgruppe in Formel I benachbart ist, bilden und R3 und R4 zusammen aus der Gruppe aus -CH2CH2CH2CH2-, -CH2CH2CH2CH2CH2- und -CH2CH2OCH2CH2- ausgewählt sind.
- Verbindung nach Anspruch 1, worin R3 und R4 zusammen einen Ring einschließlich des α-Kohlenstoffatoms, das der Carbonylgruppe in Formel I benachbart ist, bilden und R3 und R4 zusammen aus der Gruppe: -CH2CH2-, -CH2CH2CH2-, -CH2CH2CH2CH2-, -CH2CH2CH2CH2CH2-, -CH2CH2CH2CH2CH2CH2-, -CH2OCH2CH2-, -CH2CH2OCH2CH2-, -CH2CH2OCH2CH2CH2-, -CH2N(H)CH2CH2-, -CH2CH2N(H)CH2CH2-, -CH2CH2N(H)CH2CH2CH2-, -CH2SCH2CH2-, -CH2CH2SCH2CH2-, -CH2CH2SCH2CH2CH2-, -CH2OCH2CH2OCH2-, -CH2N(H)CH2CH2N(H)CH2- und -CH2SCH2CH2SCH2- ausgewählt sind, wobei die terminalen Kohlenstoffe beide an das α-Kohlenstoffatom, das der Carbonylgruppe in Formel I benachbart ist, gebunden sind.
- Verbindung nach Anspruch 1, worin die Verbindung die folgende Formel besitzt:worin n für 1, 2 oder 3 steht.

- Verbindung nach Anspruch 12, worin R3 und R4 zusammengenommen einschließlich des α-Kohlenstoffatoms, das der Carbonylgruppe in Formel I benachbart ist, einen Ring bilden und R3 und R4 zusammen aus der Gruppe -CH2CH2CH2CH2- und -CH2CH2CH2CH2CH2- ausgewählt sind.
- Verbindung nach Anspruch 1, worin die Verbindung die folgende Formel besitzt:worin n für 1, 2 oder 3 steht.

- Verbindung nach Anspruch 1, worin die Verbindung aus der folgenden Gruppe ausgewählt ist: 2-Methyl-2-(6,7,8,9-tetrahydro-dibenzofuran-3-sulfonylamino)propionsäure; N-Hydroxy-2-methyl-2-(6,7,8,9-tetrahydro-dibenzofuran-3-sulfonylamino)-propionamid; 1-(6,7,8,9-Tetrahydro-dibenzofuran-3-sulfonylamino)cyclopentancarbonsäure; 2-(5,6,7,8,9,9a-Hexahydro-4bH-10-oxa-benzo(a)azulen-2-sulfonylamino)-2-methylpropionsäure; N-Hydroxy-2-methyl-2-(6,7,8,9-tetrahydro-5H-10-oxa-benzo[a]azulen-2-sulfonylamino)-propionamid; 2-(2,3-Dihydro-1H-8-oxa-cyclopenta[a]inden-6-sulfonylamino)-2-methyl-propionsäure; 2-(2,3-Dihydro-1H-8-oxa-cyclopenta[a]inden-6-sulfonylamino)-N-hydroxy-2-methyl-propionamid; 1-(2,3-Dihydro-1H-8-oxa-cyclopenta[a]inden-6-sulfonylamino)-cyclopentancarbonsäure; 1-(2,3-Dihydro-1H-8-oxa-cyclopenta[a]inden-6-sulfonylamino)-cyclopentancarbonsäurehydroxyamid; 1-(6,7,8,9-Tetrahydro-dibenzofuran-3-sulfonylamino)cyclopentancarbonsäurehydroxyamid; 1-(6,7,8,9-Tetrahydro-5H-10-oxa-benzo[a]azulen-2-sulfonylamino)-cyclopentancarbonsäure; 1-(6,7,8,9-Tetrahydro-5H-10-oxa-benzo[a]azulen-2-sulfonylamino)-cyclopentancarbonsäurehydroxyamid; 2-(5,6,7,8,9,10-Hexahydro-11-oxa-cycloocta[a]inden-2-sulfonylamino)-2-methylpropionsäure; 2-(5,6,7,8,9,10-Hexahydro-11-oxa-cycloocta[a]inden-2-sulfonylamino)-N-hydroxy-2-methyl-propionamid; 1-(5,6,7,8,9,10-Hexahydro-11-oxa-cycloocta[a]inden-2-sulfonylamino)-cylcopentancarbonsäure und 1-(5,6,7,8,9,10-Hexahydro-11-oxa-cycloocta[a]inden-2-sulfonylamino)-cyclopentacarbonsäurehydroxyamid.
- Verbindung nach Anspruch 1, worin die Verbindung aus der folgenden Gruppe ausgewählt ist: 2-(5a,6,7,8,9,9a-Hexahydro-dibenzofuran-3-sulfonylamino)-2-methyl-propionsäure; 2-(5a,6,7,8,9,9a-Hexahydro-dibenzofuran-3-sulfonylamino)-N-hydroxy-2-methyl-propionamid; 1-(5a,6,7,8,9,9a-Hexahydro-dibenzofuran-3-sulfonylamino)-cyclopentancarbonsäure und 1-(5a,6,7,8,9,9a-Hexahydro-dibenzofuran-3-sulfonylamino)-cyclopentancarbonsäurehydroxyamid.
- Verbindung nach Anspruch 1, worin die Verbindung aus der folgenden Gruppe ausgewählt ist: 1-(Dibenzofuran-3-sulfonylamino)cyclohexancarbonsäure; 1-(Dibenzofuran-3-sulfonylamino)cyclohexancarbonsäurehydroxyamid; 2-(Dibenzofuran-3-sulfonylamino)-2-methylpropionsäure; 2-(Dibenzofuran-3-sulfonylamino)-N-hydroxy-2-methylpropionamid; 4-(Dibenzofuran-3-sulfonylamino)-tetrahydro-pyran-4-carbonsäurehydroxyamid; 1-(7-Brom-dibenzofuran-2-sulfonylamino)cyclopentancarbonsäurehydroxyamid und 2-(7-Chlor-dibenzofuran-2-sulfonylamino)-N-hydroxy-2-methyl-propionamid.
- Verbindung der Formel Ioder ein pharmazeutisch akzeptables Salz hiervon, worin X ausgewählt ist aus OH und NHOH, d für 1 oder 2 steht; R1 ausgewählt ist aus der Gruppe:
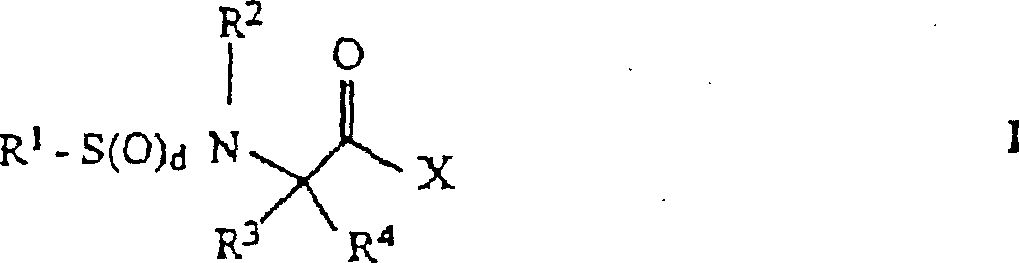 worin Y ausgewählt ist aus O, S, S(O)d (worin d für 1 oder 2 steht), CH2, C(=O) und NRq (worin Rq für H, C1-C6-Alkyl oder C1-C6-Alkylphenyl steht); R5 ausgewählt ist aus der Gruppe aus H, C1-C10-Alkyl, CF3, CONH2, Halogen, CN, COOH, C1-C4-Alkoxy, CHO, NO2, OH, (CH2 pOH, (CH2)pNH2, Ar und NH2, p für 0 bis 3 steht und Ar ausgewählt ist aus der Gruppe aus (a) Phenyl, (b) Phenyl, das mit C1-C4-Alkyl, C1-C4-Alkoxy, Halogen, NH2, NO2, CN, COOH, CONH2, CF3 oder COOR6 (worin R6 für C1-C10-Alkyl steht) substituiert ist, und (c) Heteroaryl; R2 ausgewählt ist aus der Gruppe aus (a) Wasserstoff, (b) C1-C4-Alkyl, (c) Benzyl und (d) Benzyl, das mit einem oder mehreren Substituenten aus C1-C4-Alkyl, C1-C4-Alkoxy, F, Cl, Br, I, NH2, NO2, CN, Carboxy und CO2R7 (worin R7 für H oder C1-C4-Alkyl steht) substituiert ist; und R3 und R4 jeweils unabhängig voneinander aus der Gruppe aus C1-C20-Alkyl, C3-C10-Cycloalkyl, Phenyl, Phenyl, das mit C1-C4-Alkyl, C1-C4-Alkoxy, Halogen, NH2, NO2, CN, COOH, CO2R7 oder CF3 substituiert ist, C3-C10-Heterocyclus und Heteroaryl ausgewählt sind.
worin Y ausgewählt ist aus O, S, S(O)d (worin d für 1 oder 2 steht), CH2, C(=O) und NRq (worin Rq für H, C1-C6-Alkyl oder C1-C6-Alkylphenyl steht); R5 ausgewählt ist aus der Gruppe aus H, C1-C10-Alkyl, CF3, CONH2, Halogen, CN, COOH, C1-C4-Alkoxy, CHO, NO2, OH, (CH2 pOH, (CH2)pNH2, Ar und NH2, p für 0 bis 3 steht und Ar ausgewählt ist aus der Gruppe aus (a) Phenyl, (b) Phenyl, das mit C1-C4-Alkyl, C1-C4-Alkoxy, Halogen, NH2, NO2, CN, COOH, CONH2, CF3 oder COOR6 (worin R6 für C1-C10-Alkyl steht) substituiert ist, und (c) Heteroaryl; R2 ausgewählt ist aus der Gruppe aus (a) Wasserstoff, (b) C1-C4-Alkyl, (c) Benzyl und (d) Benzyl, das mit einem oder mehreren Substituenten aus C1-C4-Alkyl, C1-C4-Alkoxy, F, Cl, Br, I, NH2, NO2, CN, Carboxy und CO2R7 (worin R7 für H oder C1-C4-Alkyl steht) substituiert ist; und R3 und R4 jeweils unabhängig voneinander aus der Gruppe aus C1-C20-Alkyl, C3-C10-Cycloalkyl, Phenyl, Phenyl, das mit C1-C4-Alkyl, C1-C4-Alkoxy, Halogen, NH2, NO2, CN, COOH, CO2R7 oder CF3 substituiert ist, C3-C10-Heterocyclus und Heteroaryl ausgewählt sind.
- Verbindung der Formel Ioder ein pharmazeutisch akzeptables Salz hiervon, worin X aus OH und NHOH ausgewählt ist, d für 1 oder 2 steht; a für 1 bis 10 steht; b für 1 bis 10 steht, h 0 oder 1 ist; R1 ausgewählt ist aus der Gruppe
 worin Y ausgewählt ist aus O, S, S(O)d (worin d für 1 oder 2 steht), CH2, C(=O) und NRq (worin Rq für H, C1-C6-Alkyl oder C1-C6-Alkylphenyl steht); R5 ausgewählt ist aus der Gruppe aus H, C1-C10-Alkyl, CF3, CONH2, Halogen, CN, COOH, C1-C4-Alkoxy, CHO, NO2, OH, (CH2)pOH, (CH2)pNH2, Ar und NH2, p für 0 bis 3 steht und Ar ausgewählt ist aus der Gruppe aus (a) Phenyl, (b) Phenyl, das mit C1-C4-Alkyl, C1-C4-Alkoxy, Halogen, NH2, NO2, CN, COOH, CONH2, CF3 oder COOR6 (worin R6 für C1-C10-Alkyl steht) substituiert ist, und (c) Heteroaryl; R2 ausgewählt ist aus der Gruppe aus (a) Wasserstoff, (b) C1-C4-Alkyl, (c) Benzyl und (d) Benzyl, das mit einem oder mehreren Substituenten aus C1-C4-Alkyl, C1-C4-Alkoxy, F, Cl, Br, I, NH2, NO2, CN, Carboxy und CO2R7 (worin R7 für H oder C1-C4-Alkyl steht) substituiert ist; und Z aus der Gruppe O, S und NR8 (worin R8 ausgewählt ist aus H und C1-C3-Alkyl) ausgewählt ist.
worin Y ausgewählt ist aus O, S, S(O)d (worin d für 1 oder 2 steht), CH2, C(=O) und NRq (worin Rq für H, C1-C6-Alkyl oder C1-C6-Alkylphenyl steht); R5 ausgewählt ist aus der Gruppe aus H, C1-C10-Alkyl, CF3, CONH2, Halogen, CN, COOH, C1-C4-Alkoxy, CHO, NO2, OH, (CH2)pOH, (CH2)pNH2, Ar und NH2, p für 0 bis 3 steht und Ar ausgewählt ist aus der Gruppe aus (a) Phenyl, (b) Phenyl, das mit C1-C4-Alkyl, C1-C4-Alkoxy, Halogen, NH2, NO2, CN, COOH, CONH2, CF3 oder COOR6 (worin R6 für C1-C10-Alkyl steht) substituiert ist, und (c) Heteroaryl; R2 ausgewählt ist aus der Gruppe aus (a) Wasserstoff, (b) C1-C4-Alkyl, (c) Benzyl und (d) Benzyl, das mit einem oder mehreren Substituenten aus C1-C4-Alkyl, C1-C4-Alkoxy, F, Cl, Br, I, NH2, NO2, CN, Carboxy und CO2R7 (worin R7 für H oder C1-C4-Alkyl steht) substituiert ist; und Z aus der Gruppe O, S und NR8 (worin R8 ausgewählt ist aus H und C1-C3-Alkyl) ausgewählt ist.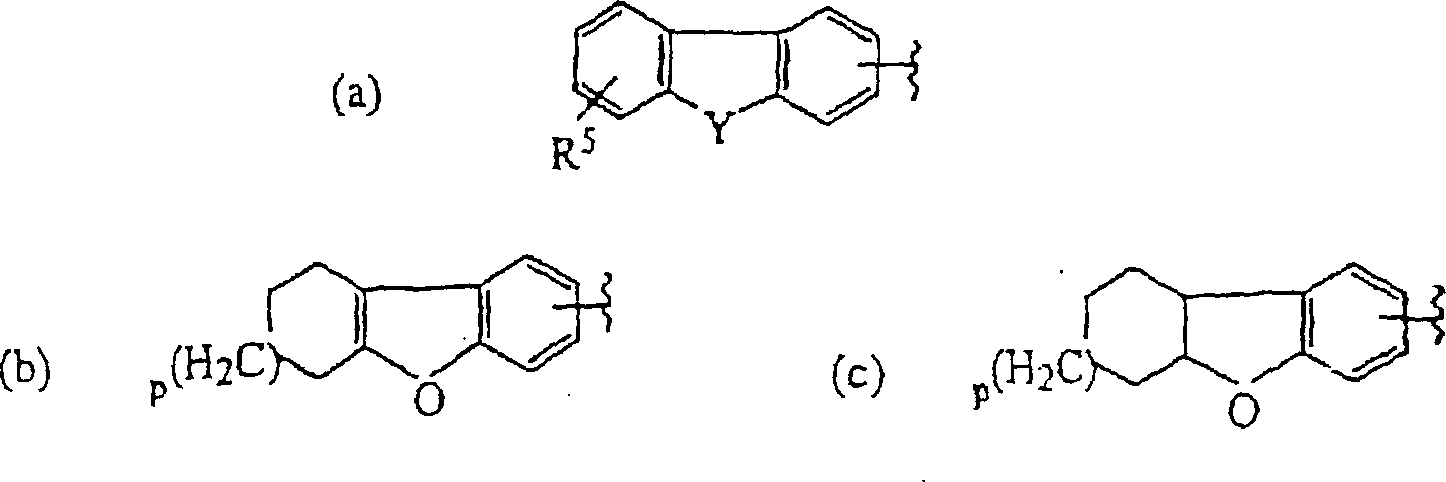
- Pharmazeutische Formulierung, die eine Verbindung nach einem der Ansprüche 1 bis 19 im Gemisch mit einem Verdünnungsmittel, Träger oder Streckmittel hierfür umfasst.
- Verwendung einer Verbindung gemäß einem der Ansprüche 1 bis 19 zur Herstellung von Pharmazeutika zur Hemmung einer Matrixmetalloproteinase bei einem eine Matrixmetalloproteinasehemmung erfordernden Patienten.
- Verwendung einer Verbindung nach einem der Ansprüche 1 bis 19 zur Herstellung von Pharmazeutika zur Behandlung von Arthritis, Multipler Sklerose, Atherosklerose, Restanose, Entzündungen, Schmerzen, Aortaaneurysma, Herzversagen, Osteoporose, Periodontiumerkrankungen, Hornhautgeschwürbildung, Verbrennungen, Dekubitus, Krebs, Nierenerkrankungen oder Linksherzdilatation.
- Verwendung einer Verbindung nach einem der Ansprüche 1 bis 19 zur Herstellung von Pharmazeutika zur Heilung von Wunden.
- Verwendung einer Verbindung nach einem der Ansprüche 1 bis 19 zur Herstellung von Pharmazeutika zur Behandlung von Autoimmunerkrankungen oder Entzündungserkrankungen, die von einer Gewebeinvasion durch Leukozyten abhängen.
- Verwendung einer Verbindung nach einem der Ansprüche 1 bis 19 zur Herstellung von Pharmazeutika zur Behandlung von akuten und chronischen neurodegenerativen Störungen, die aus der Gruppe von Schlaganfall, Kopftrauma, Rückenmarkverletzungen, Alzheimer-Erkrankung, Amyotropher Lateralsklerose, Gehirnamyloidangiopathie, AIDS, Parkinson-Erkrankung, Chorea Huntington, Prionerkrankungen, Myasthenia gravis und Duchenne-Krankheit ausgewählt sind.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14966099P | 1999-08-18 | 1999-08-18 | |
| US149660P | 1999-08-18 | ||
| PCT/US2000/021884 WO2001012592A2 (en) | 1999-08-18 | 2000-08-10 | Hydroxamic acid compounds useful as matrix metalloproteinase inhibitors |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE60008548D1 DE60008548D1 (de) | 2004-04-01 |
| DE60008548T2 true DE60008548T2 (de) | 2004-08-05 |
Family
ID=22531286
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE60008548T Expired - Fee Related DE60008548T2 (de) | 1999-08-18 | 2000-08-10 | Hydroxamsäurederivate als matrix-metalloproteinase-inhibitoren |
Country Status (18)
| Country | Link |
|---|---|
| US (1) | US6677355B1 (de) |
| EP (1) | EP1210326B1 (de) |
| JP (1) | JP2003507362A (de) |
| AR (1) | AR030160A1 (de) |
| AT (1) | ATE260251T1 (de) |
| AU (1) | AU6764400A (de) |
| BR (1) | BR0013390A (de) |
| CA (1) | CA2378332A1 (de) |
| CO (1) | CO5300407A1 (de) |
| DE (1) | DE60008548T2 (de) |
| DK (1) | DK1210326T3 (de) |
| ES (1) | ES2216938T3 (de) |
| MX (1) | MXPA01013324A (de) |
| PE (1) | PE20010486A1 (de) |
| PT (1) | PT1210326E (de) |
| TR (4) | TR200202211T2 (de) |
| UY (1) | UY26302A1 (de) |
| WO (1) | WO2001012592A2 (de) |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1613269B1 (de) | 2003-04-04 | 2015-02-25 | Incyte Corporation | Zusammensetzungen, verfahren und kits in zusammenhang mit her-2 spaltung |
| GB0326546D0 (en) * | 2003-11-14 | 2003-12-17 | Amersham Plc | Inhibitor imaging agents |
| US7638513B2 (en) * | 2004-06-02 | 2009-12-29 | Schering Corporation | Compounds for the treatment of inflammatory disorders |
| PE20060426A1 (es) * | 2004-06-02 | 2006-06-28 | Schering Corp | DERIVADOS DE ACIDO TARTARICO COMO INHIBIDORES DE MMPs, ADAMs, TACE Y TNF-alfa |
| US7488745B2 (en) * | 2004-07-16 | 2009-02-10 | Schering Corporation | Compounds for the treatment of inflammatory disorders |
| US7504424B2 (en) * | 2004-07-16 | 2009-03-17 | Schering Corporation | Compounds for the treatment of inflammatory disorders |
| KR20070032787A (ko) | 2004-07-16 | 2007-03-22 | 쉐링 코포레이션 | 염증 질환 치료용 히단토인 유도체 |
| PE20071240A1 (es) * | 2006-01-17 | 2008-01-14 | Schering Corp | Compuestos derivados de hidantoina para el tratamiento de trastornos inflamatorios |
| SI2343286T1 (sl) | 2006-10-28 | 2015-05-29 | Methylgene Inc. | Derivati dibenzo(b,f)(1,4) oksazepina kot inhibitorji histonske deacetilaze |
| AR066412A1 (es) * | 2007-05-04 | 2009-08-19 | Wyeth Corp | Derivados de dibenzofurano y dibenzotiofeno, composiciones farmaceuticas que los contienen y usos en patologias tales como trastornos oseos, crecimiento de tumores, diabetes y obesidad. |
| US8859529B2 (en) | 2008-09-24 | 2014-10-14 | Merck Sharp & Dohme Corp. | Compounds for the treatment of inflammatory disorders |
| AR073741A1 (es) | 2008-09-24 | 2010-12-01 | Schering Corp | Derivados heterociclicos de hidantoina, composiciones farmaceuticas que los contienen y uso de los mismos en el tratamiento de enfermedades inflamatorias, tales como psoriasis o artritis |
| US8569336B2 (en) | 2008-11-10 | 2013-10-29 | Ling Tong | Compounds for the treatment of inflammatory disorders |
| EP2356111A1 (de) | 2008-11-10 | 2011-08-17 | Schering Corporation | Verbindungen zur behandlung entzündlicher erkrankungen |
| BRPI0922938A2 (pt) * | 2008-12-05 | 2017-06-06 | Intermed Discovery Gmbh | inibidores de acúmulo de proteína hif-1 |
| US20210393632A1 (en) | 2018-10-04 | 2021-12-23 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Egfr inhibitors for treating keratodermas |
| CN113336670B (zh) * | 2021-05-28 | 2023-06-02 | 河南大学 | 一种轴手性芴胺-苯酚类衍生物及其制备方法 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ZA98376B (en) * | 1997-01-23 | 1998-07-23 | Hoffmann La Roche | Sulfamide-metalloprotease inhibitors |
| PL334846A1 (en) * | 1997-01-23 | 2000-03-27 | Hoffmann La Roche | Sulphamidic inhibitors of metaloproteases |
| US6376506B1 (en) | 1997-01-23 | 2002-04-23 | Syntex (U.S.A.) Llc | Sulfamide-metalloprotease inhibitors |
| ATE263147T1 (de) | 1997-08-08 | 2004-04-15 | Pfizer Prod Inc | Derivate von aryloxyarylsulfonylamino hydroxyaminsäuren |
-
2000
- 2000-08-10 DK DK00955435T patent/DK1210326T3/da active
- 2000-08-10 US US10/049,544 patent/US6677355B1/en not_active Expired - Fee Related
- 2000-08-10 AT AT00955435T patent/ATE260251T1/de not_active IP Right Cessation
- 2000-08-10 DE DE60008548T patent/DE60008548T2/de not_active Expired - Fee Related
- 2000-08-10 TR TR2002/02211T patent/TR200202211T2/xx unknown
- 2000-08-10 CA CA002378332A patent/CA2378332A1/en not_active Abandoned
- 2000-08-10 BR BR0013390-6A patent/BR0013390A/pt not_active IP Right Cessation
- 2000-08-10 AU AU67644/00A patent/AU6764400A/en not_active Abandoned
- 2000-08-10 TR TR2002/02163T patent/TR200202163T2/xx unknown
- 2000-08-10 TR TR2002/02164T patent/TR200202164T2/xx unknown
- 2000-08-10 ES ES00955435T patent/ES2216938T3/es not_active Expired - Lifetime
- 2000-08-10 EP EP00955435A patent/EP1210326B1/de not_active Expired - Lifetime
- 2000-08-10 JP JP2001516893A patent/JP2003507362A/ja active Pending
- 2000-08-10 WO PCT/US2000/021884 patent/WO2001012592A2/en active IP Right Grant
- 2000-08-10 PT PT00955435T patent/PT1210326E/pt unknown
- 2000-08-10 MX MXPA01013324A patent/MXPA01013324A/es active IP Right Grant
- 2000-08-10 TR TR2002/00410T patent/TR200200410T2/xx unknown
- 2000-08-17 UY UY26302A patent/UY26302A1/es not_active Application Discontinuation
- 2000-08-17 PE PE2000000833A patent/PE20010486A1/es not_active Application Discontinuation
- 2000-08-17 AR ARP000104249A patent/AR030160A1/es not_active Application Discontinuation
- 2000-08-17 CO CO00061938A patent/CO5300407A1/es not_active Application Discontinuation
Also Published As
| Publication number | Publication date |
|---|---|
| ES2216938T3 (es) | 2004-11-01 |
| TR200202211T2 (tr) | 2002-11-21 |
| UY26302A1 (es) | 2000-10-31 |
| AR030160A1 (es) | 2003-08-13 |
| DE60008548D1 (de) | 2004-04-01 |
| MXPA01013324A (es) | 2002-07-02 |
| WO2001012592A2 (en) | 2001-02-22 |
| CO5300407A1 (es) | 2003-07-31 |
| TR200202163T2 (tr) | 2002-11-21 |
| BR0013390A (pt) | 2002-04-30 |
| EP1210326B1 (de) | 2004-02-25 |
| US6677355B1 (en) | 2004-01-13 |
| PT1210326E (pt) | 2004-07-30 |
| TR200200410T2 (tr) | 2002-06-21 |
| ATE260251T1 (de) | 2004-03-15 |
| CA2378332A1 (en) | 2001-02-22 |
| PE20010486A1 (es) | 2001-04-20 |
| WO2001012592A3 (en) | 2001-07-05 |
| TR200202164T2 (tr) | 2002-11-21 |
| AU6764400A (en) | 2001-03-13 |
| JP2003507362A (ja) | 2003-02-25 |
| DK1210326T3 (da) | 2004-06-21 |
| EP1210326A2 (de) | 2002-06-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE60008548T2 (de) | Hydroxamsäurederivate als matrix-metalloproteinase-inhibitoren | |
| DE69925840T2 (de) | Tace inhibitoren | |
| DE69822839T2 (de) | Derivate von aryloxyarylsulfonylamino hydroxyaminsäuren | |
| DE69729007T2 (de) | Hydroxamsäure- und carbonsäure-derivate mit mmp und tnf hemmender wirkung | |
| DE69915634T2 (de) | Tricyclische sulfonamide und ihre derivate als inhibitoren von matrix-metalloproteinasen | |
| DE60003863T2 (de) | Dioxocyclopentylhydroxamsäure | |
| DE69730151T2 (de) | Cyclische sulfonderivate | |
| DE69917124T2 (de) | Cyclobutyl-Aryloxysulfonylamin-Hydroxamsäurederivate | |
| DE60013302T2 (de) | Cystein protease inhibitore | |
| DE69831233T2 (de) | Barbitursaure derivaten mit antimetastatischer und antitumorischer wirkung | |
| DE69827236T2 (de) | Matrix-metalloproteinase-hemmer | |
| DE3207033C2 (de) | Amidinverbindungen, Verfahren zu ihrer Herstellung und diese Verbindungen enthaltende pharmazeutische Mittel | |
| DE69832268T2 (de) | Peptidyl2-amino-1hydroxyalkansulfonsäuren als zysteinprotease-inhibitoren | |
| UA53786C2 (uk) | Похідні біциклічних гідроксамових кислот, фармацевтична композиція (варіанти), спосіб інгібування (варіанти) | |
| DE60012883T2 (de) | 2,3,4,5-tetrahydro-1h-[1,4]benzodiazepin-3-hydroxamsäure als matrix metalloproteinaseinhibitoren | |
| DE69934094T2 (de) | N-hydroxyformamid-derivate | |
| WO1995005358A1 (de) | Verwendung von phenolen und phenolderivaten als arzneimittel mit fibrinogensenkender wirkung | |
| EP0157267B1 (de) | Substituierte Benzopyrane, Verfahren zu ihrer Herstellung sowie ihre Verwendung in Arzneimitteln | |
| US5403952A (en) | Substituted cyclic derivatives as novel antidegenerative agents | |
| DE69925914T2 (de) | Succinamid hemmstoffe des interleukin-1 beta konvertierenden enzyms | |
| DE69819206T2 (de) | Matrixmetalloproteinaseinhibitoren | |
| DE69734764T2 (de) | Eine amidgruppe tragende schwefelhaltige verbindungen, verfahren zu ihrer herstellung, deren verwendung in medikamenten und sie enthaltende pharmazeutische zubereitungen | |
| DE60005818T2 (de) | 3-(arylsulfonylamino)-tetrahydrofuran-3-carbonsäure-hydroxamide | |
| EP1005449B1 (de) | 3-aryl-succinamido-hydroxamsäuren, prozesse zu ihrer herstellung und diese substanzen enthaltende medikamente | |
| DE69908756T2 (de) | Matrix-metalloproteinase-inhibitoren |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition | ||
| 8339 | Ceased/non-payment of the annual fee |