-
Die
vorliegende Erfindung betrifft Melanocortinrezeptoragonisten und
genauer gesagt Piperazinderivate als Melanocortinrezeptoragonisten,
die zur Behandlung oder Prävention
von Erkrankungen und Störungen
brauchbar sind, welche auf die Aktivierung von Melanocortinrezeptoren
ansprechen.
-
Von
Pro-Opiomelanocortin (POMC) abgeleiteten Peptiden ist bekannt, dass
sie die Nahrungsaufnahme beeinflussen. Mehrere unabhängige Hinweise
unterstützen
die Meinung, dass die G-Protein-gekuppelten Rezeptoren
(GPCRs) der Melanocortinrezeptorfamilie (MC-R), von denen mehrere
im Gehirn exprimiert werden, Ziele der von POMC abgeleiteten Peptide
sind, die bei der Kontrolle der Nahrungsaufnahme und deren Metabolismus
beteiligt sind.
-
Hinweise
für die
Beteiligung von MC-R bei der Obesität umfassen: i) Die Agoutimaus
(Avy), die ektopisch einen Antagonisten
von MC-1R, MC-3R und MC-4R exprimiert, ist fettleibig, was zeigt,
dass die Blockierung der Wirkung dieser drei MC-Rs zur Hyperphagie
und metabolischen Störungen
führen
kann, ii) die MC-4R Knockout Maus (Huzar et al., Cell, 88: 131–141, 1997)
wiederholt den Phänotyp
der Agouti-Maus
und diese Mäuse
sind fettleibig, iii) der cyclische Heptapeptid MC-1R, MC-3R, MC-4R
und MC-5R Melanotonin-II-Agonist (MT-II), der intracerebroventrikulär (ICV)
in Nager injiziert wird, verringert die Nahrungsaufnahme in mehreren Tierfütterungsmodellen
(NPY, ob/ob, Agouti, gefastet), während ICV injizierter SHU-9119
(MC-3R, MC-4R Antagonist, MC-1R und MC-5R Agonist) diese Wirkung
umkehrt und eine Hyperphagie auslösen kann, und iv) von der chronischen
intraperitonealen Behandlung von fettleibigen Zucker-Ratten mit
einem α-NDP-MSH
Derivat (HP228) wurde berichtet, dass sie MC-1R, MC-3R, MC-4R und
MC-5R aktiviert und die Nahrungsaufnahme und die Körpergewichtszunahme über eine
Periode von 12 Wochen hemmt.
-
Es
wurden bisher 5 MC-Rs identifiziert und diese werden in bestimmten
Geweben exprimiert. MC-1R wurde ursprünglich durch die dominante
Zunahme von Funktionsmutationen am Extensionslokus charakterisiert,
die die Fellfarbe durch die Kontrolle der Umwandlung von Phäomelanin
zu Eumelanin durch die Kontrolle der Tyrosinase beeinflussen. MC-1R
wird vorwiegend in Melanocyten exprimiert. MC-2R wird in der Nebennierendrüse exprimiert
und repräsentiert
den ACTH Rezeptor. MC-3R wird im Gehirn, Darm und der Plazenta exprimiert
und kann bei der Kontrolle der Nahrungsaufnahme und Thermogenese
beteiligt sein. MC-4R wird einzigartig im Gehirn exprimiert und
von dessen Inaktivierung wurde gezeigt, dass sie Obesität verursacht.
(A. Kask et al., "Selective
Antagonist for the melanocortin-4-receptor (HS014) increases food intake
in free-feeding rats, Biochem. Biophys. Res. Commun., 245: 90–93, 1998),
MC-5R wird in vielen Geweben exprimiert, einschließlich weißem Fett,
Plazenta und exokrinen Drüsen.
Es wird auch eine geringe Expression im Gehirn beobachtet. Die MC-5R
Knockout Maus zeigt eine verringerte Lipidbildung der Sebumdrüsen (Chen
et al., Cell, 91: 789–798,
1997).
-
MC-4R
scheint auch eine Rolle bei anderen physiologischen Funktionen zu
spielen, nämlich
bei der Kontrolle des Beischlafverhaltens, der Erektion und des
Blutdrucks. Eine Erektionsstörung
bezeichnet den medizinischen Zustand der Unfähigkeit, eine Peniserektion
zu erreichen, die für
einen erfolgreichen Sexualverkehr ausreichend ist. Der Ausdruck "Impotenz" wird oft zur Beschreibung
dieses häufigen
Zustands verwendet. Es wurden synthetische Melanocortinrezeptoragonisten
gefunden, um Erektion bei Männern
mit psychogener Erektionsstörung
zu initiieren (H. Wessells et al., "Synthetic Melanotropic Peptide Initiates
Erections in Men With Psychogenic Erectile Dysfunction: Double-Blind,
Placebo Controlled Crossover Study", J. Urol., 160: 389–393, 1998). Die Aktivierung
der Melanocortinrezeptoren des Gehirns scheint eine normale Stimulierung der
sexuellen Erregung zu verursachen. Eine Evidenz für die Beteiligung
des MC-R bei einer Sexualstörung bei
Männern
und/oder Frauen ist in WO 00/74679 A beschrieben.
-
Diabetes
ist eine Erkrankung, bei der die Fähigkeit eines Säugers gestört ist,
die Blutglucosespiegel im Blut zu regulieren, da der Säuger eine
verringerte Fähigkeit
zur Umwandlung von Glucose in Glycogen zur Lagerung in Muskel- und
Leberzellen aufweist. Bei Typ I Diabetes wird diese verringerte
Fähigkeit
zur Lagerung von Glucose durch eine verringerte Insulinbildung verursacht. "Typ II Diabetes" oder "nicht-Insulin-abhängiger Diabetes
mellitus" (NIDDM)
ist die Form von Diabetes, die auf einer deutlichen Resistenz gegenüber dem stimulierenden
oder regulatorischen Effekt von Insulin auf Glucose und dem Lipidmetabolismus
in den hauptsächlichen
Insulin-sensitiven Geweben beruht, wie Muskel, Leber und Fettgewebe.
Die Resistenz gegenüber Insulinreaktionsfähigkeit
führt zu
einer nicht ausreichenden Insulinaktivierung der Aufnahme, Oxidation
und Lagerung von Glucose im Muskel und einer unangemessenen Insulinrepression
der Lipolyse in Fettgewebe und der Glucosebildung und Sekretion
in der Leber. Wenn diese Zellen gegenüber Insulin unempfindlich werden,
versucht der Körper,
dies durch die Bildung von anormal hohen Mengen an Insulin zu kompensieren
und es entsteht eine Hyperinsulinämie. Eine Hyperinsulinämie ist
mit Bluthochdruck und einem erhöhten
Körpergewicht
assoziiert. Da Insulin bei der Förderung
der zellulären
Aufnahme von Glucose, Aminosäuren
und Triglyceriden aus dem Blut durch Insulin-empfindliche Zellen
beteiligt ist, kann eine Insulinunempfindlichkeit zu erhöhten Mengen
an Triglyceriden und LDL führen,
die Risikofaktoren bei cardiovaskulären Erkrankungen sind. Die
Konstellation der Symptome, die eine Hyperinsulinämie in Kombination
mit Bluthochdruck, erhöhtem Körpergewicht,
erhöhten
Triglyceriden und erhöhtem
LDL umfasst, ist als Syndrom X bekannt.
-
Spiropiperidin-
und Piperidinderivate werden in
US 6 294 534 B1 , WO 01/70337 A, WO 00/74679
A und WO 01/70708 A als Agonisten der Melanocortinrezeptoren beschrieben,
die zur Behandlung von Erkrankungen und Störungen verwendet werden können, wie
Obesität,
Diabetes und Sexualstörung.
-
In
Anbetracht der ungelösten
Defizienzen bei der Behandlung von verschiedenen Erkrankungen und Störungen,
wie sie oben beschrieben sind, ist es ein Ziel der vorliegenden
Erfindung, neue Piperazinderivate bereitzustellen, die als Melanocortinrezeptoragonisten
brauchbar sind, um Obesität,
Diabetes und Sexualstörung
beim Mann und der Frau zu behandeln.
-
Kurze Zusammenfassung
der Erfindung
-
Die
vorliegende Erfindung betrifft eine Verbindung neuer Piperazinderivate
als Melanocortinrezeptoragonisten, wie sie in Formel I gezeigt ist
oder ein pharmazeutisch annehmbares
Salz oder Stereoisomer hiervon, worin
L und L
1 unabhängig für Wasserstoff
oder zusammen für
Oxo stehen,
T steht für
R unabhängig steht
für
Wasserstoff,
Hydroxy, Cyano, Nitro, Halogen, C
1-C
8 Alkyl, C
1-C
8 Alkoxy, C
1-C
4 Halogenalkyl, (D)C(O)R
9, (D)C(O)OR
9, (D)C(O)SR
9, (D)C(O)-Heteroaryl,
(D)C(O)-Heterocyclyl, (D)C(O)N(R
9)
2, (D)N(R
9)
2, (D)NR
9COR
9, (D)NR
9CON(R
9)
2, (D)NR
9C(O)OR
9, (D)NR
9C(R
9)=N(R
9), (D)NR
9C(=NR
9)-N(R
9)
2, (D)NR
9SO
2R
9, (D)NR
9SO
2N(R
9)
2, (D)NR
9(CH
2)
n-Heterocyclyl,
(D)NR
9(CH
2)
n-Heteroaryl, (D)OR
9,
OSO
2R
9, (D)[O]
q(C
3-C
7 Cycloalkyl),
(D)[O]
q(CH
2)
n-Aryl, (D)[O]
q(CH
2)
n-Heteroaryl, (D)[O]
q(CH
2)
n-Heterocyclyl, worin
Heterocyclyl ein Heterocyclyl ausschließt, das einen einzelnen Stickstoff
enthält,
wenn q für
1 steht, für
(D)SR
9, (D)SOR
9,
(D)SO
2R
9 oder (D)SO
2N(R
9)
2,
worin
C
1-C
8 Alkyl, C
1-C
8 Alkoxy, C
3-C
7 Cycloalkyl,
Aryl, Heterocyclyl und Heteroaryl wahlweise mit einem bis fünf Substituenten
substituiert sind, die unabhängig
aus R
8 ausgewählt sind,
R
1 unabhängig steht
für
Wasserstoff,
CONH(C
1-C
8 Alkyl),
C
1-C
8 Alkyl, (D)-Phenyl,
(D)-C
3-C
7 Cycloalkyl
oder Oxo, mit der Maßgabe, dass
Oxo nicht an denselben Kohlenstoff gebunden ist, der an den Stickstoff
gebunden ist, der eine Amidbindung bildet,
R
3 unabhängig für Aryl oder
Thienyl steht,
worin Aryl und Thienyl wahlweise mit einem bis
drei Substituenten substituiert sind, die aus der Gruppe ausgewählt sind,
welche besteht aus
Cyano, Halogen, C
1-C
8 Alkyl, (D)-C
3-C
7 Cycloalkyl, C
1-C
4 Alkoxy, C
1-C
4 Halogenalkyl und C
1-C
4 Halogenalkyloxy,
R
4 unabhängig steht
für
Wasserstoff,
C
1-C
8 Alkyl, C(O)R
9, C(O)OR
9, C
3-C
7 Cycloalkyl oder
(CH
2)
nO(C
1-C
8 Alkyl), worin
n für 2
bis 8 steht,
R
8 jeweils unabhängig steht
für
Wasserstoff,
Halogen, Oxo, N(R
10)
2,
C
1-C
8 Alkyl, (D)-C
3-C
7 Cycloalkyl,
C
1-C
4 Halogenalkyl,
C
1-C
4 Alkoxy, Heteroaryl,
Hydroxy, Heterocyclyl, worin Heterocyclyl ein Heterocyclyl ausschließt, das
einen einzelnen Stickstoff enthält,
für Phenyl,
(D)-COR
9, (D)C(O)OR
9,
(D)OR
9, (D)OCOR
9,
(D)OCO
2R
9, (D)SR
9, (D)SOR
9 oder (D)SO
2R
9, worin Aryl,
Heteroaryl, Heterocyclyl, Alkyl oder Cycloalkyl wahlweise mit ein
bis drei Substituenten substituiert sind, die aus der Gruppe ausgewählt sind,
die besteht aus Oxo, C
1-C
8 Alkyl,
N(R
10)
2, OR
10, SR
10 und CO
2R
10,
R
9 jeweils unabhängig ausgewählt ist aus
Wasserstoff,
C
1-C
8 Alkyl, C
1-C
4 Halogenalkyl,
(D)-C
3-C
7 Cycloalkyl,
(D)-Aryl, worin Aryl für
Phenyl oder Naphthyl steht, (D)-Heteroaryl oder (D)-Heterocyclyl,
worin Heterocyclyl ein Heterocyclyl ausschließt, das einen einzelnen Stickstoff
enthält
und worin
Aryl, Heteroaryl, Heterocyclyl, Alkyl oder Cycloalkyl
wahlweise mit einem bis drei Substituenten substituiert sind, die
aus der Gruppe ausgewählt
sind, die besteht aus Oxo, C
1-C
8 Alkyl,
N(R
10)
2, OR
10, SR
10 und CO
2R
10,
R
10 jeweils unabhängig steht für
Wasserstoff,
C
1-C
8 Alkyl, C(O)-C
1-C
8 Alkyl, Aryl
oder C
3-C
7 Cycloalkyl,
R
11 jeweils unabhängig steht für
Wasserstoff,
C
1-C
8 Alkyl, (D)-Aryl,
(D)-Heteroaryl, (CH
2)
nN(R
8)
2, (CH
2)
nNR
8C(O)C
1-C
4 Alkyl, (CH
2)
nNR
8SO
2-C
1-C
4 Alkyl,
(CH
2)
nSO
2N(R
8)
2,
(CH
2)
n[O]
q-C
1-C
8 Alkyl,
(CH
2)
n[O]
q(CH
2)
nNR
8COR
8, (CH
2n[O]
q(CH
2)
nN-R
8SO
2R
8, (CH
2)
n[O]
q-Heterocyclyl
oder (CH
2)
n[O]
q(C
1-C
8 Alkyl)heterocyclyl
und worin n für
2 bis 8 steht,
R
12 jeweils unabhängig steht
für
Wasserstoff,
C
1-C
8 Alkyl, (D)-Phenyl,
C(O)-C
1-C
8 Alkyl,
C(O)-Phenyl, SO
2C
1-C
8 Alkyl oder SO
2-Phenyl,
D
für eine
Bindung oder -(CH
2)
n-
steht,
n für
0 bis 8 steht,
p für
0 bis 5 steht,
q für
0 bis 1 steht und
r für
1 bis 2 steht.
-
Die
erfindungsgemäßen Verbindungen
sind zur Prävention
oder Behandlung von Obesität
oder Diabetes mellitus bei einem Säuger brauchbar, die die Verabreichung
einer therapeutisch wirksamen Menge der Verbindung der Formel I
umfasst.
-
Die
erfindungsgemäßen Verbindungen
sind auch zur Prävention
oder Behandlung einer männlichen oder
weiblichen Sexualstörung
bei einem Säuger,
genauer gesagt einer Erektionsstörung
brauchbar, die die Verabreichung einer therapeutisch wirksamen Menge
der Verbindung der Formel I umfasst.
-
Ebenfalls
im Umfang der vorliegenden Erfindung befindet sich eine pharmazeutische
Zusammensetzung oder Formulierung, die einen pharmazeutischen Träger und
zumindest eine Verbindung der Formel I oder pharmazeutisch annehmbare
Salze oder Stereoisomere hiervon umfasst.
-
Die
vorliegende Erfindung umfasst ferner ein Verfahren zur Herstellung
einer pharmazeutischen Zusammensetzung oder Formulierung, die eine
Verbindung der Formel I oder die pharmazeutisch annehmbaren Salze
oder Stereoisomere hiervon und einen pharmazeutisch annehmbaren
Träger
enthält.
-
Die
vorliegende Erfindung umfasst ferner ein Verfahren zur Herstellung
einer Verbindung der Formel I.
-
Detaillierte Beschreibung
der Erfindung
-
Die
vorliegende Erfindung betrifft Melanocortinrezeptoragonisten und
insbesondere Piperazinderivate, die Melanocortinrezeptoragonisten
sind. Die erfindungsgemäßen Verbindungen
sind zur Behandlung oder Prävention
von Erkrankungen und Störungen
brauchbar, die auf die Aktivierung von Melano cortinrezeptoren ansprechen,
wie Fettsucht, Diabetes und Sexualstörung, einschließlich Erektionsstörung und
weiblicher Sexualstörung.
-
Eine
Ausführungsform
der vorliegenden Erfindung ist eine Verbindung der Formel I
oder ein pharmazeutisch annehmbares
Salz oder Stereoisomer hiervon, worin
L und L
1 unabhängig für Wasserstoff
oder zusammen für
Oxo stehen,
T steht für
R unabhängig steht
für
Wasserstoff,
Hydroxy, Cyano, Nitro, Halogen, C
1-C
8 Alkyl, C
1-C
8 Alkoxy, C
1-C
4 Halogenalkyl, (D)C(O)R
9, (D)C(O)OR
9, (D)C(O)SR
9, (D)C(O)-Heteroaryl,
(D)C(O)-Heterocyclyl, (D)C(O)N(R
9)
2, (D)N(R
9)
2, (D)NR
9COR
9, (D)NR
9CON(R
9)
2, (D)NR
9C(O)OR
9, (D)NR
9C(R
9)=N(R
9), (D)NR
9C(=NR
9)-N(R
9)
2, (D)NR
9SO
2R
9, (D)NR
9SO
2N(R
9)
2, (D)NR
9(CH
2)
n-Heterocyclyl,
(D)NR
9(CH
2)
n-Heteroaryl, (D)OR
9,
OSO
2R
9, (D)[O]
q(C
3-C
7 Cycloalkyl),
(D)[O]
q(CH
2)
n-Aryl, (D)[O]
q(CH
2)
n-Heteroaryl, (D)[O]
q(CH
2)
n-Heterocyclyl, worin
Heterocyclyl ein Heterocyclyl ausschließt, das einen einzelnen Stickstoff
enthält,
wenn q für
1 steht, für
(D)SR
9, (D)SOR
9,
(D)SO
2R
9 oder (D)SO
2N(R
9)
2,
worin
C
1-C
8 Alkyl, C
1-C
8 Alkoxy, C
3-C
7 Cycloalkyl,
Aryl, Heterocyclyl und Heteroaryl wahlweise mit einem bis fünf Substituenten
substituiert sind, die unabhängig
aus R
8 ausgewählt sind,
R
1 unabhängig steht
für
Wasserstoff,
CONH(C
1-C
8 Alkyl),
C
1-C
8 Alkyl, (D)-Phenyl,
(D)-C
3-C
7 Cycloalkyl
oder Oxo, mit der Maßgabe, dass
Oxo nicht an denselben Kohlenstoff gebunden ist, der an den Stickstoff
gebunden ist, der eine Amidbindung bildet,
R
3 unabhängig für Aryl oder
Thienyl steht,
worin Aryl und Thienyl wahlweise mit einem bis
drei Substituenten substituiert sind, die aus der Gruppe ausgewählt sind,
welche besteht aus Cyano, Halogen, C
1-C
8 Alkyl, (D)-C
3-C
7 Cycloalkyl, C
1-C
4 Alkoxy, C
1-C
4 Halogenalkyl und C
1-C
4 Halogenalkyloxy,
R
4 unabhängig steht
für
Wasserstoff,
C
1-C
8 Alkyl, C(O)R
9, C(O)OR
9, C
3-C
7 Cycloalkyl oder
(CH
2)
nO(C
1-C
8 Alkyl), worin
n für 2
bis 8 steht,
R
8 jeweils unabhängig steht
für
Wasserstoff,
Halogen, Oxo, N(R
10)
2,
C
1-C
8 Alkyl, (D)-C
3-C
7 Cycloalkyl,
C
1-C
4 Halogenalkyl,
C
1-C
4 Alkoxy, Heteroaryl,
Hydroxy, Heterocyclyl, worin Heterocyclyl ein Heterocyclyl ausschließt, das
einen einzelnen Stickstoff enthält,
für Phenyl,
(D)-COR
9, (D)C(O)OR
9,
(D)OR
9, (D)OCOR
9,
(D)OCO
2R
9, (D)SR
9, (D)SOR
9 oder (D)SO
2R
9, worin Aryl,
Heteroaryl, Heterocyclyl, Alkyl oder Cycloalkyl wahlweise mit ein
bis drei Substituenten substituiert sind, die aus der Gruppe ausgewählt sind,
die besteht aus Oxo, C
1-C
8 Alkyl,
N(R
10)
2, OR
10, SR
10 und CO
2R
10,
R
9 jeweils unabhängig ausgewählt ist aus
Wasserstoff,
C
1-C
8 Alkyl, C
1-C
4 Halogenalkyl,
(D)-C
3-C
7 Cycloalkyl,
(D)-Aryl, worin Aryl für
Phenyl oder Naphthyl steht, (D)-Heteroaryl oder (D)-Heterocyclyl,
worin Heterocyclyl ein Heterocyclyl ausschließt, das einen einzelnen Stickstoff
enthält
und worin
Aryl, Heteroaryl, Heterocyclyl, Alkyl oder Cycloalkyl
wahlweise mit einem bis drei Substituenten substituiert sind, die
aus der Gruppe ausgewählt
sind, die besteht aus Oxo, C
1-C
8 Alkyl,
N(R
10)
2, OR
10, SR
10 und CO
2R
10,
R
10 jeweils unabhängig steht für
Wasserstoff,
C
1-C
8 Alkyl, C(O)-C
1-C
8 Alkyl, Aryl
oder C
3-C
7 Cycloalkyl,
R
11 jeweils unabhängig steht für
Wasserstoff,
C
1-C
8 Alkyl, (D)-Aryl,
(D)-Heteroaryl, (CH
2)
nN(R
8)
2, (CH
2)
nNR
8C(O)C
1-C
4 Alkyl, (CH
2)
nN-R
8SO
2-C
1-C
4 Alkyl,
(CH
2)
nSO
2N(R
8)
2,
(CH
2)
n[O]
q-C
1-C
8 Alkyl,
(CH
2)
n[O]
q(CH
2)
nNR
8COR
8, (CH
2n[O]
q(CH
2)
nN-R
8SO
2R
8, (CH
2)
n[O]
q-Heterocyclyl
oder (CH
2)
n[O]
q(C
1-C
8 Alkyl)heterocyclyl
und worin n für
2 bis 8 steht,
R
12 jeweils unabhängig steht
für
Wasserstoff,
C
1-C
8 Alkyl, (D)-Phenyl,
C(O)-C
1-C
8 Alkyl,
C(O)-Phenyl, SO
2C
1-C
8 Alkyl oder SO
2-Phenyl,
D
für eine
Bindung oder -(CH
2)
n-steht,
n
für 0 bis
8 steht,
p für
0 bis 5 steht,
q für
0 bis 1 steht und
r für
1 bis 2 steht.
-
Die
erfindungsgemäße Verbindung
ist wie oben angegeben, worin R3 für Phenyl
steht, das wahlweise mit Chlor, Brom, Fluor, Iod, Methoxy, Benzyloxy
oder Methyl para-substituiert ist.
-
Das
bevorzugte R3 steht für Phenyl, das mit Chlor, Fluor
oder Methoxy para-substituiert ist.
-
Die
erfindungsgemäße Verbindung
ist wie oben angegeben worin R4 für Wasserstoff
steht.
-
Die
erfindungsgemäße Verbindung
ist wie oben angegeben, worin -(CH
2)
n-T steht für
worin
* für ein
chirales Kohlenstoffatom mit einer R oder S Konfiguration steht.
-
Die
erfindungsgemäße Verbindung
ist wie oben angegeben, worin L und L1 zusammen
für Oxo
stehen und das chirale Kohlenstoffatom die R Konfiguration aufweist.
-
Die
bevorzugte Ausführungsform
der vorliegenden Erfindung liefert eine Verbindung der Formel II
oder pharmazeutisch annehmbare
Salze oder Stereoisomere hiervon.
-
Eine
weitere bevorzugte Ausführungsform
der vorliegenden Erfindung liefert eine Verbindung der Formel III
oder pharmazeutisch annehmbare
Salze oder Stereoisomere hiervon.
-
Eine
weitere bevorzugte Ausführungsform
der vorliegenden Erfindung liefert eine Verbindung der Formel IV
oder pharmazeutisch annehmbare
Salze oder Stereoisomere hiervon.
-
Eine
weitere bevorzugte Ausführungsform
der vorliegenden Erfindung liefert eine Verbindung der Formel V
oder pharmazeutisch annehmbare
Salze oder Stereoisomere hiervon.
-
Die
erfindungsgemäße Verbindung
ist wie oben in Formel II bis V angegeben, worin
p für 0 bis
5 steht,
n für
0 bis 8 steht,
q für
0 bis 1 steht,
D für
eine Bindung oder -(CH2)n-
steht,
R unabhängig
steht für
Wasserstoff,
Hydroxy, Cyano, Nitro, Halogen, C1-C8 Alkyl, C1-C8 Alkoxy, C1-C4 Halogenalkyl, (D)C(O)R9, (D)C(O)OR9, (D)C(O)SR9, (D)C(O)-Heteroaryl,
(D)C(O)-Heterocyclyl, (D)C(O)N(R9)2, (D)N(R9)2, (D)NR9COR9, (D)NR9CON(R9)2, (D)NR9C(O)OR9, (D)NR9C(R9)=N(R9), (D)NR9C(=NR9) N(R9)2,
(D)NR9SO2R9, (D)NR9SO2N(R9)2,
(D)NR9(CH2)n-Heterocyclyl, (D)NR9(CH2)n-Heteroaryl, (D)OR9, OSO2R9,
(D)[O]q(C3-C7 Cycloalkyl), (D)[O]q(CH2)n-Aryl, (D)[O]q(CH2)n-Heteroaryl,
(D)[O]q(CH2)n-Heterocyclyl,
worin Heterocyclyl ein Heterocyclyl ausschließt, das einen einzelnen Stickstoff
enthält,
wenn q für
1 steht, für
(D)SR9, (D)SOR9,
(D)SO2R9 oder (D)SO2N(R9)2,
worin
C1-C8 Alkyl, C1-C8 Alkoxy, C3-C7 Cycloalkyl,
Aryl, Heterocyclyl und Heteroaryl wahlweise mit einem bis fünf Substituenten
substituiert sind, die unabhängig
aus R8 ausgewählt sind,
R8 jeweils
unabhängig
steht für
Wasserstoff,
Halogen, Oxo, N(R10)2,
C1-C8 Alkyl, (D)-C3-C7 Cycloalkyl,
C1-C4 Halogenalkyl,
C1-C4 Alkoxy, Heteroaryl,
Hydroxy, Heterocyclyl, worin Heterocyclyl ein Heterocyclyl ausschließt, das
einen einzelnen Stickstoff enthält,
für Phenyl,
(D)-COR9, (D)C(O)OR9,
(D)OR9, (D)OCOR9,
(D)OCO2R9, (D)SR9, (D)SOR9 oder (D)SO2R9, worin Aryl,
Heteroaryl, Heterocyclyl, Alkyl oder Cycloalkyl wahlweise mit ein
bis drei Substituenten substituiert sind, die aus der Gruppe ausgewählt sind,
die besteht aus Oxo, C1-C8 Alkyl,
N(R10)2, OR10, SR10 und CO2R10,
R9 jeweils unabhängig ausgewählt ist aus
Wasserstoff,
C1-C8 Alkyl, C1-C4 Halogenalkyl,
(D)-C3-C7 Cycloalkyl,
(D)-Aryl, worin Aryl für
Phenyl oder Naphthyl steht, Heteroaryl oder Heterocyclyl, worin
Heterocyclyl ein Heterocyclyl ausschließt, das einen einzelnen Stickstoff
enthält
und worin
Aryl, Heteroaryl, Heterocyclyl, Alkyl oder Cycloalkyl
wahlweise mit einem bis drei Substituenten substituiert sind, die
aus der Gruppe ausgewählt
sind, die besteht aus Oxo, C1-C8 Alkyl,
N(R10)2, OR10, SR10 und CO2R10, und
R10 jeweils unabhängig steht für
Wasserstoff,
C1-C8 Alkyl, C(O)-C1-C8 Alkyl, Aryl
oder C3-C7 Cycloalkyl.
-
Die
erfindungsgemäße Verbindung
ist wie oben in Formel IV angegeben, worin R10 für Wasserstoff oder
C1-C8 Alkyl steht.
-
Die
am meisten bevorzugte Verbindung der vorliegenden Erfindung ist
die im folgenden angegebene Verbindung
-
-
-
Ebenfalls
von der vorliegenden Erfindung umfasst wird eine pharmazeutische
Zusammensetzung oder Formulierung, die einen pharmazeutischen Träger und
zumindest eine Verbindung der Formel I oder ein pharmazeutisch annehmbares
Salz oder Stereoisomer hiervon umfasst. Die pharmazeutische Zusammensetzung oder
Formulierung kann wahlweise ferner einen zweiten Wirkstoff umfassen,
der aus der Gruppe ausgewählt ist,
welche besteht aus einem Insulinsensitizer, einem Insulinmimetikum,
einem Sulfonylharnstoff, einem α-Glucosidaseinhibitor,
einem HMG-CoA Reduktaseinhibitor, einem sequestrierenden Cholesterinsenker,
einem β-3-adrenergen
Rezeptoragonist, einem Neuropeptid Y Antagonisten, einem Phosphodiester
V Inhibitor und einem α-2-adrenergen
Rezeptorantagonisten.
-
Ein
weiterer Aspekt der vorliegenden Erfindung ist ein Verfahren zur
Herstellung einer pharmazeutischen Zusammensetzung, die eine Verbindung
der Formel I oder ein pharmazeutisch annehmbares Salz oder Stereoisomer
hiervon, wie dies oben angegeben ist, und einen pharmazeutisch annehmbaren
Träger
umfasst.
-
Ein
weiterer Aspekt der vorliegenden Erfindung sind die Verbindungen
der Formel I zur Verwendung in einem Verfahren zur Prävention
oder Behandlung von Obesität
oder Diabetes mellitus bei einem Säuger, das die Verabreichung
einer therapeutisch wirksamen Menge der Verbindung der Formel I
umfasst.
-
Ein
weiterer Aspekt der vorliegenden Erfindung sind die Verbindungen
der Formel I zur Verwendung in einem Verfahren zur Prävention
oder Behandlung einer männlichen
oder weiblichen Sexualdysfunktion bei einem Säuger, genauer gesagt der männlichen
oder weiblichen Sexualstörung,
das die Verabreichung einer therapeutisch wirksamen Menge der Verbindung
der Formel I umfasst.
-
Ein
weiterer Aspekt der vorliegenden Erfindung ist ein Verfahren zur
Herstellung einer Verbindung der Formel I
oder eines pharmazeutisch
annehmbaren Salzes oder Stereoisomers hiervon, worin
-CLL
1-(CH
2)
n-T
steht für
worin R
1 für Wasserstoff,
C
1-C
8 Alkyl, Boc,
CBZ, Phenyl, FMOC oder (C
1-C
8 Alkyl)phenyl
steht,
Q für
folgenden Rest steht
und
R, R
1,
R
3, R
4, R
10, p und r wie für Formel I definiert sind,
wobei
das Verfahren die folgenden Schritte umfasst
- a)
Umsetzung einer Verbindung der Strukturformel 1 mit CH2CH=C(O)ORa, worin Ra für Wasserstoff
oder C1-C8 Alkyl
steht und X für
Halogen steht in Gegenwart eines Katalysators und einer Base in
einem geeigneten organischen Lösemittel
unter Bildung der Verbindung der Formel 2
- b) reduktive Aminierung der Verbindung der Formel 2 in Gegenwart
eines Amins bei sauren Bedingungen unter Bildung einer Verbindung
der Formel 3
- c) Cyclisierung der Verbindung der Formel 3 durch eine Michael-Addition
unter Bildung einer Verbindung der Formel 4 oder von Stereoisomeren
hiervon
- d) Kupplung der Verbindung der Formel 4 oder von Stereoisomeren
hiervon, worin Ra der Verbindung 4 für H steht,
mit einer Verbindung der Formel 5 worin Ra der
Verbindung 5 für
C1-C8 Alkyl steht,
unter Bildung einer Verbindung der Formel 6 und
- e) Kupplung der Verbindung der Formel 6, worin Ra für H steht,
mit einer Verbindung der Struktur unter Bildung der Verbindung
der Formel I.
-
Das
erfindungsgemäße Verfahren
ist wie oben angegeben, worin
in Schritt (a) für 2-Brombenzaldehyd
steht.
-
Das
erfindungsgemäße Verfahren
ist wie oben angegeben, worin CH2CH=C(O)OR
in Schritt (a) für Methylacrylat
steht.
-
Das
erfindungsgemäße Verfahren
ist wie oben angegeben, worin der Katalysator in Schritt (a) aus
der Gruppe ausgewählt
ist, die besteht aus: Pd(Ph3P)2Cl2, Pd(Ph3P)4Cl2, Pd(Ph3P)4, Pd(Ph3P)2Cl2/CuI, Pd(OAc)2/Ph3P-Bu4NBr, Pd(Ph3P)4Cl2/H2 und
Pd(OAc)2/P(O-Tol)3 und
worin die Base in Schritt (a) für
NH3 steht, worin R für Wasserstoff oder C1-C8 Alkyl steht.
-
Das
erfindungsgemäße Verfahren
ist wie oben angegeben, worin das Amin in Schritt (b) aus der Gruppe
ausgewählt
ist, die besteht aus Benzylamin, α-Methylbenzylamin
und Boc-NH2.
-
Das
erfindungsgemäße Verfahren
ist wie oben angegeben, worin der Schritt (b) ferner die Reduktion einer
Zwischenproduktiminverbindung in Gegenwart eines Reduktionsmittels
umfasst, wobei das Reduktionsmittel aus der Gruppe ausgewählt ist,
die besteht aus NaCNBH3, Na(OAc)3BH, NaBH4/H+ und einer Kombination aus Et3SiH
und TFA in CH3CN oder CH2Cl2.
-
Das
erfindungsgemäße Verfahren
ist wie oben angegeben, worin das Stereoisomer der Verbindung der
Formel 4 in Schritt (c) eine Verbindung der Formel 4a ist
-
-
Das
erfindungsgemäße Verfahren
ist wie oben angegeben, worin die Verbindung der Formel 4a durch asymmetrische
Hydrierung einer Verbindung der folgenden Strukturformel hergestellt
wird
-
-
Das
erfindungsgemäße Verfahren
ist wie oben angegeben, worin der Michael-Additionsschritt (c) unter basischen
Aufarbeitungsbedingungen ausgeführt
wird.
-
Das
erfindungsgemäße Verfahren
ist wie oben angegeben, worin der Schritt (e) ferner die Schutzgruppenanbringung
oder -abspaltung an der Verbindung der Formel (4) an NR1 umfasst.
-
Ein
weiterer Aspekt der vorliegenden Erfindung ist ein Verfahren zur
Herstellung einer Verbindung der Formel I
oder eines pharmazeutisch
annehmbaren Salzes oder Stereoisomers hiervon, worin
-CLL
1-(CH
2)
n-T
steht für
Q für folgenden Rest steht
R, R
1,
R
3, R
4, p und r
wie in Formel I definiert sind, und
R
11 jeweils
unabhängig
für Wasserstoff
oder C
1-C
8 Alkyl
steht,
das die folgenden Schritte umfasst
- a)
Veresterung einer Verbindung der Formel 1 mit einem Alkohol RaOH unter Bildung einer Verbindung der Formel
2 worin Ra für C1-C4 Alkyl oder (D)-Phenyl
steht
- b) Umsetzung einer Verbindung der Formel 2 mit R11COR11 unter Bildung einer Verbindung der Formel
3 worin R11 unabhängig für Wasserstoff
oder C1-C4 Alkyl
steht,
- c) Umsetzung einer Verbindung der Formel 3 mit einer Aktivierungsgruppe
unter Bildung einer Verbindung der Formel 4 worin A für eine Aktivierungsgruppe steht,
- d) Desoxygenierung der Verbindung der Formel 4 durch Hydrierung
unter Bildung einer Verbindung der Formel 5
- e) wahlweise Umsetzung der Verbindung der Formel 5 mit einer
anorganischen Base unter Bildung einer Verbindung der Formel 6 worin HA für ein saures
und M für
ein univalentes Kation steht,
- f) Auftrennung der Verbindung der Formel 5 oder Formel 6 unter
Bildung einer chiralen Verbindung der Formel 7 worin M für Wasserstoff steht und Ra' für H oder
Ra steht,
- g) Kupplung der Verbindung der Formel 7 mit einer Verbindung
der Formel 8 unter Bildung einer Verbindung
der Formel 9 und
- h) Kupplung der Verbindung der Formel 9 mit einer Verbindung
der Formel unter Bildung einer Verbindung
der Formel I.
-
Ein
weiterer Aspekt der vorliegenden Erfindung ist ein Verfahren zur
Herstellung einer Verbindung der Formel I
oder eines pharmazeutisch
annehmbaren Salzes oder Stereoisomers hiervon, worin
-CLL
1-(CH
2)
n-T
steht für
Q für folgenden Rest steht
R, R
1,
R
3, R
4, p und r
wie in Formel I definiert sind und
R
11 jeweils
unabhängig
für Wasserstoff
oder C
1-C
8 Alkyl
steht,
das die folgenden Schritte umfasst:
- a)
Umsetzung einer Verbindung der Formel 1 worin X für Halogen steht und R11 unabhängig
für Wasserstoff
oder C1-C4 Alkyl
steht, mit CNCH2CO2Ra, worin Ra für C1-C8 Alkyl oder Benzyl
steht unter Bildung einer Verbindung der Formel 2
- b) Schutzgruppenanbringung an der Verbindung der Formel 2 unter
Bildung der Verbindung der Formel 3
- c) Hydrierung der Verbindung der Formel 3 unter Bildung einer
Verbindung der Formel 4
- d) Kupplung der Verbindung der Formel 4, worin Ra' für Wasserstoff
oder Ra steht, mit einer Verbindung der Formel
5 unter Bildung einer Verbindung
der Formel 6
- e) Kupplung der Verbindung der Formel 6 mit einer Verbindung
der Formel unter Bildung einer Verbindung
der Formel I.
-
In
der vorliegenden Anmeldung haben die folgenden Ausdrücke die
angegebenen Bedeutungen:
-
Der
Ausdruck "Alkyl" steht, falls nichts
anderes angegeben ist, für
die Alkylgruppen mit einer angegebenen Anzahl an Kohlenstoffatomen
mit einer entweder geraden oder verzweigten gesättigten Konfiguration. Beispiele
für "Alkyl" umfassen unter anderem
Methyl, Ethyl, n-Propyl, Isopropyl, n-Butyl, Isobutyl, sek-Butyl und
t-Butyl, Pentyl, Hexyl, Neopentyl, Isopentyl und dergleichen. Alkyl
kann, wie dies oben definiert ist, optional mit einer angegebenen
Anzahl an Substituenten substituiert sein, wie dies in den oben
angegebenen Ausführungsformen
angegeben ist.
-
Der
Ausdruck "Alkenyl" steht für eine Kohlenwasserstoffkette
mit einer spezifizierten Anzahl an Kohlenstoffatomen mit einer entweder
geraden oder verzweigten Konfiguration und mit mindestens einer
Kohlenstoff-Kohlenstoff-Doppelbindung, die an jeder Stelle entlang
der Kette auftreten kann, wie Ethenyl, Propenyl, Butenyl, Pentenyl,
Vinyl, Alkyl, 2-Butenyl und dergleichen. Wie oben definiertes Alkenyl
kann mit einer angegebenen Anzahl an Substituenten substituiert
sein, wie dies in der oben angegebenen Ausführungsform beschrieben ist.
-
Der
Ausdruck "Halogenalkyl" steht für eine Alkylgruppe
mit der angegebenen Anzahl an Kohlenstoffatomen, die mit 1 bis 5
Halogenatomen substituiert ist, welche aus F, Br, Cl und I ausgewählt sind.
Ein Beispiel für
eine Halogenalkylgruppe ist Trifluormethyl.
-
Der
Ausdruck "Alkoxy" steht für eine Alkylgruppe
mit einer angegebenen Anzahl an Kohlenstoffatomen, die über eine
Sauerstoffbrücke
gebunden sind, wie Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, tert-Butoxy,
Pentoxy und dergleichen. Wie oben definiertes Alkoxy kann wahlweise
mit einer angegebenen Anzahl an Substituenten substituiert sein,
wie dies in der oben angegebenen Ausführungsform beschrieben ist.
-
Der
Ausdruck "Cycloalkyl" bezieht sich auf
einen Ring, der sich aus 3 bis 7 Methylengruppen zusammensetzt,
die jeweils wahlweise mit anderen Kohlenwasserstoffsubstituenten
substituiert sein können.
Beispiele für
Cycloalkyl umfassen unter anderem Cyclopropyl, Cyclobutyl, Cyclopentyl,
Cyclohexyl, Cycloheptyl und dergleichen. Wie oben definiertes Cycloalkyl
kann wahlweise mit einer angegebenen Anzahl an Substituenten substituiert
sein, wie dies in der oben angegebenen Ausführungsform beschrieben ist.
-
Der
Ausdruck "Halogen" steht für Fluor,
Chlor, Brom und Iod.
-
Der
Ausdruck "Halogenalkoxy" steht für eine Halogenalkylgruppe
mit der angegebenen Anzahl an Kohlenstoffatomen, die über eine
Sauerstoffbrücke
gebunden sind, wie OCF3. Wie oben definiertes "Halogenalkyloxy" kann wahlweise mit
einer angegebenen Anzahl an Substituenten substituiert sein, wie
dies in der oben angegebenen Ausführungsform beschrieben ist.
-
Der
Ausdruck "Aryl" steht für Phenyl,
Naphthyl, Anthracenyl, Phenanthrenyl und dergleichen, das wahlweise
mit der angegebenen Anzahl an Substituenten substituiert ist, wie
dies in der obigen Ausführungsform
beschrieben ist.
-
Der
Ausdruck "Heteroaryl" bezieht sich auf
einen monocyclischen oder bicyclischen aromatischen Ring mit 5 bis
10 Kohlenstoffatomen, die 1 bis 4 Heteroatome enthalten, welche
ausgewählt
sind aus O, N oder S und das Heteroaryl ist wahlweise mit einer
angegebenen Anzahl an Substituenten substituiert, wie dies in der
obigen Ausführungsform
beschrieben ist. Beispiele für
Heteroaryl sind unter anderem Furanyl, Thienyl, Thiazolyl, Imidazolyl,
Isoxazolyl, Oxazolyl, Pyrazolyl, Pyrrolyl, Pyrazinyl, Pyridyl, Pyrimidyl
und Purinyl, Cinnolinyl, Benzothienyl, Benzotriazolyl, Benzoxazolyl,
Chinolin, Isochinolin und dergleichen.
-
Das "Heterocyclyl" ist als monocyclischer,
bicyclischer oder tricyclischer Ring mit 5 bis 14 Kohlenstoffatomen
definiert, der gesättigt
oder teilweise gesättigt
ist und 1 bis 4 Heteroatome enthält,
die aus N, O oder S ausgewählt
sind. Das "Heterocyclyl" umfasst "Stickstoff-enthaltendes
Heterocyclyl", das
1 bis 4 Stickstoffatome enthält
und wahlweise ferner ein anderes Heteroatom enthält, das aus O oder S ausgewählt ist.
Wie oben definiertes Heterocyclyl kann wahlweise mit einer angegebenen
Anzahl an Substituenten substituiert sein, wie dies in der oben
angegebenen Ausführungsform
beschrieben ist.
-
Wie
hierin verwendet umfasst ein Säuger
einen Menschen und einen Warmblüter,
wie eine Katze, einen Hund und dergleichen.
-
Der
Ausdruck "Zusammensetzung" oder "Formulierung", wie bei pharmazeutischer
Zusammensetzung oder Formulierung, soll ein Produkt umfassen, das
die Wirkstoffe und die inerten Inhaltsstoffe enthält, die den
Träger
ausmachen. Demnach umfassen die pharmazeutischen Zusammensetzungen
der vorliegenden Erfindung jede Zusammensetzung, die durch Mischen
einer erfindungsgemäßen Verbindung
(einer Verbindung der Formel I) und eines pharmazeutisch annehmbaren
Trägers
hergestellt wird.
-
Der
Ausdruck "pharmazeutisch" meint, wenn er hierin
als Adjektiv verwendet wird, dass es im wesentlichen für den empfangenden
Säuger
unschädlich
ist.
-
Der
Ausdruck "Einheitsdosierungsform" bezieht sich auf
physikalisch getrennte Einheiten, die als einmalige Dosierungen
für den
Menschen oder andere Tiere, wie Warmblüter, geeignet sind, wobei jede
Einheit eine vorbestimmte Menge an Wirkstoff (Verbindung der Formel
I), die zur Herstellung des gewünschten
therapeutischen Effekts berechnet wurde, zusammen mit einem geeigneten
pharmazeutischen Träger
enthält.
-
Der
Ausdruck "Behandlung" oder "Prävention", wie er hierin verwendet
wird, umfasst die allgemein anerkannten Bedeutungen, das heißt Prävention,
Verhinderung, Zurückdrängung, Linderung,
Besserung, Verlangsamung, Stoppen oder Umkehrung der Progression
oder Schwere eines pathologischen Zustands oder der Leiden hierdurch,
wie dies hierin beschrieben ist.
-
"Erektionsstörung" ist eine Störung, die
das Versagen eines männlichen
Säugers
umfasst, eine Erektion, Ejakulation oder beides zu erreichen. Symptome
der Erektionsstörung
umfassen eine Unfähigkeit
eine Erektion zu erreichen oder aufrechtzuerhalten, ein Ejakulationsversagen,
eine frühe
Ejakulation und eine Unfähigkeit,
einen Orgasmus zu erreichen. Eine Zunahme der Erektionsstörung ist
oft mit dem Alter assoziiert und sie wird im allgemeinen durch eine
physische Erkrankung verursacht oder ist eine Nebenwirkung einer Arzneimittelbehandlung.
-
"Weibliche Sexualstörung" umfasst ohne Beschränkung Zustände, wie
fehlendes Sexualverlangen und verwandte Luststörungen, gehemmten Orgasmus,
Gleitmittelschwierigkeiten und Vaginismus.
-
Da
bestimmte erfindungsgemäße Verbindungen
einen sauren Rest (beispielsweise Carboxy) enthalten, kann die Verbindung
der Formel I als pharmazeutisches Basenadditionssalz hiervon verwendet
werden. Solche Salze umfassen jede, die von anorganischen Basen
stammen, wie Ammonium und Alkali- und Erdalkalimetallhydroxide,
-carbonate, -bicarbonate und dergleichen, wie auch Salze, die von
basischen organischen Aminen stammen, wie aliphatischen und aromatischen
Aminen, aliphatischen Diaminen, Hydroxyalkaminen und dergleichen.
-
Da
bestimmte Verbindungen der Erfindung einen basischen Rest (beispielsweise
Amino) enthalten, kann die Verbindung der Formel I auch als pharmazeutisches
Säureadditionssalz
vorkommen. Solche Salze umfassen Sulfat, Pyrosulfat, Bisulfat, Sulfit,
Bisulfit, Phosphat, Monohydrogenphosphat, Dihydrogenphosphat, Metaphosphat,
Pyrophosphat, Chlorid, Bromid, Iodid, Acetat, Propionat, Decanoat,
Caprylat, Acrylat, Formfiat, Isobutyrat, Heptanoat, Propiolat, Oxalat,
Malonat, Succinat, Suberat, Sebacat, Fumarat, Maleat, 2-Butin-1,4-dioat,
3-Hexin-2,5-dioat, Benzoat, Chlorbenzoat, Hydroxybenzoat, Methoxybenzoat,
Phthalat, Xylolsulfonat, Phenylacetat, Phenylpropionat, Phenylbutyrat,
Citrat, Lactat, Hippurat, β-Hydroxybutyrat,
Glycolat, Maleat, Tartrat, Methansulfonat, Propansulfonat, Naphthalin-1-sulfonat, Naphthalin-2-sulfonat,
Mandelat und dergleichen. Die bevorzugte Salzform einer Verbindung
der Formel I ist ein Säureadditionssalz,
genauer gesagt das Hydrochloridsalz.
-
Einige
der hierin beschriebenen Verbindungen können als Tautomere vorkommen,
wie Keto-Enol-Tautomere.
Die einzelnen Tautomere wie auch Gemische hiervon werden vom Umfang
der vorliegenden Erfindung umfasst.
-
Brauchbarkeit
-
Die
Verbindungen der Formel I sind als Melanocortinrezeptormodulatoren
wirksam, insbesondere als Agonisten des humanen MC-4 Rezeptors.
Als Melanocortinrezeptoragonisten sind die Verbindungen der Formel
I zur Behandlung von Erkrankungen, Störungen oder Zuständen brauchbar,
die auf die Aktivierung eines oder mehrerer Melanocortinrezeptoren
ansprechen, wie unter anderem MC-1, MC-2, MC-3, MC-4 und MC-5. Erkrankungen,
Störungen
oder Zustände,
die auf eine Behandlung mit einem MC-4 Agonisten ansprechen, umfassen
die oben erwähnten
und die in WO 00/74679 a beschriebenen, deren Beschreibung hiermit
eingeführt
ist. Insbesondere umfassen Erkrankungen, Störungen oder Zustände, die
auf die Behandlung mit einem MC-4 Agonisten ansprechen, Obesität oder Diabetes
mellitus, männliche
oder weibliche Sexualstörung,
genauer gesagt Erektionsstörung.
-
Bei
der Beschreibung der verschiedenen Aspekte der vorliegenden Verbindungen
der Formel I werden die Ausdrücke "A Domäne", "B Domäne" und "C Domäne" im folgenden verwendet.
Dieses Domänenkonzept ist
im folgenden gezeigt:
-
-
Die
folgenden Listen liefern einige Beispiele für die "A Domäne", "B
Domäne" und "C Domäne" der Verbindung der
Formel I. Diese Listen dienen nur zur Erläuterung und soll nicht beschränkend wirken.
-
-
-
-
Formulierung
-
Die
Verbindung der Formel I wird vorzugsweise in einer Einheitsdosierungsform
vor der Verabreichung formuliert. Demnach umfasst die vorliegende
Erfindung auch eine pharmazeutische Zusammensetzung, die eine Verbindung
der Formel I und einen geeigneten pharmazeutischen Träger enthält.
-
Die
vorliegenden pharmazeutischen Zusammensetzungen werden durch bekannte
Verfahren mittels gut bekannter und leicht verfügbarer Inhaltsstoffe hergestellt.
Bei der Herstellung der erfindungsgemäßen Formulierungen wird der
Wirkstoff (eine Verbindung der Formel I) gewöhnlich mit einem Träger gemischt
oder mit einem Träger
verdünnt
oder in einem Träger
eingeschlossen, der in Form einer Kapsel, eines Sachets, eines Papiers
oder eines anderen Behälters
vorliegen kann. Wenn der Träger
als Verdünnungsmittel
dient, kann dies ein festes, halbfestes oder flüssiges Material sein, das als
Vehikel, Hilfsstoff oder Medium für den Wirkstoff dient. Daher
können
die Zusammensetzungen vorliegen in Form von Tabletten, Pillen, Pulvern,
Lonzetten, Sachets, Cachets, Elixieren, Suspensionen, Emulsionen,
Lösungen,
Sirupen, Aerosolen (als Feststoff oder in einem flüssigen Medium),
Weich- und Hartgelatinekapseln, Zäpfchen, sterilen injizierbaren
Lösungen
und sterilen verpackten Pulvern.
-
Einige
Beispiele für
geeignete Träger,
Hilfsstoffe und Verdünnungsmittel
sind unter anderem Lactose, Glucose, Saccharose, Sorbit, Mannit,
Stärkearten,
Akaziengummi, Calciumphosphat, Alginate, Traganth, Gelatine, Calciumsilicat,
mikrokristalline Cellulose, Polyvinylpyrrolidon, Cellulose, Wasser,
Sirup, Methylcellulose, Methyl- und Propylhydroxybenzoate, Talkum,
Magnesiumstearat und Mineralöl.
Die Formulierungen können zusätzlich enthalten
Gleitmittel, Netzmittel, Emulgier- und Suspendiermittel, Konservierungsstoffe,
Süßstoffe oder
Geschmacksstoffe. Die erfindungsgemäßen Zusammensetzungen können so
formuliert werden, dass sie eine schnelle, anhaltende oder verzögerte Freisetzung
des Wirkstoffs nach der Verabreichung an den Patienten bereitstellen.
-
Dosierung
-
Die
spezifisch verabreichte Dosis wird durch die einzelnen Umstände bestimmt,
die jede Situation umgeben. Diese Umstände umfassen den Verabreichungsweg,
die medizinische Vorgeschichte des Empfängers, den pathologischen Zustand
oder das Symptom, das behandelt wird, die Schwere der zu behandelnden
Zustände/Symptome
und das Alter und Geschlecht des Empfängers. Zusätzlich ist es verständlich,
dass die verabreichte therapeutische Dosis durch einen Arzt in Anbetracht
der relevanten Umstände
bestimmt wird.
-
Im
allgemeinen beträgt
eine minimale Tagesdosis einer Verbindung der Formel I etwa 1, 5,
10, 15 oder 20 mg. Typischerweise beträgt eine wirksame Maximaldosis
etwa 500, 100, 60, 50 oder 40 mg. Die geeignete Dosis kann gemäß der Standardpraxis
in der Medizin durch "Dosistitration" am Patienten bestimmt
werden, die die anfängliche
Verabreichung einer geringen Dosis der Verbindung und dann die graduelle
Erhöhung
der Dosis umfasst, bis der gewünschte
therapeutische Effekt beobachtet wird.
-
Verabreichungsweg
-
Die
Verbindungen können
auf eine Vielzahl an Wegen verabreicht werden, einschließlich auf
oralem, rektalem, transdermalem, subkutanem, topischem, intravenösem, intramuskulärem oder
intranasalem Weg.
-
Kombinationstherapie
-
Die
Verbindungen der Formel I können
in Kombination mit anderen Arzneimitteln verwendet werden, die bei
der Behandlung der Erkrankungen oder Zustände verwendet werden, für die die
Verbindungen der Formel I brauchbar sind. Solche anderen Arzneimittel
können über einen
Weg und in einer Menge, die hierfür herkömmlich verwendet werden, gleichzeitig
oder sequenziell mit einer Verbindung der Formel I verabreicht werden.
Wenn eine Verbindung der Formel I gleichzeitig mit einem oder mehreren
anderen Arzneimitteln verwendet wird, ist eine pharmazeutische Zusammensetzung
bevorzugt, die diese anderen Arzneimittel zusammen mit der Verbindung
der Formel I enthält.
Demnach umfassen die pharmazeutischen Zusammensetzungen der vorliegenden
Erfindung die, welche auch einen oder mehrere andere Wirkstoffe
zusätzlich
zu einer Verbindung der Formel I enthalten. Beispiele für solche
anderen Wirkstoffe, die mit einer Verbindung der Formel I entweder getrennt
oder in denselben pharmazeutischen Zusammensetzungen verabreicht
werden, umfassen unter anderem:
- (a) Insulinsensitizer
einschließlich
(i) PPARγ Agonisten,
wie die Glitazone (beispielsweise Troglitazon, Pioglitazon, Englitazon,
MCC-555, BRL49653 und dergleichen) und Verbindungen, die in WO 97/27857
A, WO 97/28115 A, WO 97/28137 A und WO 97/27847 A beschrieben sind,
(ii) Biguanide, wie Metformin und Phenformin,
- (b) Insulin oder Insulinmimetika,
- (c) Sulfonylharnstoffe, wie Tolbutamid und Glipizid,
- (d) α-Glucosidaseinhibitoren
(wie Acarbose),
- (e) Cholesterinsenker, wie (i) HMG-CoA Reduktaseinhibitoren
(Lovastatin, Simvastatin und Pravastatin, Fluvastatin, Atorvastatin
und andere Statine), (ii) Sequestriermittel (Cholestyramin, Colestipol
und Dialkylaminoalkylderivate eines quervernetzten Dextrans), (iii)
Nicotinoylalkohol, Nicotinsäure
oder ein Salz hiervon, (iv) Agonisten für den Proliferator-Aktivator-Rezeptor α, wie Fenobrinsäurederivate
(Gemfibrozil, Clofibrat, Fenofibrat und Benzafibrat), (v) Inhibitoren
der Cholesterinabsorption, wie β-Sitosterol und Acyl-CoA:Cholesterinacyltransferaseinhibitoren,
wie Melinamid, (vi) Probucol, (vii) Vitamin E und (viii) Thyromimetika,
- (f) PPARδ Agonisten,
wie die, welche in WO 97/28149 A beschrieben sind,
- (g) Antiobesitätsverbindungen,
wie Fenfluramin, Dexfenfluramin, Phentermin, Sibutramin, Ortistat
und β-3-adrenerge Rezeptoragonisten,
- (h) Ernährungsverhalten
modifizierende Mittel, wie Neuropeptid Y Antagonisten (beispielsweise
Neuropeptid Y5) wie dies in WO 97/19682 A, WO 97/20820 A, WO 97/20821
A, WO 97/20822 A und WO 97/20823 A beschrieben ist,
- (i) PPARα Agonisten,
wie dies in WO 97/36579 A beschrieben ist,
- (j) PPARγ Antagonisten,
wie dies in WO 97/10813 A beschrieben ist,
- (k) Serotoninwiederaufnahmeinhibitoren, wie Fluoxetin und Sertralin,
- (l) Wachstumshormonsekretionsmittel, wie MK-0677 und
- (m) Mittel, die zur Behandlung der männlichen und/oder weiblichen
Sexualstörung
brauchbar sind, wie Phosphodiester V Inhibitoren, einschließlich Sildenafil
und ICI-351 und α-2
adrenerge Rezeptorantagonisten, einschließlich Phentolaminmesylat und
Dopaminrezeptoragonisten, wie Apomorphin.
-
Biologische Tests
-
A. Bindungstest
-
Der
radioaktive Bindungstest wird zur Identifizierung von kompetitiven
Inhibitoren der 125I-NDP-α-MSH Bindung an klonierte
humane MCRs mittels Membranen aus stabil transfizierten, humanen,
embryonalen 293-Nierenzellen (HEK 293) verwendet.
-
HEK
293 Zellen, die mit Melanocortinrezeptoren des Menschen oder der
Ratte transfiziert sind, werden entweder als adhärente Monolagen oder als Suspensionskultur
angezogen. Die Monoschichtzellen werden in Rollflaschenkulturen
bei 37°C
und 5% CO2/Luftatmosphäre in einem 3:1 Gemisch aus
Dulbecco's modifiziertem
Eagle Medium (DMEM) und Ham F12 angezogen, worin 25 mM L-Glucose,
100 Einheiten/ml Penicillin G 100 μg/ml Streptomycin, 250 ng/ml
Amphotericin B und 300 μg/ml
Genticin enthalten sind und das mit 5% fetalem Rinderserum supplementiert
ist. Die Monoschichtzellen werden an eine Suspensionskultur angepasst
(Berg et al., Biotechniques, Band 14, Nr. 6, 1993) und werden entweder
in Kreiskolben oder Schüttelkolben
(37°C und
7,5% CO2/Luftüberschichtung) in einem modifizierten
DMEF/F12 Medium angezogen, worin 0,1 mM CaCl2,
2% Pferdeserum und 100 μg/ml
Natriumheparin enthalten sind, um eine Zell-Zell-Aggregation zu vermeiden.
Die Zellen werden durch Zentrifugation geerntet, mit PBS gewaschen
und die Pellets werden bei –80°C gefroren
bis zu den Membranpräparationen
gelagert.
-
Die
Zellpellets werden in 10 Volumina an Membranpräparationspuffer (das heißt 1 g Pellt
in 10 ml Puffer) resuspendiert, der die folgende Zusammensetzung
aufweist: 50 mM Tris pH 7,5 bei 4°C,
250 mM Saccharose, 1 mM MgCl2, Complete® EDTA-freie
Proteaseinhibitortablette (Boehringer Mannheim) und 24 μg/ml DNase
I (Sigma, St. Louis, MO). Die Zellen werden mit einem motorgetriebenen
Homogenisator mit 20 Schlägen homogenisiert
und das Homogenat wird bei 38 000 × g bei 4°C für 40 Minuten zentrifugiert.
Die Pellets werden in Membranpräparationspuffer
bei einer Konzentration von 2,5–7,5
mg/ml resuspendiert und 1 ml Aliquots der Membranhomogenate werden
schnell in flüssigem
Stickstoff eingefroren und dann bei –80°C gelagert.
-
Die
Lösungen
einer Verbindung der Formel I (300 pmol/l bis 30 μmol/l) oder
unmarkiertes NDP-α-MSH (1 pmol/l bis
100 nmol/l) werden zu 150 μl
Membranbindungspuffer unter Bildung der Endkonzentrationen (in Klammern
angegeben) zugegeben. Der Membranbindungspuffer hat die folgende
Zusammensetzung: 25 mM HEPES pH 7,5, 10 mM CaCl2,
0,3% BSA. 150 μl
Membranbindungspuffer, worin 0,5–5,0 μg Membranprotein enthalten ist,
werden gefolgt von 50 nmol/l an 125I-NDP-α-MSH auf
eine Endkonzentration von 100 pmol/l zugegeben. Zusätzlich werden
50 μl an
SPA Kügelchen
(5 mg/ml) zugegeben und das entstehende Gemisch wird kurz gerührt und
für 10
Stunden bei RT inkubiert. Die Radioaktivität wird in einem Wallac Trilux
Mikrotiterplattenscintillationszähler
quantifiziert. Die in Kompetitionstests erhaltenen HK50 Werte
werden unter Verwendung der Cheng-Prusoff Gleichung Ki =
HK50/(1 + D/Kd)
in Affinitätskonstanten
(Ki Werte) umgewandelt.
-
B. Funktionstest
-
Es
werden funktionelle Zell-basierte Tests entwickelt, um Agonisten
und Antagonisten zu unterscheiden.
-
Agonisttest:
HEK 293 Zellen, die stabil einen humanen Melanocortinrezeptor exprimieren
(siehe beispielsweise Yang et al., Mol. Endocinol., 11 (3): 274–280, 1997)
werden mittels Tripsin/EDTA Lösung
(0,25%, Life Technologies, Rockville, MD) von den Gewebekulturflaschen
abgelöst.
Die Zellen werden durch Zentrifugation gesammelt und in DMEM (Life
Technologies, Rockville, MD) resuspendiert, das mit 1% L-Glutamin
und 0,5% fetalem Rinderserum supplementiert ist. Die Zellen werden
gezählt
und auf 4,5 × 105 pro ml verdünnt.
-
Es
wird eine Verbindung der Formel I in Dimethylsulfoxid (DMSO) (Endkonzentration
3 × 10–5 bis
3 × 10–10 M)
verdünnt
und 0,05 Volumen der Lösung
der Verbindung werden zu 0,95 Volumina der Zellsuspension gegeben,
wobei die DMSO Endkonzentration 0,5% beträgt. Nach der Inkubation bei
37°C/5%
CO2 für
5 Stunden werden die Zellen durch die Zugabe von Luciferinlösung (50
mM Tris, 1 mM MgCl2, 0,2% Triton-X 100,
5 mM DTT, 500 μmol/l
Coenzym A, 150 μmol/l
ATP und 440 μmol/l
Luciferin) lysiert, um die Aktivität des Reportergens Luciferase
zu quantifizieren, die eine indirekte Messung der intrazellulären cAMP
Produktion ist.
-
Die
Luciferaseaktivität
wird aus dem Zelllysat mittels eines Wallac Victor 2 Luminometers
gemessen. Die Menge an Lichtproduktion, die von einer Verbindung
der Formel I stammt, wird mit der Menge an Licht verglichen, die
in Reaktion auf NDP-α-MSH
gebildet wird, welcher als 100% Agonist definiert wird, um eine
relative Effizienz einer Verbindung zur erhalten. Die EK50 wird als die Verbindungskonzentration
definiert, die zu einer halbmaximalen Stimulation führt, wenn
sie mit dem eigenen Maximalniveau der Stimulierung verglichen wird.
-
Antagonisttest:
Die Antagonistaktivität
wird als die Fähigkeit
einer Verbindung definiert, um die Lumenproduktion in Reaktion auf
NDP-α-MSH
zu blockieren. Die Konzentrations-Reaktionskurven werden für NDP-α-MSH in Abwesenheit
und Gegenwart einer fixierten Konzentration einer Lösung einer
Verbindung der Formel I erzeugt (10 × Ki aus
den Bindungstests). Suspensionen von MCR-exprimierenden Zellen werden
hergestellt und mit NDP-α-MSH
und Verbindungslösungen
für 5 Stunden
inkubiert, wie dies oben beschrieben ist. Der Test wird durch die
Zugabe von Luciferinreagenz und Lichtbildung quantifiziert. Die
Antagonistenstärke wird
aus der Rechtsverschiebung des EK50 Werts
in Abwesenheit einer Verbindung der Formel I mittels der folgenden
Gleichung bestimmt: Kb = Konzentration des
Antagonisten/[EK50'/EK50) – 1].
-
cAMP Akkumulationstest
mit ganzen Zellen
-
Verbindungspräparation
-
Im
Agonisttest werden die Verbindungen als 10 mM und NDP-α-MSH (Kontrolle)
als 33,3 μM
Stammlösungen
in 100% DMSO hergestellt. Diese werden seriell in 100% DMSO verdünnt. Die
Verbindungsplatte wird weiter 1:200 in Verbindungsverdünnungspuffer
verdünnt
(HBSS-092, 1 mM Ascorbinsäure,
1 mM IBMX, 0,6% DMSO, 0,1% BSA). Die Endkonzentration reicht von
10 μM bis
100 pM für
die Verbindung und 33,33 nM bis 0,3 pM für die Kontrolle in 0,5% DMSO.
Es erfolgt ein Transfer von 20 μl
aus dieser Platte in 4 PET Platten mit 96 Vertiefungen (alle Tests
werden zweifach für
jeden Rezeptor ausgeführt).
-
Zellkultur und Zellstimulierung
-
HEK
293 Zellen, die stabil mit dem MC3R und MC4R transfiziert sind,
werden in DMEM angezogen, das 10% FBS und 1% Antibiotikum/Antimykotikumlösung enthält. Am Tag
des Tests werden die Zellen mit Enzym-freier Dissoziationslösung abgelöst und in
Zellpuffer (HBSS-092, 0,1% BSA, 10 mM HEPES) mit 1 × 106 Zellen/ml resuspendiert. Es werden 40 μl Zellen/Vertiefung
zu den PET Platten mit 96 Vertiefungen gegeben, worin 20 μl verdünnte Verbindung
und Kontrolle enthalten sind. Man inkubiert bei 37°C in einem
Wasserbad für
20 Minuten. Man stoppt den Test durch die Zugabe von 50 μl Stoppuffer
(50 mM Na-Acetat, 0,25% Triton X-100).
-
Bindungstests mit radioaktiven
Liganden
-
Es
werden Bindungstests mit radioaktiven Liganden in SPA Puffer (50
mM Natriumacetat, 0,1% BSA) ausgeführt. Die Kügelchen, der Antikörper und
der radioaktive Ligand werden in SPA Puffer unter Bereitstellung
von ausreichend Volumen für
jede Platte mit 96 Vertiefungen verdünnt. Zu jeder gestoppten Testvertiefung werden
100 μl eines
Cocktails zugegeben, der 33,33 μl
Kügelchen,
33,33 μl
Antikörper
und 33,33 Mikroliter 125I-cAMP enthält. Dies
führt zu
einer Endkonzentration von 6,3 mg/ml Kügelchen, 0,65% Anti-Ziege-Antikörper und
61 pM an 125I-cAMP (enthält 25000–30000 cpm) in einem schließlichen
Testvolumen von 210 μl.
Die Platten werden in einem Wallac MicroBeta Zählgerät nach einer Inkubation von
12 Stunden ausgezählt.
-
Die
Daten werden in pmol cAMP mittels einer Standardkurve umgewandelt,
die unter denselben Bedingungen gemessen wurde. Die Daten werden
mittels einer Activity Base Software analysiert, um Agoniststärken (EK50)
und Daten einer prozentualen relativen Wirksamkeit gegenüber NDP-α-MSH zu generieren.
-
C. In vivo Nahrungsaufnahmemodelle
-
1) Tägliche Nahrungsaufnahme
-
Männlichen
Long-Evans Ratten injiziert man intracerebroventrikulär (ICV)
eine Testverbindung in 5 μl einer
50% Propylenglycol/künstlichen
Cerebrospinalflüssigkeit
eine Stunde nach dem Einsetzen des Dunkelzyklus (12 Stunden). Die
Nahrungsaufnahme wird durch die Subtraktion des Nahrungsgewichts
bestimmt, das nach 24 Stunden vom Nahrungsgewicht direkt vor der
ICV Injektion verbleibt.
-
2) Akutkalorimetrie
-
Männlichen
Long-Evans-Ratten verabreicht man eine Testverbindung durch subkutane
Injektion, intramuskuläre
Injektion, intravenöse
Injektion, intraperitoneale Injektion, ICV Injektion oder durch
orale Verabreichung zwischen 0 und 5 Stunden nach dem Einsetzen
des Dunkelzyklus. Die Ratten werden in eine Kalorimetriekammer gegeben
und das Volumen des verbrauchten Sauerstoff oder des ausgeatmeten
Kohlendioxids wird für
jede Stunde für
24 Stunden gemessen. Die Nahrungsmittelaufnahme wird für die 24
Stundenperiode wie in C1) beschrieben gemessen. Die lokomotorische
Aktivität
wird gemessen, wenn die Ratte eine Reihe an Infrarotlaserstrahlen
unterbricht, wenn sie im Kalorimeter ist. Diese Messungen erlauben
die Berechnung des Energieverbrauchs, des Respirationsquotienten
und der Energiebilanz.
-
3) Nahrungsaufnahme in
diätinduzierten übergewichtigen
Ratten
-
Männlichen
C57/B16J Mäusen,
die auf einer Hochfettdiät
(60% Kalorien aus Fett) für
6,5 Monate seit der 4 Lebenswoche gehalten wurden, wird intraperitoneal
eine Verbindung der Formel I verabreicht. Die Nahrungsaufnahme und
das Körpergewicht
werden über
einen Zeitraum von 8 Tagen gemessen. Die biochemischen Parameter
bezüglich
Obesität,
einschließlich
Leptin, Insulin, Triglycerid, Fettsäure, Cholesterin und Serumglucosespiegel
werden bestimmt.
-
D. Ratten Ex Copula Test
-
Sexuell
reife, männliche
Caesarian Derived Sprague Dawley (CD) Ratten (über 60 Tage alt) werden verwendet,
wobei das anbindende Ligament operativ entfernt wurde, um eine Zurückziehung
des Penis in die Penisvorhaut während
der ex copula Evaluierungen zu verhindern. Die Tiere erhalten freien
Zugang zu Nahrung und Wasser und werden unter einem normalen Licht/Dunkel-Zyklus
gehalten. Die Studien werden während
dem Lichtzyklus ausgeführt.
-
1) Konditionierung auf
eine Rückenlagenzwangshaltung
für Ex
Copula Reflextests
-
Die
Konditionierung dauert etwa 4 Tage. Am Tag 1 werden die Tiere in
einen abgedunkelten Behälter gegeben
und dort für
15 bis 30 Minuten gehalten. Am Tag 2 werden die Tiere in eine Rückenlagenposition
im Behälter
für 15
bis 30 Minuten gezwungen. Am Tag 3 werden die Tiere in die Rückenlagenposition
gezwungen, wobei die Penisvorhaut für 15 bis 30 Minuten zurückgezogen
wird. Am Tag 4 werden die Tiere in die Rückenlagenposition gezwungen,
wobei die Penisvorhaut zurückgezogen
wird, bis Penisreaktionen beobachtet werden. Einige Tiere erfordern
zusätzliche
Konditionierungstage, bevor sie vollkommen an die Verfahren akklimatisiert
sind, wobei Tiere, die nicht ansprechen, aus der weiteren Evaluierung
ausgeschlossen werden. Nach jeder Behandlung oder Evaluierung erhalten
die Tiere eine Behandlung, um eine positive Verstärkung sicherzustellen.
-
2) Ex Copula Reflextest
-
Die
Ratten werden sanft in eine Rückenlagenposition
gezwungen, wobei der anteriore Torso in einen Zylinder mit einer
passenden Größe gegeben
wird, um eine normale Kopf und Pfotenbeischlafposition zu erlauben.
Für eine
400–500
g Ratte beträgt
der Durchmesser des Zylinders etwa 8 cm. Der untere Torso und die hinteren
Gliedmaßen
werden mit einem nicht-adhäsiven
Material (Vetrap) festgehalten. Ein zusätzliches Stück Vetrap mit einem Loch darin,
durch das die Peniseichel geführt
wird, wird über
dem Tier festgemacht, um die Penisvorhaut in einer zurückgezogenen
Position zu halten. Die Penisreaktionen werden beobachtet, die typischerweise
ex Copulu Genitalreflextests genannt werden. Typischerweise wird
eine Reihe an Peniserektionen beobachtet, die spontan innerhalb
weniger Minuten nach der Vorhautzurückziehung auftreten. Die Typen
der normalen reflexogenen Erektionsreaktionen umfassen Elongation,
Verdickung, Tassenbildung und Verdrehung. Eine Elongation wird als
Ausdehnung des Peniskörpers
definiert. Eine Verdickung ist eine Dilatation der Peniseichel.
Eine Tasse wird als intensive Erektion definiert, bei der das distale
Ende der Peniseichel für
einen Moment aufklappt, um eine Tasse zu bilden. Eine Verdrehung
ist eine Dorsiflexion des Peniskörpers.
-
Es
werden Grundlinien und/oder Trägerevaluierungen
ausgeführt,
um zu bestimmen, wie und ob ein Tier reagieren wird. Einige Tiere
haben eine lange Zeit bis zur ersten Reaktion, während andere ins gesamt nicht
ansprechen. Während
dieser Grundlinienevaluierung werden die Latenz bis zur ersten Reaktionszeit,
die Anzahl und der Typ der Reaktionen aufgezeichnet. Der Testzeitrahmen
beträgt
15 Minuten nach der ersten Reaktion.
-
Nach
einem Minimum von 1 Tag zwischen den Evaluierungen wird denselben
Tieren eine Verbindung der Formel I mit 20 mg/kg verabreicht und
sie werden auf Penisreflexe untersucht. Alle Evaluierungen werden mit
Video aufgezeichnet und später
ausgewertet. Die Daten werden gesammelt und mittels eines paarweisen gebündelten
2 t-Tests werden vergleichende Grundlinien und/oder Trägerevaluierungen
mit Arzneimittelevaluierungen für
einzelne Tiere analysiert. Es werden Gruppen mit minimal 4 Tieren
verwendet, um die Variabilität zu
verringern.
-
Positive
Referenzkontrollen werden in jede Studie eingebaut, um die Validität sicherzustellen.
Die Tiere können
in Abhängigkeit
der Art der auszuführenden
Studie durch mehrere Verabreichungswege die Dosis erhalten. Die
Verabreichungswege umfassen intravenös (i.v.), intraperitoneal (i.p.),
subkutan (s.c.) und intracerebral-ventrikulär (i.c.y.).
-
E. Modelle der weiblichen
Sexualstörung
-
Nagertests,
die für
die sexuelle Empfänglichkeit
der Weibchen relevant sind, umfassen das Verhaltensmodell der Lordose
und direkte Beobachtungen der Kopulationsaktivität. Es existiert auch ein urethrogenitales
Reflexmodell in betäubten
Ratten mit Spinaltranssektion zur Messung des Orgasmus sowohl bei
männlichen
als auch weiblichen Ratten. Diese und andere etablierte Tiermodelle
für die
weibliche Sexualstörung sind
in McKenna et al., Am. J. Physiol., (Regulatory Integrative Comp.
Physiol 30): R 1276-R1285, 1991, McKenna et al., Pharm. Bioch. Behav.,
40: 151–156,
1991 und Takahashi et al., Brain Res. 359: 194–207, 1985, beschrieben.
-
Herstellung der erfindungsgemäßen Verbindungen
-
Die
Herstellung der erfindungsgemäßen Verbindungen
kann durch sequentielle oder konvergente Synthesewege ausgeführt werden.
Der Fachmann erkennt, dass im allgemeinen die 3 Domänen einer
Verbindung der Formel I über
Amidbindungen verbunden sind. Die B und C Domänen werden wahlweise über eine reduzierte
oder teilweise reduzierte Amidbindung verbunden (beispielsweise über reduktive
Aminierung). Der Fachmann kann daher leicht mehrere Routen und Verfahren
zur Verbindung der drei Domänen
mittels Standardpeptidkupplungsreaktionsbedingungen entwerten.
-
Der
Ausdruck "Standardpeptidkupplungsreaktionsbedingungen" meint die Kupplung
einer Carbonsäure
mit einem Amin mittels eines Säureaktivierungsmittels,
wie EDC, Dicyclohexylcarbodiimid und Benzotriazol-1-yloxytris(dimethylamino)phosphoniumhexafluorphosphat
in einem inerten Lösemittel,
wie DCM, in Gegenwart eines Katalysators, wie HOBT. Die Verwendung
von Schutzgruppen für
Amin- und Carbonsäuren
zur Erleichterung der gewünschten
Reaktion und zur Minimierung der unerwünschten Reaktionen ist gut
dokumentiert. Die Bedingungen, die zur Entfernung von Schutzgruppen
erforderlich sind, die vorkommen können, kann man in Greene et
al., Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., New
York, NY 1991 finden.
-
Cbz,
Boc und Fmoc Schutzgruppen werden ausgiebig bei der Synthese verwendet
und ihre Entfernungsbedingungen sind dem Fachmann gut bekannt. Beispielsweise
kann die Entfernung der Cbz Gruppen durch eine katalytische Hydrierung
mit Wasserstoff in Gegenwart eines Edelmetalls oder des sen Oxids,
wie Palladium auf Aktivkohle in einem protischen Lösemittel,
wie Ethanol, erreicht werden. In Fällen, bei denen die katalytische
Hydrierung durch das Vorkommen einer anderen potentiell reaktiven
Funktionalität
kontraindiziert ist, kann die Entfernung von Cbz auch durch die
Behandlung mit einer Lösung
aus Bromwasserstoff in Essigsäure
oder durch die Behandlung mit einem Gemisch aus TFA und Dimethylsulfid
erreicht werden. Die Entfernung der Boc Schutzgruppen wird in einem
Lösemittel,
wie Methylenchlorid, Methanol oder Ethylacetat mit einer starken
Säure,
wie TFA oder HCl oder Chlorwasserstoffgas ausgeführt.
-
Die
Verbindungen der Formel I können,
wenn sie als Diastereomerengemisch vorkommen, in die Diastereomerenpaare
der Enantiomeren durch fraktionierte Kristallisation aus einem geeigneten
Lösemittel,
wie Methanol, Ethylacetat oder einem Gemisch hiervon getrennt werden.
Das so erhaltene Enantiomerenpaar kann in einzelne Stereoisomere
durch herkömmliche
Mittel mittels einer optisch aktiven Säure als Auftrennungsmittel
getrennt werden. Alternativ dazu kann jedes Enantiomer einer Verbindung
der Formel I durch eine stereospezifische Synthese mittels optisch
reiner Ausgangsmaterialien oder Reagenzien mit bekannter Konfiguration
erhalten werden.
-
Die
erfindungsgemäßen Verbindungen
können
gemäß dem Verfahren
der folgenden Schemata und Beispiele hergestellt werden, die weitere
Details zur Herstellung der erfindungsgemäßen Verbindungen erläutern. Die
in den Beispielen dargestellten Verbindungen sollen aber nicht so
gesehen werden, dass sie die einzige Gruppe sind, die als vorliegende
Erfindung betrachtet wird.
-
In
den folgenden Schemata, Präparationen
und Beispielen haben verschiedene Reagenziensymbole und Abkürzungen
die folgenden Bedeutungen:
- BINAP
- 2,2'-Bis(diphenylphosphino)-1,1'-binapthyl
- Boc
- t-Butoxycarbonyl
- Cbz
- Benzyloxycarbonyl
- DCM
- Dichlormethan
- DEAD
- Diethylazodicarboxylat
- DIAD
- Diisopropylazodicarboxylat
- DIPEA
- Diisopropylethylamin
- DMAP
- 4-Dimethylaminopyridin
- DMF
- N,N-Dimethylformamid
- DMSO
- Dimethylsulfoxid
- Äqu.
- Äquivalente
- EDC
- 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid × HCl
- ESI-MS
- Elektronensprayionenmassenspektroskopie
- Et
- Ethyl
- EtOAc
- Ethylacetat
- FMOC
- 9-Fluorenylmethylcarbamat
- HATU
- O-(7-Azabenzotriazol-1-yl)-N,N,N',N'-tetramethyluroniumhexafluorphosphat
- HOAT
- 1-Hydroxy-7-azabenzotriazol
- HOBT
- 1-Hydroxybenzotriazolhydrat
- HPLC
- Hochleistungsflüssigchromatographie
- HRMS
- Hochauflösungsmassenspektroskopie
- h
- Stunde
- LRMS
- gering auflösende Massenspektrometrie
- Me
- Methyl
- Ms
- Methansulfonyl
- NMM
- 4-Methylmorpholin
- Pd2(dba)3
- Tris(dibenzylidenaceton)dipalladium-(0)
- Ph
- Phenyl
- Phe
- Phenylalanin
- Pr
- Propyl
- RT
- Raumtemperatur
- TBAF
- Tetrabutylammoniumfluorid
- TBS
- tert-Butyldimethylsilyl
- TFA
- Trifluroessigsäure
- TEA
- Triethylamin
- THF
- Tetrahydrofuran
- Tic
- 1,2,3,4-Tetrahydroisochinolin-3-carbonsäure
- TLC
- Dünnschichtchromatographie
-
Reaktionsschema
1: Kupplungsverfahren Verfahren
1
-
-
-
-
-
Im
Kupplungsverfahren 1 wird eine geeignete A Domäne (beispielsweise Piperazin)
an die B Domäne (beispielsweise
D-Boc-p-Cl-Phe-OH) in Gegenwart von EDC/HOBt gekuppelt, wonach eine
Boc-Schutzgruppenabspaltung
erfolgt. Die gekuppelte AB Verbindung wird dann an eine geeignete
C Domäne
gekuppelt, wonach eine Boc-Schutzgruppenabspaltung und Salzbildung
erfolgt. Alternativ dazu kann die schließliche Verbindung ohne Schutzgruppenabspaltungsschritt
erhalten werden, wenn die C Domäne
nicht mit der Boc Gruppe geschützt
ist.
-
Im
Kupplungsverfahren 2 wird eine geeignete A Domäne (beispielsweise Piperazin)
an eine geeignete BC Domäne
in Gegenwart von HATU gekuppelt, wonach eine Schutzgruppenabspaltung
der Boc Gruppe und eine Salzbildung erfolgt. Alternativ dazu kann
die schließliche
Verbindung ohne Schutzgruppenabspaltungsschritt erhalten werden,
wenn die BC Domäne
nicht mit der Boc Gruppe geschützt
ist.
-
Im
Kupplungsverfahren 3 wird eine geeignete AB Domäne an eine geeignete C Domäne in Gegenwart von
EDC/HOBt gekuppelt, wonach eine Abspaltung der Boc Gruppe und eine
Salzbildung erfolgt.
-
Im
Kupplungsverfahren 4 wird eine geeignete BC Domäne an eine geeignete A Domäne in Gegenwart von
EDC/HOBT gekuppelt, wonach eine Abspaltung der Boc Gruppe und eine
Salzbildung erfolgt. Alternativ dazu kann die schließliche Verbindung
ohne Schutzgruppenabspaltungsschritt erhalten werden, wenn die C Domäne nicht
mit der Boc Gruppe geschützt
ist.
-
Im
Kupplungsverfahren 5 wird eine geeignete AB Domäne an eine geeignete C Domäne in Gegenwart von
HATU gekuppelt, wonach eine Abspaltung der Boc Gruppe und eine Salzbildung
erfolgt.
-
Zur
Kupplung von A mit Boc-B kann EDC/HOAT, EDC/HOBT oder DCC/HOBT verwendet
werden.
-
Im
allgemeinen kann das Ausgangsmaterial des Boc-geschützten Piperazins
(A Domäne)
in Gegenwart von TFA/CH2Cl2,
HCl/EtOAc, HCl/Dioxan oder HCl in MeOH/Et2O
mit oder ohne Kationenfänger,
wie Dimethylsulfid (DMS) von der Schutzgruppe befreit werden, bevor
es dem Kupplungsverfahren unterzogen wird. Es kann vor dem Kupplungsverfahren
in die freie Base umgewandelt werden oder in manchen Fällen als
Salz verwendet werden.
-
Ein
geeignetes Lösemittel,
wie CH2Cl2, DMF,
THF oder ein Gemisch der obigen Lösemittel kann für das Kupplungsverfahren
verwendet werden. Geeignete Basen umfassen Triethylamin (TEA), Diisopropylethylamin
(DIPEA), N-Methylmorpholin, Collidin oder 2,6-Lutidin. Es ist keine
Base erforderlich, wenn EDC/HOBt verwendet wird.
-
Nachdem
die Umsetzung vollständig
ist, kann im allgemeinen das Reaktionsgemisch mit einem geeigneten
organischen Lösemittel,
wie EtOAc, CH2Cl2 oder
Et2O verdünnt werden, das dann mit geeigneten
wässrigen
Lösungen
gewaschen wird, wie Wasser, HCl, NaHSO4,
Bicarbonat, NaH2PO4,
Phosphatpuffer (pH 7), Kochsalzlösung
oder jede Kombination hiervon. Das Reaktionsgemisch kann konzentriert
werden und dann zwischen einem geeigneten organischen Lösemittel
und einer wässrigen
Lösung
aufgeteilt werden. Das Reaktionsgemisch kann dann konzentriert werden
und ohne wässriger
Aufarbeitung einer Chromatographie unterzogen werden.
-
Die
Schutzgruppe, wie Boc oder Cbz, Fmoc, CF3CO
und H2/Pd-C kann in Gegenwart von TFA/CH2Cl2, HCl/EtOAc,
HCl/Dioxan, HCl in MeOH/Et2O, NH3/MeOH oder TBAF mit oder ohne einem Kationenfänger, wie Thioanisol,
Ethanthiol und Dimethylsulfid (DMS) abgespalten werden. Die von
den Schutzgruppen befreiten Amine können als entstehendes Salz
verwendet werden oder werden durch Lösen in CH2Cl2 und Waschen mit wässrigem Bicarbonat oder wässrigem
NaOH in die freie Base umgewandelt. Die von den Schutzgruppen befreiten
Amine können
auch durch Ionenaustauschchromatographie in die freie Base umgewandelt
werden.
-
Die
Verbindungen der vorliegenden Erfindung können als Salze, wie als TFA-,
Hydrochlorid- oder Succinatsalze unter Verwendung von bekannten
Standardverfahren hergestellt werden.
-
Reaktionsschema zur Herstellung
der "A Domäne"
-
Die
A Domänen
der vorliegenden Erfindung können
im allgemeinen aus im Handel erhältlichen
Ausgangsmaterialien über
bekannte chemische Transformationen hergestellt werden. Die Herstellung
der A Domäne
der erfindungsgemäßen Verbindung
ist im folgenden Reaktionsschema erläutert. Reaktionsschemata
der "A Domäne" Reaktionsschema
2: Buchwald
X = Halogen und Q = Aryl.
-
Wie
in Reaktionsschema 2 gezeigt, kann die "A Domäne" der erfindungsgemäßen Verbindungen durch die
Kupplung des Halogen-substituierten Aryl 1 (X-Q) mit Piperazinen
2 in Gegenwart von Tris(dibenzylidenaceton)dipalladium (Pd
2(dba)
3), 1,1'-Bi[(2-Diphenylphosphinen)naphthalin]
(BINAP) und Natrium-t-butoxid (NaO-t-Bu) oder Cäsiumcarbonat (Cs
2CO
3) in einem organischen Lösemittel, wie Toluol bei einer
geeigneten Temperatur hergestellt werden. Detailliertere Beispiele
für die
Herstellung der A Domäne
sind im folgenden beschrieben. Reaktionsschema
3: SnAr
EWG = Elektronenziehende Gruppe
-
Wie
in Reaktionsschema 3 gezeigt, kann die "A Domäne" der erfindungsgemäßen Verbindungen durch Erhitzen
der geeignet substituierten Fluorarylverbindungen 4 und Piperazine
2 pur oder mit einem geeigneten Lösemittel und mit oder ohne
einer geeigneten Base hergestellt werden.
-
Reaktionsschema
4: SNAr, gefolgt von Buchwald
-
Wie
in Reaktionsschema 4 gezeigt, kann die "A Domäne" der erfindungsgemäßen Verbindungen durch Erhitzen
von 9-Brom-2-fluorbenzol 6 mit verschiedenen Alkoholen (R9-OH) in Gegenwart von NaH unter Bildung
von ortho-substituierten Brombenzolen 7 hergestellt werden, die
dann, wie oben in Reaktionsschema 4 gezeigt, Buchwald-Bedingungen
unterzogen werden.
-
Reaktionsschema 5:
-
Durch
Kupfer vermittelte O-Arylierung von 2-Bromphenol mit Arylborsäuren, gefolgt
von Buchwald.
-
-
A
steht für
Aryl oder Heteroaryl.
-
Wie
in Reaktionsschema 5 gezeigt, kann die "A Domäne" der erfindungsgemäßen Verbindungen durch Erhitzen
von 2-Bromphenol 9 mit verschiedenen Aryl- und Heteroarylboraten
(X-OH) in Gegenwart von Cu(OAc)
2 und Pyridin
unter Bildung von ortho-substituierten Brombenzolen 10 hergestellt
werden, die dann Buchwald-Bedingungen unterzogen werden können. Reaktionsschema
6: Benzylamine 6A.
Nitrilreduktion
A = SO
2R
9,
SO
2N(R
9)
2, C(O)R
9, C(O)OR
9, C(O)SR
9, C(O)N(R
9)
2 und dergleichen.
-
Wie
in Reaktionsschema 6A gezeigt, kann die "A Domäne" der erfindungsgemäßen Verbindungen durch die
Reduktion des Nitrils von (2-Cyanophenyl)piperazin 12 zum entsprechenden
Benzylamin 13 entweder mit NaBH
4 und TFA
oder H
2 und Raney Nickel hergestellt werden.
Benzylamin 13 kann in andere Benzylaminderivate 14 mittels verschiedener
dem Fachmann bekannter Verfahren umgewandelt werden. 6B.
Aus Benzylalkohol über
Mitsunobu oder über
Mesylat
A = saures Heteroaryl, Azid, Imid und dergleichen
B
= basisches Heteroaryl, Heterocyclyl und dergleichen
-
Wie
in Reaktionsschema 6B gezeigt, kann die "A Domäne" der erfindungsgemäßen Verbindungen durch die
Hydrolyse des Nitrils von (2-Cyanophenyl)piperazin 12 in die entsprechende
Carbonsäure
15 mit KOH, gefolgt von der Reduktion zum Benzylalkohol 16 mit BH
3-THF hergestellt werden. Der Benzylalkohol
16 kann in die Benzylamine 17 entweder mittels Mitsunobu-Bedingungen
oder durch Aktivierung des Alkohols als Mesylat, gefolgt durch eine
nukleophile Verdrängung
umgewandelt werden. Reaktionsschema
7: Derivate von 1-Boc-4-(2-aminophenyl)piperazin
A = SO
2R
9,
SO
2N(R
9)
2, C(O)R
9, C(O)OR
9, C(O)SR
9, C(O)N(R
9)
2 und dergleichen.
-
Wie
in Reaktionsschema 7 gezeigt, kann die "A Domäne" der erfindungsgemäßen Verbindungen aus 1-Boc-4-(2-Aminophenyl)piperazin
19 hergestellt werden, das aus 4-(2-Nitrophenyl)piperazin 18 durch
eine Boc-Schätzung,
gefolgt von einer Nitroreduktion hergestellt wird. 1-Boc-4-(2-Aminophenyl)piperazin
19 kann in andere Anilinderivate 20 mittels verschiedener dem Fachmann
bekannter Verfahren umgewandelt werden. Sulfonamide 21 können aus
1-Boc-4-(2-Aminophenyl)piperazin 19 durch die Umsetzung mit verschiedenen Sulfonylchloriden
hergestellt werden. Die entstehenden Sulfonamide 21 können dann
mit NaH oder K2CO3 in DMF
gefolgt von einer Alkylierung mit verschiedenen Alkylhalogeniden
(R9X) unter Bildung der alkylierten Sulfonamide
22 deprotoniert werden. 1-Boc-4-(2-Aminophenyl)piperazin kann auch mit
verschiedenen Säurechloriden
unter Bildung der Acetamide 23 acyliert werden. Die Acetamide 23
können
mit BH3-THF unter Bildung von Alkylaminen
24 reduziert werden, die in andere Aminderivate 25 mittels verschiedener
dem Fachmann bekannter Verfahren umgewandelt werden können.
-
Reaktionsschema
8: Derivate von 2-(N-Boc-Piperazin-1-yl)benzaldehyd
-
Wie
in Reaktionsschema 8 gezeigt kann die "A Domäne" der erfindungsgemäßen Verbindungen durch die
Reduktion des Nitrils von (2-Cyanophenyl)piperazin 12 zum entsprechenden
Aldehyd 26 mit DIBAL hergestellt werden. Das Aldehyd 26 kann zu
den Benzylaminen 27 durch reduktive Aminierung mit verschiedenen Aminen,
einschließlich
Stickstoff-enthaltenden Heterocyclen, umgewandelt werden. Diese
Benzylamine 27 können
in andere Aminderivate mittels verschiedener dem Fachmann bekannter
Verfahren umgewandelt werden.
-
Der
Aldehyd 26 kann mit verschiedenen Organolithiumreagenzien (einschließlich lithiierter
Aryl- und Heteroarylgruppen) unter Bildung der Alkohole 28 umgesetzt
werden. Der Alkohol kann unter Bildung der Ketone 29 oxidiert oder
durch Bartondesoxygenierung unter Bildung der Verbindung 30 entfernt
werden.
-
Reaktionsschema
9: Derivate von 1-Boc-4-(2-hydroxyphenyl)piperazin
-
Wie
in Reaktionsschema 9 gezeigt, kann die "A Domäne" der erfindungsgemäßen Verbindungen durch die
Behandlung von 1-Boc-(2-Hydroxyphenyl)piperazin 31 mit einer Base
und einem Alkylhalogenid (RX) hergestellt werden oder Mitsunobu-Bedingungen
mit R
9OH unter Bildung der ortho-substituierten
Arylpiperazine 32 unterzogen werden. Reaktionsschema
10: Derivate von 1-Boc-4-(2-Carboxyphenyl)piperazin
A = Heterocyclyl, N(R
9)
2, OR
9 oder SR
9 und dergleichen.
-
Wie
dies in Reaktionsschema 10 gezeigt ist, kann die "A Domäne" der erfindungsgemäßen Verbindungen
durch Hydrolyse des Nitrils von (2-Cyanophenyl)piperazin 12 zur
entsprechenden Carbonsäure
15 mit KOH, gefolgt von der Umwandlung in andere Carbonsäurederivate
33 mittels verschiedener dem Fachmann bekannter Verfahren hergestellt
werden.
-
Reaktionsschema
11: Tetrazole
-
Wie
in Reaktionsschema 11 gezeigt kann die "A Domäne" der erfindungsgemäßen Verbindungen durch die
Umsetzung des Nitrils von (2-Cyanophenyl)piperazin 12 mit Tributylzinnazid
unter Bildung der Tetrazole 34 hergestellt werden. Die Tetrazole
können
ferner in die Verbindung 35 mittels verschiedener dem Fachmann bekannter
Verfahren umgewandelt werden.
-
Die
vorliegende Erfindung liefert auch ein neues Verfahren zur Herstellung
bestimmter Zwischenprodukte und/oder erfindungsgemäßer Verbindungen,
wie dies in den Reaktionsschemata 12 bis 14 gezeigt ist.
-
-
Wie
in Reaktionsschema 12 gezeigt wird eine konvergente Synthese eines
Schlüsselzwischenproduktisoindolins
(5) über
eine Heck-Kupplung, gefolgt von einer reduktiven Aminierung, einer
Ringcyclisierung und einer Auftrennung entwickelt. Ebenfalls werden
alternative asymmetrische Ansätze
einschließlich
asymmetrsicher Michael-Addition und asymmetrischer Hydrierung entwickelt,
um die erfindungsgemäßen Verbindungen und/oder
Zwischenprodukte hiervon herzustellen.
-
Wie
dies in Reaktionsschema 12 gezeigt ist, können die Isoindolinverbindungen
der vorliegenden Erfindung aus 2-Halogenbenzaldehyd 1 oder einem
substituierten Analogon hiervon hergestellt werden. Das bevorzugte
Ausgangsmaterial ist 2-Brombenzaldehyd oder ein substituiertes Analogon
hiervon. Eine durch Pd vermittelte Heck-Kupplung von 2-Brombenzaldehyden
1 mit beispielsweise Methylacrylat liefert alpha, beta-ungesättigte Methylester
2, die einer reduktiven Aminierung unter Bildung von Aminen 3 unterzogen
werden (oder Carbamaten, worin R1 beispielsweise
für Boc
steht). Es sind verschiedene Heck-Kupplungsreagenzien und Bedingungen
zur Bewirkung der Kupplungsreaktion geeignet. Geeignete Katalysatoren
und Liganden umfassen Pd(OAc)2/PPh3, Pd(OAc)PPh3/BU4NBr, Pd(PPh3)2Cl2/CUI, Pd(OAc)2/P(O-Tol)3. Geeignete
Lösemittel oder
Lösemittelsysteme
für die
Heck-Kupplungsreaktion umfassen DMF, Toluol und Ethylacetat. Die
bevorzugtere Base ist Triethylamin.
-
Eine
reduktive Aminierung der Aldehydfunktionalität der Verbindung 2 zu Aminen
wird in guten Ausbeuten durch die Umsetzung mit Benzylamin oder
alpha-Methylbenzylamin unter sauren Bedingungen erreicht, wonach
eine in situ Reduktion der entstehenden Imine mit NaCNBH3 bei etwa pH 5 erfolgt. Andere Reduktionsmittel
umfassen Na(OAc)3BH und NaBH4/H
können
auch zur Bewirkung der Reduktion der entstehenden Imine verwendet
werden. Interessanterweise cyclisieren die entstehenden Amine unmittelbar
zu den Isoindolinverbindungen unter denselben Säurebedingungen wie für die Reduktion.
Die direkte Herstellung der Verbindung 4 kann auch durch die Verwendung
von BocNH2 anstelle von Benzylamin im reduktiven
Aminierungsschritt bewirkt werden. Das Screening der verschiedenen
Reduktionsmittel zeigt, dass die Kombination von Et3SiH
und TFA in CH3CN das bevorzugte Verfahren
zur Bewirkung der reduktiven Aminierung mittels BocNH2 darstellt.
-
Die
N-Boc-Isoindolincarbonsäure
5 kann auch aus der Verbindung 3 als Carbamat durch eine intramolekulare
Michael-Addition und Esterhydrolyse hergestellt werden. Die Auftrennung
der Isoindolincarbonsäuren 4
durch Kristallisation ergibt eine enantiomerenreine Verbindung 5.
-
Es
wurden auch 2 alternative asymmetrsche Ansätze zur Synthese der Isoindolincarbonsäure 5 entwickelt,
das heißt
asymmetrische Michael-Additionen und asymmetrische Hydrierung. Im
asymmetrischen Michael-Additionsansatz wird das alpha-Methylbenzylamin
als chirales Auxiliar verwendet, um die Enantioselektivität zu induzieren.
Im asymmetrischen Hydrierungsansatz kann die Verbindung 4' stereoselektiv in
die Verbindung 5 in Gegenwart von chiralen Liganden umgewandelt
werden.
-
Schließlich wird
die Kupplung der Isoindoline 5 mit dem Teil der "B Domäne", das heißt D-Cl-Phe, unter Bildung
der Verbindung 6 ("BC" Teil) durch Standardaminosäurekupplungsreaktionen
erreicht, wie beispielsweise durch die Verwendung von EDC oder EDCl
oder anderer geeigneter Aktivierungsmittel in Gegenwart von Dimethylaminopyridin
(DMAP). Das Produkt (6) wird dann mit einem Teil der "A Domäne" unter Bildung der MC4R
Zielagonistverbindung der Formel I durch die dem Fachmann bekannten
Kupplungsreaktionen gekuppelt.
-
Vorzugsweise
wird das Isoindol oder ein anderer Teil der "C Domäne" an das Stück der gekuppelten "AB Domäne" unter Bildung der
Verbindung der Formel I gekuppelt. Reaktionsschema
13:
M = Li
+, K
+,
Na
+ -
Wie
in Reaktionsschema 13 gezeigt, können
m-Tyrosinester oder Analoga, einschließlich substituierter Analoga
hiervon, durch die Bildung des Säurehalogenids
gefolgt von der nukleophilen Verdrängung des Halogenids durch
die Alkoxygruppe eines Alkohols verestert werden, das heißt Methanol
oder Ethanol. Wenn Thionylchlorid oder eine andere Halogenidquelle
verwendet wird, kann das Produkt als Säureadditionssalz (2) verwendet
werden. Der entstehende Ester (2) wird einer Pictet-Spengler-Reaktion
durch Erhitzen mit einem geeigneten Keton oder Aldehyd unter Rückflussbedingungen
unterzogen. Beispielsweise kann ein unsubstituiertes Isochinolinrückgrad (3)
zuerst durch die Verwendung von Formaldehyd in der Pictet-Spengler-Reaktion gebildet
werden. Andererseits kann ein Gem-substituiertes Isochinolin, worin
R11 für
Methyl steht, durch die Verwendung von Aceton als Ketonquelle und
Lösemittel
gebildet werden. Andere weniger reaktive Substituenten können als
R11 Gruppe zur Durchführung der vorliegenden Erfindung
substituiert werden.
-
Das
Isochinolinprodukt (3) kann vorzugsweise als Säureadditionssalz isoliert werden.
Wenn m-Tyrosin als
Ausgangsmaterial verwendet wird, wird die freie Hydroxylgruppe zuerst
durch Schützung/Aktivierung
mit einer guten Abgangsgruppe, wie beispielsweise Umsetzung mit
Trifluormethansulfonsäureanhydrid
oder Methansulfonsäure
unter Bildung des Triflats oder Mesylats in Gegenwart einer Base
entfernt. Das Triflat ist eine bevorzugte Gruppe, die zur Vorbereitung
der Verbindung (3) zur Desoxygenierung aufgrund des besonderen elektronenziehenden
Effekts des Trifluormethansubstituenten verwendet wird. Die Desoxygenierungsreaktion wird
durch Hydrierung bei Drücken
von etwa 50 psi ausgeführt.
Das Produkt (4) kann als Säureadditionssalz isoliert
werden. Das Produkt (4) kann unter basischen Bedingungen unter Bildung
des Säuresalzes
isoliert werden. Geeignete Basen für die obige Hydrolyse umfassen
wässriges
Natriumhydroxid, Kaliumhydroxid und Natriumlithiumhydroxid. Die
Umsetzung wird vorzugsweise in einem Gemisch aus wässrigen
und organischen Lösemitteln
ausgeführt.
Eine Exothermie während
der Zugabe der Base kann reguliert werden (das heißt auf weniger
als 35°C),
um eine Überhitzung
oder "durchgehende
Reaktionen" zu vermeiden.
Das Reaktionsprodukt kann durch eine wässrige Aufarbeitung isoliert
werden. Alternativ dazu kann das gesamte Gemisch konzentriert und
mit organischen Lösemitteln
unter Bildung des gewünschten
Produkts (6) nach der Kristallisation gewaschen werden.
-
Das
Produkt (6) wird dann mit einem Substrat der "B Domäne", wie beispielsweise 4-Chlor-D-phenylalanin umgesetzt,
wie dies vorher im Experimentalteil beschrieben ist. Das entstehende "BC Kombinationsprodukt" wird dann mit einem
Stück der "A Domäne" unter Bildung der
entsprechenden Verbindung der Formel I umgesetzt. Alternativ kann
das Produkt (6) mit einem Kombinationsprodukt der "AB Domänen" unter Bildung einer
Verbindung der Formel I umgesetzt werden.
-
Der
Fachmann erkennt, dass bestimmte Schutzgrupenanbringungen und Schutzgruppenabspaltungen
der Zwischenprodukte in Reaktionsschema 13 unter Bildung des Carbamats,
substituierten Amins oder freien Amins am Isochinolylstickstoff
möglich
sind und im Schutzumfang der Erfindung enthalten sind. Falls nichts
anderes angegeben ist, sind dem Fachmann Reagenzien und Verfahren
zur Bewirkung der hierin beschriebenen Reaktionen bekannt und können in
allgemeinen Referenztexten gefunden werden, wie in Advanced Organic
Chemistry von J. March, 5. Ausgabe, Wiley Interscience Publishers,
New York, NY und den hierin angegebenen Literaturangaben.
-
In
einem alternativen Verfahren kann das Isochinolinprodukt, das heißt die Verbindung
(3) oder (5) einschließlich
ihrer N-geschützten
Analoga durch die Umsetzung mit einem chiralen Trennmittel aufgetrennt
werden, wie beispielsweise L-Weinsäure, Dehydroabietylamin oder
andere in der Technik bekannte chirale Trennmittel.
-
Alternativ
dazu können
asymmetrische Analoga von Produkt (6) durch die Verwendung von asymmetrischen
Ausgangsmaterialien hergestellt werden. Beispielsweise kann L-Dopa
anstelle von m-Tyrosinester
in Reaktionen verwendet werden, die im wesentlichen zu denen ähnlich sind,
die in Reaktionsschema 13 und in den Beispielen beschrieben und
erläutert
sind, um das asymmetrische Analogon der Verbindung (6) zu erhalten.
-
Tetrahydroisochinolinessigsäurederivate
können
hergestellt und verwendet werden, wie dies im folgenden in Reaktionsschema
14 beschrieben ist:
-
-
Wie
in Reaktionsschema 14 gezeigt, wird eine Verbindung der Formel 10a,
worin X für
Halogen steht, vorzugsweise für
Brom oder Chlor und R und R11 wie vorher
definiert sind, und die im Handel erhältlich ist oder aus im Handel
erhältlichen
Ausgangsmaterialien hergestellt werden können, mit Cyanomethylethylacetat
unter Bildung einer Verbindung der Formel 10b umgesetzt. Die Verbindung
der Formel 10b kann wie die Verbindung 10c mit einer geeigneten
Schutzgruppe (Pg) geschützt
werden und dann Hydrierungsbedingungen, einschließlich beispielsweise
einer asymmetrischen Hydrierung unter Bildung einer Verbindung der
Formel 10d unterzogen werden, die chiral sein kann (in Abhängigkeit
der Hydrierungsbedingungen, das heißt asymmetrischer gegenüber nicht-asymmetrischer
Hydrierung). Die Verbindung der Formel 10d oder ein Stereoisomer
hiervon wird mit einem Stück
der B Domäne,
wie beispielsweise 4-Chlor-D-Phe unter Bildung eines BC Stücks (10e) umgesetzt.
Die Verbindung der Formel 10e wird dann mit einem Stück der A
Domäne
unter Bildung einer Verbindung der Formel I umgesetzt. Die Details
der spezifischen Reaktionsschritte sind zu den Reaktionen, die hierin
und im Beispielteil beschrieben sind, ähnlich oder analog. Ferner
ist dem Fachmann bekannt, dass solche Zwischenproduktreaktionen,
wie Hydrolyse und Schutzgruppenabspaltung erforderlich sein können, um optimale
Ausbeuten bei bestimmten Schritten des gezeigten Schemas zu erreichen.
Dem Fachmann sind auch weitere Manipulationen bekannt, wie N-Alkylierung
oder N-Acylierung und Alkylierungen des Benzolrings, um andere Verbindungen
der Formel I zu erhalten.
-
Im
Folgenden werden die detaillierten Beispiele der Herstellung einer
A Domäne
beschrieben.
-
Präparation
1A (Buchwald mittels NaOtBu) (3R)-3-Methyl-(2-methylthiophenyl)piperazin
-
Es
werden 2-Bromthioanisol (300 mg, 1,48 mmol), (R)-2-Methylpiperazin
(185 mg, 1,85 mmol), Pd2(dba)3 (32
mg, 0,35 mmol), BINAP (41 mg, 0,66 mmol), Natrium-t-butoxid (200
mg, 2,08 mmol) und wasserfreies Toluol (3 ml) in einem 15 ml Rundbodenkolben
vereinigt. Die Atmosphäre
in dem Kolben wird evakuiert und mit Stickstoff (3×) gespült. Das
Gemisch wird in ein Ölbad
abgesenkt und auf 100°C
erhitzt. Nach dem Erhitzen für
etwa 1,2 Stunden wird das Gemisch gekühlt, mit Ethylacetat (100 ml)
verdünnt,
durch Celite filtriert und zu einem rohen Öl (285 mg) konzentriert. Das Öl wird auf
eine Kationenaustauschsäule
gegeben und die Säule
wird mit Methanol (100 ml) und dann mit 2 M Ammoniak/Methanol (100
ml) gewaschen. Die basische Methanollösung wird zu einem Öl (250 mg)
konzentriert. Das Öl
wird weiter durch Blitzchromatographie mittels 19:1 Dichlormethan:
0,5 M Ammoniak/Methanol als Eluent unter Bildung des schließlichen
Produkts (160 mg, 58%) als Öl
gereinigt.
LRMS (ESI+): 223,0 (M + 1).
-
Präparation
2A (Buchwald mittels CsCO
3) 4-(2-Diethylcarbamoylphenyl)piperazin
-
Es
werden HOBT (2,72 g, 10,08 mmol), DIPEA (3,52 ml, 20,16 mmol), 2-Brombenzoesäure (4,08
g, 10,08 mmol) und Diethylamin (2,08 ml, 10,08 mmol) in DCM (100
ml) gelöst
und bei RT für
etwa 30 Minuten gerührt.
EDCl (3,86 g, 10,08 mmol) wird zugegeben und das Gemisch wird bei
RT für
etwa 16 Stunden gerührt. Die
Reaktion wird zu einem Öl
konzentriert und das Öl
wird mittels Säulenchromatographie
unter Bildung von 2-Brom-N,N-diethylbenzamid (3,35 g, 68%) als gelbes Öl gereinigt.
-
Piperazin
(489 mg, 4,8 mmol), 2-Brom-N,N-diethylbenzamid (1 g, 3,95 mmol),
Pd2(dba)3 (235 mg,
0,2 mmol), BINAP (442 mg, 0,6 mmol) und Cäsiumcarbonat (3 g, 5,53 mmol)
werden zusammen in Toluol (20 ml) gemischt. Das Gemisch wird entgast
und für
etwa 72 Stunden auf 100°C
erhitzt. Das Gemisch wird mit Ether (100 ml) verdünnt und über Celite
filtriert. Das Filtrat wird konzentriert und dann einer Chromatographie
auf Silicagel unter Bildung der Titelverbindung (480 mg, 47%) als
braunes Öl
unterzogen.
LRMS (ESI+): 262,2 (M + 1).
-
Präparation
3A 1-Boc-4-(2-Piperazin-1-yl-benzoyl)piperazin
-
Boc
geschütztes
Piperazin (849 mg, 4,56 mmol) wird in DCM (20 ml) gelöst und Triethylamin
(2,54 ml, 18,2 mmol) wird zugegeben. Zu der gerührten Lösung wird ortho-Brombenzoylchlorid
(2 g, 9,11 mmol) mittels einer Spritze unter Stickstoff gegeben.
Das System wird für
etwa 12 Stunden bei RT gerührt.
Die Reaktion wird mit Wasser gewaschen, getrocknet, filtriert und
konzentriert. Der Rückstand
wird unter Bildung von 1-Boc-4-(2-Brom)piperazin (1,48 g, 8,85 mmol)
als weißer
Schaum einer Chromatographie auf Silicagel unterzogen. 1-Boc-4-(2-Brombenzoyl)piperazin
wird auf ähnliche
Weise zu Präparation
1A an Piperazin gekuppelt.
LRMS (ESI+): 375,2 (M + 1)
-
Präparation
4A 1-(2-Methoxy-5-nitrophenyl)piperazin
-
1-(2-Methoxy-5-nitrophenyl)piperazin
wird auf ähnliche
Weise zu Präparation
1A hergestellt, mit der Ausnahme, dass Piperazin an 2-Brom-1-methoxy-4-nitrobenzol
gekuppelt wird.
LRMS (ESI+): 238,4 (M + 1).
-
Präparation
5A 1-(2-Methyl-6-nitrophenyl)piperazin
-
1-(2-Methyl-6-nitrophenyl)piperazin
wird auf ähnliche
Weise zu Präparation
1A hergestellt, mit der Ausnahme, dass Piperazin an 2-Brom-1-methyl-3-nitrobenzol
gekuppelt wird.
LRMS (ESI+): 222,4 (M + 1).
-
Präparation
6A 1-(2-Isopropoxyphenyl)piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
1A hergestellt, mit der Ausnahme, dass Piperazin an 1-Brom-2-isopropoxybenzol
gekuppelt wird.
LRMS (ESI+): 221,4 (M + 1).
-
Präparation
7A 1-(2-Isopropylphenyl)piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
1A hergestellt, mit der Ausnahme, dass Piperazin an 1-Brom-2-isopropylbenzol
gekuppelt wird.
LRMS (ESI+): 205,4 (M + 1).
-
Präparation
8A 1-(2-Isopropyl-5-methylphenyl)piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
1A hergestellt, mit der Ausnahme, dass Piperazin an 1-Brom-S-methyl-2-isopropylbenzol
gekuppelt wird.
1H NMR (CDCl3): δ 7,05–7,00 (m,
1H), 6,85–6,75
(m, 2H), 3,95 (s, 1H), 3,10–3,00
(m, 4H), 2,95–2,90
(m, 4H), 2,30 (s, 3H), 1,25–1,20
(m, 6H).
-
Präparation
9A 1-(2-Cyclohexylphenyl)piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
1A hergestellt, mit der Ausnahme, dass Piperazin an 1-Brom-2-cyclohexylbenzol
gekuppelt wird.
LRMS (ESI+): 245,1 (M + 1).
-
Präparation
10A 1-[2-(1,1-Difluorethyl)phenyl]piperazin
-
Eine
Lösung
aus Diethylaminoschwefeltrifluorid (560 mg, 3,47 mmol, 3 Äquivalente)
und 2-Bromacetophenon
(230 mg, 1,16 mmol, 1,0 Äquivalente)
wird für
etwa 72 Stunden auf 40°C
erhitzt. Die Lösung
wird mit CH2Cl2 verdünnt und
mit gesättigtem
Natriumbicarbonat, Wasser und Kochsalzlösung gewaschen, über Na2SO4 getrocknet,
filtriert und konzentriert. Eine Reinigung durch Blitzchromatographie
(35 g SiO2, linearer Gradient 0–10% Ethylacetat/Hexan,
30 ml/Minute, über
30 Minuten) ergibt etwa 125 mg (0,57 mmol, 49%) an 2-(1,1-Difluorethyl)-1-brombenzol.
GC/MS
(EI): 220 (M + H).
-
2-(1,1-Difluorethyl)-1-brombenzol
wird auf ähnliche
Weise zu Präparation
1A an Piperazin gekuppelt.
LRMS (ESI+): 227,2 (M + 1).
-
Präparation
11A (S)-1-{2-[1-(tert-Butyldimethylsilanyloxy)ethyl]phenyl}piperazin
-
Zu
einem 25 ml Kolben, der (S)-(–)-2-Brom-alpha-methylbenzylalkohol
(200 mg, 1,0 mmol), tert-Butyldimethylsilylchlorid
(165 mg, 1,1 mmol) und Imidazol (203 mg, 3,0 mmol) enthält und mit
Stickstoff gewaschen ist werden 5 ml Dimethylformamid gegeben. Nach
dem Rühren über Nacht
wird das Gemisch mit gesättigtem Natriumbicarbonat
gestoppt, mit Ethylacetat verdünnt,
mit NaH2PO4, gesättigtem
Natriumbicarbonat, Wasser und Kochsalzlösung gewaschen, getrocknet
(Na2SO4), filtriert
und konzentriert. Eine Reinigung durch Blitzchromatographie (10
g SiO2, linearer Gradient 0–10% Ethylacetat/Hexan,
30 ml/Minute über
30 Minuten) ergibt etwa 260 mg (0,82 mmol, 82%) an (5)-[1-(2-Bromphenyl)ethoxy]-tert-butyldimethylsilan
als farbloses Öl.
GC/MS
(EI): 315 (M).
-
Das
(5)-[1-(2-Bromphenyl)ethoxy]-tert-butyldimethylsilan wird auf ähnliche
Weise zu Präparation
1A an Piperazin gekuppelt.
LRMS (ESI+): 321,5 (M + 1).
-
Präparation
12 A (R)-1-{2-[1-(tert-Butyldimethylsilanyloxy)ethyl]phenyl}piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
11A hergestellt, mit der Ausnahme, dass (R)-(–)-2-Brom-α-methylbenzylalkohol verwendet
wird.
LRMS (ESI+): 321,3 (M + 1).
-
Präparation
13 A (2R)-3-Ethyl-1-(2-methylthiophenyl)piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
1A hergestellt.
LRMS (ESI+): 237,1 (M + 1).
-
Präparation
14 A (2S)-3-Methyl-1-(2-methylthiophenyl)piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
1A hergestellt.
-
Präparation
15 A 1-(2-Ethylphenyl)piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
1A hergestellt.
LRMS (ESI+): 191,2 (M + 1).
-
Präparation
16A (2R)-2-Methyl-1-(2-methylthiophenyl)piperazin
-
(2R)-4-Benzyl-2-methyl-1-(2-methylthiophenyl)piperazin
wird mit 26% Ausbeute aus ortho-Bromthioanisol
und (R)-3-Methyl-1-benzylpiperazin auf ähnliche Weise zu Präparation
1A hergestellt.
LRMS (ESI+): 223,2 (M + 1)
-
(2R)-4-Benzyl-2-methyl-1-(2-thiomethylphenyl)piperazin
(24 mg, 0,077 mmol) wird in 1,2-Dichlorethan (4
ml) gelöst
und in einem Eisbad gekühlt.
Zu der gekühlten
Lösung
wird 1-Chlorethylchlorformiat
(38 μl,
50 mg, 0,35 mmol) in einer Portion gegeben. Die Lösung wird
mit einer Stickstoffatmosphäre
bedeckt und dann auf 50°C
erhitzt. Nach dem Rühren
bei 50°C
für etwa
1,25 Stunden wird die Lösung
unter verringertem Druck konzentriert und dann in Methanol (6 ml)
gelöst.
Die methanolische Lösung
wird mit einer Stickstoffatmosphäre bedeckt
und kann über
Nacht bei RT rühren.
Die Lösung
wird unter Bildung von etwa 21 mg eines rohen Öls konzentriert. Eine Blitzchromatographie
(10% 0,5 M NH3/Methanol in DCM als Eluent)
ergibt die schließliche Titelverbindung
(14 mg, 82%).
LRMS (ESI+): 223,2 (M + 1).
-
Präparation
17A (2S)-2-Methyl-1-(2-methylthiophenyl)piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
16A hergestellt.
LRMS (ESI+): 223,2 (M + 1).
-
Präparation
18A 1-[2-(2-Methylpropan-1-sulfonyl)phenyl]piperazin
-
Zu
einer Lösung
aus 2-Brombenzolthiol (10,0 g, 52,8 mmol, 1,0 Äquivalente) in DMF (250 ml)
werden K2CO3 (17,5
g, 126,7 mmol, 2,4 Äquivalente)
und Isobutyliodid (7,3 ml, 63,36 mmol, 1,2 Äquivalente) gegeben. Die Reaktion
wird auf etwa 40°C
erwärmt
und über
Nacht gerührt.
Das Gemisch wird mit EtOAc (300 ml) verdünnt und mit Wasser (100 ml)
und Kochsalzlösung
(100 ml) gewaschen. Die organische Phase wird mit EtOAc (2×) extrahiert.
Die vereinigten organischen Extrakte werden getrocknet (Na2SO4), filtriert
und unter Bildung von 1-Brom-2-isobutylsulfanylbenzol (12,94 g,
52,8 mmol, 100%) konzentriert, das im nächsten Schritt ohne weitere
Reinigung verwendet wird.
GCMS (EI): 244,0.
-
Zu
einer Lösung
aus 1-Brom-2-isobutylsulfanylbenzol (8,0 g, 32,6 mmol, 1,0 Äquivalente)
in DCM (100 ml) bei 0°C
werden CaCO3 (13,05 g, 130,4 mmol, 4,0 Äquivalente)
und MCPBA (28,1 g, 81,5 mmol, 2,5 Äquivalente) gegeben. Das Gemisch
wird für
etwa 30 Minuten gerührt
und durch ein Kissen aus Celite filtriert. Die Lösung wird mit Natriumbisulfit
(2×) und
5 N NaOH (2×)
gewaschen. Die organische Phase wird getrocknet (Na2SO4), filtriert und konzentriert. Eine Reinigung
durch Blitzchromatographie (250 g SiO2,
linearer Gradient, 40 ml/min, 10%–40% EtOAc/Hexan für etwa 33
Minuten) ergibt 1-Brom-2-(2-methylpropan-1-sulfonyl)benzol (7,4
g, 26,6 mmol, 82%).
GCMS (EI): 276,0.
-
1-Brom-2-(2-methylpropan-1-sulfonyl)benzol
wird auf ähnliche
Weise zu Präparation
1A an Piperazin gekuppelt.
LRMS (ESI+): 283,06 (M + 1).
-
Präparation
19A (SNAr) 1-(2-Aminosulfonylphenyl)piperazin
-
Zu
einem 50 ml fassenden Kolben, der 2-Fluorbenzolsulfonamid (200 mg,
1,14 mmol, 1 Äquivalent) und
Piperazin (245 mg, 2,84 mmol, 2,5 Äquivalente) enthält, werden
20 ml Dioxan gegeben. Die Lösung
wird für
etwa 4 Stunden auf 100°C
erhitzt. Weiteres Piperazin (200 mg, 2,32 mmol, 2 Äquivalente)
wird zugegeben und die Lösung
wird für
weitere 72 Stunden auf 100°C
erhitzt. Die Lösung
wird zu einem Öl
konzentriert und in 30 ml an 0,1 M pH 7,0 Phosphatpuffer gelöst. Die
wässrige
Lösung
wird mit CH2Cl2 (3 × 30 ml)
extrahiert. Die vereinigten organischen Extrakte werden über Na2SO4 getrocknet und
unter Bildung von etwa 275 mg (1,14 mmol, 100%) der Titelverbindung
konzentriert.
LRMS (ESI+): 242,1 (M + H).
-
Präparation
20A 1-Boc-4-(3-Chlor-2-cyanophenyl)piperazin
-
Zu
einer Lösung
aus N-Boc-Piperazin (2,02 g, 11,0 mmol) in DMSO (20 ml) wird 2-Fluor-6-chlorbenzonitril
(1,55 g, 10 mmol) und Kaliumcarbonat (1,52 g, 11 mmol) gegeben.
Das Gemisch wird bei 80°C
für etwa 48
Stunden gerührt.
Das Gemisch wird auf RT gekühlt
und mit Diethylether (200 ml) verdünnt. Die Lösung wird mit 1 N HCl (2 × 20 ml)
gewaschen, H2O (3 × 20 ml) und Kochsalzlösung (20
ml) werden zugegeben und dann über
Natriumsulfat getrocknet und zu einem gelben Öl konzentriert. Eine Reinigung
durch Blitzchromatographie (4:1 Hexan/Ethylacetat) ergibt die Titelverbindung
(2,5 g, 86%) als farbloses Öl.
1H NMR (CDCl3) δ 7,40–7,50 (m,
1H), 7,10–7,20
(m, 1H), 6,80–6,90
(m, 1H), 3,70 (s, 4H), 3,20 (s, 4H), 1,48 (s, 9H).
TLC (SiO2): 0,48 (4:1 Hexan/Ethylacetat).
-
Präparation
21A 1-Boc-4-(3-Chlor-2-dimethylaminomethylphenyl)piperazin
-
Natriumborhydrid
(1,2 g, 31,4 mmol) wird in THF (20 ml) gelöst und TFA (2,42 ml, 31,4 mmol)
in THF (20 ml) wird tropfenweise bei 0°C zugegeben und die Reaktion
wird für
etwa 30 Minuten gerührt. 1-Boc-4-(3-Chlor-2-cyanophenyl)piperazin
(2,0 g, 6,3 mmol) wird in THF (20 ml) gelöst und tropfenweise zu einer
Lösung
bei 0°C
gegeben und die Reaktion wird für
etwa 24 Stunden gerührt.
Die Reaktion wird vorsichtig mit H2O gestoppt
und Ethylacetat (200 ml) wird zugegeben. Das Gemisch wird mit H2O (3 × 25
ml) und Kochsalzlösung
(25 ml) gewaschen und über
MgSO4 getrocknet. Die Lösemittel werden im Vakuum entfernt
und das rohe Reaktionsgemisch wird in Acetonitril (7 ml) gelöst. Formalin
(1,6 ml, 59,2 mmol) wird gefolgt von Natriumcyanoborhydrid (0,26
g, 7,4 mmol) bei 0°C
zugegeben. Die Reaktion wird auf RT erwärmt und für etwa eine Stunde gerührt. Die
Reaktion wird mit H2O gestoppt und Ethylacetat
(100 ml) wird zugegeben. Die Lösung wird
mit gesättigtem
NaHCO3 (2 × 10 ml) gewaschen und über MgSO4 getrocknet. Eine Reinigung durch Silicagelchromatographie
(1:1 Hexan/Ethylacetat) ergibt die Titelverbindung als gelbes Öl (180 mg,
13%).
1H NMR (CDCl3): δ 7,10–7,15 (m,
2H), 6,92–6,98
(m, 1H), 3,68 (s, 2H), 3,50–3,60
(m, 4H), 2,90–2,97
(m, 4H), 2,25 (s, 6H), 1,48 (s, 9H).
TLC (SiO2):
0,28 (1:1 Hexan/Ethylacetat)
-
Präparation
22A 1-Boc-4-(2-cyanophenyl)-[1,4]diazepan
-
Zu
einer Lösung
aus 1-Boc-Homopiperazin (2,18 g, 11,0 mmol) in DMSO (20 ml) werden
2-Fluorbenzonitril
(1,21 g, 1,08 ml, 10 mmol) und Kaliumcarbonat (1,52 g, 11 mmol)
gegeben. Das Gemisch wird bei 80°C für etwa 48
Stunden gerührt.
Das Gemisch wird auf RT gekühlt
und mit Diethylether (200 ml) verdünnt. Die Lösung wird mit 1 N HCl (2 × 20 ml),
H2O (3 × 20
ml) und Kochsalzlösung
(20 ml) gewaschen, über
Natriumsulfat getrocknet und zu einem gelben Öl konzentriert. Eine Reinigung
durch Blitzchromatographie (3:1 Hexan/Ethylacetat) ergibt die Titelverbindung
(1,1 g, 36%) als farbloses Öl.
1H NMR (CDCl3): δ 7,49 (dd,
J = 6,7, 1,7 Hz, 1H), 7,38 (td, J = 7,3, 1,7 Hz, 1H), 6,92 (d, J
= 8,5 Hz, 1H), 6,83 (t, J = 7,4 Hz, 1H), 3,63–3,66 (m, 2H), 3,46–3,57 (m,
6H), 2,01–2,10
(m, 2H), 1,40–1,45
(m, 9H).
TLC (SiO2): Rf =
0,38 (3:1 Hexan/Ethylacetat).
-
Präparation
23A 1-Boc-4-(2-Dimethylaminomethylphenyl)-[1,4]diazepan
-
Eine
Lösung
aus 1-Boc-4-(2-Cyanophenyl)-[1,4]diazepan (600 mg, 2,0 mmol) und
Raney Nickel (50% Dispersion in H2O, 1 ml)
in Methanol (50 ml) wird unter Wasserstoff (1 atm) für etwa 16
Stunden gerührt.
Formalin (2 ml) wird zugegeben und die Lösung wird für weitere 24 Stunden gerührt. Das
Gemisch wird durch Celite filtriert. Der Filterkuchen wird mit Methanol
(100 ml) gewaschen und das Filtrat wird zu einem klaren Öl konzentriert.
Eine Reinigung durch Blitzchromatographie (1% Methanol/Ethylacetat)
ergibt die Titelverbindung (285 mg, 55%) als farbloses Öl.
1H NMR (CDCl3): δ 7,39 (d,
J = 7,4 Hz, 1H), 7,19 (t, J = 7,4 Hz, 1H), 7,03–7,10 (m, 2H), 3,53–3,64 (m,
4H), 3,52 (s, 2H), 3,04–3,08
(m, 4H), 2,25 (s, 6H), 1,88–1,94
(m, 2H), 1,49 (s, 9H).
TLC (SiO2):
Rf = 0,40 (Ethylacetat).
-
Präparation
24A (SNAr dann Buchwald) 1-(2-Cyclohexyloxyphenyl)piperazin
-
NaH
(8,4 g, 210 mmol, 60% in Mineralöl)
wird in DMF (40 ml) aufgeschlämmt
und auf etwa 65°C
erhitzt. Zu der Aufschlämmung
wird Cyclohexanol (7 g, 69,9 mmol) gelöst in DMF (50 ml) gegeben.
Das Gemisch wird bei 65°C
für etwa
1 Stunde gerührt.
Ortho-Fluorbrombenzol (9,2 ml, 83,9 mmol) wird tropfenweise in DMF (10
ml) zugegeben und das Gemisch wird bei 65°C für etwa 16 Stunden gerührt und
mit Wasser gestoppt und mit DCM verdünnt. Das Gemisch wird zu einem öligen Feststoff
konzentriert und zwischen Wasser und 1/1 EtOAc/Hexan extrahiert.
Die organische Phase wird getrocknet, filtriert und konzentriert.
Eine Chromatographie auf Silicagel (EtOAc/Hexan) ergibt 1-Brom-2-cyclohydroxybenzol
(6,13 g, 34%) als gelbes Öl. 1-Brom-2-cyclohexyloxybenzol
wird mittels der in Präparation
1A beschriebenen Buchwald-Chemie an das Piperazin gekuppelt.
LRMS
(ESI+): 261,1 (M + 1).
-
Präparation
25A 1-(2-Cycloheptyloxyphenyl)piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
24A hergestellt, mit der Ausnahme, dass Cycloheptanol verwendet
wird.
LRMS (ESI+): 275,2 (M + 1).
-
Präparation
26A 1-[2-(3,3-Dimethylcyclohexyloxyphenyl)]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
24A hergestellt, mit der Ausnahme, dass 3,3-Dimethylcyclohexanol
verwendet wird.
LRMS (ESI+): 289,2 (M + 1).
-
Präparation
27A 1-(2-Cyclopentyloxyphenyl)piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
24A hergestellt, mit der Ausnahme, dass Cyclopentanol verwendet
wird.
LRMS (ESI+): 247,1 (M + 1).
-
Präparation
28A 1-[2-(Tetrahydrothiopyran-3-yloxy)phenyl]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
24A hergestellt, mit der Ausnahme, dass Tetrahydrothiopyran-3-ol
verwendet wird.
LRMS (ESI+): 279,2 (M + 1).
-
Präparation
29A 1-(2-(Tetrahydropyran-3-yloxy)phenyl]piperazin
-
3-Hydroxytetrahydropyran
wird gemäß Herbert
C. Brown, J. V. N. Vara Prasad und Sheng-Hsu Zee, J. Org. Chem.
50 (10), 1985, 1582–1589,
hergestellt. Die Verbindung wird mit ortho-Fluorbrombenzol gemäß der Buchwald
Kupplung auf ähnliche
Weise zu Präparation
24A unter Bildung der Titelverbindung hergestellt.
LRMS (ESI+):
263,1 (M + 1).
-
Präparation
30A 1-[2-(1,1-Dioxohexahydro-1λ
6-thiopyran-4-yloxy)phenyl]piperazin
-
Ortho-Fluorbrombenzol
wird mit Tetrahydrothiopyran-3-ol unter Bildung von 4-(2-Bromphenoxy)tetrahydrothiopyran
auf ähnliche
Weise zu Präparation
24A hergestellt. 4-(2-Bromphenoxy)tetrahydrothiopyran (1,94 g, 7,10
mmol) wird in DCM (70 ml) gegeben und Calciumcarbonat (2,84 g, 28,41
mmol) wird zugegeben. Zu diesem auf 0°C gekühlten Gemisch in einem Eisbad
wird meta-Chlorperoxybenzoesäure
(6,13 g, 17,75 mmol 50%) portionsweise gegeben, während die
Temperatur überwacht
wird. Das Gemisch kann sich auf RT erwärmen und wird für etwa 15
Minuten gerührt.
Das Gemisch wird über
Celite filtriert und mit Natriumbisulfitlösung (2 × 250 ml) und Natriumbicarbonat
(2 × 250
ml) gewaschen. Das Gemisch wird dann zu einem Öl konzentriert. Eine Chromatographie
(EtOAc/Hexan) ergibt 4-(2-Bromphenoxy)tetrahydrothiopyran-1,1-dioxid
(2,2 g, quantitativ) als gelben Feststoff. 4-(2-Bromphenoxy)tetrahydrothiopyran-1,1-dioxid
wird mittels der in Präparation
1A beschriebenen Buchwald Chemie unter Bildung der Titelverbindung
an das Piperazin gekuppelt.
LRMS (ESI+): 311,1 (M + 1).
-
Präparation
31A (o-Arylierung
des 2-Bromphenols gefolgt von Buchwald) 1-[2-(Pyridin-3-yloxy)phenyl]piperazin
-
2-Bromphenol
(355 mg, 2,05 mmol), 3-Pyridylborsäure (500 mg, 4,1 mmol), Kupferacetat
(745 mg, 4,1 mmol) und Pyridin (3,3 ml, 41 mmol) werden zu Dichlormethan
(41 ml) gegeben und für
etwa 48 Stunden unter Luft gerührt.
Die Reaktion wird mit Wasser (50 ml) verdünnt und die Phasen werden getrennt.
Die organische Phase wird mit 5 N NaOH gewaschen. Die organische
Phase wird konzentriert und auf Silicagel (MeOH/Dichlormethan) unter
Bildung von 3-(2-Bromphenoxy)pyridin (30 mg, 6%) als gelbes Öl chromatographiert.
MS
gefunden 249,1 M + 1.
-
3-(2-Bromphenoxy)pyridin
wird mittels der in Präparation
1A beschriebenen Buchwald Chemie unter Bildung der Titelverbindung
an das Piperazin gekuppelt.
LRMS (ESI+): 256,1 (M + 1).
-
Präparation
32A 1-(2-Phenoxyphenyl)piperazin
-
Ein
Gemisch aus Phenylborsäure
(5,12 g, 42 mmol), 2-Bromphenol (3,55 g, 21 mmol), Cu(OAc)2 (7,63 g, 42 mmol), Pyridin (8 ml, 103 mmol)
und 4 Å Molekularsiebe
(2,1 g) in CH2Cl2 wird
bei RT über Nacht
gerührt. Das
Gemisch wird mit CH2Cl2 verdünnt, durch
Celite filtriert, mit 1 M NaOH und Kochsalzlösung gewaschen und getrocknet.
Eine Entfernung des Lösemittels
ergibt 1-Brom-2-phenoxybenzolkristalle (1,40 g, 27%).
LRMS
(ESI+): 248 (M + 1).
-
1-Brom-2-phenoxybenzol
wird mittels der in Präparation
1A beschriebenen Buchwald Chemie unter Bildung der Titelverbindung
an das Piperazin gekuppelt.
LRMS (ESI+) 255 (M + 1).
-
Präparation
33A 1-(2-m-Tolyloxyphenyl)piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
32A hergestellt, mit der Ausnahme, dass 3-Methylphenylborsäure verwendet
wird.
LRMS (ESI+): 269 (M + 1).
-
Präparation
34A 1-(2-p-Tolyloxyphenyl)piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
32A hergestellt, mit der Ausnahme, dass 4-Methylphenylborsäure verwendet
wird.
LRMS (ESI+): 269 (M + 1).
-
Präparation
35A 1-[2-(3-Chlorphenoxy)phenyl]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
32A hergestellt, mit der Ausnahme, dass 3-Chlorphenylborsäure verwendet
wird.
LRMS (ESI+): 289 (M + 1).
-
Präparation
36A 1-[2-(3-Methoxyphenoxy)phenyl]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
32A hergestellt, mit der Ausnahme, dass 3-Methoxyphenylborsäure verwendet
wird.
LRMS (ESI+): 285 (M + 1).
-
Präparation
37A (Benzylamin aus Nitrilreduktion) 1-Boc-4-(2-Aminomethylphenyl)piperazin
-
Zu
einer Lösung
aus (2-Cyanophenyl)piperazin (2,4 g, 12,78 mmol) in TNF und H2O (25 ml, 1:1) wird K2CO3 (3,9 g, 28,12 mmol) gegeben. Die Lösung kann
für etwa
10 Minuten bei RT rühren.
Boc-Anhydrid (3,1 g,
14,06 mmol) wird dann zugegeben und die Reaktion kann für 1 h rühren. Das
Reaktionsgemisch wird mit EtOAc (100 ml) verdünnt und mit gesättigtem
NaHCO3 (100 ml) und Kochsalzlösung (100
ml) gewaschen. Die organische Phase wird unter Bildung von 3,2 g
an 1-Boc-4-(2-Cyanophenyl)piperazin (88%) zur Trockne konzentriert.
Zu einer Lösung
aus Natriumborhydrid (2,1 g, 56,03 mmol) in THF (25 ml) bei 0°C wird TFA
(4,3 ml, 56,03 mmol) tropfenweise gegeben. 1-Boc-4-(2-Cyanophenyl)piperazin
(3,2 g, 11,21 mmol) wird dann langsam bei RT zugegeben. Die Reaktion
kann für
etwa 12 Stunden bei RT rühren.
Die Reaktion wird mit H2O gestoppt, fünffach mit
EtOAc verdünnt
und mit Kochsalzlösung
gewaschen. Die organische Phase wird unter Bildung von etwa 1,0
g an 1-Boc-4-(2-Aminomethylphenyl)piperazin (30%) zur Trockne konzentriert.
MS
(ESI+) 292,1 (M + 1).
-
Präparation
38A 1-Boc-4-(2-Dimethylaminomethylphenyl)piperazin
-
1-Boc-4-(2-Aminomethylphenyl)piperazin
(2,0 g, 6,86 mmol) wird in CH3CN (15 ml)
gelöst
und auf etwa 0°C
gekühlt.
Wässriges
Formaldehyd (37 Gewichtsprozent in H2O)
(7,56 ml) wird zu der kalten Lösung gefolgt
von der Zugabe von Natriumcyanoborhydrid (2,15 g, 34,32 mmol) gegeben.
Das Reaktionsgemisch kann bei 0°C
für etwa
5 Minuten rühren
und kann sich dann natürlich
auf Raumtemperatur erwärmen.
Das Gemisch wird dann zur Trockne konzentriert. Der entstehende
Rückstand
wird in EtOAc (100 ml) aufgenommen und mit gesättigter NaHCO3 Lösung (100
ml) und Kochsalzlösung
(100 ml) gewaschen. Die organische Phase wird unter Bildung von
etwa 2,2 g des rohen Materials zur Trockne konzentriert.
MS
(ESI+) 320,2 [M + 1].
-
Präparation
39A 1-Boc-4-[2-(Methansulfonylaminomethyl)phenyl]piperazin
-
1-Boc-4-(2-Aminomethylphenyl)piperazin
(2,09 g, 7,18 mmol) wird in Methylenchlorid (50 ml) gelöst, auf
0°C gekühlt und
mit Triethylamin (1,5 ml, 10,8 mmol) gefolgt von Methansulfonylchlorid
(0,67 ml, 8,61 mmol) behandelt. Das entstehende Gemisch wird für etwa 3
Stunden bei RT gerührt,
dann mit Ether (200 ml) verdünnt
und mit Wasser (50 ml), gesättigtem
wässrigem
Natriumbicarbonat (50 ml) und Kochsalzlösung (50 ml) gewaschen und
dann über
wasserfreiem Magnesiumsulfat getrocknet. Eine Konzentration unter
verringertem Druck gefolgt von einer Silicagelchromatographie (30%
Ethylacetat in Hexan) ergibt die Titelverbindung (2,07 g, 78%) als
klares Öl.
1H NMR (CDCl3): δ 7,25–7,40 (m,
2H), 7,00–7,15
(m, 2H), 4,40 (s, 1H), 3,55–3,65
(m, 4H), 2,80–2,95
(m, 4H), 2,75 (s, 3H), 1,60 (s, 9H).
TLC (SiO2):
Rf = 0,50 (50% EtOAc/Hexan).
-
Präparation
40A 1-Boc-4-[2-(Acetylaminomethyl)phenyl]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
39A hergestellt, mit der Ausnahme, dass Essigsäureanhydrid an Stelle von Methansulfonylchlorid
verwendet wird.
1H NMR (CDCl3): δ 7,45–7,55 (m,
2H), 7,05–7,15
(m, 2H), 6,20 (s, 1H), 4,45–4,50
(m, 2H), 3,55–3,65
(m, 4H), 2,75–2,90
(m, 4H), 2,05 (s, 3H), 1,60 (s, 9H).
TLC (SiO2):
Rf = 0,15 (50% EtOAc/Hexan).
-
Präparation
41A 1-Boc-4-[2-(Benzolsulfonylaminomethyl)phenyl]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
39A hergestellt, mit der Ausnahme, dass Benzolsulfonylchlorid an
Stelle von Methansulfonylchlorid verwendet wird.
1H
NMR (CDCl3): δ 6,90–7,90 (m, 9H), 5,75–5,85 (m,
1H), 4,15–4,25
(m, 2H), 3,50–3,60
(m, 4H), 2,60–2,75
(m, 4H), 1,20–1,55
(m, 9H).
TLC (SiO2): Rf =
0,85 (100% EtOAc).
-
Präparation
42A 1-Boc-4-[2-(Ethansulfonylaminomethyl)phenyl]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
39A hergestellt, mit der Ausnahme, dass Ethansulfonylchlorid an
Stelle von Methansulfonylchlorid verwendet wird.
1H
NMR (CDCl3): δ 7,05–7,35 (m, 4H), 4,35–4,45 (m,
2H), 3,70–3,80
(m, 5H), 2,85–2,90
(m, 6H), 1,25–1,50
(m, 12H).
TLC (SiO2): Rf =
0,85 (100% EtOAc).
-
Präparation
43A 1-Boc-4-[2-(Propan-2-sulfonylaminomethyl)phenyl]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
39A hergestellt, mit der Ausnahme, dass Isopropylsulfonylchlorid
an Stelle von Methansulfonylchlorid verwendet wird.
1H NMR (CDCl3): δ 7,00–7,35 (m,
4H), 4,45–4,50
(m, 1H), 3,75–3,85
(m, 4H), 2,90–3,00
(m, 4H), 1,95–2,25
(m, 8H), 1,20–1,55
(m, 10H).
-
Präparation
44A 1-Boc-4-[2-(Isobutyrylaminomethyl)phenyl]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
39A hergestellt, mit der Ausnahme, dass Isobutyrylchlorid an Stelle
von Methansulfonylchlorid und Diisopropylethylamin als Base verwendet
wird.
1H NMR (CDCl3): δ 7,34–7,41 (m,
2H), 7,14–7,22
(m, 2H), 6,39–6,47
(m, 1H), 4,53–4,58
(m, 2H), 2,78–2,95
(m, 4H), 2,76–2,87
(m, 4H), 1,43–1,54
(s, 9H), 1,15–1,21
(m, 6H).
-
Präparation
45A [2-(Propionylaminomethyl)phenyl]piperazin
-
1-Boc-4-(2-Aminomethylphenyl)piperazin
(0,75 g, 2,6 mmol) wird in Methylenchlorid (20 ml) gelöst, mit DIPEA
(2,3 ml, 13 mmol) behandelt und auf etwa 0°C gekühlt. Propionylchlorid (0,20
ml, 2,34 mmol) wird zugegeben und das Gemisch wird für etwa 1
Stunde bei 0°C
gerührt
und anschließend über Nacht
bei Raumtemperatur gerührt.
Das Gemisch wird mit Ethylacetat (400 ml) verdünnt, mit Wasser (45 ml), gesättigtem wässrigem
Natriumbicarbonat (45 ml) und Kochsalzlösung (45 ml) gewaschen und
dann über
wasserfreiem Natriumsulfat getrocknet. Eine Konzentration unter
verringertem Druck gefolgt von einer Silicagelchromatographie (50%
Ethylacetat in Hexan) ergibt ein Öl, das in Methylenchlorid (10
ml) gelöst
wird. Das Gemisch wird mit TFA (10 ml) für etwa 1,5 Stunden gerührt. Das
Gemisch wird unter verringertem Druck konzentriert und der Rückstand
wird in Wasser (25 ml) aufgenommen. Natriumhydroxid (1,0 g, 25 mmol)
und Ethylacetat (25 ml) werden zugegeben und das Gemisch wird für etwa 45
Minuten gerührt.
Die organische Phase wird gesammelt und die wässrige Phase wird mit Ethylacetat
(45 ml) extrahiert. Die vereinigten organischen Fraktionen werden mit
Wasser (20 ml) und Kochsalzlösung
(20 ml) gewaschen und dann über
wasserfreiem Natriumsulfat getrocknet. Das Lösemittel wird unter verringertem
Druck unter Bildung der Titelverbindung (0,26 g, 40%) als klares Öl konzentriert.
1H NMR (CDCl3): δ 6,99–7,43 (m,
5H), 6,46–6,71
(bs, 1H), 4,46–4,72
(s, 2H), 2,79–3,23
(m, 8H), 2,14–2,43
(m, 2H), 1,07–1,38
(m, 3H).
-
Alternativ
dazu wird die Titelverbindung in folgendem Verfahren hergestellt:
Etwa
0,40 g (1,37 mmol) 1-Boc-4-(2-Aminomethylphenyl)piperazin, 0,11
ml (1,51 mmol) Propionsäure,
0,22 g (1,64 mmol) HOBt, 0,31 g (1,64 mmol) EDC und 0,24 ml (1,37
mmol) DIEA werden in 30 ml THF unter Stickstoff gemischt und über Nacht
bei RT gerührt.
Die Reaktion wird zur Trockne konzentriert und Ethylacetat wird
zugegeben. Das Gemisch wird mit gesättigtem Bicarbonat und Kochsalzlösung gewaschen
und dann mit Natriumsulfat getrocknet. Der Rückstand wird durch Blitzchromatographie
unter Elution mit 1:1 Hexan/Ethylacetat unter Bildung von etwa 0,41
g (86% Ausbeute) gereinigt. Das Material wird mittels TFA/DCM unter
Bildung von 4-[2-(Propionylaminomethyl)phenyl]piperazin von den
Schutzgruppen befreit.
LRMS (ESI+): 248 (M + 1).
-
Präparation
46A 4-{2-[(2,2-Dimethylpropionylamino)methyl]phenyl}piperazin
-
1-Boc-4-(2-Aminomethylphenyl)piperazin
(0,75 g, 2,6 mmol) wird in Methylenchlorid (20 ml) gelöst. DIPEA
(2,3 ml, 13 mmol) wird zugegeben und das Gemisch wird auf etwa 0°C gekühlt. Die
Lösung
wird mit Trimethylacetylchlorid (0,28 g, 0,28 ml, 2,3 mmol) behandelt
und für
etwa 1 Stunde bei 0°C
gerührt.
Die Lösung wird
auf RT erwärmt
und über
Nacht gerührt.
Das Gemisch wird mit Ethylacetat (400 ml) verdünnt, mit Wasser (60 ml) und
gesättigtem
wässrigem
Natriumbicarbonat (60 ml) und Kochsalzlösung (60 ml) gewaschen und dann über wasserfreiem
Natriumsulfat getrocknet. Die Lösung
wird unter verringertem Druck konzentriert und mittels Silicagelchromatographie
(80% Ethylacetat in Hexan) unter Bildung eines klaren Öls gereinigt,
das nacheinander in reiner TFA (5 ml) für etwa 1 Stunde gerührt wird.
Das Lösemittel
wird unter verringertem Druck verdampft und der Rückstand
wird in Wasser (30 ml) aufgenommen. Natriumhydroxid (1 g, 25 mmol)
und Ethylacetat (30 ml) werden zugegeben und das Gemisch wird für etwa 45
Minuten gerührt.
Die organische Phase wird gesammelt und die wässrige Phase wird mit Ethylacetat
(60 ml) extrahiert. Die vereinigten organischen Fraktionen werden
mit Wasser (45 ml) und Kochsalzlösung
(30 ml) gewaschen und dann über
wasserfreiem Natriumsulfat getrocknet. Eine Konzentration unter
verringertem Druck ergibt die Titelverbindung (0,54 g, 75%) als
klares Öl.
1H NMR (CDCl3): δ 6,97–7,36 (m,
4H), 6,63–6,86
(bs, 1H), 4,47–4,65
(m, 2H), 2,66–3,24
(m, 8H), 1,18 (s, 9H).
-
Präparation
47A 4-[2-(Benzoylaminomethyl)phenyl]piperazin
-
1-Boc-4-(2-Aminomethylphenyl)piperazin
(0,47 g, 1,6 mmol) wird in Methylenchlorid (20 ml) gelöst. DIPEA
(1,5 ml, 8,5 mmol) wird zugegeben und das Gemisch wird auf etwa
0°C gekühlt. Das
Gemisch wird mit Benzoylchlorid (0,20 g, 0,16 ml, 1,4 mmol) behandelt.
Das entstehende Gemisch wird für
etwa 1 Stunde bei 0°C
gerührt
und dann auf RT erwärmt
und über
Nacht gerührt.
Das Gemisch wird mit Ethylacetat (500 ml) verdünnt, mit Wasser (45 ml), gesättigtem
wässrigem
Natriumbicarbonat (45 ml) und Kochsalzlösung (45 ml) gewaschen und
dann über
wasserfreiem Natriumsulfat getrocknet. Die Lösung wird unter verringertem
Druck konzentriert und mittels Silicagelchromatographie (50% Ethylacetat
in Hexan) unter Bildung eines klaren Öls gereinigt, das nacheinander
in reinem TFA (5 ml) für
etwa 1 Stunde gerührt
wird. Das Lösemittel
wird unter verringertem Druck unter Bildung der Titelverbindung
als klares Öl
(0,30 g, 100%) eingedampft.
1H NMR
(CDCl3) δ 7,32–7,84 (m,
9H), 4,71–4,86
(m, 2H), 3,42–3,65
(m, 4H), 3,24–3,42
(m, 4H).
-
Präparation
48A 1-Boc-4-{2-[(methansulfonylmethylamino)methyl]phenyl}piperazin
-
Zu
einer gerührten
Suspension aus Natriumhydrid (60% in Öl, 113 mg, 2,82 mmol) in THF
(20 ml) bei 0°C
unter Stickstoff wird eine Lösung
aus 1-Boc-4-[2-(Methansulfonylaminomethyl)phenyl]piperazin (0,99
g, 2,68 mmol) in THF (5 ml) gegeben. Das Gemisch wird für etwa 1
Stunde bei Raumtemperatur gerührt.
Es wird dann auf 0°C
rückgekühlt und
mit Methyliodid (0,184 ml, 2,95 mmol) behandelt. Nach dem Rühren für etwa 20 Stunden
wird das Reaktionsgemisch mit Ether (150 ml) verdünnt und
dann durch die Zugabe von gesättigtem wässrigem
Ammoniumchlorid (50 ml) gestoppt. Die organische Phase wird abgetrennt,
mit Wasser (50 ml) und Kochsalzlösung
(50 ml) gewaschen und dann über
Magnesiumsulfat getrocknet. Eine Konzentration unter verringertem
Druck gefolgt von einer Silicagelchromatographie (30% Ethylacetat
in Hexan) ergibt die Titelverbindung (0,96 g, 94%) als klares Öl.
1H NMR (CDCl3): δ 7,45–7,55 (m,
1H), 6,95–7,35
(m, 3H), 4,45 (s, 2H), 3,45–3,60
(m, 4H), 3,05 (s, 3H), 2,75–2,90
(m, 4H), 2,75 (s, 3H), 1,60 (s, 9H).
TLC (SiO2):
Rf = 0,70 (50% EtOAc/Hexan).
-
Präparation
49A 1-Boc-4-{2-[(Benzylmethansulfonylamino)methyl]phenyl}piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
48A hergestellt, mit der Ausnahme, dass Benzylbromid verwendet wird.
1H NMR (CDCl3): δ 7,70–7,75 (m,
1H), 7,25–7,55
(m, 8H), 4,75 (s, 2H), 4,50 (s, 2H), 3,45–3,60 (m, 4H), 3,05 (s, 3H),
2,75–2,90
(m, 4H), 1,65 (s, 9H).
TLC (SiO2):
Rf = 0,70 (50% EtOAc/Hexan).
-
Präparation
50A 1-Boc-4-{2-[(Ethylmethansulfonylamino)methyl]phenyl}piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
48A hergestellt, mit der Ausnahme, dass Ethyliodid verwendet wird.
TLC
(SiO2): Rf = 0,25
(30% EtOAc/Hexan).
-
Präparation
51A 1-Boc-4-{2-[(Acetylmethylamino)methyl]phenyl}piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
48A ausgehend von 1-Boc-4-[2-(Acetylaminomethyl)phenyl]piperazin
(0,58 g, 1,7 mmol) hergestellt. Die Titelverbindung (0,36 g, 60%)
wird als klares Öl
erhalten.
TLC (SiO2): Rf =
0,33 (66% Ethylacetat in Hexan).
-
Präparation
52A 1-Boc-4-{2-[(Acetylbenzylamino)methyl]phenyl}piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
51A hergestellt, mit der Ausnahme, dass Benzylbromid verwendet wird.
TLC
(SiO2): Rf = 0,20
(66% Ethylacetat in Hexan).
-
Präparation
53A 1-Boc-4-{2-[Acetylethylamino)methyl]phenyl}piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
51A hergestellt, mit der Ausnahme, dass Ethyliodid verwendet wird.
TLC
(SiO2): Rf = 0,35
(66% Ethylacetat in Hexan).
-
Präparation
54A (Benzylamin aus Benzylalkohol mittels Mitsunobu) 1-Boc-4-(2-[1,2,4]triazol-1-ylmethylphenyl)piperazin
-
Schritt
1: 1-Boc-4-(2-Carboxyphenyl)piperazin
-
Zu
einer Lösung
aus 1-(2-Cyanophenyl)piperazin (7,5 g, 40 mmol) in 100 ml absolutem
Ethanol werden 200 ml an 25% wässrigem
KOH gegeben. Die Lösung
wird am Rückfluss
für etwa
48 Stunden erhitzt und dann auf etwa 0°C gekühlt. Die Lösung wird mit 180 ml an 5 M
HCl angesäuert
und dann wird festes NaHCO3 zugegeben um
den pH der Lösung
auf etwa 10 zu bringen. Nach der Konzentration im Vakuum zur Entfernung von
60 ml des Lösemittels
werden Dioxan (300 ml), NaHCO3 (12,7 g,
120 mmol) und Boc2O (11,4 g, 52,2 mmol) zugegeben.
Die Lösung
wird über
Nacht gerührt
und dann mit 5 M HCl auf etwa pH 1 angesäuert. Nach der Auftrennung
wird die wässrige
Lösung
mit EtOAc (3×)
extrahiert. Die vereinigten organischen Lösungen werden mit Wasser (2×) und Kochsalzlösung gewaschen
und dann getrocknet (Na2SO4),
filtriert und unter Bildung der Titelverbindung konzentriert.
LRMS
(ES–):
305,2 (M – 1).
-
Schritt
2: 1-Boc-4-(2-Hydroxymethylphenyl)piperazin
-
Zu
einer Lösung
aus 1-Boc-4-(2-Carboxyphenyl)piperazin aus Schritt 1 in 340 ml THF
bei 0°C
wird BH3-THF (120 ml einer 1 M Lösung in
THF) gegeben. Das Kühlbad
wird entfernt und die Lösung
wird über Nacht
gerührt.
Die Lösung
wird auf etwa 0°C
gekühlt
und dann werden 60 ml an 2 M NaOH gefolgt von EtOAc und Kochsalzlösung zugegeben.
Nach der Auftrennung wird die wässrige
Lösung
mit EtOAc (3×)
extrahiert. Die vereinigten organischen Lösungen werden mit Wasser (2×) und Kochsalzlösung gewaschen
und dann getrocknet (Na2SO4),
filtriert und unter Bildung von etwa 11,2 g (38,3 mmol, 96%) der
Titelverbindung konzentriert.
LRMS (ESI+): 393,2 [M + 1].
-
Schritt 3:
-
Zu
einer Lösung
aus 1-Boc-4-(Hydroxymethylphenyl)piperazin (300 mg, 1,02 mmol, 1,0 Äquivalente), 1,2,4-Triazol
(104 mg, 1,53 mmol, 1,5 Äquivalente),
Triphenylphosphin (535 mg, 2,04 mmol, 2,0 Äquivalente) und THF bei 0°C unter Stickstoff
wird DEAD (0,321 ml, 2,04 mmol, 2,0 Äquivalente) so langsam gegeben,
so dass die Temperatur der Reaktion nicht über 10°C ansteigt. Nachdem die Zugabe vollständig ist,
wird das Eisbad entfernt und das Reaktionsgemisch wird bei RT über Nacht
gerührt.
Methanol wird zugegeben und das Gemisch wird für etwa 15 Minuten gerührt. Das
Gemisch wird dann konzentriert. Eine Reinigung durch Blitzchromatographie
(35 g SiO2, linearer Gradient 50–70% EtOAc/Hexan
für 15
Minuten und 70% EtOAc für
18 Minuten) ergibt die Boc geschützte
Titelverbindung (200 mg, 0,5 mmol, 57%).
LRMS (ESI+): 344,1
(M + 1).
-
Präparation
55A 1-Boc-4-(2-Tetrazol-2-ylmethylphenyl)piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
54A hergestellt, mit der Ausnahme, dass Tetrazol verwendet wird.
LRMS
(ESI+): 289,1 (M-Boc).
-
Präparation
56A 1-Boc-4-(2-Imidazol-1-ylmethylphenyl)piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
54A hergestellt, mit der Ausnahme, dass Imidazol verwendet wird.
LRMS
(ESI+): 343,2 (M + 1).
-
Präparation
57A 1-Boc-4-(2-Azidomethylphenyl)piperazin
-
1-Boc-4-(2-Hydroxymethylphenyl)piperazin
(4,59 g, 15,7 mmol) wird in Toluol (75 ml) gelöst. Triphenylphosphin (8,3
g, 31,6 mmol) wird gefolgt von Zinkazidpyridinsalz (3,61 g, 11,72
mmol) zugegeben. Diisopropylazodicarboxylat (6,27 ml, 31,6 mmol)
wird tropfenweise zugegeben und die Lösung wird bei RT für etwa 12
Stunden gerührt.
Das Gemisch wird unter verringertem Druck konzentriert und mittels
Silicachromatographie (12% Ethylacetat in Hexan) unter Bildung der
Titelverbindung (1,89 g, 51%) als Öl gereinigt.
1H
NMR (CDCl3): δ 7,35–7,05 (m, 4H), 4,45 (s, 2H),
3,60–3,50
(m, 4H), 2,85–2,75
(m, 4H), 1,50 (s, 9H).
-
Präparation
58A 1-Boc-4-[2-(4-Methoxycarbonyl-[1,2,3]triazol-1-ylmethyl)phenyl]piperazin
-
1-Boc-4-(2-Azidomethylphenyl)piperazin
(0,25 g, 0,79 mmol) wird in deuteriertem Chloroform (3 ml) gelöst. Methylpropiolat
(0,35 ml, 3,9 mmol) wird zugegeben und das Gemisch wird am Rückfluss
für etwa
4 Stunden erhitzt und dann auf RT gekühlt. Das Gemisch wird unter
verringertem Druck konzentriert und mittels Silicachromatographie
(50% Ethylacetat in Hexan) unter Bildung der Titelverbindung (0,155
g, 49%) als Öl
gereinigt.
1H NMR (CDCl3): δ 7,35–7,05 (m,
4H), 5,75 (s, 2H), 3,95 (s, 3H), 3,55–3,45 (m, 4H), 2,80–2,70 (m,
4H), 3,80–3,85
(m, 1H), 1,50 (s, 9H).
-
Präparation
59A 1-Boc-4-[2-(4-tert-Butyl-[1,2,3]triazol-1-ylmethyl)phenyl]piperazin
-
In
einem verschlossenen Röhrchen
wird 4-(2-Azidomethylphenyl)piperazin-1-carbonsäure-tert-butylester (0,366 g, 1,15 mmol) in Toluol
(5 ml) gelöst.
3,3-Dimethyl-1-butin (0,7 ml, 5,64 mmol) wird zugegeben und das
Gemisch wird am Rückfluss
für etwa
48 Stunden erhitzt und dann auf RT gekühlt. Das Gemisch wird unter
verringertem Druck konzentriert und mittels Silicachromatographie
(50% Ethylacetat in Hexan) unter Bildung der Titelverbindung (0,212
g, 60%) als Öl
gereinigt.
1H NMR (CDCl3) δ 7,35–7,05 (m,
4H), 5,75 (s, 2H), 3,60–3,45
(m, 4H), 2,80–2,70
(m, 4H), 1,50 (s, 9H), 1,35 (s, 9H).
-
Präparation
60A (Benzylamin aus Benzylalkohol über Mesylat) 1-Boc-4-[2-(3R-Dimethylaminopyrrolidin-1-ylmethyl)phenylpiperazin
-
Zu
einer Lösung
aus 1-Boc-4-(2-Hydroxymethylphenyl)piperazin (300 mg, 1,03 mmol,
1,0 Äquivalente),
Triethylamin (0,17 ml, 1,2 mmol, 1,2 Äquivalente), DMAP (6 mg, 0,05
mol, 0,05 Äquivalente)
in CH2Cl2 (10 ml)
wird Methansulfonylchlorid (0,085 ml, 1,1 mmol, 1,1 Äquivalente)
gegeben. Die Lösung
wird bei RT unter N2 für etwa 2 Stunden gerührt. Eine
Lösung
aus 3R-3-(Dimethylamino)pyrrolidin (0,63 ml, 5,0 mmol, 5,0 Äquivalente)
in THF (3 ml) wird zugegeben und das Gemisch kann bei RT über Nacht
rühren.
Das Gemisch wird mit CH2Cl2 (10
ml) verdünnt
und mit gesättigtem
wässrigem
NaHCO3 (15 ml) und Kochsalzlösung (15
ml) gewaschen. Die wässrigen
Phasen werden mit CH2Cl2 (3×) extrahiert.
Die vereinigten organischen Extrakte werden getrocknet (Na2SO4), filtriert
und konzentriert. Eine Reinigung mittels Blitzchromatographie (35
g SiO2, 40 ml/min, linearer Gradient 0–10% an
2,0 M NH3 in MeOH/CH2Cl2 für
25 Minuten und 10% 2,0 M NH3 in MeOH/CH2Cl2 für 7 Minuten)
ergibt die Titelverbindung als weißen Feststoff (280 mg, 0,72
mmol, 72%).
LRMS (ESI+): 389,2 [M + 1].
-
Präparation
61A 1-Boc-4-[2-(3S-Dimethylaminopyrrolidin-1-ylmethyl)phenylpiperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
60A hergestellt, mit der Ausnahme, dass 3S-3-(Dimethylamino)pyrrolidin
verwendet wird.
LRMS (ESI+): 389,2 (M + 1).
-
Präparation
62A 1-Boc-4-(2-Pyrrolidin-1-ylmethylphenyl)piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
60A hergestellt, mit der Ausnahme, dass Pyrrolidin verwendet wird.
LRMS
(ESI+): 246,1 (M + 1).
-
Präparation
63A 1-Boc-4-[2-(2-Methylimidazol-1-ylmethyl)phenyl]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
60A hergestellt, mit der Ausnahme, dass 2-Methylimidazol verwendet
wird.
LRMS (ESI+): 357,2 (M + 1).
-
Präparation
64A 1-Boc-4-[2-(2-Ethylimidazol-1-ylmethyl)phenyl]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
60A hergestellt, mit der Ausnahme, dass 2-Isopropylimidazol verwendet
wird.
LRMS (ESI+): 371,3 (M + 1).
-
Präparation
65A 1-Boc-4-[2-(2-Ethylimidazol-1-ylmethyl)phenyl]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
60A hergestellt, mit der Ausnahme, dass 2-Ethylimidazol verwendet
wird.
LRMS (ESI+): 385,2 (M + 1).
-
Präparation
66A 1-Boc-4-[2-(2-Methylsulfanylimidazol-1-ylmethyl)phenyl]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
60A hergestellt, mit der Ausnahme, dass 2-Ethylsulfanyl-1H-imidazol
verwendet wird.
LRMS (ESI+): 403,3 (M + 1).
-
Präparation
67A 1-Boc-4-(5-Methyl-2-pyrrolidin-1-ylmethylphenyl)piperazin
-
Zu
einer Lösung
aus 2-Brom-4-methylanilin (558 mg, 3,0 mmol) in 30 ml Acetonitril
wird Tetrafluorborat (600 μl
einer 54% Lösung
in Et2O, 4,35 mmol) gegeben. Die Lösung wird
auf etwa 0°C
gekühlt
und t-Butylnitrit (55 μl,
4,62 mmol) wird zugegeben. Nach dem Rühren für etwa 45 Minuten wird die
Lösung
zu einer Lösung aus
CuCN (800 mg, 8,93 mmol) und NaCN (1,47 g, 30 mmol) in 30 ml Wasser,
das auf 0°C
gekühlt
ist, mittels einer Spritze gegeben. Das Kühlbad wird entfernt. Nach dem
Rühren über Nacht
wird die wässrige
Lösung
mit Et2O (2×) extrahiert. Die vereinigten
organischen Lösungen
werden mit 1 M HCl, gesättigtem
Natriumbicarbonat, Wasser und Kochsalzlösung gewaschen und dann getrocknet
(Na2SO4), filtriert
und konzentriert. Das Material wird auf 3 g Silicagel adsorbiert
und mittels Silicagelblitzchromatographie (4 × 15 cm Säule, 5–20 Et2O/Pentan, über 48 min
bei 35 ml/min) unter Bildung von etwa 320 mg (1,63 mmol, 54%) an
2-Brom-4-methylbenzonitril als farbloses Öl gereinigt.
GC/MS (EI):
195.
-
2-Brom-4-methylbenzonitril
wird mittels der Präparation
1A Buchwald Chemie unter Bildung von 4-(2-Cyano-5-methylphenyl)piperazin
an das Piperazin gekuppelt. 4-(2-Cyano-5-methylphenyl)piperazin
wird in 4-(2-Hydroxymethyl-5-methylphenyl)piperazin auf ähnliche
Weise zu Präparation
54A Schritte 1 und 2 umgewandelt. 4-(2-Hydroxymethyl-5-methylphenyl)piperazin
wird in die Titelverbindung auf ähnliche
Weise zu Präparation
60A umgewandelt, mit der Ausnahme, dass Pyrrolidin an Stelle von
Mesylat verwendet wird.
LRMS (ESI+): 360,3 (M + 1).
-
Präparation
68A 1-Boc-4-(5-Isopropyl-2-pyrrolidin-1-ylmethylphenyl)piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
67A hergestellt, mit der Ausnahme, dass 2-Brom-4-isopropylanilin
als Ausgangsmaterial verwendet wird.
LRMS (ESI+): 388,3 (M
+ 1).
-
Präparation
69A 1-Boc-4-(2-Dimethylaminomethyl-5-trifluormethylphenyl)piperazin
-
Zu
einer Lösung
aus Piperazin (13,7 g, 159 mmol) in 20 ml DMSO wird 2-Fluor-4-trifluormethylbenzonitril
(10 g, 52,9 mmol) gegeben. Nach dem Rühren über Nacht wird die Lösung mit
200 ml EtOAc verdünnt, mit
Wasser und Kochsalzlösung
gewaschen und dann getrocknet (Na2SO4), filtriert und unter Bildung von etwa 13,0
g (51,1 mmol, 96%) an 4-(2-Cyano-5-trifluormethylphenyl)piperazin
konzentriert.
LRMS (ESI+): 256,1 [M + 1].
-
Die
Titelverbindung wird aus 4-(2-Cyano-5-trifluormethylphenyl)piperazin
auf die gleiche Weise wie in Präparation
67A beschrieben hergestellt, mit der Ausnahme, dass Dimethylamin
zum Ersetzen des Mesylats verwendet wird.
LRMS (ESI+): 388,1
[M + 1].
-
Präparation
70A 1-Boc-4-(2-Pyrrolidin-1-ylmethyl-5-trifluormethylphenyl)piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise wie in Präparation
69A beschrieben, hergestellt, mit der Ausnahme, dass Pyrrolidin
verwendet wird um Mesylat zu ersetzen.
LRMS (ESI+): 414,3 (M
+ 1).
-
Präparation
71A 1-Boc-4-(2-Pyrrolidin-1-ylmethyl-4-trifluormethylphenyl)piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise wie in Präparation
70A beschrieben, synthetisiert, mit der Ausnahme, dass 2-Fluor-5-trifluormethylbenzonitril
als Ausgangsmaterial verwendet wird.
LRMS (ESI+): 414,3 (M
+ 1).
-
Präparation
72A 1-Boc-4-(2-Pyrrolidin-1-ylmethyl-6-trifluormethylphenyl)piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise wie in Präparation
70A beschrieben, synthetisiert, mit der Ausnahme, dass 2-Fluor-3-trifluormethylbenzonitril
als Ausgangsmaterial verwendet wird.
LRMS (ESI+): 414,3 (M
+ 1).
-
Präparation
73A 1-Boc-4-(2-Pyrrolidin-1-ylmethyl-3-trifluormethylphenyl)piperazin
-
4-(2-Cyano-3-trifluormethylphenyl)piperazin
wird auf ähnliche
Weise zu 4-(2-Cyano-5-trifluormethylphenyl)piperazin wie oben beschrieben
hergestellt, mit der Ausnahme, dass 2-Fluor-6-trifluormethylbenzonitril als Ausgangsmaterial
verwendet wird. Zu einer Lösung
aus 4-(2-Cyano-3-trifluormethylphenyl)piperazin
(1,35 g, 5,29 mmol, 1,0 Äquivalente)
in Dioxan (40 ml) wird eine Lösung
aus DIBAL in Heptan (1,0 M in Heptan, 13,2 ml, 13,22 mmol, 2,5 Äquivalente)
gegeben. Das entstehende Gemisch wird bei RT für etwa 3 Tage gerührt. Das
Gemisch wird mittels einer Spritze zu 0,5 M Rochelle Salz gegeben
und für
etwa 2 Stunden gerührt. NaHCO3 (1,3 g, 15,9 mmol, 3,0 Äquivalente) und Di-tert-Butyldicarbonat (1,7
g, 7,29 mmol, 1,5 Äquivalente) werden
zugegeben und das Gemisch wird bei RT über Nacht gerührt. Das
Gemisch wird zwischen EtOAc (100 ml) und Kochsalzlösung (50
ml) aufgeteilt. Die organische Phase wird abgetrennt und die wässrige Phase
wird mit EtOAc (2×)
extrahiert. Die vereinigten organischen Extrakte werden mit H2O und Kochsalzlösung gewaschen und dann getrocknet
(Na2SO4), filtriert
und konzentriert. Eine Reinigung durch Blitzchromatographie (120
g SiO2, 40 ml/min, linearer Gradient 0–25% EtOAc/Hexan
für 10
Minuten und 25% EtOAc/Hexan für
23 Minuten) ergibt N-Boc-4-(2-Formyl-3-trifluormethylphenyl)piperazin
(637 mg, 1,77 mmol, 35%).
LRMS (ESI+): 359,1 [M + 1].
-
Zu
einer Lösung
aus N-Boc-4-(2-Formyl-3-trifluormethylphenyl)piperazin (358 mg,
1 mmol, 1,0 Äquivalente)
in MeOH (10 ml) wird Pyrrolidin (0,093 ml, 1,1 mmol, 1,1 Äquivalente)
gegeben. Das Gemisch wird am Rückfluss über Nacht
erhitzt. Die Reaktion wird auf etwa 0°C gekühlt und NaBH4 auf
Aluminiumoxid (10 Gewichtsprozent auf basischem Aluminiumoxid, 570
mg, 1,5 mmol, 1,5 Äquivalente)
wird zugeben. Nachdem die Zugabe vollständig ist, wird das Eisbad entfernt
und das Gemisch wird bei RT für
etwa 2 Stunden gerührt. Das
Gemisch wird durch Celite filtriert, mit Methanol gewaschen und
konzentriert. Die Lösung
wird mit EtOAc (50 ml) verdünnt
und mit gesättigtem
NaHCO3 und Kochsalzlösung gewaschen. Die wässrigen
Phasen werden mit EtOAc (2×)
extrahiert. Die vereinigten organischen Extrakte werden getrocknet
(Na2SO4), filtriert
und konzentriert. Eine Reinigung durch Blitzchromatographie (35
g, SiO2, 40 ml/min, linearer Gradient 0–10% MeOH/CH2Cl2 für 25 Minuten
und 10% MeOH/CH2Cl2 für 7 Minuten)
ergibt die Titelverbindung (298 mg, 0,72 mmol, 72%).
LRMS (ESI+):
414,3 (M + 1).
-
Präparation
74A (Derivate von 1-Boc-4-(2-aminophenyl)piperazin) 1-Boc-4-(2-Aminophenyl)piperazin
-
Zu
einer Lösung
aus N-(2-Nitrophenyl)piperazin (30 g, 145 mmol) und Triethylamin
(28,3 ml, 203 mmol) in 600 ml CH2Cl2 wird Boc2O (38
g, 174 mmol) gegeben. Nach dem Rühren über Nacht
wird die Lösung mit
gesättigtem
wässrigem
Natriumbicarbonat und Kochsalzlösung
gewaschen und dann ge trocknet (Na2SO4), filtriert und unter Bildung eines orangen Öls konzentriert.
Zu einer Lösung
des Öls
in 2 l Ethanol werden 6 g an 5% Pd/C gegeben. Nach dem Schütteln unter
60 psi H2 über Nacht wird die Lösung filtriert
und unter Bildung von etwa 39 g (140 mmol, 97%) an 1-Boc-4-(2-Aminophenyl)piperazin
als brauner Feststoff konzentriert.
LRMS: 278,1 (M + 1).
-
Präparation
75A 1-Boc-4-(2-Dimethylaminophenyl)piperazin
-
Zu
einer Lösung
aus 1-Boc-4-(2-Nitrophenyl)piperazin (500 mg, 1,63 mmol, 1,0 Äquivalente)
in IPA (20 mL) wird Formaldehyd (3,3 ml, 37% Lösung in H2O,
4,07 mmol, 2,5 Äquivalente)
und 10% Pd/C (125 mg, 25 Gewichtsprozent) gegeben. Das Gemisch wird
unter Wasserstoff bei 60 psi über
Nacht geschüttelt.
Das Gemisch wird filtriert und mit CH2Cl2 verdünnt.
Die wässrige
Lösung
wird abgetrennt und die organische Lösung wird getrocknet (Na2SO4), durch ein
Kissen aus Celite filtriert und konzentriert. Eine Reinigung durch
Blitzchromatographie (35 g SiO2, 40 ml/min,
linearer Gradient 0–15%
ETOAc/Hexan für
20 Minuten und 15% EtOAc/Hexan für
13 Minuten) ergibt etwa 480 mg (1,57 mmol, 97%) der Titelverbindung
als Feststoff.
LRMS (ESI+): 306,2 (M + 1).
-
Präparation
76A 1-Boc-4-[2-(Isobutylamido)phenyl]piperazin
-
Zu
einer Lösung
aus 1-Boc-4-(2-aminophenyl)piperazin (2,77 g, 10 mmol) Triethylamin
(2,8 ml, 20 mmol) und DMAP (70 mg, 0,57 mmol) in 50 ml CH2Cl2 wird Isobutyrylchlorid
(1,15 ml, 11 mmol) gegeben. Nach dem Rühren über Nacht wird gesättigtes
wässriges
Natriumbicarbonat zugegeben und die Lösung wird konzentriert. Die
Lösung
wird mit EtOAc verdünnt,
mit 1 M HCl, Wasser, gesättigtem
wässri gem
Natriumbicarbonat und Kochsalzlösung
gewaschen und dann getrocknet (Na2SO4), filtriert und unter Bildung von etwa
3,29 g (9,4 mmol, 94%) der Titelverbindung konzentriert.
LRMS:
348,2 (M + 1).
-
Präparation
77A 1-Boc-4-[2-(3-methylbutyrylamino)phenyl]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
76A hergestellt, mit der Ausnahme, dass Isovalerylchlorid an Stelle
von Isobutyrylchlorid verwendet wird.
LRMS (ESI+): 362,2 (M
+ 1).
-
Präparation
78A 1-Boc-4-(2-Isobutylaminophenyl)piperazin
-
Zu
einer Lösung
aus 1-Boc-4-(2-Isobutylaminophenyl)piperazin (2,72 g, 7,8 mmol)
in 50 ml THF wird BH3-THF (24 ml einer 1
M Lösung
in THF, 24 mmol) gegeben. Nach dem Rühren für etwa 1 Stunden bei 60°C wird die
Lösung
auf RT gekühlt
und dann werden 25 ml an 1 M NaOH zugegeben. Nach dem Rühren für etwa 2
Stunden werden Kochsalzlösung
und EtOAc zugegeben. Die organische Lösung wird mit Wasser (2×) und Kochsalzlösung gewaschen
und dann getrocknet (Na2SO4),
filtriert und konzentriert. Eine Reinigung durch Blitzchromatographie
(Biotage 40 L Säule,
0 bis 30% EtOAc/Hexan linearer Gradient über 48 min bei 35 ml/min) ergibt
etwa 2,35 g (7,05 mmol, 90%) der Titelverbindung.
LRMS: 334,2
(M + 1).
-
Präparation
79A 1-Boc-4-(2-Methansulfonylaminophenyl)piperazin
-
Zu
einer Lösung
aus 1-Boc-4-(2-Aminophenyl)piperazin (5,55 g, 20 mmol) und Triethylamin
(5,6 ml, 40 mmol) in 200 ml CH2Cl2 wird Methansulfonylchlorid (1,55 ml, 20
mmol) gegeben. Nach dem Rühren
für etwa 4
Stunden wird die Lösung
konzentriert und der Rückstand
wird in 200 ml EtOAc gelöst.
Die Lösung
wird mit 1 M HCl (2×),
Wasser und Kochsalzlösung
gewaschen und dann getrocknet (Na2SO4), filtriert und unter Bildung von etwa
6,68 g (18,8 mmol, 94%) der Titelverbindung als brauner Feststoff
konzentriert.
LRMS: 356,1 (M + 1).
-
Präparation
80A 1-Boc-4-[2-(3,3-Dimethylureido)phenyl]piperazin
-
Zu
einer Lösung
aus 1-Boc-4-(2-Aminophenyl)piperazin (270 mg, 1,0 mmol) und Et3N (400 μl,
2,89 mmol) in 10 ml CH2Cl2 wird
Dimethylcarbamylchlorid (135 μl,
1,48 mmol) gegeben. Nach dem Rühren
für etwa 1
Stunde wird DMAP (10 mg) zugegeben. Nach dem Rühren für etwa 3 Tage werden weitere
800 μl Et3N und 270 μl Dimethylcarbamylchlorid zugegeben.
Nach dem Rühren über Nacht
wird die Lösung
mit EtOAc verdünnt,
mit 1 M HCl (2×),
gesättigtem
Natriumbicarbonat, Wasser und Kochsalzlösung gewaschen und dann getrocknet
(Na2SO4), filtriert
und konzentriert. Eine Reinigung durch Silicagelchromatographie
(35 g SiO2, 20 bis 50% EtOAc/Hexan über 30 Minuten
bei 35 ml/min) ergibt etwa 20 mg (0,057 mmol, 6%) der Titelverbindung als
weißen
Feststoff.
LRMS: 349,2 (M + 1).
-
Präparation
81A 1-Boc-4-[2-(3-Isopropylureido)phenyl]piperazin
-
Zu
einer Lösung
aus 1-Boc-4-(2-Aminophenyl)piperazin (270 mg, 1,0 mmol) in 10 ml
THF wird Isopropylisocyanat (90 μl,
1,46 mmol) gegeben. Nach dem Rühren
für etwa
1 Stunde werden weitere 90 μl
Isopropylisocyanat zugegeben. Nach dem Rühren für weitere 3 Tage werden 290 μl Isopropylisocyanat
zugegeben Nach dem Rühren über Nacht
wird die Lösung
konzentriert. Eine Reinigung durch Silicagelchromatographie (35
g, SiO2, 20 bis 50% EtOAc/Hexan über 30 Minuten
bei 35 ml/min) ergibt 240 mg (0,66 mmol, 66%) der Titelverbindung
als weißen
Feststoff.
LRMS: 363,2 (M + 1).
-
Präparation
82A 1-Boc-4-[2-(Isobutylmethansulfonylamino)phenyl]piperazin
-
Zu
einer Lösung
aus 1-Boc-4-(2-Methansulfonylaminophenyl)piperazin (1,07 g, 3,0
mmol) in 50 ml DMF wird NaH (240 mg, einer 60% Dispersion in Öl, 6 mmol)
gegeben. Nach dem Rühren
für etwa
15 Minuten bei RT wird Isobutyliodid (420 μl, 3,65 mmol) zugegeben und
die Lösung
wird auf 60°C
erwärmt.
Nach dem Rühren
bei 60°C über Nacht
wird die Reaktion mit gesättigtem
wässrigem
Ammoniumchlorid gestoppt und mit EtOAc verdünnt. Die Lösung wird zweimal mit Wasser
und Kochsalzlösung
gewaschen und dann getrocknet (Na2SO4), filtriert und konzentriert. Eine Reinigung
durch Blitzchromatographie (40 M Biotage Säule, 10–30% linearer Gradient EtOAc/Hexan, über 45 min
bei 35 ml/min) ergibt etwa 1,07 g (2,6 mmol, 87%) der Titelverbindung
als weißen
Schaum.
LRMS: 412,3 (M + 1).
-
Präparation
83A 1-Boc-4-[2-(Methylmethansulfonylamino)phenyl]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
82A hergestellt, mit der Ausnahme, dass Methyliodid und K2CO3 an Stelle von
NaH als Base verwendet werden.
LRMS (ESI+): 370,2 (M + 1).
-
Präparation
84A 1-Boc-4-[2-(Ethylmethansulfonylamino)phenyl]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
82A hergestellt, mit der Ausnahme, dass Ethyliodid und K2CO3 an Stelle von
NaH als Base verwendet werden.
LRMS (ESI+): 384,2 (M + 1).
-
Präparation
85A 1-Boc-4-[2-(n-Butylmethansulfonylamino)phenyl]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
82A hergestellt, mit der Ausnahme, dass n-Butyliodid verwendet wird.
LRMS
(ESI+): 412,2 (M + 1).
-
Präparation
86A 1-Boc-4-[2-(2-Ethylbutyl)methansulfonylamino)phenyl]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
82A hergestellt, mit der Ausnahme, dass 1-Brom-2-ethylbutan verwendet
wird.
LRMS (ESI+): 440,2 (M + 1).
-
Präparation
87A 1-Boc-4-[2-(Cyclohexylmethylmethansulfonylamino)phenyl]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
82A hergestellt, mit der Ausnahme, dass Brommethylcyclohexan verwendet
wird.
LRMS (ESI+): 452,2 (M + 1).
-
Präparation
88A 1-Boc-4-[2-(Cyclobutylmethylmethansulfonylamino)phenyl]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
82A hergestellt, mit der Ausnahme, dass Brommethylcyclobutan verwendet
wird.
LRMS (ESI+): 424,1 (M + 1).
-
Präparation
89A 1-Boc-4-[2-(Cyclopropylmethylmethansulfonylamino)phenyl]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
82A hergestellt, mit der Ausnahme, dass Brommethylcyclopropan verwendet
wird.
LRMS (ESI+): 410,1 (M + 1).
-
Präparation
90A 1-Boc-4-{2-[Methansulfonyl-(3-methylbutyl)amino]phenyl}piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
82A hergestellt, mit der Ausnahme, dass 1-Iod-3-methylbutan verwendet
wird.
LRMS (ESI+): 426,2 (M + 1).
-
Präparation
91A 1-Boc-4-[2-(1,1-Dioxo-2-isothiazolidinyl)phenyl]piperazin
-
Zu
einer Lösung
aus N-Boc-4-(2-Aminophenyl)piperazin (555 mg, 2,0 mmol) und Et3N (837 μl,
6 mmol) in 20 ml CH2Cl2 wird
3-Chlorpropansulfonylchlorid (255 μl, 2,1 mmol) gegeben. Nach dem
Rühren
für etwa
30 Minuten wird das Gemisch mit gesättigtem wässrigem Natriumbicarbonat gestoppt,
mit EtOAc verdünnt,
mit 1 M HCl, Wasser und Kochsalzlösung gewaschen und dann getrocknet
(Na2SO4), filtriert
und konzentriert. Eine Reinigung mittels Silicagelchromatographie
(35 g SiO2, 10 bis 30% EtOAc/Hexan über 30 min
bei 35 ml/min) ergibt etwa 781 mg (1,87 mmol, 93%) an N-Boc-4-[2-(3-Chlorpropan-1-sulfonylamino)phenyl]piperazin
als weißen
Feststoff.
LRMS (ESI+): 418,1 [M + 1].
-
Zu
einer Lösung
aus N-Boc-4-[2-(3-Chlorpropylamino)phenyl]piperazin (593 mg, 1,42
mmol) in 140 ml DMF wird NaH (567 mg einer 60% Dispersion in Öl, 14 mmol)
gegeben. Nach dem Rühren
für etwa
1 Stunde wird das Gemisch mit gesättigtem wässrigem Natriumbicarbonat gestoppt,
mit EtOAc verdünnt,
mit Wasser und Kochsalzlösung
gewaschen und dann getrocknet (Na2SO4), filtriert und unter Bildung von etwa
740 mg an N-Boc-4-[2-(1,1-Dioxoisothiazolidin-2-yl)phenyl]piperazin
konzentriert.
LRMS (ESI+): 382,1 [M + 1].
-
Präparation
92A 1-Boc-4-(2-Ethansulfonylaminophenyl)piperazin
-
Zu
einer Lösung
aus 1,0 g (4,4 mmol) an 1-Boc-4-(2-Aminophenyl)piperazin und 1,1
ml (6,6 mmol) Triethylamin in 12 ml DCM werden 0,63 ml, (6,6 mmol)
Ethansulfonylchlorid gegeben und das Gemisch wird bei RT für etwa 16
Stunden gerührt.
Das Gemisch wird mit Ethylacetat verdünnt und einmal mit 10% wässrigem Natriumbisulfat
und dann einmal mit gesättigtem
wässrigem
Natriumbicarbonat gewaschen. Die organische Portion wird getrocknet
(Na2SO4), filtriert
und im Vakuum konzentriert. Eine Silicagelchromatographie (Biotage, 40%
Ethylacetat/Hexan) des Rückstands
ergibt etwa 0,73 g (45%) der Titelverbindung.
LRMS (ESI–): 368
(M – 1).
-
Präparation
93A 1-Boc-4-(2-n-Butansulfonylaminophenyl)piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
92A hergestellt, mit der Ausnahme, dass n-Butansulfonylchlorid verwendet
wird.
LRMS (ESI+): 398 (M + 1).
-
Präparation
94A 1-Boc-4-[2-(Propan-2-sulfonylamino)phenyl]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
92A hergestellt, mit der Ausnahme, dass Propan-2-sulfonylchlorid
verwendet wird.
LRMS (ESI+): 384,3 (M + 1).
-
Präparation
95A 1-Boc-4-(2-Benzolsulfonylaminophenyl)piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
92A hergestellt, mit der Ausnahme, dass Benzolsulfonylchlorid verwendet
wird.
LRMS (ESI+): 418,1 (M + 1).
-
Präparation
96A 1-Boc-4-(2-Phenylmethansulfonylaminophenyl)piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
92A hergestellt, mit der Ausnahme, dass α-Toluolsulfonylchlorid verwendet
wird.
LRMS (ESI+): 432 (M + 1).
-
Präparation
97A 1-Boc-(2-Piperazin-1-ylphenyl)-N,N-dimethylsulfonimid
-
Zu
einer 0°C
wasserfreien Methylenchloridlösung
(10 ml) aus 1-Boc-4-(2-Aminophenyl)piperazin (1,0 g, 3,61 mmol)
und TEA (0,60 ml, 4,33 mmol) wird Dimethylsulfamoylchlorid (0,46
ml, 4,33 mmol) gegeben. Das Bad wird nach 5 Minuten entfernt und
die Reaktion wird unter einer Stickstoffatmosphäre für etwa 3 Tage gerührt und
für 1 Tag
am Rückfluss
erhitzt. Das Gemisch wird mit Methylenchlorid und 1 N HCl verdünnt. Die
abgetrennte wässrige
Phase wird mit Methylenchlorid (2×) extrahiert. Die vereinigten
organischen Phasen werden getrocknet (Natriumsulfat), filtriert
und unter Bildung eines rohen Öls
konzentriert. Eine Silicagelchromatographie (0 bis 5% Methanol in
Methylenchlorid) ergibt etwa 0,2 g (14%) des schließlichen
Produkts.
LRMS (ESI+): 385,3.
-
Präparation
98A 1-Boc-4-[2-(Acetylisobutylamino)phenyl]piperazin
-
Zu
einer Lösung
aus N-Boc-4-(2-Isobutylaminophenyl)piperazin (333 mg, 1,0 mmol,
1,0 Äquivalente), Et3N (0,42 ml, 3,0 mmol, 3,0 Äquivalente)
und DMAP (6 mg, 0,05 mol, 0,05 Äquivalente)
in DCM (10 ml) wird Essigsäureanhydrid
(0,14 ml, 1,5 mmol, 1,5 Äquivalente)
gegeben. Das Gemisch wird bei RT über Nacht gerührt. Die
Reaktion wird mit DCM (50 ml) verdünnt und mit gesättigtem
wässrigem
NaHCO3 (25 ml) und Kochsalzlösung (25
ml) gewaschen. Die organische Phase wird abgetrennt und die wässrige Phase
wird mit DCM (2×)
extrahiert. Die vereinigten organischen Extrakte werden getrocknet
(Na2SO4), filtriert
und unter Bildung der Titelverbindung (375 mg, 1,0 mmol, 100%) konzentriert.
LRMS
(ESI+): 376,18 (M + 1).
-
Präparation
99A 1-Boc-4-[2-(Isobutylmethoxycarbonylamino)phenyl]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
98A hergestellt, mit der Ausnahme, dass Methylchlorformiat an Stelle
von Essigsäureanhydrid
verwendet wird.
LRMS (ESI+): 392,2 (M + 1).
-
Präparation
100A 1-Boc-4-[2-(Isobutylisopropoxycarbonylamino)phenyl]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
98A hergestellt, mit der Ausnahme, dass Isopropylchlorformiat an
Stelle von Essigsäureanhydrid
verwendet wird.
LRMS (ESI+): 420,26 (M + 1).
-
Präparation
101A 1-Boc-4-[2-(Isobutylisobutoxycarbonylamino)phenyl]piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
98A hergestellt, mit der Ausnahme, dass Isobutylchlorformiat an
Stelle von Essigsäureanhydrid
verwendet wird.
LRMS (ESI+): 434,27 (M + 1).
-
Präparation
102A 1-Boc-4-{2-[2,2-Dimethylpropoxycarbonyl)isobutylamino]phenyl}piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
98A hergestellt, mit der Ausnahme, dass Neopentylchlorformiat an
Stelle von Essigsäureanhydrid
verwendet wird.
LRMS (ESI+): 448,32 (M + 1).
-
Präparation
103A 4-{2-[(1-Methyl-1H-imidazolylmethyl)amino]phenyl}piperazin
-
Zu
einer Lösung
aus 1-Boc-4-(2-Aminophenyl)piperazin (554 mg, 2,0 mmol, 1,0 Äquivalente)
in Methanol wird (1-Methyl-1H-imidazol-2-carbaldehyd (220 mg, 2,0
mmol, 1,0 Äquivalente)
gegeben. Das Gemisch wird am Rückfluss
für etwa
1 Stunde erhitzt und dann auf etwa 0°C gekühlt. Natriumborhydrid auf Aluminiumoxid
(10 Gewichtsprozent auf basischem Aluminiumoxid, 1,13 g, 3,0 mmol,
1,5 Äquivalente)
wird zugegeben. Die Lösung
wird auf RT erwärmt
und dann über
Nacht gerührt.
Das Reaktionsgemisch wird durch Celite filtriert und dann konzentriert.
Eine Reinigung durch Blitzchromatographie (35 g SiO2,
40 ml/min, linearer Gradient, 0–8%
MeOH/CH2Cl2 für 25 Minuten
und dann 8% MeOH für
7 Minuten) ergibt die Boc geschützte
Titelverbindung (176 mg, 0,47 mmol, 24%).
LRMS (ESI+): 372,3
[M + 1].
-
Präparation
104A 2-(N-Boc-Piperazin-1-yl)benzaldehyd
-
Zu
einer Lösung
aus 1-(2-Cyanophenyl)piperazin (375 mg, 2,0 mmol) in 15 ml Dioxan
wird DIBAL-H (6 ml einer 1 M Lösung
in Heptan, 6 mmol) gegeben. Nach dem Rühren bei RT für etwa 48
Stunden wird die Lösung
mittels einer Spritze in 20 ml an 0,5 M Rochelle Salz überführt. Nach
dem Rühren
für etwa
2 Stunden werden NaHCO3 (636 mg, 6 mmol)
und Boc2O (567 mg, 2,6 mmol) zugegeben.
Nach dem Rühren über Nacht werden
EtOAc und Kochsalzlösung
zugegeben. Nach der Auftrennung wird die wässrige Lösung mit EtOAc (3×) extrahiert.
Die vereinigten organischen Phasen werden mit Wasser und Kochsalzlösung gewaschen
und dann getrocknet (Na2SO4),
filtriert und konzentriert. Eine Reinigung mittels Blitzchromatographie
(35 g SiO2, linearer Gradient 10–20% EtOAc/Hexan über 30 min
bei 35 ml/min ergibt etwa 436 mg (1,50 mmol, 75%) der Titelverbindung
als gelbes Öl.
LRMS
(ESI+): 291,1 (M + 1).
-
Präparation
105A 1-Boc-4-(2-Pyrrolidin-1-ylmethylphenyl)piperazin
-
Zu
einer Lösung
aus 2-(N-Boc-Piperazin-1-yl)benzaldehyd (400 mg, 1,4 mmol) in Pyrrolidin
(0,33 ml, 4 mmol) wird Titanisopropoxid (1,2 ml, 4 mmol) gegeben
und das Gemisch wird bei RT unter einer Stickstoffatmosphäre gerührt. Nach
etwa 30 Minuten wird das Gemisch mit Ethanol (4 ml) verdünnt. Natriumborhydrid (106
mg, 2,8 mmol) wird zugegeben und das Gemisch wird für etwa 16
Stunden gerührt.
Wasser (2 ml) wird zugegeben und die entstehende Suspension wird
filtriert. Der Filterkuchen wird mit Methanol (5 ml) gewaschen und
das Filtrat wird zur Trockne konzentriert. Eine Reinigung mittels
Blitzchromatographie (1:1 Hexan/Ethylacetat) ergibt die Titelverbindung
(470 mg, 96%) als farbloses Öl.
1H NMR (CDCl3): δ 7,40–7,44 (m,
1H), 7,19–7,26
(m, 1H), 7,01–7,08
(m, 2H), 3,68 (s, 2H), 3,55 (t, J = 4,5 Hz, 4H), 2,92–2,95 (m,
4H), 2,53 (m, 4H), 1,75 (m, 4H), 1,49 (s, 9H).
TLC (SiO2): Rf = 0,28 (50%
EtOAc/Hexan).
-
Präparation
106A 1-Boc-4-(2-Piperidin-1-ylmethylphenyl)piperazin
-
Die
Titelverbindung wird auf dieselbe Weise wie in Präparation
105A beschrieben, hergestellt, mit der Ausnahme, dass Piperidin
verwendet wird.
1H NMR (CDCl3): δ 7,38
(d, J = 7,6 Hz, 1H), 7,20–7,26
(m, 1H), 7,03–7,08
(m, 2H), 3,54–3,57
(m, 4H), 3,50 (s, 2H), 2,92–2,95
(m, 4H), 2,40 (m, 4H), 1,23–1,59
(m, 15H).
TLC (SiO2): Rf =
0,52 (50% EtOAc/Hexan).
-
Präparation
107A 1-Boc-4-(2-Diethylaminomethylphenyl)piperazin
-
Die
Titelverbindung wird auf dieselbe Weise wie in Präparation
105A beschrieben hergestellt, mit der Ausnahme, dass Diethylamin
verwendet wird.
1H NMR (CDCl3): 7,54 (d, J = 7,3 Hz, 1H), 7,19–7,26 (m,
1H), 7,03–7,11
(m, 2H), 3,63 (s, 2H), 3,56 (t, J = 4,4 Hz, 4H), 2,88 (t, J = 4,6
Hz, 4H), 2,54 (q, J = 7,2 Hz, 4H), 1,49 (s, 9H), 1,03 (t, J = 7,2
Hz, 6H).
TLC (SiO2): Rf =
0,36 (50% EtOAc/Hexan).
-
Präparation
108A 1-Boc-4-(2-di-n-Butylaminomethylphenyl)piperazin
-
Die
Titelverbindung wird auf dieselbe Weise wie in Präparation
105A beschrieben hergestellt, mit der Ausnahme, dass Dibutylamin
verwendet wird.
1H NMR (CDCl3): δ 7,54–7,57 (m,
1H), 7,01–7,26
(m, 3H), 3,58 (s, 2H), 3,49–3,53
(m, 4H), 2,85–2,90
(m, 4H), 2,38 (t, J = 7,3 Hz, 4H), 1,40–1,50 (m, 13H), 0,84 (t, J
= 7,3 Hz, 6H).
TLC (SiO2): Rf = 0,70 (80% EtOAc/Hexan).
-
Präparation
109A 1-Boc-4-(2-Morpholin-4-ylmethylphenyl)piperazin
-
Zu
einer Lösung
aus 1-Boc-4-(2-Formylphenyl}piperazin (500 mg, 1,7 mmol) in Methanol
(10 ml) wird Morpholin (348 mg, 4,0 mmol) und Natriumcyanoborhydrid
(315 mg, 5 mmol) gegeben und das Gemisch wird für etwa 24 Stunden gerührt. Das
Gemisch wird mit Ethylacetat (100 ml) verdünnt und mit gesättigter
NaHCO3 Lösung
(10 ml), Wasser (10 ml) und Kochsalzlösung (10 ml) gewaschen. Die
organische Phase wird über
wasserfreiem Natriumsulfat getrocknet und konzentriert. Eine Silicagelchromatographie
(1:1 Hexan/Ethylacetat) ergibt die Titelverbindung als farbloses Öl (182 mg,
30%).
1H NMR (CDCl3): δ 7,38–7,40 (m,
1H), 7,21–7,27
(m, 1H), 7,04–7,09
(m, 2H), 3,67 (t, J = 4,4 Hz, 4H), 3,53–3,55 (s, 6H), 2,92–2,95 (m,
4H), 2,46–2,49
(m, 4H), 1,49 (s, 9H).
TLC (SiO2):
Rf = 0,44 (50% EtOAc/Hexan).
-
Präparation
110A 1-Boc-4-[2-(Isopropylaminomethyl)phenyl]piperazin
-
Die
Titelverbindung wird auf dieselbe Weise wie in Präparation
105A beschrieben, hergestellt, mit der Ausnahme, dass Isopropylamin
verwendet wird.
1H NMR (CDCl3): δ 7,30–7,00 (m,
4H), 3,80 (s, 2H), 3,60–3,45
(m, 4H), 2,95–2,85
(m, 4H), 2,85–2,80
(m, 1H), 1,50 (m, 9H), 1,10–1,00
(m, 6H).
TLC (SiO2): Rf =
0,10 (25% Ethylacetat/Hexan).
-
Präparation
111A 1-Boc-4-{2-[(Acetylisopropylamino)methyl]phenyl}piperazin
-
1-Boc-4-[2-(Isopropylaminomethyl)phenyl]piperazin
(0,325 g, 0,975 mmol) wird in Tetrahydrofuran (5 ml) gelöst und auf
etwa 0°C
gekühlt.
TEA (0,54 ml, 3,9 mmol) wird zugegeben gefolgt von der tropfenweisen Zugabe
von Acetylchlorid (0,2 ml, 2,93 mmol). Die Lösung kann sich auf RT erwärmen. Die
Lösemittel
werden unter verringertem Druck entfernt und das entstehende Öl wird mittels
Silicachromatographie (Ethylacetat) unter Bildung der Titelverbindung
(0,650 g, 82%) als Öl
gereinigt.
1H NMR (CDCl3) δ 7,25–6,95 (m,
4H), 4,65 (s, 1H), 4,45 (s, 1H), 3,70–3,50 (m, 4H), 2,90–2,80 (m,
4H), 1,50 (s, 9H), 1,30–1,20
(m, 1H), 1,10–1,00
(m, 6H).
-
Präparation
112A 1-Boc-4-[2-(Isopropylmethansulfonylaminomethyl)phenyl]piperazin
-
Die
Titelverbindung wird auf dieselbe Weise wie in Präparation
111A beschrieben, hergestellt, mit der Ausnahme, dass Methansulfonylchlorid
an Stelle von Acetylchlorid verwendet wird.
1H
NMR (CDCl3): δ 7,70–7,60 (m, 1H), 7,25–6,95 (m,
3H), 4,45 (s, 2H), 3,65 (s, 3H), 2,95–2,80 (m, 8H), 1,50 (s, 9H),
1,30–1,15
(m, 1H), 1,10–1,00
(m, 6H).
-
Präparation
113A 1-Boc-4-{2-[Hydroxy-(1-methyl-1H-imidazol-2-yl)methyl]phenyl}piperazin
-
Zu
einer Lösung
aus 1-Methylimidazol (350 μl,
4,4 mmol) in 15 ml THF bei –78°C wird n-BuLi
(1,5 ml einer 1,6 M Lösung
in Hexan, 2,4 mmol) gegeben. Nach dem Rühren für etwa 30 Minuten wird die
Lösung
auf etwa 0°C
erwärmt
und dann für
etwa 15 Minuten gerührt.
Das Gemisch wird dann auf etwa –78°C gekühlt. Eine Lösung aus
N-Boc-4-(2-Formylphenyl)piperazin (580 mg, 1,0 mmol) in 5 ml THF
wird mittels einer Spritze zugegeben. Die Lösung kann sich langsam auf
RT über
Nacht erwärmen.
Nach der Zugabe von gesättigtem wässrigem
NH4Cl und Kochsalzlösung wird die Lösung mit
EtOAc (2×)
extrahiert. Die vereinigten organischen Lösungen werden getrocknet (Na2SO4), filtriert
und konzentriert. Eine Reinigung durch Silicagelchromatographie
(35 g SiO2, 0 bis 10% an 0,2 M NH3 in MeOH/CH2Cl2 über
30 min bei 35 ml/min) ergibt etwa 592 mg (1,59 mmol, 79%) des Alkohols
als farbloses Öl.
LRMS
(ESI+): 373,2 (M + 1).
-
Präparation
114A 1-Boc-4-[2-(1-Methyl-1H-imidazol-2-carbonyl)phenyl]piperazin
und 1-Boc-4-{2-[Acetoxy-(1H-imidazol-2-yl)methyl]phenyl}piperazin
-
Zu
einer Lösung
aus 1-Boc-4-{2-[Hydroxy-(1H-imidazol-2-yl)methyl]phenyl}piperazin
(200 mg, 0,734 mmol) und Natriumbicarbonat (185 mg, 2,2 mmol) in
8 ml CH2Cl2 wird
Dess-Martin-Periodan (467 mg, 1,1 mmol) gegeben. Nach dem Rühren für etwa 1
Stunde werden 2 ml gesättigtes
wässriges
Natriumbicarbonat und 2 ml an 0,5 M Na2S2O3 zugegeben. Nach
dem Rühren
für etwa
1 Stunde wird die Lösung
mit CH2Cl2 verdünnt und
mit Wasser und Kochsalzlösung
gewaschen und dann getrocknet (Na2SO4), filtriert und konzentriert. Eine Reinigung
durch Silicagelblitzchromatographie (35 g SiO2,
0–5% an
0,2 M NH3 in MeOH/CH2Cl2) linearer Gradient über 30 min bei 35 ml/min) ergibt
etwa 53 mg (0,14 mmol, 19%) des Ketons {LRMS (ESI+): 371,2 [M +
1]} und etwa 114 mg (0,28 mmol, 37%) des Acetats {LRMS (ESI+): 415,2
[M + 1]}.
-
Präparation
115A 1-Boc-4-[2-(1-Methyl-1H-imidazol-2-ylmethyl)phenyl]piperazin
-
Zu
einer Lösung
aus 1-Boc-4-{2-[Hydroxy-(1H-imidazol-2-yl)methyl]phenyl}piperazin
(93 mg, 0,25 mmol) in 5 ml THF wird NaH (30 mg, 0,75 mmol) gegeben.
Nach dem Rühren
für etwa
45 Minuten wird CS2 (75 μl, 1,25 mmol) zugegeben. Nach
dem Rühren
für etwa
30 Minuten werden 5 ml THF gefolgt von MeI (78 μl, 1,25 mmol) zugegeben. Nach
dem Rühren
für etwa
1 Stunde wird gesättigtes
wässriges
NH4Cl und Kochsalzlösung zugegeben. Die Lösung wird
mit EtOAc (2×)
extrahiert. Die vereinigten organischen Lösungen werden getrocknet (Na2SO4), filtriert
und konzentriert. Eine Reinigung durch Silicagelchromatographie
(35 g, SiO2, 0 bis 5% MeOH/CH2Cl2) über
30 Minuten bei 35 ml/min) ergibt etwa 97 mg (0,21 mmol, 84%) des
Xanthats als gelbes Öl.
LRMS
(ESI+): 463,2 (M + 1).
-
Zu
einer Lösung
des Xanthats (90 mg, 0,195 mmol) und Bu3SnH
(260 μl,
0,967 mmol) in 2 ml Toluol bei 80°C
wird AIBN (50 μl
einer 0,4 M Lösung
in Toluol, 0,02 mmol) gegeben. Weitere 50 μl der AIBN Lösung werden alle 2 bis 3 Stunden
für 8 Stunden
zugegeben. Nach dem Rühren über Nacht
werden weitere 50 μl
der AIBN Lösung
zugegeben. Nach dem Rühren
für etwa
8 weitere Stunden wird die Lösung
konzentriert und durch Celite mit CH2Cl2 filtriert. Eine Reinigung mittels Silicagelchromatographie
(35 g SiO2, 0 bis 5% an 0,2 M NH3 in MeOH/CH2Cl2 über
30 Minuten bei 35 ml/min) ergibt etwa 46 mg (0,13 mmol, 66%) des
desoxygenierten Produkts als farbloses Öl.
LRMS (ESI+): 357,2
(M + 1).
-
Präparation
116A 1-Boc-4-(2-Thiazol-2-ylmethylphenyl)piperazin
-
Thiazol
wird mit n-Butyllithium lithiiert und mit N-Boc-4-(2-Formylphenyl)piperazin
auf ähnliche
Weise zu Präparation
113A umgesetzt. Der entstehende Alkohol wird auf ähnliche
Weise zu Präparation
115A unter Bildung der schließlichen
Verbindung desoxygeniert.
LRMS (ESI+): 360,1 (M + 1).
-
Präparation
117A 1-Boc-4-[2-(2-Methyl-2H-[1,2,4]triazol-3-ylmethyl)phenyl]piperazin
-
1-Methyltetrazol
wird mit n-Butyllithium lithiiert und mit N-Boc-4-(2-Formylphenyl)piperazin
auf ähnliche
Weise zu Präparation
113A umgesetzt. Der entstehende Alkohol wird auf ähnliche
Weise zu Präparation 115A
unter Bildung der schließlichen
Verbindung umgesetzt.
LRMS (ESI+): 358,3 (M + 1).
-
Präparation
118A 1-Boc-4-(2-Isobutoxyphenyl)piperazin
-
Zu
einer Lösung
aus 1-Boc-4-(2-Hydroxyphenyl)piperazin (560 mg, 2,0 mmol) in 10
ml DMF wird K2CO3 (835
mg, 6 mmol) gegeben. Nach dem Rühren
für etwa
5 Minuten wird Isobutyliodid (350 μl, 3 mmol) zugegeben Nach dem
Rühren über Nacht
bei 60°C
wird die Lösung
auf etwa 80°C
erwärmt.
Nach dem Rühren für etwa 4
Stunden wird die Lösung
auf RT gekühlt,
mit EtOAc verdünnt,
mit Wasser und Kochsalzlösung
gewaschen und dann getrocknet (Na2SO4), filtriert und konzentriert. Eine Reinigung
durch Silicagelchromatographie (35 g SiO2,
10 bis 30% EtOAc/Hexan, über
45 Minuten bei 35 ml/min) ergibt 418 mg (1,25 mmol, 62%) an N-Boc-4-(2-Isobutoxyphenyl)piperazin
als farbloses Öl.
LRMS
(ESI+): 335,1 [M + 1].
-
Präparation
119A 1-Boc-4-[2-(1-Methyl-1H-imidazol-2-ylmethoxy)phenyl]piperazin
-
Zu
einer Lösung
aus 1-Boc-4-(2-Hydroxyphenyl)piperazin (556 mg, 2,0 mmol, 1,0 Äquivalente),
(1-Methyl-1H-imidazol-2-yl)methanol (448 mg, 4,0 mmol, 2,0 Äquivalente),
Triphenylphosphin (1,04 g, 4,0 mmol, 2,0 Äquivalente) und THF bei 0°C unter Stickstoff
wird DEAD (0,629 ml, 4,0 mmol, 2,0 Äquivalente) langsam gegeben,
so dass die Temperatur der Reaktion nicht über 10°C steigt. Nachdem die Zugabe
vollständig
ist wird das Eisbad entfernt und das Gemisch wird bei RT über Nacht
gerührt.
Methanol wird zugegeben und das Gemisch wird für etwa 15 Minuten gerührt und
dann konzentriert. Eine Reinigung durch Blitzchromatographie (35 g,
SiO2, 40 ml/min, linearer Gradient 0–8% an 2,0
M NH3 in MeOH/CH2Cl2 für
25 Minuten und 8% an 2,0 M NH3 in MeOH für 7 Minuten)
ergibt die Titelverbindung (279 mg, 0,75 mmol, 37%).
LRMS (ESI+):
373,3 (M + 1).
-
Präparation
120A 1-Boc-4-(2-Benzyloxyphenyl)piperazin
-
Die
Titelverbindung wird auf ähnliche
Weise zu Präparation
118A hergestellt, außer
dass Benzylbromid verwendet wird, LRMS (ESI+): 369,1 (M + 1).
-
Präparation
121A 1-Boc-4-(2-Carboxyphenyl)piperazin
-
Zu
einer Lösung
aus 1-(2-Cyanophenyl)piperazin (37,45 g, 200 mmol) in 500 ml absolutem
Ethanol werden 1000 ml an 25% wässrigem
KOH gegeben. Die Lösung
wird am Rückfluss
für etwa
72 Stunden erhitzt und dann auf etwa 0°C gekühlt. Die Lösung wird mit 890 ml an 5 M
HCl angesäuert
und dann wird festes NaHCO3 zugegeben um
den pH der Lösung
auf etwa 8 zu bringen. NaHCO3 (12,7 g, 120
mmol) und Boc2O (11,4 g, 52,2 mmol) werden
zugegeben und das Gemisch wird über
Nacht gerührt
und dann mit 5 M HCl auf etwa pH 1 eingestellt. Nach der Zugabe
von EtOAc und Kochsalzlösung
wird die wässrige
Lösung
abgetrennt und mit EtOAc (2×)
extrahiert. Die vereinigten organischen Lösungen werden mit Wasser (2×) und Kochsalzlösung gewaschen
und dann getrocknet (Na2SO4),
filtriert und konzentriert. Das Material wird durch Umkristallisation
aus EtOAc/Hexan unter Bildung von 49,8 g (162 mmol, 81%) der Titelverbindung
gereinigt.
LRMS (ESI–):
305,2 (M – 1).
-
Präparation
122A (2-Piperazin-1-yl-phenyl)piperazin-1-yl-methanon
-
Das
1-Boc-4-(2-Carboxyphenyl)piperazin (1 g, 3,26 mmol), Piperidin (278
mg, 3,26 mmol), EDCl (625 mg, 3,26 mmol) und DMAP (50 mg, katalytisch)
werden in DCM (20 ml) gelöst
und bei RT für
etwa 12 Stunden gerührt.
Das Gemisch wird mit Wasser gewaschen, getrocknet, filtriert und
konzentriert. Der entstehende Schaum wird in DCM (10 ml) aufgenommen
und TFA (5 ml) wird zugegeben und das Gemisch wird bei RT für etwa 2
Stunden gerührt.
Die Reaktion wird konzentriert und einer SCX Ionenaustauschchromatographie
unterzogen, wonach eine Silicagelchromatographie unter Bildung des
schließlichen
Produkts (868 mg, 71%) als weißer
Schaum erfolgt.
LRMS (ESI+): 274,1 (M + 1).
-
Präparation
123A 1-Boc-4-[2-(2H-Tetrazol-5-yl)phenyl]piperazin
-
Eine
Lösung
aus 4-(2-Cyanophenyl)piperazin (1,7 g, 9,0 mmol, 1,0 Äquivalente)
in Azidotributylzinn (5,0 g, 15 mmol, 1,5 Äquivalente) wird bei 80°C für etwa 5
Tage gerührt.
Eine Reinigung durch SCX (10 g) Ionenaustauschchromatographie ergibt
rohes 4-(2-(-Tetrazol-5-yl-phenyl)piperazin.
LRMS (ESI+): 231,0
(M + 1).
-
Zu
einer Lösung
aus 4-[2-(2H-Tetrazol-5-yl-phenyl)piperazin (1,8 g, 7,7 mmol, 1,0 Äquivalente), NaHCO3 (978 mg, 9,2 mmol, 1,2 Äquivalente), DMAP (94 mg, 0,77
mmol, 0,1 Äquivalente)
in H2O: Dioxan (1:1) wird (Boc)2O
(1,6 g, 7,7 mmol, 1,0 Äquivalente)
gegeben. Das entstehende Gemisch wird bei RT über Nacht gerührt und
dann mit 1,0 M HCl neutralisiert und mit EtOAc (3×) extrahiert.
Die vereinigten organischen Extrakte werden getrocknet (Na2SO4), filtriert
und konzentriert. Eine Reinigung mittels Blitzchromatographie (35
g SiO2, linearer Gradient, 40 ml/min, 0–10% an
2,0 M NH3 in MeOH/CH2Cl2 für
20 Minuten und 10% an 2,0 M NH3 in MeOH/H2Cl2 für 13 Minuten)
ergibt die Titelverbindung (798 mg, 2,41 mmol, 32%).
LRMS (ESI+):
331,1 (M + 1).
-
Präparation
124A 4-[2-(2-Isobutyl-2H-tetrazol-5-yl)phenyl]piperazin
und 4-[2-(1-Isobutyl-1H-tetrazol-5-yl)phenyl]piperazin
-
Zu
einer Lösung
aus N-Boc-4-[2-(2N-Tetrazol-5-yl-phenyl)piperazin (330 mg, 1,0 mmol,
1,0 Äquivalente)
in DMF (10 ml) werden K2CO3 (331
mg, 2,4 mmol, 2,4 Äquivalente)
und Isobutyliodid (0,14 ml, 1,2 mmol, 1,2 Äquivalente) gegeben. Das Gemisch
wird bei RT über
Nacht gerührt.
Das Gemisch wird mit Ethylacetat (50 ml) verdünnt und mit H2O
(20 ml) und Kochsalzlösung
(20 ml) gewaschen. Die wässrigen
Phasen werden mit EtOAc (2×)
extrahiert. Die vereinigten organischen Extrakte werden getrocknet
(Na2SO4), filtriert
und unter Bildung von etwa 370 mg (96%, 0,96 mmol) eines Gemisches
(60:40 gemäß NMR) an
Boc geschützten
Titelverbindungen konzentriert, wobei das 2H substituierte Tetrazol
bevorzugt wird.
LRMS (ESI): 387,2 (M + 1).
-
Zu
einer Lösung
des Gemisches der Boc-geschützten
Verbindungen von oben (360 mg, 0,93 mmol, 1,0 Äquivalente) in CH2Cl2 (10 ml) werden TFA (5 ml) und DMS (0,25
ml) gegeben. Das entstehende Gemisch wird bei RT für etwa 2
Stunden gerührt.
Das Reaktionsgemisch wird konzentriert und mittels SCX (10 g) Ionenaustauschsäulenchromatographie
unter Bildung eines Gemisches der Titelverbindungen (240 mg, 0,84
mmol, 90%) gereinigt.
LRMS (ESI+): 287,1 (M + 1).
-
C Domänen-Präparationen
-
Die
geschützten
Aminosäurederivate
die den B und C Domänen
entsprechen sind in vielen Fällen
im Handel erhältlich.
Andere Aminosäurederivate
können
gemäß bekannter
Literaturverfahren (Siehe R. M. Williams, Synthesis of Oprically
Active α-Aminoacids,
Pergamon Press: Oxford, 1989) hergestellt werden. Im folgenden wird
die Herstellung der C-Domänen
gezeigt.
-
Präparation
1C 1-Methoxycarbonylmethyl-1,3-dihydroisoindol-2-carbonsäure-tert-butylester
-
Schritt A: (2-Brombenzyl)carbaminsäure-tert-butylester
-
Zu
einem Gemisch aus 125,0 g (561,8 mmol) an 2-Brombenzylaminhydrochlorid
und 170,7 g (1236,0 mmol) Kaliumcarbonat in 300 ml an 50% THF/Wasser
werden 134,9 g (618,0 mmol) an Di-tert-Butyldicarbonat in vier Portionen über 20 Minuten
gegeben. Das Gemisch wird bei RT für etwa 16 Stunden gerührt und
dann mit 300 ml Ethylacetat und 300 ml Wasser verdünnt. Die
organische Portion wird abgetrennt und die wässrige Portion wird jeweils
dreimal mit 200 ml Ethylacetat extrahiert. Die vereinigten Ethylacetatportionen
werden einmal mit 250 ml an 10% wässrigem Natriumbisulfat gewaschen.
Die organische Portion wird getrocknet (MgSO4),
filtriert und unter Bildung von etwa 161 g der Schritt A Verbindung
zur Trockne konzentriert.
-
Schritt B: 3-[2-(tert-Butoxycarbonylaminomethyl)phenyl]arylsäuremethylester
-
Zu
einer Verbindung von Schritt A (161,0 g, 561,8 mmol) in DMF (800
ml) werden Methylacrylat (58,0 g, 674 mmol), TEA (170,5 g, 1685,4
mmol) und Dichlorbis(triphenylphosphin)palladium(II) (7,9 g, 11,2
mmol) gegeben. Das Gemisch wird für etwa 32 Stunden auf 80°C erhitzt.
Das Gemisch wird abgekühlt,
mit 1000 ml EtOAc verdünnt
und mit 10% wässrigem
Natriumbisulfat gewaschen. Die wässrige
Portion wird dreimal mit EtOAc extrahiert und die vereinigten organischen
Bestandteile werden getrocknet (Na2SO4) und zur Trockne konzentriert. Der Rückstand
wird in einer kleinen Menge an DCM gelöst und durch 7 Inch Silicagel
in einer 2 l Glasnutsche unter Elution mit 25% EtOAc/Hexan filtriert.
Der Eluent wird zur Trockne konzentriert und aus EtOAc/Hexan unter
Bildung von etwa 116,9 g (71%) der Schritt B Verbindung umkristallisiert.
-
Schritt C:
-
Zu
einer 0°C
Lösung
aus dem Material von Schritt B (116,9 g, 401,2 mmol) in DCM (800
ml) werden 200 ml TFA tropfenweise über 15 Minuten gegeben. Nach
der Entfernung des Kühlbades
wird das Gemisch für
etwa 2,5 Stunden gerührt
und dann zur Trockne konzentriert. Der Rückstand wird in 500 ml DCM
gelöst und
gesättigtes
wässriges
Natriumbicarbonat wird langsam zugegeben bis das Gemisch leicht
basisch ist. Die organische Portion wird abgetrennt und die wässrige Portion
wird zweimal mit DCM extrahiert. Die vereinigten organischen Portionen
werden getrocknet (Na2SO4)
und zur Trockne konzentriert. Der Rückstand wird in 800 ml DCM
gelöst
und DIPEA (57,0 g, 441,4 mmol) wird zugegeben. Zu dem Gemisch wird
Di-tert-Butyldicarbonat (96,3 g, 441,4 mmol) in fünf Portionen über 45 Minuten
gegeben und dann bei RT für
16 Stunden gerührt.
Das Gemisch wird mit 10% wässrigem
Natriumbisulfat gewaschen und die organische Portion wird abgetrennt
und die wässrige
Portion wird zweimal mit DCM extrahiert. Die vereinigten organischen
Extrakte werden getrocknet (Na2SO4) und zur Trockne konzentriert. Der entstehende
Rückstand
wird in kleinen Mengen an DCM gelöst und durch 7 Inch Silicagel
unter Elution mit 25% EtOAc/Hexan in einer 2 l Glasnutsche filtriert.
Der Eluent wird zur Trockne konzentriert und die Enantiomere werden
durch Chiralchromatographie getrennt. Das zuerst eluierende Isomer
wird als Isomer Nr. 1 markiert und das als zweites eluierende Isomer
wird als Isomer Nr. 2 markiert, was etwa 52,6 g (45%) der schließlichen
Verbindung (Isomer 2) ergibt.
EIS-MS 292 [M + 1].
-
Präparation
2C 1-Carboxymethyl-1,3-dihydroisoindol-2-carbonsäure-tert-butylester
-
Zu
1-Methoxycarbonylmethyl-1,3-dihydroisoindol-2-carbonsäure-tert-butylester
(52,6 g, 180,5 mmol) in MeOH (500 ml) wird 1 N NaOH (199 ml, 199,0
mmol) gegeben. Das Gemisch wird bei RT für etwa 48 Stunden gerührt und
dann zur Trockne konzentriert. Der entstehende Rückstand wird in Wasser (300
ml) gelöst
und mit Diethylether (2×)
extrahiert. Die wässrige
Portion wird mit 10% wässrigem
Natriumbisulfat auf pH 2 angesäuert
und mit EtOAc extrahiert. Die vereinigten organischen Extrakte werden
getrocknet (MgSO4) und unter Bildung von
49,8 g der schließlichen
Verbindung (99%) zur Trockne konzentriert.
EIS-MS 276 [M – 1]
-
Präparation
3C (2-Isopropyl-2,3-dihydro-1H-isoindol-1-yl)essigsäure
-
Schritt A: (2,3-Dihydro-1H-isoindol-1-yl)essigsäuremethylester
-
Zu
der in Präparation
C1 (11,75 g, 40,41 mmol) in DCM (50 ml) hergestellten Präparation
C1 wird TFA (50 ml) tropfenweise gegeben. Nach etwa 2 Stunden wird
das Gemisch zur Trockne konzentriert und der entstehende Rückstand
wird zwischen gesättigtem
wässrigem
Natriumbicarbonat (200 ml) und EtOAc (300 ml) aufgeteilt. Die organische
Portion wird abgetrennt und die wässrige Phase wird mit DCM (4 × 500 ml)
extrahiert. Die vereinigten DCM Extrakte werden vereinigt, getrocknet
(Na2SO4) und unter
Bildung von etwa 3,97 g (51%) zur Trockne konzentriert.
-
Schritt B: (2-Isopropyl-2,3-dihydro-1H-isoindol-1-yl)essigsäuremethylester
-
Zu
der aus Schritt A (0,50 g, 2,61 mmol) in Dichlorethan (46 ml) erhaltenen
Verbindung werden Aceton (1,76 ml, 24,01 mmol) und Natriumtriacetoxyborhydrid
(2,48 g, 11,74 mmol) gegeben. Nach 6 Stunden wird das Gemisch mit
1,0 N NaOH (100 ml) verdünnt
und die organische Portion wird abgetrennt. Die wässrige Phase
wird mit DCM (3 × 100
ml) extrahiert. Die vereinigten DCM Extrakte werden getrocknet (MgSO4) und unter Bildung von etwa 0,60 g (99%)
zur Trockne konzentriert.
EIS-MS 235 [M + 1].
-
Schritt C:
-
Zu
der Verbindung von Schritt B (0,53 g, 2,30 mmol) in MeOH (5,1 ml)
wird 1,0 N NaOH (2,53 ml, 2,53 mmol) gegeben. Nach zwei Tagen wird
die Lösung
zur Trockne konzentriert. Der entstehende Rückstand wird mit 1,0 N HCl
und Wasser verdünnt
und wird auf eine starke Kationenaustauschersäule gegeben. Das Harz wird
mit Wasser, THF/Wasser (1:1) und dann Wasser gewaschen. Das Produkt
wird vom Harz mit Pyridin/Wasser (1:9) eluiert. Der Eluent wird
unter Bildung von etwa 0,43 g (85%) der schließlichen Verbindung zur Trockne konzentriert.
EIS-MS 220 [M + 1].
-
Präparation
4C (2-Methyl-2,3-dihydro-1H-isoindol-1-yl)essigsäure
-
Schritt A: (2-Methyl-2,3-dihydro-1H-isoindol-1-yl)essigsäuremethylester
-
Die
Verbindung von Präparation
C1 wird mit TFA auf ähnliche
Weise zu Präparation
3C von Schritt A von den Schutzgruppen befreit. Zu der von den Schutzgruppen
befreiten Verbindung (0,50 g, 2,61 mmol) in Dichlorethan (46 ml)
werden 37% wässrige
Formaldehydlösung
(1,80 ml, 24,01 mmol) und Natriumtriacetoxyborhydrid (2,48 g, 11,74
mmol) gegeben. Nach 3 Tagen wird das Gemisch mit 1,0 N NaOH (100
ml) verdünnt. Die
organische Portion wird abgetrennt und die wässrige Phase wird mit DCM (3 × 100 ml)
extrahiert. Die vereinigten DCM Extrakte werden getrocknet (Na2SO4) und zur Trockne
konzentriert. Der entstehende Rückstand wird
durch Blitzchromatographie (SiO2, Elution
mit 100% EtOAc) unter Bildung von etwa 0,43 g (79%) des alkylierten
Isoindols gereinigt.
EIS-MS 206 [M + 1]
-
Schritt B:
-
Zu
der Verbindung von Schritt A (0,34 g, 1,66 mmol) in MeOH (3,7 ml)
wird 1,0 N NaOH (1,82 ml, 1,82 mmol) gegeben. Nach 2 Tagen wird
die Lösung
zur Trockne konzentriert. Der entstehende Rückstand wird mit 1,0 N HCl
und Wasser verdünnt
und wird dann auf ein starkes Kationenaustauscherharz gegeben. Das
Harz wird mit Wasser, THF/Wasser (1:1) und Wasser gewaschen und
das Produkt wird aus dem Harz mit Pyridin/Wasser (1:9) eluiert.
Der Eluent wird unter Bildung von etwa 0,31 g (98%) der schließlichen
Verbindung zur Trockne konzentriert.
EIS-MS 192 [M + 1].
-
-
Die
obige Verbindung wird aus Boc-L-Tic-OH wie in Präparation 6C unten beschrieben
hergestellt, mit der Ausnahme, dass das Weinrebamid durch ein ähnliches
Verfahren zu dem in Synthesis, 676, 1983 beschriebenen, hergestellt
wird.
-
-
Boc-D-Tic-OH
(14,9 g, 53,7 mmol), Methoxymethylaminhydrochlorid (5,24 g, 53,7
mmol), EDC (11,3 g, 59,1 mmol), HOBT (7,98 g, 59,1 mmol), DIEA (9,83
ml, 59,1 mmol) und THF (500 ml) werden vereinigt und das entstehende
Gemisch wird für
etwa 18 Stunden bei RT unter Stickstoff gerührt. Das Reaktionsgemisch wird
konzentriert und der Rückstand
wird in Ethylacetat aufgenommen. Das entstehende Gemisch wird mit
1 M HCl, gesättigtem
NaHCO3 und Kochsalzlösung gewaschen, und dann durch
Filtration durch ein Phasentrennpapier getrocknet. Die Entfernung
des Lösemittels
ergibt einen Rückstand,
der auf Silicagel mittels (1:1 Ethylacetat/Hexan) unter Bildung
von etwa 12,3 g an Boc-D-Tic-NMeOMe
(Weinrebamid) chromatographiert wird.
-
Lithiumaluminiumhydrid
(1,0 M in THF, 5,1 ml, 5,00 mmol) wird langsam zu dem oben hergestellten Weinrebamid
(1,28 g, 4,00 mmol) in THF (35 ml) bei 0°C gegeben. Das Reaktionsgemisch
wird bei 0°C
für etwa
15 Minuten gerührt.
Wässriges
KHSO4 (970 mg in 20 ml H2O)
wird langsam gefolgt von Diethylether zugegeben. Die organische
Phase wird abgetrennt und die wässrige
Phase wird mit Diethylether extrahiert. Die organischen Phasen werden
vereinigt und mit wässriger
1 M HCl, gesättigtem
wässrigem
NaHCO3 und Kochsalzlösung gewaschen, das dann über Na2SO4 getrocknet wird.
Eine Entfernung des Lösemittels
ergibt etwa 780 mg der schließlichen
Verbindung. MS: MH+ 262.
-
Präparation
7C (2-Butyl-2,3-dihydro-1H-isoindol-1-yl)essigsäuremethylester
-
Die
Verbindung von Präparation
C1 wird mit TFA auf ähnliche
Weise zu Präparation
3C von Schritt A von den Schutzgruppen befreit. Zu der von den Schutzgruppen
befreiten Verbindung (0,50 g, 2,61 mmol) und Butyraldehyd (2,16
ml, 24,01 mmol) in Dichlorethan (46 ml) wird Natriumtriacetoxyborhydrid
(2,48 g, 11,74 mmol) gegeben. Nach der Umsetzung für etwa 3
Stunden wird das Gemisch mit 1,0 N NaOH (100 ml) verdünnt und
aufgeteilt. Die wässrige
Phase wird mit DCM (3 × 75
ml) extrahiert. Die DCM Phasen werden vereinigt, über Natriumsulfat
getrocknet, filtriert und unter verringertem Druck unter Bildung
eines braunen Rückstands konzentriert.
Der Rückstand
wird durch Silicagelchromatographie (Eluent: Ethylacetat/Hexan (1:3))
gereinigt. Die gereinigten Fraktionen werden vereinigt und unter
Bildung der Titelverbindung als braunes Öl konzentriert. (0,51 g, 77%).
MS
ES 249,2 (M + H).
-
Präparation
8C (2-Butyl-2,3-dihydro-1H-isoindol-1-yl)essigsäure
-
Zu
einer Lösung
die die Verbindung 7C (0,47 g, 1,89 mmol) in Methanol (4,2 ml) enthält, wird
1,0 N NaOH (2,08 ml, 2,08 mmol) gegeben. Nach der Umsetzung für etwa 2
Stunden wird die Lösung
unter verringertem Druck konzentriert. Der Rückstand wird mit 1,0 N HCl
verdünnt
und Wasser wird auf ein starkes Kationenaustauscherharz gegeben.
Das Harz wird mit Wasser und THF/Wasser (1:1) gewaschen und das
Produkt wird aus dem Harz mit Pyridin/Wasser (1:9) eluiert. Die
Pyridinwaschlösungen
werden unter verringertem Druck konzentriert und mit Aceton unter
Bildung der Titelverbindung als braune Feststoffe (0,28 g, 64%)
azeotrop destilliert.
MS ES 234,9 (M + H).
-
-
Schritt A:
-
Zu
einer Lösung
aus N-Boc-4-Fluor-D-Phe (2,37 g, 8,366 mmol) in Methanol werden
3 ml konzentrierte Schwefelsäure
gegeben. Das Gemisch wird am Rückfluss über Nacht
erhitzt und dann im Vakuum konzentriert.
MS (M + 1) 198,1
-
Schritt B:
-
Zu
einem eisgekühlten
Gemisch aus 1,65 g (8,367 mmol) der Verbindung von Schritt A, werden
1,353 ml Pyridin und Ethylchlorformiat (0,848 ml, 8,869 mmol) langsam
unter Rühren
für etwa
30 Minuten gegeben, wobei sich ein weißer Feststoff bildet. Das Gemisch
wird zwischen Wasser und Ethylacetat aufgeteilt. Die wässrige Phase
wird mit EtOAc (2×)
extrahiert. Die vereinigte organische Lösung wird über MgSO4 getrocknet, filtriert
und im Vakuum unter Bildung von etwa 2,17 g eines gelben Öls (96%)
konzentriert.
MS M + 1 270,1.
-
Schritt C:
-
Eine
Gemisch, das 2,17 g (8,06 mmol) der Verbindung von Schritt B, Paraformaldehyd
(0,254 g, 8,46 mmol) und 10 ml einer 3:1 Eisessig/konz. Schwefelsäure enthält wird
bei RT für
etwa 48 Stunden gerührt.
Das Gemisch wird zwischen Wasser und Ethylacetat aufgeteilt. Die
wässrige
Phase wird mit EtOAc (3×)
extrahiert. Die vereinigte EtOAc Lösung wird über Magnesiumsulfat getrocknet,
filtriert und im Vakuum konzentriert. Das gewünschte Produkt wird durch Säulenchromatographie
unter Elution mit 25% EtOAc in Hexan unter Bildung von etwa 1,31
g (58%) eines farblosen Öls
gereinigt.
MS: M + 1 282,1
-
Schritt D:
-
Eine
Lösung
von 1,31 g (4,656 mmol) des Materials von Schritt C in 20 ml an
5 N HCl wird am Rückfluss
für etwa
24 Stunden erhitzt. Die Lösung
wird im Vakuum konzentriert. Der entstehende weiße Feststoff wird mit Ether
unter Bildung von etwa 0,87 g (81%) gewaschen.
MS M + 1 196,1.
-
Schritt E:
-
Zu
einer Lösung
aus 0,87 g (3,755 mmol) des Materials von Schritt D in 20 ml 1:1
Dioxan/Wasser werden Di-t-Butyldicarbonat (0,901 g, 4,131 mmol)
und 2,355 ml (16,90 mmol) TEA gegeben. Das Gemisch kann bei RT über Nacht
rühren.
Das Gemisch wird mit EtOAc verdünnt
und die abgetrennte wässrige
Phase wird mit EtOAc (3×)
extrahiert. Die vereinigte organische Lösung wird über Magnesiumsulfat getrocknet,
filtriert und im Vakuum unter Bildung von etwa 0,64 g (58%) der
schließlichen
Verbindung konzentriert.
MS M – 1 294,1.
-
-
Schritt A:
-
Gemäß dem Verfahren
von Präparation
28C, Schritt A und 1,0 g (5,58 mmol) an α-Methyl-DL-phenylalanin, werden etwa 1,4 g des
Esters hergestellt.
MS M + 1 194,1
-
Schritt B:
-
Gemäß dem Verfahren
von Präparation
28C, Schritt B und 1,08 g (5,59 mmol) des Materials von Schritt
A, werden etwa 1,48 g (100%) des Produkts hergestellt.
MS M
+ 1 266,1
-
Schritt C:
-
Gemäß dem Verfahren
von Präparation
28C, Schritt C und 1,48 g (5,59 mmol) des Materials von Schritt
B, werden etwa 1,55 g (100%) des Produkts hergestellt.
MS M
+ 1 278,1
-
Schritt D:
-
Gemäß dem Verfahren
von Präparation
28C, Schritt D und 1,55 g (5,59 mmol) des Materials von Schritt
C, werden etwa 1,33 g des Produkts hergestellt.
MS M + 1 192,1
-
Schritt E:
-
Gemäß dem Verfahren
von Präparation
28C, Schritt E und 1,33 g (5,84 mmol) des Materials von Schritt
D, werden etwa 1,70 g (100%) der schließlichen Verbindung hergestellt.
MS
M + 1 292,2.
-
-
Schritt A:
-
Gemäß dem Verfahren
von Präparation
28C, Schritt A und 2,0 g (11,16 mmol) α-Methyl-D-phenylalanin werden etwa 2,15 g des
Esters hergestellt.
MS M + 1 194,1.
-
Schritt B:
-
Gemäß dem Verfahren
von Präparation
28C, Schritt B und 2,15 g (11,16 mmol) des Materials von Schritt
A, werden etwa 1,46 g (49%) des Produkts hergestellt.
MS M
+ 1 266,1
-
Schritt C:
-
Gemäß dem Verfahren
von Präparation
28C, Schritt C und 1,46 g (5,503 mmol) des Materials von Schritt
B, werden etwa 0,74 g (48%) des Produkts hergestellt.
MS M
+ 1 278,1.
-
Schritt D:
-
Gemäß dem Verfahren
von Präparation
28C, Schritt D und 0,74 g (2,67 mmol) des Materials von Schritt
C, werden etwa 0,54 g (89%) des Produkts hergestellt.
MS M
+ 1 192,1.
-
Schritt E:
-
Gemäß dem Verfahren
von Präparation
28C, Schritt E und 0,54 g (2,37 mmol) des Materials von Schritt
D, werden etwa 0,54 g (78%) der schließlichen Verbindung hergestellt.
MS
M + 1 292,2.
-
-
Schritt A:
-
Gemäß dem Verfahren
von Präparation
28C, Schritt A und 0,65 g (1,95 mmol) N-Boc-4-trifluormethyl-D-phenylalanin werden
etwa 0,48 g des Esters hergestellt.
MS M + 1 248,0.
-
Schritt B:
-
Gemäß dem Verfahren
von Präparation
28C, Schritt B und 0,48 g (1,95 mmol) des Materials von Schritt
A, werden etwa 0,60 g (96%) des Produkts hergestellt.
MS M
+ 1 320,1
-
Schritt C:
-
Gemäß dem Verfahren
von Präparation
28C, Schritt C und 0,6 g (1,879 mmol) des Materials von Schritt
B, werden etwa 0,37 g (59%) des Produkts hergestellt.
MS M
+ 1 332,1.
-
Schritt D:
-
Gemäß dem Verfahren
von Präparation
28C, Schritt D und 0,37 g (1,117 mmol) des Materials von Schritt
C, werden etwa 0,11 g (35%) des Produkts hergestellt.
MS M
+ 1 246,1.
-
Schritt E:
-
Gemäß dem Verfahren
von Präparation
28C, Schritt E und 1,11 g (0,391 mmol) des Materials von Schritt
D, werden etwa 0,234 g (> 100%)
der schließlichen
Verbindung hergestellt.
MS M – 1 344,1.
-
Präparation
13C Lithium-(2-tert-butoxycarbonyl-1,2,3,4-tetrahydroisochinolin-1-yl)acetat
-
Schritt 1: (1,2,3,4-Tetrahydroisochinolin-1-yl)essigsäuremethylester
-
Zu
einer Lösung
aus 100,4 g (52 mmol) an Boc-Tetrahydroisochinolin-1-essigsäure (100,4
g, 520,0 mmol) in 200 ml Methanol werden 400 ml an 2,3 M HCl in
Methanol gegeben. Das Gemisch wird über Nacht gerührt und
im Vakuum konzentriert. Der Rückstand
wird in Ethylacetat gelöst
und mit gesättigtem
Natriumbicarbonat und Kochsalzlösung
gewaschen und dann getrocknet (Na2SO4) und im Vakuum unter Bildung von etwa 109,5
g (100%) der Titelverbindung konzentriert.
EIS-MS: 206 (M +
1).
-
Schritt 2: 1-Methoxycarbonylmethyl-3,4-dihydro-1H-isochinolin-2-carbonsäure-tert-butylester
-
Zu
einer 0°C
Lösung
des Materials von Schritt 1 (50,5 g, 240,0 mmol) in 250 ml trockenem
THF wird Di-tert-butyldicarbonat (59,3 g, 270,0 mmol) in 50 ml tropfenweise
gegeben. Nach dem Rühren
für etwa
45 Minuten wird das Gemisch im Vakuum konzentriert. Der Rückstand
wird in Ethylacetat gelöst,
mit gesättigtem Natriumbicarbonat
und Kochsalzlösung
gewaschen und dann getrocknet (Na2SO4) und im Vakuum konzentriert. Eine Chromatographie
des Rückstands
ergibt beide Enantiomere der Titelverbindung.
EIS-MS: 306 (M
+ 1).
-
Schritt 3:
-
Zu
einer Lösung
des Materials von Schritt 2 (10,2 g, 33,4 mmol) in 220 ml Dioxan
wird eine Lösung aus
Lithiumhydroxidmonohydrat (1,67 g, 39,8 mmol) in 110 ml Wasser portionsweise
gegeben, um die Temperatur unter 30°C zu halten. Das Gemisch wird
für etwa
16 Stunden gerührt
und im Vakuum unter Bildung von etwa 11,2 g der schließlichen
Verbindung konzentriert.
EIS-MS: 292 (M + 1).
-
Präparation
14C Lithium-(2-methyl-1,2,3,4-tetrahydroisochinolin-1-yl)acetat
-
Schritt 1: (1,2,3,4-Tetrahydroisochinolin-1-yl)essigsäuremethylester
-
Das
Material von Präparation
13C Schritt 2 (9,98 g, 32,7 mmol) wird mit 500 ml kaltem 4 M HCl/Dioxan gemischt
und bei RT für
etwa eine Stunde gerührt.
Das Gemisch wird im Vakuum konzentriert. Der Rückstand wird in Ethylacetat
gelöst
und dann mit gesättigtem
Natriumbicarbonat und Kochsalzlösung
gewaschen. Die organische Portion wird getrocknet (Na2SO4), filtriert und im Vakuum unter Bildung
von etwa 6,9 g (100%) der Titelverbindung konzentriert.
EIS-MS:
206 (M + 1).
-
Schritt 2: (2-Methyl-1,2,3,4-tetrahydroisochinolin-1-yl)essigsäuremethylester
-
Zu
einer Lösung
des Materials von Schritt 1 (6,71 g, 32,0 mmol) in 175 ml Dichlorethan
wird 37 wässriges
Formaldehyd (22,6 ml, 300 mmol) gegeben. Nach etwa 10 Minuten wird
Natriumtriacetoxyborhydrid (31,2 g, 147,0 mmol) in 2 oder 3 g Portionen
unter einigem Kühlen
zur Aufrechterhaltung der Umgebungstemperatur zugegeben. Das Gemisch
wird für
etwa 16 Stunden gerührt
und DCM und Wasser werden zugegeben. Das Gemisch wird mit 5 N Natriumhydroxid
auf pH 9–10
eingestellt. Die organische Phase wird abgetrennt, mit Kochsalzlösung gewaschen
und dann getrocknet (Na2SO4)
und im Vakuum konzentriert. Eine Chromatographie (Silicagel, 5%
(2 N Ammoniak in Methanol)/DCM) des Rückstands ergibt etwa 6,9 g
(96%) der Titelverbindung.
EIS-MS: 220 (M + 1).
-
Schritt 3:
-
Zu
einer Lösung
des Materials von Schritt 2 (4,45 g, 18,9 mmol) in 120 ml Dioxan
wird Lithiumhydroxidmonohydrat (1,02 g, 22,7 mmol) in 65 ml Wasser
in Portionen gegeben um die Temperatur unter 30°C zu halten. Nach etwa 16 h
wird das Gemisch im Vakuum unter Bildung von etwa 8,12 g der schließlichen
Verbindung konzentriert.
EIS-MS: 206 (M + 1).
-
Präparation
15C 1,1-Dimethyl-6-methoxy-1,2,3,4-tetrahydroisochinolin-3-carbonsäureethylester
-
Zu
einer Lösung
des Triflatsalzes aus 1,1-Dimethyl-1,2,3,4-tetrahydroisochinolin-3-carbonsäureethylester
(1,5 g, 3,76 mmol, 1,0 Äquivalente)
in MeOH (20 ml) und CH2Cl2 (2
ml) bei 0°C
wird eine Lösung
aus (Trimethylsilyl)diazomethan (2,0 M in Hexan, 3,7 ml, 2,0 Äquivalente)
gegeben. Das entstehende Gemisch wird auf RT erwärmt und über Nacht gerührt und
dann wird die Lösung
konzentriert. Eine Reinigung durch Blitzchromatographie (125 g SiO2 linearer Gradient, 40 ml/min, 1:1 EtOAc/Hexan
für 33
Minuten) ergibt etwa 900 mg der schließlichen Verbindung (96%).
LRMS
(Elektrospray): 250,2 (M + 1).
-
"A-Domäne" und "B Domäne" Kombination Präparation
1AB und 2AB 1-(D-p-Cl-Phe)-4-(2-Methansulfonylphenyl)piperazin
und 1-(D-p-Cl-Phe)-4-(2-Methansulfinylphenyl)piperazin
-
Im
Handel erhältliches
1-(2-Methylthiophenyl)piperazin wird an Boc-p-Cl-D-Phe-OH auf eine
Weise die im wesentlichen ähnlich
zu der im Kupplungsverfahren 1 beschriebenen ist, gekuppelt. Zu
einer Lösung des
Kupplungsprodukts (100 mg, 0,204 mmol) in 5 ml CH2Cl2, die auf –78°C gekühlt ist, wird m-Chlorperbenzoesäure (49
mg, 0,204 mmol) gegeben. Nach dem Rühren für etwa 30 Minuten wird die
Reaktion mit 1 M Na2S2O3 gestoppt und mit CH2Cl2 extrahiert. Die vereinigten organischen
Lösungen
werden mit gesättigtem Natriumbicarbonat
gewaschen, getrocknet (Na2SO4),
filtriert und konzentriert. Eine Reinigung mittels Blitzchromatographie
(10 g SiO2, linearer Gradient 0–10% Methanol/CH2Cl2, 30 ml/Minute, über 30 Minuten)
ergibt etwa 46 mg (0,090 mmol, 43%) des Sulfoxids und 60 mg (0,115
mmol, 56%) des Sulfons. Jedes hiervon wird getrennt auf im wesentlichen ähnliche
Weise zu der im Kupplungsverfahren 1 beschriebenen von den Schutzgruppen
befreit
-
-
1-(2-Nitrophenyl)piperazin
(3,13 g, 15,1 mmol), Boc-D-4-Chlorphenylalanin (4,52 g, 15,1 mmol),
EDC (3,19 g, 16,6 mmol), HOBT (2,21 g, 16,7 mmol) und DIEA (2,63
ml, 15,1 mmol) werden zu THF gegeben. Das entstehende Gemisch wird über Nacht
bei RT unter Stickstoff gerührt.
Das Reaktionsgemisch wird dann im Vakuum konzentriert. Der Rückstand
wird in Ethylacetat aufgenommen und mit 1 M HCl gewaschen, mit NaHCO3 und Kochsalzlösung verdünnt und dann mit Na2SO4 getrocknet.
Eine Entfernung des Lösemittels
ergibt einen Rückstand,
der auf einer normalen Phase (Ethylacetat/Hexan 1:1) unter Bildung
von etwa 6,8 g der Boc-geschützten
Verbindung chromatographiert wird.
-
Die
Boc geschützte
Verbindung (6,88 g, 14,1 mmol) wird in 4 M HCl/Dioxan (230 ml) gelöst und das entstehende
Gemisch wird bei RT für
etwa eine Stunde gerührt.
Das Gemisch wird im Vakuum unter Bildung von etwa 5,1 g der schließlichen
Verbindung konzentriert.
-
-
Das
Piperazingemisch aus Präparation
54A (6,99 g, 28,76 mmol), N-Boc-D-Cl-Phe (8,624 g, 28,76 mmol),
HATU (10,94 g, 28,76 mmol) und DIEA (25,05 ml, 143,8 mmol) in 160
ml DCM wird bei RT über
Nacht gerührt.
Das Gemisch wird zwischen Wasser und CH2Cl2 aufgeteilt. Die wässrige Phase wird mit CH2Cl2 (2×) extrahiert.
Die vereinigte organische Lösung
wird über
MgSO4 getrocknet, filtriert und im Vakuum
konzentriert. Der Rückstand
wird durch eine Silicagelsäule
mittels 10% MeOH in EtOAc unter Bildung des Boc geschützten Produkts
gereinigt. Die Boc geschützte
Verbindung wird mit 1:1 TFA/CH2Cl2 behandelt. Das Gemisch wird bei RT für etwa 2
Stunden gerührt
und dann im Vakuum unter Bildung der schließlichen Verbindung (13,9 g,
74%) konzentriert.
MS M + 1 425,2.
-
Präparation
5AB 1-(D-p-Cl-Phe)-4-1-[(2-Aminosulfonyl)phenyl]piperazin
-
1-[(2-Aminosulfonyl)phenyl]piperazin
aus Präparation
19A wird an Boc-p-Cl-D-Phe-OH gemäß Schutzgruppenabspaltung und
HCl Salzbildung auf ähnliche
Weise zu dem Kupplungsverfahren 1, Schritte 1 und 4, gekuppelt.
-
"B Domäne" und "C-Domäne" Kombination Präparation
1BC N-Boc-D-Tic-D-p-Cl-Phe-OH
-
Schritt 1:
-
Das
HCl Salz von H-D-p-Cl-Phe-OMe (35,8 g, 129 mmol) wird in Wasser
(200 ml) gelöst.
Ethylacetat (200 ml) wird zugegeben, gefolgt von der Zugabe einer
gesättigen
Natriumbicarbonatlösung.
Das Gemisch wird für
etwa 5 Minuten gerührt
und dann wird die organische Phase abgetrennt, mit Wasser (200 ml)
gewaschen und über
Magnesiumsulfat getrocknet. Eine Konzentration des Gemisches unter verringertem
Druck ergibt einen weißen
Feststoff (32,2 g). Der Feststoff wird dann in Methylenchlorid (200
ml), D-Boc-Tic (35,8 g, 129 mmol) und 4-Dimethylaminopyridin (75
mg) gelöst.
Das Gemisch wird auf 0°C
gekühlt
und EDC (24,7 g, 129 mmol) wird in zwei Portionen zugegeben. Nach
dem Rühren
für etwa
20 Minuten wird das Eisbad entfernt und die Lösung kann sich auf RT erwärmen. Die
Lösung
wird für
etwa 4 Stunden gerührt
und dann mit Wasser (400 ml) verdünnt. Die organische Phase wird
mit Wasser (3×)
gewaschen, über
Magnesiumsulfat getrocknet und unter verringertem Druck unter Bildung
eines klaren Öls
(70 g) konzentriert. Eine Säulenchromatographie (35%
Ethylacetat/Heptan) ergibt etwa 55,6 g des Zwischenprodukts Boc-D-Tic-D-p-Cl-Phe-OMe
(85%).
1H NMR (DMSO) (Zwei Rotamere
werden beobachtet) δ 8,26
(d, 1H), 8,19 (d, 0,5H), 7,24 (d, 2H), 7,00–7,19 (m, 8H), 4,68 (m, 0,5H),
4,20–4,60
(m, 4,5H), 3,58 (s, 3H), 3,51 (s, 1,5H), 2,77–3,10 (m, 6H), 1,42 (s, 3H),
1,21 (s, 9H).
MS (ES) 473,0 (M+), 471,1
(M–).
-
Schritt 2:
-
Die
Verbindung von Schritt 1 (54,3 g, 114 mmol) wird in Methanol (170
ml) gelöst.
Die Lösung
wird mit einem Eisbad auf 0°C
gekühlt
und 1 N NaOH (290 ml) wird tropfenweise zugegeben. Nach kräftigem Rühren für etwa 20
Minuten wird das Gemisch auf etwa 25°C erwärmt. Die Lösung wird unter verringertem
Druck unter Bildung eines gelben Öls konzentriert. Das Öl wird in
Wasser (200 ml) gelöst
und der pH wird auf etwa 1 eingestellt. Ethylacetat (200 ml) wird
zugegeben und die organische Phase wird abgetrennt und über Magnesiumsulfat
getrocknet. Eine Konzentration der organischen Bestandteile ergibt
etwa 46,3 g der schließlichen
Verbindung.
1H NMR (DMSO) (Zwei Rotamere
werden beobachtet) δ 7,98
(d, 1H), 7,82 (d, 0,5H), 6,90–7,41
(m, 16H), 4,20–4,70
(m, 8,5H), 2,60–3,20
(m, 8,5H), 1,32–1,41
(m, 19H).
MS (ES) 459,1 m/z (M+), 457,1
(M–).
-
-
Die
obige Verbindung wird mittels N-Boc-L-Tic-OH wie in Präparation
1BC beschrieben, hergestellt.
1H NMR
(DMSO) (Zwei Rotamere werden beobachtet) δ 7,98 (d, 1H), 7,72 (d, 0,5H),
6,90–7,41
(m, 16H), 4,0–4,70
(m, 8,5H), 2,60–3,20
(m, 8,5H), 1,32–1,41
(m, 19H).
MS (ES) 459,1 m/z (M+), 457,1
(M–).
-
Präparation
3BC Lithium-2-[(2-tert-butoxycarbonyl-1,2,3,4-tetrahydroisochinolin-3-ylmethyl)amino]-3-(4-chlorphenyl)propionat
-
Schritt A:
-
3-{[2-(4-Chlorphenyl)-1-methoxycarbonylethylamino]methyl}-3,4-dihydro-1H-isochinolin-2-carbonsäure-tert-butylester
-
Zu
einer 0°C
Lösung
aus 4-Cl-D-Phe-OMe (6,27 g, 25,1 mmol) und Natriumacetat (8,23 g,
100,0 mmol) in 850 ml trockenem MeOH wird der Aldehyd von Präparation
6C (9,8 g, 37,6 mmol) in 50 ml MeOH gegeben. Das Gemisch wird für etwa 15
Minuten gerührt
und dann wird Natriumcyanoborhydrid (2,37 g, 37,6 mmol) zugegeben.
Das Kühlbad
wird entfernt und die Reaktion wird für 16 Stunden bei RT gerührt. Das
Gemisch wird zur Trockne konzentriert und der entstehende Rückstand
wird in Wasser und 1 ml an 1 M HCl aufgenommen. Das Gemisch wird
mit EtOAc extrahiert und die organischen Bestandteile werden mit
gesättigtem Natriumbicarbonat
und Kochsalzlösung
gewaschen und dann getrocknet (Na2SO4) und zur Trockne konzentriert. Der entstehende
Rückstand
wird mittels Blitzchromatographie (SiO2,
Elution mit 2:1 Hexan/EtOAc) unter Bildung von etwa 8,62 g (75%)
gereinigt.
EIS-MS 459 [M + 1].
-
Schritt B:
-
Zu
einer 12°C
Lösung
des Materials von Schritt A (1,11 g, 2,42 mmol) in Dioxan (15 ml)
wird eine Lösung
aus Lithiumhydroxid (0,10 g, 2,42 mmol) in Wasser (7,5 ml) gegeben.
Das Gemisch wird für
etwa 16 Stunden gerührt
und dann zur Trockne unter Bildung von etwa 1,08 g (100%) der schließlichen
Verbindung konzentriert. EIS-MS 445 [M + 1].
-
Präparation
4BC Lithium-2-[(2-tert-butoxycarbonyl-1,2,3,4-tetrahydroisochinolin-3-ylmethyl)amino]-3-(4-chlorphenyl)propionat
-
Die
obige Verbindung wird auf ähnliche
Weise zu der Präparation
3BC oben hergestellt, mit der Ausnahme, dass der Aldehyd von Präparation
5C verwendet wird.
-
Präparation
5BC Präparation
von Lithium-2-[(2-tert-butoxycarbonyl-1,2,3,4-tetrahydroisochinolin-3-ylmethyl)methylamino]-3-(4-chlorphenyl)propionat
-
Schritt A:
-
Zu
einer Lösung
aus 3-{[2-(4-Chlorphenyl)-1-methoxycarbonylethylamino]methyl]-3,4-dihydro-1H-isochinolin-2-carbonsäure-tert-butylester
aus Präparation
3BC Schritt A (0,60 g, 1,31 mmol) in wasserfreiem Methanol wird
Natriumacetat (0,54 g, 6,54 mmol) gegeben. Die Lösung wird mit 3–4 Tropfen
Eisessig auf pH 5–6 gebracht.
Wässriges
Formaldehyd (37 Gewichtsprozent, 0,49 ml) wird dann zugegeben. Die
Lösung
wird unter eine Stickstoffatmosphäre gegeben und auf 0°C gekühlt. Nach
etwa 15 Minuten wird Natriumcyanoborhydrid (0,25 g, 3,92 mmol) zugegeben
und in die Reaktion mit wasserfreiem Methanol (5 ml) gewaschen.
Das Gemisch wird bei RT über
Nacht gerührt
und dann im Vakuum konzentriert und in wässrigem Natriumbicarbonat und
Ethylacetat rekonstituiert. Nach der Trennung der Phasen wird die
wässrige
Phase mit Ethylacetat (2×) extrahiert
und alle organischen Bestandteile werden vereinigt, getrocknet (Magnesiumsulfat),
filtriert und zu einem opaquen weißen Öl (0,64 g) konzentriert. Eine
Chromatographie (0–20%
Ethylacetat in Hexan) ergibt etwa 0,6 g des methylierten Produkts
als klares Öl
(97%).
MS (m/z, ES+): 473,2.
-
Schritt B:
-
Eine
Lösung
aus LiOH × H2O (0,05 g, 1,27 mmol) in destilliertem Wasser
(4 ml) wird zu einer Lösung des
Materials von Schritt A in 1,4-Dioxan (8 ml) gegeben und die Reaktion
wird leicht in einem Eiswasserbad gekühlt. Das Gemisch wird unter
einer Stickstoffatmosphäre
bei RT über
Nacht gerührt.
Zusätzliche
1,5 Äquivalente
an LiOH × H2O (0,08 g) werden als wässrige Lösung (4 ml) zugegeben und das
Gemisch wird bei RT über
das Wochenende gerührt.
Das Gemisch wird konzentriert und dann mit THF vereinigt und um
das Material zu trocknen konzentriert (3×). Der entstehende Schaum
wird bei RT über
Nacht in einem Vakuumofen unter Bildung von etwa 0,67 g der schließlichen
Verbindung als weißer
Schaum (114%) getrocknet.
MS (m/z, ES+): 459,2.
-
Präparation
6BC Lithium-2-[(2-tert-butoxycarbonyl-1,2,3,4-tetrahydroisochinolin-3-ylmethyl)-(2-methoxyethyl)amino]-3-(4-chlorphenyl)propionat
-
Schritt A:
-
Zu
einer Lösung
aus Methoxyacetaldehyd (0,15 g, 2,03 mmol), 3-{[2-(4-Chlorphenyl)-1-methoxycarbonylethylamino]methyl}-3,4-dihydro-1H-isochinolin-2-carbonsäure-tert-butylester
aus Präparation
3BC Schritt C (0,31 g, 0,68 mmol) in Acetonitril wird Natriumtriacetoxyborhydrid
(0,72 g, 3,38 mmol) gegeben. Nach dem Rühren über Nacht unter einer Stickstoffatmosphäre bei RT
wird zusätzliches
Acetaldehyd (0,25 g) in Acetonitril gelöst und Natriumtriacetoxyborhydrid
(0,21 g) wird zugegeben und das Gemisch wird für etwa 8,5 Stunden gerührt. Das
Gemisch wird bei RT mit 5 N NaOH (5 ml) gestoppt. Die wässrige Phase
wird aus der organischen abgetrennt und mit Ethylacetat (4×) extrahiert.
Die vereinigten organischen Bestandteile werden mit einer Kochsalzlösung gewaschen,
dann getrocknet, filtriert und konzentriert. Eine Chromatographie
(Gradient Ethylacetat in Hexan, 0 bis 12%) ergibt etwa 0,23 g an 3-{[[2-(4-Chlorphenyl)-1-methoxycarbonylethyl]-(2-methoxyethyl)amino]methyl}-3,4-dihydro-1H-isochinolin-2-carbonsäure-tert-butylester
als gelbes Öl
(70%).
MS (m/z, ES+): 517,2.
-
Schritt B:
-
Zu
einer Lösung
des Materials von Schritt A in 1,4-Dioxan wird eine Lösung aus
Lithiumhydroxidmonohydrat (0,05 g, 1,11 mmol) in destilliertem Wasser
(2 ml) gegeben. Das Gemisch wird über Nacht bei RT gerührt und
dann zu einem weißen
Rückstand
konzentriert. Die Zugabe von THF und Konzentration (3×) ergibt Lithiumcarboxylat
als Schaum. Der Schaum wird über
Nacht unter Vakuum unter Bildung von etwa 0,25 g der rohen Feststoffe
(109%) getrocknet.
MS (m/z, ES+): 503,3.
-
Präparation
7BC 1-{[1-Carboxy-2-(4-chlorphenyl)ethylcarbamoyl]methyl}-1,3-dihydroisoindol-2-carbonsäure-tert-butylester
-
Schritt A:
-
Zu
einer Suspension aus 4-Cl-D-Phe-OMe Hydrochlorid (40,4 g, 161,5
mmol) in DCM (250 ml) wird gesättigtes
wässriges
Natriumbicarbonat (250 ml) gegeben und das Gemisch wird bei RT für etwa 1
Stunde gerührt.
Die organische Portion wird abgetrennt und die wässrige Portion wird mit DCM
(2×) extrahiert.
Die vereinigten organischen Portionen werden getrocknet (Na2SO4) und zur Trockne
konzentriert. Zu dem freien Amin in DCM (400 ml) bei 0°C wird 1-Carboxymethyl-1,3-dihydroisoindol-2-carbonsäure-tert-butylester aus
Präparation
2C (Isomer 2, 44,8 g, 161,5 mmol), EDC (31,0 g, 161,5 mmol) und
4-DMAP (2,0 g, 16,1
mmol) gegeben. Das Gemisch wird bei 0°C für etwa 30 Minuten gerührt während das
Kühlbad
entfernt wird und das Gemisch wird für weitere 5 Stunden bei RT
gerührt.
Das Gemisch wird dann mit gesättigtem
wässrigem
Natriumbicarbonat (200 ml) und 10% wässrigem Natriumbisulfat (200
ml) gewaschen und dann getrocknet (Na2SO4) und unter Bildung von etwa 76,4 g (100%)
des Esters zur Trockne konzentriert.
EIS-MS 471 [M – 1].
-
Schritt B:
-
Zu
dem Ester von Schritt A (76,4 g, 161,5 mmol) in MeOH (760 ml) wird
1 N NaOH (242,0 ml, 242,0 mmol) gegeben und das Gemisch wird bei
50°C für 4 Stunden
erhitzt und dann für
weitere 16 Stunden bei RT gerührt.
Nach der Konzentration zur Trockne wird der entstehende Rückstand
in 500 ml Wasser aufgenommen und mit Diethylether (2×) gewaschen.
Die wässrige
Portion wird mit 10% wässrigem
Natriumbisulfat auf pH 2 angesäuert
und mit EtOAc (4 × 200
ml) extrahiert. Die vereinigten organischen Extrakte werden getrocknet (MgSO4) und zur Trockne konzentriert. Der entstehende
Feststoff wird in Hexan suspendiert, filtriert und unter Bildung
von etwa 67,7 g (91%) der schließlichen Verbindung getrocknet.
EIS-MS
457 [M – 1].
-
Präparation
8BC 3-(4-Chlorphenyl)-2-[(1,1-dimethyl-1,2,3,4-tetrahydroisochinolin-3-carbonyl)amino]propionsäuremethylester
-
Zu
einer Lösung
aus 1,1-Dimethyl Tic (240 mg, 1,17 mmol), 4-Cl-D-Phe-OMe (322 mg,
1,28 mmol), HOBT (197 mg, 1,46 mmol) und DIPEA (0,81 ml, 44,68 mmol)
in DCM/DMF (1:1) wird EDC (280 mg, 1,46 mmol) gegeben. Das entstehende
Gemisch wird bei RT über
Nacht gerührt.
Das Gemisch wird dann mit EtOAc (100 ml) verdünnt, mit gesättigtem
wässrigem
NaHCO3 und Kochsalzlösung gewaschen und dann getrocknet (Na2SO4) und zur Trockne
konzentriert. Eine Reinigung und Trennung der Diastereomere durch
Blitzchromatographie (35 g SiO2 linearer
Gradient, 40 ml/min 10–50%
EtOAc/Hexan für
25 Minuten und 50% EtOAc/Hexan für
7 Minuten) ergibt die schließliche
Verbindung.
LRMS (ESI+): 401,1 (M + H).
-
Präparation
9BC 3-(4-Chlorphenyl)-2-[(1,1-dimethyl-1,2,3,4-tetrahydroisochinolin-3-carbonyl)amino]propionsäure
-
Zu
der Verbindung von Präparation
8BC (5,95 g, 14,88 mmol) in einem 1:1 Gemisch an THF/H2O
(50 ml) wird Lithiumhydroxidhydrat (0,75 g, 17,87 mmol) gegeben.
Das Gemisch wird bei RT für
etwa 18 Stunden gerührt.
Das Gemisch wird dann zur Trockne konzentriert. Der entstehende
Rückstand
wird in Wasser (50 ml) gelöst,
mit 1 N HCl (25 ml) basisch gemacht und mit Et2O
(100 ml) gewaschen. Die wässrige
Phase wird unter Bildung von etwa 6,18 g der schließlichen
Verbindung (98%) zur Trockne eingedampft.
LRMS (EIS+): 387
[M + 1].
-
Präparation
10BC 1-{[1-Carboxy-2-(4-methoxyphenyl)ethylcarbamoyl]methyl}-1,3-dihydroisoindol-2-carbonsäure-tert-butylester
-
Schritt 1:
-
Zu
einer Lösung
aus p-Methoxy-D-Phe-OMe (1,72 g, 8,23 mmol) gelöst in THF (45 ml) und 1-Carboxymethyl-1,3-dihydroisoindol-2-carbonsäure-tert-butylester
(2,51 g, 9,05 mmol) werden HOBT (1,22 g, 9,05 mmol), EDC (1,73 g,
9,05 mmol) und DIPEA (1,6 ml, 9,05 mmol) gegeben. Die Reaktion wird über Nacht
bei RT gerührt
und dann konzentriert. Das Gemisch wird mit 1 M HCl, verdünntem NaHCO3 und Kochsalzlösung gewaschen und dann mit
Natriumsulfat getrocknet. Das Gemisch wird auf Silicagel unter Elution
mit 3% an 2 M NH3 in MeOH/CH2Cl2 unter Bildung von etwa 2,58 g als weiße Feststoffe
chromatographiert.
Masse: MH+ 469.
-
Schritt 2:
-
Der
weiße
Feststoff von Schritt 1 (2,58 g, 5,5 mmol) wird in Dioxan (37 ml)
gelöst
und Lithiumhydroxidhydrat (0,35 g, 8,3 mmol) gelöst in H2O
(19 ml) wird zugegeben. Das Gemisch wird für etwa 2,5 Stunden bei RT gerührt und
dann konzentriert. Ethylacetat wird zugegeben und das Gemisch wird
mit 1 M HCl behandelt, das dann mit Kochsalzlösung gewaschen wird und unter
Bildung von 2,56 g der schließlichen
freien Säure konzentriert
wird. LRMS (ESI+): 455 (M + 1).
-
Präparation
11 BC 1-[1-Carboxy-2-(4-chlorphenyl)ethylcarbamoyl]-1,3-dihydroisoindol-2-carbonsäure-tert-butylester
-
Schritt 1:
-
Etwa
2,0 g (7,60 mmol) an (R,S)-Boc-1,3-Dihydro-2-isoindolcarbonsäure werden
in 100 ml THF gelöst und
etwa 2,28 g (9,12 mmol) an 4-Cl-D-Ph-Methylester HCl, 1,25 g (9,12
mmol) HOBT, 1,75 g (9,12 mmol) EDC und 1,6 ml (9,12 mmol) DIEA werden
zugegeben. Das Gemisch wird über
Nacht bei RT gerührt,
zur Trockne konzentriert, mit 1 M HCl, verdünntem NaHCO3 und
Kochsalzlösung
gewaschen und dann über
Natriumsulfat getrocknet. Das Material wird auf Silicagel unter
Elution mit Ethylacetat/Hexan 1:2 unter Bildung von etwa 1,05 g
des Isomers 1 und etwa 0,82 g des Isomers 2 und etwa 1,61 g des
Gemisches der Isomere 1 und 2 chromatographiert.
Masse: NH+ 459.
-
Schritt 2:
-
Etwa
0,82 g (1,79 mmol) des Isomers 2, das in Schritt 1 erhalten wurde,
werden in 11 ml Dioxan gelöst und
0,11 g (2,68 mmol) an LiOH-Hydrat in 5,5 ml H2O
werden zugegeben. Das Gemisch wird für etwa 4 Stunden bei RT gerührt und
dann zur Trockne konzentriert. Ethylacetat wird zugegeben und die
Lösung
wird mit 1 M HCl und Kochsalzlösung
gewaschen und dann unter Bildung von etwa 0,75 g der freien Säure zur
Trockne konzentriert.
Masse: 445 (MH+).
-
Beispiel Kupplungsverfahren
1 1-(D-TIC-4-Cl-D-Phe)-4-(2-Methylphenyl)piperazin,
HCl
-
Schritt 1:
-
Zu
einer Lösung
aus N-Boc-4-Cl-D-Phe (200 mg, 0,67 mmol, 1,0 Äquivalente), 1-(2-Methylphenyl)piperazin
(140 mg, 0,79 mmol, 1,2 Äquivalente),
HOBT (113 mg, 0,84 mmol, 1,25 Äquivalente),
DIPEA (290 μl, 1,66
mmol, 2,5 Äquivalente),
CH2Cl2 (4 ml) und
DMF (1 ml) wird EDC (160 mg, 0,84 mmol, 1,25 Äquivalente) gegeben. Die Lösung wird
bei RT über
Nacht gerührt.
Die Lösung
wird mit Ethylacetat (30 ml) verdünnt und mit gesättigtem
wässrigem
Natriumbicarbonat, 0,05 M Phosphatpuffer (pH 7, 2×) und Kochsalzlösung gewaschen
und dann über
Na2SO4 getrocknet,
filtriert und konzentriert. Eine Reinigung durch Blitzchromatographie (10
g SiO2, linearer Gradient 0–10% Methanol/CH2Cl2, 30 ml/Minute über 15 Minuten)
ergibt etwa 293 mg (96%) an 1-(N-Boc-4-Ci-D-Phe)-4-(2-Methylphenyl)piperazin.
LRMS
(ESI+): 458,2 (M + H).
-
Schritt 2:
-
Zu
einer Lösung
aus 1-(N-Boc-4-Cl-D-Phe)-4-(2-Methylphenyl)piperazin (293 mg, 0,64
mmol), CH2Cl2 (2
ml) und DMS (0,5 ml) wird TFA (2 ml) gegeben. Nach dem Rühren für etwa 1
Stunde wird die Lösung
aus Heptan (3×)
azeotrop destilliert. Der Rückstand
wird in CH2Cl2 gelöst und mit
gesättigtem
Natriumbicarbonat gewaschen. Die wässrige Lösung wird mit CH2Cl2 (2×)
extrahiert. Die vereinigten organischen Lösungen werden über Na2SO4 getrocknet,
filtriert und unter Bildung von etwa 200 mg (87%) an 1-(4-Cl-D-Phe)-4-(2-Methylphenyl)piperazin
konzentriert.
-
Schritt 3:
-
Zu
einer Lösung
aus 1-(4-Cl-D-Phe)-4-(2-Methylphenyl)piperazin (60 mg, 0,17 mmol,
1,0 Äquivalente),
N-Boc-D-TIC (56 mg, 0,20 mmol, 1,2 Äquivalente), HOBT (28 mg, 0,21
mmol, 1,25 Äquivalente),
DIPEA (73 μl,
0,42 mmol, 2,5 Äquivalente),
CH2Cl2 (2 ml) und
DMF (0,5 ml) wird EDC (40 mg, 0,21 mmol, 1,25 Äquivalente) gegeben. Die Lösung wird
bei RT über
Nacht gerührt.
Die Lösung
wird mit Ethylacetat verdünnt
und mit gesättigtem
wässrigem
Natriumbicarbonat, 1 M NaHSO4 und Kochsalzlösung gewaschen
und dann über Na2SO4 getrocknet,
filtriert und konzentriert. Eine Reinigung durch Blitzchromatographie
(10 g SiO2, linearer Gradient 0–100% EtOAc/CH2Cl2, 30 ml/min über 30 Minuten)
ergibt etwa 81 mg (0,13 mmol, 77%) an 1-(N-Boc-D-TIC-4-Cl-D-Phe)-4-(2-Methylphenyl)piperazin.
LRMS
(ESI+): 617,2 (M + H).
-
Schritt 4:
-
Zu
einer Lösung
aus 1-(N-Boc-D-TIC-4-Cl-D-Phe)-4-(2-Methylphenyl)piperazin (81 mg,
0,13 mmol), CN2Cl2 (2
ml) und DMS (0,5 ml) wird TFA (2 ml) gegeben. Nach dem Rühren für etwa 1
Stunde wird die Lösung aus
Heptan (3×)
azeotrop destilliert. Der Rückstand
wird in CH2Cl2 gelöst und mit
gesättigtem
wässrigem
Natriumbicarbonat gewaschen. Die wässrige Lösung wird mit CH2Cl2 (2×)
extrahiert. Die vereinigten organischen Lösungen werden über Na2SO4 getrocknet,
filtriert und konzentriert. Der Rückstand wird in 5% Methanol/Et2O gelöst
und mit 1 M HCl in Et2O aufgefällt. Der
Niederschlag wird mit Et2O (2×) unter
Bildung von etwa 64 mg (0,12 mmol, 92%) der Titelverbindung gewaschen.
HRMS
(ESI+) berechnet für
C30H34ClN4O2: 517,2370. Gefunden:
517,2383 (M + H).
-
Kupplungsverfahren
2 1-(D-TIC-4-Cl-D-Phe)-4-(2-Methoxy-5-nitrophenyl)piperazin,
HCl
-
Schritt 1:
-
Zu
einer Lösung
aus N-Boc-D-TIC-4-Cl-D-Phe-OH (348 mg, 0,76 mmol, 1,2 Äquivalente),
1-(2-Methoxy-5-nitrophenyl)piperazin
(150 mg, 0,63 mmol, 1,0 Äquivalente),
HOAT (108 mg, 0,79 mmol, 1,25 Äquivalente),
2,6-Lutidin (0,37 ml, 3,18 mmol, 5,0 Äquivalente), CH2Cl2 (8 ml) und DMF (2 ml) wird HATU (300 mg,
0,79 mmol, 1,25 Äquivalente)
gegeben. Nach dem Rühren
bei RT über
Nacht wird die Lösung
mit Ethylacetat verdünnt
und mit 1 M HCl (2×),
gesättigtem
Natriumbicarbonat und Kochsalzlösung
gewaschen und dann über Na2SO4 getrocknet,
filtriert und konzentriert. Eine Reinigung durch Blitzchromatographie
(10 g SiO2, linearer Gradient 0–5% Methanol/CH2Cl2, 30 ml/Minute, über 20 Minuten)
ergibt etwa 392 mg (0,58 mmol, 91%) an 1-(N-Boc-D-TIC-4-Cl-D-Phe)-4-(2-Methoxy-5-nitrophenyl)piperazin.
LRMS
(ESI+): 678,5 (M + H).
-
Schritt 2:
-
Zu
einer Lösung
aus 1-(N-Boc-D-TIC-4-Cl-D-Phe)-4-(2-Methoxy-5-nitrophenyl)piperazin
(53 mg, 0,078 mmol) in CH2Cl2 (2
ml) und DMS (0,2 ml) wird TFA (1 ml) gegeben. Nach dem Rühren für etwa 2
Stunden wird die Lösung
aus Heptan (2×)
azeotrop destilliert. Der Rückstand
wird in CH2Cl2 gelöst und mit
gesättigtem Natriumbicarbonat
gewaschen, Die wässrige
Lösung
wird mit CH2Cl2 (3×) extrahiert.
Die vereinigten organischen Lösungen
werden über
Na2SO4 getrocknet,
filtriert und konzentriert. Eine Reinigung durch Blitzchromatographie
(10 g SiO2, linearer Gradient 0–10% Methanol/CH2Cl2 30 ml/Minute, über 30 Minuten)
ergibt D-TIC-4-Cl-D-Phe-4-(2-Methoxy-5-nitrophenyl)piperazin. Der
Feststoff wird in CH2Cl2 gelöst und mit
1 M HCl in Et2O ausgefällt. Die Lösung wird unter Bildung von
etwa 40 mg (0,065 mmol, 84%) der Titelverbindung konzentriert.
HRMS
(ESI+) berechnet für
C30H34ClN4O2: 578,2170 Gefunden:
578,2157 (M + H).
-
Kupplungsverfahren
3 1-(D-TIC-4-Cl-D-Phe)-4-(2-Methansulfonylphenyl)piperazin,
HCl
-
Schritt 1:
-
Zu
einer Lösung
aus 1-(D-p-Cl-Phe)-4-(2-Methansulfinylphenyl)piperazin (Präparation
1AB) (168 mg, 0,39 mmol, 1,0 Äquivalente),
N-Boc-D-TIC (132 mg, 0,47 mmol, 1,2 Äquivalente), HOBT (69 mg, 0,49
mmol, 1,25 Äquivalente),
DIPEA (0,17 ml, 1,0 mmol, 2,5 Äquivalente),
CH2Cl2 (5 ml) und
DMF (2 ml) wird EDC (95 mg, 0,49 mmol, 1,25 Äquivalente) gegeben. Die Lösung wird
bei RT über
Nacht gerührt.
Die Lösung
wird mit EtOAc verdünnt
und mit gesättigtem
wässrigem
NaHCO3 und Kochsalzlösung gewaschen und dann über Na2SO4 getrocknet,
filtriert und konzentriert. Eine Reinigung durch Blitzchromatographie
(35 g SiO2, 40 ml/min linearer Gradient
40–60%
EtOAc/Hexan über
15 min und 60% EtOAc/Hexan für
18 Minuten) ergibt 1-(N-Boc-D-TIC-4-Cl-D-Phe)-4-(2-Methansulfonylphenyl)piperazin
(256 mg 0,39 mmol, 96%).
LRMS (ESI+): 681,2 (M + H).
-
Schritt 2:
-
Zu
einer Lösung
aus 1-(N-Boc-D-TIC-4-Cl-D-Phe)-4-(2-Methansulfonylphenyl)piperazin
(240 mg, 0,35 mmol), CH2Cl2 (2
ml) und DMS (0,25 ml) wird TFA (2 ml) gegeben. Nach dem Rühren für etwa 2
Stunden wird die Lösung
aus Heptan azeotrop (3×)
destilliert. Der Rückstand
wird in CH2Cl2 gelöst und mit
gesättigtem
Natriumbicarbonat gewaschen. Die wässrige Phase wird mit CH2Cl2 (2×) extrahiert.
Die vereinigten organischen Extrakte werden getrocknet (Na2SO4), filtriert
und konzentriert. Der Rückstand
wird in 5% MeOH/Et2O gelöst und mit 1 M HCl in Et2O ausgefällt.
Die Niederschläge
werden mit Et2O (2×) unter Bildung des Chloridsalzes (191
mg, 0,31 mmol, 88%) der Titelverbindung gewaschen.
HRMS (ESI+)
berechnet für
C30H34ClSN4O4: 581,1989. Gefunden:
581,1995.
-
Kupplungsverfahren
4 1-(D-TIC-4-Cl-D-Phe)-1-(5-Isopropyl-2-pyrrolidin-1-ylmethylphenyl)piperazin,
3HCl
-
Schritt 1:
-
1-Boc-4-(5-Isopropyl-2-pyrrolidin-1-ylmethylphenyl)piperazin
(162 mg, 0,42 mmol, 1,0 Äquivalente) wird
mit TFA von den Schutzgruppen befreit und mittels SCX Ionenaustauschchromatographie
in die freie Base umgewandelt. Zu einer Lösung aus dem von den Schutzgruppen
befreiten Piperazin, der BC Domäne von
Präparation
3BC (245 mg, 0,54 mmol, 1,3 Äquivalente),
HOBT (68 mg, 0,50 mmol, 1,2 Äquivalente),
Et3N (140 μl, 1,0 mmol, 2,4 Äquivalente),
CH2Cl2 (4 ml) und
DMF (4 ml) wird 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimidhydrochlorid
(96 mg, 0,5 mmol, 1,2 Äquivalente)
gegeben. Die Lösung
wird bei RT über
Nacht gerührt. Die
Lösung
wird mit Ethylacetat (30 ml) verdünnt und mit gesättigtem
Natriumbicarbonat, Wasser und Kochsalzlösung gewaschen und dann über Na2SO4 getrocknet,
filtriert und konzentriert. Eine Reinigung durch Blitzchromatographie
(35 g SiO2, linearer Gradient 0–10% an
2 M NH3 in Methanol/CH2Cl2, 35 ml/Minute, über 30 Minuten) ergibt etwa
250 mg (0,35 mmol, 84%) an 2-Boc-3-({1-(4-Chlorbenzyl)-2-[4-(5-isopropyl-2-pyrrolidin-1-ylmethylphenyl)piperazin-1-yl]-2-oxo-ethylamino}methyl)-3,4-dihydro-1H-isochinolin.
LRMS
(ESI+): 714,2 (M + H).
-
Schritt 2:
-
Zu
einer Lösung
der Verbindung von Schritt 1 (240 mg, 0,035 mmol) in CH2Cl2 (2 ml) und DMS (0,2 ml) wird TFA (1 ml)
gegeben. Nach dem Rühren
für etwa
2 Stunden wird die Lösung
aus Heptan (2×)
azeotrop destilliert. Der Rückstand
wird in CH2Cl2 gelöst und mit
gesättigtem
Natriumbicarbonat gewaschen. Die wässrige Lösung wird mit CH2Cl2 (3×)
extrahiert. Die vereinigten organischen Lösungen werden über Na2SO4 getrocknet,
filtriert und konzentriert. Eine Reinigung durch Blitzchromatographie
(35 g SiO2, linearer Gradient 0–10% an
2 M NH3 in Methanol/CH2Cl2, 35 ml/Minute über 30 Minuten) ergibt die
Titelverbindung. Der Feststoff wird in CH2Cl2 gelöst
und mit 1 M HCl in Et2O ausgefällt. Die
Lösung
wird unter Bildung von etwa 235 mg (0,325 mmol, 93%) der Titelverbindung
konzentriert.
HRMS (ESI+) berechnet für C37H49ClN5O: 614,3626,
Gefunden: 614,3627 (M + H).
-
Kupplungsverfahren
5 N-{1-(4-Chlorbenzyl)-2-oxo-2-[4-(2-pyrrolidin-1-ylmethylphenyl)piperazin-1-yl]ethyl}-2-(2-methyl-2,3-dihydro-1H-isoindol-1-yl)acetamid,
2HCl
-
Bei
Raumtemperatur wird zu einer gerührten
Lösung
aus 2-Amino-3-(4-chlorphenyl)-1-[4-((2-pyrrolidin-1-yl)methylphenyl)piperazin-1-yl]propan-1-on
(0,49 g, 1,15 mmol), (2-Methyl-2,3-dihydro-1H-isoindol-1-yl)essigsäure (0,17 g, 1,15 mmol) und
HATU (0,43 g, 1,15 mmol) in DCM N,N-Diisopropylethylamin (0,40 ml,
2,31 mmol) gegeben. Nach etwa 1 Stunde wird die Lösung unter
verringertem Druck konzentriert und der Rückstand wird durch Silicagelchromatographie
(Eluent: 5–10%
an 2,0 M NH3 in MeOH)/DCM) gereinigt. Die gereinigten
Fraktionen werden vereinigt und unter Bildung der Boc geschützten Verbindung
als gelber Film (0,15 g, 22%) konzentriert..
LRMS (ESI+): 600,2
(M + H).
-
Zu
einem Kolben, der (R)-N-{1-(4-Chlorbenzyl)-2-oxo-2-[4-((2-pyrrolidin-1-yl)methylphenyl)piperazin-1-yl]ethyl}-2-(2-methyl-2,3-dihydro-1H-isoindol-1-yl)acetamid
enthält,
wird 1,0 N HCl (7 ml) gegeben. Nach etwa 1 Stunde wird die Lösung bei –78°C verfestigt
und der Feststoff wird unter Bildung der Titelverbindung als lilafarbene
Feststoffe (0,10 g) lyophilisiert.
LRMS (ESI+): 600,2 (M +
H).
-
Beispiele 1–83
-
Die
Verbindungen der Beispiele 1–83
werden aus einem geeigneten A Domänen Piperazin gemäß einem
im wesentlichen ähnlichen
Kupplungsverfahren wie in den Verfahren 1–5 beschrieben, hergestellt.
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
Beispiele 84–85
-
Die
Verbindungen der Beispiele 84–85
werden aus einem geeigneten A Domänen Piperazin gemäß einem
im wesentlichen ähnlichen
Kupplungsverfahren wie in den Verfahren 1–5 beschrieben, hergestellt.
-
-
-
Beispiel 86
-
Das
Beispiel 86 wird gemäß einem
im wesentlichen ähnlichen
Kupplungsverfahren wie im Verfahren 2 beschrieben, hergestellt.
-
-
Beispiele 87–100
-
Die
Verbindungen der Beispiele 87–100
werden aus einem geeigneten A Domänen Piperazin gemäß einem
im wesentlichen ähnlichen
Kupplungsverfahren wie in den Verfahren 1–5 beschrieben, hergestellt.
-
-
-
-
-
Beispiele 101–102
-
Die
Verbindungen der Beispiele 101 und 102 werden aus einem geeigneten
A Domänen
Piperazin gemäß einem
im wesentlichen ähnlichen
Kupplungsverfahren wie in den Verfahren 1–5 beschrieben, hergestellt.
-
-
-
Beispiele 103–146
-
Die
Verbindungen der Beispiele 103–146
werden aus einem geeigneten A Domänen Piperazin gemäß einem
im wesentlichen ähnlichen
Kupplungsverfahren wie in den Verfahren 1–5 beschrieben, hergestellt.
-
-
-
-
-
-
-
-
-
-
-
-
Beispiele 147–148
-
Die
Verbindungen der Beispiele 147–148
werden aus einem geeigneten A Domänen Piperazin gemäß einem
im wesentlichen ähnlichen
Kupplungsverfahren wie in den Verfahren 1–5 beschrieben, hergestellt.
-
-
-
Beispiele
149–150 N-(1-(4-Chlorbenzyl)-2-{4-[2-(2-isobutyl-2H-tetrazol-5-yl)phenyl]piperazin-1-yl}-2-oxoethyl)-2-(2,3-dihydro-1H-isoindol-1-yl)acetamid
und N-(1-(4-Chlorbenzyl)-2-{4-[2-(1-isobutyl-1H-tetrazol-5-yl)phenyl]piperazin-1-yl}-2-oxoethyl)-2-(2,3-dihydro-1H-isoindol-1-yl)acetamid
-
Das
Gemisch aus 4-[2-(2-Isobutyl-2N-tetrazol-5-yl)phenyl]piperazin und
4-[2-(1-Isobutyl-1H-tetrazol-5-yl)phenyl]piperazin
(60:40 gemäß NMR, wobei
das 2H substituierte Tetrazol bevorzugt ist, 230 mg, 0,8 mmol, 1,0 Äquivalente)
wird auf ähnliche
Weise wie im Kupplungsverfahren 2 beschrieben, gekuppelt. Die Regioisomere
werden mittels Silicagelchromatographie getrennt. Die getrennten
Verbindungen werden mittels TFA gefolgt von einer Reinigung und
HCl Salzbildung von den Schutzgruppen befreit.
2H substituiertes
Tetrazol: HRMS (ES+) berechnet für
C34H40N8O2Cl: 627,2963. Gefunden: 627,2946.
1H
substituiertes Tetrazol: HRMS (ES+) berechnet für C34H40N8O2Cl:
627,2963. Gefunden: 627,2961.
-
Beispiel
151 6-Hydroxy-1,1-dimethyl-1,2,3,4-tetrahydroisochinolin-3-carbonsäure-{1-(4-chlorbenzyl)-2-oxo-2-[4-(2-[1,2,4]triazol-1-ylmethylphenyl)piperazin-1-yl]ethyl}amid
-
1-Boc-4-(2-[1,2,4]triazol-1-ylmethylphenyl)piperazin
wird von den Schutzgruppen befreit und an Boc-D-p-Cl-Phe-OH auf ähnliche
Weise zu dem Kupplungsverfahren 1 gekuppelt. Das gekuppelte Produkt wird
von den Schutzgruppen befreit und als Chloridsalz hergestellt. Zu
einer Lösung
des Chloridsalzes (1,16 g, 2,52 mmol), 6-Hydroxy-1,1-dimethyl-1,2,3,4-tetrahydroisochinolin-3-carbonsäure (714
mg), DIEA (1,75 ml), HOBt (408 mg) und DMAP (62 mg) in 2,52 ml CH2Cl2 wird EDC (579
mg) gegeben. Nach dem Rühren über Nacht
wird das Gemisch mit EtOAc extrahiert, mit Wasser, gesättigtem
Bicarbonat und Kochsalzlösung
gewaschen und dann über
Na2SO4 getrocknet,
filtriert und zur Trockne eingedampft. Das Gemisch wird mit 5% MeOH/EtOAc
chromatographiert. Die Diastereomere werden auf einer Waters Symmetry
C18 Säule
80:20 bis 50:50 Wasser (0,05% TFA) Acetonitril über 40 Minuten getrennt, wobei
bei 230 nm detektiert wird.
LRMS (ESI+): 628,3 (M + 1).
-
Beispiel
152 1-(D-TIC-4-Cl-D-Phe)-4-(2-Methansulfonatphenyl)piperazin
-
Zu
einer Lösung
aus 1-(N-Boc-D-TIC-4-Cl-D-Phe)-4-(2-Hydroxyphenyl)piperazin (150
mg, 0,242 mmol) und Et3N (50 ml, 0,36 mmol)
in 6 ml CH2Cl2 die
auf 0°C
gekühlt
ist, wird Methansulfonylchlorid (19 μl, 0,24 mmol) gegeben. Nach
dem Rühren
für 2 Stunden
wird die Reaktion mit gesättigtem
Natriumbicarbonat gestoppt und mit CH2Cl2 extrahiert. Die vereinigten organischen
Lösungen
werden mit 1 M HCl, gesättigtem Natriumbicarbonat
und Kochsalzlösung
gewaschen, getrocknet (Na2SO4),
filtriert und konzentriert. Das Produkt wird ohne weitere Reinigung
mit TFA gemäß dem im
Kupplungsverfahren 1 Schritt 4, beschriebenen Verfahren von den
Schutzgruppen befreit.
HRMS (ESI+) berechnet für C30H34ClN4O5S: 597,1938. Gefunden: 597,1954 (M + H).
-
Beispiel
153 1-(D-TIC-4-Cl-D-Phe)-4-(2-Aminophenyl)piperazin
-
Eine
Lösung
aus 1-(N-Boc-D-Tic-4-Cl-D-Phe)-4-(2-nitrophenyl)piperazin (260 mg,
0,4 mmol), PtO2 (70 mg) in 30 ml Isopropanol
wird in einem Parr Hydriergerät
unter 45 psi H2 für etwa 1 Stunde geschüttelt. Die Lösung wird
durch Celite filtriert und unter Bildung von etwa 263 mg (0,4 mmol,
100%) des Amins konzentriert, das ohne weitere Reinigung verwendet
wird. Das Amin wird mit TFA gemäß dem im
Kupplungsverfahren 1, Schritt 4 beschriebenen Verfahren, von den
Schutzgruppen befreit.
HRMS (ESI+) berechnet für C29H33ClN5O2: 518,2323. Gefunden: 518,2338 (M + H).
-
Beispiel
154 1-(D-TIC-4-Cl-D-Phe)-4-(2-Sulfonamid)piperazin
-
Zu
einer Lösung
aus 1-(N-Boc-D-TIC-4-Cl-D-Phe)-4-(2-Aminophenyl)piperazin (120 mg,
0,19 mmol) und Et3N (27 μl, 0,19 mmol) in 6 ml CH2Cl2 die auf 0°C gekühlt ist,
wird Methansulfonylchlorid (15 μl,
0,19 mmol) gegeben. Nach dem Rühren
für 2 Stunden
wird die Reaktion mit gesättigtem
Natriumbicarbonat gestoppt und mit CH2Cl2 extrahiert. Die vereinigten organischen
Lösungen
werden mit 1 M HCl, gesättigtem
Natriumbicarbonat und Kochsalzlösung
gewaschen, dann getrocknet (Na2SO4), filtriert und konzentriert. Das Produkt
wird ohne weitere Reinigung mit TFA gemäß dem im Kupplungsverfahren
1 Schritt 4, beschriebenen Verfahren von den Schutzgruppen befreit.
HRMS
(ESI+) berechnet für
C30H35ClN5O4S: 596,2098. Gefunden:
596,2104 (M + H).
-
Beispiel
155 N-[2-(4-{3-D-Chlorphenyl)-2D-[(1,2,3,4-tetrahydroisochinolin-3-ylmethyl)amino]propionyl}piperazin-1-yl]methansulfonamidtrihydrochlorid
-
Schritt 1:
-
1-(2-Nitrophenyl)piperazin
(3,13 g, 15,1 mmol) wird mit Boc-D-4-chlorphenylalanin (4,52 g,
15,1 mmol) in Anwesenheit von EDC/HOBt gekuppelt. Das rohe Produkt
wird auf Silicagel (EtOAc/Hexan 1:1) unter Bildung der gelben Feststoffe
(6,88 g) chromatographiert.
Masse: MH+ 489.
-
Schritt 2:
-
Oben
hergestellter {1-4-Chlorphenyl)-2-[4-2-nitrophenyl)piperazin-1-yl]-2-oxoethyl}-carbaminsäure-tert-butylester
(6,88 g, 14,1 mmol) wird mit 4 M HCl in Dioxan (230 ml) gemischt
und bei RT für
etwa eine Stunde gerührt
und dann unter Bildung von gelben Feststoffen (5,1 g) konzentriert.
Masse:
MH+ 389.
-
Schritt 3:
-
Oben
hergestelltes 2-Amino-3-(4-chlorphenyl)-1-[4-(2-nitrophenyl)piperazin-1-yl]propan-1-on-hydrochlorid (2,5
g, 5,88 mmol) und NaOAc (1,7 g, 20,7 mmol) werden in MeOH (175 ml)
gelöst
und in einem Eiswasserbad gekühlt.
Das Aldehyd von Präparation
6C (2,02 g, 7,7 mmol) wird zugegeben und für mehrere Minuten gerührt und
dann wird NaBH3CN (0,48 g, 7,6 mmol) zugegeben.
Das Gemisch wird bei RT über
Nacht gerührt.
Weiteres NaOAc (0,57 g, 7,0 mmol), das Aldehyd (0,67 g, 2,6 mmol)
und NaBH3CN (0,16 g, 2,5 mmol) werden mit
einem angefügten
Tauchbad zugegeben. Das Gemisch wird bei RT für etwa 4 Stunden gerührt und dann
zur Trockne eingedampft. Dann werden 1 M HCl und EtOAc zugegeben,
gefolgt von Waschen mit NaHCO3 und Kochsalzlösung und
Trocknen über
Na2SO4. Eine Entfernung
des Lösemittels
ergibt einen Rückstand,
der auf Silicagel (2% MeOH/CH2Cl2) unter Bildung von gelben Feststoffen (2,53
g) chromatographiert wird.
Masse: MH+ 634.
-
Schritt 4:
-
Oben
hergestellter 3-({1-(4-Chlorbenzyl)-2-[4-(2-nitrophenyl)piperazin-1-yl]-2-oxo-ethylamino}methyl)-3,4-1H-isochinolin-2-carbonsäure-tert-butylester
(2,5 g, 3,94 mmol) wird in CH2Cl2 (10 ml) gelöst und auf 0°C gekühlt. TEA
(0,4 g, 4,0 mmol) und Boc-Anhydrid (0,86 g, 3,94 mmol) das in CH2Cl2 (10 ml) gelöst ist,
wird zu dem Gemisch tropfenweise gegeben. Zusätzliches TEA (0,4 g, 4,0 mmol)
wird zugegeben und das Gemisch wird für etwa 1,5 Stunden gerührt. Das
Gemisch wird zur Entfernung von Et3N konzentriert
und CH2Cl2 wird
zugegeben. Das Gemisch wird über
das Wochenende gerührt.
Zusätzliches
DMAP (0,096 g, 0,70 mmol) und TEA (0,4 g, 4,0 mmol) werden zugegeben
und das Gemisch wird für
etwa 5 Stunden gerührt.
Das Gemisch wird zur Trockne eingedampft und mit Ethylacetat/Hexan
(2:8) unter Bildung von etwa 1,06 g des Produkts chromatographiert.
Masse:
MH+ 734.
-
Schritt 5:
-
Oben
hergestellter 3-[tert-Butoxycarbonyl-{1-(4-chlorbenzyl)-2-[4-2-nitrophenyl)piperazin-1-yl]-2-oxo-ethyl}amino)methyl]-3,4-dihydro-1H-isochinolin-2-carbonsäure-tert-butylester
(0,50 g, 0,68 mmol) wird in Isopropylalkohol (100 ml) gelöst und Pt2O (0,13 g, 0,59 mmol) wird zugegeben. Die
Hydrierung wird auf einem Parr Schüttler bei 45 psi für etwa eine
Stunde bei RT ausgeführt.
Das Gemisch wird filtriert und unter Bildung eines weißen Feststoffs
(0,46 g) zur Trockne eingedampft.
Masse: MH+ 704.
-
Schritt 6:
-
Oben
hergestellter 3-({2-[4-(2-Aminophenyl)piperazin]-1-yl]-(4-chlorbenzyl)-2-oxo-ethyl]-tert-butoxycarbonylamino]methyl)-3,4-dihydro-1H-isochinolin-2-carbonsäure-tert-butylester
(0,46 g, 0,65 mmol) wird in CH2Cl2 (10 ml) gelöst. Das Gemisch wird mit einem
Eisbad unter Stickstoff gekühlt
und dann wird TEA (0,13 g, 1,31 mmol) gefolgt von einer langsamen
Zugabe von MsCl (0,075 g 0,65 mmol) in CH2Cl2 (1 ml) zugegeben. Nach etwa 30 Minuten
wird eine zusätzliche
Menge an MsCl (0,025 g, 0,22 mmol) zugegeben. Das Gemisch wird gekühlt, mit
Ethylacetat verdünnt,
mit gesättigtem
Na2CO3 extrahiert,
mit Kochsalzlösung
gewaschen, getrocknet und im Vakuum eingedampft. Das Material wird
auf einer Ionenaustauschchromatographie (0,35 g) chromatographiert.
Masse:
MH+ 782.
-
Schritt 7:
-
Oben
hergestellter 3-[tert-Butoxycarbonyl-{1-(4-chlorbenzyl)-2-[4-(2-methansulfonylaminophenyl)piperazin-1-yl]-2-oxo-ethyl}amino)methyl]-3,4-dihydro-1H-isochinolin-2-carbonsäure-t-butylester
(0,35 g, 0,65 mmol) wird mit 4 M HCl in Dioxan (30 ml) bei RT für etwa eine
Stunde gerührt.
Das Gemisch wird zur Trockne eingedampft und gesättigtes Natriumbicarbonat wird
zugegeben. Das Gemisch wird dann mit Ethylacetat extrahiert, mit
Kochsalzlösung
gewaschen und getrocknet. Das Material wird auf Silicagel mittels
5% MeOH/CH2Cl2 chromatographiert.
Der Rückstand
wird in Methanol (40 ml) gelöst
und 2 M HCl in Ether (3 ml) wird zugegeben, der dann unter Bildung von
etwa 0,23 g der schließlichen
Verbindung zur Trockne eingedampft wird.
Berechnete exakte
Masse: 582,2305. Gefundene exakte Masse: 582,2286.
-
Beispiel
156 2-(4-{3-D-(4-Chlorphenyl)-2-D-[(1,2,3,4-tetrahydroisochinolin-3-ylmethyl)amino]propionyl}piperazin-1-yl)benzolsulfonamidtrihydrochlorid
-
Der
Aldehyd von Präparation
6C wird mit 2-{4-[2-Amino-3-D-(4-chlorphenyl)propionyl]piperazin-1-yl}benzolsulfonamidhydrochlorid
gemäß dem in
Beispiel 158, Schritt 3 und dann Schritt 7 beschriebenen Verfahren,
umgesetzt. Eine Schutzgruppenabspaltung der Boc-Gruppe in Anwesenheit
von 4 M HCl/Dioxan ergibt die Titelverbindung.
-
Exakte
Masse berechnet: 568,2419. Gefunden: 568,2158.
-
Beispiel
157 3-(4-Chlorphenyl)-2-[methyl-(1,2,3,4-tetrahydroisochinolin-3-ylmethyl)amino]-1-[4-(2-pyrrolidin-1-ylmethylphenyl)piperazin-1-yl]propan-1-on-tetrahydrochlorid
-
Schritt A:
-
Eine
4 M Lösung
aus HCl in Dioxan (20 ml) wird zu einer Lösung aus 4-(2-Pyrrolidin-1-ylmethylphenyl)piperazin-1-carbonsäure-t-butylester
(2,01 g, 5,82 mmol) gegeben. Die Lösung wird bei RT über Nacht
unter Stickstoff gerührt
und dann zur Entfernung des Dioxans konzentriert. Es wird Diethylether
zugegeben und die Lösung
wird konzentriert (2×).
Diethylether wird zugegeben und das Produkt wird durch Unterdruckfiltration isoliert
und dann mit Diethylether gewaschen. Eine Vakuumtrocknung bei 50°C über Nacht
ergibt 1-(2-Pyrrolidin-1-ylmethylphenyl)piperazin-2HCl (1,62 g,
87,6%).
MS (M/z, ES+): 246,1.
-
Schritt B:
-
Lithium-2-[(2-tert-butoxycarbonyl-1,2,3,4-tetrahydroisochinolin-3-ylmethyl)methylamino]-3-(4-chlorphenyl)propionat
(0,59 g, 1,27 mmol), die Verbindung von Schritt A (0,27 g, 0,85
mmol), EDC (0,24 g, 1,27 mmol) und HOBt (0,17 g, 1,27 mmol) werden
vereinigt und in wasserfreiem DMF (5 ml) gelöst. DIPEA wird zugegeben (440 μl, 2,54 mmol)
und die Reaktion wird unter Stickstoff über Nacht bei Raumtemperatur
gerührt.
Die Reaktion wird konzentriert und in CH2Cl2 wieder hergestellt und dann mit NaHCO3 verdünnt.
Nach der Abtrennung der organischen Phase wird die wässrige Phase
mit CH2Cl2 (2×) extrahiert.
Die vereinigten organischen Bestandteile werden getrocknet (Na2SO4), filtriert
und konzentriert. Eine Chromatographie (EtOAc bis 5% MeOH/EtOAc)
ergibt etwa 100 mg an 3-[({1-(4-Chlorbenzyl)-2-oxo-2-[4-(2-pyrrolidin-1-ylmethylphenyl)piperazin-1-yl]ethyl}methylamino)methyl]-3,4-dihydro-1H-isochinolin-2-carbonsäure-t-butylester.
MS (m/z, ES+): 686,4.
-
Schritt C:
-
Das
Material von Schritt B wird in einer 4 M Lösung aus HCl in Dioxan (30
ml) aufgenommen. Die Reaktion wird bei RT über Nacht unter Stickstoff
gerührt.
Das Gemisch wird zur Entfernung des Dioxans konzentriert und der
entstehende Film wird mit Diethylether behandelt und dann konzentriert
(2×).
Eine Behandlung mit Diethylether, Isolierung durch Unterdruckfiltration
und Trocknen bei RT unter Vakuum ergibt etwa 0,103 g der schließlichen
Verbindung als gelbe Feststoffe (97%).
MS (m/z, ES+): 586,3.
-
Beispiel
158 3-(4-Chlorphenyl)-2-[(2-methoxyethyl)-(1,2,3,4-tetrahydroisochinolin-3-ylmethyl)amino]-1-[4-(2-pyrrolidin-1-ylmethylphenyl)piperazin-1-yl]propan-1-on-tetrahydrochlorid
-
Es
wird 4-(2-Pyrrolidin-1-ylmethylphenyl)piperazin-1-carbonsäure-t-butylester
von den Schutzgruppen befreit und dann wird das entstehende Aminhydrochlorid
(0,10 g, 0,30 mmol) mit Lithium-2-[(2-tert-butoxycarbonyl-1,2,3,4-tetrahydroisochinolin-3-ylmethyl)-(2-methoxyethyl)amino]-3-(4-chlorphenyl)propionat
(0,23 g, 0,45 mmol) gekuppelt. Das Gemisch wird unter Bildung des
rohen gekuppelten Produkts chromatographiert [MS (m/z, ES+): 730,4],
das unter Bildung von etwa 0,068 g der schließlichen Verbindung als braune
Feststoffe von den Schutzgruppen befreit wird.
MS (m/z, ES+):
630,3.
-
Beispiel
159 (R)-N-{1-(4-Chlorbenzyl)-2-oxo-2-[4-(2-([1,2,4]triazol-1-yl)methylphenyl)piperazin-1-yl]ethyl}-2-(2-isopropyl-2,3-dihydro-1H-isoindol-1-yl)acetamid
(Isomer 1)
-
Zu
einer Lösung
aus 2-Amino-3-(4-chlorphenyl)-1-[4-(2-[1,2,4]triazol-1-ylmethylphenyl)piperazin-1-yl]propan-1-on-trifluoracetylcarboxylatsalz
(0,30 g, 0,55 mmol), (2-Isopropyl-2,3-dihydro-1H-isoindol-1-yl)essigsäure (0,12
g, 0,55 mmol), HATU (0,21 g, 0,55 mmol) in DCM wird DIPEA (0,19
ml, 1,13 mmol) gegeben. Nach etwa 3 Stunden wird die Lösung durch
Silicagelchromatographie (Eluent: 3% an 2,0 M NH3 in MeOH/DCM)
gereinigt. Die gereinigten Fraktionen werden vereinigt und unter
verringertem Druck unter Bildung der schließlichen Verbindung als weißer Schaum
(0,06 g, 18%) konzentriert.
ES MS 626,3 (M + H).
-
Beispiel
160 (R)-N-{1-(4-Chlorbenzyl)-2-oxo-2-[4-(2-([1,2,4]triazol-1-yl)methylphenyl)piperazin-1-yl]ethyl}-2-(2-isopropyl-2,3-dihydro-1H-isoindol-1-yl)acetamiddihydrochloridsalz
(Isomer 1)
-
In
einen Kolben, der (R)-N-{1-(4-Chlorbenzyl)-2-oxo-2-[4-(2-([1,2,4]triazol-1-yl)methylphenyl)piperazin-1-yl]ethyl}-2-(2-isopropyl-2,3-dihydro-1H-isoindol-1-yl)acetamid
(Beispiel 162) enthält,
wird 1,0 N HCl (5 ml) gegeben. Nach etwa einer Stunde wird die Lösung bei –78°C verfestigt
und der Feststoff wird unter Bildung von etwa 0,06 g der schließlichen
Verbindung als hellbraune Feststoffe lyophilisiert.
ES MS 626,3
(M + H).
-
Beispiel
161 2-(2-Butyl-2,3-dihydro-1H-isoindol-1-yl)-N-{1-(4-chlorbenzyl)-2-oxo-2-[4-(2-([1,2,4]triazol-1-yl)methylphenyl)piperazin-1-yl]ethyl}acetamid
(Isomer 1)
-
Zu
einer in Präparation
4AB (0,30 g, 0,45 mmol) hergestellten Lösung, (2-Butyl-2,3-dihydro-1H-isoindol-1-yl)essigsäure (Präparation
8C) (0,10 g, 0,45 mmol), HATU (0,17 g, 0,45 mmol) in DCM (5,1 ml)
wird DIPEA (0,16 ml, 0,91 mmol) gegeben. Nach etwa 3 Stunden wird
die Lösung
durch Silicagelchromatographie (Eluent: 2–4% an 2,0 M NH3 in
MeOH)/DCM) gereinigt. Die gereinigten Fraktionen werden vereinigt
und unter verringertem Druck unter Bildung von etwa 0,07 g der schließlichen
Verbindung als nicht ganz weißer
Schaum (26%) konzentriert.
ES MS 640,3 (M + H).
-
Beispiel
162 2-(2-Butyl-2,3-dihydro-1H-isoindol-1-yl)-N-{1-(4-chlorbenzyl)-2-oxo-2-[4-(2-([1,2,4]triazol-1-yl)methylphenyl)piperazin-1-yl]ethyl}acetamiddihydrochloridsalz
(Isomer 1)
-
In
einen Kolben, der 2-(2-Butyl-2,3-dihydro-1H-isoindol-1-yl)-N-{1-(4-chlorbenzyl)-2-oxo-2-[4-(2-([1,2,4]triazol-1-yl)methylphenyl)piperazin-1-yl]ethyl}acetamid
(Beispiel 166) (0,07 g, 0,11 mmol) enthält, wird 1,0 N HCl (5 ml) gegeben.
Nach etwa einer Stunde wird die Lösung bei –78°C verfestigt und der Feststoff
wird unter Bildung von etwa 0,06 g der schließlichen Verbindung als grüne Feststoffe
lyophilisiert.
ES MS 640,3 (M + H).
-
Beispiele 163–166
-
Die
Beispiele 163–166
werden folgendermaßen
hergestellt. Das Gemisch aus 4AB-2TFA Salzen oder 4AB-HCl Salzen
(Präparation
4AB) (1,0 Äquivalente),
N-Boc-substituiertes-D-Tic-OH oder N-Boc-substituiertes-DL-Tic-OH (1,0 Äquivalente),
HATU (1,0 Äquivalente)
und DIEA (5,0–10,0 Äquivalente)
in DCM wird bei RT über
Nacht gerührt.
Das Gemisch wird zwischen Wasser und CH2Cl2 aufgeteilt. Die wässrige Phase wird mit CH2Cl2 (2×) extrahiert.
Die vereinigte organische Lösung
wird über
MgSO4 getrocknet, filtriert und im Vakuum
konzentriert. Das Gemisch wird durch eine Silicagelsäule mittels
10 MeOH in EtOAc unter Bildung des N-Boc Produkts gereinigt.
-
Das
N-Boc Produkt wird mit 5 ml gesättigtem
HCl in EtOAc gemischt und bei RT über Nacht gerührt. Diethylester
wird zugegeben und der entstehende weiße Feststoff wird filtriert
und mit Ether (3×)
unter Bildung der schließlichen
Verbindung als HCl Salz gewaschen. Beispiel
163 3-Methyl-1,2,3,4-tetrahydroisochinolin-3-carbonsäure-{1-(4-chlorbenzyl)-2-oxo-2-[4-(2-[1,2,4]-triazol-ylmethylphenyl)piperazin-1-yl]ethyl}amid,
HCl Salz
MS M + 1 598,2 (64%) Beispiel
164 7-Fluor-1,2,3,4-tetrahydroisochinolin-3-carbonsäure-{1-(4-chlorbenzyl)-2-oxo-2-[4-(2-[1,2,4]-triazol-1-ylmethylphenyl)piperazin-1-yl]ethyl}amid,
HCl Salz
MS M + 1 602,2 (86%) Beispiel
165 7-Trifluormethyl-1,2,3,4-tetrahydroisochinolin-3-carbonsäure-{1-(4-chlorbenzyl)-2-oxo-2-[4-(2-[1,2,4]triazol-1-yl-methylphenyl)piperazin-1-yl]ethyl}amid,
HCl Salz
MS M + 1 652,2 (10%) Beispiel
166 3-Methyl-1,2,3,4-tetrahydroisochinolin-3-carbonsäure-{1-(4-chlorbenzyl)-2-oxo-2-[4-(2-[1,2,4]-triazol-1-ylmethylphenyl)piperazin-1-yl]ethyl}amid,
HCl Salz
MS M + 1 598,3 (58%)
-
Beispiel
167 1,2,3,4-Tetrahydroisochinolin-3-carbonsäure-[2-{4-[2-isobutylmethansulfonylamino)phenyl]piperazin-1-yl}-1-(4-methoxybenzyl)-2-oxo-ethyl]amid,
2HCl Salz (Isomer 2)
-
Die
obige Verbindung wird aus der A Domäne 98A (Präparation 98A) und der BC Domäne aus Präparation
11 BC gemäß dem Verfahren
das im wesentlichen ähnlich
zu dem Kupplungsverfahren 2 ist, hergestellt.
LRMS (ESI+):
648,3 (M + H)
-
Beispiel
168 2-(2,3-Dihydro-1H-isoindol-1-yl)-N-(1-(4-fluorbenzyl)-2-{4-[2-(isobutylmethansulfonylamino)phenyl]piperazin-1-yl}-2-oxo-ethyl)acetamid,
HCl Salz
-
Die
obige Verbindung wird im wesentlichen gemäß dem ähnlichen Verfahren das in Beispiel
167 beschrieben ist, hergestellt.
MS M + 1 636,3 (90%).
-
Beispiel
169 3-(4-Chlorphenyl)-1-[4-(2-dimethylaminomethylphenyl)piperazin-1-yl]-2-[methyl-(1,2,3,4-tetrahydroisochinolin-3-ylmethyl)amino]propan-1-on-trihydrochloridsalz
-
Die
Boc geschützte
Verbindung von Beispiel 90 (0,19 g, 0,29 mmol) wird in MeOH gelöst und unter
N2 bei Raumtemperatur gerührt. NaOAc
(0,12 g, 1,5 mmol) wird zu dem Gemisch gefolgt von wässrigem
HCHO (0,11 ml, 1,5 mmol) gegeben. Das Gemisch wird bei RT für etwa 30
Minuten gerührt.
NaBH3CN (0,06 g, 88 mmol) in MeOH (2 ml)
wird tropfenweise bei 0°C
zugegeben. Das Gemisch wird bei RT für etwa eine Stunde gerührt. Das
Gemisch wird konzentriert, in EtOAc aufgenommen und mit verdünntem NaHCO3 und Kochsalzlösung gewaschen. Das Gemisch
wird über
Na2SO4 getrocknet
und das Lösemittel
wird verdampft. Der entstehende Rückstand wird durch Blitzchromatographie
(Silicagel, 6% 2 M NH3/MeOH/CH2Cl2) unter Bildung von etwa 0,3 g an Boc-geschützter Aminverbindung
(2) als weißer
Feststoff gereinigt. Masse: MH+ 660.
-
Zur
oben erhaltenen Verbindung (0,18 g) werden 4 M HCl/Dioxan 815 ml)
gegeben und das Gemisch wird bei RT für etwa 20 Minuten gerührt. Das
Gemisch wird zur Trockne eingedampft und mit Et2O
unter Bildung von etwa 0,24 g der schließlichen Verbindung als weißer Feststoff
(92%) behandelt.
LC-MS: MH+ 560, Exakt
berechnete Masse: 560.3156, Gefunden: 560,3170.
-
Beispiel
170 1-(D-TIC-4-Cl-D-Phe)-4-[(2-(1-S-Hydroxyethyl)phenyl]piperazin
-
Das
A Domänen
Piperazin von Präparation
11A wird an Boc-D-TIC-4-Cl-D-Phe-OH auf eine Weise gekuppelt, die
im wesentlichen ähnlich
zu der im Kupplungsverfahren 2 beschriebenen, ist. Zu einer Lösung des geschützten Produkts
(100 mg, 0,131 mmol) in 2 ml CH2Cl2 wird 1 Tropfen H2O
und 1 ml TFA gegeben. Nach dem Rühren
bei RT für
3 Stunden wird die Lösung
aus Heptan (3×)
azeotrop destilliert. Zu einer Lösung
des Rückstands
in THF bei 0°C
wird 1 ml TF-Pyr gegeben. Nach dem Rühren über Nacht wird die Lösung mit CH2Cl2 verdünnt, mit
gesättigtem
Natriumbicarbonat (2×)
und Kochsalzlösung
gewaschen und dann getrocknet (Na2SO4), filtriert und konzentriert. Nach der
Reinigung durch Blitzchromatographie (10 g SiO2,
linearer Gradient 0–10%
Methanol/CH2Cl2,
30 ml/Minute über
30 Minuten) wird das Produkt in CH2Cl2 gelöst
und mit 1 M HCl in Et2O unter Bildung von
etwa 63 mg (0,11 mmol, 82%) der schließlichen Verbindung ausgefällt.
HRMS
(Elektrospray) berechnet für
C31H36ClN4O3: 547,2476. Gefunden:
C 547,2485 (M + H).
-
Beispiel
171 1-(D-TIC-4-Cl-D-Phe)-4-[(2-(1-R-Hydroxyethyl)phenyl]piperazin
-
Das
A Domänen
Piperazin aus Präparation
12A wird an Boc-D-TIC-4-Cl-D-Phe-OH gekuppelt und auf eine Weise
die im wesentlichen ähnlich
zu der in Beispiel 171 oben beschriebenen ist, von den Schutzgruppen befreit.
HRMS
(Elektrospray) berechnet für
C31H36ClN4O3: 547,2476. Gefunden:
547,2480 (M + H).
-
Die
folgenden Beispiele 172–174
werden aus einem geeigneten substituierten A Domänen Piperazin gemäß einem
im wesentlichen ähnlichen
Kupplungsverfahren wie im Kupplungsverfahren 1 beschrieben, hergestellt.
-
Beispiel
172 Isochinolin-3-carbonsäure-{1-(4-chlorbenzyl)-2-oxo-2-[4-(2-[1,2,4]triazol-1-ylmethylphenyl)piperazin-1-yl]ethyl}amid
-
Die
obige Verbindung wird gemäß dem Kupplungsverfahren
1 hergestellt.
Gefunden: MS (ESI): 580,2 (M + H).
-
Beispiel
173 Isochinolin-3-carbonsäure-(1-(4-chlorbenzyl)-2-oxo-2-{4-[2-propinoylaminomethyl)phenyl]piperazin-1-yl}ethyl)amid
-
Die
obige Verbindung wird gemäß dem Kupplungsverfahren
1 hergestellt.
Gefunden: MS (ESI): 584,3 (M + H).
-
Beispiel
174 Isochinolin-3-carbonsäure-(1-(4-chlorbenzyl)-2-{4-[2-N-isobutylhydrazino)phenyl]piperazin-1-yl}-2-oxoethyl)amid
-
Die
obige Verbindung wird gemäß dem Kupplungsverfahren
1 hergestellt.
Gefunden: MS (ESI): 648,0 (M + H).
-
Herstellung
der neuen C-Domänenteile Heck-Kupplung
-
Präparation PP1
-
Synthese der Verbindung
(2a) durch eine Heck-Kupplung aus 2-Brombenzaldehyd (1a) mit Methylacrylat (Pd(OAc)2/PPh3 als Katalysator):
-
Ein
Gemisch aus 2-Brombenzaldehyd (1a) (24,5 g, 132 mmol) Methylacrylat
(17,9 ml, 199 mmol), Pd(OAc)2 (590 mg, 2,65
mmol, 2 Molprozent), PPh3 (1,39 g, 5,30
mmol, 4 Molprozent) und Et3N (46 ml, 331 mmol)
wird bei 80°C
für 15
h gerührt.
Eine große
Menge des gelben Feststoffs wird nach der nach der vollständigen Reaktion
gebildet. Das Gemisch wird auf RT gekühlt, konzentriert und mit H2O (200 ml) gemischt Der organische Feststoff
wird durch Filtration gesammelt und dann auf ein Kissen aus Silicagel
(25g) (EtOAc/Hexan 1:1) unter Bildung eines dunkelgelben Feststoffs
gegeben. Der Feststoff wird durch Kristallisation (100 ml EtOAc
Bodenschicht, 120 ml Hexan obere Schicht) unter Bildung von 17,57
g (70%) (100% rein gemäß NMR) eines
ersten Kristallisats und 5,23 g (21%) (95% gemäß NMR) eines zweiten Kristallisats
der Verbindung 2a gereinigt.
-
Präparation PP2
-
Synthese der Verbindung
(2a) gemäß einer
Heck Kupplung aus 2-Brombenzaldehyd (1a) mit Methylacrylat (R=H)
(Pd(OAc)2/P(O-Tolyl)3 als
Katalysator):
-
Die
Verbindung 1a (9,998 g, 54,04 mmol) wird in Toluol (20 ml) bei RT
gelöst.
Methylacrylat (5,996 g, 69,65 mmol, 1,29 Äquivalente), NEt3 (15
ml), Pd(OAc)2 und P(O-Tolyl)3 werden
nacheinander zugegeben und das Gemisch wird unter Rückfluss
gerührt.
Nach 2 Stunden kann sich das Reaktionsgemisch auf RT abkühlen. Dann
wird der ausgefällte
Katalysator durch Filtration entfernt. Der Katalysator wird mit
Toluol (2 × 10
ml) gewaschen und die Filtrate werden unter verringertem Druck zur
Trockne konzentriert. Das restliche Öl wird unter Vakuum übers Wochenende
unter Bildung eines rohen Feststoffs (11,449 g) getrocknet. Der
Feststoff wird mit Isopropanol (25 ml) aufgenommen und über Nacht
bei RT gerührt.
Dann wird der Niederschlag filtriert und mit Isopropanol (5 ml)
gewaschen. Der nasse Kuchen (8,240 g) wird über Nacht bei RT unter Bildung
eines hochreinen 2-Carboxaldehydmethylcinnamats mit 74% Ausbeute
(7,627 g, 40,1 mmol) getrocknet.
-
Präparation PP3
-
Heck Kupplung der Verbindung
1b und Methylacrylat zur Bildung der Verbindung 2b (R = 5-OMe):
-
Ein
Gemisch aus 2-Brom-5-methoxybenzaldehyd (1b) (4,5 g, 20,9 mmol,
Aldrich), Methylacrylat (2,7 g, 1,5 Äquivalente, 2,83 ml), Et3N (7,4 g, 3,5 Äquivalente, 10,2 ml), Pd(OAc)2 (93 mg, 0,02 Äquivalente) und P(O-Tol)3 wird gerührt und über 2–3 Tage auf 80°C erhitzt.
Das Reaktionsgemisch wird auf RT gekühlt und zwischen EtOAc (50
ml) und Kochsalzlösung
(50 ml) aufgeteilt. Die wässrige
Phase wird mit EtOAc (2 × 50
ml) extrahiert. Die vereinigte organische Phase wird mit Kochsalzlösung (1 × 50 ml)
gewaschen, über
MgSO4 getrocknet, filtriert und unter Bildung
eines gelbbraunen Öls
(5,01 g, 109%) konzentriert. Dieses rohe Öl wird in einem heißen Lösemittel
aus Hexan/EtOAc (80 ml/15 ml) unter Bildung der Verbindung 2b als
blassgelber Feststoff (3,5 g, 76%) gereinigt.
-
Präparation PP4
-
Heck-Kupplung der Verbindung
1C und Methylacrylat unter Bildung der Verbindung 2c (R = 4,5-OMe):
-
Zu
einer Lösung
der Verbindung 1c (906 mg, 3,70 mmol) in Toluol (2 ml) wird Pd(OAc)2 (17 mg, 0,074 mmol, 2 Molprozent), P(O-Tolyl)3 (45 mg, 0,148 mmol, 4 Molprozent), Methylacrylat
(0,5 ml, 5,55 mmol) und Et3N (1,5 ml, 11,1
mmol) gegeben. Das Gemisch wird bei 80°C für 21 h gerührt, auf RT gekühlt und
mit H2O (40 ml) gemischt. Die organischen
Verbindungen werden mit EtOAc (50 ml) extrahiert, mit Kochsalzlösung (40 ml)
gewaschen, getrocknet (Na2SO4)
und konzentriert. Der Rückstand
wird durch Blitzchromatographie unter Bildung von 466 mg (47%) der
gewonnenen Verbindung 1 c gefolgt von 450 mg (49%) der Verbindung
2c (4,5-OMe) gereinigt.
-
Präparation PP5
-
Heck-Kupplung der Verbindung
1d und Methylacrylat unter Bildung der Verbindung 2d (R = 5-NO2):
-
Das
Verfahren ist dasselbe wie das für
die Verbindung 2c, wobei 82% der Verbindung 2d nach der Reinigung
gebildet werden.
-
Präparation
PP6 Reduktive
Aminierung
-
Reduktive
Aminierung der Verbindung (2a) mit Benzylamin unter Bildung von
Isoindolin (10a). Zu einer Lösung
der Verbindung 2a (11,27 g, 59,2 mmol) in ClCH2CH2Cl (60 ml) wird BnNH2 (6,47
ml, 59,2 mmol) gefolgt von HOAc (5,1 ml, 89 mmol) gegeben. Das Gemisch
wird bei RT für
1 h gerührt.
NaCNBH3 (5,58 g, 88,8 mmol) und MeOH (30
ml) werden dann zu der obigen Lösung
gegeben. Das entstehende Gemisch wird bei RT für weitere 2 h gerührt und
mit gesättigter
NaHCO3 Lösung
(150 ml) gestoppt. Das Gemisch wird mit EtOAc (2 × 100 ml)
extrahiert und die vereinigten organischen Phasen werden mit Kochsalzlösung (150
ml) gewaschen, getrocknet (Na2SO4) und unter Bildung von 15,3 g des rohen
Produkts der Verbindung 10a konzentriert, das in der nächsten Hydrolysereaktion
ausgeführt
wird.
-
-
Eintopfverfahren aus 2-Carboxaldehydmethylcinnamat
zur Bildung des gewünschten
cyclisierten Isoindolinprodukts mittels NaBH3CN:
-
2-Carboxyaldehydmethylcinnamat
der Verbindung 2a (3,254 g, 17,1 mmol) wird in einem 1:1 MeOH:PhCH3 Gemisch (20 ml) bei RT gelöst. R-(+)-Phenethylamin
(2,073 g, 17,1 mmol) wird zugegeben und die Lösung wird unter Rückfluss
für 2 Stunden
erhitzt. HPLC zur Verfahrenskontrolle zeigt an, dass die Iminbildung vollständig ist.
Dann werden AcOH (2,055 g, 34,2 mmol) und NaBH3CN
(2,15 g, 34,2 mmol) nacheinander bei RT zugegeben und das Reaktionsgemisch
wird mit einem Wasserbad gekühlt.
Das Reaktionsgemisch wird über
Nacht nachgerührt.
Wasser (10 ml), MeOH (20 ml) und 37% HCl (2,8 ml) werden nacheinander
zugegeben und die organische Phase wird extrahiert. Die wässrige Phase
wird mit PhCH3 (10 ml) gewaschen. Dann wird
die wässrige
Phase mit 5 N NaOH (20 ml) basisch gemacht und MeOH wird zur teilweisen
Entfernung des MeOH konzentriert. Eine Extraktion mit EtOAc (2 × 25 ml)
wird ausgeführt.
Die vereinigten organischen Phasen werden über MgSO4 getrocknet,
filtriert und mit EtOAc (10 ml) gewaschen. Die Filtrate werden unter
verringertem Druck konzentriert und das restliche Öl wird unter
Vakuum über
Nacht bei RT unter Bildung des gewünschten cyclisierten Isoindolinprodukts
10b mit 92% Ausbeute (4,642 g, 15,7 mmol) getrocknet. Die prozentualen
HPLC Flächen
zeigen an, dass die 2 Diastereomere in einem 55:45 Verhältnis hergestellt
werden. Die 1H NMR bestätigt dieses Ergebnis durch
Integration der Methylgruppe des Phenethylsubstituenten.
-
Anmerkung:
Die Heck oder Heck-Typ Kupplung wird in Toluol mit einem leichten Überschuß an Methylacrylat
ausgeführt,
das durch Destillation vor der MeOH und der R-(+)-Phenethylaminzugabe
entfernt wird.
-
-
Reduktive Aninierung der
Verbindung (2a) mit t-Butylcarbamat unter Bildung der Verbindung
(11a):
-
Zu
einer Lösung
des Aldehyds der Verbindung 2a (238 mg, 1,25 mmol) in CH3Cn (8 ml) wird t-Butylcarbamat (439 mg, 3,75 mmol) gefolgt
von Triethylsilan (0,6 ml, 3,75 mmol) und TFA (0,19 ml, 2,5 mmol)
gegeben. Das Gemisch wird bei RT über Nacht gerührt, mit
gesättigter
NaHCO3 Lösung
(20 ml) gestoppt und mit EtOAc (2 × 30 ml) extrahiert. Die vereinigten
organischen Phasen werden mit Kochsalzlösung (30 ml) gewaschen, getrocknet
(Na2SO4) und konzentriert.
Der Rückstand
wird durch Blitzchromatographie (Hexan/EtOAc 3:1) unter Bildung
von 317 mg (87%) der Verbindung 11a gereinigt.
-
-
Reduktive Aminierung der
Verbindung 2b mit t-Butylcarbamat zur Bildung der Verbindung 11b:
-
Ein
Gemisch des Aldehyds der Verbindung 2b (600 mg, 2,72 mmol), Et3SiH (955 mg, 3 Äquivalente, 1,31 ml), TFA (620
mg, 2 Äquivalente,
420 μl),
t-Butylcarbamat (980 mg, 3 Äquivalente)
in Acetonitril (15 ml) werden bei Raumtemperatur für 2 Tage
gerührt.
Das Lösemittel
wird auf einem Rotationsverdampfer entfernt und der rohe Rückstand
wird auf einer Blitzsäule
(100 g SiO2, 7:1 → 6:1 Hexan/EtOAc) gereinigt.
Es werden 307 mg gutes gewünschtes
Produkt der Verbindung 11b (35%) gewonnen, wobei 195 mg des Produkts
mit Aldehyd SM (22%) verunreinigt sind.
-
-
Reduktive Aminierung der
Verbindung (2c) mit t-Butylcarbamat zur Bildung der Verbindung (11c):
-
Zu
einer Lösung
des Aldehyds der Verbindung 2c (411 mg, 1,64 mmol) in CH3Cn (10 ml) wird t-Butylcarbamat (580 mg, 4,93 mmol) gefolgt
von Triethylsilan (0,8 ml, 4,93 mmol) und TFA (0,25 ml, 3,28 mmol)
gegeben. Das Gemisch wird bei RT über Nacht gerührt, mit
gesättigter
NaHCO3 Lösung
(30 ml) gestoppt und mit EtOAc (2 × 30 ml) extrahiert. Die vereinigten
organischen Phasen werden mit Kochsalzlösung (30 ml) gewaschen, getrocknet
(Na2SO4) und konzentriert.
Der Rückstand
wird durch Blitzchromatographie (Hexan/EtOAc 3:1, Hexan/EtOAc 1:1)
unter Bildung von 535 mg (93%) der Verbindung 11c gereinigt.
-
-
Zu
einer Lösung
der Verbindung 2d (1,02 g, 4,34 mg) in CH2Cl2/CH3Cn (1:1, 24
ml) werden BocNH2 (1,5 g, 13,02 mmol), Et3SiH (2,1 ml, 13,02 mmol) und TFA (0,067
ml, 8,67 mmol) gegeben. Das Gemisch wird bei RT für 7 h gerührt. Während der
Reaktion bildet sich ein Niederschlag. Das Reaktionsgemisch wird
mit gesättigter
NaHCO3 Lösung
(30 ml) gestoppt und mit CH2Cl2 (40
ml) verdünnt.
Die organische Phase wird mit Kochsalzlösung (30 ml) gewaschen, getrocknet
(Na2SO4) und konzentriert.
Der Rückstand
wird durch Blitzchromatographie (Hexan/EtOAc 3:1, dann CH2Cl2/EtOAc 10:1)
unter Bildung von 2,08 g eines gelben Feststoffs gereinigt, der
noch BocNH2 enthält. Das Produkt ist nicht das
gewünschte
Boc-Carbamat der Verbindung 14c. Das LC-MS Ergebnis zeigt, dass
das Produkt das Schiff-Basenzwischenprodukt
ist.
-
Zu
dem obigen Produkt (420 mg) in CH2Cl2 (10 ml) werden Et3SiH
(1 ml) und TFA (0,4 ml) gegeben. Das Gemisch wird bei RT für 1 h gerührt und
eine kleine Menge der Probe wird zur NMR verwendet. Eine NMR Analyse
zeigt, dass das Ausgangsmaterial verbraucht ist und das Produkt
die Verbindung 14c ist. Dann wird TFA (0,7 ml) zu dem obigen Gemisch
gegeben und die entstehende Lösung
wird bei RT für
weitere 5 h gerührt und
konzentriert. Der Rückstand
wird in EtOAc (20 ml) gelöst
und mit H2O (10 ml) gewaschen. Die wässrige Phase
wird mit gesättigtem
NaHCO3 (30 ml) basisch gemacht und die organischen
Verbindungen werden mit CH2Cl2 (2 × 25 ml)
extrahiert. Die vereinigten organischen Phasen werden mit Kochsalzlösung (20
ml) gewaschen, getrocknet (Na2SO4) und unter Bildung von 218 mg der cyclisierten
Verbindung 14c konzentriert.
-
-
Kondensation der Verbindung
2a mit α-Methylbenzylamin
zur Bildung des Imins der Verbindung 9:
-
2-Carboxaldehydmethylcinnamat
der Verbindung 2a (0,897 g, 4,72 mmol) wird in MeOH (10 ml) bei RT
gelöst.
R-(+)-Phenethylamin (0,577 g, 4,76 mmol) wird zugegeben und die
Lösung
wird unter Rückfluss
für 2 Stunden
erhitzt. Eine HPLC Verlaufskontrolle zeigt an, dass die Iminbildung
vollständig
ist. Das Lösemittel wird
auf einem Rotationsverdampfer verdampft und das entstehende Öl wird bei
RT unter Vakuum über
Nacht getrocknet. Die Schiffsche Base der Verbindung 9 wird beinahe
quantitativ erhalten (1,412 g, 4,81 mmol).
-
Präparation
PP13 Michael-Addition
-
Die
Verbindung aus α-Methylbenzylamin
wird als Hilfsmittel aufgetragen. Wie oben gezeigt ergibt die Eintopfreaktion
des Aldehyds der Verbindung 2a und α-Methylbenzylamin 90% der Verbindung
10b in einem Verhältnis
von 1,2:1.
-
Schrittweise Reduktion,
Aminierung und Cyclisierung:
-
Eine
Kondensation des Aldehyds der Verbindung 2a mit α-Methylbenzylamin in Acetonitril,
Methanol, Methanol/Toluol (1:1) oder Toluol ergibt das Imin der
Verbindung 9 in ausgezeichneter Ausbeute. Eine Reduktion des Imins
wird anfänglich
bei RT mit NaCNBH3/HOAc ausgeführt. Als
Ergebnis wird ein schlechtes ee Verhältnis (1,2:1) erhalten, ähnlich zu
dem vorher beschriebenen Eintopfverfahren. Aber wenn die Reaktion
mit NaBH4/TFA bei RT ausgeführt wird,
wird das Verhältnis
auf 2:1 verbessert. Durch Absenken der Reaktionstemperatur auf –78°C wird das
Verhältnis
auf 5 bis 6:1 verbessert.
-
Präparation PP14
-
Cyclisierung von t-Butylcarbamat
(11a):
-
Der
N-Boc-Isoindolinmethylester der Verbindung 12 wird original aus
der Verbindung 11a durch Schutzgruppenabspaltung des Boc mit TFA
gefolgt von einer basischen Aufarbeitung und Schutzgruppenanbringung
mit einer Boc Gruppe synthetisiert. Dieses Verfahren wurde durch
ein Einschrittverfahren gewaltig verbessert.
-
-
In
einem 3 Liter fassenden Dreihalsrundbodenkolben, der mit einem Stickstoffeinlass,
einem Thermoelement und einem mechanischen Rührer ausgestattet ist, wird
eine Lösung
aus 160 g (1,15 mol) K2CO3 in 180
ml Wasser bei RT gerührt.
Festes BOC Anhydrid (120 g, 0,55 mol) wird in einer Portion zur
Bildung einer halbfesten Lösung
zugegeben. Zu dem Reaktionsgemisch wird eine Lösung des rohen Aminoesterausgangsmaterials,
87 g (0,46 mol) in 120 ml THF langsam mit einer derartigen Geschwindig keit
gegeben, dass die innere Temperatur unter 35°C gehalten wird. Es wird ein
mildes Aufschäumen
beobachtet. Das Reaktionsgemisch wird für 18 Stunden bei RT gerührt. Eine
Analyse eines Reaktionsaliquots mittels NMR (DMSO6)
zeigt das gewünschte
Produkt. Die Reaktion wird mit Kochsalzlösung verdünnt und das Produkt wird mit
EtOAc extrahiert. Die organische Phase wird über Na2SO4 getrocknet, filtriert und unter Bildung
eines dunklen Öls,
150,1 g, > 100% Ausbeute,
konzentriert. Das rohe Material wird im nächsten Schritt aufgenommen.
-
-
In
einem 3 Liter fassenden Dreihalsrundbodenkolben, der mit einem mechanischen
Rühre,
einem Thermoelement und einem Rückflusskühler ausgestattet
ist, wird eine Lösung
aus 150 g (etwa 0,46 mol) des rohen N-BOC Esterausgangsmaterials
in 750 ml Methanol bei RT gerührt.
Zu der Lösung
werden 750 ml Wasser gegeben und das trübe Gemisch wird kräftig gerührt. Festes
LiOH, 25 g (1,03 mol) wird in kleinen Portionen so zugegeben, dass
die innere Temperatur unter 45°C
bleibt. Nachdem die Reaktion vollständig ist, wird die Reaktion über Nacht
bei RT gerührt
und es entwickelt sich eine dunkelgrüne Farbe. Nach 18 Stunden wird
die Reaktion unter Bildung eines dicken Halb-Feststoffs konzentriert.
Das rohe Produkt wird in EtOAc gelöst und mit 1 N HCl schnell
gewaschen, gefolgt von zwei Kochsalzwaschschritten. Die organische
Phase wird mit Na2SO4 getrocknet,
filtriert und unter Bildung von 81 g eines dunkelgrünen Feststoffs
konzentriert. Die wässrigen
Phasen werden vereinigt und mit Methylenchlorid rückextrahiert, über Na2SO4 getrocknet,
filtriert und unter Bildung von 6 g eines dunkelgrünen Feststoffs
konzentriert. Beide Feststoffe werden unter Bildung von 87 g des
gewünschten
Produkts vereinigt, was mittels NMR (DMSO6)
bestätigt
wird.
-
-
Synthese der Verbindung
14B:
-
Die
N-Boc Verbindung 11 b (200 mg, 0,62 mmol) wird in CH2Cl2 (1,0 ml) gelöst. Die klare hellgelbe Lösung wird
auf 0°C
gekühlt.
Dann wird TFA (710 mg, 10 Äquivalente,
500 μl)
mittels einer Spritze zugegeben. Das Kühlbad wird entfernt und die
klare hellbraune Lösung
wird bei RT über
Nacht gerührt.
Eine TLC (3:1 Hexan/EtOAc, UV) bestätigt die Vollständigkeit
der Reaktion. Das TFA wird auf einem Rotationsverdampfer entfernt.
EtOAc wird zugegeben und wieder konzentriert (zweimal). Der rohe
Rückstand
wird zwischen EtOAc (10–15
ml) und gesättigtem
NaHCO3 (10–15 ml) aufgeteilt. Die wässrige Phase
wird mit EtOAc (2 × 10
ml) extrahiert. Die vereinigte organische Phase wird über MgSO4 getrocknet, filtriert und unter Bildung
eines hellbraunen nassen Feststoffs (212 mg, 138%) konzentriert.
Eine NMR (CD3OD) bestätigt das gewünschte Isoindolin 14b.
Dieses rohe Isoindolin wird im nächsten
Schutzgruppenschritt ohne weitere Reinigung verwendet.
-
Präparation PP18
-
Synthese der Verbindung
12b:
-
Zu
einem Gemisch des Isoindolins 14b (190 mg, 0,859 mmol), K2CO3 (189 mg, 1,5 Äquivalente)
in einem Lösemittel
aus 1:1 THF/H2O (1,0 ml) bei RT wird BOC2O (210 mg, 1,1 Äquivalente) gegeben. Das Reaktionsgemisch
wird bei RT über
Nacht gerührt.
Eine TLC (3:1 Hexan/EtOAc, UV) zeigt die Vollständigkeit der Reaktion an. Das
Gemisch wird mit EtOAc (15 ml) verdünnt und mit H2O
(1 × 20
ml) gewaschen. Die wässrige Phase
wird mit EtOAc (1 × 20
ml) verdünnt.
Die vereinigten organischen Phasen werden mit Kochsalzlösung gewaschen
(1 × 20
ml), über
MgSO4 getrocknet, filtriert und unter Bildung
eines klaren braunen Öls
(340 mg, 123%) konzentriert. Dieses rohe Öl wird auf einer präparativen
TLC Platte (2 × 1000 μm, Lösemittel
2:1,5:0,5 CHCl3/Hexan/EtOAc) unter Bildung
der Verbindung 12b als klares gelbes Öl (190 mg, 69%) gereinigt. 1H und 13C NMR (CDCl3) werden erhalten.
-
Verfahren PP19
-
Synthese der Verbindung
12d (5-NO2) durch Boc-Schutzgruppenanbringung.
-
Die
Verbindung wird gemäß demselben
Verfahren wie für
die Verbindung 12b beschrieben, hergestellt.
-
-
Das
Imin der Verbindung 9 (1,412 g, 4,81 mmol) wird in wasserfreiem
THF (10 ml) bei RT gelöst
und TFA (5 ml) wird zugegeben. Die schwarze Lösung wird dann auf –78°C (Trockeneisbad)
gekühlt
und NaBH4 (0,893 g, 23,6 mmol, 5 Äquivalente)
wird in 2 Portionen über
5 Minuten zugegeben. Dann wird das Reaktionsgemisch bei –78°C für 3 Stunden
gerührt
und kann sich bei RT über
Nacht langsam erwärmen.
Wasser (20 ml), Cyclohexan (10 ml) und EtOAc (20 ml) werden nacheinander
zugegeben und die organische Phase wird extrahiert und verworfen.
Die wässrige
Phase wird mit 5 N NaOH (20 ml) basisch gemacht und zweimal mit einem
2:1 EtOAc/PhCH3 Gemisch (30 ml) extrahiert.
Die vereinigten organischen Phasen werden über MgSO4 getrocknet,
filtriert und mit EtOAc (10 ml) gewaschen. Die Filtrate werden unter
verringertem Druck konzentriert und das restliche Öl wird unter
Vakuum über
Nacht bei RT unter Bildung des gewünschten cyclisierten Isoindolinprodukts
10b (1,273 g, 4,31 mmol) mit 91,4% Ausbeute getrocknet. Eine prozentuale
HPLC zeigt an, dass die 2 Diastereomere in einem 84:16 Verhältnis (68%)
hergestellt werden. Eine 1H NMR bestätigt dieses
Ergebnis durch Integration der Methylgruppe des Phenethylsubstituenten.
-
-
N-Boc
Methylester der Verbindung 11a (36,3 g, 0,125 mmol) wird in THF
(250 ml) gelöst
und die Lösung
wird auf etwa 0°C
gekühlt.
Eine Lösung
aus Kaliumbis(trimethylsilyl)amid (1,24 g, 0,05 mol Äquivalente) wird
langsam mittels einer Spritze unter einer Stickstoffatmosphäre zugegeben.
Die Temperatur wird während der
Zugabe um etwa 8 Grad erhöht.
Das Kühlbad
wird entfernt und die Lösung
wird bei RT für
30–45
Minuten gerührt.
Die klare braune Lösung
wird in einen Trenntrichter, der etwa 100 ml gesättigter HN4Cl
enthält,
gegossen. Die Phasen werden getrennt. Die wässrige Phase wird mit EtOAc
(2 × 50
ml) extrahiert. Die vereinigten organischen Phasen werden mit Kochsalzlösung (1 × 100 ml)
gewaschen, über
Na2SO4 getrocknet,
filtriert und auf einem Rotationsverdampfer zu einem klaren gelben Öl (37,3
g) konzentriert. Dieses rohe Öl
wird auf einer Blitzsäule
(600 g SiO2) mit einem Gradientenlösemittel
von 6:1 Hexan/EtOAc (2,1 l), 5:1 Hexan/EtOAc (1,2 l), 4:1 Hexan/EtOAc
(1,5 l) gereinigt, um die Verbindung 12a als reines gelbes Öl (34,5
g, 95%) zu erhalten.
-
-
Zu
einer Lösung
der Verbindung 11c (535 mg, 1,52 mmol) in THF (10 ml) wird KHMDS
(0,5 M in Toluol, 0,1 ml, 0,05 mmol, 2 Molprozent) gegeben. Das
Gemisch wird bei RT für
20 min gerührt,
mit gesättigter
NH4Cl Lösung
(20 ml) gestoppt und mit EtOAc (20 ml) verdünnt. Die organische Phase wird
abgetrennt, mit Kochsalzlösung
(20 ml) gewaschen, getrocknet (Na2SO4) und konzentriert. Der Rückstand
wird durch ein Kissen aus Silicagel (EtOAc/CH2Cl2 1:10) unter Bildung von 530 mg (99%) der
Verbindung 12c als nicht ganz weißer Feststoff filtriert.
-
Präparation
PP22 Schutzgruppenabspaltungen:
-
Hydrolyse der Verbindung
10a (R = Bn) zur Bildung der Verbindung 14a:
-
Zu
einer Lösung
der rohen Verbindung 10a (15,3 g, 54,4 mmol) in MeOH (100 ml) wird
Pd(OH)2/C (Pearlman's Katalysator, 1,02 g, 6 Molprozent)
in einer Parr-Schüttlerflasche
gegeben. Die Suspension wird unter 30 psi H2 Druck über Nacht
in dem Parr-Schüttler
geschüttelt
und durch ein Kissen aus Celite filtriert. Das Filtrat wird unter
Bildung von 10,1 g der rohen Verbindung 14a als braunes Öl konzentriert.
(Das Verfahren ist dasselbe für
das Methylbenzylaminisoindolinsubstrat 10b).
-
-
In
einer typischen Reaktion wird ein Gemisch des Isoindolinesters 12a
(92 mg, 0,316 mmol) mit 1:1 MeOH/H2O (2
ml) mit LiOH (15 mg, 2 Äquivalente)
bei RT über
Nacht behandelt. Das Gemisch wird mit CH2Cl2 (5 ml) und Wasser (5 ml) verdünnt. Der
pH des Reaktionsgemisches wird mit 10% NaHSO4 Lösung auf
pH 1–3 eingestellt.
Die Phasen werden getrennt. Die wässrige Phase wird mit CH2Cl2 (1 × 10 ml)
extrahiert. Die vereinigten organischen Phasen werden über Na2SO4 getrocknet,
filtriert und unter Bildung der Verbindung 16a als blassgelber Schaum
(76 mg, 87%) konzentriert. NMR (CDCl3) zeigt
ein sauberes gewünschtes
Säureprodukt.
-
Es
ist bekannt, dass die Reaktion mehr als 6 Stunden dauern muss. Der
rohe Schaum kann durch Aufschlämmung
in warmem Hexan gereinigt werden und wird dann zur Bildung des hellbraunen
Feststoffs filtriert. Eine Hydrolyse mittels KOH (2–5 Äquivalente)
mit 1:1 MeOH/H2O über Nacht erzielt dasselbe
Ergebnis.
-
Präparation
PP24 Auftrennung:
-
Reinigung des partiell
aufgetrennten Isoindolincarbonsäuremethylesters:
-
Eine
Lösung
des rohen Materials (97,62 g) Isoindolincarbonsäuremethylester in CH2Cl2 (350 ml) wird mit
1 M HCl (400 ml, 200 ml) extrahiert. Die vereinigten wässrigen
Portionen werden mit CH2Cl2 (4 × 250 ml) gewaschen
und dann mit K2CO3 Lösung (85
g in 150 ml Wasser) basisch gemacht. Das Gemisch wird mit CH2Cl2 (6 × 100 ml)
extrahiert und die vereinigten organischen Extrakte werden getrocknet
(Na2SO4) und unter Bildung
eines partiell abgetrennten Isoindolincarbonsäuremethylesters als Öl (33,2
g) konzentriert. 60% ee gemäß chiraler
CE.
-
-
Auftrennung des partiell
aufgetrennten Isoindolincarbonsäuremethylesters:
-
Eine
Lösung
des partiell aufgetrennten Isoindolincarbonsäuremethylesters (33,24 g, 0,174
mol) in EtOH (130 ml) wird langsam mit einer Lösung aus Dibenzoyl-L-weinsäure (56,06
g, 0,156 mol) in EtOH (200 ml) behandelt. Die Lösung wird mit Produkt angeimpft
und bei RT für
4 Stunden gerührt.
Das reine Produkt wird durch Filtration gesammelt, mit EtOH (30
ml) gewaschen und zu nicht ganz weißen Kristallen (60,49 g) getrocknet.
96,5% ee gemäß chiraler
CE.
-
-
Auftrennung der N-BOC
Isoindolincarbonsäure:
-
Eine
Lösung/Aufschlämmung der
razemischen N-BOC-Isoindolincarbonsäure (114,5 g, 0,413 mol) in EtOAc
(1000 ml) wird langsam mit Triethylamin (28,8 ml, 0,206 mol), gefolgt
von (S)-(–)-α-Methylbenzylamin behandelt.
Die Lösung
wird mit dem Produkt angeimpft und bei RT über Nacht gerührt. Das
Produkt wird durch Filtration gesammelt, mit EtOAc (200 ml) gewaschen
und zu einem weißen
Pulver (62,98 g) getrocknet. 97,6% ee gemäß chiraler CE.
-
Asymmetrische
Hydrierungsverfahren: Teil
I: Synthese des Z-Isomers (Vorläufer
der asymmetrischen Hydrierung) Schema
P1
-
Präparation PP27
-
Das
Z-Isomer der Verbindung 5 wird wie in Schema P1 angegeben, synthetisiert.
Von der Verbindung 5 wird gemäß HPLC und 1H NMR gezeigt, dass sie ein einzelnes Isomer
ist. Die Doppelstrangstereochemie ist aus vergleichenden NOE Daten
mittels des beschriebenen E-Isomers (Schema P1) abgeleitet. Die
beste chirale Induktion wird unter Verwendung der Verbindung 8/Ferrotan/MeOH-THF
erreicht. In Hinblick auf die Umwandlung der Verbindung 9 in die
Verbindung 10, das eine formale asymmetrische Synthese des Isoindolens der
Verbindung 10 darstellen würde,
wird dies mittels Super Hydrid-BF3 × OEt2 erreicht. Jedoch ist das Produkt ein Gemisch
der Verbindung 10 und der entsprechenden DeBOC (Schutzgruppen sind
abgespalten) Verbindung.
-
Präparation PP28
-
Verbindung 2 (Schema P1)
-
Phthalsäureanhydrid
(751,5 g, 5,014 mol), Kaliumacetat (498 g, 5,014 mol) und Essigsäureanhydrid (1
l) werden zusammen unter Stickstoff gerührt. Das Gemisch wird langsam
auf 145–150°C erwärmt und
für 10
Minuten gerührt
und dann bei 140°C
für 20
Minuten. Das Gemisch kann sich langsam auf 80°C über 1 Stunde abkühlen. Drei
Volumina Wasser werden zugegeben, wobei ein Feststoff ausfällt. Nach
der Filtration wird der filtrierte Feststoff mit warmem Wasser gewaschen
und so trocken wie möglich
für 30
Minuten abgenutscht. Der Feststoff wird dann entsprechend mit Ethanol
und Aceton gewaschen. Falls erwünscht
kann eine weitere Reinigung durch Aufschlämmen des Feststoffs in Aceton
bei Raumtemperatur für
15 Minuten und dann Filtration erreicht werden. Ein Trocknen im
Vakuum bei 50°C
für 20
Stunden ergibt die Verbindung 2 als nicht ganz weißen Feststoff,
470 g (48%) mit einer NMR Reinheit von etwa 90%.
-
Präparation PP29
-
Verbindung 3 (Schema P1)
-
Die
Verbindung 2 (470 mg, 2,47 mol) wird zu einem gerührten wässrigen
Ammoniak (470 ml konz. NH3 in 4,7 l Wasser)
gegeben. Das entstehende Gemisch wird bei Raumtemperatur für 1 Stunde
gerührt
und dann filtriert. Der filtrierte Feststoff wird mit Wasser gewaschen.
Das vereinigte wässrige
Filtrat und die Waschschritte werden vorsichtig mit 6 M wässrigem
HCl (2,35 l) angesäuert.
Der Niederschlag wird durch Filtration entfernt und im Vakuum bei
50°C unter
Bildung der Verbindung 3 als gelber Feststoff, 259 g (52%) getrocknet.
-
Präparation PP30
-
Verbindung 4 (Schema P1)
-
Die
Verbindung 3 (511 g, 2,7 mol) wird in Toluol aufgeschlämmt (10
Vol.) Thionylchlorid (385 g, 3,24 mol) wird über 10 Minuten zu dem gerührten Gemisch
gegeben, das dann am Rückfluss
für 1,5
Stunden erhitzt wird. Eine 1H NMR Analyse
zeigt etwa 80% Umwandlung zum Säurechlorid.
Dann wird DMF (3,7 ml) zugegeben und das Gemisch wird am Rückfluss
für weitere
3 Stunden erhitzt. Das entstehende Gemisch kann sich auf 35°C abkühlen und
Methanol (1,27 l) wird mit einer derartigen Geschwindigkeit zugegeben,
dass die Reaktionstemperatur bei 30–35°C gehalten wird. Das Reaktionsgemisch
wird bei dieser Temperatur für
weitere 15 Minuten gehalten und dann im Vakuum unter Bildung der
Verbindung 4 als brauner Feststoff, 536 g (quantitativ), konzentriert.
-
Präparation PP31
-
Verbindung 5 (Schema P1)
-
Die
Verbindung 4 (750 g, 3,65 mol) wird in Acetonitril (15 l) gelöst. Das
gerührte
Gemisch wird auf 0–5°C gekühlt und
DMAP (624 g, 5,11 mol) wird in einer Portion zugegeben. Nach 10
Minuten wird Boc-Anhydrid (1115 g, 5,11 mol) in einer Portion zugegeben:
Es bildet sich eine leichte Exothermie, die von einer Gasentwicklung
begleitet wird. Das Gemisch wird bei Raumtemperatur für 5 Stunden
gerührt
und dann im Vakuum konzentriert. Der Rückstand wird in EtOAc gelöst und jeweils
mit 10% wässriger
Zitronensäure,
gesättigtem wässrigem
Na2CO3 und Wasser
gereinigt. Nach dem Trocknen ergibt eine Konzentration der organischen
Bestandteile einen dicken Sirup. Dieses Material wird durch ein
Kissen aus Silicagel (1,5 g) unter Elution mit 1:1 EtOAc Hexan gegeben.
Die Verbindung 5 wird als dunkler Feststoff isoliert, 619 g (55%).
Eine vorsichtige Chromatographie auf Silicagel unter Elution mit
20% EtOAc – Hexan
ergibt die Verbindung 5 als flauschigen weißen Feststoff.
-
Schema
P2 Teil
II: Synthese des E-Isomers (Vorläufer
der asymmetrischen Hydrierung)
-
Präparation PP32
-
Das
E-Isomer der Verbindung 8 (Schema P2) wird wie in Schema P2 gezeigt,
hergestellt.
-
Präparation PP33
-
Verbindung 7 (Schema P2)
-
Die
Verbindung 7 wird gemäß dem Verfahren
von Einhorn et al., Synth. Commun. 2001, 31 (5), 741–748, hergestellt.
-
Präparation PP34
-
Verbindung 8 (Schema P2)
-
Die
Verbindung 7 (15,00 g, 60,7 mmol) und Methyl(triphenylphosphoranyliden)acetat
(41,40 g, 121,3 mmol) werden in Toluol (150 ml) aufgeschlämmt. Das
Gemisch wird am Rückfluss
gerührt
und die Reaktion der Verbindung 7 wird mittels GC verfolgt. Nach
1,5 Stunden ist die Reaktion gemäß GC vollständig. Nach
dem Kühlen
auf Raumtemperatur wird das Gemisch filtriert. Der Feststoff auf
dem Filter wird mit Toluol gewaschen, bis er farblos ist. Die vereinigten
Filtrat/Waschschritte werden im Vakuum unter Bildung eines hellbraunen
Feststoffs konzentriert. Dieses Material wird auf Silicagel gegeben
und auf Silicagel (1 kg) unter Elution mit 10% EtOAc – Hexan
chromatographiert. Die Verbindung 8 wird als weißes oder blassgelbes Pulver,
5,52 g (30%) isoliert.
-
Schema
P3 Asymmetrische
Hydrierung:
-
Präparation PP35
-
Das
Screenen der chiralen Hydrierungsbedingungen zeigt, dass die beste
chirale Induktion mittels der Verbindung 8/Ferrotan/MeOH-THF erreicht
wird. In Hinblick auf die Umwandlung der Verbindung 9 in die Verbindung
10, was eine formale asymmetrische Synthese des Isoindolins 10 aufbaut,
wird dies mittels Super Hydrid-BF3 × OEt2 erreicht. Jedoch ist das Produkt ein Gemisch
der Verbindung 10 und der entsprechenden De-BOC-Verbindung (abgespaltene
Schutzgruppen).
-
Schema
P4 Kupplung
des chiralen Isoindolins mit d-4-Chlorphenylalanin mittels des Tartratsalzes:
-
Präparation PP36
-
Verbindung 15 (Schema
P4)
-
Das
Tartratsalz der Verbindung 14 (58,00 g, 100,27 mmol) wird in Wasser
(580 ml) aufgeschlämmt. Festes
NaHCO3 (25,27 g, 300,8 mmol) wird vorsichtig
zugegeben. BOC Anhydrid (22,98 g, 105,28 mmol) wird in einer Portion
zugegeben und der Fortschritt der Reaktion wird durch Umkehrphasen
HPLC aufgezeichnet. Nach 1 Stunde wird zusätzliches BOC Anhydrid (2,18
g, 10,00 mmol) zugegeben. Die Reaktion ist nach 3 Stunden vollständig (gemäß HPLC).
Das Gemisch wird mit EtOAc (2 × 250
ml) extrahiert. Die vereinigten organischen Extrakte werden mit
Wasser (250 ml) gewaschen und getrocknet (MgSO4).
Eine Filtration und Konzentration im Vakuum ergibt die Verbindung
15 als klares hellbraunes Öl
(31,33 g), das mit einer kleinen Menge an t-BuOH und BOC Anhydrid
kontaminiert ist. Dieses Material wird direkt in der nächsten Reaktion
verwendet.
-
Präparation PP37
-
Verbindung 16 (Schema
P4)
-
Der
Ester der Verbindung 15 (29,21 g, 100,26 mmol) wird in 3:1 THF – Wasser
(100 ml) gelöst.
LiOH (6,00 g, 250,65 mmol) wird in einer Portion zu der gerührten Reaktion
gegeben. Nach 17 Stunden wird das Gemisch zur Trockne gebracht und
der Rückstand
wird in Wasser (500 ml) gelöst.
EtOAc (250 ml) wird zugegeben und festes NaHSO4 wird
zu dem gerührten
Gemisch gegeben, bis der pH 3 ist. Die organische Phase wird abgetrennt
und die wässrige
Phase wird mit EtOAc (250 ml) extrahiert. Die vereinigten EtOAc
Phasen werden getrocknet (MgSO4). Eine Filtration
und Konzentration im Vakuum ergibt die Säure 16 als hellbraunen Feststoff,
27,10 g (97%).
-
Schema
P5 Rrom α-Methylbenzylaminsalz:
-
Die
verwendete Chemie wird in Schema P5 gezeigt. Zwei Protokolle werden
verwendet: Verfahren A verwendet die isolierte Verbindung 16, Verfahren
B verwendet eine Lösung
der Verbindung 16, die aus dem aufgetrennten Salz 19 abgeleitet
ist.
-
Präparation PP38
-
Verbindung 17 (Schema
P5, Verfahren A)
-
Die
Säure 16
(24,18 g, 87,2 mmol) und D-Chlorphenylalaninhydrochlorid (21,81
g, 87,2 mmol) werden in CH2Cl2 (100
ml) und DMF (25 ml) gelöst.
Das Gemisch wird bei Umgebungstemperatur gerührt. HOBT (13,55 g, 100,3 mmol)
und Hunig's Base
(45,6 ml, 33,81 g, 261,6 mmol) werden zugegeben. HATU (38,13 g, 100,3
mmol) wird in einer Portion zugegeben (es entwickelt sich eine schnelle
Exothermie auf 50°C).
Das Gemisch wird für
90 Minuten gerührt
und dann mit EtOAc (750 ml) verdünnt.
Das entstehende Gemisch wird mit Wasser, 5% KHSO4,
Kochsalzlösung
und entsprechendem gesättigtem
NaHCO3 gewaschen und dann getrocknet. Eine
Filtration und Konzentration im Vakuum ergibt die rohe Verbindung
17 als braunen Schaum. Das Produkt wird durch Chromatographie auf
Silicagel (1 kg) unter Elution mit 1:1 EtOAc – Hexan gereinigt. Der Ester
17 wird als hellbraunes Pulver isoliert, 38,85 g (94%).
-
Präparation PP39
-
Verbindung 17 (Schema
P5, Verfahren B)
-
Aufgetrenntes
Salz 19 (96,27 g, 232,5 mmol) wird zwischen Wasser (500 ml) und
CH2Cl2 (250 ml)
aufgeteilt. Festes KHSO4 wird portionsweise
zugegeben, bis der pH 2,5 beträgt.
Die organische Phase wird abgetrennt und die wässrige Phase wird mit CH2Cl2 (150 ml) extrahiert.
Die vereinigten organischen Phasen werden getrocknet (MgSO4) und dann filtriert. Zu dieser Lösung wird
4-Chlor-D-phenylalanin
(58,1 g, 232,5 mmol), HOBT (34,57 g, 255,8 mmol), Hunig's Base (93,2 ml,
69,13 g, 534,9 mmol) und schließlich
HATU (97,26 g, 255,8 mmol) gegeben. Das entstehende Gemisch wird
bei Raumtemperatur für
18,5 Stunden gerührt
und dann auf ein Kissen aus Silicagel (1 kg) gegeben. Dies wird
mit 1:1 EtOAc – Hexan
gewaschen, bis kein weiteres Produkt eluiert. Der Ester der Verbindung
17 wird als pinkfarbener Schaum, 101,79 g (93%) isoliert: enthält etwa
1% nicht umgesetzte Verbindung 16.
-
Präparation PP40
-
Verbindung 18 (Schema
P5)
-
Der
Ester der Verbindung 17 (38,64 g, 81,7 mmol) wird in 3:1 THF – Wasser
(200 ml) gelöst.
LiOH (2,15 g, 89,9 mmol) wird zu dem Gemisch gegeben, das bei Raumtemperatur
für 2 Stunden
gerührt
wird. Das Lösemittel
wird dann im Vakuum entfernt und der übrige Feststoff wird in Wasser
(600 ml) aufgenommen. Dieser wird mit MTBE (250 ml) extrahiert.
Die wässrige
Phase wird abgetrennt und mit EtOAc (250 ml) gerührt und festes KHSO4 wird portionsweise zugegeben, bis der pH
3 beträgt.
Die Phasen werden abgetrennt und die wässrige Phase wird mit EtOAc
(250 ml) extrahiert. Die vereinigten organischen Phasen werden über MgSO4 getrocknet. Eine Filtration und Konzentration
im Vakuum ergibt die saure Verbindung 18 als hellpinkfarbenen Schaum,
38,41 g (35,71 g korrigiert für
das restliche Lösemittel,
95%).
-
Präparation
PP41 Schritt
1: Veresterung
-
In
einem 22 l fassenden Vierhalsrundbodenkolben, der mit einem Rückflusskühler, einem
Thermoelement und einem Stickstoffeinlass ausgestattet ist, wird
eine Aufschlämmung
aus 1000 g (5,4 mol) an m-Tyrosin in 10 l an 2B-3 EtOH auf 5°C gekühlt. Zu
der Aufschlämmung
werden 350 ml (12,4 mol) Thionylchlorid tropfenweise mittels eines
Zugabetrichters bei einer solchen Geschwindigkeit gegeben, dass
die Reaktionstemperatur unter 20°C
liegt. Nachdem die Zugabe vollständig
ist, wird die Reaktion auf Rückflusstemperatur
erhitzt und für
18 Stunden gerührt.
Die Reaktion wird auf ein Drittel ihres Volumens konzentriert und
8 l MTBE werden zugegeben. Die entstehende dicke Aufschlämmung wird
für 14
Stunden in einem Rotationsverdampfer bei RT gerührt. Der entstehende Feststoff
wird auf einem Filterkissen isoliert und bei 40°C für 48 Stunden unter Bildung
von 1288 g (95%) getrocknet. NMR (DMSOd6)
zeigt das gewünschte
Material.
-
Präparation
PP42 Schritt
2: Pictet-Spengler
-
In
einem 22 l fassenden Vierhalsrundbodenkolben, der mit einem mechanischen
Rührer,
einem Thermoelement und einem Rückflusskühler an
der Spitze des Soxhlet Extraktionsgeräts mit 4° Sieben ausgestattet ist, wird
eine Halb-Lösung
aus m-Tyrosinethylesterhydrochlorid 1288 g (5,26 mol) in 13 l Aceton
auf Rückflusstemperatur
erhitzt. Das Kondensat wird durch die Siebe zur Entfernung des Wassers
filtriert. Die Reaktion wird kräftig
am Rückfluss
für 48
Stunden gerührt.
Eine NMR Probe in DMSOd6t zeigt die Abwesenheit des Ausgangsmaterials.
Die Reaktion wird auf RT gekühlt
und unter Bildung eines nicht ganz weißen Feststoffs konzentriert,
1411 g (94%).
-
Präparation
PP43 Schritt
3: Triflatbildung:
-
In
einem 22 l fassenden Vierhalsrundbodenkolben, der mit einem Rückflusskühler, einem
mechanischen Rührer,
einem Stickstoffeinlass und einem Thermoelement ausgestattet ist,
werden 1240 g (4,35 mol) des Ausgangsmaterialsalzes in 12,4 l Methylenchlorid
auf 4°C
gekühlt.
Zu dem Gemisch werden 1452 ml (10,4 mol) Triethylamin gegeben und
in die Lösung
gerührt.
Trifluormethansulfonsäure,
1472 ml (5,22 mol) werden tropfenweise zu der Reaktion bei einer
derartigen Geschwindigkeit zugegeben, dass die innere Temperatur
unter 10°C
beträgt.
Das Eisbad wird entfernt und die Reaktion wird auf RT erwärmt und
für 18
Stunden gerührt. Die
Reaktion wird zu einem Öl
konzentriert und dann in 4 l EtOAc gelöst und wieder zu einem Öl konzentriert, um
das überschüssige Trifluormethansulfonsäureanhydrid
zu entfernen. Der rohe Rückstand
wird in 4 l EtOAc gelöst
und mit Wasser und gesättigter
Natriumbicarbonatlösung
gewaschen. Die organische Phase wird isoliert und mit Natriumsulfat
getrocknet, filtriert und unter Bildung von 1720 g (> 100%) eines rohen
dunklen Öls konzentriert,
das ohne weitere Reinigung verwendet wird.
-
Präparation
PP44 Schritt
4: Desoxygenierung
-
Eine
Lösung
aus 1720 g (4,35 mol) des rohen Ausgangsmaterials in 14 l Aceton
wird in einen 10 Gallonen fassenden Edelstahlautoklaven gegeben.
Zu der Lösung
wird eine Aufschlämmung
aus 5% Pd/C in 1,2 l Toluol gegeben. Das Reaktionsgemisch wird evakuiert
und mit H2 Gas bei 50 psi zweimal gewaschen.
Die Reaktion wird über
Nacht bei 50°C
mit H2 bei 50 psi gerührt. Ein Probealiquot zeigt
an, dass keine Reaktion stattgefunden hat. Das Gemisch wird filtriert
und zu einem dicken Öl
konzentriert und Reaktionsbedingungen unterzogen. Nach 18 Stunden
zeigt eine NMR eines Probenaliquots die Abwesenheit des Ausgangsmaterials. Das
Reaktionsgemisch wird filtriert und das Filtrat wird unter Bildung
von 1581 g eines nicht ganz weißen
Feststoffs (95%) konzentriert.
-
Präparation
PP45 Schritt
5: Hydrolyse/Salzbildung
-
Zu
einem 2 l fassenden Dreihalskolben, der mit einem mechanischen Rührer, einem
Thermoelement und einem Stickstoffeinlass ausgestattet ist, wird
ein Gemisch aus 700 g (1,83 mol) des Trifluormethansulfonsäuresalzausgangsmaterials
gegeben. Eine Lösung
aus 427 g (1,83 mol) des Ausgangsmaterials der freien Base in 13,3
l THF wird zugegeben, gefolgt von 700 ml Wasser. Die Halb-Lösung wird
kräftig
bei RT gerührt. Zu
dem Reaktionskolben werden 43,7 g (1,83 mol) an festem LiOH in kleinen
Por tionen bei einer derartigen Geschwindigkeit gegeben, dass die
innere Temperatur unter 35°C
bleibt. Die Reaktion wird für
18 Stunden bei RT gerührt
und zu einem dicken Öl
konzentriert. THF (4 l) wird zugegeben und die Halb-Lösung wird
konzentriert. Dies wird mit Toluol wiederholt und der Halb-Feststoff
wird unter Hausvakuum auf einem Rotationsverdampfer unter Rühren für 18 Stunden
gegeben, wobei sich 650 g eines rohen Feststoffs bilden. Der Feststoff wird
in EtOAc wieder aufgeschlämmt,
filtriert und unter Bildung von 525 g (68%) des Lithiumsalzes als
nicht ganz weißer
Feststoff getrocknet.
-
Präparation
PP46 Schritt
6: Kupplung
-
Festes
D-Chlorphenylalanin, 446 g (1,78 mol), wird zu der halbfesten Lösung gefolgt
von 20 g (0,162 mol) DMAP gegeben. Das entstehende Gemisch wird
für 15
Minuten gerührt
und dann wird festes EDCl (1-(3-Dimethylaminopropyl)-3-ethylcarbodiimidhydrochlorid),
390 g, (2,03 mol) zugegeben. Das Reaktionsgemisch wird auf 80°C erhitzt
und für
18 Stunden gerührt.
Eine Dünnschichtchromatographie
(1:1 EtOAc:Hexan) zeigt die Anwesenheit von sehr wenig Ausgangsmaterial.
Die Reaktion wird auf RT gekühlt
und unter Bildung eines dicken Öls
konzentriert. Das rohe Öl
wird in EtOAc gelöst
und mit Wasser und Kochsalzlösung
gewaschen. Die Lösung
wird mit Natriumsulfat getrocknet, filtriert und unter Bildung eines
dicken Öls,
426 g, konzentriert. Das rohe Öl
wird in mehreren Schritten mittels einer Waters Präp 500 Chromatographieapparatur chromatographiert.
Der Eluent besteht aus einem Gradientensystem, 5%–80% EtOAc
in Heptan bei einer Flussrate von 240 ml/min über 38 Minuten. Die zwei Diastereomere
werden getrennt und unter Bildung von 119,04 g für den oberen Fleck und 111,3
g für den
unteren Fleck isoliert. Eine Bestätigung für beide gewünschten Diasteroemere wird
mittels NMR (DMSO6) erreicht.
-
Präparation
PP47 Auftrennung
des Tetrahydroisochinolincarbonsäureethylesters
unter Bildung des L-Weinsäuresalzes:
-
Herstellung der freien
Base:
-
Ein
razemisches Gemisch aus Tetrahydroisochinolincarbonsäure (7,43
g) in EtOAc (60 ml) wird mit gesättigter
NaHCO3 Lösung
(60 ml) und gesättigter
Na2CO3 Lösung (10
ml) behandelt. Das Gemisch wird gerührt und die Phasen werden getrennt.
Die organische Phase wird getrocknet (Na2SO4) und unter Bildung der entsprechenden freien
Base als Öl
(4,85 g) konzentriert.
-
Auftrennung:
-
Ein
Gemisch der obigen freien Base (467 mg, 2,0 mmol) und L-Weinsäure (300
mg, 2,0 mmol) in Aceton (4 ml) wird bei RT über Nacht gerührt. Das
Titel-L-Weinsäuresalz
wird durch Filtration gesammelt, mit Aceton gewaschen (etwa 2 ml)
und zu einem weißen
Pulver (367 mg) getrocknet. 100% ee gemäß chiraler CE.
-
Präparation
PP48 Auftrennung
der N-BOC-Tetrahydroisochinolincarbonsäure
-
2-{2-[(tert-Butyl)oxycarbonyl]-1,2,3,4-tetrahydroisochinolyl}essigsäuredehydroabietylaminsalz:
-
Razemische
2-{2-[(tert-Butyl)oxycarbonyl]-1,2,3,4-tetrahydroisochinolyl}essigsäure (30,15
g, 103,5 mmol) wird in i-PA (300 ml) gelöst. Dehydroabietylamin (22,11
g, 52,7 mmol eines Gemisches mit 68 Gewichtsprozent) wird zu der
Lösung
gegeben, die dann auf einem Multiarmschüttler für 63 h geschüttelt wird.
Die entstehende dicke Paste wird filtriert und mit i-PA (50 ml,
25 ml) gewaschen. Trocknen in einem 50°C Vakuumofen ergibt einen weißen Feststoff
(27,73 g, 52% ee gemäß chiraler
CE Analyse). Das Produkt wird in i-PA (266 ml) wieder aufgeschlämmt und
auf einem Multiarmschüttler
für 23,5
h geschüttelt.
Die dicke Aufschlämmung
wird filtriert und mit kaltem i-PA (50 ml, 30 ml) gewaschen. Der
Kuchen wird in einem 50°C
Vakuumofen getrocknet und das Produkt wird als weißer Feststoff
(23,63 g, 40% Ausbeute, 94% ee gemäß chiraler CE Analyse) erhalten.
-
Schema
P6 Asymmetrische
Hydrierung:
-
Präparation PP49
-
Das
Enamin der Verbindung 21 (Schema P6) wird als Substrat für asymmetrische
Hydrierungsscreeningstudien hergestellt. Es wird als etwa 10:1 Gemisch
mit dem Imin der Verbindung 22 gebildet. Das Enamin (21) kann mit
einer NH Schutzgruppe versehen sein, wie mit einer Boc Schutzgruppe.
Die entstehende Verbindung 23 kann einer asymmetrischen Hydrierung
zur Bildung der Essigsäure
oder des Methylacetat substituierten Isochinolins unterzogen werden,
was wie vorher beschrieben, in eine Verbindung der Formel I umgewandelt
werden kann.
-
Präparation
PP50 Verbindung
21 (Schema P6) Herstellung
wie in W. Sobotka et al., J. Org. Chem., 1965, 30, 3667 beschrieben Schema
P7 Synthese
des Gem-Dimethyl TIC:
-
Präparation PP51
-
Die
chirale Synthese des Gem-Dimethyl TIC mittels L-DOPA als Ausgangsmaterial
an Stelle von Tyrosin wird erfolgreich in der Pictet-Spengler Reaktion
mit L-DOPA und Aceton gezeigt. Das Produkt ist ein Gemisch des Ausgangsmaterials
24 und des Produkts 25 (Hauptkomponente). Das Produkt wird mittels
allgemeiner Isolierungsverfahren isoliert. Ein alternatives Isolierungsverfahren
ist die Umsetzung des Gemisches (24 und 25) mit BOC Anhydrid, wobei
das weniger gehinderte N-H in Verbindung 24 zur bevorzugten BOC
Schutzgruppenanbringung der Verbindung 24 führt, wobei eine schnelle Abtrennung
der Verbindung 25 erfolgen kann. Die Chemie für den Rest der Sequenz beispielsweise,
Desoxygenierungsreaktion wird hierin gezeigt.









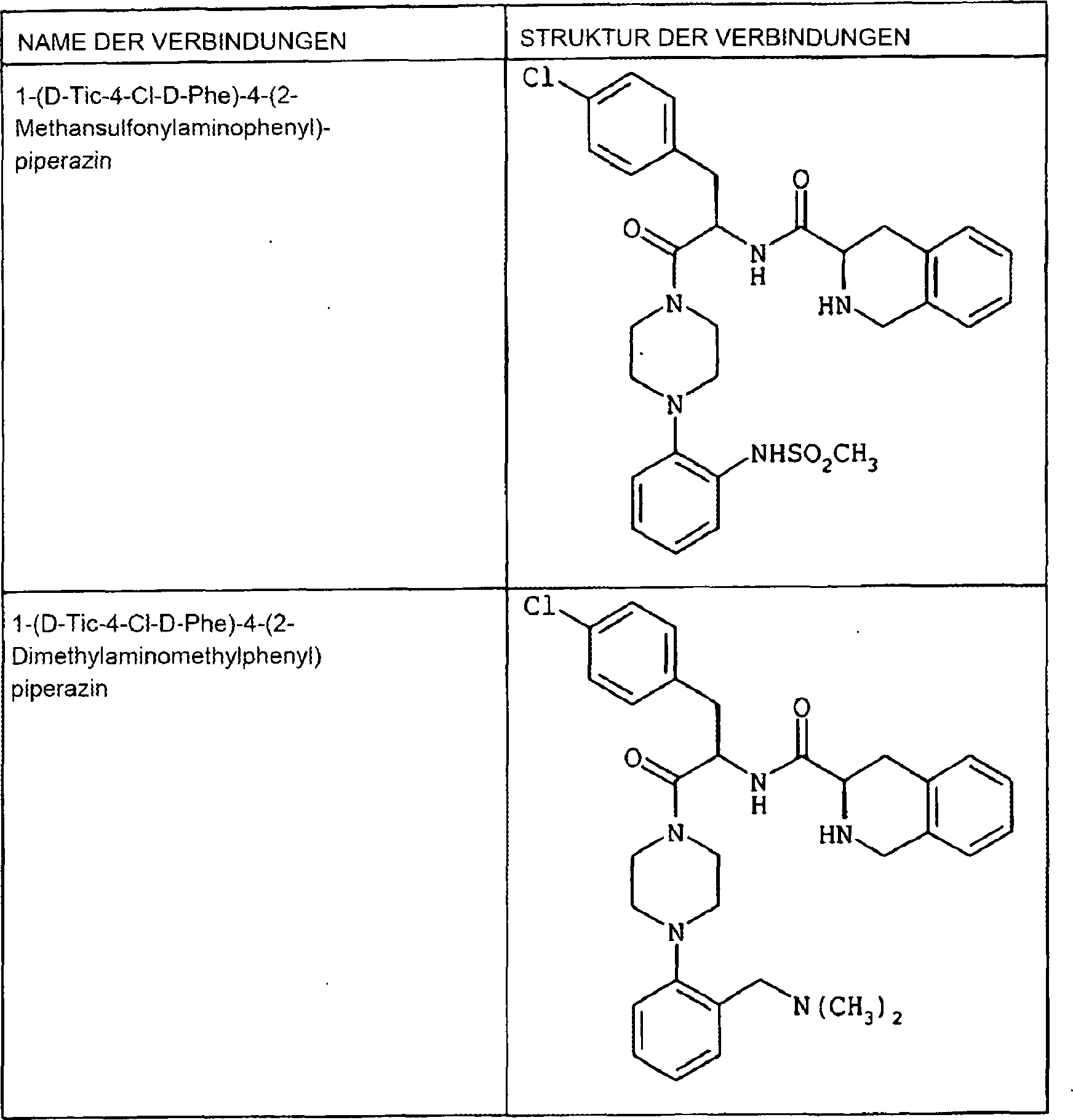








 und
und





 R, R1, R3, R4, p und r wie in Formel I definiert sind, und
R, R1, R3, R4, p und r wie in Formel I definiert sind, und
 worin Ra für C1-C4 Alkyl oder (D)-Phenyl steht
worin Ra für C1-C4 Alkyl oder (D)-Phenyl steht





 und
und


 R, R1, R3, R4, p und r wie in Formel I definiert sind und
R, R1, R3, R4, p und r wie in Formel I definiert sind und






























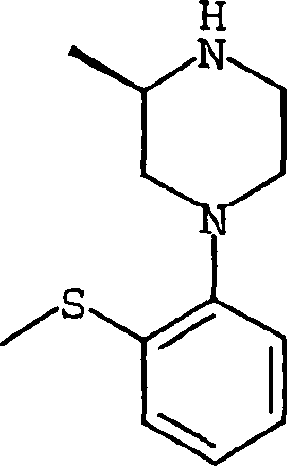












































































































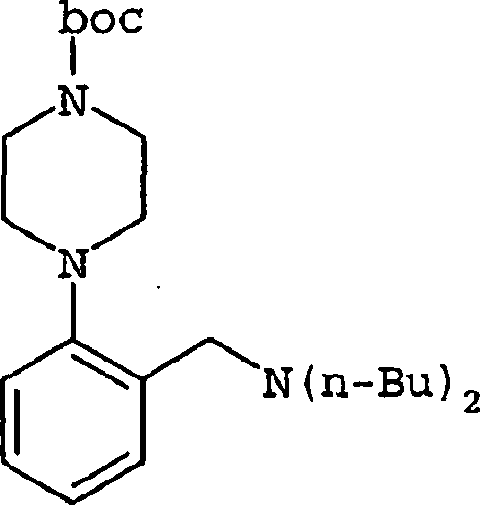














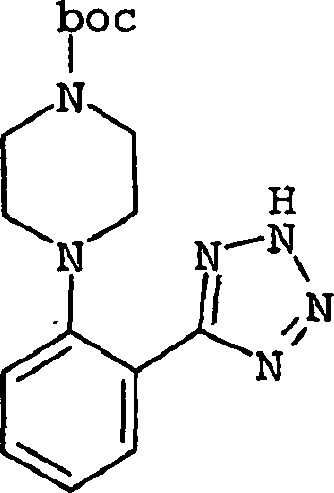






























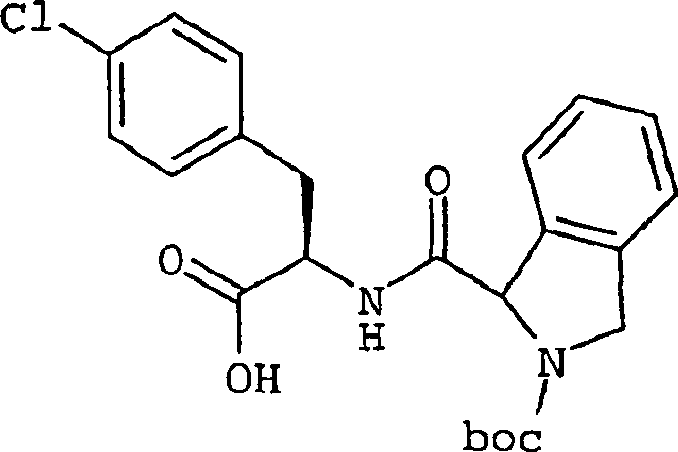
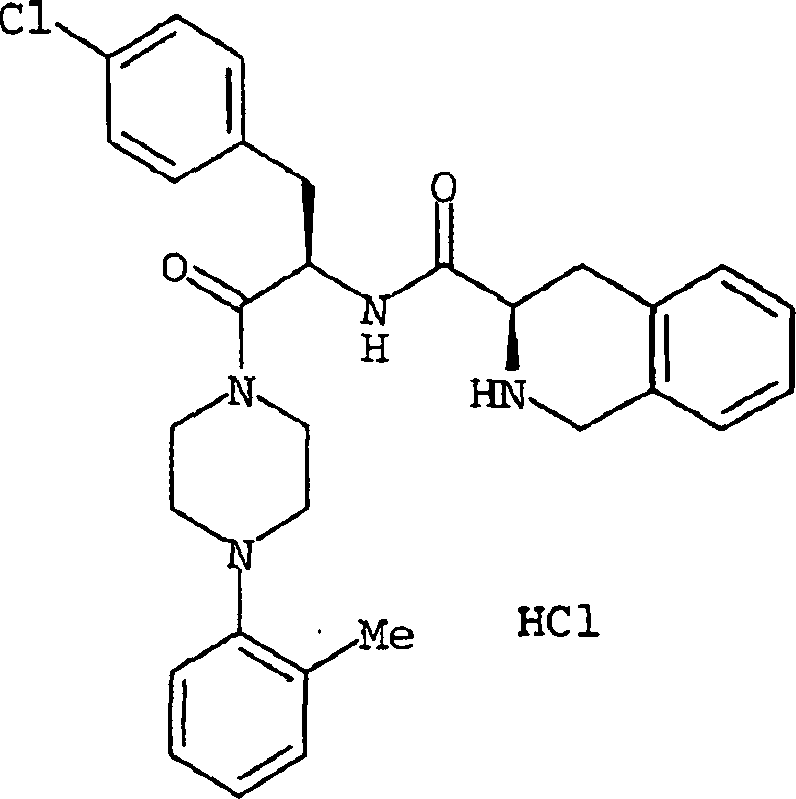
















































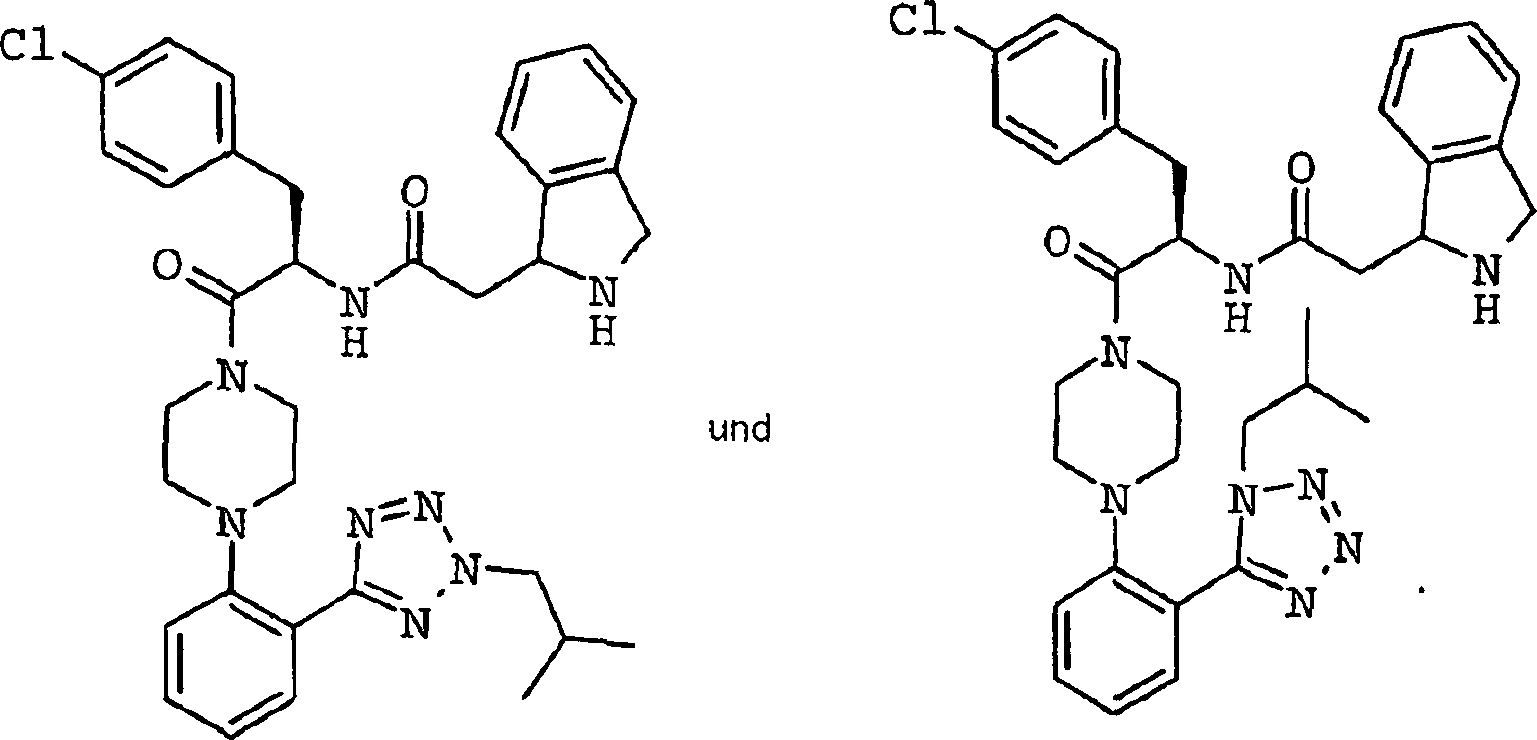












 MS M + 1 602,2 (86%) Beispiel 165 7-Trifluormethyl-1,2,3,4-tetrahydroisochinolin-3-carbonsäure-{1-(4-chlorbenzyl)-2-oxo-2-[4-(2-[1,2,4]triazol-1-yl-methylphenyl)piperazin-1-yl]ethyl}amid, HCl Salz
MS M + 1 602,2 (86%) Beispiel 165 7-Trifluormethyl-1,2,3,4-tetrahydroisochinolin-3-carbonsäure-{1-(4-chlorbenzyl)-2-oxo-2-[4-(2-[1,2,4]triazol-1-yl-methylphenyl)piperazin-1-yl]ethyl}amid, HCl Salz MS M + 1 652,2 (10%) Beispiel 166 3-Methyl-1,2,3,4-tetrahydroisochinolin-3-carbonsäure-{1-(4-chlorbenzyl)-2-oxo-2-[4-(2-[1,2,4]-triazol-1-ylmethylphenyl)piperazin-1-yl]ethyl}amid, HCl Salz
MS M + 1 652,2 (10%) Beispiel 166 3-Methyl-1,2,3,4-tetrahydroisochinolin-3-carbonsäure-{1-(4-chlorbenzyl)-2-oxo-2-[4-(2-[1,2,4]-triazol-1-ylmethylphenyl)piperazin-1-yl]ethyl}amid, HCl Salz MS M + 1 598,3 (58%)
MS M + 1 598,3 (58%)




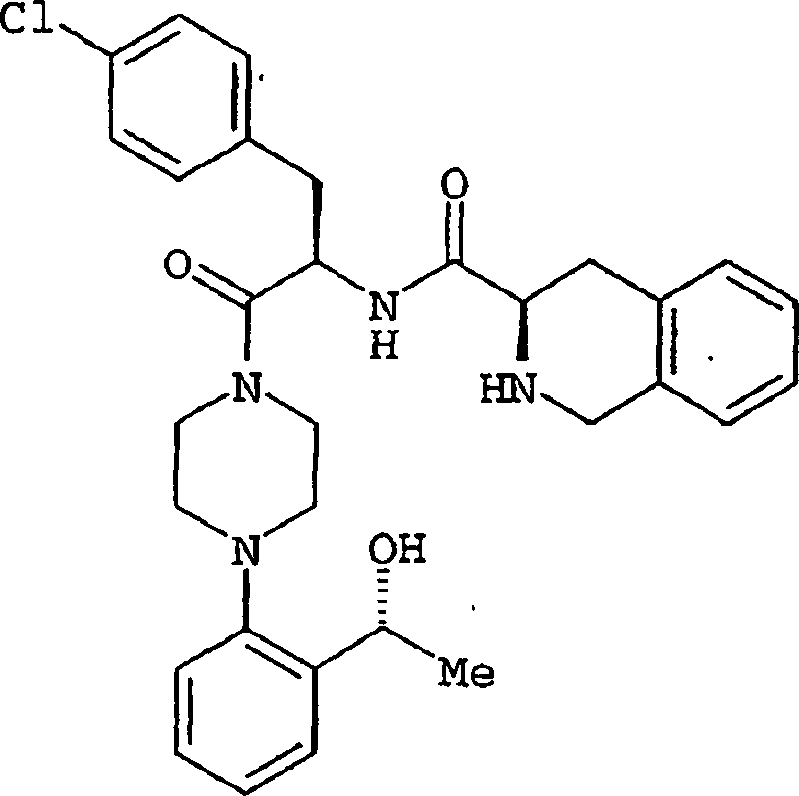


































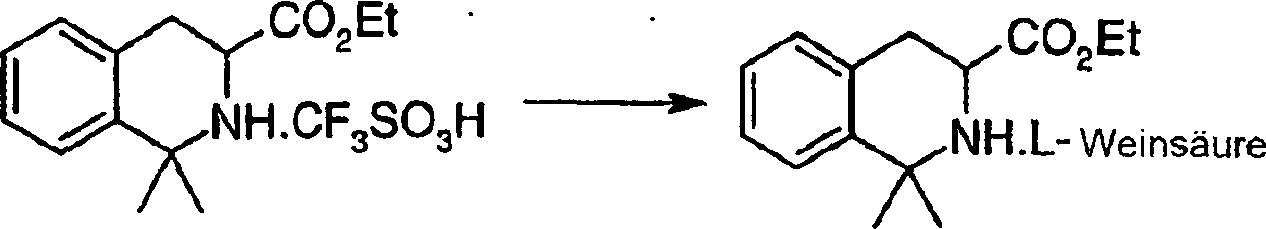



 R unabhängig steht für Wasserstoff, Hydroxy, Cyano, Nitro, Halogen, C1-C8 Alkyl, C1-C8 Alkoxy, C1-C4 Halogenalkyl, (D)C(O)R9, (D)C(O)OR9, (D)C(O)SR9, (D)C(O)-Heteroaryl, (D)C(O)-Heterocyclyl, (D)C(O)N(R9)2, (D)N(R9)2, (D)NR9COR9, (D)NR9CON(R9)2, (D)NR9C(O)OR9, (D)NR9C(R9)=N(R9), (D)NR9C(=NR9)N(R9)2, (D)NR9SO2R9, (D)NR9SO2N(R9)2, (D)NR9(CH2)n-Heterocyclyl, (D)NR9(CH2)n-Heteroaryl, (D)OR9, OSO2R9, (D)[O]q(C3-C7 Cycloalkyl), (D)[O]q(CH2)n-Aryl, (D)[O]q(CH2)n-Heteroaryl, (D)[O]q(CH2)n-Heterocyclyl, worin Heterocyclyl ein Heterocyclyl ausschließt, das einen einzelnen Stickstoff enthält, wenn q für 1 steht, für (D)SR9, (D)SOR9, (D)SO2R9 oder (D)SO2N(R9)2, worin C1-C8 Alkyl, C1-C8 Alkoxy, C3-C7 Cycloalkyl, Aryl, Heterocyclyl und Heteroaryl wahlweise mit einem bis fünf Substituenten substituiert sind, die unabhängig aus R8 ausgewählt sind, R1 unabhängig steht für Wasserstoff, CONH(C1-C8 Alkyl), C1-C8 Alkyl, (D)-Phenyl, (D)-C3-C7 Cycloalkyl oder Oxo, mit der Maßgabe, dass Oxo nicht an denselben Kohlenstoff gebunden ist, der an den Stickstoff gebunden ist, der eine Amidbindung bildet, R3 unabhängig für Aryl oder Thienyl steht, worin Aryl und Thienyl wahlweise mit einem bis drei Substituenten substituiert sind, die aus der Gruppe ausgewählt sind, welche besteht aus Cyano, Halogen, C1-C8 Alkyl, (D)-C3-C7 Cycloalkyl, C1-C4 Alkoxy, C1-C4 Halogenalkyl und C1-C4 Halogenalkyloxy, R4 unabhängig steht für Wasserstoff, C1-C8 Alkyl, C(O)R9, C(O)OR9, C3-C7 Cycloalkyl oder (CH2)nO(C1-C8 Alkyl), worin n für 2 bis 8 steht, R8 jeweils unabhängig steht für Wasserstoff, Halogen, Oxo, N(R10)2, C1-C8 Alkyl, (D)-C3-C7 Cycloalkyl, C1-C4 Halogenalkyl, C1-C4 Alkoxy, Heteroaryl, Hydroxy, Heterocyclyl, worin Heterocyclyl ein Heterocyclyl ausschließt, das einen einzelnen Stickstoff enthält, für Phenyl, (D)-COR9, (D)C(O)OR9, (D)OR9, (D)OCOR9, (D)OCO2R9, (D)SR9, (D)SOR9 oder (D)SO2R9, worin Aryl, Heteroaryl, Heterocyclyl, Alkyl oder Cycloalkyl wahlweise mit ein bis drei Substituenten substituiert sind, die aus der Gruppe ausgewählt sind, die besteht aus Oxo, C1-C8 Alkyl, N(R10)2, OR10, SR10 und CO2R10, R9 jeweils unabhängig ausgewählt ist aus Wasserstoff, C1-C8 Alkyl, C1-C4 Halogenalkyl, (D)-C3-C7 Cycloalkyl, (D)-Aryl, worin Aryl für Phenyl oder Naphthyl steht, (D)-Heteroaryl oder (D)-Heterocyclyl, worin Heterocyclyl ein Heterocyclyl ausschließt, das einen einzelnen Stickstoff enthält und worin Aryl, Heteroaryl, Heterocyclyl, Alkyl oder Cycloalkyl wahlweise mit einem bis drei Substituenten substituiert sind, die aus der Gruppe ausgewählt sind, die besteht aus Oxo, C1-C8 Alkyl, N(R10)2, OR10, SR10 und CO2R10, R10 jeweils unabhängig steht für Wasserstoff, C1-C8 Alkyl, C(O)-C1-C8 Alkyl, Aryl oder C3-C7 Cycloalkyl, R11 jeweils unabhängig steht für Wasserstoff, C1-C8 Alkyl, (D)-Aryl, (D)-Heteroaryl, (CH2)nN(R8)2, (CH2)nNR8C(O)C1-C4 Alkyl, (CH2)nNR8SO2-C1-C4 Alkyl, (CH2)nSO2N(R8)2, (CH2)n[O]q-C1-C8 Alkyl, (CH2)n[O]q(CH2)nNR8COR8, (CH2n[O]q(CH2)nN-R8SO2R8, (CH2)n[O]q-Heterocyclyl oder (CH2)n[O]q(C1-C8 Alkyl)heterocyclyl und worin n für 2 bis 8 steht, R12 jeweils unabhängig steht für Wasserstoff, C1-C8 Alkyl, (D)-Phenyl, C(O)-C1-C8 Alkyl, C(O)-Phenyl, SO2C1-C8 Alkyl oder SO2-Phenyl, D für eine Bindung oder -(CH2)n-steht, n für 0 bis 8 steht, p für 0 bis 5 steht, q für 0 bis 1 steht und r für 1 bis 2 steht.
R unabhängig steht für Wasserstoff, Hydroxy, Cyano, Nitro, Halogen, C1-C8 Alkyl, C1-C8 Alkoxy, C1-C4 Halogenalkyl, (D)C(O)R9, (D)C(O)OR9, (D)C(O)SR9, (D)C(O)-Heteroaryl, (D)C(O)-Heterocyclyl, (D)C(O)N(R9)2, (D)N(R9)2, (D)NR9COR9, (D)NR9CON(R9)2, (D)NR9C(O)OR9, (D)NR9C(R9)=N(R9), (D)NR9C(=NR9)N(R9)2, (D)NR9SO2R9, (D)NR9SO2N(R9)2, (D)NR9(CH2)n-Heterocyclyl, (D)NR9(CH2)n-Heteroaryl, (D)OR9, OSO2R9, (D)[O]q(C3-C7 Cycloalkyl), (D)[O]q(CH2)n-Aryl, (D)[O]q(CH2)n-Heteroaryl, (D)[O]q(CH2)n-Heterocyclyl, worin Heterocyclyl ein Heterocyclyl ausschließt, das einen einzelnen Stickstoff enthält, wenn q für 1 steht, für (D)SR9, (D)SOR9, (D)SO2R9 oder (D)SO2N(R9)2, worin C1-C8 Alkyl, C1-C8 Alkoxy, C3-C7 Cycloalkyl, Aryl, Heterocyclyl und Heteroaryl wahlweise mit einem bis fünf Substituenten substituiert sind, die unabhängig aus R8 ausgewählt sind, R1 unabhängig steht für Wasserstoff, CONH(C1-C8 Alkyl), C1-C8 Alkyl, (D)-Phenyl, (D)-C3-C7 Cycloalkyl oder Oxo, mit der Maßgabe, dass Oxo nicht an denselben Kohlenstoff gebunden ist, der an den Stickstoff gebunden ist, der eine Amidbindung bildet, R3 unabhängig für Aryl oder Thienyl steht, worin Aryl und Thienyl wahlweise mit einem bis drei Substituenten substituiert sind, die aus der Gruppe ausgewählt sind, welche besteht aus Cyano, Halogen, C1-C8 Alkyl, (D)-C3-C7 Cycloalkyl, C1-C4 Alkoxy, C1-C4 Halogenalkyl und C1-C4 Halogenalkyloxy, R4 unabhängig steht für Wasserstoff, C1-C8 Alkyl, C(O)R9, C(O)OR9, C3-C7 Cycloalkyl oder (CH2)nO(C1-C8 Alkyl), worin n für 2 bis 8 steht, R8 jeweils unabhängig steht für Wasserstoff, Halogen, Oxo, N(R10)2, C1-C8 Alkyl, (D)-C3-C7 Cycloalkyl, C1-C4 Halogenalkyl, C1-C4 Alkoxy, Heteroaryl, Hydroxy, Heterocyclyl, worin Heterocyclyl ein Heterocyclyl ausschließt, das einen einzelnen Stickstoff enthält, für Phenyl, (D)-COR9, (D)C(O)OR9, (D)OR9, (D)OCOR9, (D)OCO2R9, (D)SR9, (D)SOR9 oder (D)SO2R9, worin Aryl, Heteroaryl, Heterocyclyl, Alkyl oder Cycloalkyl wahlweise mit ein bis drei Substituenten substituiert sind, die aus der Gruppe ausgewählt sind, die besteht aus Oxo, C1-C8 Alkyl, N(R10)2, OR10, SR10 und CO2R10, R9 jeweils unabhängig ausgewählt ist aus Wasserstoff, C1-C8 Alkyl, C1-C4 Halogenalkyl, (D)-C3-C7 Cycloalkyl, (D)-Aryl, worin Aryl für Phenyl oder Naphthyl steht, (D)-Heteroaryl oder (D)-Heterocyclyl, worin Heterocyclyl ein Heterocyclyl ausschließt, das einen einzelnen Stickstoff enthält und worin Aryl, Heteroaryl, Heterocyclyl, Alkyl oder Cycloalkyl wahlweise mit einem bis drei Substituenten substituiert sind, die aus der Gruppe ausgewählt sind, die besteht aus Oxo, C1-C8 Alkyl, N(R10)2, OR10, SR10 und CO2R10, R10 jeweils unabhängig steht für Wasserstoff, C1-C8 Alkyl, C(O)-C1-C8 Alkyl, Aryl oder C3-C7 Cycloalkyl, R11 jeweils unabhängig steht für Wasserstoff, C1-C8 Alkyl, (D)-Aryl, (D)-Heteroaryl, (CH2)nN(R8)2, (CH2)nNR8C(O)C1-C4 Alkyl, (CH2)nNR8SO2-C1-C4 Alkyl, (CH2)nSO2N(R8)2, (CH2)n[O]q-C1-C8 Alkyl, (CH2)n[O]q(CH2)nNR8COR8, (CH2n[O]q(CH2)nN-R8SO2R8, (CH2)n[O]q-Heterocyclyl oder (CH2)n[O]q(C1-C8 Alkyl)heterocyclyl und worin n für 2 bis 8 steht, R12 jeweils unabhängig steht für Wasserstoff, C1-C8 Alkyl, (D)-Phenyl, C(O)-C1-C8 Alkyl, C(O)-Phenyl, SO2C1-C8 Alkyl oder SO2-Phenyl, D für eine Bindung oder -(CH2)n-steht, n für 0 bis 8 steht, p für 0 bis 5 steht, q für 0 bis 1 steht und r für 1 bis 2 steht.







 worin R1 für Wasserstoff, C1-C8 Alkyl, Boc, CBZ, Phenyl, FMOC oder (C1-C8 Alkyl)phenyl steht, Q für folgenden Rest steht
worin R1 für Wasserstoff, C1-C8 Alkyl, Boc, CBZ, Phenyl, FMOC oder (C1-C8 Alkyl)phenyl steht, Q für folgenden Rest steht und R, R1, R3, R4, R10, p und r wie in Anspruch 1 definiert sind, wobei das Verfahren die folgenden Schritte umfasst a) Umsetzung einer Verbindung der Strukturformel 1
und R, R1, R3, R4, R10, p und r wie in Anspruch 1 definiert sind, wobei das Verfahren die folgenden Schritte umfasst a) Umsetzung einer Verbindung der Strukturformel 1 mit CH2CH=C(O)ORa, worin Ra für Wasserstoff oder C1-C8 Alkyl steht und X für Halogen steht in Gegenwart eines Katalysators und einer Base in einem geeigneten organischen Lösemittel unter Bildung der Verbindung der Formel 2
mit CH2CH=C(O)ORa, worin Ra für Wasserstoff oder C1-C8 Alkyl steht und X für Halogen steht in Gegenwart eines Katalysators und einer Base in einem geeigneten organischen Lösemittel unter Bildung der Verbindung der Formel 2 b) reduktive Aminierung der Verbindung der Formel 2 in Gegenwart eines Amins bei sauren Bedingungen unter Bildung einer Verbindung der Formel 3
b) reduktive Aminierung der Verbindung der Formel 2 in Gegenwart eines Amins bei sauren Bedingungen unter Bildung einer Verbindung der Formel 3 c) Cyclisierung der Verbindung der Formel 3 durch eine Michael-Addition unter Bildung einer Verbindung der Formel 4 oder von Stereoisomeren hiervon
c) Cyclisierung der Verbindung der Formel 3 durch eine Michael-Addition unter Bildung einer Verbindung der Formel 4 oder von Stereoisomeren hiervon d) Kupplung der Verbindung der Formel 4 oder von Stereoisomeren hiervon, worin Ra der Verbindung 4 für H steht, mit einer Verbindung der Formel 5
d) Kupplung der Verbindung der Formel 4 oder von Stereoisomeren hiervon, worin Ra der Verbindung 4 für H steht, mit einer Verbindung der Formel 5 worin Ra der Verbindung 5 für C1-C8 Alkyl steht, unter Bildung einer Verbindung der Formel 6
worin Ra der Verbindung 5 für C1-C8 Alkyl steht, unter Bildung einer Verbindung der Formel 6 und e) Kupplung der Verbindung der Formel 6, worin Ra für H steht, mit einer Verbindung der Struktur
und e) Kupplung der Verbindung der Formel 6, worin Ra für H steht, mit einer Verbindung der Struktur unter Bildung der Verbindung der Formel I.
unter Bildung der Verbindung der Formel I.
 Q für folgenden Rest steht
Q für folgenden Rest steht R, R1, R3, R4, p und r wie in Anspruch 1 definiert sind, und R11 unabhängig für Wasserstoff oder C1-C8 Alkyl steht, wobei das Verfahren die folgenden Schritte umfasst a) Veresterung einer Verbindung der Formel 1
R, R1, R3, R4, p und r wie in Anspruch 1 definiert sind, und R11 unabhängig für Wasserstoff oder C1-C8 Alkyl steht, wobei das Verfahren die folgenden Schritte umfasst a) Veresterung einer Verbindung der Formel 1 mit einem Alkohol RaOH unter Bildung einer Verbindung der Formel 2
mit einem Alkohol RaOH unter Bildung einer Verbindung der Formel 2 worin Ra für C1-C4 Alkyl oder (D)-Phenyl steht b) Umsetzung einer Verbindung der Formel 2 mit R11 COR11 unter Bildung einer Verbindung der Formel 3
worin Ra für C1-C4 Alkyl oder (D)-Phenyl steht b) Umsetzung einer Verbindung der Formel 2 mit R11 COR11 unter Bildung einer Verbindung der Formel 3 worin R11 unabhängig für Wasserstoff oder C1-C4 Alkyl steht, c) Umsetzung einer Verbindung der Formel 3 mit einer Aktivierungsgruppe unter Bildung einer Verbindung der Formel 4
worin R11 unabhängig für Wasserstoff oder C1-C4 Alkyl steht, c) Umsetzung einer Verbindung der Formel 3 mit einer Aktivierungsgruppe unter Bildung einer Verbindung der Formel 4 worin A für eine Aktivierungsgruppe steht, d) Desoxygenierung der Verbindung der Formel 4 durch Hydrierung unter Bildung einer Verbindung der Formel 5
worin A für eine Aktivierungsgruppe steht, d) Desoxygenierung der Verbindung der Formel 4 durch Hydrierung unter Bildung einer Verbindung der Formel 5 e) wahlweise Umsetzung der Verbindung der Formel 5 mit einer anorganischen Base unter Bildung einer Verbindung der Formel 6
e) wahlweise Umsetzung der Verbindung der Formel 5 mit einer anorganischen Base unter Bildung einer Verbindung der Formel 6 worin HA für ein saures und M für ein univalentes Kation steht, f) Auftrennung der Verbindung der Formel 5 oder Formel 6 unter Bildung einer chiralen Verbindung der Formel 7
worin HA für ein saures und M für ein univalentes Kation steht, f) Auftrennung der Verbindung der Formel 5 oder Formel 6 unter Bildung einer chiralen Verbindung der Formel 7 worin M für Wasserstoff steht und Ra' für H oder Ra steht, g) Kupplung der Verbindung der Formel 7 mit einer Verbindung der Formel 8
worin M für Wasserstoff steht und Ra' für H oder Ra steht, g) Kupplung der Verbindung der Formel 7 mit einer Verbindung der Formel 8 unter Bildung einer Verbindung der Formel 9
unter Bildung einer Verbindung der Formel 9 und h) Kupplung der Verbindung der Formel 9 mit einer Verbindung der Formel
und h) Kupplung der Verbindung der Formel 9 mit einer Verbindung der Formel unter Bildung einer Verbindung der Formel I.
unter Bildung einer Verbindung der Formel I.
 Q für folgenden Rest steht
Q für folgenden Rest steht R, R1, R3, R4, p und r wie in Anspruch 1 definiert sind und R11 unabhängig für Wasserstoff oder C1-C8 Alkyl steht, wobei das Verfahren die folgenden Schritte umfasst: a) Umsetzung einer Verbindung der Formel 1
R, R1, R3, R4, p und r wie in Anspruch 1 definiert sind und R11 unabhängig für Wasserstoff oder C1-C8 Alkyl steht, wobei das Verfahren die folgenden Schritte umfasst: a) Umsetzung einer Verbindung der Formel 1 worin X für Halogen steht und R11 unabhängig für Wasserstoff oder C1-C4 Alkyl steht, mit CNCH2CO2Ra, worin Ra für C1-C8 Alkyl oder Benzyl steht unter Bildung einer Verbindung der Formel 2
worin X für Halogen steht und R11 unabhängig für Wasserstoff oder C1-C4 Alkyl steht, mit CNCH2CO2Ra, worin Ra für C1-C8 Alkyl oder Benzyl steht unter Bildung einer Verbindung der Formel 2 b) Schutzgruppenanbringung an der Verbindung der Formel 2 unter Bildung der Verbindung der Formel 3
b) Schutzgruppenanbringung an der Verbindung der Formel 2 unter Bildung der Verbindung der Formel 3 c) Hydrierung der Verbindung der Formel 3 unter Bildung einer Verbindung der Formel 4
c) Hydrierung der Verbindung der Formel 3 unter Bildung einer Verbindung der Formel 4 d) Kupplung der Verbindung der Formel 4, worin Ra' für Wasserstoff oder Ra steht, mit einer Verbindung der Formel 5
d) Kupplung der Verbindung der Formel 4, worin Ra' für Wasserstoff oder Ra steht, mit einer Verbindung der Formel 5 unter Bildung einer Verbindung der Formel 6
unter Bildung einer Verbindung der Formel 6 e) Kupplung der Verbindung der Formel 6 mit einer Verbindung der Formel
e) Kupplung der Verbindung der Formel 6 mit einer Verbindung der Formel unter Bildung einer Verbindung der Formel I.
unter Bildung einer Verbindung der Formel I.