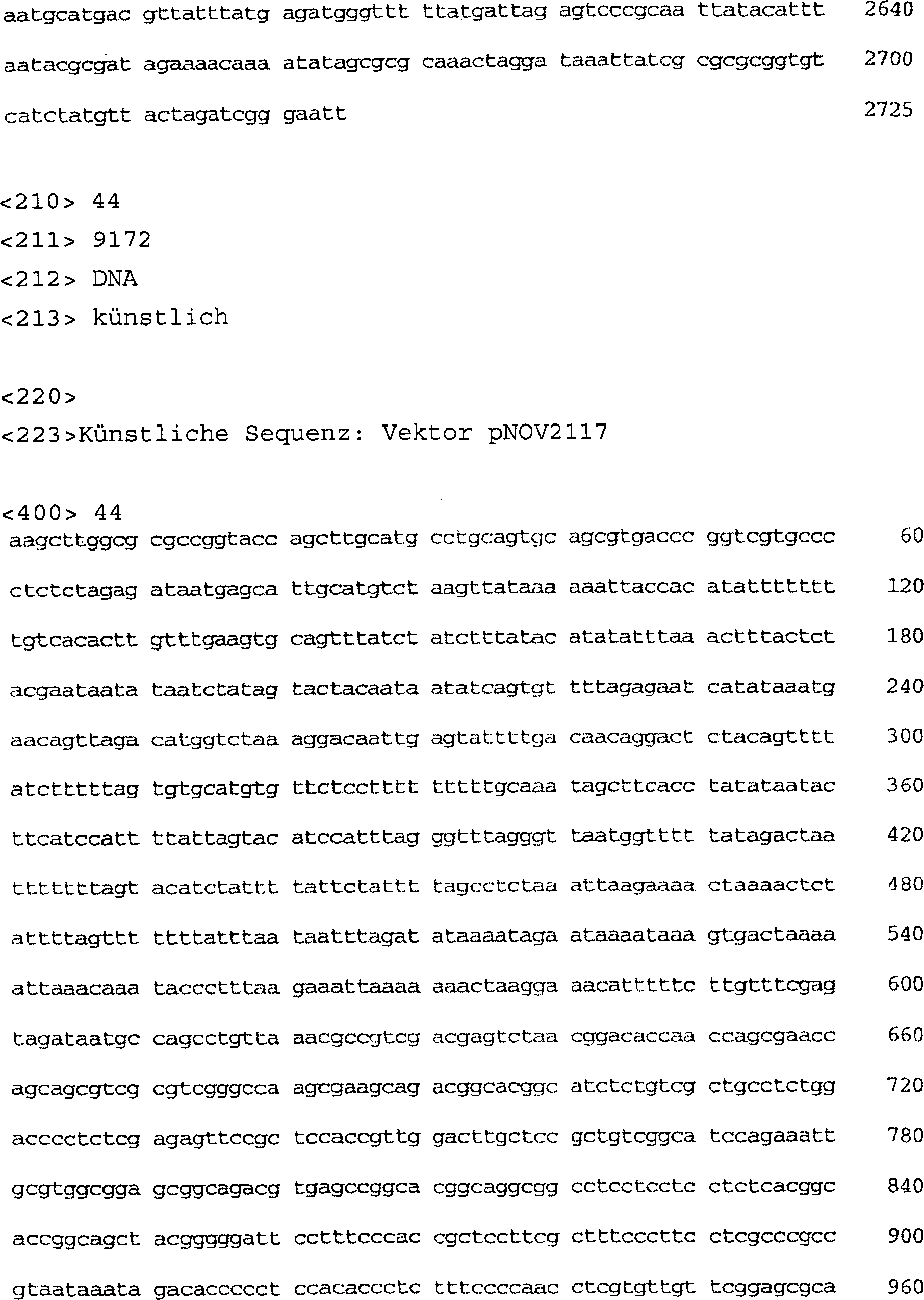
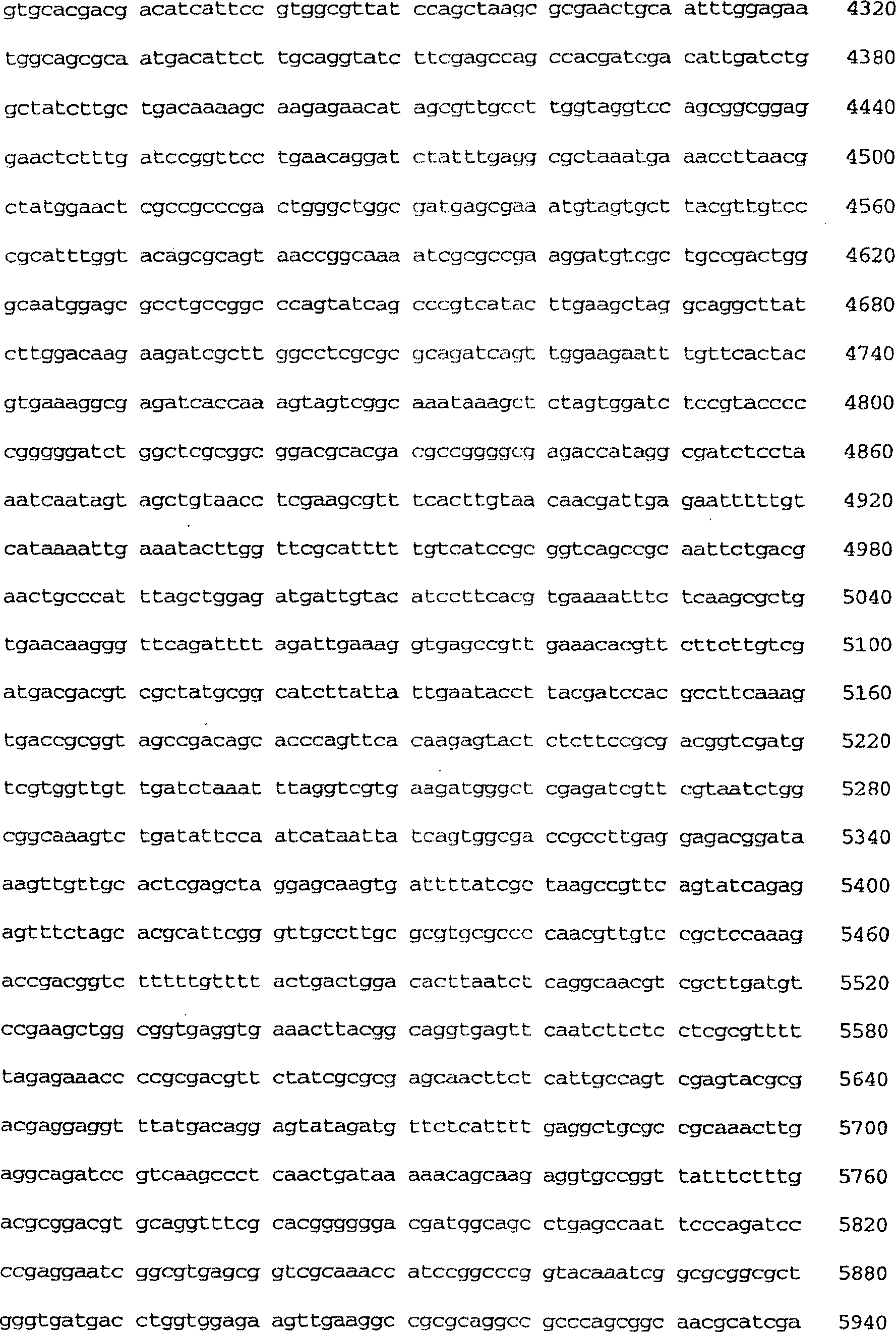
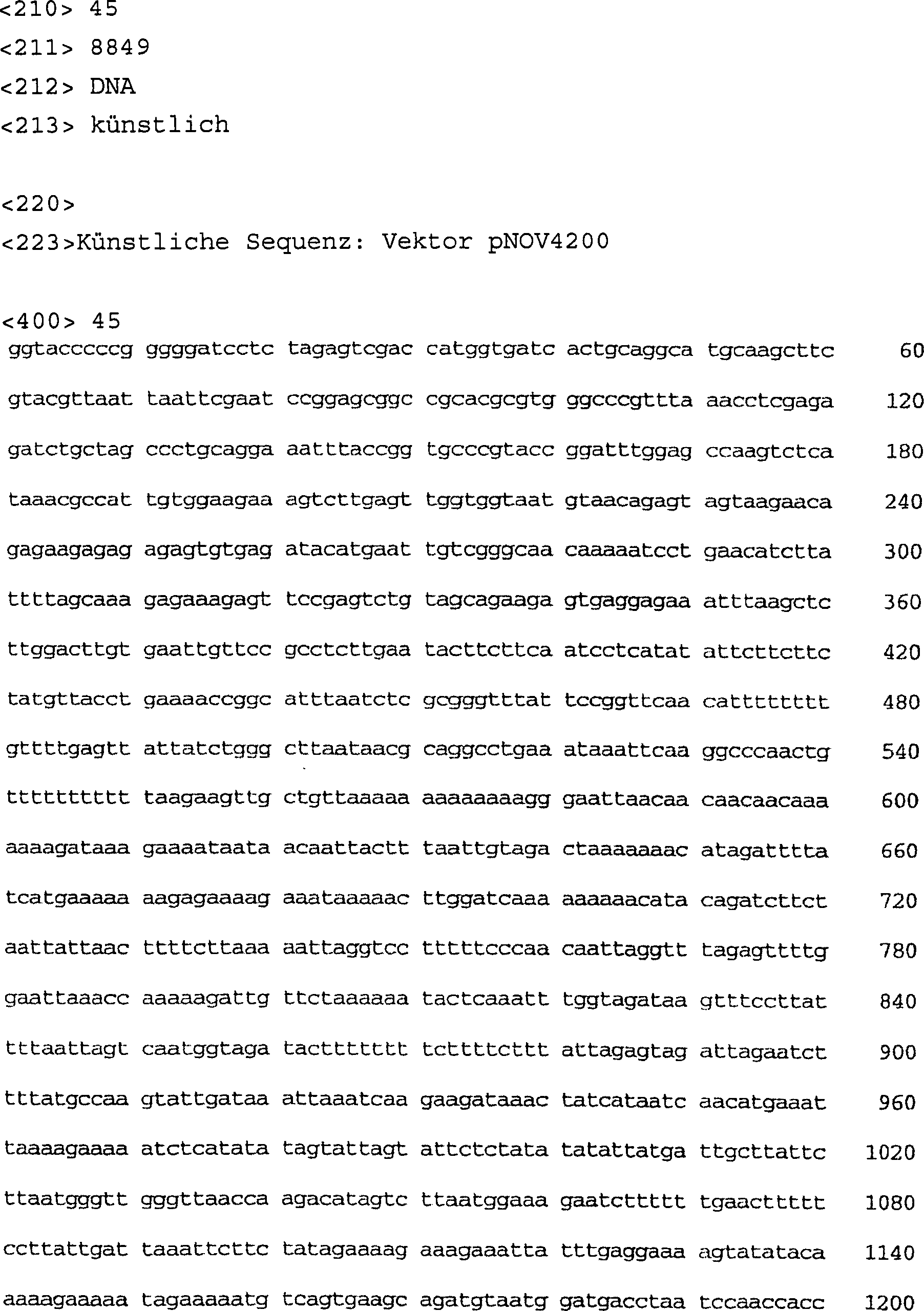
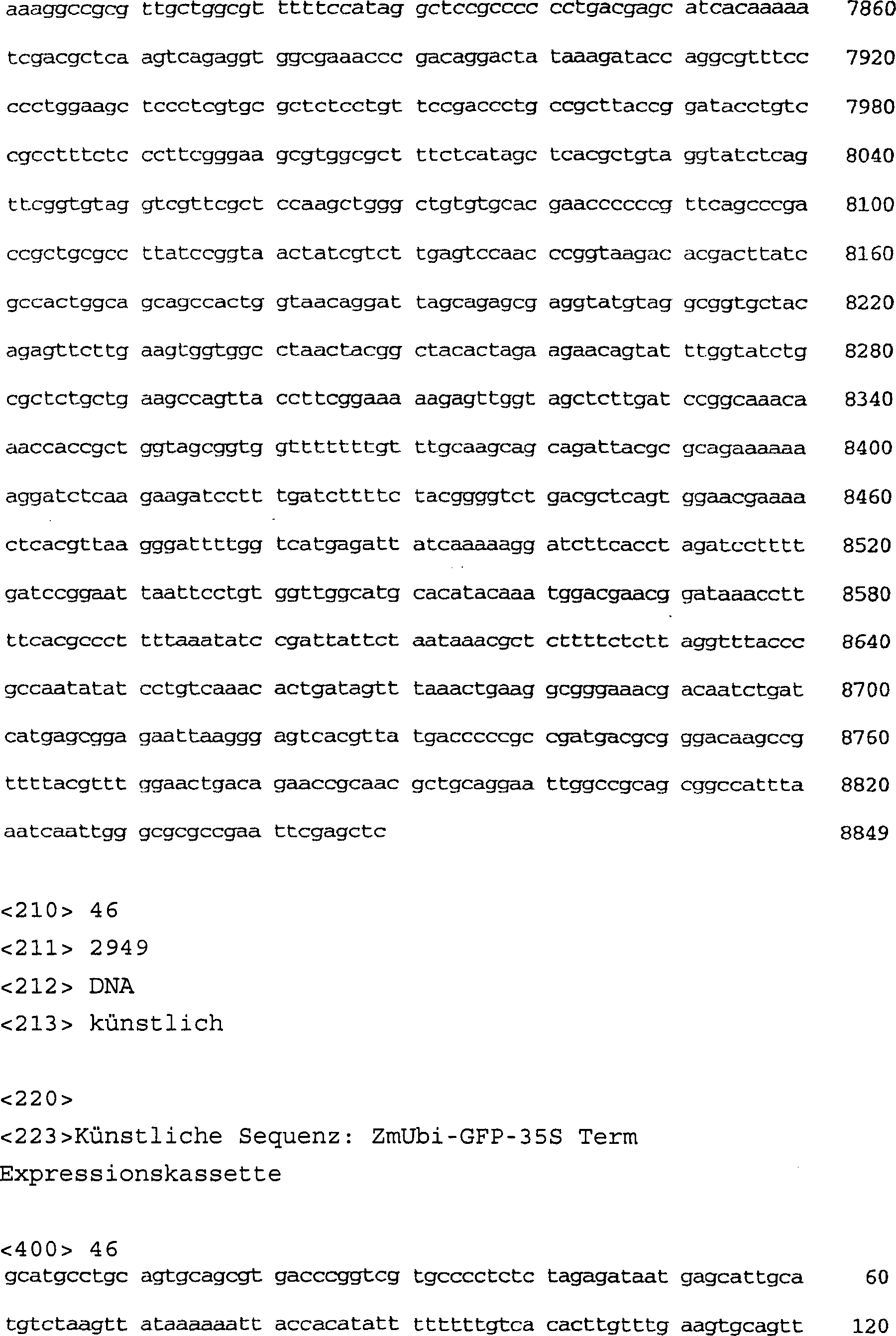
-
Die
vorliegende Erfindung betrifft neue DNA-Sequenzen, die als Promotoren
für die
Transkription damit verbundener Nucleotidsequenzen in Pflanzen dienen.
Genauer betrifft diese Erfindung neue Promotoren, die einer damit
verbundenen Nucleotidsequenz von Interesse eine konstitutive Expression
verleihen.
-
Im
Gebiet der Landwirtschaft gibt es einen ständigen Bedarf, Pflanzen nach
Wunsch zu verändern.
Ein Weg, dies zu erzielen, ist die Verwendung moderner gentechnischer
Verfahren. Beispielsweise kann durch Einschleusung eines Gens von
Interesse in eine Pflanze, diese Pflanze spezifisch modifiziert
werden, um einen gewünschten
phänotypischen
Marker zu exprimieren. Hierfür
werden Pflanzen am häufigsten
mit einem heterologen Gen transformiert, welches eine Promotorregion,
eine kodierende Region und einen Beendigungsbereich umfasst. Wenn
ein heterologes Gen für
die Expression in Pflanzen genetisch hergestellt wird, ist die Auswahl
eines Promotors ein kritischer Faktor. Während beabsichtigt werden kann,
bestimmte Gene nur in Reaktion auf bestimmte Stimuli zu exprimieren
oder diese auf spezifische Zellen oder Gewebe zu beschränken, werden
andere Gene wünschenswerterweise
konstitutiv exprimiert, d. h. in der ganzen Pflanze zu allen Zeitpunkten
und in den meisten Geweben und Organen. In der Vergangenheit ist
der 35S-Promotor
des Blumenkohl-Mosaik-Virus (CaMV 35S Promotor) verbreitet für die konstitutive
Expression heterologer Gene in Pflanzen verwendet worden. Es gibt
jedoch Umstände,
unter denen es wünschenswert
ist, alternative Promotoren zu verwenden. Daher ist es ein Hauptziel
der vorliegende Erfindung, solche alternativen Promotoren für die Expression
einer Nucleotidsequenz von Interesse in Pflanzen bereitzustellen.
Die Erfindung stellt auch rekombinante DNA-Moleküle, Expressionsvektoren und
transgene Pflanzen bereit, die die Vektoren der vorliegenden Erfindung
umfassen.
-
Die
vorliegende Erfindung stellt daher bereit: eine DNA-Sequenz, die
in der Lage ist, die Expression einer damit verbundenen Nucleotidsequenz
zu steuern, wobei die DNA-Sequenz aus dem Genom von Cestrum yellow
leaf curling virus (CmYLCV) erhältlich
ist. Bevorzugt ist eine DNA-Sequenz, die aus den CmYLCV-Promotor-Transkript
vollständiger
Länge erhältlich ist
und die Nucleotidsequenz umfasst, die in SEQ ID Nr. 1 wiedergegeben
ist.
-
Insbesondere
sind DNA-Sequenzen bereitgestellt, worin
- – diese
DNA-Sequenz die Nucleotidsequenz umfasst, die in SEQ ID Nr. 2 wiedergegeben
ist
- – diese
DNA-Sequenz die Nucleotidsequenz umfasst, die in SEQ ID Nr. 3 wiedergegeben
ist
- – diese
DNA-Sequenz die Nucleotidsequenz umfasst, die in SEQ ID Nr. 4 wiedergegeben
ist
- – diese
DNA-Sequenz die Nucleotidsequenz umfasst, die in SEQ ID Nr. 5 wiedergegeben
ist
- – diese
DNA-Sequenz die Nucleotidsequenz umfasst, die in SEQ ID Nr. 6 wiedergegeben
ist
- – die
DNA-Sequenz unter stringenten Bedingungen mit irgendeiner der SEQ
ID Nr. 1, SEQ ID Nr. 2, SEQ ID Nr. 3, SEQ ID Nr. 4, SEQ ID Nr. 5
oder SEQ ID Nr. 6 hybridisiert, insbesondere worin die DNA-Sequenz, die
oben erwähnt
worden ist, die Nucleotidsequenz umfasst, die in SEQ ID Nr. 19 oder
SEQ ID Nr. 20 wiedergegeben ist.
-
Die
Erfindung stellt ferner DNA-Sequenzen bereit, die eine konsekutive
Abfolge von mindestens 50 Nucleotiden umfassen, vorzugsweise ungefähr zwischen
400 Basen und ungefähr
650 Basen, mehr bevorzugt zwischen ungefähr 200 Basen und ungefähr 400 Basen
und am meisten bevorzugt von ungefähr 350 Basen Länge der
SEQ ID Nr. 1, worin diese DNA-Sequenzen in der Lage sind, die Expression
einer damit verbundenen Nucleotidsequenz zu steuern.
-
In
einer besonderen Ausführungsform
der Erfindung hat diese konsekutive Abfolge von mindestens 50 Nucleotiden,
vorzugsweise von zwischen ungefähr
400 Basen und ungefähr
650 Basen, mehr bevorzugt von zwischen ungefähr 200 Basen und ungefähr 400 Basen
und am meisten bevorzugt von ungefähr 350 Basen Länge mindestens
75%, vorzugsweise 80%, mehr bevorzugt 90% und am meisten bevorzugt
95% Sequenzidentität
mit einer entsprechenden konsekutiven Abfolge von mindestens 50
Nucleotiden, vorzugsweise zwischen ungefähr 400 Basen und ungefähr 650 Basen,
mehr bevorzugt zwischen ungefähr
200 Basen und ungefähr
400 Basen, und am meisten bevorzugt ungefähr 350 Basen Länge von
SEQ ID Nr. 1 aufweist. Die Erfindung stellt ferner rekombinante
DNA-Moleküle
bereit, die einen Transkriptionspromotorbereich voller Länge umfassen,
welcher aus Cestrum yellow leaf curling virus isoliert wurde. Zusätzlich stellt
die Erfindung rekombinante DNA-Moleküle und DNA-Expressionskassetten
bereit, die eine DNA-Sequenz gemäß der Erfindung umfassen,
die operativ mit einer Nucleotidsequenz von Interesse verbunden
ist. Die Erfindung stellt auch DNA-Expressionsvektoren bereit, die
diese rekombinante DNA bzw. Expressionskassetten umfassen.
-
Insbesondere
werden rekombinante DNA-Moleküle
und DNA-Expressionskassetten bereitgestellt, wobei die Nucleotidsequenz
von Interesse eine kodierende Sequenz umfasst, insbesondere worin
- – die
kodierende Sequenz eine gewünschte
phänotypische
Eigenschaft kodiert
- – die
kodierende Sequenz ein selektierbares oder screenfähiges Markergen
kodiert
- – die
kodierende Sequenz ein Protein kodiert, das eine Antibiotika-Resistenz,
Virusresistenz, Insektenresistenz, Krankheitsresistenz oder Resistenz
gegenüber
anderen Seuchen, Herbizidtoleranz, verbesserten Nährwert,
verbesserte Leistungsfähigkeit
in einem industriellen Verfahren oder veränderte reproduktive Fähigkeiten
kodiert.
- – die
kodierende Sequenz ein Protein kodiert, welches einen positiven
Selektionsvorteil an Zellen verleiht, die mit dieser kodierenden
Sequenz transformiert worden sind
- – die
kodierende Sequenz ein Protein kodiert, welches Zellen einen metabolischen
Vorteil verleiht, die mit dieser kodierenden Sequenz transformiert
worden sind, welcher darin besteht, dass sie in der Lage sind, eine
Verbindung zu metabolisieren, wobei die Verbindung Mannose oder
Xylose oder ein Derivat oder ein Vorläufer davon ist, oder ein Substrat
des Proteins, oder das in der Lage ist, durch Zellen, die mit dieser kodierenden
Sequenz transformiert worden sind, in ein solches Substrat metabolisiert
werden zu können
- – die
kodierende Sequenz ein Enzym kodiert, das aus der Gruppe ausgewählt ist,
die aus Xyloisomerasen, Phosphomannoisomerase, Mannose-6-phosphat-Isomerase,
Mannose-1-phosphat-Isomerase,
Phosphomannomutase, Mannoseepimerase, Mannose- oder Xylosephosphatase,
Mannose-6-phosphatase, Mannose-1-phosphatase oder Mannose- oder
Xylosepermease besteht
- – die
kodierende Sequenz eine Phosophomannoisomerase kodiert
- – die
kodierende Region nicht translatierbar ist
- – die
nicht translatierbare kodierende Region aus einem viralen Gen stammt,
insbesondere aus TSWV, genauer gesagt aus dem TSWV NP-Gen
- – die
kodierende Sequenz kommerziell wertvolle Enzyme oder Metabolite
in der Pflanze kodiert
- – die
kodierende Sequenz in Gegensinn-Richtung vorliegt.
-
Die
Erfindung stellt auch DNA-Expressionsvektoren bereit, die eine DNA-Sequenz
umfassen, oder ein rekombinantes DNA-Molekül, das oben erwähnt worden
ist. In einer besonderen Ausführungsform
der Erfindung ist dieser DNA-Expressionsvektor pNOV2819 oder pNOV2819.
Des Weiteren werden DNA-Expressionsvektoren bereitgestellt, die
eine erste DNA-Sequenz gemäß der Erfindung
umfassen, die operativ mit einer Nucleotidsequenz von Interesse
verbunden ist, und eine zweite DNA-Sequenz gemäß der Erfindung, operativ verbunden
mit einer Nucleotidsequenz von Interesse. In einer spezifischen
Ausführungsform
der Erfindung sind die oben erwähnten
DNA-Expressionsvektoren in der Lage, die Expression eines viralen
Genoms zu verändern.
-
In
einer noch spezifischeren Ausführungsform
umfasst der oben erwähnte
DNA-Expressionsvektor eine erste DNA-Sequenz, die in der Lage ist,
ein Sinn-RNA-Fragment dieses viralen Genoms oder ein Teil davon
zu exprimieren, und eine zweite DNA-Sequenz, die in der Lage ist,
in einer Zelle ein Gegensinn-DNA-Fragment dieses viralen Genoms
oder einen Teil davon zu exprimieren, worin dieses Sinn-RNA-Fragment
und das Gegensinn-RNA-Fragment in der Lage sind, eine doppelsträngige RNA
zu bilden.
-
Es
werden Expressionvektoren, die oben erwähnt worden sind, bereitgestellt,
worin
- – das
Virus ausgewählt
ist aus der Gruppe, die aus Tospoviren, Potyviren, Potexviren, Tobamoviren,
Luteoviren, Cucumoviren, Bromoviren, Closteoviren, Tombusviren und
Furoviren besteht,
- – die
DNA-Sequenzen eine Nucleotidsequenz umfassen, die von einem viralen
Hüllproteingen,
einem viralen Nucleocapsidproteingen, einem viralen Replicasegen
oder einem viralen Bewegungsproteingen oder Teilen davon stammt,
- – die
DNA-Sequenz aus dem Tomatenbronzefleckenvirus (TSWV) stammt,
- – die
DNA von einem Nucleocapsid-Proteingen stammt.
-
Die
Erfindung stellt ferner bereit Wirtszellen, die stabil mit einer
DNA-Sequenz, einem rekombinanten DNA-Molekül oder einem DNA-Expressionsvektor
gemäß der Erfindung
transformiert worden sind. Insbesondere, worin
- – die Wirtszelle
ein Bakterium ist,
- – die
Wirtszelle eine Pflanzenzelle ist,
- – die
Wirtszelle eine Pflanzenzelle ist, die ausgewählt ist aus der Gruppe bestehend
aus Zellen von Reis, Mais, Weizen, Gerste, Roggen, Süßkartoffeln,
Süßmais, Bohnen,
Erbsen, Chicoree, Salat, Kohl, Blumenkohl, Brokkoli, Rüben, Radieschen,
Spinat, Spargel, Zwiebel, Knoblauch, Paprika, Sellerie, Squash-Kürbis, Hanf,
Zucchini, Äpfel,
Birnen, Quitten, Melonen, Pflaumen, Kirschen, Pfirsischen, Nektarinen,
Aprikosen, Erdbeeren, Trauben, Himbeeren, Brombeeren, Ananas, Avocado,
Papaya, Mango, Bananen, Sojabohnen, Tomaten, Hirse, Zuckerrüben, Zuckerrohr,
Sonnenblumen, Raps, Klee, Tabak, Karotten, Baumwolle, Alfalfa, Kartoffeln,
Auberginen, Gurken, Kresse (Arabidopsis thaliana) und Gehölzpflanzen,
wie beispielsweise Koniferen und Laubbäumen, aber insbesonder Reis,
Mais, Weizen, Gerste, Kohl, Blumenkohl, Paprika, Kürbis, Melone,
Sojabohnen, Tomaten, Zuckerrüben,
Sonnenblumen oder Baumwolle, Reis, Mais, Weizen, Sorgum bicolor,
Knaulgras, Zuckerrüben
und Sojabohnenzellen
- – die
Wirtszelle ist eine Pflanzenzelle einer dicotyledonen Pflanze
- – die
Wirtszelle ist eine Pflanzenzelle einer dicotyledonen Pflanze ausgewählt aus
der Gruppe bestehend aus Sojabohne, Baumwolle, Tabak, Zuckerrübe und Ölsaatraps
- – die
Wirtszelle ist eine Pflanze ausgewählt aus monocotyledonen Pflanzen
- – die
Wirtszelle ist eine Pflanze ausgewählt aus einer Monocotyledonenpflanze
ausgewählt
aus der Gruppe bestehend aus Mais, Weizen, Hirse, Roggen, Hafer,
Rasengras, Reis und Gerste.
-
Zusätzlich werden
Pflanzen und deren Nachkommen, einschließlich Samen, bereitgestellt,
die stabil mit einer DNA-Sequenz, einem rekombinanten DNA-Molekül oder einem
DNA-Expressionsvektor gemäß der Erfindung
transformiert sind. Insbesondere, worin die Pflanze ausgewählt ist
aus der Gruppe bestehend aus Reis, Mais, Weizen, Gerste, Roggen,
Süßkartoffel,
Süßmais, Bohnen,
Erbsen, Chicoree-Salat, Kohl, Blumenkohl, Brokkoli, Rüben, Radieschen,
Spinat, Spargel, Zwiebeln, Knoblauch, Paprika, Sellerie, Kürbis, Hanf,
Zucchini, Apfel, Birne, Quitte, Melone, Pflaume, Kirsche, Pfirsisch,
Nektarine, Aprikose, Erdbeeren, Trauben, Himbeeren, Brombeeren,
Ananas, Avocado, Papaya, Mango, Bananen, Sojabohnen, Tomaten, Hirse,
Zuckerrohr, Zuckerrüben,
Sonnenblumen, Raps, Klee, Tabak, Karotten, Baumwolle, Alfalfa, Kartoffeln,
Auberginen, Gurken, Arabidopsis thaliana und Gehölzpflanzen wie Koniferen und
Laubbäumen,
aber insbesondere Reis, Mais, Weizen, Gerste, Kohl, Blumenkohl,
Paprika, Kürbis,
Melonen, Sojabohnen, Tomaten, Zuckerrüben, Sonnenblumen oder Baumwolle,
Reis, Mais, Weizen, Sorghum bicolor, Knaulgras, Zuckerrüben und
Sojabohnen.
-
Die
vorliegende Erfindung offenbart ferner
- – die Verwendung
der DNA-Sequenz gemäß der Erfindung,
um eine Nucleotidsequenz von Interesse zu exprimieren.
- – ein
Verfahren zum Herstellen einer DNA-Sequenz gemäß der Erfindung, worin die
DNA durch eine Polymerase-Kettenreaktion hergestellt wird, worin
mindestens ein Oligonucleotid, das verwendet wird, eine Sequenz
aus Nucleotiden umfasst, welche eine konsekutive Abfolge von 15,
18, 20, 22, 24 oder mehr Basenpaaren der SEQ ID Nr. 1 darstellen.
-
Um
eine klares und konsistentes Verständnis der Beschreibung und
der Ansprüche
sicherzustellen, werden die folgenden Definitionen bereitgestellt:
CmYLCV:
Cestrum yellow leaf curling virus.
-
DNA-Shuffling:
DNA-Shuffling ist ein Verfahren, um schnell, leicht und wirksam
Rearrangements, vorzugsweise zufallsgemäß in einem DNA-Molekül einzuführen, oder
um Austausche von DNA-Sequenzen zwischen zwei oder mehr DNA-Molekülen, vorzugsweise
zufallsgemäß zu erzeugen.
Das DNA-Molekül,
das aus DNA-Shuffling resultiert, ist ein geshuffeltes DNA-Molekül, das ein
nicht natürlich
auftretendes DNA-Molekül ist,
welches von mindestens einem Matrizen-DNA-Molekül stammt.
-
Expression:
betrifft die Transkription und/oder Translation eines endogenen
Gens oder eines Transgens in Pflanzen. Im Fall von Gegensinn-Konstrukten
kann Expression beispielsweise allein die Transkription der Gegensinn-DNA
betreffen.
-
„Funktional äquivalente
Sequenz" bezieht
sich auf eine DNA-Sequenz, die eine Promotoraktivität besitzt,
die im Wesentlich ähnlich
der des CmYLCV-Transkriptpromotors vollständiger Länge oder Teilen davon ist und
welche unter stringenten Hybridisierungsbedingungen mit diesen Promotorsequenzen
hybridisiert.
-
Gen:
betrifft eine kodierende Sequenz und assoziierte regulatorische
Sequenz, wobei die kodierende Sequenz in RNA umgeschrieben wird,
wie beispielsweise in mRNA, rRNA, rRNA, snRNA, Sinn-RNA oder Gegensinn-RNA.
Beispiele für
regulatorische Sequenzen sind Promotorsequenzen, 5'- und 3'-nichttranslatierte Sequenzen
und Beendigungssequenzen. Weitere Elemente, die vorhanden sein können, sind
beispielsweise Introns.
-
Gen
von Interesse: bezieht sich auf jedes Gen, welches, wenn es in eine
Pflanze überführt wird,
der Pflanze eine gewünschte
Charakteristik verleiht, wie beispielsweise eine Antibiotika-Resistenz,
Virusresistenz, Insektenresistenz, Krankheitsresistenz oder Resistenz
gegenüber
anderen Seuchen, Herbizidtoleranz, verbesserte Nährwert-Gehalte, verbesserte
Leistungsfähigkeit
in einem industriellen Verfahren oder veränderte Reproduktionsfähigkeit.
Das „Gen
von Interesse" kann
auch eines sein, das in Pflanzen zur Herstellung kommerziell wertvoller
Enzyme oder Metabolite in die Pflanze eingeschleust worden ist.
-
„Heterolog" bedeutet, so wie
es hier verwendet wird, von unterschiedlichem natürlichem
oder von synthetischem Ursprung. Beispielsweise, wenn eine Wirtszelle
mit einer Nucleinsäuresequenz
transformiert ist, die in einer nicht transformierten Wirtszelle
nicht auftritt, wird von dieser Nucleinsäuresequenz, bezogen auf die
Wirtszelle als heterolog gesprochen. Die transformierende Nucleinsäure kann
einen heterologen Promotor, eine heterologe kodierend Sequenz, oder
eine heterologe Beendigungssequenz umfassen. Alternativ dazu kann
die transformierende Nucleinsäure
vollständig
heterolog sein oder sie kann irgendeine mögliche Kombination aus heterologen
und endogenen Nucleinsäuresequenzen
umfassen.
-
Leader-Bereich:
ein Bereich in einem Gen zwischen der Transkriptions-Startstelle
und der Translations-Startstelle.
-
Markergen:
bezieht sich auf ein Gen, welches eine selektierbare oder screenfähige Markierung
kodiert.
-
Operativ
gebunden an/verbunden mit: eine regulatorische DNA wird als „operativ
gebunden an" oder „verbunden
mit" einer DNA-Sequenz
beschrieben, die für
eine RNA oder ein Protein kodiert, wenn die zwei Sequenzen so angeordnet
sind, dass die regulatorische DNA-Sequenz die Expression der kodierenden DNA-Sequenz
beeinflusst.
-
Pflanze:
bezieht sich auf irgendeine Pflanze, insbesondere auf Samenpflanzen.
-
Pflanzenzelle:
strukturelle und physiologische Einheit der Pflanze, umfassend einen
Protoplasten und eine Zellwand. Die Pflanzenzelle kann in Form einer
isolierten Einzelzelle oder einer kultivierten Zelle vorliegen,
oder als Teil einer höheren
organisierten Einheit, wie beispielsweise einem Pflanzengewebe oder
einem Pflanzenorgan.
-
Pflanzenmaterial:
bezieht sich auf Blätter,
Stämme,
Wurzeln, Blüten
oder Blütenteile,
Früchte,
Pollen, Pollenröhrchen,
Eier, Embryosäcke,
Eierzellen, Zygoten, Embryonen, Samen, Stecklinge, Zellen oder Gewebekulturen
oder irgendeinen anderen Teil oder Produkt einer Pflanze.
-
Promotor:
bezieht sich auf eine DNA-Sequenz, die die Transkription einer damit
verbundenen DNA-Sequenz initiiert. Die Promotorregion kann auch
Elemente einschließen,
die als Regulatoren der Genexpression dienen, wie beispielsweise
Aktivatoren, Enhancer und/oder Repressoren und kann alles oder einen Teil
der 5' nicht-translatierten
Leadersequenz einschließen.
-
Rekombinantes
DNA-Molekül:
eine Kombination aus DNA-Sequenzen, die unter Verwendung der rekombinanten
DNA-Technologie miteinander verbunden sind.
-
Rekombinante
DNA-Technologie: Verfahren, die verwendet werden, um DNA-Sequenzen,
wie beispielsweise in Sambrook et al., 1989, Cold Spring Harbor,
NY: Cold Spring Harbor Laboratory Press, miteinander zu verbinden.
-
Screenfähiges Markergen:
bezieht sich auf ein Gen, dessen Expression einer transformierten
Zelle keinen Selektionsvorteil verleiht, aber dessen Expression
die transformierte Zelle von nicht transformierten Zellen phänotypisch
unterscheidet.
-
Selektierbares
Markergen: bezieht sich auf ein Gen, dessen Expression in einer
Pflanzenzelle der Zelle einen Selektionsvorteil verleiht. Der Selektionsvorteil,
der von den Zellen besessen wird, die mit dem selektierbaren Markergen
transformiert sind, kann auf deren Fähigkeit zurückzuführen sein, in Gegenwart eines
negativen Selektionsmittels zu wachsen, wie beispielsweise einem
Antibioticum oder einem Herbizid, verglichen mit dem Wachstum nicht
transformierter Zellen. Der Selektionsvorteil, den transformierte
Zellen besitzen, verglichen mit den nicht transformierten Zellen,
kann auch auf ihrer verstärkten
oder neuen Fähigkeit
beruhen, eine zusätzliche
Verbindung zu verwenden, wie beispielsweise einen Nährstoff,
einen Wachstumsfaktor oder eine Energiequelle. Ein selektierbares Markergen
verleiht einem Gen oder einer Kombination von Genen, deren Expression
in einer Pflanzenzelle in Gegenwart des Selektionsmittels, verglichen
mit der Abwesenheit des Selektionsmittels, der transformierten Pflanzenzelle
eine positive Wirkung verleiht und der nicht transformierten Pflanzenzelle
negative Wirkung, beispielsweise bezüglich des Wachstums und verleiht
so der transformierten Pflanzenzelle einen positiven Selektionsvorteil.
-
Sequenzidentität: der Prozentsatz
der Sequenzidentität
wird unter Verwendung von Computerprogrammen bestimmt, die auf dynamischen
Programmalgorithmen beruhen. Computerprogramme, die im Schutzbereich
der vorliegenden Erfindung bevorzugt sind, schließen das
BLAST (Basic Local Alignment Search Tool)-Rechercheprogramm ein,
welches entworfen worden ist, um alle erhältlichen Sequenzdatenbanken
zu nutzen, unabhängig
davon, ob der Suchbegriff ein Protein oder eine DNA betrifft. Die
Version BLAST 2.0 (Gaped BLAST) dieses Recherchewerkzeugs ist über das
Internet öffentlich
zugänglich
gemacht worden (gegenwärtig
http:://www.ncbi.nlm.nih.gov/BLAST/). Es verwendet einen heuristischen
Algorithmus, welcher lokal sucht, im Gegensatz zu globalen Anlagerungen,
und ist daher in der Lage, Verwandtschaften zwischen Sequenzen nachzuweisen,
die nur isolierte Bereiche teilen. Die in einer BLAST-Recherche
zugeordneten Werte besitzen eine gut definierte statistische Interpretierbarkeit.
Diese Programme werden vorzugsweise mit optionalen Parametern laufen
gelassen, die den vorgegebenen Werten hinzugefügt werden.
-
Transformation:
bezieht sich auf die Einschleusung einer Nucleinsäure in eine
Zelle. Insbesondere betrifft es die stabile Integration eines DNA-Moleküls in das
Genom eines Organismus von Interesse.
-
TSWV: Tomatenbronzefleckenvirus
-
Die
vorliegende Erfindung betrifft DNA-Sequenzen, die aus dem Genom
des Cestrum yellow leaf curling virus (CmYLCV) erhältlich sind.
Bevorzugt sind DNA-Sequenzen, die aus dem CmYLCV Transkriptpromotor
vollständiger
Länge gewinnbar
sind, welche in der Lage sind, die Expression einer damit verbundenen
Nucleinsäure
von Interesse anzutreiben. DNA-Sequenzen, die funktionale und/oder
strukturelle Äquivalente
davon umfassen, sind ebenfalls durch die Erfindung eingeschlossen.
Die vorliegende Erfindung betrifft daher DNA-Sequenzen, die als
Promotoren der Transkription von damit verbundenen Nucleotidsequenzen
dienen. Die Promotorregion kann ebenfalls Elemente einschließen, die
als Regulatoren der Genexpression dienen, wie beispielsweise Aktivatoren,
Enhancer und/oder Repressoren und kann die 5' nicht-translatierte Leadersequenz der
transkribierten mRNA einschließen.
-
In
einer bevorzugten Ausführungsform
der Erfindung verleiht diese DNA-Sequenz einer damit verbundenen
Nucleotidsequenz eine konstitutive Expression. Die konstitutive
Expression bedeutet, dass die Nucleotidsequenz von Interesse zu
allen Zeitpunkten und in den meisten Geweben und Organen exprimiert
wird. Wenn sie mit GUS- oder einem CAT-Reportergen in transienten
Expressionsassays getestet wird, verleiht die erfindungsgemäße DNA-Sequenz
ein hohes Niveau der Expression des GUS- oder CAT-Reportergens.
Daher ist die DNA-Sequenz gemäß der Erfindung
nützlich
für die
Expression einer damit verbundenen Nucleotidsequenz von Interesse
auf hohem Niveau, welche vorzugsweise eine kodierende Sequenz ist.
Es ist dem Fachmann bekannt, dass die damit verbundene kodierende
Sequenz von Interesse in Sinn- oder Gegensinnrichtung exprimiert
werden kann. Des Weiteren kann die kodierende Sequenz von Interesse
heterologen oder homologen Ursprungs sein, bezogen auf die zu transformierende
Pflanze. Im Fall einer homologen kodierenden Sequenz ist die DNA-Sequenz
gemäß der Erfindung
nützlich
für die
ectopische Expression dieser Sequenz. In einer besonderen Ausführungsform
der Erfindung unterdrückt
die kodierende Sequenz von Interesse unter Kontrolle einer DNA-Sequenz
gemäß der Erfindung
dessen eigene Expression und die der Originalkopie des Gens durch
einen Prozess, der als Co-Suppression bezeichnet wird.
-
Die
Promotoren der vorliegenden Erfindung können aus genomischer DNA des
Cestrum yellow leaf curling virus (CmYLCV) gewonnen werden, welches
beispielsweise aus infizierten Chilenischen Hammerstrauch-Pflanzen
isoliert werden kann. CmYLCV infizierte Chilenischer Hammerstrauch-Pflanzen werden durch
ihre mosaikartigen Blätter
und eine Missbildung in Form von zerknitterten Blättern identifiziert.
Diese DNA wird dann sequenziert und analysiert. Ein Sequenzvergleich
mit Sequenzen in der Datenbank zeigt, dass die genomische Organisation
von CmYLCV mit der eines Mitglieds der Caulimovirus-Familie verwandt
ist. CmYLCV enthält
8 offene Leseraster. Ein Sequenzvergleich der CmYLCV-Genprodukte zeigt,
dass das Gen I-Produkt in intrazelluläre Bewegungen involviert ist:
Gen A kodiert ein kleines Protein unbekannter Funktion, Gen B kodiert
den Aphid-Transmissionsfaktor;
Gen C kodiert ein Virion-assoziiertes Protein; das Gen IV ist das Haupt-Capsidprotein;
Gen V kodiert eine Reverse Transkriptase und das Gen VI aktiviert
in trans die Translation viraler Gene des Transkripts vollständiger Länge. Von
besonderem Interesse ist der sogenannte CmYLCV Transkript-Promotor
voller Länge.
-
Es
ist dem Fachmann bekannt, dass verschiedene Bereiche des CmYLCV-Transkriptpromotors
voller Länge
erfindungsgemäß verwendet
werden können.
Eine besondere Ausführungsform
der Erfindung ist der CmYLCV-Transkriptpromotorbereich vollständiger Länge, welcher
in SEQ ID Nr. 1 gezeigt ist, welcher als CmpD-Promotor bezeichnet
wird. Diese Sequenz enthält
350 bp des CmYLCV-Transkriptpromotors vollständiger Länge und 320 bp des CmYLCV-Transkript
5' nicht-translatierten Leadersequenz
vollständiger
Länge. Diese
Sequenz wird durch Sequenzieren der genomischen CmYLCV-DNA gewonnen.
Der Promotor und die Leadersequenzen werden jeweils durch einen
Vergleich der Sequenzen in der Datenbank identifiziert. Eine Variante
der SEQ ID Nr. 1, die als CmpE bezeichnet wird, in der die CmYLCV-Transkript
5' nicht-translatierte Leadersequenz
318 bp anstelle 320 bp lang ist, ist in SEQ ID Nr. 19 gezeigt.
-
Eine
bevorzugte Ausführungsform
der Erfindung ist die DNA-Sequenz, die in SEQ ID Nr. 2 wiedergegeben
ist, die als CmpC-Promotor bezeichnet wird. Der CmpC-Promotor ist
ein Fragment der Sequenz, welches in SEQ ID Nr. 1 gezeigt wird,
und er enthält
346 bp des CmYLCV-Transkriptpromotors vollständiger Länge, was der Base 5 bis 350
der SEQ ID Nr. 1 entspricht. Diese DNA-Sequenz ist durch PCR mit
genomischer DNA von Cestrum yellow leaf curling virus oder von Cestrum
yellow leaf curling virus-infizierten Chilenischen Hammerstrauch-Pflanzen
unter Verwendung des Vorwärtsprimers
Cmp1 (SEQ ID Nr. 13) und des Umkehrprimers CmpC2 (SEQ ID Nr. 14)
erhältlich.
Die vermutete TATA-Box des CmpC-Promotors reicht von der Base 308
bis zur Base 315 der SEQ ID Nr. 2. Der CmpC-Promotor enthält mindestens
3 vermutete Enhancer-Bereiche.
Enhancer-Bereich 1 mit der Nucleotidsequenz CAAT reicht von Base
232 bis zur Base 235 der SEQ ID Nr. 2 und Verstärkerbereich 2 ebenfalls mit
der Nucleotidsequenz CAAT reicht von der Base 243 bis Base 246 der
SEQ ID Nr. 2. Enhancer-Bereich 3 reicht von der Base 246 bis Base
253 der SEQ ID Nr. 2 und hat die Nucleotidsequenz TGACGTAA.
-
Eine
andere bevorzugte Ausführungsform
der Erfindung ist die DNA, die in SEQ ID Nr. 3 gezeigt ist, welche
als CmpS-Promotor
bezeichnet wird. Der CmpS-Promotor ist ebenfalls ein Fragment der
Sequenz, welche in SEQ ID Nr. 1 gezeigt wird und enthält 346 bp
der SEQ ID Nr. 2 plus 54 bp des Leaderbereichs des CmYLCV-Transkriptpromotors
voller Länge.
Der Leaderbereich ist eine Nucleotidsequenz, die der kodierenden
Region vorangeht, welche transkribiert wird, aber nicht in Protein
translatiert wird. Es ist dem Fachmann bekannt, dass der Leaderbereich
regulatorische Elemente mit wichtigen Funktionen der Genexpression
enthalten kann. Der CmpS-Promotor
ist durch PCR mit genomischer DNA des Cestrum yellow leaf curling
virus oder aus Cestrum yellow leaf curling virus-infizierten Chilenischen
Hammerstrauch-Pflanzen unter Verwendung des Vorwärtsprimers Cmp1 (SEQ ID Nr.
13) und des Umkehrprimers CmpS2 (SEQ ID Nr. 15) erhältlich. Der
CmpS-Promotor enthält mindestens
2 vermutete Enhancer-Bereiche in den Leaderbereich. Enhancer-Bereich
4 (GAGAGA) liegt zwischen Base 354 und Base 359 der SEQ ID Nr. 3
und Enhancer-Bereich 5 (GAGAGAGA) liegt zwischen Base 368 und Base
375 der SEQ ID Nr. 3.
-
Noch
eine weitere bevorzugte Ausführungsform
der Erfindung sind die Sequenzen, die in SEQ ID Nr. 4 wiedergegeben
sind, welche als CmpL-Promotor bezeichnet wird. Wie die oben genannten
Promotoren ist der CmpL-Promotor ein Fragment der Sequenz, die in
SEQ ID Nr. 1 gezeigt ist, und enthält die 346 bp der SEQ ID Nr.
2 plus 288 bp des CmYLCV-Transkript 5' nicht-translatierten Leadersequenz
vollständiger
Länge.
Der CmpL-Promotor ist durch PCR mit genomischer DNA von Cestrum
yellow leaf curling virus oder aus Cestrum yellow leaf curling virus-infizierten
Chilenischen Hammerstrauch-Pflanzen unter Verwendung des Vorwärtsprimers
Cmp1 (SEQ ID Nr. 13) und des Umkehrprimers CmpL2 (SEQ ID Nr. 16)
erhältlich.
Eine Variante von SEQ ID Nr. 4, welche als CmpF bezeichnet wird,
in dem die CmYLDV-Transkript 5' nicht-translatierte
Leadersequenz vollständiger
Länge 286
pb anstelle von 280 bp Länge
beträgt,
ist in SEQ ID Nr. 20 gezeigt.
-
Es
ist dem Fachmann bekannt, dass die Nucleotidsequenzen, die in SEQ
ID Nr. 1, 2, 3, 4, 19 und 20 gezeigt sind, an ihren 5'- und 3'-Enden mit homologen
oder heterologen Nucleotidsequenzen verlängert werden können. Beispielsweise
kann das 5'-Ende
der SEQ ID Nr. 1, 2, 3, 4, 19 und 20 durch alle oder einen Teil der
Nucleotide verlängert
werden, die in SEQ ID Nr. 6 gezeigt sind. SEQ ID Nr. 6 wird natürlich stromaufwärts der
SEQ ID Nr. 2, 3, 4 und 20 gefunden und enthält einen Teil des ORF VI von
CmYLCV (Base 1 bis Base 100 der SEQ ID Nr. 6), wie auch des CmYLCV-Transkriptpromotors
vollständiger
Länge (Base
101 bis Base 104 der SEQ ID Nr. 6). Gleichermaßen können die 3'-Enden der SEQ ID Nr. 2, 3, 4 und 20
durch alle oder einen Teil der Nucleotide verlängert werden, die SEQ ID Nr.
5 gezeigt sind, welche die Nucleotidsequenz darstellen, die natürlicherweise
am 3'-Ende der SEQ
ID Nr. 4 und 20 gefunden wird, und einen Teil der CmYLCV-Transkript-Leadersequenz
vollständiger
Länge umfasst.
-
Wie
oben beschrieben worden ist, können
die erfindungsgemäßen DNA-Sequenzen
beispielsweise durch PCR mit genomischer DNA aus Cestrum yellow
leaf curling virus oder aus Cestrum yellow leaf curling virus-infizierten
Chilenischen Hammerstrauch-Pflanzen
gewonnen werden. Alternativ dazu können die erfindungsgemäßen DNA-Sequenzen
aus irgendeinem anderen Virus der Caulimovirus-Familie gewonnen
werden, welche Homologe der DNA-Sequenz der Erfindung umfassen,
indem Sequenz-spezifischer
Primer verwendet werden. Es ist dem Fachmann klar, dass auf Grundlage
der Nucleotidsequenzen, die in SEQ ID Nr. 1 bis SEQ ID Nr. 6 gezeigt
sind, und in SEQ ID Nr. 19 und 20 jede Primerkombination von Interesse
ausgewählt werden
kann, um mittels PCR DNA-Fragmente verschiedener Längen zu
amplifizieren, die erfindungsgemäß verwendet
werden können.
Die Erfindung schließt
Fragmente ein, die von den CmYLCV-Transkriptpromotoren vollstäniger Länge stammen,
welche erfindungsgemäß arbeiten,
d. h. sie sind in der Lage, eine damit verbundene Nucleotidsequenz
einer Expression zu ermöglichen.
-
Dies
kann durch Herstellen von Promotorfragmenten getestet werden, indem
sie mit einem selektierbaren oder screenfähigen Markergen fusioniert
werden und indem die fusionierten Konstrukte auf die Bewahrung der
Promotoraktivität
hin untersucht werden. Solche Untersuchungen liegen innerhalb des
Allgemeinwissens eines Fachmanns auf dem Gebiet. Bevorzugte DNA-Fragmente
der Erfindung haben mindestens ungefähr 50 Basen, vorzugsweise ungefähr 400 Basen
bis ungefähr
650 Basen, mehr bevorzugt ungefähr
200 Basen und ungefähr
400 Basen und am meisten bevorzugt ungefähr 350 Basen Länge.
-
Es
ist dem Fachmann auf dem Gebiet auch bekannt, dass Mutationen, Insertionen,
Deletionen und/oder Substitutionen einer oder mehrerer Nucleotide
in die DNA-Sequenzen der SEQ ID Nr. 1, 2, 3, 4, 5 und 6 oder längeren oder
kürzeren
Fragmenten, die aus dieser Sequenzinformation stammen, indem bekannte Verfahren
in dem Gebiet verwendet werden, eingeschleust werden können. Zusätzlich kann
eine nicht modifizierte oder modifizierte Nucleotidsequenz der vorliegenden
Erfindung durch Shuffling der erfindungsgemäßen Sequenz variiert werden.
Um einen Funktionstest einer von varianten DNA-Sequenzen gemäß der Erfindung durchzuführen, wird
die Sequenz von Interesse operativ mit einem selektierbaren oder
screenfähigen
Markergen verbunden, und die Expression des Markergens wird in transienten
Expressionstests mit Protoplasten getestet, oder in stabil transformierten
Pflanzen. Es ist dem Fachmann bekannt, dass DNA-Sequenzen in der Lage
sind, die Expression einer damit verbundenen Nucleotidsequenz anzutreiben,
welche in modularer Form aufgebaut ist. Demgemäß können die Expressionsniveaus
von kürzeren
DNA-Fragmenten verschieden sein als die vom längsten Fragment, und sie können untereinander
auch verschieden sein. Beispielsweise wird die Deletion eines herunterregulierenden
stromaufwärts
gelegenen Elements zu einem Anstieg der Expressionsmengen der damit
verbundenen Nucleotidsequenz führen,
während
die Deletion eines heraufregulierenden Elements die Expressionsniveaus
der damit verbundenen Nucleotidsequenz absenken wird. Es ist dem
Fachmann auch bekannt, dass die Deletion von entwicklungsspezifischen
oder gewebespezifischen Elementen zu zeitlich oder räumlich veränderten
Expressionsprofilen der damit verbundenen Nucleotidsequenz führen. Von der
vorliegenden Erfindung sind auch funktionale Äquivalente der Promotoren der
vorliegenden Erfindung umfasst, d. h. Nucleotidsequenzen, die unter
stringenten Bedingungen mit irgendeiner der SEQ ID Nr. 1, SEQ ID Nr.
2, SEQ ID Nr. 3, SEQ ID Nr. 4, SEQ ID Nr. 5 oder SEQ ID Nr. 6 hybridisieren,
beispielsweise die Sequenzen, die in SEQ ID Nr. 19 und SEQ ID Nr.
20 gezeigt sind. Eine stringente Hybridisierung wird bei einer Temperatur von
65°C, vorzugsweise
60°C und
am meisten bevorzugt 55°C
in Zitrat-gepufferter Salzlösung
(SSC) doppelter Stärke
(2×) durchgeführt, welche
0,1% SDS enthält,
gefolgt vom Spülen
des Trägers
bei der gleichen Temperatur mit einem Puffer, welcher eine reduzierte
SSC-Konzentration hat. Eine derartig reduzierte Konzentration von
Puffern ist typischerweise SSC mit einem Zehntel der Stärke (0,1 × SSC),
welche 0,1% SDS enthält, vorzugsweise
0,2 × SSC
enthaltend 0,1% SSC, und am meisten bevorzugt SSC halber Stärke (0,5 × SSC), welche
0,1% SDS enthält.
So können
funktionale Äquivalente
des gesamten oder eines Teils des CmYLCV Transkriptpromotors der
vollständigen
Länge aus
anderen Organismen durch Hybridisieren irgendeiner der SEQ ID Nr.
1, SEQ ID Nr. 2, SEQ ID Nr. 3, SEQ ID Nr. 4, SEQ ID Nr. 5, SEQ ID
Nr. 6, SEQ ID Nr. 19 oder SEQ ID Nr. 20 mit genomischer DNA isoliert
werden, welche aus einem Organismus von Interesse isoliert worden ist,
insbesondere aus einem anderen Caulimovirus. Der Fachmann weiß, wie vorzugehen
ist, um solche Sequenzen zu finden, da es viele Wege gibt, die in
dem Fachgebiet bekannt sind, um homologe Sequenzen in anderen Organismen
zu identifizieren. Solche neuen identifizierten DNA-Moleküle können dann
sequenziert werden und die Sequenz kann mit irgendeiner von SEQ
ID Nr. 1, SEQ ID Nr. 2, SEQ ID Nr. 3, SEQ ID Nr. 4, SEQ ID Nr. 5,
SEQ ID Nr. 6, SEQ ID Nr. 19 oder SEQ ID Nr. 20 verglichen werden
und bezüglich
der Promotoraktivität
getestet werden. Innerhalb des Schutzbereichs der vorliegenden Erfindung
sind DNA-Moleküle
mit mindestens 75%, vorzugsweise 80%, mehr bevorzugt 90% und am
meisten bevorzugt 95% Sequenzidentität mit der Nucleotidsequenz
in irgendeiner der SEQ ID Nr. 1, 2, 3, 4 oder 6 über eine Länge von mindestens 50 Nucleotiden.
Der Prozentsatz an Sequenzidentität wird unter Verwendung von
Computerprogrammen bestimmt, die auf dynamischen Programmalgorithmen
beruhen. Computerprogramme, die innerhalb des Schutzbereichs der
Erfindung bevorzugt sind, schließen die BLAST (Basic Local
Alignment Search Tool)-Rechercheprogramme
ein, die entworfen worden sind, um alle verfügbaren Sequenzdatenbanken,
unabhängig
davon, ob die Suche ein Protein oder eine DNA ist, zu nutzen. Die
Version BLAST 2.0 (Gapped BLAST) dieses Recherchetools ist öffentlich übers Internet
zugänglich
gemacht worden (gegenwärtig
http://www.ncbi.nlm.nih.gov/BLAST/). ES verwendet einen heuristischen
Algorithmus, welcher lokal sucht, statt in globalen Anlagerungen,
und ist daher in der Lage, Verwandtschaften zwischen Sequenzen nachzuweisen,
die nur isolierte Bereiche teilen. Die durch eine BLAST-Recherche
zugeschriebenen Werte ermöglichen
eine wohl definierte statistische Interpretation. Diese Programme
werden vorzugsweise mit optionalen Parametern, die für die vorgegebenen
Werte eingesetzt werden, laufen gelassen.
-
Es
ist ein weiteres Ziel der vorliegenden Erfindung, rekombinante DNA-Moleküle bereitzustellen,
die eine DNA-Sequenz gemäß der Erfindung
umfassen, welche operativ mit einer Nucleotidsequenz von Interesse verbunden
ist. Die Nucleotidsequenz von Interesse kann beispielsweise eine
ribosomale RNA, eine Gegensinn-RNA oder irgendeine andere Art von
RNA kodieren, welche nicht in ein Protein translatiert wird. In
einer anderen bevorzugten Ausführungsform
der Erfindung wird die Nucleotidsequenz von Interesse in ein Proteinprodukt
translatiert. Die Nucleotidsequenz, die mit der Promotorsequenz
assoziiert ist, kann homologen oder heterologen Ursprungs sein,
bezogen auf die zu transformierende Pflanze. Die Sequenz kann auch
vollständig oder
teilweise synthetisch sein. Unabhängig vom Ursprung wird die
damit verbundene DNA-Sequenz in der transformierten Pflanze gemäß den Expressionseigenschaften
des Promotors exprimiert, mit dem sie verbunden ist. Im Falle homologer Nucleotidsequenzen,
die mit der Promotorsequenz assoziiert sind, ist der Promotor gemäß der Erfindung
für die
ectopische Expression dieser homologen Sequenz nützlich. Ectopische Expression
bedeutet, dass die Nucleotidsequenz, die mit der Promotorsequenz
assoziiert ist, in Geweben und Organen exprimiert wird, und/oder
zu Zeitpunkten, an denen diese Sequenz nicht exprimiert wird, unter
Kontrolle ihres eigenen Promotors. In einer besonderen Ausführungsform
der Erfindung wird die Expression einer Nucleotidsequenz, die mit
der Promotorsequenz assoziiert ist, ihre eigene Expression unterdrücken und
das der Originalkopie des Gens, durch ein Verfahren, welches durch
einen Prozess, der als Co-Suppression bezeichnet wird.
-
In
einer bevorzugten Ausführungsform
der Erfindung kann die vorhandene Nucleotidsequenz auch ein Protein
kodieren, von dem man wünscht,
dass es in der Pflanze zu allen Zeitpunkten und in den meisten Geweben
und Organen exprimiert wird. Solche Nucleotidsequenzen kodieren
vorzugsweise Proteine, die eine gewünschte phänotypische Markierung an die
verbundene Pflanze verleihen. Beispiele sind Nucleotidsequenzen,
die Proteine kodieren, die eine Antibiotikaresistenz verleihen,
eine Virusresistenz, Insektenresistenz, Krankheitsresistenz oder
Resistenz gegenüber
anderen Seuchen, Herbizidtoleranz, verbesserte Nährwerte, verbesserte Leistungsfähigkeit
in einem industriellen Verfahren oder veränderte reproduktive Eigenschaften. Die
verbundene Nucleotidsequenz kann auch eine sein, die in Pflanzen
zu Herstellung kommerziell wertvoller Enzyme oder Metabolite in
der Pflanze eingeschleust worden ist. Von der vorliegenden Erfindung
umfasst sind auch selektierbare oder screenfähige Markergene, d. h. Gene,
die eine DNA-Sequenz der Erfindung umfassen, welche operativ mit
einer kodierenden Region verbunden ist, die eine selektierbare oder
screenfähige Markierung
kodiert.
-
Beispiele
selektierbarer oder screenfähiger
Markergene sind unten beschrieben. Für einige Zielarten können verschiedene Antibiotika-
oder Herbizidselektionsmarker bevorzugt sein. Die Selektionsmarker,
die routinegemäß in der
Transformation verwendet werden, schließen das nptII-Gen ein, welches
gegenüber
Kanamycin, Paromomycin, Geneticin oder verwandten Antibiotika Resistenz
verleiht (Vieira and Messing, 1982, Gene 19: 259-268; Bevan et al.,
1983, Nature 304: 184-187) das bakterielle aadA-Gen (Goldschmidt-Clermont,
1991, Nucl. Acids Res. 19: 4083-4089), welches eine Aminoglycosid
3'-Adenylyltransferase
kodiert, und Resistenz gegenüber
Streptomycin und Spectinomycin verleiht, das hph-Gen, welches eine
Resistenz gegenüber
dem Antibioticum Hygromycin verleiht (Blochlinger and Diggelmann,
1984, Mol. Cell. Biol. 4: 2929-2931), und das dhfr-Gen, welches
gegenüber
Methotrexat (Bourouis and Jarry, 1983, EMBO J. 2: 1099-1104) Resistenz
verleiht. Andere Marker, die zu verwenden sind, schließen ein
Phosphinothricinacetyltransferasegen ein, welches Resistenz gegenüber dem
Herbizid Phosphinothricin verleiht (White et al., 1990, Nucl. Acids
Res. 18: 1062; Spencer et al., 1990, Theor. Appl. Genet. 79: 625-631),
eine mutiertes EPSP-Synthasegen,
welches ein Glyphosatresistenz kodiert (Hinchee et al., 1988, Bio/Technology
6: 915-922), eine mutierte Acetolactatsynthase (ALS)-Gen, welches
Imidazolion- oder Sulonylharnstoffresistenz verleiht (Lee et al.,
1988, EMBO J. 7: 1241-1248), ein mutiertes psbA-Gen, welches eine
Atrazinresistenz verleiht (Smeda et al., 1993, Plant Physiol. 103:
911-917), oder ein mutiertes Protoporphyrinogenoxidasegen, das in
EP 0 769 059 beschrieben
ist. Die Identifizierung der transformierten Zellen kann ebenfalls
durch die Expression eines screenfähigen Markergens, wie beispielsweise
Genen erzielt werden, die für
Chloramphenicolacetyltransferase (CAT), β-Glucuronidase (GUS) Luciferase
(LUC) und grünes
fluoreszierendes Protein (GFP) oder irgendein anderes Protein, das der
transformierten Zelle ein phänotypisch
eindeutiges Merkmal verleiht. Selektionsmarker, die zur positiven Selektion
führen,
wie beispielsweise Phosphomannoseisomerasegen, wie in Patentanmeldung
WO 93/05163 beschrieben,
werden ebenfalls verwendet. Andere Gene, die für die positive Selektion verwendet
werden, sind beschrieben in
WO
94/20627 und kodieren Xyloisomerasen und Phosphomanno-Isomerasen,
wie Mannose-6-phosphatisomerase und Mannose-1-phosphatisomerase;
Phosphomannomutase; Mannoseepimerasen, wie solche, die Kohlenhydrate
umwandeln in Mannose oder Mannose in Kohlenhydrate, wie z. B. Glucose
oder Galactose; Phosphatasen wie Mannose oder Xylosephosphatase,
Mannose-6-phosphatase und Mannose-1-phosphatase, und Permeasen,
die an dem Transport von Mannose beteiligt sind oder einem Derivativ, oder
einem Vorläufer
davon in die Zelle. Das die Toxizität der Verbindung gegenüber Zellen
reduzierende Mittel ist typischerweise en Glucosederivat, wie Methyl-3-O-glucose
oder Phloridizin. Transformierte Zellen werden ohne Beschädigung oder
Abtöten
der nicht transformierten Zellen der Population und ohne gleichzeitige
Einschleusung von Antibiotika- und Herbizidresistenzgenen hergestellt.
Wie in
WO 93/05163 beschrieben,
ist zusätzlich
zur Tatsache, dass der Bedarf für
Antibiotika- und Herbizidresistenzgene eliminiert worden ist, gezeigt worden,
dass das positive Selektionsverfahren häufig viel effizienter ist als
die traditionelle Negativselektion.
-
In
einer bevorzugten erfindungsgemäßen Ausführungsform
sind die erfindungsgemäßen Promotoren operativ
mit der kodierenden Region des E. coli manA-Gens (Joersbo and Okkels,
1996, Plant Cell Reports 16: 219-221, Negrotto et al., 2000, Plant
Cell Reports 19: 798-803) verbunden, welche eine Phosphomannoseisomerase
(PMI) kodiert, und mit einem Nos (Nopalinsynthetase)-Terminator,
welcher aus Agrobacterium tumefaciens T-DNA (Depicker et al., J.
Mol. Appl. Genet. 1 (6), 561-573 (1982)) gewonnen wird. Der Fachmann auf
dem Gebiet weiß,
dass der Nos-Terminator durch irgendeinen anderen Terminator substituiert
werden kann, welcher in Pflanzen funktioniert. Der Promotor der
Erfindung kann ein CmpD-, CmpC-, CmpS-, CmpL-, CmpB-, CmpA-, CmpE-
oder CmpF-Promotor
sein, welcher in den SEQ ID Nr. 1 bis 6 und 19 bis 20 wiedergegeben
ist, oder irgendeinen Promotor, der davon abstammt. In einer mehr
bevorzugten Ausführungsform
der Erfindung ist der Promotor der CmpS-Promotor, welcher in SEQ
ID Nr. 3 wiedergegeben ist.
-
Die
erfindungsgemäßen Promotoren
können
ebenfalls verwendet werden, um durch operative Verbindung der Promotoren
der Erfindung mit kodierenden Regionen an Genen von Viren, die kontrolliert
werden sollen, eine Resistenz oder Toleranz gegenüber Viren
zu verleihen.
-
Für die Viruskontrolle
von negativ-strängigen
Viren, wie beispielsweise dem Tomatenbronzefleckenvirus (TSWV),
können
für Gensequenzen
das virale Nucleocapsidprotein (NP) und das Bewegungsprotein (MP) verwendet
werden, und bei Viren, die zur Alpha-artigen Übergruppe an Viren gehören, wie
beispielsweise TMV und CMV, können
für die
Gensequenzen RNA-abhängige
RNA-Polymerase (RdRp) und Bewegungsprotein (MP) verwendet werden.
Für Viren,
die zur Picorna-artigen Gruppe an Viren gehören, einschließlich Potyviren, kann
jede virale Sequenz mit den Promotoren der Erfindung verbunden werden.
Schließlich
können
bei allen anderen einschließlich
DNA-Viren, RNA-abhängige
RNA-Polymerase-/(RdRp) und Replicase-assoziierte Gensequenzen verwendet
werden. Derartige Konstrukte für
die Viruskontrolle können
hergestellt werden, wie in Beispiel 8 und Beispiel 9 der
WO 00/68374 exemplarisch
angegeben, konstruiert werden. Es liegt innerhalb der allgemeinen
Kenntnisse eines Fachmanns auf dem Gebiet, die Promotoren, die in
WO 00/68374 offenbart sind,
durch Promotoren der vorliegenden Erfindung auszutauschen. Nach
der Lehre der
WO 00/68374 weiß der Fachmann
ebenfalls, wie Konstrukte herzustellen sind, die Promotoren der
Erfindung umfassen, welche operativ mit viralen Sequenzen, die oben
erwähnt
worden sind, verbunden werden, um eine Resistenz oder Toleranz gegenüber dem
Virus zu verleihen.
-
Statt
doppelsträngige
virale RNA in transgenen Pflanzen zu exprimieren, wie in
WO 0068374 offenbart ist,
kann die virale RNA auch als nicht translatierbare RNA in Form eines
plus-Strang einer
viralen RNA-Moleküls
exprimiert werden, wie beispielsweise beschrieben in
US 558302 oder in den Beispielen 12
und 13 der vorliegenden Erfindung.
-
Es
ist ein weiteres Ziel der Erfindung, rekombinante Expressionsvektoren
bereitzustellen, die eine DNA-Sequenz der Erfindung umfassen, welche
mit einer verbundenen Nucleotidsequenz von Interesse fusioniert
ist. In diesen Vektoren kann die fremde DNA in einen Polylinkerbereich
inseriert werden, so dass diese exogenen Sequenzen in einer geeigneten
Wirtszelle exprimiert werden können,
welche beispielsweise bakteriellen oder pflanzlichen Ursprungs sind.
Beispielsweise erlaubt das Plasmid pBI101, welches aus dem binären Agrobacterium
tumefaciens Vektor pBIN19 stammt, die Klonierung und das Testen
von Promotoren unter Verwendung des beta-Glucuronidase (GUS)-Expressionssignal
(Jefferson et al., 1987, EMBO J. 6: 3901-3907). Die Größe des Vektors
beträgt
12,2 kb. Er hat einen Niedrigkopie-RK2-Replikationsursprung und
verleiht sowohl an Bakterien als auch an Pflanzen Kanamycinresistenz.
Es gibt zahlreiche andere Expressionsvektoren, die dem Fachmann
bekannt sind, die erfindungsgemäß verwendet
werden können.
-
Es
ist ein weiteres Ziel der vorliegenden Erfindung, transgene Pflanzen
bereitzustellen, welche die erfindungsgemäßen rekombinanten DNA-Sequenzen
umfassen. Die Erfindung betrifft daher Pflanzenzellen, Pflanzen,
die aus solchen Zellen stammen, Pflanzenmaterial, die Nachkommen
und die Samen, die aus solchen Pflanzen stammen, und landwirtschaftliche
Produkte mit verbesserten Eigenschaften, die durch irgendeines der
Transformationsverfahren, die unten beschrieben sind, gewonnen werden.
Pflanzen, die erfindungsgemäß transformiert
sind, können
monocotyledon oder dicotyledon sein und schließen ein, aber sind nicht beschränkt auf,
Reis, Mais, Weizen, Gerste, Roggen, Süßkartoffeln, Süßmais, Bohnen,
Erbsen, Chicoree, Salat, Kohl, Blumenkohl, Brokkoli, Zuckerrübe, Radieschen,
Spinat, Spargel, Zwiebeln, Knoblauch, Paprika, Sellerie, Kürbis, Kürbis, Hanf,
Zucchini, Apfel, Birne, Quitte, Melone, Pflaume, Kirsche, Pfirsisch,
Nektarine, Aprikose, Erdbeere, Traube, Himbeere, Brombeere, Ananas,
Avocado, Papaya, Mango, Banane, Soyabohne, Tomate, Hirse, Zuckerrohr,
Zuckerrübe,
Sonnenblumen, Raps, Klee, Tabak, Karotten, Baumwolle, Alfalfa, Kartoffeln, Auberginen,
Gurken, Arabidopsis thaliana und Holzpflanzen wie Koniferen und
Laubbäume.
Bevorzugte Pflanzen, die transformiert werden sollen, sind Reis,
Mais, Weizen, Gerste, Kohl, Blumenkohl, Paprika, Kürbis, Melone,
Soyabohnen, Tomaten, Zuckerrüben,
Sonnenblumen oder Baumwolle, aber insbesondere Reis, Mais, Weizen,
Zuckerhirse, Knaulgras, Zuckerrüben
und Soyabohnen. Die rekombinanten DNA-Sequenzen der Erfindung können in
eine Pflanzenzelle durch eine Anzahl wohl bekannter Verfahren eingeschleust
werden. Fachleute auf dem Gebiet werden erkennen, dass die Auswahl
eines solchen Verfahrens von der Art der Pflanze, die für die Transformation
ins Visier genommen wird, abhängt,
d. h. von Monocotyledonen oder Dicotyledonen. Geeignete Verfahren
zur Transformierung von Pflanzenzellen schließen die Mikroinjektion (Crossway et
al., 1986, Bio Techniques 4: 320-334), die Elektroporation (Riggs
and Bates, 1986, Proc. Natl. Acad. Sci., USA 83: 5602-5606), Agrobacterium-vermittelte Transformation
(Hinchee et al., 1988, Bio/Technology 6: 915-922;
EP 0 853 675 ), direkten Gentransfer
(Paszkowski et al., 1984, EMBO J. 3: 2717-2722) und ballistische
Partikelbeschleunigung unter Verwendung von Vorrichtungen, die von
Agracetus, Inc., Madison, Wisconsin und Dupont, Inc., Wilmington,
Delaware erhältlich
sind (siehe z. B.
US Patent Nr.
4,945,050 und McCabe et al., 1988, Bio/Technology 6: 923-926).
Die zu transformierenden Zellen können differenzierte Blattzellen, embryogene
Zellen oder irgendeine andere Art von Zelle sein. Bei der direkten
Transformation von Protoplasten kann die Aufnahme von exogenem genetischen
Material in einen Protoplasten durch die Verwendung eines chemischen
Mittels oder eines elektrischen Felds verstärkt werden. Das exogene Material
kann dann in das nukleäre
Genom integriert werden. Die vorhergehende Arbeit wurde an dicotyledonen
Tabakpflanzen durchgeführt,
was dazu führte,
dass die fremde DNA in Nachkommenpflanzen eingebaut und transferiert
wurde (Paszkowski et al., 1984, EMBO J. 3: 2712-2722; Potrykus et
al., 1985, Mol. Gen. Genet 199: 169-177). Monocotyledonen-Protoplasten,
z. B. von Einkorn, einjähriges
Weidelgras, Mais und schwarzen mexikanischen Süßmais werden durch dieses Verfahren
transformiert. Eine zusätzliche
bevorzugte Ausführungsform ist
die Protoplastentransformationsmethode für Mais, wie offenbart in
EP 0 292 435 , und in
EP 0 846 771 . Für die Maistransformation
siehe auch Koziel et al., 1993, Bio/Technology 11: 194-200. Die
Transformation von Reis kann durch direkte Gentransfer-Techniken unter Verwendung
von Protoplasten oder durch Partikelbombardierung durchgeführt werden.
Die Protoplasten-vermittelte
Transformation wird für
Japonica-Arten und Indica-Arten (Zhang et al., 1988, Plant Cell
Rep., 7: 379-384; Shimamoto et al., 1989, nature 338: 274-276; Datta et
al., 1990, Bio/Technology 8: 736-740) beschrieben. Beide oben beschriebenen
Typen sind auch routinegemäß unter
Verwendung von Partikelbombardierung transformierbar (Christou et
al., 1991, Bio/Technology 9: 957-962). Die Patentanmeldung Nr.
EP 0 332 581 beschreibt
Techniken für
die Erzeugung, Transformation und Regeneration von Süßpflanzen-Protoplasten. Diese
Techniken ermöglichen
die Transformation aller Pooideae-Pflanzen einschließlich Knäuelgras
und Weizen. Außerdem
wird die Weizentransformation in der Patentanmeldung Nr.
EP 0 674 715 und von Weeks
et al., 1993 (Plant Physiol. 102: 1077-1084) beschrieben. Der so konstruierte
Pflanzen-Expressionsvektor
kann beispielsweise in die Calli von Reis gemäß einem konventionellen Pflanzen-Transformationsverfahren
eingeschleust werden, und die Differenzierung von Wurzeln und Blättern wird
daraus induziert, und dann kann sie in einen Blumentopf zur Kultivierung überführt werden,
wodurch der transformierte Reis gewonnen wird.
-
Die
Pflanzen, die aus der Transformation mit den DNA-Sequenzen oder
Vektoren der vorliegenden Erfindung gewonnen werden, exprimieren
eine Nucleotidsequenz von Interesse in der ganzen Pflanze und in
den meisten Geweben und Organen.
-
Die
genetischen Eigenschaften, die in die transgenen Pflanzen durch
Rekombination eingeführt
werden, welche oben beschrieben wurden, werden durch sexuelle Reproduktion
oder vegetatives Wachstum weitergegeben und können dadurch in den Nachkommenpflanzen
bewahrt und propagiert werden. Im Allgemeinen macht diese Bewahrung
und Propagierung von bekannten landwirtschaftlichen Methoden Gebrauch,
die entwickelt wurden, um spezifische Zwecke zu erfüllen, wie
beispielsweise die Bodenbearbeitung, das Säen oder Ernten. Spezialisierte
Verfahren wie Hydrokulturen oder Treibhaustechnologien können ebenfalls
angewandt werden. Von den vorteilhaften genetischen Eigenschaften
der transgenen Pflanzen gemäß der Erfindung
kann ferner in der Pflanzenzucht Gebrauch gemacht werden, die auf
die Entwicklung von Pflanzen mit verbesserten Eigenschaften, wie
beispielsweise Toleranz gegenüber
Seuchen, Herbiziden oder Stress, auf verbesserte Nährwerte,
erhöhte
Ausbeute oder verbesserte Struktur, welche einen geringeren Verlust
aufgrund Lagerung oder Bruch zielen. Die verschiedenen Zuchtschritte
werden durch gut definierte menschliche Eingriffe gekennzeichnet,
wie beispielsweise die Auswahl der zu kreuzenden Linien, die direkte
Bestäubung der
Parentallinien oder die Auswahl geeigneter Nachkommenpflanzen. Abhängig von
den gewünschten
Eigenschaften werden verschiedene Zuchtmaßnahmen durchgeführt. Die
relevanten Techniken sind in dem Fachgebiet gut bekannt und schließen ein,
sind aber nicht begrenzt auf, Hybridisierung, Inzucht, Rückkreuzungszüchtung,
Multilinienzüchtung,
Mischung von Sorten, Interspezies- Hybridisierung, Aneuploidie-Techniken,
etc. Hybridisierungstechniken schließen auch die Sterilisierung
von Pflanzen ein, um sterile männliche
oder weibliche Pflanzen hervorzubringen, indem mechanische, chemische
oder biochemische Mittel verwendet werden. Eine Kreuzbestäubung einer
männlichen
sterilen Pflanze mit Pollen einer anderen Linie sichert, dass das
Genom der männlichen
sterilen, aber der weiblichen fruchtbaren Pflanze gleichmäßig die
Eigenschaften beider Parentallinien gewinnt. So können die
transgenen Pflanzen nach der Erfindung für die Zucht verbesserter Pflanzenlinien
verwendet werden, beispielsweise den Anstieg der Wirksamkeit konventioneller
Verfahren, wie beispielsweise die Herbizid- oder Pestizidbehandlung,
oder ermöglichen,
auf dieses Verfahren, aufgrund ihrer modifizierten genetischen Eigenschaften
zu verzichten. Alternativ dazu können
neue Futterpflanzen mit verbesserter Stresstoleranz gewonnen werden,
die aufgrund ihrer optimierten genetischen „Ausrüstung" ein Ernteprodukt von besserer Qualität hervorbringen
als Produkte, die nicht in der Lage waren, vergleichsweise ungünstige Entwicklungsbedingungen
zu tolerieren.
-
Es
ist ein weiteres Ziel der vorliegenden Erfindung, DNA-Sequenzen
bereitzustellen, die verwendet werden können, um eine Nucleotidsequenz
von Interesse in einem gewünschten
Organismus zu exprimieren. Dieser Organismus kann ein Bakterium,
eine Pflanze oder irgendein anderer Organismus von Interesse sein.
-
Des
Weiteren ermöglicht
die Offenbarung der SEQ ID Nr. 1 bis 6 einem Fachmann auf dem Gebiet, Oligonucleotide
für die
Polymerase-Kettenreaktion zu entwerfen, welche versucht, DNA-Fragmente
aus Matrizen zu amplifizieren, welche eine Sequenz von Nucleotiden
umfassen, die durch irgeneine kontinuierliche Sequenz von 15, und
vorzugsweise 20 bis 30 oder mehr Basenpaaren in SEQ ID Nr. 1, 2,
3, 4, 5 oder 6 gekennzeichnet ist. Diese Nucleotide umfassen eine
Sequenz von Nucleotiden, die 15 und vorzugsweise 20 bis 30 oder
mehr Basenpaare der SEQ ID Nr. 1, 2, 3, 4, 5 oder 6 darstellen.
Polymerase-Kettenreaktion, die unter Verwendung mindestens eines
solchen Oligonucleotids durchgeführt
werden, und ihre Amplifikationsprodukte bilden andere Ausführungsformen
der vorliegenden Erfindung. Kurze
Beschreibung der Sequenzen in der Sequenzliste
| SEQ
ID Nr. 1 | CmpD |
| SEQ
ID Nr. 2 | CmpC |
| SEQ
ID Nr. 3 | CmpS |
| SEQ
ID Nr. 4 | CmpL |
| SEQ
ID Nr. 5 | CmpB |
| SEQ
ID Nr. 6 | CmpA |
| SEQ
ID Nr. 7 | S1
Vorwärtsprimer |
| SEQ
ID Nr. 8 | S2
Umkehrprimer |
| SEQ
ID Nr. 9 | GUS
Vorwärsprimer |
| SEQ
ID Nr. 10 | GUS
Umkehrprimer |
| SEQ
ID Nr. 11 | CAT
Vorwärtsprimer |
| SEQ
ID Nr. 12 | CAT
Umkehrprimer |
| SEQ
ID Nr. 13 | Cmp1
Vorwärsprimer |
| SEQ
ID Nr. 14 | CmpC2
Umkehrprimer |
| SEQ
ID Nr. 15 | CmpS2
Umkehrprimer |
| SEQ
ID Nr. 16 | CmpL2
Umkehrprimer |
| SEQ
ID Nr. 17 | PA
Vorwärtsprimer |
| SEQ
ID Nr. 18 | PA
Umkehrprimer |
| SEQ
ID Nr. 19 | CmpE |
| SEQ
ID Nr. 20 | CmpF |
| SEQ
ID Nr. 21 | Nos-Terminator
Vorwärtsprimer |
| SEQ
ID Nr. 22 | Nos-Terminator
Umkehrprimer |
| SEQ
ID Nr. 23 | TSWV
NP-Gen Vorwärtsprimer |
| SEQ
ID Nr. 24 | TSWV
NP-Gen Umkehrprimer |
| SEQ
ID Nr. 25 | CmYLCV-Promotor
Vorwärtsprimer |
| SEQ
ID Nr. 26 | CmYLCV-Promotor
Umkehrprimer |
| SEQ
ID Nr. 27 | AGLINK
Multiklonierungsstelle |
| SEQ
ID Nr. 28 | BIGLINK
Multiklonierungsstelle |
| SEQ
ID Nr. 29 | Cmpf-B-Primer |
| SEQ
ID Nr. 30 | CmpS-B-Primer |
| SEQ
ID Nr. 31 | pNOV2803;
binärer
Vektor, enthält
eine Promotor-lose PMI-nos-Terminator-Expressions-Kassette |
| SEQ
ID Nr. 32 | pNOV2803;
Vektor für
die Transformation mittels Biolistic, enthält eine Promotorlose PMI-nos-Terminator-Expressions-Kassette |
| SEQ
ID Nr. 33 | CmpMr
Umkehrprimer |
| SEQ
ID Nr. 34 | CmpMf
Vorwärtsprimer |
| SEQ
ID Nr. 35 | CmpMrs
Umkehrprimer |
| SEQ
ID Nr. 36 | CmpMfs
Vorwärtsprimer |
| SEQ
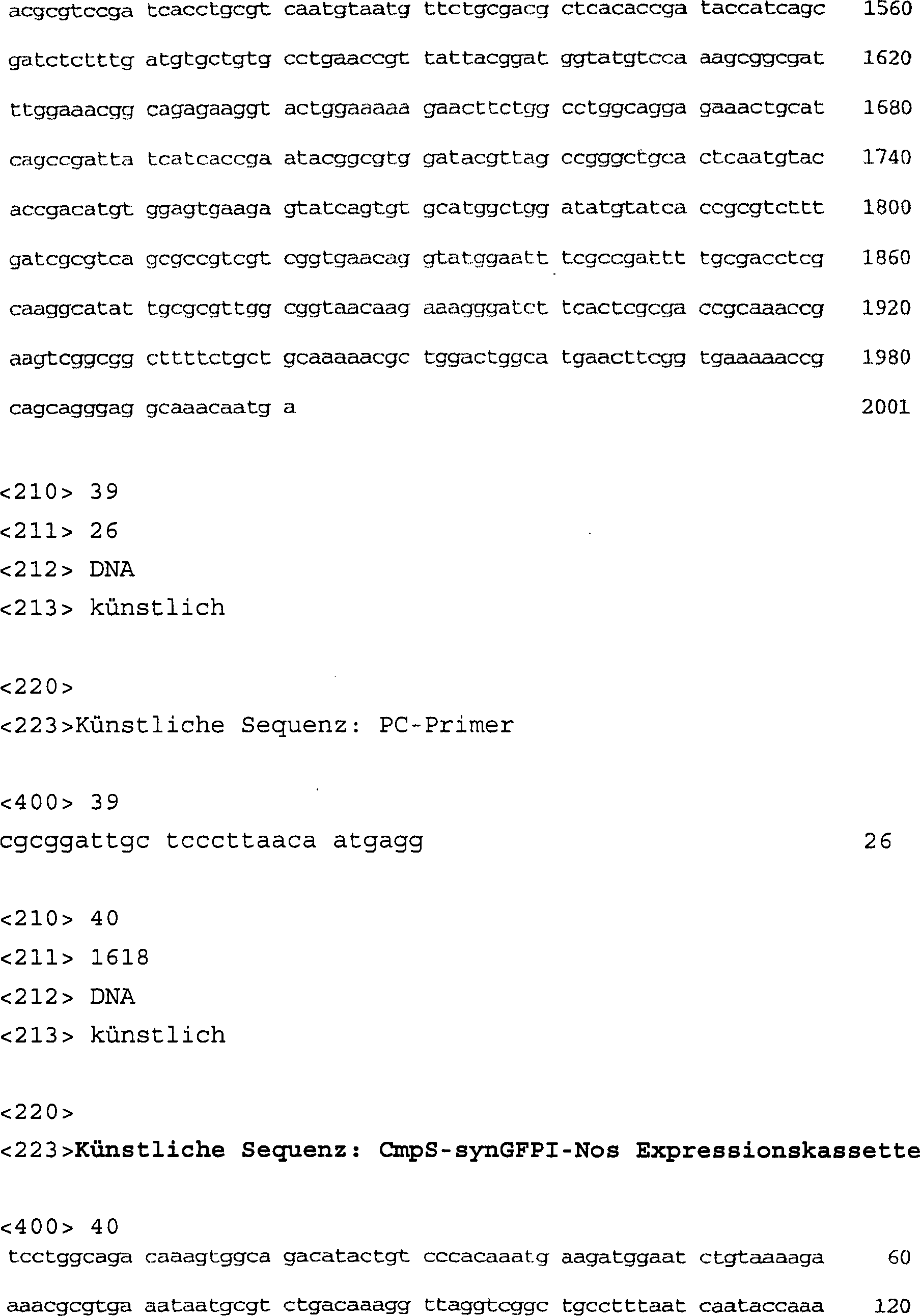
ID Nr. 37 | synGFPI;
Pflanze, die ein optimiertes GFP-Gen mit Intron hat |
| SEQ
ID Nr. 38 | GIG;
GUS-Gen mit Intron |
| SEQ
ID Nr. 39 | Cmp-C
Umkehrprimer |
| SEQ
ID Nr. 40 | CmpS-synGFPI-nos
Expressions-Kassette |
| SEQ
ID Nr. 41 | CmpS-GIG-nos
Expressions-Kassette |
| SEQ
ID Nr. 42 | CmpC-synGFPI-nos
Expressions-Kassette |
| SEQ
ID Nr. 43 | CmpC-GIG-nos
Expressions-Kassette |
| SEQ
ID Nr. 44 | pNOV2117-Vektor |
| SEQ
ID Nr. 45 | pNOV4200-Vektor |
| SEQ
ID Nr. 46 | ZmUbi-GFP-35S
Term Expressions-Kassette |
| SEQ
ID Nr. 47 | ZmUbi-GIG-nos
Expressions-Kassette |
| SEQ
ID Nr. 48 | Ubg3(At)-synGFPI-nos
Expressions-Kassette |
| SEQ
ID Nr. 49 | Ubg3(At)-GIG-nos
Expressions-Kassette |
-
Beispiele
-
Standard-Verfahren
für die
DNA-Rekombination und molekulare Klonierungstechniken, die hier
verwendet werden, sind im Gebiet gut bekannt und werden beispielsweise
beschrieben in Sambrook et al., 1989, „Molecular Cloning", Cold Spring Harbor,
Cold Spring Harbor Laboratory Press, NY, und von Ausubel et al., 1994, „Current
protocols in molecular biology",
John Wiley and Sons, New York.
-
Beispiel 1: Virus-Klonierung
-
1. DNA Extraktion.
-
Virus-Genom-DNA
wird aus Cestrum yellow leaf curling virus-infizierten Chilenischen
Hammerstrauch-Pflanzen
extrahiert. Fünf
Gramm der infizierten Blätter
werden in 30 ml Mahlpuffer (0,2 M Tris, pH 7,0, 0,02 M EDTA, 2 M
Harnstoff) für
60 sec. bei maximaler Geschwindigkeit in einem Brinkman Polytron
Homogenisator mit einer PT10-Probe homogenisiert und leicht bei
4°C über Nacht
mit Triton X-100 (2% Endkonzentration) geschüttelt. Virus wird aus dem rohen
Homogenat durch Zentrifugation bei niedriger Geschwindigkeit (10.000
UpM für
20 min. in einem Sorvall SS-34-Rotor) gereinigt und aus dem gewonnenen Überstand
mit Hilfe der Hochgeschwindigkeits-Zentrifugation (27.000 UpM für 2 h in
einem Beckman SW-28-Rotor) durch ein Saccharosekissen (3 ml 15%
Saccharose). Das Virus-haltige Pellet wird dann in 0,1 M Tris, pH
7,4, 2,5 mM MgCl2 resuspendiert. DNase I
(Sigma) und RNase A (Sigma) werden bis zu einer Konzentration von
jeweils 10 mg/ml zugegeben. Nach 30 min. bei 37°C wird die Reaktion durch die
Zugabe von EDTA zu 10 mM gestoppt. Virus-DNA wird aus CmYLCV-Partikeln
durch Behandlung mit Protease K isoliert (E. Merck, 0,5 mg/ml Endkonzentration)
in Gegenwart von 1% SDS bei 65°C
für 15
min. Die virale DNA wird dann durch Phenolextraktion und Ethanolpräzipitation
gereinigt. Die Virus-DNA, die in dem ersten Ethanolpräzipitat
enthalten ist, wird in Wasser gelöst.
-
2. DNA-Amplifizierung und Klonierung.
-
100
ng der erhaltenen DNA werden als Matrize für die PCR-Amplifikation in
einem 50 μl
Reaktionsvolumen enthaltend 10 μM
jedes Primers, 25 mM jedes dNTPs, 5 μl des pfu-Reaktionspuffers (Stratagene)
und 2,5 Einheiten der pfu-Turbo-DNA-Polymerase (Stratagene) verwendet.
Der S1-Vorwärtsprimer
(gaccacaaacatcagaag, SEQ ID Nr. 7) und der S2-Umkehrprimer (caaacttattgggtaatc,
SEQ ID Nr. 8) werden für
die PCR-Reaktion verwendet. Die Zyklusparameter der PCR sind: 1 × (94°C für 1 min.);
30 × (94°C für 1 min.,
50°C für 1 min.,
72°C für 1 min.),
1 × (72°C für 10 min.).
Jedes einzelne DNA-Fragment wird in pPCR-ScriptTM Amp
SK (+) Plasmid (Stratagene) nach den Anweisungen des Herstellers
kloniert.
-
Beispiel 2: DNA-Sequenzierung
-
Die
Sequenzierung der DNA-Virusklone wird unter Verwendung des automatisierten
ABI PRISM 377-DNA-Sequenziergeräts
(Perkin Elmer) und des ABI PRISM dRhodaminterminator-Zyklus-Sequenzierkits (Perkin
Elmer) nach den Anweisungen des Herstellers durchgeführt. Der
beschriebene Si-Primer und S2-Primer (Beispiel 1) wie auch der universale
M13-20 und die Umkehrprimer werden für die Sequenzierungsreaktionen
verwendet.
-
Beispiel 3: Herstellung von Plasmiden
für die
transiente Expression
-
Alle
PCR-Reaktionen, die mit der Pfu-Polymerase (Stratagene) durchgeführt werden,
sind wie in Beispiel 1 beschrieben.
-
1. Amplifizierung der Reportergene.
-
Die
beta-Glucuronidase (GUS) und die Chloramphenicolacetlytransferase
(CAT)-Reportergene
werden amplifiziert. Für
die GUS-Gen-Amplifikation
werden GUS-Vorwärts
(cagggtaccactaaaatcacc, SEQ ID Nr. 9) und GUS-Umkehr (aggggatccccaattcccc,
SEQ ID Nr. 10)-Oligonucleotide bei der Anlagerungstemperatur von
60°C verwendet.
CAT-Vorwärts
(aggggtaccatcgatatggag, SEQ ID Nr. 11) und CAT-Umkehr (ttaggatccgccctgccac,
SEQ ID Nr. 12)-Oligonucleotide
werden bei 62°C
angelagert. Die Vorwärtsprimer
werden so entworfen, dass sie eine KpnI-Erkennungssequenz und die Umkehrprimer
eine BamHI-Erkennungssequenz enthalten.
-
2. CmYLCV-Promotor-Amplifikation.
-
Drei
verschiedene Promotorfragmente werden ausgewählt. CmpC (SEQ ID Nr. 2), CmpS
(SEQ ID Nr. 3) und CmpL (SEQ ID Nr. 4). Cmp1-Vorwärtsprimer
(cttctagacaaagtggcagac, SEQ ID Nr. 13) wird für alle drei Amplifikationen
bei der Anlagerungstemperatur von 52°C verwendet. Er ist gegenüber der
Originalsequenz modifiziert (in fett gezeigt), so dass er eine XbaI-Erkennungsstelle
enthält.
Der CmpC2-Umkehrprimer (ttggtaccttaacaatgaggg, SEQ ID Nr. 14) wird
für die
CmpC-Fragment-Amplifizierung
verwendet und der CmpS2-Umkehrprimer (ctacttctaggtaccttgctc, SEQ
ID Nr. 15) wird für
das CmpS-Fragment
verwendet. Beide sind modifiziert, so dass sie eine KpnI-Erkennungsstelle
enthalten. Der CmpL2-Umkehrprimer (ttggtaccttaacaatgaggg, SEQ ID
Nr. 16) wird für
die CmpL-Fragment-Amplifizierung
verwendet und ist so modifiziert, dass er eine ClaI-Erkennungsstelle
enthält.
-
3. Polyadenylierungs-Signal-Amplifizierung.
-
Die
Polyadenylierungs-Signalfragmente werden aus dem infektiösen Klon
des Blumenkohl-Mosaikvirus (CaMV) (Franck et al., Cell 21 (1), 285-294,
1980) amplifiziert. PA (Polyadenylierungs-Signal-Amplifizierung) Vorwärts-(ggggatccccagtctctctc,
SEQ ID Nr. 17) und PA-Umkehr (gtgaatcgagctcggta, SEQ ID Nr. 18)-Oligonucleotide
werden in der PCR verwendet und bei 60°C angelagert. Der Vorwärtsprimer
ist so entworfen, dass er eine BamHI-Erkennungsstelle und der Umkehrprimer
eine EcoRI-Erkennungsstelle
enthält.
-
4. Hergestellte Konstrukte
-
- – pCmpCG
(CmpC Promotorfragment + GUS-Gen + polyA)
- – pCmpSG
(CmpS Promotorfragment + GUS-Gen + polyA)
- – pCmpCC
(CmpC Promotorfragment + CAT-Gen + polyA)
- – pCmpSC
(CmpS Promotorfragment + CAT-Gen + polyA)
- – pCmpLC
(CmpL Promotorfragment + CAT-Gen + polyA)
-
Die
Kassetten, die das Promotorfragment, die Reportergene und das Polyadenylierungssignal
enthalten, werden in den pUC19- Vektor
(Stratagene) inseriert, der mit XbaI und EcoRI geschnitten ist.
-
5. Konstrukte, die für einen Vergleich verwendet
werden
-
- – pCapG
(Cap-Promotorfragment + GUS-Gen + polyA)
- – pCapSG
(CapS-Promotorfragment + GUS-Gen + polyA)
- – pCapC
(Cap-Promotorfragment + CAT-Gen + polyA)
-
Promotorfragmente,
die in diesem Konstrukten enthalten sind, werden aus CaMV (Franck
et al., Cell 21 (1), 285-294, 1980, siehe GenBank Zugangsnummer
V00141) gewonnen. GUS-, CAT- und polyA-Fragmente sind identisch
mit denen, die für
die CmYLCV-Konstrukte
verwendet werden. Cap entspricht dem Fragment von der Base –227 bis
zur Base +33 der TATA-Box (Base 7175 bis Base 7438 des CaMV-Genoms,
siehe GenBank Zugangsnummer V00141) und CapS entspricht dem Fragment
von der Base –227
bis zur Base +82 von der TATA-Box (Base 7175 bis Base 7486 des CaMV-Genoms,
siehe GenBank Zugangsnummer V00141). Diese Positionen entsprechen
ungefähr
denen, die für
die CmYLCV-Fragmente
ausgewählt
werden.
-
Beispiel 4: Transiente Expressionsexperimente
-
1. Suspensionskulturen und Protoplastenpräparierung
-
Orychophragmus violaceus.
-
Suspensionskulturen
werden in 40 ml MS-Medium (Murashige and Skoog, Physiol Plant 15,
474-497, 1962) einschließlich
100 mg/ml Inositol, 2% Saccharose und 0,1 mg/ml 2,4 D aufbewahrt.
Die Protoplasten werden aus 4 bis 5 Tage alten Kulturen isoliert.
Die Zellwände
werden bei 26°C
für 1 h
in 0,1% Pectolyase Y23 (Seishin Pharmaceutical Co., Japan), 1% Zellulase
Onozuka R10 (Yakult Honsha Co., Japan), 0,4 M D-Mannitol und 0,1%
MES, pH 5,5, verdaut. Die Protoplasten werden durch ein 50 um Sieb
gefiltert und zweimal mit Electroporationslösung (EP) (10 mM HEPES, 150
mM NaCl, 5 mM CaCl2, 0,2 M Mannitol, pH
7,1) gewaschen.
-
Nicotiana plumbaginifolia.
-
Die
Pflanzen werden keimfrei auf RPM2-Medium (Blonstein et al., Mol
Gen Genet 211, 252-259, 1988) plus 7 g/l Bacto Agar, pH 5,6, gehalten.
Für die
Protoplasetenpräparation
werden die Blätter
abgeschnitten und über
Nacht bei 26°C
in einer Lösung
aus 0,5% Driselase (Fluka), 0,25 mM PVP 10 (Polyvinylpyrrolidon
MW 10000), 3,85 mM CaCl2, 6 mg/l NAA, 2
mg/l BAP, und 0,5 M Saccharose, pH 5,7, inkubiert. Die Protoplasten werden
durch ein 100 μm
Sieb filtriert. Eine Saccharoselösung
(0,6 M Saccharose, 0,1 MES und 15 mM CaCl2, pH
5,7) wird zu der Protoplastesuspension für den ersten Reinigungsschritt
zugegeben, und die Suspension wird mit W5-Lösung (150 mM NaCl, 125 mM CaCl2, 5 mM KCl, 6 mM Glucose; Menczel et al.,
Theor Appl Genet 59, 191-195, 1981) überlagert. Protoplasten werden
dann einmal mit W5-Lösung
und schließlich
mit der EP-Lösung
gewaschen.
-
Oryza sativa.
-
Protoplasten
werden aus einer morphogenen Reissuspensionskultur hergestellt,
die aus O. sativa cv. Nipponbare etabliert wurde (Datta et al.,
1990, Bio/Technology 8: 736-740) wie beschrieben hergestellt.
-
2. Transientes Expressionsexperiment.
-
Die
Transfektion durch Electroporation von 2 × 106 Orychophragmus
violaceus-Protoplaseten
in 0,66 ml EP-Puffer wird durch Entladen eines 960 μF Kapazitätsgeräts über einen
Weg von 4 mm einer Protoplastensuspension durchgeführt. Der
Kondensator wird mit 450 Volt beladen. Die Electroporation wird
in Gegenwart von 5–10 μg Plasmid-DNA
durchgeführt,
und dann werden die Protoplasten 16 bis 24 Stunden bei 25°C kultiviert.
Die Transfektion von 2 × 106 Nicotiana plumbagenifolia-Protoplasten
in einer 0,3 ml Suspension wird in Gegenwart von 0,3 ml PEG (40%
Polyetylenglycol 6000) und 5–10 μg Plasmid-DNA
durchgeführt.
Die Protoplasten werden in 0,4 ml K3-Medium (Godall et al., Methods
Enzymol 181, 148-161, 1990) für
16 bis 24 Stunden bei 25°C
durchgeführt,
und hierzu wird 10 ml W5-Puffer vor der Ernte zugegeben.
-
Die
Proteinextrakte werden durch mindestens drei Zyklen aus Einfrieren
und Auftauen hergestellt und durch Zentrifugieren gereinigt.
-
Beispiel 5: CAT- und GUS-Tests
-
Zum
Nachweis der CAT-Genexpression wird ein Doppel-Antikörper-Sandwich (DAS)-ELISA unter
Verwendung eines CAT-ELISA-Kits
(Boehringer) nach den Anweisungen des Herstellers durchgeführt. Die CAT-Aktivität wird durch
einen Dynex MRXII-Apparat
gemessen. Proben für
den GUS-Test werden in GUS-Puffer
(0,5 M NaPO4, pH 7, 0,01 M EDTA, 0,1% Triton-X-100,
0,1% Sarkosyl) verdünnt.
Die Reaktion wird in Gegenwart einer gleichen Menge GUS-Reaktionspuffers
(100 ml/l 10 × GUS-Puffer, 200 mg/l
BSA, 705 mg/l 4-Methylumbelliferylglucuronid) bei 37°C durchgeführt und
mit 2 M Ammediol gestoppt. Die Aktivität wird in einem Titertek Fluoroskan
II gemessen.
-
Sowohl
die CAT als auch die GUS-Testergebnisse werden auf einen Standard-Klonwert
normalisiert. Die Ergebnisse, die aus zehn verschiedenen Experimenten
gewonnen wurden, zeigen, dass die verschiedenen CmYLCV-Promotorfragmente
als stark aktive Promotoren dienen. Transiente Expression in N. plumbaginifolia-Protoplasten
GUS-Reportergen
| CmpCG | 100 |
| CmpSG | 86,9
+/– 20% |
| CapG | 9
+/– 20% |
| CaSG | 38,9
+/– 15% |
CAT-Reportergen
| CmpCC | 100 |
| CmpSC | 78
+/– 20% |
| CapSC | 89,5
+/– 20% |
| CmpLC | 9
+/– 15% |
Transiente Expression in O. violaceus-Protoplasten GUS-Reportergen
| CmpCG | 100 |
| CmpSG | 84,6
+/– 15% |
| CapG | 9,6
+/– 15% |
| CapSG | 40,7
+/– 15% |
CAT-Reportergen
| CmpCC | 100 |
| CmpSC | 255
+/– 20% |
| CapSC | 546
+/– 15% |
| CmpLC | 10
+/– 20% |
Transiente Expression in O. sativa-Protoplasten GUS-Reportergen
| CmpCG | 100 |
| CmpSG | 84,6
+/– 15% |
| CapG | 9,6
+/– 15% |
| CapSG | 40,7
+/– 15% |
-
Beispiel 6: Herstellung von Lösungen und
Medien für
die Pflanzenregenerierung und Transformation
-
Die
Kulturmedien GM, CIM und SIM sind die Medien, die von Valvekens
et al. beschrieben werden (1988, Proc. Natl. Acad. Sci. USA. 85:
5536-5540).
-
Das
Kulturmedium GM enthält
die Mineralsalze nach Murashige und Skoog (1962, Physiol. Plant.
15: 473-497), 1,0 mg/l Thiamin (Stock 1 mg/ml) 0,5 mg/l Pyridoxin
HCl (Stock 1 mg/ml), 0,5 mg/l Nicotinsäure (Stock 1 mg/ml), 0,5 g/l
2-(N-Morpholino)ethansulfonsäure
(MES), 10 g/l Saccharose, 8 g/l Agar, wobei der pH-Wert mit 1 N
KOH auf 5,8 eingestellt ist. CIM enthält die Mineralsalze und Vitamine
des B5-Mediums (Garnborg et al., 1968, Exp. Cell Res. 50: 151-158),
0,5 g/l 2-(N-Morpholino)ethansulfonsäure (MES), 20 g/l Glucose, 0,5
mg/l 2,4-Dichlorphenoxyessigsäure
(2,4-D) (Stock 10 mg/ml in DMSO), 0,5 mg/l Kinetin (Stock 5 mg/ml
in DMSO), pH 5,8. Festes CIM-Medium enthält 8 g/l Agar. SIM enthält die Mineralsalze
und Vitamine von B5-Medium (Garnborg et al., 1968, siehe oben),
0,5 g/l 2-(N-Morpholino)ethansulfonsäure (MES), 20 g/l Glucose,
5 mg/l N6-(2-Isopentenyl)adenin (2iP) (Stock 20 mg/ml in DMSO),
0,15 mg/l Indol-3-essigsäure
(IAA) (Stock 1,5 mg/ml in DMSO), 8 g/l Agar, pH 5,8. SIM V750 K100
ist SIM-Medium, das mit 750 mg/l Vancomycin und 100 mg/l Kanamycin
ergänzt
ist. SIM V500 K100 ist SIM-Medium, das mit 500 mg/l Vancomycin und
100 mg/l Kanamycin supplementiert ist. GM K50 ist GM-Medium, das
mit 50 mg/l Kanamycin supplementiert ist.
-
Die
Kulturmedien werden alle durch Autoclavieren (20 min., 121°C) sterilisiert.
Die Vitamine werden in Wasser gelöst und vor dem Autoclavieren
zum Medium zugegeben. Die Hormone werden in Dimethylsulfoxid (DMSO)
gelöst.
Die Antibiotica werden in Wasser gelöst und durch Filtrieren sterilisiert
(0,22 μm).
Die Hormone und Antibiotica werden nach dem Autoclavieren und Abkühlen des
Mediums auf 65°C
zugegeben. In allen Fällen
werden 9 cm Petrischalen (Falcon, 3003) verwendet, außer für GM und
GM K50, welche für
gewöhnlich
in 15 cm Petrischalen (Falcon, 3025) geschüttet werden.
-
Platten
mit festem Medien werden vor der Verwendung in einer Sterilbank
(Laminar-flow) getrocknet, um das Kondensat zu entfernen.
-
Beispiel 7: Kresse-Stamm und Wachstumsbedingungen
-
Arabidopsis
thaliana-Samen Ecotype Columbia (Co1-o) Wildtyp werden von Lehle
Seeds, USA (1102 South Industrial Blvd. Suite D, Round Rock TX 78681,
USA) bezogen. Die Pflanzen werden bei 22°C in einem 16/8 Stunden Licht/Dunkel-Zyklus
in Töpfen
in der Mischung aus 4 Teilen Sand, 4 Teilen Gartenboden und 1 Teil
Agrilit gezüchtet.
-
Beispiel 8: Agrobacterium-Stamm und Kultur
-
Vektorplasmide
werden in den Empfänger-Agrobacterium
tumefaciens Stamm LBA4404 (Clontech) durch triparentales Kreuzen
gemäß dem Protokoll,
das von Walkerpeach and Velten beschrieben ist („Agrobacterium-mediates gene
transfer to plant cells: Cointegrate and binary vector systems” in: Plant
Molecular Biology Manual, B1: 1-19, 1994. Eds.: S. B. Gelvin, R.
A., Schilperoort, Kluvers Acad. Publishers) eingeschleust. Der Mobilisierungsstamm,
der verwendet wird, ist E. coli HB101, welcher das Konjugationsplasmid
pRK2013 (Ditta et al., 1980, Broad host range DNA cloning system
from Gram-negative bacteria. Construction of gene bank of Rhizobium
meliloti. Proc. Natl. Acad. Sci. USA 77: 7347-7351). Agrobacterien,
die für
die Wurzeltransformation verwendet werden, werden auf LB-Medium (Sambrock
et al., 1989, „Molecular
Cloning", Cold Spring Harbor,
Cold Spring Harbor Laboratory Press, NY), ohne Antibiotica bei 28°C und 200
UpM gezüchtet.
-
Beispiel 9: Samensterilisierung
-
Die
Samen werden in 70% EtOH/0,05% Tween 20 für 1 Minute in ein 2 ml Eppendorfröhrchen gegeben.
70% EtOH/0,05% Tween 20 wird mit einer Pipette entfernt und für 15 Minuten
durch 5% NaOCl/0,05% Tween 20 ersetzt. Die Samen werden regelmäßig geschüttelt. Die
Lösung
wird unter sterilen Bedingungen entfernt und die Samen werden in
sterilem destilliertem Wasser 3 Mal für 10 Minuten jeweils gewaschen.
Bei dem letzten Waschschritt werden die Samen in 0,5–1 ml Wasser
gelassen. Die Samen können
direkt verwendet werden oder für
zwei bis drei Wochen bei 4°C
gelagert werden. Sterilisierte Samen (20–30) werden mit Pinzetten auf
GM-Medium in 15 cm Petrischalen transferiert. Die Sämlinge werden
auf vertikal platzierten Platten in einer Wachstumskammer (22°C; 16/8 Stunden
Licht-/Dunkel-Zyklus) gezüchtet.
-
Beispiel 10: Transformation von Wurzelexplantaten
von Arabidopsis thaliana
-
Wurzeln
der drei Wochen alten Sämlinge
werden in dem Transformationsverfahren verwendet. Die Wurzeln sollten
nicht grün
oder braun sein. Grüne
Teile der Sämlinge
werden mit einem Skalpell und Pinzetten entfernt. Die verbleibenden
Wurzeln werden gesammelt und ungefähr 5 gesamte Wurzelsysteme
werden pro Platte festes CIM-Medium platziert. Die Wurzeln werden
vorsichtig auf die Oberfläche
der Platte gepresst, um einen vollständigen Kontakt mit dem Medium
zu gewährleisten,
aber sie sollten nicht in den Agar getaucht werden. Die Wurzeln
werden für
drei Tage in einer Wachstumskammer (22°C; 16/8 Stunden Licht-/Dunkel-Zyklus) inkubiert.
Die Wurzeln werden dann auf eine sterile Petrischale mit Filterpapier überführt, welches
mit flüssigem
CIM-Medium angefeuchtet ist, und mit einem Skalpell in 0,5–1 cm Stücke zerschnitten.
Die Wurzelexplantate werden dann in ein steriles 50 ml Falconröhrchen überführt, welches
10 ml flüssiges
CIM-Medium enthält. Hierzu
werden 0,5 ml einer über
Nacht gezüchteten
Agrobacterium-Kultur (OD 0,6–1)
hinzugegeben und für 1–2 Minuten
unter leichtem Schütteln
inkubiert. Die Flüssigkeit
wird aus dem Röhrchen
durch sterile Metallrahmen geschüttet
(50-er Netz, Sigma, S-0895), welche mit Pinzetten festgehalten werden.
Die Wurzeln bleiben für
gewöhnlich
an der Wand des Röhrchens,
nahe dem Rand. Dann werden die Wurzelexplantate in eine sterile
Petrischale mit einem Filterpapier überführt und kurz trocken getupft,
um den Überschuss
an Flüssigkeit zu
entfernen. Die Wurzelexplantate werden auf Platten mit festem CIM-Medium
gegeben und für
2 Tage unter gedämpftem
Licht (1,5–2
Klux) in einer Wachstumskammer inkubiert. Leichte Spuren von Überwachsen
mit Agrobacterium sollten nach dem Cokultivierungszeitraum sichtbar
sein. Wurzelexplantate werden dann in sterile 50 ml Falconröhrchen mit
20 ml flüssigem
CIM-Medium überführt, welches
mit 1.000 mg/l Vancomycin ergänzt
ist. Die Falconröhrchen
werden dann leicht gevortext, um die Agrobacterien zu entfernen.
Die Flüssigkeit wird
wie oben erwähnt
aus dem Röhrchen
ausgeschüttet
und die Explantate werden kurz auf Filterpapier trocken getupft.
Die Explantate werden dann auf Platten überführt, die SIM V750 K100 Medium
enthalten. Die Wurzeln sollten mit dem Medium in engem Kontakt sein.
Die Explantate werden in einer Wachstumskammer unter normalen Bedingungen
für 1 Woche
inkubiert und dann auf SIM V500 K100 Medium überführt und für eine weitere Woche inkubiert.
Dann wird die Menge an Vancomycin auf 250 mg/l reduziert. Die ersten
Schösslinge
sollten am Ende der dritten Woche der Kultivierung auf SIM-Medium
auftreten. Die Wurzeln werden abgeschnitten, wenn sie 0,3–0,5 cm
lang sind, und alle restlichen Calli werden entfernt, und die Schösslinge
werden auf 15 cm Platten überführt, die
GM K50-Medium enthalten. Maximal 3 Schösslinge werden auf jede Platte gesetzt.
Um mehr Schösslinge
zu erhalten, können
die verbleibenden Wurzelexplantate auf frische SIM-Platten überführt werden,
die mit 125 mg/l Vancomycin und 100 mg/l Kanamycin ergänzt sind,
für zusätzliche
zwei Wochen. Schösslinge
mit Wurzeln können
in Boden überführt werden
und es kann ihnen ermöglicht
werden, dass sich der Samen festsetzt. Die Schösslinge, die keine Wurzeln
entwickeln, werden in Magentagefäße überführt (einer
pro Gefäß), welches
GM-Medium enthält,
um in vitro Samen zu produzieren.
-
Die
Samen individueller transgener Pflanzen werden auf GM K50-Medium
in einer Wachstumskammer für
2 Wochen keimen gelassen. Phänotypisch
normale Kanamycin-resistente Sämlinge,
die echte grüne Blätter und
ein verzweigtes Wurzelsystem bilden, werden für weitere Analysen ausgewählt.
-
Beispiel 11: Histochemischer β-Glucuronidase
(GUS)-Test
-
In
vitro gezüchtete
Sämlinge
oder Pflanzen, die in Boden gezüchtet
werden, werden in GUS-Tests verwendet. Entweder gesamte Sämlinge oder
herausoperierte Organe werden in eine GUS-Färbelösung getaucht. Die GUS-Färbelösung enthält 1 mM
5-Brom-4-chlor-3-indoylglucuronid (X-Gluc, Duchefa, 20 mM Stock in
DMSO), 100 mM Na-Phosphatpuffer pH 7,0, 10 mM EDTA, pH 8,0, und
0,1% Triton X100. Die Gewebeproben werden bei 37°C für 1–16 Stunden inkubiert. Falls
notwendig können
Proben durch mehrere Waschschritte mit 70% Ethanol geklärt werden,
um Chlorophyll zu entfernen.
-
Beispiel 12: Konstruktion des Tabak-Transformationsvektors
pZU627
-
Alle
PCR-Reaktionen werden mit Platin Pfx DNA-Polymerase (Life Technologies)
nach den Anweisungen des Herstellers durchgeführt.
-
1. NOS-Terminator-Amplifizierung
und Klonierung
-
Der
NOS-Terminator wird aus pBIN19 (Bevan et al. 1984) unter Verwendung
des NOS-Terminator-Vorwärtsprimers
(SEQ ID Nr. 21) und des NOS-Terminator-Umkehrprimers (SEQ ID Nr.
22) amplifiziert. Der Vorwärtsprimer
wird modifiziert, um eine PstI-Schnittstelle zu enthalten, und der
Umkehrprimer wird modifiziert, um eine HindIII-Schnittstelle zu
enthalten. Das NOS-Terminator-Amplifikationsprodukt wird in ein PstI/HindIII-Fragment
an die gleichen Stellen von pBluescript (Stratagene) kloniert, welches
zum Plasmid pZU400A führt.
-
2. Amplifizierung des Tomatenbronzefleckenvirus
Nucleocapsidproteins (TSWV NP)-Gens, Amplifikation und Klonierung
-
Um
ein nicht-translatierbares TSWV NP-Gen zu gewinnen, werden modifizierte
TSWV NP-Genprimer verwendet, um das TSWV NP-Gen zu amplifizieren.
Der TSWV NP-Gen-Vorwärtsprimer
führt BamHI,
eine SphI ein und einen Start- und einen Stopkodon (SEQ ID Nr. 23)
ein. Der Umkehrprimer ist modifiziert, um eine PstI-Schnittstelle (SEQ
ID Nr. 24) einzuschleusen. Das Amplifikationsprodukt wird als BamHI/PstI-Fragment
in die BamHI/PstI-Schnittstelle von pZU400A stromaufwärts von
und in gleicher Richtung wie der NOS-Terminator kloniert. Im resultierenden
Klon pZU400b wird die PstI-Schnittstelle zwischen dem TSWV NP-Gen
und dem NOS-Terminator unter Verwendung einer T4 DNA-Polymerase-Behandlung
entfernt (Life Technologies), indem die Anweisungen des Herstellers
befolgt werden. Vom resultierenden Klon, welcher als pZU400C bezeichnet
wird, werden das TSWV NP-Gen und der NOS-Terminator als ein SphI-/HindIII-Fragment
in die SphI-/HindIII-Schnittstelle des Shuttle-Vektors pZO1560 kloniert,
was zum Plasmid pZU400 führt.
pZO1560 ist ein Derivat von pBluescript, bei dem die ursprüngliche
Multiklonierungsschnittstelle durch die AGLINK-Multiklonierungsstelle ersetzt ist (SEQ
ID Nr. 27).
-
3. CmYLCV-Promotor-Amplifizierung
und Klonierung
-
Der
CmYLCV virale Promotor wird unter Verwendung des CmYLCV-Promotor-Vorwärtsprimers
(SEQ ID Nr. 25) und des CmYLCV-Promotor-Umkehrprimers
(SEQ ID Nr. 26) amplifiziert. Der Vorwärtsprimer wird modifiziert,
um so eine SstI-Schnittstelle
zu enthalten, und der Umkehrprimer wird modifiziert, um eine PstI-Schnittstelle
zu enthalten. Der amplifizierte virale Promotor von CmYLCV wird
als SstI-/PstI-Fragment
in gleiche Richtung wie und stromaufwärts des TSWV-N-Gens, in die
SstI-/PstI-Schnittstelle von pZU400 kloniert. Dies führt zum
Klon pZU625, welcher eine virale Genkassette enthält, die
eine nicht translatierbare TSWV NP-RNA herstellt.
-
4. Klonierung der viralen
Genkassette in pVictorHiNK
-
Die
virale Kassette wird als AscI-/PacI-Fragment aus pZU625 in die AscI-/PacI-Schnittstelle
von pVictorHiNK-Vektor pZU494 kloniert. Dies führt zu Pflanzentransformations-Vektor
pZU627. pVictorHiNK ist ein binärer
Vektor, welcher einen Replikationsursprung (ORI) umfasst, der aus
Pseudomonas aeruginosa-Plasmid pVS1 stammt, welches dafür bekannt
ist, in A. tumefaciens sehr stabil zu sein (Itoh et al., 1984. Plasmid
11: 206-220; Itoh and Haas, 1985. Gene 36; 27-36). Der pVS1 ORI
ist nur in Agrobacterium funktional und kann durch das Helferplasmid
pRK2013 von E. coli in A. tumefaciens mit Hilfe einer triparenteralen
Kreuzungsprozedur mobilisiert werden (Ditta et al., 1980. Proc.
Natl. Acad. Sci. USA 77: 7347-7351). Diese binäre Vektor umfasst einen ColEI-Replikationsursprung,
welche in E. coli funktional ist und aus pUC19 (Yannisch-Perron
et al., 1985. Gene 33: 103-119) stammt. Für die Bewahrung in E. coli
und A. tumefaciens enthält
dieser Vektor ein bakterielles Resistenzgen gegenüber Spectomycin
und Streptomycin, welche durch ein 0,93 kb-Gen des Transposons Tn7
(Fling et al., 1985. Nucl. Acid Res. 13: 7095) kodiert wird, welches
als Selektionsmarker für die
Bewahrung des Vektors in E. coli und A. tumefaciens dient. Das Gen
wird für
effiziente bakterielle Expression mit dem tac-Promotor fusioniert
(Amman et al., 1983. Gene 25: 167-178). Die rechten und linken T-DNA Grenzfragmente
von 1,9 kb und 0,9 kb, die jeweils die 24 bp Grenzwiederholungen
umfassen, stammen aus dem Ti-Plasmid des Nopalin-artigen A. tumefaciens-Stamms
pTil37 (Yadev et al., 1982, Proc. Natl. Acad. Sci. USA. 79: 6322-6326).
Anschließend
wird die T-DNA- Region
zwischen den Grenzen durch Deletieren der von M13 stammenden Sequenzen
modifiziert, und um die Klonierungs-Flexibilität zu verbessern, wird die BIGLINK-Multiklonierungsstelle
(SEQ ID Nr. 28) eingeschleust, welche zu pVictorHiNK führt.
-
PvictorHiNK
enthält
eine NPTII-Genkassette zur Selektion von Transformanten während des
Pflanzen-Transformationsprozesses. Diese Genkassette enthält einen
NOS-Promotor, das NPTII-Gen und den NOS-Terminator, welcher aus
A. tumefaciens gewonnen ist.
-
Beispiel 13: Pflanzentransformation und
Durchmusterung der Resistenz
-
1. Transformation der binären Vektoren
in Pflanzenmaterial
-
Verfahren
zum Transfer binärer
Vektoren in Pflanzenmaterial sind gut etabliert und einem Fachmann auf
dem Gebiet bekannt. Variationen in den Verfahren sind vorhanden,
beispielsweise aufgrund der Unterschiede der verwendeten Agrobacterienstämme, der
verschiedenen Quellen des Explantatmaterials, der Unterschiede der
Regenerationssysteme, die davon abhängen und aufgrund des Cultivars
der verwendeten Pflanzenspezies. Der binäre Vektor pZU627 wird in Pflanzentransformationsexperimenten
gemäß den folgenden
Verfahren verwendet. pZU627 wird durch triparenterales Kreuzen auf
einen Acceptor-Agrobacterium tumefaciens-Stamm überführt, gefolgt von einer Southern-Blot-Analyse der Ex-Conjuganten
zur Überprüfung des
richtigen Transfers des Konstrukts in den Acceptorstamm, die Inoculierung
und die Co-Kultivierung von axenischem Explantatmaterial mit dem
rekombinanten Agrobacterium tumefaciens-Stamm, dem selektiven Abtöten des
Agrobacterium tumefaciens-Stamms unter Verwendung der geeigneten
Antibiotika, die Auswahl transformierter Zellen durch Züchten auf
Selektionsmedien, die Kanamycin enthalten, den Transfer von Pflänzchen in
den Boden, das Untersuchen hinsichtlich der Unversehrtheit der integrierten
T-DNA durch Southern-Blot-Analyse
an isolierter chromosomaler DNA der Pflanze.
-
2. Resistenz von Pflanzen
gegen TSWV-Infektionen
-
Transformierte
Pflanzen werden im Treibhaus unter Standard-Quarantäne-Bedingungen gezüchtet, um
jegliche Infektion durch Pathogene zu verhindern. Im Vier-Blatt-Stadium
werden die Pflanzen mit TSWV durch mechanische Inokulation infiziert.
Gewebe aus Pflanzen, die systemisch mit TSWV infiziert sind, werden in
5 Volumen eiskaltem Inokulationspuffer gemahlen (10 mM Phosphatpuffer
ergänzt
durch 1% Na2SO3)
und in Gegenwart von Carborundumpulver auf die ersten zwei voll
ausgedehnten neuen Blätter
gerieben. Die inokulierten Pflanzen werden auf die Entwicklung von
Symptomen während
der drei Wochen nach der Inokulation überwacht. Pflanzen, die pZU627-Sequenzen
enthalten, zeigen keine TSWV-Symptome, während nicht transformierte
Kontrollpflanzen innerhalb von 7 Tagen nach Inokulation schwere
systemische TSWV-Symptome zeigen. Resistente Pflanzen werden selbst
bestäubt
und die Samen geerntet. Die Pflanzennachkommen werden hinsichtlich
der Segregierung des inserierten Gens analysiert und anschließend hinsichtlich
der Resistenz gegen TSWV-Infektion, wie oben beschrieben, erneut
durchmustert.
-
Beispiel 14: Konstruktion von CmpS-Phosphomannoseisomerase-nos-Konstrukten für die Pflanzentransformation
-
Der
CmpS-Promotor wird PCR-amplifiziert, indem die Cmpf-B (cgc gga tcc
tgg cag aca aag tgg cag a; SEQ ID Nr. 29) und CmpS-B (cgc gga tcc
tac ttc tag gct act tg, SEQ ID Nr. 30) Primer mit flankierendem
BamHI-Schnittstellen verwendet werden. Das erhaltene PCR-Fragment
wird in die BamHI-Schnittstelle
des pBluescript KS (+) kloniert, um pNOV4211 zu bilden.
-
Um
einen binären
Vektor für
die Transformation über
Agrobacterium tumefaciens zu kreieren, wird der CmpS-Promotor aus
pNOV4211 unter Verwendung von BamHI ausgeschnitten und stromaufwärts des PMI-Gens
in ein mit BamHI verdautes pNOV2804 (SEQ ID Nr. 31) inseriert, und
der erhaltene Vektor wird als pNOV2819 bezeichnet. pNOV2804 (SEQ
ID Nr. 31) ist ein binärer
Vektor mit einem VS1-Replikationsursprung, einer Kopie des Agrobacterium
virG-Gens im Hintergrund und einer promotorlosen PMI-Nos-Terminator
Expressionskassette, welche zwischen den linken und rechten Grenzen
der T-DNA liegt. PMI (Phosphomannosisomerase) ist die kodierende
Region des E. coli manA-Gens (Joersbo and Okkels, 1996, Plant Cell
Reports 16: 219-221, Negrotto et al., 2000, Plant Cell Reports 19:
798-803). Der Nos (Nopalinsynthase)-Terminator wird aus Agrobacterium
tumefaciens T-DNA (Depicker et al., 1982, J. Mol. Appl. Genet. 1
(6), 561-573) gewonnen. Die Phosphomannoseisomerase kodierende Region
und der Nos-Terminator
sind in den Nt 290 bis Nt 1465 und Nt 1516 bis Nt 1789 von pNOV2804
(SEQ ID Nr. 31) lokalisiert. Um einen Vektor für die biolistische Transformation
zu erzeugen, wird der CmpS-Promotor aus pNOV4211 unter Verwendung
von BamHI ausgeschnitten und in ein mit BamHI verdautes pNOV3604
(SEQ ID Nr. 32) inseriert, um pNOV2820 zu bilden, wodurch eine PMI-Epressionskassette
erzeugt wird, die durch den CmpS-Promotor
angetrieben wird. pNOV3604 ist ein Vektor zum Herstellen biolistischer
Fragmente und beruht auf pUC19 mit einem ColEI-Replikationsursprung
und einem beta-Lactamasegen, welches eine Ampicillin-Resistenz verleiht.
Wie pNOV2804, enthält auch
pNOV3604 eine promotorlose PMI-Nos-Terminator-Expressionskassette (siehe oben). Die
Phosphomannoseisomerase kodierende Region und der Nos-Terminator
sind in den Nt 42 bis Nt 1217 und Nt 1268 bis Nt 1541 von pNOV3604
(SEQ ID Nr. 32) lokalisiert.
-
Der
binäre
Vektor pNOV2819 wird für
die Transformation über
Agrobacterium tumefaciens verwendet, und der Vektor pNOV2820 wird
für die
biolistische Transformation verwendet.
-
Beispiel 15: Konstruktion von CmpC-GUS-Plasmid
für die
stabile Expression in planta.
-
1. Konstruktion des binären Vektors
-
Der
Vektor pCambia 1302 (Cambia, Canberra, Australia; Roberts, C. S.,
Rajagopal, S., Smith, L., Nguyen, T., Ynag, W., Nugroho, S., Ravi,
K. S. Cao, M. L., Vijayachandra, K., et al. A comprehensive set
of modular vectors for advanced manipulation and efficient transformation
plants. Presented at the Rockefeller Foundation Meeting of the International
Program an Rice Biotechnology, Malacca, Malaysia, 15.-19. September
1997) wird ausgewählt
und für
die Experimente modifiziert: Der CaMV 35S-Promotor vor dem GFP-Gen wird
durch Verdau der Position 9788 mit HindIII-Endonuclease entfernt
und an der Position 1 durch NcoI-Endonuclease und der Vektor wird
an den zwei kompatiblen Enden relegiert. Der Verdau mit XhoI-Endonuclease an
den Positionen 7613 und BstXI an der Position 9494 schneiden das
Hygromycin-Gen und den CaMV 35S-Promotor
davor aus. Die 35S-Promotorsequenzen werden aus dem Vektor eliminiert,
um jedes Risiko einer auf Homologie beruhenden Genabschaltung auszuschließen. Das
bar-Gen, welches durch den 1'-Promotor
angetrieben wird (Mengiste et al., Plant J, 1997, 12 (4): 945-948)
wird in die EcoRI-Schnittstelle
an Position 9737 als Selektionsmarker eingeschleust. Der erhaltene
Vektor wird als pCamBar bezeichnet.
-
2. Hergestellte Konstrukte
-
Die
Kassetten CmpCG und CapG, die in Beispiel 3 beschrieben sind, werden
in den pCamBar-Vektor zwischen der XbaI-Schnittstelle an Position 9764 und der
EcoRI-Schnittstelle an Position 9737 inseriert, um pCamBarCmpCG
zu erhalten bzw. das pCamBarCapG-Plasmid. Die zwei Konstrukte werden
verwendet, um Agrobacterium tumefaciens-Zellen zu transformieren.
-
Beispiel 16: Stabile Expression in Arabidopsis
thaliana
-
1. Pflanzenproduktion und Transfektion
-
Arabidopsis
thaliana (Columbia 0) wt-Samen werden ausgesät, für 3 Tage bei 4°C in der
Dunkelheit Kälte-behandelt,
um die Keimung zu synchronisieren und in den Zuchtraum überführt (22°C/24 h Licht).
Gekeimte Pflanzen werden infiltriert, wenn sie eine maximale Anzahl
an nicht geöffneten
Knospen produziert haben. Die Infiltration wird gemäß dem Protokoll
durchgeführt,
das beschrieben ist in Clough and Bent, Plant J. 16: 735-743, 1987.
-
2. Auswahl der transgenen
Pflanzen
-
Sämlinge,
die aus Samen der T1-Generation gewonnen werden, werden ausgewählt, indem
sie mit dem BASTA-Herbizid (Plüss-Staufer AG/SA) (150
mg/l) fünf
Tage dreimal besprüht
werden. Zwanzig pCamBarCmpCG und neun pCamBarCapG-resistente Pflanzen
werden nach Selektion eingesammelt. Sie werden unter den beschriebenen
Bedingungen gezüchtet,
um die Samen der T2-Generation zu gewinnen. Diese Samen werden dem
gleichen Verfahren unterzogen, wie oben für die wt beschrieben wurde,
und die T2-Sämlinge werden
durch BASTA-Besprühen
ausgewählt,
und die resistenten Mutanten werden hinsichtlich der GUS-Genexpression analysiert.
-
3. Histochemische GUS-Expression
in stabil transformierten Arabidopsis-Pflanzen
-
Histochemische
GUS-Färbung
wird wie im Beispiel 11 beschrieben, durchgeführt. Die Analyse wird in 15
ml Kultur-Röhrchen durch
Tränken
der transformierten Sämlinge
in X-gluc-Lösung,
die Ausübung
von einem Druck von 130 mBar für
10 min. und Inkubation bei 37°C über Nacht
durchgeführt.
Die Ergebnisse, die mit Arabidopsis-Pflanzen gewonnen werden, die mit
dem pCamBarCmpCG-Promotor transformiert sind, weisen eine konstitutive
Expression des GUS-Reportergens an, welches vom CmpC-Promotor in
allen vegetativen Organen angetrieben wird.
-
4. Quantitative enzymatische GUS-Tests
in stabil transformierten Arabidopsispflanzen
-
A. Probenpräparierung
-
Vier
Pflanzen der transgenen Linie und ein Blatt jeder dieser Pflanzen
werden für
den Test ausgewählt. Die
vier Blätter
werden im gleichen Eppendorfröhrchen
eingesammelt, mit flüssigem
Stickstoff eingefroren und in ein feines Pulver mit einmal Mörsern gemahlen.
Dieses wird mit 150 μl
GUS-Puffer (0,05 M NaPO4, pH 7,0, 0,01 M
EDTA, 0,1% Triton-X-100, 0,1% Sarkosyl), 5 min. bei 37°C inkubiert
und in einer Microzentrifuge für
5 min. bei maximaler Geschwindigkeit heruntergeschleudert. Der Überstand
wird in ein neues Eppendorf-Röhrchen überführt und
diese Proben werden für
den Enzymtest verwendet. Das Protein in diesen Pflanzenextrakten
wird gemäß dem Bradford-Verfahren
(Bradford, M. M. Anal. Biochem. 72: 248-254, 1976) unter Verwendung
von BSA als Standard abgeschätzt.
Das Vorhandensein des Promotors und des GUS-Gens in den mutierten
Linien wird mittels PCR-Analyse unter Verwendung der extrahierten
Proben als Matrize durchgeführt.
-
B. Fluorometrischer GUS-Test
-
sDer
fluorometrische GUS-Test wird wie in Beispiel 5 beschrieben durchgeführt. Zur
Detektion der GUS-Genexpression werden Proben in GUS-Puffer verdünnt. Die
Reaktion wird in Gegenwart des gleichen Volumens an GUS-Reaktionspuffer
bei 37°C
durchgeführt
und mit 2 M Ammediol gestoppt. Die Aktivität wird in einem Titertek Fluoroskan
II gemessen.
-
Fünfzehn von
zwanzig ausgewählten
Linien, die mit pCamBarCmpCG transformiert sind und acht von neun
Linien, die mit pCamBarCapG transformiert sind, zeigen GUS-Enzymaktivität in den
Blättern.
Das Niveau der Aktivität
in mehreren CmpCG-transformierten
Linien ist vergleichbar mit CapG. Jedoch sind die Ergebnisse bei
verschiedenen transgenen Linien aufgrund der Verschiedenheit ihrer
Genotypen variabel (Anzahl an Loci und Anzahl an transgenen Insertionen
pro Locus).
-
Beispiel 17: Konstruktion von CmpC-Promotorvarianten
-
Alle
PCR-Reaktionen werden mit Pfu-Polymerase (Stratagene), wie in Beispiel
1 beschrieben, durchgeführt.
-
1. DNA-Amplifizierung und Klonierung.
-
Zwei
Varianten, pCmpMG und pCmpMsG des pCmpCG-Plasmid (Beispiel 3) werden
hergestellt. Die zuerst genannte mit deletierter Sequenz „GTGGTCCC" in dem CmpC-Promotor
und letztere ist mutiert und hat die Sequenz „GTGCTCGC".
-
Um
pCmpMG zu gewinnen, werden drei PCR-Produkte amplifiziert:
CmpM1
unter Verwendung des Cmp1-Vorwärtsprimers
(siehe Beispiel 3, SEQ ID Nr. 13), der CmpMr-Umkehrprimer (CCATCGTGGTATTTGGTATTG,
SEQ ID Nr. 33) und das pCmpCG-Plasmid als Matrize.
-
CmpM2
unter Verwendung des CmpMf-Vorwärtsprimers
(CAATACCAAATACCACGATGG; SEQ ID Nr. 34), des CmpC2-Umkehrprimers (siehe
Beispiel 3; SEQ ID Nr. 14) und von pCmpCG-Plasmid als Matrize.
-
CmpM
unter Verwendung des Cmp1-Vorwärtsprimers,
des CmpC2-Umkehrprimers
und einer äquimolaren
Mischung von CmpM1 und CmpM2 PCR-Produkten als Mischung.
-
Um
pCmpMsG zu gewinnen, werden drei PCR-Produkte amplifiziert:
CmpMs1
unter Verwendung des Cmp1-Vorwärtsprimers,
des CmpMrs-Umkehrprimers
(CGTGGTAGCGAGCACTTTGGT, SEQ ID Nr. 35) und des pCmpCG-Plasmids als
Matrize.
-
CmpMs2
unter Verwendung von CmpMfs-Vorwärtsprimer
(AAGTGCTCGCTACCACGATGG, SEQ ID Nr. 36), des Cmp2-Umkehrprimers und des pCmpCG-Plasmids
als Matrize.
-
CmpsM
unter Verwendung des Cmp1-Vorwärtsprimers,
des Cmp2-Umkehrprimers
und einer äquimolaren
Mischung von CmpMs1 und CmpMs2-PCR-Produkten als Matrize.
-
Um
die pCmpMG- und die pCmpMsG-Plasmide zu gewinnen, wird das ursprüngliche
pCmpCG-Plasmid mit XbaI- und BamHI-Endonucleasen geschnitten, um das CmpC-Fragment
zu eliminieren, und um an dessen Stelle CmpM und die CmpMs PCR-Produkte zu inserieren.
-
Beispiel 18: Transiente Expressionsexperimente
mit pCmpM- und pCmpMs-Konstrukten
-
1. Suspensionskulturen und
Protoplastenpräparation
-
Suspensionskulturen
und Protoplasten werden wie im Beispiel 4 beschrieben, hergestellt.
-
2. GUS-Tests
-
Die
Transfektion und Proteinextrakt-Präparierung wird wie in Beispiel
4 beschrieben, durchgeführt, und
der GUS-Test wird wie in Beispiel 5 beschrieben, durchgeführt.
-
Die
GUS-Testergebnisse werden aus dem Durchschnitt von zehn verschiedenen
Experimenten gewonnen und mit dem Standard- Klonwert normalisiert. Die Deletion
der „GTGGTCCC"-Sequenz im CmpCG-Konstrukt
reduziert die Expression des GUS-Reportergens um ungefähr 50% der
GUS-Reportergenexpression von CmpC in allen Protoplastenspezies
(siehe unten). Die Wirkung, die durch die „GTGCTCGC"-Mutation eingeführt wird, hängt von der Pflanzenspezies
ab. In O. violaceus- und O. sativa-Protoplasten ist das Niveau der GUS-Genexpression
auf 65,2% bzw. 73,6% gesenkt. In N. plumbaginifolia-Protoplasten
ist die CmpMs-Promotoraktivität ähnlich der
des CmpM-Promotors. N. plumbaginifolia
| CmpCG | 100 |
| CmpMG | 52
+/– 12% |
| CmpMsG | 53,7
+/– 16% |
| CapG | 28,3
+/– 14% |
O. violaceus
| CmpCG | 100 |
| CmpMG | 55,2
+/– 12% |
| CmpMsG | 73,6
+/– 10% |
| CapG | 5
+/– 1% |
O. sativa
| CmpCG | 100 |
| CmpMG | 59,6
+/– 21% |
| CmpMsG | 65,2
+/– 20% |
| CapG | 42,5
+/– 16% |
-
Beispiel 19: Konstruktion von Pflanzen-Transformationsvektoren,
die 5'-Promotorfragmente
enthalten, die operativ mit GFP oder GUS-Reportergenen verbunden
sind
-
1. Promotoramplifikation und Klonierung
-
Ein
chimäres
Gen wird konstruiert, das eine DNA-Sequenz vom CmYLCV Transkriptpromotor
voller Länge
(SEQ ID Nr. 1) einschließt,
welches mit dem Pflanzen-optimierten GFP mit Intron (synGFPI, SEQ
ID Nr. 37) oder dem GUS-Reportergen mit Intron (GIG; SEQ ID Nr.
38)-Sequenz fusiert ist. Das GIG-Gen enthält das ST-LS1-Intron aus Solanum
tuberosum in den Nucleotiden 385 bis Nucleotid 576 der SEQ ID Nr.
38 (erhalten von Dr. Stanton Gelvin, Purdue University, und beschrieben
in Narasimhulu et al., 1996, Plant Cell, 8: 873-886). SynGFPI ist
ein Pflanzen-optimiertes GFP-Gen, in das das ST-LS1-Intron eingeschleust
ist (Nt 278 bis Nt 465 der SEQ ID Nr. 37). Bezüglich der Promotoren wird CmYLCV
Genom-DNA als Matrize für
die Polymerase-Kettenreaktion (PCR) verwendet. Genspezifische Primer
werden entworfen, um die DNA-Sequenz zu amplifizieren. Ein Genfragment,
das CmpC entspricht (SEQ ID Nr. 2) wird unter Verwendung des Cmpf-B
Vorwärtsprimers
(SEQ ID Nr. 29) in Kombination mit dem 5'- bis 3'-Primer CGCGGATTGCTCCCTTAACAATGAGG (SEQ ID
Nr. 39) als Umkehrprimer isoliert. Ein Genfragment, das dem CmpS
(SEQ ID Nr. 3) entspricht, wird unter Verwendung des Cmpf-B (SEQ
ID Nr. 29)-Vorwärtsprimers
in Kombination mit dem CmpS-B Umkehrprimer (SEQ ID Nr. 30) isoliert.
Alle drei Primer enthalten die BamHI-Restriktionsenzym-Erkennungsstelle
CGCGGA an ihrem 5'-Ende.
Alle PCR-Reaktionen werden mit Pfu-Polymerase nach Empfehlungen
der Hersteller (Stratagene) durchgeführt. Ein Thermocycler (DNA
Engine, MJResearch, Inc. Waltham, MA USA) wird verwendet, um die
Promotorfragmente unter Verwendung der folgenden PCR-Bedingungen
zu amplifizieren [(94°C:10 s):(94°C:10 s/56°C:30 s/72°C:1 min) × 20]:(72°C:1,5 m).
-
Nach
dem Restriktionsverdau mit BamHI werden die Promotorfragmente in
die auf pUC basierenden Vektoren mit dem GIG-Gen oder mit synGFPI-Gen
ligiert, um die Promotor-Reporter-Genfusionen
zu erzeugen, die operativ mit einem Nos-Terminator verbunden sind.
-
2. Hergestellte Promotor-Reporterkassetten
-
- – CmpS-Promotorfragment
+ synGFPI-Gen + nos (SEQ ID Nr. 40)
- – CmpS-Promotorfragment
+ GIG-Gen + nos (SEQ ID Nr. 41)
- – CmpC-Promotorfragment
+ synGFPI-Gen + nos (SEQ ID Nr. 42)
- – CmpC-Promotorfragment
+ GIG-Gen + nos (SEQ ID Nr. 43)
-
| SEQ ID Nr. |
Promotor |
Gen (synGFPI
oderG) GIG) |
Terminator |
| 40 |
Nt 1 bis
Nt 402 |
Nt 411
bis Nt 1331 |
Nt 1343
bis Nt 1618 |
| 41 |
Nt 1 bis
Nt 405 |
Nt 425
bis Nt 2425 |
Nt 2479
bis Nt 2751 |
| 42 |
Nt 1 bis
Nt 354 |
Nt 380
bis Nt 1292 |
Nt 1304
bis Nt 1577 |
| 43 |
Nt 1 bis
Nt 354 |
Nt 399
bis Nt 2399 |
Nt 2453
bis Nt 2725 |
-
3. Subklonierung in einen Agrobacterium-Binärvektor
-
Die
Promotor-Reporterkassetten (SEQ ID Nr. 40 bsi 43), die das Promotorfragment,
das Reportergen und den Nos-Terminator enthalten, werden in den
binären
Vektor pNOV2117 für
Mais und den binären
Vektor pNOV4200 für
die Tomatentransformation inseriert. pNOV2117 (SEQ ID Nr. 44) ist
ein binärer
Vektor mit einem VS1-Replikationsursprung, einer Kopie des Agrobacterium
virG-Gens im Hintergrund, und einer Mais-Ubiquitin-Promotor-PMI-Gen Nos-Terminator
Expressionskassette, zwischen den linken und rechten Grenzen der T-DNA.
PMI (Phosphomannoseisomerase) liegt die kodierende Region des E.
coli manA-Gens (Joersbo and Okkels, 1996, Plant Cell Reports 16:
219-221, Negrotto et al., 2000, Plant Cell Reports 19: 798-803).
Der Nos (Nopalinsynthase)-Terminator wird aus Agrobacterium tumefaciens
T-DNA (Depicker et al., 1982, J. Mol. Appl. Genet. 1 (6), 561-573)
gewonnen. Der Mais-Upiquitin-Promotor, die Phosphomannoseisomerase
kodierende Region und der Nos-Terminator liegen bei Nt 31 bis Nt
2012, Nt 2109 bis Nt 3212 bzw. Nt 3273 bis 3521 von pNOV2117 (SEQ
ID Nr. 44). Die Reporter-Promotorkassetten werden nahe dem rechten
Rand inseriert. Die selektierbare Marker-Expressionskassette in
den binären
Vektoren liegt am nächsten
am linken Rand. Für
die Tomatentransformation wird pNOV4200 SEQ ID Nr. 45) als binärer Vektor
verwendet, welcher den VS1-Replikationsursprung enthält, eine
Kopie des Agrobacterium virG-Gens im Hintergrund, und eine Hygromycin-selektierbare Markerkassette
zwischen den linken und rechten Randsequenzen. Die Hygromycin-selektierbare Markerkassette
umfasst den Arabidopsis-Ubiquitin 3-Promotor (Ubq3(At), Callis et
al., J. Biol. Chem. 265: 12486-12493 (1990)), welcher operativ mit
dem Gen verbunden ist, welches die Hygromycin-Resistenz kodiert (hier
bezeichnet als "HYG", synthetisches Hygromycin
B Phosphotransferasegen aus E. coli, Patent:
JP 1986085192-A1 30.
April 1986) und den Nos-Terminator
(Depicker et al., J. Mol. Appl. Genet. 1 (6), 561-573 (1982). Der
Arabidopsis-Ubiquitin-Promotor, das HYG-Gen und der Nos-Terminator liegen an
Nt 162 bis Nt 11494, Nt 1897 bis NT 2939 bzw. Nt 2939 bis Nt 3236
von pNOV4200 (SEQ ID Nr. 45). Die Reporter-Promotorkassetten (SEQ
ID Nr. 40 bis 43) werden nahe dem rechten Rand inseriert. Die selektierbare
Marker-Expressionskassette in den binären Vektoren ist nahe dem linken
Rand.
-
Unten
gezeigt sind die Richtungen des selektierbaren Markers und der Promtor-Reporterkassetten
in den binären
Vektorkonstrukten.
-
4. Binäre
Vektorkonstrukte:
-
- – NOV
4215 (RB CmpS Promotorfragment + synGFPI-Gen + Nos – ZmUbi
+ PMI-Gen + Nos LB)
- – NOV
4216 (RB Nos + synGFPI-Gen + SmpS Promotorfragment – ZmUbi
+ PMI-Gen + Nos LB)
- – NOV
4217 (RB CmpS Promotorfragment + synGFPI-Gen + Nos – Ubq3(At)
+ HYG-Gen + Nos LB)
- – NOV
4220 (RB CmpS Promotorfragment + GIG-Gen + Nos – Ubq3(At) + HYG-Gen + Nos
LB)
- – NOV
4224 (RB CmpS Promotorfragment + GIG-Gen + Nos – ZmUbi + HYG-Gen + Nos LB)
- – NOV
4226 (RB CmpS Promotorfragment + synGFPI-Gen + Nos – ZmUbi
+ PMI-Gen + Nos LB)
-
Zusätzliche
Kassetten werden mit bekannten Promotoren hergestellt, die zum Vergleich
verwendet werden. Hierfür
wird eine Promotorkassette ZmUbi-GFP-35S Term (SEQ ID Nr. 46), welche
den Mais-Ubiquitin-Promotor (Christensen et al. Plant Molec. Biol.
12: 619-632 (1989)) operativ verbunden mit synGFP und dem 35S-Terminator
(35S Term) umfasst, hergestellt und eine Promotorkassette, die den
Mais-Ubiquitin-Promotor umfasst, der operativ mit dem GIG-Gen und
dem Nos-Terminator verbunden ist (NOV 3612, SEQ ID Nr. 47), wird
ebenfalls hergestellt. Der ZmUbi-Promotor, das GFP-Gen und der 35S-Terminator
liegen bei Nt 1 bis Nt 2019, Nt 2020 bis Nt 2751 bzw. Nt 2876 bis
Nt 2949 der SEQ ID Nr. 46. Der ZmUbi-Promotor, das GIG-Gen und der
Nos-Terminator liegen an Nt 1 bis Nt 1982, Nt 2015 bis Nt 4015 bzw.
Nt 4069 bis Nt 4341 der SEQ ID Nr. 47. Die ZmUbi-GIG-Nos-Kassette
(SEQ ID Nr. 47) wird in einen auf pUC basierenden Vektor kloniert.
Die ZmUbi-GFP-Nos-Kassette (SEQ ID Nr. 46) wird in den binären Vektor
pNOV2117 für
die Transformation von Mais, der oben beschrieben wurde, kloniert.
Promotorkassetten, die den Arabidopsis-Ubiquitin-3-Promotor (Ubq3(At)),
operativ verbunden mit dem synGFPI und dem Nos-Terminator (SEQ ID
Nr. 48) und den Arabidopsis-Ubiquitin-3-Promotor operativ mit dem
GIG-Gen und dem Nos-Terminator (SEQ ID Nr. 49) umfassen, werden
hergestellt. Der Ubiquitin-3-Promotor, synGFPI und der Nos-Terminator
sind lokalisiert bei Nt 1 bis Nt 1332, Nt 1738 bis Nt 2658 bzw.
Nt 2670 bis Nt 2943 der SEQ ID Nr. 48. Der Ubiquitin-3-Promotor,
das GIG-Gen und der Nos-Terminator sind von Nt 1 bis Nt 1335, Nt
1746 bis Nt 3746 bzw. Nt 3800 bis Nt 4072 der SEQ ID Nr. 49 zu finden.
Wie oben beschrieben wurde, werden die Promotorkassetten in den
binären
Vektor pNOV4200 kloniert, welcher eine durch Hygromycin selektierbaren
Markerkassette für
die Transformation von Tomaten enthält. Unten sind die Richtungen
des selektierbaren Markers und der Promotor-Reporterkassetten in
den binären
Vektorkonstrukten gezeigt.
-
5. Binäre
Vektorkonstrukte
-
- – NOV2110
(RB ZmUbi-Promotor + synGFP-Gen + 35S Term – ZmUbi + PMI-Gen + Nos LB)
- – NOV4209
(RB Ubq3(At)-Promotor + synGFPI-Gen + Nos – Ubq3(At) + HYG-Gen + Nos
LB)
- – NOV4208
(RB Ubq3(At)-Promotor + GUS-Intron – GUS-Gen + Nos – Ubq3(At)
+ HYG-Gen + Nos LB)
-
Beispiel 20: Agrobacterium-vermittelte
Transformation von Mais
-
Die
Transformation unreifer Maisembryonen wird im Wesentlichen, wie
in Negrotto et al. (2000) Plant Cell Reports 19: 798-803 beschrieben,
durchgeführt.
In diesem Beispiel sind alle Medienbestandteile, wie in Negrotto
et al., siehe oben, beschrieben. Jedoch können verschiedene Medienbestandteile,
die in der Literatur beschrieben sind, substituiert werden.
-
1. Transformationsplasmide
und Selektionsmarker
-
Die
Gene, die für
die Transformation verwendet werden, werden in einen Vektor kloniert,
der für
die Transformation von Mais geeignet ist, wie in Beispiel 19 beschrieben.
Vektoren, die verwendet werden, enthalten das Phosphomannoseisomerase
(PMI)-Gen (Negrotto et al. (2000) Plant Cell Reports 19: 798-803).
-
2. Herstellung von Agrobacterium
tumefaciens
-
Der
Agrobacteriumstamm LBA4404 (pSB1), welcher das Pflanzentransformationsplasmid
enthält, wird
auf YEP (Hefeextrakt (5 g/l), Pepton (10 g/l), NaCl (5 g/l), 15
g/l Agar, pH 6,8) Festmedium für
2 bis 4 Tage bei 28°C
gezüchtet.
Ungefähr
0,8 × 109 Agrobacterien werden in LS-inf Medium,
das mit 100 μM
Acetosyringon (As) (Negrotto et al., (2000) Plant Cell Rep 19: 798-803)
ergänzt
ist, suspendiert. Die Bakterien werden in diesem Medium für 30–60 Minuten
prä-induziert.
-
3. Inokulierung
-
Unreife
Embryonen von A188 oder anderen geeigneten Mais-Gentypen werden aus den 8–12 Tage alten
Kolben in Flüssig-LS-inf + 100 μM As ausgeschnitten.
Die Embryonen werden einmal mit frischem Infektionsmedium gespült. Die
Agrobacteriumlösung
wird dann zugefügt
und die Embryonen werden für
30 Sekunden gevortext und es wird ihnen ermöglicht, sich für 5 Minuten
mit den Bakterien zu setzen. Die Embryonen werden dann mit der Scutellum-Seite
aufwärts
auf LSAs-Medium überführt und
in der Dunkelheit für
zwei bis drei Tage kultiviert. Anschließend werden zwischen 20 und
25 Embryonen pro Petrischale auf LSDc-Medium überführt, welches mit Cefotaxim
(250 mg/l) und Silbernitrat (1,6 mg/l) ergänzt ist und in der Dunkelheit
für 10 Tage
bei 28°C
kultiviert.
-
4. Selektion der tranfsformierten
Zellen und Regenerierung transformierter Pflanzen
-
Unreife
Embryonen, die embryogene Calli herstellen, werden auf LDS1MO.5S-Medium überführt. Die Kulturen
werden auf diesem Medium für
6 Wochen mit einem Subkulturschritt nach 3 Wochen ausselektioniert. Überlebende
Calli werden entweder auf LSD1MO.5S-Medium oder in Reg1-Medium überführt, um
aufgestockt zu werden. Nach der Kultivierung im Licht (16 Stunden
Licht/8 Stunden Dunkel-Rhythmus), werden die grünen Gewebe dann auf Reg2-Medium
ohne Wachstumsregulatoren überführt und
für 1–2 Wochen
inkubiert. Die Pflänzchen
werden in Magenta GA-7-Kästen überführt (Magenta
Corp, Chicago III.), welche Reg3-Medium enthalten, und im Licht
gezüchtet.
Pflanzen, die PCR-positiv für
die Promotor-Reporterkassette sind, werden in den Boden überführt und
im Treibhaus gezüchtet.
-
Beispiel 21: Stabile Transformation von
Tomaten
-
Die
Transformation von Tomaten wird im Wesentlichen wie in Meissner
et al., The Plant Journal 12: 1465-1472, 1997, durchgeführt.
-
1. Transformationsplasmide und Selektionsmarker
-
Die
Gene, die für
die Transformation verwendet werden, werden in den binären Vektor
kloniert, der für die
Tomatentransformation geeignet ist, wie in Beispiel 19 beschrieben.
Vektoren enthalten das Hygromycin-Resistenzgen zur Selektion.
-
2. Herstellung von Agrobacterium
tumefaciens
-
Agrobacterium-Stamm
GV3101, welcher das Pflanzentransformations-Plasmid enthält, wird
auf LB-(Hefeextrakt
(5 g/l), Pepton (10 g/l), NaCl (5 g/l), 15 g/l Agar, pH 6,8) Flüssigmedium
für 2 Tage
bei 22°C gezüchtet. Ungefähr 0,8 × 109 Agrobacterien werden in KCMS Inoculationsmedium
(MS-Salze (4,3 g/l), Myo-Inositol, Thiamin (1,3 μg/ml), 2,4-D (0,2 μg/ml), Kinetin
0,1 μg/ml
und 3% Saccharose, pH 5,5), welches mit 100 μM Acetocyringon ergänzt ist,
suspendiert. Die Bakterien werden in diesem Medium für 60 Minuten prä-induziert.
-
3. Sterilisierung von Samen
und Keimung
-
Die
Samen des Micro-Tom-Cultivars der Tomate (Meissner et al., The Plant
Journal 12: 1465-1472, 1997) und der ZTV-840-Cultivar werden mit einer 20% Bleichelösung mit
Tween für
20 min. getränkt.
Die Samen werden dann dreimal mit sterilem Wasser gewaschen und
auf TSG-Medium ausplattiert (1/2 MS-Salze, 1% Saccharose, 1% PhytoAgar,
pH 5,8) in Magentakästen
(Magenta Corp, Chicago III) zur Keimung. die Cotyledonen werden
ausgeschnitten und die Cotyledonenpetiolen werden 7 bis 10 Tage
nach der Keimung in 5 mm Segmente zerschnitten.
-
4. Inoculierung
-
Sieben
bis zehn Tage alte Codyledonen und Cotyledonenpetiolensegmente werden
für 24
h auf Tabak BY-2 Feederzellplatten (MS-Salze, Myo-Inisitol 0,1 mg/ml,
Caseinhydrosylat 0,2 mg/ml, Thiamin-HCl 1,3 μg/ml, 3% Saccharose, pH 5,5)
vor der Inoculierung mit Agrobacterium inkubiert.
-
Die
Cotyledonen werden für
5 Minuten in Flüssig-KCMS-Agrobacteriumsuspension
getaucht. Zwischen 20 und 25 Cotyledonen werden dann mit der abaxialen
Seite aufwärts
auf die gleichen Feederzellplatten überführt und in Dunkelheit für zwei bis
drei Tage kultiviert.
-
5. Selektion transformierter
Cotyledonen und Petiolen und Regenerierung transformierter Pflanzen
-
Die
Cotyledonen und Petiolenfragmente werden zwei bis drei Tage nach
der Inoculierung mit Agrobacterium bei Licht auf Selektionsmedium
TSM-1 (4,3 g/l MS-Salze, 1 × B5-Vitamine,
2% Saccharose, 1% Glucose, 1 μg/ml
Zeatin, 0,05 μg/ml
IAA, 5 μg/ml
Hygromicin und 250 μg/ml
Carenicillin, pH 5,8) überführt. Die
Cotyledonen und Petiolensegemente, die embryogene Calli produzieren,
werden auf TSM-2-Medium (MS-Salze 4,3
g/l, 1 × MS-Vitamine,
2% Saccharose, 1 μg/ml
Zeatin, 5 μg/ml
Hygromycin und 250 μg/ml
Carbenicillin, pH 5,8) überführt. Überlebende
Calli, die Pflänzchen
bilden, werden in Magenta GA-7-Kästen überführt (Magenta Corp,
Chicago III.), welche TRM-Medium (4,3 g/l MS-Salze, 1 × MS-Vitamine, 2% Saccharose,
pH 5,8) enthalten und bei Licht gezüchtet. Nach der Wurzelbildung
werden die primären
Transformanten in Boden überführt.
-
Beispiel 22: Fluorimetrischer Test mit
grün fluoreszierendem
Protein (GFP)
-
Zum
Nachweis der synGFP-Genexpression wird ein fluorimetrischer Test
durchgeführt.
-
Blattscheibchen
stabil transformierten Maises und Tomaten werden auf Trockeneis
eingefroren. Die gefrorenen Gewebe werden gut gemahlen und mit 300 μl GFP-Extraktionspuffer
(10 mM Tris-HCl (pH 7,5), 100 mM NaCl, 1 mM MgCl2,
10 mM DTT und 0,1% Sarcosyl) gemischt. Die Extrakte werden von den
Blatt-Trümmern
durch Zentrifugieren getrennt, gefolgt vom Transfer in eine 96 Well
Black-Plate mit durchsichtigem Boden (Costar 3615). Die relativen
Fluoreszenzeinheiten (RFU) werden durch einen Spectra Fluor Plus
Plate Reader bei 465 nm Anregung und 512 nm Emission gemessen. Die
GFP-Aktivität
wird anhand des Gesamtproteins normalisiert, welches mit Hilfe des
BCA-Tests (gemäß Pierce)
gemessen wird.
-
Beispiel 23: Expressionsanalyse von stabil
transformiertem Mais und Tomatenpflanzen
-
1. Transgene Zea-Mais To-Pflanzen
-
GFP-Expression
ist unter Kontrolle des CmpS-Promotors (NOV4215, NOV4216) verglichen
mit dem ZmUbi-Promotor (NOV2110) 10 bis 20 Mal höher. Diese Ergebnisse werden
aus Blattproben von 42 unabhängigen
To-Linien gewonnen, die insgesamt 93 To-Pflanzen darstellen.
-
2. GFP Elisa-Test
-
Ein
quantitativer Sandwich-Immunassay wird zum Nachweis von grün fluoreszierendem
Protein (GFP) durchgeführt.
Zwei kommerziell verfügbare
polyklonale Antikörper
werden verwendet. Der Ziege-Anti-GFP IAP (Rockland, #600-1-1-215)
ist immunaffinitäts-gereinigt
und der Kaninchen-Anti-GFP- Antikörper (Torreg
Pines Biolabs, #TP401) ist Protein A gereinigt. Das GFP-Protein
in dem Pflanzenextrakt wird mit Hilfe des Kaninchen-Antikörpers auf
die Festphase der Microtiterplatte gefangen. Dann wird ein „Sandwich" zwischen dem Festphasen-Kaninchenantikörper, dem
GFP-Protein und dem sekundären
Ziegenantikörper
gebildet, welche in dem Well vorhanden sind. Nach einem Waschschritt,
wodurch nicht gebundener Sekundär-Antikörper entfernt
wird, wird der gebundene Antikörper
unter Verwendung eines mit Alkalischer Phosphatase markierten Antikörpers nachgewiesen.
Das Substrat für
das Enzym wird hinzugegeben und die Farbentwicklung wird durch Ablesen
der Absorption jedes Wells gemessen. Die Ablesung wird dann an eine
Standardkurve angepasst, um die GFP-Konzentration gegenüber der
Absorption aufzutragen. synGFP Reportergen
| Kassette
im binären
Vektor | GFP-Aktivität (Durchschnitt) RFU/mf
Protein | ELISA
(Durchschnitt) ng GFP/mg Protein |
| NOV4215
(CmpS) | 4722,3 | 657,1 |
| NOV4216
(CmpS) | 2795,8 | 246,1 |
| NOV2110
(ZmUbi) | 272,0 | 41,4 |
-
2. Lycopersicon esculentum
To-Pflanzen
-
Das
CmpS-Fragment von CmYLCV-Promotor (NOV 4217) führt zu einem synGFP-Expressionsniveau,
das wesentlich höher
ist, als das Expressionsniveau des Ubq3-Promotors von Arabidopsis
(NOV4209).
-
synGFP-Reportergen
-
Die
Ergebnisse werden durch das Untersuchen der Intensität der synGFP-Fluoreszenz
durch Mikroskopie in dem Callus und den primären Schösslingen 5 unabhängiger stabiler
transformierter Callus-Linien, die mit der NOV4217-Kassette transformiert
sind, und 2 unabhängig
stabil transformierten Callus-Linien, die mit der NOV4209-Kassette
transformiert sind, gewonnen.
| Kassette
im binären
Vektor (Promotor) | Callus
ID | GFP
Fluoreszenz-Intensität |
| NOV4217
(CmpS) | NOV4217-1 | +++++ |
| NOV4217
(CmpS) | NOV4217-2 | ++++ |
| NOV4217
(CmpS) | NOV4217-3 | ++++ |
| NOV4217
(CmpS) | NOV4217-4 | +++ |
| NOV4217
(CmpS) | NOV4217-5 | +++ |
| NOV4209
(Ubq3 (At)) | NOV4209-1 | + |
| NOV4209
(Ubq3 (At)) | NOV4290-2 | + |
-
Gus-Intron GUS-Reportergen
-
Die
Ergebnisse, die aus der Untersuchung der Intensität der GUS-Färbung in
11 unabhängigen
stabil transformierten Callus-Linien
gewonnen werden, welche die CmpS-Promotor-Reporterkassetten (Linien NOV4220-1
bis NOV4220-11) enthalten. Zwei stabil transformierte Callus-Linien,
die CmpC-Promotorfragment von CmYLCV-Promotor (NOV4224-1, NOV4224-2)
werden untersucht. Zum Vergleich werden zwei stabil transformierte
Callus-Linien, die den Arabidopsis Ubiquitin 3 (Ubq3)-Promotor (NOV4208-1,
NOV4208-2) enthalten, untersucht.
| Kassette
im binären
Vektor (Promotor | Callus
ID | GFP
Fluoreszenz-Intensität |
| NOV4220
(CmpS) | NOV4220-1 | ++++ |
| NOV4220
(CmpS) | NOV4220-2 | +++ |
| NOV4220
(CmpS) | NOV4220-3 | ++++ |
| NOV4220
(CmpS) | NOV4220-4 | +++++ |
| NOV4220
(CmpS) | NOV4220-5 | +++++ |
| NOV4220
(CmpS) | NOV4220-6 | +++ |
| NOV4220
(CmpS) | NOV4220-7 | ++++ |
| NOV4220
(CmpS) | NOV4220-8 | +++++ |
| NOV4220
(CmpS) | NOV4220-9 | ++++ |
| NOV4220
(CmpS) | NOV4220-10 | +++ |
| NOV4220
(CmpS) | NOV4220-11 | ++++ |
| NOV4224
(CmpC) | NOV4224-1 | ++++ |
| NOV4224
(CmpC) | NOV4224-2 | +++ |
| NOV4208
(Ubq3 (At)) | NOV4208-1 | + |
| NOV4208
(Ubq3 (At)) | NOV4208-2 | ++ |
-
Beispiel 24: Transiente Expressionsexperimente
in Mais
-
1. Embryonengewinnung
-
Unreife
Embryonen von A188 oder anderen geeigneten Maisgenotypen werden
aus 8–12
Tage alten Kolben in Flüssigkeit
2DG4 + 0,5 d (Duncan's
D Medium modifiziert, damit es 20 mg/l Glucose enthält, ergänzt durch
10 g/l Mannose) ausgeschnitten und für 1 bis 2 Tage kultiviert.
-
2. Plasmolyse
-
Die
unreifen Embryonen werden in 12% Saccharoselösung für 3–4 Stunden vor der Bombardierung inkubiert.
Die Embryonen werden in 8–10
mm Abständen
auf einer Platte arrangiert.
-
3. Bombardierung
-
Die
Transfektion mittels Partikelbombardierung von Maisembryonen wird
in Gegenwart von 5 μg
Plasmid-DNA bei 650 psi durchgeführt,
wie beschrieben (Wright et al., 2001, Plant Cell Reports, im Druck).
Im Falle, dass die Referenz „im
Druck" nicht ausreicht,
sind hier die aus dem Artikel entnommenen Details:
Das DNA-Fragment
wird auf Gold-Mikroträger
(< 1 μm) präzipitiert,
wie im DuPont Biolistics Manual beschrieben. Die Gene werden unter
Verwendung des PDS-1000He-Biolistic- Geräts
in die Zellen des Zielgewebes verabreicht. Die Einstellungen des
Geräts
sind wie folgt: 8 mm zwischen der Rupture-Disk und dem Macrocarrier, 10
mm zwischen dem Macrocarrier und dem Stopping-Screen und 7 cm zwischen
dem Stopping-Screen und dem Ziel. Die Platten werden zweimal mit
DNA beschichteten Partikeln unter Verwendung von 650 psi-Rupture-Disks beschossen.
Um die Gewebeschäden
der Schockwelle des Heliumblasts zu minimieren, wird ein Edelstahlnetz,
mit 200 Öffnungen
pro linearem horizontalen und vertikalen Inch (McMaster-Carr, New
Brunswick, NJ) zwischen den Stopping-Screen und das Zielgewebe gegeben.
Die Embryonen werden auf den Platten für 48 Stunden in Dunkelheit
bei 25°C
kultiviert. Proteinextrakte werden durch Lysieren von Zellen in GUS-Lysispuffer
hergestellt und mittels Zentrifugieren geklärt.
-
Beispiel 25: GUS-Tests
-
Zur
Durchführung
der GUS-Genexpression werden histochemische und Chemilumineszenztests durchgeführt.
-
1. Histochemische β-Glucuronidase (GUS)-Test
-
Maisembryonen
und Tomaten der in vitro Callus-Linien werden in den GUS-Tests verwendet.
Entweder werden gesamte Embryonen oder kleine Teile eines Callus
in die GUS-Färbelösung getunkt.
Die GUS-Färbelösung enthält 1 mM
5-Brom-4-chlor-3-indolylglucuronid
(X-Gluc, Duchefa, 20 mM Stock in DMSO), 100 mM Na-Phosphatpuffer
pH 7,0, 10 mM EDTA pH 8,0 und 0,1% Triton X100. Die Gewebeproben
werden bei 37°C für 1–16 Stunden
inkubiert. Falls notwendig, werden die Proben mittels mehrerer Waschschritte
mit 70% EtOH geklärt,
um Chorophyll zu entfernen.
-
2. β-Glucuronidase
(GUS) Chemillumineszenz-Test
-
Für die quantitative
Analyse der GUS-Expression wird der GUS-Chemilumineszenz-Test unter Verwendung
des GUS-Light-Kit (Tropix. Inc) nach den Anweisungen des Herstellers
durchgeführt.
Die Aktivität wird
mit einem Luminometer gemessen.
-
Die
GUS-Ergebnisse mit dem Konstrukt pCmpS und pCmpMG (Beispiel 17)
werden mit einem Vergleichsklon-Wert (pNOV3612, Beispiel 19) normalisiert.
Die Ergebnisse, die aus zwei verschiedenen Experimenten gewonnen
werden, zeigen, dass die 8 bp Deletion des CmYLCV-Promotors (pCmpMG,
Beispiel 17) verglichen mit pCmpS, nicht signifikant die Expressionsniveaus
der GUS-Reporter beeinflusst. Transiente Expression in Z. Mais-Embryonen GUS-Aktivität 48 bzw. 72 Stunden nach der
Transfektion
| Konstrukt ID | GUS Aktivität RFU/mg
Protein |
| 48
h | 72
h |
| pCmpS | 103,7 | 51,7 |
| pCmpMG | 81,4 | 70,8 |
| ZmUbi
(pNOV3612) | 52,6 | 70,2 |
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-