-
Hintergrund
der Erfindung
-
Eines der Hauptziele der Pflanzengentechnik
ist es, Pflanzen zu erhalten, die verbesserte Merkmale oder Eigenschaften
haben. Diese Merkmale oder Eigenschaften umfassen Virusresistenz,
Insektenresistenz, Herbizidresistenz, erhöhte Stabilität und einen
verbesserten Nährwert,
um einige zu nennen. Jüngste
Fortschritte in der Gentechnik haben die Inkorporation von präselektierten
Genen in Pflanzenzellen ermöglicht,
um der Pflanze der Wahl die gewünschten
Qualitäten
zu verleihen. Das eingeführte
Gen, das heißt „Transgen", wird
dann in den Zellen der regenerierten Pflanze exprimiert, so dass
die Pflanze die Eigenschaft oder das Merkmal, das durch das Transgen
kodiert wird, aufweist.
-
Um ein Transgen in einer Pflanzenzelle
zu exprimieren, muß das
geeignete regulatorische Signal in der geeigneten Lokation in Bezug
auf das Transgen vorhanden sein. Diese regulatorischen Signale umfassen im
Allgemeinen eine Promotorregion, eine 5' untranslatierte Leadersequenz
und eine 3' Polyadenylierungssequenz. Die Promotorregion beeinflusst
die Rate, mit welcher das RNA-Produkt des Transgens und das resultierende
Proteinprodukt des Transgens gemacht wird. Die Promotoraktivität kann auch
von der Anwesenheit verschiedener anderer, cis-wirkender, regulatorischer
Elemente abhängen,
die, in Verbindung mit zellulären Faktoren,
die Stärke,
Spezifität
und die Transkriptionsinitiationsstelle bestimmen (für eine Review
siehe Zawel und Reinberg, Curr. Opin. Cell Biol., 4, 488 (1992)).
Starke Promotoren sind in der Lage, die RNA-Synthese auf einer höheren Rate
zu steuern relativ zu schwachen Promotoren. Konstitutive Promotoren
steuern die RNA-Produktion in vielen oder allen Zelltypen.
-
Der Blumenkohlmosaikvirus 35S-Promotor
(CaMV35S für
Englisch cauliflower mosaic virus 35S promoter) ist ein starker,
konstitutiver Promotor in Pflanzen (Odell et al., Nature, 313, 810
(1985); Jensen et al., Nature, 321, 669 (1986); Jefferson et al.,
EMBO J., 6, 3901 (1987); Kay et al., Science, 236, 1299 (1987);
Sanders et al., Nucl. Acids, Res., 4, 1543 (1987)). Dieses wurde
durch die Detektion beträchtlicher
Level von Reportergenproteinen oder mRNAs in Extrakten, die aus
den Blättern,
Stämmen,
Wurzeln und Blüten
von transgenen Pflanzen vorbereitet wurden, gezeigt. Als ein Resultat
findet der CaMV35S-Promoter in dem Gebiet der Pflanzengentechnik
eine weite Anwendung. Obwohl der CaMV35S-Promoter ein starker, konstitutiver
Promoter in Assays, die Zellextrakte involvieren, zu sein scheint,
zeigen detaillierte histologische Analysen der Reportergenprodukte,
die auf der Zell- und
Gewebeebene detektierbar sind, einen ziemlich hohen Grad in der Expressionsvariabilität der Genprodukte
in Pflanzengeweben.
-
CaMV ist ein Caulimovirus, eine Untergruppe
der Pararetroviren, die ikosaedrische Kapside haben und nur Dikotyledonen
infizieren, obwohl der CaMV35S-Promoter ein starker Promoter in
Monokotyledonen ist. Der Zuckerrohr Bacilliformvirus (SCBV für Sugarcane
bacilliform virus), der Commelina Yellow Mottle Virus (CoYMV) und
der Rice Tungro Bacilliform Virus (RTBV) sind Badnaviren, eine Untergruppe
der Pararetroviren, die bacilliforme Kapside haben und hauptsächlich Monokotyledonen
infizieren. Ein Promotorfragment, das aus dem CoYMV isoliert wurde,
verleiht ein gewebespezifisches Expressionsmuster, das unterschiedlich
zu dem Muster ist, das durch den CaMV35S-Promoter verliehen wird.
Transformierte Tabakpflanzen, die den CoYMV-Promotor verbunden mit
dem β-Glukuronidasereportergen
(„GUS";
uidA) enthielten, zeigten, dass, während der CoYMV-Promotor in
allen Organen aktiv ist, die β-Glukuronidaseaktivität hauptsächlich im
Phloem, den phloemassoziierten Zellen und dem axialen Parenchym
der Wurzeln, Stämme,
Blätter
und Blüten
vorkommt (Medberry et al., Plant Cell, 4, 185 (1992); Medberry und
Olszewski, Plant J., 3, 619 (1993)). Im Gegensatz dazu ist der CaMV35S-Promoter
in fast allen Zelltypen aktiv (Medberry et al., Plant Cell, 4, 185, (1992);
Medberry und Olszewski, Plant J., 3, 619 (1993)). Darüber hinaus
ist der CoYMV-Promotor in Tabaksuspensionszellen 30% so aktiv und
in Maissuspensionszellen bis zu 25% so aktiv verglichen mit einem
duplizierten CaMV35S-Promoter (Medberry et al., Plant Cell, 4, 185
(1992)).
-
Transgener Reis, der den RTBV-Promotor
verbunden mit dem GUS-Gen
enthält,
zeigt eine starke, phloemspezifische Promotoraktivität. Dieses
war in Übereinstimmung
mit der Expression dieses Promotors in Reisprotoplasten. Der RTBV-Promotor
zeigte jedoch nur schwache Aktivität in Maisprotoplasten (Bhattacharyya-Pakrasi
et al., Plant J., 4, 71 (1993); Yin et al., Plant J., 7, 969 (1995)).
Im Gegensatz dazu zeigt der entsprechende CaMV-Promoter eine starke
Promotoraktivität
in Protoplasten und in fast allen Geweben transgener Pflanzen (von
Hohn und Fütterer
reviewed, Curr. Opin. Genet. Dev., 2, 90 (1992)).
-
Was folglich benötigt wird ist ein hochexprimierter,
konstitutiver Promotor, um Transgene in fertilen, transgenen monokotylen
und dikotylen Pflanzen zu exprimieren.
-
Zusammenfassung der Erfindung
-
Die vorliegende Erfindung stellt
ein isoliertes und aufgereinigtes DNA-Molekül bereit, das ein präselektiertes
DNA-Segment umfasst, das einen Zuckerrohr Bacilliformvirus (ScBV)
-promotor oder eine biologisch aktive Untereinheit davon umfasst,
die einen konstitutiv hohen Expressionslevel von dem wirksam verbundenen,
präselektierten
DNA-Segmenten in beiden, monokotylen und dikotylen Pflanzen, Pflanzengeweben, Pflanzenteilen
oder Pflanzenzellen verleihen. Während
die Nukleotidsequenz des Genoms von ScBV bekannt ist (Bouhida et
al., J. Gen. Virol., 74, 1 (1993)), war die Lokation eines Promotors
für die
virale RNA über
die gesamte Genomlänge,
sogar nach Nukleotidsequenzvergleichen des ScBV-Genoms mit Promotorsequenzen naher
verwandter Viren wie CoYMV und RTBV, nicht offensichtlich. Überraschenderweise
ist der ScBV-Promotor in vielen Zelltypen ein starker und konstitutiver
Promotor ungleich der starken, gewebespezifischen Expression, die
für die
CoMYV- und RTBV-Promotoren beobachtet werden. Eine bevorzugte Ausführungsform der
Erfindung ist ein präselektiertes
DNA-Segment, das einen ScBV-Promotor umfasst, der SEQ ID NO: 3 umfasst,
das heißt
ein präselektiertes
DNA-Segment, das den Nukleotidpositionen 5999–7420 des ScBV-Genoms entspricht.
Wie hierin unten beschrieben verleiht der ScBV-Promotor eine konstitutive
und vaskuläre
Genexpression in A. sativa und A. thaliana. Der ScBV-Promotor kann
folglich für
konstitutive und gewebespezifische, pflanzliche und nicht-pflanzliche
Genexpression in Monokotyledonen und Dikotyledonen eingesetzt werden. Zum
Beispiel kann der ScBV-Promotor mit Genen verbunden sein, die Getreiden
eine Resistenz gegen Schädlinge
verleihen, die die Getreidegräser
am Stengel attackieren, beispielsweise Blattläuse, die den barley yellow dwarf
virus übertragen,
oder die eine gewebespezifische Resistenz an dikotyle Wirte verleihen,
in welchen Organe wie die Wurzel durch Pathogene befallen werden,
beispielsweise die soybean cyst nematode.
-
Wie hierin verwendet umfasst „ScBV"
irgendeinen nicht-verpackten, bacilliformen, DNA-enthaltenden Badnavirus,
der in der Lage ist, Saccharum oder verwandte Gattungen zu infizieren.
Weitere charakteristische Eigenschaften von Badnaviren sind von
Lockhart und Olszewski in The Encyclopedia of Virology, Webster
und Granoff (eds.), Academic Press, New York, NY (1994) beschrieben.
-
Wie hierin verwendet bedeutet der
Begriff „ScBV-Promotor"
eine Nukleotidsequenz, die, wenn diese Sequenz wirksam mit einem
präselektierten
DNA-Segment verbunden ist, das ein Protein, ein RNA-Transkript oder eine
Mischung davon kodiert, zu der Expression des verbundenen, präselektierten
DNA-Segments führt, das
heißt
der kodierten RNA und/oder dem kodierten Protein. Ein bevorzugter
ScBV-Promotor hat wenigstens etwa 60%, vorzugsweise wenigstens etwa
80%, besonders vorzugsweise wenigstens etwa 90% und sogar besonders
vorzugsweise wenigstens etwa 95% Nukleotidsequenzidentität mit SEQ
ID NO: 3, SEQ ID NO: 4 oder SEQ ID NO: 5. Eine weitere, bevorzugte
Ausführungsform
der Erfindung ist ein ScBV-Promotor, der die minimale Zahl an zusammenhängenden
Nukleotiden umfasst, welche die RNA-Transkription iniziieren.
-
Wie hierin verwendet bedeutet „biologisch
aktiv", dass der Promotor wenigstens etwa 0,1%, vorzugsweise wenigstens
etwa 10% und besonders vorzugsweise wenigstens etwa 25% der Aktivität des ScBV-Promotors
hat, umfassend SEQ ID NO: 3, SEQ ID NO: 4 oder SEQ ID NO: 5. Die
Aktivität
eines Promotors kann durch Verfahren, die in Fachkreisen gut bekannt
sind, bestimmt werden. Siehe zum Beispiel (Medberry et al., Plant
Cell, 4, 185 (1992); Medberry et al., The Plant J., 3, 619 (1993);
Sambrook et al., In: Molecular Cloning: A Laboratory Manual (1989);
McPherson et al., U.S. Patent No. 5,164,316.
-
Darüber hinaus wird eine Expressionskassette
bereitgestellt, die ein erstes präselektiertes DNA-Segment umfasst,
das einen ScBV-Promotor umfasst, der in einer Wirtszelle funktionell
ist und wirksam mit einem zweiten präselektierten DNA-Segment verbunden
ist, das ein Protein, ein RNA-Transkript
oder eine Kombination davon kodiert. Eine bevorzugte Wirtszelle
ist eine Pflanzenzelle, beispielsweise eine monokotyle oder dikotyle
Zelle. Eine weitere bevorzugte Ausführungsform der Erfindung ist
eine Expressionskassette, die einen ScBV-Promotor umfasst, der wirksam
mit einem selektierbaren Markergen verbunden ist. Noch eine weitere bevorzugte
Ausführungsform
der Erfindung ist eine Expressionskassette, die einen ScBV-Promotor umfasst, der
SEQ ID NO: 3 umfasst.
-
Die Erfindung stellt auch Verfahren
zur Selektion stabiler, genetischer Transformanden von transformierten
Pflanzenzellen und Verfahren zur Herstellung fertiler, transgener
Pflanzen aus den besagten Pflanzenzellen bereit. Das Verfahren zur
Herstellung von transformierten Pflanzenzellen umfasst die Einführung eines rekombinanten
DNA-Segments in regenerierbare Pflanzenzellen, das ein erstes präselektiertes
DNA-Segment umfasst, welches einen ScBV-Promotor wirksam verbunden
mit einem zweiten präselektierten
DNA-Segment umfasst, um transformierte Zellen zu gewinnen. Dann
wird eine transformierte Zellinie identifiziert oder selektiert.
Exemplarische Transformationsverfahren umfassen die Verwendung von
Mikroprojektilbombardements, um ein präselektiertes DNA-Segment, das
ein phänotypisch
beobachtbares oder detektierbares Merkmal kodiert, das wirksam mit
dem ScBV-Promotor verbunden ist, in regenerierbare, monokotyle Pflanzenzellen
einzuführen.
Eine bevorzugte Ausführungsform
der Erfindung ist ein Verfahren, wobei die Expression des rekombinanten
DNA-Segments in den transformierten Zellen den transformierten Zellen
ein phänotypisches
Kennzeichen verleiht wie eine Herbizid- oder Antibiotikumresistenz.
-
Wie hierin verwendet bezieht sich
der Begriff „rekombinantes
DNA-Segment" auf
eine Nukleinsäure, das
heißt
auf eine DNA, die aus irgendeiner geeigneten Gewebequelle gewonnen
oder isoliert worden ist und aus dem Verdund mit anderen Zellkomponenten
isoliert worden ist, wie Nukleinsäuren oder Proteinen. Die DNA
kann anschließend
in vitro chemisch verändert
werden, so dass ihre Sequenz nicht natürlich vorkommt oder natürlich vorkommenden
Sequenzen entspricht, die nicht so positioniert sind wie sie in
einem Genom positioniert sein würden,
das nicht mit exogener DNA transformiert worden ist, so dass sie
sequenziert, repliziert und/oder exprimiert werden kann.
-
Ein bevorzugtes, isoliertes, rekombinantes
DNA-Segment umfasst ein erstes, präselektiertes DNA-Segment, das
einen in Pflanzenzellen funktionellen ScBV-Promotor umfasst, der
wirksam mit einem zweiten, präselektierten
DNA-Segment verbunden ist, das ein selektierbares Markergen umfasst.
Ein weiteres, bevorzugtes, isoliertes, rekombinates DNA-Segment
umfasst ein zweites, präselektiertes
DNA-Segment, das einem Gen entspricht, welches bereits in dem Pflanzengenom
vorhanden ist, oder einem Gen entspricht, welches normalerweise
nicht in dem Pflanzengenom vorhanden ist und die der Pflanze einen
agronomisch nützlichen
Phänotyp
verleihen, beispielsweise eine Schädlingsresistenz. Wenn das präselektierte
DNA-Segment normalerweise
in dem Pflanzengenom vorhanden ist, muß es nicht oder nicht hoch
exprimiert sein. Folglich wird das präselektierte DNA-Segment eingeführt, um
die Expression des Proteins oder RNA-Transkripts zu verändern, die
durch das präselektierte
DNA-Segment in Zellen der Pflanze kodiert werden.
-
Die Erfindung stellt auch ein Verfahren
zur Herstellung einer fertilen, transgenen Pflanze bereit. Das Verfahren
umfasst das Einführen
eines rekombinanten DNA-Segments, das ein erstes, präselektiertes DNA-Segment
umfasst, das einen ScBV-Promotor wirksam verbunden mit einem zweiten
präselektierten DNA-Segment
umfasst, in regenerierbare Pflanzenzellen, um regenerierbare transformierte
Zellen zu gewinnen. Eine Population transformierter Zellen wird
selektiert oder identifiziert und eine fertile, transgene Pflanze daraus
regeneriert. Das rekombinante DNA-Segment wird durch einen vollständigen,
sexuellen Zyklus der besagten transgenen Pflanze zu deren Nachkommenschaft übertragen,
so dass es von den Pflanzennachkommen exprimiert wird. Folglich
stellt die Erfindung auch eine transgene Pflanze und Samen, andere
Pflanzenteile, Gewebe und Pflanzennachkommen, die davon abstammen,
bereit.
-
Die transgenen Pflanzen der Erfindung
umfassen, sind aber nicht darauf beschränkt, eine transgene T0- oder
R0-Pflanze, das heißt
die erste Pflanze, die aus den transformierten Pflanzenzellen regeneriert
wurde, eine transgene T1- oder R1-Pflanze, das heißt die Pflanze
der ersten Nachkommengeneration und die Nachkommenpflanzen weiterer
Generationen, die davon abstammen, welche das rekombinante DNA-Segment
umfassen und exprimieren. Um das rekombinante DNA-Segment in regenerierbare,
monokotyle Zellen einzuführen,
kann das Mikroprojektilbombardement verwendet werden, während Agrobacterium-vermittelter DNA-Transfer
verwendet werden kann, um die rekombinante DNA in regenerierbare,
dikotyle Zellen einzuführen.
-
Es wird auch eine transformierte,
monokotyle oder dikotyle Pflanze bereitgestellt, deren Zellen ein
rekombinantes DNA-Segment umfassen, das ein erstes, präselektiertes
DNA-Segment umfasst, das einen Zuckerrohr Bacilliformviruspromotor
umfasst, der wirksam mit einem zweiten, präselektierten DNA-Segment verbunden
ist. Das zweite, präselektierte
DNA-Segment wird
in den transformierten Zellen in einer Menge exprimiert, die unterschiedlich
zu der Menge in den Zellen einer Pflanze ist, in der sich die Zellen
nur durch die Abwesenheit des rekombinanten DNA-Segments von den
transformierten Zellen unterscheiden. Solche Zellen können in
einigen Fällen
untransformierte Zellen desselben Teils der transformierten oder
transgenen Pflanze umfassen. Das zweite, präselektierte DNA-Segment wird
exprimiert, um die transformierte Pflanze oder einen Teil davon
von der entsprechenden untransformierten Pflanze oder einem Teil
davon identifizierbar zu machen.
-
Das rekombinante DNA-Segment wird
durch einen vollständigen,
normalen, sexuellen Zyklus der transformierten Pflanze auf die nächste Generation übertragen.
-
Es wird auch ein Verfahren bereitgestellt,
das die Gewinnung einer Nachkommenschaft von einer fertilen, transgenen
Pflanze umfasst, die durch das Verfahren, welches oben beschrieben
wird, erhalten wurde.
-
Wie hierin verwendet meinen die Begriffe „transgen"
oder „transformiert"
in Bezug auf eine Pflanzenzelle, einen Pflanzenteil (einschließlich den
Samen), ein Pflanzengewebe oder eine Pflanze, eine Pflanzenzelle,
einen Pflanzenteil, ein Pflanzengewebe oder eine Pflanze, die ein
isoliertes, aufgereinigtes, präselektiertes DNA-Segment
umfassen, das in das Genom einer Pflanzenzelle, eines Pflanzenteils,
eines Pflanzengewebes oder einer Pflanze durch ein „gentechnisches"
Transformationsverfahren eingeführt
worden ist. Das heißt
das Genom einer transgenen Pflanzenzelle, eines Pflanzenteils, eines
Pflanzengewebes oder einer Pflanze ist durch wenigstens ein präselektiertes
DNA-Segment erweitert worden. Die Begriffe „wildtyp", „nativ"
oder „nicht transgen"
beziehen sich auf eine untransformierte Pflanzenzelle, Pflanzenteil,
Pflanzengewebe oder Pflanze, das heißt eine, wo das Genom nicht
durch die Anwesenheit eines präselektierten
DNA-Segments verändert worden
ist.
-
Die Transformation von Pflanzen gemäß der Erfindung
kann im Wesentlichen mittels irgendeinem der verschiedenen Verfahren
durchgeführt
werden, die für
jene verfügbar
sind, die auf dem Gebiet der Molekularbiologie der Pflanzen Fachkenntnisse
ahebn. Diese umfassen, sind aber nicht darauf beschränkt, das
Mikroprojektilbombardement, die Mikroinjektion, die Elektroporation
von Protoplasten oder Zellen, die partielle Zellwände umfassen,
den Silikoncarbidfiber-vermittelten DNA-Transfer und den Agrobacterium-vermittelten DNA-Transfer.
Pflanzen, die bei der Anwendung der Erfindung brauchbar sind, umfassen,
sind aber nicht darauf beschränkt,
Hafer, Weizen, Soja, Mais, Tabak, Reis, Gerste, Kartoffel, Tomate,
Salat, Ölsamenraps,
Baumwolle, Flacks, Zuckerrüben,
Sorghum, Sonnenblumen, Alfalfa, Hirse und Roggen.
-
Kurze Beschreibung der Figuren
-
1 stellt
die abgeleitete Aminosäuresequenz
(SEQ ID NO: 1) des Zuckerrohr Bacilliformvirus dar.
-
2 stellt
die Nukleotidsequenz (SEQ ID NO: 2) des Zuckerrohr Bacilliformvirus
dar.
-
3 stellt
die Expressionskassetten dar, die einen ScBV-Promotor umfassen,
der zur Pflanzentransformation brauchbar ist.
-
4 stellt
(A) eine Karte von Konstrukten dar, die verwendet wurden, um die
Aktivität
des ScBV-Promotors in A. sativa zu testen. Die resultierenden pMON755i-Derivate,
die die ScBV-Kassetten 1, 2 oder 3 enthalten, wurden mit pScBV-1,
pScBV-2 bzw. pScBV-3 bezeichnet; und (B) eine Karte der Reportergenregion
in dem binären
Vektor, der verwendet wurde, um die Promotoraktivität der ScBV-3-Kassette
in A. thaliana zu testen. Die ScBV-3-Kassette wurde in die SalI-
und XbaI-Stelle von pOCA101 kloniert. Die Klonierungsschritte führten zu
der Entfernung der SpeI/StuI und der SalI/XhoI Stellen.
-
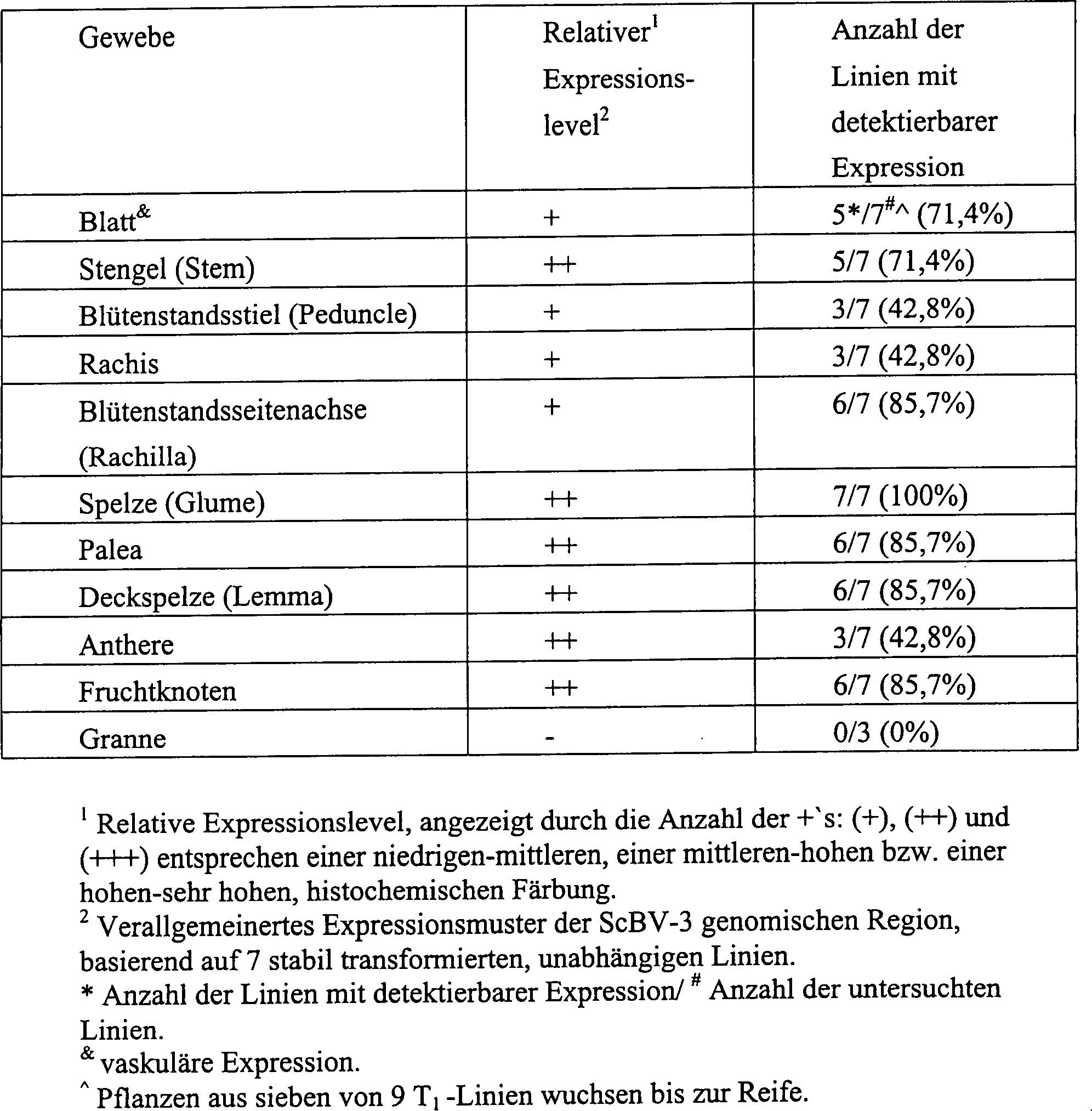
5 A.
sativa- und A. thaliana-Gewebe gefärbt auf GUS-Aktivität: (A) A.
sativa-Antheren; (B) A. sativa-Fruchtknoten; (C) A. thaliana-Blütenorgane;
(D) A. sativa-Ärchen;
(E) A. sativa-Stengel; (F) A. sativa-Blatt; (G) A. thaliana-Blatt.
-
Detaillierte Beschreibung
der Erfindung
-
Das Einführen von exogenen Genen (Transgenen)
in Pflanzen, um eine fertile, transgene Pflanze mit verbesserten,
agronomischen Eigenschaften bereitzustellen, hat das Potential zur
Langzeitverbesserung und der Expansion der Agrikultur weltweit.
Die vorliegende Erfindung stellt in Pflanzen eine konstitutive und
starke Expression von Transgenen bereit, die beispielsweise Substanzen
kodieren, die zu veränderten,
agronomischen oder physiologischen Merkmalen führen. Solche transgenen Pflanzen
und die davon gewonnenen Samen können
dieses Merkmal sexuell auf ihre Nachkommen übertragen. Exemplarische Merkmale
für transgene
Pflanzen umfassen eine erhöhte
Stresstoleranz, Schädlingsresistenz,
Krankheitsresistenz (zum Beispiel Bakterien, Viren und Pilze), verbesserte
Erträge,
einen verbesserten Nährstoffwert
und verbesserte Kornzusammensetzung oder -qualität.
-
Um eine konstitutive und starke Expression
von Transgenen in Pflanzen, Pflanzenzellen, Pflanzengeweben oder
Pflanzenteilen bereitzustellen, wird ein Zuckerrohr Bacilliformviruspromotor,
der beispielsweise SEQ ID NO: 3 umfaßt, wirksam mit einem definierten
Transgen verbunden, das heißt
einem präselektierten DNA-Segment,
und in regenerierbare Pflanzenzellen eingeführt. Die resultierende, regenerierte, transgene Pflanze
exprimiert das Protein oder RNA-Transkript, das von dem präselektierten
DNA-Segment kodiert wird, auf einem hohen und gleichmäßigen Level,
vorzugsweise in allen Geweben und Zellen der transformierten Pflanze.
Eine bevorzugte ScBV-Promotorsequenz ist zwischen den Nukleotiden
5999 und 7420 (SEQ ID NO: 3) des ScBV-Genoms (SEQ ID NO: 2) lokalisiert.
Es wird vorzugsweise eine 5' untranslatierte Leadersequenz mit dem
Promotor gekoppelt. Die Leadersequenz kann von dem ScBV-Genom selbst
oder von einer anderen Quelle als dem ScBV sein, beispielsweise
von dem ersten Intron der Mais-Alkoholdehydrogenase und den 5' untranslatierten
Leadersequenzen des Petunien-HSP70 (Winter et al., Mol. Gen. Genet.,
211, 315 (1988)); des Soya-HSP17,9 (Raschke et al., J. Mol. Biol.,
199, 549 (1988)); oder des Mais-HSP70 (Rochester et al., EMBO J.,
5, 541 (1986)).
-
I. Rezipientenzellen
-
Die vorliegende Erfindung setzt Rezipientenzellen
ein, die empfänglich
für die
Transformation und die darauffolgende Regeneration in stabil transformierte,
fertile Pflanzen sind. Für
zum Beispiel die Transformation von Monokotyledonen können unreife
Embryonen, meristematisches Gewebe, gametisches Gewebe, embryogene
Suspensionskulturen oder embryogenes Kallusgewebe als eine Quelle
für Rezipientenzellen
eingesetzt werden, die bei der Ausführung der Erfindung brauchbar
sind. Bevorzugte Rezipientenzellen für die Transformation von Hafer
sind Haferkalluskulturen, die von unreifen Embryonen iniziiert wurden.
Um eine solche Kultur von Hafenezipientenzellen bereitzustellen,
werden unreife Haferembryonen geschält und sterilisiert. Die Embryonen
werden in Flüssigmedien über Nacht
inkubiert, dann herausgeschnitten und auf feste Medien mit der Skutellumseite
nach unten plaziert, um eine Kalluskultur zu initiieren. Ein bevorzugtes
festes Medium zur Initiation einer Kalluskultur ist MS2D (siehe
Torbert et al., Plant Cell Reports, 14, 635 (1995)).
-
Zur Transformation von Dikotyledonen
können
Organ- und Gewebekulturen als eine Quelle für Rezipientenzellen eingesetzt
werden. Folglich können
Gewebe, beispielsweise Blätter,
Samen und Wurzeln, von Dikotyledonen eine Quelle für Rezipientenzellen
bereitstellen, die in der Ausführung
der Erfindung brauchbar ist.
-
Die kultivierten, empfänglichen
Rezipientenzellen werden vorzugsweise auf festen Unterlagen angezogen.
Nährstoffe
werden den Kulturen in Form von Medien bereitgestellt und die Umweltbedingungen
der Kulturen werden kontrolliert. Medien und Umweltbedingungen,
die das Wachstum von regenerierbaren Pflanzenkulturen unterstützen, sind
im Stand der Technik gut bekannt.
-
II. DNA-Sequenzen
-
Es kann so gut wie jede DNA-Komposition
für die Übertragung
auf die Rezipienten-Pflanzenzellen verwendet werden, um letzten
Endes fertile, transgene Pflanzen gemäß der vorliegenden Erfindung
zu produzieren. Das DNA-Segment oder Gen, das für die zelluläre Einführung ausgewählt wurde,
wird häufig
ein Protein kodieren und kann in den resultierenden, transformierten
Zellen exprimiert werden, um zu einem screenbaren oder selektierbaren
Merkmal zu führen
und/oder um der regenerierten Pflanze einen verbesserten Phänotyp zu
verleihen. Das DNA-Segment oder Gen, das für die zelluläre Einführung ausgewählt worden
ist, kann auch eine antisense-RNA
kodieren, das heißt
ein Komplement eines prädeterminierten
DNA-Moleküls oder
eines Teils davon, die in einer untransformierten Pflanzenzelle
exprimiert werden. Die Transkription einer antisense-RNA supprimiert
die Expression der komplementären
RNA, beispielsweise von einer, welche eine unerwünschte Eigenschaft kodiert.
Folglich kann ein präselektiertes
DNA-Segment in Form
von Vektoren und Plasmiden oder linearen DNA-Fragmenten, die in einigen Fällen nur
das in der Pflanze zu exprimierende DNA-Element enthalten, und dergleichen
eingesetzt werden. Dieses mag jedoch nicht immer der Fall sein und
die vorliegende Erfindung umfasst auch transgene Pflanzen, die nicht
exprimierte Transgene enthalten. Exemplarische DNA-Sequenzen werden
in den Tabellen 1, 2 und 3 in Weising et al. bereitgestellt (Ann.
Rev. Genet., 22, 421 (1988)), und in Lundquist et al. (U.S. Patent
No. 5, 484, 956), wobei beide davon hierin als Referenz enthalten
sind.
-
Für
bestimmte Ausführungsformen
wird in Erwägung
gezogen, dass jemand wünschen
könnte,
replikationskompetente, virale Vektoren in der Pflanzentransformation
einzusetzen, wie solche, die zur Maistransformation eingesetzt werden,
um das präselektierte
DNA-Segment in die Pflanzen zu transferieren. Solche Vektoren umfassen
zum Beispiel die wheat dwarf virus (WDV) „shuttle"-Vektoren wie pW1-11
und PW1-GUS (Ugaki et al., Nucl. Acid Res., 19, 391 (1991)). Diese
Vektoren sind zur autonomen Replikation sowohl in Maiszellen als
auch in E. coli in der Lage und können als solche eine erhöhte Sensitivität zur Detektion
von DNA-übertragenen
gegenüber
transgenen nicht-Maispflanzenzellen bereitstellen.
-
Eine replizierender Vektor kann auch
zur Übertragung
von Genen nützlich
sein, die von DNA-Sequenzen transponierbarer Elemente wie Ac, Ds
oder Mu flankiert werden, da diese Elemente aktiv die Integration der
gewünschten
DNA begünstigen
würden
und daher die Häufigkeit
einer stabilen Transformation erhöhen. Es wird auch in Erwägung gezogen,
dass transponierbare Elemente nützlich
wären,
um DNA-Fragmente einzuführen,
denen Elemente fehlen, die zur Selektion und Erhaltung des Plasmidvektors
in Bakterien nötig
sind, beispielsweise Antibiotikaresistenzgene oder DNA-Replikationsursprünge.
-
Eine DNA, die zur Einführung in
Pflanzenzellen brauchbar ist, umfasst, dass sie von irgendeiner
Quelle abgeleitet oder aus irgendeiner Quelle isoliert wurde, die
anschließend
durch eine Struktur, Größe und/oder Funktion
charakterisiert, chemisch verändert
und später
in Pflanzen eingeführt
werden könnte.
Ein Beispiel für eine
solche DNA, die aus einer Quelle „isoliert" wurde, wäre eine
brauchbare DNA-Sequenz, die aus der besagten Quelle mit chemischen
Mitteln, beispielsweise durch die Verwendung von Restriktionsendonukleasen, ausgeschnitten
oder entfernt wurde, so dass sie weiter manipuliert werden kann,
beispielsweise separiert oder amplifiziert mit zum Beispiel einer
Polymeryasekettenreaktion (PCR für
Englisch polymerase chain reaction), zur Verwendung in der Erfindung
durch die Methodologie der Gentechnik. Die Wiedergewinnung oder
Isolation eines gegebenen DNA-Fragments
aus einem Restriktionsverdau kann die Auftrennung des Verdaus auf
einem Polyakrylamid- oder Agarosegel durch Elektrophorese, die Identifikation
des Fragments von Interesse durch einen Vergleich seiner Mobilität gegenüber der
Mobilität
eines Marker-DNA-Fragments bekannten Molekulargewichts, das Entfernen
der Gelsektion, die das gewünschte
Fragment enthält
und die Trennung des Gels von der DNA einsetzen. Siehe Lawn et al.,
Nucleic Acids Res., 9, 6103 (1981) und Goedel et al., Nucleic Acids Res.,
8, 4057 (1980). Folglich ist die DNA „isoliert", insofern sie von
wenigstens einer kontaminierenden Nukleinsäure frei ist, mit der sie normalerweise
in der natürlichen
RNA- oder DNA-Quelle assoziiert ist und vorzugsweise im wesentlichen
von irgendeiner anderen Säugetier-RNA
oder -DNA frei ist. Die Phrase „frei von wenigstens einer
kontaminierenden Quellennukleinsäure,
mit der sie normalerweise assoziiert ist" schließt den Fall mit ein, wo die
Nukleinsäure
in die Quelle oder natürliche
Zelle zurückgeführt wird,
sie aber in einer unterschiedlichen, chromosomalen Lage ist oder
anderweitig von Nukleinsäuresequenzen,
die normalerweise nicht in der Quellenzelle gefunden werden, flankiert
wird.
-
Ein Beispiel für eine DNA, die sich von einer
Quelle „ableitet",
wäre eine
DNA-Sequenz oder ein DNA-Segment, die oder das als ein brauchbares
Fragment innerhalb eines gegebenen Organismus identifiziert wird
und welche oder welches dann chemisch in einer im wesentlichen reinen
Form synthetisiert wird. Deshalb umfasst eine „rekombinante oder präselektierte
DNA" vollständig
synthetische DNA-Sequenzen, semisynthetische DNA- Sequenzen, DNA-Sequenzen, die aus biologischen
Quellen isoliert wurden und DNA-Sequenzen, die sich von RNA ableiten,
sowie Mischungen davon.
-
Die eingeführte DNA umfasst, ist aber
nicht darauf beschränkt,
DNA von Pflanzengenen und nicht-pflanzlichen Genen wie solche von
Bakterien, Hefen, Tieren oder Viren. Darüber hinaus ist es innerhalb des
Bereichs der Erfindung, ein präselektiertes
DNA-Segment aus einem gegebenen Pflanzengenotyp zu isolieren und
anschließend
mehrfache Kopien des präselektierten
DNA-Segments in denselben Genotyp einzuführen, zum Beispiel um die Produktion
eines gegebenen Genprodukts zu steigern. Die eingeführte DNA
kann modifizierte Gene, Teile von Genen oder chimäre Gene
umfassen, einschließlich
Genen desselben oder eines anderen Planzengenotyps. Der Begriff „chimäres Gen"
oder „chimäre DNA"
wird als ein Gen oder eine DNA-Sequenz oder ein DNA-Segment definiert,
umfassend wenigstens zwei DNA-Sequenzen oder Segmente aus Spezien,
die unter natürlichen
Bedingungen die DNA nicht kombinieren oder deren DNA-Sequenzen oder DNA-Segmente
auf eine Weise positioniert oder verbunden sind, die normalerweise
nicht in dem nativen Genom der untransformierten Pflanze auftritt.
-
Die eingeführte DNA, die zur Transformation
hierin verwendet wurde, kann zirkulär oder linear, doppelsträngig oder
einzelsträngig
sein. Im allgemeinen ist die DNA in Form von chimärer DNA
wie Plasmid-DNA, die auch kodierende Regionen enthalten kann, die
von regulatorischen Sequenzen flankiert werden, welche die Expression
der rekombinanten DNA, die in der resultierenden Pflanze vorhanden
ist, begünstigen.
-
Im allgemeinen wird die eingeführte DNA
relativ klein sein, das heißt
kleiner als etwa 30 kb um jede Anfälligkeit gegenüber einem
physikalischen, chemischen oder enzymatischen Abbau zu minimieren,
von welchen bekannt ist, dass sie mit der Größe der DNA zunehmen. Wie oben
erwähnt
wird die Anzahl der Proteine, der RNA-Transkripte oder der Mischungen
davon, die von den DNA-Molekülen
kodiert werden, welche in das Pflanzengenom eingeführt werden,
vorzugsweise präselektiert
und definiert, bis sich zum Beispiel eins bis etwa 5–10 solcher
Produkte der eingeführten
DNAs gebildet haben können.
-
A. Vorbereitung einer Expressionskassette
-
Eine Expressionskassette gemäß der Erfindung
kann ein rekombinantes DNA-Molekül
umfassen, das ein präselektiertes
DNA-Segment enthält, welches
wirksam mit einem ScBV-Promotor verbunden und funktionell in einer
Wirtszelle, vorzugsweise einer Pflanzenzelle ist. Vorzugsweise ist
die Expressionskassette selbst chimär, das heißt die Kassette umfasst DNA
von wenigstens zwei verschiedenen Spezies oder DNA von derselben
Spezien, die in einer Weise verbunden oder assoziiert ist, wie sie
in dem „nativen"
oder Wildtyp der Spezien nicht vorkommt.
-
1. DNA-Moleküle gemäß der Erfindung,
die den ScBV-Promotor umfassen
-
Ein Promotor ist eine DNA-Region,
die die Genexpression reguliert. Promotorregionen werden typischerweise
in der flankierenden DNA-Sequenz stromaufwärts von der kodierenden Sequenz
in Viren sowie prokaryotischen und eukaryotischen Zellen gefunden.
Eine Promotorsequenz sorgt für
die Regulation der Transkription der stromabwärts gelegenen Gensequenz und
umfasst typischerweise von etwa 50 bis etwa 2000 Nukleotidbasenpaare.
Promotorsequenzen können
auch regulatorische Sequenzen wie Enhancersequenzen enthalten, die
den Level der Genexpression beeinflussen. Einige isolierte Promotorsequenzen
können
für die
Genexpression von heterologen DNAs sorgen, das heißt einer
DNA, die unterschiedlich zu der nativen oder homologen DNA ist.
Es ist auch bekannt, dass Promotorsequenzen stark oder schwach oder
induzierbar sind. Ein starker Promotor sorgt für einen hohen Level der Genexpression,
während
ein schwacher Promotor für
einen sehr niedrigen Level der Genexpression sorgt. Ein induzierbarer
Promotor ist ein Promotor, der für
ein An- und Abschalten der Genexpression als Antwort auf ein exogen
zugegebenes Mittel oder einen Umwelt- oder Entwicklungsstimulus
sorgt. Promotoren können
auch für
eine gewebespezifische oder entwicklungsbedingte Regulation sorgen.
Eine isolierte Promotorsequenz, die ein starker Promotor für heterologe DNAs
ist, ist vorteilhaft, weil sie für
einen ausreichenden Level an Genexpression sorgt, um eine einfache
Detektion und Selektion transformierter Zellen zu ermöglichen
und für
einen hohen Level an Genexpression sorgt, wenn dieser gewünscht wird.
-
Das DNA-Molekül der Erfindung umfasst ein
präselektiertes
DNA-Segment, das
einen Zuckerrohr Bacilliformvirus (ScBV) -promotor umfasst. ScBV
ist ein Pararetrovirus. Im Allgemeinen haben Pararetroviren einen
Promotor, der die Transkription eines RNA-Transkripts steuert, das
als beides, als eine Vorlage für
die Replikation des viralen Genoms und als mRNA dient. Während der
Replikation der zirkulären,
doppelsträngigen, viralen
DNA bindet das 3'-Ende der Wirts-tRNA nahe dem 5'-Ende des ScBV-Transkripts,
um die DNA-Synthese durch die viruskodierte reverse Transkriptase
zu primen. Folglich sind die ScBV-Promotoren im Allgemeinen 5' zu
der tRNA-Bindungsstelle
und 3' zu der 5'-Hälfte
des dritten offenen Leserahmens (ORF III) in dem viralen Genom positioniert.
Promotoren von ScBV-Isolaten können
durch die Aufreinigung von Virion und/oder viraler DNA mittels der
Methodologie beschrieben in Bouhida et al. (supra) vorbereitet und
die Promotorregion kloniert werden, wobei Verfahren verwendet werden,
die im Stand der Technik gut bekannt sind, beispielsweise das Screenen
einer DNA-Expressionsbibliothek,
die durch die Ligation des ScBV viralen DNA-Fragments mit einem screenbaren Markergen
erstellt wurde. Als Alternative kann ein ScBV-Promotor von der viralen
DNA unter Verwendung eines degenerierten Primers, zum Beispiel BADNAT
(Lockhart und Olszewski, in: Proceedings of INIPAB Conference on
Breeding Banana and Plantains for Pest and Disease, pp. 105–113 (1994))
und eines Primers, der an eine konservierte Region des viralen Genoms
hybridisiert, zum Beispiel der tRNA- Bindungsstelle oder der DNA, die die
virale Replikase kodiert, amplifiziert werden.
-
Eine bevorzugte Ausführungsform
der Erfindung ist ein isoliertes und aufgereinigtes DNA-Molekül, das ein
präselektiertes
DNA-Segment umfasst, das einen ScBV-Promotor umfasst, der SEQ ID
NO: 3, SEQ ID NO: 4 oder SEQ ID NO: 5 oder eine davon abweichende
Nukleotidsequenz (siehe unten) umfasst. Es wird auch bevorzugt,
dass die ScBV-Promotor-enthaltende DNA Sequenzen ausschließt, die
die Translation des RNA-Transkripts, das durch den ScBV-Promotor
generiert wird, inhibieren oder damit interferieren, zum Beispiel,
wenn das RNA-Transkript einen hohen Grad an Sekundärstruktur
hat oder potentielle Start (ATG) -kodons kodiert.
-
Ein präselektiertes DNA-Segment kann
mit dem ScBV-Promotor mittels Standardverfahren kombiniert werden,
wie sie in Sambrook et al. (In: Molecular Cloning: A Laboratory
Manual, Cold Spring Harbor (1989)) beschrieben werden. Kurz, das
präselektierte
DNA-Segment kann stromabwärts
von dem Promotor unter Verwendung von Restriktionsenzymen subkloniert
werden, um sicherzustellen, dass die DNA in der richtigen Orientierung
in Bezug auf den Promotor inseriert wird, so dass die DNA exprimiert
werden kann. Ist das präselektierte
DNA-Segment einmal wirksam mit dem Promotor verbunden, kann die
so gebildete Expresionskassette in ein Plasmid oder in andere Vektoren
subkloniert werden.
-
2. Varianten des DNA-Moleküls gemäß der Erfindung
-
Nukleinsäuremoleküle, die Nukleotidsequenzvarianten
des ScBV-Promotors
kodieren, umfassend SEQ ID NO: 3, SEQ ID NO: 4 oder SEQ ID NO: 5
und dergleichen, können
durch eine Auswahl von Verfahren, die im Stand der Technik bekannt
sind, vorbereitet werden. Diese Verfahren umfassen, sind aber nicht
darauf beschränkt,
die Isolierung aus einer natürlichen
Quelle (im Falle von natürlich
auftretenden Nukleotidsequenzvarianten, zum Beispiel von anderen
Isolaten des ScBV, die aus infiziertem Pflanzenmaterial isoliert
wurden) oder die Vorbereitung mittels Oligonukleotid-vermittelter
(oder sie directed) Mutagenese, PCR-Mutagenese und Kassettenmutagenese einer
früher
vorbereiteten, varianten oder einer nicht-varianten Version des ScBV-Promotors.
Oligonukleotid-vermittelte Mutagenese ist ein bevorzugtes Verfahren
zur Vorbereitung von Nukleotidsubstitutionsvarianten des ScBV-Promotors.
Diese Technik ist im Stand der Technik wohlbekannt, wie durch Adelman
et al., DNA, 2, 183 (1983) beschrieben. Kurz, die ScBV-Promotor-DNA
wird durch die Hybridisierung eines Oligonukleotids verändert, das
die gewünschte
Mutation zu einer DNA-Vorlage kodiert, wobei die Vorlage die einzelsträngige Form
eines Plasmids oder Bakteriophagen ist, das oder der die unveränderte oder
native DNA-Sequenz des ScBV-Promotors enthält. Nach der Hybridisierung
wird eine DNA-Polymerase verwendet, um einen gesamten, zweiten,
komplementären
Strang von der Vorlage zu synthetisieren, was folglich zur Inkorporation
des Oligonukleotidprimers führt
und die ausgewählte
Veränderung
in dem ScBV-Promotor kodieren wird.
-
Im allgemeinen werden Oligonukleotide
von wenigstens 25 Nukleotiden Länge
verwendet. Ein optimales Nukleotid wird 12 bis 15 Nukleotide haben,
die vollständig
komplementär
zu der Vorlage auf jeder Seite des Nukleotids oder der Nukleotide
sind, das oder die die Mutation kodiert oder kodieren. Dieses stellt
sicher, dass die Oligonukleotide richtig mit dem einzelsträngigen DNA-Vorlagemolekül hybridisieren
werden. Die Oligonukleotide sind fertig synthetisiert, wobei Techniken
verwendet werden, die im Stand der Technik bekannt sind, wie diese,
beschrieben durch Crea et al., Proc. Natl. Acad. Sci. U.S.A., 75,
5765, (1978).
-
Die DNA-Vorlage kann durch jene Vektoren
generiert werden, die sich entweder von Bakteriophage-M13-Vektoren
(die kommerziell erhältlichen
M13mp18 und M13mp19 Vektoren sind geeignet) ableiten oder jenen
Vektoren, die einen einzelsträngigen
Phagenreplikationsursprung enthalten, wie er von Viera et al., Meth.
Enzymol., 153, 3 (1987) beschrieben wird. Auf diese Weise kann die
DNA, die mutiert werden soll, in einen dieser Vektoren inseriert
werden, um eine einzelsträngige
Vorlage zu generieren. Die Herstellung der einzelsträngigen Vorlage
wird in den Abschnitten 4.21–4.41
in Sambrook et al., Molecular Cloning: A Laboratory Manual (Cold
Spring Harbor Laboratory Press, N. Y. 1989) beschrieben.
-
Alternativ kann die einzelsträngige DNA-Vorlage
durch die Denaturierung doppelsträngiger Plasmid (oder anderer)
-DNA unter Verwendung von Standardtechniken generiert werden.
-
Für
die Veränderung
der nativen DNA-Sequenz wird das Oligonukleotid an die einzelsträngige Vorlage unter
geeigneten Hybridisationsbedingungen hybridisiert. Ein DNA-polymerisierendes
Enzym, gewöhnlich
das Klenow-Fragment der DNA-Polymerase I, wird dann zugegeben, um
den komplementären
Strang zu der Vorlage zu synthetisieren, wobei das Oligonukleotid
als ein Primer für
die Synthese verwendet wird. Folglich wird ein Heteroduplexmolekül gebildet,
so dass ein Strang der DNA die mutierte Form des ScBV-Promotors
kodiert und der andere Strang (die ursprüngliche Vorlage) die native,
unveränderte
Sequenz des ScBV-Promotors kodiert. Dieses Heteroduplexmolekül wird dann
in eine geeignete Wirtszelle transformiert, gewöhnlich einen Prokaryoten wie
E. coli JM101. Nachdem die Zellen gewachsen sind, werden sie auf
Agaroseplatten ausplattiert und gescreent, wobei der Oligonukleotidprimer
mit 32-Phosphat radiomarkiert wurde, um die bakteriellen Kolonien,
die die mutierte DNA enthalten, zu identifizieren. Die mutierte
Region wird dann entfernt und in einen geeigneten Vektor zur Proteinherstellung
untergebracht, im allgemeinen ein Expressionsvektor von der Art,
die typischerweise für
die Transformation eines geeigneten Wirts eingesetzt wird.
-
Das Verfahren, das direkt oben beschrieben
wurde, kann so modifiziert werden, dass ein Homoduplexmolekül erzeugt
wird, worin beide Stränge
des Plasmids die Mutationen) enthalten. Die Modifikationen sind
wie folgt: das einzelsträngige
Oligonukleotid wird an die einzelsträngige Vorlage wie oben beschrieben
angelagert. Eine Mischung aus drei Desoxyribonukleotiden, Desoxyriboadenosin
(dATP), Desoxyriboguanosin (dGTP) und Desoxyribothymidin (dTTP)
wird mit einem modifizierten Thiodesoxyribocytosin, genannt dCTP-(aS)
(welches von der Amersham Cooperation erhältlich ist) kombiniert. Diese
Mischung wird zu dem Vorlage-Oligonukleotidkomplex
zugegeben. Auf die Zugabe einer DNA-Polymerase zu dieser Mischung
wird ein zweiter Strang der DNA identisch zu der Vorlage, mit Ausnahme
für die
mutierte Base, generiert. Zusätzlich
wird dieser neue DNA-Strang dCTP-(aS) anstelle von dCTP enthalten,
welches dazu dient, den DNA-Strang vor dem Verdau mit Restriktionsendonukleasen
zu schützen.
-
Nachdem der Vorlagenstrang der doppelsträngigen Heteroduplex
mit einem geeigneten Restriktionsenzym genickt worden ist, kann
der Vorlagenstrang mit der Exo-III-Nuklease oder einer anderen geeigneten Nuklease
hinter der Region, die die zu mutagenisierende(n) Stelle(n) enthält, verdaut
werden. Diese Reaktion wird dann angehalten, um ein Molekül zu hinterlassen,
das nur teilweise einzelsträngig
ist. Ein vollständige, doppelsträngige DNA-Homoduplex
wird dann unter Verwendung der DNA-Polymerase in Anwesenheit aller vier
Desoxyribonukleotidtriphosphate, ATP und der DNA-Ligase gebildet.
Dieses Homoduplexmolekül
kann dann in eine geeignete Wirtszelle wie E. coli JM101 transformiert
werden.
-
Die Ausführungsformen der Erfindung
umfassen ein isoliertes und aufgereinigtes DNA-Molekül, das ein
präselektiertes
DNA-Segment umfasst, das einen ScBV-Promotor umfasst, der SEQ ID
NO: 3 oder Nukleotidsequenzvarianten von SEQ ID NO: 3 umfasst, die
nicht die biologische Aktivität
des Promotors reduzieren.
-
3. Bevorzugte, präselektierte
DNA-Segmente
-
Eine bevorzugte Ausführungsform
der Erfindung stellt ein Verfahren zur Einführung eines präselektierten
DNA-Segments in fertile Pflanzen bereit, das, wenn das präselektierte
DNA-Segment von dem ScBV-Promotor in der Pflanze exprimiert wird,
der Pflanze eine gewünschte,
agronomische Eigenschaft verleiht. Solche DNA-Segmente oder „Gene"
werden zum Beispiel in Lundquist et al. (U.S. Patent No. 5,484,956),
Lundquist et al. (U.S. Patent No. 5,508,468), Dobres (internationale
Anmeldung PCT/US95/11231) und von K. Weising et al. (Ann. Rev. Genet.,
22, 421 (1988), siehe Tabellen 1, 2 und 3) offenbart, wobei davon
alle hierin durch Referenz enthalten sind. Die vorliegende Erfindung
ist jedoch im Bereich nicht auf die präselektierten DNA-Segmente beschränkt, die
eine gewünschte,
agronomische Eigenschaft kodieren, da viele andere präselektierte
DNA-Segmente, die Proteine oder RNA-Transkripte kodieren, den Pflanzen
wünschenswerte
Merkmale verleihen, innerhalb des Bereichs der Erfindung sind.
-
Bevorzugte, agronomische Eigenschaften,
die durch das präselektierte
DNA-Segment kodiert werden, umfassen, sind aber nicht darauf beschränkt, Insektenresistenz
oder -toleranz, Herbizidresistenz oder -toleranz, Krankheitsresistenz
oder -toleranz (zum Beispiel Resistenz gegenüber Viren oder Pilzpathogenen), Stresstoleranz
(erhöhte
Salztoleranz), verbesserter Nährwert
oder gesteigerte Erträge.
Genetische Studien haben zum Beispiel gezeigt, dass eine Pflanze,
um der Infektion mit einem bestimmten Pflanzenpathogen zu widerstehen,
ein Resistenz (R) -Gen haben muss, das direkt oder indirekt mit
einem einzigen Avirulenz (avr) -Gen interagiert, das in dem Genom
des Pathogens vorhanden ist. Folglich kann die Einführung eines
präselektierten
DNA-Segments, das ein R-Gen umfasst, in eine Pflanze, der das R-Gen
fehlt, dieser Pflanze gegenüber
einem Pathogen, welches das entsprechende avr-Gen exprimiert, eine
Resistenz verleihen.
-
Eine gesteigerte Resistenz gegenüber Pilzinfektionen
kann durch die Einführung
eines präselektierten DNA-Segments
in eine Pflanze erhalten werden, das ein mit der Pathogenese zusammenhängendes
(PR für englisch
pathogenesis related) Protein kodiert. PR-Proteine sind Proteine,
die von Getreiden als Antwort auf eine Infektion durch einige pathogene
Pilze synthetisiert werden (Scott, Australasian Plant Path., 23,
154 (1994)).
-
Eine gesteigerte Resistenz gegenüber viralen
Infektionen kann durch die Einführung
eines präselektierten
DNA-Segments in eine Pflanze erhalten werden, das ein virales Hüllprotein
kodiert. Nelson et al. (Bio/technol., 6, 403 (1988)) offenbart zum
Beispiel, dass die Expression des Tabakmosaikvirus (TMV) -hüllproteins
in einer Tomatenpflanze der Pflanze Toleranz gegenüber dem
TMV und dem Tomatenmosaikvirus (ToMV), einem Virus verwandt mit
dem TMV, verleiht. Clark et al. (Internationale Anmeldung PCT/EP92/03001) offenbart,
dass die Expression des Maize Dwarf Mosaic Virus-Hüllproteins
in Mais zu Pflanzen führt,
die verringerte Erkrankungssymptome aufzeigen, wenn sie dem Virus
exponiert werden.
-
Vaeck et al. (Nature, 328, 33 (1987))
offenbart, dass die Expression des Bacillus thurigenesis (Bt) -endotoxingens
in Tabak, diese Pflanzen toleranter gegenüber einem Insektenbefall machte.
Lundquist et al. (U.S. Patent No. 5,484,956) offenbart, dass die
Expression des Gens, welches das Bt-Endotoxin kodiert, transgenem
Mais eine Insektenresistenz verleiht.
-
Darüber hinaus wird vorgesehen,
dass mehr als ein präselektiertes
DNA-Segment in eine Pflanze eingeführt werden kann. Zum Beispiel
kann ein Plasmid, das ein selektierbares Markergen (siehe unten)
und ein Gen, das eine Resistenz gegenüber einem bestimmten Virus
verleiht, beispielsweise dem Barley Yellow Dwarf Virus, in regenerierbare
Pflanzenellen eingeführt
werden.
-
4. Optionale Sequenzen in
der Expressionskassette
-
Die Expressionskassette kann optional
auch andere DNA-Sequenzen enthalten.
-
a. Markergene
-
Um das Vermögen zu verbessern Transformanden
zu identifizieren, mag jemand wünschen
ein Markergen, das selektiert und gescreent werden kann, als oder
zusätzlich
zu dem exprimierbaren, präselektierten DNA-Segment einzusetzen. „Markergene"
sind Gene, die Zellen, die das Markergen exprimieren, einen abweichenden
Phänotyp
verleihen und folglich erlauben, solche transformierten Zellen von
Zellen, die keinen Marker haben, zu unterscheiden. Solche Gene können entweder
einen selektierbaren oder screenbaren Marker kodieren, abhängig davon,
ob der Marker ein Merkmal verleiht, auf welches man mit chemischen
Mitteln ‚selektieren’ kann,
das heißt
durch die Verwendung eines selektiven Agenzes (zum Beispiel einem
Herbizid, einem Antibiotikum oder dergleichen) oder ob der Marker
einfach ein Merkmal ist, das man durch Beobachtung und Testen identifiziert
kann, das heißt
durch ‚Screenen’ (zum
Beispiel die β-Glucuronidase).
Natürlich
sind im Stand der Technik viele Beispiele für geeignete Markergene bekannt
und können
in der Ausführung
der Erfindung eingesetzt werden.
-
In den Begriffen selektierbare und
screenbare Markergene sind auch Gene umfasst, die einen „sekretierbaren
Marker" kodieren, dessen Sekretion als ein Mittel zur Identifizierung
oder Selektion von transformierten Zellen detektiert werden kann.
Beispiele umfassen Marker, die ein sekretierbares Antigen kodieren,
das durch eine Antikörperinteraktion
identifiziert werden kann, oder sogar sekretierbare Enzyme, die
durch ihre katalytische Aktivität
detektiert werden können.
Sekretierbare Proteine fallen in eine Anzahl von Klassen, einschließlich kleiner,
diffusionsfähiger
Proteine, die beispielsweise durch einen ELISA detektierbar sind;
kleine aktive Enzyme, die in der extrazellulären Lösung detektierbar sind (zum
Beispiel α-Amylase, β-Lactamase, Phosphinotrizinacetyltransferase);
und Proteine, die in der Zellwand inseriert oder verankert sind
(zum Beispiel Proteine, die eine Leadersequenz umfassen, wie diejenige,
die in der Expressionseinheit von Extensin oder Tabak-PR-S gefunden
wird).
-
Elemente der vorliegenden Offenbarung
sind im Detail durch die Verwendung bestimmter Markergene veranschaulicht,
jedoch werden im Licht dieser Offenbarung zahlreiche andere, mögliche,
selektierbare und/oder screenbare Markergene jenen, die auf diesem
Gebiet Fachkenntnisse haben, zusätzlich
zu den Nachstehenden offensichtlich sein. Folglich ist zu verstehen, dass
die folgenden Diskussion vielmehr exemplarisch als vollständig ist.
Im Licht der hierin offenbarten Techniken und der allgemeinen rekombinanten
Techniken, die im Stand der Technik bekannt sind, macht die vorliegende
Erfindung die Einführung
eines jeden Gens, einschließlich
der von Markergenen, in eine Rezipientenzelle möglich, um ein transformiertes
Monokotyledon zu bilden.
-
1. Selektierbare Marker
-
Mögliche
selektierbare Marker zur Verwendung in Verbindung mit der vorliegenden
Erfindung umfassen, sind aber nicht darauf beschränkt, ein
neo-Gen (Potrykus et al., Mol. Gen. Genet., 199, 183 (1985)), das eine
Kanamyzinresistenz kodiert und durch die Verwendung von Kanamyzin,
G418 und dergleichen selektiert werden kann; das npt II-Gen, das
eine Paromomycinresistenz kodiert; das hyg-Gen, das eine Hygromycin-B-Resistenz kodiert;
ein bar-Gen, das eine Bialaphosresistenz kodiert; ein Gen, das ein
verändertes
EPSP-Synthaseprotein kodiert (Hinchee et al., Biotech., 6, 915,
(1988)), das folglich eine Glyphosatresistenz verleiht; ein Nitrilasegen
wie bxn von Klebsiella ozaenae, das eine Resistenz gegenüber Bromoxynil
verleiht (Stalker et al., Science, 242, 419 (1988)); ein mutiertes
Acetolactatsynthasegen (ALS), das eine Resistenz gegenüber Imidazolinon,
Sulfonylharnstoff und anderen ALS-inhibierenden Chemikalien verleiht
(Europäische
Patentanmeldung 154,204,1985); ein Methotrexatresistenz-DHFR-Gen (Thillet
et al., J. Biol. Chem., 263, 12500 (1988)); ein Dalapondehalogenasegen,
das eine Resistenz gegenüber
dem Herbizid Dalapon verleiht; oder ein mutiertes Anthranilatsynthasegen,
das eine Resistenz gegenüber
5-Methyltryptophan verleiht. Wo ein mutiertes EPSP-Synthasegen eingesetzt
wird, kann ein zusätzlicher
Vorteil durch die Inkorporation eines geeigneten Chloroplastentransitpeptids,
CTP (Europäische
Patentanmeldung 0,218,571, 1987), realisiert werden. Siehe auch
Tabelle 1 von Lundquist et al. (U.S. Patent No. 5,484,956).
-
Eine veranschaulichende Ausführungsform
eines selektierbaren Markergens, das geeignet ist verwendet zu werden,
um Transformanden zu selektieren, ist das Gen, das das Enzym Phosphinotricinacetyltransferase
kodiert wie das bar-Gen (siehe Somers et al., supra (1992)). Das
Enzym Phosphinotricinacetyltransferase (PAT) inaktiviert den aktiven
Bestandteil in dem Herbizid Bialaphos, Phosphinotricin (PPT). PPT inhibiert
die Glutaminsynthase (Murakami et al., Mol. Gen. Genet., 205, 42
(1986); Twell et al., Plant Physiol., 91,1270 (1989)) was eine rasche
Akkumulation von Ammoniak und den Zelltod hervorruft.
-
2. Screenbare Marker
-
Screenabre Marker, die eingesetzt
werden können,
umfassen, sind aber nicht darauf beschränkt, ein β-Glucuronidase oder uidA-Gen
(GUS), das ein Enzym kodiert, für
welches verschiedene, chromogene Substrate bekannt sind; ein R-Lokus-Gen,
das ein Produkt kodiert, welches die Produktion von Anthocyaninpigmenten
(rote Farbe) in Pflanzengeweben reguliert (Dellaporta et al., in
Chromosome Structure and Function, pp. 263–282 (1988)); ein β-Lactamasegen
(Sutcliffe, PNAS USA, 75, 3737 (1978)), das ein Enzym kodiert, für welches
verschiedene chromogene Substrate bekannt sind (zum Beispiel PADAC,
ein chromogenes Cephalosporin), ein xylE-Gen (Zukowsky et al., PNAS
USA, 80, 1101 (1983)), das eine Catecholdioxygenase kodiert, die
chromogene Catechole konvertieren kann; ein α-Amylasegen (Ikuta et al., Biotech.,
8, 241 (1990)); ein Thyrosinasegen (Katz et al., J. Genet. Microbiol.,
129, 2703 (1983), das ein Enzym kodiert, welches zur Oxidation von
Thyrosin zu DOPA und Dopaquinon in der Lage ist, welches wiederum
kondensiert, um die einfach zu detektierende Verbindung Melanin
zu bilden; ein β-Galactosidasegen,
das ein Enzym kodiert, für
das es chromogene Substrate gibt; ein Luziferase (lux) -Gen (Ow
et al., Science, 234, 856 (1986)), das eine Biolumineszenzdetektion
ermöglicht;
oder sogar ein Aequoringen (Prasher et al., Biochem. Biophys. Res.
Comm., 126, 1259 (1985)), das in der kalziumsensitiven Biolumineszenzdetektion
eingesetzt werden kann, oder ein grün fluoreszierendes Proteingen
(Niedz et al., Plant Cell Reports, 14, 403 (1995)).
-
Ein weiterer screenbarer Marker,
der für
die Verwendung in der vorliegenden Erfindung in Erwägung gezogen
wird, ist die Leuchtkäferluziferase,
die von dem lux-Gen kodiert wird. Die Anwesenheit des lux-Gens in
transformierten Zellen kann unter Verwendung von zum Beispiel einem
Röntgenfilm,
einer Szintillationszählung,
einer Fluoreszenzspektrophotometrie, von Schwachlichtvideokameras,
photonenzählenden
Kameras oder einer Multiwell-Luminometrie detektiert werden. Es
ist auch vorgesehen, dass dieses System für das Screenen von Populationen
auf Biolumineszenz wie auf Gewebekulturplatten oder sogar für das Screenen ganzer
Pflanzen weiter entwickelt werden kann.
-
b. Andere Sequenzen
-
Transkriptionsenhancer oder Duplikationen
von Enhancern können
verwendet werden, um die Expression von einem bestimmten Promotor
zu steigern. Beispiele für
solche Enhancer umfassen, sind aber nicht darauf beschränkt, Elemente
des CaMV35S-Promotors und des Octopinsynthasegens (Last et al.,
U.S. Patent No. 5,290,924, herausgegeben am 1. März 1994). Es wird dargelegt,
dass die Verwendung eines Enhancerelements wie dem ocs-Element und insbesondere
viele Kopien des Elements bewirken werden die Transkriptionslevel
von den benachbarten Promotoren zu steigern, wenn es in dem Kontext
der Pflanzentransformation angewendet wird.
-
Da die DNA-Sequenz, die zwischen
der Transkriptionsinitiationsstelle und dem Start der kodierenden Sequenz
inseriert wurde, das heißt
die untranslatierte Leadersequenz, die Genexpression beeinflussen
kann, kann man auch eine bestimmte Leadersequenz verwenden. Bevorzugte
Leadersequenzen umfassen jene, die Sequenzen umfassen, die ausgewählt wurden,
um eine optimale Expression des zugeteilten Gens zu steuern, das
heißt
eine bevorzugte Konsensusleadersequenz zu umfassen, die die mRNA-Stabilität steigern
oder aufrechterhalten kann und eine unpassende Initiation der Translation
verhindert (Joshi, Nucl. Acid Res., 15, 6643 (1987)). Solche Sequenzen
sind jenen, die auf diesem Gebiet Fachkenntnisse haben, bekannt.
Einige Leadersequenzen jedoch, zum Beispiel die Leadersequenz des
RTBV, haben einen hohen Grad an Sekundärstruktur, von der man erwarten
wird, dass sie die mRNA-Stabilität
und/oder die Translation der mRNA vermindert. Folglich werden Leadersequenzen,
die keinen hohen Grad an Sekundärstruktur
haben oder die einen hohen Grad an Sekundärstruktur haben, wobei die
Sekundärstruktur
nicht die mRNA-Stabilität
inhibiert und/oder die Translation vermindert, oder Leadersequenzen,
die sich von Genen ableiten, die in Pflanzen hoch exprimiert, werden
besonders bevorzugt.
-
Regulatorische Elemente wie das Adh-Intron
I (Callis et al., Genes Develop., 1, 1183 (1987)), das Saccharosesynthase-Intron
(Vasil et al., Plant Physiol., 91, 5175 (1989)) oder das TMV-Omega-Element
(Gallie et al., The Plant Cell, 1, 301 (1989)) können auch wo gewünscht umfasst
sein. Weitere solche regulatorische Elemente, die in der Praxis
der Erfindung nützlich
sind, sind jenen, die auf diesem Gebiet Fachkenntnisse haben, bekannt.
-
Zusätzlich können Expressionskassetten konstruiert
und eingesetzt werden, um das Genprodukt des präselektierten DNA-Segments auf
ein intrazelluläres
Kompartiment innerhalb der Pflanzenzelle zu richten oder ein Protein
an die extrazelluläre
Umgebung zu dirigieren. Dieses kann im Allgemeinen durch das Verbinden
einer DNA-Sequenz, die eine Transit- oder Signalpeptidsequenz kodiert,
mit der kodierenden Sequenz des präselektierten DNA-Segments erreicht
werden. Das resultierende Transit- oder Signalpeptid wird das Protein
zu einem bestimmten, intrazellulären
bzw. extrazellulären
Bestimmungsort transportieren und kann dann posttranslational entfernt
werden. Transit- oder Signalpeptide wirken, indem sie den Transport
von Proteinen über
intrazelluläre
Membranen, zum Beispiel die Membranen der Vakuole, Vesikel, Plastiden
und Mitochondrien, erleichtern, während Signalpeptide die Proteine über die
extrazelluläre
Membran leiten. Durch die Erleichterung des Transports des Proteins
in Kompartimente innerhalb oder außerhalb der Zelle können diese Sequenzen
die Akkumulation des Genprodukts erhöhen.
-
Das Transit- oder Signalpeptid, das
von dem präselektierten
DNA-Segment kodiert
wird, kann vielmehr zu einer bestimmte Organelle wie den Chloroplasten
dirigiert werden, als dass es zum Cytoplasma dirigiert wird. Folglich
kann die Expressionskassette ferner eine Chloroplasten-Transitpeptid-kodierende
DNA-Sequenz umfassen, die wirksam zwischen einem ScBV-Promotor und
dem präselektierten
DNA-Segment eingebunden ist (für
einen Review über
plastid targeting Peptide siehe Heijne et al., Eur. J. Biochem.,
180, 535 (1989); Keegstra et al., Ann. Rev. Plant Physiol. Plant
Mol. Biol., 40, 471 (1989)). Dieses wird beispielhaft durch die
Verwendung des rbcS (RuBISCO)-Transitpeptids erläutert, das Proteine spezifisch
zu den Plastiden dirigiert. Siehe zum Beispiel Glassman et al.,
U.S. Patent No. 5,258,300.
-
Es kann nützlich sein, DNA selbst innerhalb
einer Zelle zielgerichtet zu dirigieren. Es kann zum Beispiel nützlich sein,
eine eingeführte,
präselektierte
DNA zu dem Nukleus zu dirigieren, da dieses die Transformationsfrequenz
steigern kann. Innerhalb des Nukleus selbst wäre es nützlich, ein Gen zielgerichtet
zu dirigieren, um eine site spezifiic Integration zu erreichen.
Es wäre
zum Beispiel nützlich,
ein durch Transformation eingeführtes
Gen dazu zu bringen, ein existierendes Gen in der Zelle zu replazieren.
-
Wenn die Expressionskassette in eine
Pflanzenzelle eingeführt
werden soll, kann die Expressionskassette auch optional eine 3'-untranslatierte
pflanzenregulatorische DNA-Sequenz umfassen, die als ein Signal wirkt,
die Transkription zu terminieren und die Polyadenylierung der resultierenden
mRNA vorsieht. Die 3'-untranslatierte, regulatorische DNA-Sequenz
umfasst vorzugsweise von etwa 300 bis 1000 Nukleotidbasenpaare und
enthält
pflanzliche Transkriptions- und Translationsterminationssequenzen.
Bevorzugte 3'-Elemente leiten sich von jenen des Nopalinsynthasegens
von Agrobacterium tumefaciens (Bevan et al., Nucl. Acid Res., 11, 369
(1983)), dem Terminator für
das T7-Transkript des Octopinsynthasegens von Agrobacterium tumefaciens und
dem 3'-Ende des Proteaseinhibitor I- oder II-Gens aus Kartoffel oder Tomate ab, obwohl
andere 3'-Elemente jenen, die auf diesem Gebiet Fachkenntnisse haben,
bekannt sind, auch eingesetzt werden können. Diese 3' untranslatierten,
regulatorischen Sequenzen können
wie in An, Methods in Enzymology, 153, 292 (1987) beschrieben, erhalten
werden oder sind bereits in Plasmiden von kommerziellen Quellen
wie Clontech, Palo Alto und California erhältlich. Diese 3' untranslatierten,
regulatorischen Sequenzen können
wirksam mit dem 3'-Terminus des präselektierten DNA-Segments verbunden
werden.
-
Es kann auch eine Expressionskassette
in einen Expressionsvektor wie ein Plasmid eingeführt werden.
Plasmidvektoren umfassen zusätzlich
DNA-Sequenzen, die
eine einfache Selektion, Amplifikation und Transformation der Expressionskassette
in prokaryotischen und eukaryotischen Zellen, zum Beispiel Vektoren die
sich von pUC, pSK, pGEM, pSP oder pBS ableiten, bereitstellen. Folglich
umfassen zusätzliche
DNA-Sequenzen Replikationsursprünge,
um eine autonome Replikation des Vektors bereitzustellen, selektierbare Markergene,
die vorzugsweise Antibiotika- oder Herbizidresistenzen kodieren,
einzigartige Multiple Cloning Sites, die mehrere Stellen vorsehen
DNA-Sequenzen oder Gene zu inserieren, die in der Expressionskassette kodiert
werden, und Sequenzen, die die Transformation von prokaryotischen
und eukaryotischen Zellen steigern.
-
Ein weiterer Vektor, der für die Expression
in beiden, pflanzlichen und prokaryotischen Zellen, brauchbar ist,
ist das binäre
Ti-Plasmid (wie in Schilperoort et al. offenbart, U.S. Patent No.
4,940,838, herausgegeben am 10. Juli 1990), wie es durch den Vektor
pGA582 exemplarisch erläutert
wird. Dieser binäre
Ti-Plasmid-Vektor ist früher
durch An charakterisiert worden und von Dr. An erhältlich (siehe
Methods in Enzymology, 153, 292 (1987)). Dieser binäre Ti-Vektor
kann in prokaryotischen Bakterien wie E. coli und Agrobacterium
repliziert werden. Die Agrobacterium-Plasmidvektoren können verwendet
werden, um die Expressionskassette in Pflanzenzellen zu transferieren.
Der binäre
Ti-Vektor umfasst vorzugsweise den rechten und linken Rand der Nopalin
T-DNA, um eine effiziente Pflanzenzell-Transformation bereitzustellen, ein
selektierbares Markergen, einzigartige Multiple Cloning Sites in
den T-Randregionen, den colE1-Replikationsursprung
und ein weites Wirtsspektrumsreplikon des. Die binären Ti-Vektoren,
die eine Expressionskassette gemäß der Erfindung tragen,
können
verwendet werden, um beide, prokaryotische und eukaryotische Zellen,
zu transformieren, werden aber vorzugsweise verwendet, um Pflanzenzellen
zu transformieren.
-
III. DNA-Übertragung
-
Die Expressionskassette oder der
Expressionsvektor wird dann in eine Rezipientenzelle eingeführt, um
eine transformierte Zelle zu bilden. Für die Einführung einer Expressionskassette
in eine Pflanzenzelle wird die Häufigkeit
des Auftretens, dass eine Pflanzenzelle DNA erhält, als niedrig angenommen.
Darüber
hinaus werden höchstwahrscheinlich
nicht alle Rezipientenzellen, die ein DNA-Segment oder eine DNA-Sequenz
erhalten, zu einer transformierten Zelle führen, worin die DNA stabil
in das Pflanzengenom integriert und/oder exprimiert wird. Einige
könnten
nur eine anfängliche
oder transiente Genexpression zeigen. Jedoch könnten bestimmte Zellen von
praktisch jeder Pflanze stabil transformiert werden und diese Zellen
zu transgenen Pflanzen regeneriert werden.
-
Ein präselektiertes DNA-Segment kann
in Pflanzenzellen oder Geweben oder in prokaryotische oder eukaryotische
nicht-pflanzliche Zellen durch derzeitig erhältliche Verfahren übertragen
werden, die die Protoplastentransformation, das Tungston Whiskers
(Coffee et al., U.S. Patent No. 5,302,523, herausgegeben am 12.
April 1994), direkt durch Mikroorganismen mit infektiösen Plasmiden,
infektiösen
Viren, die Verwendung von Liposomen, die Mikroinjektion durch mechanische
oder Laserstrahlverfahren, durch ganze Chromosomen oder Chromosomenfragmente,
die Elektroporation, die Silikonkarbitfibern und das Mikroprojetilbombardement umfassen,
aber nicht darauf beschränkt
sind. Eine bevorzugte Ausführungsform
der Erfindung erreicht die Einführung
eines präselektierten
DNA-Segments in monokotyle Zellen durch Transformationsverfahren,
die besonders effektiv für
Monokotyledonen sind und das Mikroprojetilbombardement (siehe Lundquist
et al., U.S. Patent No. 5,538,877) umfassen, aber nicht darauf beschränkt sind.
-
Es wurde gezeigt, dass die Einführung und
Expression fremder Gene in dikotyle (breitblättrige) Pflanzen, wie Tabak,
Kartoffel und Alfalfa unter Verwendung der tDNA des tumorinduzierienden
(Ti) Plasmids von Agrobacterium tumefaciens möglich ist (siehe zum Beispiel
Umbeck, U.S. Patent No. 5,004,863, und die internationale Anmeldung
PCT/US93/02480). Unter Verwendung rekombinanter DNA-Techniken und
Bakteriengenetik kann eine breite Auswahl fremder DNAs in die tDNA
in Agrobacterium inseriert werden. Im Anschluss an die Infektion
durch das Bakterium, das das rekombinante Ti-Plasmid enthält, wird
die fremde DNA in das Wirtspflanzenchromosom inseriert, wobei folglich
eine genmanipulierte Zelle und letztendlich eine genmanipulierte Pflanze
hergestellt wird. Ein zweiter Ansatz ist es, wurzelinduzierende
(Ri für
Englisch Root inducing) Plasmide als Genvektoren einzuführen.
-
Dikotyledonen sind für die Transformation
durch Agrobacterium empfänglich.
Kürzlich
wurde gezeigt, dass Reis und Mais, welche Monokotylen sind, ebenso
für die
Transformation durch Agrobacterium empfänglich sind. Viele andere,
wichtige, monokotyle Getreidepflanzen jedoch, einschließlich Weizen,
Gerste, Hafer, Sorghum, Hirse und Roggen sind bis jetzt noch nicht
erfolgreich durch Agrobacterium transformiert worden. Das Ti-Plasmid
jedoch könnte
in der Zukunft so manipuliert werden, um als ein Vektor für diese
anderen monokotylen Pflanzen zu fungieren. Zusätzlich könnte es unter Verwendung des
Ti-Plasmids als ein Modellsystem möglich sein, artifiziell Transformationsvektoren
für diese
Pflanzen zu konstruieren. Ti-Plasmide könnten auch in monokotyle Pflanzen
durch artifizielle Verfahren eingeführt werden, wie durch eine
Mikroinjektion oder eine Fusion von monokotylen Protoplasten und
bakteriellen Spheroplasten, die die T-Region enthalten, die dann
in die pflanzliche, nukleäre
DNA integriert werden kann.
-
Andere Transformationsverfahren für Dikotyledonen
umfassen das Blattscheibenverfahren von Horsch et al. (Science,
227, 1229 (1985)) und wie von Fry et al. (Plant Cell Reports, 6,
321 (1987)) für
Brassica napus adaptiert.
-
IV. Herstellung und Charakterisierung
von stabil transgenen Pflanzen
-
Nach der Bewerkstelligung der Übertragung
eines präselektierten
DNA-Segments auf die Rezipientenzellen durch irgendeines der oben
diskutierten Verfahren, involviert der nächste Schritt der Erfindung
im Allgemeinen die Identifizierung der transformierten Zellen für die weitere
Kultivierung und Pflanzenregeneration. Wie oben erwähnt kann
man, um das Vermögen
zu verbessern Transformanden zu identifizieren, ein selektierbares
oder screenbares Markergen als oder zusätzlich zu dem exprimierbaren,
präselektierten
DNA-Segment einsetzen. In diesem Fall würde man dann im Allgemeinen
die potentiell transformierte Zellpopulation durch die Exposition
der Zellen zu einem selektiven Agens oder zu selektiven Agenzien
untersuchen oder man würde
die Zellen auf das gewünschte
Markergenmerkmal screenen.
-
A. Selektion
-
Eine exemplarische Ausführungsform
der Verfahren zur Identifizierung transformierter Zellen involviert die
Exposition bombardierter Kulturen mit einem selektiven Agens wie
einem metabolischen Inhibitor, einem Antibiotikum, einem Herbizid
oder dergleichen, wie es hierin oben beschrieben wird. Zellen, die
transformiert worden sind und die ein Markergen stabil integriert
haben, das ihnen Resistenz gegenüber
dem verwendeten, selektiven Agens verleiht, werden wachsen und sich
in Kultur teilen. Sensitive Zellen werden nicht für eine weitere
Kultivierung zugänglich
sein.
-
Es wird ferner in Erwägung gezogen,
dass Kombinationen von screenbaren und selektierbaren Markern zur
Identifikation transformierter Zellen verwendet werden. In einigen
Zell- oder Gewebetypen könnte
ein Selektionsagens wie das Antibiotikum Kanamyzin oder G418 eine
entweder nicht ausreichend selektive Abtötung bereitstellen, um deutlich
transformierte Zellen zu identifizieren, oder könnte eine erhebliche, nicht-selektive
Inhibierung von Transformanden und Nicht-Transformanden gleichermaßen verursachen,
was folglich dazu führen
könnte,
dass die Selektionstechnik fehl schlägt. Es wird vorgeschlagen,
dass die Selektion mit einer wachstumsinhibierenden Verbindung wie
einem Antibiotikum, mit Konzentrationen unter jenen, die eine 100%
Inhibition verursachen, gefolgt von dem Screenen des wachsenden
Gewebes auf die Expression eines screenbaren Markergens wie gus
(beta-Glucuronidase) oder lux (Luciferase) einem ermöglichen
würde,
Transformanden von Zellen oder Gewebetypen wiederzugewinnen, die
für die
Selektion alleine nicht zugänglich sind.
Deshalb können
Kombinationen von Selektion und Screening die Indentifikation von
Transformanden in einer breiteren Auswahl von Zellen und Gewebetypen
ermöglich.
-
B. Regeneration und Samenherstellung
-
Zellen, die die Exposition mit dem
selektiven Agens überleben,
oder Zellen, die in einer Screening-Untersuchung positiv gezählt wurden,
können
in Medien, die die Regeneration der Pflanzen unterstützen, kultiviert
werden. Den transformierten Zellen, die durch Selektion oder Screening
identifiziert wurden und in einem geeigneten Medium, das die Regeneration
unterstützt,
kultiviert wurden, wird dann ermöglicht
werden, sich zu reifen Pflanzen zu regenerieren. Nachdem die Pflanzen
das Stadium der Trieb- und Wurzelentwicklung erreicht haben, können sie
für weiteres
Wachstum und Testen in ein Gewächshaus
transferiert werden.
-
Reife Pflanzen werden dann von Zellinien
erhalten, die identifiziert werden, wenn sie das präselektierte
DNA-Segment exprimieren. Wenn möglich
werden die regenerierten Pflanzen selbst bestäubt. Zusätzlich werden Pollen, die von
den regenerierten Pflanzen erhalten wurden, mit samengezogenen Pflanzen
agronomisch wichtiger Pflanzengenotypen gekreuzt. In einigen Fällen wird
Pollen von Pflanzen dieser Genotypen verwendet, um die regenerierten
Pflanzen zu bestäuben.
Das Merkmal wird genetisch durch die Auswertung der Merkmalssegregation
in der Nachkommenschaft der ersten und der späteren Generationen charakterisiert. Die
Erblichkeit und Expression in Pflanzen von Merkmalen, die in Gewebekultur
selektiert wurden, sind von besonderer Wichtigkeit, falls die Merkmale
kommerziell brauchbar sein sollen.
-
Regenerierte Pflanzen können wiederholt
mit anderen Pflanzengenotypen gekreuzt werden, um das präselektierte
DNA-Segment in das Genom der anderen Pflanzen einzukreuzen (für Englisch
introgress). Dieser Prozess wird als Rückkreuzungskonversion bezeichnet.
Wenn eine ausreichende Anzahl von Kreuzungen mit dem wiederauftretenden
Elter vervollständigt
worden sind, um ein Produkt des Rückkreuzungskonversionsprozesses
herzustellen, welches im wesentlichen mit dem wiederaufgetretenen
Elter isogen ist, mit Ausnahme für
die Anwesenheit des eingeführten
präselektierten
DNA-Segments, wird die Pflanze wenigstens einmal selbst bestäubt, um
eine homozygote, rückkreuzungskonvertierte
Pflanze herzustellen, die das präselektierte
DNA-Segment enthält.
Die Nachkommen dieser Pflanzen sind eine echte Züchtung.
-
Alternativ werden Samen von transformierten
Pflanzen, die von transformierten Gewebekulturen regeneriert wurden,
auf dem Feld gezogen und selbst bestäubt, um echte Züchtungspflanzen
zu generieren.
-
Sind die echten Züchtungslinien einmal selektiert,
werden Testkreuzungen gemacht und es wird Hybridsame hergestellt.
Die Nachkommen von diesen Pflanzen werden echte Züchtungslinien.
Die Testkreuzungshybriden und Züchtungspopulationen
werden in mehreren verschiedenen Anordnungen auf dem Feld angepflanzt.
Ein Evaluationsschema ist, die Populationen von Hybridpflanzen,
die das präselektierte
DNA-Segment enthalten, an vielen verschiedenen Orten wachsen zu
lassen und die Leistung der Pflanzen an diesen verschiedenen Orten
zu messen. Informationen werden gewonnen, sowie andere Messungen
der Pflanzengesundheit, der Überlegenheit
und der Lebensfähigkeit
werden gemacht. Die Informationen bezüglich der Leistung dieser Hybriden
einhergehend mit der Leistung von nicht-transformierten Hybriden
wird verglichen.
-
Auf die Identifikation der besseren
Leistung der transgenen Pflanzen sind die Elternselektionen besser und
es wird eine Inzuchtlinie mittels konventioneller Zuchttechniken
hergestellt. Die Hybridpflanzen, die ein oder mehrere Eltern haben,
die das präselektierte
DNA-Segment enthalten, werden in kommerziellen Tests und Evaluationsprogrammen
getestet und die Leistung dokumentiert. Dieses Testen schließt die Evaluation der
Leistungsmerkmale ein, durchgeführt über einen
weiten geographischen Bereich, sowie die Verwendung der dedizierten
Merkmale, um einen Leistungsvorteil und infolge dessen, den Wert
aufzuzeigen.
-
Ein zusätzlicher Vorteil der Expression
des präselektierten
DNA-Segments ist
die überlegene
Leistung der elterlichen Linien bei der Produktion von Hybriden.
-
C. Charakterisierung
-
Um die Anwesenheit des oder der präselektierten
DNA-Segment oder Segmente oder „transgens" oder „transgene"
in den regenerierten Pflanzen zu bestätigen, kann eine Auswahl von
Untersuchungen durchgeführt
werden. Solche Nachweise schließen
zum Beispiel ein „molekularbiologische"
Nachweise, die jenen, welche auf diesem Gebiet Fachkenntnisse haben,
gut bekannt sind, wie Southern und Northern Blotting und PCR; „biochemische"
Nachweise wie die Detektion der Anwesenheit eines Proteinprodukts,
zum Beispiel durch immunologische Mittel (ELISA's und Western Blots)
oder durch die enzymatische Funktion; Pflanzenteilnachweise wie
Blatt- oder Wurzelnachweise; und auch durch die Analyse des Phänotyps der
gesamten regenerierten Pflanze.
-
1. DNA-Integration, RNA-Expression
und Vererbung
-
Genomische DNA kann aus Kalluszellinien
oder jedem Pflanzenteil isoliert werden, um die Präsenz des
präselektierten
DNA-Segments zu bestimmen, wobei Techniken verwendet werden, die
jenen, die auf diesem Gebiet Fachkenntnisse haben, gut bekannt sind.
Bitte nehmen sie zur Kenntnis, dass die intakte Sequenz nicht immer
vorhanden sein muß,
vermutlich aufgrund von Rearrangements oder Deletionen von Sequenzen in
der Zelle.
-
Die Präsenz von DNA-Elementen, die
mittels der Verfahren gemäß Erfindung
eingeführt
wurden, kann durch die Polymerasekettenreaktion (PCR für Englisch
polymerase chain reaction) bestimmt werden. Unter Verwendung dieser
Technik werden diskrete DNA-Fragmente amplifiziert und mittels Gelelektrophorese
detektiert. Diese Art von Analyse erlaubt einem zu bestimmen, ob
ein präselektiertes
DNA-Segment in einem stabilen Transformanden vorhanden ist, aber
es weißt
nicht die Integration des eingeführten,
präselektierten DNA-Segments
in das Wirtszellgenom nach. Zusätzlich
ist es nicht möglich
unter Verwendung von PCR-Techniken zu bestimmen, ob die Transformanden
exogene Gene haben, die an verschiedene Stellen in das Genom eingeführt wurden,
d. h. ob die Transformanden von einem unabhängigen Ursprung sind. Es wird
in Erwägung gezogen,
dass es unter Verwendung von PCR-Techniken möglich wäre, Fragmente der genomischen
DNA des Wirts zu klonen, die zu einem eingeführten, präselektierten DNA-Segment benachbart
sind.
-
Eine positive Probe für die DNA-Integration
in das Wirtszellgenom und die unabhängigen Identitäten der
Transformanden können
unter Verwendung der Southern Hybridisationstechnik bestimmt werden.
Unter Verwendung dieser Technik können spezifische DNA-Sequenzen,
die in das Wirtszellgenom eingeführt
worden sind und die Wirts-DNA-Sequenzen flankieren, identifiziert
werden. Infolge dessen dient das Southern-Hybridisationsmuster für einen
gegebenen Transformanden als ein Identifikationskennzeichen dieses
Transformanden. Zusätzlich
ist es mittels Southern Hybridisation möglich, die Anwesenheit von
eingeführten,
präselektierten
DNA-Segmenten in DNA hohen Molekulargewichts zu demonstrieren, d.
h. zu bestätigen,
dass das eingeführte,
präselektierte
DNA-Segment in das
Wirtszellgenom integriert worden ist. Die Technik der Southern-Hybridisation
stellt Informationen bereit, die unter Verwendung von PCR erhalten
wird, zum Beispiel die Anwesenheit eines präselektierten DNA-Segments, aber demonstriert
auch die stabile Integration in das Genom und kennzeichnet jeden
individuellen Transformanden.
-
Es wird in Erwägung gezogen, dass man unter
Verwendung der Techniken der Dot- oder Slot-Blot-Hybridisierung,
welche Modifikationen der Southern-Hybridisationstechniken sind,
die selbe Information erhalten kann, die aus der PCR abgeleitet
wird, zum Beispiel die Anwesenheit eines präselektierten DNA-Segments.
-
Beides, die PCR und Southern Hybridisationstechniken,
können
verwendet werden, um die Übertragung
eines präselektierten
DNA-Segments auf die Nachkommenschaft zu demonstrieren. In den meisten
Fällen
wird das kennzeichnende Southern-Hybridisationsmuster für einen
gegebenen Transformanden in der Nachkommenschaft als eines oder
mehrere, mendelsche Gene segregieren (Spencer et al., Plant Mol.
Biol., 18, 201 (1992); Laursen et al., Plant Mol. Biol., 24, 51
(1994)), wobei die stabile Vererbung des Gens angezeigt wird.
-
Während
DNA-Analysetechniken unter Verwendung von DNA durchgeführt werden
können
die aus jedem Teil einer Pflanze isoliert worden ist, mag RNA nur
in bestimmten Zellen oder Gewebetypen exprimiert sein und es wird
infolge dessen nötig
sein, RNA für
die Analyse aus diesen Geweben vorzubereiten. PCR-Techniken können auch
zur Detektion und Quantifizierung von RNA verwendet werden, die
von den eingeführten,
präselektierten
DNA-Segmenten hergestellt wurden. Bei dieser Anwendung der PCR ist
es als erstes nötig,
die RNA revers in DNA zu transkribieren, wobei Enzyme wie die reverse
Transkriptase verwendet werden, und dann die DNA unter Verwendung
konventioneller PCR-Techniken zu amplifizieren. In den meisten Fällen werden
PCR-Techniken, obwohl nützlich,
nicht die Integrität
des RNA-Produkts demonstrieren. Weitere Informationen über die
Beschaffenheit des RNA-Produkts kann durch Northern Blotting erhalten
werden. Diese Technik wird die Anwesenheit einer RNA-Spezies demonstrieren
und Informationen über
die Integrität
dieser RNA ergeben. Die An- oder Abwesenheit einer RNA-Spezies kann
auch unter Verwendung von Dot- oder Slot-Blot-Northern-Hybridisierungen
bestimmt werden. Diese Techniken sind Modifikationen des Northern Blottings
und werden nur die An- oder Abwesenheit einer RNA-Spezies demonstrieren.
-
2. Genexpression
-
Während
Southern Blotting und PCR verwendet werden können um das in Frage gestellte,
präselektierte
DNA-Segment zu detektieren, stellen sie keine Informationen dahingehend
bereit, ob das präselektierte DNA-Segment
exprimiert sein wird. Die Expression kann durch die spezifische
Identifizierung des Proteinprodukts der eingeführten, präselektierten DNA-Segmente oder die
Evaluation der phänotypischen
Veränderungen,
die durch ihre Expression herbeigeführt werden, evaluiert werden.
-
Untersuchungen zur Herstellung und
Indentifizierung spezifischer Proteine kann sich physikalisch-chemische,
strukturelle, funktionelle oder andere Eigenschaften der Proteine
zu Nutze machen. Einmalige, physikalischchemische oder strukturelle
Eigenschaften erlauben die Proteine zu separieren und durch elektrophoretische
Verfahren zu identifizieren, wie native oder denaturierende Gelelektrophorese
oder isoelektrische Fokussierung oder chromographische Techniken
wie Ionenaustauscher oder Gelausschlußchromatographie. Spezifische
Antikörper
können
verwendet werden, um die einzigartigen Strukturen von Proteinen
mittels Aufmachungen wie einer ELISA-Untersuchung zu detektieren,
zum Beispiel um npt II zu detektieren. Kombinationen dieser Ansätze können eingesetzt
werden, um eine noch höhere
Spezifität
zu erhalten, die dem Western Blotting, bei welchem Antikörper verwendet
werden, die an individuelle Genprodukte binden, die durch elektrophoretische
Techniken aufgetrennt worden sind. Zusätzliche Techniken können eingesetzt
werden, um unumschränkt
die Identität
des Produkts von Interesse zu bestätigen, wie die Evaluation durch
eine Aminosäuresequenzierung
in Folge einer Aufreinigung. Obwohl diese Verfahren zu den am häufigsten
eingesetzten Verfahren gehören,
können
andere Verfahren zusätzlich
verwendet werden.
-
Es können auch Untersuchungsverfahren
verwendet werden, um die Expression von Proteinen durch ihre Funktionalität zu identifizieren,
insbesondere die Fähigkeit
von Enzymen spezifische, chemische Reaktionen zu katalysieren, die
spezifische Substrate und Produkte miteinbeziehen. Diese Reaktionen
können
durch die Bereitstellung und Quantifizierung des Substratverlusts
oder der Generierung von Produkten dieser Reaktionen von physikalischen
oder chemischen Verfahren gefolgt sein.
-
Die Expression eines Genprodukts
ist sehr häufig
durch die Evaluation der phänotypischen
Ergebnisse ihrer Expression bestimmt. Diese Untersuchungen können auch
viele Formen annehmen, einschließlich, aber nicht darauf beschränkt, der
Analyse der Veränderungen
in der chemischen Zusammensetzung, Morphologie oder der physiologischen
Eigenschaften der Pflanze. Die chemische Zusammensetzung kann durch
die Expression der präselektierten
DNA-Segmente verändert
werden, die Proteine kodieren, die die Pigmentierung von Pflanzenteilen
betreffen und können
phänotypisch
oder durch ein Produkt detektiert werden, das vermehrt wird, wenn
das Protein, das durch das präselektierte
DNA-Segment kodiert ist, exprimiert wird, was durch eine Hochleistungs-Flüssigchromatographie
oder einen ELISA (zum Beispiel npt II) analysiert werden kann.
-
D. Etablierung der eingeführten DNA
in andere Pflanzenvarietäten
-
Fertile, transgene Pflanzen können dann
in einem konventionellen Pflanzenkreuzungsprogramm verwendet werden,
um das präselektierte
DNA-Segment in die
gewünschten
Linien oder Varietäten
zu inkorporieren.
-
Im Allgemeinen wird der kommerzielle
Wert der transformierten Pflanze, die hierin hergestellt wird, am höchsten sein,
falls das präselektierte
DNA-Segment in viele verschiedene Hybridkombinationen inkorporiert werden
kann. Ein Landwirt baut typischerweise mehrere Hybriden an, basierend
auf Unterschieden in der Reife, der Standabilität und anderen agronomischen
Merkmalen. Außerdem
muss der Landwirt basierend auf seiner oder ihrer geographischen
Lage eine Hybride auswählen,
da Hybriden, die zu einer Region adaptiert sind, generell nicht
zu anderen Regionen adaptiert sind, aufgrund der Unterschiede in
solchen Merkmalen wie Reife, Krankheits-, Dürre- und Insektenresistenz.
Als solches ist es notwendig das Gen in eine große Anzahl parentaler Linien
zu inkorporieren, sodass viele Hybridkombinationen hergestellt werden
können,
die das präselektierte
DNA-Segment enthalten.
-
Pflanzenkreuzung und die Techniken
und Fertigkeiten, die verlangt sind, um Gene von einer Linie oder Varietät zu einer
Anderen zu Transferrieren, sind jenen, die auf diesem Gebiet Fachkenntnisse
haben, gut bekannt. Folglich kann die Einführung eines präselektierten
DNA-Segments, vorzugsweise in Form von rekombinanter DNA, in irgendeine
andere Linie oder Varietät
durch diese Zuchtverfahren bewerkstelligt werden.
-
E. Verwendung transgener
Pflanzen
-
Es wird erwartet, dass die transgenen
Pflanzen, die hierin hergestellt werden, nützlich für eine Vielfalt kommerzieller
Zwecke und Forschungszwecke nützlich
sind. Transgene Pflanzen können
für die
Verwendung in der traditionellen Landwirtschaft kreiert werden,
um Merkmale zu besitzen, die für
den Erzeuger von Vorteil sind (zum Beispiel agronomische Merkmale
wie die Resistenz gegenüber
einem Wasserdefizit, eine Schädlingsresistenz,
eine Herbizidresistenz oder ein gesteigerter Ertrag), die für den Konsumenten
des von der Pflanze geernteten Getreides von Vorteil sind (zum Beispiel
ein verbesserter Nährwert
in der menschlichen Nahrung oder im Tierfutter) oder die für den Essensverarbeiter
von Vorteil sind (zum Beispiel verbesserte Verarbeitungsmerkmale).
In solchen Anwendungen werden die Pflanzen im Allgemeinen für die Verwendung
ihres Getreides in Lebensmitteln für Menschen oder Tiere angebaut.
Andere Teile der Pflanzen jedoch, einschließlich der Stengel, den Schalen,
den vegetativen Teilen und dergleichen, können auch einen Nutzen haben,
einschließlich
einer Verwendung als Tiersilage oder für dekorative Zwecke. Häufig werden
chemische Getreidebestandteile für
Nahrungsmittel und industrielle Verwendung extrahiert und es können transgene
Pflanzen kreiert werden, die gesteigerte oder veränderte Level
solcher Komponenten haben.
-
Transgene Pflanzen können auch
in der kommerziellen Herstellung von Proteinen oder anderen Verbindungen
Nutzen finden, wo die Verbindung von Interesse aus Pflanzenteilen,
Samen oder dergleichen extrahiert oder aufgereinigt wird. Zellen
oder Gewebe von diesen Pflanzen können kultiviert, in vitro gewachsen oder
fermentiert werden, um solche Moleküle herzustellen.
-
Transgene Pflanzen können auch
in kommerziellen Züchtungsprogrammen
verwendet werden oder können
mit Pflanzen verwandter Getreidearten gekreuzt oder gezüchtet werden.
Die Verbesserungen, die von dem präselektierten DNA-Segment kodiert
werden, können
zum Beispiel von Zellen der einen Pflanzenspezies zu Zellen der
anderen Pflanzenspezies, zum Beispiel durch Protoplastenfusion, übertragen
werden.
-
Die transgenen Pflanzen können viele
Verwendungen in der Forschung oder Züchtung haben, einschließlich der
Kreation neuer, mutanter Pflanzen durch Insertionsmutagenese, um
vorteilhafte Mutanten zu identifizieren, die später durch traditionelle Mutation
und Selektion kreiert werden könnten.
Ein Beispiel wäre die
Einführung
einer rekombinanten DNA-Sequenz,
die ein transponierbares Element kodiert, das für die Generierung einer genetischen
Variation verwendet werden kann. Die Verfahren gemäß der Erfindung
können auch
verwendet werden, um Pflanzen zu kreieren, die einmalige „Signatursequenzen"
oder andere Markersequenzen haben, die verwendet werden können, um
geschützte
Linien oder Varietäten
zu identifizieren.
-
Die Erfindung wird durch die folgenden
Beispiele weiter beschrieben werden.
-
Beispiel I
-
Expressionskassetten für die Pflanzentransformation
-
Um eine Expressionskassette, die
einen ScBV-Promotor umfasst, werden drei Regionen des ScBV-Genoms
mittls der Polymerasekettenreaktion (PCR für Englisch polymerase chain
reaction) amplifiziert. Die Regionen sind stromaufwärts der
offenen Leseramen und stromabwärts
einer putativen tRNA-Bindungsstelle und umfassen keine AUG-Codons.
Die PCR setzt einen BamHI-linearisierten pScBV-20 als die Vorlage ein
(Bouhida et al., J. Gen. Virol., 74, 1 (1993)). Die Primer-Paare,
die in der PCR eingesetzt werden, sind in Tabelle 1 dargestellt.
Die Amplifikationsreaktionen ergaben sich in den folgenden Regionen
des ScBV-Genoms, das amplifiziert wurde: Nukleotidpositionen 5999–7205 (amplifiziert
mit dem Primer SCBV-PRO FORW-5999, SEQ ID NO: 6 und dem Primer SCBV-PRO
REV-7205, SEQ ID NO: 7), 5999–7299
(amplifiziert mit dem Primer SCBV-PRO FORW-5999, SEQ ID NO: 6 und dem Primer SCBV-PRO
REV-7299, SEQ ID NO: 8) und 5999–7420 (amplifiziert mit dem
Primer SCBV-PRO FORW-5999, SEQ ID NO: 6 und dem Primer SCBV-PRO
REV-7420, SEQ ID NO: 9) (entsprechend dem Nummerierungssystem in
Bouhida et al., supra). SCBV-PRO
FORW-5999 wurde so synthetisiert, dass er eine PstI-Stelle umfasste.
ScBV-PRO REV-7205, SCBV-PRO REV-7299 und SCBV-PRO REV-7420 wurden
so synthetisiert, dass sie eine StuI-Stelle umfassten. Nach der
Amplifikation wurden die PCR-Produkte mit PstI und StuI verdaut
und dann Gel-aufgereinigt.
-
Um Expressionsvektoren für Monokotyledonen
vorzubereiten, wurden die Gelfragmente mit pMON755i ligiert (Medberry
und Olszewski, Plant J., 3, 619 (1993)), der mit Pst1 und Stu1 verdaut
wurde, um das CaMV35S-Promotorfragment
zu entfernen. pMON755i wurde von pMON755 abgeleitet (Medberry et
al., The Plant Cell, 4, 185 (1992)), indem ein modifizierten Mais-Alkoholdehydrogenase
ersten Intronsegments 5' zu dem GUS-Gen inseriert wurde. Die resultierenden
Plasmide wurden mit ScBV-1 (5999–7205), ScBV-2 (5999–7299) und
ScBV-3 (5999–7240)
bezeichnet.
-
-
Um Expressionsvektoren für die Agrobakterium-vermittelte
Transformation vorzubereiten, wird ein binärer Expressionsvektor konstruiert.
ScBV-3 wurde mit PstI und StuI verdaut und ein 1,4 kb ScBV-enthaltendes
Fragment an pBluescriptKS+ ligiert (Stratagene,
La Jolla, CA), der mit SpeI verdaut worden ist, die überhängenden
Enden mit dem Klenow-Fragment aufgefüllt wurden und dann mit Pst1
verdaut wurde. Aus dem resultierenden Konstrukt wurde ein XhoI-XbaI-Fagment
isoliert und mit pOCA101 ligiert, das mit Sal1 und Xba1 verdaut
worden ist. pOCA101 ist ein Derivat von pOCA28 (Medberry et al.,
Nucl. Acids Res., 18, 5505 (1990)), in welchem der Polylinker durch
ein HindIII-EcoRI-Fragment ersetzt wird, welches das GUS-Gen enthält. pOCA28
(Olszewski et al., Nucl. Acids Res., 16, 10765 (1988)) hat ein Smr/Spcr-Gen von pHP45omega
(Prenti und Kritsch, Gene, 28, 303 (1984)), das ein BglII/SmaI-Fragment
ersetzt, das eine Tetrazyklinresistenz enthält.
-
Um die Transkriptionsstartstelle
eines ScBV-Promotors zu kartieren, wird ein markierter Primer oder ein
markiertes DNA-Fragment und RNA, die von dem ScBV-Promotor gebildet
wurde, in einer Primer-Extensionsreaktion und/oder einer S1-Nukleasereaktion
eingesetzt. Ein nützlicher
Primer, um die Transkriptionsstartstelle eines ScBV-Promotors zu
kartieren, ist in Tabelle 1 gezeigt (MAP-SCBV-GUS: SEQ ID NO: 10).
-
Beispiel II
-
Monokotyledonen- (Hafer)
Transformation mit ScBV-Prhomotor-Konstrukten
-
Transiente Expressionsanalysen in
Mais (unter Verwendung von Black Mexican Sweet-Zellen, „BMS"-Zellen)
und Haferssuspensionskulturen zeigten, dass ScBV-3 die höchsten Expressionslevel
ergab. Insbesondere in BMS-Zellen verlieh ScBV-3 eine Expression,
die zu etwa 25 und 800 mal mehr unabhängigen Ereignissen führte, die
GUS-exprimierende Zellen hervorbrachten, als die Expression die
von ScBV-2 bzw. pScBV-1 verliehen wird.
-
Unreife Haferembryonen (Avena sativa
L. Varietät
GP-1) wurden verwendet, um Kallus zu initiieren. Bröckliger,
embryogener Kallus wurde visuell unter Verwendung eines low power
microscopes (6,6x) selektiert und alle zwei Wochen auf MS2D-Medium,
das MS-Salze enthält
(Torbert et al., supra) subkultiviert. Gewebe, das sich von dem
Kallus ableitet, wurde auf festes MS2D-Medium, das 0,2 M Sorbitol
und 0,2 M Mannitol enthielt, als eine Osmotikum-Vorbehandlung für 4 Stunden
vor dem Mikroprojektilbombardement, wie es von Vain et al. beschrieben
wird (Plant Cell Reports, 12, 84 (1993)), ausplattiert.
-
Im Allgemeinen können entweder Wolfram- (1,1
Mikron; M-17; Biorad Laboratories, Hercules, CA) oder Gold- (1,0
Mikron; M-17; Biorad Laboratories, Hercules, CA) Partikel für das Mikropartikelbombardement eingesetzt
werden. Es werden etwa 60 mg trockene Wolfram- oder Goldpartikel
in 1 ml 100% Ethanol in ein Mikrotube gegeben. Das Tube wird kräftig für 1–2 Minuten
gevortext oder unter Verwendung eines Standardaufsatzes bei niedriger
Leistung für
30 Sekunden sonifiziert. Das Vortexen wird dreimal wiederholt. Dann
wird das Mikrotube einer Zentrifugation bei 10000 rpm für eine Minute
unterworfen. Der Überstand
wird entfernt und 1 ml steriles, destilliertes Wasser zugegeben,
die Partikel resuspendiert, zentrifugiert und der Überstand
entfernt. Dieses Verfahren wird einmal mehr wiederholt. Die Partikel
werden dann in 1 ml steriles, destilliertes Wasser resuspendiert.
Fünfzig
Mikroliter, genug 4–8
Bombardements, werden unter Vortexen in Mikrotubes aliquotiert.
Wolfram- oder Goldaliquots werden bei –20°C aufbewahrt.
-
Zu einem einzigen 50 ml Partikelaliquot
wird unter kontinuierlicher Bewegung das folgende inder folgenden
Reihenfolge zugegeben: 5 μl
DNA (1 μg/μl), 50 μl 2,5 M CaCl2 und 20 μl
0,1 M Spermidin (freie Base, tissue culture grade, Sigma Chemical
Co.). Die Mischung wird für
drei Minuten gevortext, einer Zentrifugation bei 10000 rpm für 10 Sekunden
unterworfen und der Überstand
entfernt. Die DNA-beschichteten Partikel werden mit 250 μl 100% Ethanol
durch kurzes Vortexen gewaschen, dann einer Zentrifugation unterworfen
und der Überstand
entfernt. Die Partikel werden dann in 60 μl 100% Ethanol resuspendiert.
Dann werden 5–10 μl der Suspension
auf das Zentrum eines Macrocarriers aufgetragen. Der Suspension wird
in einer erschütterungsfreien
Umgebung mit geringer Luftfeuchtigkeit für etwa 1 Minute erlaubt zu
trocknen.
-
Etwa 800 mg wurden in die Mitte einer
Petrischale gegeben. Die Schale wurde 5 cm unter die Halteplatte
positioniert und das Gewebe mit Goldpartikel, die mit pSCBV-3 und
einem Plasmid beschichtet waren, das den npt II-Selektionsmarker
für Pflanzen
verbunden mit dem CaMV35S-Promotor enthielt (pH 24, siehe Torbert
et al., supra) (0,625 μg/Bombardement)
unter Verwendung des Biolistic® PDS-1000/He
Partikelübertragungssystems
(BioRad Laboratories, Hercules, CA), das entsprechend den Angaben
des Herstellers betrieben wurde, bombardiert.
-
Das Gewebe blieb über Nacht auf dem Osmotikummedium
(MS2D plus 0,4 M Osmotikum) und wurde auf MS2D Erhaltungsmedien
für 7 Tage
bei 20°C
im Dunkeln transferiert. Das transformierte Gewebe wurde auf Selektionsmedium
transferriert, das 50 mg/L Paromomyzin enthielt und das mit 0,35%
low EEO Typ I Agarose (Sigma Chemical Co.) verfestigt wurde, und
alle zwei Wochen subkultiviert (Torbert et al., supra). Wachsende
Kolonien wurden nach etwa 6–8
Wochen isoliert und es wurde ihnen gestattet bis zu etwa vier weitere Wochen
zu wachsen. Es wurden Triebe in Triebregenerationsmedium regeneriert
(MS-Salze plus Thiamin-HCL, 20 g/L Saccharose, 2 mg/L NAA, 0,2 mg/L,
BAP, 50 mg/L Paromomycin, pH 5,8, verfestigt mit 0,35% low EEO Typ
I Agarose). Wurzeln wurden in Wurzelregenerationsmedium regeneriert
(MS-Salze plus Thiamin-HCL verfestigt mit 0,35% niedrig EEO Typ
I Agarose). Die Pflanzen wurden dann in Erde eingesetzt und bis
zur Reife herangezogen.
-
-
In stabil transformierten Geweben
von A. sativa verlieh ScBV-3 den vegetativen Organen und in besonderem
Maße dem
Stengel (5E) eine konstitutive
GUS-Expression. Im Blatt jedoch verlieh ScBV-3 ein Expressionsmuster,
das meistens, aber nicht ausschließlich auf das vaskuläre Gewebe
(5F) begrenzt ist. ScBV-3 führte auch
zu einer konstitutiven GUS-Expression in den Blütenorganen wie der Palea, dem
Lemma und dem Fruchtknoten (5D und 5B).
-
Regenerierte Pflanzen (T0-Generation)
wurden auf die GUS-Aktivität
gescreent. Es wurden 56 Pflanzen und in einigen Fällen viele
Pflanzen von demselben Kallus wiedergewonnen. Pflanzen, die sich
von demselben Kallus ableiten, wurden als eine Linie definiert.
Da Pflanzen, die sich von demselben Kallus ableiten, potentiell
unabhängige
Ereignisse repräsentieren
könnten,
wurden Daten von Pflanzen, die von einem individuellen Kallus wiedergewonnen
wurden, zusammengefasst. Die Hafergewebe wurden von Hand von den Pflanzen
mit Blütenorganen
und grünem,
vegetativem Gewebe geschnitten.
-
Es wurden 23 stabil transformierte
unabhängige
Linien auf die GUS-Expression
analysiert. Die Pflanzengewebe wurden mit einem GUS-histochemischen Färbepuffer
in der Anwesenheit von 1 : 5000 (v/v) -Lösung des Detergens Silwet L-77
(OSI Specialitites, Charleston, WV) gefärbt und für 20 min. einem Vakuum unterworfen.
Die Gewebe wurden für
8–48 Stunden
bei Raumtemperatur gefärbt,
für 24
Stunden entfärbt
und in 70% Ethanol für
die Analyse aufbewahrt. Die GUS-Expression wurde unter einem Disektinn
Mmikroskop gezählt.
Die Ergebnisse der GUS-Analyse sind in Tabelle 2 dargestellt. Eine
GUS-Aktivität
wurde in beinahe allen getesteten Organen von A. sativa detektiert
und Linien mit einer GUS-Aktivität
in einem Organ tendierten dazu, eine Aktivität in allen der getesteten Organen
zu haben. Stengel, Palea, Deckspelze (Lemma) und Fruchtknoten hatten
die höchsten
Expressionslevel. Darüber
hinaus hatten von den 23 getesteten, unabhängigen Linien über 82%
detektierbare Level einer GUS-Expression in Blatt, Stengel, Blütenstandsstiel
(Peduncle), Rachis, Spelze (Glume), Blütenstandsseitenachse (Rachilla),
Palea, Deckspelze (Lemma), Anthere und Fruchknoten. Die Npt II-Level
in den Pflanzenzellen, -Teilen oder -Geweben wurden durch einen
NPTII -ELISA-Assay (5'-3', Boulder, CO) bestimmt, wie es auch in
Torbert et al. (supra) beschrieben wird. Npt II wird in allen Linien,
die GUS exprimieren, detektiert.
-
Es wurden A. sativa-Blümchen von
reifen Pflanzen (kein vegetatives Gewebe) gesammelt. T1-Samen wurden
durch Selbstbestäubung
der T0-Pflanzen hergestellt. Diese Samen wurden geschält und dann
durch das Einweichen zuerst in 95% Ethanol für 30 Sekunden und dann in 2,5%
Bleiche und 1–2
Tropfen Tween-20/100 ml für
5 Minuten gefolgt von dem dreimaligen Waschen mit sterilem Wasser
sterilisiert. Die Samen wurden in Magenta-Boxen auf MS-Medium, dem
Hormone fehlten, zum Auskeimen gebracht, für 10–12 Tage angezogen und dann
auf Erde transferiert. Vor dem Transfer auf die Erde wurden die
ausgeschnittenen Wurzeln der T1-Pflanzen
einer Färbung
für die
GUS-Genexpression wie oben beschrieben unterworfen. Die A. sativa-Nachkommen
(T1)-Pflanzen und die T1-Setzlinge
wurden einer Analyse auf die GUS-Genexpression unterworfen.
-
Tabelle
3. GUS-Genexpression in A. sativa T
1-Setzlingen
-
Beide, Samen und T1-Pflanzen, zeigten
eine GUS-Aktivität,
die folglich demonstrierte, dass das GUS-Gen erblich war. Die Ergebnisse
der GUS-Analyse der T1-Pflanzen ist in Tabelle 4 dargestellt. Relativ
zu den T0- Pflanzen
schien der Expressionslevel in den T1-Pflanzen in den meisten Organen
reduziert (Tabelle 2 und Tabelle 4). Dieses Ergebnis deutet an,
dass die Expression in der Nachkommenschaft reduziert ist.
-
Tabelle
4. GUS-Genexpression in reifen A. sativa T
1-Pflanzen
-
Beispiel III
-
Dicotyledonen (Arabidopsis)
-Transformation mit ScBV-Promotorkonstrukten
-
Ein Expressionsvektor, der einen
ScBV-Promotor verbunden mit dem GUS-Gen umfasst, wurde in Arabidopsis
thaliana (Columbia ecotype) mittels Vakuuminfiltration wie im wesentlichen
von van Hoof und Green, Plant J., 10, 415 (1996); Bechthold et al.,
C. r. Acad. Sci., 316, 1194 (1993) beschrieben mit den folgenden
Modifikationen transformiert: 200 μl/L Silwet L-33 wurden in der
Infiltrationslösung
eingesetzt und der Agrobakterium tumefaciens-Stamm C58C1 (pMP 90) wurde als ein nicht-eukaryotischer
Wirt für
den Expressionsvektor verwendet.
-
Samen von Pflanzen, die dem Vakuuminfiltrationsverfahren
unterworfen wurden, (bezeichnet T1-Samen)
wurden auf Kanamyzin-haltigen Platten ausgesät, wie von Feldmann beschrieben
(In: „Methods
in Arabidopsis Research" (C. Koncz, N. -H. Chua, und J. Schell,
Eds.). World Scientific Publishing Co., Singapore. pp. 274–289 (1992)).
Individuelle Pflanzen (T1-Pflanzen) die das
Kanamyzin-Markergen trugen, wurden auf Erde transferiert. Gewebe
von diesen Pflanzen wurden angefärbt,
um die GUS-Aktivität
wie oben für
A. sativa-Gewebe beschrieben zu lokalisieren, mit Ausnahme dass
das Silwet L-77-Detergens in einer 1 : 10000 -Verdünnung (v/v)
verwendet wurde und die Färbelösung in
das Gewebevakuum infiltriert wurde, indem es einem Vakuum für fünf Minuten
unterworfen wurde. Die Samen der T1-Pflanzen
(T2-Pflanzen) wurden gesammelt und die Nachkommenpflanzen
(T2-Pflanzen) ebenfalls auf eine GUS-Aktivität gefärbt.
-
Die Tabellen 5 und 6 zeigen das verallgemeinerte
Expressionsmuster des ScBV-Promotors in T1-bzw. T2-A. thaliana-Pflanzen. Es wurden 29 T1-Pflanzen
untersucht, die von 7 infiltrierten Pflanzen abgeleitet wurden.
Alle transformierten Setzlinge, die von einer einzigen, infiltrierten
Pflanze abgeleitet wurden, wurden einer Linie zugeteilt.
-
In A. thaliana-Blüten ist das Expressionsmuster
meistens vaskulär
und dennoch zeigten die Staubblätter
in einigen Blüten
eine konstitutive GUS-Genexpression
(5C). Wenn die Wurzeln von A. sativa
gefärbt wurden,
wurde in 44% der untersuchten Linien eine GUS-Expression detektiert,
während
in 86% der untersuchten Linien für
A. thaliana-Wurzeln eine konstitutive GUS-Genexpression gefunden
wurde.
-
Tabelle
5. GUS-Genexpression in A. thaliana T
1-Pflanzen
-
Tabelle
6. GUS-Genexpression in A. thaliana T
1-Pflanzen
-
Beispiel IV
-
Einführung eines ScBV-Promotorkonstrukts
in Sojazellen, Regeneration transformierter R0-Pflanzen und Nachkommenherstellung
-
Es werden Sojaexplanate aus Meristemen,
die aus den embryonischen Achsen unreifer Samen ausgeschnitten wurden,
abgeleitet. Vor der Transformation werden die Meristemexplanate
in einem hoch zytokininenthaltenden Medium (Barwhale et al., Planta,
176, 473 (1986)) über
Nacht vorinkubiert. Der Boden einer 60 mm Petrischale wurde mit
1% Wasseragar gefüllt.
Die Embryonen wurden oberflächensterilisiert
und auf dem Agar in der Petrischale ausplattiert.
-
Eine Menge von 1–3 Mikrometer Goldkügelchen
(Alfa Chemical Co.) wurden mit Polylysin vorbeschichtet, indem sie
in 0,02% Polylysin gespült
wurden und an der Luft getrocknet. Es wird ein linearisierter Expressionsvektor,
der eine ScBV-Promotorsequenz, zum Beispiel SEQ ID NO: 3, wirksam
verbunden mit einem präselektierten
DNA-Segment umfasst, vorbereitet. Vorzugsweise umfasst der Expressionsvektor
ferner ein Markergen, wie das neo-Gen (APT 3'II). Es werden 225
Mikrogramm des Expressionsvektors in wässriger Lösung zu 35 mg beschichteten
Goldkügelchen
zugegeben und dann schrittweise 22 μl 10 mM Na2HP O4 und 22 μl 10 mM CaCl2 zugegeben, die ein feines Präzipitat
bilden, währen
die Lösung
unter N2 getrocknet wird. Im Allgemeinen
werden 1,0–0,001
mg DNA pro mg Goldkügelchen
vorbereitet. Die getrockneten, präzipitatbeschichteten Kügelchen
werden dann in 100% Ethanol resuspendiert und auf 2,0 Millimeter
plastikbschichteten, aluminisierten Mylarblättern etwa 9 mm auf 11 mm ausgelegt.
Die beschichteten Kügelchen
werden so aufgetragen, dass sie eine letztendliche Dichte von 0,2
mg/cm2 auf dem Mylarträgerblatt ergeben. Im Allgemeinen wird
das Trägerblatt
mit 0,05–40
mg beschichteter Kügelchen
pro Quadratzentimeter beladen.
-
Es wird ein Vakuum von 500 mm Quecksilber
angewendet. Eine 24 kV-Entladung von dem 2 Mikrofaradkondensator
wird durch die Elektroden, welche die Partikel auf den Sojaembryo
beschleunigen, entladen. Die bombardierten Embryos werden von der
Zieloberfläche
entfernt und auf Platten für
Organogeneseverfahren ausplattiert, zum Beispiel siehe Barwhale
et al., Planta, 176, 473 (1986).
-
Die Explanate werden in der Dunkelheit
auf MS-Basismedium wie von Barwhale et al. (supra) modifiziert,
ausplattiert. Im Anschluß an
eine ein-bis zweiwöchige Inkubation
in der Dunkelheit werden die Gewebe auf dasselbe Basismedium transferiert,
das einen niederen Level an Zytochinin (1,7 Mikromolar) enthält, um die
Triebverlängerung
zu begünstigen.
Die Triebe werden bei einer Höhe
von 0,5–1,0
cm geerntet. Drei bis acht Triebe werden pro Explanat in 2–4 Monaten
wiedergewonnen.
-
Nachdem die Triebe eine Höhe von 0,5–1,0 cm
erreicht haben, werden sie auf die Wurzeln von keimenden, etwa zehn
Tage alten Sojasetzlingen gepfropft. Vor dem Pfropfen werden sie
auf ½ MS-Medium
für eine
Woche abgehärtet.
Sobald ausreichend Prlanzengewebe erhalten wird, werden die Gewebe
auf die Anwesenheit des Markergens hin untersucht. Falls zum Beispiel
das neo-Gen als ein Markergen verwendet wird, wird die APH 3'II-Aktivität in den
Pflanzengeweben untersucht.
-
Die Anwesenheit des präselektierten
DNA-Moleküls
von Interesse, zum Beispiel eines, das ein virales Hüllprotein
kodiert, wird durch Southern Blot-Analyse detektiert. Zehn Mikrogramm
genomischer DNA des Pflanzengewebes werden mit einem geeigneten
Restriktionsenzym verdaut. Die DNA wird dann Phenol:Chlorophorm-extrahiert
und mit Ammoniumazetat und Ethanol präzipitiert. Die verdaute DNA
wird auf einem Agarosegel durch Elektrophorese fraktioniert. Es
wird eine 32P-markierte Sonde eingesetzt,
die wenigstens mit einem Teil des DNA-Moleküls von Interesse übereinstimmt.
Nach dem Waschen des Filters, werden die hybridisierenden DNA-Fragmente
durch eine Autoradiographie sichtbar gemacht. Auf diese Weise werden
Pflanzen aufgezeigt, die das DNA-Molekül von Interesse tragen. Eine
Pflanze, deren Gewebe eine markergen enzymatische Aktivität zeigen
oder die bei einer Southern Blot-Analyse für ein DNA-Molekül von Interesse
oder für
das Markergen positiv sind, werden zur Reife herangezogen. Die Pflanze
wird selbst bestäubt
und die Samen gewonnen. Es werden Setzlinge eingesetzt um Pflanzen
zu generieren, deren Blätter
auf die Markergenassoziierte, enzymatische Aktivität oder durch
Southern Blot-Analyse auf das DNA-Molekül von Interesse hin untersucht
werden.
-
Beispiel V
-
Einführung eines ScBV-Promotorkonstrukts
in Tabakzellen, Regeneration von transformierten R0-Pflanzen und
Nachkommenherstellung
-
Es werden Tabak-(Nicotiani tabacum
var. samsun) Blattscheiben mit einem Durchmesser von etwa 6 mm von
Oberflächen-sterilisierten
Tabakblättern
entnommen. Diese werden auf MS 104 Agarmedium für 2 Tage kultiviert, um eine
partielle Zellwandbildung an den verwundeten Oberflächen zu
begünstigen.
Sie werden dann in einer Kultur von A. tumefaciens-Zellen untergetaucht,
die ein Plasmid, das einen ScBV-Promotor wirksam verbunden mit einem
präselektierten
DNA-Segment hat, das zum Beispiel GUS kodiert, und eine weitere
Expressionskassette, die eine Kanamyzinresistenz kodiert, und pMP90RK
enthalten ein Helferplasmid, das über Nacht in Luria broth bei
28°C gezogen
wird, und behutsam geschüttelt.
Die Zellen werden aus der Bakteriensuspension entfernt, trocken
geblottet und umgedreht auf Filterpapier inkubiert, das über „Pflege
(für Englisch
nurse)" -Kulturen von Tabakzellen plaziert wurde, wie von Horsch
beschrieben (in vitro, 16, 103 (1980)). Nach zwei oder drei Tagen
werden die Scheiben auf Petrischalen transferiert, die MS-Medien
mit 500 μg/ml
Carbenicillin und keine Pflegekultur enthielten.
-
Kontrollgewebe wird unter Verwendung
von A. tumefaciens-Zellen erzeugt, die das Helferplasmid pMP90RK
und einen unterschiedlichen Pflanzentransformationsvektor, pMON505
enthielten, der eine T-DNA- Region
mit einem NOS-NPTII/NOS-Kanamyzinresistenzgen und ein selektierbares
Markergen enthält.
-
Innerhalb von 10 Tagen nach dem Transfer
auf die MS-Medien, tauchen aktiv wachsende Kallusgewebe an der Peripherie
aller Scheiben, an beiden, der Kontrolle und den transformierten
Platten, auf. Transformierte Tabakpflanzen werden dann durch die
Regeneration aus den oben beschriebenen, transformierten Blattscheiben
mittels dem Verfahren hergestellt, das von Horsch et al. beschrieben
wurde (Science, 227, 1229 (1985)). Die erhaltenen transformierten
Pflanzen enthalten die Expressionskassette, die den ScBV-Promotor mit
dem β-Glucuronidasegen
fusioniert enthält.
-
Dasselbe Verfahren wird verwendet,
um transformierten Tabak mit einem CaMV35S-Promotor zu erhalten,
der zu demselben, präselektierten
DNA-Segment, das heißt
GUS-fusioniert ist.
-
Transformierte Pflanzen, die das
GUS-Gen enthalten, das entweder durch den ScBV-Promotor oder den
CaMV35S-Promotor angetrieben wird, werden unter Verwendung eines
histologischen Färbeverfahrens untersucht,
um die GUS-Aktivität
in den transformierten Zellen zu bestimmen. Die Ergebnisse dieser
Untersuchungen mit Pflanzen, die mit ScBV/GUS/NOS transformiert
wurden, werden mit den Ergebnissen desselben Untersuchungen verglichen,
der mit Pflanzen durchgeführt
wurde, die mit CaMV35S/GUS/NOS transformiert wurden.
-
Die histochemische Untersuchung der
Tabakpflanzen, die die ScBV/GUS/NOS- und die CaMV35S/GUS/NOS- Konstrukte
enthielten, involvierte eine Prüfung
der Pflanzenorgan- und/oder Pflanzengewebeschnitte der transformierten
Pflanzen, um die GUS-Aktivität
zu bestimmen. Die Gewebe- oder Organschnitte der transformierten
Pflanze wurden unter Verwendung einer Rasierklinge vorbereitet,
um die Pflanzengewebe mit der freien Hand in Schnitte dünner als
0,5 mm Dicke zu sezieren. Die Schnitte werden dann in einen Überschuß X-Gluc-Lösung gegeben,
so dass der Schnitt vollkommen bedeckt war. Das Ansetzen eines Vakuums
an die Schnitte kann bei der Penetration der X-Gluc-Lösung helfen.
Eine 50 ml X-Gluc-Lösung
wird vorbereitet indem 25 ml 0,2 M Na3PO4-Puffer, pH 7,9, 24,0 ml dH2O,
0,25 ml 0,1 M K3[Fe(Cn)6],
0,25 ml 0,1 M K4[Fe(Cn)6]
und 0,5 ml 1 M EDTA, pH 7,0 kombiniert werden. Zu dieser Lösung werden
50 mg X-Gluc (5-Brom-4-chlor-3-indolyl-β-glucuronid) zugegeben, die
von Research Organics (Cleveland, Ohio) erhalten wurden, und unter
rühren
gelöst.
Die Lösung
wurde dann vorzugsweise durch Filtration sterilisiert. Die Schnitte in
der X-Gluc-Lösung
werden dann für
2–4 Stunden
auf 37°C
gestellt. Es muss Vorsicht walten, um das Verdampfen der Lösung zu
verhindern. Nach der Inkubationsperiode werden die Schnitte mit
Phosphatpuffer oder destilliertem H2O gespült und dann
umgehend mit einem Stereomikroskop oder einem Compound-Mikroskop untersucht.
Falls es eine Interferenz von den Pigmenten gibt, kann das Gewebe
in FAA-Lösung
(85 ml 50% Ethanol, 5 ml Eisessig und 10 ml Formalin) für 24 Stunden
fixiert werden. Probleme mit phenolischen Verbindungen können durch
die Zugabe von Natriummetabisulfit auf 20 mM zu der Färbelösung kurz
vor dem Färben gemildert
werden. Ein positiver Test auf das Vorhandensein einer GUS-Aktivität wird durch
eine blaue Koloration angezeigt, die in dem Gewebe des untersuchten
Pflanzenschnittes auftaucht.
-
Eine histologische Färbeuntersuchung
wird an Schnitten durchgeführt,
die mit dem β-Glucuronidasegen
transformiert wurden, das entweder von dem CaMV35S-Promotor oder
dem ScBV-Promotor angetrieben wird. Ein typisches Färbeprofil
wurde für
den CaMV35S-Promotor, der das GUS-Gen antreibt, mit Färbungen in
einigen Geweben und keinen Färbungen
in anderen Geweben innerhalb einer einzigen, transgenen Pflanze beobachtet.
Gewebe jedoch von einer transformierten Pflanze mit dem ScBV-Promotor
angetriebenen GUS-Gen zeigten, dass die transformierte Pflanze einen
angemessenen, vorzugsweise viel höheren GUS-Expressionslevel
und ein gleichmäßigeres
Expressionsmuster überall
im Gewebe und den Zellen aufzeigt, als er in Geweben von Pflanzen,
die mit CaMV35S/GUS transformiert wurden, beobachtet wurde. Dieses
wird durch die vorherrschende, blaue Kolloration durch den gesamten
Schnitt hindurch dargestellt.
-
Die Verteilung der Expression und
die Anzahl der erhaltenen, hochexprimierenden, transgenen Pflanzen
zeigen, dass der ScBV-Promotor in der Gewebeverteilung und der Gleichmäßigkeit
der Expression überlegen
ist, wenn er mit den CaMV/GUS-Pflanzen verglichen wird. Mehr als
90% der ScBV/GUS-enthaltenden, transformierten Pflanzen zeigen eine
angemessene, vorzugsweise sehr starke GUS-Expression und dass die Färbung gleichmäßig von
Pflanze zu Pflanze und Gewebe zu Gewebe ist. Diese Färbung ist
durchwegs genauso gut in den ScBV-enthaltenden Pflanzen wie jene
in den CaMV/GUS-Pflanzen.
-
Um einen weiteren Beweis bereitzustellen,
dass der ScBV-Promotor ein starker und konstitutiv exprimierender
Promotor ist, werden transgene Nicotiana tabacum-Pflanzen, die die
Konstrukte enthalten, auf eine GUS-Aktivität hin unter Verwendung der
fluorimetrischen Untersuchung von Jefferson et al. (EMBO J., 6,
3901 (1987)) in extrahierten Blättern,
Blüten,
Stämmen
und Wurzeln untersucht. Die Inkubationen werden bei 37°C für 15 Minuten
durchgeführt.
Ein 1 g viertes Internodienblatt, eine Blüte, eine 4 cm lange Stengelsektion
oder Wurzeln werden ausgeschnitten. Das ausgewählte Gewebe wird durch das
Gefrieren in flüssigem
Stickstoff, das Zerreiben mit einem Mörser und Pistill und das Resuspendieren
in 1 ml 0,1 M K3P O4, pH 7,8, mM EDTA, 10 mM DT, 0,8 mM PMSF
und 5% Glycerin resuspendiert. Die fluorogene Reaktion wurde in
2 mM 4-Methyl-umbelliferylglukuronid
durchgeführt.
Die Fluoreszenz wird unter Verwendung eines Hoechst DNA-Fluorometers
(Modell TKO 100) gemessen. Die Proteinkonzentration der Pflanzenextrakte
wurde durch eine Bradford-Untersuchung
bestimmt.
-
Der CaMV35S-Promotor ergibt einen
willkürlichen
Expressionslevel von „1"
für jedes
analysierte Gewebe und daraufhin wird der relative Expressionslevel
des ScBV-Promotors bestimmt. Es wurde gefunden, dass die GUS-Expression,
die mit dem ScBV-Promotor erhalten wird, der, die mit dem CaMV35S-Promotor
erhalten wird, überlegen
ist.
-
Die Erfindung ist nicht auf die exakten
Details, die oben gezeigt und beschrieben werden, beschränkt, denn
es sollte sich verstehen, dass viele Variationen und Modifikationen
gemacht werden könnten,
während die
Erfindung innerhalb des Geistes und Bereiches der Erfindung, die
durch die Ansprüche
definiert wird, bleibt.
-
-
-
-
-
-
-
-
-
-
-
-
-