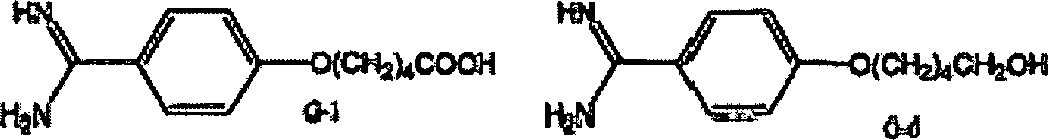-
Beschreibung
der Erfindung
-
Die
Erfindung bezieht sich auf die Behandlung von Tumorerkrankungen,
wie beispielsweise Krebs.
-
Krebs
ist eine Erkrankung, die durch unkontrolliertes Wachstum abnormer
Zellen gekennzeichnet ist. Die abnormen Zellen arbeiten nicht mehr
wie normale Zellen, sie proliferieren und zerstören gesundes Gewebe.
-
Lungenkrebs
ist die häufigste
krebsbedingte Todesursache und die am zweithäufigsten auftretende Krebsart
bei Männern
und Frauen. Schätzungen
zufolge kommt es allein im Jahr 2000 in den USA zu 164.000 Neuerkrankungen.
Während
die Lungenkrebsrate bei Männern
in den USA zurückgeht,
steigt sie bei Frauen weiterhin an. Lungenkrebs kann tödlich sein.
Laut der American Lung Association ist zu erwarten, dass im Jahr 2000
schätzungsweise
156.900 Amerikaner an dieser Krebserkrankung versterben.
-
Krebs,
der in der Lunge beginnt, wird abhängig vom mikroskopischen Zellbild
in zwei Hauptarten unterteilt: nicht-kleinzelliger Lungenkrebs und
kleinzelliger Lungenkrebs. Der nicht-kleinzellige Lungenkrebs (Plattenepithelkarzinom,
Adenokarzinom und großzellig-anaplastisches
Bronchialkarzinom) breitet sich im Allgemeinen langsamer auf andere
Organe aus als der kleinzellige Lungenkrebs. Der kleinzellige Lungenkrebs ist
nicht so häufig;
er ist für
etwa 20% aller Lungenkrebserkrankungen verantwortlich.
-
Zu
den weiteren Krebsarten zählen
Gehirnkrebs, Brustkrebs, Gebärmutterhalskrebs,
Darmkrebs, Magenkrebs, Nierenkrebs, Leukämie, Leberkrebs, Lymphoma,
Eierstockkrebs, Bauchspeicheldrüsenkrebs,
Prostatakrebs, Mastdarmkrebs, Sarkoma, Hautkrebs, Hodenkrebs und
Gebärmutterkrebs.
Diese Krebsarten werden genau wie Lungenkrebs manchmal mit Chemotherapie
behandelt.
-
Zu
den derzeit verwendeten Chemotherapeutika gehören Paclitaxel, Docetaxel,
Tamoxifen, Vinorelbin, Gemcitabin, Cisplatin, Etopsid, Topotecan,
Irinotecan, Anastrozol, Rituximab, Trastuzumab, Fludarabin, Cyclophosphamid,
Gentuzumab, Carboplatin, Interferon und Doxorubicin. Die am häufigsten
eingesetzte antineoplastische Substanz ist Paclitaxel, welches allein
oder in Kombination mit anderen Chemotherapeutika wie beispielsweise
5-FU, Doxorubicin, Vinorelbin, Cytoxan und Cisplatin verabreicht
wird.
-
Darstellung
der Erfindung
-
Wir
haben erfunden, dass die Kombination des Antipsychotikums Chlorpromazin
und der antiprotozoalen Substanz Pentamidin eine bedeutende antiproliferative
Aktivität
gegen Krebszellen entwickelt. Die Struktur- und funktionellen Analoge
beider Verbindungen sind bekannt, und jedes dieser Analoge kann
in der antiproliferativen Kombination dieser Erfindung verwendet
werden. Auch bekannt sind die Metabolite von Chlorpromazin und Pentamidin.
Viele dieser Metabolite haben mit der Ausgangsverbindung eine biologische
Aktivität
oder mehrere biologische Aktivitäten
gemeinsam und können
daher entsprechend ebenso in der erfindungsgemäßen antiproliferativen Kombination
eingesetzt werden.
-
Die
Erfindung ermöglicht
eine Methode zur Behandlung von Patienten mit Krebs oder anderen
Tumorarten, indem dem Patienten Chlorpromazin und Pentamidin zusammen
oder einzeln in einem Abstand von nicht mehr als 14 Tagen in einer
Menge verabreicht werden, die ausreicht, das Wachstum des Tumors
zu hemmen.
-
Vorzugsweise
werden die beiden Verbindungen einzeln in einem Abstand von nicht
mehr als zehn Tagen gegeben, besser noch in einem Abstand von nicht
mehr als fünf
Tagen und idealerweise innerhalb von vierundzwanzig Stunden oder
sogar gleichzeitig. Bei der entsprechend einer der unten beschriebenen
erfindungsgemäßen Methoden
behandelten Krebsart kann es sich um Lungenkrebs (Plattenepithelkarzinom,
Adenokarzinom oder großzellig-anaplastisches
Bronchialkarzinom), Gehirnkrebs, Brustkrebs, Gebärmutterhalskrebs, Darmkrebs,
Magenkrebs, Nierenkrebs, Leukämie,
Leberkrebs, Lymphoma, Eierstockkrebs, Bauchspeicheldrüsenkrebs,
Prostatakrebs, Mastdarmkrebs, Sarkoma, Hautkrebs, Hodenkrebs oder
Gebärmutterkrebs handeln.
-
In
jeder der Behandlungen wird jede einzelne Verbindung, die Teil der
Kombination ist, dem Patienten vorzugsweise als Teil einer pharmazeutischen
Zusammensetzung verabreicht, die auch einen pharmazeutisch akzeptablen
Träger
enthält.
Chlorpromazin wird vorzugsweise in einer Dosierung von 10 bis 2500
Milligramm und Pentamidin vorzugsweise in einer Dosierung von 1
bis 1000 Milligramm gegeben. Die bevorzugten Verabreichungsformen
beinhalten die intravenöse
und intramuskuläre
Route, die Inhalation und die orale Einnahme.
-
Die
antiproliferativen Kombinationen der Erfindung können Teil einer pharmazeutischen
Packung sein. Vorzugsweise werden Chlorpromazin und Pentamidin zusammen
oder einzeln formuliert und zwar in individuellen Dosierungen.
-
Für den Fachmann
auf diesem Gebiet, versteht es sich, dass die Verbindungen auch
nützlich
in Formulierungen in Form ihrer Salze sind. Beispielsweise, wie
hierin beschrieben, zeigt das Isethionatsalz von Pentamidin synergistische
antiproliferative Aktivität,
wenn es mit Chlorpromazin kombiniert wird. Weitere Salze von Pentamidin
sind: das Platinsalz, das Dihydrochloridsalz und das Dimethansulfonatsalz
(siehe, zum Beispiel, Mongiardo et al., Lancet 2:108, 1989). Ähnlich umfassen
die Chlorpromazinsalze beispielsweise die Hydrochlorsalze und die
Maleatsalze.
-
Diese
Erfindung beinhaltet auch Methoden zur Identifizierung von Verbindungen
für die
Behandlung eines Patienten mit einem Neoplasma (Tumor). Die Methode
umfasst die Schritte der Kontaktaufnahme der Krebszellen in vitro
mit (i) Pentamidin oder Chlorpromazin und (ii) einer Testverbindung
sowie die Bestimmung, ob die Krebszellen langsamer wachsen als (a)
die mit Chlorpromazin oder Pentamidin, aber nicht mit der Testverbindung
in Kontakt gebrachten Zellen und (b) Krebszellen, die zwar mit der
Testverbindung aber nicht mit Chlorpromazin oder Pentamidin in Kontakt
gebracht wurden. Eine Testverbindung, die, wenn sie mit Chlorpromazin
oder Pentamidin kombiniert wird, die Zellproliferation senkt, dies
aber in Abwesenheit von Chlorpromazin oder Pentamidin nicht bewirkt,
ist eine hilfreiche Verbindung für
die Behandlung eines Patienten mit einem Neoplasma.
-
Unabhängig davon,
wo die Chemotherapie erfolgt, kann auch eine Kombinationstherapie
angeboten werden: zu Hause, in der Arztpraxis, in der Klinik, in
der Ambulanz oder in einem Krankenhaus. Mit der Behandlung wird
im Allgemeinen im Krankenhaus begonnen, damit der Arzt die Therapiewirkung
eng überwachen
und bei Bedarf diese Therapie entsprechend anpassen kann. Die Dauer
der Kombinationstherapie hängt von
der Art der zu behandelnden Krebsart ab sowie dem Alter und dem
Zustand des Patienten, dem Stadium und der Krankheitsart, welche
bei dem Patienten vorliegen, und auch davon, wie der Körper des
Patienten auf die Therapie reagiert. Die Medikamentenverabreichung
kann in unterschiedlichen Intervallen erfolgen (z.B. täglich, wöchentlich
oder monatlich), und die Gabe jeder einzelnen Verbindung ist individuell festlegbar.
Möglich
ist es, die Kombinationstherapie in Zyklen bestehend aus Einnahmezeiten
und einnahmefreien Zeiten zu verabreichen, sodass der Körper des
Patienten die Chance erhält,
gesunde neue Zellen zu bilden und seine Stärke wiederzuerlangen.
-
Abhängig von
der Krebsart und dem Entwicklungsstadium kann die Kombinationstherapie
dazu verwendet werden, Krebs zu behandeln, die Metastasierung sowie
das Krebszellenwachstum zu verlangsamen, Krebszellen, die sich ausgehend
vom ursprünglichen
Tumor bereits in andere Körperteile
ausgebreitet haben, abzutöten
oder zu hemmen, die krebsbedingten Symptome zu verbessern oder Krebs
von Anfang an zu verhindern. Auch wird die Kombinationstherapie
eingesetzt, um Patienten ein komfortableres Leben zu ermöglichen,
indem Krebszellen, die Schmerzen oder Unbehagen hervorrufen, entfernt
werden.
-
Weitere
Charakteristika und Vorteile der Erfindung werden durch die nachfolgende
detaillierte Beschreibung und aus den Ansprüchen offensichtlich.
-
Detaillierte
Beschreibung der Erfindung
-
Wir
haben erfunden, dass die Kombination des Antipsychotikums Chlorpromazin
und der antiprotozoalen Substanz Pentamidin eine bedeutende antiproliferative
Aktivität
gegen Krebszellen entwickelt. Konzentrationen, die maximale antiproliferative
Aktivität
gegen Krebszellen zeigten, waren für normale Zellen nicht toxisch.
Somit ist diese Medikamentenkombination hilfreich bei der Behandlung
von Krebs und anderen Tumorarten.
-
Basierend
auf bekannten Eigenschaften, die sowohl Chlorpromazin als auch Pentamidin
und ihren Analogen und Metaboliten gemeinsam sind, ist es wahrscheinlich,
dass strukturverwandte Verbindungen durch Chlorpromazin und/oder
Pentamidin in der antiproliferativen Kombination der Erfindung ersetzt
werden können.
-
Der
am häufigsten
verordnete Wirkstoff aus der Phenothiazin-Familie ist Chlorpromazin
mit der Struktur:
-
-
Chlorpromazin
ist derzeit erhältlich
in den folgenden Verabreichungsformen: Tabletten, Kapseln, Suppositorien,
orale Konzentrate und Sirups und Formulierungen zur Injektion.
-
Chlorpromazin
bewirkt eine starke alpha-Adrenorezeptorenblockade und führt möglicherweise
zu orthostatischer Hypotension. Auch weist diese Verbindung eine
mäßige anticholinerge
Aktivität
auf, die sich in gelegentlicher Mundtrockenheit, verschwommener
Sicht, Harnretention und Konstipation zeigt. Chlorpromazin erhöht die Prolaktinsekretion
aufgrund seiner Dopaminrezeptor-Hemmwirkung in der Hypophyse und
dem Hypothalamus.
-
Chlorpromazin
wird leicht über
den Gastrointestinaltrakt resorbiert. Seine Bioverfügbarkeit
variiert aufgrund des beträchtlichen
First-Pass-Metabolismus der Leber. Flüssige Konzentrationen haben
vermutlich eine höhere
Bioverfügbarkeit
als Tabletten. Durch Nahrung wird die Bioverfügbarkeit anscheinend nicht
nachhaltig beeinflusst. Mithilfe von i.m.-Gaben wird ein Großteil der
First-Pass-Wirkung umgangen und eine höhere Plasmakonzentration erreicht.
Der Wirkungseintritt nach einer i.m.-Injektion setzt gewöhnlich nach
15 bis 30 Minuten ein und nach einer oralen Einnahme nach 30 bis
60 Minuten. Bei rektal verabreichtem Chlorpromazin dauert es gewöhnlich länger als
bei oral verabreichtem, bis die Wirkung eintritt.
-
Chlorpromazinmetabolite
-
Da
Chlorpromazin einer extensiven metabolischen Umwandlung zu einer
Reihe von Metaboliten unterliegt, die therapeutisch wirksam sein
können,
ist es möglich,
diese Metabolite anstelle von Chlorpromazin in der antiproliferativen
Kombination dieser Erfindung einzusetzen. Der Metabolismus für die Erzeugung
von Chlorpromazinprodukten, zum Beispiel, die oxidative N-Demethylierung
für den
Erhalt des entsprechenden primären
und sekundären
Amins, die aromatische Oxidation für den Erhalt eines Phenols,
die N-Oxidation für
den Erhalt des N-Oxids, die S-Oxidation für den Erhalt des Sulfoxids
oder Sulfons, die oxidative Deaminierung der Aminopropylseitenkette
für den
Erhalt von Phenothiazinkernen und die Glucuronidation der phenolischen
Hydroxygruppen und der tertiären
Aminogruppe, um ein quartäres
Ammoniumglucuronid zu erhalten.
-
In
weiteren Beispielen für
Chlorpromazinmetabolite, die nützlich
in der antiproliferativen Kombination der Erfindung sind, kann jede
der Positionen 3, 7 und 8 von Phenothiazin unabhängig mit einem Hydroxyl- oder Methoxylanteil
substituiert werden.
-
Pentamidin
-
Pentamidin
wird derzeit zur Behandlung von Infektionen mit Pneumocystis carinii,
Leishmania donovani, Trypanosoma brucei, T. gambiense und T. rhodesiense
verwendet. Die Struktur von Pentamidin ist:
-
-
Verfügbar ist
es als Formulierung für
die Injektion oder Inhalation. Für
die Injektion ist Pentamidin als nicht-pyrogenes, lyophilisiertes
Produkt verpackt. Nach der Rekonstitution wird es als intramuskuläre oder
intravenöse
Injektion verabreicht.
-
Pentamidinisethionat
ist ein weißes
kristallines Pulver, löslich
in Wasser und Glycerin, unlöslich
in Ether, Aceton und Chloroform. Chemisch betrachtet handelt es
sich um 4,4'-diamidino-diphenoxypentan di(β-hydroxyethansulfonat).
Die Molekularformel lautet: C23H36N4O10S2; und das Molekulargewicht beträgt 592,68.
-
Der
Wirkmechanismus von Pentamidin ist noch nicht vollständig aufgeklärt. Invitro-Studien
mit Säugetiergeweben
und der Protozoe Crithidia oncopelti deuten darauf hin, dass der
Wirkstoff mit dem Kernmetabolismus interferiert und die Synthese
von DNA, RNA, Phopsholipiden und Proteinen hemmt.
-
Es
ist auch nur wenig über
die Pharmakokinetik des Wirkstoffes bekannt. Bei sieben Patienten,
die mit täglichen
Dosen i.m. von 4 mg/kg Pentamidin über 10 bis 12 Tage behandelt
wurden, lagen die Plasmakonzentrationen zwischen 0,3 und 0,5 μg/ml. Die
Patienten schieden noch weitere sechs bis acht Wochen nach Beendigung
der Therapie abnehmende Mengen an Pentamidin mit dem Urin aus.
-
Die
Verteilung von Pentamidin im Gewebe wurde an Mäusen untersucht, denen eine
einmalige intraperitoneale Injektion an Pentamidin von 10 mg/kg
verabreicht wurde. Am höchsten
war die Konzentration in den Nieren, gefolgt von der Konzentration
in der Leber. Bei Mäusen
wurde Pentamidin unverändert
ausgeschieden und zwar primär über die
Nieren und in geringen Mengen über
den Stuhl. Das Mengenverhältnis
der Ausscheidung in Urin und Stuhl (4:1) blieb über den Zeitraum der Studien
konstant.
-
Pentamidinmetabolite
-
Pentamidinmetabolite
sind auch in der erfindungsgemäßen antiproliferativen
Kombination hilfreich. Pentamidin wird im Körper rasch zu mindestens sieben
primären
Metaboliten verstoffwechselt. Einige dieser Metabolite haben eine
oder mehrere Aktivitäten
wie Pentamidin. Es ist möglich,
dass manche Pentamidinmetabolite eine antiproliferative Wirkung
zeigen, wenn sie mit Chlorpromazin oder einem Analog davon kombiniert werden.
-
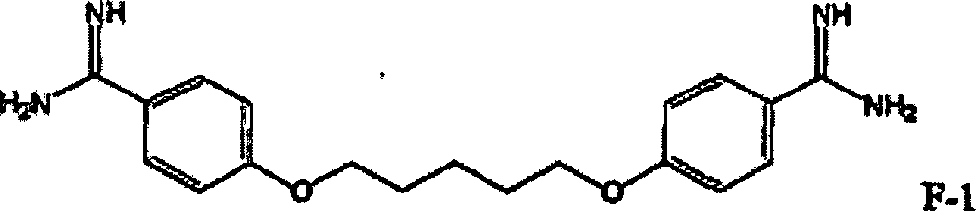
Unten
sind sieben Pentamidinmetabolite dargestellt.
-
-
-
Therapie
-
Die
Kombinationen von erfindungsgemäßen Verbindungen
sind hilfreich bei der Behandlung von Neoplasmen. Die Kombinationstherapie
kann allein oder in Verbindung mit einer anderen Therapie (z.B.
Chirurgie, Bestrahlung, Chemotherapie, biologische Therapie) eingesetzt
werden. Zusätzlich
dazu kann eine Person mit einem großen Risiko ein Neoplasma zu
entwickeln (z.B. eine Person, die genetisch prädisponiert ist oder kürzlich ein
Neoplasma hatte) eine prophylaktische Therapie erhalten, um die
Tumorbildung zu hemmen oder zu verzögern.
-
Die
Dosierung und Häufigkeit
der Verabreichung einer jeden Komponente kann unabhängig kontrolliert
werden. Beispielsweise wird eine Verbindung oral dreimal pro Tag
gegeben, während
eine zweite Verbindung intramuskulär einmal pro Tag verabreicht
wird. Die Verbindungen können
auch zusammen formuliert werden, sodass eine Verabreichung beide
Substanzen enthält.
Formulierungen und Dosierungen sind unten beschrieben.
-
Formulierung
der pharmazeutischen Zusammensetzungen
-
Die
Verabreichung jeder einzelnen der Verbindungen aus der Kombination
kann in einer beliebig geeigneten Form erfolgen, die zu einer Konzentration
der Verbindung führt,
welche kombiniert mit der jeweils anderen Verbindung am Zielort
spezifisch antineoplastisch wirkt. Die Verbindung kann in einer
beliebigen Menge in jeder geeigneten Trägersubstanz enthalten sein
und liegt im Allgemeinen in einer Menge von 1–95 Gew% bezogen auf das Gesamtgewicht
der Zusammensetzung vor. Die Zusammensetzung kann in einer Dosierungsform
angeboten werden, die für
die orale, parenterale (z.B. intravenöse, intramuskuläre), rektale,
kutane, nasale, vaginale, Inhalations-, Haut- (Patch) oder Augenverabreichungsroute
geeignet ist. Somit ist die Verbindung in Form von z.B. Tabletten,
Kapseln, Pillen, Pulvern, Granulaten, Suspensionen, Emulsionen,
Lösungen, Gels,
einschließlich
Hydrogels, Pasten, Salben, Cremes, Pflastern, über Drenches, Zulieferungsvorrichtungen, Suppositorien,
Einläufe,
Injektionslösungen,
Implantate, Sprays oder Aerosole verabreichbar. Die pharmazeutische
Zusammensetzung kann gemäß konventioneller
pharmazeutischer Praxis (siehe, z.B. Remington: The Science and
Practice of Pharmacy, (19th ed.) ed. A.R. Gennaro, 1995, Mack Publishing
Company, Easton, PA. und Encyclopedia of Pharmaceutical Technology,
eds. J. Swarbrick and J.C. Boylan, 1988–1999, Marcel Dekker, New York,
formuliert werden.
-
Die
erfindungsgemäßen pharmazeutischen
Zusammensetzungen können
so formuliert werden, dass die aktive Verbindung im Wesentlichen
sofort nach der Verabreichung freigesetzt wird oder zu einem beliebigen
vorbestimmten Zeitpunkt oder einem Zeitabschnitt nach der Verabreichung.
Die letzteren Typen der Zusammensetzungen sind allgemein bekannt
als Formulierungen mit kontrollierter Freisetzung, welche (i) Formulierungen
beinhalten, die eine im Wesentlichen konstante Konzentration des
Wirkstoffes im Körper über einen
längeren
Zeitraum hervorrufen; (ii) Formulierungen, die nach einer vorbestimmten
Lag-Zeit zu einer wesentlich konstanten Wirkstoffkonzentration im
Köper über einen
längeren
Zeitraum führen;
(iii) Formulierungen, die die Wirkstoffkonzentration über einen
vorbestimmten Zeitraum beibehalten, indem sie eine relativ konstante
Wirkstoffkonzentration im Körper
aufrechterhalten mit begleitender Minimierung der unerwünschten
Nebeneffekte assoziiert mit Fluktuationen im Plasmalevel des aktiven
Wirkstoffs (sägeblattähnliches kinetisches Muster);
(iv) Formulierungen, die die Wirkstoffaktivität lokalisieren durch z.B. räumliche
Platzierung der Zusammensetzung mit kontrollierter Freisetzung in
der Nähe
vom oder im kranken Gewebe oder Organ; und (v) Formulierungen, die
die Wirkstoffaktivität
zielgerichtet platzieren, durch die Verwendung von Trägerstoffen
oder chemischen Derivaten, um so den Wirkstoff an einen bestimmten
Zielzelltyp zu liefern.
-
Die
Verabreichung von Verbindungen in der Form von Formulierungen mit
kontrollierter Freisetzung wird besonders in den Fällen bevorzugt,
in denen die Verbindung entweder allein oder in Kombination, einen (i)
engen therapeutischen Index (d.h. der Unterschied zwischen der Plasmakonzentration,
die zu schädlichen Nebenwirkungen
oder toxischen Reaktionen führt
und der Plasmakonzentration, die einen therapeutischen Effekt hervorruft,
ist gering; im Allgemeinen ist der therapeutische Index, TI, definiert
als das Verhältnis
der mittleren letalen Dosis (LD50) zur mittleren
effektiven Dosis (ED50)); (ii) ein enges
Resorptionsfenster im Gastrointestinaltrakt, oder (iii) eine sehr
kurze biologische Halbwertszeit besitzt, sodass eine häufige Dosierung über den
Tag verteilt erforderlich ist, um die Plasmakonzentration auf einem
therapeutischen Level zu halten.
-
Es
kann eine beliebige Strategie aus einer Reihe von Strategien angewandt
werden, um eine Formulierung mit kontrollierter Freisetzung zu erhalten,
in welcher die Rate der Freisetzung die Stoffwechselrate der eingesetzten
Verbindung übersteigt.
In einem Beispiel wird eine kontrollierte Freisetzung erhalten durch
die geeignete Auswahl an zahlreichen Formulierungsparametern und
Inhaltsstoffen, einschließlich
z.B. verschiedener Typen von Zusammensetzungen mit kontrollierter
Freisetzung und Coatings. Daher wird die Wirksubstanz mit geeigneten
Hilfsstoffen in einer pharmazeutischen Zusammensetzung formuliert,
die bei Verabreichung an den Organismus die aktive Substanz auf
kontrollierte Weise freisetzt. Zu den Beispielen gehören Tabletten
mit einer Einzeleinheit oder mehreren Einheiten oder Kapselzusammensetzungen, Öllösungen,
Suspensionen, Emulsionen, Mikrokapseln, Mikrosphären (Mikrokügelchen), Nanopartikel, Patches
und Liposomen.
-
Festdosierungsformen
für die
orale Verwendung
-
Zu
den Formulierungen für
die orale Verwendung gehören
Tabletten, die den/die aktiven Inhaltsstoff(e) in einer Mischung
mit nicht-toxischen pharmazeutisch akzeptablen Hilfsstoffen enthalten.
Bei diesen Hilfsstoffen kann es sich beispielsweise um inerte Verdünner oder
Füllstoffe
(z.B. Sucrose, Sorbitol, Zucker, Mannitol, mikrokristalline, Stärken, einschließlich Kartoffelstärke, Kalziumcarbonat,
Natriumchlorid, Laktose, Kalziumphosphat, Kalziumsulfat oder Natriumphosphat)
handeln, granulierende und auflösende
Agenzien (z.B. Zellulosederivate, einschließlich mikrokristalline Zellulose,
Stärken,
einschließlich
Kartoffelstärke,
Croscarmellose-Natrium, Algiante oder Alginsäure); Bindungsagenzien (z.B.
Sucrose, Glucose, Sorbitol, Akazia, Alginsäure, Natriumalginat, Gelatine,
Stärke,
vorgelierte Stärke,
mikrokristalline Zellulose, Magnesium-Aluminium-Silikat, Carboxymethylzellulose-Natrium,
Methylzellulose, Hydroxypropylmethylzellulose, Ethylzellulose, Polyvinylpyrrolidon
oder Polyethylenglykol) und Lubrikantien, Gleitstoffe und Antiadhäsiva (z.B.
Magnesiumstearat, Zinkstearat, Stearinsäure, Siliziumdioxide, hydrogenierte
Pflanzenöle
oder Talkum). Weitere pharmazeutisch akzeptable Trägerstoffe
sind Farb-, Geschmacksstoffe, Weichmacher, Feuchthaltemittel, Puffer
und ähnliche.
-
Die
Tabletten können
nicht überzogen
oder optional mithilfe einer der bekannten Techniken überzogen sein,
um den Zerfall und die Resorption im Gastrointestinaltrakt zu verzögern und
dadurch eine dauerhafte Wirkung über
einen längeren
Zeitraum zu gewährleisten.
Das Coating kann entsprechend angepasst werden, um die aktive Wirksubstanz
nach einem vorbestimmten Muster (z.B. um eine Formulierung mit kontrollierter
Freisetzung zu erhalten) freizusetzen, oder es kann so angepasst
werden, dass die aktive Substanz erst nach der Magenpassage (darmresistentes
Coating) freigesetzt wird. Bei dem Coating kann es sich um einen
Zuckerüberzug,
einen Filmüberzug
(z.B. basierend auf Hydroxypropylmethylzellulose, Methylzellulose,
Methylhydroxyethylzellulose, Hydroxypropylzellulose, Carboxymethylzellulose,
Acrylatcopolymere, Polyethylenglykole und/oder Polyvinylpyrrolidon)
oder ein darmresistentes Coating (z.B. basierend auf Methacrylsäurecopolymer, Zelluloseacetatphthalat,
Hydroxypropylmethylzellulosephthalat, Hydroxypropylmethylzelluloseacetatsuccinat, Polyvinylacetatphthalat,
Schellack und/oder Ethylzellulose) handeln. Des Weiteren kann ein
die Freisetzung zeitlich verzögerndes
Material wie z.B. Glycerylmonostearat oder Glyceryldistearat eingesetzt
werden.
-
Die
Festtablettenzusammensetzungen können
ein Coating enthalten, welches so angepasst ist, dass es die Zusammensetzung
vor unerwünschten
chemischen Veränderungen
(z.B. chemischer Abbau vor der Freisetzung der aktiven Wirksubstanz)
schützt.
Das Coating kann auf die Festdosierungsform in ähnlicher Weise wie in der Encyplopedia
of Phamaceutical Technology, supra, beschrieben, angewandt werden.
-
Die
beiden Wirkstoffe können
in einer Tablette zusammen oder getrennt formuliert werden. In einem Beispiel
ist der erste Wirkstoff in der Tablette enthalten und der zweite
außen
auf der Tablette aufgebracht, sodass ein wesentlicher Anteil des
zweiten Wirkstoffs vor dem ersten Wirkstoff freigesetzt wird.
-
Formulierungen
für die
orale Verwendung können
auch als Kautabletten oder als Hartgelatinekapseln angeboten werden,
in denen der aktive Inhaltsstoff mit einem inerten festen Verdünner (z.B.
Kartoffelstärke, Laktose,
mikrokristalline Zellulose, Kalziumcarbonat, Kalziumphosphat oder
Kaolin) vermischt wird oder als Weichgelatinekapseln, in denen der
aktive Inhaltsstoff mit Wasser oder einem Ölmedium beispielsweise Erdnussöl, flüssigem Paraffin
oder Olivenöl
gemischt wird. Pulver und Granulate können unter Verwendung der oben
unter Tabletten und Kapseln erwähnten
Inhaltsstoffe auf konventionelle Weise hergestellt werden, z.B. mit
einem Mixer, einer Flüssigbettvorrichtung
oder einer Sprühtrockenanlage.
-
Orale Dosierungsformen
mit kontrollierter Freisetzung
-
Zusammensetzungen
mit kontrollierter Freisetzung für
die orale Anwendung können
z.B. so aufgebaut sein, dass sie die aktive Substanz entsprechend
der Kontrolle ihrer Auflösung
und Diffusion freisetzen.
-
Die über Auflösung und
Diffusion kontrollierte Freisetzung kann mithilfe eines geeigneten
Coatings einer Tabletten-, Kapsel-, Pellet- oder Granulatformulierung
der Zusammensetzung oder durch den Einschluss der betreffenden Zusammensetzung
in einer geeigneten Matrix erreicht werden. Das Coating einer kontrollierten
Freisetzung kann eine oder mehrere der oben erwähnten Coating-Substanzen beinhalten
und/oder z.B. Schellack, Bienenwachs, Glycowachs, Castorwachs, Carnaubawachs,
Stearylalkohol, Glycerylmonostearat, Glyceryldistearat, Glycerinpalmitostearat,
Ethylzellulose, Acrylharz, dl-Polymilchsäure, Zelluloseacetatbutyrat, Polyvinylchlorid,
Polyvinylacetat, Vinylpyrrolidon, Polyethylen, Polymethacrylat,
Methylmethacrylat, 2-Hydroxymethacraylat,
Methacrylat-Hydrogele, 1,3Butylenglycol, Ethylenglycolmethacrylat
und/oder Polyethylenglycole. In einer Matrixformulierung mit kontrollierter
Freisetzung kann das Matrixmaterial auch z.B. hydrierte Methylzellulose,
Carnaubawachs und Stearylalkohol, Carbopol 934, Silikon, Glyceryltristearat,
Methylacrylat-methylmethacrylat, Polyvinylchlorid, Polyethylen und/oder
halogenierten Fluorkohlenstoff enthalten.
-
Eine
Zusammensetzung mit kontrollierter Freisetzung, die eine oder mehrere
Verbindungen der beanspruchten Kombinationen enthält, kann
auch in Form einer schwimmenden Tablette oder Kapsel (d.h. eine
Tablette oder Kapsel, die nach der Einnahme über einen bestimmten Zeitraum
auf dem Mageninhalt schwimmend verbleibt) dargereicht werden. Eine
schwimmende Tablettenformulierung der Verbindung(en) kann hergestellt
werden durch Granulierung einer Mischung aus einem Wirkstoff(en)
mit Hilfsstoffen und 20–75%
w/w Hydrokolloiden, wie beispielsweise Hydroxyethylzellulose, Hydroxypropylzellulose
und Hydroxypropylmethylzellulose. Die erhaltenen Granulate können dann
in Tabletten gepresst werden. Bei Kontakt mit dem Magensaft bildet
die Tablette um ihre Oberfläche
herum eine beträchtliche
wasserundurchdringliche Gelbarriere. Diese Gelbarriere trägt dazu
bei, eine Dichte unter eins beizubehalten, weshalb die Tablette
auf dem Magensaft schwimmend verbleibt.
-
Flüssigkeiten
für die
orale Verabreichung
-
Pulver,
dispergierbare Pulver oder Granulate, die für die Herstellung einer wässrigen
Suspension durch Zugabe von Wasser geeignet sind, stellen für die orale
Verabreichung bequeme Dosierungsformen dar. Die Formulierung in
Form einer Suspension liefert den aktiven Inhaltstoff in einer Mischung
mit einem Dispersions- oder Befeuchtungsagens, einem Suspensionsagens
und einem oder mehreren Konservierungsstoffen. Geeignete Suspensions-
oder Befeuchtungsagenzien sind beispielsweise natürlich vorkommende
Phosphatide (z.B. Lecithin oder Kondensationsprodukte von Ethylenoxid
mit einer Fettsäure,
einem langkettigen aliphatischen Alkohol oder einem partiellen Ester
einer Fettsäure)
und ein Hexitol oder ein Hexitolanhydrid (z.B. Polyoxyethylenstearat,
Polyoxyethylensorbitolmonooleat, Polyoxyethylensorbitanmonooleat
und ähnliche).
Zu den geeigneten Suspensionsagenzien gehören zum Beispiel Natrium-Carboxymethylzellulose,
Methylzellulose, Natriumalginat und ähnliche.
-
Parenterale
Zusammensetzungen
-
Die
pharmazeutische Zusammensetzung kann auch parenteral durch Injektion,
Infusion oder Implantation (intravenös, intramuskulär, subkutan
oder ähnliche)
in Dosierungsformen, Formulierungen oder über geeignete Zufuhrsysteme
oder Implantate, die konventionelle nicht toxische pharmazeutisch
akzeptable Träger und
Adjuvantien enthalten, verabreicht werden. Die Formulierung und
Herstellung solcher Zusammensetzungen sind dem Fachmann auf diesem
Gebiet der pharmazeutischen Formulierungen gut bekannt. Spezifische Formulierungen
finden sich in Remington: The Science and Practice of Pharmacy,
supra.
-
Zusammensetzungen
für die
parenterale Verwendung werden in Einheiten-Dosierungsformen (z.B. in Einzeldosis-Ampullen)
oder in Vials, die verschiedene Dosen enthalten und in denen ein
geeignetes Konservierungsmittel zugegeben werden kann (siehe unten),
angeboten. Die Zusammensetzung kann in Form einer Lösung, einer
Suspension oder einer Emulsion, eines Infusionssystems oder eines
Zufuhrsystem über
die Implantation sein, oder sie kann in Form eines trockenen Pulvers,
das mit Wasser oder einem anderen geeigneten Vehikel vor Gebrauch
rekonstituiert werden kann, dargereicht werden. Abgesehen von dem/den
aktiven Wirkstoff(en) enthält
die Zusammensetzung möglicherweise
geeignete parenteral akzeptable Träger und/oder Hilfsstoffe. Der
aktive Wirkstoff/die aktiven Wirkstoffe sind in Mikrosphären, Mikrokapseln,
Nanopartikeln, Liposomen oder ähnlichen
Medien für
die kontrollierte Freisetzung einschließbar. Des Weiteren kann die
Zusammensetzung Agenzien für
die Suspension, Solubilisierung, pH-Einstellung und/oder Dispersion enthalten.
-
Wie
oben angegeben, kann die erfindungsgemäße pharmazeutische Zusammensetzung
in einer Form angeboten werden, die für die sterile Injektion geeignet
ist. Um eine solche Zusammensetzung herzustellen, wird ein geeigneter
aktiver Wirkstoff (oder Wirkstoffe) in einem parenteralen akzeptablen
flüssigen
Vehikel aufgelöst
oder suspendiert. Zu den akzeptablen Vehikeln, die verwendbar sind,
zählen
Wasser, Wasser, das durch Zugabe einer entsprechenden Menge von
Hydrochlorsäure,
Natriumhydroxid oder einem entsprechenden Puffer auf einen geeigneten
pH-Wert eingestellt wurde, 1,3-Butanediol,
Ringer-Lösung
und isotonische Natriumchloridlösung.
Die wässrige
Formulierung kann auch einen oder mehrere Konservierungsstoffe (z.B.
Methyl-, Ethyl-, n-Propyl-p-Hydroxybenzoat) enthalten. In Fällen, in
denen eine der Verbindungen nur schlecht oder wenig in Wasser löslich ist,
besteht die Möglichkeit,
ein Agens, das die Auflösung
verstärkt
oder ein Solubilisierungsmittel beizugeben oder ein Lösungsmittel
zu verwenden, das 10–60%
w/w Propylenglycol oder ähnliches enthält.
-
Parenterale
Zusammensetzungen mit kontrollierter Freisetzung
-
Parenterale
Zusammensetzungen mit kontrollierter Freisetzung können in
Form von wässrigen
Suspensionen, Mikrosphären,
Mikrokapseln, magnetischen Mikrosphären, Öllösungen oder Suspensionen dargereicht
werden. Alternativ dazu wird der aktive Wirkstoff (die aktiven Wirkstoffe)
in biokompatible Träger,
Liposomen, Nanopartikel, Implantate oder Infusionssysteme eingeschlossen.
-
Materialien
für die
Verwendung in der Herstellung von Mikrosphären und/oder Mikrokapseln sind
z.B. biologisch abbaubare/erodierbare Polymere wie Polyglaktin,
Poly-(isobutylcyanoacrylat), Poly(2-hydroxyethyl-L-Glutamin und
Poly(-Milchsäure).
Zu den biokompatiblen Trägerstoffen,
die zur Formulierung einer parenteralen Formulierung mit kontrollierter
Freisetzung eingesetzt werden können,
zählen
Kohlenhydrate (z.B. Dextrane), Proteine (z.B. Albumin), Lipoproteine
oder Antikörper.
-
Bei
den Materialien zur Verwendung in Implantaten kann es sich um nicht
biologisch abbaubare (z.B. Polydimethylsiloxan) oder biologisch
abbaubare (z.B. Poly(caprolacton), Poly(-Milchsäure), Poly(-Glykolsäure) oder
Poly(orthoester) handeln.
-
Rektale Zusammensetzungen
-
Zu
den geeigneten Dosierungsformen für die rektale Verabreichung
einer Zusammensetzung gehören Suppositorien
(Emulsions- oder Suspensionsarten) und rektale Gelatinekapseln (Lösungen oder
Suspensionen). In einer typischen Suppositorienformulierung ist
der aktive Wirkstoff (die aktiven Wirkstoffe) mit einer geeigneten
pharmazeutisch akzeptablen Suppositorienbasis wie Kakaobutter, esterifizierten
Fettsäuren,
glycerinierter Gelatine und verschiedenen wasserlöslichen
oder dispergierbaren Basissubstanzen wie Polyethylenglycolen und
Polyoxyethylensorbitan-Fettsäureestern
kombiniert. Möglich
ist es, unterschiedliche Additive, Verstärker oder oberflächenaktive
Agenzien mit einzuschließen.
-
Zusammensetzungen
für die
Inhalation
-
Typische
Dosierungsformen für
die Inhalation beinhalten Nasensprays und Areosole. In einer typischen
nasalen Formulierung wird der aktive Inhaltsstoff (die aktiven Inhaltsstoffe)
in einem geeigneten Vehikel aufgelöst oder dispergiert. Die pharmazeutisch
akzeptablen Vehikel und Hilfsstoffe (ebenso wie andere pharmazeutisch
akzeptable Materialien, die in der Zusammensetzung vorliegen, wie
beispielsweise Verdünner, Verstärker, Geschmacksstoffe
und Konservierungsstoffe) werden gemäß konventioneller pharmazeutischer Praxis
ausgewählt,
welche dem Fachmann auf dem Gebiet der Formulierung von Pharmazeutika
bekannt sind.
-
Perkutane
und topische Zusammensetzungen
-
Die
pharmazeutischen Zusammensetzungen können auch für die perkutane Absorption
topisch auf die Haut appliziert werden und zwar in Dosierungsformen
oder Formulierungen, die konventionelle nicht toxische pharmazeutisch
akzeptable Trägerstoffe
und Hilfsstoffe, einschließlich
Mikrosphären
und Liposomen, enthalten. Die Formulierungen beinhalten Cremes,
Salben, Lotionen, Linimente, Gele, Hydrogele, Lösungen, Suspensionen, Sticks
(Stifte), Sprays, Pasten, Pflaster und andere Arten transdermaler
Zufuhrsysteme. Die pharmazeutisch akzeptablen Trägerstoffe oder Hilfsstoffe
können
auch Emulgatoren, Antioxidanzien, Puffer, Konservierungsmittel,
Feuchthaltemittel, Penetrationsverstärker, Komplexbildner, Gelbildner,
Salbenbasen, Parfüms
und hautschützende
Agenzien umfassen.
-
Beispiele
für Emulgatoren
sind natürlich
vorkommende Gummis (z.B. Gummi acacia oder Gummi tragacanth) und
natürlich
vorkommende Phosphatide (z.B. Sojabohnenlecithin und Sorbitanmonooleatderivate). Zu
den Beispielen für
Antioxidantien gehören
butyliertes Hydroxyanisol (BHA) Ascorbinsäure und Derivate davon, Tocopherol
und Derivate davon, butyliertes Hydroxyanisol und Cystein. Beispiele
für Konservierungsstoffe sind
Parabene, wie beispielsweise Methyl- oder Propyl-p-hydroxybenzoat und
Benzoalkoniumchlorid. Beispiele für Feuchthaltemittel sind Glycerin,
Propylenglycol, Sorbit und Harnstoff. Beispiele für Penetrationsverstärker sind
Propylenglycol, DMSO, Triethanolamin, N,N-Dimethylacetamid, N,N,-Dimethylformamid,
2-Pyrrolidon und Derivate davon, Tetrahydrofurfurylalkohol und Azon.RTM.
Beispiele für
Komplexbildner sind Natrium-EDTA, Zitronensäure und Phophorsäure. Beispiele
für Gelbildner
sind Carbopol, Zellulosederivate, Bentonite, Alginate, Gelatine
und Polyvinylpyrrolidon. Beispiele für Salbenbasen sind Bienenwachs,
Paraffin, Cetylpalmitat, Pflanzenöle, Sorbitanester der Fettsäuren (Span),
Polyethylenglycole und Kondensationsprodukte von Sorbitanestern
von Fettsäuren
und Ethylenoxid (z.B. Polyoxyethylensorbitanmonooleat (Tween)).
-
Die
oben beschriebenen pharmazeutischen Zusammensetzungen für die topische
Applikation auf der Haut sind auch in Verbindung mit einer topischen
Applikation auf oder nahe dem zu behandelnden Körperteil möglich. Die Zusammensetzungen
können
für die
direkte Applikation oder für
die Einbringung in die relevante(n) Öffnung(en) des Körpers (z.B.
rektal, urethral, vaginal oder orale Öffnungen) entsprechend angepasst werden.
Sie können
auch mittels spezieller Wirkstoffzulieferungssysteme wie Dressings
oder alternativ Pflastern, Pads, Schwämmen, Strips oder anderen Formen
von geeignetem Material aufgebracht werden.
-
Perkutane
und topische Zusammensetzungen mit kontrollierter Freisetzung
-
Es
gibt verschiedene Methoden zur Kontrolle des Verhältnisses
von Freisetzung und transdermaler Wirkstoffpenetration beinhaltend:
membranvermittelte Systeme, adhäsive
diffusionskontrollierte Systeme, Systeme vom Matrixdispersionstyp
und Mikroreservoirsysteme. Eine perkutane und/oder topische Zusammensetzung
mit kontrollierter Freisetzung kann durch Verwendung einer geeigneten
Mischung aus den oben erwähnten
Methoden erhalten werden.
-
In
einem membranvermittelten System befindet sich die aktive Substanz
in einem Reservoir, welches vollständig in einem schmalen Bereich
eingeschlossen ist, der aus einem für den Wirkstoff undurchdringlichen Laminat,
wie beispielsweise einem metallischen Kunststofflaminat und einer
das Verhältnis
kontrollierenden Polymermembran, wie beispielsweise einer mikroporösen oder
nicht-porösen
Polymermembran (z.B. Ethylen-vinylacetat-Copolymer) besteht. Die
aktive Komponente wird somit nur durch die das Verhältnis kontrollierende
Polymermembran freigesetzt. Die aktive Substanz kann in dem Wirkstoffreservoir
entweder dispergiert in einer festen Polymermatrix oder suspendiert
in einem viskosen flüssigen
Medium wie beispielsweise als Silikonflüssigkeit vorliegen. Auf der
externen Oberfläche
der Polymermembran ist eine dünne
Schicht eines adhäsiven
Polymers aufgebracht, um einen engen Kontakt zwischen transdermalem
System und Hautoberfläche zu
erreichen. Bei dem adhäsiven
Polymer handelt es sich vorzugsweise um ein hypoallergenes Polymer,
welches mit dem aktiven Wirkstoff kompatibel ist.
-
In
einem adhäsiven
diffusionskontrollierten System wird ein Reservoir des aktiven Wirkstoffs
durch direkte Dispersion des aktiven Wirkstoffs in einem adhäsiven Polymer
und anschließendem
Ausbreiten des den aktiven Wirkstoff enthaltenden Adhäsivs auf
einem flachen Bogen der im Wesentlichen wirkstoffundurchlässigen metallischen
Kunststoffgrundlage gebildet, um so eine dünne Wirkstoffreservoirschicht
zu formen. Ein Matrixsystem vom Dispersionstyp ist dadurch charakterisiert,
dass ein Reservoir der aktiven Substanz gebildet wird durch im Wesentlichen
homogene Dispersion der aktiven Substanz in einer hydrophilen oder
lipophilen Polymermatrix und anschließendem Formen des den aktiven
Wirkstoff enthaltenden Polymers zu einer Scheibe mit einem wesentlich
gut definierten Oberflächenbereich
und einer gut definierten Dicke. Das adhäsive Polymer wird um die Peripherie
verteilt, um so einen Adhäsivstreifen
um die Scheibe zu bilden.
-
In
einem Mikroreservoirsystem wird das Reservoir der aktiven Substanz
gebildet, indem zuerst die Wirkstoffbestbestandteile in einer wässrigen
Lösung
eines wasserlöslichen
Polymers suspendiert werden und dann die Wirkstoffsuspension in
einem lipophilen Polymer dispergiert wird, um eine Vielzahl an mikroskopischen
Sphären
(Kügelchen)
von Wirkstoffreservoiren zu bilden.
-
Dosierungen
-
Die
Dosierung einer jeden Komponente der beanspruchten Kombination hängt von
verschiedenen Faktoren ab, einschließlich der Verabreichungsmethode,
der zu behandelnden Krankheit, ob die Krankheit behandelt oder verhindert
werden soll sowie dem Alter, dem Gewicht und der Gesundheit der
zu behandelnden Person.
-
Die
Verbindungen werden vorzugsweise in einer Menge von etwa 0,1–30 mg/kg
Körpergewicht
pro Tag verabreicht und idealerweise in einer Menge von 0,5–15 mg/kg
Körpergewicht
pro Tag. Wie oben beschrieben, kann die in Frage kommende Verbindung
oral in Form von Tabletten, Kapseln, Elixieren oder Sirup oder rektal
in Form von Suppositorien verabreicht werden. Die parenterale Verabreichung
einer Verbindung wird in geeigneter Weise durchgeführt in Form
einer Salzlösung
oder mit der in Liposomen inkorporierten Verbindung. In Fällen, in
denen die Verbindung selbst nicht ausreichend löslich ist, kann ein Lösungsvermittler,
wie beispielsweise Ethanol, angewandt werden. Zu Anschauungszwecken
sind unten die Dosierungen für
Chlorpromazin und Pentamidin beschrieben. Der Fachmann wird erkennen,
dass, wenn eine zweite Verbindung als Ersatz für entweder Chlorpromazin oder
Pentamidin eingesetzt wird, die korrekte Dosierung sowohl anhand
der Untersuchung der Wirksamkeit der Verbindung in den Zellproliferationsassays
als auch durch Bestimmung der Toxizität beim Menschen ermittelt werden
kann.
-
Orale Verabreichung
-
Die
Dosierung für
Chlorpromazin zur oralen Verabreichung für den systemischen Einsatz
beträgt
normalerweise etwa 1 mg bis 1000 mg pro verabreichter Dosis (vorzugsweise
etwa 5 mg bis 500 mg und idealerweise etwa 10 mg bis 300 mg) ein-
bis zehnmal täglich
(vorzugsweise ein- bis fünfmal
täglich)
für einen
Tag bis zu einem Jahr, kann aber auch dem Patienten zeitlebens gegeben
werden. Da die erfindungsgemäßen Kombinationen
primär
eher als Zytostatika anstatt als zytotoxische Agenzien fungieren
und eine niedrige Toxizität
besitzen, kann in vielen Fällen
eine Langzeitgabe angezeigt sein. Es können Dosierungen von bis zu
8 g pro Tag erforderlich sein.
-
Die
Dosierung für
Pentamidin beträgt
normalerweise etwa 0,1 mg bis 300 mg pro verabreichter Dosis (vorzugsweise
etwa 1 mg bis 100 mg) ein bis viermal täglich für einen Tag oder ein Jahr und
wie Chlorpromazin kann es zeitlebens eingenommen werden. Die Verabreichung
kann auch in Zyklen erfolgen, in denen Zeiträume mit eingeschlossen sind,
in denen Pentamidin nicht gegeben wird. Dieser Zeitraum kann beispielsweise etwa
einen Tag, eine Woche, einen Monat oder ein Jahr und mehr betragen.
-
Rektale Verabreichung
-
Für Zusammensetzungen,
die für
die rektale Anwendung zur Krankheitsprävention angepasst wurden, wird
vorzugsweise eine etwas größere Menge
einer Verbindung eingesetzt. Somit beträgt die Dosierung für Chlorpromazin
normalerweise 5 mg bis 2000 mg pro Dosis (vorzugsweise etwa 10 mg
bis 1000 mg, idealerweise etwa 25 mg bis 500 mg), in einer Verabreichung
von ein bis viermal täglich.
Die Behandlungsdauer ist wie für
die orale Einnahme beschrieben. Die Dosierung für Pentamidin entspricht der
für oral
verabreichtes Pentamidin beschriebenen.
-
Parenterale
Verabreichung
-
Zur
intravenösen
oder intramuskulären
Verabreichung von Chlorpromazin wird eine Dosierung von 0,1 mg/kg
bis etwa 100 mg/kg Körpergewicht
pro Tag empfohlen, bevorzugt wird eine Dosis von 1 mg/kg bis etwa 25
mg/kg und idealerweise eine Dosis von 1 mg/kg bis 10 mg/kg.
-
Jede
Komponente wird normalerweise täglich
für bis
zu 6 bis 12 Monate oder länger
verabreicht. Es mag wünschenswert
sein, eine Verbindung über
einen Zeitraum von ein bis drei Stunden zu geben; dieser Zeitraum
kann bis auf 24 Stunden oder länger
ausgedehnt werden. Wie für
die orale Verabreichung beschrieben, kann es dabei Zeitabschnitte
von etwa einem Tag bis zu einem Jahr oder länger geben, in denen mindestens einer
der Wirkstoffe nicht gegeben wird.
-
Inhalation
-
Zur
Inhalation wird Chlorpromazin in einer Dosierung von 1 mg bis zu
1000 mg täglich,
vorzugsweise in einer Dosierung von etwa 10 mg bis 500 mg täglich gegeben.
Die Dosis für
Pentamidin beträgt
etwa 10 mg bis 1000 mg, vorzugsweise 30 mg bis 600 mg täglich.
-
Perkutane
Verabreichung
-
Zur
topischen Verabreichung einer jeden einzelnen Komponente wird vorzugsweise
eine Dosierung von 1 mg bis zu etwa 5 g ein- bis zehnmal täglich über den
Zeitraum von einer Woche bis zu 12 Monaten eingesetzt.
-
Die
folgenden Beispiele dienen dazu, die Erfindung zu veranschaulichen.
Sie sind nicht dazu gedacht, den Erfindungsgedanken auf irgendeine
Weise einzuschränken.
-
Beispiel 1: Herstellung
der Chlorpromazin/Pentamidinisethionat-Verdünnungsmatrix
-
Es
wurden Stocklösungen
mit Chlorpromazin- und Pentamidinisethionat (Sigma Katalognummer C8138
beziehungsweise P0547) in Dimethylsulfoxid (DMSO) in Konzentrationen
von 11,25 mM beziehungsweise 6,74 mM angesetzt. Eine achtfache Stocklösung (128 μM) einer
jeden einzelnen Verbindung wurde in Dulbeco's Modified Eagle Medium (DMEM) (Gibco
11995–040),
enthaltend 10% fetales Rinderserum (FBS), 200 mM L-Glutamin und
1 % Antibiotikum/Antimykotikum-Lösung
hergestellt. Daraus wurde eine 2-fache Verdünnungsreihe in DMEM erstellt.
Diese Reihe ergab 9 Konzentrationsstufen im Bereich von 64 μM bis 240
nM und eine Konzentration von 0 M. Die Matrix mit dem Gemisch der
Verbindungen wurde hergestellt durch Befüllen von Spalten (Columns)
einer Mikrotiterplatte mit 384 Wells mit der Verdünnungsreihe
von Chlorpromazin (erste Spalte: 32 μM, zweite Spalte: 16 μM, dritte
Spalte: 8 μM,
vierte Spalte: 4 μM,
fünfte
Spalte: 2 μM,
sechste Spalte: 1 μm,
siebte Spalte: 500 nM, achte Spalte: 250 nM, neunte Spalte: 125
nM und zehnte Spalte: keine Verbindung) und dem Befüllen der
Reihen (Rows) mit der Verdünnungsreihe
von Pentamidin (erste Reihe: 32 μM,
zweite Reihe: 16 μM,
dritte Reihe: 8 μM,
vierte Reihe: 4 μM,
fünfte
Reihe: 2 μM,
sechste Reihe: 1 μm,
siebte Reihe: 500 nM, achte Reihe: 250 nM, neunte Reihe: 125 nM
und zehnte Reihe: keine Verbindung) mit einem 16-Kanal-Pipettor
(Finnipette). Die Platte mit der Mischung aus Verbindungen lieferte
4fache Konzentrationen einer jeden Verbindung, die auf die Assayplatten
transferiert wird. Die Verdünnungsmatrix
enthielt 100 verschiedene Punkte – 81 Wells mit unterschiedlichen
Mengen an Chlorpromazin und Pentamidin sowie eine 10-Punkt-Verdünnungsreihe
(2fach) für
jede einzelne Verbindung.
-
Beispiel 2: Assay für antiproliferative
Aktivität
-
Die
Matrix mit den verdünnten
Verbindungen wurde mithilfe der A549 Bromodeoxyuridin (BrdU9)-Cyblot-Methode
untersucht. Fünfundvierzig
Mikroliter einer Suspension enthaltend A549 Lungenadenokarzinomzellen
(ATCC Nr.CCL-185) wurden in eine weiße opaque sterile zellkulturbehandelte
384-Well-Mikrotiterplatte aus Polystyrol (NalgeNunc Nr. 164610)
unter Verwendung eines Multidrop (Labsystems) gegeben, um eine Dichte
von 3000 Zellen pro Well zu erhalten. Die Matrix mit den verdünnten Verbindungen
wurde mithilfe eines 16-Kanal-Pipettors (Finnpipette) transferiert.
Zusätzlich
wurden zu jeder Platte Kontrollwells mit Paclitaxel (Endkonzentration
4,6 μM),
Podophyllotoxin (9,6 μM)
und Quinacrin (8,5 μM)
angelegt. Jedes der Experimente wurde in dreifacher Ausführung durchgeführt.
-
Nach
einer Inkubation von 48 Stunden bei 37 °C wurde BrdU zu jedem Well in
einer Konzentration von 10 μM
hinzugegeben. Nach 16 Stunden wurde das Medium abgesaugt und die
Zellen durch Zugabe von 70 % Ethanol und Phosphat gepufferter Salzlösung (PBS)
bei Raumtemperatur für
1 Stunde fixiert. Das Fixierungmittel wurde abgesaugt und 2N HCL
mit Tween 20 (Polyoxyethylensorbitanmonolaurat zu jedem Well hinzugegeben
und die Platten für
20 Minuten bei Raumtemperatur inkubiert. Die HCL wurde mit einer
Lösung
an 2N NaOH neutralisiert und die Zellen zweimal mit Hanks Balanced
Salt Solution (HBSS) gewaschen und einmal mit PBS enthaltend 0,5
% Rinderserumalbumin (BSA) und 0,1 % Tween 20. Die Waschlösung wurde
entfernt und Maus-Anti-BrdU-Primärantikörper (PharMingen
Nr. 555627) 1:1000 in PBS enthaltend BSA, Tween 20 sowie der sekundäre Antikörper 1:2000
(Amersham Nr.NA931) verdünnt.
Der sekundäre
Antikörper
erkennt den Maus-Antikörper
und wird mit dem Enzym Meerrettich-Peroxidase (HRP) konjugiert.
Nach einer Stunde Inkubation wurde die Lösung entfernt und die Zellen
mit PBS gewaschen. Nach dem PBS-Waschvorgang wurde das Substrat
HRP (welches Luminol, Wasserstoffperoxid und einen Verstärker wie
beispielsweise para-Jodphenol enthält) zu jedem Well hinzugegeben.
Die Platten wurden mithilfe eines LJL Analyst gelesen. Alle Absaugvorgänge sowie
die Waschungen mit PBS und HBSS wurden mithilfe eines TECAN Power
Washer 384 durchgeführt.
Die von jedem Well abgegebene Lichtmenge ist ein Hinweis auf das
Ausmaß an
DNA-Synthese, die in dem Well stattgefunden hat. Die verminderte
Lichtabgabe bedeutet somit eine antiproliferative Wirkung der Verbindung.
-
Die
Lumineszenz für
jede einzelne Position in der Chlorpromazin/Pentamidin-Verdünnungsmatrix
wurde unterteilt in die Lumineszenzwerte für A549 Zellen, die nur mit
DMSO behandelt wurden, und lieferten Antiproliferationsverhältnisse
für jede
Einzelposition in der Chlorpromazin/Pentamidin-Verdünnungsmatrix.
Auch für
Paclitaxel, Podophyllotoxin und Quinacrin wurden Antiproliferationsverhältnisse
berechnet und zu Vergleichzwecken verwendet.
-
Tabelle – Antiproliferationsverhältnisse Pentamidinkonzentrationen
(μM)
-
Chlorpromazinkonzentrationen
(μM) (Beschriftung
Tabelle links)
-
In
einer Konzentration von 4,0 μM
erzielt Pentamidinisethionat allein ein Antiproliferationsverhältnis von
3,9 und dieses steigt bei einer Verdopplung der Konzentration auf
8,0 μM auf
4,9 an. Viermikromolares Chlorpromazin ergibt ein Verhältnis von
2,9, welches durch Verdopplung der Konzentration auf 8,0 μM nicht weiter
ansteigt. Wenn 4,0 μM
Pentamidin in Kombination mit 4,0 μM Chlorpromazin (8,0 μM Gesamtverbindungsspezies)
getestet wird, wird ein Antiproliferationsverhältnis von 9,7 erreicht.
-
In
einer anderen Analyse wird die Potenz der jeweiligen Einzelverbindung
durch Anwesenheit einer anderen Verbindung verlagert. Das maximale
mit Pentamidin allein erreichte Proliferationsverhältnis betrug
4,9 (bei 8,0 mM); beobachtet wurde dieses, wenn 1 μM Pentamidin
mit Chlorpromazin mit einer Konzentration von 125 nM kombiniert
wurde, wodurch die Gesamtwirkstoffspezies, die zum Erreichen dieses
Effektes erforderlich war, deutlich gesenkt wurde.
-
Wir
zeigten, dass die Kombination von Chlorpromazin- und Pentamidinisethionat
das Wachstum von A549-Zellen verhindert ohne diese zu töten. A549-Zellen
wurden als Subkonfluenz in 6-Well-Mikrotiterplatten gegeben und
mit 4 μM
eines jeden Wirkstoffs für
72 Stunden behandelt. Das Medium wurde ausgetauscht und die Zellen
für sieben
Tage kultiviert (mit einem Mediumwechsel); die Zellen wurden dann
ausgezählt
und ihre Lebensfähigkeit
bestimmt. Während
die unbehandelten Zellen nach sieben Tagen bis zur Konfluenz gewachsen
sind, wuchsen die mit Chlorpromazin/Pentamidinisethionat behandelten
Zellen nicht; ihre Dichte blieb aber nahe dem Wert, zu dem sie eingesetzt
wurden. Darüber
hinaus waren 95% der mit Chlorpromazin/Pentamidinisethionat behandelten
Zellen noch lebensfähig,
nur geringfügig
weniger als für
die unbehandelten Zellen zu beobachten war.
-
Die
Kombinationen haben wenig bis gar keinen Effekt auf die Lebensfähigkeit
von normalen Lungenfibroblasten (MRC9). Die Kontrollexperimente,
die dies belegen, sind unten beschrieben. MRC9-Zellen wurden in
384-Well-Mikrotiterplatten gegeben, um zu konfluieren. Eine Chlorpromazin/Pentamindisethionat-Verdünnungsmatrix
wurde zugegeben, um überlappende
2-fache Verdünnungsreihen
in einer 12 × 12
Matrix zu erhalten, wobei 16 μM
einer jeden Komponente die Maximaldosis darstellte. Die Zellen wurden
mit den Verbindungen für
48 Stunden inkubiert. Danach wurden 6 μl Alamar Blue Vitalitätsfarbstoff
zu jedem Well gegeben. Die Zellen wurden für weitere 4 Stunden mit dem
Farbstoff inkubiert. Die reduzierte Farbstoffmenge wurde durch Fluoreszenz
mithilfe eines Analyst AD bestimmt. Die nicht fluoreszierende (oxidierte)
Form der Farbe wird durch die lebenden Zellen zu einer fluoreszierenden
Form reduziert. Somit ist das Verhältnis der Fluoreszenz der Wells,
in denen keine Verbindung enthalten war, im Vergleich zu denen,
die eine Verbindung enthielten, proportional zum Ausmaß des durch
die Verbindungen hervorgerufenen Zelltods. In diesem Experiment traten
bis zu einer Konzentration von 16 μM jeder Einzelverbindung keine
toxischen Effekte auf, wohingegen antiproliferative Effekte bei
niedrigeren Konzentrationen beobachtet wurden.
-
Die
antiproliferative Wirkung, die bei A459-Zellen gezeigt wurde, kann
in ähnlicher
Weise unter Verwendung anderer Krebszelllinien, wie beispielsweise
MCF7 Mamma-Adenokarzinom, PA-1 Teratokarzinom des Ovars, HT29 kolorektales
Adenokarzinom, H1299 großzelliges
Karzinom, U-2 OS Osteosarkom, U-373 MG Glioblastom, Hep-3B hepatozelluläres Karzinom,
BT-549 Mammakarzinom, T-24 Blasenkrebs, C-33A Zervixkarzinom, HT-3
metastatisches Zervixkarzinom, SiHa squamöses Zervixkarzinom, CaSki epidermoides
Zervixkarzinom, NCI-H292 mukoepidermoides Karzinom, NCI-2030, nicht-kleinzelliges
Lungenkarzinom, HeLa epitheliales Zervikaladenokarzinom, KB epitheliales
Mundkarzinom, HT 1080 epitheliales Fibrosarkom, Saos-2 epitheliales
Osteosarkom, PC3 epitheliales Prostata-Adenokarzinom, SW480 kolorektales Karzinom, CCL-228
und MS-751 epidermoide Zervixkarzinom-Zelllinien demonstriert werden.
Die Spezifität
kann unter Verwendung von Zellen wie beispielsweise NHLF-Lungenfibroblasten,
NHDF dermale Fibroblasten, HMEC Mammaepithelzellen, PrEC Prostataepithelzellen,
HRE Nierenepithelzellen, NHBE Bronchialepithelzellen, CoSmC glatte
Muskelzellen des Kolons, CoEC Endothelzellen des Kolons, NHEK Epidermiskeratozyten
und Knochenmarkszellen als Kontrollzellen getestet werden.