-
Die
vorliegende Erfindung betrifft eine bei variabler Temperatur härtbare Zusammensetzung,
das Verfahren zur Herstellung davon, ein Verfahren zum Härten davon,
ein gehärtetes
reines Harz, ein Pre-preg, ein Composite und einen geformten Artikel,
und ein Verfahren zum Auswählen
eines Temperatur-Zeit-Profils zum Härten einer bei variabler Temperatur
härtbaren
Zusammensetzung. Insbesondere betrifft die vorliegende Erfindung
eine bei niedriger Temperatur oder eine bei variabler Temperatur
rasch-härtende
härtbare
Harzzusammensetzung, umfassend ein reaktives Thermoplast-Harz und
einen reaktiven Katalysator, das gehärtete Harz, Pre-preg, Composite,
geformte Produkt, und ein Verfahren zur Herstellung, zum Vor-Härten und
Nach-Härten davon,
und ein Verfahren zum Auswählen
eines Temperatur-Zeit-Profils zum Härten einer bei variabler Temperatur
härtbaren
Zusammensetzung.
-
Für die Herstellung
technischer Materialien (engineering grade materials), wie etwa
Composites und Adhäsive,
gekennzeichnet durch anspruchsvolle mechanische Eigenschaften, werden
härtbare
Harzzusammensetzungen, umfassend eine Thermoplast-Komponente und eine
wärmehärtbare Komponente
in Kombination, typischerweise in einem Autoklaven bei erhöhter Temperatur
und erhöhtem
Druck, während
eines ausreichenden Zeitraums gehärtet, um zu ermöglichen,
dass die Reaktion zu einer Zunahme von Molekulargewicht und Glasübergangstemperatur
(Tg) führt.
Das Härten
muss während
eines ausreichenden Zeitraums ausgeführt werden, damit sich diese
und andere mechanische Eigenschaften entwickeln können.
-
Es
gibt zwei wichtige Möglichkeiten
bei der Anwendung von Härten
bei niedriger Temperatur (LTC, low temperature curing), zuerst die
Verwendung von kostengünstigen
Werkzeugmaterialien, welche ein geringes Gewicht aufweisen, d.h.
Ersetzen von Stahl durch Aluminium. Dies ist möglich, da LTC bedeutet, dass
Unterschiede der thermischen Ausdehnungskoeffizienten zwischen dem
Composite und Aluminium nicht so kritisch wie bei hoher Temperatur
sind. Zweitens sind Toleranzen im Hinblick auf kleinere Dimensionen
erzielbar, was sehr wichtig für
das Konstruieren von Mehrkomponentenstrukturen hoher Qualität und für das Zusammenfügen von
Komponententeilen ist, wobei Spannungen vermieden werden, aber gutes
Zusammenpassen bereitgestellt wird.
-
Härten bei
niedriger Temperatur erfordert jedoch verlängerte (extended) Zeiträume zum
Härten.
In Fällen,
in denen Härten
in einem Autoklav durchgeführt
wird, oder bei der Produktion von gehärteten Teilen bei einer hohen
Turnover-Rate, können
die verlängerten
Zeiträume,
welche zum Härten
erforderlich sind, nicht akzeptierbar sein. In einem solchen Fall,
kann es lediglich eine Frage des Erhöhens der Härttemperatur sein, dies ist
jedoch nicht in allen Fällen
möglich,
oder es ist mit einigen Katalysatoren nicht wirksam.
-
Es
besteht ein Bedarf an einer Thermoplast-Harz-enthaltenden Zusammensetzung, welche
bei niedrigen Temperaturen gehärtet
werden kann. Eine Anzahl von Katalysatoren zur Härtung bei niedriger Temperatur
sind zweckmässig
in anderen Systemen, wie etwa Imidazol- und Harnstoff-basierte Härtungs-Katalysatoren bei
niedriger Temperatur. Jedoch wurde beim Testen dieser in der vorliegenden
Zusammensetzung gefunden, dass diese schlechtere Eigenschaften ergeben,
und selbst bei verlängertem
Nach-Härten
die Glasübergangstemperatur
nicht ausreichend entwickelten. Es besteht ebenfalls ein Bedarf
an einem Katalysator, welcher für rasches
Härten
wirksam ist.
-
Wir
haben nun überraschend
gefunden, dass eine Thermoplast-wärmehärtbare Harz-enthaltende Zusammensetzung
bereitgestellt werden kann, welche bei variablen Temperaturen und
während
variabler Härtungszeiträume härtbar ist
und gehärtete
Produkte mit akzeptierbaren Eigenschaften bereitstellt. Die Zusammensetzung
der Erfindung ist beispielsweise bei niedriger Temperatur härtbar, bei
welcher noch stärker überraschend
gefunden wurde, dass das gehärtete
Produkt vergleichbare oder bessere Eigenschaften im Vergleich zu
herkömmlichen
gehärteten
Zusammensetzungen, aufweist. Darüberhinaus
wurde gefunden, dass die Verwendung der Härtung bei niedriger Temperatur
in einem neuen Verfahren, umfassend ein Vor-Härten und ein Nach-Härten zu
weiteren Vorteilen im Hinblick auf Processing und Eigenschaften
führt.
Wir haben darüberhinaus
gefunden, dass die Verwendung einer Härtung bei hoher Temperatur
in einem einzigen Schritt (stage) zu erwünschtem raschen Härten und
erwünschtem
Turnover führt.
-
Gemäss einem
breitesten Aspekt der vorliegenden Erfindung wird ein Verfahren
zum Herstellen und Vor-Härten einer
härtbaren
Zusammensetzung bereitgestellt, welche wenigstens eine polyaromatische
Verbindung mit reaktiven Endgruppen, wenigstens ein wärmehärtbares
Harz und einen reaktiven Katalysator umfasst, wobei der Katalysator
eine Lewis-Säure
mit Aminfunktionalität
umfasst, welche die reaktiven Endgruppen des Thermoplasten mit dem
wärmehärtbaren
Harz bei einer Vor-Härtungs-Temperatur im Bereich
von 60 bis 150°C
koordiniert, und
wobei das wenigstens eine wärmehärtbare Harz
ein Epoxy-Harz ist, und
wobei die wenigstens eine polyaromatische
Verbindung wenigstens ein Polyarylsulfon umfasst, welches über Etherbindungen
verknüpfte,
sich wiederholende Einheiten umfasst, und optional zusätzlich über Thioether-Bindungen
verknüpfte,
sich wiederholende Einheiten umfasst, wobei die Einheiten ausgewählt sind
aus der Gruppe bestehend aus
-(PhSO2Ph)n
und optional zusätzlich
-(Ph)a
wobei
Ph gleich Phenylen ist und n = 1 bis 2 ist und eine gebrochene Zahl
sein kann, a = 1 bis 3 ist und eine gebrochene Zahl sein kann, und
wenn a über
1 ist, die Phenylene linear durch eine einzelne chemische Bindung
oder eine andere bivalente Gruppe als -SO2-
verbunden sind, oder sie miteinander verschmolzen sind, vorausgesetzt,
dass die sich wiederholende Einheit -(PhSO2Ph)n- in dem wenigstens einen Polyarylsulfon
immer in einem solchen Anteil vorliegt, dass durchschnittlich wenigstens
zwei der Einheiten -(PhSO2Ph)n-
in einer Sequenz in jeder Polymerkette vorhanden sind,
dadurch
gekennzeichnet, dass das Verfahren umfasst:
Vermischen der
entsprechenden reaktiven polyaromatischen Komponente und der wärmehärtbaren
Komponente wie hierin zuvor definiert, optional in Gegenwart von
Lösungsmittel,
Entfernen
von verbleibendem Lösungsmittel
durch Verdampfung bei 50 bis 200°C
und anschliessend Zumischen des Katalysators und Vor-Härten in
einem ersten Schritt durch Erwärmen
der Zusammensetzung in einem ersten Temperaturschritt im Bereich
von 60 bis 150°C
unter Verwendung von erhöhtem
Druck von bis zu 10 bar, um Verformungseffekte entweichender Gase
zu beschränken
oder um Hohlraumbildung zu beschränken.
-
Vorzugsweise
umfasst das Verfahren zusätzlich
Nach-Härten in
einem zweiten Schritt durch Erwärmen
der vorgehärteten
Zusammensetzung auf eine Temperatur im Bereich von 150 bis 200°C bei atmospärischem
Druck, um die Glasübergangstemperatur
des Produkts zu verbessern. Vorzugsweise handelt es sich um ein
solches Verfahren, bei welchem Nach-Härten ohne Verwendung eines
Autoklaven oder Formen oder Werkzeugen durchgeführt wird, welche bei der Vor-Härtungs-Reaktion eingesetzt
wurden.
-
Vorzugsweise
hat der Katalysator die Formel: LXn·R,wobei
LXn eine Lewis-Säure
und R ein Amin ist. Vorzugsweise ist L ausgewählt aus Gruppen IIb, IIIb,
VIII des Periodensystems der Elemente, und X ist Halogen.
-
Bevorzugte
Katalysatoren umfassen BF3, AlF3,
FeF3, ZnF2 als Lewis-Säure-Komponente
und primäres oder
sekundäres
aliphatisches oder aromatisches Amin, wie etwa Monoethylamin (mea),
Dimethylamin (dma), Benzylamin (bea) oder Piperidin.
-
Es
wird angenommen, dass der Lewis-Säure-Katalysator als Komplex
oder als äquivalente
Form vorliegt, welcher imstande ist die reaktiven Endgruppen des
Thermoplast-Harzes mit einem wärmehärtbaren
Harz zu koordinieren. Es wurde gefunden, dass die bestimmten Lewis-Säure-Komplexe
der Erfindung sowohl eine reaktive Funktion als auch eine moderierende (moderating)
Funktion aufweisen, wobei die Vor-Härtungs-Reaktion
die erforderliche Selektivität
aufweist. Ohne an diese Theorie gebunden zu sein, wird angenommen, dass
zwei mögliche
Reaktionen in der härtbaren
Zusammensetzung ablaufen können – die Reaktion
zwischen reaktiven Gruppen der entsprechenden Thermoplasten und
den wärmehärtbaren
Harz-Komponenten, und die Reaktion zwischen entsprechenden Gruppen
von nur einem von diesen, beispielsweise die Reaktion zwischen Amingruppe
und einer Epoxy-Gruppe, oder die Reaktion zwischen einer Epoxy-Gruppe
und einer Hydroxyl-Gruppe, abgeleitet von der ringöffnenden-Reaktion
einer anderen Epoxy-Gruppe.
Die Reaktion zwischen Thermoplast und wärmehärtbarem Harz würde Kettenwachstum
genannt werden, während
die Selbstreaktion, beispielsweise des wärmehärtbaren Harzes einfach eine
Mono-Phasenreaktion
sein würde,
in diesem Fall Veretherung.
-
Es
wird angenommen, dass Kettenwachstum die katalytischen Reaktionen
in der Zusammensetzung der vorliegenden Erfindung dominiert und
fördert
die Kettenverlängerungsreaktion
zwischen Anteilen mit niedrigem Molekulargewicht des Thermoplast-Polymers.
Als Ergebnis käme
ein kontrollierter Aufbau der Gesamtverteilung des Molekulargewichts
des Polymers heraus, was zu Phasentrennung führen würde.
-
Die
Zusammensetzung kann zusätzliche
Komponenten, welche auf dem technischen Gebiet herkömmlich sind,
umfassen. Vorzugsweise umfasst die Zusammensetzung einen oder mehrere
zusätzliche
Katalysatoren oder Härtungsmittel.
-
Das
zusätzliche
Härtungsmittel
wird geeigneterweise aus jeglichen bekannten Härtungsmitteln gewählt, beispielsweise
wie in EP-A-0 311 349, EP-A-486 197 (EP-A-913 101 67.1), EP-A-0
365 168 oder in WO-A-95/33785
(PCT/GB95/01303) offenbart, wie etwa eine Aminoverbindung mit einem
Molekulargewicht von bis zu 500 pro Aminogruppe, beispielsweise
ein aromatisches Amin oder ein Guanidin-Derivat. Besondere Beispiele
sind 3,3'- und 4-,4'-Diaminodiphenylsulfon
(erhältlich
als "DDS" von kommerziellen
Quellen), Methylendianilin,bis(4-amino-3,5-dimethylphenyl)-1,4-diisopropylbenzol
(erhältlich
als EPON 1062 von Shell Chemical Co); Bis(4-aminophenyl)-1,4-diisopropylbenzol
(erhältlich
als EPON 1061 von Shell Chemical Co); 4-Chlorophenyl-N,N-dimethylharnstoff,
z. B. Monuron; 3,4-Dichlorophenyl-N,N-dimethylharnstoff, z. B. Diuron und
Dicyandiamid (erhältlich
als "Amicure CG
1200" von Pacific
Anchor Chemical). Andere Standard-Epoxy-Härtungsmittel, wie etwa aliphatische
Diamine, Amide, Carbonsäureanhydride,
Carbonsäuren
und Phenole können – falls
gewünscht – verwendet
werden.
-
Es
wird beispielsweise in WO-A-99/43731 (PCT/GB99/00540) angegeben,
und wie in EP-A-0 311 349 oder in WO-A-95/33785 (PCT/GB95/01303)
beschrieben, dass für
die Reaktion von Epoxy-Harz-Komponente/Härtungsmittel
auch ein Katalysator, typischerweise eine Lewis-Säure oder
eine Lewis-Base, verwendet werden kann. Die vorliegende Erfindung unterscheidet
sowohl nach der Auswahl der Lewis-Säure,
eine moderierende Komplex-Funktion aufweisend, wie oben beschrieben,
als auch nach der Wahl von reaktiver polyaromatischer Komponente.
-
Vorzugsweise
umfasst die polyaromatische Verbindung Polyethersulfon, stärker bevorzugt
eine Kombination aus Polyethersulfon und Polyethersulfon-verknüpften sich
wiederholenden Einheiten, in welchen die Phenylengruppe meta- oder
para- ist und vorzugsweise para ist, und wobei die Phenylene linear
durch eine einzige chemische Bindung oder eine bivalente Gruppe,
die nicht Sulfon ist, verknüpft
sind oder miteinander verschmolzen (fused together) sind. Unter "gebrochene Zahl" wird der Durchschnittswert
für eine
bestimmte Polymerkette verstanden, die Einheiten mit verschiedenen
Werten für
n oder a enthält.
-
Zusätzlich sind – wie ebenfalls
diskutiert – in
dem wenigstens einen Polyarylsulfon die relativen Anteile der sich
wiederholenden Einheiten derart, dass im Durchschnitt wenigstens
zwei Einheiten (PhSO2Ph)n in
unmittelbar wechselseitiger Reihenfolge in jeder Polymerkette vorhanden
sind, und vorzugsweise im Bereich 1:99 bis 99:1, insbesondere 10:90
bis 90:10 entsprechend. Das Verhältnis
liegt typischerweise im Bereich 25 bis 50 (Ph)3,
Balance (PhSO2Ph)n.
In bevorzugten Polyarylsulfonen sind die Einheiten:
- I X Ph SO2Ph X Ph SO2Ph
("PES") und
- II X (Ph)a X Ph SO2Ph
("PES")
wobei
X gleich O oder S ist und sich von Einheit zu Einheit unterscheiden
kann; das Verhältnis
ist I zu II, vorzugsweise zwischen 10:90 und 80:20 liegt, insbesondere
zwischen 10:90 und 55:45.
-
Die
bevorzugten relativen Anteile der sich wiederholenden Einheiten
des Polyarylsulfons können
als Gewichtsprozent SO2-Gehalt, definiert
als hundert mal (Gewicht von SO2)/(Gewicht
der durchschnittlichen sich wiederholenden Einheit), ausgedrückt werden.
Der bevorzugte SO2-Gehalt ist wenigstens
22%, vorzugsweise 23% bis 25%. Wenn a = 1, entspricht dies dem PES/PEES-Verhältnis von
wenigstens 20:80, vorzugsweise im Bereich 35:65 bis 65:35.
-
Die
obengenannten Anteile beziehen sich nur auf die genannten Einheiten.
Zusätzlich
zu solchen Einheiten kann das Polyarylsulfon bis zu 50, insbesondere
bis zu 25 Mol-% anderer sich wiederholender Einheiten enthalten:
die bevorzugten Bereiche des SO2-Gehalts
(falls verwendet) gelten dann für
das gesamte Polymer. Solche Einheiten können beispielsweise die Formel: R-Ph-A-Ph-R haben,
wie hier zuvor definiert, in welcher A eine direkte Verknüpfung, Sauerstoff,
Schwefel, -CO- oder ein zweiwertiges Kohlenwasserstoffradikal ist. Wenn
das Polyarylsulfon das Produkt einer nukleophilen Synthese ist,
können
seine Einheiten beispielsweise von einem oder mehreren Bisphenolen
und/oder entsprechenden Bisthiolen oder Phenol-thiolen abgeleitet werden,
ausgewählt
aus Hydrochinon, 4,4'-Dihydroxybiphenyl,
Resorcinol, Dihydroxynaphthalin (2,6 und andere Isomere), 4,4'-Dihydroxybenzophenon,
2,2'-Di-(4-hydroxyphenyl)propan
und -methan.
-
Wenn
ein Bis-thiol verwendet wird, kann es in situ gebildet werden, das
heisst, ein Dihalid, wie beispielsweise weiter unten beschrieben,
kann mit einem Alkalisulfid oder einem Polysulfid oder Thiosulfat
zur Reaktion gebracht werden.
-
Andere
Beispiele für
solche zusätzlichen
Einheiten haben die Formel: -Ph-Q(Ar-Q')n-Ph in
welcher Q und Q',
welche gleich oder verschieden sein können, CO oder SO2 sind;
Ar ein zweiwertiges aromatisches Radikal ist; und n gleich 0, 1,
2 oder 3 ist, vorausgesetzt, dass n nicht Null ist, wenn Q gleich
SO2 ist. Ar ist vorzugsweise wenigstens
ein zweiwertiges aromatisches Radikal, ausgewählt aus Phenylen, Biphenylen
oder Terphenylen. Besondere Einheiten haben die Formel: -Ph-Q-[-(-Ph-)m-Q'-]n-Ph wobei m gleich 1, 2 oder 3 ist. Wenn
das Polymer das Produkt einer nukleophilen Synthese ist, können solche
Einheiten von einem oder mehreren Dihaliden abgeleitet sein, beispielsweise
ausgewählt
aus 4,4'-Dihalobenzophenon,
4,4'-Bis(4-Chlorophenyl-sulfonyl) biphenyl,
1,4,Bis(4-halobenzoyl)benzol und 4,4'-Bis(4-halobenzoyl)biphenyl.
-
Sie
können
selbstverständlich
teilweise von den entsprechenden Bisphenolen abgeleitet sein.
-
Die
polyaromatisch Verbindung kann das Produkt einer nukleophilen Synthese
von Halophenolen und/oder Halothiophenolen sein. In jeder nukleophilen
Synthese kann das Halogen, wenn es Chlor oder Brom ist, durch die
Gegenwart eines Kupferkatalysators aktiviert sein.
-
Eine
solche Aktivierung ist häufig
nicht erforderlich, wenn das Halogen durch eine Elektronenabziehende
Gruppe aktiviert ist. In jedem Fall ist Fluorid für gewöhnlich stärker aktiv
als Chlorid. Jede nukleophile Synthese der polyaromatischen Verbindung
wird vorzugsweise in Gegenwart eines oder mehrerer Alkalimetallsalze,
wie etwa KOH, NaOH oder K2CO3,
in bis zu 10% molarem Überschuss über der
Stöchiometrie
ausgeführt.
-
Wie
zuvor erwähnt,
enthält
die wenigstens eine polyaromatische Verbindung reaktive Endgruppen und/-oder Seitengruppen.
Endgruppen können
durch eine Reaktion von Monomeren oder durch nachfolgende Umwandlung
von Produktpolymeren vor oder nach Isolierung erhalten werden. Vorzugsweise
haben die Gruppen die Formel -A'-Y,
wobei A' eine zweiwertige
Kohlenwasserstoffgruppe, vorzugsweise aromatisch ist, und Y eine
Gruppe ist, die mit Epoxidgruppen oder mit Härtungsmittel oder mit ähnlichen
Gruppen an anderen Polymermolekülen
reaktionsfähig
ist. Beispiele für
Y sind Gruppen, die aktiven Wasserstoff bereitstellen, insbesondere
OH, NH2, NHR' oder -SH, wobei R' eine Kohlenwasserstoffgruppe ist, enthaltend
bis zu 8 Kohlenstoffatome, oder eine andere Vernetzungsreaktivität bereitstellen,
insbesondere Epoxy, (Meth)acrylat, Cyanat, Isocyanat, Acetylen oder
Ethylen, wie in Vinyl, Allyl oder Maleimid, Anhydrid, Oxazolin und
Monomeren, enthaltend eine Sättigung.
Bevorzugte Endgruppen umfassen Amin und Hydroxyl.
-
Das
anzahlgemittelte Molekulargewicht der polyaromatischen Verbindung
liegt geeigneterweise im Bereich von 2 000 bis 60 000. Ein zweckmässiger Unterbereich
ist über
9 000, insbesondere über
10 000, beispielsweise 11 000 bis 25 000, oder ist unter 9 000,
insbesondere im Bereich von 3 000 bis 11 000, beispielsweise 3 000
bis 9 000, [..] und strukturell, sowie durch chemische Wechselwirkung
erhöht
Zähigkeit
durch Vergleich mit der des wärmehärtbaren
Harzes allein durch Bereitstellen von Zonen des zähen Thermoplasten
zwischen vernetzten wärmehärtbaren
Zonen.
-
Das
wärmehärtbare Polymer
ist vorzugsweise ein Epoxy-Harz,
welches abgeleitet ist von dem Mono- oder Poly-Glycidyl-Derivat von einer oder mehreren
der Gruppe von Verbindungen, bestehend aus aromatischen Diaminen,
aromatischen monoprimären
Aminen, Aminophenolen, mehrwertigen Phenolen, mehrwertigen Alkoholen,
Polycarbonsäuren
und dergleichen, oder einem Gemisch von diesen.
-
Vorzugsweise
umfasst das wärmehärtbare Polymer
wenigstens einen Epoxy-Harz-Precursor, der bei Umgebungstemperatur
flüssig
ist, wie beispielsweise in EP-A-0 311 349, EP-A-0 365 168, EP-A-486197 (EP-A-91310167.1) oder
in WO-A-95/33785 (PCT/GB95/01303) offenbart.
-
Ein
Epoxy-Harz kann ausgewählt
werden aus N,N,N',N'-Tetraglycidyl-diaminodiphenylmethan
(z.B. "MY 9663", "MY 720" oder "MY 721", das von Ciba-Geigy
vertrieben wird), mit einer Viskosität von 10 bis 20 Pa s bei 50°C; (MY 721
ist eine Version mit geringerer Viskosität als MY 720 und ist für höhere Verwendungstemperaturen
bestimmt); N,N,N',N'-Tetraglycidyl-bis-(4-aminophenyl)-1,4-diisopropylbenzol
(z.B. Epon 1071, vertrieben von Shell Chemical Co.), Viskosität 18 bis
22 Poise bei 110°C;
N,N,N',N'-Tetraglycidyl-bis-(4-amino-3,5-dimethylphenyl)-1,4-diisopropylbenzol,
(z.B. Epon 1072, vertrieben von Shell Chemical Co.), Viskosität 30 bis
40 Poise bei 110°C;
Triglycidylether von p-Aminophenol
(z.B. "MY 0510", vertrieben von
Ciba-Geigy), Viskosität 0,55 bis
0,85 Pa s bei 25°C;
vorzugsweise mit einer Viskosität
von 8 bis 20 Pa s bei 25°C;
vorzugsweise bildet dies wenigstens 25% der verwendeten Epoxy-Komponenten;
Diglycidylether von Materialien auf der Basis von Bisphenol A, wie
etwa 2,2-Bis(4,4'-dihydoxyphenyl)propan
(z.B. "DE R 661,
vertrieben von Dow, oder "Epikote
828", vertrieben
von Shell), und Novolak-Harze, mit einer Viskosität von vorzugsweise
8 bis 20 Pa s bei 25°C;
Glycidylether von Phenol-Novolak-Harzen (z.B. "DEN 431" oder "DEN 438", vertrieben von Dow), von welchen Variationen
in der niederen Viskositätsklasse
in der Herstellung von Zusammensetzungen gemäss der Erfindung bevorzugt
sind; Diglycidyl-1,2-phthalat, z.B. GLY CEL A-100; Diglycidylderivat
von Dihydroxydiphenylmethan (Bisphenol F), (z.B. "PY 306", vertrieben von
Ciba-Geigy), das zur niederen Viskositätsklasse gehört. Andere
Epoxy-Harz-Precursor umfassen cycloaliphatische Verbindungen, wie
etwa 3',4'-Epoxycyclohexyl-3,-4-epoxycyclohexancarboxylat
(z.B. "CY 179", vertrieben von
Ciba Geigy) und jene im "Bakelit"-Bereich von Union
Carbide Corporation.
-
Das
wärmehärtbare Polymer
ist geeigneterweise das Produkt von wenigstens teilweisem Härten eines Harz-Precursors unter
Verwendung eines Härtungsmittels
und optional eines Katalysators.
-
Der
Gewichtsanteil von Thermoplast-Komponente in der Zusammensetzung
liegt typischerweise im Bereich von 5 bis 100%, vorzugsweise 5 bis
90%, insbesondere 5 bis 50%, beispielsweise 5 bis 40%. In einer besonders
vorteilhaften Ausführungsform
der Erfindung kann der Gewichtsanteil von Thermoplast-Komponente
im Bereich von 10 bis 30%, für
eine gewünschte
Haftmenge, in der endgültigen
Zusammensetzung ausgewählt
werden.
-
Die
entsprechenden Komponenten können
in jeglichen Mengen vorhanden sein, welche für die Reaktion davon geeignet
sind. Vorzugsweise sind die wärmehärtbare Komponente
und die Thermoplast-Harz-Komponente in Mengen vorhanden, jeweils
15 bis 75 Gewichtsteile und in geeigneter Stöchiometrie.
-
Vorzugsweise
sind das Thermoplast-Harz oder das Thermoplast-Harz und die zusätzliche
reaktive Amin-enthaltende
Komponente (DDS) in einer Menge von 35 bis 55 Gewichtsteile, stärker bevorzugt
40 bis 50 Gewichtsteile vorhanden, und die Epoxy-Komponente ist
in einer Menge von 45 bis 75 Gewichtsteile, vorzugsweise 50 bis
60 Gewichtsteile vorhanden.
-
Der
Lewis-Säure-Katalysator
ist in einer katalytisch wirksamen Menge im Bereich von 0,1 bis
5,0 Gewichtsteilen vorhanden, in Abhängigkeit von der Wahl des Katalysators.
Stärker
bevorzugt ist der Katalysator in einer Menge von 0,2 bis 3,0 Gewichtsteilen
vorhanden.
-
Eine
Harzzusammensetzung ist besonders geeignet zur Fabrikation von Strukturen,
umfassend tragende (loadbearing) oder schlagfeste (impact resistant)
Strukturen. Zu diesem Zweck kann sie ein Verstärkungsmittel, wie etwa Fasern
enthalten. Fasern können
als kurze oder geschnittene (chopped) Fasern, typischerweise mit
einer mittleren Faserlänge
von nicht mehr als 2 cm, beispielsweise etwa 6 mm, zugegeben werden.
Alternativ und vorzugsweise sind die Fasern kontinuierlich und können beispielsweise
unidirektional-abgelegte Fasern oder ein Gewebe sein, d.h., das
Compositematerial umfasst ein Prepreg. Kombinationen aus sowohl
kurzen und/oder geschnittenen Fasern wie auch kontinuierlichen Fasern
können
verwendet werden. Die Fasern können
beschlichtet oder unbeschlichtet sein. Fasern können typischerweise mit einer
Konzentration von 5 bis 35 Gewichtsprozent, vorzugsweise wenigstens
20 Gewichtsprozent zugegeben werden. Für strukturelle Anwendungen
ist bevorzugt, eine kontinuierliche Faser, beispielsweise Glas oder
Carbon, mit insbesondere 30 bis 70 Volumenprozent, stärker bevorzugt
50 bis 70 Volumenprozent zu verwenden.
-
Die
Faser kann organisch sein, insbesondere aus steifen Polymeren sein,
wie etwa Polyparaphenylenterephthalamid, oder anorganisch sein.
Von anorganischen Fasern können
Glasfasern, wie etwa "E" oder "S" verwendet werden, oder Aluminiumoxid,
Zirkonoxid, Siliziumcarbid, andere Verbindungen Keramiken (compound
ceramics) oder Metalle. Eine sehr geeignete Verstärkungsfaser
ist Kohlenstofffaser, insbesondere als Graphitfaser. Graphitfasern,
die sich als besonders zweckmässig
in der Erfindung erwiesen haben, sind jene, die von Amoco unter
der Handelsbezeichnung T650-35, T650-42 und T300 geliefert werden;
jene, die von Toray unter der Handelsbezeichnung T800-HB geliefert
werden; und jene, die von Hercules unter der Handelsbezeichnung
AS4, AU4, IM8 und IM7 geliefert werden.
-
Organische
Fasern oder Kohlenstofffasern sind vorzugsweise unbeschlichtet oder
mit einem Material beschlichtet, das mit der Zusammensetzung gemäss der Erfindung
in dem Sinne kompatibel ist, dass es in der flüssigen Precursor-Zusammensetzung
löslich
ist, ohne nachteilige Reaktion oder Bindung sowohl an die Faser
als auch an die wärmehärtbare/thermoplastische
Zusammensetzung gemäss
der Erfindung. Insbesondere Kohlenstoff- oder Graphitfasern, die
unbeschlichtet sind, oder mit einem Epoxy-Harz-Precursor oder einem Thermoplast,
wie etwa Polyarylsulfon, beschlichtet sind, sind bevorzugt. Anorganische
Fasern sind vorzugsweise mit einem Material beschlichtet, das sowohl
an die Faser als auch an die Polymerzusammensetzung bindet; Beispiele
sind die Organo-Silan-Kupplungsmittel, die bei Glasfasern angewendet
werden.
-
Die
Zusammensetzung kann beispielsweise herkömmliche Zähigkeit verbessernde Mittel
enthalten, wie etwa flüssige
Gummen/Kautschuke mit reaktiven Gruppen, Aggregate wie etwa Glasperlen,
Gummi/Kautschukpartikel und mit Gummi/Kautschuk beschichtete Glasperlen,
Füllmittel,
wie etwa Polytetrafluorethylen, Silica, Graphit, Bornitrid, Mika,
Talk und Vermiculit, Pigmente, Keimbildungsmittel und Stabilisatoren
wie etwa Phosphate. Die Gesamtheit solcher Materialien und eines
jeglichen Faserverstärkungsmittels
in der Zusammensetzung sollte wenigstens 20 Volumenprozent betragen,
als ein Prozentsatz des Gesamtvolumens des Gemischs polyaromatische
Verbindung/wärmehärtbares
Harz. Die Prozentsätze
von Fasern und solcher anderer Materialien werden berechnet auf
der Basis der gesamten Zusammensetzung nach Härtung bei den hier weiter unten
definierten Temperaturen.
-
In
einem weiteren Aspekt der Erfindung wird ein Verfahren zur Herstellung
einer Zusammensetzung, wie hierin zuvor definiert, bereitgestellt,
umfassend Vermischen der entsprechenden polyaromatischen Komponente
und der wärmehärtbaren
Komponente, wie hierin zuvor definiert, und anschliessend Zumischen
des Katalysators.
-
Vorzugsweise
wird die Zusammensetzung in der Form einer härtbaren Harzzusammensetzung,
wie hierin zuvor definiert, verwendet, hergestellt durch Vermischen
der polyaromatischen Verbindung, des wärmehärtbaren Precursors und (in
irgendeinem Schritt) irgendein Faserverstärkungsmittel und andere Materialien. Ein
Lösungsmittel
kann vorhanden sein. Das Lösungsmittel
und der Anteil davon werden gewählt,
so dass das Gemisch aus polyaromatischer Verbindung und wärmehärtbarem
Harz-Precursor wenigstens eine stabile Emulsion bilden, vorzugsweise
eine stabile, scheinbar einphasige Lösung. Das Gewichtsverhältnis Lösungsmittel
zu polyaromatische Verbindung liegt geeigneterweise im Bereich von
5:1 bis 20:1. Vorzugsweise wird ein Gemisch von Lösungsmitteln
verwendet, beispielsweise eines aus einem halogenierten Kohlenwasserstoff und
einem Alkohol, in einem Verhältnis
geeigneterweise im Bereich von 99:1 bis 85:15. Geeigneterweise sollten
die Lösungsmittel
in einem solchen Gemisch unterhalb von 100°C bei einem Druck von 1 atm
sieden und sollten in den verwendeten Anteilen wechselseitig mischbar
sein. Als Alternativ können
die polyaromatische Verbindung und das wärmehärtbare Harz oder der Precursor
durch Heissschmelzen und/oder Hochschermischen (high shear mixing)
zusammengebracht werden.
-
Das
Gemisch wird gerührt,
bis es ausreichend homogen ist. Danach wird jegliches Lösungsmittel durch Verdampfung
entfernt, um eine Harzzusammensetzung zu erhalten. Verdampfung erfolgt
geeigneterweise bei 50 bis 200°C,
und wenigstens am Ende (final stages) kann sie unterhalb von Atmosphärendruck
erfolgen, beispielsweise im Bereich von 13,33 Pa bis 1333 Pa (0,1
bis 10 mm Hg). Die Harzzusammensetzung enthält vorzugsweise bis zu 5 Gewichtsprozent
flüchtiges
Lösungsmittel,
um Fliessen zu unterstützen,
bei Verwendung um Fasern zu imprägnieren.
Das restliche Lösungsmittel
wird bei Kontakt mit den warmen Walzen der Imprägniermaschine entfernt.
-
Nach
Entfernung von restlichem Lösungsmittel
wird der reaktive Katalysator eine kurze Zeit vor Giessen und Härten oder
unmittelbar vor Giessen und Härten
zugegeben.
-
Die
Harzzusammensetzung, welche möglicherweise
bereits vorhandenes oder neu-hinzugefügtes, flüchtiges Lösungsmittel enthält, kann
beispielsweise als ein Adhäsiv
oder zur Beschichtung von Oberflächen oder
zum Herstellen fester Strukturen durch Giessen, möglicherweise
in einem geschäumten
Zustand, verwendet werden. Eine Verstärkung auf der Basis kurzer
Fasern kann in die Zusammensetzung vor Härten davon eingearbeitet werden.
Vorzugsweise wird eine faserverstärkte Zusammensetzung hergestellt
durch in Kontakt bringen von im Wesentlichen kontinuierlicher Faser
mit einer solchen Harzzusammensetzung. Das resultierende imprägnierte
Faserverstärkungsmittel
kann allein oder zusammen mit anderen Materialien, beispielsweise
einer weiteren Menge desselben Polymers oder eines anderen Polymers
oder Harz- Precursors oder
eines Gemisches, verwendet werden, um einen geformten Artikel zu
bilden. Diese Technik wird weiter ausführlich in EP-A-56703, 102158
und 102159 beschrieben.
-
In
einem weiteren Aspekt der Erfindung wird ein Verfahren zum Härten einer
härtbaren
Zusammensetzung, wie hierin zuvor definiert, bereitgestellt.
-
Die
härtbare
Harzzusammensetzung der Erfindung kann auf bekannte Weise gehärtet werden.
Geeigneterweise wird die Zusammensetzung in Form einer Harzlösung oder
einer stabilen -emulsion, wie hierin zuvor beschrieben, auf eine
geeignete Form oder ein geeignetes Werkzeug zur Herstellung einer
Platte, eines Prepregs oder dergleichen, übertragen, wobei die Form oder
das Werkzeug auf eine gewünschte
Entgasungs-Temperatur vorgewärmt
wurden.
-
Die
stabile Emulsion ist kombiniert mit jeglichen Verstärkungsmaterialien,
Zähigkeit
verbessernden Materialien, Füllmaterialien,
Keimbildungsmaterialien oder -mitteln oder dergleichen, und die
Temperatur wird erhöht,
um Härten
zu initiieren.
-
Geeigneterweise
wird Härten
ausgeführt
bei erhöhter
Temperatur bis zu 200°C,
vorzugsweise im Bereich von 60 bis 200°C, stärker bevorzugt bei etwa 70
bis 190°C,
und unter Verwendung von erhöhtem
Druck, um Verformungseffekte entweichender Gase zu beschränken oder
um Hohlraumbildung zu beschränken,
geeigneterweise bei einem Druck von bis zu 10 bar, vorzugsweise
im Bereich von 3 bis 7 bar abs. Geeigneterweise wird die Härt-Temperatur
erreicht durch Erwärmen
auf bis zu 5°C/Min.,
beispielsweise 2°C
bis 3°C/Min. und
wird gehalten während
des erforderlichen Zeitraums von bis zu 18 Stunden, vorzugsweise
bis zu 9 Stunden, stärker
bevorzugt bis zu 6 Stunden, beispielsweise 3 bis 4 Stunden. Druckentlastung
findet immer statt und Temperaturreduktion erfolgt durch Kühlen bei
bis zu 5°C/Min.,
beispielsweise bis zu 3°C/Min.
Nach-Härtung
bei Temperaturen im Bereich von 150°C bis 200°C kann bei atmosphärischem
Druck durchgeführt
werden, unter Anwendung geeigneter Wärmeraten, um die Glasübergangstemperatur
des Produktes oder anderweitig (otherwise) zu verbessern.
-
Härten kann
in einem einzigen Schritt oder in zwei Schritten erfolgen, in Abhängigkeit
von der ausgewählten
Härt-Temperatur
und den Erfordernissen von Verarbeitung (processing) und Produkt.
Beispielsweise kann Härten
in einem Autoklaven während
der gesamten Prozedur, oder nur während des Vorhärtungs-Zeitraums
durchgeführt
werden. In einer ersten Ausführungsform
umfasst das Verfahren zum Härten
einer Zusammensetzung, wie hierin zuvor definiert, in einem einzelnen
Schritt, Behandeln der Zusammensetzung mit erhöhter Temperatur und erhöhtem Druck,
während
eines Zeitraums von über
einer Stunde, wobei die Temperatur im Bereich von 150 bis 200°C, vorzugsweise
170 bis 190°C
während
eines Zeitraums im Bereich von 4 bis 7 Stunden, liegt.
-
In
einer weiteren Ausführungsform
umfasst das Verfahren zum Härten
einer Zusammensetzung, wie hierin zuvor definiert, Vor-Härten bei
einer erhöhten
Temperatur und einem erhöhten
Druck, während
eines Zeitraums von über
einer Stunde, wobei die Vor-Härtungs-Temperatur
im Bereich von 60 bis 150°C,
vorzugsweise 70 bis 145°C,
stärker
bevorzugt 80 bis 135°C
liegt.
-
Die
Vor-Härtung
wird vorzugsweise durch anfängliches
Verwenden einer Zeit-Temperatur-Rampe (ramp) erzielt, was auf dem
technischen Gebiet bekannt ist, um eine gewünschte Vor-Härtungs-Temperatur
zu erzielen.
-
Es
wurde überraschend
gefunden, dass unter diesen Bedingungen der Vor-Härtung die
Zusammensetzung selektiv reagiert, um Kettenverlängerungsreaktionen, wie hierin
zuvor beschrieben, zu fördern.
-
Die
Vor-Härtung
wird geeigneterweise während
eines Zeitraums von 1 bis 18 Stunden, vorzugsweise von 10 bis 18
Stunden, stärker
bevorzugt von 12 bis 5 Stunden ausgeführt.
-
In
einem weiteren Aspekt der Erfindung wird eine vorgehärtete Zusammensetzung
oder ein Prepreg eines polyaromatischen Thermoplasten und eines
wärmehärtbaren
Harzes bereitgestellt, umfassend einen kettenverlängerten
Thermoplasten, wie hierin zuvor definiert, mit einem anzahlgemittelten
Molekukargewicht im Bereich von 3.000 bis 30.000, nicht-umgesetztes
wärmehärtbares
Harz, wie hierin zuvor definiert, und einen Katalysator, wie hierin
zuvor definiert, und optional eine Verstärkung, wobei die gehärtete Zusammensetzung eine Glasübergangstemperatur
im Bereich von 50 bis 70°C
aufweist, und wobei das nicht-umgesetzte wärmehärtbare Harz in einer Menge
von bis zu 50 Gewichtsteile, beispielsweise 30 bis 50 Gewichtsteile
vorhanden ist.
-
Die
gehärtete
Zusammensetzung ist überraschenderweise
geeignet zur Nach-Härtungs-Reaktion
des verbleibenden wärmehärtbaren
Harzes in der Form einer Selbstreaktion (self reaction). Es wurde
gefunden, dass bei einigen herkömmlichen
Zusammensetzungen, weiteres Härten
nicht zu einer Verbesserung der Eigenschaften des Materials führt.
-
In
einem weiteren Aspekt der Erfindung wird ein Verfahren bereitgestellt
zum Nach-Härten
eines vor-gehärteten Harzes
oder Pre-pregs, wie hierin zuvor definiert, bei einer erhöhten Temperatur,
während
eines Zeitraums über
eine Stunde, wobei die Temperatur im Bereich von 150 bis 200°C, vorzugsweise
170 bis 190°C
liegt.
-
Nach-Härten kann
bei Umgebungsdruck oder einem erhöhtem Druck erfolgen und erfolgt
vorzugsweise bei Umgebungsdruck oder bei leicht erhöhtem Druck,
was die Verwendung eines Autoklaven nicht erfordert.
-
Ohne
sich auf eine Theorie festlegen zu wollen, wird angenommen, dass
der Katalysator während
der Nach-Härtung Selbstreaktion
fördert,
wie etwa eine Veretherungsreaktion, beispielsweise mit einem wärmehärtbaren
Epoxy-Harz, was die thermischen und Umwelteigenschaften des Harzes
festlegt. Diese können Compositematerialen
mit herausragenden mechanischen Eigenschaften, eine verbesserte
Umweltbeständigkeit
gegenüber
den Materialien des Stands der Technik und herausragende thermische
Eigenschaften herstellen. Es ist besonders überraschend, dass die Zusammensetzungen
der Erfindungen für
verschiedenene Vor-Härtungs-
und Nach-Härtungs-Reaktionen
unter geeigneten Bedingungen geeignet sind, wobei die entsprechenden
Reaktionen bei jenen Bedingungen selektiv sind und eine Produktarchitektur
bereitstellen, die gut definiert ist, und gut kontrolliert ist und
mit spezifischen vorteilhaften Eigenschaften verbunden ist.
-
Die
Nach-Härtungs-Reaktion
wird ausgeführt
unter Verwendung einer herkömmlichen
auf dem technischen Gebiet bekannten Zeit-Temperatur-Rampe und beginnend
bei einer Temperatur, welche den Tg-Wert (Tg = Glasübergangstemperatur)
der vor-gehärteten
Zusammensetzung oder des Prepregs nicht überschreitet. Die Nach-Härtung wird
geeigneterweise während
eines Zeitraums von 1 bis 8 Stunden, vorzugsweise 1 bis 5 Stunden,
stärker
bevorzugt von 1 bis 3 Stunden ausgeführt.
-
Es
wurde überraschend
gefunden, dass die vorgehärtete
Zusammensetzung oder das Prepreg der Erfindung eine ausreichende
dimensionelle Stabilität
aufweist, um Nach-Härten
ohne Verwendung eines Autoklaven oder von Formen oder Werkzeugen,
welche in der Anfangsreaktion verwendet werden, zu ermöglichen. Dies
führt zu
der Freiheit, Nach-Härten
bei höheren
Temperaturen durchzuführen,
ohne dass das Belegen von Autoklavenplatz während weiterer Zeiträume erforderlich
wäre und
ohne die Notwendigkeit Formen oder Werkzeuge mit weiter höheren Temperaturen
zu behandeln.
-
Es
ist ein besonderer Vorteil der Erfindung, dass Härtung bei niedriger Temperatur
die Verwendung von Werkzeugen und Formen für Composite ermöglicht,
welche ohne Weiteres und kostengünstig
hergestellt werden können,
und geeignet sind den niedrigeren Härtungs-Temperaturen standzuhalten,
welche beim Vor-Härten verwendet
wurden.
-
In
einem weiteren Aspekt der Erfindung wird ein Verfahren zum Härten einer
härtbaren
Zusammensetzung, wie hierin zuvor definiert, bereitgestellt, wobei
die Zusammensetzung in einem Compositewerkzeug oder einer -form
in dem Vor-Härtungsschritt
geformt wird und anschliessend aus dem Werkzeug oder der Form zur
Nach-Härtung
entfernt wird.
-
Eine
weitere Methode umfasst das Bilden einer unvollständig gehärteten Zusammensetzung
zu einem Film, beispielsweise durch Formpressen, Extrudieren, Schmelzguss
oder Bandguss, Laminieren solcher Filme auf Faserverstärkungsmittel
in der Form von beispielsweise einer Vliesmatte aus relativ kurzen
Fasern, eines gewebten Tuchs oder im Wesentlichen kontinuierlicher
Faser unter Temperatur- und Druckbedingungen, die ausreichend sind,
um das Gemisch fliessen zu lassen und die Fasern zu imprägnieren,
und das Härten
des erhaltenen Laminats.
-
Lagen
von imprägniertem
Faserverstärkungsmittel,
das insbesondere durch die Methode von einem oder mehreren von EP-A-56703,
102158 und 102159 hergestellt wurde, können durch Wärme und
Druck zusammenlaminiert werden, beispielsweise durch einen Autoklaven,
durch Vakuum oder Formpressen oder durch erwärmte Walzen, bei einer Temperatur über der
Härtungs-Temperatur
des wärmehärtbaren
Harzes, oder, wenn Härtung
bereits stattgefunden hat, über
der Glasübergangstemperatur
des Gemisches, geeigneterweise bei wenigstens 180°C und typischerweise
bei bis zu 200°C
und bei einem Druck von insbesondere über 1 bar, vorzugsweise im
Bereich von 1 bis 10 bar.
-
Das
erhaltene mehrlagige Laminat kann anisotrop sein, wobei die Fasern
kontinuierlich und unidirektional sind, im Wesentlichen parallel
zueinander orientiert sind, oder quasi-isotrop in jeder Lage sind,
wobei die Fasern in einem Winkel, geeigneterweise 45°, wie bei
den meisten quasi-isotropen Laminaten, aber möglicherweise beispielsweise
30° oder
60° oder
90° oder
dazwischen, zu jenen in den darüber
und darunter liegenden Lagen orientiert sind. Orientierungen zwischen
anisotrop und quasi-isotrop und eine Kombination aus Laminaten können verwendet
werden. Geeignete Laminate enthalten wenigstens 4, vorzugsweise
wenigstens 8 Lagen. Die Anzahl von Lagen hängt von der Anwendung für das Laminat
ab, beispielsweise der erforderlichen Festigkeit (strength), und
Laminate, die 32 oder noch mehr, beispielsweise mehrere hundert
Lagen enthalten, können
wünschenswert
sein. las können
Aggregate, wie oben erwähnt,
in interlaminaren Bereichen vorliegen. Gewebte Stoffe sind ein Beispiel
für quasi-isotrope
oder zwischen anisotrop und quasi-isotrop.
-
Es
wurde auch gefunden, dass die gehärteten Materialien eine genau
definierte kontinuierliche Morphologie aufweisen. Die Materialien
weisen auch aussergewöhnliche
Bruchzähigkeitseigenschaften
(fracture toughness) auf (die sich auf Composite übertragen
lasen). Diese Werte sind höher
als das herkömmliche
bei hoher Temperatur gehärtete
Material und werden erreicht durch Verwendung geringerer Mengen
Thermoplast, beispielsweise bis zu 15% weniger Thermoplast. Ein
weiteres überraschendes
Merkmal sind Lösungsmittelbeständigkeits-Eigenschaften der
Materialien. In Dichlormethan bei Raumtemperatur eingetauchte (immersed)
reine Harzproben absorbieren weniger als 0,5% nach 100 Tagen. Das
herkömmliche
bei hoher Temperatur gehärtete
Harz würde
mehr als 4% nach einem solchen Zeitraum absorbiert haben.
-
In
einem weiteren Aspekt der Erfindung wird ein gehärtetes reines Harz bereitgestellt,
umfassend einen kettenverlängerten
polyaromatischen, durch Reaktion von reaktiven Endgruppen innerhalb
eines wärmehärtbaren
Netzwerkes verankerten Thermoplasten, wobei das Harz einen Tg-Wert
höher als
150°C, beispielsweise
im Bereich von 150 bis 185°C,
stärker
bevorzugt im Bereich von 170 bis 185°C aufweist, wobei bis zu 100%
der wärmshärtbaren
Komponente in der Reaktion verbraucht wird, beispielsweise 60 bis
90% verbraucht wird.
-
In
einem weiteren Aspekt der Erfindung wird ein Verfahren für ein Design
für einen
Härtungszyklus, zum
Härten
einer Zusammensetzung, wie hierin zuvor definiert, in einem einzelnen
Härtschritt,
oder in einem Vor-Härtschritt
und einem Nach-Härtschritt,
bereitgestellt. Das Design für
einen Härtungszyklus
kann ausgewählt
werden unter Verwendung bekannter Prinzipien, welche in diesem Fall
für die
Zusammensetzungen der Erfindung gelten, und welche durch eine exponentielle
Beziehung von Zeit und Temperatur dargestellt sind.
-
Tabelle – anschauliche
Härtungszyklen
-
Das
Verfahren für
ein Design für
einen Härtungszyklus
kann Gesichtspunkte umfassen, wie etwa die Gelzeit der Zusammensetzung,
das Risiko von Vakuum-Bag-Creep oder Bruch (rupture) bei Strukturen
innerhalb des Autoklaven während
längerer
Zeiträume,
Autoklavenvorbereitungszeit, insbesondere in Industrien, welche
hohe Produktionsraten erfordern, und Autoklavenoptimierung in Fällen, in
denen das Autoklavieren verhindert, dass andere Komponenten gleichzeitig
autoklaviert werden.
-
In
einem weiteren Aspekt der Erfindung wird ein Composite bereitgestellt,
umfassend ein nachgehärtetes
Pre-preg, wie hierin zuvor definiert. Das Composite kann in der
Form eines geformten Artikels bereitgestellt werden.
-
In
einem weiteren Aspekt der Erfindung wird die Verwendung eines Werkzeugs
oder einer -form aus einem Composite in einem Verfahren, wie hierin
zuvor definiert, bereitgestellt.
-
In
einem weiteren Aspekt der Erfindung wird die Verwendung einer Zusammensetzung,
eines gehärteten
Harzes, eines Composites oder eines geformten Produktes, wie hierin
zuvor definiert, im Bereich der Luft- und Raumfahrt, Seefahrt oder
Bauwirtschaft als ein Composite oder Klebstoff (adhesive), oder
bei der Herstellung eines Luft-, Land- oder Wasserfahrzeugs, einem
Gebäude
oder kommerziellem Produkt oder einer Komponente davon, bereitgestellt.
-
Die
Erfindung wird nun, in nicht einschränkender Weise, unter Bezugnahme
auf die folgenden Beispiele veranschaulicht.
-
BEISPIEL 1 – HÄRTBARE ZUSAMMENSETZUNGEN
-
Die
verwendeten Epoxy-Verbindungen waren die folgenden:
- MY0510
- – Trifunktionales Epoxy auf
Aminophenol basierend
- PY306
- – ein difunktionales Epoxy
auf Oligomeren von Bisphenol F basierend
-
Das
hauptsächlich
verwendete Härtungsmittel
war 3,3'-Diaminodiphenylsulfon,
welches mit-gehärtet wurde
mit einer Anzahl von LTC-Katalysatoren, diese waren die folgenden:
BF3 (mea), BF3 (dma),
BF3 (Benzylamin), BF3 (Piperidin)
Diuron, Chlorotoluron, Fenuron, CA150
Curamid CN
DICY
-
Das
Härtungsmittel
ist kommerziell erhältlich
als DDES (3,3'-Bis(diaminodiphenylether)sulfon.
Der Thermoplast, welcher zur Verbesserung der Zähigkeit des Systems verwendet
wurde, basiert auf einem Copolymer mit 40:60 PES:PEES – Copolymer
mit primärem
Amin als Endgruppe, synthetisiert durch Umsetzen von 1 Mol DCDPS
mit zwei Molen m-Aminophenol unter Verwendung von Kaliumcarbonat
als der Katalysator und Sulfolan als das Reaktionslösungsmittel.
-
BEISPIEL 2 – HERSTELLUNG
DER ZUSAMMENSETZUNGEN X99 + 0,5 BF3(dma)
-
Harzformulierungen
wurden hergestellt durch Erwärmen
der zwei Epoxy-Verbindungen. Die Temperatur der Epoxy-Verbindungen
durfte 60°C
nicht übersteigen.
Das themoplastische Harz, zuvor in einer kleinen Menge Dichlormethan
aufgelöst,
wurde danach zugegeben. Sobald die Harze erwärmt waren und deren Viskosität reduziert
war, wurde das Diaminodiphenylsulfon danach zugegeben. Das Lösungsmittel
wurde danach bei 60°C
entfernt. Das DDS wurde durch kräftiges
Rühren
dispergiert. Vor dem Vor-Härten
wurde der Härtungs-Katalysator
bei niedriger Temperatur, BF3(dma), zugegeben
und innerhalb des Harzes sorgfältig
dispergiert, was als reine Harzplatten in eine Form, unter Verdampfung
von Lösungsmittel,
gegossen wurde. Die Proben wurden danach bei 85°C während eines Zeitraums von 1
bis 18 Stunden gehärtet.
Die Proben wurden danach durch FTIR charakterisiert, um den verbleibenden
Epoxidgehalt und die Menge gebildeten Ethers in den Systemen zu
bestimmen. Eine begrenzte Anzahl dieser Proben wurde danach bei
175°C während eines Zeitraums
von 2 Stunden nach-gehärtet.
Um die Temperaturfluktuation des nur Heizens des Ofens auf 175°C zu eliminieren,
wurde eine Rampen-Rate von 2°C
pro Minute eingestellt, und verschiedene andere Effekte bezogen
auf die Rampenrate wurden untersucht.
-
BEISPIEL 2.1 – HERSTELLUNG
DER PROBEN ZUR BESTIMMUNG VON Tg (GLASÜBERGANGSTEMPERATUR)
-
Die
für die
FTIR-Untersuchung verwendeten Proben wurden auch zur Bestimmung
des Tg-Wertes (Glasübergangstemperatur)
des entsprechenden Systems verwendet. In einigen Fällen bedeutete
dies die Verwendung von DSC (im Fall von flüssigen oder weichen Materialien).
In Fällen,
in denen Testproben bei Raumtemperatur hart (hard) waren, wurde
Tortional-Rheometrie verwendet. Probemuster wurden aus den FTIR-Proben
ausgeschnitten, deren Abmessungen 5 cm Länge, 1 cm Breite und < 2 mm Dicke betrugen.
In einigen Fällen
wurde Dynamic-Mechanical-Thermal-Analysis
(DMTA) zur Bestimmung der Glasübergangstemperatur
(Tg) des Materials verwendet.
-
BEISPIEL 2.2 -VERWENDETE
FTIR-AUSSTATTUNG
-
Standard-FTIR-Ausstattung
wurde verwendet, mit welcher man Spektren überlappen lassen konnte und
die Menge Ether und Epoxid aus der Peakintensität berechnen konnte. Für die Ethergruppen
wurde der Peak bei 1115 cm–1 bestimmt und für das Epoxid
wurde der Peak bei 912 cm–1 bestimmt.
-
BEISPIEL 2.3 – MECHANISCHE
EIGENSCHAFTEN DES REINEN HARZES
-
Ein
Screen wurde mit einem reinen Harz ausgeführt, um eine Anzahl mechanischer
Eigenschaften zu bestimmen.
-
Reine
Harzplatten (6'' × 4'' × 3 mm)
wurden entsprechend Beispiel 2 hergestellt.
-
BEISPIEL 2.4 – MORPHOLOGIE
DES REINEN HARZES
-
Proben
von unter 2.3 hergestellten Platten wurden durch TEM (transmission
electron microscopy) untersucht, um die Morphologie des entsprechenden
Systems zu bestimmen.
-
BEISPIEL 2.5 – LÖSUNGSAUFNAHME-EXPERIMENTE
-
Gehärtete Proben
wurden als reine Harzscheiben mit einem Durchmesser von etwa 2'' und einer Dicke von etwa 3 mm hergestellt.
Die reinen Harzproben wurden vor-getrocknet, vor Eintauchen (immersion)
in Lösungsmittel
bei 135°C
während
eines Zeitraums von etwa 6 Stunden.
-
BEISPIEL 3.1 – HERSTELLUNG
DES ZUSAMMENSETZUNGEN X99 + 0,5 BF3(mea)
-
Die
Formulierungen wurden unter Verwendung der Methode von Beispiel
2 hergestellt.
-
BEISPIEL 3.1.1 – MECHANISCHE
EIGENSCHAFTEN DES REINEN HARZES
-
Für die obige
Formulierung wurden Platten mit 6'' × 4'' × 3
mm hergestellt durch Härten
der Harzsysteme bei 85°C
während
eines Zeitraums von 18 Stunden. Nach Kühlen wurden die Platten in
einen frei stehenden Umluftofen platziert: und bei 175°C während eines
Zeitraums von 2 Stunden gehärtet
unter Verwendung einer Rampen-Rate von 2°C pro Minute.
-
Die
vollständig
gehärteten
Platten wurden danach ausgewertet unter Verwendung der folgenden Tests;
Bruchzähigkeit
(fracture toughness) (G1c)
Bruchfestigkeit (fracture strength)
(K1c)
Modul (modulus)
Dehngrenze (tensile yield strength)
Duktilitätsfaktor
(ductility factor)
Tabelle I gibt ausführlich die Ergebnisse der Bewertung
der mechanischen Eigenschaften des reinen Harzes an und umfasst
die Ergebnisse eines herkömmlichen
bei hoher Temperatur gehärteten
Systems. Tabelle
I
- * Bei 175°C während eines Zeitraums von 3
Stunden gehärtet
(20% Amin-endständiger
Thermoplast)
- ** Bei 85°C
während
eines Zeitraums von 18 Stunden gehärtet und bei 175°C während eines
Zeitraums von 2 Stunden nach-gehärtet
(20% Amin-endständiger Thermoplast)
- *** Bei 85°C
während
eines Zeitraums von 18 Stunden gehärtet und bei 175°C während eines
Zeitraums von 2 Stunden nach-gehärtet
(23% Amin-endständiger Thermoplast)
-
Wie
aus Tabelle I ersichtlich ist, produzieren die herkömmlichen
bei hoher Temperatur gehärteten
Systeme die Art von Brucheigenschaften von reinem Harz, welche typisch
für diese
Menge (level) an Thermoplast sind. Wenn jedoch dasselbe Material
während
eines Zeitraums von 18 Stunden bei 85°C vor-gehärtet wird, und danach während eines
Zeitraums von 2 Stunden bei 175°C
nach-gehärtet
wird, sind die Brucheigenschaften beträchtlich geringer.
-
Wenn
dasselbe Material bei 85°C
wiederholt gehärtet
wird und danach bei 175°C
nach-gehärtet
wird, aber ein Katalysator (BF3(mea)) bei niedriger Temperatur eingearbeitet
wird, nehmen die Brucheigenschaften zu und das Material scheint
tatsächlich
eine höhere
Zähigkeit
aufzuweisen als das herkömmliche
HTC-System. Dies
nimmt weiter zu, wenn zusätzlicher
Thermoplast zur Formulierung zugegeben wird.
-
BEISPIEL 3.1.2 MORPHOLOGIE
DES REINEN HARZES (TRANSMISSIONSELEKTRONENMIKROSKOPIE) – TEM
-
Anlage
I enthält
die TEM-Mikrographen für
alle in Tabelle I angegebenen Formulierungen.
-
Im
Fall des X99** liegt kein sichtbarer Nachweis für eine Morphologie einer getrennten
Phase vor. Die Brucheigenschaften des reinen Harzes deuten darauf
hin, dass das System ein Zweiphasen-Trennungssystem eingegangen
ist. Im Fall von X99* ist die Grösse
der Morphologie der getrennten Phase unterhalb der Detektionsgrenze
der TEM-Technik. Jedoch weisen die Brucheigenschaften des Materials
tatsächlich
darauf hin, dass ein Zweiphasensystem vorliegt.
-
Bei
Beobachtung X99** + 0,5 BF3(mea) und X99TB*** + 0,5 BF3(mea) kann
gesehen werden, dass in beiden Proben eine co-kontinuierliche Morphologie
existiert.
-
BEISPIEL 3.1.3 FTIR
-
Proben
wurden hergestellt, wie unter Beispiel 2 ausführlich beschrieben und danach
unter Verwendung von FTIR charakterisiert, um die Menge an verbleibendem
Epoxy und die produzierte Menge Ether zu bestimmen als Funktion
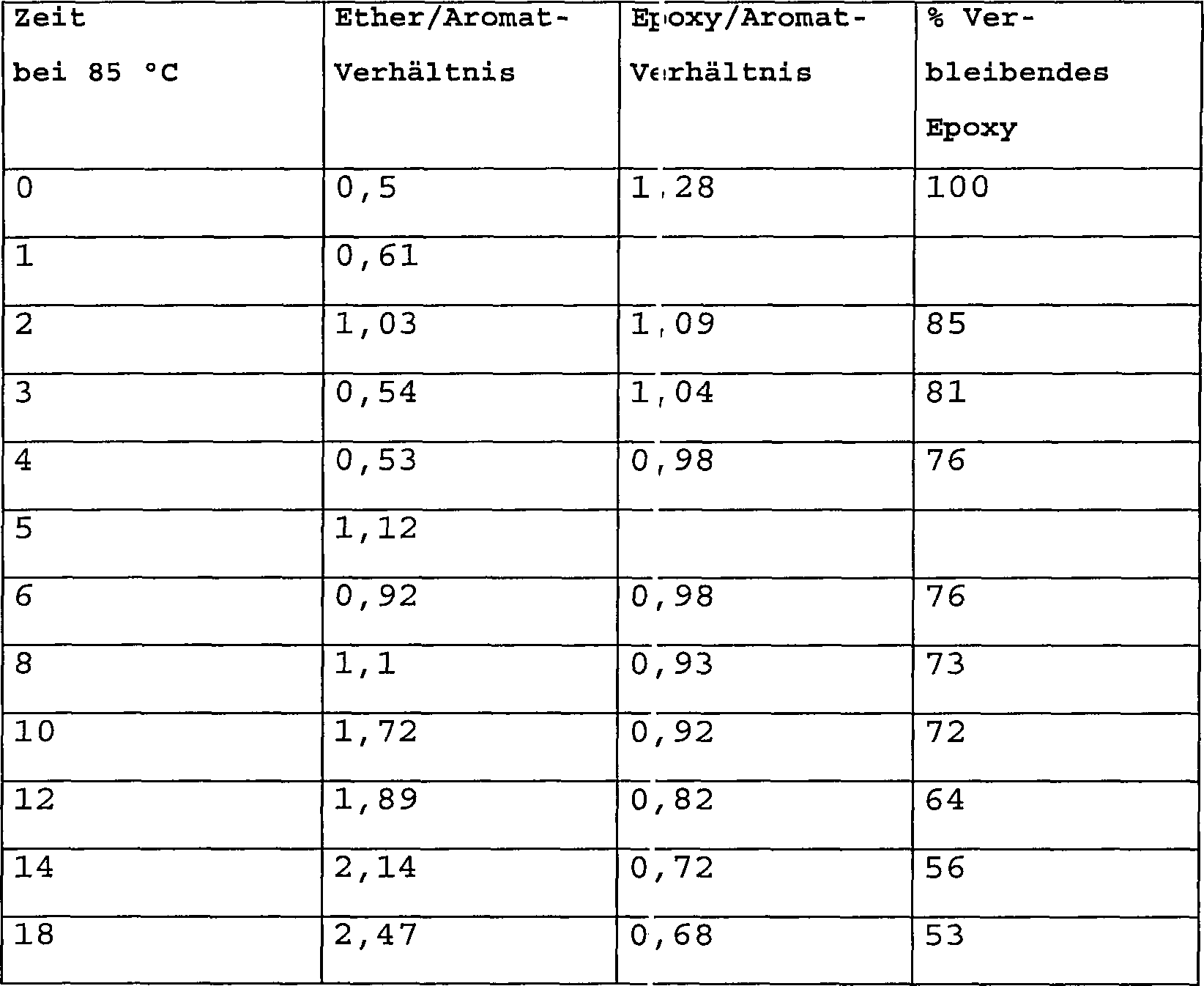
der Zeit, wobei die Ergebnisse davon in den folgenden Tabellen aufgeführt sind: Tabelle
II – X99
(Bei 85°C
während
eines Zeitraums von x Stunden gehärtet)
-
Bei
175°C während eines
Zeitraums von 2 Stunden nach-gehärtete Proben
von Tabelle II unter Verwendung einer Rampen-Rate von 2°C/Min.
-
-
Tabelle
III – X99
(Bei 175°C
während
eines Zeitraums von 3 Stunden gehärtet)
-
Tabelle
IV – X99
+ 0,5 BF3(mea) Bei 85°C
gehärtet
als Funktion der Zeit.
-
Bei
175°C während eines
Zeitraums von 2 Stunden nach-gehärtete
Proben von Tabelle IV unter Verwendung einer Rampen-Rate von 2°C/Min..
-
-
Die
Proben wurden bei verschiedenen Temperaturen vorgehärtet und
die Ergebnisse werden in den I und II gezeigt.
-
Als
Hinweis auf eine Umsetzung wurden Isotherm-Sweep und Gelzeit bestimmt,
und die Ergebnisse werden in den IIIa und IIIb gezeigt.
-
BEISPIEL 3.1.4 Glasübergangstemperaturen
Tg von sowohl vor-gehärteten
als auch nach-gehärteten
Systemen
-
Proben
zur Bestimmung von Tg wurden hergestellt – wie unter 2.1 beschrieben – und wurden
unter Verwendung von entweder DSC oder Tortional-Rheometrie in Abhängigkeit
von der physikalischen Natur der Testproben bestimmt.
-
Die
HTC-Version von X99 wurde bei 175°C
während
eines Zeitraums von 3 Stunden gehärtet und Tg davon wurde bestimmt
unter Verwendung von [both] Tortional-Rheometrie. Der Wert für die Tg
lag im Bereich von 170°C
bis 180°C – durch
DMTA, und 170°C
bis 180°C,
unter Verwendung des Wertes für
G', durch Tortional
Rheometrie-Methoden (tortional rheometrics).
-
X99
und X99 + 0,5 BF3(mea) wurden vor-gehärtet bei 85°C als Funktion der Zeit und
Tg-Daten wurden gesammelt. Diese Proben wurden auch bei 175°C während eines
Zeitraums von 2 Stunden nach-gehärtet
unter Verwendung einer Rampen-Rate von 2°C/Min. Tabelle V enthält die Tg-Daten
für sowohl
die vor-gehärteten als
auch die nach-gehärteten
Testproben.
-
-
[...]
wurde vor-gehärtet
bei 85°C
als Funktion der Zeit und Tg-Daten wurden gesammelt. Diese Proben wurden
auch bei 175°C
während
eines Zeitraums von 2 Stunden nach-gehärtet unter Verwendung einer
Rampen-Rate von
2°C/Min.
-
Wie
aus der obigen Tabelle ersichtlich ist, hatte die Zugabe des 0,5
BF3(mea) einen signifikanten Effekt auf die Tg des vor-gehärteten Systems
und hatte auch einen geringen Effekt auf die Tg des nach-gehärteten Systems,
welche immer noch im Bereich von 175°C liegt, wie für das HTC-gehärtete System
beobachtet wurde.
-
Proben
wurden auch vor-gehärtet
in einem Bereich von Temperaturen als Funktion der Zeit und Tg-Daten gesammelt.
Die Ergebnisse werden in Tabelle VI gezeigt.
-
-
BEIPIEL 3.1.5 – Lösungsmittelaufnahme
betreffend X99 + 0,6 BF3(mea)
-
IV zeigt
Ergebnisse für
Dichlormethan bei 25°C – X99 +
0,6 BF3(mea).
-
IV umfasst
auch die Ergebnisse hinsichtlich der Lösungsmittelaufnahme für das bei
175°C während eines
Zeitraums von 3 Stunden gehärtete
X99-System. Wie aus den in dieser Figur gezeigten Ergebnissen ersichtlich
ist, produzierten die Zugabe des BF3(mea) und die Inklusion des
vor-gehärteten
Systems (hold) eine nach-gehärtete
Harzprobe, welche eine aussergewöhnliche
Beständigkeit
(resistance) gegenüber
Dichlormethan bei 25°C
aufweist.
-
V zeigt
Ergebnisse für
Wasser bei 25°C – X99 +
0,5 BF3(mea).
-
In ähnlicher
Weise zeigt VI Ergebnisse für Methyl-Ethyl-Keton
(MEK) bei 25°C – X99 +
0,5 BF3(mea).
-
Die
Lösungsmittelaufnahme
bei allen drei Lösungsmitteln
war für
das – X99TB
+ 0,5 BF3(mea) identisch. Lösungsmittelaufnahme
wurde auch mit Dichlormethan, Methylethylketon und Wasser für Scheiben (discs)
und Prepregs untersucht, umfassend verschiedene Prepreg-Lay-Ups:
[0]16, [(0,90)4]s, [(+45,0, –45,90)2]s
und mit kommerziellen Härtungssystemen
bei niedriger Temperatur unter Verwendung eines Curimid-Katalysators
verglichen. Es wurde gefunden, dass die Erfindung in allen Fällen eine
geringere Lösungsmittelaufnahme
zeigt und zusätzlich
Beständigkeit
gegenüber
MEK zeigt, welches scheinbar das Curimid-Vergleichssystem chemisch
angreift.
-
BEIPIEL 3.2 – HERSTELLUNG
DER ZUSAMMENSETZUNGEN X99 + alternative Bortrifluorid-Katalysatoren
-
Die
in Betracht gezogenen alternativen BF3-Katalysatoren waren die folgenden;
BF3(dma)
BF3(bea)
BF3(pip)
-
BEISPIEL 3.2.1 – Mechanische
Eigenschaften der reinen Harze
-
Unter
Verwendung der Methode von Beispiel 2 wurden vollständig gehärtete Platten
(panels) bewertet unter Verwendung der in Beispiel 3.1.1. angegebenen
Tests; Tabelle VII gibt die Ergebnisse der Bewertung der mechanischen
Eigenschaften des reinen Harzes an und umfasst auch die Ergebnisse
von Beispiel 3.1.1. und auch die eines herkömmlich bei hoher Temperatur
gehärteten
Systems. Tabelle
VII
- * Bei 175°C während eines Zeitraums von 3
Stunden gehärtet
(20% Amin-endständiger
Thermoplast)
- ** Bei 85°C
während
eines Zeitraums von 18 Stunden gehärtet und bei 175°C während eines
Zeitraums von 2 Stunden nach-gehärtet
(20% Amin-endständiger Thermoplast)
-
Wie
aus der obigen Tabelle ersichtlich ist, sind im Fall aller drei
verschiedenen BF3-Katalysatortypen die Brucheigenschaften des reinen
Harzes gleich.
-
Es
wurden auch Tests durchgeführt,
welche die nach-gehärteten Systeme
der Erfindung mit unterschiedlichen Mengen Thermoplast, von bis
zu 35 Gewichtsprozent, vergleichen. Es wurde gefunden, dass Zähigkeit
(toughness) bei einer höheren
Rate über
15 Gewichtsprozent Thermoplast zunimmt, und dies weist darauf hin,
dass ein Umsetzungseffekt mit dem Katalysator stattfindet, zusätzlich zur
erwarteten Zunahme aufgrund des zusätzlichen Zähigkeitverbessernden Thermoplasten.
Es wurde gefunden, dass bei dieser Menge sich die Zähigkeitsverbesserung
auch als besser als in herkömmlichen
Härtungssystemen
bei hoher Temperatur erweist.
-
BEISPIEL 3.2.2. Morphologie
des reinen Harzes (Transmissionselektronenmikroskopie) – TEM
-
Anhang
6 enthält
die TEM-Micrographen für
alle in Tabelle VII einzeln aufgeführten Formulierungen. Beim
Beobachten von X99** + 0,5 BF3(mea), X99** + 0,5 BF3(dma) und BF3(pip)
kann man sehen, dass in allen drei Proben eine co-kontinuierliche
Morphologie existiert.
-
BEISPIEL 3.2.3 FTIR
-
Proben
wurden hergestellt wie ausführlich
in Beispiel 2 beschrieben und danach unter Verwendung von FTIR charakterisiert,
um die Menge von verbleibendem Epoxy und die Menge von produziertem
Ether als Funktion der Zeit zu bestimmen, wobei die Ergebnisse davon
in den folgenden Tabellen angegeben sind.
-
Tabelle
VIII – X99
+ 0,5 BF3(dma) (Bei 85°C
während
eines Zeitraums von x Stunden gehärtet)
-
Tabelle
IX – X99
+ 0,5 BF3(bea) (Bei 85°C
während
eines Zeitraums von x Stunden gehärtet)
-
BEISPIEL 3.2.4 Tg-Daten
von sowohl vor-gehärteten
als auch nach-gehärteten
Systemen
-
Proben
zur Bestimmung von Tg wurden hergestellt – wie unter 2.1 beschrieben – und wurden
unter Verwendung von entweder DSC oder Tortional-Rheometrie in Abhängigkeit
von der physikalischen Natur der Testproben bestimmt.
-
VIII stellt die Tg-Werte der vor-gehärteten Systeme
dar, unter Verwendung der Bortrifluorid-Katalysatoren zur Härtung von X99. Die Tg-Werte
sind als Funktion der Vor-Härtungs-Zeit
in Stunden dargestellt.
-
Wie
aus VIII ersichtlich ist, produzieren
nach etwa 12 Stunden alle der BF3-basierten Katalysatoren Tg-Werte
der vorgehärteten
Systeme über
60°C.
-
IX stellt
die Tg-Werte der nach-gehärteten
Systeme der in VIII beschriebenen Materialien
dar. Wie aus dieser Figur ersichtlich ist, produziert jeder der
BF3-Systeme aufgrund der Nach-Härtung
Tg-Werte über 170°C.
-
VERGLEICHSBEISPIEL 3.3 – HERSTELLUNG
VON ZUSAMMENSETZUNGEN
-
Mit Curamid CN (Imidazol)
gehärtetes
X99
-
BEISPIEL 3.3.1 – Mechanische
Eigenschaften der reinen Harze
-
Unter
Verwendung der Methode von Beispiel 2 wurden vollständig gehärtete Platten
bewertet unter Verwendung der in Beispiel 3.1.1. angegebenen Tests;
Tabelle X gibt die Ergebnisse der Bewertung der mechanischen Eigenschaften
des reinen Harzes an und umfasst auch die Ergebnisse von Beispiel
3.1.1. und die eines herkömmlich
bei hoher Temperatur gehärteten
Systems. Tabelle
X
- * Bei 175°C während eines Zeitraums von 3
Stunden gehärtet
(20% Amin-endständiges
KM180)
- ** Bei 85°C
während
eines Zeitraums von 18 Stunden gehärtet und bei 175°C während eines
Zeitraums von 2 Stunden nach-gehärtet
(20% Amin-endständiges KM180)
-
Die
Ergebnisse der obigen Tabelle zeigen, dass die Zugabe des Curamid
CN zu einer Verbesserung der Zähigkeit
des LTC-X99-Systems führt,
welche jedoch immer noch nur halb so gross wie die des mit 0,55 BF3(mea)
gehärteten
X99-Systems ist.
-
3.3.2 Morphologie von
mit Curamid CN gehärtetem
X99
-
Der
TEM-Mikrograph in Anhang 7 zeigt deutlich, dass es unter Verwendung
dieser bestimmten Technik, einen sichtbaren Hinweis auf ein Zweiphasensystem
gibt, welches verschieden von dem ist, welches für das mit einer der beschriebenen
BF3-Lewis-Säuren
gehärteten
X99-System beobachtet worden ist.
-
BEISPIEL 3.3.3 FTIR
-
Proben
wurden hergestellt wie ausführlich
in Beispiel 2 beschrieben und danach unter Verwendung von FTIR charakterisiert,
um die Menge von verbleibendem Epoxy und die Menge von produziertem
Ether, als Funktion der Zeit zu bestimmen, wobei die Ergebnisse
davon in den folgenden Tabellen angegeben sind.
-
Tabelle
XI – X99
+ 0,5 Curamid CN (Bei 85°C
während
eines Zeitraums von x Stunden gehärtet)
-
Tabelle
XII – X99
+ 1,0 Curamid CN (Bei 85°C
während
eines Zeitraums von x Stunden gehärtet)
-
Der
unmittelbare Unterschied zwischen den Daten in den beiden Tabellen
oben, verglichen mit den Daten in den Tabellen II, III, VIII und
IX (X99 mit BF3-basierten
Katalysatoren gehärtet)
liegt in der Menge an Ether, welche produziert wird. Diese ist fast
doppelt so hoch in dem Curamid-System. Das Auftreten grösserer Mengen
von Ether weist darauf hin, dass die Epoxy-Baugruppen mit einander
reagiert haben. Dies führt
zu einer hohen Vernetzung (crosslinked network).
-
Die X und XI zeigen
die Menge des Verbrauchs von Epoxid und die Menge von Ether – generiert
aus den Daten in Tabelle XI beziehungsweise Tabelle XII.
-
X zeigt
Ergebnisse für
den Verbrauch von Epoxid als Funktion der Zeit bei 85°C.
-
XI zeigt
Ergebnisse für
die Bildung von Ether als Funktion der Zeit bei 85°C.
-
BEISPIEL 3.3.4 Tg-Daten
von sowohl vor-gehärteten
als auch nach-gehärteten
Systemen
-
Proben
zur Bestimmung von Tg wurden hergestellt – wie unter 2.3 beschrieben – und wurden
unter Verwendung von entweder DSC oder Tortional-Rheometrie in Abhängigkeit
von der physikalischen Natur der Testproben bestimmt.
-
Tabelle
VIII stellt die Tg-Werte der vor-gehärteten Systeme für das mit
0,5 und 1,0 pbw Curamid CN gehärteten
X99-Systems dar.
-
-
Tabelle
XIV stellt die Tg-Werte der nach-gehärteten Systeme für das mit
0,5 und 1,0 pbw Curamid CN gehärteten
X99-Systems dar.
-
-
Wie
aus den beiden Tabellen oben ersichtlich ist, führen beide Mengen Curamid CN
zu Tg-Werten der vor-gehärteten Systeme über 30°C. Jedoch
steigen die Tg-Werte
ihrer nach-gehärteten
Systeme nicht über 160°C. Der mögliche Grund
dafür liegt
darin, dass die Imidazole über
Veretherungsreaktionen reagieren, d.h. ohne Aminverbrauch und Kettenwachstum
und daher sehr starke Vernetzungen (crosslinked networks) produzieren.
Wenn dies unter den Bedingungen der Vor-Härtung
auftritt, wird das System rasch sehr unbeweglich, und ist buchstäblich „eingefroren" im Hinblick auf
weitere chemische Reaktionen. Während
das System nach-härtet,
dauert der „eingefrorene" Zustand an und verhindert
so einen nur minimalen Anstieg des Tg-Wertes. Dies ist das genaue
Gegenteil von dem, was im Fall von BF3(mea) gefunden wurde, wo Vor-Härten zu einem
Tg-Wert über
50°C führt, jedoch
mit der Minimalmenge an Veretherung. Aufgrund von Nach-Härtung ist es
immer noch verhältnismässig beweglich
(mobile) und kann weiterreagieren um viel höhere Tg-Werte des nach-gehärteten Systems
zu liefern.
-
BEISPIEL 3.4 VERGLEICHSBEISPIEL – ALTERNATIVE
KATALYSATOREN FÜR
CURAMID CN
-
Eine
Reihe Imidazol- und Harnstoff-basierter Katalysatoren bei niedriger
Temperatur wurden verglichen, um zu sehen, ob sie sich wie der Curamid
CN-Katalysator oder wie die Bortrifluorid-Katalysatoren verhielten.
-
Die
ausgewählten
Katalysatoren waren die folgenden:
Die alternativen Katalysatoren
wurden zuerst durch FTIR untersucht, um die Rate an Verbrauch von
Epoxid und die Bildung von Ether festzustellen.
-
Das
FTIR zeigte, dass jeder der Harnstoff-basierten Katalysatoren sich
genau gleich wie die Imidazol-Katalysatoren
verhalten, und zwar darin, dass sie bei niedrigen Temperaturen sehr
rasch reagieren, um Epoxid zu verbrauchen, jedoch deren Mechanismus
dabei so ist, dass Veretherung erzeugt wird. Dies führt zu einer
starken Vernetzung, unter Vor-Härtungs-Bedingungen und führt zu einer
schlechten Übersetzung
hoher Tg-Werte während
Nach-Härtung.
-
Diese
Bemerkungen werden bestätigt
durch den folgenden Datensatz, abgeleitet von der Härtung von X99
mit den obigen alternativen Harnstoff-basierten Katalysatoren.
-
Tg-Werte
des vor-gehärteten
Systems des X99-Systems, gehärtet
mit Diuron, Chlorotoluron, Fenuron und CA150.
-
XII zeigt Ergebnisse für Tg-Werte der vor-gehärteten Systeme
als Funktion der Zeit bei 85°C.
-
Ähnlich zeigt XIII Ergebnisse für Tg-Werte der nach-gehärteten Systeme
der obigen vor-gehärteten
Systeme.
-
BEISPIEL 4 – HERSTELLUNG
VON HARZ FÜR
HEISSSCHMELZIMPRÄGNIERUNG
-
Die
Epoxyharze MY0510 und Rutapox 0158 wurden zusammengemischt und erwärmt. Als
die Harztemperatur 100 bis 110°C
erreichte, wurde mit einer langsamen, portionsweisen Zugabe von
Thermoplast-Polymer von Beispiel 2 begonnen, unter kräftigem Bewegen,
um Klumpenbildung zu verhindern. Das Erwärmen des Gemisches wurde unter
Rühren
fortgesetzt bis 130 bis 135°C,
bis alles Polymer aufgelöst
war, etwa 30 bis 45 Minuten lang. Das Gemisch wurde danach auf 75°C gekühlt und
der LTC-Katalysator, BF3·mea wurde portionsweise unter
wirksamem Rühren
zugegeben. Mischen wurde ein paar weitere Minuten lang fortgesetzt
bis der Katalysator aufgelöst
war. Vorgesiebtes 3,3'-DDS
wurde portionsweise unter Mischen 5 bis 10 Minuten lang zugegeben,
bis das Harz homogen war. Das Harz wurde vom Gemisch getrennt (drained),
sofort auf Abkühlplatten
gekühlt
und in einem Gefrierschrank bei – 18°C aufbewahrt.
-
Die
Epoxy-Harz-Precursor umfassten 0,05% eines Silikonöl-Entschäumer/Luftfreisetzungsmittel,
wie etwa Foamkill.
-
Das
Harz war geeignet für
Heissschmelzimprägnierung
zur Bildung von Composites, welche getestet wurden und wobei gefunden
wurde, dass sie äquivalente
Eigenschaften oder bessere Eigenschaften als jene, welche herkömmliche
Katalysatoren verwenden, aufweisen.