CN1266032C - 氧化物材料、氧化物薄膜的制造方法以及使用该材料的元件 - Google Patents
氧化物材料、氧化物薄膜的制造方法以及使用该材料的元件 Download PDFInfo
- Publication number
- CN1266032C CN1266032C CNB018174132A CN01817413A CN1266032C CN 1266032 C CN1266032 C CN 1266032C CN B018174132 A CNB018174132 A CN B018174132A CN 01817413 A CN01817413 A CN 01817413A CN 1266032 C CN1266032 C CN 1266032C
- Authority
- CN
- China
- Prior art keywords
- film
- oxide
- bso
- bit
- sio
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images























Classifications
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B13/00—Oxygen; Ozone; Oxides or hydroxides in general
- C01B13/14—Methods for preparing oxides or hydroxides in general
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B82—NANOTECHNOLOGY
- B82Y—SPECIFIC USES OR APPLICATIONS OF NANOSTRUCTURES; MEASUREMENT OR ANALYSIS OF NANOSTRUCTURES; MANUFACTURE OR TREATMENT OF NANOSTRUCTURES
- B82Y30/00—Nanotechnology for materials or surface science, e.g. nanocomposites
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B13/00—Oxygen; Ozone; Oxides or hydroxides in general
- C01B13/14—Methods for preparing oxides or hydroxides in general
- C01B13/32—Methods for preparing oxides or hydroxides in general by oxidation or hydrolysis of elements or compounds in the liquid or solid state or in non-aqueous solution, e.g. sol-gel process
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B33/00—Silicon; Compounds thereof
- C01B33/20—Silicates
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G23/00—Compounds of titanium
- C01G23/003—Titanates
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G25/00—Compounds of zirconium
- C01G25/006—Compounds containing, besides zirconium, two or more other elements, with the exception of oxygen or hydrogen
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G29/00—Compounds of bismuth
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G29/00—Compounds of bismuth
- C01G29/006—Compounds containing, besides bismuth, two or more other elements, with the exception of oxygen or hydrogen
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G35/00—Compounds of tantalum
- C01G35/006—Compounds containing, besides tantalum, two or more other elements, with the exception of oxygen or hydrogen
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L21/00—Processes or apparatus adapted for the manufacture or treatment of semiconductor or solid state devices or of parts thereof
- H01L21/02—Manufacture or treatment of semiconductor devices or of parts thereof
- H01L21/02104—Forming layers
- H01L21/02107—Forming insulating materials on a substrate
- H01L21/02109—Forming insulating materials on a substrate characterised by the type of layer, e.g. type of material, porous/non-porous, pre-cursors, mixtures or laminates
- H01L21/02112—Forming insulating materials on a substrate characterised by the type of layer, e.g. type of material, porous/non-porous, pre-cursors, mixtures or laminates characterised by the material of the layer
- H01L21/02123—Forming insulating materials on a substrate characterised by the type of layer, e.g. type of material, porous/non-porous, pre-cursors, mixtures or laminates characterised by the material of the layer the material containing silicon
- H01L21/02142—Forming insulating materials on a substrate characterised by the type of layer, e.g. type of material, porous/non-porous, pre-cursors, mixtures or laminates characterised by the material of the layer the material containing silicon the material containing silicon and at least one metal element, e.g. metal silicate based insulators or metal silicon oxynitrides
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L21/00—Processes or apparatus adapted for the manufacture or treatment of semiconductor or solid state devices or of parts thereof
- H01L21/02—Manufacture or treatment of semiconductor devices or of parts thereof
- H01L21/02104—Forming layers
- H01L21/02107—Forming insulating materials on a substrate
- H01L21/02109—Forming insulating materials on a substrate characterised by the type of layer, e.g. type of material, porous/non-porous, pre-cursors, mixtures or laminates
- H01L21/02112—Forming insulating materials on a substrate characterised by the type of layer, e.g. type of material, porous/non-porous, pre-cursors, mixtures or laminates characterised by the material of the layer
- H01L21/02123—Forming insulating materials on a substrate characterised by the type of layer, e.g. type of material, porous/non-porous, pre-cursors, mixtures or laminates characterised by the material of the layer the material containing silicon
- H01L21/02142—Forming insulating materials on a substrate characterised by the type of layer, e.g. type of material, porous/non-porous, pre-cursors, mixtures or laminates characterised by the material of the layer the material containing silicon the material containing silicon and at least one metal element, e.g. metal silicate based insulators or metal silicon oxynitrides
- H01L21/02161—Forming insulating materials on a substrate characterised by the type of layer, e.g. type of material, porous/non-porous, pre-cursors, mixtures or laminates characterised by the material of the layer the material containing silicon the material containing silicon and at least one metal element, e.g. metal silicate based insulators or metal silicon oxynitrides the material containing more than one metal element
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L21/00—Processes or apparatus adapted for the manufacture or treatment of semiconductor or solid state devices or of parts thereof
- H01L21/02—Manufacture or treatment of semiconductor devices or of parts thereof
- H01L21/02104—Forming layers
- H01L21/02107—Forming insulating materials on a substrate
- H01L21/02109—Forming insulating materials on a substrate characterised by the type of layer, e.g. type of material, porous/non-porous, pre-cursors, mixtures or laminates
- H01L21/02112—Forming insulating materials on a substrate characterised by the type of layer, e.g. type of material, porous/non-porous, pre-cursors, mixtures or laminates characterised by the material of the layer
- H01L21/02172—Forming insulating materials on a substrate characterised by the type of layer, e.g. type of material, porous/non-porous, pre-cursors, mixtures or laminates characterised by the material of the layer the material containing at least one metal element, e.g. metal oxides, metal nitrides, metal oxynitrides or metal carbides
- H01L21/02197—Forming insulating materials on a substrate characterised by the type of layer, e.g. type of material, porous/non-porous, pre-cursors, mixtures or laminates characterised by the material of the layer the material containing at least one metal element, e.g. metal oxides, metal nitrides, metal oxynitrides or metal carbides the material having a perovskite structure, e.g. BaTiO3
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L21/00—Processes or apparatus adapted for the manufacture or treatment of semiconductor or solid state devices or of parts thereof
- H01L21/02—Manufacture or treatment of semiconductor devices or of parts thereof
- H01L21/02104—Forming layers
- H01L21/02107—Forming insulating materials on a substrate
- H01L21/02225—Forming insulating materials on a substrate characterised by the process for the formation of the insulating layer
- H01L21/0226—Forming insulating materials on a substrate characterised by the process for the formation of the insulating layer formation by a deposition process
- H01L21/02282—Forming insulating materials on a substrate characterised by the process for the formation of the insulating layer formation by a deposition process liquid deposition, e.g. spin-coating, sol-gel techniques, spray coating
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L21/00—Processes or apparatus adapted for the manufacture or treatment of semiconductor or solid state devices or of parts thereof
- H01L21/02—Manufacture or treatment of semiconductor devices or of parts thereof
- H01L21/02104—Forming layers
- H01L21/02107—Forming insulating materials on a substrate
- H01L21/02296—Forming insulating materials on a substrate characterised by the treatment performed before or after the formation of the layer
- H01L21/02318—Forming insulating materials on a substrate characterised by the treatment performed before or after the formation of the layer post-treatment
- H01L21/02356—Forming insulating materials on a substrate characterised by the treatment performed before or after the formation of the layer post-treatment treatment to change the morphology of the insulating layer, e.g. transformation of an amorphous layer into a crystalline layer
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L21/00—Processes or apparatus adapted for the manufacture or treatment of semiconductor or solid state devices or of parts thereof
- H01L21/02—Manufacture or treatment of semiconductor devices or of parts thereof
- H01L21/04—Manufacture or treatment of semiconductor devices or of parts thereof the devices having at least one potential-jump barrier or surface barrier, e.g. PN junction, depletion layer or carrier concentration layer
- H01L21/18—Manufacture or treatment of semiconductor devices or of parts thereof the devices having at least one potential-jump barrier or surface barrier, e.g. PN junction, depletion layer or carrier concentration layer the devices having semiconductor bodies comprising elements of Group IV of the Periodic System or AIIIBV compounds with or without impurities, e.g. doping materials
- H01L21/30—Treatment of semiconductor bodies using processes or apparatus not provided for in groups H01L21/20 - H01L21/26
- H01L21/31—Treatment of semiconductor bodies using processes or apparatus not provided for in groups H01L21/20 - H01L21/26 to form insulating layers thereon, e.g. for masking or by using photolithographic techniques; After treatment of these layers; Selection of materials for these layers
- H01L21/314—Inorganic layers
- H01L21/316—Inorganic layers composed of oxides or glassy oxides or oxide based glass
- H01L21/31691—Inorganic layers composed of oxides or glassy oxides or oxide based glass with perovskite structure
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/02—Amorphous compounds
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/30—Three-dimensional structures
- C01P2002/34—Three-dimensional structures perovskite-type (ABO3)
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/50—Solid solutions
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/70—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data
- C01P2002/72—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data by d-values or two theta-values, e.g. as X-ray diagram
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/70—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data
- C01P2002/77—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data by unit-cell parameters, atom positions or structure diagrams
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/80—Crystal-structural characteristics defined by measured data other than those specified in group C01P2002/70
- C01P2002/85—Crystal-structural characteristics defined by measured data other than those specified in group C01P2002/70 by XPS, EDX or EDAX data
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/60—Particles characterised by their size
- C01P2004/64—Nanometer sized, i.e. from 1-100 nanometer
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/80—Particles consisting of a mixture of two or more inorganic phases
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/40—Electric properties
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S502/00—Catalyst, solid sorbent, or support therefor: product or process of making
- Y10S502/514—Process applicable either to preparing or to regenerating or to rehabilitating catalyst or sorbent
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S502/00—Catalyst, solid sorbent, or support therefor: product or process of making
- Y10S502/525—Perovskite
Abstract
本发明提供了在钙钛矿型或层状钙钛矿型晶格结构的氧化物中固溶含有选自Si、Ge、Sn的1种以上的IVA族元素的催化物质的氧化物材料。该钙钛矿型或层状钙钛矿型晶格结构的氧化物材料在低温下可结晶化,能够维持或改善这些氧化物材料的特性。
Description
技术领域
本发明涉及氧化物材料、氧化物薄膜的制造方法以及使用该材料的元件,详细涉及可用于作为非易失性存储器的强电介质存储器的氧化物材料、氧化物薄膜的制造方法以及使用该材料的元件。
背景技术
近年来,随着移动电话、笔记本电脑、掌上电脑等便携式终端的发展,强电介质存储器正受到关注。强电介质存储器能够使书写输入速度加快、实现大容量,所以对能够同时处理含有影像、声音等大量数据的大型媒体器材特别有用,并且存储数据不需电力,因而可以实现低耗电。
强电介质存储器是利用强电介质的极化特性,通过外部电场自由地控制极化的方向,能够以“1”或“0”的二进制保存数据,同时也能够在断电时保存数据。
但是,只有4-256K的小容量存储器被商品化。目前,由于强电介质材料自身存在的问题阻碍了强电介质存储器向Mbit级别的大容量化的发展。
现在,常用的钛酸锆酸铅(PZT;PbZrxTi1-xO3)钙钛矿型强电介质材料(ABO3)、钽酸锶铋(SBT;SrBi2Ta2O9)及掺杂了La的高亮度钛酸铋(BIT;Bi4Ti3O12)等层状结构的强电介质材料(BiAm-1BmO3m+3)一般都需要在600~800℃的高温下长时间烧结(中村孝:信学技报,电子情报通信学会,ED97-208(1998)p25-32;惠下隆他:信学技报,电子情报通信学会,ED98-242(1999)p21-26;山口正树:《有关在硅基板上形成钛酸铋薄膜的评估和研究》,芝浦工业大学博士学位论文,(1998)p39-47)。象这样的高温长时间结晶化,不仅可以充分显现强电介质自身的特性,还会如下所述的那样在用强电介质制作元件的工艺中,例如,SiO2钝化和电容器加工等过程中,可以尽可能改善强介电特性的劣化。
因而,把用这些强电介质材料制作的强电介质电容器与半导体元件组合形成强电介质存储器时,由于强电介质材料形成需要很高的结晶化温度,所以必须将强电介质电容器与晶体管分开制作,这样就会造成制作工艺复杂化,组合所用的电极材料受到限制,强电介质存储器难以高密度集成化等问题。
通常,在强电介质存储器中使用强电介质材料的薄膜。形成过程中,由于简便和忽略了各批之间的质量差而进行很好的批量生产,使用溶胶-凝胶法。
在溶胶-凝胶法中,考虑到强电介质材料膜的组成中含有挥发性较高的铅和铋等成分,为了提高结晶性同时将成膜后的膜组成的差异控制在最小范围,通常在溶胶凝胶原料溶液中加入10%左右过量的挥发性较大的组分。
但是,铅和铋这样的成分的过量加入会导致最终形成的强电介质薄膜的组成分布不均。此外,膜中的组成差异促使异相的产生(例如,BIT、SBT的烧绿石相和荧光相等),很难得到所希望的强电介质的单一相。
而且,在强电介质存储器的制造中,如上所述,由于强电介质材料的结晶化温度较高,因而在高温烧结过程中需要具有足够耐久性的电极材料。
例如,PbZrO3反强电介质与PbTiO3强电介质的固溶体PZT能够在比较低的温度下烧结,因而对电极材料造成的负担较小。但是,为确保实际应用中不可欠缺的残留极化值,烧结温度必须达到600~750℃(中村孝:信学技报,电子情报通信学会,ED97-208(1998)p25-32),对电极材料造成的负担也不小。总之,在标准Pt电极上形成PZT薄膜时,由于反复翻转,极化值急剧劣化,明显出现所谓的膜疲劳。
因此,经常使用价高而加工困难的Ir、IrOz2等对强电介质的疲劳具有良好控制性的氧化物电极或Pt/IrO2等和氧化物电极的复杂的复合电极。
另一方面,具有代表性的铋层状结构的强电介质材料SBT(SrBi2Ta2O9:m=2),由于其在Pt电极上经1012次的反复翻转也不疲劳,因此受到了人们的关注,目前正积极开展对它的实用化探讨。
但是,如果使SBT形成为薄膜状,则出现粗大的粒子低密度聚集,只能够得到劣化的表面组织(K.Aizawa,等,Jpn.J.Appl.Phys.,39(2000)p1191-1193),目前还无法实现高密度集成化(薄膜化)。
虽然SBT的薄膜化的P-E磁滞形状非常良好,残留极化值Pr较低,为7~10μC/cm2,可用于现在已经商品化的强电介质电容器容量读取型存储器,但没有富余的极化特性,尚未有实用化的特性。
此外,降低SBT结晶化的温度较困难。即:为了SBT薄膜化,尝试了800℃的高温烧结或在650℃左右的相对低温下的5小时的长时间烧结(泽田佳宏等:信学技报,电子情报通信学会、ED98-240(1999)p9-14),或两者结合的2个阶段的烧结(林慎一郎等:信学技报,电子情报通信学会、ED98-241(1999)p15-19)。但是,因受热对电极材料产生的负担不是PZT的比,这种材料应用于实际还有很大的问题。
近年,提出了通过掺杂La降低其结晶化(烧结)温度的方法。该方法中使用的材料BIT(Bi4Ti3O12:m=3)正受到注目。这种材料与SBT一样,无铋层状结构,疲劳特性良好,转变温度(Tc)高达675℃,具有常温下非常稳定的材料特性。
但是,即使这种材料也必须经历650℃下1小时的受热过程。因而,对电极材料的负担很大(B.H Park,B.S.Kang,S.D.Bu,T.W.Noh,J.Lee和W.Jo,Nature 401(1999)p682)。
这种强电介质存在的最大课题是容易形成巨大粒子(中村孝:《具有浮置栅构造的强电介质存储器的研究》、京都大学博士学位论文(1998)p118-140),与SBT一样难以薄膜化。
要使强电介质薄膜元件的高密度集成化、低电压驱动化,必须使强电介质自身极端的薄膜化。
但是,由于强电介质的表面c的不良,不能形成具有良好重现性的100nm以下的薄膜。即使能形成100nm以下的膜厚,最终强介电特性也会急剧劣化(青木克裕等:信学技报,电子情报通信学会、ED98-245(1999)p43-49))。
强电介质薄膜的表面组织的劣化,不论是溶胶凝胶法还是MOCVD法等成膜法,都会从下部电极(例如,铂电极)上、即强电介质薄膜的最下端结晶化,在上面聚集成凸出的组织。另外,不仅下部电极材料与强电介质的相容性较差,还由于强电介质的结晶化仅依靠铂电极的催化性,所以强电介质的结晶化初期晶核的形成密度较低。因此,要使强电介质形成100nm以下的薄膜,强电介质薄膜不能完全覆盖下部电极,成长为岛状。其结果是,形成非常混乱的表面组织,得到的强电介质薄膜的漏电电流密度增加。此外,众所周知,以有机金属原料为原始材料的强电介质薄膜,膜中残留了大量的碳,这也是造成漏电电流密度增大的一个原因。
另外,这些强电介质材料在还原氛围气下的强介电特性劣化(Y.Shimamoto,等,Appl Phys Lett.,70(1997)p1-2)。
例如,强电介质材料用于电容器时,通常进行以臭氧TEOS等为强电介质电容器的保护膜的SiO2钝化。此时,形成于铂上部电极的强电介质电容器处于氢氛围气中。于是,在铂上部电极的催化作用下活化的氢原子使强电介质还原,强电介质的结构被破坏,导致强介电特性大幅下降。
其原因是,强电介质是离子结合性较强的材料(一般认为离子键是较强的结合,但离子产生的攻击却非常弱),因劣化的表面组织受攻击的实际面积较大。
因此,为使结构遭到破坏的强电介质材料的特性恢复,在SiO2钝化后再次在氧氛围气下通过烧结等方法进行强电介质材料的再氧化。
但是,这种氧化给元件带来多余的受热过程,同时,暂时劣化的强介电特性即使再度氧化也不能完全恢复。
总之目前的情况是,虽然对很多的强电介质材料进行了研究,但无论哪种材料均存在如下诸多问题:1)需要加入过量的挥发性强的铅、铋等成分,2)很难得到强电介质的单一相,3)结晶化温度高,4)膜中存在过量的碳残渣,5)100nm以下的薄膜化很困难,6)在氢等还原氛围气下会分解等,因此强电介质薄膜元件的高密度集成化尚未实现。
代表性的钙钛矿型(ABO3)强电介质材料PZT会在常规的铂电极上产生极化翻转疲劳,铋层状结构强电介质材料(BiAm-1BmO3m+3)SBT及BIT由于表面组织劣化,很难薄膜化等,因此,目前有望用于存储器的强电介质都分别存在这样那样的问题。
此外,不仅是强电介质,可作为SrRuO3的钙钛矿型电极材料及次生代DRAM用高电容率表层氧化膜的(Ba,Sr)TiO3和SrTiO3等钙钛矿型氧化物材料也有上述同样的问题。
发明的揭示
本发明提供了一种氧化物材料,它由钙钛矿型或层状钙钛矿型晶格结构的氧化物以及固溶在所述氧化物中的催化物质形成,所述氧化物由ABO3表示,式中,A是选自Pb2+、Ca2+、Sr2+和Ba2+中的一种或多种的离子,B是选自Ti4+和Zr4+中的一种或多种的离子,或者所述氧化物由(Bi2O2)2+(Am-1BmO3m+1)2-表示,式中,A是选自Ca2+、Sr2+、Ba2+、Bi3+、Y3+及La3+中的一种或多种的离子,B是选自Ti4+、Zr4+、Nb5+、Ta5+和V5+中的一种或多种的离子,m是2或3;所述催化物质选自WSiO5、V2SiO7、ZrSiO4、Sb2SiO5、Fe2SiO5、La2SiO5、Bi2SnO5、CuSiO3、ZnSiO3、MnSiO3、Bi2SiO5、Y2SiO5、Bi2GeO5、Bi4Si3O12和Bi4Ge3O12。
本发明还提供了氧化物材料的制作方法,该方法是使选自Pb、Ca、Sr、Ba、Bi、La、Ti、Zr、Nb、Ta和V的2种以上的金属的醇盐或有机酸盐水解反应而成的钙钛矿型或层状钙钛矿型晶格结构的氧化物形成用凝胶溶液,与选自Zn、Fe、Y、Sc、Sb、Cr、Bi、Cu、Mn、Zr、Ti、Mo、W、V、La、Ce、Pr、Nd、Pm、Sm、Eu、Gd、Tb、Dy、Ho、Er、Tm、Yb及Lu的1种以上的金属和选自Si、Ge、Sn的1种以上的IVA族金属的金属醇盐或有机酸盐水解反应而成的催化物质形成用凝胶溶液在无水状态下混合,再将该混合物涂布在基板上,烧结制成氧化物材料,其中,所述钙钛矿型或层状钙钛矿型晶格结构的氧化物形成用凝胶溶液与催化物质形成用凝胶溶液的比例R为0.1、0.2、0.33或0.4。
本发明提供了氧化物形成用混合溶液,该溶液由形成钙钛矿型或层状钙钛矿型晶格结构的氧化物材料的缩聚反应物与形成复合氧化物材料的缩聚反应物的无水混合物形成,钙钛矿型或层状钙钛矿型晶格结构的氧化物由ABO3表示,式中,A是选自Pb2+、Ca2+、Sr2+和Ba2+中的一种或多种的离子,B是选自Ti4+和Zr4+中的一种或多种的离子,或者所述氧化物由(Bi2O2)2+(Am-1BmO3m+1)2-表示,式中,A是选自Ca2+、Sr2+、Ba2+、Bi3+、Y3+及La3+中的一种或多种的离子,B是选自Ti4+、Zr4+、Nb5+、Ta5+和V5+中的一种或多种的离子,m是2或3;所述复合氧化物材料选自WSiO5、V2SiO7、ZrSiO4、Sb2SiO5、Fe2SiO5、La2SiO5、Bi2SnO5、CuSiO3、ZnSiO3、MnSiO3、Bi2SiO5、Y2SiO5、Bi2GeO5、Bi4Si3O12和Bi4Ge3O12。
本发明还提供了半导体元件,其构成是在基板上形成导电性材料膜,在该导电性材料膜上再形成上述氧化物材料膜获得被氧化物材料膜覆盖的基板,或进一步在该基板上形成上部电极。或者该半导体元件的构成是,在半导体基板上形成上述氧化物材料膜以及导电性材料膜,然后在该导电性材料膜的两侧的上述半导体基板的表面形成一对杂质扩散层。
附图的简要说明
图1显示宽容系数与原子数的关系。
图2显示BSO溶液、BIT溶液以及BSO-BIT混合溶液的差示热分析(TG-DTA)结果。
图3显示BIT-BOS混合溶液以及BOS-BIT的EDX图谱。
图4为BSO溶胶凝胶溶液与PZT溶胶凝胶溶液混合形成的原料溶液的状态的原理图。
图5为使用BIT-BOS混合溶液成膜时,(a)临时烧结后的氧化物材料膜的XRD图谱,(b)结晶后的氧化物材料膜的XRD图谱,(c)BSO溶液与BIT溶液交替涂布成膜时的结晶后的氧化物材料膜的XRD图谱。
图6(a)为将BIT-BSO的混合溶液涂布在铂电极上时的状态的显示图,(b)是BSO溶液和BIT溶液交替涂布时的状态的显示图。
图7(a)为图6(a)得到的氧化物材料膜的磁滞特性图,(b)为图6(b)得到的氧化物材料膜的磁滞特性图。
图8为氧化物膜的结晶化过程的状态说明图。
图9为PZT溶胶凝胶溶液的配制流程图。
图10为BSO溶胶凝胶溶液的配制流程图。
图11为SBT溶胶凝胶溶液的配制流程图。
图12为使用加入了水分的原料溶液进行结晶化时的氧化物材料膜的XRD图谱。
图13为图12的氧化物材料膜的磁滞特性图。
图14为BIT以及本发明的BSO-BIT(R=0.4)的XRD图谱。
图15表示Bi4Ti3O12以及本发明的添加了Bi2SiO5的Bi4Ti3O12(R=0.4)的结晶化过程。
图16为SrBi2Ta2O9以及添加了Bi2SiO5的SBT(R=0.33)薄膜、PbZr0.52Ti0.48O3以及添加了Bi2SiO5的PbZr0.52Ti0.48O3(R=0.1)薄膜的XRD图谱。
图17为添加了Bi2SiO5的Bi4Ti3O12(R=0.4)、添加了Bi2SiO5的SrBi2Ta2O9(R=0.33)以及添加了Bi2SiO5的PbZr0.52Ti0.48O3(R=0.1)薄膜的D-E磁滞图谱。
图18为添加了Bi2SiO5的Bi4Ti3O12(R=0.4)、添加了Bi2SiO5的SrBi2Ta2O9(R=0.33)以及添加了Bi2SiO5的PbZr0.52Ti0.48O3(R=0.1)薄膜的膜疲劳特性图谱。
图19为BIT的结晶构造图。
图20为BSO的结晶构造图。
图21为BSO和BIT的超晶格构造图。
图22为本发明的Pt/BIT/Pt电容器在含有3%H2的N2中于400℃进行退火时的还原耐性。
图23为本发明的BIT+Bi4Si3O12、BIT+BSO薄膜以及BIT薄膜的XRD图谱的比较图。
图24为本发明的膜厚100nm的Bi4Ge3O12-Bi4Ti3O12强电介质薄膜的D-E磁滞特性图。
图25为本发明的膜厚100nm的Bi4Ge3O12-Bi4Ti3O12强电介质薄膜的1010次极化翻转后的膜疲劳特性图。
图26为本发明的膜厚25nm~100nm的添加了BSO的BIT以及添加了BSO的SBT的P-E磁滞特性图,其横坐标轴表示外加电场。
图27为本发明的膜厚25nm~100nm的添加了LSO的BIT以及添加了LSO的SBT的P-E磁滞特性图,其横坐标轴表示外加电场。
图28为本发明的膜厚10nm的添加了BSO的BLT的P-E磁滞特性图,其横坐标轴表示外加电场。
图29为本发明的添加了LSO的BIT薄膜的R=0.4(BSO=0.2)的BIT膜的XRD图谱。
图30为本发明的添加了LSO的BIT薄膜以R=0.2(BSO=0)、R=0.4(BSO=0.2)、R=0.8(BSO=0.6)、R=1.6(BSO=1.4)、R=3.2(BSO=3.0)的变化时的磁滞特性变化图。
图31为本发明的添加了BSO的PZT薄膜的R=0.025~0.2的PZT膜的XRD图谱。
图32为图31所示薄膜的磁滞特性图。
图33表示各种催化物质与BIT的固溶体薄膜的强介电特性。
图34表示各种催化物质与SBT的固溶体薄膜的强介电特性。
图35表示各种催化物质与PZT的固溶体薄膜的强介电特性。
图36表示BSO-BST(R=0.2)的薄膜的电特性。
图37表示BSO-Bi2Sr2Ca2Cu3Ox(Bi2223)结晶的XRD图谱。
图38为使用本发明的氧化物材料制作的元件的简单剖面图。
图39为本发明的BIT-BSO结晶的XRD图谱。
图40表示使用了图39的氧化物材料膜的元件的二极管特性。
图41为本发明的SBT-BSO结晶的XRD图谱。
图42以及图43表示使用了图41的氧化物材料膜制成的晶体管的晶体管特性。
实施发明的最佳方式
本发明的氧化物材料由在钙钛矿型或层状钙钛矿型晶格结构的氧化物中含有选自Si、Ge、Sn的1种以上的IV族元素的催化物质固溶而成。这种结构维持并改善了钙钛矿型或层状钙钛矿型晶格结构本身的特性的同时,也实现了在特别低的温度下的结晶化。
本发明中,主要由于钙钛矿型或层状钙钛矿型晶格结构使钙钛矿型或层状钙钛矿型晶格结构的氧化物中包含强电介质、超导氧化物等具有各种特性的化合物。例如,ABO3、(Bi2O2)2+(Am-1BmO3m+1)2、LnBa2Cu3O7、Z2Ba2Can-1CunO2n+4或ZBa2Can-1CunO2n+3等通式表示的化合物。式中,A是选自Li+、Na+、K+、Pb2+、Ca2+、Sr2+、Ba2+、Bi3+、Y3+、Mn3+及La3+的1种或2种以上的离子,B是选自Ru3+、Fe3+、Ti4+、Zr4+、Cu4+、Nb5+、Ta5+、V5+、W6+及Mo6+的1种或2种以上的离子,Ln是选自Y、La、Ce、Pr、Nd、Pm、Sm、Eu、Gd、Tb、Dy、Ho、Er、Tm、Yb及Lu的1种或2种以上的离子,Z是选自Bi、T1、Hg的1种或2种以上的离子,m是大于1的自然数,n是1~5的自然数。
本发明的催化物质中含有选自Si、Ge、Sn的1种以上的IV族金属。这种催化物质在氧化物材料的结晶过程的加热时能起到加快钙钛矿型或层状钙钛矿型晶格结构的氧化物形成的反应速度、促成晶核生成、降低结晶能量等催化作用,并能稳定存在。但当晶核在其表面形成时,其催化功能也就结束。此后,降温时,硅酸盐或二价锗酸盐与钙钛矿型或层状钙钛矿型晶格结构的氧化物混合成一体形成固溶体。总之,催化物质起到在低温促使钙钛矿型或层状钙钛矿型晶格结构的氧化物形成的催化作用,同时它也是氧化物材料。
具体来讲,这种催化物质是比构成固溶体的钙钛矿型或层状钙钛矿型晶格结构的氧化物更能促成低温下结晶化的物质。钙钛矿型或层状钙钛矿型晶格结构的氧化物是具有良好共格性和层状结构的物质。特别是具有层状结构时,因为其表面积较大,近年来作为催化物质受到关注(尾崎萃,催化机能《第四章催化剂的制作方法,4.1多孔质的制作方法(1986)p74-78》)。
具体包括选自CaO、BaO、PbO、ZnO、SrO、MgO、FeO、Fe2O3、B2O3、Al2O3、In2O3、Y2O3、Sc2O3、Sb2O3、Cr2O3、Bi2O3、Ga2O3、CuO2、MnO2、ZrO2、TiO2、MoO3、WO3、V2O5及稀土元素的氧化物的1种以上的氧化物和选自SiO2、GeO2及SnO2等的1种以上的IV族金属氧化物形成的复合氧化物材料等。
复合氧化物材料具体包括X2SiO5、X4Si3O12、X2GeO5、X4Ge3O12、X2SnO5或X4Sn3O12(式中,X表示Ca2+、Ba2+、Pb2+、Zn2+、Sr2+、Mg2+、Fe2+、Fe3+、B3+、Al3+、In3+、Y3+、Sc3+、Sb3+、Cr3+、Bi3+、Ga3+、Cu4+、Mn4+、Zr4+、Ti4+、Mo6+、W6+、V5+、La3+、Ce3+、Pr3+、Nd3+、Pm3+、Sm3+、Eu3+、Gd3+、Tb3+、Dy3+、Ho3+、Er3+、Tm3+、Yb3+、Lu3+等)表示的材料等。
进一步包括Bi2SiO5、La2SiO5、Y2SiO5、Bi4Si3O12、La4Si3O12、Y4Si3O12、Bi2GeO5、La2GeO5、Y2GeO5、Bi4Ge3O12、La4Ge3O12、Y4Ge3O12、Bi2SnO5、La2SnO5、Y2SnO5、Bi4Sn3O12、La4Sn3O12、Y4Sn3O12或2种以上的混合体Bi2-xLaxSiO5、La4-xYxSi3O12等。
例如,铋氧化物与硅氧化物的1∶1的复合氧化物Bi2SiO5是在400℃左右的低温下结晶化的氧化物材料(M.Yamagushi等.,Proceedings of the 2000 12th IEEEInternational Symposium on Application of Ferroelectrics.,2(2001)p629-632))。Bi2SiO5与后述的所有氧化物几乎都有良好的共格性,可选择性地促使氧化物在低温下完成结晶化。此外,Bi2SiO5中的SiO2是4位配位,因而非常稳定。
本发明的氧化物材料是固溶体,具有在结晶相的晶格点的原子不规则地与其他原子取代,或者在晶格间隙有其他原子按照统计分布原则进入,即在结晶相中溶入了其他物质形成了混合相的构造。众所周知,层状钙钛矿型晶格结构的氧化物材料具有在某些结晶相中混入了其他物质的结晶相的晶体结构,并不是所谓的固溶体(Carlos A.Pazde Araujo:强电介质存储器尖端工程、科学论坛、(1999)p35-43)。因此,本发明的氧化物以含有IV族元素的催化物质与钙钛矿型或层状钙钛矿型晶格结构的氧化物材料组成的固溶体状态存在,例如,构成钙钛矿型或层状钙钛矿型晶格结构的氧化物的晶格中的阳离子位置含有Si4+、Ge4+或Sn4+的氧化物材料,更好的是构成钙钛矿型或层状钙钛矿型晶格结构的氧化物的氧八面体的中心含有Si4+、Ge4+或Sn4+的材料。本发明的氧化物材料并不要求含有IV族元素的催化物质与钙钛矿型或层状钙钛矿型晶格结构的氧化物材料完全固溶,但最好是晶格中的阳离子的位置或氧八面体的中心含有Si4+、Ge4+或Sn4+的材料。
构成钙钛矿型或层状钙钛矿型晶格结构的氧化物材料的晶格中的阳离子或氧八面体的中心离子被Si4+、Ge4+或Sn4+取代时,对其取代程度没有特别的限定,例如,为Bi4Ti3O12时,出现由于Ti被0.1个Si取代形成Bi4Ti2.9Si0.1O12而使结晶化温度下降等差异,Ti被0.5个Si取代形成Bi4Ti2.5Si0.5O12可进一步改善电特性,因此每1分子可被0.1个以上或进一步被0.5个其他原子取代。另外,从其他方面考虑,钙钛矿型或层状钙钛矿型晶格结构的氧化物与含有IV元素的催化物质一般以1∶0.01~5左右的摩尔比,更好是以1∶0.01~0.8左右、1∶0.01~0.6左右、1∶0.01~0.5左右的摩尔比固溶。构成钙钛矿型或层状钙钛矿型晶格结构的氧化物的晶格中的阳离子或氧八面体的中心离子最好以约50%以下、约40%以下、约35%以下的比例被Si4+、Ge4+或Sn4+取代。
一般来讲,为ABO3型钙钛矿氧化物材料时,作为结晶存在的条件,晶格对氧八面体的各构成离子的离子半径有所限定。用宽容系数t来表达这一概念,宽容系数t=(RA+RO)/(2(RB+RO))必须在0.8<t<1.02的范围内。
对于(Bi2O2)2+(Am-1BmO3m+1)2-、铋层状钙钛矿型晶格结构的材料,以宽容系数t表示的对构成钙钛矿型晶格的离子的限定在m=2时,0.81<t<0.93;(m=3时,0.83<t<0.91);m=4时,0.85<t<0.89;m=5时,0.86<t<0.87,随着m的数值增大,t的允许范围越来越窄(竹中正:应用电子物性分科会研究报告,应用物理学会应用电子物性分科会,456(1994)1-8)。由于没有记载m=3时的宽容系数,根据m=2、4、5的最小和最大宽容系数,如图1所示用外差法求算比例关系得到m=3时的t的范围。
在氧八面体的中心被Si4+取代时,PZT、SBT、BIT的晶格的宽容系数t分别为PZT:t=1.10(Å)、SBT:t=1.09(Å)、BIT:t=1.04(Å),并不完全在允许范围内。分别使用O2-:1.26(Å)、Pb2+:1.33(Å)、Sr2+:1.32(Å)、Bi3+:1.17(Å)、La3+:1.17(Å)、Ti4+:0.75(Å)、Zr4+:0.84(Å)、Ta5+:0.78(Å)、Si4+:0.40(Å)的离子半径和Shannon(1976)、Prewitt(1969,70)的经验离子半径,为PZT这样的固溶体时,由于Zr/Ti=0.52/0.48,因此以0.84×0.52+0.75×0.48=1.18(Å)表示的平均值作为离子半径。
由此可知,钙钛矿型晶格结构、层状钙钛矿型晶格结构的氧化物材料的晶格,在氧八面体的中心(B侧)被Si4+取代时,几乎所有的宽容系数t都在允许范围以外,可知Si4+在B侧非常困难。这一事实在美国专利第5519234号也有明确叙述,即,SBT中的B侧离子可被Ti、Ta、Hf、W、Nb或Zr等金属取代,但未记载作为半金属的Si4+可进行取代。
为在钙钛矿型或层状钙钛矿型晶格结构的晶格点进行取代,必须具有SiO6 8-这样的6配位的八面体结构,但IV族元素的配位数是4,例如,SiO2(SiO4 4-)这样的四面体构造。但是,在地球600km地下金属层中的20Gpa(200000大气压)以上的高压下,存在大量GaSiO3、MgSiO3等钙钛矿型晶格结构的材料(S.K.Saxena等.,Science,《Stability of Perovskite(MgSiO3)in the Earth’s Mantle,274(1996)p1357-1359),除了这样的高压环境以外,还没有发现存在IV族元素(Si、Ge、Sn)的6配位结构。
尽管如此,本发明根据上述结构,在晶格中导入最强的共价键,即结晶相的晶格中的原子被除C、Pb以外(C的离子半径过小,Pb的离子半径过大)的lV族元素(Si、Ge、Sn)以不规则的方式取代,或者是IV族元素按照统计分布原则进入晶格间隙,钙钛矿型晶格结构的氧化物材料能够防止对活性氢(H+)和外加电场(e-)等离子作用的过程具有非常强的反应性的离子键的反应性。其结果是能够防止在氢还原氛围下钙钛矿型晶格结构被破坏。
钙钛矿型晶格结构、层状钙钛矿型晶格结构的氧化物的晶核生成必须有大量的热能,但成长过程中的热能可以较低(T.Kijima,S.Satoh,H.Matsunaga和M.Koba《Ultra-Thin Fatigue-Free Bi4Ti3O12 Films for Ferroelectric Memories》,Jpn.J.Appl.Phys,35(1996),p1246-1250))。因此,可利用催化物质促进含有构成氧化物材料的元素的原料生成氧化物材料的反应,同时由于能降低氧化物材料的晶核生成时的活化能,因而得以降低结晶化温度。
具体地讲,固溶体结晶化时的烧结温度可根据最终目标氧化物的特性、各溶液的组成等进行适当调节。通常,该烧结温度比用钙钛矿型或层状钙钛矿型晶格结构的氧化物形成用溶胶凝胶溶液形成薄膜时的烧结温度低,例如,为PbZr0.52Ti0.48O3(PZT)时,烧结温度为450℃~600℃左右,为Bi4Ti3O12(BIT)以及Bi4-xLaxTi 3 O 12 (BIT)时,烧结温度为500℃~650℃左右,为SrBi2Ta2O9(SBT)时,烧结温度为500℃~650℃左右。
利用含有IV族元素的催化物质,能够促进构成钙钛矿型或层状钙钛矿型晶格结构的氧化物的原料的分解,除去膜中的有机金属原料中的C。
本发明的氧化物材料为含有IV族元素的催化物质和钙钛矿型或层状钙钛矿型晶格结构的氧化物的固溶体,它们最好在氧化物材料中混为一体。通常,钙钛矿型或层状钙钛矿型晶格结构的氧化物仅利用作为下部电极的导电膜(一般为Pb)的催化能结晶化,所以晶核的形成密度较低。因此,晶核持续成长最终成膜时其上形成突出的杂乱的表面组织。膜厚在100nm以下时,外加电场形成漏电电流,使氧化物的特性不能充分显现。但是,由于上述材料混在一起,可在膜中的任何位置形成晶核,因此可以使氧化物材料的薄膜化达到致密化。
此外,在含有IV族元素的催化物质的表面选择性地生成钙钛矿型或层状钙钛矿型晶格结构的氧化物(例如,BIT等)。其结果是两者几乎完全固溶,能够得到钙钛矿的单一相。但是,有时两者并不完全固溶仍残留含有IV族元素的催化物质,在这种情况下得到的氧化物材料仍然具有与钙钛矿的单一相同等的良好特性。例如,钙钛矿型或层状钙钛矿型晶格结构的氧化物材料为强电介质时,由于强电介质的极化造成变位,与强电介质的极化一样催化材料也发生极化,被动的强电介质发挥作用不会造成强电介质的劣化。
本发明中,含有IV族元素的催化物质在结晶化过程的加热时,促进钙钛矿型或层状钙钛矿型晶格结构的氧化物的晶核的生成,此时稳定存在。例如,SiO2(或GeO2、SnO2)为4配位,非常稳定。晶核在其表面生成后,降温时由于硅酸盐或二价锗酸盐与钙钛矿型或层状钙钛矿型晶格结构的氧化物间的热膨胀差,膜中有很大的压缩应力。在这种压缩应力的作用下,钙钛矿型或层状钙钛矿型晶格结构的氧化物的晶格中硅等被取代。根据含有IV族元素的催化物质与钙钛矿型或层状钙钛矿型晶格结构的氧化物的组合方式,例如,使Bi4Ti3Ol2与Y2O3-SiO2固溶,同时取代钙钛矿型或层状钙钛矿型晶格结构的氧化物的所谓A侧离子和B侧离子,最终能够得到(Bi,Y)4(Ti,Si)3O12,获得具有各种的特性,例如,具有强介电特性的氧化物材料。
以下参照附图对本发明的氧化物材料进行详细说明。
实施例1:Bi2SiO5(BSO)的催化性
在基板上形成的铂电极上通过旋转涂敷法涂布BSO溶胶凝胶溶液,除去加热板(400℃)上的有机成分,制得100nm的BSO薄膜。
然后,将形成了BSO薄膜的附有铂的基板在1%氨水溶液中浸泡。此时,BSO薄膜中的Bi3+在氨水溶液中变成Bi(OH)3,其结果是在水溶液中造成H+过剩,pH5呈弱酸性。
分别使用10重量%的Bi4Ti3O12(BIT)溶液、10重量%的BSO溶液以及BIT-BSO混合溶液(1摩尔BIT与0.4摩尔BSO的混合溶液:R=0.4),进行差示热分析(TG-DTA),其结果如图2所示。
由图2可知,BSO溶液在350℃的低温下结晶化。BIT溶液在600℃附近出现非常宽的结晶峰,可知结晶化较难。BIT-BSO混合溶液首先被认为是BSO溶液,在350℃附近出现持续的峰,在400℃左右出现明确的结晶峰。这是由于BSO首先结晶化,在BSO的表面BIT以低温结晶化。
比较图2的TG图,BIT溶液直至800℃才缓慢地减少重量。另一方面,BIT-BSO混合溶液在400℃就有较大的重量减少,此后几乎没有变化。这是由于BIT-BSO混合溶液中BSO起到氧催化剂的作用,在400℃以下的温度,充分分解有机金属,使金属离子转化成强电介质,碳成分转化成CO2。
如图3所示,分析由BIT-BSO混合溶液以及BIT溶液制成的薄膜的EDX(Energy Dispersive X-ray)分析,可知使用BIT-BSO的混合溶液时,材料中几乎没有碳元素的残渣。
如上所述,通过使用含有IV族元素的催化物质(BSO),层状钙钛矿型晶格结构的氧化物(BIT)不仅可促进有机金属的分解,还能够使氧化物材料在低温下结晶化。
实施例2:溶胶凝胶法的制膜
首先,混合5重量%的BIT溶胶凝胶溶液与1重量%的BSO溶胶凝胶溶液,配制BIT-BSO混合溶液(R=0.4)。如图4所示,此时的混合溶液中各种成分以混合状态存在。
在铂电极上进行以下一系列的操作,1)以旋转涂敷法涂布上述混合溶液(500rpm、5秒钟→4000rpm、20秒钟)、2)干燥(大气中,50℃下2分钟)、3)暂时烧结(大气中,400℃5分钟),重复操作4次,使膜厚达到100nm。这时的膜如图5(a)所示,只有BOS处于结晶化状态,如图6(a)所示,非晶态的BIT包围了结晶化的BOS
接着,进行4)烧结(结晶化)(600℃,15分钟,1kg/cm2的氧中的RTA),在氧化物材料膜上形成铂上部电极。
此后,进行5)焊后的退火(500℃,5分钟,2kg/cm2的氧中的RTA)。
测定结晶化后的氧化物材料膜的XRD,结果如图5(b)所示,BSO结晶的反射峰完全消失。
测定所得氧化物材料膜的磁滞特性,结果如图7(a)所示,可以确认有良好的磁滞特性。
为了进行比较,分别配制5重量%的BIT溶胶凝胶溶液与1重量%的BSO溶胶凝胶溶液,如图6(b)所示,BIT每层14nm,BSO每层7nm,交替形成9层,总膜厚约100nm,除此之外,采用与上述1)~3)同样的方法成膜。将整个膜的组成调节为R=0.4。
进行上述4)和5)完成结晶化的氧化物材料膜与以往的BIT膜不同,可确认BIT层在500℃结晶化。结晶化温度的降低是由于对BSO结晶起到催化作用。测定XRD,其结果如图5(c)所示,BSO与BIT的结晶峰混在一起,未形成单层。
测定所得氧化物材料膜的磁滞特性,其结果如图7(b)所示,不能确认该膜具有磁滞特性。
上述结果表示BIT-BSO薄膜中BSO在层状钙钛矿型晶格结构中发生结晶结构的变化。
总之,上述结果意味着BSO结晶中的SiO4 4-四面体转变成了SiO6 8-八面体结构,最终转变成含有30%以上的作为B侧离子的Si的Bi4(Ti,Si)3O12。此结构形成时,Si的配位数由4转变为6的同时,离子半径也由0.04nm转变成0.054nm。由于Si在大气环境中的离子半径过小而不能形成钙钛矿型晶格结构,但地球的金属层的20GPa的压缩应力下存在钙钛矿型晶格结构的氧化物(Irifune,T.和Ringwood,A.E.,Phase thansformations in subducted oceanic crust和buoyancyrelationships at depths of 600-800km in the mantle.Earth Planet.Sci.Lett.117,10l-110(1993).,Surendra,K.S.等,Stability of Perovskite(MgSiO3)in the Earth’s Mantls.Science.274,1357-1359(1996).,Dubrovinsky,L.S.等,Experimental和Theoreticalidentification of new high-pressure phase of silica.Nature.388,362-365(1997)),因此可以认为以上是由于BIT-BSO薄膜中产生了很大的压缩应力的结果。
BSO与BIT之间的热膨胀系数的差异对产生这样大的压缩应力起到了很大的作用。即,使用BSO溶胶凝胶和BIT溶胶凝胶的混合溶液,使BIT结晶包围在BSO结晶的周围,结晶化退火后从高温冷却至室温的过程中,BIT给BSO附加了几乎将其挤碎(使结晶结构产生变化)的压缩应力。BSO的主要组成物质是SiO2,要与石英(Si)的热膨胀系数没有大的差别,使BSO的热膨胀系数小到几乎可以忽略,计算BIT的热膨胀系数(Suubarao,E.C.,Ferroelectricity in Bi4Ti3O12和Its Solid Solutions.Phys.Rev.122,804-807(1961).),采用文献中的杨式弹性模量(Nagatsuma,K.,Ito,Y.,Jyomura,S.,Takeuchi,H.和Ashida,S.,Piezoelecteicity,inFERROELECTRICITY和RELATED PHENOMENA.4,167-176(Gordon和BreachScience Publishers,London,1985).Jaffe,B.,Cook,W.R.和Jaffe,H.,Non-PerovskiteOxide Piezoelectrics和Ferroelectrics,in PIEZOELECTRIC CERAMICS.70-74(ACADEMIC PRESS,NEW YORK,1971)),计算因BIT结晶的收缩产生的压缩应力(Ishiwara,H.,Sato,T.& Sawaoka,Epitaxial growth of strain-free Gefilms on Sisubstrates by solid phase epitaxy at ultrahigh pressure.Appl.Phys.Lett.61,1951-1953(1992)),不考虑铂电极的收缩带来的应力,在BSO结晶施加12GPa左右的压缩应力。
另一方面,为图6(b)所示的三明治结构时,由于两者界面的滑行效果释放了压缩应力,因此BIT-BSO的膜内没有产生结构变化。
与图5(c)所示的层叠结构的BIT结晶相比,从图5(b)所示的BSO-BIT结晶的XRD图谱可以确认有0.3~1的很大的峰位移。由于该峰位移值可知,BIT-BSO结晶与BIT相比出现约8%的体积收缩。目前已知,要产生这样大的体积收缩必须要施加20~30GPa的压缩应力(J.Haines等.,Physcial Review B,58(1998)2909-2912;J.Haines和J.M.Leger,Physical Review B,55(1997)11144-11154;B.B.Karki等.,Physical Review B,55(1997)3465-3471)。
总之,BIT-BSO结构如图8所示,在结晶化过程中的加热时,BSO使BIT在低温下结晶化,冷却时由于BSO与BIT的热膨胀差异,形成Bi4(Ti,Si)3O12(BSO-BIT)固溶体。Si原子形成钙钛矿型晶格结构时,由于Si的强共价结合性,形成具有很大的共价结合性的钙钛矿型结晶,因而强电介质对氢产生了前所未有的高耐受性。
溶胶凝胶溶液
溶胶凝胶法中,为形成上述氧化物薄膜所使用的溶液最好是金属醇盐、有机酸盐、无机盐等溶于醇类等有机溶剂形成的溶液。最好有一定的蒸气压,这样在加热回流的过程中就容易得到高纯度产品,还要易溶于有机溶剂,在与水反应时生成羟基化凝胶或沉淀,经氧化氛围气中的烧结可生成金属氧化物,考虑上述情况,最好使用金属醇盐。此外,配制这些溶胶凝胶溶液的有机溶剂需互相易混合,且它们都是无水溶剂,因此相互混合不应促进缩聚,考虑到这些情况最好使用正丁醇、正丙醇和2-甲氧基乙醇等醇类。
构成金属醇盐的金属元素只要是能够构成含有IV族元素的催化物质和钙钛矿型或层状钙钛矿型晶格结构的氧化物的元素即可,对其无特别限定,例如,作为碱金属的K、Li、Na,作为碱土金属的Ba、Ca、Mg、Sr,III族的A1、B、In,IV族的Si、Ge、Sn,V族的P、Sb、Bi,作为过渡元素的Y、Ti、Zr、Nb、Ta、V、W,作为镧系元素的La、Ce、Nd等。
使用金属醇盐作为起始原料时,通过部分水解控制缩聚反应。如下所示,[n=分子长(大小)]可以控制,对反应有利。即,通过加入已知量的水可以控制金属醇盐的缩聚反应本身。
1)水解:

2)2分子的缩聚:
链状缩聚:
采用旋转涂敷法、刮刀法和喷雾法等方法,将以上得到的缩聚物形成的溶胶凝胶溶液涂布在基板等上得到薄膜。
本实施例中,形成PZT的金属醇盐和羧酸金属盐系中,作为Pb(铅)的起始原料使用了乙酸铅((CH3CO2)2Pb·3H2O)、作为Ti(钛)的起始原料使用了四异丙氧基钛(((CH3)2CHO)4Ti)、作为Zr(锆)的起始原料使用了四正丁氧基锆((CH3(CH2)3O)4Zr)、作为溶剂使用了2-甲氧基乙醇(CH3O(CH2)2OH)。
图9为PZT强电介质薄膜形成用溶胶凝胶溶液的合成流程图。上述化学式中,Pb、Zr、Ti的醇盐取代Si混合缩聚(部分水解),通过氧原子结合各元素获得所谓的PZT强电介质形成用溶胶凝胶溶液。
如上所述,为了能够通过调整添加水量对水解缩聚物的聚合度进行调节,首先将作为结晶水存在的(CH3CO2)2Pb·3H2O中的3H2O除去,即加热回流使其与作为溶剂的CH3O(CH2)2OH一起共沸蒸去。经过上述蒸馏过程得到的粘性液体的化学结构是(CH3CO2)2Pb·3H2O中的1个乙酸基(CH3CO2-)取代成2-甲氧基乙氧基(CH3O(CH2)2O-)为CH3CO2PbO(CH2)2OCH3·XH2O(X<0.5)。取代反应生成了乙酸(CH3CO2H)以及乙酸与CH3O(CH2)2OH的酯(CH3CO2(CH2)2OCH3和水(H2O))。
接着,将((CH3)2CHO)4Ti溶于CH3O(CH2)2OH中进行以下的醇取代反应,没有溶剂存在时,以1.4倍体存在的((CH3)2CHO)4Ti的异丙氧基((CH3)2CHO-)的一部分或全部被2-甲氧基乙氧基取代。
另外,使(CH3(CH2)3O)4Zr溶于CH3O(CH2)2OH时也进行同样的醇取代反应。
将这三种溶液混合,加入经过分子测定的水控制水解反应,得到作为更大分子的PZT强电介质薄膜形成用溶胶凝胶溶液。此时,考虑目标强电介质材料等的组成,适当调节各元素的醇盐的混合比。
在溶液中存在以下聚合物。

采用与上述PZT强电介质薄膜形成用凝胶同样的水解以及缩聚反应,形成图10所示的BSO凝胶。
相对于溶胶凝胶法,还有称作为有机金属分解法(MOD法)的方法。这种方法将金属醇盐、有机酸盐溶于甲苯(C6H5CH3)、二甲苯(C6H4(CH3)2)等有机溶剂中,将该溶液涂布于基板上,经热分解形成氧化物薄膜。这种方法没有添加任何水,因而加入溶液的金属醇盐和有机酸盐以原有的形态存在,不会发生缩聚反应。但出现象SBT那样的配位基交换反应而存在缩聚体。
例如,如图11所示配制SBT强电介质薄膜形成用溶液,即所谓的MOD溶液,羧酸盐的羰基(-CO-)位于正中,Bi元素与Ta元素交替进入形成凝胶结构,在此空隙中存在Sr,被认为发生了缩聚,属于本发明的缩聚凝胶(溶胶凝胶溶液)的范畴。
如上所述,将抑制水解使其缩聚获得的强电介质薄膜形成用BIT、SBT、PZT溶液与抑制水解使其缩聚获得的含有IV族元素的催化物质形成用BSO溶液在室温下分别混合。考虑各溶液的组成、目标氧化物材料的特性和烧结温度等,可适当调节强电介质形成用凝胶溶液与催化物质形成用溶液的混合比。
例如,将以正丁醇(比重:0.813)为溶剂的Bi2SiO5(BSO)溶胶凝胶溶液以R=0.4的比例与以正丙醇(比重:0.79)为溶剂的Bi4Ti3O12(BIT)溶胶凝胶溶液混合而成的溶液作为原料溶液时,各自的浓度均为10wt%,溶剂中所含的BSO和BIT的克数是1升中BSO:1000×0.813=813g、BIT:1000×00.79=790g。这里,除去BSO与BIT中的氧后1摩尔的分子量分别是BSO:446.0455、BIT:979.56。各自的摩尔浓度为BSO:1.8摩尔/升、BIT:0.81摩尔/升。然后,将两者混合调制出R=0.4的混合溶胶凝胶溶液,由于是1份的BIT与0.4份的BSO混合,所以可在150ml的BSO溶胶凝胶溶液中加入850ml的BIT溶胶凝胶溶液。
各种溶液最好使用经过缩聚反应的溶液。这样各溶液均为无水状态,即使混合也不会发生水解反应,能够成为非常稳定的原料溶液。
溶胶凝胶溶液的水分状态
将与上述方法同样配制的5重量%的BIT溶胶凝胶溶液和1重量%的BSO溶胶凝胶溶液的混合溶液装入密闭容器中,再将该容器放入洁净室的气流室中,打开气栓1小时后关闭气栓,再过24小时后就形成了与上述条件相同的图6(a)所示的薄膜。
如果有意识地使混合溶胶凝胶溶液中吸入大气中的水分后形成膜,则形成图12所示的电介质烧绿石层,BSO并没有起到作用。其结果如图13所示,薄膜的强介电特性非常差。
其原因是,空气中的水分导致水解反应的产生,混合溶胶凝胶溶液中的BSO与BIT之间的网络不能独立存在,这样不能保持图4所示的结构。因此,含有IV族元素的催化物质形成用溶胶凝胶溶液与强电介质形成用溶胶凝胶溶液在无水状态下混合,即,使借助氧元素结合的IV族元素的溶胶与借助氧元素结合的强电介质构成元素的溶胶在同一溶液中分散而独立存在的溶液作为薄膜形成用原料溶液是非常重要的。
实施例3
BSO-Bi4Ti3O12(BIT)、BSO-SrBi2Ta2O9(SBT)及BSO-PbZr0.52Ti0.48O3(PZT)固溶体薄膜的特性
与实施例2同样,使抑制水解完成了缩聚反应的强电介质薄膜形成用BIT、SBT、PZT溶液与抑制水解完成了缩聚反应的电介质材料薄膜形成用BSO溶液分别在室温下混合。强电介质形成用混合凝胶中的BSO凝胶含有率对应于BIT、SBT、PZT的摩尔浓度比R=0.4、0.33、0.1。各原料凝胶使用化学理论量组成。
接着,通过旋转涂敷法在Pt/Ti/SiO2/Si基板上涂布上述溶胶凝胶混合溶液,在以下的成膜条件下成膜,膜厚全部为100nm。
[强电介质薄膜形成条件]
①旋转涂敷(500rpm,5秒钟→4000rpm,20秒钟)、②干燥(大气中,50℃,2分钟)、③临时烧结(大气中,400℃,5分钟)的一系列操作,反复进行4次,接着进行④烧结(结晶化)(400-700℃,10分钟,氧中的RTA)。
图14是上述过程中形成的膜厚100nm的以往的BIT与本发明的BSO-BIT(R=0.4)的XRD图谱。
本发明的添加了BSO的BIT在500℃下也显示出良好的结晶性。另一方面,不含BSO的BIT即使在700℃下也不能得到BIT的单一层,可知电介质烧绿石层(Bi2Ti2O7)与BIT混在一起。
由以上可知,与以往的BIT比较,本发明的添加了BSO的BIT能在约200℃的低温下完成结晶化。
此外,对以往的BIT和本发明的添加了BSO的BIT的表面进行观察时,以往的BIT在600℃也几乎没有出现结晶峰,而且膜表面非常杂乱,与此相比,本发明的添加了BSO的BIT具有致密平滑的膜表面。
在通过两者的TEM剖面图观察铂与强电介质的界面时,可以看到以往的BIT存在厚达5nm左右的非晶态层,而本发明的添加了BSO的BIT的界面情况良好,不存在非晶态层。
由此可见,两者的结晶成长的机理完全不同。
如图15(a)所示,以往的BIT的BIT结晶的初期晶核全都只在与铂电极的交界面生成,它沿着上部电极和铂界面成长,所以成长后的膜表面形成向上突出的杂乱表面组织。
另一方面,如图15(b)所示,本发明的添加了BOS的BIT在结晶化前的非晶态状态形成时的5个界面(如形成条件所显示的在Pt基板上涂布4层,各层与Pt电极的界面共计5层)全部有BIT结晶初期晶核生成。特别是在与Pt电极的界面没有非晶态的存在。所以认为由于此原因能够形成致密平滑的表面组织。这点对在450℃这样的低温下开始结晶化的BSO也非常有利(Kijima,T.& Matsunaga,H.Preparation of Bi4Ti3O12 Thin film on Si(100)Substrate Using Bi2SiO5 Buffer Layerand Its Electric Characterization.Jpn.J.Appl.Phys.37,p5171-5173(1998))。
如图16的XRD图谱所示,在550℃下烧结10分钟的添加了BSO的SBR(R=0.33)、在450℃烧结10分钟的添加了BSO的PZT(R=0.1)与BIT一样,结晶化温度的降低和表面组织得到大幅度改善。以往的PZT则在与铂电极的界面形成低电容率的非晶态层。
接着,在得到的各种强电介质薄膜上形成直径100μmφ的铂上部电极,进行P-V磁滞特性的测定。
其结果是,本发明的添加了BSO的BIT、SBT、PZT强电介质薄膜与以往的BIT、SBT、PZT强电介质薄膜相比,不仅可以在150~200℃左右的低温下进行烧结,显现出图17所示的良好的磁滞特性。另一方面,以往的BIT、SBT、PZT强电介质薄膜的膜厚为100nm,不能确认因其表面组织造成漏电特性劣化导致磁滞。
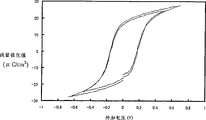
500℃的添加了BSO的BIT的强介电特性是残留极化值Pr=17μC/cm2、矫顽电场Ec=95kV/cm;600℃的添加了BSO的BIT的强介电特性是残留极化值Pr=21μC/cm2、矫顽电场Ec=95kV/cm。550℃的添加了BSO的SBT的强介电特性是Pr=7μC/cm2、Ec=50kV/cm;600℃的添加了BSO的SBT的强介电特性是Pr=11μC/cm2、Ec=60kV/cm。450℃的添加了BSO的PZT的强介电特性是Pr=20μC/cm2、Ec=45kV/cm;550℃的添加了BSO的PZT的强介电特性是Pr=25μC/cm2、Ec=38kV/cm。
由上述结果可知,本发明的添加了BSO的强电介质材料通过降低结晶化温度和大幅度地改善表面组织,没有改变原有强电介质材料的特性,并最大限度地使其发挥效果。
疲劳特性的评估结果如图18所示。分别施加外加电压3V、频率100kHz的脉冲电场,进行极化翻转1010次,所有强电介质电容器的极化值的下降均在3%以下,显示出良好的疲劳特性。出现这种结果是由于良好的结晶性、平滑的膜表面、不含异相的良好界面等。特别是众所周知以往的PZT在Pt电极上会出现疲劳,但本发明的添加了BSO的PZT得到了与BIT同样的良好的疲劳特性。
通过考察本发明与以往的差别可见不仅结晶成长的机理不同,结晶构造本身也不同。
从两者的TEM剖面图的观察可知,本发明的添加了BSO的BIT与以往的BIT相比,发生了B侧离子取代以外的变化。
在以往的BIT中,BIT的c轴长为32Å或更小,与图19的容量值很一致。此外,由图可见BIT在横向上为规则联系的结构。
另一方面,本发明的添加了BSO的BIT的BIT的c轴缩短为31Å。且可确认与以往的BIT不同,图中横向没有连续的结构,部分上下交错,形成在交错部分混入了BSO的复杂构造。BSO的c轴长为15Å或更小,与图20的容量值几乎一致。
根据以上可知,本发明的添加了BSO的BIT的结构如图21所示,这说明BSO促进了BIT的结晶化。结晶化的交错反映了硅酸盐的成长机理,此结构增大了硅酸盐的表面积,发挥了良好的作用。
目前实际使用的多结晶Pt基板不存在表面完全水平没有凹凸的基板,其上部形成的强电介质薄膜上也存在很多的凹凸。因此,结晶化时在任何地方生成的BIT初期晶核均成长,邻接的几乎都不能形成一致地连续成长,结晶未连接的部分就妨碍了结晶的成长。另一方面,本发明的结构不管基板中存在多少凹凸差异,都能够吸收这些差异使强电介质结晶连续成长。
如图20所示的结晶结构,即使含有电介质BSO也能够得到良好的强介电特性,其说明如下。
一般,所用基板与其上部的薄膜的晶格匹配完全一致几乎是不可能的,两者的晶格匹配差异使薄膜中存在晶格变形和应力。本发明所用的强电介质是被称为变位型的强电介质,极化时变位由此产生应力。
另一方面,如图20所示,BSO是Bi2O3层与SiO2层的层叠结构。即,氧化硅层为氧化硅的四面体构造沿与氧化铋层水平的方向连接的结构。氧化硅是良好的压电材料,外部压力产生极化。非共格性是造成膜中应力产生的原因,氧化硅极化后(BSO极化后),BSO对BIT的强介电特性没有损害,能够有效地发挥良好的特性。
本发明的Pt/BIT/Pt电容器在含有3%的H2的N2中于400℃进行退火处理,如图22所示显现出良好的还原耐受性。这是由于BIT中的B侧Ti的一部分被添加的BSO中的Si取代,BIT的共价结合性增强。BIT的c轴长的变化是B侧取代以及与BSO固溶的结果。
本发明的BSO与BIT的固溶体薄膜中因含有大量的Si元素而显现出明显的差异。众所周知,一般当有大量的Si元素从Si基板扩散混入强电介质膜时,即使混入的量不是非常多,Si的扩散也是造成漏电电流密度增大的原因。而且,一般来讲,对于被Pt等金属电极覆盖的Si基板,为了提高Pt与Si基板之间的紧密性,用厚度200nm左右的热氧化SiO2膜覆盖,且SiO2膜非常稳定。因此,只要制作强电介质薄膜时的烧结温度不是很高,Si就很难在强电介质膜中扩散。现在已知,以往的BIT的烧结温度虽然为700℃,但Si没有扩散到强电介质薄膜中。
本发明的BSO与BIT的固溶体薄膜中虽然含有大量的Si,但确认其具有非常优异的强介电特性,上述膜中的Si并不是从基板中扩散出来的,经分析得知是BSO中的Si,由此很容易判断本发明与以往技术的巨大差异。
BIT-BSO固溶体薄膜以及BIT-Bi4Si3O12固溶体薄膜的特性比较
10重量%浓度的BIT溶胶凝胶溶液和10重量%浓度的Bi4Si3O12溶胶凝胶溶液以R=0.33比例混合制成混合溶液,作为BIT-Bi4Si3O12固溶体形成用溶液。
接着,通过旋转涂敷法在Pt/Ti/SiO2/Si基板上涂布上述溶胶凝胶溶液,在以下成膜条件下制成膜厚为100nm的强电介质薄膜。
[强电介质薄膜的形成条件]
①旋转涂敷(500rpm,5秒钟→4000rpm,20秒钟)、②干燥(大气中,150℃,2分钟)、③临时烧结(大气中,400℃,5分钟)的一系列操作,重复操作4次,接着进行④烧结(结晶化)(600℃,10分钟,氧中的RTA)。
此时的XRD图谱如图23所示。将上述实施例制得的R=0.4的BIT+BSO与以往的BIT的XRD图谱进行比较。本发明的添加了Bi4Si3O12的BIT以及BIT+BSO的烧结温度为600℃,以往的BIT为700℃。
与以往的BIT相比较,本发明的添加了Bi4Si3O12的BIT以及BIT+BSO的所有的峰均在高角侧。与以往的BIT相比较,添加了Bi4Si3O12的BIT以及BIT+BSO的所有结晶均以8%(2%×2%×2%)左右的比例压缩呈紧缩结构,这一现象表示在膜中存在相当量的压缩应力。
据已报道的SnO2、GeO2等高温下压缩实验结果显示(B.B.Karki等,PhysicalReview B,″Ab initio studies of high-pressure structural transformations in silica″55(1997)p3456-3471:J.Haines等,Physical Review B,″Phase transitions inruthenium dioxide up to 40 Gpa:Mechanisum for the rutile-to-fluorite phasetransformation and a model for the high-pressure behavior of stishovite SiO2″48(1993)p13344-13350:J.Haines等,Physical Review B,″X-ray diffraction study ofthe high-pressure:Relationships between structure types and implications for otherrutile-type dioxides″55(1997)p11144-11154)),当强电介质结晶的体积被压缩了8%时,必须在整个膜上外加25~30Gpa的压缩应力,本发明的薄膜上施加了同样大的压缩应力。
在测定添加了Bi4Si3O12的BIT的磁滞特性时,与BIT+BSO相比,虽然磁滞形状有一些劣化,但得到了以往的BIT薄膜根本没有的非常好的矩型磁滞特性。并且,在评估添加了Bi4Si3O12的BIT的膜疲劳特性时,经1010次的极化翻转,没有出现任何膜疲劳现象。
在上述实施例中,结晶化温度降低了约200℃,Bi2SiO5(BSO)的结晶化温度降到400℃,且BSO晶格中的Bi氧化物层与ABO3或(Bi2O2)2+(Am-1BmO3m+1)组成的强电介质钙钛矿及铋层状结构的强电介质材料的钙钛矿层及拟钙钛矿层的晶格匹配均良好,这是将BSO作为晶核的强电介质钙钛矿型及层状钙钛矿型晶格结构的强电介质结晶化的结果。
众所周知,Si-O的共价结合性强,Si不仅不是金属元素,而且对于钙钛矿构成元素的取代要使用离子半径非常接近的元素。由于相对于Ti4+的0.75Å,Si4+的0.26Å非常小,所以虽然本来Si即使作为B侧离子的取代候补也很难,但是通过强电介质材料的B侧离子被Si4+取代,解决了目前强电介质薄膜中存在的问题。
含有Ge的催化物质的使用
本实施例中,对B侧离子不是被Si而是被其同族元素Ge取代的强电介质,即B侧不是被Si4+而是被Ge4+取代的强电介质进行评估。
使用涂敷了100nm的Pt的Si基板,将Bi4Ge3O12与含有16%的Bi4Ti3O12溶胶凝胶溶液以R=0.4的比例混合,按以下成膜条件在上述基板上形成100nm的薄膜。
[强电介质薄膜的形成条件]
①旋转涂敷(500rpm,5秒钟→4000rpm,20秒钟)、②干燥(大气中,150℃,2分钟)、③临时烧结(大气中,400℃,5分钟)的一系列操作,重复操作4次,接着进行④烧结(结晶化)(550℃,30分钟,氧中的RTA)。
此时,进行膜表面的AFM观察,凹凸最严重部分的RMAX为2.001nm,表示膜整体平滑性的Ra为1.5022nm,说明该膜具有良好的表面平滑性。
接着,形成Pt上部电极,使用上部Pt和下部Pt评估本发明的膜厚100nm的Bi4Ge3O12-Bi4Ti3O12强电介质薄膜的强介电特性,其结果如图24所示。该膜的D-E的磁滞特性良好,Pr=19μC/cm2。
评估疲劳特性,其结果如图25所示。重复进行1010次的极化翻转,几乎没有发现膜疲劳,显示膜具有良好的疲劳特性。
由此可知Ge4+具有与Si4+同样的B侧离子取代作用。因而,可与Bi2SiO5一样使用Bi2GeO5。Bi2SiO5(BSO)与Bi4Ti3O12(BIT)以及Bi2SiO5(BSO)与SrBi2Ta2O9(SBT)的固溶体分别用以正丙醇为溶剂的LSO溶胶凝胶溶液、以R=0.4添加了BIT的溶胶凝胶溶液、以R=0.33添加了SBT的溶胶凝胶溶液,通过旋转涂敷法形成膜厚为25nm、50nm和100nm的膜。
同时,用以R=0.4添加了BSO的Bi3.25La0.75Ti3O12(BIT)溶胶溶液制作膜厚10nm的超薄膜。
[强电介质薄膜形成条件]
①旋转涂敷(500rpm,5秒钟→4000rpm,20秒钟)、②干燥(大气中,150℃,2分钟)、③临时烧结(大气中,400℃,5分钟)的一系列操作,重复操作4次,接着进行④烧结(结晶化)(BIT:550℃,SBT:600℃,30分钟,氧中的RTA)。
膜厚25nm时涂布1次,膜厚50nm时涂布2次,膜厚100nm时涂布4次。10nm的BLT薄膜涂1层,涂布时将转速设定为7000rpm。
接着,与上述实施例一样,形成上部Pt电极(100μm),评估P-E的磁滞特性。如图26以及图27所示,得到良好的磁滞特性。
如图26所示,在共通的500kV/cm的外加电场下的磁滞特性是BIT的Pr=15~18μC/cm2、Ec=~100kV/cm;SBT的Pr=~11μC/cm2、Ec=60~70kV/cm,有良好的一致性。
如图27所示,横轴为外加电压时,膜厚100nm、50nm、25nm各自对应的矫顽电压分别为BIT:1V、0.5V、0.25V,SBT:0.7V、0.35V、0.2V,随膜厚的变化而变化。
10nm的BSO-BLT的磁滞特性见图28。在外加电压0.5V下饱和,Pr=16μC/cm2、Ec=100kV/cm,在0.7V的外加电压下Pr=17μC/cm2、Ec=120kV/cm。这说明利用厚10nm的BSO-BLT可提供能够在0.5V的驱动电压下使用的存储元件。
这是因为使用本发明后,在临时烧结阶段膜中存在的所有界面都产生晶核,其中的最上部的界面能够有效用于强电介质薄膜的结晶化,因此,通过薄膜化能够获得最重要的致密且平坦的表面组织。
含有IV族元素的催化物质的固溶量的变化对强介电特性的影响
在BIT溶胶凝胶溶液中添加作为基础的以R=0.2添加了LSO的溶胶凝胶溶液,再添加BSO凝胶,制得5种R=0.2(BSO=0)、R=0.4(BSO=0.2)、R=0.8(BSO=0.6)、R=1.6(BSO=1.4)、R=3.2(BSO=3.0)的混合溶胶凝胶溶液。
在以下成膜条件下,通过旋转涂敷法在Pt/Ti/SiO2/Si基板上涂敷上述混合溶胶凝胶溶液,形成强电介质薄膜。
烧结条件为500℃、10分钟,整体膜厚为100nm。
[强电介质薄膜的形成条件]
①涡旋混合(500rpm下5秒钟→4000rpm20秒钟)、②干燥(大气中150℃下2分钟)、③反烧结(大气中400℃5分钟)的一系列过程,反复操作4次、接着进行④烧结(结晶化)(500、600、700℃10分钟,氧中的RTA)。
如图29所示,由R=0.4(BSO=0.2)的BIT膜的XRD图谱可确认在基板温度为500℃时也具有良好的结晶性。
如图30所示,在500℃结晶化的5种BIT电容器的磁滞特性表现出随着R的增大Pr反而减小。
以往,在强电介质中加入电介质时,多数情况下随着添加数%会导致不良影响。这是由于多数电介质比强电介质的比电容率小,外加电压几乎全部加在增加的数%的电介质层,不能发挥出良好的强介电特性。
但是,在本发明中,当强电介质中添加BSO或LSO时,能够使强电介质材料的特性最大限度地发挥出来,解决目前强电介质中存在的问题。
此外,超过基本的强电介质的摩尔浓度,以R=1.6添加BSO、LSO,可以发挥出Ec与Pr不同的各种强介电特性。
R=3.2时,几乎全是电介质,虽然没有显现强介电性,但形成了比电容率约为200的强电介质膜。
结合上述实施例的超薄膜化,利用本发明由良好的强电介质制作DRAM用电容器材料、超微晶体管用高电容率表层氧化膜材料,根据不同目的获得具有任意的介电特性的薄膜。
即,如上述实施例所述,超过基本强电介质的摩尔浓度,如本发明所述添加BSO、LSO时,可以控制Ec以及Pr的任意值,这也证明了固溶体的形成。
对应于PZT(Zr/Ti=52/48),测定以R=0.025、0.05、0.1、0.2添加了BSO时的强介电特性。
以下条件作为成膜条件。
①旋转涂敷(500rpm,5秒钟→4000rpm,20秒钟)、②干燥(大气中,150℃,2分钟)、③临时烧结(大气中,400℃,5分钟)的一系列操作,重复上述操作4次,接着进行④烧结(结晶化)(500℃,15分钟,1kg/cm2的氧中的RTA)。然后,形成上部铂电极,再进行⑤焊后退火(500℃,5分钟,1kg/cm2或2kg/cm2氧中的RTA)。
据图31所示的XRD图谱,以R=0.025添加BSO都显现出良好的结晶性。
而且,可以看到无论任何情况均有良好的表面组织,这一结果只能是因为在整个膜中生成了高密度的PZT晶核。
接着,测定磁滞特性。如图32所示,与BIT一样,随添加量的不同显现出各种磁滞特性。这一现象说明PZT与BSO形成了固溶体。
最后,在临时烧结温度为300℃时,不使BSO结晶化,作为只除去了有机成分的非晶态BSO使用。
对于铋系强电介质,BSO中的Bi2O3层可以直接作为铋系层状结构的强电介质的Bi2O3层的一部分被活用,而对于钙钛矿型强电介质PZT,有必要在A侧导入Bi。
比起某种结晶系向不同晶系转变,由非晶态向结晶系的A侧导入Bi较为容易。非晶态是指构成原子间呈现无序状态,充分发挥PZT选择性地结晶化的效果。此外,比较结晶,非晶态的表面积大,非晶态也起到了很大的催化作用。
实施例4
本实施例中,由各种催化物质与以往的强电介质BIT、SBT及PZT形成固溶体薄膜。
使用5重量%浓度的强电介质薄膜形成用溶胶凝胶溶液,3重量%浓度的催化物质形成用溶胶凝胶溶液,混合两者作为原料溶液。接着,通过旋转涂敷法在以下的成膜条件下,在Pt/Ti/SiO2/Si基板上涂布上述溶胶凝胶溶液形成膜厚20nm的薄膜。各自的混合摩尔比是BIT的R=0.4、SBT的R=0.33、PZT的R=0.2。
[薄膜形成条件]
①旋转涂敷(500rpm,5秒钟→4000rpm,20秒钟)、②干燥(大气中,150℃,2分钟)、③临时烧结(大气中,400℃,5分钟)的一系列操作,重复上述操作4次,使膜厚达到100nm。接着,进行④烧结(结晶化)(600℃,15分钟,1kg/cm2的氧中的RTA),形成上部铂电极,此后,进行⑤焊后退火处理(500℃,5分钟,1kg/cm2或2kg/cm2的氧中的RTA)。
此时,得到的薄膜是整体膜厚为70nm的结晶膜,如图33~图35所示,具有良好的强介电特性。
实施例5
以R=0.2的比例,在5重量%浓度的BST溶胶凝胶溶液中混合1重量%浓度的BSO溶胶凝胶溶液作为原料溶液。接着,在以下的成膜条件下通过旋转涂敷法,在Pt/Ti/SiO2/Si基板上涂布上述溶胶凝胶溶液形成20nm的薄膜。
[强电介质薄膜的形成条件]
①旋转涂敷(500rpm,5秒钟→6500rpm,20秒钟)、②干燥(大气中,150℃,2分钟)、③临时烧结(大气中,400℃,5分钟)。接着,进行④烧结(结晶化)(600℃,15分钟,1kg/em2的氧中的RTA)。
此时,可以确认得到的膜厚20nm的薄膜是非常良好的结晶膜,其电特性如图36所示。比电容率约为600与容量值相对应。
接着,以R=0.4的比例混合2重量%浓度的Bi2223溶胶凝胶溶液与0.3重量%浓度的BSO溶胶凝胶溶液作为原料溶液。
在以下的成膜条件下通过旋转涂敷法,在自然氧化膜去除后的Si(100)基板上涂布上述溶胶凝胶溶液形成20nm的薄膜。
[超导薄膜的形成条件]
①旋转涂敷(500rpm,5秒钟→4000rpm,20秒钟)、②干燥(大气中,150℃,2分钟)、③临时烧结(大气中,400℃,5分钟),接着进行④烧结(结晶化)(700℃,1分钟,1kg/cm2的氧中的RTA),形成上部电极,此后进行⑤焊后退火处理(500℃,5分钟,1kg/cm2的氧中的RTA)。
此时,如图37所示,得到的膜厚15nm的薄膜为Bi2223单一相构成的良好的结晶膜。
BST作为取代次生代DRAM的SiO2的表层氧化物材料受到人们的期待,已经持续了10年以上的研究(Kazuhide Abe和Shuichi Komatsu,Jpn.J.Appl.Phys.,33(1994)5297-5300),Ba0.5Sr0.5TiO3(BST)钙钛矿型强电介质材料以及Ba2Sr2Ca2Cu3OX(Bi2223)超导氧化物材料通过使用固体酸催化物质,因此可作为具有良好特性的薄膜使用。
实施例6
如图38(a)所示,在1.8nm厚的覆盖了非晶态Si3N4的Si基板上,通过溶胶凝胶法形成BSO-BIT薄膜,膜厚为150nm。
与以上一样,使BIT溶胶凝胶溶液与BSO溶胶凝胶溶液的混合溶液(R=0.4)经过以下处理,即①旋转涂敷(500rpm,5秒钟→4000rpm,20秒钟)、②干燥(大气中,150℃,2分钟)、③临时烧结(大气中,400℃,5分钟)的一系列操作,重复操作3次,接着进行④烧结(结晶化)(500℃,30分钟,1kg/cm2的氧中的RTA),形成上部铂电极。
如图39所示,得到的BIT-BSO结晶薄膜显示良好的结晶性,得到图40所示的良好的二极管特性。
接着,将除去了自然氧化膜的单晶Si(100)作为基板,在其上涂布SBT溶胶凝胶溶液与BSO溶胶凝胶溶液的混合溶液(R=0.33),即经过①旋转涂敷(500rpm,5秒钟→4000rpm,20秒钟)、②干燥(大气中,150℃,2分钟)、③临时烧结(大气中,400℃,5分钟)的一系列操作,重复上述操作3次,接着进行④烧结(结晶化)(600℃,30分钟,1kg/cm2的氧中的RTA)。
此时,如图41所示,显现出良好的结晶性。
制作如图38(b)所示的A1/BSO-SBT/Si晶体管,如图42和43所示,显现出良好的晶体管特性。
此外,本发明的氧化物材料可作为光变频器、超声波传感器、红外线直线运动感应式传感器、DRAM和MMIC用电容器、强电介质装置或半导体装置的构成的一部分,运用于集成回路中。例如,强电介质元件作为非易失性存储器的容量部或作为FET的栅极使用,组合形成表层绝缘膜、源极/漏极等,也可作为MFMIS-FET、MFS-FET等使用。
运用薄膜制作技术将本发明的氧化物材料用于基板或电极时,通常隔着或不隔着导电膜在基板上形成氧化物材料薄膜。可使用的基板包括硅、锗等元素半导体,GaAs、ZnSe等化合物半导体等半导体基板,Pt等金属基板,蓝宝石基板,MgO基板,SrTiO3、SrTiO3、BaTiO3、玻璃基板等绝缘性基板等。其中,较好的是硅基板,更好的是硅单晶基板。
基板上可形成的导电膜包括Pt、Ir、Au、Al、Ru等金属或合金,IrO2、RuO2等氧化物导电体,TiN、TaN等氮化物导电体等的单层膜或层叠膜。通常对作为电极和配线使用的导电性材料没有特定的限定。导电膜的膜厚一般为100nm~200nm。
导电膜与基板之间还可形成绝缘层及粘结层等中间层,绝缘层可以由SiO2、Si3N4等形成。粘结层只要能够确保基板与导电膜或绝缘层与导电膜的粘结强度即可,对其材料没有特别的限定,可使用钽和钛等高熔点金属。中间层可通过热氧化法、CVD法、溅射法、真空蒸镀法、MOCVD法等各种方法形成。
此外,本发明的氧化物材料最好通过溶胶凝胶法形成,但也可以用MOCVD法、激光磨蚀法、溅射法等各种方法形成。
产业上利用的可能性
本发明中,由固体酸催化物质和作为催化活性物质的钙钛矿型或层状钙钛矿型晶格结构的氧化物材料形成固溶体,在钙钛矿型或层状钙钛矿型晶格结构的氧化物材料中导入了最强的共价结合性,可防止活性氢(H+)、外加电场(e-)等离子作用过程中的反应性。由此,可防止钙钛矿型或层状钙钛矿型晶格结构的氧化物材料的氢劣化,同时也可防止钙钛矿型或层状钙钛矿型晶格结构的氧化物材料中的组成分布不均,使其能够应用于存储器元件。
在固体催化物质的作用下,钙钛矿型或层状钙钛矿型晶格结构的氧化物材料能够在低温下有效结晶化。而且,由于形成了固溶体,所以减轻了结晶粒子之间晶格的不一致,能够形成致密平滑的界面以及表面。这样不仅不改变钙钛矿型或层状钙钛矿型晶格结构的氧化物材料的特性,还可将其作用最大限度地发挥。即,将钙钛矿型或层状钙钛矿型晶格结构的氧化物材料用于电子元件时,能够改善元件的漏电特性,得到良好的磁滞特性。此外,由于具有良好的结晶性、平滑的膜表面和不含异相的良好界面等,因而能够得到良好的疲劳特性。
特别是当钙钛矿型或层状钙钛矿型晶格结构的氧化物材料的晶格中或氧八面体中的阳离子的位置含有Si4+、Ge4+、Sn4+时,上述效果更显著。
此外,本发明通过混合使用催化形成用溶胶凝胶溶液与钙钛矿型或层状钙钛矿型晶格结构的氧化物形成用溶胶凝胶溶液,能够简便、高效地制作上述具有低温结晶化、平滑界面和膜表面等特性的钙钛矿型或层状钙钛矿型晶格结构的氧化物材料。
Claims (6)
1.氧化物材料,其特征在于,它由钙钛矿型或层状钙钛矿型晶格结构的氧化物以及固溶在所述氧化物中的催化物质形成,所述氧化物由ABO3表示,式中,A是选自Pb2+、Ca2+、Sr2+和Ba2+中的一种或多种的离子,B是选自Ti4+和Zr4+中的一种或多种的离子;所述催化物质选自WSiO5、V2SiO7、ZrSiO4、Sb2SiO5、Fe2SiO5、La2SiO5、CuSiO3、ZnSiO3、MnSiO3、Bi2SiO5、Y2SiO5、Bi2GeO5、Bi4Si3O12和Bi4Ge3O12,其中,所述钙钛矿型或层状钙钛矿型晶格结构的氧化物与催化物质的摩尔比为1∶0.01至1∶5。
2.如权利要求1所述的氧化物材料,其特征在于,构成所述钙钛矿型晶格结构的氧化物材料氧八面体的中心含有Si4+或Ge4+。
3.权利要求1的氧化物材料的制作方法,其特征在于,使选自Pb、Ca、Sr、Ba、Ti和Zr的2种以上的金属的醇盐或有机酸金属盐水解反应而成的钙钛矿型或层状钙钛矿型晶格结构的氧化物形成用凝胶溶液,与选自Zn、Fe、Y、Sb、Bi、Cu、Mn、Zr、W、V和La的1种以上的金属和选自Si或Ge的1种以上的IVA族金属的金属醇盐或有机酸金属盐水解反应而成的催化物质形成用凝胶溶液在无水状态下混合,其中,所述催化物质选自WSiO5、V2SiO7、ZrSiO4、Sb2SiO5、Fe2SiO5、La2SiO5、CuSiO3、ZnSiO3、MnSiO3、Bi2SiO5、Y2SiO5、Bi2GeO5、Bi4Si3O12和Bi4Ge3O12;再将该混合物涂布在基板上,烧结制成氧化物材料,其中,所述钙钛矿型或层状钙钛矿型晶格结构的氧化物形成用凝胶溶液与催化物质形成用凝胶溶液的比例R为0.1、0.2、0.33或0.4。
4.被氧化物材料膜覆盖的基板,其特征在于,在基板上形成导电性材料膜,在该导电性材料膜上形成权利要求1~2中任一项所述的氧化物材料的膜。
5.半导体元件,其特征在于,在基板上依次形成下部电极、权利要求1~2中任一项所述的氧化物材料的膜和上部电极。
6.半导体元件,其特征在于,在半导体基板上形成权利要求1~2中任一项所述的氧化物材料的膜和导电性材料膜,然后在该导电性材料膜的两侧的所述半导体基板的表面形成一对杂质扩散层。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2000316910 | 2000-10-17 | ||
| JP316910/2000 | 2000-10-17 | ||
| JP2001030170 | 2001-02-06 | ||
| JP30170/2001 | 2001-02-06 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN 200610004826 Division CN1803584A (zh) | 2000-10-17 | 2001-09-03 | 氧化物材料、其制造方法以及使用该氧化物材料的半导体元件 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1469842A CN1469842A (zh) | 2004-01-21 |
| CN1266032C true CN1266032C (zh) | 2006-07-26 |
Family
ID=26602247
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB018174132A Expired - Fee Related CN1266032C (zh) | 2000-10-17 | 2001-09-03 | 氧化物材料、氧化物薄膜的制造方法以及使用该材料的元件 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US7205256B2 (zh) |
| EP (1) | EP1338555A4 (zh) |
| JP (1) | JP4210743B2 (zh) |
| KR (1) | KR100570576B1 (zh) |
| CN (1) | CN1266032C (zh) |
| TW (1) | TWI290907B (zh) |
| WO (1) | WO2002032809A1 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106006710A (zh) * | 2016-05-24 | 2016-10-12 | 合肥工业大学 | 一种β-NaYF4:Yb/Tm@ZnO核壳纳米颗粒的制备方法 |
Families Citing this family (72)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4245116B2 (ja) * | 2001-06-13 | 2009-03-25 | セイコーエプソン株式会社 | セラミックスの原料液、セラミックス膜および強誘電体メモリ装置 |
| US7008669B2 (en) * | 2001-06-13 | 2006-03-07 | Seiko Epson Corporation | Ceramic and method of manufacturing the same, dielectric capacitor, semiconductor device, and element |
| JP4051567B2 (ja) | 2001-09-05 | 2008-02-27 | セイコーエプソン株式会社 | 強誘電体メモリ装置 |
| KR100723399B1 (ko) * | 2002-08-06 | 2007-05-30 | 삼성전자주식회사 | 비스무트 티타늄 실리콘 산화물, 비스무트 티타늄 실리콘산화물 박막 및 그 제조방법 |
| CN1983464B (zh) * | 2002-10-24 | 2010-12-08 | 精工爱普生株式会社 | 强电介质膜、强电介质电容器、强电介质存储器、压电元件、半导体元件 |
| JP3791614B2 (ja) | 2002-10-24 | 2006-06-28 | セイコーエプソン株式会社 | 強誘電体膜、強誘電体メモリ装置、圧電素子、半導体素子、圧電アクチュエータ、液体噴射ヘッド及びプリンタ |
| JP4601896B2 (ja) * | 2002-10-30 | 2010-12-22 | 富士通セミコンダクター株式会社 | 半導体装置及びその製造方法 |
| US6953768B2 (en) * | 2002-11-26 | 2005-10-11 | Teck Cominco Metals Ltd. | Multi-component catalyst system for the polycondensation manufacture of polyesters |
| JP4609621B2 (ja) * | 2002-12-24 | 2011-01-12 | セイコーエプソン株式会社 | 強誘電体キャパシタの製造方法 |
| KR100513724B1 (ko) * | 2002-12-24 | 2005-09-08 | 삼성전자주식회사 | 강유전성 박막 및 그 제조방법 |
| KR20040070564A (ko) * | 2003-02-04 | 2004-08-11 | 삼성전자주식회사 | 강유전체 커패시터 및 그 제조방법 |
| JP2004311924A (ja) * | 2003-03-26 | 2004-11-04 | Seiko Epson Corp | 強誘電体キャパシタおよびその製造方法、強誘電体メモリ、圧電素子。 |
| JP4720969B2 (ja) * | 2003-03-28 | 2011-07-13 | セイコーエプソン株式会社 | 強誘電体膜、圧電体膜、強誘電体メモリ及び圧電素子 |
| JP4572361B2 (ja) * | 2003-03-28 | 2010-11-04 | セイコーエプソン株式会社 | 強誘電体膜の製造方法、強誘電体キャパシタおよびその製造方法、強誘電体メモリならびに圧電素子 |
| EP1624468A1 (en) * | 2003-04-30 | 2006-02-08 | Asahi Glass Company Ltd. | Liquid composition for forming ferroelectric thin film and method for forming ferroelectric thin film |
| KR20060095876A (ko) * | 2003-07-28 | 2006-09-04 | 아사히 가라스 가부시키가이샤 | 강유전체 박막 형성용 액상 조성물 및 강유전체 박막의제조방법 |
| JP2005255468A (ja) * | 2004-03-11 | 2005-09-22 | Tokyo Ohka Kogyo Co Ltd | 常誘電性あるいは強誘電性のBi系誘電体薄膜形成用塗布液、およびBi系誘電体薄膜 |
| US7208401B2 (en) * | 2004-03-12 | 2007-04-24 | Hewlett-Packard Development Company, L.P. | Method for forming a thin film |
| JP4968654B2 (ja) * | 2004-03-29 | 2012-07-04 | シャープ株式会社 | 酸化物材料、強誘電体材料及びそれを用いた電子デバイス |
| KR100655894B1 (ko) * | 2004-05-06 | 2006-12-08 | 서울옵토디바이스주식회사 | 색온도 및 연색성이 우수한 파장변환 발광장치 |
| KR100658700B1 (ko) | 2004-05-13 | 2006-12-15 | 서울옵토디바이스주식회사 | Rgb 발광소자와 형광체를 조합한 발광장치 |
| KR100665299B1 (ko) * | 2004-06-10 | 2007-01-04 | 서울반도체 주식회사 | 발광물질 |
| US8308980B2 (en) * | 2004-06-10 | 2012-11-13 | Seoul Semiconductor Co., Ltd. | Light emitting device |
| KR100665298B1 (ko) * | 2004-06-10 | 2007-01-04 | 서울반도체 주식회사 | 발광장치 |
| AU2005286166B2 (en) * | 2004-09-23 | 2012-01-12 | Element Six (Pty) Ltd | Coated abrasive materials and method of manufacture |
| US7544574B2 (en) * | 2005-10-11 | 2009-06-09 | Intermolecular, Inc. | Methods for discretized processing of regions of a substrate |
| KR100729113B1 (ko) * | 2005-10-17 | 2007-06-14 | 주식회사 엘지화학 | 신규 스트론튬 란타늄 이트륨 함유 복합 산화물 및 이를이용한 이온 전도체 |
| TWI340126B (en) | 2005-10-19 | 2011-04-11 | Lg Chemical Ltd | Composite oxides comprising strontium, lantanium, tungsten and ionic conductors using the same |
| KR101258397B1 (ko) * | 2005-11-11 | 2013-04-30 | 서울반도체 주식회사 | 구리 알칼리토 실리케이트 혼성 결정 형광체 |
| JP2007145672A (ja) * | 2005-11-29 | 2007-06-14 | Seiko Epson Corp | 複合金属酸化物用原料組成物 |
| JP5156188B2 (ja) * | 2005-12-14 | 2013-03-06 | 公益財団法人国際超電導産業技術研究センター | 厚膜テープ状re系(123)超電導体の製造方法 |
| KR101055772B1 (ko) * | 2005-12-15 | 2011-08-11 | 서울반도체 주식회사 | 발광장치 |
| KR100875443B1 (ko) | 2006-03-31 | 2008-12-23 | 서울반도체 주식회사 | 발광 장치 |
| KR101258227B1 (ko) * | 2006-08-29 | 2013-04-25 | 서울반도체 주식회사 | 발광 소자 |
| JP5253895B2 (ja) * | 2007-06-08 | 2013-07-31 | 富士フイルム株式会社 | 強誘電体膜、圧電素子、及び液体吐出装置 |
| US20080302658A1 (en) * | 2007-06-08 | 2008-12-11 | Tsutomu Sasaki | Oxide body, piezoelectric device, and liquid discharge device |
| WO2009025469A2 (en) * | 2007-08-22 | 2009-02-26 | Seoul Semiconductor Co., Ltd. | Non stoichiometric tetragonal copper alkaline earth silicate phosphors and method of preparing the same |
| KR101055769B1 (ko) * | 2007-08-28 | 2011-08-11 | 서울반도체 주식회사 | 비화학양론적 정방정계 알칼리 토류 실리케이트 형광체를채택한 발광 장치 |
| JP5355148B2 (ja) * | 2008-03-19 | 2013-11-27 | キヤノン株式会社 | 圧電材料 |
| DE102009030205A1 (de) * | 2009-06-24 | 2010-12-30 | Litec-Lp Gmbh | Leuchtstoffe mit Eu(II)-dotierten silikatischen Luminophore |
| KR101055762B1 (ko) * | 2009-09-01 | 2011-08-11 | 서울반도체 주식회사 | 옥시오소실리케이트 발광체를 갖는 발광 물질을 채택한 발광 장치 |
| EP2448658B1 (en) | 2009-06-26 | 2014-10-01 | BL Technologies, Inc. | Non-braided, textile-reinforced hollow fiber membrane |
| CN102139231B (zh) * | 2010-02-02 | 2013-05-01 | 中国石油化工股份有限公司 | 一种氧化硅改性的二氧化钛成型载体的制备方法 |
| JP5754619B2 (ja) * | 2010-03-02 | 2015-07-29 | セイコーエプソン株式会社 | 液体噴射ヘッド、液体噴射装置、圧電素子、超音波センサー及び赤外センサー |
| CN101792312A (zh) * | 2010-03-10 | 2010-08-04 | 天津大学 | SrTiO3陶瓷电介质材料及其电容器的制备方法 |
| CN102858691B (zh) * | 2010-04-28 | 2015-09-23 | 株式会社村田制作所 | 具有阴离子控制的介电性质的钙钛矿材料、薄膜电容器器件及其制造方法 |
| US8187705B2 (en) * | 2010-07-15 | 2012-05-29 | Silberline Manufacturing Company, Inc. | Manganese vanadium tantalum oxide and pigments having a black metallic effect coated with the same |
| JP5841156B2 (ja) * | 2011-09-13 | 2016-01-13 | 東芝三菱電機産業システム株式会社 | 酸化膜成膜方法および酸化膜成膜装置 |
| US9321014B2 (en) | 2011-12-16 | 2016-04-26 | Bl Technologies, Inc. | Hollow fiber membrane with compatible reinforcements |
| US9643129B2 (en) | 2011-12-22 | 2017-05-09 | Bl Technologies, Inc. | Non-braided, textile-reinforced hollow fiber membrane |
| CN103289689B (zh) * | 2012-02-28 | 2015-07-08 | 海洋王照明科技股份有限公司 | 锰铬共掺杂氧化锆发光材料、制备方法及其应用 |
| US9217721B2 (en) * | 2012-02-29 | 2015-12-22 | Ohio State Innovation Foundation | No sensor and sensor systems |
| CN103421498B (zh) * | 2012-05-14 | 2016-04-13 | 海洋王照明科技股份有限公司 | 镱钕共掺杂铋酸盐玻璃上转换发光材料、制备方法及应用 |
| JP6067524B2 (ja) | 2013-09-25 | 2017-01-25 | 株式会社東芝 | 半導体装置および誘電体膜 |
| US10115456B2 (en) | 2016-01-14 | 2018-10-30 | Council Of Scientific & Industrial Research | Multi-states nonvolatile opto-ferroelectric memory material and process for preparing the same thereof |
| KR102600473B1 (ko) * | 2016-06-09 | 2023-11-13 | 삼성디스플레이 주식회사 | 조명장치 |
| US10378123B2 (en) | 2016-10-31 | 2019-08-13 | Quest Integrated, Llc | Single-crystal perovskite solid solutions with indifferent points for epitaxial growth of single crystals |
| JP6956189B2 (ja) * | 2017-01-25 | 2021-11-02 | 中国科学院上海光学精密机械研究所 | ドープされた酸化ガリウム結晶材料、その製造方法及び使用 |
| WO2018232303A1 (en) * | 2017-06-16 | 2018-12-20 | Mcpeak Kevin Michael | Metal-semiconductor-metal plasmonic device and absorber and method for making the same |
| CN107935590B (zh) * | 2017-12-08 | 2021-02-05 | 安阳工学院 | 微波烧结制备Aurivillius相SrBiFeCoTiO材料的方法及制备的产品 |
| CN108465475A (zh) * | 2018-04-04 | 2018-08-31 | 东莞市石鼓污水处理有限公司 | 一种WO3-ZrO2光催化污水处理复合膜的制备方法 |
| CN108706971B (zh) * | 2018-06-26 | 2021-01-05 | 桂林电子科技大学 | 一种具有大压电应变记忆特性的无铅铁电陶瓷材料及其制备方法 |
| CN109745992B (zh) * | 2018-12-04 | 2021-06-25 | 信阳师范学院 | 一种高光催化活性单相铁电纳米材料及其制备方法 |
| CN109652823B (zh) * | 2018-12-27 | 2020-10-16 | 景德镇陶瓷大学 | 一种高性能质子导体陶瓷膜反应器电解池阳极材料 |
| JP7230579B2 (ja) * | 2019-02-21 | 2023-03-01 | 三菱マテリアル株式会社 | 強誘電体膜の製造方法 |
| CN109904498B (zh) * | 2019-02-28 | 2021-03-23 | 武汉理工大学 | 一种用于低温固体氧化物燃料电池的矿物材料电解质 |
| CN110817860A (zh) * | 2019-11-14 | 2020-02-21 | 中国科学院青岛生物能源与过程研究所 | 一种二氧化锡/石墨炔复合物界面层及其制备和应用 |
| CN110690021B (zh) * | 2019-11-18 | 2020-11-06 | 国网湖南省电力有限公司 | 一种掺杂钙钛矿型镧锰氧化物防冰材料及其制备方法、应用 |
| KR102346432B1 (ko) * | 2020-06-01 | 2021-12-31 | 동의대학교 산학협력단 | 강유전체를 이용하여 충전 효율을 개선한 디지털 콘덴서의 구조 및 제조방법 |
| CN113371760A (zh) * | 2020-06-29 | 2021-09-10 | 贵州大学 | 类钙钛矿结构铁酸铋材料及其制备方法和应用 |
| CN114574204B (zh) * | 2022-03-30 | 2023-05-02 | 中国科学院长春光学精密机械与物理研究所 | 一种近紫外激发的led用红色荧光粉及制备方法 |
| CN115155603A (zh) * | 2022-08-04 | 2022-10-11 | 中国石油大学(北京) | 一种双金属元素共掺杂的镧基钙钛矿氧化物催化剂及其制备方法和应用 |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1167235A (en) * | 1966-09-14 | 1969-10-15 | Ca Atomic Energy Ltd | Ferroelectric Semiconductors |
| GB1331551A (en) | 1971-04-19 | 1973-09-26 | Matsushita Electric Ind Co Ltd | Dielectric ceramic composition |
| JPS547070B2 (zh) * | 1973-07-30 | 1979-04-03 | ||
| GB2097778B (en) * | 1981-05-06 | 1984-11-21 | Toyoda Chuo Kenkyusho Kk | Barium titanate composition |
| JPS58212102A (ja) * | 1982-06-03 | 1983-12-09 | 株式会社デンソー | 正特性磁器半導体の製造方法 |
| DE3234224A1 (de) * | 1982-09-15 | 1984-03-15 | Siemens AG, 1000 Berlin und 8000 München | Dielektrische keramikmasse |
| US4863883A (en) * | 1986-05-05 | 1989-09-05 | Cabot Corporation | Doped BaTiO3 based compositions |
| IT1270828B (it) | 1993-09-03 | 1997-05-13 | Chon Int Co Ltd | Processo per la sintesi di polveri ceramiche cristalline di composti di perovskite |
| JP2642876B2 (ja) | 1994-08-11 | 1997-08-20 | 工業技術院長 | チタン酸鉛系誘電体薄膜 |
| JP3129175B2 (ja) | 1995-11-27 | 2001-01-29 | 三菱マテリアル株式会社 | (Ba,Sr)TiO3薄膜コンデンサの製造方法 |
| JP3681844B2 (ja) | 1995-11-30 | 2005-08-10 | 京セラ株式会社 | 誘電体薄膜及びセラミックコンデンサ |
| CA2281123A1 (en) * | 1998-09-03 | 2000-03-03 | Gaz De France | Thermally stable, highly active perovskite catalysts for complete oxidation at high temperatures, and the process for their preparation |
| US6495878B1 (en) * | 1999-08-02 | 2002-12-17 | Symetrix Corporation | Interlayer oxide containing thin films for high dielectric constant application |
| TW200306664A (en) * | 2002-02-12 | 2003-11-16 | Matsushita Electric Ind Co Ltd | Ferroelectric capacitor device |
| JP2005159308A (ja) * | 2003-11-05 | 2005-06-16 | Seiko Epson Corp | 強誘電体膜、強誘電体キャパシタ、および強誘電体メモリ |
-
2001
- 2001-09-03 EP EP01961325A patent/EP1338555A4/en not_active Ceased
- 2001-09-03 US US10/399,576 patent/US7205256B2/en not_active Expired - Fee Related
- 2001-09-03 JP JP2002536000A patent/JP4210743B2/ja not_active Expired - Fee Related
- 2001-09-03 CN CNB018174132A patent/CN1266032C/zh not_active Expired - Fee Related
- 2001-09-03 KR KR1020037005316A patent/KR100570576B1/ko not_active IP Right Cessation
- 2001-09-03 WO PCT/JP2001/007619 patent/WO2002032809A1/ja not_active Application Discontinuation
- 2001-09-03 TW TW090121793A patent/TWI290907B/zh not_active IP Right Cessation
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106006710A (zh) * | 2016-05-24 | 2016-10-12 | 合肥工业大学 | 一种β-NaYF4:Yb/Tm@ZnO核壳纳米颗粒的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN1469842A (zh) | 2004-01-21 |
| WO2002032809A1 (fr) | 2002-04-25 |
| US7205256B2 (en) | 2007-04-17 |
| KR100570576B1 (ko) | 2006-04-13 |
| JP4210743B2 (ja) | 2009-01-21 |
| EP1338555A1 (en) | 2003-08-27 |
| TWI290907B (en) | 2007-12-11 |
| JPWO2002032809A1 (ja) | 2004-09-30 |
| US20040136891A1 (en) | 2004-07-15 |
| KR20030055285A (ko) | 2003-07-02 |
| EP1338555A4 (en) | 2004-12-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1266032C (zh) | 氧化物材料、氧化物薄膜的制造方法以及使用该材料的元件 | |
| CN1644496A (zh) | 陶瓷及其制造方法、以及电介质电容器、半导体装置及元件 | |
| CN1296283C (zh) | 陶瓷膜及其制造方法和半导体装置及压电元件 | |
| CN100386291C (zh) | 铌酸钾钠系无铅压电陶瓷及其制备方法 | |
| CN102077376B (zh) | 压电体元件和其制造方法 | |
| CN1179232A (zh) | 具有混合层状超点阵材料的集成电路及用于制备该电路的前体溶液 | |
| CN1929038A (zh) | 复合氧化物层压体、复合氧化物层压体的制造方法、装置 | |
| CN1807346A (zh) | 前驱体组合物及其制造方法、喷墨涂布用墨液 | |
| CN1873926A (zh) | 铁电体层的制造方法及电子设备的制造方法 | |
| CN1269215C (zh) | 铁电存储装置及其制造方法 | |
| CN1165380A (zh) | 铁电元件及其制备方法 | |
| CN1645617A (zh) | 铁电体膜、铁电存储器、以及压电元件 | |
| CN1797771A (zh) | 铁电膜、铁电膜的制造方法、铁电电容器、以及铁电存储器 | |
| CN101714579B (zh) | 铁电体电容器、铁电体电容器的制造方法、铁电体存储器 | |
| Li et al. | Ferroelectric and piezoelectric properties of La-modified lead-free (Bi0. 5Na0. 5) TiO3–(Bi0. 5K0. 5) TiO3–SrTiO3 thin films | |
| CN1076850C (zh) | 强电介质薄膜及其制造方法 | |
| JPH07252664A (ja) | ゾルーゲル法による強誘電体膜の形成方法、キャパシタの製造方法、その原料溶液の調製方法及びその原料溶液 | |
| CN1924083A (zh) | 绝缘性靶材及其制造方法、绝缘性复合氧化膜及装置 | |
| CN1803584A (zh) | 氧化物材料、其制造方法以及使用该氧化物材料的半导体元件 | |
| CN1830811A (zh) | Bi类电介质薄膜形成用涂布液以及Bi类电介质薄膜 | |
| CN100346464C (zh) | 陶瓷膜及其制造方法、强电介质电容器及其制造方法 | |
| CN1311541C (zh) | 陶瓷膜及其制造方法、强电介质电容器及其制造方法 | |
| CN1921112A (zh) | 电介质膜电容器及其制造方法 | |
| CN1990419A (zh) | 复合金属氧化物用原料组合物 | |
| Yu et al. | Preparation, structure, and properties of 0.3 Pb (Zn1/3Nb2/3) O3-0.7 PbTiO3 thin films on LaNiO3/YSZ/Si substrates |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20060726 Termination date: 20150903 |
|
| EXPY | Termination of patent right or utility model |