CN112996497A - 补体因子d抑制剂的形态形式 - Google Patents
补体因子d抑制剂的形态形式 Download PDFInfo
- Publication number
- CN112996497A CN112996497A CN201980074140.4A CN201980074140A CN112996497A CN 112996497 A CN112996497 A CN 112996497A CN 201980074140 A CN201980074140 A CN 201980074140A CN 112996497 A CN112996497 A CN 112996497A
- Authority
- CN
- China
- Prior art keywords
- compound
- crystalline form
- isolated crystalline
- xrpd
- isolated
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 102000003706 Complement factor D Human genes 0.000 title claims description 43
- 108090000059 Complement factor D Proteins 0.000 title claims description 43
- 239000003112 inhibitor Substances 0.000 title description 114
- 229940126214 compound 3 Drugs 0.000 claims abstract description 306
- 238000000034 method Methods 0.000 claims description 163
- 239000007787 solid Substances 0.000 claims description 84
- 239000003814 drug Substances 0.000 claims description 79
- 239000002904 solvent Substances 0.000 claims description 68
- 230000001404 mediated effect Effects 0.000 claims description 54
- 150000001875 compounds Chemical class 0.000 claims description 51
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 39
- 239000008194 pharmaceutical composition Substances 0.000 claims description 34
- 230000008569 process Effects 0.000 claims description 19
- 238000004519 manufacturing process Methods 0.000 claims description 17
- 238000001694 spray drying Methods 0.000 claims description 6
- 150000001413 amino acids Chemical class 0.000 claims description 5
- 238000001144 powder X-ray diffraction data Methods 0.000 claims 21
- 230000001225 therapeutic effect Effects 0.000 abstract description 19
- 229940076722 Complement factor D inhibitor Drugs 0.000 abstract description 2
- 238000000634 powder X-ray diffraction Methods 0.000 description 199
- 239000000203 mixture Substances 0.000 description 157
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 102
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 98
- 239000000543 intermediate Substances 0.000 description 85
- 239000000463 material Substances 0.000 description 85
- 208000035475 disorder Diseases 0.000 description 82
- -1 2-methyl THF Chemical compound 0.000 description 74
- 239000000523 sample Substances 0.000 description 71
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 68
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 66
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 58
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 57
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 52
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 51
- 239000000243 solution Substances 0.000 description 49
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 43
- 238000006243 chemical reaction Methods 0.000 description 42
- 238000004458 analytical method Methods 0.000 description 37
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 36
- 238000002474 experimental method Methods 0.000 description 33
- 239000003153 chemical reaction reagent Substances 0.000 description 32
- 208000000733 Paroxysmal Hemoglobinuria Diseases 0.000 description 31
- 102100036050 Phosphatidylinositol N-acetylglucosaminyltransferase subunit A Human genes 0.000 description 31
- 201000003045 paroxysmal nocturnal hemoglobinuria Diseases 0.000 description 31
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 30
- 238000000113 differential scanning calorimetry Methods 0.000 description 30
- 208000002780 macular degeneration Diseases 0.000 description 28
- 230000000877 morphologic effect Effects 0.000 description 27
- 230000024203 complement activation Effects 0.000 description 26
- 230000000295 complement effect Effects 0.000 description 26
- 229960002224 eculizumab Drugs 0.000 description 26
- 206010064930 age-related macular degeneration Diseases 0.000 description 25
- 238000001907 polarising light microscopy Methods 0.000 description 25
- 239000000047 product Substances 0.000 description 24
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 23
- 230000001351 cycling effect Effects 0.000 description 23
- 239000008213 purified water Substances 0.000 description 22
- 229960005486 vaccine Drugs 0.000 description 22
- 230000015572 biosynthetic process Effects 0.000 description 21
- 230000008859 change Effects 0.000 description 20
- 229940125904 compound 1 Drugs 0.000 description 20
- 239000002002 slurry Substances 0.000 description 20
- 201000010099 disease Diseases 0.000 description 19
- 206010025135 lupus erythematosus Diseases 0.000 description 19
- 239000012296 anti-solvent Substances 0.000 description 18
- 235000019439 ethyl acetate Nutrition 0.000 description 18
- 238000004128 high performance liquid chromatography Methods 0.000 description 18
- 238000001179 sorption measurement Methods 0.000 description 18
- 239000003795 chemical substances by application Substances 0.000 description 17
- 238000005859 coupling reaction Methods 0.000 description 17
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 17
- 208000035913 Atypical hemolytic uremic syndrome Diseases 0.000 description 16
- 229940079593 drug Drugs 0.000 description 16
- 238000001704 evaporation Methods 0.000 description 16
- 230000008020 evaporation Effects 0.000 description 16
- 239000010410 layer Substances 0.000 description 16
- RDTRHBCZFDCUPW-KWICJJCGSA-N 2-[(4r,7s,10s,13s,19s,22s,25s,28s,31s,34r)-4-[[(2s,3r)-1-amino-3-hydroxy-1-oxobutan-2-yl]carbamoyl]-34-[[(2s,3s)-2-amino-3-methylpentanoyl]amino]-25-(3-amino-3-oxopropyl)-7-[3-(diaminomethylideneamino)propyl]-10,13-bis(1h-imidazol-5-ylmethyl)-19-(1h-indol Chemical compound C([C@H]1C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CSSC[C@@H](C(N[C@H](C(=O)N[C@H](C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)NCC(=O)N[C@@H](CC=2NC=NC=2)C(=O)N1)C(C)C)C(C)C)=O)NC(=O)[C@@H](N)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(N)=O)C1=CN=CN1 RDTRHBCZFDCUPW-KWICJJCGSA-N 0.000 description 15
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 15
- 230000008878 coupling Effects 0.000 description 15
- 238000010168 coupling process Methods 0.000 description 15
- 239000002552 dosage form Substances 0.000 description 15
- 230000037361 pathway Effects 0.000 description 15
- 239000012047 saturated solution Substances 0.000 description 15
- 208000023275 Autoimmune disease Diseases 0.000 description 14
- 208000035143 Bacterial infection Diseases 0.000 description 14
- 102000004127 Cytokines Human genes 0.000 description 14
- 108090000695 Cytokines Proteins 0.000 description 14
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 14
- 150000001408 amides Chemical class 0.000 description 14
- 208000022362 bacterial infectious disease Diseases 0.000 description 14
- 229940125782 compound 2 Drugs 0.000 description 14
- 239000012065 filter cake Substances 0.000 description 14
- 238000001914 filtration Methods 0.000 description 14
- 238000010922 spray-dried dispersion Methods 0.000 description 14
- 241000606768 Haemophilus influenzae Species 0.000 description 13
- 230000000694 effects Effects 0.000 description 13
- 238000009472 formulation Methods 0.000 description 13
- 229910052757 nitrogen Inorganic materials 0.000 description 13
- IEDVJHCEMCRBQM-UHFFFAOYSA-N trimethoprim Chemical compound COC1=C(OC)C(OC)=CC(CC=2C(=NC(N)=NC=2)N)=C1 IEDVJHCEMCRBQM-UHFFFAOYSA-N 0.000 description 13
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 12
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 12
- 239000013543 active substance Substances 0.000 description 12
- 230000002860 competitive effect Effects 0.000 description 12
- 238000002425 crystallisation Methods 0.000 description 12
- 230000008025 crystallization Effects 0.000 description 12
- 229940047650 haemophilus influenzae Drugs 0.000 description 12
- 238000003801 milling Methods 0.000 description 12
- 239000012044 organic layer Substances 0.000 description 12
- 230000004044 response Effects 0.000 description 12
- 238000003756 stirring Methods 0.000 description 12
- 238000003786 synthesis reaction Methods 0.000 description 12
- 238000001757 thermogravimetry curve Methods 0.000 description 12
- 210000001519 tissue Anatomy 0.000 description 12
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 11
- 150000002148 esters Chemical class 0.000 description 11
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 11
- 239000002245 particle Substances 0.000 description 11
- 235000002639 sodium chloride Nutrition 0.000 description 11
- 108091023037 Aptamer Proteins 0.000 description 10
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 10
- 208000004451 Membranoproliferative Glomerulonephritis Diseases 0.000 description 10
- 210000004027 cell Anatomy 0.000 description 10
- 238000007906 compression Methods 0.000 description 10
- 230000006835 compression Effects 0.000 description 10
- 238000010438 heat treatment Methods 0.000 description 10
- 230000003902 lesion Effects 0.000 description 10
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 10
- 108090000623 proteins and genes Proteins 0.000 description 10
- 238000000746 purification Methods 0.000 description 10
- 229960001082 trimethoprim Drugs 0.000 description 10
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 9
- 239000003246 corticosteroid Substances 0.000 description 9
- 230000028709 inflammatory response Effects 0.000 description 9
- 238000010898 silica gel chromatography Methods 0.000 description 9
- 229960005404 sulfamethoxazole Drugs 0.000 description 9
- JLKIGFTWXXRPMT-UHFFFAOYSA-N sulphamethoxazole Chemical compound O1C(C)=CC(NS(=O)(=O)C=2C=CC(N)=CC=2)=N1 JLKIGFTWXXRPMT-UHFFFAOYSA-N 0.000 description 9
- 239000003826 tablet Substances 0.000 description 9
- 230000008685 targeting Effects 0.000 description 9
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 8
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- 102100022133 Complement C3 Human genes 0.000 description 8
- ULGZDMOVFRHVEP-RWJQBGPGSA-N Erythromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 ULGZDMOVFRHVEP-RWJQBGPGSA-N 0.000 description 8
- 229910016860 FaSSIF Inorganic materials 0.000 description 8
- 229910005429 FeSSIF Inorganic materials 0.000 description 8
- 208000032759 Hemolytic-Uremic Syndrome Diseases 0.000 description 8
- 239000012317 TBTU Substances 0.000 description 8
- 229910052782 aluminium Inorganic materials 0.000 description 8
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 8
- 230000000052 comparative effect Effects 0.000 description 8
- 230000004154 complement system Effects 0.000 description 8
- 230000006378 damage Effects 0.000 description 8
- 229910052805 deuterium Inorganic materials 0.000 description 8
- 238000004108 freeze drying Methods 0.000 description 8
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 8
- 201000006417 multiple sclerosis Diseases 0.000 description 8
- 208000008338 non-alcoholic fatty liver disease Diseases 0.000 description 8
- 206010053219 non-alcoholic steatohepatitis Diseases 0.000 description 8
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 8
- 238000001556 precipitation Methods 0.000 description 8
- 230000000069 prophylactic effect Effects 0.000 description 8
- 235000018102 proteins Nutrition 0.000 description 8
- 102000004169 proteins and genes Human genes 0.000 description 8
- 238000002411 thermogravimetry Methods 0.000 description 8
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 7
- 241000588650 Neisseria meningitidis Species 0.000 description 7
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 7
- 239000003242 anti bacterial agent Substances 0.000 description 7
- 230000001363 autoimmune Effects 0.000 description 7
- MQTOSJVFKKJCRP-BICOPXKESA-N azithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)N(C)C[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 MQTOSJVFKKJCRP-BICOPXKESA-N 0.000 description 7
- 230000027455 binding Effects 0.000 description 7
- 230000003115 biocidal effect Effects 0.000 description 7
- 239000002775 capsule Substances 0.000 description 7
- WMEMLXDTLKSUOD-OGCOPIPOSA-N chembl436844 Chemical compound C([C@H]1C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CSSC[C@@H](C(N[C@H](C(=O)N[C@@H](CC=2C3=CC=CC=C3N(C)C=2)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)NCC(=O)N[C@@H](C)C(=O)N1)C(C)C)=O)NC(=O)[C@@H](NC(C)=O)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(N)=O)C1=CN=CN1 WMEMLXDTLKSUOD-OGCOPIPOSA-N 0.000 description 7
- 238000001035 drying Methods 0.000 description 7
- 239000000706 filtrate Substances 0.000 description 7
- 239000011521 glass Substances 0.000 description 7
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 7
- 230000002757 inflammatory effect Effects 0.000 description 7
- 238000002844 melting Methods 0.000 description 7
- 230000008018 melting Effects 0.000 description 7
- 238000001000 micrograph Methods 0.000 description 7
- 239000002105 nanoparticle Substances 0.000 description 7
- 239000000843 powder Substances 0.000 description 7
- GPTONYMQFTZPKC-UHFFFAOYSA-N sulfamethoxydiazine Chemical compound N1=CC(OC)=CN=C1NS(=O)(=O)C1=CC=C(N)C=C1 GPTONYMQFTZPKC-UHFFFAOYSA-N 0.000 description 7
- 229960002229 sulfametoxydiazine Drugs 0.000 description 7
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 6
- 208000016323 C3 glomerulonephritis Diseases 0.000 description 6
- 206010061218 Inflammation Diseases 0.000 description 6
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 6
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 6
- 241000193998 Streptococcus pneumoniae Species 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 6
- CLZISMQKJZCZDN-UHFFFAOYSA-N [benzotriazol-1-yloxy(dimethylamino)methylidene]-dimethylazanium Chemical compound C1=CC=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 CLZISMQKJZCZDN-UHFFFAOYSA-N 0.000 description 6
- 239000013060 biological fluid Substances 0.000 description 6
- 239000000969 carrier Substances 0.000 description 6
- 230000015556 catabolic process Effects 0.000 description 6
- 230000001684 chronic effect Effects 0.000 description 6
- 238000006731 degradation reaction Methods 0.000 description 6
- 208000022401 dense deposit disease Diseases 0.000 description 6
- 208000010706 fatty liver disease Diseases 0.000 description 6
- 108020001507 fusion proteins Proteins 0.000 description 6
- 102000037865 fusion proteins Human genes 0.000 description 6
- 239000012535 impurity Substances 0.000 description 6
- 230000001965 increasing effect Effects 0.000 description 6
- 208000027866 inflammatory disease Diseases 0.000 description 6
- 230000004054 inflammatory process Effects 0.000 description 6
- 230000005764 inhibitory process Effects 0.000 description 6
- 239000011859 microparticle Substances 0.000 description 6
- 208000008795 neuromyelitis optica Diseases 0.000 description 6
- 210000000056 organ Anatomy 0.000 description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 6
- 239000011347 resin Substances 0.000 description 6
- 229920005989 resin Polymers 0.000 description 6
- 239000011780 sodium chloride Substances 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M sodium hydroxide Inorganic materials [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 239000007921 spray Substances 0.000 description 6
- 239000007858 starting material Substances 0.000 description 6
- 229940031000 streptococcus pneumoniae Drugs 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 6
- PAQZWJGSJMLPMG-UHFFFAOYSA-N 2,4,6-tripropyl-1,3,5,2$l^{5},4$l^{5},6$l^{5}-trioxatriphosphinane 2,4,6-trioxide Chemical compound CCCP1(=O)OP(=O)(CCC)OP(=O)(CCC)O1 PAQZWJGSJMLPMG-UHFFFAOYSA-N 0.000 description 5
- MUSGYEMSJUFFHT-UWABRSFTSA-N 2-[(4R,7S,10S,13S,19S,22S,25S,28S,31S,34R)-34-[[(2S,3S)-2-[[(2R)-2-amino-3-(4-hydroxyphenyl)propanoyl]amino]-3-methylpentanoyl]amino]-4-[[(2S,3S)-1-amino-3-methyl-1-oxopentan-2-yl]-methylcarbamoyl]-25-(3-amino-3-oxopropyl)-7-(3-carbamimidamidopropyl)-10-(1H-imidazol-5-ylmethyl)-19-(1H-indol-3-ylmethyl)-13,17-dimethyl-28-[(1-methylindol-3-yl)methyl]-6,9,12,15,18,21,24,27,30,33-decaoxo-31-propan-2-yl-1,2-dithia-5,8,11,14,17,20,23,26,29,32-decazacyclopentatriacont-22-yl]acetic acid Chemical compound CC[C@H](C)[C@H](NC(=O)[C@H](N)Cc1ccc(O)cc1)C(=O)N[C@H]1CSSC[C@H](NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](Cc2cnc[nH]2)NC(=O)[C@H](C)NC(=O)CN(C)C(=O)[C@H](Cc2c[nH]c3ccccc23)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](Cc2cn(C)c3ccccc23)NC(=O)[C@@H](NC1=O)C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)C(N)=O MUSGYEMSJUFFHT-UWABRSFTSA-N 0.000 description 5
- VHRSUDSXCMQTMA-PJHHCJLFSA-N 6alpha-methylprednisolone Chemical compound C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2[C@@H](O)C[C@]2(C)[C@@](O)(C(=O)CO)CC[C@H]21 VHRSUDSXCMQTMA-PJHHCJLFSA-N 0.000 description 5
- 208000029574 C3 glomerulopathy Diseases 0.000 description 5
- 108010008165 Etanercept Proteins 0.000 description 5
- 208000004930 Fatty Liver Diseases 0.000 description 5
- 206010019708 Hepatic steatosis Diseases 0.000 description 5
- 239000004201 L-cysteine Substances 0.000 description 5
- 235000013878 L-cysteine Nutrition 0.000 description 5
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 5
- 108020004459 Small interfering RNA Proteins 0.000 description 5
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 5
- 229920002472 Starch Polymers 0.000 description 5
- 210000001744 T-lymphocyte Anatomy 0.000 description 5
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 5
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 5
- 208000027418 Wounds and injury Diseases 0.000 description 5
- 230000004913 activation Effects 0.000 description 5
- 239000008186 active pharmaceutical agent Substances 0.000 description 5
- 108010081667 aflibercept Proteins 0.000 description 5
- 238000013019 agitation Methods 0.000 description 5
- RITAVMQDGBJQJZ-FMIVXFBMSA-N axitinib Chemical compound CNC(=O)C1=CC=CC=C1SC1=CC=C(C(\C=C\C=2N=CC=CC=2)=NN2)C2=C1 RITAVMQDGBJQJZ-FMIVXFBMSA-N 0.000 description 5
- 229960004099 azithromycin Drugs 0.000 description 5
- 210000004369 blood Anatomy 0.000 description 5
- 239000008280 blood Substances 0.000 description 5
- 239000000872 buffer Substances 0.000 description 5
- 238000012512 characterization method Methods 0.000 description 5
- 208000019425 cirrhosis of liver Diseases 0.000 description 5
- 108010027437 compstatin Proteins 0.000 description 5
- 238000001816 cooling Methods 0.000 description 5
- 229960001334 corticosteroids Drugs 0.000 description 5
- 206010012601 diabetes mellitus Diseases 0.000 description 5
- 239000003085 diluting agent Substances 0.000 description 5
- 206010013023 diphtheria Diseases 0.000 description 5
- 238000004090 dissolution Methods 0.000 description 5
- 229960000403 etanercept Drugs 0.000 description 5
- 208000006454 hepatitis Diseases 0.000 description 5
- 208000014674 injury Diseases 0.000 description 5
- 239000000314 lubricant Substances 0.000 description 5
- 210000004379 membrane Anatomy 0.000 description 5
- 239000012528 membrane Substances 0.000 description 5
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 5
- 229960004584 methylprednisolone Drugs 0.000 description 5
- 229950009686 mirococept Drugs 0.000 description 5
- MQQNFDZXWVTQEH-UHFFFAOYSA-N nafamostat Chemical compound C1=CC(N=C(N)N)=CC=C1C(=O)OC1=CC=C(C=C(C=C2)C(N)=N)C2=C1 MQQNFDZXWVTQEH-UHFFFAOYSA-N 0.000 description 5
- 229950009865 nafamostat Drugs 0.000 description 5
- 239000012299 nitrogen atmosphere Substances 0.000 description 5
- 208000027134 non-immunoglobulin-mediated membranoproliferative glomerulonephritis Diseases 0.000 description 5
- 239000000825 pharmaceutical preparation Substances 0.000 description 5
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 230000002265 prevention Effects 0.000 description 5
- 238000001953 recrystallisation Methods 0.000 description 5
- 206010039073 rheumatoid arthritis Diseases 0.000 description 5
- 150000003839 salts Chemical class 0.000 description 5
- 210000003491 skin Anatomy 0.000 description 5
- 235000019698 starch Nutrition 0.000 description 5
- 231100000240 steatosis hepatitis Toxicity 0.000 description 5
- 238000003860 storage Methods 0.000 description 5
- 239000000829 suppository Substances 0.000 description 5
- 230000008646 thermal stress Effects 0.000 description 5
- 102000003390 tumor necrosis factor Human genes 0.000 description 5
- FFTVPQUHLQBXQZ-KVUCHLLUSA-N (4s,4as,5ar,12ar)-4,7-bis(dimethylamino)-1,10,11,12a-tetrahydroxy-3,12-dioxo-4a,5,5a,6-tetrahydro-4h-tetracene-2-carboxamide Chemical compound C1C2=C(N(C)C)C=CC(O)=C2C(O)=C2[C@@H]1C[C@H]1[C@H](N(C)C)C(=O)C(C(N)=O)=C(O)[C@@]1(O)C2=O FFTVPQUHLQBXQZ-KVUCHLLUSA-N 0.000 description 4
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 4
- OJPQVYZLQGRFSO-UHFFFAOYSA-N 4-bromo-2-methylpyrimidine Chemical compound CC1=NC=CC(Br)=N1 OJPQVYZLQGRFSO-UHFFFAOYSA-N 0.000 description 4
- 101100268548 Caenorhabditis elegans apl-1 gene Proteins 0.000 description 4
- 208000011038 Cold agglutinin disease Diseases 0.000 description 4
- 206010009868 Cold type haemolytic anaemia Diseases 0.000 description 4
- 108010028780 Complement C3 Proteins 0.000 description 4
- 102100031506 Complement C5 Human genes 0.000 description 4
- 102100035432 Complement factor H Human genes 0.000 description 4
- GKQLYSROISKDLL-UHFFFAOYSA-N EEDQ Chemical compound C1=CC=C2N(C(=O)OCC)C(OCC)C=CC2=C1 GKQLYSROISKDLL-UHFFFAOYSA-N 0.000 description 4
- 238000005033 Fourier transform infrared spectroscopy Methods 0.000 description 4
- 239000007821 HATU Substances 0.000 description 4
- 208000035186 Hemolytic Autoimmune Anemia Diseases 0.000 description 4
- 206010019663 Hepatic failure Diseases 0.000 description 4
- 102000003814 Interleukin-10 Human genes 0.000 description 4
- 108090000174 Interleukin-10 Proteins 0.000 description 4
- 108090000978 Interleukin-4 Proteins 0.000 description 4
- 102000004388 Interleukin-4 Human genes 0.000 description 4
- 239000002136 L01XE07 - Lapatinib Substances 0.000 description 4
- 239000002176 L01XE26 - Cabozantinib Substances 0.000 description 4
- XNRVGTHNYCNCFF-UHFFFAOYSA-N Lapatinib ditosylate monohydrate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1.CC1=CC=C(S(O)(=O)=O)C=C1.O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 XNRVGTHNYCNCFF-UHFFFAOYSA-N 0.000 description 4
- 208000005777 Lupus Nephritis Diseases 0.000 description 4
- KSPIYJQBLVDRRI-UHFFFAOYSA-N N-methylisoleucine Chemical compound CCC(C)C(NC)C(O)=O KSPIYJQBLVDRRI-UHFFFAOYSA-N 0.000 description 4
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 4
- NCDNCNXCDXHOMX-UHFFFAOYSA-N Ritonavir Natural products C=1C=CC=CC=1CC(NC(=O)OCC=1SC=NC=1)C(O)CC(CC=1C=CC=CC=1)NC(=O)C(C(C)C)NC(=O)N(C)CC1=CSC(C(C)C)=N1 NCDNCNXCDXHOMX-UHFFFAOYSA-N 0.000 description 4
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 4
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 4
- 238000010521 absorption reaction Methods 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 230000009471 action Effects 0.000 description 4
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 description 4
- 230000003092 anti-cytokine Effects 0.000 description 4
- 206010003246 arthritis Diseases 0.000 description 4
- 230000008901 benefit Effects 0.000 description 4
- 239000003054 catalyst Substances 0.000 description 4
- 229960002412 cediranib Drugs 0.000 description 4
- 229960003719 cefdinir Drugs 0.000 description 4
- RTXOFQZKPXMALH-GHXIOONMSA-N cefdinir Chemical compound S1C(N)=NC(C(=N\O)\C(=O)N[C@@H]2C(N3C(=C(C=C)CS[C@@H]32)C(O)=O)=O)=C1 RTXOFQZKPXMALH-GHXIOONMSA-N 0.000 description 4
- MYSWGUAQZAJSOK-UHFFFAOYSA-N ciprofloxacin Chemical compound C12=CC(N3CCNCC3)=C(F)C=C2C(=O)C(C(=O)O)=CN1C1CC1 MYSWGUAQZAJSOK-UHFFFAOYSA-N 0.000 description 4
- 230000007882 cirrhosis Effects 0.000 description 4
- 230000007850 degeneration Effects 0.000 description 4
- 201000001981 dermatomyositis Diseases 0.000 description 4
- 239000003937 drug carrier Substances 0.000 description 4
- 229960003276 erythromycin Drugs 0.000 description 4
- AEUTYOVWOVBAKS-UWVGGRQHSA-N ethambutol Chemical compound CC[C@@H](CO)NCCN[C@@H](CC)CO AEUTYOVWOVBAKS-UWVGGRQHSA-N 0.000 description 4
- 239000007789 gas Substances 0.000 description 4
- 238000006703 hydration reaction Methods 0.000 description 4
- 229910052739 hydrogen Inorganic materials 0.000 description 4
- 229960000598 infliximab Drugs 0.000 description 4
- WOSKHXYHFSIKNG-UHFFFAOYSA-N lenvatinib Chemical compound C=12C=C(C(N)=O)C(OC)=CC2=NC=CC=1OC(C=C1Cl)=CC=C1NC(=O)NC1CC1 WOSKHXYHFSIKNG-UHFFFAOYSA-N 0.000 description 4
- 208000007903 liver failure Diseases 0.000 description 4
- 231100000835 liver failure Toxicity 0.000 description 4
- 208000018191 liver inflammation Diseases 0.000 description 4
- 235000019359 magnesium stearate Nutrition 0.000 description 4
- 238000000386 microscopy Methods 0.000 description 4
- FABPRXSRWADJSP-MEDUHNTESA-N moxifloxacin Chemical compound COC1=C(N2C[C@H]3NCCC[C@H]3C2)C(F)=CC(C(C(C(O)=O)=C2)=O)=C1N2C1CC1 FABPRXSRWADJSP-MEDUHNTESA-N 0.000 description 4
- 229910052763 palladium Inorganic materials 0.000 description 4
- 229920001223 polyethylene glycol Polymers 0.000 description 4
- 229960004618 prednisone Drugs 0.000 description 4
- 239000003755 preservative agent Substances 0.000 description 4
- 238000003825 pressing Methods 0.000 description 4
- 108090000765 processed proteins & peptides Proteins 0.000 description 4
- 230000005855 radiation Effects 0.000 description 4
- 229960002633 ramucirumab Drugs 0.000 description 4
- FNHKPVJBJVTLMP-UHFFFAOYSA-N regorafenib Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=C(F)C(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 FNHKPVJBJVTLMP-UHFFFAOYSA-N 0.000 description 4
- WDZCUPBHRAEYDL-GZAUEHORSA-N rifapentine Chemical group O([C@](C1=O)(C)O/C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)\C=C\C=C(C)/C(=O)NC=2C(O)=C3C(O)=C4C)C)OC)C4=C1C3=C(O)C=2\C=N\N(CC1)CCN1C1CCCC1 WDZCUPBHRAEYDL-GZAUEHORSA-N 0.000 description 4
- 229960000311 ritonavir Drugs 0.000 description 4
- NCDNCNXCDXHOMX-XGKFQTDJSA-N ritonavir Chemical compound N([C@@H](C(C)C)C(=O)N[C@H](C[C@H](O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1SC=NC=1)CC=1C=CC=CC=1)C(=O)N(C)CC1=CSC(C(C)C)=N1 NCDNCNXCDXHOMX-XGKFQTDJSA-N 0.000 description 4
- FSYKKLYZXJSNPZ-UHFFFAOYSA-N sarcosine Chemical group C[NH2+]CC([O-])=O FSYKKLYZXJSNPZ-UHFFFAOYSA-N 0.000 description 4
- 239000000741 silica gel Substances 0.000 description 4
- 229910002027 silica gel Inorganic materials 0.000 description 4
- 239000010703 silicon Substances 0.000 description 4
- 229910052710 silicon Inorganic materials 0.000 description 4
- 150000003384 small molecules Chemical group 0.000 description 4
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 4
- 239000008107 starch Substances 0.000 description 4
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 4
- 229960001796 sunitinib Drugs 0.000 description 4
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 4
- 230000009885 systemic effect Effects 0.000 description 4
- 229940124597 therapeutic agent Drugs 0.000 description 4
- 238000002560 therapeutic procedure Methods 0.000 description 4
- 230000007704 transition Effects 0.000 description 4
- GUXHBMASAHGULD-SEYHBJAFSA-N (4s,4as,5as,6s,12ar)-7-chloro-4-(dimethylamino)-1,6,10,11,12a-pentahydroxy-3,12-dioxo-4a,5,5a,6-tetrahydro-4h-tetracene-2-carboxamide Chemical compound C1([C@H]2O)=C(Cl)C=CC(O)=C1C(O)=C1[C@@H]2C[C@H]2[C@H](N(C)C)C(=O)C(C(N)=O)=C(O)[C@@]2(O)C1=O GUXHBMASAHGULD-SEYHBJAFSA-N 0.000 description 3
- HMLGSIZOMSVISS-ONJSNURVSA-N (7r)-7-[[(2z)-2-(2-amino-1,3-thiazol-4-yl)-2-(2,2-dimethylpropanoyloxymethoxyimino)acetyl]amino]-3-ethenyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid Chemical compound N([C@@H]1C(N2C(=C(C=C)CSC21)C(O)=O)=O)C(=O)\C(=N/OCOC(=O)C(C)(C)C)C1=CSC(N)=N1 HMLGSIZOMSVISS-ONJSNURVSA-N 0.000 description 3
- HOPFEUJFXKXWNN-UHFFFAOYSA-N 1-(5-bromo-1h-indazol-3-yl)ethanone Chemical compound C1=C(Br)C=C2C(C(=O)C)=NNC2=C1 HOPFEUJFXKXWNN-UHFFFAOYSA-N 0.000 description 3
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 3
- ZADWXFSZEAPBJS-JTQLQIEISA-N 1-methyl-L-tryptophan Chemical group C1=CC=C2N(C)C=C(C[C@H](N)C(O)=O)C2=C1 ZADWXFSZEAPBJS-JTQLQIEISA-N 0.000 description 3
- YOETUEMZNOLGDB-UHFFFAOYSA-N 2-methylpropyl carbonochloridate Chemical compound CC(C)COC(Cl)=O YOETUEMZNOLGDB-UHFFFAOYSA-N 0.000 description 3
- 108010003529 Alternative Pathway Complement C3 Convertase Proteins 0.000 description 3
- 241000894006 Bacteria Species 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- 201000004569 Blindness Diseases 0.000 description 3
- 241000282465 Canis Species 0.000 description 3
- 208000024172 Cardiovascular disease Diseases 0.000 description 3
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 description 3
- 108010067641 Complement C3-C5 Convertases Proteins 0.000 description 3
- 102000016574 Complement C3-C5 Convertases Human genes 0.000 description 3
- 102000016550 Complement Factor H Human genes 0.000 description 3
- 108010053085 Complement Factor H Proteins 0.000 description 3
- 102100030886 Complement receptor type 1 Human genes 0.000 description 3
- 229910017488 Cu K Inorganic materials 0.000 description 3
- 208000016192 Demyelinating disease Diseases 0.000 description 3
- 206010012689 Diabetic retinopathy Diseases 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- 206010018370 Glomerulonephritis membranoproliferative Diseases 0.000 description 3
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 3
- 102000051628 Interleukin-1 receptor antagonist Human genes 0.000 description 3
- 108700021006 Interleukin-1 receptor antagonist Proteins 0.000 description 3
- 108010065805 Interleukin-12 Proteins 0.000 description 3
- 102000013462 Interleukin-12 Human genes 0.000 description 3
- 108010002350 Interleukin-2 Proteins 0.000 description 3
- 102000000588 Interleukin-2 Human genes 0.000 description 3
- 108010065637 Interleukin-23 Proteins 0.000 description 3
- 102000013264 Interleukin-23 Human genes 0.000 description 3
- 239000003798 L01XE11 - Pazopanib Substances 0.000 description 3
- 239000002137 L01XE24 - Ponatinib Substances 0.000 description 3
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 3
- 208000016604 Lyme disease Diseases 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- 206010063837 Reperfusion injury Diseases 0.000 description 3
- RAHZWNYVWXNFOC-UHFFFAOYSA-N Sulphur dioxide Chemical compound O=S=O RAHZWNYVWXNFOC-UHFFFAOYSA-N 0.000 description 3
- 238000006069 Suzuki reaction reaction Methods 0.000 description 3
- 206010052779 Transplant rejections Diseases 0.000 description 3
- 206010046851 Uveitis Diseases 0.000 description 3
- 108010059993 Vancomycin Proteins 0.000 description 3
- 206010047115 Vasculitis Diseases 0.000 description 3
- 229960002964 adalimumab Drugs 0.000 description 3
- 239000000654 additive Substances 0.000 description 3
- AVKUERGKIZMTKX-NJBDSQKTSA-N ampicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 AVKUERGKIZMTKX-NJBDSQKTSA-N 0.000 description 3
- 230000003321 amplification Effects 0.000 description 3
- 229960004238 anakinra Drugs 0.000 description 3
- 230000000844 anti-bacterial effect Effects 0.000 description 3
- 239000002260 anti-inflammatory agent Substances 0.000 description 3
- 230000003110 anti-inflammatory effect Effects 0.000 description 3
- 230000000845 anti-microbial effect Effects 0.000 description 3
- 208000006673 asthma Diseases 0.000 description 3
- 229960003005 axitinib Drugs 0.000 description 3
- 239000011324 bead Substances 0.000 description 3
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 3
- 229960000397 bevacizumab Drugs 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- AFZFFLVORLEPPO-UVYJNCLZSA-N cefditoren pivoxil Chemical compound S([C@@H]1[C@@H](C(N1C=1C(=O)OCOC(=O)C(C)(C)C)=O)NC(=O)\C(=N/OC)C=2N=C(N)SC=2)CC=1\C=C/C=1SC=NC=1C AFZFFLVORLEPPO-UVYJNCLZSA-N 0.000 description 3
- AZZMGZXNTDTSME-JUZDKLSSSA-M cefotaxime sodium Chemical compound [Na+].N([C@@H]1C(N2C(=C(COC(C)=O)CS[C@@H]21)C([O-])=O)=O)C(=O)\C(=N/OC)C1=CSC(N)=N1 AZZMGZXNTDTSME-JUZDKLSSSA-M 0.000 description 3
- 239000003610 charcoal Substances 0.000 description 3
- 239000000460 chlorine Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- AGOYDEPGAOXOCK-KCBOHYOISA-N clarithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@](C)([C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)OC)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 AGOYDEPGAOXOCK-KCBOHYOISA-N 0.000 description 3
- KDLRVYVGXIQJDK-AWPVFWJPSA-N clindamycin Chemical compound CN1C[C@H](CCC)C[C@H]1C(=O)N[C@H]([C@H](C)Cl)[C@@H]1[C@H](O)[C@H](O)[C@@H](O)[C@@H](SC)O1 KDLRVYVGXIQJDK-AWPVFWJPSA-N 0.000 description 3
- 238000002648 combination therapy Methods 0.000 description 3
- 239000002178 crystalline material Substances 0.000 description 3
- 229910000071 diazene Inorganic materials 0.000 description 3
- 238000011038 discontinuous diafiltration by volume reduction Methods 0.000 description 3
- 239000006185 dispersion Substances 0.000 description 3
- 230000002349 favourable effect Effects 0.000 description 3
- 229910052731 fluorine Inorganic materials 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 229920000159 gelatin Polymers 0.000 description 3
- 235000019322 gelatine Nutrition 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 230000002068 genetic effect Effects 0.000 description 3
- 239000008103 glucose Substances 0.000 description 3
- 229960001743 golimumab Drugs 0.000 description 3
- 238000000227 grinding Methods 0.000 description 3
- 238000001631 haemodialysis Methods 0.000 description 3
- 230000000322 hemodialysis Effects 0.000 description 3
- 230000036571 hydration Effects 0.000 description 3
- XXSMGPRMXLTPCZ-UHFFFAOYSA-N hydroxychloroquine Chemical compound ClC1=CC=C2C(NC(C)CCCN(CCO)CC)=CC=NC2=C1 XXSMGPRMXLTPCZ-UHFFFAOYSA-N 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 230000028993 immune response Effects 0.000 description 3
- 208000017169 kidney disease Diseases 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- DMJNNHOOLUXYBV-PQTSNVLCSA-N meropenem Chemical compound C=1([C@H](C)[C@@H]2[C@H](C(N2C=1C(O)=O)=O)[C@H](O)C)S[C@@H]1CN[C@H](C(=O)N(C)C)C1 DMJNNHOOLUXYBV-PQTSNVLCSA-N 0.000 description 3
- 229960004023 minocycline Drugs 0.000 description 3
- 229960003702 moxifloxacin Drugs 0.000 description 3
- 206010028417 myasthenia gravis Diseases 0.000 description 3
- 229940015638 narsoplimab Drugs 0.000 description 3
- 201000008383 nephritis Diseases 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 238000003199 nucleic acid amplification method Methods 0.000 description 3
- 229960002450 ofatumumab Drugs 0.000 description 3
- 239000003921 oil Substances 0.000 description 3
- 235000019198 oils Nutrition 0.000 description 3
- 239000003960 organic solvent Substances 0.000 description 3
- 244000052769 pathogen Species 0.000 description 3
- 229960000639 pazopanib Drugs 0.000 description 3
- CUIHSIWYWATEQL-UHFFFAOYSA-N pazopanib Chemical compound C1=CC2=C(C)N(C)N=C2C=C1N(C)C(N=1)=CC=NC=1NC1=CC=C(C)C(S(N)(=O)=O)=C1 CUIHSIWYWATEQL-UHFFFAOYSA-N 0.000 description 3
- 229940056360 penicillin g Drugs 0.000 description 3
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 3
- 239000010452 phosphate Substances 0.000 description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 3
- 239000006187 pill Substances 0.000 description 3
- 229940124733 pneumococcal vaccine Drugs 0.000 description 3
- PHXJVRSECIGDHY-UHFFFAOYSA-N ponatinib Chemical compound C1CN(C)CCN1CC(C(=C1)C(F)(F)F)=CC=C1NC(=O)C1=CC=C(C)C(C#CC=2N3N=CC=CC3=NC=2)=C1 PHXJVRSECIGDHY-UHFFFAOYSA-N 0.000 description 3
- 229960001131 ponatinib Drugs 0.000 description 3
- 229910000027 potassium carbonate Inorganic materials 0.000 description 3
- 201000008171 proliferative glomerulonephritis Diseases 0.000 description 3
- 230000002685 pulmonary effect Effects 0.000 description 3
- 238000010791 quenching Methods 0.000 description 3
- ZADWXFSZEAPBJS-UHFFFAOYSA-N racemic N-methyl tryptophan Natural products C1=CC=C2N(C)C=C(CC(N)C(O)=O)C2=C1 ZADWXFSZEAPBJS-UHFFFAOYSA-N 0.000 description 3
- 208000023504 respiratory system disease Diseases 0.000 description 3
- JQXXHWHPUNPDRT-WLSIYKJHSA-N rifampicin Chemical compound O([C@](C1=O)(C)O/C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)\C=C\C=C(C)/C(=O)NC=2C(O)=C3C([O-])=C4C)C)OC)C4=C1C3=C(O)C=2\C=N\N1CC[NH+](C)CC1 JQXXHWHPUNPDRT-WLSIYKJHSA-N 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 238000010956 selective crystallization Methods 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 3
- 229940055944 soliris Drugs 0.000 description 3
- IVDHYUQIDRJSTI-UHFFFAOYSA-N sorafenib tosylate Chemical compound [H+].CC1=CC=C(S([O-])(=O)=O)C=C1.C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 IVDHYUQIDRJSTI-UHFFFAOYSA-N 0.000 description 3
- SKIVFJLNDNKQPD-UHFFFAOYSA-N sulfacetamide Chemical compound CC(=O)NS(=O)(=O)C1=CC=C(N)C=C1 SKIVFJLNDNKQPD-UHFFFAOYSA-N 0.000 description 3
- SEEPANYCNGTZFQ-UHFFFAOYSA-N sulfadiazine Chemical compound C1=CC(N)=CC=C1S(=O)(=O)NC1=NC=CC=N1 SEEPANYCNGTZFQ-UHFFFAOYSA-N 0.000 description 3
- 208000011580 syndromic disease Diseases 0.000 description 3
- 230000002195 synergetic effect Effects 0.000 description 3
- BNWCETAHAJSBFG-UHFFFAOYSA-N tert-butyl 2-bromoacetate Chemical compound CC(C)(C)OC(=O)CBr BNWCETAHAJSBFG-UHFFFAOYSA-N 0.000 description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- 229950009054 tesidolumab Drugs 0.000 description 3
- MYPYJXKWCTUITO-LYRMYLQWSA-N vancomycin Chemical compound O([C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1OC1=C2C=C3C=C1OC1=CC=C(C=C1Cl)[C@@H](O)[C@H](C(N[C@@H](CC(N)=O)C(=O)N[C@H]3C(=O)N[C@H]1C(=O)N[C@H](C(N[C@@H](C3=CC(O)=CC(O)=C3C=3C(O)=CC=C1C=3)C(O)=O)=O)[C@H](O)C1=CC=C(C(=C1)Cl)O2)=O)NC(=O)[C@@H](CC(C)C)NC)[C@H]1C[C@](C)(N)[C@H](O)[C@H](C)O1 MYPYJXKWCTUITO-LYRMYLQWSA-N 0.000 description 3
- MYPYJXKWCTUITO-UHFFFAOYSA-N vancomycin Natural products O1C(C(=C2)Cl)=CC=C2C(O)C(C(NC(C2=CC(O)=CC(O)=C2C=2C(O)=CC=C3C=2)C(O)=O)=O)NC(=O)C3NC(=O)C2NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(CC(C)C)NC)C(O)C(C=C3Cl)=CC=C3OC3=CC2=CC1=C3OC1OC(CO)C(O)C(O)C1OC1CC(C)(N)C(O)C(C)O1 MYPYJXKWCTUITO-UHFFFAOYSA-N 0.000 description 3
- 230000004393 visual impairment Effects 0.000 description 3
- 230000004580 weight loss Effects 0.000 description 3
- 210000004885 white matter Anatomy 0.000 description 3
- UGOMMVLRQDMAQQ-UHFFFAOYSA-N xphos Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 UGOMMVLRQDMAQQ-UHFFFAOYSA-N 0.000 description 3
- OCXAGXCMZACNEC-CTWZREHQSA-N (1R,3S,5R)-2-[2-[3-acetyl-5-(2-methylpyrimidin-5-yl)indazol-1-yl]acetyl]-N-(6-bromo-3-methylpyridin-2-yl)-5-methyl-2-azabicyclo[3.1.0]hexane-3-carboxamide Chemical compound C(C)(=O)C1=NN(C2=CC=C(C=C12)C=1C=NC(=NC=1)C)CC(=O)N1[C@@H]2C[C@@]2(C[C@H]1C(=O)NC1=NC(=CC=C1C)Br)C OCXAGXCMZACNEC-CTWZREHQSA-N 0.000 description 2
- JDXCOXKBIGBZSK-PSNKNOTQSA-N (2S)-2-[[(2S)-2-[[(2S)-1-[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S,5S,8S,11S,14S,22S)-22-acetamido-11-benzyl-8-(3-carbamimidamidopropyl)-5-(2-carboxyethyl)-3,6,9,12,16,23-hexaoxo-2-propan-2-yl-1,4,7,10,13,17-hexazacyclotricosane-14-carbonyl]-methylamino]-3-carboxypropanoyl]amino]-3,3-dimethylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-3-(1H-pyrrolo[2,3-b]pyridin-3-yl)propanoyl]amino]-4-carboxybutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]pyrrolidine-2-carbonyl]amino]-2-cyclohexylacetyl]amino]-6-[3-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[[(4S)-4-carboxy-4-(hexadecanoylamino)butanoyl]amino]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]propanoylamino]hexanoic acid Chemical compound CCCCCCCCCCCCCCCC(=O)N[C@@H](CCC(=O)NCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCC(=O)NCCCC[C@H](NC(=O)[C@@H](NC(=O)[C@@H]1CCCN1C(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](Cc1c[nH]c2ncccc12)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)N(C)C(=O)[C@@H]1CC(=O)NCCCC[C@H](NC(C)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](Cc2ccccc2)C(=O)N1)C(C)(C)C)C1CCCCC1)C(O)=O)C(O)=O JDXCOXKBIGBZSK-PSNKNOTQSA-N 0.000 description 2
- FGZKEQDXWUHLMD-MOROJQBDSA-N (2s,3s,4r,5r)-n-[6-(2-aminoethylamino)-6-oxohexyl]-5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolane-2-carboxamide Chemical compound O[C@@H]1[C@H](O)[C@@H](C(=O)NCCCCCC(=O)NCCN)O[C@H]1N1C2=NC=NC(N)=C2N=C1 FGZKEQDXWUHLMD-MOROJQBDSA-N 0.000 description 2
- VCOPTHOUUNAYKQ-WBTCAYNUSA-N (3s)-3,6-diamino-n-[[(2s,5s,8e,11s,15s)-15-amino-11-[(6r)-2-amino-1,4,5,6-tetrahydropyrimidin-6-yl]-8-[(carbamoylamino)methylidene]-2-(hydroxymethyl)-3,6,9,12,16-pentaoxo-1,4,7,10,13-pentazacyclohexadec-5-yl]methyl]hexanamide;(3s)-3,6-diamino-n-[[(2s,5s,8 Chemical compound N1C(=O)\C(=C/NC(N)=O)NC(=O)[C@H](CNC(=O)C[C@@H](N)CCCN)NC(=O)[C@H](C)NC(=O)[C@@H](N)CNC(=O)[C@@H]1[C@@H]1NC(N)=NCC1.N1C(=O)\C(=C/NC(N)=O)NC(=O)[C@H](CNC(=O)C[C@@H](N)CCCN)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CNC(=O)[C@@H]1[C@@H]1NC(N)=NCC1 VCOPTHOUUNAYKQ-WBTCAYNUSA-N 0.000 description 2
- SOVUOXKZCCAWOJ-HJYUBDRYSA-N (4s,4as,5ar,12ar)-9-[[2-(tert-butylamino)acetyl]amino]-4,7-bis(dimethylamino)-1,10,11,12a-tetrahydroxy-3,12-dioxo-4a,5,5a,6-tetrahydro-4h-tetracene-2-carboxamide Chemical compound C1C2=C(N(C)C)C=C(NC(=O)CNC(C)(C)C)C(O)=C2C(O)=C2[C@@H]1C[C@H]1[C@H](N(C)C)C(=O)C(C(N)=O)=C(O)[C@@]1(O)C2=O SOVUOXKZCCAWOJ-HJYUBDRYSA-N 0.000 description 2
- WDLWHQDACQUCJR-ZAMMOSSLSA-N (6r,7r)-7-[[(2r)-2-azaniumyl-2-(4-hydroxyphenyl)acetyl]amino]-8-oxo-3-[(e)-prop-1-enyl]-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@@H]3N(C2=O)C(=C(CS3)/C=C/C)C(O)=O)=CC=C(O)C=C1 WDLWHQDACQUCJR-ZAMMOSSLSA-N 0.000 description 2
- IPYWNMVPZOAFOQ-NABDTECSSA-N (6r,7r)-7-[[(2z)-2-(2-amino-1,3-thiazol-4-yl)-2-(carboxymethoxyimino)acetyl]amino]-3-ethenyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid;trihydrate Chemical compound O.O.O.S1C(N)=NC(C(=N\OCC(O)=O)\C(=O)N[C@@H]2C(N3C(=C(C=C)CS[C@@H]32)C(O)=O)=O)=C1 IPYWNMVPZOAFOQ-NABDTECSSA-N 0.000 description 2
- MINDHVHHQZYEEK-UHFFFAOYSA-N (E)-(2S,3R,4R,5S)-5-[(2S,3S,4S,5S)-2,3-epoxy-5-hydroxy-4-methylhexyl]tetrahydro-3,4-dihydroxy-(beta)-methyl-2H-pyran-2-crotonic acid ester with 9-hydroxynonanoic acid Natural products CC(O)C(C)C1OC1CC1C(O)C(O)C(CC(C)=CC(=O)OCCCCCCCCC(O)=O)OC1 MINDHVHHQZYEEK-UHFFFAOYSA-N 0.000 description 2
- JRMGHBVACUJCRP-BTJKTKAUSA-N (z)-but-2-enedioic acid;4-[(4-fluoro-2-methyl-1h-indol-5-yl)oxy]-6-methoxy-7-(3-pyrrolidin-1-ylpropoxy)quinazoline Chemical compound OC(=O)\C=C/C(O)=O.COC1=CC2=C(OC=3C(=C4C=C(C)NC4=CC=3)F)N=CN=C2C=C1OCCCN1CCCC1 JRMGHBVACUJCRP-BTJKTKAUSA-N 0.000 description 2
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 2
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 2
- SNUSZUYTMHKCPM-UHFFFAOYSA-N 1-hydroxypyridin-2-one Chemical compound ON1C=CC=CC1=O SNUSZUYTMHKCPM-UHFFFAOYSA-N 0.000 description 2
- 229940031767 13-valent pneumococcal conjugate vaccine Drugs 0.000 description 2
- SPXOTSHWBDUUMT-UHFFFAOYSA-N 138-42-1 Chemical compound OS(=O)(=O)C1=CC=C([N+]([O-])=O)C=C1 SPXOTSHWBDUUMT-UHFFFAOYSA-N 0.000 description 2
- RTQWWZBSTRGEAV-PKHIMPSTSA-N 2-[[(2s)-2-[bis(carboxymethyl)amino]-3-[4-(methylcarbamoylamino)phenyl]propyl]-[2-[bis(carboxymethyl)amino]propyl]amino]acetic acid Chemical compound CNC(=O)NC1=CC=C(C[C@@H](CN(CC(C)N(CC(O)=O)CC(O)=O)CC(O)=O)N(CC(O)=O)CC(O)=O)C=C1 RTQWWZBSTRGEAV-PKHIMPSTSA-N 0.000 description 2
- LXBGSDVWAMZHDD-UHFFFAOYSA-N 2-methyl-1h-imidazole Chemical compound CC1=NC=CN1 LXBGSDVWAMZHDD-UHFFFAOYSA-N 0.000 description 2
- HJBLUNHMOKFZQX-UHFFFAOYSA-N 3-hydroxy-1,2,3-benzotriazin-4-one Chemical compound C1=CC=C2C(=O)N(O)N=NC2=C1 HJBLUNHMOKFZQX-UHFFFAOYSA-N 0.000 description 2
- XXJWYDDUDKYVKI-UHFFFAOYSA-N 4-[(4-fluoro-2-methyl-1H-indol-5-yl)oxy]-6-methoxy-7-[3-(1-pyrrolidinyl)propoxy]quinazoline Chemical compound COC1=CC2=C(OC=3C(=C4C=C(C)NC4=CC=3)F)N=CN=C2C=C1OCCCN1CCCC1 XXJWYDDUDKYVKI-UHFFFAOYSA-N 0.000 description 2
- QDPVYZNVVQQULH-UHFFFAOYSA-N 4-amino-5-fluoro-3-[6-(4-methylpiperazin-1-yl)-1H-benzimidazol-2-yl]-1H-quinolin-2-one 2-hydroxypropanoic acid hydrate Chemical compound O.CC(O)C(O)=O.C1CN(C)CCN1C1=CC=C(N=C(N2)C=3C(NC4=CC=CC(F)=C4C=3N)=O)C2=C1 QDPVYZNVVQQULH-UHFFFAOYSA-N 0.000 description 2
- NEDJTEXNSTUKHW-UHFFFAOYSA-N 5-bromo-2-methylpyrimidine Chemical compound CC1=NC=C(Br)C=N1 NEDJTEXNSTUKHW-UHFFFAOYSA-N 0.000 description 2
- BKLJUYPLUWUEOQ-UHFFFAOYSA-N 6-bromopyridin-2-amine Chemical compound NC1=CC=CC(Br)=N1 BKLJUYPLUWUEOQ-UHFFFAOYSA-N 0.000 description 2
- TZCYLJGNWDVJRA-UHFFFAOYSA-N 6-chloro-1-hydroxybenzotriazole Chemical compound C1=C(Cl)C=C2N(O)N=NC2=C1 TZCYLJGNWDVJRA-UHFFFAOYSA-N 0.000 description 2
- GSDSWSVVBLHKDQ-UHFFFAOYSA-N 9-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-2,3-dihydro-7H-[1,4]oxazino[2,3,4-ij]quinoline-6-carboxylic acid Chemical compound FC1=CC(C(C(C(O)=O)=C2)=O)=C3N2C(C)COC3=C1N1CCN(C)CC1 GSDSWSVVBLHKDQ-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 2
- 208000009304 Acute Kidney Injury Diseases 0.000 description 2
- 206010001052 Acute respiratory distress syndrome Diseases 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- YUWPMEXLKGOSBF-GACAOOTBSA-N Anecortave acetate Chemical compound O=C1CC[C@]2(C)C3=CC[C@]4(C)[C@](C(=O)COC(=O)C)(O)CC[C@H]4[C@@H]3CCC2=C1 YUWPMEXLKGOSBF-GACAOOTBSA-N 0.000 description 2
- FYEHYMARPSSOBO-UHFFFAOYSA-N Aurin Chemical compound C1=CC(O)=CC=C1C(C=1C=CC(O)=CC=1)=C1C=CC(=O)C=C1 FYEHYMARPSSOBO-UHFFFAOYSA-N 0.000 description 2
- 241000193738 Bacillus anthracis Species 0.000 description 2
- 108010077805 Bacterial Proteins Proteins 0.000 description 2
- 208000009299 Benign Mucous Membrane Pemphigoid Diseases 0.000 description 2
- 241000589968 Borrelia Species 0.000 description 2
- 108010065839 Capreomycin Proteins 0.000 description 2
- 241001060848 Carapidae Species 0.000 description 2
- JFPVXVDWJQMJEE-QMTHXVAHSA-N Cefuroxime Chemical compound N([C@@H]1C(N2C(=C(COC(N)=O)CS[C@@H]21)C(O)=O)=O)C(=O)C(=NOC)C1=CC=CO1 JFPVXVDWJQMJEE-QMTHXVAHSA-N 0.000 description 2
- 229920001661 Chitosan Polymers 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 208000005590 Choroidal Neovascularization Diseases 0.000 description 2
- 206010060823 Choroidal neovascularisation Diseases 0.000 description 2
- 208000002691 Choroiditis Diseases 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 2
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 description 2
- 108010078015 Complement C3b Proteins 0.000 description 2
- 108090000056 Complement factor B Proteins 0.000 description 2
- 102000003712 Complement factor B Human genes 0.000 description 2
- 102100035436 Complement factor D Human genes 0.000 description 2
- 208000011990 Corticobasal Degeneration Diseases 0.000 description 2
- 206010011715 Cyclitis Diseases 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- 229930195709 D-tyrosine Natural products 0.000 description 2
- 125000002849 D-tyrosine group Chemical group [H]N([H])[C@@]([H])(C(=O)[*])C([H])([H])C1=C([H])C([H])=C(O[H])C([H])=C1[H] 0.000 description 2
- 108020004414 DNA Proteins 0.000 description 2
- 108010013198 Daptomycin Proteins 0.000 description 2
- FMTDIUIBLCQGJB-UHFFFAOYSA-N Demethylchlortetracyclin Natural products C1C2C(O)C3=C(Cl)C=CC(O)=C3C(=O)C2=C(O)C2(O)C1C(N(C)C)C(O)=C(C(N)=O)C2=O FMTDIUIBLCQGJB-UHFFFAOYSA-N 0.000 description 2
- 206010012305 Demyelination Diseases 0.000 description 2
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 2
- 229940124846 Diphtheria-Tetanus-Pertussis vaccine Drugs 0.000 description 2
- 208000006926 Discoid Lupus Erythematosus Diseases 0.000 description 2
- 208000005189 Embolism Diseases 0.000 description 2
- 241000196324 Embryophyta Species 0.000 description 2
- 206010014989 Epidermolysis bullosa Diseases 0.000 description 2
- 206010016654 Fibrosis Diseases 0.000 description 2
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 2
- 208000001034 Frostbite Diseases 0.000 description 2
- IECPWNUMDGFDKC-UHFFFAOYSA-N Fusicsaeure Natural products C12C(O)CC3C(=C(CCC=C(C)C)C(O)=O)C(OC(C)=O)CC3(C)C1(C)CCC1C2(C)CCC(O)C1C IECPWNUMDGFDKC-UHFFFAOYSA-N 0.000 description 2
- CEAZRRDELHUEMR-URQXQFDESA-N Gentamicin Chemical compound O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N CEAZRRDELHUEMR-URQXQFDESA-N 0.000 description 2
- 108010072051 Glatiramer Acetate Proteins 0.000 description 2
- 206010018364 Glomerulonephritis Diseases 0.000 description 2
- 206010070476 Haemodialysis complication Diseases 0.000 description 2
- 206010018910 Haemolysis Diseases 0.000 description 2
- 101000997835 Homo sapiens Tyrosine-protein kinase JAK1 Proteins 0.000 description 2
- 241000725303 Human immunodeficiency virus Species 0.000 description 2
- 208000010159 IgA glomerulonephritis Diseases 0.000 description 2
- 206010021263 IgA nephropathy Diseases 0.000 description 2
- 229940076838 Immune checkpoint inhibitor Drugs 0.000 description 2
- 108060003951 Immunoglobulin Proteins 0.000 description 2
- 108010005716 Interferon beta-1a Proteins 0.000 description 2
- 108010005714 Interferon beta-1b Proteins 0.000 description 2
- 102000003815 Interleukin-11 Human genes 0.000 description 2
- 108090000177 Interleukin-11 Proteins 0.000 description 2
- 102000003816 Interleukin-13 Human genes 0.000 description 2
- 108090000176 Interleukin-13 Proteins 0.000 description 2
- JUZNIMUFDBIJCM-ANEDZVCMSA-N Invanz Chemical compound O=C([C@H]1NC[C@H](C1)SC=1[C@H](C)[C@@H]2[C@H](C(N2C=1C(O)=O)=O)[C@H](O)C)NC1=CC=CC(C(O)=O)=C1 JUZNIMUFDBIJCM-ANEDZVCMSA-N 0.000 description 2
- 208000033463 Ischaemic neuropathy Diseases 0.000 description 2
- 229940116839 Janus kinase 1 inhibitor Drugs 0.000 description 2
- 239000002138 L01XE21 - Regorafenib Substances 0.000 description 2
- 108090001090 Lectins Proteins 0.000 description 2
- 102000004856 Lectins Human genes 0.000 description 2
- GSDSWSVVBLHKDQ-JTQLQIEISA-N Levofloxacin Chemical compound C([C@@H](N1C2=C(C(C(C(O)=O)=C1)=O)C=C1F)C)OC2=C1N1CCN(C)CC1 GSDSWSVVBLHKDQ-JTQLQIEISA-N 0.000 description 2
- TYMRLRRVMHJFTF-UHFFFAOYSA-N Mafenide Chemical compound NCC1=CC=C(S(N)(=O)=O)C=C1 TYMRLRRVMHJFTF-UHFFFAOYSA-N 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- RJQXTJLFIWVMTO-TYNCELHUSA-N Methicillin Chemical compound COC1=CC=CC(OC)=C1C(=O)N[C@@H]1C(=O)N2[C@@H](C(O)=O)C(C)(C)S[C@@H]21 RJQXTJLFIWVMTO-TYNCELHUSA-N 0.000 description 2
- 101100268066 Mus musculus Zap70 gene Proteins 0.000 description 2
- 108010000123 Myelin-Oligodendrocyte Glycoprotein Proteins 0.000 description 2
- 102100023302 Myelin-oligodendrocyte glycoprotein Human genes 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- 229930193140 Neomycin Natural products 0.000 description 2
- 208000008589 Obesity Diseases 0.000 description 2
- UOZODPSAJZTQNH-UHFFFAOYSA-N Paromomycin II Natural products NC1C(O)C(O)C(CN)OC1OC1C(O)C(OC2C(C(N)CC(N)C2O)OC2C(C(O)C(O)C(CO)O2)N)OC1CO UOZODPSAJZTQNH-UHFFFAOYSA-N 0.000 description 2
- 206010034277 Pemphigoid Diseases 0.000 description 2
- 201000005702 Pertussis Diseases 0.000 description 2
- YGYAWVDWMABLBF-UHFFFAOYSA-N Phosgene Chemical compound ClC(Cl)=O YGYAWVDWMABLBF-UHFFFAOYSA-N 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 208000003971 Posterior uveitis Diseases 0.000 description 2
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 2
- 206010037211 Psychomotor hyperactivity Diseases 0.000 description 2
- 108091008103 RNA aptamers Proteins 0.000 description 2
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 2
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 2
- 208000033626 Renal failure acute Diseases 0.000 description 2
- 208000007135 Retinal Neovascularization Diseases 0.000 description 2
- 201000007737 Retinal degeneration Diseases 0.000 description 2
- 108010077895 Sarcosine Proteins 0.000 description 2
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- 101000942680 Sus scrofa Clusterin Proteins 0.000 description 2
- 206010051379 Systemic Inflammatory Response Syndrome Diseases 0.000 description 2
- 229940032047 Tdap vaccine Drugs 0.000 description 2
- 206010043376 Tetanus Diseases 0.000 description 2
- 229920001615 Tragacanth Polymers 0.000 description 2
- 239000007983 Tris buffer Substances 0.000 description 2
- 208000037386 Typhoid Diseases 0.000 description 2
- 102100033438 Tyrosine-protein kinase JAK1 Human genes 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 208000036142 Viral infection Diseases 0.000 description 2
- 208000000208 Wet Macular Degeneration Diseases 0.000 description 2
- 239000012391 XPhos Pd G2 Substances 0.000 description 2
- JMNXSNUXDHHTKQ-QVMSTPCGSA-N [(3r,6r)-6-[(3s,5r,7r,8r,9s,10s,13r,14s,17r)-3-[3-(4-aminobutylamino)propylamino]-7-hydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]-2-methylheptan-3-yl] hydrogen sulfate;(2s)-2-hydroxypropanoic ac Chemical compound C[C@H](O)C(O)=O.C([C@@H]1C[C@H]2O)[C@@H](NCCCNCCCCN)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@H](C)CC[C@H](C(C)C)OS(O)(=O)=O)[C@@]2(C)CC1 JMNXSNUXDHHTKQ-QVMSTPCGSA-N 0.000 description 2
- SRXKIZXIRHMPFW-UHFFFAOYSA-N [4-[6-[amino(azaniumylidene)methyl]naphthalen-2-yl]oxycarbonylphenyl]-(diaminomethylidene)azanium;methanesulfonate Chemical compound CS([O-])(=O)=O.CS([O-])(=O)=O.C1=CC(N=C([NH3+])N)=CC=C1C(=O)OC1=CC=C(C=C(C=C2)C([NH3+])=N)C2=C1 SRXKIZXIRHMPFW-UHFFFAOYSA-N 0.000 description 2
- 208000002223 abdominal aortic aneurysm Diseases 0.000 description 2
- 230000001154 acute effect Effects 0.000 description 2
- 201000011040 acute kidney failure Diseases 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 239000000556 agonist Substances 0.000 description 2
- 230000000735 allogeneic effect Effects 0.000 description 2
- LKCWBDHBTVXHDL-RMDFUYIESA-N amikacin Chemical compound O([C@@H]1[C@@H](N)C[C@H]([C@@H]([C@H]1O)O[C@@H]1[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O1)O)NC(=O)[C@@H](O)CCN)[C@H]1O[C@H](CN)[C@@H](O)[C@H](O)[C@H]1O LKCWBDHBTVXHDL-RMDFUYIESA-N 0.000 description 2
- LSQZJLSUYDQPKJ-NJBDSQKTSA-N amoxicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=C(O)C=C1 LSQZJLSUYDQPKJ-NJBDSQKTSA-N 0.000 description 2
- 229960000723 ampicillin Drugs 0.000 description 2
- 229960001232 anecortave Drugs 0.000 description 2
- 208000007502 anemia Diseases 0.000 description 2
- 230000003172 anti-dna Effects 0.000 description 2
- 229940121363 anti-inflammatory agent Drugs 0.000 description 2
- 208000007474 aortic aneurysm Diseases 0.000 description 2
- 229940120638 avastin Drugs 0.000 description 2
- RROBIDXNTUAHFW-UHFFFAOYSA-N benzotriazol-1-yloxy-tris(dimethylamino)phosphanium Chemical compound C1=CC=C2N(O[P+](N(C)C)(N(C)C)N(C)C)N=NC2=C1 RROBIDXNTUAHFW-UHFFFAOYSA-N 0.000 description 2
- 230000008033 biological extinction Effects 0.000 description 2
- 230000007321 biological mechanism Effects 0.000 description 2
- 229960000074 biopharmaceutical Drugs 0.000 description 2
- 229960000106 biosimilars Drugs 0.000 description 2
- 208000002352 blister Diseases 0.000 description 2
- 238000006795 borylation reaction Methods 0.000 description 2
- PPKJUHVNTMYXOD-PZGPJMECSA-N c49ws9n75l Chemical compound O=C([C@@H]1N(C2=O)CC[C@H]1S(=O)(=O)CCN(CC)CC)O[C@H](C(C)C)[C@H](C)\C=C\C(=O)NC\C=C\C(\C)=C\[C@@H](O)CC(=O)CC1=NC2=CO1.N([C@@H]1C(=O)N[C@@H](C(N2CCC[C@H]2C(=O)N(C)[C@@H](CC=2C=CC(=CC=2)N(C)C)C(=O)N2C[C@@H](CS[C@H]3C4CCN(CC4)C3)C(=O)C[C@H]2C(=O)N[C@H](C(=O)O[C@@H]1C)C=1C=CC=CC=1)=O)CC)C(=O)C1=NC=CC=C1O PPKJUHVNTMYXOD-PZGPJMECSA-N 0.000 description 2
- 229960001292 cabozantinib Drugs 0.000 description 2
- ONIQOQHATWINJY-UHFFFAOYSA-N cabozantinib Chemical compound C=12C=C(OC)C(OC)=CC2=NC=CC=1OC(C=C1)=CC=C1NC(=O)C1(C(=O)NC=2C=CC(F)=CC=2)CC1 ONIQOQHATWINJY-UHFFFAOYSA-N 0.000 description 2
- HFCFMRYTXDINDK-WNQIDUERSA-N cabozantinib malate Chemical compound OC(=O)[C@@H](O)CC(O)=O.C=12C=C(OC)C(OC)=CC2=NC=CC=1OC(C=C1)=CC=C1NC(=O)C1(C(=O)NC=2C=CC(F)=CC=2)CC1 HFCFMRYTXDINDK-WNQIDUERSA-N 0.000 description 2
- OSGAYBCDTDRGGQ-UHFFFAOYSA-L calcium sulfate Chemical compound [Ca+2].[O-]S([O-])(=O)=O OSGAYBCDTDRGGQ-UHFFFAOYSA-L 0.000 description 2
- 238000004364 calculation method Methods 0.000 description 2
- 239000004202 carbamide Substances 0.000 description 2
- FPPNZSSZRUTDAP-UWFZAAFLSA-N carbenicillin Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)C(C(O)=O)C1=CC=CC=C1 FPPNZSSZRUTDAP-UWFZAAFLSA-N 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 229960004841 cefadroxil Drugs 0.000 description 2
- NBFNMSULHIODTC-CYJZLJNKSA-N cefadroxil monohydrate Chemical compound O.C1([C@@H](N)C(=O)N[C@H]2[C@@H]3N(C2=O)C(=C(CS3)C)C(O)=O)=CC=C(O)C=C1 NBFNMSULHIODTC-CYJZLJNKSA-N 0.000 description 2
- MLYYVTUWGNIJIB-BXKDBHETSA-N cefazolin Chemical compound S1C(C)=NN=C1SCC1=C(C(O)=O)N2C(=O)[C@@H](NC(=O)CN3N=NN=C3)[C@H]2SC1 MLYYVTUWGNIJIB-BXKDBHETSA-N 0.000 description 2
- 229960001139 cefazolin Drugs 0.000 description 2
- 229960002129 cefixime Drugs 0.000 description 2
- 229960004261 cefotaxime Drugs 0.000 description 2
- 229960002580 cefprozil Drugs 0.000 description 2
- 229940036735 ceftaroline Drugs 0.000 description 2
- ZCCUWMICIWSJIX-NQJJCJBVSA-N ceftaroline fosamil Chemical compound S([C@@H]1[C@@H](C(N1C=1C([O-])=O)=O)NC(=O)\C(=N/OCC)C=2N=C(NP(O)(O)=O)SN=2)CC=1SC(SC=1)=NC=1C1=CC=[N+](C)C=C1 ZCCUWMICIWSJIX-NQJJCJBVSA-N 0.000 description 2
- NMVPEQXCMGEDNH-TZVUEUGBSA-N ceftazidime pentahydrate Chemical compound O.O.O.O.O.S([C@@H]1[C@@H](C(N1C=1C([O-])=O)=O)NC(=O)\C(=N/OC(C)(C)C(O)=O)C=2N=C(N)SC=2)CC=1C[N+]1=CC=CC=C1 NMVPEQXCMGEDNH-TZVUEUGBSA-N 0.000 description 2
- UNJFKXSSGBWRBZ-BJCIPQKHSA-N ceftibuten Chemical compound S1C(N)=NC(C(=C\CC(O)=O)\C(=O)N[C@@H]2C(N3C(=CCS[C@@H]32)C(O)=O)=O)=C1 UNJFKXSSGBWRBZ-BJCIPQKHSA-N 0.000 description 2
- 229960004755 ceftriaxone Drugs 0.000 description 2
- VAAUVRVFOQPIGI-SPQHTLEESA-N ceftriaxone Chemical compound S([C@@H]1[C@@H](C(N1C=1C(O)=O)=O)NC(=O)\C(=N/OC)C=2N=C(N)SC=2)CC=1CSC1=NC(=O)C(=O)NN1C VAAUVRVFOQPIGI-SPQHTLEESA-N 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 235000010980 cellulose Nutrition 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 229960005395 cetuximab Drugs 0.000 description 2
- WIIZWVCIJKGZOK-RKDXNWHRSA-N chloramphenicol Chemical compound ClC(Cl)C(=O)N[C@H](CO)[C@H](O)C1=CC=C([N+]([O-])=O)C=C1 WIIZWVCIJKGZOK-RKDXNWHRSA-N 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 229960003405 ciprofloxacin Drugs 0.000 description 2
- 239000007979 citrate buffer Substances 0.000 description 2
- 229960002626 clarithromycin Drugs 0.000 description 2
- 229960002227 clindamycin Drugs 0.000 description 2
- 229960004287 clofazimine Drugs 0.000 description 2
- WDQPAMHFFCXSNU-BGABXYSRSA-N clofazimine Chemical compound C12=CC=CC=C2N=C2C=C(NC=3C=CC(Cl)=CC=3)C(=N/C(C)C)/C=C2N1C1=CC=C(Cl)C=C1 WDQPAMHFFCXSNU-BGABXYSRSA-N 0.000 description 2
- LQOLIRLGBULYKD-JKIFEVAISA-N cloxacillin Chemical compound N([C@@H]1C(N2[C@H](C(C)(C)S[C@@H]21)C(O)=O)=O)C(=O)C1=C(C)ON=C1C1=CC=CC=C1Cl LQOLIRLGBULYKD-JKIFEVAISA-N 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 229940034568 cometriq Drugs 0.000 description 2
- 239000004074 complement inhibitor Substances 0.000 description 2
- 102000006834 complement receptors Human genes 0.000 description 2
- 108010047295 complement receptors Proteins 0.000 description 2
- 239000013256 coordination polymer Substances 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 201000003278 cryoglobulinemia Diseases 0.000 description 2
- 208000004921 cutaneous lupus erythematosus Diseases 0.000 description 2
- MGNCLNQXLYJVJD-UHFFFAOYSA-N cyanuric chloride Chemical compound ClC1=NC(Cl)=NC(Cl)=N1 MGNCLNQXLYJVJD-UHFFFAOYSA-N 0.000 description 2
- VMKJWLXVLHBJNK-UHFFFAOYSA-N cyanuric fluoride Chemical compound FC1=NC(F)=NC(F)=N1 VMKJWLXVLHBJNK-UHFFFAOYSA-N 0.000 description 2
- DOAKLVKFURWEDJ-QCMAZARJSA-N daptomycin Chemical compound C([C@H]1C(=O)O[C@H](C)[C@@H](C(NCC(=O)N[C@@H](CCCN)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@H](C)C(=O)N[C@@H](CC(O)=O)C(=O)NCC(=O)N[C@H](CO)C(=O)N[C@H](C(=O)N1)[C@H](C)CC(O)=O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](CC(N)=O)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)CCCCCCCCC)C(=O)C1=CC=CC=C1N DOAKLVKFURWEDJ-QCMAZARJSA-N 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 229960003957 dexamethasone Drugs 0.000 description 2
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 2
- RAABOESOVLLHRU-UHFFFAOYSA-N diazene Chemical compound N=N RAABOESOVLLHRU-UHFFFAOYSA-N 0.000 description 2
- AJDPNPAGZMZOMN-UHFFFAOYSA-N diethyl (4-oxo-1,2,3-benzotriazin-3-yl) phosphate Chemical compound C1=CC=C2C(=O)N(OP(=O)(OCC)OCC)N=NC2=C1 AJDPNPAGZMZOMN-UHFFFAOYSA-N 0.000 description 2
- 238000004455 differential thermal analysis Methods 0.000 description 2
- 238000009792 diffusion process Methods 0.000 description 2
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 2
- 239000007884 disintegrant Substances 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- AVAACINZEOAHHE-VFZPANTDSA-N doripenem Chemical compound C=1([C@H](C)[C@@H]2[C@H](C(N2C=1C(O)=O)=O)[C@H](O)C)S[C@@H]1CN[C@H](CNS(N)(=O)=O)C1 AVAACINZEOAHHE-VFZPANTDSA-N 0.000 description 2
- 238000007876 drug discovery Methods 0.000 description 2
- 239000000428 dust Substances 0.000 description 2
- AEOCXXJPGCBFJA-UHFFFAOYSA-N ethionamide Chemical compound CCC1=CC(C(N)=S)=CC=N1 AEOCXXJPGCBFJA-UHFFFAOYSA-N 0.000 description 2
- FIRHQRGFVOSDDY-UHFFFAOYSA-N ethyl 1-hydroxytriazole-4-carboxylate Chemical compound CCOC(=O)C1=CN(O)N=N1 FIRHQRGFVOSDDY-UHFFFAOYSA-N 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 229960000556 fingolimod Drugs 0.000 description 2
- KKGQTZUTZRNORY-UHFFFAOYSA-N fingolimod Chemical compound CCCCCCCCC1=CC=C(CCC(N)(CO)CO)C=C1 KKGQTZUTZRNORY-UHFFFAOYSA-N 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 201000005206 focal segmental glomerulosclerosis Diseases 0.000 description 2
- 231100000854 focal segmental glomerulosclerosis Toxicity 0.000 description 2
- 230000003325 follicular Effects 0.000 description 2
- 239000012634 fragment Substances 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 229960003170 gemifloxacin Drugs 0.000 description 2
- ZRCVYEYHRGVLOC-HYARGMPZSA-N gemifloxacin Chemical compound C1C(CN)C(=N/OC)/CN1C(C(=C1)F)=NC2=C1C(=O)C(C(O)=O)=CN2C1CC1 ZRCVYEYHRGVLOC-HYARGMPZSA-N 0.000 description 2
- 235000001727 glucose Nutrition 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 230000008588 hemolysis Effects 0.000 description 2
- 208000007475 hemolytic anemia Diseases 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 229960004171 hydroxychloroquine Drugs 0.000 description 2
- 229960001001 ibritumomab tiuxetan Drugs 0.000 description 2
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 2
- 208000026278 immune system disease Diseases 0.000 description 2
- 239000012274 immune-checkpoint protein inhibitor Substances 0.000 description 2
- 102000018358 immunoglobulin Human genes 0.000 description 2
- 239000003018 immunosuppressive agent Substances 0.000 description 2
- 229940125721 immunosuppressive agent Drugs 0.000 description 2
- 230000001024 immunotherapeutic effect Effects 0.000 description 2
- 239000007943 implant Substances 0.000 description 2
- CGIGDMFJXJATDK-UHFFFAOYSA-N indomethacin Chemical compound CC1=C(CC(O)=O)C2=CC(OC)=CC=C2N1C(=O)C1=CC=C(Cl)C=C1 CGIGDMFJXJATDK-UHFFFAOYSA-N 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 229940005319 inlyta Drugs 0.000 description 2
- 210000005007 innate immune system Anatomy 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 229960004461 interferon beta-1a Drugs 0.000 description 2
- 229960003161 interferon beta-1b Drugs 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 208000012947 ischemia reperfusion injury Diseases 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 229960004891 lapatinib Drugs 0.000 description 2
- 239000002523 lectin Substances 0.000 description 2
- 229960003784 lenvatinib Drugs 0.000 description 2
- 229940064847 lenvima Drugs 0.000 description 2
- 201000011486 lichen planus Diseases 0.000 description 2
- OJMMVQQUTAEWLP-KIDUDLJLSA-N lincomycin Chemical compound CN1C[C@H](CCC)C[C@H]1C(=O)N[C@H]([C@@H](C)O)[C@@H]1[C@H](O)[C@H](O)[C@@H](O)[C@@H](SC)O1 OJMMVQQUTAEWLP-KIDUDLJLSA-N 0.000 description 2
- ZEKZLJVOYLTDKK-UHFFFAOYSA-N lomefloxacin Chemical compound FC1=C2N(CC)C=C(C(O)=O)C(=O)C2=CC(F)=C1N1CCNC(C)C1 ZEKZLJVOYLTDKK-UHFFFAOYSA-N 0.000 description 2
- 208000018769 loss of vision Diseases 0.000 description 2
- 231100000864 loss of vision Toxicity 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- 210000002540 macrophage Anatomy 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 239000000155 melt Substances 0.000 description 2
- 201000008350 membranous glomerulonephritis Diseases 0.000 description 2
- 229960002260 meropenem Drugs 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 229920000609 methyl cellulose Polymers 0.000 description 2
- 239000001923 methylcellulose Substances 0.000 description 2
- 235000010981 methylcellulose Nutrition 0.000 description 2
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 2
- 229960003085 meticillin Drugs 0.000 description 2
- GTGQRSIMEUWHPA-ZBJAFUORSA-M mezlocillin sodium Chemical compound [Na+].N([C@@H](C(=O)N[C@H]1[C@H]2SC([C@@H](N2C1=O)C([O-])=O)(C)C)C=1C=CC=CC=1)C(=O)N1CCN(S(C)(=O)=O)C1=O GTGQRSIMEUWHPA-ZBJAFUORSA-M 0.000 description 2
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- DDHVILIIHBIMQU-YJGQQKNPSA-L mupirocin calcium hydrate Chemical compound O.O.[Ca+2].C[C@H](O)[C@H](C)[C@@H]1O[C@H]1C[C@@H]1[C@@H](O)[C@@H](O)[C@H](C\C(C)=C\C(=O)OCCCCCCCCC([O-])=O)OC1.C[C@H](O)[C@H](C)[C@@H]1O[C@H]1C[C@@H]1[C@@H](O)[C@@H](O)[C@H](C\C(C)=C\C(=O)OCCCCCCCCC([O-])=O)OC1 DDHVILIIHBIMQU-YJGQQKNPSA-L 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- HPNSFSBZBAHARI-RUDMXATFSA-N mycophenolic acid Chemical compound OC1=C(C\C=C(/C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-RUDMXATFSA-N 0.000 description 2
- LBWFXVZLPYTWQI-IPOVEDGCSA-N n-[2-(diethylamino)ethyl]-5-[(z)-(5-fluoro-2-oxo-1h-indol-3-ylidene)methyl]-2,4-dimethyl-1h-pyrrole-3-carboxamide;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C LBWFXVZLPYTWQI-IPOVEDGCSA-N 0.000 description 2
- GPXLMGHLHQJAGZ-JTDSTZFVSA-N nafcillin Chemical compound C1=CC=CC2=C(C(=O)N[C@@H]3C(N4[C@H](C(C)(C)S[C@@H]43)C(O)=O)=O)C(OCC)=CC=C21 GPXLMGHLHQJAGZ-JTDSTZFVSA-N 0.000 description 2
- 229960004927 neomycin Drugs 0.000 description 2
- 229960000808 netilmicin Drugs 0.000 description 2
- 208000015122 neurodegenerative disease Diseases 0.000 description 2
- 229910052759 nickel Inorganic materials 0.000 description 2
- 229960003301 nivolumab Drugs 0.000 description 2
- OGJPXUAPXNRGGI-UHFFFAOYSA-N norfloxacin Chemical compound C1=C2N(CC)C=C(C(O)=O)C(=O)C2=CC(F)=C1N1CCNCC1 OGJPXUAPXNRGGI-UHFFFAOYSA-N 0.000 description 2
- 235000020824 obesity Nutrition 0.000 description 2
- 208000015200 ocular cicatricial pemphigoid Diseases 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 210000004248 oligodendroglia Anatomy 0.000 description 2
- 238000005580 one pot reaction Methods 0.000 description 2
- 108010006945 oritavancin Proteins 0.000 description 2
- UWYHMGVUTGAWSP-JKIFEVAISA-N oxacillin Chemical compound N([C@@H]1C(N2[C@H](C(C)(C)S[C@@H]21)C(O)=O)=O)C(=O)C1=C(C)ON=C1C1=CC=CC=C1 UWYHMGVUTGAWSP-JKIFEVAISA-N 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- UOZODPSAJZTQNH-LSWIJEOBSA-N paromomycin Chemical compound N[C@@H]1[C@@H](O)[C@H](O)[C@H](CN)O[C@@H]1O[C@H]1[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](N)C[C@@H](N)[C@@H]2O)O[C@@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)N)O[C@@H]1CO UOZODPSAJZTQNH-LSWIJEOBSA-N 0.000 description 2
- MQHIQUBXFFAOMK-UHFFFAOYSA-N pazopanib hydrochloride Chemical compound Cl.C1=CC2=C(C)N(C)N=C2C=C1N(C)C(N=1)=CC=NC=1NC1=CC=C(C)C(S(N)(=O)=O)=C1 MQHIQUBXFFAOMK-UHFFFAOYSA-N 0.000 description 2
- 108010027737 peginterferon beta-1a Proteins 0.000 description 2
- 229960002621 pembrolizumab Drugs 0.000 description 2
- 210000001539 phagocyte Anatomy 0.000 description 2
- 229940127557 pharmaceutical product Drugs 0.000 description 2
- BPLBGHOLXOTWMN-MBNYWOFBSA-N phenoxymethylpenicillin Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)COC1=CC=CC=C1 BPLBGHOLXOTWMN-MBNYWOFBSA-N 0.000 description 2
- 239000008363 phosphate buffer Substances 0.000 description 2
- 229950010773 pidilizumab Drugs 0.000 description 2
- 229960001539 poliomyelitis vaccine Drugs 0.000 description 2
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- 231100000654 protein toxin Toxicity 0.000 description 2
- 238000010926 purge Methods 0.000 description 2
- IPEHBUMCGVEMRF-UHFFFAOYSA-N pyrazinecarboxamide Chemical compound NC(=O)C1=CN=CC=N1 IPEHBUMCGVEMRF-UHFFFAOYSA-N 0.000 description 2
- 229960003876 ranibizumab Drugs 0.000 description 2
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 229960004836 regorafenib Drugs 0.000 description 2
- 230000010410 reperfusion Effects 0.000 description 2
- 230000000241 respiratory effect Effects 0.000 description 2
- 230000004258 retinal degeneration Effects 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 229910052703 rhodium Inorganic materials 0.000 description 2
- 229960001225 rifampicin Drugs 0.000 description 2
- 229960004641 rituximab Drugs 0.000 description 2
- 229940043230 sarcosine Drugs 0.000 description 2
- 229960002930 sirolimus Drugs 0.000 description 2
- 208000017520 skin disease Diseases 0.000 description 2
- 239000001632 sodium acetate Substances 0.000 description 2
- 235000017281 sodium acetate Nutrition 0.000 description 2
- 239000001509 sodium citrate Substances 0.000 description 2
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 2
- 238000000527 sonication Methods 0.000 description 2
- 238000002336 sorption--desorption measurement Methods 0.000 description 2
- 229960000268 spectinomycin Drugs 0.000 description 2
- UNFWWIHTNXNPBV-WXKVUWSESA-N spectinomycin Chemical compound O([C@@H]1[C@@H](NC)[C@@H](O)[C@H]([C@@H]([C@H]1O1)O)NC)[C@]2(O)[C@H]1O[C@H](C)CC2=O UNFWWIHTNXNPBV-WXKVUWSESA-N 0.000 description 2
- 101150081011 ssl7 gene Proteins 0.000 description 2
- 229940090374 stivarga Drugs 0.000 description 2
- 229960005322 streptomycin Drugs 0.000 description 2
- 238000009662 stress testing Methods 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- 229960004306 sulfadiazine Drugs 0.000 description 2
- 238000004441 surface measurement Methods 0.000 description 2
- 238000001356 surgical procedure Methods 0.000 description 2
- 229940034785 sutent Drugs 0.000 description 2
- 208000006379 syphilis Diseases 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 108010089019 telavancin Proteins 0.000 description 2
- BVCKFLJARNKCSS-DWPRYXJFSA-N temocillin Chemical compound N([C@]1(OC)C(N2[C@H](C(C)(C)S[C@@H]21)C(O)=O)=O)C(=O)C(C(O)=O)C=1C=CSC=1 BVCKFLJARNKCSS-DWPRYXJFSA-N 0.000 description 2
- UTNUDOFZCWSZMS-YFHOEESVSA-N teriflunomide Chemical compound C\C(O)=C(/C#N)C(=O)NC1=CC=C(C(F)(F)F)C=C1 UTNUDOFZCWSZMS-YFHOEESVSA-N 0.000 description 2
- 229960000814 tetanus toxoid Drugs 0.000 description 2
- 150000003573 thiols Chemical class 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- 229960004089 tigecycline Drugs 0.000 description 2
- 229960000707 tobramycin Drugs 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- 229960005267 tositumomab Drugs 0.000 description 2
- 238000002054 transplantation Methods 0.000 description 2
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 2
- 201000008827 tuberculosis Diseases 0.000 description 2
- 229940094060 tykerb Drugs 0.000 description 2
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 2
- 201000008297 typhoid fever Diseases 0.000 description 2
- 229960003165 vancomycin Drugs 0.000 description 2
- 235000015112 vegetable and seed oil Nutrition 0.000 description 2
- 239000008158 vegetable oil Substances 0.000 description 2
- 230000009385 viral infection Effects 0.000 description 2
- 229940036061 zaltrap Drugs 0.000 description 2
- 229960002760 ziv-aflibercept Drugs 0.000 description 2
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- OIXVKQDWLFHVGR-GQTDVWSESA-N (2r,3s,4r,5r,6r)-5-amino-2-(aminomethyl)-6-[(1r,2r,3s,4r,6s)-4,6-diamino-2-[(2s,3r,4s,5r)-4-[(3r,4r,5s,6s)-3-amino-6-(aminomethyl)-4,5-dihydroxyoxan-2-yl]oxy-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-3-hydroxycyclohexyl]oxyoxane-3,4-diol;sulfuric acid Chemical compound OS(O)(=O)=O.N[C@@H]1[C@@H](O)[C@H](O)[C@H](CN)OC1O[C@H]1[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](N)C[C@@H](N)[C@@H]2O)O[C@@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CN)O2)N)O[C@@H]1CO OIXVKQDWLFHVGR-GQTDVWSESA-N 0.000 description 1
- NZKFUBQRAWPZJP-BXKLGIMVSA-N (2s,3r,4s,5s,6r)-4-amino-2-[(1s,2s,3r,4s,6r)-4,6-diamino-3-[(2r,3r,5s,6r)-3-amino-6-(aminomethyl)-5-hydroxyoxan-2-yl]oxy-2-hydroxycyclohexyl]oxy-6-(hydroxymethyl)oxane-3,5-diol;sulfuric acid Chemical compound OS(O)(=O)=O.OS(O)(=O)=O.OS(O)(=O)=O.OS(O)(=O)=O.OS(O)(=O)=O.N[C@@H]1C[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N.N[C@@H]1C[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N NZKFUBQRAWPZJP-BXKLGIMVSA-N 0.000 description 1
- LJRDOKAZOAKLDU-UDXJMMFXSA-N (2s,3s,4r,5r,6r)-5-amino-2-(aminomethyl)-6-[(2r,3s,4r,5s)-5-[(1r,2r,3s,5r,6s)-3,5-diamino-2-[(2s,3r,4r,5s,6r)-3-amino-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-6-hydroxycyclohexyl]oxy-4-hydroxy-2-(hydroxymethyl)oxolan-3-yl]oxyoxane-3,4-diol;sulfuric ac Chemical compound OS(O)(=O)=O.N[C@@H]1[C@@H](O)[C@H](O)[C@H](CN)O[C@@H]1O[C@H]1[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](N)C[C@@H](N)[C@@H]2O)O[C@@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)N)O[C@@H]1CO LJRDOKAZOAKLDU-UDXJMMFXSA-N 0.000 description 1
- YGWZXQOYEBWUTH-RQJHMYQMSA-N (2s,4r)-4-fluoro-1-[(2-methylpropan-2-yl)oxycarbonyl]pyrrolidine-2-carboxylic acid Chemical compound CC(C)(C)OC(=O)N1C[C@H](F)C[C@H]1C(O)=O YGWZXQOYEBWUTH-RQJHMYQMSA-N 0.000 description 1
- LITBAYYWXZOHAW-XDZRHBBOSA-N (2s,5r,6r)-6-[[(2r)-2-[(4-ethyl-2,3-dioxopiperazine-1-carbonyl)amino]-2-phenylacetyl]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid;(2s,3s,5r)-3-methyl-4,4,7-trioxo-3-(triazol-1-ylmethyl)-4$l^{6}-thia-1-azabicyclo[3.2.0]hept Chemical compound C([C@]1(C)S([C@H]2N(C(C2)=O)[C@H]1C(O)=O)(=O)=O)N1C=CN=N1.O=C1C(=O)N(CC)CCN1C(=O)N[C@H](C=1C=CC=CC=1)C(=O)N[C@@H]1C(=O)N2[C@@H](C(O)=O)C(C)(C)S[C@@H]21 LITBAYYWXZOHAW-XDZRHBBOSA-N 0.000 description 1
- KMEGBUCIGMEPME-LQYKFRDPSA-N (2s,5r,6r)-6-[[(2r)-2-amino-2-phenylacetyl]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid;(1r,4s)-3,3-dimethyl-2,2,6-trioxo-2$l^{6}-thiabicyclo[3.2.0]heptane-4-carboxylic acid Chemical compound O=S1(=O)C(C)(C)[C@H](C(O)=O)C2C(=O)C[C@H]21.C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 KMEGBUCIGMEPME-LQYKFRDPSA-N 0.000 description 1
- OQANPHBRHBJGNZ-FYJGNVAPSA-N (3e)-6-oxo-3-[[4-(pyridin-2-ylsulfamoyl)phenyl]hydrazinylidene]cyclohexa-1,4-diene-1-carboxylic acid Chemical compound C1=CC(=O)C(C(=O)O)=C\C1=N\NC1=CC=C(S(=O)(=O)NC=2N=CC=CC=2)C=C1 OQANPHBRHBJGNZ-FYJGNVAPSA-N 0.000 description 1
- SGKRLCUYIXIAHR-AKNGSSGZSA-N (4s,4ar,5s,5ar,6r,12ar)-4-(dimethylamino)-1,5,10,11,12a-pentahydroxy-6-methyl-3,12-dioxo-4a,5,5a,6-tetrahydro-4h-tetracene-2-carboxamide Chemical compound C1=CC=C2[C@H](C)[C@@H]([C@H](O)[C@@H]3[C@](C(O)=C(C(N)=O)C(=O)[C@H]3N(C)C)(O)C3=O)C3=C(O)C2=C1O SGKRLCUYIXIAHR-AKNGSSGZSA-N 0.000 description 1
- OWFJMIVZYSDULZ-PXOLEDIWSA-N (4s,4ar,5s,5ar,6s,12ar)-4-(dimethylamino)-1,5,6,10,11,12a-hexahydroxy-6-methyl-3,12-dioxo-4,4a,5,5a-tetrahydrotetracene-2-carboxamide Chemical compound C1=CC=C2[C@](O)(C)[C@H]3[C@H](O)[C@H]4[C@H](N(C)C)C(=O)C(C(N)=O)=C(O)[C@@]4(O)C(=O)C3=C(O)C2=C1O OWFJMIVZYSDULZ-PXOLEDIWSA-N 0.000 description 1
- NWXMGUDVXFXRIG-WESIUVDSSA-N (4s,4as,5as,6s,12ar)-4-(dimethylamino)-1,6,10,11,12a-pentahydroxy-6-methyl-3,12-dioxo-4,4a,5,5a-tetrahydrotetracene-2-carboxamide Chemical compound C1=CC=C2[C@](O)(C)[C@H]3C[C@H]4[C@H](N(C)C)C(=O)C(C(N)=O)=C(O)[C@@]4(O)C(=O)C3=C(O)C2=C1O NWXMGUDVXFXRIG-WESIUVDSSA-N 0.000 description 1
- OAPVUSSHCBRCOL-KBHRXELFSA-N (4s,4as,5as,6s,12ar)-7-chloro-4-(dimethylamino)-1,6,10,11,12a-pentahydroxy-3,12-dioxo-4a,5,5a,6-tetrahydro-4h-tetracene-2-carboxamide;hydrochloride Chemical compound Cl.C1([C@H]2O)=C(Cl)C=CC(O)=C1C(O)=C1[C@@H]2C[C@H]2[C@H](N(C)C)C(=O)C(C(N)=O)=C(O)[C@@]2(O)C1=O OAPVUSSHCBRCOL-KBHRXELFSA-N 0.000 description 1
- ZFHMQZAREIBMCO-BAFYGKSASA-N (6r)-4-chloro-5-thia-1-azabicyclo[4.2.0]oct-2-en-8-one Chemical compound C1=CC(Cl)S[C@@H]2CC(=O)N21 ZFHMQZAREIBMCO-BAFYGKSASA-N 0.000 description 1
- MMRINLZOZVAPDZ-LSGRDSQZSA-N (6r,7r)-7-[[(2z)-2-(2-amino-1,3-thiazol-4-yl)-2-methoxyiminoacetyl]amino]-3-[(1-methylpyrrolidin-1-ium-1-yl)methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid;chloride Chemical compound Cl.S([C@@H]1[C@@H](C(N1C=1C([O-])=O)=O)NC(=O)\C(=N/OC)C=2N=C(N)SC=2)CC=1C[N+]1(C)CCCC1 MMRINLZOZVAPDZ-LSGRDSQZSA-N 0.000 description 1
- GMVPRGQOIOIIMI-UHFFFAOYSA-N (8R,11R,12R,13E,15S)-11,15-Dihydroxy-9-oxo-13-prostenoic acid Natural products CCCCCC(O)C=CC1C(O)CC(=O)C1CCCCCCC(O)=O GMVPRGQOIOIIMI-UHFFFAOYSA-N 0.000 description 1
- QKDHBVNJCZBTMR-LLVKDONJSA-N (R)-temafloxacin Chemical compound C1CN[C@H](C)CN1C(C(=C1)F)=CC2=C1C(=O)C(C(O)=O)=CN2C1=CC=C(F)C=C1F QKDHBVNJCZBTMR-LLVKDONJSA-N 0.000 description 1
- WHTVZRBIWZFKQO-AWEZNQCLSA-N (S)-chloroquine Chemical compound ClC1=CC=C2C(N[C@@H](C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-AWEZNQCLSA-N 0.000 description 1
- XUBOMFCQGDBHNK-JTQLQIEISA-N (S)-gatifloxacin Chemical compound FC1=CC(C(C(C(O)=O)=CN2C3CC3)=O)=C2C(OC)=C1N1CCN[C@@H](C)C1 XUBOMFCQGDBHNK-JTQLQIEISA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- NCCJWSXETVVUHK-ZYSAIPPVSA-N (z)-7-[(2r)-2-amino-2-carboxyethyl]sulfanyl-2-[[(1s)-2,2-dimethylcyclopropanecarbonyl]amino]hept-2-enoic acid;(5r,6s)-3-[2-(aminomethylideneamino)ethylsulfanyl]-6-[(1r)-1-hydroxyethyl]-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid Chemical compound C1C(SCC\N=C/N)=C(C(O)=O)N2C(=O)[C@H]([C@H](O)C)[C@H]21.CC1(C)C[C@@H]1C(=O)N\C(=C/CCCCSC[C@H](N)C(O)=O)C(O)=O NCCJWSXETVVUHK-ZYSAIPPVSA-N 0.000 description 1
- PFKFTWBEEFSNDU-UHFFFAOYSA-N 1,1'-Carbonyldiimidazole Substances C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 1
- JYEUMXHLPRZUAT-UHFFFAOYSA-N 1,2,3-triazine Chemical compound C1=CN=NN=C1 JYEUMXHLPRZUAT-UHFFFAOYSA-N 0.000 description 1
- QKDHBVNJCZBTMR-UHFFFAOYSA-N 1-(2,4-difluorophenyl)-6-fluoro-7-(3-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid Chemical compound C1CNC(C)CN1C(C(=C1)F)=CC2=C1C(=O)C(C(O)=O)=CN2C1=CC=C(F)C=C1F QKDHBVNJCZBTMR-UHFFFAOYSA-N 0.000 description 1
- DZRIAAFSBYXRSH-UHFFFAOYSA-N 1-(3-hydroxytriazol-4-yl)ethanone Chemical compound CC(=O)C1=CN=NN1O DZRIAAFSBYXRSH-UHFFFAOYSA-N 0.000 description 1
- OOSZCNKVJAVHJI-UHFFFAOYSA-N 1-[(4-fluorophenyl)methyl]piperazine Chemical compound C1=CC(F)=CC=C1CN1CCNCC1 OOSZCNKVJAVHJI-UHFFFAOYSA-N 0.000 description 1
- VPBYZLCHOKSGRX-UHFFFAOYSA-N 1-[2-chloro-4-(6,7-dimethoxyquinazolin-4-yl)oxyphenyl]-3-propylurea Chemical compound C1=C(Cl)C(NC(=O)NCCC)=CC=C1OC1=NC=NC2=CC(OC)=C(OC)C=C12 VPBYZLCHOKSGRX-UHFFFAOYSA-N 0.000 description 1
- SPMVMDHWKHCIDT-UHFFFAOYSA-N 1-[2-chloro-4-[(6,7-dimethoxy-4-quinolinyl)oxy]phenyl]-3-(5-methyl-3-isoxazolyl)urea Chemical compound C=12C=C(OC)C(OC)=CC2=NC=CC=1OC(C=C1Cl)=CC=C1NC(=O)NC=1C=C(C)ON=1 SPMVMDHWKHCIDT-UHFFFAOYSA-N 0.000 description 1
- DOJNLTSZOHKYAA-UHFFFAOYSA-N 1-diethylphosphoryloxy-4-nitrobenzene Chemical compound CCP(=O)(CC)OC1=CC=C([N+]([O-])=O)C=C1 DOJNLTSZOHKYAA-UHFFFAOYSA-N 0.000 description 1
- OOWSDKUFKGVADH-UHFFFAOYSA-N 1-diphenylphosphoryloxy-2,3,4,5,6-pentafluorobenzene Chemical compound FC1=C(F)C(F)=C(F)C(F)=C1OP(=O)(C=1C=CC=CC=1)C1=CC=CC=C1 OOWSDKUFKGVADH-UHFFFAOYSA-N 0.000 description 1
- MVXMDWWPPYZUGR-UHFFFAOYSA-N 1-hydroxy-2-phenylbenzimidazole Chemical compound N=1C2=CC=CC=C2N(O)C=1C1=CC=CC=C1 MVXMDWWPPYZUGR-UHFFFAOYSA-N 0.000 description 1
- DGIBHCWBCOAPDN-UHFFFAOYSA-N 1-hydroxy-6-(trifluoromethyl)benzotriazole Chemical compound C1=C(C(F)(F)F)C=C2N(O)N=NC2=C1 DGIBHCWBCOAPDN-UHFFFAOYSA-N 0.000 description 1
- HZPVTKPGROKWRL-UHFFFAOYSA-N 1-hydroxy-6-nitrobenzotriazole Chemical compound C1=C([N+]([O-])=O)C=C2N(O)N=NC2=C1 HZPVTKPGROKWRL-UHFFFAOYSA-N 0.000 description 1
- FPIRBHDGWMWJEP-UHFFFAOYSA-N 1-hydroxy-7-azabenzotriazole Chemical compound C1=CN=C2N(O)N=NC2=C1 FPIRBHDGWMWJEP-UHFFFAOYSA-N 0.000 description 1
- FQKFPGMGQXQHLP-UHFFFAOYSA-N 1-hydroxytriazole Chemical compound ON1C=CN=N1 FQKFPGMGQXQHLP-UHFFFAOYSA-N 0.000 description 1
- BSXPDVKSFWQFRT-UHFFFAOYSA-N 1-hydroxytriazolo[4,5-b]pyridine Chemical compound C1=CC=C2N(O)N=NC2=N1 BSXPDVKSFWQFRT-UHFFFAOYSA-N 0.000 description 1
- YFYIKMFTXUYSEC-UHFFFAOYSA-N 1-hydroxytriazolo[4,5-c]pyridine Chemical compound N1=CC=C2N(O)N=NC2=C1 YFYIKMFTXUYSEC-UHFFFAOYSA-N 0.000 description 1
- MCTWTZJPVLRJOU-UHFFFAOYSA-N 1-methyl-1H-imidazole Chemical compound CN1C=CN=C1 MCTWTZJPVLRJOU-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- DVDYICDUKVJRHA-UHFFFAOYSA-N 1-methyl-3-phenyl-1,2,4-triazin-1-ium-5-olate Chemical compound C[N+]1=CC([O-])=NC(C=2C=CC=CC=2)=N1 DVDYICDUKVJRHA-UHFFFAOYSA-N 0.000 description 1
- BBGYASXSZUVYLQ-UHFFFAOYSA-N 1-nitro-2-[(2-nitrophenoxy)-phenylphosphoryl]oxybenzene Chemical compound [O-][N+](=O)C1=CC=CC=C1OP(=O)(C=1C=CC=CC=1)OC1=CC=CC=C1[N+]([O-])=O BBGYASXSZUVYLQ-UHFFFAOYSA-N 0.000 description 1
- 238000004293 19F NMR spectroscopy Methods 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- QMNWYGTWTXOQTP-UHFFFAOYSA-N 1h-triazin-6-one Chemical compound O=C1C=CN=NN1 QMNWYGTWTXOQTP-UHFFFAOYSA-N 0.000 description 1
- JVSFQJZRHXAUGT-UHFFFAOYSA-N 2,2-dimethylpropanoyl chloride Chemical compound CC(C)(C)C(Cl)=O JVSFQJZRHXAUGT-UHFFFAOYSA-N 0.000 description 1
- AEQDJSLRWYMAQI-UHFFFAOYSA-N 2,3,9,10-tetramethoxy-6,8,13,13a-tetrahydro-5H-isoquinolino[2,1-b]isoquinoline Chemical compound C1CN2CC(C(=C(OC)C=C3)OC)=C3CC2C2=C1C=C(OC)C(OC)=C2 AEQDJSLRWYMAQI-UHFFFAOYSA-N 0.000 description 1
- UJUQLGSPTXBVFD-UHFFFAOYSA-N 2-(3-acetyl-5-bromoindazol-1-yl)acetic acid Chemical compound C(C)(=O)C1=NN(C2=CC=C(C=C12)Br)CC(=O)O UJUQLGSPTXBVFD-UHFFFAOYSA-N 0.000 description 1
- ACTOXUHEUCPTEW-BWHGAVFKSA-N 2-[(4r,5s,6s,7r,9r,10r,11e,13e,16r)-6-[(2s,3r,4r,5s,6r)-5-[(2s,4r,5s,6s)-4,5-dihydroxy-4,6-dimethyloxan-2-yl]oxy-4-(dimethylamino)-3-hydroxy-6-methyloxan-2-yl]oxy-10-[(2s,5s,6r)-5-(dimethylamino)-6-methyloxan-2-yl]oxy-4-hydroxy-5-methoxy-9,16-dimethyl-2-o Chemical compound O([C@H]1/C=C/C=C/C[C@@H](C)OC(=O)C[C@@H](O)[C@@H]([C@H]([C@@H](CC=O)C[C@H]1C)O[C@H]1[C@@H]([C@H]([C@H](O[C@@H]2O[C@@H](C)[C@H](O)[C@](C)(O)C2)[C@@H](C)O1)N(C)C)O)OC)[C@@H]1CC[C@H](N(C)C)[C@@H](C)O1 ACTOXUHEUCPTEW-BWHGAVFKSA-N 0.000 description 1
- TWYCZJMOEMMCGC-UHFFFAOYSA-N 2-[4-(6,7-dimethoxyquinazolin-4-yl)oxyphenyl]-n-(1-propan-2-ylpyrazol-4-yl)acetamide Chemical compound C=12C=C(OC)C(OC)=CC2=NC=NC=1OC(C=C1)=CC=C1CC(=O)NC=1C=NN(C(C)C)C=1 TWYCZJMOEMMCGC-UHFFFAOYSA-N 0.000 description 1
- NWXMGUDVXFXRIG-UHFFFAOYSA-N 2-carbamoyl-4-(dimethylazaniumyl)-6,10,11,12a-tetrahydroxy-6-methyl-3,12-dioxo-4,4a,5,5a-tetrahydrotetracen-1-olate Chemical compound C1=CC=C2C(O)(C)C3CC4C(N(C)C)C(=O)C(C(N)=O)=C(O)C4(O)C(=O)C3=C(O)C2=C1O NWXMGUDVXFXRIG-UHFFFAOYSA-N 0.000 description 1
- ABFPKTQEQNICFT-UHFFFAOYSA-M 2-chloro-1-methylpyridin-1-ium;iodide Chemical compound [I-].C[N+]1=CC=CC=C1Cl ABFPKTQEQNICFT-UHFFFAOYSA-M 0.000 description 1
- VMWLBGPLCQOZCV-UHFFFAOYSA-N 2-cyano-2-ethoxyiminoacetic acid Chemical compound CCON=C(C#N)C(O)=O VMWLBGPLCQOZCV-UHFFFAOYSA-N 0.000 description 1
- GQWLWXRYRKNILE-UHFFFAOYSA-N 2-diphenylphosphoryloxy-5-nitropyridine Chemical compound N1=CC([N+](=O)[O-])=CC=C1OP(=O)(C=1C=CC=CC=1)C1=CC=CC=C1 GQWLWXRYRKNILE-UHFFFAOYSA-N 0.000 description 1
- XVNKJDGEOJZCEL-UHFFFAOYSA-N 2-hydroxytetrazole Chemical compound ON1N=CN=N1 XVNKJDGEOJZCEL-UHFFFAOYSA-N 0.000 description 1
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 1
- SJPJHLBDGCNLAL-UHFFFAOYSA-N 3-(dimethylamino)-1-(4-nitrophenyl)propan-1-one Chemical compound CN(C)CCC(=O)C1=CC=C([N+]([O-])=O)C=C1 SJPJHLBDGCNLAL-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- GRGLUGAZUQACSG-UHFFFAOYSA-N 3-(naphthalen-2-ylsulfonylmethyl)triazolo[4,5-b]pyridine Chemical compound C1=CC=CC2=CC(S(=O)(CN3C4=NC=CC=C4N=N3)=O)=CC=C21 GRGLUGAZUQACSG-UHFFFAOYSA-N 0.000 description 1
- NHFDRBXTEDBWCZ-NTEUORMPSA-N 3-[2,4-dimethyl-5-[(e)-(2-oxo-1h-indol-3-ylidene)methyl]-1h-pyrrol-3-yl]propanoic acid Chemical compound OC(=O)CCC1=C(C)NC(\C=C\2C3=CC=CC=C3NC/2=O)=C1C NHFDRBXTEDBWCZ-NTEUORMPSA-N 0.000 description 1
- CGKYNYPOXYSJFN-UHFFFAOYSA-N 3-[[3,3-dimethylbutan-2-yloxy(methyl)phosphoryl]oxy-methylphosphoryl]oxy-2,2-dimethylbutane Chemical compound CC(C)(C)C(C)OP(C)(=O)OP(C)(=O)OC(C)C(C)(C)C CGKYNYPOXYSJFN-UHFFFAOYSA-N 0.000 description 1
- KLDLRDSRCMJKGM-UHFFFAOYSA-N 3-[chloro-(2-oxo-1,3-oxazolidin-3-yl)phosphoryl]-1,3-oxazolidin-2-one Chemical compound C1COC(=O)N1P(=O)(Cl)N1CCOC1=O KLDLRDSRCMJKGM-UHFFFAOYSA-N 0.000 description 1
- AEJUQXPVZOLXOO-UHFFFAOYSA-N 3-hydroxytriazolo[4,5-c]pyridine Chemical compound C1=NC=C2N(O)N=NC2=C1 AEJUQXPVZOLXOO-UHFFFAOYSA-N 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- LZPWAYBEOJRFAX-UHFFFAOYSA-N 4,4,5,5-tetramethyl-1,3,2$l^{2}-dioxaborolane Chemical compound CC1(C)O[B]OC1(C)C LZPWAYBEOJRFAX-UHFFFAOYSA-N 0.000 description 1
- BWGRDBSNKQABCB-UHFFFAOYSA-N 4,4-difluoro-N-[3-[3-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)-8-azabicyclo[3.2.1]octan-8-yl]-1-thiophen-2-ylpropyl]cyclohexane-1-carboxamide Chemical compound CC(C)C1=NN=C(C)N1C1CC2CCC(C1)N2CCC(NC(=O)C1CCC(F)(F)CC1)C1=CC=CS1 BWGRDBSNKQABCB-UHFFFAOYSA-N 0.000 description 1
- MGWGWNFMUOTEHG-UHFFFAOYSA-N 4-(3,5-dimethylphenyl)-1,3-thiazol-2-amine Chemical compound CC1=CC(C)=CC(C=2N=C(N)SC=2)=C1 MGWGWNFMUOTEHG-UHFFFAOYSA-N 0.000 description 1
- NZBKIOJQXNGENQ-UHFFFAOYSA-N 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholin-4-ium Chemical class COC1=NC(OC)=NC([N+]2(C)CCOCC2)=N1 NZBKIOJQXNGENQ-UHFFFAOYSA-N 0.000 description 1
- CYDQOEWLBCCFJZ-UHFFFAOYSA-N 4-(4-fluorophenyl)oxane-4-carboxylic acid Chemical compound C=1C=C(F)C=CC=1C1(C(=O)O)CCOCC1 CYDQOEWLBCCFJZ-UHFFFAOYSA-N 0.000 description 1
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 1
- WZRJTRPJURQBRM-UHFFFAOYSA-N 4-amino-n-(5-methyl-1,2-oxazol-3-yl)benzenesulfonamide;5-[(3,4,5-trimethoxyphenyl)methyl]pyrimidine-2,4-diamine Chemical compound O1C(C)=CC(NS(=O)(=O)C=2C=CC(N)=CC=2)=N1.COC1=C(OC)C(OC)=CC(CC=2C(=NC(N)=NC=2)N)=C1 WZRJTRPJURQBRM-UHFFFAOYSA-N 0.000 description 1
- NRGZTHQFAQCJCQ-UHFFFAOYSA-N 4-nitrophenyl hydrogen phenylphosphonate Chemical compound C=1C=CC=CC=1P(=O)(O)OC1=CC=C([N+]([O-])=O)C=C1 NRGZTHQFAQCJCQ-UHFFFAOYSA-N 0.000 description 1
- 101710131943 40S ribosomal protein S3a Proteins 0.000 description 1
- PIJUKNMCRGCJAQ-UHFFFAOYSA-N 5-chloro-1-hydroxytriazole Chemical compound ON1N=NC=C1Cl PIJUKNMCRGCJAQ-UHFFFAOYSA-N 0.000 description 1
- MJZJYWCQPMNPRM-UHFFFAOYSA-N 6,6-dimethyl-1-[3-(2,4,5-trichlorophenoxy)propoxy]-1,6-dihydro-1,3,5-triazine-2,4-diamine Chemical compound CC1(C)N=C(N)N=C(N)N1OCCCOC1=CC(Cl)=C(Cl)C=C1Cl MJZJYWCQPMNPRM-UHFFFAOYSA-N 0.000 description 1
- JWOSPQYKRZZPPB-UHFFFAOYSA-N 6-chloro-1-hydroxy-2-phenylbenzimidazole Chemical compound N=1C2=CC=C(Cl)C=C2N(O)C=1C1=CC=CC=C1 JWOSPQYKRZZPPB-UHFFFAOYSA-N 0.000 description 1
- HWTDMFJYBAURQR-UHFFFAOYSA-N 80-82-0 Chemical compound OS(=O)(=O)C1=CC=CC=C1[N+]([O-])=O HWTDMFJYBAURQR-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 208000002874 Acne Vulgaris Diseases 0.000 description 1
- 206010056508 Acquired epidermolysis bullosa Diseases 0.000 description 1
- 229940124962 ActHIB Drugs 0.000 description 1
- 241000251468 Actinopterygii Species 0.000 description 1
- 208000004142 Acute Retinal Necrosis Syndrome Diseases 0.000 description 1
- 241000701242 Adenoviridae Species 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 208000032671 Allergic granulomatous angiitis Diseases 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 229910000809 Alumel Inorganic materials 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 108010089414 Anaphylatoxins Proteins 0.000 description 1
- 241000606646 Anaplasma Species 0.000 description 1
- WZPBZJONDBGPKJ-UHFFFAOYSA-N Antibiotic SQ 26917 Natural products O=C1N(S(O)(=O)=O)C(C)C1NC(=O)C(=NOC(C)(C)C(O)=O)C1=CSC(N)=N1 WZPBZJONDBGPKJ-UHFFFAOYSA-N 0.000 description 1
- 208000003343 Antiphospholipid Syndrome Diseases 0.000 description 1
- 101100248451 Arabidopsis thaliana RICE2 gene Proteins 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- 206010003694 Atrophy Diseases 0.000 description 1
- 241000271566 Aves Species 0.000 description 1
- 108010074708 B7-H1 Antigen Proteins 0.000 description 1
- 102000008096 B7-H1 Antigen Human genes 0.000 description 1
- 241000193830 Bacillus <bacterium> Species 0.000 description 1
- GUBGYTABKSRVRQ-DCSYEGIMSA-N Beta-Lactose Chemical compound OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-DCSYEGIMSA-N 0.000 description 1
- 229940124899 Biothrax Drugs 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- 229940124900 Boostrix Drugs 0.000 description 1
- 241000588832 Bordetella pertussis Species 0.000 description 1
- 241001148604 Borreliella afzelii Species 0.000 description 1
- 241000589969 Borreliella burgdorferi Species 0.000 description 1
- 241001148605 Borreliella garinii Species 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 206010006482 Bronchospasm Diseases 0.000 description 1
- DCMBUMJVTHRUGX-UHFFFAOYSA-N C(#N)C1=CC=C(C=C1)OP(OC1=CC=C(C=C1)C#N)(=O)C1=CC=CC=C1 Chemical compound C(#N)C1=CC=C(C=C1)OP(OC1=CC=C(C=C1)C#N)(=O)C1=CC=CC=C1 DCMBUMJVTHRUGX-UHFFFAOYSA-N 0.000 description 1
- 108010074051 C-Reactive Protein Proteins 0.000 description 1
- 102100032752 C-reactive protein Human genes 0.000 description 1
- 238000011357 CAR T-cell therapy Methods 0.000 description 1
- CVMJCZOEMPYFAJ-WAYWQWQTSA-N CC1(C)C=C(\C=C/C(=O)C=[N+]=[N-])C(C)(C)N1[O] Chemical compound CC1(C)C=C(\C=C/C(=O)C=[N+]=[N-])C(C)(C)N1[O] CVMJCZOEMPYFAJ-WAYWQWQTSA-N 0.000 description 1
- 210000004366 CD4-positive T-lymphocyte Anatomy 0.000 description 1
- 108010071134 CRM197 (non-toxic variant of diphtheria toxin) Proteins 0.000 description 1
- 241000714198 Caliciviridae Species 0.000 description 1
- 201000005488 Capillary Leak Syndrome Diseases 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 102000014914 Carrier Proteins Human genes 0.000 description 1
- 102100024974 Caspase recruitment domain-containing protein 8 Human genes 0.000 description 1
- 108090000426 Caspase-1 Proteins 0.000 description 1
- UQLLWWBDSUHNEB-CZUORRHYSA-N Cefaprin Chemical compound N([C@H]1[C@@H]2N(C1=O)C(=C(CS2)COC(=O)C)C(O)=O)C(=O)CSC1=CC=NC=C1 UQLLWWBDSUHNEB-CZUORRHYSA-N 0.000 description 1
- KEJCWVGMRLCZQQ-YJBYXUATSA-N Cefuroxime axetil Chemical compound N([C@@H]1C(N2C(=C(COC(N)=O)CS[C@@H]21)C(=O)OC(C)OC(C)=O)=O)C(=O)\C(=N/OC)C1=CC=CO1 KEJCWVGMRLCZQQ-YJBYXUATSA-N 0.000 description 1
- 229930186147 Cephalosporin Natural products 0.000 description 1
- 208000018380 Chemical injury Diseases 0.000 description 1
- 208000010445 Chilblains Diseases 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 206010008631 Cholera Diseases 0.000 description 1
- 208000017667 Chronic Disease Diseases 0.000 description 1
- 208000006344 Churg-Strauss Syndrome Diseases 0.000 description 1
- 208000032544 Cicatrix Diseases 0.000 description 1
- HZZVJAQRINQKSD-UHFFFAOYSA-N Clavulanic acid Natural products OC(=O)C1C(=CCO)OC2CC(=O)N21 HZZVJAQRINQKSD-UHFFFAOYSA-N 0.000 description 1
- 241000193449 Clostridium tetani Species 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 102000055157 Complement C1 Inhibitor Human genes 0.000 description 1
- 108700040183 Complement C1 Inhibitor Proteins 0.000 description 1
- 108010034753 Complement Membrane Attack Complex Proteins 0.000 description 1
- 108010060123 Conjugate Vaccines Proteins 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 206010010996 Corneal degeneration Diseases 0.000 description 1
- 241000711573 Coronaviridae Species 0.000 description 1
- 239000000055 Corticotropin-Releasing Hormone Substances 0.000 description 1
- 241000186227 Corynebacterium diphtheriae Species 0.000 description 1
- 241000606678 Coxiella burnetii Species 0.000 description 1
- 208000011231 Crohn disease Diseases 0.000 description 1
- 208000025962 Crush injury Diseases 0.000 description 1
- 208000019707 Cryoglobulinemic vasculitis Diseases 0.000 description 1
- 229910017541 Cu-K Inorganic materials 0.000 description 1
- 108010069514 Cyclic Peptides Proteins 0.000 description 1
- 102000001189 Cyclic Peptides Human genes 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 1
- 229930105110 Cyclosporin A Natural products 0.000 description 1
- 108010036949 Cyclosporine Proteins 0.000 description 1
- 201000003883 Cystic fibrosis Diseases 0.000 description 1
- 241000701022 Cytomegalovirus Species 0.000 description 1
- DYDCUQKUCUHJBH-UWTATZPHSA-N D-Cycloserine Chemical compound N[C@@H]1CONC1=O DYDCUQKUCUHJBH-UWTATZPHSA-N 0.000 description 1
- DYDCUQKUCUHJBH-UHFFFAOYSA-N D-Cycloserine Natural products NC1CONC1=O DYDCUQKUCUHJBH-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 229940124888 DECAVAC Drugs 0.000 description 1
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N DMSO-d6 Substances [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 1
- 102000053602 DNA Human genes 0.000 description 1
- 238000012287 DNA Binding Assay Methods 0.000 description 1
- MQJKPEGWNLWLTK-UHFFFAOYSA-N Dapsone Chemical compound C1=CC(N)=CC=C1S(=O)(=O)C1=CC=C(N)C=C1 MQJKPEGWNLWLTK-UHFFFAOYSA-N 0.000 description 1
- 229940124902 Daptacel Drugs 0.000 description 1
- 208000001490 Dengue Diseases 0.000 description 1
- 206010012310 Dengue fever Diseases 0.000 description 1
- 201000004624 Dermatitis Diseases 0.000 description 1
- 206010012442 Dermatitis contact Diseases 0.000 description 1
- 206010012688 Diabetic retinal oedema Diseases 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- RWSOTUBLDIXVET-UHFFFAOYSA-N Dihydrogen sulfide Chemical compound S RWSOTUBLDIXVET-UHFFFAOYSA-N 0.000 description 1
- 102100025012 Dipeptidyl peptidase 4 Human genes 0.000 description 1
- QXNVGIXVLWOKEQ-UHFFFAOYSA-N Disodium Chemical compound [Na][Na] QXNVGIXVLWOKEQ-UHFFFAOYSA-N 0.000 description 1
- 208000000059 Dyspnea Diseases 0.000 description 1
- 206010013975 Dyspnoeas Diseases 0.000 description 1
- 208000019878 Eales disease Diseases 0.000 description 1
- 241000605282 Ehrlichia ewingii Species 0.000 description 1
- 206010014561 Emphysema Diseases 0.000 description 1
- 208000037487 Endotoxemia Diseases 0.000 description 1
- 208000018428 Eosinophilic granulomatosis with polyangiitis Diseases 0.000 description 1
- 241000283073 Equus caballus Species 0.000 description 1
- 206010015150 Erythema Diseases 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 238000005079 FT-Raman Methods 0.000 description 1
- 241000282324 Felis Species 0.000 description 1
- 241000192125 Firmicutes Species 0.000 description 1
- 241000710781 Flaviviridae Species 0.000 description 1
- UIOFUWFRIANQPC-JKIFEVAISA-N Floxacillin Chemical compound N([C@@H]1C(N2[C@H](C(C)(C)S[C@@H]21)C(O)=O)=O)C(=O)C1=C(C)ON=C1C1=C(F)C=CC=C1Cl UIOFUWFRIANQPC-JKIFEVAISA-N 0.000 description 1
- 206010055690 Foetal death Diseases 0.000 description 1
- QZJIMDIBFFHQDW-LMLSDSMGSA-N Fosfomycin tromethamine Chemical compound C[C@@H]1O[C@@H]1P(O)([O-])=O.OCC([NH3+])(CO)CO QZJIMDIBFFHQDW-LMLSDSMGSA-N 0.000 description 1
- 241000589602 Francisella tularensis Species 0.000 description 1
- JRZJKWGQFNTSRN-UHFFFAOYSA-N Geldanamycin Natural products C1C(C)CC(OC)C(O)C(C)C=C(C)C(OC(N)=O)C(OC)CCC=C(C)C(=O)NC2=CC(=O)C(OC)=C1C2=O JRZJKWGQFNTSRN-UHFFFAOYSA-N 0.000 description 1
- 208000034826 Genetic Predisposition to Disease Diseases 0.000 description 1
- 229930182566 Gentamicin Natural products 0.000 description 1
- 208000008069 Geographic Atrophy Diseases 0.000 description 1
- 208000003736 Gerstmann-Straussler-Scheinker Disease Diseases 0.000 description 1
- 206010072075 Gerstmann-Straussler-Scheinker syndrome Diseases 0.000 description 1
- 208000007465 Giant cell arteritis Diseases 0.000 description 1
- 208000010412 Glaucoma Diseases 0.000 description 1
- 102000006395 Globulins Human genes 0.000 description 1
- 108010044091 Globulins Proteins 0.000 description 1
- 208000022461 Glomerular disease Diseases 0.000 description 1
- 208000024869 Goodpasture syndrome Diseases 0.000 description 1
- 201000005569 Gout Diseases 0.000 description 1
- 201000005708 Granuloma Annulare Diseases 0.000 description 1
- AIJTTZAVMXIJGM-UHFFFAOYSA-N Grepafloxacin Chemical compound C1CNC(C)CN1C(C(=C1C)F)=CC2=C1C(=O)C(C(O)=O)=CN2C1CC1 AIJTTZAVMXIJGM-UHFFFAOYSA-N 0.000 description 1
- 208000035895 Guillain-Barré syndrome Diseases 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 108700035897 Haemophilus influenzae HibTITER Proteins 0.000 description 1
- 229940124855 Haemophilus influenzae vaccine Drugs 0.000 description 1
- 108010050195 Haemophilus influenzae-type b polysaccharide-Neisseria meningitidis outer membrane protein conjugate vaccine Proteins 0.000 description 1
- 208000030836 Hashimoto thyroiditis Diseases 0.000 description 1
- 208000000616 Hemoptysis Diseases 0.000 description 1
- 206010069440 Henoch-Schonlein purpura nephritis Diseases 0.000 description 1
- 241000711549 Hepacivirus C Species 0.000 description 1
- 241000700739 Hepadnaviridae Species 0.000 description 1
- 241000700721 Hepatitis B virus Species 0.000 description 1
- 241000709721 Hepatovirus A Species 0.000 description 1
- 206010019860 Hereditary angioedema Diseases 0.000 description 1
- 241000700586 Herpesviridae Species 0.000 description 1
- 101000761247 Homo sapiens Caspase recruitment domain-containing protein 8 Proteins 0.000 description 1
- 101000908391 Homo sapiens Dipeptidyl peptidase 4 Proteins 0.000 description 1
- 101000998146 Homo sapiens Interleukin-17A Proteins 0.000 description 1
- 101000661807 Homo sapiens Suppressor of tumorigenicity 14 protein Proteins 0.000 description 1
- 101000666896 Homo sapiens V-type immunoglobulin domain-containing suppressor of T-cell activation Proteins 0.000 description 1
- 241000701085 Human alphaherpesvirus 3 Species 0.000 description 1
- 241000701044 Human gammaherpesvirus 4 Species 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 229940124915 Infanrix Drugs 0.000 description 1
- 102000037984 Inhibitory immune checkpoint proteins Human genes 0.000 description 1
- 108091008026 Inhibitory immune checkpoint proteins Proteins 0.000 description 1
- 102100023915 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 108010064593 Intercellular Adhesion Molecule-1 Proteins 0.000 description 1
- 102100037877 Intercellular adhesion molecule 1 Human genes 0.000 description 1
- 102000019223 Interleukin-1 receptor Human genes 0.000 description 1
- 108050006617 Interleukin-1 receptor Proteins 0.000 description 1
- 102000013691 Interleukin-17 Human genes 0.000 description 1
- 108050003558 Interleukin-17 Proteins 0.000 description 1
- 102100033461 Interleukin-17A Human genes 0.000 description 1
- 206010022557 Intermediate uveitis Diseases 0.000 description 1
- 241000764238 Isis Species 0.000 description 1
- UETNIIAIRMUTSM-UHFFFAOYSA-N Jacareubin Natural products CC1(C)OC2=CC3Oc4c(O)c(O)ccc4C(=O)C3C(=C2C=C1)O UETNIIAIRMUTSM-UHFFFAOYSA-N 0.000 description 1
- 229940121730 Janus kinase 2 inhibitor Drugs 0.000 description 1
- 229940123241 Janus kinase 3 inhibitor Drugs 0.000 description 1
- 102000001399 Kallikrein Human genes 0.000 description 1
- 108060005987 Kallikrein Proteins 0.000 description 1
- 229940124919 Kinrix Drugs 0.000 description 1
- 201000006165 Kuhnt-Junius degeneration Diseases 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 239000004166 Lanolin Substances 0.000 description 1
- OJMMVQQUTAEWLP-UHFFFAOYSA-N Lincomycin Natural products CN1CC(CCC)CC1C(=O)NC(C(C)O)C1C(O)C(O)C(O)C(SC)O1 OJMMVQQUTAEWLP-UHFFFAOYSA-N 0.000 description 1
- QPJBONAWFAURGB-UHFFFAOYSA-L Lobenzarit disodium Chemical compound [Na+].[Na+].[O-]C(=O)C1=CC=CC=C1NC1=CC(Cl)=CC=C1C([O-])=O QPJBONAWFAURGB-UHFFFAOYSA-L 0.000 description 1
- 208000008771 Lymphadenopathy Diseases 0.000 description 1
- 208000001344 Macular Edema Diseases 0.000 description 1
- 206010025415 Macular oedema Diseases 0.000 description 1
- 208000035719 Maculopathy Diseases 0.000 description 1
- SBDNJUWAMKYJOX-UHFFFAOYSA-N Meclofenamic Acid Chemical compound CC1=CC=C(Cl)C(NC=2C(=CC=CC=2)C(O)=O)=C1Cl SBDNJUWAMKYJOX-UHFFFAOYSA-N 0.000 description 1
- ZRVUJXDFFKFLMG-UHFFFAOYSA-N Meloxicam Chemical compound OC=1C2=CC=CC=C2S(=O)(=O)N(C)C=1C(=O)NC1=NC=C(C)S1 ZRVUJXDFFKFLMG-UHFFFAOYSA-N 0.000 description 1
- 102000018697 Membrane Proteins Human genes 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- 229940124904 Menactra Drugs 0.000 description 1
- 229940124951 Menveo Drugs 0.000 description 1
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 1
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 description 1
- 206010049567 Miller Fisher syndrome Diseases 0.000 description 1
- 206010060880 Monoclonal gammopathy Diseases 0.000 description 1
- 208000010164 Multifocal Choroiditis Diseases 0.000 description 1
- 101100069392 Mus musculus Gzma gene Proteins 0.000 description 1
- 241000186366 Mycobacterium bovis Species 0.000 description 1
- 241000186362 Mycobacterium leprae Species 0.000 description 1
- 241000187479 Mycobacterium tuberculosis Species 0.000 description 1
- 208000009525 Myocarditis Diseases 0.000 description 1
- FOFDIMHVKGYHRU-UHFFFAOYSA-N N-(1,3-benzodioxol-5-ylmethyl)-4-(4-benzofuro[3,2-d]pyrimidinyl)-1-piperazinecarbothioamide Chemical compound C12=CC=CC=C2OC2=C1N=CN=C2N(CC1)CCN1C(=S)NCC1=CC=C(OCO2)C2=C1 FOFDIMHVKGYHRU-UHFFFAOYSA-N 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- GXCLVBGFBYZDAG-UHFFFAOYSA-N N-[2-(1H-indol-3-yl)ethyl]-N-methylprop-2-en-1-amine Chemical compound CN(CCC1=CNC2=C1C=CC=C2)CC=C GXCLVBGFBYZDAG-UHFFFAOYSA-N 0.000 description 1
- 150000001204 N-oxides Chemical class 0.000 description 1
- CMWTZPSULFXXJA-UHFFFAOYSA-N Naproxen Natural products C1=C(C(C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-UHFFFAOYSA-N 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 241000604969 Neorickettsia sennetsu Species 0.000 description 1
- 206010056677 Nerve degeneration Diseases 0.000 description 1
- 208000012902 Nervous system disease Diseases 0.000 description 1
- 208000025966 Neurological disease Diseases 0.000 description 1
- 244000061176 Nicotiana tabacum Species 0.000 description 1
- 235000002637 Nicotiana tabacum Nutrition 0.000 description 1
- 102400001111 Nociceptin Human genes 0.000 description 1
- 108090000622 Nociceptin Proteins 0.000 description 1
- ULTJSTZEJQGQBD-UHFFFAOYSA-N ON1N=NC=C1CC(C)=O Chemical compound ON1N=NC=C1CC(C)=O ULTJSTZEJQGQBD-UHFFFAOYSA-N 0.000 description 1
- OUXCBXHYDBLCDA-UHFFFAOYSA-N OP(=O)OC1=CC=C([N+]([O-])=O)C=C1 Chemical compound OP(=O)OC1=CC=C([N+]([O-])=O)C=C1 OUXCBXHYDBLCDA-UHFFFAOYSA-N 0.000 description 1
- 241000712464 Orthomyxoviridae Species 0.000 description 1
- 206010068786 Overlap syndrome Diseases 0.000 description 1
- 206010033733 Papule Diseases 0.000 description 1
- 208000002774 Paraproteinemias Diseases 0.000 description 1
- 208000030852 Parasitic disease Diseases 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- 206010073286 Pathologic myopia Diseases 0.000 description 1
- 229940124908 Pediarix Drugs 0.000 description 1
- 229940124909 PedvaxHIB Drugs 0.000 description 1
- 241000721454 Pemphigus Species 0.000 description 1
- 229930195708 Penicillin V Natural products 0.000 description 1
- 229940124910 Pentacel Drugs 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 239000004264 Petrolatum Substances 0.000 description 1
- 206010057249 Phagocytosis Diseases 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical class [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 241000709664 Picornaviridae Species 0.000 description 1
- 206010035664 Pneumonia Diseases 0.000 description 1
- 208000000474 Poliomyelitis Diseases 0.000 description 1
- 241000700625 Poxviridae Species 0.000 description 1
- 229940124950 Prevnar 13 Drugs 0.000 description 1
- 208000002158 Proliferative Vitreoretinopathy Diseases 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 1
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 1
- 206010037151 Psittacosis Diseases 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- 208000006193 Pulmonary infarction Diseases 0.000 description 1
- 206010037457 Pulmonary vasculitis Diseases 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 206010063897 Renal ischaemia Diseases 0.000 description 1
- 241000702247 Reoviridae Species 0.000 description 1
- 208000013616 Respiratory Distress Syndrome Diseases 0.000 description 1
- 201000001949 Retinal Vasculitis Diseases 0.000 description 1
- 206010038848 Retinal detachment Diseases 0.000 description 1
- 206010057430 Retinal injury Diseases 0.000 description 1
- 206010038910 Retinitis Diseases 0.000 description 1
- 208000007014 Retinitis pigmentosa Diseases 0.000 description 1
- 206010038915 Retinitis viral Diseases 0.000 description 1
- 206010038933 Retinopathy of prematurity Diseases 0.000 description 1
- 206010038934 Retinopathy proliferative Diseases 0.000 description 1
- 241000711931 Rhabdoviridae Species 0.000 description 1
- 241000293871 Salmonella enterica subsp. enterica serovar Typhi Species 0.000 description 1
- 206010039705 Scleritis Diseases 0.000 description 1
- 206010040047 Sepsis Diseases 0.000 description 1
- 208000009714 Severe Dengue Diseases 0.000 description 1
- 108010079723 Shiga Toxin Proteins 0.000 description 1
- 208000037549 Shiga toxin-associated hemolytic uremic syndrome Diseases 0.000 description 1
- 241000700584 Simplexvirus Species 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- BCKXLBQYZLBQEK-KVVVOXFISA-M Sodium oleate Chemical compound [Na+].CCCCCCCC\C=C/CCCCCCCC([O-])=O BCKXLBQYZLBQEK-KVVVOXFISA-M 0.000 description 1
- 229920002125 Sokalan® Polymers 0.000 description 1
- 239000004187 Spiramycin Substances 0.000 description 1
- 206010041660 Splenomegaly Diseases 0.000 description 1
- 241000191967 Staphylococcus aureus Species 0.000 description 1
- 206010061372 Streptococcal infection Diseases 0.000 description 1
- 241000194017 Streptococcus Species 0.000 description 1
- 241000193996 Streptococcus pyogenes Species 0.000 description 1
- 208000006011 Stroke Diseases 0.000 description 1
- DQDZQHMCPDUUPC-UHFFFAOYSA-N Sulfadimethoxine sodium Chemical compound [Na+].COC1=NC(OC)=CC([N-]S(=O)(=O)C=2C=CC(N)=CC=2)=N1 DQDZQHMCPDUUPC-UHFFFAOYSA-N 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 102100037942 Suppressor of tumorigenicity 14 protein Human genes 0.000 description 1
- 206010042742 Sympathetic ophthalmia Diseases 0.000 description 1
- 208000031932 Systemic capillary leak syndrome Diseases 0.000 description 1
- 229940124930 TENIVAC Drugs 0.000 description 1
- 229940124929 TYPHIM Vi Drugs 0.000 description 1
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 1
- 108010053950 Teicoplanin Proteins 0.000 description 1
- 206010043189 Telangiectasia Diseases 0.000 description 1
- 239000004098 Tetracycline Substances 0.000 description 1
- 229920002807 Thiomer Polymers 0.000 description 1
- 206010043561 Thrombocytopenic purpura Diseases 0.000 description 1
- 201000007023 Thrombotic Thrombocytopenic Purpura Diseases 0.000 description 1
- HJLSLZFTEKNLFI-UHFFFAOYSA-N Tinidazole Chemical compound CCS(=O)(=O)CCN1C(C)=NC=C1[N+]([O-])=O HJLSLZFTEKNLFI-UHFFFAOYSA-N 0.000 description 1
- 239000004012 Tofacitinib Substances 0.000 description 1
- 241000710924 Togaviridae Species 0.000 description 1
- 201000005485 Toxoplasmosis Diseases 0.000 description 1
- GSNOZLZNQMLSKJ-UHFFFAOYSA-N Trapidil Chemical compound CCN(CC)C1=CC(C)=NC2=NC=NN12 GSNOZLZNQMLSKJ-UHFFFAOYSA-N 0.000 description 1
- 208000030886 Traumatic Brain injury Diseases 0.000 description 1
- 241000589884 Treponema pallidum Species 0.000 description 1
- 229940124923 Tripedia Drugs 0.000 description 1
- 206010052568 Urticaria chronic Diseases 0.000 description 1
- 102100038282 V-type immunoglobulin domain-containing suppressor of T-cell activation Human genes 0.000 description 1
- 241000607626 Vibrio cholerae Species 0.000 description 1
- 206010047663 Vitritis Diseases 0.000 description 1
- 206010052428 Wound Diseases 0.000 description 1
- 241000289690 Xenarthra Species 0.000 description 1
- 241000607479 Yersinia pestis Species 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- KPFBUSLHFFWMAI-HYRPPVSQSA-N [(8r,9s,10r,13s,14s,17r)-17-acetyl-6-formyl-3-methoxy-10,13-dimethyl-1,2,7,8,9,11,12,14,15,16-decahydrocyclopenta[a]phenanthren-17-yl] acetate Chemical compound C1C[C@@H]2[C@](CCC(OC)=C3)(C)C3=C(C=O)C[C@H]2[C@@H]2CC[C@](OC(C)=O)(C(C)=O)[C@]21C KPFBUSLHFFWMAI-HYRPPVSQSA-N 0.000 description 1
- AIAXOZVCSOIQTJ-UHFFFAOYSA-N [O-][N+](C1=CC=CC=C1OP(O)=O)=O Chemical compound [O-][N+](C1=CC=CC=C1OP(O)=O)=O AIAXOZVCSOIQTJ-UHFFFAOYSA-N 0.000 description 1
- FPQVGDGSRVMNMR-JCTPKUEWSA-N [[(z)-(1-cyano-2-ethoxy-2-oxoethylidene)amino]oxy-(dimethylamino)methylidene]-dimethylazanium;tetrafluoroborate Chemical compound F[B-](F)(F)F.CCOC(=O)C(\C#N)=N/OC(N(C)C)=[N+](C)C FPQVGDGSRVMNMR-JCTPKUEWSA-N 0.000 description 1
- GPDHNZNLPKYHCN-DZOOLQPHSA-N [[(z)-(1-cyano-2-ethoxy-2-oxoethylidene)amino]oxy-morpholin-4-ylmethylidene]-dimethylazanium;hexafluorophosphate Chemical compound F[P-](F)(F)(F)(F)F.CCOC(=O)C(\C#N)=N/OC(=[N+](C)C)N1CCOCC1 GPDHNZNLPKYHCN-DZOOLQPHSA-N 0.000 description 1
- SORGEQQSQGNZFI-UHFFFAOYSA-N [azido(phenoxy)phosphoryl]oxybenzene Chemical compound C=1C=CC=CC=1OP(=O)(N=[N+]=[N-])OC1=CC=CC=C1 SORGEQQSQGNZFI-UHFFFAOYSA-N 0.000 description 1
- 230000001594 aberrant effect Effects 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 229960000583 acetic acid Drugs 0.000 description 1
- 125000000218 acetic acid group Chemical group C(C)(=O)* 0.000 description 1
- FHEAIOHRHQGZPC-KIWGSFCNSA-N acetic acid;(2s)-2-amino-3-(4-hydroxyphenyl)propanoic acid;(2s)-2-aminopentanedioic acid;(2s)-2-aminopropanoic acid;(2s)-2,6-diaminohexanoic acid Chemical compound CC(O)=O.C[C@H](N)C(O)=O.NCCCC[C@H](N)C(O)=O.OC(=O)[C@@H](N)CCC(O)=O.OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 FHEAIOHRHQGZPC-KIWGSFCNSA-N 0.000 description 1
- 229940049505 achromycin v Drugs 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 206010000496 acne Diseases 0.000 description 1
- 208000015228 acquired partial lipodystrophy Diseases 0.000 description 1
- 229940119059 actemra Drugs 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 208000024340 acute graft versus host disease Diseases 0.000 description 1
- 229940102614 adacel Drugs 0.000 description 1
- 210000005006 adaptive immune system Anatomy 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 208000013228 adenopathy Diseases 0.000 description 1
- 108700010877 adenoviridae proteins Proteins 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 208000011341 adult acute respiratory distress syndrome Diseases 0.000 description 1
- 201000000028 adult respiratory distress syndrome Diseases 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 229960002833 aflibercept Drugs 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 230000004520 agglutination Effects 0.000 description 1
- 229940006546 albon Drugs 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- JMLGXYWHNOKLBE-HOTXNYTESA-A alicaforsen sodium Chemical compound [Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](COP([O-])(=S)O[C@@H]2[C@H](O[C@H](C2)N2C(N=C(N)C=C2)=O)COP([O-])(=S)O[C@@H]2[C@H](O[C@H](C2)N2C3=C(C(NC(N)=N3)=O)N=C2)COP([O-])(=S)O[C@@H]2[C@H](O[C@H](C2)N2C3=NC=NC(N)=C3N=C2)COP([O-])(=S)O[C@@H]2[C@H](O[C@H](C2)N2C3=NC=NC(N)=C3N=C2)COP([O-])(=S)O[C@@H]2[C@H](O[C@H](C2)N2C(N=C(N)C=C2)=O)COP([O-])(=S)O[C@@H]2[C@H](O[C@H](C2)N2C(N=C(N)C=C2)=O)COP([O-])(=S)O[C@@H]2[C@H](O[C@H](C2)N2C(N=C(N)C=C2)=O)COP([O-])(=S)O[C@@H]2[C@H](O[C@H](C2)N2C3=C(C(NC(N)=N3)=O)N=C2)CO)[C@@H](OP([O-])(=S)OC[C@@H]2[C@H](C[C@@H](O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OP([O-])(=S)OC[C@@H]2[C@H](C[C@@H](O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OP([O-])(=S)OC[C@@H]2[C@H](C[C@@H](O2)N2C(N=C(N)C=C2)=O)OP([O-])(=S)OC[C@@H]2[C@H](C[C@@H](O2)N2C3=NC=NC(N)=C3N=C2)OP([O-])(=S)OC[C@@H]2[C@H](C[C@@H](O2)N2C(NC(=O)C(C)=C2)=O)OP([O-])(=S)OC[C@@H]2[C@H](C[C@@H](O2)N2C(N=C(N)C=C2)=O)OP([O-])(=S)OC[C@@H]2[C@H](C[C@@H](O2)N2C(N=C(N)C=C2)=O)OP([O-])(=S)OC[C@@H]2[C@H](C[C@@H](O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OP([O-])(=S)OC[C@@H]2[C@H](C[C@@H](O2)N2C(NC(=O)C(C)=C2)=O)OP([O-])(=S)OC[C@@H]2[C@H](C[C@@H](O2)N2C(N=C(N)C=C2)=O)OP([O-])(=S)OC[C@@H]2[C@H](C[C@@H](O2)N2C3=NC=NC(N)=C3N=C2)O)C1 JMLGXYWHNOKLBE-HOTXNYTESA-A 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229960000711 alprostadil Drugs 0.000 description 1
- 229960004821 amikacin Drugs 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 229960003022 amoxicillin Drugs 0.000 description 1
- 229940090588 amoxil Drugs 0.000 description 1
- 229940043312 ampicillin / sulbactam Drugs 0.000 description 1
- 238000002399 angioplasty Methods 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 235000021120 animal protein Nutrition 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 230000002924 anti-infective effect Effects 0.000 description 1
- 229940124599 anti-inflammatory drug Drugs 0.000 description 1
- 230000001064 anti-interferon Effects 0.000 description 1
- 230000003460 anti-nuclear Effects 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 230000001494 anti-thymocyte effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 230000009830 antibody antigen interaction Effects 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- VLAXZGHHBIJLAD-UHFFFAOYSA-N arsphenamine Chemical compound [Cl-].[Cl-].C1=C(O)C([NH3+])=CC([As]=[As]C=2C=C([NH3+])C(O)=CC=2)=C1 VLAXZGHHBIJLAD-UHFFFAOYSA-N 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 239000010425 asbestos Substances 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 239000000305 astragalus gummifer gum Substances 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 208000010668 atopic eczema Diseases 0.000 description 1
- 230000037444 atrophy Effects 0.000 description 1
- 208000032013 atypical susceptibility to 1 hemolytic uremic syndrome Diseases 0.000 description 1
- 229940057415 aubagio Drugs 0.000 description 1
- 229940098164 augmentin Drugs 0.000 description 1
- AUJRCFUBUPVWSZ-XTZHGVARSA-M auranofin Chemical compound CCP(CC)(CC)=[Au]S[C@@H]1O[C@H](COC(C)=O)[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O AUJRCFUBUPVWSZ-XTZHGVARSA-M 0.000 description 1
- 229960005207 auranofin Drugs 0.000 description 1
- 230000006472 autoimmune response Effects 0.000 description 1
- 230000005784 autoimmunity Effects 0.000 description 1
- 229950006867 avacincaptad pegol Drugs 0.000 description 1
- 229940062316 avelox Drugs 0.000 description 1
- 229940003504 avonex Drugs 0.000 description 1
- XOEMATDHVZOBSG-UHFFFAOYSA-N azafenidin Chemical compound C1=C(OCC#C)C(Cl)=CC(Cl)=C1N1C(=O)N2CCCCC2=N1 XOEMATDHVZOBSG-UHFFFAOYSA-N 0.000 description 1
- 229960002170 azathioprine Drugs 0.000 description 1
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 1
- 229960003623 azlocillin Drugs 0.000 description 1
- JTWOMNBEOCYFNV-NFFDBFGFSA-N azlocillin Chemical compound N([C@@H](C(=O)N[C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C=1C=CC=CC=1)C(=O)N1CCNC1=O JTWOMNBEOCYFNV-NFFDBFGFSA-N 0.000 description 1
- 229960003644 aztreonam Drugs 0.000 description 1
- WZPBZJONDBGPKJ-VEHQQRBSSA-N aztreonam Chemical compound O=C1N(S([O-])(=O)=O)[C@@H](C)[C@@H]1NC(=O)C(=N/OC(C)(C)C(O)=O)\C1=CSC([NH3+])=N1 WZPBZJONDBGPKJ-VEHQQRBSSA-N 0.000 description 1
- 229940065181 bacillus anthracis Drugs 0.000 description 1
- 229960001212 bacterial vaccine Drugs 0.000 description 1
- 229940028420 bactroban Drugs 0.000 description 1
- 238000000498 ball milling Methods 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 210000002469 basement membrane Anatomy 0.000 description 1
- 229960003270 belimumab Drugs 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 229940022836 benlysta Drugs 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- 235000012216 bentonite Nutrition 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- FYXKZNLBZKRYSS-UHFFFAOYSA-N benzene-1,2-dicarbonyl chloride Chemical compound ClC(=O)C1=CC=CC=C1C(Cl)=O FYXKZNLBZKRYSS-UHFFFAOYSA-N 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- 229960004217 benzyl alcohol Drugs 0.000 description 1
- 229910052790 beryllium Inorganic materials 0.000 description 1
- ATBAMAFKBVZNFJ-UHFFFAOYSA-N beryllium atom Chemical compound [Be] ATBAMAFKBVZNFJ-UHFFFAOYSA-N 0.000 description 1
- AEMOLEFTQBMNLQ-QIUUJYRFSA-N beta-D-glucuronic acid Chemical compound O[C@@H]1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-QIUUJYRFSA-N 0.000 description 1
- 108010051210 beta-Fructofuranosidase Proteins 0.000 description 1
- 229940021459 betaseron Drugs 0.000 description 1
- 229940090821 bexsero Drugs 0.000 description 1
- 229940087430 biaxin Drugs 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 108091008324 binding proteins Proteins 0.000 description 1
- 239000012620 biological material Substances 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 1
- 229940057194 bleph-10 Drugs 0.000 description 1
- 230000008499 blood brain barrier function Effects 0.000 description 1
- 210000000601 blood cell Anatomy 0.000 description 1
- 210000001218 blood-brain barrier Anatomy 0.000 description 1
- 229910021538 borax Inorganic materials 0.000 description 1
- 238000005271 boronizing Methods 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 230000007885 bronchoconstriction Effects 0.000 description 1
- 235000021162 brunch Nutrition 0.000 description 1
- 201000004781 bullous keratopathy Diseases 0.000 description 1
- 208000000594 bullous pemphigoid Diseases 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 235000011132 calcium sulphate Nutrition 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 229940063703 capastat Drugs 0.000 description 1
- 229960004602 capreomycin Drugs 0.000 description 1
- 229960003669 carbenicillin Drugs 0.000 description 1
- 150000001718 carbodiimides Chemical class 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 230000002612 cardiopulmonary effect Effects 0.000 description 1
- 229940097644 cedax Drugs 0.000 description 1
- QYIYFLOTGYLRGG-GPCCPHFNSA-N cefaclor Chemical compound C1([C@H](C(=O)N[C@@H]2C(N3C(=C(Cl)CS[C@@H]32)C(O)=O)=O)N)=CC=CC=C1 QYIYFLOTGYLRGG-GPCCPHFNSA-N 0.000 description 1
- 229960005361 cefaclor Drugs 0.000 description 1
- 229960000603 cefalotin Drugs 0.000 description 1
- 229960004350 cefapirin Drugs 0.000 description 1
- FLKYBGKDCCEQQM-WYUVZMMLSA-M cefazolin sodium Chemical compound [Na+].S1C(C)=NN=C1SCC1=C(C([O-])=O)N2C(=O)[C@@H](NC(=O)CN3N=NN=C3)[C@H]2SC1 FLKYBGKDCCEQQM-WYUVZMMLSA-M 0.000 description 1
- 229960004069 cefditoren Drugs 0.000 description 1
- 229960002100 cefepime Drugs 0.000 description 1
- 229940088508 cefizox Drugs 0.000 description 1
- GCFBRXLSHGKWDP-XCGNWRKASA-N cefoperazone Chemical compound O=C1C(=O)N(CC)CCN1C(=O)N[C@H](C=1C=CC(O)=CC=1)C(=O)N[C@@H]1C(=O)N2C(C(O)=O)=C(CSC=3N(N=NN=3)C)CS[C@@H]21 GCFBRXLSHGKWDP-XCGNWRKASA-N 0.000 description 1
- 229960005090 cefpodoxime Drugs 0.000 description 1
- WYUSVOMTXWRGEK-HBWVYFAYSA-N cefpodoxime Chemical compound N([C@H]1[C@@H]2N(C1=O)C(=C(CS2)COC)C(O)=O)C(=O)C(=N/OC)\C1=CSC(N)=N1 WYUSVOMTXWRGEK-HBWVYFAYSA-N 0.000 description 1
- LTINZAODLRIQIX-FBXRGJNPSA-N cefpodoxime proxetil Chemical compound N([C@H]1[C@@H]2N(C1=O)C(=C(CS2)COC)C(=O)OC(C)OC(=O)OC(C)C)C(=O)C(=N/OC)\C1=CSC(N)=N1 LTINZAODLRIQIX-FBXRGJNPSA-N 0.000 description 1
- 229960000484 ceftazidime Drugs 0.000 description 1
- 229960004086 ceftibuten Drugs 0.000 description 1
- 229960005229 ceftiofur Drugs 0.000 description 1
- ZBHXIWJRIFEVQY-IHMPYVIRSA-N ceftiofur Chemical compound S([C@@H]1[C@@H](C(N1C=1C(O)=O)=O)NC(=O)\C(=N/OC)C=2N=C(N)SC=2)CC=1CSC(=O)C1=CC=CO1 ZBHXIWJRIFEVQY-IHMPYVIRSA-N 0.000 description 1
- 229960001991 ceftizoxime Drugs 0.000 description 1
- NNULBSISHYWZJU-LLKWHZGFSA-N ceftizoxime Chemical compound N([C@@H]1C(N2C(=CCS[C@@H]21)C(O)=O)=O)C(=O)\C(=N/OC)C1=CSC(N)=N1 NNULBSISHYWZJU-LLKWHZGFSA-N 0.000 description 1
- 229960002620 cefuroxime axetil Drugs 0.000 description 1
- 230000005779 cell damage Effects 0.000 description 1
- 208000037887 cell injury Diseases 0.000 description 1
- 230000006037 cell lysis Effects 0.000 description 1
- 238000002659 cell therapy Methods 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 201000005667 central retinal vein occlusion Diseases 0.000 description 1
- AVGYWQBCYZHHPN-CYJZLJNKSA-N cephalexin monohydrate Chemical compound O.C1([C@@H](N)C(=O)N[C@H]2[C@@H]3N(C2=O)C(=C(CS3)C)C(O)=O)=CC=CC=C1 AVGYWQBCYZHHPN-CYJZLJNKSA-N 0.000 description 1
- 229940124587 cephalosporin Drugs 0.000 description 1
- 150000001780 cephalosporins Chemical class 0.000 description 1
- VUFGUVLLDPOSBC-XRZFDKQNSA-M cephalothin sodium Chemical compound [Na+].N([C@H]1[C@@H]2N(C1=O)C(=C(CS2)COC(=O)C)C([O-])=O)C(=O)CC1=CC=CS1 VUFGUVLLDPOSBC-XRZFDKQNSA-M 0.000 description 1
- 229960003115 certolizumab pegol Drugs 0.000 description 1
- 210000003467 cheek Anatomy 0.000 description 1
- AZFBLLCNOQPJGJ-DVAZEERGSA-N chembl1332607 Chemical compound O([C@](C1=O)(C)O\C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)/C=C\C=C(C)/C(=O)N2)C)OC)C(C(=C3O)C)=C1C(C1=N4)=C3C(O)=C2C1=NC14CCN(CC(C)C)CC1 AZFBLLCNOQPJGJ-DVAZEERGSA-N 0.000 description 1
- DDTDNCYHLGRFBM-YZEKDTGTSA-N chembl2367892 Chemical compound CC(=O)N[C@H]1[C@@H](O)[C@H](O)[C@H](CO)O[C@H]1O[C@@H]([C@H]1C(N[C@@H](C2=CC(O)=CC(O[C@@H]3[C@H]([C@H](O)[C@H](O)[C@@H](CO)O3)O)=C2C=2C(O)=CC=C(C=2)[C@@H](NC(=O)[C@@H]2NC(=O)[C@@H]3C=4C=C(O)C=C(C=4)OC=4C(O)=CC=C(C=4)[C@@H](N)C(=O)N[C@H](CC=4C=C(Cl)C(O5)=CC=4)C(=O)N3)C(=O)N1)C(O)=O)=O)C(C=C1Cl)=CC=C1OC1=C(O[C@H]3[C@H]([C@@H](O)[C@H](O)[C@H](CO)O3)NC(C)=O)C5=CC2=C1 DDTDNCYHLGRFBM-YZEKDTGTSA-N 0.000 description 1
- 230000035605 chemotaxis Effects 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 229960005091 chloramphenicol Drugs 0.000 description 1
- 239000012320 chlorinating reagent Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 229960003677 chloroquine Drugs 0.000 description 1
- WHTVZRBIWZFKQO-UHFFFAOYSA-N chloroquine Natural products ClC1=CC=C2C(NC(C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-UHFFFAOYSA-N 0.000 description 1
- 210000003161 choroid Anatomy 0.000 description 1
- 208000017760 chronic graft versus host disease Diseases 0.000 description 1
- 208000020832 chronic kidney disease Diseases 0.000 description 1
- 208000024376 chronic urticaria Diseases 0.000 description 1
- 229960001265 ciclosporin Drugs 0.000 description 1
- DHSUYTOATWAVLW-WFVMDLQDSA-N cilastatin Chemical compound CC1(C)C[C@@H]1C(=O)N\C(=C/CCCCSC[C@H](N)C(O)=O)C(O)=O DHSUYTOATWAVLW-WFVMDLQDSA-N 0.000 description 1
- 229960002436 cladribine Drugs 0.000 description 1
- 229940088530 claforan Drugs 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 229940063193 cleocin Drugs 0.000 description 1
- 229960003326 cloxacillin Drugs 0.000 description 1
- 229940047766 co-trimoxazole Drugs 0.000 description 1
- 239000002817 coal dust Substances 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 229940121394 complement c5 inhibitor Drugs 0.000 description 1
- 239000002720 complement component C5 inhibitor Substances 0.000 description 1
- 230000001010 compromised effect Effects 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 229940031670 conjugate vaccine Drugs 0.000 description 1
- 208000010247 contact dermatitis Diseases 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 239000012050 conventional carrier Substances 0.000 description 1
- 229940038717 copaxone Drugs 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- IDLFZVILOHSSID-OVLDLUHVSA-N corticotropin Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)NC(=O)[C@@H](N)CO)C1=CC=C(O)C=C1 IDLFZVILOHSSID-OVLDLUHVSA-N 0.000 description 1
- 229960000258 corticotropin Drugs 0.000 description 1
- 229940010466 cosentyx Drugs 0.000 description 1
- 239000007822 coupling agent Substances 0.000 description 1
- 239000006059 cover glass Substances 0.000 description 1
- 229940111134 coxibs Drugs 0.000 description 1
- DYNHJHQFHQTFTP-UHFFFAOYSA-N crenolanib Chemical compound C=1C=C2N(C=3N=C4C(N5CCC(N)CC5)=CC=CC4=CC=3)C=NC2=CC=1OCC1(C)COC1 DYNHJHQFHQTFTP-UHFFFAOYSA-N 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 229940021392 cubicin Drugs 0.000 description 1
- 239000003255 cyclooxygenase 2 inhibitor Substances 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 229960003077 cycloserine Drugs 0.000 description 1
- 229930182912 cyclosporin Natural products 0.000 description 1
- 229960002488 dalbavancin Drugs 0.000 description 1
- 108700009376 dalbavancin Proteins 0.000 description 1
- 229960000860 dapsone Drugs 0.000 description 1
- 229960005484 daptomycin Drugs 0.000 description 1
- 229940088965 declomycin Drugs 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 229920006237 degradable polymer Polymers 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 229940027008 deltasone Drugs 0.000 description 1
- 229960002398 demeclocycline Drugs 0.000 description 1
- 208000025729 dengue disease Diseases 0.000 description 1
- 230000000779 depleting effect Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000003795 desorption Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 201000011190 diabetic macular edema Diseases 0.000 description 1
- ZOCHARZZJNPSEU-UHFFFAOYSA-N diboron Chemical compound B#B ZOCHARZZJNPSEU-UHFFFAOYSA-N 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 229960001259 diclofenac Drugs 0.000 description 1
- DCOPUUMXTXDBNB-UHFFFAOYSA-N diclofenac Chemical compound OC(=O)CC1=CC=CC=C1NC1=C(Cl)C=CC=C1Cl DCOPUUMXTXDBNB-UHFFFAOYSA-N 0.000 description 1
- 229960001585 dicloxacillin Drugs 0.000 description 1
- YFAGHNZHGGCZAX-JKIFEVAISA-N dicloxacillin Chemical compound N([C@@H]1C(N2[C@H](C(C)(C)S[C@@H]21)C(O)=O)=O)C(=O)C1=C(C)ON=C1C1=C(Cl)C=CC=C1Cl YFAGHNZHGGCZAX-JKIFEVAISA-N 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 238000001938 differential scanning calorimetry curve Methods 0.000 description 1
- LDCRTTXIJACKKU-ONEGZZNKSA-N dimethyl fumarate Chemical compound COC(=O)\C=C\C(=O)OC LDCRTTXIJACKKU-ONEGZZNKSA-N 0.000 description 1
- 229960004419 dimethyl fumarate Drugs 0.000 description 1
- QLZHNIAADXEJJP-UHFFFAOYSA-L dioxido-oxo-phenyl-$l^{5}-phosphane Chemical compound [O-]P([O-])(=O)C1=CC=CC=C1 QLZHNIAADXEJJP-UHFFFAOYSA-L 0.000 description 1
- ASMQGLCHMVWBQR-UHFFFAOYSA-M diphenyl phosphate Chemical compound C=1C=CC=CC=1OP(=O)([O-])OC1=CC=CC=C1 ASMQGLCHMVWBQR-UHFFFAOYSA-M 0.000 description 1
- 239000001177 diphosphate Substances 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 229960005097 diphtheria vaccines Drugs 0.000 description 1
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 1
- 208000037765 diseases and disorders Diseases 0.000 description 1
- KDQPSPMLNJTZAL-UHFFFAOYSA-L disodium hydrogenphosphate dihydrate Chemical compound O.O.[Na+].[Na+].OP([O-])([O-])=O KDQPSPMLNJTZAL-UHFFFAOYSA-L 0.000 description 1
- NWOYIVRVSJDTLK-YSDBFZIDSA-L disodium;(2s,5r,6r)-6-[[(2r)-2-amino-2-phenylacetyl]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylate;(1r,4s)-3,3-dimethyl-2,2,6-trioxo-2$l^{6}-thiabicyclo[3.2.0]heptane-4-carboxylate Chemical compound [Na+].[Na+].O=S1(=O)C(C)(C)[C@H](C([O-])=O)C2C(=O)C[C@H]21.C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C([O-])=O)(C)C)=CC=CC=C1 NWOYIVRVSJDTLK-YSDBFZIDSA-L 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 229940111539 doribax Drugs 0.000 description 1
- 229960000895 doripenem Drugs 0.000 description 1
- 229960003722 doxycycline Drugs 0.000 description 1
- 229940088679 drug related substance Drugs 0.000 description 1
- 208000011325 dry age related macular degeneration Diseases 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 210000005069 ears Anatomy 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 229960002549 enoxacin Drugs 0.000 description 1
- IDYZIJYBMGIQMJ-UHFFFAOYSA-N enoxacin Chemical compound N1=C2N(CC)C=C(C(O)=O)C(=O)C2=CC(F)=C1N1CCNCC1 IDYZIJYBMGIQMJ-UHFFFAOYSA-N 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 210000003979 eosinophil Anatomy 0.000 description 1
- 210000002615 epidermis Anatomy 0.000 description 1
- 201000011114 epidermolysis bullosa acquisita Diseases 0.000 description 1
- 206010015037 epilepsy Diseases 0.000 description 1
- 229960002770 ertapenem Drugs 0.000 description 1
- 231100000321 erythema Toxicity 0.000 description 1
- 210000003743 erythrocyte Anatomy 0.000 description 1
- 229960000285 ethambutol Drugs 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 229960002001 ethionamide Drugs 0.000 description 1
- LCFXLZAXGXOXAP-QPJJXVBHSA-N ethyl (2e)-2-cyano-2-hydroxyiminoacetate Chemical compound CCOC(=O)C(=N\O)\C#N LCFXLZAXGXOXAP-QPJJXVBHSA-N 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 229940077362 extavia Drugs 0.000 description 1
- 210000001723 extracellular space Anatomy 0.000 description 1
- 201000001155 extrinsic allergic alveolitis Diseases 0.000 description 1
- 229940107247 factive Drugs 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 230000004129 fatty acid metabolism Effects 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 230000003176 fibrotic effect Effects 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 230000009969 flowable effect Effects 0.000 description 1
- 229940072686 floxin Drugs 0.000 description 1
- 229960004369 flufenamic acid Drugs 0.000 description 1
- LPEPZBJOKDYZAD-UHFFFAOYSA-N flufenamic acid Chemical compound OC(=O)C1=CC=CC=C1NC1=CC=CC(C(F)(F)F)=C1 LPEPZBJOKDYZAD-UHFFFAOYSA-N 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 239000012025 fluorinating agent Substances 0.000 description 1
- 229940124307 fluoroquinolone Drugs 0.000 description 1
- IJJVMEJXYNJXOJ-UHFFFAOYSA-N fluquinconazole Chemical compound C=1C=C(Cl)C=C(Cl)C=1N1C(=O)C2=CC(F)=CC=C2N=C1N1C=NC=N1 IJJVMEJXYNJXOJ-UHFFFAOYSA-N 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 229940089936 fortaz Drugs 0.000 description 1
- 229960000308 fosfomycin Drugs 0.000 description 1
- YMDXZJFXQJVXBF-STHAYSLISA-N fosfomycin Chemical compound C[C@@H]1O[C@@H]1P(O)(O)=O YMDXZJFXQJVXBF-STHAYSLISA-N 0.000 description 1
- 229940118764 francisella tularensis Drugs 0.000 description 1
- 229940048400 fucidin Drugs 0.000 description 1
- 229960004675 fusidic acid Drugs 0.000 description 1
- IECPWNUMDGFDKC-MZJAQBGESA-N fusidic acid Chemical compound O[C@@H]([C@@H]12)C[C@H]3\C(=C(/CCC=C(C)C)C(O)=O)[C@@H](OC(C)=O)C[C@]3(C)[C@@]2(C)CC[C@@H]2[C@]1(C)CC[C@@H](O)[C@H]2C IECPWNUMDGFDKC-MZJAQBGESA-N 0.000 description 1
- 229940072360 garamycin Drugs 0.000 description 1
- 238000004868 gas analysis Methods 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 229960003923 gatifloxacin Drugs 0.000 description 1
- XUBOMFCQGDBHNK-UHFFFAOYSA-N gatifloxacin Chemical compound FC1=CC(C(C(C(O)=O)=CN2C3CC3)=O)=C2C(OC)=C1N1CCNC(C)C1 XUBOMFCQGDBHNK-UHFFFAOYSA-N 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000007897 gelcap Substances 0.000 description 1
- QTQAWLPCGQOSGP-GBTDJJJQSA-N geldanamycin Chemical compound N1C(=O)\C(C)=C/C=C\[C@@H](OC)[C@H](OC(N)=O)\C(C)=C/[C@@H](C)[C@@H](O)[C@H](OC)C[C@@H](C)CC2=C(OC)C(=O)C=C1C2=O QTQAWLPCGQOSGP-GBTDJJJQSA-N 0.000 description 1
- JIYMVSQRGZEYAX-CWUUNJJBSA-N gemifloxacin mesylate Chemical compound CS(O)(=O)=O.C1C(CN)C(=N/OC)/CN1C(C(=C1)F)=NC2=C1C(=O)C(C(O)=O)=CN2C1CC1 JIYMVSQRGZEYAX-CWUUNJJBSA-N 0.000 description 1
- 229960002518 gentamicin Drugs 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 229960003776 glatiramer acetate Drugs 0.000 description 1
- 230000001434 glomerular Effects 0.000 description 1
- 229960003180 glutathione Drugs 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- 208000035474 group of disease Diseases 0.000 description 1
- 208000019622 heart disease Diseases 0.000 description 1
- 208000014951 hematologic disease Diseases 0.000 description 1
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 1
- 238000009957 hemming Methods 0.000 description 1
- 231100000283 hepatitis Toxicity 0.000 description 1
- 208000005252 hepatitis A Diseases 0.000 description 1
- 229940124724 hepatitis-A vaccine Drugs 0.000 description 1
- 229940124736 hepatitis-B vaccine Drugs 0.000 description 1
- 229940124737 hepatitis-C vaccine Drugs 0.000 description 1
- MCAHMSDENAOJFZ-BVXDHVRPSA-N herbimycin Chemical compound N1C(=O)\C(C)=C\C=C/[C@H](OC)[C@@H](OC(N)=O)\C(C)=C\[C@H](C)[C@@H](OC)[C@@H](OC)C[C@H](C)[C@@H](OC)C2=CC(=O)C=C1C2=O MCAHMSDENAOJFZ-BVXDHVRPSA-N 0.000 description 1
- 229930193320 herbimycin Natural products 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- 150000002391 heterocyclic compounds Chemical class 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 238000005570 heteronuclear single quantum coherence Methods 0.000 description 1
- 238000000589 high-performance liquid chromatography-mass spectrometry Methods 0.000 description 1
- 229940048921 humira Drugs 0.000 description 1
- 208000013653 hyalitis Diseases 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical compound [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 229910000037 hydrogen sulfide Inorganic materials 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 208000022098 hypersensitivity pneumonitis Diseases 0.000 description 1
- 229960001680 ibuprofen Drugs 0.000 description 1
- 229960002240 iloprost Drugs 0.000 description 1
- HIFJCPQKFCZDDL-ACWOEMLNSA-N iloprost Chemical compound C1\C(=C/CCCC(O)=O)C[C@@H]2[C@@H](/C=C/[C@@H](O)C(C)CC#CC)[C@H](O)C[C@@H]21 HIFJCPQKFCZDDL-ACWOEMLNSA-N 0.000 description 1
- 229960002411 imatinib Drugs 0.000 description 1
- 229960003685 imatinib mesylate Drugs 0.000 description 1
- YLMAHDNUQAMNNX-UHFFFAOYSA-N imatinib methanesulfonate Chemical compound CS(O)(=O)=O.C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 YLMAHDNUQAMNNX-UHFFFAOYSA-N 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 230000000899 immune system response Effects 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 229910052738 indium Inorganic materials 0.000 description 1
- APFVFJFRJDLVQX-UHFFFAOYSA-N indium atom Chemical compound [In] APFVFJFRJDLVQX-UHFFFAOYSA-N 0.000 description 1
- 229960000905 indomethacin Drugs 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 210000004969 inflammatory cell Anatomy 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 229940074383 interleukin-11 Drugs 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 229940054114 invanz Drugs 0.000 description 1
- 239000001573 invertase Substances 0.000 description 1
- 235000011073 invertase Nutrition 0.000 description 1
- 230000000622 irritating effect Effects 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 208000037906 ischaemic injury Diseases 0.000 description 1
- 208000028867 ischemia Diseases 0.000 description 1
- 229960003350 isoniazid Drugs 0.000 description 1
- QRXWMOHMRWLFEY-UHFFFAOYSA-N isoniazide Chemical compound NNC(=O)C1=CC=NC=C1 QRXWMOHMRWLFEY-UHFFFAOYSA-N 0.000 description 1
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 1
- 229940011051 isopropyl acetate Drugs 0.000 description 1
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 1
- 238000010902 jet-milling Methods 0.000 description 1
- 208000018934 joint symptom Diseases 0.000 description 1
- 229960000318 kanamycin Drugs 0.000 description 1
- 229930027917 kanamycin Natural products 0.000 description 1
- SBUJHOSQTJFQJX-NOAMYHISSA-N kanamycin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N SBUJHOSQTJFQJX-NOAMYHISSA-N 0.000 description 1
- 229930182823 kanamycin A Natural products 0.000 description 1
- 229940090589 keflex Drugs 0.000 description 1
- 201000006370 kidney failure Diseases 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 229940039717 lanolin Drugs 0.000 description 1
- 235000019388 lanolin Nutrition 0.000 description 1
- 229960000681 leflunomide Drugs 0.000 description 1
- VHOGYURTWQBHIL-UHFFFAOYSA-N leflunomide Chemical compound O1N=CC(C(=O)NC=2C=CC(=CC=2)C(F)(F)F)=C1C VHOGYURTWQBHIL-UHFFFAOYSA-N 0.000 description 1
- 229960003376 levofloxacin Drugs 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 229940033683 lincocin Drugs 0.000 description 1
- 229960005287 lincomycin Drugs 0.000 description 1
- MPVGZUGXCQEXTM-UHFFFAOYSA-N linifanib Chemical compound CC1=CC=C(F)C(NC(=O)NC=2C=CC(=CC=2)C=2C=3C(N)=NNC=3C=CC=2)=C1 MPVGZUGXCQEXTM-UHFFFAOYSA-N 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 229960002422 lomefloxacin Drugs 0.000 description 1
- 229960001977 loracarbef Drugs 0.000 description 1
- JAPHQRWPEGVNBT-UTUOFQBUSA-N loracarbef Chemical compound C1([C@H](C(=O)N[C@@H]2C(N3C(=C(Cl)CC[C@@H]32)C([O-])=O)=O)[NH3+])=CC=CC=C1 JAPHQRWPEGVNBT-UTUOFQBUSA-N 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 229920001684 low density polyethylene Polymers 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- 239000004702 low-density polyethylene Substances 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 230000002934 lysing effect Effects 0.000 description 1
- 201000010230 macular retinal edema Diseases 0.000 description 1
- 229960003640 mafenide Drugs 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 238000002595 magnetic resonance imaging Methods 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- WJEOLQLKVOPQFV-UHFFFAOYSA-N masitinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3SC=C(N=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 WJEOLQLKVOPQFV-UHFFFAOYSA-N 0.000 description 1
- 229940103196 maxaquin Drugs 0.000 description 1
- 229940021422 maxipime Drugs 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 229960003803 meclofenamic acid Drugs 0.000 description 1
- 229960001929 meloxicam Drugs 0.000 description 1
- 229940032713 merrem Drugs 0.000 description 1
- 210000003975 mesenteric artery Anatomy 0.000 description 1
- 208000030159 metabolic disease Diseases 0.000 description 1
- CPMDPSXJELVGJG-UHFFFAOYSA-N methyl 2-hydroxy-3-[N-[4-[methyl-[2-(4-methylpiperazin-1-yl)acetyl]amino]phenyl]-C-phenylcarbonimidoyl]-1H-indole-6-carboxylate Chemical compound OC=1NC2=CC(=CC=C2C=1C(=NC1=CC=C(C=C1)N(C(CN1CCN(CC1)C)=O)C)C1=CC=CC=C1)C(=O)OC CPMDPSXJELVGJG-UHFFFAOYSA-N 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- AMJWLACDVOGEHM-UHFFFAOYSA-N methyl-[(4-nitrophenyl)methyl]phosphinic acid Chemical compound CP(=O)(CC1=CC=C(C=C1)[N+](=O)[O-])O AMJWLACDVOGEHM-UHFFFAOYSA-N 0.000 description 1
- XLSZMDLNRCVEIJ-UHFFFAOYSA-N methylimidazole Natural products CC1=CNC=N1 XLSZMDLNRCVEIJ-UHFFFAOYSA-N 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- VAOCPAMSLUNLGC-UHFFFAOYSA-N metronidazole Chemical group CC1=NC=C([N+]([O-])=O)N1CCO VAOCPAMSLUNLGC-UHFFFAOYSA-N 0.000 description 1
- HPNSFSBZBAHARI-UHFFFAOYSA-N micophenolic acid Natural products OC1=C(CC=C(C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-UHFFFAOYSA-N 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 210000000274 microglia Anatomy 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 229940110254 minocin Drugs 0.000 description 1
- 229960001156 mitoxantrone Drugs 0.000 description 1
- ZAHQPTJLOCWVPG-UHFFFAOYSA-N mitoxantrone dihydrochloride Chemical compound Cl.Cl.O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO ZAHQPTJLOCWVPG-UHFFFAOYSA-N 0.000 description 1
- 238000002625 monoclonal antibody therapy Methods 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- BMLIZLVNXIYGCK-UHFFFAOYSA-N monuron Chemical compound CN(C)C(=O)NC1=CC=C(Cl)C=C1 BMLIZLVNXIYGCK-UHFFFAOYSA-N 0.000 description 1
- 230000004660 morphological change Effects 0.000 description 1
- 210000004400 mucous membrane Anatomy 0.000 description 1
- 229960003128 mupirocin Drugs 0.000 description 1
- 229930187697 mupirocin Natural products 0.000 description 1
- 229940052202 myambutol Drugs 0.000 description 1
- 229960000951 mycophenolic acid Drugs 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- FEKRFYZGYUTGRY-UHFFFAOYSA-N n'-ethylmethanediimine Chemical compound CCN=C=N FEKRFYZGYUTGRY-UHFFFAOYSA-N 0.000 description 1
- OMTIGRBXFBATPI-UHFFFAOYSA-N n'-methylmethanediimine Chemical compound CN=C=N OMTIGRBXFBATPI-UHFFFAOYSA-N 0.000 description 1
- MDBAAYRCFODAFZ-UHFFFAOYSA-N n'-phenylmethanediimine Chemical compound N=C=NC1=CC=CC=C1 MDBAAYRCFODAFZ-UHFFFAOYSA-N 0.000 description 1
- UPXAZUFKXWLNMF-UHFFFAOYSA-N n'-propan-2-ylmethanediimine Chemical compound CC(C)N=C=N UPXAZUFKXWLNMF-UHFFFAOYSA-N 0.000 description 1
- QHHHYLFZGYIBCX-UHFFFAOYSA-N n,n'-bis[(2,2-dimethyl-1,3-dioxolan-4-yl)methyl]methanediimine Chemical compound O1C(C)(C)OCC1CN=C=NCC1OC(C)(C)OC1 QHHHYLFZGYIBCX-UHFFFAOYSA-N 0.000 description 1
- ONDPWWDPQDCQNJ-UHFFFAOYSA-N n-(3,3-dimethyl-1,2-dihydroindol-6-yl)-2-(pyridin-4-ylmethylamino)pyridine-3-carboxamide;phosphoric acid Chemical compound OP(O)(O)=O.OP(O)(O)=O.C=1C=C2C(C)(C)CNC2=CC=1NC(=O)C1=CC=CN=C1NCC1=CC=NC=C1 ONDPWWDPQDCQNJ-UHFFFAOYSA-N 0.000 description 1
- ZESIAEVDVPWEKB-ORCFLVBFSA-N n-[(2s)-4-amino-1-[[(2s,3r)-1-[[(2s)-4-amino-1-oxo-1-[[(3s,6s,9s,12s,15r,18s,21s)-6,9,18-tris(2-aminoethyl)-3-[(1r)-1-hydroxyethyl]-12,15-bis(2-methylpropyl)-2,5,8,11,14,17,20-heptaoxo-1,4,7,10,13,16,19-heptazacyclotricos-21-yl]amino]butan-2-yl]amino]-3-h Chemical compound OS(O)(=O)=O.OS(O)(=O)=O.CC(C)CCCCC(=O)N[C@@H](CCN)C(=O)N[C@H]([C@@H](C)O)CN[C@@H](CCN)C(=O)N[C@H]1CCNC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCN)NC(=O)[C@H](CCN)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@H](CCN)NC1=O.CCC(C)CCCCC(=O)N[C@@H](CCN)C(=O)N[C@H]([C@@H](C)O)CN[C@@H](CCN)C(=O)N[C@H]1CCNC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCN)NC(=O)[C@H](CCN)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@H](CCN)NC1=O ZESIAEVDVPWEKB-ORCFLVBFSA-N 0.000 description 1
- 229960000515 nafcillin Drugs 0.000 description 1
- MHWLWQUZZRMNGJ-UHFFFAOYSA-N nalidixic acid Chemical compound C1=C(C)N=C2N(CC)C=C(C(O)=O)C(=O)C2=C1 MHWLWQUZZRMNGJ-UHFFFAOYSA-N 0.000 description 1
- 229960002009 naproxen Drugs 0.000 description 1
- CMWTZPSULFXXJA-VIFPVBQESA-N naproxen Chemical compound C1=C([C@H](C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-VIFPVBQESA-N 0.000 description 1
- 229960005027 natalizumab Drugs 0.000 description 1
- 208000021971 neovascular inflammatory vitreoretinopathy Diseases 0.000 description 1
- 210000005036 nerve Anatomy 0.000 description 1
- AGFWIZQEWFGATK-UNZHCMSXSA-N netilmicin sulfate Chemical compound OS(O)(=O)=O.OS(O)(=O)=O.OS(O)(=O)=O.OS(O)(=O)=O.OS(O)(=O)=O.O([C@@H]1[C@@H](N)C[C@H]([C@@H]([C@H]1O)O[C@@H]1[C@@H]([C@@H](NC)[C@@](C)(O)CO1)O)NCC)[C@H]1OC(CN)=CC[C@H]1N.O([C@@H]1[C@@H](N)C[C@H]([C@@H]([C@H]1O)O[C@@H]1[C@@H]([C@@H](NC)[C@@](C)(O)CO1)O)NCC)[C@H]1OC(CN)=CC[C@H]1N AGFWIZQEWFGATK-UNZHCMSXSA-N 0.000 description 1
- ZBGPYVZLYBDXKO-HILBYHGXSA-N netilmycin Chemical compound O([C@@H]1[C@@H](N)C[C@H]([C@@H]([C@H]1O)O[C@@H]1[C@]([C@H](NC)[C@@H](O)CO1)(C)O)NCC)[C@H]1OC(CN)=CC[C@H]1N ZBGPYVZLYBDXKO-HILBYHGXSA-N 0.000 description 1
- 230000004770 neurodegeneration Effects 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 210000002569 neuron Anatomy 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- JCXJVPUVTGWSNB-UHFFFAOYSA-N nitrogen dioxide Inorganic materials O=[N]=O JCXJVPUVTGWSNB-UHFFFAOYSA-N 0.000 description 1
- PULGYDLMFSFVBL-SMFNREODSA-N nociceptin Chemical compound C([C@@H](C(=O)N[C@H](C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CO)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(O)=O)[C@@H](C)O)NC(=O)CNC(=O)CNC(=O)[C@@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 PULGYDLMFSFVBL-SMFNREODSA-N 0.000 description 1
- 229960001180 norfloxacin Drugs 0.000 description 1
- 229940064764 noroxin Drugs 0.000 description 1
- 210000001331 nose Anatomy 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 229960003347 obinutuzumab Drugs 0.000 description 1
- 229960001699 ofloxacin Drugs 0.000 description 1
- 229940046166 oligodeoxynucleotide Drugs 0.000 description 1
- 230000014207 opsonization Effects 0.000 description 1
- 210000001328 optic nerve Anatomy 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 229940022146 orbactiv Drugs 0.000 description 1
- 229960004534 orgotein Drugs 0.000 description 1
- 108010070915 orgotein Proteins 0.000 description 1
- VHFGEBVPHAGQPI-MYYQHNLBSA-N oritavancin Chemical compound O([C@@H]1C2=CC=C(C(=C2)Cl)OC=2C=C3C=C(C=2O[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O[C@@H]2O[C@@H](C)[C@H](O)[C@@](C)(NCC=4C=CC(=CC=4)C=4C=CC(Cl)=CC=4)C2)OC2=CC=C(C=C2Cl)[C@@H](O)[C@H](C(N[C@@H](CC(N)=O)C(=O)N[C@H]3C(=O)N[C@H]2C(=O)N[C@@H]1C(N[C@H](C1=CC(O)=CC(O)=C1C=1C(O)=CC=C2C=1)C(O)=O)=O)=O)NC(=O)[C@@H](CC(C)C)NC)[C@H]1C[C@](C)(N)[C@@H](O)[C@H](C)O1 VHFGEBVPHAGQPI-MYYQHNLBSA-N 0.000 description 1
- 229960001607 oritavancin Drugs 0.000 description 1
- PWTROOMOPLCZHB-BHYQHFGMSA-N oritavancin bisphosphate Chemical compound OP(O)(O)=O.OP(O)(O)=O.O([C@@H]1C2=CC=C(C(=C2)Cl)OC=2C=C3C=C(C=2O[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O[C@@H]2O[C@@H](C)[C@H](O)[C@@](C)(NCC=4C=CC(=CC=4)C=4C=CC(Cl)=CC=4)C2)OC2=CC=C(C=C2Cl)[C@@H](O)[C@H](C(N[C@@H](CC(N)=O)C(=O)N[C@H]3C(=O)N[C@H]2C(=O)N[C@@H]1C(N[C@@H](C1=CC(O)=CC(O)=C1C=1C(O)=CC=C2C=1)C(O)=O)=O)=O)NC(=O)[C@@H](CC(C)C)NC)[C@H]1C[C@](C)(N)[C@@H](O)[C@H](C)O1 PWTROOMOPLCZHB-BHYQHFGMSA-N 0.000 description 1
- 238000012261 overproduction Methods 0.000 description 1
- 229960001019 oxacillin Drugs 0.000 description 1
- 229950007318 ozogamicin Drugs 0.000 description 1
- LSQZJLSUYDQPKJ-UHFFFAOYSA-N p-Hydroxyampicillin Natural products O=C1N2C(C(O)=O)C(C)(C)SC2C1NC(=O)C(N)C1=CC=C(O)C=C1 LSQZJLSUYDQPKJ-UHFFFAOYSA-N 0.000 description 1
- 125000000636 p-nitrophenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)[N+]([O-])=O 0.000 description 1
- 239000003002 pH adjusting agent Substances 0.000 description 1
- 230000036407 pain Effects 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 208000003154 papilloma Diseases 0.000 description 1
- 208000029211 papillomatosis Diseases 0.000 description 1
- 229960001914 paromomycin Drugs 0.000 description 1
- 208000036274 partial acquired susceptibility to lipodystrophy Diseases 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 230000007310 pathophysiology Effects 0.000 description 1
- 229960005492 pazopanib hydrochloride Drugs 0.000 description 1
- 229960001291 peginterferon beta-1a Drugs 0.000 description 1
- 239000003961 penetration enhancing agent Substances 0.000 description 1
- 229960001639 penicillamine Drugs 0.000 description 1
- 229940056367 penicillin v Drugs 0.000 description 1
- 238000005897 peptide coupling reaction Methods 0.000 description 1
- 208000008494 pericarditis Diseases 0.000 description 1
- 229940066827 pertussis vaccine Drugs 0.000 description 1
- 229940066842 petrolatum Drugs 0.000 description 1
- 235000019271 petrolatum Nutrition 0.000 description 1
- 229950003203 pexelizumab Drugs 0.000 description 1
- 230000008782 phagocytosis Effects 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- HCTVWSOKIJULET-LQDWTQKMSA-M phenoxymethylpenicillin potassium Chemical compound [K+].N([C@H]1[C@H]2SC([C@@H](N2C1=O)C([O-])=O)(C)C)C(=O)COC1=CC=CC=C1 HCTVWSOKIJULET-LQDWTQKMSA-M 0.000 description 1
- 229960002895 phenylbutazone Drugs 0.000 description 1
- VYMDGNCVAMGZFE-UHFFFAOYSA-N phenylbutazonum Chemical compound O=C1C(CCCC)C(=O)N(C=2C=CC=CC=2)N1C1=CC=CC=C1 VYMDGNCVAMGZFE-UHFFFAOYSA-N 0.000 description 1
- 239000002587 phosphodiesterase IV inhibitor Substances 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- 210000000608 photoreceptor cell Anatomy 0.000 description 1
- 108091008695 photoreceptors Proteins 0.000 description 1
- 125000005545 phthalimidyl group Chemical group 0.000 description 1
- 229960002292 piperacillin Drugs 0.000 description 1
- 229940104641 piperacillin / tazobactam Drugs 0.000 description 1
- WCMIIGXFCMNQDS-IDYPWDAWSA-M piperacillin sodium Chemical compound [Na+].O=C1C(=O)N(CC)CCN1C(=O)N[C@H](C=1C=CC=CC=1)C(=O)N[C@@H]1C(=O)N2[C@@H](C([O-])=O)C(C)(C)S[C@@H]21 WCMIIGXFCMNQDS-IDYPWDAWSA-M 0.000 description 1
- 229960002702 piroxicam Drugs 0.000 description 1
- QYSPLQLAKJAUJT-UHFFFAOYSA-N piroxicam Chemical compound OC=1C2=CC=CC=C2S(=O)(=O)N(C)C=1C(=O)NC1=CC=CC=N1 QYSPLQLAKJAUJT-UHFFFAOYSA-N 0.000 description 1
- 229940072689 plaquenil Drugs 0.000 description 1
- 230000033885 plasminogen activation Effects 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 229940007060 plegridy Drugs 0.000 description 1
- 208000008423 pleurisy Diseases 0.000 description 1
- 229940033515 pneumovax 23 Drugs 0.000 description 1
- 229920000724 poly(L-arginine) polymer Polymers 0.000 description 1
- 229920000447 polyanionic polymer Polymers 0.000 description 1
- 229940068917 polyethylene glycols Drugs 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 239000002675 polymer-supported reagent Substances 0.000 description 1
- 239000004926 polymethyl methacrylate Substances 0.000 description 1
- 229940031937 polysaccharide vaccine Drugs 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 229940014329 polysulfated glycosaminoglycan Drugs 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- DWHGNUUWCJZQHO-ZVDZYBSKSA-M potassium;(2s,5r,6r)-6-[[(2r)-2-amino-2-(4-hydroxyphenyl)acetyl]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid;(2r,3z,5r)-3-(2-hydroxyethylidene)-7-oxo-4-oxa-1-azabicyclo[3.2.0]heptane-2-carboxylate Chemical compound [K+].[O-]C(=O)[C@H]1C(=C/CO)/O[C@@H]2CC(=O)N21.C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=C(O)C=C1 DWHGNUUWCJZQHO-ZVDZYBSKSA-M 0.000 description 1
- 201000011461 pre-eclampsia Diseases 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 229940087661 priftin Drugs 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 208000011511 primary membranoproliferative glomerulonephritis Diseases 0.000 description 1
- 229940027836 primaxin Drugs 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 230000006785 proliferative vitreoretinopathy Effects 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229960003415 propylparaben Drugs 0.000 description 1
- GMVPRGQOIOIIMI-DWKJAMRDSA-N prostaglandin E1 Chemical compound CCCCC[C@H](O)\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1CCCCCCC(O)=O GMVPRGQOIOIIMI-DWKJAMRDSA-N 0.000 description 1
- XEYBRNLFEZDVAW-UHFFFAOYSA-N prostaglandin E2 Natural products CCCCCC(O)C=CC1C(O)CC(=O)C1CC=CCCCC(O)=O XEYBRNLFEZDVAW-UHFFFAOYSA-N 0.000 description 1
- 201000006301 pulmonary embolism and infarction Diseases 0.000 description 1
- 208000005069 pulmonary fibrosis Diseases 0.000 description 1
- 238000004537 pulping Methods 0.000 description 1
- 229960005206 pyrazinamide Drugs 0.000 description 1
- JUJWROOIHBZHMG-UHFFFAOYSA-O pyridinium Chemical compound C1=CC=[NH+]C=C1 JUJWROOIHBZHMG-UHFFFAOYSA-O 0.000 description 1
- 125000002112 pyrrolidino group Chemical group [*]N1C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 108010071077 quinupristin-dalfopristin Proteins 0.000 description 1
- 229940052337 quinupristin/dalfopristin Drugs 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 229940038850 rebif Drugs 0.000 description 1
- 229940044601 receptor agonist Drugs 0.000 description 1
- 239000000018 receptor agonist Substances 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 230000007115 recruitment Effects 0.000 description 1
- 230000000306 recurrent effect Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 229940116176 remicade Drugs 0.000 description 1
- 230000004264 retinal detachment Effects 0.000 description 1
- 230000004283 retinal dysfunction Effects 0.000 description 1
- 230000002207 retinal effect Effects 0.000 description 1
- 208000004644 retinal vein occlusion Diseases 0.000 description 1
- 229910052895 riebeckite Inorganic materials 0.000 description 1
- 229940063639 rifadin Drugs 0.000 description 1
- 229960002599 rifapentine Drugs 0.000 description 1
- 229960003040 rifaximin Drugs 0.000 description 1
- NZCRJKRKKOLAOJ-XRCRFVBUSA-N rifaximin Chemical compound OC1=C(C(O)=C2C)C3=C4N=C5C=C(C)C=CN5C4=C1NC(=O)\C(C)=C/C=C/[C@H](C)[C@H](O)[C@@H](C)[C@@H](O)[C@@H](C)[C@H](OC(C)=O)[C@H](C)[C@@H](OC)\C=C\O[C@@]1(C)OC2=C3C1=O NZCRJKRKKOLAOJ-XRCRFVBUSA-N 0.000 description 1
- 229950007943 risankizumab Drugs 0.000 description 1
- 229940081561 rocephin Drugs 0.000 description 1
- 238000002390 rotary evaporation Methods 0.000 description 1
- HNMATTJJEPZZMM-BPKVFSPJSA-N s-[(2r,3s,4s,6s)-6-[[(2r,3s,4s,5r,6r)-5-[(2s,4s,5s)-5-[acetyl(ethyl)amino]-4-methoxyoxan-2-yl]oxy-6-[[(2s,5z,9r,13e)-13-[2-[[4-[(2e)-2-[1-[4-(4-amino-4-oxobutoxy)phenyl]ethylidene]hydrazinyl]-2-methyl-4-oxobutan-2-yl]disulfanyl]ethylidene]-9-hydroxy-12-(m Chemical compound C1[C@H](OC)[C@@H](N(CC)C(C)=O)CO[C@H]1O[C@H]1[C@H](O[C@@H]2C\3=C(NC(=O)OC)C(=O)C[C@@](C/3=C/CSSC(C)(C)CC(=O)N\N=C(/C)C=3C=CC(OCCCC(N)=O)=CC=3)(O)C#C\C=C/C#C2)O[C@H](C)[C@@H](NO[C@@H]2O[C@H](C)[C@@H](SC(=O)C=3C(=C(OC)C(O[C@H]4[C@@H]([C@H](OC)[C@@H](O)[C@H](C)O4)O)=C(I)C=3C)OC)[C@@H](O)C2)[C@@H]1O HNMATTJJEPZZMM-BPKVFSPJSA-N 0.000 description 1
- 210000003296 saliva Anatomy 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- 201000000306 sarcoidosis Diseases 0.000 description 1
- 210000004761 scalp Anatomy 0.000 description 1
- 231100000241 scar Toxicity 0.000 description 1
- 230000037387 scars Effects 0.000 description 1
- 201000000980 schizophrenia Diseases 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 239000013049 sediment Substances 0.000 description 1
- 239000008299 semisolid dosage form Substances 0.000 description 1
- IPQVTOJGNYVQEO-CXZNLNCXSA-N sennoside A Natural products O=C(O)c1cc(O)c2C(=O)c3c(O[C@H]4[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O4)cccc3[C@@H]([C@H]3c4c(c(O)cc(C(=O)O)c4)C(=O)c4c(O[C@H]5[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O5)cccc34)c2c1 IPQVTOJGNYVQEO-CXZNLNCXSA-N 0.000 description 1
- IPQVTOJGNYVQEO-AIFLABODSA-N sennoside B Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1OC1=CC=CC2=C1C(=O)C1=C(O)C=C(C(O)=O)C=C1[C@H]2[C@H]1C2=CC(C(O)=O)=CC(O)=C2C(=O)C2=C(O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O3)O)C=CC=C21 IPQVTOJGNYVQEO-AIFLABODSA-N 0.000 description 1
- 229940004991 sennoside b Drugs 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 238000007493 shaping process Methods 0.000 description 1
- 229950010077 sifalimumab Drugs 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 229920002545 silicone oil Polymers 0.000 description 1
- 229960003323 siltuximab Drugs 0.000 description 1
- 229960003600 silver sulfadiazine Drugs 0.000 description 1
- UEJSSZHHYBHCEL-UHFFFAOYSA-N silver(1+) sulfadiazinate Chemical compound [Ag+].C1=CC(N)=CC=C1S(=O)(=O)[N-]C1=NC=CC=N1 UEJSSZHHYBHCEL-UHFFFAOYSA-N 0.000 description 1
- 238000010583 slow cooling Methods 0.000 description 1
- 229940126586 small molecule drug Drugs 0.000 description 1
- 239000000779 smoke Substances 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- ORVLUIMCZUPAPB-LBTQIPEASA-M sodium (4S,4aS,5aS,6S,12aR)-4-(dimethylamino)-1,6,10,11,12a-pentahydroxy-6-methyl-3,12-dioxo-4,4a,5,5a-tetrahydrotetracene-2-carboxamide dioxido(oxo)phosphanium phosphenic acid Chemical compound [Na+].O[P+]([O-])=O.O[P+]([O-])=O.O[P+]([O-])=O.O[P+]([O-])=O.O[P+]([O-])=O.[O-][P+]([O-])=O.CN(C)[C@H]1[C@@H]2C[C@H]3C(=C(O)c4c(O)cccc4[C@@]3(C)O)C(=O)[C@]2(O)C(O)=C(C(N)=O)C1=O.CN(C)[C@H]1[C@@H]2C[C@H]3C(=C(O)c4c(O)cccc4[C@@]3(C)O)C(=O)[C@]2(O)C(O)=C(C(N)=O)C1=O.CN(C)[C@H]1[C@@H]2C[C@H]3C(=C(O)c4c(O)cccc4[C@@]3(C)O)C(=O)[C@]2(O)C(O)=C(C(N)=O)C1=O.CN(C)[C@H]1[C@@H]2C[C@H]3C(=C(O)c4c(O)cccc4[C@@]3(C)O)C(=O)[C@]2(O)C(O)=C(C(N)=O)C1=O.CN(C)[C@H]1[C@@H]2C[C@H]3C(=C(O)c4c(O)cccc4[C@@]3(C)O)C(=O)[C@]2(O)C(O)=C(C(N)=O)C1=O ORVLUIMCZUPAPB-LBTQIPEASA-M 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 235000011083 sodium citrates Nutrition 0.000 description 1
- 229940074545 sodium dihydrogen phosphate dihydrate Drugs 0.000 description 1
- BBMHARZCALWXSL-UHFFFAOYSA-M sodium dihydrogenphosphate monohydrate Chemical compound O.[Na+].OP(O)([O-])=O BBMHARZCALWXSL-UHFFFAOYSA-M 0.000 description 1
- 239000000176 sodium gluconate Substances 0.000 description 1
- 235000012207 sodium gluconate Nutrition 0.000 description 1
- 229940005574 sodium gluconate Drugs 0.000 description 1
- 239000001540 sodium lactate Substances 0.000 description 1
- 235000011088 sodium lactate Nutrition 0.000 description 1
- 229940005581 sodium lactate Drugs 0.000 description 1
- RYYKJJJTJZKILX-UHFFFAOYSA-M sodium octadecanoate Chemical compound [Na+].CCCCCCCCCCCCCCCCCC([O-])=O RYYKJJJTJZKILX-UHFFFAOYSA-M 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 235000011008 sodium phosphates Nutrition 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 235000010339 sodium tetraborate Nutrition 0.000 description 1
- HJHVQCXHVMGZNC-JCJNLNMISA-M sodium;(2z)-2-[(3r,4s,5s,8s,9s,10s,11r,13r,14s,16s)-16-acetyloxy-3,11-dihydroxy-4,8,10,14-tetramethyl-2,3,4,5,6,7,9,11,12,13,15,16-dodecahydro-1h-cyclopenta[a]phenanthren-17-ylidene]-6-methylhept-5-enoate Chemical compound [Na+].O[C@@H]([C@@H]12)C[C@H]3\C(=C(/CCC=C(C)C)C([O-])=O)[C@@H](OC(C)=O)C[C@]3(C)[C@@]2(C)CC[C@@H]2[C@]1(C)CC[C@@H](O)[C@H]2C HJHVQCXHVMGZNC-JCJNLNMISA-M 0.000 description 1
- ILJOYZVVZZFIKA-UHFFFAOYSA-M sodium;1,1-dioxo-1,2-benzothiazol-3-olate;hydrate Chemical compound O.[Na+].C1=CC=C2C(=O)[N-]S(=O)(=O)C2=C1 ILJOYZVVZZFIKA-UHFFFAOYSA-M 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 238000000935 solvent evaporation Methods 0.000 description 1
- 229960000487 sorafenib tosylate Drugs 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 235000010356 sorbitol Nutrition 0.000 description 1
- 235000014347 soups Nutrition 0.000 description 1
- 229960004954 sparfloxacin Drugs 0.000 description 1
- DZZWHBIBMUVIIW-DTORHVGOSA-N sparfloxacin Chemical compound C1[C@@H](C)N[C@@H](C)CN1C1=C(F)C(N)=C2C(=O)C(C(O)=O)=CN(C3CC3)C2=C1F DZZWHBIBMUVIIW-DTORHVGOSA-N 0.000 description 1
- 229950007213 spartalizumab Drugs 0.000 description 1
- 229940010329 spectracef Drugs 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 229960001294 spiramycin Drugs 0.000 description 1
- 235000019372 spiramycin Nutrition 0.000 description 1
- 229930191512 spiramycin Natural products 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 230000035882 stress Effects 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 229960002317 succinimide Drugs 0.000 description 1
- 229960002673 sulfacetamide Drugs 0.000 description 1
- ZZORFUFYDOWNEF-UHFFFAOYSA-N sulfadimethoxine Chemical compound COC1=NC(OC)=CC(NS(=O)(=O)C=2C=CC(N)=CC=2)=N1 ZZORFUFYDOWNEF-UHFFFAOYSA-N 0.000 description 1
- 229940091629 sulfamylon Drugs 0.000 description 1
- 229960001940 sulfasalazine Drugs 0.000 description 1
- NCEXYHBECQHGNR-UHFFFAOYSA-N sulfasalazine Natural products C1=C(O)C(C(=O)O)=CC(N=NC=2C=CC(=CC=2)S(=O)(=O)NC=2N=CC=CC=2)=C1 NCEXYHBECQHGNR-UHFFFAOYSA-N 0.000 description 1
- OPYGFNJSCUDTBT-PMLPCWDUSA-N sultamicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(=O)OCOC(=O)[C@H]2C(S(=O)(=O)[C@H]3N2C(C3)=O)(C)C)(C)C)=CC=CC=C1 OPYGFNJSCUDTBT-PMLPCWDUSA-N 0.000 description 1
- 230000036561 sun exposure Effects 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 229940053017 sylvant Drugs 0.000 description 1
- 229940020707 synercid Drugs 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 229960003879 tedizolid Drugs 0.000 description 1
- XFALPSLJIHVRKE-GFCCVEGCSA-N tedizolid Chemical compound CN1N=NC(C=2N=CC(=CC=2)C=2C(=CC(=CC=2)N2C(O[C@@H](CO)C2)=O)F)=N1 XFALPSLJIHVRKE-GFCCVEGCSA-N 0.000 description 1
- 229940036731 teflaro Drugs 0.000 description 1
- 229960001608 teicoplanin Drugs 0.000 description 1
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 1
- 108010041283 teixobactin Proteins 0.000 description 1
- 208000009056 telangiectasis Diseases 0.000 description 1
- ONUMZHGUFYIKPM-MXNFEBESSA-N telavancin Chemical compound O1[C@@H](C)[C@@H](O)[C@](NCCNCCCCCCCCCC)(C)C[C@@H]1O[C@H]1[C@H](OC=2C3=CC=4[C@H](C(N[C@H]5C(=O)N[C@H](C(N[C@@H](C6=CC(O)=C(CNCP(O)(O)=O)C(O)=C6C=6C(O)=CC=C5C=6)C(O)=O)=O)[C@H](O)C5=CC=C(C(=C5)Cl)O3)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](NC(=O)[C@@H](CC(C)C)NC)[C@H](O)C3=CC=C(C(=C3)Cl)OC=2C=4)O[C@H](CO)[C@@H](O)[C@@H]1O ONUMZHGUFYIKPM-MXNFEBESSA-N 0.000 description 1
- 229960005240 telavancin Drugs 0.000 description 1
- GSSIWSIRBWAZHG-ACOPVEIWSA-N telavancin hydrochloride Chemical compound Cl.O1[C@@H](C)[C@@H](O)[C@](NCCNCCCCCCCCCC)(C)C[C@@H]1O[C@H]1[C@H](OC=2C3=CC=4[C@H](C(N[C@H]5C(=O)N[C@H](C(N[C@@H](C6=CC(O)=C(CNCP(O)(O)=O)C(O)=C6C=6C(O)=CC=C5C=6)C(O)=O)=O)[C@H](O)C5=CC=C(C(=C5)Cl)O3)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](NC(=O)[C@@H](CC(C)C)NC)[C@H](O)C3=CC=C(C(=C3)Cl)OC=2C=4)O[C@H](CO)[C@@H](O)[C@@H]1O GSSIWSIRBWAZHG-ACOPVEIWSA-N 0.000 description 1
- 229960004576 temafloxacin Drugs 0.000 description 1
- 229960001114 temocillin Drugs 0.000 description 1
- 206010043207 temporal arteritis Diseases 0.000 description 1
- CBPNZQVSJQDFBE-HGVVHKDOSA-N temsirolimus Chemical compound C1C[C@@H](OC(=O)C(C)(CO)CO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CCC2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 CBPNZQVSJQDFBE-HGVVHKDOSA-N 0.000 description 1
- 229960003676 tenidap Drugs 0.000 description 1
- LXIKEPCNDFVJKC-QXMHVHEDSA-N tenidap Chemical compound C12=CC(Cl)=CC=C2N(C(=O)N)C(=O)\C1=C(/O)C1=CC=CS1 LXIKEPCNDFVJKC-QXMHVHEDSA-N 0.000 description 1
- 229940061354 tequin Drugs 0.000 description 1
- 229960000331 teriflunomide Drugs 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- TUGDLVFMIQZYPA-UHFFFAOYSA-N tetracopper;tetrazinc Chemical compound [Cu+2].[Cu+2].[Cu+2].[Cu+2].[Zn+2].[Zn+2].[Zn+2].[Zn+2] TUGDLVFMIQZYPA-UHFFFAOYSA-N 0.000 description 1
- 229960002180 tetracycline Drugs 0.000 description 1
- 229930101283 tetracycline Natural products 0.000 description 1
- 235000019364 tetracycline Nutrition 0.000 description 1
- 150000003522 tetracyclines Chemical class 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- KTAVBOYXMBQFGR-MAODNAKNSA-J tetrasodium;(6r,7r)-7-[[(2z)-2-(2-amino-1,3-thiazol-4-yl)-2-methoxyimino-1-oxidoethylidene]amino]-3-[(2-methyl-5,6-dioxo-1h-1,2,4-triazin-3-yl)sulfanylmethyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate;heptahydrate Chemical compound O.O.O.O.O.O.O.[Na+].[Na+].[Na+].[Na+].S([C@@H]1[C@@H](C(N1C=1C([O-])=O)=O)NC(=O)\C(=N/OC)C=2N=C(N)SC=2)CC=1CSC1=NC(=O)C([O-])=NN1C.S([C@@H]1[C@@H](C(N1C=1C([O-])=O)=O)NC(=O)\C(=N/OC)C=2N=C(N)SC=2)CC=1CSC1=NC(=O)C([O-])=NN1C KTAVBOYXMBQFGR-MAODNAKNSA-J 0.000 description 1
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 1
- 238000002076 thermal analysis method Methods 0.000 description 1
- 238000005382 thermal cycling Methods 0.000 description 1
- AYEKOFBPNLCAJY-UHFFFAOYSA-O thiamine pyrophosphate Chemical compound CC1=C(CCOP(O)(=O)OP(O)(O)=O)SC=[N+]1CC1=CN=C(C)N=C1N AYEKOFBPNLCAJY-UHFFFAOYSA-O 0.000 description 1
- OTVAEFIXJLOWRX-NXEZZACHSA-N thiamphenicol Chemical compound CS(=O)(=O)C1=CC=C([C@@H](O)[C@@H](CO)NC(=O)C(Cl)Cl)C=C1 OTVAEFIXJLOWRX-NXEZZACHSA-N 0.000 description 1
- 229960003053 thiamphenicol Drugs 0.000 description 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 description 1
- 229960004659 ticarcillin Drugs 0.000 description 1
- OHKOGUYZJXTSFX-KZFFXBSXSA-N ticarcillin Chemical compound C=1([C@@H](C(O)=O)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)C=CSC=1 OHKOGUYZJXTSFX-KZFFXBSXSA-N 0.000 description 1
- 229940111100 tice bcg Drugs 0.000 description 1
- 229960005053 tinidazole Drugs 0.000 description 1
- NLVFBUXFDBBNBW-PBSUHMDJSA-N tobramycin Chemical compound N[C@@H]1C[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N NLVFBUXFDBBNBW-PBSUHMDJSA-N 0.000 description 1
- 229960003989 tocilizumab Drugs 0.000 description 1
- 229960001350 tofacitinib Drugs 0.000 description 1
- UJLAWZDWDVHWOW-YPMHNXCESA-N tofacitinib Chemical compound C[C@@H]1CCN(C(=O)CC#N)C[C@@H]1N(C)C1=NC=NC2=C1C=CN2 UJLAWZDWDVHWOW-YPMHNXCESA-N 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- GYDJEQRTZSCIOI-LJGSYFOKSA-N tranexamic acid Chemical compound NC[C@H]1CC[C@H](C(O)=O)CC1 GYDJEQRTZSCIOI-LJGSYFOKSA-N 0.000 description 1
- 229960000401 tranexamic acid Drugs 0.000 description 1
- 229960000363 trapidil Drugs 0.000 description 1
- 230000008733 trauma Effects 0.000 description 1
- 230000009529 traumatic brain injury Effects 0.000 description 1
- 229940079342 trecator Drugs 0.000 description 1
- YNDXUCZADRHECN-JNQJZLCISA-N triamcinolone acetonide Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]1(C)C[C@@H]2O YNDXUCZADRHECN-JNQJZLCISA-N 0.000 description 1
- 229960002117 triamcinolone acetonide Drugs 0.000 description 1
- BSVBQGMMJUBVOD-UHFFFAOYSA-N trisodium borate Chemical compound [Na+].[Na+].[Na+].[O-]B([O-])[O-] BSVBQGMMJUBVOD-UHFFFAOYSA-N 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 229960000497 trovafloxacin Drugs 0.000 description 1
- WVPSKSLAZQPAKQ-CDMJZVDBSA-N trovafloxacin Chemical compound C([C@H]1[C@@H]([C@H]1C1)N)N1C(C(=CC=1C(=O)C(C(O)=O)=C2)F)=NC=1N2C1=CC=C(F)C=C1F WVPSKSLAZQPAKQ-CDMJZVDBSA-N 0.000 description 1
- DYNZICQDCVYXFW-AHZSKCOESA-N trovafloxacin mesylate Chemical compound CS(O)(=O)=O.C([C@H]1[C@@H]([C@H]1C1)N)N1C(C(=CC=1C(=O)C(C(O)=O)=C2)F)=NC=1N2C1=CC=C(F)C=C1F DYNZICQDCVYXFW-AHZSKCOESA-N 0.000 description 1
- 229940055820 trovan Drugs 0.000 description 1
- 229940035144 trumenba Drugs 0.000 description 1
- 208000017271 typical hemolytic-uremic syndrome Diseases 0.000 description 1
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 1
- 229940079023 tysabri Drugs 0.000 description 1
- 229940020930 unasyn Drugs 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 230000003827 upregulation Effects 0.000 description 1
- 230000002485 urinary effect Effects 0.000 description 1
- 229960003824 ustekinumab Drugs 0.000 description 1
- 238000003828 vacuum filtration Methods 0.000 description 1
- 229940072335 vancocin Drugs 0.000 description 1
- 229940054969 vantin Drugs 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 210000000264 venule Anatomy 0.000 description 1
- 229940020733 vibativ Drugs 0.000 description 1
- 229940118696 vibrio cholerae Drugs 0.000 description 1
- 230000004304 visual acuity Effects 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
- 229940104152 vivotif Drugs 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
- 229920001285 xanthan gum Polymers 0.000 description 1
- 239000000230 xanthan gum Substances 0.000 description 1
- 235000010493 xanthan gum Nutrition 0.000 description 1
- 229940082509 xanthan gum Drugs 0.000 description 1
- KGPGQDLTDHGEGT-JCIKCJKQSA-N zeven Chemical compound C=1C([C@@H]2C(=O)N[C@H](C(N[C@H](C3=CC(O)=C4)C(=O)NCCCN(C)C)=O)[C@H](O)C5=CC=C(C(=C5)Cl)OC=5C=C6C=C(C=5O[C@H]5[C@@H]([C@@H](O)[C@H](O)[C@H](O5)C(O)=O)NC(=O)CCCCCCCCC(C)C)OC5=CC=C(C=C5)C[C@@H]5C(=O)N[C@H](C(N[C@H]6C(=O)N2)=O)C=2C(Cl)=C(O)C=C(C=2)OC=2C(O)=CC=C(C=2)[C@H](C(N5)=O)NC)=CC=C(O)C=1C3=C4O[C@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@@H]1O KGPGQDLTDHGEGT-JCIKCJKQSA-N 0.000 description 1
- 229940072251 zithromax Drugs 0.000 description 1
- 229930195724 β-lactose Natural products 0.000 description 1
Images




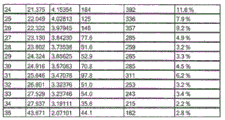







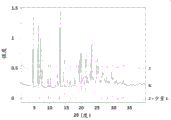




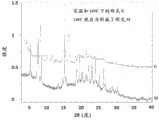






























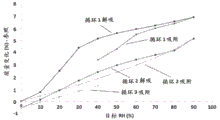
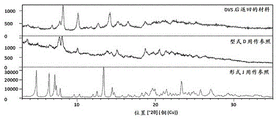
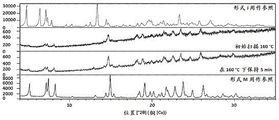




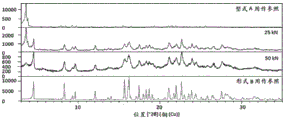







Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicinal Preparation (AREA)
Abstract
本发明提供了用于治疗应用的补体因子D抑制剂化合物3的稳定的高度结晶形式。
Description
相关申请的交叉引用
本申请要求2018年9月25日提交的美国申请号62/736,294;2018年11月8日提交的美国申请号62/757,565;2018年11月13日提交的美国申请号62/760,520;和2019年1月25日提交的美国申请号62/796,776的优先权权益。这些申请中的每一个的全部以引用方式并入本文。
技术领域
本发明提供了补体因子D抑制剂(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-羧酰胺的有利的分离的形态形式。
背景技术
补体系统是先天免疫系统的一部分,其不适应于宿主生命过程中的变化,但是被适应性免疫系统所募集和使用。例如,其协助或补充抗体和吞噬细胞清除病原体的能力。这一复杂的调控途径允许对病原生物体产生快速反应,同时保护宿主细胞不受破坏。超过三十种蛋白质和蛋白质片段组成补体系统。这些蛋白质通过调理作用(增强抗原的吞噬作用)、趋化作用(吸引巨噬细胞和嗜中性粒细胞)、细胞裂解(使外源细胞的膜破坏)和凝集(使病原体簇集和结合在一起)起作用。
补体系统有三种途径:经典途径、替代途径和凝集素途径。补体因子D在补体级联的替代途径的激活中起着早期和核心作用。替代补体途径的激活始于C3蛋白质内硫酯键的自发水解,产生C3(H2O),其与因子B缔合而形成C3(H2O)B复合物。补体因子D的作用是切割C3(H2O)B复合物内的因子B而形成Ba和Bb。Bb片段仍与C3(H2O)缔合而形成替代途径C3转化酶C3(H2O)Bb。另外,由任何C3转化酶生成的C3b也与因子B缔合而形成C3bB,因子D切割而生成后期替代途径C3转化酶C3bBb。替代途径C3转化酶的此后一形式可在所有三种所定义补体途径内提供重要的下游扩增,最终导致补体级联途径中其他因子的募集和组装,包括C5断裂为C5a和C5b。C5b在因子C6、C7、C8和C9组装成膜攻击复合物中起作用,所述复合物可通过裂解细胞来破坏病原细胞。
补体的功能障碍或过度激活已经与某些自身免疫性、炎性和神经变性疾病以及缺血再灌注损伤和癌症关联起来。例如,补体级联的替代途径的激活有助于C3a和C5a(二者均为强效过敏毒素)的产生,C3a和C5a也在许多炎性病症中起作用。因此,在一些情况下,期望的是减少补体途径(包括替代补体途径)的响应。由补体途径介导的病症的一些实例包括年龄相关性黄斑变性(AMD)、阵发性睡眠性血红蛋白尿症(PNH)、多发性硬化症和类风湿性关节炎。
年龄相关性黄斑变性(AMD)是工业化国家中视力丧失的主要原因。基于多项遗传学研究,有证据表明补体级联与黄斑变性之间存在关联。在编码补体因子H的基因中具有突变的个体具有五倍增加的黄斑变性风险,且在其他补体因子基因中具有突变的个体也具有增加的AMD风险。具有突变的因子H的个体也有着增高的C反应蛋白水平,C反应蛋白是炎症的标记物。在因子H未充分发挥作用的情况下,补体级联的替代途径受到过度激活,导致细胞损伤。
阵发性睡眠性血红蛋白尿症(PNH)是非恶性血液系统病症,其特征为缺乏一些表面蛋白质的造血干细胞和子代成熟血细胞的扩增。PNH红细胞不能够调节它们的表面补体激活,这导致PNH的典型标志-补体介导的血管内贫血的慢性激活。美国目前仅有一种产品(抗-C5单克隆抗体依库珠单抗(eculizumab))已批准用于治疗PNH。然而,用依库珠单抗治疗的许多患者仍患贫血,且许多患者继续需要输血。另外,依库珠单抗治疗需要终身静脉内注射。
其他补体介导的病症包括归类于成分3肾小球病(C3G)的那些病症。C3G是最近定义的由致密沉积物病(DDD)和C3肾小球肾炎(C3GN)组成的实体,涵盖了一系列慢性肾脏疾病,其中替代补体途径和末端补体途径的活性升高导致仅由补体C3组成而无免疫球蛋白(Ig)的肾小球沉积物。
免疫复合物膜增生性肾小球肾炎(IC-MPGN)是一种肾病,其与C3G共有许多临床、病理、遗传和实验室特征,因此可被认为是C3G的姐妹疾病。在大多数患有IC-MPGN的患者中,鉴定了潜在的疾病或病症—最常见的是感染、自身免疫疾病或单克隆丙种球蛋白病,其中肾病是继发性的。患有特发性IC-MPGN的患者可具有低C3和正常C4水平,类似于在C3G中观察到的那些,以及与异常的替代途径活性相关的许多相同遗传或获得性因子。虽然目前的假设表明大多数IC-MPGN可归因于经典途径的过度活性,但具有低C3和正常C4的那些患者可能具有替代途径的显著过度活性。具有低C3和正常C4的IC-MPGN患者可受益于替代途径抑制。
已经与补体级联关联的其他病症包括非典型溶血性尿毒症综合征(aHUS)、溶血性尿毒症综合征(HUS)、腹主动脉瘤、血液透析并发症、溶血性贫血或血液透析、视神经脊髓炎(NMO)、重症肌无力(MG)、脂肪肝、非酒精性脂肪性肝炎(NASH)、肝脏炎症、肝硬化、肝衰竭、皮肌炎和肌萎缩性侧索硬化。
因子D由于其在替代补体途径中的早期和必要作用,以及其在经典途径和凝集素补体途径内的信号放大中的潜在作用,其是用于抑制或调控补体级联的有吸引力的靶标。因子D的抑制有效地中断该途径并减弱膜攻击复合物的形成。
标题为“Indole compounds or analogues thereof useful for the treatmentof age-related macular degeneration”的Novartis PCT专利公布WO2012/093101描述了某些因子D抑制剂。另外的因子D抑制剂描述于Novartis PCT专利公布WO2012093101、WO2013/164802、WO2013/192345、WO2014/002051、WO2014/002052、WO2014/002053、WO2014/002054、WO2014/002057、WO2014/002058、WO2014/002059、WO2014/005150、WO2014/009833、WO2014/143638、WO2015/009616、WO2015/009977、WO2015/066241和WO2016088082中。
另外的补体因子D抑制剂描述于美国专利号9,598,446、9,643,986、9,663,543、9,695,205、9,732,103、9,732,104、9,758,537、9,796,741、9,828,396、10,000,516、10,005,802、10,011,612、10,081,645、10,087,203、10,092,584、10,100,072、10,138,225、10,189,869、10,106,563、10,301,336和10,287,301;国际公布号WO2019/028284、WO2018/160889、WO2018/160891、WO2018/160892、WO2017/035348、WO2017/035349、WO 2017/035351、WO2017/035352、WO 2017/035353、WO 2017/035355、WO2017/035357、WO2017/035360、WO2017/035361、WO2017//035362、WO2017/035415、WO2017/035401、WO2017/035405、WO2017/035413、WO2017/035409、WO2017/035411、WO2017/035417、WO2017/035408WO2015/130784、WO2015/130795、WO2015/130806、WO2015/130830、WO2015/130838、WO2015/130842、WO2015/130845和WO2015/130854;以及美国专利公布号US 2016-0361329、US 2016-0362432、US2016-0362433、US 2016-0362399、US 2017-0056428、US 2017-0057950、US 2017-0057993、US 2017-0189410、US 2017-0226142、US 2017-0260219、US 2017-0298084、US 2017-0298085、US 2018-0022766、US 2018-0022767、US 2018-0072762、US 2018-0030075、US2018-0169109、US 2018-0177761、US 2018-0179185、US 2018-0179186、US 2018-0179236、US 2018-0186782、US 2018-0201580、US 2019-0031692、US 2019-0048033、US 2019-0144473和US 2019-0211033中,这些专利全部由Achillion Pharmaceuticals,Inc所拥有。
考虑到由有害的免疫或炎性反应引起的各种各样的医学病症,有益的是提供可以增加治疗活性和/或稳定性的另外的有利的化合物及其形式以实现有利递送。
发明内容
已经发现化合物3((1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-羧酰胺)可以仅以几种表现出有利性质的高度纯化的形态形式制备。现在发现存在化合物3的几种形态形式,包括形式A、形式B和形式M。这些形态形式有利于治疗功效且有利于药物制剂的制造。化合物3公开于转让给Achillion Pharmaceuticals的PCT申请WO2017035353中。

如实施例2中所讨论,27种独特溶剂和多种结晶技术促使发现了四种稳定形式:形式A、形式B、形式J和形式M。特别地,发现形式A在80℃下高度稳定,且纯度未降低并且得到充分表征(实施例6和实施例7)。对化合物3形式A进行的涉及重结晶技术的另外的研究导致形式B、形式M和形式J的形成。这些形式得到表征(参见例如实施例9和实施例10)。形式B是高度稳定的,并且当该材料在40℃/75%RH、80℃或环境条件下储存时,通过XRPD或HPLC未观察到变化(实施例10)。
因此,本发明总体上提供了化合物3的分离的形态形式,含有这种形态形式的药物组合物,使用所述分离的形态形式抑制或降低宿主中酶因子D活性的方法,以及使用本文所述的形态形式治疗患有阵发性睡眠性血红蛋白尿症(PNH)或C3肾小球病(C3G)的宿主,以及制备这种形态形式的方法。在一个实施方案中,形态形式是形式A。在一个实施方案中,形态形式是形式B。在一个实施方案中,形态形式是形式M。
在本发明的一个方面中,化合物3的形态形式的特征在于以下附图中的2θ值+/-0.5°、0.4°或0.3°2θ。在本发明的一个方面中,化合物3的形态形式的特征在于以下附图中的2θ值+/-0.2°2θ。在本发明的一个方面中,化合物3的形态形式的特征在于以下附图中的2θ值+/-0.1°2θ。因此,当XRPD峰被讨论为或描绘为+/-0.2°2θ值时,应当理解替代实施方案是+/-0.3°2θ、+/-0.4°2θ和+/-0.4°2θ。
在本发明的一个方面中,化合物3的形态形式的特征在于附图中其代表性XRPD图谱的至少三个2θ值。在本发明的一个方面中,化合物3的形态形式的特征在于附图中其代表性XRPD图谱的至少四个2θ值。在本发明的一个方面中,化合物3的形态形式的特征在于附图中其代表性XRPD图谱的至少五个、六个、七个或八个2θ值。
在本发明中,XRPD图谱不用于鉴定化合物的化学结构,而是用于区分化合物的固体形式。因此,在一些情况下,仅有几个或甚至一个特征峰可以将具有已知化学结构的化合物的一种形式与另一种形式区分开。
在一个实施方案中,提供的化合物3的形态形式用于产生用于药物级药物剂型的高纯度材料。例如,提供的形态形式可用于喷雾干燥分散体技术中,以产生高纯度的无定形化合物3。
还提供了用于合成化合物1、化合物2和化合物3的改进的合成方法。化合物1通常公开于转让给Achillion Pharmaceuticals的PCT申请WO2015130795中。化合物2和化合物3通常公开于转让给Achillion Pharmaceuticals的PCT申请WO2017035353中。

附图说明
图1提供了如实施例2中所述的几种化合物3形式的XRPD(X射线粉末衍射)图谱。x轴是测得的2θ(度),且y轴是测得的强度(计数)。示出了形式A、B、C、D、H和I。
图2是如实施例2中所述用Cu-Kα收集的化合物3形式B的XRPD。XRPD条件如以下所列出:Bravais类型:原始单斜;a 10.651;b
10.651;b 6.672;c
6.672;c 19.447;α[度]:90;β[度]:93.32;γ[度]:90;体积[
19.447;α[度]:90;β[度]:93.32;γ[度]:90;体积[ /晶胞]:1,380.9;手性内容物:手性;消光符号:P 1 21 1;和空间群:P21(4)。x轴是测得的2θ(度),且y轴是测得的强度(计数)。
/晶胞]:1,380.9;手性内容物:手性;消光符号:P 1 21 1;和空间群:P21(4)。x轴是测得的2θ(度),且y轴是测得的强度(计数)。
图3是如实施例2中所述的化合物3形式A的XRPD。x轴是测得的2θ(度),且y轴是测得的强度(计数)。
图4A和图4B是与图3中的XRPD图相对应的峰强度表。在一个实施方案中,化合物3形式A的特征在于具有至少3、4、5或6个相对强度为至少为10%的2θ值+/-0.2 2θ。
图5提供了如实施例2中所述的几种化合物3形式的XRPD(X射线粉末衍射)图谱。x轴是测得的2θ(度),且y轴是测得的强度(计数)。示出了形式B、A、C,和形式F/形式C的混合物。
图6是如实施例2中所述的化合物3形式B的DSC和TGA图。x轴是测得的温度(℃),左y轴是测得的热流(W/g),且右y轴是测得的重量(百分比)。
图7是如实施例2中所述的化合物3形式C的DSC和TGA图。x轴是测得的温度(℃),左y轴是测得的热流(W/g),且右y轴是测得的重量(百分比)。
图8提供了如实施例2中所述的在EtOAc浆液中在50C热应力之前和之后的化合物3的形式B和在乙腈浆液中在室温下的形式E的XRPD图谱。
图9提供了如实施例2中所述的在50C热应力之后的化合物3的形式C、形式G以及形式F和形式C的混合物的XRPD图谱。
图10提供了如实施例2中所述的在50C热应力之前和之后的化合物3的形式G的XRPD图谱。
图11是如实施例2中所述的化合物3形式E的DSC和TGA图。x轴是测得的温度(℃),左y轴是测得的热流(W/g),且右y轴是测得的重量(百分比)。
图12提供了如实施例2中所述的几种化合物3形式的XRPD(X射线粉末衍射)图谱。x轴是测得的2θ(度),且y轴是测得的强度(计数)。示出了形式J、形式K和带有少量形式L杂质的形式J。
图13是如实施例2中所述的化合物3形式J的XRPD。XRPD条件如以下所列出:Bravais类型:原始单斜;a 4.802;b
4.802;b 27.440;c
27.440;c 24.660;α[度]:90;β[度]:91.48;γ[度]:90;体积[
24.660;α[度]:90;β[度]:91.48;γ[度]:90;体积[ /晶胞]:3,248.3;手性内容物:手性;消光符号:P 1 21 1;和空间群:P21(4)。x轴是测得的2θ(度),且y轴是测得的强度(计数)。
/晶胞]:3,248.3;手性内容物:手性;消光符号:P 1 21 1;和空间群:P21(4)。x轴是测得的2θ(度),且y轴是测得的强度(计数)。
图14是化合物3形式J的DSC和TGA图。如实施例2中所述,x轴是测得的温度(℃),左y轴是测得的热流(W/g),且右y轴是测得的重量(百分比)。
图15提供了如实施例2中所述的化合物3形式J(带有少量形式L杂质)的XRPD图谱,相比形式J(带有少量形式L杂质)经受了50C热应力后所得形式G的XRPD。
图16是如实施例2中所述的化合物3形式G的DSC和TGA图。x轴是测得的温度(℃),左y轴是测得的热流(W/g),且右y轴是测得的重量(百分比)。
图17提供了如实施例2中所述的化合物3形式G的XRPD,相比形式G经受了160C热应力后所得形式M的XRPD。
图18是如实施例2中所述的化合物3形式M的XRPD。示出了XRPD条件。
图19是如实施例2中所述的化合物3形式M的DSC和TGA图。x轴是测得的温度(℃),左y轴是测得的热流(W/g),且右y轴是测得的重量(百分比)。
图20提供了如实施例2中所述的几种化合物3形式的XRPD(X射线粉末衍射)图谱。x轴是测得的2θ(度),且y轴是测得的强度(计数)。当在室温下混合形式B和形式M时,保留了两种形式。当在50C下混合形式B和形式M时,形成了形式M。
图21是如实施例2中所述的化合物3形式A的DSC和TGA图。x轴是测得的温度(℃),左y轴是测得的热流(W/g),且右y轴是测得的重量(百分比)。
图22提供了如实施例2中所述的化合物3形式A和所公开的材料的XRPD(X射线粉末衍射)图谱。x轴是测得的2θ(度),且y轴是测得的强度(计数)。
图23是如实施例2中所述的化合物3形式A的熔融实验图像。
图24是如实施例2中所述的化合物3和所公开的材料的DSC和TGA图。x轴是测得的温度(℃),左y轴是测得的热流(W/g),且右y轴是测得的重量(百分比)。
图25是无定形化合物3的DVS等温线图。x轴是测得的RH(百分比),且y轴是测得的质量变化(百分比)。实施例3给出了与该实验相对应的数据。
图26是比较了化合物2形式I与无定形化合物2的XRPD(X射线粉末衍射)叠加图。
图27是从化合物2形式I的热台显微镜法分析中收集的图像。该图像示出了形态形式的双折射特征。
图28是如实施例6中所述的化合物3形式A的PLM显微照片。
图29是如实施例6中所述的化合物3形式A的TG/DTA图。X轴是测得的温度(摄氏度)。右x轴是测得的重量损失(百分比),且左x轴是测得的DTA(μV)。
图30是如实施例6中所述的化合物3形式A的DVS等温线图。样品在25℃下运行。x轴是测得的目标RH(百分比),且y轴是测得的质量变化(百分比)。
图31是如实施例6中所述的化合物3形式A的DVS动力学图。右y轴是测得的目标RH(百分比),且左y轴是测得的质量变化(百分比)。X轴是测得的时间(分钟)。
图32是如实施例7中所述的化合物3形式A的XRPD分析水合后研究。x轴是测得的2θ(度),且y轴是强度。
图33提供了如实施例7中所述的研磨2分钟和5分钟后化合物3形式A的XRPD分析。x轴是测得的2θ(度),且y轴是强度。
图34A提供了如实施例7中所述的研磨2分钟后化合物3形式A的PLM显微照片。
图34B提供了如实施例7中所述的研磨5分钟后化合物3形式A的PLM显微照片。
图35A提供了如实施例7中所述的2.5KN和5.0KN压缩后化合物3形式A的XRPD分析。x轴是测得的2θ(度),且y轴是强度。
图35B是如实例7中所述的2.5KN压缩后化合物3形式A的PLM显微照片。
图35C是如实例7中所述的5.0KN压缩后化合物3形式A的PLM显微照片。
图36是如实施例7中所述的在选定介质(FaSSIF、FeSSIF、FaSSGF、pH 4柠檬酸盐和pH 6.8磷酸盐)中搅拌后化合物3形式A的XRPD叠加图。x轴是测得的2θ(度),且y轴是强度。
图37提供了如实施例9中所述的形式N和形式O以及在温度循环实验后分离的形式B和形式J的XRPD叠加图。x轴是测得的2θ(度),且y轴是强度。
图38是如实施例9中所述的分离的型式N的TG/DTA热谱图。X轴是测得的温度(摄氏度)。右x轴是测得的重量损失(百分比),且左x轴是测得的DTA(μV)。
图39是如实施例9中所述的分离的型式O的TG/DTA热谱图。X轴是测得的温度(摄氏度)。右x轴是测得的重量损失(百分比),且左x轴是测得的DTA(μV)。
图40提供了如实施例9中所述的由温度循环产生的选定固体(包括型式P)的XRPD叠加图。x轴是测得的2θ(度),且y轴是强度。
图41A是如实施例10中所述的化合物3形式B的DVS等温线图。x轴是测得的目标RH(百分比),且y轴是测得的质量变化(百分比)。
图41B是如实施例10中所述的化合物3形式B的DVS动力学图。右y轴是测得的目标RH(百分比),且左y轴是测得的质量变化(百分比)。X轴是测得的时间(分钟)。
图42提供了如实施例10中所述的将化合物3形式B的样品加热至250℃时的VT-XRPD衍射图。x轴是测得的2θ(度),且y轴是强度。
图43是如实施例10中所述的化合物3形式B的PLM显微照片。
图44A是如实施例10中所述的化合物3形式J的DVS等温线图。x轴是测得的目标RH(百分比),且y轴是测得的质量变化(百分比)。
图44B是如实施例10中所述的化合物3形式J的DVS动力学图。右y轴是测得的目标RH(百分比),且左y轴是测得的质量变化(百分比)。X轴是测得的时间(分钟)。
图44C提供了如实施例10中所述的化合物3形式J的DVS后XRPD分析。x轴是测得的2θ(度),且y轴是强度。
图44D提供了如实施例10中所述的将化合物3形式J的样品加热至170℃时的VT-XRPD衍射图的叠加图。x轴是测得的2θ(度),且y轴是强度。
图44E是如实施例10中所述的化合物3形式J的PLM显微照片。
图45是如实施例9和实施例10中所述的多晶型物研究的流程图。每个数字代表一种结晶技术,所述技术可提供箭头所示的形式、型式或无定形材料。编号的结晶技术如下:1)在2-丙醇、甲基乙基酮、2-甲基THF、1-丁醇、2-丙醇/庚烷(95:5%v/v)和2-丙醇/水(70:30%v/v);2)在50℃下干燥24小时,干燥2-丙醇/水(70:30%v/v)或冻干(1,4-二噁烷);3)在丙酮和2-丙醇/庚烷(70:30%v/v)中进行温度循环或将反溶剂(庚烷)中加入饱和MBK溶液中;4)在乙腈中进行温度循环;5)加热至120℃;6)加热至160℃;7)在50℃下干燥24小时;8)在甲醇中进行温度循环;9)在丙酮中进行温度循环持续2.5天;10)在乙醇/水(40:60%v/v)中进行温度循环;11)将反溶剂(水)加入饱和乙醇:水(40:60%v/v)溶液中,或将反溶剂(庚烷)加入饱和2-丙醇/庚烷(70:30%v/v)中;以及12)在50℃下干燥约24小时。
图46是如实施例11中所述的竞争性浆液实验的流程图。条件如上所述。条件1:70%2-丙醇/30%庚烷(%v/v),环境温度,搅动2天。条件2:庚烷,60℃,搅动5天和12天。条件3:丙酮,环境温度和40℃,搅动2天。条件4:乙腈,环境温度和60℃,搅动2天。条件5:乙腈,环境温度,搅动5天。条件6:丙酮,环境温度,搅动5天。条件7:丙酮,环境温度和40℃,1周和12天(使用化合物3的饱和溶液)。条件8:水,60℃,搅动5天和12天(使用化合物3的饱和溶液)。条件9:50%2-丙醇/50%庚烷(%v/v),60℃,12天(主要为无定形的,有A的迹象(使用化合物3的饱和溶液)。
图47提供了如实施例12中所述的研磨实验后的XRPD衍射图的叠加图。x轴是测得的2θ(度),且y轴是强度。
图48A提供了如实施例13中所述的压缩化合物3形式A和形式B的混合物后的XRPD衍射图的叠加图。x轴是测得的2θ(度),且y轴是强度。
图48B提供了如实施例13中所述的压缩化合物3形式B后的XRPD衍射图的叠加图。x轴是测得的2θ(度),且y轴是强度。
图49是如实施例14中所述的化合物3形式A和形式B的混合物的DSC热谱图。x轴是测得的温度(摄氏度),且y轴是归一化的热流(W/g)。
图50是因子D抑制剂化合物1、化合物2和化合物3。
图51是化合物3形式A的XRPD衍射图。
图52是化合物3形式B的XRPD衍射图。
图53是化合物3形式G的XRPD衍射图。
图54是化合物3形式J的XRPD衍射图。
图55是化合物3形式M的XRPD衍射图。
具体实施方式
无法预先预测化合物是否以多于一种固体形式存在,或者如果确实存在一种或多种固体形式任一固体形式的各种性质可能是什么,或者这些性质是否有利于治疗剂型或是否有利于制造要求,是否满足制药规范和/或是否有利于有利制剂。作为一个实例,药物利托那韦(ritonavir)在呈一种多晶型形式时具有活性,而在呈另一种形式时无活性,并且该无活性形式更稳定。
化合物的固体形式可以通过分析方法来表征,所述分析方法如X射线粉末衍射图谱(XRDP或PXRD)、热重分析(TGA)、TGA联合IR废气分析、差示扫描量热法(DSC)、熔点、FT-拉曼光谱法、动态蒸汽吸附(DVS)、偏振光显微镜法(PLM)或本领域中已知的其他技术。
化合物3
化合物3的多晶型物研究促使在至少13种已鉴定形式中发现了三种优良的形态形式(形式A、B和M)。化合物3形式的特征在于图1至图24和图51至图55中所示的XRPD图谱。

形式A
形式A的特征在于图3或图51中所示的XRPD图谱或与图3或图51中所示的图谱基本上相似的XRPD图谱。在一个实施方案中,分离的化合物3形式A的特征在于图37中的DSC。
在一个实施方案中,化合物3形式A的特征在于包括以下的XRPD图谱:
a)包括或选自3.73、9.3、11.7、9.5、7.6、6.7、6.0、5.7、5.6、5.4和4.2+/-0.2°2θ的2θ值;
b)选自3.73、9.3、11.7、9.5、7.6、6.7、6.0、5.7、5.6、5.4和4.2+/-0.2°2θ的至少两个、三个或四个2θ值;
c)选自3.73、9.3、11.7、9.5、7.6、6.7、6.0、5.7、5.6、5.4和4.2+/-0.2°2θ的至少五个、六个或七个2θ值;
d)选自3.73、9.3、11.7、9.5、7.6、6.7、6.0、5.7、5.6、5.4和4.2+/-0.2°2θ的至少八个或九个2θ值;
e)至少包括或选自3.73、9.3、11.7、9.5、7.6、6.7、6.0、5.7、5.6、5.4和4.2+/-0.2°2θ的2θ值;或
f)选自3.73、9.3、11.7、9.5、7.6、6.7、6.0、5.7、5.6、5.4和4.2+/-0.2°2θ的至少一个2θ值。
在一个实施方案中,分离的化合物3形式A的特征在于具有在110℃与120℃之间的Tg。可以使用选择性结晶来制备形式A。如下文更详细地描述,所述方法可以通过任选地在一种或多种包含形式A的晶种的存在下在提供形式A的结晶的条件下处理包含一种或多种合适的溶剂和化合物3的溶液来进行。在一个实施方案中,形式A是高度稳定的,具有长的保存期限和最小程度的降解。
本发明至少包括化合物3形式A的以下实施方案:
a)一种化合物3的分离的结晶形式A,其特征在于包括选自3.7±0.2°、9.3±0.2°、11.7±0.2°、9.5±0.2°、7.6±0.2°、6.7±0.2°、6.0±0.2°、5.7±0.2°、5.6±0.2°、5.4±0.2°和4.2±0.2°的至少三个2θ值的XRPD图谱;
b)实施方案(a)的化合物3的分离的结晶形式A,其特征在于包括选自3.7±0.2°、9.3±0.2°、11.7±0.2°、9.5±0.2°、7.6±0.2°、6.7±0.2°、6.0±0.2°、5.7±0.2°、5.6±0.2°、5.4±0.2°和4.2±0.2°的至少四个2θ值的XRPD图谱;
c)实施方案(a)或(b)的化合物3的分离的结晶形式A,其特征在于至少包括3.7±0.2°的2θ值的XRPD图谱;
d)实施方案(a)、(b)或(c)的化合物3的分离的结晶形式A,其特征在于至少包括9.3±0.2°的2θ值的XRPD图谱;
e)实施方案(a)-(d)中任一项的化合物3的分离的结晶形式A,其中XRPD图谱具有图3的特征2θ值;
f)一种药物组合物,所述药物组合物包含实施方案(a)-(e)中任一项的化合物3的分离的结晶形式A,所述分离的结晶形式处于药学上可接受的赋形剂中,用于固体剂量递送;
g)一种治疗补体因子D介导的病症的方法,所述方法包括向有需要的受试者施用治疗有效量的根据实施方案(a)-(e)中任一项的化合物3的分离的结晶形式A或其药物组合物,所述分离的结晶形式任选地处于药学上可接受的赋形剂中,用于固体剂量递送;
h)(g)的实施方案的方法,其中所述受试者是人;
i)实施方案(a)-(e)中任一项的化合物3的分离的结晶形式A,所述分离的结晶形式用于治疗有需要的受试者的补体因子D介导的病症,所述分离的结晶形式任选地处于药学上可接受的赋形剂中,用于固体剂量递送;
j)实施方案(i)的化合物3的分离的结晶形式A,其中所述受试者是人;
k)实施方案(a)-(e)中任一项的化合物3的分离的结晶形式A或其药物组合物在制造用于治疗有需要的受试者的补体因子D介导的病症的药物中的用途,所述分离的结晶形式任选地处于药学上可接受的赋形剂中,用于固体剂量递送;
l)实施方案(k)的用途,其中所述受试者是人;
m)以上实施方案中任一项,其中所述峰替代地在±0.4°内;并且
n)以上实施方案中任一项,其中所述峰替代地在±0.3°内。
在另一个实施方案中,本发明至少包括化合物3形式A的以下实施方案:
a)一种化合物3的分离的结晶形式A,其特征在于包括选自2.6±0.4°、3.6±0.4°和3.8±0.4°的至少一个2θ值的XRPD图谱;
b)实施方案(a)的化合物3的分离的结晶形式A,其特征在于包括选自2.6±0.4°、3.6±0.4°和3.8±0.4°的至少两个2θ值的XRPD图谱;
c)实施方案(a)的化合物3的分离的结晶形式A,其特征在于包括选自2.6±0.4°、3.6±0.4°和3.8±0.4°的2θ值的XRPD图谱;
d)实施方案(a)或(b)的化合物3的分离的结晶形式A,其特征在于至少包括2.6±0.2°的2θ值的XRPD图谱;
e)实施方案(a)-(d)中任一项的化合物3的分离的结晶形式A,其特征在于至少包括3.6±0.2°的2θ值的XRPD图谱;
f)实施方案(a)-(e)中任一项的化合物3的分离的结晶形式A,其中XRPD图谱具有图51的特征2θ值;
g)一种药物组合物,所述药物组合物包含实施方案(a)-(f)中任一项的化合物3的分离的结晶形式A,所述分离的结晶形式处于药学上可接受的赋形剂中,用于固体剂量递送;
h)一种治疗补体因子D介导的病症的方法,所述方法包括向有需要的受试者施用治疗有效量的根据实施方案(a)-(f)中任一项的化合物3的分离的结晶形式A或其药物组合物,所述分离的结晶形式任选地处于药学上可接受的赋形剂中,用于固体剂量递送;
i)(h)的实施方案的方法,其中所述受试者是人;
j)实施方案(a)-(f)中任一项的化合物3的分离的结晶形式A,所述分离的结晶形式用于治疗有需要的受试者的补体因子D介导的病症,所述分离的结晶形式任选地处于药学上可接受的赋形剂中,用于固体剂量递送;
k)实施方案(j)的化合物3的分离的结晶形式A,其中所述受试者是人;
l)实施方案(a)-(f)中任一项的化合物3的分离的结晶形式A或其药物组合物在制造用于治疗有需要的受试者的补体因子D介导的病症的药物中的用途,所述分离的结晶形式任选地处于药学上可接受的赋形剂中,用于固体剂量递送;
m)实施方案(l)的用途,其中所述受试者是人。
形式B
形式B的特征在于图2或图52中所示的XRPD图谱或与图2或图52中所示的图谱基本上相似的XRPD图谱。在一个实施方案中,分离的化合物3形式B的特征在于图21中的DSC。在一个实施方案中,分离的化合物3形式B的特征在于在差示扫描量热分析中在大约176℃处具有宽泛的吸热特征。在一个实施方案中,分离的化合物3形式B的特征在于在VT-XRPD分析中在150℃与250℃之间熔融。在一个实施方案中,形式B是高度稳定的,具有长的保存期限和最小程度的降解。
本发明至少包括化合物3形式B的以下实施方案:
a)一种化合物3的分离的结晶形式B,其特征在于包括选自16.2±0.4°、15.7±0.4°、4.5±0.4°、22.6±0.4°、17.4±0.4°、22.0±0.4°、8.3±0.4°、16.1±0.4°、21.1±0.4°、18.7±0.4°、18.3±0.4°、23.9±0.4°和27.5±0.4°的至少三个2θ值的XRPD图谱;
b)实施方案(a)的化合物3的分离的结晶形式B,其特征在于包括选自16.2±0.4°、15.7±0.4°、4.5±0.4°、22.6±0.4°、17.4±0.4°、22.0±0.4°、8.3±0.4°、16.1±0.4°、21.1±0.4°、18.7±0.4°、18.3±0.4°、23.9±0.4°和27.5±0.4°的至少四个2θ值的XRPD图谱;
c)实施方案(a)的化合物3的分离的结晶形式B,其特征在于包括选自16.2±0.4°、15.7±0.4°、4.5±0.4°、22.6±0.4°、17.4±0.4°、22.0±0.4°、8.3±0.4°、16.1±0.4°、21.1±0.4°、18.7±0.4°、18.3±0.4°、23.9±0.4°和27.5±0.4°的至少四个2θ值的XRPD图谱;
d)实施方案(a)的化合物3的分离的结晶形式B,其特征在于包括选自16.2±0.4°、15.7±0.4°、4.5±0.4°、22.6±0.4°、17.4±0.4°、22.0±0.4°、8.3±0.4°、16.1±0.4°、21.1±0.4°、18.7±0.4°、18.3±0.4°、23.9±0.4°和27.5±0.4°的至少五个2θ值的XRPD图谱;
e)实施方案(a)的化合物3的分离的结晶形式B,其特征在于包括选自16.2±0.4°、15.7±0.4°、4.5±0.4°、22.6±0.4°、17.4±0.4°、22.0±0.4°、8.3±0.4°、16.1±0.4°、21.1±0.4°、18.7±0.4°、18.3±0.4°、23.9±0.4°和27.5±0.4°的至少六个2θ值的XRPD图谱;
f)实施方案(a)的化合物3的分离的结晶形式B,其特征在于包括选自16.2±0.4°、15.7±0.4°、4.5±0.4°、22.6±0.4°、17.4±0.4°、22.0±0.4°、8.3±0.4°、16.1±0.4°、21.1±0.4°、18.7±0.4°、18.3±0.4°、23.9±0.4°和27.5±0.4°的所有2θ值的XRPD图谱;
g)实施方案(a)-(e)中任一项的化合物3的分离的结晶形式B,其特征在于至少包括16.2±0.4°的2θ值的XRPD图谱;
h)实施方案(a)-(e)中任一项的化合物3的分离的结晶形式B,其特征在于至少包括15.7±0.4°的2θ值的XRPD图谱;
i)实施方案(a)-(e)中任一项的化合物3的分离的结晶形式B,其中XRPD图谱具有图52的特征2θ值;
j)实施方案(a)-(h)中任一项的化合物3的分离的结晶形式B,其中每个2θ值在0.3°内;
k)实施方案(a)-(h)中任一项的化合物3的分离的结晶形式B,其中每个2θ值在0.2°内;
l)一种药物组合物,所述药物组合物包含实施方案(a)-(k)中任一项的化合物3的分离的结晶形式B,所述分离的结晶形式处于药学上可接受的赋形剂中,用于固体剂量递送;
m)一种治疗补体因子D介导的病症的方法,所述方法包括向有需要的受试者施用治疗有效量的根据实施方案(a)-(k)中任一项的化合物3的分离的结晶形式B或其药物组合物,所述分离的结晶形式任选地处于药学上可接受的赋形剂中,用于固体剂量递送;
n)(m)的实施方案的方法,其中所述受试者是人;
o)实施方案(a)-(k)中任一项的化合物3的分离的结晶形式B,所述分离的结晶形式用于治疗有需要的受试者的补体因子D介导的病症,所述分离的结晶形式任选地处于药学上可接受的赋形剂中,用于固体剂量递送;
p)实施方案(o)的化合物3的分离的结晶形式B,其中所述受试者是人;
q)实施方案(a)-(k)中任一项的化合物3的分离的结晶形式B或其药物组合物在制造用于治疗有需要的受试者的补体因子D介导的病症的药物中的用途,所述分离的结晶形式任选地处于药学上可接受的赋形剂中,用于固体剂量递送;
r)实施方案(q)的用途,其中所述受试者是人。
可以使用选择性结晶来制备形式B。如下文更详细地描述,所述方法可以通过任选地在一种或多种包含形式B的晶种的存在下在提供形式B的结晶的条件下处理包含一种或多种合适的溶剂和化合物3的溶液来进行。在一个实施方案中,通过在丙酮和2-丙醇/庚烷中温度循环无定形化合物3来制备形式B。
形式M
形式M的特征在于图18或图55中所示的XRPD图谱或与图18或图55中所示的图谱基本上相似的XRPD图谱。在一个实施方案中,分离的化合物3形式M的特征在于图19中的DSC。在一个实施方案中,分离的化合物3形式M的特征在于在差示扫描量热分析中在大约205℃处具有宽泛的吸热特征。在一个实施方案中,形式M是高度稳定的,具有长的保存期限和最小程度的降解。
本发明至少包括化合物3形式M的以下实施方案:
a)一种化合物3的分离的结晶形式M,其特征在于包括选自15.0±0.4°、7.5±0.4°、23.8±0.4°、7.2±0.4°、19.1±0.4°、5.2±0.4°、8.3±0.4°、26.2±0.4°、22.8±0.4°、21.7±0.4°和24.9±0.4°的至少三个2θ值的XRPD图谱;
b)实施方案(a)的化合物3的分离的结晶形式M,其特征在于包括选自15.0±0.4°、7.5±0.4°、23.8±0.4°、7.2±0.4°、19.1±0.4°、5.2±0.4°、8.3±0.4°、26.2±0.4°、22.8±0.4°、21.7±0.4°和24.9±0.4°的至少四个2θ值的XRPD图谱;
c)实施方案(a)的化合物3的分离的结晶形式M,其特征在于包括选自15.0±0.4°、7.5±0.4°、23.8±0.4°、7.2±0.4°、19.1±0.4°、5.2±0.4°、8.3±0.4°、26.2±0.4°、22.8±0.4°、21.7±0.4°和24.9±0.4°的至少四个2θ值的XRPD图谱;
d)实施方案(a)的化合物3的分离的结晶形式M,其特征在于包括选自15.0±0.4°、7.5±0.4°、23.8±0.4°、7.2±0.4°、19.1±0.4°、5.2±0.4°、8.3±0.4°、26.2±0.4°、22.8±0.4°、21.7±0.4°和24.9±0.4°的至少五个2θ值的XRPD图谱;
e)实施方案(a)的化合物3的分离的结晶形式M,其特征在于包括选自15.0±0.4°、7.5±0.4°、23.8±0.4°、7.2±0.4°、19.1±0.4°、5.2±0.4°、8.3±0.4°、26.2±0.4°、22.8±0.4°、21.7±0.4°和24.9±0.4°的至少六个2θ值的XRPD图谱;
f)实施方案(a)的化合物3的分离的结晶形式M,其特征在于包括选自15.0±0.4°、7.5±0.4°、23.8±0.4°、7.2±0.4°、19.1±0.4°、5.2±0.4°、8.3±0.4°、26.2±0.4°、22.8±0.4°、21.7±0.4°和24.9±0.4°的所有2θ值的XRPD图谱;
g)实施方案(a)-(e)中任一项的化合物3的分离的结晶形式M,其特征在于至少包括15.0±0.4°的2θ值的XRPD图谱;
h)实施方案(a)-(e)中任一项的化合物3的分离的结晶形式M,其特征在于至少包括7.5±0.4°的2θ值的XRPD图谱;
i)实施方案(a)-(e)中任一项的化合物3的分离的结晶形式M,其中XRPD图谱具有图55的特征2θ值;
j)实施方案(a)-(h)中任一项的化合物3的分离的结晶形式M,其中每个2θ值在0.3°内;
k)实施方案(a)-(h)中任一项的化合物3的分离的结晶形式M,其中每个2θ值在0.2°内;
l)一种药物组合物,所述药物组合物包含实施方案(a)-(k)中任一项的化合物3的分离的结晶形式M,所述分离的结晶形式处于药学上可接受的赋形剂中,用于固体剂量递送;
m)一种治疗补体因子D介导的病症的方法,所述方法包括向有需要的受试者施用治疗有效量的根据实施方案(a)-(k)中任一项的化合物3的分离的结晶形式M或其药物组合物,所述分离的结晶形式任选地处于药学上可接受的赋形剂中,用于固体剂量递送;
n)(m)的实施方案的方法,其中所述受试者是人;
o)实施方案(a)-(k)中任一项的化合物3的分离的结晶形式M,所述分离的结晶形式用于治疗有需要的受试者的补体因子D介导的病症,所述分离的结晶形式任选地处于药学上可接受的赋形剂中,用于固体剂量递送;
p)实施方案(o)的化合物3的分离的结晶形式M,其中所述受试者是人;
q)实施方案(a)-(k)中任一项的化合物3的分离的结晶形式M或其药物组合物在制造用于治疗有需要的受试者的补体因子D介导的病症的药物中的用途,所述分离的结晶形式任选地处于药学上可接受的赋形剂中,用于固体剂量递送;
r)实施方案(q)的用途,其中所述受试者是人。
可以使用选择性结晶来制备形式M。如下文更详细地描述,所述方法可以通过任选地在一种或多种包含形式M的晶种的存在下在提供形式M的结晶的条件下处理包含一种或多种合适的溶剂和化合物3的溶液来进行。在一个实施方案中,通过在丙酮中温度循环无定形化合物3大约2.5天来制备形式M。
形式G
形式G的特征在于图10或图53中所示的XRPD图谱或与图10或图53中所示的图谱基本上相似的XRPD图谱。在一个实施方案中,分离的化合物3形式G的特征在于图16中的DSC。
本发明至少包括化合物3形式G的以下实施方案:
a)一种化合物3的分离的结晶形式G,其特征在于包括选自15.0±0.4°、8.0±0.4°、16.3±0.4°、14.8±0.4°、7.3±0.4°、16.1±0.4°、4.1±0.4°、13.1±0.4°、10.2±0.4°和5.1±0.4°的至少三个2θ值的XRPD图谱;
b)实施方案(a)的化合物3的分离的结晶形式G,其特征在于包括选自15.0±0.4°、8.0±0.4°、16.3±0.4°、14.8±0.4°、7.3±0.4°、16.1±0.4°、4.1±0.4°、13.1±0.4°、10.2±0.4°和5.1±0.4°的至少四个2θ值的XRPD图谱;
c)实施方案(a)的化合物3的分离的结晶形式G,其特征在于包括选自15.0±0.4°、8.0±0.4°、16.3±0.4°、14.8±0.4°、7.3±0.4°、16.1±0.4°、4.1±0.4°、13.1±0.4°、10.2±0.4°和5.1±0.4°的至少四个2θ值的XRPD图谱;
d)实施方案(a)的化合物3的分离的结晶形式G,其特征在于包括选自15.0±0.4°、8.0±0.4°、16.3±0.4°、14.8±0.4°、7.3±0.4°、16.1±0.4°、4.1±0.4°、13.1±0.4°、10.2±0.4°和5.1±0.4°的至少五个2θ值的XRPD图谱;
e)实施方案(a)的化合物3的分离的结晶形式G,其特征在于包括选自15.0±0.4°、8.0±0.4°、16.3±0.4°、14.8±0.4°、7.3±0.4°、16.1±0.4°、4.1±0.4°、13.1±0.4°、10.2±0.4°和5.1±0.4°的至少六个2θ值的XRPD图谱;
f)实施方案(a)的化合物3的分离的结晶形式G,其特征在于包括选自15.0±0.4°、8.0±0.4°、16.3±0.4°、14.8±0.4°、7.3±0.4°、16.1±0.4°、4.1±0.4°、13.1±0.4°、10.2±0.4°和5.1±0.4°的所有2θ值的XRPD图谱;
g)实施方案(a)-(e)中任一项的化合物3的分离的结晶形式G,其特征在于至少包括15.0±0.4°的2θ值的XRPD图谱;
h)实施方案(a)-(e)中任一项的化合物3的分离的结晶形式G,其特征在于至少包括8.0±0.4°的2θ值的XRPD图谱;
i)实施方案(a)-(e)中任一项的化合物3的分离的结晶形式G,其中XRPD图谱具有图53的特征2θ值;
j)实施方案(a)-(h)中任一项的化合物3的分离的结晶形式G,其中每个2θ值在0.3°内;
k)实施方案(a)-(h)中任一项的化合物3的分离的结晶形式G,其中每个2θ值在0.2°内;
l)一种药物组合物,所述药物组合物包含实施方案(a)-(k)中任一项的化合物3的分离的结晶形式G,所述分离的结晶形式处于药学上可接受的赋形剂中,用于固体剂量递送;
m)一种治疗补体因子D介导的病症的方法,所述方法包括向有需要的受试者施用治疗有效量的根据实施方案(a)-(k)中任一项的化合物3的分离的结晶形式G或其药物组合物,所述分离的结晶形式任选地处于药学上可接受的赋形剂中,用于固体剂量递送;
n)(m)的实施方案的方法,其中所述受试者是人;
o)实施方案(a)-(k)中任一项的化合物3的分离的结晶形式G,所述分离的结晶形式用于治疗有需要的受试者的补体因子D介导的病症,所述分离的结晶形式任选地处于药学上可接受的赋形剂中,用于固体剂量递送;
p)实施方案(o)的化合物3的分离的结晶形式G,其中所述受试者是人;
q)实施方案(a)-(k)中任一项的化合物3的分离的结晶形式G或其药物组合物在制造用于治疗有需要的受试者的补体因子D介导的病症的药物中的用途,所述分离的结晶形式任选地处于药学上可接受的赋形剂中,用于固体剂量递送;
r)实施方案(q)的用途,其中所述受试者是人。
形式J
形式J的特征在于图13或图54中所示的XRPD图谱或与图13或图54中所示的图谱基本上相似的XRPD图谱。在一个实施方案中,分离的化合物3形式J的特征在于图14中的DSC。
本发明至少包括化合物3形式J的以下实施方案:
a)一种化合物3的分离的结晶形式J,其特征在于包括选自13.4±0.4°、4.8±0.4°、6.4±0.4°、7.2±0.4°、23.3±0.4°和7.4±0.4°的至少三个2θ值的XRPD图谱;
b)实施方案(a)的化合物3的分离的结晶形式J,其特征在于包括选自13.4±0.4°、4.8±0.4°、6.4±0.4°、7.2±0.4°、23.3±0.4°和7.4±0.4°的至少四个2θ值的XRPD图谱;
c)实施方案(a)的化合物3的分离的结晶形式J,其特征在于包括选自13.4±0.4°、4.8±0.4°、6.4±0.4°、7.2±0.4°、23.3±0.4°和7.4±0.4°的至少四个2θ值的XRPD图谱;
d)实施方案(a)的化合物3的分离的结晶形式J,其特征在于包括选自13.4±0.4°、4.8±0.4°、6.4±0.4°、7.2±0.4°、23.3±0.4°和7.4±0.4°的至少五个2θ值的XRPD图谱;
e)实施方案(a)的化合物3的分离的结晶形式J,其特征在于包括选自13.4±0.4°、4.8±0.4°、6.4±0.4°、7.2±0.4°、23.3±0.4°和7.4±0.4°的至少六个2θ值的XRPD图谱;
f)实施方案(a)的化合物3的分离的结晶形式J,其特征在于包括选自13.4±0.4°、4.8±0.4°、6.4±0.4°、7.2±0.4°、23.3±0.4°和7.4±0.4°的所有2θ值的XRPD图谱;
g)实施方案(a)-(e)中任一项的化合物3的分离的结晶形式J,其特征在于至少包括13.4±0.4°的2θ值的XRPD图谱;
h)实施方案(a)-(e)中任一项的化合物3的分离的结晶形式J,其特征在于至少包括4.8±0.4°的2θ值的XRPD图谱;
i)实施方案(a)-(e)中任一项的化合物3的分离的结晶形式J,其中XRPD图谱具有图54的特征2θ值;
j)实施方案(a)-(h)中任一项的化合物3的分离的结晶形式J,其中每个2θ值在0.3°内;
k)实施方案(a)-(h)中任一项的化合物3的分离的结晶形式J,其中每个2θ值在0.2°内;
l)一种药物组合物,所述药物组合物包含实施方案(a)-(k)中任一项的化合物3的分离的结晶形式J,所述分离的结晶形式处于药学上可接受的赋形剂中,用于固体剂量递送;
m)一种治疗补体因子D介导的病症的方法,所述方法包括向有需要的受试者施用治疗有效量的根据实施方案(a)-(k)中任一项的化合物3的分离的结晶形式J或其药物组合物,所述分离的结晶形式任选地处于药学上可接受的赋形剂中,用于固体剂量递送;
n)(m)的实施方案的方法,其中所述受试者是人;
o)实施方案(a)-(k)中任一项的化合物3的分离的结晶形式J,所述分离的结晶形式用于治疗有需要的受试者的补体因子D介导的病症,所述分离的结晶形式任选地处于药学上可接受的赋形剂中,用于固体剂量递送;
p)实施方案(o)的化合物3的分离的结晶形式J,其中所述受试者是人;
q)实施方案(a)-(k)中任一项的化合物3的分离的结晶形式J或其药物组合物在制造用于治疗有需要的受试者的补体因子D介导的病症的药物中的用途,所述分离的结晶形式任选地处于药学上可接受的赋形剂中,用于固体剂量递送;
r)实施方案(q)的用途,其中所述受试者是人。
在一个实施方案中,提供了一种药物组合物,所述药物组合物包含分离的化合物3形态形式A、形式B或形式M和药学上可接受的赋形剂。
在本发明的一个方面中,提供了一种用于治疗由补体因子D介导的病症的方法,例如,提供了治疗阵发性睡眠性血红蛋白尿症(PNH)或C3肾小球病(C3G)的方法,所述方法包括向有需要的宿主施用治疗有效量的化合物3的分离的形式。在一个实施方案中,所述形式选自形式A、形式B或形式M。
在本发明的一个方面中,提供了一种用于治疗选自形式II膜性增生性肾小球性肾炎(MPGNII)、非酒精性脂肪性肝炎(NASH)、脂肪肝、肝脏炎症、肝硬化、肝衰竭、皮肌炎和肌萎缩性侧索硬化的病症的方法,所述方法包括向有需要的宿主施用治疗有效量的化合物3的分离的形式。在一个实施方案中,所述形式选自形式A、形式B或形式M。
在本发明的一个方面中,提供了一种用于治疗选自多发性硬化、关节炎、呼吸系统疾病、心血管疾病、COPD、类风湿性关节炎、非典型溶血性尿毒症综合征和典型溶血性尿毒症综合征的病症的方法,所述方法包括向有需要的宿主施用治疗有效量的化合物3的分离的形式。在一个实施方案中,所述形式选自形式A、形式B或形式M。
在本发明的一个方面中,提供了一种用于治疗选自膜性肾小球性肾炎、年龄相关性黄斑变性(AMD)、视网膜变性和形式I糖尿病或其并发症的病症的方法,所述方法包括向有需要的宿主施用治疗有效量的化合物3的分离的形式。在一个实施方案中,所述形式选自形式A、形式B或形式M。
在一个实施方案中,提供了一种治疗患有补体因子D介导的病症的患者的治疗方法,所述方法包括向有需要的患者施用有效量的化合物3形式A和C5抑制剂。在一个实施方案中,化合物3形式A和C5抑制剂具有重叠的治疗效果。在一个实施方案中,提供了一种治疗患有补体因子D介导的病症的患者的治疗方法,所述方法包括向有需要的患者施用有效量的化合物3形式A和依库丽单抗。在一个实施方案中,化合物3形式A和依库丽单抗具有重叠的治疗效果。在一个实施方案中,提供了一种治疗患有补体因子D介导的病症的患者的治疗方法,所述方法包括向有需要的患者施用化合物3形式A和拉鲁珠单抗。在一个实施方案中,化合物3形式A和拉鲁珠单抗具有重叠的治疗效果。例如,治疗效果可以是组合或协同抑制。
在一个实施方案中,化合物3结晶形式A和C5抑制剂的AUC重叠。
在一个实施方案中,C5抑制剂是依库丽单抗。在一个实施方案中,C5抑制剂是拉鲁珠单抗。在一个实施方案中,C5抑制剂是小分子。在另一个实施方案中,C5抑制剂是靶向C5的多克隆抗体。在另一个实施方案中,C5抑制剂是适体。
在一个实施方案中,提供了一种治疗患有补体因子D介导的病症的患者的治疗方法,所述方法包括向有需要的患者施用有效量的化合物3形式B和C5抑制剂。在一个实施方案中,化合物3形式B和C5抑制剂具有重叠的治疗效果。在一个实施方案中,提供了一种治疗患有补体因子D介导的病症的患者的治疗方法,所述方法包括向有需要的患者施用有效量的化合物3形式B和依库丽单抗。在一个实施方案中,化合物3形式B和依库丽单抗具有重叠的治疗效果。在一个实施方案中,提供了一种治疗患有补体因子D介导的病症的患者的治疗方法,所述方法包括向有需要的患者施用化合物3形式B和拉鲁珠单抗。在一个实施方案中,化合物3形式B和拉鲁珠单抗具有重叠的治疗效果。例如,治疗效果可以是组合或协同抑制。
在一个实施方案中,化合物3结晶形式B和C5抑制剂的AUC重叠。
在一个实施方案中,C5抑制剂是依库丽单抗。在一个实施方案中,C5抑制剂是拉鲁珠单抗。在一个实施方案中,C5抑制剂是小分子。在另一个实施方案中,C5抑制剂是靶向C5的多克隆抗体。在另一个实施方案中,C5抑制剂是适体。
在一个实施方案中,提供了一种治疗患有补体因子D介导的病症的患者的治疗方法,所述方法包括向有需要的患者施用有效量的化合物3形式M和C5抑制剂。在一个实施方案中,化合物3形式M和C5抑制剂具有重叠的治疗效果。在一个实施方案中,提供了一种治疗患有补体因子D介导的病症的患者的治疗方法,所述方法包括向有需要的患者施用有效量的化合物3形式M和依库丽单抗。在一个实施方案中,化合物3形式M和依库丽单抗具有重叠的治疗效果。在一个实施方案中,提供了一种治疗患有补体因子D介导的病症的患者的治疗方法,所述方法包括向有需要的患者施用化合物3形式M和拉鲁珠单抗。在一个实施方案中,化合物3形式M和拉鲁珠单抗具有重叠的治疗效果。例如,治疗效果可以是组合或协同抑制。
在一个实施方案中,化合物3结晶形式M和C5抑制剂的AUC重叠。
在一个实施方案中,C5抑制剂是依库丽单抗。在一个实施方案中,C5抑制剂是拉鲁珠单抗。在一个实施方案中,C5抑制剂是小分子。在另一个实施方案中,C5抑制剂是靶向C5的多克隆抗体。在另一个实施方案中,C5抑制剂是适体。
化学说明和术语
使用标准命名法描述化合物。除非另外定义,否则在本文中使用的所有技术和科学术语均具有与本发明所属领域的技术人员通常理解的含义相同的含义。
术语“一”和“一种”不表示量的限制,而是表示存在至少一个所提及项目。术语“或”意指“和/或”。除非另外指出,否则值的范围的叙述仅意图起到单独地提及落在该范围内的每一个单独的值的简略方法的作用,并且每一个单独的值并入本说明书中就好像其在本文中被单独地陈述一样。所有范围的端点均包括在所述范围内并可独立地合并。
除非本文另外指出或与上下文明显矛盾,否则本文所述的所有方法都可以按任何合适的顺序进行。除非另外要求,否则使用任何以及所有实例或示例性语言(例如,“如”)仅意图说明而不对本发明的范围施加限制。
“氘代”和“氘代”意指氢被氘替代,使得氘以自然丰度存在且因此被“富集”。50%的富集意指指定位置处不是氢,而是氘,含量为50%。为了清楚起见,确认的是如本文所用的术语“富集”不表示相对于自然丰度的富集百分比。在其他实施方案中,在一个或多个指定氘代位置处将存在至少80%、至少90%或至少95%的氘富集。在其他实施方案中,在指出的一个或多个指定氘代位置处将存在至少96%、至少97%、至少98%或至少99%的氘富集。在没有相反指示的情况下,本文所述化合物的指定位置中的氘富集为至少90%。
“剂型”意指活性剂的施用单元。剂型的非限制性实例包括片剂、胶囊剂、凝胶帽、注射剂、混悬剂、液体剂、静脉注射液、乳剂、乳霜剂、软膏剂、栓剂、可吸入形式、透皮形式等。
“药物组合物”是包含至少一种活性剂(如本文所公开的一种活性化合物的化合物或盐)和至少一种其他物质(如载体)的组合物。药物组合物任选地含有多于一种活性剂。“药物组合”或“联合疗法”是指施用至少两种活性剂(并且在一个实施方案中,三种或四种或更多种活性剂),所述至少两种活性剂可组合成单一剂型或任选地根据这些活性剂一起使用来治疗病症的说明以分开的剂型一起提供。
术语“载体”意指提供形态形式的稀释剂、赋形剂或媒介物。
“药学上可接受的赋形剂”意指通常安全、充分无毒并且既不在生物学上也不在其他方面不合期望的可用于制备药物组合物/组合的赋形剂。如本申请中所用的“药学上可接受的赋形剂”包括一种和多于一种这样的赋形剂。
“患者”或“宿主”是需要治疗的人类或非人类动物,包括但不限于猿猴、禽类、猫科动物、犬科动物、牛科动物、马科动物或猪。医学治疗可以包括对现有疾患(如疾病或病症)的治疗,或预防性或诊断性治疗。在特定实施方案中,患者或宿主是人类患者。在替代实施方案中,治疗患者如宿主以预防本文所述的病症或疾病。
如本文所用,术语“分离的”是指呈基本上纯的形式的材料。分离的化合物不具有会实质性影响该化合物的性质的另一成分。在特定实施方案中,分离的形式是至少60%、70%、80%、90%、95%、98%或99%纯的。
药物制剂
可以使用达到期望治疗结果的任何合适的方法,将本文所述的分离的形态形式以有效量施用至宿主以治疗本文所述的任何病症。当然,分离的形态形式的施用量和时间将取决于所治疗的宿主、指导医学专家的说明、暴露的时程、施用方式、特定活性化合物的药代动力学性质以及开药医师的判断。因此,由于宿主之间的变化性,下面给出的剂量是一个指引,医师可以滴定化合物的剂量,以实现医师认为适合宿主的治疗。在考虑期望治疗程度时,医师可以平衡各种因素,如宿主的年龄和体重、既往疾病的存在以及其他疾病的存在。
有效量的如本文所述的形态形式或者与另一活性剂组合或与另一活性剂交替地或在另一活性剂之前、与其同时或在其之后的本文所述的形态形式可以以足以(a)抑制通过补体途径介导的病症(包括炎性、免疫(包括自身免疫)病症或补体因子D相关病症)的进展;(b)引起炎性、免疫(包括自身免疫)病症或补体因子D相关病症的消退;(c)引起炎性、免疫(包括自身免疫)病症或补体因子D相关病症的治愈;或者抑制或防止炎性、免疫(包括自身免疫)病症或补体因子D相关病症的发展的量使用。因此,有效量的本文所述的形态形式或组合物当施用至患者时将提供足够量的活性剂以提供临床益处。
药物组合物可以在医疗装置、栓剂、口腔或舌下中配制为任何药学上有用的形式,例如丸剂、胶囊剂、片剂、透皮贴剂、皮下贴剂、干粉剂、吸入制剂、医疗器械、栓剂、经颊或舌下制剂。一些剂量形式如片剂和胶囊剂被细分为合适规格的单位剂量,其含有适宜量的活性成分,例如有效量以实现期望目的。
本文所述的形态形式的治疗有效剂量将由健康护理从业者根据患者的状况、体型和年龄以及递送途径来确定。在某些实施方案中,药物组合物为在单位剂型中含有约0.1mg至约2000mg、约10mg至约1000mg、约100mg至约800mg或约200mg至约600mg的活性化合物和任选地约0.1mg至约2000mg、约10mg至约1000mg、约100mg至约800mg或约200mg至约600mg的另外的活性剂的剂型。实例为具有至少约0.5、1、1.5、2、2.5、3、3.5、4、4.5、5、10、15、20、25、50、75、100、150、200、250、300、350、400、450、500、550、600、650、700、750、800、900、1000、1100、1200、1250、1300、1400、1500或1600mg的活性化合物的剂型。在一个实施方案中,剂型具有至少约1mg、5mg、10mg、25mg、50mg、75mg、100mg、200mg、400mg、500mg、600mg、1000mg、1200mg或1600mg的活性化合物。剂型可以,例如,按照需要,一天一次(q.d.)、一天两次(b.i.d.)、一天三次(t.i.d.)、一天四次(q.i.d.)、每隔一天一次(Q2d)、每隔两天一次(Q3d)施用,或以提供本文所述的病症的治疗的任何剂量时间表施用。
本文公开的或如本文所述使用的分离的形态形式可以在含有常规药学上可接受的载体的剂量单位制剂中通过口服、局部、肠胃外、通过吸入或喷射、舌下、经由植入物(包括眼植入物)、透皮、经由经颊施用、直肠、肌内、吸入、动脉内、颅内、真皮下、腹膜内、皮下、经鼻、舌下、或直肠或者通过其他方式施用。
根据目前公开的方法,用于施用的口服剂型可以是任何期望形式,其中形态形式作为固体是稳定的。在某些实施方案中,分离的形态形式以固体微粒或纳米颗粒的形式递送。当通过吸入施用时,分离的形态形式可以是具有任何期望粒径的多个固体颗粒或液滴的形式,所述粒径为例如约0.01、0.1或0.5至约5、10、20或更大的微米,且任选地约1至约2微米。本发明中公开的分离的形态形式例如当通过口服途径施用时具有良好的药代动力学和药效动力学性质。
药物制剂可以包含处于任何药学上可接受的载体中的本文所述的分离的形态形式。
颗粒可以使用相转化方法从如本文所述的形态形式形成。在这种方法中,将形态形式溶解在合适的溶剂中,并将溶液倒入强非溶剂(non-solvent)中以使得化合物在有利的条件下自发地产生微粒或纳米颗粒。该方法可以用于产生广泛大小范围的纳米颗粒,包括,例如,纳米颗粒至微粒,其通常具有窄的粒度分布。
在替代实施方案中,对形态形式进行研磨工艺,包括但不限于手工研磨、转子研磨、球磨和喷射研磨,以获得微粒和纳米颗粒。
在一个实施方案中,颗粒在约0.1nm至约10000nm之间、约1nm至约1000nm之间、约10nm与1000nm之间、约1与100nm之间、约1与10nm之间、在约1与50nm之间、在约100nm与800nm之间、在约400nm与600nm之间、或约500nm。在一个实施方案中,微粒不大于约0.1nm、0.5nm、1.0nm、5.0nm、10nm、25nm、50nm、75nm、100nm、150nm、200nm、250nm、300nm、400nm、450nm、500nm、550nm、600nm、650nm、700nm、750nm、800nm、850nm、900nm、950nm、1000nm、1250nm、1500nm、1750nm或2000nm。
载体包括赋形剂和稀释剂并且必须是足够高纯度且足够低毒性以使得它们适于施用至被治疗的患者。载体可以是惰性的或者其可具有其自身的药学有益效果。与化合物结合使用的载体的量足以为施用每单位剂量的化合物提供实际的材料量。
载体的类别包括,但不限于粘合剂、缓冲剂、着色剂、稀释剂、崩解剂、乳化剂、调味剂、助流剂、润滑剂、防腐剂、稳定剂、表面活性剂、压片剂和润湿剂。一些载体可能被列在超过一个类别中,例如植物油在一些制剂中可用作润滑剂而在其他制剂中用作稀释剂。示例性的药学上可接受载体包括糖、淀粉、纤维素、黄芪胶粉末、麦芽、明胶、滑石和植物油。可在药物组合物中包含任选的活性剂,其不实质性地干扰本发明的化合物的活性。
取决于预期的施用方式,药物组合物可以是分离的形态形式处于稳定状态下的固体形式或半固体剂型,例如片剂、栓剂、丸剂、胶囊、粉末等,优选适合单次施用精确剂量的单位剂型。组合物将包括有效量的所选药物与药学上可接受的载体的组合,此外,还可包括其他药剂、佐剂、稀释剂、缓冲剂等。
因此,本公开的组合物可以作为药物制剂施用,包括适合口服(包括口腔和舌下)、直肠、鼻、局部、肺、阴道的那些,或者以适于通过吸入或吹入施用的形式施用。优选的施用方式是使用方便的日剂量方案(其可根据痛苦程度进行调整)的口服施用。对于固体组合物,常规的无毒固体载体包括,例如,药物级的甘露醇、乳糖、淀粉、硬脂酸镁、糖精钠、滑石、纤维素、葡萄糖、蔗糖、碳酸镁等。
在另一个实施方案中,使用渗透增强剂赋形剂,包括聚合物,如:聚阳离子(壳聚糖及其季铵衍生物、聚-L-精氨酸、胺化明胶);聚阴离子(N-羧甲基壳聚糖、聚丙烯酸)和硫醇化聚合物(羧甲基纤维素-半胱氨酸、聚卡波非-半胱氨酸、壳聚糖-硫代丁基脒、壳聚糖-硫代乙醇酸、壳聚糖-谷胱甘肽缀合物)。
对于口服施用,组合物一般将采取片剂、丸剂、胶囊、粉末等形式。片剂和胶囊是优选的口服施用形式。用于口服使用的片剂和胶囊可包括一种或多种常用的载体如乳糖和玉米淀粉。通常还添加润滑剂,如硬脂酸镁。通常,本公开的组合物可与口服的、无毒、药学上可接受的惰性载体组合,如乳糖、淀粉、蔗糖、葡萄糖、甲基纤维素、硬脂酸镁、磷酸二钙、硫酸钙、甘露醇、山梨糖醇等。此外,当需要或必要时,也可将合适的粘合剂、润滑剂、崩解剂和着色剂掺入混合物中。合适的粘合剂包括淀粉、明胶、天然糖(如葡萄糖或β-乳糖)、玉米甜味剂、天然和合成树胶(如阿拉伯树胶、黄蓍胶或海藻酸钠)、羧甲基纤维素、聚乙二醇、蜡等。这些剂型中使用的润滑剂包括油酸钠、硬脂酸钠、硬脂酸镁、苯甲酸钠、乙酸钠、氯化钠等。崩解剂包括但不限于淀粉、甲基纤维素、琼脂、膨润土、黄原胶等。
除活性化合物或其盐之外,药物制剂还可包含其他添加剂,如pH调节添加剂。特别地,可用的pH调节剂包括酸(如盐酸)、碱或缓冲剂(如乳酸钠、乙酸钠、磷酸钠、柠檬酸钠、硼酸钠或葡萄糖酸钠)。此外,制剂可以含有抗微生物防腐剂。可用的抗微生物防腐剂包括对羟基苯甲酸甲酯、对羟基苯甲酸丙酯和苄醇。当将制剂置于设计用于多剂量使用的小瓶中时,通常采用抗微生物防腐剂。可以使用本领域中众所周知的技术将本文所述的药物制剂冻干。
还提供了药物制剂,所述药物制剂提供了本文所描述的化合物的受控释放,包括通过使用可降解的聚合物,如本领域中已知的。
在一个实施方案中,以下组合部分中描述的另外的治疗剂以药学上可接受的盐,例如以下描述的盐的形式施用。
适合用于直肠施用的制剂通常作为单位剂量栓剂存在。它们可以通过将所公开的活性化合物与一种或多种常规固体载体(例如,可可脂)混合并随后使所得混合物成型而制备。
适合用于局部施用至皮肤的制剂优选采取软膏、乳膏、洗剂、糊剂、凝胶、喷雾剂、气溶胶或油的形式,其保持分离的形态形式的稳定性。。可以使用的载体包括矿脂、羊毛脂、聚乙二醇、醇类、透皮促进剂及其两种或更多种的组合。
适合用于透皮施用的制剂可以作为适应于保持与接受者的表皮密切接触延长的时间段的离散贴片存在。在一个实施方案中,微针贴片或装置提供用于递送药物跨越生物组织(特别地皮肤)或至生物组织中。微针贴片或装置允许以临床相关的速率跨越皮肤或其他组织屏障递送药物或者递送至皮肤或其他组织屏障中,而具有最小的或没有对组织的损伤、疼痛或刺激。
适合用于施用至肺的制剂可以通过广泛的被动呼吸驱动的或主动能力驱动的单剂量/多剂量干粉吸入器(DPI)递送。最常用于呼吸系统递送的装置包括雾化器、定量吸入器和干粉吸入器。几种类型的雾化器是可用的,包括喷射雾化器、超声雾化器和振动网孔雾化器。合适的肺递送装置的选择取决于如药物及其制剂的性质、作用的位点和肺的病理生理的参数。
在喷雾干燥分散体(SDD)中使用形态形式来制备纯度提高的化合物
在一个实施方案中,本文所述的形态形式用于产生喷雾干燥分散体(SDD),其被施用至有需要的患者。通过首先将无定形形式的化合物3转化为优选的形态形式,然后将其重新溶解并制成SDD,可获得更高纯度的API。在这种方法中,将形态形式溶解在有机溶剂如丙酮、二氯甲烷、甲醇、乙醇或它们的混合物(例如90:10、80:20或50:50的DCM与甲醇)或另一种合适的有机溶剂或它们的混合物中。溶液通过由压缩气体流驱动的微粉化喷嘴泵送,且所得的气溶胶悬浮在加热的空气旋流器中,从而允许溶剂从微滴蒸发而形成颗粒。微粒和纳米颗粒可以使用这一方法获得。
在一个实施方案中,如本文所述的形态形式作为喷雾干燥分散体(SDD)施用至有需要的患者。在另一个实施方案中,本发明提供了包含本发明的形态形式和如本文中限定的一种或多种药学上可接受的赋形剂的喷雾干燥分散体(SDD)。在另一个实施方案中,SDD包含本发明的形态形式和另外的治疗剂。在进一步的实施方案中,SDD包含本发明的形态形式、另外的治疗剂和一种或多种药学上可接受的赋形剂。在另一个实施方案中,任何所描述的喷雾干燥分散体可以包衣以形成包衣片剂。在替代的实施方案中,喷雾干燥分散体被配制成片剂,但是未包衣的。
活性化合物用于治疗所选病症的用途
在一个方面,有效量的如本文所述的形态形式或组合物用于治疗作为炎性或免疫疾患的医学病症、通过补体级联(包括功能异常的级联)介导的病症(包括补体因子D相关病症或替代补体途径相关病症)、不利地影响细胞参与正常补体活性或对正常补体活性作出反应的能力的细胞病症或异常、或对于医学治疗(如手术或其他医疗过程)或药物或生物药物施用、输血或其他同种异体组织或流体施用的不希望的补体介导的反应。
在一个实施方案中,提供了一种用于治疗C3肾小球肾炎(C3G)的方法,所述方法包括施用有效量的本文所述的形态形式,任选地以药学上可接受的组合物的形式。
在一个实施方案中,提供了一种用于治疗阵发性睡眠性血红蛋白尿症(PNH)的方法,所述方法包括施用有效量的本文所述的形态形式,任选地以药学上可接受的组合物的形式。
在另一个实施方案中,提供了一种用于治疗宿主的与湿式或干式年龄相关性黄斑变性(AMD)的方法,所述方法包括施用有效量的本文所述的形态形式,任选地以药学上可接受的组合物的形式。
在另一个实施方案中,提供了一种用于治疗宿主的类风湿性关节炎的方法,所述方法包括施用有效量的本文所述的形态形式,任选地以药学上可接受的组合物的形式。
在另一个实施方案中,提供了一种用于治疗宿主的多发性硬化或肌萎缩性侧索硬化的方法,所述方法包括施用有效量的本文所述的形态形式,任选地以药学上可接受的组合物的形式。
在另一个实施方案中,提供了一种用于治疗宿主的II型膜增生性肾小球性肾炎(MPGN II)的方法,所述方法包括施用有效量的本文所述的形态形式,任选地以药学上可接受的组合物的形式。
在另一个实施方案中,提供了一种用于治疗宿主的非酒精性脂肪性肝炎(NASH)的方法,所述方法包括施用有效量的本文所述的形态形式,任选地以药学上可接受的组合物的形式。
在另一个实施方案中,提供了一种用于治疗宿主的脂肪肝、肝脏炎症、肝硬化或肝衰竭的方法,所述方法包括施用有效量的本文所述的形态形式,任选地以药学上可接受的组合物的形式。
在另一个实施方案中,提供了一种用于治疗宿主的皮肌炎的方法,所述方法包括施用有效量的本文所述的形态形式,任选地以药学上可接受的组合物的形式。
在另一个实施方案中,提供了一种用于治疗宿主的关节炎或COPD的方法,所述方法包括施用有效量的本文所述的形态形式,任选地以药学上可接受的组合物的形式。
在另一个实施方案中,提供了一种用于治疗宿主的呼吸系统疾病或心血管疾病的方法,所述方法包括施用有效量的本文所述的形态形式,任选地以药学上可接受的组合物的形式。
在另一个实施方案中,提供了一种用于治疗宿主的非典型或典型溶血性尿毒症综合征的方法,所述方法包括施用有效量的本文所述的形态形式,任选地以药学上可接受的组合物的形式。
在另一个实施方案中,提供了一种用于治疗宿主的膜性增生性肾小球性肾炎或年龄相关性黄斑变性(AMD)的方法,所述方法包括施用有效量的本文所述的形态形式,任选地以药学上可接受的组合物的形式。
在另一个实施方案中,提供了一种用于治疗宿主的I型糖尿病或其并发症的方法,所述方法包括施用有效量的本文所述的形态形式,任选地以药学上可接受的组合物的形式。
如本文所公开的,任选地呈药学上可接受的组合物的形态形式也可用于与第二药剂组合(以相同或不同剂型)联合施用或与第二药剂交替施用,以用于改善或减轻第二药剂的副作用。
提供了另一个实施方案,其包括将有效量的形态形式,任选地以药学上可接受的组合物的形式施用至宿主以治疗可得益于局部或局部递送的眼、肺、胃肠道或其他病症。
在本发明的其他实施方案中,本文所提供的形态形式可以用于治疗或预防宿主的由补体因子D或补体途径的补体C3扩增环的过量或有害量介导的病症。作为实例,本发明包括治疗或预防由抗体-抗原相互作用,免疫或自身免疫性病症的成分或缺血性损伤诱导的补体相关病症的方法。本发明还提供了减少由因子D介导或影响的炎症或免疫反应,包括自身免疫反应的方法。
在一个实施方案中,所述病症选自脂肪肝和源自脂肪肝的疾患如非酒精性脂肪性肝炎(NASH)、肝脏炎症、肝硬化和肝衰竭。在本发明的一个实施方案中,提供了一种用于通过施用有效量的如本文所述的形态形式或组合物治疗宿主的脂肪肝疾病的方法。
在另一个实施方案中,本文所述的形态形式或组合物用于在手术或其他医疗过程之前或期间调节免疫反应。一种非限制性实例是结合急性或慢性移植物抗宿主病使用,该疾病是同种异体组织移植导致的常见并发症,且也可以由于输血而发生。
在一个实施方案中,本发明提供了通过向有需要的受试者施用有效量的本文所述的形态形式或组合物来治疗或预防皮肌炎的方法。
在一个实施方案中,本发明提供了通过向有需要的受试者施用有效量的本文所述的形态形式或组合物来治疗或预防肌萎缩性侧索硬化的方法。
在一个实施方案中,本发明提供了通过向有需要的受试者施用有效量的本文所述的形态形式或组合物来治疗或预防腹主动脉瘤、血液透析并发症、溶血性贫血或血液透析的方法。
在一个实施方案中,本发明提供了通过向有需要的受试者施用有效量的如本文所述的形态形式或组合物来治疗或预防C3肾小球病(glomurenopathy)的方法。在一个实施方案中,所述病症选自致密沉积物病(DDD)和C3肾小球肾炎(C3GN)。
在一个实施方案中,本发明提供了通过向有需要的受试者施用有效量的如本文所述的形态形式或组合物来治疗或预防IC-MPGN的方法。
在一个实施方案中,本发明提供了通过向有需要的受试者施用有效量的如本文所述的形态形式或组合物来治疗或预防阵发性睡眠性血红蛋白尿症(PNH)的方法。
在一个实施方案中,本发明提供了通过向有需要的受试者施用有效量的如本文所述的形态形式或组合物来治疗或预防年龄相关性黄斑变性(AMD)的方法。
在一个实施方案中,本发明提供了通过向有需要的受试者施用有效量的如本文所述的形态形式或组合物来治疗或预防类风湿性关节炎的方法。
在一个实施方案中,本发明提供了通过向有需要的受试者施用有效量的如本文所述的形态形式或组合物来治疗或预防多发性硬化的方法。
在一个实施方案中,本发明提供了通过向有需要的受试者施用有效量的如本文所述的形态形式或组合物来治疗或预防重症肌无力的方法。
在一个实施方案中,本发明提供了通过向有需要的受试者施用有效量的如本文所述的形态形式或组合物来治疗或预防非典型溶血性尿毒症综合征(aHUS)的方法。
在一个实施方案中,本发明提供了通过向有需要的受试者施用有效量的如本文所述的形态形式或组合物来治疗或预防视神经脊髓炎(NMO)的方法。
在另一个实施方案中,本发明提供了通过向有需要的受试者施用有效量的如本文所述的形态形式或组合物来治疗或预防如下所述的病症的方法,所述病症包括:玻璃体炎、结节病、梅毒、结核病或莱姆病;视网膜脉管炎、Eales病、结核病、梅毒或弓形体病;视神经视网膜炎、病毒性视网膜炎或急性视网膜坏死;水痘带状疱疹病毒、单纯疱疹病毒、巨细胞病毒、EB病毒、扁平苔癣或登革热相关疾病(例如,出血性登革热);伪装(Masquerade)综合征、接触性皮炎、创伤诱导的炎症、UVB诱导的炎症、湿疹、环状肉芽肿或痤疮。
在另一个实施方案中,所述病症选自:湿式(渗出性)AMD、干式(非渗出性)AMD、脉络膜视网膜变性、脉络膜新生血管(CNV)、脉络膜炎、RPE功能的损失、视力损失(包括视敏度或视野的损失)、由于AMD的视力损失、响应于光暴露的视网膜损伤、视网膜变性、视网膜脱离、视网膜功能障碍、视网膜新生血管(RNV)、早产儿视网膜病变、病理性近视或RPE变性;假晶状体大疱性角膜病、症状性黄斑变性相关病症、视神经变性、光感受器退化、视锥细胞退化、光感受细胞的损失、扁平部睫状体炎、巩膜炎、增生性玻璃体视网膜病变或眼脉络膜小疣(ocular drusen)的形成;慢性荨麻疹、Churg-Strauss综合征、冷凝集素病(CAD)、皮质基底节变性(CBD)、冷球蛋白血症、睫状体炎、布鲁赫氏膜的损伤、德戈斯病、糖尿病血管病变、肝酶升高、内毒素血症、大疱性表皮松解或获得性大疱性表皮松解;特发性混合型冷球蛋白血症、过度血尿素氮-BUN、局灶性节段性肾小球硬化、Gerstmann-Straussler-Scheinker病、巨细胞动脉炎、痛风、哈勒沃登-施帕茨病、桥本氏甲状腺炎、过敏性紫癜肾炎或异常尿沉渣;肝炎、甲型肝炎、乙型肝炎、丙型肝炎或人免疫缺陷病毒(HIV);更一般地例如选自以下的病毒感染:黄病毒科、逆转录病毒、冠状病毒科、痘病毒科、腺病毒科、疱疹病毒科、杯状病毒科、呼肠孤病毒科、小RNA病毒科、披膜病毒科、正粘病毒科、弹状病毒科或嗜肝DNA病毒科;脑膜炎奈瑟氏菌、产志贺毒素大肠杆菌溶血性尿毒症综合征(STEC-HUS)、溶血性尿毒症综合征(HUS)、链球菌或链球菌感染后肾小球肾炎。
在进一步的实施方案中,所述病症选自青光眼、糖尿病性视网膜病、水疱性皮肤病(blistering cutaneous disease)(包括大疱性类天疱疮、天疱疮和大疱性表皮松解症)、眼瘢痕性类天疱疮、葡萄膜炎、成人黄斑变性、糖尿病性视网膜病变(diabetic retinopa)色素性视网膜炎、黄斑水肿、糖尿病性黄斑水肿、白塞氏葡萄膜炎、多灶性脉络膜炎、Vogt-Koyangi-Harada综合征、中间葡萄膜炎(imtermediate uveitis)、鸟枪弹样视网膜-脉络膜炎、交感性眼炎、眼瘢痕性类天疱疮(ocular dicatricial pemphigoid)、眼天疱疮、非动脉缺血性视神经病变(nonartertic ischemic optic neuropathy)、术后炎症和视网膜静脉阻塞或视网膜中央静脉阻塞(CVRO)。
在一些实施方案中,补体介导的疾病包括眼科疾病(包括早期或新生血管性年龄相关性黄斑变性和地图样萎缩)、自身免疫性疾病(包括关节炎、类风湿性关节炎)、呼吸系统疾病、心血管疾病。在其他实施方案中,本发明的化合物适合用于治疗与脂肪酸代谢相关的疾病和病症,包括肥胖症和其他代谢病症。
可以通过如本文所述的形态形式或组合物治疗或预防的补体介导的病症包括但不限于:遗传性血管水肿、毛细血管渗漏综合征、溶血性尿毒症综合征(HUS)、神经病症、格林-巴利综合征、中枢神经系统的疾病和其他神经退行性疾患、肾小球性肾炎(包括膜性增生性肾小球性肾炎)、SLE肾炎、增生性肾炎、肝纤维化、组织再生和神经再生或Barraquer-Simons综合征;败血症的炎性效应、全身炎性反应综合症(SIRS)、不适当的或不希望的补体激活的病症、IL-2治疗期间白介素-2诱导的毒性、炎性病症、自身免疫性疾病的炎症、系统性红斑狼疮(SLE)、狼疮性肾炎(lupus nephritides)、关节炎、免疫复合物病症和自身免疫性疾病、系统性狼疮或红斑狼疮;局部缺血/再灌注损伤(I/R损伤)、心肌梗死、心肌炎、缺血后再灌注状况、球囊血管成形术、动脉粥样硬化、心肺转流或肾分流术中的泵后综合征、肾缺血、主动脉重建后的肠系膜动脉再灌注、抗磷脂综合征、自身免疫性心脏病、局部缺血-再灌注损伤、肥胖症或糖尿病;阿尔茨海默氏痴呆、中风、精神分裂症、创伤性脑损伤、创伤、帕金森氏病、癫痫、移植排斥、妊娠丢失的预防、生物材料反应(例如在血液透析、在设备中)、超急性同种异体移植排斥、异种移植排斥、移植、银屑病、烧伤、热损伤(包括烧伤或冻伤)、或挤压伤;哮喘、过敏症、急性呼吸窘迫综合征(ARDS)、囊性纤维化、成人呼吸窘迫综合征、呼吸困难、咳血、慢性阻塞性肺病(COPD)、肺气肿、肺栓塞和梗塞、肺炎、纤维化粉尘病、惰性粉尘和矿物质(例如,硅、煤尘、铍和石棉)、肺纤维化、有机粉尘疾病、化学损伤(由于刺激性气体和化学品,例如,氯、光气、二氧化硫、硫化氢、二氧化氮、氨和盐酸)、烟雾损伤、热损伤(例如,烧伤、冻伤)、支气管收缩、过敏性肺炎、寄生虫病、肺出血肾炎综合征(抗肾小球基底膜肾炎)、肺脉管炎、寡免疫脉管炎或免疫复合物相关炎症。
在另外的替代实施方案中,如本文所述的形态形式或组合物用于治疗自身免疫性病症。
补体途径增强抗体和吞噬细胞从机体清除微生物和受损细胞的能力。它是先天免疫系统的部分且在健康个体中是必要过程。抑制补体途径会降低身体的免疫系统反应。因此,本发明的目的是通过向有需要的受试者施用有效剂量的如本文所述的形态形式或组合物来治疗自身免疫性病症。
在一个实施方案中,所述自身免疫性病症由补体系统的活性引起。在一个实施方案中,所述自身免疫性病症由替代补体途径的活性引起。在一个实施方案中,所述自身免疫性病症由经典补体途径的活性引起。在另一个实施方案中,所述自身免疫性病症由不直接与补体系统相关的作用机制引起,例如T-淋巴细胞的过度增殖或细胞因子的过度产生。
在一个实施方案中,如本文所述的形态形式或组合物用于治疗狼疮。狼疮的非限制性实例包括红斑狼疮、皮肤狼疮、盘状红斑狼疮、冻疮样红斑狼疮、红斑狼疮-扁平苔藓重叠综合征。
红斑狼疮是包括全身性和皮肤病症的一大类疾病。该疾病的全身性形式可以具有皮肤以及全身的表现。但是,也存在仅涉及皮肤而没有全身牵累的疾病形式。例如,SLE是主要在女性中发生的病因学未知的炎性病症,且特征在于关节症状、蝶形红斑、复发性胸膜炎、心包炎、广义腺病、脾肿大以及CNS牵连和进行性肾衰竭。大多数患者(超过98%)的血清包含抗核抗体,包括抗DNA抗体。高滴度的抗DNA抗体本质上对于SLE是特异性的。对于这一疾病的常规治疗是施用皮质类固醇或免疫抑制剂。
存在三种形式的皮肤狼疮:慢性皮肤狼疮(也称为盘状红斑狼疮或DLE)、亚急性皮肤狼疮和急性皮肤狼疮。DLE是主要影响皮肤的外形损伤性慢性病症,其具有表现出红斑、毛囊栓塞、鳞屑、毛细管扩张和萎缩症的明显局限的斑点和斑块。该疾患通常通过日晒促成,且早期病变为直径5至10mm且表现出毛囊栓塞的红斑性、圆形鳞屑性丘疹。DLE病变最通常出现在面颊、鼻子、头皮和耳朵上,但它们也可以普遍存在于躯干的上部、四肢的伸肌面和嘴的粘膜上。如果不经治疗,中心病变萎缩且留下疤痕。与SLE不同,针对双链DNA的抗体(例如,DNA结合测试)在DLE中几乎总是不存在的。
多发性硬化是据信为T淋巴细胞依赖性的自身免疫性脱髓鞘病症。MS一般表现出复发-缓解性病程或慢性进展性病程。MS的病因学是未知的,但是,病毒感染、遗传倾向性、环境和自身免疫性全部表现为造成所述病症。MS患者中的病变包含主要T淋巴细胞介导的小胶质细胞和浸润性巨噬细胞的浸润物。CD4+T淋巴细胞是这些病变处存在的主要细胞类型。MS病变的标志是斑块,在MRI扫描中看到的与通常白质明显分界的脱髓鞘区域。MS斑块的组织学表现在疾病的不同阶段是变化的。在活跃病变中,血脑屏障受损,由此允许血清蛋白溢出到细胞外空间中。炎性细胞可以在血管周围袖套(perivascular cuff)中和整个白质中观察到。CD4+T-细胞,尤其是Th1,在斑块边缘处积聚在毛细血管后微静脉周围且也散布在白质中。在活跃病变中,也观察到粘附分子和淋巴细胞和单核细胞活化的标记物如IL2-R和CD26的上调。活跃病变中的脱髓鞘不伴随少突细胞的破坏。相反,在该疾病的慢性期中,病变特征在于少突细胞的损失且因此血液中髓鞘少突胶质细胞糖蛋白(MOG)抗体的存在。
糖尿病可以指1型或2型糖尿病。在一个实施方案中,如本文所述的形态形式或组合物以有效的剂量提供以治疗患有1型糖尿病的患者。在一个实施方案中,如本文所述的形态形式或组合物以有效的剂量提供以治疗患有2型糖尿病的患者。
1型糖尿病是自身免疫性疾病。当机体对抗感染的系统(免疫系统)攻击身体的部分时产生自身免疫性疾病。在1型糖尿病的情况下,胰腺然后产生很少或不产生胰岛素。
在一些实施方案中,本发明提供了通过向有需要的受试者施用有效量的如本文所述的形态形式或组合物来治疗或预防IC-MPGN的方法。
在一些实施方案中,本发明提供了通过向有需要的受试者施用有效量的如本文所述的形态形式或组合物来治疗或预防阵发性睡眠性血红蛋白尿症(PNH)的方法。
在一些实施方案中,本发明提供了通过向有需要的受试者施用有效量的如本文所述的形态形式或组合物来治疗或预防年龄相关性黄斑变性(AMD)的方法。
在一些实施方案中,本发明提供了通过向有需要的受试者施用有效量的如本文所述的形态形式或组合物来治疗或预防黄斑营养不良的方法。
在一些实施方案中,本发明提供了通过向有需要的受试者施用有效量的如本文所述的形态形式或组合物来治疗或预防克罗恩氏病的方法。
在一些实施方案中,本发明提供了通过向有需要的受试者施用有效量的如本文所述的形态形式或组合物来治疗或预防哮喘(TH2)或哮喘(非TH2)的方法。
在一些实施方案中,本发明提供了通过向有需要的受试者施用有效量的如本文所述的形态形式或组合物来治疗或预防糖尿病性视网膜病的方法。
在一些实施方案中,本发明提供了通过向有需要的受试者施用有效量的如本文所述的形态形式或组合物来治疗或预防选自急性肾损伤(AKI)、特发性膜性肾病、IgA肾病(IgAN)狼疮性肾炎(LN)和原发性局灶性节段性肾小球硬化的肾病病症的方法。
在一些实施方案中,本发明提供了通过向有需要的受试者施用有效量的如本文所述的形态形式或组合物来治疗或预防先兆子痫的方法。
联合疗法
在一个实施方案中,如本文所述的形态形式或组合物可以与有效量的至少一种另外的治疗剂组合地、或者与其交替地、或者在至少一种另外的治疗剂之后、同时或之前提供,例如,用于治疗本文中列出的病症。用于这种联合疗法的第二活性剂的非限制性实例在下面提供。
在一个实施方案中,如本文所述的形态形式或组合物可以与至少一种另外的补体系统抑制剂或具有不同的生物作用机制的第二活性化合物组合地或交替地提供。
在非限制性实施方案中,如本文所述的形态形式或组合物可以与蛋白酶抑制剂、可溶性补体调节剂、治疗性抗体(单克隆或多克隆)、补体成分抑制剂、受体激动剂或siRNA一起提供。
在其他实施方案中,本文所述的形态形式可以与针对肿瘤坏死因子(TNF)的抗体或受体融合蛋白组合或交替地施用,所述抗体包括但不限于英夫利昔单抗(Remicade)、阿达木单抗、赛妥珠单抗、戈利木单抗,所述受体融合蛋白如依那西普(Embrel)。
在另一个实施方案中,如本文所述的形态形式可以与抗CD20抗体组合或交替地施用,所述抗CD20抗体包括但不限于利妥昔单抗(Rituxan)、阿达木单抗(Humira)、奥法木单抗(Arzerra)、托西莫单抗(Bexxar)、奥滨尤妥珠单抗(Gazyva)或替伊莫单抗(Zevalin)。
在替代实施方案中,如本文所述的形态形式可以与抗IL6抗体组合或交替地施用,所述抗IL6抗体包括但不限于托珠单抗(tocilizumab)(Actemra)和司妥昔单抗(siltuximab)(Sylvant)。
在替代实施方案中,如本文所述的形态形式可以与IL17抑制剂组合或交替地施用,所述IL17抑制剂包括但不限于secukibumab(Cosentyx)。
在替代实施方案中,如本文所述的形态形式可以与p40(IL12/IL23)抑制剂组合或交替地施用,所述p40(IL12/IL23)抑制剂包括但不限于优特克单抗(ustekinumab)(Stelara)。
在替代实施方案中,如本文所述的形态形式可以与IL23抑制剂组合或交替地施用,所述IL23抑制剂包括但不限于risankizumab。
在替代实施方案中,如本文所述的形态形式可以与抗干扰素α抗体组合或交替地施用,所述抗干扰素α抗体例如但不限于西法木单抗(sifalimumab)。
在替代实施方案中,如本文所述的形态形式可以与激酶抑制剂组合或交替地施用,所述激酶抑制剂例如但不限于JAK1/JAK3抑制剂,例如但不限于托法替尼(Xelianz)。在替代实施方案中,如本文所述的形态形式可以与JAK1/JAK2抑制剂组合或交替地施用,所述JAK1/JAK2抑制剂例如但不限于baracitibib。
在一个替代实施方案中,如本文所述的形态形式可以与抗VEGF剂组合或交替施用,所述抗VEGF剂例如但不限于:阿柏西普( Regeneron Pharmaceuticals);兰尼单抗(
Regeneron Pharmaceuticals);兰尼单抗( Genentech和Novartis);哌加他尼(
Genentech和Novartis);哌加他尼( OSI Pharmaceuticals和Pfizer);贝伐单抗(Avastin;Genentech/Roche);拉帕替尼(Tykerb);舒尼替尼(Sutent);阿西替尼(Inlyta);帕唑帕尼;索拉非尼(Nexavar);帕纳替尼(Inclusig);瑞格非尼(Stivarga);卡博替尼(Abometyx;Cometriq);vendetanib(Caprelsa);雷莫芦单抗(Cyramza);乐伐替尼(Lenvima);ziv-阿柏西普(Zaltrap);西地尼布(Recentin);醋酸阿奈可他(anecortane acetate)、乳酸角鲨胺和皮质类固醇。
OSI Pharmaceuticals和Pfizer);贝伐单抗(Avastin;Genentech/Roche);拉帕替尼(Tykerb);舒尼替尼(Sutent);阿西替尼(Inlyta);帕唑帕尼;索拉非尼(Nexavar);帕纳替尼(Inclusig);瑞格非尼(Stivarga);卡博替尼(Abometyx;Cometriq);vendetanib(Caprelsa);雷莫芦单抗(Cyramza);乐伐替尼(Lenvima);ziv-阿柏西普(Zaltrap);西地尼布(Recentin);醋酸阿奈可他(anecortane acetate)、乳酸角鲨胺和皮质类固醇。
在另一个实施方案中,如本文所述的形态形式可以与免疫检查点抑制剂组合或交替地施用。检查点抑制剂的非限制性实例包括抗PD-1或抗PDL1抗体,例如,纳武单抗(Opdivo)、派姆单抗(Keytruda)、皮地立珠单抗(pidilizumab)、AMP-224(AstraZeneca和MedImmune)、PF-06801591(Pfizer)、MEDI0680(AstraZeneca)、PDR001(Novartis)、REGN2810(Regeneron)、SHR-12-1(江苏恒瑞医药公司和Incyte公司)、TSR-042(Tesaro)和PD-L1/VISTA抑制剂CA-170(Curis Inc.)、阿特朱单抗、度伐单抗和KN035,或抗CTLA4抗体,例如伊匹单抗、Tremelimumab、AGEN1884和AGEN2041(Agenus)。
可以与本文所述的活性化合物组合使用的活性剂的非限制性实例是:
蛋白酶抑制剂:血浆来源的C1-INH浓缩物,例如 (Sanquin)、
(Sanquin)、 (CSL Behring,Lev Pharma)和
(CSL Behring,Lev Pharma)和 重组人C1-抑制剂,例如
重组人C1-抑制剂,例如 利托那韦(
利托那韦( Abbvie,Inc.);
Abbvie,Inc.);
可溶性补体调节剂:可溶性补体受体1(TP10)(Avant Immunotherapeutics);sCR1-sLex/TP-20(Avant Immunotherapeutics);MLN-2222/CAB-2(MilleniumPharmaceuticals);Mirococept(Inflazyme Pharmaceuticals);
治疗性抗体:依库珠单抗/艾库组单抗(Soliris)(Alexion Pharmaceuticals);培克珠单抗(Alexion Pharmaceuticals);奥法木单抗(Genmab A/S);TNX-234(Tanox);TNX-558(Tanox);TA106(Taligen Therapeutics);Neutrazumab(G2 Therapies);抗备解素(Novelmed Therapeutics);HuMax-CD38(Genmab A/S);
补体组分抑制剂:坎普他汀(Compstatin)/POT-4(Potentia Pharmaceuticals);ARC1905(Archemix);4(1MEW)APL-1,APL-2(Appelis);CP40/AMY-101,PEG-Cp40(Amyndas);
补体C3或CAP C3转化酶靶向分子:TT30(CR2/CFH)(Alexion);TT32(CR2/CR1)(Alexion Pharmaceuticals);萘莫司他(FUT-175,Futhan)(Torri Pharmaceuticals);Bikaciomab,NM9308(Novelmed);CVF,HC-1496(InCode)ALXN1102/ALXN1103(TT30)(Alexion Pharmaceuticals);rFH(Optherion);H17 C3(C3b/iC3b)(EluSysTherapeutics);Mini-CFH(Amyndas)Mirococept(APT070);sCR1(CDX-1135)(Celldex);CRIg/CFH;抗CR3,抗MASP2,抗C1s和抗C1n分子:Cynryze(ViroPharma/Baxter);TNT003(True North);OMS721(Omeros);OMS906(Omeros);和Imprime PGG(Biothera);
可以与本文所述的形态形式或组合物组合或交替地使用的另外的非限制性实例包括以下。




在一个实施方案中,如本文所述的形态形式或组合物可以与抑制代谢所施用的蛋白酶抑制剂的酶的化合物一起提供。在一个实施方案中,形态形式或组合物可以与利托那韦一起提供。
在一个实施方案中,如本文所述的形态形式或组合物可以与补体C5抑制剂或C5转化酶抑制剂组合提供。在另一个实施方案中,如本文所述的形态形式或组合物可以与依库珠单抗(一种针对补体因子C5且由Alexion Pharmaceuticals以商品名Soliris制造和销售的单克隆抗体)组合提供。依库珠单抗已经由U.S.FDA批准用于治疗PNH和aHUS。
在一个实施方案中,如本文所述的形态形式或组合物可以与抑制补体因子D的化合物一起提供。在本发明的一个实施方案中,如本文所述的形态形式或组合物可以与以下文献中描述的化合物组合或交替地使用:标题为“Compounds useful in the complement,coagulate and kallikrein pathways and method for their preparation”的BiocrystPharmaceuticals的美国专利号6,653,340描述了作为因子D的强效抑制剂的稠合双环化合物;Biocyst Pharmaceuticals的美国专利申请US2019/0142802描述了开链因子D抑制剂;标题为“Indole compounds or analogues thereof useful for the treatment of age-related macular degeneration”的Novartis的PCT专利公布WO2012/093101描述了某些因子D抑制剂;Novartis的PCT专利公布WO2013/164802、WO2013/192345、WO2014/002051、WO2014/002052、WO2014/002053、WO2014/002054、WO2014/002057、WO2014/002058、WO2014/002059、WO2014/005150、WO2014/009833、WO2014/143638、WO2015/009616、WO2015/009977、WO2015/066241;标题为“Open chain prolyl urea-related modulators of androgenreceptor function”的Bristol-Myers Squibb的PCT专利公布WO2004/045518;标题为“Amide derivatives and nociceptin antagonists”的日本烟草公司的PCT专利公布WO1999/048492;标题为“CCK and/or gastrin receptor ligands”的Ferring B.V.和Yamanouchi Pharmaceutical Co.LTD.的PCT专利公布WO1993/020099;标题为“Methodsand compositions for the treatment of glomerulonephritis and otherinflammatory diseases”的Alexion Pharmaceuticals的PCT专利公布WO1995/029697;或标题为“Alkyne Compounds for Treatment of Complement Mediated Disorders”的Achillion Pharmaceuticals提交的PCT专利申请号PCT/US2015/017523和美国专利申请号14/631,090;标题为“Amide Compounds for Treatment of Complement MediatedDisorders”的PCT专利申请号PCT/US2015/017538和美国专利申请号14/631,233;标题为“Amino Compounds for Treatment of Complement Mediated Disorders”的PCT专利申请号PCT/US2015/017554和美国专利申请号14/631,312;标题为“Carbamate,Ester,andKetone Compounds for Treatment of Complement Mediated Disorders”的PCT专利申请号PCT/US2015/017583和美国专利申请号14/631,440;标题为“Aryl,Heteroaryl,andHeterocyclic Compounds for Treatment of Complement Mediated Disorders”的PCT专利申请号PCT/US2015/017593和美国专利申请号14/631,625;标题为“Ether Compoundsfor Treatment of Complement Mediated Disorders”的PCT专利申请号PCT/US2015/017597和美国专利申请号14/631,683;标题为“Phosphonate Compounds for Treatmentof Complement Mediated Disorders”的PCT专利申请号PCT/US2015/017600和美国专利申请号14/631,785;以及标题为“Compounds for Treatment of Complement MediatedDisorders”的PCT专利申请号PCT/US2015/017609和美国专利申请号14/631,828。
在一个实施方案中,本发明提供了通过向有需要的受试者施用有效量的如本文所述的形态形式或组合物与抗VEGF剂的组合来治疗或预防年龄相关性黄斑变性(AMD)的方法。抗VEGF剂的非限制性实例包括,但不限于阿柏西普( RegeneronPharmaceuticals);兰尼单抗(
RegeneronPharmaceuticals);兰尼单抗( Genentech和Novartis);哌加他尼(
Genentech和Novartis);哌加他尼( OSI Pharmaceuticals和Pfizer);贝伐单抗(Avastin;Genentech/Roche);拉帕替尼(Tykerb);舒尼替尼(Sutent);阿西替尼(Inlyta);帕唑帕尼;索拉非尼(Nexavar);帕纳替尼(Inclusig);瑞格拉非尼(Stivarga);卡博替尼(Abometyx;Cometriq);vendetanib(Caprelsa);雷莫芦单抗(Cyramza);乐伐替尼(Lenvima);ziv-阿柏西普(Zaltrap);西地尼布(Recentin);醋酸阿奈可他、乳酸角鲨胺和皮质类固醇,包括但不限于曲安奈德。
OSI Pharmaceuticals和Pfizer);贝伐单抗(Avastin;Genentech/Roche);拉帕替尼(Tykerb);舒尼替尼(Sutent);阿西替尼(Inlyta);帕唑帕尼;索拉非尼(Nexavar);帕纳替尼(Inclusig);瑞格拉非尼(Stivarga);卡博替尼(Abometyx;Cometriq);vendetanib(Caprelsa);雷莫芦单抗(Cyramza);乐伐替尼(Lenvima);ziv-阿柏西普(Zaltrap);西地尼布(Recentin);醋酸阿奈可他、乳酸角鲨胺和皮质类固醇,包括但不限于曲安奈德。
在一个实施方案中,本发明提供了通过向有需要的受试者施用有效量的如本文所述的形态形式或组合物与抗因子H或抗因子B剂的组合来治疗或预防年龄相关性黄斑变性(AMD)的方法,所述抗因子H或抗因子B剂选自抗FB siRNA(Alnylam);FCFD4514S(Genentech/Roche)用于CFB和CFD的SOMAmer(SomaLogic);TA106(AlexionPharmaceuticals);5C6和AMY-301(Amyndas)。
在一个实施方案中,本发明提供了通过向有需要的受试者施用有效量的如本文所述的形态形式或组合物与补体系统的另外抑制剂或具有不同的生物作用机制的另一活性化合物来治疗或预防阵发性睡眠性血红蛋白尿症(PNH)的方法。
在一个实施方案中,本发明提供了通过将有效量的如本文所述的形态形式或组合物与补体系统的另外的抑制剂或通过不同的作用机制发挥作用的活性剂组合或交替地施用至有需要的受试者来治疗或预防多发性硬化的方法。在另一个实施方案中,本发明提供了通过将有效量的如本文所述的形态形式或组合物与皮质类固醇组合或交替地施用至有需要的受试者来治疗或预防多发性硬化的方法。皮质类固醇的实例包括,但不限于强的松、地塞米松、甲泼尼龙和甲基强的松龙。在一个实施方案中,如本文所述的形态形式或组合物与例如,选自以下的至少一种抗多发性硬化药物组合:Aubagio(特立氟胺)、Avonex(干扰素β-1a)、Betaseron(干扰素β-1b)、Copaxone(醋酸格拉替雷)、Extavia(干扰素β-1b)、Gilenya(芬戈莫德)、Lemtrada(阿仑珠单抗)、Novantrone(米托蒽醌)、Plegridy(聚乙二醇干扰素β-1a)、Rebif(干扰素β-1a)、Tecfidera(富马酸二甲酯)、Tysabri(那他珠单抗)、Solu-Medrol(甲基强的松龙)、高剂量口服Deltasone(泼尼松)、H.P.Acthar Gel(ACTH)或它们的组合。
在另外的替代实施方案中,如本文所述的形态形式或组合物可以与依库珠单抗组合提供以治疗PNH、aHUS、STEC-HUS、ANCA-脉管炎、AMD、CAD、C3肾小球病(例如DDD或C3GN)、慢性溶血、视神经脊髓炎或移植排斥。在一个实施方案中,如本文所述的形态形式或组合物可以与坎普他汀或坎普他汀衍生物组合提供以治疗PNH、aHUS、STEC-HUS、ANCA-脉管炎、AMD、CAD、C3肾小球病(例如DDD或C3GN)、慢性溶血、视神经脊髓炎或移植排斥。在一个实施方案中,另外的药剂是补体成分抑制剂,例如但不限于坎普他汀/POT-4(PotentiaPharmaceuticals);ARC1905(Archemix);4(1MEW)APL-1,APL-2(Appelis);CP40/AMY-101,PEG-Cp40(Amyndas);PDGF抑制剂,例如但不限于索拉非尼甲苯磺酸盐;甲磺酸伊马替尼(STI571);舒尼替尼苹果酸;帕纳替尼(AP24534);阿西替尼;伊马替尼(STI571);尼达尼布(BIBF 1120);帕唑帕尼盐酸盐(GW786034HCl);多韦替尼(TKI-258,CHIR-258);Linifanib(ABT-869);Crenolanib(CP-868596);马赛替尼(AB1010);Tivozanib(AV-951);二磷酸莫沙替尼(AMG-706);Amuvatinib(MP-470);TSU-68(SU6668,Orantinib);CP-673451;Ki8751;替拉替尼;PP121;帕唑帕尼;KRN 633;多韦替尼(TKI-258)二乳酸;MK-2461;Tyrphostin(AG1296);多韦替尼(TKI258)乳酸盐;番泻苷B;舒尼替尼AZD2932和曲匹地尔;抗因子H或抗因子B剂,例如抗FB siRNA(Alnylam);FCFD4514S(Genentech/Roche)用于CFB和CFD的SOMAmer(SomaLogic);TA106(Alexion Pharmaceuticals);5C6和AMY-301(Amyndas);补体C3或CAP C3转化酶靶向分子,例如但不限于TT30(CR2/CFH)(Alexion);TT32(CR2/CR1)(Alexion Pharmaceuticals);萘莫司他(FUT-175,Futhan)(Torri Pharmaceuticals);Bikaciomab,NM9308(Novelmed);CVF,HC-1496(InCode)ALXN1102/ALXN1103(TT30)(Alexion Pharmaceuticals);rFH(Optherion);H17 C3(C3b/iC3b)(EluSysTherapeutics);Mini-CFH(Amyndas)Mirococept(APT070);sCR1(CDX-1135)(Celldex);CRIg/CFH,抗CR3,抗MASP2,抗C1s或抗C1n分子,例如但不限于Cynryze(ViroPharma/Baxter);TNT003(True North);OMS721(Omeros);OMS906(Omeros);和Imprime PGG(Biothera)。
在一个实施方案中,如本文所述的形态形式或组合物可以与非甾体抗炎药组合提供用于治疗狼疮。
在一个实施方案中,如本文所述的形态形式或组合物可以与皮质类固醇组合提供用于治疗狼疮。
皮质类固醇在一个实施方案中,如本文所述的形态形式或组合物可以与贝利木单抗(Benlysta)组合提供用于治疗狼疮。
在一个实施方案中,如本文所述的活性化合物或其盐或组合物可以与羟化氯喹(Plaquenil)组合提供用于治疗狼疮。
在一个实施方案中,如本文所述的形态形式或组合物可以与西法木单抗组合提供用于治疗狼疮。
在一个实施方案中,如本文所述的形态形式或组合物可以与OMS721(Omeros)组合提供用于治疗补体介导的病症。在一个实施方案中,如本文所述的形态形式或组合物可以与OMS906(Omeros)组合提供用于治疗补体介导的病症。在一个实施方案中,所述补体介导的病症是,例如,血栓性血小板减少性紫癜(TTP)或aHUS。
在一个实施方案中,如本文所述的形态形式或组合物可以与抗炎剂、免疫抑制剂或抗细胞因子剂组合提供用于治疗或预防响应于药物或生物治疗药物的施用(例如过继T细胞疗法(ACT)如CAR T细胞疗法,或单克隆抗体疗法)的细胞因子或炎性反应。在一个实施方案中,如本文所述的形态形式或组合物可以与皮质类固醇(例如强的松、地塞米松、甲泼尼龙和甲基强的松龙)和/或靶向例如IL-4、IL-10、IL-11、IL-13和TGFβ的抗细胞因子化合物组合提供。在一个实施方案中,如本文所述的形态形式或组合物可以与抗细胞因子抑制剂组合提供,所述抗细胞因子抑制剂包括但不限于阿达木单抗、英夫利昔单抗、依那西普、普特皮(protopic)、依法利珠单抗、阿法西普(alefacept)、阿那白滞素、司妥昔单抗、secukibumab、优特克单抗、戈利木单抗和托珠单抗或它们的组合。可以与如本文所述的形态形式或组合物组合使用的另外的抗炎剂包括但不限于非甾体抗炎药(NSAID);细胞因子抑制性抗炎药(CSAID);CDP-571/BAY-10-3356(人源化抗TNFα抗体;Celltech/Bayer);cA2/英夫利昔单抗(嵌合抗TNFα抗体;Centocor);75kd TNFR-IgG/依那西普(75kD TNF受体-IgG融合蛋白;Immunex);55kd TNF-IgG(55kD TNF受体-IgG融合蛋白;Hoffmann-LaRoche);IDEC-CE9.1/SB 210396(非消耗性灵长类化抗CD4抗体;IDEC/SmithKline);DAB 486-IL-2和/或DAB 389-IL-2(IL-2融合蛋白;Seragen);抗-Tac(人源化抗IL-2Rα;Protein DesignLabs/Roche);IL-4(抗炎细胞因子;DNAX/Schering);IL-10(SCH 52000;重组IL-10、抗炎细胞因子;DNAX/Schering);IL-4;IL-10和/或IL-4激动剂(例如,激动剂抗体);IL-1RA(IL-1受体拮抗剂;Synergen/Amgen);阿那白滞素( /Amgen);TNF-bp/s-TNF(可溶性TNF结合蛋白);R973401(IV型磷酸二酯酶抑制剂);MK-966(COX-2抑制剂);伊洛前列素,来氟米特(抗炎和细胞因子抑制剂);氨甲环酸(纤溶酶原激活的抑制剂);T-614(细胞因子抑制剂);前列腺素E1;替尼达普(非甾体抗炎药);萘普生(非甾体抗炎药);美洛昔康(非甾体抗炎药);布洛芬(非甾体抗炎药);吡罗昔康(非甾体抗炎药);双氯芬酸(非甾体抗炎药);吲哚美辛(非甾体抗炎药);柳氮磺胺吡啶;咪唑硫嘌呤;ICE抑制剂(白介素-1β转化酶的抑制剂);zap-70和/或lck抑制剂(酪氨酸激酶zap-70或lck的抑制剂);TNF-转化酶抑制剂;抗IL-12抗体;抗IL-18抗体;白介素-11;白介素-13;白介素-17抑制剂;金;青霉胺;氯喹;苯丁酸氮芥;羟基氯喹;环孢菌素;环磷酰胺;抗胸腺细胞球蛋白;抗CD4抗体;CD5-毒素;口服施用的肽和胶原蛋白;氯苯扎利二钠(lobenzarit disodium)、细胞因子调节剂(CRAB)HP228和HP466(Houghten Pharmaceuticals,Inc.);ICAM-1反义硫代磷酸酯寡脱氧核苷酸(ISIS2302;Isis Pharmaceuticals,Inc.);可溶性补体受体1(TP10;TCell Sciences,Inc.);强的松;奥古蛋白(orgotein);多硫酸糖胺聚糖;米诺环素;抗IL2R抗体;海洋和植物脂质(鱼和植物种子脂肪酸);金诺芬;苯基丁氮酮;甲氯芬那酸;氟芬那酸;静脉内免疫球蛋白;齐留通;阿扎立平;霉酚酸(RS-61443);他克莫司(FK-506);西罗莫司(雷帕霉素);氨普立糖(盐酸氨普立糖);克拉屈滨(2-氯脱氧腺苷)。
/Amgen);TNF-bp/s-TNF(可溶性TNF结合蛋白);R973401(IV型磷酸二酯酶抑制剂);MK-966(COX-2抑制剂);伊洛前列素,来氟米特(抗炎和细胞因子抑制剂);氨甲环酸(纤溶酶原激活的抑制剂);T-614(细胞因子抑制剂);前列腺素E1;替尼达普(非甾体抗炎药);萘普生(非甾体抗炎药);美洛昔康(非甾体抗炎药);布洛芬(非甾体抗炎药);吡罗昔康(非甾体抗炎药);双氯芬酸(非甾体抗炎药);吲哚美辛(非甾体抗炎药);柳氮磺胺吡啶;咪唑硫嘌呤;ICE抑制剂(白介素-1β转化酶的抑制剂);zap-70和/或lck抑制剂(酪氨酸激酶zap-70或lck的抑制剂);TNF-转化酶抑制剂;抗IL-12抗体;抗IL-18抗体;白介素-11;白介素-13;白介素-17抑制剂;金;青霉胺;氯喹;苯丁酸氮芥;羟基氯喹;环孢菌素;环磷酰胺;抗胸腺细胞球蛋白;抗CD4抗体;CD5-毒素;口服施用的肽和胶原蛋白;氯苯扎利二钠(lobenzarit disodium)、细胞因子调节剂(CRAB)HP228和HP466(Houghten Pharmaceuticals,Inc.);ICAM-1反义硫代磷酸酯寡脱氧核苷酸(ISIS2302;Isis Pharmaceuticals,Inc.);可溶性补体受体1(TP10;TCell Sciences,Inc.);强的松;奥古蛋白(orgotein);多硫酸糖胺聚糖;米诺环素;抗IL2R抗体;海洋和植物脂质(鱼和植物种子脂肪酸);金诺芬;苯基丁氮酮;甲氯芬那酸;氟芬那酸;静脉内免疫球蛋白;齐留通;阿扎立平;霉酚酸(RS-61443);他克莫司(FK-506);西罗莫司(雷帕霉素);氨普立糖(盐酸氨普立糖);克拉屈滨(2-氯脱氧腺苷)。
在具体实施方案中,如本文所述的形态形式或组合物可以与皮质类固醇组合提供用于治疗或预防响应于药物或生物治疗药物的施用的细胞因子或炎性反应。在另一个实施方案中,如本文所述的形态形式或组合物可以与依那西普(etarnercept)组合提供用于治疗或预防响应于药物或生物治疗药物的施用的细胞因子或炎性反应。在另一个实施方案中,如本文所述的形态形式或组合物可以与托珠单抗组合提供用于治疗或预防响应于药物或生物治疗药物的施用的细胞因子或炎性反应。在另一个实施方案中,如本文所述的形态形式或组合物可以与依那西普和托珠单抗组合提供用于治疗或预防响应于药物或生物治疗药物的施用的细胞因子或炎性反应。在另一个实施方案中,如本文所述的形态形式或组合物可以与英夫利昔单抗组合提供用于治疗或预防响应于药物或生物治疗药物的施用的细胞因子或炎性反应。在另一个实施方案中,如本文所述的形态形式或组合物可以与戈利木单抗组合提供用于治疗或预防响应于药物或生物治疗药物的施用的细胞因子或炎性反应。
C5抑制剂
本文提供了治疗受试者的因子D介导的病症的方法,所述方法包括将有效量C5抑制剂与有效量的化合物3的形态形式组合或交替地施用至受试者。在某些实施方案中,因子D介导的病症是PNH。
C5抑制剂在本领域中是已知的。在一个实施方案中,C5抑制剂是靶向C5的单克隆抗体。在一个实施方案中,C5抑制剂是依库丽单抗(SolirisTM Alexion Pharmaceuticals,New Haven,CT,参见例如美国专利号9,352,035)。在一个实施方案中,C5抑制剂是拉鲁珠单抗。在一个实施方案中,C5抑制剂是小分子药物。在另一个实施方案中,C5抑制剂是抗体。在另一个实施方案中,C5抑制剂是靶向C5的多克隆抗体。在另一个实施方案中,C5抑制剂是适体。
在一些实施方案中,C5抑制剂可以是但不限于:重组人微型抗体,例如 (单克隆抗体,Adienne Pharma and Biotech,Bergamo,Italy;参见美国专利号7,999,081);coversin(小动物蛋白,Volution Immuno-pharmaceuticals,Geneva,Switzerland;参见例如Penabad等人Lupus,2012,23(12):1324-6);LFG316(单克隆抗体,Novartis,Basel,Switzerland和Morphosys,Planegg,Germany;参见美国专利号8,241,628和8,883,158);ARC-1905(PEG化RNA适体,Ophthotech,Princeton,NJ和New York,NY;参见Keefe等人,Nature Reviews Drug Discovery,9,537-550);RA101348和RA101495(大环肽,Ra Pharmaceuticals,Cambridge,MA);SOBI002(亲和体,Swedish Orphan Biovitrum,Stockholm,Sweden);ALN-CC5(Si-RNA,Alnylam Pharmaceuticals,Cambridge,MA);ARC1005(适体,Novo Nordisk,Bagsvaerd,Denmark);SOMAmer(适体,SomaLogic,Boulder,Co);SSL7(细菌蛋白毒素,参见例如Laursen等人Proc.Natl.Acad.Sci.U.S.A.,107(8):3681-6);MEDI7814(单克隆抗体,MedImmune,Gaithersburg,MD);玫红酸三羧酸;玫红酸三羧酸衍生物(Aurin Biotech,Vancouver,BC,参见美国专利申请公布2013/003592);RG6107(抗C5循环抗体,Roche Pharmaceuticals,Basel,Switzerland);拉鲁珠单抗(ALXN1210)和ALXN5500(单克隆抗体,Alexion Pharmaceuticals,New Haven,CT);TT30(融合蛋白,Alexion Pharmaceuticals,New Haven,CT);REGN3918(单克隆抗体,Regeneron,Tarrytown,NY);ABP959(依库丽单抗生物类似物,Amgen,Thousand Oaks,CA);或它们的组合。
(单克隆抗体,Adienne Pharma and Biotech,Bergamo,Italy;参见美国专利号7,999,081);coversin(小动物蛋白,Volution Immuno-pharmaceuticals,Geneva,Switzerland;参见例如Penabad等人Lupus,2012,23(12):1324-6);LFG316(单克隆抗体,Novartis,Basel,Switzerland和Morphosys,Planegg,Germany;参见美国专利号8,241,628和8,883,158);ARC-1905(PEG化RNA适体,Ophthotech,Princeton,NJ和New York,NY;参见Keefe等人,Nature Reviews Drug Discovery,9,537-550);RA101348和RA101495(大环肽,Ra Pharmaceuticals,Cambridge,MA);SOBI002(亲和体,Swedish Orphan Biovitrum,Stockholm,Sweden);ALN-CC5(Si-RNA,Alnylam Pharmaceuticals,Cambridge,MA);ARC1005(适体,Novo Nordisk,Bagsvaerd,Denmark);SOMAmer(适体,SomaLogic,Boulder,Co);SSL7(细菌蛋白毒素,参见例如Laursen等人Proc.Natl.Acad.Sci.U.S.A.,107(8):3681-6);MEDI7814(单克隆抗体,MedImmune,Gaithersburg,MD);玫红酸三羧酸;玫红酸三羧酸衍生物(Aurin Biotech,Vancouver,BC,参见美国专利申请公布2013/003592);RG6107(抗C5循环抗体,Roche Pharmaceuticals,Basel,Switzerland);拉鲁珠单抗(ALXN1210)和ALXN5500(单克隆抗体,Alexion Pharmaceuticals,New Haven,CT);TT30(融合蛋白,Alexion Pharmaceuticals,New Haven,CT);REGN3918(单克隆抗体,Regeneron,Tarrytown,NY);ABP959(依库丽单抗生物类似物,Amgen,Thousand Oaks,CA);或它们的组合。
在一个实施方案中,C5抑制剂是重组人微型抗体,例如 是由Adienne Pharma and Biotech开发的完全人重组抗体C5。在美国专利号7,999,081中描述了
是由Adienne Pharma and Biotech开发的完全人重组抗体C5。在美国专利号7,999,081中描述了
在一个实施方案中,C5抑制剂是coversin。Coversin是一种重组蛋白,它来源于在非洲钝缘蜱(Ornithodoros moubata)的唾液中发现的蛋白,当前由Akari Therapeutics以重组蛋白开发。Coversin描述于Penabad等人Lupus 2012,23(12):1324-6。
在一个实施方案中,C5抑制剂是Tesidolumab/LFG316。Tesidolumab是由Novartis和Morphosys开发的单克隆抗体。Tesidolumab描述于美国专利号8,241,628和8,883,158。
在一个实施方案中,C5抑制剂是ARC-1905。ARC-1905是由Ophthotech开发的PEG化RNA适体。ARC-1905描述于Keefe等人Nature Reviews Drug Discovery,9:537-550。
在一个实施方案中,C5抑制剂是RA101348。RA101348是由Ra Pharmaceuticals开发的大环肽。
在一个实施方案中,C5抑制剂是RA101495。RA101495是由Ra Pharmaceuticals开发的大环肽。
在一个实施方案中,C5抑制剂是SOBI002。SOBI002是由瑞典的Orphan Biovitrum开发的亲和体。
在一个实施方案中,C5抑制剂是ARC1005。ARC1005是由Novo Nordisk开发的适体。
在一个实施方案中,C5抑制剂是用于C5的SOMAmer。SOMAmer是由SomaLogic开发的适体。
在一个实施方案中,C5抑制剂是SSL7。SSL7是描述于Laursen等人Proc.Natl.Acad.Sci.U.S.A.,107(8):3681-6中的细菌蛋白毒素。
在一个实施方案中,C5抑制剂是MEDI7814。MEDI7814是由MedImmune开发的单克隆抗体。
在一个实施方案中,C5抑制剂是玫红酸三羧酸。在另一个实施方案中,C5抑制剂是玫红酸三羧酸衍生物。这些玫红酸衍生物是由Aurin Biotech开发的,且进一步描述于美国专利申请公布号2013/003592)中。
在一个实施方案中,C5抑制剂是RG6107/SKY59。RG6107/SKY59是由RochePharmaceuticals开发的抗C5循环抗体。
在一个实施方案中,C5抑制剂是拉鲁珠单抗(ALXN1210)。在另一个实施方案中,C5抑制剂是ALXN5500。ALXN1210和ALXN5500是由Alexion Pharmaceuticals开发的单克隆抗体。
在一个实施方案中,C5抑制剂是TT30。TT30是由Alexion Pharmaceuticals开发的融合蛋白。
在一个实施方案中,C5抑制剂是ABP959。ABP959是由Amgen开发的依库丽单抗生物类似物单克隆抗体。
在一个实施方案中,C5抑制剂是抗C5 siRNA。抗C5 siRNA由AlnylamPharmaceuticals开发。
在一个实施方案中,C5抑制剂是 是由Adienne Pharma开发的抗体。
是由Adienne Pharma开发的抗体。
在一个实施方案中,C5抑制剂是avacincaptad pegol/ avacincaptadpegol是由Opthotech开发的适体。
avacincaptadpegol是由Opthotech开发的适体。
在一个实施方案中,C5抑制剂是SOBI005。SOBI005是由瑞典的Orphan Biovitrum开发的蛋白。
在一个实施方案中,C5抑制剂是ISU305。ISU305是由ISU ABXIS开发的单克隆抗体。
在一个实施方案中,C5抑制剂是REGN3918。REGN3918是由Regeneron开发的单克隆抗体。
在另一个实施方案中,如本文所述的形态形式或组合物可以与ABP959(一种针对补体因子C5且由Amgen制造和销售的单克隆抗体)组合提供。在另一个实施方案中,如本文所述的形态形式或组合物可以与BOWo8o(一种针对补体因子C5且由EpirusBiopharmaceuticals制造和销售的单克隆抗体)组合提供。在另一个实施方案中,如本文所述的形态形式或组合物可以与SB12(一种针对补体因子C5且由Samsung Bioepis制造和销售的单克隆抗体)组合提供。
C3抑制剂
本文提供了治疗受试者的因子D介导的病症的方法,所述方法包括将有效量C3抑制剂与有效量的化合物3的形态形式组合或交替地施用至受试者。在某些实施方案中,因子D介导的病症是PNH。
在一个实施方案中,C3抑制剂是小分子。在另一个实施方案中,C3抑制剂是靶向C3的多克隆抗体。在另一个实施方案中,C3抑制剂是靶向C3的单克隆抗体。在另一个实施方案中,C3抑制剂是适体。
C3抑制剂在本领域中是已知的。在一个实施方案中,本发明的形态形式或组合物与坎普他汀和/或坎普他汀类似物组合或交替地施用。坎普他汀和坎普他汀类似物是已知的,且发现它们是有用的C3抑制剂,参见美国专利号9,056,076;8,168,584;9,421,240;9,291,622;8,580,735;9371365;9,169,307;8,946,145;7,989,589;7,888,323;6,319,897;和美国专利申请公布号2016/0060297;2016/0015810;2016/0215022;2016/0215020;2016/0194359;2014/0371133;2014/0323407;2014/0050739;2013/0324482;和2015/0158915。在一个实施方案中,坎普他汀类似物具有氨基酸序列ICVVQDWGHHCRT(SEQ.ID.NO.1)。在另一个实施方案中,C3抑制剂是坎普他汀类似物。在一个实施方案中,坎普他汀类似物是4(1MeW)/APL-1,其具有序列Ac-ICV(1-mW)QDWGAHRCT(SEQ.ID.NO.2),其中Ac是乙酰基且1-mW是1-甲基色氨酸。在另一个实施方案中,坎普他汀类似物是Cp40/AMY-101,其具有氨基酸序列yICV(1mW)QDW-Sar-AHRC-mI(SEQ.ID.NO.3),其中y是D-酪氨酸,1mW是1-甲基色氨酸,Sar是肌氨酸,且mI是N-甲基异亮氨酸。在另一个实施方案中,坎普他汀类似物是PEG-Cp40,其具有氨基酸序列PEG-yICV(1mW)QDW-Sar-AHRC-mI(SEQ.ID.NO.4),其中PEG是聚乙二醇(40kDa),y是D-酪氨酸,1mW是1-甲基色氨酸,Sar是肌氨酸,且mI是N-甲基异亮氨酸。在另一个实施方案中,坎普他汀类似物是4(1MeW)POT-4。4(1MeW)POT-4由Potentia开发。
在另一个实施方案中,坎普他汀类似物是AMY-201。AMY-201由AmyndasPharmaceuticals开发。
在一个实施方案中,C3抑制剂是H17。H17是由EluSys Therapeutics开发的人源化单克隆抗体。H17描述于Paixao-Cavalcante等人J.Immunol.2014,192(10):4844-4851。
在一个实施方案中,C3抑制剂是mirococept。Mirococept是由InflazymePharmaceuticals开发的基于CR1的蛋白。
在一个实施方案中,C3抑制剂是sCR1。sCR1是由Celldex开发的CR1蛋白的可溶形式。
在一个实施方案中,C3抑制剂是TT32。TT32是由Alexion Pharmaceuticals开发的基于CR-1的蛋白。
在一个实施方案中,C3抑制剂是HC-1496。HC-1496是由InCode开发的重组肽。
在一个实施方案中,C3抑制剂是CB 2782。CB 2782是由Catalyst Biosciences开发的来源于人膜型丝氨酸蛋白酶1(MTSP-1)的新型蛋白酶。
在一个实施方案中,C3抑制剂是APL-2。APL-2是由Apellis Pharmaceuticals开发的APL-1的PEG化型式。
补体成分的泛抑制剂
本文提供了用于治疗PNH的方法,所述方法包括与本发明的化合物组合或交替地施用补体成分的泛抑制剂。补体成分的泛抑制剂在本领域中是已知的。在一个实施方案中,抑制剂是FUT-175。
用于预防性或伴随的抗细菌疗法的组合
在本发明的一个方面中,提供了治疗有需要的宿主的方法,所述方法包括在施用用于本文所述的任何病症的形态形式或组合物之前施用有效量的预防性抗细菌疫苗。在本发明的另一方面中,提供了用于治疗有需要的宿主的方法,所述方法包括在施用用于本文所述的任何病症的形态形式或组合物之前施用有效量的预防性抗细菌药物,如药品。在本发明的一个方面中,提供了用于治疗有需要的宿主的方法,所述方法包括在施用用于本文所述的任何病症的形态形式或组合物之后施用有效量的抗细菌疫苗。在本发明的另一方面中,提供了用于治疗有需要的宿主的方法,所述方法包括在施用用于本文所述的任何病症的形态形式或组合物之后施用有效量的抗细菌药物,如药品。在一个实施方案中,所述病症是PNH、C3G或aHUS。在一个实施方案中,宿主接受了器官或其他组织或生物流体移植。在一个实施方案中,宿主也施用依库珠单抗。
在本发明的一个方面中,如本文所述的形态形式或组合物在针对细菌感染的疫苗的预防性施用后附随地施用至受试者。在一个实施方案中,所述补体介导的病症是PNH、C3G或aHUS。在一个实施方案中,受试者接受了器官或其他组织或生物流体移植。在一个实施方案中,受试者也施用依库珠单抗。
在本发明的一个方面中,如本文所述的形态形式或组合物与针对细菌感染的疫苗的预防性施用同时施用至受试者。在一个实施方案中,所述补体介导的病症是PNH、C3G或aHUS。在一个实施方案中,受试者接受了器官或其他组织或生物流体移植。在一个实施方案中,受试者也施用依库珠单抗。
在本发明的一个方面中,如本文所述的形态形式或组合物施用至受试者,且在形态形式的施用期中,将针对细菌感染的疫苗施用至受试者。在一个实施方案中,所述补体介导的病症是PNH、C3G或aHUS。在一个实施方案中,受试者接受了器官或其他组织或生物流体移植。在一个实施方案中,受试者也施用依库珠单抗。
在本发明的一个方面中,受试者在因子D抑制剂施用期间连同抗生素化合物组合施用如本文所述的形态形式或组合物。在一个实施方案中,所述补体介导的病症是PNH、C3G或aHUS。在一个实施方案中,受试者接受了器官或其他组织或生物流体移植。在一个实施方案中,受试者也施用依库珠单抗。
在本发明的一个方面中,如本文所述的形态形式或组合物在针对细菌感染的疫苗的预防性施用后且在因子D抑制剂施用期间连同抗生素化合物组合施用至受试者。在一个实施方案中,所述补体介导的病症是PNH或aHUS。在一个实施方案中,受试者接受了器官或其他组织或生物流体移植。在一个实施方案中,受试者也施用依库珠单抗。在一个实施方案中,在接受如本文所述的形态形式或组合物之前,受试者针对由脑膜炎奈瑟氏菌细菌引起的细菌感染接种疫苗。在一个实施方案中,受试者针对由流感嗜血杆菌细菌引起的细菌感染接种疫苗。在一个实施方案中,流感嗜血杆菌是流感嗜血杆菌血清型B(Hib)。在一个实施方案中,受试者针对由肺炎链球菌引起的细菌感染接种疫苗。在一个实施方案中,受试者针对由细菌脑膜炎奈瑟氏菌、流感嗜血杆菌或肺炎链球菌或者脑膜炎奈瑟氏菌、流感嗜血杆菌或肺炎链球菌中的一种或多种的组合引起的细菌感染接种疫苗。在一个实施方案中,受试者针对由细菌脑膜炎奈瑟氏菌、流感嗜血杆菌和肺炎链球菌引起的细菌感染接种疫苗。
在其他实施方案中,受试者针对由选自格兰氏阴性菌的细菌引起的细菌感染接种疫苗。在一个实施方案中,受试者针对由选自格兰氏阳性菌的细菌引起的细菌感染接种疫苗。在一个实施方案中,受试者针对由细菌脑膜炎奈瑟氏菌、流感嗜血杆菌或肺炎链球菌(Streptococcus pneunemoniae)或者脑膜炎奈瑟氏菌、流感嗜血杆菌或肺炎链球菌中的一种或多种与以下(但不限于此)的一种或多种的组合引起的细菌感染接种疫苗:炭疽杆菌、百日咳博尔德氏杆菌、破伤风梭菌、白喉棒状杆菌、贝氏柯克斯体、结核分枝杆菌、伤寒沙门氏菌、霍乱弧菌、嗜粒细胞无形体(Anaplasma phagocytophilum)、伊氏埃立克体(Ehrlichia ewingii)、查菲埃立克体、犬埃立克体、Neorickettsia sennetsu、麻风分枝杆菌、伯氏疏螺旋体、梅奥疏螺旋体(Borrelia afzelii)、埃氏疏螺旋体、伽氏疏螺旋体、牛分枝杆菌、金黄色葡萄球菌、酿脓链球菌、苍白密螺旋体、土拉热弗朗西丝菌、鼠疫耶尔森菌。
在一个实施方案中,受试者接种选自以下(但不限于此)的一种或多种疫苗:活伤寒菌苗(Vivotif Berna Vaccine,PaxVax)、伤寒Vi多糖疫苗(Typhim Vi,Sanofi)、23价肺炎球菌疫苗PCV13(Pneumovax 23,Merck)、7价肺炎球菌疫苗PCV7(Prevnar,Pfizer)、13价肺炎球菌疫苗PCV13(Prevnar 13,Pfizer)、流感嗜血杆菌b结合(prp-t)疫苗(ActHIB,Sanofi;Hibrix,GSK)、流感嗜血杆菌b结合(hboc)疫苗(HibTITER,Neuron Biotech)、流感嗜血杆菌b结合(prp-omp)疫苗(PedvaxHIB,Merck)、流感嗜血杆菌b结合(prp-t)疫苗/脑膜炎球菌结合疫苗(MenHibrix,GSK)、流感嗜血杆菌b结合(prp-t)疫苗/脑膜炎球菌结合疫苗/乙型肝炎疫苗(Comvax,Merck)、脑膜炎球菌多糖菌苗(Menomune A/C/Y/W-135,Sanofi)、脑膜炎球菌结合疫苗/白喉CRM197结合物(Menveo,GSK;Menactra,Sanofi)、B型脑膜炎球菌疫苗(Bexsero,GSK;Trumenba,Pfizer)、炭疽吸附疫苗(Biothrax,EmergentBiosolutions)、破伤风类毒素(Te Anatoxal Berna,Hendricks Regional Health)、卡介苗(Bacillus Calmette and Guérin)、膀胱内活疫苗(TheraCys,Sanofi;Tice BCG,Organon)、口服霍乱活疫苗(Vachora,Sanofi;Dukoral,SBL Vaccines;ShanChol,ShanthaBiotec;Micromedex,Truven Health)、破伤风类毒素和白喉吸附疫苗(Tdap;Decavac,Sanofi;Tenivac,Sanofi;td,Massachusetts Biological Labs)、白喉-破伤风-百日咳疫苗(DTap;Daptacel,Sanofi;Infanrix,GSK;Tripedia,Sanofi)、白喉-破伤风-百日咳/脊髓灰质炎疫苗(Kinrix,GSK;Quadracel,Sanofi)、白喉-破伤风-百日咳/乙型肝炎/脊髓灰质炎疫苗(Pediarix,GSK)、白喉-破伤风-百日咳/脊髓灰质炎b型流感嗜血杆菌疫苗(Pentacel,Sanofi)和/或白喉和百日咳疫苗(Tdap;Boostrix,GSK;Adacel,Sanofi)或它们的组合。
如上所述,接受本发明化合物以治疗病症的受试者在本文所述的因子D抑制剂之外预防性地施用抗生素化合物。在一个实施方案中,受试者在施用活性化合物期间施用抗生素化合物以减少细菌感染的发生。用于与本文所述的因子D抑制剂同时施用的抗生素化合物可以是可用于预防或减少细菌感染的效应的任何抗生素。抗生素在本领域中是众所周知的且包括但不限于阿米卡星(Amikin)、庆大霉素(Garamycin)、卡那霉素(Kantrex)、新霉素(Neo-Fradin)、奈替米星(Netromycin)、妥布霉素(Nebcin)、巴龙霉素(Humatin)、链霉素、壮观霉素(Trobicin)、格尔德霉素、除莠霉素、利福昔明(Xifaxan)、氯碳头孢(Lorabid)、厄他培南(Invanz)、多利培南(Doribax)、亚胺培南/西司他丁(Primaxin)、美罗培南(Merrem)、头孢羟氨苄(Duricef)、头孢唑啉(Ancef)、头孢噻啶/头孢噻吩(Keflin)、头孢力新(Keflex)、头孢克洛(Distaclor)、头孢羟唑(Mandol)、头孢西丁(Mefoxin)、头孢丙烯(Cefzil)、头孢呋辛(Ceftin,Zinnat)、头孢克肟(Cefspan)、头孢地尼(Omnicef,Cefdiel)、头孢妥仑(Spectracef,Meiact)、头孢哌酮(Cefobid)、头孢噻肟(Claforan)、头孢泊肟(Vantin)、头孢他啶(Fortaz)、头孢布烯(Cedax)、头孢唑肟(Cefizox)、头孢曲松(Rocephin)、头孢吡肟(Maxipime)、头孢洛林酯(Teflaro)、头孢吡普(Zeftera)、替考拉宁(Targocid)、万古霉素(Vancocin)、特拉万星(Vibativ)、达巴万星(Dalvance)、奥利万星(oritavancin)(Orbactiv)、克林霉素(Cleocin)、林可霉素(Lincocin)、达托霉素(Cubicin)、阿奇霉素(Zithromax,Sumamed,Xithrone)、克拉霉素(Biaxin)、地红霉素(Dynabac)、红霉素(Erythocin,Erythroped)、罗红霉素、醋竹桃霉素(Tao)、泰利霉素(Ketek)、螺旋霉素(Rovamycine)、氨曲南(Azactam)、呋喃唑酮(Furoxone)、呋喃妥英(Macrodantin,Macrobid)、利奈唑酮(Zyvox)、泼斯唑来、雷得唑来、特地佐利、阿莫西林(Novamox,Amoxil)、氨苄青霉素(Principen)、阿洛西林、羧苄青霉素(Geocillin)、氯唑西林(Tegopen)、双氯西林(Dynapen)、氟氯西林(Floxapen)、美洛西林(Mezlin)、甲氧西林(Staphcillin)、萘夫西林(Unipen)、苯唑西林(Prostaphlin)、青霉素G(Pentids)、青霉素V(Veetids)(Pen-Vee-K)、哌拉西林(Pipracil)、青霉素G(Pfizerpen)、替莫西林(Negaban)、替卡西林(Ticar)、阿莫西林/克拉维酸(Augmentin)、氨苄青霉素/舒巴坦(Unasyn)、哌拉西林/他唑巴坦(Zosyn)、替卡西林/克拉维酸(Timentin)、杆菌肽、粘菌素(Coly-Mycin-S)、多粘菌素B、环丙沙星(Cipro,Ciproxin,Ciprobay)、依诺沙星(Penetrex)、加替沙星(Tequin)、吉米沙星(Factive)、左氧氟沙星(Levaquin)、洛美沙星(Maxaquin)、莫西沙星(Avelox)、萘啶酸(NegGram)、诺氟沙星(Noroxin)、氧氟沙星(Floxin,Ocuflox)、曲伐沙星(Trovan)、格帕沙星(Raxar)、司帕沙星(Zagam)、替马沙星(Omniflox)、磺胺米隆(Sulfamylon)、磺乙酰胺(Sulamyd,Bleph-10)、磺胺嘧啶(Micro-Sulfon)、磺胺嘧啶银(Silvadene)、磺胺地索辛(Di-Methox,Albon)、磺胺甲噻二唑(Thiosulfil Forte)、磺胺甲噁唑(Gantanol)、对氨基苯磺酰胺、柳氮磺胺吡啶(Azulfidine)、磺胺异噁唑(Gantrisin)、甲氧苄氨嘧啶-磺胺甲噁唑(Co-trimoxazole)(TMP-SMX)(Bactrim,Septra)、磺胺柯衣定(sulfonamidochrysoidine)(Prontosil)、地美环素(Declomycin)、强力霉素(Vibramycin)、米诺环素(Minocin)、氧四环素(Terramycin)、四环素(Sumycin,AchromycinV,Steclin)、氯法齐明(Lamprene)、氨苯砜(Avlosulfon)、卷曲霉素(Capastat)、环丝氨酸(Seromycin)、乙胺丁醇(Myambutol)、乙硫异烟胺(Trecator)、异烟肼(I.N.H.)、吡嗪酰胺(Aldinamide)、利福平(Rifadin,Rimactane)、利福布汀(Mycobutin)、利福喷汀(Priftin)、链霉素、胂凡纳明(Salvarsan)、氯霉素(Chloromycetin)、磷霉素(Monurol,Monuril)、夫西地酸(Fucidin)、甲硝哒唑(Flagyl)、莫匹罗星(Bactroban)、平板霉素、奎奴普丁/达福普汀(Synercid)、甲砜霉素、替加环素(Tigacyl)、替硝唑(Tindamax Fasigyn)、甲氧苄氨嘧啶(Proloprim,Trimpex)和/或泰斯巴汀(teixobactin)或它们的组合。
在一个实施方案中,受试者施用选自以下的预防性抗生素:头孢菌素(例如,头孢曲松或头孢噻肟)、氨苄青霉素-舒巴坦、青霉素G、氨苄青霉素、氯霉素、氟喹诺酮、氨曲南、左氧氟沙星、莫西沙星、吉米沙星、万古霉素、克林霉素、头孢唑啉、阿奇霉素、美罗培南、头孢洛林、替加环素、克拉霉素、莫西沙星、甲氧苄氨嘧啶/磺胺甲噁唑、头孢呋辛酯、环丙沙星、利福平、米诺环素、螺旋霉素和头孢克肟,或者其两种或更多种的组合。
实施例
实施例1
方案1.化合物3的合成

向中间体10和中间体33在DMF中的溶液中加入N,N-二异丙基乙胺。在保持反应温度的同时加入TBTU。将反应加温至室温,并搅拌4-8小时。用水稀释反应,并通过离心收集形成的所得固体。将该固体用水洗涤两次,然后溶解在DCM中,并用Siliabondthiol树脂和活性炭处理以除去基于Pd的杂质。通过过滤除去树脂和活性炭,并用MeOH/DCM洗涤。将滤液蒸发至干,并将残余物通过硅胶色谱法使用甲醇/DCM纯化。合并纯级分并蒸发至干。
实施例2.化合物3的多晶型物实验
表1.化合物3的多晶型物研究,起始材料是无序的化合物3,除非另外指明。




*图谱成功建立索引
FE:快速蒸发
ET:升高温度快速蒸发
SE:缓慢蒸发
VR:体积缩减
SC:缓慢冷却
RT ppt:环境温度沉淀
CP:急剧沉淀
SVD:固体-蒸汽扩散
Roto-vap:旋转蒸发
表1中条件的程序在下面讨论。
快速蒸发(FE)
制备化合物3和目标溶剂/溶剂体系的溶液。过滤样品,并将样品置于环境条件下直至干燥。
缓慢蒸发(SE)
制备化合物3和目标溶剂/溶剂体系的溶液。过滤样品。将样品用打有5个孔的铝箔覆盖。将样品置于环境条件下直至干燥。
体积缩减(VR)
制备化合物3和目标溶剂/溶剂体系的溶液。过滤样品,并将样品置于环境条件下,但不允许完全干燥。监测样品中溶液内固体的生成。
升高温度(ET)体积缩减(VR)
在升高的温度下,制备化合物3和目标溶剂/溶剂体系的溶液。在一定温度下过滤样品。发生蒸发,但不允许样品完全干燥。监测样品中溶液内固体的生成。
浆液–环境温度(RT)或升高温度(ET)
制备化合物3和目标溶剂/溶剂体系的溶液。固体在溶液中持续存在。在环境温度(RT)或升高温度(ET)下将样品放在搅拌板上。监测样品,以确保固体在制浆过程中持续存在。经由真空过滤收集固体,并将固体在环境条件下干燥。
缓慢冷却(SC)
制备化合物3和目标溶剂/溶剂体系的溶液。将样品在升高温度下搅拌。监测溶液以确保固体在整个搅拌过程中持续存在。在于指定温度下平衡的小瓶中在一定温度下过滤样品。关闭热源,并允许样品缓慢冷却至环境温度。
环境温度(RT)沉淀(Ppt)
在环境温度下制备化合物3在目标溶剂中的饱和溶液。将该溶液过滤到环境温度反溶剂中,或将反溶剂加入化合物3溶液中。监测样品中是否有固体生成的迹象。
急剧沉淀(CP)
在升高温度下制备化合物3在目标溶剂中的饱和溶液。将该溶液过滤到保持在较低温度下的反溶剂中。监测样品中是否有固体生成的迹象。
相对湿度(RH)压力
将化合物3放入小瓶中,将小瓶密封在装有饱和盐溶液的室中。将样品在这些相对湿度下放置几天,然后检查其是否有形态差异的迹象。
固体蒸汽扩散(SVD)
将化合物3放入小瓶中,将小瓶密封在装有有机溶剂的室中。将样品在这些相对湿度下放置几天,然后检查其是否有形态变化的迹象。
加热(Δ)
制备化合物3的饱和溶液或尝试从结晶生成的固体。将少量样品放入设置为指定温度的烘箱中。通过显微镜监测样品的结晶迹象。
声波处理
利用带有3mm探针的Cole-Parmer超声波处理器(型号CP130)进行探针超声处理。设置为:幅度40,脉冲2秒。将溶液超声处理五次,然后密封并置于环境条件下。
仪器技术
以下方法尚未验证该化合物是否符合21CFR 211.165(e)。
X射线粉末衍射(XRPD)
大多数XRPD图谱是用PANalytical X'Pert PRO MPD衍射仪使用长细聚焦源和镍滤波器所产生的Cu Kα辐射入射束收集的。衍射仪使用对称Bragg-Brentano几何学进行配置。在分析之前,先对硅样本(NIST SRM 640e)进行分析,以核实观察到的Si 111峰位置与NIST认证的位置一致。将样品的样本制备为以硅零背景衬底为中心的薄圆形层。防散射狭缝(SS)用于使空气产生的背景降至最小。用入射光束和衍射光束的索勒狭缝使轴向发散的宽展降至最小。用位于离样品240mm的扫描位置灵敏检测器(X'Celerator)和数据收集器软件v.2.2b收集衍射图谱。
用PANalytical X'Pert PRO MPD衍射仪使用Optix长细聚焦源产生的Cu辐射入射光束收集一些XRPD图谱。使用椭圆梯度多层反射镜使Cu KαX射线聚焦穿过样本并到达检测器。在分析之前,先对硅样本(NIST SRM 640e)进行分析,以核实观察到的Si 111峰位置与NIST认证的位置一致。将样品的样本夹在3μm厚的膜之间,用透射几何学进行分析。使用光束阻挡器(beam-stop)、短防散射伸出部和防散射刀口使空气产生的背景降至最小。用入射光束和衍射光束的索勒狭缝使轴向发散的宽展降至最小。用位于离样本240mm的扫描位置灵敏检测器(X'Celerator)和数据收集器软件v.2.2b收集衍射图谱。
差示扫描量热法(DSC)
使用TA Instruments 2920或Q2000差示扫描量热仪进行DSC。使用NIST可追踪的铟金属进行温度校准。将样品放入铝DSC盘中,盖上盖,并准确纪录重量。将配置为样品盘的称过重的铝盘放在小室的参照侧上。热谱图上的方法代码是开始温度和结束温度以及加热速率的缩写;例如,-30-250-10表示“以10℃/min的速率从-30℃到250℃”。下表汇总了在每个图像中针对盘配置所使用的缩写:
缩写(注释中) 含义
T0C Tzero卷边盘
HS 盖子密封
HSLP 盖子密封并打有激光针孔
T0HSLP Tzero盘,盖子密封并打有激光针孔
C 盖子卷边
NC 盖子未卷边
热重(TG)分析
使用TA Instruments Q5000 IR热重分析仪进行TG分析。使用镍和AlumelTM进行温度校准。将各样品放在铝盘中。将样品气密密封,刺穿盖子,然后插入TG熔炉中。在氮气下加热熔炉。热谱图上的方法代码是开始温度和结束温度以及加热速率的缩写;例如,00-350-10表示“以10℃/min的速率从25℃到350℃”。
热台(HS)显微镜法
使用配备有SPOT InsightTM彩色数码相机的Leica DM LP显微镜上的Linkam热台(FTIR 600)进行热台显微镜法。使用USP熔点标准进行温度校准。将样品放在盖玻片上,将第二张盖玻片放在样品的上面。随着台的加热,使用带正交偏光镜和一级红色补偿器的20×物镜视觉观察各样品。使用SPOT软件(v.4.5.9)捕获图像。
质子(1H)溶液核磁共振(NMR)光谱法
用Agilent DD2-400光谱仪获得溶液的NMR光谱。通过将各样品溶解在含有DMSO-d6的TMS中来制备样品。
还进行了赋形形式研究(表2)。
表2.化合物3形态形式的赋形形式研究

表3化合物3形式的表征

表4化合物3形式的另外的表征



表5.室温下的蒸汽应力实验

表6.化合物3的相互转化实验


*图谱成功建立索引
表7.尝试生成另外的化合物3形式E
起始材料:无序化合物3

*图谱成功建立索引
表8.尝试生成另外的化合物3形式B和M
起始材料:无序化合物3

*图谱成功建立索引
与无序材料一起生成了化合物3的13种独特的XRPD图谱。索引了三种形式的XRPD图谱(形式B、J和M),表明已分离出单相。此外,四种形式(形式C、F、H和I)的结晶性较差。形式A和E的XRPD图谱指示二维结构。
化合物3形式E和形式J的热分析指示,这些形式是溶合的/水合的。温和加热(大约50℃)形式E和形式J/形式L产生了形式G。
对形式A和形式G以及形式B进行了竞争性浆液实验。在乙酸乙酯中形式B似乎是在环境温度和升高(大约50℃)温度下都更稳定的形式。对形式B和M进行的相同实验表明,形式M在升高温度(大约50℃)下最稳定。在环境温度下,经过近一周的浆化后,形式B和形式M都存在。这表明这两种形式之间可能存在对映体系,其转变温度接近环境温度。
化合物3似乎形成溶剂合物和水合物。在分析过程中生成的高度溶合/水合形式全部干燥成单一固体形式,形式G。在升高温度(大约160℃)下加热形式G生成了形式M。形式B和形式M在环境温度下长时间浆化且未转化,表明可能存在转变温度接近环境温度的对映体系。形式B和形式M均成功放大(大约250mg)。
化合物3形成高度稳定的形式A。
化合物3的形态形式由图1至图24中提供的XRPD和DSC图谱表征。
实施例3:化合物3的应力测试
化合物3具有以下与其稳定性有关的特征:


实施例4:化合物2的应力测试
化合物2具有以下与其稳定性有关的特征:


实施例5:无定形化合物2的ICH稳定性
储存在带盖的铝容器中的硅胶双LDPE袋中的化合物2具有表9中所描述的以下稳定性特征。
表9.ICH稳定性研究期间化合物2的稳定性特征

实施例6.化合物3形式A的物理表征
通过XRPD、可变湿度XRPD(VH-XRPD)、可变温度XRPD(VH-XRPD)、偏振光显微镜法、热重/差热分析(TG/DTA)、差示扫描量热法(DSC)和动态蒸汽吸附(DVS)全面表征化合物3形式A的样品。XRPD、TG和DSC分析结果与实施例5的表3和表4中讨论的形式A的结果相当。下面讨论了其他方法。还通过1HNMR、HSQC NMR、HPLC和HPLC-MS对该材料进行了表征。
可变湿度X射线粉末衍射(VH-XRPD)
在配备有湿度箱的Philips X’Pert Pro多功能衍射仪上进行VT-XRPD分析。使用40kV/40mA发电机组,用以Bragg-Brentano几何学(步长0.008°2θ)运行的Cu K辐射(
 α1:α2比=0.5)在4与35.99°2θ之间扫描样品。表10详细列出了所使用的湿度程序。VH-XRPD显示在0%与90%RH之间无形式变化。
α1:α2比=0.5)在4与35.99°2θ之间扫描样品。表10详细列出了所使用的湿度程序。VH-XRPD显示在0%与90%RH之间无形式变化。
表10:化合物3形式A的VH-XRPD湿度程序

可变温度X射线粉末衍射(VT-XRPD)
在配备有温度箱的Philips X’Pert Pro多功能衍射仪上进行VT-XRPD分析。使用40kV/40mA发电机组,用以Bragg-Brentano几何学(步长0.008°2θ)运行的Cu K辐射(
 α1:α2比=0.5)在4与35.99°2θ之间扫描样品。表11详细列出了用于形式A的温度程序。使用VT-XRPD,显示化合物3形式A在125℃-170℃之间熔融。样品冷却后未观察到重结晶事件。
α1:α2比=0.5)在4与35.99°2θ之间扫描样品。表11详细列出了用于形式A的温度程序。使用VT-XRPD,显示化合物3形式A在125℃-170℃之间熔融。样品冷却后未观察到重结晶事件。
表11:化合物3形式A的VT-XRPD湿度程序
| 温度(℃) | 加热速率(℃/分钟) | 扫描时间 |
| 30 | 0 | 初始扫描 |
| 40 | 10 | 初始和保持5min后 |
| 50 | 10 | 初始和保持5min后 |
| 125 | 10 | 初始和保持5min后 |
| 170 | 10 | 初始和保持5min后 |
| 30 | -10 | 初始和保持5min后 |
| 100 | 10 | 初始和保持5min后 |
| 145 | 10 | 初始和保持5min后 |
| 30 | -10 | 初始和保持5min后 |
偏振光显微镜法(PLM)
使用配备有交叉偏振镜和Motic相机的Olympus BX50显微镜确定结晶度(双折射)的存在。使用Motic Images Plus 2.0捕获图像。除非另外指出,否则所有图像均使用20x物镜记录。所有样品均使用硅油制备,并在完成PLM分析之前盖上盖玻片。PLM分析显示双折射附聚物(图28)。
热重/差热分析(TG/DTA)
将大约5mg的材料称重到敞开的铝盘中,并装入联立的热重/差热分析仪(TG/DTA)中并保持在室温下。然后将样品以10℃/min的速率从20℃加热到400℃,在此期间记录样品重量的变化以及任何差热事件(DTA)。氮气用作吹扫气体,流速为300cm3/min。
DTA迹线显示出多个较弱的吸热事件,起始温度为约139℃和154℃,这可能与材料的熔融有关。注意到在209℃至296℃范围内还可能发生宽泛的吸热事件,这可能与样品分解有关。TG迹线表明,在大约200℃下质量损失为0.8%,这与在250℃以上在降解之前的水分流失(0.3当量水)有关(图29)。
动态蒸汽吸附(DVS)
将大约10-20mg的样品放入网状蒸汽吸附天平盘中,并装入Surface MeasurementSystems的DVS本征动态蒸汽吸附天平中。使样品以10%增量经受40%至90%相对湿度(RH)的升高曲线,在25℃下在每一步骤下维持样品直至达到稳定重量(dm/dt 0.004%,最小步长30分钟,最大步长500分钟)。在完成吸附循环后,使用相同程序将样品干燥至0%RH,然后再进行第二次吸附循环返回至40%RH。进行两个循环。绘制在吸附/解吸附循环期间的重量变化的图形,从而允许确定样品的吸湿性质。然后对保留的任何固体进行XRPD分析。
将大约10mg的样品放入网状蒸汽吸附天平盘中,并装入Surface MeasurementSystems的DVS-1动态蒸汽吸附天平中。使样品以10%增量经受40%至90%相对湿度(RH)的升高曲线,在25℃下在每一步骤下维持样品直至达到稳定重量(dm/dt 0.004%,最小步长30分钟,最大步长500分钟)。在完成吸附循环后,使用相同程序将样品干燥至0%RH,然后再进行第二次吸附循环返回至40%RH。进行两个循环。绘制在吸附/解吸附循环期间的重量变化的图形,从而允许确定样品的吸湿性质。然后对保留的任何固体进行XRPD分析。
DVS分析发现该材料具有吸湿性,在90%Rh下的总吸收率为6.9%(2.5当量的水)。在50%-60%Rh之间,观察到3.0%(1.0当量的水)的急剧吸收。从两个解吸附循环中,都注意到该材料损失了在较高湿度下吸附的所有水分(图30)。DVS动力学图(图31)显示在50%-60%RH之间质量急剧增加,其中一当量的水被材料吸收。这可能表明形成了水合形式。然而,VH-XRPD证实在吸收1.0当量水后未发生形式变化,且未观察到化合物3的水合形式。在第二次吸附循环中注意到了这种急剧的质量增加。
KF分析返回0.5%水的平均值(超过3次进样)。该值与在环境RH下从DVS分析生成的数据一致。
实施例7.化合物3形式A的稳定性研究
化合物3形式A的初始稳定性显示该材料在80℃下稳定,且经HPLC(相对面积)测定纯度未降低。在40℃/75%RH(未加盖)下储存的材料主要产生形式A。从该样品中未观察到纯度变化。重复进行40℃/75%RH稳定性研究,从加盖和未加盖的样品瓶中获得了弱结晶(WC)形式A材料。将形式A在25℃/60%RH下储存,从未加盖的小瓶中得到了弱结晶形式A,从加盖的小瓶中得到了形式A材料。冻干的化合物3在加盖或未加盖的小瓶中于40℃/75%RH或25℃/60%RH储存后未显示重结晶。
使用4种不同的aw 2-丙醇/水溶剂体系对化合物3形式A进行水合研究。将大约20mg化合物3形式A称入四个2mL玻璃小瓶中。将适当体积的溶剂体系加入固体中,并在环境条件下将样品搅动约48小时。48小时后,使用离心过滤分离固体,并通过XRPD分析。使化合物3形式A在选定2-丙醇/水体系中浆化的水合研究显示,随着水活度增加,结晶度降低。(图32)
表12:化合物3形式A的水合
| 水活度(a<sub>w</sub>) | XRPD结果 |
| 0.354 | 形式A |
| 0.526 | 形式A |
| 0.704 | 主要为无定形的 |
| 0.910 | 主要为无定形的 |
使用 Evolution SUPER均质机研磨化合物3形式A材料。将大约50mg化合物3形式A称入2个塑料2mL珠磨小瓶中,并将5x 2.4min金属珠粒放入各小瓶中。使用以下方法研磨样品1:RPM=5000;循环=2x 60秒;暂停=10秒(两次循环之间)。使用上述方法研磨样品2,但第二次循环为5x 60秒而不是2x 60秒。研磨后,从每个样品中提取一个子样品,并通过XRPD和PLM进行分析。重复上述程序,并通过XRPD和DSC分析对所得样品进行分析。
Evolution SUPER均质机研磨化合物3形式A材料。将大约50mg化合物3形式A称入2个塑料2mL珠磨小瓶中,并将5x 2.4min金属珠粒放入各小瓶中。使用以下方法研磨样品1:RPM=5000;循环=2x 60秒;暂停=10秒(两次循环之间)。使用上述方法研磨样品2,但第二次循环为5x 60秒而不是2x 60秒。研磨后,从每个样品中提取一个子样品,并通过XRPD和PLM进行分析。重复上述程序,并通过XRPD和DSC分析对所得样品进行分析。
下表13详细列出了对化合物3形式A进行的研磨实验的结果。通过XRPD分析确定研磨形式A主要返回的是无定形材料,但在PLM分析中观察到了双折射,表明该材料可能由小的结晶颗粒组成(图33和图34A至图34B是初始研磨过程的XRPD和PLM结果)。
表13.化合物3形式A研磨结果

使用specac压机压缩化合物3形式A材料。将大约125mg化合物3形式A称入20mL玻璃小瓶中。然后将材料转移到IR模具中,并压至2.5KN持续大约5秒。将所得材料(轻轻研磨,使返回的盘破裂,并)通过XRPD和PLM进行分析。然后将材料放回模具中,并压至5.0KN(持续大约5秒)。将所得材料轻轻研磨,使返回的盘破裂,并通过XRPD和PLM进行分析。对于2.5KN和5.0KN实验使用不同批次的固体以重复该程序详细信息。对压制后返回的材料完成XRPD分析和PLM分析。表14列出了化合物3形式的压缩研究结果。所有样品均返回形式A材料(经XRPD测定),且由小的双折射颗粒组成而无清晰形态(经PLM测定)(图35A至图35C)。
表14:化合物3形式A压缩结果

还测试了化合物3形式A在选定介质中的溶解度,且结果示于表15。经测量在选定介质(FaSSIF、FeSSIF、FaSSGF、pH 4柠檬酸盐和pH 6.8磷酸盐)中的溶解度为<0.01mg/mL。该研究的XRPD结果示于图36中。
表15.化合物3形式A溶解度结果
| 介质 | XRPD结果 | HPLC结果(mg/mL) |
| FaSSIF | 形式A | <0.01 |
| FeSSIF | 形式A | <0.01 |
| FaSSGF | 形式A | <0.01 |
| pH 4柠檬酸盐缓冲液 | 形式A | 未检测到 |
| pH 6.8磷酸盐缓冲液 | 形式A | <0.01 |
通过将柠檬酸钠(987mg)和柠檬酸(1.28g)溶解在100mL H2O中来制备pH 4柠檬酸盐缓冲液。将pH调节至4。通过将磷酸氢二钠二水合物(873mg)和磷酸二氢钠一水合物(708mg)溶解在100mL H2O中来制备pH 6.8磷酸盐缓冲液。将pH调节至6.8。用在H2O(0.25L)中的氢氧化钠(108mg)、氯化钠(1.55g)和磷酸二氢钠二水合物(1.12g)制备FaSSIF介质,并将pH调节至6.5。将FaSSIF/FeSSIF/FaSSGF(0.56g)溶解在缓冲液中并混合至乳白色。通过将氢氧化钠(1.01g)、氯化钠(2.96g)和冰醋酸(2.97g)溶解在H2O(0.25L)中来制备FeSSIF介质,并将pH调节至pH 5。将FaSSIF/FeSSIF/FaSSGF(2.81g)溶解在缓冲液中并充分混合。通过将FaSSIF/FeSSIF/FaSSGF(0.06g)溶解在溶于H2O(1L)的氯化钠(2.00g)溶液中并充分混合来制备FaSSGF培养基。
实施例8.化合物3形式A的溶剂溶解度
使用32种溶剂体系进行溶剂溶解度筛选。将大约360mg化合物3形式A称入20mL玻璃小瓶中,并使用18mL的1,4-二噁烷溶解。然后将0.5mL溶液分配到34个2mL玻璃小瓶中(每瓶大约10mg)。然后将化合物3形式A溶液冷冻(在-50℃下),并使用Lablyo小型冷冻干燥机通过冻干进行干燥。冻干后,取出子样品并通过XRPD进行分析。通过冻干成功地使材料成为无定形的。
将冻干产生的材料用于溶解度评估。通过溶剂添加技术来估计溶解度。如下完成溶解度研究:
·将每种溶剂体系以5体积等分试样加入适当的小瓶中,直至已加入100体积或直至API溶解;
·在每次加入之间,将样品加热至40℃,以检查在升高温度下的溶解;
·如果加入了100体积的溶剂而没有溶解,则溶解度被计算为低于该点。
使用离心过滤分离未观察到溶解的样品,并通过XRPD分析固体。观察到溶解的样品在环境条件下蒸发以返回固体。在适用情况下,
通过XRPD分析固体。表16详细列出了溶解度评估后的蒸发实验的观察结果和XRPD结果。溶剂蒸发后回收的固体完全是形式A。
冻干的化合物3形式A在研究的大多数溶剂体系中显示出高溶解度(≥200mg/mL):1,4-二噁烷、1-丁醇、2-甲基四氢呋喃、95%2-丙醇/5%水((%v/v)计算aw=0.5);丙酮、乙腈、95%乙腈/5%水((%v/v)计算aw=0.4);氯仿、二氯甲烷、二甲亚砜、乙酸乙酯(干)、乙醇、乙酸异丙酯、甲醇、甲基乙基酮、甲基异丁基酮、N,N'-二甲基甲酰胺、N,N'-二甲基乙酰胺、四氢呋喃和95%甲醇/5%水((%v/v)计算aw=0.2)。在某些溶剂(2-丙醇、95%2-丙醇/5%庚烷(%v/v)、70%2-丙醇/30%庚烷(%v/v)和甲苯)中,在升高温度下观察到溶解(≥200mg/mL)。
含有庚烷(50%2-丙醇/50%庚烷(%v/v)和50%乙醇/50%庚烷(%v/v))的溶剂体系混合物显示出略低的溶解度(>100mg/mL)。常见的反溶剂如正庚烷和叔丁基甲基醚、乙醚、水以及溶剂/水混合物(50%2-丙醇/50%水((%v/v)aw=0.7)和40%乙醇/60%水((%v/v)计算aw=0.7))显示出<10mg/mL的低溶解度。
表16:溶解度评估后蒸发的观察结果和XRPD结果


实施例9.化合物3形式A的多晶型物研究
将大约960mg化合物3形式A称入20mL玻璃小瓶中,并使用12mL的1,4-二噁烷溶解。然后将0.5mL溶液分配到24个1.5mL HPLC玻璃小瓶中(每瓶大约40mg)。然后将化合物3形式A溶液冷冻(在-50℃下),并使用Lablyo小型冷冻干燥机通过冻干进行干燥。冻干后,取出子样品并通过XRPD进行分析。通过冻干成功地使材料成为无定形的。
使用24种不同的溶剂体系和四种不同的结晶技术完成了多晶型物研究:温度循环、急剧冷却反溶剂添加和溶剂蒸发。下面将对每一种技术进行描述。
温度循环
将适当体积的溶剂(溶剂体系详见表17)加入冻干的固体中。将样品在环境温度与40℃(4小时循环)之间进行温度循环,持续约72小时。在72小时的温度循环后进行观察。将另外的溶剂加入样品中以产生可流动浆液,并且将浆液在40℃下搅动另外的18小时。XRPD分析了固体的子样品(在适用情况下)。使用热风枪加热样品帮助溶解,以确保产生饱和溶液。注射器过滤饱和溶液以除去任何潜在的晶种材料(作为预防措施),并分为三种不同的结晶条件:冷却、蒸发和反溶剂添加(每种条件下的饱和溶液的体积可见表17)。
表17.温度循环实验中用于各条件的溶剂体积


在温度循环后,从大多数溶剂体系中返回了形式A。发现了两种新型式,从40%乙醇/60%水(%v/v)中分离出的型式N和从甲醇中分离出的型式O。还分离出了两种先前见过的形式,从丙酮中分离出的形式B和从乙腈中分离出的形式J。图37示出了分离的型式N和O以及分离的形式B和形式J的XRPD衍射图。形式B和形式J的表征对应于实施例5的表3和表4中所述的表征。具体地,对于形式B,DT迹线显示了一个吸热事件,其起始温度为约180℃,与材料的熔融有关(SSCI报告起始温度=约176℃)。TG迹线显示该材料在大约270℃以上降解。对于形式J,当加热至约120℃时,TG迹线显示质量损失为3.5重量%(0.5当量乙腈),且材料在250℃以上降解。
型式N用TG/DTA表征。DT迹线(图38)显示了复杂的微弱热事件,其起始温度为约135℃,可能与材料的熔融有关。从TG迹线看,在降解之前(大约270℃)未观察到质量损失,这表明该材料是无水的。型式O的特征是在DT迹线中可以看到的一个吸热事件,其起始温度为180℃(峰值190℃),与材料的熔融有关(图39)。从TG迹线看,在降解之前(大约270℃)未观察到质量损失,这表明该材料是无水的。
表18详细列出了72小时温度循环后的观察结果和XRPD结果。
表18.72小时温度循环产生的观察结果和XRPD结果


急剧冷却
将温度循环实验产生的化合物3的饱和溶液放入冰箱中,使其急剧冷却至4℃。约4天后,进行观察,并使用离心过滤分离产生的固体,且通过XRPD进行分析。在无固体回收的情况下,将溶液放入冰箱(-20℃)中14天。通过离心过滤分离产生的任何固体,并通过XRPD进行分析(表19)。
表19详细列出了急剧冷却实验返回的观察结果和XRPD结果。在沉淀出结晶材料的地方,通过XRPD分析观察到形式A材料。从急剧冷却实验中未观察到新的形式。将该材料在-20℃下储存14天后也进行XRPD分析,但14天后未观察到新的形式。
表19:在4℃下储存约96小时后的观察结果和XRPD结果


反溶剂添加
对由温度循环实验产生的化合物3的饱和溶液完成反溶剂添加。在环境条件下储存时,从所选溶剂体系中观察到固体沉淀(在完成反溶剂添加之前)。通过XRPD分析沉淀的固体,并通过缓慢加热以及加入最少量的溶剂将其重新溶解。将适当的反溶剂以50μL等分试样加入各饱和溶液中,直至观察到沉淀,或者总共加入了1mL反溶剂。使用离心过滤分离有固体沉淀的样品,并通过XRPD进行分析。将保留有澄清溶液的样品放入冰箱(4℃)中,以诱导沉淀,持续约48小时。使用离心过滤分离有固体沉淀的样品,并通过XRPD进行分析(表20A和表20B)。
表20A和表20B列出了通过将反溶剂加入化合物3的饱和溶液中返回的观察结果和XRPD结果。从返回结晶材料的大多数样品中返回了形式A材料。从70%2-丙醇/30%庚烷(%v/v)和40%乙醇/60%水(%v/v)返回了型式L和M的混合物。从反溶剂添加实验中未观察到新的形式。
表20A.反溶剂观察结果和XRPD结果



*=8天后在环境条件下沉淀的固体
表20B.所选溶剂的其他反溶剂观察结果和XRPD结果


蒸发
由温度循环实验产生的化合物3的饱和溶液在环境条件下蒸发。大约4天后,进行观察,并通过XRPD分析回收的固体(表21)。
从环境条件下的蒸发实验返回的固体的观察结果和XRPD结果可见于下表21。发现从蒸发实验返回的结晶材料仅是形式A,但回收的大多数固体都是无定形的。
表21:蒸发观察结果和XRPD结果


将由热循环、冷却和蒸发实验产生的样品在50℃下干燥约24小时,以评估任何去溶合/脱水作用。根据温度循环实验,干燥后3个样品发生转化:
·乙腈样品从形式J转化为无定形的;
·40%乙醇/60%水(%v/v)从形式N转化为形式P;以及
·甲醇样品从形式O转化为无定形的。
型式P的XRPD图谱示于图40中。
将冷却和蒸发分离出的样品在50℃下干燥约24小时后,未见显著变化。
从这四种技术中,形式A是产生的主要形式。在分别在丙酮和乙腈溶剂体系中进行温度循环实验后,观察到B和形式J。在40%乙醇:60%水(%v/v)和甲醇溶剂体系中观察到三种新的型式(型式N、O和P)。型式N、O和P的固态性质很差,且从任何其他结晶实验中都未观察到。通过在环境条件下从70%2-丙醇/30%庚烷(%v/v)和40%乙醇:60%水(%v/v)饱和溶液中沉淀观察到型式L和M的混合物。从叔丁基甲基醚的蒸发中返回了弱的结晶型式E。表22A是多晶型物研究中使用的四种技术的结果的汇总,且表22B是观察到的形式的性质的比较。
表22A.结晶技术的结果的汇总


WC=弱结晶;*在环境条件下沉淀的固体
表22B.分离的形式和型式的比较

实施例10.形式B和形式J的多晶型物研究
形式B
完成了对形式B的多晶型物研究。形式B的产生是通过对在125μL丙酮中的无定形化合物3(形式B)进行温度循环并在环境条件下干燥约24小时来完成的。
通过XRPD、TG/DTA、DSC、DVS、VT-XRPD和PLM来表征形式B。XRPD、TG和DSC分析结果与实施例8的表23和表24中讨论的形式B的结果相当。还通过1HNMR、HPLC和FT-IR对该材料进行了表征。
通过DVS分析发现形式B材料具有轻微的吸湿性,并且在达到90%RH时吸收率为0.26重量%(0.1当量水)。根据等温线图(图41A),等温线为形式1,表明可逆地吸附在颗粒表面上。根据动力学图(图41B),未观察到明显的质量增加或减少,表明没有发生重结晶事件。VT-XRPD显示该材料在150℃与250℃之间熔融。冷却样品后未观察到重结晶(图42)并且PLM分析显示形式B与双折射棒状形态一致(图43)。
当该材料在40℃/75%RH、80℃或环境条件下储存时,根据XRPD或HPLC纯度未观察到变化。形式B在所选pH 4、pH 6.8、FaSSIF、FeSSIF和FaSSGF介质中的溶解性较差。
形式J
完成了对形式J的多晶型物研究。形式J的产生是通过对在乙腈中的无定形化合物3(形式B)进行温度循环并在环境条件下干燥约24小时来完成的。表22中给出了每一步骤的确切程序的观察结果和XRPD结果。
表22.形式J的形成过程


通过XRPD、TG、DCS、DVS、1HNMR、HPLC、PLM和FT-IR表征形式J。XRPD、TG和DSC结果与实施例8的表3和表4中的结果相当。1HNMR、FT-IR和HPLC是形式J特征性的。
DVS分析显示该材料为吸湿形式,在第一吸附循环中吸收率为6.9重量%(2.5当量水),在第二吸附循环中吸收量率5.2重量%(1.8当量水)。在分析的开始和结束之间存在2.2重量%的差异(损失0.3重量%乙腈),这可能是由于样品内有乙腈所致。观察到形式1等温线,指示可逆地吸附在颗粒表面上。根据动力学图,初始质量损失为5重量%(0.8当量乙腈)表明样品已干燥/除去了过量的乙腈。DVS后的XRPD分析返回了型式D(图44A至图44C)。
尽管XRPD图谱指示了形式J,但VT-XRPD结果指示形式J是乙腈半溶剂合物,当加热至170℃时其通过形式G去溶合为形式M(图44D)。
PLM分析显示形式J由双折射棒状形态组成(图44E)。
图45是实施例9和实施例10中描述的多晶型物研究的图。
实施例11.竞争性浆液
分别称量约10mg的形式A、形式B和形式J,并将这些固体合并到一个小瓶中。然后加入适当体积的所选溶剂以形成浆液。然后将浆液在环境温度或升高温度下搅动约48小时。搅动后,使用离心过滤分离固体,并收集XRPD分析结果。下表23详细列出了所选的溶剂体系和使用的体积。
将溶剂再次引入(体积按照表24)样品中,并且将样品再搅动72小时,因为在搅动48小时后没有从实验中返回单一的多晶型物形式。使用离心过滤分离固体,并收集XRPD分析结果。
使用上述程序,在60℃下于2-丙醇和2-丙醇/庚烷(70/30%v/v)中重复样品。将样品搅动24小时,溶剂体积可见于下表24。没有明确的多态形式被确定为最稳定的形式。通过竞争性浆化分离单一形式似乎取决于溶剂,因为形式B和J分别是从丙酮和乙腈产生的。在这些溶剂体系中长时间浆化会导致这两种形式均转化为形式A。在72小时内形式B(最初在丙酮中观察到)转化为形式A表明形式A可能是最稳定的形式。形式A和形式B均可从不同溶剂体系的竞争性浆化实验中分离出来,但都不是在特定温度下唯一占主导的形式。
表24:用于初始竞争性浆液实验的输入溶剂、温度和体积


进行了另一个竞争性浆液实验。将约20mg的形式A和形式B称入单独的小瓶中。合并固体,并加入0.5mL过滤的饱和化合物3溶液(使用所选溶剂)以形成浆液。表25详细列出了为竞争性浆液实验选择的溶剂体系和温度。将浆液在环境温度和升高温度下搅动(在恒温振荡器中)约2周。使用离心过滤分离子样品,并收集1周和12天后返回的所有固体的XRPD分析结果。表25详细列出了其他竞争性浆液实验的溶剂和温度,且表26详细列出了XRPD结果。从形式A和形式B在丙酮、水和庚烷中的浆液中回收形式B。除了在无定形样品中有对应于形式A的少量峰之外,没有从任何所测溶剂体系中回收到单相的形式A。这表明形式B可能是最具热力学稳定性的形式。在特定温度下搅动2周后,从所有其他溶剂体系中返回了形式A和B的混合物。图46是竞争性浆液实验的图。
表25.用于其他初始竞争性浆液实验的输入溶剂、温度和体积

表26.其他竞争性浆液实验的结果


*=12天后的XRPD结果
表27A是形式A、形式B和形式J的性质的汇总,并且表27B是形式A、形式B和形式J的稳定性和溶解度的汇总。
表27A.实施例9和实施例10的多晶型物研究中形式A、形式B和形式J的性质


表27B.实施例9和实施例10的多晶型物研究中形式A、形式B和形式J的溶解度和稳定性


实施例12.碾磨研究
将10mg适当的化合物3材料(详见表28)称入单独的HPLC小瓶中。然后使用研杵研磨固体。通过XRPD分析所得材料(图47)。结果示于表28。从使用各形式的混合物的样品中,回收形式的混合物。在使用所有3种形式的情况下未返回形式A,但有可能形式A原本已经很小的粒径通过研磨进一步减小,因此没有被XRPD分析检测到。研磨后的形式B没有改变形式。
表28.研磨研究中的材料的组成
| 样品编号 | 形式 | XRPD结果 |
| 1 | A+B | 混合物A+B |
| 2 | A+J | 混合物A+J |
| 3 | A+B+J | 混合物B+J |
| 4 | B | B |
实施例13.压缩研究
使用specac IR压机将形式A+形式B和形式B的混合物压缩。将总计约100mg(50mgA+50mg B和100mg B)称入单独的2mL玻璃小瓶中。然后将固体放入IR模具中,并在25kN和50kN下压制约10-15秒。压至25kN后,将材料从模具中取出并稍微研磨。取子样品并收集XRPD分析。然后将材料放回模具中,并压至50kN。压至50kN后,将材料从模具中取出并稍微研磨,然后收集XRPD分析结果。压缩研究的结果示于表29中,而XRPD结果示于图48A和图48B中。在压至25kN后,两个样品均观察到从白色到灰白色的很小的颜色变化。形式B在压力下没有变化。当压缩形式A和形式B材料的混合物时,没有观察到单一形式,但是当在混合物上施加50kN时,观察到结晶度降低。
表29.压缩研究结果

实施例14.形式A+形式B混合物的DSC分析
将约5mg的形式A和形式B的混合物称入铝DSC盘中,并用穿孔的铝盖非气密密封。然后将样品盘装入Seiko DSC6200(配备有冷却器)中。一旦获得稳定的热流反应,就以10℃/min的扫描速率将样品和参照加热至250℃,并监控所得热流反应。氮气用作吹扫气体,流速为50cm3/min。在DSC的加热循环内观察到两个吸热事件。第一次吸热事件的起始温度为约140℃(形式A熔融转变),而第二次吸热事件的起始温度为约180℃(形式B熔融转变)。DSC示于图49中。
实施例15.中间体3的制造工艺描述
在以下方案2中,使boc-tans-4-氟脯氨酸(中间体1)在甲基咪唑(NMI)和甲烷磺酰氯(MsCl)存在下与2-氨基-6-溴吡啶(中间体2)反应。通过HPLC监测反应完成,并且将产物用二氯甲烷萃取并用二氯甲烷/正庚烷沉淀。将产物在真空盘式干燥机中在真空下干燥。
方案2.中间体3的合成

工艺描述如下:在N2气氛下,在25±5℃下,向反应器中装入DCM(干)(20.0体积)和boc-trans-4-氟脯氨酸(1.0w/w)。将反应物料冷却至0±5℃并搅拌15分钟。在0±5℃下向反应器中缓慢装入1-甲基咪唑(0.88w/w),并搅拌15分钟(观察到温度从0.8℃变化到3.8℃)。在0±5℃下将甲烷磺酰氯(0.59w/w)缓慢加入反应器中,并搅拌60分钟。在0±5℃下加入2-氨基-6-溴吡啶(0.74w/w),将物料温度保持在0℃至25±5℃之间,并搅拌直至反应完成。反应完成后,向反应器中装入纯化水(10.0体积),搅拌20分钟,并分离各层。将水层用DCM(20体积)萃取,然后用另外的(10体积)DCM萃取。合并有机层,并用10%HCl溶液(纯化水9.0w/w,HCl 1.0w/w)、碳酸氢钠溶液(纯化水9.5w/w,NaHCO3 0.5w/w)和盐水(纯化水9.0w/w,NaCl 1.0w/w)洗涤。将有机层在40℃以下在真空下浓缩,直至观察不到馏出物。向反应器中装入DCM(2体积)和正庚烷(6体积),并搅拌30分钟。将反应物料冷却至25±5℃并搅拌1小时。将该物料通过Nutsche过滤器过滤,并用正庚烷(2体积)洗涤滤饼。将滤饼在过滤器上真空干燥40-50分钟,同时保持Nutsche过滤器处于抽吸状态。将该材料在真空盘式干燥机中在25±5℃下干燥2小时,然后在30±5℃下干燥6小时或直至达到水含量(验收标准:水含量:NMT 1.0%)。将产物储存在受控温度下。
表29包含最近两个批次的试剂数量和产品产量的实例。
表29.两个批次的中间体3的规模和产量

实施例16.中间体4的制造工艺描述
在以下方案3中,将中间体3用含盐酸的二噁烷水解。通过HPLC监测反应中中间体3的消耗。将反应混合物用NaHCO3处理,并将产物用二氯甲烷萃取。通过蒸馏除去二氯甲烷,并用二氯甲烷和庚烷代替以沉淀产物。通过过滤分离后,将湿饼用庚烷洗涤,然后在真空盘式干燥机中真空干燥。
方案3.中间体4的合成

工艺描述如下:在氮气气氛下,在20±5℃下将含HCl(4M)的二噁烷(6体积)和中间体3(1.0w/w)装入反应器中,并在20±5℃下将物料搅拌4小时。反应完成后,向反应器中装入DCM(20.0体积),并搅拌15分钟。将NaHCO3溶液(纯化水22.5w/w,NaHCO3 2.5w/w)缓慢加入反应物料中并将pH调节至7-8并搅拌20分钟。分离各层,并用DCM(20.0体积x2次)萃取水层。将有机层在40℃以下浓缩,并在50℃以下与正庚烷(5体积)共蒸发。向反应器中装入DCM(2体积)和正庚烷(6体积),并搅拌40-60分钟。过滤所得物料,并用正庚烷(2体积)洗涤滤饼。将滤饼抽真空并干燥2小时。将该材料在真空盘式干燥机中于25±5℃干燥2小时,然后于45±5℃干燥12小时或直至达到水含量。
表30包含最近两个批次的试剂数量和产品产量的实例。
表30.两个批次的中间体4的规模和产量

实施例17.中间体10的制造工艺描述
在以下方案4中,以3-乙酰基-5-溴吲唑(中间体5)开始,分三个步骤制造中间体10。将中间体5用溴乙酸叔丁酯烷基化,然后与原位生成的2-甲基嘧啶-5-硼酸酯(中间体8)偶联,产生中间体9。然后将叔丁酯水解,得到2-(3-乙酰基-5-溴-1H-吲唑-1-基)乙酸(10),然后将其真空干燥。该路线由以下示出的方案中的步骤3-5组成。
方案4.中间体10的合成

中间体6的合成
在方案4的步骤1中,在碳酸钾存在下,将3-乙酰基5-溴吲唑(中间体5)用DMF中的溴乙酸叔丁酯烷基化。通过HPLC监测反应中起始材料的转化。将产物用纯化水沉淀并通过过滤分离。用纯化水、乙酸乙酯和庚烷洗涤后,将产物在真空盘式干燥机中真空干燥。
工艺描述如下:在氮气气氛下,在25±5℃下,向容器中装入1-(5-溴-1H-吲唑-3-基)乙-1-酮(1.0w/w)和DMF(7.0体积)并搅拌15分钟。然后向容器中装入碳酸钾(1.15w/w)并搅拌15分钟。在30±10℃下将溴乙酸叔丁酯缓慢加入(0.97w/w)反应物料中。将反应物料温度升高至50±5℃并在50±5℃下搅拌1小时。将反应物料冷却至25±5℃,并缓慢加入纯化水(21体积)。将获得的固体搅拌1小时。将反应物料过滤,并用纯化水(3体积)洗涤床。将湿滤饼与纯化水(10体积)一起搅拌15分钟。过滤所得滤饼,并用水(3体积)洗涤。如果样品不合格,则将湿饼加入乙酸乙酯(10体积)和正庚烷(10体积)的混合物中。将物料在25±5℃下搅拌1小时。过滤物料,并用乙酸乙酯(0.1体积)和正庚烷(0.9体积)的混合物洗涤滤饼。将滤饼在25±5℃下干燥2小时,然后在真空盘式干燥机中在50±5℃下干燥12小时。
表31包含最近两个批次的试剂数量和产品产量。
表31.两个批次的中间体6的规模和产量

中间体9的合成

在流程4的步骤2中,在钯催化剂(Pd(dppf)Cl2)存在下,将5-溴-2-甲基嘧啶与双联频哪醇基二硼烷在1,4-二噁烷中反应。通过HPLC监测反应中起始材料的转化。加入中间体6,并通过HPLC监测偶联反应中中间体6的消耗。提取和碳处理后,加入L-半胱氨酸以清除钯。硫醇树脂也可以用于清除钯。一旦达到可接受的Pd水平,将产物混合物用木炭处理,然后将产物用MTBE和庚烷沉淀。通过过滤分离后,将湿饼用MTBE和庚烷洗涤,然后在真空盘式干燥机中真空干燥。
工艺描述如下:在氮气气氛下,在25±5℃下,向反应器中装入1,4-二噁烷(20体积)和5-溴-2-甲基嘧啶(1.0w/w)。向反应器中装入双联频哪醇基二硼烷(1.47w/w)和乙酸钾(1.7w/w),并将该物料在25±5℃下搅拌15分钟。使用氮气将反应物料脱气30分钟。向反应器中装入Pd(dppf)Cl2.DCM(0.093w/w)。将物料温度升高至85℃-90±5℃,并搅拌物料直至达到起始嘧啶含量。反应完成后,将物料冷却至25±5℃。向容器中装入中间体6(1.63w/w)和K2CO3(2.39w/w)。向容器中装入纯化水(1.0w/w)。将反应物料在25±5℃下搅拌15分钟。使用氮气将反应物料脱气30分钟。将物料温度升高至85℃-90±5℃,并搅拌物料直至达到起始材料含量。反应完成后,将物料冷却至25±5℃。将该容器装入乙酸乙酯(30.0体积),并冷却至15±5℃。在15±5℃下缓慢加入纯化水(20.0w/w)。向容器中装入活性炭(0.15w/w)。将物料温度升高至25±5℃并搅拌30分钟。通过硅藻土床过滤物料,并用乙酸乙酯(4.5体积)洗涤该床。分离各层,并用乙酸乙酯(10.0体积)萃取水层。将有机层装回到反应器中。向反应器中装入氯化钠溶液(纯化水20.0w/w和NaCl 1.0w/w),并分离各层。向有机层中加入5%L-半胱氨酸溶液(纯化水20.0w/w和L-半胱氨酸1.0w/w),并搅拌15分钟。分离各层。将浓缩后的有机层样品提交QC以确定Pd含量。向有机层中加入5%L-半胱氨酸溶液(纯化水20.0w/w和L-半胱氨酸0.6w/w),并搅拌15分钟。分离各层。向有机层中加入纯化水(20.0w/w),并搅拌30分钟。分离各层。向有机层中加入纯化水(20.0w/w),并搅拌30分钟。分离各层。向有机层中加入活性炭(0.1w/w),并搅拌60分钟。通过硅藻土床过滤物料,并用乙酸乙酯(3.0体积)洗涤该床。将滤液在55℃以下在真空下浓缩,直至观察不到馏出物。在55℃以下在真空下将滤液与正庚烷(2.0体积)共蒸发,直至观察不到馏出物。向容器中装入MTBE(7.0体积),将温度升高至45±5℃,并搅拌60分钟。缓慢加入正庚烷(3.0体积),并在45±5℃下搅拌60分钟。将物料冷却至10±5℃并搅拌60分钟。通过Nutsche过滤器过滤物料,并用MTBE(1.0体积)和正庚烷(3.0体积)的混合物洗涤滤饼。如果样品未通过验收标准,则重复纯化步骤。将湿滤饼加入正庚烷(5.0体积)和MTBE(5.0体积)的混合物中,并在25±5℃下搅拌40分钟。过滤物料,并用正庚烷(3.0体积)和MTBE(1.0体积)混合物洗涤滤饼。将材料在真空盘式干燥机中在25±5℃下干燥2小时,然后在50±5℃下干燥8小时,直至达到所需的水含量(验收标准:水含量:NMT 5.0%)。
表32包含最近五个批次的试剂数量和产品产量。
表32.五个批次的中间体9的规模和产量

中间体9可用于化合物1或化合物3的合成。例如,在化合物1或化合物3的合成中,中间体9是通过一锅式钯催化的Miyaura硼基化/Suzuki交叉偶联反应从中间体6合成的。使4-溴-2-甲基嘧啶(7)与双联(频哪醇基)二硼反应,得到硼酸酯8。在催化剂Pd(ddpf)Cl2存在下,中间体6与硼酸酯8进行Suzuki反应,生成偶联产物中间体9。
可以在含溴的试剂、含氯化物的试剂、含碘化物的试剂、含有机三氟甲磺酸酯的试剂或它们的任何组合之间进行这种一锅式Miyaura硼基化/Suzuki偶联。如Molander等人(Journal of Organic Chemistry,2012,72,8678-8688)所述,该反应还可以利用替代Suzuki催化剂(包括但不限于如Molander等人所定义的XPhos-Pd-G1、XPhos-Pd-G2、XPhos或CataCXium)进行。在一个实施方案中,该反应利Suzuki催化剂XPhos-Pd-G1和XPhos或XPhos-Pd-G2和XPhos进行。除双联(频哪醇基)二硼之外,硼基化试剂还可以选自但不限于频哪醇硼烷或双硼酸。

中间体10的合成
在方案4的步骤3中,将中间体9用含三氟乙酸的二氯甲烷水解。通过HPLC监测反应中中间体9的消耗。将反应混合物用NaHCO3处理以减少RRT 0.86处的杂质,其通过HPLC监测。通过过滤分离产物,将湿滤饼用水和MTBE洗涤,然后在真空盘式干燥机中真空干燥。
工艺描述如下:在氮气气氛下,在25±5℃下,向容器中装入中间体9(1.0w/w)和DCM(7.0体积),并将物料搅拌15分钟。将反应物料冷却至15±5℃。在15±5℃下将三氟乙酸(7.49w/w)缓慢加入反应物料中,并在15±5℃下搅拌10-15分钟。将物料温度升高至35±5℃,并在35±5℃下搅拌2小时。在55℃以下浓缩反应物料以除去DCM和三氟乙酸。向容器中装入DCM(10.0体积),并搅拌以获得澄清溶液。在20±5℃下将上述物料加入NaHCO3溶液(纯化水15.0w/w,NaHCO3 1.5w/w)中。将物料在25±5℃下搅拌15分钟。检查物料的pH,如果需要,加入更多的NaHCO3溶液(验收标准:pH 7-8)。向NaHCO3溶液中缓慢加入(纯化水10.0w/w,NaHCO3 1.0w/w),搅拌40分钟或直至达到RRT 0.86处的杂质的含量。在25±5℃下向上述物料中加入HCl(0.8-2.0w/w)以将pH调节至2-3。检查物料的pH。将反应物料搅拌15分钟。将物料离心,并用纯化水(5.0体积)洗涤滤饼。将物料旋转干燥3-4小时。如果样品不符合验收标准,则将湿饼加入MTBE(15.0体积)中,并在25±5℃下搅拌20-30分钟。将物料离心,并用MTBE(3.0体积)洗涤滤饼。将材料在25±5℃下干燥2小时,然后在真空盘式干燥机中在55±5℃下干燥8小时。
表33包含最近四个批次的试剂数量和产品产量。
表33.四个批次的中间体10的规模和产量

实施例18.化合物1、化合物2和化合物3的制造工艺描述
在以下方案5中,在含TBTU/DIPEA的DMF存在下,使中间体4与中间体10偶联,生成化合物1原料药。通过HPLC监测偶联反应中中间体10的消耗。萃取到乙酸乙酯中后,将溶液用硅胶、木炭和碳酸钾水溶液和 硫醇树脂(如有需要)处理,直至达到可接受的氟和Pd水平。通过蒸馏除去乙酸乙酯,并用IPA替换,产生结晶化合物1。加入庚烷以帮助分离产物。通过过滤分离后,将湿饼用IPA和庚烷的混合物洗涤,然后在VTD中真空干燥。
硫醇树脂(如有需要)处理,直至达到可接受的氟和Pd水平。通过蒸馏除去乙酸乙酯,并用IPA替换,产生结晶化合物1。加入庚烷以帮助分离产物。通过过滤分离后,将湿饼用IPA和庚烷的混合物洗涤,然后在VTD中真空干燥。
方案5.化合物1的大规模合成

工艺描述如下:在氮气气氛下,在25±5℃下,向反应器中装入中间体10(1.0w/w)和DMF(7.0体积)。然后向反应器中装入中间体4(0.922w/w),搅拌10分钟,然后将所得物料冷却至10±5℃。接下来,向反应器中装入TBTU(1.35w/w),并在10±5℃下将N,N-二异丙基乙胺(LR)(2.06w/w)缓慢加入反应物料中,并搅拌20分钟。将温度升高至25±5℃,并搅拌6小时。向反应器中装入乙酸乙酯(35体积),并搅拌15分钟。缓慢加入纯化水(25体积),并搅拌15分钟。分离各层,用乙酸乙酯(15w/w)萃取水层。合并有机层,并用纯化水(20.0体积x2次)洗涤。
蒸发少量样品(100mL)中的溶剂,并将样品提交QC以通过19FNMR确定19F峰(验收标准:经19F NMR测定,在-145至-150ppm处不存在氟峰)。检查水层的pH(验收标准:pH 7-8),如果样品不符合验收标准,则连续洗涤水层。向有机层中加入硫酸钠(0.25w/w)和硅胶(60-120)(0.5w/w),并将所得物料在25±5℃下搅拌30分钟。(在一些批次中,省略了含硅胶的浆液)。使反应物料通过硅胶玻璃色谱柱(40kg,相对于使用的20kg中间体10),并用EtOAc洗涤该柱。向含有API的级分中加入活性炭(0.05w/w),并将物料温度升高至35±5℃,持续60-70分钟。然后将物料冷却至25±5℃,并通过硅藻土过滤。用乙酸乙酯(2.0w/w)洗滤饼涤。
如果Pd含量不合适,则向滤液中加入 硫醇(0.25w/w)和硅胶(60-120)(0.25w/w),并搅拌8小时或直至达到Pd含量。
硫醇(0.25w/w)和硅胶(60-120)(0.25w/w),并搅拌8小时或直至达到Pd含量。
然后将物料过滤并用乙酸乙酯(2.0w/w)洗涤。使滤液/溶液先后通过5.0μ筒式过滤器,和0.2μ筒式过滤器。用乙酸乙酯(1.0w/w)冲洗管线,并在45℃以下浓缩滤液,直至观察不到馏出物。在55℃以下将滤液与IPA(2.5w/w)共蒸发,直至观察不到馏出物。将IPA(7体积)先后通过5.0μ筒式过滤器和0.2μ筒式过滤器装入容器中,并将物料温度升高至60±5℃并搅拌6小时(API在此步骤下结晶)。在60±5℃下将正庚烷(3体积)先后通过5.0μ筒式过滤器和0.2μ筒式过滤器缓慢加入容器中,并搅拌2小时。将物料温度冷却至25±5℃并搅拌2-3小时。将物料离心,并用经滤筒过滤(先后用5.0μ筒式过滤器和0.2μ筒式过滤器)的IPA(2.35体积)和正庚烷(2.5体积)混合物洗涤滤饼。
如果材料未通过验收标准,则将其溶解在DCM中,蒸发,然后再次先后通过5.0μ筒式过滤器和0.2μ筒式过滤器向容器中装入IPA,将物料温度升高至60±5℃并搅拌6小时。在60±5℃下将正庚烷(3体积)先后通过5.0μ筒式过滤器和0.2μ筒式过滤器缓慢加入容器中,并搅拌2小时。将物料温度冷却至25±5℃并搅拌2-3小时。将物料离心,并用经滤筒过滤(先后用5.0μ筒式过滤器和0.2μ筒式过滤器)的IPA(2.35体积)和正庚烷(2.5体积)混合物洗涤滤饼。
将材料在25±5℃下干燥2小时,然后在真空盘式干燥机中在55±5℃下干燥16小时。将盘式干燥机冷却至25±5℃,将物料研磨,并使用10号筛网过筛。
表34包含最近三个批次的试剂数量和产品产量的实例。
通过分别用适当取代的杂芳基和吡咯烷取代中间体10和4,可以将上述工艺应用于化合物2和化合物3。

一旦形成,就可以使用上述方法将化合物3制成形态形式A、B或M。然后可以将该形态形式溶解在丙酮、DCM或乙醇或其混合物中,以用于喷雾干燥分散体中。喷雾干燥分散体所得材料将是无定形的,但比原始合成的化合物3具有更高的纯度。可以用实现期望效果的任何挥发性溶剂或挥发性溶剂的混合物来进行这种有利的程序。例如,可以将化合物3溶解在丙酮和DCM的90:10、80:20或50:50混合物中。在另一个实施方案中,将化合物3溶解在丙酮和乙醇的90:10、80:20或50:50混合物中。在另一个实施方案中,将化合物3溶解在DCM和乙醇的90:10、80:20或50:50混合物中。
表34.三个批次的化合物1的规模和产量

在一个实施方案中,用于使吡咯烷与吲唑片段(例如中间体4与中间体10)连接的酰胺偶联试剂是二酰亚胺,例如:二环己基碳二亚胺(DCC)、二异丙基碳二亚胺(DIC)、N-环己基,N'-异丙基碳二亚胺(CIC)、N-叔丁基,N'-甲基碳二亚胺(BMC)、N-叔丁基,N'-乙基碳二亚胺(BEC)、N,N'-二环戊基碳二亚胺(CPC)、双[[4-(2,2-二甲基-1,3-二噁茂基)]甲基]碳二亚胺(BDDC)、N-乙基,N-苯基碳二亚胺(PEC)、N-苯基,N-异丙基碳二亚胺(PIC)、N-(3-二甲基氨基丙基)-N'-乙基碳二亚胺*HCl或N-(3-二甲基氨基丙基)-N'-乙基碳二亚胺。
在一个实施方案中,二酰亚胺与加入剂结合使用,例如:1-羟基苯并三唑(HOBt)、1-羟基苯并三唑-6-磺酰胺基甲基树脂HCl(HOBt-6-磺酰胺基甲基树脂HCl)、1-羟基-6-硝基苯并三唑(6-硝基-HOBt)、6-三氟甲基-1-羟基苯并三唑(6-CF3-HOBt)、6-氯-1-羟基苯并三唑(6-Cl-HOBt)、羟基-3,4-二氢-4-氧代-1,2,3-苯并三嗪(HOOBt)、N-羟基琥珀酰亚胺(HOSu)、1-羟基-7-氮杂-1H-苯并三唑(HOAt)、4-氮杂-1-羟基苯并三唑(4-HOAt)、5-氮杂-1-羟基苯并三唑(5-HOAt)、6-氮杂-1-羟基苯并三唑(6-HOAt)、3,4-二氢-3-羟基-4-氧代-1,2,3-苯并三嗪(HODhbt)、3-羟基-4-氧代-3,4-二氢-5-氮杂苯并-1,2,3-三氮烯(HODhat)、3-羟基-4-氧代-3,4-二氢-5-氮杂苯并-1,3-二氮烯(HODhad)、N-羟基-5-降冰片烯-内-2,3-二羧基酰亚胺(HONB)、1-羟基-1H-1,2,3-三唑、5-氯-1-羟基-1H-1,2,3-三唑、5-乙酰基-1-羟基-1H-1,2,3-三唑、1-(1-羟基-1H-1,2,3-三唑-5-基)丙-2-酮、乙基-1-羟基-1H-1,2,3-三唑-4-羧酸酯(HOCt)、1-羟基-1H-1,2,3,5-四唑、1-羟基-2吡啶酮(HOPy)、N-羟基-2-苯基苯并咪唑(HOBI)、N-羟基吲哚-2-酮(HOI)、6-氯-N-羟基-2-苯基苯并咪唑(6-CL-HOBI)、2-氰基-2-(羟基亚氨基)乙酸乙酯(Oxyma)或4-(N,N-二甲基氨基)吡啶)(DMAP)。
在另一个实施方案中,用于使吡咯烷与吲唑片段(例如中间体4与中间体10)连接的酰胺偶联试剂是活性酯,例如:对硝基苯基活性酯、2,4,5-三氯苯基活性酯、五氟活性酯、邻苯二甲酰亚胺基活性酯、N-琥珀酰亚胺活性酯、N-羟基-5-降冰片烯-内-2,3-二甲酰亚胺活性酯或4-氧代-3,4-二氢苯并三嗪基活性酯。
在另一个实施方案中,用于使吡咯烷与吲唑片段(例如中间体4与中间体10)连接的酰胺偶联试剂是氯化剂,例如:特戊酰氯、邻苯二甲酰氯、亚硫酰氯、草酰氯、光气、CC、DMCT、TPP、四甲基-a-氯烯胺或BTC。
在另一个实施方案中,用于使吡咯烷与吲唑片段(例如中间体4与中间体10)连接的酰胺偶联试剂是氟化剂,例如:氰尿酰氟(CF)、2-氟-1-乙基吡啶鎓四硼酸酯(FEP)、2-氟-1-乙基吡啶鎓六氯锑酸酯(FEPH)、TFFH、BTFFH、2-氟-1,3-二甲基咪唑啉鎓六氟磷酸酯(FIP)、HEFFH、DMFH、1,2-二乙基-3,3-四亚甲基氟甲脒六氟磷酸酯(DEFFH)、1,2-二甲基-3,3-四亚甲基氟甲脒六氟磷酸酯(DMFFH)或PTF。
在另一个实施方案中,用于使吡咯烷与吲唑片段(例如中间体4与中间体10)连接的酰胺偶联试剂是磷试剂,例如:苯并三唑-1-基氧基-三(二甲氨基)-鏻六氟磷酸酯(BOP)、苯并三唑-1-基氧基-三吡咯烷子基-鏻六氟磷酸酯(PyBOP)、溴-三吡咯烷子基-鏻六氟磷酸酯(PyBrOP)、7-氮杂-苯并三唑-1-基氧基-三吡咯烷子基鏻六氟磷酸酯(PyAOP)、乙基氰基(羟基亚氨基)乙酸根-O2)-三-(1-吡咯烷基)-鏻六氟磷酸酯(PyOxim)、3-(二乙氧基-磷酰氧基)-1,2,3-苯并[d]三嗪-4(3H)-酮(DEPBT)、BrOP、PyCloP、PyBroP、CloP、AOP、[(7-氮杂苯并三唑-1-基)氧基]三(吡咯烷子基)鏻六氟磷酸酯(PyAOP)、PyNOP、[[6-(三氟甲基)苯并三唑-1-基]氧基]-三(吡咯烷子基)鏻六氟磷酸酯(PyFOP)、[4-硝基-6-(三氟甲基)苯并三唑-1-基)-氧基]三(吡咯烷子基)鏻六氟磷酸酯(PyFNBOP)、(6-氯-苯并三唑-1-基氧基)三(吡咯烷子基)鏻六氟磷酸酯(PyCloK)、(五氟苯氧基)三(吡咯烷子基)鏻六氟磷酸酯(PyPOP)、(吡啶基-2-硫代)三(吡咯烷子基)鏻六氟磷酸酯(PyTOP)、(五氟苯氧基)三(吡咯烷子基)鏻六氟磷酸酯(PyDOP)或[(3,4-二氢-4-氧代-5-氮杂苯并-1,2,3-三嗪-3-基]三(吡咯烷子基)六氟磷酸酯(PyDAOP)。
在另一个实施方案中,用于使吡咯烷与吲唑片段(例如中间体4与中间体10)连接的酰胺偶联剂是铵或铀-亚铵试剂,例如:2-(1H-苯并三唑-1-基)-N,N,N’,N’-四甲基铵四氟硼酸酯/六氟磷酸酯(TBTU/HBTU)、(2-(6-氯-1H-苯并三唑-1-基)-N,N,N’,N’-四甲基铵六氟磷酸酯(HCTU)、N-[(5-氯-1H-苯并三唑-1-基)-二甲氨基-吗啉代]-脲六氟磷酸N-氧化物(HDMC)、2-(7-氮杂-1H-苯并三唑-1-基)-N,N,N’,N’-四甲基铵四氟硼酸酯/六氟磷酸酯(TATU/HATU)、1-[1-(氰基-2-乙氧基-2-氧代亚乙基氨基氧基)-二甲基氨基-吗啉代]-脲六氟磷酸酯(COMU)、2-(1-氧基-吡啶-2-基)-1,1,3,3-四甲基异硫脲四氟硼酸酯(TOTT)、四甲基氟甲脒六氟磷酸酯(TFFH)、TDTU、HDTU、TDATU、HDATU、TPTU、HPTU、TSTU、HSTU、TPFTU、HPFTU、N-CF3-TBTU、N-CF3-HBTU、N-HATU、N-TATU、N-HATTU、HOTT、TOTU、HOTU、HTODC、HTODeC、HTOPC、TNTU、TPhTU BTCFH、HBPyU、HAPyU、HDPyU、HPyOPfp、HPySPfp、HAPyTU、HPyONP、HPyOTCp、HBPipU、HAPipU、TOPPipU、CIP、HBMDU、HAMDU、CPP、HBMTU、HAMTU、HBPTU、HAPTU、HBM2PyU、HAM2PyU、HBM2PipU、HAM2PipU、HBE2PyU、HAE2PyU、HBE2PipU、HAE2PipU、HBTeU、HATeU、DMCH、HDMB、HDMA、HDMC、4-HDMA、6-HDMFB、HDMPfp、HDMP、HDTMA、HDTMB、HDMODC、HDMODcC、HDMOPC、HDmPyODC、HMPyODC、HDmPyODeC、HDmPyOC、HMPyOC、BOMI、BDMP、AOMP、BPMP、FOMP、SOMP或DOMP。
在另一个实施方案中,用于使吡咯烷与吲唑片段(例如,中间体4与中间体10)连接的酰胺偶联试剂是有机磷试剂,例如:DECP、DEPB、DEPC、DPPA、MPTA、MPTO、2-5-二氧代吡咯烷-1-基二苯基磷酸酯、NDPP、FNDPP、Cpt-Cl、BMP-Cl、DEBP、BDP、双(2-硝基苯基)苯基膦酸酯、(5-硝基-吡啶基)二苯基次膦酸酯、DPOOP、BIODPP、ADP、BDOP、ADOP、BDTP、ADTP、DPPCl、FDPP、DEBPO、DOBPO、DOPBT、DEPBT、BOP-Cl、T3P、DEPAT、DPPAT、4-氧代苯并[d][1,2,3]三嗪3(4H)-基膦酸二苯基酯、DOEPBI、DOPPBI、DPPBI、三(4-硝基苯基)膦酸酯、乙基-双(2-硝基苯基)膦酸酯、三嘧啶-2-基磷酸酯、CDPOP、CDPP、二嘧啶-2-基苯基膦酸酯、双(4-硝基苯基)苯基膦酸酯、双(4-氰基苯基)苯基膦酸酯、4-硝基苯基苯基苯基膦酸酯、3-硝基苯基苯基苯基膦酸酯、4-硝基苯基甲基(苯基)膦酸酯、4-硝基苯基甲氧基甲基(苯基)次膦酸酯、4-硝基苯基-二甲基次膦酸酯、4-硝基苯基二乙基次膦酸酯、FDMP、PyDPP或TFMS-DEP。
在另一个实施方案中,用于使吡咯烷与吲唑片段(例如,中间体4与中间体10)连接的酰胺偶联试剂是有机磷试剂,例如:1-((萘-2-基磺酰基)甲基)-1H-苯并-[d][1,2,3]三唑(NBs)、3-((萘-2-基磺酰基)甲基)-3H-[1,2,3]-三唑并[4,5-b]吡啶(NAs)、4-硝基苯磺酸1H-苯并[d][1,2,3]三唑-1-基酯(4-NBs)、4-硝基苯磺酸3H-[1,2,3]三唑并[4,5-b]吡啶-3基酯(4-NAs)、4-甲基苯磺酸1H-苯并[d][1,2,3]三唑-1基酯(TBs)、4-甲基苯磺酸3H-[1,2,3]三唑并[4,5-b]吡啶-3-基酯(TAs)、2-硝基苯磺酸1H-苯并[d][1,2,3]三唑-1-基酯(2-NBs)、2-硝基苯磺酸3H-[1,2,3]三唑并[4,5-b]吡啶-3-基酯(2-NAs)、DNBs、DNAs、HCSP、HCSCP、PFNB、SMDOP、SPDOP和MSOxm。
在另一个实施方案中,用于使吡咯烷与吲唑片段(例如,中间体4与中间体10)连接的酰胺偶联试剂是三嗪试剂,例如:DMCT、DMTMM、4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基-吗啉鎓四氟硼酸酯(TBCR1)、1-(4,6-二甲氧基-1,3,5-三嗪-2-基)-1-甲基-哌啶鎓四氟硼酸酯(TBCR2)、1-(4,6-二甲氧基-1,3,5-三嗪-2-基)喹啉鎓四氟硼酸酯(TBCR3)或TBCR4。
在另一个实施方案中,用于使吡咯烷与吲唑片段(例如,中间体4与中间体10)连接的酰胺偶联试剂是吡啶鎓试剂,例如:PS-EDC、PS-DCC、PS-TBTU、PS-DCT、PS-HOBt、PS-SO2HOBt、PS-HOSu、PS-IIDQ或PS-EEDQ。
在另一个实施方案中,用于使吡咯烷与吲唑片段(例如,中间体4与中间体10)连接的酰胺偶联试剂是聚合物负载试剂,例如:mukaiyama试剂、2-溴-3-乙基-4-甲基噻唑鎓四氟硼酸酯(BEMT)、2-溴-1-乙基吡啶鎓四氟硼酸酯(BEP)、FEP、2-溴-1-乙基吡啶鎓六氯锑酸酯(BEPH)或2-氟-1-乙基吡啶鎓六氯锑酸酯(FEPH)。
在另一个实施方案中,酰胺偶联试剂选自:N-乙氧基羰基-2-乙氧基-1,2-二氢喹啉(EEDQ)、2-丙膦酸酐(T3P)、4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉鎓盐(DMTMM)、双三氯甲基碳酸酯或“三光气(BTC)、或1,1’-羰基二咪唑(CDI)。
使用和制备这些试剂的方法在本领域中是众所周知的,例如Peptide CouplingReagents,More than a Letter Soup by El-Faham,等人,Chem..Rev.2011,111,6557-602提供了若干实验方案。该论文以引用方式并入。
上述试剂可用于化合物1、化合物2或化合物3的合成。
这些偶联试剂取代的非限制性实例在下面显示,用于化合物1合成的最后一步。
方案6.化合物1与替代偶联试剂的合成

T3P
将中间体10(10.8g,34.7mmol)和中间体4(10.0g,34.7mmol)溶解在DMF(70mL)中。在10±5℃下加入DIPEA(5当量),并在10±5℃下搅拌5分钟。通过保持温度5-10℃缓慢加入T3P(50%,DMF溶液,1.3当量),并在25±5℃下搅拌2小时。通过硅胶色谱法纯化,得到18.5g化合物1(92%)。
TBTU
将中间体10(10.8g,34.7mmol)和中间体4(10.0g,34.7mmol)溶解在DMF(70mL)中。在10±5℃下加入DIPEA(5当量),并在10±5℃下搅拌5分钟。通过保持温度5-10℃缓慢加入TBTU(1.3当量),并在25±5℃下搅拌2小时。通过硅胶色谱法纯化,得到17.9g化合物1(89%)。
HATU
将中间体10(10.8g,34.7mmol)和中间体4(10.0g,34.7mmol)溶解在DMF(70mL)中。在10±5℃下加入DIPEA(5当量),并在10±5℃下搅拌5分钟。通过保持温度5-10℃缓慢加入HATU(1.3当量),并在25±5℃下搅拌2小时。通过硅胶色谱法纯化,得到18.3g化合物1(91%)。
MsCl
将中间体10(10.8g,34.7mmol)和中间体4(10.0g,34.7mmol)溶解在DCM(200mL)中。在10±5℃下加入咪唑(5当量),并在10±5℃下搅拌5分钟。通过保持温度5-10℃缓慢加入MsCl(1.3当量),并在25±5℃下搅拌2小时。通过硅胶色谱法纯化,得到15.7g化合物1(78%)。
HOBt,EDC
将中间体10(10.8g,34.7mmol)和中间体4(10.0g,34.7mmol)溶解在DMF(70mL)中。在10±5℃下加入DIPEA(5当量),并在10±5℃下搅拌5分钟。通过保持温度5-10℃缓慢加入HOBt(1.3当量)和EDC(1.3当量),并在25±5℃下搅拌2小时。通过硅胶色谱法纯化,得到15.1g化合物1(75%)。
EEDQ
将中间体10(10.8g,34.7mmol)和中间体4(10.0g,34.7mmol)溶解在甲苯(80mL)中。在10±5℃下加入EEDQ(1.2当量),并在保持温度在60至65℃下搅拌16小时。通过硅胶色谱法纯化,得到17.1g化合物1(85%)。
IBCF
将中间体10(10.8g,34.7mmol)和TEA(6当量)溶解在THF(250mL)中,并在-25±5℃下加入IBCF(氯甲酸异丁酯,1.3当量),并在-25±5℃下搅拌1小时。在-25±5℃下加入中间体4(10.0g,34.7mmol),随后在-25±5℃下加入TEA。将反应在25±5℃下搅拌3-4小时。通过硅胶色谱法纯化,得到5.44g化合物1(27%)。
乙基氰基乙醛酸肟
将中间体10(10.8g,34.7mmol)和中间体4(10.0g,34.7mmol)溶解在DMF(70mL)中,并在25±5℃下加入乙基氰基乙醛酸肟(1.3当量)和EDC.HCl(1.3当量)。将反应在25±5℃下搅拌12小时。通过硅胶色谱法纯化,得到14.7g化合物1(73%)。
氰尿酰氯
将中间体10(10.8g,34.7mmol)和氰尿酰氯(1.3当量)溶解在DCM(100ml)中,并在15±5℃下加入TEA(25当量),并在25±5℃下搅拌3小时。加入中间体4,并在25±5℃下搅拌3小时。通过硅胶色谱法纯化,得到8.1g化合物1(40%)。
实施例19:在喷雾干燥分散体中使用形态形式A的代表性实例
在搅拌下将化合物3形态形式A溶解在丙酮中。搅拌混合物直至溶解。然后使用合适的喷雾干燥机用该喷雾溶液将混合物喷雾干燥,并将所得喷雾干燥产物收集在合适的容器中。然后将喷雾干燥的产物在合适的干燥机中干燥。可以将相似的方法用于形式B或形式M,并且可以将溶剂替换为其他挥发性溶剂,如DCM或乙醇,或它们的混合物。
实施例20.对应于图51至图55的XRPD峰表
下表35中显示了从图51中识别出的峰。
表35化合物3形式A的XRPD峰


下表36中显示了从图52中识别出的峰。
表36化合物3形式B的XRPD峰


下表37中显示了从图53中识别出的峰。
表37化合物3形式G的XRPD峰

下表38中显示了从图54中识别出的峰。
表38化合物3形式J的XRPD峰


下表39中显示了从图55中识别出的峰。
表38化合物3形式M的XRPD峰


已经参考本发明的实施方案描述了本说明书。然而,本领域普通技术人员将理解,在不脱离如权利要求书所阐述的本发明的范围的情况下,可以进行各种修改和改变。因此,本说明书应被认为是说明性的而不是限制性的,并且所有这样的修改旨在包括在本发明的范围内。
Claims (33)
1.一种化合物3的分离的结晶形式B:

所述分离的结晶形式的特征在于包括选自16.2+0.4°、15.7+0.4°、4.5+0.4°、22.6+0.4°、17.4+0.4°、22.0+0.4°、8.3+0.4°、16.1+0.4°、21.1+0.4°、18.7+0.4°、18.3+0.4°、23.9+0.4°和27.5+0.4°的至少三个2θ值的粉末X射线衍射(PXRD)图谱。
2.如权利要求1所述的分离的结晶形式,其中所述PXRD图谱包括选自16.2+0.4°、15.7+0.4°、4.5+0.4°、22.6+0.4°、17.4+0.4°、22.0+0.4°、8.3+0.4°、16.1+0.4°、21.1+0.4°、18.7+0.4°、18.3+0.4°、23.9+0.4°和27.5+0.4°的至少四个2θ值。
3.如权利要求1所述的分离的结晶形式,其中所述PXRD图谱包括选自16.2+0.4°、15.7+0.4°、4.5+0.4°、22.6+0.4°、17.4+0.4°、22.0+0.4°、8.3+0.4°、16.1+0.4°、21.1+0.4°、18.7+0.4°、18.3+0.4°、23.9+0.4°和27.5+0.4°的至少五个2θ值。
4.如权利要求1所述的分离的结晶形式,其中所述PXRD图谱包括选自16.2+0.4°、15.7+0.4°、4.5+0.4°、22.6+0.4°、17.4+0.4°、22.0+0.4°、8.3+0.4°、16.1+0.4°、21.1+0.4°、18.7+0.4°、18.3+0.4°、23.9+0.4°和27.5+0.4°的至少六个2θ值。
5.如权利要求1所述的分离的结晶形式,其中所述PXRD图谱包括选自16.2+0.4°、15.7+0.4°、4.5+0.4°、22.6+0.4°、17.4+0.4°、22.0+0.4°、8.3+0.4°、16.1+0.4°、21.1+0.4°、18.7+0.4°、18.3+0.4°、23.9+0.4°和27.5+0.4°的至少七个2θ值。
6.如权利要求1-5中任一项所述的分离的结晶形式,其中所述PXRD图谱至少包括16.2+0.4°的2θ值。
7.如权利要求1-6中任一项所述的分离的结晶形式,其中所述PXRD图谱至少包括15.7+0.4°的2θ值。
8.一种化合物3的分离的结晶形式A:

所述分离的结晶形式的特征在于包括选自2.6+0.4°、3.6+0.4°和3.8+0.4°的至少一个2θ值的粉末X射线衍射(PXRD)图谱。
9.如权利要求8所述的分离的结晶形式,其中所述PXRD图谱包括选自2.6+0.4°、3.6+0.4°和3.8+0.4°的至少两个2θ值。
10.如权利要求8所述的分离的结晶形式,其中所述PXRD图谱包括选自2.6+0.4°、3.6+0.4°和3.8+0.4°的2θ值。
11.如权利要求8所述的分离的结晶形式,其中所述PXRD图谱包括选自9.3+0.4°、11.7+0.4°、9.5+0.4°、7.6+0.4°、6.7+0.4°、6.0+0.4°、5.7+0.4°、5.6+0.4°、5.4+0.4°和4.2+0.4°的至少六个2θ值。
12.如权利要求8所述的分离的结晶形式,其中所述PXRD图谱包括选自9.3+0.4°、11.7+0.4°、9.5+0.4°、7.6+0.4°、6.7+0.4°、6.0+0.4°、5.7+0.4°、5.6+0.4°、5.4+0.4°和4.2+0.4°的至少七个2θ值。
13.如权利要求8-12中任一项所述的分离的结晶形式,其中所述PXRD图谱至少包括2.6+0.4°的2θ值。
14.如权利要求8-13中任一项所述的分离的结晶形式,其中所述PXRD图谱至少包括3.6+0.4°的2θ值。
15.一种化合物3的分离的结晶形式M:

所述分离的结晶形式的特征在于包括选自15.0+0.4°、7.5+0.4°、23.8+0.4°、7.2+0.4°、19.1+0.4°、5.2+0.4°、8.3+0.4°、26.2+0.4°、22.8+0.4°、21.7+0.4°和24.9+0.4°的至少三个2θ值的粉末X射线衍射(PXRD)图谱。
16.如权利要求15所述的分离的结晶形式,其中所述PXRD图谱包括选自15.0+0.4°、7.5+0.4°、23.8+0.4°、7.2+0.4°、19.1+0.4°、5.2+0.4°、8.3+0.4°、26.2+0.4°、22.8+0.4°、21.7+0.4°和24.9+0.4°的至少四个2θ值。
17.如权利要求15所述的分离的结晶形式,其中所述PXRD图谱包括选自15.0+0.4°、7.5+0.4°、23.8+0.4°、7.2+0.4°、19.1+0.4°、5.2+0.4°、8.3+0.4°、26.2+0.4°、22.8+0.4°、21.7+0.4°和24.9+0.4°的至少五个2θ值。
18.如权利要求15所述的分离的结晶形式,其中所述PXRD图谱包括选自15.0+0.4°、7.5+0.4°、23.8+0.4°、7.2+0.4°、19.1+0.4°、5.2+0.4°、8.3+0.4°、26.2+0.4°、22.8+0.4°、21.7+0.4°和24.9+0.4°的至少六个2θ值。
19.如权利要求15所述的分离的结晶形式,其中所述PXRD图谱包括选自15.0+0.4°、7.5+0.4°、23.8+0.4°、7.2+0.4°、19.1+0.4°、5.2+0.4°、8.3+0.4°、26.2+0.4°、22.8+0.4°、21.7+0.4°和24.9+0.4°的至少七个2θ值。
20.如权利要求15-19中任一项所述的分离的结晶形式,其中所述PXRD图谱至少包括15.0+0.4°的2θ值。
21.如权利要求15-19中任一项所述的分离的结晶形式,其中所述PXRD图谱至少包括7.5+0.4°的2θ值。
22.如权利要求1-21中任一项所述的分离的结晶形式,其中所述峰在+0.3° 2θ内。
23.如权利要求1-21中任一项所述的分离的结晶形式,其中所述峰在+0.2° 2θ内。
24.一种药物组合物,所述药物组合物包含如权利要求1-23中任一项所述的分离的结晶形式,所述分离的结晶形式处于药学上可接受的赋形剂中,用于固体剂量递送。
25.一种用于治疗补体因子D介导的病症的方法,所述方法包括向有需要的受试者施用治疗有效量的根据权利要求1-23中任一项所述的分离的结晶形式或其药物组合物,所述分离的结晶形式任选地处于药学上可接受的赋形剂中,用于固体剂量递送。
26.如权利要求25所述的方法,其中所述受试者是人。
27.如权利要求1-23中任一项所述的分离的结晶形式,所述分离的结晶形式用于治疗有需要的受试者的补体因子D介导的病症,所述分离的结晶形式任选地处于药学上可接受的赋形剂中,用于固体剂量递送。
28.如权利要求27所述的分离的结晶形式,其中所述受试者是人。
29.如权利要求1-23中任一项所述的分离的结晶形式或其药物组合物在制造用于治疗有需要的受试者的补体因子D介导的病症的药物中的用途,所述分离的结晶形式任选地处于药学上可接受的赋形剂中,用于固体剂量递送。
30.如权利要求29所述的用途,其中所述受试者是人。
31.一种用于制备无定形化合物3的工艺:

所述工艺包括将化合物3形态形式A溶解在溶剂中并进行喷雾干燥以形成无定形化合物3。
32.一种用于制备无定形化合物3的工艺:

所述工艺包括将化合物3形态形式B溶解在溶剂中并进行喷雾干燥以形成无定形化合物3。
33.一种用于制备无定形化合物3的工艺:

所述工艺包括将化合物3形态形式M溶解在溶剂中并进行喷雾干燥以形成无定形化合物3。
Applications Claiming Priority (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862736294P | 2018-09-25 | 2018-09-25 | |
| US62/736,294 | 2018-09-25 | ||
| US201862757565P | 2018-11-08 | 2018-11-08 | |
| US62/757,565 | 2018-11-08 | ||
| US201862760520P | 2018-11-13 | 2018-11-13 | |
| US62/760,520 | 2018-11-13 | ||
| US201962796776P | 2019-01-25 | 2019-01-25 | |
| US62/796,776 | 2019-01-25 | ||
| PCT/US2019/053012 WO2020069024A1 (en) | 2018-09-25 | 2019-09-25 | Morphic forms of complement factor d inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN112996497A true CN112996497A (zh) | 2021-06-18 |
Family
ID=69949508
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201980074140.4A Pending CN112996497A (zh) | 2018-09-25 | 2019-09-25 | 补体因子d抑制剂的形态形式 |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US11807627B2 (zh) |
| EP (1) | EP3856164A4 (zh) |
| JP (1) | JP2022502500A (zh) |
| KR (1) | KR20210093855A (zh) |
| CN (1) | CN112996497A (zh) |
| AU (1) | AU2019346464A1 (zh) |
| BR (1) | BR112021005506A2 (zh) |
| CA (1) | CA3114039A1 (zh) |
| CO (1) | CO2021005140A2 (zh) |
| MX (1) | MX2021003425A (zh) |
| WO (1) | WO2020069024A1 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113195472A (zh) * | 2018-12-17 | 2021-07-30 | 艾其林医药公司 | 用于治疗补体介导的病症的靶向给药 |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017035409A1 (en) | 2015-08-26 | 2017-03-02 | Achillion Pharmaceuticals, Inc. | Aryl, heteroaryl, and heterocyclic compounds for treatment of immune and inflammatory disorders |
| WO2017035405A1 (en) | 2015-08-26 | 2017-03-02 | Achillion Pharmaceuticals, Inc. | Amino compounds for treatment of immune and inflammatory disorders |
| WO2017035401A1 (en) | 2015-08-26 | 2017-03-02 | Achillion Pharmaceuticals, Inc. | Amide compounds for treatment of immune and inflammatory disorders |
| AU2018227849B2 (en) | 2017-03-01 | 2022-04-28 | Achillion Pharmaceuticals, Inc. | Aryl, heteroary, and heterocyclic pharmaceutical compounds for treatment of medical disorders |
| EP3589287B1 (en) | 2017-03-01 | 2022-09-14 | Achillion Pharmaceuticals, Inc. | Macrocyclic compounds for treatment of medical disorders |
| US11814363B2 (en) | 2018-09-06 | 2023-11-14 | Achillion Pharmaceuticals, Inc. | Morphic forms of danicopan |
| JP7443375B2 (ja) | 2018-09-06 | 2024-03-05 | アキリオン ファーマシューティカルズ, インコーポレーテッド | 医学的障害の治療のための大環状化合物 |
| US11807627B2 (en) | 2018-09-25 | 2023-11-07 | Achillon Pharmaceuticals, Inc. | Morphic forms of complement factor D inhibitors |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012093101A1 (en) * | 2011-01-04 | 2012-07-12 | Novartis Ag | Indole compounds or analogues thereof useful for the treatment of age-related macular degeneration (amd) |
| US20170066783A1 (en) * | 2015-08-26 | 2017-03-09 | Achillion Pharmaceuticals, Inc. | Aryl, Heteroaryl, and Heterocyclic Compounds for Treatment of Medical Disorders |
Family Cites Families (112)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9206757D0 (en) | 1992-03-27 | 1992-05-13 | Ferring Bv | Novel peptide receptor ligands |
| US6074642A (en) | 1994-05-02 | 2000-06-13 | Alexion Pharmaceuticals, Inc. | Use of antibodies specific to human complement component C5 for the treatment of glomerulonephritis |
| US5628984A (en) | 1995-07-31 | 1997-05-13 | University Of North Carolina At Chapel Hill | Method of detecting lung disease |
| CA2248772C (en) | 1996-03-13 | 2007-06-12 | John D. Lambris | Novel peptides which inhibit complement activation |
| AU7808198A (en) | 1997-06-03 | 1998-12-21 | Biocryst Pharmaceuticals, Inc. | Novel compounds useful in the complement, coagulat and kallikrein pathways and method for their preparation |
| AU754716B2 (en) | 1998-03-26 | 2002-11-21 | Japan Tobacco Inc. | Amide derivatives and nociceptin antagonists |
| JP2002524463A (ja) | 1998-09-09 | 2002-08-06 | メタバシス・セラピューティクス・インコーポレイテッド | フルクトース−1,6−ビスホスファターゼの新規な芳香族インヒビター |
| KR20000047461A (ko) | 1998-12-29 | 2000-07-25 | 성재갑 | 트롬빈 억제제 |
| AU6868900A (en) | 1999-09-03 | 2001-04-10 | Ajinomoto Co., Inc. | Novel processes for preparing oxazepine derivatives |
| US7528165B2 (en) | 2001-12-13 | 2009-05-05 | National Health Research Institutes | Indole compounds |
| ITMI20021527A1 (it) | 2002-07-11 | 2004-01-12 | Consiglio Nazionale Ricerche | Anticorpi anti componente c5 del complemento e loro uso |
| NZ537853A (en) | 2002-07-16 | 2007-02-23 | Amura Therapeutics Ltd | Inhibitors of cathepsin K and related cysteine protesases of the CA clan |
| US9415102B2 (en) | 2002-09-06 | 2016-08-16 | Alexion Pharmaceuticals, Inc. | High concentration formulations of anti-C5 antibodies |
| AU2003275075A1 (en) | 2002-09-20 | 2004-04-08 | The Trustees Of The University Of Pennsylvania | Compstatin analogs with improved activity |
| FR2845382A1 (fr) | 2002-10-02 | 2004-04-09 | Sanofi Synthelabo | Derives d'indazolecarboxamides, leur preparation et leur utilisation en therapeutique |
| EP1567487A4 (en) | 2002-11-15 | 2005-11-16 | Bristol Myers Squibb Co | OPEN-CHAINED, PROLYL-FROSTED MODULATORS OF ANDROGEN RECEPTOR FUNCTION |
| AU2003902946A0 (en) | 2003-06-12 | 2003-06-26 | Fujisawa Pharmaceutical Co., Ltd. | Dpp-iv inhibitor |
| US7482376B2 (en) | 2003-07-03 | 2009-01-27 | 3-Dimensional Pharmaceuticals, Inc. | Conjugated complement cascade inhibitors |
| BRPI0506629A (pt) | 2004-02-10 | 2007-05-02 | Univ Colorado | inibição do fator b, a via alternativa do sistema complemento e métodos relacionados |
| MXPA06010756A (es) | 2004-03-24 | 2006-12-15 | Jerini Ag | Nuevos compuestos para la inhibicion de la angiogenesis y su uso. |
| US7417063B2 (en) | 2004-04-13 | 2008-08-26 | Bristol-Myers Squibb Company | Bicyclic heterocycles useful as serine protease inhibitors |
| US20080075728A1 (en) | 2004-07-20 | 2008-03-27 | Walter Newman | Combination Therapies Of Hmgb And Complement Inhibitors Against Inflammation |
| US8168584B2 (en) | 2005-10-08 | 2012-05-01 | Potentia Pharmaceuticals, Inc. | Methods of treating age-related macular degeneration by compstatin and analogs thereof |
| EP2377877B1 (en) | 2005-11-28 | 2018-03-28 | The Trustees Of The University Of Pennsylvania | Potent compstatin analogs |
| EP1976829A2 (en) | 2005-12-12 | 2008-10-08 | Genelabs Technologies, Inc. | N-(6-membered aromatic ring)-amido anti-viral compounds |
| DK2359834T5 (en) | 2006-03-15 | 2017-02-06 | Alexion Pharma Inc | Treatment of paroxysmal nocturnal hemoglobinuria patients with a complement inhibitor |
| EP2108642A1 (en) | 2006-10-17 | 2009-10-14 | Kyowa Hakko Kirin Co., Ltd. | Jak inhibitor |
| RU2470918C2 (ru) | 2007-01-15 | 2012-12-27 | Сантен Фармасьютикал Ко., Лтд. | НОВЫЕ ПРОИЗВОДНЫЕ ИНДОЛА, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ IκB КИНАЗЫ β |
| HUE026001T2 (en) | 2007-02-05 | 2016-04-28 | Apellis Pharmaceuticals Inc | Compstatin analogues for use in the treatment of the inflammatory condition of the respiratory system |
| DK2238142T3 (da) | 2007-12-24 | 2012-10-08 | Janssen R & D Ireland | Makrocykliske indoler som hepatitis C-virusinhibitorer |
| WO2009091826A2 (en) | 2008-01-14 | 2009-07-23 | The Board Of Regents Of The University Of Texas System | Compositions and methods related to a human cd19-specific chimeric antigen receptor (h-car) |
| EP2837388A1 (en) | 2008-08-05 | 2015-02-18 | Novartis AG | Compositions and methods for antibodies targeting complement protein C5 |
| GB2469471B (en) | 2009-04-14 | 2015-01-14 | Skype | Optimising communications |
| HUE035378T2 (en) | 2009-05-01 | 2018-05-02 | Univ Pennsylvania | Compstatin with peptide framework and C-terminal modifications |
| US9291622B2 (en) | 2009-05-21 | 2016-03-22 | Apellis Pharmaceuticals, Inc. | Complement assays and uses thereof |
| CA2777013A1 (en) | 2009-10-16 | 2011-04-21 | Mochida Pharmaceutical Co., Ltd. | Marker associated with non-alcoholic steatohepatitis |
| EA201290286A1 (ru) | 2009-11-05 | 2013-01-30 | Алексион Кембридж Корпорейшн | Лечение пароксизмальной ночной гемоглобинурии, гемолитических анемий и патологических состояний с вовлечением внутрисосудистого и внесосудистого гемолиза |
| EP2966075B1 (en) | 2009-12-31 | 2019-02-27 | Hutchison Medipharma Limited | Certain triazolopyridines, compositions thereof and their use in the treatment of cancer. |
| DK2558446T5 (da) | 2010-04-16 | 2019-12-09 | Ac Immune Sa | Nye forbindelser til behandling af sygdomme associerede med amyloid- eller amyloidlignende proteiner |
| WO2011163394A2 (en) | 2010-06-22 | 2011-12-29 | Apellis Pharmaceuticals, Inc. | Compstatin analogs for treatment of neuropathic pain |
| WO2012006599A2 (en) | 2010-07-09 | 2012-01-12 | Apellis Pharmaceuticals, Inc. | Compstatin analogs for treatment of rhinosinusitis and nasal polyposis |
| ES2915265T3 (es) | 2011-05-11 | 2022-06-21 | Apellis Pharmaceuticals Inc | Análogos de compstatina reactivos con células, de acción prolongada o dirigidos y usos de los mismos |
| HUE029326T2 (hu) | 2011-05-27 | 2017-02-28 | Achillion Pharmaceuticals Inc | Szubsztituált alifánok, ciklofánok, heterafánok, heterofánok, hetero-heterafánok és metallocének, amelyek alkalmasak HCV fertõzések kezelésére |
| JP2014518223A (ja) | 2011-06-20 | 2014-07-28 | アルツハイマーズ・インスティテュート・オブ・アメリカ・インコーポレイテッド | 化合物とその治療用途 |
| MX363606B (es) | 2011-06-22 | 2019-03-28 | Apellis Pharmaceuticals Inc | Uso de inhibidores del complemento en trastornos crónicos. |
| US20130035392A1 (en) | 2011-08-01 | 2013-02-07 | Mcgeer Patrick L | Selective inhibition of the membrane attack complex of complement and C3 convertase by low molecular weight components of the aurin tricarboxylic acid synthetic complex |
| HUE047375T2 (hu) | 2011-09-07 | 2020-04-28 | Univ Pennsylvania | Javított farmakokinetikai tulajdonságokkal rendelkezõ compstatin analógok |
| NZ742004A (en) | 2012-05-03 | 2019-04-26 | Kala Pharmaceuticals Inc | Pharmaceutical nanoparticles showing improved mucosal transport |
| US9056874B2 (en) | 2012-05-04 | 2015-06-16 | Novartis Ag | Complement pathway modulators and uses thereof |
| SG11201406973PA (en) | 2012-05-04 | 2014-12-30 | Novartis Ag | Complement pathway modulators and uses thereof |
| RS61755B1 (sr) | 2012-06-18 | 2021-05-31 | Omeros Corp | Kompozicije i postupci za inhibiciju masp-1 i/ili masp-2 i/ili masp-3 za tretman različitih bolesti i poremećaja |
| EA201590053A1 (ru) | 2012-06-20 | 2015-08-31 | Новартис Аг | Модуляторы пути системы комплемента и их применение |
| US9579360B2 (en) | 2012-06-20 | 2017-02-28 | The Trustees Of The University Of Pennsylvania | Methods of treating or preventing periodontitis and diseases associated with periodontitis |
| AU2013282768A1 (en) | 2012-06-28 | 2015-01-22 | Novartis Ag | Pyrrolidine derivatives and their use as complement pathway modulators |
| JP6273274B2 (ja) | 2012-06-28 | 2018-01-31 | ノバルティス アーゲー | 補体経路モジュレーターおよびその使用 |
| JP6155332B2 (ja) | 2012-06-28 | 2017-06-28 | ノバルティス アーゲー | ピロリジン誘導体、および補体経路調節因子としてのその使用 |
| EP2867227B1 (en) | 2012-06-28 | 2018-11-21 | Novartis AG | Complement pathway modulators and uses thereof |
| CN104603126B (zh) | 2012-06-28 | 2017-05-31 | 诺华股份有限公司 | 吡咯烷衍生物及其作为补体途径调节剂的用途 |
| WO2014002052A1 (en) | 2012-06-28 | 2014-01-03 | Novartis Ag | Pyrrolidine derivatives and their use as complement pathway modulators |
| ITMI20121156A1 (it) | 2012-06-29 | 2013-12-30 | Consiglio Nazionale Ricerche | Metodo di elaborazione di immagini di tomografia a coerenza ottica |
| WO2014002059A1 (en) | 2012-06-29 | 2014-01-03 | Novartis Ag | CRYSTALLINE FORMS OF 1-(2-((1R,3S,5R)-3-(((R)-1-(3-chloro-2-fluorophenyl)ethyl)carbamoyl)-2-azabicyclo[3.1.0]hexan-2-yl)-2-oxoethyl)-1Hpyrazolo[3,4-c]pyridine-3-carboxamide |
| WO2014005150A1 (en) | 2012-06-29 | 2014-01-03 | Novartis Ag | Crystalline forms of l-(2-((lr,3s,5r)-3-( (2 -fluoro-3 - (trifluoromethoxy) phenyl) carbamoyl) - 2 -azabicycl o [3.1.0] hexan- 2 -yl) - 2 -oxoethyl) - 5 -methyl - 1h - pyrazolo [3, 4 -c] pyridine - 3 - carboxami de and salts thereof |
| JP6238980B2 (ja) | 2012-07-12 | 2017-11-29 | ノバルティス アーゲー | 補体経路モジュレーターおよびその使用 |
| SG11201500377UA (en) | 2012-09-10 | 2015-02-27 | Hoffmann La Roche | 6-amino acid heteroaryldihydropyrimidines for the treatment and prophylaxis of hepatitis b virus infection |
| WO2014078734A2 (en) | 2012-11-15 | 2014-05-22 | Apellis Pharmaceuticals, Inc. | Cell-reactive, long-acting, or targeted compstatin analogs and related compositions and methods |
| DK2920201T3 (da) | 2012-11-15 | 2020-04-14 | Apellis Pharmaceuticals Inc | Langtidsvirkende compstatin-analoger og relaterede sammensætninger og fremgangsmåder |
| US9773428B2 (en) | 2012-12-11 | 2017-09-26 | Fluidity Software, Inc. | Computerized system and method for teaching, learning, and assessing step by step solutions to stem problems |
| EP2948480A4 (en) | 2013-01-23 | 2016-12-07 | Musc Found For Res Dev | NATURAL ANTIBODY TARGETING CONSTRUCTS AND USES THEREOF |
| EP2970269B1 (en) | 2013-03-14 | 2017-04-19 | Novartis AG | 2-(1h-indol-4-ylmethyl)-3h-imidazo[4,5-b]pyridine-6-carbonitrile derivatives as complement factor b inhibitors useful for the treatment of ophthalmic diseases |
| US10308687B2 (en) | 2013-03-15 | 2019-06-04 | Apellis Pharmaceuticals, Inc. | Cell-penetrating compstatin analogs and uses thereof |
| JO3425B1 (ar) | 2013-07-15 | 2019-10-20 | Novartis Ag | مشتقات البابيريدينيل-اندول واستخدامها كعامل متمم لمثبطات b |
| WO2015009977A1 (en) | 2013-07-18 | 2015-01-22 | Novartis Ag | Aminomethyl-biaryl derivatives as complement factor d inhibitors and uses thereof |
| JP2016169161A (ja) | 2013-07-19 | 2016-09-23 | 大日本住友製薬株式会社 | 新規イミダゾピリジン化合物 |
| JP6211701B2 (ja) | 2013-08-07 | 2017-10-11 | アレクシオン ファーマシューティカルズ, インコーポレイテッド | 非典型溶血性尿毒症症候群のバイオマーカータンパク質 |
| WO2015054569A1 (en) | 2013-10-10 | 2015-04-16 | Viropharma Holdings Limited | Methods of inhibiting the alternative pathway of complement immune system activation and compositions used therein |
| WO2015066241A1 (en) | 2013-10-30 | 2015-05-07 | Novartis Ag | 2-benzyl-benzimidazole complement factor b inhibitors and uses thereof |
| IL301427A (en) | 2014-02-25 | 2023-05-01 | Achillion Pharmaceuticals Inc | Aryl, heteroaryl, and heterocyclic compounds for the treatment of complement-mediated disorders |
| WO2016088082A1 (en) | 2014-12-05 | 2016-06-09 | Novartis Ag | Amidomethyl-biaryl derivatives complement factor d inhibitors and uses thereof |
| WO2017024318A1 (en) | 2015-08-06 | 2017-02-09 | Dana-Farber Cancer Institute, Inc. | Targeted protein degradation to attenuate adoptive t-cell therapy associated adverse inflammatory responses |
| WO2017035361A1 (en) | 2015-08-26 | 2017-03-02 | Achillion Pharmaceuticals, Inc. | Disubstituted compounds for the treatment of medical disorders |
| WO2017035409A1 (en) | 2015-08-26 | 2017-03-02 | Achillion Pharmaceuticals, Inc. | Aryl, heteroaryl, and heterocyclic compounds for treatment of immune and inflammatory disorders |
| AR105808A1 (es) | 2015-08-26 | 2017-11-08 | Achillion Pharmaceuticals Inc | Compuestos de amida para el tratamiento de trastornos médicos |
| WO2017035401A1 (en) | 2015-08-26 | 2017-03-02 | Achillion Pharmaceuticals, Inc. | Amide compounds for treatment of immune and inflammatory disorders |
| WO2017035355A1 (en) | 2015-08-26 | 2017-03-02 | Achillion Pharmaceuticals, Inc. | Ether compounds for treatment of medical disorders |
| ES2908479T3 (es) | 2015-08-26 | 2022-04-29 | Achillion Pharmaceuticals Inc | Compuestos para el tratamiento de trastornos inmunitarios e inflamatorios |
| WO2017035415A1 (en) | 2015-08-26 | 2017-03-02 | Achillion Pharmaceuticals, Inc. | Alkyne compounds for treatment of immune and inflammatory disorders |
| WO2017035405A1 (en) | 2015-08-26 | 2017-03-02 | Achillion Pharmaceuticals, Inc. | Amino compounds for treatment of immune and inflammatory disorders |
| WO2017035362A1 (en) | 2015-08-26 | 2017-03-02 | Achillion Pharmaceuticals, Inc. | Use of complement pathway inhibitor compounds to mitigate adoptive t-cell therapy associated adverse immune responses |
| WO2017035352A1 (en) | 2015-08-26 | 2017-03-02 | Achillion Pharmaceuticals, Inc. | Carbamate, ester, and ketone compounds for treatment of medical disorders |
| WO2017035413A2 (en) | 2015-08-26 | 2017-03-02 | Achillion Pharmaceuticals, Inc. | Carbamate, ester, and ketone compounds for treatment of immune and inflammatory disorders |
| WO2017035417A1 (en) | 2015-08-26 | 2017-03-02 | Achillion Pharmaceuticals, Inc. | Phosphonate compounds for treatment of immune and inflammatory disorders |
| AR105809A1 (es) | 2015-08-26 | 2017-11-08 | Achillion Pharmaceuticals Inc | Compuestos para el tratamiento de trastornos médicos |
| WO2017035351A1 (en) | 2015-08-26 | 2017-03-02 | Achillion Pharmaceuticals, Inc. | Amino compounds for treatment of medical disorders |
| WO2017035418A1 (en) | 2015-08-26 | 2017-03-02 | Achillion Pharmaceuticals, Inc. | Disubstituted compounds for treatment of immune and inflammatory disorders |
| WO2017035411A1 (en) | 2015-08-26 | 2017-03-02 | Achillion Pharmaceuticals, Inc. | Ether compounds for treatment of immune and inflammatory disorders |
| WO2017035357A1 (en) | 2015-08-26 | 2017-03-02 | Achillion Pharmaceuticals, Inc. | Phosphonate compounds for treatment of medical disorders |
| WO2017035348A1 (en) | 2015-08-26 | 2017-03-02 | Achillion Pharmaceuticals, Inc. | Alkyne compounds for treatment of medical disorders |
| EP3386504A4 (en) | 2015-12-11 | 2019-05-22 | Lifesci Pharmaceuticals, Inc. | THERAPEUTIC INHIBITOR COMPOUNDS |
| TWI791423B (zh) | 2016-01-14 | 2023-02-11 | 美商卡默森屈有限公司 | C3腎絲球病變之治療方法 |
| WO2017127761A1 (en) | 2016-01-20 | 2017-07-27 | Vitrisa Therapeutics, Inc. | Compositions and methods for inhibiting factor d |
| TW202222786A (zh) | 2016-02-01 | 2022-06-16 | 美商百歐克斯製藥公司 | 苯并吡唑化合物及其類似物 |
| CN109414441A (zh) | 2016-06-27 | 2019-03-01 | 艾其林医药公司 | 治疗医学障碍的喹唑啉和吲哚化合物 |
| JOP20170154B1 (ar) | 2016-08-01 | 2023-03-28 | Omeros Corp | تركيبات وطرق لتثبيط masp-3 لعلاج أمراض واضطرابات مختلفة |
| WO2018160891A1 (en) | 2017-03-01 | 2018-09-07 | Achillion Pharmaceutical, Inc. | Pharmaceutical compounds for treatment of medical disorders |
| EP3589287B1 (en) | 2017-03-01 | 2022-09-14 | Achillion Pharmaceuticals, Inc. | Macrocyclic compounds for treatment of medical disorders |
| AU2018227849B2 (en) | 2017-03-01 | 2022-04-28 | Achillion Pharmaceuticals, Inc. | Aryl, heteroary, and heterocyclic pharmaceutical compounds for treatment of medical disorders |
| US20190038623A1 (en) | 2017-08-02 | 2019-02-07 | Achillion Pharmaceuticals, Inc. | Therapeutic regimens for treatment of paroxysmal nocturnal hemoglobinuria |
| US20200262897A1 (en) | 2017-10-04 | 2020-08-20 | Alexion Pharmaceuticals, Inc. | Dosage and administration of anti-c5 antibodies for treatment of patients with membranoproliferative glomerulonephritis |
| US20190359645A1 (en) | 2018-05-03 | 2019-11-28 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotides comprising carbocyclic nucleotide |
| US11807627B2 (en) | 2018-09-25 | 2023-11-07 | Achillon Pharmaceuticals, Inc. | Morphic forms of complement factor D inhibitors |
| WO2020109343A1 (en) | 2018-11-29 | 2020-06-04 | F. Hoffmann-La Roche Ag | Combination therapy for treatment of macular degeneration |
| JP7471300B2 (ja) | 2018-12-17 | 2024-04-19 | アキリオン ファーマシューティカルズ, インコーポレーテッド | 補体介在性疾患を治療するための的を絞った投与 |
-
2019
- 2019-09-25 US US17/279,767 patent/US11807627B2/en active Active
- 2019-09-25 WO PCT/US2019/053012 patent/WO2020069024A1/en unknown
- 2019-09-25 AU AU2019346464A patent/AU2019346464A1/en not_active Abandoned
- 2019-09-25 BR BR112021005506-1A patent/BR112021005506A2/pt not_active Application Discontinuation
- 2019-09-25 JP JP2021540397A patent/JP2022502500A/ja active Pending
- 2019-09-25 MX MX2021003425A patent/MX2021003425A/es unknown
- 2019-09-25 CN CN201980074140.4A patent/CN112996497A/zh active Pending
- 2019-09-25 CA CA3114039A patent/CA3114039A1/en active Pending
- 2019-09-25 EP EP19864090.6A patent/EP3856164A4/en active Pending
- 2019-09-25 KR KR1020217010839A patent/KR20210093855A/ko unknown
-
2021
- 2021-04-22 CO CONC2021/0005140A patent/CO2021005140A2/es unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012093101A1 (en) * | 2011-01-04 | 2012-07-12 | Novartis Ag | Indole compounds or analogues thereof useful for the treatment of age-related macular degeneration (amd) |
| US20170066783A1 (en) * | 2015-08-26 | 2017-03-09 | Achillion Pharmaceuticals, Inc. | Aryl, Heteroaryl, and Heterocyclic Compounds for Treatment of Medical Disorders |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113195472A (zh) * | 2018-12-17 | 2021-07-30 | 艾其林医药公司 | 用于治疗补体介导的病症的靶向给药 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3856164A4 (en) | 2022-07-13 |
| EP3856164A1 (en) | 2021-08-04 |
| BR112021005506A2 (pt) | 2021-06-15 |
| CO2021005140A2 (es) | 2021-04-30 |
| WO2020069024A1 (en) | 2020-04-02 |
| CA3114039A1 (en) | 2020-04-02 |
| KR20210093855A (ko) | 2021-07-28 |
| US11807627B2 (en) | 2023-11-07 |
| AU2019346464A1 (en) | 2021-04-08 |
| MX2021003425A (es) | 2021-07-16 |
| JP2022502500A (ja) | 2022-01-11 |
| US20210332026A1 (en) | 2021-10-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11807627B2 (en) | Morphic forms of complement factor D inhibitors | |
| US11649229B2 (en) | Amide compounds for treatment of immune and inflammatory disorders | |
| US10660876B2 (en) | Amino compounds for treatment of medical disorders | |
| US11001600B2 (en) | Disubstituted compounds for treatment of medical disorders | |
| US10961252B2 (en) | Quinazoline and indole compounds to treat medical disorders | |
| US10906887B2 (en) | Amino compounds for treatment of immune and inflammatory disorders | |
| US10138225B2 (en) | Amide compounds for treatment of medical disorders | |
| US10385097B2 (en) | Ether compounds for treatment of medical disorders | |
| US11814363B2 (en) | Morphic forms of danicopan | |
| CN110603252A (zh) | 用于治疗医学障碍的芳基、杂芳基和杂环药物化合物 | |
| TW201718611A (zh) | 用於治療醫學病症之膦酸酯化合物 | |
| WO2017035352A1 (en) | Carbamate, ester, and ketone compounds for treatment of medical disorders | |
| JP2022519924A (ja) | 補体媒介性障害の治療のための医薬化合物 | |
| JP2021535112A (ja) | 補体d因子の医学的障害の治療のための医薬化合物 | |
| JP2021536510A (ja) | 医学的障害の治療のための大環状化合物 | |
| JP2024069504A (ja) | 補体d因子の医学的障害の治療のための医薬化合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination |