CN113195472A - 用于治疗补体介导的病症的靶向给药 - Google Patents
用于治疗补体介导的病症的靶向给药 Download PDFInfo
- Publication number
- CN113195472A CN113195472A CN201980086227.3A CN201980086227A CN113195472A CN 113195472 A CN113195472 A CN 113195472A CN 201980086227 A CN201980086227 A CN 201980086227A CN 113195472 A CN113195472 A CN 113195472A
- Authority
- CN
- China
- Prior art keywords
- compound
- dosage form
- grain
- dose
- complement
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 230000000295 complement effect Effects 0.000 title claims abstract description 61
- 238000011282 treatment Methods 0.000 title claims description 106
- 230000001404 mediated effect Effects 0.000 title description 22
- 238000012377 drug delivery Methods 0.000 title description 2
- 230000000694 effects Effects 0.000 claims abstract description 103
- 230000037361 pathway Effects 0.000 claims abstract description 49
- 238000000034 method Methods 0.000 claims abstract description 42
- 239000002552 dosage form Substances 0.000 claims abstract description 39
- 230000024203 complement activation Effects 0.000 claims abstract description 38
- 229940125904 compound 1 Drugs 0.000 claims description 232
- 208000034332 Body integrity dysphoria Diseases 0.000 claims description 129
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 60
- 239000006186 oral dosage form Substances 0.000 claims description 52
- 208000035475 disorder Diseases 0.000 claims description 49
- 239000003814 drug Substances 0.000 claims description 46
- 230000002060 circadian Effects 0.000 claims description 44
- -1 2- (3-acetyl-5- (2-methylpyrimidin-5-yl) -1H-indazol-1-yl) acetyl Chemical group 0.000 claims description 42
- 150000003839 salts Chemical class 0.000 claims description 35
- 208000004451 Membranoproliferative Glomerulonephritis Diseases 0.000 claims description 31
- 208000000733 Paroxysmal Hemoglobinuria Diseases 0.000 claims description 27
- 102100036050 Phosphatidylinositol N-acetylglucosaminyltransferase subunit A Human genes 0.000 claims description 27
- 201000003045 paroxysmal nocturnal hemoglobinuria Diseases 0.000 claims description 27
- 206010064930 age-related macular degeneration Diseases 0.000 claims description 20
- 206010018370 Glomerulonephritis membranoproliferative Diseases 0.000 claims description 17
- 229910052799 carbon Inorganic materials 0.000 claims description 16
- 208000016323 C3 glomerulonephritis Diseases 0.000 claims description 15
- 208000022401 dense deposit disease Diseases 0.000 claims description 14
- 208000002780 macular degeneration Diseases 0.000 claims description 12
- 208000008338 non-alcoholic fatty liver disease Diseases 0.000 claims description 12
- 206010053219 non-alcoholic steatohepatitis Diseases 0.000 claims description 12
- 201000010099 disease Diseases 0.000 claims description 11
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 11
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 claims description 10
- 208000017169 kidney disease Diseases 0.000 claims description 9
- 208000003556 Dry Eye Syndromes Diseases 0.000 claims description 8
- 206010013774 Dry eye Diseases 0.000 claims description 8
- 208000022461 Glomerular disease Diseases 0.000 claims description 8
- 241000282412 Homo Species 0.000 claims description 7
- 230000001363 autoimmune Effects 0.000 claims description 7
- 208000019425 cirrhosis of liver Diseases 0.000 claims description 7
- 208000004930 Fatty Liver Diseases 0.000 claims description 6
- 206010019663 Hepatic failure Diseases 0.000 claims description 6
- 206010019708 Hepatic steatosis Diseases 0.000 claims description 6
- 208000010706 fatty liver disease Diseases 0.000 claims description 6
- 208000006454 hepatitis Diseases 0.000 claims description 6
- 231100000835 liver failure Toxicity 0.000 claims description 6
- 208000007903 liver failure Diseases 0.000 claims description 6
- 208000018191 liver inflammation Diseases 0.000 claims description 6
- 231100000240 steatosis hepatitis Toxicity 0.000 claims description 6
- 230000007882 cirrhosis Effects 0.000 claims description 5
- OCXAGXCMZACNEC-CTWZREHQSA-N (1R,3S,5R)-2-[2-[3-acetyl-5-(2-methylpyrimidin-5-yl)indazol-1-yl]acetyl]-N-(6-bromo-3-methylpyridin-2-yl)-5-methyl-2-azabicyclo[3.1.0]hexane-3-carboxamide Chemical compound C(C)(=O)C1=NN(C2=CC=C(C=C12)C=1C=NC(=NC=1)C)CC(=O)N1[C@@H]2C[C@@]2(C[C@H]1C(=O)NC1=NC(=CC=C1C)Br)C OCXAGXCMZACNEC-CTWZREHQSA-N 0.000 claims description 4
- 208000008069 Geographic Atrophy Diseases 0.000 claims description 4
- 201000006165 Kuhnt-Junius degeneration Diseases 0.000 claims description 4
- 208000022873 Ocular disease Diseases 0.000 claims description 4
- 201000007737 Retinal degeneration Diseases 0.000 claims description 4
- 208000000208 Wet Macular Degeneration Diseases 0.000 claims description 4
- 230000007613 environmental effect Effects 0.000 claims description 4
- 238000002360 preparation method Methods 0.000 claims description 4
- 230000004258 retinal degeneration Effects 0.000 claims description 4
- 201000008171 proliferative glomerulonephritis Diseases 0.000 claims description 3
- 239000008186 active pharmaceutical agent Substances 0.000 claims description 2
- 229940088679 drug related substance Drugs 0.000 claims description 2
- 230000005764 inhibitory process Effects 0.000 abstract description 114
- 230000036470 plasma concentration Effects 0.000 abstract description 74
- 230000003285 pharmacodynamic effect Effects 0.000 abstract description 41
- 238000001727 in vivo Methods 0.000 abstract description 6
- 241000544061 Cuculus canorus Species 0.000 abstract 1
- 239000003112 inhibitor Substances 0.000 description 83
- 210000002966 serum Anatomy 0.000 description 53
- 238000003556 assay Methods 0.000 description 35
- 150000001875 compounds Chemical class 0.000 description 34
- 206010018910 Haemolysis Diseases 0.000 description 32
- 230000008588 hemolysis Effects 0.000 description 31
- 239000000902 placebo Substances 0.000 description 24
- 229940068196 placebo Drugs 0.000 description 24
- 210000004369 blood Anatomy 0.000 description 23
- 239000008280 blood Substances 0.000 description 23
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 22
- 238000004458 analytical method Methods 0.000 description 20
- 230000002949 hemolytic effect Effects 0.000 description 19
- 102000003712 Complement factor B Human genes 0.000 description 18
- 108090000056 Complement factor B Proteins 0.000 description 18
- 239000000203 mixture Substances 0.000 description 14
- 108090000623 proteins and genes Proteins 0.000 description 14
- 208000029574 C3 glomerulopathy Diseases 0.000 description 13
- 208000027134 non-immunoglobulin-mediated membranoproliferative glomerulonephritis Diseases 0.000 description 13
- 235000018102 proteins Nutrition 0.000 description 13
- 102000004169 proteins and genes Human genes 0.000 description 13
- 210000003743 erythrocyte Anatomy 0.000 description 12
- 102100035436 Complement factor D Human genes 0.000 description 11
- 239000013543 active substance Substances 0.000 description 11
- 229940079593 drug Drugs 0.000 description 11
- 238000000338 in vitro Methods 0.000 description 10
- 210000002700 urine Anatomy 0.000 description 10
- 108090000059 Complement factor D Proteins 0.000 description 9
- 230000004913 activation Effects 0.000 description 9
- 230000001174 ascending effect Effects 0.000 description 9
- 239000000872 buffer Substances 0.000 description 9
- 230000005714 functional activity Effects 0.000 description 9
- 239000008194 pharmaceutical composition Substances 0.000 description 9
- 210000004027 cell Anatomy 0.000 description 8
- 231100000673 dose–response relationship Toxicity 0.000 description 8
- 230000001965 increasing effect Effects 0.000 description 8
- 239000007788 liquid Substances 0.000 description 8
- 230000004044 response Effects 0.000 description 8
- RDTRHBCZFDCUPW-KWICJJCGSA-N 2-[(4r,7s,10s,13s,19s,22s,25s,28s,31s,34r)-4-[[(2s,3r)-1-amino-3-hydroxy-1-oxobutan-2-yl]carbamoyl]-34-[[(2s,3s)-2-amino-3-methylpentanoyl]amino]-25-(3-amino-3-oxopropyl)-7-[3-(diaminomethylideneamino)propyl]-10,13-bis(1h-imidazol-5-ylmethyl)-19-(1h-indol Chemical compound C([C@H]1C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CSSC[C@@H](C(N[C@H](C(=O)N[C@H](C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)NCC(=O)N[C@@H](CC=2NC=NC=2)C(=O)N1)C(C)C)C(C)C)=O)NC(=O)[C@@H](N)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(N)=O)C1=CN=CN1 RDTRHBCZFDCUPW-KWICJJCGSA-N 0.000 description 7
- 230000008859 change Effects 0.000 description 7
- 239000000499 gel Substances 0.000 description 7
- 208000027866 inflammatory disease Diseases 0.000 description 7
- 230000003389 potentiating effect Effects 0.000 description 7
- 238000005070 sampling Methods 0.000 description 7
- 108091023037 Aptamer Proteins 0.000 description 6
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 6
- 230000000875 corresponding effect Effects 0.000 description 6
- 230000003247 decreasing effect Effects 0.000 description 6
- 229960002224 eculizumab Drugs 0.000 description 6
- 108090000765 processed proteins & peptides Proteins 0.000 description 6
- 230000009467 reduction Effects 0.000 description 6
- 239000007909 solid dosage form Substances 0.000 description 6
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 5
- 241000283973 Oryctolagus cuniculus Species 0.000 description 5
- 108020004459 Small interfering RNA Proteins 0.000 description 5
- 230000004154 complement system Effects 0.000 description 5
- 230000006378 damage Effects 0.000 description 5
- 230000004064 dysfunction Effects 0.000 description 5
- 208000014674 injury Diseases 0.000 description 5
- 238000012417 linear regression Methods 0.000 description 5
- 239000000546 pharmaceutical excipient Substances 0.000 description 5
- 230000002829 reductive effect Effects 0.000 description 5
- 150000003384 small molecules Chemical class 0.000 description 5
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 5
- 208000035913 Atypical hemolytic uremic syndrome Diseases 0.000 description 4
- 208000023275 Autoimmune disease Diseases 0.000 description 4
- 108010010803 Gelatin Proteins 0.000 description 4
- 208000032759 Hemolytic-Uremic Syndrome Diseases 0.000 description 4
- 102000003855 L-lactate dehydrogenase Human genes 0.000 description 4
- 108700023483 L-lactate dehydrogenases Proteins 0.000 description 4
- 206010063837 Reperfusion injury Diseases 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 208000027418 Wounds and injury Diseases 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 239000002775 capsule Substances 0.000 description 4
- PIBARDGJJAGJAJ-NQIIRXRSSA-N danicopan Chemical compound C(C)(=O)C1=NN(C2=CC=C(C=C12)C=1C=NC(=NC1)C)CC(=O)N1[C@@H](C[C@H](C1)F)C(=O)NC1=NC(=CC=C1)Br PIBARDGJJAGJAJ-NQIIRXRSSA-N 0.000 description 4
- 239000003085 diluting agent Substances 0.000 description 4
- 238000009472 formulation Methods 0.000 description 4
- 239000008273 gelatin Substances 0.000 description 4
- 229920000159 gelatin Polymers 0.000 description 4
- 235000019322 gelatine Nutrition 0.000 description 4
- 235000011852 gelatine desserts Nutrition 0.000 description 4
- 238000001631 haemodialysis Methods 0.000 description 4
- 230000000322 hemodialysis Effects 0.000 description 4
- 230000004054 inflammatory process Effects 0.000 description 4
- 230000028709 inflammatory response Effects 0.000 description 4
- 230000002401 inhibitory effect Effects 0.000 description 4
- 229940007667 lnp023 Drugs 0.000 description 4
- 238000005259 measurement Methods 0.000 description 4
- 206010028417 myasthenia gravis Diseases 0.000 description 4
- 238000011084 recovery Methods 0.000 description 4
- 239000000523 sample Substances 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 230000002459 sustained effect Effects 0.000 description 4
- ZRFXAOXSSXSMRI-UHFFFAOYSA-N 2-ethylpentanamide Chemical compound CCCC(CC)C(N)=O ZRFXAOXSSXSMRI-UHFFFAOYSA-N 0.000 description 3
- 108010003529 Alternative Pathway Complement C3 Convertase Proteins 0.000 description 3
- 102100022133 Complement C3 Human genes 0.000 description 3
- 206010070476 Haemodialysis complication Diseases 0.000 description 3
- 206010061218 Inflammation Diseases 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 208000002223 abdominal aortic aneurysm Diseases 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 3
- 150000001412 amines Chemical class 0.000 description 3
- 208000007474 aortic aneurysm Diseases 0.000 description 3
- GIXWDMTZECRIJT-UHFFFAOYSA-N aurintricarboxylic acid Chemical class C1=CC(=O)C(C(=O)O)=CC1=C(C=1C=C(C(O)=CC=1)C(O)=O)C1=CC=C(O)C(C(O)=O)=C1 GIXWDMTZECRIJT-UHFFFAOYSA-N 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 239000000090 biomarker Substances 0.000 description 3
- 239000011575 calcium Substances 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 229940125782 compound 2 Drugs 0.000 description 3
- 108010027437 compstatin Proteins 0.000 description 3
- 229940121428 danicopan Drugs 0.000 description 3
- 238000009826 distribution Methods 0.000 description 3
- 230000008030 elimination Effects 0.000 description 3
- 238000003379 elimination reaction Methods 0.000 description 3
- 238000011156 evaluation Methods 0.000 description 3
- 239000012634 fragment Substances 0.000 description 3
- 208000007475 hemolytic anemia Diseases 0.000 description 3
- 230000002757 inflammatory effect Effects 0.000 description 3
- 208000012947 ischemia reperfusion injury Diseases 0.000 description 3
- RENRQMCACQEWFC-UGKGYDQZSA-N lnp023 Chemical compound C1([C@H]2N(CC=3C=4C=CNC=4C(C)=CC=3OC)CC[C@@H](C2)OCC)=CC=C(C(O)=O)C=C1 RENRQMCACQEWFC-UGKGYDQZSA-N 0.000 description 3
- 206010025135 lupus erythematosus Diseases 0.000 description 3
- 239000011777 magnesium Substances 0.000 description 3
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 208000015122 neurodegenerative disease Diseases 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 244000052769 pathogen Species 0.000 description 3
- 239000013641 positive control Substances 0.000 description 3
- 230000002269 spontaneous effect Effects 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 231100000419 toxicity Toxicity 0.000 description 3
- 230000001988 toxicity Effects 0.000 description 3
- FGZKEQDXWUHLMD-MOROJQBDSA-N (2s,3s,4r,5r)-n-[6-(2-aminoethylamino)-6-oxohexyl]-5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolane-2-carboxamide Chemical compound O[C@@H]1[C@H](O)[C@@H](C(=O)NCCCCCC(=O)NCCN)O[C@H]1N1C2=NC=NC(N)=C2N=C1 FGZKEQDXWUHLMD-MOROJQBDSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- 206010001052 Acute respiratory distress syndrome Diseases 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- 108010089414 Anaphylatoxins Proteins 0.000 description 2
- FYEHYMARPSSOBO-UHFFFAOYSA-N Aurin Chemical compound C1=CC(O)=CC=C1C(C=1C=CC(O)=CC=1)=C1C=CC(=O)C=C1 FYEHYMARPSSOBO-UHFFFAOYSA-N 0.000 description 2
- 108010077805 Bacterial Proteins Proteins 0.000 description 2
- 238000011357 CAR T-cell therapy Methods 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- 241001060848 Carapidae Species 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 2
- 229940076722 Complement factor D inhibitor Drugs 0.000 description 2
- 102000004127 Cytokines Human genes 0.000 description 2
- 108090000695 Cytokines Proteins 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- 238000002965 ELISA Methods 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- 208000001034 Frostbite Diseases 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 206010018364 Glomerulonephritis Diseases 0.000 description 2
- 208000035895 Guillain-Barré syndrome Diseases 0.000 description 2
- 101000661807 Homo sapiens Suppressor of tumorigenicity 14 protein Proteins 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 108060003951 Immunoglobulin Proteins 0.000 description 2
- 102000000588 Interleukin-2 Human genes 0.000 description 2
- 108010002350 Interleukin-2 Proteins 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 2
- 108090001090 Lectins Proteins 0.000 description 2
- 102000004856 Lectins Human genes 0.000 description 2
- 208000005777 Lupus Nephritis Diseases 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- 206010049567 Miller Fisher syndrome Diseases 0.000 description 2
- 240000003492 Neolamarckia cadamba Species 0.000 description 2
- 206010028980 Neoplasm Diseases 0.000 description 2
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 2
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- 108091008103 RNA aptamers Proteins 0.000 description 2
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 2
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 2
- RAHZWNYVWXNFOC-UHFFFAOYSA-N Sulphur dioxide Chemical compound O=S=O RAHZWNYVWXNFOC-UHFFFAOYSA-N 0.000 description 2
- 102100037942 Suppressor of tumorigenicity 14 protein Human genes 0.000 description 2
- 206010051379 Systemic Inflammatory Response Syndrome Diseases 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 2
- 208000026935 allergic disease Diseases 0.000 description 2
- 230000003321 amplification Effects 0.000 description 2
- 208000007502 anemia Diseases 0.000 description 2
- 230000000692 anti-sense effect Effects 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 210000000601 blood cell Anatomy 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 201000011510 cancer Diseases 0.000 description 2
- 150000001768 cations Chemical class 0.000 description 2
- WMEMLXDTLKSUOD-OGCOPIPOSA-N chembl436844 Chemical compound C([C@H]1C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CSSC[C@@H](C(N[C@H](C(=O)N[C@@H](CC=2C3=CC=CC=C3N(C)C=2)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)NCC(=O)N[C@@H](C)C(=O)N1)C(C)C)=O)NC(=O)[C@@H](NC(C)=O)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(N)=O)C1=CN=CN1 WMEMLXDTLKSUOD-OGCOPIPOSA-N 0.000 description 2
- 208000020832 chronic kidney disease Diseases 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 230000009089 cytolysis Effects 0.000 description 2
- 201000001981 dermatomyositis Diseases 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 238000007876 drug discovery Methods 0.000 description 2
- 201000000523 end stage renal failure Diseases 0.000 description 2
- 108020001507 fusion proteins Proteins 0.000 description 2
- 102000037865 fusion proteins Human genes 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- 230000028993 immune response Effects 0.000 description 2
- 210000000987 immune system Anatomy 0.000 description 2
- 102000018358 immunoglobulin Human genes 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 208000028867 ischemia Diseases 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- 239000002523 lectin Substances 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 150000002739 metals Chemical class 0.000 description 2
- QPJVMBTYPHYUOC-UHFFFAOYSA-N methyl benzoate Chemical compound COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 201000006417 multiple sclerosis Diseases 0.000 description 2
- 201000008383 nephritis Diseases 0.000 description 2
- 230000004770 neurodegeneration Effects 0.000 description 2
- 229910017604 nitric acid Inorganic materials 0.000 description 2
- 238000003199 nucleic acid amplification method Methods 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 230000008506 pathogenesis Effects 0.000 description 2
- 230000001575 pathological effect Effects 0.000 description 2
- 239000008188 pellet Substances 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 231100000654 protein toxin Toxicity 0.000 description 2
- 230000010410 reperfusion Effects 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 229940055944 soliris Drugs 0.000 description 2
- 101150081011 ssl7 gene Proteins 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 230000001629 suppression Effects 0.000 description 2
- 208000011580 syndromic disease Diseases 0.000 description 2
- 229940095064 tartrate Drugs 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 238000012384 transportation and delivery Methods 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- 235000015112 vegetable and seed oil Nutrition 0.000 description 2
- 239000008158 vegetable oil Substances 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- OTGQTQBPQCRNRG-UHFFFAOYSA-N (2-carbamimidoyl-1-benzothiophen-6-yl) thiophene-2-carboxylate Chemical compound C1=C2SC(C(=N)N)=CC2=CC=C1OC(=O)C1=CC=CS1 OTGQTQBPQCRNRG-UHFFFAOYSA-N 0.000 description 1
- JDXCOXKBIGBZSK-PSNKNOTQSA-N (2S)-2-[[(2S)-2-[[(2S)-1-[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S,5S,8S,11S,14S,22S)-22-acetamido-11-benzyl-8-(3-carbamimidamidopropyl)-5-(2-carboxyethyl)-3,6,9,12,16,23-hexaoxo-2-propan-2-yl-1,4,7,10,13,17-hexazacyclotricosane-14-carbonyl]-methylamino]-3-carboxypropanoyl]amino]-3,3-dimethylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-3-(1H-pyrrolo[2,3-b]pyridin-3-yl)propanoyl]amino]-4-carboxybutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]pyrrolidine-2-carbonyl]amino]-2-cyclohexylacetyl]amino]-6-[3-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[[(4S)-4-carboxy-4-(hexadecanoylamino)butanoyl]amino]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]propanoylamino]hexanoic acid Chemical compound CCCCCCCCCCCCCCCC(=O)N[C@@H](CCC(=O)NCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCC(=O)NCCCC[C@H](NC(=O)[C@@H](NC(=O)[C@@H]1CCCN1C(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](Cc1c[nH]c2ncccc12)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)N(C)C(=O)[C@@H]1CC(=O)NCCCC[C@H](NC(C)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](Cc2ccccc2)C(=O)N1)C(C)(C)C)C1CCCCC1)C(O)=O)C(O)=O JDXCOXKBIGBZSK-PSNKNOTQSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- LDMOEFOXLIZJOW-UHFFFAOYSA-N 1-dodecanesulfonic acid Chemical compound CCCCCCCCCCCCS(O)(=O)=O LDMOEFOXLIZJOW-UHFFFAOYSA-N 0.000 description 1
- HCSBTDBGTNZOAB-UHFFFAOYSA-N 2,3-dinitrobenzoic acid Chemical compound OC(=O)C1=CC=CC([N+]([O-])=O)=C1[N+]([O-])=O HCSBTDBGTNZOAB-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- MGWGWNFMUOTEHG-UHFFFAOYSA-N 4-(3,5-dimethylphenyl)-1,3-thiazol-2-amine Chemical compound CC1=CC(C)=CC(C=2N=C(N)SC=2)=C1 MGWGWNFMUOTEHG-UHFFFAOYSA-N 0.000 description 1
- 241000251468 Actinopterygii Species 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- 208000003343 Antiphospholipid Syndrome Diseases 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- 241000271566 Aves Species 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 208000019838 Blood disease Diseases 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 206010006482 Bronchospasm Diseases 0.000 description 1
- RENRQMCACQEWFC-BPARTEKVSA-N CCOC1C[C@@H](C(C=C2)=CC=C2C(O)=O)N(CC(C2=C(C(C)=C3)NC=C2)=C3OC)CC1 Chemical group CCOC1C[C@@H](C(C=C2)=CC=C2C(O)=O)N(CC(C2=C(C(C)=C3)NC=C2)=C3OC)CC1 RENRQMCACQEWFC-BPARTEKVSA-N 0.000 description 1
- 101100268548 Caenorhabditis elegans apl-1 gene Proteins 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 201000005488 Capillary Leak Syndrome Diseases 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 1
- 208000018380 Chemical injury Diseases 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 description 1
- 206010053567 Coagulopathies Diseases 0.000 description 1
- 108010028780 Complement C3 Proteins 0.000 description 1
- 108010067641 Complement C3-C5 Convertases Proteins 0.000 description 1
- 102000016574 Complement C3-C5 Convertases Human genes 0.000 description 1
- 108010034753 Complement Membrane Attack Complex Proteins 0.000 description 1
- 229940122127 Complement factor B inhibitor Drugs 0.000 description 1
- 229940124073 Complement inhibitor Drugs 0.000 description 1
- 208000011231 Crohn disease Diseases 0.000 description 1
- 108010069514 Cyclic Peptides Proteins 0.000 description 1
- 102000001189 Cyclic Peptides Human genes 0.000 description 1
- 201000003883 Cystic fibrosis Diseases 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- RWSOTUBLDIXVET-UHFFFAOYSA-N Dihydrogen sulfide Chemical compound S RWSOTUBLDIXVET-UHFFFAOYSA-N 0.000 description 1
- 208000000059 Dyspnea Diseases 0.000 description 1
- 206010013975 Dyspnoeas Diseases 0.000 description 1
- 238000012286 ELISA Assay Methods 0.000 description 1
- 238000008157 ELISA kit Methods 0.000 description 1
- 206010014561 Emphysema Diseases 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 206010016654 Fibrosis Diseases 0.000 description 1
- 208000024869 Goodpasture syndrome Diseases 0.000 description 1
- 102000001554 Hemoglobins Human genes 0.000 description 1
- 108010054147 Hemoglobins Proteins 0.000 description 1
- 208000000616 Hemoptysis Diseases 0.000 description 1
- 206010019860 Hereditary angioedema Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 1
- 206010022822 Intravascular haemolysis Diseases 0.000 description 1
- 102000001399 Kallikrein Human genes 0.000 description 1
- 108060005987 Kallikrein Proteins 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- 244000178870 Lavandula angustifolia Species 0.000 description 1
- 235000010663 Lavandula angustifolia Nutrition 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 102000018697 Membrane Proteins Human genes 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- UEZVMMHDMIWARA-UHFFFAOYSA-N Metaphosphoric acid Chemical compound OP(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-N 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- 208000009525 Myocarditis Diseases 0.000 description 1
- GXCLVBGFBYZDAG-UHFFFAOYSA-N N-[2-(1H-indol-3-yl)ethyl]-N-methylprop-2-en-1-amine Chemical compound CN(CCC1=CNC2=C1C=CC=C2)CC=C GXCLVBGFBYZDAG-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 208000012902 Nervous system disease Diseases 0.000 description 1
- 208000025966 Neurological disease Diseases 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 208000008589 Obesity Diseases 0.000 description 1
- 241000238890 Ornithodoros moubata Species 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 208000030852 Parasitic disease Diseases 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 206010057249 Phagocytosis Diseases 0.000 description 1
- YGYAWVDWMABLBF-UHFFFAOYSA-N Phosgene Chemical compound ClC(Cl)=O YGYAWVDWMABLBF-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- 206010035664 Pneumonia Diseases 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- 206010037211 Psychomotor hyperactivity Diseases 0.000 description 1
- 208000006193 Pulmonary infarction Diseases 0.000 description 1
- 206010037457 Pulmonary vasculitis Diseases 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 206010062237 Renal impairment Diseases 0.000 description 1
- 206010063897 Renal ischaemia Diseases 0.000 description 1
- 208000013616 Respiratory Distress Syndrome Diseases 0.000 description 1
- 206010040047 Sepsis Diseases 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 208000006011 Stroke Diseases 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-N Sulfurous acid Chemical compound OS(O)=O LSNNMFCWUKXFEE-UHFFFAOYSA-N 0.000 description 1
- 208000031932 Systemic capillary leak syndrome Diseases 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 206010052779 Transplant rejections Diseases 0.000 description 1
- 208000030886 Traumatic Brain injury Diseases 0.000 description 1
- 206010047112 Vasculitides Diseases 0.000 description 1
- 206010047115 Vasculitis Diseases 0.000 description 1
- SRXKIZXIRHMPFW-UHFFFAOYSA-N [4-[6-[amino(azaniumylidene)methyl]naphthalen-2-yl]oxycarbonylphenyl]-(diaminomethylidene)azanium;methanesulfonate Chemical group CS([O-])(=O)=O.CS([O-])(=O)=O.C1=CC(N=C([NH3+])N)=CC=C1C(=O)OC1=CC=C(C=C(C=C2)C([NH3+])=N)C2=C1 SRXKIZXIRHMPFW-UHFFFAOYSA-N 0.000 description 1
- 230000001594 aberrant effect Effects 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 229940022663 acetate Drugs 0.000 description 1
- IPBVNPXQWQGGJP-UHFFFAOYSA-N acetic acid phenyl ester Natural products CC(=O)OC1=CC=CC=C1 IPBVNPXQWQGGJP-UHFFFAOYSA-N 0.000 description 1
- 208000015228 acquired partial lipodystrophy Diseases 0.000 description 1
- 210000005006 adaptive immune system Anatomy 0.000 description 1
- 208000011341 adult acute respiratory distress syndrome Diseases 0.000 description 1
- 201000000028 adult respiratory distress syndrome Diseases 0.000 description 1
- 230000004520 agglutination Effects 0.000 description 1
- 230000004931 aggregating effect Effects 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910000272 alkali metal oxide Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- AEMOLEFTQBMNLQ-BKBMJHBISA-N alpha-D-galacturonic acid Chemical compound O[C@H]1O[C@H](C(O)=O)[C@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-BKBMJHBISA-N 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 238000002399 angioplasty Methods 0.000 description 1
- 235000021120 animal protein Nutrition 0.000 description 1
- 230000003171 anti-complementary effect Effects 0.000 description 1
- 239000003146 anticoagulant agent Substances 0.000 description 1
- 229940127219 anticoagulant drug Drugs 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 159000000032 aromatic acids Chemical class 0.000 description 1
- 206010003246 arthritis Diseases 0.000 description 1
- 239000010425 asbestos Substances 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 201000004988 autoimmune vasculitis Diseases 0.000 description 1
- 229950006867 avacincaptad pegol Drugs 0.000 description 1
- 229940000201 avapro Drugs 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 210000002469 basement membrane Anatomy 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 229910052790 beryllium Inorganic materials 0.000 description 1
- ATBAMAFKBVZNFJ-UHFFFAOYSA-N beryllium atom Chemical compound [Be] ATBAMAFKBVZNFJ-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 239000012620 biological material Substances 0.000 description 1
- 230000007321 biological mechanism Effects 0.000 description 1
- 229960000106 biosimilars Drugs 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- SXDBWCPKPHAZSM-UHFFFAOYSA-N bromic acid Chemical compound OBr(=O)=O SXDBWCPKPHAZSM-UHFFFAOYSA-N 0.000 description 1
- 230000007885 bronchoconstriction Effects 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- FATUQANACHZLRT-KMRXSBRUSA-L calcium glucoheptonate Chemical compound [Ca+2].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O FATUQANACHZLRT-KMRXSBRUSA-L 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- 230000002612 cardiopulmonary effect Effects 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000006037 cell lysis Effects 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 208000015114 central nervous system disease Diseases 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- ZAIPMKNFIOOWCQ-UEKVPHQBSA-N cephalexin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@@H]3N(C2=O)C(=C(CS3)C)C(O)=O)=CC=CC=C1 ZAIPMKNFIOOWCQ-UEKVPHQBSA-N 0.000 description 1
- 238000006243 chemical reaction Methods 0.000 description 1
- 230000035605 chemotaxis Effects 0.000 description 1
- XTEGARKTQYYJKE-UHFFFAOYSA-N chloric acid Chemical compound OCl(=O)=O XTEGARKTQYYJKE-UHFFFAOYSA-N 0.000 description 1
- 229940005991 chloric acid Drugs 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- KVSASDOGYIBWTA-UHFFFAOYSA-N chloro benzoate Chemical compound ClOC(=O)C1=CC=CC=C1 KVSASDOGYIBWTA-UHFFFAOYSA-N 0.000 description 1
- VDANGULDQQJODZ-UHFFFAOYSA-N chloroprocaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1Cl VDANGULDQQJODZ-UHFFFAOYSA-N 0.000 description 1
- 229960002023 chloroprocaine Drugs 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 230000035602 clotting Effects 0.000 description 1
- 239000002817 coal dust Substances 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 239000004074 complement inhibitor Substances 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 230000002074 deregulated effect Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 150000001991 dicarboxylic acids Chemical class 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 229940043237 diethanolamine Drugs 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- FPAFDBFIGPHWGO-UHFFFAOYSA-N dioxosilane;oxomagnesium;hydrate Chemical compound O.[Mg]=O.[Mg]=O.[Mg]=O.O=[Si]=O.O=[Si]=O.O=[Si]=O.O=[Si]=O FPAFDBFIGPHWGO-UHFFFAOYSA-N 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-N diphosphoric acid Chemical compound OP(O)(=O)OP(O)(O)=O XPPKVPWEQAFLFU-UHFFFAOYSA-N 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- VFNGKCDDZUSWLR-UHFFFAOYSA-N disulfuric acid Chemical compound OS(=O)(=O)OS(O)(=O)=O VFNGKCDDZUSWLR-UHFFFAOYSA-N 0.000 description 1
- POULHZVOKOAJMA-UHFFFAOYSA-M dodecanoate Chemical compound CCCCCCCCCCCC([O-])=O POULHZVOKOAJMA-UHFFFAOYSA-M 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 239000000428 dust Substances 0.000 description 1
- 238000010410 dusting Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 208000028208 end stage renal disease Diseases 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 206010015037 epilepsy Diseases 0.000 description 1
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 1
- 229940012017 ethylenediamine Drugs 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 201000001155 extrinsic allergic alveolitis Diseases 0.000 description 1
- 231100000562 fetal loss Toxicity 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 238000002825 functional assay Methods 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 208000035474 group of disease Diseases 0.000 description 1
- 208000019622 heart disease Diseases 0.000 description 1
- 208000014951 hematologic disease Diseases 0.000 description 1
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 1
- 208000018706 hematopoietic system disease Diseases 0.000 description 1
- 208000006750 hematuria Diseases 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 229910000037 hydrogen sulfide Inorganic materials 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 208000022098 hypersensitivity pneumonitis Diseases 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 210000005007 innate immune system Anatomy 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- ICIWUVCWSCSTAQ-UHFFFAOYSA-N iodic acid Chemical class OI(=O)=O ICIWUVCWSCSTAQ-UHFFFAOYSA-N 0.000 description 1
- 230000000622 irritating effect Effects 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 230000000155 isotopic effect Effects 0.000 description 1
- 238000011862 kidney biopsy Methods 0.000 description 1
- 230000005977 kidney dysfunction Effects 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-N lactobionic acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-N 0.000 description 1
- 229940070765 laurate Drugs 0.000 description 1
- 239000001102 lavandula vera Substances 0.000 description 1
- 235000018219 lavender Nutrition 0.000 description 1
- 230000031700 light absorption Effects 0.000 description 1
- 238000012886 linear function Methods 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 230000005923 long-lasting effect Effects 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- 230000002934 lysing effect Effects 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- 235000012054 meals Nutrition 0.000 description 1
- 210000004379 membrane Anatomy 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 210000003975 mesenteric artery Anatomy 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 229940095102 methyl benzoate Drugs 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 230000003232 mucoadhesive effect Effects 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- ACTNHJDHMQSOGL-UHFFFAOYSA-N n',n'-dibenzylethane-1,2-diamine Chemical compound C=1C=CC=CC=1CN(CCN)CC1=CC=CC=C1 ACTNHJDHMQSOGL-UHFFFAOYSA-N 0.000 description 1
- 229950009865 nafamostat Drugs 0.000 description 1
- 125000005609 naphthenate group Chemical group 0.000 description 1
- 210000005036 nerve Anatomy 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- JCXJVPUVTGWSNB-UHFFFAOYSA-N nitrogen dioxide Inorganic materials O=[N]=O JCXJVPUVTGWSNB-UHFFFAOYSA-N 0.000 description 1
- 235000020824 obesity Nutrition 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-M octanoate Chemical compound CCCCCCCC([O-])=O WWZKQHOCKIZLMA-UHFFFAOYSA-M 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 230000014207 opsonization Effects 0.000 description 1
- 229940126701 oral medication Drugs 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 229940039748 oxalate Drugs 0.000 description 1
- 208000036274 partial acquired susceptibility to lipodystrophy Diseases 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 239000013610 patient sample Substances 0.000 description 1
- 210000001539 phagocyte Anatomy 0.000 description 1
- 230000008782 phagocytosis Effects 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 229940049953 phenylacetate Drugs 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 208000011511 primary membranoproliferative glomerulonephritis Diseases 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 201000001474 proteinuria Diseases 0.000 description 1
- 201000006301 pulmonary embolism and infarction Diseases 0.000 description 1
- 208000005069 pulmonary fibrosis Diseases 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 229940005657 pyrophosphoric acid Drugs 0.000 description 1
- 238000004445 quantitative analysis Methods 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 229950007085 ravulizumab Drugs 0.000 description 1
- 230000007115 recruitment Effects 0.000 description 1
- 239000013074 reference sample Substances 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 238000000611 regression analysis Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 229910052895 riebeckite Inorganic materials 0.000 description 1
- 201000000980 schizophrenia Diseases 0.000 description 1
- 229940116351 sebacate Drugs 0.000 description 1
- CXMXRPHRNRROMY-UHFFFAOYSA-L sebacate(2-) Chemical compound [O-]C(=O)CCCCCCCCC([O-])=O CXMXRPHRNRROMY-UHFFFAOYSA-L 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000000779 smoke Substances 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- TYFQFVWCELRYAO-UHFFFAOYSA-L suberate(2-) Chemical compound [O-]C(=O)CCCCCCC([O-])=O TYFQFVWCELRYAO-UHFFFAOYSA-L 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 229950009054 tesidolumab Drugs 0.000 description 1
- CBXCPBUEXACCNR-UHFFFAOYSA-N tetraethylammonium Chemical compound CC[N+](CC)(CC)CC CBXCPBUEXACCNR-UHFFFAOYSA-N 0.000 description 1
- QEMXHQIAXOOASZ-UHFFFAOYSA-N tetramethylammonium Chemical compound C[N+](C)(C)C QEMXHQIAXOOASZ-UHFFFAOYSA-N 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 238000011287 therapeutic dose Methods 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 231100000155 toxicity by organ Toxicity 0.000 description 1
- 230000007675 toxicity by organ Effects 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 230000008733 trauma Effects 0.000 description 1
- 230000009529 traumatic brain injury Effects 0.000 description 1
- 229940070710 valerate Drugs 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
Images























Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
Landscapes
- Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Nutrition Science (AREA)
- Physiology (AREA)
- Hematology (AREA)
- Diabetes (AREA)
- Gastroenterology & Hepatology (AREA)
- Ophthalmology & Optometry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
本文所述的剂量和方法提供了抑制旁路途径补体活性的所需药代动力学(PK)和药效学(PD)特征,例如,在约120 mg至200 mg BID的剂量下对体内AP活性的抑制达至少85%或更高,其提供了至少约65 ng/ml的血浆水平C谷,这提供了足够的AP抑制以降低补体突破的风险。另外,本文所述的剂型提供了显著低的Cmax,从而提供了额外的安全裕度和更好的给药灵活性。
Description
相关申请的交叉引用
本申请要求2018年12月17日提交的美国临时申请第62/780,573号和2019年7月22日提交的美国临时申请第62/877,193号的权益。这些申请通过引用以其整体并入。
技术领域
本发明属于施用小分子补体因子D (fD)抑制剂- (1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺的有利剂型和方法的领域-根据人临床试验,所述剂型和方法在人中提供了用于治疗补体介导的病症的特定的药代动力学和药效学特性。一方面,公开了提供所需药代动力学和药效学特征(包括C谷)的剂型。
背景技术
当免疫系统不能以正常方式运行时,就会发生免疫病症。炎症是保护性响应,其通常涉及免疫细胞、免疫系统、血管和分子介质。多种医学病症是由有害的免疫或炎性响应引起的,或者细胞无法对正常的免疫或炎性过程做出响应。
补体系统是先天免疫系统的一部分,其不适应宿主生命过程中的变化,但被适应性免疫系统募集和使用。例如,其有助于或补充抗体和吞噬细胞清除病原体的能力。这种复杂的调节途径允许对病原生物体快速响应,同时保护宿主细胞免受破坏。超过三十种蛋白质和蛋白质片段构成了补体系统。这些蛋白质通过调理作用(增强抗原的吞噬作用)、趋化作用(吸引巨噬细胞和中性粒细胞)、细胞裂解(破裂外源细胞膜)和凝集反应(使病原体聚集和结合在一起)起作用。
补体系统具有三种途径:经典途径、旁路途径和凝集素途径。补体因子D在补体级联的旁路途径的激活中起早期和核心作用。旁路补体途径的激活是由C3中硫酯键的自发水解产生C3(H2O)引发的,所述C3(H2O)与因子B缔合形成C3(H2O)B复合物。补体因子D用于裂解C3(H2O)B复合物中的因子B,形成Ba和Bb。Bb片段仍然与C3(H2O)缔合以形成旁路途径C3转化酶C3(H2O)Bb。另外,由任何C3转化酶产生的C3b也与因子B缔合形成C3bB,该因子D将其裂解以产生后期旁路途径C3转化酶C3bBb。旁路途径C3转化酶的该后一种形式可以在所有三个定义的补体途径中提供重要的下游放大,最终导致补体级联途径中其他因子的募集和组装,包括C5裂解为C5a和C5b。C5b在将因子C6、C7、C8和C9组装成膜攻击复合物中起作用,所述膜攻击复合物可通过裂解细胞来破坏致病细胞。
补体的功能障碍或过度激活与某些自身免疫性疾病、炎性疾病和神经变性疾病以及缺血再灌注损伤和癌症相关。例如,补体级联旁路途径的激活有助于C3a和C5a的产生,这两者都是强效过敏毒素,它们也在许多炎性病症中起作用。因此,在一些情况下,需要降低补体途径(包括旁路补体途径)的响应。
阵发性睡眠性血红蛋白尿症(Paroxysmal nocturnal hemoglobinuria) (PNH)是非恶性血液病症,其特征在于造血干细胞和后代成熟血细胞的放大,所述造血干细胞和后代成熟血细胞缺乏一些表面蛋白。PNH红细胞不能调节其表面补体激活,这产生了PNH的典型标志—补体介导的血管内贫血的慢性激活。目前,只有一种产品,即抗C5单克隆抗体依库珠单抗,在美国获批用于治疗PNH。然而,许多接受依库珠单抗治疗的患者仍然贫血,并且许多患者继续需要输血。另外,依库珠单抗治疗需要终生静脉内注射。因此,对开发补体途径的新型抑制剂存在未满足的需求。
与补体级联相关的其他病症包括但不限于非典型溶血性尿毒症综合征(aHUS)、溶血性尿毒症综合征(HUS)、C3肾小球病(C3G)或C3肾小球肾炎(C3GN)、腹主动脉瘤、血液透析并发症、溶血性贫血或血液透析、神经脊髓炎(NMO)、重症肌无力(MG)、脂肪肝、非酒精性脂肪性肝炎(NASH)、肝脏炎症、肝硬化、肝衰竭、皮肌炎、肌萎缩侧索硬化、年龄相关性黄斑变性(AMD)、多发性硬化症、类风湿性关节炎以及对生物治疗剂(例如CAR T细胞疗法)做出响应的细胞因子或炎性响应。
由于因子D在补体旁路途径中的早期和重要作用以及其在经典和凝集素补体途径中的信号放大中的潜在作用,因此其是抑制或调节补体级联的有吸引力的靶标。因子D的抑制有效地中断了途径并削弱了膜攻击复合物的形成。虽然已经进行了开发因子D的抑制剂的初步尝试,但目前还没有临床批准的小分子因子D抑制剂。因子D抑制剂化合物的实例描述于以下公开内容中。
名称为“Indole compounds or analogues thereof useful for the treatmentof age-related macular degeneration”的Novartis PCT专利公布WO2012/093101描述了某些因子D抑制剂。其他因子D抑制剂描述于Novartis PCT专利公布:WO2012093101、WO2013/164802、WO2013/192345、WO2014/002051、WO2014/002052、WO2014/002053、WO2014/002054、WO2014/002057、WO2014/002058、WO2014/002059、WO2014/005150、WO2014/009833、WO2014/143638、WO2015/009616、WO2015/009977、WO2015/066241和WO2016088082中。
其他补体因子D抑制剂描述于Achillion Pharmaceuticals, Inc拥有的专利提交:美国专利第9,598,446号;第9,643,986号;第9,663,543号;第9,695,205号;第9,732,103号;第9,732,104号;第9,758,537号;第9,796,741号;第9,828,396号;第10,000,516号;第10,005,802号;第10,011,612号;第10,081,645号;第10,087,203号;第10,092,584号;第10,100,072号;第10,138,225号;第10,189,869号;第10,106,563号;第10,301,336号和第10,287,301号;国际公布第WO2019/028284号;第WO2018/160889号;第WO2018/160891号;第WO2018/160892号;第WO2017/035348号;第WO2017/035349号;第WO 2017/035351号;第WO2017/035352号;第WO 2017/035353号;第WO 2017/035355号;第WO2017/035357号;第WO2017/035360号;第WO2017/035361号;第WO2017//035362号;第WO2017/035415号;第WO2017/035401号;第WO2017/035405号;第WO2017/035413号;第WO2017/035409号;第WO2017/035411号;第WO2017/035417号;第WO2017/035408号;第WO2015/130784号;第WO2015/130795号;第WO2015/130806号;第WO2015/130830号;第WO2015/130838号;第WO2015/130842号;第WO2015/130845号和第WO2015/130854号以及美国专利公布第US2016-0361329号;第US 2016-0362432号;第US 2016-0362433号;第US 2016-0362399号;第US 2017-0056428号;第US 2017-0057950号;第US 2017-0057993号;第US 2017-0189410号;第US 2017-0226142号;第US 2017-0260219号;第US 2017-0298084号;第US 2017-0298085号;第US 2018-0022766号;第US 2018-0022767号;第US 2018-0072762号;第US2018-0030075号;第US 2018-0169109号;第US 2018-0177761号;第US 2018-0179185号;第US 2018-0179186号;第US 2018-0179236号;第US 2018-0186782号;第US 2018-0201580号;第US 2019-0031692号;第US 2019-0048033号;第US 2019-0144473号和第US 2019-0211033号中。
名称为“Therapeutic Inhibitory Compounds”的Lifesci Pharmaceuticals PCT专利公布WO2017/098328描述了在中心核心杂环中具有变化的各种因子D抑制剂。PCT专利公布WO2018/015818也称为“Therapeutic Inhibitory Compounds”,描述了不具有环状中央核心的因子D抑制剂。
名称为“Compounds useful in the complement, coagulation and kallikreinpathways and method for their preparation”的Biocryst Pharmaceuticals美国专利第6653340号描述了作为因子D的抑制剂的稠合双环化合物。由于因子D的抑制剂BCX1470缺乏特异性和半衰期短,因此停止了其开发。
名称为“Methods and compositions for the treatment ofglomerulonephritis and other inflammatory diseases”的Alexion PharmaceuticalsPCT专利公布WO1995/029697公开了针对补体途径的C5的抗体,其用于治疗肾小球肾炎和涉及补体系统的病理性激活的炎性疾患。Alexion Pharmaceutical的抗C5抗体依库珠单抗(Soliris®)和雷武珠单抗(ravulizumab)-cwvz (Ultomiris®)是目前市场上唯一的补体特异性抗体,也是唯一获批的阵发性睡眠性血红蛋白尿症(PNH)的治疗。
与补体介导的病症治疗相关的一个特殊困难是给药间隔期间抗补体活性的持续时间,以及在接受下一个疗程之前发生突破性溶血(breakthrough hemolysis)的可能性。例如,超过17天的依库珠单抗给药间隔可能与PNH患者发生突破性溶血的更大风险相关联。Nakayama 等人,Bio. Pharm. Bull. 2018; 39(2), 285-288。
鉴于由有害的免疫或炎性响应引起的多种医学病症,本发明的目的是提供治疗患有补体介导的病症的患者的剂量和方法,所述剂量和方法提供用于抑制旁路补体的所需药代动力学和药效学特征,所述药代动力学和药效学特征在治疗剂量的施用之间是持久的。
发明内容
本发明属于施用小分子补体因子D (fD)抑制剂- (1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1;参见下面的结构)的有利口服剂型和方法的领域,根据人临床试验,所述口服剂型和方法提供了随时间推移保持药物的有效性,同时最大限度地减小副作用的药代动力学C谷。C谷定义为给药间隔结束时的平均血浆浓度或最低平均血浆浓度。剂量间隔定义为刚好在重复剂量之前的点。例如,如果剂量是一天两次(BID),则剂量间隔是当天服用第一剂量与服用第二剂量之间的时间段,或第二剂量与第二天第一剂之间的时间段。

化合物1
本发明之所以重要是因为补体级联的临界性和敏感性赋予其攻击和破坏其发觉是外来的、患病的或受感染的细胞的作用。如果剂量太高,可能会引起不希望的副作用,诸如抑制抗感染的能力或引起器官毒性。如果剂量太低,药物不能有效抵消旁路补体途径系统的过度活动或功能失调性活性。
令人惊讶地发现,化合物1在人中表现出昼夜代谢模式,这意味着其在白天的代谢明显高于夜间。这在给人施用药物之前是无法预测的。例如,表6提供了四种剂量(40 mgBID、80 mg BID、120 mg BID和200 mg BID,持续14天)下的C(0)和C(12)的平均值(ng/mL)。对于四种剂量方案中的每一种,早晨测量的C(0)明显高于晚上测量的C(12),事实上几乎是晚上测量的两倍。令人惊讶且出乎意料的是,这与结构非常相似的补体因子D抑制剂(化合物2(下面),(2S,4R)-1-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴吡啶-2-基)-4-氟吡咯烷-2-甲酰胺)形成对比,所述化合物2在向人施用时不表现出昼夜代谢模式。

化合物2
已发现,在每日两次约100 mg至200 mg,更具体地每日两次约120 mg至180 mg,甚至更具体地每日两次约120 mg至150 mg,或每日两次约150 mg至约200 mg,以及在一个实施方案中每日两次约120 mg的化合物1的口服剂量下,在剂量间隔结束时提供最佳最低平均血浆浓度(C谷),使得剂量在一天中任何时间不降低至低于其EC90有效性。具体而言,发现了可将两个昼夜最低平均血浆浓度(C谷)中的较低者维持在既定EC90之上的给药方案。如下文进一步描述的建模建立了介于67 ng/mL至88 ng/mL之间的EC90,将提供约90%的AP活性抑制。在离体旁路途径活性测定中从健康受试者血清的多递增给药研究(MAD;下文进一步描述)得出的数据确定,在约为80 ng/mL的最低平均血浆浓度(C谷)下可实现约90%的AP活性抑制(参见表6和表7);在约为150 ng/mL的最低平均血浆浓度(C谷)下可实现高于95%的AP活性抑制(参见表6和表7)。令人惊讶的是,约为80 ng/mL的最低平均血浆浓度(C谷)可在120 mgBID给药方案的昼夜周期中,在较低的C谷水平下达到。
因此,作为下文详细描述的人临床试验的结果,令人惊讶地确定了口服给药方案,所述口服给药方案可以指导补体因子D (fD)抑制剂化合物1或其药学上可接受的盐的安全且有效的长期施用,所述化合物1或其药学上可接受的盐可用于治疗受试者,所述受试者患有补体功能障碍或过度激活,例如但不限于阵发性睡眠性血红蛋白尿症(PNH)、C3肾小球病(C3G)诸如致密沉积物病(DDD)和C3肾小球肾炎(C3GN),以及免疫复合物膜性增生性肾小球肾炎(IC-MPGN)。本文所述的剂量和方法提供了抑制旁路途径补体活性的所需药代动力学(PK)和药效学(PD)特征,例如,在120 mg BID至200 mg BID的剂量下,体内对AP活性的抑制至少为85%、至少为90%、至少为95%或更多,所述剂量提供了约65 ng/mL至450 ng/mL的最低平均血浆浓度(C谷),该浓度高至足够避免补体突破(complement breakthrough)。
在一些实施方案中,提供的剂量提供了约50 ng/mL至450 ng/mL的最小平均血浆浓度(C谷)。在一些实施方案中,提供的剂量提供了约75 ng/mL至160 ng/mL的最小平均血浆浓度(C谷)。在一些实施方案中,提供的剂量提供了约80 ng/mL至150 ng/mL的最小平均血浆浓度(C谷)。
在一些实施方案中,提供的剂量在BID给药方案的昼夜周期中,在较低的C谷水平下提供至少约65 ng/mL的最低平均血浆浓度(C谷)。在一些实施方案中,提供的剂量在BID给药方案的昼夜周期中,在较低的C谷水平下提供至少约80 ng/mL的最低平均血浆浓度(C谷)。在一些实施方案中,提供的剂量在BID给药方案的昼夜周期中,在较低的C谷水平下提供至少约90 ng/mL的最低平均血浆浓度(C谷)。在一些实施方案中,提供的剂量在BID给药方案的昼夜周期中,在较低的C谷水平下提供至少约100 ng/mL的最低平均血浆浓度(C谷)。在一些实施方案中,提供的剂量在BID给药方案的昼夜周期中,在较低的C谷水平下提供至少约125 ng/mL的最低平均血浆浓度(C谷)。在一些实施方案中,提供的剂量在BID给药方案的昼夜周期中,在较低的C谷水平下提供至少约150 ng/mL的最低平均血浆浓度(C谷)。在一些实施方案中,提供的剂量在BID给药方案的昼夜周期中,在较低的C谷水平下提供约65 ng/mL至150 ng/mL的最低平均血浆浓度(C谷)。
此外,当以120 mg BID和更高剂量(例如,200 mg BID)给药时,化合物1实现了接近完全和持续的旁路途径(AP)抑制,其中如通过AP溶血和AP Wieslab测定所测量的,在120mg BID时在最低平均血浆浓度(C谷)下的平均值>90%,在200 mg BID时约>95%(见表7)。在化合物1暴露相对较低的情况下有效地实现了C谷,其中对于120 mg BID观察到小于约1000ng/mL的Cmax,对于200 mg BID观察到小于2000 ng/mL的Cmax。在给药之间维持对AP的抑制的能力允许有利的给药方案,所述给药方案对患者来说是安全和方便的,降低了与无效C谷相关的突破性溶血风险和与过度Cmax相关的毒性。
一方面,对于补体介导的病症的治疗,以在治疗期间提供约50 ng/ml至200 ng/ml的的最低平均血浆浓度(C谷)的口服剂型和施用方案提供化合物1。在一些实施方案中,以在治疗期间提供约70 ng/mL至170 ng/mL的最低平均血浆浓度(C谷)的剂型和施用方案提供化合物1。在一些实施方案中,以在治疗期间提供约75 ng/mL至160 ng/mL的最低平均血浆浓度(C谷)的剂型和施用方案提供化合物1。在一些实施方案中,C谷至少约为50 ng/ml、至少约为60 ng/mL、至少约为65 ng/mL、至少约为70 ng/mL、至少约为75 ng/mL、至少约为80 ng/mL、至少约为85 ng/mL、至少约为90 ng/mL、至少约为95 ng/mL、至少约为100 ng/mL、至少约为105 ng/mL、至少约为110 ng/mL、至少约为115 ng/mL、至少约为120 ng/mL、至少约为125 ng/mL、至少约为130 ng/mL、至少约为135 ng/mL、至少约为140 ng/mL、至少约为145ng/mL或至少约为150 ng/mL。在一个实施方案中,C谷至少约为100 ng/mL。在一些实施方案中,C谷小于约170 ng/ml、小于约150 ng/ml、小于约125 ng/mL、小于约115 ng/mL、小于约110 ng/mL、小于约105 ng/mL、小于约100 ng/mL、小于约95 ng/mL或小于约90 ng/mL。已经发现,保持约为50 ng/mL的C谷浓度在该浓度下提供了85%的AP抑制,而约为67-88 ng/mL的C谷提供了大于90%的AP抑制。在一些实施方案中,以在治疗过程中提供约50 ng/mL至约200ng/mL的C谷浓度和小于约1000 ng/ml的最大平均血浆浓度(Cmax)的剂型和施用方案提供化合物1。在一些实施方案中,以在治疗过程中提供约70 ng/mL至约170 ng/mL的C谷浓度和小于约1000 ng/ml的最大平均血浆浓度(Cmax)的剂型和施用方案提供化合物1。在一些实施方案中,以在治疗过程中提供约100 ng/mL的C谷浓度和小于约1000 ng/ml的Cmax的剂型和施用方案提供化合物1。在一些实施方案中,以在治疗过程中提供约125 ng/mL的C谷浓度和小于约1000 ng/ml的最大平均血浆浓度(Cmax)的剂型和施用方案提供化合物1。在一些实施方案中,以在治疗过程中提供约150 ng/mL的C谷浓度和小于约1000 ng/ml的最大平均血浆浓度(Cmax)的剂型和施用方案提供化合物1。在一些实施方案中,以在治疗过程中提供约175 ng/mL的C谷浓度和小于约1000 ng/ml的最大平均血浆浓度(Cmax)的剂型和施用方案提供化合物1。在一个实施方案中,以120mg BID的剂型施用化合物1。在一个实施方案中,补体介导的病症是PNH。在一个实施方案中,补体介导的病症是3CG或IC-MPGN。
一方面,对于补体介导的病症的治疗,以在治疗期间提供约225 ng/ml至450 ng/ml的的最低平均血浆浓度(C谷)的口服剂型和施用方案提供化合物1。在一些实施方案中,以在治疗期间提供约240 ng/mL至400 ng/mL的最低平均血浆浓度(C谷)的剂型和施用方案提供化合物1。在一些实施方案中,C谷至少约为225 ng/ml、至少约250 ng/mL、至少约275 ng/mL、至少约300 ng/mL、至少约325 ng/mL、至少约350 ng/mL、至少约375 ng/mL、至少约400ng/mL、至少约425 ng/mL或至少约450 ng/mL。在一个实施方案中,C谷至少约为300 ng/mL。在一些实施方案中,C谷小于约450 ng/ml、小于约425 ng/ml、小于约400 ng/mL、小于约375ng/mL、小于约350 ng/mL、小于约325 ng/mL、小于约300 ng/mL、小于约275 ng/mL或小于约250 ng/mL。在一些实施方案中,以在治疗过程中提供约225 ng/mL至约450 ng/mL的C谷浓度和小于约2000 ng/ml的最大平均血浆浓度(Cmax)的剂型和施用方案提供化合物1。在一些实施方案中,以在治疗过程中提供约240 ng/mL至约400 ng/mL的C谷浓度和小于约2000 ng/ml的最大平均血浆浓度(Cmax)的剂型和施用方案提供化合物1。在一些实施方案中,以在治疗过程中提供约320 ng/mL的C谷浓度和小于约2000 ng/ml的Cmax的剂型和施用方案提供化合物1。在一些实施方案中,以在治疗过程中提供约400 ng/mL的C谷浓度和小于约2000 ng/ml的最大平均血浆浓度(Cmax)的剂型和施用方案提供化合物1。在一个实施方案中,以200 mgBID施用化合物1。在一个实施方案中,补体介导的病症是PNH。在一个实施方案中,补体介导的病症是3CG或IC-MPGN。
一方面,以本文所述的特定剂量施用化合物1。在一些实施方案中,施用化合物1使得单剂量提供如本文所述的特定PK血液分布。在一些实施方案中,施用于受试者的剂量为约25 mg至约275 mg。在一些实施方案中,所施用的剂量为约40 mg至约160 mg。在一些实施方案中,所施用的剂量至少约为40 mg、至少约为50 mg、至少约为60 mg、至少约为75 mg、至少约为90 mg、至少约为100 mg、至少约为125 mg、至少约为150 mg、至少约为160 mg、至少约为170 mg、至少约为175 mg、至少约为180 mg、至少约为190 mg、至少约为200 mg、至少约为210 mg、至少约为220 mg、至少约为230 mg、至少约为240 mg、至少约为250 mg、至少约为260 mg或至少约为275 mg。在一些实施方案中,在治疗期间以每天两次120mg的剂量施用化合物1。在一些实施方案中,在治疗期间以每天两次150 mg的剂量施用化合物1。在一些实施方案中,在治疗期间以每天两次175 mg的剂量施用化合物1。在一些实施方案中,在治疗期间以每天两次200 mg的剂量施用化合物1。在一些实施方案中,在治疗期间以每天两次220mg的剂量施用化合物1。在一些实施方案中,在治疗期间以每天两次240 mg的剂量施用化合物1。在一些实施方案中,在治疗期间以每天一次240 mg的剂量施用化合物1。
因此,本文提供的某些实施方案包括但不限于:
A) BID口服剂型,其包含对降低补体D途径的活性有效的量的(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)或其药学上可接受的盐,所述BID口服剂型在人血浆中提供为约65 ng/mL至95 ng/mL的两种不同的昼夜C谷水平中的较低者。
B) BID口服剂型,其包含对降低补体D途径的活性有效的量的(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)或其药学上可接受的盐,所述BID口服剂型在人血浆中提供约为90 ng/mL+/- 10%的两种不同的昼夜C谷水平中的较低者。
C) 口服剂型,其包含对降低旁路补体D途径的活性有效的量的(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)或其药学上可接受的盐,所述口服剂型在人血浆中提供为约65 ng/mL至95 ng/mL的C谷水平。
D) 口服剂型,其包含对降低补体D途径的活性有效的量的(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)或其药学上可接受的盐,所述口服剂型在人血浆中提供约为90 ng/mL+/- 10%的C谷水平。
E) BID口服剂型,其包含对降低补体D途径的活性有效的量的(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)或其药学上可接受的盐,所述BID口服剂型在人血浆中提供至少为65 ng/mL的两种不同的昼夜C谷水平中的较低者。
F) 口服剂型,其包含对降低补体D途径的活性有效的量的(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)或其药学上可接受的盐,所述口服剂型在人中提供至少为65 ng/mL的人血浆C谷。
G) BID口服剂型,其包含对降低补体D途径的活性有效的量的(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)或其药学上可接受的盐,所述BID口服剂型在人血浆中提供约为100 ng/mL+/- 10%的两种不同的昼夜C谷水平中的较低者。
H) 口服给药方案,其包含对降低补体D途径的活性有效的量的(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)或其药学上可接受的盐,所述口服给药方案在人血浆中提供至少为100 ng/mL+/- 10%的C谷。
I) 上述A)至H)中任一项的口服给药方案,其中所述剂型包含约100 mg至200 mg。
J) 上述I)的口服剂量,其包含约120 mg。
K) 上述I)的口服剂量,其包含约200 mg。
L) 上述A)至K)中任一项的口服剂型,其中在患有阵发性睡眠性血红蛋白尿症(PNH)的患者中测量人血浆中的C谷水平。
M) 上述A)至K)中任一项的口服剂型,其中在患者中测量人血浆中的C谷水平,所述患者患有选自脂肪肝诸如非酒精性脂肪性肝炎(NASH)、肝脏炎症、肝硬化或肝衰竭的病症。
N) 上述A)至K)中任一项的口服剂型,其中在患者中测量人血浆中的C谷水平,所述患者患有选自肌萎缩侧索硬化、类风湿性关节炎、补体旁路途径(AP)相关肾病、组分3肾小球病(C3G)病症、C3肾小球肾炎(C3GN)、致密沉积物病(DDD)、膜性增生性肾小球肾炎(MPGN)和免疫复合物膜性增生性肾小球肾炎(IC-MPGN)的病症。
O) 上述A)至K)中任一项的口服剂型,其中在患者中测量人血浆中的C谷水平,所述患者患有选自肌萎缩侧索硬化、类风湿性关节炎、补体旁路途径(AP)相关肾病和肾小球病的病症。
P) 上述A)至K)中任一项的口服剂型,其中在患者中测量人血浆中的C谷水平,所述患者患有选自年龄相关性黄斑变性(AMD)、视网膜变性、眼部疾病、地图样萎缩、早期或新生血管性年龄相关性黄斑变性、自身免疫性干眼病和环境性干眼病的病症。
Q) 一种治疗患有补体D相关病症的患者的方法,所述方法包括施用有效量的上述A)至K)中任一项的口服剂型。
R) 上述Q)的方法,其中所述患者患有阵发性睡眠性血红蛋白尿症(PNH)。
S) 上述Q)的方法,其中患者患有选自脂肪肝诸如非酒精性脂肪性肝炎(NASH)、肝脏炎症、肝硬化或肝衰竭的病症。
T) 上述Q)的方法,其中患者患有选自肌萎缩侧索硬化、类风湿性关节炎、补体旁路途径(AP)相关肾病、组分3肾小球病(C3G)病症、C3肾小球肾炎(C3GN)、致密沉积物病(DDD)、膜性增生性肾小球肾炎(MPGN)和免疫复合物膜性增生性肾小球肾炎(IC-MPGN)的病症。
U) 上述Q)的方法,其中患者患有选自肌萎缩侧索硬化、类风湿性关节炎、补体旁路途径(AP)相关肾病和肾小球病的病症。
V) 上述Q)的方法,其中患者患有选自年龄相关性黄斑变性(AMD)、视网膜变性、眼部疾病、地图样萎缩、早期或新生血管性年龄相关性黄斑变性、自身免疫性干眼病和环境性干眼病的病症。
W) 上述Q)至V)中任一项的方法,其中将所述剂型施用一个月或以上。
X) 上述Q)至V)中任一项的方法,其中将所述剂型施用至少6个月。
Y) 上述A)至K)中任一项的口服剂型,其提供了小于约2000 ng/mL的Cmax。
Z) 上述A)至K)中任一项的口服剂型,其提供了小于约1000 ng/mL的Cmax。
AA) 上述A)至K)中任一项的口服剂型,其用于治疗患有补体D相关病症的患者。
BB) 一种制备用于治疗患有补体D相关病症的患者的药物的方法,所述方法包括制备上述A)至K)中任一项的口服剂型。
CC) 上述BB)的口服剂型用于治疗上述R)至V)中任一项所列的任何病症。
DD) 上述CC)的方法,其用于治疗上述R)至V)中所列的任何病症。
附图简述
图1是表示在施用40 mg、80 mg和120 mg的单剂量的化合物1或安慰剂后通过AP溶血测量的AP活性百分比的图。第1至第3组在第1天第0小时被施予单剂量的化合物1。y轴代表相对于对照的AP活性的抑制百分比。x轴代表从第一次施用化合物1后的时间。
图2是表示化合物1的血浆浓度与通过AP溶血测定评价的血清AP活性的抑制之间的关系的PK-PD分析的图。y轴代表AP活性的抑制百分比。x轴代表化合物1的血浆浓度。
图3是表示两个端点之间AP活性的百分比的线性回归的图。
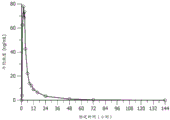
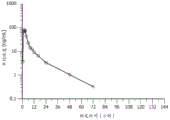
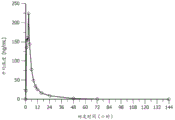
图4A和图4B是表示对于40 mg的化合物1剂量的从0小时至144小时(第7天)的平均血浆化合物I的浓度-时间曲线(线性[图4A]和半对数[图4B]标度)的图。
图5A和图5B是表示对于80 mg的化合物1剂量的从0至144小时(第7天)的平均血浆化合物1的浓度-时间曲线(线性[图5A]和半对数[图5B]标度)的图。
图6A和图6B是表示对于120 mg的化合物1剂量的从0至144小时(第7天)的平均血浆化合物1的浓度-时间曲线(线性[图6A]和半对数[图6B]标度)的图。
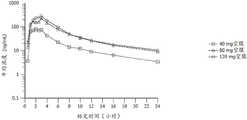
图7A和图7B是表示剂量比例分析的从0至24小时的平均血浆化合物I的浓度-时间曲线图(线性标度[图7A]和半对数标度[图7B])的图。
图8A和图8B是表示通过AP Wieslab测定评估的按随机治疗组计的在给药后24小时内(图8A)和随时间推移(图8B)的平均旁路途径功能活性(相对于阳性对照的百分比)的图。
图9A和图9B是表示按随机治疗组计的在给药后24小时内(图9A)和随时间推移(图9B)的的Bb血浆浓度相对于基线的平均改变的图。
图10表示稳态建模的图形显示,其表明90%的AP活性的抑制(IC90)在C谷约为67ng/mL时实现。
图11表示每天两次以120 mg施用的化合物1的预计稳态的图形显示。
图12是将化合物1和Danicopan的多递增剂量抑制与S型模型拟合的图形显示。y轴是对旁路途径的抑制百分比。x轴是以ng/mL为单位的药物血浆浓度。化合物1的模拟90%抑制水平为88 ng/mL。Danicopan的模拟90%抑制水平为235 ng/mL。
图13是服用40 mg、80 mg、120 mg或200 mg的化合物1达14天的患者的旁路途径溶血的平均减少的图形显示。y轴是相对于基线的AP溶血百分比。x轴是治疗的天数,其中第1天是给药的第一天。每天在0小时时收集数据点。40 mg BID研究中的两名受试者因非安全相关原因而停药,第3名受试者因在第7天漏服剂量而从分析中移除。
图14是在空腹的受试者(n=6)中以240 mg的单剂量、在进食的受试者(n=6)中以120 mg的单剂量施用的化合物1以及安慰剂(n=4)的平均血清旁路途径溶血的图形显示。在从第1天(给药前)至第7天的预定时间点收集的样品中评估血清AP溶血。x轴是以小时为单位的时间,y轴是AP活性百分比。
图15是在空腹的受试者(n=6)中以240 mg的单剂量、在进食的受试者(n=6)中以120 mg的单剂量施用化合物1后以及施用安慰剂(n=4)后的前24小时内,如通过ELISA测定法对所有受试者测定的平均血浆Bb浓度的图形显示。水平虚线网格线表示:正常值上限(1.42 µg/ml);正常值下限(0.48 µg/ml);和定量下限(LLOQ) (0.33 µg/ml)。x轴是以小时为单位的时间,y轴是以µg/mL为单位测量的平均血浆Bb浓度。
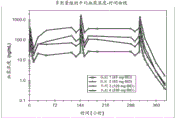
图16是以40 mg BID (n=8)、80 mg BID (n=7)、120 mg BID (n=8)、200 mg BID(n=8)施用的化合物1以及安慰剂BID (n=14)的平均血浆浓度的图形显示。在从第1天(给药前)至第16天的预定时间点收集的样品中评估每个队列的血浆浓度。x轴是以小时为单位的时间,y轴是以ng/mL为单位的平均血浆浓度。
图17是以40 mg BID (n=8)、80 mg BID (n=7)、120 mg BID (n=8)、200 mg BID(n=8)施用的化合物1以及安慰剂BID (n=14)的平均血清旁路途径的溶血活性的图形显示。显示了每个队列的完整的21天时间过程的活性。在第1天(0小时至12小时)、第7天(0小时至12小时)和第14天(0小时至16小时)进行了密集采样;在指定的日期于0小时时(早晨的PK谷)收集所有其他血清样品。x轴是以天为单位的时间,y轴是AP活性%。
图18是以40 mg BID (n=8)、80 mg BID (n=7)、120 mg BID (n=8)、200 mg BID(n=8)施用的化合物1以及安慰剂BID (n=14)的平均血清旁路途径的溶血活性的图形显示。在第1天(0小时至12小时)进行了密集采样。x轴是以小时为单位的时间,y轴是AP活性百分比。
图19是以40 mg BID (n=8)、80 mg BID (n=7)、120 mg BID (n=8)、200 mg BID(n=8)施用的化合物1以及安慰剂BID (n=14)的平均血清旁路途径溶血活性的图形显示。在第7天(0小时至12小时)进行了密集采样。x轴是以小时为单位的时间,y轴是AP活性百分比。
图20是以40 mg BID (n=8)、80 mg BID (n=7)、120 mg BID (n=8)、200 mg BID(n=8)施用的化合物1以及安慰剂BID (n=14)的平均血清旁路途径的溶血活性的图形显示。在第14天(0小时至12小时)进行了密集采样。x轴是以小时为单位的时间,y轴是AP活性百分比。
图21A是根据血浆化合物1的浓度和相应血清样品中AP Wieslab活性的抑制百分比进行的PK-PD评估的图形显示。使用简单的Emax模型进行非线性回归分析。最佳拟合值和95%置信区间以ng/mL为单位。x轴是以ng/mL为单位测量的化合物1血浆浓度,y轴是以百分比测量的AP活性的抑制百分比。
图21B是根据血浆化合物1的浓度和相应血清样品中AP Wieslab活性的抑制百分比进行的PK-PD评估的图形显示。使用四参数S型模型进行非线性回归分析。最佳拟合值和95%置信区间以ng/mL为单位。x轴是以ng/mL为单位测量的化合物1血浆浓度,y轴是以百分比测量的AP活性的抑制百分比。
具体实施方式
补体的功能障碍或过度激活与某些自身免疫性疾病、炎性疾病和神经变性疾病以及缺血再灌注损伤和癌症相关。例如,补体级联的旁路途径的激活有助于C3a和C5a的产生,这两者都是强效过敏毒素,它们也在许多炎性病症中起作用。因此,在一些情况下,需要降低补体途径(包括旁路补体途径)的响应。已发现,在每日两次约100 mg至200 mg,更具体地每日两次约120 mg至180 mg,甚至更具体地每日两次约120 mg至150 mg,或每日两次约150mg至约200 mg,以及在一个实施方案中每日两次约120 mg的化合物1的口服剂量下,在剂量间隔结束时提供最佳最低平均血浆浓度(C谷),使得剂量在一天中任何时间不降低至低于其EC90有效性。具体而言,发现了可将两个昼夜最低平均血浆浓度(C谷)中的较低者维持在既定EC90之上的给药方案。如下文进一步描述的建模建立了介于 67 ng/mL至88 ng/mL之间的EC90,将提供约90%的AP活性抑制。按本文所述的给药曲线施用化合物1导致对AP活性的强效抑制,在仅65ng/mL至95ng/mL的C谷下导致超过85%的抑制。具体地,本发明包括口服剂型,例如固体剂型或基于液体凝胶的剂型,所述口服剂型包含具有以下结构的化合物1:

或其药学上可接受的组合物、盐、同位素类似物或前药,所述口服剂型被施用于受试者(优选为人)以用于治疗由补体功能障碍或过度激活介导的病症(“补体介导的病症”),使得获得本文所述的特定PK和/或PD血液分布。已发现,给人受试者服用化合物1至为约65ng/mL至95 ng/mL的平均血浆浓度水平提供了对AP的强效抑制。制备化合物1的方法描述于PCT 公布第开2017/035353号(通过引用并入本文)。
定义
“如本文中所用,AUC”(量*时间/体积)意指血浆浓度-时间曲线下的面积。
如本文中所用,“AUC(0-inf)”(量*时间/体积)意指从时间零至无限时间的血浆浓度-时间曲线下面积。
如本文中所用,“AUC(0-24)”(量*时间/体积)意指从时间零至给药后24小时的血浆浓度时间曲线下面积。
如本文中所用,“AUEC”是指药效(PD)效应曲线下的面积。
如本文中所用,“AUEC0-12”意指从0小时至12小时的AUEC。
如本文中所用,“%AUEC0-12”意指作为其最大值的百分比的AUEC0-12。
如本文中所用,“A405”意指在405 nM处的光吸收率(Optical Absorbance)。
如本文中所用,“BID”是指“每日两次(bis en die)”或每天两次。
如本文中所用,“Cmax”(量/体积)意指最大(峰值)血浆药物浓度。
如本文中所用,“C谷”(量/体积)意指剂量间隔结束时的平均血浆浓度或最小平均血浆浓度。
如本文中所用,“CSR”意指临床研究报告。
如本文中所用,“持续时间0-12”意指从0小时至12小时的药效(PD)作用的持续时间。
如本文中所用,“EC50 (或EC90)”意指50% (或90%)的有效浓度。
如本文中所用,“最低平均血浆浓度”意指C谷。
如本文所用,“tmax”(时间)意指药物施用后达到最大(峰值)血浆浓度的时间。
如本文中所用,“%CV”意指变异系数。
如本文中所用,“AP”意指旁路补体途径。
如本文中所用,“Bb”意指补体因子B的Bb片段。
如本文中所用,“ELISA”意指酶联免疫吸附测定。
如本文中所用,“FD”意指补充因子D。
如本文中所用,“MAD”意指多递增剂量。
如本文中所用,“N”意指患者总数。
如本文中所用,“PD”意指药效学。
如本文中所用,“PK”意指药代动力学。
如本文中所用,“SAD”意指单一递增剂量。
如本文中所用,“SD”意指标准偏差。
适用于本发明的药物组合物/组合的术语“载体”是指与化合物I一起提供的稀释剂、赋形剂或媒介物。
“剂型”意指活性剂的施用单位。剂型的实例包括片剂、胶囊剂、混悬剂、液体剂、乳剂、颗粒剂、球剂(spheres)、颊用形式(buccal)、舌下剂(sublingual)、凝胶剂、粘膜剂(mucosal)等。
“患者”或“宿主”或“受试者”是需要治疗或预防(包括但不限于通过调节补体因子D途径)本文具体描述的任何病症的人或非人动物。通常宿主是人。“患者”或“宿主”或“受试者”还指例如哺乳动物、灵长类动物(例如,人)、牛、绵羊、山羊、马、狗、猫、兔、大鼠、小鼠、鱼、鸟等。
如本文中所用,术语“药学上可接受的盐”是指在合理的医学判断范围内,适用于与受试者(例如,人受试者)接触而没有过度毒性、刺激、过敏响应等,与合理的益处/风险比相称,并且对其预期用途有效的那些盐,以及在可能的情况下,目前公开的主题的化合物的两性离子形式。
因此,术语“盐”是指本公开主题的化合物的相对无毒的无机和有机酸加成盐。这些盐可以在化合物的最终分离和纯化期间原位制备,或通过将纯化的化合物以其游离碱的形式与适合的有机或无机酸单独反应以及分离由此形成的盐来制备。药学上可接受的碱加成盐可用金属或胺诸如碱金属和碱土金属氢氧化物或有机胺形成。用作阳离子的金属的实例包括但不限于钠、钾、镁、钙等。合适的胺的实例包括但不限于N,N'-二苄基乙二胺、氯普鲁卡因、胆碱、二乙醇胺、乙二胺、N-甲基葡糖胺和普鲁卡因。
盐可由无机酸硫酸、焦硫酸、重硫酸、亚硫酸、重亚硫酸、硝酸、磷酸、磷酸一氢酸、磷酸二氢酸、偏磷酸、焦磷酸、氯酸、溴酸、碘酸诸如盐酸、硝酸、磷酸、硫酸、氢溴酸、氢碘酸、磷酸等制备。代表性盐包括氢溴酸盐、盐酸盐、硫酸盐、重硫酸盐、硝酸盐、乙酸盐、草酸盐、戊酸盐、油酸盐、棕榈酸盐、硬脂酸盐、月桂酸盐、硼酸盐、苯甲酸盐、乳酸盐、磷酸盐、甲苯磺酸盐、柠檬酸盐、马来酸盐、富马酸盐、琥珀酸盐、酒石酸盐、萘酸盐、甲磺酸盐、葡庚糖酸盐、乳糖酸盐、月桂基磺酸盐和羟乙基磺酸酯等。盐也可以由有机酸,诸如脂族单羧酸和二羧酸、苯基取代的链烷酸、羟基链烷酸、链烷二酸、芳族酸、脂族和芳族磺酸等制备。代表性盐包括乙酸盐、丙酸盐、辛酸盐、异丁酸盐、草酸盐、丙二酸盐、琥珀酸盐、辛二酸盐、癸二酸盐、延胡索酸盐、马来酸盐、扁桃酸盐、苯甲酸盐、氯苯甲酸盐、甲基苯甲酸盐、二硝基苯甲酸盐、邻苯二甲酸盐、苯磺酸盐、甲苯磺酸盐、苯乙酸盐、柠檬酸盐、乳酸盐、马来酸盐、酒石酸盐、甲磺酸盐等。药学上可接受的盐可包括基于碱金属和碱土金属(诸如钠、锂、钾、钙、镁等)的阳离子,以及无毒的铵、季铵和胺阳离子,包括但不限于铵、四甲铵、四乙铵、甲胺、二甲胺、三甲胺、三乙胺、乙胺等。还设想了氨基酸的盐,诸如精氨酸盐、葡糖酸盐、半乳糖醛酸盐等。参见,例如,Berge等人,J. Pharm. Sci., 1977, 66, 1-19,其通过引用并入本文。
“药物组合物”是包含至少一种活性剂和至少一种其他物质诸如载体的组合物。“药物组合”是至少两种活性剂的组合,所述至少两种活性剂可以组合成单一剂型,或者以单独的剂型一起提供,并说明所述活性剂将一起用于治疗本文所述的任何病症。
“药学上可接受的赋形剂”意指可用于制备药物组合物/组合,通常是安全的、无毒的,并且在生物学上和其他方面都适合于向宿主(通常是人)施用的赋形剂。在一些实施方案中,使用对于兽医用途是可接受的赋形剂。
除非另有说明,否则在整个说明书和权利要求书中,给定的化学式或名称应涵盖所有光学和立体异构体,以及其中存在此类异构体和混合物的外消旋混合物。
在下文和本文中的一般描述中,无论何时使用涉及化合物1的任何术语,都应该理解为包括药学上可接受的盐或组合物,除非另有说明或与本文不一致。
如本文所预期的并且出于本文公开范围的目的,本文所述的所有范围包括出现在确定范围内的任何和所有数值。例如,如本文所设想的,1至10或1与10之间的范围将包括数值1、2、3、4、5、6、7、8、9、10及其分数。
(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-
(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺的剂型
本发明提供了特定剂型和使用所述剂型治疗患有补体介导的病症的受试者的方法,所述剂型提供补体因子D (fD)抑制剂(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)的血液分布范围。
一方面,向患有补体介导的病症的受试者施用化合物1,使得单剂量的化合物1提供如本文所述的特定的PK和/或PD血液分布。在一些实施方案中,施用化合物1使得在治疗期间,在BID给药方案的昼夜周期中,为约50 ng/mL至约200 ng/mL的最低平均血浆浓度(C谷)维持在较低的C谷水平之上。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,使为约75 ng/mL至约125 ng/mL的最低平均血浆浓度(C谷)维持在较低的C谷水平之上。在一些实施方案中,C谷至少约为50 ng/mL、至少约为55 ng/mL、至少约为60 ng/mL、至少约为65 ng/mL、至少约为70 ng/mL、至少约为75 ng/mL、至少约为80 ng/mL、至少约为85 ng/mL、至少约为90 ng/mL、至少约为95 ng/mL、至少约为100 ng/mL、至少约为105 ng/mL、至少约为110 ng/mL、至少约为115 ng/mL、至少约为120 ng/mL、至少约为125 ng/mL、至少约为130 ng/mL、至少约为135 ng/mL、至少约为140 ng/mL、至少约为145ng/mL、至少约为150 ng/mL、至少约为155 ng/mL、至少约为160 ng/mL、至少约为165 ng/mL、至少约为170 ng/mL、至少约为175 ng/mL、至少约为180 ng/mL、至少约为185 ng/mL、至少约为190 ng/mL、至少约为195 ng/mL或至少约为200 ng/mL。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,使小于约150 ng/mL的最低平均血浆浓度(C谷)维持在较低的C谷水平之上。在一些实施方案中,C谷小于约50 ng/mL、小于约55ng/mL、小于约60 ng/mL、小于约65 ng/mL、小于约70 ng/mL、小于约75 ng/mL、小于约80ng/mL、小于约85 ng/mL、小于约90 ng/mL、小于约95 ng/mL、小于约100 ng/mL、小于约110ng/mL、小于约120 ng/mL、小于约130 ng/mL、小于约140 ng/mL、小于约150 ng/mL、小于约160 ng/mL、小于约175 ng/mL、小于约190 ng/mL或小于约200 ng/mL。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,约为125 ng/mL的C谷维持在较低的C谷水平之上。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,至少约为100 ng/mL的C谷维持在较低的C谷水平之上。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,至少约为85 ng/mL的C谷维持在较低的C谷水平之上。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,至少约为67 ng/mL的C谷维持在较低的C谷水平之上。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,至少约为50 ng/mL的C谷维持在较低的C谷水平之上。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,至少为50 ng/mL +/- 10%的C谷维持在较低的C谷水平之上。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,至少为67 ng/mL +/- 10%的C谷维持在较低的C谷水平之上。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,至少为85 ng/mL +/- 10%的C谷维持在较低的C谷水平之上。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,为100 ng/mL +/- 10%的C谷维持在较低的C谷平之上。已发现,维持约为50 ng/mL的C谷浓度提供>85%的AP抑制,约为88ng/mL的C谷提供>90%的AP抑制。
一方面,化合物1以剂型提供,例如以诸如固体剂型或基于液体凝胶的剂型等口服剂型提供,所述剂型提供最大平均血浆浓度(Cmax)。在一些实施方案中,以提供为300 ng/mL至至少约3000 ng/mL的最大平均血浆浓度(Cmax)的剂型提供化合物1。在一些实施方案中,Cmax至少约为300 ng/mL、至少约为325 ng/mL、至少约为350 ng/mL、至少约为375 ng/mL、至少约为400 ng/mL、至少约为425 ng/mL、至少约为450 ng/mL、至少约为475 ng/mL、至少约为500 ng/mL、至少约为525 ng/mL、至少约为550 ng/mL、至少约为575 ng/mL、至少约为600ng/mL、至少约为625 ng/mL、至少约为650 ng/mL、至少约为675 ng/mL、至少约为700 ng/mL、至少约为725 ng/mL、至少约为750 ng/mL、至少约为775 ng/mL、至少约为800 ng/mL、至少约为825 ng/mL、至少约为850 ng/mL、至少约为875 ng/mL、至少约为900 ng/mL、至少约为925 ng/mL、至少约为950 ng/mL、至少约为975 ng/mL、至少约为1000 ng/mL、至少约为1150 ng/mL、至少约为1200 ng/mL、至少约为1250 ng/mL、至少约为1300 ng/mL、至少约为1350 ng/mL、至少约为1400 ng/mL、至少约为1450 ng/mL、至少约为1500 ng/mL、至少约为1550 ng/mL、至少约为1600 ng/mL、至少约为1650 ng/mL、至少约为1700 ng/mL、至少约为1750 ng/mL、至少约为1800 ng/mL、至少约为1850 ng/mL、至少约为1900 ng/mL、至少约为1950 ng/mL、至少约为2000 ng/mL、至少约为2050 ng/mL、至少约为2100 ng/mL、至少约为2150 ng/mL、至少约为2200 ng/mL、至少约为2250 ng/mL、至少约为2300 ng/mL、至少约为2350 ng/mL、至少约为2400 ng/mL、至少约为2450 ng/mL、至少约为2500 ng/mL、至少约为2550 ng/mL、至少约为2600 ng/mL、至少约为2650 ng/mL、至少约为2700 ng/mL、至少约为2750 ng/mL、至少约为2800 ng/mL、至少约为2850 ng/mL、至少约为2900 ng/mL、至少约为2950 ng/mL或至少约为3000 ng/mL。在一些实施方案中,Cmax小于约2500 ng/mL.在一些实施方案中,Cmax小于约2000 ng/mL。在一些实施方案中,Cmax小于约1500 ng/mL。在一些实施方案中,Cmax小于约1000 ng/mL。在一些实施方案中,Cmax小于约500 ng/mL。在一些实施方案中,Cmax小于约2500 ng/mL +/- 10%。在一些实施方案中,Cmax小于约2000 ng/mL +/- 10%。在一些实施方案中,Cmax小于约1500 ng/mL +/- 10%。在一些实施方案中,Cmax小于约1000ng/mL +/- 10%。在一些实施方案中,Cmax小于约500 ng/mL +/- 10%。
一方面,化合物1以剂型提供,例如以诸如固体剂型或基于液体凝胶的剂型等口服剂型提供,所述剂型提供如本文所述的AUC(0-24)。在一些实施方案中,化合物1以剂型提供,所述剂型提供约6000 ng*hr/mL至约20000 ng*hr/mL的AUC(0-24)。在一些实施方案中,化合物1以剂型提供,所述剂型提供至少约为6000 ng*hr/mL、至少约为6500 ng*hr/mL、至少约为7000 ng*hr/mL、至少约为7500 ng*hr/mL、至少约为8000 ng*hr/mL、至少约为8500 ng*hr/mL、至少约为9000 ng*hr/mL、至少约为9500 ng*hr/mL、至少约为10000 ng*hr/mL、至少约为10500 ng*hr/mL、至少约为11000 ng*hr/mL、至少约为11500 ng*hr/mL、至少约为12000 ng*hr/mL、至少约为12500 ng*hr/mL、至少约为13000 ng*hr/mL、至少约为13500ng*hr/mL、至少约为14000 ng*hr/mL、至少约为14500 ng*hr/mL、至少约为15000 ng*hr/mL、至少约为15500 ng*hr/mL、至少约为16000 ng*hr/mL、至少约为16500 ng*hr/mL、至少约为17000 ng*hr/mL、至少约为17500 ng*hr/mL、至少约为18000 ng*hr/mL、至少约为18500 ng*hr/mL、至少约为19000 ng*hr/mL、至少约为19500 ng*hr/mL或至少约为20000ng*hr/mL的AUC(0-24)。在一些实施方案中,AUC(0-24)至少约为1450 ng*hr/mL。在一些实施方案中,AUC(0-24)小于约8000 ng*hr/mL。在一些实施方案中,AUC(0-24)至少约为8000 ng*hr/mL+/- 10%。在一些实施方案中,AUC(0-24)小于约8000 ng*hr/mL +/- 10%。
对每个采样的12小时给药间隔计算另外两个药效学活性测量值:持续时间(0-12)和AUEC(0-12) (作用曲线下面积)。持续时间(0-12)表示12小时给药间隔期间AP抑制达到90%或更高的时间;12小时的最大值表示在整个间隔内持续90%或更大的抑制。AUEC(0-12)表示在间隔期间AP抑制随时间的积分幅度。另外,%AUEC(0-12)将AUEC(0-12)表示为在间隔内的最大AP抑制的百分比(100%抑制× 12小时)。
一方面,化合物1以剂型提供,例如以诸如固体剂型或充液胶囊或基于液体凝胶的剂型等口服剂型提供,所述剂型提供如本文所述的持续时间(0-12)。一方面,向患有补体介导的病症的受试者施用化合物1,导致为10小时至12小时的最小平均持续时间(0-12)。在一些实施方案中,化合物1以剂型提供,所述剂型提供至少约为10小时、至少约为10.25小时、至少约为10.5小时、至少约为10.75小时、至少约为11小时、至少约为11.25小时、至少约为11.5小时、至少约为11.75小时、至少约为12小时的持续时间(0-12)。在一些实施方案中,化合物1以剂型提供,所述剂型提供至少约为12小时的持续时间(0-12)。
一方面,化合物1以剂型提供,例如以诸如固体剂型或充液胶囊或基于凝胶的剂型等口服剂型提供,所述剂型提供如本文所述的最大血浆浓度%AUEC(0-12)的时间。一方面,向患有补体介导的病症的受试者施用化合物1,导致为80%至100%的最小平均%AUEC(0-12)。一方面,向患有补体介导的病症的受试者施用化合物1,导致为90%至100%的最小平均%AUEC(0-12)。在一些实施方案中,化合物1以剂型提供,所述剂型提供至少约为80%、至少约为85%、至少约为90%、至少约为95%或者至少约为100%的%AUEC(0-12)。
一方面,化合物1以剂型提供,例如以诸如固体剂型或充液胶囊或基于凝胶的剂型等口服剂型提供,所述剂型提供如本文所述的最大血浆浓度的时间(tmax)。在一些实施方案中,化合物1以剂型提供,所述剂型提供约0.5小时至3小时的tmax。在一些实施方案中,化合物1以剂型提供,所述剂型提供至少约为3小时、至少约为2.75小时、至少约为2.5小时、至少约为2.25小时、至少约为2.0小时、至少约为1.75小时、至少约为1.5小时、至少约为1.25小时、至少约为1小时、至少约为0.75小时或至少约为0.5小时的tmax。在一些实施方案中,化合物1以剂型提供,所述剂型提供至少约为2小时的tmax。在一些实施方案中,化合物1以剂型提供,所述剂型提供至少约为1小时的tmax。
在一些实施方案中,施用化合物1,使得单剂量提供如本文所述的特定PK和/或PD血液特性。在一些实施方案中,施用于受试者的剂量为约25 mg至约275 mg。在一些实施方案中,施用的剂量至少约为25 mg、至少约为30 mg、至少约为35 mg、至少约为40 mg、至少约为45 mg、至少约为50 mg、至少约为55 mg、至少约为60 mg、至少约为65 mg、至少约为70mg、至少约为75 mg、至少约为80 mg、至少约为85 mg、至少约为90 mg、至少约为95 mg、至少约为100 mg、至少约为110 mg、至少约为120 mg、至少约为130 mg、至少约为140 mg、至少约为150 mg、至少约为160 mg、至少约为170 mg、至少约为180 mg、至少约为190 mg、至少约为200 mg、至少约为210 mg、至少约为220 mg、至少约为230 mg、至少约为240 mg、至少约为250 mg、至少约为260 mg、至少约为270mg或至少约为275 mg。在一些实施方案中,以至少约为120mg的单剂量施用化合物1。在一些实施方案中,以至少约为240 mg的单剂量施用化合物1。在一些实施方案中,以120 mg +/-10%的单剂量施用化合物1。在一些实施方案中,在治疗期间每天施用化合物1一次。在一些实施方案中,在治疗期间每天施用化合物1两次(BID)。在一些实施方案中,在治疗期间每天施用化合物1至少两次。在一些实施方案中,在治疗期间每天施用化合物1三次或更多次。在一些实施方案中,每天以至少约为120 mg的单剂量施用化合物1两次。在一些实施方案中,每天以至少约为150 mg的单剂量施用化合物1两次。在一些实施方案中,每天以至少约为175 mg的单剂量施用化合物1两次。在一些实施方案中,每天以至少约为200 mg的单剂量施用化合物1两次。在一些实施方案中,每天以至少约为210 mg的单剂量施用化合物1两次。在一些实施方案中,每天以至少约为225 mg的单剂量施用化合物1两次。在一些实施方案中,每天以至少约为250 mg的单剂量施用化合物1两次。在一些实施方案中,每天施用化合物1两次,每次剂量间隔大约12小时。在一些实施方案中,每天施用化合物1两次,每次剂量间隔约12小时,持续28天。在一些实施方案中,每天施用化合物1两次,持续至少约4周、至少约5周、至少约6周或至少约12周。在一些实施方案中,每天施用化合物1两次,持续至少约3周、至少约6周或至少约9周。
药物制备
化合物1可作为纯化学品施用。或者,化合物1可作为药物组合物施用,如本文所述,所述药物组合物包含对于需要这种治疗的宿主(通常是人)有效的量的化合物1。因此,本公开提供了纯净的药物组合物,其包含以一定量的化合物或药学上可接受的盐存在的化合物1的剂型以及至少一种药学上可接受的载体,以实现本文所述的血液分布范围。药物组合物可包含化合物或盐作为唯一的活性剂,或者在替代实施方案中,包含所述化合物和至少一种另外的活性剂。药物组合物还可包含一定摩尔比的活性化合物和另外的活性剂。
载体包括赋形剂和稀释剂,并且必须具有足够高的纯度和足够低的毒性,以使其适合施用于向接受治疗的患者施用。载体可以是惰性的,或者其可具有其自身的药物益处。与化合物1结合使用的载体的量足以为每单位剂量的化合物的施用提供物质的实际量。
载体类别包括但不限于粘合剂、缓冲剂、着色剂、稀释剂、崩解剂、乳化剂、调味剂、助流剂、润滑剂、防腐剂、稳定剂、表面活性剂、压片剂和润湿剂。一些载体可被列于不止一个类别中,例如植物油可在一些制剂中用作润滑剂,而在另一些制剂中用作稀释剂。示例性药学上可接受的载体包括糖、淀粉、纤维素、黄蓍胶粉、麦芽、明胶;滑石粉和植物油。药物组合物中可包含任选的活性剂,其基本上不干扰本发明化合物的活性。
可配制用于口服施用的药物组合物/组合。这些组合物可包含实现所需结果的任何量的活性化合物,例如0.1重量% (wt.%)至99重量% (wt.%)的化合物,通常至少约为5重量%的化合物。一些实施方案包含至少约25重量%至至少约50重量%或至少约5重量%至至少约75重量%的化合物。
治疗方法
如本文中所设想的,提供如本文所述的PK和/或PD血液分布的剂型可用于治疗补体介导的病症。通过达到和/或维持本文所述的某些血液分布,可抑制旁路补体途径,提供可用于治疗由有缺陷或过度活跃的补体反应导致的病症的给药方案。
在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,为约50 ng/mL至约200 ng/mL的最低平均血浆浓度(C谷)维持在较低的C谷水平之上。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,使为约75ng/mL至约125 ng/mL的最低平均血浆浓度(C谷)维持在较低的C谷水平之上。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,至少约为50 ng/mL的C谷维持在较低的C谷水平之上。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,约为67 ng/mL的C谷维持在较低的C谷水平之上。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,约为85 ng/mL的C谷维持在较低的C谷水平之上。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,约为100 ng/mL的C谷维持在较低的C谷水平之上。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,约为125 ng/mL的C谷维持在较低的C谷水平之上。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,至少为50 ng/mL +/- 10%的C谷维持在较低的C谷水平之上。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,至少为67 ng/mL +/- 10%的C谷维持在较低的C谷水平之上。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,至少为85 ng/mL +/- 10%的C谷维持在较低的C谷水平之上。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,至少为100 ng/mL +/- 10%的C谷维持在较低的C谷水平之上。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,至少为125 ng/mL +/- 10%的C谷维持在较低的C谷水平之上。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,为50 ng/mL +/- 10%的C谷维持在较低的C谷水平之上。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,67 ng/mL +/- 10%的C谷维持在较低的C谷水平之上。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,85 ng/mL +/- 10%的C谷维持在较低的C谷水平之上。在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,100 ng/mL +/- 10%的C谷维持在较低的C谷水平之上。
在一些实施方案中,施用化合物1,使得在治疗期间达到约10小时至约12小时的持续时间(0-12)。在一些实施方案中,施用化合物1,使得在治疗期间达到约11小时至约12小时的持续时间(0-12)。在一些实施方案中,施用化合物1,使得在治疗期间达到至少10小时的持续时间(0-12)。在一些实施方案中,施用化合物1,使得在治疗期间达到至少11小时的持续时间(0-12)。在一些实施方案中,施用化合物1,使得在治疗期间达到至少12小时的持续时间(0-12)。
在一些实施方案中,施用化合物1,使得在治疗期间达到约80%至约100%的%AEUC(0-12)。在一些实施方案中,施用化合物1,使得在治疗期间达到约90%至约100%的%AEUC(0-12)。在一些实施方案中,施用化合物1,使得在治疗期间达到至少为80%的%AEUC(0-12)。在一些实施方案中,施用化合物1,使得在治疗期间达到至少为90%的%AEUC(0-12)。在一些实施方案中,施用化合物1,使得在治疗期间达到至少为100%的%AEUC(0-12)。
在一些实施方案中,施用化合物1,使得在治疗期间达到小于约3000 ng/mL的Cmax。在一些实施方案中,施用化合物1,使得在治疗期间达到小于约2000 ng/mL的Cmax。在一些实施方案中,施用化合物1,使得在治疗期间达到小于约1000 ng/mL的Cmax。在一些实施方案中,施用化合物1,使得在治疗期间达到为约600 ng/mL至约1700 ng/mL的Cmax。在一些实施方案中,施用化合物1,使得在治疗期间达到小于约3000 ng/mL +/- 10%的Cmax。在一些实施方案中,施用化合物1,使得在治疗期间达到小于约2000 ng/mL +/- 10%的Cmax。在一些实施方案中,施用化合物1,使得在治疗期间达到小于约1000 ng/mL +/- 10%的Cmax。在一些实施方案中,施用化合物1,使得在治疗期间达到小于约600 ng/mL +/- 10%的Cmax。在一些实施方案中,施用化合物1,使得在治疗期间达到为800 ng/mL +/- 10%至1500 ng/mL +/- 10%的Cmax。
在一些实施方案中,施用化合物1,使得在治疗期间达到为约6000 ng*hr/mL至20000 ng*hr/mL的AUC(0-24)。在一些实施方案中,施用化合物1,使得在治疗期间达到至少约为6000 ng*hr/mL的AUC(0-24)。
在一些实施方案中,提供了用于治疗与宿主的补体级联功能障碍相关联的病症的方法,所述方法包括施用化合物1以实现如本文所述的特定PK和/或PD血液分布。在一些实施方案中,提供了用于抑制受试者的旁路补体途径激活的方法,所述方法包括施用化合物1以实现如本文所述的特定PK和/或PD血液分布。在一些实施方案中,提供了调节受试者的因子D活性的方法,所述方法包括施用化合物1以实现如本文所述的特定 PK和/或PD血液分布。
在一些实施方案中,所述病症是阵发性睡眠性血红蛋白尿症(PNH)。一方面,以足够的量向患有PNH的受试者施用化合物1,其中在治疗过程中,乳酸脱氢酶(LDH)水平(血管内溶血的生物标志物)从施用前测量的基线LDH水平相对于基线降低至少约30%、至少约40%、至少约50%、至少约60%、至少约70%、至少约80%或超过80%。一方面,以足够的量向患有PNH的受试者施用化合物1,其中在治疗过程中,血红蛋白水平从施用前测量的基线水平相对于基线升高至少约10%、至少约15%、至少约20%、至少约25%或超过25%。
在一些实施方案中,所述病症是膜性增生性肾小球肾炎(MPGN)。MPGN是影响肾脏肾小球或过滤器的疾病。直到最近,当潜在病因可鉴定时,膜性增生性肾小球肾炎(MPGN)在临床上被归类为原发性MPGN、特发性MPGN或继发性MPGN。主要根据电子致密沉积物的超微结构外观和位置,将原发性MPGN进一步分为三种类型—I型、II型和III型。然而,临床和组织病理学方案都存在问题,因为它们都不是基于疾病的发病机制。对补体在MPGN发病机制中的作用的更好理解导致提议重新分类为免疫球蛋白介导的疾病(由经典补体途径驱动的)和非免疫球蛋白介导的疾病(由旁路补体途径驱动的)。这种重新分类导致了改进的诊断临床算法,并出现了一组新的疾病,称为C3肾小球病,最好的代表是致密沉积物病(DDD)和C3肾小球肾炎(C3GN)。
在一些实施方案中,所述病症是C3肾小球病。C3肾小球病是一组导致肾脏功能障碍的相关疾患。C3肾小球病的主要特征包括尿中的蛋白质水平高(蛋白尿)、尿中带血(血尿)、尿量减少、血液中蛋白质水平低以及身体许多部位肿胀。受影响的个体的血液中的补体组分3(或C3)的水平可能特别低。与C3肾小球病相关的肾脏问题往往会随着时间的推移而恶化。大约一半的受影响的个体在他们的诊断后10年内发展为终末期肾病(ESRD)。ESRD是危及生命的疾患,其会阻止肾脏有效地过滤身体中的液体和废物。
在一些实施方案中,所述疾病是致密沉积物病(DDD)。在一些实施方案中,所述疾病是C3肾小球肾炎(C3GN)。
在一些实施方案中,所述疾病是免疫复合物膜性增生性肾小球肾炎(IC-MPGN))。IC-MPGN是肾脏疾病,其与C3G共有许多临床、病理、遗传和实验室特征。高达40%的IC-MPGN患者没有可鉴定的潜在病因,被认为患有特发性IC-MPGN。患有特发性IC-MPGN的受试者可具有低的C3水平和正常的C4水平,类似于在C3G中观察到的那些,以及许多与异常旁路途径的活性相关的相同遗传或获得性因素。那些具有低的C3和正常的C4的受试者很可能具有显著的旁路途径的过度活性。C3Nef在MPGN 1型患者中的存在被确认为与其在C3GN患者中的存在一样频繁。在IC-MPGN患者中发现了编码旁路途径蛋白(包括 fH和fI)的基因的突变。尽管在肾活检中存在IgG、IgM和C1q (在一小部分病例中)的免疫阳性荧光染色,但大约46%的IC-MPGN病例表现出降低的C3水平和正常的C4水平。这些数据表明旁路途径在IC-MPGN中失调。
在一些实施方案中,施用化合物1,使得LDH水平在治疗期间从高于或等于正常值上限(ULN)的1.5倍的水平降低。在一些实施方案中,施用化合物1,使得Hgb水平在治疗期间升高。
可由本文所述的化合物1或其盐或组合物治疗或预防的其他病症还包括但不限于:
(i) 腹主动脉瘤、血液透析并发症、溶血性贫血或血液透析、神经脊髓炎(NMO)、脂肪肝、非酒精性脂肪性肝炎(NASH)、肝脏炎症、肝硬化、肝衰竭、皮肌炎、肌萎缩侧索硬化、年龄相关性黄斑变性(AMD)、类风湿性关节炎以及响应于生物治疗剂(例如CAR T细胞疗法)的细胞因子或炎性反应、阵发性睡眠性血红蛋白尿症(PNH)、遗传性血管性水肿、毛细血管渗漏综合征、非典型溶血性尿毒症综合征(aHUS)、溶血性尿毒症综合征(HUS)、腹主动脉瘤、血液透析并发症、溶血性贫血或血液透析;
(ii) 重症肌无力(MG)、多发性硬化、神经病学病症、格林巴利综合征(GuillainBarre Syndrome)、中枢神经系统疾病和其他神经变性疾患、肾小球肾炎(包括膜性增生性肾小球肾炎)、SLE肾炎、增生性肾炎、肝纤维化、组织再生和神经再生或巴勒奎尔-西蒙斯综合征(Barraquer-Simons Syndrome);
(iii) 脓毒症、全身炎性响应综合征(SIRS)、不适当或不良补体激活的病症、IL-2治疗期间的白介素2诱导的毒性、炎性病症、自身免疫性疾病的炎症、系统性红斑狼疮(SLE)、克罗恩病(Crohn’s disease)、类风湿性关节炎、炎性肠病、狼疮性肾炎、关节炎、免疫复合物病症和自身免疫性疾病、系统性狼疮或红斑狼疮的炎性作用;
(iv) 缺血/再灌注损伤(I/R损伤)、心肌梗塞、心肌炎、缺血再灌注后疾患、球囊血管成形术、动脉粥样硬化、心肺旁路术或肾旁路术中的泵后综合征、肾局部缺血、主动脉重建后肠系膜动脉再灌注、抗磷脂综合征、自身免疫性心脏病、缺血再灌注损伤、肥胖或糖尿病;
(v) 阿尔茨海默痴呆症、中风、精神分裂症、创伤性脑损伤、创伤、帕金森病(Parkinson's disease)、癫痫、移植排斥、胎儿丢失的预防、生物材料反应(例如,在血液透析、植入物中)、超急性同种异体移植排斥、异种移植排斥、移植、银屑病、烧伤、热损伤,包括烧伤或冻伤;
(vi) 哮喘、过敏、急性呼吸窘迫综合征(ARDS)、囊性纤维化、成人呼吸窘迫综合征、呼吸困难、咳血、慢性阻塞性肺病(COPD)、肺气肿、肺栓塞和梗塞、肺炎、纤维性粉尘病(纤维性粉尘病)、惰性粉尘和矿物质(例如,硅、煤尘、铍和石棉)、肺纤维化、有机尘病、化学损伤(由于刺激性气体和化学品,例如氯、光气、二氧化硫、硫化氢、二氧化氮、氨和盐酸)、烟雾损伤、热损伤(例如,烧伤、冻伤)、支气管收缩、过敏性肺炎、寄生虫病、古德帕斯彻氏综合征(Goodpasture's Syndrome)(抗肾小球基底膜肾炎)、肺血管炎、寡免疫性血管炎(Pauci-immune vasculitis)或免疫复合物相关炎症。
组合疗法
在一些实施方案中,化合物1可以与至少一种另外的治疗剂组合或交替提供,例如用于治疗本文所列的病症。在一些实施方案中,化合物1可以与补体系统的至少一种另外的抑制剂或具有不同生物学作用机制的第二活性化合物(例如,C5抑制剂、C3抑制剂、补体因子B抑制剂或泛补体抑制剂)组合或交替施用以实现如本文所述的特定PK和/或PD血液分布。
C5抑制剂
C5抑制剂是本领域已知的。在一些实施方案中,C5抑制剂是靶向C5的单克隆抗体。在一些实施方案中,C5抑制剂是依库珠单抗(SolirisTM Alexion Pharmaceuticals, NewHaven, CT,参见例如美国专利第9,352,035号)。在一些实施方案中,C5抑制剂是雷武珠单抗-cwvz (UltomirisTM Alexion Pharmaceuticals, New Haven, CT.)。
在一些实施方案中,C5抑制剂可以是但不限于:重组人微型抗体,例如Mubodina®(单克隆抗体,Adienne Pharma and Biotech, Bergamo, Italy;参见美国专利第7,999,081号)、coversin (小动物蛋白,Volution Immuno-pharmaceuticals, Geneva,Switzerland;参见例如Penabad等人Lupus, 2012, 23(12):1324-6)、LFG316 (单克隆抗体,Novartis, Basel, Switzerland和Morphosys, Planegg, Germany;参见美国专利第8,241,628号和第8,883,158号); ARC-1905 (聚乙二醇化的RNA适体,Ophthotech,Princeton, NJ and New York, NY;参见Keefe等人,Nature Reviews Drug Discovery,9, 537-550)、RA101348和RA101495 (大环肽,Ra Pharmaceuticals, Cambridge, MA)、SOBI002 (亲和体(affibody),Swedish Orphan Biovitrum, Stockholm, Sweden)、ALN-CC5 (Si-RNA, Alnylam Pharmaceuticals, Cambridge, MA)、ARC1005 (适体,NovoNordisk, Bagsvaerd, Denmark)、SOMAmers (适体,SomaLogic, Boulder, Co)、SSL7 (细菌蛋白毒素,参见,例如Laursen等人Proc. Natl. Acad. Sci. U.S.A., 107(8):3681-6)、MEDI7814 (单克隆抗体,MedImmune, Gaithersburg, MD)、金精三羧酸、金精三羧酸衍生物(Aurin Biotech, Vancouver, BC,参见美国专利申请公布2013/003592)、RG6107 (抗C5循环抗体,Roche Pharmaceuticals, Basel, Switzerland)、ALXN1210和ALXN5500 (单克隆抗体,Alexion Pharmaceuticals, New Haven, CT)、TT30 (融合蛋白,AlexionPharmaceuticals, New Haven, CT)、REGN3918 (单克隆抗体,Regeneron, Tarrytown,NY)、ABP959 (依库珠单抗生物仿制药,Amgen, Thousand Oaks, CA)或其组合。
在一些实施方案中,C5抑制剂是重组人微型抗体,例如Mubodina®。Mubodina®是由Adienne Pharma and Biotech开发的完全人源重组抗体C5。Mubodina®描述于美国专利第7,999,081号中。
在一些实施方案中,C5抑制剂是coversin。Coversin是重组蛋白,其源自在毛白钝 缘蜱(Ornithodoros moubata)的唾液中发现的蛋白质,所述蛋白质目前由AkariTherapeutics开发为重组蛋白。Coversin描述于Penabad等人 Lupus 2012, 23(12):1324-6中。
在一些实施方案中,C5抑制剂是特度鲁单抗(Tesidolumab)/LFG316. 特度鲁单抗是Novartis和Morphosys开发的单克隆抗体。特度鲁单抗描述于美国专利第8,241,628号和第8,883,158号中。
在一些实施方案中,C5抑制剂是ARC-1905。ARC-1905 是Ophthotech开发的聚乙二醇化的RNA适体。ARC-1905描述于Keefe等人Nature Reviews Drug Discovery, 9:537-550中。
在一些实施方案中,C5抑制剂是RA101348。RA101348是Ra Pharmaceuticals开发的大环肽。
在一些实施方案中,C5抑制剂是RA101495。RA101495是Ra Pharmaceuticals开发的大环肽。
在一些实施方案中,C5抑制剂是SOBI002。SOBI002是Swedish Orphan Biovitrum开发的亲和体。
在一些实施方案中,C5抑制剂是ARC1005。ARC1005是Novo Nordisk开发的适体。
在一些实施方案中,C5抑制剂是C5的SOMAmers。SOMAmers是SomaLogic开发的适体。
在一些实施方案中,C5抑制剂是SSL7。SSL7是Laursen等人Proc. Natl. Acad.Sci. U.S.A., 107(8):3681-6中描述的细菌蛋白质毒素。
在一些实施方案中,C5抑制剂是MEDI7814。MEDI7814是MedImmune开发的单克隆抗体。
在一些实施方案中,C5抑制剂是金精三羧酸。在另一个实施方案中,C5抑制剂是金精三羧酸衍生物。这些金精衍生物由Aurin Biotech开发,并在美国专利申请公布第2013/003592号中进一步描述)。
在一些实施方案中,C5抑制剂是RG6107/SKY59。RG6107/SKY59是由RochePharmaceuticals开发的抗C5循环抗体。
在一些实施方案中,C5抑制剂是ALXN1210。在另一个实施方案中,C5抑制剂是ALXN5500。ALXN1210和ALXN5500是Alexion Pharmaceuticals开发的单克隆抗体。
在一些实施方案中,C5抑制剂是TT30。TT30是Alexion Pharmaceuticals开发的融合蛋白。
在一些实施方案中,C5抑制剂是ABP959。ABP959是Amgen开发的依库珠单抗生物仿制药单克隆抗体。
在一些实施方案中,C5抑制剂是抗C5 siRNA。抗C5 siRNA是由AlnylamPharmaceuticals开发的。
在一些实施方案中,C5抑制剂是Erdigna®。Erdigna®是Adienne Pharma开发的抗体。
在一些实施方案中,C5抑制剂是培戈-阿伐普他(avacincaptad pegol)/Zimura®。培戈-阿伐普他是Opthotech开发的适体。
在一些实施方案中,C5抑制剂是SOBI005。SOBI005是Swedish Orphan Biovitrum开发的蛋白质。
在一些实施方案中,C5抑制剂是ISU305。ISU305是ISU ABXIS开发的单克隆抗体。
在一些实施方案中,C5抑制剂是REGN3918。REGN3918是Regeneron开发的单克隆抗体。
C3抑制剂
本文提供了用于治疗受试者的PNH的方法,所述方法包括向受试者施用有效量的C3抑制剂与有效量的选自式I或式II的CFD抑制剂的组合或交替施用有效量的所述C3抑制剂和有效量的所述CFD抑制剂。
C3抑制剂是本领域已知的。在一些实施方案中,化合物1与康普他汀(compstatin)和/或康普他汀类似物组合或交替施用。康普他汀和康普他汀类似物是已知的并且被发现是有用的C3抑制剂,参见美国专利第9,056,076号、第8,168,584号、第9,421,240号、第9,291,622号、第8,580,735号、第9371365号、第9,169,307号、第8,946,145号、第7,989,589号、第7,888,323号、第6,319,897号以及美国专利申请公布第2016/0060297号、第2016/0015810号、第2016/0215022号、第2016/0215020号、第2016/0194359号、第2014/0371133号、第2014/0323407号、第2014/0050739号、第2013/0324482号和第2015/0158915号。在又一实施方案中,康普他汀类似物是4(1MeW)POT-4. 4(1MeW)POT-4由Potentia开发。在又一实施方案中,康普他汀类似物是AMY-201。AMY-201是由Amyndas Pharmaceuticals开发的。
在一些实施方案中,化合物1可以与C3抑制剂组合,所述抑制剂包含但不限于:H17(单克隆抗体,EluSys Therapeutics, Pine Brook, NJ)、米考西普(基于CR1的蛋白)、sCR1(基于CR1的蛋白,Celldex, Hampton, NJ)、TT32 (基于CR-1的蛋白,AlexionPharmaceuticals, New Haven, CT)、HC-1496 (重组肽)、CB 2782 (酶,CatalystBiosciences, South San Francisco, CA)、APL-2 (peg化的合成周期肽,ApellisPharmaceuticals, Crestwood, KY)或其组合。
在一些实施方案中,C3抑制剂是H17。H17是EluSys Therapeutics正在开发的人源化单克隆抗体。H17描述于Paixao-Cavalcante等人J. Immunol. 2014, 192(10):4844-4851中。
在一些实施方案中,C3抑制剂是米考西普。米考西普是由InflazymePharmaceuticals开发的基于CR1的蛋白。
在一些实施方案中,C3抑制剂是sCR1。 sCR1是由Celldex开发的CR1蛋白的可溶性形式。
在一些实施方案中,C3抑制剂是TT32。TT32是Alexion Pharmaceuticals开发的基于CR-1的蛋白。
在一些实施方案中,C3抑制剂是HC-1496。HC-1496是InCode开发的重组肽。
在一些实施方案中,C3抑制剂是CB 2782。CB 2782是源自Catalyst Biosciences开发的人膜型丝氨酸蛋白酶1 (MTSP-1)的新型蛋白酶。
在一些实施方案中,C3抑制剂是APL-2。APL-2是Apellis Pharmaceuticals开发的APL-1的聚乙二醇化形式。
补体因子B (CFB)抑制剂
CFB抑制剂是本领域已知的。在一些实施方案中,化合物1可以与CFB抑制剂组合,所述CFB抑制剂包括但不限于:抗-FB SiRNA (Alnylam Pharmaceuticals, Cambridge,MA)、TA106 (单克隆抗体,Alexion Pharmaceuticals, New Haven, CT)、LNP023 (小分子,Novartis, Basel, Switzerland)、SOMAmers (适体,SomaLogic, Boulder, CO)、拜卡西奥单抗(bikaciomab)(Novelmed Therapeutics, Cleveland, OH)、complin (例如,Kadam等人, J. Immunol. 2010, DOI:10.409/jimmunol.10000200)、Ionis-FB-LRx (缀合有配体的反义药物, Ionis Pharmaceuticals, Carlsbad, CA)或其组合。在另一个实施方案中,可与如本文所述的化合物1组合的CFB抑制剂包括在PCT/US17/39587中公开的那些。在另一个实施方案中,可与如本文所述的化合物1组合的CFB抑制剂包括在PCT/US17/014458中公开的那些。在另一个实施方案中,可与如本文所述的化合物1组合的CFB抑制剂包括在美国专利申请公布第2016/0024079号(授予Novartis AG)中公开的那些。在一些实施方案中,CFB抑制剂是

在一些实施方案中,CFB抑制剂是抗FB siRNA。抗FB siRNA是AlnylamPharmaceuticals开发的。
在一些实施方案中,CFB抑制剂是TA106。TA106是Alexion Pharmaceuticals开发的单克隆抗体。
在一些实施方案中,CFB抑制剂是LNP023。LNP023是Novartis开发的CFB的小分子抑制剂。LNP023和相关抑制剂描述于Maibaum等人Nat. Chem. Biol. 2016, 12:1105-1110中。
在一些实施方案中,CFB抑制剂是complin。Complin是肽在Kadam等人J. Immunol.2010 184(12):7116-24中描述的肽抑制剂。
在一些实施方案中,CFB抑制剂是Ionis-FB-LRx。Ionis-FB-LRx是由IonisPharmaceuticals开发的缀合有配体的反义药物。
补体成分的泛抑制剂(Pan-inhibitor)
补体成分的泛抑制剂是本领域已知的。在一些实施方案中,所述抑制剂是FUT-175。
实施例
I期单一递增剂量研究
单一递增剂量(SAD)研究是化合物1的首次人体研究。主要目的是证明化合物1(一种经口施用的补体因子D抑制剂)的单一递增口服剂量在健康的志愿者中的安全性和耐受性。次要目的包括评价药代动力学(PK)特性以及化合物1 PK与药效学(PD)特征之间的关系,即补体旁路途径(AP)活性的抑制(PK/PD)。
在4个剂量组中对18名健康志愿者进行给药和评价,其中10名受试者接受安慰剂(表1)。第一剂量组具有6名活性剂受试者和6名安慰剂受试者;随后的组各自具有6名活性剂受试者和2名安慰剂受试者。给药后对每组随访28天。直到第28天的最后一次预定随访,对所有受试者的安全性进行监测。血液和尿液样品在从第1天至第4天的预定时间点收集以确定化合物1的血浆和尿液浓度,并从第1天至第7天的预定时间点收集以确定补体相关活性。测定了通过AP Wieslab测定法测量的血清AP活性以及血浆Bb、因子D水平和几种其他PD生物标志物的结果。
表1. ACH228-001中的剂量组(SAD研究)
| 组 | 剂量(mg) | 方案 | 活性剂(N) | 安慰剂(N) | 空腹对比进食 |
| 1 | 40 mg或PBO | 单剂量 | 6 | 6 | 空腹 |
| 2 | 80 mg或PBO | 单剂量 | 6 | 2 | 空腹 |
| 3 | 120 mg或PBO | 单剂量 | 6 | 2 | 空腹 |
PBO=安慰剂
材料和方法
材料
兔红细胞(RBC)、不含Ca++和Mg++ (GVB0)的明胶佛罗那缓冲液(gelatin veronalbuffer)和100 mM MgCl2 - 100 mM EGTA (MgEGTA)购自Complement Technology, Inc,(Tyler, TX)。 GVB0-MgEGTA缓冲液是通过将GVB0和100 mM MgEGTA以9:1的比率混合制备的。RBC在购买的两周内使用;每天在测定前通过在800 x g和4℃下离心3分钟收集细胞,并将其重悬于等体积的新鲜冷GVB0 - MgEGTA中至密度为5×108个细胞/mL。本研究中使用的商业ELISA试剂盒的信息包含在下面的每个特定测定中。
人血清/血浆的制备、储存和递送
对于人血清的制备,通过标准临床程序将静脉血收集到Gold Top SST真空采血管(vacutainer) (BD Vacutainer)中。在室温下凝固30分钟或更长时间后,将管以约1,300 xg在4℃下离心15分钟以将血清与凝块分离。然后将血清等分到预冷的冷冻小瓶(50-200 µl/小瓶)中,于-80℃下储存。每个小瓶仅解冻一次并用于每次补体相关测定,丢弃剩余样品而不进一步使用。为了制备血浆,将静脉血收集到Lavender Top, K2EDTA真空采血管中,并轻轻倒转几次,使血液样品与抗凝剂完全混合。将管置于冰浴中30分钟,然后在4℃下以约1300 x g离心15分钟以将血浆与血细胞分离。将血浆在预冷的低温储存管中分成等分试样,并在采集血样后一小时内储存在-80℃的冰箱中。对于递送,将含有血清或血浆的管在干冰存在的情况下进行运送,而无需额外的冻融循环。
血清AP溶血测定
离体血清AP溶血测定在96孔微量滴定板中进行,所述96孔微量滴定板分为两部分,一部分用于溶血,并且另一部分用于血清背景颜色扣除。简言之,将兔红细胞(RBC)的悬浮液在4℃下以800 x g离心3分钟,并去除上清液以除去RBC碎片。用含有10 mM MgEGTA的预冷明胶佛罗那缓冲液(GVB0)轻轻重新悬浮RBC细胞沉淀,使其密度为5 x 108个细胞/mL。其他对照包括缓冲液、自发性兔RBC裂解、100% RBC裂解(于水中)和正常血清对照也与测试样品一起设置。将血清置于37℃水浴中直至大部分解冻,并立即转移到冰浴。20%的血清用GVB0-MgEGTA缓冲液制备,并在测试前保持在冰浴。通过将50 µl 20%的血清与等体积的GVB0-MgEGTA缓冲液混合,然后快速加入20 µl RBC悬浮液来在96孔板中进行反应。将板在微量滴定板振荡器上振荡5秒,然后在37℃下孵育30分钟。在孵育期间的中途,如上所述将板再振荡5秒。在孵育结束时,将板在4℃下以800 x g离心3分钟,以沉淀未裂解的兔RBC。从所有孔中小心取出80 µl上清液并转移至透明平底微量滴定板中,用于在MolecularDevices Spectramax Plus读板器中测量405 nm处的光吸收率(A405)。如下相对于NHS参考样品配方的活性计算每个孔中的AP溶血:
溶血(%) = ((Hm – Bk) – (Sp – Bf)) / ((NH - BkNH) – (Sp – Bf)) × 100%
其中Hm = 溶血孔中的A405,Bk = 对应背景孔中的A405,Sp = 自发溶血孔中的平均A405,Bf = 仅有缓冲液的孔中的平均A405,NH = 参考NHS孔中的平均A405,以及BkNH = 相应背景孔中的平均A405。
PK-PD分析
使用以下两个模型,利用时间匹配的血浆化合物1的浓度值和血清AP抑制值,通过利用GraphPad Prism (La Jolla, CA)进行的非线性回归来评价PK-PD关系:
1. 简单的Emax模型,用于确定50%有效浓度(EC50):
Y = E0 + Emax × X / (EC50 + X)
其中X =血浆化合物1的浓度,Y =相应血清样品中的抑制百分比,E0 = 0%抑制(给药前AP活性)和Emax = 100%抑制。
2. 四参数S型模型,用于确定EC50和90%的有效浓度(EC90):
Y = E0 + (Emax - E0) / (1 + 10^((LogEC50 - X) × HillSlope))
其中X、Y、E0和 Emax定义如上(Emax模型),并且HillSlope = 斜率因子或斜坡斜率(Hill slope)。
药效学和药代动力学结果
用AP溶血测量的AP活性
对于所有受试者,在从第1天至第7天的特定方案确定的时间点收集血液样品,以使用AP溶血测定和AP Wieslab测定来测定离体血清AP活性。
对于施用了40 mg、80 mg或120 mg的单剂量的化合物1的所有受试者观察到AP活性的强效抑制(图1)。具体而言,所有组中的受试者在化合物1给药后均实现了对AP活性的完全抑制。在第1组中,根据AP溶血测定,大约90%或更高的抑制在给药后维持了至少4小时。在第2组和第3组中,大约90%或更高的抑制在给药后维持了至少8小时。相反地,在安慰剂受试者中未观察到AP活性的显著变化(图1)。使用简单的Emax模型分析通过溶血测定法测定的所有受试者的血浆化合物1浓度(PK)与血清AP活性之间的关系(图2)。根据该分析,化合物1的EC50值为11.1 ng/mL。
另外,对于第3组中化合物1-治疗的受试者和安慰剂受试者,通过本文所述的AP溶血测定法测量的所有血清样品中的AP活性与AP Wieslab数据密切相关(相关系数,r =0.83)(图3)。使用以下简单的Emax剂量响应模型,分析了参加SAD研究的所有给药受试者的血浆化合物1的浓度与通过溶血测定测量的血清AP抑制之间的关系:Y=E0+Emax • X/(EC50+X),其中E0=未抑制的AP活性(0%的抑制)和Emax=完全抑制的AP活性(100%)。总样品数为306。在给药后的特定时间点收集血清样品,并用AP Wieslab和AP溶血测定法评估以测定AP活性。将每个时间点时的AP活性以来自同一受试者的给药前AP活性(100%)作归一化。通过线性回归(Prism软件,GraphPad, La Jolla, CA)分析两个终点之间的关系。
对于包含组1至组3 SAD PK群体的18名受试者,总共306个血浆样品(在给药前和给药后0.5小时、1小时、1.5小时、2小时、2.5小时、3小时、4小时、6小时、8小时、10小时、12小时、16小时、24小时[第2天]、48小时[第3天]、72小时[第4天]和144小时[第7天]从每名受试者收集的17个样品)可用于血浆中的PK测定。
在包含PK群体的所有18名受试者中,给药前血浆化合物1的浓度低于LLOQ (即,<0.100 ng/mL)。给药后化合物1被迅速吸收,其中所有18名受试者在评价的第一个给药后采样时间点(即,给药后0.5小时)具有可测量的血浆化合物1的浓度。随机化治疗组的平均血浆化合物1的浓度相对时间曲线的图分别显示于图4A (40 mg)、图5A (80mg)、图6A (120mg)(线性标度)和图4B (40 mg)、图5B (80 mg)和图6B (120 mg)(半对数标度)中,汇总统计数据提供于表2中。最大平均血浆浓度(78.03 ± 19.65 ng/mL在40 mg组中,224.05 ±108.27 ng/mL在80 mg组中,以及282.83 ± 89.15 ng/mL在120 mg组中)发生在给药后1至3小时之间。基于平均浓度曲线,在单剂量施用至多120 mg后,血浆化合物1的浓度似乎以剂量比例的方式增加。在所有剂量组中,在给药后72小时的时间点最后一次检测到血浆化合物1的浓度。
评价化合物1的单一递增剂量以确定所研究的40 mg至120 mg的剂量范围内的血浆和尿液PK参数。每个标称采样时间点时的血浆和尿液化合物1的浓度的汇总统计数据按随机治疗组呈现在表2中。
血浆化合物1的Cmax和AUC随着施用的化合物1的剂量(即,40 mg、80 mg和120 mg)增加而成比例地增加。在40 mg和120 mg剂量组中,血浆化合物1的PK参数Cmax和AUC的变化性通常较低且相似,其中Cmax的几何CV为30% (对于40 mg组)或31% (对于120 mg组),并且AUC的几何CV的范围为23%至32%。在80 mg剂量组中,与40 mg和120 mg剂量组相比,Cmax(几何CV为51%)和AUC(几何CV的范围为50%至53%)的变化性要大得多。在所研究的40 mg至120mg的剂量范围内,观察到化合物1的快速吸收,其具有相似的中位tmax(1.50小时至2.75小时)值。在所研究的剂量范围内,平均终末消除半衰期(t½ term)值的范围为约9.92小时至13.90小时。
PK群体中包含的所有受试者(n = 18;每6名受试者随机接受40 mg、80 mg和120mg的化合物1)被纳入剂量比例分析。血浆化合物1的Cmax和AUC0-inf 值的几何平均值示于表2中。用于评估剂量比例的平均血浆化合物1-相对-时间曲线(从0至24小时)示于图7A (线性标度)和图7B (半对数标度)。
化合物1的肾排泄低,其中几何平均剂量恢复为未改变的化合物1 (范围为所施用的剂量的0.17%至0.26%)。各剂量组间的肾清除率值也相似,其中几何平均值的范围为0.16L/h至0.19 L/h。
表2. 血浆和尿液中化合物1的药代动力学参数估计值的总结

缩写:Aeu =尿液中排泄的化合物I的总量;Aeu(%) =表示为剂量的百分比的尿液中排泄的化合物I的总量;AUC0-12 =从施用时至具有可定量的浓度的给药后12小时的血浆浓度-时间曲线下面积;AUC0-24 =从施用时至具有可定量的浓度的给药后24小时的血浆浓度-时间曲线下面积;AUC0-inf =外推至无穷大的血浆浓度-时间曲线下面积;AUClast =从施用时至具有可定量的浓度的最后一次给药后的血浆浓度-时间曲线下面积;CL/F = 表观口服药物清除率;CLr =总化合物I的肾清除率;Cmax =最大血浆浓度;GCV% =几何百分比变异系数;GM =几何平均值;h =小时;L = 升;mL =毫升;ng =纳克;SD =标准偏差;t½ term =表观终末消除半衰期;tmax =达到最大血浆浓度的时间;Vz/F =表观分布体积;λz =终末消除率常数。
通过AP Wieslab测定测量的旁路途径功能活性
一直至第7天(以天为单位报告的)和给药后24小时内(通过随机治疗组和整体)的平均补体AP功能活性(相对于阳性对照的百分比)的图形显示分别见于图8B和图8A。如通过Wieslab测定所评估的,对于施用了40 mg、80 mg或120 mg的单剂量的化合物1的所有受试者均观察到对AP功能活性的强效抑制。
早在给药后0.5小时,对于所有化合物1治疗的受试者均注意到>80%的对AP功能活性的抑制(或相对于阳性对照≤20%的AP功能活性结果),所有受试者在给药后2小时实现了> 90%的对AP功能活性的抑制。在给药后1.5小时(在40 mg剂量组中)至3小时(在80 mg和120 mg剂量组中),达到了AP功能活性的最低点;对于40 mg、80 mg和120 mg剂量组,平均(± SD) AP功能活性分别降至最低水平1.25 (± 1.153)%、0.13 (± 0.097)%和0.09 (±0.102)%。对于40 mg剂量组,90%或更高的平均抑制在给药后维持了至少6小时。对于所研究的较高剂量组(即,120 mg和240 mg),90%或更高的平均抑制维持了更长的时间段(即,给药后持续至少10小时)。
Bb血浆浓度
一直至第7天(以天为单位报告的)和给药后24小时内(通过随机治疗组和整体)的平均血浆Bb浓度的图形显示可分别见于图9B和图9A。如图9A和图9B所示,相对于基线和安慰剂治疗组,所有评价的化合物1剂量组在单剂量施用后Bb血浆浓度均显著降低。尽管在单剂量组中评价的受试者数量相对较少(n=6),但观察到的变化幅度似乎是剂量依赖性的。在基线时,40 mg、80 mg和120 mg剂量组的Bb血浆浓度在测定的正常参考范围内(即,0.49 µg/mL至1.42 µg/mL);40 mg、80 mg和120 mg剂量组的平均(± SD)基线值分别为0.80 (±0.195) µg/mL、0.78 (±0.128) µg/mL和0.70 (±0.095) µg/mL。Bb血浆浓度在给药后6小时(在40 mg剂量组中)至8小时(在80 mg和120 mg剂量组中)达到最低点。给药后6小时,40mg剂量组中的平均(± SD) Bb血浆浓度已降至0.51 (±0.091) µg/mL。给药后8小时,平均Bb血浆浓度对于80 mg剂量组已降至0.48 (±0.056) µg/mL,并且对于120 mg剂量组降至0.40 (±0.029) µg/mL。对于80 mg和120 mg剂量组,平均Bb血浆浓度分别到给药后10小时(对于80 mg剂量组)和给药后24小时(第2天)(对于120 mg剂量组)增加到高于LLN(以用于测定),到给药后48小时(第3天)恢复至基线值。在研究期间,受试者在Bb血浆浓度方面都未出现治疗突发异常。
基于离体AP抑制测定,制备了比较基于药物血浆浓度的AP的抑制百分比的稳态模型。稳态建模的图形显示如图10和图11所示。图10显示用约为67 ng/mL的C谷实现了90%的抑制(IC90)。因此,预计化合物1在体内标谷浓度下可实现85%的抑制。图11显示,每天施用120mg化合物1两次的预计稳态提供了约50 ng/mL的C谷,实现了对AP活性的高于85%的抑制,而Cmax低于约350 ng/mL。因此,化合物1被证明是强效的,同时仍具有改善的安全裕度(safetymargin),这可以允许更好的给药灵活性。
总结
本文报道的采用AP溶血测定的离体AP功能测定,证明了在SAD的所有队列中施用化合物I后,均快速实现了对AP途径的完全或接近完全的抑制。使用简单的Emax模型的PK/PD分析揭示了与约为10 ng/mL的EC50的明确剂量响应关系。药效学结果表明,化合物1 (一种fD抑制剂)通过范围为40 mg至120 mg的单剂量施用实现了对补体的AP的强效抑制。评价血清AP活性的离体Wieslab测定表明在化合物1的单剂量施用后几乎立即几乎完全抑制了AP活性。早在给药后0.5小时,对于所有化合物1治疗的受试者,注意到>80%的对AP活性的抑制,其中所有受试者在给药后2小时均实现了>90%的对AP活性的抑制。在给药后1.5小时(在40 mg剂量组中)至3小时(在80 mg和120 mg剂量组中)达到AP活性的最低点。对于所研究的较高剂量组(即,80 mg和120 mg),与所研究的较低的40 mg剂量组(至少6小时)相比,90%或更高的平均抑制在给药后维持了更长的时间段(至少10小时)。使用Emax模型,利用来自所有剂量队列的所有时间匹配的血浆PK数据和血清AP活性抑制数据进行定量分析,得到为11ng/mL的EC50。另外,单剂量施用后,Bb血浆浓度(AP活性的体内生物标志物)显著降低,表明AP活性在体内受到抑制。尽管在单剂量组中评价的受试者数量相对较少(n=6),但观察到的变化幅度似乎是剂量依赖性的。Bb血浆浓度在给药后6小时(在40 mg剂量组中)至8小时(在80 mg和120 mg剂量组中)达到最低值。Bb血浆浓度逐渐从最低点恢复,40 mg剂量组恢复较快,所有剂量组在给药后48小时(第3天)恢复至基线值。对于评价fD血清浓度、C3血清浓度、C4血清浓度和血清总CP功能所进行的其他补体测定,与基线和安慰剂相比,在单剂量施用高达120 mg化合物1后随着时间推移没有显著变化。
具有两个额外的单剂量队列进行的化合物1的多递增剂量(MAD)研究
具有两个额外的单剂量队列进行的化合物1的多递增剂量(MAD)研究也已完成。主要目的是证明多递增口服剂量的化合物1 (一种口服施用的补体因子D抑制剂)在健康志愿者中的安全性和耐受性。次要目的包括评价药代动力学(PK)分布以及化合物1 PK与药效学(PD)特征之间的关系,即补体旁路途径(AP)的活性的抑制(PK/PD)。在MAD研究中,在于空腹状态下施用化合物1 (40 mg、80 mg、120 mg和200 mg BID)的四个剂量组中持续14天评价了化合物1的多剂量药代动力学和药效学(表3)。每组中8名受试者接受活性药物;第一剂量组(40 mg)中8名受试者中接受安慰剂,而在其他三个剂量组中的每一组中,2名受试者接受安慰剂。以240 mg (空腹的受试者)和120 mg (进食的受试者)的单剂量向两个额外队列施用化合物1 (表3)。两个单剂量队列各自包括6名接受活性药物的受试者和两名接受安慰剂的受试者。在给药前的选定日(在局部低谷)以及在第1天、第7天和第14天以0.5小时至2小时的间隔收集用于PK和PD评估的血浆和血清样品以用于更详细的评估。
表3. 具有额外的单剂量队列的化合物1 MAD研究中的剂量组
| 组 | 剂量(mg) | 方案 | 活性剂(N) | 安慰剂(N) | 空腹对比进食 |
| S1 | 240 mg或PBO | 单剂量 | 6 | 2 | 空腹 |
| S2 | 120 mg或PBO | 单剂量 | 6 | 2 | 进食 |
| 1 | 40 mg或PBO | 14天BID | 8 | 8 | 空腹 |
| 2 | 80 mg或PBO | 14天BID | 8 | 2 | 空腹 |
| 3 | 120 mg或PBO | 14天BID | 8 | 2 | 空腹 |
| 4 | 200 mg或PBO | 14天BID | 8 | 2 | 空腹 |
a - 两名第1组受试者在第6天之前因非治疗突发不良事件的原因停药;从第1天至第3天可获得PK和PD结果。
2个额外剂量队列的化合物1的单剂量药代动力学
240 mg单剂量的药代动力学结果与来自SAD研究的数据一致,并证明在40 mg至240 mg的单剂量范围内,暴露(Cmax,AUC)与剂量成比例增加(表4)。将与中等脂肪餐一起给予的单一120 mg剂量与在空腹状态下给予的相同剂量(SAD研究)进行比较,进食状态下的平均Cmax和AUC值分别低2.2%和10.8%。该结果表明可以在进食或空腹状态下以化合物1给药,而在全身性暴露方面没有临床上显著的差异。
表4. 240 mg (空腹)和120 mg (进食)的单剂量施用后化合物1的药代动力学参数的总结

2个额外剂量队列的化合物1的单剂量药效学
来自化合物1的MAD临床研究中额外单剂量队列的药效学结果表明对补体AP活性的持续剂量依赖性抑制。在从第1天(给药前)至第7天的预定时间点收集的样品中评估血清AP溶血活性。240mg剂量组中的离体血清AP抑制比之前在高达120mg的剂量下进行的SAD研究中观察到的延长更长时间(图14)。单剂量的化合物1施用后,血清AP溶血活性显示出快速抑制。活性剂受试者当中的平均给药前(第1天,0小时)活性为123.02% (240 mg)和110.43%(120 mg)。 240 mg组中的活性从1.5小时至16小时降低到10%或更低,而120 mg组中的活性从2小时至6小时降低到10%或更低。到48小时时,活性恢复到接近给药前水平。相比之下,安慰剂受试者的AP活性与为109.52%的给药前值相比几乎没有变化,其中在所有时间点中最小值为81.46%。 240 mg剂量的所有受试者在给药两小时内实现了90%或更高的抑制,并且在给药后至少12小时内平均抑制持续在90%或更高。另外,作为体内AP抑制的指征,血浆Bb显示出剂量依赖性降低;化合物1的抑制效应在240 mg剂量组中比在较低剂量下更长,在给药后至少16小时内保持在其最低点附近(图15)。安慰剂受试者在给药后在AP活性或血浆Bb浓度方面未显示显著的变化,并且活性剂受试者在化合物1施用后在血清CP活性或血清因子D、C3或C4浓度方面未显示显著的变化。
化合物1的多剂量药代动力学
在40 mg、80 mg、120 mg和200 mg多剂量队列中进行初始剂量后,平均暴露(Cmax和AUC0-inf)在第1天以线性、剂量成比例的方式增加。当以每日重复两次(BID)的方案给予每个剂量时,观察到稳态暴露的大于剂量比例的增加。Cmax和AUC两者的从第1天(第一剂)至第7/14天(稳态)的平均积累随着剂量的增加,从1.7增加至4.7,突出了化合物1的非线性PK行为。对于所有4个剂量组,第14天最后一次给药后洗掉后的平均t1/2term在9.0小时至11.9小时的范围内,其量级与单剂量施用后观察到的量级相似。随着剂量的增加,未能观察到该半衰期延长的趋势。
图16是四个剂量组的平均浓度图,并且表5总结了所有组的平均多剂量药代动力学参数。
稳定状态下给药前血浆浓度有一致的趋势,早晨比夜晚高,一些早晨的谷约为夜晚的谷的两倍。40 mg、80 mg、120 mg和200 mg BID组的平均给药前浓度在为15.6 ng/mL-23.5 ng/mL、45.9 ng/mL-83.7 ng/mL、80.6 ng/mL-153 ng/mL和247 ng/mL-392 ng/mL的相应范围内(表6)。
表5. 重复剂量施用14天后化合物1的药代动力学参数的总结

表6. 重复剂量施用后化合物1的平均给药前稳态(谷值)血浆浓度(ng/mL)

化合物1的多剂量药效学
用化合物1 BID给药14天的患者中AP溶血的平均减少
旁路途径溶血的平均减少如图13所示。在研究持续期间,120 mg和200 mg BID患者组的AP溶血均减少了90%以上。平均而言,这种减少超过95% (参见图13)。
多剂量队列中的血清AP溶血活性
在从第1天(给药前)至第21天的预定时间点,在从所有队列收集的样品中评估血清AP溶血活性。在第1天(0小时至12小时)、第7天(0小时至12小时)和第14天(0小时至16小时)进行密集采样;在指定日期的0小时(早晨的PK谷)时收集所有其他血清样品。显示了每个队列在第1-21天(图17)、第1天(图18)、第7天(图19)和第14天(图20)的完整的时间过程中的活性。还在预定的时间点测量了通过AP Wieslab测定法离体测量的血清补体AP活性、血清总补体经典途径(CP)活性、血浆补体Bb浓度以及血清FD、C3和C4浓度。
多剂量ACH-5228施用后,血清AP活性显示出快速且剂量依赖性的抑制。每个队列中活性剂受试者间的平均给药前(第1天,0小时)AP活性的范围为69.43% (40 mg)至73.29%(80 mg)。在第7天和第14天(稳态PK条件),给药后的活性降低至4.00% (40 mg)和0.00%(第2组至第4组)的最小值。然而,在40 mg BID和80 mg BID时,这种AP抑制伴随着在PK谷值(0小时和12小时时间点)时的显著恢复。相反,在120 mg BID时,平均AP活性在第7天在从0小时至10小时以及在第14天在从0小时至12小时的所有时间点时均保持低于10%。在200 mgBID时,平均AP活性在第7天和第14天在从0小时至12小时的所有时间点时均保持低于10%。到第17天,即最后一次给药后三天,所有剂量的AP活性都恢复到大约给药前水平。相比之下,安慰剂受试者中的AP活性与为84.97%的给药前值相比几乎没有变化,其中所有时间点间的最小值为76.84%。
旁路途径抑制百分比的S型模型
将如通过利用患者样品进行的离体AP溶血测定法测量的旁路途径的总抑制百分比拟合至S型剂量响应模型,以确定化合物1的IC90。对于化合物1,建模的IC90药物血浆浓度为88 ng/mL(见图12)。这比第一代因子D抑制剂Danicopan强2.5倍以上,Danicopan在235ng/mL时实现90%的抑制(见图12)。另外,通过口服120 mg BID给药,在稳态C谷下实现了高于95%的平均旁路途径抑制。
在PK谷值时(0小时和12小时)的AP抑制
表7显示了在第7天和第14天的0小时和12小时时间点时的AP溶血活性的经计算的抑制百分比,以及从第6天至第14天的所有0小时和12小时时间点(代表在稳态PK的条件下的PK谷值)共同的AP溶血活性的经计算的抑制百分比。在40 mg BID和80 mg BID时,在0小时和12小时时间点观察到部分抑制。在120 mg BID时,所有0小时时间点时的抑制超过90%(97.2%的抑制),并且所有12小时时间点也几乎如此(87.8%的抑制)。在200 mg BID时,所有0小时和12小时时间点时的抑制超过90% (分别为97.6%和95.1%)。这些结果与AP Wieslab测定得出的结果一致,其中在120 mg BID和200 mg BID时,所有0小时和12小时时间点时的经计算的抑制百分比超过90%。
表7. 0小时和12小时时间点(PK谷值):第7天、第14天和第6天至第14天时的多剂量队列中对血清AP溶血活性的抑制

a. 第7天0小时和12小时(早晨和夜晚PK谷值)的AP抑制率(%)。
b. 第14天0小时和12小时(早晨和夜晚PK谷值)的AP抑制率(%)。
c. 第6天、第7天、第8天、第9天、第13天和第14天0小时(早晨的PK谷值)以及第7天和第14天的12小时(夜晚PK谷值)的AP抑制(%)。
多剂量队列中AP溶血抑制的积分幅度和持续时间
将每个时间点时的多剂量个体的血清AP活性以同一个体的给药前活性(第1天,0小时,定义为100%)作归一化。将每个时间点时的对这种给药前归化一化的AP活性的抑制计算为相对于给药前活性的降低(即,100% -给药前归一化的活性)。这些AP抑制值用于计算以下两个PD参数,以定量第7天和第14天的AP抑制的积分幅度和持续时间。
首先,将从0小时至12小时的AP抑制的积分幅度计算为从0小时至12小时的效应曲线下面积(AUEC) (AUEC0-12)。通过线性梯形法计算 AUEC0-12,其中将从0小时至12小时的相邻时间点之间的线性函数下的面积相加。另外,将%AUEC0-12计算为以其间隔的最大可能值作归一化的AUEC0-12;100%的最大%AUEC0-12表明从0小时至12小时连续完全(100%)抑制。在40 mg BID和80 mg BID时,低于90%的经计算的%AUEC0-12值反映了在剂量之间观察到的AP溶血活性的显著恢复。在120 mg BID和200 mg BID时,平均%AUEC0-12值(第7天和第14天,组合分析)分别为98.2%和99.1%,表明在整个12小的时期间几乎完全抑制AP (表8)。这些值与从AP Wieslab测定得出的结果一致,其中在120 mg BID和200 mg BID时,%AUEC0-12值(第7天和第14天,组合分析)分别为98.6%和99.3%。
表8. 多剂量队列中从0小时至12小时的AP溶血抑制的积分幅度(AUEC0-12):第7天和第14天

a. %AUEC0-12被定义为0小时至12小时的12小时给药间隔内的效应曲线下面积(AUEC),表示为整个间隔内的最大抑制的百分比。%AUEC0-12是使用在整个间隔中的时间点确定的AP活性抑制来计算的。
b. 组合分析包括两个观察(第7天和第14天) × 每个队列中的N个个体。
其次,AP抑制的持续时间计算为0小时与12小时之间流逝的时间(持续时间0-12),在此期间AP抑制为90%或更高。在需要时通过线性插值估计具有90%或更高的抑制的开始和结束时间点。12小时的最大持续时间0-12值表明从0小时至12小时连续90%或更高的抑制。在40 mg BID和80 mg BID时,经计算的持续时间0-12值(第7天和第14天,组合分析)分别为2.3小时和7.6小时,反映了在剂量之间观察到的AP溶血活性的显著恢复。在120 mg BID和200mg BID时,平均持续时间0-12值(第7天和第14天,组合分析)分别为11.3小时和 11.7小时,接近12小时的最大可能值(表9)。这些值与从AP Wieslab测定得出的发现结果一致,在120mg BID和200 mg BID时,持续时间0-12值(第7天和第14天,组合分析)分别为11.6小时和11.9小时。
表9. 多剂量队列中从0小时至12小时的AP溶血抑制的持续时间(持续时间0-12):第7天和第14天

a. 持续时间0-12被定义为从0小时至12小时的流逝时间,在此期间血清AP抑制为90%或更高。
b. 组合分析包括两个观察(第7天和第14天) × 每个队列中的N个个体。
多剂量队列中血浆ACH-5228浓度与血清AP抑制之间的PK-PD关系
使用两种模型(图21A和图21B)在所有时间点和多剂量队列中的个体之间评价时间匹配的血浆化合物1的浓度(PK)与血清AP溶血活性(PD)的抑制之间的PK-PD关系。使用简单的Emax模型,通过非线性回归分析数据以确定EC50,并且为了更近似地估计有效活性所需的浓度,使用S型模型来确定EC50和EC90。
利用两种模型的分析证明了给药后AP活性的浓度依赖性抑制。使用Emax模型的分析得出的EC50值为19.1 ng/mL,其中95%置信区间(CI)为18.1 ng/mL至20.1 ng/mL。使用S型模型的分析得出的EC50和EC90值为24.6 ng/mL和88.1 ng/mL,其中围绕EC90的95% CI为82.5ng/mL至94.0 ng/mL。这些值与AP Wieslab测定得出的结果一致,所述AP Wieslab测定使用Emax模型产生为14.3 ng/mL的EC50值,以及使用S型模型产生分别为19.2 ng/mL和66.9 ng/mL的EC50和EC90值。
总结
在1期SAD研究中单剂量化合物1施用后,使用AP溶血测定确认了对血清AP活性的快速且几乎完全的抑制。在240 mg空腹的受试者中在给药后1.5小时至16小时以及在120mg进食的受试者中在给药后2小时至6小时,平均给药前活性降低至10%或更低。
来自1期MAD研究中的四个多剂量队列的结果,化合物1在化合物1施用后表现出对AP溶血活性的快速和剂量依赖性抑制。40 mg BID (第1组)和80 mg BID (第2组)时的化合物1实现了部分至几乎完全的AP抑制,但在每个12小时的给药期结束前显著恢复。120 mgBID (第3组)和200 mg BID (第4组)时的化合物1实现了在12小时的给药期的大部分时间内维持的基本上完全的AP抑制,其中AP活性在第7天和第14天(代表稳态PK条件)在12小时的给药期中的10至12小时内维持低于10%。为本研究开发的两个计算的PD参数(持续时间0-12和%AUEC0-12)进一步描述了120 mg和200 mg BID时的化合物1对AP的实质性和持续的抑制。
评价了多剂量队列间的血浆化合物1的浓度(PK)与血清AP溶血抑制(PD)之间的PK-PD关系。使用简单的Emax模型的分析得出的EC50值为19.1 ng/mL。使用S型模型的进一步分析得出分别为24.6 ng/mL和88.1 ng/mL的EC50和EC90值。在对离体AP Wieslab结果的平行分析中获得了可比较的结果,所述平行分析使用Emax模型得出为14.3 ng/mL的EC50值,并且使用S型模型得出分别为19.2 ng/mL和66.9 ng/mL的EC50和EC90值。
已参考本发明的实施方案描述了本说明书。然而,本领域的一般技术人员应理解,在不脱离随附权利要求书中所阐明的本发明的范围的情况下,可以作出各种修改和改变。因此,本说明书应被视为说明性的而非限制性的,并且所有此类修改都旨在包括在本发明的范围内。
Claims (30)
1.一种BID口服剂型,其包含对降低补体D途径活性有效的量的(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)或其药学上可接受的盐,所述BID口服剂型在人血浆中提供为约65 ng/mL至95 ng/mL的两种不同的昼夜C谷水平中的较低者。
2.一种BID口服剂型,其包含对降低补体D途径活性有效的量的(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)或其药学上可接受的盐,所述BID口服剂型在人血浆中提供约为90 ng/mL+/- 10%的两种不同的昼夜C谷水平中的较低者。
3.一种口服剂型,其包含对降低旁路补体D途径活性有效的量的(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)或其药学上可接受的盐,所述口服剂型在人血浆中提供为约65 ng/mL至95 ng/mL的C谷水平。
4.一种口服剂型,其包含对降低补体D途径活性有效的量的(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)或其药学上可接受的盐,所述口服剂型在人血浆中提供约为90 ng/mL+/- 10%的C谷水平。
5.一种BID口服剂型,其包含对降低补体D途径活性有效的量的(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)或其药学上可接受的盐,所述BID口服剂型在人血浆中提供至少为65 ng/mL的两种不同的昼夜C谷水平中的较低者。
6.一种口服剂型,其包含对降低补体D途径活性有效的量的(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)或其药学上可接受的盐,所述口服剂型在人中提供至少为65 ng/mL的人血浆C谷。
7.一种BID口服剂型,其包含对降低补体D途径活性有效的量的(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)或其药学上可接受的盐,所述BID口服剂型在人血浆中提供约为100 ng/mL+/- 10%的两种不同的昼夜C谷水平中的较低者。
8.一种口服剂型,其包含对降低补体D途径的活性有效的量的(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)或其药学上可接受的盐,所述口服剂型在人血浆中提供至少为100 ng/mL+/- 10%的C谷。
9.如权利要求1至8中任一项所述的口服给药方案,其中所述剂型包含约100 mg至200mg。
10.如权利要求9所述的口服剂量,其包含约120 mg。
11.如权利要求9所述的口服剂量,其包含约200 mg。
12.如权利要求1-11中任一项所述的口服剂型,其中在患有阵发性睡眠性血红蛋白尿症(PNH)的患者中测量人血浆中的所述C谷水平。
13.如权利要求1-11中任一项所述的口服剂型,其中在患者中测量人血浆中的所述C谷水平,所述患者患有选自脂肪肝诸如非酒精性脂肪性肝炎(NASH)、肝脏炎症、肝硬化或肝衰竭的病症。
14.如权利要求1-11中任一项所述的口服剂型,其中在患者中测量人血浆中的所述C谷水平,所述患者患有选自肌萎缩侧索硬化、类风湿性关节炎、补体旁路途径(AP)相关肾病、组分3肾小球病(C3G)病症、C3肾小球肾炎(C3GN)、致密沉积物病(DDD)、膜性增生性肾小球肾炎(MPGN)和免疫复合物膜性增生性肾小球肾炎(IC-MPGN)的病症。
15.如权利要求1-11中任一项所述的口服剂型,其中在患者中测量人血浆中的所述C谷水平,所述患者患有选自肌萎缩侧索硬化、类风湿关节炎、补体旁路途径(AP)相关肾病和肾小球病的病症。
16.如权利要求1-11中任一项所述的口服剂型,其中在患者中测量人血浆中的所述C谷水平,所述患者患有选自年龄相关性黄斑变性(AMD)、视网膜变性、眼部疾病、地图样萎缩、早期或新生血管性年龄相关性黄斑变性、自身免疫性干眼病和环境性干眼病的病症。
17.一种用于治疗患有补体D相关病症的患者的方法,所述方法包括施用有效量的如权利要求1-11所述的口服剂型。
18.如权利要求17所述的方法,其中所述患者患有阵发性睡眠性血红蛋白尿症(PNH)。
19.如权利要求17所述的方法,其中所述患者患有选自脂肪肝诸如非酒精性脂肪性肝炎(NASH)、肝脏炎症、肝硬化或肝衰竭的病症。
20.如权利要求17所述的方法,其中所述患者患有选自肌萎缩侧索硬化;类风湿性关节炎、补体旁路途径(AP)相关肾病、组分3肾小球病(C3G)病症、C3肾小球肾炎(C3GN)、致密沉积物病(DDD)、膜性增生性肾小球肾炎(MPGN)和免疫复合物膜性增生性肾小球肾炎(IC-MPGN)的病症。
21.如权利要求17所述的方法,其中所述患者患有选自肌萎缩侧索硬化;类风湿性关节炎、补体旁路途径(AP)相关肾病和肾小球病的病症。
22.如权利要求17所述的方法,其中所述患者患有选自年龄相关性黄斑变性(AMD)、视网膜变性、眼部疾病、地图样萎缩、早期或新生血管性年龄相关性黄斑变性、自身免疫性干眼病和环境性干眼病的病症。
23.如权利要求17-22所述的方法,其中将所述剂型施用一个月或以上。
24.如权利要求17-22中任一项所述的方法,其中将所述剂型施用至少6个月。
25.如权利要求1至11中任一项所述的口服剂型,其提供了小于约2000 ng/mL的Cmax。
26.如权利要求1至11中任一项所述的口服剂型,其提供了小于约1000 ng/mL的Cmax。
27.如权利要求1-11中任一项所述的口服剂型,其用于治疗患有补体D相关病症的患者。
28.一种用于制备治疗患有补体D相关病症的患者的药物的方法,所述方法包括制备如权利要求1至11中任一项所述的口服剂型。
29.如权利要求28所述的口服剂型,其用于治疗权利要求18-22中所列的任何病症。
30.如权利要求29所述的方法,其用于治疗权利要求18-22中所列的任何病症。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862780573P | 2018-12-17 | 2018-12-17 | |
| US62/780,573 | 2018-12-17 | ||
| US201962877193P | 2019-07-22 | 2019-07-22 | |
| US62/877,193 | 2019-07-22 | ||
| PCT/US2019/066999 WO2020131974A1 (en) | 2018-12-17 | 2019-12-17 | Targeted dosing for the treatment of complement mediated disorders |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN113195472A true CN113195472A (zh) | 2021-07-30 |
Family
ID=71102328
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201980086227.3A Pending CN113195472A (zh) | 2018-12-17 | 2019-12-17 | 用于治疗补体介导的病症的靶向给药 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US20220079943A1 (zh) |
| EP (1) | EP3898617A4 (zh) |
| JP (1) | JP7471300B2 (zh) |
| KR (1) | KR20210104071A (zh) |
| CN (1) | CN113195472A (zh) |
| AU (1) | AU2019406830A1 (zh) |
| BR (1) | BR112021011727A2 (zh) |
| CA (1) | CA3123583A1 (zh) |
| MX (1) | MX2021007329A (zh) |
| WO (1) | WO2020131974A1 (zh) |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017035405A1 (en) | 2015-08-26 | 2017-03-02 | Achillion Pharmaceuticals, Inc. | Amino compounds for treatment of immune and inflammatory disorders |
| WO2017035401A1 (en) | 2015-08-26 | 2017-03-02 | Achillion Pharmaceuticals, Inc. | Amide compounds for treatment of immune and inflammatory disorders |
| WO2018160891A1 (en) | 2017-03-01 | 2018-09-07 | Achillion Pharmaceutical, Inc. | Pharmaceutical compounds for treatment of medical disorders |
| EP3589287B1 (en) | 2017-03-01 | 2022-09-14 | Achillion Pharmaceuticals, Inc. | Macrocyclic compounds for treatment of medical disorders |
| WO2018160889A1 (en) | 2017-03-01 | 2018-09-07 | Achillion Pharmaceuticals, Inc. | Aryl, heteroary, and heterocyclic pharmaceutical compounds for treatment of medical disorders |
| US20190038623A1 (en) | 2017-08-02 | 2019-02-07 | Achillion Pharmaceuticals, Inc. | Therapeutic regimens for treatment of paroxysmal nocturnal hemoglobinuria |
| US11814363B2 (en) | 2018-09-06 | 2023-11-14 | Achillion Pharmaceuticals, Inc. | Morphic forms of danicopan |
| WO2020051532A2 (en) | 2018-09-06 | 2020-03-12 | Achillion Pharmaceuticals, Inc. | Macrocyclic compounds for the treatment of medical disorders |
| KR20210093855A (ko) | 2018-09-25 | 2021-07-28 | 아칠리온 파르마세우티칼스 인코포레이티드 | 보체 인자 d 억제제의 형태체 형태 |
| MX2022014275A (es) * | 2020-05-12 | 2022-12-07 | Alexion Pharma Inc | Uso de inhibidores del factor d del complemento solos o junto con anticuerpos anti-c5 para el tratamiento de la hemoglobinuria paroxistica nocturna. |
| JP2024518787A (ja) * | 2021-05-10 | 2024-05-02 | アレクシオン ファーマシューティカルズ, インコーポレイテッド | 全身型重症筋無力症の処置のための補体因子d阻害剤の使用 |
| WO2024076999A2 (en) * | 2022-10-04 | 2024-04-11 | Engrail Therapeutics, Inc. | Gabaa receptor modulators and uses thereof |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103402996A (zh) * | 2011-01-04 | 2013-11-20 | 诺瓦提斯公司 | 可用于治疗年龄相关性黄斑变性(amd)的吲哚化合物或其类似物 |
| CN108024992A (zh) * | 2015-08-26 | 2018-05-11 | 艾其林医药公司 | 用于治疗医学障碍的芳基、杂芳基和杂环化合物 |
| WO2018160891A1 (en) * | 2017-03-01 | 2018-09-07 | Achillion Pharmaceutical, Inc. | Pharmaceutical compounds for treatment of medical disorders |
| CN111163767A (zh) * | 2017-08-02 | 2020-05-15 | 艾其林医药公司 | 治疗阵发性睡眠性血红蛋白尿症的治疗方案 |
| CN112996497A (zh) * | 2018-09-25 | 2021-06-18 | 艾其林医药公司 | 补体因子d抑制剂的形态形式 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018160889A1 (en) * | 2017-03-01 | 2018-09-07 | Achillion Pharmaceuticals, Inc. | Aryl, heteroary, and heterocyclic pharmaceutical compounds for treatment of medical disorders |
| WO2020109343A1 (en) * | 2018-11-29 | 2020-06-04 | F. Hoffmann-La Roche Ag | Combination therapy for treatment of macular degeneration |
-
2019
- 2019-12-17 US US17/414,057 patent/US20220079943A1/en active Pending
- 2019-12-17 BR BR112021011727-0A patent/BR112021011727A2/pt unknown
- 2019-12-17 CA CA3123583A patent/CA3123583A1/en active Pending
- 2019-12-17 WO PCT/US2019/066999 patent/WO2020131974A1/en unknown
- 2019-12-17 JP JP2021534700A patent/JP7471300B2/ja active Active
- 2019-12-17 KR KR1020217021046A patent/KR20210104071A/ko unknown
- 2019-12-17 AU AU2019406830A patent/AU2019406830A1/en active Pending
- 2019-12-17 EP EP19897806.6A patent/EP3898617A4/en active Pending
- 2019-12-17 CN CN201980086227.3A patent/CN113195472A/zh active Pending
- 2019-12-17 MX MX2021007329A patent/MX2021007329A/es unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103402996A (zh) * | 2011-01-04 | 2013-11-20 | 诺瓦提斯公司 | 可用于治疗年龄相关性黄斑变性(amd)的吲哚化合物或其类似物 |
| CN108024992A (zh) * | 2015-08-26 | 2018-05-11 | 艾其林医药公司 | 用于治疗医学障碍的芳基、杂芳基和杂环化合物 |
| WO2018160891A1 (en) * | 2017-03-01 | 2018-09-07 | Achillion Pharmaceutical, Inc. | Pharmaceutical compounds for treatment of medical disorders |
| CN111163767A (zh) * | 2017-08-02 | 2020-05-15 | 艾其林医药公司 | 治疗阵发性睡眠性血红蛋白尿症的治疗方案 |
| CN112996497A (zh) * | 2018-09-25 | 2021-06-18 | 艾其林医药公司 | 补体因子d抑制剂的形态形式 |
Non-Patent Citations (1)
| Title |
|---|
| CLAIRE L. HARRIS, ET AL.: "Developments in anti-complement therapy; from disease to clinical trial", 《MOLECULAR IMMUNOLOGY》, vol. 102, pages 89 - 119, XP085471606, DOI: 10.1016/j.molimm.2018.06.008 * |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2020131974A1 (en) | 2020-06-25 |
| BR112021011727A2 (pt) | 2021-08-31 |
| JP2022514269A (ja) | 2022-02-10 |
| EP3898617A4 (en) | 2022-08-17 |
| JP7471300B2 (ja) | 2024-04-19 |
| CA3123583A1 (en) | 2020-06-25 |
| US20220079943A1 (en) | 2022-03-17 |
| MX2021007329A (es) | 2021-09-21 |
| EP3898617A1 (en) | 2021-10-27 |
| KR20210104071A (ko) | 2021-08-24 |
| AU2019406830A1 (en) | 2021-07-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7471300B2 (ja) | 補体介在性疾患を治療するための的を絞った投与 | |
| US20220339169A1 (en) | Methods of treating or selecting a treatment for a subject resistant to tnf inhibitor using a nlrp3 antagonist | |
| ES2795667T3 (es) | Composiciones para inhibir la activación del complemento dependiente de MASP-2 | |
| KR102679441B1 (ko) | 조혈 줄기 세포 이식과 관련된 이식편대숙주병 및/또는 미만성 폐포 출혈 및/또는 정맥 폐쇄 질환의 치료 및/또는 예방 방법 | |
| JP2021181471A (ja) | ピペリジニルインドール誘導体の新規な使用 | |
| US20190038623A1 (en) | Therapeutic regimens for treatment of paroxysmal nocturnal hemoglobinuria | |
| EP3021833B2 (en) | Methods and formulations which allow the modulation of immune responses related to the administration of a biopharmaceutical drug | |
| KR102615565B1 (ko) | 리실 옥시다제 유사 2 억제제의 용도 | |
| TW202214690A (zh) | 用於治療各種疾病和病症的抑制masp-3的組合物和方法 | |
| EA018129B1 (ru) | Полипептиды, вариабельные домены антитела и антагонисты | |
| KR20230066126A (ko) | 고농도 항-c5 항체 제형 | |
| JP2012505160A (ja) | 炎症を処置する方法 | |
| EA026959B1 (ru) | Набор, содержащий агент, устраняющий sap, и анти-sap антитело, и способ лечения или профилактики амилоидной болезни | |
| US20220160820A1 (en) | Modulators of complement activity | |
| US11446376B2 (en) | Methods for treating heart failure using glucagon receptor antagonistic antibodies | |
| AU2017376884A1 (en) | Stable aqueous anti-C5 antibody composition | |
| US20210215714A1 (en) | Complement alternative pathway-associated nephropathy biomarkers | |
| EP3110847A1 (en) | Antibodies to matrix metalloproteinase 9 and methods of use thereof | |
| JP2017109987A (ja) | ErbB4+炎症性マクロファージによって媒介される疾患の治療方法 | |
| KR20220104201A (ko) | 조혈 줄기 세포 이식과 관련된 특발성 폐렴 증후군 (ips) 및/또는 모세혈관 누출 증후군 (cls) 및/또는 생착 증후군 (es) 및/또는 체액 과부하 (fo)를 치료 및/또는 예방하는 방법 | |
| AU2015201676A1 (en) | Anti-C5a antibodies and methods for using the antibodies | |
| WO2019222331A1 (en) | Treating solid tumors with bromodomain inhibitors | |
| WO2024148269A2 (en) | Oral dosage forms and oral dosing regimens for treatment of complement mediated diseases | |
| US20220073600A1 (en) | Methods for treating disease using psmp antagonists | |
| TW200920364A (en) | Method for reducing amyloid deposition, amyloid neurotoxicity, and microgliosis with (-)-nilvadipine enantiomer |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination |