CN1084324C - (甲基)丙烯酸的烷基酯的连续制备方法和所使用的设备 - Google Patents
(甲基)丙烯酸的烷基酯的连续制备方法和所使用的设备 Download PDFInfo
- Publication number
- CN1084324C CN1084324C CN96113098A CN96113098A CN1084324C CN 1084324 C CN1084324 C CN 1084324C CN 96113098 A CN96113098 A CN 96113098A CN 96113098 A CN96113098 A CN 96113098A CN 1084324 C CN1084324 C CN 1084324C
- Authority
- CN
- China
- Prior art keywords
- reactor
- reaction zone
- methyl
- acid
- reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 title claims abstract description 19
- 238000002360 preparation method Methods 0.000 title claims abstract description 9
- 125000005907 alkyl ester group Chemical group 0.000 title abstract description 5
- 238000006243 chemical reaction Methods 0.000 claims abstract description 109
- 238000000034 method Methods 0.000 claims abstract description 52
- 230000032050 esterification Effects 0.000 claims abstract description 28
- 238000005886 esterification reaction Methods 0.000 claims abstract description 28
- 239000007788 liquid Substances 0.000 claims abstract description 27
- 239000003054 catalyst Substances 0.000 claims abstract description 18
- 239000003377 acid catalyst Substances 0.000 claims abstract description 7
- 125000004432 carbon atom Chemical group C* 0.000 claims abstract description 6
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 53
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 claims description 43
- 239000000203 mixture Substances 0.000 claims description 36
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 34
- 125000005250 alkyl acrylate group Chemical group 0.000 claims description 33
- 239000002253 acid Substances 0.000 claims description 25
- 239000012074 organic phase Substances 0.000 claims description 24
- NIXOWILDQLNWCW-UHFFFAOYSA-N Acrylic acid Chemical compound OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 claims description 20
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims description 20
- 239000011541 reaction mixture Substances 0.000 claims description 19
- LDHQCZJRKDOVOX-UHFFFAOYSA-N 2-butenoic acid Chemical compound CC=CC(O)=O LDHQCZJRKDOVOX-UHFFFAOYSA-N 0.000 claims description 16
- 238000010523 cascade reaction Methods 0.000 claims description 14
- 239000000463 material Substances 0.000 claims description 11
- 239000000047 product Substances 0.000 claims description 11
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 claims description 9
- 239000000376 reactant Substances 0.000 claims description 9
- 229910052799 carbon Inorganic materials 0.000 claims description 7
- 230000003197 catalytic effect Effects 0.000 claims description 5
- 239000012043 crude product Substances 0.000 claims description 4
- 125000000217 alkyl group Chemical group 0.000 claims description 3
- 239000012530 fluid Substances 0.000 claims description 3
- 150000007513 acids Chemical class 0.000 claims description 2
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 claims description 2
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 claims 1
- 230000008569 process Effects 0.000 abstract description 9
- 230000002378 acidificating effect Effects 0.000 abstract 1
- 150000002148 esters Chemical class 0.000 description 32
- 238000009835 boiling Methods 0.000 description 30
- 150000001875 compounds Chemical class 0.000 description 22
- CQEYYJKEWSMYFG-UHFFFAOYSA-N butyl acrylate Chemical compound CCCCOC(=O)C=C CQEYYJKEWSMYFG-UHFFFAOYSA-N 0.000 description 14
- 239000006227 byproduct Substances 0.000 description 14
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 12
- 238000000926 separation method Methods 0.000 description 12
- 238000004821 distillation Methods 0.000 description 10
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 8
- 239000003795 chemical substances by application Substances 0.000 description 7
- 150000001983 dialkylethers Chemical class 0.000 description 7
- 238000004064 recycling Methods 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 6
- 230000000694 effects Effects 0.000 description 5
- 238000002474 experimental method Methods 0.000 description 5
- -1 oxygen ester Chemical class 0.000 description 5
- 230000009466 transformation Effects 0.000 description 5
- DURPTKYDGMDSBL-UHFFFAOYSA-N 1-butoxybutane Chemical compound CCCCOCCCC DURPTKYDGMDSBL-UHFFFAOYSA-N 0.000 description 4
- 238000009833 condensation Methods 0.000 description 4
- 230000005494 condensation Effects 0.000 description 4
- 238000000354 decomposition reaction Methods 0.000 description 4
- 238000007599 discharging Methods 0.000 description 4
- 235000019439 ethyl acetate Nutrition 0.000 description 4
- 239000007789 gas Substances 0.000 description 4
- 238000006116 polymerization reaction Methods 0.000 description 4
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N acetic acid Substances CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- NWVVVBRKAWDGAB-UHFFFAOYSA-N p-methoxyphenol Chemical compound COC1=CC=C(O)C=C1 NWVVVBRKAWDGAB-UHFFFAOYSA-N 0.000 description 3
- 238000003672 processing method Methods 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- AZQWKYJCGOJGHM-UHFFFAOYSA-N 1,4-benzoquinone Chemical compound O=C1C=CC(=O)C=C1 AZQWKYJCGOJGHM-UHFFFAOYSA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 230000035508 accumulation Effects 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 230000006399 behavior Effects 0.000 description 2
- 230000000052 comparative effect Effects 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 239000012535 impurity Substances 0.000 description 2
- 239000000178 monomer Substances 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 2
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- GEYOCULIXLDCMW-UHFFFAOYSA-N 1,2-phenylenediamine Chemical compound NC1=CC=CC=C1N GEYOCULIXLDCMW-UHFFFAOYSA-N 0.000 description 1
- WJFKNYWRSNBZNX-UHFFFAOYSA-N 10H-phenothiazine Chemical compound C1=CC=C2NC3=CC=CC=C3SC2=C1 WJFKNYWRSNBZNX-UHFFFAOYSA-N 0.000 description 1
- RBTBFTRPCNLSDE-UHFFFAOYSA-N 3,7-bis(dimethylamino)phenothiazin-5-ium Chemical compound C1=CC(N(C)C)=CC2=[S+]C3=CC(N(C)C)=CC=C3N=C21 RBTBFTRPCNLSDE-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 1
- VQTUBCCKSQIDNK-UHFFFAOYSA-N Isobutene Chemical group CC(C)=C VQTUBCCKSQIDNK-UHFFFAOYSA-N 0.000 description 1
- 125000000218 acetic acid group Chemical group C(C)(=O)* 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 238000004891 communication Methods 0.000 description 1
- 239000007859 condensation product Substances 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 238000005194 fractionation Methods 0.000 description 1
- 230000008676 import Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 229960000907 methylthioninium chloride Drugs 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 150000002989 phenols Chemical group 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 230000000630 rising effect Effects 0.000 description 1
- DCKVNWZUADLDEH-UHFFFAOYSA-N sec-butyl acetate Chemical compound CCC(C)OC(C)=O DCKVNWZUADLDEH-UHFFFAOYSA-N 0.000 description 1
- 238000001577 simple distillation Methods 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 239000012808 vapor phase Substances 0.000 description 1
- 238000003809 water extraction Methods 0.000 description 1
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/03—Preparation of carboxylic acid esters by reacting an ester group with a hydroxy group
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/08—Preparation of carboxylic acid esters by reacting carboxylic acids or symmetrical anhydrides with the hydroxy or O-metal group of organic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/34—Esters of acyclic saturated polycarboxylic acids having an esterified carboxyl group bound to an acyclic carbon atom
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
在通过让(甲基)丙烯酸和具有1-8个碳原子的一元链烷醇在均匀的、液态的无溶剂相中,在升高的温度下和在酸性酯化催化剂存在下进行反应来连续制备(甲基)丙烯酸的烷基酯的方法和装置中,将(甲基)丙烯酸、链烷醇和酸催化剂被连续进给反应区,反应区由至少两个串联的反应区的级联反应区组成,和一个反应区的液态排出料流形成下一个反应区的进给料流。级联反应区具有2-4个在空间上相互分开的反应区。
Description
本发明涉及通过让(甲基)丙烯酸和具有1-8个碳原子的一元链烷醇在均匀的、液态的无溶剂相中,在升高的温度下和在酸性酯化催化剂存在下进行反应来连续制备(甲基)丙烯酸的烷基酯的方法,其中(甲基)丙烯酸、链烷醇和酸催化剂被连续进给反应区,在一段停留时间之后,通过精馏,经过安装在反应区上的精馏区的顶部分离出所形成的、作为至少一种共沸混合物的一个组分的(甲基)丙烯酸烷基酯,该混合物-除(甲基)丙烯酸烷基酯外-还由水或水和作为另一组分的起始链烷醇组成,所得到的馏出物被分成至少一个含有(甲基)丙烯酸烷基酯的有机相和至少一个含水的水相,一部分含有(甲基)丙烯酸烷基酯的有机相经精馏区的顶部回用,和如果需要,水被循环至反应区,从剩余的含有(甲基)丙烯酸烷基酯的有机相分离出(甲基)丙烯酸烷基酯,和一部分反应混合物连续从反应区排出。
术语(甲基)丙烯酸按已知方式表示丙烯酸或甲基丙烯酸。(甲基)丙烯酸的烷基酯一般是已知的,而且是十分重要的,例如用作制备水性聚合物分散液(例如用作粘合剂)的起始单体。
通过让(甲基)丙烯酸和具有1-8个碳原子的一元链烷醇在均匀的液态相中,在升高的温度下和在给质子催化剂存在下进行反应来连续制备(甲基)丙烯酸的烷基酯的方法是已知的,并且例如描述在DE-A 14 68 932,22 26829和22 52 334中。这些是典型的平衡反应,其中(甲基)丙烯酸和特定的链烷醇转化转化成相应的酯的反应主要受平衡常数的限制。因此,对于经济的操作程序,未转化的起始原料必须从所得到的酯分离出来并回用至反应区。一般,从未转化的(甲基)丙烯酸分离所得到的酯已证明特别困难,因为它们的沸点一般非常接近。已经有人建议了提高(甲基)丙烯酸至相应的酯的转化率的各种措施,例如,使用相对于(甲基)丙烯酸来说更多地摩尔过量的链烷醇,借助于有机夹带剂(它形成合适的共沸混合物)除去反应的水或在反应过程中用合适的溶剂萃取所得到的酯。然而,这些方法具有缺点:必须回收大量过量的链烷醇或必须分离夹带剂或萃取剂。还有,大量过量的链烷醇促进副产物二烷基醚的形成。
现在还已知在未转化的(甲基)丙烯酸的沸点和所得到的烷基酯的沸点之间的差别能够通过如下措施得以增大:在至少一种低沸点含水共沸混合物中引入所得到的(甲基)丙烯酸的烷基酯,该混合物除了含有(甲基)丙烯酸烷基酯和水外还含有起始链烷醇,和通过连续精馏从包含未转化的(甲基)丙烯酸的反应区将(甲基)丙烯酸烷基酯(作为至少一种这样的共沸混合物的一个组分)除去,从而使(甲基)丙烯酸烷基酯与未转化的(甲基)丙烯酸分离。当连续进行上述操作程序时,以馏出物形式获得的含水共沸混合物被分成至少一种含有(甲基)丙烯酸烷基酯的有机相和至少一种含水的水相。一部分含有(甲基)丙烯酸烷基酯的有机相经连通精馏区的顶部加以回用,为的是提高精馏分离效果(精馏回流比)。
如果含有所需要的酯、并在该新型方法的过程中连续经过精馏区的顶部从反应区分离出的该含水共沸混合物不是多相非共沸混合物,该共沸混合物不会在其冷凝后自发地分成含水相和有机相。然而,这一分离能够通过简单方式实现,例如通过用水萃取在共沸混合物中所含有的链烷醇和精馏分离所得到的水/链烷醇。链烷醇最好被回用至反应区,优选经过所连通的精馏区的顶部,为的是提高精馏分离效果。
(甲基)丙烯酸烷基酯按本身已知的方式从含有(甲基)丙烯酸烷基酯的多余有机相中分离出来。在低级C1-C2链烷醇的酯化中,在酯化过程中以副产物形式形成的反应水通常足以形成含水共沸混合物的组合物(含水共沸混合物的合适组成例如描述在Azeotropic Data-III,Advances in Chemistry,Series 116,American Chemical Society,Washington,D.C.(1973))和因而作为含水共沸混合物的一个组分同时和连续地从酯化平衡中除去。然而,对于链长更长的链烷醇来说情况不是这样,在这些情况下超出在精馏过程中形成的反应水的附加水必须引入反应区。这可通过将在分离(以馏出物形式获得的)含水共沸混合物过程中产生的合适份额的含水相回用至酯化部分来实现。由于水趋向于对实际的酯化反应的转化率有影响作用,含水相的回用优选经过所连通的精馏区的顶部进行。如果经过所连通的精馏区的顶部精馏分离出的含水共沸混合物含有作为附加组分的起始链烷醇,这可按本身已知的方式从在共沸馏出物被分成有机相和含水相之后部分回用后保留下来的多余的有机和含水相中分离出来,并回用至反应区。由于起始链烷醇(两反应物中的一种)直接参与酯化反应,这一回用最好通过直接的途径来进行。
GB-1017622公开了制备丙烯酸正丁酯的合适方法。作为酯化条件,GB-1017622建议起始链烷醇与起始酸的摩尔比为2.3-5,反应温度<100℃和催化活性的硫酸或有机磺酸的含量为>0.05-<5wt%,以反应物的总重量为基础计。这一方法的缺点是需要大量过量的起始链烷醇,它会促进不需要的二烷基醚的形成,以及在上述条件下以丙烯酸的用量为基础计的丙烯酸正丁酯的产率不完全令人满意。
德国专利2,552,987公开了通过让丙烯酸与具有1-4个碳原子的一元链烷醇按摩尔比1(链烷醇)∶1(丙烯酸)-2(链烷醇)∶1(丙烯酸),在均匀的、液态的无溶剂相中,在升高的温度下和在作为催化剂的硫酸或有机磺酸存在下进行反应来连续制备丙烯酸的烷基酯的方法,其中丙烯酸、链烷醇和酸催化剂被连续进给反应区,在几个小时的停留时间后所得到的丙烯酸烷基酯通过精馏经过安装在反应区上的精馏塔的顶部(塔顶压力为0.1-1atm),作为至少一种共沸混合物(除了丙烯酸烷基酯外,该混合物还包括水或水和作为另一组分的起始链烷醇)的一个组分被分离出来,所得到的馏出物I被分成一个含有所得到的丙烯酸酯的有机相和一个含水相,一部分有机相经精馏区的顶部回用,和如果需要,一部分含水相经精馏区的顶部回用,为的是保持含水共沸混合物的组成,烷基酯按本身已知的方法从剩余的有机相分离出来和一部分反应混合物连续从反应区排出并通过蒸馏除去高沸点化合物,所得到的馏出物II被回用到反应区。
德国专利2,552,987的主要目的是避免从起始链烷醇形成不需要的醚。然而,德国专利2,552,987的方法的缺点是,尽管来自反应混合物的排出物进行了蒸馏处理和所得到的馏出物回用到反应区,但以所使用的丙烯酸为基础计算的丙烯酸烷基酯的产率仍不能令人满意。所实现的二烷基醚副产物的形成量的减少程度也不能完全令人满意。此外,在实施例中所需要的停留时间不能令人满意。这对于时空产率来说也是如此。这可能是由于酸性酯化催化剂的浓度低造成的。
因而有人建议(德国专利申请195 10 891.4)在高浓度的酸性酯化催化剂存在下进行相应的酯化方法,它会促进在其它副产物的酯化中所形成的含氧酯的再分解,因而,对于给定的停留时间,提高了以所使用的(甲基)丙烯酸为基础计算的产率。
本发明的目的是提供一种用于制备(甲基)丙烯酸的烷基醚的酯化方法,该方法能够获得酯的最佳产率和同时最大程度地减少二烷基醚副产物的形成。
我们发现这一目的可通过连续制备(甲基)丙烯酸的烷基酯的方法来实现,这是通过让(甲基)丙烯酸和具有1-8个碳原子的一元链烷醇在均匀的、液态的无溶剂相中,在升高的温度下和在酸性酯化催化剂存在下进行反应来连续制备(甲基)丙烯酸的烷基酯的方法,其中(甲基)丙烯酸、链烷醇和酸催化剂被连续进给反应区,在一段停留时间之后,通过精馏、经过安装在反应区上的精馏区的顶部,分离出所形成的、作为至少一种共沸混合物的一个组分的(甲基)丙烯酸烷基酯,该混合物-除(甲基)丙烯酸烷基酯外-还由水或水和作为另一组分的起始链烷醇组成,所得到的馏出物被分成至少一个含有(甲基)丙烯酸烷基酯的有机相和至少一个含水的水相,一部分含有(甲基)丙烯酸烷基酯的有机相经精馏区的顶部回用,和如果需要,水被循环至反应区,从剩余的含有(甲基)丙烯酸烷基酯的有机相分离出(甲基)丙烯酸烷基酯,和一部分反应混合物连续从反应区排出,其中反应区由至少两个串联的(优选连续操作的)级联反应区组成,并且一个反应区的液态排出料流形成下一个反应区的进给料流。
在这一方法中,酸性酯化催化剂的重量(以具体的反应区中含有的酯化混合物的量为基础计算)沿着级联反应区逐渐提高,归因于酸性酯化催化剂的较低的挥发性。这导致酯化反应和再分解反应的空间分离并得以减少二烷基醚的形成。然而,沿着级联反应区这一酸的分布型式(profile)也可以通过向各反应区中外加其它的酸得以实现。
在一个实施方案中,一个反应区的液体输出料流借助于泵通入下一反应区。在又一实施方案中,排出料流也借助于溢流被引入下一反应区,或流入精馏塔的较低部分,不含低沸点化合物的精馏混合物从此处流入下一反应区(它与精馏塔保持液体联通)。
在本发明的一个理想的实施方案中,在第一个反应区中催化活性的酸的含量是0.1-5wt%(以其中所含有的反应混合物为基础计)的硫酸(或等摩尔量的有机磺酸,优选甲烷磺酸,苯磺酸,十二烷基苯磺酸或对-甲苯磺酸或者有机磺酸和硫酸的混合物)。在最后一个反应区中相应的酸含量优选是>5-20wt%。在级联反应区中反应物的总停留时间一般是1-20小时。
各反应区是空间上相互分离的反应器,它们的数目从成本上考虑最好是≥2和≤4。如果一个以上的反应区在一个和同一反应器(例如通过使用隔板)中产生,则反应区的数目也可以大于4。
在本新颖方法的一个可能实施方案中,只有一个精馏塔安装在整个反应区上,该塔的液体回流物仅仅连通于第一反应区。对于剩下的反应区,只有一个连通于精馏塔的上升蒸汽通道。在再一实施方案中,第一反应区作为停留容器(初步反应器)进行操作,而没有连通于精馏塔的通道。优选地,第一反应区在气体侧即不连接于下游的反应区也不连接于精馏区。精馏塔的顶部压力优选是0.1-1大气压,常压是特别优选的。
在各反应区中反应混合物的温度通常对应于具体的反应混合物在设定的压力下的沸点,即它通常沿着级联反应区逐渐提高,这对于本方法的目的来说是有利的。在第一反应区中,温度通常是≥80℃和≤125℃。在级联反应区的最后一个反应区中,它应该是≤135℃,为的是抑制作为付反应的不需要的聚合反应。
转化率通常是≥95mol%,以(甲基)丙烯酸的用量为基础计。第一反应区优选以高达≥90mol%的转化率进行操作。当停留反应器(初步反应器)用作第一反应区时,其中的反应混合物一般不沸腾。这里同样地,建议的反应温度是80-125℃。当停留反应器用作第一反应区时,一般达到平衡值的约80%的转化率。有利的是,反应区这样设计,以使停留时间沿着级联反应区从反应区到反应区下降。
在本发明的理想的实施方案中,所使用的链烷醇/(甲基)丙烯酸摩尔比是≥1和一般是低于5。1∶1至3∶1的比例是特别有利的。1.1∶1-1.8∶1的比例是极其有利的。特别有利的是使用硫酸和/或有机磺酸作为酯化催化剂。
一般,从反应区、优选从最后的反应区连续排出2.5wt%(以所获得的酯的量为基础计)的反应混合物,为的是限制高沸点的不可分解的副产物的量。
有利的是,一部分料流从反应区级联的最后反应区回用至第一反应区和如果需要,回用至其它反应区。酸性酯化催化剂与高沸点化合物一道排出并连续加入到反应混合物中。本工艺方法的稳定剂也可同时排出,这样它的含量固定在稳态的值。这会得到所需酸性酯化催化剂浓度的稳态条件。与此同时,这一循环免除了催化剂的后处理和减少新鲜催化剂的需要量。
由于所使用的丙烯酸含有少量的乙酸,除了二烷基醚以外,还形成副产物形式的乙酸烷基酯。这两种副产物都流过所连通的精馏塔的顶部和在蒸馏分离链烷醇/酯过程中保留在链烷醇中,它最好被回用至起始混合物中。结果,两种杂质积累在返回的料流中。借助于以下事实确定稳态浓度:在某种程度上,这些低沸点化合物进入所得到的酯中。取决于酯的纯度要求,也需要从链烷醇循环中除去一些杂质。
在链烷醇/酯分离的下游的简单蒸馏塔中,酯能够通过蒸馏与工艺方法的稳定剂分离,并能够在它们的原位用贮存稳定剂处理。优选地,被酯化的酸是丙烯酸和/或被酯化的链烷醇是C1-C4链烷醇,尤其正丁醇。
本新颖的方法特别优选用于制备丙烯酸正丁酯。
在根据本发明设置为级联反应区的反应区中形成的蒸汽被连续进给精馏区,如上所述。对于经过顶部从中分离的和含有目标酯的含水共沸混合物,可以分为两种情况。如果它是多相非共沸混合物,例如与制备丙烯酸正丁酯时的情况一样,共沸混合物在它冷凝后自发分成含水相和有机相。含水相通常主要由水和少量链烷醇组成,而有机相一般主要由所得到的酯和链烷醇组成。为了提高精馏分离效果,适当比例的有机相经精馏区的顶部回用。
为了保持含水共沸混合物的组成,适当比例的含水相被回用至反应区,优选经过所连通的精馏区的顶部。所存在的链烷醇能够从未回用部分的含水相中分离出来(例如通过抽提(如用空气))并被回用至反应区。回用优选直接进行。排出所得到的基本纯净的水。所形成的(甲基)丙烯酸烷基酯按本身已知的方法,例如根据DE-A25 52 987,从未回用部分的有机相中分离出来。例如,剩余的有机相被进给下游的精馏塔,链烷醇以纯净的形式或作为由酯和链烷醇组成的共沸混合物形式经过顶部分离出来。如此分离的链烷醇(以纯净形式或以共沸物形式)优选被回用至反应区。回用优选直接进行。
这一精馏塔的底液主要由所需要的酯和少量比这种酯有更低和更高沸点的副产物组成。这一精馏塔的底液因而也称作粗(甲基)丙烯酸烷基酯。低沸点副产物尤其是二烷基醚和起始链烷醇的乙酸酯,因为(甲基)丙烯酸用链烷醇的酯化通常从粗(甲基)丙烯酸开始。这主要由C3/C4起始化合物如丙烯或异丁烯的催化气相氧化反应产生的,少量乙酸形成为副产物(参见DE-A44 36 243)。高沸点副产物例如是α,β-单烯属不饱和目标酯的齐聚物和聚合物。
在下游低沸点化合物精馏塔中,低沸点副产物通常经过顶部与粗(甲基)丙烯酸烷基酯分离,在所需要的纯(甲基)丙烯酸烷基酯能够经过下游的高沸点化合物精馏塔中的顶部分离出来之前。高沸点化合物精馏塔的底液(该液体含有高沸点副产物)最好被回用至反应区,优选直接进行。
在一个具体的实施方案中,如果乙酸酯从返回的丁醇液流分离出来,它按以上所述进行,在分离任何夹带的液滴之后通过侧边泄出将酯从用来回收丁醇的精馏塔中排出并被冷凝得到纯的酯。在冷凝中,将贮存稳定剂加入到其中(例如氢醌单甲醚)。在这一实施方案中,用于丁醇回收的精馏塔的底部排出物(主要包括丙烯酸烷基酯)最好被回用至用于分解少量存在的高沸点化合物和用于回收纯酯的级联反应区的最后反应区中。
在两种情况下,从级联反应区的这一最后反应区排出(优选连续地)少量的反应混合物,为的是保持稳态和避免高沸点化合物在其中的积累。按同时排出的酸催化剂的量向该反应区中加入酸催化剂。
通常,从反应区中排出的含水共沸混合物不含起始的酸,当正确地确定精馏分离条件时。然而,如果不是这种情况,通过用水萃取,该酸能够与链烷醇一起被分离出来,萃取物然后按本身已知的方法通过精馏进一步分离。在本新颖的方法中,优选的是,酯化反应与精馏分离和萃取都是在常规量的普通聚合抑制剂存在下进行。一般来说,使用0.01-0.1wt%(以α,β-单烯属不饱和单体的量为基础计)合适的聚合抑制剂。它们最好在所连通的精馏塔的顶部和在链烷醇/酯分离塔的顶部加进去。合适的聚合抑制剂的例子是酚类化合物,如氢醌和氢醌单甲醚,以及对苯醌,吩噻嗪,甲基蓝,亚苯基二胺和/或空气。
与现有技术的方法相比,本新颖方法突出表现在:明显减少操作步骤和分离操作的次数,较短的停留时间,较高产率的所需酯(以所使用的起始酸为基础计)较少量的醚和减少了液体相从反应区的排出量;以及突出表现在以下事实:不再需要处理来自反应区的排出物。后者和提高的产率都归因于这样一种事实:在反应区中建立的稳态在动力学上受到控制。较高沸点的含氧酯类(例如烷氧基丙酸酯或酰氧基丙酸酯)很可能起着关键的作用。
在一理想的实施方案中,整个工艺方法是用总共4个分离塔进行操作,用于除去高沸点化合物和低沸点化合物。这些是用来经过顶部分出酯的塔,丁醇回收塔,用来从返回的丁醇液流中分出乙酸酯的乙酸酯塔和用来从排出的反应水中分出残留的丁醇的汽提塔。
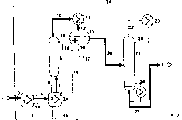
本发明还涉及用来进行所描述的连续制备(甲基)丙烯酸烷基酯的方法的装置,它具有第一反应器(1),该反应器设有反应物的进料管(3)和它的顶部经管道(5)连接于精馏塔(6)的下部,精馏塔的顶部经一个冷凝器连接于一个具有粗产物出料管(20)的分离器(13),其中第一反应器(1)的下端经管道(4)连接于至少一个其它反应器(2)的下端,该反应器(2)的顶部经管道(8)连接于精馏塔(6)的下部。
优选地,至少一个其它反应器(2)的上部经管道(7)连接于第一反应器(1)的上部。
优选地,精馏塔(6)的下部经管道(9)连接于第一反应器(1)的顶部。
优选地,分离器(13)的下部连接于精馏塔(6)的顶部和连接于至少一个其它反应器(2)的顶部。
优选地,分离器(13)连接于另外一个精馏塔(21)。
本发明还涉及进行所描述的连续制备(甲基)丙烯酸烷基酯的方法的装置,它具有第一反应器(1),该反应器设有反应物的进料管(3)和它(作为停留容器)形成第一反应区,其中第一反应器(1)具有连接于至少一个其它反应器(2)的液体侧连接管路(4),反应器(2)的顶部经管道(8)连接于精馏塔(6)的下部,精馏塔(6)的顶部经一个冷凝器连接于一个具有粗产物出料管道(20)的分离器(13)。
优选地,至少一个其它反应器(2)经管道(7)连接于第一反应器(1)。
优选地,精馏塔(6)的下部经管道(9)连接于其它反应器(2)当中的任何一个的顶部。
优选地,分离器(13)的下部连接于精馏塔(6)的顶部和连接于其它反应器(2)中的任何一个。
优选地,分离器(13)连接于其它精馏塔(21)。
从所描述的实施例并参考附图,本发明的进一步的细节和优点将变得清楚明白。在此时应该再次强调,所有以上和以下的叙述内容对于制备丙烯酸正丁酯的方法来说尤其适用,即丁醇是正丁醇。
实施例1
下面参考图1叙述本实施例。5mol/h的丙烯酸(AA)和7mol/h的丁醇(BuOH)和作为酯化催化剂的硫酸的混合物经过管道3进给反应器1,反应器1具有强制循环蒸发器1A。硫酸的量是2-3wt%,以反应混合物为基础计。反应在120℃和1大气压和在2.5小时的停留时间内进行。气态反应产物从反应器1的顶部经过管道5进给精馏塔。反应混合物的一部分液流(液流4)通过循环泵(未画出)从反应器1的下部连续泵抽到一个具有强制循环蒸发器2A的第二反应器2的下部。第二反应器在130℃的较高温度下和在约10wt%的较高硫酸浓度下进行操作。反应器2具有连接于反应器1的溢流管7,借助于它可在反应器1中保持恒定的催化剂浓度。反应器2同样具有连接于精馏塔6的下部的气体侧管道8。它的流出液经过管道9流入反应器1。在反应器2中发生随后的反应和所形成的氧化化合物(米歇尔(Michael)加合物)的就地分解反应。两反应器1和2在它们的下端经过管道4相互连接,形成新型的反应区,构成级联反应区。从这一反应区连续流入蒸馏塔6的气体在其中进行精馏,从这一蒸馏塔6的顶部经管道10流出的含水共沸混合物(它含有所要制备的酯并由丙烯酸丁酯、丁醇和水组成)被进给冷凝器11并在其中冷凝。冷凝物经管道12流入分离器13。在分离器中,共沸混合物被分成含水相和有机相。含水相(主要包括水和少量丁醇)通过管道14泄出和经管道15部分地加入到蒸馏塔6的顶部,进行精馏分离操作以及在其中与上升的蒸汽形成共沸物。另一部分含水相经管道16加入到反应器2的顶部。再一部分经管道17排出,用来抽提出丁醇。在实验中,从冷凝器通过管道14流出的回流物的一半量经过管道15加入到蒸馏塔中和一半经过管道16加入到反应器2中。有机回流物从分离器13经过管道18进给蒸馏塔6的顶部,为的是抑制丙烯酸。含水的回流液加以温度调节,而有机回流液加以液面控制。在反应器2的下部设置的管道19用未排泄保留下来的残余物。
主要由丁醇和丙烯酸丁酯组成的有机相仅仅含有876ppm(ppm数据全部按重量计算)的二丁基醚。它连续从分离器13经管道20排出和作为进给料引入另一塔21(它在实验中是泡罩塔形式)中,为的是回收丁醇。在这一丁醇回收塔21中,剩余的丁醇(有水和部分丙烯酸丁酯的残余物)经过顶部通过管道22排出并被加入到冷凝器23中。从后者,剩余的丁醇经过管道24加入到反应物的混合物中,反应物混合物通过管道3导入反应器1。一部分馏出液从分离器23经过管道25加入到塔21的顶部,作为用来提高精馏分离效果的回流液。
不含高沸点化合物的粗丙烯酸丁酯(仅含有低沸点化合物的残余物和含有99.5wt%的丙烯酸丁酯)从丁醇回收塔21经过管道27排出。在随后除去低沸点化合物和高沸点化合物之后,有可能获得纯度为>99.9wt%的纯丙烯酸丁酯。丁醇回收塔21在400毫巴下进行操作。该纯酯仅仅含有598ppm的二丁基醚。借助于热交换器26在这一塔的底部确立115℃的温度。
在同样条件下的第二实验,仅仅减少丁醇过量的程度(摩尔比1.3∶1),导致液流20中的醚减少至623ppm和在纯产物的量上提高374ppm。以丙烯酸为基础计算的丙烯酸丁酯的产率,在第一实验中是理论值的98.8%和在第二实验中是理论值的97.3%。
对比实施例
为了对比,仅仅使用一个反应区进行已知的酯化方法。在本对比实施例中,将4mol/h丙烯酸、4mol/h新鲜丁醇和1.6mol/h的回用的丁醇(来自丁醇回收的馏出物)加入到循环蒸发器(2.5升,装填水准2升)中。停留时间是2.5小时。酯化反应在9.7wt%的硫酸存在下于126℃进行。反应器连接于泡罩塔(30个塔板),该塔用15ml/h的氢醌单甲醚(MeHQ)丁醇溶液加以稳定。通过向塔中添加340ml/h作为回流液(共沸夹带剂)的水,使所形成的丙烯酸丁酯经过顶部排出。还有,175ml/h有机回流液被加入到塔中以便抑制丙烯酸。
剩余的有机相(610ml/h)作为进给料被连续加入到第二泡罩塔中,与在实施例中用来回收丁醇(BuOH)的新型方法一样。在丁醇塔(处在115℃下)的底部获得含有1295ppm二丁基醚的粗酯。1940ppm的醚存在于供给BuOH塔的进给料中。该塔用MeHQ(在丁醇中2%)加以稳定。
在除去低沸点化合物和高沸点化合物之后,获得纯度为99.9wt%的纯酯。按丙烯酸计算的产率是理论值的98.4%。
实施例2
参考图2叙述本实施例。这里,与图1中同样参考符号用来表示相同的或对应的部件。
5mol/h丙烯酸(AA)和5.5mol/h正丁醇(BuOH)和作为酯化催化剂的硫酸的混合物经进料管3加入到具有强制循环蒸发器1A的反应器1(停留反应器)。BuOH/AA的摩尔比是1.1∶1。硫酸的量是2-3wt%(以反应混合物为基础计算)。反应器1中的反应是在120℃下和在3.5小时的停留时间下进行的。液体反应混合物的一部分液流连续从反应器1经过管道4通入第二反应器2。第二反应器在130℃的较高温度下和在约10wt%的较高硫酸浓度下进行操作。反应器2具有连接于反应器1的液体回用管道7,借助于回用管道在反应器1中保持恒定的催化剂浓度。反应器2具有连接于蒸馏塔6的下部的气体侧管道8。它的流出液经过管道9流入反应器2。在反应器2中发生随后的反应和所形成的氧化化合物(未歇尔(Michael)加合物)的就地分解反应。两反应器1和2经过管道4相互连接,形成新型的反应区,构成级联反应区。
反应混合物按类似第一个实施例中描述的纯酯的分离方法加以处理。在除去低沸点化合物和高沸点化合物之前,在丁醇回收塔21的下端排出的粗酯含有134ppm的二丁基醚。纯酯含有99.9%的丙烯酸丁酯。丙烯酸丁酯的产率是理论值的98.1%(以丙烯酸为基础计算)。
Claims (26)
1、通过让(甲基)丙烯酸和具有1-8个碳原子的一元链烷醇在均匀的、液态的无溶剂相中,在升高的温度下和在酸性酯化催化剂存在下进行反应来连续制备(甲基)丙烯酸的烷基酯的方法,其中(甲基)丙烯酸、链烷醇和酸催化剂被连续进给反应区,在一段停留时间之后,通过精馏、经过安装在反应区上的精馏区的顶部,分离出所形成的、作为至少一种共沸混合物的一个组分的(甲基)丙烯酸烷基酯,该混合物-除(甲基)丙烯酸烷基酯外-还由水或水和作为另一组分的起始链烷醇组成,所得到的馏出物被分成至少一个含有(甲基)丙烯酸烷基酯的有机相和至少一个含水的水相,一部分含有(甲基)丙烯酸烷基酯的有机相经精馏区的顶部回用,和如果需要,水被循环至反应区,从剩余的含有(甲基)丙烯酸烷基酯的有机相分离出(甲基)丙烯酸烷基酯,和一部分反应混合物连续从反应区排出,其中反应区由至少两个串联的反应区的级联反应区组成和一个反应区的液态排出料流形成下一个反应区的进给料流。
2、根据权利要求1所要求的方法,其中联级反应区具有2-4个在空间上相互分开的反应区。
3、根据权利要求1所要求的方法,其中从反应区上升的蒸汽被加入到各精馏区,而精馏区的液体回流液仅仅循环至第一反应区。
4、根据权利要求1-3中任何一项所要求的方法,其中第一反应区在气体侧既不连接于下游反应区也不连接于精馏区。
5、根据权利要求1-3中任何一项所要求的方法,其中在第一反应区中的温度是80-125℃和在最后反应区中的温度是低于135℃。
6、根据权利要求1-3中任何一项所要求的方法,其中精馏塔的顶部压力是0.1-1大气压。
7、根据权利要求1-3中任何一项所要求的方法,其中反应物在级联反应区中的总停留时间是1-20小时。
8、根据权利要求1-3中任何一项所要求的方法,其中所使用的链烷醇/(甲基)丙烯酸的摩尔比是≥1。
9、根据权利要求8所要求的方法,其中所使用的和加入到第一反应区的链烷醇/(甲基)丙烯酸摩尔比是1∶1-3∶1。
10、根据权利要求1-3中任何一项所要求的方法,其中硫酸或有机磺酸用作酯化催化剂。
11、根据权利要求10所要求的方法,其中所使用的有机磺酸是甲烷磺酸,苯磺酸,十二烷基苯磺酸或对-甲苯磺酸。
12、根据权利要求1-3中任何一项所要求的方法,其中催化活性酸在第一反应区中的含量,以其中所含有的反应混合物为基础,是0.1-5重量%的硫酸或等摩尔量的有机磺酸。
13、根据权利要求1-3中任何一项所要求的方法,其中催化活性酸在最后反应区中的含量,以其中所含有的反应混合物为基础,是5-20重量%的硫酸或等摩尔量的有机磺酸。
14、根据权利要求1-3中任何一项所要求的方法,其中所要酯化的酸是丙烯酸。
15、根据权利要求1-3中任何一项所要求的方法,其中所要酯化的链烷醇是C1-C4链烷醇。
16、根据权利要求15所要求的方法,其中所要酯化的链烷醇是正丁醇。
17、用来进行根据权利要求1-3中任何一项所要求的连续制备(甲基)丙烯酸烷基酯的方法的装置,它具有第一反应器(1),该反应器设有反应物的进料管(3)和它的顶部经管道(5)连接于精馏塔(6)的下部,精馏塔的顶部经一个冷凝器连接于一个具有粗产物出料管(20)的分离器(13),其中第一反应器(1)的下端经管道(4)连接于至少一个其它反应器(2)的下端,该反应器(2)的顶部经管道(8)连接于精馏塔(6)的下部。
18、根据权利要求17所要求的装置,其中至少一个其它反应器(2)的上部经过管道(7)连接于第一反应器(1)的上部。
19、根据权利要求17所要求的装置,其中精馏塔(6)的下部经过管道(9)连接于第一反应器(1)的顶部。
20、根据权利要求17所要求的装置,其中分离器(13)的下部连接于精馏塔(6)的顶部和连接于至少一个其它反应器(2)的顶部。
21、根据权利要求17所要求的装置,其中分离器(13)连接于其它精馏塔(21)。
22、用来进行根据权利要求1-3中任何一项所要求的连续制备(甲基)丙烯酸烷基酯的方法的装置,它具有第一反应器(1),该反应器设有反应物的进料管(3)和它作为停留容器形成第一反应区,其中第一反应器(1)具有连接于至少一个其它反应器(2)的液体侧连接管路(4),反应器(2)的顶部经管道(8)连接于精馏塔(6)的下部,精馏塔(6)的顶部经一个冷凝器连接于一个具有粗产物出料管道(20)的分离器(13)。
23、根据权利要求22所要求的装置,其中至少一个其它反应器(2)经管道(7)连接于第一反应器(1)。
24、根据权利要求22所要求的装置,其中精馏塔(6)的下部经管道(9)连接于其它反应器(2)当中的任何一个的顶部。
25、根据权利要求22所要求的装置,其中分离器(13)的下部连接于精馏塔(6)的顶部和连接于其它反应器(2)中的任何一个。
26、根据权利要求22所要求的装置,其中分离器(13)连接于其它精馏塔(21)。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE19536178.4 | 1995-09-28 | ||
| DE19536178A DE19536178A1 (de) | 1995-09-28 | 1995-09-28 | Verfahren und Vorrichtung zur kontinuierlichen Herstellung von Alkylestern der (Meth)acrylsäure |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1150142A CN1150142A (zh) | 1997-05-21 |
| CN1084324C true CN1084324C (zh) | 2002-05-08 |
Family
ID=7773485
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN96113098A Expired - Fee Related CN1084324C (zh) | 1995-09-28 | 1996-09-27 | (甲基)丙烯酸的烷基酯的连续制备方法和所使用的设备 |
Country Status (10)
| Country | Link |
|---|---|
| US (2) | US5811574A (zh) |
| EP (1) | EP0765859B1 (zh) |
| JP (1) | JPH09110789A (zh) |
| KR (1) | KR970015558A (zh) |
| CN (1) | CN1084324C (zh) |
| CA (1) | CA2185821A1 (zh) |
| CZ (1) | CZ273696A3 (zh) |
| DE (2) | DE19536178A1 (zh) |
| MX (1) | MX9604237A (zh) |
| SG (1) | SG72705A1 (zh) |
Families Citing this family (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE19604252A1 (de) * | 1996-02-06 | 1997-08-07 | Basf Ag | Verfahren und Vorrichtung zur kontinuierlichen Herstellung von Alkylestern der (Meth)acrylsäure |
| DE19648746A1 (de) * | 1996-11-25 | 1998-05-28 | Basf Ag | Verfahren zur Herstellung von (Meth)acrylsäureestern |
| ATE228109T1 (de) * | 1997-05-20 | 2002-12-15 | Union Carbide Chem Plastic | Verfahren zur raffination von butylacrylat |
| WO1998052903A1 (en) * | 1997-05-20 | 1998-11-26 | Union Carbide Chemicals & Plastics Technology Corporation | Processes for conducting equilibrium-limited reactions |
| CA2252748A1 (en) * | 1997-11-17 | 1999-05-17 | Rohm And Haas Company | Process for preparing alkyl (meth) acrylates |
| DE19851983A1 (de) | 1998-11-11 | 2000-05-18 | Basf Ag | Verfahren zur kontinuierlichen Herstellung von Alkylestern der (Meth)acrylsäure |
| JP2003521478A (ja) * | 1999-06-17 | 2003-07-15 | ユニオン・カーバイド・ケミカルズ・アンド・プラスティックス・テクノロジー・コーポレイション | 平衡律速反応を行う方法 |
| US6482976B1 (en) | 1999-06-17 | 2002-11-19 | Union Carbide Chemicals & Plastics Technology Corporation | Processes for conducting equilibrium-limited reactions |
| JP4558870B2 (ja) * | 1999-11-08 | 2010-10-06 | 株式会社日本触媒 | 塔式処理方法および装置 |
| DE10062754A1 (de) * | 2000-12-15 | 2002-06-20 | Mg Technologies Ag | Verfahren zum Herstellen eines Esters der Acrylsäure oder Methacrylsäure |
| DE10127941A1 (de) | 2001-06-08 | 2002-05-29 | Basf Ag | Verfahren zur Herstellung von (Meth)acrylsäureestern |
| DE10138630A1 (de) * | 2001-08-13 | 2003-02-27 | Basf Ag | Verfahren zur Herstellung von Rein-(Meth)acrylsäure und Methacrylsäureestern |
| WO2004078679A2 (en) * | 2003-02-28 | 2004-09-16 | Union Carbide Chemicals & Plastics Technology Corporation | Process for conducting equilibrium-limited reactions |
| FR2876375B1 (fr) * | 2004-10-12 | 2007-02-02 | Arkema Sa | Procede de preparation d'esters ou d'anydrides (meth) acryliques |
| DE102005010588A1 (de) * | 2005-03-08 | 2006-05-24 | Basf Ag | Verfahren zur Herstellung von Alkylestern der (Meth)acrylsäure |
| DE102005010587A1 (de) * | 2005-03-08 | 2006-06-08 | Basf Ag | Verfahren zur Herstellung von Alkylestern der (Meth)acrylsäure |
| DE102005023976A1 (de) * | 2005-05-20 | 2006-11-23 | Röhm Gmbh | Verfahren zur Umesterung |
| DE102005043719A1 (de) * | 2005-09-13 | 2007-03-15 | Röhm Gmbh | Vorrichtung und Verfahren für kontinuierlich durchgeführte Gleichgewichtsreaktionen |
| US8765217B2 (en) | 2008-11-04 | 2014-07-01 | Entrotech, Inc. | Method for continuous production of (meth)acrylate syrup and adhesives therefrom |
| US8329079B2 (en) | 2009-04-20 | 2012-12-11 | Entrochem, Inc. | Method and apparatus for continuous production of partially polymerized compositions and polymers therefrom |
| CN102249913B (zh) * | 2011-05-17 | 2013-11-06 | 上海华谊丙烯酸有限公司 | 一种制备丙烯酸丁酯的方法 |
| KR101601938B1 (ko) | 2013-08-30 | 2016-03-09 | 주식회사 엘지화학 | (메트)아크릴산의 연속 회수 방법 및 회수 장치 |
| FR3024143B1 (fr) * | 2014-07-28 | 2016-07-15 | Arkema France | Procede perfectionne de fabrication de (meth)acrylates d'alkyle |
| EP3212606B1 (en) * | 2014-10-31 | 2019-11-20 | Dow Global Technologies Llc | Process for in situ water removal from an oxidative esterification reaction using a coupled reactor-distillation system |
| CN110997619B (zh) * | 2017-08-17 | 2023-06-27 | 巴斯夫欧洲公司 | 连续制备丙烯酸正丁酯或丙烯酸异丁酯的方法 |
| JP2023537201A (ja) | 2020-06-29 | 2023-08-31 | ベーアーエスエフ・エスエー | 物質交換プロセスを実施するためのデバイス |
| WO2024089254A1 (en) | 2022-10-28 | 2024-05-02 | Basf Se | Process for the manufacture of a c4-olefin-derived chemical of interest, in particular citral, from renewably-sourced ethanol |
| WO2024089252A1 (en) | 2022-10-28 | 2024-05-02 | Basf Se | Process for the manufacture of a propylene-derived chemical of interest, in particular an acrylic ester, from renewably-sourced ethanol |
| CN116236805A (zh) * | 2023-03-03 | 2023-06-09 | 中建安装集团有限公司 | 一种高效生产甲基丙烯酸甲酯的装置与方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2252334A1 (de) * | 1972-10-10 | 1974-05-22 | Nippon Catalytic Chem Ind | Verfahren zur kontinuierlichen herstellung von acrylsaeureestern oder methacrylsaeureestern |
| DE2552987A1 (de) * | 1975-11-26 | 1977-06-02 | Hoechst Ag | Verfahren zur kontinuierlichen herstellung aetherfreier acrylsaeurealkylester |
| DE3308879A1 (de) * | 1982-03-17 | 1983-09-29 | Nippon Kayaku K.K., Tokyo | Verfahren zur herstellung von acrylsaeure- oder methacrylsaeureestern |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB770551A (en) * | 1953-08-27 | 1957-03-20 | Celanese Corp | Continuous esterification process |
| GB1017522A (en) * | 1963-09-26 | 1966-01-19 | Distillers Co Yeast Ltd | Improvements in or relating to the production of butyl acrylate |
| DE1468932B2 (de) * | 1965-09-23 | 1976-01-22 | Röhm GmbH, 6100 Darmstadt | Verfahren zur kontinuierlichen herstellung von niedermolekularen aliphatischen acrylsaeureestern |
| DE2226829A1 (de) * | 1972-06-02 | 1973-12-20 | Knapsack Ag | Verfahren zur herstellung von acrylsaeureestern |
| DE3146191A1 (de) * | 1981-11-21 | 1983-05-26 | Röhm GmbH, 6100 Darmstadt | Verfahren zum kontinuierlichen verestern von methacrylsaeure |
| DE69313068T2 (de) | 1992-10-22 | 1998-03-05 | Sumitomo Chemical Co | Verfahren zur Herstellung von Methylmethacrylat |
| DE19510891A1 (de) * | 1995-03-24 | 1996-09-26 | Basf Ag | Verfahren zur kontinuierlicheen Herstellung von Alkylestern der (Meth)acrylsäure |
| FR2740919B1 (fr) * | 1995-11-07 | 1997-11-28 | Gec Alsthom T & D Sa | Dispositif de refroidissement pour ligne de transport electrique enterree |
-
1995
- 1995-09-28 DE DE19536178A patent/DE19536178A1/de not_active Withdrawn
-
1996
- 1996-09-12 US US08/713,208 patent/US5811574A/en not_active Expired - Lifetime
- 1996-09-17 CA CA002185821A patent/CA2185821A1/en not_active Abandoned
- 1996-09-18 CZ CZ962736A patent/CZ273696A3/cs unknown
- 1996-09-23 MX MX9604237A patent/MX9604237A/es unknown
- 1996-09-25 KR KR1019960042412A patent/KR970015558A/ko not_active Application Discontinuation
- 1996-09-26 EP EP96115454A patent/EP0765859B1/de not_active Expired - Lifetime
- 1996-09-26 DE DE59606209T patent/DE59606209D1/de not_active Expired - Lifetime
- 1996-09-26 JP JP8255012A patent/JPH09110789A/ja not_active Withdrawn
- 1996-09-27 SG SG1996010745A patent/SG72705A1/en unknown
- 1996-09-27 CN CN96113098A patent/CN1084324C/zh not_active Expired - Fee Related
-
1998
- 1998-07-21 US US09/119,649 patent/US5900125A/en not_active Expired - Lifetime
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2252334A1 (de) * | 1972-10-10 | 1974-05-22 | Nippon Catalytic Chem Ind | Verfahren zur kontinuierlichen herstellung von acrylsaeureestern oder methacrylsaeureestern |
| DE2552987A1 (de) * | 1975-11-26 | 1977-06-02 | Hoechst Ag | Verfahren zur kontinuierlichen herstellung aetherfreier acrylsaeurealkylester |
| DE3308879A1 (de) * | 1982-03-17 | 1983-09-29 | Nippon Kayaku K.K., Tokyo | Verfahren zur herstellung von acrylsaeure- oder methacrylsaeureestern |
Also Published As
| Publication number | Publication date |
|---|---|
| KR970015558A (ko) | 1997-04-28 |
| US5811574A (en) | 1998-09-22 |
| SG72705A1 (en) | 2000-05-23 |
| EP0765859A1 (de) | 1997-04-02 |
| DE19536178A1 (de) | 1997-04-03 |
| MX9604237A (es) | 1997-03-29 |
| EP0765859B1 (de) | 2000-12-13 |
| DE59606209D1 (de) | 2001-01-18 |
| CZ273696A3 (cs) | 1998-05-13 |
| JPH09110789A (ja) | 1997-04-28 |
| CA2185821A1 (en) | 1997-03-29 |
| US5900125A (en) | 1999-05-04 |
| CN1150142A (zh) | 1997-05-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1084324C (zh) | (甲基)丙烯酸的烷基酯的连续制备方法和所使用的设备 | |
| JP4376057B2 (ja) | アルキル(メタ)アクリレートの連続的製造方法 | |
| CN1137875C (zh) | (甲基)丙烯酸烷基酯的连续制备方法 | |
| WO2006019495A1 (en) | Improved process for production of organic acid esters | |
| CN1066710C (zh) | (甲基)丙烯酸烷基酯的连续制备方法 | |
| US5990343A (en) | Process for producing butyl acrylate | |
| CN1084325C (zh) | (甲基)丙烯酸与一种链烷醇的酯化 | |
| CN1095827C (zh) | (甲基)丙烯酸与链烷醇的酯化方法 | |
| CN103221379A (zh) | 丙烯酸酯生产方法 | |
| EP0255773A2 (en) | Continuous process for production of methacrylic acid ester of C1 to C4 aliphatic alcohol | |
| US6992209B2 (en) | Methods of forming alpha, beta-unsaturated acids and esters | |
| CN1095826C (zh) | (甲基)丙烯酸与链烷醇的酯化方法 | |
| CN1830944A (zh) | 制备(甲基)丙烯酸烷基酯的方法 | |
| CN1244189A (zh) | (甲基)丙烯酸与链烷醇的酯化方法 | |
| US6482976B1 (en) | Processes for conducting equilibrium-limited reactions | |
| US5206421A (en) | Method for producing 4-hydroxybutyl (meth)acrylate | |
| US20040267045A1 (en) | Processes for producing (meth)acrylic acid compound | |
| MXPA96006244A (en) | Esterification of acid (met) acrylic with an ftaa | |
| JP2003521478A (ja) | 平衡律速反応を行う方法 | |
| JP2024506199A (ja) | 高純度アルキルアクリレートを製造するための改善された方法 | |
| KR0122429B1 (ko) | 불포화 카르복실산 에스테르류의 개량된 제조방법 및 이의 새로운 제조장치(공정) | |
| JPS6320415B2 (zh) | ||
| AU2022395238A1 (en) | Method for producing 2-octyl (meth)acrylate | |
| KR20240050334A (ko) | 고순도 부틸 아크릴레이트의 개선된 제조 방법 | |
| TW202302513A (zh) | 藉由甲基丙烯酸甲酯之催化水解而連續製備甲基丙烯酸之新穎方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |