CN1031088C - 形成功能沉积膜的方法 - Google Patents
形成功能沉积膜的方法 Download PDFInfo
- Publication number
- CN1031088C CN1031088C CN89100622.2A CN89100622A CN1031088C CN 1031088 C CN1031088 C CN 1031088C CN 89100622 A CN89100622 A CN 89100622A CN 1031088 C CN1031088 C CN 1031088C
- Authority
- CN
- China
- Prior art keywords
- film
- deposited film
- gas release
- gas
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images










Classifications
-
- C—CHEMISTRY; METALLURGY
- C23—COATING METALLIC MATERIAL; COATING MATERIAL WITH METALLIC MATERIAL; CHEMICAL SURFACE TREATMENT; DIFFUSION TREATMENT OF METALLIC MATERIAL; COATING BY VACUUM EVAPORATION, BY SPUTTERING, BY ION IMPLANTATION OR BY CHEMICAL VAPOUR DEPOSITION, IN GENERAL; INHIBITING CORROSION OF METALLIC MATERIAL OR INCRUSTATION IN GENERAL
- C23C—COATING METALLIC MATERIAL; COATING MATERIAL WITH METALLIC MATERIAL; SURFACE TREATMENT OF METALLIC MATERIAL BY DIFFUSION INTO THE SURFACE, BY CHEMICAL CONVERSION OR SUBSTITUTION; COATING BY VACUUM EVAPORATION, BY SPUTTERING, BY ION IMPLANTATION OR BY CHEMICAL VAPOUR DEPOSITION, IN GENERAL
- C23C16/00—Chemical coating by decomposition of gaseous compounds, without leaving reaction products of surface material in the coating, i.e. chemical vapour deposition [CVD] processes
- C23C16/44—Chemical coating by decomposition of gaseous compounds, without leaving reaction products of surface material in the coating, i.e. chemical vapour deposition [CVD] processes characterised by the method of coating
- C23C16/50—Chemical coating by decomposition of gaseous compounds, without leaving reaction products of surface material in the coating, i.e. chemical vapour deposition [CVD] processes characterised by the method of coating using electric discharges
- C23C16/511—Chemical coating by decomposition of gaseous compounds, without leaving reaction products of surface material in the coating, i.e. chemical vapour deposition [CVD] processes characterised by the method of coating using electric discharges using microwave discharges
-
- C—CHEMISTRY; METALLURGY
- C23—COATING METALLIC MATERIAL; COATING MATERIAL WITH METALLIC MATERIAL; CHEMICAL SURFACE TREATMENT; DIFFUSION TREATMENT OF METALLIC MATERIAL; COATING BY VACUUM EVAPORATION, BY SPUTTERING, BY ION IMPLANTATION OR BY CHEMICAL VAPOUR DEPOSITION, IN GENERAL; INHIBITING CORROSION OF METALLIC MATERIAL OR INCRUSTATION IN GENERAL
- C23C—COATING METALLIC MATERIAL; COATING MATERIAL WITH METALLIC MATERIAL; SURFACE TREATMENT OF METALLIC MATERIAL BY DIFFUSION INTO THE SURFACE, BY CHEMICAL CONVERSION OR SUBSTITUTION; COATING BY VACUUM EVAPORATION, BY SPUTTERING, BY ION IMPLANTATION OR BY CHEMICAL VAPOUR DEPOSITION, IN GENERAL
- C23C16/00—Chemical coating by decomposition of gaseous compounds, without leaving reaction products of surface material in the coating, i.e. chemical vapour deposition [CVD] processes
- C23C16/44—Chemical coating by decomposition of gaseous compounds, without leaving reaction products of surface material in the coating, i.e. chemical vapour deposition [CVD] processes characterised by the method of coating
- C23C16/448—Chemical coating by decomposition of gaseous compounds, without leaving reaction products of surface material in the coating, i.e. chemical vapour deposition [CVD] processes characterised by the method of coating characterised by the method used for generating reactive gas streams, e.g. by evaporation or sublimation of precursor materials
- C23C16/452—Chemical coating by decomposition of gaseous compounds, without leaving reaction products of surface material in the coating, i.e. chemical vapour deposition [CVD] processes characterised by the method of coating characterised by the method used for generating reactive gas streams, e.g. by evaporation or sublimation of precursor materials by activating reactive gas streams before their introduction into the reaction chamber, e.g. by ionisation or addition of reactive species
-
- C—CHEMISTRY; METALLURGY
- C23—COATING METALLIC MATERIAL; COATING MATERIAL WITH METALLIC MATERIAL; CHEMICAL SURFACE TREATMENT; DIFFUSION TREATMENT OF METALLIC MATERIAL; COATING BY VACUUM EVAPORATION, BY SPUTTERING, BY ION IMPLANTATION OR BY CHEMICAL VAPOUR DEPOSITION, IN GENERAL; INHIBITING CORROSION OF METALLIC MATERIAL OR INCRUSTATION IN GENERAL
- C23C—COATING METALLIC MATERIAL; COATING MATERIAL WITH METALLIC MATERIAL; SURFACE TREATMENT OF METALLIC MATERIAL BY DIFFUSION INTO THE SURFACE, BY CHEMICAL CONVERSION OR SUBSTITUTION; COATING BY VACUUM EVAPORATION, BY SPUTTERING, BY ION IMPLANTATION OR BY CHEMICAL VAPOUR DEPOSITION, IN GENERAL
- C23C16/00—Chemical coating by decomposition of gaseous compounds, without leaving reaction products of surface material in the coating, i.e. chemical vapour deposition [CVD] processes
- C23C16/44—Chemical coating by decomposition of gaseous compounds, without leaving reaction products of surface material in the coating, i.e. chemical vapour deposition [CVD] processes characterised by the method of coating
- C23C16/52—Controlling or regulating the coating process
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Materials Engineering (AREA)
- Mechanical Engineering (AREA)
- Metallurgy (AREA)
- Organic Chemistry (AREA)
- Physics & Mathematics (AREA)
- Plasma & Fusion (AREA)
- Chemical Vapour Deposition (AREA)
Abstract
向成膜空间引入成膜材料化合物,如果需要引入含能控制沉积膜价电子的元素的化合物,形成半导体功能沉积膜,每一化合物处于气态或至少一种化合物是激活态,同时形成激发态氢原子,与成膜空间的气态的或激活空间中的激活态化合物发生化学反应,在基片上形成沉积膜,其中用一微波回路中与两个阻抗匹配回路结合的共振腔中的等离子体产生室产生的微波等离子,从氢气或氢气与惰性气体的混合气体中形成激发态氢原子,并且其激发态受到控制。
Description
本发明涉及形成含有第四族原子作为主要组成原子的功能沉积膜,或含有硅原子和第四族原子作为主要组成原子的功能沉积膜的改进了的方法,该功能沉积膜可专门作半导体器件的光导元件和用在电子摄象技术,象输入线路传感器,摄象装置,光电动势装置或类似装置的光敏器件。
更具体地说,本发明涉及在基片上有效地形成功能沉积膜的改进了的方法,这是通过用氢气或氢气与惰性气体组成的混合气体被微波等离子体造成处于激发态的激发的氢原子,并使所述激发态的氢原子与成膜原料气体或单独激活的成膜原料气体在成膜室中接触,从而产生化学反应,同时控制氢原子的激发态来进行的,微波等离子体是在一微波迥路中置于与两个阻抗匹配迥路结合的共振腔中的等离子体产生室中产生的。
对功能沉积膜,特别是半导体沉积薄膜的形成,已经存在考虑到与所要求的电学、物理性质以及其应用相适应的成膜方法。
例如,已经试用过等离子体化学汽相沉积(CVD)法,活性溅射法、离子镀法,光CVD法,热诱导的CVD法,MO CVD法,MBE法等,它们中有几个因适于形成要求的半导体装置已被用于工业生产中。
但是,即使在已被广泛采用的等离子体CVD方法中,考虑到制造所需半导体装置,产生的沉积膜的电学和物理性质也不是非常满意的,有时等离子体稳定性及形成沉积膜的重复性不好,此外它可以造成生产率的显著降低。
为克服这些问题,例如在日本专利公开60-41047中已提出了一个增加膜沉积速率的方法,从而显著地用加氢基CVD法(HR-CVD法)改进了沉积形成高质量的第四族半导体膜的生产率。
进一步,作为有效地用大约2.45京赫(109Hz)的微波形成高密度等离子体的手段,在例如日本专利公开55-141729和57-133636中提出了把电磁铁绕在共振腔上设计来建立电子迥旋共振(ECR)条件的方法,以及在学术会议等也报告了用高密度等离子体形成各类半导体薄膜的方法。
顺便提一下,在上述HR-CVD法中,虽然激活态的氢原子(氢基)对控制形成的沉积膜的性质和均匀性有重要的作用,但对于形成沉积膜时大量并均匀地控制氢原子的激发态,并在激发态控制下在形成沉积膜时控制化学反应,从而有选择地并稳定地控制形成的沉积膜的性质没有充分地研究,仍留下了几个要解决的问题。
另一方面,在使用ECR的微波等离子体CVD装置中也有几个问题。即:为建立ECR条件,在等离子体产生室中的压强必须保持在低于大约10-3乇,这样对形成沉积膜的压强有了限制:在这种水平的压强下,增大了气体分子平均自由程(大约为1米),由此成膜原料气体扩散到微波输入窗口附近,在那里被还原和反应,使沉积膜粘附在微波输入窗口或共振腔内壁上,从而使放电不稳定;基片上的膜被粘附膜的碎片及散射物沾污。进一步还指出了几个问题,即,在等离子体产生室中产生的等离子体沿电磁铁的发散的磁场扩散到成膜室。基片在成膜室中置于密度较高的等离子体中。因此,形成的沉积膜易于被带电粒子损伤等等,这就限制了改进要形成的膜的性质。在制作半导体器件过程的逐层沉积膜的步骤中,由于带电粒子造成的损伤,边界特性常常被降低等等,这就难以改进半导体器件的特性。
本发明的主要目的是克服上述形成沉积膜的现有技术方法中的各种问题,并提供一个包括周期表的第四族原子作为主要组成原子的所需的功能沉积膜,或包括硅原子和所述第四族原子作为主要组成原子的所需的功能沉积膜,它具有面积大,满意的均匀性,好的稳定性和重复性等优点,对制造高质量的半导体装置是有效的。
本发明者为实现本发明的目的对克服形成沉积膜的现有技术中的上述各种问题进行了认真的研究,从而认识到可以通过置于微波迥路中与两个阻抗匹配迥路结合的共振腔中的等离子体产生室,并用氢气或由氢气与一种惰性气体组成的气体混合物进行微波等离子体放电,以好的重复性稳定地和有效地提供处于可选择的激发态中的氢原子。
本发明是在上述认识的基础上进一步研究而实现的,它包括下述两个实施例。
根据本发明的第一实施例(以下称作“第一发明”)涉及一种形成包含属于周期表第四族的原子作为主要组成原子的功能沉积膜的方法,它向形成沉积膜的基片所在的成膜空间引入一种包含周期表第四族元素的化合物作为成膜原料,以及,如果需要,引入一种能控制沉积膜价电子的作为组成成分的元素,每一种都处于气态,或处于至少一种化合物已在与膜形成空间分开的激活空间中预先激活的状态,同时形成激发态氢原子,与处于气态或在一不同于成膜空间的激活空间中处于激活态的至少一种化合物发生化学反应,并且把它们引入成膜空间,在成膜空间在基片上形成沉积膜,其中通过置于一微波迥路中的与两个阻抗匹配迥路结合的共振腔中的等离子体产生室中产生的微波等离子体,从氢气或氢气与一种惰性气体的混合气体形成激发态的氢原子,并且氢原子的激发态是受到控制的。
根据本发明的第二个实施例(以下称作“第二发明”)涉及到一个包含笫四族原子和硅原子作为主要组成原子的功能沉积膜的形成方法,其特征描述如下:
第二发明涉及到一种形成沉积膜的方法,它向在其上形成沉积膜的基片所在的膜形成空间引入一种含有硅的化合物(1)和一种用下面的一般公式(I)表示的化合物(2)作成膜原料,以及,如果需要的话,引入一种含有一种能控制沉积膜价电子的元素的化合物(3)作为组成成分,每一种都处于气态或处于化合物(1)(2)(3)中至少一种已在一与成膜空间分开的激活空间中预先激活的状态,同时形成激发态氢原子,它与处于气态或在不同于成膜空间的激活空间中处于激活态的至少一种化合物进行化学反应,并把它们引入成膜空间,从而在置于其中的基片上形成沉积膜,其中通过置于微波迥路中的与两个阻抗匹配迥路结合的共振腔中的等离子体产生室产生的微波等离子体,由氢气或氢气与一种惰性气体的气体混合物形成激发态的氢原子,并且氢原子的激发态受到控制:
AaBb……(1)其中A代表一种除硅以外属于周期表第四族的元素,B代表从氢(H),卤素(X)和烃族中选出的一种材料,a代表一个等于或整数倍于B的价数的正整数,b代表一个正整数。
根据本发明,可以以显著改进了的成膜速率,以好的重复性,稳定有效地形成含有第四族原子作为主要组成原子或含有硅原子和第四族原子作为主要组成原子的,具有均匀的膜质量,均匀的膜厚度,各种极好的性质以及好的膜质量的功能沉积膜。
进一步,与通常方法比较,根据本发明,在每个形成功能沉积膜的方法中生产率得到显著改进,可以实现有效地大规模生产功能沉积膜。
而且,根据本发明,在其上形成沉积膜的基片的温度可以比现有技术方法降低,可以通过控制氢原子的激发态和引入成膜原料气体的量等等,容易地,稳定地控制膜的质量。
图1是简略说明适于根据本发明用微波等离子体CVD方法形成功能沉积膜的实用方法的装置的结构实例的透视图;
图2(a),2(b),2(c)分别是用在图1装置中的气体释放环的简图;
图3(1),3(2)分别是说明在根据本发明用微波等离子体CVD方法形成功能沉积膜的方法中,膜沉积速率对基片和金属网组件之间距离的关系的图;
图4(1)、4(2)分别说明根据本发明用微波等离子体CVD方法形成功能沉积膜的方法中,形成沉积膜的沉积速率相对于基片和金属网组件之间角度的差别的图;
图5(1),5(2)分别是说明形成的沉积膜厚度分布对于根据本发明实行微波等离子体CVD方法的装置中,图2(a)到2(c)的气体释放环的气体释放孔的直径增大率的关系的图;
图6(1)、6(2)分别是说明形成的沉积膜厚度的分布对于根据本发明实行微波等离子体CVD方法的装置中图2(a)到2(c)的气体释放环的气体释放孔的间隔减小率的关系的图;以及
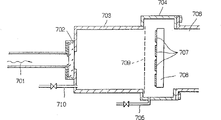
图7是通常的微波等离子体CVD装置的结构的剖面图。
在本发明中,是通过用发射的分光光度测定法测量Hα和Hβ辐射强度作为氢的激发态,控制供给共振腔的微波功率、阻抗匹配条件、氢气流动率或由氢气和惰性气体组成的气体混合物的流动率以及总压力等因素中的一个或多个而实现对氢原子激发态的控制的。
微波迥路中与两个阻抗匹配迥路相结合的共振腔中的阻抗匹配迥路,是一个约束体,它置于改变空腔长度的活动圆柱,并置于微波波导管和共振腔之间的连接部分,阻抗匹配条件通过调节这个约束体来控制。
阻抗匹配迥路可以是一个改变空腔长度的活动圆柱和一个E-H调谐器或一个三抽头调谐器。
进一步,等离子体产生室是由一金属网组件和一可渗透微波的钟罩组成,并用金属网组件连到成膜空间,激发态的氢原子由金属网组件引入成膜空间。
另一方面,基片与金属网表面成30°或更小的角度,并与金属网表面相距100毫米,气态或激活态的化合物由置于金属网表面和基片之间的气体释放装置引入成膜空间。
气体释放装置放置得使它环绕着基片,使得从每一气体释放孔释放的气体量均匀,这是通过从引入气体释放装置的一侧向着最后释放孔逐渐减小气体释放孔的间隔,或逐渐增大气体释放孔的孔径,或者使释放孔至少在基片平面均匀分布并从引入气体释放装置的一侧向着中央部分逐渐增大每一气体释放孔的大小来实现的。
当用根据本发明的方法形成所需的功能沉积膜时,向成膜室引入一种包含第四族元素的化合物,或如上描述的化合物(1)和(2),以及如果必要,引入一种含有能控制沉积膜价电子的元素作为组成成分的化合物,并且以气态或激活态分开引入其激发态受到控制的氢原子,在它们之间产生化学反应,从而在基片上形成第四族半导体薄膜或第四族系列的半导体薄膜。在这种情况下,任何所述半导体薄膜的结晶特性,氢含量等可以得到稳定的重复性极好的控制。
在本发明中的原子氢的激发态是从氢气或氢气与一种惰性气体的气体混合物的微波等离子体中观察到的光辐射来决定的。特别,从微波等离子体来的光辐射是用发射的分光光度测定法测量的,激发态是从原子氢(H*)的辐射线中,属于Hα的在656毫微米处的辐射线和属于Hβ的在486毫微米处的辐射线的强度比来确定的,控制下述参数中至少一个参数,即供给共振腔的微波功率、阻抗匹配条件、氢气流率或由氢气和惰性气体组成的气体混合物的流率以及总压强,以得到要求的强度比。
在本发明中,控制氢原子的激发态使强度比Hα/Hβ控制在1/1到1000/1较好,控制在10/1到500/1更好。
在上述强度比的范围内,基片的种类和温度的结合特别是膜质量等等的一个决定因素,可以通过适当结合它们两者而形成所需质量和性质的沉积膜。因此,在本发明中必须采用具有能测量上述强度比感度的辐射分光光度计。
在本发明中,因为微波等离子体的监视器就全部气体的流动方向来看,位于含有第四族元素的化合物或化合物(1)和(2)的气体释放装置的上游,所以实质上没有沉积膜在监视器上形成,从膜形成开始到结束都能进行稳定的监视。
当含有笫四族元素的化合物用于第一发明时,较好的是选取那些自发产生的,能与激发态氢原子进行分子碰撞并发生化学反应,从而对在基片上形成沉积膜作出贡献的化学物质。若它们不与激发态氢原子发生所要求的反应,或是它们存在的通常状态没有足够高的活性,那就必须将第四族元素的化合物激活到能与激发态的氢原子起化学反应的状态,使用的激活能不应在膜形成前或形成中完全分离第四族元素,从而使第四族元素的化合物达到能与激发态的氢原子起化学反应的激发状态。使用能形成这种激发态的化合物作为可用于本发明方法的化合物之一。
在第一发明中,作为含有第四族元素的化合物,特别可以使用那些对电子吸引力大的原子,原子团或与碳原子成键的极性基团,硅原子或锗原子。例如,可用那些诸如直链或环形硅烷化合物的含硅和卤素的化合物,其中氢原子部分地或完全地被卤素原子取代。作为具体的例子,可以提出:用分子式SiuY2u+2表示的直链硅卤化物,其中u是一个大于或等于1的整数,Y是从F,Cl,Br和I中选的至少一个元素;用分子式SivY2v表示的环形硅卤化物,其中v代表大于或等于3的整数,Y与上面的意义相同;以及用分子式SiuHxYy表示的直链或环形链化合物,其中u和Y与上面的意义相同,且x+y=2u或2u+2。
具体地,作为例子可提出气态的或易气化态的SiF4,(SiF2)5,(SiF2)6,(SiF2)4,Si2F6,Si3F8,SiHF3,SiH2F2,Si2H2F4,Si2H3F3,Sicl4,(Sicl2)5,(SiBr)4,(SiBr2)5,Si2cl6,Si2Br6,SiHcl3,SiHBr3,SiHI3、Si2cl3F3等。
这些硅化合物可以单独使用,也可以两种或多种合在一起使用。
作为含碳和卤素的化合物,可以使用直链或环形烃化合物中的氢原子部分地或全部地被卤素原子取代的化合物。具体地,作为例子可以提出:由分子式CuY2u+2表示的直链碳卤化物,其中u是大于或等于1的整数,Y代表从F,Cl,Br和I中选出的至少一种元素;由分子式CvY2v表示的环形碳卤化物,其中v代表大于或等于3的整数,Y与上面的意义相同;一种由分子式CuHxYy表示的直链或环形化合物,其中u和Y与上面有相同的意义且x+y=2u或2u+2。
具体地,作为例子可以提出气态的或易于气化态的CF4,(CF2)5,(CF2)6,(CF2)4,C2F6,C3F8,CHF3,CH2F2,Ccl4,(CCl2)5,CBr4,(CBr2)5,C2cl6,C2Br6,CHcl3,CHI3,C2Cl3F3等等。
这些碳化合物可以单独使用也可以两种或多种合在一起使用。
作为含锗和卤素的化合物,可以使用在直链或环形氢化锗化合物中氢原子被卤素原子部分地或全部地替代的化合物。具体地,作为例子可提出:由分子式GeuY2u+2表示的直链锗卤化物,其中u代表大于或等于1的整数,Y代表从F,cl,Br和I中选出的至少一种元素;由分子式GevY2v表示的环形锗卤化物,其中V代表大于或等于3的整数,y的意义与上面相同;由分子式GeuHxYy表示的直链或环形复合物,其中u和Y与上面有同样意义,x+y=2u或2u+2。
具体地,作为例子可以提出气态的或易于气化态的GeF4,(GeF2)5,(GeF2)6,(GeF2)4,Ge2F6,Ge3F8,GeHF3,GeH2F2,Ge2H2F4,Ge2H3F3,Gecl4,(Gecl2)5,GeBr4,(GeBr2)5,Ge2Cl6,Ge2Br6,GeHCl3,GeHBr3,GeHI3,GeCl3F3等等。
在第一发明的方法中,作为控制价电子的元素的,可以提出:第三族元素,如B,Al,Ga,In,Tl等作为P-型杂质较好,第五族元素,如N,P,As,Sb,Bi等等作为n-型杂质较好。特别地,B,Ga,P和Sb最好。这类杂质的量根据待形成的沉积膜所要求的电学和光学性质来适当确定。
用在正常温度和正常压力下处于气态的或在成膜条件下至少容易气化的那些化合物作为引入这种杂质的原料。作为引入这种杂质的原料,具体地可以提出PH3,P2H4,PF3,PF5,PCl3,AsH3,AsF3,AsF5,Ascl3,SbH3,SbH5,BiH3,BF3,BCl3,BBr3,B2H6,B4H10,B5H9,B5H11,B6H10,B6H12,AlCl3等等。
含有杂质原子的化合物可以单独使用或者两种或多种结合起来使用。
引入的杂质物质可以直接以气态引入成膜空间,或者与含第四族元素等的化合物混合后引入成膜空间,也可以在激活空间里激活后引入成膜空间。可以适当选择加热,光照,放电等方法来激动引入杂质的物质,并如上所述地使用。
作为用于第二发明的化合物(1)和(2)以及含有能控制沉积膜价电子的元素作为组成成分的化合物(3),希望从与激发态氢原子进行分子碰撞并发生化学反应,从而在基片所在空间中自发形成化学物质,对在基片上形成沉积膜作贡献的那些化合物中选择。
如果它们在通常的存在状态下不按所要求的与激发态氢原子反应或没有很强的活性,就必须把化合物(1)和(2)变成激发态使其能与激发态氢原子反应,所用激活能不应在成膜前或成膜时造成Si或上面描述的一般公式中的A完全分解。可以形成这种激发态的化合物被用作本发明的方法中使用的化合物(1)和(2)。
在第二发明中,作为能有效地用作化合物(1)和化合物(2)的那些化合物可以提出下列一些。
在根据本发明得到具有半导体性质的第四族元素沉积膜的情况,作为含硅化合物(1)可以使用氢原子被卤素原子部分地或全部地取代的直链或环形硅烷化合物。具体地,可以提出:由分子式SiuY2u+2表示的直链硅卤化物,其中u是大于或等于1的整数,Y代表从F,cl,Br和I中选出的至少一种元素;由分子式SivY2v表示的环形硅卤化物,其中v是大于或等于3的整数,Y与上面的意义相同;由分子式SiuHxYy表示的直链或环形化合物,其中u和Y与上面的意义相同且x+y=2u或2u+2。
具体地可提出作例子的是气态的或易于气化态的SiH4,SiF4,(SiF2)5,(SiF2)6,(SiF2)4,Si2F6Si3F8,SiHF3,SiH2F2,Si2H2F4,Si2H3F3,SiCl4,(SiCl2)5,SiBr4,(SiBr2)5,Si2Cl6,SiHCl3,SiHBr2,SiH2Cl2,Si2Cl3F3等等。
再有,作为化合物(2)中的A,可以提出属于周期表第四族元素的那些元素,特别是Ge,C,Sn,Pb。作为含有这些元素的化合物(2),可以提出:含锗化合物,例如直链锗烷或由分子式GeuY2u+2表示的锗卤化物,其中u是大于或等于1的整数,Y代表从F,Cl,Br和I中选出的至少一种元素;由分子式GevY2v表示的环形锗烷或锗卤化物,其中v是大于或等于3的整数,Y与上面意义相同;由分子式GeuHxYy表示的直链或环形锗化合物,其中u是大于或等于1的整数,Y是从F,Cl,Br和I选出的至少一种元素且x+y=2u或2u+2;以及具有烷基团的有机锗化合物等,具体地作为例子可提出GeH4,Ge2H6,Ge3H8,n-Ge4H10,t-Ge4H10,GeH6,Ge5H10,GeH3F,GeH3Cl,GeH2F2,Ge(CH3)4,Ge(C2H5)4,Ge(C6H5)4,Ge(CH3)2F2,GeF2,GeF4,GeS等等。
此外,作为含碳化合物,可使用:直链或环形烃化合物,其中氢原子被卤素原子部分地或全部地取代,例如,分子式为CuY2u+2的直链碳卤化物,其中u是大于或等于1的整数,Y代表从H,F,Cl,Br和I中选出的至少一种元素,例如CH4,C2H6,C3H8,n-C4H10,C5H12,C2H4,C3H8,C4H8,C5H10,C2H2,C4H6;由分子式CvY2v表示的直链碳卤化物,其中v是大于或等于3的整数,Y与上面的意义相同;由分子式CuHxYy表示的直链或环形碳化合物,其中u是大于或等于1的整数,Y代表从F,Cl,Br和I中选出的至少一种元素,且x+y=2u或2u+2。后两种化合物的例子是CF4,(CF2)5,(CF2)6,(CF2)4,C2F6,C3F8,CHF3,CH2F2,CCl4,(CCl2)5,CBr4,(CBr2)5,C2Cl6,C2Cl3F3等等。
此外,作为含锡的化合物,例如可以提出SnH4,SnCl4,SnBr4,Sn(CH4),Sn(C2H5)4,Sn(C3H7)4,Sn(C4H9)4,Sn(OCH3)4,Sn(OC2H5)4,Sn(i-OC3H7)4,Sn(t-OC4H9)4等等。作为含铅的化合物,例如可提出Pb(CH3)4,Pb(C2H5)4,Pb(C4H9)4等等。
对化合物(1)和(2),上述原料可以单独使用,如果需要也可两种或多种结合起来使用。
在根据第二发明的方法中,对含有控制价电子的元素作为组成成分的化合物(3),较好地是选择那些在常温常压下是气态的或者至少在形成沉积膜的条件下是气态的并且能够在适当的气化装置中易于气化的化合物。
作为用于本发明的方法的化合物(3),在得到所需第四族沉积膜情况下,可以提出那些含有周期表第三族和第五族元素的化合物。具体地可提出:BX3,B2H6,B4H10,B5H9,B5H11,B6H10,B(CH3)3,B(C2H5)3,B6H12,AlX3,Al(CH3)2Cl,Al(CH3)3,Al(OCH3)3Al(CH3)Cl2,Al(C2H5)3,Al(OC2H5)3,Al(CH3)3Cl3,Al(i-C4H9)5,Al(i-C3H7)3,Al(C3H7)3,Al(OC4H9)3,GaX3,Ga(OCH3)3,Ga(OC2H5)3,Ga(OC3H7)3,Ga(OC4H9)3,Ga(CH3)3,Ga2H6,GaH(C2H5)2,Ga(OC2H5)(C2H5)2,In(CH3)3,In(C3H7)3,In(C4H9)3等等作为含有第三族元素的化合物;NH3,HN3,N2H5N3,N2H4,NH4N3,PX3,P(OCH3)3,P(OC2H5)3,P(C2H7)3,P(OC4H9)3,P(CH3)3,P(C2H5)3,P(C3H7)3,P(C4H9)3,P(OCH3)3,P(OC2H5)3,P(OC3H7)3,P(OC3H9)3,P(SCN)3,P2H4,PH3,AsH3,AsX3,As(OCH3)3,As(OC2H5)3,As(OC3H7)3,As(OC4H9)3,As(CH3)3,As(C2H5)3,As(C6H5)3,SbX3,Sb(OCH3)3,Sb(OC2H5)3,Sb(OC3H7)3,Sb(OC4H9)3,Sb(CH3)3,Sb(CH7)3,Sb(C4H9)3等等作为含有第五族元素的化合物。
在上面,X代表卤族元素(F,Cl,Br,I)。
上面描述的原料可以单独使用或者两种或多种结合在一起使用。
在上述原料于常温常压下是气态时,引入成膜空间或激活空间的量由质量流量控制器来控制。在材料是液态时,用Ar或He这类惰性气体或氢气作为气体载体使材料气化,如果需要也可以用一个能控制温度的扩散器使材料气化。在原料是固态时,用Ar或He这类惰性气体或氢气作为气体载体并用一个热升华炉使材料气化,引入量主要由控制气体载体的流量和温度来控制。
在第一发明中,所用的激发态氢原子是在成膜空间中形成沉积膜的同时引入该空间的,从而与化合物或含有以第四族元素作为待形成的沉积膜的主要成分的激发态化合物起化学反应。结果,与通常情况相比,具有所要求功能的主要由第四族原子组成的半导体沉积膜很容易在基片温度较低情况下在其上形成。同样,用在第二发明中的激发态氢原子是在成膜空间中形成沉积膜的同时送入该空间的,从而与含有以组成元素作为形成沉积膜的主要成份的化合物(1)和(2)和/或激发态化合物(1)和/或激发态化合物(2)起化学反应。因此,与通常情况相比,具有所要求功能的第四族沉积膜在基片温度较低情况下,比较容易地在所需基片上形成。
为了在与成膜空间分开的激活空间中预先激活含第四族元素的化合物或化合物(1)和(2)以及能够控制价电子的化合物,诸如加热,光照和放电的激活能可以作为用在激活空间的能源。
具体地可以提出:电阻加热,红外加热等热能,诸如激光束,汞灯光,卤素灯光等光能,诸如微波、射频、低频和直流放电电能。这些激活能可以单独用于激活区也可以两种或多种结合起来使用。为有效地利用激活能的作用,也可以结合使用催化作用。
在本发明中,用氢气或氢气与一种惰性气体的气体混合物来形成激发态氢原子。如果不能使微波等离子体稳定或者仅用氢气不能产生等离子体,则适当混以惰性气体是有效的。
作为用于本发明的惰性气体,较好地可以提出He,Ne,ArKr,Xe和Rn。
现在来说明,用于本发明的在微波迥路中与两个阻抗匹配迥路相结合的共振腔结构形成微波等离子体的方法。
为作比较,首先说明至今一直使用的形成微波等离子的方法。图7是已知微波CVD装置结构的剖面简图。
图7中分别表示出一个方型波导管701,微波输入窗702,等离子体产生室703,成膜室704,供气管705,710,排气口706,待加工物707,物体支架708和金属网组件709。
如图7所示,该装置包括等离子体产生室703和使用等离子体的成膜室704,两者用金属网组件709分开,并控制其穿透使得微波和带电粒子不能直接进入成膜室704。等离子体产生室703有共振腔结构,通过方型波导管701的微波由诸如石英(SiO2),铝陶瓷(Al2O3),特夫隆(teflon)等介电材料构成的微波输入窗702送入等离子体产生室。待加工物707放在成膜室704中,该室有供气管705和抽空等离子体产生室703和成膜室704的排气口706。
这样构成的等离子体产生装置一启动,微波从方型波导管701输入到等离子体产生室,氢气等在通过气体引入口710引入后被微波的电场能转变成等离子体,形成大量处于激发态的氢原子。激发态的氢原子通过金属网组件709进入成膜室704,在那里它们与从供气管705来的气体碰撞,并产生化学反应,从而在待加工物707上形成沉积膜。
但是在使用具有上述结构的通常的微波等离子体产生装置中,如果方型波导管701与作为共振腔的等离子体产生室703夹紧,则由于输入阻抗不匹配,会存在大部分微波电场能被反射的问题,因而不能有效地利用能量。
作为此问题的一种解决办法,已使用了一种把电磁铁绕共振腔排列得到ECR(电子迥路共振)的方法(参看日本专利公开55-141729)。但是因为在这个方法中需要高达875高斯的磁通量密度,所以装置很大也很重。而且该室被设计成通常是在真空中构成共振腔。由此,假如由放电产生等离子体,则因等离子体折射率小于1,它就不再起共振腔的作用(参见电气学会编“放电手册”,第4部分,第二章,298页)。此外,在由电磁铁形成静磁场的情况,因为线圈加热使电流改变,所以在抑制这种改变的同时,不仅需相当长时间来稳定地准备ECR条件(即高到875高斯的磁通量密度),而且如果偏离ECR条件,就会降低微波吸收率,并且在电场稳定之前,难以改进电场的使用效率。
因此本发明者发现下述设计是克服上述问题的有效手段,即设计出一种不论等离子体存在与否及其密度如何都能起共振腔作用的结构,并在共振腔中安置一个钟罩作等离子体产生室用来激发TM膜。
具体地,在共振腔中,安装一个改变空腔长度的活动园柱体,方型波导管和园柱共振腔互相夹紧使它们的轴互相垂直,如图1所示。而且为了进行阻抗匹配,较好地是使用在方型波导管和共振腔之间的连接部分的一个约束体,或者一个与改变空腔长度的活动园柱结合安置的E-H调谐器,或者三抽头调谐器中的任何一个。
放在共振腔中用来形成等离子体的钟罩能让微波透过,并由能保持气密的材料做成,例如所谓新陶瓷,诸如石英(SiO2),铝土陶瓷(Al2O3),氮化硼(BN),氮化硅(Si3N4 ),碳化硅(SiC),氧化铍-(BeO),氧化镁(MgO)和氧化锆(ZrO2)等。
改变空腔长度的活动园柱在引入微波的一侧对着钟罩放置,即在大气压一侧。因此,由于可以在大气中改变空腔长度来实现阻抗匹配,所以空腔长度可以容易地按等离子体存在与否或由于等离子体密度的改变造成的空腔谐振条件的改变来调整,从而保证以好的重复性和稳定性产生微波等离子体。
在本发明中,金属网组件置于钟罩和成膜空间之间,起形成腔共振条件的一个端板的作用,因而网状组件的直径小于λ/2较好,更好是小于管中所用微波波长的1/4(λ/4)。
金属网组件形如金属规,即一个打了很多方形的或园形的孔的薄金属板,它可由金属材料做成,如Al,Fe,Ni,Ti,No,W,Pt,Au,Ag和不锈钢,或表面用上述金属溅镀,汽相沉积等处理过的玻璃,陶瓷等,或金属复合材料。
而且,为把在钟罩中形成的激发态氢原子有效并均匀地引入成膜空间,需要改变金属网组件的孔分布及孔直径。整个孔隙率等于或大于10%较好,等于或大于20%更好,最好是等于大于30%。
为使本发明中基片上形成的沉积膜的厚度均匀,性质一致,考察了基片与金属网组件之间的间距和金属网组件相对于水平轴的角度,由此得到下述结果。
图3(1)给出了两个典型例子,每一个都说明形成沉积膜的沉积率对于基片与金属网组件之间的距离的关系,其中曲线a用“o”点表示,曲线b用“·”点组成,它们分别在表1(1)所示的成膜条件(A)和(B)下得到。
图4(1)代表当基片与金属网组件的角度变化时,沉积在基片上的,以沉积率差的形式表示出的膜的厚度分布。这是在成膜条件(A)下,基片与金属网组件之间的距离为40毫米(曲线C用“▲”表示)和80毫米(曲线d用“△”表示)时得到的。
图3(2)示出两个典型例子,每一个都说明形成沉积膜的沉积速率对于基片与金属网组件之间的距离的关系。其中曲线a用“o”点表示,曲线b用“·”点表示,它们分别在表1(2)所示的成膜条件(A)和(B)下得到。图4(2)代表当基片与金属网组件之间的角度变化时,沉积在基片上以沉积速率差的形式表示出的膜的厚度分布。这是在成膜条件(B)下,基片与金属网组件之间的距离为30毫米(曲线C,用“▲”表示)和70毫米(曲线d,用“△”表示)时得到的。
从图3(1)和3(2)可以看到,当基片和金属网组件距离增大时,沉积速率趋于迅速减小。特别在成膜条件(A)下,若距离超过100毫米,几乎不能形成沉积膜。在成膜条件(B)下,在距离超过100毫米时,形成的沉积膜特性也很差,不适于实际应用。此外,从图4(1)和4(2)看到,若基片和金属网组件的夹角超过30度时,对所有位置上的基片,膜厚度的分布迅速增加,相应地,膜性质的分布也急骤变化,从而显著降低其均匀性。
表1(1)项目 成膜条件 (A) (B)基片温度 220℃含第四族元素的化合物 Si2F6 10sccm组成原料气体的氢原子 H2 50sccm
Ar 150sccm成膜时压强 0.02乇 0.04乇金属网组件 穿孔的铝板 (100毫米直径)
孔径6毫米
孔隙度45%
孔分布均匀对含第四族元素的化合物 等距分布在8个位置上,2毫米的气体释放装置 孔径的环状释放环,离金属网
组件10毫米微波输入功率 250瓦 350瓦
表1(2)
(A) (B)基片温度 250℃ 250℃化合物(1) Si2F6 10sccm Si2F6 10sccm化合物(2)* GeF410sccm GeF4 10sccm组成原料气体的 H2 10sccm H2 10sccm氢原子 Ar 200sccm Ar 200sccm成膜时的压强 0.05乇 0.15乇金属网组件 孔径:6毫米 孔径:6毫米
均匀分布 均匀分布
孔隙率:45% 孔隙率:30%
铝:120毫米直径 铝:120毫米直径对化合物(1)和(2) 环向释放环 环向释放环的气体释放装置 孔径:2毫米×8 孔径:2毫米×8
(等距) (等距)
距金属网10毫米 距金属网10毫米微波输入功率 250瓦 300瓦
*)化合物(2)用10%的氦气稀释。
在几种条件下对形成其它第四族半导体薄膜还进行过与上述相同的考察,在每一情况都得到实质上相同的结果。
因此在本发明中,基片和金属网组件的距离定为100毫米或更近较好,最好是70毫米或更近,基片和金属网组件的水平轴之间的夹角30度或更小些较好,最好是20度或更小些,这些是把膜厚度的性质与分布及膜性质均匀性保持在±5%内的必要条件。
此外还对本发明中用于第四族元素化合物或化合物(1)和(2)的以及如果必要,用于含有能控制价电子的元素作为组成成分的化合物的气体释放装置进行了进一步的考察,以改进膜厚度分布和膜性质的均匀性。
在本发明中较好的压力范围介于流体技术中所说的粘滞流动和分子流动之间的区域,这时不能使用属于分子流动范围的传导计算公式。因此在本发明中作了下述实验,注意了孔径大小,气体释放装置上的气体释放孔的距离和分布,由此得到了图5(1),5(2)和图6(1),6(2)的结果。
图5(1)说明在表1(1)的成膜条件(A)下形成沉积膜的结果,基片距离为40毫米,用了如图2(a)所示的气体释放环201。图5(2)则说明在表1(2)的成膜条件(A)下形成沉积膜的结果,基片距离30毫米,气体释放环201如图2(a)所示。
在图2(a)所示的气体释放环201中,8个释放孔201a-201d,201a′-201d′等距配置,其中孔径从最靠近箭头(→)方向的释放孔201a、201a′向下游的孔201d,201d逐渐增加。图5(1)和5(2)说明了当使用孔径变化率为0-80%的每一气体释放环时形成的沉积膜厚度分布的变化。
从结果中可以看到,虽然在孔径增加率从0变到40%时膜厚度分布得到了改进,若它超过40%,膜厚度分布增大了,如果它超过了60%,膜厚度分布比孔径不变时(孔径增加率0%)还大。膜性质实质上与膜厚度分布相关。在其它成膜条件下,趋势实质上也是一样。
因此在本发明中,较好地是使孔径增加率在0-50%之间,在20-40%之间更好。
然后,图6(1)和6(2)是在与上面同样的成膜条件下,用图2(6)所示气体释放环202的实验结果。
在图2(b)的气体释放环中,环上有8个相同孔径的释放孔202a-202d,202a′-202d′其相互间距相对于释放孔202a到202a′间的距离逐渐减少,用减少率从0-70%变化的每一气体释放环形成的沉积膜厚度分布的改变示于图6(1)和6(2)。
可以看到虽然当孔径间距减少率从0-40%变化时,膜厚度分布改进了,但若超过40%,膜厚度分布增加了,若超过50%,膜厚度分布进一步增加到孔径距离不变(孔径间距减少率0%)的情况。
膜的性质实质上表示出与膜厚度分布相关。在其它成膜条件下,趋势也实质上一样。
因此在本发明中,孔径间距减少率设在0-50%较好,从20-40%更好。
而且还用图2(c)的气体释放环203进行了与图5(1),5(2)所示同样的确定关系的实验。在图2(c)所示气体释放环203中,释放孔是均匀分布的,孔径从203a到203d依次增大。用不同增加率的气体释放环得到的膜厚度分布和膜的性质的改变趋势实质上与图5(1)和5(2)所示结果一样。
因此在本发明中气体释放孔如图2(c)所示分布时,孔径增加率在0-40%范围变化较好,在10-30%范围内更好。
在本发明中,成膜阶段的内部压强可以适当地根据成膜情况决定,该成膜情况取决于从氢气或氢气和惰性气体组成的气体混合物稳定地形成微波等离子体的条件,取决于所选的含第四族元素的化合物或化合物(1)和(2)及能控制价电子的化合物(3)的状态和种类,以及要求的沉积膜的性质等。内部压强设在100乇-1×10-4乇较好,10乇-5×10-4乇更好,最好是1乇-1×10-3乇。
用本发明的方法,可以在任何结晶性的基片上形成所需结晶性沉积膜,不论其非晶体或晶体性质如何。
在本发明中,为了建立稳定的腔共振条件,采用连续振荡方式,所用功率的起伏范围在30%范围内较好。
用本发明的方法,有很好的可控制性、稳定性和可重复性的激发态的氢原子可以用一个微波回路中与两个阻抗匹配回路结合的共振腔的微波等离子体形成,从而显著地改进了激发态氢原子和成膜原料化合物反应的可控制性,具有所需结晶性和氢含量等的第四族元素半导体薄膜能以好的均匀性,高效率和好的重复性形成。
将对一个适于实施本发明方法的沉积膜形成装置的典型实施例进行描述,但应指出,本发明并不仅限于这种沉积膜形成装置。
图1是说明适于实施本发明方法的沉积膜形成装置的结构简图。
在图1中,园柱形共振腔101包括一个微波等离子体产生室钟罩102。一个金属网组件103,一个改变腔长度的活动园柱104,一个方波导管108和一个约束器110等主要组成元件。一个磷青铜作的弹簧105用来改进活动园柱104和园柱共振腔101的接触,以防止反常放电。可用马达106来移动改变腔长的活动圆柱104,一个变速齿轮107朝着钟罩102。一个E-H调谐器或三抽头调谐器109组成在本发明构成一微波回路的阻抗匹配回路,它是用来与作为另一阻抗匹配回路的改变共振长度的活动园柱102配对作阻抗匹配用的。约束器110以同样方式构成阻抗线路之一,它与改变腔长度的活动园柱102成对使用。
约束器110成对地装在方波导管108和园柱共振腔110间的连接部分的右边和左边,且配接得使它们能沿方波导管108的园柱表面在纵向互相独立地稍作移动,并用未示出的磷青铜弹簧保持与园柱共振腔101的接触。
一个释放气体输入管111来的氢气或氢气与惰性气体的气体混合物的释放孔通过金属网组件并指向钟罩102的内部。输入钟罩的氢气等被加到共振腔101上的微波转换成等离子体以形成激发态氢原子等。然后通过金属网组件103进入成膜空间116。成膜空间的压强用压强计125测量。
在成膜空间116,把形成沉积膜原料气体的释放环放在基片118和基片支架119之间。
激活空间114在必要时用来预激活从供气管120引入的成膜原料气体,在激活空间114周围放置了能产生热光,放电能量的激活能的装置115。
在引入预激活成膜原料气体时,输运管117的直径和其材料应能维持该气体的激活态。
在气体释放环112上有如图2所说明的气体释放孔113。
引入成膜空间116的成膜原料气体等依图中箭头所示方向用未示出的抽气泵抽出。
出口121用于安放微波等离子体监视器,其上连看一个光探测器122。光探测器122用石英光纤123连到未示出的分光光度计进行发射光度分析。124是一个在成膜空间侧面监视等离子体的监测孔。
参考下述例子将更详细地描述根据本发明的沉积膜形成方法。但应该指出本发明不仅限于这些例子。
例1
首先一个直径150的玻璃基片118(商牌号:7059,康宁玻璃公司制造)放在成膜空间116的基片支架119上,用未示出的抽气泵将成膜空间压强降到1×10-6乇。然后加热基片支架。用未示出的基片温度控制器使基片表面温度定在220℃。
当基片表面温度确定后,从未示出的气体源通过气体输入管111向石英钟罩102引入20sccm的氢气和100sccm的氩气。然后用自动压强控制器将成膜空间116中压强控制在0.1乇。
然后从未示出的连续振荡型微波振荡器通过方形波导管118向共振腔101输入微波。此后很快用马达106和变速齿轮107把改变空腔长度的活动圆柱104调节到由未示出的微波线路中的功率监视器测量的反射功率/入射功率比值为最小的位置,进一步调节约束器110的张开程度使反射功率/入射功率之比为最小。然后重复细调改变腔长度的活动圆柱104的位置和约束器110的张开程度,使反射功率一入射功率比最小,使入射功率/反射功率代表的有效入射功率放置在200瓦。
在这个例子中,通过孔121监测到的氢原子激发态发射线Hα/Hp的强度比是120。
基片108与金属网组件103的距离为40毫米且二者相互平行。由直径150毫米带孔的铝板用作金属网组件103,孔的直径均为8毫米,孔隙率为80%且均匀分布。所用气体释放环有如图2(a)所示构造,其中相应于201a和201a′的孔径为1.5毫米,孔径增加比为30%。
然后把10sccm的Si2F6气体和5sccm的BF3(用SiF4稀释到百万分之200(200ppm))的混合物从未示出的气体源通过供气管120输入并从气体释放环112进入成膜空间116,在这个情况下,自动压强控制器把成膜空间中的压强维持在0.1乇。BF3还起着掺杂剂的作用。
很快在60分钟内于基片上形成由激发态的氢原子和Si2F4,BF3起化学反应形成4.2微米厚的Si∶H∶F∶B膜(1-1号样品)。
冷却后取出基片,用-6英吋的n+Si(110)芯片代替它,以上述同样步骤,除了改变氢气流量到150sccm,微波输入功率到350瓦,成膜空间压强到0.02乇,基片温度到280℃,形成沉积膜。在这种情况下Hα/Hp强度比是40(1-2号样品)。
当用X射线衍射仪和电子射线衍射仪(RHEED)得到的每一沉积膜样品的膜厚度分布和其结晶性时,指出每一样品膜厚度分布在±3%之内,1-1号样品是非晶膜,而1-2号样品是在平行于基片表面有几乎(110)取向的外处延膜。
每一样品被放入真空沉积装置中,用250微米间隙的梳型铝电极和用电阻加热的2毫米直径的点状电极作汽相沉积,电导率(δ)(δp:光导电率,δd:暗电导率)及空穴迁移率(μh)用万,德。波尔方法测量。1-1号样品的δp/δd的值为4.5×105(δp:为AM-1照射下的值),对1-2样品整个表面内μh的值是450±13cm2/伏秒,其特性分布大约为±3%。而且在用热伏打功率测量检查其传导类型表明,它们都是p-型。
而且当用SIMS测量氢含量时,在1-1号样品中原子百分比为9%,在1-2号样品中原子百分比为0.08%。
从前述可以发现,结晶性很容易按照本发明来控制。
例2
除把微波输入功率从200瓦和350瓦调到150瓦和250瓦及用红外加热炉作激活能产生装置加热置于供气管120中的石英管包围的激活空间114到700℃之外,以与例1同样的步骤形成沉积膜(2-1号及2-2号样品)。
对2-1号样品,成膜吋Hα/Hβ为155,对2-2号样品是60。
当用例1同样的步骤鉴定得到的沉积膜时,虽然微波输入功率降低了,但沉积率却增加了约10%,并得到与例1样品基本相同的性质,如表2所示。
例3
除了基片改用玻璃基片(商用名:7059号,康宁玻璃公司制造),设置成膜空间压强为0.06乇,基片温度250℃外,在例1中样品1-2形成的同样步骤和同样形成条件下形成沉积膜,成膜时Hα/Hp为70。
当用X-射线衍射仪和电子衍射仪(RHEED)对沉积膜测量其膜厚度分布及结晶性时,证明该膜厚度分布在±3%之内,它是-多晶膜,在平行于基片表面的取向几乎为(110),平均颗粒大小为1.2微米。氢含量的原子百分比为1.2%。
空穴迁移率测定为45±1。3cm2/伏·秒,传导类型是p-型。
例4-例6
除了改变成膜材料化合物和一部分成膜条件如表3所示外,在例1中同样步骤和成膜条件下形成沉积膜。
当测定所得沉积膜性质时,结果如表4所示它们是膜厚度和膜性质分布在±3%之内的高质量膜。
例7
首先,把直径150毫米的玻璃基片(商用品:7059号,康宁玻璃公间制造)放在成膜空间116的基片支架119上,成膜空间的压强用未示出的抽气机抽到1×10-6乇。然后,加热基片支架用未示出的基片温度控制器使基片表面温度设置在230℃。
当基片表面温度确定后,把20sccm氢气和200sccm氩气从未示出的储气罐通过气体输入管111送到石英钟罩102。随后用未示出的自动压强控制器把成膜空间116的压强控制在0.2乇。
然后,从未示出的连续型微波振荡器通过方形波导管118把微波引到共振腔101。不久之后,用与达106和变速齿轮107,把改变腔长的活动圆柱调节到由未示出的微波回路中的功率监视器测量的反射功率/入射功率比值为最小的位置,然后调节约束器110的张开程度使反射功率/入射功率之比为最小,然后重复细调改变腔长的活动圆柱104的位置和约束器110的张开程度,使反射功率/入射功率比最小,使入射功率/反射功率代表的有效入射功率放置在350瓦。
在该例中,通过孔121监测到的氢原子激发态发射线Hα/Hp的强度比是200。
基片118与金属网组件103距离为40毫米并相互平行。由直径156毫米的带孔铝板用作金属网组件103,孔径约为8毫米,以孔隙率50%均匀分布。所用气体释放环有如图2(a)所示构造,其中相应于201a和201a′的孔径为1.5毫米,孔径增加比为30%。
随后把10sccm的Si2F6气体和用氢气稀释到10%的GeF4气体从未示出的气体源通过供气管120输入,并通过气体释放环112送入成膜空间116。在这种情况下,成膜空间116中的压强被自动压强控制器维持在0.2乇。
不久,在激发态的氢原子和Si2F6,GeF4气体起化学反应,在60分钟内在基片上形成7.5微米厚的膜。基片冷却后取出作为7-1号样品。
除了用一个6英吋n+Si(110)芯片作基片且氢气流量为10sccm,Ar气流量为50sccm,成膜空间压强为0.01乇,带孔板的孔隙率为30%外,用上述同样的步骤形成沉积膜,强度比Hα/Hp为40(称作7-3号样品)。用X射线衍射仪和电子射线衍射仪(RHEED)测量得到每一沉积膜的膜厚度分布和其结晶性。每一样品都有好的均匀性,并指出7-1号样品是非晶形,7-2号样品是多晶膜,7-3号样品是在平行于基片表面有几乎(110)取向的外延膜。
此外,把每一样品切下一部分,用SIMS分析其组成。氢的含量按7-1号,7-2号,7-3号样品的次序逐减。
每一样品放于真空沉积装置中,通过电阻加热的直径2毫米的铝点状电极进行汽相沉积,用万·德·波尔方法测量空穴迁移率(μh)。在整个样品表面,特性分布误差在±3%之内,结果见表5。
从此结果可以看到根据本发明很容易控制沉积膜的结晶性。
例8
在下述条件下按例1同样的步骤形成一种Si∶Ge∶H∶F膜。除使GeF4气体流量为5sccm外在与制作例7样品1同样的条件下形成膜。(称为8-1号样品)
然后除分别设GeF4气体流量为7sccm,12sccm和15sccm外,在同样的方式分3次制作膜(称为8-2,8-3,8-4号样品)。
用X射线衍射仪和电子射线衍射仪(RHEED)鉴定每一得到的沉积膜样品的膜厚度分布及其结晶性,说明了每一样品的均匀性很好并都是非晶膜。
其次,从每一样品上切下一部分,用SIMS分析其硅和锗成份比。随后再用可见光谱仪测每一样品的吸收谱以定出光能带隙。进而将每一样品置于真空沉积室,用梳型间隙电极(隙宽250微米,宽5毫米)进行汽相沉积。测量10伏电压下的暗导电率,然后用AM-1光(100毫瓦/平方厘米)照射决定光导电率并由此决定暗导电率与光导电率之比。每一性质在整个平面中都在±3%之内。上述结果见表6。从结果发现,硅锗成份比可通过变化气体流量有选择地变化,光能带隙也能控制得形成有所需性质的沉积膜。
从结果可以看到,沉积膜的光学膜性质可以容易地按本发明来控制。
例9
除使氢气流量为50sccm和微波输入功率为300瓦外,在如下述条件下,按例7同样的步骤形成-Si∶C∶H∶F膜。用一个150毫米直径的玻璃基片(7059号)用作基片,把流量为20sccm的Si2F6气体和流量为10sccm的CH4气体送入成膜空间,成膜空间压强为0.2乇,基片温度为260℃。用与例1同样的气体释放环,金属网组件和基片位置,形成膜的时间为60分钟,
然后除了增加用氦气稀释到10%的10sccm的BF3外,用如上同样的步骤形成膜。在每一情况下,膜形成时Hα/Hp比是180(称为9-1号、9-2号样品)。
然后除了使氢气流量为100sccm及内压强0.03乇外,以与9-2号样品同样的步骤形成膜(称为9-3号样品)。
对每一这样得到的样品鉴定其结晶性和氢含量,进而用例7中同样的方式测热伏打功率以判断导电类型。分别从每一样品中切下一部分,放入真空沉积室,用梳型铝间隙电极(隙宽250微米,宽5毫米)进行汽相沉积。然后测量在10伏下的暗导电率,再在AM-1光(100毫瓦/平方厘米)照射下测光导电率,由此决定暗导电率和光导电率比值。在整个平面上每一样品性质在±3%之内。所得的结果见表7。
从上述结果可知,沉积膜的传导类型和电性质能按本发明容易地控制。
例10
除了把微波输入功率从350瓦变到200瓦及用激活能量产生装置红外加热炉把供气管120上面的石英管构成的激活空间114加热到700℃外,采用与例7同样的步骤形成沉积膜。
首先用7059号玻璃作基片并使氢气流量为100sccm,内压强为0.05乇条件下形成膜(称为10-1号样品)。然后,用6吋n+Si芯片作基片,使氢气流量为20sccm,成形空间压强为0.01乇(称为10-2号样品)条件下,形成另一个膜。
当这样得到的沉积膜以例7中同样的方式鉴定时,虽然微波输入功率减小,沉积率没有减少,得到了与例7中基本相同的性质,结果示于表8。
例11
除了改变成膜原料化合物并如表9所示改变一部分成膜条件。在与例7同样的成膜条件和同样步骤下形成沉积膜。
这样得到的膜以例9中同样的步骤鉴定,得到如表10所示的结果。
例12
除使氢气流量为50sccm,微波输入功率为200瓦,及下述条件下,以例7中同样的步骤形成Si∶C∶H∶F膜。
流量为20sccm的SiF4气体和流量为5sccm的SiH4气体的混合物用作第一原料气体,流量为10sccm的CF4作为第二原料气体,它们被引入成膜空间,成膜空间压强为0.03乇,基片温度为200℃。金属网组件的孔隙率为30%气体释放环和基片部分与例1中的一样。成膜时的Hα/Hβ比值是100(称为12-1号、12-2号样品)。
按例9中同样的步骤鉴定得到的样品,得到表11中所示的结果。
每一性质都表明该平面内的分布均匀性在±3%。
表2
表3
| 例 | 原料化合物及流率 | 成膜条件的改变 |
| 4 | GeF4 10sccmPF5(用氦稀释到500ppm)5sccmH2/Ar 30/150sccm | 微波功率 250WHα/Hβ= 140玻璃基片;#7059压强 0.08Torr基片温度 180℃ |
| 5 | GeF4/Ge(450℃)10SccmBF3(用氦气稀释到800ppm)5sccmH2/Ar 100/150sccm | 微波功率 250WHα/Hβ= 200n+Si 晶片压强 0.03Torr基片温度 250℃ |
| CH3F 15sccmH2/He 250/50sccm | 微波功率 350WHα/Hβ= 35非掺杂硅晶片压强 0.005torr基片温度 300℃ |
表4
| 例 | 膜质量 | 氢含量 | 膜性质 |
| 4 | 非晶Ge∶H∶F∶P | 21原子百分比% | μe1.5cm2/v.secn-型 |
| 5 | 外延Ge∶H∶F∶B | 0.05原子百分比% | μn3500cm2/v.secp-型 |
| 6 | 类钻石C∶H∶F | 0.08原子百分比% | 莫氏硬度>9 |
表5
| 样品号 | 7-1 | 7-2 | 7-3 |
| 膜质量 | Si∶Ge∶H∶F | Si∶Ge∶H∶F | Si∶Ge∶H∶F |
| 结晶性 | 非晶型 | 多晶型 | 外延型 |
| 氢含量 | 10.5% | 3.5% | 0.5% |
| 空穴迁移率 | 0.8cm2/v.sec | 65cm2/v.sec | 1200cm2/v.sec |
| 传导类型 | 本征 | 本征 | 本征 |
表6
| 样品号 | 8-1 | 8-2 | 8-3 | 8-4 |
| GeF4流率 | 5sccm | 7sccm | 12sccm | 15sccm |
| Si∶Ge 成分比(原子百分数) | 86∶14 | 79∶21 | 70∶30 | 61∶39 |
| 光能带隙 | 1.65eV | 1.60eV | 1.53eV | 1.44eV |
| δp/δd | 4.0×105 | 9.2×104 | 3.3×104 | 1.8×104 |
表7
| 样品号 | 9-1 | 9-2 | 9-3 |
| 膜质量 | Si∶C∶H∶F | Si∶C∶H∶F | Si∶C∶H∶F |
| 结晶型 | 非晶型 | 非晶型 | 多晶型 |
| 氢含量 | 12.5% | 10.8% | 4.5% |
| δp/δd | 5.5/×104 | 6.2×104 | 3.9×102 |
| 传导类型 | 本征 | p-型 | p-型 |
表8
| 样品号 | 10-1 | 10-2 |
| 膜质量 | Si∶Ge∶H∶F | Si∶Ge∶H∶F |
| 结晶型 | 多晶型 | 外延型 |
| 氢含量 | 10.5% | 3.5% |
| 空穴迁移率 | 62cm2/v.sec | 1220cm2/v.sec |
| 传导类型 | 本征 | 本征 |
表9
| 样品号 | 原料气体及流率 | 成膜条件 |
| 11-1 | SiF4 30sccmC2H2 5 ″H2 20 ″Ar 200 ″ | 微波功率 280W引入在激活室中用射频放电(10W)激活的SiF4 |
| 11-2 | SiH2F2 30sccmSnF4 20 ″H2 40 ″Ar 200 ″ | 微波功率 200W压强 0.65Torr |
| 11-3 | SiH2Cl230sccmSnCl4 10 ″H2 20 ″Ar 200 ″B2H6 10 ″ | 微波功率 280W压强 0.15Torr |
表10
| 样品号 | 11-1 | 11-2 | 11-3 |
| 膜质量 | Si∶C∶H∶F | Si∶Sn∶H∶F | Si∶Sn∶H∶F |
| 结晶型 | 非晶 | 非晶 | 非晶 |
| 氢含量 | 9.8% | 7.6% | 8.1% |
| δp/δα | 2.5×104 | 3.8×102 | 2.8×102 |
| 传导类型 | 本征 | 本征 | p-型 |
表11
| 样品号 | 12-1 | 12-2 |
| 膜质量 | Si∶C∶H∶F | Si∶C∶H∶F |
| 结晶性 | 多晶 | 外延 |
| 氢含量 | 2.5% | 0.02% |
| δp/δd | 8.5×102 | 3.2 |
| 传导类型 | 本征 | 本征 |
Claims (18)
1.一种形成功能沉积膜的方法,通过向在其上形成沉积膜的基片所在的成膜空间引入作为成膜原料的含有属于周期表第四族元素的化合物,以及,如果需要,引入含有能控制沉积膜价电子的元素作为组成成分的化合物,形成含有周期表第四族原子作为主要组成原子的功能沉积膜,每一化合物成气态或者至少化合物是在与所述成膜空间分开的激活空间中预激活的状态,同时形成激发态氢原子,它们与至少一种所述气态的或者在不同于所述成膜空间的激活空间中处于激活态的并被引入成膜空间的化合物起化学反应,从而在所述基片上形成沉积膜,其特征在于:
所述激发态氢原子是用一微波回路中的与两个阻抗匹配回路相结合的共振腔中的等离子体产生室中产生的微波等离子体,从氢气或氢气与一惰性气体组成的气态混合物形成的,所述等离子体产生室包括一个金属网组件和可渗透微波的钟罩,并通过所述金属网组件与所述成膜空间连在一起,而且氢原子的激发态是受到控制的。
2.根据权利要求1的形成功能沉积膜的方法,其中氢原子的激发态是通过用发射分光光度测量方法测量氢原子激发态的发射强度比Hα/Hβ来控制的,并控制供给其共振腔的微波功率,阻抗匹配条件,氢气流率或氢气与惰性气体的气态混合物的流率以及总压强中的至少一个参数,以获得所述强度比的所需值。
3.根据权利要求1或2的形成功能沉积膜的方法,其中在微波回路中与两个阻抗匹配回路相结合的共振腔中的阻抗匹配回路是置于改变空腔长度的活动圆柱或置于波导管与共振腔连接部分的约束装置,通过调整这些装置来控制阻抗匹配回路。
4.根据权利要求3的形成功能沉积膜的方法,其中阻抗匹配回路是改变共振腔的活动圆柱和一个E—H调谐器或一个三抽头调谐器。
5.根据权利要求1的形成功能沉积膜的方法,其中激发态的氢原子通过金属网组件引入成膜空间。
6.根据权利要求1的形成功能沉积膜的方法,其中基片相对于金属网表面水平轴成30°度角或更小,并与所述金属网表面相距在100毫米以内,把气态或激活态化合物从置于所述金属网表面和所述基片之间的气体释放装置引入成膜空间。
7.根据权利要求6的形成功能沉积膜的方法,其中气体释放装置呈环形安装在基片四周,气体释放孔之间的间隔从引入气体一侧向着所述气体释放装置的最后释放孔逐渐减小,使从各气体释放孔来的气体释放量均匀。
8.根据权利要求6的形成功能沉积膜的方法,其中气体释放装置呈环形安装在基片四周,气体释放孔的孔径从引入气体一侧向着所述气体释放装置的最后释放孔逐渐增加,使从各气体释放孔来的气体释放量均匀。
9.根据权利要求6的形成功能沉积膜的方法,其中气体释放装置的气体释放孔至少在基片平面内均匀分布,各气体释放孔的孔径从引入气体一侧向着所述气体释放装置的中央部分逐渐增加,使从各气体释放孔来的气体释放量均匀。
10.一种形成功能沉积膜的方法,通过向在其上形成沉积膜的基片所在的成膜空间引入含有硅的化合物(1)和用下述公式(1)表示的作为成膜原料的化合物(2),以及,如果需要,引入含有能控制沉积膜价电子的元素作为组成成分的化合物(3),形成含有硅原子和周期表第四族原子作为主要组成原子的功能沉积膜,每一化合物成气态或者成至少一种化合物是在与所述成膜空间分开的激活空间中预激活的状态,同时形成激发态的氢原子,与处于所述气态或在不同于所述属膜空间的激活空间中处于激活态的并被引入成膜空间的所述化合物(1),(2)和(3)的至少一种起化学反应,从而在所述基片上形成沉积膜,其特征在于:
所述激发态氢原子是用一微波回路中的与两个阻抗匹配回路相结合的共振腔中的等离子体产生室中产生的微波等离子体,从氢气或氢气与一种惰性气体组成的气态混合物形成的,所述等离子体产生室包括一个金属网组件和可渗透微波的钟罩,并通过所述金属网组件与所述成膜空间连在一起,而且氢原子的激发态是受到控制的:
AaBb ……(I)其中A代表除硅以外的属于周期表第四族的元素,B代表从氢(H),卤素(X)和烃中选的一种成分,a代表一个等于B的价数或该价数整数倍的正整数,b代表一个正整数。
11.根据权利要求10的形成功能沉积膜的方法,其中氢原子的激发态是通过用发射分光光度测量方法测量氢原子激发态的发射强度比Hα/Hβ,以及控制供给共振的微波功率,阻抗匹配条件,氢气流率或氢气与惰性气体的气态混合物的流量以及总压强中的至少一个参数来控制的,以获得所述预期的强度比。
12.根据权利要求10或11的形成功能沉积膜的方法,其中在微波回路中与两个阻抗匹配回路相结合的共振腔中的阻抗匹配回路是置于改变空腔长度的活动圆柱或置于波导管与共振腔连接部分的约束装置,通过调整这些装置来控制匹配状态。
13.根据权利要求12的形成功能沉积膜的方法,其中阻抗匹配回路是改变共振腔的活动圆柱和一个E—H调谐器或一个三抽头调谐器。
14.根据权利要求10的形成功能沉积膜的方法,其中激发态氢原子是通过金属网组件引入成膜空间的。
15.根据权利要求10的形成功能沉积膜的方法,其中基片相对于金属网表面水平轴成30度角或更小,并与所述金属网表面相距100毫米以内,气态或激活态化合物(1)、(2)和(3)从置于所述金属网表面和所述基片之间的气体释放装置引入成膜空间。
16.根据权利要求15的形成功能沉积膜的方法,其中气体释放装置呈环形安放在基片四周,气体释放孔的间隔从引入气体一侧向着所述气体释放装置的最后释放孔逐渐减小,使从各气体释放孔来的气体释放量均匀。
17.根据权利要求15的形成功能沉积膜的方法,其中气体释放装置呈环形放置在基片四周,气体释放孔的孔径从引入气体的一侧向着所述气体释放装置的最后释放孔逐渐增加,使从各气体释放孔来的气体释放量均匀。
18.根据权利要求15的形成功能沉积膜的方法,其中气体释放装置的气体释放孔至少在基片平面均匀分布,各气体释放孔的孔径从引入气体一侧向着所述气体释放装置的中央部分逐渐增加,使从各气体释放孔来的气体释放量均匀。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP21799/1988 | 1988-02-01 | ||
| JP2179888A JPH01198479A (ja) | 1988-02-01 | 1988-02-01 | マイクロ波プラズマcvd法による堆積膜形成法 |
| JP21798/88 | 1988-02-01 | ||
| JP2179988A JPH01198480A (ja) | 1988-02-01 | 1988-02-01 | マイクロ波プラズマcvd法による堆積膜形成法 |
| JP21798/1988 | 1988-02-01 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1039680A CN1039680A (zh) | 1990-02-14 |
| CN1031088C true CN1031088C (zh) | 1996-02-21 |
Family
ID=26358903
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN89100622.2A Expired - Fee Related CN1031088C (zh) | 1988-02-01 | 1989-02-01 | 形成功能沉积膜的方法 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US4908330A (zh) |
| EP (1) | EP0334000B1 (zh) |
| CN (1) | CN1031088C (zh) |
| DE (1) | DE68917500T2 (zh) |
Families Citing this family (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0242182B1 (en) * | 1986-04-14 | 1993-06-30 | Canon Kabushiki Kaisha | Process for forming deposited film |
| US5221643A (en) * | 1989-02-21 | 1993-06-22 | Siemens Aktiengesellschaft | Method for producing polycrystalline semiconductor material by plasma-induced vapor phase deposition using activated hydrogen |
| US5214002A (en) * | 1989-10-25 | 1993-05-25 | Agency Of Industrial Science And Technology | Process for depositing a thermal CVD film of Si or Ge using a hydrogen post-treatment step and an optional hydrogen pre-treatment step |
| JPH0560242A (ja) * | 1991-08-28 | 1993-03-09 | Japan Atom Energy Res Inst | セラミツクス製真空容器及びその製造方法 |
| US5204272A (en) * | 1991-12-13 | 1993-04-20 | United Solar Systems Corporation | Semiconductor device and microwave process for its manufacture |
| US5665640A (en) * | 1994-06-03 | 1997-09-09 | Sony Corporation | Method for producing titanium-containing thin films by low temperature plasma-enhanced chemical vapor deposition using a rotating susceptor reactor |
| WO1995034092A1 (en) * | 1994-06-03 | 1995-12-14 | Materials Research Corporation | A method of nitridization of titanium thin films |
| US5628829A (en) * | 1994-06-03 | 1997-05-13 | Materials Research Corporation | Method and apparatus for low temperature deposition of CVD and PECVD films |
| US5975912A (en) * | 1994-06-03 | 1999-11-02 | Materials Research Corporation | Low temperature plasma-enhanced formation of integrated circuits |
| US6161500A (en) | 1997-09-30 | 2000-12-19 | Tokyo Electron Limited | Apparatus and method for preventing the premature mixture of reactant gases in CVD and PECVD reactions |
| US6313017B1 (en) | 1999-01-26 | 2001-11-06 | University Of Vermont And State Agricultural College | Plasma enhanced CVD process for rapidly growing semiconductor films |
| US6173673B1 (en) | 1999-03-31 | 2001-01-16 | Tokyo Electron Limited | Method and apparatus for insulating a high power RF electrode through which plasma discharge gases are injected into a processing chamber |
| US6743700B2 (en) * | 2001-06-01 | 2004-06-01 | Semiconductor Energy Laboratory Co., Ltd. | Semiconductor film, semiconductor device and method of their production |
| US7199027B2 (en) * | 2001-07-10 | 2007-04-03 | Semiconductor Energy Laboratory Co., Ltd. | Method of manufacturing a semiconductor film by plasma CVD using a noble gas and nitrogen |
| JP5072157B2 (ja) * | 2001-09-27 | 2012-11-14 | 株式会社半導体エネルギー研究所 | 半導体装置の作製方法 |
| JP2003293128A (ja) * | 2002-04-05 | 2003-10-15 | Canon Inc | 堆積膜形成方法 |
| US7205187B2 (en) * | 2005-01-18 | 2007-04-17 | Tokyo Electron Limited | Micro-feature fill process and apparatus using hexachlorodisilane or other chlorine-containing silicon precursor |
| DE102005024041A1 (de) | 2005-05-25 | 2006-11-30 | City Solar Ag | Verfahren zur Herstellung von Silicium aus Halogensilanen |
| DE102006043929B4 (de) * | 2006-09-14 | 2016-10-06 | Spawnt Private S.À.R.L. | Verfahren zur Herstellung von festen Polysilanmischungen |
| KR20100093347A (ko) * | 2009-02-16 | 2010-08-25 | 엘지전자 주식회사 | 태양전지, 태양전지의 제조방법 및 제조장치, 박막 증착방법 |
| JP5993720B2 (ja) | 2011-11-30 | 2016-09-14 | キヤノン株式会社 | 電子写真感光体、プロセスカートリッジおよび電子写真装置 |
| JP6071439B2 (ja) | 2011-11-30 | 2017-02-01 | キヤノン株式会社 | フタロシアニン結晶の製造方法、および電子写真感光体の製造方法 |
| JP5827612B2 (ja) | 2011-11-30 | 2015-12-02 | キヤノン株式会社 | ガリウムフタロシアニン結晶の製造方法、及び該ガリウムフタロシアニン結晶の製造方法を用いた電子写真感光体の製造方法 |
| JP2017010009A (ja) | 2015-06-24 | 2017-01-12 | キヤノン株式会社 | 電子写真感光体、プロセスカートリッジおよび電子写真装置 |
| DE102015115355B3 (de) * | 2015-09-11 | 2017-01-19 | Schunk Kohlenstofftechnik Gmbh | Heizkammer zum Analysieren von Fremdstoffgehalten in Proben |
| US10095137B2 (en) | 2016-04-04 | 2018-10-09 | Canon Kabushiki Kaisha | Electrophotographic photosensitive member, method of producing electrophotographic photosensitive member, process cartridge, and electrophotographic image forming apparatus |
| PT109387B (pt) * | 2016-05-13 | 2021-12-14 | Inst Superior Tecnico | Processo e sistema para a produção seletiva de nanoestruturas bidimensionais autónomas utilizando tecnologia plasma |
| JP6978858B2 (ja) | 2016-06-21 | 2021-12-08 | キヤノン株式会社 | 電子写真感光体、電子写真感光体の製造方法、該電子写真感光体を有するプロセスカートリッジおよび電子写真装置 |
| CN106770144B (zh) * | 2017-03-06 | 2024-04-09 | 清华大学 | 一种基于氢等离子体的固体样品化学蒸气发生进样方法 |
| KR102058865B1 (ko) * | 2018-04-12 | 2019-12-24 | (주)아이엠 | 초가속 열소재를 이용한 발열 디바이스 및 이의 제조방법 |
| US11320754B2 (en) | 2019-07-25 | 2022-05-03 | Canon Kabushiki Kaisha | Process cartridge and electrophotographic apparatus |
| US11573499B2 (en) | 2019-07-25 | 2023-02-07 | Canon Kabushiki Kaisha | Process cartridge and electrophotographic apparatus |
| US12000062B1 (en) | 2019-07-30 | 2024-06-04 | United States Of America As Represented By The Secretary Of The Air Force | Method for the deposition of monocrystalline or polycrystalline tin alloys on crystallographcially mis-matched or amorphous substrates |
| JP7449151B2 (ja) | 2020-04-21 | 2024-03-13 | キヤノン株式会社 | 電子写真感光ドラム |
| JP7444691B2 (ja) | 2020-04-21 | 2024-03-06 | キヤノン株式会社 | 電子写真感光体の製造方法 |
| JP7483477B2 (ja) | 2020-04-21 | 2024-05-15 | キヤノン株式会社 | 電子写真感光ドラム、プロセスカートリッジおよび電子写真画像形成装置 |
| CN114457323B (zh) * | 2022-04-12 | 2022-08-02 | 成都纽曼和瑞微波技术有限公司 | 一种反应腔装置及微波等离子体气相沉积系统 |
Family Cites Families (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS55131175A (en) * | 1979-03-30 | 1980-10-11 | Toshiba Corp | Surface treatment apparatus with microwave plasma |
| CA1159012A (en) * | 1980-05-02 | 1983-12-20 | Seitaro Matsuo | Plasma deposition apparatus |
| US4421592A (en) * | 1981-05-22 | 1983-12-20 | United Technologies Corporation | Plasma enhanced deposition of semiconductors |
| JPS5871369A (ja) * | 1981-10-20 | 1983-04-28 | Konishiroku Photo Ind Co Ltd | 感光体の製造装置 |
| JPS59193265A (ja) * | 1983-03-14 | 1984-11-01 | Stanley Electric Co Ltd | プラズマcvd装置 |
| JPS6063376A (ja) * | 1983-09-14 | 1985-04-11 | Canon Inc | 気相法堆積膜製造装置 |
| JPS59103331A (ja) * | 1983-09-21 | 1984-06-14 | Hitachi Ltd | プラズマ処理装置 |
| US4657777A (en) * | 1984-12-17 | 1987-04-14 | Canon Kabushiki Kaisha | Formation of deposited film |
| US4566403A (en) * | 1985-01-30 | 1986-01-28 | Sovonics Solar Systems | Apparatus for microwave glow discharge deposition |
| JPS61202438A (ja) * | 1985-03-06 | 1986-09-08 | Ulvac Corp | グロ−放電安定化方法 |
| JPH07101751B2 (ja) * | 1985-03-28 | 1995-11-01 | キヤノン株式会社 | 光起電力素子の製造方法 |
| JPS61276977A (ja) * | 1985-05-30 | 1986-12-06 | Canon Inc | 堆積膜形成法 |
| JPS6277479A (ja) * | 1985-09-30 | 1987-04-09 | Shimadzu Corp | プラズマcvd法による薄膜形成方法 |
| JPS62116775A (ja) * | 1985-11-15 | 1987-05-28 | Canon Inc | プラズマcvd装置 |
| JPH0821542B2 (ja) * | 1985-12-24 | 1996-03-04 | キヤノン株式会社 | 機能性堆積膜の製造法 |
| JPH084071B2 (ja) * | 1985-12-28 | 1996-01-17 | キヤノン株式会社 | 堆積膜形成法 |
| US4673589A (en) * | 1986-02-18 | 1987-06-16 | Amoco Corporation | Photoconducting amorphous carbon |
| US4832778A (en) * | 1987-07-16 | 1989-05-23 | Texas Instruments Inc. | Processing apparatus for wafers |
| US4837113A (en) * | 1987-07-16 | 1989-06-06 | Texas Instruments Incorporated | Method for depositing compound from group II-VI |
-
1989
- 1989-01-27 US US07/302,245 patent/US4908330A/en not_active Expired - Lifetime
- 1989-01-31 DE DE68917500T patent/DE68917500T2/de not_active Expired - Lifetime
- 1989-01-31 EP EP89101644A patent/EP0334000B1/en not_active Expired - Lifetime
- 1989-02-01 CN CN89100622.2A patent/CN1031088C/zh not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| US4908330A (en) | 1990-03-13 |
| DE68917500T2 (de) | 1994-12-15 |
| EP0334000A2 (en) | 1989-09-27 |
| DE68917500D1 (de) | 1994-09-22 |
| EP0334000B1 (en) | 1994-08-17 |
| CN1039680A (zh) | 1990-02-14 |
| EP0334000A3 (en) | 1989-11-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1031088C (zh) | 形成功能沉积膜的方法 | |
| CN1016449B (zh) | 用微波等离子体化学沉积工艺形成主要含ii和vi族原子的实用沉积膜的工艺 | |
| CN1082569C (zh) | 微波等离子体处理装置及其处理方法 | |
| CN1201408C (zh) | 薄膜多晶太阳能电池及其形成方法 | |
| CN1160483C (zh) | 薄膜制备方法和淀积设备 | |
| JP6046237B2 (ja) | マイクロ波プラズマ化学気相成長装置 | |
| JPH08239295A (ja) | 結晶製造方法及び結晶製造装置 | |
| WO2013087702A2 (en) | Large area optical quality synthetic polycrystalline diamond window | |
| CN100338744C (zh) | 形成氧化物薄膜的方法及其装置 | |
| CN1260009A (zh) | 涂层工件的制造方法、方法的应用及其装置 | |
| CN1670917A (zh) | 制造化合物半导体的方法和制造半导体器件的方法 | |
| CN1016519B (zh) | 用微波等离子化学气相沉积法形成以iii和v族原子为主组分的功能沉积膜的方法 | |
| CN1132962C (zh) | 淀积膜形成系统和方法 | |
| CN111223761A (zh) | 一种沉积多晶硅表面颗粒质量改善方法 | |
| CN1029135C (zh) | 汽相淀积金刚石的装置 | |
| CN1053229C (zh) | 利用磁场的微波增强型cvd系统和方法 | |
| JP2019112288A (ja) | 炭化ケイ素部材および半導体製造装置用部材 | |
| CN88101737A (zh) | 汽相淀积金刚石的方法及装置 | |
| KR20030090650A (ko) | 부품 제조 방법 및 진공 처리 시스템 | |
| CN1023239C (zh) | 利用分别生成的多种活性气体制备大面积沉积膜的装置 | |
| JP2000096239A (ja) | 誘導結合型プラズマcvd方法及びそのための誘導結合型プラズマcvd装置 | |
| JPH05339730A (ja) | ダイヤモンド被膜の形成方法 | |
| JP3857446B2 (ja) | SiC成形体 | |
| JP2013038099A (ja) | 気相成長装置 | |
| JP2002037669A (ja) | 炭化珪素材、耐プラズマ部材及び半導体製造用装置 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C15 | Extension of patent right duration from 15 to 20 years for appl. with date before 31.12.1992 and still valid on 11.12.2001 (patent law change 1993) | ||
| OR01 | Other related matters | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |