CN101912421A - 涉及痘病毒和癌的方法及组合物 - Google Patents
涉及痘病毒和癌的方法及组合物 Download PDFInfo
- Publication number
- CN101912421A CN101912421A CN2010101583381A CN201010158338A CN101912421A CN 101912421 A CN101912421 A CN 101912421A CN 2010101583381 A CN2010101583381 A CN 2010101583381A CN 201010158338 A CN201010158338 A CN 201010158338A CN 101912421 A CN101912421 A CN 101912421A
- Authority
- CN
- China
- Prior art keywords
- cell
- virus
- polypeptide
- tumor
- vaccinia virus
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images













Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/66—Microorganisms or materials therefrom
- A61K35/76—Viruses; Subviral particles; Bacteriophages
- A61K35/768—Oncolytic viruses not provided for in groups A61K35/761 - A61K35/766
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/19—Cytokines; Lymphokines; Interferons
- A61K38/21—Interferons [IFN]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/86—Viral vectors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N7/00—Viruses; Bacteriophages; Compositions thereof; Preparation or purification thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/24011—Poxviridae
- C12N2710/24111—Orthopoxvirus, e.g. vaccinia virus, variola
- C12N2710/24121—Viruses as such, e.g. new isolates, mutants or their genomic sequences
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/24011—Poxviridae
- C12N2710/24111—Orthopoxvirus, e.g. vaccinia virus, variola
- C12N2710/24132—Use of virus as therapeutic agent, other than vaccine, e.g. as cytolytic agent
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/24011—Poxviridae
- C12N2710/24111—Orthopoxvirus, e.g. vaccinia virus, variola
- C12N2710/24141—Use of virus, viral particle or viral elements as a vector
- C12N2710/24143—Use of virus, viral particle or viral elements as a vector viral genome or elements thereof as genetic vector
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/24011—Poxviridae
- C12N2710/24111—Orthopoxvirus, e.g. vaccinia virus, variola
- C12N2710/24161—Methods of inactivation or attenuation
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/24011—Poxviridae
- C12N2710/24111—Orthopoxvirus, e.g. vaccinia virus, variola
- C12N2710/24161—Methods of inactivation or attenuation
- C12N2710/24162—Methods of inactivation or attenuation by genetic engineering
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Zoology (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Wood Science & Technology (AREA)
- Virology (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Immunology (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- Biomedical Technology (AREA)
- General Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Biochemistry (AREA)
- Mycology (AREA)
- Oncology (AREA)
- Plant Pathology (AREA)
- Gastroenterology & Hepatology (AREA)
- Molecular Biology (AREA)
- Physics & Mathematics (AREA)
- Biophysics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
本发明涉及利用改变的痘病毒,包括经改造后可产生更有效的治疗性剂的痘苗病毒,治疗癌和癌细胞的方法和组合物。这种痘病毒经改造后变成减毒株,或者其影响正常细胞的能力减弱。在某些实施方案中,方法和组合物包括携带突变的痘病毒,这种突变可使痘病毒在宿主内激发抗病毒反应的能力减弱或消失。携带这些突变及其他突变的痘病毒可用于更有效地治疗癌症。
Description
本申请是国际申请PCT/US2003/025141,国际申请日2003年8月11日,中国国家阶段申请号03823719.9,名称“涉及痘病毒和癌的方法及组合物”的发明专利申请的分案申请。
发明背景
1.发明领域
本发明涉及肿瘤学和病毒学领域。更具体地说,本发明涉及包含一个或多个突变从而使其特别适于癌症治疗的痘病毒,尤其是指痘苗病毒。
2.相关领域的描述
正常组织的内环境稳定是一种细胞增殖和细胞死亡之间高度平衡的过程。细胞增殖和细胞死亡之间失去平衡就会致癌(Solyanik等,1995;Stokke等,1997;Mumby和Walter,1991;Natoli等,1998;Magi-Galluzzi等,1998)。例如,宫颈癌、肾癌、肺癌、胰腺癌、结肠癌和脑癌只是可能发生的多种癌中的几种(Erlandsson,1998;Kolmel,1998;Mangray和King,1998;Gertig和Hunter,1997;Mougin等,1998)。事实上,癌症的发生率相当高,仅在美国每年就有超过500,000的人死于癌症。
细胞增殖和细胞死亡的维持至少部分是由原癌基因和肿瘤抑制基因调节的。原癌基因或肿瘤抑制基因可编码诱导细胞增殖的蛋白质(如sis、erbB、src、ras和myc)、抑制细胞增殖的蛋白质(如Rb,p16,p19,p21,p53,NF1和WT1)或调节细胞调亡的蛋白质(如bcl-2)(Ochi等,1998;Johnson和Hamdy,1998;Liebermann等,1998)。但是,基因重排或这些原癌基因和肿瘤抑制基因的突变都会导致原癌基因转化为致癌的癌基因或使肿瘤抑制因子变成无活性的多肽。一般来说单一点突变就足以完成这种转化。例如,肿瘤抑制蛋白p53上的点突变就可以导致其完全失去野生型p53的功能(Vogelstein和Kinzler,1992)。
到目前为止,只有几种有效的方法用于治疗多种常见类型的癌。对于单个个体来说,选用何种治疗措施要根据诊断结果、疾病分期以及其他因素如年龄、性别及患者的健康状况。最常用的癌症治疗方法是手术、放疗和化疗。手术在肿瘤的诊断和治疗中扮演主要角色。通常活检和去除癌性生长物都需要通过手术方法。但是,如果肿瘤已经转移和扩散,则手术就不可能使患者痊愈,因此需要采用其他治疗措施。放疗、化疗和免疫治疗是手术治疗措施的替代方法(Mayer,1998;Ohara,1998;Ho等,1998)。放疗是指通过高能射线精确照射以摧毁癌细胞,与手术很相似,主要用于治疗未转移的局域化的癌细胞。放疗的副作用包括皮肤刺激、吞咽困难、口干、恶心、腹泻、脱发和无力(Curran,1998;Brizel,1998)。
化疗是指利用抗癌药治疗癌症,是另一种癌症治疗模式。一种特定抗癌药的疗效通常由于药物难以进入实体瘤内而受到限制(el-Kareh和Secomb,1997)。化疗策略的基础在于肿瘤组织可以生长,其中抗肿瘤药物的靶位是快速分裂的癌细胞。最常用的化疗方法包括多种抗癌药的联合化疗,这种方法被证实可以提高多种癌的反应率(美国专利5,824,348;美国专利5,633,016和美国专利5,798,339,本文已纳入作为参考)。化疗药物的主要副作用是这些药物也可以影响正常组织细胞,最可能受影响的是那些在某些情况下快速分裂的细胞(如骨髓、胃肠道、生殖系统和毛囊)。化疗药物的其他毒性效应包括口疮、吞咽困难、口干、恶心、腹泻、呕吐、疲乏、出血、脱发和感染。
免疫治疗是癌症研究中一个快速发展的领域,是另一种治疗某些类型癌的方法。从理论上说,免疫系统被刺激后可将肿瘤细胞认定为异源物质,从而将它们作为摧毁的靶位。但不幸的是,这时的免疫系统发生的反应一般都不足以阻止大多数肿瘤的生长。但是,最近研究的热点集中在免疫治疗领域以开发出增强或补充免疫系统自然防卫机制的方法。目前正在研究或正在使用的免疫治疗方法有免疫佐剂(如牛结核菌、恶性疟原虫、二硝基氯苯和芳香化合物)(美国专利5,801,005;美国专利5,739,169;Hui和Hashimoto,1998;Christodoulides等,1998)、细胞因子治疗(如干扰素(IL-1,GM-CSF和TNF)(Bukowski等,1998;Davidson等,1998;Hellstrand等,1998)以及基因治疗(如TNF,IL-1,IL-2,p53)(Qin等,1998;Austin-Ward和Villaseca,1998;美国专利5,830,880和美国专利5,846,945)和单克隆抗体(如抗神经节苷脂GM2,抗-HER-2,抗-p185)(Pietras等,1998;Hanibuchi等,1998;美国专利5,824,311)。这些方法虽然表现出了一定的前景,但是取得成功的例子还是有限的。
复制选择性的瘤溶解病毒具有可用于癌症治疗的前景(Kirn等,2001)。这些病毒可通过直接的复制依赖性和/或病毒基因表达依赖性瘤溶解效应引起肿瘤细胞的死亡(Kirn等,2001)。另外,该病毒还可以增强诱导宿主内细胞介导的抗肿瘤免疫反应(Todo等,2001;Sinkovics等,2000)。这些病毒经改造后还可以在肿瘤内表达治疗性目的基因以增强抗肿瘤效应(Hermiston,2000)。
但是这种治疗方法还存在一些主要的限制。虽然某些种类的病毒已被证明具有一定程度的肿瘤选择性,但是这种新方法还需要将瘤溶解病毒改造和/或增强其肿瘤选择性以使其安全性达到最高。当通过静脉注射时以及当具有潜在毒性的治疗基因插入到这些病毒中以增强其抗肿瘤能力时这种选择性尤其重要;基因的表达也要只限于正常组织。另外,通过其他机制如诱导抗肿瘤免疫反应或靶向到肿瘤相关血管以增强抗肿瘤能力也是值得赞许的。
因此,需要找到一种更有效的并且毒性较低的癌症治疗措施。使用瘤溶解病毒进行治疗是一个克服上述缺点的领域。因此,本发明的目的在于解决这些问题。
发明概述
本发明基于以下发现:痘病毒经改造后可以1)产生出对不同细胞群或不同类型的组织有不同影响的物质和/或2)产生出具有更高感染性并且通过从感染细胞内释放出来而能够感染其他细胞的痘病毒。本发明的痘病毒,如痘苗病毒的Copenhagen株,可以和其他治疗措施(如化疗)产生协同效应,通过靶向到肿瘤的血管产生有益的疗效而不会对抗或抑制TNF和/或INF通路。需要说明的是痘苗病毒的任何病毒株都可以使用。在某些实施方案中,痘苗病毒的Copenhagen株或其衍生物是优选的。
在一些实施方案中,痘病毒对靶细胞具有较高的毒性或治疗效应,而对于其他的非靶细胞来说是相对无毒的,这是通过抗病毒反应将非靶细胞区分开来。这种病毒可用于在靶细胞内表达痘病毒或异源肽或多肽,从而导致这些细胞发生致命性感染。特别需要注意的是对于非靶细胞或非靶组织来说,痘病毒是“减毒的”,即该病毒的毒力是减弱的、降低的、下降的、被抑制的或被去除的,包括其介导宿主内源性抗病毒反应的能力。因此,本发明涉及包含痘苗病毒的组合物和方法,其中的痘苗病毒经过改造,使其可用于处理细胞或组织,这些细胞或组织产生抗病毒反应的能力已被消除,而对于可诱导产生有效的抗病毒反应的正常细胞或组织来说,这些病毒是无效的。本发明的方法特别优选使用这种痘苗病毒来治疗癌细胞或肿瘤。
本发明的病毒被认为是可以溶解肿瘤的并且其安全性得到提高和/或可被正常组织加速清除,其机制在于与非正常细胞(如癌细胞或癌组织)相对应的正常细胞具有产生抗病毒反应的能力(如拥有、产生和/或诱导免疫反应)。正常细胞或组织具有这种能力,而癌细胞或癌组织通常不表达、或低水平表达可诱导或参与抗病毒反应的细胞蛋白。这种细胞蛋白包括干扰素、TNF、趋化因子、细胞因子以及其他因子。在正常细胞或组织内,激发抗病毒免疫反应能力下降的减毒病毒可以很容易地被清除;但是在非正常细胞和/或组织内抗病毒反应减弱,因此即使是减毒的病毒也不能被有效清除。本发明某些实施方案的基础就是本文所讨论的病毒是一种经改善的治疗方式,如增强其在正常细胞内的安全性、降低其毒性,因此这些病毒对正常细胞的影响要小于非正常细胞。因此,减毒病毒在癌细胞内更易复制和表达基因,其中对干扰素的诱导或反应减弱或缺失。
本发明的组合物和方法涉及痘病毒。下面所讨论的病毒包含在本发明的组合物和方法中。虽然许多实施方案利用的是痘苗病毒,但是其他痘病毒也可以用同样的方式制备或使用,因此,涉及痘苗病毒的实施方案也适用于具有相同基因的其他痘病毒或其他病毒。
本发明涉及在其病毒基因组内含有一个或多个突变的经改造的痘苗病毒。突变可通过重组基因工程方法引入到病毒内,如随机突变或将病毒重复传代。利用重组基因工程将病毒基因组(或病毒基因组前体)改造的病毒被称为“重组”病毒。突变可以是一个或多个核苷酸残基的缺失或替代。突变可以在基因内(包括编码序列和非编码序列,如转录控制序列),也可以在其他位置。编码区的突变可产生具有缺失、插入或替代的同源肽或多肽。核酸突变也可以导致移码突变从而产生出截短的肽或多肽,或者氨基酸序列发生改变的肽或多肽。
在本发明的某些实施方案中,痘苗病毒是减毒的,这就需要改造病毒的基因组以使其毒力减弱。突变可影响具有不同功能的多肽。下列类型的多肽中的一种或多种都可以发生突变:1)干扰素调节多肽;2)补体控制多肽;3)TNF或趋化因子调节多肽;4)丝氨酸蛋白酶抑制因子;5)IL-1β调节多肽;6)非感染性EEV形式的多肽;以及7)可抑制感染性病毒从细胞内释放的病毒多肽(抗感染性病毒形式的多肽)。另外,痘苗病毒的A41L或C11R(或其他痘病毒的相应多肽)也可以发生突变。每一类多肽都是直接或间接具有特定功能的多肽。该类多肽的突变不是排他性的,因为一个多肽可能不仅仅具有一种特定的功能。
干扰素调节多肽是指具有影响细胞内干扰素诱导或活化通路活性的痘病毒多肽。干扰素参与某些细胞或生物的抗病毒机制。痘病毒表达可抑制这种机制的干扰素调节多肽。特别需要说明的是这些多肽可抑制、降低或消除这种特殊的抗病毒反应。这些多肽可直接或间接调节、影响、干扰、抑制、降低、改变或消除干扰素的活性或功能。干扰素α、β和γ是干扰素调节多肽的靶位。干扰素调节多肽还可以细分为可特异性结合干扰素的多肽;这种多肽可被称为干扰素结合多肽。B18R是一种可溶性的痘苗病毒多肽,是一种干扰素结合多肽,尤其可与IFNα/β特异性结合。B8R是可与干扰素γ特异性结合的另外一种痘苗病毒多肽。干扰素调节多肽包括而不限于B18R,在其他病毒株、如痘苗病毒的Copenhagen株中被称为B19R,痘苗病毒的B8R,B13R,vC12L,A53R和E3L,以及具有相似活性或特征的其他病毒多肽。干扰素调节多肽还可以非排他性地分为优先调节IFNα和/或β通路的一类(包括痘苗病毒的B18R、B8R、B13R和vC12L)和调节IFNγ通路的一类(包括痘苗病毒的B8R、B13R和vC12L)。任何具有免疫抑制功能的其他多肽也包括在内。
补体控制多肽是指参与抑制补体介导的细胞杀伤和/或病毒灭活的痘病毒多肽。病毒病原的清除机制包括杀伤被感染的细胞或者通过补体依赖的机制灭活宿主内的病毒颗粒。特别需要说明的是这些多肽可抑制、降低或消除这种特殊的抗病毒反应。这些多肽可直接或间接调节、影响、干扰、抑制、降低、改变或消除补体的活性或功能。补体控制多肽包括而不限于痘苗病毒的VCP,也被称为C3L或C21L,以及具有该特性或功能的其他多肽(术语“功能”和“活性”可互换)。
TNF调节多肽是指具有影响细胞免疫反应和炎症反应活性的痘病毒多肽,这些免疫反应或炎症反应可通过TNF受体灭活。这种反应包括诱导细胞调亡。痘病毒表达这些TNF调节多肽作为中和TNF介导的病毒清除和/或病毒感染细胞清除的方式。这些多肽具有特异性结合及抑制细胞外TNF的功能,从而抑制病毒的清除。需要特别说明的是这些多肽可抑制、降低或消除这种特殊的抗病毒反应。这些TNF调节多肽可直接或间接调节、影响、干扰、抑制、降低、改变或消除该机制的活性或功能。因此可使病毒的感染继续进行、病毒的毒力增强。TNF调节多肽包括而不限于痘苗病毒的A53R和B28R以及具有相似活性或特性的其他多肽。
丝氨酸蛋白酶抑制因子(SPI)是指能抑制丝氨酸蛋白酶的痘病毒多肽。这种多肽被称为serpins。这些多肽可通过其SPI活性抑制调亡诱导分子引起的调亡,因此即使在存在抗病毒调亡诱导细胞因子、fas、粒酶或其他调亡刺激因子的情况下也允许病毒复制。SPI包括而不限于痘苗病毒的B13R和B22R以及具有相似活性或特性的其他多肽。
IL-1β调节因子是指能直接或间接影响IL-1激发的抗病毒反应的痘病毒多肽。IL-1可直接作用于B细胞,促使其增殖和合成免疫球蛋白。IL-1还可以作为一种启动因子使B细胞对IL-5产生反应。IL-1可刺激NK细胞和成纤维细胞、胸腺细胞、胶质母细胞的增殖和活化。IL-18调节因子是指能直接或间接影响IL-18激发的抗病毒反应的痘病毒多肽。IL-18可诱导IFNγ和/或诱导T细胞和NK细胞的活化。这些IL-1β或IL-18调节因子可直接或间接调节、影响、干扰、抑制、降低、改变或消除该机制的活性或功能。需要特别说明的是这些调节多肽可抑制、降低或消除这种特殊的抗病毒反应。IL-1β调节多肽包括而不限于痘苗病毒的B13R和B15R以及具有相似活性或特性的其他多肽。需要说明的是突变的其他IL-1调节因子也是本发明的一部分。IL-18调节因子包括而不限于vC12L和其他多肽。
抑制感染性病毒从细胞内释放的病毒多肽、抗感染性EEV形式的多肽是指直接使感染性EEV形式的痘苗病毒不产生的病毒多肽。例如,约束或抑制EEV形式从细胞膜释放的多肽是抑制抗感染性EEV形式的多肽。参与调节病毒EEV形式的多肽包括而不限于痘苗病毒的A34R和B5R以及可影响痘病毒EEV形式形成的各种其他蛋白质。A34R上的密码子151突变使赖氨酸替换成天冬氨酸(K151D突变)的突变可使A34R蛋白抑制EEV形式的病毒到达细胞膜的能力下降。
本发明痘病毒内的其他突变包括编码C11R、病毒EGF样蛋白、A41L、B7R、N1L和/或vCKBP的基因上的突变,这些蛋白可能具有趋化因子结合活性(美国专利5,871,740和Seet等,2001,本文已纳入作为参考)。对于vCKBP的描述见美国专利5,871,740和Seet等,2001,本文都已纳入作为参考。另外需要说明的是,本发明的病毒还可以具有病毒基因组的缺失突变以便于插入异源核酸序列。这些缺失突变可以发生在非必须区,或者与辅助病毒或宿主细胞互补的必须区。
在某些实施方案中,痘病毒、特别是痘苗病毒在编码干扰素调节多肽的第一基因上至少包含一个突变从而产生出至少缺乏第一干扰素调节功能的病毒。在其他的实施方案中,突变发生在编码可直接结合干扰素的干扰素调节蛋白的基因上。需要说明的使干扰素结合多肽可以是B8R和/或B18R。
其他比较常见的类型也可以应用。突变也可以发生在下列一种或多种多肽上:1)抑制免疫反应组分的分泌型病毒因子(如TNF和其他细胞因子;趋化因子,补体级联蛋白;干扰素α/β和γ;白细胞介素如IL-1和IL18;A41L;N1L;vC12L和C11R);2)可抑制调亡的细胞内病毒因子(如丝氨酸蛋白酶抑制因子)和/或抑制免疫活化的细胞内病毒因子(如B13R、B22R和B7R);以及3)可抑制感染性病毒从细胞内释放的病毒多肽。这些种类的突变是非排他性的,因为一种蛋白可能不仅仅具有一种特定的功能。
在整个申请文件中术语“缺乏X功能的病毒”是指该病毒至少缺乏蛋白质X的一种功能。如果蛋白质X在正常情况下具有两种功能,那么缺乏X功能的病毒就是指该病毒至少缺乏多肽X的两种功能之一。功能的缺失可通过各种方式达到,其中包括编码多肽X的核酸、或者参与其表达的核酸区相对于具有功能性多肽X的病毒来说是突变的。另外,这个术语并不意味着该病毒缺乏所有的X功能,但是在其基因组内有突变从而使多肽所具有的X功能1)不再表达或者2)对于功能X来说不再具有活性(多肽仍可以具有其他的完整功能)。
本发明的痘苗病毒(或其他痘病毒)在下列7类基因中的一类或多类基因中可以具有修饰或突变:1)编码干扰素调节多肽的基因(包括而不限于B8R、B18R、B13R、E3L和/或vC12L),这些基因上的突变可导致病毒至少缺乏一种干扰素调节功能;2)编码补体控制多肽的基因(包括而不限于VCP),该基因的突变可导致病毒至少缺乏一种补体调节功能;3)编码TNF调节多肽的基因(包括而不限于A53R和B28R),这些基因的突变可导致病毒至少缺乏一种TNF调节功能;4)编码丝氨酸蛋白酶抑制因子的基因(包括而不限于B13R、B22R和/或K2L),这些基因的突变可导致病毒至少缺乏一种丝氨酸蛋白酶抑制因子的功能;5)编码IL-1β调节多肽的基因(包括而不限于B15R),这些基因的突变可导致病毒至少缺乏一种IL-1β调节多肽的功能;6)编码多肽的基因(包括而不限于B5R和/或A34R),这些基因的突变可导致感染性EEV形式的痘苗病毒增多;或7)C11R、vCKBP、B7R、N1L和/或A41L。
本发明的其他痘苗病毒也可以发生突变使病毒缺乏vC12L IL-18调节功能。另外,这种病毒还可以在上述7类基因的任何一类上发生突变。
需要说明的是本发明的病毒可在一类多肽的不止一个基因上发生突变,因此,病毒基因组可在同一类多肽上发生1、2、3、4、5或更多个多肽的突变而使其缺乏该类多肽的功能。需要进一步说明的是,本发明的病毒也可在不止一类多肽上发生突变。因此,病毒基因组可在编码1、2、3、4、5、6、7或8类多肽的基因上发生突变从而使病毒缺乏编码多肽的相应功能。另外,本发明的病毒可在同一类多肽及不同类多肽的多个基因上发生突变。本申请中上面所讨论的基因及其编码多肽是突变的优选靶位,通过这些突变可使本发明的痘苗病毒缺乏那些特定的多肽及相应的功能。而且,有关痘苗病毒的任何突变不需要额外的试验就可以用于其他痘病毒。
可以被突变或者可以使其至少一种功能丧失的特异性痘病毒多肽包括而不限于:A34R、A41L、A53R、B5R、B7R、B8R、B13R、B15R、B18R、B22R、B28R、B29R、C11R、E3L、K2L、N1L、vC12L及vCKBP。因此,本发明的痘病毒可在编码这些相应的痘苗病毒多肽的基因中的一个或多个上引入突变。
在本发明的痘苗病毒中,痘苗病毒可引入使A34R的至少一个功能丧失的突变和使下列多肽中的1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16或17个多肽的至少一个功能丧失的突变:A41L、A53R、B5R、B7R、B8R、B13R、B15R、B18R、B22R、B28R、B29R、C11R、E3L、K2L、N1L、vC12L和/或vCKBP。
在本发明的其他痘苗病毒中,痘苗病毒可引入使A41LR的至少一个功能丧失的突变和使下列多肽中的1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16或17个多肽的至少一个功能丧失的突变:A34R、A53R、B5R、B7R、B8R、B13R、B15R、B18R、B22R、B28R、B29R、C11R、E3L、K2L、N1L、vC12L和/或vCKBP。在本发明的其他痘苗病毒中,痘苗病毒可引入使A53R的至少一个功能丧失的突变和使下列多肽中的1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16或17个多肽的至少一个功能丧失的突变:A34R、A41L、B5R、B7R、B8R、B13R、B15R、B18R、B22R、B28R、B29R、C11R、E3L、K2L、N1L、vC12L和/或vCKBP。
在本发明的其他痘苗病毒中,痘苗病毒可引入使B5R的至少一个功能丧失的突变和使下列多肽中的1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16或17个多肽的至少一个功能丧失的突变:A34R、A41L、A53R、B7R、B8R、B13R、B15R、B18R、B22R、B28R、B29R、C11R、E3L、K2L、N1L、vC12L和/或vCKBP。
在本发明的其他痘苗病毒中,痘苗病毒可引入使B7R的至少一个功能丧失的突变和使下列多肽中的1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16或17个多肽的至少一个功能丧失的突变:A34R、A41L、A53R、B5R、B8R、B13R、B15R、B18R、B22R、B28R、B29R、C11R、E3L、K2L、N1L、vC12L和/或vCKBP。
在本发明的其他痘苗病毒中,痘苗病毒可引入使B8R的至少一个功能丧失的突变和使下列多肽中的1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16或17个多肽的至少一个功能丧失的突变:A34R、A41L、A53R、B5R、B7R、B13R、B15R、B18R、B22R、B28R、B29R、C11R、E3L、K2L、N1L、vC12L和/或vCKBP。
在本发明的其他痘苗病毒中,痘苗病毒可引入使B13R的至少一个功能丧失的突变和使下列多肽中的1、2、3、4、5、6、7、8、9、10、11、12、13、14、15或16个多肽的至少一个功能丧失的突变:A34R、A41L、A53R、B5R、B7R、B8R、B15R、B18R、B22R、B28R、B29R、C11R、E3L、K2L、N1L、vC12L和/或vCKBP。
在本发明的其他痘苗病毒中,痘苗病毒可引入使B15R的至少一个功能丧失的突变和使下列多肽中的1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16或17个多肽的至少一个功能丧失的突变:A34R、A41L、A53R、B5R、B7R、B8R、B13R、B18R、B22R、B28R、B29R、C11R、E3L、K2L、N1L、vC12L和/或vCKBP。
在本发明的其他痘苗病毒中,痘苗病毒可引入使B18R的至少一个功能丧失的突变和使下列多肽中的1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16或17个多肽的至少一个功能丧失的突变:A34R、A41L、A53R、B5R、B7R、B8R、B13R、B15R、B22R、B28R、B29R、C11R、E3L、K2L、N1L、vC12L和/或vCKBP。
在本发明的其他痘苗病毒中,痘苗病毒可引入使B22R的至少一个功能丧失的突变和使下列多肽中的1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16或17个多肽的至少一个功能丧失的突变:A34R、A41L、A53R、B5R、B7R、B8R、B13R、B15R、B18R、B28R、B29R、C11R、E3L、K2L、N1L、vC12L和/或vCKBP。
在本发明的其他痘苗病毒中,痘苗病毒可引入使B28R的至少一个功能丧失的突变和使下列多肽中的1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16或17个多肽的至少一个功能丧失的突变:A34R、A41L、A53R、B5R、B7R、B8R、B13R、B15R、B18R、B22R、B29R、C11R、E3L、K2L、N1L、vC12L和/或vCKBP。
在本发明的其他痘苗病毒中,痘苗病毒可引入使B29R(也被称为C23L)的至少一个功能丧失的突变和使下列多肽中的1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16或17个多肽的至少一个功能丧失的突变:A34R、A41L、A53R、B5R、B7R、B8R、B13R、B15R、B18R、B22R、B28R、C11R、E3L、K2L、N1L、vC12L和/或vCKBP。
在本发明的其他痘苗病毒中,痘苗病毒可引入使C11R的至少一个功能丧失的突变和使下列多肽中的1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16或17个多肽的至少一个功能丧失的突变:A34R、A41L、A53R、B5R、B7R、B8R、B13R、B15R、B18R、B22R、B28R、B29R、E3L、K2L、N1L、vC12L和/或vCKBP。
在本发明的其他痘苗病毒中,痘苗病毒可引入使E3L的至少一个功能丧失的突变和使下列多肽中的1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16或17个多肽的至少一个功能丧失的突变:A34R、A41L、A53R、B5R、B7R、B8R、B13R、B15R、B18R、B22R、B28R、B29R、C11R、K2L、N1L、vC12L和/或vCKBP。
在本发明的其他痘苗病毒中,痘苗病毒可引入使K2L的至少一个功能丧失的突变和使下列多肽中的1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16或17个多肽的至少一个功能丧失的突变:A34R、A41L、A53R、B5R、B7R、B8R、B13R、B15R、B18R、B22R、B28R、B29R、C11R、E3L、N1L、vC12L和/或vCKBP。
在本发明的其他痘苗病毒中,痘苗病毒可引入使N1L的至少一个功能丧失的突变和使下列多肽中的1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16或17个多肽的至少一个功能丧失的突变:A34R、A41L、A53R、B5R、B7R、B8R、B13R、B15R、B18R、B22R、B28R、B29R、C11R、E3L、K2L、vC12L和/或vCKBP。
在本发明的其他痘苗病毒中,痘苗病毒可引入使vC12L的至少一个功能丧失的突变和使下列多肽中的1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16或17个多肽的至少一个功能丧失的突变:A34R、A41L、A53R、B5R、B7R、B8R、B13R、B15R、B18R、B22R、B28R、B29R、C11R、E3L、K2L、N1L和/或vCKBP。
在本发明的其他痘苗病毒中,痘苗病毒可引入使vCKBP的至少一个功能丧失的突变和使下列多肽中的1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16或17个多肽的至少一个功能丧失的突变:A34R、A41L、A53R、B5R、B7R、B8R、B13R、B15R、B18R、B22R、B28R、B29R、C11R、E3L、K2L、N1L和/或vC12L。
在本发明的某些实施方案中,Copenhagen株或Western Reserve病毒株被突变以制备本发明的痘苗病毒。这些病毒株还可以在上述7类多肽的一类或多类上被进一步突变。
本发明的痘苗病毒还可以包含缺乏B8R、B18R、B13R或vC12L干扰素调节功能的病毒;缺乏B8R干扰素调节功能的病毒;缺乏B13R干扰素调节功能的病毒;缺乏B8R和B13R干扰素调节功能的病毒;缺乏B8R、B13R和vC12L干扰素调节功能并且还缺乏B28或A53R干扰素调节功能或者二者都缺乏的病毒;缺乏B8R、B13R和vC12L干扰素调节功能并且还缺乏B18R干扰素调节功能的病毒;缺乏B8R、B13R、B18R和vC12L干扰素调节功能并且还缺乏B28和/或A53R干扰素调节功能的病毒;缺乏至少一种干扰素调节功能和VCP补体调节功能的病毒;缺乏至少一种干扰素调节功能和A53R、B28R和/或vCKBP TNF调节功能的病毒;缺乏至少一种干扰素调节功能和B13R、B22R和/或K2L丝氨酸蛋白酶功能的病毒;缺乏至少一种干扰素调节功能和B13R和/或B15R IL-1β调节功能的病毒;缺乏至少一种干扰素调节功能和包含A34R或B5R的突变使感染性EEV形式的痘苗病毒产率提高的病毒;缺乏C11R、vCKBP、B7R、N1L和/或A41L功能的病毒;或者缺乏上述功能的任意组合的病毒。
其他的痘苗病毒具有导致病毒缺乏vC12L干扰素调节功能的突变。这种病毒还可以包含上述7类多肽中一类或多类多肽上发生的突变。在某些实施方案中,病毒缺乏B8R、B13R和/或B18R功能,而在另一些实施方案中,病毒还可以缺乏B15R IL-1β调节功能,和/或上述的其他任意功能,如vCKBP和/或B13R和/或B29R TNF调节功能。
在本发明的某些实施方案中,痘病毒用于注射给生物体,其中病毒包含在药用组合物中。本发明的组合物还包含干扰素(α、β和/或γ)和/或抗肿瘤药物,如抗体、化疗多肽或编码治疗性肿瘤多肽的核酸。
本发明的方法包括使用本文所述的任意痘病毒。许多实施方案涉及通过给予癌细胞或癌症患者以有效量的痘苗病毒来治疗癌细胞或癌症患者。在某些实施方案中,痘苗病毒不能表达如下多肽中的至少一种:
a)功能性第一干扰素调节多肽;
b)功能性补体控制多肽;
c)功能性TNF调节多肽;
d)功能性丝氨酸蛋白酶抑制因子;
e)功能性IL-1β调节多肽;
f)功能性非感染性EEV形式的多肽;
g)功能性A41L、B7R、N1L或vCKBP趋化因子结合多肽或C11R EGF样多肽。
需要特别注意的是那些缺乏a)-g)功能性多肽中一种以上多肽的病毒。“功能性多肽”是指具有特定功能的多肽;例如,缺乏功能性干扰素调节多肽的病毒是指基因组被修饰从而使病毒缺乏干扰素调节功能的病毒(与此相对的是缺乏所有干扰素调节多肽的所有干扰素调节功能)。病毒可以包含突变以表达突变的多肽,但是所得到的多肽是突变的因而不再具有野生型的功能。
在本发明的某些方法中,癌细胞就是肿瘤细胞。另外,本发明的组合物可在体外或体内给予细胞。因此,癌细胞可位于肿瘤患者体内。患者可以是实体瘤患者。在这种情况下,实施方案还可以包括对患者实施手术,如切除部分或全部肿瘤。病毒组合物可在手术前、手术后或手术的同时给予患者。在另外的实施方案中,患者还可以被直接注射、内窥镜注射、气管内注射、瘤内注射、静脉注射、损伤部位注射、肌肉注射、腹腔内注射、区域注射、经皮注射、局部注射、动脉内注射、膀胱内注射或皮下注射。病毒组合物可注射1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20或更多次,可以每1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24小时注射一次,或者每1、2、3、4、5、6、7天注射一次,或者每1、2、3、4、5周注射一次;或者每1、2、3、4、5、6、7、8、9、10、11、12个月注射一次。
治疗癌症的方法还可以包括对患者实施化疗或放疗,并且可以实施多次。化疗药物包括而不限于顺铂(CDDP)、卡铂、丙卡巴肼、氮芥、环磷酰胺、喜树碱、异环磷酰胺、美法仑、苯丁酸氮芥、bisulfan、亚硝脲、放线菌素、柔红霉素、阿霉素、博来霉素、plicomycin、丝裂霉素、依托泊甙(VP16)、他莫昔芬、泰索帝、紫杉醇、反铂(transplatinum)、5-氟尿嘧啶、长春新碱、长春花碱、氨甲蝶呤、吉西他滨、奥沙利铂、依立替康、托泊替康或上述药物的类似物或衍生物。放疗包括而不限于X射线照射、紫外线照射、γ-照射、电子束辐射或微波。另外,细胞或患者可注射蛋白酶或肽酶以提高细胞产生感染性EEV形式的病毒的能力。肽酶或蛋白酶可以包含在含病毒的药用组合物中。另外,还可以给予细胞或患者微管稳定剂,其中包括而不限于紫杉烷,这也是本发明方法的一部分。需要特别说明的是本文所讨论的任何痘病毒或经修饰的痘病毒,包括Western Reserve和Copenhagen痘苗病毒株(以及其衍生物)都可以联合使用。例如,Copenhagen痘苗病毒株可与紫杉醇联合使用以获得对癌细胞或癌症患者的治疗效果。
在某些实施方案中,给予病毒组合物的癌细胞可以是膀胱、血液、骨、骨髓、脑、乳腺、结肠、食管、胃肠道、头、肾、肝、肺、鼻咽、宫颈、卵巢、胰腺、前列腺、皮肤、胃、睾丸、舌或子宫细胞。
在本发明的其他实施方案中,减毒的痘苗病毒还可以包含编码异源治疗多肽的核酸序列。在本发明的不同实施方案中,异源治疗多肽可以是肿瘤抑制因子、免疫调节因子、血管生成抑制剂、抗血管多肽、细胞毒性多肽、调亡诱导因子、药物前体活化酶或细胞增殖抑制多肽。
本发明的方法可以包括使用减毒痘苗病毒的IHD-J株或包含A34R的K151D突变的减毒痘苗病毒。另外,为了制备对补体或补体级联损伤耐受性更高的痘苗病毒,可以从过量表达至少一种人补体抑制蛋白的细胞系内制备病毒。补体抑制蛋白可以是CD55、CD46或CD59。
在某些特殊实施方案中包含治疗患者癌症的方法,该方法包括给予患者有效量的药用组合物,其中含有重组痘苗病毒,该病毒内编码B8R、B18R或vC12L的基因发生了突变,导致该病毒缺乏B8R、B18R或vC12L干扰素调节功能。“有效量”的药用组合物一般定义为足以达到可检测的及可重复的改善、减轻、缓解或限制疾病,如癌或其症状的药物量。含102、103、104、105、106、107、108、109、1010、1011、1012、1013、1014、1015、1016、1017、1018、1019、1020、1025或更多病毒颗粒或pfu的组合物可以分1、2、3、4、5、6、7、8、9、10或更多次注射给患者。
需要进一步说明的是减毒痘苗病毒缺乏B8R、B18R或vC12L干扰素调节功能,并且在编码B13R的基因上拥有突变,这样就会使病毒缺乏B13R干扰素调节功能。在其他的实施方案中,减毒痘苗病毒缺乏B8R、vC12L和B13R干扰素调节功能,而在其他的实施方案中病毒缺乏B8R、B18R或vC12L干扰素调节功能中的至少两种。需要特别注意的是减毒痘苗病毒缺乏B8R、B18R和vC12L干扰素调节功能。另外,缺乏B8R、B18R或vC12L干扰素调节功能的重组痘苗病毒还可以缺少下列功能中的至少一种:
a)补体控制多肽功能;
b)TNF-调节功能;
c)丝氨酸蛋白酶抑制因子功能;
d)IL-1β调节因子功能;
e)抗感染性EEV形成功能;或者
f)A41L、B7R、N1L和/或vCKBP趋化因子调节功能或C11R EGF样功能。
本发明的其他实施方案包括通过使癌细胞与减毒痘苗病毒接触从而杀伤癌细胞的方法,其中减毒痘苗病毒包含突变,该突变可使病毒抑制干扰素、趋化因子、细胞因子、补体或中和抗体介导的抗病毒反应的能力下降。某些实施方案包括在编码B8R、B13R、B18R或vC12L的核酸序列上具有突变的减毒痘苗病毒。需要进一步说明的是这些病毒可以缺乏上述自然段中所讨论的a)-f)中的一种或多种功能。
在本发明的其他实施方案中包含治疗癌症患者肿瘤的方法,该方法包括将治疗有效量的含减毒痘苗病毒和增强减毒痘苗病毒抗肿瘤活性药物的组合物与肿瘤接触,从而有效地治疗肿瘤。可以增强减毒痘苗病毒抗肿瘤活性的药物可以是干扰素、蛋白酶、肽酶、微管稳定剂、化疗、放疗、基因治疗、免疫治疗或免疫调节治疗。
除了治疗方法外,本发明还涉及制备增强型EEV形式的痘苗病毒的方法,该方法包括:a)用痘苗病毒感染过量表达补体抑制蛋白的人细胞系;b)从感染细胞内分离EEV形式的痘苗病毒。“增强型EEV形式的痘苗病毒”是指比野生型痘苗病毒的EEV形式具有更强的或更高的耐受病毒降解机制能力的EEV形式的痘苗病毒。
在某些方法中,痘苗病毒在编码A34R的基因上具有突变。在某些情况下,突变可导致K151D突变。其他的实施方案包含补体抑制蛋白CD55、CD46或CD59。还有一些实施方案的人细胞系过量表达不止一种补体抑制蛋白。
除了方法以外,通过制备增强型EEV形式的痘苗病毒的方法而得到的组合物也是本发明的一部分。至少包含10%、15%、20%、25%、30%、40%、50%、60%、70%、80%、90%或100%增强型EEV形式的痘苗病毒的组合物都包括在本发明中。
同样,本发明还涉及制备增强型EEV形式的痘苗病毒所用的人源细胞系。这种细胞系可被痘苗病毒感染、含有痘苗病毒或痘苗病毒表达构建体,过量表达至少一种补体抑制多肽。补体抑制多肽可以是CD55、CD46或CD59。宿主细胞可被至少缺乏下列一种功能的痘苗病毒感染:
a)干扰素调节功能;
b)补体控制多肽功能;
c)TNF-调节功能;
d)丝氨酸蛋白抑制因子功能;
e)IL-1β调节因子功能;
f)功能性抗感染的EEV形式的多肽;或
g)功能性A41L、B7R、N1L或vCKBP趋化因子结合多肽或C11R EGF样多肽。
本发明的其他方法包括治疗精微的残存肿瘤的方法,该方法包括:i)确定患者具有可切除的肿瘤;(ii)切除肿瘤;以及(iii)使肿瘤床(tumer bed)与痘苗病毒接触,这些痘苗病毒在编码下列多肽的基因上至少拥有一个突变:A34R、A41L、A53R、B5R、B7R、B8R、B13R、B15R、B18R、B22R、B28R、B29R、C11R、E3L、K2L、N1L、vC12L和/或vCKBP。
其他方法包括治疗携带肿瘤的患者,该方法包括:(i)手术暴露肿瘤;以及(ii)使所述的肿瘤与缺乏A34R、A41L、A53R、B5R、B7R、B8R、B13R、B15R、B18R、B22R、B28R、B29R、C11R、E3L、K2L、N1L、vC12L和/或vCKBP功能的减毒痘苗病毒接触。
在其他的实施方案中,治疗荷瘤患者的方法包括在一段时期内用减毒痘苗病毒灌注肿瘤。而在另外一些实施方案中,涉及抑制荷瘤患者的肿瘤发生转移的方法,该方法包括给予患者减毒痘苗病毒,从而在患者体内达到治疗效果。整个申请中所用的术语“治疗效果”是指通过治疗可使患者的健康状况改善或增强的情况,其中包括癌前病变和癌症的治疗。这些效果的非穷尽性例子包括延长患者一定时间的生存期、降低或延缓疾病的恶性进展、肿瘤缩小、转移延迟、癌细胞或肿瘤细胞的增殖速度降低以及由疾病所造成的疼痛减轻。
本发明的其他方面涉及治疗患者多药耐药肿瘤的方法,该方法包括i)给予患者减毒的痘苗病毒以及ii)对患者进行化疗或放疗,从而在患者体内产生疗效。另外,还涉及使患者体内无法手术切除的肿瘤变成可切除的肿瘤的方法,该方法包括给予患者有效量的减毒痘苗病毒,然后切除部分或全部肿瘤。另外,本发明还涉及治疗对化疗或放疗耐受的肿瘤患者的方法,该方法包括给予患者减毒痘苗病毒并对患者进行化疗或放疗。
需要特别说明的是上述有关特殊方法或组合物的任一实施方案也可以通过本发明的其他方法和组合物来实现。
当单词“一个”或“一种”与权利要求和/或说明书中的术语“包含”合用时是指“一个”,但是也可以指“一个或多个”、“至少一个”和“一个或一个以上”。
本发明的其他目的、特征和优点通过下面的详细描述可以变得更加明晰。但是应当理解的是详细描述和阐述本发明特殊实施方案的特殊实施例只是以说明的方式提供,因为本领域的技术人员根据这些详细描述可以很容易地理解包括在本发明的精神和范围内的各种变化和修改。
附图简述
下面的附图是本说明书的一部分,可用于进一步证明本发明的某些方面。通过参考这些附图中的一个或多个再结合本文对特殊实施方案的详细描述可以更好地理解本发明。
图1A和1B举例说明了痘苗病毒株在(图1A)A2780人卵巢癌细胞系和(图1B)HCT116人结肠癌细胞系内的复制情况(x-轴是病毒株,y-代表噬菌斑形成单位/ml+/-S.E.)。
图2举例说明了痘苗病毒株在正常人支气管上皮细胞(NHBE)内的复制情况(x-轴代表病毒株,y轴代表噬菌斑形成单位/ml+/-S.E.)。
图3举例说明了痘苗病毒株在癌细胞(A2780)和正常细胞(NHBE)内的爆发比例(爆发比例)(x轴代表病毒株,y代表爆发比例)。
图4举例说明了痘苗病毒株在癌细胞(HCT116)和正常细胞(NHBE)内的爆发比例(爆发比例)(x轴代表病毒株,y代表爆发比例)。爆发比例是指肿瘤细胞内的PFU与正常细胞或非肿瘤细胞内的PFU之比。
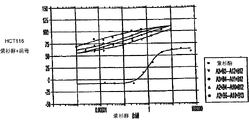
图5A和5B显示的是图6、7、8、9中的数据所进行的典型等效线图解法分析结果,表明痘苗病毒(Copenhagen株)和紫杉醇在HCT116(图5B)和LNCaP(图5A)细胞系内有协同作用。
图6显示的是利用紫杉醇和痘苗病毒联合治疗后所进行的典型MTS分析所得到HCT116细胞增殖数据。
图7显示的是利用紫杉醇和痘苗病毒联合治疗后所进行的典型MTS分析所得到HCT116细胞增殖数据。
图8显示的是利用紫杉醇和痘苗病毒联合治疗后所进行的典型MTS分析所得到LNCaP细胞增殖数据。
图9显示的是利用紫杉醇和痘苗病毒联合治疗后所进行的典型MTS分析所得到LNCaP细胞增殖数据。
图10说明了一个典型试验,其中IFN耐药的和IFN敏感的细胞用WR或WR-B18R(-)处理+/-IFN处理(感染后5小时用IFNα处理)。
说明性实施方案的描述
本发明涉及用于治疗癌症的瘤溶解痘病毒。痘病毒经过改造使其可以更好地或更有效地杀伤癌细胞和/或对非癌细胞的毒性更低或损伤更小。更具体地说,痘病毒可以被突变以修饰其基因产物,使其可以更好地感染宿主和癌细胞。
I.痘病毒
在正常情况下病毒可被免疫调节分子如干扰素(-α、-β、-γ)和肿瘤坏死因子-α(TNF)灭活、抑制或清除(Moss、1996)。病毒感染后宿主组织和炎症/免疫细胞通常可分泌这些分子。这些分子具有直接的抗病毒效应,和/或通过补充和/活化炎症细胞和淋巴细胞产生间接的效应。由于这些免疫清除机制的重要性,因此病毒经进化后可表达抑制这些细胞因子/趋化因子和干扰素诱导和/或功能的基因产物。例如,痘苗病毒(VV;以及其他一些痘病毒)可编码结合并抑制CC趋化因子(如RANTES,嗜酸细胞活化趋化因子,MIP-1-α)的分泌蛋白vCKBP(B29R)(Alcami等,1998)。某些VV病毒株还可以表达可结合并灭活TNF的分泌型病毒蛋白(如Lister A53R)(Alcami等,1999)。大多数痘病毒株具有编码结合并抑制干扰素α/β(如B18R)或干扰素-γ(B8R)的分泌蛋白的基因。vC12L是一种IL-18结合蛋白,可抑制IL-18诱导IFN-γ分泌和NK细胞/细胞毒T细胞活化。
大多数痘病毒的毒力研究都是在小鼠体内进行的。这些蛋白中的许多,但不是全部(如B18R在小鼠体内是无活性的)在小鼠体内是有活性的。当这些蛋白具有抗鼠靶细胞因子的活性时,这些基因的缺失将导致其毒力减弱、安全性提高,产生出这些基因缺失或功能突变的VV突变株。另外,对这些突变株的炎症反应/免疫反应及对这些突变株的清除通常要比表达抑制蛋白的亲本病毒株高。例如,痘病毒分泌蛋白的T1/35kDa家族(趋化因子结合/抑制蛋白)的缺失可导致侵润到病毒感染组织内的白细胞显著增加(Graham等,1997)。VV内vC12L基因的缺失可导致病毒在接种到小鼠鼻内后的滴度/毒性下降;另外,NK细胞和细胞毒T细胞的活性随IFN-γ的诱导而升高(Smith等,2000)。粘液瘤病毒T7基因(可结合IFN-γ和多种趋化因子)的缺失可导致病毒毒力下降,毒性模型内的组织炎症/侵润显著增高(Upton等,1992;Mossman等,1996)。粘液瘤病毒M-T2基因的缺失也可以导致病毒在兔模型内的毒力下降(Upton等,1991)。B18R抗干扰素-α/-β基因产物的缺失也可以导致病毒对IFN介导的清除的敏感性升高,在正常组织内的滴度降低、毒力下降(Symons等,1995;Colamonici等,1995;Alcami等,2000)。总之,这些病毒基因产物具有降低抗病毒免疫反应和减少炎症细胞侵润到病毒感染组织内的功能。通过缺失/突变使蛋白功能丧失可导致病毒在宿主组织内的毒力降低和/或原炎症特性升高。
细胞因子和趋化因子具有很强的抗肿瘤活性效应(Vicari等,2002;Homey等,2002)。这些效应可直接作用于肿瘤细胞(如TNF),或者通过对非肿瘤细胞的作用间接发挥效应。后者的例子是TNF,它是通过对肿瘤相关血管的毒性而发挥抗肿瘤活性的;这样可导致肿瘤失去血液供应而发生坏死。另外,趋化因子可补充(在某些情况下可活化)免疫效应细胞,如中性粒细胞、嗜酸性粒细胞、巨噬细胞和/或淋巴细胞。这些免疫效应细胞可通过多种机制杀伤肿瘤。这些机制包括表达抗肿瘤的细胞因子(如TNF),表达fas配基,表达穿孔素和粒酶,补充自然杀伤细胞等。炎症反应最终可诱导出全身的肿瘤特异性免疫反应。最后,许多这些细胞因子(如TNF)或趋化因子可协同化疗或放疗措施来破坏肿瘤。
通过全身给予重组的免疫刺激蛋白产生临床疗效是不可行的,因为1)全身给药会产生严重的毒性反应以及2)刺激局部侵润和抗肿瘤效应需要在肿瘤组织内的局部表达。我们需要的是找到方法使这些分子在肿瘤块内达到局部高浓度,而循环中的浓度达到最低。病毒可以经改造后使其表达细胞因子或趋化因子基因以增强其效应。这些基因从复制选择性载体中的表达与从非复制选择性载体中的表达相比具有潜在的优势。从复制病毒中的表达可使其在肿瘤块内达到局部较高的浓度;另外,复制病毒有助于通过肿瘤细胞的破坏/溶解及在原炎症环境中释放肿瘤抗原来诱导抗肿瘤免疫反应。但是这种方法也有几个不足。如果病毒在局部高浓度表达,携带毒性基因的复制完整型病毒(即使是肿瘤特异性的)就有可能释放到环境中,从而引起人们对其安全性的严重关注。因而,基因组内表达有强原炎症基因的病毒可能会对患者和公众产生安全性风险。另外,病毒大小限制了利用病毒如腺病毒来表达多个基因和/或大基因;这些分子在联合使用时的效率肯定更高。最后,现在使用的许多瘤溶解病毒表达抗炎症蛋白,因此这些病毒将会在其感染的肿瘤组织内诱导出炎症环境。结果会抑制抗肿瘤免疫反应、抗血管效应和化疗/放疗敏感性的诱导。
A.痘苗病毒
1.干扰素调节多肽
干扰素-α/-β可通过几种机制阻断病毒的复制。干扰素-γ直接的病毒抑制效应较弱,但是它可以通过几种机制诱导细胞介导的免疫反应。病毒经进化后可表达中和干扰素抗病毒效应的分泌型基因产物。例如,痘苗病毒(及其他痘病毒)编码可分别结合干扰素-γ和-α/-β的B8R和B18R(Smith等,1997;Symons等,1995;Alcami等,2000)。另外的一种可抑制干扰素产生的痘苗病毒基因产物是caspase-1抑制因子B13R,它可以抑制干扰素-γ-诱导因子IL-18的活化。干扰素调节蛋白包括而不限于B18R,在其他的病毒株如痘苗病毒的Copenhagen株中被称为B19R;B8R;B13R;vC12L;A53R;E3L以及其他具有相似活性或特性的病毒多肽。IFN调节多肽还可以非排他性地分为优先调节IFN α和/或β通路的一类(包括痘苗病毒的B18R、B8R、B13R或vC12L)和调节IFNγ通路的一类(包括痘苗病毒的B8R、B13R或vC12L)。
癌细胞通常对干扰素耐受。多种机制参与其中。这些机制包括ras转导通路的活化(如ras突变,上游生长因子受体过量表达/突变等),这是癌细胞的一个常见特征,会导致PKR抑制。另外,肿瘤组织内的淋巴细胞通常通过各种机制被抑制,如肿瘤细胞产生IL-10和fas-L。由于淋巴细胞是干扰素-γ产生的主要来源,因此淋巴细胞的抑制会导致肿瘤内干扰素-γ的分泌减少。因而干扰素无法对肿瘤组织产生效应。另外,干扰素自身具有抗肿瘤效应。例如,IFN-γ可增强MHC I类分子相关抗原的提呈;这将使CTL可以更有效地杀伤肿瘤细胞。例如,IFN-α/β可阻止肿瘤组织内的血管形成,从而抑制肿瘤的生长。
2.补体控制多肽
病毒克隆被清除的主要机制是通过补体依赖的机制杀伤宿主内被感染的细胞或肌体内的病毒颗粒。被感染的细胞死亡后就不可能再产生有感染性的病毒了。另外,在调亡过程中细胞内可释放降解DNA的酶。这些酶可使病毒DNA降解,使病毒灭活。调亡可被各种机制诱导,其中包括活化的补体与补体膜攻击复合物的结合。痘病毒如痘苗病毒经进化后可表达基因产物抑制补体介导的病毒和/病毒感染细胞的清除。因此这些基因可阻止调亡的发生,抑制通过补体依赖的机制清除病毒,因此病毒感染得以进行,病毒的毒力升高。例如,痘苗病毒补体控制蛋白(VCP;如C21L)可阻止补体介导的细胞杀伤和/或病毒灭活(Isaacs等,1992)。VCP还具有抗炎症效应,因为它的表达可使侵润到病毒感染组织内的白细胞减少。补体控制多肽包括而不限于VCP,也被称为C3L或C21L。
癌细胞通常过量表达细胞抗补体蛋白;这将使癌细胞在受补体攻击+/-肿瘤特异性抗体时得以存活(Caragine等,2002;Durrant等,2001;Andoh等,2002)。因此,那些由于其自身对补体介导的杀伤具有耐受性的优先作用于肿瘤细胞的药物对各种人源肿瘤具有选择性和较高的活性(Durrant等,2001)。另外,癌细胞的标志之一就是失去了正常的调亡机制(Gross等,1999)。对调亡的耐受促使其形成肿瘤并且对抗肿瘤药物,如免疫治疗药物、化疗药物和放疗耐受(Eliopoulos等,1995)。调亡抑制可由前调亡分子(如bax)功能的丢失、抗调亡分子(如bcl-2)表达水平的增加/功能的增强以及补体敏感性的丧失所介导。
3.TNF调节多肽
在各种病毒颗粒清除机制中有一种机制就是通过诱导如上所述的调亡杀伤宿主内被感染的细胞。调亡可由各种机制所介导,其中包括TNF和淋巴毒素α(LTα)与细胞TNF受体的结合,这种结合可激发细胞内的信号级联反应。TNF受体被活化后可参与免疫反应和炎症反应的调节以及诱导细胞调亡(Wallach等,1999)。
痘病毒的各种病毒株,包括某些痘苗病毒株,经进化后可表达基因产物抑制TNF介导的病毒和/和病毒感染细胞的清除。这些基因所编码的蛋白质通过结合及隔绝细胞内的TNF来抑制炎症反应和TNF的调亡诱导活性,结果导致病毒的清除被抑制。由于病毒不能被清除,因此病毒的感染得以继续,病毒的毒力增强。许多痘病毒家族都可以表达分泌型病毒TNF受体(vTNFR)。例如,有几种痘病毒可编码vTNFRs,如粘液瘤病毒(T2蛋白),牛痘病毒株和痘苗病毒株,如Lister可编码CrmB、CrmC(A53R)、CrmD、CrmE、B28R蛋白和/或其等价物。这些vTNFRs具有抑制TNF介导的细胞杀伤和/或病毒灭活的功能(Saraiva和Alcami,2001)。TNF调节蛋白包括而不限于A53R、B28R(这个蛋白是存在的,但是在痘苗病毒的Copenhagen株中可能是无活性的)以及具有相似活性或特性的其他多肽。
癌细胞的一个标志是异常的基因表达,这可导致其对多种生长调节分子机制的敏感性丧失,如对TNF抗肿瘤活性的敏感性。因此,对于病毒在肿瘤微环境内的传播来说病毒免疫调节机制是不需要的。
4.丝氨酸蛋白酶抑制剂
病毒颗粒被清除的一个主要机制是诱导宿主内被感染的细胞调亡。感染细胞死亡后就无法继续产生有感染性的病毒了。另外,在调亡过程中细胞可以释放出降解DNA的酶。这些酶可降解病毒DNA使病毒灭活。各种机制都可以诱导调亡,其中包括细胞因子(如肿瘤坏死因子)的结合、细胞毒T细胞或fas配基结合而导致的粒酶表达;级联活化是常见的调亡通路的关键部分。病毒经进化后可表达基因产物抑制细胞内由某些分子所诱导的信号级联反应,这些分子包括fas配基或肿瘤坏死因子(TNF)/TNF相关分子(如腺病毒的E310.4/14.5、14.7基因(Wold等,1994);腺病毒的E1B-19kD(Boyd等,1994);牛痘病毒的crmA;痘苗病毒的B13R)(Dobbelstein等,1996;Kettle等,1997)。这些基因产物可抑制调亡诱导分子所诱导的调亡,因此即使存在抗病毒调亡诱导细胞因子、fas、粒酶或其他调亡刺激因子,病毒的复制也可以进行。
VV SPI-2/B13R与牛痘病毒CrmA高度同源;SPI-1(VV)与CrmA的同源性较低(Dobbelstein等,1996)。这些蛋白是serpins(丝氨酸蛋白酶抑制剂),CrmA和SPI-2都具有阻止各种形式的调亡的功能。例如对白细胞介素-1β-转化酶(ICE)和粒酶的抑制可阻止感染细胞的调亡。这些蛋白可抑制IL-18的活化进而降低IL-18所诱导的IFN-γ释放。因而IFN-γ对细胞介导的免疫反应的免疫刺激效应被抑制(Kettle等,1997)。SPIs包括而不限于B13R、B22R以及其他具有相似活性或特性的多肽。
癌细胞的标志之一就是失去了正常的调亡机制(Gross等,1999)。对调亡的耐受促使其形成肿瘤并且对抗肿瘤药物,如免疫治疗药物、化疗药物和放疗耐受(Eliopoulos等,1995)。调亡抑制可由前调亡分子(如bax)功能的丢失、抗调亡分子(如bcl-2)表达水平的增加/功能的增强所介导。
5.IL-1β-调节多肽
IL-1β是一种具有局部和全身生物学活性的因子。已有的研究结果表明IL-1β和IL-1α之间的功能差异只有几种。IL-1β的许多生物学活性是用描述IL-1的许多不同缩写来描述的。除了在猪细胞内无活性外IL-1无种属特异性。IL-1的某些生物学活性是通过诱导其他介质的合成来间接介导的,其中包括ACTH(促肾上腺皮质激素)、PGE2(前列腺素E2)、PF4(血小板因子-4)、CSF(集落刺激因子)、IL-6和IL-8。IL-1的合成可被其他细胞因子所诱导,其中包括TNF-α、IFN-α、IFN-β和IFN-γ以及细菌内毒素、病毒、丝裂原和抗原。IL-1的主要生物学活性是刺激T辅助细胞,该细胞被诱导后可分泌IL-2并表达IL-2受体。病毒感染巨噬细胞后可产生大量的IL-1抑制因子,这些抑制因子支持T细胞成熟缺陷患者体内细胞的机会感染和转化。IL-1可直接作用于B细胞,促使其增殖并合成免疫球蛋白。IL-1也可以作为启动因子使B细胞对IL-5发生反应。IL-1可刺激NK细胞、成纤维细胞、胸腺细胞、胶质母细胞的增殖和活化。
病毒蛋白阻断IL-1β的合成是病毒抑制或消除IL-1激发的系统抗病毒反应的一种策略。研究发现可有效阻断IL-1功能的结合蛋白也可由牛痘病毒的基因编码,这些蛋白的活性与B15R相似。痘苗病毒还可以编码被称为B8R的另外一种蛋白,该蛋白的功能类似于细胞因子的受体(Alcami和Smith,1992;Spriggs等,1992)。IL-1调节多肽包括而不限于B13R、B15R以及其他具有相似活性或特性的多肽。
癌细胞的一个标志是异常的基因表达,这可导致其对多种生长调节分子机制的敏感性丧失,如对IL-1抗肿瘤活性的敏感性。因此,对于病毒在肿瘤微环境内的传播来说病毒免疫调节机制是不需要的。
6.EEV形式
病毒传播到转移瘤部位、甚至固体瘤内的效率一般不高(Heise等,1999)。静脉注射的病毒通常会被抗体(如腺病毒)(Kay等,1997)和/或补体系统(如HSV)(Ikeda等,1999)清除或灭活。除了这些免疫反应介导的机制之外,这些病毒的生物分布也会导致大多数病毒沉淀在正常组织而不是肿瘤组织内。例如,静脉注射腺病毒主要集中在肝脏和脾脏内;只有低于0.1%的病毒可以进入肿瘤内,即使是免疫缺陷小鼠(Heise等,1999)。因此,虽然在免疫缺陷小鼠肿瘤模型内用极高剂量的病毒可以达到适度的抗肿瘤效果,但是静脉注射途径的效率极低,并且对病毒的效果有严重影响。
痘苗病毒可在固体瘤内复制并引起肿瘤的坏死。另外,胸苷激酶缺失突变可使病毒具有感染肿瘤块和卵巢组织的能力,并且在小鼠肿瘤模型系统内可很好地表达标记基因(Gnant等,1999)。但是,由于这些研究一般都是基于5天后标记基因表达的情况来确定肿瘤的变化,因此还不清楚病毒是否可以在肿瘤/卵巢组织内良好地沉淀、表达基因和复制(Puhlmann等,2000)。无论通过何种机制,不含其他目的基因的病毒所得到的抗肿瘤效果都是无统计学意义的(Gnant等,1999)。相反,瘤内注射病毒可以得到显著的抗肿瘤效果(McCart等,2000)。因此,如果提高静脉注射到肿瘤内的病毒量则可以改善静脉注射的效果。
痘苗病毒在细胞内的复制可产生胞内病毒(IMV,胞内成熟病毒;IEV,胞内包装病毒)和胞外病毒(EEV,胞外包装病毒;CEV,细胞相关的胞外病毒)(Smith等,1998)。野生型痘苗病毒株复制后所产生的病毒中有约99%的是IMV。该病毒形式在环境中相对稳定,因此是个体之间传播的主要形式;但是,该形式的病毒在感染宿主内无法有效传播,因为病毒无法从细胞内有效释放以及对补体和/或抗体中和反应的敏感性。与此相反的是,EEV可释放到细胞外基质中,这种形式的病毒一般只占病毒产量的约1%(Smith等,1998)。EEV负责病毒在感染宿主内的传播,在宿主外则容易被降解。更重要的是,EEV进化出了几种机制来避免其在血液内被中和。首先,EEV对补体相对耐受(Vanderplasschen等,1998);这一特性归功于宿主细胞将补体抑制剂插入到外膜包被内以及痘苗病毒可将补体控制蛋白(VCP)分泌到细胞外环境中。其次,EEV与IMV相比对中和抗体的耐受性相对要高(Smith等,1997)。EEV还可以在感染后比IMV更早(如4-6小时)释放(IMV只在细胞死亡过程中或死亡后释放),因此EEV形式的病毒传播更快(Blasco等,1993)。
但是,不幸的是野生型痘苗病毒只能产生极少量的EEV。另外,用痘苗病毒治疗(即病毒的输入剂量)到目前为止只限于细胞内病毒。常规的痘苗病毒(VV)制备和纯化方法都可以导致EEV被灭活(Smith等,1998),病毒的制备一般用非人源细胞系;非人源细胞来源的EEV不能耐受补体介导的清除作用(EEV所产生的补体抑制蛋白具有种属特异性)。因此,痘苗病毒的疗效受IMV形式的病毒对中和反应的相对敏感性及病毒在实体瘤组织内的传播效率低下的限制;这种传播一般是指从一个细胞到临近的细胞内。IMF通过血流或淋巴系统传播到远端肿瘤组织内的效率也不高。
因此,EEV形式的痘苗病毒天然具有优于目前所用形式(IMV)的痘苗病毒的特征;EEV适于在实体瘤的局部及其临近或远端肿瘤位点内快速而有效地传播。由于EEV对补体效应相对耐受,因此当EEV从同一物种的细胞内制备时,通过血管内注射后该病毒形式在血液中的稳定性要比标准方法制备的痘苗病毒(只含IMV)高、并且维持活性的时间要更长(Smith等,1998)。由于EEV对抗体介导的中和反应耐受,因此该病毒形式通过血管内注射后在血液中维持活性的时间要比标准方法制备的痘苗病毒(只含IMV)长(Vanderplasschen等,1998)。这个特性对于在中和抗体升高的情况下需要重复注射时特别重要;所有被认可的抗癌疗法都需要重复注射。因此,痘苗病毒及其他痘病毒的EEV形式能高效地将治疗性病毒及其携带的基因通过血流转移到肿瘤内,与标准的痘病毒制备物相比其系统效应会提高。最后,病毒在大众中传播的危险会显著降低,因为EEV在体外是很不稳定的。参与病毒EEV形式调节的多肽包括而不限于A34R、B5R以及可影响痘病毒EEV形式产生的其他各种蛋白。A34R上的密码子151突变可使赖氨酸残基变成天冬氨酸残基(K151D),这样A34R蛋白就不能将EEV形式的病毒定位到细胞膜上。B5R是一种可结合补体的EEV膜结合多肽。A34R的完全缺失可导致EEV的释放增加,但是病毒的感染性会大大降低,而K151D突变可增加EEV的释放,同时也能保持释放病毒的感染活性。B5R的序列与VCP(抗补体)同源,但是还未发现它具有补体抑制活性。
一种鉴定增强型EEV形式的方法简述如下。EEV稀释到冷MEM中,与含活性血清或热灭活血清(56℃,30min,对照)的冷MEM混合(1∶1)(血清的最终稀释度为1/10、1/20或1/30)。在7℃孵育75分钟后,样品在冰上冷却,将mAb 5B4/2F2加到新鲜的EEV样品中中和所有的污染物(IMV和破裂的EEV)。然后使病毒颗粒与RK13细胞在冰上结合1小时,将补体和未结合的病毒颗粒洗去,2天后计数形成的噬菌斑。噬菌斑的数量越多说明对补体的耐受性约高。Vanderplasschen等,PNAS 1998;95(13):7544-7549,本文已纳入作为参考。描述分离EEV形式的痘苗病毒的经典方法参见Blasco等,1992(本文已纳入作为参考)。
7.其他多肽
其他病毒免疫调节多肽包括可结合免疫反应的其他介质的多肽和/或调节免疫反应相关分子通路的多肽。例如,趋化因子结合多肽如B29R(在痘苗病毒的Copenhagen株中虽然存在该蛋白,但是无活性)、C23L、vCKBP、A41L以及具有相似活性或特性的多肽。其他痘苗病毒蛋白,如痘苗病毒生长因子(如C11L)是一种病毒EGF样生长因子,在本发明的某些实施方案中也是修饰的靶位。可被称为病毒免疫调节因子的其他多肽包括而不限于B7R、N1L或具有提高痘病毒毒力活性或特征的其他多肽。
8.痘苗病毒诱导的细胞融合
在本发明的某些实施方案中,A56R或K2L编码核酸的修饰、缺失或突变可导致细胞与细胞的融合或VV感染所诱导的syncitia形成。痘苗病毒诱导的细胞融合常常会增强VV的抗肿瘤效应,因为VV可在肿瘤内传播。由于细胞融合而导致的肿瘤内病毒传播通常可使病毒逃过中和抗体和免疫反应的攻击。这些基因中的一个或两个都发生突变的VV可以更有效地杀伤和感染临近的未感染细胞(即“旁观者效应”),这样就可以提高病毒的局部抗肿瘤效应。
B.其他痘病毒
痘苗病毒是痘病毒科、脊椎动物痘病毒、正痘病毒属的一员。正痘病毒属与脊椎动物痘病毒亚科的其他属相比其成员具有更高的同源性,正痘病毒属包括11个不同但是又密切相关的种,其中有痘苗病毒、天花病毒(天花致病原)、牛痘病毒、水牛痘病毒、猴痘病毒、鼠痘病毒和马痘病毒以及其他病毒(见Moss,1996)。如本文所描述,本发明的某些实施方案可以延伸到正痘病毒属以及副痘病毒属、禽痘病毒属、羊痘病毒属、兔痘病毒属、猪痘病毒属软疣痘病毒属和亚塔痘病毒属的其他成员。痘病毒家族的一个属通常用血清学方式来定义,其中包括在试验动物体内的中和反应和交叉反应。正痘病毒属的各个成员及Chordovirinae亚科的其他成员利用免疫调节分子,如本文所描述的那些调节分子来抑制宿主的免疫反应。因此,本文所描述的发明并不限于痘苗病毒,而是适用于多种病毒。
C.病毒制备
痘苗病毒可用Earl和Moss描述的方法制备,参见Ausbel等,《分子生物学前沿方法》16.15.1-16.18.10页,本文已纳入作为参考。
II.蛋白和核酸组合物
本发明涉及适用于研究和治疗癌细胞和癌症患者的痘病毒。本发明涉及经改造后与野生型相比含有一个或多个突变的痘病毒,尤其是痘苗病毒,这样的病毒就具有了可用于治疗癌细胞的优良特性,并且对非癌细胞来说是低毒的或无毒的。下文以实施例的方式描述了实现本发明方法和组合物的各种方法。提供了利用重组DNA技术制备突变病毒的背景技术。
A.蛋白质性组合物
在某些实施方案中,本发明涉及制备缺乏一种或多种功能性多肽或蛋白的方法和/或制备能更好释放的特殊形式病毒,如具有感染性的EEV形式病毒的方法。在其他的实施方案中,本发明涉及痘病毒以及和作为药用制剂一部分的蛋白质性组合物联合使用。
本文所用的术语“蛋白质”或“多肽”是指至少含一个氨基酸残基的分子。在某些实施方案中使用的是野生型的蛋白质或多肽,而在本发明的多数实施方案中,病毒蛋白或多肽是缺失的或者是经过修饰的,这样就可以使病毒更适用于治疗癌细胞或癌症患者。上述术语在本文中可以交互使用。“修饰蛋白”或“修饰多肽”是指化学结构相对于野生型蛋白或多肽发生改变的蛋白或多肽。在某些实施方案中,修饰蛋白或多肽至少具有一个改变的活性或功能(假设蛋白或多肽有多种活性或功能)。相对于野生型蛋白或多肽的活性或功能来说,改变的活性或功能可以是以某些其他方式(如特异性)降低、消失、消除、增强、提高或改变的活性或功能。需要特别说明的是修饰蛋白或多肽的一种活性或功能发生改变但是却保留了野生型蛋白或多肽的其他活性或功能。另外,修饰蛋白可以是完全失去功能的,或者其编码核酸序列被改变从而使其根本不再表达该多肽、因移码突变而表达截短的多肽或者表达不同的氨基酸序列。
在某些实施方案中,突变蛋白或多肽的长度可以包含但不限于5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100、110、120、130、140、150、160、170、180、190、200、210、220、230、240、250、275、300、325、350、375、400、425、450、475、500、525、550、575、600、625、650、675、700、725、750、775、800、825、850、875、900、925、950、975、1000、1100、1200、1300、1400、1500、1750、2000、2250、2500或更多个氨基酸残基,以及上述数值之间的任何数目的氨基酸残基。需要说明的是多肽可通过截短而发生突变,使其比相应的野生型多肽短。
本文所用的术语“氨基分子”是指任何氨基酸、氨基酸衍生物或氨基酸类似物,这是本领域技术人员所熟知的。在某些实施方案中,蛋白分子的残基是连续的,其间无任何非氨基分子将氨基分子残基的序列中断。在其他的实施方案中,序列可包含一个或多个非氨基分子基序。在一些特殊的实施方案中,蛋白分子的残基序列可被一个或多个非氨基分子基序中断。
相应的,术语“蛋白质性组合物”包含氨基分子序列,该序列内至少含有一个含20个天然合成蛋白质中常见氨基酸的序列,或者至少含有一个修饰的或不常见的氨基酸。
蛋白组合可用本领域技术人员所熟知的任何技术制备,其中包括通过标准的分子生物学技术表达蛋白质、多肽或肽,从天然材料中分离蛋白化合物,或者用蛋白材料化学合成。各种基因的核苷酸和蛋白质、多肽和肽序列已经被鉴定出来,可在本领域技术人员所熟知的电子数据库中找到。一个这样的数据库是生物技术信息基因库国家中心(National Center for Biotechnology Information’s Genbank)和GenPept数据库(http://www.ncbi.nlm.nih.gov/)。这些已知基因的编码区可用本文所描述的方法或本领域技术人员所熟知的其他方法扩增和/或表达。
1.功能方面
当本申请提及病毒蛋白或多肽的功能或活性时,除非特别说明,是指该病毒蛋白或多肽在生理条件下的活性或功能。例如,干扰素调节多肽是指可直接或间接影响至少一种干扰素及其活性的多肽。多肽可直接或间接诱导、增强、提高、增加、消除、减弱、降低、抑制或屏蔽干扰素的活性。在某些实施方案中,直接影响干扰素的例子包括可特异性结合干扰素的干扰素调节多肽。确定哪个分子具有这种活性可通过本领域技术人员熟知的试验方法实现。例如,将编码调节干扰素或其突变体的产物的基因转移到可诱导出干扰素活性的细胞内,然后与转移这种基因的细胞进行比较,通过确定其对干扰素反应水平的不同就可以确定哪些分子具有干扰素调节功能。
需要特别说明的是调节因子可以是影响蛋白质性组合物表达的分子,这些蛋白质性组合物参与靶分子通路,如通过结合干扰素编码的转录子。确定哪个分子是干扰素、IL-1β、TNF或其他具有疗效的分子的调节因子可通过本领域技术人员所熟知的方法实现,本文中已经描述了一些,例如利用天然的和/或重组的病毒蛋白。
2.病毒多肽的突变体
本发明的多肽氨基酸序列的改变可以是替代、插入或缺失突变体。与野生型相比,编码病毒多肽的基因发生突变可影响1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100、110、120、130、140、150、160、170、180、190、200、210、220、230、240、250、275、300、325、350、375、400、425、450、475、500或更多个多肽的非连续或连续氨基酸。痘苗病毒编码的各种多肽可通过参考Rosel等,1986,Goebel等,1990和Genbank登录号NC_001559来确定,本文都已纳入作为参考。
缺失突变体缺少天然或野生型蛋白的一个或多个残基。每个残基都可以缺失,一个区(如催化区或结合区)的全部或部分也可以缺失。终止密码子被引入(通过替代或插入)到核酸编码序列中时可产生出截短的蛋白。插入突变一般是指在多肽的非终止点上插入残基,包括插入一个免疫原性表位或者仅仅插入一个或多个残基。也可以制备末端添加产物,这被称为融合蛋白。
替代突变体一般是指在蛋白质的一个或多个位点上将一个氨基酸替换成另一个氨基酸,这样可以使多肽的一个或多个特性发生改变而不会丢失其他的功能或活性。替代可以是保守替代,即一个氨基酸用另外一个具有相似形状或电荷的氨基酸替代。保守替代是本领域所熟知的,例如其中包括如下交换:丙氨酸到丝氨酸;精氨酸到赖氨酸;天冬酰胺到谷氨酰胺或组氨酸;天冬氨酸到谷氨酸;半胱氨酸到丝氨酸;谷胺酰胺到天冬酰胺;谷氨酸到天冬氨酸;甘氨酸到脯氨酸;组氨酸到天冬酰胺或谷胺酰胺;异亮氨酸到亮氨酸或缬氨酸;亮氨酸到缬氨酸或异亮氨酸;赖氨酸到精氨酸;蛋氨酸到亮氨酸或异亮氨酸;苯丙氨酸到酪氨酸、亮氨酸或蛋氨酸;丝氨酸到苏氨酸;苏氨酸到丝氨酸;色氨酸到酪氨酸;酪氨酸到色氨酸或苯丙氨酸;以及缬氨酸到异亮氨酸或亮氨酸。另外,替代也可以是非保守的,这样多肽的功能或活性会受到影响。非保守的替代一般是指用一个化学结构不同的残基来替代另一个残基,如极性氨基酸或带电荷的氨基酸替代非极性氨基酸或不带电荷的氨基酸,反之亦然。
术语“功能等价的密码子”用于本文中是指编码相同氨基酸的密码子,如编码精氨酸或丝氨酸的密码子有6个,另外也指编码生物等价氨基酸的密码子(见下表1)。
表1
密码子列表

还应当理解的是氨基酸和核酸序列还可以包含附加的残基,如附加的N-或C-末端氨基酸或者5’或3’序列,这在本文所描述的上述序列中是必需的,只要该序列复合上述标准,包括保持所表达蛋白的生物学活性。添加末端序列特别适合于那些包含各种非编码序列的核酸序列,其中非编码序列位于编码区的5’或3’端,或者那些包含各种内部序列,即内含子的核酸序列,内含子是大家所熟知的包含在基因内的序列。
下面的讨论是基于蛋白质的氨基酸发生交换而产生等价的或者是改进的第二代分子。例如,蛋白质内的某些氨基酸被其他氨基酸所替代,但是蛋白质与某些结构,如抗体上的抗原结合区或底物分子上的结合位点的结合能力并没有显著下降。由于蛋白质的相互结合能力和性质决定了蛋白质的生物功能活性,因此某些氨基酸替代可在蛋白质序列内或者其DNA编码序列内发生,但是却可以产生出具有相似特性的蛋白质。因此发明者认为在基因的DNA序列上进行的各种改变可以使其生物功能或活性不丢失,如下面所讨论的。表1显示的是编码特定氨基酸的密码子。
在进行这些改变时,需要考虑氨基酸的水性指数。氨基酸水性指数对蛋白质生物功能的重要性在本领域中是被普遍认知的(Kyte和Doolittle,1982)。氨基酸的相对水性特征可影响蛋白质的二级结构,从而也就决定了蛋白质与其他分子,如酶、底物、受体、DNA、抗体、抗原等的相互作用。
还应当理解的是在本领域中相似氨基酸的替代可在亲水性指数的基础上有效进行。美国专利4,554,101阐明一个蛋白质的局部最大平均亲水性指数是由其相邻的氨基酸所决定的,并且与蛋白质的生物学特性相关,本文已纳入作为参考。如美国专利4,554,101所详细描述的,下列亲水性指数值被赋予了各个氨基酸残基:精氨酸(+3.0);赖氨酸(+3.0);天冬氨酸(+3.0±1);谷氨酸(+3.0±1);丝氨酸(+0.3);天冬酰胺(+0.2);谷胺酰胺(+0.2);甘氨酸(0);苏氨酸(-0.4);脯氨酸(-0.5±1);丙氨酸(-0.5);组氨酸*-0.5);半胱氨酸(-1.0);蛋氨酸(-1.3);缬氨酸(-1.5);亮氨酸(-1.8);异亮氨酸(-1.8);酪氨酸(-2.3);苯丙氨酸(-2.5);色氨酸(-3.4)。
应当理解的是一个氨基酸被另外一个具有相似亲水性指数的氨基酸替代后可以得到一个生物等价和免疫等价的蛋白质。在这种替代中,亲水性指数值差异在±2之内的氨基酸替代是优选的,差异在±1之内的氨基酸替代是更优选的,差异在±0.5之内的氨基酸替代是尤其优选的。
如上面所指出的,氨基酸替代一般都是根据氨基酸侧链取代基的相对相似性来进行,如其疏水性、亲水性、电荷、侧链长度等。考虑了上述各种特征的典型替代是本领域技术人员所熟知的,其中包括:精氨酸和赖氨酸;谷氨酸和天冬氨酸;丝氨酸和苏氨酸;谷胺酰胺和天冬酰胺;以及缬氨酸、亮氨酸和异亮氨酸。
III.核酸分子
A.编码天然蛋白或修饰蛋白的多核苷酸
本发明涉及可从细胞中分离的多核苷酸,该多核苷酸可表达全长的或部分的蛋白或多肽。在本发明的某些实施方案中,涉及被特异性突变的病毒基因组,这种突变可使病毒缺乏某些功能性病毒多肽。多核苷酸可编码含全部或部分病毒氨基酸序列的肽或多肽,这些多核苷酸也可以被改造使之不编码这种病毒多肽,或者编码至少有一个功能或活性降低、消除或缺失的病毒多肽。重组蛋白可从产生活性蛋白的表达细胞内纯化。基因组及痘苗病毒编码区的定义见于Rosel等,1986,Goebel等,1990和/或Genbank登录号NC_00159,本文都已纳入作为参考。
本文所用的术语“DNA节段”是指从特定物种的基因组DNA中分离出来的游离形式的DNA分子。因此,编码多肽的DNA节段是指含野生型、多态性或突变多肽编码序列的DNA节段,可以从哺乳动物或人的基因组总DNA内分离或纯化。多肽、比多肽短的DNA节段以及重组载体,如质粒、粘粒、噬菌体、病毒等也包含在术语“DNA节段”内。
本申请所使用的术语“痘病毒多核苷酸”是指编码痘病毒多肽的核酸分子,可从基因组总核酸中分离。同样,“痘苗病毒多核苷酸”是指编码痘苗病毒多肽的核酸分子,可从基因组总核酸中分离。“痘病毒基因组”或“痘苗病毒基因组”是指在存在或不存在辅助病毒的情况下可使宿主细胞产出病毒颗粒的核酸分子。与野生型病毒相比,基因组可以是重组突变的,也可以不是。
术语“cDNA”是指以信使RNA(mRNA)为模板制备的DNA。与基因组DNA或基因组非或部分处理的RNA模板来源的DNA多聚物不同的是,利用cDNA的优点在于cDNA主要包含相应蛋白的编码序列。有时也需要使用全部或部分基因组序列,比如要获得最佳表达需要非编码区时或者非编码区如内含子是反义策略的靶位时。
还需要说明的是一个给定物种的特定多肽可由天然突变体代表,该突变体的核酸序列有些许不同,但是所编码的蛋白是相同的(见上述表1)。
同样,含游离的或纯化的野生型或突变多肽基因的多核苷酸是指包含野生型或突变多肽编码序列的DNA节段,在某些情况下也包含调节序列,这些调节序列是从天然基因或蛋白编码序列中分离出来的。在这种情况下,术语“基因”也可以简单地指功能蛋白、多肽或肽编码单位(包括正确转录、翻译后修饰或定位所需要的任何序列)。如本领域技术人员所理解的,这个功能术语包含可表达或适于表达蛋白质、多肽、结构域、肽、融合蛋白和突变体的基因组序列、cDNA序列和人工基因小片段。编码全长的或部分的天然或修饰多肽的核酸含有编码该多肽的全部或部分的连续核酸序列,其长度可以是:10、20、30、40、50、60、70、80、90、100、110、120、130、140、150、160、170、180、190、200、210、220、230、240、250、260、270、280、290、300、310、320、330、340、350、360、370、380、390、400、410、420、430、440、441、450、460、470、480、490、500、510、520、530、540、550、560、570、580、590、600、610、620、630、640、650、660、670、680、690、700、710、720、730、740、750、760、770、780、790、800、810、820、830、840、850、860、870、880、890、900、910、920、930、940、950、960、970、980、990、1000、1010、1020、1030、1040、1050、1060、1070、1080、1090、1095、1100、1500、2000、2500、3000、3500、4000、4500、5000、5500、6000、6500、7000、7500、8000、9000、10000或更多个核苷酸、核苷或碱基对。
本发明的某些特殊实施方案涉及游离的DNA节段以及含DNA插入序列的重组载体,其中DNA插入序列编码野生型或突变的痘病毒多肽或肽,这些多肽或肽的氨基酸序列内包含与天然多肽一致或基本一致的连续氨基酸序列。因此,游离的DNA节段或含DNA节段的载体可编码抑制或降低TNF活性的TNF调节因子或TNF调节多肽。术语“重组的”可与多肽联用或作为特异性多肽的名称,一般是指在体外经改造的核酸分子编码的多肽或这种分子的复制产物所编码多肽。
本发明的其他实施方案涉及游离的DNA节段以及含DNA插入序列的重组载体,其中DNA插入序列编码多肽或肽,这些多肽或肽的氨基酸序列内包含与多肽一致或基本一致的连续氨基酸序列。
本发明所用的核酸片段不论其编码序列的长度如何都可以和其他的核酸序列连接,如启动子、多聚腺苷酸信号、额外的限制性内切酶位点、多克隆位点、其他编码序列等,这样其全长的变化就会相当大。因此需要说明的是几乎任何长度的核酸片段都可以使用,但是优选的核酸片段长度受制备方法及其在重组DNA技术中的用途限制。
需要说明的是本发明的核酸构建体可编码任何物种的全长多肽,也可以编码截短的多肽,如截短的痘苗病毒多肽,这时编码区的转录子代表了截短的版本。截短的转录子被翻译成截短的蛋白。另外,核酸序列可编码带有其他异源编码序列的全长多肽序列,这样有利于多肽的纯化、运输、分泌、转录后修饰或多肽的疗效如靶向性和活性。如上面所讨论的,修饰多肽编码序列可添加上标签或其他异源多肽,其中“异源的”是指与修饰多肽不一样的多肽。
在一个非限定性例子中,一个或多个核酸构建体可制备成包含与特定基因,如B18R基因一致或互补的一段连续核苷酸的构建体。核酸构建体可以至少含有20、30、40、50、60、70、80、90、100、110、120、130、140、150、160、170、180、190、200、250、300、400、500、600、700、800、900、1,000、2,000、3,000、4,000、5,000、6,000、7,000、8,000、9,000、10,000、15,000、20,000、30,000、50,000、100,000、250,000、500,000、750,000到1,000,000核苷酸,或者更长的构建体,甚至可以达到和包含染色体的长度(包括所有的中间长度和中间范围),假设核酸构建体如酵母人工染色体是本领域技术人员所熟知。很容易理解的是本文所用的“中间长度”和“中间范围”是指包含上述数值或上述数值之间的任意长度或范围(即包含这些数值和这些数值之间的所有整数值)。
本发明所用的DNA节段包含生物功能等价的修饰多肽和肽,如修饰的白树素毒素。这种序列的产生可能是密码子简并性和功能等价的结果,已知在天然状态下核酸序列及其编码的蛋白序列都可能发生。另外,功能等价的蛋白或肽可利用重组DNA技术制备,其中可根据修饰氨基酸的特性人工改变蛋白的结构。通过位点特异性突变技术可将人为设计的改变引入到蛋白序列内,如可以提高蛋白质的抗原性、降低蛋白质对受体的体内毒性效应、或提高含蛋白的治疗药物的疗效。
本发明的某些实施方案涉及游离的DNA节段和序列内包含本文所描述的序列(和/或参考文献所描述的序列)来源的连续核酸序列的重组载体。但是,这种序列可被突变以制备活性不同于野生型蛋白的蛋白产物。
还应当理解的是本发明并不局限于这些已知序列的特定核酸序列和氨基酸序列。重组载体和游离DNA节段可以包含不同序列,包括痘病毒编码区自身、在基础编码区上具有特定改变或修饰的编码区,或者可以编码包含痘病毒编码区在内的更长的多肽、或编码具有突变氨基酸序列的生物等价蛋白或肽。
本发明的DNA节段包含生物等价的痘病毒蛋白和肽。这种序列的产生可能是密码子简并性和功能等价的结果,已知在天然状态下核酸序列及其编码的蛋白序列都可能发生。另外,功能等价的蛋白或肽可利用重组DNA技术制备,其中可根据修饰氨基酸的特性人工改变蛋白的结构。通过位点特异性突变技术可将人为设计的改变引入到蛋白序列内,如可以提高蛋白质的抗原性。
B.诱导痘病毒多核苷酸发生突变
在各种实施方案中,痘病毒多核苷酸都可以被改变或突变。改变或突变包括插入、缺失、点突变、颠换等,结果可导致某些通路或分子机制的调节、活化和/或灭活,或者改变基因产物的功能、定位或表达,特别是使基因产物失去功能。编码全长或部分痘病毒的多核苷酸的突变可用各种标准的突变方法完成(Sambrook等,1989)。突变是生物的量或结构发生改变的过程。突变包括单个基因的核苷酸序列的修饰、基因或整个染色体的阻断。单个基因的改变可以是点突变的结果,其中包括DNA序列内单个核苷酸的去除、添加或替代,也可以是插入或缺失多个核苷酸的结果。
突变可在接触化学或物理突变剂后被诱导。这些突变诱导剂包括离子辐射、紫外线和化学物质的不同组合,如烷化剂和多环芳香烃,这些物质都可以直接或间接(一般发生在经代谢生物转化后)与核酸发生相互作用。当被影响的DNA复制或修饰时这种诱导剂所诱导的DNA损伤可导致碱基序列的改变,从而发生突变。也可以通过特殊的靶向性方法进行位点特异性突变。
1.随机突变
a)插入突变
插入突变是指插入一段已知的DNA片段使基因灭活。由于插入了某类DNA片段,因此所产生的突变一般是丧失功能的突变而不是获得功能的突变。但是,通过插入使DNA片段获得功能的突变也有几个例子。插入突变已在细菌和果蝇中取得了很大成功(Cooley等,1988),现在已经成为谷物突变(Arabidopsis;(Marks等,1991;Koncz等,1990)和金鱼草突变(Sommer等,1990)的有效工具。插入突变可通过标准的分子生物学方法实现。
b)化学诱变
化学诱变具有某些优势,如能发现所有突变中哪个对表型的影响最大,操作简单而且成本低。大多数化学致癌物都可以使DNA发生突变。苯并[a]芘、N-乙酰氧基-2-乙酰氨基芴和aflotoxin B1可引起细菌和哺乳动物细胞DNA的GC到TA的颠换。苯并[a]芘还可以引起替代如AT到TA的替代。N-硝基化合物可引起GC到AT的转换。接触n-亚硝脲后胸腺嘧啶O4位置上的烷基化可导致TA到CG的转换。
c)辐射突变
离子辐射可使生物分子降解。吸收额外能量可导致离子和自由基的形成以及某些共价键的断裂。不同分子之间以及同一分子的不同晶体形式之间对辐射损伤的敏感度不同。依赖于总的积聚剂量以及剂量率(如果存在自由基,它们所引起的分子损伤大小依赖于其自然扩散率和作用时间)。使样品尽可能冷却可减轻或控制损伤的程度。离子辐射引起的DNA损伤程度一般与剂量率成正比。
在本发明中,术语“离子辐射”是指含粒子或光子的辐射,这些粒子或光子具有足够的能量,或者可以产生足够的能力引起离子化(获得或失去电子)。一种典型的和优选的离子辐射是X射线。特定细胞或特定分子发生突变所需要的离子辐射的剂量一般依赖于细胞或分子的特性以及所要突变靶位的特性。确定有效辐射剂量的方法是本领域所熟知的。
d)体外筛选突变
利用易错PCR也可以引入随机突变。通过在多个含不同模板稀释度的管内进行PCR可提高突变的几率。
一个特别有用的突变技术是丙氨酸筛选突变,其中可以有多个残基分别被丙氨酸替代,这样就可以了解失去侧链相互作用后的效应,并且使大规模干扰蛋白构象的危险降到最低(Cunningham等,1989)。
体外筛选饱和诱变为大量获得结构-功能信息提供了一种快速的方法,其中包括:(i)确定调节配基结合特异性的残基,(ii)根据这些可使活性保持的氨基酸以及可使活性丧失的氨基酸所处的位置可以更好地了解配基的结合活性,(iii)评价一个活性位点或蛋白子域的整体可塑性,(iv)确定可使结合活性增强的氨基酸替代。
2.位点特异性突变
结构指导的位点特异性突变代表了分析和改造蛋白-配基相互作用的一个有效工具(Wells,1996;Braisted等,1996)。该技术可通过在目的DNA内引入一个或多个核苷酸序列的改变来制备和检测序列突变体。
位点特异性突变利用编码DNA序列目的突变及其临近的足够数目的未修饰核苷酸的特异性寡核苷酸序列。在这种方法中,引物序列的长度和复杂性要足以在要交叉的缺失连接的两侧形成稳定的双螺旋。优选的引物长度约为17到25个核苷酸,其中交叉的连接序列两侧约为5到10个残基。
该方法一般利用即有单链形式又有双链形式的噬菌体载体。用于位点特异性突变的载体包括载体如M13噬菌体。这些噬菌体载体都是商业化的,其使用方法都是本领域技术人员所熟知的。双链质粒也常被用于位点特异性突变,利用该载体可以省却将目的基因从噬菌体转移到质粒的步骤。
一般来说,需要首先获得单链载体或者使双链载体的双链退火,载体的序列内包含一个编码目的蛋白或基因元件的DNA序列。然后使合成的携带目的突变序列的寡核苷酸引物与单链DNA制备物复性,在选择杂交条件时要考虑发生错配的程度。杂合的产物利用DNA聚合酶如大肠杆菌聚合酶I(Klenow片段)完成含突变的DNA链的合成。这样就形成了异源双螺旋,其中一条链编码原始的非突变序列,另一条链带有目的突变。然后用这种异源双螺旋分子转化合适的宿主细胞,如大肠杆菌,筛选含重组载体的克隆,其中重组载体内带有突变序列。
一个蛋白质上某个残基在功能上的完整信息以及信息的内容可通过饱和突变来获得,其中要检测所有的19个氨基酸替代。这种方法的缺点是多残基饱和突变的算法是令人畏惧的(Warren等,1996,Zeng等,1996;Burton和Barbas,1994;Yelton等,1995;Hilton等,1996)。需要研究数百个、甚至数千个位点特异性的突变。但是,经改进的技术可使突变的制备和筛选变得更快速简捷。见美国专利5,798,208和5,830,650对“草率”突变的描述。位点特异性突变的其他方法见美国专利5,220,007;5,284,760;5,354,670;5,366,878;5,389,514;5,635,377和5,789,166的描述。
C.载体
为了在痘病毒基因组内引入突变,天然和修饰多肽可由包含在载体内的核酸分子编码。本文所用的术语“载体”是指携带核酸分子的载体,其中载体内插入的外源核酸分子可通过载体被导入到可以复制载体的细胞内。核酸序列是“外源性的”是指对于载体导入的细胞来说该核酸是外源的,或者该序列虽然与细胞内的序列同源但是其在宿主细胞核酸内的位置通常不包含该核酸。载体包括质粒、粘粒、病毒(噬菌体、动物病毒和植物病毒)以及人工染色体(如YACs)。本领域的技术人员可以通过标准的重组技术构建这种载体,这些方法在Sambrook等,(1989)和Ausubel等,1994中有描述,本文已纳入作为参考。除了可以编码修饰多肽如白树素以外,载体还可以编码未修饰的多肽序列,如标记物或靶向性分子。可以编码这种融合蛋白的载体包括pIN载体(Inouye等,1985)、编码一组组氨酸的载体和用于制备谷胱甘肽S-转移酶(GST)可溶性融合蛋白的pGEX载体以利于以后的纯化和分离或裂解。靶向性分子是一种可以使修饰多肽特异性地定位到特定器官、组织、细胞或受体其他位置的分子。
术语“表达载体”是指含有核酸序列的载体,其中的核酸序列至少编码部分能被转录的基因产物。在某些情况下,RNA分子随后被翻译成蛋白、多肽或肽。在另外一些情况下,这些序列不翻译,而是转录出反义分子或核酶。表达载体含有各种“调控序列”,调控序列是指操作连接的编码序列在特定宿主内转录和翻译所需要的核酸序列。除了控制转录和翻译的调控序列外,载体和表达载体还可以包含具有其他功能的核酸序列,这些序列将在下文描述。
1.启动子和增强子
“启动子”是一种调控序列,是核酸序列上一个控制转录起始和速率的区域。可以包含调节蛋白和分子结合的元件如RNA聚合酶及其他转录因子。术语“操作位置的”、“操作连接的”、“控制下的”和“转录控制下的”是指相对于核酸序列来说启动子位于正确的功能位置和/或方向,可以调控该序列的转录起始和/或表达。启动子可以连接有“增强子”,也可以不连接,增强子是指参与核酸序列转录活化的顺式调节序列。
启动子可以是与某个基因或序列天然相连的序列,如通过分离位于编码片段和/或外显子上游的5’非编码序列而得到的启动子。这种启动子被称为“内源性的”。同样,增强子也可以是与某个序列天然相连、位于该序列上游或下游的序列。另外,将编码核酸片段置于重组的或异源的启动子控制之下也有某些优势,重组的或异源的启动子是指在天然状态下该序列不是与核酸序列相联系的。重组的或异源的增强子也是指在天然状态下不是与核酸序列相联系的增强子。这种启动子或增强子包括其他基因的启动子或增强子、从其他原核细胞、病毒或真核细胞中分离出的启动子或增强子以及非“天然的”启动子或增强子,即含有不同转录调节区和/或改变表达的突变的不同元件。除了可以通过合成方法制备启动子和增强子的核酸序列外,还可以利用重组克隆和/或核酸扩增技术,其中包括PCRTM,结合本文所描述的组合物来制备序列(见美国专利4,683,202,5,928,906,本文都已纳入作为参考)。另外,需要说明的是可控制无核细胞器如线粒体、叶绿体内的序列转录和/或表达的调控序列也可以使用。
很显然,利用启动子和/或增强子来有效地控制DNA节段在细胞、细胞群和生物体内的表达是十分重要的。分子生物学领域的技术人员一般都熟悉如何使用启动子和增强子,知道如何选择细胞以表达蛋白,例如Sambrook等,(1989)所描述的,本文已纳入作为参考。所使用的启动子可以是组成性的、组织特异性的、可诱导的启动子和/或可在适当的条件下使被导入的DNA节段高水平表达的启动子,例如在大规模制备重组蛋白和/或肽时。启动子可以是异源的,也可以是内源性的。
表2列举了一些可用于本发明中调节基因表达的元件/启动子。这个列表并没有将调控表达的所有可能的元件都包括在内,而仅仅是举了其中的一些例子。表3给出了可诱导元件的一些例子,这些元件是核酸序列上可被特异性刺激因子活化的区域。



组织特异性启动子或元件的鉴定及其活性特征的分析是本领域技术人员所熟知的。这种区域的例子包括人LIMK2基因(Nomoto等,1999)、生长抑素受体2基因(Kraus等,1998)、鼠附睾视黄酸结合基因(Lareyre等,1999)、人CD4(Zhao-Emonet等,1998),鼠α2(XI)胶原(Tsumaki,等,1998)、D1A多巴胺受体基因(Lee,等,1997)、胰岛素样生长因子II(Wu等,1997)、人血小板内皮细胞粘附分子-1(Almendro等,1996)、以及SM22α启动子。
可用于本发明的启动子还有dectin-1和dectin-2启动子。其他可用于本发明的病毒启动子、细胞启动子/增强子以及可诱导启动子/增强子列在表2和表3中。其他任何的启动子/增强子组合(如每个真核细胞启动子数据库EPDB)都可以用于驱动编码寡糖处理酶、蛋白折叠辅助蛋白、选择性标记蛋白或异源目的蛋白的结构基因的表达。另外,用于肿瘤基因治疗(表4)或可以靶向到肿瘤(表5)的组织特异性启动子也可用于调控本发明的核酸分子。
表4:
癌症基因治疗的候选组织特异性启动子
| 组织特异性启动子 | 启动子在其中有活性的肿瘤 | 启动子在其中有活性的正常细胞 |
| 癌胚抗原(CEA)* | 大多数结肠癌;50%的肺癌;40-50%的胃癌;大多数胰腺癌;多种乳腺癌 | 结肠粘膜;胃粘膜;肺上皮;小汗腺;睾丸细胞 |
| 前列腺特异抗原(PSA) | 大多数前列腺癌 | 前列腺上皮 |
| 血管紧张肽(VIP) | 大多数非小细胞肺癌 | 神经元;淋巴细胞;肥大细胞;嗜酸性粒细胞 |
| 表面活化蛋白A(SP-A) | 许多非腺癌细胞 | II型肺细胞;Clara |
| 人achaete-scute类似物(hASH) | 大多数小细胞肺癌 | 肺内的神经内分泌细胞 |
| 粘蛋白-1(MUC1)** | 大多数腺癌(任何组织起源的) | 乳腺腺上皮细胞以及呼吸道、胃肠道和泌尿生殖道腺上皮细胞 |
| 甲胎蛋白 | 大多数肝细胞癌;在许多睾丸癌内可能有活性 | 肝细胞(在特定条件下);睾丸细胞 |
| 白蛋白 | 大多数肝细胞癌 | 肝细胞 |
| 酪氨酸酶 | 大多数黑色素瘤 | |
| 酪氨酸结合蛋白(TRP) | 大多数黑色素瘤 | 黑色素细胞;星形胶质细胞;Schwann细胞;某些神经元 |
| 角蛋白14 | 据推测在许多鳞状细胞癌内有活性(如头颈癌) | 角质细胞 |
| EBV LD-2 | 头颈部的多种鳞状细胞癌 | 上消化道的角质细胞 |
| 神经胶质原纤维酸性蛋白(GFAP) | 许多星形细胞瘤 | 星形胶质细胞 |
| 髓磷脂碱蛋白(MBP) | 许多胶质瘤 | 少突胶质细胞 |
| 睾丸特异性的血管紧张素转化蛋白(睾丸特异性ACE) | 在许多睾丸癌内可能有活性 | Spermatazoa |
| 骨钙素 | 在许多骨肉瘤内可能有活性 | 成骨细胞 |
表5:
用于靶向道肿瘤的组织特异性候选启动子
| 启动子 | 启动子在其中有活性的肿瘤 | 启动子在其中有活性的正常细胞 |
| E2F-调节启动子 | 几乎所有的肿瘤 | 增生的细胞 |
| HLA-G | 许多结肠癌;许多黑色素瘤;可能在许多其他肿瘤内有活性 | 淋巴细胞;单核细胞;精母细胞;滋养细胞 |
| FasL | 大多数黑色素瘤;许多胰腺癌;大多数星形胶质瘤;在许多其他癌内也可能有活性 | 活化的白细胞;神经元;内皮细胞;角化细胞;免疫优势组织内的细胞;肺、卵巢、肝和前列腺内的某些细胞 |
| Myc-调节启动子 | 大多数肺癌(包括小细胞肺癌和非小细胞肺癌);大多数结肠癌 | 增生的细胞(只限于某些类型的细胞);乳腺上皮细胞(包括非增生性的细胞) |
| MAGE-1 | 许多黑色素瘤;某些非细胞肺癌;某些乳腺癌 | 睾丸细胞 |
| VEGF | 70%的肿瘤(在许多肿瘤中可以结构性地过量表达) | 新血管形成部位的细胞(但是在肿瘤内部可能有活性,表达是瞬时的,强度很弱并且是非结构性的) |
| bFGF | 据推测可能在许多不同种类的肿瘤中有活性,因为bFGF的表达可被局部缺血所诱导 | 缺血部位的细胞(但是在肿瘤内部可能有活性,表达是瞬时的,强度很弱并且是非结构性的) |
| COX-2 | 大多数结肠癌;许多肺癌;可能在许多其他癌中也有活性 | 炎症部位的细胞 |
| IL-10 | 大多数结肠癌;许多肺癌;头颈部的许多鳞状细胞癌;可能在许多其他癌中也有活性 | 白细胞 |
| GRP78/BiP | 据推测可能在许多不同种类的肿瘤中有活性,因为GRP7S的表达可被肿瘤特异性的条件所诱导 | 缺血部位的细胞 |
| Egr-1的CarG元件 | 被离子辐射所诱导,因此可以想象在大多数受辐射的肿瘤内有活性 | 受离子辐射的细胞;白细胞 |
2.起始信号和内部核糖体结合位点
编码序列的有效翻译还需要特异性的起始信号。这些信号包括ATG起始密码子或其临近序列。包含ATG起始密码子的外源翻译控制信号也需要提供。本领域的技术人员能够很容易地确定这一点并提供必须的信号。大家都知道起始密码子必须位于目的编码序列阅读框架内才能确保整个插入序列被翻译。外源性的翻译控制信号可以是天然的,也可以是合成的。插入合适的转录增强元件可以提高表达的效率。
在本发明的某些实施方案中,使用内部核糖体进入位点(IRES)可以使多个基因或多个顺反子信使RNA表达。IRES元件可以使核糖体绕过5’甲基化帽子依赖性翻译的扫描模式在内部位点开始翻译(Pelletier和Sonenberg,1988)。小核糖核酸病毒家族的两个成员(脊髓灰质炎病毒和脑心肌炎病毒)来源的IRES元件以及哺乳动物信使RNA来源的IRES(Macejak和Sarnow,1991)。IRES元件可以被连接到异源开放阅读框架上。多个开放阅读框架可以一起转录,其中每个阅读框架都被IRES隔开,转录出一条多顺反子信使RNA。利用IRES元件每个开放阅读框架都可以有核糖体结合从而可以高效翻译。利用单个启动子/增强子转录出一条信使RNA可以使多个基因都得到有效表达(见美国专利5,925,565和5,935,819,本文已纳入作为参考)。
3.多克隆位点
载体可包含多克隆位点(MCS),MCS是指包含多个限制性酶切位点的核酸序列,通过标准的重组技术每个酶切位点都可以用于消化载体(见Carbonelli等,1999,Levenson等,1998,和Cocea,1997,本文已纳入作为参考)。“限制性内切酶消化”是指利用内切酶催化裂解核酸分子的过程,其中内切酶只在核酸分子的特定位置上切割。很多这样的限制性内切酶都可以买到。这种内切酶的使用方法是本领域技术人员所熟知的。载体通常被在MCS内切割的限制性内切酶线性化或片段化从而可以使外源序列连接到载体上。“连接“是指两个核酸片段之间形成磷酸二酯键的过程,其中两个核酸片段可以是彼此不相临近的。涉及限制性内切酶和连接反应的技术是重组技术领域的技术人员所熟知的。
4.剪接位点
大多数被转录出来的真核RNA分子都会发生RNA剪接以去除初级转录子内的内含子。含基因组真核序列的载体需要供体和/或受体剪接位点以保证转录子被正确处理,然后表达蛋白(见Chandler等,1997,本文已纳入作为参考)。
5.终止信号
本发明的载体或构建体一般都包含至少一个终止信号。“终止信号”或“终止子”由参与RNA聚合酶所转录的转录子特异性终止的DNA序列组成。因此,在某些实施方案中,需要结束RNA转录子合成的终止信号。终止子是在体内获得理想的信使RNA表达水平所必须的。
在真核系统中,终止子区域还可以包含允许新转录子发生位点特异性裂解以暴露多聚腺苷酸信号的特异性DNA序列。这种序列可向特异的内源性聚合酶发出信号,使之将一段约200A的残基(poly A)添加到转录子的3’末端。经这种polyA尾巴修饰的RNA分子更稳定,其翻译的效率也更高。因此,在其他涉及真核生物的实施方案中,优选包含RNA裂解信号的终止子,更优选的是该终止子信号可促进信使RNA的多聚腺苷酸化。终止子和/或多聚腺苷酸位点元件可提高信使RNA的表达水平和/或使核糖体阅读过表达盒进入其他序列的可能降到最低。
本发明所用的终止子包括本文所描述的或者本领域技术人员所熟知的转录子的任何已知的终止子,其中包括而不限于基因的终止序列,如牛生长激素终止子,或者病毒的终止序列,如SV40终止子。在某些实施方案中,可转录或可翻译的序列可能缺少终止信号,如由于序列截短。
6.多聚腺苷酸信号
基因表达,特别是真核基因表达一般都包含多聚腺苷酸信号以使转录子正确多聚腺苷酸化。据信多聚腺苷酸信号的性质不是成功实现本发明的关键,任何这样的序列都可以使用。优选的实施方案包括SV40多聚腺苷酸信号和/或牛生长激素多聚腺苷酸信号,已知这些多聚腺苷酸信号在各种靶细胞内都有功能,并且使用方便。多聚腺苷酸化可提高转录子的稳定性,或者可以促进胞浆转运。
7.复制起点
为了在宿主细胞内扩增载体,载体内需包含一个或多个复制起始位点(通常被称为“ori”),这是一种复制在此处开始的特异性核酸序列。另外,如果宿主细胞是酵母,可以使用自主复制序列(ARS)。
8.选择和筛选标记
在本发明的某些实施方案中,可以通过在表达载体内添加一个标记而便于含本发明核酸构建体的细胞在体外或体内被鉴定。这种标记可以使细胞具有可鉴定的表型从而便于鉴定含表达载体的细胞。一般来说,筛选标记是一种可提供筛选特性的序列。阳性筛选标记是指在标记物存在的情况下可被筛选的标记,而负性标记是指在标记物存在的情况下不被筛选的标记。阳性筛选标记的例子有耐药标记。
一般来说,包含药物筛选标记有助于转化子的克隆和鉴定,例如提供新霉素嘌罗霉素、潮霉素、DHFR、GPT、zeocin和组胺醇耐药性的基因都是有用的筛选标记。除了能提供一种表型以便于区分转化子的标记外,其他类型的标记,包括筛选标记,也包括在本发明的范围之内,如GFP,其筛选的基础是比色分析。另外,可筛选的酶如单纯疱疹病毒胸苷激酶(tk)或氯霉素乙酰转移酶(CAT)也可以使用。本领域的技术人员了解如何使用免疫标记,可能结合FACS分析。用何种标记被认为是不重要的,只要该标记可与编码基因产物的核酸同时表达就可以。选择和筛选标记的其他例子是本领域技术人员所熟知的。
D.宿主细胞
本文所用的术语“细胞”、“细胞系”和“细胞培养物”可以互用。所有这些术语都包括其子代,可以是任何代和所有代的细胞。应当理解的是并不是所有的子代细胞都是一致的,因为可能会发生定向的或随机的突变。在用于表达异源核酸序列时,“宿主细胞”是指原核细胞或真核细胞,包括任何可被转化的生物,只要它能够复制载体和/或表达载体所编码的异源基因。宿主细胞可以而且已经被用作载体或病毒(如果不能表达外源多肽则不能作为载体)的受体。宿主细胞可以是“被转染的”或“被转化的”是指外源核酸,如修饰蛋白编码序列,被转移或导入到宿主细胞内的过程。转化细胞包括原代受体细胞及其子代细胞。
根据所要得到的结果是复制载体或是表达部分或全部载体编码核酸序列,宿主细胞可以是原核细胞或真核细胞,其中包括酵母细胞、昆虫细胞和哺乳动物细胞。许多细胞系和细胞培养物都可用作宿主细胞,可从美国模式培养物保藏所(ATCC)获得,这是收集和保存活体培养物和遗传材料的组织(www.atcc.org)。本领域的技术人员根据载体的结构及所要得到的结果能够确定选择何种合适的宿主。例如,质粒和粘粒可被导入到原核宿主细胞内以复制大量的载体。作为宿主细胞用于载体复制和/或表达的细菌包括DH5α、JM109和KC8,以及许多商业化的细菌宿主,如CompetentCells和SOLOPACK TM Gold Cells( La Jolla,CA)。另外,细菌细胞如大肠杆菌LE392也可以作为宿主细胞复制噬菌体。合适的酵母细胞包括酿酒酵母、啤酒酵母和毕赤酵母。
La Jolla,CA)。另外,细菌细胞如大肠杆菌LE392也可以作为宿主细胞复制噬菌体。合适的酵母细胞包括酿酒酵母、啤酒酵母和毕赤酵母。
用于复制和/或表达载体的真核宿主细胞的例子包括HeLa、NIH3T3、Jurkat、293、Cos、CHO、Saos和PC12。各种类型的细胞和生物来源的宿主细胞都可以得到,这是本领域技术人员所熟知的。同样,病毒载体可用于真核宿主细胞,也可用于原核宿主细胞,特别是能复制或表达载体的宿主细胞。
某些载体可包含调控序列以使其既可以在原核细胞,又可以在真核细胞内复制和/或表达。本领域的技术人员还了解培养上述宿主细胞并使其中的载体复制的条件。还清楚和了解大规模制备载体以及载体编码的核酸及其同源多肽、蛋白或肽的技术和条件。
E.表达系统
许多表达系统至少包含上述部分或全部组合物。原核和真核表达系统可用于本发明以制备核酸序列或其同源的多肽、蛋白和肽。许多这样的系统都已商品化,并且在许多地方都可以购买。
昆虫细胞/杆状病毒系统可使异源核酸片段高水平地表达蛋白质,如美国专利5,871,986、4,879,236所描述,本文已纳入作为参考,这种系统可以购买到,如 的
的 2.0杆状病毒表达系统和
2.0杆状病毒表达系统和 的BACPACK TM杆状病毒表达系统除了本发明描述的表达系统以外,其他表达系统的例子有的COMPLETECONTROL TM可诱导哺乳动物表达系统,该系统包含一个合成的蜕皮素可诱导受体,以及该公司的pET表达系统,这是一种大肠杆菌表达系统。另外一种可诱导表达系统是的T-REX TM(可被四环素调节的表达)系统,这是一种含全长CMV启动子的可诱导哺乳动物表达系统。
的BACPACK TM杆状病毒表达系统除了本发明描述的表达系统以外,其他表达系统的例子有的COMPLETECONTROL TM可诱导哺乳动物表达系统,该系统包含一个合成的蜕皮素可诱导受体,以及该公司的pET表达系统,这是一种大肠杆菌表达系统。另外一种可诱导表达系统是的T-REX TM(可被四环素调节的表达)系统,这是一种含全长CMV启动子的可诱导哺乳动物表达系统。 还有一种酵母表达系统,被称为毕赤酵母表达系统,该系统可在嗜甲醇的毕赤酵母内高水平表达重组蛋白。本领域的技术人员知道如何构建一个载体,如表达构建体,来制备核酸序列或其同源的多肽、蛋白或肽。
还有一种酵母表达系统,被称为毕赤酵母表达系统,该系统可在嗜甲醇的毕赤酵母内高水平表达重组蛋白。本领域的技术人员知道如何构建一个载体,如表达构建体,来制备核酸序列或其同源的多肽、蛋白或肽。
F.核酸检测
本文所描述的核酸序列除了可用于表达痘病毒蛋白、多肽和/或肽之外,还有很多其他用途。例如,可作为探针或引物用于涉及核酸杂交的实施方案中。也可用于本发明的诊断或筛选方法中。本发明包含编码痘病毒或痘病毒多肽调节因子的核酸的检测方法。
1.杂交
利用13到100个核苷酸长度的探针或引物,优选17到100个核苷酸长度的探针或引物,在本发明的某些方面可达到1-2kb或更长,可以形成稳定而特异的双螺旋分子。具有20个以上的连续碱基是互补序列的分子一般是优选的,可提高所得到的杂合分子的稳定性和/或特异性。通常来说,优选的用于杂交的核酸分子含有一段或多段含20到30个核苷酸的互补序列,如果需要甚至可以更长。这样的片段很容易制备,例如通过化学方法直接合成这样的片段,或通过将所选择的序列导入到重组载体内以制备这样的片段。
相应的,本发明的核苷酸序列可用于和互补的DNA和/或RNA片段形成特异性的双螺旋分子,或从样品中扩增DNA或RNA的引物。根据应用的目的不同,人们可以在不同的杂交条件下利用不同特异性的探针或引物来发现或扩增靶序列。
在需要高特异性的应用中,人们一般希望在相对高严格的条件下杂交。例如,相对低的盐浓度和/或相对较高的温度,如约0.02M到0.10M NaCl、约50℃到70℃的条件。这种高严格条件不允许探针或引物和模板或靶序列之间有错误配对,特别适于分离特异性基因或检测特异性的mRNA转录子。一般来说,逐渐提高甲酰胺的加入量可以使杂交条件更严格。
对于某些用途来说,如位点特异性突变,优选较低的严格条件。在这种条件下,即使杂交链的序列不是完全互补也可以发生杂交,但是会在一个或多个位置上发生碱基错配。通过提高盐的浓度和/或降低温度可以使杂交条件的严格程度降低。例如,中等严格程度的杂交条件是约0.1到0.25M NaCl、约37℃到55℃的温度,而低严格条件是指约0.15M到0.9M的盐浓度和约20℃到55℃的温度。根据所希望得到的结果可以很容易的调整杂交条件。
在其他实施方案中,杂交可在如下条件下进行:50mM Tris-HCl(pH 8.3)、75mMKCl、3mM MgCl2、1.0mM二硫苏糖醇,温度约在20℃到37℃之间。其他可用的杂交条件包括约10mM Tris-HCl(pH 8.3)、50mM KCl、1.5mM MgCl2,温度约在40℃到72℃之间。
在某些实施方案中,在本发明的具有确定序列的核酸上连接一些合适的分子,如标记物来进行杂交是有好处的。很多适宜的指示剂都是本领域所熟知的,其中包括荧光的、放射性、酶催化的指示剂或其他配基,如亲和素/生物素,这些都可以被检测。在优选的实施方案中,可以用荧光标记物或酶标记物,如尿素酶、碱性磷酸酶或过氧化物酶来代替放射性标记物或其他影响环境的试剂。在使用酶标记物的例子中,可使用比色指示剂底物通过可见的或分光光度的检测方式来检测与样品中互补核酸的杂交情况。
一般来说,本文所描述的探针或引物可作为试剂用于溶液杂交,如PCRTM中以检测相应基因的表达情况,或者用于固相杂交的实施方案中。在固相杂交的实施方案中,被检测的DNA(或RNA)被吸附或固定在基质或表面上。然后使这种被固定的单链核酸与所选择的探针在适当的条件下杂交。杂交条件的选择要根据特定的环境(如靶核酸的G+C含量、类型、核酸的来源、杂交探针的大小等)。对于某种特定用途来说如何选择最佳的杂交条件是本领域技术人员所熟知的。洗涤杂交的分子去除非特异性结合的探针分子以后,通过确定结合标记物的量来检测和/或定量杂交的情况。典型的固相杂交方法见于美国专利5,843,663、5,900,481和5,919,626的描述。可用于实现本发明的其他杂交方法见于美国专利5,849,481、5,849,486和5,851,772。这些文献的相关部分以及本说明书中这部分所涉及的其他参考文献都以参考文献的方式被纳入到本文中。
2.核酸的扩增
用作扩增模板的核酸可根据标准的方法从细胞、组织和其他样品中分离(Sambrook等,1989)。在某些实施方案中,分析是用完整细胞和组织匀浆物和液体生物样品进行,不需要纯化模板核酸。核酸可以是基因组DNA或分离的或全细胞RNA。如果用RNA为模板,则需要首先将其反转录成互补的DNA。
本文所用的术语“引物”包括可以模板依赖的方式启动起始核酸合成的任何核酸序列。一般来说,引物是长度为10到20和/或30bp的寡核苷酸,但是也可以使用更长的序列。引物可以双链和/或单链的形式提供,但是优选单链形式的引物。
可与本文所鉴定的基因序列相应的核酸特异性杂交的引物对与模板核酸在允许特异性杂交的条件下接触。根据使用目的的不同,可以选择高严格杂交条件从而只允许引物与其完全互补的序列杂交。在其他的实施方案中,可以降低的杂交条件的严格性以使与引物序列有一个或多个错配碱基的核酸也可以被扩增。杂交后,模板-引物复合物与一种或多种具有促进模板依赖的核酸合成的酶接触。经过多轮扩增,也被称为“循环”,就可以制备出足量的扩增产物。
然后检测和定量扩增产物。在某些用途中,检测以可见的方式进行。另外,检测也可以是通过测定掺入的放射性标记物或荧光标记物所产生的化学发光、放射活性来间接确定产物的量,甚至可以通过一个能发出电脉冲信号和/或热脉冲信号的系统来检测(Bellus,1994)。
许多模板依赖的方法都可用于扩增存在于给定模板样品中的寡核苷酸序列。一个最为大家所熟知的扩增方法就是多聚酶链式反应(被称为PCRTM),本方法的详细描述见于美国专利4,683,195、4,683,202和4,800,159以及Innis等,1988,本文都已完整纳入作为参考。
确定所要扩增的mRNA的量可通过反转录酶PCRTM扩增方法。将RNA反转录成cDNA的方法是大家所熟知的(见Sambrook等,1989)。另外一些反转录方法利用热稳定性的DNA聚合酶。这些方法描述于WO 90/07641。聚合酶链式反应的方法是本领域所熟知的。RT-PCR的经典方法见美国专利5,882,864的描述。
另外一种扩增方法是连接酶链式反应(“LCR”),描述于欧洲专利申请320308,本文已完整纳入作为参考。美国专利4,883,750描述了一种与LCR类似的方法用于探针对与靶序列的结合。基于PCRTM和寡核苷酸连接酶分析(OLA)的方法也可以使用,见美国专利5,912,148的描述。
可用于实现本发明的其他靶核酸序列扩增方法描述于美国专利5,843,650、5,846,709、5,846,783、5,849,546、5,849,497、5,849,547、5,858,652、5,866,366、5,916,776、5,922,574、5,928,905、5,928,906、5,932,451、5,935,825、5,939,291和5,942,391,英国专利申请2202328和PCT申请PCT/US89/01025,本文都已完整纳入作为参考。
PCT申请PCT/US87/00880所描述的Q beta复制酶也可以作为本发明的扩增方法。在该方法中,复制RNA序列在存在RNA聚合酶的情况下被加到样品中,该复制RNA序列含有一个与靶序列互补的区。聚合酶就会拷贝该复制序列,从而可以检测这种复制序列。
一种恒温扩增方法也可用于扩增本发明的核酸,其中限制性内切酶和连接酶被用于完成靶分子的扩增,靶分子内限制性酶切位点的一条链上含有核苷5’-[α-硫代]-三磷酸(Walker等,1992)。美国专利5,916,779所描述的链错位扩增(SDA)是另外一种可以恒温扩增核酸的方法,其中包括多轮的链错位和合成,即切口平移。
其他的核酸扩增方法包括转录扩增系统(TAS),如基于核酸序列的扩增(NASBA)和3SR(Kwoh等,1989;PCT申请WO 88/10315,本文已完整纳入作为参考)。欧洲专利申请329822描述了一种核酸扩增方法,其中包括循环合成单链RNA(“ssRNA”)、ssDNA和双链DNA(dsDNA),该方法也可用于本发明。
PCT专利申请WO 89/06700(本文已完整纳入作为参考)描述了核酸序列扩增方案,该方案的基础是启动子区/引物序列与单链靶DNA(“ssDNA”)杂交,然后转录出该序列的多个RNA拷贝。该方案不用多个循环,即从合成的RNA转录子中不产生新的模板。其他的扩增方法有“RACE”和“one-sided PCR”(Frohman,1990;Ohara等,1989)。
3.核酸的检测
扩增以后需要将扩增产物与模板和/或剩余的引物分开。在一个实施方案中,扩增产物用标准方法通过琼脂糖、琼脂糖-丙稀酰胺或聚丙稀酰胺凝胶电泳分开(Sambrook等,1989)。分开的扩增产物从凝胶上切下并洗脱出来以便于进行下一步的操作。使用低熔点琼脂糖凝胶可将分开的条带通过加热凝胶然后提取而获得核酸。
核酸也可以用本领域熟知的色谱技术分离。有许多种色谱技术可用于实现本发明,其中包括吸附层析、分配层析、离子交换层析、羟磷灰石结晶层析、分子排阻层析、反向层析、柱层析、纸层析、薄层色谱、以及气相色谱和HPLC。
在某些实施方案中,可将扩增产物显影。一个典型的显影方法是用溴乙锭染色,然后在紫外灯下观察。另外,如果扩增产物已经被标记上放射性或荧光标记核苷酸,分离出的扩增产物可使X-射线胶片曝光或者在适当的激发光下显影。
在一个实施方案中,扩增产物被分离出来后用标记的核酸探针与扩增的标记序列接触。探针最好连接上发光基团,但也可以进行放射性标记。在另外的实施方案中,探针连接有结合物如抗体或生物素或其他带有可检测基序的结合物。
在特定的实施方案中,可通过DNA印迹技术与标记探针杂交而检测。该技术涉及本领域技术人员所熟知的DNA印迹(见Sambrook等,1989)。上述技术的一个例子见于美国专利5,279,721的描述,本文已纳入作为参考,其中描述了自动化的电泳和核酸转移装置和方法。该装置不需要在外面处理凝胶就可以进行电泳和杂交,特别适于实现本发明的方法。
可用于实现本发明的其他核酸检测方法描述于美国专利5,840,873、5,843,640、5,843,651、5,846,708、5,846,717、5,846,726、5,846,729、5,849,487、5,853,990、5,853,992、5,853,993、5,856,092、5,861,244、5,863,732、5,863,753、5,866,331、5,905,024、5,910,407、5,912,124、5,912,145、5,919,630、5,925,517、5,928,862、5,928,869、5,929,227、5,932,413和5,935,791,本文都已纳入作为参考。
4.其他测定方法
在本发明的范围内其他一些方法也可用于遗传筛选,如检测基因组DNA、cDNA和/或RNA样品中是否存在突变。可用于检测点突变的方法包括变性梯度凝胶电泳(“DGGE”)、限制性片段长度多态性分析(“RFLP”)、化学或酶催化裂解方法、PCRTM扩增出的靶区域直接测序(见上)、单链构象多态性分析(“SSCP”)以及本领域所熟知的其他方法。
一种筛选点突变的方法是基于RNA酶可裂解RNA/DNA或RNA/DNA异源双链上错配的碱基对。本文所用的术语“错配”是指双链RNA/RNA、RNA/DNA或DNA/DNA分子上含有一个或多个未配对或错误配对核苷酸的区域。因此,这个定义包括了由于插入/缺失突变以及单个碱基或多个碱基点突变而导致的错配。
美国专利4,946,773描述了一种RNA酶A错配裂解分析方法,该方法是使单链DNA或RNA检测样品与RNA探针接触,然后用RNA酶A处理核酸双螺旋,为了检测错配的碱基,RNA酶A处理的单链产物根据大小的不同在电泳上分开,然后与经同样处理的对照双链相比较。包含对照双螺旋中未见的小片段(裂解产物)的样品标记为阳性。
其他研究人员也描述了RNA酶I在错配试验中的应用。使用RNA酶I检测错配的方法描述于Promega Biotech的文献中。Promega出品了一种包含RNA酶I的试剂盒,据报道这种RNA酶I可以裂解已知的4个错配碱基中的3个。其他一些报道显示利用MutS蛋白或其他DNA修饰酶也可以检测单碱基错配。
可用于实现本发明的检测缺失、插入或替代突变的其他方法描述于美国专利5,849,483、5,851,770、5,866,337、5,925,525和5,928,870,本文都已完整纳入作为参考。
G.基因转移方法
适于核酸转移以表达本发明的组合物的方法被认为可以包括能将核酸(如DNA,包括病毒载体和非病毒载体)导入到细胞器、细胞、组织或生物体内的任何方法,如本文所描述的和本领域技术人员所熟知的方法。这些方法包括而不限于通过注射(美国专利5,994,624、5,981,274、5,945,100、5,780,448、5,736,524、5,702,932、5,656,610、5,589,466和5,580,859,本文已纳入作为参考)、包括微注射(Harlan和Weintraub,1985;美国专利5,789,215,本文已纳入作为参考);电穿孔(美国专利5,384,253,本文已纳入作为参考);钙磷酸盐沉淀(Graham和Van Der Eb,1973;Chen和Okayama,1987;Rippe等,1990);DEAE-葡聚糖、然后用聚乙二醇(Gopal,1985);直接的声波负载(Fechheimer等,1987);脂质体介导的转染(Nicolau和Sene,1982;Fraley等,1979;Nicolau等,1987;Wong等,1980;Kaneda等,1989;Kato等,1991);微发射炮击(PCT专利申请WO 94/09699和95/06128;美国专利5,610,042、5,322,783、5,563,055、5,550,318、5,538,877和5,538,880,本文已纳入作为参考);碳化硅纤维搅拌(Kaeppler等,1990;美国专利5,302,523和5,464,765,本文已纳入作为参考);土壤杆菌介导的转化(美国专利5,591,616和5,563,055,本文已纳入作为参考);或PEG-介导的原生质体转化(Omirulleh等,1993;美国专利4,684,611和4,952,500,本文已纳入作为参考);脱水/抑制介导的DNA摄取(Potrykus等,1985)直接转移基因。通过使用这些技术,细胞器、细胞、组织或生物体可被稳定转化或瞬时转化。
H.脂质组分和基序
在某些实施方案中,本发明涉及包含与核酸、氨基酸分子如肽或其他小分子化合物相连接的一个或多个脂质的组合物。在本文所讨论的所有实施方案中,分子既可以是痘病毒多肽或痘病毒多肽调节因子,如编码全长或部分痘病毒多肽的核酸,也可以是编码全长或部分痘病毒多肽调节因子的氨基酸分子。脂质是不溶于水、能用有机溶剂提取的物质。本文所特别指出的化合物以外的化合物可以被本领域的技术人员理解为脂质,也包括在本发明的组合物和方法之内。脂质成分和非脂质成分可通过共价键或非共价键彼此结合。
脂质可以是天然的,也可以是人工合成的(即人工设计并制备的)。但是,脂质通常是一种生物物质。生物脂质是本领域所熟知的,包括中性脂肪、磷脂、磷酸甘油酯、类固醇、萜烯、溶血脂质(lysolipids)、鞘糖脂、糖脂、硫苷脂、含有醚和酯连接脂肪酸的脂质、可聚合的脂质、以及上述脂质的组合物。
与脂质相关的核酸分子或氨基酸分子、如肽可被分散在含脂质溶液中、用脂质溶解、乳化、混合、与脂质连接、共价结合、以悬液的形式悬浮于脂质中、以及以其他方式与脂质相联系。脂质或脂质/本发明的痘病毒相关组合物并不局限于任何特定结构的分子。例如,可以简单地分散在溶液中,可能形成不同大小和形状的凝集物。在其他的例子中,可以双层的结构存在,如微胶粒或萎陷的结构。在其他的非限定性例子中,也可以考虑lipofectamine(Gibco BRL)-痘病毒或Superfect(Qiagen)-痘病毒复合物。
在某些实施方案中,脂质组合物可以包含约1%、约2%、约3%、约4%、约5%、约6%、约7%、约8%、约9%、约10%、约11%、约12%、约13%、约14%、约15%、约16%、约17%、约18%、约19%、约20%、约21%、约22%、约23%、约24%、约25%、约26%、约27%、约28%、约29%、约30%、约31%、约32%、约33%、约34%、约35%、约36%、约37%、约38%、约39%、约40%、约41%、约42%、约43%、约44%、约45%、约46%、约47%、约48%、约49%、约50%、约51%、约52%、约53%、约54%、约55%、约56%、约57%、约58%、约59%、约60%、约61%、约62%、约63%、约64%、约65%、约66%、约67%、约68%、约69%、约70%、约71%、约72%、约73%、约74%、约75%、约76%、约77%、约78%、约79%、约80%、约81%、约82%、约83%、约84%、约85%、约86%、约87%、约88%、约89%、约90%、约91%、约92%、约93%、约94%、约95%、约96%、约97%、约98%、约99%、约100%或上述数值之间任何范围的特定脂质、脂类或非脂质组分,如药物、蛋白、糖、核酸或本文所描述的以及本领域技术人员所熟知的其他物质。在一个非限定性例子中,脂质组合物包含约10%到20%中性脂质、约33%到34%的脑苷脂和约1%的胆固醇。在另一个非限定性例子中,脂质体包含约4%到12%的萜烯,其中约1%的微胶粒是番茄红素,另外约有3%到11%的脂质体包含其他萜烯;约10%到35%的磷脂酰胆碱以及约1%的药物。因此,需要说明的是本发明的组合物包含任何组合或任何百分比的脂质、脂类或其他组分。
IV.药用制剂、给药途径和治疗方案
在本发明的一个实施方案中,提供了一种通过给予经修饰的痘病毒,如痘苗病毒治疗过度增殖性疾病、如癌症的方法。可用该方法治疗的肿瘤包括肺癌、头颈癌、乳腺癌、胰腺癌、前列腺癌、肾癌、骨癌、睾丸癌、宫颈癌、胃肠癌、淋巴瘤、肺内的癌前病变、结肠癌、黑色素瘤、膀胱癌以及可被治疗的其他癌或肿瘤。
一般来说,有效量的药用组合物是指足够量的可重复达到改善、减轻、缩小或抑制疾病及其症状的组合物。也可以用更严格的定义,其中包括消除、根除或治愈疾病。
患者最好具有适当的骨髓功能(定义为外周粒细胞绝对值>2,000/mm3,血小板绝对值为100,000/mm3)、适当的肝功能(胆红素<1.5mg/dl)和适当的肾功能(肌氨酸酐<1.5mg/dl)。
A.给药途径
利用本发明的方法和组合物杀死细胞、抑制细胞生长、抑制肿瘤转移、缩小肿瘤或组织以及反转或降低肿瘤细胞恶性表型一般是通过使过度增殖的细胞与治疗性化合物如多肽或编码多肽的表达构建体接触。给药途径可根据损伤的部位及损伤的程度不同而不同,如真皮内注射、透皮注射、胃肠外途径、静脉注射、肌肉注射、鼻内给药、皮下注射、局部给药、经皮注射、气管内给药、腹腔注射、动脉注射、膀胱内给药、瘤内注射、吸入给药、灌注、灌洗、直接注射或口服给药及制剂
为了改善血管疾病的治疗效果,可以使血管细胞与治疗性化合物接触。与癌症治疗或诊断相关的任何制剂和给药途径都适用于血管疾病。
对于不连续的易接近的实体瘤来说,采用瘤内注射或注射到肿瘤血管内的方式给药是特别合适的。局部、区域或系统给药也可以采用。对于>4cm的肿瘤来说,注射的量约为4-10ml(优选10ml),而对于<4cm的肿瘤来说,注射的量约为1-3ml(优选3ml)。多点注射给药时单次剂量包含约0.1-0.5ml的体积。对于病毒颗粒来说最好通过多点注射来给药,点与点之间的间隔约1cm。
在进行手术干预的情况下,本发明可作为预处理措施使无法进行手术的肿瘤变成可被切除的肿瘤。另外,本发明还可以在手术的时候和/或手术以后使用以治疗残存的或转移的肿瘤。例如,被切除的肿瘤床可注射或灌注含痘病毒多肽或痘病毒的制剂,其中的痘病毒包含突变,该突变可使痘病毒能够更有效地治疗癌或癌细胞。灌注可在手术切除后持续进行,例如通过在手术部位植入一个导管。本发明也包含周期性的术后治疗措施。
在合适的情况下可进行连续给药,例如当肿瘤被切除后处理肿瘤床以去除残存的微小肿瘤时。优选通过注射器或导管给药。这种连续给药可持续约1-2小时到2-6小时、约6-12小时、约12-24小时、约1-2天、约1-2周或更长时间。一般来说,通过持续灌注方式所给予的治疗性组合物的剂量与单点注射或多点注射所给予的剂量等价,并且在灌注期间可以进行适当调整。还需要说明的是四肢灌注方式也可用于注射本发明的治疗性组合物,尤其是用于治疗黑色素瘤和肉瘤时。
治疗方案可以不同,要根据肿瘤的类型、肿瘤的部位、疾病的进展情况以及患者的健康状况和年龄而定。显然,某些类型的肿瘤需要更积极的治疗措施,而同时某些患者可能无法耐受较繁重的治疗过程。临床医生最适宜根据治疗性制剂的效力和毒性(如果有)来作决定。
在某些实施方案中,被处理的肿瘤可能是无法被切除的,或者至少最初是无法被切除的。通过治疗性病毒构建体的处理可能能够提高肿瘤的可切除性,可能由于肿瘤的边界发生皱缩,或者由于去除了肿瘤某些侵入的部分。经过处理后就可能能够切除了。切除后可进一步采用其他处理措施来消除肿瘤部位残存的微小肿瘤组织。
处理原发肿瘤或切除后肿瘤床的过程通常包括多次注射。一般来说,治疗原发肿瘤需要在2周内注射6次。2周的治疗方案可重复进行1、2、3、4、5、6或更多次。在治疗期间,需要对整个治疗方案不断进行评价和调整。
治疗方案包括不同的“单位剂量”。单位剂量定义为预先确定的治疗性组合物的量。药物使用剂量、给药途径和剂型的选择是临床医学领域的技术人员所熟知的。单位剂量并不一定只通过一次注射来完成,也可以在一定的期间内持续输注。本发明的单位剂量为了方便用病毒构建体的噬菌斑形成单位(pfu)来表示。另外,根据病毒种类及其可得到的滴度不同,可给予患者或患者的细胞1到100、10到50、100-1000、或者高达约1×104、1×105、1×106、1×107、1×108、1×109、1×1010、1×1011、1×1012、1×1013、1×1014、或1×1015或更多的有感染性的病毒颗粒(vp)。
B.可注射的组合物和制剂
本发明中将编码全长或部分痘病毒基因组的表达构建体转移到肿瘤或肿瘤细胞内的优选方法是通过瘤内注射。但是,本文所描述的药用组合物也可以通过其他方式给药,如胃肠外途径、静脉注射、真皮内注射、肌肉注射、透真皮注射、甚至腹腔注射,如美国专利5,543,158、5,641,515和5,399,363所描述(本文都已完整纳入作为参考)。
核酸构建体可用注射器或其他任何可注射溶液的方法注射,只要表达构建体能够通过特定孔径的注射针头就可以。最近所描述的一种无针注射系统(美国专利5,846,233)含有一个安装在安瓿腔内的喷嘴用于吸取溶液,一个动力装置用于将液体推出喷嘴转移到注射部位。在基因治疗中也可以用注射器系统将预先确定的精确量的溶液以任意深度注射到多个部位(美国专利5,846,225)。
活性化合物的游离碱溶液或药用盐溶液可通过在水中与适宜的表面活性剂,如羟丙基纤维素混合而制备。分散剂可用甘油、液体聚乙烯甘油或其混合物、或油来制备。在普通储存和使用条件下,这些制剂都要加入防腐剂以阻止微生物的生长。适于注射的药用制剂包括无菌水溶液或分散剂,以及在使用前可临时制备成无菌注射溶液或分散剂的粉末(美国专利5,466,468,本文已完整纳入作为参考)。无论何种情况,制剂都必须是无菌的,并且其流动性要足以能够通过注射器。在制造和储存条件下必须是稳定的,并且要防止微生物,如细菌和真菌的污染。载体可以是含水、乙醇、多元醇(如甘油、丙二醇和液体聚乙烯甘油等)、上述物质的适当混合物和/或植物油的溶剂或分散介质。可以通过包被,如卵磷脂,使颗粒在分散剂中保持所需的直径以及通过使用表面活性剂使制剂维持合适的流动性。可通过加入各种抗细菌和抗真菌物质,如对羟基苯甲酸酯类(parabens)、氯代丁醇、酚、山梨酸、硫柳汞等来防止微生物的生长。在许多情况下,加入一些等渗物质,如糖或氯化钠是优选的。可通过在组合物中加入吸收延迟物质,如单硬脂酸铝和明胶使可注射组合物的吸收缓慢。
对于通过胃肠外途径给药的水溶液来说,如果需要溶液应是缓冲的,液体稀释剂应首先用足量的盐或糖调节成等渗的。这些水溶液特别适于静脉注射、肌肉注射、皮下注射、瘤内注射和腹腔注射。本领域的技术人员根据本说明书的描述可以选择应用这种水溶液所需要的无菌水溶剂。例如,一个剂量的药物可溶解到1ml等渗的氯化钠溶液中,然后添加到1000ml皮下补液溶液中或注射到输注的适当部位(见“Remington’s Pharmaceutical Sciences”,第15版,1035-1038页和1570-1580页)。要根据被治疗的个体的情况来调整注射的剂量。不论在任何情况下都负责给药的人来确定不同个体所需要的合适剂量。而且对于给人注射来说,制剂必须符合FDA生物标准办公室所制定的无菌、热源、一般安全性和纯度标准。
无菌注射液可通过使溶于合适溶剂中的所需量的活性化合物与上述各种其他组分混合、然后通过过滤除菌来制备。一般来说,分散剂可通过将各种无菌活性成分加入到含基础分散介质和所需要的上述各种其他组分的无菌载体内而制备。如果是用于制备无菌注射液的无菌粉末,优选的制备方法是真空干燥和冷冻干燥技术,该技术可从上述过滤除菌的溶液中制备出活性成分和其他所需成分的粉末。
本文所描述的组合物可制备成中性制剂和盐制剂。药用盐包括酸加盐(与蛋白质的游离氨基形成的盐),可用无机酸制备,如盐酸和磷酸,也可以用有机酸制备,如乙酸、醋浆草酸、酒石酸、扁桃酸等。与游离羧基形成的盐也可以来源于无机碱,如氢氧化钠、氢氧化钾、氢氧化铵、氢氧化钙和氢氧化铁,或者有机碱如异丙胺、三甲胺、组胺、普如卡因等。制成制剂以后,溶液就可以以剂型相配的方式、以治疗有效量注射。各种剂型都可以很容易地给药,如可注射溶液、缓释胶囊等。
本文所用的术语“载体”包含各种溶剂、分散介质、载体、包被物、稀释剂、抗细菌和抗真菌药物、等渗和吸收延迟物质、缓冲液、载体溶液、悬液、胶体等。利用这种介质和试剂制备药用活性物质的方法是本领域所熟知的。除了那些与活性组分不相容的常规介质或试剂以外,其在治疗性组合物中的应用也是本发明所涉及的。补充的活性组分也可以添加到组合物中。
术语“药用的”是指给人体注射以后不会产生变态反应或类似不良反应的分子实体和组合物。含有蛋白性活性组分的水性组合物的制备方法是本领域所熟知的。一般来说,这种组合物应制备成可注射的溶液或悬液,或者在注射前可制备成溶液或悬液的固体形式。
C.联合治疗
本发明的化合物和方法可用于治疗过度增殖性疾病,其中包括癌症和动脉粥样硬化。为了提高本发明的组合物,如减毒痘苗病毒的治疗效率,这些组合物可与其他能有效治疗这些疾病的药物联合使用。例如,肿瘤可用本发明的治疗性化合物联合其他的肿瘤治疗措施来治疗,如抗癌药或手术。
各种联合治疗措施都可以使用;例如,减毒痘病毒如痘苗病毒是“A”,第二种抗癌措施是“B”:
A/B/A B/A/B B/B/A A/A/B A/B/B B/A/A A/B/B/B B/A/B/B
B/B/B/A B/B/A/B A/A/B/B A/B/A/B A/B/B/A B/B/A/A
B/A/B/A B/A/A/B A/A/A/B B/A/A/A A/B/A/A A/A/B/A
将本发明的治疗性表达构建体给予患者可按照第二种治疗措施的通用方法进行,并且需要考虑痘病毒治疗措施的毒性。如果需要,可重复进行几个治疗周期。还需要说明的是,各种标准的治疗措施以及手术干预都可以和本文所描述的癌症或肿瘤治疗措施联合使用。
1.抗癌治疗措施
“抗癌”药是对生物体内的肿瘤能产生消极影响的药物,如杀伤癌细胞,诱导癌细胞的调亡、降低癌细胞的生长速度、抑制转移的发生或转移的数目、缩小肿瘤、抑制肿瘤的生长、减少肿瘤或癌细胞的血供、激发抗癌细胞或肿瘤的免疫反应、阻止或抑制肿瘤的发展、或者延长癌症患者的生存期。抗癌药包括生物药(生物治疗)、化疗药和放疗药。最常见的情况是这些组合物各以有效量联合使用以杀伤或抑制细胞的增殖。这个过程包括细胞与表达构建体和药物或多个因子同时接触。这可以通过使细胞与一种包含两种药物的组合物或药用制剂接触,或者使细胞与两种不同的组合物或制剂同时接触,其中一种组合物含表达构建体,另一种组合物含第二药物。
肿瘤细胞对化疗药和放疗药耐受是临床肿瘤学领域遇到的一个主要问题。目前癌症研究的一个目标就是通过结合基因治疗找到提高化疗和放疗疗效的方法。例如,单纯疱疹病毒-胸苷激酶(HS-tK)基因用逆转录病毒载体系统转移到脑肿瘤内后可成功地诱导出肿瘤对抗病毒药物更昔洛韦的敏感性(Culver等,1992)。在本发明中,除了其他一些前调亡调节因子或细胞周期调节因子之外,痘病毒治疗同样可联合化疗、放疗、免疫治疗以及其他生物干预措施。
另外,基因治疗可在其他药物治疗前或治疗后数分钟到数周内进行。在其他药物和表达构建体分别给予细胞的实施方案中,两种治疗措施实施的时间间隔一般不能太长,这样才能保证药物和表达构建体对细胞产生协同效应。在这种情况下,两种治疗措施最好在约12-24小时内分别给予细胞,更优选的是在6到12小时内。但是在某些情况下,人们希望将治疗的间隔时间延长,比如不同的治疗措施之间间隔数天(2、3、4、5、6或7)到数周(1、2、3、4、5、6、7或8)。
a.化疗
癌症的治疗措施还包括与化疗和放疗联合的各种治疗措施。例如,联合化疗包括顺铂(CDDP)、卡铂、丙卡巴肼、氮芥、环磷酰胺、喜树碱、异环磷酰胺、美法仑、苯丁酸氮芥、白消安、亚硝脲、放线菌素、柔红霉素、阿霉素、博来霉素、plicomycin、丝裂霉素、依托泊甙(VP16)、他莫昔芬、雷洛昔芬、雌激素受体结合药、紫杉醇、吉西他滨、新霉酰胺、法尼基蛋白转移酶抑制剂、反铂、5-氟尿嘧啶、长春新碱、长春花碱、氨甲蝶呤、Temazolomide(DTIC的液态形似)、或上述药物的类似物或衍生物。化疗药与生物治疗联合被称为生物化疗。
b.放疗
可引起DNA损伤并被广泛应用的其他因素包括大家所熟知的射线、X射线和/或直接给予肿瘤细胞放射性同位素。本发明涉及的其他形式的DNA损伤因子还包括微波和紫外线辐射。最可能的情况是所有这些因子都可以对DNA、DNA的前体、DNA的复制和修复、染色体的装配和维持造成广泛的损伤。X射线的使用剂量范围为每日50-200伦琴持续一段时间(3到4周)到单次剂量2000-6000伦琴。放射性同位素的剂量使用范围很宽,要根据放射性同位素的半衰期、所激发的辐射的强度和类型以及肿瘤细胞的摄取量而定。
术语“接触”和“暴露于”在本文中用于细胞时是指治疗性构建体和化疗或放疗药转移到靶细胞或者与靶细胞直接并排放置的过程。为了达到杀死细胞或抑制细胞生长的目的,两种治疗措施都要以有效量给予细胞以便于杀死细胞或阻止其分裂。
c.免疫治疗
一般来说,免疫治疗依赖于利用免疫效应细胞和分子靶向并摧毁癌细胞。例如,免疫效应因子可以是肿瘤细胞表面某些标记物特异性的抗体。抗体本身可以单独作为治疗效应因子或者可以激活其他细胞来影响细胞的杀伤。抗体也可以被连接到药物或毒素(化疗药物、放射性核苷酸、蓖麻毒蛋白A链、霍乱毒素、百日咳毒素等)上,只作为一种靶向性药物。另外,效应因子可以是携带能与肿瘤细胞靶位直接或间接相互作用的表面分子的淋巴细胞。效应细胞包括细胞毒T细胞和NK细胞。联合治疗,即直接的细胞毒活性和某种痘病毒多肽的抑制或降低活性可以为癌症的治疗提供有益的帮助。
免疫治疗还可以作为联合治疗的一部分。联合治疗的常用方法将在下面讨论。在免疫治疗的一个方面,肿瘤细胞必须携带可作为靶位的某种标记,即在其他大多数细胞上不存在的标记。肿瘤细胞上存在多种标记物,这些标记物都可以作为本发明的适宜靶位。常见的肿瘤标记物包括癌胚抗原、前列腺特异性抗原、泌尿系统肿瘤相关抗原、胎儿抗原、酪氨酸酶(p97)、gp68、TAG-72、HMFG、Sialyl Lewis抗原、MucA、MucB、PLAP、雌激素受体、层粘连蛋白受体、erb B和p155。免疫治疗的另一个方面涉及利用免疫刺激效应产生抗癌效应。免疫刺激分子包括:细胞因子如IL-2、IL-4、IL-12、GM-CSF、IFNγ,趋化因子如MIP-1、MCP-1、IL-8,以及生长因子如FLT3配基。联合使用免疫刺激分子,无论是以蛋白的形式,还是肿瘤抑制因子如mda-7的基因转移,都可以增强抗肿瘤效应(Ju等,2000)。
正如以前所讨论的,目前正在研究或正在应用的免疫治疗措施有免疫佐剂(如牛型结核菌、恶性疟原虫、二硝基氯苯和芳香族化合物)(美国专利5,801,005;美国专利5,739,169;Hui和Hashimoto,1998;Christodoulides等,1998)、细胞因子治疗(如干扰素α、β和γ;IL-1,GM-CSF和TNF)(Bukowski等,1998;Davidson等,1998;Hellstrand等,1998)、基因治疗(如TNF,IL-1,IL-2,p53)(Qin等,1998;Austin-Ward和Villaseca,1998;美国专利5,830,880和5,846,945)以及单克隆抗体(如抗神经节苷脂GM2,抗HER-2,抗-p185)(Pietras等,1998;Hanibuchi等,1998;美国专利5,824,311)。赫赛汀(Herceptin)(曲妥单抗)是一种嵌合的(鼠-人)单克隆抗体,能够阻断HER2-neu受体。它具有抗肿瘤活性,已被批准用于治疗恶性肿瘤(Dillman,1999)。赫赛汀和化疗药物联合治疗癌症已被证明比单独的治疗措施更有效。因此,本发明涉及利用一种或多种抗癌治疗措施与本文所描述的痘病毒相关治疗方法联合使用以治疗肿瘤的方法。
i)被动免疫治疗
现在已经有许多治疗癌症的被动免疫疗法。大致可以分为以下几类:单独注射抗体;注射连接有毒素或化疗药的抗体;注射连接有放射性同位素的抗体;注射抗独特型抗体;以及净化骨髓中的肿瘤细胞。
被动免疫治疗优选人源单克隆抗体,因为它们在患者体内的毒副作用很低或没有。但是,这样的抗体很少,并且到目前为止只适用于瘤内注射,因此其应用受到限制。抗神经节苷脂抗原的人源单克隆抗体已被用于注射到再发的皮肤黑色素瘤患者的肿瘤内(Iri e和Morton,1986)。经过每日或每周的瘤内注射后,10位患者中有6位患者的肿瘤生长受到抑制。在另外的试验中,通过瘤内注射两种人源单克隆抗体获得了中等程度的疗效(Irie等,1989)。
较好的是注射一种以上的抗两种不同抗原的单克隆抗体,甚至多种抗原的抗体。治疗方法还包括给予淋巴因子或其他免疫增强剂,如Bajorin等,(1988)的描述。有关人源单克隆抗体的研究进展会在本说明书的其他部分作进一步的详细描述。
ii)主动免疫治疗
主动免疫治疗注射的是抗原性肽、多肽或蛋白质,或者自身的或同种异体的肿瘤细胞组合物或“疫苗”,一般需要和不同的细菌佐剂联合使用(Ravindranath和Morton,1991;Morton等,1992;Mitchell等,1990;Mitchell等,1993)。对于黑色素瘤的免疫治疗来说,那些激发出高滴度IgM反应的患者通常比未激发出或激发的IgM抗体滴度比较低的患者生存率更高(Morton等,1992)。IgM抗体一般都是瞬时抗体,但是抗神经节苷脂和抗糖类的抗体好像是例外。
iii)获得性免疫治疗
获得性免疫治疗是指在体外分离出患者血液中的淋巴细胞或肿瘤侵润淋巴细胞,在体外用淋巴因子如IL-2活化或者转导肿瘤坏死基因,然后再输注到患者体内(Rosenberg等,1988;1989)。为达到这个目的,需要给动物或患者注射免疫有效量的活化的淋巴细胞和本文所描述的含佐剂的抗原肽组合物。活化的淋巴细胞最好是患者自身的易于从血液或肿瘤标本中分离并在体外被活化(或“扩增”)的细胞。这种形式的免疫治疗已对几例黑色素瘤和肾癌产生了抑制作用,但是发生反应的患者比未发生反应的患者明显要少。
d.基因
在另外一个实施方案中,第二治疗措施是基因治疗,其中治疗性多核苷酸在注射减毒痘病毒之前、之后或同时注射。痘病毒与编码下列基因产物之一的载体一起转移可对靶组织产生联合的抗癌效应。另外,痘病毒可改造成含治疗性多核苷酸的病毒载体。本发明包含多种蛋白质,其中一些将在下文描述。表7列举了本发明所涉及的可作为基因治疗靶位的各种基因。
i)细胞增殖诱导因子
诱导细胞增殖的蛋白质根据功能的不同可分为不同的类型。这些蛋白质的一个共同点就是其调节细胞增殖的能力。例如,一种形式的PDGF,sis癌基因,就是一种分泌型的生长因子。癌基因很少来源于编码生长因子的基因,到目前为止,sis是唯一已知的天然癌基因生长因子。在本发明的一个实施方案中,一种特殊的细胞增殖诱导因子的反义mRNA被用于阻止该细胞增殖诱导因子的表达。
蛋白质FMS、ErbA、ErbB和neu是生长因子受体。这些受体的突变可导致其调节功能的丧失。例如,Neu受体蛋白跨膜区的一个点突变可导致neu癌基因的产生。ErbA癌基因来源于甲状腺激素的胞内受体。被修饰的癌基因ErbA受体被认为可与内源性的甲状腺激素受体竞争,引起增殖失控。
最大的一类癌基因包括信号转导蛋白(如Src、Abl和Ras)。Src蛋白是胞浆蛋白酪氨酸激酶,在某些情况下它可以从原癌基因转化成癌基因,这是其酪氨酸残基527突变的结果。相反,在一个例子中,GTP酶蛋白ras从原癌基因转化成癌基因是其序列上的12位氨基酸从缬氨酸转化成甘氨酸的结果,这种突变可导致ras的GTP酶活性下降。
蛋白Jun、Fos和Myc可作为转录因子对核功能直接产生影响。
ii)细胞增殖的抑制因子
肿瘤抑制癌基因的功能是抑制过度的细胞增殖。这些基因的灭活可使其抑制活性丧失,结果导致增殖失控。下面将要描述肿瘤抑制因子p53、p16和C-CAM。
除了上面所描述的p53之外,另外一个细胞增殖抑制因子是p16。真核细胞周期的转换主要被细胞周期素依赖的激酶或CDK激发。一种CDK,细胞周期素依赖的激酶4(CDK4)可调节细胞通过G1期。这个酶的活性可在G1晚期使Rb磷酸化。CDK4的活性受活化亚单位-D型周期素和抑制亚单位-p16INK4的调节,这两个亚单位是能够特异性结合并抑制CDK的蛋白质,因此可以调节Rb的磷酸化(Serrano等,1993;Serrano等,1995)。由于p16INK4蛋白是CDK4的抑制因子(Serrano,1993),因此这个基因的缺失可使CDK的活性升高,导致Rb蛋白的过度磷酸化。已知p16还可以调节CDK6的功能。
p16INK4属于新发现的一类CDK-蛋白,该类CDK蛋白还包括p16B、p19、p21WAF1和p27KIP1。p16INK4基因位于染色体上的9p21区,这是一个多种类型的肿瘤经常发生缺失的染色体区。p16INK4基因在人源肿瘤细胞系中常常发生纯合的缺失和突变。这些现象说明p16INK4基因是一种肿瘤抑制基因。但是这种解释受到了挑战,因为已经观察到p16INK4基因在原发的未经培养的肿瘤中发生突变的几率比培养的细胞要低的多(Caldas等,1994;Cheng等,1994;Hussussian等,1994;Kamb等,1994;Kamb等,1994;Mori等,1994;Okamoto等,1994;Nobori等,1994;Orlow等,1994;Arap等,1995)。通过转染质粒表达载体恢复野生型p16INK4的功能可降低某些人源癌细胞系的克隆形成能力(Okamoto,1994;Arap、1995)。
可用于本发明的其他基因包括Rb、APC、DCC、NF-1、NF-2、WT-1、MEN-I、MEN-II、zac1、p73、VHL、MMAC1/PTEN、DBCCR-1、FCC、rsk-3、p27、p27/p16融合蛋白、p21/p27融合蛋白、抗血栓基因(如COX-1、TFPI)、PGS、Dp、E2F、ras、myc、neu、raf、erb、fms、trk、ret、gsp、hst、abl、E1A、p300、参与血管形成的基因(如VEGF、FGF、血小板凝血酶敏感蛋白、BAI-1、GDAIF或其受体)和MCC。
iii)细胞调亡调节因子
调亡或程序性细胞死亡是正常胚胎发育、维持成年组织内环境稳定、以及抑制肿瘤形成的一个基本过程(Kerr等,1972)。Bcl-2家族的蛋白和ICE样蛋白酶已被证明在其他系统中是重要的调节因子和调亡效应因子。Bcl-2蛋白是一种与滤泡淋巴瘤有关的蛋白,在受到不同调亡刺激信号后在控制调亡和增加细胞的存活中扮演主要角色(Bakhshi等,1985;Cleary和Sklar,1985;Cleary等,1986;Tsujimoto等,1985;Tsujimoto和Croce,1986)。现在已经认识到进化保守的Bcl-2蛋白是相关蛋白家族的一个成员,这个家族可被分类为死亡激动剂或死亡拮抗剂。
Bcl-2被发现以后被证明可抑制各种刺激信号所激发的细胞死亡。现在已经清楚Bcl-2细胞死亡调节蛋白家族具有相似的结构和序列同源性。这个家族的不同成员或者具有与Bcl-2相同的功能(如BclXL、BclW、BclS、Mcl-1、A1、Bfl-1),或者与Bcl-2的功能相反,可促进细胞的死亡(如Bax、Bak、Bik、Bim、Bid、Bad、Harakiri)。
e.手术
大约60%的癌症患者都做过不同类型的手术,其中包括为了预防、诊断或分期、治愈和缓解的目的而进行的手术。以治愈为目的的手术可结合其他治疗措施,如本发明的治疗措施、化疗、放疗、激素治疗、基因治疗、免疫治疗和/或其他治疗方法。
以治愈为目的的手术包括切除肿瘤,其中癌组织的全部或部分被物理去除、切掉和/或毁坏。肿瘤切除是指通过物理方法至少去除肿瘤的一部分。除了切除肿瘤以外,手术治疗措施还包括激光手术、冷冻手术、电手术和显微手术(Mohs手术)。本发明还可用于配合浅表癌、癌前病变或少量正常组织的去除。
全部或部分癌细胞、癌组织或肿瘤被切除以后,在体内可能会形成空腔。本发明的治疗措施可通过灌注、直接注射或局部使用来实施,并配合以其他的抗癌治疗措施。这种治疗措施可重复进行,如每1、2、3、4、5、6或7天、每1、2、3、4和5周、每1、2、3、4、5、6、7、8、9、10、11或12月进行一次。这种治疗措施还可以选择不同剂量。
f.其他因子
其他因子也可与本发明联合使用以改善治疗的效果。这些因子包括免疫调节因子、影响细胞表面受体和GAP连接表达上调的因子、细胞增殖抑制和分化因子、细胞粘附抑制因子、增加过度增殖细胞对调亡诱导因子敏感性的因子以及其他生物因子。免疫调节因子包括肿瘤坏死因子;干扰素α、β和γ;IL-2及其他细胞因子;F42K及其他细胞因子类似物;或MIP-1、MIP-1β、MCP-1、RANTES及其他趋化因子。细胞表面受体或其配基如Fas/Fas配基、DR4或DR5/TRAIL(Apo-2配基)的表达上调可通过其对过度增殖细胞的自分泌或旁分泌效应提高本发明的调亡诱导能力。提高增加GAP连接的数目增强细胞内的信号水平可提高对临近过度增殖细胞群的抗过度增殖效应。在其他的实施方案中,细胞增殖抑制和分化因子可与本发明联合使用以改善本发明的抗过度增殖效应。细胞粘附抑制因子也可以提高本发明的疗效。细胞粘附抑制因子的例子有病灶粘附激酶(FAK)抑制因子和洛伐他汀。本发明还包括能提高过度增殖细胞对调亡的敏感性的其他因子,如抗体c225,这些因子也可与本发明联合以改善治疗的效果。
Apo2配基(Apo2L,也叫TRAIL)是肿瘤坏死因子(TNF)细胞因子家族的一个成员。TRAIL可激活多种类型的癌细胞发生快速调亡,但是对正常细胞没有毒性。各种组织中都有TRAIL mRNA。大多数正常细胞对TRAIL的细胞毒活性耐受,说明存在保护细胞不发生TRAIL诱导的调亡的机制。TRAIL的第一个被发现的受体被称为死亡受体4(DR4),含有胞浆“死亡域”;DR4可转导TRAIL携带的调亡信号。其他可结合TRAIL的受体也被鉴定出来。其中一个受体被称为DR5,含有一个与DR4非常相似的死亡域和调亡信号。DR4和DR5mRNA在多种正常组织和肿瘤细胞系内都有表达。最近,诱骗受体如DcR1和DcR2被证明可通过DR4和DR5阻止TRAIL诱导的调亡。因此,这些诱骗受体代表了一种新型的对前调亡细胞因子在细胞表面的直接调节敏感性的机制。这些抑制性受体在正常组织内优先表达说明TRAIL可作为抑制抗癌因子诱导癌细胞的调亡,而对正常细胞起保护作用(Marsters等,1999)。
利用细胞毒性化疗药物进行肿瘤的治疗取得了很多成功,但是,化疗的后果之一就是药物耐药表型的出现/获得以及多药耐药现象的出现。耐药现象的出现依然是治疗这类肿瘤所遇到的主要问题,因此需要采用其他的方法,如基因治疗。
可与化疗、放疗或生物治疗联合应用的其他治疗措施包括温热疗法,该方法是将患者的组织置于高温下(可达106°F)。外部或内部发热装置可用于局部、区域或全身温热治疗。局部温热治疗是指只对一个小的区域加热,如肿瘤部位。外部加热通常是通过体外的装置发射高频微波靶向到肿瘤。内部加热是利用一个无菌的探针,如一个细的热金属丝或充满热水的中空细管、植入的微波天线或射频电极。
患者的器官或四肢可被加热以进行区域治疗,该疗法利用能产生高能量的装置进行,如磁体。另外,可以抽出患者的一部分血液,加热后再输注到能内部加热的区域。全身温热疗法在癌扩散到全身的情况下也可以使用。热水套、热蜡、感应线圈和热房可用于此目的。
激素疗法也可与本发明或上述其他癌症治疗措施联合使用。激素可被用于降低某些激素的表达水平或阻断某些激素的效应以治疗某些类型的肿瘤,如乳腺癌、前列腺癌、卵巢癌或宫颈癌。这种疗法通常与至少一种其他治疗措施联合使用,作为辅助治疗措施或降低转移的风险。
表6
癌基因



实施例
下面的实施例用于证明本发明的优选实施方案。本领域的熟练技术人员应该知道,下面实施例中所描述的技术代表了发明者发现的技术,它们在本发明的实践中发挥了很好的作用,因此被认为构成了实现本发明的优选实施模式。但是,本领域的那些熟练技术人员根据本说明书可以对本文所描述的特定实施方案作出修改,只要不脱离本发明的精神和范围一样可以得到类似的或相同的结果。
实施例1:
痘苗病毒在细胞系内的扩增
本实施例评价了含16种不同常规痘苗病毒实验室株/突变株(和兔痘病毒及其他痘病毒)的一组病毒:Copenhagen、Dairen、Evans、USSR、Tashkent、Tian Tan、WR、IHD-J、IHD-W、Lister、NYCBOH、Patwadangar、King和WR突变株B8R、B18R、B13R。评价了这些病毒株在癌细胞和正常细胞内的复制能力。优选的病毒应该是在癌细胞内相对高复制而在正常细胞内弱复制的病毒(即在肿瘤和正常细胞之间有较大的治疗比或指数)。共检测了两个人源肿瘤细胞系:A2780结肠癌细胞系和HCT116结肠癌细胞系(美国模式培养物保藏所)。正常细胞包括正常的人支气管上皮细胞(NHBE)。为了测定病毒的细胞致病效应,使增殖的细胞生长到70%汇合时(含2%FBS的DMEM)转染0.001到10感染单位(MOI)的病毒。5到6天后培养皿用MTT(Promega)染色,然后测定吸光度值。正常细胞在生长到完全汇合后使其停止增殖,随后用含0.2%FBS的DMEM培养。通过细胞周期分析和细胞计数来确定增殖是否停止。每个样品测定4次,试验至少重复两次。在病毒复制试验中,细胞生长(37℃、5%CO2、含2%FBS、NHBE细胞生长因子的DMEM,如Heise等,(2000)所描述)到70%汇合后用1或10感染单位(MOI,每个细胞感染的病毒量)的病毒感染。在培养基中孵育3小时后换液。48小时后(以前的数据表明痘苗病毒的复制在这个时间点达到峰值)收获细胞和上清用于病毒滴度分析。经3轮冷冻和融解后获得细胞裂解物,随后在超声/水浴中进行30秒的脉冲。然后通过蔗糖垫纯化病毒,上清液和裂解物的系列稀释物用BSC-1细胞测定病毒的滴度(纯化和滴度测定方法见Alcami和Smith(1995)的描述)。
典型的结果显示在图1-4中。痘苗病毒Copenhagen株在两个肿瘤细胞系内的复制水平都等于或超过本试验中的其他所有病毒(图1A和1B;y-轴代表噬菌斑形成单位/ml+/-S.E.)。相反,在正常人源细胞中Copenhagen株的毒力比其他病毒都弱(图2)。最后,所有病毒在癌细胞和正常细胞内爆发的比值显示在图3(A2780:NHBE)和图4(HCT116:NHBE)中。Copenhagen株在A2780(P<0.001)和HCT116(p<0.001)中的爆发比例都明显升高。Copenhagen株在两株细胞内的爆发比例分别约为20,000和30,000,而其他病毒的爆发比例都低于5000,最少的低于2000(如Lister和Wyeth株)。同样,体外细胞致病效应分析发现Copenhagen对A2780和HCT116的肿瘤细胞杀伤活性明显高于或等于Lister株和NYCBOH株。
实施例2:
痘苗病毒/紫杉醇联合治疗
虽然有些病毒如腺病毒和HSV已被用于和化疗药联用,但是痘苗病毒还没有进行过这样的试验(包括Copenhagen株)。痘苗病毒经改造后表达前药活化酶(如胸苷激酶),与相对无毒的前药联合应用时可通过前药活化基因产物活化前药而使其变成有毒性的药物(Puhlmann等,2000)。
被批准用于治疗癌症患者的标准细胞毒化疗药物与痘苗病毒之间的协同作用是痘病毒,包括痘苗病毒和特异性的Copenhagen株的一个优越特性。紫杉醇(a.k.a.紫杉醇)在美国和欧洲已被批准用于治疗癌症患者。痘苗病毒Copenhagen株与紫杉醇联合处理HCT116和LNCaP癌细胞系。等效线图解法作图和分析结果表明痘苗病毒和紫杉醇有协同效应(图5)。等效线图解结果得自MTS试验数据,如 Aqueous非放射性的细胞增殖分析(Catalog#G5421,Promega Corp.,Madison,WI),如图6、7、8和9中所显示的细胞存活率。如果没有协调效应,也没有拮抗作用,图5中所有的数据点都位于代表数据点预期位置的线以下,因此这些数据点说明了痘苗病毒和紫杉醇对这些癌细胞系杀伤作用的协同效应。
Aqueous非放射性的细胞增殖分析(Catalog#G5421,Promega Corp.,Madison,WI),如图6、7、8和9中所显示的细胞存活率。如果没有协调效应,也没有拮抗作用,图5中所有的数据点都位于代表数据点预期位置的线以下,因此这些数据点说明了痘苗病毒和紫杉醇对这些癌细胞系杀伤作用的协同效应。
本方法包括在96孔培养板中培养细胞系,培养条件为5%CO2、37°、含10%FCS的DMEM。细胞生长到约50%汇合时用含2%FCS的DMEM换液,然后用紫杉醇(剂量范围为3×10-8到3×104nM,对数增量)和/或病毒(VV Copenhagen株,MOI为每个细胞10-6到105病毒颗粒)处理。细胞分为四组,分别用1)空白处理,2)只用病毒处理,3)只用紫杉醇处理,4)病毒处理后再用紫杉醇处理。细胞共感染6天,然后进行MTS分析(Promega,WI,USA,参见厂家的说明书)。细胞用紫杉醇共处理6天,然后进行MTS分析(参见厂家的说明书)。进行联合处理的细胞感染病毒的方式与病毒单独处理组一样,然后立刻再用紫杉醇处理(固定比例的病毒∶紫杉醇)。然后按照上述方法分析细胞。MTS细胞存活率数据表示为对照细胞存活率的百分比,如图6、7、8和9中所示。协同效应评价的数据分析方法(等效线图解法作图和分析)见Nielsen等(1997、1998)的描述。简言之,制作剂量效应曲线,计算出每个细胞系(与未处理细胞比较)每个细胞(即,以及所有的试验条件)的EC50值。每种药物检测9个浓度(见上),单独使用或联合使用。每个试验包括VV/紫杉醇的4个不同稀释比例(3、33、333、3333)。制作等效线图以确定是否存在协调效应或拮抗作用。每个数据点代表3个重复的样品。
实施例3:
痘苗病毒注射导致的肿瘤抑制
在C57B/6小鼠腹侧皮下注射悬浮于100μl PBS中的5×105CMT-64细胞或106CMT-93细胞使其形成小鼠皮下肿瘤异种移植物。在第一组试验中,携带B8R和B18R突变的WR株痘苗病毒注射到具有免疫力的C57/B6小鼠腹侧皮下CMT-93肿瘤内(估计基线肿瘤大小为40-100μl),剂量为104到108病毒颗粒,悬浮于40μl液体中,分别于1、3和5天注射。在肿瘤的中心刺一个针孔,在肿瘤的四个象限中分别刺出一个针孔。当针头退出时约有1/4的液体进入到每个针孔内。对照组用补骨脂素-UV灭活的病毒和PBS处理,处理方式与治疗组一样。每两周测量一次肿瘤的大小,得到二维数据,肿瘤的体积用如下公式计算:(长)×(宽)×(宽)×(3.14/6)。
通过与注射溶剂和灭活病毒的对照组肿瘤相比可以发现治疗组出现了明显的抗肿瘤效应,生存期延长,并且可以观察到剂量-效应关系。10只用108和1010B8R突变VV处理的小鼠中有8只的肿瘤被完全抑制,在整个试验过程中处于无瘤状态(共3个月)。10只接收相同剂量B18R注射的小鼠中有9只的肿瘤被完全抑制,在同样的试验周期内处于无瘤状态。而在对照组中只有1只小鼠的肿瘤被完全抑制。平均生存期为2周,处理后24天处死所有的小鼠。108到1010颗粒治疗组的小鼠生存期明显延长(KM生存期分析;时序检验;p<0.01)。观察动物的总体状况(如活动情况,皮毛起皱情况),每周称重2次;结果未发现明显的体重变化或总体状况改变。
然后在CMT-64鼠肿瘤异种移植模型上利用B18R和对照溶剂实施相同的处理/注射方案。虽然没有观察到完全的反应,但是由于肿瘤生长而需要处死动物所需的延迟时间,在B18R处理组和补骨脂素-UV灭活病毒组之间有显著差异(平均值约为2周对4周;p<0.05,时序检验,Kaplan-Meier分析由于肿瘤生长而需要处死动物的时间)。
实施例4:
有关鼠肿瘤异种移植物的EEV-增强效应
在具有免疫力的BALB/c小鼠腹侧皮下注射悬浮于100μl PBS中的5×105JC鼠乳腺癌细胞使其形成小鼠皮下肿瘤异种移植物。肿瘤一旦达到可注射的尺寸(基线肿瘤大小40-100μl),就将Western Reserve(无IHD-J突变的WR)VV、IHD-J突变的WR(A34R/K151D突变)病毒或PBS注射到皮下JC肿瘤内。病毒剂量为每天1010病毒颗粒,悬浮于40μl PBS中,分别于1、3和5天注射(每个治疗组含8只小鼠)。PBS对照组用相同的方式处理。在肿瘤的中心刺一个针孔,在肿瘤的四个象限中分别刺出一个针孔。当针头退出时约有1/4的液体进入到每个针孔内。对照组用PBS处理,处理方式与治疗组一样。每两周测量一次肿瘤的大小,得到二维数据,肿瘤的体积用如下公式计算:(长)×(宽)×(宽)×(3.14/6)。
与PBS处理组和Western Reserve处理组相比,IHD-J处理组出现了明显的抗肿瘤效应(肿瘤生长延迟和肿瘤缩小),由于肿瘤生长而需要处死动物的时间(即生存期)明显延长。根据Kaplan-Meier生存期分析,通过时序检验(用于比较存活曲线)发现,IHD-J组由于肿瘤生长而需要处死动物的时间(生存期)比PBS(p<0.05)和WR(p<0.05)组明显延长。两个对照组的平均存活时间为2周,而IHD-J组的平均存活时间为4.5周。治疗组内的所有小鼠都未出现明显的系统毒性反应,也没有动物因处理而死亡。IHD-J突变可显著提高抗肿瘤效应(与无IHD-J突变的WR病毒相比),并且毒性效应没有升高。肿瘤可在皮下生长,长到直径2-6mm时可开始处理。
实施例5:
干扰素-结合基因的缺失及对IFN耐受性癌细胞的特异性相对于正常细胞有提高
利用试验来分析缺失干扰素(IFN)结合基因的痘病毒与野生型对照痘病毒相比是否具有更高的肿瘤特异性,在正常细胞内的复制能力是否减弱。比较WR(WesternReserve)和B18R基因(B18R)缺失的WR突变株在存在或缺少IFN-α(1,000单位/ml,体外感染后5小时加入)的情况下的复制情况(图10)。试验细胞为正常人支气管上皮细胞(NHBE,Clonetics Corp.,USA)、C33A人宫颈癌细胞(ATCC)和HCT116人结肠癌细胞(ATCC)。在预试验中,这些细胞在病毒感染前用IFN(5,000单位/ml)预处理24小时以确定其对干扰素抗病毒复制效应的敏感性。和预想的一样,两种病毒对干扰素预处理抑制效应的敏感性是一样的。NHBE和HCT116细胞是干扰素敏感性的,因为IFN预处理后病毒的复制明显减少;C33A细胞是干扰素耐受性的,因为IFN预处理对病毒的复制没有明显影响。
癌细胞用含10%FCS的DMEM培养;正常细胞用厂商说明书描述的含血清添加物的DMEM培养。当细胞生长到70%汇合时,每个细胞用10病毒感染单位(moi)的病毒感染。感染后5小时,IFN或者以1,000单位/ml的浓度添加到培养基中(添加IFN的细胞),或者不添加IFN(不添加IFN的细胞)。感染后48小时收获细胞及其裂解物,然后分离痘苗病毒病测定病毒的滴度(噬菌斑形成单位的确定)(用BS-C-1细胞测定滴度,如Tscharke等,2002所描述,本文已纳入作为参考)。如图10所示,在IFN存在的情况下,含有B18R突变的痘苗病毒在两株IFN敏感细胞,包括NHBE,内的复制被明显抑制(p<0.01,斯氏t-检验,与不含IFN的相比)。相反,这个突变病毒在IFN耐受C33A癌细胞内的复制不被抑制。正如所预料的,野生型WR不受IFN处理的影响,因为该病毒能产生功能性B18R基因产物(P=0.8)。利用其他的IFN耐受癌细胞系可得到相似的结果。因此,与野生型病毒相比,IFN结合基因B18R的缺失可导致治疗指数的增加,保护正常细胞不受损伤。
在鼠肿瘤异种移植模型上也证明了B8R和B18R上的IFN结合突变可产生抗肿瘤效应。由B18R和/或B8R缺失导致的相对安全性的提高在小鼠体内是被低估的,因为鼠的靶IFN分子的亲和力显著降低(Symons等,2002)。其他一些试验是在兔或非人灵长类动物体内进行的,注射方式为真皮内注射,方法见Symons等的描述,所得到的结果与文献报道的兔试验结果一致(与野生型对照病毒相比,B8R突变的痘苗病毒可增加炎症细胞的积聚,从而可以加速病毒从皮肤损伤部位的清除)。因此,抗肿瘤效应和从正常细胞加速病毒清除的效应在体外和体内都得到证实(Symons等)。
实施例6:
预演性实施例显示TNF调节功能丧失所引起的效应
含有和不含有一个或多个细胞因子或趋化因子抑制基因缺失/灭活的病毒对(如野生型WR及其B29R/vCKBP缺失的突变株)用于感染具有免疫力的小鼠(C57B/6或BALB/c)体内的鼠肿瘤(如CMT-93、CMT-64或JC)。这种比较只在病毒基因产物抑制鼠细胞因子/趋化因子的程度与抑制人细胞因子/趋化因子的程度一样时才有效。静脉注射和/或瘤内注射病毒(病毒颗粒为105到1010,注射1到6次)后检测肿瘤组织和正常组织内病毒复制的情况(不同时间点得到的噬菌斑形成单位)、CC趋化因子水平(如免疫组化染色或ELISA分析)、细胞因子水平(如免疫组化(IHC)染色或ELIS分析)以及炎症细胞浸润的情况(H和E或IHC染色)。另外,还要测定两种病毒的抗肿瘤效应(Kaplan-Meier肿瘤抑制/存活曲线)和毒性(体重下降情况;血液学指标;血清生化指标)。结果将是突变病毒1)抗肿瘤效应提高,肿瘤内的炎症诱导反应加强(如免疫效应细胞增多);在正常组织内的复制能力和毒性与野生型病毒一样或降低(如肝、脾、肺和/或脑);3)对肿瘤特异性的细胞免疫反应的诱导增强或维持原水平。最后,化疗和/或放疗联合突变病毒比联合野生型病毒所达到的抗肿瘤效应有望得到提高。还可以在兔或非人灵长类动物体内进行毒性试验以进一步确定本发明病毒的特征(Tscharke等,2002,本文已纳入作为参考)。
实施例7:
预演性实施例显示IFN调节功能丧失所引起的效应
含有和不含有一个或多个干扰素结合多肽缺失/灭活的病毒对(如野生型WR痘苗病毒及其B18R缺失的突变株)用于感染具有免疫力的小鼠体内的肿瘤,如上述实施例所讨论的,其靶IFN分子可被痘苗病毒基因产物有效结合。需要指出的是,由于鼠干扰素与人干扰素相比对VV多肽具有一定的耐受性,因此在小鼠体内得到的效果可能比人体内的效果弱。如上所述,静脉注射和/或瘤内注射病毒后检测肿瘤组织和正常组织内的病毒复制和扩散情况。另外,还要测定两种病毒的抗肿瘤效应和毒性。预期结果是突变病毒在正常组织内的复制能力和毒性降低(如肝、脾、肺和/或脑),但是在肿瘤内依然可以复制并诱导肿瘤坏死。野生型痘苗病毒(如WR)在正常组织和肿瘤组织内的复制/诱导坏死的能力差异较小。另外,与盐水处理的对照肿瘤和野生型病毒处理的对照肿瘤相比,用突变病毒处理的肿瘤血管形成能力下降(如在等量的血管标记物上通过H和E染色,CD31的免疫组化染色)。最后,化疗和/或放疗联合突变病毒比联合野生型病毒所达到的免疫介导的抗肿瘤效应和疗效有望得到提高。还可以在兔或非人灵长类动物体内进行毒性试验以进一步确定本发明病毒的特征(Tscharke等,2002,本文已纳入作为参考)。
实施例8:
预演性实施例显示丝氨酸蛋白酶抑制因子功能丧失所引起的效应
如上所述,含有和不含有抗调亡基因缺失/灭活的病毒对用于感染具有免疫力的小鼠体内的肿瘤。病毒包括而不限于表达或不表达B13R(SPI-2)的痘苗病毒。如上面所描述,静脉注射病毒后检测肿瘤组织和正常组织内病毒的复制和扩散情况。另外,在瘤内和/或静脉注射病毒后按照上述方法评价突变病毒的抗肿瘤效应和毒性。预期结果是突变病毒在正常组织内的复制能力和毒性下降,和/或在正常组织内被清除的速度快于肿瘤组织。野生型病毒在正常组织和肿瘤组织内的复制能力和毒性的差异(如果有)较小。瘤内、腹腔内、静脉或其他途径注射病毒以后,突变病毒的抗肿瘤效应有望能达到或超过野生型病毒的抗肿瘤效应。联合化疗研究用的是同一个模型系统。小鼠分别接受对照药(安慰剂)、单独的化疗药、单独的病毒(野生型或突变型)或病毒加化疗药处理。突变病毒加化疗药的疗效有望高于单独的化疗药和野生型病毒加化疗药的疗效。病毒联合放疗也可能得到同样的结果。
实施例9:
预演性实施例显示补体调节功能丧失所引起的效应
如上所述,含有和不含有VCP基因缺失/灭活的病毒对用于感染具有免疫力的小鼠体内的肿瘤。如上面所描述,静脉或其他途径注射病毒后检测肿瘤组织和正常组织内病毒的复制和扩散情况。另外,在瘤内和/或静脉注射病毒后按照上述方法评价突变病毒的抗肿瘤效应和毒性。预期结果是突变病毒在正常组织内的复制能力和毒性下降,和/或在正常组织内被清除的效率高于肿瘤组织。野生型病毒在正常组织和肿瘤组织内的复制能力和毒性的差异(如果有)较小。瘤内、腹腔内、静脉或其他途径注射病毒以后,突变病毒的抗肿瘤效应有望能达到或超过野生型病毒的抗肿瘤效应。联合化疗研究用的是同一个模型系统。例如,小鼠分别接受对照药(安慰剂)、单独的化疗药、单独的病毒(野生型或突变型)或病毒加化疗药处理。突变病毒加化疗药的疗效有望高于单独的化疗药和野生型病毒加化疗药的疗效。肿瘤靶向性的单克隆抗体联合病毒也可能得到同样的结果。
*********
根据本说明书,本说明书和权利要求书所描述的所有组合物和方法无需特别的试验就可以制备和实施。本发明的组合物和方法是利用优选实施方案来描述的,但是,本领域的技术人员在不背离本发明的概念、精神和范围的情况下,可以对本发明的组合物和/或方法以及本文所描述的方法的步骤或步骤的顺序作出改动。更具体地说,本文所描述的因子可用某些化学和物理相关的因子代替,但是可以得到相同或相似的结果。对于本领域技术人员来说很明显的是,所有这种相似的替代和修改都被认为包含在权利要求所定义的本发明的精神、范围和概念之内。
参考文献
下面的文献都已特地纳入本文作为参考。
美国专利4,554,101
美国专利4,683,195
美国专利4,683,202
美国专利4,684,611
美国专利4,800,159
美国专利4,879,236
美国专利4,883,750
美国专利4,946,773
美国专利4,952,500
美国专利5,220,007
美国专利5,279,721
美国专利5,284,760
美国专利5,302,523
美国专利5,322,783
美国专利5,354,670
美国专利5,366,878
美国专利5,384,253
美国专利5,389,514
美国专利5,399,363
美国专利5,464,765
美国专利5,466,468
美国专利5,538,877
美国专利5,538,880
美国专利5,543,158
美国专利5,550,318
美国专利5,563,055
美国专利5,580,859
美国专利5,589,466
美国专利5,591,616
美国专利5,610,042
美国专利5,633,016
美国专利5,635,377
美国专利5,641,515
美国专利5,656,610
美国专利5,702,932
美国专利5,736,524
美国专利5,739,169
美国专利5,780,448
美国专利5,789,166
美国专利5,789,215
美国专利5,798,208
美国专利5,798,339
美国专利5,801,005
美国专利5,824,311
美国专利5,824,348
美国专利5,830,650
美国专利5,830,880
美国专利5,840,873
美国专利5,843,640
美国专利5,843,650
美国专利5,843,651
美国专利5,843,663
美国专利5,846,225
美国专利5,846,233
美国专利5,846,708
美国专利5,846,709
美国专利5,846,717
美国专利5,846,726
美国专利5,846,729
美国专利5,846,783
美国专利5,846,945
美国专利5,849,481
美国专利5,849,483
美国专利5,849,486
美国专利5,849,487
美国专利5,849,497
美国专利5,849,546
美国专利5,849,547
美国专利5,851,770
美国专利5,851,772
美国专利5,853,990
美国专利5,853,992
美国专利5,853,993
美国专利5,856,092
美国专利5,858,652
美国专利5,861,244
美国专利5,863,732
美国专利5,863,753
美国专利5,866,331
美国专利5,866,337
美国专利5,866,366
美国专利5,871,740
美国专利5,871,986
美国专利5,882,864
美国专利5,900,481
美国专利5,905,024
美国专利5,910,407
美国专利5,912,124
美国专利5,912,145
美国专利5,912,148
美国专利5,916,776
美国专利5,916,779
美国专利5,919,626
美国专利5,919,630
美国专利5,922,574
美国专利5,925,517
美国专利5,925,525
美国专利5,925,565
美国专利5,928,862
美国专利5,928,869
美国专利5,928,870
美国专利5,928,905
美国专利5,928,906
美国专利5,928,906
美国专利5,929,227
美国专利5,932,413
美国专利5,932,451
美国专利5,935,791
美国专利5,935,819
美国专利5,935,825
美国专利5,939,291
美国专利5,942,391
美国专利5,945,100
美国专利5,981,274
美国专利5,994,624
Alcami和Smith,Cell.,71(1):153-67,1992.
Alcami等,Sem.Virol.,5:419-427,1998.
Alcami等,Virology,74(23):11230-9,2000.
Almendro等,J.Immunol.,157(12):5411-5421,1996.
Andoh等,Cancer Immunol.Immunother.,50(12):663-72,2002.
Angel等,Mol.Cell.Biol.,7:2256,1987.
Angel等,Cell,49:729,1987b.
Angel等,Mol.Cell.Biol.,7:2256,1987a.
Arap等,Cancer Res.,55(6):1351-1354,1995.
Atchison和Perry,Cell,46:253,1986.
Atchison和Perry,Cell,48:121,1987.
Austin-Ward和Villaseca,Rev.Med.Chil.,126(7):838-45,1998.
Ausubel等,Current Protocols in Molecular Biology,John,Wiley & Sons,Inc,New York,1994.
Bajorin等,J.Clin.Oncol.,6(5):786-92,1988.
Bakhshi等,Cell.,41(3):899-906,1985.
Banerji等,Cell.,27(2Pt 1):299-308,1981.
Banerji等,Cell.,33(3):729-740,1983.
Bellus,J.Macromol.Sci.Pure Appl.Chem.,A31(1):1355-1376,1994.
Berkhout等,Cell,59:273-282,1989.
Blanar等,EMBO J.,8:1139,1989.
Blasco和Moss,J.Virology,66(7):4170-4179,1992.
Blasco等,J.Virology,67(6):3319-3325,1993.
Bodine和Ley,EMBO J.,6:2997,1987.
Boshart等,Cell,41:521,1985.
Bosze等,EMBO J.,5(7):1615-1623,1986.
Boyd等,Cell.,79:341-351,1994.
Braddock等,Cell,58:269,1989.
Braisted和Wells,Proc.Natl.Acad. Sci.USA,93(12):5688-5692,1996.
Brizel,Semin.Radiat.Oncol.,8(4):237-246,1998.
Bukowski等,Clin.Cancer Res.,4(10):2337-47,1998.
Bulla和Siddiqui,J.Virol.,62:1437,1986.
Burton和Barbas,Adv.Immunol.,57:191-280,1994.
Caldas等,Nat.Genet.,8(1):27-32,1994.
Campbell和Villarreal,Mol.Cell.Biol.,8:1993,1988.
Campere和Tilghman,Genes and Dev.,3:537,1989.
Campo等,Nature,303:77,1983.
Caragine等,Cancer Res.,62(4):1110-5,2002.
Carbonelli等,FEMS Microbiol.Lett.,177(1):75-82,1999.
Celander和Haseltine,J.Virology,61:269,1987.
Celander等,J.Virology,62:1314,1988.
Chandler等,Cell,33:489,1983.
Chandler等,Proc.Natl.Acad.Sci.USA,94(8):3596-601,1997.
Chang等,Mol.Cell.Biol.,9:2153,1989.
Chatterjee等,Proc Natl.Acad Sci.U.S.A.,86:9114,1989.
Chen和Okayama,Mol.Cell Biol.,7(8):2745-2752,1987.
Cheng等,Cancer Res.,54(21):5547-5551,1994.
Choi等,Cell,53:519,1988.
Christodoulides等,Microbiology,144(Pt 11):3027-37,1998.
Cleary和Sklar,Proc.Natl.Acad.Sci.USA,(21):7439-7443,1985.
Cleary等,J.Exp.Med.,164(1):315-320,1986.
Cocea,Biotechniques,23(5):814-816,1997.
Cohen等,J.Cell.Physiol.,5:75,1987.
Colamonici等,J.Biol.Chem.,270:15974-15978,1995.
Cooley等,Science,239(4844):1121-1128,1988.
Costa等,Mol.Cell.Biol.,8:81,1988.
Cripe等,EMBO J.,6:3745,1987.
Culotta和Hamer,Mol.Cell.Biol.,9:1376,1989.
Culver等,Science,256(5063):1550-1552,1992.
Cunningham和Wells,Science,244(4908):1081-1085,1989
Curran,Semin.Radiat.Oncol.,8(4Suppl 1):2-4,1998.
Dandolo等,J.Virology,47:55-64,1983.
Davidson等,J.Immunother.,21(5):389-98,1998.
De Villiers等,Nature,312(5991):242-246,1984.
Deschamps等,Science,230:1174-1177,1985.
Dillman,Cancer Biother.Radiopharm.,14(1):5-10,1999.
Dobbelstein和Shenk,J.Virology,70:6479-6485,1996.
Durrant和Spendlove,Curr.opin.Investig.Drugs,2(7):959-66,2001.
Edbrooke等,Mol.Cell.Biol.,9:1908,1989.
Edlund等,Science,230:912-916,1985.
Eliopoulos等,Oncogene,11(7):1217-28,1995.
el-Kareh和Secomb,Crit.Rev.Biomed.Eng.,25(6):503-571,1997.
Erlandsson,Cancer Genet.Cytogenet.,104(1):1-18,1998.
欧洲专利申请320308
欧洲专利申请329822
Feng和Holland,Nature,334:6178,1988.
Firak和Subramanian,Mol.Cell.Biol.,6:3667,1986.
Foecking和Hofstetter,Gene,45(1):101-105,1986.
Fraley等,Proc.Natl.Acad.Sci.USA,76:3348-3352,1979.
Frohman,PCR Protocols:A Guide To Methods And Applications,AcademicPress,N.Y.,1990.
Fujita等,Cell,49:357,1987.
GB申请2202328
Genbank登录号NC_001559
Gertig等,Semin.Cancer Biol.,8(4):285-98,1998.
Gilles等,Cell,33:717,1983.
Gloss等,EMBO J.,6:3735,1987.
Gnant等,Cancer Res.,59(14):3396-403,1999.
Godbout等,Mol.Cell.Biol.,8:1169,1988.
Goebel等,Virology,179(1):247-66和517-63,1990.
Goodbourn和Maniatis,Proc.Natl.Acad.Sci.USA,85:1447,1988.
Goodbourn等,Cell,45:601,1986.
Gopal,Mol.Cell Biol.,5:1188-1190,1985.
Graham和Van Der Eb,Virology,52:456-467,1973.
Graham等,Virology,229(1):12-24,1997.
Greene等,Immunology Today,10:272,1989
Gross等,Genes Dev.,13(15):1899-911,1999.
Grosschedl和Baltimore,Cell,41:885,1985.
Hanibuchi等,Int.J.Cancer,78(4):480-5,1998.
Harland和Weintraub,J.Cell Biol.,101:1094-1099,1985.
Haslinger和Karin,Proc.Natl.Acad.Sci.USA,82:8572,1985.
Hauber和Cullen,J.Virology,62:673,1988.
Heise等,Cancer Gene Ther.,6(6):499-504,1999.
Hellstrand等,Acta Oncol.,37(4):347-353,1998.
Hen等,Nature,321:249,1986.
Hensel等,Lymphokine Res.,8:347,1989.
Hermiston,J.Clin.lnvest.,105:1169-1172,2000.
Herr和Clarke,Cell.,45:461,1986.
Hilton等,J.Biol.Chem.,271(9):4699-4708,1996.
Hirochika等,J.Virolology,61:2599,1987.
Hirsch等,Mol.Cell.Biol.,10:1959,1990.
Ho等,Environ Health Perspect,106(5):1219-1228,1998.
Holbrook等,Virology,157:211,1987.
Homey等,Nature.Rev.Immunol.,2:175-184,2002.
Horlick和Benfield,Mol.Cell.Biol.,9:2396,1989.
Huang等,Cell.,27:245,1981.
Hug等,Mol.Cell.Biol.,8:3065,1988.
Hui和Hashimoto,Infect.Immun.,66(11):5329-36,1998.
Hussussian等,Nat.Genet.,8(1):15-21,1994.
Hwang等,Mol.Cell.Biol.,10:585,1990.
Ikeda等,Nat.Med.,5(8):881-7,1999.
Imagawa等,Cell,51:251,1987.
Imbra和Karin,Nature,323:555,1986.
Imler等,Mol.Cell.Biol,7:2558,1987.
Imperiale和Nevins,Mol.Cell.Biol.,4:875,1984.
Innis等,Proc.Natl.Acad.Sci.USA,85(24):9436-9440,1988.
Inouye和Inouye,Nucleic Acids Res.,13:3101-3109,1985.
Irie和Morton,Proc.Natl.Acad.Sci.USA,83(22):8694-8698,1986.
Irie等,Lancet..,1(8641):786-787,1989.
Isaacs等,Proc.Natl.Acad.Sci.USA,89(2):628-32,1992.
Jakobovits等,Mol.Cell.Biol.,8:2555,1988.
Jameel和Siddiqui,Mol.Cell.Biol.,6:710,1986.
Jaynes等,Mol.Cell.Biol.,8:62,1988.
Johnson和Hamdy,Oncol.Rep.,5(3):553-7,1998.
Johnson等,Mol.Cell.Biol.,9:3393,1989.
Ju等,J.Neuropathol.Exp.Neurol.,59(3):241-50,2000.
Kadesch和Berg,Mol.Cell.Biol.,6:2593,1986.
Kaeppler等,Plant Cell Reports,9:415-418,1990.
Kamb等,Nat.Genet.,8(1):23-2,1994.
Kaneda等,Science,243:375-378,1989.
Karin等,Mol.Cell.Biol.,7:606,1987.
Karin等,Mol.Cell.Biol.,7:606,1987.
Katinka等,Cell,20:393,1980.
Kato et al,J.Biol.Chem.,266:3361-3364,1991.
Kawamoto等,Mol.Cell.Biol.,8:267,1988.
Kay等,Proc.Natl.Acad.Sci.USA,94(9):4686-91,1997.
Kerr等,Br.J.Cancer,26(4):239-257,1972.
Kettle等,J.Gen.Virology,78:677-685,1997.
Kiledjian等,Mol.Cell.Biol.,8:145,1988.
Kirn等,Nat.Med.,7(7):781-787,2001.
Klamut等,Mol.Cell.Biol.,10:193,1990.
Koch等,Mol.Cell.Biol.,9:303,1989.
Kolmel,J.Neurooncol.,38(2-3):121-5,1998.
Koncz等,EMBO J.,9(5):1337-1346,1990.
Kraus等,FEBS Lett.,428(3):165-170,1998.
Kriegler和Botchan,Eukaryotic Viral Vectors,Gluzman(ed),Cold SpringHarbor:Cold Spring Harbor Laboratory,NY,1982.
Kriegler和Botchan,Mol.Cell.Biol.,3:325,1983.
Kriegler等,Cell,38:483,1984.
Kriegler等,Cell,53:45,1988.
Kuhl等,Cell,50:1057,1987.
Kunz等,Nucl.Acids Res.,17:1121,1989.
Kwoh等,Proc.Nat.Acad.Sci.USA,86:1173,1989.
Kyte和Doolittle,J.Mol.Biol.,57(1):105-32,1982.
Lareyre等,J.Biol.Chem.,274(12):8282-8290,1999.
Larsen等,Proc Natl.Acad.Sci.USA.,83:8283,1986.
Laspia等,Cell,59:283,1989.
Latimer等,Mol.Cell.Biol.,10:760,1990.
Lee等,DNA Cell.Biol.,16(11):1267-75,1997.
Lee等,Nature,294:228,1981.
Lee等,Nucleic Acids Res.,12:4191-206,1984.
Levenson等,Hum Gene Ther.20;9(8):1233-1236,1998.
Levinson等,Nature,295:79,1982.
Liebermann,Oncogene,17(10):1189-94,1998.
Lin等,Mol.Cell.Biol.,10:850,1990.
Luria等,EMBO J.,6:3307,1987.
Lusky和Botchan,Proc.Natl.Acad.Sci.USA,83:3609,1986.
Lusky等,Mol.Cell.Biol.3:1108,1983.
Macejak和Sarnow,Nature,353:90-94,1991.
Magi-Galluzzi等,Anal.Quant.Cytol.Histol.,20(5):343-50,1998.
Majors和Varmus,Proc.Natl.Acad.Sci.USA,80:5866,1983.
Mangray和King,Front Biosci.,3:D1148-60,1998.
Marks等,Symp.Soc.Exp.Biol.,45:77-87,1991.
Marsters等,Recent Prog Horm Res,54:225-234,1999.
Mayer等,Radiat.Oncol.Investig,6(6):281-288,1998.
McCart等,Gene Ther.,7(14):1217-23,2000.
McNeall等,Gene,76:81,1989.
Miksicek等,Cell,46:203,1986.
Mitchell等,Ann.NY Acad.Sci.,690:153-166,1993.
Mitchell等,J.Clin.Oncol.,8(5):856-869,1990.
Mordacq和Linzer,Genes and Dev.,3:760,1989.
Moreau等,Nucl.Acids Res.,9:6047,1981.
Mori等,Cancer Res.,54(13):3396-3397,1994.
Morton等,Arch.Surg.,127:392-399,1992.
Moss,Fields Virology,Fields(ed.),Lippincott-Raven Publ,Phila.,3:3637,2672,1996.
Moss,Fields Virology,Fields(ed.),Lippincott-Raven Publ,Phila.,3:3637-2672,1996.
Mossman等,Virology,215(1):17-30,1996.
Mougin等,Ann.Biol.Clin.,(Paris)56(1):21-8,1998.
Muesing等,Cell,48:691,1987.
Mumby和Walter,Cell Regul.,2(8):589-98,1991.
Natoli等,Biochem.Pharmacol.,56(8):915-20,1998.
Ng等,Nuc.Acids Res.,17:601,1989.
Nicolau和Sene,Biochim.Biophys.Acta,721:185-190,1982.
Nicolau等,Methods Enzymol.,149:157-176,1987.
Nielsen等,Cancer Gene 7herapy,4(6):S12,1997.
Nielsen等,Clin.Cancer Res.,4(4):835-846,1998.
Nobori等,Nature,368(6473):753-6,1994.
Nomoto等,Gene,236(2):259-71,1999.
Ochi等,Am.J.Gastroenterol.,93(8):1366-8,1998.
Ochi等,Am.J.Gastroenterol.,93(8):1366-1368,1998.
Ohara等,Proc.Natl.Acad.Sci.USA,86:5673-5677,1989.
Ohara,Gan To Kagaku Ryoho,25(6):823-8,1998.
Okamoto等,Proc.Natl.Acad.Sci.USA,1(23):11045-11049,1994.
Omirulleh等,Plant Mol.Biol.,21(3):415-428,1993.
Ondek等,EMBO J.,6:1017,1987.
Orlow等,Cancer Res.,54(11):2848-2851,1994.
Ornitz等,Mol.Cell.Biol.,7:3466,1987.
Palmiter等,Nature,300:611,1982.
PCT申请PCT/US87/00880
PCT申请PCT/US89/01025
PCT申请WO 88/10315
PCT申请WO 89/06700
PCT申请WO 90/07641
PCT申请WO 94/09699
PCT申请WO 95/06128
Pech等,Mol.Cell.Biol.,9:396,1989.
Pelletier和Sonenberg,Nature,334(6180):320-325,1988.
Perez-Stable和Constantini,Mol.Cell.Biol.,10:1116,1990.
Picard和Schaffner,Nature,307:83,1984.
Pietras等,Oncogene,17(17):2235-49,1998.
Pietras等,Oncogene,17(17):2235-49,1998.
Pinkert等,Genes and Dev.,1:268,1987.
Ponta等,Proc.Natl.Acad.Sci.USA,82:1020,1985.
Porton等,Mol.Cell.Biol.,10:1076,1990.
Potrykus等,Mol.Gen.Genet.,199:183-188,1985.
Puhlmann,等,Cancer Gene Ther.,7(1):66-73,2000.
Qin等,Proc.Natl.Acad.Sci.USA,95(24):14411-14416,1998.
Queen和Baltimore,Cell,35:741,1983.
Quinn等,Mol.Cell.Biol.,9:4713,1989.
Ravindranath和Morton,Intern.Rev.Immunol.,7:303-329,1991.
Redondo等,Science,247:1225,1990.
Reisman和Rotter,Mol.Cell.Biol.,9:3571,1989.
Remington’s Pharmaceutical Sciences第15版,第1035-1038和1570-1580页
小Resendez等,Mol.Cell.Biol.,8:4579,1988.
Ripe等,Mol.Cell.Biol.,9:2224,1989.
Rippe等,Mol.Cell.Biol.,10:689-695,1990.
Rittling等,Nuc.Acids Res.,17:1619,1989.
Rosel等,J.Viroiogy,60(2):436-449,1986.
Rosen等,Cell,41:813,1988.
Rosenberg等,Ann.Surg.210(4):474-548,1989.
Rosenberg等,N.Engl.J.Med.,319:1676,1988.
Sakai等,Genes and Dev.,2:1144,1988.
Sambrook等,In Molecular Cloning:A Laboratory Manual,第二版,ColdSpring Harbor Laboratory,Cold Spring Harbor,New York,1989.
Saraiva和Alcami,J.Virology,75(1):226-33,2001.
Satake等,J.Virology,62:970,1988.
Schaffner等,J.Mol.Biol.,201:81,1988.
Searle等,Mol.Cell.Biol.,5:1480,1985.
Seet等,Proc.Natl.Acad.Sci.USA,98(16):9008-13,2001.
Serrano等,Nature,366:704-707,1993.
Serrano等,Science,267(5195):249-252,1995.
Sharp和Marciniak,Cell,59:229,1989.
Shaul和Ben-Levy,EMBO J.,6:1913,1987.
Sherman等,Mol.Cell.Biol.,9:50,1989.
Sinkovics和Horvath,J.Clin.Viro.,16:1-15,2000.
Sleigh和Lockett,J.EMBO,4:3831,1985.
Smith和Vanderplasschen,Adv.Exp.Med.Biol.,440:395-414,1998.
Smith等,Immunol.Rev.,159:137-154,1997.
Solyanik等,Cell.Prolif.,28(5):263-78,1995.
Sommer等EMBO J.,9(3):605-613,1990.
Spalholz等,Cell,42:183,1985.
Spandau和Lee,J.Virology,62:427,1988.
Spandidos和Wilkie,EMBO J.,2:1193,1983.
Spriggs等,Cell,71(1):145-52,1992.
Stephens和Hentschel,Biochem.J.,248:1,1987.
Stokke等,Gell.Prolif.,30(5):197-218,1997.
Stuart等,Nature,317:828,1985.
Sullivan和Peterlin,Mol.Cell.Biol.,7:3315,1987.
Swartzendruber和Lehman,J.Cell.Physiology,85:179,1975.
Symons等,Cell,81:551-560,1995.
Takebe等,Mol.Cell.Biol.,8:466,1988.
Tavernier等,Nature,301:634,1983.
Taylor和Kingston,Mol.Cell.Biol.,10:165,1990a.
Taylor和Kingston,Mol.Cell.Biol.,10:176,1990b.
Taylor等,J.Biol.Chem.,264:15160,1989.
Thiesen等,J.Virology,62:614,1988.
Todo等,Cancer Res.,61:153-161,2001.
Treisman,Cell,42:889,1985.
Tronche等,Mol.Biol.Med.,7:173,1990.
Trudel和Constantini,Genes and Dev.6:954,1987.
Tsujimoto和Croce,等,Proc.Natl.Acad.Sci.USA,.83(14):5214-5218,1986.
Tsujimoto等,Science,228(4706):1440-1443,1985.
Tsumaki等,J.Biol.Chem,273(36):22861-4,1998.
Tyndell等,Nuc.Acids.Res.,9:6231,1981.
Upton等,Virology,184(1):370-82,1991.
Vanderplasschen等,Proc.Natl.Acad.Sci.USA,95(13):7544-9,1998.
Vannice和Levinson,J.Virology,62:1305,1988.
Vasseur等,Proc Natl.Acad.Sci.USA.,77:1068,1980.
Vicari和Caus,Cytokine Growth Factor Rev.,13:143-154,2002.
Vogelstein和Kinzler,Cell,70(4):523-6,1992.
Walker等,Proc.Natl.Acad.Sci.USA,89:392-3961992.
Wallach等,The cytokine network and immune functions,Theze(ed.),Oxford Univ.Press,Oxford,UK,51-84,1999.
Wang和Calame,Cell,47:241,1986.
Warren等,Biochemistry,35(27):8855-8862,1996.
Weber等,Cell,36:983,1984.
Weinberger等Mol.Cell.Biol.,8:988,1984.
Winoto和Baltimore,Cell 59:649,1989.
Wold等,Trendds Microbiol.,2:437-443,1994.
Wong等,Gene,10:87-94,1980.
Wu和Wu,J.Biol.Chem,262:4429-4432,1987.
Yelton等,J.Immunol.,155(4):1994-2004,1995.
Yutzey等Mol.Cell.Biol.,9:1397,1989.
Zeng等,Biochemistry,35(40):13157-13164,1996.
Zhao-Emonet等,Biochim.Biophys.Acta,1142(2-3):109-119,1998.
Claims (15)
1.具有A34R基因内或B5R基因内突变的痘苗病毒用于制造治疗癌症的药物的用途,所述痘苗病毒能使感染性EEV形式的痘苗病毒产量增加。
2.如权利要求1所述的用途,所述癌症是精微的残存癌症。
3.如权利要求1所述的用途,所述突变在A34R多肽内造成K151D取代。
4.如权利要求1、2或3所述的用途,所述痘苗病毒是Copenhagen株或WesternReserve株。
5.如权利要求1所述的用途,所述痘苗病毒是减毒病毒。
6.如权利要求5所述的用途,所述痘苗病毒具有B8R基因内或B18R基因内的灭活突变。
7.如权利要求1所述的用途,将所述药物配制成瘤内给药或静脉内给药的制剂。
8.如权利要求7所述的用途,将所述药物配制成向实体瘤内给药的制剂。
9.如权利要求1所述的用途,所述癌是膀胱、血、骨、骨髓、脑、乳腺、结肠、食管、胃肠道、头、肾、肝、肺、鼻咽、颈、卵巢、胰腺、前列腺、皮肤、胃、睾丸、舌或子宫癌。
10.如权利要求5所述的用途,所述减毒痘苗病毒还含有编码异源治疗性多肽的核酸。
11.如权利要求10所述的用途,所述异源治疗性多肽是肿瘤抑制因子、免疫调节因子、血管形成抑制因子、抗血管多肽、细胞毒性多肽、调亡诱导因子、前药活化酶或细胞增殖抑制多肽。
12.一种制造增强型EEV形式的痘苗病毒的方法,所述方法包括:
a)用痘苗病毒感染过量表达补体抑制蛋白的人细胞系;
b)从被感染的细胞内分离EEV形式的痘苗病毒。
13.如权利要求12所述的方法,所述痘苗病毒具有A34R蛋白编码基因内的突变,该突变导致K151D突变。
14.如权利要求12所述的方法,所述补体抑制蛋白是CD55、CD46或CD59。
15.一种用于生产增强型EEV形式的痘苗病毒的人细胞系,所述细胞系含有痘苗病毒并过量表达至少一种选自CD55、CD46和CD59的补体抑制蛋白。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US40285702P | 2002-08-12 | 2002-08-12 | |
| US60/402,857 | 2002-08-12 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN038237199A Division CN1754002B (zh) | 2002-08-12 | 2003-08-11 | 涉及痘病毒和癌的组合物及制备方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN101912421A true CN101912421A (zh) | 2010-12-15 |
Family
ID=31715904
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN2010101583381A Pending CN101912421A (zh) | 2002-08-12 | 2003-08-11 | 涉及痘病毒和癌的方法及组合物 |
| CN038237199A Expired - Lifetime CN1754002B (zh) | 2002-08-12 | 2003-08-11 | 涉及痘病毒和癌的组合物及制备方法 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN038237199A Expired - Lifetime CN1754002B (zh) | 2002-08-12 | 2003-08-11 | 涉及痘病毒和癌的组合物及制备方法 |
Country Status (10)
| Country | Link |
|---|---|
| US (3) | US20060099224A1 (zh) |
| EP (4) | EP2269619A1 (zh) |
| JP (2) | JP4796299B2 (zh) |
| KR (3) | KR20130012595A (zh) |
| CN (2) | CN101912421A (zh) |
| AU (2) | AU2003258168B2 (zh) |
| CA (2) | CA2494844C (zh) |
| ES (1) | ES2444699T3 (zh) |
| HK (1) | HK1130196A1 (zh) |
| WO (1) | WO2004014314A2 (zh) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105658795A (zh) * | 2013-08-22 | 2016-06-08 | 匹兹堡大学联邦系统高等教育 | 免疫溶瘤疗法 |
| CN112313339A (zh) * | 2018-01-05 | 2021-02-02 | 渥太华医院研究所 | 修饰的牛痘载体 |
| CN113454231A (zh) * | 2018-12-21 | 2021-09-28 | 渥太华医院研究所 | 修饰的正痘病毒载体 |
| CN113840914A (zh) * | 2019-05-14 | 2021-12-24 | 国立大学法人鸟取大学 | 诱导细胞融合的痘苗病毒及其应用 |
Families Citing this family (58)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7285526B2 (en) | 1995-07-14 | 2007-10-23 | Meiogen Biotechnology Corporation | Interferon antagonists useful for the treatment of interferon related diseases |
| GB0109515D0 (en) * | 2001-04-17 | 2001-06-06 | Neg Micon As | A method for transporting a set of large longitudinal items, a package system to be used by the method and use of such a package system |
| EP1281767A3 (en) * | 2001-07-31 | 2003-05-28 | Aladar A. Szalay | Light emitting microorganisms and cells for diagnosis and therapy of tumors |
| EP2269619A1 (en) | 2002-08-12 | 2011-01-05 | Jennerex Biotherapeutics ULC | Methods and compositions concerning poxviruses and cancer |
| DE602004018927D1 (de) * | 2003-06-18 | 2009-02-26 | Genelux Corp | Modifizierte rekombinante vacciniaviren, verwendungen davon |
| CN101137748B (zh) * | 2005-03-07 | 2011-12-14 | 罗巴斯研究机构 | 粘液瘤病毒与雷帕霉素的组合在治疗性处理中的应用 |
| US8980246B2 (en) | 2005-09-07 | 2015-03-17 | Sillajen Biotherapeutics, Inc. | Oncolytic vaccinia virus cancer therapy |
| CA2621982C (en) * | 2005-09-07 | 2017-11-28 | Jennerex Biotherapeutics Ulc | Systemic treatment of metastatic and/or systemically-disseminated cancers using gm-csf-expressing poxviruses |
| DE602006010902D1 (de) * | 2005-11-24 | 2010-01-14 | Aicuris Gmbh & Co Kg | Parapocken-viren in kombination mit klassischen zytotoxischen chemotherapeutika als biochemotherapie zur behandlung von krebs |
| GB0605761D0 (en) * | 2006-03-22 | 2006-05-03 | Imp Innovations Ltd | Anti-inflammatory proteins and improved vaccines |
| DK2029756T3 (en) * | 2006-06-20 | 2017-02-13 | Transgene Sa | PROCEDURE FOR PREPARING POXVIRA AND POXVIRUS COMPOSITIONS |
| US20100086522A1 (en) * | 2006-07-18 | 2010-04-08 | Ottawa Health Research Institute | Disparate suicide carrier cells for tumor targeting of promiscuous oncolytic viruses |
| EP2426142A3 (en) | 2006-10-16 | 2012-06-13 | Genelux Corporation | Modified vaccinia virus strains for use in a diagnostic and therapeutic method |
| KR20080084528A (ko) * | 2007-03-15 | 2008-09-19 | 제네렉스 바이오테라퓨틱스 인크. | 종양살상형 백시니아 바이러스 암 치료 |
| WO2008156655A2 (en) * | 2007-06-15 | 2008-12-24 | Genelux Corporation | Vaccinia virus vectors encoding a sodium-dependent transporter protein for imaging and/or treatment of tumors |
| EP2173368A1 (en) * | 2007-07-18 | 2010-04-14 | Genelux Corporation | Use of a chemotherapeutic agent in the preparation of a medicament for treating or ameliorating an adverse side effect associated with oncolytic viral therapy |
| WO2009052426A2 (en) * | 2007-10-17 | 2009-04-23 | The Ohio State University | Oncolytic virus |
| US8450106B2 (en) * | 2007-10-17 | 2013-05-28 | The Ohio State University Research Foundation | Oncolytic virus |
| WO2009054996A2 (en) * | 2007-10-25 | 2009-04-30 | Genelux Corporation | Systems and methods for viral therapy |
| WO2009082594A2 (en) * | 2007-11-26 | 2009-07-02 | Fox Chase Cancer Center | Secreted/cell bound poxvirus proteins and methods of use thereof as vaccines and anti-viral agents |
| CA2706750A1 (en) * | 2007-11-27 | 2009-06-04 | Ottawa Health Research Institute | Amplification of cancer-specific oncolytic viral infection by histone deacetylase inhibitors |
| WO2009143610A1 (en) * | 2008-05-27 | 2009-12-03 | Oncolytics Biotech Inc. | Modulating interstitial pressure and oncolytic viral delivery and distribution |
| WO2010124393A1 (en) * | 2009-04-30 | 2010-11-04 | National Research Council Of Canada | Anticancer agent comprising a yatapoxvirus mutant and uses thereof |
| ES2848650T3 (es) | 2009-09-14 | 2021-08-11 | Sillajen Biotherapeutics Inc | Terapia combinada contra el cáncer con virus vaccinia oncolítico |
| EP2501405A4 (en) * | 2009-11-20 | 2013-12-04 | Inviragen Inc | COMPOSITION, METHOD, AND USES OF POX VIRUS ELEMENTS IN VACCINATE CONSTRUCTS |
| US20110288545A1 (en) * | 2010-04-22 | 2011-11-24 | Old Dominion University Research Foundation | Method and Device for Ablation of Cancer and Resistance to New Cancer Growth |
| CN103429258B (zh) * | 2011-01-04 | 2016-03-09 | 新罗杰公司 | 通过施用溶瘤痘苗病毒生成针对肿瘤抗原的抗体和生成肿瘤特异性补体依赖性细胞毒性 |
| JP6415977B2 (ja) | 2011-04-15 | 2018-10-31 | ジェネラックス・コーポレイションGenelux Corporation | 弱毒化ワクシニアウイルスのクローン株およびその使用方法 |
| CN102747103A (zh) * | 2011-04-20 | 2012-10-24 | 中国疾病预防控制中心性病艾滋病预防控制中心 | 基于a52l基因和b8r基因双缺失的复制型痘苗病毒的艾滋病疫苗 |
| KR101370620B1 (ko) | 2011-08-05 | 2014-03-06 | 한국생명공학연구원 | 다람쥐폭스바이러스를 유효성분으로 포함하는 암 예방 및 치료용 약학적 조성물 |
| PL2739293T3 (pl) | 2011-08-05 | 2020-11-16 | Sillajen Biotherapeutics, Inc. | Sposoby i kompozycje wytwarzania wirusa krowianki |
| FI20115914L (fi) * | 2011-09-16 | 2013-03-17 | Oncos Therapeutics Ltd | Muunnettu onkolyyttinen virus |
| WO2013138522A2 (en) | 2012-03-16 | 2013-09-19 | Genelux Corporation | Methods for assessing effectiveness and monitoring oncolytic virus treatment |
| WO2013158265A1 (en) | 2012-04-20 | 2013-10-24 | Genelux Corporation | Imaging methods for oncolytic virus therapy |
| WO2014047350A1 (en) * | 2012-09-20 | 2014-03-27 | Morningside Technology Ventures Ltd. | Oncolytic virus encoding pd-1 binding agents and uses of the same |
| US20140140959A1 (en) | 2012-10-05 | 2014-05-22 | Aladar A. Szalay | Energy Absorbing-Based Diagnostic and Therapeutic Methods Employing Nucleic Acid Molecules Encoding Chromophore-Producing Enzymes |
| US10335484B2 (en) | 2013-01-08 | 2019-07-02 | Humabs Biomed Sa | Methods of generating robust passive and active immune responses |
| KR101540319B1 (ko) | 2013-10-01 | 2015-07-30 | 한국생명공학연구원 | cyb5R3 유전자 또는 단백질을 유효성분으로 포함하는 암 예방 및 치료용 약학적 조성물 |
| US10238700B2 (en) | 2014-01-02 | 2019-03-26 | Genelux Corporation | Oncolytic virus adjunct therapy with agents that increase virus infectivity |
| CN106413726B (zh) | 2014-04-10 | 2020-07-24 | 特朗斯吉有限公司 | 痘病毒溶瘤载体 |
| CN106794208B (zh) * | 2014-07-17 | 2021-06-01 | 西密歇根大学信托董事会 | 用于处理癌细胞的组合物及其方法 |
| ES2795010T3 (es) | 2015-06-19 | 2020-11-20 | Sillajen Inc | Composiciones y métodos para la embolización viral |
| EP3347460A4 (en) * | 2015-09-08 | 2019-04-10 | Sillajen, Inc. | MODIFIED ONCOLYTIC VACCINE VIRUSES EXPRESSING CYTOKINE AND CARBOXYESTERASE AND METHODS OF USE THEREOF |
| MX2019001919A (es) * | 2016-08-19 | 2019-09-04 | Sementis Ltd | Vacunas virales. |
| EP3515419A4 (en) | 2016-09-21 | 2020-06-10 | Stephen H. Thorne | HIGH MOBILITY GROUP 1 BOX MUTANT |
| WO2018091680A1 (en) | 2016-11-18 | 2018-05-24 | Transgene Sa | Cowpox-based oncolytic vectors |
| JP7274138B2 (ja) * | 2017-11-30 | 2023-05-16 | アステラス製薬株式会社 | Scr欠失ワクシニアウイルス |
| EP3717000A1 (en) * | 2017-12-01 | 2020-10-07 | Gerd Sutter | Immuno-modulated replication-efficient vaccinia virus strain |
| MX2020007010A (es) | 2018-01-05 | 2020-12-10 | Ottawa Hospital Res Inst | Vectores modificados de orthopoxvirus. |
| SG11202103097QA (en) | 2018-09-26 | 2021-04-29 | Astellas Pharma Inc | Cancer therapy by combination use of oncolytic vaccinia virus and immune checkpoint inhibitor, and pharmaceutical composition and combination medicine for use in the cancer therapy |
| US11685904B2 (en) | 2019-02-14 | 2023-06-27 | Ignite Immunotherapy, Inc. | Recombinant vaccinia virus and methods of use thereof |
| EP4011453A4 (en) | 2019-08-09 | 2023-09-27 | KM Biologics Co., Ltd. | ONCOLYTIC VACCINE VIRUS |
| WO2021071534A1 (en) * | 2019-10-08 | 2021-04-15 | Icell Kealex Therapeutics | Mutant vaccinia viruses and use thereof |
| US11110165B2 (en) | 2019-10-10 | 2021-09-07 | Ricardo Rosales Ledezma | Therapeutic vaccine for the treatment of papillomavirus lesions |
| EP4045657A4 (en) * | 2019-10-16 | 2023-11-15 | KaliVir Immunotherapeutics, Inc. | MODIFIED EXTRACELLULAR ENVELOPED VIRUS |
| US20230002740A1 (en) * | 2019-12-12 | 2023-01-05 | Ignite Immunotherapy, Inc | Variant oncolytic vaccinia virus and methods of use thereof |
| IL308018A (en) | 2021-04-30 | 2023-12-01 | Kalivir Immunotherapeutics Inc | Oncolytic viruses for different MHC expression |
| WO2023128672A1 (ko) * | 2021-12-29 | 2023-07-06 | 재단법인 아산사회복지재단 | 외피외 바이러스 생산이 증가한 신규한 백시니아 바이러스 변이주 |
Family Cites Families (152)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US563055A (en) | 1896-06-30 | Sand-pump | ||
| US4554101A (en) | 1981-01-09 | 1985-11-19 | New York Blood Center, Inc. | Identification and preparation of epitopes on antigens and allergens on the basis of hydrophilicity |
| US5833975A (en) * | 1989-03-08 | 1998-11-10 | Virogenetics Corporation | Canarypox virus expressing cytokine and/or tumor-associated antigen DNA sequence |
| NL8200523A (nl) | 1982-02-11 | 1983-09-01 | Univ Leiden | Werkwijze voor het in vitro transformeren van planteprotoplasten met plasmide-dna. |
| US4879236A (en) | 1984-05-16 | 1989-11-07 | The Texas A&M University System | Method for producing a recombinant baculovirus expression vector |
| US4957858A (en) | 1986-04-16 | 1990-09-18 | The Salk Instute For Biological Studies | Replicative RNA reporter systems |
| US4883750A (en) | 1984-12-13 | 1989-11-28 | Applied Biosystems, Inc. | Detection of specific sequences in nucleic acids |
| US4683202A (en) | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| US4683195A (en) | 1986-01-30 | 1987-07-28 | Cetus Corporation | Process for amplifying, detecting, and/or-cloning nucleic acid sequences |
| US4946773A (en) | 1985-12-23 | 1990-08-07 | President And Fellows Of Harvard College | Detection of base pair mismatches using RNAase A |
| DE3785591T2 (de) | 1986-01-10 | 1993-09-02 | Amoco Corp | Kompetitiver homogener test. |
| US4800159A (en) | 1986-02-07 | 1989-01-24 | Cetus Corporation | Process for amplifying, detecting, and/or cloning nucleic acid sequences |
| AU622104B2 (en) | 1987-03-11 | 1992-04-02 | Sangtec Molecular Diagnostics Ab | Method of assaying of nucleic acids, a reagent combination and kit therefore |
| IL86724A (en) | 1987-06-19 | 1995-01-24 | Siska Diagnostics Inc | Methods and kits for amplification and testing of nucleic acid sequences |
| US5824311A (en) | 1987-11-30 | 1998-10-20 | Trustees Of The University Of Pennsylvania | Treatment of tumors with monoclonal antibodies against oncogene antigens |
| AU622426B2 (en) | 1987-12-11 | 1992-04-09 | Abbott Laboratories | Assay using template-dependent nucleic acid probe reorganization |
| EP0359789B1 (en) | 1988-01-21 | 1993-08-04 | Genentech, Inc. | Amplification and detection of nucleic acid sequences |
| US4952500A (en) | 1988-02-01 | 1990-08-28 | University Of Georgia Research Foundation, Inc. | Cloning systems for Rhodococcus and related bacteria |
| CA1340807C (en) | 1988-02-24 | 1999-11-02 | Lawrence T. Malek | Nucleic acid amplification process |
| WO1989009284A1 (en) | 1988-03-24 | 1989-10-05 | University Of Iowa Research Foundation | Catalytic hybridization systems for the detection of nucleic acid sequences based on their activity as cofactors in catalytic reactions in which a complementary labeled nucleic acid probe is cleaved |
| US5932413A (en) | 1988-04-01 | 1999-08-03 | Celebuski; Joseph Eugene | DNA probe assay using neutrally charged probe strands |
| US5858652A (en) | 1988-08-30 | 1999-01-12 | Abbott Laboratories | Detection and amplification of target nucleic acid sequences |
| US5151509A (en) * | 1988-12-16 | 1992-09-29 | United States Of America | Gene encoding serine protease inhibitor |
| US4932207A (en) | 1988-12-28 | 1990-06-12 | Sundstrand Corporation | Segmented seal plate for a turbine engine |
| US5856092A (en) | 1989-02-13 | 1999-01-05 | Geneco Pty Ltd | Detection of a nucleic acid sequence or a change therein |
| US5703055A (en) | 1989-03-21 | 1997-12-30 | Wisconsin Alumni Research Foundation | Generation of antibodies through lipid mediated DNA delivery |
| US5284760A (en) | 1989-04-03 | 1994-02-08 | Feinstone Stephen M | Techniques for producing site-directed mutagenesis of cloned DNA |
| US5925525A (en) | 1989-06-07 | 1999-07-20 | Affymetrix, Inc. | Method of identifying nucleotide differences |
| US5302523A (en) | 1989-06-21 | 1994-04-12 | Zeneca Limited | Transformation of plant cells |
| US7705215B1 (en) | 1990-04-17 | 2010-04-27 | Dekalb Genetics Corporation | Methods and compositions for the production of stably transformed, fertile monocot plants and cells thereof |
| US5550318A (en) | 1990-04-17 | 1996-08-27 | Dekalb Genetics Corporation | Methods and compositions for the production of stably transformed, fertile monocot plants and cells thereof |
| US5073627A (en) | 1989-08-22 | 1991-12-17 | Immunex Corporation | Fusion proteins comprising GM-CSF and IL-3 |
| US5322783A (en) | 1989-10-17 | 1994-06-21 | Pioneer Hi-Bred International, Inc. | Soybean transformation by microparticle bombardment |
| US5484956A (en) | 1990-01-22 | 1996-01-16 | Dekalb Genetics Corporation | Fertile transgenic Zea mays plant comprising heterologous DNA encoding Bacillus thuringiensis endotoxin |
| US5149797A (en) | 1990-02-15 | 1992-09-22 | The Worcester Foundation For Experimental Biology | Method of site-specific alteration of rna and production of encoded polypeptides |
| US5220007A (en) | 1990-02-15 | 1993-06-15 | The Worcester Foundation For Experimental Biology | Method of site-specific alteration of RNA and production of encoded polypeptides |
| US5466468A (en) | 1990-04-03 | 1995-11-14 | Ciba-Geigy Corporation | Parenterally administrable liposome formulation comprising synthetic lipids |
| DE69112207T2 (de) | 1990-04-05 | 1996-03-28 | Roberto Crea | "walk-through"-mutagenese. |
| US5849481A (en) | 1990-07-27 | 1998-12-15 | Chiron Corporation | Nucleic acid hybridization assays employing large comb-type branched polynucleotides |
| US5645987A (en) | 1990-09-21 | 1997-07-08 | Amgen Inc. | Enzymatic synthesis of oligonucleotides |
| US5798339A (en) | 1990-12-17 | 1998-08-25 | University Of Manitoba | Treatment method for cancer |
| US5384253A (en) | 1990-12-28 | 1995-01-24 | Dekalb Genetics Corporation | Genetic transformation of maize cells by electroporation of cells pretreated with pectin degrading enzymes |
| US5399363A (en) | 1991-01-25 | 1995-03-21 | Eastman Kodak Company | Surface modified anticancer nanoparticles |
| EP0575491B1 (en) * | 1991-03-07 | 2003-08-13 | Virogenetics Corporation | Genetically engineered vaccine strain |
| CA2105277C (en) | 1991-03-07 | 2006-12-12 | William I. Cox | Genetically engineered vaccine strain |
| GB9105383D0 (en) * | 1991-03-14 | 1991-05-01 | Immunology Ltd | An immunotherapeutic for cervical cancer |
| WO1993004169A1 (en) | 1991-08-20 | 1993-03-04 | Genpharm International, Inc. | Gene targeting in animal cells using isogenic dna constructs |
| US5846717A (en) | 1996-01-24 | 1998-12-08 | Third Wave Technologies, Inc. | Detection of nucleic acid sequences by invader-directed cleavage |
| US5610042A (en) | 1991-10-07 | 1997-03-11 | Ciba-Geigy Corporation | Methods for stable transformation of wheat |
| US5849486A (en) | 1993-11-01 | 1998-12-15 | Nanogen, Inc. | Methods for hybridization analysis utilizing electrically controlled hybridization |
| DK0612248T3 (da) | 1991-11-15 | 2003-12-08 | Smithkline Beecham Corp | Præparat, der indeholder cisplatin og topotecan som antitumormiddel |
| US5846708A (en) | 1991-11-19 | 1998-12-08 | Massachusetts Institiute Of Technology | Optical and electrical methods and apparatus for molecule detection |
| WO1993013216A1 (en) | 1991-12-24 | 1993-07-08 | The President And Fellows Of Harvard College | Site-directed mutagenesis of dna |
| US20020146702A1 (en) | 1992-01-31 | 2002-10-10 | Vielkind Juergen R. | Nucleic acid molecule associated with prostate cancer and melanoma immunodetection and immunotherapy |
| ES2210239T3 (es) | 1992-04-01 | 2004-07-01 | The Johns Hopkins University School Of Medicine | Metodo para detectar acidos nucleicos de mamiferos aislados en heces y reactivos para el mismo. |
| US5843640A (en) | 1992-06-19 | 1998-12-01 | Northwestern University | Method of simultaneously detecting amplified nucleic acid sequences and cellular antigens in cells |
| DE69334225D1 (de) | 1992-07-07 | 2008-07-31 | Japan Tobacco Inc | Verfahren zur transformation einer monokotyledon pflanze |
| US5702932A (en) | 1992-07-20 | 1997-12-30 | University Of Florida | Microinjection methods to transform arthropods with exogenous DNA |
| BR9306802A (pt) | 1992-07-27 | 1998-12-08 | Pioneer Hi Bred Int | Processo independente de genótipos para produção de planta de soja transgénica e processo de regeneração de plantas de soja a partir de nodos cotiledonais |
| DE4228457A1 (de) | 1992-08-27 | 1994-04-28 | Beiersdorf Ag | Herstellung von heterodimerem PDGF-AB mit Hilfe eines bicistronischen Vektorsystems in Säugerzellen |
| US5389514A (en) | 1992-08-28 | 1995-02-14 | Fox Chase Cancer Center | Method for specifically altering the nucleotide sequence of RNA |
| US5861244A (en) | 1992-10-29 | 1999-01-19 | Profile Diagnostic Sciences, Inc. | Genetic sequence assay using DNA triple strand formation |
| GB9222888D0 (en) | 1992-10-30 | 1992-12-16 | British Tech Group | Tomography |
| US5846945A (en) | 1993-02-16 | 1998-12-08 | Onyx Pharmaceuticals, Inc. | Cytopathic viruses for therapy and prophylaxis of neoplasia |
| US5801005A (en) | 1993-03-17 | 1998-09-01 | University Of Washington | Immune reactivity to HER-2/neu protein for diagnosis of malignancies in which the HER-2/neu oncogene is associated |
| US5658751A (en) | 1993-04-13 | 1997-08-19 | Molecular Probes, Inc. | Substituted unsymmetrical cyanine dyes with selected permeability |
| US5279721A (en) | 1993-04-22 | 1994-01-18 | Peter Schmid | Apparatus and method for an automated electrophoresis system |
| GB9311386D0 (en) | 1993-06-02 | 1993-07-21 | Pna Diagnostics As | Nucleic acid analogue assay procedures |
| US5846709A (en) | 1993-06-15 | 1998-12-08 | Imclone Systems Incorporated | Chemical process for amplifying and detecting nucleic acid sequences |
| US5543158A (en) | 1993-07-23 | 1996-08-06 | Massachusetts Institute Of Technology | Biodegradable injectable nanoparticles |
| FR2708288B1 (fr) | 1993-07-26 | 1995-09-01 | Bio Merieux | Procédé d'amplification d'acides nucléiques par transcription utilisant le déplacement, réactifs et nécessaire pour la mise en Óoeuvre de ce procédé. |
| US5969094A (en) * | 1993-10-12 | 1999-10-19 | Emory University | Anti-paramyxovirus screening method and vaccine |
| US5925517A (en) | 1993-11-12 | 1999-07-20 | The Public Health Research Institute Of The City Of New York, Inc. | Detectably labeled dual conformation oligonucleotide probes, assays and kits |
| GB2284208A (en) | 1993-11-25 | 1995-05-31 | Pna Diagnostics As | Nucleic acid analogues with a chelating functionality for metal ions |
| JP2935950B2 (ja) | 1993-12-03 | 1999-08-16 | 株式会社山田製作所 | ステアリングシャフト及びその製造装置 |
| DE69432919T2 (de) | 1993-12-28 | 2004-05-27 | Tanabe Seiyaku Co., Ltd. | Verfahren für den Nachweis spezifischer Polynukleotiden |
| US5928905A (en) | 1995-04-18 | 1999-07-27 | Glaxo Group Limited | End-complementary polymerase reaction |
| US5851770A (en) | 1994-04-25 | 1998-12-22 | Variagenics, Inc. | Detection of mismatches by resolvase cleavage using a magnetic bead support |
| US5656465A (en) * | 1994-05-04 | 1997-08-12 | Therion Biologics Corporation | Methods of in vivo gene delivery |
| US6093700A (en) * | 1995-05-11 | 2000-07-25 | Thomas Jefferson University | Method of inducing an immune response using vaccinia virus recombinants encoding GM-CSF |
| CN1152946A (zh) | 1994-05-28 | 1997-06-25 | 特普尼尔医学有限公司 | 生产核酸的拷贝 |
| US5656610A (en) | 1994-06-21 | 1997-08-12 | University Of Southern California | Producing a protein in a mammal by injection of a DNA-sequence into the tongue |
| US5942391A (en) | 1994-06-22 | 1999-08-24 | Mount Sinai School Of Medicine | Nucleic acid amplification method: ramification-extension amplification method (RAM) |
| FR2722208B1 (fr) | 1994-07-05 | 1996-10-04 | Inst Nat Sante Rech Med | Nouveau site interne d'entree des ribosomes, vecteur le contenant et utilisation therapeutique |
| US5849483A (en) | 1994-07-28 | 1998-12-15 | Ig Laboratories, Inc. | High throughput screening method for sequences or genetic alterations in nucleic acids |
| DE69528706T2 (de) | 1994-08-19 | 2003-06-12 | Pe Corp Ny Foster City | Gekoppeltes ampflikation- und ligationverfahren |
| GB9506466D0 (en) * | 1994-08-26 | 1995-05-17 | Prolifix Ltd | Cell cycle regulated repressor and dna element |
| US5599668A (en) | 1994-09-22 | 1997-02-04 | Abbott Laboratories | Light scattering optical waveguide method for detecting specific binding events |
| US5871986A (en) | 1994-09-23 | 1999-02-16 | The General Hospital Corporation | Use of a baculovirus to express and exogenous gene in a mammalian cell |
| ATE340866T1 (de) | 1994-10-28 | 2006-10-15 | Gen Probe Inc | Zusammensetzungen und verfahren für die gleichzeitige detektion und quantifizierung von einer mehrheit spezifischer nuklein säure sequenzen |
| US5736524A (en) | 1994-11-14 | 1998-04-07 | Merck & Co.,. Inc. | Polynucleotide tuberculosis vaccine |
| US5935825A (en) | 1994-11-18 | 1999-08-10 | Shimadzu Corporation | Process and reagent for amplifying nucleic acid sequences |
| US5599302A (en) | 1995-01-09 | 1997-02-04 | Medi-Ject Corporation | Medical injection system and method, gas spring thereof and launching device using gas spring |
| US5866337A (en) | 1995-03-24 | 1999-02-02 | The Trustees Of Columbia University In The City Of New York | Method to detect mutations in a nucleic acid using a hybridization-ligation procedure |
| IE80468B1 (en) | 1995-04-04 | 1998-07-29 | Elan Corp Plc | Controlled release biodegradable nanoparticles containing insulin |
| US5843650A (en) | 1995-05-01 | 1998-12-01 | Segev; David | Nucleic acid detection and amplification by chemical linkage of oligonucleotides |
| US5929227A (en) | 1995-07-12 | 1999-07-27 | The Regents Of The University Of California | Dimeric fluorescent energy transfer dyes comprising asymmetric cyanine azole-indolenine chromophores |
| WO1998004689A1 (en) | 1995-07-31 | 1998-02-05 | Urocor, Inc. | Biomarkers and targets for diagnosis, prognosis and management of prostate disease |
| US5916779A (en) | 1995-09-21 | 1999-06-29 | Becton, Dickinson And Company | Strand displacement amplification of RNA targets |
| KR19990063653A (ko) | 1995-09-29 | 1999-07-26 | 크리스토퍼 엘. 와이트, 스코트 지. 홀퀴스트, 스티븐 엘. 말라스카 | 케모킨 억제제 |
| US5866331A (en) | 1995-10-20 | 1999-02-02 | University Of Massachusetts | Single molecule detection by in situ hybridization |
| US5780448A (en) | 1995-11-07 | 1998-07-14 | Ottawa Civic Hospital Loeb Research | DNA-based vaccination of fish |
| DE19541450C2 (de) * | 1995-11-07 | 1997-10-02 | Gsf Forschungszentrum Umwelt | Genkonstrukt und dessen Verwendung |
| US5789166A (en) | 1995-12-08 | 1998-08-04 | Stratagene | Circular site-directed mutagenesis |
| US5612473A (en) | 1996-01-16 | 1997-03-18 | Gull Laboratories | Methods, kits and solutions for preparing sample material for nucleic acid amplification |
| US5851772A (en) | 1996-01-29 | 1998-12-22 | University Of Chicago | Microchip method for the enrichment of specific DNA sequences |
| US5928906A (en) | 1996-05-09 | 1999-07-27 | Sequenom, Inc. | Process for direct sequencing during template amplification |
| US5739169A (en) * | 1996-05-31 | 1998-04-14 | Procept, Incorporated | Aromatic compounds for inhibiting immune response |
| US5939291A (en) | 1996-06-14 | 1999-08-17 | Sarnoff Corporation | Microfluidic method for nucleic acid amplification |
| US5912124A (en) | 1996-06-14 | 1999-06-15 | Sarnoff Corporation | Padlock probe detection |
| US5853990A (en) | 1996-07-26 | 1998-12-29 | Edward E. Winger | Real time homogeneous nucleotide assay |
| US5945100A (en) | 1996-07-31 | 1999-08-31 | Fbp Corporation | Tumor delivery vehicles |
| US5928870A (en) | 1997-06-16 | 1999-07-27 | Exact Laboratories, Inc. | Methods for the detection of loss of heterozygosity |
| US5849546A (en) | 1996-09-13 | 1998-12-15 | Epicentre Technologies Corporation | Methods for using mutant RNA polymerases with reduced discrimination between non-canonical and canonical nucleoside triphosphates |
| US5981274A (en) | 1996-09-18 | 1999-11-09 | Tyrrell; D. Lorne J. | Recombinant hepatitis virus vectors |
| US5853992A (en) | 1996-10-04 | 1998-12-29 | The Regents Of The University Of California | Cyanine dyes with high-absorbance cross section as donor chromophores in energy transfer labels |
| US5853993A (en) | 1996-10-21 | 1998-12-29 | Hewlett-Packard Company | Signal enhancement method and kit |
| US5900481A (en) | 1996-11-06 | 1999-05-04 | Sequenom, Inc. | Bead linkers for immobilizing nucleic acids to solid supports |
| US5905024A (en) | 1996-12-17 | 1999-05-18 | University Of Chicago | Method for performing site-specific affinity fractionation for use in DNA sequencing |
| US5846225A (en) | 1997-02-19 | 1998-12-08 | Cornell Research Foundation, Inc. | Gene transfer therapy delivery device and method |
| JP4191255B2 (ja) * | 1997-02-21 | 2008-12-03 | オックソン セラピュティックス リミテッド | ケモカイン類に結合する可溶性ワクシニアウイルスタンパク質 |
| US5846729A (en) | 1997-02-27 | 1998-12-08 | Lorne Park Research, Inc. | Assaying nucleotides in solution using a fluorescent intensity quenching effect |
| US5849497A (en) | 1997-04-03 | 1998-12-15 | The Research Foundation Of State University Of New York | Specific inhibition of the polymerase chain reaction using a non-extendable oligonucleotide blocker |
| US5846726A (en) | 1997-05-13 | 1998-12-08 | Becton, Dickinson And Company | Detection of nucleic acids by fluorescence quenching |
| US5928869A (en) | 1997-05-30 | 1999-07-27 | Becton, Dickinson And Company | Detection of nucleic acids by fluorescence quenching |
| US5919626A (en) | 1997-06-06 | 1999-07-06 | Orchid Bio Computer, Inc. | Attachment of unmodified nucleic acids to silanized solid phase surfaces |
| US5866366A (en) | 1997-07-01 | 1999-02-02 | Smithkline Beecham Corporation | gidB |
| US5916776A (en) | 1997-08-27 | 1999-06-29 | Sarnoff Corporation | Amplification method for a polynucleotide |
| US5935791A (en) | 1997-09-23 | 1999-08-10 | Becton, Dickinson And Company | Detection of nucleic acids by fluorescence quenching |
| IL135507A0 (en) | 1997-10-09 | 2001-05-20 | Pro Virus Inc | Treatment of neoplasms with viruses |
| US20030044384A1 (en) * | 1997-10-09 | 2003-03-06 | Pro-Virus, Inc. | Treatment of neoplasms with viruses |
| US5994624A (en) | 1997-10-20 | 1999-11-30 | Cotton Incorporated | In planta method for the production of transgenic plants |
| US5932451A (en) | 1997-11-19 | 1999-08-03 | Incyte Pharmaceuticals, Inc. | Method for unbiased mRNA amplification |
| US6177076B1 (en) * | 1997-12-09 | 2001-01-23 | Thomas Jefferson University | Method of treating bladder cancer with wild type vaccinia virus |
| EP1390046A4 (en) * | 1999-04-15 | 2005-04-20 | Wellstat Biologics Corp | TREATMENT OF NEOPLASMS WITH VIRUSES |
| CA2375189C (en) | 1999-05-28 | 2010-02-09 | The Government Of The United States Of America | A combined growth factor-deleted and thymidine kinase-deleted vaccinia virus vector |
| EP1227828A1 (en) | 1999-11-12 | 2002-08-07 | Oncolytics Biotech, Inc. | Viruses for the treatment of cellular proliferative disorders |
| US20020048777A1 (en) | 1999-12-06 | 2002-04-25 | Shujath Ali | Method of diagnosing monitoring, staging, imaging and treating prostate cancer |
| WO2001060156A1 (en) | 2000-02-14 | 2001-08-23 | Gary R. Davis, M.D., L.L.C. | Neutralizing antibody and immunomodulatory enhancing compositions |
| US20040091995A1 (en) * | 2001-06-15 | 2004-05-13 | Jeffrey Schlom | Recombinant non-replicating virus expressing gm-csf and uses thereof to enhance immune responses |
| DE10134955C1 (de) * | 2001-07-23 | 2003-03-06 | Infineon Technologies Ag | Anordnung von Gräben in einem Halbleitersubstrat, insbesondere für Grabenkondensatoren |
| US20030206886A1 (en) * | 2002-05-03 | 2003-11-06 | University Of Medicine & Dentistry Of New Jersey | Neutralization of immune suppressive factors for the immunotherapy of cancer |
| EP2269619A1 (en) | 2002-08-12 | 2011-01-05 | Jennerex Biotherapeutics ULC | Methods and compositions concerning poxviruses and cancer |
| DE602004018927D1 (de) * | 2003-06-18 | 2009-02-26 | Genelux Corp | Modifizierte rekombinante vacciniaviren, verwendungen davon |
| US20050207974A1 (en) | 2004-03-17 | 2005-09-22 | Deng David X | Endothelial cell markers and related reagents and methods of use thereof |
| CA2621982C (en) * | 2005-09-07 | 2017-11-28 | Jennerex Biotherapeutics Ulc | Systemic treatment of metastatic and/or systemically-disseminated cancers using gm-csf-expressing poxviruses |
| GB0519303D0 (en) | 2005-09-21 | 2005-11-02 | Oxford Biomedica Ltd | Chemo-immunotherapy method |
| WO2008043576A1 (en) | 2006-10-13 | 2008-04-17 | Medigene Ag | Use of oncolytic viruses and antiangiogenic agents in the treatment of cancer |
| EP2426142A3 (en) | 2006-10-16 | 2012-06-13 | Genelux Corporation | Modified vaccinia virus strains for use in a diagnostic and therapeutic method |
| KR20080084528A (ko) | 2007-03-15 | 2008-09-19 | 제네렉스 바이오테라퓨틱스 인크. | 종양살상형 백시니아 바이러스 암 치료 |
| US8312249B1 (en) | 2008-10-10 | 2012-11-13 | Apple Inc. | Dynamic trampoline and structured code generation in a signed code environment |
| FR2957821B1 (fr) | 2010-03-24 | 2014-08-29 | Inst Francais Du Petrole | Nouvelle zone de regeneration du catalyseur divisee en secteurs pour unites catalytiques regeneratives |
-
2003
- 2003-08-11 EP EP10181845A patent/EP2269619A1/en not_active Ceased
- 2003-08-11 AU AU2003258168A patent/AU2003258168B2/en not_active Expired
- 2003-08-11 CN CN2010101583381A patent/CN101912421A/zh active Pending
- 2003-08-11 KR KR1020137001466A patent/KR20130012595A/ko active Search and Examination
- 2003-08-11 CN CN038237199A patent/CN1754002B/zh not_active Expired - Lifetime
- 2003-08-11 EP EP10181820A patent/EP2269618A1/en not_active Withdrawn
- 2003-08-11 US US10/524,932 patent/US20060099224A1/en not_active Abandoned
- 2003-08-11 CA CA2494844A patent/CA2494844C/en not_active Expired - Lifetime
- 2003-08-11 JP JP2004528045A patent/JP4796299B2/ja not_active Expired - Lifetime
- 2003-08-11 KR KR1020117028101A patent/KR20120002613A/ko not_active Application Discontinuation
- 2003-08-11 CA CA2818693A patent/CA2818693C/en not_active Expired - Lifetime
- 2003-08-11 ES ES08167984.7T patent/ES2444699T3/es not_active Expired - Lifetime
- 2003-08-11 KR KR1020057002384A patent/KR101170653B1/ko active IP Right Grant
- 2003-08-11 EP EP08167984.7A patent/EP2044948B1/en not_active Expired - Lifetime
- 2003-08-11 WO PCT/US2003/025141 patent/WO2004014314A2/en active Application Filing
- 2003-08-11 EP EP03785197A patent/EP1578396A4/en not_active Withdrawn
-
2007
- 2007-08-14 US US11/838,757 patent/US8329164B2/en not_active Expired - Lifetime
-
2009
- 2009-10-06 HK HK09109228.6A patent/HK1130196A1/xx not_active IP Right Cessation
-
2010
- 2010-01-29 AU AU2010200333A patent/AU2010200333B2/en not_active Ceased
- 2010-11-26 JP JP2010263090A patent/JP2011078427A/ja active Pending
-
2012
- 2012-11-13 US US13/675,953 patent/US8986674B2/en not_active Expired - Lifetime
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105658795A (zh) * | 2013-08-22 | 2016-06-08 | 匹兹堡大学联邦系统高等教育 | 免疫溶瘤疗法 |
| CN105658795B (zh) * | 2013-08-22 | 2020-07-03 | 匹兹堡大学联邦系统高等教育 | 免疫溶瘤疗法 |
| CN112313339A (zh) * | 2018-01-05 | 2021-02-02 | 渥太华医院研究所 | 修饰的牛痘载体 |
| CN113454231A (zh) * | 2018-12-21 | 2021-09-28 | 渥太华医院研究所 | 修饰的正痘病毒载体 |
| CN113661246A (zh) * | 2018-12-21 | 2021-11-16 | 渥太华医院研究所 | 修饰的正痘病毒载体 |
| CN113840914A (zh) * | 2019-05-14 | 2021-12-24 | 国立大学法人鸟取大学 | 诱导细胞融合的痘苗病毒及其应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| ES2444699T3 (es) | 2014-02-26 |
| AU2010200333A1 (en) | 2010-02-18 |
| KR20050074436A (ko) | 2005-07-18 |
| KR20120002613A (ko) | 2012-01-06 |
| US20130183271A1 (en) | 2013-07-18 |
| EP1578396A2 (en) | 2005-09-28 |
| JP4796299B2 (ja) | 2011-10-19 |
| WO2004014314A3 (en) | 2005-09-29 |
| US8986674B2 (en) | 2015-03-24 |
| CN1754002B (zh) | 2010-09-08 |
| EP1578396A4 (en) | 2007-01-17 |
| JP2011078427A (ja) | 2011-04-21 |
| KR20130012595A (ko) | 2013-02-04 |
| EP2044948A1 (en) | 2009-04-08 |
| US20060099224A1 (en) | 2006-05-11 |
| HK1130196A1 (en) | 2009-12-24 |
| EP2269618A1 (en) | 2011-01-05 |
| CA2818693A1 (en) | 2004-02-19 |
| EP2044948B1 (en) | 2013-10-30 |
| KR101170653B1 (ko) | 2012-08-03 |
| US20090004723A1 (en) | 2009-01-01 |
| WO2004014314A2 (en) | 2004-02-19 |
| CA2494844A1 (en) | 2004-02-19 |
| EP2269619A1 (en) | 2011-01-05 |
| US8329164B2 (en) | 2012-12-11 |
| AU2010200333B2 (en) | 2012-09-13 |
| JP2006506974A (ja) | 2006-03-02 |
| CA2818693C (en) | 2016-05-17 |
| AU2003258168A1 (en) | 2004-02-25 |
| AU2003258168B2 (en) | 2009-10-29 |
| CN1754002A (zh) | 2006-03-29 |
| CA2494844C (en) | 2013-10-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1754002B (zh) | 涉及痘病毒和癌的组合物及制备方法 | |
| CN101351213B (zh) | 使用表达gm-csf的痘病毒系统性治疗转移性癌和/或全身散布性癌 | |
| KR20090122293A (ko) | 종양 용해 백시니아 바이러스 암 치료법 | |
| AU2012244210B2 (en) | Systemic treatment of metastatic and/or systemically-disseminated cancers using gm-csf-expressing poxviruses | |
| AU2012261681B2 (en) | Methods and compositions concerning poxviruses and cancer | |
| KIRN et al. | Patent 2621982 Summary |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C12 | Rejection of a patent application after its publication | ||
| RJ01 | Rejection of invention patent application after publication |
Application publication date: 20101215 |