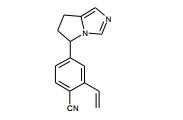
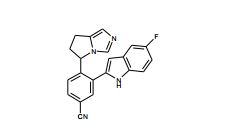
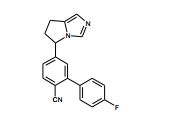
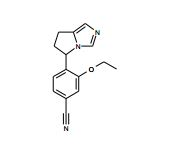
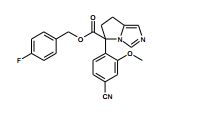
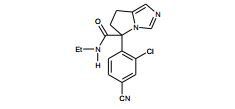
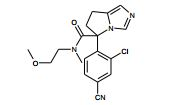
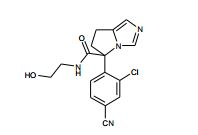
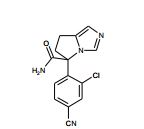
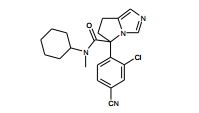
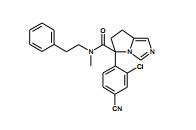
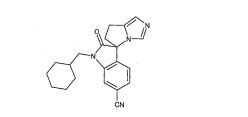
[001] A presente invenção se refere a novos derivados de imidazol que usados como inibidores de aromatasi e sintase de aldosterona bem como para tratamento de um distúrbio ou doença mediada por sintase ou aromatase de aldosterona.
[002] A presente invenção fornece um composto de Fórmula (I)
[003] na qual
[004] n é 1, ou 2, ou 3;
[005] R é hidrogênio, (C1-C7) alquila, ou (C1-C7) alquenila, as referidas (C1-C7) alquila e (C1-C7) alquenila sendo opcionalmente substituídas por um a cinco substituintes independentemente selecionados do grupo consistindo em -O-Rs θ -N(Rs)(R9), em que Rs θ R9 são independentemente selecionados do grupo consistindo em hidrogênio, (C1-C7) alquila, acila, arila e heteroarila, cada dos quais é também opcionalmente substituído por um a quatro substituintes independentemente selecionados do grupo consistindo em halo, (Ci- C7) alcóxi e (C1-C7) alquila; ou
[006] R é -C(0)0-Rio, ou -C(O)N(RH)(RI2), em que Rw, R11 e R12 são selecionados independentemente do grupo consistindo em hidrogênio, (C1-C7) alquila, (C3-Cs) cicloalquila, arila, aril-(Ci-C7) alquila, (C1-C7) haloalquila e heteroarila, cada dos quais é também opcionalmente substituído por um a quatro substituintes independentemente selecionados do grupo consistindo em halo, hidroxila, (C1-C7) alcóxi, (OC7) alquila, e arila, em que Rn e Rw tomados juntamente com 0 átomo de nitrogênio ao qual eles são ligados opcionalmente formam um anel de 3 a 8 membros;
[007] Ri R2, R3, R4, e R5 são selecionados independentemente do grupo consistindo em hidrogênio, (C1-C7) alquenila, (C1-C7) alquila, (C3- Ce) cicloalquila, halo, ciano, nitro, H2N--, (C1-C7) haloalquila, (C1-C7) alcóxi, (C3-C8) cicloalcóxi, arilóxi, arila, heretoarila, -C(0)ORio, e - N(Ri3)(Ri4), referida (C1-C7) alquila, (C1-C7) alquenila, (C1-C7) alcóxi, arila e heteroarila sendo também opcionalmente substituído por um a três substituintes selecionados a partir de (C1-C7) alquila, hidroxila, halo, (C1-C7) alcóxi, nitro, ciano, (C1-C7) dialquilamiπo, (C1-C7) alcóxi- (C1-C7) alquil--, e (C1-C7) haloalquila, a referida R10 tendo 0 mesmo significado como definido acima, os referidos Rw e Ru são independentemente selecionados do grupo consistindo em hidrogênio, (C1-C7) alquila, (C3-C8) cicloalquila, (C1-C7) haloalquila, (C1-C7) haloalcóxi, arila e ciano, com a condição de que não mais de que três RL R2 R3 R4 E R5 sejam simultaneamente hidrogênio;
[008] qual eles membros;
[009] a 6 membros contendo 0 ou 1 heteroátomo selecionado de O, N, ou S;
[0010] Re e R7 são independentemente hidrogênio, hidroxila, (Ci- C7) alquila, (C1-C7) alcóxi, fenila, ou benzila, em que fenila e benzila são opcionalmente substituídos por um a quatro substituintes independentemente selecionados do grupo consistindo em halo, (Ci- C7) alcóxi e (C1-C7) alquila:
[0011] quando Re e R7 são ligados a um mesmo átomo de carbono, eles opcionalmente formam uma porção (A) representada pela seguinte estrutura:
[0012] em que Ra e Rb são independentemente hidrogênio, (C1-C7) alquila, (C1-C7) alcóxi, acila, -COOR15 ou -COR15, referidos R15 sendo hidrogênio, (C1-C7) alquila, (C1-C7) haloalquila, arila, ou -NH2; ou
[0013] quando Re e R7 são ligados a um mesmo átomo de carbono, eles tomados juntamente com 0 referido átomo de carbono, opcionalmente formam um anel de 3 a 8 membros; ou
[0014] um sal farmaceuticamente aceitável deste; ou um isômero ótico deste; ou uma mistura de isômeros óticos.
[0015] Preferivelmente, a presente invenção fornece um composto de Fórmula (I), em que R é hidrogênio, (C1-C4) alquila, (C1-C4) alquenila, -C(0)0-Rio, ou -C(O)N(Rn)(Ri2), os referidos (C1-C4) alquila e (C1-C4) alquenila são opcionalmente substituídos por um a três substituintes independentemente selecionados a partir de hidroxila, (C1-C4) alcóxi, halo, -NH2, ou (C1-C4) dialquilamino;
[0016] em que Rw, Rn θ R12 são independentemente hidrogênio, (C1-C4) alquila, (C6-Cio) aril-(Ci-C4) alquil—, (C3-C8) cicloalquila, ou (Ci- C4) alquenila, cada dos quais é opcionalmente substituído por um ou três substituintes independentemente selecionados a partir de halo, hidroxila, ou (C1-C4) alcóxi; em que Rn e Rw tomados juntamente com 0 átomo de nitrogênio ao qual eles são ligados opcionalmente formam um anel de 3 a 8 membros;
[0017] Ri R2 R3, R4, θ Rs sã° independentemente selecionados a partir de hidrogênio, halo, ciano, --NH2, (C1-C4) dialquilamino, (C1-C4) alcóxi, (C1-C4) alquenila, (C1-C4) alquila, (C1-C4) haloalquila, (Ce-Cw) arila, ou heteroarila de 5 a 9 membros, os referidos (C1-C4) alcóxi, (Ci- C4) alquenila, (C1-C4) alquila e (C6-Cw) arila sendo opcionalmente substituídos por um a três substituintes independentemente selecionados a partir de halo, (C1-C4) alcóxi, (C1-C4) alquila, --NH2, ciano, nitro, (C1-C4) alcóxi-(Ci-C4) alquil-, ou (C1-C4) haloalquila, com a condição de que não mais de que três de Ri, R2, R3, R4, θ Rs sejam simultaneamente hidrogênio; R e Ri tomados juntamente, opcionalmente formam um anel de 5 a 6 membros contendo 0 ou 1 heteroátomo selecionado a partir de O, N, ou S;
[0018] Re e R7 são independentemente hidrogênio, (C1-C4) alquila, (C3-C8) cicloalquila, (C1-C4) alcóxi, fenila, ou benzila, as referidas fenila e benzila são opcionalmente substituídas por um ou três substituintes independentemente selecionados a partir de halo, (C1-C4) alquila, ou (C1-C4) alcóxi;
[0019] quando R6 e R7 são ligados a um mesmo átomo de carbono, eles opcionalmente formam uma porção (A) descrita acima, em que Ra e Rb são independentemente hidrogênio, ou (C1-C4) alquila, ou Ra e Rb tomados juntamente com 0 referido átomo de carbono opcionalmente formam um anel de 3 a 8 membros, ou um sal farmaceuticamente aceitável deste; ou um isômero ótico deste; ou um mistura de isômeros óticos.
[0020] Em uma modalidade, a presente invenção fornece um Composto de Fórmula (II)
[0021] em que
[0022] R, Ri, R2, R3, R4- RΔ- RΘ e R7 têm os mesmos significado daqueles definidos para a Fórmula (I) acima,
[0023] ou
[0024] um sal farmaceuticamente aceitável deste; ou um isômero ótico deste; ou uma mistura de isômeros óticos deste.
[0025] Preferivelmente, a presente invenção fornece o composto de Fórmula (II), em que R é hidrogênio, (C1-C4) alquila, (C1-C4) alquenila, -C(0)0-Rw, ou -C(O)N(Rn)(Ri2), referida (C1-C4) alquila e (C1-C4) alquenila são opcionalmente substituídos por um a três substituintes independentemente selecionados a partir de hidroxila, (C1-C4) alcóxi, halo, -NH2, ou (C1-C4) dialquilamiπo;
[0026] em que R10, Rn θ R12 são independentemente hidrogênio, (C1-C4) alquila, (C6-Cio) aril-(Ci-C4) alquil-, (C3-C8) cicloalquila, ou (Ci- C4) alquenila, cada dos quais é opcionalmente substituído por um ou três substituintes independentemente selecionados a partir de halo, hidroxila, ou (C1-C4) alcóxi; em que Rn e RI2 tomados juntamente com 0 átomo de nitrogênio ao qual eles são ligados opcionalmente formam um anel de 3 a 8 membros;
[0027] Ri, R2, R3, R4, θ Rs são independentemente selecionados a partir de hidrogênio, halo, ciano, —NH2, (C1-C4) dialquilamiπo, (C1-C4) alcóxi, (C1-C4) alquenila, (C1-C4) alquila, (C1-C4) haloalquila, (C6-Cio) arila, ou heteroarila de 5 a 9 membros, os referidos (C1-C4) alcóxi, (Ci- C4) alquenila, (C1-C4) alquila e (CΘ-CW) arila sendo opcionalmente substituídos por um ou três substituintes independentemente selecionados a partir de halo, (C1-C4) alcóxi, (C1-C4) alquila, --NH2, ciano, nitro, (C1-C4) alcóxi-(Ci-C4) alquil-, ou (C1-C4) haloalquila, com a condição de que não mais de que três de Ri, R2, R3, R4, θ Rs sejam simultaneamente hidrogênio; R e Ri tomados juntamente, opcionalmente formam um anel de 5 a 6 membros contendo 0 ou 1 heteroátomo selecionado a partir de O, N, ou S;
[0028] RΘ e R7 são independentemente hidrogênio, (C1-C4) alquila, (C3-C8) cicloalquila, (C1-C4) alcóxi, fenila, ou benzil, as referidas fenila e benzila são opcionalmente substituídas por um ou três substituintes independentemente selecionados a partir de halo, (C1-C4) alquila, ou (C1-C4) alcóxi;
[0029] quando Re e R7 são ligados a um mesmo átomo de carbono, eles opcionalmente formam uma porção (A) descrita acima, em que Ra e Rb são independentemente hidrogênio, ou (C1-C4) alquila, ou Ra e Rb tomados juntamente com 0 referido átomo de carbono opcionalmente formam um anel de 3 a 8 membros; ou um sal farmaceuticamente aceitável deste; ou um isômero ótico deste; ou um mistura de isômeros óticos.
[0030] Em outra modalidade, a presente invenção fornece um Composto de Fórmula (III)
[0031] em que
[0032] R, Ri, R2, R3, R4, Rs, Re e R7 têm 0 mesmo significado daquele definido pela Fórmula (I) acima, ou
[0033] um sal farmaceuticamente aceitável deste; ou um isômero ótico deste; ou uma mistura de isômeros óticos deste.
[0034] Preferivelmente, a presente invenção fornece um composto de Fórmula (III), em que R é hidrogênio, (C1-C4) alquila, (C1-C4) alquenila, -C(0)0-Rio, ou -C(O)N(Rn)(Rw), as referidas (C1-C4) alquila e (C1-C4) alquenila são opcionalmente substituídos por um a três substituintes independentemente selecionados a partir de hidroxila, (C1-C4) alcóxi, halo, -NH2, ou (C1-C4) dialquilamino;
[0035] em que Rw, R11 e Rw são independentemente hidrogênio, (C1-C4) alquila, (C6-Cw) aril-(Ci-C4) alquil-, (C3-C8) cicloalquila, ou (Ci- C4) alquenila, cada dos quais é opcionalmente substituído por um ou três substituintes independentemente selecionados a partir de halo, hidroxila, ou (C1-C4) alcóxi; em que Rn e R12 tomados juntamente com 0 átomo de nitrogênio ao qual eles são ligados opcionalmente formam um anel de 3 a 8 membros;
[0036] Ri R2, R3, R4, e Rs são independentemente selecionados a partir de hidrogênio, halo, ciano, -NH2, (C1-C4) dialquilamino, (C1-C4) alcóxi, (C1-C4) alquenila, (C1-C4) alquila, (C1-C4) haloalquila, (Ce-Cw) arila, ou heteroarila de 5 a 9 membros, as referidas (C1-C4) alcóxi, (Ci- C4) alquenila, (C1-C4) alquila e (C6-Cio) arila sendo opcionalmente substituídas por um ou três substituintes independentemente selecionados a partir de halo, (C1-C4) alcóxi, (C1-C4) alquila, --NH2, ciano, nitro, (C1-C4) alcóxi-(Ci-C4) alquil-, ou (C1-C4) haloalquila, com a condição de que não mais de que três de Ri, R2, R3, R4, e Rs sejam simultaneamente hidrogênio; R e Ri tomados juntamente, opcionalmente formam um anel de 5 a 6 membros contendo 0 ou 1 heteroátomo selecionado a partir de O, N, ou S;
[0037] Re e R7 são independentemente hidrogênio, (C1-C4) alquila, (C3-C8) cicloalquila, (C1-C4) alcóxi, fenila, ou benzila, as referidas fenila e benzila são opcionalmente substituídas por um ou três substituintes independentemente selecionados a partir de halo, (C1-C4) alquila, ou (C1-C4) alcóxi;
[0038] quando Re e R7 são ligados a um mesmo átomo de carbono, eles opcionalmente formam uma porção (A) descrita acima, em que Ra e Rb são independentemente hidrogênio, ou (C1-C4) alquila, ou Ra e Rb tomados juntamente com 0 referido átomo de carbono opcionalmente formam um anel de 3 a 8 membros; ou um sal farmaceuticamente aceitável deste; ou um isômero ótico deste; ou um mistura de isômeros óticos.
[0039] Em outra modalidade, a presente invenção fornece um composto de acordo com a Fórmula (IV)
[0040] em que
[0041] R, R1, R2, R3, R4, R5, R6 e R7 têm os mesmos significados daqueles definidos para Fórmula (I) acima , ou
[0042] sais farmaceuticamente aceitáveis deste; ou um isômero ótico deste; ou um mistura de isômeros óticos deste.
[0043] Preferivelmente, a presente invenção fornece um composto de Fórmula (IV), em que R é hidrogênio, (C1-C4) alquila, (C1-C4) alquenila, -C(0)0-Rw, ou -C(O)N(Rn)(Ri2), referida (C!-C4) alquila e (C1-C4) alquenila são opcionalmente substituídos por um a três substituintes independentemente selecionados a partir de hidroxila, (C1-C4) alcóxi, halo, -NH2, ou (C1-C4) dialquilamino;
[0044] em que R10, Rn θ R12 são independentemente hidrogênio, (C1-C4) alquila, (C6-Cio) aril-(Ci-C4) alquil-, (C3-C8) cicloalquila, ou (Ci- C4) alquenila, cada dos quais é opcionalmente substituído por um ou três substituintes independentemente selecionados a partir de halo, hidroxila, ou (C1-C4) alcóxi; em que Rn e RI2 tomados juntamente com 0 átomo de nitrogênio ao qual eles são ligados, opcionalmente formam um anel de 3 a 8 membros;
[0045] Ri, R2, R3, R4, θ R5 são independentemente selecionados a partir de hidrogênio, halo, ciano, —NH2, (C1-C4) dialquilamino, (C1-C4) alcóxi, (C1-C4) alquenila, (C1-C4) alquila, (C1-C4) haloalquila, (Ce-Cio) arila, ou heteroarila de 5 a 9 membros, as referidas (C1-C4) alcóxi, (Ci- C4) alquenila, (C1-C4) alquila e (Ce-C-io) arila sendo opcionalmente substituídas por um ou três substituintes independentemente selecionados a partir de halo, (C1-C4) alcóxi, (C1-C4) alquila, —NH2, ciano, nitro, (Ci-C4) alcóxi-(Ci-C4) alquil-, ou (Ci-C4) haloalquila, com a condição de que não mais de que três de Ri, R2, R3, R4, θ Rs sejam simultaneamente hidrogênio; R e Ri tomados juntamente, opcionalmente formam um anel de 5 a 6 membros contendo 0 ou 1 heteroátomo selecionado a partir de O, N, ou S;
[0046] RΘ e R7 são independentemente hidrogênio, (Ci-C4) alquila, (C3-C8) cicloalquila, (Ci-C4) alcóxi, fenila, ou benzila, as referidas fenila e benzila são opcionalmente substituídas por um ou três substituintes independentemente selecionados a partir de halo, (Ci-C4) alquila, ou (Ci-C4) alcóxi;
[0047] quando RΘ e R7 são ligados a um mesmo átomo de carbono, eles opcionalmente formam uma porção (A) descrita acima, em que Ra e Rb são independentemente hidrogênio, ou (Ci-C4) alquila, ou Ra e Rb tomados juntamente com 0 referido átomo de carbono opcionalmente formam um anel de 3 a 8 membros; ou um sal farmaceuticamente aceitável deste; ou um isômero ótico deste; ou um mistura de isômeros óticos.
[0048] Para os propósitos de interpretação desta especificação, as seguintes definições aplicar-se-ão e onde quer que apropriado, os termos usados no singular também incluirão 0 plural e vice-versa.
[0049] Como aqui empregado, 0 termo "alquila" se refere a uma porção de hidrocarboneto ramificada ou não ramificada totalmente saturada. Preferivelmente a alquila compreende 1 a 6 átomos de carbono, mais preferivelmente 1 a 16 átomos de carbono, 1 a 10 átomos de carbono, 1 a 7 átomos de carbono, ou 1 a 4 átomos de carbono. Exemplos representativos de alquila incluem, porém não são limitados a, metila, etila, n-propila, /sopropila, n-butila, sec-butila, /sobutila, terc-butila, n-pentila, isopentila, neopentila, n-hexila, 3- metilaxila, 2,2- dimetilpentila, 2,3-dimetilpentila, n-heptila, n-octila, n- nonila, n- decila e os similares.
[0050] Como aqui empregado, o termo "alcóxi" se refere a alquil- O-, em que alquila é definida aqui acima. Exemplos representativos de alcóxi incluem, porém não são limitados a, metóxi, etóxi, propóxi, 2- propóxi, butóxi, terc-butóxi, pentilóxi, hexilóxi, ciclopropilóxi-, cicloexilóxi- e os similares. Como aqui empregado, o termo "alcóxi inferior" se refere aos grupos alcóxi tendo cerca de 1 a 7, preferivelmente cerca de 1 a 4 carbonos.
[0051] Como aqui empregado, o termo "acila" se refere a um grupo R-C(O)- de 1 a 10 átomos de carbono de uma configuração linear, ramificada, ou cíclica ou uma combinação destas, ligados a uma estrutura origem através de uma funcionalidade de carbolina. Tal grupo pode ser saturado ou insaturado, e alifático ou aromático. Preferivelmente, R no resíduo de acila é alquila, ou alcóxi, ou arila, ou heteroarila. Também preferivelmente, um ou mais carbonos no resíduo de acila podem ser substituídos por nitrogêgio, oxigênio ou enxofre contanto que o ponto de ligação à origem permaneça na carbonila. Exemplos incluem porém não são limitados a, acetila, benzoila, propionila, isobutirila, t-butoxicarbonila, benziloxicarbonila e os similares. A acila inferior se refere a uma acila contendo um a quatro carbonos.
[0052] Como aqui empregado, o termo "cicloalquila" se refere a grupos de hidrocarboneto saturados ou insaturados monocíclicos, bicíclicos ou tricíclicos opcionalmente substituídos de 3 a 12 átomos de carbono, cada dos quais pode ser substituído por um ou mais substituintes, tais como alquila, halo, oxo, hidróxi, alcóxi, alcanoíla, acilamino, carbamoíla, alquilamino, dialquilamino, tiol, alquiltio, nitro, ciano, carbóxi, alcoxicarbonila, sulfonila, sulfonamido, sulfamoíla, heterociclila e os similares. Grupos exemplares de hidrocarboneto monocíclico incluem, porém não são limitados a, ciclopropila, ciclobutila, ciclopentila, ciclopentenila, cicloexila e cicloexenila e os similares. Grupos exemplares de hidrocarboneto bicíclico incluem bornila, indila, hexaidroindila, tetraidronaftila, decaidronaftila, biciclo[2,1,1 Jhexila, biciclo[2,2,1 ]heptila, biciclo[2,2,1 Jheptenila, 6,6- dimetilbiciclo[3,1,1 ]heptila, 2,6,6-trimetilbiciclo[3,1,1 ]heptila, biciclo[2,2,2]octila e os similares. Grupos exemplares de hidrocarboneto tricíclico incuem adamantila e os similares.
[0053] Como aqui empregado, o termo "cicloalcóxi" se refere a grupos --O—cicloalquila.
[0054] O termo "arila" se refere a grupos hidrocarboneto aromáticos monocíclicos ou bicíclicos tendo 6 a 20 átomos de carbono na porção de anel. Preferivelmente, a arila é uma (Ce-Cw) arila. Exemplos não limitados incluem fenila, bifenila, naftila ou tetraidronaftila, cada dos quais pode opcionalmente ser substituído por 1 a 4 substituintes, tais como alquila, triflúormetila, cicloalquila, halogênio, hidróxi, alcóxi, acila, alquil-C(O)-O--, aril-O—, heteroaril-O--, amino, HS--, alquil-S—, aril-S—, nitro, ciano, carbóxi, alquil-O-C(O)--, carbamoíla, alquil-S(O)—, sulfonila, sulfonamido, heterociclila e os similares, em que R é independentemente hidrogênio, alquila, arila, heteroarila, aril-alquil—, heteroaril-alquil— e os similares.
[0055] Além disso, o termo "arila" como usado aqui, se refere a um substituinte aromático que pode ser um anel aromático singular, ou anéis aromáticos múltiplos que são fundidos um ao outro, ligados covalentemente, ou ligados a um grupo comum tal como uma porção de metilano ou etilano. O grupo de ligação comum também pode ser uma carbonila, como em benzofenona, ou oxigênio como em difeniléter, ou nitrogênio como em difenilamina.
[0056] Como aqui empregado, o termo "carbamoíla" se refere a H2NC(O)-, alquil-NHC(O)-, (alquil)2NC(O)-, aril-NHC(O)-, alquil(aril)- NC(O)-, heteroaril-NHC(O)-, alquil(heteroaril)-NC(O)-, aril-alquil- NHC(O)-, alquil(aril-alquil)-NC(O)- e os similares.
[0057] Como aqui empregado, o termo "sulfonila" se refere a R- SO2--, em que R é hidrogênio, alquila, arila, hereoarila, aril-alquila, heteroaril-alquila, aril-O—, heteroaril-O--, alcóxi, arilóxi, cicloalquila, ou heterociclila.
[0058] Como aqui empregado, 0 termo "sulfonamido" se refere a alquil-S(O)2-NH-, aril-S(O)2-NH-, aril-alquil-S(O)2-NH-, heteroaril-S(O)2- NH-, heteroaril-alquil-S(O)2-NH-, alquil-S(O)2-N(alquil)-, aril-S(O)2- N(alquil)-, aril-alquil-S(O)2-N(alquil)-, heteroaril-S(O)2-N(alquil)-, heteroaril-alquil-S(O)2-N(alquil)- e os similares.
[0059] Como aqui empregado, 0 termo "heterociclila" ou "heterociclo" se refere a um grupo cíclico opcionalmente substituído, totalmente saturado ou insaturado, aromático ou não aromático, por exemplo, que é um sistema de anel de 4 a 7 membros monocíclicos, 7 a 12 membros bicíclicos ou 10 a 15 membros tricíclicos, que tem pelo menos um heteroátomo em pelo menos um anel contendo átomo de carbono. Cada anel do grupo heterocíclico contendo um heteroátomo pode ter 1, 2 ou 3 heteroátomos selecionados a partir de átomos de nitrogênio, átomos de oxigênio e átomos de enxofre, onde os heteroátomos de nitrogênio e enxofre podem também opcionalmente ser oxidados. O grupo heterocíclico pode ser ligado a um heteroátomo ou átomo de carbono.
[0060] Grupos exemplares heterocíclicos monocíclicos incluem pirrolidinila, pirrolila, pirazolila, oxetanila, pirazolinila, imidazolila, imidazolinila, imidazolidinila, triazolila, oxazolila, oxazolidinila, isoxazolinila, isoxazolila, tiazolila, tiadiazolila, tiazolidinila, isotiazolila, isotiazolidinila, furila, tetraidrofurila, tienila, oxadiazolila, piperidinila, piperazinila, 2-oxopiperazinila, 2-oxopiperidinila, 2-oxopirrolodinila, 2- oxoazepinila, azepinila, 4-piperidonila, piridila, pirazinila, pirimidinila, piridazinila, tetraidropiranila, morfolinila, tiamorfolinila, sulfóxido de tiamorfolinila, sulfona de tiamorfolinila, 1,3-dioxolano e tetraidro-1,1- dioxotienila, 1,1,4-trioxo-1,2,5-tiadiazolidin-2-ila e os similares.
[0061] Grupos exemplares heterocíclicos bicíclicos incluem indolila, diidroidolila, benzotiazolila, benzoxazinila, benzoxazolila, benzotienila, benzotiazinila, quinuclidinila, quinolinila, tetraidroquinolinila, decaidroquinolinila, isoquinolinila, tetraidroisoquinolinila, decaidroisoquinolinila, benzimidazolila, benzopiranila, indolizinila, benzofurila, cromonila, coumarinila, benzopiranila, cinnolinila, quinoxalinila, indazolila, pirrolopiridila, furopiridinila (tal como furo[2,3-c]piridinila, furo[3,2-b]-piridinila] ou furo[2,3-b]piridiniI), diidroisoindolila, 1,3-dioxo-1,3-diidroisoindol-2-ila, diidroquinazolinila (tal como 3,4-diidro-4-oxo-quinazolinila), ftalazinila e os similares.
[0062] Grupos exemplares heterocíclicos tricíclicos incluem carbazolila, dibenzoazepinila, ditienoazepinila, benzindolila, fenantrolinila, acridinila, fenantridinila, fenoxazinila, fenotiazinila, xantenila, carbolinila e os similares.
[0063] O termo "heterociclila" também se refere a grupos heterocíclicos como definido aqui, substituído com 1, 2 ou 3 substituintes selecionados dos grupos consistindo nos seguintes: (a) alquila; (b) hidroxila (ou hidróxi protegido); (c) halo; (d) oxo, isto é, =0; (e) amino, alquilamino ou dialquilamino; (f) alcóxi; (g) cicloalquila; (h) carbóxi; (i) heterocicloóxi, em que heterocicloóxi denota um grupo heterociclico ligado através de uma ponte de oxigênio; 0) alquil-O-C(O)—; (k) mercapto; (l) nitro; (m) ciano; (n) sulfamoila ou sulfonamide; (o) arila; (p) alquil-C(O)-O--; (q) aril-C(O)-O—; (r) aril-S—; (s) arilóxi; (t) alquil-S--; (u) formila, isto é, HC(O)--; (v) carbamoila; (w) aril-alquil—; e (x) arila substituída com alquila, cicloalquila, alcóxi, hidróxi, amino, alquil-C(O)-NH--, alquilamino, dialquilamino ou halogênio.
[0064] Como aqui empregado, o termo "sulfamoila" se refere a H2NS(O)2-, alquil-NHS(O)2-, (alquil)2NS(O)2-, aril-NHS(O)2-, alquil(aril)- NS(O)2-, (aril)2NS(O)2-, heteroaril-NHS(O)2-, aralquil-NHS(O)2-, heteroaralquil-NHS(O)2- e os similares.
[0065] Como aqui empregado, o termo "arilóxi" se refere tanto a um grupo --O-arila quanto a um --O- heteroarila, em que arila e heteroarila são aqui definidos.
[0066] Como aqui empregado, o termo "heteroarila" se refere a um sistema de anel de 5 a 14 membros monocíclicos ou bicíclicos ou policíclico fundido, tendo 1 a 8 heteroátomos selecionados a partir de N, O ou S. Preferivelmente, a heteroarila é um sistema de anel de 5 a 10 ou 5 a 7 membros. Grupos típicos de heteroarila incluem 2- ou 3- tienila, 2- ou 3-furila, 2- ou 3-pirrolila, 2-, 4-, ou 5-imidazolila, 3-, 4-, ou 5- pirazolila, 2-, 4-, ou 5-tiazolila, 3-, 4-, ou 5-isotiazolila, 2-, 4-, ou 5- oxazolila, 3-, 4-, ou 5-isoxazolila, 3- ou 5-1,2,4-triazolila, 4- ou 5-1,2, 3- triazolila, tetrazolila, 2-, 3-, ou 4-piridila, 3- ou 4-piridazinila, 3-, 4- , ou 5-pirazinila, 2-pirazinila, 2-, 4-, ou 5-pirimidinila.
[0067] O termo "heteroarila" também se refere a um grupo no qual um anel heteroaromático é fundido a um ou mais anéis arila, cicloalifático, ou heterociclila, onde o radical ou ponto de ligação está sobre o anel heteroaromático. Exemplos não limitantes incluem porém não são limitados a 1-, 2-, 3-, 5-, 6-, 7-, ou 8- indolizinila, 1-, 3-, 4-, 5-, 6-, ou 7-isoindolila, 2-, 3-, 4-, 5-, 6-, ou 7-indolila, 2-, 3-, 4-, 5-, 6-, ou 7- indazolila, 2-, 4-, 5-, 6-, 7-, ou 8- purinila, 1-, 2-, 3-, 4-, 6-, 7-, 8-, ou 9- quinolizinila, 2-, 3-, 4-, 5-, 6-, 7-, ou 8-quinoliila, 1-, 3-, 4-, 5-, 6-, 7-, ou 8-isoquinoliila, 1-, 4-, 5-, 6-, 7-, ou 8-ftalazinila, 2-, 3-, 4-, 5-, ou 6- naftiridinila, 2-, 3- , 5-, 6-, 7-, ou 8-quinazolinila, 3-, 4-, 5-, 6-, 7-, ou 8- cinolinila, 2-, 4-, 6-, ou 7-pteridinila, 1-, 2-, 3-, 4-, 5-, 6-, 7-, ou 8-4aH carbazolila, 1-, 2-, 3-, 4-, 5-, 6-, 7-, ou 8-carbazolila, 1-, 3-, 4-, 5-, 6-, 7-, 8-, ou 9-carbolinila, 1-, 2-, 3-, 4-, 6-, 7-, 8-, 9-, ou 1O-fenantridinila, 1- , 2-, 3-, 4-, 5-, 6-, 7-, 8-, ou 9-acridinila, 1-, 2-, 4-, 5-, 6-, 7-, 8-, ou 9- perimidinila, 2-, 3-, 4-, 5-, 6-, 8-, 9-, ou 10-fenatrolinila, 1-, 2-, 3-, 4-, 6-, 7-, 8-, ou 9-fenazinila, 1-, 2-, 3-, 4-, 6-, 7-, 8-, 9-, ou 10-fenotiazinila, 1-, 2-, 3-, 4-, 6-, 7-, 8-, 9-, ou 10-fenoxazinila, 2-, 3-, 4-, 5-, 6-, ou I-, 3-, 4-, 5-, 6-, 7-, 8-, 9-, ou 10- benzisoquinolinila, 2-, 3-, 4-, ou tieno[2,3- b]furanila, 2-, 3-, 5-, 6-, 7-, 8-, 9-, 10 -, ou 11-7H-pirazino[2,3- c]carbazolila,2-, 3-, 5-, 6-, ou 7-2H- furo[3,2-b]-piranila, 2-, 3-, 4-, 5-, 7-, ou 8-5H-pirido[2,3-d]-o-oxazinila, 1-, 3-, ou 5-1H-pirazolo[4,3-d]- oxazolila, 2-, 4-, ou 5-4H-imidazo[4,5-d] tiazolila, 3-, 5-, ou 8- pirazino[2,3-d]piridazinila, 2-, 3-, 5-, ou 6- imidazo[2,1-b] tiazolila, 1-, 3-, 6-, 7-, 8-, ou 9-furo[3,4-c]cinolinila, 1-, 2-, 3-, 4-, 5-, 6-, 8-, 9-, 10, ou 11- 4H-pirido[2,3-c]carbazolila, 2-, 3-, 6-, ou 7-imidazo[1,2- b][1,2,4]triazinila, 7-benzo[b]tienila, 2-, 4-, 5-, 6-, ou 7-benzoxazolila, 2- , 4-, 5-, 6-, ou 7-benzimidazolila, 2-, 4-, 4-, 5-, 6-, ou 7-benzotiazolila, 1- , 2-, 4-, 5-, 6-, 7-, 8-, ou 9- benzoxapinila, 2-, 4-, 5-, 6-, 7-, ou 8- benzoxazinila, 1-, 2-, 3-, 5-, 6-, 7-, 8-, 9-, 10-, ou 11 -1 H-pirrolo[1,2- b][2]benzazapinila. Grupos heteroarila típicos fundidos incluem, porém não são limitados a 2-, 3-, 4-, 5-, 6-, 7-, ou 8-quinolinila, 1-, 3-, 4-, 5-, 6- , 7-, ou 8-isoquinolinila, 2-, 3-, 4-, 5-, 6-, ou 7-indolila, 2-, 3-, 4-, 5-, 6-, ou 7-benzo[b]tienila, 2-, 4-, 5- , 6-, ou 7-benzoxazolila, 2-, 4-, 5-, 6-, ou 7-benzimidazolila, 2-, 4-, 5-, 6-, ou 7-benzotiazolila.
[0068] Um grupo heteroarila pode ser mono-, bi-, tri-, ou policíclico, preferivelmente mono-, bi-, ou tricíclico, mais preferivelmente mono- ou bicíclico.
[0069] Como aqui empregado, o termo "halogênio" ou "halo" se refere a flúor, cloro, bromo, e iodo.
[0070] Como aqui empregado, o termo "acilamino" se refere a acil- NH-, em que "acila" é aqui definido.
[0071] Como aqui empregado, o termo "alcoxicarbonila" se refere a alcóxi-C(O)--, em que alcóxi é aqui definido.
[0072] Como aqui empregado, o termo "alcanoíla" se refere a alquil-C(O)--, em que alquila é aqui definido.
[0073] Como aqui empregado, o termo "alquenila" se refere a um grupo hidrocarboneto linear ou ramificado tendo 2 a 20 átomos de carbono e que contém pelo menos uma ligação dupla. Os grupos alquenila preferivelmente têm cerca de 2 a 8 átomos de carbono.
[0074] Como aqui empregado, o termo "haloalquila" se refere a uma alquila como definido aqui, que é substituída por um ou mais grupos halo como definido aqui. Preferivelmente a haloalquila pode ser monohaloalquila, dihaloalquila ou polihaloalquila incluindo peraloalquila. A monohaloalquila pode ter um iodo, bromo, cloro ou flúor no grupo alquila. Grupos dihaloalquila e polihaloalquila podem ter dois ou mais dos mesmos átomos de halo ou uma combinação de diferentes grupos halo na alquila. Preferivelmente, a polihaloalquila contém até 12, 10, ou 8, ou 6, ou 4, ou 3, ou 2 grupos halo. Exemplos não limitantes de haloalquila incluem flúormetila, diflúormetila, triflúormetila, clorometila, diclorometila, triclorometila, pentaflúoretila, heptaflúorpropila, diflúorclorometila, dicloroflúormetila, diflúoretila, diflúorpropila, dicloroetila e dicloropropila. Uma peraloalquila se refere a uma alquila tendo todos os átomos de hidrogênio substituídos por átomos de halo.
[0075] Como aqui empregado, o termo "haloalcóxi" se refere a haloalquil-O--, em que haloalquila é aqui definido.
[0076] Como aqui empregado, o termo "alquilamino" se refere a alquil-NH—, em que alquila é aqui definido.
[0077] Como aqui empregado, o termo "dialquilamino" se refere a (alquil)(alquil)N—, em que alquila é aqui definido.
[0078] Como aqui empregado, o termo "isômeros" se refere a compostos diferentes que têm a mesma Fórmula molecular. Também como aqui empregado, o termo "um isômero ótico" se refere a qualquer das várias configurações éstereoisoméricas que podem existir para um determinado composto da presente invenção e incluem isômeros geométricos. Entende-se que um substituinte pode ser ligado a um centro quiral de um átomo de carbono. Portanto, a invenção inclui enantiômeros, diastereômeros ou racematos do composto. "Enantiômeros" são pares de éstereoisômeros que são imagens de espelho uma da outra que não podem ser sobrepostas. Uma mistura de 1:1 de um par de enantiômeros é uma mistura "racêmica". O termo é usado para designa uma mistura racêmica onde apropriado. "Diastereoisômeros" são éstereoisômeros que têm pelo menos dois átomos assimétricos, porém que não são imagens de espelho um do outro. A éstereoquímica absoluta é especificada de acordo com o sistema Cahn- Ingold- Prelog R-S. Quando um composto é um enantiômero puro a éstereoquímica em cada carbono quiral pode ser especificada por R ou S. Os compostos resolvidos cuja a configuração absoluta é desconhecida pode ser designada (+) ou (-) dependendo da direção (dextro- ou levorotatoria) em que eles giram o plano de luz polarizada no comprimento de onda da linha D de sódio. Adicionalmente, os compostos resolvidos cuja a configuração absoluta é desconhecida podem ser designados por tempo de retenção (tr) de cromatografia líquida de alta pressão (HPLC) usando um absorvente quiral. Certos dos compostos descritos aqui contêm um ou mais centros assimétricos e pode desse modo dar origem a enantiômeros, diastereômeros, e outras formas éstereoisoméricas que podem ser definidas, em termos de éstereoquímica absoluta, como (/?)- ou (S)-. A presente invenção destina-se a incluir todos os tais possíveis isômeros, incluindo misturas racêmicas, formas oticamente puras e misturas intermediária. Isômeros (/?) e (S) oticamente ativos podem ser preparados usando síntons quirais ou reagentes quirais, ou resolvidos usando técnicas convencionais. Se o composto contem uma ligação dupla, o substituinte pode ser de configuração E ou Z. Se o composto contém uma cicloalquila dissubstituída, o substituinte cicloalquila pode ter uma configuração cis ou trans. Pretende-se também que todas as formas tautoméricas sejam incluídas.
[0079] Como aqui empregado, o termo "sais farmaceuticamente aceitáveis" se refere a sais que retêm a eficácia e propriedades biológicas dos compostos desta invenção e, que não são biologicamente ou de outro modo indesejáveis. Em muitos casos, os compostos da presente invenção são capazes de fomar sais de ácido e/ou base em virtude da presença de grupos de amino e/ou carboxila ou grupos similares a este. Os sais de adição de ácido farmaceuticamente aceitáveis podem ser formados com ácidos inorgânicos e ácidos orgânicos. Ácidos inorgânicoa dos quais sais podem ser derivados incluem, por exemplo, ácido clorídrico, ácido bromídrico, ácido sulfúrico, ácido nítrico, ácido forfórico, e os similares. Ácidos orgânicos dos quais sais podem ser derivados incluem, por exemplo, ácido acético, ácido propriônico, ácido glicólico, ácido pirúvico, ácido oxálico, ácido maléico, ácido malônico, ácido succínico, ácido fumárico, ácido tartárico, ácido cítrico, ácido benzóico, ácido cinámico, ácido mandélico, ácido metanossulfônico, ácido etanossulfônico, ácido p- toluenossulfônico, ácido salicílico, e os similares. Sais de adição de base farmaceuticamente aceitáveis podem ser formados com bases inorgânicas e orgânicas. Bases inorgânicas das quais sais podem ser derivados incluem, por exemplo, sódio, potássio, lítio, amónio, cálcio, magnésio, ferro, zinco, cobre, manganês, alumínio, e os similares; particularmente preferidos são os sais de amónio, potássio, sódio, cálcio e magnésio. Bases orgânicas das quais sais podem ser derivados incluem, por exemplo, aminas primárias, secundárias e terciárias, aminas substituídas incluindo aminas substituídas de ocorrência natural, aminas cíclicas, resinas de permuta de íon básicas, e as similares, especificamente tais como isopropilamina, trimetilamina, dietilamina, trietilamina, tripropilamina, e etanolamina. Os sais farmaceuticamente aceitáveis da presente invenção podem ser sintetizados a partir de um composto origem, uma porção básica ou acídica, por métodos químicos convencionais. Geralmente, tais sais podem ser preparados por reação de formas de ácido livre destes compostos com uma quantidade estequiométrica de uma base apropriada (tais como hidróxido, carbonato, bicarbonato de Na, Ca, Mg, ou K , ou os similares), ou por reação de formas de bases livres destes compostos com uma quantidade estequiométrica de um ácido apropriado. Tais reações são tipicamente realizadas em água ou em solvente orgânico, ou em uma mistura dos dois. Geralmente, meios não aquosos tipo éter, acetato de etila, etanol, isopropanol, ou acetonitrila são preferidos, onde praticados. Listas de sais adequados adicionais podem ser encontradas, por exemplo, em Remington's Pharmaceutical Sciences, 20a edição, Mack Publishing Company, Easton, Pa., (1985), que é aqui incorporada por referência.
[0080] Como aqui empregado, o termo "veículo farmaceuticamente aceitável" inclui qualquer e todos os solventes, meios de dispersão, revestimentos, tensoativos, antioxidantes, conservantes (por exemplo, agentes antibacterianos, agentes antifúngicos), agentes isotônicos, agentes de retardo da absorção, sais, conservantes, fármacos, estabilizadores de fármacos, aglutinantes, excipientes, agentes de desintegração, lubrificantes, agentes adoçantes, agentes flavorizantes, corantes, tais como materiais e combinações destes, como será conhecido por alguém versado na técnica (vide, por exemplo, Remington's Pharmaceutical Sciences, 18a edição. Mack Printing Company, 1990, pp. 1289- 1329, incorporada aqui por referência). Exceto a medida em que qualquer veículo convencional é incompatível com ingredientes ativos, seu uso nas composições farmacêuticas e terapêuticas é contemplado.
[0081] O termo "quantidade terapeuticamente eficaz" de um composto da presente invenção se refere a uma quantidade de um composto da presente invenção que eliciará a resposta biológica ou médica de um indivíduo, ou melhorará sintomas, tornará mais lenta ou retardará a progressão da doença, ou prevenirá uma doença, etc. Em uma modalidade preferida, a "quantidade eficaz" se refere à quantidade que inibe ou reduz a expressão de sintase ou aromatase de aldosterona.
[0082] Como aqui empregado, o termo "indivíduo" se refere a um animal. Preferivelmente, o animal é um mamífero. Um indivíduo também se refere a por exemplo, primatas (por exemplo, seres humanos), vacas, ovelhas, cabras, cavalos, cães, gatos, coelhos, ratos, camundongos, peixes, pássaros e os similares. Em uma modalidade preferida, o indivíduo é um ser humano.
[0083] Como aqui empregado, o termo "um distúrbio" ou "uma doença" se refere a qualquer insanidade ou anormalidade de função; um estado mental ou físico mórbido. Vide Dorland's Illustrated Medical Dictionary, (W.B. Saunders Co. 27a ed. 1988).
[0084] Como aqui empregado, o termo "inibição" ou "inibindo" se refere à redução ou supressão de uma determinada condição, sintoma, ou doença, ou um decréscimo na atividade de linha de base de uma atividade ou processo biológico. Preferivelmente, a condição é devido à expressão anormal de sintase ou aromatase de aldosterona e a atividade ou processo biológico está associada com a expressão anormal de sintase ou aromatase de aldosterona.
[0085] Como aqui empregado, o termo "tratar" ou "tratamento" de qualquer doença ou distúrbio se refere em uma modalidade, à melhora da doença ou distúrbio (isto é, interrupção ou redução do desenvolvimento da doença ou pelo menos um dos sintomas clínicos desta). Em outra modalidade "tartar" ou "tratamento" se refere à melhora de pelo menos um parâmetro físico, que não pode ser discernível pelo paciente. Em ainda outra modalidade, "tratar" ou "tratamento" se refere à modulação da doença ou distúrbio, ou fisicamente, (por exemplo, estabilização de um sintoma discernível), fisiologicamente, (por exemplo, estabilização de um parâmetro físico), ou ambos. Em ainda outra modalidade, "tratar" ou "tratamento" se refere a prevenir ou atrasar o início ou desenvolvimento ou progressão da doença ou distúrbio.
[0086] Como aqui empregado, o termo "anormal" se refere a uma atividade ou característica que difere de uma atividade ou característica normal.
[0087] Como aqui empregado, o termo "atividade anormal" se refere a uma atividade que difere da atividade da proteína ou gene nativo ou tipo silvestre, ou que difere da atividade do gene ou proteína em um indivíduo sadio. A atividade anormal pode ser mais forte ou mais fraca do que a atividade normal. Em uma modalidade, a "atividade anormal" inclui a anormal (ou super- ou sub-) produção de mRNA transcrito de um gene. Em outra modalidade, a "atividade anormal" inclui a anormal (ou super- ou sub-) produção de polipeptídeo de um gene. Em outra modalidade, a atividade anormal se refere a um nível de um mRNA ou polipeptídeo que é diferente de um nível normal do referido mRNA ou polipeptídeo por cerca de 15%, cerca de 25%, cerca de 35%, cerca de 50%, cerca de 65%, cerca de 85%, cerca de 100% ou mais. Preferivelmente, o nível anormal do mRNA ou polipeptídeo pode ser superior ou inferior ao nível normal do referido mRNA ou polipeptídeo. Todavia outra modalidade, a atividade anormal se refere à atividade funcional da proteína que é diferente a partir da atividade normal da proteína tipo silvestre, devido a mutações no gene correspondente. Preferivelmente, a atividade anormal pode ser mais forte ou mais fraca do que a atividade normal. As mutações podem ser na região de codificação do gene ou regiões de não codificação tais como as regiões promotoras transcricionais. As mutações podem ser substituições, deleções, inserções.
[0088] Como aqui empregado, o termo "um, uma (a)" "um, uma (an)," "o,a" e termos similares usados no contexto da presente invenção (especialmente no contexto das reivindicações) devem ser construídos para abrander tanto o singular quanto o plural, a menos que de outro modo indicado aqui ou claramente contradito pelo contexto. Menção de faixas de valores aqui são meramente destinadas a servir como um método sintetizado de referir-se individualmente a cada valor separado incluindo-se na faixa. A menos que de outro modo indicado aqui, cada valor individual é incorporado na especificação como se ele fosse individualmente mencionado aqui. Todos os métodos descritos aqui podem ser realizados em qualquer ordem adequada a menos que de outro modo indicado aqui ou de outro modo claramente contradito pelo contexto. O uso de qualquer e todos os exemplos, ou linguagem exemplares (por exemplo, "tal como") fornecidos aqui é pretendido meramente para melhor iluminar a invenção e não apresenta uma limitação sobre o escopo da invenção de outro modo reivindicado. Nenhuma linguagem na especificação deve ser construída como indicando qualquer elemento essencial não- reivindicado para a prática da invenção.
[0089] Qualquer átomo de carbono assimétrico nos compostos da presente invenção pode estar presente na configuração (/?), (S) ou (R,S), preferivelmente na configuração (R) ou (S). Substituintes a átomos com ligações insaturadas podem, se possível, estar presente na forma cis-(Z)- ou trans(E). Portanto, os compostos da presente invenção podem estar na forma de um dos possíveis isômeros ou misturas destes, por exemplo, isômeros geométricos substancialmene puros (c/s ou trans), diastereômeros, isômeros óticos (antípodas), racematos ou misturas destes.
[0090] Quaisquer misturas de isômeros podem ser separadas com base nas diferenças fisico químicas dos constituintes, nos isômeros geométricos ou óticos puros, diastereômeros, racematos, por exemplo, por cromatografia e/ou cristalização fracionai.
[0091] Quaisquer racematos resultantes de produtos finais ou intermediários podem ser resolvidas nos antípodas óticos por métodos conhecidos, por exemplo, por separação dos sais diastereoméricos destes, obtido com um ácido ou base oticamente ativo, e liberando o composto acídico ou básico oticamente ativo. Em particular, a porção de imidazolila pode desse modo ser empregada para resolver os compostos da presente invenção em seus antípodas óticos, por exemplo, por cristalização fracionai de um sal formado com um ácido oticamente ativo, por exemplo, ácido tartárico, ácido dibenzoiltartárico, ácido diacetiltartárico, ácido di-O,O'-p-toluoil tartárico, ácido mandélico, ácido málico ou ácido camfor-10-sulfônico. Produtos racêmicos podem também ser resolvidos por cromatografia quiral, por exemplo, cromatografia líquida de alta pressão (HPLC) usando um adsorvente quiral.
[0092] Finalmente, compostos da presente invenção são ou obtidos na forma livre, como um sal destes, ou como derivados de pro- fármacos destes.
[0093] Quando um grupo básico está presente nos compostos da presente invenção, os compostos podem ser convertidos em sais de adição de ácido destes, em particular, sais de adição de ácido com a porção de imidazolila da estrutura, preferivelmente sais farmaceuticamente aceitáveis destes. Estes são formados, com ácidos inorgânicos ou ácidos orgânicos. Os ácidos inorgânicos adequados incluem, porém não estão limitados a, ácido clorídrico, ácido sulfúrico, um ácido fosfórico ou halídrico. Os ácidos orgânicos adequados incluem, porém não estão limitados a, ácidos carboxílicos, tais como ácidos (Ci-C4)alcanocarboxílico que, por exemplo, são não substituídos ou substituídos por halogênio, por exemplo, ácido acético, tal como ácidos dicarboxílicos saturados ou insaturados, por exemplo, ácido oxálico, succínico, maléico ou fumárico, tal como ácidos hidroxicarboxílico, por exemplo, ácido glicólico, lático, málico, tartárico ou cítrico, tais como aminoácidos, por exemplo, ácido aspártico ou glutâmico, sulfônicos orgânicos, tais como ácidos (Ci- C4)alquilsulfônicos, por exemplo, ácido metanossulfônico; ou ácidos arilsulfônicos que são não substituídos ou substituídos, por exemplo, por halogênio. São preferidos os sais formados com ácido clorídrico, ácido metanossulfônico e ácido maléico.
[0094] Quando um grupo acídico está presente nos compostos da presente invenção, os compostos podem ser convertidos em sais com bases farmaceuticamente aceitáveis. Tais sais incluem sais de metal de álcali, similares aos sais de sódio, lítio e potássio; sais de metal alcalino terroso, similares aos sais de cálcio e magnésio; saus de amónios com bases orgânicas, por exemplo, sais de trimetilamina, sais de dietilamina, sais de fns(hidroximetil)metilamina, sais de dicicloexilamina e sais de /V-metil-D-glucamina; sais com aminoácidos similares à arginina, lisina e os similares. Os sais podem ser formados usando métodos convencionais, vantajosamente na presença de um solvente etéreo ou alcoólico, tal como um alcanol inferior. A partir das soluções do último, os sais podem ser precipitados com éteres, por exemplo, éter de dietila. Os sais resultantes podem ser convertidos nos compostos por tratamento com ácidos. Estes ou outros sais podem também ser usados por purificação dos compostos obtidos.
[0095] Quando tanto um grupo básico quanto um grupo ácido estão presentes na mesma molécula, os compostos da presente invenção podem também formar sais internos.
[0096] A presente invenção também fornece pro-fármacos dos compostos da presente invenção que convertem-se in vivo nos compostos da presente invenção. Um pro-fármaco é um composto ativo ou inativo que é modificado quimicamente através de ação fisiológica in vivo, tal como hidrólise, metabolismo e os similares, em um composto desta invenção seguindo a administração do pro-fármaco ao indivíduo. A adequabilidade e as técnicas envolvidas na preparação e uso de pro-fármacos são bem-conhecidos por aqueles versados na técnica. Pro-fármacos podem ser conceitualmente dividido em duas categorias não em duas categorias não-exclusivas, pro-fármacos bioprecursorores e pro-fármacos veículos. Observe, The Practice Medicinal Chemistry, Capítuos 31-32 (Ed. Wermuth, Academic Press, San Diego, Calif., 2001). Geralmente, pro-fármacos bioprecursorores são os compostos que são inativos ou têm baixa atividade em comparação ao correspondente composto de fármaco ativo, que contém um ou mais grupos protetores e são convertidos em uma forma ativa por metabolismo ou solvólise. Tanto a forma de fármaco ativo quanto quaisquer produtos metabólicos liberados devem aceitavelmente ter toxicidade aceitavelmente baixa. Tipicamente, a formação de compostos de fármaco ativo envolve uma reação ou processo metabólico que é um dos seguintes tipos: 1. Reações oxidativas, tais como oxidação de álcool, carbonila, e funções de ácido, hidroxiação de carbonos alifáticos, hidroxiação de átomos de carbono cíclicos, oxidação de átomos de carbono aromáticos, oxidação de ligações duplas de carbono-carbono, oxidação de grupos funcionais contendo nitrogênio, oxidação de silício, fósforo, arsênico, e enxofre, N-desalquilação oxidativa, desalquilação de O e S oxidativas, desaminação oxidativa, bem como outras reações oxidativas. 2. Reações oxidativas, tais como redução de grupos carbonila, redução de grupos alcoólicos e ligações duplas de carbono- carbono, redução de grupos de funções contendo nitrogênio, e outras reações de redução. 3. Reações sem mudança no estado de oxidação, tal como hidrólise de ésteres e éteres, clivagem hidrolítica de ligações únicas de carbono-nitrogênio, clivagem hidrolítica de heterociclos não- aromáticos, hidratação e desidratação de ligações múltiplas, novas ligações atômicas que resultam a partir de reações de desidratação, desalogenação hidrolítica, remoção de molécula de haleto de hidrogênio, e outras tais reações.
[0097] Os pro-fármacos veículos são compostos de fármaco que contêm uma porção de transporte, por exemplo, que melhora a absorção e/ou liberação localizada para síteo (s) de ação. Desejavelmente para um tal pro-fármaco veículo, a ligação entre a porção de fármaco e a porção de transporte é uma ligação covalente, o pro-fármaco é inativo ou menos ativo do que o composto de fármaco, e qualquer porção de transporte liberada é aceitavelmente não tóxico. Para pro-fármacos onde a porção de transporte é destinada a realçar a absorção, tipicamente a liberação da porção de transporte deve ser rápida. Em outros casos, é desejável utilizar uma porção que forneça lenta liberação, por exemplo, certos polímeros ou outras porções, tais como ciclodextrinas. Observe, Cheng e outros, US20040077595, Pedido Número de Série 10/656,838, incorporado aqui por referência. Tais pro-fármacos veículos são freqüentemente vantajosos para fármacos oralmente administrados. Pro-fármacos veículos podem, por exemplo, ser usados para melhorar uma ou mais das seguintes propriedades: lipofilicidade aumentada, duração aumentada de efeitos farmacológicoss, especificidade do sítio aumentada, toxicidade diminuída e reações adversas, e/ou melhora em formulação de fármaco (por exemplo, estabilidade, solubilidade em água, supressão de uma propriedade organoléptica ou fisioquímica indesejável). Por exemplo, a lipofiliciade pode ser aumentda por ésterificação de gupos hidróxi com ácidos carboxílicos lipofílicos, ou de grupos de ácido carboxílico com álcoois, por exemplo, álcoois alifáticos. Wermuth, The Practice de Medicinal Chemistry, Capítulo 31-32, Ed. Werriuth, Academic Press, San Diego, Calif., 2001.
[0098] Pro-fármacos exemplares são, por exemplo, ésteres de ácidos carboxílicos livres e derivados de S-acila e O-acila de tióis, álcoois ou fenóis, em que a acila tem um significado como aqui definido. São preferidos os derivados de éster farmaceuticamente aceitáveis convertíveis por solvólise sob condições fisiológicas para o ácido carboxílico de origem, por exemplo, ésteres de alquila inferior, ésteres de cicloalquila, ésteres de alquenila inferior, ésteres de benzila, ésteres de alquila inferior mono- ou dissubstituída, tal como os ésteres de ω-(amino, mono- ou dialquilamiπo inferior, carbóxi, alcoxicarbonil inferior)-alquila inferior, os ésteres de oc-( di-alcanoilóxi inferior, alcoxicarbonila inferior ou alquilaminocarbonila inferior)-alquila inferior, tai como o éster de pivaloiloximetila e os similares convencionalmente usados na técnica. Além disso, aminas têm sido mascaradas como derivados de arilcarboniloximetila substituída que são clivados por ésterases in vivo liberando o fármaco livre e formaldeído (Bundgaard, J. Med. Chem. 2503 (1989)). Além disso, fármacos contendo um grupo NH acídico, tal como imidazol, imida, indol e os similares, têm sido mascarados com grupos N-aciloximetila (Bundgaard, Design of Prodrugs, Elsevier (1985)). Grupos hidróxi têm sido mascarados como ésteres e éteres. A EP 039.051 (Sloan e Little) descreve pro-fármacos de ácido hidróxâmico de base Mannich, sua preparação e uso.
[0099] Em vista da ligação próxima entre os compostos, os compostos na forma de seus sais e os pro-fármacos, qualquer referência aos compostos da presente invenção deve ser entendida como referindo-se também aos correspondendes pro-fármacos dos compostos da presente invenção, como apropriado e conveniente.
[00100] Além disso, os compostos da presente invenção, incluindo seus sais, podem também ser obtidos na forma de seus hidratos, ou incluir outros solventes usados para sua cristalização.
[00101] Os compostos da presente invenção têm propriedades farmacológicas valiosas. Os compostos da presente invenção são úteis como inibidores de sintase de aldosterona. A sintase de aldosterona (CYP11B2) é uma enzima P450 de citocromo mitocondrial que cataliza a última etapa de produção de aldosterona no córtex adrenal, isto é, a conversão de 11-deoxicorticosterona em aldosterona. A síntese de aldosterona foi demonstrada ser expressa em todos os tecidos cardiovasculares tal como células do coração, cordão umbilical, artérias mesentéricas e pulmonares, aorta, endotélio e vasculares. Além disso, a expressão de sintase de aldosterona está intimamente correlacionada com produção de aldosterona em células. Observou-se que elevações de atividades de aldosterona ou níveis de aldosterona induzem diferentes doenças tais como insuficiência cardíaca congestiva, fibrose cardíaca ou miocardiana, insuficiência renal, hipertensão, arritmia ventricular e outros efeitos adversos, etc., e que a inibição de aldosterona ou sintase de aldosterona seriam métodos terapêuticos úteis. Observe, por exemplo, Ulmschenider e outro "Development e evaluation of a pharmacofore model for inibitors os aldosterone synthase(CYP11B2)," Bioorganic & Medicinal Chemistry Letters, 16: 25-30 (2006); Bureik e outro , "Development of test systems for the discovery of selective human aldosteron synthase (CYP11B2) e 11β-hidroxilase (CYP11B1) inibitors, discovery of a new lead compound for the therapy of congestive heart failure, myocardial fibrosis e hipertension," Moleculare e Cellular Endocrinology, 217: 249- 254 (2004); Bos e outro , "Inhibition of catechnolamine-induced cardiac fibrosis by an aldosteron antagonist," J. Cardiovascular Pharmacol, 45(1): 8-13 (2005); Jaber e Madias, "Progression of chronic kidney disease: can it be prevented ou arrested?" Am. J. Med. 118(12): 1323- 1330 (2005); Khan e Movahed, "The role of aldosterone e aldosterone- receptor antagonists in heart failure," Rev. Cardiovasc Med., 5(2): 71- 81 (2004); Struthers, "Aldosterone in heart failure: pathophysiology e treatment," Cyrr. Heart Fail., 1(4): 171-175( 2004); Harris e Rangan, "Retardation of kidney failure - applying principles to practice," Ann. Acad. Med. Singapore, 34(1): 16-23 (2005); Arima, "Aldosterone e the kidney: rapid regulation of renal microcirculation," Steroids, online publication November 2005; Brown, "Aldosterone e end-organ damage," Curr. Opin. Nephrol Hypertens, 14:235-241 (2005); Grandi, "Antihypertensive therapy: role of aldosteron antagonists," Curr. Pharmaceutical Design, 11: 2235-2242 (2005); Declayre e Swynghedauw, "Molecular mechanisms of myocardial remodeling: the role of aldosterone," J. Mol. Cell. Cardiol., 34: 1577-1584 (2002). Conseqüentemente, os compostos da presente invenção como inibidores de sintase de aldosterona, são também úteis para o tratamento de um distúrbio ou doença mediada por sintase de aldosterona ou responsiva à inibição de sintase de aldosterona. Em particular, os compostos da presente invenção como sintase de inibidores de aldosterona são úteis para o tratamento de um distúrbio ou doença caracterizada por sintase anormal de aldosterona atividade. Preferivelmente, os compostos da presente invenção são também úteis para o tratamento de um distúrbio ou doença selecionada a partir de hipocalemia, hipertensão, insuficiência cardíaca congestiva, fibrilação atrial, insuficiência renal, em particular, insuficiência renal crônica, restenose, aterosclerose, síndrome X, obesidade, nefropatia, infarto pós-miocardiano, doenças cardíacas coronarianas, inflamação, formação aumentada de colágeno, fibrose tal como fibrose cardíaca ou miocardiana e remodelagem após hipertensão e disfunção endotelial.
[00102] Além disso, os compostos das presentes invenções são úteis como inibidores de aromatase. Aromatase é uma enzima P450 de citocromo, ela desempenha um papel central na biossíntese extragonadal de estrogênios tais como estradiol, estrona e estrol, e é amplamente distribuída em tecido muscular e de adipose (Longcope C, Pratt J H, Schneider S H, Fineberg S E, 1977, J. Clin. Endocrinol. Metab. 45:1134- 1145). Um aumento em atividade de aromatase foi confirmada estar associada com distúrbios ou doenças dependentes de estrogênio. Conseqüentemente, os compostos da presente invenção são também úteis para o tratamento de um distúrbio ou doença caracterizada por expressão anormal de aromatase. Preferivelmente, os compostos da presente invenção são úteis para o tratamento de um distúrbio ou doença dependente de estrogênio. Mais preferivelmente, os compostos da presente invenção são úteis para o tratamento de um distúrbio ou doença dependente de estrogênio selecionada a partir de ginecomastia, osteoporose, câncer de próstata, endometriose, fibróides uterinos, sangramento uterino disfuncional, hiperplasia endometrial, doença ovariana policística, infertilidade, doença de mama fibrocística, câncer de mama e mastopatia fibrocística.
[00103] Adicionalmente, a presente invenção fornece: - um composto da presente invenção para uso como um medicamento; - o uso de um composto da presente invenção para a preparação de uma composição farmacêutica para o retardo de progressão e/ou tratamento de um distúrbio ou doença mediada por sintase de aldosterona, ou responsiva à inibição de sintase de aldosterona, ou caracterizada por atividade anormal ou expressão de sintase de aldosterona. - o uso de um composto da presente invenção para a preparação de uma composição farmacêutica para o retardo de progressão e/ou tratamento de um distúrbio ou doença mediada por aromatase, ou responsiva à inibição de aromatase, ou caracterizada por atividade anormal ou expressão de aromatase. - o uso de um composto da presente invenção para a preparação de uma composição farmacêutica para o retardo de progressão e/ou tratamento de um distúrbio ou doença selecionada a partir de hipocalemia, hipertensão, insuficiência cardíaca congestiva, fibrilação atrial, insuficiência renal, em particular, insuficiência renal crônica, restenose, aterosclerose, síndrome X, obesidade, nefropatia, infarto pós-miocardiano, doenças cardíacas coronarianas, formação aumentada de colágeno, fibrose, tais como fibrose cardíaca ou miocardiana e remodelagem após hipertensão e disfunção endotelial. - o uso de um composto da presente invenção para a preparação de uma composição farmacêutica para o retardo de progressão e/ou tratamento de um distúrbio ou doença selecionada a partir de ginecomastia, osteoporose, câncer de próstata, endometriose, fibróides uterinos, sangramento uterino disfuncional, hiperplasia endometrial, doença ovariana policística, infertilidade, doença de mama fibrocística, câncer de mama e mastopatia fibrocística.
[00104] Os compostos de Fórmulas (l)-(IV) podem ser preparados para os procedimentos descritos nas seguintes seções.
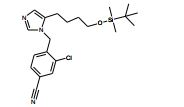
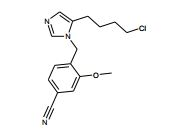
[00105] Geralmente, os compostos de Fórmula (II) podem ser preparados de acordo com Esquema 1, que contém 13 etapas. ESQUEMA 1
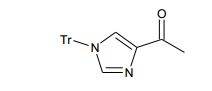
[00106] Como para as etapas individuais no Esquema acima, etapa 1 envolve a introdução de um grupo de proteção adequado no N1 do imidazol de (V), preferivelmente trifenilmetila, reagindo (V) com um reagente adequado tal como cloreto de trifenilmetila, na presença de piridina. A etapa 2 envolve a redução do ácido carboxílico com um reagente de redução adequado, preferivelmente complexo de BH3*THF. A Etapa 3 envolve a proteção do álcool resultante a partir da etapa 2 como um éter de silila, preferivelmente como éter de t- butildimetilsilila, com um reagente adequado tal como cloreto de t- butildimetilsilila na presença de uma base adequada, preferivelmente EtsN ou imidazol, e um solvente aprótico, preferivelmente DMF ou CH2CI2 para fornecer (VI).
[00107] Altemativamente (VI) podem ser preparados a partir de (V) por uma seqüência de quatro etapas. Na etapa 1 (V) é convertido no correspondente éster de metila em reação com metanol na presença de um ácido, preferivelmente HCI. A etapa 2 envolve a proteção de N1 do imidazol, preferivelmente com trifenilmetila, em reação com cloreto de trifenilmetila na presença de uma base adequada, preferivelmente EtsN. A Etapa 3 envolve a redução do éster formado na etapa 1 em reação com um reagente de redução adequado, preferivelmente LÍAIH4, em um solvente aprótico, preferivelmente THF. A etapa 4 envolve a proteção da porção de álcool resultante como um éter de silila como descrito na etapa 3 do parágrafo precedente para fornecer (VI).
[00108] A etapa 4 envolve a reação de um (VI) com 0 reagente de alquilação apropriado (VII), tal como X = Br, em um solvente aprótico, preferivelmente CH3CN para fornecer (VIII). Agentes de alquilação (VII) ou (IX) podem ser preparados por tratamento do correspondente derivado de éster de ácido fenilacético ou tolueno com um agente de bromação adequado, por exemplo, NBS, na presença de um iniciador de radical adequado, tal como AIBN ou peróxido de benzoíla. Alternativamente, agentes de alquilação (VII) podem ser gerados por conversão de um benzilálcool substituído no correspondente haleto por tratamento com, por exemplo, CBr4 e PPha.
[00109] A etapa 6 envolve a reação de (VIII) com uma base adequada, preferivelmente LHMDS, e reagente eletrofílico adequado, preferivelmente cianometilformiato ou clorometilformiato. A etapa 7 envolve a remoção do grupo de proteção de t-butildimetilsilila no tratamento com ácido, preferivelmente HCI, para fornecer éster (X).
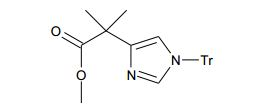
[00110] Alternativamente (X) podem ser preparados por alquilação de (VI) com um reagente de alquilação apropriado (IX), preferivelmente onde X = Br, mostrado na etapa 5 seguida por remoção do grupo de proteção de silila como descrito na etapa 7.
[00111] A etapa 8 envolve a conversão de álcool (X) no grupo de partida adequado, preferivelmente mesilato, reagindo (X) com cloreto de metanossulfonila na presença de uma base adequada, preferivelmente EtsN, e um solvente aprótico, preferivelmente CH2CI2. A etapa 9 envolve a alquilação intramolecular na reação do mesilato a partir da etapa 8 com uma base adequada, preferivelmente EtsN, em um solvente aprótico polar, preferivelmente DMF ou CH3CN, para fornecer compostos de Fórmula (I) onde R = CO2alquila.
[00112] Adicionalmente, compostos a partir da etapa 9 onde R = CO2alquila, podem ser tratados com um alcóxido de metal adequado, preferivelmente hidróxido de lítio em um solvente, por exemplo, H2O e THF, para fornecer compostos a partir da etapa 10 onde R = CO2H. A etapa 11 envolve descarboxilação dos compostos, onde R = CO2H em aquecimento em um solvente adequado, preferivelmente DMSO, para fornecer compostos a partir da etapa 12 onde R = H.
[00113] Compostos adicionais de Fórmula (I) podem ser preparados a partir da conversão de carboxilico ácido (I), onde R = CO2H, no correspondente cloreto de ácido no tratamento com um reagente de cloração adequado, preferivelmente cloreto de oxalila, em um solvente aprótico, preferivelmente CH2CI2. O cloreto de ácido obtido é então reagido com 0 apropriado nucleófilo, preferivelmente um álcool ou uma amina, na presença de uma base adequada para fornecer compostos de Fórmula (I) onde R = CO2R10 ou CO2NR11NR12 (etapa 12).
[00114] Alternativamente, os compostos de Fórmula (II) podem ser preparados de acordo com Esquema 2, que contém quatro etapas. ESQUEMA 2
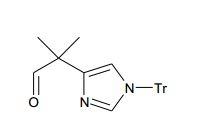
[00115] Como para as etapas individuais no Esquema 2 acima, etapa 1 envolve redução do éster carboxílico conhecido (XI) no correspondente aldeído (XII) no tratamento com um reagente de redução adequado, preferivelmente DIBAL-H, e um solvente aprótico, preferivelmente CH2CI2. A etapa 2 envolve a reação de aldeído (XII) com um apropriado reagente organometálico (XIII), preferivelmente onde M = Li, MgBr, ou MgCI, para fornecer álcool (XIV). Os reagentes organometálicos (XIII) são obtidos a partir de fontes comerciais ou gerados sob condições padrão pela ação de uma base forte, por exemplo, n-BuLi.
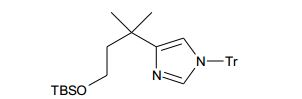
[00116] A Etapa 3 envolve a conversão da porção de álcool em (XIV) em um grupo de partida, preferivelmente mesilato, na reação de (XIV) com cloreto metanossulfonila, e uma base adequada, preferivelmente EtsN, em um solvente, preferivelmente CH2CI2. A etapa 4 envolve a alquilação de N3 intramolecular do imidazol sob aquecimento do mesilato preparado na etapa 3 em um solvente aprótico polar, preferivelmente CH3CN ou DMF para fornecer os compostos de Fórmula (II).
[00117] Altemativamente, os compostos de Fórmula (II) podem ser preparados a partir de outros compostos de Fórmula (II), onde Ri, R2, ou R3 representa um halogênio ou pseudo-halogênio, por exemplo, brometo ou triflato por acoplamento catalisado por paládio ou cobre da alquila, alquenila, ou ácido arilborônico, éster borônico, ou boroxina; organoestanano; organozinco; alcóxido de metal; álcool; amida; ou os similares pra produzir um correspondente análogo de alquila, cicloalquila, arila, heteroarila, alcóxi, arilóxi, ou acilamino. Estas transformações envolvem a conversão de compostos de Fórmula (II) onde Ri, R2, e/ou R3 podem ser iguais a um halogênio ou pseudohalogênio, tais como Br, a compostos de Fórmula (II) onde Ri, R2, e/ou R3 podem ser alquila ou arila por acoplamento transversal Suzuki com um ácido borônico, ou os similares, na presença de um catalisador, preferivelmente Pd(PPh3)4, uma base, preferivelmente hidróxido de potássio e carbonato de sódio, para fornecer compostos de Fórmula (II). Compostos adicionais de Fórmula (II) são preparados a partir de compostos existentes de Fórmula (II) por manipulação independente de radicals R, Ri, R2, R3, R4, e Rs por métodos conhecidos por aqueles versados na técnica, tais como, por exemplo, redução de um grupo nitro em uma anilina ou redução de um éster em um álcool.
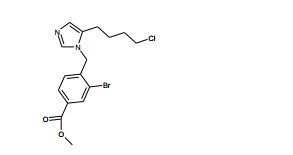
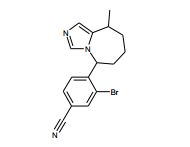
[00118] Alternativamente, os compostos de Fórmula (II) podem ser preparados de acordo com Esquema 3, que contém três etapas. ESQUEMA 3
[00119] Como para as etapas individuais em Esquema 3, a etapa 1 envolve a alquilação de N3 de imidazol (XV) com eletrófilos (VII) para fornecer (XVI). A etapa 2 envolve a conversão do álcool de (XVI) em um grupo de partida, preferivelmente cloreto, em reação com um reagente de cloração adequado, preferivelmente cloreto de tionila. A Etapa 3 envolve a alquilação intramolecular na reação do cloreto resultante a partir da etapa 2 com uma base, preferivelmente LDA, para fornecer compostos da Fórmula (II) onde R = H.
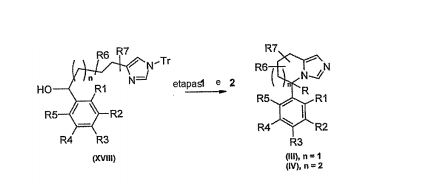
[00120] Geralmente, compostos de Fórmula (III) ou (IV) podem ser preparados de acordo com Esquema 4 por analogia à ciclização descrita acima nas etapa 2 e 3 em Esquema 3 para a preparação de (II), por exemplo, por conversão de um álcool (XVII) no grupo de partida adequado, preferivelmente no cloreto gerado por tratamento com SOCI2, seguida por desprotonação com base forte, tais como t- BuOK, LDA, ou LHMDS, ou os similares, para realizar a ciclização do ânion resultante no grupo de partida. ESQUEMA 4
[00121] Alternativamente, compostos de Fórmula (III) ou (IV) podem ser preparados de acordo com Esquema 5, por conversão de um álcool secundário (XVII11) no grupo de partida adequado, por exemplo, cloreto ou mesilato (etapa 1), e ciclização intramolecular subseqüente (etapa 2) por analogia às etapas 3 e 4 de Esquema 2 acima. ESQUEMA 5
[00122] Adicionalmente, compostos de Fórmula (III) ou (IV) são preparados a partir de compostos existentes de Fórmula (III) ou (IV) por manipulação independente de radicals R, Ri, R2, R3, R4, θ R5 por métodos conhecidos por aqueles versados na técnica, tais como, por exemplo, redução de um grupo nitro em uma anilina ou redução de um éster em um álcool, por exemplo, compostos de Fórmula (III) ou (IV) podem ser preparados a partir de outros compostos de Fórmula (III) ou (IV), onde Ri, R2, ou R3 representa um halogênio ou pseudo-halogênio, por exemplo, brometo ou triflato por acoplamento catalisado por paládio ou cobre de an alquila, alquenila, ou arilborônicoácido, éster borônico, ou boroxina; organoestanano; organozinco; alcóxido de metal; álcool; amida; ou os similares para produzir um correspondente análogo de alquila, cicloalquila, arila, heteroarila, alcóxi, arilóxi, ou acilamino. Estas transformações envolvem a conversão de compostos de Fórmula (III) ou (IV) onde Ri, R2, e/ou R3 podem ser iguais a um halogênio ou pseudohalogênio, tais como Br, a compostos de Fórmula (III) ou (IV) onde Ri, R2, e/ou R3 podem ser alquila ou arila por acoplamento transversal Suzuki com um ácido borônico, ou os similares, na presença de um catalisador, preferivelmente Pd(PPh3)4, uma base, preferivelmente hidróxido de potássio e carbonato de sódio, para fornecer compostos de Fórmula (III) ou (IV). Compostos adicionais de Fórmula (III) ou (IV) são gerados por tratamento de compostos (III) ou (IV) se R=H com uma base forte, por exemplo, LHMDS, seguida por um eletrófilo adequado, por exemplo, iodeto de metila ou brometo de alila para fornecer compostos de Fórmula (III) ou (IV) onde R não é igual a H.
[00123] Adicionalmente, compostos de Fórmula (I) são gerados a partir de compostos existentes de Fórmula (I) onde R e Ri não são iguais a H e R e Ri pode ser reagido para formar compostos onde R e Ri juntos compreendem um anel.
[00124] Álcoois intermediários (XVII) são preparados por desproteção de um éter de silila (XIX), preferivelmente um éter de TBS, sob, por exemplo, condições acídicas ou por redução do éster análogo (XX), preferivelmente com NaBFU, de acordo com Esquema 6. ESQUEMA 6
[00125] Éteres (XIX) e ésteres (XX) são gerados por A/-alqu ilação de imidazóis adequadamente protegidos (XXI) ou (XXII), respectivamente, utilizando um eletrófilo adequado (VII) de acordo com Esquema 7. ESQUEMA 7
[00126] Os intermediários de imidazol N-protegidos (XXI) e (XXII) são preparados de acordo com Esquema 8. Ésterificação de ácido (XXIII) com um álcool, preferivelmente metanol ou etanol, sob condições acídicas seguida por proteção do nitrogênio de imidazol, preferivelmente como o análogo de N-tritila fornece (XXII), com Re e R? igual a hidrogênio, redução de (XXII) para o álcool por um agente de redução adequado, preferivelmente NaBH4, seguida por proteção como o éter de TBS fornece (XXI). Ésteres (XXII) onde Re e R? não são ambos hidrogênio são gerados por alquilação de ésteres (XXII) com um eletrófilo adequado, por exemplo, um benzilbrometo, sob condições básicas. Conversão do éster (XXII) em éter (XXI) com Re e R? não ambos hidrogênio pode ser realizada por redução e proteção do álcool resultante por analogia ao acima. Substituintes Re e/ou Rz não iguais a hidrogênio podem ser introduzidos no carbono adjacente para o imidazol por tratamento de éster (XXV) com uma base adequada, por exemplo, LDA, e eletrófilo, tais como iodeto de metila. Ésteres (XXII) onde R? iguala-se a H podem ser gerados por olefinação Wittig de cetonas (XXIV) por analogia aos métodos descritos em Bioorg. Med. Chem. 2004, 12(9), 2273. Redução da porção olefínica subseqüente com um agente de redução adequado, tal como hidrogênio, utilizando um catalisador de paládio produz éster (XXII). Os ésteres (XXV) são produzidos por alquilações de ésteres (XXV) onde Re e/ou R7 são hidrogênio sob condições básicas na presença de um eletrófilo adequado, por exemplo, iodeto de metila. Homologação de éster (XXV) em éter (XXI) pode ser obtida por redução com um reagente adequado, tal como LAH, seguida por oxidação para 0 aldeído, tratamento do aldeído com 0 ilato gerado a partir de cloreto de trifenilfosfônio de metoximetila para produzir 0 aldeído homólogo. A redução do aldeído e proteção do álcool subseqüente produz éteres (XXI).ESQUEMA 8

[00127] Compostos de Fórmula (IV) onde R7 é definido acima, são preparados a partir de aldeído ou cetona (XXIV), por olefinação Wittig utilizando um sal de fosfônio adequadamente substituído, por exemplo, brometo de trifenilfosfônio de 3-(ferc-butildimetilsililóxi)propila na presença de uma base, preferivelmente n-BuLi de acordo com Esquema 9. A redução das olefinas resultantes produz 0 éter saturado (XXI) com RΘ igual a hidrogênio, que pode ser N-alquilado com um brometo (VII) por analogia à etapa 4 descrita no Esquema 1 para a conversão de (VI) a (VIII). ESQUEMA 9
[00128] Adicionalmente, substituinte Re não igual a hidrogênio pode ser introduzido em compostos de Fórmula (IV) de acordo com Esquema 10 por conversão de éster (XXII) em olefina (XXVI) por um processo de três etapas: 1) redução para o álcool primário, 2) oxidação Swern para o aldeído e 3) conversão para a olefina (XXVI) por olefinação Wittig. Metátese cruzada da olefina (XXVI) com enona (XXVII) utilizando catalização de segunda geração de Grubbs fornece enona (XXVIII), que sofre adição de conjugado mediada por cobre com um nucleófilo adequado, tal como um reagente de alquilzinco para fornecer cetona saturada (XXIX). Redução de (XXIX) com um reagente adequado, tal como NaBH4, fornece álcool secundário (XVIII). ESQUEMA 10
[00129] Geralmente, enantiômeros dos compostos da presente invenção podem ser preparados por métodos conhecidos por aqueles versados na técnica para resolver misturas racêmicas, tal como por formação e recristalização de sais diastereoméricos ou por cromatografia quiral ou separação por HPLC utilizando fases estacionárias quirais.
[00130] Em compostos de partida e intermediários que são convertidos nos compostos da invenção de uma maneira descrita aqui, os grupos funcionais presentes, tais como grupos amino, tiol, carboxila e hidróxi, são opcionalmente protegidos por grupos de proteção convencionais que são comuns em química orgânica preparativa. Grupos protegidos de amino, tiol, carboxila e hidroxila são aqueles que podem ser convertidos sob condições amenas nos grupos livres de amino, tiol, carboxila e hidroxila sem a estrutura molecular ser destruída ou outras reações colaterais indesejadas ocorrerem.
[00131] O propósito de introduzir grupos de proteção é proteger os grupos funcionais a partir de reações indesejadas com componentes de reação sob as condições usadas para realizar uma transformação química desejada. A necessidade e escolha de grupos de proteção para uma reação particular é conhecida por aqueles versados na técnica e depende da natureza do grupo funcional a ser protegido (grupo hidroxila, grupo amino, etc.), a estrutura e estabilidade da molécula da qual o substituinte é uma parte e condições de reação.
[00132] Grupos de proteção bem-conhecidos que atendem à estas condições e sua introdução e remoção são descritos, por exemplo, em McOmie, "Protective groups in organic chemistry", Plenum Press, London, NY (1973); e Greene e Wuts, "Protective groups in Organic Synthesis", John Wilay e Sons, Inc., NY (1999).
[00133] As reações acima mencionadas são realizadas de acordo com métodos padrão, na presença ou ausência de diluente, preferivelmente, tal como são intertes aos reagentes e são solventes destes, de catalisadores, agentes de condensação ou outros referidos, respectivamente e/ou atmosferas inertes, em baixas temperaturas , temperatura ambiente ou temperaturas elevadas, preferivelmente em ou próximo ao ponto de ebulição dos solventes usados, e uma pressão atmosférica ou superatmosférica. Os preferidos solventes, catalisadores e condições de reação são mencionados nos exemplos ilustrativos anexos.
[00134] A invenção também inclui qualquer variante dos presentes processos, em que um produto intermediário obtenível em qualquer estágio desta é usado como material de partida e as etapas restantes são realizadas, ou em que os materiais de partida são formados in situ sob condições de reação, ou em que os componentes de reação são usados na forma de seus sais ou antípodas oticamente puros .
[00135] Compostos da invenção e intermediários podem também ser convertidos um no outro de acordo com métodos geralmente conhecidos por si próprios.
[00136] Em outro aspecto, a presente invenção fornece uma composição farmacêutica compreendendo um composto da presente invenção e um veículo farmaceuticamente aceitável. A composição farmacêutica pode ser Formulada para rotinas particulares de administração tais como administração oral, administração parenteral, e administração retal, etc. Em adição, as composições farmacêuticas da presente invenção podem ser preparadas em uma forma sólida incluindo cápsulas, comprimidos, pílulas, grânulos, pós ou supositórios, ou em uma forma líquida incluindo soluções, suspensões ou emulsões. As composições farmacêuticas podem ser submetidas a operações farmacêuticas convencionais tais como ésterilização e/ou podem conter diluentes inertes convencionais, agentes lubrificantes, ou agentes de tamponamento, bem como adjuvantes, tais como conservantes, estabilizantes, agentes umectantes, emulsificantes e tampões, etc.
[00137] Preferivelmente, as composições farmacêuticas são comprimidos e cápsulas de gelatinaa compreendendo o ingrediente ativo juntamente com a) diluentes, por exemplo, lactose, dextrose, sacarose, manitol, sorbitol, celulose e/ou glicina; b) lubrificantes, por exemplo, sílica, talco, ácido esteárico, seu sal de magnésio ou cálcio e/ou polietilanoglicol; para comprimidos também c) aglutinantes, por exemplo, silicato de alumínio de magnésio, pasta de amido, gelatina, tragacanto, metilcelulose, carboximetilcelulose de sódio e/ou polivinilpirrolidona; se desejado d) desintegrantes, por exemplo, amidos, ágar, ácido algínico ou seu sal de sódio, ou misturas efervescentes; e/ou e) absorventes, corantes, flavorizantes e adoçantes.
[00138] Comprimidos podem ser ou revestidos por película ou revestidos por entérico de acordo com métodos conhecidos na técnica.
[00139] Composições adequadas para administração oral incluem uma quantidade eficaz de um composto da invenção na forma de comprimidos, pastilhas, suspensões aquosas ou oleosas, pós dispersíveis ou grânulos, emulsão, cápsulas duras ou macias, ou xaropes ou elixires. Composições destinadas ao uso oral são preparadas de acordo com qualquer método conhecido na técnica para a fabricação de composições farmacêuticas e tais composições podem conter um ou mais agentes selecionados a partir do grupo que consiste em agentes adoçantes, agentes flavorizantes, agentes corantes e agentes conservantes a fim de fornecer preparações farmaceuticamente elegantes e palatáveis. Comprimidos contêm o ingrediente ativo em mistura com excipientes farmaceuticamente aceitáveis não tóxicos que são adequados para a fabricação de comprimidos. Estes excipientes são, por exemplo, diluentes inertes, tais como carbonato de cálcio, carbonato de sódio, lactose, fosfato de cálcio ou fosfato de sódio; agentes de granulação e desintegrantes, por exemplo, amido de milho, ou ácido algínico; agentes de ligação, por exemplo, amido, gelatina ou acácia; e agentes lubrificantes, por exemplo, estearato de magnésio, ácido esteárico ou talco. Os comprimidos são não revestidos ou revestidos por técnicas conhecidas para retardar a desintegração e absorção no trato gastrointestinal e desse modo fornecem uma ação sustentada durante ou maior período. Por exemplo, um material de retardo do tempo tal como monoestearato de glicerila ou diestearato de glicerila pode ser empregado. Formulações para uso oral podem ser apresentadas como cápsulas de gelatina dura em que o ingrediente ativo é misturado com um diluente sólido inerte, por exemplo, carbonato de cálcio, fosfato de cálcio ou caulim, ou como cápsulas macias de gelatina em que o ingrediente ativo é misturado com água ou um meio oleoso, por exemplo, óleo de amendoim, parafina líquida ou óleo de oliva.
[00140] As composições injetáveis são preferivelmente soluções ou suspensões isotônicas aquosas, e supositórios são vantajosamente preparados a partir de emulsões ou suspensões graxas. As referidas composições podem ser ésterilizadas e/ou contêm adjuvantes, tais como agentes conservantes, estabilizantes, umectantes ou emulsificantes, promotores de solução, sais para regular a pressão osmótica e/ou tampões. Além disso, eles podem também conter outras substâncias terapeuticamente valiosas. As referidas composições são preparadas de acordo com métodos convencionais de mistura, granulação ou revestimentos, respectivamente, e contêm cerca de 0,1 - 75%, preferivelmente cerca de 1 - 50%, do ingrediente ativo.
[00141] Composições adequadas para aplicação transdérmica incluem uma quantidade eficaz de um composto da invenção com veículo. Veículos vantajosos incluem solventes farmacologicamente aceitáveis absorvíveis para ajutar a passagem através da pele do hospedeiro. Por exemplo, dispositivos transdérmicos são na forma de uma bandagem que compreende um membro de reforço, um reservatório contendo o composto opcionalmente protegidos com veículos, opcionalmente uma barreira de controle da taxa para liberar o composto da pele do hospedeiro em uma taxa controlada e predeterminada durante um período de tempo prolongado, e significa prender o dispositivo à pele.
[00142] Composições adequadas para aplicação tópica, por exemplo, à pele e olhos, incluem soluções aquosas, suspensões, ungüentos, cremes, géis ou formulações vaporizáveis, por exemplo, para liberação por aerossol ou os similares. Tais sistemas de liberação tópica serão em particular apropriados para aplicação dérmica, por exemplo, para o tratamento de câncer de pele, por exemplo, para uso profilático em cremes solares, loções, sprays e os similares. Eles são desse modo particularmente adaptados para uso em formulações tópicas, incluindo cosméticos, formulações bem-conhecidas na técnica. Tais podem conter solubilizantes, estabilizantes, agentes de realce da tonicidade, tampões e conservantes.
[00143] A presente invenção também fornece composições farmacêuticas anidrosas e formas de dosagem compreendendo os compostos da presente invenção como ingredientes ativos, visto que a água pode facilitar a degradação de alguns compostos. Por exemplo, a adição de água (por exemplo, 5%) é amplamente aceita nas técnicas farmacêuticas como um meio de estimular a estocagem a longo prazo a fim de determinar características tais como vida de prateleira ou a estabilidade de formulações em tempo prolongado. Observe, por exemplo, Jens T. Carstensen, Drug Stability: Principles & Practice, 2a Ed., Marcel Dekker, NY, N.Y., 1995, pp. 379-80. Na verdade, água e calor aceleram a decomposição de alguns compostos. Desse modo, o efeito de água sobre uma formulação pode ser de maior significância visto que a umectação e/ou umidade são comumente encontradas durante a fabricação, manipulação, principalmente encontrada durante a fabricação, manipulação, empacotamento, estocagem, expedição, e uso de formulações.
[00144] Composições farmacêuticas anidrosas e formas de dosagem da invenção podem ser preparadas usando ingredientes contendo baixa umectação ou anidroso e condições de baixa umectação ou baixa umidade. Composições farmacêuticas e formas de dosagem que compreendem lactose e pelo menos um ingrediente ativo que compreende uma amina primária ou secundária são preferivelmente anidrosas se contato substancial com umectação (moisture) e/ou umidade durante fabricação, empacotamento, e/ou estocagem é esperado.
[00145] Uma composição farmacêutica anidrosa mostrou ser preparada e estocada de modo que sua natureza anidrosa seja mantida. Conseqüentemente, composições anidrosas são preferivelmente empacotadas usando materiais conhecidos para prevenir a exposição à água de modo que elas possam ser incluídas em kits de formulações adequados. Exemplos de empacotamento adequado incluem, porém não são limitados a, lâminas hermeticamente seladas, plásticos, recipientes de dose unitária (por exemplo, frasconetes), pacotes de empolas, e pacotes de faixas.
[00146] A invenção também fornece composições farmacêuticas e formas de dosagem que compreendem um ou mais agentes que reduz a taxa para as quais o composto da presente invenção como um ingrediente ativo decomporse-a. Tais agentes, que são referidos aqui como "estabilizantes," incluem, porém não são limitados a, antioxidantes tais como ácido ascórbico, tampões de pH , ou tampões de sal, etc.
[00147] As composições farmacêuticas contêm uma quantidade terapeuticamente eficaz de um composto da invenção como definido acima, ou sozinha ou em uma combinação com um ou mais agentes terapêuticos, por exemplo, tal como uma dose terapêutica eficaz como reportado na técnica. Tais agentes terapêuticos incluem pelo menos um ou dois ou mais selecionados a partir dos seguintes grupos : (i) antagonista de receptor de angiotensina II ou um sal farmaceuticamente aceitável deste, (ii) inibidor de HMG-Co-A reductase ou um sal farmaceuticamente aceitável deste, (iii) inibidor de enzima conversora de angiotensina (ACE) ou um sal farmaceuticamente aceitável deste, (iv) bloqueador de canal de cálcio (CCB) ou um sal farmaceuticamente aceitável deste, (v) enzima conversora de angiotensina dual / inibidor de endopeptidase neutra (ACE/NEP) ou um sal farmaceuticamente aceitável deste, (vi) antagonista de endotelina ou um sal farmaceuticamente aceitável deste, (vii) inibidor de renina ou um sal farmaceuticamente aceitável deste, (viii) diurético ou um sal farmaceuticamente aceitável deste, (ix) Uma mímica de ApoA-l ; (x) um agente antidiabético ; (xi) um agente redutor de obesidade; (xii) um bloqueador de receptor de aldosterona; (xiii) um bloqueador de receptor de endotelina; (xiv)um inibidor de CETP; (xiv) m inibidor de bomba de membrana de Na-K-ATPase; (xvi) um bloqueador de receptor beta-adrenérgico ou um bloqueador de receptor alfa-adrenérgico; (xvii) um inibidor de endopeptidase neutra (NEP); e (xviii) um agente inotrópico.
[00148] Um antagonista de receptor de angiotensina II ou um sal farmaceuticamente aceitável deste é entendido ser um ingrediente ativo que liga-se ao subtipo de receptor ATi de receptor de angiotensina II porém não resulta em ativação do receptor. Como uma conseqüência da inibição do receptor ATi , estes antagonistas podem, por exemplo, ser empregados como anti-hipertensivos ou para tratar insuficiência cardíaca congestiva.
[00149] A classe de antagonistas de receptor ATi compreende compostos tendo características estruturais divergentes, os essencialmente preferidos são aqueles não peptídeos . Por exemplo, menção pode ser feita dos compostos que são selecionados a partir do grupo que consiste em valsartan, losartan, candesartan, eprosartan, irbesartan, saprisartan, tasosartan, telmisartan, o composto com a designação E-1477 da seguinte Fórmula
[00150] o composto com a designação SC-52458 da seguinte Fórmula
[00151] e o composto com a designação ZD-8731 da seguinte Fórmula
[00152] ou, em cada caso, um sal farmaceuticamente aceitável deste.
[00153] Antagonista de receptor ATi preferidos são aqueles agentes que foram comercializados, mais preferido é valsartan ou um sal farmaceuticamente aceitável deste.
[00154] Inibidores de HMG-Co-A reductase (também chamados inibidores de reductase de beta-hidróxi-beta-metilglutaril-co-enzima-A) são entendidos ser aqueles agentes ativos que podem ser usados para reduzir os níveis de lipídios incluindo colésterol em sangue.
[00155] A classe de inibidores de HMG-Co-A reductase comprende compostos tendo características estruturais divergentes. Por exemplo, menção pode ser feita dos compostos que são selecionados a partir do grupo que consiste em atorvastatina, cerivastatina, compactina, dalvastatina, diidrocompactina, fluindostatina, fluvastatina, lovastatina, pitavastatina, mevastatina, pravastatina, rivastatina, simvastatina, e velostatina, ou, em cada caso, um sal farmaceuticamente aceitável deste.
[00156] Inibidores de HMG-Co-A reductase preferidos são aqueles agentes que foram comercializados, mais preferido é fluvastatina e pitavastatina ou, em cada caso, um sal farmaceuticamente aceitável deste.
[00157] A interrupção da degradação enzimática de angiotensina I para angiotensina II com os assim chamados inibidores de ACE (também chamados inibidores de enzima conversora de angiotensina) é uma variante bem-sucedida para a regularização da pressão sangüínea e desse modo também torna disponível um método terapêutico para o tratamento de insuficiência cardíaca congestiva.
[00158] A classe de inibidores de ACE compreende compostos tendo características estruturais divergentes. Por exemplo, menção pode ser feita dos compostos que são selecionados a partir do grupo que consiste em alacepril, benazepril, benazeprilat, captopril, ceronapril, cilazapril, delapril, enalapril, enaprilat, fosinopril, imidapril, lisinopril, moveltopril, perindopril, quinapril, ramipril, spirapril, temocapril, e trandolapril, ou, em cada caso, um sal farmaceuticamente aceitável deste.
[00159] Inibidores de ACE preferidos são aqueles agentes que foram comercializados, mais preferidos são benazepril e enalapril.
[00160] A classe de CCBs essencialmente compreende diidropiridinas (DHPs) e não-DHPs tais como CCBs tipo diltiazem e tipo verapamil.
[00161] Um CCB útil na referida combinação é preferivelmente um representativo de DHP selecionado a partir do grupo que consiste em amlodipina, felodipina, riosidina, isradipina, lacidipina, nicardipina, nifedipina, niguldipina, niludipina, nimodipina, nisoldipina, nitrendipina, e nivaldipina, e é preferivelmente um representativo de não-DHP selecionado a partir do grupo que consiste em flunarizina, prenilamina, diltiazem, fendilina, galopamil, mibefradil, anipamil, tiapamil e verapamil, e em cada caso, um sal farmaceuticamente aceitável deste. Todos estes CCBs são terapeuticamente usados, por exemplo, como fármacos anti-hipertensivo, antiangina do peito ou antiarrítimicos.
[00162] CCBs preferidos compreendem amlodipina, diltiazem, isradipina, nicardipina, nifedipina, nimodipina, nisoldipina, nitrendipina, e verapamil, ou, por exemplo, dependente do CCB específico, um sal farmaceuticamente aceitável deste. Especialmente preferidos como DHP é amlodipina ou um sal farmaceuticamente aceitável, especialmente o besilato, deste. Um representativo especialmente preferido de não-DHPs é verapamil ou um sal farmaceuticamente aceitável, especialmente o cloridrato, deste.
[00163] Uma enzima conversora de angiotensina dual preferida / inibidor de endopetidase neutra (ACE/NEP) é, por exemplo, omapatrilato (conforme EP 629627), fasidotril ou fasidotrilato, ou, se apropriado, um sal farmaceuticamente aceitável deste.
[00164] Um antagonista de endotelina preferido é, por exemplo, bosentan (conforme EP 526708 A), além disso, tezosentan (conforme WO 96/19459), ou em cada caso, um sal farmaceuticamente aceitável deste.
[00165] Inibidores de renina adequado inclui compostos tendo características estruturais diferentes. Por exemplo, menção pode ser feita de compostos que são selecionados a partir do grupo que consiste em ditekiren (nome químico: [1S-[1R*,2R*,4R*(1R*,2R*)]]-1- [(1,1 -d i meti latóxi)carbon i l]-L-prol i l-L-f eni lalan i l-N-[2-h id róxi-5-meti I-1 -(2- metilpropil)-4-[[[2-metil-1 -[[(2- piridinilmetil)amino]carbonil]butil]amino]carbonil]hexil]-N-alfa-metil-L- histidinamida); terlakiren (nome químico: [R-(R*,S*)]-N-(4- morfolinilcarbonil)-L-fenilalanil-N-[1-(cicloexilmetil)-2-hidróxi-3-(1- metilatóxi)-3-oxopropil]-S-metil-L-cisteineamida); e zankiren (nome químico: [1 S-[ 1 R*[R*(R*)],2S*,3R*]]-N-[1 -(cicloexi I meti l)-2,3-d i id róxi-5- metilhexil]-alfa-[[2-[[(4-metil-1-piperazinil)sulfonil]metil]-1-oxo-3- fenilpropil]-amino]-4-tiazolpropanamida), preferivelmente, em cada caso, o sal de cloridrato deste, SPP630, SPP635 e SPP800 como desenvolvido por Speedel.
[00166] Inibidor de renina preferido da presente invenção inclui RO 66-1132 e RO 66-1168 de Fórmula (A) e (B)
[00167] respectivamente, ou um sal farmaceuticamente aceitável deste
[00168] Em particular, a presente invenção se refere ao inibidor de renina que é uma amida de ácido δ-amino-y-hidroxi-ω-aril-alcanoico derivado da Fórmula (C)
[00169] em que Ri é halogênio, Ci-ehalogenalquila, Ci-ealcóxi-Ci- ealquilóxi ou Ci-ealcóxi-Ci-ealquila; R2 é halogênio, Ci-4alquila ou Ci- 4alcóxi; R3 e R4 são Cs-βalquila independentemente ramificada; e Rs é cicloalquila, Ci-ealquila, Ci-ehidroxialquila, Ci-6alcóxi-Ci-salquila, Ci- 6alcanoilóxi-Ci-6alquila, Ci-eaminoalquila, Ci-ealquilamino-Ci-ealquila, Ci-6dialquilamino-Ci-6alquila, Ci-ealcanoilamino-Ci-ealquila, HO(O)C-Ci- 6alquila, Ci-βalquil-O-(O)C-Ci-6alquila, H2N-C(O)-Ci-ealquila, Ci-salquil- HN-C(O)-Ci-ealquila ou (Ci-6alquil)2N-C(O)-Ci-6alquila; ou um sal farmaceuticamente aceitável deste.
[00170] Como uma alquila, Ri pode ser linear ou ramificado e preferivelmente compreende 1 a 6 átomos de C, especialmente 1 ou 4 átomos de C. Exemplos são metila, etila, n- e i-propila, n-, i- e t-butila, pentila e hexila.
[00171] Como uma halogenalquila, Ri pode ser linear ou ramificado e preferivelmente compreende 1 a 4 átomos de C, especialmente 1 ou 2 átomos de C. Exemplos são flúormetila, diflúormetila, triflúormetila, clorometila, diclorometila, triclorometila, 2-cloroetila e 2,2,2-triflúoretila.
[00172] Como um alcóxi, Ri e R2 podem ser lineares ou ramificados e preferivelmente compreendem 1 a 4 átomos de C. Exemplos são metóxi, etóxi, n- e i-propilóxi, n-, i- e t-butilóxi, pentilóxi e hexilóxi.
[00173] Como uma alcoxialquila, Ri pode ser linear ou ramificado. O grupo alcóxi preferivelmente compreende 1 a 4 e especialmente 1 ou 2 átomos de C, e 0 grupo alquila preferivelmente compreende 1 a 4 átomos de C. Exemplos são metoximetila, 2-metoxietila, 3- metoxipropila, 4-metoxibutila, 5-metoxi pentila, 6-metoxihexila, etoximetila, 2-etoxietila, 3-etoxipropila, 4-etoxibutila, 5-etoxipentila, 6- etoxihexila, propiloximetila, butiloximetila, 2-propiloxietila e 2- butiloxietila.
[00174] Como um Ci-ealcóxi-Ci-ealquilóxi, Ri pode ser linear ou ramificado. O grupo alcóxi preferivelmente compreende 1 a 4 e especialmente 1 ou 2 átomos de C, e 0 grupo alquilóxi preferivelmente compreende 1 a 4 átomos de C. Exemplos são metoximetilóxi, 2- metoxietilóxi, 3-metoxipropilóxi, 4-metoxibutilóxi, 5-metoxipentilóxi, 6- metoxihexilóxi, etoximetilóxi, 2-etoxietilóxi, 3-etoxipropilóxi, 4- etoxibutilóxi, 5-etoxi pentilóxi, 6-etoxihexilóxi, propiloximetilóxi, butiloximetilóxi, 2-propiloxietilóxi e 2-butiloxietilóxi.
[00175] Em uma modalidade preferida, Ri é metóxi- ou etóxi-Ci- 4alquilóxi, e R2 θ preferivelmente metóxi ou etóxi. Particularmente preferidos são compostos de Fórmula (III), em que Ri é 3- metoxipropilóxi e R2 é metóxi.
[00176] Como uma alquila ramificada, R3 e R4 preferivelmente compreendem 3 a 6 átomos de C. Exemplos são i-propila, i- e t-butila, e isômeros ramificados de pentila e hexila. Em uma modalidade preferida, R3 e R4 em compostos de Fórmula (C) são em cada caso i- propila.
[00177] Como uma cicloalquila, Rs pode preferivelmente compreender 3 a 8 átomos de carbono de anel, 3 ou 5 sendo especialmente preferidos. Alguns exemplos são ciclopropila, ciclobutila, ciclopentila, cicloexila e ciclooctila. A cicloalquila pode ser opcionalmente protegida e ser substituída por um ou mais substituintes, tais como alquila, halo, oxo, hidróxi, alcóxi, amino, alquilamino, dialquilamiπo, tiol, alquiltio, nitro, ciano, heterociclila e os similares.
[00178] Como uma alquila, Rs pode ser linear ou ramificado na forma de alquila e preferivelmente compreende 1 a 6 átomos de C. Exemplos de alquila são listados aqui acima. Metila, etila, n- e i- propila, n-, i- e t-butila são preferidas.
[00179] Como uma Ci-shidroxialquila, Rs pode ser linear ou ramificado e preferivelmente compreende 2 a 6 átomos de C. Alguns exemplos são 2-hidroxietila, 2-hidroxipropila, 3-hidroxipropila, 2-, 3- ou 4-hidroxibutila, hidroxipentila e hidroxihexila.
[00180] Como uma Ci-ealcóxi-Ci-ealquila, Rs pode ser linear ou ramificado. O grupo alcóxi preferivelmente compreende 1 a 4 átomos de C e o grupo alquila preferivelmente 2 a 4 átomos de C. Alguns exemplos são 2-metoxietila, 2-metoxipropila, 3-metoxipropila, 2-, 3- ou 4-metoxibutila, 2-etoxietila, 2-etoxipropila, 3-etoxipropila, e 2-, 3- ou 4- etoxibutila.
[00181] Como uma Ci-ealcanoilóxi-Ci-ealquila, Rs pode ser linear ou ramificado. O grupo alcanoilóxi preferivelmente compreende 1 a 4 átomos de C e o grupo alquila preferivelmente 2 a 4 átomos de C. Alguns exemplos são formiloximetila, formiloxietila, acetiloxietila, propioniloxietila e butiroiloxietila.
[00182] Como uma Ci-eaminoalquila, Rs pode ser linear ou ramificado e preferivelmente compreende 2 a 4 átomos de C. Alguns exemplos são 2-aminoetila, 2- ou 3-aminopropila e 2-, 3- ou 4- aminobutila.
[00183] Como Ci-ealquilamino-Ci-ealquila e Ci-6dialquilamino-Ci- βalquila, Rs pode ser linear ou ramificado, o grupo de alquilamino preferivelmente compreende grupos Ci-4alquila e o grupo alquila tem preferivelmente 2 a 4 átomos de C. Alguns exemplos são 2- metilaminoetila, 2-dimetilaminoetila, 2-etilaminoetila, 2-etilaminoetila, 3-metilaminopropila, 3-dimetilaminopropila, 4-metilaminobutila e 4- dimetilaminobutila.
[00184] Como uma HO(O)C-Ci-ealquila, Rs pode ser linear ou ramificado e o grupo alquila preferivelmente compreende 2 a 4 átomos de C. Alguns exemplos são carboximetila, carboxietila, carboxipropila e carboxibutila.
[00185] Como uma Ci-6alquil-O-(O)C-Ci-6alquila, Rs pode ser linear ou ramificado, e os grupos alquila preferivelmente compreendem independentemente um do outro 1 a 4 átomos de C. Alguns exemplos são metoxicarbonilmetila, 2-metoxicarbonilatila, 3- metoxicarbonilpropila, 4-metóxi-carbonilbutila, etoxicarbonilmetila, 2- etoxicarbonilatila, 3-etoxicarbonilpropila, e 4-etoxicarbonilbutila.
[00186] Como uma H2N-C(O)-Ci-6alquila, Rs pode ser linear ou ramificado, e o grupo alquila preferivelmente compreende 2 a 6 átomos de C. Alguns exemplos são carbamidometila, 2-carbamidoetila, 2- carbamido-2,2-dimetilatila, 2- ou 3-carbamidopropila, 2-, 3- ou 4- carbamidobutila, 3-carbamido-2-metilpropila, 3-carbamido-1,2- dimetilpropila, 3-carbamido-3-etilpropila, 3-carbamido-2,2- dimetilpropila, 2-, 3-, 4- ou 5-carbamidopentila, 4-carbamido-3,3- ou - 2,2-dimetilbutila. Preferivelmente, Rs é 2-carbamido-2,2-dimetilatila.
[00187] Conseqüentemente, preferidos são amida de ácido δ-amino- y-hidróxi-o-aril-alcanóico derivadas de Fórmula (C) tendo a Fórmula
[00188] em que Ri é 3-metoxipropilóxi; R2 é metóxi; e R3 θ R4 são isopropila; ou um sal farmaceuticamente aceitável deste; quimicamente definido como 2(S),4(S),5(S),7(S)-N-(3-amino-2,2-dimetil-3-oxopropil)- 2,7-di(1-metilatil)-4-hidróxi-5-amino-8-[4-metóxi-3-(3-metóxi- propóxi)fenil]-octanamida, também conhecido como aliskiren.
[00189] O termo "aliskiren", se não definido especificamente, deve ser entendido tanto como a base livre quanto como um sal desta, especialmente um sal farmaceuticamente aceitável deste, mais preferivelmente um sal de hemi-fumarato deste.
[00190] Um diurético é, por exemplo, um derivado de tiazida selecionada a partir do grupo que consiste em clorotiazida, hidroclorotiazida, metilclotiazida, e clorotalidon. O mais preferido é hidroclorotiazida.
[00191] Uma mímica de ApoA-l é, por exemplo, peptídeo de D4F, especialmente de Fórmula D-W-F-K-A-F-Y-D-K-V-A-E-K-F-K-E-A-F
[00192] Um agente antidiabético inclui realçadores de secreção de insulina que são ingrediente ativos que têm a propriedade de promover a secreção de insulina a partir de células β pancreáticas. Exemplo de realçadores de secreção de insulina é um derivado de biguanida, por exemplo, metformina ou, se apropriado, um sal farmaceuticamente aceitável deste, especialmente 0 cloridrato deste. Outros realçadores de secreção de insulina incluem sulfoniluréias (SU), especialmente aquelas que promovem a secreção de insulina a partir de células β pancreáticas transmitindo sinais de secreção de insulina por meio de receptores de SU na membrana celular, incluindo (porém não são limitados a) tolbutamida; clorpropamida; tolazamida; acetoexamida; 4- cloro-N-[(1-pirolidinilamino)carbonil]-benzensulfonamida (glicopiramida); glibenclamida (gliburida); gliclazida; 1-butil-3- metanililuréia; carbutamida; glibonurida; glipizida; gliquidona; glisoxepid; glibutiazol; glibuzol; gliexamida; glimidina; glipinamida; fenbutamida; e tolilciclamida, ou sais farmaceuticamente aceitáveis deste.
[00193] Realçadores de secreção de insulina além disso incluem realçadores de curta ação de secreção de insulina, tais como o derivado de fenilalanina nateglinida [N-(trans-4-isopropilcicloexil- carbonil)-D-fenilalanina] (conforme. EP 196222 e EP 526171) da Fórmula
[00194] e repaglinida [(S)-2-etóxi-4-{2-[[3-metil-1-[2-(1- piperidinil)fenil]butil]amino]-2-oxoetil}benzoicoácido], Repaglinida é descrita na EP 589874, EP 147850 A2, em particular Exemplo 11 na página 61, e EP 207331 A1. Ela pode ser administrada na forma como ela é comercializada, por exemplo, sob a marca comercial NovoNorm®; diidrato de (2S)-2-benzil-3-(cis-hexaidro-2- isoindolinlicarbonil)-propionato de cálcio (mitiglinida - conforme EP 507534); além disso representativos da nova geração de Slls tais como glimepirida (conforme EP 31058); em forma de sal livre ou farmaceuticamente aceitáveis. O termo nateglinida igualmente compreende modificações de cristal tal como descrito na EP 0526171 B1 ou US 5,488,510, respectivamente, a matéria objeto da qual, especialmente com respeito à identificação, fabricação e caracterização de modificações de cristal, é aqui incorporada por referência a este pedido, especialmente a matéria objeto de reivindicações 8 a 10 de referida Patente dos Estados Unidos (referindo-se à modificação de cristal de forma H) bem como a correspondente referência à modificação de cristal tipo B na EP 196222 B1 a matéria objeto da qual, especialmente com respeito à identificação, fabricação e caracterização da modificação de cristal de forma B. Preferivelmente, na presente invenção, o tipo B ou H, mais preferivelmente o tipo H, é usado. Nateglinida pode ser administrada na forma como ela é comercializada por exemplo, sob a marca comercial STARLIX®.
[00195] Realçadores de secreção de insulina igualmente incluem inibidores de DPP-IV realçador da secreção de longa ação de insulina , GLP-1 e agonistas de GLP-1.
[00196] DPP-IV é responsável pela inativação de GLP-1. Mais particularmente, DPP-IV gera um antagonista de receptor de GLP-1 e desse modo encurta a resposta fisiológica a GLP-1. GLP-1 é um estimulador maior de secreção de insulina pancreática e tem efeitos benéficos diretos sobre disposição de glicose.
[00197] O inibidor de DPP-IV pode ser peptídico ou, preferivelmente, não peptídico. Inibidores de DPP-IV são em cada caso genericamente e especificamente descrito por exemplo, em WO 98/19998, DE 196 16 486 A1, WO 00/34241 e WO 95/15309, em cada caso em particular nas reivindicações de composto e nos produtos finais dos exemplos de trabalho, a matéria objeto dos produtos finais, as preparações farmacêuticas e as reivindicações são pelo presente incorporados no presente pedido por referência a estas publicações. Preferidos são aqueles compostos que são especificamente descritos no Exemplo 3 de WO 98/19998 e Exemplo 1 de WO 00/34241, respectivamente.
[00198] GLP-1 é uma proteína insulinotrópica que foi descrita, por exemplo, por W.E. Schmidt e outro em Diabetologia, 28, 1985, 704-707 e em US 5.705.483.
[00199] O termo "agonistas de GLP-1" usado aqui significa variantes e análogos de GLP-1 (7-36)NH2 que são descritos em particular em US 5.120.712, US 5.118666, US 5.512.549, WO 91/11457 e por C. Orskov e outro em J. Biol. Chem. 264 (1989) 12826. O termo "agonistas de GLP-1" compreende especialmente compostos similares a GLP-1 (7-37), em cujo composto a funcionalidade de amida de terminal carbóxi de Arg36 é deslocada com Gly para a posição 37a da molécula de GLP-1 (7-36)NH2 e variantes e análogos deste incluindo GLN9-GLP-1(7-37), D-GLN9-GLP-1(7-37), acetila LYS9-GLP-1(7-37), LYS18-GLP-1(7-37) e, em particular, GLP-1 (7-37)OH, VAL8-GLP-1(7- 37), GLY8-GLP-1(7-37), tR8-GLP-1 (7-37), MET8-GLP-1(7-37) e 4- imidazopropionil-GLP-1. Preferência especial é também dada ao análogo de agonista de GLP exendina-4, descrito por Greig e outro em Diabetologia 1999, 42, 45-50.
[00200] Um realçador de sensibilidade à insulina restaura a função receptora de insulina prejudicada para reduzir a resistência à insulina e conseqüentemente realçar a sensibilidade à insulina.
[00201] Um realçador de sensibilidade à insulina apropriado é, por exemplo, um derivado de tiazolidinadiona hipoglicêmico apropriado (glitazona).
[00202] Uma Glitazona Apropriada É, Por Exemplo, (S)-((3,4-diidro-2- (Fenil-metil)-2h-1-benzopiran-6-il)Metil-tiazolidina-2,4-diona (Englitazona), 5-{[4-(3-(5-metil-2-fenil-4-oxazolil)-1-oxopropil)-fenil]- Metil}-tiazolidina-2,4-diona (Darglitazona), 5-{[4-(1-metil- Cicloexil)Metóxi)-fenil]Metil}-tiazolidina-2,4-diona (Ciglitazona), 5-{[4-(2- (1 -indolil)Etóxi)Fenil]Metil}-tiazolidina-2,4-diona (Drf2189), 5-{4-[2-(5- Metil-2-fenil-4-oxazolil)-etóxi)]Benzil}-tiazolidina-2,4-diona (Bm- 13,1246), 5-(2-naftilsulfonil)-tiazolidina-2,4-diona (Ay-31637), Bis{4- [(2,4-dioxo-5-tiazolidinil)Metil]Fenil}Metano (Ym268), 5-{4-[2-(5-metil-2- Fenil-4-oxazolil)-2-hidroxietóxi]Benzil}-tiazolidina-2,4-diona (Ad-5075), 5-[4-(1-fenil-1-ciclopropanocarbonilamino)-benzil]-tiazolidina-2,4-diona (Dn-108) 5-{[4-(2-(2,3-d I Idroi Ndol-1 -i L)Etóxi)Fen I L]Metil}-tiazol Id Ina-2,4- Diona, 5-[3-(4-cloro-fenil])-2-propinil]-5-fenilsulfonil)Tiazolidina-2,4- Diona, 5-[3-(4-clorofenil])-2-propinil]-5-(4-flúorfenil-sulfonil)Tiazolidina- 2,4-diona, 5-{[4-(2-(Metil-2-piridinil-amino)-etóxi)Fenil]Metil}-tiazolidina- 2,4-diona (Rosiglitazona), 5-{[4-(2-(5-etil-2-piridil)Etóxi)Fenil]- Metil}Tiazolidina-2,4-diona (Pioglitazona), 5-{[4-((3,4-diidro-6-hidróxi- 2,5,7,8-tetrametil-2h-1-benzopiran-2-il)Metóxi)-fenil]-metil}-tiazolidina- 2,4-diona (Troglitazona), 5-[6-(2-flúor-benzilóxi)Naftalen-2-ilmetil]- Tiazolidina-2,4-diona (Mcc555), 5-{[2-(2-naftil)-benzoxazol-5-il]- Metil}Tiazolidina-2,4-diona (T-174) E 5-(2,4-dioxotiazolidin-5-ilmetil)-2- Metóxi-n-(4-triflúormetil-benzil)Benzamida (Krp297). Preferidos São Pioglitazona, Rosiglitazona E Troglitazona.
[00203] Outros agentes antidiabéticos incluem, moduladores da trilha de sinalização de insulina, como inibidores de proteína tirosina fosfatases (PTPases), compostos miméticos de molécula não pequena antidiabética e inibidores de glutamina-fructose-6-fosfato amidotransferase (GFAT); compostos que influenciam uma produção de glicose hepática desregulada, como inibidores de glucose-6- fosfatase (G6Pase), inibidores de fructose-1,6-bisfosfatase (F-1,6- BPase), inibidores de glicogênio fosforilase (GP), antagonistas de receptor de glucagon e inibidores de carboxicinase fosfoenolpiruvato (PEPCK); inibidores de cinase de piruvato desidrogenase (PDHK); inibidores de esvaziamento gástrico; insulina; inibidores de GSK-3; agonistas de receptor de retinóide X (RXR); agonistas de Beta-3 AR; agonistas de proteínas de não acoplamento (UCPs); agonistas de PPARytipo não glitazona; agonistas de PPARa/ PPARy duais; compostos contendo vanádio antidiabéticos; hormônios de incretina, tipo peptídeo-1 similar a glucagon (GLP-1) e agonistas de GLP-1; antagonistas de receptor de imidazolina de células beta; miglitol; e antagonistas c^-adrenérgicos; em que os ingredientes ativos estão presentes em cada caso em forma livre ou na forma de um sal farmaceuticamente aceitável.
[00204] Um agente redutor de obesidade inclui inibidores de lipase tais como orlistat e supressores de apetite tais como sibutramina, fentermina.
[00205] Um bloqueador de receptor de aldosterona inclui espironolactona e eplerenona.
[00206] Um bloqueador de receptor de endotelina inclui bosentan, etc.
[00207] Um inibidor de CETP se refere a um composto que inibe o transporte mediado por proteína de transferência de éster de cholésterila (CETP) de vários ésteres de cholésterila e triglicerídeos a partir de HDL para LDL e VLDL. Tal atividade de inibição de CETP é facilmente determinada por aqueles versados na técnica de acordo com ensaios padrão (por exemplo, Patente dos Estados Unidos No. 6.140.343). Os inibidores de CETP incluem aqueles descritos em Patente dos Estados Unidos No. 6.140.343 e Patente dos Estados Unidos No. 6.197.786. Inibidores de CETP descritos nestas patentes incluem compostos, tal como éster de etila de ácido [2R,4S]4-[(3,5-bis- triflúormetil-benzil)-metoxicarbonil- amino]-2-etil-6-triflúormetil-3,4- diidro-2H-quinolina-1-carboxílico, que é também conhecido como torcetrapib. Inibidores de CETP são também descritos em Patente dos Estados Unidos No. 6.723.752, que inclui um número de inibidores de CETP incluindo (2R)-3-{[3-(4-Cloro-3-etil-fenóxi)-fenil]-[[3-(1,1,2,2- tetraflúor-etóxi)-fenil]-metil]-amino}-1,1,1 -triflúor-2-propanol. Inibidores de CETP também incluem aqueles descritos em Pedido de Patente dos Estados Unidos número de série 10/807.838 depositado em 23 de março de 2004. Patente dos Estados Unidos No. 5.512.548 descreve certos derivados de polipeptídeo tendo atividade como Inibidores de CETP, também certos derivados de rosenonolactona inibidores de CETP e análogos de éster de cholésterila contendo fosfato são descritos em J. Antibiot., 49(8): 815- 816 (1996), e Bioorg. Med. Chem. Lett.; 6:1951-1954 (1996), respectivamente. Além disso, os inibidores de CETP também incluem aqueles descritos em W02000/017165, W02005/095409 e W02005/097806.
[00208] Um inibidor de Na_K-ATPase pode ser usado para inibir a permuta de Na e K através de membrana celulares. Tal inibidor pode ser por exemplo, digoxina.
[00209] Um bloqueador de receptor beta-adrenérgico inclui porém não está limitado a: esmolol especialmente o cloridrato deste; acebutolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.857.952; alprenolol, que pode ser preparado como descrito em Pedido de Patente Holandês No. 6.605.692; amosulalol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 4.217.305; arotinolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.932.400; atenolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.663.607 ou 3.836.671; befunolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.853.923; betaxolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 4.252.984; bevantolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.857.981; bisoprolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 4.171.370; bopindolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 4.340.541; bucumolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.663.570; bufetolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3. 723.476; bufuralol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.929.836; bunitrolol, que pode ser preparado como descrito em Patente dos Estados Unidos Nos. 3.940, 489 e 3.961.071; buprandolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.309.406; cloridrato de butiridina, que pode ser preparado como descrito em Patente Francesa No. 1.390.056; butofilolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 4.252.825; carazolol, que pode ser preparado como descrito em Patente Alemã No. 2.240.599; carteolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.910.924; carvedilol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 4.503.067; celiprolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 4.034.009; cetamolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 4.059.622; cloranolol, que pode ser preparado como descrito em Patente Alemã No. 2.213.044; dilavalol, que pode ser preparado como descrito em Clifton e outro , Journal de Medicinal Chemistry, 1982, 25, 670; epanolol, que pode ser preparado como descrito em Pedido de Publicação de Patente Européia No. 41.491; indenolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 4.045.482; labetalol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 4.012.444; levobunolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 4. 463.176; mepindolol, que pode ser preparado como descrito em Seeman e outro , Helv. Chim. Acta, 1971, 54, 241; metipranolol, que pode ser preparado como descrito em Pedido de Patente Tchecoslovaco No. 128.471; metoprolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.873.600; moprolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.501.7691; nadolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.935.267; nadoxolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.819.702; nebivalol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 4.654.362; nipradilol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 4.394.382; oxprenolol, que pode ser preparado como descrito em Patente Britânica No. 1.077.603; perbutolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.551.493; pindolol, que pode ser preparado como descrito em Patente Suíça Nos. 469.002 e 472.404; practolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.408.387; pronetalol, que pode ser preparado como descrito em Patente Britânica No. 909.357; propranolol, que pode ser preparado como descrito em Patente dos Estados Unidos Nos. 3.337.628 e 3.520.919; sotalol, que pode ser preparado como descrito em Uloth e outro , Journal de Medicinal Chemistry, 1966, 9, 88; sufinalol, que pode ser preparado como descrito em Patente Alemã No. 2.728.641; talindol, que pode ser preparado como descrito em Patente dos Estados Unidos Nos. 3.935.259 e 4.038.313; tertatolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.960.891; tilisolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 4.129.565; timolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.655.663; toliprolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.432.545; e xibenolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 4.018.824.
[00210] Um bloqueador de receptor alfa-adrenérgico inclui porém não é limitado a: amosulalol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 4.217.307; arotinolol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.932.400; dapiprazol, que pode ser preparado como descrito em Patente dos Estados Unidos No. 4.252.721; doxazosina, que pode ser preparado como descrito em Patente dos Estados Unidos No. 4.188.390; fenspirida, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.399.192; indoramina, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.527.761; labetolol, que pode ser preparado como descrito acima; naftopidil, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.997.666; nicergolina, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.228.943; prazosina, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.511.836; tamsulosina, que pode ser preparado como descrito em Patente dos Estados Unidos No. 4.703.063; tolazolina, que pode ser preparado como descrito em Patente dos Estados Unidos No. 2.161.938; trimazosina, que pode ser preparado como descrito em Patente dos Estados Unidos No. 3.669.968; e yohimbine, que pode ser isolado a partir de fontes naturais de acordo com métodos bem-conhecidos por aqueles versados na técnica.
[00211] Os peptídeos natriuréticos constituem a família de peptídeos que incluem os peptídeos atrial (ANP), derivados do cérebro (BNP) e natriuréticos tipo C (CNP). A vasodilatação de efeito de peptídeos natriuréticos, natriurese, diurese, liberação diminuída de aldosterona, crescimento celular diminuído, e inibição do sistema nervoso simpático e o sistema renina-angiotensina-aldosterona indicando seu envolvimento na regulação da pressão sangüínea e de equilíbrio de sódio e água. A endopeptidase neutra 24. 11 inibidores de (NEP) impedem a degradação de peptídeos natriuréticos e eliciam ações farmacológicas potencialmente benéficas no controle de diversos distúrbios cardiovasculares. Um inibidor de NEP útil na referida combinação é um agente selecionado a partir do grupo representado por candoxatril, sinorfan, SCH 34826 e SCH 42495.
[00212] Um agente inotrópico é selecionado a partir do grupo que consiste em: digoxina, digitoxina, digitalis, dobutamina, dopamina, epinefrina, milrinona, amrinona e norepinefrina, etc.
[00213] Um composto da presente invenção pode ser administrada ou simultaneamente, antes ou após os outros ingredientes ativos, ou separadamente pela mesma ou diferente rotina de administração ou juntos na mesma formulação farmacêutica.
[00214] Além disso, as combinações como descrito acima podem ser administradas a um indivíduo por meio de administração simultânea, separada ou seqüencial (uso). A administração simultânea (uso) pode ocorrer na forma de uma combinação fixa com dois ou três ou mais ingredientes ativos, ou por administração simultânea de dois ou três ou mais compostos que são formulados independentemente. A administração (uso) seqüencial preferivelmente significa a administração de um (ou mais) compostos ou ingredientes ativos de uma combinação em um ponto do tempo, outros compostos ou ingrediente ativos em um diferente ponto do tempo, isto é, de uma maneira em etapas cronológicas, preferivelmente de modo que a combinação mostre mais eficiência do que os compostos sozinhos administrados independentemente (especialmente mostrando sinergismo). Administração separada (uso) preferivelmente significa a administração dos compostos ou ingredientes ativos da combinação independentemente um do outro em diferentes pontos do tempo, preferivelmente significando que dois, ou três ou mais compostos são administrados de modo que nenhuma sobreposição de níveis sangüíneos mensuráveis de ambos os compostos esteja presente de uma maneira coincidente (ao mesmo tempo).
[00215] Além disso, as combinações de dois ou três ou mais de administração seqüencial, separada e simultânea são possíveis, preferivelmente de modo que a combinação composto-fármacos mostre um efeito terapêutico associado que exceda o efeito encontrado quando a combinação composto-fármacos é usada independentemente em intervalos de tempo tão grandes que nenhum efeito mútuo sobre sua eficiência terapêutica possa ser encontrado, um efeito sinérgico sendo especialmente preferido.
[00216] Alternativamente, as composições farmacêuticas contêm uma quantidade terapeuticamente eficaz de um composto da invenção como acima definido, ou sozinho ou em uma combinação com um ou mais agentes terapêuticos, por exemplo, cada em uma dose terapêutica eficaz como reportado na técnica, selecionada a partir do grupo que consiste em um antiestrogênio; um antiandrogênio; um agonista de gonadorelina; um inibidor de topoisomerase I; um inibidor de topoisomerase II; um agente ativo de microtúbulo; um agente de alquilação; um agente antimetabólito antineoplásico; um composto de platina; um composto alvejando/diminuindo uma atividade de proteína ou lipídeo cinase ou uma atividade de proteína ou lipídeo fosfatase, um composto antiangiogênico; um composto que induz processos de diferenciação celular; anticorpos monoclonais; um inibidor de ciclooxigenase; um bisfosfonato; um inibidor de heparanase; um modificador de resposta biológica; um inibidor de isoformas oncogênicas de Ras; um inibidor de telomerase; um inibidor de protease, um inibidor metaloproteinase matriz, um inibidor de metionina aminopeptidase; um inibidor de proteassoma; agentes que alvejam, diminuem ou inibem a atividade de Flt-3; um inibidor de HSP90; anticorpos antiproliferatives; um inibidor HDAC; um composto que alveja, diminui ou inibe a atividade/função de serina/treonina mTOR cinase; um antagonista de receptor de somatostatina; um composto anti-leucêmico; métodos de danificação de célula de tumor; um aglutinante de EDG; um inibidor de ribonucleotídeo reductase; um inibidor de S-adenosilmetionina decarboxilase; um anticorpo monoclonal de VEGF ou VEGFR; terapia fotodinâmica; um ésteróide angiostático; um implante contendo corticosteróides; um antagonista de receptor AT1; e um inibidor de ACE.
[00217] Adicionalmente, a presente invenção fornece: - uma composição farmacêutica ou combinação da presente invenção para uso como um medicamento; - o uso de uma composição farmacêutica ou combinação da presente invenção para o retardo de progressão e/ou tratamento de um distúrbio ou doença mediada por sintase de aldosterona, ou responsiva à inibição de sintase de aldosterona, ou caracterizada por atividade anormal ou expressão de sintase de aldosterona. - o uso de uma composição farmacêutica ou combinação da presente invenção para o retardo de progressão e/ou tratamento de um distúrbio ou doença mediada por aromatase, ou responsiva à inibição de aromatase, ou caracterizada por atividade anormal ou expressão de aromatase. - o uso de uma composição farmacêutica ou combinação da presente invenção para o retardo de progressão e/ou tratamento de um distúrbio ou doença selecionada a partir de hipocalemia, hipertensão, insuficiência cardíaca congestiva, fibrilação atrial, insuficiência renal, em particular, insuficiência renal crônica, restenose, aterosclerose, síndrome X, obesidade, nefropatia, infarto pós-miocardiano, doenças cardíacas coronarianas, formação aumentada de colágeno, fibrose tais como fibrose cardíaca ou miocardiana e remodelagem após hipertensão e disfunção endotelial. - o uso de uma composição farmacêutica ou combinação da presente invenção para o retardo de progressão e/ou tratamento de um distúrbio ou doença selecionada a partir de ginecomastia, osteoporose, câncer de próstata, endometriose, fibróides uterinos, sangramento uterino disfuncional, hiperplasia endometrial, doença ovariana policística, infertilidade, doença de mama fibrocística, câncer de mama e mastopatia fibrocística.
[00218] A composição ou combinação farmacêutica da presente invenção pode ser em dosagem unitária de cerca de 1-1000 mg de ingredientes ativos para um indivíduo de cerca de 50-70 kg, preferivelmente cerca de 5-500 mg de ingredientes ativos. A dosagem terapeuticamente eficaz de um composto, a composição farmacêutica, ou as combinações deste, é dependente da espécie de indivíduo, o peso corporal, idade e condição individual, um distúrbio ou doença ou a gravidade desta que está sendo tratada. Um médico, clínico ou veterinário versado pode facilmente determinar a quantidade eficaz de cada dos ingredientes ativos necessários para prevenir, tratar ou inibir o progresso do distúrbio ou doença.
[00219] As propriedades de dosagem acima citadas são demonstráveis em testes in vitro e in vivo usando vantajosamente mamíferos, por exemplo, camundongos, ratos, cães, macacos ou órgãos, tecidos e preparações. Os compostos da presente invenção podem ser aplicados in vitro na forma de soluções, por exemplo, preferivelmente soluções aquosas, e in vivo ou enteralmente, parenteralmente, vantajosamente intravenosamente, por exemplo, como uma suspensão ou em solução aquosa. A dosagem in vitro pode variar entre cerca de concentrações de 10-3 molar e 10-9 molar. Uma quantidade terapeuticamente eficaz in vivo pode variar dependendo da rotina de administração, entre cerca de 0,1 - 500 mg/kg, preferivelmente entre cerca de 1 -100 mg/kg.
[00220] As atividades de um composto de acordo com a presente invenção podem ser avaliadas pelos seguintes métodos in vitro & in vivo métodos bem descritos na técnica. Observe, Fieber, A e outro (2005), "Aldosterone Syntase Inhibitor Ameliorates Angiotensina II- Induced Organ Damage," Circulation, 111:3087-3094; Observe, também Stresser DM, Turner SD, McNamara J, et al (2000), "A high- throughput screen to identify inhibitors de aromatase (CYP19)," Anal Biochenr, 284:427-30. Todas as referências aqui citadas são incorporadas por referência em sua totalidade.
[00221] Em particular, as atividades inibidoras de aromatase e aldosterona sintase in vitro podem ser determinadas pelos seguintes ensaios.
[00222] A linhagem de célula NCI-H295R de carcinoma adrenocortical humano é obtida a partir da American Type Culture Collection (Manassas, VA). Suplemento de insulina/transferrina/selênio (ITS)-A (100x), DMEM/F-12, antibiótico/antimicótico (100x), e soro de bezerro fetal (FCS) são comprados a partir de Gibco (Grand Island, NY). Contas de ensaio (SPA) de proximidade de cintilação de PVT anticamundongo beads e placas de 96 cavidades NBS são obtidas a partir de Amersham (Piscataway, NJ) e Corning (Acton, MA), respectivamente. Placas de base plana de 96 cavidades pretas sólidas são compradas de Costar (Corning, NY). Aldosterona e angiotensina (Ang II) são compradas a partir de Sigma (St. Louis, MO). D-[1,2,6,7- 3H(N)]aldosterona foi adquirida de PerkinElmer (Boston, MA). Soro de Nu foi um produto de BD Biosciences (Franklin Lakes, NJ). O sistema de regeneração de NADPH, dibenzilfluoresceína (DBF), e human aromatase supersomes® são obtidos de Gentest (Woburn, MA).
[00223] Para avaliação in vitro de atividade de aldosterona, células NCI-H295R de carcinoma adrenocortical humano são semeadas em placas de 96 cavidades NBS e a densidade de 25.000 células/cavidade em 100 pl de um meio de crescimento contendo DMEM/F12 suplementado com 10% FCS, 2,5% de soro de Nu, 1 pg de ITS/ml, e 1x antibiótico/antimicótico. O meio é mudado após a cultura durante 3 dias a 37 °C sob uma atmosfera de 5% CO2/ 95% ar. No dia seguinte, as células são enxagüadas com 100 pl de DMEM/F12 e incubadas com 100 pl de meio de tratamento medium contendo 1 pM Ang II e um composto em diferentes concentrações em cavidades quadruplicadas a 37 °C durante 24 horas. Ao término da incubação, 50 pl de meio são retirados de cada cavidade para avaliação de produção de aldosterona por um RIA usando anticorpos monoclonais antialdosterona.
[00224] Avaliação de atividade de aldosterona pode também ser realizada usando um formato de placa de 96 cavidades. Cada amostra de teste é incubada com 0,02 pCi de D-[1,2,6,7-3H(N)]aldosterona e 0,3 pg de anticorpo antialdosterona em salina tamponada por fosfato (PBS) contendo 0,1% de Triton X-100, 0,1% de albumina de soro bovino, e 12% de glicerol em um volume total de 200 pl em temperatura ambiente durante 1 hora. Contas PVT SPA anticamundongo (50 pl) são então adicionadas a cada cavidade e incubadas durante a noite em temperatura ambiente antes da contagem em uma registradora de placa de Microbeta. A quantidade de aldosterona em cada amostra é calculada por comparação com uma curva padrão gerada usando quantidades conhecidas quantities de hormônio.
[00225] Para avaliar atividade de aromatase atividade, o ensaio de aromatase humana é realizado em placas de base plana de 96 cavidades de acordo com um protocolo publicado (Stresser e outro, 2000) com modificações menores. Em síntese, 10 pl de um sistema de regeneração de NADPH contendo 2,6 mM de NADP+, 6,6 mM de glicose 6-fosfato, 6,6 mM de MgCh, e 0,8 U/ml de glicose-6-fosfato desidrogenase em 50 mM de fosfato de potássio, pH 7,4, é pré- incubado com o composto teste em uma concentração desejada a 30 °C por 10 minutos em um volume total de 100 pl. Posteriormente, 4 pmols de aromatase humana, 20 pg de proteína microssômica de controle, e 4 pM de DBF em 100 pl de 50 mM de fosfato de potássio, pH 7,4, são adicionados a cada cavidade e incubados a 30 °C por 90 minutos. A reação é terminada pela adição de 75 pl de NaOHa 2 N a cada cavidade. Após 2 horas, o produto, fluoresceína, é avaliado por um fluorímetro usando comprimentos de onda de excitação e emissão de 485 e 538 nm, respectivamente.
[00226] As curvas de resposta de concentração total do composto teste são realizadas pelo menos 3 vezes. Os valores de ICso são derivados usando um programa de ajuste de curva de quadrados pelo menos não linear a partir de Microsoft XLfit.
[00227] As atividades inibidoras in vivo para sintase de aldosterona e aromatase podem ser determinadas pelos seguintes ensaios.
[00228] Os compostos teste (isto é, sintase potencial de inibidores de aldosterona) são apresentados in vivo em um modelo de rato consciente de hiperaldosteronismo secundário agudo. Os ratos tipo silvestre são instrumentados com cânulas venosas e arteriais de cronicamente de demora, que são exteriorizados através de um sistema téter/giratório. Os ratos de ambulatório são alojados em gaiolas especializadas para permitir amostragem de sangue e administração de fármaco parenteral sem perturbar os animais. Angiotensina II é continuamente infundida intravenosamente em um nível suficiente para elevar a concentração de aldosterona de plasma (PAC) por ~200-vezes a 1-5 nM. Este aumento de PAC é sustentado em um nível estável durante pelo menos 8 a 9 horas. Os compostos de teste são administrados p.o. (por meio de gavagem oral) ou parenteralmente (por meio do catetér arterial) após uma hora de infusão de angiotensina II em um tempo quando PAC tiver aumentado para um nível de estado estável. As amostras de sangue arterial são coletadas antes e em vários momentos (até 24 horas) após a administração de agente de teste para determinação posterior de concentração de agente de teste e PAC. A partir destas avaliações, vários parâmetros podem ser derivados, por exemplo, 1) início e duração de redução de PAC para o agente de teste, 2) parâmetros farmacocinéticos do agente de teste tal como meia-vida, depuração, volume de distribution, e biodisponibilidade, 3) resposta de dose/PAC, concentração de dose/agente teste, e ligações de concentração de agente de teste /resposta de PAC, e 4) potências de dose e concentração e eficácia do agente de teste. Um composto teste bem- sucedido diminui PAC em de um modo dependente da dose e do tempo na faixa de dose de cerca de 0,01 a cerca de 10 mg/kg i.a. ou p.o. TABELA 1. ATIVIDADES INIBIDORAS DOS COMPOSTOS





ABREVIAÇÕES DCM: diclorometano DIBAL: hidreto de diisobutilalumínio DMAP: N,/V-dimetilaminopiridina DME: dimetoxietano DMF: /V,/V-dimetilformamida DMSO: dimetilsulfóxido ESI: ionização de eletrovaporização h: horas HPLC: cromatografia líquida de alta pressão HRMS: espectrometria de massa de alta resolução IPA: álcool isopropílico IV: espectroscopia de infravermelho KHMDS: hexametildisilazida de potássio LAH: hidreto de alumínio de lítio LCMS: cromatografia líquida/espectrometria de massa LDA: diisoproilamida de lítio LHMDS: hexametildisilazida de lítio min: minutos EM: espectrometria de massa NBS: A/-bromossucinimida RMN: ressonância magnética nuclear PS -PPh3-Pd(0): complexo de paládio trifenilfosfina sustentado por polímero TBSCI: cloreto de terc-butildimetilsilila TFA: ácido triflúoracético THF: tetraidrofurano TMEDA: tetrametilatilanodiamina TBS: dimetilsilila de terc-butila TMSCI: cloreto de trimetilsilila TLC: cromatografia de camada fina Tr: tritila tr: tempo de retenção TMEDA: diamina de tetrametilatilano
[00229] Os seguintes exemplos destinam-se a ilustrar a invenção e não devem ser interpretados como sendo limitações sobre ela. As temperaturas são mencionadas em graus centígrados. Se não mencionada de outra maneira, todas as evaporações são realizadas sob pressão reduzida, preferivelmente entre cerca de 15 mm Hg e 100 mm Hg (= 20-133 mbar). A estrutura de produtos finais, intermediários e materiais de partida é confirmada por métodos analíticos padrões, por exemplo, características espectroscópicas e microanálises, por exemplo, MS, IR, RMN. As abreviações empregadas são aquelas convencionais na técnica. Foi descoberto que os compostos nos seguintes exemplos possuem valores de ICso na faixa de cerca de 0,1 nM a cerca de 10,000 nM para ambas, aromatase e sintase de aldosterona. EXEMPLO 1 BROMETO DE BENZILA A. 4-BROMOMETIL-3-CLOROBENZONITRILO (CAS N° 21924-83-4)
[00230] 3-Cloro-4-metilbenzonitrila (2,34 g, 15,4 mmols), NBS (3,0 g, 16,9 mmols) e peróxido de benzoíla (0,37 g, 1,54 mmosl) são absorvidos em tetracloreto de carbono (50 mL, 0,3M) e refluxados até a reação ser concluída por TLC. A mistura é em seguida deixada esfriar para à temperatura ambiente e é filtrada. O filtrado é concentrado e purificado por meio de cromatografia de coluna instantânea (0-5% EtOAc/hexanos) para fornecer 4-bromometil-3- clorobenzonitrila como um sólido branco. HRMS (ESI) m/z 229,9133 (229,9193 calculado para C8H6CIBrN, M+H).
[00231] Similarmente preparados são os seguintes compostos de toluenos correspondentes:
[00232] 4-Bromometil-3-flúorbenzonitrila (cas n° 105942-09-4)
[00233] 4-Bromometil-2-bromobenzonitrila (cas n° 89892-38-6)
[00234] 4-Bromometil-3-metoxibenzonitrila (cas n° 104436-60-4)
[00235] 4-Bromometil-3-nitrobenzonitrila (cas n° 223512-70-7)
[00236] Éster de metila de ácido 3-Bromo-4-bromometilbenzóico (cas n° 78946-25-5) B. 4-BROMOMETIL-3-TRIFLÚORMETILBENZONITRILO
[00237] 4-Metil-3-triflúormetilbenzonitrila é bromado com NBS de acordo com o exemplo 1A para fornecer 4-bromometil-3- triflúormetilbenzonitrila. 1H RMN (400 MHz, CDCh) δ (ppm) 7,94 (s, 1H), 7,85 (d, J = 8,1 Hz, 1H), 7,76 (d, J= 8,1 Hz, 1H), 4,63 (s, 2H). C. 4-BROMOMETIL-3-TRIFLÚORMETOXIBENZONITRILA
[00238] A uma mistura de CuBr2 (25,5 g, 114 mmols) em CH3CN (500 mL) a 0 °C é adicionado nitrito de f-butila (17,7 mL, 148 mmols). Em seguida 4-amino-3-triflúormetoxibenzonitrila (20,0 g, 99,0 mmols) é adicionado em 4 porções durante urn periodo de 10 minutos. A mistura é deixada aquecer para à temperatura ambiente e agitar durante a noite . O solvente é removido e 0 resíduo é dividido entre Et2O e 1M HCI. a fase aquosa é também extraída com Et2O e as camadas orgânicas combinadas são secadas (Na2SO4) e concentradas. O resíduo sólido é em seguida triturado com hexanos para fornecer 4- Bromo-3-triflúormetoxibenzonitrila como um sólido cristalino amarelo. 1H RMN (400 MHz, CDCh) δ (ppm) 7,47 (dd, J = 8,0, 2,0 Hz, 1 H), 7,59 (m, 1H), 7,80 (d, J = 8,0 Hz, 1H).
[00239] Uma mistura de 4-bromo-3-triflúormetoxibenzonitrila (10,0 g, 37,6 mmols), K2COs (15,6 g, 113 mmols), trimetilboroxina (5,5 mL, 39,5 mmols) e DMF (150 mL) é desgaseificada durante 10 min com nitrogênio antes de Pd(PPh3)4 (4,34 g, 3,76 mmols) ser adicionado, a mistura é em seguida selada e aquecida para 120 °C durante 14 h. A mistura é em seguida concentrada e então dividida entre Et2O e solução de salmoura a 50%. A fase aquosa é também extraída com Et2O e as camadas orgânicas combinadas são secadas (Na2SO4) e concentradas. O resíduo é purificado por meio de cromatografia instantânea (10% EtOAc/hexanos) para fornecer 4-metil-3- triflúormetoxibenzonitrila. H RMN (400 MHz, CDCh) δ(ppm) 2,40 (s, 3H), 7,40 (d, J = 7,7 Hz, 1H), 7,50 (s, 1 H), 7,51 (dd, J = 7,7, 1,5 Hz, 1 H). MS (ESI) m/z 202,1.
[00240] 4-Metil-3-Triflúormetoxibenzonitrila é bromado com NBS de acordo com exemplo 1A para fornecer 4-bromometil-3- triflúormetoxibenzonitrila. 1H RMN (400 MHz, CDCh) δ (ppm) 4,51 (s, 2H), 7,55 (br s, 1H), 7,59 (d, J = 8,0z, 1 H), 7,64 (d, J = 8,0 Hz, 1 H). D. 4-BROMOMETIL-2,5-DIMETOXIBENZQNITRILO
[00241] Por analogia às etapas delineadas em J. Med. Chem. 1976, 19(12), 1400-1404. 2,5-dimetóxi-4-metilbenzaldeído (14,8 g, 82,2 mmols) é dissolvido em piridina (300 mL) e a ele é adicionado cloridrato de hidroxilamina (6,8 g, 98,6 mmols). A suspensão é aquecida a 105 nC por 2 h. Anidrido acético (15,5 mL, 164 mmols) é então adicionado à reação e a agitação é continuada por mais 2 h. A solução é evaporada até a secura e dividida entre EtOAc e NaHCOs aquoso saturado. A fração orgânica é secada (Na2SO4) e evaporada para fornecer um sólido amarelo que é absorvido em hexanos e filtrado para fornecer 2,5-dimetóxi-4-metilbenzonitrilo como um sólido branco, (cas n° 51267-09-5) MS (ESI) m/z 178,2 (M + H).
[00242] 2,5-Dimetóxi-4-metilbenzonitrila (4,06 g, 21,5 mmols) é bromado com NBS de acordo com o exemplo 1A para fornecer 4- bromometil-2,5-dimetoxibenzonitrila. 1H RMN (400 MHz, CDCh) δ (ppm) 6,95 (s, 1H), 6,91 (s, 1H), 4,43 (s, 2H), 3,84 (s, 3H), 3,80 (s, 3H).
[00243] Similarmente preparado é o seguinte:
[00244] 4-Bromometil-3-bromobenzonitrila (cas n° 89892-39-7). 1H RMN (400 MHz, CDCh)δ(ppm) 7,87 (d, 1H, J= 1,2 Hz), 7,60 (dd, 1H, J = 7,6, 1,2 Hz), 7,57 (d, 1H, J= 7,6 Hz), 4,58 (s, 2H).
D. 2-BROMOMETIL-4'-FLLJORBIFENILA (CAS N° 791078-01-8)
[00245] A uma mistura de ácido 4-flúorfenilborônico (2,5 g, 13,4 mmols), álcool de 2-bromobenzila (2,81 g, 20,1 mmols) e Pd(PPh3)4 (0,25 g, 0,216 mmols) em DME (20 mL) é adicionado na solução aquosa de Na2COs (11,5 mL, 2,7 M, 31 mmols). A mistura é aquecida para 115 °C em vaso selado durante a noite. A reação é deixada resfriar para a temperatura ambiente e é diluída com EtOAc e água. A camada aquosa é extraída também com EtOAc (2X). As camadas orgânicas combinadas são lavadas com água, NH4CI saturado, salmoura e secadas sobre Na2SO4. Após concentração, 0 resíduo resultante é purificado por cromatografia instantânea (hexano/C^CL) para fornecer (4'-flúorbifenil-2-il)-metanol como óleo, (cas n° 773871- 75-3) 1H RMN (400 MHz, CDCh) δ (ppm) 7,46-7,43 (m, 1H), 7,32-7,22 (m, 4H), 7,18-7,16 (m, 1H), 7,04-6,99 (m, 2H), 4,48 (d, J = 5,5 Hz, 2H), 1,68 (brs, 1H).
[00246] A uma solução de (4'-flúorbifenil-2-il)-metanol (2,58 g, 12,8 mmols) em CH2CI2 (100 mL), tetrabrometo de carbono (7,40 g, 22,3 mmols) é adicionado. A solução é resfriada para 0 °C e em seguida trifenilfosfina (7,53 g, 28,7 mmols) é adicionada em porções. A reação é agitada a 0 °C por 1,5h e em seguida à temperatura ambiente por 90 h antes do solvente ser removido. O resíduo é dividido entre Et2θ e água e em seguida filtrado. As camadas são separadas e as camadas aquosas são extraídas com Et2θ. As camadas orgânicas combinadas são lavadas com água, salmoura e secadas sobre Na2SO4. Após concentração, o resíduo é purificado por cromatografia instantânea (hexano) para fornecer 2-bromometil-4'-flúorbifenila como um óleo. (Uma preparação alternativa aparece em J. Med. Chem. 2004, 47(22), 5441) 1H RMN (400 MHz, CDCh) □ 7,53-7,50 (m, 1H), 7,43-7,31 (m, 4H), 7,24-7,21 (m, 1H), 7,15-6,80 (m, 2H), 4,42 (s, 2H). E. ÉSTER DE METILA DE ÁCIDO BROMO-(3-FLÚOR-4- METOXIFENIDACÉTICO
[00247] Ácido (3-flúor-4-metoxifenil)acético (5,0 g, 27,1 mmols) é dissolvido em MeOH (100 mL). A ele é adicionado H2SO4 (5 mL) concentrado e a solução é aquecida até 0 refluxo por 2 h. Naquele ponto, a solução é evaporada até a secura e absorvida em EtOAc. A solução é lavada com NaHCOs aquoso saturado, secada (Na2SO4) e evaporada para fornecer éster de metila de ácido (3-flúor-4- metoxifenil)acético (casn0 588-14-7) como um óleo amarelo. MS (ESI) m/z 199,3 (M + H).
[00248] O éster de metila de ácido (3-flúor-4-metoxifenil)acético (5,16 g, 26,0 mmols) é dissolvido em tetracloreto de carbono (300 mL) juntamente com NBS (5,56 g, 31,3 mmols) e peróxido de benzoíla (0,63 g, 2,60 mmols) e refluxado por 2 h. A solução é então deixada esfriar parà temperatura ambiente e é filtrada. O filtrado é evaporado e 0 resíduo purificado por cromatografia de coluna instantânea (EtOAc/hexanos 5:95—>EtOAc/hexanos 2:8) para fornecer éster de metila de ácido bromo-(3-flúor-4-metoxifenil)acético como um óleo amarelo. 1H RMN (CDCh) δ 7,37 (d, J= 12 Hz, 1H), 7,26-7,23 (m, 1H), 6,93 (t, J= 8 Hz, 1H), 5,31 (s, 1H), 3,91 (s, 3H), 3,81 s, 3H). EXEMPLO 2 INTERMEDIÁRIOS DE IMIDAZOL SUBSTITUÍDOS A. 1-TRITIL-4-CARBOXALDEÍDO-1H-IMIDAZOL (CAS N°33016-47-6)
[00249] De acordo com procedimento delineado em J. Med. Chem. 2002, 45(1), 177, para imidazol-4-carboxaldeído (15,0 g, 156,2 mmols) em DMF (300 mL) é adicionado trietilamina (43,8 ml, 312 mmols) seguido por cloreto de tritila (44,4 g, 159,0 mmols). A mistura de reação é agitada em temperatura ambiente durante 18 h antes do solvente ser removido em vácuo. O resultante sólido é dissolvido em diclorometano e lavado com bicarbonato de sódio e água. A fase orgânica é concentrada em vácuo para fornecer o material desejado como um sólido. B. 1-(1-TRITIL-1H-IMIDAZOL-4-IL)ETANOL (CAS N°62256-50-2)
[00250] A 1-tritil-4-carboxaldeído-1H-imidazol (11,7 g, 34,6 mmols) em tF (250ml) a 0 °C é adicionado brometo de metilmagnésio (12,6 mL, 38 mmols, 3,0 M em éter de dietila). A mistura de reação é agitada a 15 °C por 4 h antes do saciamento com água (10 mL), seguido por cloreto de amónio aquoso. A reação é extraída em acetato de etila e lavada com 30 mL bicarbonato de sódio aquoso saturado. O solvente orgânico é removido em vácuo. Cromatografia (sílica-gel, acetato de etila:hexanos, 1:1 a 1:0) produz o produto desejado. MS (ESI) m/z 355 (M + H). (preparado similarmente em J. Med. Chem. 1977, 20(5), 721) C. 1- (1-TRITIL-1H-IMIDAZOL-4-IDETANONA (CAS N°116795-55-2)
[00251] A 1-(1-tritil-1 H-imidazol-4-il)etanol (8,06 g, 22,7 mmols) em dioxano (400 mL) é adicionado dióxido de manganês (9,9 g, 113,8 mmols). A mistura de reação é aquecida para 90 °C e agitada por 18 h. A reação é deixada resfriar à temperatura ambiente e filtrada através de terra diatomácea. O solvente filtrado é removido em vácuo para produzir o produto. MS (ESI) m/z 353 (M + H) (preparado similarmente em Bioorg. Med. Chem. 2004, 12(9), 2251.) D. ÁCIDO (1-TRITIL-1H-IMIDAZOL-4-IL)ACÉTICO (CAS N° 168632- 03-9)
[00252] Cloreto de tritila (51 g, 0,18 mol) é adicionado a uma suspensão de cloridrato de ácido (1H-imidazol-4-il)acético (25 g, 0,15 mol) em piridina (500 mL). Esta é agitada à temperatura ambiente por 16 h, no término do qual MeOH (150 mL) é adicionado. Esta solução é agitada à temperatura ambiente por 1 h. Solventes são evaporados e o resíduo é absorvido em CH2CI2 e lavados com 1 M de solução de ácido cítrico aquoso (2X) e salmoura. A fase orgânica é secada em Na2SO4 anidroso e evaporada para fornecer um resíduo pegajoso que quando absorvido em éter de dietila e evaporado forneceu 0 produto como um sólido branco que é usado sem outra purificação. MS (ESI) m/z 368,9 (M + H) (Procedimento adaptado de J. Org. Chem. 1993, 58, 4606, também preparado no W02003013526) E. 2-(1-TRITIL-1H-IMIDAZOL-4-IL)ETANOL (CAS N° 127607-62-9)
[00253] Ácido (1-tritil-1H-imidazol-4-il)Acético (65 G, 0,17 Mol) É Suspenso Em Thf (400 Ml) E Resfriado A 0 °c. A Isto É Adicionado Solução De Bh3 Thf (350 Ml, 1,0 M). A Solução Clara Obtida É Agitada A 0 °c Por 30 Min Antes Do Aquecimento Parà Temperatura Ambiente Até Que Lcms Indique 0 Término Da Reação. A Solução É Resfriada Novamente A 0 °c E Extinguida Cuidadosamente Com Água (250 Ml). A Solução Resultante É Diluída Com Etoac (300 Ml) E Transferida Com Um Funil Separador E A Camada Aquosa É Extraída Com Etoac. A Fase Orgânica É Secada Sobre Na2so4 Anidroso E Evaporado Para Fornecer Um Resíduo Pegajoso Que É Absorvido Em Etanolamina (800 Ml) E Aquecido A 90 °c Por 2 H. A Reação É Transferida Para Um Funil Separador, Diluída Com Etoac (1 L) E Lavada Com Água (3 X 600 Ml). A Fase Orgânica É Secada Sobre Na2so4 Anidroso E Evaporado Para Fornecer 2-(1-tritil-1H-imidazol-4-il)-etanol Como Um Sólido Branco Que É Usado Como Está, Sem Outra Purificação. Ms (Esi) M/Z 354,8 (M + H) (Preparado Por Método Alternativo Em J. Med. Chem. 1996, 39(19), 3806) F.4-[2-(Terc-butildimetilsilanilóxi)Etil]-1-tritil-1h- Imidazol

[00254] 2-(1-Tritil-1H-imidazol-4-il)etanol (20 g, 56,5 mmols) é dissolvido em CH2CI2 (500 mL). A este é adicionado imidazol (11,5 g, 169 mmols) e ferc-butildimetilsililcloreto (10,2 g, 67,8 mmols). A solução é agitada à temperatura ambiente até que LCMS indique que a reação está concluída. A solução é dividida entre CH2CI2 e NaHCOs aquoso saturado. A camada orgânica é lavada também com NaHCOs aquoso saturado e salmoura. A fase orgânica é secada sobre Na2SO4 anidroso e evaporado para fornecer um óleo que é purificado por meio de coluna de cromatografia instantânea (EtOAc/hexanos 3:7) para fornecer 3-[2-(ferc-butildimetilsilanilóxi)etil]-1 -tritil-1 H-imidazol como um sólido branco. MS (ESI) m/z 469,3 (M + H). G. ÉSTER ÁCIDO 4-[(1-TRITIL-1H-IMIDAZOL-4-IL)]PROPANÓICO DE METILA (CASN0 102676-60-8)
[00255] A uma suspensão branca de ácido 3-(1H-imidazol-4- il)propiônico (5 g, 35,7 mmols) em MeOH (140 mL) é adicionado em gotas HCI/Dioxano (4M, 29 mL, 116 mmols). A solução clara resultante é lentamente aquecida até à temperatura ambiente e agitada durante a noite. A mistura de reação é concentrada em vácuo e secada em uma bomba de vácuo elevado para fornecer um óleo.
[00256] A uma solução de cloridrato de éster de metila de ácido 3- (1H-imidazol-4-il)propiônico (6,8 g, 35,7 mmols) em CH3CN (160 mL) é adicionado cloreto de tritila (11,0 g, 39,5 mmols) em porção a 0 °C e seguida por trietilamina (40 mL). A mistura branca em suspensão é agitada à temperatura ambiente durante a noite. O solvente é evaporado e 0 resíduo é suspenso em 200 mL H2O-gelo e agitado por 1 h. O sólido é coletado e secado sob uma bomba de vácuo elevado para fornecer um sólido branco, (preparado em J. Med. Chem. 1996, 39(6), 1220.) H. ÉSTER DE METILA DE ÁCIDO (1-TRITIL-1H-IMIDAZOL-4- IDACÉTICO (CASN0 145133-11-5)
[00257] Preparado a partir do correspondente ácido de acordo com o procedimento G acima, (preparado em US5140034) I. 4-[3-ÍTERC-BUTILDIMETILSILANILÓXI)PROPIL1-1-TRITIL-1H- IMIDAZOL
[00258] A uma suspensão de LAH (1,0 g, 26,4 mmols) em THF (80 mL) a 0 °C é adicionado éster de metila de ácido 3-(1 -tritil-1 H-imidazol- 4-il)propanóico (6,76 g, 17,1 mmols) em porção. Então a mistura resultante é agitada à temperatura ambiente durante a noite. A reação é extinguida com água, 15% hidróxido de sódio, e água, então diluída com cloreto de metilano e filtrado. O precipitado no filtro é lavado com cloreto de metilano. O filtrado é evaporado até a secura para fornecer o composto bruto.
[00259] A uma solução do composto bruto acima (7,46 g, 20,3 mmols) em DMF (60 mL) à temperatura ambiente é adicionado imidazol (2,07 g, 30,4 mmols), cloreto de ferc-butildimetilsilila (3,5 g, 23,2 mmols) e seguido por DMAP (70 mg). A mistura é agitada à temperatura ambiente durante a noite. A mistura é dividida entre EtOAc e salmoura. A camada orgânica é lavada com salmoura, secado sobre Na2SO4 e concentrado para fornecer o composto desejado. J. ÉSTER DE ETILA DE ÁCIDO 3-(1-TRITIL-1H-IMIDAZOL-4- IDBUTÍRICO (CASN0 698367-52-1)
[00260] O éster título é preparado de acordo com a estratégia descrita em Bioorg. Med. Chem. 2004, 12(9), 2273. A uma suspensão de NaH (dispersão a 60% em óleo mineral, 1,7 g, 42,5 mmols) em THF (10 mL) à temperatura ambiente é adicionado em gotas trietilfosfonoacetato (8,53 mL, 42,6 mmols). A esta mistura é lentamente adicionada a solução de 1 -(1-tritil-1 H-imidzaol-4-il)etanona (10 g, 28,4 mmols) em THF (100 mL). A mistura resultante é aquecida em refluxo por 3 h. A mistura de reação é despejado sobre gelo e extraído com EtOAc. A camada orgânica é lavada em salmoura, secada sobre Na2SO4 anidroso e concentrado para fornecer um sólido bruto.
[00261] A uma solução desgaseificada de éster de etila de ácido 3- (1-tritil-1H-imidazol-4-il)but-2-enóico (5 g, 11,8 mmols) em etanol (100 mL) em um frasco Parr é adicionado paládio em carbono a 5% (0,5 g). O frasco é purgado com nitrogênio, evacuado, e gás hidrogênio (103,42 Kpa (15 psi)) adicionado. O frasco é colocado sobre uma aparato de hidrogenação Parr e agitado por 18 h. O hidrogênio é evacuado e o frasco purgado com gás nitrogênio . A mistura de reação é em seguida filtrada por meio de terra diatomácea e a solução líquida clara coletada e o solvente removido em vácuo para fornecer o óleo bruto, o qual é submetido à cromatografia instantânea (sílica-gel) eluindo com MeOH:CH2Cl2 para produzir o composto desejado. K. ÉSTER DE METILA DE ÁCIDO 2,2-DIMETIL-2-(1-TRITIL-1H- IMIDAZOL-4-ID-PROPIÔNICO
[00262] A uma solução de éster de metila de ácido (1 -tritil-1 H- imidazol-4-il)acético (10 g, 26,2 mmols) em THF (150 mL) a 0 °C é adicionado NaH em pó (dispersão a 60% em óleo mineral, 3,15 g, 78,8 mmols). A suspensão é agitada a 0 °C por 0,5 h, em seguida é adicionado CH3I (4 mL, 64,1 mmols). A mistura resultante é aquecida até à temperatura ambiente e agitada durante a noite. A uma mistura de suspensão é adicionado Florisil (2,5 g), e 0 sólido é removido por filtragem por meio de uma almofada de celito. O filtrado é concentrado e 0 resíduo é dividido entre EtOAc e salmoura, e a camada orgânica é lavada em salmoura, secado em Na2SO4 anidroso e concentrado para fornecer 0 óleo bruto, 0 qual é submetido à cromatografia instantânea (sílica-gel) eluindo com MeOH:CH2Cl2 para produzir 0 composto desejado. L. 2,2-DIMETIL-2-(1-TRITIL-1H-IMIDAZOL-4-IL)PROPIONALDEÍDO (CASN0 64464-49-9)
[00263] A uma solução de éster de metila de ácido 2,2-dimetil-2-(1 - tritil-1 H-imidazol-4-il)propiônico (4,2 g, 10,2 mmols) em THF (40 mL) a 0 °C é adicionado LAH (600mg, 15,8 mmols). A suspensão resultante é agitada a 0 °C por 2 h. A reação é finalizada com água, 15% hidróxido de sódio, e água, em seguida diluída com cloreto de metilano e filtrada. O precipitado no filtro é lavado com cloreto de metilano. O filtrado é evaporado até secura para fornecer o composto bruto.
[00264] A uma solução do composto bruto acima (3,83 g, 10,0 mmols) em CH2CI2 (50 mL) à temperatura ambiente é adicionado periodinano de Dess-Martin em porção. A solução clara resultante é agitada à temperatura ambiente for 2 h. A reação é finalizada com 1 N Na2S20s aquoso, saturado aquoso NaHCOs aquoso saturado, e extraída com CH2CI2. A camada orgânica é lavada em salmoura, secada em Na2SO4 anidroso e concentrada para fornecer 0 óleo bruto, 0 qual é submetido a cromatografia instantânea (sílica-gel) eluindo com MeOH:CH2Cl2 para produzir 0 composto desejado, (preparado por um método alternativo em Bioorg. Med. Chem. 2004, 12(9), 2251.) M. 3,3-DIMETIL-3-(1-TRITIL-1H-IMIDAZOL-4-IL)BUTAN-1-OL
[00265] A uma suspensão de cloreto de metoximetila de trifenilfosfônio (11,0 g, 32,1 mmols) em THF(15 mL) à temperatura ambiente é adicionado f-BuOK/THF (1,0 M, 30 mL, 30 mmols). A mistura resultante é agitada à temperatura ambiente cerca de 10 min em seguida é adicionada a solução de 2,2-dimetil-2-(1 -tritil-1 H- imidazol-4-il)propionaldeído (3,6 g, 9,5 mmols) em THF (70 mL). A mistura é agitada à temperatura ambiente durante a noite. A reação é finalizada por NH4CI saturado, e a mistura é dividida entre EtOAc e salmoura. A camada orgânica é lavada em salmoura, secada e concentrada para fornecer um óleo, 0 qual é submetido a cromatografia instantânea (sílica-gel) eluindo com MeOH:CH2Cl2 para produzir o composto desejado.
[00266] Ao composto acima (1,04g, 2,55 mmols) em 10% de H2O- THF (22 mL) à temperatura ambiente é adicionado resina de TsOH. A mistura é agitada à temperatura ambiente por 2 h. A mistura de reação é filtrada da resina e lavada com CH2CI2. A camada orgânica é neutralizada e lavada com salmoura, secada e concentrado para fornecer 0 composto bruto.
[00267] Ao composto bruto acima em THF (10 mL) a 0 °C é adicionado LAH (150 mg, 3,95 mmols) e a mistura é agitada a 0 °C por 30 min. A reação é extinguida com água, hidróxido de sódio a 15%, e água, em seguida diluída com cloreto de metilano e filtrada. O precipitado no filtro é lavado com cloreto de metilano. O filtrado é evaporado até a secura para fornecer um óleo, 0 qual é submetido à cromatografia instantânea (sílica-gel) eluindo com MeOH:CH2Cl2 para produzir 0 composto desejado. N. 4-[3-(TERC-BUTILDIMETILSILANILÓXI)-1,1-DIMETIL-PROPIL]-1- TRITIL-1H-IMIDAZOL
[00268] A uma solução de 3,3-dimetil-3-(1 -tritil-1 H-imidazol-4- il)butan-1-ol (0,87 g, 2,2 mmols) em CH2CI2 (10 mL) à temperatura ambiente é adicionado imidazol (200 mg, 2,94 mmols), cloreto de terc- butildimetilsilila (350 mg, 2,32 mmols). A mistura é agitada à temperatura ambiente durante a noite. A mistura é dividida entre EtOAc e salmoura. A camada orgânica é lavada com salmoura, secada em Na2SO4 e concentrada para fornecer um óleo, 0 qual é submetido à cromatografia instantânea (sílica-gel) eluindo com MeOH:CH2Cl2 para produzir 0 composto desejado. O. (E E Z)-4-[4-TE/?C-BUTIL-DIMETIL-SILANIL0XI)-BUT-1-ENIL]-1- TRITIL-1H-IMIDAZOL
[00269] a 3-(terc-butildimetilsilanilóxi)propil-1-brometo é convertido em sal de trifenila fosfônio de acordo com literatura precedente (Tetrahedron Letters 1997, 38(20), 3647-3650). Ao brometo (25 g, 95 mmols) em tolueno (200 mL) é adicionado trifenilfosfina (40 g, 158 mmols). A mistura de reação é agitada a 105 °C por 18 h. A mistura é em seguida deixada esfriar para a temperatura ambiente pelo curso de uma hora. O sólido branco é filtrado, lavado com hexano (50 mL), em seguida lavado com acetato de etila, e secado em vácuo por 24 h.
[00270] A brometo de [3-(terc- butildimetilsilanilóxi)propil]trifenilfosfônio (35,5 g, 68,9 mmols) é adicionado THF (300 mL) anidroso por meio de cânula. Esta suspensão é resfriada para -78 °C e n-butillítio em hexanos (2,5 M, 30 mL, 75 mmols) é adicionado por meio de seringa. A mistura é deixada em agitação por 20 minutos a -78 °C antes de uma solução de 1 -tritil-4- carboxaldeído-1H-imidazol (20,0 g, 59,1 mmols) em THF (300 mL) ser adicionado por meio de cânula. A mistura é deixada aquecer para temperatura ambiente durante 30 minutos, em seguida agitada por mais 3,5 h à temperatura ambiente. A reação é extinguida por adição de metanol (20 mL) seguida por cloreto de amónio saturado aquoso. A mistura de reação é então dividida entre acetato de etila e bicarbonato de sódio aquoso saturado. A camada orgânica é concentrada a vácuo para fornecer o produto bruto. Purificação por cromatografia (sílica-gel, acetato de etila:hexanos 0:1 a 3:2) fornece o produto como um sólido branco, uma mistura de isômeros cis e trans. MS (ESI) m/z 495 (M + H) P. (E E Z)-4-[4-7ERC-BUTILDIMETILSILANIL0XI)-1-METIL-BUT-1- ENIL1-1-TRITIL-1H-IMIDAZOL

[00271] O composto do título é preparado a partir do exemplo 2C de uma maneira similar a preparação O acima. MS (ESI) m/z 509 (M + H) Q. 4-[4-fTERC-BUTILDIMETILSILANIL0XI)BUTIL]-1-TRITIL-1H- IMIDAZOL
[00272] A uma mistura de E- e Z-4-[4-íerc-butildimetilsilanilóxi)but-1 - enil]-1-tritil-1H-imidazol (7,4 g, 14,9 mmols) em etanol desgaseificado em um frasco Parr é adicionado 5% de paládio sobre carbono (0,1 g). O frasco é purgado com nitrogênio, evacuado, e adicionado gás hidrogênio ((206,84 (30 psi)). O frasco é colocado em um aparato de hidrogenação Parr e agitado por 18 horas. O hidrogênio é evacuado e o frasco purgado com gás nitrogênio. A mistura de reação é então filtrada por meio de terra diatomácea e a solução líquida clara coletada e o solvente removido a vácuo para fornecer o produto como um sólido branco. MS (ESI) m/z 497 (M + H) R. 4-[4-f7ERC-BUTILDIMETILSILANILÓXI)-1-METILBUTIL]-1-TRITIL- 1H-IMIDAZOL
[00273] O composto do título é preparado a partir de uma mistura de E- e Z-4-[4-ferc-butildimetilsilanilóxi)-1 -metilbut-1 -enil]-1 -tritil-1 H- imidazol em uma maneira similar a preparação Q acima. MS (ESI) m/z 511 (M + H) EXEMPLO 3 A.4-{5-[2-7~E/?C-BUTILDIMETILSILANILÓXI)ETIL]IMIDAZOL-1- ILMETILI-3-CLOROBENZONITRILA
[00274] 4-[2-fferc-Butildimetilsilanilóxi)etil]-1 -tritil-1 H-imidazol (3,98 g, 8,5 mmols) e 4-bromometil-3-clorobenzonitrila (2,93 g, 12,7 mmols) são dissolvidos em MeCN (40 mL) e aquecido a 80 °C por 5 h. Após resfriamento para temperatura ambiente MeOH (40 mL) e EÍ2NH (7 mL) são em seguida adicionados e a solução é aquecida a 70 °C por 1 h. A solução é evaporada a secura e o resíduo purificado por meio de cromatografia de coluna instantânea (acetona/C^CL 1:3 MeOH/ CH2CI2 5:95) para fornecer 4-{5-[2-terc-butil-dimetilsilanilóxi)etil]- imidazol-1-ilmetil}-3-clorobenzonitrila como um óleo. MS (ESI) m/z 376,3, 378,3 (M + H). B. ÉSTER DE METILA DE ÁCIDO {5-Í2-7ERC-BUTILDIMETILSILA- NILÓXI)ETILllMIDAZOL-1-IL}-(2-CLORO-4-CIANOFENIL) ACÉTICO
[00275] 4-{5-[2-ferc-Butildimetilsilanilóxi)etil]imidazol-1-ilmetil}-3- clorobenzonitrila (1,7 g, 4,52 mmols) é dissolvido em THF (30 mL) anidroso e agitado a -78 °C antes que uma solução THF de LHMDS (8,1 mL, 1,0 M) seja adicionada. Após 15 min, cianoformiato de metila (0,38 mL, 4,74 mmols) é adicionado e a solução é deixada a -78 DC por 2 h. O excesso de LHMDS é extingüido com NH4CI saturado aquoso e a mistura é deixada aquecer para temperatura ambiente. A mistura é então diluída com EtOAc e lavada com NH4CI (2X) saturado aquoso. O orgânico é secado (Na2SO4) e evaporado. O resíduo bruto é purificado por meio de cromatografia de coluna instantânea (EtOAc/hexanos 1:1 -> EtOAc) para fornecer éster de metila de ácido {5-[2-ferc-butildimetilsilanilóxi)etil]-imidazol-1 -il}-(2-cloro-4- cianofenil)acético como um óleo. MS (ESI) m/z 434,3, 436,3 (M + H). C. ÉSTER DE METILA DE ÁCIDO 5-(2-CLORO-4-CIANOFENIL)-6,7- DIIDRO-5H-PIRROLO[1,2-C]IMIDAZOL-5-CARBOXÍLICO
[00276] Éster de metila de ácido {5-[2-íerc-Butildimetilsilanilóxi)etil]- imidazol-1-il}-(2-cloro-4-cianofenil)-acético (2,8 g, 6,46 mmols) em THF (25 mL) é resfriada a 0 °C antes que uma solução de HCI em 1,4- Dioxano (10 mL, 4,0 M, 40 mmols) seja adicionada. Após a conclusão da reação como julgado por LCMS, a solução é dividida entre EtOAc e NaHCOs saturado aquoso. A camada orgânica é secada (Na2SO4) e evaporada para fornecer 0 álcool bruto, Éster de metila de ácido (2- cloro-4-cianofenil)-[5-(2-hidroxietil)imidazol-1-il]-acético que é usado sem outra purificação. MS (ESI) m/z 320,1,322,1 (M + H).
[00277] O éster de metila de ácido (2-cloro-4-cianofenil)-[5-(2- hidroxietil)imidazol-1 -il]acético bruto (2,06 g, 6,46 mmols) é dissolvido em CH2CI2 (25 mL) e agitado a 0 °C antes que EtsN (1,4 mL, 9,69 mmols) e cloreto de metanossulfonila (0,6 mL, 7,75 mmols) sejam adicionado. Após a conclusão da reação, a solução é dividida entre CH2CI2 e NaHCOs saturado aquoso. A camada orgânica é secada (Na2SO4) e evaporada para fornecer éster de metila de ácido (2-cloro- 4-cianofenil)-[5-(2-metanosul-foniloxietil)imidazol-1 -il]-acético bruto que é usado sem outra purificação. MS (ESI) m/z 398,2, 400,2 (M + H).
[00278] O éster de metila de ácido (2-cloro-4-cianofenil)-[5-(2- metanosulfoniloxietil)imidazol-1-il]-acético bruto (2,56 g, 6,45 mmols) é dissolvido em DMF (50 mL) seco e a isso é adicionado K2COa(2,67 g, 19,4 mmols), Nal (2,9 g, 19,4 mmols) e EtsN (2,7 mL, 19,4 mmols), a reação é agitada a 80 °C por 2 h antes de ser concentratada até a secura. O resíduo é então diluído com EtOAc e lavado com água. A camada orgânica é secada (Na2SO4) e evaporada para fornecer um resíduo bruto que é purificado por meio de coluna de cromatografia instantânea (Acetona/ CH2CI2 1:3) para fornecer éster de metila de ácido 5-(2-cloro-4-cianofenil)-6,7-diidro-5H-pirrolo[1,2-c]imidazol-5- carboxílico como um óleo. MS (ESI) m/z 302,2, 304,2 (M + H). 1H RMN (400 MHz, CDCI3) δ ppm 2,64-2,76 (m, 2 H), 2,97-3,06 (m, 1 H), 3,84 (s, 3 H), 3,86-3,93 (m, 1 H), 6,56 (d, J = 8,1 Hz, 1 H), 6,87 (s, 1 H), 7,50 (obs d, J = 8,1 Hz, 1 H), 7,52 (s, 1H), 7,73 (s, 1 H). D. ÁCIDO 5-(2-CLORO-4-CIANOFENIL)-6,7-DIIDRO-5H-PIRROLO[1,2- C1I-MIDAZOL-5-CARBOXÍLICO

[00279] Éster de metila de ácido 5-(2-cloro-4-cianofenil)-6,7-diidro- 5H-pirrolo[1,2-c]imidazol-5-carboxílico (0,6 g, 2,0 mmols) é dissolvido em THF/água 3:2 (20 mL) e a isso é adicionado LiOH (0,17 g, 4,0 mmols). A mistura é agitada à temperatura ambiente por 2 h antes de ser neutralizada a pH 6 com 1M HCI. A solução é evaporada até a secura para fornecer ácido, ácido 5-(2-cloro-4-cianofenil)-6,7-diidro-5H- pirrolo[1,2-c]imidazol-5-carboxíli-co como um sólido. MS (ESI) m/z 288,2, 290,2 (M + H); 1H RMN (400 MHz, MeOD) (sal de amónio) δ ppm 2,64-2,74 (m, 1 H), 2,77-2,86 (m, 1 H), 2,94-3,02 (m, 1 H), 3,74 (ddd, J= 13,1, 9,1, 8,0 Hz, 1 H), 6,74 (d, J = 8,1 Hz, 1 H), 6,85 (s, 1 H), 7,59 (dd, J = 8,1, 1,5 Hz, 1 H), 7,84 (d, J = 1,8 Hz, 1 H), 7,91 (s, 1 H). E. 3-CLQRO-4-(6,7-DIIDRO-5H-PIRRQLQ[1,2-C]IMIDAZOL-5-IL)BENZONI- TRILA
[00280] O ácido 5-(2-cloro-4-cianofenil)-6,7-diidro-5H-pirrolo[1,2- c]imidazol-5-carboxílico (0,02g, 70 pmols) é dissolvido em DMSO (2 mL) e EtsN (0,2 mL) e aquecido a 100 °C por 2 h. A solução é evaporada até a secura e o resíduo purificado por meio de HPLC de fase reversa (5-100% MeCN/água w/ 0,1% TFA) para fornecer 3-cloro- 4-(6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-il)-benzonitrila como um sólido branco. MS (ESI) m/z 244,2, 246,2 (M + H); 1H RMN (400 MHz, CDCh) (sal de TFA) δ ppm 2,60-2,73 (m, 1 H), 3,08-3,20 (m, 2 H), 3,22-3,36 (m, 1 H), 6,04 (dd, J = 7,6, 5,8 Hz, 1 H), 6,91 (d, J = 8,1 Hz, 1 H), 7,24 (s, 1 H), 7,61 (d, J = 8,1 Hz, 1 H), 7,81 (d, J = 1,5 Hz, 1 H), 8,53 (s, 1 H).
[00281] Similarmente preparados são os seguintes compostos <Tabela 2): Tabela 2. Compostos de Fórmula (II)
[00282] Resolução de enantiômeros do Composto do título é obtida por HPLC quiral usando coluna ChiralPak IA com uma fase móvel de EtOAc:hexano a 70% para fornecer enantiômero A (tr = 22,4 min) e enantiômero B (tr = 41,9 min).
2) Éster de metila de ácido (R) e (Sj-5-(2-Cloro-4-cianofenil)-6,7- diidro-5H-pirrolo[1,2-c]imidazol-5-carboxilico
[00283] Resolução de enantiômeros do Composto do título é obtida por HPLC quiral usando coluna ChiralPak AS com uma fase móvel de IPA:hexano a 15% para fornecer enantiômero A (tr = 51,8 min) e enantiômero B (tr = 63,2 min).
3) (R) e (S)- 4-(6,7-Diidro-5H-pirrolo[1,2-c]imidazol-5-il)-3- metoxibenzonitrila
[00284] Resolução de enantiômeros do Composto do título é obtida por HPLC quiral usando coluna ChiralPak AS-H com uma fase móvel de EtOH:MeCN a 1% para fornecer enantiômero A (tr = 16,7 min) e enantiômero B (tr = 25,7 min).
4) Éster de metila de ácido (R) e (S)-5-(4-Ciano-2-metoxifenil)-6,7- diidro-5H-pirrolo[1,2-c]imidazol-5-carboxilico
[00285] Resolução de enantiômeros do Composto do título é obtida por HPLC quiral usando coluna ChiralPak AD com uma fase móvel de IPA:hexano a 30% para fornecer enantiômero A (tr = 31,6 min) e enantiômero B (tr = 41,7 min).
5) (R) e (S)-4-(6,7-Diidro-5H-pirrolo[1,2-c]imidazol-5-il)-3- flúorbenzonitrila
[00286] Resolução de enantiômeros do Composto do título é obtida por HPLC quiral usando coluna ChiralPak AS-H com uma fase móvel de MeCN para fornecer enantiômero A (tr = 16,7 min) e enantiômero B (tr = 22,5 min).
6) Éster de metila de ácido (R) e (S)-5-(4-Ciano-2-flúorfenil)-6,7- diidro-5H-pirrolo[1,2-c]imidazol-5-carboxílico
[00287] Resolução de enantiômeros do Composto do título é obtida por HPLC quiral usando coluna ChiralPak AS com uma fase móvel de IPA:hexano a 20% para fornecer enantiômero A (tr = 61,4 min) e enantiômero B (tr = 73,8 min).
7) (R) e (S)-3-Bromo-4-(6,7-diidro-5H-pirrolo[1,2-c]imidazol-5- il)benzonitrila
[00288] Resolução de enantiômeros do Composto do título é obtida por HPLC quiral usando coluna ChiralPak AS-H com uma fase móvel de IPA:hexano a 25% para fornecer enantiômero A (tr = 44,0 min) e enantiômero B (tr = 66,0 min).
8) (R) e (S)-4-(6,7-Diidro-5H-pirrolo[1,2-c]imidazol-5-il)-3- triflúormetoxibenzonitrila
[00289] Resolução de enantiômeros do Composto do título é obtida por HPLC quiral usando coluna ChiralPak AS-H com uma fase móvel de IPA:heptano a 10% para fornecer enantiômero A (tr = 53,4 min) e enantiômero B (tr = 59,4 min). EXEMPLO 4 A. 4-(6,7-DIIDRO-5H-PIRROLO[1,2-C]IMIDAZOL-5-IL)-3- METILBENZO-NI- TRILA
[00290] A uma solução de 3-bromo-4-(6,7-diidro-5H-pirrolo[1,2- c]imidazol-5-il)benzonitrila (0,100 g, 0,347 mmols) fornecido no Exemplo 3 (tabela 2) e trimetilboroxina (0,145 g, 1,04 mmols) em DME (3 mL), solução aquosa de Na2COs (0,69 mL, 2 M) e KOH (0,17 mL, 2 M) são adicionados. Depois de desgaseificado com nitrogênio, Pd(PPhs)4 (0,040 g, 0,035 mmols) é adicionado. A mistura é aquecida em um reator de microondas a 130 °C 1,5 h. Naquele ponto LCMS mostrou consumo do material de partida. A solução é diluída com acetato de etila e bicarbonato de sódio saturado. A camada aquosa resultante é extraída novamente com acetato de etila (3X). As camadas orgânicas combinadas são lavadas com salmoura e secadas em sulfato de sódio anidroso. Após a concentração o produto bruto é filtrado através de um filtro de 0,45pm e então purificada por HPLC preparativa (0% por 5 minutos e 0 - 34% acetonitrila com 0,1% TFA em 17 minutos). MS (ESI) m/z 224,0 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 2,33-2,42 (m, 1 H), 2,43 (s, 3 H), 2,84-2,95 (m, 2 H), 3,07-3,18 (m, 1 H), 5,54 (dd, J = 8,1,4,8 Hz, 1 H), 6,65 (d, J = 8,1 Hz, 1 H), 6,83 (s, 1 H), 7,32 (s, 1 H), 7,41 (d, J = 8,1 Hz, 1 H), 7,50 (s, 1 H). Resolução dos enantiômeros (R) e (S) do composto do título é obtida por HPLC quiral usando coluna ChiralPak AS-H e 9:1 hexano/EtOH para fornecer enantiômero A (tr = 84 min) e enantiômero B (tr = 104 min).
[00291] Similarmente preparados são os seguintes: 1) 4-(6,7-Diidro-5H-pirrolo[1,2-c]imidazol-5-il)-3- vinilbenzonitrila. MS (ESI) m/z 236,2 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 2,36-2,51 (m, 1 H), 2,83-2,96 (m, 2 H), 3,05-3,18 (m, 1 H), 5,57 (d, J = 11,6 Hz, 1 H), 5,63 (dd, J = 8,1,4,5 Hz, 1 H), 5,77 (d, J = 17,2 Hz, 1 H), 6,69 (d, J = 8,1 Hz, 1 H), 6,85 (s, 1 H), 6,90 (dd, J = 17,2, 11,1 Hz, 1 H), 7,34 (s, 1 H), 7,48 (dd, J = 8,1, 1,5 Hz, 1 H), 7,78 (d, J = 1,5 Hz, 1 H). Resolução de enantiômeros (R) e (S) do composto do título é obtida por HPLC quiral usando coluna ChiralPak AS-H com uma fase móvel de 4:1 Heptano/IPA para fornecer enantiômero A (tr = 32,3 min) e enantiômero B (tr = 58,2 min).
2) 4-(6,7-Diidro-5H-pirrolo[1,2-c]imidazol-5-il)-3-((E)- propenil) benzonitrila.
[00292] MS (ESI) m/z 250,1 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,97 (d, J = 6,6 Hz, 3 H), 2,37-2,49 (m, 1 H), 2,82-2,95 (m, 2 H), 3,04-3,18 (m, 1 H), 5,62 (dd, J = 8,0, 4,7 Hz, 1 H), 6,16-6,30 (m, 1 H), 6,53 (d, J = 15,4 Hz, 1 H), 6,66 (d, J = 8,1 Hz, 1 H), 6,84 (s, 1 H), 7,32 (s, 1 H), 7,42 (d, J = 9,3 Hz, 1 H), 7,71 (s, 1 H).
[00293] A partir de 2-Bromo-4-(6,7-diidro-5H-pirrolo[1,2-c]imidazol- 5-il)benzonitrila fornecida no exemplo 3 (tabela 2) os seguintes são preparados por analogia ao exemplo 4A: 1) 4-(6,7-Diidro-5H-pirrolo[1,2-c]imidazol-5-il)-2- vinilbenzonitrila MS (ESI) m/z 236,1 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 2,43-2,56 (m, 1 H), 2,85-3,01 (m, 2 H), 3,05-3,17 (m, 1 H), 5,35 (dd, J = 7,8, 5,6 Hz, 1 H), 5,57 (d, J = 11,1 Hz, 1 H), 5,90 (d, J = 17,4 Hz, 1 H), 6,84 (s, 1 H), 7,01-7,10 (m, 2 H), 7,31 (s, 1 H), 7,35 (d, J = 1,3 Hz, 1 H), 7,63 (d, J = 8,1 Hz, 1 H).
2) 4-(6,7-Diidro-5H-pirrolo[1,2-c]imidazol-5-il)-2-metilbenzonitrila
[00294] MS (ESI) m/z 224,1 (M + H); 1H RMN (400 MHz, CD3CN) (sal de TFA) δ ppm 2,51 (s, 3 H), 2,52-2,61 (m, 1 H), 2,94-3,20 (m, 3 H), 5,65 (app t, J = 6,9 Hz, 1 H), 7,19 (s, 1 H), 7,18-7,23 (obs m, 1 H), 7,26 (s, 1 H), 7,70 (d, J = 7,8 Hz, 1 H), 8,23 (s, 1 H).
B. 6-(6,7-DIIDRO-5H-PIRROLO[1 ^-CHMIDAZOL-δ-ILM'-FLLJOR- BIFENIL-3-CARBONITRILA
[00295] A uma solução c]imidazol-5-il)benzonitrila (0,100 g, 0,347 mmols) e ácido 4- flúorfenilborônico (0,158 g, 1,04 mmols) em DME (2 mL), solução aquosa de Na2COs (0,69 mL, 2 M) e KOH (0,17 mL, 2 M) são adicionados. Após desgaseificado com nitrogênio, Pd(PPh3)4 (0,040 g, 0,035 mmols) é adicionado. A mistura é aquecida em um reator de microondas a 130 °C, for 20 minutos. LCMS mostrou consumo de material de partida. A solução é então diluída com acetato de etila e bicarbonato de sódio saturado aquoso. A camada resultante aquosa é extraída novamente com acetato de etila (3X). As camadas orgânicas combinadas são lavadas com salmoura e secadas em sulfato de sódio anidroso. Após concentração, o produto bruto é purificado por cromatografia instantânea (Hexano/EtOAc, e em seguida 10% de MeOH/DCM) seguida por HPLC preparativa (0 - 50% acetonitrila com 0,1% TFA em 21 minutos). Após concentração e baseamento livre, 6- (6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-il)-4’-flúorbifenil-3-carbonitrila é obtido como um pó branco. MS (ESI) m/z 304,0 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 2,38-2,54 (m, 1 H), 2,76-3,03 (m, 3 H), 5,33 (app t, J = 6,7 Hz, 1 H), 6,79 (s, 1 H), 7,01 (d, J = 8,1 Hz, 1 H), 7,15 - 7,24 (m, 3 H), 7,29 (dd, J = 13,4, 5,1 Hz, 2 H), 7,54 - 7,65 (m, 2 H).
[00296] Similarmente preparados são os seguintes: 1) 6-(6,7-Diidro-5H-pirrolo[1,2-c]imidazol-5-il)-4'- metoxibifenil-3-carbonitrila MS (ESI) m/z 316,1 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 2,60 - 2,73 (m, 1 H), 2,96-3,10 (m, 2 H), 3,12 - 3,24 (m, 1 H), 3,88 (s, 3 H), 5,63 (app t, J = 7,5 Hz, 1 H), 7,03 (d, J = 8,6 Hz, 2 H), 7,06 (d, J = 8,6 Hz, 1 H), 7,14 (s, 1 H), 7,21 (d, J = 8,6 Hz, 2 H), 7,65 - 7,70 (m, 1 H), 7,66 (s, 1 H), 8,32 (br s, 1 H).
2) éster de ferc-butila de ácido 2-[5-Ciano-2-(6,7-diidro-5H- pirrolo[1,2-c]imidazol-5-il)fenil]-5-flúorindol-1 -carboxílico. MS (ESI) m/z 443,2 (M + H)
3) 4-(6,7-Diidro-5H-pirrolo[ 1,2-c]imidazol-5-il)-3-(5-flúor-1H- indol-2-il)benzonitrila. MS (ESI) m/z 343,1 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 2,50-2,61 (m, 1 H), 2,86-3,04 (m, 2 H), 3,06-3,17 (m, 1 H), 5,77 (dd, J = 8,0, 5,4 Hz, 1 H), 6,56 (d, J = 1,5 Hz, 1 H), 6,81 (s, 1 H), 6,94 (d, J = 8,3 Hz, 1 H), 7,01 (app dt, J = 9,1,2,5 Hz, 1 H), 7,09 (s, 1 H), 7,30 (dd, J = 9,3, 2,3 Hz, 1 H), 7,38 (dd, J = 8,8, 4,3 Hz, 1 H), 7,57 (dd, J = 8,2, 1,6 Hz, 1 H), 7,85 (d, J = 1,5 Hz, 1 H), 9,93 (br s, 1 H).
[00297] A partir de 2-Bromo-4-(6,7-diidro-5H-pirrolo[1,2-c]imidazol- 5-il)benzonitrila fornecida no exemplo 3 (tabela 2) o seguinte é preparado por analogia ao exemplo 4B:
[00298] 5-(6,7-Diidro-5H-pirrolo[1,2-c]imidazol-5-il)-4'-flúorbifenil-2- carbonitrila. MS (ESI) m/z 304,1 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 2,48-2,62 (m, 1 H), 2,89-3,05 (m, 2 H), 3,08-3,20 (m, 1 H), 5,42 (dd, J = 7,7, 5,9 Hz, 1 H), 6,95 (s, 1 H), 7,04 (app t, J = 9,0 Hz, 2 H), 7,13-7,21 (m, 3 H), 7,46 (dd, J = 8,7, 5,2 Hz, 2 H), 7,54 (s, 1 H).
C. 4-(6,7-DIIDRO-5H-PIRROLO[1,2-C]IMIDAZOL-5-IL)-3-(/V- PROPIQBEM- ZONITRILA
[00299] A suspensão de 4-(6,7-Diidro-5H-pirrolo[1,2-c]imidazol-5-il)- 3-((E)-propenil)benzonitrila (0,150 g, 0,602 mmols), 20% (peso/peso) de paládio sobre carbono (0,040 g), THF (15 mL), e EtOH (15 mL) é agitada sob uma atmosfera de hidrogênio (1 atm) por 60 h. A suspensão é filtrada e o filtrado concentrado. O resíduo é então purificado por cromatografia instantânea (Hexano/EtOAc, e em seguida 10% de MeOH/EtOAc) para fornecer 4-(6,7-Diidro-5H-pirrolo[1,2- c]imidazol-5-il)-3-(n-propil)benzonitrila como um sólido branco. MS (ESI) m/z 252,1 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,04 (t, J = 7,3 Hz, 3 H), 1,63-1,77 (m, 2 H), 2,33-2,45 (m, 1 H), 2,62-2,80 (m, 2 H), 2,92 (app t, J = 7,1 Hz, 2 H), 3,04-3,19 (m, 1 H), 5,58 (dd, J = 7,8, 5,6 Hz, 1 H), 6,73 (d, J = 8,1 Hz, 1 H), 6,81 (s, 1 H), 7,25 (s, 1 H), 7,41 (d, J = 8,1 Hz, 1 H), 7,51 (s, 1 H).
[00300] Similarmente preparados são os seguintes: 1) 4-(6,7-Diidro-5H-pirrolo[1,2-c]imidazol-5-il)-3- etilbenzonitrila. MS (ESI) m/z 238,1 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,34 (t, J = 7,6 Hz, 3 H), 2,34-2,47 (m, 1 H), 2,68-2,88 (m, 2 H), 2,89-2,97 (m, 2 H), 3,07-3,19 (m, 1 H), 5,59 (dd, J = 7,8, 5,1 Hz, 1 H), 6,72 (d, J = 8,1 Hz, 1 H), 6,84 (s, 1 H), 7,28 (s, 1 H), 7,43 (dd, J = 8,1, 1,8 Hz, 1 H), 7,55 (d, J = 1,3 Hz, 1 H). Resolução de enantiômeros (R) e (SJ do composto do título é obtido por HPLC quiral usando coluna ChiralPak AS-H e 4:1 heptano//-PrOH para fornecer enantiômero A (tr = 28,1 min) e enantiômero B (tr = 43,1 min).
2) 4-(6,7-Diidro-5H-pirrolo[1,2-c]imidazol-5-il)-2- etilbenzonitrila. MS (ESI) m/z 238,1 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,28 (t, J = 7,5 Hz, 3 H), 2,37-2,55 (m, 1 H), 2,78-3,01 (m, 4 H), 3,02-3,18 (m, 1 H), 5,26-5,40 (m, 1 H), 6,82 (br s, 1 H), 6,99 (d, J = 7,1 Hz, 1 H), 7,05 (br s, 1 H), 7,30 (br s, 1 H), 7,60 (d, J = 7,8 Hz, 1 H).
D. 4-(6,7-DIIDRO-5H-PIRROLO[1,2-C]IMIDAZOL-5-IL)-3- ETOXIBENZONI- TRILA
[00301] A solução de 3-bromo-4-(6,7-diidro-5H-pirrolo[1,2- c]imidazol-5-il)benzonitrila (0,090 g, 0,312 mmols) fornecido no exemplo 3 (tabela 2), Pd2(dba)3 (0,029 g, 0,031 mmols), BINAP (0,039 g, 0,062 mmols), Cs2CO3 (0,204 g, 0,625 mmols), EtOH (0,091 mL, 1,54 mmols), e DME (4 mL) é aquecido em um reator de microondas a 135 °C por 1,5 h. Naquele ponto LCMS mostrou consumo do material de partida. A mistura é filtrada e em seguida o filtrado é concentrado. O resíduo é em seguida purificado por cromatografia instantânea (0-10% MeOH/DCM) e fornece 4-(6,7-Diidro-5H-pirrolo[1,2-c]imidazol-5-il)-3- etoxibenzonitrila. MS (ESI) m/z 254,1 (M + H); 1H RMN (400 MHz, MeOD) (sal de TFA) δ ppm 1,32 (t, J = 6,9 Hz, 3 H), 2,69-2,82 (m, 1 H), 3,07-3,22 (m, 3 H), 4,02-4,22 (m, 2 H), 5,92-6,03 (m, 1 H), 7,27 (d, J = 7,8 Hz, 1 H), 7,31 (s, 1 H), 7,36 (dd, J = 7,8, 1,5 Hz, 1 H), 7,43 (d, J = 1,3 Hz, 1 H), 8,68 (s, 1 H).
[00302] Similarmente preparados são os seguintes:
[00303] 3-Butóxi-4-(6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-il) benzonitrila. MS (ESI) m/z 282,1 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,01 (t, J = 7,3 Hz, 3 H), 1,46-1,59 (m, 2 H), 1,78-1,88 (m, 2 H), 2,38-2,49 (m, 1 H), 2,77-2,93 (m, 2 H), 3,02-3,15 (m, 1 H), 4,06 (app t, J = 6,3 Hz, 2 H), 5,67 (dd, J = 8,2, 4,2 Hz, 1 H), 6,66 (d, J = 7,8 Hz, 1 H), 6,82 (s, 1 H), 7,13 (d, J = 1,3 Hz, 1 H), 7,17 (dd, J = 7,8, 1,3 Hz, 1 H), 7,36 (s, 1 H).
[00304] A partir de 2-Bromo-4-(6,7-diidro-5H-pirrolo[1,2-c]imidazol- 5-il)benzonitrila fornecido no exemplo 3 (tabela 2) o seguinte é preparado por analogia ao exemplo 4D: 1) 4-(6,7-Diidro-5H-pirrolo[1,2-c]imidazol-5-il)-2- metoxibenzonitrila. MS (ESI) m/z 240,1 (M + H); 1H RMN (400 MHz, CD3CN) (sal de TFA) δ ppm 2,97-3,17 (m, 4 H), 3,91 (s, 3 H), 5,65 (app t, J = 7,2 Hz, 1 H), 6,90 (dd, J = 8,0, 1,4 Hz, 1 H), 7,01 (d, J = 1,3 Hz, 1 H), 7,19 (s, 1 H), 7,65 (d, J = 8,1 Hz, 1 H), 8,23 (s, 1 H).
2) 4-(6,7-Diidro-5H-pirrolo[ 1,2-c]imidazol-5-il)-2-pirrolidin-1 - il-benzonitrila. MS (ESI) m/z 279,1 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,97- 2,03 (m, 4 H), 2,44-2,58 (m, 1 H), 2,85-2,98 (m, 2 H), 2,99- 3,13 (m, 1 H), 3,53-3,60 (m, 4 H), 5,25 (dd, J = 7,7, 5,9 Hz, 1 H), 6,26 (d, J = 1,3 Hz, 1 H), 6,38 (dd, J = 8,1, 1,5 Hz, 1 H), 6,85 (s, 1 H), 7,42 (d, J = 8,1 Hz, 1 H), 7,44 (s, 1 H).
E. 3-FLÚOR-4-(5-METIL-6,7-DIIDRO-5H-PIRRQLO[1,2-CllMIDAZOL- 5-IL) BENZONITRILA
[00305] A solução de 4-(6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-il)-4- flúorbenzonitrila (0,050 g, 0,220 mmols) fornecido no exemplo 3 (tabela 2), 18-coroa-6 (0,006 g, 0,022 mmols), e THF (2 mL) é resfriada para - 78 °C. KHMDS (0,66 mL, 0,5 M) é então adicionado. Após 20 min Mel (0,07 mL, 1,10 mmols) é adicionado. Após 2,5 h a solução é diluída com NaHCOs saturado aquoso e DCM. A camada aquosa é ainda extraída com DCM (3 x 20 mL). As camadas orgânicas combinadas são secadas (Na2SO4), filtradas e concentradas. O resíduo é purificado por meio de cromatografia instantânea (50-100% EtOAc/hexanos) para fornecer 3-flúor-4-(5-metil-6,7-diidro-5H-pirrolo[1,2-c]imidazol-5- il)benzonitrila como um sólido branco. MS (ESI) m/z 242,0 (M + H); 1H RMN (400 MHz, CDCh) δ Qppm 2,01 (s, 3 H), 2,49-2,62 (m, 1 H), 2,73- 2,99 (m, 3 H), 6,35 (app t, J = 8,0 Hz, 1 H), 6,84 (s, 1 H), 7,31 (d, J = 8,1 Hz, 1 H), 7,40 (d, J = 10,9 Hz, 1 H), 7,55 (s, 1 H). F. 3-FLÚOR-4-(7-METILANO-6,7-DIIDRO-5H-PIRROLO[1,2-C] IMIDAZOL- 5-IDBENZONITRILA

[00306] 3-flúor-4-(5-iodo-imidazol-1-ilmetil)benzonitrila
[00307] Uma mistura de 4-iodo-1-tritil-1 H-imidazol (17,1 g, 39,2 mmols) e 4-bromometil-3-flúorbenzonitrila (9,65 g, 45,08 mmols) em 150 mL de acetonitrila seca é agitada à temperatura ambiente por 7 dias. Após concentração, o resíduo é misturado com metanol, e aquecido até o refluxo por 1,5 h. O solvente é subsequentemente removido e o resíduo é tratado com 1M HCI (300 mL). A suspensão resultante é filtrada e lavada com HCI (1M). A solução combinada é ajustada para PH 9-10 por solução de NaHCOs saturado. A precipitação resultante é coletada por filtração, secada em forno a vácuo. MS (ESI) m/z (M + H) 328,1.
[00308] 3-f I úor-4-[ 1 -(5-iodo-imidazol-1 -i l)-but-3-en i I] benzonitrila
[00309] LDA (1,5 M em tF, 20,85 mL, 31,7 mmols) é adicionado em gotas a uma suspensão de 3-flúor-4-(5-iodo-imidazol-1- ilmetil)benzonitrila (7,98 g, 24,4 mmols) em 150 mL de THF seco a -78 °C. Após 1 h a esta temperatura, brometo de alila (2,09 mL, 24,4 mmols) é adicionado lentamente, e a mistura resultante é agitada a -78 °C por 3 h. A reação é finalizada com solução de NH4CI saturado, e extraída com CH2CI2. Os extratos combinados são lavados com salmoura e secados em Na2SO4 anidroso. Após filtração e concentração, 0 resíduo é purificado por coluna instantânea. MS (ESI) m/z (M + H) 368,0.
[00310] Uma mistura de 3-flúor-4-[1-(5-iodo-imidazol-1-il)-but-3- enil]benzonitrila (2,14 g, 5,83 mmols), PS-PPhs-Pd (0,06 mmols) e EtsN (4,0 mL, 29,15 mmols) em 20 mL de DMF é aquecida a 150 °C em microondas por 1 h. Após filtração e evaporação, o resíduo é na maioria das vezes dissolvido por solução de HCI a 1 M. A mistura preta resultante é filtrada, e a solução é subsequentemente ajustado para PH 9-10 por solução de NaHCOs saturado. A mistura resultante é extraída com CH2CI2, e os extratos combinados são lavados com salmoura e secado em Na2SO4 anidroso. Após filtração e concentração, 0 resíduo é purificado por coluna instantânea. MS (ESI) m/z 240,3 (M + H); 1H RMN (400 MHz, CDCh) δ 7,37-7,33 (m, 2H), 7,32 (s, 1H), 7,18 (s, 1H), 6,82 (t, J = 8,0 Hz, 1H), 5,71 (dd, J = 12,0 Hz, 4,0 Hz, 1H), 5,35 (t, J = 4,0 Hz, 1H), 4,98 (t, J = 4,0 Hz, 1H), 3,83-3,76 (m, 1H), 3,05-2,99 (m, 1H). Resolução de enantiômeros do composto do título é obtida por HPLC quiral usando coluna ChiralPak IA com uma fase móvel de EtOH-hexanos (20%, v/v) para fornecer enantiômero A (tr = 21 min) e enantiômero B (tr = 25 min). G. 5'-[2-FLLJOR^-CIANO-FENILl-δ'.e'-DIIDROESPIRO [CICLOPROPANO-1,7'-PIRROLO[1,2-C]IMIDAZOL]
[00311] A solução de TFA (0,234 mL, 3 mmols) em 0,6 mL de CH2CI2 seco é adicionado em gotas a uma solução de Et2Zn (1M em Hexanos, 3,06 mL, 3,06 mmols) a 0 °C. A suspensão resultante é subsequentemente tratada com uma solução de CH2I2 (0,246 mL, 3,06 mmols) em CH2CI2 (0,4 mL). Após 2 h a 0 °C, a solução de 3-flúor-4-(7- metilano-6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-il)benzonitrila (333,8 mg, 1,392 mmols) em CH2CI2 é adicionado. Após uma noite, a reação é finalizada com solução de NaHCOs saturado, e extraída com CH2CI2 (20 mL x 4). Os extratos combinados são lavados com salmoura, secado em Na2SO4 anidroso. Após filtração e evaporação, 0 resíduo é purificado por cromatografia e produz 112 mg de óleo. MS (ESI) m/z 254,2 (M + H). 1H RMN (400,3 MHz, CDCh): δ 8,93 (s, 1H), 7,54-7,52 (m, 1H), 7,45 (d, J = 8,00 Hz, 1H), 7,35 (m, 1H), 6,83 (s, 1H), 6,27 (brs, 1H), 3,22 (m, 1H), 2,61 (d, J= 12 Hz, 1H), 1,33-1,19 (m, 2H), 1,16-1,14 (m, 1H), 0,90-0,85 (m, 1H). EXEMPLO 5 A. ÉSTER DE METILA DE ÁCIDO 5-(3-FLÚOR-4-METOXIFENIL)-6,7- DIIDRO-5H-PIRROLO[1,2-C]IMIDAZOL-5-CARBOXÍLICO
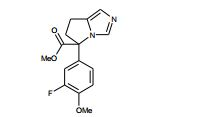
[00312] Éster de metila de ácido 4-[2-(ferc-Butil-dimetilsilanilóxi)etil]- 1-tritil-1H-imidazol (6,5 g, 14,0 mmols) e bromo-(3-flúor-4- metoxifenil)acético (5,8 g, 21,0 mmols) são agitadas em 60 mL MeCN à temperatura ambiente por 2 dias. Naquele ponto MeOH (50 mL) e EÍ2NH (50 mL) são então adicionados e a solução resultante aquecida a 75 °C por 0,5 h. A solução é evaporada e 0 resíduo purificado por meio cromatografia de coluna instantânea (EtOAc/DCM 1:9—>EtOAc/DCM 1:1) para fornecer éster de metila de ácido {5-[2- fferc-butildimetilsilanilóxi)etil]-imidazol-1 -il}-(3-flúor-4- metoxifenil)acético como um óleo. MS (ESI) m/z 423,3 (M + H).
[00313] Éster de metila de ácido {5-[2-(fe/'c-butildimetilsilanilóxi)etil]- imidazol-1-il}-(3-flúor-4-metoxifenil)-acético (3,1 g, 7,34 mmols) é dissolvido em THF (100 mL) e resfriado até 0 °C e então HCI em dioxano (11 mL, 4,0 M) é adicionado. Após 2 h a solução é evaporada até a secura e o álcool resultante, éster de metila de ácido (3-flúor-4- metoxifenil)-[5-(2-hidróxi-etil)-imidazol-1-il]acético, é usado sem outra purificação. MS (ESI) m/z 309,2 (M + H).
[00314] Éster de metila de ácido (3-flúor-4-metoxifenil)-[5-(2- hidroxietil)imidazol-1 -il]-acético (2,26 g, 7,34 mmols) é dissolvido em DCM (100 mL) e resfriado até 0 °C antes que EtsN (5,1 mL, 36,7 mmols) e cloreto de metanossulfonila (0,7 mL, 8,81 mmols) sejam adicionados. Após 1 h, a solução é transferida para um funil separador e dividida entre DCM e NaHCOs aquoso saturado. O orgânico é secado (Na2SÜ4) e evaporado para fornecer um resíduo bruto que é purificdo por meio cromatografia de coluna instantânea (DCM—>MeOH/DCM 5:95) para fornecer o mesilato intermediário puro como óleo amarelo. MS (ESI) m/z 387,2 (M + H).
[00315] O mesilato acima (1,8 g, 4,66 mmols) é dissolvido em THF seco (50 mL) e resfriado até -78 °C. A esta solução é adicionado LHMDS (5,6 mL, 1,0M THF). A solução é então deixada aquecer gradualmente à temperatura ambiente durante 12 h. A solução é então dividida entre EtOAc e NH4CI aquoso saturado. A camada orgânica é secada (Na2SO4) e concentrada. O resíduo é purificado por meio de cromatografia de coluna instantânea (DCM—>MeOH/DCM 5:95) para fornecer éster de metila de ácido 5-(3-flúor-4-metoxifenil)-6,7-diidro- 5H-pirrolo[1,2-c]imidazol-5-carboxílico. MS (ESI) m/z 291,2 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 2,64-2,86 (m, 3 H), 3,31-3,40 (m, 1 H), 3,79 (s, 3 H), 3,84 (s, 3 H), 6,62-6,68 (m, 1 H), 6,76 (s, 1 H), 6,80-6,89 (m, 2 H), 7,72 (s, 1 H). B. 5-(3-FLÚOR-4-METOXIFENIL)-6,7-DIIDRO-5H-PIRROLO[1,2-CllMI- DA-ZOL
[00316] Éster de metila de ácido 5-(3-flúor-4-metóxi-fenil)-6,7-diidro- 5H-pirrolo[1,2-c]imidazol-5-carboxílico (0,530 g, 1,82 mmols) é dissolvido em THF/água (20 mL) (3:2) e a uma solução é adicionado LiOH (0,230 g, 9,58 mmols). Após 1 h, a solução é levada para pH 4-5 com 1 M de HCI e então evaporado até a secura para fornecer ácido 5- (3-flúor-4-metóxi-fenil)-6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-carboxílico que é usado sem outra purificação. MS (ESI) m/z 277,2 (M + H).
[00317] O ácido acima (0,500 g, 1,81 mmols) é dissolvido em DMSO (8,0 mL) e EtsN (3,0 mL) e aquecido em um reator de microondas a 200 °C por 0,5 h. A solução é evaporada até a secura e purificada por meio de cromatografia de coluna instantânea (MeOH/DCM 5:95—>MeOH/DCM 1:9) para fornecer 5-(3-flúor-4-metóxi- fenil)-6,7-diidro-5H-pirrolo[1,2-c]imidazol como um sólido amarelo pálido. MS (ESI) m/z 233,3 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 2,44-2,58 (m, 1 H), 2,81-3,11 (m, 3 H), 3,91 (s, 3 H), 5,25 (app t, J = 6,8 Hz, 1 H), 6,82-6,91 (m, 3 H), 6,95 (app t, J = 8,6 Hz, 1 H), 7,35 (s, 1 H). EXEMPLO 5A A. 4-(6,7-DIIDRO-5H-PIRROLO[1,2-C]IMIDAZOL-5-IL)-3,5- DIFLÚORBEN- ZONITRILA
[00318] Éster de metila de ácido 3-(1-Tritil-7H-imidazol-4- il)propiônico (7,75 g, 19,5 mmols), J. Org. Chem. 2000, 65, 2229-2230, é dissolvido em DCM (300 mL) e resfriado até -78 °C. A este é adicionado uma solução de tolueno de DIBAL (21,0 mL, 1,5 M). Após 2 h MeOH (20 mL) é adicionado seguido por solução aquosa saturada de sal Rochelle (50 mL). A mistura é dividida entre DCM e solução aquosa saturada de sal Rochelle. A fase orgânica é secada (Na2SO4) e evaporada. O resíduo bruto é purificado por meio de cromatografia de coluna instantânea (MeOH/DCM 1:99 —> MeOH/DCM 5:95) para fornecer o aldeído, 3-(1 -tritil-7H-imidazol-4-il)propionaldeído como uma goma amarela (cas n° 184030-88-4). 1H RMN (CDCh) δ 9,83 (s, 1H), 7,38-7,31 (m, 10H), 7,16-7,12 (m, 6H), 6,58 (s, 1H), 2,90 (t, J = 8,0 Hz, 2H), 2,83-2,79 (m, 2H).
[00319] Diisopropilamina (0,53 mL, 3,75 mmols) é dissolvida em 5 mL THF e resfriada até - 78 °C. A esta é adicionado n-BuLi (2,3 mL, 1,6M em hexanos). Após 15 min, a solução de 3,5-diflúorbenzonitrila (0,52 g, 3,75 mmols) e THF (5 mL) são adicionados e a solução mantida naquela temperatura por 0,5 h antes que seja adicionada uma solução de 3-(1-tritil-7H-imidazol-4-il)propionaldeído e THF (20 mL). Após 2 h, a solução é diluída com EtOAc e finalizada com NH4CI saturado aquoso. A mistura é então dividida entre EtOAc e NH4CI aquoso saturado. O orgânico é secado (Na2SO4) e evaporado. O resíduo é purificado por meio de cromatografia de coluna instantânea (EtOAc/hexanos 2:8—>EtOAc) para fornecer 3,5-diflúor-4-[1-hidróxi-3- (1 -tritil-1 H-imidazol-4-il)-propil]benzonitrila como um pó amarelo. MS (ESI) m/z 506,2 (M + H).
[00320] 3,5-diflúor-4-[1-hidróxi-3-(1-tritil-1 H-imidazol-4-il)- propil]benzonitrila (0,105 g, 0,21 mmols) é dissolvido em DMF (5 mL) e resfriado até 0 °C. A este é adicionado EtsN (0,043 mL, 0,31 mmols) e cloreto de metanossulfonila (0,019 mL, 0,25 mmols). Após 2 h, LCMS mostrou a presença de mesilato intermediário [MS (ESI) m/z 584,3 (M + H)]. A solução é evaporada até a secura e dissolvida em DMF (5 mL), ao qual é adicionado K2CO3 (0,086 g, 0,62 mmols) e Nal (0,093 g, 0,62 mmols) e a mistura é aquecida até 90 °C por 0,5 h. A solução é então evaporada, dissolvida em MeCN e filtrada. O resíduo bruto é purificado por meio de cromatografia de coluna instantânea (MeOH/DCM 1:99 —► MeOH/DCM 1:9) para fornecer 4-(6,7-diidro-5H- pirrolo[1,2-c]imidazol-5-il)-3,5-diflúorbenzonitrila. MS (ESI) m/z 246,2 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 2,72-2,85 (m, 1 H), 2,93- 3,05 (m, 1 H), 3,08-3,21 (m, 2 H), 5,75 (app t, J = 7,2 Hz, 1 H), 6,79 (s, 1 H), 7,18-7,41 (m, 3 H). B. 4-(6,7-DIIDRO-5H-PIRROLO[1,2-C]IMIDAZOL-5-IL)-3-FLÚOR-5- METOXI-BENZONITRILA

[00321] Condições usadas para síntese de 3-flúor-4-[1-hidroxi-3-(1- tritil-1H-imidazol-4-il)-propil]-5-metoxibenzonitrila são similares ao procedimento acima para a síntese de 3,5-diflúor-4-[1 -hidroxi-3-(1 -tritil- 1H-imidazol-4-il)-propil]benzonitrila. MS (ESI) m/z 537,2 (M + H).
[00322] 3-flúor-4-[1-hidroxi-3-(1 -tritil-1 H-imidazol-4-il)-propil]-5- metoxibenzonitrila (0,283 g, 0,55 mmols) é dissolvido em DCM (5 mL) ao qual cloreto de tionila (0,12mL, 1,64 mmols) é adicionado. A solução é aquecida até o refluxo por 1 h antes da reação ser deixada resfriar à temperatura ambiente e dividida entre DCM e NaHCOs aquoso saturado. O orgânico é secado (Na2SO4) e evaporado.
[00323] O cloreto bruto é dissolvido em DMF (5 mL), ao qual é adicionado K2CO3 (0,243 g, 1,76 mmols) e Nal (0,25 g, 1,66 mmols) e a mistura é aquecida a 120 °C por 0,5 h em um microondas. A solução é então evaporada, dissolvida em MeCN e filtrada. O resíduo bruto é purificado por meio de cromatografia de coluna instantânea (MeOH/DCM 1:99 MeOH/DCM 1:9) e finalmente por HPLC para fornecer 4-(6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-il)-3-flúor-5- metoxibenzonitrila como um sal de TFA. MS (ESI) m/z 258,3 (M + H); 1H RMN (400 MHz, CDCh) (sal de TFA) δ ppm 2,79-2,93 (m, 1 H), 3,05-3,19 (m, 2 H), 3,19-3,31 (m, 1 H), 3,86 (s, 3 H), 6,05 (app t, J = 7,6 Hz, 1 H), 7,03 (s, 1 H), 7,07 (s, 1 H), 7,10 (dd, J = 9,6, 1,3 Hz, 1 H), 8,32 (s, 1 H). EXEMPLO 6 A. 4-(6,7-DIIDRO-5H-PIRROLO[1,2-C]IMIDAZOL-5-IL)-3- NITROBENZONI- TRILO
[00324] 3-[2-(ferc-Butildimetilsilanilóxi)etil]-1 -tritil-1 H-imidazol (1,00 g, 2,13 mmols) e 4-bromometil-3-nitrobenzonitrila (casn0 223512-70-7, preparado em WO9919301) (0,77 g, 3,20 mmols), preparados por meio do procedimento geral de brominação benzílica descrito anteriormente, são dissolvidos em MeCN (11 mL) e agitados à temperatura ambiente por 15 h. Naquele momento a solução é diluída com MeOH (5 mL) e EÍ2NH (1 mL) e então aquecida a 70 °C por 1,5 h. A solução é então evaporada até a secura e o resíduo purificado por meio de cromatografia de coluna instantânea (20-100% EtOAc/hexanos) para fornecer 4-{5-[2-(terc- butiIdimetiIsilanilóxi)etiI]imidazol-1 -iImetil}-3-nitrobenzonitrila como um óleo. MS (ESI) m/z 387,0 (M + H).
[00325] 4-{5-[2-(terc-Butildimetilsilanilóxi)etil]imidazol-1 -ilmetil}-3- nitrobenzonitrila (0,540 g, 1,40 mmols) é dissolvido em THF (8 mL) e MeOH (2 mL) e resfriada até 0 °C. Então uma solução de dioxano de HCI (1,75 mL, 4,0 M, 7 mmols) é adicionada. Após 0,5 h, a solução é concentrada.
[00326] O resíduo bruto, 4-[5-(2-hidroxietil)imidazol-1 -ilmetil]-3- nitrobenzonitrila, é então absorvido em CH2CI2 (10 mL), resfriado para 0 °C, e tratado com EtsN (0,58 mL, 4,19 mmols). A esta solução é adicionado cloreto de metanossulfonila (0,13 mL, 1,68 mmols). Após 0,5 h a solução é diluída com CH2CI2 (10 mL) e NaHCOs aquoso saturado (20 mL). A camada orgânica é ainda extraída com CH2CI2 (3 X 20 mL) e as camadas combinadas são secadas (Na2SÜ4) e concentradas para fornecer éster de 2-[3-(4-ciano-2-nitrobenzil)-3H- imidazol-4-il]etila de ácido metanossulfônico bruto. Uma suspensão de resíduo de éster de 2-[3-(4-ciano-2-nitrobenzil)-3H-imidazol-4-il]-etila de ácido metanossulfônico, DMF (17 mL), iodeto de sódio (0,630 g, 4,19 mmols), EtaN (0,58 mL, 4,19 mmols), e carbonato de potássio (0,580 g, 4,19 mmols) é aquecida para 60 °C por 1 hora e em seguida 75 °C por mais 2 h. Naquele ponto a suspensão é concentrada e diluída com CH2CI2 (20 mL) e NaHCOs aquoso saturado (20 mL). A camada orgânica é novamente extraída com CH2CI2 (3 X 20 mL) e as camadas combinadas são secadas (Na2SO4). O resíduo é então purificado por meio de HPLC (fase reversa, CH3CN/H2O) para fornecer 4-(6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-il)-3-nitrobenzonitrila como um sólido. MS (ESI) m/z 254,9 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 2,48-2,57 (m, 1 H), 2,81-2,92 (m, 1 H), 2,92-3,01 (m, 1 H), 3,35-3,47 (m, 1 H), 6,03 (dd, J = 8,6, 3,5 Hz, 1 H), 6,80 (d, J = 8,3 Hz, 1 H), 6,89 (s, 1 H), 7,36 (s, 1 H), 7,81 (dd, J = 7,8, 1,8 Hz, 1 H), 8,41 (d, J = 1,8 Hz, 1 H). B. 3-AMINO-4-(6,7-DIIDRO-5H-PIRROLO[1,2-C]IMIDAZOL-5-IL) BENZONI-TRILA
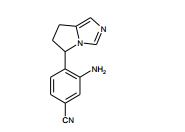
[00327] 4-(6,7-Diidro-5H-pirrolo[1,2-c]imidazol-5-il)-3- nitrobenzonitrila (0,054 g, 0,212 mmols) é dissolvido em THF (2 mL) e EtOH (2 mL) e 5% de paládio sobre carbono (úmido) (15 mg) é então adicionado. A mistura é colocada sob uma atmosfera de hidrogênio durante a noite. A mistura é então filtrada e concentrada. O resíduo é purificado por meio de HPLC (fase reversa, CH3CN/H2O) para fornecer 3-amino-4-(6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-il)benzonitrila como um sólido. MS (ESI) m/z 225,0 (M + H); 1H RMN (400 MHz, CH3CN) (sal de TFA) δ ppm 2,45-2,57 (m, 1 H), 2,99-3,16 (m, 3 H), 4,47 (br s, 2 H), 5,68-5,75 (m, 1 H), 6,69 (d, J = 7,8 Hz, 1 H), 6,98 (d, J = 1,5 Hz, 1 H), 7,09 (d, J = 1,8 Hz, 1 H), 7,21 (s, 1 H), 8,32 (s, 1 H). C. 1-[5-CIANO-2-(6,7-DIIDRO-5H-PIRROLO[1.2-C1IMIDAZOL-5-IL) FENIL1-3-ETILURÉIA
[00328] A uma solução de 3-Amino-4-(6,7-diidro-5H-pirrolo[1,2- c]imidazol-5-il)benzonitrila (0,090 g, 0,420 mmols), fornecida como o exemplo 6B, e DMF (5 mL) é adicionado etilisocianato (0,040 mL, 0,510 mmols). A solução é aquecida para 70 °C em um reator de microondas por 1,5 h seguida por aquecimento a 70 °C em um banho de óleo durante a noite. A solução é então concentrada e o resíduo purificado por meio de cromatografia instantânea (1-10% MeOH/CH2Cl2) para fornecer 1-[5-Ciano-2-(6,7-diidro-5H-pirrolo[1,2- c]imidazol-5-il)fenil]-3-etiluréia. MS (ESI) m/z 296,1 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,16 (t, J = 7,2 Hz, 3 H), 2,47-2,62 (m, 1 H), 2,82-3,04 (m, 2 H), 3,05-3,17 (m, 1 H), 3,21-3,33 (m, 2 H), 5,63 (app t, J = 6,8 Hz, 1 H), 5,80 (br s, 1 H), 6,82 (s, 1 H), 6,91 (d, J = 8,1 Hz, 1 H), 7,30 (dd, J = 8,1, 1,5 Hz, 1 H), 7,39 (s, 1 H), 7,53 (br s, 1 H), 7,97 (d, J = 1,3 Hz, 1 H). D. N-[5-CIANO-2-(6,7-DIIDRO-5H-PIRROLO[1.2-C1IMIDAZOL-5-IL)- FENIL1-BUTIRAMIDA
[00329] Uma solução de DMF (3 mL) de 3-Amino-4-(6,7-diidro-5H- pirrolo[1,2-c]imidazol-5-il)benzonitrila (0,095 g, 0,42 mmols), fornecida como no exemplo 6B, é tratada com EtsN (0,09 mL, 0,64 mmols) e cloreto de butirila (0,05 mL, 0,51 mmols) à temperatura ambiente. Após 1 h a solução é concentrada. Purificação por meio de HPLC preparativa fornece N-[5-Ciano-2-(6,7-diidro-5H-pirrolo[1,2-c]imidazol- 5-il)-fenil]-butiramida. MS (ESI) m/z 295,1 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,01 (t, J = 7,3 Hz, 3 H), 1,67-1,80 (m, 2 H), 2,35 (t, J = 7,5 Hz, 2 H), 2,49-2,61 (m, 1 H), 2,88-3,01 (m, 2 H), 3,05-3,18 (m, 1 H), 5,56 (dd, J = 8,0, 5,9 Hz, 1 H), 6,84 (s, 1 H), 6,97 (d, J = 8,1 Hz, 1 H), 7,40 (s, 1 H), 7,45 (dd, J = 8,1, 1,5 Hz, 1 H), 7,70 (br s, 1 H), 7,91 (s, 1 H). EXEMPLO 7 A.5-(4l-FLÚORBIFENIL-2-IL)-6,7-DIIDRO-5H-PIRROLO[1,2-C] IMIDAZOL
[00330] A uma suspensão de 2-(1-tritil-1H-imidazol-4-il)etanol (0,65 g, 1,80 mmols), acetonitrila (9 mL) e CH2CI2 (12 mL) é adicionada uma solução de 2-bromometil-4'-flúorbifenila (0,478 g, 1,80 mmols) em CH2CI2 (2 mL). A mistura resultante é agitada à temperatura ambiente durante a noite. A solução é concentrada e 0 resíduo é absorvido em MeOH e aquecido até 75 °C por 3,5 h. A solução é então concentrada e 0 resíduo é dividido entre CH2CI2 e NaHCOsa 5% aquoso. A camada aquosa é então extraída com CH2CI2 (3X). As camadas orgânicas combinadas são então lavadas com salmoura e secadas em Na2SO4. O resíduo é então purificado por cromatografia instantânea (CH2Ch/MeOH) para fornecer 2-[3-(4'-flúorbifenil-2-ilmetil)-3H-imidazol- 4-il]etanol. MS (ESI) m/z 297,1 (M + H).
[00331] A uma solução de 2-[3-(4'-flúorbifenil-2-ilmetil)-3H-imidazol- 4-il]etanol (0,341 g, 1,15 mmols) em CH2CI2 (10 mL) é adicionado, cloreto de tionila (0,11 mL, 1,50 mmols) a 0 °C. A mistura é então aquecida até 0 refluxo por 3h antes que 0 solvente seja removido e 0 resíduo secado sob pressão reduzida para fornecer 5-(2-cloroetil)-1-(4'- flúorbifenil-2-ilmetil)-1H-imidazol. O resíduo é absorvido em THF (60 mL). TMEDA (0,71 ml, 4,72 mmols) é adicionado, seguido por uma solução de hexano/THF de LDA (2,62 mL, 1,8 M) a -78 °C. A mistura resultante é agitada a -78 °C por 5 h. O excesso de LDA é então extingüido por adição de NH4CI saturado. A mistura é então diluída com CH2CI2 e água. A camada orgânica é separada e lavada com água, salmoura e secada sobre Na2SO4. Após concentração, 0 resíduo é purificado por cromatografia instantânea (C^CL/MeOH) para fornecer 5-(4'-flúorbifenil-2-il)-6,7-diidro-5H-pirrolo[1,2-c]imidazol. MS (ESI) m/z 279,1 (M + H). Resolução de enantiômeros (R) e (S) do composto do título é obtido por HPLC quiral usando coluna de ChiralPak AD e 13% IPA:hexano para fornecer enantiômero A (tr = 9,6 min) e enantiômero B (tr = 12,6 min). Por enantiômero B: 1H RMN (400 MHz, CDCI3) (sal de HCI) δ ppm 2,68-2,79 (m, 1 H), 2,95-3,06 (m, 2 H), 3,13-3,22 (m, 1 H), 5,55 (app t, J = 7,6 Hz, 1 H), 6,96 (dd, J = 7,6, 1,5 Hz, 1 H), 7,13 (s, 1 H), 7,18 (app t, J = 8,6 Hz, 2 H), 7,27-7,32 (m, 2 H), 7,33-7,38 (m, 1 H), 7,40-7,49 (m, 2 H), 8,01 (s, 1 H).
[00332] Similarmente preparado é 0 seguinte:
[00333] 5-Bifenil-2-il-6,7-diidro-5H-pirrolo[1,2-c]imidazol. MS (ESI) m/z 261,3 (M + H). Resolução de enantiômeros (R) e (S) do composto do título é obtida por HPLC quiral usando coluna de ChiralPak AD e 13% IPA:hexano para fornecer enantiômero A (tr = 9,1 min) e enantiômero B (tr = 12,4 min). Por enantiômero B: 1H RMN (400 MHz, MeOD) (sal de TFA) δ ppm 2,65-2,74 (m, 1 H), 3,00 (ddd, J = 15,6, 10,8, 8,5 Hz, 2 H), 3,09- 3,19 (m, 1 H), 5,67-5,73 (m, 1 H), 7,08-7,14 (m, 1 H), 7,27 (s, 1 H), 7,32- 7,41 (m, 3 H), 7,42-7,50 (m, 5 H), 8,66 (s, 1 H).
EXEMPLO 8 A. ÉSTER DE ISOPROPILA DE ÁCIDO 5-(4-CIANO-2-FLÚORFENIL)- 6,7-DIIDRO-5H-PIRROLO[1,2-C]IMIDAZOL-5-CARBOXÍLICO
[00334] A uma solução de éster de metila de ácido 5-(4-ciano-2- flúorfenil)-6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-carboxílico (0,186 g, 0,652 mmols) em THF (15 mL), uma solução aquosa de LiOH (0,065 mL, 2 M) é adicionada. A solução é agitada à temperatura ambiente por 20 min, tempo no qual LCMS mostrou somente material de partida. Naquele momento mais H2O (1,5 mL) é adicionada. Após mais 1h 0 material de partida foi consumido. A solução é então neutralizada para pH 5-6 com 1 N de HCI e evaporado até a secura. O ácido bruto, ácido 5-(4-ciano-2-flúor-fenil)-6,7-diidro-5H-pirrolo[1,2-c]imidazol-5- carboxílico é usado sem outra purificação. MS (ESI) m/z 272,0 (M + H).
[00335] ácido 5-(4-Ciano-2-flúorfenil)-6,7-diidro-5H-pirrolo[1,2- c]imidazol-5-carboxílico (0,15 g, 0,279 mmols) é suspenso em CH2CI2 (3 mL) e DMF (0,01 ml) e resfriado para 0 °C. A solução de cloreto de oxalila (0,31 mL, 2 M) em CH2CI2 é adicionado em gotas. Após 1h /- PrOH (3 mL) é adicionado. Após 2h EtsN é adicionado até tornar-se básico e a solução é concentrada. A suspensão resultante é dissolvida em CH2CI2, lavada com NaHCO3/H2O (1:1) saturado aquoso e salmoura, então secado em Na2SO4. Após concentração, 0 resíduo é purificado por HPLC (2-38% MeCN/H20 contendo 0,1% de TFA) para fornecer éster de isopropila de ácido 5-(4-ciano-2-flúorfenil)-6,7-diidro- 5H-pirrolo[1,2-c]imidazol-5-carboxílico como um sólido. MS (ESI) m/z 314,0 (M + H); 1H RMN (400 MHz, CD3CN) (sal de TFA) δ ppm 1,22 (d, J = 6,06 Hz, 3 H), 1,23 (d, J = 6,06 Hz, 3 H), 3,01-3,18 (m, 3 H), 3,38- 3,45 (m, 1 H), 5,08-5,18 (m, 1 H), 7,20 (s, 1 H), 7,34 (app t, J = 8,08 Hz, 1 H), 7,62-7,68 (m, 2 H) 8,52 (s, 1 H).
[00336] Similarmente preparados são os seguintes: 1) Éster de isopropila de ácido 5-(2-Cloro-4-cianofenil)-6,7- diidro-5H-pirrolo[1,2-c]imidazol-5-carboxílico. MS (ESI) m/z 330,2, 332,2 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,23 (d, J = 6,3 Hz, 3 H), 1,27 (d, J = 6,3 Hz, 3 H), 2,63- 2,77 (m, 2 H), 2,94-3,07 (m, 1 H), 3,81-3,93 (m, 1 H), 5,09-5,22 (m, 1 H), 6,54 (d, J = 8,1 Hz, 1 H), 6,89 (s, 1 H), 7,50 (dd, J = 8,2, 1,6 Hz, 1 H), 7,58 (s, 1 H), 7,73 (d, J = 1,5 Hz, 1 H).
2) Éster de isopropila de ácido 5-(4-Ciano-2-metoxifenil)- 6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-carboxílico. MS (ESI) m/z 326,3 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,18 (d, J = 6,3 Hz, 3 H), 1,23 (d, J = 6,3 Hz, 3 H), 2,55-2,64 (m, 1 H), 2,65-2,76 (m, 1 H), 2,97 (ddd, J = 15,4, 9,4, 2,8 Hz, 1 H), 3,61-3,71 (m, 1 H), 3,88 (s, 3 H), 5,04- 5,14 (m, 1 H), 6,57 (d, J = 8,1 Hz, 1 H), 6,83 (s, 1 H), 7,14 (d, J = 1,3 Hz, 1 H), 7,21 (dd, J = 8,0, 1,4 Hz, 1 H), 7,58 (s, 1 H).

3) Éster de 2-isopropoxietila de ácido 5-(2-Cloro-4- cianofenil)-6,7-diidro-5H-pirrolo[ 1,2-c]imidazol-5-carboxílico. MS (ESI) m/z 374,2, 376,2 (M + H); 1H RMN (400 MHz, CDCh) (sal de HCI) δ ppm 1,08 (d, J = 6,1 Hz, 3 H), 1,10 (d, J = 6,1 Hz, 3 H), 2,89-3,02 (m, 2 H), 3,17-3,28 (m, 1 H), 3,52-3,59 (m, 1 H), 3,61 (app t, J = 4,5 Hz, 2 H), 3,88-3,96 (m, 1 H), 4,25-4,33 (m, 1 H), 4,57-4,65 (m, 1 H), 6,73 (d, J = 8,1 Hz, 1 H), 7,29 (s, 1 H), 7,61 (s, 1 H), 7,81 (s, 1 H), 8,69 (s, 1 H), 10,6 (br s, 1 H). Resolução de enantiômeros (R) e (S) do composto do título é obtida por HPLC quiral usando a coluna ChiralPak IA com uma fase móvel de 2:8 IPA/hexano para fornecer enantiômero A (tr = 23,9 min) e enantiômero B (tr = 38,6 min).

4) Éster de 2-isopropoxietila de ácido 5-(4-Ciano-2- metoxifenil)-6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-carboxílico. MS (ESI) m/z 370,2 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,08 (d, J = 6,1,3 H), 1,10 (d, J = 6,1, 3 H), 2,58-2,78 (m, 2 H), 2,92-3,03 (m, 1 H), 3,44- 3,58 (m, 3 H), 3,62-3,73 (m, 1 H), 3,90 (s, 3 H), 4,14-4,23 (m, 1 H), 4,35-4,44 (m, 1 H), 6,57 (d, J = 8,1 Hz, 1 H), 6,84 (s, 1 H), 7,15 (d, J = 1,3 Hz, 1 H), 7,21 (dd, J = 7,8, 1,3 Hz, 1 H), 7,69 (s, 1 H).
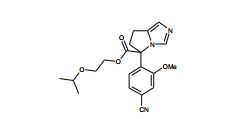
5) Éster de 4-flúorbenzila de ácido 5-(2-Cloro-4-cianofenil)- 6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-carboxílico. MS (ESI) m/z 396,1, 398,1 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 2,68-2,80 (m, 2 H), 2,99-3,09 (m, 1 H), 3,84-3,91 (m, 1 H), 5,22 (ABq, J = 11,9 Hz, 2 H), 6,56 (d, J = 8,3 Hz, 1 H), 6,92 (s, 1 H), 7,01-7,08 (m, 2 H), 7,23-7,28 (m, 2 H), 7,50 (dd, J = 8,2, 1,6 Hz, 1 H), 7,57 (s, 1 H), 7,70 (d, J = 1,5 Hz, 1 H). Resolução de enantiômeros (R) e (S) do composto do título é obtida por HPLC quiral usando Coluna ChiralPak IA e 3:7 /- PrOH/hexanos para fornecer enantiômero A (tr = 39,2 min) e enantiômero B (tr = 60,3 min).

6) Éster de 4-flúorbenzila de ácido 5-(4-Ciano-2- metoxifenil)-6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-carboxílico. MS (ESI) m/z 392,1 (M + H); 1H RMN (400 MHz, CDCh) δppm 2,59 (ddd, J = 13,0, 8,1, 3,0 Hz, 1 H), 2,65-2,75 (m, 1 H), 2,92-3,00 (m, 1 H), 3,55 (s, 3 H), 3,61-3,71 (m, 1 H), 5,15 (ABq, J = 11,9 Hz, 2 H), 6,58 (d, J = 7,8 Hz, 1 H), 6,81 (s, 1 H), 7,00-7,08 (m, 3 H), 7,20 (dd, J = 8,0, 1,4 Hz, 1 H), 7,23-7,29 (m, 2 H), 7,47 (s, 1 H).
EXEMPLO 9 A. (4-FLÚORBENZIDMETILAMIDA DE ÁCIDO 5-(4-CIANO-2- METOXIFENIL)-6,7-DIIDRO-5H-PIRROLO[1,2-C]IMIDAZOL-5- CARBOXÍLICO
[00337] Ácido 5-(4-ciano-2-metoxifenil)-6,7-diidro-5H-pirrolo[1,2- c]imidazol-5-carboxílico bruto (0,168 g, 0,59 mmols) é suspenso em CH2CI2 (3 mL) e resfriado para 0 °C. A este é adicionado DMF (0,2 mL) seguido pela adição de uma solução de cloreto de oxalila de CH2CI2 (0,6 mL, 2,0 M). Após 2 h, 4-flúor-/V-metilbenzilamina (0,23 mL, 1,78 mmols) é adicionado. A reação é agitada por mais 2 h , evaporada até a secura, e dividida entre EtOAc e NaHCOs aquoso saturado. A fase orgânica é secada (Na2SO4), filtrada, e concentrada. O resíduo bruto é purificado por meio de cromatografia de coluna instantânea (2-5% MeOH/ CH2CI2) para fornecer (4-flúorbenzil)metilamida de ácido 5-(4- ciano-2-metoxifenil)-6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-carboxílico como um sólido amarelo. MS (ESI) m/z 405,2 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 2,60 (s, 3 H), 2,76-2,88 (m, 2 H), 2,95-3,04 (m, 1 H), 3,57-3,69 (m, 1 H), 3,74 (s, 3 H), 4,43 (d, J = 14,4 Hz, 1 H), 4,58- 4,70 (m, 1 H), 6,79 (s, 1 H), 6,86-6,94 (m, 1 H), 6,96-7,05 (m, 2 H), 7,10 (s, 1 H), 7,19-7,25 (m, 3 H), 7,55 (s, 1 H). Resolução de enantiômeros (R) e (S) do composto do título é obtida por HPLC quiral usando coluna ChiralPak AD com uma fase móvel de 6:4 EtOH/hexano para fornecer enantiômero A (tr = 30,2 min) e enantiômero B (tr = 36,0 min).
[00338] Similarmente preparados são os seguintes: 1) etilamida de ácido 5-(2-Cloro-4-cianofenil)-6,7-diidro-5H- pirrolo[1,2-c]imidazol-5-carboxílico. MS (ESI) m/z 315,0, 317,0 (M + H); 1H RMN (400 MHz, CDCh) (sal de TFA) δ ppm 1,09 (t, J = 7,2 Hz, 3 H), 2,72-2,81 (m, 1 H), 2,84 (m, 1 H), 3,15-3,27 (m, 2 H), 3,32-3,42 (m, 1 H), 4,12-4,22 (m, 1 H), 6,81 (d, J = 8,1 Hz, 1 H), 7,11 (s, 1 H), 7,53 (obs d, J = 7,3 Hz, 1 H), 7,54 (s, 1H), 7,78 (s, 1 H) 9,52 (s, 1 H).
2) Metilamida de ácido 5-(2-Cloro-4-cianofenil)-6,7-diidro- 5H-pirrolo[1,2-c]imidazol-5-carboxílico. MS (ESI) m/z 301,0, 303,0 (M + H); 1H RMN (400 MHz, CDCh) (sal de TFA) δ ppm 2,74-2,79 (obs m, 1 H), 2,80 (d, J = 4,6 Hz, 3 H), 2,85-2,94 (m, 1 H), 3,19-3,28 (m, 1 H), 4,13-4,22 (m, 1 H), 6,83 (d, J = 8,1 Hz, 1 H), 7,11 (s, 1 H), 7,53 (dd, J = 8,2, 1,4 Hz, 1 H), 7,62 (br s, 1 H), 7,77 (d, J = 1,5 Hz, 1 H), 9,48 (s, 1 H).
3) Dimetilamida de ácido 5-(2-Cloro-4-cianofenil)-6,7-diidro- 5H-pirrolo[1,2-c]imidazol-5-carboxílico. MS (ESI) m/z 315,0, 317,0 (M + H); 1H RMN (400 MHz, CDCh) (sal de TFA) δ ppm 2,73 (s, 3 H), 3,10 (s, 3 H), 3,17-3,22 (m, 2 H), 3,35-3,44 (m, 2 H), 3,52-3,62 (m, 2 H), 7,15 (s, 1 H), 7,42 (d, J = 8,1 Hz, 1 H), 7,72 (dd, J = 8,2, 1,6 Hz, 1 H), 7,84 (d, J = 1,5 Hz, 1 H), 8,61 (s, 1 H).
4) 3-Cloro-4-[5-(morfolino-4-carbonil)-6,7-diidro-5H- pirrolo[1,2-c]imidazol-5-il]benzonitrila. MS (ESI) m/z 356,9, 358,9 (M + H); 1H RMN (400 MHz, CDCh) (sal de TFA) δ ppm 2,97-3,64 (m, 8 H), 3,74 (br s, 4 H), 7,15 (s, 1 H), 7,39-7,53 (m, 1 H), 7,71 (d, J = 7,3 Hz, 1 H), 7,85 (s, 1 H), 8,66 (s, 1 H).
5) (2-metoxietil)metilamida de ácido 5-(2-Cloro-4- cianofenil)-6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-carboxilico. MS (ESI) m/z 359,0, 361,0 (M + H); 1H RMN (400 MHz, CDCh) (sal de HCI) δ ppm 2,78 (s, 3H), 2,98-3,12 (m, 1 H), 3,13-3,40 (m, 6 H), 3,42-3,75 (m, 1 H), 7,09 (s, 1 H), 7,62 (brs, 1 H), 7,73 (s, 1 H), 8,46 (s, 1H).
6) (4-flúorbenzil)metilamida de ácido 5-(2-Cloro-4- cianofenil)-6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-carboxilico. MS (ESI) m/z 409,2, 411,2 (M + H). MS (ESI) m/z 405,1 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 2,61 (brs, 3 H), 2,64-2,86 (m, 2 H), 2,98-3,10 (m, 1 H), 3,97-4,12 (m, 1 H), 4,37-4,51 (m, 1 H), 4,68-4,84 (m, 1 H), 6,63- 6,77 (m, 1 H), 6,91 (s, 1 H), 7,01 (app t, J = 8,6 Hz, 2 H), 7,15-7,26 (m, 2 H), 7,48-7,58 (m, 2 H), 7,73 (br s, 1 H).
7) (2-flúorbenzil)metilamida de ácido 5-(4-Ciano-2- metoxifenil)-6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-carboxilico. MS (ESI) m/z 405,1 (M + H); 1H RMN (400 MHz, CDCh) δD ppm 2,68 (br s, 3 H), 2,72-2,92 (m, 2 H), 3,00 (app dd, J = 13,6, 7,8 Hz, 1 H), 3,57-3,89 (m, 1 H), 3,67 (s, 3 H), 4,49-4,82 (m, 2 H), 6,77 (s, 1 H), 6,91 (br s, 1 H), 6,99-7,18 (m, 3 H), 7,18-7,32 (m, 2 H), 7,43 (br s, 1 H), 7,54 (br s, 1 H).
8) (3-flúorbenzil)metilamida de ácido 5-(4-Ciano-2- metoxifenil)-6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-carboxilico. MS (ESI) m/z 405,1 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 2,63 (br s, 3 H), 2,73-2,84 (m, 1 H), 2,84-2,93 (m, 1 H), 2,97-3,07 (m, 1 H), 3,59-3,73 (m, 1 H), 3,82 (br s, 3 H), 4,44 (d, J = 14,4 Hz, 1 H), 4,76 (m, 1 H), 6,80 (s, 1 H), 6,92 (d, J = 7,8 Hz, 1 H), 6,96-7,07 (m, 2 H), 7,10-7,17 (m, 1 H), 7,19-7,35 (m, 3 H), 7,58 (s, 1 H).

9) (4-metoxibenzil)metilamida de ácido 5-(2-Cloro-4- cianofenil)-6,7-diidro-5H-pirrolo[ 1,2-c]imidazol-5-carboxilico. MS (ESI) m/z 421,1, 423,1 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 2,57 (br s, 3 H), 2,60-2,77 (m, 2 H), 2,95-3,06 (m, 1 H), 3,79 (s, 3 H), 4,04-4,18 (m, 1 H), 4,28-4,40 (m, 1 H), 4,71-4,87 (m, 1 H), 6,54-6,68 (m, 1 H), 6,78-6,90 (m, 4 H), 7,10-7,20 (m, 1 H), 7,38-7,53 (m, 2 H), 7,72 (br s, 1 H). Resolução de enantiômeros (R) e (S) do composto do título é obtido por HPLC quiral usando uma coluna ChiralPak IA e CH3CN para fornecer enantiômero A (tr = 25,9 min) e enantiômero B (tr = 40,3 min).

10) isobutilmetilamida de ácido 5-(2-Cloro-4-cianofenil)-6,7- diidro-5H-pirrolo[1,2-c]imidazol-5-carboxilico. MS (ESI) m/z 357,1. 359,1 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 0,87-0,93 (m, 6 H), 1,95-2,12 (m, 1 H), 2,59-2,80 (m, 2 H), 2,69 (s, 3 H), 2,96-3,06 (m, 1 H), 3,06-3,18 (m, 1 H), 3,31-3,46 (m, 1 H), 3,95-4,12 (m, 1 H), 6,61- 6,75 (m, 1 H), 6,88 (s, 1 H), 7,53 (dd, J = 8,2, 1,6 Hz, 1 H), 7,60 (br s, 1 H), 7,75 (s, 1 H).
11) etilamida de ácido 5-(2-Bromo-4-cianofenil)-6,7-diidro- 5H-pirrolo[1,2-c]imidazol-5-carboxilico. MS (ESI) m/z 359,0, 361,0(M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,17 (t, J = 7,2 Hz, 3 H), 2,54- 2,68 (m, 1 H), 2,86 (ddd, J = 13,4, 8,1, 2,5 Hz, 1 H), 2,99 (ddd, J = 15,5, 9,0, 1,8 Hz, 1 H), 3,33-3,45 (m, 2 H), 3,85-3,97 (m, 1 H), 5,57 (app t, J = 5,4 Hz, 1 H), 6,60 (d, J = 8,1 Hz, 1 H), 6,86 (s, 1 H), 7,53 (s, 1 H), 7,57 (dd, J = 8,2, 1,6 Hz, 1 H), 7,96 (d, J = 1,8 Hz, 1 H).
12) cicloexilmetilamida de ácido 5-(2-Bromo-4-cianofenil)- 6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-carboxilico. MS (ESI) m/z 427,2, 429,2 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 0,80-0,97 (m, 2 H), 1,06-1,29 (m, 3 H), 1,44-1,58 (m, 1 H), 1,57-1,76 (m, 5 H), 2,56-2,70 (m, 1 H), 2,83-2,94 (m, 1 H), 2,94-3,03 (m, 1 H), 3,08-3,25 (m, 2 H), 3,83-3,96 (m, 1 H), 5,63 (t, J = 5,7 Hz, 1 H), 6,63 (d, J = 8,3 Hz, 1 H), 6,85 (s, 1 H), 7,49 (s, 1 H), 7,57 (dd, J = 8,2, 1,6 Hz, 1 H), 7,96 (d, J = 1,5 Hz, 1 H).
13) (2,2,2-triflúoretil)amida de ácido 5-(2-Bromo-4- cianofenil)-6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-carboxilico. MS (ESI) m/z 413,0, 415,0 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 2,56-2,67 (m, 1 H), 2,87 (ddd, J = 13,3, 8,1, 2,5 Hz, 1 H), 3,01 (ddd, J = 15,9, 8,8, 1,6 Hz, 1 H), 3,78-3,92 (m, 1 H), 3,94-4,04 (m, 1 H), 4,05-4,20 (m, 1 H), 6,48 (d, J = 8,3 Hz, 1 H), 6,65 (s, 1 H), 7,05 (br s, 1 H), 7,41 (s, 1 H), 7,48 (dd, J = 8,1, 1,5 Hz, 1 H), 7,94 (d, J = 1,5 Hz, 1 H).

14) (2,2-dimetoxietil)amida de ácido 5-(2-Cloro-4- cianofenil)-6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-carboxilico. MS (ESI) m/z 375,1, 377,1 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 2,57-2,70 (m, 1 H), 2,81 (ddd, J = 13,3, 8,0, 2,4 Hz, 1 H), 2,95-3,05 (m, 1 H), 3,30-3,36 (m, 1 H), 3,36 (s, 3 H), 3,38 (s, 3 H), 3,58 (ddd, J = 13,8, 6,4, 4,8 Hz, 1 H), 3,84-3,94 (m, 1 H), 4,39 (t, J = 4,9 Hz, 1 H), 5,89 (t, J = 5,7 Hz, 1 H), 6,63 (d, J = 8,1 Hz, 1 H), 6,85 (s, 1 H), 7,54 (dd, J = 8,1, 1,8 Hz, 1 H), 7,64 (s, 1 H), 7,76 (d, J = 1,5 Hz, 1 H).

15) (2-hidroxietil)amida de ácido 5-(2-Cloro-4-cianofenil)- 6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-carboxilico. MS (ESI) m/z 331,2, 333,2 (M + H); 1H RMN (400 MHz, DMSO-cfe) δ ppm 2,52-2,62 (m, 1 H), 2,88 (dd, J = 13,8, 8,7 Hz, 1 H), 3,05-3,15 (m, 1 H), 3,21-3,30 (m, 1 H), 3,34-3,49 (m, 2 H), 3,71-3,84 (m, 1 H), 4,64 (t, J = 5,3 Hz, 2 H), 6,45 (d, J = 8,1 Hz, 1 H), 6,74 (s, 1 H), 7,64 (s, 1 H), 7,85 (dd, J = 8,2, 1,6 Hz, 1 H), 8,02 (t, J = 5,6 Hz, 1 H), 8,16 (d, J = 1,5 Hz, 1 H).
16) Amida de ácido 5-(2-Cloro-4-cianofenil)-6,7-diidro-5H- pirrolo[1,2-c]imidazol-5-carboxilico. MS (ESI) m/z 287,3, 289,3 (M + H); 1H RMN (400 MHz, DMSO-cfe) δ ppm 2,53-2,66 (m, 2 H), 2,84-2,94 (m, 1 H), 3,69-3,78 (m, 1 H), 6,53 (d, J = 8,1 Hz, 1 H), 6,74 (s, 1 H), 7,57 (s, 1 H), 7,64 (s, 1 H), 7,71 (s, 1 H), 7,84 (dd, J = 8,2, 1,6 Hz, 1 H), 8,17 (d, J = 1,5 Hz, 1 H).
17) 3-Cloro-4-[5-(piperidina-1-carbonil)-6,7-diidro-5H- pirrolo[1,2-c]imidazol-5-il]benzonitrila. MS (ESI) m/z 355,3, 357,3 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,20 (br s, 2 H), 1,60 (br s, 4 H), 2,58-2,75 (m, 2 H), 2,96-3,07 (m, 1 H), 3,13-3,42 (m, 3 H), 3,84-4,02 (m, 1 H), 3,99-4,12 (m, 1 H), 6,71 (d, J = 7,8 Hz, 1 H), 6,87 (s, 1 H), 7,54 (dd, J = 8,2, 1,6 Hz, 1 H), 7,56 (s, 1 H), 7,76 (d, J = 1,5 Hz, 1 H).
18) 3-Metóxi-4-[5-(piperidina-1-carbonil)-6,7-diidro-5H- pirrolo[1,2-c]imidazol-5-il]benzonitrila. MS (ESI) m/z 351,1 (M + H); 1H RMN (400 MHz, CDCh) δQ ppm 1,58 (br s, 4 H), 1,88 (br s, 2 H), 2,72- 2,89 (m, 2 H), 2,95-3,06 (m, 1 H), 3,32 (br s, 4 H), 3,59-3,74 (m, 1 H), 3,92 (s, 3 H), 6,77 (s, 1 H), 6,87-6,99 (m, 1 H), 7,19 (d, J = 1,5 Hz, 1 H), 7,24-7,30 (m, 1 H), 7,54 (s, 1 H).
19) Cicloexilmetilamida de ácido 5-(2-Cloro-4-cianofenil)- 6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-carboxilico. MS (ESI) m/z 383,3, 385,3 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,24-1,86 (m, 10 H), 2,54 (s, 3 H), 2,56-2,78 (m, 2 H), 2,93-3,11 (m, 1 H), 3,15-3,32 (m, 1 H), 3,98-4,16 (m, 1 H), 6,63 (d, J = 8,1 Hz, 1 H), 6,86 (s, 1 H), 7,44- 7,60 (m, 2 H), 7,74 (s, 1 H).
20) Metilfenetilamida de ácido 5-(2-Cloro-4-cianofenil)-6,7- diidro-5H-pirrolo[1,2-c]imidazol-5-carboxilico. MS (ESI) m/z 405,3, 407,3 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 2,56 (brs, 3 H), 2,60- 2,76 (m, 2 H), 2,79-3,25 (m, 3 H), 3,55 (d, J = 10,4 Hz, 1 H), 3,67-3,83 (m, 1 H), 3,98-4,13 (m, 1 H), 6,62 (d, J = 7,8 Hz, 1 H), 6,88 (s, 1 H), 7,13-7,36 (m, 5 H), 7,44 (br s, 1 H), 7,50 (d, J = 7,8 Hz, 1 H), 7,76 (br s, 1 H).
21) (3-metoxibenzil)metilamida de ácido 5-(4-Ciano-2- metoxifenil)-6,7-diidro-5H-pirrolo[ 1,2-c]imidazol-5-carboxilico. MS (ESI) m/z 417,4 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 2,62 (br s, 3 H), 2,76-2,88 (m, 1 H), 2,88-2,99 (m, 1 H), 2,98-3,09 (m, 1 H), 3,58-3,73 (m, 1 H), 3,80 (s, 3 H), 3,82 (br s, 3 H), 4,43-4,72 (m, 2 H), 6,75-6,89 (m, 4 H), 6,95 (d, J = 7,6 Hz, 1 H), 7,10-7,19 (m, 1 H), 7,24 (d, J = 9,9 Hz, 2 H), 7,62 (brs, 1 H).
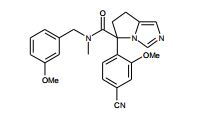
22) (4-metoxibenzil)metilamida de ácido 5-(4-Ciano-2- metoxifenil)-6,7-diidro-5H-pirrolo[ 1,2-c]imidazol-5-carboxilico. MS (ESI) m/z 417,1 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 2,59 (br s, 3 H), 2,71-2,92 (m, 2 H), 2,94-3,06 (m, 1 H), 3,58-3,70 (m, 1 H), 3,75 (brs, 3 H), 3,80 (s, 3 H), 4,36-4,48 (m, 1 H), 4,54-4,72 (m, 1 H), 6,78 (s, 1 H), 6,81-6,95 (m, 3 H), 7,11 (br s, 1 H), 7,14-7,27 (m, 3 H), 7,53 (br s, 1 H). Resolução de enantiômeros (R) e (S) do composto do título é obtido por HPLC quiral usando ChiralPak AS-H coluna com uma fase móvel de 3:7 de EtOH/Heptano para fornecer enantiômero A (tr = 28,9 min) e enantiômero B (tr = 92,3 min).
23) (2-metoxibenzil)metilamida de ácido 5-(4-Ciano-2- metoxifenil)-6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-carboxilico. MS (ESI) m/z 417,1 (M + H);
B. 1,2,6',7'-TETRAIDRO-2-OXO-1-(2,2,2-TRIFLLJORETIL) ESPIROÍ3H- INDOL-S.δ'-rδHlPIRROLOri^-CIIMIDAZOLI-e-CARBONITRILA
[00339] Uma suspensão de (2,2,2-triflúoretil)amida de ácido 5-(2- Bromo-4-cianofenil)-6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-carboxilico (0,168 g, 0,405 mmols), fornecido no exemplo 9, Cui (0,004 g, 0,021 mmols), N, N'-dimetilatilano diamina (0,005 mL, 0,042 mmols), CS2CO3 (0,198 g, 0,609 mmols), e THF (15 mL) é aquecido a 110 °C por 60 h. A mistura é então filtrada e concentrada. O resíduo é purificado por meio de cromatografia instantânea (EtOAc/hexanos) para fornecer 1,2,6',7'-Tetraidro-2-oxo-1 -(2,2,2-triflúoretil) espiro[3H-indol-3,5'- [5H]pirrolo[1,2-c]imidazol]-6-carbonitrila. MS (ESI) m/z 333,1 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 2,85-2,96 (m, 1 H), 3,03-3,18 (m, 2 H), 3,27-3,39 (m, 1 H), 4,20-4,34 (m, 1 H), 4,43-4,57 (m, 1 H), 6,86 (s, 1 H), 7,04 (s, 1 H), 7,30 (s, 1 H), 7,35 (d, J = 7,8 Hz, 1 H), 7,52 (dd, J = 7,6, 1,3 Hz, 1 H).
[00340] Similarmente preparados são os seguintes: 1) 1 -(C icloexi I meti I)-1,2,6',7'-tetraidro-2-oxoespiro[3H-indol- 3,5'-[5H]pirrolo[1,2-c]imidazol]-6-carbonitrila. MS (ESI) m/z 347,1 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 0,99-1,14 (m, 2 H), 1,21 (d, J = 9,3 Hz, 2 H), 1,61-1,72 (m, 4 H), 1,73-1,89 (m, 3 H), 2,79-2,91 (m, 1 H), 2,99-3,15 (m, 2 H), 3,27-3,37 (m, 1 H), 3,58 (dd, J = 7,5, 2,7 Hz, 2 H), 6,84 (s, 1 H), 7,02 (s, 1 H), 7,16 (d, J = 0,8 Hz, 1 H), 7,28 (d, J = 7,6 Hz, 1 H), 7,42 (dd, J = 7,7, 1,4 Hz, 1 H).
2) 1 -Etil-1,2,6',7'-tetraidro-2-oxoespiro[3H-indol-3,5'-[5H]pir- rolo[1,2-c]imida-zol]-6-carbonitrila. MS (ESI) m/z 279,1 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,34 (t, J = 7,3 Hz, 3 H), 2,77-2,89 (m, 1 H), 2,99-3,15 (m, 2 H), 3,27 - 3,38 (m, 1 H), 3,73-3,90 (m, 2 H), 6,85 (s, 1 H), 7,06 (s, 1 H), 7,19 (s, 1 H), 7,29 (d, J = 7,6 Hz, 1 H), 7,43 (dd, J = 7,6, 1,3 Hz, 1 H).
EXEMPLO 10 ÁCIDO 5-(4-ETOXICARBONIL-2-FLÚORFENIL)-6,7-DIIDRO-5H- PIRRO-LO[1,2-C]IMIDAZOL-5-CARBOXÍLICO
[00341] A uma solução de éster de metila de ácido 5-(4-ciano-2- flúorfenil)-6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-carboxílico (0,250 g, 0,877 mmols) em THF/EtOH (2:1, 21 mL) é adicionada uma solução aquosa de LiOH (0,88 mL, 2 M). Após 2,5 h a solução é concentrada e o resíduo resultante é purificado por HPLC (2-23% MeCN/água contendo 0,1% de TFA). O composto do título, ácido 5-(4-etoxicarbonil- 2-flúorfenil)-6,7-diidro-5H-pirrolo[ 1,2-c]imidazol-5-carboxílico, é isolado como um produto menor. MS (ESI) m/z 319,0 (M + H). EXEMPLO 11 4-(5-HIDROXIMETIL-6,7-DIIDRO-5H-PIRROLOri,2-CllMIDAZOL-5-IL)- 3-METOXIBENZONITRILA
[00342] Éster de metila de ácido 5-(4-Ciano-2-metoxifenil)-6,7- diidro-5H-pirrolo[1,2-c]imidazol-5-carboxílico (0,100 g, 0,336 mmols) é dissolvido em THF (3 mL) e resfriada para -10 °C. A solução de THF de boroidreto de lítio (0,08 mL, 2 M) é então adicionado. A solução é removida a partir de banho frio e deixada aquecer para temperatura ambiente antes de ser aquecida para 40 °C, tempo no qual boroidreto de lítio adicional (0,08 mL, 2 M) é adicionado. A solução é mantida naquela temperatura por 1,5 h antes de ser deixada resfriar para temperatura ambiente e ser finalizada por adição de 1 M de HCI. Seguindo a preparação aquosa o resíduo é aquecido para 80 °C em etanolamina/CHsCN (1:3) por 1 h. A solução é então concentrada e o resíduo purificado por meio de HPLC preparativa (fase reversa; 5- 100% CH3CN/H2O e 0,1% de TFA). Conversão para base livre forneceu 4-(5-hidroximetil-6,7-diidro-5H-pirrolo[1,2-c]imidazol-5-il)-3- metoxibenzonitrila como um sólido branco. MS (ESI) m/z 270,1 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 2,55-2,67 (m, 1 H), 2,78-2,96 (m, 3 H), 3,91 (s, 3 H), 4,10 (d, J = 11,6 Hz, 1 H), 4,46 (d, J = 11,6 Hz, 1 H), 6,57 (d, J = 8,3 Hz, 1 H), 6,73 (s, 1 H), 7,13-7,18 (obs m, 1 H), 7,16 (s, 1 H), 7,52 (s, 1 H). EXEMPLO 12 A. ÉSTER DE METILA DE ÁCIDO 3-[3-(2-BROMOBENZIL)-3H- IMIDAZOL-4-IL1PROPIÔNICO
[00343] A uma solução de éster de metila de ácido 3-(1 -tritil-1 H- imidazol-4-il)propanóico (5,0 g, 12,6 mmols) em CH3CN (80 mL) é adicionada uma solução de 2-bromobenzilbrometo (2,84 g, 11,4 mmols) em CH3CN (20 mL). A solução clara resultante é agitada à temperatura ambiente durante 0 final de semana. A mistura é concentrada e 0 óleo restante é absorvido em 85 mL de metanol e aquecido para 70 °C por 2 h. Então a mistura é concentrada para fornecer um óleo amarelo, que é absorvido em EtOAc e lavada com NaHCOs aquoso saturado e salmoura, secada em Na2SO4 anidroso, e concentrada para produzir um óleo viscoso. A mistura de reação bruta é submetida à cromatografia instantânea (sílica-gel) eluindo com MeOH:CH2Cl2 para fornecer o composto desejado. MS (ESI) m/z 323 (M + H). B. 3-[3-(2-BROMOBENZIL)-3H-IMIDAZOL-4-IL]PROPAN-1-OL
[00344] A uma solução de éster de metila de ácido 3-[3-(2- bromobenzil)-3H-imidazol-4-il]propiônico (1,89 g, 5,85 mmols) em MeOH a 0 °C é adicionado NaBH4 e a mistura resultante é agitada a 0 °C por 1 h e aquecida para temperatura ambiente. A mistura é agitada à temperatura ambiente por 3 h e é finalizada com NH4CI aquoso saturado, e é adjustado 0 pH para 7 com Na2COs aquoso saturado. Então a mistura de reação é concentrada e extraída por CH2CI2. A camada orgânica é lavada com NaCI aquoso meio saturado, secado em Na2SO4 anidroso, e concentrada para produzir um sólido branco. MS (ESI) m/z 295 (M + H). C. 1-(2-BROMOBENZIL)-5-(3-CLOROPROPIL)-1H-IMIDAZOL
[00345] A uma solução de SOCI2 (0,55 mL, 7,54 mmols) em CH2CI2 (15 mL) a 0 °C é adicionado 3-[3-(2-bromobenzil)-3H-imidazol-4- il]propan-1-ol em porções e a suspensão branca resultante é refluxada durante 1,5 h. A mistura é resfriada com gelo e coletada para fornecer um sólido de cor de camurça, que é dividido entre CH2CI2 e NaHCOs aquoso saturado. A camada orgânica é lavada com salmoura, secada em Na2SO4 anidroso, e concentrada para fornecer um óleo. MS (ESI) m/z 313 (M + H). D.5-(2-BROMOFENIL)-5,6,7,8,-TETRAIDRO-IMIDAZO[1,5-A] PIRIDINA
[00346] A uma suspensão de 1-(2-bromobenzil)-5-(3-cloropropil)- 1H-imidazol (0,662 g, 2,12 mmols) e TMEDA (0,683 mL, 4,55 mmols) em THF (10 mL) a -78 °C é adicionado LDA (1,8 M em heptano/THF/etilbenzeno, 2,5 mL, 4,5 mmols) e a solução clara amarela é agitada a -78 °C. Após 3,5 h, a reação é finalizada por NH4CI aquoso saturado, e a mistura é dividida entre EtOAc e NaHCOs aquoso saturado. A camada orgânica é lavada com salmoura, secada e concentrada para fornecer um óleo, que é submetido a cromatografia instantânea (sílica-gel) eluindo com MeOH:CH2Cl2 para produzir 0 composto desejado. MS (ESI) m/z 277 (M + H).
[00347] Similarmente preparado é:
[00348] 5-(4-Bromofenil)-5,6,7,8-tetraidro-imidazo[1,5-a]piridina. MS (ESI) m/z 277 (M + H).
[00349] O seguinte composto é preparado de uma maneira similar usando LHMDS (1,0 M em THF) como base no lugar de LDA para a ciclização: 5-(2-Bromo-4-flúorfenil)-5,6,7,8-tetraidro-imidazo[1,5- a]piridina. MS (ESI) m/z 295 (M + H).
[00350] A partir do material descrito no exemplo 2J acima, o procedimento acima forneceu o material ciclizado: 5-(2-Bromofenil)-8- metil-5,6,7,8-tetraidro-imidazo[1,5-a]piridina. MS (ESI) m/z 291 (M + H).
EXEMPLO 13 A. 5-(2-TIOFEN-2-IL-FENIL)-5,6,7,8-TETRAIDRO-IMIDAZO[1,5-A] PIRIDINA
[00351] A uma solução de 5-(2-bromofenil)-5,6,7,8,-tetraidro- imidazo[1,5-a]piridina, exemplo 12D (120 mg, 0,43 mmols) em DME (2 mL) é adicionado ácido de tiofeno-2-borônico (166 mg, 1,3 mmols), Na2Cθ3 aquoso (2M, 1,0 mL, 2,0 mmols), e Pd(PPhs)4 (20 mg). A mistura de reação é refluxada durante a noite. A mistura é dividida entre EtOAc e 1M de NaOH. A camada orgânica é lavada por NaHCOs saturado, secada e concentrada para fornecer um óleo. A mistura de reação bruta é submetida a cromatografia instantânea (sílica-gel) eluindo com acetona:hexano para produzir o composto desejado. MS (ESI) m/z 280 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,58-1,74 (m, 1 H), 1,85-2,01 (m, 2 H), 2,14-2,27 (m, 1 H), 2,74-2,94 (m, 2 H), 5,45 (dd, J = 8,6, 5,1 Hz, 1 H), 6,81 (s, 1 H), 7,00-7,05 (m, 2 H), 7,06 (s, 1 H), 7,10 (dd, J = 5,3, 3,5 Hz, 1 H), 7,31-7,37 (m, 2 H), 7,39 (dd, J = 5,2, 1,1 Hz, 1 H), 7,41-7,46 (m, 1 H).
B. 5-(2-TIOFEN-3-IL-FENIL)-5,6,7,8-TETRAIDROIMIDAZO[1,5-A] PIRIDINA
[00352] Depois disso, de uma maneira similar, é também preparado o seguinte composto exceto para o procedimento de acoplamento Suzuki:
[00353] A uma solução de 5-(2-bromofenil)-5,6,7,8, - tetraidroimidazo[1,5-a]piridina (95 mg, 0,343 mmols) em DME (2 mL) é adicionado ácido de tiofeno-3-borônico (53 mg, 0,414 mmols), Na2CO3 aquoso (2M, 0,5 mL, 1,0 mmols), e Pd(PPh3)4 (20 mg). A mistura de reação é conduzida sob irradiação de microondas a 120 °C por 20 min. A mistura é dividida entre EtOAc e salmoura. A camada orgânica é lavada por salmoura, secada em Na2SO4 e concentrada para fornecer um óleo. A mistura de reação bruta é submetida a cromatografia instantânea eluindo com acetona:hexano para produzir o composto desejado. MS (ESI) m/z 280 (M + H).
[00354] Similarmente preparados são os seguintes: 1) 5-Bifenil-2-il-5,6,7,8-tetraidroimidazo[1,5-a]piridina. MS (ESI) m/z 275 (M + H).
2) 5-(2-Furan-2-il-fenil)-5,6,7,8-tetraidroimidazo[1,5-a] piridina. MS (ESI) m/z 264 (M + H).
[00355] Os seguintes compostos são preparados de uma maneira similar exceto usando resina de PS-PPh3-Pd(0) em lugar de Pd(PPhs)4 para a reação de acoplamento Suzuki: 3) 5-(4’-Flúorbifenil-2-il)-5,6,7,8-tetraidroimidazo[1,5-a] piridina. MS (ESI) m/z 293 (M + H); 1H RMN (400 MHz, MeOD) δ ppm 1,55-1,76 (m, 1 H), 1,86-2,07 (m, 2 H), 2,06-2,26 (m, 1 H), 2,68-3,01 (m, 2 H), 5,32 (dd, J = 8,6, 5,1 Hz, 1 H), 6,78 (s, 1 H), 6,99-7,09 (m, 1 H), 7,17-7,29 (m, 3 H), 7,29-7,38 (m, 1 H), 7,38-7,51 (m, 4 H).
[00356] Similarmente, seguindo o mesmo procedimento de acoplamento Suzuki é preparado: 4) 5-(4’-TrifIúormetiIbifenil-2-il)-5,6,7,8-tetraidroimidazo[ 1,5- a]piridina. MS (ESI) m/z 343 (M + H).
5) 5-(4’-Metoxi bifeni l-2-i l)-5,6,7,8-tetraidroimidazo[ 1,5-a] piridina. MS (ESI) m/z 305 (M + H).
6) 5-(2’-Clorobifenil-2-il)-5,6,7,8-tetraidroimidazo[1,5-a] piridina. MS (ESI) m/z 309, 311 (M + H).
7) 5-(3’-Clorobifenil-2-il)-5,6,7,8-tetraidroimidazo[1,5-a] piridina. MS (ESI) m/z 309, 311 (M + H); 1H RMN (400 MHz, MeOD) δ ppm 1,47-1,74 (m, 1 H), 1,84-2,03 (m, 2 H), 2,08-2,23 (m, 1 H), 2,71- 2,99 (m, 2 H), 5,30 (dd, J = 8,6, 5,3 Hz, 1 H), 6,78 (s, 1 H), 7,00-7,10 (m, 1 H), 7,24 (s, 1 H), 7,27-7,39 (m, 2 H), 7,41-7,47 (m, 3 H), 7,47- 7,53 (m, 2 H).
8) 5-(4’-Clorobifenil-2-il)-5,6,7,8-tetraidro-imidazo[1,5-a] piridina. MS (ESI) m/z 309, 311 (M + H); 1H RMN (400 MHz, MeOD) δ ppm 1,51-1,70 (m, 1 H), 1,81-2,01 (m, 2 H), 2,03-2,21 (m, 1 H), 2,66- 2,96 (m, 2 H), 5,29 (dd, J = 8,5, 5,2 Hz, 1 H), 6,75 (s, 1 H), 6,95-7,07 (m, 1 H), 7,19 (s, 1 H), 7,25-7,32 (m, 1 H), 7,34-7,44 (m, 4 H), 7,44- 7,54 (m, 2 H).
9) 5-(3’, 5’-Diclorobifenil-2-il)-5,6,7,8-tetraidroimidazo[1,5-a] piridina. MS (ESI) m/z 343, 345, 347 (M + H).
10) 5-(3’, 5’-D if lúorb ifen i l-2-i l)-5,6,7,8-tetraid roimidazo[ 1,5- ajpiridina. MS (ESI) m/z 311 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,49-1,70 (m, 1 H), 1,76-1,90 (m, 1 H), 1,90-2,00 (m, 1 H), 2,02-2,16 (m, 1 H), 2,68-2,83 (m, 1 H), 2,88 (dt, J = 16,2, 5,1 Hz, 1 H), 5,15 (dd, J = 9,1, 5,1 Hz, 1 H), 6,81 (s, 1 H), 6,82-6,91 (m, 3 H), 7,05 (s, 1 H), 7,06-7,10 (m, 1 H), 7,20-7,26 (m, 1 H), 7,32-7,41 (m, 2 H).
11) 5-(3’,5’-Dimetilbifenil-2-il)-5,6,7,8-tetraidroimidazo[1,5- a]piridina. MS (ESI) m/z 303 (M + H).
12) 5-(3’-C loro-4’-f lúorbifeni l-2-i l)-5,6,7,8-tetraidroi m idazo [1,5-a]piridina. MS (ESI) m/z 327,329 (M + H).
13) 5-(3’,4’-Diclorobifenil-2-il)-5,6,7,8-tetraidroimidazo[1,5-a] piridina.
[00357] MS (ESI) m/z 343, 345, 347 (M + H).
14) 5-(4’-1sopropi I bifeni l-2-i l)-5,6,7,8-tetraidroimidazo[ 1,5,-a] piridina.
[00358] MS (ESI) m/z 317 (M + H).
15) 5-(2’-F I úorbifeni l-2-i l)-5,6,7,8-tetraidroi m idazo[ 1,5-a] piridina.
[00359] MS (ESI) m/z 293 (M + H).
16) 5-(2’-Metoxi bifeni l-2-i l)-5,6,7,8-tetraidroi m idazo[ 1,5-a] piridina.
[00360] MS (ESI) m/z 305 (M + H).
17) /V,/V-Dimetil-[2’-(5,6,7,8-tetraidroimidazo[1,5-a] pi rid i n-5- il)-bifenil-4-il]amina. MS (ESI) m/z 318 (M + H).
18) 5-(2’-Trif lúormeti I bifen i l-2-i l)-5,6,7,8-tetraid roi m idazo [1,5-a]piridina. MS (ESI) m/z 343 (M + H).
19) 5-(2’-Meti I bifeni l-2-i l)-5,6,7,8-tetraidroimidazo[ 1,5-a] piridina. MS (ESI) m/z 289 (M + H).
20) 5-(2’-Etoxi bifeni l-2-i l)-5,6,7,8-tetraidroimidazo[ 1,5-a] piridina.
[00361] MS (ESI) m/z 319 (M + H).
21) 5-(2’-Metoximetil-bifenil-2-il)-5,6,7,8-tetraidroimidazo [1,5-a] pi rid i na. MS (ESI) m/z 319 (M + H).
[00362] Por analogia aos compostos descritos acima os seguintes são preparados a partir de 5-(2-Bromo-4-flúorfenil)-5,6,7,8-tetraidro- imidazo[1,5-a]piridina, fornecido como exemplo 12D: 1) 5-(5-F lúorb ifen i l-2-i l)-5,6,7,8-tetraid roimidazo[ 1,5- a]piridina. MS (ESI) m/z 293 (M + H). Resolução de enantiômeros (R) e (S) do composto do título é obtida por HPLC quiral usando coluna ChiralPak AD com uma fase móvel de IPA/hexanos para fornecer enantiômero A e enantiômero B. Por enantiômero B: 1H RMN (400 MHz, CDCh) δ ppm 1,42-1,67 (m, 1 H), 1,76-1,88 (m, 1 H), 1,88-1,97 (m, 1 H), 2,01-2,13 (m, 1 H), 2,65-2,80 (m, 1 H), 2,86 (dt, J = 15,9, 5,1 Hz, 1 H), 5,13 (dd, J = 9,5, 4,9 Hz, 1 H), 6,78 (s, 1 H), 6,97-7,02 (m, 1 H), 7,02-7,10 (m, 3 H), 7,28-7,36 (m, 2 H), 7,36-7,49 (m, 3 H).
2) 5-(5,4'-Dif lúorbifeni l-2-i l)-5,6,7,8-tetraidroimidazo[ 1,5- a]piridina. MS (ESI) m/z 311 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,33-1,60 (m, 1 H), 1,66-1,79 (m, 1 H), 1,79-1,91 (m, 1 H), 1,90-2,03 (m, 1 H), 2,60-2,73 (m, 1 H), 2,79 (dt, J = 16,2, 4,6 Hz, 1 H), 5,01 (dd, J = 9,6, 4,8 Hz, 1 H), 6,72 (s, 1 H), 6,89 (dt, J = 9,1, 1,5 Hz, 1 H), 6,92- 7,00 (m, 3 H), 7,01-7,13 (m, 2 H), 7,16-7,24 (m, 2 H).
3) 5-(5,2'-Diflúorbifenil-2-il)-5,6,7,8-tetraidroimidazo[1,5-a] piridina. MS (ESI) m/z 311 (M + H).
4) 5-(2'-Cloro-5-flúorbifenil-2-il)-5,6,7,8-tetraidroimidazo[1,5- ajpiridina. MS (ESI) m/z 327 (M + H).
5) 5-(4-flúor-2-tiofen-3-il-fenil)-5,6,7,8-tetraidroimidazo[1,5- a]piridina. MS (ESI) m/z 299 (M + H): 1H RMN (400 MHz, CDCh) δ ppm 1,58-1,68 (m, 1 H), 1,74-2,02 (m, 2 H), 2,05-2,22 (m, 1 H), 2,67-2,82 (m, 1 H), 2,88 (dt, J = 16,2, 4,8 Hz, 1 H), 5,24 (dd, J = 9,2, 4,9 Hz, 1 H), 6,80 (s, 1 H), 6,99-7,06 (m, 4 H), 7,08 (dd, J = 4,9, 1,1 Hz, 1 H), 7,21 (dd, J = 2,9, 1,1 Hz, 1 H), 7,43 (dd, J = 4,8, 3,0 Hz, 1 H).
[00363] O seguinte composto é preparado de uma maneira similar usando o procedimento de acoplamento Suzuki modificado:
[00364] A uma solução de 5-(2-bromofenil)-5,6,7,8, - tetraidroimidazo[1,5-a]piridina (89 mg, 0,321 mmols) em dioxano (2 mL) é adicionado éster cíclico de 1,3-propanodiol de ácido de piridina- 3-borônico (105 mg, 0,644 mmols), K3PO4 (105 mg, 0,707 mmols), e resina de PS-PPh3Pd (0) (0,13 mmols/g, 100 mg). A mistura de reação é realizada em um microondas a 130 °C por 20 min. A mistura é dividida entre EtOAc e salmoura. A camada orgânica é lavada com salmoura, secada em Na2SO4 e concentrada para fornecer um óleo. A mistura de reação bruta é submetida a cromatografia instantânea (sílica-gel) eluindo com MeOH:CH2Cl2 para produzir o composto desejado. MS (ESI) m/z 275 (M + H).
[00365] Similarmente preparados são os seguintes: 1) 5-(2-Piridin-4-il-fenil)-5,6,7,8-tetraidroimidazo[1,5-a] piridina. MS (ESI) m/z 275 (M + H)
2) 2’-(5,6,7,8-Tetraidroimidazo[1,5-a]piridina-5-il)bifenil-2- carbonitrila. MS (ESI) m/z 300 (M + H).
[00366] Resolução de enantiômeros do composto do título é obtida por HPLC quiral usando uma coluna ChiralPak AD com uma fase móvel de 25% de IPA/Hexano para fornecer enantiômero A (tr = 5,3 min) e enantiômero B (tr = 8,1 min). Por enantiômero B: 1H RMN (400 MHz, DMSO-cfe) δ ppm 1,34-1,59 (m, 1 H), 1,64-1,87 (m, 2 H), 1,90- 2,12 (m, 1 H), 2,72 (t, J = 6,2 Hz, 2 H), 5,23 (dd, J = 8,0, 5,2 Hz, 1 H), 6,65 (s, 1 H), 6,81-6,95 (m, 1 H), 7,12 (s, 1 H), 7,20-7,28 (m, 1 H), 7,34- 7,40 (m, 2 H), 7,40-7,44 (m, 3 H), 7,44-7,52 (m, 2 H).
[00367] Similiarmente resolvidos são os seguintes: 1) (R) e (S)-5-(2’-Clorobifenil-2-il)-5,6,7,8- tetraidroimidazo[1,5-a]pirid ina
[00368] Coluna ChiralPak AD com uma fase móvel de 20% de IPA/Hexano para fornecer enantiômero A (tr = 7,0 min) e enantiômero B (tr = 8,8 min). 2) (/?) e (S)-5-(2’-Triflúormetilbifenil-2-il)-5,6,7,8- tetraidroimidazo[1,5-a]pirid ina
[00369] Coluna ChiralPak AS com uma fase móvel de 10% de IPA/Hexano para fornecer enantiômero A (tr = 15 min) e enantiômero B (tr = 21 min). Por enantiômero B: 1H RMN (400 MHz, CDCh) δ ppm 1,45-1,54 (m, 1 H), 1,78-1,90 (m, 1 H), 1,90-1,98 (m, 1 H), 1,99-2,09 (m, 1 H), 2,67-2,81 (m, 1 H), 2,81-2,92 (m, 1 H), 4,74 (dd, J= 10,1,4,5 Hz, 1 H), 6,78 (s, 1 H), 7,08 (s, 1 H), 7,16 (d, J = 7,6 Hz, 1 H), 7,20-7,28 (m, 1 H), 7,30-7,43 (m, 3 H), 7,52 (t, J = 7,6 Hz, 1 H), 7,58 (t, J = 7,1 Hz, 1 H), 7,80 (d, J = 7,6 Hz, 1 H). E. 5-(2-CICLOPROPILFENIL)-5,6,7,8-TETRAIDROIMIDAZO[1,5-A] PIRIDINA
[00370] A partir de 5-(2-bromofenil)-5,6,7,8,-tetraidro-imidazo[1,5- a]piridina, exemplo 12D, é convertido o composto do título de acordo com o método descrito em Tet. Lett. 2002, 43, 6987. MS (ESI) m/z 239 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 0,78 (br s, 2 H), 1,02 (br s, 2 H), 1,79 (br s, 1 H), 2,00 (br s, 3 H), 2,31 (br s, 1 H), 2,84-2,97 (m, 2 H), 5,83 (dd, J = 7,6, 5,3 Hz, 1 H), 6,79 (d, J = 7,6 Hz, 1 H), 6,85 (s, 1 H), 7,09-7,13 (m, 2 H), 7,15 (t, J = 7,3 Hz, 1 H), 7,23 (t, J = 7,3 Hz, 1 H).
[00371] Similarmente preparado a partir de 5-(4-Bromofenil)-5,6,7,8- tetraidro-imidazo[1,5-a]pirid ina é: 5-(4-Ciclopropi Ifeni l)-5,6,7,8- tetraidroimidazo[1,5-a]piridina. MS (ESI) m/z 239 (M + H); 1H RMN (400 MHz, MeOD) δ ppm 0,68 (br s, 2 H), 0,96 (br s, 2 H), 1,71-1,83 (m, 1 H), 1,87-2,04 (m, 3 H), 2,22-2,33 (m, 1 H), 2,81-2,94 (m, 2 H), 5,26 (dd, J = 7,8, 5,1 Hz, 1 H), 6,74 (s, 1 H), 7,00 (d, J = 8,3 Hz, 2 H), 7,09 (d, J = 8,3 Hz, 2 H), 7,14 (br s, 1 H). F. 5-BIFENIL-2-IL-8-METIL-5,6,7,8-TETRAIDRO-IMIDAZO[1,5-A] PIRIDINA
[00372] A uma solução de 5-(2-bromofenil)-8-metil-5,6,7,8-tetraidro- imidazo[1,5-a]piridina (573 mg, 1,97 mmols) em DME (10 mL) é adicionado ácido fenilborônico (480 mg, 3,94 mmols), Na2CO3 aquoso (2M, 4 mL, 8,0 mmols), e Pd(PPh3)4 (500 mg). A mistura de reação é agitada até o refluxo durante a noite. A mistura é dividida entre EtOAc e salmoura. A camada orgânica é lavada por salmoura, secada em Na2SO4 e concentrada para fornecer um óleo, que é submetido à cromatografia instantânea (sílica-gel) eluindo com MeOH:CH2Cl2 para produzir o composto desejado. MS (ESI) m/z 289 (M + H)
[00373] Resolução de éstereoisômeros do composto do título é obtida por HPLC quiral usando uma coluna ChiralPak AD com uma fase móvel de 15% de IPA/Hexano para fornecer isômero A (tr = 7,8 min), isômero B (tr = 9,9 min), isômero C (tr = 17 min) e isômero D (tr = 24 min). Por isômero B: 1H RMN (400 MHz, CDCh) δ ppm 1,35 (d, J = 6,8 Hz, 3 H), 1,45-1,58 (m, 1 H), 1,63-1,74 (m, 1 H), 1,84-2,00 (m, 2 H), 2,88-3,00 (m, 1 H), 5,41 (t, J = 5,4 Hz, 1 H), 6,68 (d, J = 7,3 Hz, 1 H), 6,86 (s, 1 H), 7,15 (s, 1 H), 7,20-7,51 (m, 8 H). EXEMPLO 14 A.6-[5-(3-HIDROXIPROPIL)-IMIDAZOL-1-ILMETIL]BIFENIL-3- CARBO-NITRILA
[00374] A uma solução de 3-bromo-4-[5-(3-hidroxipropil)-imidazol-1- ilmetil]benzonitrila (1,02 g, 3,2 mmols) que é preparada por alquilação de imidazol a partir do exemplo 2I com brometo descrito no exemplo 1C seguida por remoção do grupo de proteção de silila por analogia a primeira etapa do exemplo 3C em DME (10 mL) é adicionado ácido fenilborônico (586 mg, 4,8 mmols), Na2COs aquoso (2M, 4,8 mL, 9,6 mmols), e resina de PS-PPh3-Pd (0) (0,13 mmols/g, 1 g). A mistura de reação é mantida em um microondas a 130 °C por 20 min. A mistura é dividida entre EtOAc e salmoura. A camada orgânica é lavada com salmoura, secada em Na2SO4 e concentrada para fornecer óleo. B.6-[5-(3-CLOROPROPIL)-IMIDAZOL-1-ILMETIL]BIFENIL-3- CARBONI-TRILA
[00375] A uma solução de SOCh (0,382 mL, 5,24 mmols) em CH2CI2 (20 mL) a 0 °C é adicionado 6-[5-(3-hidroxipropil)imidazol-1- ilmetil]bifenil-3-carbonitrila em porções e a suspensão branca resultante é refluxada por 1,5 h. A mistura é resfriada em gelo e dividida entre CH2CI2 e NaHCOs aquoso saturado. A camada orgânica é lavada com salmoura, secada em Na2SO4 anidroso e concentrada para fornecer uma espuma oleosa. C. 6-(5,6,7,8-TETRAIDROIMIDAZO[1,5-A]PIRIDIN-5-IL)-BIFENIL-3- CARBO-NITRILA
[00376] A uma solução de 6-[5-(3-cloropropil)imidazol-1- ilmetil]bifenil-3-carbonitrila (1,0 g, 3,0 mmols) em THF (16 mL) a 0 °C é adicionado t-BuOK/THF (1,0 M, 6 mL, 6,0 mmols). A mistura é aquecida até à temperatura ambiente e agitada por 2 h. A mistura de reação é finalizada por NH4CI aquoso saturado e dividida entre CH2CI2 e salmoura. A camada orgânica é lavada por salmoura, secada em Na2SO4 anidroso e concentrada para fornecer 0 óleo bruto, que é submetido a cromatografia instantânea (sílica-gel) eluindo com MeOH:CH2Cl2 para produzir 0 composto desejado. MS (ESI) m/z 300 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,50-1,66 (m, 1 H), 1,73- 1,83 (m, 1 H), 1,83-1,95 (m, 1 H), 1,95-2,16 (m, 1 H), 2,68-2,95 (m, 2 H), 5,28 (dd, J = 8,5, 5,2 Hz, 1 H), 6,83 (s, 1 H), 7,01-7,15 (m, 2 H), 7,27-7,35 (m, 2 H), 7,43-7,54 (m, 3 H), 7,58 (s, 1 H), 7,62 (d, J = 8,3 Hz, 1 H). Resolução de enantiômeros do composto do título é obtida por HPLC quiral usando a coluna ChiralPak AD com uma fase móvel de 20% de IPA/Hexano. EXEMPLO 15 A. 3-BROMO-4-(5,6,7,8-TETRAIDRO-IMIDAZO[1.5-A1PIRIDIN-5-IL) BENZO-NITRILA

[00377] 4-{5-[3-Hidroxipropil]imidazol-1-ilmetil}-3-bromobenzonitrila é convertido no cloreto correspondente de acordo com 0 exemplo 14B. O cloreto resultante é tratado com uma base, tal como t-BuOK como no exemplo 14C acima, para realizar ciclização para 0 composto do título. MS (ESI) m/z 302,1, 304,1 (M + H); 1H RMN (400 MHz, MeOD) δ ppm 1,74 - 1,85 (m, 2 H), 2,08 - 2,17 (m, 1 H), 2,34 - 2,43 (m, 1 H), 2,82 - 2,91 (m, 1 H), 2,93 - 3,00 (m, 1 H), 5,88 (t, J = 5,6 Hz, 1 H), 6,75 (d, J = 8,1 Hz, 1 H), 6,82 (d, J = 1,0 Hz, 1 H), 7,34 (s, 1 H), 7,69 (dd, J = 8,1, 1,5 Hz, 1 H), 8,09 (d, J = 1,5 Hz, 1 H).
[00378] Similarmente preparados são os seguintes: 1) 3-flúor-4-(5,6,7,8-tetraidro-imidazo[1,5-a] pi rid i n-5- il)benzonitrila MS (ESI) m/z 242,1 (M + H); 1H RMN (400 MHz, MeOD) (sal de HCI) δ ppm 1,88-1,98 (m, 1 H), 2,02-2,11 (m, 1 H), 2,14-2,24 (m, 1 H), 2,39-2,47 (m, 1 H), 2,92-3,00 (m, 1 H), 3,02-3,09 (m, 1 H), 5,83 (dd, J = 9,1, 5,3 Hz, 1 H), 7,37-7,39 (m, 1 H), 7,42 (dd, J = 10,4, 8,8 Hz, 1 H), 7,67 (dd, J = 6,8, 2,0 Hz, 1 H), 7,86-7,90 (m, 1 H), 8,65 (s, 1 H). Resolução de enantiômeros (R) e (S) do composto do título é obtido por HPLC quiral usando coluna ChiralPak AS-H e 30 IPA:hexano para fornecer enantiômero A (tr = 24 min) e enantiômero B (tr = 30 min).

2) 3-Cloro-4-(5,6,7,8-tetraidro-imidazo[1,5-a]piridin-5- il)benzonitrila MS (ESI) m/z 258,1, 260,1 (M + H); 1H RMN (400 MHz, MeOD) δ ppm 1,74-1,85 (m, 2 H), 2,07-2,17 (m, 1 H), 2,33-2,44 (m, 1 H), 2,81-2,91 (m, 1 H), 2,91-3,00 (m, 1 H), 5,92 (d, J = 5,3 Hz, 1 H), 6,76 (dd, J = 8,1 Hz, 1 H), 6,82 (s, 1 H), 7,35 (s, 1 H), 7,65 (dd, J = 8,1, 1,5 Hz, 1 H), 7,89-7,95 (m, 1 H).
3) 3-Metóxi-4-(5,6,7,8-tetraidro-imidazo[1,5-a]pirid in-5- il)benzonitrila MS (ESI) m/z 254,1 (M + H); 1H RMN (400 MHz, MeOD) (sal de HCI) δ ppm 1,85-1,95 (m, 1 H), 1,95-2,04 (m, 1 H), 2,18-2,28 (m, 1 H), 2,31-2,41 (m, 1 H), 2,99 (s, 2 H), 3,88-3,94 (m, 3 H), 5,85- 5,92 (m, 1 H), 7,09 (d, J = 7,8 Hz, 1 H), 7,32-7,41 (m, 2 H), 7,48 (s, 1 H), 8,53 (s, 1 H).
B. 3-METIL-4-(5,6,7,8-TETRAIDRO-IMIDAZO[1.5-A1PIRIDIN-5-IL) BENZO-NITRILA
[00379] 3-Bromo-4-(5,6,7,8-tetraidro-imidazo[1,5-a]pirid in-5- il)benzonitrila, preparado acima, é convertido para o tolueno correspondente pelo método descrito em Tet. Lett. 2000, 41, 6237. MS (ESI) m/z 238,1 (M + H); 1H RMN (400 MHz, MeOD) δ ppm 1,77-1,89 (m, 2 H), 1,91-2,00 (m, 1 H), 2,29-2,39 (m, 1 H), 2,45 (s, 3 H), 2,86- 2,93 (m, 2 H), 5,71 (t, J = 6,1 Hz, 1 H), 6,77 (d, J = 8,1 Hz, 1 H), 6,81 (s, 1 H), 7,29 (s, 1 H), 7,49 (d, J = 8,1 Hz, 1 H), 7,61 (s, 1 H). C. 3-CICLOPROPIL-4-(5,6,7,8-TETRAIDRO-IMIDAZO[1,5-AlPIRIDIN- 5-IDBENZONITRILA
[00380] 3-Bromo-4-(5,6,7,8-tetraidro-imidazo[1,5-a]pirid in-5- il)benzonitrila, preparado acima, é convertido para o ciclopropano correspondente pelo método descrito em Tet. Lett. 2002, 43, 6987. MS (ESI) m/z 264,1 (M + H); 1H RMN (400 MHz, MeOD) δ ppm 0,69-0,80 (m, 2 H), 0,94-1,06 (m, 2 H), 1,69-1,80 (m, 2 H), 1,93-2,04 (m, 2 H), 2,24-2,34 (m, 1 H), 2,76-2,86 (m, 2 H), 5,95 (d, J = 6,1 Hz, 1 H), 6,64- 6,72 (m, 2 H), 7,15 (s, 1 H), 7,35 (s, 1 H), 7,39 (d, J = 8,1 Hz, 1 H).
EXEMPLO 17
ÉSTER DE METILA DE ÁCIDO 5-(4-CIANO-FENIL)-5,6,7,8- TETRAIDRO-IMIDAZOri,5-AlPIRIDINA-5-CARBOXÍLICO.
[00381] 1H RMN (400 MHz, CDCh) δ ppm 1,67-1,77 (m, 2 H), 2,10- 2,17 (m, 1 H), 2,78-2,88 (m, 3 H), 3,83 (s, 3 H), 6,87 (s, 1 H), 7,11 (d, J = 8,3 Hz, 2 H), 7,46 (s, 1 H), 7,64 (d, J = 8,3 Hz, 2 H).
EXEMPLO 18 4-((R)-1-BROMO-5,6,7,8-TETRAIDROIMIDAZO[1,5-A]PIRIDIN-5- IDBENZONITRILA
[00382] A (R)-4-(5,6,7,8-Tetraidroimidazo[1,5-a]iridine-5-il) benzonitrila (1,2 g, 5,4 mmols) em acetonitrila (50 mL) a 0 °C é adicionado NBS ( 0,96 g, 5,4 mmols). A reação é agitada até completo consumo do material de partida (cerca de 2 h) antes da concentração sob pressão reduzida. O resíduo é dividido entre CH2CI2 e salmoura. A fase orgânica separada é lavada com salmoura fresca (2x), secada (Na2SO4), e concentrada sob pressão reduzida. O resíduo é purificado por cromatografia instantânea eluindo com 2,5% MeOH em CH2CI2. A espuma resultante é diluída com CH2CI2 e HCI (g) é borbulhado através, durante 10 min. O produto é isolado por concentração sob pressão reduzida. 1H RMN (400 MHz, MeOD) (sal de HCI) δ ppm 1,88- 1,99 (m, 1 H), 2,01-2,17 (m, 2 H), 2,34-2,50 (m, 1 H), 2,80-2,95 (m, 2 H), 5,64 (dd, J = 8,6, 5,1 Hz, 1 H), 7,50 (d, J = 8,3 Hz, 2 H), 7,80 (d, J = 8,3 Hz, 2 H), 8,54 (s, 1 H).
EXEMPLO 18
A. 3-[3-(2-BROMOBENZIL)-3H-IMIDAZOL-4-IL]-3-METILBUTAN-1-OL
[00383] A uma solução de 4-[3-(terc-butildimetilsilanilóxi)-1,1- dimetilpropil]-1-tritil-1H-imidazol (650 mg, 1,27 mmols) em CH3CN (10 mL) à temperatura ambiente é adicionada a solução de brometo de 2- bromobenzila (420 mg, 1,68 mmols) em CH3CN (10 mL). A solução resultante é agitada a 80 °C por 6 h. A mistura é resfriada para temperatura ambiente e dietilamina (2 mL) é adicionada, seguida por MeOH (10 mL) e a mistura é aquecida a 75 °C por 1 h e agitada à temperatura ambiente durante a noite. À mistura de reação é adicionado HCI/Dioxano (4 M, 5 mL, 20 mmols) e agitada à temperatura ambiente for 1 h. A mistura é neutralizada com NaHCOs aquoso saturado e concentrada. O resíduo é dividido entre CH2CI2 e salmoura. A camada orgânica é lavada com salmoura, secada em Na2SO4 e concentrada para fornecer um óleo, que é submetido à cromatografia instantânea (sílica-gel) eluindo com MeOH:CH2Cl2 para produzir 0 composto desejado. MS (ESI) m/z 323 (M + H). B. 1-(2-BROMOBENZIL)-5-(3-CLORO-1,1-DIMETILPROPIL)-1H- IMIDAZOL

[00384] Por analogia ao exemplo 14B, 0 composto do título é preparado a partir do álcool 18A. MS (ESI) m/z 341 (M + H). C. 5-(2-BROMOFENIL)-8,8-DIMETIL-5,6,7,8-TETRAIDRO-IMIDAZO [1,5-AlPIRIDINA
[00385] O composto 16B é ciclizado de acordo com 0 procedimento descrito em 14C. MS (ESI) m/z 305 (M + H). D. 5-BIFENIL-2-IL-8,8-DIMETIL-5,6,7,8-TETRAIDROIMIDAZO[1,5-A] PIRI-DINA
[00386] O composto do título é preparado a partir do brometo 16C pelo método descrito no exemplo 13A. MS (ESI) m/z 303 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,40 (s, 3 H), 1,44 (s, 3 H), 1,59-1,74 (m, 1 H), 1,76-1,92 (m, 1 H), 2,12-2,32 (m, 2 H), 5,19-5,35 (m, 1 H), 6,94-7,05 (m, 1 H), 7,22 (s, 1 H), 7,29-7,34 (m, 2 H), 7,34-7,40 (m, 1 H), 7,40-7,52 (m, 5 H), 7,89 (s, 1 H). EXEMPLO 19 A. 6-(5-METIL-5,6,7,8-TETRAIDROIMIDAZO[1.5-A1PIRIDIN-5-IL) BIFENIL-3-CARBONITRILA
[00387] A uma solução de 6-(5,6,7,8-tetraidroimidazo[1,5-a]piridin-5- il)bifenil-3-carbonitrila (53 mg, 0,177 mmols) em THF (2 mL) a -40 °C é adicionado LHMDS (1,0 M em THF, 0,27 mL, 0,27 mmols). A solução marrom resultante é agitada a -30 °C por 0,5 h. A uma mistura de reação é adicionado iodeto de metila (0,017 mL, 0,272 mmols) e a mistura é aquecida até à temperatura ambiente e agitada durante a noite. A mistura de reação é finalizada por NH4CI aquoso saturado e a reação é dividida entre CH2CI2 e salmoura. A camada orgânica é lavada com salmoura, secada em Na2SO4 anidroso e concentrada para fornecer 0 óleo bruto, 0 qual é submetido a cromatografia instantânea (sílica-gel) eluindo com MeOH:CH2Cl2 para produzir 0 composto desejado. MS (ESI) m/z 314 (M + H).
[00388] Similarmente preparado é:
[00389] 6-(5-alil-5,6,7,8-tetraidroimidazo[1,5-a]piridina-5-il)bifenil-3- carbonitrila.
[00390] 6-(5-alil-5,6,7,8-tetraidroimidazo[1,5-a]piridina-5-il)bifenil-3- carbonitrila é preparado a partir de 6-(5,6,7,8-tetraidroimidazo[1,5- a]piridin-5-il)bifenil-3-carbonitrila e brometo de alila por analogia a 19A acima. MS (ESI) m/z 340 (M + H); 1H RMN (400 MHz, CDCh) δ ppm (sal de HCI) 1,73-1,98 (m, 2 H), 2,02-2,16 (m, 1 H), 2,23-2,38 (m, 1 H), 2,54-2,65 (m, 1 H), 2,68-2,83 (m, 1 H), 2,86-3,02 (m, 1 H), 3,00-3,14 (m, 1 H), 5,11 (d, J = 17,2 Hz, 1 H), 5,23 (d, J = 10,1 Hz, 1 H), 5,44- 5,69 (m, 1 H), 6,87 (d, J = 7,3 Hz, 1 H), 6,92 (s, 1 H), 7,02-7,12 (m, 1 H), 7,13-7,22 (m, 1 H), 7,27-7,39 (m, 3 H), 7,45 (d, J = 2,0 Hz, 1 H), 7,71 (dd, J = 8,5, 1,9 Hz, 1 H), 8,31 (s, 1 H). B. 6-(5-/V-PROPIL-5,6,7,8-TETRAIDRO-IMIDAZO[1,5-A]PIRIDINA-5-IL) BI-FENIL-3-CARBONITRILA
[00391] A uma solução de 6-(5-alil-5,6,7,8-tetraidroimidazo[1,5- a]piridina-5-il)bifenil-3-carbonitrila (35 mg, 0,103 mmols) em EtOH (2 mL) à temperatura ambiente são adicionados H2NNH2/THF (1,0 M, 15 mL, 15 mmols) e CUSO4 aquoso saturado (0,1 mL). A mistura de reação é agitada à temperatura ambiente durante a noite. A mistura é concentrada e dissolvida em EtOAc, que é lavada com água, secada em Na2SO4 anidroso e concentrada para fornecer 0 óleo bruto, que é submetido a cromatografia instantânea (sílica-gel) eluindo com MeOH:CH2Cl2 para produzir 0 composto desejado. MS (ESI) m/z 342 (M + H); 1H RMN (400 MHz, CDCh) δ ppm (sal de HCI) 0,95 (t, J = 7,2 Hz, 3 H), 1,21-1,33 (m, 2 H), 1,80-1,90 (m, 2 H), 1,90-2,02 (m, 1 H), 2,09-2,37 (m, 3 H), 2,59-2,68 (m, 1 H), 2,71-2,85 (m, 1 H), 6,81-6,88 (m, 1 H), 6,90-6,98 (m, 2 H), 7,19-7,25 (m, 1 H), 7,27-7,39 (m, 3 H), 7,43 (d, J= 1,8 Hz, 1 H), 7,71 (dd, J = 8,3, 2,0 Hz, 1 H), 8,10-8,18 (m, 1 H). EXEMPLO 20 CIS- E 77?A/VS-3-FLÚOR-4-[7-(4-FLÚORBENZIL)-5,6,7,8- TETRAIDROIMI-DAZOÍ1,5-A]PIRIDIN-5-IL]BENZONITRILA

[00392] O éster obtido no exemplo 2G (5,0g, 12,6 mmols) é secado azeotropicamente com tolueno e então dissolvidos em THF (20 mL). Esta solução é adicionada em gotas a uma solução de LHMDS (1M em hexanos, 15,7 mL, 15,7 mmols) em THF (20 mL) a -75 °C (banho de gelo seco-acetona). Após 10 min, brometo de 4-flúorbenzila (3,57 g, 18,9 mmols) é adicionado em gotas. A mistura é agitada a -75 °C por 4 h, sobre a qual ácido acético aquoso a 10% é adicionado e o banho de resfriamento é removido. Após extração com acetato de etila, a fase orgânica é secada sobre Na2SO4 e concentrada em vácuo. Purificação por cromatografia de sílica-gel (acetato de etila-hexanos, 85:15) fornecido o produto como um óleo.
[00393] O éster obtido acima (2,15 g, 4,26 mmols) é dissolvido em THF (15 mL) e resfriado para -20 °C. LiAIH4 (1M em THF, 10,7 mL, 10,7 mmols) é adicionado e o banho de resfriamento é removido. Após 1 h à temperatura ambiente, NaHCOs aquoso saturado é cuidadosamente adicionado. Após extração com acetato de etila, a fase orgânica é secada sobre Na2SO4 e concentrada em vácuo para fornecer o produto como óleo.
[00394] O álcool obtido acima (3,6 g, 7,56 mmols), TBSCI (1,25 g, 8,31 mmols), DMAP (0,092 g, 0,756 mmols) e imidazol (1,54 g, 22,6 mmols) são dissolvido em DMF (10 mL) e aquecido a 75 °C por 4 h. Após diluir com acetato de etila, a mistura é lavada três vezes com água. A fase orgânica é secada sobre Na2SO4 e concentrada a vácuo para fornecer o produto como um óleo.
[00395] O éster de silila obtido acima (2,0 g, 3,38 mmols) e brometo de 2-flúor-4-cianobenzila (0,94 g, 4,40 mmols) são dissolvidos em acetonitrila (10 mL) e refluxada durante a noite. Após resfriamento à temperatura ambiente, metanol (5 mL) é adicionado e a mistura é refluxada por 3 h, após o que, os voláteis são removidos a vácuo. O resíduo é purificado por cromatografia de sílica-gel (diclorometano- metanol, 19:1) para fornecer o produto como um óleo.
[00396] O álcool obtido acima (0,65 g, 1,70 mmols) é dissolvido em tetracloreto de carbono (5 mL) e cloreto de tionila (0,51 g, 4,29 mmols) é adicionado. A mistura é refluxada por 1,5 h, resfriada à temperatura ambiente e finalizada com NaHCOs aquoso saturado. A fase aquosa é extraída com diclorometano. A fase orgânica combinada é secada em Na2SO4 e concentrada em vácuo. Purificação por cromatografia de sílica-gel (diclorometano-metanol, 19:1) forneceu o produto como um óleo.
[00397] O cloreto obtido acima (0,46 g, 1,20 mmols) é dissolvido em THF (45 mL) e resfriado para 0 °C. Potássio terc-butóxido (1,0M em THF, 4,8 mL, 4,8 mmols) é adicionado em gotas e após mais 30 min, ácido acético aquoso a 10% é adicionado. Após extração com acetato de etila, a fase orgânica é secada sobre Na2SO4 e concentrada em vácuo. Purificação por cromatografia de sílica-gel (diclorometano- metanol, 9:1) forneceu o isômero eis limpo e uma mistura de isômeros cis e trans. O isômero trans limpo é obtido por purificação de HPLC preparativa da mistura. MS (ESI) m/z 350 (M + H); Para isômero cis, 1H RMN (400 MHz, MeOD) δ ppm 1,64-1,73 (m, 1 H), 2,05 - 2,20 (m, 2 H), 2,35 (dd, J = 15,0, 12,8 Hz, 1 H), 2,65 (d, J = 6,8 Hz, 2 H), 2,81 - 2,88 (m, 1 H), 5,40 (dd, J = 11,5, 4,9 Hz, 1 H), 6,61 (s, 1 H), 6,87 - 6,96 (m, 2 H), 7,05 (s, 1 H), 7,09 - 7,18 (m, 2 H), 7,35 (t, J = 7,6 Hz, 1 H), 7,47 - 7,57 (m, 2 H); Para isômero trans, 1H RMN (400 MHz, MeOD) δ ppm 1,85-2,13 (m, 3 H), 2,39 (dd, J = 16,0, 11,0 Hz, 1 H), 2,49-2,61 (m, 2 H), 2,87 (dd, J = 16,0, 4,7 Hz, 1 H), 5,90 (dd, J = 5,2, 3,4 Hz, 1 H), 6,40 (t, J = 7,8 Hz, 1 H), 6,71 (s, 1 H), 6,81-6,90 (m, 2 H), 6,95-7,01 (m, 2 H), 7,38 (dd, J = 8,1, 1,3 Hz, 1 H), 7,40 (s, 1 H), 7,47-7,50 (d, J = 10,2, 1,3 Hz, 1 H).
[00398] Similarmente preparado é o seguinte:
[00399] trans-3-Metóxi-4-[7-(4-flúorbenzil)-5,6,7,8-tetraidroimidazo [1,5-a]piridin-5-il]benzonitrila. MS (ESI) m/z 362 (M + H); 1H RMN (400 MHz, CDCh) (sal de TFA) δ ppm 1,88-2,12 (m, 3 H), 2,40 (dd, J = 16,0, 9,6 Hz, 1 H), 2,54 (dd, J = 13,6, 7,6 Hz, 1 H), 2,63 (dd, J = 13,6, 6,1 Hz, 1 H), 2,89 (dd, J = 16,0, 4,4 Hz, 1 H), 3,89 (s, 3 H), 5,80 (dd, J = 5,3, 3,0 Hz, 1 H), 6,26 (d, J = 7,8 Hz, 1 H), 6,82 (s, 1 H), 6,88-7,04 (m, 4 H), 7,09-7,16 (m, 2 H), 7,22 (s, 1 H).
EXEMPLO 21 A. 3-BROMO-4-{4-[4-/TE/?C-BUTILDIMETILSILANIL0XI) BUTIL1IMIDAZOL- 1-ILMETILIBENZONITRILA
[00400] A 4-[4-fferc-butildimetilsilanilóxi)butil]-1 -tritil-1 H-imidazol (3,95 g, 7,95 mmols) é adicionado acetonitrila (300 mL). A esta solução é adicionado 3-bromo-4-bromometilbenzonitrila (2,14 g, 7,78 mmols). A solução é agitada a 40 °C por 18 h. O solvente é então removido em vácuo e metanol (300 ml) é adicionado. A solução é aquecida para 55 °C e agitada por 1,5 h. Bicarbonato de sódio saturado é então adicionado e agitada for 10 min. O solvente orgânico é removido em vácuo, e o produto bruto extraído em acetato de etila e lavado com água. O solvente orgânico é removido em vácuo para fornecer o produto bruto. Cromatografia (sílica-gel, 1:0 a 1:1 a 0:1 hexanos:acetato de etila) fornece o produto puro. MS (ESI) m/z 448, 450 (M + H).
[00401] Seguindo este protocolo são também preparados: 1) 3-Cloro-4-{4-[4-(íerc-butildimetilsilanilóxi)butil]-imidazol-1 - ilmetiljbenzonitrila. MS (ESI) m/z 404, 406 (M + H).
2) 3-Metóxi-4-{4-[4-(íerc-butildimetilsilanilóxi)butil]-imidazol- 1-ilmetiljbenzonitrila. MS (ESI) m/z 400 (M + H).
3) 3-flúor-4-{4-[4-(íerc-butildimetilsilanilóxi)butil]-imidazol-1 - ilmetiljbenzonitrila. MS (ESI) m/z 388 (M + H).
4) 2-Bromo-4-{5-[4-ferc-butildimetilsilanilóxi)butil]-imidazol- 1-ilmetiljbenzonitrila. MS (ESI) m/z 448,450 (M + H).
5) 1-(2-Bromo-4-Flúorbenzil)-5-[4-ferc-butildimetilsilanilóxi) butil]-1 H-imidazol.
MS (ESI) m/z 441,443 (M + H).
6) 1 -2-Bromo-benzil-5-[4-(terc-butildimetilsilanilóxi)-butil]- 1H-imidazol. MS (ESI) m/z 423, 425 (M + H) .
7) 3-Bromo-4-{5-(4-íerc-butildimetilsilanilóxi)-1- metilbutilimidazol-1 -ilmetil-] benzonitrila. MS (ESI) m/z 462, 464 (M + H).
8) Éster de metila de ácido 3-Bromo-4-{5-[4-(ierc- butildimetilsilanilóxi)butil]-imidazol-1 -ilmetilj-benzóico. MS (ESI) m/z 481,484 (M + H).
EXEMPLO 22 3-BROMO-4-[5-(4-CLOROBUTIL)-IMIDAZOL-1-ILMETIL] BENZONITRILA
[00402] A 3-bromo-4-{4-[4-ferc-butildimetilsilaniloxibutil]-imidazol-1- ilmetilj-benzonitrila (1,09 g, 2,4 mmols) é adicionado metanol (100 ml). A esta solução é adicionado cloreto de hidrogênio, como uma solução a 4 M em dioxano (1,0 ml, 4,0 mmols). A solução é agitada 30 minutos. O solvente é então removido em vácuo para fornecer o produto como sal de cloreto de hidrogênio. O produto é extraído em cloreto de metilano e lavado com bicarbonato de sódio saturado. A fase orgânica é absorvida e o solvente removido em vácuo para fornecer o álcool intermediário como base livre. A este intermediário é adicionado cloreto de metilano (100 ml). A esta solução é adicionado cloreto de tionila (1,0 ml, 13,6 mmols) à temperatura ambiente. A solução é aquecida para 40 °C e agitada 3 h. A solução é deixada resfriar, e o solvente e cloreto de tionila excedentes são removidos em vácuo. O sólido resultante é extraído em cloreto de metilano e lavado com bicarbonato de sódio saturado. A fase orgânica é absorvida e removida em vácuo para fornecer o produto. MS (ESI) m/z 352, 354,356 (M + H).
[00403] Seguindo este protocolo é também preparado: 1) 3-Cloro-4-[5-(4-clorobutil)imidazol-1 -i I meti I] benzon itri la. MS (ESI) m/z 308, 310, 312 (M + H).
2) 3-Metóxi-4-[5-(4-clorobutil)imidazol-1 -ilmetiljbenzonitrila. MS (ESI) m/z 304, 306 (M + H).
3) 3-flúor-4-[5-(4-clorobutil)-imidazol-1-ilmetil]benzonitrila. MS (ESI) m/z 292, 294 (M + H).
4) 2-Bromo-4-[5-(4-clorobutil)imidazol-1 -iImetiI]benzonitrila. MS (ESI) m/z 352, 354, 356 (M + H).
5) 3-Bromo-4-{5-(4-cloro-1 -meti I buti l)-i m idazol-1 - ilmetiljbenzonitrila. MS (ESI) m/z 366, 368, 370 (M + H).
6) 1-(2-Bromobenzil)-5-(4-clorobutil)-1H-imidazol. MS (ESI)
7) 1-(2-Bromo-4-flúorbenzil)-5-(4-clorobutil)-1H-imidazol. MS (ESI) m/z 345, 347, 349 (M + H).
8) Éster de metila de ácido 3-Bromo-4-[5-(4- clorobutil)imidazol-1-ilmetil]benzóico.
[00404] MS (ESI) m/z 385, 387, 389 (M + H).
EXEMPLO 23 3-BROMO-4-(6,7,8,9-TETRAIDRO-5H-IMIDAZO[1,5-A]AZEPIN-5- IDBENZONITRILA
[00405] A 3-Bromo-4-[5-(4-clorobutil)imidazol-1 -i I meti I] benzon itri la (0,672 g, 1,9 mmols) é adicionado THF anidroso (40 mL). A solução é desoxigenada com nitrogênio borbulhado através dela por 15 minutos. Uma solução de f-butóxido de potássio em THF (3,41 ml, 1M, 3,41 mmols) é adicionada. A reação é deixada prosseguir por 2 h à temperatura ambiente. A reação é então finalizada por adição de metanol (3 mL) seguida por cloreto de amónio aquoso. O produto é extraído em acetato de etila e lavado com bicarbonato de sódio saturado. A camada orgânica é removida em vácuo para fornecer o produto bruto. Cromatografia (HPLC de fase reversa, gradiente 1:9 acetonitrila:água a 7:3 acetonitrila:água, durante 8 minutos, pH 2) fornece o produto puro. MS (ESI) m/z 316, 318 (M + H)
[00406] Resolução de enantiômeros (R) e (S) do composto do título é obtida por HPLC quiral usando a coluna ChiralPak IA com uma fase móvel de IPA:hexano a 20% para fornecer enantiômero A (t r = 24,0 min) e enantiômero B (tr = 29 minutos).
[00407] Similarmente preparados são os seguintes: 1) 3-flúor-4-(6,7,8,9-tetraidro-5H-imidazo[1,5-a]azepin-5- il)benzonitrila. MS (ESI) m/z 256 (M + H).
[00408] Resolução de enantiômeros (R) e (S) do composto acima é obtida por HPLC quiral usando a coluna ChiralPak IA com uma fase móvel de IPA:hexano a 20% para fornecer enantiômero A (t r = 18,2 min) e enantiômero B (tr = 20,3 minutos). Por enantiômero A: 1H RMN (400 MHz, CDCh) δ ppm 1,38-1,67 (m, 2 H), 1,72-1,93 (m, 2 H), 1,96- 2,14 (m, 1 H), 2,44-2,74 (m, 2 H), 2,97 (dd, J = 15,4, 6,3 Hz, 1 H), 5,73 (dd, J = 6,1, 2,8 Hz, 1 H), 6,80-6,94 (m, 2 H), 7,23 (s, 1 H), 7,39-7,42 (m, 1 H), 7,43 (s, 1 H). 2) 3-Cloro-4-(6,7,8,9-tetraidro-5H-imidazo[1,5-a]azepin-5- il)benzonitrila.
[00409] MS (ESI) m/z 272, 274 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,50-1,87 (m, 4 H), 1,97-2,19 (m, 1 H), 2,42-2,61 (m, 1 H), 2,67- 2,85 (m, 1 H), 2,85-3,00 (m, 1 H), 5,71 (dd, J = 7,2, 2,7 Hz, 1 H), 6,83 (s, 1 H), 6,98 (s, 1 H), 7,17 (d, J = 8,1 Hz, 1 H), 7,56 (dd, J = 8,1, 1,5 Hz, 1 H), 7,72 (d, J= 1,5 Hz, 1 H).
[00410] Resolução de enantiômeros (R) e (S) do composto acima é obtida por HPLC quiral usando a coluna ChiralPak IA com uma fase móvel de IPA:hexano a 20% para fornecer enantiômero A (t r = 24,0 min) e enantiômero B (tr = 29,0 minutos). 3) 3-Bromo-4-(9-metil-6,7,8,9-tetraidro-5H-imidazo[1,5- a]azepin-5-il)benzonitrila
[00411] MS (ESI) m/z 330, 332 (M + H).
4) 2-Bromo-4-(6,7,8,9-tetraidro-5H-imidazo[1,5-a]azepin-5- il)benzonitrila. MS (ESI) m/z 316, 318 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,39-1,57 (m, 2 H), 1,73-1,96 (m, 2 H), 2,04-2,17 (m, 1 H), 2,18-2,37 (m, 1 H), 2,47-2,73 (m, 1 H), 2,81-3,03 (m, 1 H), 5,50- 5,70 (m, 1 H), 6,90 (s, 1 H), 6,93-7,00 (m, 1 H), 7,29 (s, 1 H), 7,35 (s, 1 H), 7,65 (d, J = 8,1 Hz, 1 H).
EXEMPLO 24 5-(2-BROMOFENIL)-6,7,8,9-TETRAIDRO-5H-IMIDAZO[1,5-Al AZEPINA.
[00412] A 5-(2-bromofenil)-6,7,8,9-tetraidro-5H-imidazo[1,5-a] azepina (1,65 g, 4,75 mmols) é adicionado THF anidroso (100 mL). Nitrogênio é borbulhado através desta solução por 15 minutos e LHMDS (10 mL, 1,0 M, 10 mmols) é adicionado. A reação é deixada prosseguir por 2 h à temperatura ambiente. A reação é então finalizada por adição de metanol (5 mL) seguida por cloreto de amónio aquoso. O produto é extraído em acetato de etila e lavado com bicarbonato de sódio saturado. A camada orgânica é removida em vácuo para fornecer o produto bruto. Cromatografia (HPLC em fase reversa, gradiente 1:9 acetonitrila:água a 7:3 acetonitrila:água, durante 8 minutos, pH 2) fornece o produto puro. MS (ESI) m/z 291, 293 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,54-1,67 (m, 1 H), 1,70-1,83 (m, 1 H), 1,83-1,96 (m, 2 H), 2,12-2,23 (m, 1 H), 2,27-2,40 (m, 1 H), 2,77- 2,87 (m, 1 H), 2,91-3,01 (m, 1 H), 5,58 (dd, J = 8,5, 1,9 Hz, 1 H), 6,81 (s, 1 H), 6,82 (s, 1 H), 7,20-7,29 (m, 2 H), 7,33-7,41 (m, 1 H), 7,64 (dd, J = 8,1, 1,3 Hz, 1 H).
[00413] Similarmente preparados são os seguintes: 1) 5-(2-Bromo-4-flúorfenil)-6,7,8,9-tetraidro-5H-imidazo[1,5- a]azepina. MS (ESI) m/z 309, 311 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,54-1,63 (m, 1 H), 1,72-1,94 (m, 3 H), 2,10-2,33 (m, 2 H), 2,75- 2,97 (m, 2 H), 5,53 (d, J = 8,3 Hz, 1 H), 6,78 (s, 1 H), 6,81 (s, 1 H), 7,01-7,13 (m, 1 H), 7,16-7,28 (m, 1 H), 7,40 (dd, J = 8,1,2,5 Hz, 1 H).
2) Éster de metila de ácido 3-Bromo-4-(6,7,8,9-tetraidro-5H- imidazo[1,5-a]azepin-5-il)benzóico. MS (ESI) m/z 349, 351 (M + H).
3) 3-Metóxi-4-(6,7,8,9-tetraidro-5H-imidazo[1,5-a]azepin-5- il)benzonitrila. MS (ESI) m/z 268 (M + H).
EXEMPLO 25 4’-FLÚOR-6-(6,7,8,9-TETRAIDRO-5H-IMIDAZO[1,5-A]AZEPIN-5-IL) BIFENIL-3-CARBONITRILA
[00414] A 3-bromo-4-(6,7,8,9-tetraidro-5H-imidazo[1,5-a]azepin-5- il)benzonitrila (0,507 g, 1,59 mmols) é adicionado DME (7,0 mL). A este é adicionado carbonato de sódio aquoso a 2M (3,0 mL, 6 mmols) seguido por ácido 4-flúorfenilborônico (0,552 g, 3,97 mmols). Esta mistura é transferida para um microondas em frasconete seguro. Tetracis(trifenilfosfina)paládio(0) (0,03 g, 0,025 mmols) é adicionado, e o vaso selado. A mistura é agitada brevemente (30 segundos) e colocada em um microondas por 25 minutos a 125 °C. O frasconete é deixado esfriar e aberto. A mistura é extraída em acetato de etila e lavada com bicarbonato de sódio saturado. A camada orgânica é removida em vácuo. O material resultante é submetido à cromatografia (Sílica-gel, 1:50 a 1:25 de metanol: cloreto de metilano) para fornecer o produto desejado. MS (ESI) m/z 332 (M + H)
[00415] Resolução de enantiômeros (R) e (S) do composto do título é obtida por HPLC quiral usando a coluna ChiralPak IA com uma fase móvel de IPA:hexano a 20% para fornecer enantiômero A (t r = 17,6 min) e enantiômero B (tr = 23,0 minutos). Por enantiômero A: 1H RMN (400 MHz, CDCh) δ ppm 1,40-1,57 (m, 2 H), 1,77-1,92 (m, 2 H), 1,94- 2,04 (m, 2 H), 2,43 (dd, J = 15,0, 9,7 Hz, 1 H), 2,85 (dd, J = 15,4, 7,3 Hz, 1 H), 5,13 (dd, J = 7,5, 3,2 Hz, 1 H), 6,83 (s, 1 H), 6,92 (s, 1 H), 7,01-7,12 (m, 4 H), 7,51 (d, J = 8,3 Hz, 1 H), 7,63 (d, J = 1,8 Hz, 1 H), 7,75 (dd, J = 8,1, 1,8 Hz, 1 H).
[00416] Similarmente preparados são os seguintes: 1) 5-Bifen i l-2-i I-6,7,8,9-tetraidro-5H-i m idazo[ 1,5-a]azepina. MS (ESI) m/z 289 (M + H); 1H RMN (400 MHz, CDCh) δ Dppm 1,35- 1,56 (m, 2 H), 1,80-1,89 (m, 1 H), 1,89-1,98 (m, 1 H), 1,99-2,08 (m, 2 H), 2,28-2,41 (m, 1 H), 2,87 (dd, J = 15,3, 6,2 Hz, 1 H), 5,05-5,15 (m, 1 H), 6,80 (br s, 1 H), 6,97 (br s, 1 H), 7,03-7,10 (m, 2 H), 7,31-7,36 (m, 4 H), 7,40- 7,48 (m, 3 H).
[00417] Resolução de enantiômeros (R) e (S) do composto acima é obtida por HPLC quiral usando a coluna ChiralPak AS com uma fase móvel de IPA:hexano a 20% para fornecer enantiômero A (tr = 7,5 min) e enantiômero B (tr = 10,8 minutos). 2) 5-(4’-Flúorbifenil-2-il)6,7,8,9-tetraidro-5H-imidazo[1,5- a]azepina
[00418] MS (ESI) m/z 307 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,34-1,56 (m, 2 H), 1,81-1,99 (m, 2 H), 2,02-2,09 (m, 2 H), 2,28- 2,41 (m, 1 H), 2,89 (dd, J = 15,2, 6,6 Hz, 1 H), 4,98-5,05 (m, 1 H), 6,79 (s, 1 H), 6,90 (s, 1 H), 7,02 (d, J = 7,6 Hz, 4 H), 7,28-7,34 (m, 1 H), 7,39-7,52 (m, 3 H).
[00419] Resolução de enantiômeros (R) e (S) do composto acima é obtida por HPLC quiral usando a coluna ChiralPak AS com uma fase móvel de IPA:hexano a 15% para fornecer enantiômero A (tr = 9,3 min) e enantiômero B (tr = 11,6 minutos). 3) 5-(2’-Clorobifenil-2-il)6,7,8,9-tetraidro-5H-imidazo[1,5- a]azepina. MS (ESI) m/z 323, 325 (M + H).
4) 2’-(6,7,8,9)-Tetraidro-5H-imidazo[1,5-a]azepin-5-il)bifenil- 4-carbonitrila.
[00420] MS (ESI) m/z 314 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,33 - 1,58 (m, 2 H), 1,84 - 1,96 (m, 1 H), 1,96 - 2,16 (m, 3 H), 2,26 - 2,38 (m, 1 H), 2,92 (dd, J = 15,3, 6,7 Hz, 1 H), 4,90 - 4,97 (m, 1 H), 6,83 (s, 1 H), 6,94 (s, 1 H), 7,17 (d, J = 7,6 Hz, 2 H), 7,28 - 7,37 (m, 1 H), 7,45 - 7,57 (m, 3 H), 7,65 (d, J = 8,3 Hz, 2 H).
5) 5-(2’-Triflúormetilbifenil-2-il)6,7,8,9-tetraidro-5H-imidazo [1,5-a]azepina MS (ESI) m/z 357 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,27 - 1,39 (m, 1 H), 1,41 - 1,55 (m, 1 H), 1,86 - 1,95 (m, 1 H), 1,97 - 2,13 (m, 3 H), 2,14 - 2,25 (m, 1 H), 2,84 (dd, J = 15,4, 6,1 Hz, 1 H), 4,61 (d, J = 9,9 Hz, 1 H), 6,80 (s, 1 H), 6,87 (d, J = 7,6 Hz, 1 H), 6,98 (s, 1 H), 7,29 (d, J = 7,6 Hz, 1 H), 7,37 (t, J = 7,6 Hz, 1 H), 7,44 - 7,51 (m, 2 H), 7,56 (d, J = 3,8 Hz, 2 H), 7,78 (d, J = 8,1 Hz, 1 H).
6) 5-(3’-Nitrobifenil-2-i l)-6,7,8,9-tetraidro-5H-imidazo[1,5- ajazepina. MS (ESI) m/z 334 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,34 - 1,58 (m, 2 H), 1,84 - 1,93 (m, 1 H), 1,95 - 2,17 (m, 3 H), 2,25 - 2,36 (m, 1 H), 2,90 (dd, J = 15,3, 6,7 Hz, 1 H), 4,97 (dd, J = 9,1, 1,5 Hz, 1 H), 6,81 (s, 1 H), 6,91 (s, 1 H), 7,23 - 7,28 (m, 1 H), 7,34 - 7,38 (m, 1 H), 7,45 - 7,57 (m, 4 H), 8,07 (t, J = 1,9 Hz, 1 H), 8,18 - 8,25 (m, 1 H).
7) 5-(5-Flúorbifenil-2-il)-6,7,8,9-tetraidro-5H-imidazo[1,5- ajazepina.
[00421] MS (ESI) m/z 307 (M + H).
8) 4’-flúor-6-(9-metil-6,7,8,9-tetraidro-5H-imidazo[1,5- a]azepin-5-il)bifenil-3-carbonitrila. MS (ESI) m/z 346 (M + H).
[00422] Resolução de isômeros (/?)(/?), (R)(S), (S)(R), e (S)(S) do composto acima é obtido por HPLC quiral usando a coluna ChiralPak IA com uma fase móvel de I PA:hexano a 15% para fornecer isômero A (t r = 18,1 min), isômero B (tr = 21,8 minutos), isômero C (tr = 24,8 minutos) e isômero D (tr = 27,5 minutos). Por isômero D: 1H RMN (400 MHz, CDCh) δ ppm 1,31-1,35 (m, 1 H), 1,40 (d, J = 6,8 Hz, 3 H), 1,64- 1,77 (m, 1 H), 1,91-2,00 (m, 1 H), 2,10-2,23 (m, 2 H), 2,26-2,34 (m, 1 H), 2,51-2,60 (m, 1 H), 5,24 (d, J = 10,1 Hz, 1 H), 6,94-7,02 (m, 2 H), 7,08-7,15 (m, 3 H), 7,70 (d, J = 1,8 Hz, 1 H), 7,77 (d, J = 8,1 Hz, 1 H), 7,85 (s, 1 H), 7,89 (dd, J = 8,2, 1,6 Hz, 1 H). 9) 3-(3,5-Dimetilisoxazol-4-il)-4-(6,7,8,9-tetraidro-5H- imidazo[1,5-a]azepin-5-il)benzonitrila. MS (ESI) m/z 333 (M + H)

10) 3-Piridin-4-il-4-(6,7,8,9-tetraidro-5H-imidazo[1,5-a] azepin-5-il)benzonitrila. MS (ESI) m/z 315 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,37-1,63 (m, 2 H), 1,77-1,95 (m, 2 H), 1,96-2,10 (m, 2 H), 2,35-2,52 (m, 1 H), 2,86 (dd, J = 15,5, 7,2 Hz, 1 H), 5,04-5,13 (m, 1 H), 6,85 (s, 1 H), 6,89 (s, 1 H), 6,98-7,07 (m, 2 H), 7,58 (d, J = 8,1 Hz, 1 H), 7,64 (d, J = 1,5 Hz, 1 H), 7,82 (dd, J = 8,3, 1,8 Hz, 1 H), 8,67 (d, J = 6,1 Hz, 2 H).
11) 3-Pirid in-3-i l-4-(6,7,8,9-tetraidro-5H-imidazo[ 1,5-a] azepin-5-il)benzonitrila. MS (ESI) m/z 315 (M + H); 1H RMN (400 MHz, CDCh) (sal de HCI) δ ppm 1,41-1,63 (m, 2 H), 1,70-1,93 (m, 2 H), 1,95- 2,13 (m, 2 H), 2,29-2,54 (m, 1 H), 2,84 (dd, J = 15,4, 7,3 Hz, 1 H), 5,11 (t, J = 5,1 Hz, 1 H), 6,82 (s, 1 H), 6,90 (s, 1 H), 7,28-7,39 (m, 2 H), 7,55 (d, J = 8,3 Hz, 1 H), 7,64 (d, J = 1,5 Hz, 1 H), 7,80 (dd, J = 8,1, 1,8 Hz, 1 H), 8,39-8,53 (m, 1 H), 8,68 (t, J = 3,3 Hz, 1 H).
12) 5-(6,7,8,9-Tetraidro-5H-imidazo[1,5-a]azepin-5-il)bifenil- 2-carbonitrila. MS (ESI) m/z 314 (M + H).
EXEMPLO 26 4-(5-ALIL-6,7,8,9-TETRAIDR0-5H-IMIDAZ0π.5-/41AZEPIN-5-IL)-3- BROMOBENZONITRILA
[00423] A 4-(6,7,8,9-tetraidro-5H-imidazo[1,5-a]azepin-5-il)-3- bromo-benzonitrila (0,330 g, 1,04 mmols) é adicionado tetraidrofurano anidroso (60 mL). Nitrogênio é borbulhado através da solução por 15 minutos. LDA em THF (2,2 mL, 1,0M, 2,2 mmols) é adicionado à temperatura ambiente. A reação é deixada agitar por 15 min à temperatura ambiente. Brometo de alila (12,78 g, 90,6 mmols) é então adicionado. A reação é deixada agitar por mais 5 min. A reação é então finalizada por adição de metanol (2 mL) seguida por cloreto de amónio aquoso. O produto é extraído em acetato de etila e lavado com bicarbonato de sódio aquoso saturado. A camada orgânica é removida em vácuo para fornecer o produto bruto. Cromatografia (sílica-gel, metanol:cloreto de metilano, gradiente 0% metanol a 5% metanol durante 30 minutos fornece o produto puro. MS (ESI) m/z 356, 358 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,35 - 1,52 (m, 2 H), 1,67 - 1,77 (m, 1 H), 1,84 - 2,00 (m, 3 H), 2,82 (dd, J = 15,3, 5,7 Hz, 1 H), 3,15 - 3,41 (m, 3 H), 5,14 - 5,30 (m, 2 H), 5,67 - 5,82 (m, 1 H), 6,26 (d, J = 8,1 Hz, 1 H), 6,86 (s, 1 H), 7,45 (dd, J = 8,3, 1,8 Hz, 1 H), 7,79 (s, 1 H), 7,94 (d, J= 1,8 Hz, 1 H).
[00424] Similarmente preparados são os seguintes: 1) 3-Cloro-(5-etil-6,7,8,9-tetraidro-5H-imidazo[1,5-a]azepin- 5-il)benzonitrila. MS (ESI) m/z 300, 302 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,07 (t, J = 7,3 Hz, 3 H), 1,24-1,52 (m, 2 H), 1,60-1,79 (m, 1 H), 1,86-2,07 (m, 3 H), 2,19-2,44 (m, 1 H), 2,50-2,62 (m, 1 H), 2,84 (dd, J = 15,4, 5,8 Hz, 1 H), 3,18 (dd, J = 14,8, 5,7 Hz, 1 H), 6,21 (d, J = 8,3 Hz, 1 H), 6,88 (s, 1 H), 7,40 (dd, J = 8,5, 1,9 Hz, 1 H), 7,65 (s, 1 H), 7,71 (d, J= 1,8 Hz, 1 H).
2) 6-(5-Etil-6,7,8,9-tetraidro-5H-imidazo[1,5-a]azepin-5-il)-4’- flúorbifenil-3-carbonitrila. MS (ESI) m/z 360 (M + H).
3) 6-(5-Metil-6,7,8,9-tetraidro-5H-imidazo[1,5-a]azepin-5-il)- 4’-flúorbifenil-3-carbonitrila. MS (ESI) m/z 346 (M + H).
EXEMPLO 27 3,-METILANO-2,,3,A7,8.9-HEXAIDROESPIROriMIDAZOri,5-/11 AZEPINA-δ.l'-INDENEI-δ'-CARBONITRILA
[00425] A 4-(5-aliI-6,7,8,9-tetraidro-5H-imidazo[ 1,5-a]azepin-5-il)-3- bromobenzonitrila (0,275 g, 0,76 mmols) é adicionado DME (3,0 mL). A esta solução é adicionado carbonato de sódio aquoso a 2M (1,5 ml, 3,0 mmols) seguido por ácido 4-flúorfenilborônico (0,269 g, 1,92 mmols). Esta mistura é trasferida para um microondas em frasconete seguro. Tetracis(trifenilfosfina)paladio(0) (0,04 g, 0,034 mmols) é adicionado, e o vaso selado. A mistura é agitada brevemente (30 segundos) e colocada em um microondas por 25 minutos a 130 °C. O frasconete é deixado resfriar e aberto. A mistura é extraída com acetato de etila e lavada com bicarbonato de sódio aquoso saturado. A camada orgânica separada é concentrada em vácuo. O material resultante é submetido a dois procedimentos seqüenciais de cromatografia (1: Sílica-gel, 1:50 a 1:25 metanol:cloreto de metilano. 2: HPLC de fase reversa, gradiente 10-95% Acetonitrila:Água, pH2, durante 8 minutos) para fornecer o produto. MS (ESI) m/z 276 (M + H); 1H RMN (400 MHz, CDCh) δ ppm 1,47 - 1,61 (m, 1 H), 1,85 - 1,99 (m, 1 H), 2,01 - 2,16 (m, 3 H), 2,25 - 2,36 (m, 1 H), 2,63 (t, 1 H), 2,99 (d, J = 16,4 Hz, 1 H), 3,10 (dd, J = 15,5, 5,7 Hz, 1 H), 3,23 - 3,32 (m, 1 H), 5,21 - 5,25 (m, 1 H), 5,62 - 5,67 (m, 1 H), 6,50 (s, 1 H), 6,78 (s, 1 H), 7,54 - 7,58 (m, 1 H), 7,66 - 7,70 (m, 1 H), 7,88 (s, 1 H). EXEMPLO 28 7-BENZIL-5-(3,5-DIMETÓXI-FENIL)-6,7,8,9-TETRAIDRO-5H-IMIDAZO [1,5-A1AZEPINA

[00426] A uma solução de DMSO (3,57 g, 45,6 mmols) em diclorometano (100 mL) a -78 °C sob nitrogênio é adicionado cloreto de oxalila (2,0M em DCM, 17,1 mL, 34,2 mmols). Após 20 min a -78 °C, o álcool preparado na etapa 2 (8,41 g, 22,82 mmols) em diclorometano (30 mL) é canulado em gotas. Após 30 min a -78 °C, trietilamina (9,24 g, 91,3 mmols) é adicionada em gotas e a mistura é deixada aquecer em temperatura ambiente. A mistura é diluída com éter de dietila (800 mL) e lavada com cloreto de amónio aquoso saturado (2 x 200 mL) e salmoura (200 mL). A fase orgânica combinada é secada em sulfato de magnésio e filtrada. Após concentração em vácuo, o produto é obtido como uma goma laranja.
B. 4-BUT-3-ENIL-1-TRITIL-1H-IMIDAZQL
[00427] terc-BuOK (5,78 g, 51,53 mmols) é dissolvido em THF (80 mL) e adicionado a uma suspensão de brometo de metiltrifenilfosfônico (20,00 g, 56,00 mmols) em THF (120 mL) a 0 °C sob nitrogênio. Após 30 min a 0 °C, o aldeído obtido na etapa 3 (8,5 g) em THF (40 mL) é canulado em gotas. O banho de resfriamento é removido e após 1 h, a mistura é diluída com acetato de etila (800 mL) e lavada com cloreto de amónio aquoso saturado (400 mL) e salmoura (400 mL). A fase orgânica combinada é secada em sulfato de magnésio e filtrada. O resíduo é purificado por cromatografia instantânea de sílica-gel (eluição com hexanos-acetato de etila, 1:1) para fornecer o alceno (6,72 g) como um sólido amarelo-pálido.
C. (E)-1-(3,5-DIMETOXIFENIL)-5-(1-TRITIL-1H-IMIDAZOL-4-IL)PENT- 2-EN-1-ONA
[00428] Tolueno (100 mL) é adicionado a (PPh3)2Pd(BnCI) (0,151 g, 0,199 mmols) e cloreto de 3,5-dimetoxibenzoila (4,00 g, 19,94 mmols) sob nitrogênio, seguida com tributilvinilastanho (6,11 g, 21,93 mmols). A solução amarela é aquecida para 80 °C, para fornecer uma solução amarela-pálida. Após 1,5 h, a mistura é parcialmente concentrada e despejada sobre uma coluna de 10% em peso de KF em sílica-gel. Eluição com hexanos em seguida hexanos-acetato de etila 10 a 15 % forneceu propenona de 1-(3,5-dimetoxifenil) (3,57 g) como um óleo amarelo pálido.
[00429] A porção (1,32 g, 6,86 mmols) é adicionada a uma solução do alceno preparado acima (1,00 g, 2,74 mmols) em diclorometano (25 mL). Ácido de p-Toluenossulfônico (0,574 g, 3,018 mmols) é adicionado, a solução é desgaseificada por borbulhamento de nitrogénio por 25 min, e esta é então refluxada por 30 min. Após resfriamento, catalisador Grubbs de segunda geração (0,116 g, 0,137 mmols) é adicionado e a solução púrpura é aquecida até o refluxo. Após 45 min, a mistura é resfriada, diluída com éter e lavada com bicarbonato de sódio aquoso saturado e salmoura. Após secagem sobre MgSO4 e filtragem, sílica-gel (2 g) é adicionada e a mistura é concentrada em vácuo. O resíduo é absorvido em uma almofada de sílica-gel em um funil sinterizado, seguida por eluição com acetato de etila-hexanos, 1:1 a 4:1. O filtrado é concentrado em vácuo para fornecer um resíduo que é purificado por cromatografia instantânea de sílica-gel (hexanos-acetato de etila, 1:1a 3:7) para fornecer o produto como uma goma marrom.
D. 3-BENZIL-1-(3,5-DIMETOXIFENIL)-5-(1-TRITIL-1H-IMIDAZOL-4- IDPEN-TAN-1-ONE
[00430] Lâmina de zinco (0,556 g, 8,512 mmols) é cortada em pequenos pedaços e coberta com THF (0,5 mL) em um frasco seco sob nitrogênio. Dibromoetano (0,132 g, 0,704 mmols) é adicionado e o frasco é ligeiramente aquecido com uma pistola térmica por dois minutos, depois do que, TMSCI (0,039 g, 0,355 mmols) é adicionado. A mistura é agitada à temperatura ambiente por 10 min e brometo de benzila (passado através de um tampão de alumina, 1,20 g, 7,04 mmols) em THF (2 mL) é adicionado em gotas por 10 min. A mistura incolor é agitada por mais 2 h e uma porção (1,87 M assumida com base na produção de 85% e 3,2 mL de volume medido, 2,00 mL, 3,74 mmols) é adicionado a uma solução de CuCN (0,231 g, 2,580 mmols) e LiCI (secada a 150 °C em vácuo por 2 h, 0,234 g, 5,512 mmols) em THF (15 mL) a -78°C. A mistura é aquecida a -20°C, agitada a esta temperatura por 5 min e resfriada para -78°C. TMSCI (0,68 g, 5,28 mmols) é adicionado, seguido com a cetona preparada acima (0,620 g, 1,173 mmols) em THF (10 mL) durante 10 min para fornecer uma suspensão amarela. Após 3 h, a mistura é colocada em um banho a -25 °C (criobanho) e agitada a esta temperatura por 30 h, depois do que, é diluída com éter, extinguida com amónia aquosa-cloreto de amónio saturado aquoso a 10% v/v, e agitada vigorosamente por 15 min. A camada orgânica é lavada com salmoura. Após secagem sobre MgSO4, filtragem e concentração em vácuo, o resíduo (0,63 g) é absorvido em éter (100 mL) e agitada com HCI aquoso a 1 M por 5 min. A fase orgânica é separada, lavada com bicarbonato de sódio aquoso saturado, salmoura e secado em MgSCU. Após secagem sobre MgSO4 e filtragem, a mistura é concentrada em vácuo para fornecer o produto bruto.
E. 3-BENZIL-1-(3,5-DIMETOXIFENIL)-5-(1-TRITIL-1H-IMIDAZOL-4-IL) PEN-TAN-1-OL
[00431] A cetona bruta preparada acima (0,53 g, 0,854 mmols) é dissolvida em diclorometano (4 mL) e metanol (8 mL), e boroidreto de sódio (0,129 g, 3,415 mmols) é adicionado. Após 30 min, água (25 mL) é adicionada e os voláteis são evaporados em vácuo. A fase aquosa é extraída com diclorometano e a fase orgânica combinada é secada em MgSO4, filtrada através de um plugue de algodão e concentrada em vácuo. Purificação por cromatografia instantânea de sílica-gel (eluição com hexanos-acetato de etila, 1:1 a 3:7) produziu o álcool como uma espuma branca e uma mistura de 1:1 de diastereômeros.
F. 7-BENZIL-5-(3,5-DIMETÓXI-FENIL)-6,7,8,9-TETRAIDRO-5H-IMIDA- ZO[1,5-A]AZEPINA
[00432] O álcool preparado acima (0,150 g, 0,241 mmols) é dissolvido em diclorometano (10 mL) e cloreto de tionila (0,100 g, 0,843 mmols) é adicionado. Após 30 min, 10 gotas de bicarbonato de sódio aquoso saturado são adicionadas. A mistura é secada em MgSO4, filtrada e concentrada em vácuo. A mistura é redissolvida em acetonitrila seca e aquecida até o refluxo. Após 48 h, metanol (5 mL) é adicionado e após 2 h a mistura é evaporada até a secura em vácuo e absorvida em HCI aquoso a 1M e acetato de etila. A fase aquosa é lavada com éter. O pH é ajustado para aroximadamente 10 com hidróxido de sódio aquoso a 2M e este é extraído com diclorometano. As frações de diclorometano combinadas são secadas em MgSCX filtradas e concentradas em vácuo. O resíduo é purificado por cromatografia instantânea de sílica-gel (eluição com diclorometano- metanol, 19:1 a 9:1) para fornecer o diastereômero cis. MS (ESI) m/z 363 (M + H); 1H RMN (400 MHz, CDCh) (sal de HCI) δ ppm 1,17-1,27 (m, 1 H), 1,84 (dt, J= 14,1, 10,9 Hz, 1 H), 1,98 - 2,10 (m, 2 H), 2,25 (d, J = 14,1 Hz, 1 H), 2,53 - 2,65 (m, 3 H), 3,01 (ddd, J = 15,4, 6,3, 1,8 Hz, 1 H), 3,79 (s, 6 H), 4,89 (d, J = 10,4 Hz, 1 H), 6,44 - 6,52 (m, 3 H), 6,76 (br s, 2 H), 7,11 - 7,22 (m, 3 H), 7,25 - 7,30 (m, 2 H)
OUTRAS MODALIDADES
[00433] Outras modalidades serão evidentes para aqueles versados na técnica. Deve-se entender que a descrição detalhada anterior é fornecida para clareza apenas e é meramente exemplar. O espírito e o escopo da presente invenção não estão limitados aos exemplos acima, porém são abrangidos pelas seguintes reivindicações.