BRPI0213425B1 - composições injetáveis para a liberação controlada de composto farmacologicamente ativo, usos do referido composto e método para suas preparações - Google Patents
composições injetáveis para a liberação controlada de composto farmacologicamente ativo, usos do referido composto e método para suas preparações Download PDFInfo
- Publication number
- BRPI0213425B1 BRPI0213425B1 BRPI0213425-0A BR0213425A BRPI0213425B1 BR PI0213425 B1 BRPI0213425 B1 BR PI0213425B1 BR 0213425 A BR0213425 A BR 0213425A BR PI0213425 B1 BRPI0213425 B1 BR PI0213425B1
- Authority
- BR
- Brazil
- Prior art keywords
- acid
- active compound
- pharmacologically active
- pharmaceutically acceptable
- lipophilic
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 117
- 238000000034 method Methods 0.000 title claims abstract description 30
- 239000007972 injectable composition Substances 0.000 title claims abstract description 22
- 238000002360 preparation method Methods 0.000 title claims description 6
- 238000013270 controlled release Methods 0.000 title abstract description 5
- 239000000203 mixture Substances 0.000 claims abstract description 92
- JTSDBFGMPLKDCD-XVFHVFLVSA-N tilmicosin Chemical compound O([C@@H]1[C@@H](C)[C@H](O)CC(=O)O[C@@H]([C@H](/C=C(\C)/C=C/C(=O)[C@H](C)C[C@@H]1CCN1C[C@H](C)C[C@H](C)C1)CO[C@H]1[C@@H]([C@H](OC)[C@H](O)[C@@H](C)O1)OC)CC)[C@@H]1O[C@H](C)[C@@H](O)[C@H](N(C)C)[C@H]1O JTSDBFGMPLKDCD-XVFHVFLVSA-N 0.000 claims abstract description 47
- 229960000223 tilmicosin Drugs 0.000 claims abstract description 44
- 239000002904 solvent Substances 0.000 claims abstract description 42
- 241000124008 Mammalia Species 0.000 claims abstract description 34
- 239000004100 Oxytetracycline Substances 0.000 claims abstract description 26
- 229960000625 oxytetracycline Drugs 0.000 claims abstract description 26
- IWVCMVBTMGNXQD-PXOLEDIWSA-N oxytetracycline Chemical compound C1=CC=C2[C@](O)(C)[C@H]3[C@H](O)[C@H]4[C@H](N(C)C)C(O)=C(C(N)=O)C(=O)[C@@]4(O)C(O)=C3C(=O)C2=C1O IWVCMVBTMGNXQD-PXOLEDIWSA-N 0.000 claims abstract description 26
- 235000019366 oxytetracycline Nutrition 0.000 claims abstract description 26
- IWVCMVBTMGNXQD-UHFFFAOYSA-N terramycin dehydrate Natural products C1=CC=C2C(O)(C)C3C(O)C4C(N(C)C)C(O)=C(C(N)=O)C(=O)C4(O)C(O)=C3C(=O)C2=C1O IWVCMVBTMGNXQD-UHFFFAOYSA-N 0.000 claims abstract description 26
- 239000002244 precipitate Substances 0.000 claims abstract description 22
- 229960002464 fluoxetine Drugs 0.000 claims abstract description 17
- 229960002237 metoprolol Drugs 0.000 claims abstract description 17
- IUBSYMUCCVWXPE-UHFFFAOYSA-N metoprolol Chemical compound COCCC1=CC=C(OCC(O)CNC(C)C)C=C1 IUBSYMUCCVWXPE-UHFFFAOYSA-N 0.000 claims abstract description 16
- RTHCYVBBDHJXIQ-MRXNPFEDSA-N (R)-fluoxetine Chemical compound O([C@H](CCNC)C=1C=CC=CC=1)C1=CC=C(C(F)(F)F)C=C1 RTHCYVBBDHJXIQ-MRXNPFEDSA-N 0.000 claims abstract description 14
- 229960005224 roxithromycin Drugs 0.000 claims abstract description 9
- RXZBMPWDPOLZGW-XMRMVWPWSA-N (E)-roxithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=N/OCOCCOC)/[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 RXZBMPWDPOLZGW-XMRMVWPWSA-N 0.000 claims abstract description 7
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 claims description 81
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 claims description 78
- POULHZVOKOAJMA-UHFFFAOYSA-N dodecanoic acid Chemical compound CCCCCCCCCCCC(O)=O POULHZVOKOAJMA-UHFFFAOYSA-N 0.000 claims description 61
- 150000003839 salts Chemical class 0.000 claims description 37
- 239000005639 Lauric acid Substances 0.000 claims description 29
- 239000002253 acid Substances 0.000 claims description 28
- 239000003960 organic solvent Substances 0.000 claims description 22
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 21
- GHVNFZFCNZKVNT-UHFFFAOYSA-N Decanoic acid Natural products CCCCCCCCCC(O)=O GHVNFZFCNZKVNT-UHFFFAOYSA-N 0.000 claims description 20
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 claims description 20
- GYSCBCSGKXNZRH-UHFFFAOYSA-N 1-benzothiophene-2-carboxamide Chemical compound C1=CC=C2SC(C(=O)N)=CC2=C1 GYSCBCSGKXNZRH-UHFFFAOYSA-N 0.000 claims description 18
- 239000002202 Polyethylene glycol Substances 0.000 claims description 17
- 229920001223 polyethylene glycol Polymers 0.000 claims description 17
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 15
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 claims description 13
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 claims description 13
- 229960002722 terbinafine Drugs 0.000 claims description 13
- DOMXUEMWDBAQBQ-WEVVVXLNSA-N terbinafine Chemical compound C1=CC=C2C(CN(C\C=C\C#CC(C)(C)C)C)=CC=CC2=C1 DOMXUEMWDBAQBQ-WEVVVXLNSA-N 0.000 claims description 11
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 claims description 9
- NOYRGONWBIVLEL-SNAWJCMRSA-N (e)-1,1-dimethoxybut-2-ene Chemical compound COC(OC)\C=C\C NOYRGONWBIVLEL-SNAWJCMRSA-N 0.000 claims description 9
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 claims description 9
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 claims description 9
- 239000005642 Oleic acid Substances 0.000 claims description 9
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 claims description 9
- 235000021313 oleic acid Nutrition 0.000 claims description 9
- OYHQOLUKZRVURQ-HZJYTTRNSA-N Linoleic acid Chemical compound CCCCC\C=C/C\C=C/CCCCCCCC(O)=O OYHQOLUKZRVURQ-HZJYTTRNSA-N 0.000 claims description 8
- 235000020778 linoleic acid Nutrition 0.000 claims description 8
- OYHQOLUKZRVURQ-IXWMQOLASA-N linoleic acid Natural products CCCCC\C=C/C\C=C\CCCCCCCC(O)=O OYHQOLUKZRVURQ-IXWMQOLASA-N 0.000 claims description 7
- FCGXLCNBWYIEAA-UHFFFAOYSA-N 1,3-benzothiazol-6-ylmethanamine Chemical compound NCC1=CC=C2N=CSC2=C1 FCGXLCNBWYIEAA-UHFFFAOYSA-N 0.000 claims description 6
- 239000001089 [(2R)-oxolan-2-yl]methanol Substances 0.000 claims description 6
- 230000002035 prolonged effect Effects 0.000 claims description 6
- BSYVTEYKTMYBMK-UHFFFAOYSA-N tetrahydrofurfuryl alcohol Chemical compound OCC1CCCO1 BSYVTEYKTMYBMK-UHFFFAOYSA-N 0.000 claims description 6
- OYHQOLUKZRVURQ-NTGFUMLPSA-N (9Z,12Z)-9,10,12,13-tetratritiooctadeca-9,12-dienoic acid Chemical compound C(CCCCCCC\C(=C(/C\C(=C(/CCCCC)\[3H])\[3H])\[3H])\[3H])(=O)O OYHQOLUKZRVURQ-NTGFUMLPSA-N 0.000 claims description 5
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 claims description 5
- ZQPPMHVWECSIRJ-MDZDMXLPSA-N elaidic acid Chemical compound CCCCCCCC\C=C\CCCCCCCC(O)=O ZQPPMHVWECSIRJ-MDZDMXLPSA-N 0.000 claims 12
- TUNFSRHWOTWDNC-HKGQFRNVSA-N tetradecanoic acid Chemical compound CCCCCCCCCCCCC[14C](O)=O TUNFSRHWOTWDNC-HKGQFRNVSA-N 0.000 claims 7
- DPUOLQHDNGRHBS-UHFFFAOYSA-N Brassidinsaeure Natural products CCCCCCCCC=CCCCCCCCCCCCC(O)=O DPUOLQHDNGRHBS-UHFFFAOYSA-N 0.000 claims 4
- URXZXNYJPAJJOQ-UHFFFAOYSA-N Erucic acid Natural products CCCCCCC=CCCCCCCCCCCCC(O)=O URXZXNYJPAJJOQ-UHFFFAOYSA-N 0.000 claims 4
- 235000021314 Palmitic acid Nutrition 0.000 claims 4
- 235000021355 Stearic acid Nutrition 0.000 claims 4
- DPUOLQHDNGRHBS-KTKRTIGZSA-N erucic acid Chemical compound CCCCCCCC\C=C/CCCCCCCCCCCC(O)=O DPUOLQHDNGRHBS-KTKRTIGZSA-N 0.000 claims 4
- 229940074076 glycerol formal Drugs 0.000 claims 4
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 claims 4
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 claims 4
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 claims 4
- 239000008117 stearic acid Substances 0.000 claims 4
- 241001237728 Precis Species 0.000 claims 1
- 239000000194 fatty acid Substances 0.000 abstract description 23
- 239000003814 drug Substances 0.000 abstract description 21
- 229940079593 drug Drugs 0.000 abstract description 21
- 235000014113 dietary fatty acids Nutrition 0.000 abstract description 20
- 229930195729 fatty acid Natural products 0.000 abstract description 20
- -1 compound salt Chemical class 0.000 abstract description 18
- 150000004665 fatty acids Chemical class 0.000 abstract description 17
- 231100000419 toxicity Toxicity 0.000 abstract description 9
- 230000001988 toxicity Effects 0.000 abstract description 9
- 229920006395 saturated elastomer Polymers 0.000 abstract description 6
- 238000009472 formulation Methods 0.000 description 36
- 241000282326 Felis catus Species 0.000 description 22
- 210000001519 tissue Anatomy 0.000 description 19
- 239000000243 solution Substances 0.000 description 14
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 9
- 239000003021 water soluble solvent Substances 0.000 description 9
- 238000000502 dialysis Methods 0.000 description 8
- 238000000338 in vitro Methods 0.000 description 8
- 230000004048 modification Effects 0.000 description 8
- 238000012986 modification Methods 0.000 description 8
- 241001465754 Metazoa Species 0.000 description 7
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 7
- 238000002347 injection Methods 0.000 description 7
- 239000007924 injection Substances 0.000 description 7
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- 230000008901 benefit Effects 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- CXMXRPHRNRROMY-UHFFFAOYSA-N sebacic acid Chemical compound OC(=O)CCCCCCCCC(O)=O CXMXRPHRNRROMY-UHFFFAOYSA-N 0.000 description 6
- 241000283690 Bos taurus Species 0.000 description 5
- 230000003115 biocidal effect Effects 0.000 description 5
- 230000037396 body weight Effects 0.000 description 5
- 210000003734 kidney Anatomy 0.000 description 5
- 210000004185 liver Anatomy 0.000 description 5
- 239000011780 sodium chloride Substances 0.000 description 5
- 241000894006 Bacteria Species 0.000 description 4
- TUNFSRHWOTWDNC-UHFFFAOYSA-N Myristic acid Natural products CCCCCCCCCCCCCC(O)=O TUNFSRHWOTWDNC-UHFFFAOYSA-N 0.000 description 4
- 235000021360 Myristic acid Nutrition 0.000 description 4
- 150000007513 acids Chemical class 0.000 description 4
- 239000003242 anti bacterial agent Substances 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 210000004072 lung Anatomy 0.000 description 4
- 210000002966 serum Anatomy 0.000 description 4
- 239000007787 solid Substances 0.000 description 4
- 241000894007 species Species 0.000 description 4
- URAYPUMNDPQOKB-UHFFFAOYSA-N triacetin Chemical compound CC(=O)OCC(OC(C)=O)COC(C)=O URAYPUMNDPQOKB-UHFFFAOYSA-N 0.000 description 4
- TWJNQYPJQDRXPH-UHFFFAOYSA-N 2-cyanobenzohydrazide Chemical compound NNC(=O)C1=CC=CC=C1C#N TWJNQYPJQDRXPH-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 3
- 241000282472 Canis lupus familiaris Species 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 241000282324 Felis Species 0.000 description 3
- 241000282887 Suidae Species 0.000 description 3
- 210000000601 blood cell Anatomy 0.000 description 3
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 3
- 229910052794 bromium Inorganic materials 0.000 description 3
- 239000013078 crystal Substances 0.000 description 3
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 150000002500 ions Chemical class 0.000 description 3
- 239000003120 macrolide antibiotic agent Substances 0.000 description 3
- 229940028582 micotil Drugs 0.000 description 3
- 239000002953 phosphate buffered saline Substances 0.000 description 3
- 208000023504 respiratory system disease Diseases 0.000 description 3
- NESIVXZOSKKUDP-ARVJLQODSA-N (4r,5s,6s,7r,9r,11e,13e,15r,16r)-6-[(2r,3r,4s,5s,6r)-4-(dimethylamino)-3,5-dihydroxy-6-methyloxan-2-yl]oxy-7-[2-[(3s,5r)-3,5-dimethylpiperidin-1-yl]ethyl]-16-ethyl-4-hydroxy-15-[[(2r,3r,4r,5r,6r)-5-hydroxy-3,4-dimethoxy-6-methyloxan-2-yl]oxymethyl]-5,9,13 Chemical class OP(O)(O)=O.O([C@@H]1[C@@H](C)[C@H](O)CC(=O)O[C@@H]([C@H](/C=C(\C)/C=C/C(=O)[C@H](C)C[C@@H]1CCN1C[C@H](C)C[C@H](C)C1)CO[C@H]1[C@@H]([C@H](OC)[C@H](O)[C@@H](C)O1)OC)CC)[C@@H]1O[C@H](C)[C@@H](O)[C@H](N(C)C)[C@H]1O NESIVXZOSKKUDP-ARVJLQODSA-N 0.000 description 2
- SGKRLCUYIXIAHR-AKNGSSGZSA-N (4s,4ar,5s,5ar,6r,12ar)-4-(dimethylamino)-1,5,10,11,12a-pentahydroxy-6-methyl-3,12-dioxo-4a,5,5a,6-tetrahydro-4h-tetracene-2-carboxamide Chemical compound C1=CC=C2[C@H](C)[C@@H]([C@H](O)[C@@H]3[C@](C(O)=C(C(N)=O)C(=O)[C@H]3N(C)C)(O)C3=O)C3=C(O)C2=C1O SGKRLCUYIXIAHR-AKNGSSGZSA-N 0.000 description 2
- 241000283707 Capra Species 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- 241000283086 Equidae Species 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 239000003125 aqueous solvent Substances 0.000 description 2
- BLFLLBZGZJTVJG-UHFFFAOYSA-N benzocaine Chemical compound CCOC(=O)C1=CC=C(N)C=C1 BLFLLBZGZJTVJG-UHFFFAOYSA-N 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 231100000457 cardiotoxic Toxicity 0.000 description 2
- 230000001451 cardiotoxic effect Effects 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 150000001991 dicarboxylic acids Chemical class 0.000 description 2
- 229960003722 doxycycline Drugs 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 230000014509 gene expression Effects 0.000 description 2
- 239000001087 glyceryl triacetate Substances 0.000 description 2
- 235000013773 glyceryl triacetate Nutrition 0.000 description 2
- 229940041033 macrolides Drugs 0.000 description 2
- 239000003550 marker Substances 0.000 description 2
- 239000000401 methanolic extract Substances 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 239000012266 salt solution Substances 0.000 description 2
- 150000004671 saturated fatty acids Chemical class 0.000 description 2
- 239000011343 solid material Substances 0.000 description 2
- 238000002798 spectrophotometry method Methods 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 150000003892 tartrate salts Chemical class 0.000 description 2
- 150000003512 tertiary amines Chemical class 0.000 description 2
- 229960002622 triacetin Drugs 0.000 description 2
- 150000004670 unsaturated fatty acids Chemical class 0.000 description 2
- 235000021122 unsaturated fatty acids Nutrition 0.000 description 2
- LEBVLXFERQHONN-UHFFFAOYSA-N 1-butyl-N-(2,6-dimethylphenyl)piperidine-2-carboxamide Chemical compound CCCCN1CCCCC1C(=O)NC1=C(C)C=CC=C1C LEBVLXFERQHONN-UHFFFAOYSA-N 0.000 description 1
- VHVPQPYKVGDNFY-DFMJLFEVSA-N 2-[(2r)-butan-2-yl]-4-[4-[4-[4-[[(2r,4s)-2-(2,4-dichlorophenyl)-2-(1,2,4-triazol-1-ylmethyl)-1,3-dioxolan-4-yl]methoxy]phenyl]piperazin-1-yl]phenyl]-1,2,4-triazol-3-one Chemical compound O=C1N([C@H](C)CC)N=CN1C1=CC=C(N2CCN(CC2)C=2C=CC(OC[C@@H]3O[C@](CN4N=CN=C4)(OC3)C=3C(=CC(Cl)=CC=3)Cl)=CC=2)C=C1 VHVPQPYKVGDNFY-DFMJLFEVSA-N 0.000 description 1
- OCKGFTQIICXDQW-ZEQRLZLVSA-N 5-[(1r)-1-hydroxy-2-[4-[(2r)-2-hydroxy-2-(4-methyl-1-oxo-3h-2-benzofuran-5-yl)ethyl]piperazin-1-yl]ethyl]-4-methyl-3h-2-benzofuran-1-one Chemical compound C1=C2C(=O)OCC2=C(C)C([C@@H](O)CN2CCN(CC2)C[C@H](O)C2=CC=C3C(=O)OCC3=C2C)=C1 OCKGFTQIICXDQW-ZEQRLZLVSA-N 0.000 description 1
- 206010001488 Aggression Diseases 0.000 description 1
- 208000019901 Anxiety disease Diseases 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- CEAZRRDELHUEMR-URQXQFDESA-N Gentamicin Chemical compound O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N CEAZRRDELHUEMR-URQXQFDESA-N 0.000 description 1
- 229930182566 Gentamicin Natural products 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 206010024264 Lethargy Diseases 0.000 description 1
- GIYXAJPCNFJEHY-UHFFFAOYSA-N N-methyl-3-phenyl-3-[4-(trifluoromethyl)phenoxy]-1-propanamine hydrochloride (1:1) Chemical compound Cl.C=1C=CC=CC=1C(CCNC)OC1=CC=C(C(F)(F)F)C=C1 GIYXAJPCNFJEHY-UHFFFAOYSA-N 0.000 description 1
- 229930193140 Neomycin Natural products 0.000 description 1
- 208000021384 Obsessive-Compulsive disease Diseases 0.000 description 1
- 239000002033 PVDF binder Substances 0.000 description 1
- 229920000805 Polyaspartic acid Polymers 0.000 description 1
- 206010057190 Respiratory tract infections Diseases 0.000 description 1
- 208000000810 Separation Anxiety Diseases 0.000 description 1
- 229920002125 Sokalan® Polymers 0.000 description 1
- 239000004098 Tetracycline Substances 0.000 description 1
- NOSIYYJFMPDDSA-UHFFFAOYSA-N acepromazine Chemical compound C1=C(C(C)=O)C=C2N(CCCN(C)C)C3=CC=CC=C3SC2=C1 NOSIYYJFMPDDSA-UHFFFAOYSA-N 0.000 description 1
- 229960005054 acepromazine Drugs 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000016571 aggressive behavior Effects 0.000 description 1
- 208000012761 aggressive behavior Diseases 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 229940126575 aminoglycoside Drugs 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 239000004004 anti-anginal agent Substances 0.000 description 1
- 230000003257 anti-anginal effect Effects 0.000 description 1
- 230000000843 anti-fungal effect Effects 0.000 description 1
- 230000003276 anti-hypertensive effect Effects 0.000 description 1
- 239000003416 antiarrhythmic agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000002220 antihypertensive agent Substances 0.000 description 1
- 159000000032 aromatic acids Chemical class 0.000 description 1
- 230000006399 behavior Effects 0.000 description 1
- 229960005274 benzocaine Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 230000036765 blood level Effects 0.000 description 1
- 229960003150 bupivacaine Drugs 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 238000006243 chemical reaction Methods 0.000 description 1
- 150000003841 chloride salts Chemical class 0.000 description 1
- 235000019416 cholic acid Nutrition 0.000 description 1
- 239000002812 cholic acid derivative Substances 0.000 description 1
- 150000001842 cholic acids Chemical class 0.000 description 1
- PUFQVTATUTYEAL-UHFFFAOYSA-N cinchocaine Chemical compound C1=CC=CC2=NC(OCCCC)=CC(C(=O)NCCN(CC)CC)=C21 PUFQVTATUTYEAL-UHFFFAOYSA-N 0.000 description 1
- 229960001747 cinchocaine Drugs 0.000 description 1
- 239000013065 commercial product Substances 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 229940113088 dimethylacetamide Drugs 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-N diphosphoric acid Chemical class OP(O)(=O)OP(O)(O)=O XPPKVPWEQAFLFU-UHFFFAOYSA-N 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 108010017796 epoxidase Proteins 0.000 description 1
- 230000008686 ergosterol biosynthesis Effects 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 229960000389 fluoxetine hydrochloride Drugs 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- 229960002518 gentamicin Drugs 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 230000002706 hydrostatic effect Effects 0.000 description 1
- 230000002631 hypothermal effect Effects 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 229960004130 itraconazole Drugs 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 210000005228 liver tissue Anatomy 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- 230000007721 medicinal effect Effects 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229960004927 neomycin Drugs 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 238000002414 normal-phase solid-phase extraction Methods 0.000 description 1
- 235000015097 nutrients Nutrition 0.000 description 1
- 244000045947 parasite Species 0.000 description 1
- 230000004526 pharmaceutical effect Effects 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 150000008103 phosphatidic acids Chemical class 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 210000004180 plasmocyte Anatomy 0.000 description 1
- 239000004584 polyacrylic acid Substances 0.000 description 1
- 108010064470 polyaspartate Proteins 0.000 description 1
- 229920002981 polyvinylidene fluoride Polymers 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 210000005084 renal tissue Anatomy 0.000 description 1
- 235000003441 saturated fatty acids Nutrition 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 229940124834 selective serotonin reuptake inhibitor Drugs 0.000 description 1
- 239000012896 selective serotonin reuptake inhibitor Substances 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 229960002135 sulfadimidine Drugs 0.000 description 1
- ASWVTGNCAZCNNR-UHFFFAOYSA-N sulfamethazine Chemical compound CC1=CC(C)=NC(NS(=O)(=O)C=2C=CC(N)=CC=2)=N1 ASWVTGNCAZCNNR-UHFFFAOYSA-N 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 150000003456 sulfonamides Chemical class 0.000 description 1
- GKCBAIGFKIBETG-UHFFFAOYSA-N tetracaine Chemical compound CCCCNC1=CC=C(C(=O)OCCN(C)C)C=C1 GKCBAIGFKIBETG-UHFFFAOYSA-N 0.000 description 1
- 229960002372 tetracaine Drugs 0.000 description 1
- 235000019364 tetracycline Nutrition 0.000 description 1
- 150000003522 tetracyclines Chemical class 0.000 description 1
- 229940040944 tetracyclines Drugs 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 230000002110 toxicologic effect Effects 0.000 description 1
- 231100000759 toxicological effect Toxicity 0.000 description 1
- MWKJTNBSKNUMFN-UHFFFAOYSA-N trifluoromethyltrimethylsilane Chemical class C[Si](C)(C)C(F)(F)F MWKJTNBSKNUMFN-UHFFFAOYSA-N 0.000 description 1
- IEDVJHCEMCRBQM-UHFFFAOYSA-N trimethoprim Chemical compound COC1=C(OC)C(OC)=CC(CC=2C(=NC(N)=NC=2)N)=C1 IEDVJHCEMCRBQM-UHFFFAOYSA-N 0.000 description 1
- 229960001082 trimethoprim Drugs 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/21—Esters, e.g. nitroglycerine, selenocyanates
- A61K31/215—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids
- A61K31/235—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids having an aromatic ring attached to a carboxyl group
- A61K31/24—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids having an aromatic ring attached to a carboxyl group having an amino or nitro group
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/65—Tetracyclines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7028—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages
- A61K31/7034—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7048—Compounds having saccharide radicals and heterocyclic rings having oxygen as a ring hetero atom, e.g. leucoglucosan, hesperidin, erythromycin, nystatin, digitoxin or digoxin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/10—Alcohols; Phenols; Salts thereof, e.g. glycerol; Polyethylene glycols [PEG]; Poloxamers; PEG/POE alkyl ethers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/12—Carboxylic acids; Salts or anhydrides thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/14—Esters of carboxylic acids, e.g. fatty acid monoglycerides, medium-chain triglycerides, parabens or PEG fatty acid esters
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P23/00—Anaesthetics
- A61P23/02—Local anaesthetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/06—Antiarrhythmics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/20—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing sulfur, e.g. dimethyl sulfoxide [DMSO], docusate, sodium lauryl sulfate or aminosulfonic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/22—Heterocyclic compounds, e.g. ascorbic acid, tocopherol or pyrrolidones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/26—Carbohydrates, e.g. sugar alcohols, amino sugars, nucleic acids, mono-, di- or oligo-saccharides; Derivatives thereof, e.g. polysorbates, sorbitan fatty acid esters or glycyrrhizin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
- A61K9/0024—Solid, semi-solid or solidifying implants, which are implanted or injected in body tissue
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Molecular Biology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Anesthesiology (AREA)
- Dermatology (AREA)
- Emergency Medicine (AREA)
- Vascular Medicine (AREA)
- Urology & Nephrology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Medicinal Preparation (AREA)
Abstract
"composições injetáveis para a liberação controlada de composto farmacologicamente ativo". a presente invenção fornece composições e métodos para prolongar o tempo de liberação e reduzir a toxicidade do composto farmacologicamente ativo. os compostos compreendem um sal do composto farmacologicamente ativo com um contra-íon lipofílico e um solvente farmaceuticamente aceitável solúvel em água, combinados juntos para formar uma composição injetável. o contra-íon lipofílico pode ser um ácido graxo c~ 8~-c~ 22~ não saturado ou saturado, e preferivelmente pode ser um ácido graxo c~ 10~-c~ 18~ não saturado ou saturado. quando injetada em um mamífero, pelo menos uma porção da composição precipita e libera o composto ativo por um tempo prolongado. desse modo, a composição forma um depósito de droga de liberação lenta do composto ativo no mamífero. portanto, a presente invenção permite que alguém forneça uma administração de dose controlada do composto ativo durante um período de até 15 dias ou mesmo durante um tempo mais prolongado. muitos compostos podem ser administrados de acordo com a presente invenção incluindo, porém não limitado à, tilmicosina, oxitetraciclina, metoprolol, fluoxetina, roxitromicina, e turbinafina.
Description
Relatório Descritivo da Patente de Invenção para COMPOSIÇÕES INJETÁVEIS PARA A LIBERAÇÃO CONTROLADA DE COMPOSTO FARMACOLOGICAMENTE ATIVO, USOS DO REFERIDO COMPOSTO E MÉTODO PARA SUAS PREPARAÇÕES.
Campo da Invenção
A presente invenção refere-se a métodos e composições para prolongar o tempo de liberação e diminuir a toxicidade dos compostos farmacoligicamente ativos.
Fundamentos da Invenção
A seguinte discussão dos fundamentos da invenção é meramente fornecida para auxiliar o leitor no entendimento da invenção e não é admitido descrever ou constituir técnicas anteriores à presente invenção.
É freqüentemente desejável prolongar o tempo de liberação de uma droga injetada para aumentar sua duração de ação, ou reduzir seus efeitos tóxicos. As formulações que são facilmente solúveis no corpo são freqüentemente absorvidas rapidamente e fornecem um estouro súbito de droga disponível quando oposta a uma liberação gradual e mais desejável do produto farmacologicamente ativo. Várias tentativas têm sido feitas para fornecer compostos farmacêuticos de liberação prolongada e controlada, porém não tem sucedido da superação de todos os problemas associados à tecnologia, tal como a obtenção de tempo de liberação prolongado, eficácia e estabilidade máxima, toxicidade reduzida, reprodução máxima na preparação, e a eliminação de efeitos físicos, bioquímicos ou toxicológicos indesejáveis introduzidos por materiais de matriz indesejáveis.
A oxitetraciclina é um antibiótico amplamente empregado e útil para tratar várias infecções em mamíferos. Em particular é empregada para tratar e prevenir infecções respiratórias em animais domésticos. Há custos significantes associados com administrações repetidas através dos meios convencionais.
A tilmicosina é um antibiótico de macrolídeo com duas aminas terciárias. Ela tem uma longa meia-vida de tecido e é eficaz contra uma ampla faixa de bactérias e é empregada para tratar doenças respiratórias no gado. Em níveis elevados a tilmicosina é cardiotóxica e seu uso em espéci es sensíveis, tal como em gatos, cabras, porcos e cavalos foi evitado, quase totalmente devida a razões de segurança. O produto comercial, Micotil® (Eli Lilly & Co., Indianápolis, IN), é uma solução do sal de difosfato e está descrito na Patente U.S. N° 5.574.020. Esta formulação é eficaz em gado, porém o antibiótico é liberado rapidamente e resulta na toxicidade em muitas espécies, incluindo cães e gatos.
Sumário da Invenção
O Pedido de Patente em Série N° 60/343.625, depositado em 19 de outubro de 2001, está aqui incorporado por referência em sua totalidade, incluindo todos os gráficos e desenhos.
A presente invenção fornece composições e método para prolongar o tempo de liberação e redução da toxicidade dos compostos farmacologicamente ativos. Os compostos compreendem um sal do composto farmacologicamente ativo com um contra-íon lipofílico e um solvente solúvel em água farmaceuticamente aceitável combinados juntos para formar uma composição injetável. O contra-íon lipofílico pode ser um ácido graxo Cs-C22 saturado ou não saturado, e preferivelmente pode ser um ácido graxo Cw Cie saturado ou não saturado. Quando injetada em um mamífero, pelo menos uma porção da composição precipita e libera o composto ativo por um tempo prolongado. Desse modo, a composição forma um depósito liberando vagarosamente a droga do composto ativo no mamífero. Portanto, a presente invenção permite que alguém forneça uma administração de dose controlada do composto ativo durante um período de até 15 dias ou ainda mais prolongado. Nas modalidades preferidas, o composto farmacologicamente ativo pode ser tilmicosina, um antibiótico tal como oxitetraciclina ou doxiciclina, ou fluoxetina, roxitromicina, terbinafina, ou metoprolol, e o contra-íon lipofílico pode ser ácido decanóico, ácido láurico, ácido oléico, ácido linoléico ou ácido mirístico. Nas modalidades preferidas, o solvente farmaceuticamente aceitável é N-metil pirrolidona (NMP). Em outra modalidade, o solvente farmaceuticamente aceitável é propileno glicol (por exemplo, em cerca de 10%) em glicerol formal, com ou sem estabilizadores.
A presente invenção também fornece novos métodos de administração de composições e formulações da presente invenção para mamíferos. Os métodos fornecem composições de compostos ativos, que, se presentes em formas atualmente disponíveis, podem resultar na toxicidade 5 ao mamífero tratado. Desse modo, as formulações e métodos da presente invenção permitem que alguém administre compostos que anteriormente não foram capazes de serem amplamente empregados em espécies particulares devido às considerações de segurança. Os métodos também permitem que alguém prolongue o tempo de liberação dos compostos e forneça 10 uma dose controlada do composto ativo ao paciente tratado. Os métodos da presente invenção permitem que alguém administre o composto farmacologicamente ativo ao mamífero tratado em uma quantidade farmaceuticamente eficaz durante 4-15 dias, incluindo qualquer número específico de dias até e incluindo 15 dias, ou ainda mais. O tempo preciso dependerá de 15 várias variáveis que podem ser manipuladas para otimizar a presente invenção para uma aplicação ou composto farmacologicamente ativo particular. Preferivelmente o composto está presente no tecido tratado 4-5 dias após a injeção; e mais preferivelmente o composto está presente no tecido tratado em uma quantidade farmaceuticamente eficaz durante 6 dias, ou ainda 7 20 dias após a injeção. Em outras modalidades, pode também ser desejável manipular as variáveis a fim de prolongar os tempos de liberação até mesmo durante mais do que 15 dias.
Em um aspecto, a presente invenção fornece composições para administração de compostos farmacologicamente ativos. As composições 25 podem compreender um sal do composto farmacologicamente ativo com contra-íon lipofílico e um solvente solúvel em água farmaceuticamente aceitável, combinados juntos sob condições para formar uma composição injetável. A composição precipita e libera o composto farmacologicamente ativo por tempo prolongado quando injetado no mamífero. Em várias moda30 lidades, a composição da presente invenção pode compreender uma ampla variedade de compostos farmacologicamente ativos tal como tilmicosina, oxitetraciclina, doxiciclina, metoprolol, sulfametazina, trimetoprima, neomici-

na, estreptomicina, gentamicina, dibucaína, bupivacaína, benzocaína, tetracaína, acepromazina, itraconazol, tetraciclinas, sulfonamidas, aminoglicosídeos, ou qualquer composto farmacologicamente ativo com funcionalidades químicas e solubilidade apropriadas. O contra-íon lipofílico pode ser um ácido graxo saturado ou não saturado de qualquer número específico de carbonos entre 8 e 22, preferivelmente um ácido graxo C8-C18, e mais preferivelmente um ácido graxo C10-C18, tal como ácido láurico, ácido linoléico, ácido decanóico, e ácido mirístico. Outros contra-íons lipofílicos podem ser também empregados, por exemplo, ácidos dicarboxílicos, tal como ácido sebácico, ácidos poliméricos, tal como ácidos policarboxílicos lipofílicos, e ácidos aromáticos, tal como ácido benzóico. O veículo farmaceuticamente aceitável pode ser um solvente orgânico. Nas modalidades preferidas, o solvente pode ser pirrolidona, N-metil pirrolidona, polietileno glicol, propileno glicol, glicerol formal, éter dimetílico de isosorbida, etanol, sulfóxido de dimetila, álcool de tetraidrofurfurila, triacetina, ou qualquer combinação desses, ou outro solvente constatado ter propriedades aceitáveis similares tal como sendo não tóxico e solúvel em água.
Em outra modalidade as composições da invenção são um sal de um composto farmacologicamente ativo com um contra-íon de ácido policarboxílico e um solvente solúvel em água farmaceuticamente aceitável, combinados juntos sob condições para formar uma composição injetável que precipita quando injetada em água em temperatura ambiente ou precipita em ambientes fisiológicos (in vivo). A composição libera o composto ativo por um tempo prolongado quando injetado em um mamífero. Por ácido policarboxílico é entendido uma molécula contendo pelo menos dois grupos de carboxila. Nas modalidades preferidas, o ácido policarboxílico é ácido poliaspártico, ácido poliacrílico, ácido sebácico, ácido polissebácico, ácido polibenzóico, ou combinações destes. Por poli é entendido dois ou mais.
Em uma modalidade, o composto farmacologicamente ativo pode ser oxitetraciclina, o contra-íon lipofílico pode ser ácido láurico, e o solvente farmaceuticamente aceitável pode ser propileno glicol, polietileno gli5 col, glicerol formal, ou uma combinação apropriada desses. Em outra modalidade o composto farmacologicamente ativo pode ser tilmicosina, o contraíon lipofílico pode ser ácido láurico, e o solvente farmaceuticamente aceitável pode ser propileno glicol, polietileno glicol, glicerol formal, ou uma combinação apropriada desses. Em ainda outra modalidade, as composições podem precipitar e liberar o composto ativo por um tempo prolongado quando introduzido ou injetado em um ambiente aquoso. As composições podem também formar um depósito de droga no mamífero quando injetado, o que libera o composto por um tempo prolongado.
Em outro aspecto, a presente invenção fornece métodos de administração de um composto farmacologicamente ativo a um mamífero. Os métodos podem compreender preparar uma composição de um sal do composto farmacologicamente ativo com um contra-íon lipofílico, e um solvente solúvel em água farmaceuticamente aceitável, combinados juntos sob condições para formar uma formulação injetável, e injetar a composição no mamífero. Pelo menos uma porção da composição precipita e libera o composto farmacologicamente ativo por um tempo prolongado quando injetado no mamífero.
Em outro aspecto, a presente invenção fornece métodos para prolongar o tempo de liberação de um composto farmacologicamente ativo administrado a um mamífero. Os métodos podem compreender preparar uma formulação de um sal do composto farmacologicamente ativo com um contra-íon lipofílico, e um solvente solúvel em água farmaceuticamente aceitável, combinados juntos sob condições para formar uma formulação injetável, e injetar a composição no mamífero, pelo menos uma porção da composição precipita e libera o composto farmacologicamente ativo por um tempo prolongado após a injeção no mamífero, dessa forma prolongando o tempo de liberação do composto. A invenção pode portanto, fornecer uma dosagem controlada do composto ativo ao animal tratado.
Em ainda outro aspecto, a presente invenção fornece métodos para a fabricação de uma formulação injetável para a administração de um composto farmacologicamente ativo a um mamífero. Os métodos podem

compreender formar um sal do composto farmacologicamente ativo com um contra-íon lipofílico, fornecendo solvente farmaceuticamente aceitável solúvel em água, combinando o sal e o solvente sob condições para formar uma formulação injetável, onde pelo menos uma porção da formulação precipita e libera o composto farmacologicamente ativo por um tempo prolongado quando injetado no mamífero.
Em outro aspecto, a presente invenção fornece composições para a administração de um composto farmacologicamente ativo a um mamífero. As composições contêm um sal do composto farmacologicamente ativo com um contra-íon lipofílico e um solvente farmaceuticamente aceitável, combinados juntos para formar uma composição injetável. Pelo menos uma porção do composto farmaceuticamente ativo com contra-íon lipofílico dissolvido no solvente precipita in vivo e libera o composto ativo por um tempo prolongado quando injetado no mamífero.
A presente invenção portanto, oferece importantes vantagens sobre as formulações previamente disponíveis. A presente invenção leva em conta a liberação controlada de compostos farmacologicamente ativos para reduzir a toxicidade, particularmente em pequenos animais tal como cães e gatos. A presente invenção também oferece a vantagem de ser capaz de administrar compostos aos animais domésticos de uma maneira eficiente, por meio da qual requerendo um investimento menor no tempo e recursos do que é disponível com modos anteriores de administração de droga. O composto farmacologicamente ativo está disponível em uma formulação estável, injetável que precipita quando injetada e libera vagarosamente o composto ativo durante um período prolongado de tempo.
O sumário da invenção descrito acima não é limitante e outros aspectos e vantagens da invenção serão aparentes a partir da seguinte descrição detalhada das modalidades preferidas, bem como a partir das reivindicações.
Breve Descrição dos Desenhos
Figura 1 é uma ilustração gráfica mostrando que a oxitetraciclina preparada de acordo com a presente invenção é liberada em solução salina em uma taxa mais lenta do que aquela da droga livre. O solvente é DMSO, e o contra-íon lipofílico é ácido láurico.
Figura 2 é uma ilustração gráfica mostrando que o metoprolol preparado de acordo com a presente invenção é liberado em solução salina em uma taxa mais lenta do que aquela da droga livre. O ácido graxo e solvente são ácido láurico e N-metilpirrolidona.
Figura 3 é uma representação gráfica ilustrando que a taxa de liberação do composto farmacologicamente ativo (tilmicosina) é afetada pela extensão da cadeia do ácido graxo. Solvente: N-metil pirrolidona; contra-íon lipofílico: ácido decanóico e ácido láurico.
Figura 4 é uma representação gráfica ilustrando o efeito do solvente em cinéticos de liberação in vitro em tilmicosina. Contra-íon lipofílico: ácido di(decanóico); tilmicosina em 100 mg/ml na formulação. As abreviações são como seguem: PEG = polietileno glicol, THFA = álcool de tetraidrofurfurila, DMA = dimetil acetamida, ISO-DME= éter dimetílico de isosorbida, DMSO= sulfóxido de dimetila, NMP= N-metil pirrolidona.
Figura 5 é uma representação gráfica ilustrando que a taxa de liberação do composto farmacologicamente ativo (tilmicosina) é uma função da concentração do sal de ácido graxo. Contra-íon lipofílico: ácido decanóico.
Figura 6 é uma representação gráfica ilustrando os cinéticos de liberação in vitro de formulação de sal de ácido graxo de fluoxetina: ácido láurico.
Figura 7 é uma representação gráfica dos farmacocinéticos de cloridrato de fluoxetina (HCI) e sal de ácido graxo de fluoxetina: ácido láurico (FAS) em gatos.
Figura 8 é um plano semilogarítmico da concentração de tilmicosina no tecido do pulmão do gato durante 21 dias. Oito gatos machos e oito fêmeas foram empregados e dosados com 10 mg/kg de peso do corpo para todos os tipos de tecido.
Figura 9 é um plano semilogarítmico de concentração de tilmicosina no tecido do rim do gato durante 21 dias. Oito gatos machos e oito fê meas foram empregados e dosados com 10 mg/kg de peso do corpo para todos os tipos de tecido.
Figura 10 é um plano semilogarítmico da concentração de tilmicosina no tecido do fígado do gato durante 21 dias. Oito gatos machos e oito fêmeas foram empregados e dosados com 10 mg/kg de peso do corpo para todos os tipos de tecido.
Descrição Detalhada da Invenção
As composições da presente invenção podem ser preparadas empregando sais de compostos farmacologicamente ativos com funcionalidades básicas. Esses podem ser feitos empregando uma variedade de ácidos lipofílicos, ácidos graxos saturados ou não saturados, ácidos cólicos, ácidos fosfatídicos, ácidos dicarboxílicos tal como ácido sebácico ou qualquer ácido que, quando combinado com o composto farmacologicamente ativo, transmita o sal resultante insolúvel em água, porém solúvel em um solvente solúvel em água. Por sal, entende-se dois compostos que não são covalentemente ligados porém são quimicamente ligados através de atrações iônicas. Por miscível em água é entendido que o solvente capaz de misturar em qualquer relação em água sem separação de duas fases. Por solúvel em água é entendido que o solvente tem algum nível significante de solubilidade em soluções aquosas, por exemplo, a triacetina é considerada um solvente solúvel em água uma vez que ela é solúvel em água em uma relação de cerca de 1:14. Por um contra-íon lipofílico, entende-se uma forma iônica de uma molécula solúvel em gordura. O contra-íon lipofílico pode preferivelmente ser um ácido graxo, porém pode ser outra molécula solúvel em gordura. O contra-íon tem pelo menos uma carga oposta àquela de um grupo químico em uma oposição ao membro de sal, dessa forma causando uma atração iônica entre as duas moléculas. Por formulação injetável ou composição injetável entende-se uma formulação ou composição que possa ser puxada em uma seringa e injetada subcutaneamente, intraperitonealmente, ou intra-muscularmente em um mamífero sem causar efeitos adversos devido à presença de materiais sólidos na composição. Os materiais sólidos incluem, porém não estão limitados a, cristais, uma massa gomosa, e um gel. Por composto farmacologicamente ativo entende-se urn composto químico que causa um efeito farmacológico no mamífero tratado. Por exemplo, o efeito pode ser destruir ou prevenir o crescimento de bactérias ou parasitas, reduzir a inflamação, ou outro efeito mensurável e farmacêutico no mamífero tratado.
Pelo verbo precipitar entende-se que o composto forma um precipitado ou sólido. Um precipitado é um sólido insolúvel formado em solução em temperatura ambiente in vitro ou em um ambiente fisiológico (in vivo). O precipitado pode tomar muitas formas tal como, por exemplo, um sólido, um cristal, uma massa gomosa, ou um gel. Por quantidade farmaceuticamente eficaz, entende-se uma quantidade que exerce um efeito médico significante e mensurável no mamífero tratado, resultando no progresso voltado para curar ou prevenir a doença do indivíduo, ou aliviar ou prevenir a condição que foi a razão para o tratamento. Um solvente farmaceuticamente aceitável é um líquido que dissolve um sal do composto farmacologicamente ativo e um contra-íon lipofílico, e que é adequado para uso com seres humanos e/ou animais sem efeitos colaterais adversos indevidos (tal como toxicidade, irritação, e resposta alérgica) comensurável com uma relação de benefício/risco razoável.
As composições da presente invenção oferecem várias vantagens. As composições são composições injetáveis que contêm altas concentrações do composto ativo. Nas modalidades preferidas, o composto farmacologicamente ativo pode ser carregado na composição na faixa de 10% -60% (peso/volume). Porém a pessoa versada na técnica perceberá que esta faixa pode ser variada amplamente, dependendo da solubilidade ou insolubilidade do composto farmacologicamente ativo, o contra-íon selecionado, o solvente selecionado, a injetabilidade do produto final, e qualquer outra necessidade relevante da aplicação particular. O composto ativo pode também ser carregado com tão pouco quanto 10%, ou 5%, ou mesmo 1% e ainda fornecer um efeito útil. Similarmente, o composto ativo pode ser carregado com 70%, ou mesmo mais quando a necessidade exigir. Nenhum veículo ou excipiente exótico é requerido. As composições são facilmente filtra das, por meio das quais simplificando o processo de fabricação. Acredita-se que a exclusão da água da formulação deva transmitir maior estabilidade para as formulações, e inibir o crescimento de microorganismos. Os processos para a preparação das composições, como descrito aqui, são simples, e a administração de acordo com a presente invenção deve resultar em reações mais brandas no local de injeção devido à neutralização do composto farmacologicamente ativo.
A presente invenção fornece a capacidade de modular a taxa de liberação e tempo de liberação do composto farmacologicamente ativo. A taxa de liberação pode ser modulada variando-se a lipofilicidade e peso molecular do contra-íon empregado para fazer o sal. Por exemplo, os sais de ácido láurico de tilmicosina são freqüentemente liberados mais vagarosamente do que os sais de decanoatos. Além disso, as concentrações mais altas do sal na formulação freqüentemente produzem taxas de liberação mais vagarosas. O sal de decanoato de tilmicosina é liberado mais vagarosamente de uma formulação de sal de tilmicosina-ácido graxo de 60% do que de uma formulação de sal de tilmicosina-ácido graxo de 30%. Similarmente, como explicado aqui, outras variáveis tal como seleção de contra-íon lipofílico, seleção de solvente, concentração de sal, e outras podem ser manipuladas para prolongar ou encurtar o tempo de liberação do composto ativo ao ponto desejado. Geralmente, pode ser desejável para os sais ser com base na relação molar dos grupos carregados. Porém alguém pode sucessivamente criar sais insolúveis utilizando-se um hemi-sal ou variando-se de outro modo de uma relação de 1:1. O solvente farmaceuticamente aceitável pode ser um solvente solúvel em água ou miscível em água, e preferivelmente pode ser um solvente miscível em água. As misturas de solventes miscíveis em água e/ou solúveis em água podem também ser utilizadas. A pessoa versada na técnica perceberá que vários solventes solúveis em água podem ser misturados para otimizar o resultado para uma aplicação particular. Por exemplo, uma mistura de polietileno glicol, propileno glicol, e glicerol formal pode ser misturada em várias relações para fornecer um solvente ótimo. Em algumas modalidades, misturar em quantidades aproxima11 damente iguais pode fornecer um solvente adequado.
Em outras modalidades, as formulações da invenção contendo um sal do composto farmacologicamente ativo com um contra-íon lipofílico podem ser combinadas com a forma não salgada do ativo, a fim de fornecer uma dose inicial maior do composto ativo.
Sem desejar estarem ligadas a qualquer teoria particular, as composições injetáveis podem ser obtidas quando um sal é formado de um agente farmacologicamente ativo com um contra-íon lipofílico, e combinado com um solvente orgânico parenteral. Acredita-se que quando esta formulação é injetada em um mamífero, o solvente pode difundir para longe do local de injeção quando os fluidos do corpo aquoso difundem em direção ao local, resultando na precipitação do composto farmacologicamente ativo no mamífero tratado. O precipitado pode tomar muitas formas, por exemplo, um sólido, cristais, uma massa gomosa, ou um gel. Existirá desse modo, uma concentração do composto ativo que é liberada em uma quantidade farmacologicamente eficaz durante um período desejado de tempo. O precipitado pode atuar como um depósito de droga no mamífero resultando na liberação do composto durante um período de tempo. Os tempos de liberação podem ser obtidos em pelo menos 3 dias, pelo menos 4 dias, pelo menos 5 dias, pelo menos 6 dias, pelo menos 7 dias, ou qualquer número específico de dias até e incluindo pelo menos 15 dias, ou ainda por mais tempo, quando desejado. Por depósito de droga é entende-se uma concentração ou precipitação do composto farmacologicamente ativo, no corpo do mamífero tratado, que libera uma quantidade farmaceuticamente eficaz do composto ativo durante um tempo prolongado.
Foi mostrado que a extensão da cadeia do ácido graxo, as combinações particulares de ácidos graxos, o percentual de composto farmacologicamente ativo: sal de contra-íon lipofílico na formulação, e o solvente farmaceuticamente aceitável selecionado, todos influenciam nos cinéticos de liberação do composto farmacologicamente ativo. Desse modo, os cinéticos de liberação do composto farmacologicamente ativo podem ser convenientemente e facilmente manuseados manipulando-se essas e outras variáveis.
Foi também constatado que as formulações ficaram estáveis por ser esterilizadas por autoclave. A pessoa de versada na técnica perceberá que a presente invenção pode ser aplicada a muitos compostos farmacologicamente ativos que têm uma funcionalidade química e solubilidade apropriada. Desse modo, é considerado que a presente invenção possa ser aplicável a uma ampla variedade de compostos farmacologicamente ativos, tal como drogas, medicamentos, nutrientes, ou outros compostos desejáveis para administração a um mamífero.
A pessoa versada na técnica perceberá que algumas modificações aos métodos apresentados aqui podem ser desejáveis com base nas características particulares do composto farmacologicamente ativo envolvido. Os seguintes exemplos não limitantes apresentam ainda aplicações da presente invenção e são fornecidos com o objetivo de exemplo somente. Exemplo 1: Oxitetraciclina

A oxitetraciclina tem um grupo de amina terciária, e o sal de cloridrato de oxitetraciclina é facilmente solúvel em água. Constatou-se que a adição de um mol dos ácidos graxos a um mol de oxitetraciclina cria um sal que tem uma solubilidade menor em água porém é mais solúvel do que a oxitetraciclina inicial em N-metilpirrolidona (NMP). Quando a água é adicionada à formulação de NMP, o sal precipita.
A taxa in vitro de liberação de oxitetraciclina pode ser determinada selando-se a formulação em uma bolsa de diálise (Pierce, Rockford, IL), colocando-a em um recipiente de solução solução salina, e medindo-se a quantidade de droga na solução salina como uma função de tempo. A formulação da presente invenção foi comparada com as formulações de oxitetraciclina existentes. A Figura 1 mostra que a formulação de oxitetraciclina da presente invenção é liberada na solução salina em uma taxa substancialmente menor do que aquela da droga livre.
Uma composição de oxitetraciclina de acordo com a presente invenção foi preparada adicionando-se 0,464 grama de oxitetraciclina e 0,203 gramas de ácido láurico a 3 ml de NMP. A mistura foi agitada durante 60 minutos, resultando em uma solução clara. 1 ml desta solução foi selado em uma bolsa de diálise, e a bolsa foi suspensa em 150 ml da solução salina tamponada por fosfato, pH 7,4. As alíquotas foram removidas em vários intervalos e a concentração de oxitetraciclina foi determinada por espectrofotometria. Os resultados na Figura 1 mostram que a oxitetraciclina continuou a difundir para fora da bolsa durante mais do que 120 horas, ponto no qual somente cerca de 50% da oxitetraciclina presente foi liberada.
Exemplo 2: Metoprolol
O metoprolol é uma droga anti-hipertensiva, antianginal e antiarrítmica, da seguinte estrutura:

Seus sais de succinato e tartarato são comercialmente disponíveis sob vários nomes comerciais. Ambos esses sais, bem como a forma de base da droga, são altamente solúveis em água. A forma de base do metoprolol foi preparada a partir de sal de tartarato comercialmente disponível por procedimento padrão. Quando o grupo amina de metoprolol é neutralizado com ácido láurico, o sal resultante é fracamente solúvel em água, porém facilmente solúvel em solventes não aquosos farmaceuticamente aceitáveis. Uma composição de metoprolol de acordo com a presente invenção foi preparada adicionando-se 0,3224 grama de base de metoprolol e 0,2661 grama de ácido láurico a 2,415 ml de NMP. A mistura foi agitada durante 30 minutos, resultando em uma solução clara. Um ml desta solução foi selado em uma bolsa de diálise, e a bolsa foi suspensa em 150 ml de solução salina tamponada por fosfato, pH 7,4. As alíquotas foram removidas em vários intervalos e a concentração de metoprolol foi determinada por espectrofotometria. Os resultados da Figura 2 mostram que o metoprolol continuou a difundir para fora da bolsa durante mais do que 48 horas ao mesmo tempo em que a solução de controle da base de metoprolol (preparada dissolvendo-se 150 mg em 1,124 ml de NMP) é rapidamente difundida.
Exemplo 2A: Tilmicosina
A tilmicosina é um antibiótico na classe dos macrolídeos com a seguinte estrutura:

É eficaz contra uma faixa ampla de bactéria, e é empregada para o tratamento de doenças respiratórias em gados. A forma básica é moderadamente solúvel em soluções aquosas, ao mesmo tempo em que os sais de fosfato e cloreto são altamente solúveis. Em níveis elevados, a tilmicosina é cardiotóxica, e então não é intravenosamente administrada. Por razões de segurança, seu uso tem sido evitado quase completamente em espécies sensíveis tal como gatos, cabra, porcos, e cavalos.
Quando os dois grupos de amina de tilmicosina são neutralizados com quaisquer dos vários ácidos graxos (tal como, por exemplo, decanóico C10, láurico C12, mirístico C14, palmítico C16, esteárico C18, oléico C18, elaídico C18, linoléico C18, e erúcico C22), o sal resultante é fracamente solúvel em água, porém facilmente solúvel em solventes não aquosos farmaceuticamente aceitáveis. Quando a formulação do sal é selada em um cassete de diálise e colocada em solução salina, o sal de tilmicosina precipita, e a tilmicosina é vagarosamente liberada da bolsa. A taxa de liberação é uma função de cadeia longa do ácido graxo (Figura 3), o solvente (Figura 4) e a concentração de sal de ácido graxo de tilmicosina (Figura 5).
Exemplo 3- Sal de Tilmicosina in Vitro gramas (0,0115 mol) de tilmocosina e 0,0253 mol de vários ácidos carboxílicos (tal como, por exemplo, ácidos decanóico, láurico, linoléico, ou mirístico, em ensaios individuais) foram tomados em um frasco e feitos até um volume final de 100 ml com N-metil-pirrolidona e agitado durante 60 minutos para obter uma solução clara. As alíquotas de um ml dessas soluções foram seladas em bolsas de diálise, e as bolsas foram suspensas em frascos contendo 150 ml de solução salina tamponada por fosfato, pH 7,4. O sal foi observado precipitar na bolsa durante cerca de 1 hora. As alíquotas da solução salina foram removidas em vários intervalos e a tilmicosina foi determinada por HPLC. Os resultados com sais de ácido decanóico (C-10) e ácido láurico (C-12) na Figura 3 mostram que a tilmicosina continuou a difundir para fora da bolsa durante mais do que 120 horas. Os ácidos de extensão de cadeia mais longa causaram uma liberação mais lenta de tilmicosina. Micotil® (Eli Lilly, Indianápolis, IN), um sal de fosfato de tilmicosina, é facilmente solúvel e rapidamente difunde da bolsa.
Exemplo 4- Diíácido decanóico) de Tilmicosina em vários Solventes
As soluções de sal de di(ácido decanóico) de tilmicosina foram preparadas em vários solventes miscíveis em água, combinando 10 gramas (0,0115 móis) de tilmicosina e 0,0253 móis de ácido decanóico em vários solventes para um volume final de 100 ml. As taxas de liberação in vitro foram medidas empregando o método de diálise do Exemplo 1. Os resultados na Figura 4 mostram que a taxa de liberação varia com o solvente, porém que todos os solventes produziram uma taxa de liberação mais lenta do que aquela observada com sal de fosfato Micotil®, mostrado na Figura 3. Exemplo 5 - Efeito da Concentração de Liberação da Tilmicosina
As soluções de sal de di(ácido decanóico) de tilmicosina foram preparadas combinando-se 30 gramas (0,0345 mol) ou 60 gramas (0,0690 mol) de tilmicosina com 2 equivalentes de ácido decanóico em NMP para um volume final de 100 ml. A taxa de liberação in vitro foi medida empre gando o método de diálise do Exemplo 1, e os dados da Figura 5 mostram que as concentrações iniciais mais elevadas resultaram em uma taxa de liberação mais lenta.
Exemplo 6 - Tolmicosina in vivo
As formulações de didecanoato, dilaurato, e dimiristato de tilmicosina foram formuladas em 100 mg/ml em N-metil pirrolidona e injetada subcutaneamente em gatos atrás do pescoço em uma dosagem de 45 ou 75 mg/kg de peso do corpo. Os dados anteriores indicam que uma dosagem de 25 mg/kg do sal de fosfato de tilmicosina é fatal para gatos. Os gatos mostraram hipotermia e letargia após injeção, indicativo da biodisponibilidade da droga. A toxicidade foi constatada ser substancialmente menor em formulações com comprimentos de cadeias de ácidos graxos maiores do que C10, consistente com a liberação mais lenta da droga dessas formulações. Todos os gatos sobreviveram e comportaram-se normalmente 3 dias após a injeção. Os resultados são sumarizados na Tabela 1 seguinte.
Tabela 1: Níveis de Sangue da Tilmicosina em Intervalos de Tempo Específico
100 mg/ml de uma formulação de um sal de tilmicosina com ácido decanóico, ácido láurico, ou ácido mirístico em N-metil pirrolidona foram injetados em 9 gatos adultos saudáveis nas dosagens indicadas. As concentrações resultantes em plasma e células sanguíneas para cada gato individual em 6 horas e 2 dias são mostradas na Tabela 1.
| N° de gato | Ácido graxo | Dosagem | Células sanguíneas 6 horas (pg/ml) | Plasma 6 horas (gg/ml) | Plasma 6 horas (pg/ml) | Células sanguíneas 2 dias (gg/ml) |
| 1 | C-12 | 45 mg/kg | 5,8 | 11,3 | 0,9 | 0,5 |
| 2 | C-12 | 75 mg/kg | 5,5 | 18,0 | 1,5 | 1,5 |
| 3 | C-12 | 75 mg/kg | 2,5 | 9,9 | 0,9 | 1,0 |
| 4 | C-14 | 45 mg | 2,4 | 7,5 | 0,9 | 1,1 |
| 5 | C-14 | 75 mg/kg | 6,7 | 11,6 | 9,1 | 4,6 |
| 6 | C-14 | 75 mg/kg | 3,5 | 6,1 | 1,5 | 0,7 |
| 7 | C-10 | 45 mg/kg | 3,7 | 15,9 | ||
| 8 | C-10 | 75 MG/kg | 4,2 | 18,4 | ||
| 9 | C-10 | 45 mg/kg | 3,3 | 9,1 |
Os sais de tilmicosina foram também estudados no tecido. Uma tilmicosina: sal de ácido graxo diláurico em 10% de propileno glicol em glicerol formal em 100 mg/ml foi administrado subcutaneamente em 10 mg/kg e a 5 biodistribuição em gatos foi determinada.
Os métodos descritos aqui foram desenvolvidos para a determinação e a quantificação da tilmicosina em vários tecidos de animais e soro, particularmente soro e tecido do fígado, rim e pulmão do felino. A pessoa versada na técnica perceberá que muitas variações dos métodos descritos 10 aqui são possíveis sem afastar-se da invenção.
As amostras de tecido do fígado, rim e pulmão foram coletadas 2, 3, 4, 7, 14 e 21 dias após injetar os animais com formulação de tilmicosina em 10 mg/kg de peso do corpo. Os resultados são apresentados nas Figuras 8, 9 & 10. Foi constatado que o rim é o tecido marcador em gatos ao 15 mesmo tempo em que o fígado é o marcador em vacas e porcos. Os níveis no rim foram consistentemente mais elevados do que no fígado com Cmax (rim: 13,8 mcg/gm; Fígado: 7,3 mcg/gm) alcançados durante cerca de 48 horas em ambos tecidos. O Cmax para pulmão foi constatado ser 7,5 mcg/gm e foi observado durante cerca de 48 horas após injetar a dose. Os níveis detectáveis de tilmicosina persistiram no tecido até o dia 21 do estudo.
A eficiência da extração da droga de -98% foi obtida em uma concentração de 1 mcg/gm de tecido. O limite da detecção para timilcosina em vários tecidos felinos foi determinado ser 0,032 mcg/gm. Para soro felino, a eficiência da extração da droga de 95% foi obtida em uma faixa de concentração de 0,15 a 6 mcg/ml após fortificação. O limite da detecção foi determinado ser 0,16 mcg/ml com o prolongamento da linearidade de 0,15 a 6 mcg/ml.
Preparação de Amostras de Tecido
As amostras de tecido foram preparadas por moagem com tesouras ou um bisturi em uma toalha de papel. 10 ml de metanol foi adicionado à cada tubo e as amostras homogeneizadas separadamente durante 10 à 15 minutos. As amostras foram sonicadas no gelo durante um minuto e centrifugadas em 10.000 rpm durante 30 minutos em 4C. O extrato de metanol foi decantado em tubos de centrífuga frescos e as amostras de tecido suspensas novamente nos tubos com 10 ml de metanol e 5,0 ml_ de 100 mM de fosfato. Os tubos foram turbilhados e centrifugados em 10.000 rpm durante 30 minutos em 4C. O extrato foi decantado em tubos de centrifuga frescos e centrifugado em 500 rpms durante 10 minutos em 4C. O extrato de metanol foi adicionado a 70 ml de água em um frasco e turbilhado no frasco para misturar.
Cada grupo de extrato foi carregado para um cartucho C18 SepPak Plus® (SPE) de extração de fase sólida (Waters, Milford, MA) empregando uma bomba hidrostática ou vários vácuos para extrair os grupos através dos cartuchos. Uma vez que a amostra tenha sido completamente carregada, cada cartucho SPE foi lavado com 10 mL de água, e em seguida com 10 mL de acetonitrila 25%/água em uma taxa de fluxo menor do que 5 ml/minuto. Os cartuchos SPE foram desconectados do aparelho e completamente secados sob vácuo elevado (26 em Hg) em uma jarra de dessecação a vácuo durante 10 minutos.
O análito foi eluído dos cartuchos SPE com 5% de ácido acético/metanol. Somente os primeiros 2,0 ml_ do eluído foram coletados. Os frascos volumétricos foram invertidos e misturados, e armazenados durante a noite em 4C.
Os eluatos da amostra foram filtrados através de um filtro PVDF de 0,22 μηη, e analisado por HPLC em uma coluna de fenila de SphereClone® de 5 pm (Phenomenex, Torrance, CA).
Exemplo 7- Fluoxetina:
H
I
N.
CH3

A fluoxetina é um inibidor da recaptação de serotonina seletiva e é extensivamente empregada para tratar distúrbios fisiológicos tal como distúrbio obsessivo compulsivo em seres humanos. É mostrado que a fluoxetina é eficaz para tratar comportamento agressivo e ansiedade da separação em cachorros e comportamento de respingo de urina em gatos. A fluoxetina é formulada em 100 e 150 mg/ml como um sal de ácido láurico em 10% de propileno glicol em glicerol formal na relação de 1:1 de droga para ácido graxo. Os cinéticos de liberação in vitro foram estudados para ambas formulações em 100 mg/mL de concentração empregando a técnica de diálise descrita no Exemplo 1 e os resultados apresentados na Figura 6. A base de fluoxetina em 10% de propileno glicol em glicerol formal em 100 mg/ml é empregada como um controle na experiência. Ao mesmo tempo em que a formulação de base de fluoxetina é liberada durante cerca de 160 horas, a formulação de sal de ácido láurico de 1:1 (droga: LA) foi liberada durante 700 horas.
A formulação de sal de ácido graxo com a relação de fluoxetina para ácido láurico de 1:1,1 em 150 mg/ml da concentração foi injetada subcutaneamente em gatos em 20 e 30 mg/kg. Ao mesmo tempo dois gatos foram dosados em 1 mg/kg/dia oralmente durante 28 dias. As amostras de soro foram coletadas até o dia 42 e analisadas quanto à fluoxetina por
HPLC empregando 35% de acetonitrila em tampão de fosfato, pH 2,7 em uma coluna Ci8. O resultado sugere que uma única injeção subcutânea da formulação de sal de ácido graxo fornece concentrações de droga comparáveis com as doses orais diárias até o dia 42 (Figura 7).
A fluoxetina foi também formulada como sal de ácido linoléico em 10% de propileno glicol em glicerol formal, produzindo uma solução clara. Esta formulação foi também constatada precipitar em água.
Exemplo 8- Roxitromicina
A roxitromicina é um antibiótico na classe dos macrolídeos com a seguinte estrutura:

É eficaz contra uma faixa ampla de bactérias, e é empregado para o tratamento de doenças respiratórias em gado. O grupo amina de roxitromicina foi neutralizado com ácido linoléico em 10% de propileno em glicerol formal, resultando em uma solução clara em 200 mg/ml. Como no caso 15 de tilmicosina, o sal precipitou quando a solução foi reforçada em água.
Exemplo 9 - Terbinafina
A terbinafina é um antifúngico e a estrutura é como mostrada abaixo:
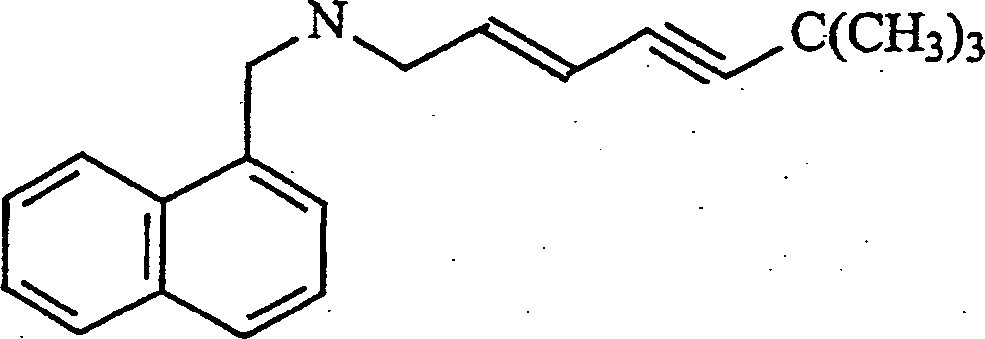
A terbinafina é um inibidor específico de epoxidase de esqualina, uma enzima importante na biossíntese de ergosterol fúngico. O grupo amina de terbinafina foi neutralizado com ácido linoléico em 10% de propileno glicol em glicerol formal, resultando em uma solução clara em 150 mg/ml. O sal resultante foi altamente insolúvel em água, e precipitado quando reforçado em água.
Ao mesmo tempo em que a invenção tem sido descrita e exemplificada em detalhes suficientes por aqueles versados nesta técnica para fazê-la e usá-la, várias alternativas, modificações, e melhoras devem ser aparentes sem afastar-se do espírito e escopo da invenção.
Alguém versado na técnica facilmente aprecia que a presente invenção é bem adaptada para realizar os objetivos e obter as finalidades e vantagens mencionadas, bem como aqueles inerentes aqui. As modificações aqui e outros usos ocorrerão por aqueles versados na técnica. Essas modificações são abrangidas no espírito da invenção e são definidas pelo escopo das reivindicações.
Será facilmente aparente por uma pessoa versada na técnica que modificações e substituições variantes possam ser feitas para a invenção divulgada aqui sem afastar-se do escopo e espírito da invenção.
Todas as patentes e publicações mencionadas no relatório descritivo são indicativas dos níveis daqueles versados na técnica à qual a invenção pertence.
A invenção ilustrativamente descrita aqui adequadamente pode ser praticada na ausência de qualquer elemento ou elementos, limitação ou limitações que não seja especificadamente divulgado aqui. Os termos e expressões que foram empregados são usados como termos de descrição e não de limitação, e não há nenhuma intenção que no uso de tais termos e expressões de excluir qualquer equivalente das características mostradas e descritas ou porções destes, porém é reconhecido que várias modificações são possíveis no escopo da invenção reivindicada. Desse modo, deve ser entendido que embora a presente invenção tenha sido especificadamente divulgada pelas modalidades preferidas e características opcionais, a modi ficação e variação dos conceitos aqui divulgados podem ser utilizadas por aqueles versados na técnica, e que tais modificações e variações são consideradas estarem no escopo desta invenção como definido pelas reivindicações anexas.
Além disso, onde as características ou aspectos da invenção estão descritos em termos de grupos Markush, aqueles versados na técnica reconhecerão que a invenção é também desse modo descrita em termos de qualquer membro individual ou subgrupo de membros do grupo Markush. Por exemplo, se X é descrito como selecionado a partir do grupo consistindo 10 em bromo, cloro, e iodo, as reivindicações para X sendo bromo e as reivindicações para X sendo bromo e cloro são totalmente descritas.
Outras modalidades são apresentadas nas seguintes reivindicações:
Claims (24)
- REIVINDICAÇÕES1. Composição, caracterizada pelo fato de que compreende:um sal compreendendo um composto farmacologicamente ativo básico e um ácido lipofílico, e um solvente orgânico farmaceuticamente aceitável miscível em água combinados juntos para formar uma composição injetável, em que o ácido lipofílico é selecionado a partir do grupo consistindo em ácido decanóico, ácido láurico, ácido mirístico, ácido palmítico, ácido esteárico, ácido oleico, ácido elaídico, ácido linoleico, ácido erúcico;o composto farmacologicamente ativo é selecionado dentre o grupo consistindo em oxitetraciclina, metoprolol, tilmicosina, fluoxetina, roxitromicina, terbinafina; e o solvente orgânico farmaceuticamente aceitável é selecionado dentre o grupo consistindo em polietileno glicol, álcool de tetraidrofurfurila, dimetilacetamida, éter dimetílico de isosorbida, sulfóxido de dimetila, N-metil pirrolidona, propilenoglicol e glicerol formal;em que a razão molar de ácido lipofílico para composto farmacologicamente ativo é de pelo menos 1;em que o sal é solúvel no solvente orgânico farmaceuticamente aceitável;em que pelo menos uma porção da composição forma um precipitado quando a composição é introduzida ou injetada em um ambiente aquoso; e em que a composição libera o composto ativo por um tempo prolongado quando introduzida ou injetada no ambiente aquoso.
- 2. Composição de acordo com a reivindicação 1, caracterizada pelo fato de que o ácido lipofílico é selecionado de um grupo consistindo dentre ácido decanóico, ácido láurico, ácido mirístico, ácido oleico e ácido linoleico.
- 3. Composição de acordo com a reivindicação 1, caracterizada pelo fato de que o solvente farmaceuticamente aceitável compreende 10%Petição 870180155523, de 27/11/2018, pág. 4/13 de propileno glicol em glicerol formal.
- 4. Composição de acordo com a reivindicação 1, caracterizada pelo fato de que o composto farmacologicamente ativo é terbinafina e o solvente farmaceuticamente aceitável compreende glicerol formal.
- 5. Composição de acordo com a reivindicação 1, caracterizada pelo fato de que o composto farmacologicamente ativo é oxitetraciclina, o contra-íon lipofílico é um ácido láurico, e o solvente orgânico farmaceuticamente aceitável é selecionado do grupo consistindo dentre polietilenoglicol, propilenoglicol e glicerol formal.
- 6. Composição de acordo com a reivindicação 5, caracterizada pelo fato de que o composto farmacologicamente ativo é tilmicosina, o contra-íon lipofílico é um ácido láurico, e o solvente orgânico farmaceuticamente aceitável é selecionado do grupo consistindo dentre polietilenoglicol, propilenoglicol e glicerol formal.
- 7. Uso de um sal compreendendo um composto farmacologicamente ativo básico e um ácido lipofílico, combinados com um solvente orgânico farmaceuticamente aceitável miscível em água, caracterizado pelo fato de que é na preparação de uma composição injetável para administração de um composto farmacologicamente ativo a um mamífero, em que o ácido lipofílico é selecionado a partir do grupo consistindo em ácido decanóico, ácido láurico, ácido mirístico, ácido palmítico, ácido esteárico, ácido oleico, ácido elaídico, ácido linoleico, ácido erúcico;o composto farmacologicamente ativo é selecionado dentre o grupo consistindo em oxitetraciclina, metoprolol, tilmicosina, fluoxetina, roxitromicina e terbinafina; e o solvente orgânico farmaceuticamente aceitável é selecionado dentre o grupo consistindo em polietileno glicol, álcool de tetraidrofurfurila, dimetilacetamida, éter dimetílico de isosorbida, sulfóxido de dimetila, N-metil pirrolidona, propilenoglicol e glicerol formal;em que a razão molar de ácido lipofílico para composto fármacologicamente ativo é de pelo menos 1;Petição 870180155523, de 27/11/2018, pág. 5/13 em que o sal é solúvel no solvente orgânico farmaceuticamente aceitável;em que pelo menos uma porção da composição forma um precipitado quando a composição é introduzida ou injetada em um ambiente aquoso, e em que a composição libera o composto ativo por um tempo prolongado quando injetada no mamífero.
- 8. Uso, de acordo com a reivindicação 7, caracterizado pelo fato de que o ácido lipofílico é selecionado de um grupo consistindo dentre ácido decanóico, ácido láurico, ácido mirístico, ácido oleico e ácido linoleico.
- 9. Uso, de acordo com a reivindicação 7, caracterizado pelo fato de que o solvente farmaceuticamente aceitável compreende 10% de propileno glicol em glicerol formal.
- 10. Uso, de acordo com a reivindicação 7, caracterizado pelo fato de que o composto farmacologicamente ativo é terbinafina e o solvente farmaceuticamente aceitável compreende glicerol formal.
- 11. Uso, de acordo com a reivindicação 7, caracterizado pelo fato de que o composto farmacologicamente ativo é oxitetraciclina, o contraíon lipofílico é um ácido láurico, e o solvente orgânico farmaceuticamente aceitável é selecionado do grupo consistindo dentre polietilenoglicol, propilenoglicol e glicerol formal.
- 12. Uso, de acordo com a reivindicação 7, caracterizado pelo fato de que o composto farmacologicamente ativo é tilmicosina, o contra-íon lipofílico é um ácido láurico, e o solvente orgânico farmaceuticamente aceitável é selecionado do grupo consistindo dentre polietilenoglicol, propilenoglicol e glicerol formal.
- 13. Uso de um sal compreendendo um composto farmacologicamente ativo básico e um ácido lipofílico, combinados com um solvente orgânico farmaceuticamente aceitável miscível em água, caracterizado pelo fato de que é na preparação de uma composição injetável para estender o tempo de liberação de um composto farmacologicamente ativo administrado a um mamífero,Petição 870180155523, de 27/11/2018, pág. 6/13 em que o ácido lipofílico é selecionado a partir do grupo consistindo em ácido decanóico, ácido láurico, ácido mirístico, ácido palmítico, ácido esteárico, ácido oleico, ácido elaídico, ácido linoleico, ácido erúcico;o composto farmacologicamente ativo é selecionado dentre o grupo consistindo em oxitetraciclina, metoprolol, tilmicosina, fluoxetina, roxitromicina e terbinafina; e o solvente orgânico farmaceuticamente aceitável é selecionado dentre o grupo consistindo em polietileno glicol, álcool de tetraidrofurfurila, dimetilacetamida, éter dimetílico de isosorbida, sulfóxido de dimetila, N-metil pirrolidona, propilenoglicol e glicerol formal;em que a razão molar de ácido lipofílico para composto fármacologicamente ativo é de pelo menos 1;em que o sal é solúvel no solvente orgânico farmaceuticamente aceitável;em que pelo menos uma porção da composição forma um precipitado quando a composição é introduzida ou injetada em um ambiente aquoso, e em que a composição libera o composto ativo por um tempo prolongado quando injetada no mamífero, através disso, estendendo o tempo de liberação do composto farmacologicamente ativo.
- 14. Uso, de acordo com a reivindicação 13, caracterizado pelo fato de que o ácido lipofílico é selecionado de um grupo consistindo dentre ácido decanóico, ácido láurico, ácido mirístico, ácido oleico e ácido linoleico.
- 15. Uso, de acordo com a reivindicação 13, caracterizado pelo fato de que o solvente farmaceuticamente aceitável compreende 10% de propileno glicol em glicerol formal.
- 16. Uso, de acordo com a reivindicação 13, caracterizado pelo fato de que o composto farmacologicamente ativo é terbinafina e o solvente farmaceuticamente aceitável compreende glicerol formal.
- 17. Uso, de acordo com a reivindicação 13, caracterizado pelo fato de que o composto farmacologicamente ativo é oxitetraciclina, o contraPetição 870180155523, de 27/11/2018, pág. 7/13 íon lipofílico é um ácido láurico, e o solvente orgânico farmaceuticamente aceitável é selecionado do grupo consistindo dentre polietilenoglicol, propilenoglicol e glicerol formal.
- 18. Uso, de acordo com a reivindicação 13, caracterizado pelo fato de que o composto farmacologicamente ativo é tilmicosina, o contra-íon lipofílico é um ácido láurico, e o solvente orgânico farmaceuticamente aceitável é selecionado do grupo consistindo dentre polietilenoglicol, propilenoglicol e glicerol formal.
- 19. Método para preparar uma composição injetável para a administração de um composto farmacologicamente ativo a um mamífero, caracterizado pelo fato de que compreende:(a) prover um sal compreendendo um composto farmacologicamente ativo básico e um ácido lipofílico, em que o ácido lipofílico é selecionado a partir do grupo consistindo em ácido decanóico, ácido láurico, ácido mirístico, ácido palmítico, ácido esteárico, ácido oleico, ácido elaídico, ácido linoleico, ácido erúcico;o composto farmacologicamente ativo é selecionado dentre o grupo consistindo em oxitetraciclina, metoprolol, tilmicosina, fluoxetina, roxitromicina e terbinafina; e (b) prover um solvente orgânico farmaceuticamente aceitável miscível em água; o solvente orgânico farmaceuticamente aceitável é selecionado dentre o grupo consistindo em polietileno glicol, álcool de tetraidrofurfurila, dimetilacetamida, éter dimetílico de isosorbida, sulfóxido de dimetila, N-metil pirrolidona, propilenoglicol e glicerol formal;(c) combinar o sal e o solvente para formar uma composição injetável;em que a razão molar de ácido lipofílico para composto fármacologicamente ativo é de pelo menos 1;em que o sal é solúvel no solvente orgânico farmaceuticamente aceitável;em que pelo menos uma porção da composição forma um preciPetição 870180155523, de 27/11/2018, pág. 8/13 pitado quando a composição é introduzida ou injetada em um ambiente aquoso; e em que a composição libera o composto ativo por um tempo prolongado quando introduzida ou injetada em um ambiente aquoso.
- 20. Método, de acordo com a reivindicação 19, caracterizado pelo fato de que o ácido lipofílico é selecionado de um grupo consistindo dentre ácido decanóico, ácido láurico, ácido mirístico, ácido oleico e ácido linoleico.
- 21. Método, de acordo com a reivindicação 19, caracterizado pelo fato de que o solvente farmaceuticamente aceitável compreende 10% de propileno glicol em glicerol formal.
- 22. Método, de acordo com a reivindicação 19, caracterizado pelo fato de que o composto farmacologicamente ativo é terbinafina e o solvente farmaceuticamente aceitável compreende glicerol formal.
- 23. Método, de acordo com a reivindicação 19, caracterizado pelo fato de que o composto farmacologicamente ativo é oxitetraciclina, o contra-íon lipofílico é um ácido láurico, e o solvente orgânico farmaceuticamente aceitável é selecionado do grupo consistindo dentre polietilenoglicol, propilenoglicol e glicerol formal.
- 24. Método, de acordo com a reivindicação 19, caracterizado pelo fato de que o composto farmacologicamente ativo é tilmicosina, o contra-íon lipofílico é um ácido láurico, e o solvente orgânico farmaceuticamente aceitável é selecionado do grupo consistindo dentre polietilenoglicol, propilenoglicol e glicerol formal.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US34362501P | 2001-10-19 | 2001-10-19 | |
| PCT/US2002/033300 WO2003034988A2 (en) | 2001-10-19 | 2002-10-18 | Injectable compositions for the controlled delivery of pharmacologically active compound |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| BR0213425A BR0213425A (pt) | 2004-12-14 |
| BRPI0213425B1 true BRPI0213425B1 (pt) | 2020-01-28 |
| BRPI0213425B8 BRPI0213425B8 (pt) | 2021-05-25 |
Family
ID=23346878
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BRPI0213425A BRPI0213425B8 (pt) | 2001-10-19 | 2002-10-18 | composições injetáveis para a liberação controlada de composto farmacologicamente ativo, usos do referido composto e método para suas preparações |
Country Status (16)
| Country | Link |
|---|---|
| US (4) | US6887487B2 (pt) |
| EP (1) | EP1446103B1 (pt) |
| JP (1) | JP4850390B2 (pt) |
| CN (2) | CN101785861B (pt) |
| AU (1) | AU2002335077B2 (pt) |
| BG (1) | BG108634A (pt) |
| BR (1) | BRPI0213425B8 (pt) |
| CA (1) | CA2463896C (pt) |
| ES (1) | ES2561108T3 (pt) |
| IL (1) | IL160282A (pt) |
| MX (1) | MXPA04003608A (pt) |
| NZ (1) | NZ531460A (pt) |
| PL (1) | PL211116B1 (pt) |
| RU (1) | RU2292871C2 (pt) |
| WO (1) | WO2003034988A2 (pt) |
| ZA (1) | ZA200401076B (pt) |
Families Citing this family (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PL211116B1 (pl) * | 2001-10-19 | 2012-04-30 | Idexx Lab | Komozycja do podawania ssakowi związku farmakologicznie czynnego i sposób wytwarzania preparatu do wstrzyknięć |
| US6946137B2 (en) * | 2001-10-19 | 2005-09-20 | Idexx Laboratories, Inc. | Methods for the controlled delivery of pharmacologically active compounds |
| US20070060604A1 (en) * | 2002-03-25 | 2007-03-15 | Council Of Scientific And Industrial Research | Antibiotic pharmaceutical composition with lysergol as bio-enhancer and method of treatment |
| US20050049210A1 (en) * | 2003-08-27 | 2005-03-03 | Idexx Laboratories, Inc. | Methods for the controlled delivery of pharmacologically active compounds |
| US7854943B2 (en) | 2004-06-24 | 2010-12-21 | Idexx Laboratories | Phospholipid gel compositions for drug delivery and methods of treating conditions using same |
| US7858115B2 (en) | 2004-06-24 | 2010-12-28 | Idexx Laboratories | Phospholipid gel compositions for drug delivery and methods of treating conditions using same |
| US7618651B2 (en) | 2004-06-24 | 2009-11-17 | Idexx Laboratories | Pharmaceutical compositions for drug delivery and methods of treating or preventing conditions using same |
| US7282487B2 (en) * | 2004-10-28 | 2007-10-16 | Idexx Laboratories | Method for treating bacterial infections in horses or pigs with tilmicosin |
| JP5144267B2 (ja) * | 2004-10-28 | 2013-02-13 | アイデックス ラボラトリーズ,インコーポレイティド | 医薬活性化合物を制御して搬送するための組成物 |
| WO2007016153A2 (en) * | 2005-07-28 | 2007-02-08 | Eli Lilly And Company | Tilmicosin formulation |
| US7754679B2 (en) * | 2005-11-16 | 2010-07-13 | Idexx Laboratories, Inc. | Pharmaceutical compositions for the administration of aptamers |
| US8114440B2 (en) * | 2005-11-16 | 2012-02-14 | Idexx Laboratories Inc. | Pharmaceutical compositions for the administration of aptamers |
| US20070196398A1 (en) * | 2006-02-17 | 2007-08-23 | Murthy Yerramilli V S | Fluoroquinolone fatty acid salt compositions |
| US7973022B2 (en) * | 2006-02-17 | 2011-07-05 | Idexx Laboratories, Inc. | Fluoroquinolone carboxylic acid salt compositions |
| US20090239891A1 (en) * | 2006-03-01 | 2009-09-24 | Shukla Atul J | Sustained Release Dosage Forms of Analgesic Medications |
| JP5591691B2 (ja) | 2007-05-22 | 2014-09-17 | アムジエン・インコーポレーテツド | 生物活性を有する融合タンパク質を作製するための組成物及び方法 |
| US20090005326A1 (en) * | 2007-06-26 | 2009-01-01 | Idexx Laboratories, Inc. | Single dose roxithromycin |
| US8828960B2 (en) * | 2007-07-17 | 2014-09-09 | Idexx Laboratories, Inc. | Amino acid vitamin ester compositions for controlled delivery of pharmaceutically active compounds |
| US8920843B2 (en) * | 2007-11-07 | 2014-12-30 | Svip5 Llc | Slow release of organic salts of local anesthetics for pain relief |
| CN101496811B (zh) * | 2008-12-24 | 2011-03-16 | 天津瑞普生物技术股份有限公司 | 一种可溶且稳定的替米考星组合物 |
| CN101690713B (zh) * | 2009-09-28 | 2011-12-28 | 普莱柯生物工程股份有限公司 | 一种喹诺酮注射液的制备方法 |
| US20110177157A1 (en) * | 2010-01-19 | 2011-07-21 | Idexx Laboratories, Inc. | Compositions for Controlled Delivery of Pharmaceutically Active Compounds |
| SG193434A1 (en) | 2011-03-16 | 2013-10-30 | Amgen Inc | Potent and selective inhibitors of nav1.3 and nav1.7 |
| ES2875957T3 (es) | 2012-12-20 | 2021-11-11 | Amgen Inc | Agonistas del receptor APJ y usos de los mismos |
| WO2014165277A2 (en) | 2013-03-12 | 2014-10-09 | Amgen Inc. | POTENT AND SELECTIVE INHIBITORS OF Nav1.7 |
| ES2836325T3 (es) | 2014-03-13 | 2021-06-24 | Neuroderm Ltd | Composiciones del inhibidor de la dopa descarboxilasa |
| US10258585B2 (en) | 2014-03-13 | 2019-04-16 | Neuroderm, Ltd. | DOPA decarboxylase inhibitor compositions |
| JP6803236B2 (ja) | 2014-06-10 | 2020-12-23 | アムジェン インコーポレイテッド | アペリンポリペプチド |
| US11318190B2 (en) | 2017-05-05 | 2022-05-03 | United States Government As Represented By The Department Of Veterans Affairs | Methods and compositions for treating liver disease |
| CN108042491B (zh) * | 2018-01-23 | 2021-02-09 | 山东迅达康兽药有限公司 | 一种替米考星纳米乳及其制备方法 |
| CN113507922A (zh) * | 2019-03-01 | 2021-10-15 | 勃林格殷格翰动物保健美国公司 | 可注射的氯舒隆组合物、其方法和用途 |
| US11213502B1 (en) | 2020-11-17 | 2022-01-04 | Neuroderm, Ltd. | Method for treatment of parkinson's disease |
| US11844754B2 (en) | 2020-11-17 | 2023-12-19 | Neuroderm, Ltd. | Methods for treatment of Parkinson's disease |
| US11331293B1 (en) | 2020-11-17 | 2022-05-17 | Neuroderm, Ltd. | Method for treatment of Parkinson's disease |
| CN113730340A (zh) * | 2021-09-17 | 2021-12-03 | 中国药科大学 | 一种注射用脂肪酸缓释组合物及其制备方法和应用 |
Family Cites Families (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB674784A (en) * | 1949-05-25 | 1952-07-02 | Chimiotherapie Lab Franc | Penicillin preparations for injection |
| GB731933A (en) * | 1951-04-28 | 1955-06-15 | Fougera And Company E | Liquid preparations of therapeutically active substances for sub-cutaneous or intramuscular injection |
| US4259331A (en) * | 1979-04-16 | 1981-03-31 | Pfizer Inc. | Oxytetracycline compositions |
| US4298375A (en) | 1979-10-01 | 1981-11-03 | Monsanto Company | 2-Substituted-5-phenyl-4-thiazolecarboxylic acids and their derivatives as safening agents |
| US4820695A (en) | 1982-09-13 | 1989-04-11 | Eli Lilly And Company | C-20-dihydro-deoxy-(cyclic amino)-derivatives of macrolide antibiotics |
| DE3248328A1 (de) * | 1982-12-28 | 1984-06-28 | Bayer Ag, 5090 Leverkusen | Schwerloesliche salze von aminoglykosiden sowie diese enthaltende formulierungen mit verzoegerter wirkstoff-freigabe |
| GB8725427D0 (en) * | 1987-10-30 | 1987-12-02 | Lilly Industries Ltd | Somatotropin formulations |
| JPH027314A (ja) | 1988-06-27 | 1990-01-11 | Asahi Denso Kk | スイッチ |
| JPH02115118A (ja) * | 1988-10-24 | 1990-04-27 | Wakunaga Pharmaceut Co Ltd | 注射剤 |
| US5574020A (en) * | 1989-09-28 | 1996-11-12 | Eli Lilly And Company | Tilmicosin formulation |
| US5658956A (en) | 1991-03-01 | 1997-08-19 | Warner-Lambert Company | Bioadhesive-wound healing compositions and methods for preparing and using same |
| GB9211268D0 (en) * | 1992-05-28 | 1992-07-15 | Ici Plc | Salts of basic peptides with carboxyterminated polyesters |
| SE9202128D0 (sv) * | 1992-07-09 | 1992-07-09 | Astra Ab | Precipitation of one or more active compounds in situ |
| NZ250465A (en) * | 1992-12-21 | 1995-10-26 | Lilly Co Eli | Bactericidal composition; contains macrolide antibiotic, use for treating aquatic species |
| US6110905A (en) | 1994-07-09 | 2000-08-29 | Norbrook Laboratories Limited | Long-acting oxytetracycline composition |
| US6333021B1 (en) * | 1994-11-22 | 2001-12-25 | Bracco Research S.A. | Microcapsules, method of making and their use |
| JP3277735B2 (ja) * | 1994-12-07 | 2002-04-22 | 富士レビオ株式会社 | ナフトエ酸誘導体の吸収促進組成物 |
| US5747058A (en) | 1995-06-07 | 1998-05-05 | Southern Biosystems, Inc. | High viscosity liquid controlled delivery system |
| US6310053B1 (en) | 1995-07-05 | 2001-10-30 | Norbrook Laboratories Limited | Long-acting oxytetracycline composition |
| US5736152A (en) | 1995-10-27 | 1998-04-07 | Atrix Laboratories, Inc. | Non-polymeric sustained release delivery system |
| US5958888A (en) * | 1996-07-02 | 1999-09-28 | Merial, Inc. | Water miscible macrolide solutions |
| US5723447A (en) | 1996-07-02 | 1998-03-03 | Rhone Merieux, Inc. | Water miscible erythromycin solutions |
| IL119029A0 (en) | 1996-08-07 | 1996-11-14 | Yeda Res & Dev | Long-acting drugs and pharamaceutical compositions comprising them |
| CA2263411A1 (en) * | 1996-08-12 | 1998-02-19 | Eyal S. Ron | Composition for pharmaceutical applications |
| SE511313C2 (sv) | 1997-01-13 | 1999-09-06 | Gs Dev Ab | Komposition med reglerad frisättning innefattande fettsyraester av diacylglycerol |
| US6630168B1 (en) * | 1997-02-20 | 2003-10-07 | Biomedicines, Inc. | Gel delivery vehicles for anticellular proliferative agents |
| US6074657A (en) * | 1997-03-20 | 2000-06-13 | Pharmacia & Upjohn Company | Administration of an injectable antibiotic in the ear of an animal |
| WO1998050045A1 (en) | 1997-05-06 | 1998-11-12 | Norbrook Laboratories Limited | Improvements in or relating to long-acting antimicrobials |
| JPH11171794A (ja) * | 1997-12-04 | 1999-06-29 | Shoji Awazu | 動脈注射製剤 |
| CO5080775A1 (es) | 1998-03-02 | 2001-09-25 | Lilly Co Eli | Tratamiento de enfermedad viral en cerdos |
| BE1011899A6 (fr) * | 1998-04-30 | 2000-02-01 | Ucb Sa | Compositions pharmaceutiques gelifiables utilisables. |
| US6239112B1 (en) * | 1998-07-09 | 2001-05-29 | Merial, Inc. | Water miscible macrolide solutions |
| US6328961B1 (en) | 1998-08-20 | 2001-12-11 | Bristol-Myers Squibb Company | Tyrissamycin antibiotic |
| US6174540B1 (en) * | 1998-09-14 | 2001-01-16 | Merck & Co., Inc. | Long acting injectable formulations containing hydrogenated caster oil |
| US6677321B1 (en) | 1999-12-09 | 2004-01-13 | Bruce Levin | Methods and compositions for treatment of inflammatory disease |
| AU781682B2 (en) * | 2000-03-20 | 2005-06-09 | Zoetis Services Llc | Sustained-release compositions for parenteral administration |
| US6726918B1 (en) | 2000-07-05 | 2004-04-27 | Oculex Pharmaceuticals, Inc. | Methods for treating inflammation-mediated conditions of the eye |
| US20030219461A1 (en) | 2000-09-12 | 2003-11-27 | Britten Nancy J. | Parenteral combination therapy for infective conditions |
| US20040033938A1 (en) | 2000-09-12 | 2004-02-19 | Britten Nancy J. | Cyclooxygenase-2 inhibitor and antibacterial agent combination for intramammary treatment of mastitis |
| PL211116B1 (pl) | 2001-10-19 | 2012-04-30 | Idexx Lab | Komozycja do podawania ssakowi związku farmakologicznie czynnego i sposób wytwarzania preparatu do wstrzyknięć |
| US6946137B2 (en) * | 2001-10-19 | 2005-09-20 | Idexx Laboratories, Inc. | Methods for the controlled delivery of pharmacologically active compounds |
| US6790867B2 (en) | 2002-05-20 | 2004-09-14 | Schering-Plough Animal Health Corporation | Compositions and method for treating infection in cattle and swine |
| US20050049210A1 (en) * | 2003-08-27 | 2005-03-03 | Idexx Laboratories, Inc. | Methods for the controlled delivery of pharmacologically active compounds |
-
2002
- 2002-10-18 PL PL368338A patent/PL211116B1/pl unknown
- 2002-10-18 RU RU2004115103/15A patent/RU2292871C2/ru active
- 2002-10-18 US US10/274,445 patent/US6887487B2/en not_active Expired - Lifetime
- 2002-10-18 AU AU2002335077A patent/AU2002335077B2/en not_active Expired
- 2002-10-18 CN CN201010124253.1A patent/CN101785861B/zh not_active Expired - Lifetime
- 2002-10-18 ES ES02802160.8T patent/ES2561108T3/es not_active Expired - Lifetime
- 2002-10-18 JP JP2003537557A patent/JP4850390B2/ja not_active Expired - Lifetime
- 2002-10-18 EP EP02802160.8A patent/EP1446103B1/en not_active Expired - Lifetime
- 2002-10-18 BR BRPI0213425A patent/BRPI0213425B8/pt not_active IP Right Cessation
- 2002-10-18 NZ NZ531460A patent/NZ531460A/en not_active IP Right Cessation
- 2002-10-18 CA CA2463896A patent/CA2463896C/en not_active Expired - Lifetime
- 2002-10-18 WO PCT/US2002/033300 patent/WO2003034988A2/en active IP Right Grant
- 2002-10-18 CN CNA028192842A patent/CN1607937A/zh active Pending
-
2003
- 2003-03-28 US US10/403,100 patent/US7033599B2/en not_active Expired - Fee Related
-
2004
- 2004-02-09 IL IL160282A patent/IL160282A/en active IP Right Grant
- 2004-02-10 ZA ZA2004/01076A patent/ZA200401076B/en unknown
- 2004-03-16 BG BG108634A patent/BG108634A/xx unknown
- 2004-04-16 MX MXPA04003608A patent/MXPA04003608A/es active IP Right Grant
-
2005
- 2005-03-25 US US11/088,922 patent/US7404964B2/en active Active
-
2007
- 2007-01-31 US US11/700,068 patent/US20070141162A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| CN101785861B (zh) | 2014-08-13 |
| US7033599B2 (en) | 2006-04-25 |
| PL211116B1 (pl) | 2012-04-30 |
| IL160282A (en) | 2009-11-18 |
| US20030130211A1 (en) | 2003-07-10 |
| EP1446103A4 (en) | 2011-01-12 |
| RU2004115103A (ru) | 2005-03-20 |
| EP1446103A2 (en) | 2004-08-18 |
| ZA200401076B (en) | 2005-06-29 |
| ES2561108T3 (es) | 2016-02-24 |
| JP4850390B2 (ja) | 2012-01-11 |
| CN101785861A (zh) | 2010-07-28 |
| US20070141162A1 (en) | 2007-06-21 |
| CA2463896C (en) | 2010-05-04 |
| CN1607937A (zh) | 2005-04-20 |
| US20050075296A1 (en) | 2005-04-07 |
| WO2003034988A2 (en) | 2003-05-01 |
| PL368338A1 (en) | 2005-03-21 |
| WO2003034988A3 (en) | 2003-07-31 |
| US20050163859A1 (en) | 2005-07-28 |
| BRPI0213425B8 (pt) | 2021-05-25 |
| EP1446103B1 (en) | 2015-12-23 |
| US7404964B2 (en) | 2008-07-29 |
| BG108634A (en) | 2005-04-30 |
| MXPA04003608A (es) | 2004-07-27 |
| US6887487B2 (en) | 2005-05-03 |
| RU2292871C2 (ru) | 2007-02-10 |
| AU2002335077B2 (en) | 2006-09-07 |
| CA2463896A1 (en) | 2003-05-01 |
| BR0213425A (pt) | 2004-12-14 |
| JP2005506992A (ja) | 2005-03-10 |
| NZ531460A (en) | 2005-05-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| BRPI0213425B1 (pt) | composições injetáveis para a liberação controlada de composto farmacologicamente ativo, usos do referido composto e método para suas preparações | |
| AU2002335077A1 (en) | Injectable compositions for the controlled delivery of pharmacologically active compound | |
| CA2522009C (en) | Methods for the controlled delivery of pharmacologically active compounds | |
| CN114177133B (zh) | 一种药物缓释载体、缓释药物组合物及其应用 | |
| US20050049210A1 (en) | Methods for the controlled delivery of pharmacologically active compounds | |
| WO2006049739A1 (en) | Method for treating bacterial infections in horses or pigs with tilmicosin | |
| PT729362E (pt) | Composicao transdermica de n-¬n-¬5-¬4-(aminoiminometil)fenil|-1oxopentil|-l-alfa-aspartil|-l-fenillalanina ou esteres e seus sais farmaceuticamente aceitaveis | |
| TW202203938A (zh) | 鹼性化學療法瘤內注射調配物 | |
| US20050148519A1 (en) | Salts of pharmacologically active compounds | |
| CN101057845B (zh) | 草乌甲素干乳剂及其制备方法与应用 | |
| Wahlberg et al. | Polymeric controlled-release amsacrine chemotherapy in an experimental glioma model | |
| JP3283543B2 (ja) | 脂溶性肝疾患治療薬とpvlaとの包接化合物 | |
| BR102021008455A2 (pt) | Composição farmacêutica e uso da mesma | |
| CN105663105A (zh) | 一种双氯芬酸钠利多卡因复方注射剂 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B06F | Objections, documents and/or translations needed after an examination request according [chapter 6.6 patent gazette] | ||
| B15K | Others concerning applications: alteration of classification |
Free format text: PARA INT.CL: A61K 9/00, A61K 31/65, A01N 37/18, A61P 25/00, A61P 9/00, A61P 31/00 Ipc: A61K 9/00 (2011.01), A61K 31/65 (2011.01), A01N 37 |
|
| B07A | Application suspended after technical examination (opinion) [chapter 7.1 patent gazette] | ||
| B09B | Patent application refused [chapter 9.2 patent gazette] | ||
| B12B | Appeal against refusal [chapter 12.2 patent gazette] | ||
| B07D | Technical examination (opinion) related to article 229 of industrial property law [chapter 7.4 patent gazette] | ||
| B07E | Notification of approval relating to section 229 industrial property law [chapter 7.5 patent gazette] | ||
| B16A | Patent or certificate of addition of invention granted [chapter 16.1 patent gazette] |
Free format text: PRAZO DE VALIDADE: 10 (DEZ) ANOS CONTADOS A PARTIR DE 28/01/2020, OBSERVADAS AS CONDICOES LEGAIS. |
|
| B16C | Correction of notification of the grant [chapter 16.3 patent gazette] |
Free format text: PRAZO DE VALIDADE: 20 (VINTE) ANOS CONTADOS A PARTIR DE 18/10/2002 OBSERVADAS AS CONDICOES LEGAIS. PATENTE CONCEDIDA CONFORME ADI 5.529/DF |
|
| B21A | Patent or certificate of addition expired [chapter 21.1 patent gazette] |
Free format text: PATENTE EXTINTA EM 18/10/2022 |