WO2017170604A1 - 吸水剤の製造方法 - Google Patents
吸水剤の製造方法 Download PDFInfo
- Publication number
- WO2017170604A1 WO2017170604A1 PCT/JP2017/012751 JP2017012751W WO2017170604A1 WO 2017170604 A1 WO2017170604 A1 WO 2017170604A1 JP 2017012751 W JP2017012751 W JP 2017012751W WO 2017170604 A1 WO2017170604 A1 WO 2017170604A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- water
- acid
- absorbing agent
- salt
- weight
- Prior art date
Links
- 239000006096 absorbing agent Substances 0.000 title claims abstract description 494
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 title claims abstract description 353
- 238000000034 method Methods 0.000 title claims abstract description 186
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 89
- 150000003839 salts Chemical class 0.000 claims abstract description 351
- 239000000178 monomer Substances 0.000 claims abstract description 217
- 239000007864 aqueous solution Substances 0.000 claims abstract description 208
- 238000006116 polymerization reaction Methods 0.000 claims abstract description 182
- 238000001035 drying Methods 0.000 claims abstract description 105
- 238000004132 cross linking Methods 0.000 claims abstract description 74
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 claims abstract description 63
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 claims abstract description 59
- 230000014759 maintenance of location Effects 0.000 claims abstract description 44
- 239000002250 absorbent Substances 0.000 claims description 165
- 239000002738 chelating agent Substances 0.000 claims description 109
- 230000002745 absorbent Effects 0.000 claims description 104
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 claims description 96
- 239000000243 solution Substances 0.000 claims description 82
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 claims description 68
- 239000001630 malic acid Substances 0.000 claims description 65
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 claims description 61
- 235000011090 malic acid Nutrition 0.000 claims description 61
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 claims description 42
- 239000000377 silicon dioxide Substances 0.000 claims description 32
- 239000004310 lactic acid Substances 0.000 claims description 21
- 235000014655 lactic acid Nutrition 0.000 claims description 21
- GDVKFRBCXAPAQJ-UHFFFAOYSA-A dialuminum;hexamagnesium;carbonate;hexadecahydroxide Chemical compound [OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[Mg+2].[Mg+2].[Mg+2].[Mg+2].[Mg+2].[Mg+2].[Al+3].[Al+3].[O-]C([O-])=O GDVKFRBCXAPAQJ-UHFFFAOYSA-A 0.000 claims description 6
- 229910001701 hydrotalcite Inorganic materials 0.000 claims description 6
- 229960001545 hydrotalcite Drugs 0.000 claims description 6
- 235000012239 silicon dioxide Nutrition 0.000 claims description 6
- 229910019142 PO4 Inorganic materials 0.000 claims description 4
- 239000010452 phosphate Substances 0.000 claims description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 claims description 4
- 238000010521 absorption reaction Methods 0.000 abstract description 86
- 239000007788 liquid Substances 0.000 abstract description 57
- 230000002829 reductive effect Effects 0.000 abstract description 29
- 239000000463 material Substances 0.000 abstract description 13
- 229920005989 resin Polymers 0.000 description 217
- 239000011347 resin Substances 0.000 description 217
- 230000000052 comparative effect Effects 0.000 description 156
- 239000002245 particle Substances 0.000 description 122
- QPCDCPDFJACHGM-UHFFFAOYSA-N N,N-bis{2-[bis(carboxymethyl)amino]ethyl}glycine Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(=O)O)CCN(CC(O)=O)CC(O)=O QPCDCPDFJACHGM-UHFFFAOYSA-N 0.000 description 90
- 229960003330 pentetic acid Drugs 0.000 description 90
- 239000000499 gel Substances 0.000 description 86
- 239000002253 acid Substances 0.000 description 64
- 229940099690 malic acid Drugs 0.000 description 63
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 57
- 239000000203 mixture Substances 0.000 description 57
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 55
- 239000000843 powder Substances 0.000 description 54
- 239000003431 cross linking reagent Substances 0.000 description 52
- 229920001223 polyethylene glycol Polymers 0.000 description 52
- 239000002202 Polyethylene glycol Substances 0.000 description 50
- 229910052757 nitrogen Inorganic materials 0.000 description 49
- 125000004386 diacrylate group Chemical group 0.000 description 47
- XEEYBQQBJWHFJM-UHFFFAOYSA-N iron Substances [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 45
- 239000000017 hydrogel Substances 0.000 description 44
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 42
- -1 nitriloacetic acid-di (methylenephosphinic acid) Nitriloacetic acid- (methylenephosphinic acid) Chemical compound 0.000 description 41
- 239000000126 substance Substances 0.000 description 40
- 230000008569 process Effects 0.000 description 39
- 229920002125 Sokalan® Polymers 0.000 description 35
- 239000004584 polyacrylic acid Substances 0.000 description 33
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 32
- 238000010298 pulverizing process Methods 0.000 description 32
- 210000002700 urine Anatomy 0.000 description 31
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 30
- 230000000704 physical effect Effects 0.000 description 30
- 238000010438 heat treatment Methods 0.000 description 29
- 238000005259 measurement Methods 0.000 description 29
- 150000001875 compounds Chemical class 0.000 description 28
- 235000013373 food additive Nutrition 0.000 description 28
- 239000002778 food additive Substances 0.000 description 28
- 239000008367 deionised water Substances 0.000 description 27
- 229910021641 deionized water Inorganic materials 0.000 description 27
- 238000005469 granulation Methods 0.000 description 27
- 230000003179 granulation Effects 0.000 description 27
- 238000006386 neutralization reaction Methods 0.000 description 27
- 229920000642 polymer Polymers 0.000 description 27
- 238000002156 mixing Methods 0.000 description 26
- 239000003795 chemical substances by application Substances 0.000 description 25
- 238000011156 evaluation Methods 0.000 description 25
- 239000013585 weight reducing agent Substances 0.000 description 25
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 24
- 238000004040 coloring Methods 0.000 description 24
- 239000011734 sodium Substances 0.000 description 24
- 229910052708 sodium Inorganic materials 0.000 description 24
- 239000002504 physiological saline solution Substances 0.000 description 23
- 239000011780 sodium chloride Substances 0.000 description 22
- 239000000654 additive Substances 0.000 description 21
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 21
- NFDRPXJGHKJRLJ-UHFFFAOYSA-N edtmp Chemical compound OP(O)(=O)CN(CP(O)(O)=O)CCN(CP(O)(O)=O)CP(O)(O)=O NFDRPXJGHKJRLJ-UHFFFAOYSA-N 0.000 description 21
- 230000006866 deterioration Effects 0.000 description 20
- 229910052742 iron Inorganic materials 0.000 description 20
- 239000007787 solid Substances 0.000 description 20
- 238000009826 distribution Methods 0.000 description 19
- 235000011121 sodium hydroxide Nutrition 0.000 description 19
- 238000012360 testing method Methods 0.000 description 18
- 239000000047 product Substances 0.000 description 17
- 230000000996 additive effect Effects 0.000 description 16
- 229960005070 ascorbic acid Drugs 0.000 description 16
- 239000003505 polymerization initiator Substances 0.000 description 16
- YQUVCSBJEUQKSH-UHFFFAOYSA-N protochatechuic acid Natural products OC(=O)C1=CC=C(O)C(O)=C1 YQUVCSBJEUQKSH-UHFFFAOYSA-N 0.000 description 15
- SOBHUZYZLFQYFK-UHFFFAOYSA-K trisodium;hydroxy-[[phosphonatomethyl(phosphonomethyl)amino]methyl]phosphinate Chemical compound [Na+].[Na+].[Na+].OP(O)(=O)CN(CP(O)([O-])=O)CP([O-])([O-])=O SOBHUZYZLFQYFK-UHFFFAOYSA-K 0.000 description 15
- 239000002211 L-ascorbic acid Substances 0.000 description 14
- 235000000069 L-ascorbic acid Nutrition 0.000 description 14
- 230000000903 blocking effect Effects 0.000 description 14
- 230000000694 effects Effects 0.000 description 14
- 238000000227 grinding Methods 0.000 description 14
- 229910052751 metal Inorganic materials 0.000 description 14
- 239000002184 metal Substances 0.000 description 14
- 238000003756 stirring Methods 0.000 description 14
- CYDQOEWLBCCFJZ-UHFFFAOYSA-N 4-(4-fluorophenyl)oxane-4-carboxylic acid Chemical compound C=1C=C(F)C=CC=1C1(C(=O)O)CCOCC1 CYDQOEWLBCCFJZ-UHFFFAOYSA-N 0.000 description 13
- 230000009467 reduction Effects 0.000 description 13
- 239000001540 sodium lactate Substances 0.000 description 13
- 235000011088 sodium lactate Nutrition 0.000 description 13
- 229940005581 sodium lactate Drugs 0.000 description 13
- 239000002904 solvent Substances 0.000 description 13
- 230000001133 acceleration Effects 0.000 description 12
- 229920006037 cross link polymer Polymers 0.000 description 12
- 230000035699 permeability Effects 0.000 description 11
- 230000035807 sensation Effects 0.000 description 11
- GLDQAMYCGOIJDV-UHFFFAOYSA-N 2,3-dihydroxybenzoic acid Chemical compound OC(=O)C1=CC=CC(O)=C1O GLDQAMYCGOIJDV-UHFFFAOYSA-N 0.000 description 10
- WXTMDXOMEHJXQO-UHFFFAOYSA-N 2,5-dihydroxybenzoic acid Chemical compound OC(=O)C1=CC(O)=CC=C1O WXTMDXOMEHJXQO-UHFFFAOYSA-N 0.000 description 10
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 10
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 10
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 10
- LBYWTNBGUBZKJM-UHFFFAOYSA-N OP(=C)=O Chemical compound OP(=C)=O LBYWTNBGUBZKJM-UHFFFAOYSA-N 0.000 description 10
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 10
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 10
- 238000006243 chemical reaction Methods 0.000 description 10
- LNTHITQWFMADLM-UHFFFAOYSA-N gallic acid Chemical compound OC(=O)C1=CC(O)=C(O)C(O)=C1 LNTHITQWFMADLM-UHFFFAOYSA-N 0.000 description 10
- 229910052698 phosphorus Inorganic materials 0.000 description 10
- 239000011574 phosphorus Substances 0.000 description 10
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 10
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 9
- 235000013372 meat Nutrition 0.000 description 9
- 230000002265 prevention Effects 0.000 description 9
- 238000004381 surface treatment Methods 0.000 description 9
- LCPVQAHEFVXVKT-UHFFFAOYSA-N 2-(2,4-difluorophenoxy)pyridin-3-amine Chemical compound NC1=CC=CN=C1OC1=CC=C(F)C=C1F LCPVQAHEFVXVKT-UHFFFAOYSA-N 0.000 description 8
- ALRHLSYJTWAHJZ-UHFFFAOYSA-N 3-hydroxypropionic acid Chemical compound OCCC(O)=O ALRHLSYJTWAHJZ-UHFFFAOYSA-N 0.000 description 8
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- 229940048053 acrylate Drugs 0.000 description 8
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 8
- 239000003638 chemical reducing agent Substances 0.000 description 8
- 238000001816 cooling Methods 0.000 description 8
- 230000007423 decrease Effects 0.000 description 8
- 238000010528 free radical solution polymerization reaction Methods 0.000 description 8
- 238000007602 hot air drying Methods 0.000 description 8
- 230000000379 polymerizing effect Effects 0.000 description 8
- CHQMHPLRPQMAMX-UHFFFAOYSA-L sodium persulfate Substances [Na+].[Na+].[O-]S(=O)(=O)OOS([O-])(=O)=O CHQMHPLRPQMAMX-UHFFFAOYSA-L 0.000 description 8
- 230000008961 swelling Effects 0.000 description 8
- JMSVCTWVEWCHDZ-UHFFFAOYSA-N syringic acid Chemical compound COC1=CC(C(O)=O)=CC(OC)=C1O JMSVCTWVEWCHDZ-UHFFFAOYSA-N 0.000 description 8
- 229920003169 water-soluble polymer Polymers 0.000 description 8
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical compound NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 7
- ODBLHEXUDAPZAU-ZAFYKAAXSA-N D-threo-isocitric acid Chemical compound OC(=O)[C@H](O)[C@@H](C(O)=O)CC(O)=O ODBLHEXUDAPZAU-ZAFYKAAXSA-N 0.000 description 7
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 7
- ODBLHEXUDAPZAU-FONMRSAGSA-N Isocitric acid Natural products OC(=O)[C@@H](O)[C@H](C(O)=O)CC(O)=O ODBLHEXUDAPZAU-FONMRSAGSA-N 0.000 description 7
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Substances CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 7
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 7
- 230000002411 adverse Effects 0.000 description 7
- 239000012298 atmosphere Substances 0.000 description 7
- 238000005342 ion exchange Methods 0.000 description 7
- 238000000691 measurement method Methods 0.000 description 7
- 239000003973 paint Substances 0.000 description 7
- 230000002441 reversible effect Effects 0.000 description 7
- 159000000000 sodium salts Chemical class 0.000 description 7
- 238000010557 suspension polymerization reaction Methods 0.000 description 7
- 239000011975 tartaric acid Substances 0.000 description 7
- 235000002906 tartaric acid Nutrition 0.000 description 7
- ODBLHEXUDAPZAU-UHFFFAOYSA-N threo-D-isocitric acid Natural products OC(=O)C(O)C(C(O)=O)CC(O)=O ODBLHEXUDAPZAU-UHFFFAOYSA-N 0.000 description 7
- 238000004448 titration Methods 0.000 description 7
- RBNPOMFGQQGHHO-UHFFFAOYSA-N -2,3-Dihydroxypropanoic acid Natural products OCC(O)C(O)=O RBNPOMFGQQGHHO-UHFFFAOYSA-N 0.000 description 6
- RBNPOMFGQQGHHO-UWTATZPHSA-N D-glyceric acid Chemical compound OC[C@@H](O)C(O)=O RBNPOMFGQQGHHO-UWTATZPHSA-N 0.000 description 6
- YDONNITUKPKTIG-UHFFFAOYSA-N [Nitrilotris(methylene)]trisphosphonic acid Chemical compound OP(O)(=O)CN(CP(O)(O)=O)CP(O)(O)=O YDONNITUKPKTIG-UHFFFAOYSA-N 0.000 description 6
- WERYXYBDKMZEQL-UHFFFAOYSA-N butane-1,4-diol Chemical compound OCCCCO WERYXYBDKMZEQL-UHFFFAOYSA-N 0.000 description 6
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 6
- 239000010419 fine particle Substances 0.000 description 6
- 230000002209 hydrophobic effect Effects 0.000 description 6
- 239000011148 porous material Substances 0.000 description 6
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 5
- AAWZDTNXLSGCEK-LNVDRNJUSA-N (3r,5r)-1,3,4,5-tetrahydroxycyclohexane-1-carboxylic acid Chemical compound O[C@@H]1CC(O)(C(O)=O)C[C@@H](O)C1O AAWZDTNXLSGCEK-LNVDRNJUSA-N 0.000 description 5
- KJTLQQUUPVSXIM-ZCFIWIBFSA-N (R)-mevalonic acid Chemical compound OCC[C@](O)(C)CC(O)=O KJTLQQUUPVSXIM-ZCFIWIBFSA-N 0.000 description 5
- PQUXFUBNSYCQAL-UHFFFAOYSA-N 1-(2,3-difluorophenyl)ethanone Chemical compound CC(=O)C1=CC=CC(F)=C1F PQUXFUBNSYCQAL-UHFFFAOYSA-N 0.000 description 5
- WHRZCXAVMTUTDD-UHFFFAOYSA-N 1h-furo[2,3-d]pyrimidin-2-one Chemical compound N1C(=O)N=C2OC=CC2=C1 WHRZCXAVMTUTDD-UHFFFAOYSA-N 0.000 description 5
- PQHYOGIRXOKOEJ-UHFFFAOYSA-N 2-(1,2-dicarboxyethylamino)butanedioic acid Chemical compound OC(=O)CC(C(O)=O)NC(C(O)=O)CC(O)=O PQHYOGIRXOKOEJ-UHFFFAOYSA-N 0.000 description 5
- SEFYJVFBMNOLBK-UHFFFAOYSA-N 2-[2-[2-(oxiran-2-ylmethoxy)ethoxy]ethoxymethyl]oxirane Chemical compound C1OC1COCCOCCOCC1CO1 SEFYJVFBMNOLBK-UHFFFAOYSA-N 0.000 description 5
- YGDVXSDNEFDTGV-UHFFFAOYSA-N 2-[6-[bis(carboxymethyl)amino]hexyl-(carboxymethyl)amino]acetic acid Chemical compound OC(=O)CN(CC(O)=O)CCCCCCN(CC(O)=O)CC(O)=O YGDVXSDNEFDTGV-UHFFFAOYSA-N 0.000 description 5
- WYMDDFRYORANCC-UHFFFAOYSA-N 2-[[3-[bis(carboxymethyl)amino]-2-hydroxypropyl]-(carboxymethyl)amino]acetic acid Chemical compound OC(=O)CN(CC(O)=O)CC(O)CN(CC(O)=O)CC(O)=O WYMDDFRYORANCC-UHFFFAOYSA-N 0.000 description 5
- CIEZZGWIJBXOTE-UHFFFAOYSA-N 2-[bis(carboxymethyl)amino]propanoic acid Chemical compound OC(=O)C(C)N(CC(O)=O)CC(O)=O CIEZZGWIJBXOTE-UHFFFAOYSA-N 0.000 description 5
- 125000006290 2-hydroxybenzyl group Chemical group [H]OC1=C(C([H])=C([H])C([H])=C1[H])C([H])([H])* 0.000 description 5
- IWTIBPIVCKUAHK-UHFFFAOYSA-N 3-[bis(2-carboxyethyl)amino]propanoic acid Chemical compound OC(=O)CCN(CCC(O)=O)CCC(O)=O IWTIBPIVCKUAHK-UHFFFAOYSA-N 0.000 description 5
- AAWZDTNXLSGCEK-UHFFFAOYSA-N Cordycepinsaeure Natural products OC1CC(O)(C(O)=O)CC(O)C1O AAWZDTNXLSGCEK-UHFFFAOYSA-N 0.000 description 5
- KJTLQQUUPVSXIM-UHFFFAOYSA-N DL-mevalonic acid Natural products OCCC(O)(C)CC(O)=O KJTLQQUUPVSXIM-UHFFFAOYSA-N 0.000 description 5
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 5
- KMTRUDSVKNLOMY-UHFFFAOYSA-N Ethylene carbonate Chemical compound O=C1OCCO1 KMTRUDSVKNLOMY-UHFFFAOYSA-N 0.000 description 5
- 235000006173 Larrea tridentata Nutrition 0.000 description 5
- 244000073231 Larrea tridentata Species 0.000 description 5
- FSVCELGFZIQNCK-UHFFFAOYSA-N N,N-bis(2-hydroxyethyl)glycine Chemical compound OCCN(CCO)CC(O)=O FSVCELGFZIQNCK-UHFFFAOYSA-N 0.000 description 5
- JYXGIOKAKDAARW-UHFFFAOYSA-N N-(2-hydroxyethyl)iminodiacetic acid Chemical compound OCCN(CC(O)=O)CC(O)=O JYXGIOKAKDAARW-UHFFFAOYSA-N 0.000 description 5
- QRNHJWLHNHODER-UHFFFAOYSA-N OP(=C)=O.OP(=C)=O.OC(=O)C#N Chemical compound OP(=C)=O.OP(=C)=O.OC(=O)C#N QRNHJWLHNHODER-UHFFFAOYSA-N 0.000 description 5
- AAWZDTNXLSGCEK-ZHQZDSKASA-N Quinic acid Natural products O[C@H]1CC(O)(C(O)=O)C[C@H](O)C1O AAWZDTNXLSGCEK-ZHQZDSKASA-N 0.000 description 5
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 5
- RUSUZAGBORAKPY-UHFFFAOYSA-N acetic acid;n'-[2-(2-aminoethylamino)ethyl]ethane-1,2-diamine Chemical compound CC(O)=O.CC(O)=O.CC(O)=O.CC(O)=O.CC(O)=O.CC(O)=O.NCCNCCNCCN RUSUZAGBORAKPY-UHFFFAOYSA-N 0.000 description 5
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 5
- 238000000149 argon plasma sintering Methods 0.000 description 5
- 239000008280 blood Substances 0.000 description 5
- 210000004369 blood Anatomy 0.000 description 5
- 229960002126 creosote Drugs 0.000 description 5
- DUYCTCQXNHFCSJ-UHFFFAOYSA-N dtpmp Chemical compound OP(=O)(O)CN(CP(O)(O)=O)CCN(CP(O)(=O)O)CCN(CP(O)(O)=O)CP(O)(O)=O DUYCTCQXNHFCSJ-UHFFFAOYSA-N 0.000 description 5
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 5
- IFQUWYZCAGRUJN-UHFFFAOYSA-N ethylenediaminediacetic acid Chemical compound OC(=O)CNCCNCC(O)=O IFQUWYZCAGRUJN-UHFFFAOYSA-N 0.000 description 5
- 230000006870 function Effects 0.000 description 5
- 229940074391 gallic acid Drugs 0.000 description 5
- 235000004515 gallic acid Nutrition 0.000 description 5
- 229960005219 gentisic acid Drugs 0.000 description 5
- 238000004128 high performance liquid chromatography Methods 0.000 description 5
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 5
- NBZBKCUXIYYUSX-UHFFFAOYSA-N iminodiacetic acid Chemical group OC(=O)CNCC(O)=O NBZBKCUXIYYUSX-UHFFFAOYSA-N 0.000 description 5
- 229960002510 mandelic acid Drugs 0.000 description 5
- 235000010746 mayonnaise Nutrition 0.000 description 5
- 239000008268 mayonnaise Substances 0.000 description 5
- MGFYIUFZLHCRTH-UHFFFAOYSA-N nitrilotriacetic acid Chemical compound OC(=O)CN(CC(O)=O)CC(O)=O MGFYIUFZLHCRTH-UHFFFAOYSA-N 0.000 description 5
- 239000003960 organic solvent Substances 0.000 description 5
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 239000011342 resin composition Substances 0.000 description 5
- 229960004889 salicylic acid Drugs 0.000 description 5
- JXOHGGNKMLTUBP-HSUXUTPPSA-N shikimic acid Chemical compound O[C@@H]1CC(C(O)=O)=C[C@@H](O)[C@H]1O JXOHGGNKMLTUBP-HSUXUTPPSA-N 0.000 description 5
- JXOHGGNKMLTUBP-JKUQZMGJSA-N shikimic acid Natural products O[C@@H]1CC(C(O)=O)=C[C@H](O)[C@@H]1O JXOHGGNKMLTUBP-JKUQZMGJSA-N 0.000 description 5
- 229940047670 sodium acrylate Drugs 0.000 description 5
- 230000002195 synergetic effect Effects 0.000 description 5
- WKOLLVMJNQIZCI-UHFFFAOYSA-N vanillic acid Chemical compound COC1=CC(C(O)=O)=CC=C1O WKOLLVMJNQIZCI-UHFFFAOYSA-N 0.000 description 5
- TUUBOHWZSQXCSW-UHFFFAOYSA-N vanillic acid Natural products COC1=CC(O)=CC(C(O)=O)=C1 TUUBOHWZSQXCSW-UHFFFAOYSA-N 0.000 description 5
- 238000009423 ventilation Methods 0.000 description 5
- UQDUUUSGRKACHF-UHFFFAOYSA-N C=P(O)=O.N#CC(=O)O Chemical compound C=P(O)=O.N#CC(=O)O UQDUUUSGRKACHF-UHFFFAOYSA-N 0.000 description 4
- DBVJJBKOTRCVKF-UHFFFAOYSA-N Etidronic acid Chemical compound OP(=O)(O)C(O)(C)P(O)(O)=O DBVJJBKOTRCVKF-UHFFFAOYSA-N 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- 239000004743 Polypropylene Substances 0.000 description 4
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 4
- 150000007513 acids Chemical class 0.000 description 4
- 229910052783 alkali metal Inorganic materials 0.000 description 4
- 210000001124 body fluid Anatomy 0.000 description 4
- 239000010839 body fluid Substances 0.000 description 4
- 229920006317 cationic polymer Polymers 0.000 description 4
- 229910001873 dinitrogen Inorganic materials 0.000 description 4
- 239000000835 fiber Substances 0.000 description 4
- 150000002978 peroxides Chemical class 0.000 description 4
- 229920001155 polypropylene Polymers 0.000 description 4
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 4
- 238000007873 sieving Methods 0.000 description 4
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 4
- 239000004094 surface-active agent Substances 0.000 description 4
- YIBXWXOYFGZLRU-UHFFFAOYSA-N syringic aldehyde Natural products CC12CCC(C3(CCC(=O)C(C)(C)C3CC=3)C)C=3C1(C)CCC2C1COC(C)(C)C(O)C(O)C1 YIBXWXOYFGZLRU-UHFFFAOYSA-N 0.000 description 4
- HRXKRNGNAMMEHJ-UHFFFAOYSA-K trisodium citrate Chemical compound [Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O HRXKRNGNAMMEHJ-UHFFFAOYSA-K 0.000 description 4
- 230000002485 urinary effect Effects 0.000 description 4
- UWFRVQVNYNPBEF-UHFFFAOYSA-N 1-(2,4-dimethylphenyl)propan-1-one Chemical compound CCC(=O)C1=CC=C(C)C=C1C UWFRVQVNYNPBEF-UHFFFAOYSA-N 0.000 description 3
- AOBIOSPNXBMOAT-UHFFFAOYSA-N 2-[2-(oxiran-2-ylmethoxy)ethoxymethyl]oxirane Chemical compound C1OC1COCCOCC1CO1 AOBIOSPNXBMOAT-UHFFFAOYSA-N 0.000 description 3
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 3
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- JZTPOMIFAFKKSK-UHFFFAOYSA-N O-phosphonohydroxylamine Chemical compound NOP(O)(O)=O JZTPOMIFAFKKSK-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- YCGNPEAWLZNXKA-UHFFFAOYSA-N [2-[bis[[hydroxy(oxido)phosphaniumylidene]methyl]amino]ethyl-[[hydroxy(oxido)phosphaniumylidene]methyl]amino]methylidene-hydroxy-oxidophosphanium Chemical compound OP(=O)=CN(C=P(O)=O)CCN(C=P(O)=O)C=P(O)=O YCGNPEAWLZNXKA-UHFFFAOYSA-N 0.000 description 3
- 229910052782 aluminium Inorganic materials 0.000 description 3
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 3
- 125000003277 amino group Chemical group 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- 230000007797 corrosion Effects 0.000 description 3
- 238000005260 corrosion Methods 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 230000018044 dehydration Effects 0.000 description 3
- 238000006297 dehydration reaction Methods 0.000 description 3
- 238000000605 extraction Methods 0.000 description 3
- 239000012467 final product Substances 0.000 description 3
- 239000003349 gelling agent Substances 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- 239000011261 inert gas Substances 0.000 description 3
- 238000007603 infrared drying Methods 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- JRKICGRDRMAZLK-UHFFFAOYSA-L peroxydisulfate Chemical compound [O-]S(=O)(=O)OOS([O-])(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 3
- 239000002994 raw material Substances 0.000 description 3
- 239000002002 slurry Substances 0.000 description 3
- 229910001220 stainless steel Inorganic materials 0.000 description 3
- 238000003860 storage Methods 0.000 description 3
- 238000010998 test method Methods 0.000 description 3
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 3
- 125000004066 1-hydroxyethyl group Chemical group [H]OC([H])([*])C([H])([H])[H] 0.000 description 2
- 229910002012 Aerosil® Inorganic materials 0.000 description 2
- 229910002016 Aerosil® 200 Inorganic materials 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- RPNUMPOLZDHAAY-UHFFFAOYSA-N Diethylenetriamine Chemical compound NCCNCCN RPNUMPOLZDHAAY-UHFFFAOYSA-N 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- UQSXHKLRYXJYBZ-UHFFFAOYSA-N Iron oxide Chemical compound [Fe]=O UQSXHKLRYXJYBZ-UHFFFAOYSA-N 0.000 description 2
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 2
- 239000004372 Polyvinyl alcohol Substances 0.000 description 2
- 239000008156 Ringer's lactate solution Substances 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 239000004809 Teflon Substances 0.000 description 2
- 229920006362 Teflon® Polymers 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- 230000032683 aging Effects 0.000 description 2
- 150000003863 ammonium salts Chemical class 0.000 description 2
- 235000010323 ascorbic acid Nutrition 0.000 description 2
- 239000011668 ascorbic acid Substances 0.000 description 2
- 150000007514 bases Chemical class 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 238000011088 calibration curve Methods 0.000 description 2
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 2
- 150000001768 cations Chemical class 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 235000010980 cellulose Nutrition 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 238000005520 cutting process Methods 0.000 description 2
- VFHVQBAGLAREND-UHFFFAOYSA-N diphenylphosphoryl-(2,4,6-trimethylphenyl)methanone Chemical compound CC1=CC(C)=CC(C)=C1C(=O)P(=O)(C=1C=CC=CC=1)C1=CC=CC=C1 VFHVQBAGLAREND-UHFFFAOYSA-N 0.000 description 2
- 238000010894 electron beam technology Methods 0.000 description 2
- 238000011049 filling Methods 0.000 description 2
- 239000006260 foam Substances 0.000 description 2
- 238000005187 foaming Methods 0.000 description 2
- 239000004088 foaming agent Substances 0.000 description 2
- 238000009775 high-speed stirring Methods 0.000 description 2
- 229920001477 hydrophilic polymer Polymers 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 239000012535 impurity Substances 0.000 description 2
- 238000009616 inductively coupled plasma Methods 0.000 description 2
- 230000000977 initiatory effect Effects 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 230000003472 neutralizing effect Effects 0.000 description 2
- 239000004745 nonwoven fabric Substances 0.000 description 2
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 229920002451 polyvinyl alcohol Polymers 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 230000005855 radiation Effects 0.000 description 2
- 230000009257 reactivity Effects 0.000 description 2
- 238000007717 redox polymerization reaction Methods 0.000 description 2
- 239000007921 spray Substances 0.000 description 2
- 239000010935 stainless steel Substances 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 210000004243 sweat Anatomy 0.000 description 2
- 239000012085 test solution Substances 0.000 description 2
- 238000005979 thermal decomposition reaction Methods 0.000 description 2
- 229940078499 tricalcium phosphate Drugs 0.000 description 2
- 229910000391 tricalcium phosphate Inorganic materials 0.000 description 2
- 235000019731 tricalcium phosphate Nutrition 0.000 description 2
- 238000001291 vacuum drying Methods 0.000 description 2
- DNIAPMSPPWPWGF-VKHMYHEASA-N (+)-propylene glycol Chemical compound C[C@H](O)CO DNIAPMSPPWPWGF-VKHMYHEASA-N 0.000 description 1
- YPFDHNVEDLHUCE-UHFFFAOYSA-N 1,3-propanediol Substances OCCCO YPFDHNVEDLHUCE-UHFFFAOYSA-N 0.000 description 1
- OOSZCNKVJAVHJI-UHFFFAOYSA-N 1-[(4-fluorophenyl)methyl]piperazine Chemical compound C1=CC(F)=CC=C1CN1CCNCC1 OOSZCNKVJAVHJI-UHFFFAOYSA-N 0.000 description 1
- IZXIZTKNFFYFOF-UHFFFAOYSA-N 2-Oxazolidone Chemical class O=C1NCCO1 IZXIZTKNFFYFOF-UHFFFAOYSA-N 0.000 description 1
- WFUGQJXVXHBTEM-UHFFFAOYSA-N 2-hydroperoxy-2-(2-hydroperoxybutan-2-ylperoxy)butane Chemical compound CCC(C)(OO)OOC(C)(CC)OO WFUGQJXVXHBTEM-UHFFFAOYSA-N 0.000 description 1
- 125000003504 2-oxazolinyl group Chemical class O1C(=NCC1)* 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- CYUZOYPRAQASLN-UHFFFAOYSA-N 3-prop-2-enoyloxypropanoic acid Chemical compound OC(=O)CCOC(=O)C=C CYUZOYPRAQASLN-UHFFFAOYSA-N 0.000 description 1
- ZMGMDXCADSRNCX-UHFFFAOYSA-N 5,6-dihydroxy-1,3-diazepan-2-one Chemical class OC1CNC(=O)NCC1O ZMGMDXCADSRNCX-UHFFFAOYSA-N 0.000 description 1
- ASTTVEMLEHKCSP-UHFFFAOYSA-N CCC(CO)(CO)CO.OC(=O)CCOC(=O)C=C.OC(=O)CCOC(=O)C=C.OC(=O)CCOC(=O)C=C Chemical compound CCC(CO)(CO)CO.OC(=O)CCOC(=O)C=C.OC(=O)CCOC(=O)C=C.OC(=O)CCOC(=O)C=C ASTTVEMLEHKCSP-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 239000004971 Cross linker Substances 0.000 description 1
- 239000004593 Epoxy Chemical class 0.000 description 1
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 206010021639 Incontinence Diseases 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 229920000881 Modified starch Polymers 0.000 description 1
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 1
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical compound OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 1
- 229920000805 Polyaspartic acid Polymers 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 108010020346 Polyglutamic Acid Proteins 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 229920001131 Pulp (paper) Polymers 0.000 description 1
- ZJCCRDAZUWHFQH-UHFFFAOYSA-N Trimethylolpropane Chemical compound CCC(CO)(CO)CO ZJCCRDAZUWHFQH-UHFFFAOYSA-N 0.000 description 1
- LXEKPEMOWBOYRF-UHFFFAOYSA-N [2-[(1-azaniumyl-1-imino-2-methylpropan-2-yl)diazenyl]-2-methylpropanimidoyl]azanium;dichloride Chemical compound Cl.Cl.NC(=N)C(C)(C)N=NC(C)(C)C(N)=N LXEKPEMOWBOYRF-UHFFFAOYSA-N 0.000 description 1
- 150000008062 acetophenones Chemical class 0.000 description 1
- 150000001252 acrylic acid derivatives Chemical class 0.000 description 1
- 238000005273 aeration Methods 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 229910000288 alkali metal carbonate Inorganic materials 0.000 description 1
- 150000008041 alkali metal carbonates Chemical class 0.000 description 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- 125000005336 allyloxy group Chemical group 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- ROOXNKNUYICQNP-UHFFFAOYSA-N ammonium peroxydisulfate Substances [NH4+].[NH4+].[O-]S(=O)(=O)OOS([O-])(=O)=O ROOXNKNUYICQNP-UHFFFAOYSA-N 0.000 description 1
- VAZSKTXWXKYQJF-UHFFFAOYSA-N ammonium persulfate Chemical compound [NH4+].[NH4+].[O-]S(=O)OOS([O-])=O VAZSKTXWXKYQJF-UHFFFAOYSA-N 0.000 description 1
- 229910001870 ammonium persulfate Inorganic materials 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 125000000751 azo group Chemical group [*]N=N[*] 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- ISAOCJYIOMOJEB-UHFFFAOYSA-N benzoin Chemical class C=1C=CC=CC=1C(O)C(=O)C1=CC=CC=C1 ISAOCJYIOMOJEB-UHFFFAOYSA-N 0.000 description 1
- 150000008366 benzophenones Chemical class 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- LWMFAFLIWMPZSX-UHFFFAOYSA-N bis[2-(4,5-dihydro-1h-imidazol-2-yl)propan-2-yl]diazene Chemical compound N=1CCNC=1C(C)(C)N=NC(C)(C)C1=NCCN1 LWMFAFLIWMPZSX-UHFFFAOYSA-N 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- HJMZMZRCABDKKV-UHFFFAOYSA-N carbonocyanidic acid Chemical compound OC(=O)C#N HJMZMZRCABDKKV-UHFFFAOYSA-N 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 239000012461 cellulose resin Substances 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 239000012986 chain transfer agent Substances 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000013522 chelant Substances 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 238000012937 correction Methods 0.000 description 1
- 238000007405 data analysis Methods 0.000 description 1
- 238000013480 data collection Methods 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 239000002781 deodorant agent Substances 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- LSXWFXONGKSEMY-UHFFFAOYSA-N di-tert-butyl peroxide Chemical compound CC(C)(C)OOC(C)(C)C LSXWFXONGKSEMY-UHFFFAOYSA-N 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- 238000010981 drying operation Methods 0.000 description 1
- 238000004993 emission spectroscopy Methods 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 239000012632 extractable Substances 0.000 description 1
- 210000003608 fece Anatomy 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 238000012685 gas phase polymerization Methods 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 229920000578 graft copolymer Polymers 0.000 description 1
- 239000003673 groundwater Substances 0.000 description 1
- LHGVFZTZFXWLCP-UHFFFAOYSA-N guaiacol Chemical class COC1=CC=CC=C1O LHGVFZTZFXWLCP-UHFFFAOYSA-N 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- 239000003230 hygroscopic agent Substances 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 229910001867 inorganic solvent Inorganic materials 0.000 description 1
- 239000003049 inorganic solvent Substances 0.000 description 1
- SURQXAFEQWPFPV-UHFFFAOYSA-L iron(2+) sulfate heptahydrate Chemical compound O.O.O.O.O.O.O.[Fe+2].[O-]S([O-])(=O)=O SURQXAFEQWPFPV-UHFFFAOYSA-L 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 1
- 230000002175 menstrual effect Effects 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- 125000005641 methacryl group Chemical group 0.000 description 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 235000019426 modified starch Nutrition 0.000 description 1
- ZIUHHBKFKCYYJD-UHFFFAOYSA-N n,n'-methylenebisacrylamide Chemical compound C=CC(=O)NCNC(=O)C=C ZIUHHBKFKCYYJD-UHFFFAOYSA-N 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- NWVVVBRKAWDGAB-UHFFFAOYSA-N p-methoxyphenol Chemical compound COC1=CC=C(O)C=C1 NWVVVBRKAWDGAB-UHFFFAOYSA-N 0.000 description 1
- 238000012856 packing Methods 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- ACVYVLVWPXVTIT-UHFFFAOYSA-N phosphinic acid Chemical compound O[PH2]=O ACVYVLVWPXVTIT-UHFFFAOYSA-N 0.000 description 1
- 125000004437 phosphorous atom Chemical group 0.000 description 1
- 238000006303 photolysis reaction Methods 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 229920002401 polyacrylamide Polymers 0.000 description 1
- 108010064470 polyaspartate Proteins 0.000 description 1
- 229920002851 polycationic polymer Polymers 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229920002643 polyglutamic acid Polymers 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 229920000166 polytrimethylene carbonate Polymers 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- USHAGKDGDHPEEY-UHFFFAOYSA-L potassium persulfate Chemical compound [K+].[K+].[O-]S(=O)(=O)OOS([O-])(=O)=O USHAGKDGDHPEEY-UHFFFAOYSA-L 0.000 description 1
- 238000003825 pressing Methods 0.000 description 1
- XOJVVFBFDXDTEG-UHFFFAOYSA-N pristane Chemical compound CC(C)CCCC(C)CCCC(C)CCCC(C)C XOJVVFBFDXDTEG-UHFFFAOYSA-N 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 238000004445 quantitative analysis Methods 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 239000007870 radical polymerization initiator Substances 0.000 description 1
- 238000004064 recycling Methods 0.000 description 1
- 239000003507 refrigerant Substances 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- UQDJGEHQDNVPGU-UHFFFAOYSA-N serine phosphoethanolamine Chemical compound [NH3+]CCOP([O-])(=O)OCC([NH3+])C([O-])=O UQDJGEHQDNVPGU-UHFFFAOYSA-N 0.000 description 1
- 238000010334 sieve classification Methods 0.000 description 1
- 238000001542 size-exclusion chromatography Methods 0.000 description 1
- 238000004513 sizing Methods 0.000 description 1
- 238000001374 small-angle light scattering Methods 0.000 description 1
- 239000000344 soap Substances 0.000 description 1
- WBHQBSYUUJJSRZ-UHFFFAOYSA-M sodium bisulfate Chemical compound [Na+].OS([O-])(=O)=O WBHQBSYUUJJSRZ-UHFFFAOYSA-M 0.000 description 1
- 229910000342 sodium bisulfate Inorganic materials 0.000 description 1
- 229940074545 sodium dihydrogen phosphate dihydrate Drugs 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 150000005846 sugar alcohols Polymers 0.000 description 1
- 239000002344 surface layer Substances 0.000 description 1
- 229920001169 thermoplastic Polymers 0.000 description 1
- 239000004416 thermosoftening plastic Substances 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 239000004034 viscosity adjusting agent Substances 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
- 229920003176 water-insoluble polymer Polymers 0.000 description 1
- 238000004383 yellowing Methods 0.000 description 1
- 238000004394 yellowing prevention Methods 0.000 description 1
Images


Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/22—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising organic material
- B01J20/26—Synthetic macromolecular compounds
- B01J20/265—Synthetic macromolecular compounds modified or post-treated polymers
- B01J20/267—Cross-linked polymers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/30—Processes for preparing, regenerating, or reactivating
- B01J20/3085—Chemical treatments not covered by groups B01J20/3007 - B01J20/3078
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61F—FILTERS IMPLANTABLE INTO BLOOD VESSELS; PROSTHESES; DEVICES PROVIDING PATENCY TO, OR PREVENTING COLLAPSING OF, TUBULAR STRUCTURES OF THE BODY, e.g. STENTS; ORTHOPAEDIC, NURSING OR CONTRACEPTIVE DEVICES; FOMENTATION; TREATMENT OR PROTECTION OF EYES OR EARS; BANDAGES, DRESSINGS OR ABSORBENT PADS; FIRST-AID KITS
- A61F13/00—Bandages or dressings; Absorbent pads
- A61F13/15—Absorbent pads, e.g. sanitary towels, swabs or tampons for external or internal application to the body; Supporting or fastening means therefor; Tampon applicators
- A61F13/53—Absorbent pads, e.g. sanitary towels, swabs or tampons for external or internal application to the body; Supporting or fastening means therefor; Tampon applicators characterised by the absorbing medium
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L15/00—Chemical aspects of, or use of materials for, bandages, dressings or absorbent pads
- A61L15/16—Bandages, dressings or absorbent pads for physiological fluids such as urine or blood, e.g. sanitary towels, tampons
- A61L15/20—Bandages, dressings or absorbent pads for physiological fluids such as urine or blood, e.g. sanitary towels, tampons containing organic materials
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L15/00—Chemical aspects of, or use of materials for, bandages, dressings or absorbent pads
- A61L15/16—Bandages, dressings or absorbent pads for physiological fluids such as urine or blood, e.g. sanitary towels, tampons
- A61L15/22—Bandages, dressings or absorbent pads for physiological fluids such as urine or blood, e.g. sanitary towels, tampons containing macromolecular materials
- A61L15/24—Macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L15/00—Chemical aspects of, or use of materials for, bandages, dressings or absorbent pads
- A61L15/16—Bandages, dressings or absorbent pads for physiological fluids such as urine or blood, e.g. sanitary towels, tampons
- A61L15/42—Use of materials characterised by their function or physical properties
- A61L15/60—Liquid-swellable gel-forming materials, e.g. super-absorbents
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/22—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising organic material
- B01J20/26—Synthetic macromolecular compounds
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/22—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising organic material
- B01J20/26—Synthetic macromolecular compounds
- B01J20/261—Synthetic macromolecular compounds obtained by reactions only involving carbon to carbon unsaturated bonds
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/22—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising organic material
- B01J20/26—Synthetic macromolecular compounds
- B01J20/265—Synthetic macromolecular compounds modified or post-treated polymers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/28—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/30—Processes for preparing, regenerating, or reactivating
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F122/00—Homopolymers of compounds having one or more unsaturated aliphatic radicals each having only one carbon-to-carbon double bond, and at least one being terminated by a carboxyl radical and containing at least one other carboxyl radical in the molecule; Salts, anhydrides, esters, amides, imides or nitriles thereof
- C08F122/10—Esters
- C08F122/12—Esters of phenols or saturated alcohols
- C08F122/20—Esters containing oxygen in addition to the carboxy oxygen
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/44—Polymerisation in the presence of compounding ingredients, e.g. plasticisers, dyestuffs, fillers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/04—Oxygen-containing compounds
- C08K5/09—Carboxylic acids; Metal salts thereof; Anhydrides thereof
- C08K5/098—Metal salts of carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L33/00—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides or nitriles thereof; Compositions of derivatives of such polymers
- C08L33/02—Homopolymers or copolymers of acids; Metal or ammonium salts thereof
Definitions
- the present invention relates to a method for producing a water-absorbing agent. More specifically, the present invention provides a method for producing a water-absorbing agent that can reduce stickiness after liquid absorption when used as a sanitary material in a water-absorbing agent having a high water absorption capacity. The present invention also relates to ingredients and uses that improve the tactile feel of the water-absorbing agent. Furthermore, the present invention relates to a water-absorbing agent having both strong urine resistance and coloration resistance with time and a method for producing the same.
- Patent Document 1 Japanese Patent No. 5415256 discloses that coloring with time is improved by adding malic acid during polymerization.
- the ⁇ -hydroxycarboxylic acid (salt) is added before the drying step in the production process of the water-absorbing agent.
- the improvement of the absorption capacity of the water-absorbing agent is usually achieved by reducing the number (density) of crosslinking points. It is considered that the decrease in the cross-linking point increases the weight average molecular weight of the polymer component that is not bonded to the polymer network (formed by the cross-linking), resulting in a deterioration in touch (stickiness).
- the present invention also provides the following items.
- a method for producing a water-absorbing agent having a centrifuge retention capacity (CRC) of 30 g / g or more including a polymerization step of an aqueous monomer solution containing acrylic acid (salt), a drying step, and a surface crosslinking step.
- a method comprising the step of adding an ⁇ -hydroxycarboxylic acid (salt) before the drying step.
- the amount of the ⁇ -hydroxycarboxylic acid (salt) to be added is 0.010 to 4.0 mol% with respect to the acrylic acid (salt). the method of.
- the drying step is preferably 120 to 250 ° C., more preferably 150 to 200 ° C., preferably 5 to 120 minutes, more preferably 10 to 90 minutes, and further preferably 15 to 60 minutes.
- Selected from the group consisting of hot air drying, vacuum drying, fluidized bed drying, infrared drying, microwave drying, drum dryer drying, drying by azeotropic dehydration with a hydrophobic organic solvent, and high-humidity drying using high-temperature steam A method according to any one of the preceding items comprising the step of performing at least one drying procedure.
- the chelating agent is iminodiacetic acid, hydroxyethyliminodiacetic acid, nitrilotriacetic acid, nitrilotripropionic acid, ethylenediaminetetraacetic acid, hydroxyethylenediaminetriacetic acid, hexamethylenediaminetetraacetic acid, diethylenetriaminepentaacetic acid (DTPA), Triethylenetetramine hexaacetic acid, trans-1,2-diaminocyclohexanetetraacetic acid, bis (2-hydroxyethyl) glycine, diaminopropanoltetraacetic acid, ethylenediamine-2-propionic acid, glycol etherdiaminetetraacetic acid, bis (2-hydroxybenzyl) ) Ethylenediaminediacetic acid, 3-hydroxy-2,2-iminodisuccinic acid, iminodisuccinic acid, methylglycinediacetic acid, ethylened
- a surface-crosslinked polyacrylic acid (salt) water-absorbing agent containing ⁇ -hydroxycarboxylic acid (salt) inside the water-absorbing agent, and having a centrifuge retention capacity (CRC) of 30 g / g or more A polyacrylic acid (salt) water-absorbing agent.
- CRC centrifuge retention capacity
- composition according to any one of the above items, wherein the ⁇ -hydroxycarboxylic acid (salt) is selected from the group consisting of malic acid (salt) and lactic acid (salt).
- a method for reducing the molecular weight of a water-absorbing agent wherein the water-containing gel is obtained by polymerization of a monomer aqueous solution containing acrylic acid (salt) capable of forming a water-absorbing agent by polymerization. And a step of producing a water-absorbing agent having a centrifuge retention capacity (CRC) of 30 g / g or more by surface crosslinking of the water-absorbent resin powder obtained by drying, and the method comprises the steps of: A method comprising the step of adding ⁇ -hydroxycarboxylic acid (salt).
- a method for reducing stickiness of a water-absorbing agent after liquid absorption comprising polymerizing an aqueous monomer solution containing acrylic acid (salt) capable of forming a water-absorbing agent by polymerization and water content obtained by polymerization
- CRC centrifuge retention capacity
- a method for reducing the viscosity of a soluble part of a water-absorbing agent at the time of liquid absorption swelling wherein the method is a polymerization of an aqueous monomer solution containing acrylic acid (salt) capable of forming a water-absorbing agent by polymerization,
- the method comprises the steps of producing a water-absorbing agent having a centrifuge retention capacity (CRC) of 30 g / g or more by drying the hydrogel obtained by polymerization and surface crosslinking of the water-absorbent resin powder obtained by drying, Adding an ⁇ -hydroxycarboxylic acid (salt) prior to the drying.
- CRC centrifuge retention capacity
- a water-absorbing agent composed of a polyacrylic acid (salt) system wherein 1 g of the water-absorbing agent contains 200 ppm of L-ascorbic acid and 0.4 ppm of divalent iron ions (0.9% Fe / L-as deteriorated and soluble after being put in 25 ml of deterioration solution composed of sodium chloride aqueous solution and placed at 37 ° C. for 16 hours, and then stirred for 10 minutes by adding physiological saline to a total amount of 200 g.
- the chelating agent is contained in an amount of 0.001 to 1.0% by weight, and the ⁇ -hydroxycarboxylic acid (salt) is contained in an amount of 0.02 to 1.5% by weight.
- the ⁇ -hydroxycarboxylic acid (salt) is lactic acid, glycolic acid, malic acid, glyceric acid, tartaric acid, citric acid, isocitric acid, mevalonic acid, quinic acid, shikimic acid, ⁇ -hydroxypropionic acid, salicylic acid.
- the chelating agent is iminodiacetic acid, hydroxyethyliminodiacetic acid, nitrilotriacetic acid, nitrilotripropionic acid, ethylenediaminetetraacetic acid, hydroxyethylenediaminetriacetic acid, hexamethylenediaminetetraacetic acid, diethylenetriaminepentaacetic acid (DTPA), Triethylenetetramine hexaacetic acid, trans-1,2-diaminocyclohexanetetraacetic acid, bis (2-hydroxyethyl) glycine, diaminopropanoltetraacetic acid, ethylenediamine-2-propionic acid, glycol etherdiaminetetraacetic acid, bis (2-hydroxybenzyl) ) Ethylenediaminediacetic acid, 3-hydroxy-2,2-iminodisuccinic acid, iminodisuccinic acid, methylglycine diacetic acid, ethylenedi
- a method for producing a polyacrylic acid (salt) water-absorbing agent comprising a step of polymerizing an aqueous monomer solution mainly composed of acrylic acid (salt) in the presence of ⁇ -hydroxycarboxylic acid (salt).
- the ⁇ -hydroxycarboxylic acid (salt) is lactic acid, glycolic acid, malic acid, glyceric acid, tartaric acid, citric acid, isocitric acid, mevalonic acid, quinic acid, shikimic acid, ⁇ -hydroxypropionic acid, salicylic acid.
- the chelating agent is iminodiacetic acid, hydroxyethyliminodiacetic acid, nitrilotriacetic acid, nitrilotripropionic acid, ethylenediaminetetraacetic acid, hydroxyethylenediaminetriacetic acid, hexamethylenediaminetetraacetic acid, diethylenetriaminepentaacetic acid (DTPA), Triethylenetetramine hexaacetic acid, trans-1,2-diaminocyclohexanetetraacetic acid, bis (2-hydroxyethyl) glycine, diaminopropanoltetraacetic acid, ethylenediamine-2-propionic acid, glycol etherdiaminetetraacetic acid, bis (2-hydroxybenzyl) ) Ethylenediaminediacetic acid, 3-hydroxy-2,2-iminodisuccinic acid, iminodisuccinic acid, methylglycine diacetic acid, ethylened
- hygroscopic fluidity improver includes at least one selected from the group consisting of silicon dioxide, phosphate, or hydrotalcite.
- composition for improving the strong urine resistance of a water-absorbing agent comprising ⁇ -hydroxycarboxylic acid (salt) and a chelating agent.
- composition for improving the color resistance with time of a water-absorbing agent comprising an ⁇ -hydroxycarboxylic acid (salt) and a chelating agent.
- a method for improving the strong urine resistance of a water-absorbing agent comprising an ⁇ -hydroxycarboxylic acid in an aqueous monomer solution containing acrylic acid (salt) capable of forming a water-absorbing agent by polymerization (salt) In the presence of a salt), drying the hydrogel obtained by the polymerization, and surface crosslinking of the water-absorbent resin powder obtained by the drying, the method comprising the steps of:
- the water-absorbing agent is composed of a physiological saline (0.9% sodium chloride aqueous solution) containing 1 g of the water-absorbing agent and 200 ppm of L-ascorbic acid and 0.4 ppm of divalent iron ions.
- the solution was poured into 25 ml of the deteriorated solution, placed at 37 ° C. for 16 hours, then added with physiological saline to a total volume of 200 g, stirred for 10 minutes, and the Fe / L-as deteriorated soluble component extracted was 35% by weight or less. Further, with the feature that YI values after coloring acceleration test is 26 or less, method.
- the water-absorbing agent is composed of a physiological saline (0.9% sodium chloride aqueous solution) containing 1 g of the water-absorbing agent and 200 ppm of L-ascorbic acid and 0.4 ppm of divalent iron ions.
- the extracted Fe / L-as deteriorated soluble content is 35 wt% or less.
- Ri further, has a feature that YI values after coloring acceleration test is 26 or less, method.
- a water-absorbing agent having a high water-absorbing ratio that can reduce stickiness after liquid absorption when used as a sanitary material has been developed.
- a water-absorbing agent having a high water-absorbing ratio that can reduce stickiness after liquid absorption when used as a sanitary material.
- an unprecedented excellent absorbency excellent urine resistance, excellent coloration resistance over time.
- a water-absorbing agent has been developed.
- FIG. 1 shows various addition amounts of ⁇ -hydroxycarboxylic acid (salt) (0 mol%, 0.165 mol%, 0.329 mol%, 0.658 mol) in the absorbents of Examples and Comparative Examples of the present invention.
- % Comparative Examples 3 to 5, Example 8, Example 3, 4, 7, Example 1
- CRC centrifuge retention capacity
- g / g centrifuge retention capacity
- soluble molecular weight Dalton
- Water absorbent resin refers to a water-swellable, water-insoluble polymer gelling agent and satisfies the following physical properties. That is, as “water swellability”, the CRC specified by ERT441.2-02 is 5 g / g or more, and as “water-insoluble”, Ext specified by ERT470.2-02 is 50% by weight or less. It refers to a polymer gelling agent that satisfies the above.
- the water-absorbing resin can be designed as appropriate according to its use and is not particularly limited, but is preferably a hydrophilic cross-linked polymer obtained by cross-linking an unsaturated monomer having a carboxyl group. Moreover, the whole amount (100 weight%) is not limited to the form which is a polymer, The water absorbing resin composition containing the additive etc. may be sufficient in the range which satisfies the said physical property (CRC, Ext).
- the water-absorbent resin in the present invention is not limited to the final product, but is an intermediate in the manufacturing process of the water-absorbent resin (for example, a water-containing gel-like crosslinked polymer after polymerization, a dried polymer after drying, a water absorption before surface crosslinking)
- water-absorbent resin for example, a water-containing gel-like crosslinked polymer after polymerization, a dried polymer after drying, a water absorption before surface crosslinking
- water-absorbent resin examples of the shape of the water absorbent resin include a sheet shape, a fiber shape, a film shape, a particle shape, and a gel shape.
- a particulate water absorbent resin is preferable.
- polyacrylic acid (salt) refers to polyacrylic acid and / or a salt thereof, and acrylic acid and / or a salt thereof (hereinafter referred to as “acrylic acid (salt)”) as a main component.
- acrylic acid (salt) acrylic acid and / or a salt thereof
- the “main component” means that the used amount (content) of acrylic acid (salt) is usually 50 to 100 mol% with respect to the entire monomer (excluding the internal crosslinking agent) used for polymerization, It is preferably 70 to 100 mol%, more preferably 90 to 100 mol%, and still more preferably substantially 100 mol%.
- Water absorbing agent in the present specification, the water-absorbing agent means an aqueous gel absorption gelling agent containing a water-absorbing resin as a main component.
- the “water absorbing agent” is sometimes referred to as “particulate water absorbing agent”.
- the aqueous liquid is not limited to water, but may be urine, blood, sweat, feces, waste liquid, moisture, steam, ice, a mixture of water and an organic solvent and / or an inorganic solvent, rainwater, groundwater, etc. If water is included, it is not restricted to a specific thing. Preferable examples include urine, menstrual blood, sweat, and other body fluids.
- the water-absorbing agent according to the present invention is suitably used as a sanitary material for absorbing an aqueous liquid.
- the water absorbent resin as a polymer is contained as a main component in the water absorbent. That is, the particulate water-absorbing agent preferably contains 60 to 100% by mass, 70 to 100% by mass, 80 to 100% by mass, 90 to 100% by mass, and other non-polymers such as water and / or inorganic fine particles, Optionally, an additive such as a valent metal cation is included.
- the preferred water content is 0.2 to 30% by mass. That is, the water-absorbing resin composition in which these components are integrated is also a category of the water-absorbing agent.
- the upper limit of the water-absorbing resin in the water-absorbing agent is 99% by mass, more preferably 97% by mass, and particularly about 95% by mass, and preferably further contains water and additives (inorganic fine particles, polyvalent metal cations) described later. .
- the water-absorbing resin that is the main component of the water-absorbing agent includes polyacrylic acid (salt) resins, polysulfonic acid (salt) resins, maleic anhydride (salt) resins, polyacrylamide resins, polyvinyl alcohol resins, poly Examples include ethylene oxide resins, polyaspartic acid (salt) resins, polyglutamic acid (salt) resins, polyalginic acid (salt) resins, starch resins, and cellulose resins, preferably polyacrylic acid (salt) resins. Is used.
- EDANA European Disposables and Nonwovens Associations
- ERT is an abbreviation for a method of measuring water-absorbent resin of the European standard (almost the world standard) (EDANA Recommended Test Methods). .
- the physical properties of the water-absorbent resin are measured according to the original ERT (revised in 2002 / known literature).
- CRC Centrifugation Retention Capacity (centrifuge retention capacity), and means the water absorption capacity of the water absorbent resin under no pressure (sometimes referred to as “water absorption capacity”).
- AAP is an abbreviation for Absorption Against Pressure, which means the water absorption capacity of a water absorbent resin under pressure.
- ERT442.2-02 describes “Absorption Under Pressure”, but has substantially the same content.
- PSD is an abbreviation for Particle Size Distribution, and means a particle size distribution of a water-absorbent resin measured by sieving classification.
- the weight average particle diameter (D50) and the logarithmic standard deviation ( ⁇ ) of the particle size distribution are described in US Pat. No. 7,638,570 “(3) Mass-Average Particle Diameter (D50) and Logical Standard Deviation ( ⁇ ). Measurement is performed in the same manner as “Particle Diameter Distribution”.
- Extractables (1-4-4) “Ext” (ERT470.2-02) “Ext” is an abbreviation for Extractables, which means the water-soluble component (water-soluble component amount) of the water-absorbent resin.
- water-soluble content is sometimes simply referred to as “soluble content”. Further, when the amount of water-soluble component is shown, the above-mentioned “water-soluble component amount” is sometimes referred to as “soluble component amount” or simply “soluble component”. Sometimes called.
- dissolved polymer refers to the amount of dissolved polymer (unit: wt%) after adding 1.0 g of water-absorbing resin to 200 ml of 0.9 wt% aqueous sodium chloride solution and stirring at 500 rpm for 16 hours.
- the amount of dissolved polymer is measured using pH titration.
- “Moisture Content” (ERT430.2-02) “Moisture Content” means the water content of the water-absorbent resin.
- the water-absorbent resin is measured at 1.0 g and the drying temperature is changed to 180 ° C.
- “Residual Monomers” (ERT410.2-02) “Residual Monomers” means the amount of monomer (monomer) remaining in the water-absorbent resin (hereinafter referred to as “residual monomer”).
- the amount of residual monomer (unit: ppm) after adding 1.0 g of water-absorbing resin to 200 ml of 0.9 wt% sodium chloride aqueous solution and stirring at 500 rpm for 1 hour.
- the amount of residual monomer is measured using high performance liquid chromatography (HPLC).
- PH (ERT400.2-02): means the pH of the water-absorbent resin.
- Liquid permeability “Liquid permeability” of the water-absorbent resin in the present invention refers to the fluidity of liquid passing between particles of the swollen gel under load or no load.
- SFC Seline
- GBP Gel Bed
- SFC refers to the liquid permeability of a 0.69 wt% sodium chloride aqueous solution to a water absorbent resin under a 2.07 kPa load, and is measured according to the SFC test method disclosed in US Pat. No. 5,669,894. .
- GBP refers to the liquid permeability of a 0.9 wt% aqueous sodium chloride solution to a water-absorbent resin under load or free swelling, and conforms to the GBP test method disclosed in International Publication No. 2005/016393. Measured.
- Water absorption speed The “water absorption rate” of the water absorbent resin in the present invention means a water absorption rate measured by “FSR” or “Vortex” (unit: second). Note that “FSR” is an abbreviation for Free Well Rate. Specific measurement methods will be described in the examples described later.
- liter may be written as “l” or “L”
- wt% may be written as “wt%” for convenience.
- D Non Detected
- This step is a step of preparing an aqueous solution containing acrylic acid (salt) as a main component (hereinafter referred to as “monomer aqueous solution”).
- monomer aqueous solution an aqueous solution containing acrylic acid (salt) as a main component
- the slurry liquid of a monomer can also be used in the range by which the water absorption performance of the water-absorbing resin obtained does not fall, in this section, monomer aqueous solution is demonstrated for convenience.
- main component means that the amount (content) of acrylic acid (salt) used is usually based on the whole monomer (excluding the internal crosslinking agent) subjected to the polymerization reaction of the water-absorbent resin. It means 50 mol% or more, preferably 70 mol% or more, more preferably 90 mol% or more (the upper limit is 100 mol%).
- acrylic acid In the present invention, acrylic acid and / or a salt thereof (hereinafter referred to as “acrylic acid (salt)”) is used as a monomer from the viewpoint of physical properties and productivity of the obtained water-absorbent resin.
- acrylic acid may be a known acrylic acid, preferably methoxyphenols, more preferably p-methoxyphenol as a polymerization inhibitor, from the viewpoint of acrylic acid polymerizability and water-absorbing resin color tone. It may contain 200 ppm or less, more preferably 10 to 160 ppm, still more preferably 20 to 100 ppm.
- impurities in acrylic acid the compounds described in US Patent Application Publication No. 2008/0161512 are also applied to the present invention.
- the “acrylic acid salt” is obtained by neutralizing the acrylic acid with the following basic composition, and as the acrylic acid salt, a commercially available acrylate salt (for example, sodium acrylate) may be used. What was obtained by neutralizing in the manufacturing plant of a water absorbing resin may be used.
- a commercially available acrylate salt for example, sodium acrylate
- the “basic composition” refers to a composition containing a basic compound, such as a commercially available sodium hydroxide aqueous solution.
- the basic compound examples include alkali metal carbonates and hydrogen carbonates, alkali metal hydroxides, ammonia, and organic amines.
- strong basicity is desired from the viewpoint of the physical properties of the obtained water-absorbent resin. That is, hydroxides of alkali metals such as sodium hydroxide, potassium hydroxide and lithium hydroxide are preferable, and sodium hydroxide is more preferable.
- neutralization As neutralization in the present invention, neutralization with respect to acrylic acid (before polymerization) or neutralization with respect to a hydrogel crosslinked polymer obtained by crosslinking polymerization of acrylic acid (after polymerization) (hereinafter referred to as “post-neutralization”) Any of these can be selected or used in combination.
- These neutralizations may be continuous or batch-type, and are not particularly limited, but are preferably continuous from the viewpoint of production efficiency and the like.
- the neutralization rate in the present invention is preferably 10 to 90 mol%, more preferably 40 to 85 mol%, still more preferably 50 to 80 mol%, particularly preferably 60 to 75 mol based on the acid group of the monomer. Mol%.
- the neutralization rate is less than 10 mol%, the water absorption ratio may be significantly reduced.
- the neutralization rate exceeds 90 mol%, a water absorbent resin having a high water absorption capacity under pressure may not be obtained.
- the above neutralization rate is the same even in the case of post-neutralization. Moreover, the said neutralization rate is applied also about the neutralization rate of the water absorbing resin as a final product.
- the neutralization rate of 75 mol% means a mixture of 25 mol% acrylic acid and 75 mol% acrylate. Moreover, this mixture may be called an acrylic acid partial neutralized material.
- the “other monomer” refers to a monomer other than the acrylic acid (salt), and can be used in combination with acrylic acid (salt) to produce a water-absorbing resin.
- the other monomer includes a water-soluble or hydrophobic unsaturated monomer.
- the compounds described in US Patent Application Publication No. 2005/0215734 (excluding acrylic acid) are also applied to the present invention.
- Internal crosslinking agent As the internal crosslinking agent used in the present invention, the compounds described in US Pat. No. 6,241,928 are also applied to the present invention. From these, one or more compounds are selected in consideration of reactivity.
- Examples of the internal crosslinking agent used in the present invention include N, N′-methylenebisacrylamide, (poly) ethylene glycol di (meth) acrylate, (poly) propylene glycol di (meth) acrylate, and (polyoxyethylene) trimethyl.
- a carboxyl group such as Tol
- a compound having two or more polymerizable unsaturated groups preferably a compound having thermal decomposability at the following drying temperature, more preferably (poly) alkylene.
- a compound having two or more polymerizable unsaturated groups having a glycol structural unit is used as an internal cross-linking agent.
- the polymerizable unsaturated group is preferably an allyl group, a (meth) acrylate group, more preferably a (meth) acrylate group.
- the (poly) alkylene glycol structural unit is preferably polyethylene glycol, and the n number is preferably 1 to 100, more preferably 6 to 50.
- the amount of the internal crosslinking agent used is preferably 0.0001 to 10 mol%, more preferably 0.001 to 1 mol%, based on the entire monomer.
- a desired water-absorbing resin can be obtained by setting the amount used within the above range. If the amount used is too small, the gel strength tends to decrease and the water-soluble content tends to increase. By appropriately adjusting the amount of the internal crosslinking agent used, the centrifugal retention capacity, soluble content, or soluble molecular weight of the resulting water-absorbing agent can be appropriately adjusted.
- a method in which a predetermined amount of an internal cross-linking agent is previously added to the monomer aqueous solution and a cross-linking reaction is performed simultaneously with the polymerization is preferably applied.
- a method of adding an internal cross-linking agent during or after polymerization and post-crosslinking a method of radical cross-linking using a radical polymerization initiator, radiation using active energy rays such as electron beams and ultraviolet rays
- a method of crosslinking and the like can also be employed. Moreover, these methods can also be used together.
- hydrophilic polymer such as starch, starch derivative, cellulose, cellulose derivative, polyvinyl alcohol, polyacrylic acid (salt), polyacrylic acid (salt) cross-linked product, preferably 50% by weight or less, more preferably Is added in an amount of 20% by weight or less, more preferably 10% by weight or less, particularly preferably 5% by weight or less (the lower limit is 0% by weight), foaming agents such as carbonates, azo compounds and bubbles, surfactants, chelating agents.
- An agent, a chain transfer agent and the like are preferably added at 5% by weight or less, more preferably 1% by weight or less, still more preferably 0.5% by weight or less (the lower limit is 0% by weight).
- the above substances may be added not only in the form added to the monomer aqueous solution, but also in the form added during polymerization, or these forms may be used in combination.
- a graft polymer or a water-absorbing resin composition eg, starch-acrylic acid polymer, PVA-acrylic acid polymer, etc.
- a graft polymer or a water-absorbing resin composition eg, starch-acrylic acid polymer, PVA-acrylic acid polymer, etc.
- each of the above substances is added when preparing an aqueous monomer solution.
- the concentration of the monomer component in the aqueous monomer solution is not particularly limited, but is preferably 10 to 80% by weight, more preferably 20 to 75% by weight, and still more preferably 30 from the viewpoint of physical properties of the water absorbent resin. ⁇ 70% by weight.
- a solvent other than water can be used in combination as required.
- the type of solvent is not particularly limited.
- the “monomer component concentration” (also referred to as “monomer concentration”) is a value determined by the following formula (1).
- the weight of the monomer aqueous solution includes the graft component and The weight of the water-absorbent resin and the hydrophobic solvent in the reverse phase suspension polymerization is not included.
- Formula (1): (Concentration of monomer component (% by weight)) (Weight of monomer component) / (Weight of monomer aqueous solution) ⁇ 100
- the polymerization initiator used in the present invention is not particularly limited because it is appropriately selected depending on the polymerization form and the like.
- the thermal decomposition type polymerization initiator, the photodecomposition type polymerization initiator, or the decomposition of these polymerization initiators is used.
- a redox polymerization initiator in combination with a reducing agent that promotes the reaction is used.
- one or more of the polymerization initiators disclosed in US Pat. No. 7,265,190 are used.
- a peroxide or an azo compound is preferably used, more preferably a peroxide, and still more preferably a persulfate.
- thermal decomposition type polymerization initiator examples include persulfate: sodium persulfate, potassium persulfate, ammonium persulfate; peroxide: hydrogen peroxide, t-butyl peroxide, methyl ethyl ketone peroxide); azo compound: azonitrile compound , Azoamidine compounds, cyclic azoamidine compounds, azoamide compounds, alkylazo compounds, 2,2′-azobis (2-amidinopropane) dihydrochloride, 2,2′-azobis [2- (2-imidazolin-2-yl) propane] dihydro And chloride.
- persulfate sodium persulfate, potassium persulfate, ammonium persulfate
- peroxide hydrogen peroxide, t-butyl peroxide, methyl ethyl ketone peroxide
- azo compound azonitrile compound , Azoamidine compounds, cyclic azoamidine compounds, azo
- photodegradable polymerization initiator examples include benzoin derivatives, benzyl derivatives, acetophenone derivatives, benzophenone derivatives, azo compounds, and the like.
- redox polymerization initiator examples include a system in which a reducing compound such as L-ascorbic acid or sodium bisulfite is used in combination with the persulfate or peroxide.
- a photodegradable polymerization initiator and a thermal decomposable polymerization initiator in combination.
- the amount of the polymerization initiator used is preferably 0.001 to 1 mol%, more preferably 0.001 to 0.5 mol%, based on the monomer.
- the amount of the reducing agent used is preferably 0.0001 to 0.02 mol% with respect to the monomer.
- the polymerization initiator may replace with the said polymerization initiator and may irradiate active energy rays, such as a radiation, an electron beam, and an ultraviolet-ray, and may implement a polymerization reaction, and these active energy rays and a polymerization initiator may be used together.
- active energy rays such as a radiation, an electron beam, and an ultraviolet-ray
- the polymerization form applied to the present invention is not particularly limited, but from the viewpoint of water absorption characteristics and ease of polymerization control, preferably spray droplet polymerization, aqueous solution polymerization, reverse phase suspension polymerization, more preferably aqueous solution polymerization. , Reverse phase suspension polymerization, more preferably aqueous solution polymerization.
- continuous aqueous solution polymerization is particularly preferable, and either continuous belt polymerization or continuous kneader polymerization is applied.
- continuous belt polymerization is disclosed in U.S. Pat. Nos. 4,893,999 and 6,241,928 and U.S. Patent Application Publication No. 2005/215734
- continuous kneader polymerization is disclosed in U.S. Pat. Nos. 6,987,151 and 6,710,141. , Respectively.
- “high temperature initiation polymerization” and “high concentration polymerization” can be mentioned.
- “High temperature initiation polymerization” means that the temperature of the aqueous monomer solution is preferably 30 ° C. or higher, more preferably 35 ° C. or higher, still more preferably 40 ° C. or higher, particularly preferably 50 ° C. or higher (the upper limit is the boiling point).
- “High concentration polymerization” means that the monomer concentration is preferably 30% by weight or more, more preferably 35% by weight or more, still more preferably 40% by weight or more, and particularly preferably 45% by weight or more. The upper limit is a saturation concentration.
- the polymerization can be carried out in an air atmosphere, but from the viewpoint of the color tone of the resulting water-absorbent resin, the polymerization is preferably carried out in an inert gas atmosphere such as nitrogen or argon.
- the oxygen concentration is preferably controlled to 1% by volume or less.
- the dissolved oxygen in the monomer aqueous solution is also preferably replaced with an inert gas (for example, dissolved oxygen; less than 1 mg / l).
- foam polymerization in which bubbles (particularly the above inert gas or the like) are dispersed in a monomer aqueous solution to perform polymerization.
- the solid concentration may be increased during the polymerization.
- the solid content increase degree is defined by the following formula (2) as an index of such solid content increase.
- a raise degree of this solid content concentration Preferably it is 1 weight% or more, More preferably, it is 2 weight% or more.
- Formula (2): (Solid content rise (wt%)) (Solid content concentration of water-containing gel after polymerization (wt%)) ⁇ (Solid content concentration of monomer aqueous solution (wt%))
- the solid content concentration of the monomer aqueous solution is a value obtained by the following formula (3), and the components in the polymerization system are the monomer aqueous solution, the graft component, the water absorbent resin, and other solids (for example, water). Insoluble fine particles, etc.) and does not include a hydrophobic solvent in reverse phase suspension polymerization.
- Formula (3): (Solid content of monomer aqueous solution (% by weight)) (weight of (monomer component + graft component + water absorbent resin + other solids)) / (weight of components in polymerization system) ⁇ 100
- ⁇ -hydroxycarboxylic acid salt
- ⁇ -hydroxycarboxylic acid salt
- ⁇ -hydroxycarboxylic acid salt
- the addition of ⁇ -hydroxycarboxylic acid reduces the molecular weight of the soluble component of the resulting water-absorbing agent, which in turn reduces stickiness and discomfort when used as a sanitary material. From these further viewpoints, it is more preferable to add ⁇ -hydroxycarboxylic acid.
- aqueous monomer solution when the ⁇ -hydroxycarboxylic acid (salt) is mixed in an aqueous monomer solution, there is no particular limitation.
- a monomer containing an ⁇ -hydroxycarboxylic acid (salt) by mixing an ⁇ -hydroxycarboxylic acid (salt) or an ⁇ -hydroxycarboxylic acid (salt) aqueous solution with the monomer or the monomer aqueous solution. Adjust the aqueous solution.
- various physical properties of the water-absorbing agent may be improved by adding, for example, 0 to 50% by weight, preferably 0 to 20% by weight, of a water-soluble resin or a water-absorbing resin with respect to the monomer.
- various foaming agents carbonates, azo compounds, bubbles, etc.
- surfactants e.g., sodium EDTA
- chelating agents e.g., sodium EDTA
- chain transfer agents such as hypophosphorous acid (salts), etc.
- Various mass properties of the water-absorbing agent may be improved by adding mass%.
- the liquid, slurry or solid (or powder) of ⁇ -hydroxycarboxylic acid (salt) is mixed as it is. It may also be mixed using a solvent.
- concentration and solvent at the time of mixing are not particularly limited, but are usually 10 to 100% by mass, preferably 20 to 100% by mass, and more preferably 30 to 100% by mass.
- the ⁇ -hydroxycarboxylic acid used in the present invention is an ⁇ -hydroxymonocarboxylic acid such as lactic acid
- the amount used is from the viewpoint of water absorption characteristics, and further from the viewpoint of reducing the molecular weight of soluble components,
- the amount is preferably 0.010 to 4.0 mol%, more preferably 0.025 to 2.5 mol%, and 0.05 to 1.0 mol%, most preferably 0.1 to 1. mol% based on the body. 0 mol%.
- ⁇ -hydroxycarboxylic acid (salt) refers to ⁇ -hydroxycarboxylic acid and / or a salt thereof, and is a hydroxycarboxylic acid having a hydroxyl group at the ⁇ -position in the molecule and / or a salt thereof.
- ⁇ acid (salt) refers to —acid and / or salt thereof.
- malic acid (salt) refers to malic acid and / or a salt thereof
- lactic acid (salt) refers to lactic acid and / or a salt thereof.
- Hydroxycarboxylic acid is a carboxylic acid having a hydroxyl group in the molecule. Lactic acid, glycolic acid, malic acid, glyceric acid, tartaric acid, citric acid, isocitric acid, mevalonic acid, quinic acid, shikimic acid, ⁇ -hydroxy Aliphatic hydroxy acids such as propionic acid, and aromatic hydroxy acids such as salicylic acid, creosote acid, vanillic acid, syringic acid, resosic acid, pyrocatechuic acid, protocatechuic acid, gentisic acid, orthoric acid, mandelic acid, gallic acid, etc. There are acids or their salts.
- the ⁇ -hydroxycarboxylic acid used in the present invention refers to a carboxylic acid in which a hydroxyl group is bonded to the carbon at the ⁇ -position in the molecule, and preferably non-polymeric ⁇ - Aliphatic hydroxycarboxylic acids such as hydroxycarboxylic acids, more preferably aliphatic ⁇ -hydroxycarboxylic acids having no cyclic structure or unsaturated groups.
- hydroxycarboxylic acids more preferably aliphatic ⁇ -hydroxycarboxylic acids having no cyclic structure or unsaturated groups.
- an aromatic ⁇ -hydroxycarboxylic acid or an ⁇ -hydroxycarboxylic acid having a cyclic structure or an unsaturated group it is not preferable because it is colored by an oxidation reaction.
- the molecular weight is preferably in the range of 40 to 2000, more preferably 60 to 1000, and particularly preferably 100 to 500.
- the ⁇ -hydroxycarboxylic acid used in the present invention has a water solubility at 20 ⁇ 5 ° C. and has a solubility in 100 g of deionized water of 1 g or more, more preferably 5 g or more, still more preferably 10 g or more, particularly preferably 20 g or more. Is preferable.
- ⁇ -hydroxycarboxylic acid salt
- lactic acid salt
- citric acid salt
- malic acid salt
- isocitric acid salt
- glyceric acid salt
- tartaric acid salt
- D-forms thereof examples include L-form and meso-form.
- the ⁇ -hydroxycarboxylic acid (salt) is lactic acid (salt), glycolic acid (salt), malic acid (salt), glyceric acid (salt), tartaric acid (salt), citric acid (salt), isocitric acid (salt) Salt), mevalonic acid (salt), quinic acid (salt), shikimic acid (salt), ⁇ -hydroxypropionic acid (salt), salicylic acid (salt), creosote acid (salt), vanillic acid (salt), shilling acid (Salt), resosic acid (salt), pyrocatechuic acid (salt), protocatechuic acid (salt), gentisic acid (salt), orceric acid (salt), mandelic acid (salt), and gallic acid (salt)
- One or more are selected.
- ⁇ -hydroxycarboxylic acid one or more of these acids (salts) may be used in combination. More preferably, malic acid (salt) and lactic acid (salt) may be used in combination as the ⁇ -hydroxycarboxylic acid (salt).
- the ⁇ -hydroxycarboxylic acid that is particularly preferably used is malic acid, lactic acid, or 2 or more, preferably 2 to 10, more preferably 2 to 6, more preferably 2 to 4 carboxyl groups in the molecule.
- One ⁇ -hydroxy polyvalent carboxylic acid is used.
- malic acid (salt), citric acid (salt), isocitric acid (salt) are particularly preferable from the viewpoint of improving water absorption characteristics and coloring, and from the viewpoint of reducing the molecular weight of soluble components.
- Tartaric acid (salt) is used.
- malic acid (salt) is used as the ⁇ -hydroxycarboxylic acid.
- the ⁇ -hydroxycarboxylic acid when it is a salt, it is preferably a monovalent salt from the viewpoint of solubility in water.
- Alkali metal salts such as lithium, potassium, sodium, ammonia salts, monovalent salts, and the like are preferable.
- An amine salt or the like is preferably used.
- ⁇ -hydroxy polyvalent carboxylic acid when ⁇ -hydroxy polyvalent carboxylic acid is used as a salt, all carboxyl groups may be used as a salt, or only a part may be used as a salt.
- post-polymerization step is a step performed after the polymerization.
- the post-polymerization process includes the gel pulverization process, drying process, pulverization process, classification process, surface treatment (crosslinking) agent mixing process, surface crosslinking (surface treatment) process, granulation process, curing process, and rehumidification process described below.
- the present invention is not limited to these.
- This step is a step of obtaining a dry polymer by drying the particulate hydrogel obtained in the polymerization step and / or the gel grinding step to a desired resin solid content.
- the resin solid content is determined from loss on drying (weight change when 1 g of water-absorbent resin is heated at 180 ° C. for 3 hours), preferably 80% by weight or more, more preferably 85 to 99% by weight, and still more preferably 90%. It is ⁇ 98% by weight, particularly preferably 92 to 97% by weight.
- the method for drying the particulate hydrous gel is not particularly limited.
- heat drying, hot air drying, reduced pressure drying, fluidized bed drying, infrared drying, microwave drying, drum dryer drying, azeotropy with a hydrophobic organic solvent examples include drying by dehydration and high-humidity drying using high-temperature steam.
- hot air drying is preferable from the viewpoint of drying efficiency, and band drying in which hot air drying is performed on a ventilation belt, hot air drying using an endless belt, or drum dryer drying is more preferable.
- the drying temperature (hot air temperature) in the hot air drying is preferably 120 to 250 ° C., more preferably 150 to 200 ° C. from the viewpoint of the color tone of the water absorbent resin and the drying efficiency.
- the drying conditions other than the drying temperature such as the speed of the hot air and the drying time, may be set as appropriate according to the moisture content and total weight of the particulate hydrous gel to be dried and the desired resin solid content, When performing band drying, various conditions described in International Publication Nos. 2006/100300, 2011/025012, 2011/025013, 2011/111657 and the like are appropriately applied.
- the desired water-absorbent resin CRC centrifuge retention capacity
- Ext water-soluble content
- color tone see [3] below
- the dried polymer obtained in the drying step is pulverized (pulverization step), adjusted to a predetermined particle size (classification step), and the water absorbent resin powder ( This is a step of obtaining a powdery water-absorbing resin before surface cross-linking for the sake of convenience as “water-absorbing resin powder”.
- Examples of the equipment used in the pulverization process of the present invention include a high-speed rotary pulverizer such as a roll mill, a hammer mill, a screw mill, and a pin mill, a vibration mill, a knuckle type pulverizer, and a cylindrical mixer. Used together.
- a high-speed rotary pulverizer such as a roll mill, a hammer mill, a screw mill, and a pin mill, a vibration mill, a knuckle type pulverizer, and a cylindrical mixer. Used together.
- the particle size adjustment method in the classification step of the present invention is not particularly limited, and examples thereof include sieve classification using JIS standard sieve (JIS Z8801-1 (2000)) and airflow classification.
- the particle size adjustment of the water-absorbing resin is not limited to the above pulverization step and classification step, but a polymerization step (especially reverse phase suspension polymerization or spray droplet polymerization), other steps (for example, granulation step, fine powder collection step) ).
- the water absorbent resin powder obtained in the present invention has a weight average particle diameter (D50) of preferably 200 to 600 ⁇ m, more preferably 200 to 550 ⁇ m, still more preferably 250 to 500 ⁇ m, and particularly preferably 350 to 450 ⁇ m.
- the ratio of particles having a particle diameter of less than 150 ⁇ m is preferably 10% by weight or less, more preferably 5% by weight or less, and further preferably 1% by weight or less.
- the ratio of particles having a particle diameter of 850 ⁇ m or more is preferably 5% or less. % By weight or less, more preferably 3% by weight or less, still more preferably 1% by weight or less.
- the lower limit of the ratio of these particles is preferably as small as possible in any case, and is preferably 0% by weight, but may be about 0.1% by weight.
- the logarithmic standard deviation ( ⁇ ) of the particle size distribution is preferably 0.20 to 0.50, more preferably 0.25 to 0.40, and still more preferably 0.27 to 0.35.
- the above-mentioned particle size is applied not only to the water-absorbing resin after surface crosslinking (hereinafter sometimes referred to as “water-absorbing resin particles” for convenience) but also to the water-absorbing resin as a final product. Therefore, in the water-absorbent resin particles, it is preferable that the surface cross-linking treatment (surface cross-linking step) is performed so as to maintain the particle size in the above range, and the particle size is adjusted by providing a sizing step after the surface cross-linking step. preferable.
- This step further increases the crosslink density on the surface layer of the water absorbent resin powder obtained through the above-described steps (part of several tens of ⁇ m from the surface of the water absorbent resin powder). It is a process of providing a part, and is composed of a mixing process, a heat treatment process, and a cooling process (optional).
- a water-absorbing resin (water-absorbing resin particles) surface-crosslinked by radical cross-linking or surface polymerization on the surface of the water-absorbing resin powder, a cross-linking reaction with a surface cross-linking agent, or the like is obtained.
- surface cross-linking agent Although it does not specifically limit as a surface crosslinking agent used by this invention, An organic or inorganic surface crosslinking agent is mentioned. Among these, an organic surface cross-linking agent that reacts with a carboxyl group is preferable from the viewpoint of the physical properties of the water-absorbing resin and the handleability of the surface cross-linking agent. Examples thereof include one or more surface cross-linking agents disclosed in US Pat. No. 7,183,456. More specifically, polyhydric alcohol compounds, epoxy compounds, haloepoxy compounds, polyvalent amine compounds or condensates thereof, oxazoline compounds, oxazolidinone compounds, polyvalent metal salts, alkylene carbonate compounds, cyclic urea compounds, etc. Can be mentioned.
- the amount of the surface cross-linking agent used is preferably 0.01 to 10 parts by weight, more preferably 0.01 to 5 parts by weight with respect to 100 parts by weight of the water absorbent resin powder. It is.
- the surface cross-linking agent is preferably added as an aqueous solution.
- the amount of water used is preferably 0.1 to 20 parts by weight, more preferably 0.1 to 100 parts by weight of the water-absorbent resin powder. 5 to 10 parts by weight.
- the amount used is preferably 10 parts by weight or less, more preferably 5 parts by weight or less, with respect to 100 parts by weight of the water-absorbent resin powder.
- each additive added in the “rehumidification step” described later is added to the surface cross-linking agent (aqueous solution) within a range of 5 parts by weight or less, or added separately in the main mixing step. You can also.
- This step is a step of mixing the water absorbent resin powder and the surface cross-linking agent.
- the method for mixing the surface cross-linking agent is not particularly limited, but a surface cross-linking agent solution is prepared in advance, and the liquid is preferably sprayed or dripped onto the water-absorbent resin powder, and more preferably sprayed. And mixing them.
- the apparatus for performing the mixing is not particularly limited, but a high-speed stirring type mixer is preferable, and a high-speed stirring type continuous mixer is more preferable.
- Heat treatment process In this step, heat is applied to the mixture discharged from the mixing step to cause a crosslinking reaction on the surface of the water absorbent resin powder.
- the apparatus for carrying out the crosslinking reaction is not particularly limited, but preferably includes a paddle dryer.
- the reaction temperature in the crosslinking reaction is appropriately set depending on the type of the surface crosslinking agent used, but is preferably 50 to 300 ° C, more preferably 100 to 200 ° C. By appropriately adjusting the heating temperature in the surface cross-linking step, the centrifugal retention capacity of the obtained water-absorbing agent can be appropriately adjusted.
- the heating time in the heat treatment step is not particularly limited, but is appropriately set according to the type, reactivity, usage amount, etc. of the surface crosslinking agent to be used, but is preferably 15 to 120 minutes, more preferably 30. ⁇ 60 minutes.
- the heating time in the surface cross-linking step it is possible to appropriately adjust the centrifugal retention capacity of the water-absorbing agent to be obtained.
- This step is an optional step that is installed as necessary after the heat treatment step.
- the apparatus for performing the cooling is not particularly limited, but is preferably an apparatus having the same specifications as the apparatus used in the heat treatment step, and more preferably a paddle dryer. It is because it can be used as a cooling device by changing the heat medium to a refrigerant.
- the water absorbent resin particles obtained in the heat treatment step are forcibly cooled to 40 to 80 ° C., more preferably 50 to 70 ° C., if necessary, in the cooling step.
- Re-humidification step involves adding the following polyvalent metal salt compound, polycationic polymer, chelating agent, inorganic reducing agent, hydroxycarboxylic acid compound to the water-absorbent resin particles obtained in the surface cross-linking step.
- the additive since the said additive is added with aqueous solution or a slurry liquid, a water absorbing resin particle is swollen with water again. For this reason, this process is referred to as a “rehumidification process”.
- the additive can be mixed with the water-absorbent resin powder simultaneously with the surface cross-linking agent (aqueous solution).
- Polyvalent metal salt and / or cationic polymer In the present invention, it is preferable to add a polyvalent metal salt and / or a cationic polymer from the viewpoint of improving the water absorption rate, liquid permeability, hygroscopic fluidity and the like of the water absorbent resin obtained.
- polyvalent metal salt and / or cationic polymer specifically, compounds disclosed in “[7] Polyvalent metal salt and / or cationic polymer” of International Publication No. 2011/040530 and the amount of use thereof Applies to the present invention.
- “Chelating agent” refers to a multidentate ligand that coordinates to a metal ion to form a chelate compound.
- the form of adding the chelating agent is not particularly limited, but in order to uniformly disperse the chelating agent in the water-absorbing agent, a form in which a liquid or powdered chelating agent is dissolved in a hydrophilic solvent such as water is added, Or the form which adds a fine powder-form chelating agent in a powder state is preferable.
- a chelating agent from the viewpoint of the color tone (coloring prevention, resistance to coloration with time) and deterioration prevention (strong urine resistance) of the water-absorbing resin obtained.
- chelating agent specifically, compounds disclosed in “[2] chelating agent” of WO 2011/040530 and the amount of use thereof are applied to the present invention.
- examples of the chelating agent include phosphorus chelating agents and aminocarboxylic acid chelating agents.
- an organic aminophosphoric acid having an amino group is preferable, a water-soluble organic aminophosphoric acid, and further a water-soluble non-polymeric organic aminophosphoric acid is used, and the amino groups per molecule thereof.
- the number of phosphate groups is preferably 1 or more, more preferably 2 or more, and especially 3 or more.
- the upper limit of the number of amino groups and phosphate groups is usually 100 or less, further 10 or less, and particularly 5 or less.
- water-soluble refers to a compound that dissolves in 100 g of water at 25 ° C. in an amount of 0.1 g or more, further 1 g or more, particularly 5 g or more.
- the molecular weight is usually in the range of 50 to 5000, preferably 100 to 1000, and more preferably 200 to 500.
- Examples of phosphorus chelating agents used include ethylenediamine-N, N′-di (methylenephosphinic acid), ethylenediaminetetra (methylenephosphinic acid), nitriloacetic acid-di (methylenephosphinic acid), nitrodiacetic acid- (methylenephosphinic acid) ), Nitriloacetic acid- ⁇ -propionic acid-methylenephosphonic acid, nitrilotris (methylenephosphonic acid), cyclohexanediaminetetra (methylenephosphonic acid), ethylenediamine-N, N′-diacetic acid-N, N′-di (methylenephosphone) Acid), ethylenediamine-N, N′-di (methylenephosphonic acid), ethylenediaminetetra (methylenephosphonic acid), polymethylenediaminetetra (methylenephosphonic acid), diethylenetriaminepenta (methylenephosphonic acid), 1-hydroxyethyl Denjihosuhon acid,
- the most preferred phosphorus-based chelating agent in the present invention is ethylenediaminetetra (methylenephosphonic acid) or a salt thereof.
- the salt include alkali metal salts such as sodium salt and potassium salt, ammonium salt, and amine salt.
- a sodium salt and potassium salt are especially preferable.
- aminocarboxylic acid chelating agent examples include iminodiacetic acid, hydroxyethyliminodiacetic acid, nitrilotriacetic acid, nitrilotripropionic acid, ethylenediaminetetraacetic acid, hydroxyethylenediaminetriacetic acid, hexamethylenediaminetetraacetic acid, diethylenetriaminepentaacetic acid (DTPA).
- DTPA diethylenetriaminepentaacetic acid
- the most preferred aminocarboxylic acid chelating agent in the present invention is diethylenetriaminepentaacetic acid (DTPA) or a salt thereof.
- the salt include alkali metal salts such as sodium salt and potassium salt, ammonium salt, and amine salt.
- a sodium salt and potassium salt are especially preferable.
- the phosphorus chelating agent used in the present invention is diethylenetriaminepentaacetic acid (DTPA) or a salt thereof, or ethylenediaminetetra (methylenephosphonic acid) (EDTMP) or a salt thereof.
- DTPA diethylenetriaminepentaacetic acid
- ETMP ethylenediaminetetra (methylenephosphonic acid)
- Inorganic reducing agent In the present invention, it is preferable to add an inorganic reducing agent from the viewpoints of color tone (anti-coloring), deterioration prevention, residual monomer reduction and the like of the water-absorbing resin obtained.
- the compounds disclosed in “[3] Inorganic reducing agent” of WO 2011/040530 and the amount used thereof are applied to the present invention.
- additives other than the above-mentioned additives can be added in order to add various functions to the water absorbent resin.
- additives include surfactants, compounds having phosphorus atoms, oxidizing agents, organic reducing agents, water-insoluble inorganic fine particles, organic powders such as metal soaps, deodorants, antibacterial agents, pulp and thermoplastics. Examples thereof include fibers.
- the surfactant is a compound disclosed in International Publication No. 2005/077500, and the water-insoluble inorganic fine particles are disclosed in International Publication No. 2011/040530 “[5] Water-insoluble inorganic fine particles”. Each of these compounds is applied to the present invention.
- the use amount (addition amount) of the additive is appropriately determined depending on the application and is not particularly limited, but is preferably 3 parts by weight or less, more preferably 1 part per 100 parts by weight of the water absorbent resin powder. Less than parts by weight. Moreover, this additive can also be added at a process different from the said process.
- a surface treatment (crosslinking) agent mixing step in addition to the steps described above, a curing step, a granulating step, a granulating step, a fine powder removing step, a fine powder reusing step, etc. It can be provided as necessary. Moreover, you may further include 1 type, or 2 or more types of processes, such as a transport process, a storage process, a packing process, and a storage process.
- the “granulating step” includes a step of performing classification and pulverization when the fine powder removing step after the surface cross-linking step and the water absorbent resin are aggregated and exceed a desired size.
- the “fine powder recycling step” includes a step of adding the fine powder as it is as in the present invention to a large water-containing gel and adding it to any step of the production process of the water absorbent resin.
- polyacrylic acid (salt) water-absorbing agent obtained by the production method according to the present invention is used when the water-absorbing agent is used in sanitary goods, particularly paper diapers.
- sanitary goods particularly paper diapers.
- the polyacrylic acid (salt) water-absorbing agent obtained by the production method according to the present invention is not particularly limited with respect to its shape, but is preferably in the form of particles.
- the physical properties of the particulate water-absorbing agent which is a preferred embodiment will be described.
- the following physical property was measured based on EDANA method unless there is particular notice.
- the CRC (centrifuge retention capacity) of the water-absorbing agent of the present invention is 30 g / g or more.
- the upper limit is not particularly limited and is preferably as high as possible, but from the viewpoint of balance with other physical properties, it is preferably 70 g / g or less, more preferably 50 g / g or less, and still more preferably 40 g / g or less.
- CRC When the CRC exceeds 70 g / g, the rate of absorbing body fluids such as urine and blood decreases, so it is not suitable for use in high water absorption rate type paper diapers.
- CRC can be controlled by an internal crosslinking agent, a surface crosslinking agent, or the like.
- the CRC (centrifuge retention capacity) of the water-absorbing agent of the present invention can be appropriately adjusted by the following “[4] Method for adjusting physical properties of polyacrylic acid (salt) -based water-absorbing agent”.
- the AAP (water absorption capacity under pressure) of the water-absorbing agent of the present invention is preferably 20 g / g or more, more preferably 22 g / g or more, still more preferably 23 g / g or more, particularly preferably 24 g / g or more, most preferably 25 g. / G or more.
- the upper limit is not particularly limited, but is preferably 30 g / g or less.
- AAP When the AAP is less than 20 g / g, the amount of return of the liquid when pressure is applied to the absorber (usually referred to as “Re-Wet”) increases, and the absorber of sanitary articles such as paper diapers Not suitable as.
- AAP can be controlled by particle size, surface cross-linking agent, and the like.
- the particle size (particle size distribution, weight average particle size (D50), logarithmic standard deviation of particle size distribution ( ⁇ )) of the water-absorbing agent of the present invention is the same as the particle size of the water-absorbing agent powder (water-absorbing resin powder) before surface crosslinking. It is controlled to become.
- the weight average particle diameter (D50) is preferably 200 to 600 ⁇ m, more preferably 200 to 550 ⁇ m, still more preferably 250 to 500 ⁇ m, and particularly preferably 350 to 450 ⁇ m.
- the ratio of particles having a particle diameter of less than 150 ⁇ m is preferably 10% by weight or less, more preferably 5% by weight or less, and further preferably 1% by weight or less.
- the ratio of particles having a particle diameter of 850 ⁇ m or more is preferably 5% or less.
- the lower limit of the ratio of these particles is preferably as small as possible in any case, and is preferably 0% by weight, but may be about 0.1% by weight.
- the logarithmic standard deviation ( ⁇ ) of the particle size distribution is preferably 0.20 to 0.50, more preferably 0.25 to 0.40, and still more preferably 0.27 to 0.35.
- the Ext (water-soluble content) of the water-absorbing agent of the present invention is usually 50% by weight or less, preferably 35% by weight or less, more preferably 25% by weight or less with respect to the upper limit.
- About a lower limit Preferably it is 10 weight% or more, More preferably, the soluble content of the said water absorbing agent is 13 weight% or more, More preferably, it is 15 weight% or more.
- the gel strength is weak and the water-absorbing agent may be inferior in liquid permeability. Furthermore, since rewetting increases, it is not suitable as an absorbent material for sanitary goods such as paper diapers. Ext can be controlled with an internal cross-linking agent or the like.
- water-soluble polymer amount and soluble fraction may be abbreviated as “water-soluble polymer amount” or “soluble component”.
- the water-soluble polymer is also referred to as “water-soluble component” or “soluble component”.
- the “soluble fraction” is a ratio (% by weight) of a soluble component in the water-absorbing agent.
- Soluble fraction should also be described as “Ext”, “water-soluble”, “soluble”, “water-soluble” or “soluble” with units of% by weight. There is also.
- the soluble content of the water-absorbing agent of the present invention is preferably 50% by weight or less, 45% by weight or less, 40% by weight or less, 35% by weight or less, 30% by weight or less, 25% by weight or less, or 20% by weight or less. 15% by weight or less and 10% by weight or less. More preferably, the upper limit is 35% by weight or less and 30% by weight or less.
- the absorption capacity such as CRC and AAP may decrease with time.
- the lower limit of the soluble content of the water-absorbing agent of the present invention is preferably 10% by weight or more, more preferably 13% by weight or more, and still more preferably 15% by weight or more. Possible combinations of the upper and lower limits of the soluble content are also included in the scope of the present invention. More preferably, the soluble content of the water-absorbing agent of the present invention is 10% by weight or more and 30% by weight or less.
- the water-absorbing article using the water-absorbing agent of the present invention may be used depending on the soluble content eluted from the absorbed water-absorbing agent. It is not preferable because a person may feel discomfort due to stickiness. On the other hand, if the lower limit of the preferred range is not reached, a large amount of the internal cross-linking agent must be used, and a sufficient water absorption ratio may not be obtained, which is not preferable.
- the soluble amount of the water-absorbing agent of the present invention can be appropriately adjusted by the following “[4] Method for adjusting physical properties of polyacrylic acid (salt) -based water-absorbing agent”.
- the water content of the water-absorbing agent of the present invention is preferably more than 0% by weight and 15% by weight or less, more preferably 1 to 13% by weight, still more preferably 2 to 10% by weight, particularly preferably Is from 2 to 9% by weight.
- a water absorbent excellent in powder characteristics for example, fluidity, transportability, damage resistance, etc.
- the residual monomer contained in the water-absorbing agent of the present invention is preferably 500 ppm or less, more preferably 400 ppm or less, and even more preferably 300 ppm or less. Although it does not specifically limit about a lower limit, Preferably it is 0 ppm, More preferably, it is about 10 ppm.
- the content of the residual monomer is within the above range, a water-absorbing agent that can reduce irritation to human skin and the like can be obtained.
- the FSR (water absorption rate) of the water-absorbing agent of the present invention is preferably 0.10 g / g / s or more, more preferably 0.15 g / g / s or more, still more preferably 0.20 g / g / s or more, particularly preferably. Is 0.25 g / g / s or more. Although it does not specifically limit about an upper limit, Preferably it is 5.0 g / g / s or less, More preferably, it is 3.0 g / g / s or less.
- FSR is less than 0.10 g / g / s, body fluids such as urine and blood may not be sufficiently absorbed and liquid leakage may occur, which is not suitable as an absorbent material for sanitary articles such as paper diapers.
- the FSR can be controlled by foam polymerization, particle size, and the like.
- Moisture absorption blocking rate “Hygroscopic blocking” refers to a phenomenon in which water-absorbing resins aggregate when absorbing moisture, and “moisture absorption blocking rate” refers to the rate of aggregation.
- the moisture absorption blocking rate is calculated by the method described in the following Example [Measurement of Physical Properties of Water Absorbing Agent] (c).
- the moisture absorption blocking rate of the water-absorbing agent of the present invention is preferably adjusted to 50% by mass or less, more preferably 30% by mass or less, and still more preferably 10% by mass or less by adding a moisture absorption fluidity improver.
- the lower limit is 0% by mass or more on the calculation principle.
- the addition amount of the hygroscopic fluidity improving agent can be adjusted in the range of 0% by mass to 1% by mass with respect to the water absorbing agent so as to achieve the above-described hygroscopic blocking rate.
- the moisture absorption blocking rate to be low, the water absorbing agent can be stably used in any working environment and user's usage conditions (for example, operating conditions of the diaper manufacturing process).
- Soluble molecular weight or “molecular weight soluble” refers to the weight average molecular weight of the soluble fraction.
- the soluble molecular weight of the water-absorbing agent of the present invention is preferably in the range of 50,000 to 1,000,000 daltons, more preferably 100,000 to 800,000 daltons, 150, 000 daltons to 700,000 daltons, 200,000 daltons to 700,000 daltons, 150,000 daltons to 600,000 daltons, 200,000 daltons to 600,000 daltons, most preferably 200,000 daltons or more Less than 550,000 daltons.
- the lower limit of the soluble molecular weight of the water-absorbing agent of the present invention is preferably 30,000, 40,000, 50,000, 75,000, 100,000, 125,000, 150,000, 175,000, 200,000, 225,000, or 250,000 daltons, and the upper limit is preferably 1,000,000, 900,000, 800,000, 700,000, 600,000, or 500,000 daltons is there. Possible combinations of the upper limit value and the lower limit value are also included in the scope of the present invention.
- the weight average molecular weight of the soluble part of the water-absorbing agent of the present invention can be calculated by the method described in the following [Measurement of physical properties of water-absorbing agent] (d).
- the soluble molecular weight of the water-absorbing agent of the present invention can be appropriately adjusted by the following “[4] Method for adjusting physical properties of polyacrylic acid (salt) -based water-absorbing agent”.
- Intrinsic viscosity of soluble component (IV) The intrinsic viscosity (IV) of the soluble component of the water-absorbing agent of the present invention is preferably 0.5 to 4.5 dl / g, more preferably 0.8 to 4.2 dl / g, 1.0 to 3.8 dl. / G, 1.5 to 4.0 dl / g, most preferably 1.5 to 3.5 dl / g.
- stickiness is reduced in the tactile sensation evaluation in the water-absorbing agent when the above-mentioned intrinsic viscosity (IV) of the soluble component is used.
- the lower limit of the intrinsic viscosity (IV) of the soluble component of the water-absorbing agent of the present invention is preferably 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9. 1.0, 1.1, 1.2, 1.3, 1.4, 1.5, 1.6, 1.7, or 1.8 dl / g, and the upper limit is preferably 5. 0, 4.9, 4.8, 4.7, 4.6, 4.5, 4.4, 4.3, 4.2, 4.1, 4.0, 3.9, 3.8, 3.7, 3.6, 3.5, 3.4, 3.3, 3.2, 3.1, or 3.0 dl / g. Possible combinations of the upper limit value and the lower limit value are also included in the scope of the present invention.
- the intrinsic viscosity (IV) of the soluble part of the water-absorbing agent of the present invention can be calculated by the method described in the following [Measurement of physical properties of water-absorbing agent] (d).
- Soluble molecular weight reduction rate (%) “Soluble content molecular weight reduction ratio (%)” means that the water-absorbing agent containing ⁇ -hydroxycarboxylic acid has a soluble molecular weight equivalent to that of the water-absorbing agent not containing ⁇ -hydroxycarboxylic acid having the same CRC. Refers to a reduced ratio to molecular weight.
- the molecular weight reduction rate (%) of the water-absorbing agent of the present invention is preferably 8% or more, 9% or more, 10% or more, 11% or more, 12% or more, 14% or more, and most preferably 15%. % Or more.
- the upper limit is not particularly limited, but is preferably 50% or less, more preferably 40% or less.
- the soluble molecular weight reduction rate (%) of the water-absorbing agent of the present invention can be calculated by the method described in the following [Measurement of physical properties of water-absorbing agent] (e).
- the molecular weight reduction rate of the soluble component of the water-absorbing agent can be used as an index of stickiness of the water-absorbing agent after liquid absorption, preferably 8% or more, 9% or more, 10% or more, 11% or more, 12% or more, When it is 14% or more, and most preferably 15% or more, it can be said that the stickiness of the water-absorbing agent is reduced.
- the stickiness of the water-absorbing agent of the present invention after liquid absorption is defined by the tactile evaluation of the dissolved soluble matter on the nonwoven fabric when the pseudo sanitary agent is absorbed. Specifically, the tactile sensation is evaluated by the method described in [Measurement of physical properties of water-absorbing agent] (f) Tactile sensation evaluation below.
- the tactile sensation evaluation is preferably 2.5 or less, 2.3 or less, 2.1 or less, 2.0 or less, 1.8 or less, and most preferably 1.5 or less, it can be said that stickiness is reduced. .
- the water-absorbing agent obtained in the present invention can be suitably used for sanitary materials such as disposable diapers, and in such a case, the water-absorbing agent can be remarkably used even under long-term storage conditions under high humidity and temperature conditions. Maintain a clean white state. Furthermore, in the Hunter Lab color system measurement using a spectroscopic color difference meter for particles exposed to an atmosphere having a temperature of 70 ⁇ 1 ° C. and a relative humidity of 65 ⁇ 1%, which is obtained by the above-described production method, the L value A time-stable color-stable water-absorbing agent having (Lightness) of at least 70, more preferably 74 or more, particularly preferably 78 or more. (Note that the upper limit of the L value is normally 100, but if it is 70, there will be no substantial problem even in actual use.)
- the yellowness before the color acceleration test (YI value / Yellow Index / European Patent No. 942014 and No. 1108745) is preferably 0-30, more preferably 0-25, 0-23, 0-20, 0. -18, more preferably 0-13, still more preferably 0-10, most preferably 0-5, and it is preferable that there is almost no yellowing. Further, even when exposed to severe high temperature and high humidity of 26.5 or less, the yellowness after the color acceleration test for 7 days at a temperature of 70 ° C. ⁇ 1 and a relative humidity of 65 ⁇ 1% as defined in the examples. Shows amazing yellowing prevention properties.
- Fe / L-as deteriorated soluble content (specified by the measurement method described in the examples) is a moisture content correction value, preferably 0 to 30% by weight, More preferably, it is controlled to 0 to 25% by weight, and further preferably 0 to 20% by weight.
- the surface tension (specified by the measurement method described in the Examples) is preferably 60 [mN / m] or more, more preferably 65 [mN / m] or more, and further preferably 67 [mN / m. m] or more, particularly preferably 70 [mN / m] or more, most preferably 72 [mN / m] or more, and there is no substantial reduction in surface tension.
- the upper limit is usually 75 [mN / m].
- the centrifugal capacity of the water-absorbing agent of the present invention is mainly determined by the amount of the internal cross-linking agent in the polymerization step, It can be adjusted as appropriate by adjusting at least one selected from the group consisting of the amount, the concentration of the monomer component, the heating temperature and the heating time in the surface crosslinking step.
- the internal cross-linking agent and the amount thereof, and the ⁇ -hydroxycarboxylic acid (salt) and the amount thereof are respectively (internal cross-linking agent of “[2] Production method of polyacrylic acid (salt) water-absorbing resin”).
- concentration of the monomer component examples include the concentrations described in “(Monomer component concentration)” in “[2] Production method of polyacrylic acid (salt) -based water absorbent resin”.
- the heating temperature and heating time in the surface cross-linking step are described in (2-6) Surface cross-linking step (heat treatment step) of “[2] Production method of polyacrylic acid (salt) water-absorbing resin” above. Examples include heating temperature and heating time.
- the soluble content of the water-absorbing agent of the present invention can be appropriately adjusted mainly by adjusting at least one selected from the group consisting of the amount of the internal crosslinking agent and the concentration of the monomer component in the polymerization step.
- the internal cross-linking agent and the amount thereof include the compounds described in (Internal cross-linking agent) in “[2] Production method of polyacrylic acid (salt) -based water absorbent resin” and the amount of use thereof.
- Examples of the concentration of the monomer component include the concentrations described in “(Monomer component concentration)” in “[2] Production method of polyacrylic acid (salt) -based water absorbent resin”.
- the soluble molecular weight of the water-absorbing agent of the present invention is mainly adjusted by adjusting at least one selected from the group consisting of the amount of the internal crosslinking agent, the amount of ⁇ -hydroxycarboxylic acid, and the concentration of the monomer component in the polymerization step. This can be adjusted as appropriate.
- the internal cross-linking agent and the amount thereof, and the ⁇ -hydroxycarboxylic acid (salt) and the amount thereof are respectively (internal cross-linking agent of “[2] Production method of polyacrylic acid (salt) water-absorbing resin”). ) And ( ⁇ -hydroxycarboxylic acid (salt)) and the amount of use thereof.
- concentration of the monomer component include the concentrations described in “(Monomer component concentration)” in “[2] Production method of polyacrylic acid (salt) -based water absorbent resin”.
- the use of the water-absorbing agent of the present invention is not particularly limited, but preferably includes absorbent use for sanitary products such as paper diapers, sanitary napkins, and incontinence pads. .
- it can be used as an absorber of high-concentration paper diapers (those with a large amount of water-absorbing agent used per paper diaper) that have been problematic in terms of odor, coloring, and the like derived from raw materials. Furthermore, a remarkable effect can be expected when used in the upper layer of the absorber.
- an absorbent material such as pulp fiber can also be used as the absorber.
- the content (core concentration) of the water-absorbing agent in the absorber is preferably 20 to 100% by weight, more preferably 40 to 100% by weight, still more preferably 50 to 100% by weight, and still more preferably 60%. -100% by weight, particularly preferably 70-100% by weight, most preferably 75-95% by weight.
- the absorbent article can maintain a clean white state. Furthermore, since the diffusibility of body fluids such as urine and blood is excellent, the amount of absorption can be improved by efficient liquid distribution.
- the method includes a polymerization step of a monomer aqueous solution containing acrylic acid (salt), a drying step, and a surface crosslinking step.
- CRC centrifuge retention capacity
- Adding an arbitrary additive before the drying step is referred to as internal addition, and adding an arbitrary additive after the drying step is sometimes referred to as external addition.
- the present invention reduces the molecular weight of soluble components that are eluted when the water absorbent absorbs and swells by internally adding ⁇ -hydroxycarboxylic acid (salt). As a result, it was found that stickiness that leads to discomfort during actual use such as paper diapers can be reduced.
- an arbitrary chelating agent for example, diethylenetriaminepentaacetic acid (DTPA) or ethylenediaminetetra (methylenephosphonic acid) (EDTMP)
- DTPA diethylenetriaminepentaacetic acid
- ETMP ethylenediaminetetra (methylenephosphonic acid)
- an ⁇ -hydroxycarboxylic acid (salt) is internally added, and from the viewpoint of color tone (anti-coloring), prevention of deterioration, etc., any chelating agent (for example, diethylenetriaminepentaacetic acid (DTPA) or its Salt or ethylenediaminetetra (methylenephosphonic acid) (EDTMP) or a salt thereof may be externally added.
- DTPA diethylenetriaminepentaacetic acid
- ETMP ethylenediaminetetra (methylenephosphonic acid)
- the centrifuge retention capacity (CRC) of the absorbent is preferably 30 g / g or more, 31 g / g or more, 32 g / g or more, 33 g / g or more, 34 g / g or more, 35 g / g or more.
- a particularly preferred centrifuge retention capacity is 30 g / g or more.
- the water-absorbing agent having a centrifuge retention capacity of less than 30 g / g under the above conditions no reduction in the molecular weight of the soluble component was observed, and therefore, stickiness after liquid absorption of the water-absorbing agent, which is the subject of the present invention, occurred. do not do.
- the water-absorbing agent has reduced stickiness after liquid absorption.
- the reduction in stickiness of the water-absorbing agent can be defined by using the molecular weight reduction rate of the soluble component of the water-absorbing agent (that is, the soluble component molecular weight reduction rate (%)) as an index.
- the water-absorbing agent has a molecular weight reduction rate of preferably 8% or more, 9% or more, 10% or more, 11% or more, 12% or more, 14% or more, and most preferably 15% or more, water absorption It can be said that the stickiness of the agent has been reduced.
- the ⁇ -hydroxycarboxylic acid (salt) is added before, during or after the polymerization step. More preferably, the ⁇ -hydroxycarboxylic acid (salt) is added before or during the polymerization step. Specifically, preferably, the ⁇ -hydroxycarboxylic acid (salt) is added to the monomer aqueous solution before polymerization. Alternatively, preferably, the ⁇ -hydroxycarboxylic acid (salt) is added to the aqueous monomer solution after the start of polymerization, and specifically, added to the aqueous monomer solution 2 minutes after the start of polymerization.
- the ⁇ -hydroxycarboxylic acid (salt) can be added at any time from the start of polymerization to the end of polymerization of the aqueous monomer solution (for example, 1 minute, 2 minutes, 3 minutes, 5 minutes after the start of polymerization, For example after 10 minutes or after 20 minutes).
- the method further includes a gel grinding step after the polymerization step and before the drying step, wherein the ⁇ -hydroxycarboxylic acid (salt) is added before or during the gel grinding step.
- the ⁇ -hydroxycarboxylic acid (salt) is added during the gel grinding step.
- the ⁇ -hydroxycarboxylic acid (salt) is added at the time of gel pulverization of the hydrogel obtained after polymerization.
- the ⁇ -hydroxycarboxylic acid (salt) is added before or during the polymerization step.
- the amount of the ⁇ -hydroxycarboxylic acid (salt) added is 0.010 to 4.0 mol% with respect to acrylic acid (salt). More preferably, the amount of the ⁇ -hydroxycarboxylic acid (salt) added is 0.025 to 2.5 mol%, 0.05 to 1.0 mol% with respect to acrylic acid (salt). Most preferably, it is 0.1 to 1.0 mol%. Although not wishing to be bound by theory, this amount is preferable from the viewpoint of water absorption characteristics and reduction of the molecular weight of soluble components.
- the lower limit of the amount of ⁇ -hydroxycarboxylic acid (salt) added is preferably 0.005, 0.006, 0.007, 0.008, 0.009, 0.010, 0.011, 0.012, 0.015, 0.02, 0.025, 0.03, 0.035, 0.040, 0.05, 0.055, 0.06, 0.065, 0.07, 0. 075, 0.08, 0.085, 0.09, 0.1, 0.11, 0.12, 0.15, or 0.2 mol%, and the upper limit is 5.0, 4.5 4.0, 3.5, 3.0, 2.5, 2.0, 1.5, 1.0, or 0.5 mol%. Possible combinations of the upper limit value and the lower limit value are also included in the scope of the present invention.
- the ⁇ -hydroxycarboxylic acid (salt) is lactic acid (salt), glycolic acid (salt), malic acid (salt), glyceric acid (salt), tartaric acid (salt), citric acid (salt).
- the ⁇ -hydroxycarboxylic acid (salt) is selected from the group consisting of malic acid (salt) and lactic acid (salt).
- the ⁇ -hydroxycarboxylic acid (salt) malic acid (salt) and lactic acid (salt) may be used in combination.
- the ⁇ -hydroxycarboxylic acid (salt) is malic acid (salt).
- the centrifuge retention capacity (CRC) of the absorbent prior to surface crosslinking is preferably greater than 34 g / g, greater than 36 g / g, greater than 38 g / g, greater than 40 g / g. Greater than 42 g / g, greater than 44 g / g, greater than 46 g / g, greater than 48 g / g, greater than 50 g / g, greater than 52 g / g, greater than 54 g / g.
- Particularly preferred absorbents have a centrifuge retention capacity (CRC) prior to surface crosslinking of greater than 34 g / g.
- the soluble amount of the water-absorbing agent is 8% to 30% by weight, 10% to 30% by weight, 13% to 30% by weight, 15% to 30% by weight. 17 wt% to 30 wt%, 10 wt% to 35 wt%, 13 wt% to 35 wt%, 15 wt% to 35 wt%, 10 wt% to 25 wt%, 13 wt% 25 wt% or less, 15 wt% or more and 25 wt% or less.
- the lower limit is 10% by weight or more, more preferably 13% by weight or more, and still more preferably 15% by weight or more.
- the upper limit is 30% by weight or less.
- the soluble content of the water-absorbing agent is 10% by weight or more and 30% by weight or less.
- the water-absorbing article using the water-absorbing agent of the present invention may be used depending on the soluble content eluted from the absorbed water-absorbing agent. It is not preferable because a person may feel discomfort due to stickiness. On the other hand, if the lower limit of the preferred range is not reached, a large amount of the internal cross-linking agent must be used, and a sufficient water absorption ratio may not be obtained, which is not preferable.
- the soluble component has a molecular weight in the range of 50,000 to 1,000,000 daltons, more preferably 100,000 to 800,000 daltons, 150 1 million daltons to 700,000 daltons, 200,000 daltons to 700,000 daltons, 150,000 daltons to 600,000 daltons, 200,000 daltons to 600,000 daltons, most preferably 200,000 daltons Above 550,000 daltons.
- the reduction in stickiness of the water-absorbing agent can be defined by using the molecular weight of the soluble component of the water-absorbing agent as an index.
- the soluble molecular weight is preferably 200,000 daltons or more and 700,000 daltons or less, and most preferably 200,000 daltons or more and 550,000 daltons or less, it can be said that the stickiness of the water-absorbing agent is reduced.
- the surface cross-linking step is performed after the drying step.
- the surface cross-linking step includes at least one of the steps described in the above “(2-6) Surface cross-linking step”.
- One preferred embodiment includes measuring the amount of iron in the raw caustic soda (NaOH).
- the following iron content is measured by a known method such as ICP (High Frequency Inductively Coupled Plasma) emission spectroscopy.
- the amount of iron in the raw material NaOH is preferably 10 ppm or less, more preferably 5 ppm or less, more preferably 2 ppm or less, and most preferably 1 ppm or less as iron oxide (Fe 2 O 3 ).
- the method further includes at least one of a classification step, a surface cross-linking step, a rehumidification step, a granulation step, a granulation step, a fine powder removal step, and a fine powder reuse step.
- the method further includes a step of reusing fine powder generated during the production of the water-absorbing agent.
- the fine powder is produced in any step after the polymerization step. More preferably, the fine powder is between a process selected from the group consisting of a gel grinding process, a drying process, a grinding process, a classification process, a surface crosslinking process, a rehumidification process, a granulation process, a granulation process, and a fine powder removal process. Or later. Even more preferably, the fines occur mainly during or after the grinding process.
- the drying step is preferably 120 to 250 ° C., more preferably 150 to 200 ° C., preferably 5 to 120 minutes, more preferably 10 to 90 minutes, even more preferably 15 to 60 minutes.
- Heat drying, hot air drying, vacuum drying, fluidized bed drying, infrared drying, microwave drying, drum dryer drying, drying by azeotropic dehydration with hydrophobic organic solvent, high humidity drying using high temperature steam Performing at least one selected drying procedure.
- a more preferable drying procedure is hot air drying using an endless belt or drum dryer drying.
- the peak temperature during the polymerization reaction of the aqueous monomer solution in the polymerization step is 85 ° C. or higher. In a more preferred embodiment, the peak temperature during the polymerization reaction of the monomer aqueous solution in the polymerization step is 90 ° C. or higher. Specifically, the peak temperature during the polymerization reaction of the aqueous monomer solution in the polymerization step is preferably 85 ° C. or higher, 86 ° C. or higher, 87 ° C. or higher, 88 ° C. or higher, 89 ° C. or higher, or 90 ° C. or higher. . As an upper limit, 100 degrees C or less, 98 degrees C or less, or 95 degrees C or less is preferable.
- the method further comprises adding a chelating agent after the drying step. This is because, by adding a chelating agent, effects such as color tone (coloring prevention) and deterioration prevention can be imparted to the water absorbing agent.
- the amount of the chelating agent added after the drying step is 0.001 to 5.0 parts by weight, 0.002 to 3.0 parts by weight with respect to 100 parts by weight of the water absorbing agent. 0.003 to 1.0 part by weight, 0.004 to 0.5 part by weight, particularly preferably 0.005 to 0.2 part by weight. Although not wishing to be bound by theory, this amount is preferable from the viewpoints of color tone (coloration prevention), deterioration prevention, and the like.
- the lower limit of the amount of the chelating agent added is preferably 0.0005, 0.001, 0.0015, 0.002, 0.0025, 0.003, 0.0035, 0.004, 0.0045.
- the upper limit is preferably 6.0, 5.5, 5.0, 4.5, 4.0, 3.5, 3. 0, 2.5, 2.0, 1.5, 1.0, 0.7, 0.6, 0.5, 0.4, 0.3, 0.2, or 0.1 parts by weight.
- Possible combinations of the upper limit value and the lower limit value are also included in the scope of the present invention.
- the chelating agent includes a phosphorus chelating agent and an aminocarboxylic acid chelating agent.
- Preferred phosphorus chelating agents are ethylenediamine-N, N′-di (methylenephosphinic acid), ethylenediaminetetra (methylenephosphinic acid), nitriloacetic acid-di (methylenephosphinic acid), nitriloacetic acid- (methylenephosphinic acid), nitriloacetic acid - ⁇ -propionic acid-methylenephosphonic acid, nitrilotris (methylenephosphonic acid), cyclohexanediaminetetra (methylenephosphonic acid), ethylenediamine-N, N'-diacetic acid-N, N'-di (methylenephosphonic acid), ethylenediamine -N, N'-di (methylenephosphonic acid), ethylenediaminetetra (methylenephosphonic acid) (EDTMP), polymethylenediaminetetra (methylenephosphonic acid), diethylenetriaminepenta (methylenephosphonic acid), 1-hydroxyethyl Nj
- the phosphorus chelating agent is selected from ethylenediaminetetra (methylenephosphonic acid) and salts thereof. More preferably, the phosphorus chelating agent is a sodium salt of ethylenediaminetetra (methylenephosphonic acid). Even more preferably, the phosphorus chelating agent is ethylenediaminetetra (methylenephosphonic acid) pentasodium.
- Preferred aminocarboxylic acid-based chelating agents are iminodiacetic acid, hydroxyethyliminodiacetic acid, nitrilotriacetic acid, nitrilotripropionic acid, ethylenediaminetetraacetic acid, hydroxyethylenediaminetriacetic acid, hexamethylenediaminetetraacetic acid, diethylenetriaminepentaacetic acid (DTPA), Triethylenetetramine hexaacetic acid, trans-1,2-diaminocyclohexanetetraacetic acid, bis (2-hydroxyethyl) glycine, diaminopropanoltetraacetic acid, ethylenediamine-2-propionic acid, glycol etherdiaminetetraacetic acid, bis (2-hydroxybenzyl) ) Selected from the group consisting of ethylenediaminediacetic acid, 3-hydroxy-2,2-iminodisuccinic acid, iminodisuccinic acid, methyl
- the aminocarboxylic acid chelating agent is a sodium salt of diethylenetriaminepentaacetic acid. Even more preferably, the aminocarboxylic acid chelating agent is diethylenetriaminepentaacetic acid trisodium.
- the chelating agent in the present invention is most preferably diethylenetriaminepentaacetic acid (DTPA) or a salt thereof, or ethylenediaminetetra (methylenephosphonic acid) (EDTMP) or a salt thereof.
- DTPA diethylenetriaminepentaacetic acid
- ETMP ethylenediaminetetra (methylenephosphonic acid)
- the method further includes a step of adding a hygroscopic fluidity improver after the drying step. This is because, for example, the fluidity and liquid permeability in a dry state can be significantly improved, and blocking during moisture absorption can be suppressed.
- the hygroscopic fluidity improver comprises at least one selected from the group consisting of silicon dioxide (silica), phosphate, or hydrotalcite.
- a water absorbent obtained by the above-described method for producing the water absorbent of the present invention described in the present specification is provided.
- an absorbent body including a water absorbent obtained by the method for producing a water absorbent of the present invention described in the present specification.
- a sanitary article including an absorbent body including a water absorbent obtained by the method for producing a water absorbent of the present invention described in the present specification is provided.
- the sanitary article can reduce the molecular weight of a soluble component that elutes when the water-absorbing agent absorbs liquid and swells. This is because stickiness that leads to discomfort can be reduced.
- the present invention provides an aqueous monomer solution mainly composed of acrylic acid (salt) in the presence of ⁇ -hydroxycarboxylic acid (salt).
- a method for producing a polyacrylic acid (salt) water-absorbing agent comprising a step of polymerizing, wherein at least one of the post-polymerization steps is performed in the presence of a chelating agent.
- the post-polymerization step is selected from the group consisting of a drying step, a surface treatment (crosslinking) agent mixing step, a surface crosslinking (surface treatment) step, a granulation step, and a curing step.
- the steps performed in the method for producing the water-absorbing agent of the present invention are performed in the order of a drying step, a treatment agent mixing step, a surface cross-linking (surface treatment) step, a granulation step, and a curing step.
- the polymerization step is performed in the presence of the chelating agent.
- the urinary resistance of the water-absorbing agent obtained thereby is further improved.
- the ratio of the chelating agent added in the polymerization step to the chelating agent added in the post-polymerization step is 1: 1 to 1:20, 1: 2 to 1:20, 1: 3 to 1:20, 1: 4 to 1:20, 1: 5 to 1:20, 1: 6 to 1:20, 1: 7 to 1:20, 1: 8 to 1:20, 1: 9 to 1:20, 1:10 to 1:20, 1:11 to 1:20, 1:12 to 1:20, 1:13 to 1:20, 1:14 to 1:20, 1:15 to 1: 20, 1:16 to 1:20, 1:17 to 1:20, 1:18 to 1:20, 1:19 to 1:20, 1: 1 to 1:19, 1: 1 to 1:18, 1: 1 to 1:17, 1: 1 to 1:16, 1: 1 to 1:15, 1: 1 to 1:14, 1: 1 to 1:13, 1: 1 to 1:12, 1: 1 to 1:11, 1: 1 to 1:10, : 1 to 1: 9, 1: 1 to 1: 8,
- the amount of the chelating agent added in the post-polymerization step is 0.005 to 0.2 parts by weight, 0.006 to 0.2 parts by weight with respect to 100 parts by weight of the water absorbing agent, 0.007 to 0.2 parts by weight, 0.008 to 0.2 parts by weight, 0.009 to 0.2 parts by weight, 0.01 to 0.2 parts by weight, 0.02 to 0.2 parts by weight, 0.03 to 0.2 parts by weight, 0.04 to 0.2 parts by weight, 0.05 to 0.2 parts by weight, 0.06 to 0.2 parts by weight, 0.07 to 0.2 parts by weight, 0.09 to 0.2 parts by weight, 0.005 to 0.09 parts by weight, 0.005 to 0.08 parts by weight, 0.005 to 0.07 parts by weight, 0.005 to 0.06 parts by weight, 0.005 to 0.05 parts by weight, 0.005 to 0.04 parts by weight, 0.005 to 0.03 parts by weight, 0.005 to 0.02 parts by weight, 0.005 to .01 parts by weight 0.005 to 0.005 to 0.00
- the content of ⁇ -hydroxycarboxylic acid (salt) added in the polymerization step is 0.02 to 1.5 parts by weight, 0 parts per 100 parts by weight of acrylic acid (salt). 0.03 to 1.5 parts by weight, 0.04 to 1.5 parts by weight, 0.05 to 1.5 parts by weight, 0.06 to 1.5 parts by weight, 0.07 to 1.5 parts by weight, 0 0.08 to 1.5 parts by weight, 0.09 to 1.5 parts by weight, 0.1 to 1.5 parts by weight, 0.2 to 1.5 parts by weight, 0.3 to 1.5 parts by weight, 0 .4 to 1.5 parts by weight, 0.5 to 1.5 parts by weight, 0.6 to 1.5 parts by weight, 0.7 to 1.5 parts by weight, 0.8 to 1.5 parts by weight, 0 1.9 to 1.5 parts by weight, 1.0 to 1.5 parts by weight, 1.1 to 1.5 parts by weight, 1.2 to 1.5 parts by weight, 1.3 to 1.5 parts by weight, 1 .4 to 1.5 parts by weight, 0.02 to 1.4 parts by weight, 0 02 to 1.3 parts by weight, 0.02 to 1.2 parts by weight, 0.02
- the step of classifying the surface-crosslinked (surface-treated) water-absorbing resin into different particle sizes, adding a chelating agent for each of the different particle sizes, and after adding the chelating agent A step of remixing a plurality of water-absorbing resins to obtain a water-absorbing agent is included.
- a water-absorbing agent having water-absorbing properties for example, water absorption capacity under pressure, centrifuge retention capacity, liquid permeability, moisture absorption blocking resistance, low coloration, etc.
- a surface-crosslinked polyacrylic acid (salt) water absorbing agent wherein ⁇ -hydroxycarboxylic acid (salt) is contained inside the water absorbing agent.
- a centrifuge retention capacity (CRC) of 30 g / g or more is provided.
- the centrifuge retention capacity (CRC) of the water absorbing agent is preferably 30 g / g or more, 31 g / g or more, 32 g / g or more, 33 g / g or more, 34 g / g or more, 35 g / g. 36 g / g or more, 37 g / g or more, 38 g / g or more, 39 g / g or more, 40 g / g or more, 41 g / g or more, 42 g / g or more, 43 g / g or more, 44 g / g or more, 45 g / g That's it.
- a particularly preferred predetermined centrifuge retention capacity is 30 g / g or more.
- the water-absorbing agent of the present invention includes a water-absorbing agent having the characteristics described in the method described in (6-1).
- the water-absorbing agent has reduced stickiness after liquid absorption.
- the reduction in stickiness of the water-absorbing agent can be defined by using the molecular weight reduction rate of the water-absorbing agent (that is, the soluble component molecular weight reduction rate (%)) as an index.
- the water-absorbing agent has a molecular weight reduction rate of preferably 8% or more, 9% or more, 10% or more, 11% or more, 12% or more, 14% or more, and most preferably 15% or more, water absorption It can be said that the stickiness of the agent has been reduced.
- the reduction in stickiness of the water-absorbing agent can be defined by using the molecular weight of the soluble component of the water-absorbing agent as an index.
- the soluble molecular weight is preferably 200,000 daltons or more and 700,000 daltons or less, and most preferably 200,000 daltons or more and 550,000 daltons or less, it can be said that the stickiness of the water-absorbing agent is reduced.
- a water-absorbing agent obtained by the method described in (6-1) is provided.
- a water absorbing agent obtained by the method described in (6-1) or an absorbent body including the water absorbing agent described in (6-3) is provided.
- a sanitary article including the absorber is provided.
- the sanitary article can reduce the molecular weight of soluble components that are eluted when the water-absorbing agent absorbs liquid and swells. This is because stickiness that leads to discomfort can be reduced.
- the present invention is a water-absorbing agent composed of a polyacrylic acid (salt) system, wherein 1 g of the water-absorbing agent is 200 ppm of L-ascorbic acid. And put into 25 ml of a deterioration solution composed of physiological saline (0.9% sodium chloride aqueous solution) containing 0.4 ppm of divalent iron ions, and left at 37 ° C. for 16 hours.
- a water-absorbing agent which is 5% by weight or less or 0% by weight, and further has a YI value of 26 or less after a color acceleration test.
- the problem caused by urine leakage and the like is that ascorbic acid is present in addition to the iron contained therein, and thus adverse effects such as synergistic deterioration and coloring of the water-absorbing agent are recognized. Such synergistic adverse effects could not be evaluated so far.
- Such an improvement is particularly effective by adding a combination of 0.001 to 1.0% by weight of a chelating agent and 0.02 to 1.5% by weight of ⁇ -hydroxycarboxylic acid (salt).
- the amount of the chelating agent present in the water-absorbing agent is preferably (1A) 0.001 to 1.0 part by weight, more preferably (1B) 0.001 to 0.5 part by weight per 100 parts by weight of the water-absorbing agent.
- Part, more preferably 0.001 to 0.25 part by weight of (1C), particularly preferably 0.001 to 0.15 part by weight of (1D), and ⁇ -hydroxycarboxylic acid (salt) present in the water-absorbing agent ) Is preferably (2A) 0.02 to 1.5 parts by weight, more preferably (2B) 0.05 to 1.0 parts by weight, and even more preferably (2C) with respect to 100 parts by weight of the water-absorbing agent. ) 0.1 to 1.0 parts by weight.
- the amount of the chelating agent present in the water absorbing agent is arbitrarily selected from the above (1A), (1B), (1C) and (1D), and the amount of the chelating agent present in the water absorbing agent is the above (2A), ( It is arbitrarily selected from 2B) and (2C).
- the combination of the amount of ⁇ -hydroxycarboxylic acid (salt) present in the water absorbing agent and the amount of chelating agent present in the water absorbing agent is an arbitrary combination of the above selected ones. It was found that using ⁇ -hydroxycarboxylic acid (salt) and a chelating agent in combination was superior to the case of adding only a simple substance, although it was not unexpected at all.
- SBS sodium bisulfite
- EDTMP ethylenediaminetetra (methylenephosphonic acid)
- the water-absorbing agent of the present invention shows a good value for the urinary resistance in the presence of iron with respect to the Fe / L-as deteriorated soluble component, which is an index newly developed in the present invention.
- the water-absorbing agent of the present invention since sodium hydrogen sulfate is not added, the problem of metal corrosion risk in the manufacturing process does not occur, and phosphoric acid is not added, so water absorption and the like deteriorate. Such a problem does not occur.
- the water-absorbing agent of the present invention contains 0.001 to 1.0% by weight, 0.002 to 1.0% by weight, 0.003 to 1.0% by weight, 0.004% of a chelating agent.
- a chelating agent -1.0 wt%, 0.005-1.0 wt%, 0.010-1.0 wt%, 0.015-1.0 wt%, 0.020-1.0 wt%, 0.025 -1.0 wt%, 0.030-1.0 wt%, 0.035-1.0 wt%, 0.040-1.0 wt%, 0.045-1.0 wt%, 0.050 -1.0 wt%, 0.055-1.0 wt%, 0.060-1.0 wt%, 0.065-1.0 wt%, 0.070-1.0 wt%, 0.075 -1.0 wt%, 0.080-1.0 wt%, 0.085-1.0 wt%, 0.090-1.0 wt%, 0.095-1.0 w
- the combination of ⁇ -hydroxycarboxylic acid (salt) and a chelating agent, which is a preferable concentration, is 0.02 to 0.005% by weight of the chelating agent added in the polymerization step, and in the post-polymerization step.
- the added chelating agent is 0.01 to 0.09% by weight and ⁇ -hydroxycarboxylic acid (salt) is 0.02 to 1% by weight.
- the effect can be further improved by further devising the addition at the time of production.
- the chelating agent added in the polymerization step is DTPA, and the chelating agent added in the post-polymerization step is also DTPA.
- the ratio of the chelating agent present in the polymerization step to the chelating agent present in the post-polymerization step is 1: 1 to 1:18.
- the amount of chelating agent added in the polymerization step is preferably 50 to 200 (mg / kg), and the amount of chelating agent added in the post-polymerization step is preferably 100 to 900 (mg / kg).
- the YI value after the color acceleration test is 26.5 or less, 26 or less, 25.5 or less, 25 or less, 24.5 or less, 24 or less, 23.5 or less, 23 or less, 22.5 or less. 22 or less, 21.5 or less, 21 or less, 20.5 or less, 20 or less, 19.5 or less, 19 or less, 18.5 or less, 18 or less, 17.5 or less, 17 or less, 16.5 or less, 16 or less, 15.5 or less, 15 or less, 14.5 or less, 14 or less, 13.5 or less, 13 or less, 12.5 or less, 12 or less, 11.5 or less, 11 or less, 10.5 or less, 10 or less 9.5 or less, 9 or less, 8.5 or less, 8 or less, 7.5 or less, 7 or less, 6.5 or less, 6 or less, 5.5 or less, 5 or less, 4.5 or less, 4 or less, 3 or less, .5 or less, 3 or less, 2.5 or less, 2 or less, 1.5 or less, 1 or less, 0.5 or less, 20 or
- the water-absorbing agent of the present invention preferably has an irregularly crushed shape because of its fixability to hydrophilic fibers such as pulp.
- the irregularly pulverized shape indicates a pulverized product, preferably a pulverized product in an aqueous solution polymerization method.
- the surface tension is 65 [mN / m] or more. More preferably, it is 67 [mN / m] or more, particularly preferably 70 [mN / m] or more, and most preferably 72 [mN / m] or more, and there is no substantial decrease in surface tension.
- the upper limit is usually 75 [mN / m].
- the neutralization rate is uniform. Without wishing to be bound by theory, this is because the water-absorbent resin of the present invention is not neutralized after polymerizing acrylic acid, but polymerizes a salt of acrylic acid (eg sodium acrylate). It is because it is obtained by.
- a salt of acrylic acid eg sodium acrylate
- the present invention provides a sanitary article including the absorber.
- the hygiene article is manufactured even after the hygroscopic agent absorbs urine, even after the hygiene article has been stored for a long period of time, with reduced discomfort felt by the user. This is because it is possible to use an appearance-like hygiene article similar to the time.
- compositions for reducing the soluble molecular weight of a water-absorbing agent comprising ⁇ -hydroxycarboxylic acid (salt).
- an agent in another aspect of the present invention, there is provided an agent, a polymerization regulator, a viscosity regulator or a performance additive for reducing the molecular weight of a water-absorbing agent, including an ⁇ -hydroxycarboxylic acid (salt).
- a polymerization regulator for reducing the molecular weight of a water-absorbing agent, including an ⁇ -hydroxycarboxylic acid (salt).
- composition etc. An active substance, a polymerization regulator, a viscosity regulator, and a performance additive, which have the same function as that of the composition of the present invention to reduce the soluble molecular weight of the water-absorbing agent and further have specific functions, are also included in the present invention. Can be included. Although not wishing to be bound by theory, it is possible to reduce the molecular weight of the water-absorbing agent by providing a composition containing ⁇ -hydroxycarboxylic acid (salt). This is because stickiness that leads to discomfort during use can be reduced.
- composition, the active substance, the polymerization regulator, the viscosity modifier, and the performance additive of the present invention are also collectively referred to as “composition etc.”.
- composition for reducing stickiness of a water-absorbing agent after liquid absorption containing ⁇ -hydroxycarboxylic acid (salt).
- composition for reducing the viscosity of a soluble part of a water-absorbing agent at the time of liquid absorption swelling including ⁇ -hydroxycarboxylic acid (salt) is provided.
- the water-absorbing resin described in (1-3) is used as the water-absorbing resin as a main component of the water-absorbing agent in the composition or the like.
- the water-absorbing agent in the composition or the like includes a water-absorbing agent having the characteristics described in the method described in (6-1).
- the ⁇ -hydroxycarboxylic acid (salt) in the composition or the like is the one described in (6-1).
- compositions and agent of the present invention in order to improve the strong urine resistance of a water-absorbing agent comprising an ⁇ -hydroxycarboxylic acid (salt) and a chelating agent A composition is provided.
- composition for improving the color resistance with time of a water-absorbing agent comprising an ⁇ -hydroxycarboxylic acid (salt) and a chelating agent.
- composition for improving the strong urinary resistance and anti-coloration with time of a water-absorbing agent comprising an ⁇ -hydroxycarboxylic acid (salt) and a chelating agent.
- Agents or performance additives that have the same function of improving urinary resistance and / or coloration resistance over time as those of the composition of the present invention, and further having specific functions can be included in the present invention.
- a composition, agent or performance additive comprising ⁇ -hydroxycarboxylic acid (salt) and a chelating agent, and / or the like; Coloring resistance with time can be improved, and as a result, discomfort after absorbing urine etc. during actual use of sanitary products can be reduced, and even after storing hygiene articles for a long time, the same as in manufacturing This is because it is possible to use a sanitary article that looks like this.
- the water-absorbing agent in the composition or the like includes the water-absorbing agent having the characteristics described in the method described in (6-2) or the water-absorbing agent (6-4).
- the ⁇ -hydroxycarboxylic acid (salt) and the chelating agent in the composition are respectively the ⁇ -hydroxycarboxylic acid (salt) and the method described in (6-2) or the water-absorbing agent in (6-4).
- ⁇ -hydroxycarboxylic acid salt for reducing stickiness of the water absorbing agent after liquid absorption.
- ⁇ -hydroxycarboxylic acid salt for reducing the viscosity of a water-soluble agent soluble component during liquid absorption swelling.
- the water-absorbing resin described in (1-3) is used as the water-absorbing resin as a main component of the water-absorbing agent in the composition or the like.
- the ⁇ -hydroxycarboxylic acid (salt) in the composition or the like is the one described in (6-1).
- an ⁇ -hydroxycarboxylic acid (salt) and a chelating agent for improving the strong urine resistance of a water absorbing agent is provided.
- ⁇ -hydroxycarboxylic acid salt
- a chelating agent for improving the color resistance of a water absorbing agent over time.
- ⁇ -hydroxycarboxylic acid salt
- a chelating agent for improving the strong urine resistance and coloration resistance with time of the water-absorbing agent.
- ⁇ -hydroxycarboxylic acid (salt) and a chelating agent provides superior coloration resistance over time and deterioration in urine resistance compared to the case where only a single substance is added. It is because it solves.
- the water-absorbing agent in the use includes the water-absorbing agent having the characteristics described in the method described in (6-2) or the water-absorbing agent (6-4).
- the ⁇ -hydroxycarboxylic acid (salt) and the chelating agent are respectively the ⁇ -hydroxycarboxylic acid (salt) and the chelating agent used in the method described in (6-2) or the water-absorbing agent in (6-4). Includes agents.
- a method for reducing the molecular weight of soluble component of a water-absorbing agent comprising absorbing water by polymerization Centrifuge retention capacity (CRC) by polymerization of monomer aqueous solution containing acrylic acid (salt) capable of forming agent, drying of hydrogel obtained by polymerization, and surface crosslinking of water absorbent resin powder obtained by drying
- CRC Polymerization Centrifuge retention capacity
- the method includes the step of producing a water-absorbing agent having an amount of 30 g / g or more, and the method includes the step of adding an ⁇ -hydroxycarboxylic acid (salt) before the drying.
- a method for reducing stickiness of a water-absorbing agent after liquid absorption which is a polymerization of an aqueous monomer solution containing acrylic acid (salt) capable of forming a water-absorbing agent by polymerization.
- the method comprises the steps of producing a water-absorbing agent having a centrifuge retention capacity (CRC) of 30 g / g or more by drying of the hydrogel obtained by the above and surface crosslinking of the water-absorbent resin powder obtained by drying,
- CRC centrifuge retention capacity
- a method for reducing the viscosity of a soluble component of a water-absorbing agent during swelling of the water-absorbing agent comprising a monomer aqueous solution containing acrylic acid (salt) capable of forming a water-absorbing agent by polymerization.
- the method provides a method comprising adding an ⁇ -hydroxy carboxylic acid (salt) prior to the drying.
- the water-absorbing agent in the method includes a water-absorbing agent having the characteristics described in the method described in (6-1).
- the ⁇ -hydroxycarboxylic acid (salt) in the composition or the like is the one described in (6-1).
- a method for improving the strong urine resistance and / or coloration resistance over time of the water-absorbing agent of the present invention includes polymerizing an aqueous monomer solution containing acrylic acid (salt) capable of forming a water-absorbing agent by polymerization in the presence of ⁇ -hydroxycarboxylic acid (salt), drying the hydrogel obtained by polymerization, And a step of producing a water-absorbing agent by surface crosslinking of the water-absorbing resin powder obtained by drying, and the method includes a step of adding a chelating agent after the polymerization, and the water-absorbing agent comprises 1 g of the water-absorbing agent.
- L-ascorbic acid was added to 25 ml of a deterioration solution composed of a physiological saline (200% sodium chloride aqueous solution) containing 200 ppm and divalent iron ions, and left at 37 ° C. for 16 hours. Add the whole liquid A method is provided wherein Fe / L-as deteriorated soluble component extracted after stirring for 10 minutes at 200 g is 35% by weight or less, and the YI value after coloration promotion test is 26 or less Is done.
- a method for improving the coloration resistance of a water-absorbing agent over time comprising the step of forming an ⁇ - of an aqueous monomer solution containing acrylic acid (salt) capable of forming a water-absorbing agent by polymerization.
- a step of adding a chelating agent after polymerization, and the water-absorbing agent is a physiological saline (0.9% sodium chloride aqueous solution) containing 1 g of the water-absorbing agent and 200 ppm of L-ascorbic acid and 0.4 ppm of divalent iron ions.
- a physiological saline (0.9% sodium chloride aqueous solution) containing 1 g of the water-absorbing agent and 200 ppm of L-ascorbic acid and 0.4 ppm of divalent iron ions.
- YI value after coloring acceleration test has the characteristic that it is 26 or less, a method is provided.
- a method for improving the strong urine resistance and coloration resistance over time of a water-absorbing agent comprising a single amount containing acrylic acid (salt) capable of forming a water-absorbing agent by polymerization Including a step of producing a water-absorbing agent by polymerizing an aqueous body solution in the presence of ⁇ -hydroxycarboxylic acid (salt), drying a hydrogel obtained by polymerization, and surface crosslinking of a water-absorbing resin powder obtained by drying, The method includes a step of adding a chelating agent after the polymerization, and the water-absorbing agent is a physiological saline solution (0.9 g) containing 1 g of the water-absorbing agent and 200 ppm of L-ascorbic acid and 0.4 ppm of divalent iron ions.
- Fe / L-as degradation that is extracted after being added to 25 ml of a deterioration solution composed of 25% sodium chloride aqueous solution and left at 37 ° C. for 16 hours, followed by addition of physiological saline to a total volume of 200 g and stirring for 10 minutes. Matter is 35 wt% or less, furthermore, YI value after coloring acceleration test has the characteristic that it is 26 or less, a method is provided.
- the water-absorbing agent in the method includes the water-absorbing agent having the characteristics described in the method described in (6-2) or the water-absorbing agent in (6-4).
- the ⁇ -hydroxycarboxylic acid (salt) and the chelating agent are respectively the ⁇ -hydroxycarboxylic acid (salt) and the chelating agent used in the method described in (6-2) or the water-absorbing agent in (6-4). Includes agents.
- liter may be written as “l” or “L”
- wt% is written as “wt%” for convenience. Further, in measurement of trace components, the value below the detection limit is expressed as “ND” (Non Detected).
- water-soluble polymer amount and soluble fraction Water-soluble polymer amount and soluble fraction
- water-soluble polymer amount is sometimes abbreviated as “water-soluble component amount” or “soluble component amount”.
- the water-soluble polymer is also referred to as “water-soluble component” or “soluble component”.
- the “soluble fraction” is a ratio (% by weight) of a soluble component in the water-absorbing agent.
- Soluble fraction should also be described as “Ext”, “water-soluble”, “soluble”, “water-soluble” or “soluble” with units of% by weight. There is also.
- the soluble fraction in the water-absorbing polymer is based on the average molecular weight of the monomer and the titration amount obtained by the above operation. It can be calculated by equation (4).
- the main component of the extracted soluble content is the extracted water-soluble polymer.
- the average molecular weight of the monomer is unknown, the average molecular weight of the monomer can be calculated using the neutralization rate obtained by titration. This neutralization rate is calculated by the following equation (5).
- Formula (5): Neutralization rate (mol%) (1 ⁇ ([NaOH] ⁇ [bNaOH]) / ([HCl] ⁇ [bHCl])) ⁇ 100
- the solid content of the water-absorbing polymer is calculated from the water content, and the amount of the predetermined water-containing gel-like crosslinked polymer is charged. Can be measured.
- the water-absorbing resin powder / water-absorbing agent in the aluminum cup was gently transferred onto a JIS standard sieve (The IIDA TESTING SIEVE / inner diameter 80 mm) having an opening of 2000 ⁇ m (8.6 mesh), and a low-tap type sieve shaker ( Using an ES-65 type sieve shaker / rotational speed of 230 rpm and impact number of 130 rpm, classification was performed for 5 seconds under conditions of a temperature of 20 to 25 ° C. and a relative humidity of 50% RH.
- JIS standard sieve The IIDA TESTING SIEVE / inner diameter 80 mm
- a low-tap type sieve shaker Using an ES-65 type sieve shaker / rotational speed of 230 rpm and impact number of 130 rpm, classification was performed for 5 seconds under conditions of a temperature of 20 to 25 ° C. and a relative humidity of 50% RH.
- the apparatus configuration is an apparatus equipped with size exclusion chromatography, a refractive index detector, a light scattering detector, and a capillary viscometer.
- the pure water used for this measurement should have sufficient impurities removed.
- the measurement is performed in a state where a sufficient amount of solvent is passed through the apparatus and the detected value baseline is stable. In particular, the measurement is performed in a state where there is no noise peak at the light scattering detector.
- the differential refractive index (dn / dc) of the polymer to be analyzed is The measurement was performed at 0.12 and the solvent refractive index was 1.33. The measurement result chart was checked, and when the light scattering intensity measurement peak contained a lot of noise, the measurement was performed again.
- Soluble fraction molecular weight reduction rate (%) The reduction rate of the weight average molecular weight of the water-soluble component when ⁇ -hydroxycarboxylic acid is added to the weight average molecular weight of the water-soluble component when ⁇ -hydroxycarboxylic acid is not added is defined as the soluble component molecular weight reduction rate. .
- the soluble component molecular weight reduction rate (%) was determined according to the following formula (7).
- a 120 mm x 400 mm SUS mesh (mesh size 850 ⁇ m) is placed on the paper diaper model absorber, and an acrylic plate having a cylinder with a diameter of 60 mm and a height of 8 cm is placed on the center, so that the entire model absorber is uniform.
- a 20 kg weight was placed on the acrylic plate so as to apply a load. From the cylindrical part, 0.9% physiological saline equivalent to the capacity of the water-absorbing agent centrifuge is charged, and after confirming that the entire amount of physiological saline has been taken into the model absorbent, It put into Unipack (size L, 480 mm x 340 mm, production Japan company), and hold
- the absorbent used as a reference for evaluation is a water-absorbing agent that does not contain ⁇ -hydroxycarboxylic acid, which is prepared by the same method so as to have an equivalent CRC with respect to the water-absorbing agent containing ⁇ -hydroxycarboxylic acid to be evaluated.
- the method for determining the ⁇ -hydroxycarboxylic acid content of the water absorbing agent is not particularly limited.
- a soluble component is extracted from the water absorbing agent, and the solution is rapidly extracted. It can quantify by analyzing using liquid chromatography. Specifically, it can be quantified by the following method.
- ⁇ -hydroxycarboxylic acid to be quantified is dissolved in a 0.9 wt% sodium chloride aqueous solution to prepare an ⁇ -hydroxycarboxylic acid solution having an arbitrary concentration.
- This solution is analyzed using the high performance liquid chromatography having the above-described configuration, and a calibration curve is prepared from the relationship between the concentration of ⁇ -hydroxycarboxylic acid and the peak area of the obtained chromatogram.
- the solution extracted from the water-absorbent resin is analyzed using high performance liquid chromatography, and the amount of ⁇ -hydroxycarboxylic acid is calculated from the relationship between the peak area of the obtained chromatogram and the calibration curve.
- the extraction efficiency from the water absorbent resin is 90%.
- the surface color (YI value (Yellow Index)) was measured with the above-mentioned spectroscopic color difference meter under the conditions of 5 g filling (filling about 60% of the equipped sample stage), room temperature (20-25 ° C.), and humidity 50 RH%. .
- the object color (L, a, b) or WB (hunter color) of other scales can be simultaneously measured by the same measuring method of the same apparatus. The larger L / WB and the smaller a / b, the lower the color and the closer to substantially white.
- a solution composed of physiological saline (0.9% sodium chloride aqueous solution) containing 200 ppm L-ascorbic acid and 0.4 ppm of divalent iron ions is prepared as a deterioration test solution.
- 2 g of iron (II) sulfate heptahydrate is dissolved in 1000 g of physiological saline to prepare an aqueous solution containing 400 ppm of iron ions.
- 1 g of an aqueous solution containing 400 ppm of iron ions and 0.2 g of L-ascorbic acid are dissolved in physiological saline to make a total amount of 1000 g.
- the Fe / L-as deteriorated soluble content is affected by the moisture content, in the present invention, it is evaluated by the value corrected for the moisture content.
- hydrogel crosslinked polymer (1) (hereinafter referred to as a hydrogel) was taken out 34 minutes after the start of the polymerization.
- the obtained hydrogel was subdivided to have a diameter of about 5 mm or less.
- This finely divided hydrogel cross-linked polymer is spread on a 50-mesh wire mesh, dried with hot air at 185 ° C. for 45 minutes, and the dried product is used using a roll mill (WML type roll crusher / Inoguchi Giken Co., Ltd.).
- a roll mill WML type roll crusher / Inoguchi Giken Co., Ltd.
- the mass average particle size (D50) is 349 ⁇ m
- the logarithmic standard deviation of particle size distribution ( ⁇ ) is 0.35.
- An irregularly crushed comparative water-absorbent resin (1) was obtained.
- the comparative water absorbent resin (1) had a centrifuge retention capacity (CRC) of 31.8 (g / g).
- Comparative water absorbent resin (1) Surface comprising 100 parts by weight of 1,4-butanediol 0.4 parts by weight, propylene glycol 0.6 parts by weight, deionized water (ion-exchanged water) 3.0 parts by weight The crosslinker solution was added and mixed uniformly. Thereafter, the mixture was heat-treated at 210 ° C. for 40 minutes for surface crosslinking.
- the mixture was pulverized until it passed through a JIS standard sieve having an opening of 850 ⁇ m to obtain comparative water absorbent resin particles (1).
- aqueous solution consisting of 0.03 part by weight of diethylenetriaminepentaacetic acid.3 sodium (DTPA.3Na) and 1.0 part by weight of deionized water (ion-exchanged water) was added to the comparative water absorbent resin particles (1) thus obtained. And uniformly granulated. Thereafter, the mixture was heat-treated at 60 ° C. for 45 minutes under no-air conditions, and then pulverized until it passed through a JIS standard sieve having an opening of 850 ⁇ m to obtain granulated comparative water absorbent resin particles (1).
- DTPA.3Na diethylenetriaminepentaacetic acid.3 sodium
- deionized water ion-exchanged water
- the above granulated comparative water-absorbent resin particles (1) (100 parts by weight) were mixed with 0.3 parts by weight of silica (product name: Leoroseal QS-20, manufactured by Tokuyama Corporation).
- silica product name: Leoroseal QS-20, manufactured by Tokuyama Corporation.
- 30 g of the comparative water-absorbing resin particles were put together with silica in a 225 mL mayonnaise bottle and mixed for 3 minutes by vibration of a paint shaker (manufactured by Toyo Seiki Co., Ltd.) to obtain a comparative water-absorbing agent (1).
- Table 1 shows CRC, soluble molecular weight and intrinsic viscosity of the obtained comparative water-absorbing agent (1).
- Comparative Example 2 In Comparative Example 1, the amount of polyethylene glycol diacrylate used was 0.093 mol% with respect to the carboxyl group-containing unsaturated monomer, and 50% apple was added simultaneously when diethylenetriaminepentaacetic acid / trisodium salt aqueous solution was added during polymerization. 10.75 g of acid aqueous solution (DL-malic acid, 50% by weight aqueous solution, manufactured by Fuso Chemical Industry Co., Ltd., food additive grade) (malic acid is 0.165 mol% based on carboxyl group-containing unsaturated monomer) Except for the addition, the same treatment as in Comparative Example 1 was performed to obtain a comparative water-absorbing agent (2). Table 1 shows CRC, soluble molecular weight, and intrinsic viscosity of the comparative water-absorbing agent (2). In addition, Table 2 shows the molecular weight reduction rate and tactile evaluation results of the comparative water-absorbing agents (1) and (2).
- the monomer aqueous solution (b ′) was cooled with stirring.
- the liquid temperature reached 39.5 ° C.
- the temperature of the monomer aqueous solution (b) rose to 76.9 ° C. due to the second stage heat of neutralization immediately after the production.
- the 48.5% by weight sodium hydroxide aqueous solution precipitates were observed, but gradually dissolved to become a transparent uniform solution.
- the polymerization reaction started 60 seconds after the monomer aqueous solution (b) was poured into the vat container.
- the polymerization reaction proceeded by expanding and foaming in all directions while generating water vapor, and then contracted to a size slightly larger than the bat-type container.
- a hydrogel crosslinked polymer hereinafter referred to as “hydrogel”.
- the water-containing gel (3) obtained by the polymerization reaction was subjected to gel pulverization using a meat chopper (HL-3225N, plate pore size: 10.0 mm / Remacom Co., Ltd.) to obtain a particulate water-containing gel (3).
- a meat chopper HL-3225N, plate pore size: 10.0 mm / Remacom Co., Ltd.
- the input amount of the hydrous gel (3) was 360 g / min, and the gel pulverization was performed while adding deionized water adjusted to 90 ° C. at 50 g / min in parallel with the addition of the hydrogel (3). .
- This finely divided hydrogel (3) is spread on a 50-mesh wire mesh, dried with hot air at 190 ° C. for 30 minutes, and the dried product is pulverized using a roll mill (WML type roll pulverizer / Inoguchi Giken Co., Ltd.). Further, by sieving with a JIS sieve having an aperture of 850 ⁇ m, 600 ⁇ m, 500 ⁇ m, 300 ⁇ m, and 150 ⁇ m, the mass average particle size (D50) is 349 ⁇ m, and the logarithmic standard deviation of particle size distribution ( ⁇ ) is 0.35. An amorphous crushed comparative water-absorbing resin (3) was obtained. The comparative water absorbent resin (3) had a centrifuge retention capacity (CRC) of 38.6 (g / g).
- CRC centrifuge retention capacity
- a surface cross-linking agent solution comprising 0.27 parts by weight of ethylene carbonate, 0.45 parts by weight of propylene glycol, and 1.8 parts by weight of deionized water (ion exchange water) with respect to 100 parts by weight of the comparative water absorbent resin (3). Added and mixed uniformly. Then, surface crosslinking was performed by heat-processing the said mixture for 40 minutes at 200 degreeC.
- the mixture was pulverized until it passed through a JIS standard sieve having an aperture of 850 ⁇ m to obtain comparative water absorbent resin particles (3).
- the granulated comparative water-absorbent resin particles (3) (100 parts by weight) were mixed with 0.3 parts by weight of silica (product name: Leoroseal QS-20, manufactured by Tokuyama Corporation).
- silica product name: Leoroseal QS-20, manufactured by Tokuyama Corporation.
- 30 g of comparative water-absorbing resin particles were put in a mayonnaise bottle with a capacity of 225 mL together with silica and mixed for 3 minutes by vibration of a paint shaker (manufactured by Toyo Seiki Co., Ltd.) to obtain a comparative water-absorbing agent (3).
- Table 1 shows the CRC, soluble molecular weight, and intrinsic viscosity of the comparative water-absorbing agent (3).
- Example 1 In Comparative Example 3, the amount of polyethylene glycol diacrylate used was 2.404 g (0.075 mol% with respect to the carboxyl group-containing unsaturated monomer), and a 50% malic acid aqueous solution was added to the monomer aqueous solution (b ′). (DL-malic acid, 50% by weight aqueous solution, Fuso Chemical Industry Co., Ltd., food additive grade) 10.8 g (malic acid is 0.658 mol% with respect to the carboxyl group-containing unsaturated monomer) was added. Except for the above, the same treatment as in Comparative Example 3 was performed to obtain a water absorbing agent (1). Table 1 shows CRC, soluble molecular weight, and intrinsic viscosity of the water-absorbing agent (1). Moreover, the moisture absorption blocking rate of the water absorbing agent (1) was 0%.
- Example 2 In Comparative Example 3, the amount of polyethylene glycol diacrylate used was 2.244 g (0.070 mol% with respect to the carboxyl group-containing unsaturated monomer), and a 50% sodium lactate aqueous solution was added to the monomer aqueous solution (b ′). The same treatment as in Comparative Example 3 was performed except that 5.4 g (sodium lactate, manufactured by Pulac) (sodium lactate was 0.394 mol% with respect to the carboxyl group-containing unsaturated monomer) was added, and the water absorbing agent (2) was obtained. Table 1 shows CRC, soluble molecular weight, and intrinsic viscosity of the water-absorbing agent (2).
- Example 3 In Example 1, the amount of polyethylene glycol diacrylate used was 2.18 g (0.068 mol% with respect to the carboxyl group-containing unsaturated monomer), and the amount of malic acid used was changed to the carboxyl group-containing unsaturated monomer.
- the water-absorbing agent (3) was obtained by carrying out the same treatment as in Example 1 except that the amount was 0.329 mol% and the silica was changed to hydrotalcite (product name DHT-6, manufactured by Kyowa Chemical Industry Co., Ltd.). It was. Table 1 shows the CRC, soluble molecular weight, and intrinsic viscosity of the water-absorbing agent (3). Table 3 shows the molecular weight reduction rate and the tactile sensation evaluation results of the comparative water-absorbing agent (3) and water-absorbing agents (1), (2) and (3).
- Comparative Example 4 In Comparative Example 3, the amount of polyethylene glycol diacrylate used was 1.122 g (0.035 mol% with respect to the carboxyl group-containing unsaturated monomer), and the surface cross-linking agent solution was diethylene glycol diglycidyl ether (trade name Denacol EX). -810 Nagase ChemteX Corporation) 0.024 parts by weight, ethylene carbonate 0.31 parts by weight, propylene glycol 0.52 parts by weight, deionized water (ion-exchanged water) 2.08 parts by weight, and the heat treatment temperature is A comparative water absorbing agent (4) was obtained by performing the same treatment as in Comparative Example 3 except that the temperature was 195 ° C. Table 1 shows the CRC, soluble molecular weight, and intrinsic viscosity of the comparative water-absorbing agent (4).
- Example 4 In Comparative Example 4, the amount of polyethylene glycol diacrylate used was 1.506 g (0.047 mol% with respect to the carboxyl group-containing unsaturated monomer), and the monomer aqueous solution (b ′) was 50% malic acid aqueous solution. (DL-malic acid, 50% by weight aqueous solution, Fuso Chemical Industry Co., Ltd., food additive grade) 5.4 g (malic acid is 0.329 mol% with respect to the carboxyl group-containing unsaturated monomer) was added. Except for the above, the same treatment as in Comparative Example 4 was performed to obtain a water absorbing agent (4). Table 1 shows CRC, soluble molecular weight and intrinsic viscosity of the water-absorbing agent (4).
- Example 5 In Comparative Example 4, the amount of polyethylene glycol diacrylate used was 1.442 g (0.045 mol% with respect to the carboxyl group-containing unsaturated monomer), and the aqueous monomer solution (b ′) was 50% aqueous sodium lactate solution. The same treatment as in Comparative Example 4 was carried out except that 4.32 g (sodium lactate, manufactured by Pulac) (sodium lactate was 0.315 mol% with respect to the carboxyl group-containing unsaturated monomer), and a water absorbing agent. (5) was obtained. Table 1 shows the CRC, soluble molecular weight and intrinsic viscosity of the water-absorbing agent (5).
- Example 6 In Comparative Example 4, the amount of polyethylene glycol diacrylate used was 1.442 g (0.045 mol% with respect to the carboxyl group-containing unsaturated monomer), and deionized water charged at the time of gel grinding was 4.32%.
- Sodium lactate aqueous solution (50% sodium lactate, manufactured by Pulac Co., Ltd. was used by adjusting the concentration with ion-exchanged water) (sodium lactate was 0.473 mol% with respect to the carboxyl group-containing unsaturated monomer) Except for the above, the same treatment as in Comparative Example 4 was performed to obtain a water absorbing agent (6).
- Table 1 shows the CRC, soluble molecular weight and intrinsic viscosity of the water-absorbing agent (6).
- Example 7 In Example 4, the amount of polyethylene glycol diacrylate used was 1.442 g (0.045 mol% with respect to the carboxyl group-containing unsaturated monomer), and the silica was tricalcium phosphate (Wako Pure Chemical Industries, Ltd.). Except for changing to CAS No. 7758-87-4), the same treatment as in Example 4 was performed to obtain a water absorbing agent (7). Table 1 shows the CRC, soluble molecular weight and intrinsic viscosity of the water-absorbing agent (7). Table 4 shows the molecular weight reduction rate and tactile evaluation results of the comparative water-absorbing agent (4) and water-absorbing agents (4) to (7).
- Comparative Example 5 In Comparative Example 3, the amount of polyethylene glycol diacrylate used was 0.801 g (0.025 mol% based on the carboxyl group-containing unsaturated monomer), and the surface cross-linking agent solution was diethylene glycol diglycidyl ether (trade name Denacol EX). -810 Nagase ChemteX Corp.) 0.017 parts by weight, ethylene carbonate 0.22 parts by weight, propylene glycol 0.36 parts by weight, deionized water (ion-exchanged water) 1.46 parts by weight, and the heat treatment temperature A comparative water absorbing agent (5) was obtained by carrying out the same treatment as in Comparative Example 3 except that the temperature was 190 ° C. Table 1 shows CRC, soluble molecular weight and intrinsic viscosity of the comparative water-absorbing agent (5).
- Example 8 In Comparative Example 5, the amount of polyethylene glycol diacrylate used was 1.026 g (0.032 mol% with respect to the carboxyl group-containing unsaturated monomer), and a 50% malic acid aqueous solution was added to the monomer aqueous solution (b ′). (DL-malic acid, 50% by weight aqueous solution, Fuso Chemical Industry Co., Ltd., food additive grade) 2.70 g (malic acid is 0.165 mol% with respect to the carboxyl group-containing unsaturated monomer) was added. Otherwise, the same treatment as in Comparative Example 5 was performed to obtain a water absorbing agent (8). Table 1 shows CRC, soluble molecular weight, and intrinsic viscosity of the water-absorbing agent (8).
- Example 9 In Comparative Example 5, the amount of polyethylene glycol diacrylate used was 1.058 g (0.033 mol% with respect to the carboxyl group-containing unsaturated monomer), and 3.60% of deionized water charged during gel pulverization was used.
- Malic acid aqueous solution (DL-malic acid, 50% by weight aqueous solution, manufactured by Fuso Chemical Industry Co., Ltd., food additive grade was used after adjusting its concentration with ion-exchanged water) (malic acid is carboxyl group-containing unsaturated) Except for 0.329 mol% with respect to the monomer, the same treatment as in Comparative Example 5 was performed to obtain a water absorbing agent (9).
- Table 1 shows the CRC, soluble molecular weight and intrinsic viscosity of the water-absorbing agent (9).
- Table 5 shows the molecular weight reduction rate and tactile sensation evaluation results of the comparative water-absorbing agent (5), water-absorbing agents (8) and (9).
- This finely divided hydrogel crosslinked polymer (6) is spread on a 50 mesh wire net and dried with hot air at 185 ° C. for 45 minutes, and the dried product is roll milled (WML type roll crusher / Inoguchi Giken Co., Ltd.). And sieving with a JIS sieve having an opening of 850 ⁇ m, 600 ⁇ m, 500 ⁇ m, 300 ⁇ m, and 150 ⁇ m, and then blended to prepare a mass average particle size (D50) of 349 ⁇ m, logarithmic standard deviation of particle size distribution ( ⁇ ) A comparatively pulverized comparative water absorbent resin (6) of 0.35 was obtained. The centrifuge retention capacity (CRC) of the comparative water absorbent resin (6) was 46.6 (g / g).
- Comparative water-absorbing resin (6) 100 parts by weight of diethylene glycol diglycidyl ether (trade name Denacol EX-810 manufactured by Nagase ChemteX Corporation) 0.024 parts by weight, ethylene carbonate 0.31 parts by weight, propylene glycol 0.52
- a surface cross-linking agent solution consisting of 2.08 parts by weight of deionized water (ion exchange water) was added and mixed uniformly. Thereafter, the mixture was subjected to surface crosslinking by heat treatment at 195 ° C. for 40 minutes.
- the mixture was pulverized until it passed through a JIS standard sieve having an aperture of 850 ⁇ m to obtain comparative water absorbent resin particles (6).
- a comparative water-absorbing agent (6) Into 100 parts by mass of the granulated comparative water-absorbent resin particles (6), 0.3 part by weight of silica (product name: Leoroseal QS-20, manufactured by Tokuyama Corporation) was mixed. For mixing, 30 g of comparative water-absorbing resin particles were put in a mayonnaise bottle with a capacity of 225 mL together with silica and mixed for 3 minutes by vibration of a paint shaker (manufactured by Toyo Seiki Co., Ltd.) to obtain a comparative water-absorbing agent (6).
- Table 1 shows CRC, soluble molecular weight, and intrinsic viscosity of the comparative water-absorbing agent (6).
- Example 10 In Comparative Example 6, the amount of polyethylene glycol diacrylate used was 5.19 g (0.041 mol% with respect to the carboxyl group-containing unsaturated monomer), and 50% malic acid aqueous solution (DL-apple) 2 minutes after the start of polymerization. Comparative Example 6 except that 10.59 g of acid, 50% by weight aqueous solution, Fuso Chemical Industry Co., Ltd., food additive grade) was added (malic acid was 0.165 mol% with respect to the carboxyl group-containing unsaturated monomer). The same treatment as that described above was performed to obtain a water absorbing agent (10). Table 1 shows CRC, soluble molecular weight and intrinsic viscosity of the water-absorbing agent (10).
- Example 11 In Comparative Example 6, the amount of polyethylene glycol diacrylate used was 5.07 g (0.04 mol% with respect to the carboxyl group-containing unsaturated monomer), and 20 minutes after the start of polymerization, the polymer-containing hydrogel (6) was taken out. The obtained hydrogel (6) was subjected to gel pulverization using a meat chopper (HL-3225N, plate pore size: 12.0 mm / Remacom Co., Ltd.) to obtain a particulate hydrous gel.
- a meat chopper HL-3225N, plate pore size: 12.0 mm / Remacom Co., Ltd.
- the amount of water-containing gel (6) charged at this time was 360 g / min, and in parallel with the water-containing gel (6), 3.6% malic acid aqueous solution (DL-malic acid, 50% by weight aqueous solution, Fuso Chemical Kogyo Co., Ltd., food additive grade was used after adjusting the concentration with ion-exchanged water), and gel pulverization was performed at 50 g / min to obtain a particulate hydrous gel. The same treatment was performed to obtain a water absorbing agent (11).
- Table 1 shows CRC, soluble molecular weight, and intrinsic viscosity of the water-absorbing agent (11).
- Table 6 shows the molecular weight reduction rate and the tactile sensation evaluation results of the comparative water-absorbing agent 6 and the water-absorbing agents 10 and 11.
- Comparative Example 7 In Comparative Example 6, the same treatment as in Comparative Example 6 was carried out except that the amount of polyethylene glycol diacrylate used was 2.53 g (0.020 mol% with respect to the carboxyl group-containing unsaturated monomer), and comparative water absorption Agent (7) was obtained. Table 1 shows CRC, soluble molecular weight and intrinsic viscosity of the comparative water-absorbing agent (7).
- Example 12 In Comparative Example 7, the amount of polyethylene glycol diacrylate used was 3.17 g (0.025 mol% with respect to the carboxyl group-containing unsaturated monomer), and 50% sodium lactate aqueous solution (sodium lactate, (Pureac Co., Ltd.) 6.46 g (sodium lactate is 0.119 mol% with respect to the carboxyl group-containing unsaturated monomer) was added, and the same treatment as in Comparative Example 7 was carried out to obtain a water absorbing agent (12). It was. Table 1 shows CRC, soluble molecular weight and intrinsic viscosity of the water-absorbing agent (12). Table 7 shows the molecular weight reduction rate and tactile evaluation results of the comparative water-absorbing agent 7 and the water-absorbing agent (12).
- a reaction vessel comprising a thermometer, a nitrogen gas introduction pipe, a lid provided with an exhaust hole, a bottom surface of 300 mm ⁇ 220 mm, and a depth of 60 mm, and a black light fluorescent lamp on the top.
- D ′ was fed and mixed and immersed in a 40 ° C. water bath to a height of 10 mm from the bottom. Nitrogen gas was introduced into this aqueous solution and deaerated for 20 minutes.
- the water-containing gel (8) obtained by the polymerization reaction was subjected to gel pulverization using a meat chopper (HL-3225N, plate pore diameter: 10.0 mm / Remacom Co., Ltd.) to obtain a particulate water-containing gel (8).
- a meat chopper HL-3225N, plate pore diameter: 10.0 mm / Remacom Co., Ltd.
- the amount of the hydrogel (8) charged was 360 g / min, and gel pulverization was performed while adding deionized water adjusted to 90 ° C. at 50 g / min in parallel with the hydrogel (8). .
- the mixture was pulverized until it passed through a JIS standard sieve having an aperture of 850 ⁇ m to obtain comparative water absorbent resin particles (8).
- comparative water-absorbent resin particles (8) Into 100 parts by mass of the granulated comparative water-absorbent resin particles (8), 0.3 part by weight of silica (product name: Leolosil QS-20, manufactured by Tokuyama Corporation) was mixed. For mixing, 30 g of comparative water-absorbent resin particles were placed in a mayonnaise bottle having a capacity of 225 mL together with silica and mixed for 3 minutes by vibration of a paint shaker (manufactured by Toyo Seiki Co., Ltd.) to obtain a comparative water-absorbing agent (8).
- Table 1 shows CRC, soluble molecular weight and intrinsic viscosity of the comparative water-absorbing agent (8).
- Example 13 In Comparative Example 8, the amount of polyethylene glycol diacrylate used was 3.29 g (0.065 mol% with respect to the carboxyl group-containing unsaturated monomer), and the monomer aqueous solution (d ′) was 50% malic acid aqueous solution. (DL-malic acid, 50% by weight aqueous solution, manufactured by Fuso Chemical Industry Co., Ltd., food additive grade) 8.58 g (malic acid is 0.329 mol% based on the carboxyl group-containing unsaturated monomer) was added. Except for the above, the same treatment as in Comparative Example 8 was performed to obtain a water absorbing agent (13). Table 1 shows CRC, soluble molecular weight, and intrinsic viscosity of the water-absorbing agent (13).
- Example 14 In Comparative Example 8, the amount of polyethylene glycol diacrylate used was 3.29 g (0.065 mol% with respect to the carboxyl group-containing unsaturated monomer), and 7.20% deionized water was added during gel pulverization.
- Malic acid aqueous solution (DL-malic acid, 50% by weight aqueous solution, manufactured by Fuso Chemical Industry Co., Ltd., food additive grade was used after adjusting its concentration with ion-exchanged water) (malic acid is carboxyl group-containing unsaturated) Except for 0.658 mol% with respect to the monomer, the same treatment as in Comparative Example 8 was performed to obtain a water absorbing agent (14).
- Table 1 shows CRC, soluble molecular weight and intrinsic viscosity of the water-absorbing agent (14).
- Table 8 shows the molecular weight reduction rate and tactile evaluation results of the comparative water-absorbing agent (8), water-absorbing agents (13) and (14).
- Comparative Example 9 In Comparative Example 8, the amount of polyethylene glycol diacrylate used was 4.05 g (0.08 mol% based on the carboxyl group-containing unsaturated monomer), and diphenyl (2,4,6-trimethylbenzoyl) phosphine oxide was used.
- a comparative water-absorbing agent (9) was obtained in the same manner as in Comparative Example 8 except that 0.1 g was changed to 0.1 g of Irgacure 819. Table 1 shows CRC, soluble molecular weight, and intrinsic viscosity of the comparative water-absorbing agent (9).
- Example 15 In Comparative Example 9, the amount of polyethylene glycol diacrylate used was 5.07 g (0.1 mol% with respect to the carboxyl group-containing unsaturated monomer), and the aqueous monomer solution (d ′) was 50% aqueous sodium lactate solution. (Sodium lactate, manufactured by Pulac) 10.3 g (sodium lactate is 0.473 mol% with respect to the carboxyl group-containing unsaturated monomer) (15) was obtained.
- Table 1 shows CRC, soluble molecular weight and intrinsic viscosity of the water-absorbing agent (15).
- Table 9 shows the molecular weight reduction rate and tactile evaluation results of the comparative water-absorbing agent (9) and water-absorbing agent (15).
- Comparative Example 10 Comparative Example 4, the amount of polyethylene glycol diacrylate used was 0.930 g (0.029 mol% with respect to the carboxyl group-containing unsaturated monomer), the heat treatment temperature was 180 ° C., and diethylenetriamine was used during granulation.
- a comparative water absorbing agent (10) was obtained by performing the same treatment as in Comparative Example 4 except that 5 acetic acid and 3 sodium were not added to the water absorbent resin particles. Table 1 shows CRC, soluble molecular weight, and intrinsic viscosity of the comparative water-absorbing agent (10).
- Example 16 In Comparative Example 10, the amount of polyethylene glycol diacrylate used was 1.026 g (0.032 mol% relative to the carboxyl group-containing unsaturated monomer), and the monomer aqueous solution (b ′) was 50% malic acid aqueous solution. (DL-malic acid, 50% by weight aqueous solution, Fuso Chemical Industry Co., Ltd., food additive grade) 1.08 g (malic acid is 0.066 mol% based on the carboxyl group-containing unsaturated monomer) was added. Except for the above, the same treatment as in Comparative Example 10 was performed to obtain a water absorbing agent (16). Table 1 shows CRC, soluble molecular weight and intrinsic viscosity of the water-absorbing agent (16).
- Table 10 shows the molecular weight reduction rate and tactile sensation evaluation results of the comparative water-absorbing agent (10) and the water-absorbing agent (16). Further, Table 14 shows the YI value, Fe / L-as deteriorated soluble matter, CRC, and AAP of the water absorbing agent (16).
- Example 17 In Comparative Example 4, 0.05 g of malic acid aqueous solution (DL-malic acid, 50 wt% aqueous solution, manufactured by Fuso Chemical Industry Co., Ltd., food additive grade) in monomer aqueous solution (b ′) (malic acid is carboxyl 0.003 mol%) was added to the group-containing unsaturated monomer, and the heat treatment temperature was 180 ° C., and the same treatment as in Comparative Example 4 was performed to obtain a water absorbing agent (17).
- Table 1 shows the CRC, soluble molecular weight and intrinsic viscosity of the water-absorbing agent (17).
- Table 14 shows the YI value, Fe / L-as deteriorated soluble matter, CRC, and AAP of the water-absorbing agent (17).
- Example 18 In Comparative Example 4, the amount of polyethylene glycol diacrylate used was 2.564 g (0.080 mol% with respect to the carboxyl group-containing unsaturated monomer), and diethylenetriaminepentaacetic acid with respect to the monomer aqueous solution (b ′). ⁇ 10.8 g of 50% malic acid aqueous solution (DL-malic acid, 50% by weight aqueous solution, manufactured by Fuso Chemical Industry Co., Ltd., food additive grade) without adding trisodium (DTPA ⁇ 3Na) aqueous solution (malic acid is carboxyl group) 0.658 mol% with respect to the contained unsaturated monomer), the heat treatment temperature is set to 180 ° C., and diethylenetriamine pentaacetic acid / trisodium is not added to the water-absorbent resin particles.
- DL-malic acid 50% by weight aqueous solution, manufactured by Fuso Chemical Industry Co., Ltd., food additive grade
- DTPA ⁇ 3Na
- Table 1 shows the CRC, soluble molecular weight and intrinsic viscosity of the water-absorbing agent (18).
- the malic acid content of the water absorbing agent (18) was 0.98% by weight with respect to the water absorbing agent.
- Table 14 shows the YI value, Fe / L-as deteriorated soluble matter, CRC, and AAP of the water absorbing agent (18).
- the obtained water-like gel (11) was obtained by continuously cutting the obtained band-shaped water-containing gel (11) in the width direction with respect to the traveling direction of the polymerization belt so that the cut length was 300 mm. .
- the hydrogel (11) had a CRC of 36.0 [g / g] and a resin solid content of 48.1% by weight.
- the water-containing gel (11) obtained above was supplied to a screw extruder and gel crushed.
- a meat chopper having a diameter of 100 mm, a hole diameter of 8 mm, a hole number of 54, a hole plate ratio of 36.1% and a thickness of 10 mm and a screw shaft having an outer diameter of 86 mm is provided. It was used.
- the hydrogel (11) was supplied at 4640 [g / min], and at the same time, water vapor was supplied at 83 [g / min].
- the temperature of the hydrogel (11) before gel pulverization was 80 ° C.
- the temperature of the pulverized gel after gel pulverization that is, the particulate hydrogel (11) was increased to 84 ° C.
- the particulate hydrogel (11) obtained in the gel pulverization step had a resin solid content of 47.5% by weight, a weight average particle size (D50) of 820 ⁇ m, and a logarithmic standard deviation ( ⁇ ) of 0.94 in the particle size distribution. .
- the particulate water-containing gel (11) is sprayed on the ventilation plate within 1 minute after the completion of the gel grinding (the temperature of the particulate water-containing gel (11) at this time is 80 ° C.) and dried at 185 ° C. for 30 minutes. And a dry polymer (11) was obtained.
- the average wind speed of the hot air was 1.0 [m / s] in the direction perpendicular to the traveling direction of the ventilation belt.
- the wind speed of the hot air was measured with a constant temperature thermal anemometer Anemomaster 6162 manufactured by Nippon Kanomax Co., Ltd.
- the entire amount of the dried polymer (11) obtained in the drying step is supplied to a three-stage roll mill and pulverized (pulverization step), and then further classified by JIS standard sieves having openings of 710 ⁇ m and 175 ⁇ m.
- a regular crushed comparative water absorbent resin (11) was obtained.
- the comparative water-absorbent resin (11) has a weight average particle diameter (D50) of 355 ⁇ m, a logarithmic standard deviation ( ⁇ ) of 0.32 in particle size distribution, CRC 48.3 [g / g], 150 ⁇ m passing particles (with an opening of 150 ⁇ m). The ratio of particles passing through the sieve) was 0.4% by weight.
- 0.025 part by weight of ethylene glycol diglycidyl ether, 0.4 part by weight of 1,4-butanediol, 0.6 part by weight of propylene glycol and 100 parts by weight of the comparative water absorbent resin (11) are removed.
- a (crosslinking) surface crosslinking agent solution consisting of 3.0 parts by weight of ionic water is uniformly mixed, and the CRC of the resulting comparative water-absorbent resin particles (11) is 38 to 39 g / g for about 30 minutes at 190 ° C. It heat-processed so that it might become. After cooling, the paint shaker test was performed, and damage equivalent to the manufacturing process was given.
- Example 19 In Comparative Example 11, the amount of polyethylene glycol diacrylate used was 0.78 parts by weight, and liquid malic acid (DL-malic acid, 50% by weight aqueous solution, manufactured by Fuso Chemical Industries, Ltd.) was used as the monomer aqueous solution (e ′). Food additive grade) Except for adding 2.2 parts by weight, the same treatment as in Comparative Example 11 was performed to obtain a water-absorbing agent (19). Table 1 shows CRC, soluble molecular weight and intrinsic viscosity of the water-absorbing agent (19). Moreover, the malic acid content of the water absorbing agent (19) was 0.29% by weight with respect to the water absorbing agent.
- DL-malic acid 50% by weight aqueous solution, manufactured by Fuso Chemical Industries, Ltd.
- the hydrogel (12) obtained above was fed to a screw extruder and gel crushed.
- a meat chopper having a diameter of 100 mm, a hole diameter of 6.4 mm, a number of holes of 83, and a perforated plate having a thickness of 10 mm and an outer diameter of the screw shaft of 86 mm was used.
- the hydrogel (12) was supplied at 4640 [g / min], and at the same time, water vapor was supplied at 83 [g / min].
- the temperature of the hydrogel (12) before gel pulverization was 80 ° C.
- the temperature of the crushed gel after gel pulverization that is, the particulate hydrogel (12) was increased to 84 ° C.
- the particulate hydrogel (12) obtained in the gel pulverization step had a resin solid content of 47.5% by weight, a weight average particle size (D50) of 820 ⁇ m, and a logarithmic standard deviation ( ⁇ ) of 0.94 in the particle size distribution. .
- the particulate water-containing gel (12) is sprayed on the ventilation plate within 1 minute after completion of the gel grinding (the temperature of the particulate water-containing gel (12) at this time is 80 ° C.), and dried at 185 ° C. for 30 minutes. And a dry polymer (12) was obtained.
- the average wind speed of the hot air was 1.0 [m / s] in the direction perpendicular to the traveling direction of the ventilation belt.
- the wind speed of the hot air was measured with a constant temperature thermal anemometer Anemomaster 6162 manufactured by Nippon Kanomax Co., Ltd.
- the entire amount of the dried polymer (12) obtained in the drying step is supplied to a three-stage roll mill and pulverized (pulverization step), and then further classified by JIS standard sieves having openings of 710 ⁇ m and 175 ⁇ m.
- a regular crushed comparative water absorbent resin (12) was obtained.
- the comparative water absorbent resin (12) has a weight average particle size (D50) of 366 ⁇ m, a logarithmic standard deviation ( ⁇ ) of 0.32 in particle size distribution, CRC 49.4 [g / g], 150 ⁇ m passing particles (mesh size 150 ⁇ m). The ratio of particles passing through the sieve) was 0.4% by weight.
- Example 20 In Comparative Example 12, the same treatment as Comparative Example 12 except that 1.8 parts by weight of DL-malic acid (powder, manufactured by Fuso Chemical Industry Co., Ltd., food additive grade) was added to the monomer aqueous solution (e ′). To obtain a water-absorbing agent (20). Table 1 shows CRC, soluble molecular weight, and intrinsic viscosity of the water-absorbing agent (20). Moreover, the malic acid content of the water absorbing agent (20) was 0.49% by weight with respect to the water absorbing agent.
- DL-malic acid powder, manufactured by Fuso Chemical Industry Co., Ltd., food additive grade
- Comparative Example 13 Comparative Example 3 except that the amount of polyethylene glycol diacrylate used in Comparative Example 3 was 2.56 g (0.08 mol% with respect to the carboxyl group-containing unsaturated monomer) and the amount of deionized water was 1511 g.
- a comparative water-absorbing agent (13) was obtained by performing the same treatment as. Table 1 shows CRC, soluble molecular weight, and intrinsic viscosity of the water-absorbing agent (13).
- Example 21 In Comparative Example 3, the amount of polyethylene glycol diacrylate used was 3.85 g (0.12 mol% with respect to the carboxyl group-containing unsaturated monomer), and the amount of deionized water was 1510 g. b ') 10.81 g of liquid malic acid (DL-malic acid, 50% by weight aqueous solution, Fuso Chemical Industry Co., Ltd., food additive grade) (malic acid is 0.1% relative to the carboxyl group-containing unsaturated monomer. Except for adding 658 mol%), the same treatment as in Comparative Example 3 was performed to obtain a water-absorbing agent (21). Table 1 shows CRC, soluble molecular weight, and intrinsic viscosity of the water-absorbing agent (21). Table 13 shows the molecular weight reduction rate and the tactile sensation evaluation results of the comparative water-absorbing agent (13) and water-absorbing agent (21).
- the monomer aqueous solution (f ′) was cooled with stirring.
- 166.80 g of a 48.5 wt% sodium hydroxide aqueous solution adjusted to 40 ° C. was added and mixed to prepare a monomer aqueous solution (f).
- the temperature of the aqueous monomer solution (f) rose to 76.9 ° C. due to the second stage neutralization heat immediately after the production.
- the 48.5% by weight sodium hydroxide aqueous solution precipitates were observed, but gradually dissolved to become a transparent uniform solution.
- the bat-type container is a hot plate (NEO HOTPLATE HI-1000 / Inoue Seieido Co., Ltd.). Was used until the surface temperature reached 40 ° C.
- the polymerization reaction started 60 seconds after the monomer aqueous solution (f) was poured into the vat container.
- the polymerization reaction proceeded by expanding and foaming in all directions while generating water vapor, and then contracted to a size slightly larger than the bat-type container.
- a hydrogel crosslinked polymer hereinafter referred to as “hydrogel”) (14) was taken out.
- the water-containing gel (14) obtained by the polymerization reaction was subjected to gel pulverization using a meat chopper (HL-3225N, plate pore size: 10.0 mm / Remacom Co., Ltd.) to obtain a particulate water-containing gel (14).
- a meat chopper HL-3225N, plate pore size: 10.0 mm / Remacom Co., Ltd.
- the amount of the hydrogel (14) charged was 230 g / min, and gel pulverization was performed while adding deionized water adjusted to 90 ° C. at 50 g / min in parallel with the hydrogel (14). .
- the particulate hydrogel (14) obtained by the above operation was spread on a stainless steel wire mesh with an opening of 850 ⁇ m and dried by aeration of hot air at 180 ° C. for 30 minutes. Subsequently, the dried polymer (14) obtained by the drying operation was pulverized using a roll mill (WML type roll pulverizer / Inoguchi Giken Co., Ltd.), and then using JIS standard sieves having openings of 710 ⁇ m and 45 ⁇ m. Classified.
- Diethylene glycol diglycidyl ether (trade name Denacol EX-810 manufactured by Nagase ChemteX Corporation) 0.024 parts by weight, ethylene carbonate 0.308 parts by weight, propylene glycol 0.515 to 100 parts by weight of the water-absorbent resin powder (14)
- a surface cross-linking agent solution (14) consisting of 2.08 parts by weight of deionized water (ion exchange water) was added and mixed uniformly. Thereafter, the mixture was subjected to a heat treatment at 180 ° C. for 40 minutes to perform surface crosslinking.
- the obtained water absorbent resin particles (14) were crushed until they passed through a JIS standard sieve having an opening of 850 ⁇ m.
- aqueous solution composed of 0.01 parts by weight of DTPA ⁇ 3Na and 1.0 parts by weight of deionized water (ion-exchanged water) was added to the obtained water-absorbing agent, and the mixture was uniformly mixed and granulated. Then, after heat-treating at 60 ° C. for 45 minutes under windless conditions, the mixture was pulverized until it passed through a JIS standard sieve having an opening of 850 ⁇ m to obtain granulated water-absorbent resin particles (14).
- silica product name: Leolosil QS-20, manufactured by Tokuyama Corporation
- 30 g of a water-absorbing resin was placed in a mayonnaise bottle having a capacity of 225 mL together with silica and mixed for 3 minutes by vibration of a paint shaker (manufactured by Toyo Seiki Co., Ltd.) to obtain a water-absorbing agent (14).
- Comparative Example 14 A comparative water-absorbing agent (14) was obtained by the method of Production Example 1. Table 14 shows the performance of the comparative water-absorbing agent (14).
- Comparative Example 15 The water absorbent resin particles (15) obtained by the method of Production Example 2 are used as a comparative water absorbent (15).
- Table 14 shows the performance of the comparative water-absorbing agent (15).
- Example 22 In Production Example 1, 30% by weight D, L-malic acid aqueous solution (DL-malic acid, 50% by weight aqueous solution, manufactured by Fuso Chemical Industries, Ltd., a food additive grade) was ion-exchanged into the monomer aqueous solution (f ′). 0.33 g) was used, and the amount of diethylenetriaminepentaacetic acid ⁇ 3 sodium (DTPA ⁇ 3Na) added during granulation was changed from 0.01 to 0.03 parts by weight. The same operation as in Production Example 1 was performed except that mixing was not performed. The water-absorbing resin particles obtained are designated as a water-absorbing agent (22). Table 14 shows the performance of the water-absorbing agent (22).
- Example 23 In Production Example 1, the addition amount of diethylenetriaminepentaacetic acid.3 sodium (DTPA.3Na) in the monomer aqueous solution (f ′) was changed from 2.48 to 9.90 g, and the monomer aqueous solution (f ′) was changed to 30.
- DTPA.3Na diethylenetriaminepentaacetic acid.3 sodium
- Example 24 In Production Example 1, the addition amount of polyethylene glycol diacrylate was changed from 0.03 mol% to 0.032 mol%, and addition of diethylenetriaminepentaacetic acid ⁇ 3sodium (DTPA ⁇ 3Na) in the monomer aqueous solution (f ′) The amount was changed from 2.48 to 9.90 g, and 30% by weight D, L-malic acid aqueous solution (DL-malic acid, 50% by weight aqueous solution, manufactured by Fuso Chemical Industry Co., Ltd.) was added to the monomer aqueous solution (f ′).
- L-malic acid aqueous solution DL-malic acid, 50% by weight aqueous solution, manufactured by Fuso Chemical Industry Co., Ltd.
- Example 25 In Production Example 1, the addition amount of polyethylene glycol diacrylate was changed from 0.03 mol% to 0.032 mol%, and a 30 wt% D, L-malic acid aqueous solution (DL) was added to the monomer aqueous solution (f ′).
- DL L-malic acid aqueous solution
- Example 26 In Production Example 2, the amount of polyethylene glycol diacrylate added was changed from 0.03 mol% to 0.032 mol%, and 30 wt% D, L-malic acid aqueous solution (DL) was added to the monomer aqueous solution (f ′). -1.65 g of malic acid, 50% by weight aqueous solution, manufactured by Fuso Chemical Industry Co., Ltd., adjusted to the concentration of food additive grade with ion-exchanged water) and added diethylenetriaminepentaacetic acid / trisodium (3 sodium) during granulation ( The same operation as in Production Example 2 was performed except that the amount of DTPA ⁇ 3Na) was changed from 0.01 to 0.03 parts by weight.
- the water-absorbing resin particles obtained are designated as a water-absorbing agent (26). Table 14 shows the performance of the water absorbing agent (26).
- Example 27 In Production Example 1, the addition amount of polyethylene glycol diacrylate was changed from 0.03 mol% to 0.045 mol%, and 30 wt% D, L-malic acid aqueous solution (DL) was added to the monomer aqueous solution (f ′). -Malic acid, 50 wt% aqueous solution, manufactured by Fuso Chemical Industry Co., Ltd., food additive grade was used after adjusting the concentration with ion-exchanged water). went. The water-absorbing resin particles obtained are designated as a water-absorbing agent (27). Table 14 shows the performance of the water absorbing agent (27).
- Example 28 In Production Example 1, the amount of polyethylene glycol diacrylate added was changed from 0.03 mol% to 0.035 mol%, and 30 wt% D, L-malic acid aqueous solution (DL) was added to the monomer aqueous solution (f ′).
- DL L-malic acid aqueous solution
- -Malic acid 50% by weight aqueous solution, Fuso Chemical Industry Co., Ltd., used a food additive grade with a concentration adjusted with ion-exchanged water (3.3 g) was added, and diethylenetriaminepentaacetic acid / trisodium (3 sodium) during granulation ( DTPA ⁇ 3Na) was changed from 0.01 to 0.03 parts by weight, and the same operation as in Production Example 1 was performed except that silica was not mixed.
- the water-absorbing resin particles obtained are designated as a water-absorbing agent (28). Table 14 shows the performance of the water-absorbing agent (28).
- Example 29 In Production Example 2, the addition amount of polyethylene glycol diacrylate was changed from 0.03 mol% to 0.035 mol%, and addition of diethylenetriaminepentaacetic acid ⁇ 3 sodium (DTPA ⁇ 3Na) in the monomer aqueous solution (f ′) The amount was changed from 2.48 to 9.90 g, and 30% by weight D, L-malic acid aqueous solution (DL-malic acid, 50% by weight aqueous solution, manufactured by Fuso Chemical Industry Co., Ltd.) was added to the monomer aqueous solution (f ′).
- L-malic acid aqueous solution DL-malic acid, 50% by weight aqueous solution, manufactured by Fuso Chemical Industry Co., Ltd.
- the food additive grade was used after adjusting its concentration with ion-exchanged water), and the amount of diethylenetriaminepentaacetic acid ⁇ 3 sodium (DTPA ⁇ 3Na) added during granulation was 0.01 to 0.03.
- the same operation as in Production Example 2 was performed except that tricalcium phosphate was mixed and changed to parts by weight.
- the water-absorbing resin particles obtained are designated as a water-absorbing agent (29).
- Table 14 shows the performance of the water absorbing agent (29).
- Example 30 In Production Example 2, the addition amount of polyethylene glycol diacrylate was changed from 0.03 mol% to 0.045 mol%, and 30 wt% D, L-malic acid aqueous solution (DL) was added to the monomer aqueous solution (f ′).
- DL L-malic acid aqueous solution
- Example 31 In Example 28, the same operation as in Example 28 was performed except that silica mixing was performed.
- the water-absorbing resin particles obtained are designated as a water-absorbing agent (31).
- Table 14 shows the performance of the water-absorbing agent (31).
- Example 32 In Production Example 1, the addition amount of polyethylene glycol diacrylate was changed from 0.03 mol% to 0.045 mol%, and the addition of diethylenetriaminepentaacetic acid ⁇ 3 sodium (DTPA ⁇ 3Na) in the monomer aqueous solution (f ′) The amount was changed from 2.48 to 9.90 g, and 30% by weight D, L-malic acid aqueous solution (DL-malic acid, 50% by weight aqueous solution, manufactured by Fuso Chemical Industry Co., Ltd.) was added to the monomer aqueous solution (f ′).
- L-malic acid aqueous solution DL-malic acid, 50% by weight aqueous solution, manufactured by Fuso Chemical Industry Co., Ltd.
- Example 33 In Production Example 1, the addition amount of polyethylene glycol diacrylate was changed from 0.03 mol% to 0.045 mol%, and 30 wt% D, L-malic acid aqueous solution (DL) was added to the monomer aqueous solution (f ′).
- DL L-malic acid aqueous solution
- Example 34 In Production Example 1, the addition amount of polyethylene glycol diacrylate was changed from 0.03 mol% to 0.055 mol%, and addition of diethylenetriaminepentaacetic acid ⁇ 3 sodium (DTPA ⁇ 3Na) in the monomer aqueous solution (f ′) The amount was changed from 2.48 to 9.90 g, and 30% by weight D, L-malic acid aqueous solution (DL-malic acid, 50% by weight aqueous solution, manufactured by Fuso Chemical Industry Co., Ltd.) was added to the monomer aqueous solution (f ′).
- L-malic acid aqueous solution DL-malic acid, 50% by weight aqueous solution, manufactured by Fuso Chemical Industry Co., Ltd.
- the food additive grade was used after adjusting its concentration with ion-exchanged water) and 12.38 g was added, and diethylenetriaminepentaacetic acid ⁇ 3 sodium (DTPA ⁇ 3Na) during granulation was changed from 0.01 to 0.03 parts by weight.
- the same operation as in Production Example 1 was carried out except that the silica was not mixed.
- the water-absorbing resin particles obtained are designated as a water-absorbing agent (34).
- Table 14 shows the performance of the water-absorbing agent (34).
- Example 35 In Production Example 1, the addition amount of polyethylene glycol diacrylate was changed from 0.03 mol% to 0.06 mol%, and 30 wt% D, L-malic acid aqueous solution (DL) was added to the monomer aqueous solution (f ′). -16.5 g of malic acid, 50% by weight aqueous solution, manufactured by Fuso Chemical Industry Co., Ltd., food additive grade adjusted in concentration with ion-exchanged water), and diethylenetriaminepentaacetic acid / trisodium (3 sodium salt) during granulation ( The same operation as in Production Example 1 was performed, except that the amount of DTPA ⁇ 3Na) was changed from 0.01 to 0.03 parts by weight, and silica was not mixed.
- the water-absorbing resin particles obtained are designated as a water-absorbing agent (35). Table 14 shows the performance of the water absorbing agent (35).
- Example 36 In Production Example 1, the addition amount of polyethylene glycol diacrylate was changed from 0.03 mol% to 0.08 mol%, and diethylenetriaminepentaacetic acid ⁇ 3 sodium (DTPA ⁇ 3Na) was added to the monomer aqueous solution (f ′).
- DTPA ⁇ 3Na diethylenetriaminepentaacetic acid ⁇ 3 sodium
- a 30% by weight D, L-malic acid aqueous solution (DL-malic acid, 50% by weight aqueous solution, manufactured by Fuso Chemical Industry Co., Ltd.) was added to the monomer aqueous solution (f ′) with ion-exchanged water.
- Example 37 In Production Example 1, the amount of polyethylene glycol diacrylate added was changed from 0.03 mol% to 0.035 mol%, and 1.65 g of a 30 wt% aqueous lactic acid solution was added to the monomer aqueous solution (f ′). The same operation as in Production Example 1 was carried out except that the addition amount of diethylenetriaminepentaacetic acid ⁇ 3 sodium (DTPA ⁇ 3Na) at the time of granulation was changed from 0.01 to 0.05 parts by weight and no silica was mixed. .
- the water-absorbing resin particles obtained are designated as a water-absorbing agent (37). Table 14 shows the performance of the water absorbing agent (37).
- the present invention it is possible to provide a water-absorbing agent having a high water absorption capacity that can reduce stickiness after liquid absorption when used as a sanitary material, or that improves both strong urine resistance and coloration resistance with time.
- the present invention can be used in various fields such as sanitary articles such as disposable diapers and sanitary napkins, as well as pet sheets and water-stopping materials.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Analytical Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Materials Engineering (AREA)
- Hematology (AREA)
- Dispersion Chemistry (AREA)
- Biomedical Technology (AREA)
- Heart & Thoracic Surgery (AREA)
- Vascular Medicine (AREA)
- Solid-Sorbent Or Filter-Aiding Compositions (AREA)
- Absorbent Articles And Supports Therefor (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Processes Of Treating Macromolecular Substances (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
Abstract
Description
(1-1)「吸水性樹脂」
本発明における「吸水性樹脂」とは、水膨潤性水不溶性の高分子ゲル化剤を指し、以下の物性を満たすものをいう。即ち、「水膨潤性」として、ERT441.2-02で規定されるCRCが5g/g以上、かつ、「水不溶性」として、ERT470.2-02で規定されるExtが50重量%以下の物性を満たす高分子ゲル化剤を指す。
本発明における「ポリアクリル酸(塩)」とは、ポリアクリル酸及び/又はその塩を指し、主成分として、アクリル酸及び/又はその塩(以下、「アクリル酸(塩)」と称する)を繰り返し単位として含み、任意成分としてグラフト成分を含む重合体を指す。
本明細書において、吸水剤とは、吸水性樹脂を主成分として含む、水性液の吸収ゲル化剤を意味する。「吸水剤」を「粒子状吸水剤」と称することもある。
ニルアルコール系樹脂、ポリエチレンオキシド系樹脂、ポリアスパラギン酸(塩)系樹脂、ポリグルタミン酸(塩)系樹脂、ポリアルギン酸(塩)系樹脂、デンプン系樹脂、セルロース系樹脂が挙げられ、好ましくはポリアクリル酸(塩)系樹脂が使用される。
「EDANA」は、欧州不織布工業会(European Disposables and Nonwovens Associations)の略称であり、「ERT」は、欧州標準(ほぼ世界標準)の吸水性樹脂の測定法(EDANA Recommended Test Methods)の略称である。本発明では、特に断りのない限り、ERT原本(2002年改定/公知文献)に準拠して、吸水性樹脂の物性を測定する。
「CRC」は、Centrifuge Retention Capacity(遠心分離機保持容量)の略称であり、吸水性樹脂の無加圧下吸水倍率(「吸水倍率」と称する場合もある)を意味する。
「AAP」は、Absorption Against Pressureの略称であり、吸水性樹脂の加圧下吸水倍率を意味する。
「PSD」は、Particle Size Distributionの略称であり、篩分級により測定される、吸水性樹脂の粒度分布を意味する。
「Ext」は、Extractablesの略称であり、吸水性樹脂の水可溶分(水可溶成分量)を意味する。
「Moisture Content」は、吸水性樹脂の含水率を意味する。
「Residual Monomers」は、吸水性樹脂中に残存する単量体(モノマー)量(以下、「残存モノマー」と称する)を意味する。
本発明における吸水性樹脂の「通液性」とは、荷重下又は無荷重下での膨潤ゲルの粒子間を通過する液の流れ性のことをいい、代表的な測定方法として、SFC(Saline
Flow Conductivity/食塩水流れ誘導性)やGBP(Gel Bed
Permeability/ゲル床透過性)がある。
本発明における吸水性樹脂の「吸水速度」とは、「FSR」又は「Vortex」(単位:秒)により測定される吸水速度を意味する。なお、「FSR」とは、Free Swell Rateの略称である。具体的な測定方法については、後述の実施例において説明する。
本明細書において、範囲を示す「X~Y」は「X以上、Y以下」を意味する。また、特に注釈のない限り、重量の単位である「t(トン)」は「Metric ton(メトリック トン)」を意味し、「ppm」は「重量ppm」又は「質量ppm」を意味する。更に、「重量」と「質量」、「重量部」と「質量部」、「重量%」と「質量%」はそれぞれ同義語として扱う。本明細書において、「単量体」と「モノマー」、「重合体」と「ポリマー」はそれぞれ同義語として扱う。また、「~酸(塩)」は「~酸及び/又はその塩」、「(メタ)アクリル」は「アクリル及び/又はメタクリル」をそれぞれ意味する。
以下に、本発明にかかわる吸水性樹脂の製造工程(2-1)~(2-10)について示す。
本工程は、アクリル酸(塩)を主成分として含む水溶液(以下、「単量体水溶液」と称する)を調製する工程である。なお、得られる吸水性樹脂の吸水性能が低下しない範囲で、単量体のスラリー液を使用することもできるが、本項では便宜上、単量体水溶液について説明を行う。
本発明では、得られる吸水性樹脂の物性及び生産性の観点から、単量体としてアクリル酸及び/又はその塩(以下「アクリル酸(塩)」と称する)が用いられる。
本発明において、「塩基性組成物」とは、塩基性化合物を含有する組成物を指し、例えば、市販の水酸化ナトリウム水溶液等が該当する。
本発明における中和として、アクリル酸に対する中和(重合前)又はアクリル酸を架橋重合して得られる含水ゲル状架橋重合体に対する中和(重合後)(以下、「後中和」と称する)の何れかを選択又は併用することができる。また、これらの中和は、連続式でもバッチ式でもよく特に限定されないが、生産効率等の観点から連続式が好ましい。
本発明において、「他の単量体」とは、上記アクリル酸(塩)以外の単量体を指し、アクリル酸(塩)と併用して吸水性樹脂を製造することができる。
本発明で使用される内部架橋剤として、米国特許第6241928号に記載された化合物が本発明にも適用される。これらの中から反応性を考慮して1種又は2種以上の化合物が選択される。
本発明において、得られる吸水性樹脂の物性向上の観点から、下記の物質を単量体水溶液の調製時に添加することもできる。
本工程において、単量体水溶液を調製する際に、上記の各物質が添加される。該単量体水溶液中の単量体成分の濃度としては特に限定されないが、吸水性樹脂の物性の観点から、好ましくは10~80重量%、より好ましくは20~75重量%、更に好ましくは30~70重量%である。上記単量体成分の濃度を適宜調整することによって、得られる吸水剤の遠心分離保持容量、可溶分量、または可溶分分子量を適宜調整することができる。
式(1):
(単量体成分の濃度(重量%))=(単量体成分の重量)/(単量体水溶液の重量)×100
本工程は、上記単量体水溶液の調製工程で得られたアクリル酸(塩)系単量体水溶液を重合させて、含水ゲル状架橋重合体(以下、「含水ゲル」と称する)を得る工程である。
本発明で使用される重合開始剤は、重合形態等によって適宜選択されるため、特に限定されないが、例えば、熱分解型重合開始剤、光分解型重合開始剤、又はこれらの重合開始剤の分解を促進する還元剤を併用したレドックス系重合開始剤等が挙げられる。具体的には、米国特許第7265190号に開示された重合開始剤のうち、1種又は2種以上が用いられる。なお、重合開始剤の取扱性や吸水性樹脂の物性の観点から、好ましくは過酸化物又はアゾ化合物、より好ましくは過酸化物、更に好ましくは過硫酸塩が使用される。
本発明に適用される重合形態としては、特に限定されないが、吸水特性や重合制御の容易性等の観点から、好ましくは噴霧液滴重合、水溶液重合、逆相懸濁重合、より好ましくは水溶液重合、逆相懸濁重合、更に好ましくは水溶液重合が挙げられる。中でも、連続水溶液重合が特に好ましく、連続ベルト重合、連続ニーダー重合の何れでも適用される。
式(2):
(固形分上昇度(重量%))=(重合後の含水ゲルの固形分濃度(重量%))-(単量体水溶液の固形分濃度(重量%))
式(3):
(単量体水溶液の固形分(重量%))=((単量体成分+グラフト成分+吸水性樹脂+その他固形物)の重量)/(重合系内の成分の重量)×100
通常、得られる吸水剤の吸水特性や色調(着色防止)等の観点から、α-ヒドロキシカルボン酸を添加することが好ましい。本発明においては、驚くべきことに、α-ヒドロキシカルボン酸を添加することによって、得られる吸水剤の可溶分分子量が低減すること、ひいては衛生材料として使用する際のべたつきおよび不快感が低減することが見出されたので、これらのさらなる観点から、α-ヒドロキシカルボン酸を添加することがより好ましい。
本明細書において、「重合後工程」とは、重合後に行われる工程である。本重合後工程は、以下に記載のゲル粉砕工程、乾燥工程、粉砕工程、分級工程、表面処理(架橋)剤混合工程、表面架橋(表面処理)工程、造粒工程、硬化工程および再加湿工程等の少なくとも1つの工程であるが、これらに限定されない。
本工程は、上記重合工程で得られた含水ゲルを、例えば、ニーダー、ミートチョッパー等のスクリュー押出し機、カッターミル等のゲル粉砕機でゲル粉砕し、粒子状の含水ゲル(以下、「粒子状含水ゲル」と称する)を得る工程である。なお、上記重合工程がニーダー重合の場合、重合工程とゲル粉砕工程が同時に実施されている。また、気相重合や逆相懸濁重合等、粒子状含水ゲルが重合過程で直接得られる場合には、該ゲル粉砕工程が実施されないこともある。
本工程は、上記重合工程及び/又はゲル粉砕工程で得られた粒子状含水ゲルを所望する樹脂固形分まで乾燥させて乾燥重合体を得る工程である。該樹脂固形分は、乾燥減量(吸水性樹脂1gを180℃で3時間加熱した際の重量変化)から求められ、好ましくは80重量%以上、より好ましくは85~99重量%、更に好ましくは90~98重量%、特に好ましくは92~97重量%である。
本工程は、上記乾燥工程で得られた乾燥重合体を粉砕(粉砕工程)し、所定範囲の粒度に調整(分級工程)して、吸水性樹脂粉末(表面架橋を施す前の、粉末状の吸水性樹脂を便宜上「吸水性樹脂粉末」と称する)を得る工程である。
本工程は、上述した工程を経て得られる吸水性樹脂粉末の表面層(吸水性樹脂粉末の表面から数10μmの部分)に、更に架橋密度の高い部分を設ける工程であり、混合工程、加熱処理工程及び冷却工程(任意)から構成される。
本発明で使用される表面架橋剤としては、特に限定されないが、有機又は無機の表面架橋剤が挙げられる。中でも、吸水性樹脂の物性や表面架橋剤の取扱性の観点から、カルボキシル基と反応する有機表面架橋剤が好ましい。例えば、米国特許7183456号に開示される1種又は2種以上の表面架橋剤が挙げられる。より具体的には、多価アルコール化合物、エポキシ化合物、ハロエポキシ化合物、多価アミン化合物又はそのハロエポキシ化合物との縮合物、オキサゾリン化合物、オキサゾリジノン化合物、多価金属塩、アルキレンカーボネート化合物、環状尿素化合物等が挙げられる。
本工程は、吸水性樹脂粉末と上記表面架橋剤を混合する工程である。該表面架橋剤の混合方法については、特に限定されないが、予め表面架橋剤溶液を作製しておき、該液を吸水性樹脂粉末に対して、好ましくは噴霧又は滴下して、より好ましくは噴霧して混合する方法が挙げられる。
本工程は、上記混合工程から排出された混合物に熱を加えて、吸水性樹脂粉末の表面上で架橋反応を起させる工程である。
本工程は、上記加熱処理工程後に必要に応じて設置される任意の工程である。
本工程は、上記表面架橋工程で得られた吸水性樹脂粒子に、下記の多価金属塩化合物、ポリカチオン性ポリマー、キレート剤、無機還元剤、ヒドロキシカルボン酸化合物からなる群から選択される少なくとも1種の添加剤を添加する工程である。
本発明において、得られる吸水性樹脂の吸水速度、通液性、吸湿流動性等の向上の観点から、多価金属塩及び/又はカチオン性ポリマーを添加することが好ましい。
「キレート剤」とは、金属イオンに配位してキレート化合物をつくる多座配位子をいう。
本発明において、得られる吸水性樹脂の色調(着色防止)、劣化防止、残存モノマー低減等の観点から、無機還元剤を添加することが好ましい。
本発明においては、上述した添加剤以外の添加剤を、吸水性樹脂に種々の機能を付加させるため、添加することもできる。該添加剤として、具体的には、界面活性剤、リン原子を有する化合物、酸化剤、有機還元剤、水不溶性無機微粒子、金属石鹸等の有機粉末、消臭剤、抗菌剤、パルプや熱可塑性繊維等が挙げられる。なお、上記界面活性剤は、国際公開第2005/075070号に開示された化合物が、また、上記水不溶性無機微粒子は、国際公開第2011/040530号の「〔5〕水不溶性無機微粒子」に開示された化合物が、それぞれ本発明に適用される。
本発明においては、上述した工程以外に、表面処理(架橋)剤混合工程、硬化工程、造粒工程、整粒工程、微粉除去工程、微粉の再利用工程等を必要に応じて設けることができる。また、輸送工程、貯蔵工程、梱包工程、保管工程等の1種又は2種以上の工程を更に含んでもよい。なお、「整粒工程」は、表面架橋工程以降の微粉除去工程や吸水性樹脂が凝集し、所望の大きさを超えた場合に分級、粉砕を行う工程を含む。また、「微粉の再利用工程」は、本発明のように微粉をそのまま添加する形態の他、大きな含水ゲルにして、吸水性樹脂の製造工程の何れかの工程に添加する工程を含む。
本発明に係る製造方法で得られるポリアクリル酸(塩)系吸水剤は、該吸水剤を衛生用品、特に紙オムツに使用する場合には、下記の(3-1)~(3-13)に掲げた物性のうち、少なくとも1つ以上、好ましくはCRCを含めた2つ以上、より好ましくはCRCを含めた3つ以上、最も好ましくは全ての物性を、所望する範囲に制御することが望まれる。これらの物性が下記の範囲を満たさない場合、本発明の効果が十分に得られず、高濃度紙オムツにおいて十分な性能を発揮しない虞がある。
本発明の吸水剤のCRC(遠心分離機保持容量)は、30g/g以上である。上限値については特に限定されず高値ほど好ましいが、他の物性とのバランスの観点から、好ましくは70g/g以下、より好ましくは50g/g以下、更に好ましくは40g/g以下である。
本発明の吸水剤のAAP(加圧下吸水倍率)は、好ましくは20g/g以上、より好ましくは22g/g以上、更に好ましくは23g/g以上、特に好ましくは24g/g以上、最も好ましくは25g/g以上である。上限値については特に限定されないが、好ましくは30g/g以下である。
本発明の吸水剤の粒度(粒度分布、重量平均粒子径(D50)、粒度分布の対数標準偏差(σζ))は、表面架橋を施す前の吸水剤粉末(吸水性樹脂粉末)の粒度と同じになるように、制御される。具体的には、重量平均粒子径(D50)として、好ましくは200~600μm、より好ましくは200~550μm、更に好ましくは250~500μm、特に好ましくは350~450μmである。また、粒子径150μm未満の粒子の割合は、好ましくは10重量%以下、より好ましくは5重量%以下、更に好ましくは1重量%以下であり、粒子径850μm以上の粒子の割合は、好ましくは5重量%以下、より好ましくは3重量%以下、更に好ましくは1重量%以下である。なお、これらの粒子の割合の下限値としては、何れの場合も少ないほど好ましく、0重量%が望まれるが、0.1重量%程度でもよい。更に、粒度分布の対数標準偏差(σζ)は、好ましくは0.20~0.50、より好ましくは0.25~0.40、更に好ましくは0.27~0.35である。なお、これらの粒度は、米国特許第7638570号やEDANA ERT420.2-02に開示されている測定方法に準じて、標準篩を用いて測定される。
本発明の吸水剤のExt(水可溶分)は、上限値については通常50重量%以下であり、好ましくは35重量%以下、より好ましくは25重量%以下である。下限値については好ましくは、10重量%以上であり、より好ましくは、前記吸水剤の可溶分量が13重量%以上、更に好ましくは、15重量%以上である。
以下、水可溶性重合体量は、「水可溶分量」または「可溶分量」と略されることがある。また、水可溶性重合体は、「水可溶分」または「可溶分」とも称される。「可溶分率」は、吸水剤中における可溶分の割合(重量%)である。「可溶分率」はまた、重量%の単位を伴って「Ext」、「水可溶分」、「可溶分」、「水可溶分量」または「可溶分量」と記載されることもある。
水性物品の実使用時、吸液した吸水剤から溶出する可溶分により、使用者がべたつきによる不快感を覚える可能性があるため好ましくない。また、好ましい範囲の下限を下回る場合、内部架橋剤を多量に使用しなければならず、十分な吸水倍率を得ることができない可能性があるため、好ましくない。
本発明の吸水剤の含水率は、好ましくは0重量%を超えて15重量%以下、より好ましくは1~13重量%、更に好ましくは2~10重量%、特に好ましくは2~9重量%である。
本発明の吸水剤に含有する残存モノマーは、安全性の観点から、好ましくは500ppm以下、より好ましくは400ppm以下、更に好ましくは300ppm以下である。下限値については特に限定されないが、好ましくは0ppm、より好ましくは10ppm程度である。
本発明の吸水剤のFSR(吸水速度)は、好ましくは0.10g/g/s以上、より好ましくは0.15g/g/s以上、更に好ましくは0.20g/g/s以上、特に好ましくは0.25g/g/s以上である。上限値については特に限定されないが、好ましくは5.0g/g/s以下、より好ましくは3.0g/g/s以下である。
「吸湿ブロッキング」とは、吸湿時に吸水性樹脂どうしが凝集する現象のことを指し、「吸湿ブロッキング率」とはその凝集の割合のことを指す。吸湿ブロッキング率は、下記実施例[吸水剤の物性測定](c)に記載の方法により算出される。本発明の吸水剤の吸湿ブロッキング率は、吸湿流動性改善剤を添加することにより、好ましくは50質量%以下、より好ましくは30質量%以下、更に好ましくは10質量%以下に調整される。下限値は算出原理上、0質量%以上となる。吸湿流動性改善剤の添加量は、前記の吸湿ブロッキング率を達成するよう、吸水剤に対して0質量%~1質量%の範囲で調整できる。理論に束縛されることを望まないが、吸湿ブロッキング率を低く制御することで、いかなる作業環境やユーザー先の使用条件(例えばおむつ製造工程の運転条件)でも安定的に吸水剤を使用できる。
「可溶分分子量」または「可溶分の分子量」とは、可溶分の重量平均分子量のことを指す。本発明の吸水剤の可溶分分子量は、好ましくは、50,000ダルトン以上1,000,000ダルトン以下の範囲内にあり、より好ましくは、100,000ダルトン以上800,000ダルトン以下、150,000ダルトン以上700,000ダルトン以下、200,000ダルトン以上700,000ダルトン以下、150,000ダルトン以
上600,000ダルトン以下、200,000ダルトン以上600,000ダルトン以下、最も好ましくは200,000ダルトン以上550,000ダルトン以下である。理論に束縛されることを望まないが、この範囲内であると吸水剤において良好なべたつき低減効果が発揮できるからである。本発明の吸水剤の可溶分分子量は、下限値が好ましくは、30,000、40,000、50,000、75,000、100,000、125,000、150,000、175,000、200,000、225,000、または250,000ダルトンであり、上限値が好ましくは、1,000,000、900,000、800,000、700,000、600,000、または500,000ダルトンである。前記上限値および下限値の可能な組み合わせもまた、本発明の範囲に包含される。本発明の吸水剤の可溶分の重量平均分子量は、下記[吸水剤の物性測定](d)に記載の方法によって算出することができる。
本発明の吸水剤の可溶分の極限粘度(IV)は、好ましくは0.5~4.5dl/g、より好ましくは、0.8~4.2dl/g、1.0~3.8dl/g、1.5~4.0dl/g、最も好ましくは、1.5~3.5dl/gである。理論に束縛されることを望まないが、上記の可溶分の極限粘度(IV)であると、吸水剤における触感評価においてべたつきが低減されるからである。本発明の吸水剤の可溶分の極限粘度(IV)は、下限値が好ましくは、0.3、0.4、0.5、0.6、0.7、0.8、0.9、1.0、1.1、1.2、1.3、1.4、1.5、1.6、1.7、または1.8dl/gであり、上限値が好ましくは、5.0、4.9、4.8、4.7、4.6、4.5、4.4、4.3、4.2、4.1、4.0、3.9、3.8、3.7、3.6、3.5、3.4、3.3、3.2、3.1、または3.0dl/gである。前記上限値および下限値の可能な組み合わせもまた、本発明の範囲に包含される。本発明の吸水剤の可溶分の極限粘度(IV)は、下記[吸水剤の物性測定](d)に記載の方法によって算出することができる。
「可溶分分子量低減率(%)」とは、α-ヒドロキシカルボン酸を含有する吸水剤の可溶分分子量が、同等のCRCを有するα-ヒドロキシカルボン酸を含有しない吸水剤の可溶分分子量に対して低減された比率のことを指す。本発明の吸水剤の可溶分分子量低減率(%)は、好ましくは、8%以上、9%以上、10%以上、11%以上、12%以上、14%以上であり、最も好ましくは15%以上である。上限値については特に限定されないが、好ましくは50%以下、より好ましくは40%以下である。前記可溶分分子量低減率(%)の上限値および下限値の可能な組み合わせもまた、本発明の範囲に包含される。本発明の吸水剤の可溶分分子量低減率(%)は、下記[吸水剤の物性測定](e)に記載の方法によって算出することができる。吸水剤の可溶分分子量低減率は、吸液後の吸水剤のべたつきの指標として使用することができ、好ましくは8%以上、9%以上、10%以上、11%以上、12%以上、14%以上であり、最も好ましくは15%以上である場合、吸水剤のべたつきが低減したといえる。
吸液後の本発明の吸水剤のべたつきは、擬似衛生剤の吸液時の不織布上の溶出可溶分の触感評価によって定義される。触感評価は、具体的には、下記[吸水剤の物性測定](f)触感評価に記載の方法で行われる。触感評価が、好ましくは、2.5以下、2.3以下、2.1以下、2.0以下、1.8以下、最も好ましくは1.5以下である場合に、べたつきが低減したと言える。
本発明で得られる吸水剤は、紙おむつ等の衛生材料向けに好適に使用できるものであり、その際、高い湿度や温度条件下での長期貯蔵状態においても著しく清浄な白い状態を維持する。更に、前述の製法によって得られる吸水剤であって、温度70±1℃、相対湿度65±1%の雰囲気に7日間曝露した粒子の分光式色差計によるハンターLab表色系測定において、L値(Lightness)が少なくとも70、さらに好ましくは74以上、特に好ましくは78以上を示す、経時色安定性吸水剤である。(なお、L値の上限は通常100であるが、70ならば実使用でも実質問題が発生しない。)
Fe/L-as劣化可溶分量(実施例に記載の測定法で規定)は、含水率補正値で、好ましくは0~30重量%、より好ましくは0~25重量%、更に好ましくは0~20重量%に制御される。
表面張力(実施例に記載の測定法で規定)は、好ましくは60[mN/m]以上、より好ましくは65[mN/m]以上、さらに好ましくは67[mN/m]以上、特に好ましくは70[mN/m]以上、最も好ましくは72[mN/m]以上であり、実質的な表面張力の低下もない。上限は通常75[mN/m]で十分である。
本発明の吸水剤の遠心分離機保持容量は主に、重合工程における内部架橋剤の量、α-ヒドロキシカルボン酸(塩)の量、単量体成分の濃度、表面架橋工程における加熱温度および加熱時間からなる群から選択される少なくとも1つを調整することによって適宜調整可能である。上記内部架橋剤およびその量、ならびに上記α-ヒドロキシカルボン酸(塩)およびその量としては、それぞれ、上記「〔2〕ポリアクリル酸(塩)系吸水性樹脂の製造方法」の(内部架橋剤)ならびに(α-ヒドロキシカルボン酸(塩))に記載された化合物およびその使用量が挙げられる。上記単量体成分の濃度としては、上記「〔2〕ポリアクリル酸(塩)系吸水性樹脂の製造方法」の(単量体成分の濃度)に記載された濃度が挙げられる。表面架橋工程における加熱温度および加熱時間としては、上記「〔2〕ポリアクリル酸(塩)系吸水性樹脂の製造方法」の(2-6)表面架橋工程における(加熱処理工程)に記載された加熱温度および加熱時間が挙げられる。
本発明の吸水剤の用途は、特に限定されないが、好ましくは紙オムツ、生理用ナプキン、失禁パッド等の衛生用品の吸収体用途が挙げられる。特に、原料由来の臭気、着色等が問題となっていた高濃度紙オムツ(紙オムツ1枚あたりの吸水剤の使用量が多いもの)の吸収体として使用することができる。更に、上記吸収体の上層部に使用される場合に、顕著な効果が期待できる。
以下に本発明の好ましい実施形態を説明する。以下に提供される実施形態は、本発明のよりよい理解のために提供されるものであり、本発明の範囲は以下の記載に限定されるべきでないことが理解される。従って、当業者は、本明細書中の記載を参酌して、本発明の範囲内で適宜改変を行うことができることは明らかである。また、本発明の以下の実施形態は単独でも使用されあるいはそれらを組み合わせて使用することができることが理解される。
本発明の1つの局面において、アクリル酸(塩)を含む単量体水溶液の重合工程、乾燥工程、および表面架橋工程を含む、遠心分離機保持容量(CRC)が30g/g以上である吸水剤の製造方法であって、該乾燥工程の前にα-ヒドロキシカルボン酸(塩)を添加する工程を含む、方法が提供される。理論に束縛されることを望まないが、高い吸水倍率を有する吸収剤の製造において、乾燥工程前の前にα-ヒドロキシカルボン酸(塩)を添加することにより、生成する重合体中の可溶分の分子量が低減し、吸液して膨潤した際に溶出する可溶分の分子量が低減することにより、粘度が低下し、その結果、衛生材料として使用する際のべたつき、不快感が軽減されるからである。
1つの局面において、本発明は、アクリル酸(塩)を主成分とする単量体水溶液をα-ヒドロキシカルボン酸(塩)の存在下に重合する工程を含む、ポリアクリル酸(塩)系吸水剤の製造方法であって、重合後工程の少なくとも1つは、キレート剤の存在下で行われる、方法が提供される。理論に束縛されることを望まないが、α-ヒドロキシカルボン酸(塩)の存在下に重合し、重合後に行われる工程の少なくとも1つが、キレート剤の存在下で行われることにより、強耐尿性と耐経時着色性を両立した吸水剤を製造できるからである。
本発明の1つの局面において、表面架橋されたポリアクリル酸(塩)系吸水剤であって
、該吸水剤内部にα-ヒドロキシカルボン酸(塩)を含み、遠心分離機保持容量(CRC)が30g/g以上である、吸水剤が提供される。
1つの局面において、本発明は、ポリアクリル酸(塩)系で構成される吸水剤であって、該吸水剤1gはL-アスコルビン酸200ppmおよび2価の鉄イオン0.4ppmを含む生理食塩水(0.9%塩化ナトリウム水溶液)で構成される劣化液25mlに投入し、37℃16時間置いた後、生理食塩水を加えて液全量を200gとして10分攪拌した後に抽出されるFe/L-as劣化可溶分が35重量%以下、30重量%以下、25重量%以下、20重量%以下、15重量%以下、10重量%以下、5重量%以下または0重量%であり、さらに、着色促進試験後のYI値が26以下である、吸水剤を提供する。尿漏れ等で問題となるのはその中に含まれる鉄分に加え、アスコルビン酸が存在することで、相乗的に吸水剤が劣化し着色する等の悪影響が認められる。このような相乗的悪影響についてはこれまで評価できなかった。本発明では、この相乗的な悪影響についてL-アスコルビン酸および2価の鉄イオンを含む生理食塩水で構成される劣化液を用いることにより初めて評価することができる系を開発し、その影響に対する効果を評価することができるようになった。この評価系により、本発明において鋭意検討した結果、アクリル酸(塩)を主成分とする単量体水溶液をα-ヒドロキシカルボン酸(塩)の存在下に重合して得られるポリアクリル酸(塩)系吸水剤にキレート剤を添加する方法によって、強耐尿性と耐経時着色性を両立した吸水剤が、これらの相乗的な悪影響を改善する効果を見出した。
本発明の1つの局面において、α-ヒドロキシカルボン酸(塩)を含む、吸水剤の可溶分分子量を低減させるための組成物が提供される。
本発明の1つの局面において、α-ヒドロキシカルボン酸(塩)およびキレート剤を含む、吸水剤の強耐尿性を改善するための組成物が提供される。
本発明の1つの局面において、吸水剤の可溶分分子量を低減させるための、α-ヒドロキシカルボン酸(塩)の使用が提供される。
本発明の1つの局面において、吸水剤の可溶分分子量を低減させる方法であって、該方法は重合により吸水剤を形成し得るアクリル酸(塩)を含む単量体水溶液の重合、重合により得られる含水ゲルの乾燥、および乾燥により得られる吸水性樹脂粉末の表面架橋により、遠心分離機保持容量(CRC)が30g/g以上である吸水剤を製造する工程を含み、該方法は、該乾燥の前にα-ヒドロキシカルボン酸(塩)を添加する工程を含む、方法が提供される。
本発明の1つの局面において、吸水剤の強耐尿性を改善するための方法であって、該方法は重合により吸水剤を形成し得るアクリル酸(塩)を含む単量体水溶液のα-ヒドロキシカルボン酸(塩)の存在下での重合、重合により得られる含水ゲルの乾燥、および乾燥により得られる吸水性樹脂粉末の表面架橋により、吸水剤を製造する工程を含み、該方法は、該重合の後にキレート剤を添加する工程を含み、該吸水剤は、該吸水剤1gをL-アスコルビン酸200ppmおよび2価の鉄イオン0.4ppmを含む生理食塩水(0.9%塩化ナトリウム水溶液)で構成される劣化液25mlに投入し、37℃16時間置いた後、生理食塩水を加えて液全量を200gとして10分間攪拌した後に抽出されるFe/L-as劣化可溶分が35重量%以下であり、さらに、着色促進試験後のYI値が26以下であるという特徴を有する、方法が提供される。
(a)遠心分離機保持容量(CRC)
本発明の吸水剤の遠心分離機保持容量(CRC)、すなわち無加圧下吸水倍率は、EDANA法(ERT441.2-02)に準拠して測定した。具体的には、吸水剤0.2gを不織布製の袋に入れた後、大過剰の0.9重量%塩化ナトリウム水溶液中に30分間浸漬して自由膨潤させ、その後、遠心分離機(250G)で水切りした後の吸水倍率(単位;g/g)として得た。
以下、水可溶性重合体量は、「水可溶分量」または「可溶分量」と略されることがある。また、水可溶性重合体は、「水可溶分」または「可溶分」とも称される。「可溶分率」は、吸水剤中における可溶分の割合(重量%)である。「可溶分率」はまた、重量%の単位を伴って「Ext」、「水可溶分」、「可溶分」、「水可溶分量」または「可溶分量」と記載されることもある。
式(4):
可溶分率(質量%)=0.1×(モノマーの平均分子量)×184.3×100×([HCl]-[bHCl])/1000/1.0/50.0
式(5):
中和率(mol%)=(1-([NaOH]-[bNaOH])/([HCl]-[bHCl]))×100
吸水性樹脂粉末/吸水剤約2gを、直径52mmのアルミカップに均一に散布した後、温度25℃、相対湿度90±5%RHに調整した恒温恒湿器(エスペック株式会社製;MODEL: SH-641)に1時間静置した。
式(6): 吸湿ブロッキング率[質量%]={W7/(W7+W8)}×100
<水可溶分の重量平均分子量(Mw)>
(試料調製)
上記水可溶分率の測定で得られた濾液を用いた。試料濃度が高い場合はポリマー成分の濃度が、0.5mg/mL程度となるように適宜GPC溶液を用いて希釈した。得られた溶液を以下に示す溶媒で4倍に希釈し、フィルター(ジーエルサイエンス社製、GLクロマトディスク、水系25A、孔径0.2μm)を通過させた。この溶液について以下の測定条件で測定を行った。
ビスコテック社製TDA302(登録商標)を用いて、測定を行った。装置構成としては、サイズ排除クロマトグラフィー、屈折率検出器、光散乱検出器及びキャピラリー粘度計を搭載した装置である。
ポンプ・オートサンプラー:ビスコテック社製GPCmax
ガードカラム:OHpak SB-G(昭和電工株式会社製)
カラム:OHpak SB-806MHQ(昭和電工株式会社製)を2本直列につないで使用
検出器:ビスコテック社製TDA302(系内温度は30℃で保持)
溶媒:リン酸2水素ナトリウム2水和物60mM・リン酸水素2ナトリウム12水和物20mM・アジ化ナトリウム400ppm水溶液(pH6.35~6.38)
流速:0.5mL/min
注入量 :100μL
α-ヒドロキシカルボン酸を加えた場合の水可溶分の重量平均分子量の、α-ヒドロキシカルボン酸を加えない場合の水可溶分の重量平均分子量に対する低減率を可溶分分子量低減率とした。可溶分分子量低減率(%)は、以下の式(7)にしたがって求めた。

木材粉砕パルプ40gと、吸水剤60gを、吸水紙を載せた400メッシュ(目開き38μm)のワイヤースクリーン上に、バッチ型空気抄造装置を用いて120mm×400mmのウェブに空気抄造した。尚、坪量は、空気抄造時間によって調整した。ついで、該ウェブを圧力下でプレスすることにより、吸収体を得た。前記吸収体に液不透過性のバックシートおよびサイドギャザーとしてポリエチレン製シートを、液透過性のトップシートとして吸水紙を備え、紙おむつ型モデル吸収体とした。
5:かなりべたつきを感じる
4:ややべたつきを感じる
3:基準
2:べたつきが少なく感じる
1:べたつきが全くない
吸水剤のα-ヒドロキシカルボン酸の定量方法は、特に限定されないが、例えば、吸水剤から可溶成分を抽出し、その溶液を高速液体クロマトグラフィーを用いて分析することにより定量することができる。具体的には、以下の方法により定量することができる。
吸水性樹脂1.0gを0.9重量%塩化ナトリウム水溶液200mLに添加し、500rpmで1時間攪拌した溶液の上澄みをシリンジフィルター(GLサイエンス社製、水系<Aタイプ>、型式25A、孔径0.45μm)を用いてろ過し、抽出溶液とした。
α-ヒドロキシカルボン酸を定量するための高速液体クロマトグラフィーの構成としては、例えば、以下のようなものを用いることができる。
ポンプ:L-7110(日立ハイテクサイエンス社製)、流量1.0mL/min
オートサンプラー:L-7200(日立ハイテクサイエンス社製)、注入量50μL
カラムオーブン:L-7300(日立ハイテクサイエンス社製)、温度設定30℃
示差屈折検出器:L-7490(日立ハイテクサイエンス社製)
カラム:Shim-pack SCR-101H(島津ジーエルシー社製)
ガードカラム:Shim-pack SCR(H)(島津ジーエルシー社製)
移動相:85%リン酸水溶液
本発明の吸水性樹脂の加圧下吸水倍率(AAP)は、EDANA法(ERT442.2-02)に準拠して測定した。
吸水剤の着色評価は、日本電色工業株式会社製の分光式色差計SZ-Σ80COLOR MEASURING SYSTEMを用いた。設定条件(反射測定/付属の粉末・ペースト試料台(内径30mm、高さ12mm/標準として粉末・ペースト用標準丸白板No.2/30Φ投光パイプ))で、吸水剤を備え付けの試料台に5g充填し(備え付け試料台の6割程度の充填)、室温(20~25℃)、湿度50RH%の条件下で上記分光式色差計にて表面色(YI値(Yellow Index))を測定した。また、同じ装置の同じ測定法によって、同時に他の尺度の物体色(L,a,b)ないしWB(ハンターカラー)も測定できる。L/WBは大きいほど、a/bは小さいほど、低着色で実質白色に近づくことを示す。
続いて、上記ペースト試料台に5gの吸水剤を充填し、温度70±1℃、相対湿度65±1%の雰囲気に調整した恒温恒湿機(エスペック株式会社製、品名:小型環境試験器、形式SH-641)中に吸水剤を充填したペースト試料台を7日間曝露した。曝露後、上記分光式色差計にて着色促進試験後の表面色(YI値(Yellow Index))を測定した。本願(例えば実施例)では、経時着色YIと記載する場合がある。
本発明に係る吸水剤のFe/L-as劣化可溶分量は、以下の手法に従って、測定を行う。
十分に洗浄された100mlのビーカーに20℃に調整された生理食塩水50mlを入れ、まず、生理食塩水の表面張力を、表面張力計(KRUSS社製のK11自動表面張力計)を用いて測定した。この測定において表面張力の値が71~75[mN/m]の範囲でなくてはならない。
シグマ型羽根を2本有する内容積10リットルのジャケット付きステンレス型双腕型ニーダーに蓋を付けて形成した反応器中で、アクリル酸425.2g、37質量%アクリル酸ナトリウム水溶液4499.5g、脱イオン水(イオン交換水)526.1g、ポリエチレングリコールジアクリレート(平均分子量523)7.41g(カルボキシル基含有不飽和単量体に対して0.06モル%)を溶解させて単量体水溶液(a)とした。次にこの単量体水溶液(a)を窒素ガス雰囲気下で、20分間脱気した。
比較例1において、ポリエチレングリコールジアクリレートの使用量をカルボキシル基含有不飽和単量体に対して0.093モル%とし、重合途中でジエチレントリアミン5酢酸・3ナトリウム塩水溶液を加える際、同時に50%リンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレード)10.75g(リンゴ酸はカルボキシル基含有不飽和単量体に対して0.165モル%)を加える以外、比較例1と同様の処理を行い、比較吸水剤(2)を得た。比較吸水剤(2)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。また、比較吸水剤(1)および(2)の可溶分分子量低減率および触感評価結果を表2に示す。
容量2リットルのポリプロピレン製容器に、アクリル酸441.62g、内部架橋剤としてポリエチレングリコールジアクリレート(分子量523)1.763g(カルボキシル基含有不飽和単量体に対して0.055モル%)、1.0重量%のジエチレントリアミン5酢酸・3ナトリウム(DTPA・3Na)水溶液2.70g(カルボキシル基含有不飽和単量体に対して0.005重量%)、48.5重量%の水酸化ナトリウム水溶液181.96g、及び脱イオン水(イオン交換水)363.9gを投入し混合させて、単量体水溶液(b’)を作製した。
比較例3において、ポリエチレングリコールジアクリレートの使用量を2.404g(カルボキシル基含有不飽和単量体に対して0.075モル%)とし、単量体水溶液(b’)に50%リンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレード)10.8g(リンゴ酸はカルボキシル基含有不飽和単量体に対して0.658モル%)を加えた以外、比較例3と同様の処理を行い、吸水剤(1)を得た。吸水剤(1)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。また、吸水剤(1)の吸湿ブロッキング率は、0%であった。
比較例3において、ポリエチレングリコールジアクリレートの使用量を2.244g(カルボキシル基含有不飽和単量体に対して0.070モル%)とし、単量体水溶液(b’)に50%乳酸ナトリウム水溶液(乳酸ナトリウム、ピューラック社製)5.4g(乳酸ナトリウムはカルボキシル基含有不飽和単量体に対して0.394モル%)を加えた以外、比較例3と同様の処理を行い、吸水剤(2)を得た。吸水剤(2)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。
実施例1において、ポリエチレングリコールジアクリレートの使用量を2.18g(カルボキシル基含有不飽和単量体に対して0.068モル%)、リンゴ酸の使用量をカルボキシル基含有不飽和単量体に対して0.329モル%とし、シリカをハイドロタルサイト(製品名DHT-6、協和化学工業株式会社製)に変更した以外、実施例1と同様の処理を行い、吸水剤(3)を得た。吸水剤(3)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。また、比較吸水剤(3)、吸水剤(1)、(2)および(3)の可溶分分子量低減率および触感評価結果を表3に示す。
比較例3において、ポリエチレングリコールジアクリレートの使用量を1.122g(カルボキシル基含有不飽和単量体に対して0.035モル%)とし、表面架橋剤溶液をジエチレングリコールジグリシジルエーテル(商品名デナコールEX-810ナガセケムテックス社製)0.024重量部、炭酸エチレン0.31重量部、プロピレングリコール0.52重量部、脱イオン水(イオン交換水)2.08重量部とし、さらに加熱処理温度を195℃とした以外、比較例3と同等の処理を行い、比較吸水剤(4)を得た。比較吸水剤(4)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。
比較例4において、ポリエチレングリコールジアクリレートの使用量を1.506g(カルボキシル基含有不飽和単量体に対して0.047モル%)とし、単量体水溶液(b’)に50%リンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレード)5.4g(リンゴ酸はカルボキシル基含有不飽和単量体に対して0.329モル%)を加えた以外、比較例4と同様の処理を行い、吸水剤(4)を得た。吸水剤(4)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。
比較例4において、ポリエチレングリコールジアクリレートの使用量を1.442g(カルボキシル基含有不飽和単量体に対して0.045モル%)とし、単量体水溶液(b’)に50%乳酸ナトリウム水溶液(乳酸ナトリウム、ピューラック社製)4.32g(乳酸ナトリウムはカルボキシル基含有不飽和単量体に対して0.315モル%)を加えた以外、比較例4と同様の処理を行い、吸水剤(5)を得た。吸水剤(5)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。
比較例4において、ポリエチレングリコールジアクリレートの使用量を1.442g(カルボキシル基含有不飽和単量体に対して0.045モル%)とし、ゲル粉砕時に投入する脱イオン水を4.32%の乳酸ナトリウム水溶液(50%乳酸ナトリウム、ピューラック社製をイオン交換水により濃度を調整して使用した)に変更した(乳酸ナトリウムはカルボキシル基含有不飽和単量体に対して0.473モル%)以外、比較例4と同様の処理を行い、吸水剤(6)を得た。吸水剤(6)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。
実施例4において、ポリエチレングリコールジアクリレートの使用量を1.442g(カルボキシル基含有不飽和単量体に対して0.045モル%)とし、シリカをリン酸3カルシウム(和光純薬工業株式会社製、CAS No.7758-87-4)に変更した以外、実施例4と同様の処理を行い、吸水剤(7)を得た。吸水剤(7)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。また、比較吸水剤(4)、吸水剤(4)~(7)の可溶分分子量低減率および触感評価結果を表4に示す。
比較例3において、ポリエチレングリコールジアクリレートの使用量を0.801g(カルボキシル基含有不飽和単量体に対して0.025モル%)とし、表面架橋剤溶液をジエチレングリコールジグリシジルエーテル(商品名デナコールEX-810ナガセケムテックス社製)0.017重量部、炭酸エチレン0.22重量部、プロピレングリコール0.36重量部、脱イオン水(イオン交換水)1.46重量部とし、さらに加熱処理温度を190℃とした以外、比較例3と同等の処理を行い、比較吸水剤(5)を得た。比較吸水剤(5)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。
比較例5において、ポリエチレングリコールジアクリレートの使用量を1.026g(カルボキシル基含有不飽和単量体に対して0.032モル%)とし、単量体水溶液(b’)に50%リンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレード)2.70g(リンゴ酸はカルボキシル基含有不飽和単量体に対して0.165モル%)を加えた以外、比較例5と同様の処理を行い、吸水剤(8)を得た。吸水剤(8)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。
比較例5において、ポリエチレングリコールジアクリレートの使用量を1.058g(カルボキシル基含有不飽和単量体に対して0.033モル%)とし、ゲル粉砕時に投入する脱イオン水を3.60%のリンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレードをイオン交換水により濃度を調整して使用した)に変更した(リンゴ酸はカルボキシル基含有不飽和単量体に対して0.329モル%)以外、比較例5と同様の処理を行い、吸水剤(9)を得た。吸水剤(9)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。また、比較吸水剤(5)、吸水剤(8)および(9)の可溶分分子量低減率および触感評価結果を表5に示す。
シグマ型羽根を2本有する内容積10リットルのジャケット付きステンレス型双腕型ニーダーに蓋を付けて形成した反応器中で、アクリル酸436.4g、37質量%アクリル酸ナトリウム水溶液4617.9g、脱イオン水(イオン交換水)402.5g、ポリエチレングリコールジアクリレート(平均分子量523)4.43g(カルボキシル基含有不飽和単量体に対して0.035モル%)を溶解させて単量体水溶液(c)とした。次にこの単量体水溶液(c)を窒素ガス雰囲気下で、20分間脱気した。
比較例6において、ポリエチレングリコールジアクリレートの使用量を5.19g(カルボキシル基含有不飽和単量体に対して0.041モル%)とし、重合開始2分後に50%リンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレード)10.59g(リンゴ酸はカルボキシル基含有不飽和単量体に対して0.165モル%)加えた以外、比較例6と同様の処理を行い、吸水剤(10)を得た。吸水剤(10)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。
比較例6において、ポリエチレングリコールジアクリレートの使用量を5.07g(カルボキシル基含有不飽和単量体に対して0.04モル%)とし、重合開始20分後、重合機から含水ゲル(6)を取り出した。得られた含水ゲル(6)をミートチョッパー(HL-3225N、プレート孔径:12.0mm/レマコム株式会社)を用いてゲル粉砕し、粒子状の含水ゲルとした。このときの含水ゲル(6)の投入量は360g/minであり、含水ゲル(6)の投入と並行して3.6%のリンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレードをイオン交換水により濃度を調整して使用した)を50g/minで添加しながらゲル粉砕を行い、粒子状の含水ゲルを得た以外は、比較例6と同様の処理を行い、吸水剤(11)を得た。吸水剤(11)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。また、比較吸水剤6、吸水剤10および11の可溶分分子量低減率および触感評価結果を表6に示す。
比較例6において、ポリエチレングリコールジアクリレートの使用量を2.53g(カルボキシル基含有不飽和単量体に対して0.020モル%)とした以外、比較例6と同様の処理を行い、比較吸水剤(7)を得た。比較吸水剤(7)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。
比較例7において、ポリエチレングリコールジアクリレートの使用量を3.17g(カルボキシル基含有不飽和単量体に対して0.025モル%)とし、重合開始2分後に50%乳酸ナトリウム水溶液(乳酸ナトリウム、ピューラック社製)6.46g(乳酸ナトリウムはカルボキシル基含有不飽和単量体に対して0.119モル%)加えた以外、比較例7と同様の処理を行い、吸水剤(12)を得た。吸水剤(12)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。また、比較吸水剤7、吸水剤(12)の可溶分分子量低減率および触感評価結果を表7に示す。
容量3リットルのポリプロピレン製容器に、アクリル酸174.57g、37質量%アクリル酸ナトリウム水溶液1847.12g、内部架橋剤としてポリエチレングリコールジアクリレート(分子量523)2.38g(カルボキシル基含有不飽和単量体に対して0.047モル%)、及び脱イオン水(イオン交換水)170.02gを投入し混合させて、単量体水溶液(d’)を作製した。
比較例8において、ポリエチレングリコールジアクリレートの使用量を3.29g(カルボキシル基含有不飽和単量体に対して0.065モル%)とし、単量体水溶液(d’)に50%リンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレード)8.58g(リンゴ酸はカルボキシル基含有不飽和単量体に対して0.329モル%)を加えた以外、比較例8と同様の処理を行い、吸水剤(13)を得た。吸水剤(13)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。
比較例8において、ポリエチレングリコールジアクリレートの使用量を3.29g(カルボキシル基含有不飽和単量体に対して0.065モル%)とし、ゲル粉砕時に投入する脱イオン水を7.20%のリンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレードをイオン交換水により濃度を調整して使用した)に変更した(リンゴ酸はカルボキシル基含有不飽和単量体に対して0.658モル%)以外、比較例8と同様の処理を行い、吸水剤(14)を得た。吸水剤(14)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。また、比較吸水剤(8)、吸水剤(13)および(14)の可溶分分子量低減率および触感評価結果を表8に示す。
比較例8において、ポリエチレングリコールジアクリレートの使用量を4.05g(カルボキシル基含有不飽和単量体に対して0.08モル%)とし、ジフェニル(2,4,6-トリメチルベンゾイル)ホスフィンオキシド0.1gをイルガキュア819 0.1gとした以外、比較例8と同様の処理を行い、比較吸水剤(9)を得た。比較吸水剤(9)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。
比較例9において、ポリエチレングリコールジアクリレートの使用量を5.07g(カルボキシル基含有不飽和単量体に対して0.1モル%)とし、単量体水溶液(d’)に50%乳酸ナトリウム水溶液(乳酸ナトリウム、ピューラック社製)10.3g(乳酸ナトリウムはカルボキシル基含有不飽和単量体に対して0.473モル%)を加えた以外、比較例8と同様の処理を行い、吸水剤(15)を得た。吸水剤(15)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。また、比較吸水剤(9)および吸水剤(15)の可溶分分子量低減率および触感評価結果を表9に示す。
比較例4において、ポリエチレングリコールジアクリレートの使用量を0.930g(カルボキシル基含有不飽和単量体に対して0.029モル%)とし、加熱処理温度を180℃とし、さらに、造粒時にジエチレントリアミン5酢酸・3ナトリウムを吸水性樹脂粒子に加えない以外、比較例4と同等の処理を行い、比較吸水剤(10)を得た。比較吸水剤(10)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。
比較例10において、ポリエチレングリコールジアクリレートの使用量を1.026g(カルボキシル基含有不飽和単量体に対して0.032モル%)とし、単量体水溶液(b’)に50%リンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレード)1.08g(リンゴ酸はカルボキシル基含有不飽和単量体に対して0.066モル%)を加えた以外、比較例10と同様の処理を行い、吸水剤(16)を得た。吸水剤(16)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。また、比較吸水剤(10)および吸水剤(16)の可溶分分子量低減率および触感評価結果を表10に示す。さらに、吸水剤(16)のYI値、Fe/L-as劣化可溶分、CRC、およびAAPを表14に示す。
比較例4において、単量体水溶液(b’)に50%リンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレード)0.05g(リンゴ酸はカルボキシル基含有不飽和単量体に対して0.003モル%)を加え、加熱処理温度を180℃とした以外、比較例4と同等の処理を行い、吸水剤(17)を得た。吸水剤(17)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。さらに、吸水剤(17)のYI値、Fe/L-as劣化可溶分、CRC、およびAAPを表14に示す。
比較例4において、ポリエチレングリコールジアクリレートの使用量を2.564g(カルボキシル基含有不飽和単量体に対して0.080モル%)とし、単量体水溶液(b’)に対してジエチレントリアミン5酢酸・3ナトリウム(DTPA・3Na)水溶液を加えず、50%リンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレード)10.8g(リンゴ酸はカルボキシル基含有不飽和単量体に対して0.658モル%)を加え、加熱処理温度を180℃とし、さらに、ジエチレントリアミン5酢酸・3ナトリウムを吸水性樹脂粒子に加えない以外、比較例4と同等の処理を行い、吸水剤(18)を得た。吸水剤(18)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。また、吸水剤(18)のリンゴ酸含有量は、吸水剤に対して0.98重量%であった。さらに、吸水剤(18)のYI値、Fe/L-as劣化可溶分、CRC、およびAAPを表14に示す。
アクリル酸300重量部、48重量%水酸化ナトリウム水溶液100重量部、ポリエチレングリコールジアクリレート(平均n数9、平均分子量523)0.61重量部、0.1重量%ジエチレントリアミン5酢酸・3ナトリウム水溶液18.4重量部、脱イオン水336.5重量部からなる単量体水溶液(e’)を作製した。
比較例11において、ポリエチレングリコールジアクリレートの使用量を0.78重量部とし、単量体水溶液(e’)に液体リンゴ酸(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレード)2.2重量部を加えた以外、比較例11と同等の処理を行い、吸水剤(19)を得た。吸水剤(19)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。また、吸水剤(19)のリンゴ酸含有量は、吸水剤に対して0.29重量%であった。
アクリル酸300重量部、48重量%水酸化ナトリウム水溶液100重量部、ポリエチレングリコールジアクリレート(平均n数9、平均分子量523)0.61重量部、0.1重量%ジエチレントリアミン5酢酸・3ナトリウム水溶液18.4重量部、脱イオン水360.2重量部からなる単量体水溶液(f’)を作製した。
比較例12において、単量体水溶液(e’)にDL-リンゴ酸(粉末、扶桑化学工業株式会社製、食品添加物グレード)1.8量部を加えた以外、比較例12と同等の処理を行い、吸水剤(20)を得た。吸水剤(20)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。また、吸水剤(20)のリンゴ酸含有量は、吸水剤に対して0.49重量%であった。
比較例3において、ポリエチレングリコールジアクリレートの使用量を2.56g(カルボキシル基含有不飽和単量体に対して0.08モル%)、及び脱イオン水の量を1511gとした以外、比較例3と同様の処理を行い、比較吸水剤(13)を得た。吸水剤(13)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。
比較例3において、ポリエチレングリコールジアクリレートの使用量を3.85g(カルボキシル基含有不飽和単量体に対して0.12モル%)、及び脱イオン水の量を1510gとし、単量体水溶液(b’)に液体リンゴ酸(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレード)10.81g(リンゴ酸はカルボキシル基含有不飽和単量体に対して0.658モル%)を加えた以外、比較例3と同様の処理を行い、吸水剤(21)を得た。吸水剤(21)のCRC、可溶分分子量および可溶分の極限粘度を表1に示す。また、比較吸水剤(13)および吸水剤(21)の可溶分分子量低減率および触感評価結果を表13に示す。










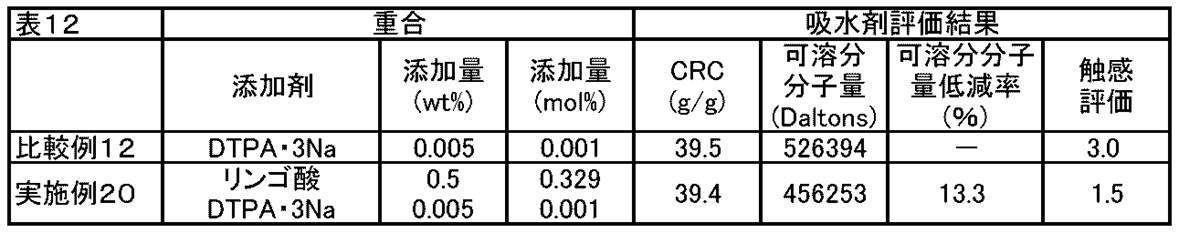
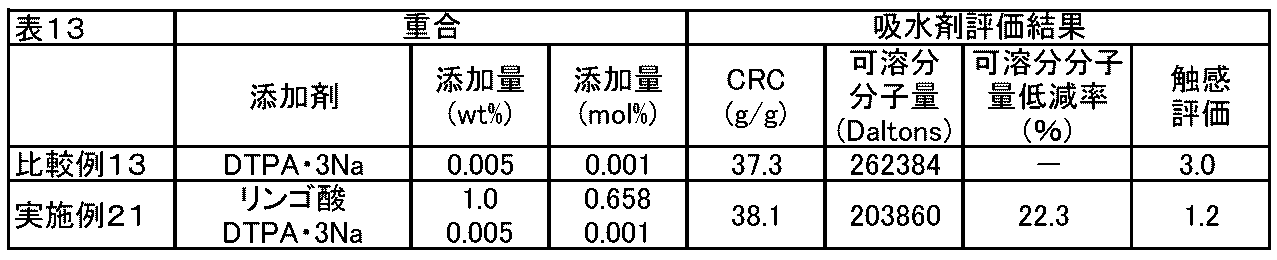
容量2リットルのポリプロピレン製容器に、アクリル酸404.82g、内部架橋剤としてポリエチレングリコールジアクリレート(分子量523)0.881g(カルボキシル基含有不飽和単量体に対して0.03モル%)、1.0重量%のジエチレントリアミン5酢酸・3ナトリウム(DTPA・3Na)水溶液2.48g、48.5重量%の水酸化ナトリウム水溶液166.8g、及び脱イオン水(イオン交換水)332.73gを投入し混合させて、単量体水溶液(f’)を作製した。
製造例1において、表面架橋を下記にしたがって行い、シリカの混合を行わないことにより、吸水性樹脂粒子(15)を得た。
製造例1の方法により比較吸水剤(14)を得た。比較吸水剤(14)の性能を表14に示す。
製造例2の方法により得られた吸水性樹脂粒子(15)を比較吸水剤(15)とする。比較吸水剤(15)の性能を表14に示す。
製造例1において、ポリエチレングリコールジアクリレートの添加量を0.03モル%から0.04モル%に変更し、造粒時のジエチレントリアミン5酢酸・3ナトリウム(DTPA・3Na)の添加量を0.01から0.1重量部に変更し、シリカの混合を行わない以外は、製造例1と同様の操作を行った。得られた吸水性樹脂粒子を比較吸水剤(16)とする。比較吸水剤(16)の性能を表14に示す。
製造例1において、単量体水溶液(f’)に30重量%のD,L-リンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレードをイオン交換水により濃度を調整して使用した)0.33gを加え、造粒時のジエチレントリアミン5酢酸・3ナトリウム(DTPA・3Na)の添加量を0.01から0.03重量部に変更し、シリカの混合を行わない以外は、製造例1と同様の操作を行った。得られた吸水性樹脂粒子を吸水剤(22)とする。吸水剤(22)の性能を表14に示す。
製造例1において、単量体水溶液(f’)のジエチレントリアミン5酢酸・3ナトリウム(DTPA・3Na)の添加量を2.48から9.90gに変更し、単量体水溶液(f’)に30重量%のD,L-リンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレードをイオン交換水により濃度を調整して使用した)0.83gを加え、造粒時のジエチレントリアミン5酢酸・3ナトリウム(DTPA・3Na)の添加量を0.01から0.03重量部に変更し、シリカの混合を行わない以外は、製造例1と同様の操作を行った。得られた吸水性樹脂粒子を吸水剤(23)とする。吸水剤(23)の性能を表14に示す。
製造例1において、ポリエチレングリコールジアクリレートの添加量を0.03モル%から0.032モル%に変更し、単量体水溶液(f’)のジエチレントリアミン5酢酸・3ナトリウム(DTPA・3Na)の添加量を2.48から9.90gに変更し、単量体水溶液(f’)に30重量%のD,L-リンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレードをイオン交換水により濃度を調整して使用した)1.65gを加え、造粒時のジエチレントリアミン5酢酸・3ナトリウム(DTPA・3Na)の添加量を0.01から0.02重量部に変更し、シリカの混合を行わない以外は、製造例1と同様の操作を行った。得られた吸水性樹脂粒子を吸水剤(24)とする。吸水剤(24)の性能を表14に示す。
製造例1において、ポリエチレングリコールジアクリレートの添加量を0.03モル%から0.032モル%に変更し、単量体水溶液(f’)に30重量%のD,L-リンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレードをイオン交換水により濃度を調整して使用した)1.65gを加え、造粒時のジエチレントリアミン5酢酸・3ナトリウム(DTPA・3Na)の添加量を0.01から0.09重量部に変更し、シリカの混合を行わない以外は、製造例1と同様の操作を行った。得られた吸水性樹脂粒子を吸水剤(25)とする。吸水剤(25)の性能を表14に示す。
製造例2において、ポリエチレングリコールジアクリレートの添加量を0.03モル%から0.032モル%に変更し、単量体水溶液(f’)に30重量%のD,L-リンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレードをイオン交換水により濃度を調整して使用した)1.65gを加え、造粒時のジエチレントリアミン5酢酸・3ナトリウム(DTPA・3Na)の添加量を0.01から0.03重量部に変更した以外は、製造例2と同様の操作を行った。得られた吸水性樹脂粒子を吸水剤(26)とする。吸水剤(26)の性能を表14に示す。
製造例1において、ポリエチレングリコールジアクリレートの添加量を0.03モル%から0.045モル%に変更し、単量体水溶液(f’)に30重量%のD,L-リンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレードをイオン交換水により濃度を調整して使用した)1.65gを加えた以外は、製造例1と同様の操作を行った。得られた吸水性樹脂粒子を吸水剤(27)とする。吸水剤(27)の性能を表14に示す。
製造例1において、ポリエチレングリコールジアクリレートの添加量を0.03モル%から0.035モル%に変更し、単量体水溶液(f’)に30重量%のD,L-リンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレードをイオン交換水により濃度を調整して使用した)3.3gを加え、造粒時のジエチレントリアミン5酢酸・3ナトリウム(DTPA・3Na)を0.01から0.03重量部に変更し、シリカの混合を行わない以外は、製造例1と同様の操作を行った。得られた吸水性樹脂粒子を吸水剤(28)とする。吸水剤(28)の性能を表14に示す。
製造例2において、ポリエチレングリコールジアクリレートの添加量を0.03モル%から0.035モル%に変更し、単量体水溶液(f’)のジエチレントリアミン5酢酸・3ナトリウム(DTPA・3Na)の添加量を2.48から9.90gに変更し、単量体水溶液(f’)に30重量%のD,L-リンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレードをイオン交換水により濃度を調整して使用した)3.3gを加え、造粒時のジエチレントリアミン5酢酸・3ナトリウム(DTPA・3Na)の添加量を0.01から0.03重量部に変更し、リン酸三カルシウムを混合した以外は、製造例2と同様の操作を行った。得られた吸水性樹脂粒子を吸水剤(29)とする。吸水剤(29)の性能を表14に示す。
製造例2において、ポリエチレングリコールジアクリレートの添加量を0.03モル%から0.045モル%に変更し、単量体水溶液(f’)に30重量%のD,L-リンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレードをイオン交換水により濃度を調整して使用した)8.25gを加え、造粒時のジエチレントリアミン5酢酸・3ナトリウム(DTPA・3Na)の添加量を0.01から0.03重量部に変更し、ハイドロタルサイト(製品名DHT-6、協和化学工業株式会社製、Mg6Al2(OH)16CO3・4H2O[一般式(1)のx=0.25、m=0.50]、体積平均粒子径0.5μm)を混合した以外は、製造例2と同様の操作を行った。得られた吸水性樹脂粒子を吸水剤(30)とする。吸水剤(30)の性能を表14に示す。
実施例28において、シリカ混合を行う以外は、実施例28と同様の操作を行った。得られた吸水性樹脂粒子を吸水剤(31)とする。吸水剤(31)の性能を表14に示す。
製造例1において、ポリエチレングリコールジアクリレートの添加量を0.03モル%から0.045モル%に変更し、単量体水溶液(f’)のジエチレントリアミン5酢酸・3ナトリウム(DTPA・3Na)の添加量を2.48から9.90gに変更し、単量体水溶液(f’)に30重量%のD,L-リンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレードをイオン交換水により濃度を調整して使用した)8.25gを加え、造粒時のジエチレントリアミン5酢酸・3ナトリウム(DTPA・3Na)の添加量を0.01から0.03重量部に変更し、シリカの混合を行わない以外は、製造例1と同様の操作を行った。得られた吸水性樹脂粒子を吸水剤(32)とする。吸水剤(32)の性能を表14に示す。
製造例1において、ポリエチレングリコールジアクリレートの添加量を0.03モル%から0.045モル%に変更し、単量体水溶液(f’)に30重量%のD,L-リンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレードをイオン交換水により濃度を調整して使用した)8.25gを加え、造粒時のジエチレントリアミン5酢酸・3ナトリウム(DTPA・3Na)の添加量を0.01から0.09重量部に変更し、シリカの混合を行わない以外は、製造例1と同様の操作を行った。得られた吸水性樹脂粒子を吸水剤(33)とする。吸水剤(33)の性能を表14に示す。
製造例1において、ポリエチレングリコールジアクリレートの添加量を0.03モル%から0.055モル%に変更し、単量体水溶液(f’)のジエチレントリアミン5酢酸・3ナトリウム(DTPA・3Na)の添加量を2.48から9.90gに変更し、単量体水溶液(f’)に30重量%のD,L-リンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレードをイオン交換水により濃度を調整して使用した)12.38gを加え、造粒時のジエチレントリアミン5酢酸・3ナトリウム(DTPA・3Na)を0.01から0.03重量部に変更し、シリカの混合を行わない以外は、製造例1と同様の操作を行った。得られた吸水性樹脂粒子を吸水剤(34)とする。吸水剤(34)の性能を表14に示す。
製造例1において、ポリエチレングリコールジアクリレートの添加量を0.03モル%から0.06モル%に変更し、単量体水溶液(f’)に30重量%のD,L-リンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレードをイオン交換水により濃度を調整して使用した)16.5gを加え、造粒時のジエチレントリアミン5酢酸・3ナトリウム(DTPA・3Na)の添加量を0.01から0.03重量部に変更し、シリカの混合を行わない以外は、製造例1と同様の操作を行った。得られた吸水性樹脂粒子を吸水剤(35)とする。吸水剤(35)の性能を表14に示す。
製造例1において、ポリエチレングリコールジアクリレートの添加量を0.03モル%から0.08モル%に変更し、単量体水溶液(f’)にジエチレントリアミン5酢酸・3ナトリウム(DTPA・3Na)を加えず、単量体水溶液(f’)に30重量%のD,L-リンゴ酸水溶液(DL-リンゴ酸、50重量%水溶液、扶桑化学工業株式会社製、食品添加物グレードをイオン交換水により濃度を調整して使用した)33.0gを加え、造粒時のジエチレントリアミン5酢酸・3ナトリウム(DTPA・3Na)の添加量を0.01から0.03重量部に変更し、シリカの混合を行わない以外は、製造例1と同様の操作を行った。得られた吸水性樹脂粒子を吸水剤(36)とする。吸水剤(36)の性能を表14に示す。
製造例1において、ポリエチレングリコールジアクリレートの添加量を0.03モル%から0.035モル%に変更し、単量体水溶液(f’)に30重量%の乳酸水溶液1.65gを加え、造粒時のジエチレントリアミン5酢酸・3ナトリウム(DTPA・3Na)の添加量を0.01から0.05重量部に変更し、シリカの混合を行わない以外は、製造例1と同様の操作を行った。得られた吸水性樹脂粒子を吸水剤(37)とする。吸水剤(37)の性能を表14に示す。
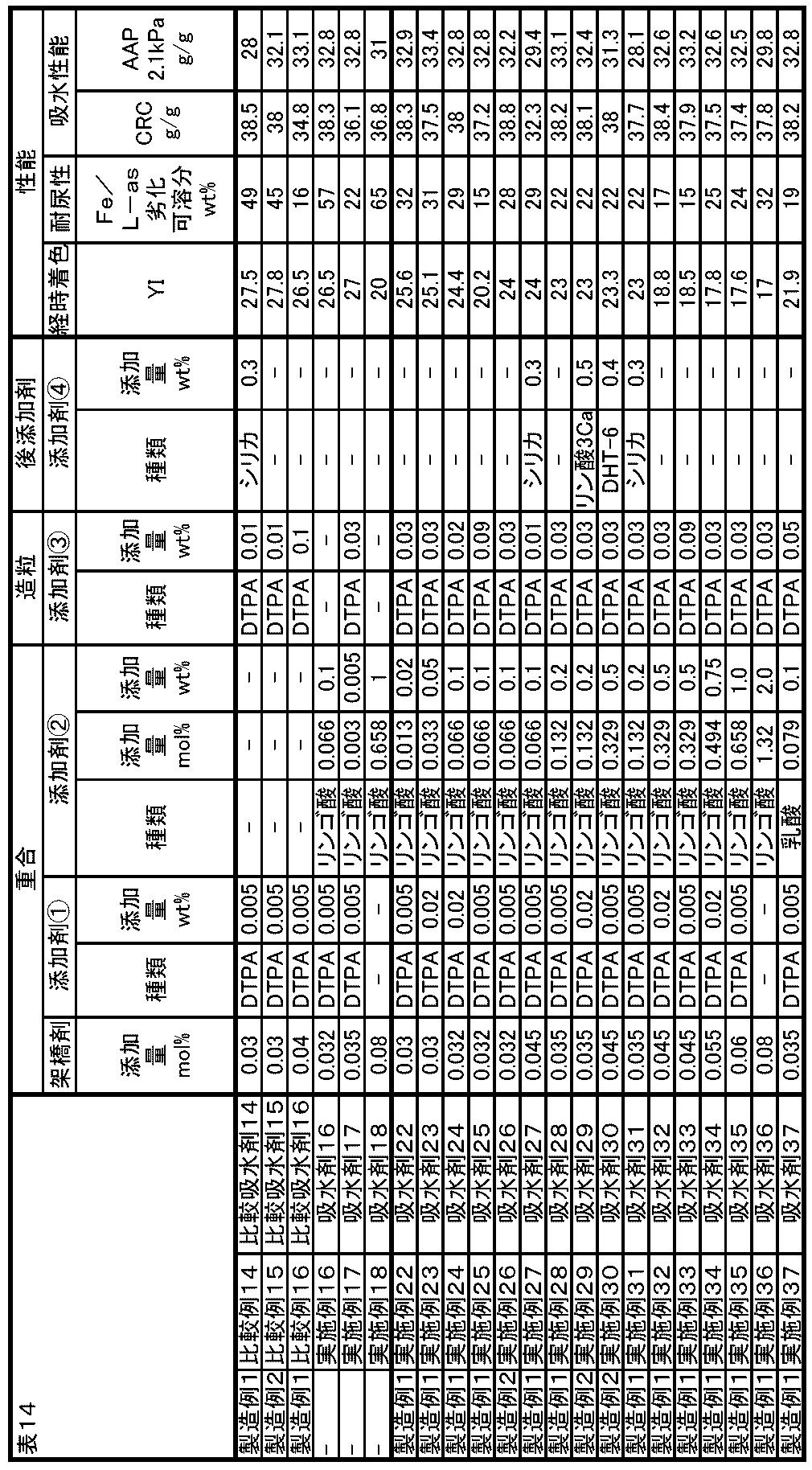
Claims (15)
- アクリル酸(塩)を含む単量体水溶液の重合工程、乾燥工程、および表面架橋工程を含む、遠心分離機保持容量(CRC)が30g/g以上である吸水剤の製造方法であって、該乾燥工程の前にα-ヒドロキシカルボン酸(塩)を添加する工程を含む、方法。
- 前記α-ヒドロキシカルボン酸(塩)は、前記重合工程の前または間に添加される、請求項1に記載の方法。
- 添加される前記α-ヒドロキシカルボン酸(塩)の量は、前記アクリル酸(塩)に対して、0.010~4.0モル%である、請求項1または2に記載の方法。
- 前記α-ヒドロキシカルボン酸(塩)は、リンゴ酸(塩)および乳酸(塩)からなる群から1つ以上選択される、請求項1~3のいずれか1項に記載の方法。
- 前記吸水剤の表面架橋前の遠心分離機保持容量(CRC)が34g/gより大きい、請求項1~4のいずれか1項に記載の方法。
- 前記吸水剤の可溶分量が10重量%以上30重量%以下である、請求項1~5のいずれか1項に記載の方法。
- 前記吸水剤の可溶分分子量が200,000ダルトン以上700,000ダルトン以下である、請求項1~6のいずれか1項に記載の方法。
- 前記重合工程における前記単量体水溶液の重合反応時のピーク温度が85℃以上である、請求項1~7のいずれか1項に記載の方法。
- 前記乾燥工程の後に、キレート剤を添加する工程をさらに含む、請求項1~8のいずれか1項に記載の方法。
- 前記乾燥工程の後に、吸湿流動性改善剤として、二酸化ケイ素、リン酸塩、またはハイドロタルサイトからなる群から選択される少なくとも1つを添加する工程をさらに含む、請求項1~9のいずれか1項に記載の方法。
- 請求項1~10のいずれか1項に記載の方法で得られた吸水剤。
- 請求項11に記載の吸水剤を含む吸収体。
- 請求項12に記載の吸収体を含む衛生物品。
- 吸水剤の可溶分分子量を低減させるための、α-ヒドロキシカルボン酸(塩)の使用。
- 前記α-ヒドロキシカルボン酸(塩)は、リンゴ酸(塩)および乳酸(塩)からなる群から1つ以上選択される、請求項14に記載の使用。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP17775141.9A EP3437730B1 (en) | 2016-03-28 | 2017-03-28 | Method for manufacturing water absorbing agent |
| US16/089,285 US11224857B2 (en) | 2016-03-28 | 2017-03-28 | Method for manufacturing water absorbing agent |
| CN201780032885.5A CN109310984A (zh) | 2016-03-28 | 2017-03-28 | 吸水剂的制造方法 |
| KR1020187031027A KR20180128462A (ko) | 2016-03-28 | 2017-03-28 | 흡수제의 제조 방법 |
| JP2018508102A JP7016798B2 (ja) | 2016-03-28 | 2017-03-28 | 吸水剤の製造方法 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016063762 | 2016-03-28 | ||
| JP2016-063762 | 2016-03-28 | ||
| JP2016-194920 | 2016-09-30 | ||
| JP2016194920 | 2016-09-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017170604A1 true WO2017170604A1 (ja) | 2017-10-05 |
Family
ID=59965760
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2017/012751 WO2017170604A1 (ja) | 2016-03-28 | 2017-03-28 | 吸水剤の製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US11224857B2 (ja) |
| EP (1) | EP3437730B1 (ja) |
| JP (1) | JP7016798B2 (ja) |
| KR (1) | KR20180128462A (ja) |
| CN (1) | CN109310984A (ja) |
| WO (1) | WO2017170604A1 (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019137833A1 (de) | 2018-01-09 | 2019-07-18 | Basf Se | Superabsorbermischungen |
| WO2019154652A1 (de) | 2018-02-06 | 2019-08-15 | Basf Se | Verfahren zur pneumatischen förderung von superabsorberpartikeln |
| JPWO2021095806A1 (ja) * | 2019-11-12 | 2021-05-20 | ||
| WO2022019218A1 (ja) * | 2020-07-22 | 2022-01-27 | 住友精化株式会社 | 吸水性樹脂組成物、吸水性樹脂組成物の製造方法、及び吸水性樹脂粒子の吸水速度低速化方法 |
| US11851346B2 (en) * | 2018-07-03 | 2023-12-26 | U.S. Peroxide, Llc | Creation of an iron product for wastewater treatment |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101570690B1 (ko) * | 2014-02-24 | 2015-11-20 | (주)호원 | 하이브리드 용접기 |
| EP3812422A4 (en) * | 2018-06-22 | 2021-07-07 | Shandong Haoyue New Materials Co., Ltd | RESIN WITH HIGH ABSORPTION AND MANUFACTURING PROCESS FOR IT |
| JP7231522B2 (ja) * | 2019-09-06 | 2023-03-01 | ユニ・チャーム株式会社 | 再生高吸水性ポリマーを製造する方法、再生高吸水性ポリマーを用いて高吸水性ポリマーを製造する方法、及び、再生高吸水性ポリマー |
| CN110987591B (zh) * | 2019-12-16 | 2022-04-05 | 苏州市纤维检验院 | 一种微波消解同时测定纺织品中的砷和汞的方法 |
| CN115066332B (zh) * | 2020-09-08 | 2024-09-13 | 株式会社Lg化学 | 聚合物膜、其制备方法和包括其的聚合物膜层合体 |
| US20220296438A1 (en) * | 2021-03-18 | 2022-09-22 | The Procter & Gamble Company | Method for producing absorbent articles comprising water-absorbing resin |
Citations (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5415256B2 (ja) | 1975-07-02 | 1979-06-13 | ||
| US4893999A (en) | 1985-12-18 | 1990-01-16 | Chemische Fabrik Stockhausen Gmbh | Apparatus for the continuous production of polymers and copolymers of water-soluble monomers |
| US5669894A (en) | 1994-03-29 | 1997-09-23 | The Procter & Gamble Company | Absorbent members for body fluids having good wet integrity and relatively high concentrations of hydrogel-forming absorbent polymer |
| EP0942014A2 (en) | 1998-03-11 | 1999-09-15 | Nippon Shokubai Co., Ltd. | Hydrophilic resin, absorbent article, and acrylic acid for polymerization |
| JP2000327926A (ja) * | 1999-05-25 | 2000-11-28 | Sanyo Chem Ind Ltd | 吸収剤組成物および吸収性物品 |
| US6241928B1 (en) | 1998-04-28 | 2001-06-05 | Nippon Shokubai Co., Ltd. | Method for production of shaped hydrogel of absorbent resin |
| EP1108745A1 (en) | 1999-12-15 | 2001-06-20 | Nippon Shokubai Co., Ltd. | Water-absorbent resin composition |
| US6710141B1 (en) | 1999-11-20 | 2004-03-23 | Basf Aktiengesellschaft | Method for continuously producing cross-linked fine-particle geleous polymerizates |
| WO2005012369A1 (ja) * | 2003-08-04 | 2005-02-10 | Sumitomo Seika Chemicals Co., Ltd. | 吸水性樹脂の製造方法 |
| WO2005016393A1 (en) | 2003-07-31 | 2005-02-24 | Kimberly-Clark Worldwide, Inc. | Absorbent materials and articles |
| WO2005075070A1 (ja) | 2004-02-05 | 2005-08-18 | Nippon Shokubai Co., Ltd. | 粒子状吸水剤及びその製造方法並びに吸水性物品 |
| US20050215734A1 (en) | 2004-03-24 | 2005-09-29 | Yorimichi Dairoku | Method for continuous production of water-absorbent resin |
| US6987151B2 (en) | 2001-09-12 | 2006-01-17 | Dow Global Technologies Inc. | Continuous polymerization process for the manufacture of superabsorbent polymers |
| JP2006055833A (ja) | 2004-03-29 | 2006-03-02 | Nippon Shokubai Co Ltd | 吸水性樹脂を主成分とする粒子状吸水剤 |
| WO2006100300A1 (de) | 2005-03-24 | 2006-09-28 | Basf Aktiengesellschaft | Verfahren zur herstellung wasserabsorbierender polymere |
| US7183456B2 (en) | 2000-09-20 | 2007-02-27 | Nippon Shokubai Co., Ltd. | Water-absorbent resin and production process therefor |
| US7265190B2 (en) | 2002-11-07 | 2007-09-04 | Nippon Shokubai Co., Ltd. | Process and apparatus for production of water-absorbent resin |
| US20080161512A1 (en) | 2005-04-07 | 2008-07-03 | Takaaki Kawano | Production Process of Polyacrylic Acid (Salt) Water-Absorbent Resin |
| US20080194863A1 (en) | 2005-09-07 | 2008-08-14 | Basf Se | Neutralization Process |
| WO2009123197A1 (ja) | 2008-03-31 | 2009-10-08 | 株式会社日本触媒 | 吸水性樹脂を主成分とする粒子状吸水剤の製造方法及びその製造装置 |
| US7638570B2 (en) | 2003-02-10 | 2009-12-29 | Nippon Shokubai Co., Ltd. | Water-absorbing agent |
| JP2010502768A (ja) * | 2006-08-31 | 2010-01-28 | 株式会社日本触媒 | 粒子状吸水剤およびその製造方法 |
| WO2011025013A1 (ja) | 2009-08-28 | 2011-03-03 | 株式会社日本触媒 | 吸水性樹脂の製造方法 |
| WO2011040530A1 (ja) | 2009-09-30 | 2011-04-07 | 株式会社日本触媒 | 粒子状吸水剤及びその製造方法 |
| JP2011517703A (ja) * | 2007-12-19 | 2011-06-16 | ビーエーエスエフ ソシエタス・ヨーロピア | 表面架橋超吸収性物質の製造方法 |
| WO2011111657A1 (ja) | 2010-03-08 | 2011-09-15 | 株式会社日本触媒 | 粒子状含水ゲル状架橋重合体の乾燥方法 |
| WO2011126079A1 (ja) | 2010-04-07 | 2011-10-13 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂粉末の製造方法及びポリアクリル酸(塩)系吸水性樹脂粉末 |
| JP2014094379A (ja) * | 2007-09-28 | 2014-05-22 | Nippon Shokubai Co Ltd | 吸水剤及びその製造方法 |
| JP2015110801A (ja) * | 2015-02-24 | 2015-06-18 | 住友精化株式会社 | 吸水性樹脂、及びその製造方法 |
| JP2016063762A (ja) | 2014-09-24 | 2016-04-28 | 千葉製粉株式会社 | アメリカンドッグ用ミックス、アメリカンドッグ、および、アメリカンドッグの製造方法 |
| JP2016194920A (ja) | 2015-03-31 | 2016-11-17 | 三菱重工業株式会社 | 緊急時プラント状況表示装置および緊急時プラント状況表示方法 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1934171B (zh) * | 2004-03-29 | 2010-09-29 | 株式会社日本触媒 | 具有不规则粉碎形状的颗粒状吸水剂 |
| CN102612379A (zh) | 2009-09-17 | 2012-07-25 | 巴斯夫欧洲公司 | 颜色稳定的超吸收剂 |
| JP6324724B2 (ja) * | 2011-11-15 | 2018-05-16 | 株式会社日本触媒 | 吸水剤組成物及びその製造方法、並びにその保管及び在庫方法 |
| JP5996664B2 (ja) | 2012-10-01 | 2016-09-21 | 株式会社日本触媒 | 多元金属化合物からなる粉塵低減剤、多元金属化合物を含む吸水剤及びその製造方法 |
| CN105916475B (zh) | 2014-01-15 | 2020-12-18 | 株式会社日本触媒 | 吸收性物品的制造方法 |
| KR20160127742A (ko) * | 2014-02-28 | 2016-11-04 | 가부시키가이샤 닛폰 쇼쿠바이 | 폴리(메트)아크릴산(염)계 입자상 흡수제 및 제조 방법 |
-
2017
- 2017-03-28 WO PCT/JP2017/012751 patent/WO2017170604A1/ja active Application Filing
- 2017-03-28 JP JP2018508102A patent/JP7016798B2/ja active Active
- 2017-03-28 CN CN201780032885.5A patent/CN109310984A/zh active Pending
- 2017-03-28 EP EP17775141.9A patent/EP3437730B1/en active Active
- 2017-03-28 KR KR1020187031027A patent/KR20180128462A/ko not_active Application Discontinuation
- 2017-03-28 US US16/089,285 patent/US11224857B2/en active Active
Patent Citations (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5415256B2 (ja) | 1975-07-02 | 1979-06-13 | ||
| US4893999A (en) | 1985-12-18 | 1990-01-16 | Chemische Fabrik Stockhausen Gmbh | Apparatus for the continuous production of polymers and copolymers of water-soluble monomers |
| US5669894A (en) | 1994-03-29 | 1997-09-23 | The Procter & Gamble Company | Absorbent members for body fluids having good wet integrity and relatively high concentrations of hydrogel-forming absorbent polymer |
| EP0942014A2 (en) | 1998-03-11 | 1999-09-15 | Nippon Shokubai Co., Ltd. | Hydrophilic resin, absorbent article, and acrylic acid for polymerization |
| US6241928B1 (en) | 1998-04-28 | 2001-06-05 | Nippon Shokubai Co., Ltd. | Method for production of shaped hydrogel of absorbent resin |
| JP2000327926A (ja) * | 1999-05-25 | 2000-11-28 | Sanyo Chem Ind Ltd | 吸収剤組成物および吸収性物品 |
| US6710141B1 (en) | 1999-11-20 | 2004-03-23 | Basf Aktiengesellschaft | Method for continuously producing cross-linked fine-particle geleous polymerizates |
| EP1108745A1 (en) | 1999-12-15 | 2001-06-20 | Nippon Shokubai Co., Ltd. | Water-absorbent resin composition |
| US7183456B2 (en) | 2000-09-20 | 2007-02-27 | Nippon Shokubai Co., Ltd. | Water-absorbent resin and production process therefor |
| US6987151B2 (en) | 2001-09-12 | 2006-01-17 | Dow Global Technologies Inc. | Continuous polymerization process for the manufacture of superabsorbent polymers |
| US7265190B2 (en) | 2002-11-07 | 2007-09-04 | Nippon Shokubai Co., Ltd. | Process and apparatus for production of water-absorbent resin |
| US7638570B2 (en) | 2003-02-10 | 2009-12-29 | Nippon Shokubai Co., Ltd. | Water-absorbing agent |
| WO2005016393A1 (en) | 2003-07-31 | 2005-02-24 | Kimberly-Clark Worldwide, Inc. | Absorbent materials and articles |
| WO2005012369A1 (ja) * | 2003-08-04 | 2005-02-10 | Sumitomo Seika Chemicals Co., Ltd. | 吸水性樹脂の製造方法 |
| WO2005075070A1 (ja) | 2004-02-05 | 2005-08-18 | Nippon Shokubai Co., Ltd. | 粒子状吸水剤及びその製造方法並びに吸水性物品 |
| US20050215734A1 (en) | 2004-03-24 | 2005-09-29 | Yorimichi Dairoku | Method for continuous production of water-absorbent resin |
| JP2006055833A (ja) | 2004-03-29 | 2006-03-02 | Nippon Shokubai Co Ltd | 吸水性樹脂を主成分とする粒子状吸水剤 |
| WO2006100300A1 (de) | 2005-03-24 | 2006-09-28 | Basf Aktiengesellschaft | Verfahren zur herstellung wasserabsorbierender polymere |
| US20080161512A1 (en) | 2005-04-07 | 2008-07-03 | Takaaki Kawano | Production Process of Polyacrylic Acid (Salt) Water-Absorbent Resin |
| US20080194863A1 (en) | 2005-09-07 | 2008-08-14 | Basf Se | Neutralization Process |
| JP2010502768A (ja) * | 2006-08-31 | 2010-01-28 | 株式会社日本触媒 | 粒子状吸水剤およびその製造方法 |
| JP2014094379A (ja) * | 2007-09-28 | 2014-05-22 | Nippon Shokubai Co Ltd | 吸水剤及びその製造方法 |
| JP2011517703A (ja) * | 2007-12-19 | 2011-06-16 | ビーエーエスエフ ソシエタス・ヨーロピア | 表面架橋超吸収性物質の製造方法 |
| WO2009123197A1 (ja) | 2008-03-31 | 2009-10-08 | 株式会社日本触媒 | 吸水性樹脂を主成分とする粒子状吸水剤の製造方法及びその製造装置 |
| WO2011025013A1 (ja) | 2009-08-28 | 2011-03-03 | 株式会社日本触媒 | 吸水性樹脂の製造方法 |
| WO2011025012A1 (ja) | 2009-08-28 | 2011-03-03 | 株式会社日本触媒 | 吸水性樹脂の製造方法 |
| WO2011040530A1 (ja) | 2009-09-30 | 2011-04-07 | 株式会社日本触媒 | 粒子状吸水剤及びその製造方法 |
| WO2011111657A1 (ja) | 2010-03-08 | 2011-09-15 | 株式会社日本触媒 | 粒子状含水ゲル状架橋重合体の乾燥方法 |
| WO2011126079A1 (ja) | 2010-04-07 | 2011-10-13 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂粉末の製造方法及びポリアクリル酸(塩)系吸水性樹脂粉末 |
| JP2016063762A (ja) | 2014-09-24 | 2016-04-28 | 千葉製粉株式会社 | アメリカンドッグ用ミックス、アメリカンドッグ、および、アメリカンドッグの製造方法 |
| JP2015110801A (ja) * | 2015-02-24 | 2015-06-18 | 住友精化株式会社 | 吸水性樹脂、及びその製造方法 |
| JP2016194920A (ja) | 2015-03-31 | 2016-11-17 | 三菱重工業株式会社 | 緊急時プラント状況表示装置および緊急時プラント状況表示方法 |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019137833A1 (de) | 2018-01-09 | 2019-07-18 | Basf Se | Superabsorbermischungen |
| WO2019154652A1 (de) | 2018-02-06 | 2019-08-15 | Basf Se | Verfahren zur pneumatischen förderung von superabsorberpartikeln |
| US11851346B2 (en) * | 2018-07-03 | 2023-12-26 | U.S. Peroxide, Llc | Creation of an iron product for wastewater treatment |
| JPWO2021095806A1 (ja) * | 2019-11-12 | 2021-05-20 | ||
| WO2021095806A1 (ja) * | 2019-11-12 | 2021-05-20 | 株式会社日本触媒 | 粒子状吸水剤およびその製造方法 |
| JP7296474B2 (ja) | 2019-11-12 | 2023-06-22 | 株式会社日本触媒 | 粒子状吸水剤およびその製造方法 |
| WO2022019218A1 (ja) * | 2020-07-22 | 2022-01-27 | 住友精化株式会社 | 吸水性樹脂組成物、吸水性樹脂組成物の製造方法、及び吸水性樹脂粒子の吸水速度低速化方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20190105633A1 (en) | 2019-04-11 |
| JPWO2017170604A1 (ja) | 2019-02-28 |
| JP7016798B2 (ja) | 2022-02-07 |
| EP3437730A1 (en) | 2019-02-06 |
| EP3437730B1 (en) | 2024-04-17 |
| US11224857B2 (en) | 2022-01-18 |
| KR20180128462A (ko) | 2018-12-03 |
| EP3437730A4 (en) | 2019-10-16 |
| CN109310984A (zh) | 2019-02-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2017170604A1 (ja) | 吸水剤の製造方法 | |
| EP3056268B1 (en) | Particulate water absorber comprising water-absorbing resin as main component and process for manufacturing same | |
| EP2112172B2 (en) | Particulate water-absorbent polymer and process for production thereof | |
| JP5785087B2 (ja) | 粒子状吸水剤及びその製造方法 | |
| EP1869119B1 (en) | Particulate water absorbing agent including polyacrylic acid (polyacrylate) based water absorbing resin as a principal component, method for production thereof, water-absorbent core and absorbing article in which the particulate water absorbing agent is used | |
| EP1903062B1 (en) | Water-absorbent resin, hydropolymer process for producing them, and uses of them | |
| EP2623198B1 (en) | Particulate water-absorbing agent and production method for the same | |
| EP2484702B1 (en) | Polyacrylic acid salt-based water absorbent resin and method for producing same | |
| EP1879930A1 (en) | Production process of polyacrylic acid (salt) water-absorbent resin | |
| JP6980398B2 (ja) | 吸水剤及びその製造方法 | |
| EP2130581A1 (en) | Granular water absorbent comprising water absorbing resin as the main component | |
| WO2014162843A1 (ja) | 吸水剤の製造方法及び吸水剤 | |
| JP2016112475A (ja) | ポリ(メタ)アクリル酸塩系吸水性樹脂を主成分とする吸水剤及びその製造方法 | |
| KR102562511B1 (ko) | 킬레이트제를 포함하는 흡수성 수지의 제조 방법 | |
| WO2020189539A1 (ja) | 粒子状吸水剤の製造方法 | |
| US20220387971A1 (en) | Particulate water-absorbing agent and method for producing the same | |
| JP7352001B2 (ja) | 吸収体、吸水剤および吸水剤の製造方法 | |
| JP6521668B2 (ja) | ポリアクリル酸(塩)系吸水性樹脂の製造方法 | |
| WO2022163849A1 (ja) | 吸水性樹脂の製造方法 | |
| JP2024068208A (ja) | 可溶化ポリマーの製造方法および吸水性樹脂の製造方法 | |
| JP2018065905A (ja) | ポリアクリル酸(塩)系吸水性樹脂粉末の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 2018508102 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20187031027 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2017775141 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2017775141 Country of ref document: EP Effective date: 20181029 |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17775141 Country of ref document: EP Kind code of ref document: A1 |