WO2016006706A1 - テトラヒドロピラ二ルメチル基を有するピリドン誘導体 - Google Patents
テトラヒドロピラ二ルメチル基を有するピリドン誘導体 Download PDFInfo
- Publication number
- WO2016006706A1 WO2016006706A1 PCT/JP2015/069976 JP2015069976W WO2016006706A1 WO 2016006706 A1 WO2016006706 A1 WO 2016006706A1 JP 2015069976 W JP2015069976 W JP 2015069976W WO 2016006706 A1 WO2016006706 A1 WO 2016006706A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- cancer
- pharmaceutically acceptable
- acceptable salt
- pyran
- Prior art date
Links
- UBQKCCHYAOITMY-UHFFFAOYSA-N pyridin-2-ol Chemical class OC1=CC=CC=N1 UBQKCCHYAOITMY-UHFFFAOYSA-N 0.000 title abstract 2
- 125000006173 tetrahydropyranylmethyl group Chemical group 0.000 title abstract 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 477
- 239000013078 crystal Substances 0.000 claims abstract description 124
- 150000003839 salts Chemical class 0.000 claims abstract description 90
- 201000010099 disease Diseases 0.000 claims abstract description 40
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 40
- -1 2-amino-5- {4-[(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl} pyridin-3-yl Chemical group 0.000 claims description 137
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 111
- 206010028980 Neoplasm Diseases 0.000 claims description 100
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 claims description 72
- 229910052802 copper Inorganic materials 0.000 claims description 72
- 239000010949 copper Substances 0.000 claims description 72
- 238000000034 method Methods 0.000 claims description 72
- 238000004519 manufacturing process Methods 0.000 claims description 60
- 238000010586 diagram Methods 0.000 claims description 58
- 201000011510 cancer Diseases 0.000 claims description 57
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 46
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 38
- 108091000080 Phosphotransferase Proteins 0.000 claims description 32
- 102000020233 phosphotransferase Human genes 0.000 claims description 32
- 230000002401 inhibitory effect Effects 0.000 claims description 29
- 229910052757 nitrogen Inorganic materials 0.000 claims description 27
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 claims description 25
- 125000005843 halogen group Chemical group 0.000 claims description 25
- 125000000217 alkyl group Chemical group 0.000 claims description 23
- 239000003814 drug Substances 0.000 claims description 23
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 19
- 239000003112 inhibitor Substances 0.000 claims description 18
- 206010059866 Drug resistance Diseases 0.000 claims description 17
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 17
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 17
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 16
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 16
- 229910019142 PO4 Inorganic materials 0.000 claims description 14
- 239000004480 active ingredient Substances 0.000 claims description 14
- 239000010452 phosphate Substances 0.000 claims description 13
- 206010027476 Metastases Diseases 0.000 claims description 12
- 230000009401 metastasis Effects 0.000 claims description 12
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 claims description 12
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 claims description 11
- 229910052805 deuterium Inorganic materials 0.000 claims description 10
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 claims description 9
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 9
- 201000005202 lung cancer Diseases 0.000 claims description 9
- 208000020816 lung neoplasm Diseases 0.000 claims description 9
- 206010006187 Breast cancer Diseases 0.000 claims description 8
- 208000026310 Breast neoplasm Diseases 0.000 claims description 8
- 206010033128 Ovarian cancer Diseases 0.000 claims description 8
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 8
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 claims description 8
- XTEGVFVZDVNBPF-UHFFFAOYSA-L naphthalene-1,5-disulfonate(2-) Chemical compound C1=CC=C2C(S(=O)(=O)[O-])=CC=CC2=C1S([O-])(=O)=O XTEGVFVZDVNBPF-UHFFFAOYSA-L 0.000 claims description 8
- 206010009944 Colon cancer Diseases 0.000 claims description 7
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical group [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 claims description 7
- 206010014733 Endometrial cancer Diseases 0.000 claims description 7
- 206010014759 Endometrial neoplasm Diseases 0.000 claims description 7
- 229910002651 NO3 Inorganic materials 0.000 claims description 7
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 claims description 7
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 7
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 claims description 7
- 229940077388 benzenesulfonate Drugs 0.000 claims description 7
- 208000029742 colonic neoplasm Diseases 0.000 claims description 7
- 206010017758 gastric cancer Diseases 0.000 claims description 7
- 201000001441 melanoma Diseases 0.000 claims description 7
- 201000011549 stomach cancer Diseases 0.000 claims description 7
- 125000006414 CCl Chemical group ClC* 0.000 claims description 6
- 208000000461 Esophageal Neoplasms Diseases 0.000 claims description 6
- 208000008839 Kidney Neoplasms Diseases 0.000 claims description 6
- 206010030155 Oesophageal carcinoma Diseases 0.000 claims description 6
- 206010060862 Prostate cancer Diseases 0.000 claims description 6
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 6
- 206010038389 Renal cancer Diseases 0.000 claims description 6
- 208000024770 Thyroid neoplasm Diseases 0.000 claims description 6
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 6
- 201000004101 esophageal cancer Diseases 0.000 claims description 6
- 208000005017 glioblastoma Diseases 0.000 claims description 6
- 230000003463 hyperproliferative effect Effects 0.000 claims description 6
- 201000010982 kidney cancer Diseases 0.000 claims description 6
- 208000032839 leukemia Diseases 0.000 claims description 6
- 201000002510 thyroid cancer Diseases 0.000 claims description 6
- 125000006415 CF Chemical group FC* 0.000 claims description 5
- 125000006417 CH Chemical group [H]C* 0.000 claims description 5
- 206010029260 Neuroblastoma Diseases 0.000 claims description 5
- 206010073071 hepatocellular carcinoma Diseases 0.000 claims description 5
- 231100000844 hepatocellular carcinoma Toxicity 0.000 claims description 5
- 201000008968 osteosarcoma Diseases 0.000 claims description 5
- 206010041823 squamous cell carcinoma Diseases 0.000 claims description 5
- XYELEXOQONCGOW-UHFFFAOYSA-N N-[6-[2-amino-5-[3-methoxy-4-(2-morpholin-4-ylethoxy)phenyl]pyridin-3-yl]pyridazin-3-yl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=CC=C(N=N1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)C1=CC(=C(C=C1)OCCN1CCOCC1)OC XYELEXOQONCGOW-UHFFFAOYSA-N 0.000 claims description 4
- 208000002495 Uterine Neoplasms Diseases 0.000 claims description 4
- 239000003937 drug carrier Substances 0.000 claims description 4
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 4
- 239000008194 pharmaceutical composition Substances 0.000 claims description 4
- 206010046766 uterine cancer Diseases 0.000 claims description 4
- RQAATFYANFZYNJ-UHFFFAOYSA-N N-[4-[2-amino-5-(1-ethylpyrazol-4-yl)pyridin-3-yl]-3-fluorophenyl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=C(C=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)F)C=1C=NN(C=1)CC RQAATFYANFZYNJ-UHFFFAOYSA-N 0.000 claims description 3
- MIJFYIHQEYKWGI-UHFFFAOYSA-N N-[4-[2-amino-5-(3,4-dimethoxyphenyl)pyridin-3-yl]phenyl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)C1=CC(=C(C=C1)OC)OC MIJFYIHQEYKWGI-UHFFFAOYSA-N 0.000 claims description 3
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 3
- 150000001975 deuterium Chemical group 0.000 claims description 3
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 3
- 201000002528 pancreatic cancer Diseases 0.000 claims description 3
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 3
- 229910052717 sulfur Inorganic materials 0.000 claims description 3
- 125000004434 sulfur atom Chemical group 0.000 claims description 3
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims description 3
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 2
- YKGMKSIHIVVYKY-UHFFFAOYSA-N dabrafenib mesylate Chemical compound CS(O)(=O)=O.S1C(C(C)(C)C)=NC(C=2C(=C(NS(=O)(=O)C=3C(=CC=CC=3F)F)C=CC=2)F)=C1C1=CC=NC(N)=N1 YKGMKSIHIVVYKY-UHFFFAOYSA-N 0.000 claims description 2
- 230000001747 exhibiting effect Effects 0.000 claims 4
- 125000001424 substituent group Chemical group 0.000 abstract description 6
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 210
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 137
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 118
- 238000006243 chemical reaction Methods 0.000 description 114
- 239000007787 solid Substances 0.000 description 94
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 80
- 239000000203 mixture Substances 0.000 description 72
- 239000000243 solution Substances 0.000 description 68
- 238000001914 filtration Methods 0.000 description 67
- 230000002829 reductive effect Effects 0.000 description 64
- 238000005160 1H NMR spectroscopy Methods 0.000 description 61
- 239000002904 solvent Substances 0.000 description 60
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 59
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 48
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 45
- 230000015572 biosynthetic process Effects 0.000 description 43
- 238000003786 synthesis reaction Methods 0.000 description 43
- 239000000543 intermediate Substances 0.000 description 41
- 238000010898 silica gel chromatography Methods 0.000 description 41
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 38
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 36
- 239000012044 organic layer Substances 0.000 description 35
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 33
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical class CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 31
- 239000007858 starting material Substances 0.000 description 31
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 30
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 30
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 30
- 229960001433 erlotinib Drugs 0.000 description 30
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 29
- 238000012360 testing method Methods 0.000 description 29
- 239000000047 product Substances 0.000 description 28
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 25
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 25
- 238000009835 boiling Methods 0.000 description 25
- 210000004027 cell Anatomy 0.000 description 25
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 24
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 24
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 23
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 22
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 22
- 238000000921 elemental analysis Methods 0.000 description 19
- 230000005764 inhibitory process Effects 0.000 description 19
- 229910000027 potassium carbonate Inorganic materials 0.000 description 19
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 18
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 18
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 18
- 239000011541 reaction mixture Substances 0.000 description 17
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 16
- 239000007864 aqueous solution Substances 0.000 description 16
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 16
- 229910000024 caesium carbonate Inorganic materials 0.000 description 16
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 16
- 239000012046 mixed solvent Substances 0.000 description 16
- FWFGVMYFCODZRD-UHFFFAOYSA-N oxidanium;hydrogen sulfate Chemical compound O.OS(O)(=O)=O FWFGVMYFCODZRD-UHFFFAOYSA-N 0.000 description 16
- 238000002360 preparation method Methods 0.000 description 16
- 239000002253 acid Substances 0.000 description 15
- 125000001037 p-tolyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])[H] 0.000 description 15
- 125000003545 alkoxy group Chemical group 0.000 description 14
- 239000002585 base Substances 0.000 description 14
- 229910052739 hydrogen Inorganic materials 0.000 description 14
- 239000012156 elution solvent Substances 0.000 description 13
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 12
- 238000003756 stirring Methods 0.000 description 12
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 11
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 11
- 235000017557 sodium bicarbonate Nutrition 0.000 description 11
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 10
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 10
- 230000000259 anti-tumor effect Effects 0.000 description 10
- 239000000706 filtrate Substances 0.000 description 10
- 239000012299 nitrogen atmosphere Substances 0.000 description 10
- 235000021317 phosphate Nutrition 0.000 description 10
- 108090000765 processed proteins & peptides Proteins 0.000 description 10
- 229920006395 saturated elastomer Polymers 0.000 description 10
- 239000000126 substance Substances 0.000 description 10
- 125000003944 tolyl group Chemical group 0.000 description 10
- YCGFHHACUCVQPI-UUWRZZSWSA-N N-[4-[2-amino-5-[4-[[(2R)-1,4-dioxan-2-yl]methoxy]-3-methoxyphenyl]pyridin-3-yl]phenyl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)C1=CC(=C(C=C1)OC[C@@H]1OCCOC1)OC YCGFHHACUCVQPI-UUWRZZSWSA-N 0.000 description 9
- 239000003054 catalyst Substances 0.000 description 9
- 239000003153 chemical reaction reagent Substances 0.000 description 9
- 230000000694 effects Effects 0.000 description 9
- 230000014509 gene expression Effects 0.000 description 9
- 150000007529 inorganic bases Chemical class 0.000 description 9
- 150000007530 organic bases Chemical class 0.000 description 9
- 238000001308 synthesis method Methods 0.000 description 9
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 8
- LVEYOSJUKRVCCF-UHFFFAOYSA-N 1,3-bis(diphenylphosphino)propane Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)CCCP(C=1C=CC=CC=1)C1=CC=CC=C1 LVEYOSJUKRVCCF-UHFFFAOYSA-N 0.000 description 8
- 0 C(C1CC234)*1C=C2C3=C1C4=C1 Chemical compound C(C1CC234)*1C=C2C3=C1C4=C1 0.000 description 8
- 239000002246 antineoplastic agent Substances 0.000 description 8
- 230000037396 body weight Effects 0.000 description 8
- 230000008859 change Effects 0.000 description 8
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 8
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 8
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 8
- 239000000758 substrate Substances 0.000 description 8
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 8
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 8
- 108700020796 Oncogene Proteins 0.000 description 7
- GPDHNZNLPKYHCN-DZOOLQPHSA-N [[(z)-(1-cyano-2-ethoxy-2-oxoethylidene)amino]oxy-morpholin-4-ylmethylidene]-dimethylazanium;hexafluorophosphate Chemical compound F[P-](F)(F)(F)(F)F.CCOC(=O)C(\C#N)=N/OC(=[N+](C)C)N1CCOCC1 GPDHNZNLPKYHCN-DZOOLQPHSA-N 0.000 description 7
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 7
- 125000001246 bromo group Chemical group Br* 0.000 description 7
- 239000000872 buffer Substances 0.000 description 7
- 239000003795 chemical substances by application Substances 0.000 description 7
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 7
- 239000002609 medium Substances 0.000 description 7
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 7
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 7
- ZDYVRSLAEXCVBX-UHFFFAOYSA-N pyridinium p-toluenesulfonate Chemical compound C1=CC=[NH+]C=C1.CC1=CC=C(S([O-])(=O)=O)C=C1 ZDYVRSLAEXCVBX-UHFFFAOYSA-N 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- 238000002054 transplantation Methods 0.000 description 7
- KGUMNRYFUFLBGA-UHFFFAOYSA-N 1,4-dihydropyridine-3-carboxamide Chemical compound NC(=O)C1=CNC=CC1 KGUMNRYFUFLBGA-UHFFFAOYSA-N 0.000 description 6
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 6
- VSXLNJTYRFDLNH-UHFFFAOYSA-N 5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxylic acid Chemical compound CC1=CC=C(C=C1)C=1C(C(=CN(C=1)CC1CCOCC1)C(=O)O)=O VSXLNJTYRFDLNH-UHFFFAOYSA-N 0.000 description 6
- 229940126062 Compound A Drugs 0.000 description 6
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 6
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 6
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 6
- 241000699670 Mus sp. Species 0.000 description 6
- 235000011054 acetic acid Nutrition 0.000 description 6
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 6
- 238000001727 in vivo Methods 0.000 description 6
- 239000002198 insoluble material Substances 0.000 description 6
- 239000002953 phosphate buffered saline Substances 0.000 description 6
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 6
- 125000005500 uronium group Chemical group 0.000 description 6
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 5
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 5
- 241000699666 Mus <mouse, genus> Species 0.000 description 5
- 102100037236 Tyrosine-protein kinase receptor UFO Human genes 0.000 description 5
- 239000000654 additive Substances 0.000 description 5
- 229940100198 alkylating agent Drugs 0.000 description 5
- 239000002168 alkylating agent Substances 0.000 description 5
- 230000012292 cell migration Effects 0.000 description 5
- 238000001816 cooling Methods 0.000 description 5
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 5
- 238000004128 high performance liquid chromatography Methods 0.000 description 5
- 229940098779 methanesulfonic acid Drugs 0.000 description 5
- 239000011259 mixed solution Substances 0.000 description 5
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 5
- 125000006239 protecting group Chemical group 0.000 description 5
- 108090000623 proteins and genes Proteins 0.000 description 5
- 229910000029 sodium carbonate Inorganic materials 0.000 description 5
- 239000012453 solvate Substances 0.000 description 5
- 229910052723 transition metal Inorganic materials 0.000 description 5
- 150000003624 transition metals Chemical class 0.000 description 5
- WLPUWLXVBWGYMZ-UHFFFAOYSA-N tricyclohexylphosphine Chemical compound C1CCCCC1P(C1CCCCC1)C1CCCCC1 WLPUWLXVBWGYMZ-UHFFFAOYSA-N 0.000 description 5
- 230000004614 tumor growth Effects 0.000 description 5
- 239000003981 vehicle Substances 0.000 description 5
- 239000011534 wash buffer Substances 0.000 description 5
- IEQAICDLOKRSRL-UHFFFAOYSA-N 2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-(2-dodecoxyethoxy)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethanol Chemical compound CCCCCCCCCCCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCO IEQAICDLOKRSRL-UHFFFAOYSA-N 0.000 description 4
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 4
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 4
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 4
- 101150018445 Axl gene Proteins 0.000 description 4
- 208000007342 Diabetic Nephropathies Diseases 0.000 description 4
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 4
- 101150022345 GAS6 gene Proteins 0.000 description 4
- 239000007995 HEPES buffer Substances 0.000 description 4
- 241001465754 Metazoa Species 0.000 description 4
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 4
- SBQHEYGRLCNAMO-UHFFFAOYSA-N N-[4-[2-amino-5-(3,4-dimethoxyphenyl)pyridin-3-yl]phenyl]-5-bromo-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)Br)CC1CCOCC1)C1=CC(=C(C=C1)OC)OC SBQHEYGRLCNAMO-UHFFFAOYSA-N 0.000 description 4
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 4
- 125000003277 amino group Chemical group 0.000 description 4
- 230000033115 angiogenesis Effects 0.000 description 4
- 125000004432 carbon atom Chemical group C* 0.000 description 4
- 229910002091 carbon monoxide Inorganic materials 0.000 description 4
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 4
- 230000005754 cellular signaling Effects 0.000 description 4
- 238000002425 crystallisation Methods 0.000 description 4
- 230000008025 crystallization Effects 0.000 description 4
- 208000033679 diabetic kidney disease Diseases 0.000 description 4
- 238000005516 engineering process Methods 0.000 description 4
- 230000032050 esterification Effects 0.000 description 4
- 238000005886 esterification reaction Methods 0.000 description 4
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 4
- 235000019253 formic acid Nutrition 0.000 description 4
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 4
- 230000012010 growth Effects 0.000 description 4
- 125000000623 heterocyclic group Chemical group 0.000 description 4
- 230000003834 intracellular effect Effects 0.000 description 4
- 230000009545 invasion Effects 0.000 description 4
- 208000017169 kidney disease Diseases 0.000 description 4
- 239000010410 layer Substances 0.000 description 4
- 239000003446 ligand Substances 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 238000004020 luminiscence type Methods 0.000 description 4
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 4
- 230000035772 mutation Effects 0.000 description 4
- XTEGVFVZDVNBPF-UHFFFAOYSA-N naphthalene-1,5-disulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1S(O)(=O)=O XTEGVFVZDVNBPF-UHFFFAOYSA-N 0.000 description 4
- 239000003960 organic solvent Substances 0.000 description 4
- IVDFJHOHABJVEH-UHFFFAOYSA-N pinacol Chemical compound CC(C)(O)C(C)(C)O IVDFJHOHABJVEH-UHFFFAOYSA-N 0.000 description 4
- 235000011056 potassium acetate Nutrition 0.000 description 4
- 229940002612 prodrug Drugs 0.000 description 4
- 239000000651 prodrug Substances 0.000 description 4
- 102000004169 proteins and genes Human genes 0.000 description 4
- 238000006467 substitution reaction Methods 0.000 description 4
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 4
- 210000001519 tissue Anatomy 0.000 description 4
- 125000002827 triflate group Chemical group FC(S(=O)(=O)O*)(F)F 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- 210000004509 vascular smooth muscle cell Anatomy 0.000 description 4
- 238000001262 western blot Methods 0.000 description 4
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 description 4
- ABDDQTDRAHXHOC-QMMMGPOBSA-N 1-[(7s)-5,7-dihydro-4h-thieno[2,3-c]pyran-7-yl]-n-methylmethanamine Chemical compound CNC[C@@H]1OCCC2=C1SC=C2 ABDDQTDRAHXHOC-QMMMGPOBSA-N 0.000 description 3
- GXXXUZIRGXYDFP-UHFFFAOYSA-N 2-(4-methylphenyl)acetic acid Chemical compound CC1=CC=C(CC(O)=O)C=C1 GXXXUZIRGXYDFP-UHFFFAOYSA-N 0.000 description 3
- PYZATJISPIITJV-UHFFFAOYSA-N 5-bromo-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxylic acid Chemical compound BrC=1C(C(=CN(C=1)CC1CCOCC1)C(=O)O)=O PYZATJISPIITJV-UHFFFAOYSA-N 0.000 description 3
- JPVNIXANASYDQI-UHFFFAOYSA-N COC=1C=C(C=CC=1OC)C=1C=C(C(=NC=1)N)B1OC(C(O1)(C)C)(C)C Chemical compound COC=1C=C(C=CC=1OC)C=1C=C(C(=NC=1)N)B1OC(C(O1)(C)C)(C)C JPVNIXANASYDQI-UHFFFAOYSA-N 0.000 description 3
- 206010018366 Glomerulonephritis acute Diseases 0.000 description 3
- 206010018367 Glomerulonephritis chronic Diseases 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 3
- 241000699660 Mus musculus Species 0.000 description 3
- ZSXGLVDWWRXATF-UHFFFAOYSA-N N,N-dimethylformamide dimethyl acetal Chemical compound COC(OC)N(C)C ZSXGLVDWWRXATF-UHFFFAOYSA-N 0.000 description 3
- MVHPGOCNSYOLJS-UHFFFAOYSA-N N-[6-[2-amino-5-(3,4-dimethoxyphenyl)pyridin-3-yl]pyridin-3-yl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=NC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)C1=CC(=C(C=C1)OC)OC MVHPGOCNSYOLJS-UHFFFAOYSA-N 0.000 description 3
- 241000283973 Oryctolagus cuniculus Species 0.000 description 3
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- 206010048955 Retinal toxicity Diseases 0.000 description 3
- 206010039491 Sarcoma Diseases 0.000 description 3
- 230000001154 acute effect Effects 0.000 description 3
- 230000000996 additive effect Effects 0.000 description 3
- 229940024606 amino acid Drugs 0.000 description 3
- 230000000903 blocking effect Effects 0.000 description 3
- 230000004709 cell invasion Effects 0.000 description 3
- 239000006285 cell suspension Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 230000002301 combined effect Effects 0.000 description 3
- 125000000753 cycloalkyl group Chemical group 0.000 description 3
- 230000007850 degeneration Effects 0.000 description 3
- 238000012217 deletion Methods 0.000 description 3
- 230000037430 deletion Effects 0.000 description 3
- NSNHWTBQMQIDCF-UHFFFAOYSA-N dihydrate;hydrochloride Chemical compound O.O.Cl NSNHWTBQMQIDCF-UHFFFAOYSA-N 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 229940071111 erlotinib 25 mg Drugs 0.000 description 3
- GDXNSFVXXGAUMU-UHFFFAOYSA-N ethyl 4-(4-methylphenyl)-3-oxobutanoate Chemical compound CCOC(=O)CC(=O)CC1=CC=C(C)C=C1 GDXNSFVXXGAUMU-UHFFFAOYSA-N 0.000 description 3
- UIKUIBBGEJWAIP-UHFFFAOYSA-N ethyl 5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxylate Chemical compound CC1=CC=C(C=C1)C=1C(C(=CN(C=1)CC1CCOCC1)C(=O)OCC)=O UIKUIBBGEJWAIP-UHFFFAOYSA-N 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 3
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 3
- 238000011580 nude mouse model Methods 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 3
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 3
- 230000026731 phosphorylation Effects 0.000 description 3
- 238000006366 phosphorylation reaction Methods 0.000 description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 3
- 231100000385 retinal toxicity Toxicity 0.000 description 3
- 239000000523 sample Substances 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 239000004055 small Interfering RNA Substances 0.000 description 3
- 230000004083 survival effect Effects 0.000 description 3
- 238000010998 test method Methods 0.000 description 3
- 229940124597 therapeutic agent Drugs 0.000 description 3
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 3
- 210000004881 tumor cell Anatomy 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- UGOMMVLRQDMAQQ-UHFFFAOYSA-N xphos Chemical group CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 UGOMMVLRQDMAQQ-UHFFFAOYSA-N 0.000 description 3
- PFNXEYVLRGZYLD-LJCGDPPZSA-N (2R)-2-[[4-bromo-2,3,5-trideuterio-6-(trideuteriomethoxy)phenoxy]methyl]-1,4-dioxane Chemical compound BrC1=C(C(=C(C(=C1[2H])[2H])OC[C@@H]1OCCOC1)OC([2H])([2H])[2H])[2H] PFNXEYVLRGZYLD-LJCGDPPZSA-N 0.000 description 2
- RCVDPBFUMYUKPB-UHFFFAOYSA-N (3,4-dimethoxyphenyl)boronic acid Chemical compound COC1=CC=C(B(O)O)C=C1OC RCVDPBFUMYUKPB-UHFFFAOYSA-N 0.000 description 2
- KMSKQZKKOZQFFG-YXRRJAAWSA-N (7S,9S)-7-[[(2R,4S,5S,6S)-4-amino-6-methyl-5-[[(2R)-2-oxanyl]oxy]-2-oxanyl]oxy]-6,9,11-trihydroxy-9-(2-hydroxy-1-oxoethyl)-4-methoxy-8,10-dihydro-7H-tetracene-5,12-dione Chemical compound O([C@H]1[C@@H](N)C[C@@H](O[C@H]1C)O[C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@@H]1CCCCO1 KMSKQZKKOZQFFG-YXRRJAAWSA-N 0.000 description 2
- MOWXJLUYGFNTAL-DEOSSOPVSA-N (s)-[2-chloro-4-fluoro-5-(7-morpholin-4-ylquinazolin-4-yl)phenyl]-(6-methoxypyridazin-3-yl)methanol Chemical compound N1=NC(OC)=CC=C1[C@@H](O)C1=CC(C=2C3=CC=C(C=C3N=CN=2)N2CCOCC2)=C(F)C=C1Cl MOWXJLUYGFNTAL-DEOSSOPVSA-N 0.000 description 2
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 2
- FPIRBHDGWMWJEP-UHFFFAOYSA-N 1-hydroxy-7-azabenzotriazole Chemical compound C1=CN=C2N(O)N=NC2=C1 FPIRBHDGWMWJEP-UHFFFAOYSA-N 0.000 description 2
- QVKJPKIMVKZJGM-UHFFFAOYSA-N 2-(4-cyclopropyl-2-fluorophenyl)acetic acid Chemical compound C1(CC1)C1=CC(=C(C=C1)CC(=O)O)F QVKJPKIMVKZJGM-UHFFFAOYSA-N 0.000 description 2
- ICSNLGPSRYBMBD-UHFFFAOYSA-N 2-aminopyridine Chemical compound NC1=CC=CC=N1 ICSNLGPSRYBMBD-UHFFFAOYSA-N 0.000 description 2
- SDTMFDGELKWGFT-UHFFFAOYSA-N 2-methylpropan-2-olate Chemical compound CC(C)(C)[O-] SDTMFDGELKWGFT-UHFFFAOYSA-N 0.000 description 2
- 125000003762 3,4-dimethoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C(OC([H])([H])[H])C([H])=C1* 0.000 description 2
- JHUUPUMBZGWODW-UHFFFAOYSA-N 3,6-dihydro-1,2-dioxine Chemical compound C1OOCC=C1 JHUUPUMBZGWODW-UHFFFAOYSA-N 0.000 description 2
- PYIVNGBQMPHEDY-QGZVFWFLSA-N 3-(4-amino-2-fluorophenyl)-5-[4-[[(2R)-1,4-dioxan-2-yl]methoxy]-3-methoxyphenyl]pyridin-2-amine Chemical compound NC1=CC(=C(C=C1)C=1C(=NC=C(C=1)C1=CC(=C(C=C1)OC[C@@H]1OCCOC1)OC)N)F PYIVNGBQMPHEDY-QGZVFWFLSA-N 0.000 description 2
- VKMFKDFUOFQDNT-UHFFFAOYSA-N 3-(4-aminophenyl)-5-(3,4-dimethoxyphenyl)pyrazin-2-amine Chemical compound NC1=CC=C(C=C1)C=1C(=NC=C(N=1)C1=CC(=C(C=C1)OC)OC)N VKMFKDFUOFQDNT-UHFFFAOYSA-N 0.000 description 2
- BYHQTRFJOGIQAO-GOSISDBHSA-N 3-(4-bromophenyl)-8-[(2R)-2-hydroxypropyl]-1-[(3-methoxyphenyl)methyl]-1,3,8-triazaspiro[4.5]decan-2-one Chemical compound C[C@H](CN1CCC2(CC1)CN(C(=O)N2CC3=CC(=CC=C3)OC)C4=CC=C(C=C4)Br)O BYHQTRFJOGIQAO-GOSISDBHSA-N 0.000 description 2
- QAOPVULVILQLPY-UHFFFAOYSA-N 3-(5-amino-3-fluoropyridin-2-yl)-5-(3,4-dimethoxyphenyl)pyridin-2-amine Chemical compound COC=1C=C(C=CC=1OC)C=1C=C(C(=NC=1)N)C1=NC=C(C=C1F)N QAOPVULVILQLPY-UHFFFAOYSA-N 0.000 description 2
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 2
- WNEODWDFDXWOLU-QHCPKHFHSA-N 3-[3-(hydroxymethyl)-4-[1-methyl-5-[[5-[(2s)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl]amino]-6-oxopyridin-3-yl]pyridin-2-yl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one Chemical compound C([C@@H](N(CC1)C=2C=NC(NC=3C(N(C)C=C(C=3)C=3C(=C(N4C(C5=CC=6CC(C)(C)CC=6N5CC4)=O)N=CC=3)CO)=O)=CC=2)C)N1C1COC1 WNEODWDFDXWOLU-QHCPKHFHSA-N 0.000 description 2
- HUTRLPYFPQSQBA-UHFFFAOYSA-N 3-bromo-5-(3,4-dimethoxyphenyl)pyridin-2-amine Chemical compound C1=C(OC)C(OC)=CC=C1C1=CN=C(N)C(Br)=C1 HUTRLPYFPQSQBA-UHFFFAOYSA-N 0.000 description 2
- CCADMPWCDJNWMQ-UHFFFAOYSA-N 3-chloro-5-(3,4-dimethoxyphenyl)pyrazin-2-amine Chemical compound ClC=1C(=NC=C(N=1)C1=CC(=C(C=C1)OC)OC)N CCADMPWCDJNWMQ-UHFFFAOYSA-N 0.000 description 2
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 2
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 2
- LMOOYAKLEOGKJR-UHFFFAOYSA-N 4-(bromomethyl)oxane Chemical compound BrCC1CCOCC1 LMOOYAKLEOGKJR-UHFFFAOYSA-N 0.000 description 2
- LLPFPLXIDMRGBA-UHFFFAOYSA-N 5-(4-bromophenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxylic acid Chemical compound BrC1=CC=C(C=C1)C=1C(C(=CN(C=1)CC1CCOCC1)C(=O)O)=O LLPFPLXIDMRGBA-UHFFFAOYSA-N 0.000 description 2
- NRTSMZFIDWZGFJ-UHFFFAOYSA-N 5-(4-cyclopropyl-2-fluorophenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxylic acid Chemical compound C1(CC1)C1=CC(=C(C=C1)C=1C(C(=CN(C=1)CC1CCOCC1)C(=O)O)=O)F NRTSMZFIDWZGFJ-UHFFFAOYSA-N 0.000 description 2
- NGUDGIXCTSONFQ-UHFFFAOYSA-N 5-(5-methylpyridin-2-yl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxylic acid Chemical compound CC=1C=CC(=NC=1)C1=CN(C=C(C1=O)C(=O)O)CC1CCOCC1 NGUDGIXCTSONFQ-UHFFFAOYSA-N 0.000 description 2
- ZYDXJCGHPMGSOG-UHFFFAOYSA-N 5-bromo-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-amine Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC(Br)=CN=C1N ZYDXJCGHPMGSOG-UHFFFAOYSA-N 0.000 description 2
- WOVKYSAHUYNSMH-RRKCRQDMSA-N 5-bromodeoxyuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(Br)=C1 WOVKYSAHUYNSMH-RRKCRQDMSA-N 0.000 description 2
- KCBWAFJCKVKYHO-UHFFFAOYSA-N 6-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-1-[[4-[1-propan-2-yl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methyl]pyrazolo[3,4-d]pyrimidine Chemical compound C1(CC1)C1=NC=NC(=C1C1=NC=C2C(=N1)N(N=C2)CC1=CC=C(C=C1)C=1N(C=C(N=1)C(F)(F)F)C(C)C)OC KCBWAFJCKVKYHO-UHFFFAOYSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- 206010003210 Arteriosclerosis Diseases 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 201000001320 Atherosclerosis Diseases 0.000 description 2
- 208000037260 Atherosclerotic Plaque Diseases 0.000 description 2
- 208000031648 Body Weight Changes Diseases 0.000 description 2
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 2
- UYCOULZBXKJNBG-AWEZNQCLSA-N COC1=C(OC[C@H]2OCCOC2)C=CC(=C1)B1OC(C(O1)(C)C)(C)C Chemical compound COC1=C(OC[C@H]2OCCOC2)C=CC(=C1)B1OC(C(O1)(C)C)(C)C UYCOULZBXKJNBG-AWEZNQCLSA-N 0.000 description 2
- 201000009030 Carcinoma Diseases 0.000 description 2
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 2
- 206010012689 Diabetic retinopathy Diseases 0.000 description 2
- BUDQDWGNQVEFAC-UHFFFAOYSA-N Dihydropyran Chemical compound C1COC=CC1 BUDQDWGNQVEFAC-UHFFFAOYSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 101150039808 Egfr gene Proteins 0.000 description 2
- 241000196324 Embryophyta Species 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 108010024636 Glutathione Proteins 0.000 description 2
- 102000005720 Glutathione transferase Human genes 0.000 description 2
- 108010070675 Glutathione transferase Proteins 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 208000007766 Kaposi sarcoma Diseases 0.000 description 2
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 2
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 2
- DPUWYVAVBSHSDS-UHFFFAOYSA-N N-(6-chloropyridazin-3-yl)-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound ClC1=CC=C(N=N1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1 DPUWYVAVBSHSDS-UHFFFAOYSA-N 0.000 description 2
- NATXQLRQRJHHRS-UHFFFAOYSA-N N-[4-(2-amino-5-bromopyridin-3-yl)phenyl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)Br NATXQLRQRJHHRS-UHFFFAOYSA-N 0.000 description 2
- HVZYRAXHTSGBOE-UHFFFAOYSA-N N-[4-[2-amino-5-(3,4-dimethoxyphenyl)pyridin-3-yl]phenyl]-5-(4-cyclopropylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C1CC1)CC1CCOCC1)C1=CC(=C(C=C1)OC)OC HVZYRAXHTSGBOE-UHFFFAOYSA-N 0.000 description 2
- PHACAAIWJAWPHA-UHFFFAOYSA-N N-[4-[2-amino-5-(3,4-dimethoxyphenyl)pyridin-3-yl]phenyl]-5-(5-methylpyridin-2-yl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C=1C(C(=CN(C=1)CC1CCOCC1)C1=NC=C(C=C1)C)=O)C1=CC(=C(C=C1)OC)OC PHACAAIWJAWPHA-UHFFFAOYSA-N 0.000 description 2
- KMPGAESRHAKXEH-UHFFFAOYSA-N N-[4-[2-amino-5-(3-fluoro-4-formylphenyl)pyridin-3-yl]phenyl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)C1=CC(=C(C=C1)C=O)F KMPGAESRHAKXEH-UHFFFAOYSA-N 0.000 description 2
- YQGUQZWOHCDPTK-UHFFFAOYSA-N N-[4-[2-amino-5-[1-(oxan-4-yl)pyrazol-4-yl]pyridin-3-yl]-3-fluorophenyl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=C(C=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)F)C=1C=NN(C=1)C1CCOCC1 YQGUQZWOHCDPTK-UHFFFAOYSA-N 0.000 description 2
- VJBZMEXUEJVLAU-UHFFFAOYSA-N N-[4-[2-amino-5-[3-fluoro-4-(morpholin-4-ylmethyl)phenyl]pyridin-3-yl]phenyl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound C1(C=2C=C(C3=CC(F)=C(CN4CCOCC4)C=C3)C=NC=2N)=CC=C(NC(=O)C=2C(=O)C(C3=CC=C(C)C=C3)=CN(C=2)CC2CCOCC2)C=C1 VJBZMEXUEJVLAU-UHFFFAOYSA-N 0.000 description 2
- 150000001204 N-oxides Chemical class 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 229920001213 Polysorbate 20 Polymers 0.000 description 2
- 201000004681 Psoriasis Diseases 0.000 description 2
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 2
- 208000017442 Retinal disease Diseases 0.000 description 2
- 206010038923 Retinopathy Diseases 0.000 description 2
- 229920002684 Sepharose Polymers 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- 108091027967 Small hairpin RNA Proteins 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 2
- 108090000190 Thrombin Proteins 0.000 description 2
- 208000007536 Thrombosis Diseases 0.000 description 2
- 229910052770 Uranium Chemical group 0.000 description 2
- 208000005475 Vascular calcification Diseases 0.000 description 2
- LXRZVMYMQHNYJB-UNXOBOICSA-N [(1R,2S,4R)-4-[[5-[4-[(1R)-7-chloro-1,2,3,4-tetrahydroisoquinolin-1-yl]-5-methylthiophene-2-carbonyl]pyrimidin-4-yl]amino]-2-hydroxycyclopentyl]methyl sulfamate Chemical compound CC1=C(C=C(S1)C(=O)C1=C(N[C@H]2C[C@H](O)[C@@H](COS(N)(=O)=O)C2)N=CN=C1)[C@@H]1NCCC2=C1C=C(Cl)C=C2 LXRZVMYMQHNYJB-UNXOBOICSA-N 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 2
- 230000003213 activating effect Effects 0.000 description 2
- 229910052783 alkali metal Inorganic materials 0.000 description 2
- 125000003282 alkyl amino group Chemical group 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 230000000340 anti-metabolite Effects 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 229940100197 antimetabolite Drugs 0.000 description 2
- 239000002256 antimetabolite Substances 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- 230000004900 autophagic degradation Effects 0.000 description 2
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 2
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical class OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 2
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 2
- 230000004579 body weight change Effects 0.000 description 2
- 229910052796 boron Inorganic materials 0.000 description 2
- 230000031709 bromination Effects 0.000 description 2
- 238000005893 bromination reaction Methods 0.000 description 2
- 238000004364 calculation method Methods 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 229960004562 carboplatin Drugs 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- YCIMNLLNPGFGHC-UHFFFAOYSA-N catechol Chemical compound OC1=CC=CC=C1O YCIMNLLNPGFGHC-UHFFFAOYSA-N 0.000 description 2
- 230000024245 cell differentiation Effects 0.000 description 2
- 230000032823 cell division Effects 0.000 description 2
- 230000010261 cell growth Effects 0.000 description 2
- 239000013592 cell lysate Substances 0.000 description 2
- 239000003638 chemical reducing agent Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 230000001684 chronic effect Effects 0.000 description 2
- 229960004316 cisplatin Drugs 0.000 description 2
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 239000000470 constituent Substances 0.000 description 2
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- WLVKDFJTYKELLQ-UHFFFAOYSA-N cyclopropylboronic acid Chemical compound OB(O)C1CC1 WLVKDFJTYKELLQ-UHFFFAOYSA-N 0.000 description 2
- WMKGGPCROCCUDY-PHEQNACWSA-N dibenzylideneacetone Chemical compound C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 WMKGGPCROCCUDY-PHEQNACWSA-N 0.000 description 2
- 125000006222 dimethylaminomethyl group Chemical group [H]C([H])([H])N(C([H])([H])[H])C([H])([H])* 0.000 description 2
- 125000000532 dioxanyl group Chemical group 0.000 description 2
- 229960004679 doxorubicin Drugs 0.000 description 2
- 229940121647 egfr inhibitor Drugs 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 229940088598 enzyme Drugs 0.000 description 2
- 108700021358 erbB-1 Genes Proteins 0.000 description 2
- CCIVGXIOQKPBKL-UHFFFAOYSA-N ethanesulfonic acid Chemical class CCS(O)(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-N 0.000 description 2
- BRYNITBKFYVFBV-UHFFFAOYSA-N ethyl 4-(4-bromophenyl)-3-oxobutanoate Chemical compound CCOC(=O)CC(=O)CC1=CC=C(Br)C=C1 BRYNITBKFYVFBV-UHFFFAOYSA-N 0.000 description 2
- PYUFYLOTFMNRSN-UHFFFAOYSA-N ethyl 4-(5-methylpyridin-2-yl)-3-oxobutanoate Chemical compound CC=1C=CC(=NC=1)CC(CC(=O)OCC)=O PYUFYLOTFMNRSN-UHFFFAOYSA-N 0.000 description 2
- NHIDERHRUULCAN-UHFFFAOYSA-N ethyl 5-(4-bromophenyl)-4-oxo-1H-pyridine-3-carboxylate Chemical compound BrC1=CC=C(C=C1)C=1C(C(=CNC=1)C(=O)OCC)=O NHIDERHRUULCAN-UHFFFAOYSA-N 0.000 description 2
- 239000012091 fetal bovine serum Substances 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 125000001153 fluoro group Chemical group F* 0.000 description 2
- 108020001507 fusion proteins Proteins 0.000 description 2
- 102000037865 fusion proteins Human genes 0.000 description 2
- 210000004211 gastric acid Anatomy 0.000 description 2
- 229960003180 glutathione Drugs 0.000 description 2
- 150000004820 halides Chemical class 0.000 description 2
- 201000011066 hemangioma Diseases 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 239000005457 ice water Substances 0.000 description 2
- 238000002513 implantation Methods 0.000 description 2
- CGIGDMFJXJATDK-UHFFFAOYSA-N indomethacin Chemical compound CC1=C(CC(O)=O)C2=CC(OC)=CC=C2N1C(=O)C1=CC=C(Cl)C=C1 CGIGDMFJXJATDK-UHFFFAOYSA-N 0.000 description 2
- 230000033001 locomotion Effects 0.000 description 2
- 238000007477 logistic regression Methods 0.000 description 2
- 230000007774 longterm Effects 0.000 description 2
- 229910001629 magnesium chloride Inorganic materials 0.000 description 2
- 230000036210 malignancy Effects 0.000 description 2
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 2
- LSEMVPNOOZDQLJ-UHFFFAOYSA-N methyl 5-bromo-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxylate Chemical compound BrC=1C(C(=CN(C=1)CC1CCOCC1)C(=O)OC)=O LSEMVPNOOZDQLJ-UHFFFAOYSA-N 0.000 description 2
- GRVDJDISBSALJP-UHFFFAOYSA-N methyloxidanyl Chemical group [O]C GRVDJDISBSALJP-UHFFFAOYSA-N 0.000 description 2
- 210000000822 natural killer cell Anatomy 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 230000002018 overexpression Effects 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 230000004962 physiological condition Effects 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 125000004193 piperazinyl group Chemical group 0.000 description 2
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 2
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 2
- 229910000160 potassium phosphate Inorganic materials 0.000 description 2
- 235000011009 potassium phosphates Nutrition 0.000 description 2
- WVUCPRGADMCTBN-UHFFFAOYSA-M potassium;3-ethoxy-3-oxopropanoate Chemical compound [K+].CCOC(=O)CC([O-])=O WVUCPRGADMCTBN-UHFFFAOYSA-M 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 238000001144 powder X-ray diffraction data Methods 0.000 description 2
- 238000011533 pre-incubation Methods 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 230000035755 proliferation Effects 0.000 description 2
- 239000012264 purified product Substances 0.000 description 2
- 238000010791 quenching Methods 0.000 description 2
- 230000000171 quenching effect Effects 0.000 description 2
- 239000011535 reaction buffer Substances 0.000 description 2
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 208000037803 restenosis Diseases 0.000 description 2
- 210000001525 retina Anatomy 0.000 description 2
- 206010039073 rheumatoid arthritis Diseases 0.000 description 2
- 230000035945 sensitivity Effects 0.000 description 2
- 239000002002 slurry Substances 0.000 description 2
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 2
- 230000021595 spermatogenesis Effects 0.000 description 2
- 208000010110 spontaneous platelet aggregation Diseases 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 230000002194 synthesizing effect Effects 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- 229960004072 thrombin Drugs 0.000 description 2
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 2
- 235000019798 tripotassium phosphate Nutrition 0.000 description 2
- 239000003656 tris buffered saline Substances 0.000 description 2
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 2
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 2
- 241000701447 unidentified baculovirus Species 0.000 description 2
- 208000019553 vascular disease Diseases 0.000 description 2
- 229960003862 vemurafenib Drugs 0.000 description 2
- GPXBXXGIAQBQNI-UHFFFAOYSA-N vemurafenib Chemical compound CCCS(=O)(=O)NC1=CC=C(F)C(C(=O)C=2C3=CC(=CN=C3NC=2)C=2C=CC(Cl)=CC=2)=C1F GPXBXXGIAQBQNI-UHFFFAOYSA-N 0.000 description 2
- 229960004528 vincristine Drugs 0.000 description 2
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 2
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 2
- 229930003231 vitamin Natural products 0.000 description 2
- 239000011782 vitamin Substances 0.000 description 2
- 235000013343 vitamin Nutrition 0.000 description 2
- 229940088594 vitamin Drugs 0.000 description 2
- VCGRFBXVSFAGGA-UHFFFAOYSA-N (1,1-dioxo-1,4-thiazinan-4-yl)-[6-[[3-(4-fluorophenyl)-5-methyl-1,2-oxazol-4-yl]methoxy]pyridin-3-yl]methanone Chemical compound CC=1ON=C(C=2C=CC(F)=CC=2)C=1COC(N=C1)=CC=C1C(=O)N1CCS(=O)(=O)CC1 VCGRFBXVSFAGGA-UHFFFAOYSA-N 0.000 description 1
- WDQLRUYAYXDIFW-RWKIJVEZSA-N (2r,3r,4s,5r,6r)-4-[(2s,3r,4s,5r,6r)-3,5-dihydroxy-4-[(2r,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-6-[[(2r,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxymethyl]oxan-2-yl]oxy-6-(hydroxymethyl)oxane-2,3,5-triol Chemical compound O[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)[C@H](O)[C@@H](CO[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)O1 WDQLRUYAYXDIFW-RWKIJVEZSA-N 0.000 description 1
- NRIYPIBRPGAWDD-UHFFFAOYSA-N (5-methylthiophen-2-yl)boronic acid Chemical compound CC1=CC=C(B(O)O)S1 NRIYPIBRPGAWDD-UHFFFAOYSA-N 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- DYLIWHYUXAJDOJ-OWOJBTEDSA-N (e)-4-(6-aminopurin-9-yl)but-2-en-1-ol Chemical compound NC1=NC=NC2=C1N=CN2C\C=C\CO DYLIWHYUXAJDOJ-OWOJBTEDSA-N 0.000 description 1
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 1
- JIHQDMXYYFUGFV-UHFFFAOYSA-N 1,3,5-triazine Chemical compound C1=NC=NC=N1 JIHQDMXYYFUGFV-UHFFFAOYSA-N 0.000 description 1
- RNHDAKUGFHSZEV-UHFFFAOYSA-N 1,4-dioxane;hydrate Chemical compound O.C1COCCO1 RNHDAKUGFHSZEV-UHFFFAOYSA-N 0.000 description 1
- BOOVIFJKQGYEON-UHFFFAOYSA-N 1-(oxan-4-yl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole Chemical compound O1C(C)(C)C(C)(C)OB1C1=CN(C2CCOCC2)N=C1 BOOVIFJKQGYEON-UHFFFAOYSA-N 0.000 description 1
- OMRFOTIFDZORHU-UHFFFAOYSA-N 1-(oxan-4-ylmethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole Chemical compound O1C(C)(C)C(C)(C)OB1C1=CN(CC2CCOCC2)N=C1 OMRFOTIFDZORHU-UHFFFAOYSA-N 0.000 description 1
- GFTXALYVNPNJLF-UHFFFAOYSA-N 1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound O=C1C(=CN(C=C1)CC1CCOCC1)C(=O)N GFTXALYVNPNJLF-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- 102100025573 1-alkyl-2-acetylglycerophosphocholine esterase Human genes 0.000 description 1
- VSNHCAURESNICA-NJFSPNSNSA-N 1-oxidanylurea Chemical compound N[14C](=O)NO VSNHCAURESNICA-NJFSPNSNSA-N 0.000 description 1
- FPIPGXGPPPQFEQ-UHFFFAOYSA-N 13-cis retinol Natural products OCC=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-UHFFFAOYSA-N 0.000 description 1
- BFPYWIDHMRZLRN-UHFFFAOYSA-N 17alpha-ethynyl estradiol Natural products OC1=CC=C2C3CCC(C)(C(CC4)(O)C#C)C4C3CCC2=C1 BFPYWIDHMRZLRN-UHFFFAOYSA-N 0.000 description 1
- ISWSIDIOOBJBQZ-KYKRLZBASA-N 2,3,4-trideuteriophenol Chemical compound [2H]c1ccc(O)c([2H])c1[2H] ISWSIDIOOBJBQZ-KYKRLZBASA-N 0.000 description 1
- PNBIYFPZODYMOO-UHFFFAOYSA-N 2-(4-bromo-2-fluorophenyl)acetic acid Chemical compound OC(=O)CC1=CC=C(Br)C=C1F PNBIYFPZODYMOO-UHFFFAOYSA-N 0.000 description 1
- AVMWJACSYAQNDG-UHFFFAOYSA-N 2-(5-methylpyridin-2-yl)-6-phenyl-1,3,6,2-dioxazaborocane Chemical compound N1=CC(C)=CC=C1B1OCCN(C=2C=CC=CC=2)CCO1 AVMWJACSYAQNDG-UHFFFAOYSA-N 0.000 description 1
- TYHTUJMBDHMENY-UHFFFAOYSA-N 2-(5-methylpyridin-2-yl)acetic acid Chemical compound CC1=CC=C(CC(O)=O)N=C1 TYHTUJMBDHMENY-UHFFFAOYSA-N 0.000 description 1
- BZHDYBZLUCKBIA-LURJTMIESA-N 2-[(2S)-1,4-dioxan-2-yl]ethanesulfonic acid Chemical compound O1[C@H](COCC1)CCS(=O)(=O)O BZHDYBZLUCKBIA-LURJTMIESA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- JEECGGXGHJUYMN-UHFFFAOYSA-N 2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine Chemical compound C1=NC(C)=CC=C1B1OC(C)(C)C(C)(C)O1 JEECGGXGHJUYMN-UHFFFAOYSA-N 0.000 description 1
- 125000005975 2-phenylethyloxy group Chemical group 0.000 description 1
- RUDHSKFULPGCLM-UHFFFAOYSA-N 2-pyrazin-2-yl-1h-benzimidazole Chemical group N=1C2=CC=CC=C2NC=1C1=CN=CC=N1 RUDHSKFULPGCLM-UHFFFAOYSA-N 0.000 description 1
- DTXVKPOKPFWSFF-UHFFFAOYSA-N 3(S)-hydroxy-13-cis-eicosenoyl-CoA Chemical compound NC1=CC=C(Cl)N=N1 DTXVKPOKPFWSFF-UHFFFAOYSA-N 0.000 description 1
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 description 1
- RUDFGVCQAZXNAJ-UHFFFAOYSA-N 3-(4-amino-2-fluorophenyl)-5-(3,4-dimethoxyphenyl)pyridin-2-amine Chemical compound C1=C(OC)C(OC)=CC=C1C1=CN=C(N)C(C=2C(=CC(N)=CC=2)F)=C1 RUDFGVCQAZXNAJ-UHFFFAOYSA-N 0.000 description 1
- BLAHKHDCYYFYCE-UHFFFAOYSA-N 3-(4-amino-2-fluorophenyl)-5-bromopyridin-2-amine Chemical compound FC1=CC(N)=CC=C1C1=CC(Br)=CN=C1N BLAHKHDCYYFYCE-UHFFFAOYSA-N 0.000 description 1
- SHIZYMRFBUULOJ-UHFFFAOYSA-N 3-(4-amino-3-fluorophenyl)-5-(3,4-dimethoxyphenyl)pyridin-2-amine Chemical compound NC1=C(C=C(C=C1)C=1C(=NC=C(C=1)C1=CC(=C(C=C1)OC)OC)N)F SHIZYMRFBUULOJ-UHFFFAOYSA-N 0.000 description 1
- IFCSWGQBKVTSFE-UHFFFAOYSA-N 3-(4-aminophenyl)-5-(3,4-dimethoxyphenyl)pyridin-2-amine Chemical compound C1=C(OC)C(OC)=CC=C1C1=CN=C(N)C(C=2C=CC(N)=CC=2)=C1 IFCSWGQBKVTSFE-UHFFFAOYSA-N 0.000 description 1
- REZBRRWYFOIXJN-UHFFFAOYSA-N 3-(4-aminophenyl)-5-bromopyridin-2-amine Chemical compound C1=CC(N)=CC=C1C1=CC(Br)=CN=C1N REZBRRWYFOIXJN-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- GJITWRFPCHXBOP-UHFFFAOYSA-N 3-bromo-5-[3-methoxy-4-(2-morpholin-4-ylethoxy)phenyl]pyridin-2-amine Chemical compound BrC=1C(=NC=C(C=1)C1=CC(=C(C=C1)OCCN1CCOCC1)OC)N GJITWRFPCHXBOP-UHFFFAOYSA-N 0.000 description 1
- AAXRJLIGAKZDSS-ZDUSSCGKSA-N 3-bromo-5-[4-[[(2S)-1,4-dioxan-2-yl]methoxy]-3-methoxyphenyl]pyridin-2-amine Chemical compound BrC=1C(=NC=C(C=1)C1=CC(=C(C=C1)OC[C@H]1OCCOC1)OC)N AAXRJLIGAKZDSS-ZDUSSCGKSA-N 0.000 description 1
- AAXRJLIGAKZDSS-CYBMUJFWSA-N 3-bromo-5-[4-[[(2r)-1,4-dioxan-2-yl]methoxy]-3-methoxyphenyl]pyridin-2-amine Chemical compound COC1=CC(C=2C=C(Br)C(N)=NC=2)=CC=C1OC[C@H]1COCCO1 AAXRJLIGAKZDSS-CYBMUJFWSA-N 0.000 description 1
- OYZOHRSCEASBER-UHFFFAOYSA-N 3-bromo-5-iodopyridin-2-amine Chemical compound NC1=NC=C(I)C=C1Br OYZOHRSCEASBER-UHFFFAOYSA-N 0.000 description 1
- KVGKDDCJSHJYCS-UHFFFAOYSA-N 3-chloro-5-iodopyrazin-2-amine Chemical compound NC1=NC=C(I)N=C1Cl KVGKDDCJSHJYCS-UHFFFAOYSA-N 0.000 description 1
- FLMNWVXAEGUVNY-UHFFFAOYSA-N 3-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(N)C=C1F FLMNWVXAEGUVNY-UHFFFAOYSA-N 0.000 description 1
- TVOJIBGZFYMWDT-UHFFFAOYSA-N 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1h-pyrazole Chemical compound O1C(C)(C)C(C)(C)OB1C1=CNN=C1 TVOJIBGZFYMWDT-UHFFFAOYSA-N 0.000 description 1
- ZANPJXNYBVVNSD-UHFFFAOYSA-N 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(N)C=C1 ZANPJXNYBVVNSD-UHFFFAOYSA-N 0.000 description 1
- QOWSWEBLNVACCL-UHFFFAOYSA-N 4-Bromophenyl acetate Chemical compound OC(=O)CC1=CC=C(Br)C=C1 QOWSWEBLNVACCL-UHFFFAOYSA-N 0.000 description 1
- XWNSFEAWWGGSKJ-UHFFFAOYSA-N 4-acetyl-4-methylheptanedinitrile Chemical compound N#CCCC(C)(C(=O)C)CCC#N XWNSFEAWWGGSKJ-UHFFFAOYSA-N 0.000 description 1
- KVCQTKNUUQOELD-UHFFFAOYSA-N 4-amino-n-[1-(3-chloro-2-fluoroanilino)-6-methylisoquinolin-5-yl]thieno[3,2-d]pyrimidine-7-carboxamide Chemical compound N=1C=CC2=C(NC(=O)C=3C4=NC=NC(N)=C4SC=3)C(C)=CC=C2C=1NC1=CC=CC(Cl)=C1F KVCQTKNUUQOELD-UHFFFAOYSA-N 0.000 description 1
- WHSIIJQOEGXWSN-RLTMCGQMSA-N 4-bromo-2,3,5-trideuterio-6-(trideuteriomethoxy)phenol Chemical compound BrC1=C(C(=C(C(=C1[2H])OC([2H])([2H])[2H])O)[2H])[2H] WHSIIJQOEGXWSN-RLTMCGQMSA-N 0.000 description 1
- 125000004800 4-bromophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Br 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- ARZPLBZSJFHUOQ-UHFFFAOYSA-N 5-(5-bromopyridin-2-yl)-1-[(4-cyanooxan-4-yl)methyl]-4-oxopyridine-3-carboxylic acid Chemical compound BrC=1C=CC(=NC=1)C1=CN(C=C(C1=O)C(=O)O)CC1(CCOCC1)C#N ARZPLBZSJFHUOQ-UHFFFAOYSA-N 0.000 description 1
- IVILGUFRMDBUEQ-UHFFFAOYSA-N 5-iodopyridin-2-amine Chemical compound NC1=CC=C(I)C=N1 IVILGUFRMDBUEQ-UHFFFAOYSA-N 0.000 description 1
- HAZMBQGWYYGRES-UHFFFAOYSA-N 6-chloro-5-fluoropyridin-3-amine Chemical compound NC1=CN=C(Cl)C(F)=C1 HAZMBQGWYYGRES-UHFFFAOYSA-N 0.000 description 1
- NKGPJODWTZCHGF-KQYNXXCUSA-N 6-thioinosinic acid Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(S)=C2N=C1 NKGPJODWTZCHGF-KQYNXXCUSA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- CYJRNFFLTBEQSQ-UHFFFAOYSA-N 8-(3-methyl-1-benzothiophen-5-yl)-N-(4-methylsulfonylpyridin-3-yl)quinoxalin-6-amine Chemical compound CS(=O)(=O)C1=C(C=NC=C1)NC=1C=C2N=CC=NC2=C(C=1)C=1C=CC2=C(C(=CS2)C)C=1 CYJRNFFLTBEQSQ-UHFFFAOYSA-N 0.000 description 1
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 108010024976 Asparaginase Proteins 0.000 description 1
- NOWKCMXCCJGMRR-UHFFFAOYSA-N Aziridine Chemical compound C1CN1 NOWKCMXCCJGMRR-UHFFFAOYSA-N 0.000 description 1
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 1
- WOVKYSAHUYNSMH-UHFFFAOYSA-N BROMODEOXYURIDINE Natural products C1C(O)C(CO)OC1N1C(=O)NC(=O)C(Br)=C1 WOVKYSAHUYNSMH-UHFFFAOYSA-N 0.000 description 1
- VGGGPCQERPFHOB-MCIONIFRSA-N Bestatin Chemical compound CC(C)C[C@H](C(O)=O)NC(=O)[C@@H](O)[C@H](N)CC1=CC=CC=C1 VGGGPCQERPFHOB-MCIONIFRSA-N 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- 206010055113 Breast cancer metastatic Diseases 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- SDDWRZFHAHXWJC-UHFFFAOYSA-N C1COCCC1CN2C=CC(=O)C(=C2)C(=O)OC3=CC=C(C=C3)Br Chemical compound C1COCCC1CN2C=CC(=O)C(=C2)C(=O)OC3=CC=C(C=C3)Br SDDWRZFHAHXWJC-UHFFFAOYSA-N 0.000 description 1
- VKXSGTOKXXENDY-UHFFFAOYSA-N CC(C=C1)=CCC1C1=CN(CC2CCOCC2)C=C(C(O)O)C1=O Chemical compound CC(C=C1)=CCC1C1=CN(CC2CCOCC2)C=C(C(O)O)C1=O VKXSGTOKXXENDY-UHFFFAOYSA-N 0.000 description 1
- RJXVWCGOXIKUTD-UHFFFAOYSA-N CC=1C(=NC=CC1)C1=CN(C=C(C1=O)C(=O)N)CC1CCOCC1 Chemical compound CC=1C(=NC=CC1)C1=CN(C=C(C1=O)C(=O)N)CC1CCOCC1 RJXVWCGOXIKUTD-UHFFFAOYSA-N 0.000 description 1
- NNJXVJIDIXWYDR-UHFFFAOYSA-N COC(C(C(C(I)=C1O)O)O)=C1OC(O)(O)O Chemical compound COC(C(C(C(I)=C1O)O)O)=C1OC(O)(O)O NNJXVJIDIXWYDR-UHFFFAOYSA-N 0.000 description 1
- MYYYRPHIWJCRLW-UHFFFAOYSA-N CS(=O)(=O)O.NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)C1=CC(=C(C=C1)OC)OC Chemical compound CS(=O)(=O)O.NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)C1=CC(=C(C=C1)OC)OC MYYYRPHIWJCRLW-UHFFFAOYSA-N 0.000 description 1
- FVLVBPDQNARYJU-XAHDHGMMSA-N C[C@H]1CCC(CC1)NC(=O)N(CCCl)N=O Chemical compound C[C@H]1CCC(CC1)NC(=O)N(CCCl)N=O FVLVBPDQNARYJU-XAHDHGMMSA-N 0.000 description 1
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- AOCCBINRVIKJHY-UHFFFAOYSA-N Carmofur Chemical compound CCCCCCNC(=O)N1C=C(F)C(=O)NC1=O AOCCBINRVIKJHY-UHFFFAOYSA-N 0.000 description 1
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 1
- GDCGPGQXISRWDO-UHFFFAOYSA-N Cc(cc1)ccc1C1=CN(CC2CCOCC2)C=C(C(Nc(cc2)ccc2-c2c(N)ncc(-c3cc(F)c(CN4CCOCC4)cc3)c2)O)C1=O Chemical compound Cc(cc1)ccc1C1=CN(CC2CCOCC2)C=C(C(Nc(cc2)ccc2-c2c(N)ncc(-c3cc(F)c(CN4CCOCC4)cc3)c2)O)C1=O GDCGPGQXISRWDO-UHFFFAOYSA-N 0.000 description 1
- FXVATMJCWRMVOG-UHFFFAOYSA-N Cc(cc1)ccc1C1=CN(CC2CCOCC2)C=C(C(Nc(cc2)ccc2C2=CC(c3cc(F)c(C=O)cc3)=CNC2N)=O)C1=O Chemical compound Cc(cc1)ccc1C1=CN(CC2CCOCC2)C=C(C(Nc(cc2)ccc2C2=CC(c3cc(F)c(C=O)cc3)=CNC2N)=O)C1=O FXVATMJCWRMVOG-UHFFFAOYSA-N 0.000 description 1
- LOCJBCFCMJSRFP-UHFFFAOYSA-N Cc(cc1)ccc1C1=CN(CC2CCOCC2)C=C(C(Nc2ccc(-c3c(NC)ncc(C(C=CC4OC)=CC4OC)c3)c(F)c2)O)C1=O Chemical compound Cc(cc1)ccc1C1=CN(CC2CCOCC2)C=C(C(Nc2ccc(-c3c(NC)ncc(C(C=CC4OC)=CC4OC)c3)c(F)c2)O)C1=O LOCJBCFCMJSRFP-UHFFFAOYSA-N 0.000 description 1
- 206010008342 Cervix carcinoma Diseases 0.000 description 1
- MKQWTWSXVILIKJ-LXGUWJNJSA-N Chlorozotocin Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](C=O)NC(=O)N(N=O)CCCl MKQWTWSXVILIKJ-LXGUWJNJSA-N 0.000 description 1
- RURLVUZRUFHCJO-UHFFFAOYSA-N Chromomycin A3 Natural products COC(C1Cc2cc3cc(OC4CC(OC(=O)C)C(OC5CC(O)C(OC)C(C)O5)C(C)O4)c(C)c(O)c3c(O)c2C(=O)C1OC6CC(OC7CC(C)(O)C(OC(=O)C)C(C)O7)C(O)C(C)O6)C(=O)C(O)C(C)O RURLVUZRUFHCJO-UHFFFAOYSA-N 0.000 description 1
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 229910021591 Copper(I) chloride Inorganic materials 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 1
- ZZZCUOFIHGPKAK-UHFFFAOYSA-N D-erythro-ascorbic acid Natural products OCC1OC(=O)C(O)=C1O ZZZCUOFIHGPKAK-UHFFFAOYSA-N 0.000 description 1
- 108020004414 DNA Proteins 0.000 description 1
- 108010092160 Dactinomycin Proteins 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- BWLUMTFWVZZZND-UHFFFAOYSA-N Dibenzylamine Chemical class C=1C=CC=CC=1CNCC1=CC=CC=C1 BWLUMTFWVZZZND-UHFFFAOYSA-N 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical class C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- 241000283074 Equus asinus Species 0.000 description 1
- BFPYWIDHMRZLRN-SLHNCBLASA-N Ethinyl estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@](CC4)(O)C#C)[C@@H]4[C@@H]3CCC2=C1 BFPYWIDHMRZLRN-SLHNCBLASA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical class NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 206010015548 Euthanasia Diseases 0.000 description 1
- 206010015719 Exsanguination Diseases 0.000 description 1
- AODJVOYNQLNOGN-UHFFFAOYSA-N FC=1C=C(C=CC=1C=O)OB(O)O Chemical compound FC=1C=C(C=CC=1C=O)OB(O)O AODJVOYNQLNOGN-UHFFFAOYSA-N 0.000 description 1
- 239000004606 Fillers/Extenders Substances 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 102100031487 Growth arrest-specific protein 6 Human genes 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- 208000017891 HER2 positive breast carcinoma Diseases 0.000 description 1
- 108010068250 Herpes Simplex Virus Protein Vmw65 Proteins 0.000 description 1
- 101000923005 Homo sapiens Growth arrest-specific protein 6 Proteins 0.000 description 1
- 101000973901 Homo sapiens Tyrosine-protein kinase Mer Proteins 0.000 description 1
- 201000009794 Idiopathic Pulmonary Fibrosis Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- XQGSVNHIIVBMPX-UHFFFAOYSA-N Improsulfan tosylate Chemical compound CC1=CC=C(S([O-])(=O)=O)C=C1.CS(=O)(=O)OCCC[NH2+]CCCOS(C)(=O)=O XQGSVNHIIVBMPX-UHFFFAOYSA-N 0.000 description 1
- PIWKPBJCKXDKJR-UHFFFAOYSA-N Isoflurane Chemical compound FC(F)OC(Cl)C(F)(F)F PIWKPBJCKXDKJR-UHFFFAOYSA-N 0.000 description 1
- 206010023421 Kidney fibrosis Diseases 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 1
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 1
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 1
- 239000002136 L01XE07 - Lapatinib Substances 0.000 description 1
- XNRVGTHNYCNCFF-UHFFFAOYSA-N Lapatinib ditosylate monohydrate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1.CC1=CC=C(S(O)(=O)=O)C=C1.O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 XNRVGTHNYCNCFF-UHFFFAOYSA-N 0.000 description 1
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 1
- 238000000134 MTT assay Methods 0.000 description 1
- 231100000002 MTT assay Toxicity 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- FQISKWAFAHGMGT-SGJOWKDISA-M Methylprednisolone sodium succinate Chemical compound [Na+].C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2[C@@H](O)C[C@]2(C)[C@@](O)(C(=O)COC(=O)CCC([O-])=O)CC[C@H]21 FQISKWAFAHGMGT-SGJOWKDISA-M 0.000 description 1
- VFKZTMPDYBFSTM-KVTDHHQDSA-N Mitobronitol Chemical compound BrC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CBr VFKZTMPDYBFSTM-KVTDHHQDSA-N 0.000 description 1
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 1
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- FXBJJYIXVDHGJQ-UHFFFAOYSA-N N-(5-bromopyrazin-2-yl)-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound BrC=1N=CC(=NC=1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1 FXBJJYIXVDHGJQ-UHFFFAOYSA-N 0.000 description 1
- YYGLURGKDYINIT-UHFFFAOYSA-N N-(5-iodopyridin-2-yl)-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound IC=1C=CC(=NC=1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1 YYGLURGKDYINIT-UHFFFAOYSA-N 0.000 description 1
- AYCPARAPKDAOEN-LJQANCHMSA-N N-[(1S)-2-(dimethylamino)-1-phenylethyl]-6,6-dimethyl-3-[(2-methyl-4-thieno[3,2-d]pyrimidinyl)amino]-1,4-dihydropyrrolo[3,4-c]pyrazole-5-carboxamide Chemical compound C1([C@H](NC(=O)N2C(C=3NN=C(NC=4C=5SC=CC=5N=C(C)N=4)C=3C2)(C)C)CN(C)C)=CC=CC=C1 AYCPARAPKDAOEN-LJQANCHMSA-N 0.000 description 1
- NDHJRDQILUOWLR-UHFFFAOYSA-N N-[4-(2-amino-5-bromopyridin-3-yl)-3-fluorophenyl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=C(C=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)F)Br NDHJRDQILUOWLR-UHFFFAOYSA-N 0.000 description 1
- VNBRGSXVFBYQNN-UHFFFAOYSA-N N-[4-[(2-amino-3-chloro-4-pyridinyl)oxy]-3-fluorophenyl]-4-ethoxy-1-(4-fluorophenyl)-2-oxo-3-pyridinecarboxamide Chemical compound O=C1C(C(=O)NC=2C=C(F)C(OC=3C(=C(N)N=CC=3)Cl)=CC=2)=C(OCC)C=CN1C1=CC=C(F)C=C1 VNBRGSXVFBYQNN-UHFFFAOYSA-N 0.000 description 1
- GEAJYTNNHUHUPD-UHFFFAOYSA-N N-[4-[2-amino-5-(3,4-dimethoxyphenyl)pyridin-3-yl]-3-fluorophenyl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound C1(C=2C=C(C3=CC=C(OC)C(OC)=C3)C=NC=2N)=CC=C(NC(=O)C=2C(=O)C(C3=CC=C(C=C3)C)=CN(C=2)CC2CCOCC2)C=C1F GEAJYTNNHUHUPD-UHFFFAOYSA-N 0.000 description 1
- JXIOZYRODOOPLU-UHFFFAOYSA-N N-[4-[2-amino-5-(3,4-dimethoxyphenyl)pyridin-3-yl]phenyl]-5-(5-methylthiophen-2-yl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C=1SC(=CC=1)C)CC1CCOCC1)C1=CC(=C(C=C1)OC)OC JXIOZYRODOOPLU-UHFFFAOYSA-N 0.000 description 1
- IPHZKBACASVMHL-UHFFFAOYSA-N N-[4-[2-amino-5-[4-[(4-methylpiperazin-1-yl)methyl]phenyl]pyridin-3-yl]phenyl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)C1=CC=C(C=C1)CN1CCN(CC1)C IPHZKBACASVMHL-UHFFFAOYSA-N 0.000 description 1
- YCXZBULDUGDETC-GDLZYMKVSA-N N-[4-[2-amino-5-[4-[[(2R)-1,4-dioxan-2-yl]methoxy]-3-methoxyphenyl]pyridin-3-yl]-3-fluorophenyl]-5-(5-bromopyridin-2-yl)-1-[(4-cyanooxan-4-yl)methyl]-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=C(C=C(C=C1)NC(=O)C=1C(C(=CN(C=1)CC1(CCOCC1)C#N)C1=NC=C(C=C1)Br)=O)F)C1=CC(=C(C=C1)OC[C@@H]1OCCOC1)OC YCXZBULDUGDETC-GDLZYMKVSA-N 0.000 description 1
- WSNUZOLBHLCQOI-UHFFFAOYSA-N N-[6-(2-amino-5-bromopyridin-3-yl)pyridin-3-yl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=NC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)Br WSNUZOLBHLCQOI-UHFFFAOYSA-N 0.000 description 1
- IIOBIZXRVAIIAJ-UHFFFAOYSA-N N-[6-[2-amino-5-(3,4-dimethoxyphenyl)pyridin-3-yl]pyridazin-3-yl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=CC=C(N=N1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)C1=CC(=C(C=C1)OC)OC IIOBIZXRVAIIAJ-UHFFFAOYSA-N 0.000 description 1
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical class CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- KCTZOTUQSGYWLV-UHFFFAOYSA-N N1C=NC=C2N=CC=C21 Chemical group N1C=NC=C2N=CC=C21 KCTZOTUQSGYWLV-UHFFFAOYSA-N 0.000 description 1
- VEQPNABPJHWNSG-UHFFFAOYSA-N Nickel(2+) Chemical compound [Ni+2] VEQPNABPJHWNSG-UHFFFAOYSA-N 0.000 description 1
- 229910021586 Nickel(II) chloride Inorganic materials 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 239000000020 Nitrocellulose Substances 0.000 description 1
- AFLXUQUGROGEFA-UHFFFAOYSA-N Nitrogen mustard N-oxide Chemical compound ClCC[N+]([O-])(C)CCCl AFLXUQUGROGEFA-UHFFFAOYSA-N 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- BZSGJAREDHFGPI-UHFFFAOYSA-N O1CCCC1.CB1OB(OB(O1)C)C Chemical compound O1CCCC1.CB1OB(OB(O1)C)C BZSGJAREDHFGPI-UHFFFAOYSA-N 0.000 description 1
- VWPUAXALDFFXJW-UHFFFAOYSA-N Oc(c(O)c(c(O)c1O)O)c1O Chemical compound Oc(c(O)c(c(O)c1O)O)c1O VWPUAXALDFFXJW-UHFFFAOYSA-N 0.000 description 1
- 102000043276 Oncogene Human genes 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 108010057150 Peplomycin Proteins 0.000 description 1
- 102000003992 Peroxidases Human genes 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- 229940122907 Phosphatase inhibitor Drugs 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 239000004153 Potassium bromate Substances 0.000 description 1
- 241000384942 Premna serratifolia Species 0.000 description 1
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 1
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 1
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- AHHFEZNOXOZZQA-ZEBDFXRSSA-N Ranimustine Chemical compound CO[C@H]1O[C@H](CNC(=O)N(CCCl)N=O)[C@@H](O)[C@H](O)[C@H]1O AHHFEZNOXOZZQA-ZEBDFXRSSA-N 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 206010038910 Retinitis Diseases 0.000 description 1
- 201000000582 Retinoblastoma Diseases 0.000 description 1
- KCOWWGWKRXKKGY-MDYNBEAQSA-N S(=O)(=O)(O)O.NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)C1=CC(=C(C=C1)OC[C@@H]1OCCOC1)OC Chemical compound S(=O)(=O)(O)O.NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)C1=CC(=C(C=C1)OC[C@@H]1OCCOC1)OC KCOWWGWKRXKKGY-MDYNBEAQSA-N 0.000 description 1
- 229920002305 Schizophyllan Polymers 0.000 description 1
- 108020004459 Small interfering RNA Proteins 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical class [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 208000000277 Splenic Neoplasms Diseases 0.000 description 1
- 108091005729 TAM receptors Proteins 0.000 description 1
- 108091077436 Tam family Proteins 0.000 description 1
- 229940123237 Taxane Drugs 0.000 description 1
- WFWLQNSHRPWKFK-UHFFFAOYSA-N Tegafur Chemical compound O=C1NC(=O)C(F)=CN1C1OCCC1 WFWLQNSHRPWKFK-UHFFFAOYSA-N 0.000 description 1
- 208000024313 Testicular Neoplasms Diseases 0.000 description 1
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 1
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 1
- 208000000728 Thymus Neoplasms Diseases 0.000 description 1
- 206010052779 Transplant rejections Diseases 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 239000013504 Triton X-100 Substances 0.000 description 1
- 229920004890 Triton X-100 Polymers 0.000 description 1
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 1
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 229940122803 Vinca alkaloid Drugs 0.000 description 1
- FPIPGXGPPPQFEQ-BOOMUCAASA-N Vitamin A Natural products OC/C=C(/C)\C=C\C=C(\C)/C=C/C1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-BOOMUCAASA-N 0.000 description 1
- 229930003268 Vitamin C Natural products 0.000 description 1
- 238000002441 X-ray diffraction Methods 0.000 description 1
- SCERXFXTNYGGCR-MDYNBEAQSA-N [N+](=O)(O)[O-].NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)C1=CC(=C(C=C1)OC[C@@H]1OCCOC1)OC Chemical compound [N+](=O)(O)[O-].NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)C1=CC(=C(C=C1)OC[C@@H]1OCCOC1)OC SCERXFXTNYGGCR-MDYNBEAQSA-N 0.000 description 1
- MNZMECMQTYGSOI-UHFFFAOYSA-N acetic acid;hydron;bromide Chemical compound Br.CC(O)=O MNZMECMQTYGSOI-UHFFFAOYSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- USZYSDMBJDPRIF-SVEJIMAYSA-N aclacinomycin A Chemical compound O([C@H]1[C@@H](O)C[C@@H](O[C@H]1C)O[C@H]1[C@H](C[C@@H](O[C@H]1C)O[C@H]1C[C@]([C@@H](C2=CC=3C(=O)C4=CC=CC(O)=C4C(=O)C=3C(O)=C21)C(=O)OC)(O)CC)N(C)C)[C@H]1CCC(=O)[C@H](C)O1 USZYSDMBJDPRIF-SVEJIMAYSA-N 0.000 description 1
- 229960004176 aclarubicin Drugs 0.000 description 1
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- FPIPGXGPPPQFEQ-OVSJKPMPSA-N all-trans-retinol Chemical compound OC\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-OVSJKPMPSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 230000009435 amidation Effects 0.000 description 1
- 238000007112 amidation reaction Methods 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- KZOWNALBTMILAP-JBMRGDGGSA-N ancitabine hydrochloride Chemical compound Cl.N=C1C=CN2[C@@H]3O[C@H](CO)[C@@H](O)[C@@H]3OC2=N1 KZOWNALBTMILAP-JBMRGDGGSA-N 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 239000003817 anthracycline antibiotic agent Substances 0.000 description 1
- 230000003432 anti-folate effect Effects 0.000 description 1
- 229940127074 antifolate Drugs 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 125000005228 aryl sulfonate group Chemical group 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 108010023337 axl receptor tyrosine kinase Proteins 0.000 description 1
- HONIICLYMWZJFZ-UHFFFAOYSA-N azetidine Chemical compound C1CNC1 HONIICLYMWZJFZ-UHFFFAOYSA-N 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical class C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- MVIOINXPSFUJEN-UHFFFAOYSA-N benzenesulfonic acid;hydrate Chemical compound O.OS(=O)(=O)C1=CC=CC=C1 MVIOINXPSFUJEN-UHFFFAOYSA-N 0.000 description 1
- 229960002537 betamethasone Drugs 0.000 description 1
- UREBDLICKHMUKA-DVTGEIKXSA-N betamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-DVTGEIKXSA-N 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000008512 biological response Effects 0.000 description 1
- YNHIGQDRGKUECZ-UHFFFAOYSA-L bis(triphenylphosphine)palladium(ii) dichloride Chemical compound [Cl-].[Cl-].[Pd+2].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-L 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 229950004398 broxuridine Drugs 0.000 description 1
- 210000005252 bulbus oculi Anatomy 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- 125000004106 butoxy group Chemical group [*]OC([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 1
- 229940127093 camptothecin Drugs 0.000 description 1
- 125000005587 carbonate group Chemical group 0.000 description 1
- UHKPOGGUWJGGID-UHFFFAOYSA-N carbonic acid;cesium Chemical compound [Cs].OC(O)=O UHKPOGGUWJGGID-UHFFFAOYSA-N 0.000 description 1
- JYYOBHFYCIDXHH-UHFFFAOYSA-N carbonic acid;hydrate Chemical compound O.OC(O)=O JYYOBHFYCIDXHH-UHFFFAOYSA-N 0.000 description 1
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 description 1
- 229960003261 carmofur Drugs 0.000 description 1
- 229960005243 carmustine Drugs 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 210000003986 cell retinal photoreceptor Anatomy 0.000 description 1
- 230000036755 cellular response Effects 0.000 description 1
- 201000010881 cervical cancer Diseases 0.000 description 1
- 229960005395 cetuximab Drugs 0.000 description 1
- 239000012295 chemical reaction liquid Substances 0.000 description 1
- VUHJZBBCZGVNDZ-TTYLFXKOSA-N chlormadinone Chemical compound C1=C(Cl)C2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)C)(O)[C@@]1(C)CC2 VUHJZBBCZGVNDZ-TTYLFXKOSA-N 0.000 description 1
- 229960003996 chlormadinone Drugs 0.000 description 1
- 229960001480 chlorozotocin Drugs 0.000 description 1
- ZYVSOIYQKUDENJ-WKSBCEQHSA-N chromomycin A3 Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@@H]1OC(C)=O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@@H](O)[C@H](O[C@@H]3O[C@@H](C)[C@H](OC(C)=O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@@H]1C[C@@H](O)[C@@H](OC)[C@@H](C)O1 ZYVSOIYQKUDENJ-WKSBCEQHSA-N 0.000 description 1
- 230000004186 co-expression Effects 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- OXBLHERUFWYNTN-UHFFFAOYSA-M copper(I) chloride Chemical compound [Cu]Cl OXBLHERUFWYNTN-UHFFFAOYSA-M 0.000 description 1
- 125000006165 cyclic alkyl group Chemical group 0.000 description 1
- 125000000392 cycloalkenyl group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- UKJLNMAFNRKWGR-UHFFFAOYSA-N cyclohexatrienamine Chemical group NC1=CC=C=C[CH]1 UKJLNMAFNRKWGR-UHFFFAOYSA-N 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 150000003946 cyclohexylamines Chemical class 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 229960000684 cytarabine Drugs 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- 229960000640 dactinomycin Drugs 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- 229960001251 denosumab Drugs 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 229960003957 dexamethasone Drugs 0.000 description 1
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 1
- 239000000032 diagnostic agent Substances 0.000 description 1
- 229940039227 diagnostic agent Drugs 0.000 description 1
- 238000002059 diagnostic imaging Methods 0.000 description 1
- ZOCHARZZJNPSEU-UHFFFAOYSA-N diboron Chemical compound B#B ZOCHARZZJNPSEU-UHFFFAOYSA-N 0.000 description 1
- BSHICDXRSZQYBP-UHFFFAOYSA-N dichloromethane;palladium(2+) Chemical compound [Pd+2].ClCCl BSHICDXRSZQYBP-UHFFFAOYSA-N 0.000 description 1
- YNHIGQDRGKUECZ-UHFFFAOYSA-N dichloropalladium;triphenylphosphanium Chemical compound Cl[Pd]Cl.C1=CC=CC=C1[PH+](C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1[PH+](C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-N 0.000 description 1
- YZDNEKBXKIZKRK-UHFFFAOYSA-N dicyclohexyl-[2-[2,4-di(propan-2-yl)phenyl]-1-propan-2-ylcyclohexa-2,4-dien-1-yl]phosphane Chemical group CC(C)C1=CC(C(C)C)=CC=C1C1=CC=CCC1(C(C)C)P(C1CCCCC1)C1CCCCC1 YZDNEKBXKIZKRK-UHFFFAOYSA-N 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical class OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 150000005332 diethylamines Chemical class 0.000 description 1
- 238000009792 diffusion process Methods 0.000 description 1
- 125000005043 dihydropyranyl group Chemical group O1C(CCC=C1)* 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 1
- 229960003668 docetaxel Drugs 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 201000006828 endometrial hyperplasia Diseases 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- 150000002118 epoxides Chemical class 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- HNGDOSBFYRVIEY-UHFFFAOYSA-N ethanesulfonic acid;hydrate Chemical compound O.CCS(O)(=O)=O HNGDOSBFYRVIEY-UHFFFAOYSA-N 0.000 description 1
- FXPHJTKVWZVEGA-UHFFFAOYSA-N ethenyl hydrogen carbonate Chemical group OC(=O)OC=C FXPHJTKVWZVEGA-UHFFFAOYSA-N 0.000 description 1
- 125000001033 ether group Chemical group 0.000 description 1
- 229960002568 ethinylestradiol Drugs 0.000 description 1
- HHFAWKCIHAUFRX-UHFFFAOYSA-N ethoxide Chemical compound CC[O-] HHFAWKCIHAUFRX-UHFFFAOYSA-N 0.000 description 1
- NAIAJPLUTCZXKC-UHFFFAOYSA-N ethyl 2-(4-bromo-2-fluorophenyl)acetate Chemical compound CCOC(=O)CC1=CC=C(Br)C=C1F NAIAJPLUTCZXKC-UHFFFAOYSA-N 0.000 description 1
- KKAFIMYLMXUFKH-UHFFFAOYSA-N ethyl 5-(4-bromophenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxylate Chemical compound BrC1=CC=C(C=C1)C=1C(C(=CN(C=1)CC1CCOCC1)C(=O)OCC)=O KKAFIMYLMXUFKH-UHFFFAOYSA-N 0.000 description 1
- UREBWPXBXRYXRJ-UHFFFAOYSA-N ethyl acetate;methanol Chemical compound OC.CCOC(C)=O UREBWPXBXRYXRJ-UHFFFAOYSA-N 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- 210000001508 eye Anatomy 0.000 description 1
- 230000003176 fibrotic effect Effects 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- DWYMPOCYEZONEA-UHFFFAOYSA-L fluoridophosphate Chemical compound [O-]P([O-])(F)=O DWYMPOCYEZONEA-UHFFFAOYSA-L 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- 239000004052 folic acid antagonist Substances 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 229960002584 gefitinib Drugs 0.000 description 1
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 230000009422 growth inhibiting effect Effects 0.000 description 1
- 230000009036 growth inhibition Effects 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 208000014829 head and neck neoplasm Diseases 0.000 description 1
- 238000003505 heat denaturation Methods 0.000 description 1
- 201000005787 hematologic cancer Diseases 0.000 description 1
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 1
- 125000004366 heterocycloalkenyl group Chemical group 0.000 description 1
- 230000036732 histological change Effects 0.000 description 1
- 102000045684 human MERTK Human genes 0.000 description 1
- GVLGAFRNYJVHBC-UHFFFAOYSA-N hydrate;hydrobromide Chemical compound O.Br GVLGAFRNYJVHBC-UHFFFAOYSA-N 0.000 description 1
- 229960000890 hydrocortisone Drugs 0.000 description 1
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- 239000012216 imaging agent Substances 0.000 description 1
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 1
- 229960002411 imatinib Drugs 0.000 description 1
- 229960003685 imatinib mesylate Drugs 0.000 description 1
- YLMAHDNUQAMNNX-UHFFFAOYSA-N imatinib methanesulfonate Chemical compound CS(O)(=O)=O.C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 YLMAHDNUQAMNNX-UHFFFAOYSA-N 0.000 description 1
- MTNDZQHUAFNZQY-UHFFFAOYSA-N imidazoline Chemical compound C1CN=CN1 MTNDZQHUAFNZQY-UHFFFAOYSA-N 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 229950008097 improsulfan Drugs 0.000 description 1
- 230000005917 in vivo anti-tumor Effects 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- JYGFTBXVXVMTGB-UHFFFAOYSA-N indolin-2-one Chemical group C1=CC=C2NC(=O)CC2=C1 JYGFTBXVXVMTGB-UHFFFAOYSA-N 0.000 description 1
- 229960000905 indomethacin Drugs 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 229960000598 infliximab Drugs 0.000 description 1
- 208000036971 interstitial lung disease 2 Diseases 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- ZCYVEMRRCGMTRW-YPZZEJLDSA-N iodine-125 Chemical compound [125I] ZCYVEMRRCGMTRW-YPZZEJLDSA-N 0.000 description 1
- 229940044173 iodine-125 Drugs 0.000 description 1
- 238000004255 ion exchange chromatography Methods 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 229960002725 isoflurane Drugs 0.000 description 1
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000000155 isotopic effect Effects 0.000 description 1
- 229960004891 lapatinib Drugs 0.000 description 1
- 239000012669 liquid formulation Substances 0.000 description 1
- 229910003002 lithium salt Inorganic materials 0.000 description 1
- 159000000002 lithium salts Chemical class 0.000 description 1
- 229960002247 lomustine Drugs 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000001165 lymph node Anatomy 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 229960004961 mechlorethamine Drugs 0.000 description 1
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical class ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 1
- FRQMUZJSZHZSGN-HBNHAYAOSA-N medroxyprogesterone Chemical compound C([C@@]12C)CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2CC[C@]2(C)[C@@](O)(C(C)=O)CC[C@H]21 FRQMUZJSZHZSGN-HBNHAYAOSA-N 0.000 description 1
- 229960004616 medroxyprogesterone Drugs 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- 230000001394 metastastic effect Effects 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- 229960003578 metenolone Drugs 0.000 description 1
- OIXMUQLVDNPHNS-UHFFFAOYSA-N methanesulfonic acid;hydrate Chemical compound O.CS(O)(=O)=O OIXMUQLVDNPHNS-UHFFFAOYSA-N 0.000 description 1
- 125000005948 methanesulfonyloxy group Chemical group 0.000 description 1
- ANJQEDFWRSLVBR-VHUDCFPWSA-N methenolone Chemical compound C1C[C@@H]2[C@@]3(C)C(C)=CC(=O)C[C@@H]3CC[C@H]2[C@@H]2CC[C@H](O)[C@]21C ANJQEDFWRSLVBR-VHUDCFPWSA-N 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- 238000006063 methoxycarbonylation reaction Methods 0.000 description 1
- MOSJHTAFHHUNRB-UHFFFAOYSA-N methyl 5-bromo-4-oxo-1h-pyridine-3-carboxylate Chemical compound COC(=O)C1=CNC=C(Br)C1=O MOSJHTAFHHUNRB-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 229960004584 methylprednisolone Drugs 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 229960005485 mitobronitol Drugs 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 150000002780 morpholines Chemical class 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- QLVWKBNYBAWIPW-UHFFFAOYSA-N n-quinolin-2-yloxybenzenesulfonamide Chemical group C=1C=C2C=CC=CC2=NC=1ONS(=O)(=O)C1=CC=CC=C1 QLVWKBNYBAWIPW-UHFFFAOYSA-N 0.000 description 1
- 229960004719 nandrolone Drugs 0.000 description 1
- NPAGDVCDWIYMMC-IZPLOLCNSA-N nandrolone Chemical compound O=C1CC[C@@H]2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 NPAGDVCDWIYMMC-IZPLOLCNSA-N 0.000 description 1
- 230000017066 negative regulation of growth Effects 0.000 description 1
- 208000025189 neoplasm of testis Diseases 0.000 description 1
- QMMRZOWCJAIUJA-UHFFFAOYSA-L nickel dichloride Chemical compound Cl[Ni]Cl QMMRZOWCJAIUJA-UHFFFAOYSA-L 0.000 description 1
- 229950010203 nimotuzumab Drugs 0.000 description 1
- 229960001420 nimustine Drugs 0.000 description 1
- VFEDRRNHLBGPNN-UHFFFAOYSA-N nimustine Chemical compound CC1=NC=C(CNC(=O)N(CCCl)N=O)C(N)=N1 VFEDRRNHLBGPNN-UHFFFAOYSA-N 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 229920001220 nitrocellulos Polymers 0.000 description 1
- OSTGTTZJOCZWJG-UHFFFAOYSA-N nitrosourea Chemical compound NC(=O)N=NO OSTGTTZJOCZWJG-UHFFFAOYSA-N 0.000 description 1
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 1
- 238000003305 oral gavage Methods 0.000 description 1
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 1
- 229960001756 oxaliplatin Drugs 0.000 description 1
- IPBPLHNLRKRLPJ-UHFFFAOYSA-N oxan-4-ylmethanamine Chemical compound NCC1CCOCC1 IPBPLHNLRKRLPJ-UHFFFAOYSA-N 0.000 description 1
- HJZXNSXKMBRTJZ-UHFFFAOYSA-N oxan-4-ylmethyl methanesulfonate Chemical compound CS(=O)(=O)OCC1CCOCC1 HJZXNSXKMBRTJZ-UHFFFAOYSA-N 0.000 description 1
- AHHWIHXENZJRFG-UHFFFAOYSA-N oxetane Chemical compound C1COC1 AHHWIHXENZJRFG-UHFFFAOYSA-N 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 229960005244 oxymetholone Drugs 0.000 description 1
- ICMWWNHDUZJFDW-DHODBPELSA-N oxymetholone Chemical compound C([C@@H]1CC2)C(=O)\C(=C/O)C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@](C)(O)[C@@]2(C)CC1 ICMWWNHDUZJFDW-DHODBPELSA-N 0.000 description 1
- ICMWWNHDUZJFDW-UHFFFAOYSA-N oxymetholone Natural products C1CC2CC(=O)C(=CO)CC2(C)C2C1C1CCC(C)(O)C1(C)CC2 ICMWWNHDUZJFDW-UHFFFAOYSA-N 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 229950003180 peplomycin Drugs 0.000 description 1
- QIMGFXOHTOXMQP-GFAGFCTOSA-N peplomycin Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCCN[C@@H](C)C=1C=CC=CC=1)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1NC=NC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C QIMGFXOHTOXMQP-GFAGFCTOSA-N 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- VLTRZXGMWDSKGL-UHFFFAOYSA-M perchlorate Inorganic materials [O-]Cl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-M 0.000 description 1
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 1
- 108040007629 peroxidase activity proteins Proteins 0.000 description 1
- 238000011170 pharmaceutical development Methods 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- TWHXWYVOWJCXSI-UHFFFAOYSA-N phosphoric acid;hydrate Chemical compound O.OP(O)(O)=O TWHXWYVOWJCXSI-UHFFFAOYSA-N 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- 150000004885 piperazines Chemical class 0.000 description 1
- NJBFOOCLYDNZJN-UHFFFAOYSA-N pipobroman Chemical compound BrCCC(=O)N1CCN(C(=O)CCBr)CC1 NJBFOOCLYDNZJN-UHFFFAOYSA-N 0.000 description 1
- 229960000952 pipobroman Drugs 0.000 description 1
- 229960001221 pirarubicin Drugs 0.000 description 1
- 108010001062 polysaccharide-K Proteins 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 229940094037 potassium bromate Drugs 0.000 description 1
- 235000019396 potassium bromate Nutrition 0.000 description 1
- 229960005205 prednisolone Drugs 0.000 description 1
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 description 1
- 238000012746 preparative thin layer chromatography Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 230000004147 pyrimidine metabolism Effects 0.000 description 1
- YAAWASYJIRZXSZ-UHFFFAOYSA-N pyrimidine-2,4-diamine Chemical group NC1=CC=NC(N)=N1 YAAWASYJIRZXSZ-UHFFFAOYSA-N 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- SBYHFKPVCBCYGV-UHFFFAOYSA-N quinuclidine Chemical compound C1CC2CCN1CC2 SBYHFKPVCBCYGV-UHFFFAOYSA-N 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 229960002185 ranimustine Drugs 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 238000007670 refining Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 201000002793 renal fibrosis Diseases 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000013557 residual solvent Substances 0.000 description 1
- 230000008261 resistance mechanism Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 230000002207 retinal effect Effects 0.000 description 1
- 229960004641 rituximab Drugs 0.000 description 1
- 239000012488 sample solution Substances 0.000 description 1
- 125000002542 secosteroid group Chemical group 0.000 description 1
- 229960003440 semustine Drugs 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 1
- 235000020183 skimmed milk Nutrition 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 201000002471 spleen cancer Diseases 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 229960001052 streptozocin Drugs 0.000 description 1
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 125000000565 sulfonamide group Chemical group 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 1
- 229960001796 sunitinib Drugs 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 229960001674 tegafur Drugs 0.000 description 1
- WFWLQNSHRPWKFK-ZCFIWIBFSA-N tegafur Chemical compound O=C1NC(=O)C(F)=CN1[C@@H]1OCCC1 WFWLQNSHRPWKFK-ZCFIWIBFSA-N 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 201000003120 testicular cancer Diseases 0.000 description 1
- 125000004853 tetrahydropyridinyl group Chemical group N1(CCCC=C1)* 0.000 description 1
- QEMXHQIAXOOASZ-UHFFFAOYSA-N tetramethylammonium Chemical class C[N+](C)(C)C QEMXHQIAXOOASZ-UHFFFAOYSA-N 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 229960001196 thiotepa Drugs 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- 229960003087 tioguanine Drugs 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 230000014616 translation Effects 0.000 description 1
- 229960000575 trastuzumab Drugs 0.000 description 1
- 229960005294 triamcinolone Drugs 0.000 description 1
- GFNANZIMVAIWHM-OBYCQNJPSA-N triamcinolone Chemical compound O=C1C=C[C@]2(C)[C@@]3(F)[C@@H](O)C[C@](C)([C@@]([C@H](O)C4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 GFNANZIMVAIWHM-OBYCQNJPSA-N 0.000 description 1
- 150000003852 triazoles Chemical group 0.000 description 1
- OVCXRBARSPBVMC-UHFFFAOYSA-N triazolopyridine Chemical compound C=1N2C(C(C)C)=NN=C2C=CC=1C=1OC=NC=1C1=CC=C(F)C=C1 OVCXRBARSPBVMC-UHFFFAOYSA-N 0.000 description 1
- YWBFPKPWMSWWEA-UHFFFAOYSA-O triazolopyrimidine Chemical group BrC1=CC=CC(C=2N=C3N=CN[N+]3=C(NCC=3C=CN=CC=3)C=2)=C1 YWBFPKPWMSWWEA-UHFFFAOYSA-O 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- 125000005951 trifluoromethanesulfonyloxy group Chemical group 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical class OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 102000003390 tumor necrosis factor Human genes 0.000 description 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 1
- 229910052721 tungsten Inorganic materials 0.000 description 1
- 229950009811 ubenimex Drugs 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- XSQUKJJJFZCRTK-UHFFFAOYSA-N urea group Chemical group NC(=O)N XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- UGGWPQSBPIFKDZ-KOTLKJBCSA-N vindesine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(N)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 UGGWPQSBPIFKDZ-KOTLKJBCSA-N 0.000 description 1
- 229960004355 vindesine Drugs 0.000 description 1
- 235000019155 vitamin A Nutrition 0.000 description 1
- 239000011719 vitamin A Substances 0.000 description 1
- 235000019154 vitamin C Nutrition 0.000 description 1
- 239000011718 vitamin C Substances 0.000 description 1
- 229940045997 vitamin a Drugs 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Images






















Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/501—Pyridazines; Hydrogenated pyridazines not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B59/00—Introduction of isotopes of elements into organic compounds ; Labelled organic compounds per se
- C07B59/002—Heterocyclic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/01—Sulfonic acids
- C07C309/02—Sulfonic acids having sulfo groups bound to acyclic carbon atoms
- C07C309/03—Sulfonic acids having sulfo groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
- C07C309/04—Sulfonic acids having sulfo groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton containing only one sulfo group
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/01—Sulfonic acids
- C07C309/28—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C309/33—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton of six-membered aromatic rings being part of condensed ring systems
- C07C309/34—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton of six-membered aromatic rings being part of condensed ring systems formed by two rings
- C07C309/35—Naphthalene sulfonic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/05—Isotopically modified compounds, e.g. labelled
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Definitions
- the present invention relates to a compound having Axl inhibitory activity or a salt thereof.
- Axl is a receptor tyrosine kinase belonging to the Tyro3-Axl-Mer (TAM) receptor tyrosine kinase family with a growth arrest specific gene 6 (Gas6) protein as a ligand, and was initially identified as a transforming gene in chronic myeloid leukemia (Non-Patent Document 1).
- the Gas6 / Axl signaling system regulates various cellular responses such as cell survival, cell division, autophagy, cell migration, angiogenesis, platelet aggregation, NK cell differentiation, etc.
- Non-patent document 2 primary colon cancer (non-patent document 3), stomach cancer (non-patent document 4), esophageal cancer (non-patent document 5), melanoma (non-patent document 6),
- Many overexpressions in cancer tissues such as ovarian cancer (Non-patent document 7), kidney cancer (Non-patent document 8), endometrial cancer (Non-patent document 9), thyroid cancer (Non-patent document 10) have been reported. . It has also been shown that the presence of Axl is greatly related to lymph node status and stage in lung cancer and ER expression in breast cancer (Non-Patent Document 11).
- Axl is immune (non-patent document 12), platelet function (non-patent document 13), spermatogenesis (non-patent document 14), vascular calcification (non-patent document 15), thrombin-induced vascular smooth muscle cells (VSMC). It has also been shown to have a role in proliferation (Non-Patent Document 16) and various kidney diseases such as acute and chronic glomerulonephritis, diabetic nephropathy and chronic allograft rejection (Non-Patent Document 17).
- Inhibitors include but are not limited to cancer (including solid tumors such as carcinomas, sarcomas and leukemias and lymphoid malignancies), as well as vascular diseases (including thrombosis, atherosclerosis and restenosis). ), Kidney disease (including but not limited to acute and chronic glomerulonephritis, diabetic nephropathy and transplant rejection), and disordered angiogenesis are critical Expected to be beneficial in the treatment of a number of diseases, including but not limited to diabetic retinopathy, retinopathy, psoriasis, rheumatoid arthritis, atheroma, Kaposi's sarcoma and hemangiomas Has been.
- cancer including solid tumors such as carcinomas, sarcomas and leukemias and lymphoid malignancies
- vascular diseases including thrombosis, atherosclerosis and restenosis.
- Kidney disease including but not limited to acute and chronic glomerulonephritis,
- Non-patent Document 24 a member of the TAM receptor tyrosine kinase family to which Axl belongs, autosomal recessive retinitis has been reported to occur due to its homozygous mutation. It has been reported that certain mutations of Mer are associated with childhood-onset rhodcone dystrophy (Non-patent Document 25).
- Examples of compounds that inhibit Axl include compounds having a sulfonamide structure (Patent Document 3), compounds having a pyrrolopyrimidine structure (Patent Documents 4 and 5), compounds having a pyridine and a pyrazine structure (Patent Document 6), and pyrazine.
- a compound having a structure (Patent Document 7), a compound having a pyrazinylbenzimidazole structure (Patent Document 8), a compound having an indolinone structure (Patent Document 9), a compound having a triazolopyridine and a triazolopyrimidine structure (Patent Document) 10), a compound having an imidazole structure (Patent Document 11), a compound having a triazole structure (Patent Document 12, Patent Document 13, Patent Document 14, Patent Document 15, Patent Document 16, Patent Document 17, Patent Document 20, Patent Document) 24, Patent Document 25, Patent Document 26, Patent Document 27, Patent 28), compounds having a pyrimidinediamine structure (Patent Document 18), compounds having a pyrimidine structure (Patent Document 19, Non-Patent Document 18, Non-Patent Document 22), compounds having a quinolinyloxyphenylsulfonamide structure (Patent Document 19) 21), compounds having a quinoline structure (Patent Document 22, Patent Document 30, Non-Patent Document 21),
- the present invention provides an Axl inhibitory compound having a high inhibitory specificity for Axl and higher safety.
- the present invention also provides a therapeutic agent for a disease caused by hyperfunction of Axl, a therapeutic agent for a disease associated with hyperfunction of Axl, and / or treatment of a disease associated with hyperfunction of Axl, comprising an Axl inhibitory compound.
- An agent such as an anticancer agent is provided.
- Example 9 (hereinafter referred to as Compound A) of Patent Document 33 has high Axl inhibitory activity, it was confirmed to cause irreversible retinal photoreceptor degeneration in a mouse long-term administration test.
- Mer which is a member of the same TAM family as Axl, the relationship between inactivation and retinal cell degeneration has been reported, and the compound of Example 9 of Patent Document 33 also has inhibitory activity against Mer.
- the present inventor considered that Mer inhibition of the compound leads to retinal toxicity.
- the present inventor has found that a compound having a structure represented by the following general formula (I) or a salt thereof has high Axl inhibitory specificity and low inhibitory activity against Mer.
- R 2 represents a hydrogen atom, a halogen atom, a C 1 -C 6 alkyl group, a C 1 -C 6 alkoxy group or a cyano group
- R 3 represents a hydrogen atom or a C 1 -C 6 alkyl group
- R 4 represents a hydrogen atom, a halogen atom or a C 1 -C 6 alkyl group
- R 5 represents a hydrogen atom or the following formula (III-1), (III-2), (III-3) or (III-4)
- R 8 and R 12 each independently represent a hydrogen atom or a deuterium atom
- R 9 represents a hydrogen atom, a halogen atom or a C 1 -C 6 alkoxy group
- R 10 is a hydrogen atom, optionally substituted heterocycloalkyl group optionally substituted C 1 have ⁇ C 6 alkyl group or a C 1 ⁇ C 6 optionally substituted by an alkyl group heterocycloalkyl group
- the sulfate crystals according to [13] which show characteristic peaks at 04, 19.42, 19.70, 20.12, 20.42 and 21.32.
- An Axl inhibitor comprising the compound according to any one of [1] to [10] or a pharmaceutically acceptable salt thereof.
- a medicament comprising as an active ingredient the compound according to any one of [1] to [10] or a pharmaceutically acceptable salt thereof.
- a disease caused by increased Axl kinase function comprising a compound according to any one of [1] to [10] or a pharmaceutically acceptable salt thereof, a disease associated with increased Axl kinase function And / or a medicament for the treatment of a disease associated with Axl kinase hyperfunction.
- a medicament for the treatment of hyperproliferative diseases comprising the compound according to any one of [1] to [10] or a pharmaceutically acceptable salt thereof as an active ingredient.
- a medicament for treating cancer comprising the compound according to any one of [1] to [10] or a pharmaceutically acceptable salt thereof as an active ingredient.
- a medicament for preventing metastasis of cancer comprising the compound according to any one of [1] to [10] or a pharmaceutically acceptable salt thereof as an active ingredient.
- a medicament for releasing drug resistance of cancer comprising as an active ingredient the compound according to any one of [1] to [10] or a pharmaceutically acceptable salt thereof.
- a medicament for inhibiting the acquisition of drug resistance in cancer comprising the compound according to any one of [1] to [10] or a pharmaceutically acceptable salt thereof as an active ingredient.
- Cancer is breast cancer, colon cancer, prostate cancer, lung cancer, gastric cancer, ovarian cancer, endometrial cancer, renal cancer, hepatocellular carcinoma, thyroid cancer, uterine cancer, esophageal cancer, squamous cell carcinoma, leukemia, osteosarcoma
- a pharmaceutical composition comprising the compound according to any one of [1] to [10] or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier.
- a disease caused by increased Axl kinase function characterized by using the compound according to any one of [1] to [10] or a pharmaceutically acceptable salt thereof, and associated with increased Axl kinase function And / or a method for treating a disease associated with increased Axl kinase function.
- a method for treating a hyperproliferative disease comprising using the compound according to any one of [1] to [10] or a pharmaceutically acceptable salt thereof.
- a method for treating cancer comprising using the compound according to any one of [1] to [10] or a pharmaceutically acceptable salt thereof.
- a method for preventing cancer metastasis comprising using the compound according to any one of [1] to [10] or a pharmaceutically acceptable salt thereof.
- a method for releasing drug resistance of cancer comprising using the compound according to any one of [1] to [10] or a pharmaceutically acceptable salt thereof.
- a method for inhibiting the acquisition of drug resistance in cancer comprising using the compound according to any one of [1] to [10] or a pharmaceutically acceptable salt thereof.
- Cancer is breast cancer, colon cancer, prostate cancer, lung cancer, gastric cancer, ovarian cancer, endometrial cancer, renal cancer, hepatocellular carcinoma, thyroid cancer, uterine cancer, esophageal cancer, squamous cell carcinoma, leukemia, osteosarcoma
- a compound represented by the above formula (I) having Axl inhibitory activity are useful as therapeutic agents, for example, anticancer agents, for diseases caused by increased Axl function, diseases associated with increased Axl function, and / or diseases associated with increased Axl function.
- FIG. 1 is a powder X-ray diffraction pattern of the crystal obtained in Example 130.
- FIG. 2 is a powder X-ray diffraction pattern of the crystal obtained in Example 131.
- the vertical axis indicates the diffraction intensity (Intensity) in units of count / second (cps)
- the horizontal axis indicates the value of the diffraction angle 2 ⁇ .
- 3 is a powder X-ray diffraction pattern of the crystal obtained in Example 132.
- the vertical axis indicates the diffraction intensity (Intensity) in units of count / second (cps), and the horizontal axis indicates the value of the diffraction angle 2 ⁇ .
- 4 is a powder X-ray diffraction pattern of the crystal obtained in Example 133.
- FIG. in the figure, the vertical axis indicates the diffraction intensity (Intensity) in units of count / second (cps), and the horizontal axis indicates the value of the diffraction angle 2 ⁇ .
- FIG. 5 is a powder X-ray diffraction pattern of the crystal obtained in Example 134.
- the vertical axis indicates the diffraction intensity (Intensity) in units of count / second (cps), and the horizontal axis indicates the value of the diffraction angle 2 ⁇ .
- 6 is a powder X-ray diffraction pattern of the crystal obtained in Example 135.
- FIG. in the figure, the vertical axis indicates the diffraction intensity (Intensity) in units of count / second (cps), and the horizontal axis indicates the value of the diffraction angle 2 ⁇ .
- FIG. 7 is a powder X-ray diffraction pattern of the crystals obtained in Example 136.
- FIG. 8 is a powder X-ray diffraction pattern of the crystal obtained in Example 137.
- the vertical axis indicates the diffraction intensity (Intensity) in units of count / second (cps), and the horizontal axis indicates the value of the diffraction angle 2 ⁇ .
- FIG. 9 is a powder X-ray diffraction pattern of the crystals obtained in Example 138.
- the vertical axis indicates the diffraction intensity (Intensity) in units of count / second (cps), and the horizontal axis indicates the value of the diffraction angle 2 ⁇ .
- 10 is a powder X-ray diffraction pattern of the crystal obtained in Example 139.
- FIG. in the figure, the vertical axis indicates the diffraction intensity (Intensity) in units of count / second (cps), and the horizontal axis indicates the value of the diffraction angle 2 ⁇ .
- FIG. 11 is a powder X-ray diffraction pattern of the crystal obtained in Example 140.
- the vertical axis indicates the diffraction intensity (Intensity) in units of count / second (cps), and the horizontal axis indicates the value of the diffraction angle 2 ⁇ .
- 12 is a powder X-ray diffraction pattern of the crystals obtained in Example 141.
- FIG. in the figure, the vertical axis indicates the diffraction intensity (Intensity) in units of count / second (cps), and the horizontal axis indicates the value of the diffraction angle 2 ⁇ .
- FIG. 13 is a powder X-ray diffraction pattern of the crystal obtained in Example 142.
- FIG. 14 is a powder X-ray diffraction pattern of the crystal obtained in Example 143.
- the vertical axis indicates the diffraction intensity (Intensity) in units of count / second (cps)
- the horizontal axis indicates the value of the diffraction angle 2 ⁇ .
- FIG. 15 is a powder X-ray diffraction pattern of the crystals obtained in Example 144.
- FIG. 16 is a powder X-ray diffraction pattern of the crystal obtained in Example 145.
- the vertical axis indicates the diffraction intensity (Intensity) in units of count / second (cps), and the horizontal axis indicates the value of the diffraction angle 2 ⁇ .
- FIG. 17 is a powder X-ray diffraction pattern of the crystal obtained in Example 146.
- FIG. 18 is a powder X-ray diffraction pattern of the crystal obtained in Example 147.
- the vertical axis indicates the diffraction intensity (Intensity) in units of count / second (cps)
- the horizontal axis indicates the value of the diffraction angle 2 ⁇ .
- 19-1 is a graph showing the combined effect of erlotinib and the Axl inhibitory compound of the present application on an HCC827 mouse subcutaneously transplanted tumor having a deletion mutation in the EGFR gene exon 19 and high sensitivity to an EGFR inhibitor.
- Symbol x indicates vehicle administration group (administration 3 times a day (Tid))
- symbol white circle indicates Compound B sulfate hydrate 50 mg / kg (administration twice a day (bid))
- symbol black circle indicates erlotinib 25 mg / kg (daily) Single dose (qd))
- symbol white triangles indicate Compound B sulfate hydrate 50 mg / kg (bid) + erlotinib 25 mg / kg (qd).
- FIG. 19-2 is a diagram showing an average value of the weight change rate of each individual in a combined test of erlotinib and the Axl inhibitory compound of the present application (the legend is the same as FIG. 19-1).
- FIG. 20-1 shows the antitumor effect of erlotinib on HCC827 mouse subcutaneously transplanted tumor.
- Symbol x indicates vehicle administration group
- symbol white circle indicates erlotinib 25 mg / kg
- symbol black circle indicates erlotinib 12.5 mg / kg
- symbol white triangle indicates erlotinib 6.25 mg / kg.
- FIG. 20-2 is a diagram showing changes in the expression level of Axl protein upon administration of erlotinib in HCC827 mouse subcutaneously transplanted tumors.
- Axl refers to a protein encoded by the Axl gene.
- Axl includes an Axl protein encoded by a full-length Axl gene or an Axl protein encoded by an Axl gene mutant (including a deletion mutant, a substitution mutant, or an additional mutant).
- Axl includes homologs derived from various animal species.
- Axl inhibitor refers to an inhibitor of the function of Axl as a tyrosine kinase.
- tumor and “cancer” are used interchangeably.
- a tumor, a malignant tumor, a cancer, a malignant neoplasm, a carcinoma, a sarcoma, etc. are collectively referred to as “tumor” or “sarcoma”.
- cancer often referred to as “cancer”.
- C 1 ⁇ C 6 “Alkyl group” means a linear or branched alkyl group having 1 to 6 carbon atoms.
- C 1 ⁇ C 6 Examples of the “alkyl group” include methyl group, ethyl group, propyl group, isopropyl group, butyl group, tert-butyl group and the like.
- alkoxy group means an alkoxy group having a linear or branched alkyl group having 1 to 6 carbon atoms.
- C 1 ⁇ C 6 Examples of the “alkoxy group” include a methoxy group, an ethoxy group, a propoxy group, an isopropoxy group, and a butoxy group.
- halogen atom include a fluorine atom, a chlorine atom, a bromine atom, and an iodine atom.
- cycloalkyl group means a cyclic alkyl group having 3 to 8 carbon atoms unless otherwise specified, and examples thereof include a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, and a cyclohexyl group.
- heterocycloalkyl group means a monovalent saturated heterocyclic group, and includes a saturated heterocyclic group having a nitrogen atom in the ring, a saturated heterocyclic group having an oxygen atom in the ring, and the like, for example, pyrrolidine, And monovalent groups derived from imidazoline, piperidine, piperazine, azetidine, morpholine, dioxane, oxetane, tetrahydropyran, and quinuclidine.
- Examples of the “cycloalkenyl group” include those having an unsaturated bond such as one or more double bonds in the above “cycloalkyl group”, and examples thereof include a cyclopentenyl group and a cyclohexenyl group.
- Examples of the “heterocycloalkenyl group” include those having an unsaturated bond such as one or more double bonds in the above “heterocycloalkyl group”.
- a tetrahydropyridinyl group and a dihydropyranyl group are Can be mentioned. Below, each substituent in Formula (I) is demonstrated.
- W, X, and Y each independently represent a nitrogen atom, C—H, C—F, or C—Cl.
- R 1 Is represented by the following formula (II-1) or (II-2) (In formula (II-1), Q and T each independently represent a nitrogen atom, C—H or C—F, T represents a nitrogen atom or C—H, and U represents a nitrogen atom or C— -H, R 6 Is a halogen atom, C 1 ⁇ C 6 Alkyl group, C 3 ⁇ C 6 A cycloalkyl group, a cyano group or a trifluoromethoxy group, in the formula (II-2), V represents a sulfur atom or an oxygen atom; 7 Is C 1 ⁇ C 6 Indicates an alkyl group.
- R 2 Is a hydrogen atom, a halogen atom, C 1 ⁇ C 6 Alkyl group, C 1 ⁇ C 6 An alkoxy group or a cyano group is shown. Where C 1 ⁇ C 6 As the alkyl group, a methyl group, an ethyl group or a propyl group is preferable, and C 1 ⁇ C 6 As an alkoxy group, a methoxy group and an ethoxy group are preferable.
- R 3 Is a hydrogen atom or C 1 ⁇ C 6 Represents an alkyl group, C 1 ⁇ C 6 As the alkyl group, a methyl group, an ethyl group or a propyl group is preferable.
- R 4 Is a hydrogen atom, a halogen atom or C 1 ⁇ C 6 Represents an alkyl group
- R 4 As a hydrogen atom, a fluorine atom, a chlorine atom, a methyl group or an ethyl group is preferable.
- R 5 Is a hydrogen atom or the following formula (III-1), (III-2), (III-3) or (III-4)
- R 8 And R 12 Each independently represents a hydrogen atom or a deuterium atom
- R 9 Is a hydrogen atom, a halogen atom or C 1 ⁇ C 6 Represents an alkoxy group
- R 10 Is a hydrogen atom, C optionally substituted by a heterocycloalkyl group 1 ⁇ C 6
- R 11 Is a hydrogen atom or C 1 ⁇ C 6 C substituted with an alkoxy group or deuterium 1 ⁇ C 6 Represents an alkoxy group
- R 13 Is optionally substituted with a heterocycloalkyl group 1 ⁇ C 6 Represents an alkyl
- R 5 Is a group represented by the formula (III-1), R 10
- the heterocycloalkyl group in is preferably a dioxanyl group, a morpholino group, a piperazinyl group or a tetrahydropyranyl group, 1 ⁇ C 6
- the alkyl group is preferably a methoxy group or an ethoxy group.
- the alkoxy group is preferably a methoxy group or an ethoxy group.
- R 5 Is a group represented by the formula (III-2), R 13
- the heterocycloalkyl group in is preferably a dioxanyl group, a morpholino group, a piperazinyl group or a tetrahydropyranyl group, and R 13 C in 1 ⁇ C 6
- the alkyl group is preferably a methyl group or an ethyl group.
- the compound represented by the general formula (I) or a pharmaceutically acceptable salt thereof is more preferably any one of the following compounds or a pharmaceutically acceptable salt thereof.
- the compound represented by the formula (I) of the present invention may have stereoisomers or optical isomers derived from asymmetric carbon atoms. These stereoisomers, optical isomers, and mixtures thereof Any of these are included in the present invention.
- the compound represented by the general formula (I) of the present invention has a basic group such as an amino group, it can be converted into a pharmaceutically acceptable salt if desired.
- salts include hydrohalides such as hydrochloride, hydrobromide and hydroiodide; inorganic acid salts such as nitrate, perchlorate, sulfate and phosphate; methanesulfone Lower alkane sulfonates such as acid salts, trifluoromethane sulfonate and ethane sulfonate; aryl sulfonates such as benzene sulfonate and p-toluene sulfonate; formic acid, acetic acid, malic acid and fumarate And organic acid salts such as succinate, citrate, tartrate, succinate and maleate; and amino acid salts such as ornithate, glutamate and aspartate; and hydrohalic acid Salts and organic acid salts are preferred.
- hydrohalides such as hydrochloride, hydrobromide and hydroiodide
- inorganic acid salts such as nitrate, perchlorate, s
- the pharmaceutically acceptable salt include alkali metal salts such as sodium salt, potassium salt and lithium salt; alkaline earth metal salts such as calcium salt and magnesium salt; inorganic salts such as ammonium salt; dibenzylamine salt , Morpholine salt, phenylglycine alkyl ester salt, ethylenediamine salt, N-methylglucamine salt, diethylamine salt, triethylamine salt, cyclohexylamine salt, dicyclohexylamine salt, N, N′-dibenzylethylenediamine salt, diethanolamine salt, N-benzyl And organic amine salts such as -N- (2-phenylethoxy) amine salt, piperazine salt, tetramethylammonium salt, and tris (hydroxymethyl) aminomethane salt.
- alkali metal salts such as sodium salt, potassium salt and lithium salt
- alkaline earth metal salts such as calcium salt and magnesium salt
- inorganic salts such as ammonium salt
- the compound represented by the general formula (I) of the present invention or a salt thereof may exist as a free form or a solvate. It may exist as a hydrate by absorbing moisture in the air.
- the solvate is not particularly limited as long as it is pharmaceutically acceptable, and specifically, a hydrate, an ethanolate, and the like are preferable.
- a nitrogen atom is present in the compound of the present invention represented by the general formula (I), it may be an N-oxide, and these solvates and N-oxides are also within the scope of the present invention. included.
- the present compound or a salt thereof includes those crystals.
- the salt includes N- [4- (2-amino-5- ⁇ 4-[(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl ⁇ pyridine-3- Yl) phenyl] -5- (4-methylphenyl) -4-oxo-1- (tetrahydro-2H-pyran-4-ylmethyl) -1,4-dihydropyridine-3-carboxamide hydrobromide, nitrate, Sulfates, phosphates, ethanesulfonates, benzenesulfonates or p-toluenesulfonates are mentioned.
- a crystal means a solid whose internal structure is made up of regularly repeated constituent atoms (or a group thereof) in a three-dimensional manner, and is distinguished from an amorphous solid that does not have such a regular internal structure. . Even crystals of the same compound may produce crystals (crystal polymorphs) having a plurality of different internal structures and physicochemical properties depending on the crystallization conditions. Any form may be sufficient and the mixture of two or more crystal polymorphs may be sufficient.
- the crystal of the present invention absorbs moisture by being left in the atmosphere, and forms a hydrate by adhering water or heating to 25 to 150 ° C. under normal atmospheric conditions. There is a case.
- the crystal of this invention may contain the solvent at the time of crystallization in the adhesion residual solvent or solvate.
- the crystal of the present invention may be represented based on powder X-ray diffraction data.
- powder X-ray diffraction is usually measured and analyzed by a technique used in this field. This can be done by the method described in the examples.
- the lattice constant of hydrates and dehydrates changes depending on the attachment and detachment of crystal water, which may change the diffraction angle (2 ⁇ ) in powder X-ray diffraction.
- the intensity of the peak may change due to a difference in crystal growth surface or the like (crystal habit).
- hydrates and dehydrates obtained therefrom are also included within the scope of the present invention.
- a more specific embodiment of the present invention is N- [4- (2-amino-5- ⁇ 4-[(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl ⁇ pyridine-3.
- a more specific embodiment of the present invention is N- [4- (2-amino-5- ⁇ 4-[(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl ⁇ pyridine-3. -Yl) phenyl] -5- (4-methylphenyl) -4-oxo-1- (tetrahydro-2H-pyran-4-ylmethyl) -1,4-dihydropyridine-3-carboxamide hydrobromide crystals About.
- a more specific embodiment of the present invention is N- [4- (2-amino-5- ⁇ 4-[(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl ⁇ pyridine-3. -Yl) phenyl] -5- (4-methylphenyl) -4-oxo-1- (tetrahydro-2H-pyran-4-ylmethyl) -1,4-dihydropyridine-3-carboxamide nitrate crystals.
- a more specific embodiment of the present invention is N- [4- (2-amino-5- ⁇ 4-[(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl ⁇ pyridine-3. -Yl) phenyl] -5- (4-methylphenyl) -4-oxo-1- (tetrahydro-2H-pyran-4-ylmethyl) -1,4-dihydropyridine-3-carboxamide sulfate crystals.
- a more specific embodiment of the present invention is N- [4- (2-amino-5- ⁇ 4-[(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl ⁇ pyridine-3. -Yl) phenyl] -5- (4-methylphenyl) -4-oxo-1- (tetrahydro-2H-pyran-4-ylmethyl) -1,4-dihydropyridine-3-carboxamide phosphate crystals.
- diffraction angles 2 ⁇ 3.74, 7.56, 8.80, 9.56, 11 ., 34, 14.56, 15.74, 23.68, 24.34 and 24.68 showing characteristic peaks N- [4- (2-amino-5- ⁇ 4-[(2R) -1, 4-Dioxane-2-ylmethoxy] -3-methoxyphenyl ⁇ pyridin-3-yl) phenyl] -5- (4-methylphenyl) -4-oxo-1- (tetrahydro-2H-pyran-4-ylmethyl)-
- the present invention relates to crystals of phosphate of 1,4-dihydropyridine-3-carboxamide.
- a more specific embodiment of the present invention is N- [4- (2-amino-5- ⁇ 4-[(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl ⁇ pyridine-3. -Yl) phenyl] -5- (4-methylphenyl) -4-oxo-1- (tetrahydro-2H-pyran-4-ylmethyl) -1,4-dihydropyridine-3-carboxamide crystals of ethanesulfonate .
- a more specific embodiment of the present invention is N- [4- (2-amino-5- ⁇ 4-[(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl ⁇ pyridine-3. -Yl) phenyl] -5- (4-methylphenyl) -4-oxo-1- (tetrahydro-2H-pyran-4-ylmethyl) -1,4-dihydropyridine-3-carboxamide crystals of benzenesulfonate .
- a more specific embodiment of the present invention is N- [4- (2-amino-5- ⁇ 4-[(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl ⁇ pyridine-3. -Yl) phenyl] -5- (4-methylphenyl) -4-oxo-1- (tetrahydro-2H-pyran-4-ylmethyl) -1,4-dihydropyridine-3-carboxamide p-toluenesulfonate Relates to crystals.
- a more specific embodiment of the present invention is N- [4- (2-amino-5- ⁇ 4-[(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl ⁇ pyridine-3. -Yl) -3-fluorophenyl] -5-methyl-4'-oxo-1 '-(tetrahydro-2H-pyran-4-ylmethyl) -1', 4'-dihydro-2,3'-bipyridine-5 '-Relates to crystals of carboxamide phosphate.
- the present invention relates to crystals of phosphate of -1 ′, 4′-dihydro-2,3′-bipyridine-5′-carboxamide.
- a more specific embodiment of the present invention is N- [4- (2-amino-5- ⁇ 4-[(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl ⁇ pyridine-3. -Yl) -3-fluorophenyl] -5-methyl-4'-oxo-1 '-(tetrahydro-2H-pyran-4-ylmethyl) -1', 4'-dihydro-2,3'-bipyridine-5 '-Relates to crystals of carboxamide sulfate.
- the present invention relates to a sulfate crystal of -1 ′, 4′-dihydro-2,3′-bipyridine-5′-carboxamide.
- a more specific embodiment of the present invention is N- [4- (2-amino-5- ⁇ 4-[(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl ⁇ pyridine-3.
- diffraction angles 2 ⁇ 9.24, 9.58, 14.00, 14.46, N- [4- (2-amino-5- ⁇ 4-[(2R) -1] showing characteristic peaks at 16.70, 17.02, 18.22, 20.24, 21.64 and 25.52.
- the compound represented by the general formula (I) of the present invention has a geometric isomer such as a cis isomer, a trans isomer, a tautomer, an optical isomer such as a d isomer, an l isomer, etc.
- the compounds of the present invention include all isomers, stereoisomers, and any ratios of these isomers and stereoisomer mixtures, unless otherwise specified. It is.
- the compound represented by the general formula (I) of the present invention may contain an unnatural proportion of atomic isotopes at one or more of the constituent atoms.
- an atomic isotope for example, deuterium ( 2 H), sometimes referred to as D in this specification.
- the present invention also relates to a compound which is converted into a compound represented by the general formula (I) which is an active ingredient of the pharmaceutical composition of the present invention by a reaction with an enzyme, gastric acid or the like under physiological conditions in vivo, that is, an enzyme In particular, oxidation, reduction, hydrolysis, etc.
- “Pharmaceutically acceptable prodrug compounds” are also included in the present invention.
- the prodrug when an amino group is present in the compound represented by the general formula (I), a compound in which the amino group is acylated, alkylated or phosphorylated (for example, the amino group is eicosanoylated).
- a carboxy group is present in the compound represented by the general formula (I)
- a compound in which the carboxy group is esterified or amidated for example, the carboxy group is ethyl esterified, phenyl esterified, carboxy Methyl esterification, dimethylaminomethyl esterification, pivaloyloxymethyl esterification, ethoxycarbonyloxyethyl esterification, amidation or methylamidated compound, etc.
- the prodrug of the compound of the present invention can be produced from the compound represented by formula (I) by a known method.
- the prodrug of the compound of the present invention is represented by the general formula (I) under physiological conditions as described in Hirokawa Shoten 1990, “Pharmaceutical Development”, Volume 7, Molecular Design, pages 163 to 198. Those that change to a compound are also included.
- typical production methods for the compound represented by formula (I) will be described.
- the compound of the present invention can be produced by various production methods, and the production methods shown below are merely examples, and the present invention should not be construed as being limited thereto.
- the substituent can be protected with an appropriate protecting group, and the kind of the protecting group is not particularly limited.
- the general formula (I) can be divided into a site A, a site B, and a site C.
- compound (B) and compound (CI) are first combined, and the complex and compound (AI) are combined, or compound (AI) and compound are first combined.
- a method in which (B) is bound and the complex is bound to compound (CI), or compound (B) and compound (CI) are first bound, and the complex and compound (A-II) are bound.
- R 31 Is a substituent necessary for bonding the compound (B) to the compound (C-I) or the compound (C-II), and represents a halogen group, a triflate group, a boronic acid or a boronic acid ester.
- R 32 Is a substituent necessary for bonding the compound (B) and the compound (CI) and represents a halogen group, a triflate group, a boronic acid, or a boronic acid ester.
- R 33 Is a substituent necessary for bonding the compound (B) and the compound (C-II), and represents boronic acid or boronic acid ester.
- Reaction Scheme 1 Compound (AI) can be synthesized by a method as shown in Reaction Scheme 1 below, but is not particularly limited.
- Reaction formula 1 [Wherein R 1 ⁇ R 3, Q, T, and U are as defined above.
- R 41 Represents a halogen group.
- R 42 Is a protecting group for carboxylic acid and C 1 -C 6 Indicates an alkyl group.
- R 43 Represents boronic acid or boronic acid ester.
- R 44 Is C 1 ⁇ C 6 Alkyl groups and cyclo-C 1 -C 6 Indicates an alkyl group.
- Compound (1d) is included in compound (1e).
- the above reaction is carried out using triphenylphosphine, tricyclohexylphosphine, dibenzylideneacetone, 1,3-bis (diphenylphosphino) propane, 2-dicyclohexylphosphino-2 ′, 4 ′, 6′-triisopropylbiphenyl. , 4,5-bis (diphenylphosphino) -9,9-dimethylxanthene and the like.
- the above reaction can be performed by using two or more kinds of the above transition metal catalysts in combination.
- the solvent examples include 1,4-dioxane, 1,2-dimethoxyethane, methanol, toluene, water, and mixed solvents thereof, and are not particularly limited.
- the reaction temperature is usually in the range from 0 ° C. to 200 ° C. or the boiling point of the solvent, but preferably in the range from 0 ° C. to the boiling point of the solvent.
- the said process can also be performed by a microwave in a sealed tube.
- the organic or inorganic base may be used in an amount of 1 to excess molar equivalents relative to compound (1a). Preferably, 1 to 5 molar equivalents may be used.
- the transition metal catalyst and the ligand may be used in an amount of 0.001 to 1 molar equivalent relative to the compound (1c). Preferably, 0.05 to 1 molar equivalent may be used.
- the boronic acid ester may be used in an amount of 1 to excess molar equivalents relative to compound (1c). Preferably, 1 to 5 molar equivalents may be used.
- Synthesis of compound (1d) The compound (1c) can be obtained by hydrolyzing with an acid such as sulfuric acid or hydrochloric acid, or an alkali such as sodium hydroxide or potassium carbonate.
- the base or acid may be used in an amount of 1 to excess molar equivalents relative to compound (1c).
- Examples of the solvent used in the reaction include methanol, ethanol, propanol, water and the like, or a mixed solvent thereof, and are not particularly limited.
- the reaction temperature is usually in the range from 0 ° C. to 200 ° C. or the boiling point of the solvent, but preferably in the range from 0 ° C. to the boiling point of the solvent.
- Synthesis of compound (1f) It can be obtained by activating (1e) to a commercially available or appropriately synthesized compound and reacting it with malonic acid monoester or a salt thereof in the presence of magnesium chloride.
- Examples of the method for activating the carboxylic acid include a method using 1,1′-carbonyldiimidazole and a method via an acid chloride.
- a base can be added as necessary, and examples of the base include triethylamine. What is necessary is just to use 1 to excess molar equivalent for a base with respect to a compound (1e). Preferably, 2 to 10 molar equivalents may be used.
- the solvent used for the reaction include tetrahydrofuran, dioxane, acetonitrile, N, N-dimethylformamide, toluene, and a mixed solvent thereof, and are not particularly limited.
- the reaction temperature is usually in the range from ⁇ 78 ° C. to 100 ° C. or the boiling point of the solvent, preferably in the range from around room temperature to 100 ° C.
- N, N-dimethylformamide dimethyl acetal may be used in an amount of 2 to an excess molar equivalent relative to compound (1f).
- the solvent used in the reaction include toluene, xylene, dichloromethane, ethanol, N, N-dimethylformamide, ethyl acetate, and the like, or a mixed solvent thereof, and is not particularly limited. Or it is carried out without solvent.
- the reaction temperature is usually in the range of 0 ° C. to 300 ° C., but preferably in the range of around room temperature to 130 ° C.
- the compound (1g) can be obtained by treating with a commercially available or appropriately synthesized compound (1h).
- the solvent used in the reaction include toluene, ethyl acetate, ethanol, 1,4-dioxane, and a mixed solvent thereof, and are not particularly limited.
- the reaction temperature is usually in the range from ⁇ 78 ° C. to 130 ° C. or the boiling point of the solvent, preferably in the range from around room temperature to the boiling point of the solvent.
- reaction is carried out by adding an organic or inorganic base such as potassium carbonate, cesium carbonate, potassium tert-butoxide or triethylamine, or an acid such as acetic acid, hydrogen chloride, hydrogen bromide or sulfuric acid as necessary.
- an organic or inorganic base such as potassium carbonate, cesium carbonate, potassium tert-butoxide or triethylamine
- an acid such as acetic acid, hydrogen chloride, hydrogen bromide or sulfuric acid as necessary.
- Compound (1h), base, or acid may be used in an amount of 1 to excess molar equivalents relative to compound (1g). Preferably, 1 to 5 molar equivalents may be used.
- Synthesis of compound (AI) It can be obtained by the same operation as the synthesis of compound (1d).
- Reaction Scheme 2 Compound (1i) can also be synthesized by a method as shown in Reaction Scheme 2 below, but is not particularly limited.
- Reaction formula 2 [Wherein R 1 , R 42 Is as defined above. R 45 Represents a halogen group, a methanesulfonyloxy group, a trifluoromethanesulfonyloxy group, or a p-toluenesulfonyloxy group.
- Compound (2c) is included in compound (1i).
- Synthesis of compound (2a) Compound (2a) is commercially available or J.I. Heterocyclic Chem. 17, 359 (1980) and Chem. Pharm. Bull. It can be synthesized from compound (1f) with reference to reported reports such as 43,450 (1995).
- Compound (2a) can be obtained by treating with compound (2b) in the presence of an organic or inorganic base such as potassium carbonate, cesium carbonate, potassium tert-butoxide or triethylamine.
- organic or inorganic base such as potassium carbonate, cesium carbonate, potassium tert-butoxide or triethylamine.
- the solvent used in the reaction include N, N-dimethylformamide, dimethyl sulfoxide, acetonitrile, and mixed solvents thereof, and are not particularly limited.
- the reaction temperature is usually in the range from 0 ° C. to 200 ° C. or the boiling point of the solvent, but preferably in the range from 0 ° C. to the boiling point of the solvent.
- Compound (2b) and the base may be used in an amount of 1 to excess molar equivalents relative to compound (2a).
- Compound (A-II) can be synthesized by a method as shown in Reaction Scheme 3 below, but is not particularly limited. Reaction formula 3 [Wherein R 42 , R 45 Is as defined above. ] Synthesis of compound (3b) Commercially available or J. Org. Med. Chem. Compound (3a), which can be easily synthesized with reference to reported examples such as 51, 5330 (2008), is obtained as compound (2b) in the presence of an organic or inorganic base such as potassium carbonate, cesium carbonate, potassium tert-butoxide or triethylamine. It can be obtained by processing.
- an organic or inorganic base such as potassium carbonate, cesium carbonate, potassium tert-butoxide or triethylamine. It can be obtained by processing.
- Examples of the solvent used in the reaction include N, N-dimethylformamide, dimethyl sulfoxide, acetonitrile, and mixed solvents thereof, and are not particularly limited.
- the reaction temperature is usually in the range from 0 ° C. to 200 ° C. or the boiling point of the solvent, but preferably in the range from 0 ° C. to the boiling point of the solvent.
- the compound (2b) and the base may be used in an amount of 1 to an excess molar equivalent relative to the compound (3a). Preferably, 1 to 5 molar equivalents may be used.
- reaction temperature is usually in the range from 0 ° C. to 200 ° C. or the boiling point of the solvent, but preferably in the range from 0 ° C. to the boiling point of the solvent.
- Compound (4b) and the base may be used in an amount of 1 to excess molar equivalents relative to compound (4a). Preferably, 1 to 5 molar equivalents may be used.
- Reaction formula 5 [Wherein R 8 , R 9 , R 12 , R 43 , R 45 , R 51 Is as defined above.
- R 61 Represents deuterium or a hydrogen atom.
- R 62 Represents a hydroxyl group and a protecting group for a hydroxyl group substituted with deuterium.
- Examples of the protecting group include an ether group typified by a tetrahydropyranyl group, a silyl group typified by a t-butyldiphenylsilyl group, an ester group typified by an acetyl group, and a carbonate group typified by a vinyl carbonate group.
- R 63 Is C substituted with deuterium 1 -C 6 Indicates an alkyl group.
- a compound in which only one hydroxyl group is protected with a tetrahydropyranyl group by treating compound (5a) with 3,4-dihydro-2H-pyran in the presence of an acid such as p-toluenesulfonic acid pyridine salt.
- an acid such as p-toluenesulfonic acid pyridine salt.
- the method for protecting the hydroxyl group is not particularly limited thereto.
- the solvent used in the reaction include dichloromethane, chloroform, toluene, and mixed solvents thereof, and are not particularly limited.
- the reaction temperature is usually in the range from 0 ° C. to 200 ° C. or the boiling point of the solvent, but preferably in the range from 0 ° C. to the boiling point of the solvent.
- the compound (4b) and the acid may be used in an amount of 0.01 to excess molar equivalents relative to the compound (5a). Preferably, 0.01 to 1 molar equivalent may be used.
- Synthesis of compound (5d) Compound (5d) can be obtained by treating compound (5b) with compound (5c) in the presence of an organic or inorganic base such as potassium carbonate, cesium carbonate, potassium tert-butoxide or triethylamine. N, N-dimethylformamide, dimethyl sulfoxide, acetonitrile, acetone, or a mixed solvent thereof may be used and is not particularly limited.
- the reaction temperature is usually in the range from 0 ° C. to 200 ° C.
- the compound (5e) and the base may be used in an amount of 1 to an excess molar equivalent relative to the compound (5b). Preferably, 1 to 5 molar equivalents may be used.
- Synthesis of compound (5e) The compound (5e) can be obtained by treating the compound (5d) with an acid such as hydrochloric acid, acetic acid, p-toluenesulfonic acid pyridine salt and the like.
- an acid such as hydrochloric acid, acetic acid, p-toluenesulfonic acid pyridine salt and the like.
- the solvent used in the reaction include ethanol, methanol, isopropanol, water, and mixed solvents thereof, and are not particularly limited.
- the reaction temperature is usually in the range from 0 ° C. to 200 ° C.
- Synthesis of compound (5f) Compound (5f) can be obtained by treating compound (5e) with a bromination reagent such as N-bromosuccinimide and potassium bromate.
- a bromination reagent such as N-bromosuccinimide and potassium bromate.
- the solvent used for the reaction include N, N-dimethylformamide, 30% hydrobromic acid-acetic acid solution, acetic acid, and mixed solvents thereof, and are not particularly limited.
- the reaction temperature is usually in the range from 0 ° C. to 200 ° C.
- the solvent examples include 1,4-dioxane, 1,2-dimethoxyethane, methanol, toluene, and mixed solvents thereof, and are not particularly limited.
- the reaction temperature is usually in the range from 0 ° C. to 200 ° C. or the boiling point of the solvent, but preferably in the range from 0 ° C. to the boiling point of the solvent.
- the said process can also be performed by a microwave in a sealed tube.
- the organic or inorganic base may be used in an amount of 1 to excess molar equivalents relative to the compound (5 g). Preferably, 1 to 5 molar equivalents may be used.
- the transition metal catalyst and the ligand may be used in an amount of 0.001 to 1 molar equivalent relative to the compound (5 g). Preferably, 0.05 to 1 molar equivalent may be used.
- the boronic acid ester may be used in an amount of 1 to excess molar equivalents relative to the compound (5 g). Preferably, 1 to 5 molar equivalents may be used.
- R 81 Represents a halogen group.
- the compound (7a) is a boronic acid or boronic acid ester compound containing the compound (4c), the compound (5h) or the compound (6c).
- Compound (7c) and compound (7d) are included in compound (CI).
- Synthesis of compound (7c) Similarly to the synthesis of compound (1c) in [Production method 1], compound (7b) was commercially available or compound (7a) synthesized by the method shown in production method 4, production method 5, and production method 6 and A compound (7c) can be obtained by performing a coupling reaction. Synthesis of compound (7d) A commercially available or appropriately synthesized compound (7c) can be used in the same manner as the synthesis of compound (5h) in [Production Method 5].
- Compound (8) can also be synthesized by the methods shown in [Production Method 9] and [Production Method 10], but is not particularly limited.
- [Production Method 9] Reaction formula 9 [Wherein R 4 , R 5 , W, X, Y, Z, R 81 Is as defined above. Compound (8) is included in Compound (B) Compound (CI) complex.
- the compound (9a) and (9b), which are commercially available or appropriately synthesized, can be used in the same manner as the synthesis of compound (1c) in [Production Method 1].
- Reaction formula 10 [Wherein R 4 , R 5 , W, X, Y, Z, R 81 Is as defined above.
- Compound (8) is included in Compound (B) Compound (CI) complex.
- a commercially available or appropriately synthesized compound (7d) and compound (10) can be used in the same manner as the synthesis of compound (1c) in [Production Method 1].
- a synthesis method for bonding the compound (B) compound (CI) complex and the compound (AI) will be described.
- Compound (B) Compound (CI) complex and compound (AI) can be bound by the following production method 11 or production method 12.
- Reaction formula 11 Although it can synthesize
- Compound (8) is included in Compound (B) Compound (CI) complex.
- It can be obtained by reacting compound (AI) with compound (8) in the presence of a condensing agent.
- the condensing agent include N- [1- (cyano-2-ethoxy-2-oxoethylideneaminooxy) dimethylamino (morpholino)] uronium hexafluorophosphate (COMU), but are not limited thereto.
- the solvent used in the reaction include N, N-dimethylformamide, dichloromethane, or a mixed solvent thereof, and are not particularly limited.
- the reaction temperature is usually in the range from ⁇ 78 ° C. to 200 ° C.
- Compound (AI) can be obtained by treating compound (12) with an acid halide reagent such as thionyl chloride or oxalyl chloride and then treating with compound (8).
- an acid halide reagent such as thionyl chloride or oxalyl chloride
- Examples of the solvent used in the reaction include dichloromethane, dichloroethane, and mixed solvents thereof, and are not particularly limited.
- the acid halide reagent may be used in an amount of 0.9 to excess molar equivalents relative to compound (AI). Preferably, 0.9 to 2 molar equivalents may be used.
- a method for synthesizing the compound (AI) compound (B) complex will be described. [Production method 13] Although it can synthesize
- Reaction formula 13 [Wherein R 1 ⁇ R 4 , W, X, Y, Z, R 31 Is as defined above. Compound (13) is included in Compound (AI) Compound (B) complex. ] Synthesis of compound (13) Compound (B) is a commercially available product or can be easily synthesized from a commercially available product by a method known in the literature. It can be obtained by the same procedure as in [Production Method 11] using compound (AI) and commercially available or appropriately synthesized compound (B). Next, a synthesis method for bonding the compound (AI), the compound (B) complex, and the compound (CI) will be described. [Production Method 14] Although it can synthesize
- Reaction formula 14 [Wherein R 1 ⁇ R 5 , W, X, Y, Z are as defined above.
- R 31 Is a halogen group, triflate group, etc.
- R 32 Represents boronic acid or boronic acid ester.
- R 31 Is boronic acid, boronic acid ester, R 32 Represents a halogen group, a triflate group or the like.
- Compound (13) is included in Compound (AI) Compound (B) complex.
- Synthesis of compound (11) Using compound (13) and commercially available or appropriately synthesized compound (CI), it can be obtained in the same manner as the synthesis of compound (1c) in [Production Method 1].
- Reaction formula 16 Although it can synthesize
- Reaction formula 16 [Wherein R 1 , R 4 ⁇ R 5 , W, X, Y, Z, R 43 Is as defined above.
- Compound (15) is included in Compound (AI) Compound (B) Compound (CI) Complex.
- Synthesis of compound (16b) The compound (15) and a commercially available or appropriately synthesized compound (16a) can be used in the same manner as the synthesis of the compound (1c) in [Production Method 1]. Next, a synthesis method for converting the halogen group of the compound (AI), the compound (B), and the compound (CI) complex into an alkyl group will be described.
- Reaction formula 17 Although it can synthesize
- Reaction formula 17 [Wherein R 2 ⁇ R 5 , Q, T, U, W, X, Y, Z, R 43 , R 44 Is as defined above.
- the compound (17a) and the compound (17b) are included in the compound (AI), the compound (B) and the compound (CI) complex.
- Synthesis of compound (17b) The compound (17a) and a commercially available or appropriately synthesized compound (1b) can be used in the same manner as the synthesis of the compound (1c) in [Production Method 1].
- bromine group is R 5 A synthesis method for converting to the above will be described.
- Reaction Method 18 Although it can synthesize
- Reaction formula 18 [Wherein R 1 ⁇ R 5 , W, X, Y, Z, R 43 Is as defined above. ]
- Synthesis of compound (18) The compound (AI) and the compound (BC) can be used in the same manner as in [Production Method 11].
- Synthesis of compound (11) The compound (18) and a commercially available or appropriately synthesized compound (7a) can be used in the same manner as the synthesis of the compound (1c) in [Production Method 1].
- Compound (18) can also be synthesized by a method such as [Production Method 19].
- Reaction formula 19 [Wherein R 1 ⁇ R 4 , R 33 , R 81 , W, X, Y, Z are as defined above. Compound (19) is included in compound (13). ] Compound (C-II) is a commercially available product or can be easily synthesized from commercially available products by methods known in the literature. The compound (19) and a commercially available or appropriately synthesized compound (C-II) can be used in the same manner as the synthesis of the compound (1c) in [Production method 1]. Compound (11) can also be produced by production method 20. [Production Method 20] Reaction formula 20 [Wherein R 1 ⁇ R 4 , R 43 , W, X, Y, Z are as defined above.
- R 201 Represents a hydrogen atom or a halogen group.
- R 202 C may form a ring 1 ⁇ C 6 Oxy-C which may form an alkylamino group or a ring 1 ⁇ C 6 An alkylamino group is shown.
- Compound (20d) is included in compound (11).
- Synthesis of compound (20b) The compound (18) and a commercially available or appropriately synthesized compound (20a) can be used in the same manner as the synthesis of compound (1c) in [Production Method 1].
- Synthesis of compound (20d) Compound (20b) can be obtained by reacting commercially available or appropriately synthesized compound (20c) in the presence of a reducing agent.
- Examples of the reducing agent to be used include sodium triacetoxyborohydride, sodium cyanoborohydride, sodium borohydride and the like.
- Examples of the solvent to be used include methanol, ethanol, water, tetrahydrofuran, dioxane, N, N-dimethylformamide, methylene chloride, acetic acid, and a mixed solvent thereof, and are not particularly limited.
- the reaction temperature is usually in the range from ⁇ 78 ° C. to 100 ° C. or the boiling point of the solvent, preferably in the range from 0 ° C. to 100 ° C.
- the Gas6 / Axl signal transduction system has various cellularity such as cell survival, cell division, autophagy, cell migration, angiogenesis, platelet aggregation, and NK cell differentiation. Since it has been reported that the response is regulated (Non-Patent Document 2), Axl inhibitors are diseases caused by Axl kinase hyperfunction, diseases associated with Axl kinase hyperfunction, and / or Axl kinase. Useful for the treatment of diseases with hyperfunction. In particular, the compounds of the present invention have high Axl inhibition specificity and do not cause retinal toxicity, and therefore can be used more safely.
- Diseases caused by increased Axl kinase function, diseases associated with increased Axl kinase function, diseases associated with increased Axl kinase function include diseases having tissues in which overexpression of Axl gene and / or protein is observed, Axl phosphorus Examples include diseases having tissues in which an increase in oxidative activity is observed. Examples of the above diseases include, for example, hyperproliferative diseases. Examples of hyperproliferative diseases include endometrial hyperplasia, thrombin-induced vascular smooth muscle cell (VSMC) proliferative benign tumor, malignant tumor (cancer). Include, but are not limited to, acute and chronic glomerulonephritis, diabetic nephropathy, and the like.
- VSMC thrombin-induced vascular smooth muscle cell
- Axl is immune (Non-patent document 12), platelet function (Non-patent document 13), spermatogenesis (Non-patent document 14), vascular calcification (Non-patent document 15), (Non-patent document 16) and various It has also been shown to have a role in kidney disease, chronic allograft rejection (Non-Patent Document 17) and Axl inhibitors include but are not limited to vascular diseases (including but not limited to thrombosis, atherosclerosis and restenosis).
- disorders in which disordered angiogenesis is critical including but not limited to diabetic retinopathy, retinopathy, psoriasis, rheumatoid arthritis, atheroma, Kaposi's sarcoma and hemangioma).
- the compounds of the present invention inhibit Axl, they are useful for the treatment of the diseases described above. More preferably, the compounds of the invention are useful for the treatment of various cancers.
- cancer examples include breast cancer, colon cancer, prostate cancer, lung cancer, gastric cancer, ovarian cancer, cervical cancer, endometrial cancer, endometrial cancer, renal cancer, hepatocellular carcinoma, thyroid cancer, esophageal cancer, squamous cell carcinoma , Leukemia, osteosarcoma, melanoma, glioblastoma, neuroblastoma, ovarian cancer, head and neck cancer, testicular tumor, colon cancer, blood cancer, retinoblastoma, pancreatic cancer, etc. It is not limited to.
- Axl shRNA has been reported to suppress the growth of breast cancer cells (Yi-Xiang et al., Cancer Res 68, 1905 (2008)). From these reports it is clear that Axl inhibitors are useful for inhibiting cell proliferation in cancer. On the other hand, it has been reported that a dominant negative mutant of Axl inhibited cell migration and invasion (Zhang et al., Cancer Res 68, 1905 (2008), Vajkoczy et al.,). PNAS 103, 5799 (2006), Holland et al., Cancer Res 65, 9294 (2005)). It has been reported that Axl-shRNA suppressed metastasis in vivo (Li et al., Oncogene 28, 3442 (2009)).
- Axl is involved in metastasis and malignancy of prostate cancer, spleen cancer, metastatic ovarian cancer, thymic cancer and the like. From these reports, it is clear that Axl inhibitors are useful for cancer metastasis, cell migration, inhibition of invasion, treatment, prevention and the like. In addition, it has been reported that an Axl inhibitor canceled imatinib resistance in gastric cancer (Mahadevan et al., Oncogene 26, 3909 (2007)).
- Axl is shown to be induced in acute myeloid leukemia in resistance to chemotherapeutic agents such as doxorubicin, VP16, cisplatin (Hong et al., Cancer Letters 268, 314 (2008)). It has been reported that Axl activation is seen in lapatinib resistance in HER-2 positive breast cancer cells (Liu et al., Cancer Res 69, 6871 (2009)). It has been reported that Axl is involved in the PLX4032 (vemurafenib) resistance mechanism (Johannessen et al., Nature 468, 968 (2010)).
- Axl inhibitors are useful for releasing drug resistance, for example, releasing resistance to various anticancer agents.
- the Axl inhibitor of the present application inhibited the resistance of the tumor to erlotinib by administration in combination with erlotinib, as shown in the test examples of the present application.
- significant induction of Axl protein was observed by administration of erlotinib, it is considered that Axl is involved in acquiring drug resistance of cancer.
- the Axl inhibitor since the sensitivity to erlotinib was recovered by using the Axl inhibitor of the present application in combination with the tumor that became resistant to erlotinib, the Axl inhibitor was effective in releasing the drug resistance of cancer. Conceivable. Furthermore, Axl has been reported to be involved in renal fibrosis such as kidney fibrosis and diabetic nephropathy (Japanese translations of publication 2005-517412), and the Axl inhibitor is the above-mentioned renal disease and other idiopathic pulmonary fibrosis.
- the Axl inhibitory activity of a compound can be measured by, for example, the method described in Test Examples of the present application, but is not limited thereto.
- the cell growth inhibitory activity can be examined using a growth inhibition test method commonly used by those skilled in the art.
- Cell growth inhibitory activity can be performed by comparing the extent of cell (eg, tumor cell) growth in the presence or absence of the test compound.
- the degree of proliferation can be examined, for example, using a test system that measures live cells.
- a method for measuring living cells for example, [ 3 H] -thymidine incorporation test, BrdU method, MTT assay and the like.
- the antitumor activity in vivo can be investigated using the antitumor test method normally used by those skilled in the art.
- various tumor cells are transplanted into mice, rats, etc., and after the engraftment of the transplanted cells is confirmed, the compound of the present invention is administered orally, intravenously, etc.
- the in vivo antitumor activity of the present invention can be confirmed by comparing the tumor growth in and the compound administration group.
- metastasis suppression activity, invasion inhibition activity, movement inhibition activity, and drug resistance release activity the relationship between Axl and each activity is reported as described in the literature cited above. It can be measured by a test method.
- the pharmaceutical composition of the present invention comprises the compound of the present invention and a pharmaceutically acceptable carrier, and is used as various injections such as intravenous injection, intramuscular injection, subcutaneous injection, or oral administration or transdermal administration. Administration can be by a variety of methods.
- a pharmaceutically acceptable carrier is a pharmaceutically acceptable material involved in transporting a compound of the present invention or a composition comprising a compound of the present invention from one organ or organ to another. (For example, excipient, diluent, additive, solvent, etc.).
- an appropriate preparation for example, oral preparation or injection
- the preparation can be prepared by various preparation preparations which are usually used.
- oral preparations examples include tablets, powders, granules, capsules, pills, troches, solutions, syrups, elixirs, emulsions, and oily or aqueous suspensions. In the case of oral administration, it may be in the free form or in the salt form.
- Aqueous preparations can be prepared by forming an acid adduct with a pharmaceutically acceptable acid or by forming an alkali metal salt such as sodium.
- stabilizers, preservatives or solubilizers can be used in the preparation.
- a solution that may contain these adjuvants and the like may be stored in a container and then prepared as a solid preparation by lyophilization or the like.
- the single dose may be stored in one container, and the multiple doses may be stored in one container.
- solid preparations include tablets, powders, granules, capsules, pills, or lozenges. These solid preparations may contain pharmaceutically acceptable additives together with the compound of the present invention. Examples of the additive include fillers, extenders, binders, disintegrants, dissolution accelerators, wetting agents, and lubricants, which are selected and mixed as necessary. And can be formulated. Examples of the liquid preparation include solutions, syrups, elixirs, emulsions, and suspensions. These liquid formulations may contain pharmaceutically acceptable additives along with the compounds of the present invention.
- the additive examples include a suspending agent or an emulsifier, and these can be selected and mixed as necessary to prepare a formulation.
- the compounds of the present invention can be used for the treatment of cancer in mammals, particularly humans.
- the dose and administration interval can be appropriately selected according to the judgment of the doctor according to the location of the disease, the height, weight, sex, or medical history of the patient.
- the dosage range is about 0.01 mg / kg body weight to about 500 mg / kg body weight, preferably about 0.1 mg / kg body weight to about 100 mg / kg body weight per day. It is.
- it is preferably administered once a day or divided into 2 to 4 times and repeated at appropriate intervals.
- the daily amount may exceed the above amount depending on the judgment of the doctor.
- the compound of the present invention may be used in combination with other antitumor agents.
- tyrosine kinase inhibitors such as erlotinib shown above, for example, antitumor antibiotics, antitumor plant components, BRM (biological response control substances), hormones, vitamins, antitumor antibodies , Other molecular target drugs, other antitumor agents, and the like.
- examples of the alkylating agent include an alkylating agent such as nitrogen mustard, nitrogen mustard N-oxide or chlorambutyl, an aziridine alkylating agent such as carbocon or thiotepa, dibromomannitol or dibromodarsi
- alkylating agent such as nitrogen mustard, nitrogen mustard N-oxide or chlorambutyl
- an aziridine alkylating agent such as carbocon or thiotepa, dibromomannitol or dibromodarsi
- examples thereof include epoxide-based alkylating agents such as Toll, carmustine, lomustine, semustine, nimustine hydrochloride, nitrosourea-based alkylating agents such as streptozocin, chlorozotocin or ranimustine, busulfan, improsulfan tosylate or dacarbazine.
- antimetabolites include, for example, purine antimetabolites such as 6-mercaptopurine, 6-thioguanine or thioinosine, and pyrimidine metabolism antagonists such as fluorouracil, tegafur, tegafur uracil, carmofur, doxyfluridine, broxuridine, cytarabine or enocytabine And antifolate inhibitors such as methotrexate or trimethrexate.
- purine antimetabolites such as 6-mercaptopurine, 6-thioguanine or thioinosine
- pyrimidine metabolism antagonists such as fluorouracil, tegafur, tegafur uracil, carmofur, doxyfluridine, broxuridine, cytarabine or enocytabine
- antifolate inhibitors such as methotrexate or trimethrexate.
- Antitumor antibiotics include, for example, anthracycline antibiotic antitumor agents such as mitomycin C, bleomycin, peplomycin, daunorubicin, aclarubicin, doxorubicin, pirarubicin, THP-adriamycin, 4′-epidoxorubicin or epirubicin, chromomycin A3 Or actinomycin D etc. are mentioned.
- Examples of the antitumor plant component include vinca alkaloids such as vindesine, vincristine or vinblastine, taxanes such as paclitaxel and docetaxel, and epipodophyllotoxins such as etoposide or teniposide.
- BRM examples include tumor necrosis factor or indomethacin.
- the hormone include hydrocortisone, dexamethasone, methylprednisolone, prednisolone, plasterone, betamethasone, triamcinolone, oxymetholone, nandrolone, metenolone, phosfestol, ethinylestradiol, chlormadinone, or medroxyprogesterone.
- vitamins include vitamin C and vitamin A.
- Antitumor antibodies and molecular targeted drugs include trastuzumab, rituximab, cetuximab, nimotuzumab, denosumab, bevacizumab, infliximab, imatinib mesylate, gefitinib, erlotinib, sunitinib, lapitib, satinibib .
- the present invention also includes a method for preventing and / or treating cancer, which comprises administering the compound of the present invention or a salt thereof.
- the present invention includes the use of the compound of the present invention, a salt thereof or a solvate thereof for producing the medicament.
- the present invention will be specifically described with reference to the following examples. However, the present invention is not limited to these examples, and is not construed as being limited in any way.
- reagents, solvents and starting materials not particularly described are readily available from commercially available sources.
- Step 2 Ethyl 5- (4-methylphenyl) -4-oxo-1- (tetrahydro-2H-pyran-4-ylmethyl) -1,4-dihydropyridine-3-carboxylate 43.0 g (195 mmol) of ethyl 4- (4-methylphenyl) -3-oxobutanoate was dissolved in 390 ml of toluene, 130 ml (976 mmol) of N, N-dimethylformamide dimethyl acetal was added, and methanol produced by Dean Stark was trapped. While stirring, the mixture was stirred at 100 ° C. for 5 hours. The reaction solution was cooled to room temperature and concentrated under reduced pressure.
- Step 3 5- (4-Methylphenyl) -4-oxo-1- (tetrahydro-2H-pyran-4-ylmethyl) -1,4-dihydropyridine-3-carboxylic acid 36.9 g (104 mmol) of ethyl 5- (4-methylphenyl) -4-oxo-1- (tetrahydro-2H-pyran-4-ylmethyl) -1,4-dihydropyridine-3-carboxylate was dissolved in 519 ml of methanol.
- Step 2 (4-Cyclopropyl-2-fluorophenyl) ethyl acetate
- a suspension of 1.95 g (7.47 mmol) of ethyl (4-bromo-2-fluorophenyl) acetate synthesized in Step 1 in 30.0 mL of toluene 0.962 g (11.2 mmol) of cyclopropylboronic acid and tricyclohexyl were added.
- Step 3 (4-Cyclopropyl-2-fluorophenyl) acetic acid 15.0 mL of methanol and 14.3 mL of 1 mol / L aqueous sodium hydroxide solution were added to a solution of 1.59 g (7.15 mmol) of ethyl (4-cyclopropyl-2-fluorophenyl) acetate synthesized in Step 2 in 30.0 mL of tetrahydrofuran. 14.3 mmol) was added and stirred at room temperature overnight. The organic solvent was distilled off under reduced pressure, water was added to the residue, and the mixture was washed with diethyl ether.
- the aqueous layer was acidified with 1N hydrochloric acid, and extracted three times with ethyl acetate.
- the organic layer was washed with saturated brine, and dried over anhydrous sodium sulfate. Filtration and concentration under reduced pressure gave 1.36 g (7.00 mmol) of the title compound as a solid.
- Step 4 5- (4-Cyclopropyl-2-fluorophenyl) -4-oxo-1- (tetrahydro-2H-pyran-4-ylmethyl) -1,4-dihydropyridine-3-carboxylic acid
- 4-cyclopropyl-2-fluorophenyl) acetic acid instead of (4-methylphenyl) acetic acid in Step 1 of Intermediate 1a
- the reaction is carried out in the same manner as in Step 1 to Step 3 of Intermediate 1a.
- the title compound was obtained as a solid.
- Steps 2 and 3 5-Methyl-4′-oxo-1 ′-(tetrahydro-2H-pyran-4-ylmethyl) -1 ′, 4′-dihydro-2,3′-bipyridine-5′-carboxylic acid Using ethyl 4- (5-methylpyridin-2-yl) -3-oxobutanoate instead of ethyl 4- (4-methylphenyl) -3-oxobutanoate in step 2 of intermediate 1a, The reaction was conducted in the same manner as in Step 2 to Step 3 to obtain the title compound as a solid.
- Step 2 Ethyl 5- (4-bromophenyl) -4-oxo-1,4-dihydropyridine-3-carboxylate To a solution of ethyl 4- (4-bromophenyl) -3-oxobutanoate 5.00 g (17.5 mmol) synthesized in Step 1 in 50.0 mL of ethanol, 1.56 g (19.3 mmol) of 1,3,5-triazine And 1.43 g (21.0 mmol) of sodium ethoxide were added and stirred at 85 ° C. for 4 hours.
- Step 3 Ethyl 5- (4-bromophenyl) -4-oxo-1- (tetrahydro-2H-pyran-4-ylmethyl) -1,4-dihydropyridine-3-carboxylate To a suspension of 0.322 g of a purified product of ethyl 5- (4-bromophenyl) -4-oxo-1,4-dihydropyridine-3-carboxylate synthesized in Step 2 in 45.0 mL of N, N-dimethylformamide After adding 0.977 g (3.00 mmol) of cesium carbonate and stirring at 80 ° C.
- Step 4 5- (4-Bromophenyl) -4-oxo-1- (tetrahydro-2H-pyran-4-ylmethyl) -1,4-dihydropyridine-3-carboxylic acid
- step 1 of intermediate 1a 4--Bromophenyl) -4-oxo-1- (tetrahydro-2H-pyran-4-ylmethyl) -1,4-dihydropyridine-3-carboxylate was reacted in the same manner as described below to give the title compound Was obtained as a solid.
- the reaction solution was cooled to room temperature, and ethyl acetate and water were added to carry out a liquid separation operation.
- the organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate.
- Step 2 (2R) -2-[( ⁇ 4-Bromo-2-[( 2 H 3 ) Methyloxy] ( 2 H 3 ) Phenyl ⁇ oxy) methyl] -1,4-dioxane 4-bromo-2-[(( 2 H 3 ) Methyloxy] ( 2 H 3 ) 2.00 g (9.57 mmol) of phenol was dissolved in 48.0 ml of N, N-dimethylformamide, 2.64 g (19.1 mmol) of potassium carbonate, (2R) -1,4-dioxane-2-ylmethyl methanesulfone 1.88 g (9.57 mmol) of acid was added and stirred with heating at 90 ° C.
- Step 1 3- (4-aminophenyl) -5- (3,4-dimethoxyphenyl) pyrazin-2-amine
- Step 1 3-Chloro-5- (3,4-dimethoxyphenyl) pyrazin-2-amine 0.510 g (2.00 mmol) of 3-chloro-5-iodopyrazin-2-amine synthesized according to the method described in the patent (WO2011 / 110545 A1), 0.363 g of (3,4-dimethoxyphenyl) boronic acid (2.
- Step 2 3- (4-Aminophenyl) -5- (3,4-dimethoxyphenyl) pyrazin-2-amine 0.0800 g (0.301 mmol) of 3-chloro-5- (3,4-dimethoxyphenyl) pyrazin-2-amine synthesized in Step 1 and 4- (4,4,5,5-tetramethyl-1,3 , 2-Dioxaborolan-2-yl) aniline 0.0990 g (0.452 mmol), [1,1′-bis (diphenylphosphino) ferrocene] palladium (II) dichloride dichloromethane complex (1: 1) 0.0246 g (0 .030 mmol) and cesium carbonate 0.294 g (0.903 mmol) were added 1,4-dioxane 2.00 mL and water 0.200 mL and stirred at 100 ° C.
- Step 2 5-Bromo-4-oxo-1- (tetrahydro-2H-pyran-4-ylmethyl) -1,4-dihydropyridine-3-carboxylic acid Refining product of methyl 5-bromo-4-oxo-1- (tetrahydro-2H-pyran-4-ylmethyl) -1,4-dihydropyridine-3-carboxylate synthesized in step 1 2.83 g of tetrahydrofuran 50.0 mL To the solution, 25.0 mL of methanol and 25.7 mL (25.7 mmol) of 1N aqueous sodium hydroxide solution were added, and the mixture was stirred at room temperature overnight.
- Step 2 N- (4- ⁇ 2-amino-5- [3-fluoro-4- (morpholin-4-ylmethyl) phenyl] pyridin-3-yl ⁇ phenyl) -5- (4-methylphenyl)- 4-oxo-1- (tetrahydro-2H-pyran-4-ylmethyl) -1,4-dihydropyridine-3-carboxamide methanesulfonate N- ⁇ 4- [2-amino-5- (3-fluoro-4-formylphenyl) pyridin-3-yl] phenyl ⁇ -5- (4-methylphenyl) -4-oxo-1- (tetrahydro-2H -Pyran-4-ylmethyl) -1,4-dihydropyridine-3-carboxamide (70.0 mg, 0.114 mmol) was suspended in dichloroethane (2.27 ml), morpholine (19.8 ⁇ l, 0.227 mmol) was added, and the mixture was
- Step 2 N- [2′-amino-5 ′-(3,4-dimethoxyphenyl) -2,3′-bipyridin-5-yl] -5- (4-methylphenyl) -4-oxo-1 -(Tetrahydro-2H-pyran-4-ylmethyl) -1,4-dihydropyridine-3-carboxamide methanesulfonate N- (2′-amino-5′-bromo-2,3′-bipyridin-5-yl) -5- (4-methylphenyl) -4-oxo-1- (tetrahydro-2H-pyran-4-ylmethyl) ) 1,4-dihydropyridine-3-carboxamide 130 mg (0.226 mmol), 3,4-dimethoxyphenylboronic acid 50 mg (0.272 mmol) in dioxane 10 ml, water 1 ml
- Example 110 N- [6- (2-Amino-5- ⁇ 3-methoxy-4- [2- (morpholin-4-yl) ethoxy] phenyl ⁇ pyridin-3-yl) pyridazin-3-yl] -5- (4-Methylphenyl) -4-oxo-1- (tetrahydro-2H-pyran-4-ylmethyl) -1,4-dihydropyridine-3-carboxamide 3-Bromo-5- ⁇ 3-methoxy-4- [2- (morpholin-4-yl) ethoxy] phenyl ⁇ pyridin-2-amine 0.245 g (0.600 mmol), bis (pinacolato) diboron 0.229 g ( 0.900 mmol), tris (dibenzylideneacetone) dipalladium (0) 0.0330 g (0.0360 mmol), tricyclohexylphosphine 0.0236 g (0.0840 mmol), potassium acetate 0.0883
- N- ⁇ 4- [2-amino-5- (3,4-dimethoxyphenyl) pyridin-3-yl] phenyl ⁇ -5-bromo-4-oxo-1- (tetrahydro-2H-pyran- 4-ylmethyl) -1,4-dihydropyridine-3-carboxamide 0.400 g (0.646 mmol), 2-methyl-5- (4,4,5,5-tetramethyl-1,3,2-dioxaborolane-2 -Yl) pyridine 0.212 g (0.969 mmol), cesium carbonate 0.631 g (1.94 m
- Example 132 N- [4- (2-amino-5- ⁇ 4-[(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl ⁇ pyridin-3-yl) phenyl]- 5- (4-Methylphenyl) -4-oxo-1- (tetrahydro-2H-pyran-4-ylmethyl) -1,4-dihydropyridine-3-carboxamide nitrate hydrate N- [4- (2-amino-5- ⁇ 4-[(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl ⁇ pyridin-3-yl) phenyl] -5-
- Example 133 N- [4- (2-amino-5- ⁇ 4-[(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl ⁇ pyridin-3-yl) phenyl]- 5- (4-Methylphenyl) -4-oxo-1- (tetrahydro-2H-pyran-4-ylmethyl) -1,4-dihydropyridine-3-carboxamide sulfate hydrate N- [4- (2-amino-5- ⁇ 4-[(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl ⁇ pyridin-3-yl) phenyl] -5
- Example 138 N- [4- (2-amino-5- ⁇ 4-[(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl ⁇ pyridin-3-yl) -3- Fluorophenyl] -5-methyl-4′-oxo-1 ′-(tetrahydro-2H-pyran-4-ylmethyl) -1 ′, 4′-dihydro-2,3′-bipyridine-5′-carboxamide phosphate Hydrate N- [4- (2-amino-5- ⁇ 4-[(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl ⁇ pyridin-3-yl) -3-fluorophenyl] -5 -Methyl-4′-oxo-1 ′-(tetrahydro-2H-pyran-4-ylmethyl) -1 ′, 4′-dihydro-2,3′-
- Example 140 N- [4- (2-amino-5- ⁇ 4-[(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl ⁇ pyridin-3-yl) -3- Fluorophenyl] -5-methyl-4′-oxo-1 ′-(tetrahydro-2H-pyran-4-ylmethyl) -1 ′, 4′-dihydro-2,3′-bipyridine-5′-carboxamide sulfate water Japanese N- [4- (2-amino-5- ⁇ 4-[(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl ⁇ pyridin-3-yl) -3-fluorophenyl] -5 -Methyl-4′-oxo-1 ′-(tetrahydro-2H-pyran-4-ylmethyl) -1 ′, 4′-dihydro-2,3′
- Example 142 N- [4- (2-amino-5- ⁇ 4-[(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl ⁇ pyridin-3-yl) -3- Fluorophenyl] -5-methyl-4′-oxo-1 ′-(tetrahydro-2H-pyran-4-ylmethyl) -1 ′, 4′-dihydro-2,3′-bipyridine-5′-carboxamide sulfate water Japanese N- [4- (2-amino-5- ⁇ 4-[(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl ⁇ pyridin-3-yl) -3-fluorophenyl] -5 -Methyl-4′-oxo-1 ′-(tetrahydro-2H-pyran-4-ylmethyl) -1 ′, 4′-dihydro-2,3
- Example 145 N- ⁇ 4- [2-amino-5- (4- ⁇ [(2R) -1,4-dioxan-2-yl] methoxy ⁇ -3-methoxyphenyl) -3-pyridyl]- 3-Fluorophenyl ⁇ -5- (5-methyl-2-pyridyl) -4-oxo-1- (tetrahydropyran-4-ylmethyl) pyridine-3-carboxamide sulfate hydrate 24.4 ml of 20% aqueous methanol was added to 2.44 g of the sulfate hydrate described in Example 139, and the mixture was stirred at 5 ° C.
- AXL human AXL intracellular domain 464-885 amino acid fusion protein and glutathione transferase in a baculovirus expression system and purified by glutathione sepharose chromatography.
- a diluted kinase solution containing 170 ng / ml of No. 08-107) was prepared and 19 ⁇ l was added to each well of a 384 well plate.
- the test compound was diluted with DMSO, and 1 ⁇ l of this diluted solution was added to each well.
- the substrate peptide FL-Peptide 30 (5FAM-KKKKEEEIFFF-CONH 2 ), Caliper Life Sciences, Catalog No. 760430) and ATP containing 1.5 ⁇ M and 10 ⁇ M, respectively, were added to each well to start the kinase reaction.
- the plate was incubated at 28 ° C.
- Inhibition (%) 100 ⁇ (1-C i / C 0 )
- C i represents the product ratio when the test compound is added
- C 0 represents the product ratio when DMSO is added instead of the test compound. From the inhibition rate data for 12 test compound concentrations, IC was determined by nonlinear regression (4 parameter logistic regression) using the following formula: 50 Asked.
- Inhibition (%) Bottom + (Top-Bottom) / (1 + ([Compound] / IC 50 ) slope ) (Test Example 2 Cell-free Mer kinase inhibitory activity) Kinase reaction buffer (100 mM HEPES (pH 7.4), 0.003% Brij-35, 0.004% Tween-20, 1 mM DTT, 10 mM MgCl 2 ), A fusion protein of Mer (human MER intracellular domain 528 to 999th amino acid and glutathione transferase) was expressed in a baculovirus expression system and purified by glutathione sepharose chromatography and ion exchange chromatography.
- a diluted kinase solution containing 20 ng / ml of Bio Inc., catalog number 08-108) was prepared and 19 ⁇ l was added to each well of a 384 well plate.
- the test compound was diluted with DMSO, and 1 ⁇ l of this diluted solution was added to each well.
- the substrate peptide FL-Peptide 27 (5FAM-EFPYDFLPAKKK-CONH 2 ), Caliper Life Sciences, catalog number 760424) and ATP 5 mM solution was prepared, and 5 ⁇ l was added to each well to initiate the kinase reaction.
- the plate was incubated at 28 ° C.
- the medium is removed the next day, 100 ⁇ l of medium is added, 37 ° C., 5% CO 2 2 Cultured for 1 day under.
- the test compound was dissolved in DMSO and diluted with a medium not added with FBS to obtain a sample solution (DMSO concentration 2%). 25 ⁇ l of medium or specimen-added medium is added to the well (DMSO concentration 0.4%), 37 ° C., 5% CO 2 Incubated for 1 hour in the presence.
- GAS6 R & D, product number: 885-GS
- wash buffer a solution obtained by diluting Triton X-100 to 0.1% with PBS (hereinafter referred to as “wash buffer”) was added, discarded with a decanter, and excess water was removed on a paper towel. Subsequently, 10% NaN was added to 10.7 mL of the wash buffer.
- washing buffer 3 And H 2 O 2 110 ⁇ l was added (hereinafter referred to as “quenching buffer”), 0.1 ml was added to the well, and the mixture was allowed to stand at room temperature for 15 minutes. Quenching buffer was discarded, 0.2 ml of wash buffer was added, discarded with a decanter, and excess water was removed on a paper towel. Skimmed milk (WAKO # 198-10605) was added to the wash buffer at a final concentration of 5% (blocking buffer), 0.25 ml was added to the well, and allowed to stand at room temperature for 1 hour.
- the blocking buffer was discarded, and anti-phospho-Axl (Y702) (D12B2) rabbit monoclonal antibody (Cell Signaling, catalog number 5724) was reacted at a concentration of 1/1000 and allowed to stand overnight at 4 ° C. Washing operation was repeated 5 times with Wash buffer, and Peroxidase Affini Pure Donkey Anti-Rabbit IgG (H + L) (Jackson ImmunoResearch, catalog number 711-035-152) was reacted at a concentration of 1/2000 at room temperature for 1 hour.
- the washing operation was performed in the same manner, and 0.05 ml of Super Signal ELISA pico chemi luminescent substrate (Thermo Scientific, catalog number 37069) was added, and the mixture was lightly stirred and incubated for 20 minutes. Thereafter, luminescence was measured with ARVO sx (Perkin ElMer), and pAxl (Y702) level was measured.
- the pAxl inhibitory activity was determined by the following formula.
- Inhibition% 100 ⁇ (A ⁇ B) ⁇ 100 / (T ⁇ B)
- B Luminescence value of a reaction solution to which a positive control compound having a concentration that suppresses almost 100% phosphorylation was added (for example, luminescence value of a reaction solution to which 1 ⁇ M of BMS-777607 was added)
- T Luminescence value of the reaction solution to which no compound was added From multiple concentrations of pAxl inhibitory activity data, GraphPad Prism 4 showed 50% inhibitory concentration (IC 50 ) Table 45 shows the intracellular Axl phosphorylation inhibitory activity IC 50 It was shown as (nM) value. (Test Example 4 Histopathological examination of the eye) 1. Animal: NOG female mouse 2.
- Drug Compound A monohydrochloride dihydrate
- Example 124 Example 128
- Example 129 3.
- Dosage preparation Compound A, Example 124 Example 128 and Example 129 were suspended in a 0.5% methylcellulose solution (0.5% MC, Wako Pure Chemical Industries, Ltd.). 4).
- Administration method 4 consecutive shots, 1 day rest, 5 consecutive throws, 2 days rest, 5 consecutive throws, 2 days rest, 5 consecutive throws, 2 days rest, 4 consecutive throws. 5.
- NIH-3T3-Axl # 7 (a cell produced by transfecting NIH3T3 cells with pLXSN retrovirus inserted with full-length Axl cDNA) into NOG female mice was blocked, and the estimated tumor volume was about 500 mm.
- Compound A shows a clear tumor regression effect at 25 and 4.2 mg / kg
- Example 124 and Example 128 suppress tumor growth almost completely during the administration period at 3.1 mg / kg
- 50 and 12.5 mg / Kg showed a clear tumor regression effect.
- Test Example 6 Examination of in vivo combined effect with erlotinib
- An HCC827 lung cancer cell having a deletion mutation in the EGFR gene exon 19 and highly sensitive to an EGFR inhibitor was treated with 5 ⁇ 10 5 using phosphate buffered saline. 7
- the cells were suspended to cells / mL, and 0.1 mL of the prepared cell suspension was transplanted subcutaneously into nude mice (female, 5 weeks old).
- erlotinib (LC Laboratories) was administered at 25 mg / kg (once a day: qd) or N- [4- (2-amino- 5- ⁇ 4-[(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl ⁇ pyridin-3-yl) -3-fluorophenyl] -5-methyl-4′-oxo-1 ′ -(Tetrahydro-2H-pyran-4-ylmethyl) -1 ', 4'-dihydro-2,3'-bipyridine-5'-carboxamide (Compound B) sulfate hydrate 50 mg / kg (twice a day: bid) was administered by oral gavage (single administration or combined administration of both agents).
- Administration is carried out at the rate of 5 days per week from the day after grouping (Day 1) (Saturday and Sunday off), Vehicle and Compound B sulfate hydrate alone administration group is Day 46, erlotinib alone administration group is Day 86, and combination administration group is Day 106. It was.
- the major axis (mm) and the minor axis (mm) of the tumor were measured over time with an electronic digital caliper, and an estimated tumor volume was calculated by the following calculation formula (1) to create a figure.
- the body weight is measured over time using an automatic balance for small animals, and the weight change rate (Body weight change%) is calculated by the following formula (2) to examine the influence of drug administration on the body weight. The latest body weight measurement result was used for dose calculation.
- Estimated tumor volume for most mice is approximately 450 mm 3 Grouping according to the tumor volume value (Day 0) at the time of reaching 52 days after tumor implantation (Day 0), erlotinib (LC Laboratories) was 25 mg / kg, 12.5 mg / kg, or 6.25 mg / kg 5 times a week Forcible oral administration was carried out at the ratio of days (Saturday and Sunday off).
- the vehicle administration group was Day 46, and the erlotinib administration group was Day 86.
- the major axis (mm) and the minor axis (mm) of the tumor were measured with electronic digital calipers over time, and the estimated tumor volume was calculated by the following formula (1) to create a diagram (FIG. 20-1).
- Cell lysate was prepared from 2-6 tumor masses in each group, the expression of AXL was confirmed by Western blot, and individual bands were quantified by ImageQuantLAS4000 (GE Healthcare) (FIG. 20-2).
- Cell lysate preparation and Western blotting were performed as follows. Lysis Buffer (Cell signaling technology, 9803) containing a phosphatase inhibitor (Roshua Diagnostics, 04 906 837 001) and a protease inhibitor (Roshua Diagnostics, 11 836 153 001). ), And the supernatant was used as a Lysate sample.
- the protein of this sample was fixed by heat denaturation with a buffer (Life technologies, NP0008 and NP0009) and subjected to Western blotting.
- a 15 ⁇ g sample was electrophoresed, transferred to a nitrocellulose membrane, and after 1 hour blocking, rabbit anti-AXL antibody (Cell signaling technology, 9803, 1/1000) or rabbit anti-Actin antibody (Santa Cruz Biotechnology, SC-1616, 1/2000) and refrigerated overnight. After washing with Tris-buffered saline, it was reacted with an HRP-labeled anti-rabbit IgG antibody (Cell signaling technology, 7074, 1/2000) at room temperature for 1 hour, washed with Tris-buffered saline, and then HRP substrate (Merck Millipore, WBLUF0500).
- mice (86/129) were gavaged with erlotinib (LC Laboratories) 25 mg / kg (once daily: qd).
- Administration is carried out at the rate of 5 days a week from the day after grouping (53 days after transplantation) (Saturday-Sunday holiday), and grouping is carried out using tumor-bearing mice whose tumors have once regressed and clearly re-grown (6 animals / group, 115 days after transplantation), Group 1: erlotinib + vehicle administration group (average estimated tumor volume 275 mm) 3 ), Group 2: erlotinib + Compound B sulfate hydrate 50 mg / kg (mean estimated tumor volume 284 mm) 3 ), Group 3: erlotinib + Compound B sulfate hydrate 25 mg / kg (mean estimated tumor volume 268 mm) 3 ) Was divided into three groups, and it was examined whether the
- Compound B sulfate hydrate was administered twice a day (bid) from the 116th day after transplantation at a rate of 5 days a week (saturday and Sunday off).
- a clear growth inhibitory effect by erlotinib was confirmed.
- group 2 the estimated tumor volume on the 116th day after transplantation, in which combined administration was started, was maintained even on the 140th day after transplantation.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Oncology (AREA)
- Physical Education & Sports Medicine (AREA)
- Endocrinology (AREA)
- Urology & Nephrology (AREA)
- Rheumatology (AREA)
- Reproductive Health (AREA)
- Neurosurgery (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Biomedical Technology (AREA)
- Diabetes (AREA)
- Dermatology (AREA)
- Neurology (AREA)
- Hematology (AREA)
- Pulmonology (AREA)
- Gastroenterology & Hepatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description
Gas6/Axlシグナル伝達系は、細胞生存(cell survival)、細胞分裂、自食作用(autophagy)、細胞移動(migration)、血管新生、血小板凝集、NK細胞分化等、多様な細胞性応答を調節していることが報告されており(非特許文献2)、原発結腸癌(非特許文献3)、胃癌(非特許文献4)、食道癌(非特許文献5)、メラノーマ(非特許文献6)、卵巣癌(非特許文献7)、腎臓癌(非特許文献8)、子宮内膜癌(非特許文献9)、甲状腺癌(非特許文献10)等の癌組織における過剰発現も多く報告されている。また、Axlの存在は、肺癌におけるリンパ節状態及びステージ並びに乳癌におけるER発現と大いに関係していることも示されている(非特許文献11)。
さらに、Axlは、免疫(非特許文献12)、血小板機能(非特許文献13)、精子形成(非特許文献14)、血管石灰化(非特許文献15)、トロンビン誘導血管平滑筋細胞(VSMC)増殖(非特許文献16)、及び種々の腎臓疾患、例えば急性及び慢性糸球体腎炎、糖尿病性腎症及び慢性同種移植拒絶反応(非特許文献17)において役割を有することも示されており、Axl阻害剤は、癌(癌腫、肉腫などの固形腫瘍及び白血病及びリンパ性悪性疾患を含む。)のみならず、血管疾患(血栓症、アテローム性動脈硬化症及び再狭窄を含むがそれらに限定されるものではない。)、腎臓疾患(急性及び慢性糸球体腎炎、糖尿病性腎症及び移植拒絶反応を含むがそれらに限定されるものではない。)、及び無秩序な血管形成が重大である疾患(糖尿病性網膜症、網膜症、乾癬、関節リウマチ、粥腫、カポジ肉腫及び血管腫を含むがそれらに限定されるものではない。)等、多数の疾患の治療に有益となることが期待されている。
一方で、Axlが属するTAMレセプターチロシンキナーゼファミリーの一員であるMerについては、そのホモ接合の突然変異によって常染色体劣性色素性網膜炎が起きることが報告されており(非特許文献24)、さらには、Merのある種の突然変異が小児期発症rodconeジストロフィーと関連していることが報告されている(非特許文献25)。
Axlを阻害する化合物としては、スルホンアミド構造を有する化合物(特許文献3)、ピロロピリミジン構造を有する化合物(特許文献4、特許文献5)、ピリジンおよびピラジン構造を有する化合物(特許文献6)、ピラジン構造を有する化合物(特許文献7)、ピラジニルベンズイミダゾール構造を有する化合物(特許文献8)、インドリノン構造を有する化合物(特許文献9)、トリアゾロピリジンおよびトリアゾロピリミジン構造を有する化合物(特許文献10)、イミダゾール構造を有する化合物(特許文献11)、トリアゾール構造を有する化合物(特許文献12、特許文献13、特許文献14、特許文献15、特許文献16、特許文献17、特許文献20、特許文献24、特許文献25、特許文献26、特許文献27、特許文献28)、ピリミジンジアミン構造を有する化合物(特許文献18)、ピリミジン構造を有する化合物(特許文献19、非特許文献18、非特許文献22)、キノリニルオキシフェニルスルホンアミド構造を有する化合物(特許文献21)、キノリン構造を有する化合物(特許文献22、特許文献30、非特許文献21)、ピリジン構造を有する化合物(特許文献23、特許文献33、非特許文献19)、ウレア構造を有する化合物(特許文献29)、2,4−ジ置換アリールアミド構造を有する化合物(非特許文献20)、セコステロイド構造を有する化合物(非特許文献23)、2環性ピリミジン構造を有する化合物(特許文献31、特許文献32、特許文献34)等が報告されている。
本発明者は、鋭意検討した結果、下記一般式(I)で表される構造を有する化合物またはその塩が、Axl阻害特異性が高く、Merに対する阻害活性が低いことを見出し、さらには、マウス長期間投与試験においても網膜毒性を惹起しないことを見出した。
すなわち、本発明は、次の[1]から[53]に関する。
[1]一般式(I)

W、X、Yは、それぞれ独立して窒素原子、C−H、C−FまたはC−Clを示し、
Zは、窒素原子、C−H、C−F、C−ClまたはC−C1~C6アルキル基を示し、
R1は、下記式(II−1)または(II−2)
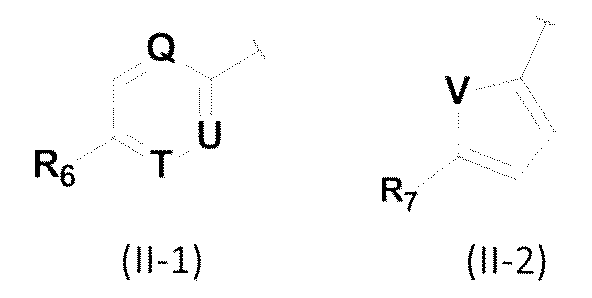
QおよびTは、それぞれ独立して、窒素原子、C−HまたはC−Fを示し、
Tは、窒素原子またはC−Hを示し、
Uは、窒素原子またはC−Hを示し、
R6は、ハロゲン原子、C1~C6アルキル基、C3~C6シクロアルキル基、シアノ基またはトリフルオロメトキシ基を示し、
式(II−2)中、
Vは、硫黄原子または酸素原子を示し、
R7は、C1~C6アルキル基を示す。)を示し、
R2は、水素原子、ハロゲン原子、C1~C6アルキル基、C1~C6アルコキシ基またはシアノ基を示し、
R3は、水素原子またはC1~C6アルキル基を示し、
R4は、水素原子、ハロゲン原子またはC1~C6アルキル基を示し、
R5は、水素原子または下記式(III−1)、(III−2)、(III−3)もしくは(III−4)

R8およびR12は、それぞれ独立して水素原子または重水素原子を示し、
R9は、水素原子、ハロゲン原子またはC1~C6アルコキシ基を示し、
R10は、水素原子、ヘテロシクロアルキル基で置換されていてもよいC1~C6アルキル基またはC1~C6アルキル基で置換されていてもよいヘテロシクロアルキル基で置換されていてもよいC1~C6アルコキシ基を示し、
R11は、水素原子またはC1~C6アルコキシ基または重水素で置換されたC1~C6アルコキシ基を示し、
式(III−2)中、
R13は、ヘテロシクロアルキル基で置換されていてもよいC1~C6アルキル基またはヘテロシクロアルキル基を示し、
式(III−3)中、
R14は、水素原子またはC1~C6アルキル基を示し、
R15は、水素原子、C1~C6アルキル基またはC1~C6アルコキシ基を示し、
R16は、水素原子またはハロゲン原子を示し、
式(III−4)中、
R17は、C1~C6アルコキシ基を示し、
R18は、C1~C6アルコキシ基を示す。)を示す。]
で表される化合物またはその薬学的に許容される塩。
[2]下記式

[3]下記式

[4]下記式
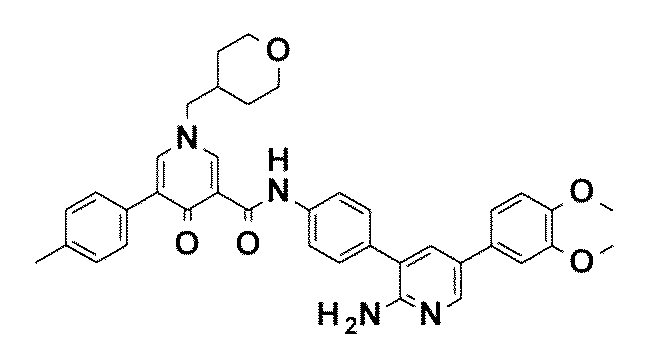
[5]下記式

[6]下記式

[7]下記式

[8]下記式

[9]下記式

[10]下記式

[11][2]に記載の化合物の臭化水素酸塩、硝酸塩、硫酸塩、リン酸塩、メタンスルホン酸塩、エタンスルホン酸塩、ベンゼンスルホン酸塩またはp−トルエンスルホン酸塩。
[12][4]に記載の化合物のメタンスルホン酸塩。
[13][5]に記載の化合物のメタンスルホン酸塩、リン酸塩、ナフタレン−1,5−ジスルホン酸塩または硫酸塩。
[14]銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.74、7.56、8.96、11.38、12.36、14.78、15.60、16.16、18.70および24.10に特徴的ピークを示す[11]に記載のメタンスルホン酸塩の結晶。
[15]銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.84、7.72、9.40、11.62、14.92、15.48、16.70、18.88、19.32および24.40に特徴的ピークを示す[11]に記載の臭化水素酸塩の結晶。
[16]銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.82、7.66、9.28、9.52、11.54、15.26、15.54、16.62、19.24および24.56に特徴的ピーク示す[11]に記載の硝酸塩の結晶。
[17]銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.74、7.56、8.92、9.58、11.36、12.38、14.68,15.64、16.06および24.38に特徴的ピーク示す[11]に記載の硫酸塩の結晶。
[18]銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.74、7.56、8.80、9.56、11.34、14.56、15.74、23.68、24.34および24.68に特徴的ピーク示す[11]に記載のリン酸塩の結晶。
[19]銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=6.72、7.90、12.02、13.40、16.90、17.88、19.00、19.80、21.26および24.18に特徴的ピーク示す[11]に記載のエタンスルホン酸塩の結晶。
[20]銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=9.22、10.60、10.82、11.10、13.40、15.78、17.50、18.66、21.02および26.10に特徴的ピーク示す[11]に記載のベンゼンスルホン酸塩の結晶。
[21]銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=4.18、5.12、13.44、14.98、16.96、17.44、18.92、19.72、20.16および23.04に特徴的ピーク示す[11]に記載のp−トルエンスルホン酸塩の結晶。
[22]銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=4.28、8.42、8.64、10.54、12.72、13.48、15.90、17.00.17.46および21.26に特徴的ピーク示す[13]に記載のリン酸塩の結晶。
[23]銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.66、6.42、7.32、9.76、11.00、12.88、18.42、19.62、20.54および24.22に特徴的ピーク示す[13]に記載の硫酸塩の結晶。
[24]銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.64、6.40、7.32、9.76、17.38、18.42、19.64、20.56、22.90および24.20に特徴的ピーク示す[13]に記載の硫酸塩の結晶。
[25]銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、同折角度2θ=3.64、6.40、7.30、9.76、17.34、18.38、19.34、20.56、21.52および22.94に特徴的ピーク示す[13]に記載の硫酸塩の結晶。
[26]銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.62、6.38、7.28、9.74、17.30、18.36、19.54、20.52、22.86および24.14に特徴的ピーク示す[13]に記載の硫酸塩の結晶。
[27]銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.64、6.40、7.30、9.76、12.86、18.40、19.62、20.54、22.92および24.20に特徴的ピーク示す[13]に記載の硫酸塩の結晶。
[28]銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.64、6.36、7.30、18.36、19.04、19.42、19.70、20.12、20.42および21.32に特徴的ピーク示す[13]に記載の硫酸塩の結晶。
[29]銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=5.62、7.18、9.22、10.36、15.56、16.40および20.86に特徴的ピーク示す[13]に記載の硫酸塩の結晶。
[30]銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=6.14、6.98、11.24、14.84、17.48、19.54、20.94、22.38、23.20および24.70に特徴的ピーク示す[13]に記載のナフタレン−1,5−ジスルホン酸塩の結晶。
[31]銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=9.24、9.58、14.00、14.46、16.70、17.02、18.22、20.24、21.64および25.52に特徴的ピーク示す[13]に記載のナフタレン−1,5−ジスルホン酸塩の結晶。
[32][1]から[10]のいずれか1に記載の化合物またはその薬学的に許容される塩を含む、Axl阻害剤。
[33][1]から[10]のいずれか1に記載の化合物またはその薬学的に許容される塩を有効成分とする医薬。
[34][1]から[10]のいずれか1に記載の化合物またはその薬学的に許容される塩を有効成分とするAxlキナーゼ機能亢進を原因とする疾患、Axlキナーゼ機能亢進と関連する疾患、および/またはAxlキナーゼ機能亢進を伴う疾患の治療のための医薬。
[35][1]から[10]のいずれか1に記載の化合物またはその薬学的に許容される塩を有効成分とする過剰増殖性疾患の治療のための医薬。
[36][1]から[10]のいずれか1に記載の化合物またはその薬学的に許容される塩を有効成分とする癌の治療のための医薬。
[37][1]から[10]のいずれか1に記載の化合物またはその薬学的に許容される塩を有効成分とする癌の転移予防のための医薬。
[38][1]から[10]のいずれか1に記載の化合物またはその薬学的に許容される塩を有効成分とする癌の薬剤耐性解除のための医薬。
[39][1]から[10]のいずれか1に記載の化合物またはその薬学的に許容される塩を有効成分とする癌の薬剤耐性獲得阻害のための医薬。
[40]癌が、乳癌、結腸癌、前立腺癌、肺癌、胃癌、卵巣癌、子宮内膜癌、腎癌、肝細胞癌、甲状腺癌、子宮癌、食道癌、扁平上皮癌、白血病、骨肉腫、メラノーマ、膠芽細胞腫、神経芽細胞腫および膵臓癌から選択されるものである[36]から[39]のいずれか1に記載の医薬。
[41][1]から[10]のいずれか1に記載の化合物またはその薬学的に許容される塩および薬学的に許容し得る担体を含有する医薬組成物。
[42][1]から[10]のいずれか1に記載の化合物またはその薬学的に許容される塩を用いることを特徴とするAxlキナーゼ機能亢進を原因とする疾患、Axlキナーゼ機能亢進と関連する疾患、および/またはAxlキナーゼ機能亢進を伴う疾患の治療方法。
[43][1]から[10]のいずれか1に記載の化合物またはその薬学的に許容される塩を用いることを特徴とする過剰増殖性疾患の治療方法。
[44][1]から[10]のいずれか1に記載の化合物またはその薬学的に許容される塩を用いることを特徴とする癌の治療方法。
[45][1]から[10]のいずれか1に記載の化合物またはその薬学的に許容される塩を用いることを特徴とする癌の転移予防方法。
[46][1]から[10]のいずれか1に記載の化合物またはその薬学的に許容される塩を用いることを特徴とする癌の薬剤耐性解除方法。
[47][1]から[10]のいずれか1に記載の化合物またはその薬学的に許容される塩を用いることを特徴とする癌の薬剤耐性獲得の阻害方法。
[48]癌が、乳癌、結腸癌、前立腺癌、肺癌、胃癌、卵巣癌、子宮内膜癌、腎癌、肝細胞癌、甲状腺癌、子宮癌、食道癌、扁平上皮癌、白血病、骨肉腫、メラノーマ、膠芽細胞腫および神経芽細胞腫から選択されるものである[44]から[49]のいずれか1に記載の方法。
[49][1]から[10]のいずれか1に記載の化合物またはその薬学的に許容される塩の、医薬製造のための使用。
[50][1]から[10]のいずれか1に記載の化合物またはその薬学的に許容される塩の、癌の治療のための使用。
[51][1]から[10]のいずれか1に記載の化合物またはその薬学的に許容される塩の、癌の転移予防のための使用。
[52][1]から[10]のいずれか1に記載の化合物またはその薬学的に許容される塩の、癌の薬剤耐性解除のための使用。
[53][1]から[10]のいずれか1に記載の化合物またはその薬学的に許容される塩の、癌の薬剤耐性獲得の阻害のための使用。
図2は、実施例131で得られた結晶の粉末X線回折図。図の縦軸は、回折強度(Intensity)をカウント/秒(cps)単位で示し、横軸は回折角度2θの値を示す。
図3は、実施例132で得られた結晶の粉末X線回折図。図の縦軸は、回折強度(Intensity)をカウント/秒(cps)単位で示し、横軸は回折角度2θの値を示す。
図4は、実施例133で得られた結晶の粉末X線回折図。図の縦軸は、回折強度(Intensity)をカウント/秒(cps)単位で示し、横軸は回折角度2θの値を示す。
図5は、実施例134で得られた結晶の粉末X線回折図。図の縦軸は、回折強度(Intensity)をカウント/秒(cps)単位で示し、横軸は回折角度2θの値を示す。
図6は、実施例135で得られた結晶の粉末X線回折図。図の縦軸は、回折強度(Intensity)をカウント/秒(cps)単位で示し、横軸は回折角度2θの値を示す。
図7は、実施例136で得られた結晶の粉末X線回折図。図の縦軸は、回折強度(Intensity)をカウント/秒(cps)単位で示し、横軸は回折角度2θの値を示す。
図8は、実施例137で得られた結晶の粉末X線回折図。図の縦軸は、回折強度(Intensity)をカウント/秒(cps)単位で示し、横軸は回折角度2θの値を示す。
図9は、実施例138で得られた結晶の粉末X線回折図。図の縦軸は、回折強度(Intensity)をカウント/秒(cps)単位で示し、横軸は回折角度2θの値を示す。
図10は、実施例139で得られた結晶の粉末X線回折図。図の縦軸は、回折強度(Intensity)をカウント/秒(cps)単位で示し、横軸は回折角度2θの値を示す。
図11は、実施例140で得られた結晶の粉末X線回折図。図の縦軸は、回折強度(Intensity)をカウント/秒(cps)単位で示し、横軸は回折角度2θの値を示す。
図12は、実施例141で得られた結晶の粉末X線回折図。図の縦軸は、回折強度(Intensity)をカウント/秒(cps)単位で示し、横軸は回折角度2θの値を示す。
図13は、実施例142で得られた結晶の粉末X線回折図。図の縦軸は、回折強度(Intensity)をカウント/秒(cps)単位で示し、横軸は回折角度2θの値を示す。
図14は、実施例143で得られた結晶の粉末X線回折図。図の縦軸は、回折強度(Intensity)をカウント/秒(cps)単位で示し、横軸は回折角度2θの値を示す。
図15は、実施例144で得られた結晶の粉末X線回折図。図の縦軸は、回折強度(Intensity)をカウント/秒(cps)単位で示し、横軸は回折角度2θの値を示す。
図16は、実施例145で得られた結晶の粉末X線回折図。図の縦軸は、回折強度(Intensity)をカウント/秒(cps)単位で示し、横軸は回折角度2θの値を示す。
図17は、実施例146で得られた結晶の粉末X線回折図。図の縦軸は、回折強度(Intensity)をカウント/秒(cps)単位で示し、横軸は回折角度2θの値を示す。
図18は、実施例147で得られた結晶の粉末X線回折図。図の縦軸は、回折強度(Intensity)をカウント/秒(cps)単位で示し、横軸は回折角度2θの値を示す。
図19−1は、EGFR遺伝子exon 19に欠失変異を有しEGFR阻害薬に高感受性を示すHCC827マウス皮下移植腫瘍に対するerlotinibと本願のAxl阻害化合物の併用効果を示した図である。シンボル×はVehicle投与群(一日3回投与(Tid))、シンボル白丸はCompound B硫酸塩水和物50mg/kg(一日2回投与(bid))、シンボル黒丸はerlotinib 25mg/kg(一日1回投与(qd))、シンボル白三角はCompound B硫酸塩水和物50mg/kg(bid)+erlotinib 25mg/kg(qd)を示す。横軸は投与開始後の日数を示す。縦軸は腫瘍径より算出した推定腫瘍体積を示す。
図19−2は、erlotinibと本願のAxl阻害化合物の併用試験における各個体の体重変化率の平均値を示す図である(凡例は図19−1と同じ)。
図20−1は、HCC827マウス皮下移植腫瘍に対するerlotinibの抗腫瘍効果効果を示した図である。シンボル×はVehicle投与群、シンボル白丸はerlotinib 25mg/kg、シンボル黒丸はerlotinib 12.5mg/kg、シンボル白三角はerlotinib 6.25mg/kgを示す。横軸は投与開始後の日数を示す。縦軸は腫瘍径より算出した推定腫瘍体積を示す。
図20−2は、HCC827マウス皮下移植腫瘍におけるerlotinib投与時のAxl蛋白質発現量変化を示す図である。
本発明において、「Axl阻害剤」とは、Axlのチロシンキナーゼとしての機能の阻害剤をいう。
本発明において、用語「腫瘍」(tumor)および「癌」(cancer)は交換可能に使用される。また、本発明において、腫瘍(tumor)、悪性腫瘍(malignant tumor)、癌(cancer)、悪性新生物(malignant neoplasm)、癌腫(carcinoma)、肉腫(sarcoma)等を総称して、「腫瘍」または「癌」と表現する場合がある。
本発明において、
「C1~C6アルキル基」とは、炭素数1~6の直鎖、分岐鎖のアルキル基を意味する。「C1~C6アルキル基」としては、例えば、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、tert−ブチル基などが挙げられる。
「C1~C6アルコキシ基」とは、炭素数1~6の直鎖、分岐鎖のアルキル基を有するアルコキシ基を意味する。「C1~C6アルコキシ基」としては、例えば、メトキシ基、エトキシ基、プロポキシ基、イソプロポキシ基、ブトキシ基などが挙げられる。
「ハロゲン原子」としては、フッ素原子、塩素原子、臭素原子、ヨウ素原子等が挙げられる。
「シクロアルキル基」とは、特に言及されない限り、炭素数3~8の環状のアルキル基を意味し、シクロプロピル基、シクロブチル基、シクロペンチル基、シクロヘキシル基等が挙げられる。
「ヘテロシクロアルキル基」とは、一価の飽和複素環基を意味し、環に窒素原子を持つ飽和複素環基、環に酸素原子を持つ飽和複素環基等が挙げられ、例えば、ピロリジン、イミダゾリン、ピペリジン、ピペラジン、アゼチジン、モルホリン、ジオキサン、オキセタン、テトラヒドロピラン、キヌクリジンから導かれる一価の基等が挙げられる。
「シクロアルケニル基」とは、上記の「シクロアルキル基」に1個以上の二重結合等の不飽和結合を有するものが挙げられ、例えば、シクロペンテニル基、シクロヘキセニル基等が挙げられる。
「ヘテロシクロアルケニル基」とは、上記の「ヘテロシクロアルキル基」に1個以上の二重結合等の不飽和結合を有するものが挙げられ、例えば、テトラヒドロピリジニル基、ジヒドロピラニル基が挙げられる。 以下に、式(I)中の各置換基について説明する。
下記一般式(I)中、

Zは、窒素原子、C−H、C−C1~C6アルコキシ基またはC−Clを示す。
R1は、下記式(II−1)または(II−2)

R2は、水素原子、ハロゲン原子、C1~C6アルキル基、C1~C6アルコキシ基またはシアノ基を示す。ここでC1~C6アルキル基としては、メチル基、エチル基またはプロピル基が好ましく、C1~C6アルコキシ基としては、メトキシ基、エトキシ基が好ましい。
R3は、水素原子またはC1~C6アルキル基を示し、C1~C6アルキル基としては、メチル基、エチル基またはプロピル基が好ましい。
R4は、水素原子、ハロゲン原子またはC1~C6アルキル基を示し、R4としては、水素原子、フッ素原子、塩素原子、メチル基またはエチル基が好ましい。
R5は、水素原子または下記式(III−1)、(III−2)、(III−3)もしくは(III−4)

R8およびR12は、それぞれ独立して水素原子または重水素原子を示し、
R9は、水素原子、ハロゲン原子またはC1~C6アルコキシ基を示し、
R10は、水素原子、ヘテロシクロアルキル基で置換されていてもよいC1~C6アルキル基またはC1~C6アルキル基で置換されていてもよいヘテロシクロアルキル基で置換されていてもよいC1~C6アルコキシ基を示し、
R11は、水素原子またはC1~C6アルコキシ基または重水素で置換されたC1~C6アルコキシ基を示し、
式(III−2)中、
R13は、ヘテロシクロアルキル基で置換されていてもよいC1~C6アルキル基またはヘテロシクロアルキル基を示し、
式(III−3)中、
R14は、水素原子またはC1~C6アルキル基を示し、
R15は、水素原子、C1~C6アルキル基またはC1~C6アルコキシ基を示し、
R16は、水素原子またはハロゲン原子を示し、
式(III−4)中、
R17は、C1~C6アルコキシ基を示し、
R18は、C1~C6アルコキシ基を示す。)を示す。
R5が、式(III−1)で表される基である場合、R10におけるヘテロシクロアルキル基はジオキサニル基、モルホリノ基、ピペラジニル基またはテトラヒドロピラニル基が好ましく、C1~C6アルキル基はメトキシ基またはエトキシ基が好ましい。R11におけるC1~C6アルコキシ基はメトキシ基またはエトキシ基が好ましい。
R5が、式(III−2)で表される基である場合、R13におけるヘテロシクロアルキル基はジオキサニル基、モルホリノ基、ピペラジニル基またはテトラヒドロピラニル基が好ましく、R13におけるC1~C6アルキル基はメチル基またはエチル基が好ましい。
また、一般式(I)で表される化合物またはその薬学的に許容される塩は次に示す化合物のいずれかまたはその薬学的に許容される塩であることがさらに好ましい。
下記式

下記式

下記式

下記式

下記式

下記式

下記式

下記式

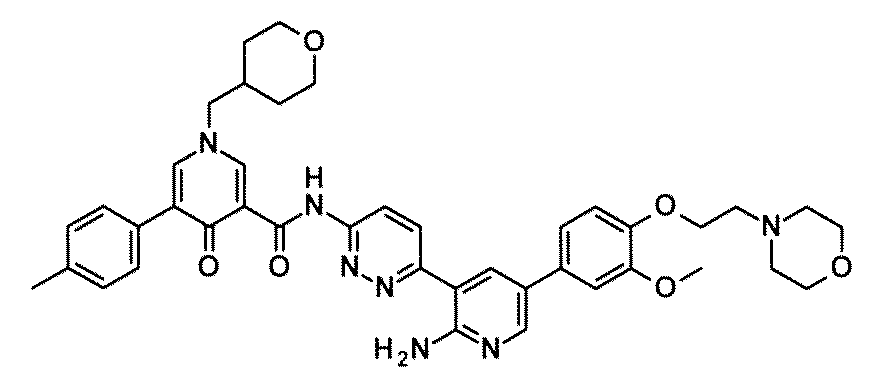
本発明の式(I)で表される化合物には、立体異性体あるいは不斉炭素原子に由来する光学異性体が存在することもあるが、これらの立体異性体、光学異性体およびこれらの混合物のいずれも本発明に含まれる。
本発明の一般式(I)で表される化合物が、アミノ基等の塩基性基を有する場合、所望により医薬的に許容される塩とすることができる。そのような塩としては、例えば塩酸塩、臭化水素塩、ヨウ化水素酸塩等のハロゲン化水素酸塩;硝酸塩、過塩素酸塩、硫酸塩、リン酸塩等の無機酸塩;メタンスルホン酸塩、トリフルオロメタンスルホン酸塩、エタンスルホン酸塩等の低級アルカンスルホン酸塩;ベンゼンスルホン酸塩、p−トルエンスルホン酸塩等のアリ−ルスルホン酸塩;ギ酸、酢酸、りんご酸、フマル酸塩、コハク酸塩、クエン酸塩、酒石酸塩、蓚酸塩、マレイン酸塩等の有機酸塩;及びオルニチン酸塩、グルタミン酸塩、アスパラギン酸塩等のアミノ酸塩;を挙げることができ、ハロゲン化水素酸塩及び有機酸塩が好ましい。
本発明の一般式(I)で表される化合物が、カルボキシ基等の酸性基を有する場合、一般的に塩基付加塩を形成することが可能である。医薬的に許容される塩としては、例えば、ナトリウム塩、カリウム塩、リチウム塩等のアルカリ金属塩;カルシウム塩、マグネシウム塩等のアルカリ土類金属塩;アンモニウム塩等の無機塩;ジベンジルアミン塩、モルホリン塩、フェニルグリシンアルキルエステル塩、エチレンジアミン塩、N−メチルグルカミン塩、ジエチルアミン塩、トリエチルアミン塩、シクロヘキシルアミン塩、ジシクロヘキシルアミン塩、N,N’−ジベンジルエチレンジアミン塩、ジエタノールアミン塩、N−ベンジル−N−(2−フェニルエトキシ)アミン塩、ピペラジン塩、テトラメチルアンモニウム塩、トリス(ヒドロキシメチル)アミノメタン塩等の有機アミン塩、等を挙げることができる。
本発明の一般式(I)で表される化合物またはその塩は、遊離体もしくは溶媒和物として存在することもある。空気中の水分を吸収すること等により水和物として存在することもある。溶媒和物としては、医薬的に許容し得るものであれば特に限定されないが、具体的には、水和物、エタノール和物等が好ましい。また、一般式(I)で表される本発明化合物中に窒素原子が存在する場合にはN−オキシド体となっていてもよく、これら溶媒和物及びN−オキシド体も本発明の範囲に含まれる。また、本化合物またはその塩にはそれらの結晶も含まれる。
より具体的には、塩としては、N−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミドの臭化水素酸塩、硝酸塩、硫酸塩、リン酸塩、エタンスルホン酸塩、ベンゼンスルホン酸塩またはp−トルエンスルホン酸塩が挙げられる。
さらには、N−{4−[2−アミノ−5−(3,4−ジメトキシフェニル)ピリジン−3−イル]フェニル}−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミドのメタンスルホン酸塩が挙げられる。
また、N−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)−3−フルオロフェニル]−5−メチル−4’−オキソ−1’−(テトラヒドロ−2H−ピラン−4−イルメチル)−1’,4’−ジヒドロ−2,3’−ビピリジン−5’−カルボキサミドのメタンスルホン酸塩、リン酸塩または硫酸塩が挙げられる。
本発明の別の態様は本発明の化合物または塩の結晶に関する。ここで、結晶とは、その内部構造が三次元的に構成原子(又はその集団)の規則正しい繰り返しでできている固体をいい、そのような規則正しい内部構造を持たない無定形固体とは区別される。
同じ化合物の結晶であっても、結晶化の条件によって、複数の異なる内部構造及び物理化学的性質を有する結晶(結晶多形)が生成することがあるが、本発明の結晶は、これら結晶多形のいずれであってもよく、2以上の結晶多形の混合物であってもよい。
本発明の結晶は、大気中に放置しておくことにより、水分を吸収し、付着水が付く場合や通常の大気条件下において25乃至150℃に加熱すること等により、水和物を形成する場合がある。さらには、本発明の結晶は付着残留溶媒または溶媒和物中に、結晶化時の溶媒を含む場合もある。
本明細書において、本発明の結晶を粉末X線回折のデータに基づき表すことがあるが、粉末X線回折は、通常、当該分野において用いられる手法により測定・解析を行えばよく、例えば、実施例に記載の方法により行うことができる。また、一般に、水和物や脱水物は結晶水の着脱によって、その格子定数が変化し、粉末X線回折における回折角(2θ)に変化を与えることがある。また、ピークの強度は、結晶の成長面等の違い(晶癖)等によって変化することもある。従って、本発明の結晶を粉末X線回折のデータに基づき表した場合、粉末X線回折におけるピークの回折角およびX線回折図が一致する結晶のほか、それらから得られる水和物および脱水物も本発明の範囲に包含される。
より具体的な本発明の一態様は、N−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミドのメタンスルホン酸塩の結晶に関する。さらに好ましくは、銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.74,7.56,8.96,11.38,12.36,14.78,15.60,16.16,18.70および24.10に特徴的ピークを示す、N−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミドのメタンスルホン酸塩の結晶に関する。当該結晶は銅のKα線(波長λ=1.54オングストローム)の照射において図1の粉末X線回折図を示す。
より具体的な本発明の一態様は、N−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミドの臭化水素酸塩の結晶に関する。さらに好ましくは、銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.84,7.72,9.40,11.62,14.92,15.48,16.70,18.88,19.32および24.40に特徴的ピークを示す、N−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミドの臭化水素酸塩の結晶に関する。当該結晶は銅のKα線(波長λ=1.54オングストローム)の照射において図2の粉末X線回折図を示す。
より具体的な本発明の一態様は、N−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミドの硝酸塩の結晶に関する。さらに好ましくは、銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.82,7.66,9.28,9.52,11.54,15.26,15.54,16.62,19.24および24.56に特徴的ピークを示すN−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミドの硝酸塩の結晶に関する。当該結晶は銅のKα線(波長λ=1.54オングストローム)の照射において図3の粉末X線回折図を示す。
より具体的な本発明の一態様は、N−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミドの硫酸塩の結晶に関する。さらに好ましくは、銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.74,7.56,8.92,9.58,11.36,12.38,14.68,15.64,16.06および24.38に特徴的ピークを示すN−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミドの硫酸塩の結晶に関する。当該結晶は銅のKα線(波長λ=1.54オングストローム)の照射において図4の粉末X線回折図を示す。
より具体的な本発明の一態様は、N−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミドのリン酸塩の結晶に関する。さらに好ましくは、銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.74,7.56,8.80,9.56,11.34,14.56,15.74,23.68,24.34および24.68に特徴的ピークを示すN−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミドのリン酸塩の結晶に関する。当該結晶は銅のKα線(波長λ=1.54オングストローム)の照射において図5の粉末X線回折図を示す。
より具体的な本発明の一態様は、N−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミドのエタンスルホン酸塩の結晶に関する。さらに好ましくは、銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=6.72,7.90,12.02,13.40,16.90,17.88,19.00,19.80,21.26および24.18に特徴的ピークを示すN−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミドのエタンスルホン酸塩の結晶に関する。当該結晶は銅のKα線(波長λ=1.54オングストローム)の照射において図6の粉末X線回折図を示す。
より具体的な本発明の一態様は、N−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミドのベンゼンスルホン酸塩の結晶に関する。さらに好ましくは、銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=9.22,10.60,10.82,11.10,13.40,15.78,17.50,18.66,21.02および26.10に特徴的ピークを示すN−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミドのベンゼンスルホン酸塩の結晶に関する。当該結晶は銅のKα線(波長λ=1.54オングストローム)の照射において図7の粉末X線回折図を示す。
より具体的な本発明の一態様は、N−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミドのp−トルエンスルホン酸塩の結晶に関する。さらに好ましくは、銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=4.18,5.12,13.44,14.98,16.96,17.44,18.92,19.72,20.16および23.04に特徴的ピークを示すN−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミドのp−トルエンスルホン酸塩の結晶に関する。当該結晶は銅のKα線(波長λ=1.54オングストローム)の照射において図8の粉末X線回折図を示す。
より具体的な本発明の一態様は、N−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)−3−フルオロフェニル]−5−メチル−4’−オキソ−1’−(テトラヒドロ−2H−ピラン−4−イルメチル) −1’,4’−ジヒドロ−2,3’−ビピリジン−5’−カルボキサミドのリン酸塩の結晶に関する。さらに好ましくは、銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=4.28,8.42,8.64,10.54,12.72,13.48,15.90,17.00,17.46および21.26に特徴的ピークを示すN−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)−3−フルオロフェニル]−5−メチル−4’−オキソ−1’−(テトラヒドロ−2H−ピラン−4−イルメチル)−1’,4’−ジヒドロ−2,3’−ビピリジン−5’−カルボキサミドのリン酸塩の結晶に関する。当該結晶は銅のKα線(波長λ=1.54オングストローム)の照射において図9の粉末X線回折図を示す。
より具体的な本発明の一態様は、N−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)−3−フルオロフェニル]−5−メチル−4’−オキソ−1’−(テトラヒドロ−2H−ピラン−4−イルメチル)−1’,4’−ジヒドロ−2,3’−ビピリジン−5’−カルボキサミドの硫酸塩の結晶に関する。さらに好ましくは、銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.66,6.42,7.32,9.76,11.00,12.88,18.42,19.62,20.54および24.22に特徴的ピークを示すN−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)−3−フルオロフェニル]−5−メチル−4’−オキソ−1’−(テトラヒドロ−2H−ピラン−4−イルメチル)−1’,4’−ジヒドロ−2,3’−ビピリジン−5’−カルボキサミドの硫酸塩の結晶に関する。当該結晶は銅のKα線(波長λ=1.54オングストローム)の照射において図10の粉末X線回折図を示す。他に好ましくは、銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.64,6.40,7.32,9.76,17.38,18.42,19.64,20.56,22.90および24.20に特徴的ピークを示すN−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)−3−フルオロフェニル]−5−メチル−4’−オキソ−1’−(テトラヒドロ−2H−ピラン−4−イルメチル)−1’,4’−ジヒドロ−2,3’−ビピリジン−5’−カルボキサミドの硫酸塩の結晶に関する。当該結晶は銅のKα線(波長λ=1.54オングストローム)の照射において図11の粉末X線回折図を示す。他に好ましくは、銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.64,6.40,7.30,9.76,17.34,18.38,19.34,20.56,21.52および22.94に特徴的ピークを示すN−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)−3−フルオロフェニル]−5−メチル−4’−オキソ−1’−(テトラヒドロ−2H−ピラン−4−イルメチル)−1’,4’−ジヒドロ−2,3’−ビピリジン−5’−カルボキサミドの硫酸塩の結晶に関する。当該結晶は銅のKα線(波長λ=1.54オングストローム)の照射において図12の粉末X線回折図を示す。他に好ましくは、銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.62,6.38,7.28,9.74,17.30,18.36,19.54,20.52,22.86および24.14に特徴的ピークを示すN−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)−3−フルオロフェニル]−5−メチル−4’−オキソ−1’−(テトラヒドロ−2H−ピラン−4−イルメチル)−1’,4’−ジヒドロ−2,3’−ビピリジン−5’−カルボキサミドの硫酸塩の結晶に関する。当該結晶は銅のKα線(波長λ=1.54オングストローム)の照射において図13の粉末X線回折図を示す。他に好ましくは、銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.64、6.40、7.30、9.76、12.86、18.40、19.62、20.54、22.92および24.20に特徴的ピークを示すN−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)−3−フルオロフェニル]−5−メチル−4’−オキソ−1’−(テトラヒドロ−2H−ピラン−4−イルメチル)−1’,4’−ジヒドロ−2,3’−ビピリジン−5’−カルボキサミドの硫酸塩の結晶に関する。当該結晶は銅のKα線(波長λ=1.54オングストローム)の照射において図14の粉末X線回折図を示す。他に好ましくは、銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.64、6.36、7.30、18.36、19.04、19.42、19.70、20.12、20.42および21.32に特徴的ピークを示すN−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)−3−フルオロフェニル]−5−メチル−4’−オキソ−1’−(テトラヒドロ−2H−ピラン−4−イルメチル)−1’,4’−ジヒドロ−2,3’−ビピリジン−5’−カルボキサミドの硫酸塩の結晶に関する。当該結晶は銅のKα線(波長λ=1.54オングストローム)の照射において図15の粉末X線回折図を示す。他に好ましくは、銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=5.62、7.18、9.22、10.36、15.56、16.40および20.86に特徴的ピークを示すN−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)−3−フルオロフェニル]−5−メチル−4’−オキソ−1’−(テトラヒドロ−2H−ピラン−4−イルメチル)−1’,4’−ジヒドロ−2,3’−ビピリジン−5’−カルボキサミドの硫酸塩の結晶に関する。当該結晶は銅のKα線(波長λ=1.54オングストローム)の照射において図16の粉末X線回折図を示す。
より具体的な本発明の一態様は、N−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)−3−フルオロフェニル]−5−メチル−4’−オキソ−1’−(テトラヒドロ−2H−ピラン−4−イルメチル)−1’,4’−ジヒドロ−2,3’−ビピリジン−5’−カルボキサミドのナフタレン−1,5−ジスルホン酸塩の結晶に関する。さらに好ましくは、銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=6.14、6.98、11.24、14.84、17.48、19.54、20.94、22.38、23.20および24.70に特徴的ピークを示すN−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)−3−フルオロフェニル]−5−メチル−4’−オキソ−1’−(テトラヒドロ−2H−ピラン−4−イルメチル)−1’,4’−ジヒドロ−2,3’−ビピリジン−5’−カルボキサミドのナフタレン−1,5−ジスルホン酸塩の結晶に関する。当該結晶は銅のKα線(波長λ=1.54オングストローム)の照射において図17の粉末X線回折図を示す。他に好ましくは、銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=9.24、9.58、14.00、14.46、16.70、17.02、18.22、20.24、21.64および25.52に特徴的ピークを示すN−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)−3−フルオロフェニル]−5−メチル−4’−オキソ−1’−(テトラヒドロ−2H−ピラン−4−イルメチル)−1’,4’−ジヒドロ−2,3’−ビピリジン−5’−カルボキサミドのナフタレン−1,5−ジスルホン酸塩の結晶に関する。当該結晶は銅のKα線(波長λ=1.54オングストローム)の照射において図18の粉末X線回折図を示す。
本発明の一般式(I)で表される化合物は、置換基の種類や組み合わせによって、シス体、トランス体等の幾何異性体、互変異性体又はd体、l体等の光学異性体等の各種異性体が存在し得るが、本発明の化合物は、特に限定していない場合はそれら全ての異性体、立体異性体及びいずれの比率のこれら異性体及び立体異性体混合物をも包含するものである。
本発明の一般式(I)で表される化合物は、構成する原子の一つまたは複数で非天然の比率の原子同位体を含むこともある。原子同位体としては、例えば、重水素(2H)、本明細書においてはDと著すことがある。)、トリチウム(3H)、ヨウ素−125(125I)または炭素−14(14C)などが挙げられる。これらの化合物は、治療又は予防剤、研究試薬、例えば、アッセイ試薬、及び診断剤、例えば、インビボ画像診断剤として有用である。一般式(I)で表される化合物のすべての同位体変種は、放射性であるかどうかにかかわらず、本発明の範囲内に含まれる。
また、本発明は、生体内における生理条件下で酵素や胃酸等による反応により本発明の医薬組成物の有効成分である一般式(I)で表される化合物に変換される化合物、すなわち、酵素的に酸化、還元、加水分解等を起こして一般式(I)で表される化合物に変化される化合物又は胃酸等により加水分解等を起こして一般式(I)で表される化合物に変化される「医薬的に許容されるプロドラッグ化合物」も本発明に包含する。
上記プロドラッグとしては、一般式(I)で表される化合物にアミノ基が存在する場合には、そのアミノ基がアシル化、アルキル化、リン酸化された化合物(例えば、そのアミノ基がエイコサノイル化、アラニル化、ペンチルアミノカルボニル化、(5−メチル−2−オキソ−1,3−ジオキソレン−4−イル)メトキシカルボニル化、テトラヒドロフラニル化、ピロリジルメチル化、ピバロイルオキシメチル化、tert−ブチル化された化合物等である)等を挙げることができ、一般式(I)で表される化合物に水酸基が存在する場合には、その水酸基がアシル化、アルキル化、リン酸化、ホウ酸化された化合物(例えば、その水酸基がアセチル化、パルミトイル化、プロパノイル化、ピバロイル化、サクシニル化、フマリル化、アラニル化、ジメチルアミノメチルカルボニル化された化合物等である。)等を挙げることができる。また、一般式(I)で表される化合物にカルボキシ基が存在する場合には、そのカルボキシ基がエステル化、アミド化された化合物(例えば、そのカルボキシ基がエチル エステル化、フェニル エステル化、カルボキシメチル エステル化、ジメチルアミノメチル エステル化、ピバロイルオキシメチル エステル化、エトキシカルボニルオキシエチル エステル化、アミド化又はメチルアミド化された化合物等である。)等が挙げられる。
本発明の化合物のプロドラッグは公知の方法によって一般式(I)で表される化合物から製造することができる。また、本発明の化合物のプロドラッグは、広川書店1990年刊「医薬品の開発」第7巻分子設計163頁~198頁に記載されているような、生理的条件で一般式(I)で表される化合物に変化するものも含まれる。
次に、一般式(I)で表される化合物の代表的な製造法について説明する。本発明の化合物は種々の製造法により製造することができ、以下に示す製造法は一例であり、本発明はこれらに限定して解釈されるべきではない。なお、反応に際しては、必要に応じて置換基を適当な保護基で保護して行うことができ、保護基の種類は特に限定されない。

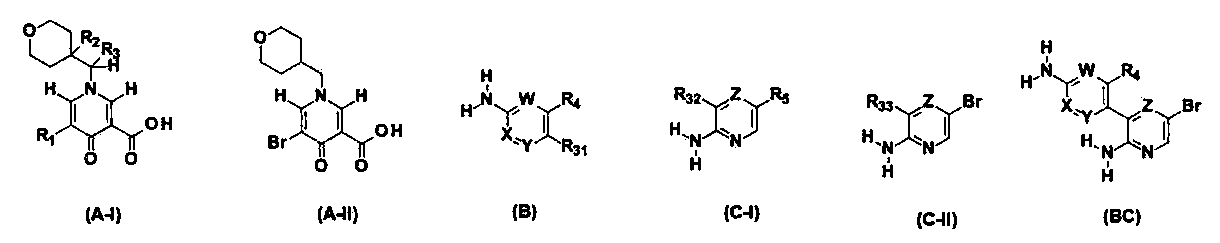
まず化合物(A−I)の合成法について説明する。
[製造法1]
化合物(A−I)は、下記反応式1のような方法で合成できるが、特に限定されない。
反応式1

化合物(1c)の合成
市販または適宜合成した化合物(1a)を、炭酸カリウム、炭酸ナトリウム、炭酸セシウム、酢酸カリウム、りん酸カリウム、tert−ブトキシド又はトリエチルアミン等の有機又は無機塩基、トリス(ジベンジリデンアセトン)−ジパラジウム(0)、テトラキス(トリフェニルフォスフィン)パラジウム(0)、ビス(トリフェニルフォスフィン)塩化パラジウム(II)、[1,1’−ビス(ジフェニルフォスフィノ)−フェロセン]塩化パラジウム(II)ジクロロメタン錯体、酢酸パラジウム(II)、沃化銅(I)塩化銅(I)等の遷移金属触媒存在下、市販の、または製造法4、製造法5、製造法6で示された方法で合成した化合物(1b)とカップリング反応を行うことにより化合物(1c)を得ることができる。また、上記反応は、トリフェニルフォスフィン、トリシクロヘキシルフォスフィン、ジベンジリデンアセトン、1,3−ビス(ジフェニルフォスフィノ)プロパン、2−ジシクロヘキシルフォスフィノ−2’,4’,6’−トリイソプロピルビフェニル、4,5−ビス(ジフェニルフォスフィノ)−9,9−ジメチルキサンテン等の配位子存在下実施することもできる。また、上記反応は、上記遷移金属触媒を2種類以上併用して行うこともできる。溶媒は、1,4−ジオキサン、1,2−ジメトキシエタン、メタノール、トルエン、水または、これらの混合溶媒が挙げられ、特に限定されない。反応温度は、通常0℃から200℃もしくは溶媒の沸点までの範囲であるが、好ましくは、0℃から溶媒の沸点までの範囲である。また上記処理は、封管中、マイクロウェーブによっても行うことができる。有機又は無機塩基は、化合物(1a)に対し、1から過剰モル当量を用いればよい。好ましくは、1から5モル当量用いればよい。遷移金属触媒や配位子は、化合物(1c)に対し、0.001から1モル当量を用いればよい。好ましくは、0.05から1モル当量用いればよい。ボロン酸エステルは、化合物(1c)に対し、1から過剰モル当量を用いればよい。好ましくは、1から5モル当量用いればよい。
化合物(1d)の合成
化合物(1c)を、硫酸、塩酸等の酸、もしくは水酸化ナトリウム、炭酸カリウム等のアルカリで加水分解処理する事で得ることができる。塩基もしくは酸は、化合物(1c)に対して1から過剰モル当量を用いればよい。反応に用いられる溶媒は、メタノール、エタノール、プロパノール、水等、または、これらの混合溶媒が挙げられ、特に限定されない。反応温度は、通常0℃から200℃もしくは溶媒の沸点までの範囲であるが、好ましくは、0℃から溶媒の沸点までの範囲である。
化合物(1f)の合成
市販または適宜合成した化合物に(1e)を活性化し、マロン酸モノエステルまたはその塩とマグネシウムクロリド存在下、反応させることで得ることができる。カルボン酸の活性化の方法としては、1,1’−カルボニルジイミダゾールを用いる方法や、酸クロリドを経由する方法を挙げることができる。反応は必要に応じて塩基を添加することができ、塩基としては、たとえばトリエチルアミン等を挙げることができる。塩基は、化合物(1e)に対して1から過剰モル当量を用いればよい。好ましくは、2から10モル当量用いればよい。反応に用いられる溶媒は、テトラヒドロフラン、ジオキサン、アセトニトリル、N,N−ジメチルホルムアミド、トルエン等、または、これらの混合溶媒が挙げられ、特に限定されない。反応温度は、通常−78℃から100℃もしくは溶媒の沸点までの範囲であるが、好ましくは、室温付近から100℃までの範囲である。
化合物(1g)の合成
化合物(1f)をN,N−ジメチルホルムアミドジメチルアセタールで処理することで得ることができる。N,N−ジメチルホルムアミドジメチルアセタールは、化合物(1f)に対して2から過剰モル当量を用いればよい。反応に用いられる溶媒は、トルエン、キシレン、ジクロロメタン、エタノール、N,N−ジメチルホルムアミド、酢酸エチル等、または、これらの混合溶媒が挙げられ、特に限定されない。もしくは無溶媒でも行われる。反応温度は、通常0℃から300℃の範囲であるが、好ましくは、室温付近から130℃までの範囲である。
化合物(1i)の合成
化合物(1g)を、市販または適宜合成した化合物(1h)で処理することで得ることができる。反応に用いられる溶媒は、トルエン、酢酸エチル、エタノール、1,4−ジオキサン等、または、これらの混合溶媒が挙げられ、特に限定されない。反応温度は、通常−78℃から130℃もしくは溶媒の沸点までの範囲であるが、好ましくは、室温付近から溶媒の沸点までの範囲である。また、上記反応は、必要に応じて、炭酸カリウム、炭酸セシウム、カリウム tert−ブトキシド又はトリエチルアミン等の有機又は無機塩基、もしくは、酢酸、塩化水素、臭化水素、硫酸等の酸を加えて行うことができる。化合物(1h)、塩基もしくは酸は、化合物(1g)に対して1から過剰モル当量を用いればよい。好ましくは、1から5モル当量用いればよい。
化合物(A−I)の合成
化合物(1d)の合成と同様の操作により得ることができる。
[製造法2]
また化合物(1i)は下記反応式2のような方法によっても合成できるが、特に限定されない。
反応式2

化合物(2a)の合成
化合物(2a)は、市販、もしくはJ.Heterocyclic Chem.17,359(1980)やChem.Pharm.Bull.43,450(1995)などの既報の報告例を参考に化合物(1f)から合成することができる。
化合物(2c)の合成
化合物(2a)を、炭酸カリウム、炭酸セシウム、カリウム tert−ブトキシド又はトリエチルアミン等の有機又は無機塩基存在下、化合物(2b)で処理することにより得ることができる。反応に用いられる溶媒は、N,N−ジメチルホルムアミド、ジメチルスルホキシド、アセトニトリルまたは、これらの混合溶媒が挙げられ、特に限定されない。反応温度は、通常0℃から200℃もしくは溶媒の沸点までの範囲であるが、好ましくは、0℃から溶媒の沸点までの範囲である。化合物(2b)、塩基は、化合物(2a)に対して1から過剰モル当量を用いればよい。好ましくは、1から5モル当量用いればよい。
次に化合物(A−II)の合成法について説明する。
[製造法3]
化合物(A−II)は、下記反応式3のような方法で合成できるが、特に限定されない。
反応式3

化合物(3b)の合成
市販、もしくはJ.Med.Chem.51,5330(2008)などの既報の報告例を参考に容易に合成できる化合物(3a)を炭酸カリウム、炭酸セシウム、カリウム tert−ブトキシド又はトリエチルアミン等の有機又は無機塩基存在下、化合物(2b)で処理することにより得ることができる。反応に用いられる溶媒は、N,N−ジメチルホルムアミド、ジメチルスルホキシド、アセトニトリルまたは、これらの混合溶媒が挙げられ、特に限定されない。反応温度は、通常0℃から200℃もしくは溶媒の沸点までの範囲であるが、好ましくは、0℃から溶媒の沸点までの範囲である。化合物(2b)、塩基は、化合物(3a)に対して1から過剰モル当量を用いればよい。好ましくは、1から5モル当量用いればよい。
化合物(A−II)の合成
[製造法1]における化合物(A−I)の合成と同様の操作により得ることができる。
次に化合物(C−I)の合成法について説明する。
R5が水素原子の場合、化合物(C−I)は、市販品であるか、市販品から文献既知の方法で容易に合成できる。
R5が水素原子以外の場合、化合物(C−I)は、下記製造法4から製造法7のような方法で合成できる。すなわち、製造法4から製造法6に示すような方法のうちいずれかを選択し、R5を構成する化合物(4c)、化合物(5h)もしくは化合物(6c)を合成したのち、これら化合物または市販化合物を用いて、製造法7で化合物(C−I)を合成することができる。ただし、特にこれらに限定されない。
[製造法4]
反応式4

化合物(4c)の合成
市販の、または適宜合成した化合物(4a)を、炭酸カリウム、炭酸セシウム、カリウム tert−ブトキシド又はトリエチルアミン等の有機又は無機塩基存在下、化合物(4b)で処理することにより得ることができる。反応に用いられる溶媒は、N,N−ジメチルホルムアミド、ジメチルスルホキシド、アセトニトリルまたは、これらの混合溶媒が挙げられ、特に限定されない。反応温度は、通常0℃から200℃もしくは溶媒の沸点までの範囲であるが、好ましくは、0℃から溶媒の沸点までの範囲である。化合物(4b)、塩基は、化合物(4a)に対して1から過剰モル当量を用いればよい。好ましくは、1から5モル当量用いればよい。
[製造法5]
反応式5
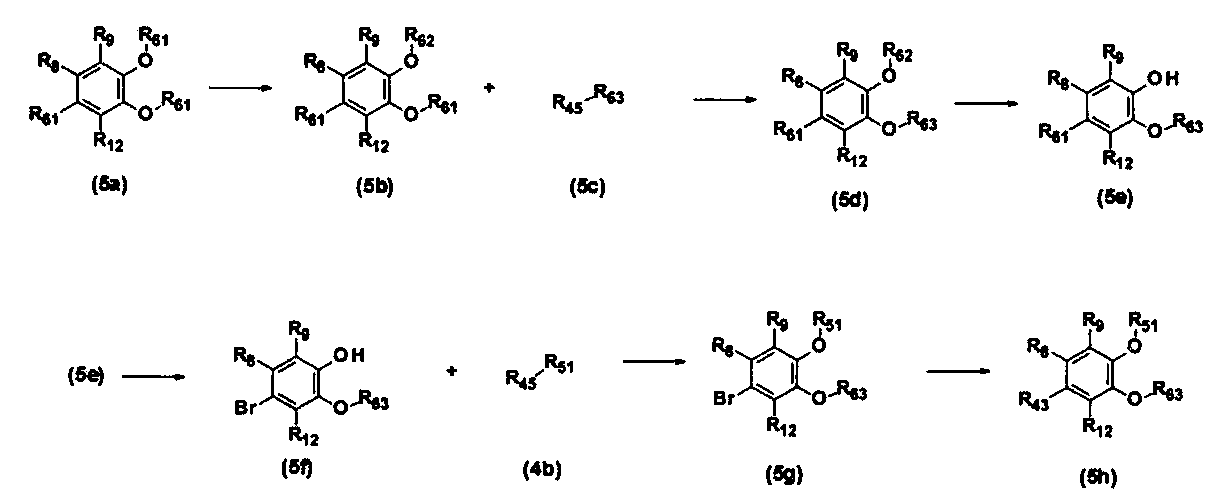
化合物(5b)の合成
市販の、または適宜合成した化合物(5a)の2つの水酸基のうち1つのみを、保護することにより化合物(5b)を得ることができる。例えば、化合物(5a)を、p−トルエンスルホン酸ピリジン塩等の酸存在下、3,4−ジヒドロ−2H−ピランで処理することにより、1つのみの水酸基をテトラヒドロピラニル基で保護した化合物(5b)を得ることができる。ただし、水酸基保護の方法は特にこれらに限定されない。反応に用いられる溶媒は、ジクロロメタン、クロロホルム、トルエンまたは、これらの混合溶媒が挙げられ、特に限定されない。反応温度は、通常0℃から200℃もしくは溶媒の沸点までの範囲であるが、好ましくは、0℃から溶媒の沸点までの範囲である。化合物(4b)、酸は、化合物(5a)に対して0.01から過剰モル当量を用いればよい。好ましくは、0.01から1モル当量用いればよい。
化合物(5d)の合成
化合物(5b)を、炭酸カリウム、炭酸セシウム、カリウム tert−ブトキシド又はトリエチルアミン等の有機又は無機塩基存在下、化合物(5c)で処理することで化合物(5d)を得ることができる。N,N−ジメチルホルムアミド、ジメチルスルホキシド、アセトニトリル、アセトンまたは、これらの混合溶媒が挙げられ、特に限定されない。反応温度は、通常0℃から200℃もしくは溶媒の沸点までの範囲であるが、好ましくは、0℃から溶媒の沸点までの範囲である。化合物(5c)、塩基は、化合物(5b)に対して1から過剰モル当量を用いればよい。好ましくは、1から5モル当量用いればよい。
化合物(5e)の合成
化合物(5d)を、塩酸、酢酸、p−トルエンスルホン酸ピリジン塩等の酸で処理することにより、化合物(5e)を得ることができる。反応に用いられる溶媒は、エタノール、メタノール、イソプロパノール、水または、これらの混合溶媒が挙げられ、特に限定されない。反応温度は、通常0℃から200℃もしくは溶媒の沸点までの範囲であるが、好ましくは、0℃から溶媒の沸点までの範囲である。化合物(4b)、酸は、化合物(5d)に対して0.01から過剰モル当量を用いればよい。好ましくは、0.01から1モル当量用いればよい。
化合物(5f)の合成
化合物(5e)をN−ブロモコハク酸イミド、臭素酸カリウム等の臭素化試薬で処理することで、化合物(5f)を得ることができる。反応に用いられる溶媒は、N,N−ジメチルホルムアミド、30%臭化水素酸−酢酸溶液、酢酸または、これらの混合溶媒が挙げられ、特に限定されない。反応温度は、通常0℃から200℃もしくは溶媒の沸点までの範囲であるが、好ましくは、0℃から溶媒の沸点までの範囲である。臭素化試薬化合物(5e)に対して0.1から過剰モル当量を用いればよい。好ましくは、0.1から5モル当量用いればよい。
化合物(5g)の合成
[製造法4]と同様の操作により得ることができる。
化合物(5h)の合成
化合物(5g)を、炭酸カリウム、炭酸ナトリウム、炭酸セシウム、酢酸カリウム、リン酸カリウム、tert−ブトキシド又はトリエチルアミン等の有機又は無機塩基、トリス(ジベンジリデンアセトン)−ジパラジウム(0)、ビス(トリフェニルフォスフィン)塩化パラジウム(II)、[1,1’−ビス(ジフェニルフォスフィノ)−フェロセン]塩化パラジウム(II)ジクロロメタン錯体、[1,1’−ビス(ジフェニルフォスフィノ)−フェロセン]塩化パラジウム(II)ジクロロメタン錯体、1,3−ビス(ジフェニルフォスフィノ)プロパン]塩化ニッケル(II)、等の遷移金属触媒、トリフェニルフォスフィン、トリシクロヘキシルフォスフィン、ジベンジリデンアセトン、1,3−ビス(ジフェニルフォスフィノ)プロパン、2−ジシクロヘキシルフォスフィノ−2’,4’,6’−トリイソプロピルビフェニル、4,5−ビス(ジフェニルフォスフィノ)−9,9−ジメチルキサンテン等の配位子存在下、ビス(ピナコラート)二ホウ素、ピナコールホウ素等のボロン酸エステルで処理することにより得ることができる。溶媒は、1,4−ジオキサン、1,2−ジメトキシエタン、メタノール、トルエンまたは、これらの混合溶媒が挙げられ、特に限定されない。反応温度は、通常0℃から200℃もしくは溶媒の沸点までの範囲であるが、好ましくは、0℃から溶媒の沸点までの範囲である。また上記処理は、封管中、マイクロウェーブによっても行うことができる。有機又は無機塩基は、化合物(5g)に対し、1から過剰モル当量を用いればよい。好ましくは、1から5モル当量用いればよい。遷移金属触媒や配位子は、化合物(5g)に対し、0.001から1モル当量を用いればよい。好ましくは、0.05から1モル当量用いればよい。ボロン酸エステルは、化合物(5g)に対し、1から過剰モル当量を用いればよい。好ましくは、1から5モル当量用いればよい。
[製造法6]
反応式6
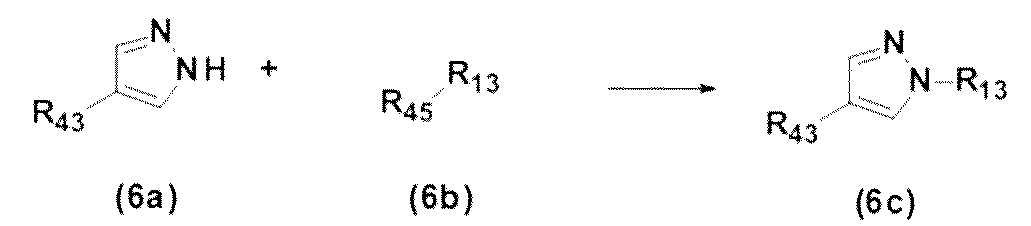
化合物(6c)の合成
市販の、または適宜合成した化合物(6a)および(6b)を用いて[製造法4]と同様の操作により得ることができる。
[製造法7]
反応式7

化合物(7c)の合成
[製造法1]における化合物(1c)の合成と同様に、化合物(7b)を、市販の、または製造法4、製造法5、製造法6で示された方法で合成した化合物(7a)とカップリング反応を行うことにより化合物(7c)を得ることができる。
化合物(7d)の合成
市販の、または適宜合成した化合物(7c)を用いて[製造法5]の化合物(5h)の合成と同様の操作により得ることができる。
次に化合物(B)化合物(C−I)複合体の合成法について説明する。
化合物(B)化合物(C−I)複合体は、[製造法8]に示すとおり、化合物(BC)の臭素基をR5に変換する方法により合成することができるが特に限定されない。
[製造法8]
反応式8

市販の、または適宜合成した化合物(BC)または化合物(8)を用いて[製造法1]の化合物(1c)の合成と同様の操作により得ることができる。
また、化合物(8)は[製造法9]や[製造法10]に示す方法によっても合成することができるが特に限定されない。
[製造法9]
反応式9

市販の、または適宜合成した化合物(9a)および(9b)を用いて[製造法1]の化合物(1c)の合成と同様の操作により得ることができる。
[製造法10]
反応式10

市販の、または適宜合成した化合物(7d)および化合物(10)を用いて[製造法1]の化合物(1c)の合成と同様の操作により得ることができる。
次に化合物(B)化合物(C−I)複合体と化合物(A−I)を結合させる合成法について説明する。
化合物(B)化合物(C−I)複合体と化合物(A−I)を結合させるには、以下の製造法11もしくは製造法12により行うことができる。
[製造法11]
下記反応式11のような方法で合成できるが、特に限定されない。
反応式11

化合物(A−I)と化合物(8)を、縮合剤存在下反応させることで得ることができる。縮合剤としては、N−[1−(シアノ−2−エトキシ−2−オキソエチリデンアミノオキシ)ジメチルアミノ(モルホリノ)]ウロニウム ヘキサフルオロフォスフェイト(COMU)等が挙げられるが、この限りではない。反応に用いられる溶媒は、N,N−ジメチルホルムアミド、ジクロロメタンまたは、これらの混合溶媒が挙げられ、特に限定されない。反応温度は、通常−78℃から200℃もしくは溶媒の沸点までの範囲であるが、好ましくは、0℃から100℃までの範囲である。縮合剤は、化合物(A−I)に対し、1から過剰モル当量を用いればよい。好ましくは、1から5モル当量用いればよい。
[製造法12]
下記反応式12のような方法で合成できるが、特に限定されない。
反応式12

化合物(A−I)を塩化チオニル、塩化オキザリル等の酸ハライド化試薬で処理することで化合物(12)を得たのち、化合物(8)で処理することで得ることができる。反応に用いられる溶媒は、ジクロロメタン、ジクロロエタンまたは、これらの混合溶媒が挙げられ、特に限定されない。酸ハライド化試薬は化合物(A−I)に対し、0.9から過剰モル当量を用いればよい。好ましくは、0.9から2モル当量用いればよい。
次に化合物(A−I)化合物(B)複合体の合成法について説明する。
[製造法13]
下記反応式13のような方法で合成できるが、特に限定されない。
反応式13
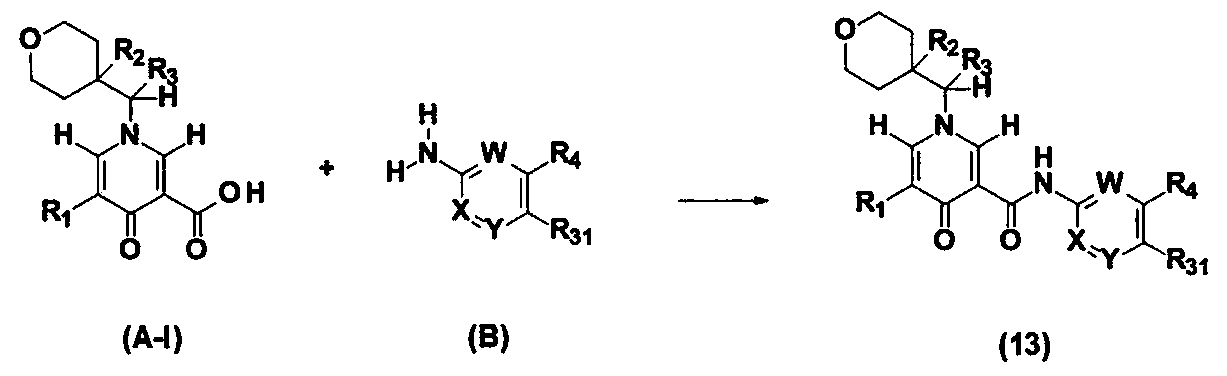
化合物(13)の合成
化合物(B)は、市販品であるか、市販品から文献既知の方法で容易に合成できる。化合物(A−I)と市販の、または適宜合成した化合物(B)を用いて[製造法11]と同様の操作により得ることができる。
次に化合物(A−I)化合物(B)複合体と化合物(C−I)を結合させる合成法について説明する。
[製造法14]
下記反応式14のような方法で合成できるが、特に限定されない。
反応式14

化合物(11)の合成
化合物(13)と市販の、または適宜合成した化合物(C−I)を用いて、[製造法1]の化合物(1c)の合成と同様の操作により得ることができる。
次に化合物(B)化合物(C−I)複合体と化合物(A−II)を結合させる合成法について説明する。
[製造法15]
下記反応式15のような方法で合成できるが、特に限定されない。
反応式15

化合物(15)の合成
化合物(A−II)と化合物(8)を用いて、[製造法11]と同様の操作により得ることができる。
次に化合物(A−II)化合物(B)化合物(C−I)複合体の臭素基をR1に変換する合成法について説明する。
[製造法16]
下記反応式16のような方法で合成できるが、特に限定されない。
反応式16

化合物(16b)の合成
化合物(15)と市販の、または適宜合成した化合物(16a)を用いて[製造法1]の化合物(1c)の合成と同様の操作により得ることができる。
次に化合物(A−I)化合物(B)化合物(C−I)複合体のハロゲン基をアルキル基に変換する合成法について説明する。
[製造法17]
下記反応式17のような方法で合成できるが、特に限定されない。
反応式17

化合物(17b)の合成
化合物(17a)と市販の、または適宜合成した化合物(1b)を用いて[製造法1]の化合物(1c)の合成と同様の操作により得ることができる。
次に化合物(A−I)と化合物(BC)を結合させたのち臭素基をR5に変換する合成法について説明する。
[製造法18]
下記反応式18のような方法で合成できるが、特に限定されない。
反応式18

化合物(18)の合成
化合物(A−I)と化合物(BC)を用いて、[製造法11]と同様の操作により得ることができる。
化合物(11)の合成
化合物(18)と市販の、または適宜合成した化合物(7a)を用いて[製造法1]の化合物(1c)の合成と同様の操作により得ることができる。
また化合物(18)は[製造法19]のような方法によっても合成することができる。
[製造法19]
反応式19

化合物(C−II)は、市販品であるか、市販品から文献既知の方法で容易に合成できる。化合物(19)と市販の、または適宜合成した化合物(C−II)を用いて[製造法1]の化合物(1c)の合成と同様の操作により得ることができる。
また、化合物(11)は製造法20によっても製造することができる。
[製造法20]
反応式20

化合物(20b)の合成
化合物(18)と市販の、または適宜合成した化合物(20a)を用いて[製造法1]の化合物(1c)の合成と同様の操作により得ることができる。
化合物(20d)の合成
化合物(20b)を還元剤存在下、市販の、または適宜合成した化合物(20c)を反応させることで得ることができる。用いる還元剤としては、水素化トリアセトキシホウ素ナトリウム、水素化シアノホウ素ナトリウム、水素化ホウ素ナトリウム等、を挙げることができる。用いる溶媒としては、メタノール、エタノール、水、テトラヒドロフラン、ジオキサン、N,N−ジメチルホルムアミド、塩化メチレン、酢酸等、または、これらの混合溶媒が挙げられ、特に限定されない。反応温度は、通常−78℃から100℃もしくは溶媒の沸点までの範囲であるが、好ましくは、0℃から100℃までの範囲である。
前述のとおり、Gas6/Axlシグナル伝達系は、細胞生存(cell survival)、細胞分裂、自食作用(autophagy)、細胞運動(migration)、血管新生、血小板凝集、NK細胞分化等、多様な細胞性応答を調節していることが報告されていることから(非特許文献2)、Axl阻害剤は、Axlキナーゼ機能亢進を原因とする疾患、Axlキナーゼ機能亢進と関連する疾患、および/またはAxlキナーゼ機能亢進を伴う疾患の治療のための有用である。
特に、本発明の化合物はAxl阻害特異性が高く、網膜毒性も引き起こさないため、より安全に使用することができる。
Axlキナーゼ機能亢進を原因とする疾患、Axlキナーゼ機能亢進と関連する疾患、Axlキナーゼ機能亢進を伴う疾患としては、Axlの遺伝子および/または蛋白質の過剰発現がみられる組織を有する疾患、Axlのリン酸化活性の上昇がみられる組織を有する疾患等が挙げられる。
上記疾患の例としては、例えば、過剰増殖性疾患が挙げられ、過剰増殖性疾患の例としては、子宮内膜増殖症、トロンビン誘導血管平滑筋細胞(VSMC)増殖良性腫瘍、悪性腫瘍(癌)、急性及び慢性糸球体腎炎、糖尿病性腎症等が挙げられるがこれらに限定されない。
さらに、Axlは、免疫(非特許文献12)、血小板機能(非特許文献13)、精子形成(非特許文献14)、血管石灰化(非特許文献15)、(非特許文献16)および種々の腎臓疾患、慢性同種移植拒絶反応(非特許文献17)において役割を有することも示されており、Axl阻害剤は血管疾患(血栓症、アテローム性動脈硬化症及び再狭窄を含むがそれらに限定されるものではない。)および無秩序な血管形成が重大である疾患(糖尿病性網膜症、網膜症、乾癬、関節リウマチ、粥腫、カポジ肉腫及び血管腫を含むがそれらに限定されるものではない。)等、多数の疾患の治療に有用である。
本発明の化合物はAxlを阻害するので、上記に記載した疾患の治療に有用である。
より好ましくは、本発明の化合物は各種癌の治療に有用である。癌としては、例えば、乳癌、結腸癌、前立腺癌、肺癌、胃癌、卵巣癌、子宮頸癌、子宮内膜癌、子宮体癌、腎癌、肝細胞癌、甲状腺癌、食道癌、扁平上皮癌、白血病、骨肉腫、メラノーマ、膠芽細胞腫、神経芽細胞腫、卵巣癌、頭頚部癌、睾丸腫瘍、大腸癌、血液癌、網膜芽細胞腫、膵臓癌等が挙げられるが、これらの癌に限定されない。
癌とAxlとの関係については、増殖阻害、転移・運動(migration)・浸潤(invasion)阻害、薬剤耐性解除の観点から各種報告がなされている。
Axlのドミナントネガティブ変異体は脳腫瘍の増殖を抑制したことが報告されている(Vajkoczy et al.,PNAS 2006,103,5799)。グリオブラストーマ患者由来組織においてAxlの発現やAxl/Gas6共発現がみられる場合、腫瘍増殖が顕著に早く進み、患者生存期間が短いことが報告されている(Hutterer et al.,Clin Cancer Res 14,130(2008))。AxlのshRNAが乳癌細胞の増殖を抑制したことが報告されている(Yi−Xiang et al.,Cancer Res 68,1905(2008))。これらの報告から、Axl阻害剤が癌における細胞増殖阻害に有用であることが明らかである。
一方で、Axlのドミナントネガティブ変異体が細胞の運動(migration)および浸潤(invasion)を阻害したことが報告されている(Zhang et al.,Cancer Res 68,1905(2008),Vajkoczy et al.,PNAS 103,5799(2006),Holland et al.,Cancer Res 65,9294(2005))。Axl−shRNAがin vivoで転移を抑制したことが報告されている(Li et al.,oncogene 28,3442(2009))。抗Axl抗体、siRNAがマウスモデルで腫瘍増殖と転移を抑制したことが報告されている(Li et al.,Oncogene 28,3442(2009),Ye et al.,Oncogene 29,5254(2010))。Axlが細胞の浸潤(invasion)を促進することが報告されている(Tai et al.,Oncogene 27,4044(2008))。Axl阻害剤であるR−428が転移性乳癌の拡散モデルを阻害したことが報告されている(Holland et al.,Cancer Res 70,1544(2010))。Axl抗体、AxlのshRNAおよびAxl阻害剤NA80x1が乳癌細胞の移動や浸潤(invasion)を阻害したことが報告されている(Yi−Xiang et al.,Cancer Res 68,1905(2008))。その他、前立腺癌、脾臓癌、転移性卵巣癌、胸腺癌等の転移や悪性化にAxlが関与することが複数報告されている。これらの報告から、Axl阻害剤が、癌の転移、細胞の運動(migration)、浸潤(invasion)の抑制、治療、予防等に有用であることが明らかである。
また、Axl阻害剤が胃癌におけるイマチニブ耐性を解除したことが報告されている(Mahadevan et al.,Oncogene 26,3909(2007))。ドキソルビシン、VP16、シスプラチン等の化学療法剤耐性において、Axlが誘導されることが急性骨髄性白血病で示されている(Hong et al.,Cancer Letters 268,314(2008))。HER−2陽性乳癌細胞におけるラパチニブ耐性においてAxlの活性化が見られることが報告されている(Liu et al.,Cancer Res 69,6871(2009))。AxlがPLX4032(vemurafenib)耐性機構に関与することが報告されている(Johannessen et al.,Nature 468,968(2010))。その他、temozolomode、カルボプラチン、ビンクリスチン耐性にAxlが関与することが報告されている(AK Keeating et al.,Mol Cancer Ther 9(5),1298(2010))。これらの報告から、Axl阻害剤が、薬剤耐性の解除、例えば、各種抗癌剤に対する耐性の解除に有用であることが明らかである。
本願のAxl阻害剤は、本願試験例に示すように、erlotinibと併用投与することにより腫瘍のerlotinibに対する耐性化を阻害した。またerlotinib投与によりAxl蛋白質の有意な誘導が観察されたことから、癌の薬剤耐性獲得にAxlが関与していると考えられる。
また、erlotinibに対し耐性をとなった腫瘍に対し、本願のAxl阻害剤を併用することにより、erlotinibへの感受性が回復したことから、癌の薬剤耐性の解除にAxl阻害剤が有効であると考えられる。
さらには、Axlは腎臓の繊維化、糖尿病性腎症等の腎疾患に関与することが報告されており(特表2005−517412)、Axl阻害剤が上記腎疾患、その他、特発性肺線維症等の繊維化疾患の治療に有用であることが明らかである。
化合物のAxl阻害活性は、例えば、本願の試験例に記載した方法により測定することができるが、これに限定されない。
細胞の増殖阻害活性は、当業者に通常用いられる増殖阻害試験法を用いて調べることができる。細胞の増殖阻害活性は、試験化合物の存在下または非存在下における細胞(例えば、腫瘍細胞)の増殖の程度を比較することによって実施することができる。増殖の程度は、例えば、生細胞を測定する試験系を用いて調べることができる。生細胞の測定方法としては、例えば、[3H]−チミジンの取り込み試験、BrdU法またはMTTアッセイ等が挙げられる。
また、in vivoでの抗腫瘍活性は、当業者に通常用いられる抗腫瘍試験法を用いて調べることができる。例えば、マウス、ラット等に各種腫瘍細胞を移植し、移植細胞の生着が確認された後に、本発明の化合物を経口投与、静脈内投与等し、数日~数週間後に、薬剤無投与群における腫瘍増殖と化合物投与群における腫瘍増殖とを比較することにより本発明のin vivoでの抗腫瘍活性を確認することができる。
その他、転移抑制活性、浸潤(invasion)阻害活性、運動(migration)阻害活性、薬剤耐性解除活性については、Axlとそれぞれの活性との関連性が報告されているとして上記に挙げた文献に記載の試験方法にて測定することができる。
本発明の医薬組成物は、本発明の化合物と薬学的に許容し得る担体を含み、静脈内注射、筋肉内注射、皮下注射等の各種注射剤として、あるいは、経口投与または経皮投与等の種々の方法によって投与することができる。薬学的に許容し得る担体とは、本発明の化合物または本発明の化合物を含む組成物を、ある器官または臓器から他の器官または臓器に輸送することに関与する、薬学的に許容される材料(例えば、賦形剤、希釈剤、添加剤、溶媒等)を意味する。
製剤の調製方法としては投与法に応じ適当な製剤(例えば、経口剤または注射剤)を選択し、通常用いられている各種製剤の調製法にて調製できる。経口剤としては、例えば、錠剤、散剤、顆粒剤、カプセル剤、丸剤、トローチ剤、溶液剤、シロップ剤、エリキシル剤、乳剤、または油性ないし水性の懸濁液等を例示できる。経口投与の場合では遊離体のままでも、塩の型のいずれでもよい。水性製剤は薬学的に許容される酸と酸付加物を形成させるか、ナトリウム等のアルカリ金属塩とすることで調製できる。注射剤の場合は製剤中に安定剤、防腐剤または溶解補助剤等を使用することもできる。これらの補助剤等を含むこともある溶液を容器に収納後、凍結乾燥等によって固形製剤として用時調製の製剤としてもよい。また、一回投与量を一の容器に収納してもよく、また複数回投与量を一の容器に収納してもよい。
固形製剤としては、例えば、錠剤、散剤、顆粒剤、カプセル剤、丸剤、またはトローチ剤が挙げられる。これらの固形製剤は、本発明の化合物とともに薬学的に許容し得る添加物を含んでもよい。添加物としては、例えば、充填剤類、増量剤類、結合剤類、崩壊剤類、溶解促進剤類、湿潤剤類または滑沢剤類が挙げられ、これらを必要に応じて選択して混合し、製剤化することができる。
液体製剤としては、例えば、溶液剤、シロップ剤、エリキシル剤、乳剤、または懸濁剤が挙げられる。これらの液体製剤は、本発明の化合物とともに薬学的に許容し得る添加物を含んでもよい。添加物としては、例えば、懸濁化剤または乳化剤が挙げられ、これらを必要に応じて選択して混合し、製剤化することができる。
本発明の化合物は、哺乳類、特にヒトの癌治療に用いることができる。投与量および投与間隔は、疾患の場所、患者の身長、体重、性別または病歴によって、医師の判断により適宜選択され得る。本発明の化合物をヒトに投与する場合、投与量の範囲は、1日当たり、約0.01mg/kg体重~約500mg/kg体重、好ましくは、約0.1mg/kg体重~約100mg/kg体重である。ヒトに投与する場合、好ましくは、1日あたり1回、あるいは2から4回に分けて投与され、適当な間隔で繰り返すのが好ましい。また、1日量は、医師の判断により必要によっては上記の量を超えてもよい。
本発明の化合物は他の抗腫瘍剤と併用して用いてもよい。上記に示したerlotinibのようなチロシンキナーゼ阻害剤(TKI)のほか、例えば、抗腫瘍抗生物質、抗腫瘍性植物成分、BRM(生物学的応答性制御物質)、ホルモン、ビタミン、抗腫瘍性抗体、他の分子標的薬、その他の抗腫瘍剤等が挙げられる。
より具体的に、アルキル化剤としては、例えば、ナイトロジェンマスタード、ナイトロジェンマスタードN−オキシドもしくはクロラムブチル等のアルキル化剤、カルボコンもしくはチオテパ等のアジリジン系アルキル化剤、ディブロモマンニトールもしくはディブロモダルシトール等のエポキシド系アルキル化剤、カルムスチン、ロムスチン、セムスチン、ニムスチンハイドロクロライド、ストレプトゾシン、クロロゾトシンもしくはラニムスチン等のニトロソウレア系アルキル化剤、ブスルファン、トシル酸インプロスルファンまたはダカルバジン等が挙げられる。
各種代謝拮抗剤としては、例えば、6−メルカプトプリン、6−チオグアニンもしくはチオイノシン等のプリン代謝拮抗剤、フルオロウラシル、テガフール、テガフール・ウラシル、カルモフール、ドキシフルリジン、ブロクスウリジン、シタラビン若しくはエノシタビン等のピリミジン代謝拮抗剤、メトトレキサートもしくはトリメトレキサート等の葉酸代謝拮抗剤等が挙げられる。
抗腫瘍性抗生物質としては、例えば、マイトマイシンC、ブレオマイシン、ペプロマイシン、ダウノルビシン、アクラルビシン、ドキソルビシン、ピラルビシン、THP−アドリアマイシン、4’−エピドキソルビシンもしくはエピルビシン等のアントラサイクリン系抗生物質抗腫瘍剤、クロモマイシンA3またはアクチノマイシンD等が挙げられる。
抗腫瘍性植物成分としては、例えば、ビンデシン、ビンクリスチン若しくはビンブラスチン等のビンカアルカロイド類、パクリタキセル、ドセタキセル等のタキサン類、またはエトポシドもしくはテニポシド等のエピポドフィロトキシン類が挙げられる。
BRMとしては、例えば、腫瘍壊死因子またはインドメタシン等が挙げられる。
ホルモンとしては、例えば、ヒドロコルチゾン、デキサメタゾン、メチルプレドニゾロン、プレドニゾロン、プラステロン、ベタメタゾン、トリアムシノロン、オキシメトロン、ナンドロロン、メテノロン、ホスフェストロール、エチニルエストラジオール、クロルマジノンまたはメドロキシプロゲステロン等が挙げられる。
ビタミンとしては、例えば、ビタミンCまたはビタミンA等が挙げられる。
抗腫瘍性抗体、分子標的薬としては、トラスツズマブ、リツキシマブ、セツキシマブ、ニモツズマブ、デノスマブ、ベバシズマブ、インフリキシマブ、メシル酸イマチニブ、ゲフィチニブ、エルロチニブ、スニチニブ、ラパチニブ、ソラフェニブ、ダサチニブ、ニロチニブ、ベムラフェニブ、チバンチニブ等が挙げられる。
その他の抗腫瘍剤としては、例えば、シスプラチン、カルボプラチン、オキサリプラチン、タモキシフェン、カンプトテシン、イホスファミド、シクロホスファミド、メルファラン、L−アスパラギナーゼ、アセクラトン、シゾフィラン、ピシバニール、プロカルバジン、ピポブロマン、ネオカルチノスタチン、ヒドロキシウレア、ウベニメクスまたはクレスチン等が挙げられる。
本発明には、本発明化合物又はその塩を投与することを特徴とする癌の予防方法及び/または治療方法も含まれる。
さらに、本発明には、前記医薬を製造するための本発明の化合物、その塩又はそれらの溶媒和物の使用も含まれる。
以下に示す実施例によって本発明を具体的に説明するが、本発明はこれらに限定されるものではなく、これらはいかなる意味においても限定的に解釈されない。また、本明細書において、特に記載のない試薬、溶媒および出発材料は、市販の供給源から容易に入手可能である。
DMF:N,N−ジメチルホルムアミド
THF:テトラヒドロフラン
HATU:N,N,N’,N’−テトラメチル−0−(7−アザベンゾトリアゾール−1−イル)ウロニウム ヘキサフルオロホスフェート
HOAt:1−ヒドロキシ−7−アザベンゾトリアゾール
DIPEA:N,N−ジイソプロピルエチルアミン
COMU:(1−シアノ−2−エトキシ−2−オキソエチリデンアミノオキシ)ジメチルアミノ−モルホリノ−カルベニウム ヘキサフルオロホスフェート
TFA:トリフルオロ酢酸
EDC・HCl:1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩
HOBt:1−ヒドロキシベンゾトリアゾール
DMAP:4−ジメチルアミノピリジン
PLC:分取用薄層クロマトグラフィー
HPLC:高速液体クロマトグラフィー
(中間体1a)5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボン酸

(4−メチルフェニル)酢酸75.0g(499mmol)をテトラヒドロフラン1.00Lに溶解し、カルボニルジイミダゾール105g(649mmol)を加え室温で40分攪拌した。反応液にマロン酸モノエチルエステル カリウム塩111g(649mmol)、無水塩化マグネシウム57.1g(599mmol)を加え、60℃で6時間加熱攪拌した。反応液を室温まで冷却し、酢酸エチル2.00Lを加え、1規定塩酸1.00Lで洗浄した。有機層を飽和重曹水、飽和食塩水で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥したのち、溶媒を減圧下留去した。得られた油状物質を減圧下トルエン共沸させ、標記化合物86.8g(収率78.8%)を油状物質として得た。
1H−NMR(CDCl3)g(47.18−7.07(4H,m),4.17(2H,q,J=7.5Hz),3.78(2H,s),3.43(2H,s),2.33(3H,s),1.26(3H,t,J=7.5Hz).
MS(ESI)m/z:221[(M+H)+].
(工程2)5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボン酸エチル
4−(4−メチルフェニル)−3−オキソブタン酸エチル43.0g(195mmol)をトルエン390mlに溶解し、N,N−ジメチルホルムアミドジメチルアセタール130ml(976mmol)を加え、ディーンスタークにより生成したメタノールをトラップしながら、100℃で5時間加熱攪拌した。反応液を室温まで冷却し、減圧下濃縮した。残渣をエタノール390mlに溶解し、テトラヒドロ−2H−ピラン−4−イルメタナミン26.0ml(215mmol)を加え、60℃で9時間加熱攪拌した。反応液を室温まで冷却し、減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(昭光サイエンティフィック社、溶出溶媒;ジクロロメタン/酢酸エチル=75/25~20/80)を用いて精製し、標記化合物36.9g(収率53.2%)をカラメル状物質として得た。
1H−NMR(CDCl3)δ:8.10(1H,d,J=2.4Hz),7.48(2H,d,J=8.0Hz),7.32(1H,d,J=2.4Hz),7.20(2H,d,J=8.0Hz),4.38(2H,q,J=7.0Hz),4.02(2H,dd,J=11.5,4.0Hz),3.74(2H,d,J=7.3Hz),3.38(2H,td,J=11.5,2.0Hz),2.08−2.00(1H,m),1.63−1.56(2H,m),1.35−1.42(5H,m).
MS(APCI)m/z:356[(M+H)+].
(工程3)5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボン酸
5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボン酸エチル36.9g(104mmol)をメタノール519mlに溶解し、1規定水酸化ナトリウム水溶液311ml(311mmol)を加え、室温で1時間攪拌した。反応液を減圧下濃縮し、残渣に1規定塩酸400mlを加え、ジクロロメタンで抽出した。有機層を無水硫酸ナトリウムで乾燥したのち、溶媒を減圧下留去した。残渣をメタノールでスラリー洗浄し、標記化合物31.6g(収率92.9%)を固体として得た。
1H−NMR(CDCl3)δ:8.46(1H,d,J=2.4Hz),7.56(1H,d,J=2.4Hz),7.48(2H,d,J=8.5Hz),7.28(2H,d,J=8.5Hz),4.03(2H,dd,J=11.3,4.0Hz),3.89(2H,d,J=7.5Hz),3.38(2H,t,J=11.3Hz),2.40(3H,s),2.16−2.04(1H,m),1.62−1.53(3H,m),1.49−1.37(2H,m).
MS(APCI)m/z:328[(M+H)+].
同様にして対応する出発原料AおよびBから表1の中間体を合成した。

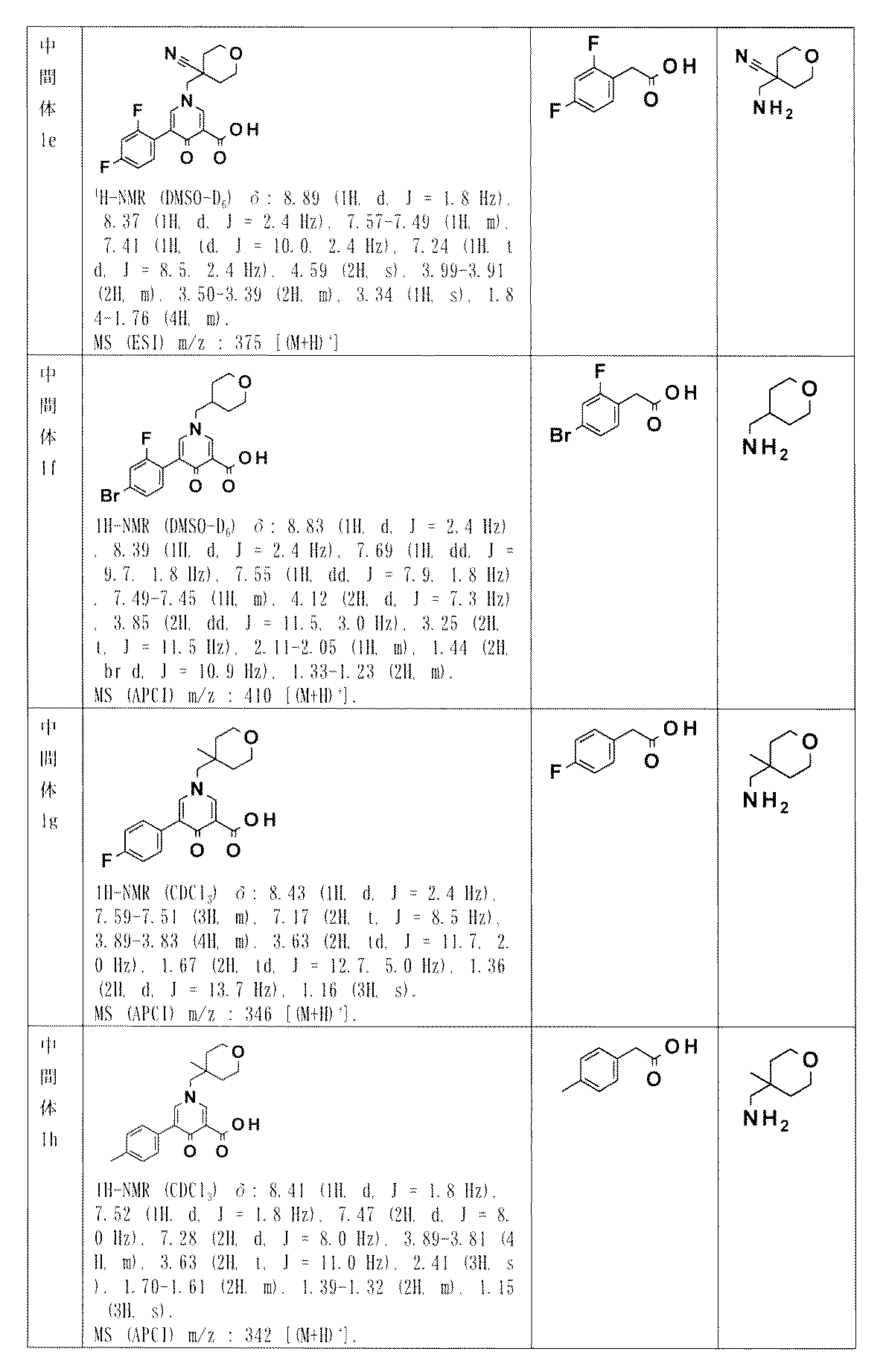


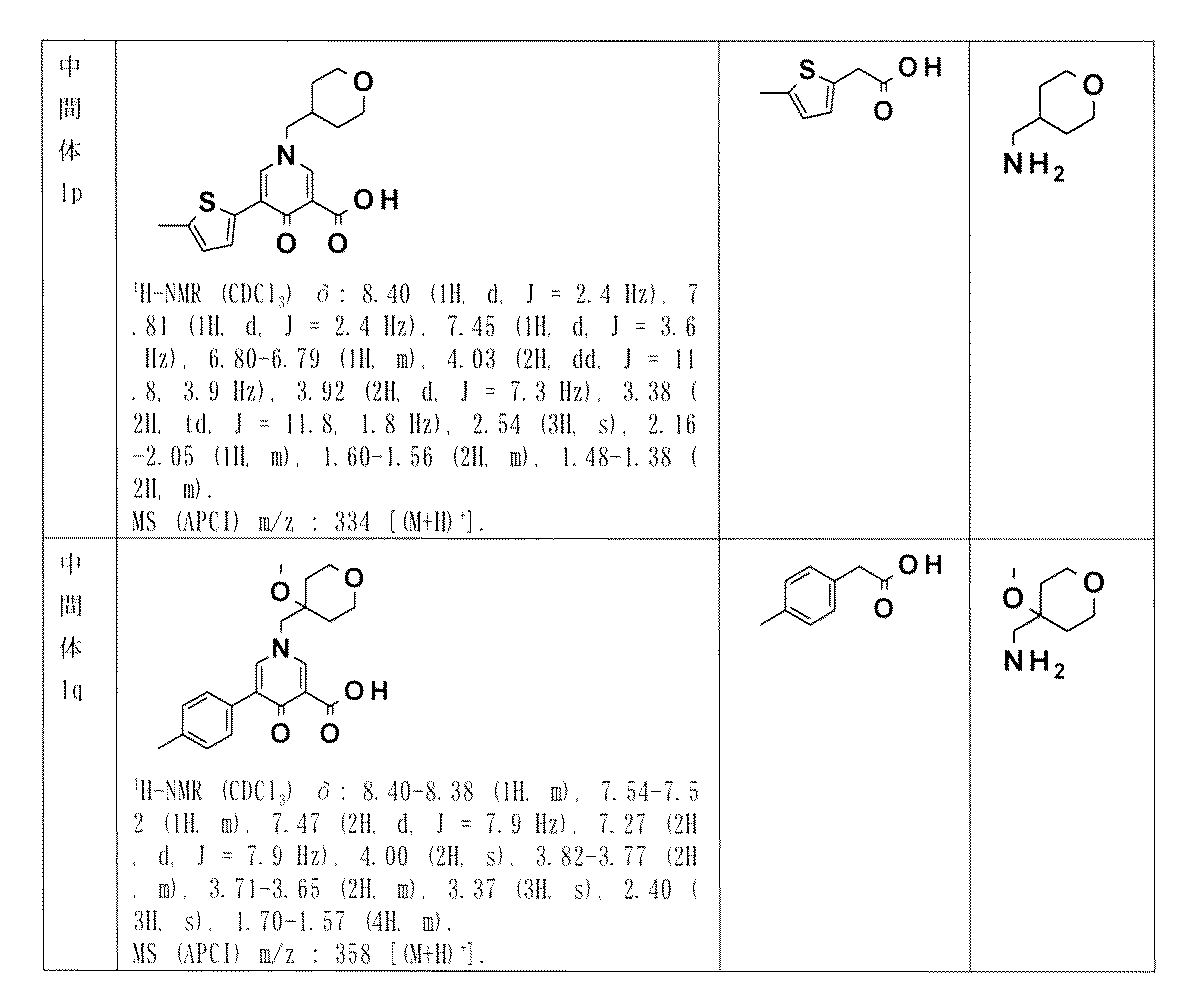

(4−ブロモ−2−フルオロフェニル)酢酸2.00g(8.58mmol)の塩化メチレン40.0mL溶液に、エタノール0.969mL(0.791g,17.2mmol)、4−ジメチルアミノピリジン0.105g(0.858mmol)、1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩(EDAC・HCl)1.97g(10.3mmol)を加え、室温で終夜撹拌した。反応液に1規定塩酸を加え、塩化メチレンで3回抽出し、有機層を飽和重曹水、飽和食塩水の順で洗浄後、無水硫酸ナトリウムで乾燥した。濾過、減圧濃縮後、得られた残渣をシリカゲルカラムクロマトグラフィー(山善社、n−ヘキサン/酢酸エチル=(100/0~87/13)で精製することで標記化合物1.96g(収率87.5%)を固体として得た。
1H−NMR(CDCl3)δ:7.27−7.24(2H,m),7.18−7.14(1H,m),4.17(2H,q,J=7.0Hz),3.62(2H,s),1.26(3H,t,J=7.0Hz).
(工程2)(4−シクロプロピル−2−フルオロフェニル)酢酸エチル
工程1で合成した(4−ブロモ−2−フルオロフェニル)酢酸エチル1.95g(7.47mmol)のトルエン30.0mL懸濁液に、シクロプロピルボロン酸0.962g(11.2mmol)、トリシクロヘキシルホスフィン0.419g(1.49mmol)、リン酸三カリウム5.55g(26.1mmol)、水6.00mLを加え、窒素置換後、酢酸パラジウム(II)0.168g(0.747mmol)を加え、100℃で2時間攪拌した。室温まで放冷後、反応液に水を加え、酢酸エチルで3回抽出し、有機層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥した。濾過、減圧濃縮後、得られた残渣をシリカゲルカラムクロマトグラフィー(山善社、n−ヘキサン/酢酸エチル=100/0~87/13)で精製し、さらにアミノシリカゲルカラムクロマトグラフィー(山善社、n−ヘキサン/酢酸エチル=100/0~87/13)で精製することで標記化合物1.59g(収率95.8%)を油状物として得た。
1H−NMR(CDCl3)δ:7.12(1H,t,J=7.9Hz),6.84−6.82(1H,m),6.76−6.72(1H,m),4.16(2H,q,J=7.0Hz),3.61(2H,s),1.90−1.83(1H,m),1.25(3H,t,J=7.0Hz),1.00−0.95(2H,m),0.70−0.66(2H,m).
MS(APCI)m/z:223[(M+H)+].
(工程3)(4−シクロプロピル−2−フルオロフェニル)酢酸
工程2で合成した(4−シクロプロピル−2−フルオロフェニル)酢酸エチル1.59g(7.15mmol)のテトラヒドロフラン30.0mL溶液に、メタノール15.0mLと1mol/L水酸化ナトリウム水溶液14.3mL(14.3mmol)を加え、室温で終夜撹拌した。有機溶媒を減圧留去後、残渣に水を加え、ジエチルエーテルで洗浄した。水層に1規定塩酸を加え酸性とした後、酢酸エチルで3回抽出し、有機層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥した。濾過、減圧濃縮することで標記化合物1.36g(7.00mmol)を固体として得た。
1H−NMR(CDCl3)δ:7.11(1H,t,J=7.9Hz),6.83(1H,dd,J=7.9,1.8Hz),6.75(1H,dd,J=11.2,1.8Hz),3.65(2H,s),1.90−1.83(1H,m),1.00−0.95(2H,m),0.70−0.66(2H,m).
(工程4)5−(4−シクロプロピル−2−フルオロフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボン酸
中間体1aの工程1の(4−メチルフェニル)酢酸の代わりに(4−シクロプロピル−2−フルオロフェニル)酢酸を用いて、以降、中間体1aの工程1から工程3までと同様に反応を行い、標記化合物を固体として得た。
1H−NMR(DMSO−D6)δ:8.80(1H,d,J=2.4Hz),8.31(1H,d,J=2.4Hz),7.34(1H,t,J=8.2Hz),7.03−6.99(2H,m),4.12(2H,d,J=7.3Hz),3.85(2H,dd,J=11.2,3.3Hz),3.24(2H,t,J=11.2Hz),2.12−1.97(2H,m),1.43(2H,br d,J=10.9Hz),1.33−1.22(2H,m),1.04−0.99(2H,m),0.77−0.73(2H,m).
MS(APCI)m/z:372[(M+H)+].
(中間体3) 5−メチル−4’−オキソ−1’−(テトラヒドロ−2H−ピラン−4−イルメチル)−1’,4’−ジヒドロ−2,3’−ビピリジン−5’−カルボン酸

(5−メチルピリジン−2−イル)酢酸0.500g(3.31mmol)のテトラヒドロフラン15.0mL溶液に、1,1’−カルボニルビス−1H−イミダゾール0.805g(4.96mmol)を加え、室温で2時間半攪拌した。別の反応容器にマロン酸モノエチルカリウム1.69g(9.92mmol)と塩化マグネシウム0.945g(9.92mmol)をテトラヒドロフラン25.0mLに懸濁し、氷冷下、トリエチルアミン2.38mL(1.74g,17.2mmol)を加え、室温で2時間半撹拌後、再び氷冷し、上記で調整した活性エステルの溶液を加え室温で終夜撹拌した。反応液に10%クエン酸水溶液を加え、酢酸エチルで5回抽出し、有機層を飽和重曹水、飽和食塩水の順で洗浄後、無水硫酸ナトリウムで乾燥した。濾過、減圧濃縮後、得られた残渣をシリカゲルカラムクロマトグラフィー(山善社、溶出溶媒;n−ヘキサン/酢酸エチル=57/43~36/64)で精製することで標記化合物0.634g(収率86.6%)を油状物として得た。
MS(APCI)m/z:222[(M+H)+].
(工程2、3)5−メチル−4’−オキソ−1’−(テトラヒドロ−2H−ピラン−4−イルメチル)−1’,4’−ジヒドロ−2,3’−ビピリジン−5’−カルボン酸
中間体1aの工程2の4−(4−メチルフェニル)−3−オキソブタン酸エチルの代わりに4−(5−メチルピリジン−2−イル)−3−オキソブタン酸エチルを用いて、中間体1aの工程2から工程3までと同様に反応を行い標記化合物を固体として得た。
1H−NMR(DMSO−D6)δ:8.86(1H,d,J=2.4Hz),8.80(1H,d,J=2.4Hz),8.53(1H,s),8.38(1H,d,J=8.5Hz),7.72(1H,dd,J=8.5,2.1Hz),4.24(2H,d,J=7.3Hz),3.84(2H,dd,J=11.2,3.3Hz),3.24(2H,t,J=11.2Hz),2.35(3H,s),2.11−2.01(1H,m),1.44(2H,br d,J=10.9Hz),1.35−1.24(2H,m).
MS(APCI)m/z:329[(M+H)+].
同様にして対応する出発原料から表2の中間体を合成した。


中間体1aの工程1の(4−メチルフェニル)酢酸の代わりに2−(4−ブロモフェニル)酢酸を用いて、以下同様の操作で反応を行い標記化合物を油状物として得た。
1H−NMR(CDCl3)δ:7.47(2H,d,J=8.5Hz),7.09(2H,d,J=8.5Hz),4.18(2H,q,J=7.1Hz),3.81(2H,s),3.46(2H,s),1.27(3H,t,J=7.1Hz).
(工程2)5−(4−ブロモフェニル)−4−オキソ−1,4−ジヒドロピリジン−3−カルボン酸エチル
工程1で合成した4−(4−ブロモフェニル)−3−オキソブタン酸エチル5.00g(17.5mmol)のエタノール50.0mL溶液に、1,3,5−トリアジン1.56g(19.3mmol)とナトリウムエトキシド1.43g(21.0mmol)を加え、85℃で4時間攪拌した。室温まで放冷後、減圧濃縮し、得られた残渣に1規定塩酸を加え酸性とした後、析出した固体を濾取、固体をアセトンで洗浄、乾燥することで標記化合物の租精製物3.73gを固体として得た。
MS(APCI)m/z:322[(M+H)+].
(工程3)5−(4−ブロモフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボン酸エチル
工程2で合成した5−(4−ブロモフェニル)−4−オキソ−1,4−ジヒドロピリジン−3−カルボン酸エチルの租精製物0.322gのN,N−ジメチルホルムアミド45.0mL懸濁液に炭酸セシウム0.977g(3.00mmol)を加え、80℃で15分撹拌後、メタンスルホン酸 テトラヒドロ−2H−ピラン−4−イルメチルエステル0.583g(3.00mmol)を加え、80℃で4時間攪拌した。室温まで放冷後、反応液に水を加え、酢酸エチルで3回抽出し、有機層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥した。濾過、減圧濃縮後、得られた残渣をシリカゲルカラムクロマトグラフィー(山善社、溶出溶媒;n−ヘキサン/酢酸エチル=57/43~36/64)で精製することで標記化合物0.234g(2工程収率36.7%)を非晶性固体として得た。
1H−NMR(DMSO−D6)δ:8.30(1H,d,J=2.4Hz),8.02(1H,d,J=2.4Hz),7.59(4H,s),4.20(2H,q,J=7.0Hz),3.92(2H,d,J=7.3Hz),3.85(2H,dd,J=10.9,3.0Hz),3.25(2H,t,J=10.9Hz),2.09−1.99(1H,m),1.43(2H,br d,J=10.9Hz),1.32−1.24(5H,m).
MS(APCI)m/z:420[(M+H)+].
(工程4)5−(4−ブロモフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボン酸
中間体1aの工程3の5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボン酸エチルの代わりに5−(4−ブロモフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボン酸エチルを用いて、以下同様の操作で反応を行い標記化合物を固体として得た。
1H−NMR(DMSO−D6)δ:8.79(1H,d,J=1.8Hz),8.45(1H,d,J=1.8Hz),7.70−7.65(4H,m),4.14(2H,d,J=7.3Hz),3.85(2H,dd,J=11.2,3.3Hz),3.24(2H,t,J=11.2Hz),2.16−2.06(1H,m),1.43(2H,br d,J=10.9Hz),1.34−1.24(2H,m).
MS(APCI)m/z:392[(M+H)+].
同様にして対応する出発原料から表3の中間体を合成した。

(2S)−2−{[2−メトキシ−4−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)フェノキシ]メチル}−1,4−ジオキサン

1H−NMR(CDCl3)δ:7.39(1H,d,J=7.9Hz),7.28(1H,s),6.89(1H,d,J=7.9Hz),4.09−4.03(2H,m),4.00−3.93(2H,m),3.89(3H,s),3.86−3.63(4H,m),3.57−3.50(1H,m),1.34(12H,s).
同様にして対応する出発原料から表4の中間体を合成した。


1H−NMR(CDCl3)δ:7.79(1H,s),7.65(1H,s),4.00(2H,d,J=7.5Hz),3.96(2H,dd,J=11.5,4.5Hz),3.35(2H,td,J=11.5,2.0Hz),2.23−2.11(1H,m),1.48(2H,d,J=11.5Hz),1.40−1.28(14H,m).
MS(APCI)m/z:293[(M+H)+].
(中間体7)(2R)−2−[({2−[(2H3)メチルオキシ]−4−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)(2H3)フェニル}オキシ)メチル}−1,4−ジオキサン

4−ブロモ−2−[(2H3)メチルオキシ](2H3)フェノール
(2H4)ベンゼン−1,2−ジオール12.0g(105mmol)をジクロロメタン105mlに懸濁させ、3,4−ジヒドロ−2H−ピラン9.53ml(105mmol)、p−トルエンスルホン酸ピリジン塩396mg(1.58mmol)を加え、室温で3時間攪拌した。反応液に飽和重曹水を加え、10分攪拌した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムにより乾燥し、減圧下溶媒を留去した。得られた油状物質をアセトン207mlに溶解し、炭酸カリウム45.8g(330mmol)、トリデュウテリオ沃化メチル14.4ml(228mmol)を加え、窒素気流下40℃で24時間加熱攪拌した。反応液を室温まで冷却したのち、キリヤマろ過し、ろ液を減圧下濃縮した。残渣にヘキサンを加え、再度キリヤマろ過したのち、ろ液を減圧下濃縮した。得られた油状物質をエタノール181mlに溶解し、p−トルエンスルホン酸ピリジン塩341mg(1.36mmol)を加え、65℃で3時間加熱攪拌した。室温まで冷却したのち、減圧下濃縮した。残渣をN,N−ジメチルホルムアミド90.0mlに溶解し、N−ブロモスクシンイミド16.1g(90.6mmol)を、氷冷下で少量ずつ加え、氷冷下で1時間攪拌した。反応液にジエチルエーテル500mlを加えたのち、8.85mol/Lチオ硫酸ナトリウム水溶液50mlを加え、反応を停止させた。有機層を無水硫酸マグネシウムで乾燥したのち減圧下濃縮した。残渣をアミノシリカゲルカラムクロマトグラフィー(昭光サイエンティフィック社、溶出溶媒;ヘキサン/酢酸エチル=90/10~55/45)を用いて精製し、標記化合物6.97g(収率28.3%)を油状物質として得た。
1H−NMR(CDCl3)δ:5.57(1H,s).
MS(CI)m/z:207[(M−H)+].
(工程2)(2R)−2−[({4−ブロモ−2−[(2H3)メチルオキシ](2H3)フェニル}オキシ)メチル]−1,4−ジオキサン
4−ブロモ−2−[(2H3)メチルオキシ](2H3)フェノール2.00g(9.57mmol)をN,N−ジメチルホルムアミド48.0mlに溶解し、炭酸カリウム2.64g(19.1mmol)、(2R)−1,4−ジオキサン−2−イルメチル メタンスルホン酸1.88g(9.57mmol)を加え90℃で4時間加熱攪拌した。反応液を室温まで冷却し、酢酸エチルを加え不溶物をろ去した。ろ液を減圧下濃縮し、残渣をシリカゲルカラムクロマトグラフィー(昭光サイエンティフィック社、溶出溶媒;ヘキサン/酢酸エチル=100/0~70/30)を用いて精製し、標記化合物1.75g(収率59.2%)を固体として得た。
1H−NMR(CDCl3)δ:4.06−3.48(9H,m).
MS(APCI)m/z:309[(M+H)+].
(工程3)(2R)−2−[({2−[(2H3)メチルオキシ]−4−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)(2H3)フェニル}オキシ)メチル}−1,4−ジオキサン
(2R)−2−[({4−ブロモ−2−[(2H3)メチルオキシ](2H3)フェニル}オキシ)メチル]−1,4−ジオキサン800mg(2.59mmol)をトルエン5.20mlに懸濁し、1,3−ビス(ジフェニルフォスフィノ)プロパン]塩化ニッケル(II)140mg(259μmol)、1,3−ビス(ジフェニルフォスフィノ)プロパン107mg(0.259mmol)、ピナコールホウ素0.744ml(5.17mmol)、トリエチルアミン1.10ml(7.76mmol)を加え、窒素気流下100℃で16時間加熱攪拌した。反応液を室温まで冷却し、酢酸エチルで希釈したのち不溶物をセライトによりろ去した。ろ液を減圧下濃縮し、残渣をシリカゲルカラムクロマトグラフィー(昭光サイエンティフィック社、溶出溶媒;ヘキサン/酢酸エチル=100/0~85/15)を用いて精製し、標記化合物840mg(91.1%)を固体として得た。
1H−NMR(CDCl3)δ:4.15−4.02(2H,m),4.00−3.93(2H,m),3.87−3.62(4H,m),3.57−3.49(1H,m),1.34(12H,s).
MS(ESI)m/z:357[(M+H)+].
(中間体8)
3−(4−アミノフェニル)−5−{4−[(2S)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−2−アミン

1H−NMR(CDCl3)δ:8.24−8.22(1H,m),7.53−7.51(1H,m),7.32−7.27(2H,m),7.07−6.99(2H,m),6.98−6.93(1H,m),6.81−6.76(2H,m),4.62−4.58(2H,m),4.14−4.01(3H,m),4.01−3.93(3H,m),3.92−3.63(7H,m),3.59−3.50(1H,m).
MS(ESI)m/z:408[(M+H)+]
(中間体9a)3−ブロモ−5−{4−[(2S)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−2−アミン

1H−NMR(DMSO−D6)δ:8.28(1H,d,J=1.8Hz),8.05(1H,d,J=2.4Hz),7.17(1H,d,J=2.4Hz),7.10(1H,dd,J=8.5,2.4Hz),6.99(1H,d,J=8.5Hz),6.28(2H,s),3.98−3.74(8H,m),3.69−3.60(2H,m),3.52−3.46(1H,m),3.42−3.36(1H,m).
MS(APCI)m/z:395[(M+H)+].
同様にして対応する出発原料から表5の中間体を合成した。

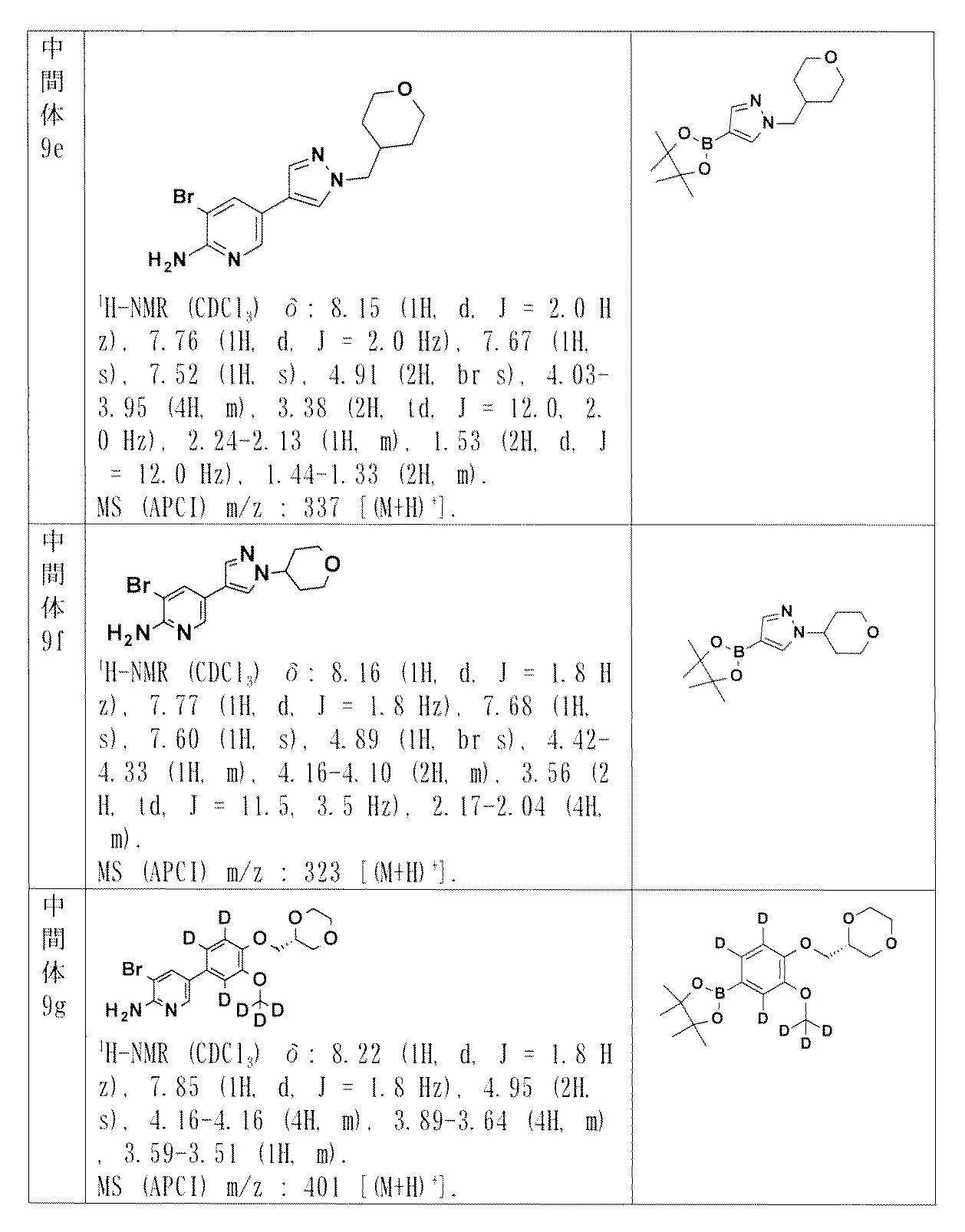
3−(4−アミノ−2−フルオロフェニル)−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−2−アミン

1H−NMR(CDCl3)δ:8.27(1H,d,J=2.4Hz),7.54(1H,d,J=2.4Hz),7.17(1H,t,J=8.0Hz),7.06−7.01(2H,m),6.95(1H,d,J=8.0Hz),6.55(1H,dd,J=8.0,2.4Hz),6.51(1H,dd,J=11.5,2.4Hz),4.52(2H,br s),4.13−3.52(12H,m).
MS(APCI)m/z:426[(M+H)+].
同様にして対応する出発原料AおよびBから表6の中間体を合成した。


1H−NMR(DMSO−D6)δ:8.20(1H,d,J=2.4Hz),7.52(1H,d,J=2.4Hz),7.18−7.11(3H,m),7.06(1H,dd,J=7.9,2.4Hz),6.98(1H,d,J=8.5Hz),6.87−6.83(1H,m),5.61(2H,s),5.30(2H,s),3.82(3H,s),3.77(3H,s).
MS(APCI)m/z:340[(M+1)+].
同様にして対応する出発原料から表7の中間体を合成した。


1H−NMR(DMSO−D6)δ:7.83(1H,d,J=5.5Hz),7.65−7.53(1H,m),6.83−6.72(3H,m),6.39(1H,d,J=6.1Hz),5.18(2H,s),5.10(2H,s),3.65(3H,s).
MS(APCI)m/z:234[(M+H)+].
同様にして対応する出発原料AおよびBから表8の中間体を合成した。

特許(WO2011/110545 A1)記載の方法に従い合成した3−クロロ−5−ヨードピラジン−2−アミン0.510g(2.00mmol)、(3,4−ジメトキシフェニル)ボロン酸0.363g(2.00mmol)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリド ジクロロメタン錯体(1:1)0.163g(0.200mmol)、炭酸セシウム1.30g(4.00mmol)に、1,4−ジオキサン7.50mLと水2.50mLを加え、80℃で4時間攪拌した。室温まで放冷後、反応液に水を加え、塩化メチレンで3回抽出し、有機層を無水硫酸ナトリウムで乾燥した。濾過、減圧濃縮後、得られた残渣をシリカゲルカラムクロマトグラフィー(山善社、クロロホルム/酢酸エチル=100/0~90/10)で精製し、さらに、アミノシリカゲルカラムクロマトグラフィー(山善社、n−ヘキサン/酢酸エチル=25/75~0/100)で精製することで標記化合物0.412g(収率77.7%)を固体として得た。
1H−NMR(DMSO−D6)δ:8.57(1H,s),7.47−7.45(2H,m),7.01(1H,d,J=7.9Hz),6.85(2H,s),3.83(3H,s),3.79(3H,s).
MS(APCI)m/z:266[(M+H)+].
(工程2)3−(4−アミノフェニル)−5−(3,4−ジメトキシフェニル)ピラジン−2−アミン
工程1で合成した3−クロロ−5−(3,4−ジメトキシフェニル)ピラジン−2−アミン0.0800g(0.301mmol)、4−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)アニリン0.0990g(0.452mmol)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリド ジクロロメタン錯体(1:1)0.0246g(0.030mmol)、炭酸セシウム0.294g(0.903mmol)に、1,4−ジオキサン2.00mLと水0.200mLを加え、100℃で4時間攪拌した。室温まで放冷後、反応液に水を加え、塩化メチレンで3回抽出し、有機層を無水硫酸ナトリウムで乾燥した。濾過、減圧濃縮後、得られた残渣をシリカゲルカラムクロマトグラフィー(山善社、酢酸エチルのみ)で精製することで標記化合物0.0940g(収率96.8%)を非晶性固体として得た。
1H−NMR(DMSO−D6)δ:8.39(1H,s),7.54−7.50(4H,m),7.01(1H,d,J=8.5Hz),6.67(2H,d,J=8.5Hz),6.00(2H,s),5.44(2H,s),3.83(3H,s),3.79(3H,s).
MS(APCI)m/z:323[(M+H)+].
(中間体14a)N−(6−クロロピリダジン−3−イル)−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミド

1H−NMR(CDCl3)δ:13.68(1H,s),8.59(1H,d,J=9.0Hz),8.48(1H,d,J=2.0Hz),7.51−7.46(4H,m),7.26(2H,d,J=8.0Hz),4.03(2H,dd,J=11.5,4.0Hz),3.87(2H,d,J=7.5Hz),3.39(2H,t,J=11.5Hz),2.40(3H,s),2.16−2.06(1H,m),1.64−1.55(2H,m),1.50−1.38(2H,m).
MS(APCI)m/z:439[(M+H)+].
同様にして対応する出発原料AおよびBから表9の中間体を合成した。
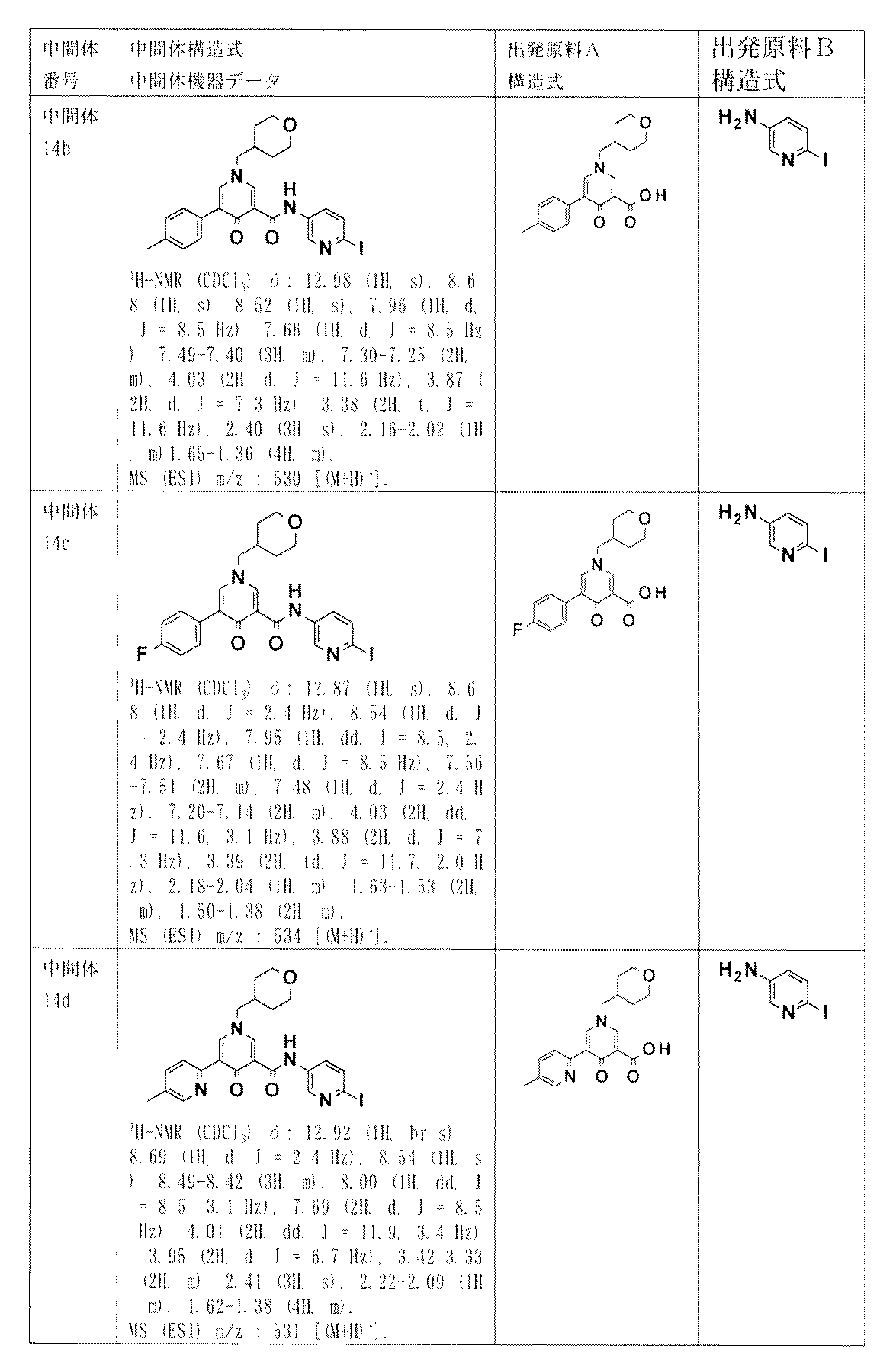


1H−NMR(CDCl3)δ:13.19(1H,s),8.53(1H,d,J=2.4Hz),8.49(1H,d,J=2.4Hz),8.15(1H,d,J=8.5Hz),7.95(1H,dd,J=8.5,2.4Hz),7.51(2H,d,J=7.9Hz),7.46(1H,d,J=2.4Hz),7.28−7.23(2H,m),4.03(2H,dd,J=11.0,3.7Hz),3.85(2H,d,J=7.3Hz),3.42−3.34(2H,m),2.40(3H,s),2.15−2.03(1H,m),1.63−1.51(2H,m),1.49−1.37(2H,m).
MS(ESI/APCI)m/z:530[(M+H)+].
同様にして対応する出発原料から表10の中間体を合成した。


1H−NMR(DMSO−D6)δ:8.36(1H,d,J=3.0Hz),7.86(1H,d,J=3.0Hz),7.09−6.97(3H,m),6.18(2H,s),3.83(3H,s),3.77(3H,s),1.32(12H,s).
同様にして対応する出発原料から表11の中間体を合成した。

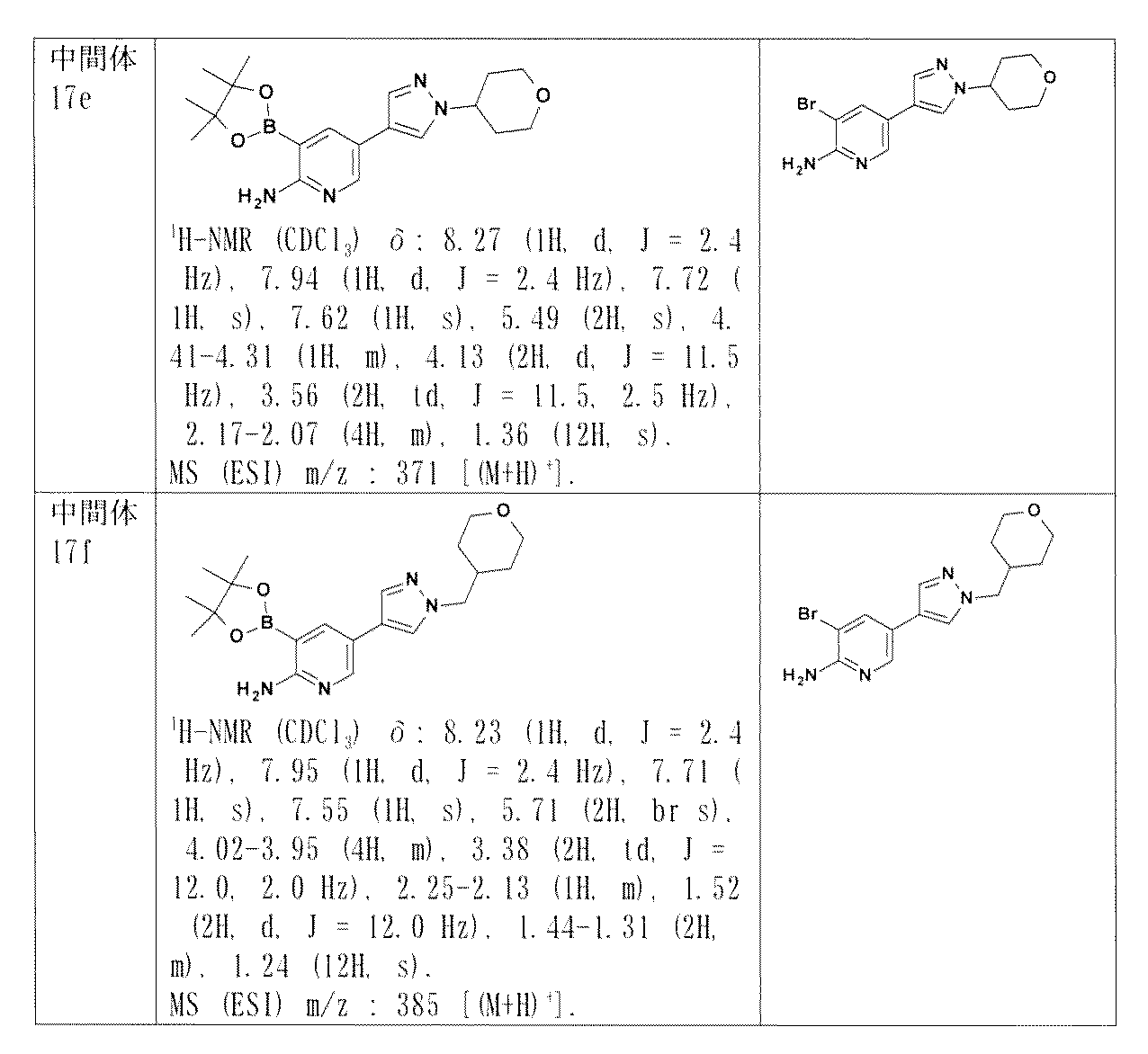
1H−NMR(CDCl3)δ:8.27(1H,d,J=2.4Hz),7.98−7.95(2H,m),7.10−7.04(2H,m),6.94(1H,d,J=7.9Hz),6.85(1H,dd,J=12.1,2.4Hz),5.99(2H,s),3.99−3.92(8H,m).
MS(APCI)m/z:341[(M+H)+].
(中間体19)5−ブロモ−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボン酸

文献(J.Med.Chem.2008,51,5330−5341)記載の方法に従い合成した5−ブロモ−4−ヒドロキシピリジン−3−カルボン酸メチル2.36g(10.2mmol)に炭酸セシウム9.94g(30.5mmol)とN,N−ジメチルホルムアミド30.0mLを加え、80℃で15分撹拌後、4−(ブロモメチル)テトラヒドロ−2H−ピラン5.46g(30.5mmol)を加え、80℃で11時間攪拌後、室温で終夜放置した。反応液に水と飽和食塩水を加え、酢酸エチルで3回抽出後、塩化メチレンで5回抽出し、有機層を合わせて無水硫酸ナトリウムで乾燥した。濾過、減圧濃縮後、得られた残渣をアミノシリカゲルカラムクロマトグラフィー(山善社、溶出溶媒;酢酸エチル/メタノール=100/0~85/15)で精製することで標記化合物2.83gの租精製物を油状物として得た。
MS(APCI)m/z:330[(M+H)+].
(工程2)5−ブロモ−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボン酸
工程1で合成した5−ブロモ−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボン酸メチルの租精製物2.83gのテトラヒドロフラン50.0mL溶液に、メタノール25.0mLと1規定水酸化ナトリウム水溶液25.7mL(25.7mmol)を加え、室温で終夜撹拌した。有機溶媒を減圧留去後、残渣に水を加え、酢酸エチルで2回洗浄した。水層に1規定塩酸を加え酸性とした後、酢酸エチルを加えた。析出した固体を濾取後、濾液を酢酸エチルで3回抽出し、有機層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥した。濾過、減圧濃縮後、得られた固体をジエチルエーテルに懸濁、濾取し、先に得られた固体と合わせて乾燥することで標記化合物1.93g(2工程収率60.0%)を固体として得た。
1H−NMR(DMSO−D6)δ:8.79(1H,d,J=1.8Hz),8.75(1H,d,J=1.8Hz),4.08(2H,d,J=7.9Hz),3.84(2H,dd,J=11.6,3.4Hz),3.23(2H,t,J=11.6Hz),2.10−2.00(1H,m),1.40(2H,br d,J=10.4Hz),1.30−1.20(2H,m).
MS(APCI)m/z:316[(M+H)+].
(中間体20)N−{4−[2−アミノ−5−(3,4−ジメトキシフェニル)ピリジン−3−イル]フェニル}−5−ブロモ−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミド

1H−NMR(DMSO−D6)δ:12.61(1H,s),8.74(1H,d,J=2.4Hz),8.63(1H,d,J=2.4Hz),8.26(1H,d,J=2.4Hz),7.82(2H,d,J=8.5Hz),7.64(1H,d,J=2.4Hz),7.55(2H,d,J=8.5Hz),7.20−7.14(2H,m),6.99(1H,d,J=8.5Hz),5.77(2H,s),4.07(2H,d,J=7.3Hz),3.87−3.83(5H,m),3.77(3H,s),3.26(2H,t,J=10.7Hz),2.11−2.02(1H,m),1.42(2H,br d,J=10.9Hz),1.33−1.23(2H,m).
MS(APCI)m/z:619[(M+H)+].
同様にして対応する出発原料から表12の中間体を合成した。


1H−NMR(DMSO−D6)δ:12.95(1H,s),8.89−8.80(3H,m),8.59(1H,d,J=8.5Hz),8.31(1H,d,J=2.4Hz),8.18(1H,dd,J=8.5,2.4Hz),7.96−7.92(1H,m),7.63(1H,d,J=2.4Hz),7.51−7.42(2H,m),7.19(1H,d,J=2.4Hz),7.11(1H,dd,J=8.5,2.4Hz),6.99(1H,d,J=8.5Hz),5.71(2H,s),4.71(2H,s),3.97−3.75(10H,m),3.69−3.59(2H,m),3.53−3.30(4H,m),1.88−1.78(4H,m).
MS(APCI)m/z:825[(M+H)+].
(実施例1)
N−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミド

1H−NMR(CDCl3)δ:12.85(1H,s),8.57(1H,d,J=2.4Hz),8.25(1H,d,J=1.8Hz),7.87(2H,d,J=8.5Hz),7.57(1H,d,J=1.8Hz),7.47(5H,dd,J=8.2,5.8Hz),7.29(2H,d,J=8.5Hz),7.08−7.02(2H,m),6.98−6.94(1H,m),4.71(2H,s),4.11−3.94(6H,m),3.93−3.79(8H,m),3.79−3.63(1H,m),3.59−3.51(1H,m),3.44−3.35(2H,m),2.41(3H,s),2.18−2.04(1H,m),1.64−1.58(2H,m),1.51−1.35(2H,m).
MS(ESI)m/z:717[(M+H)+]
同様にして対応する出発原料AおよびBから表13の最終体を合成した。




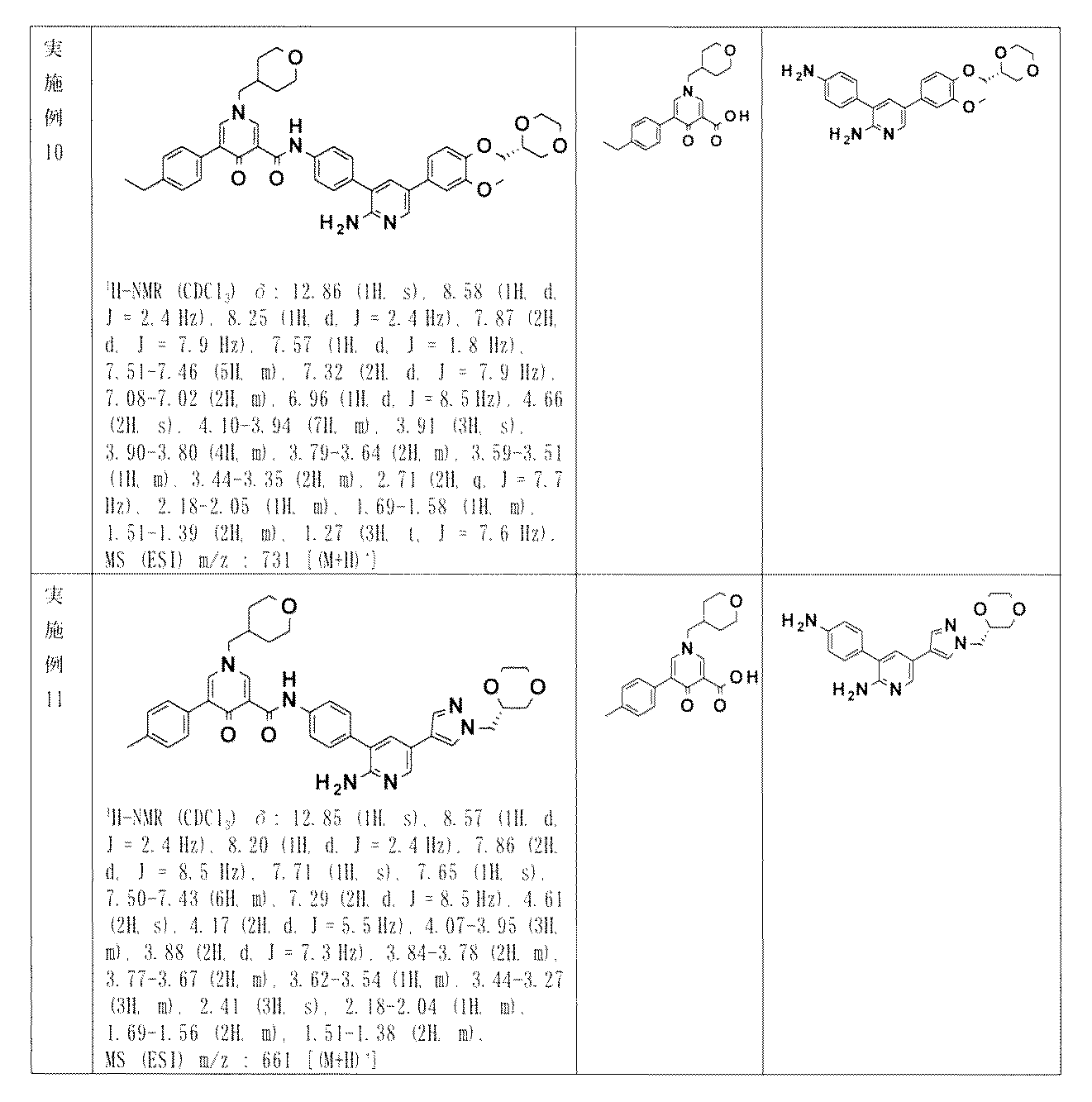



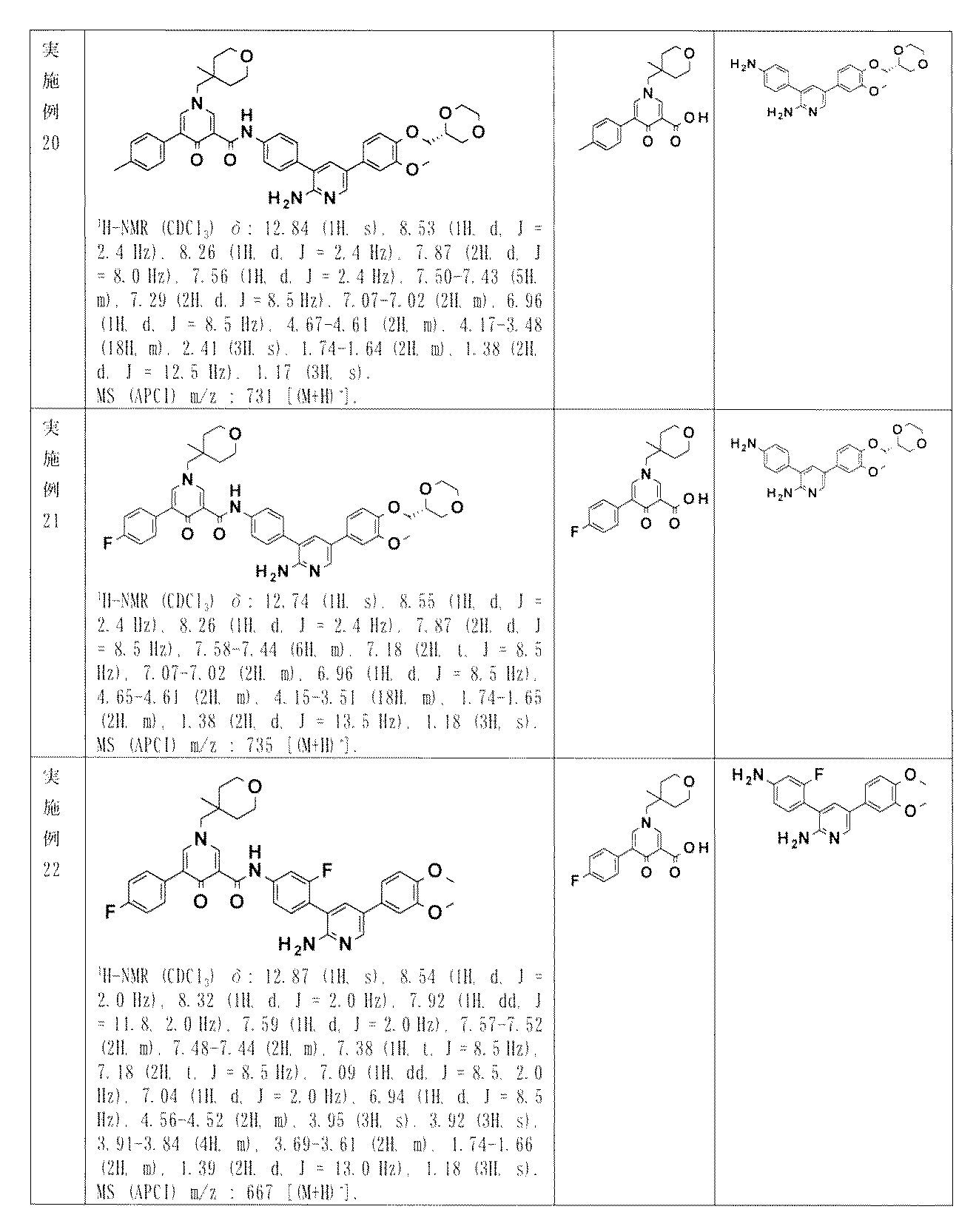


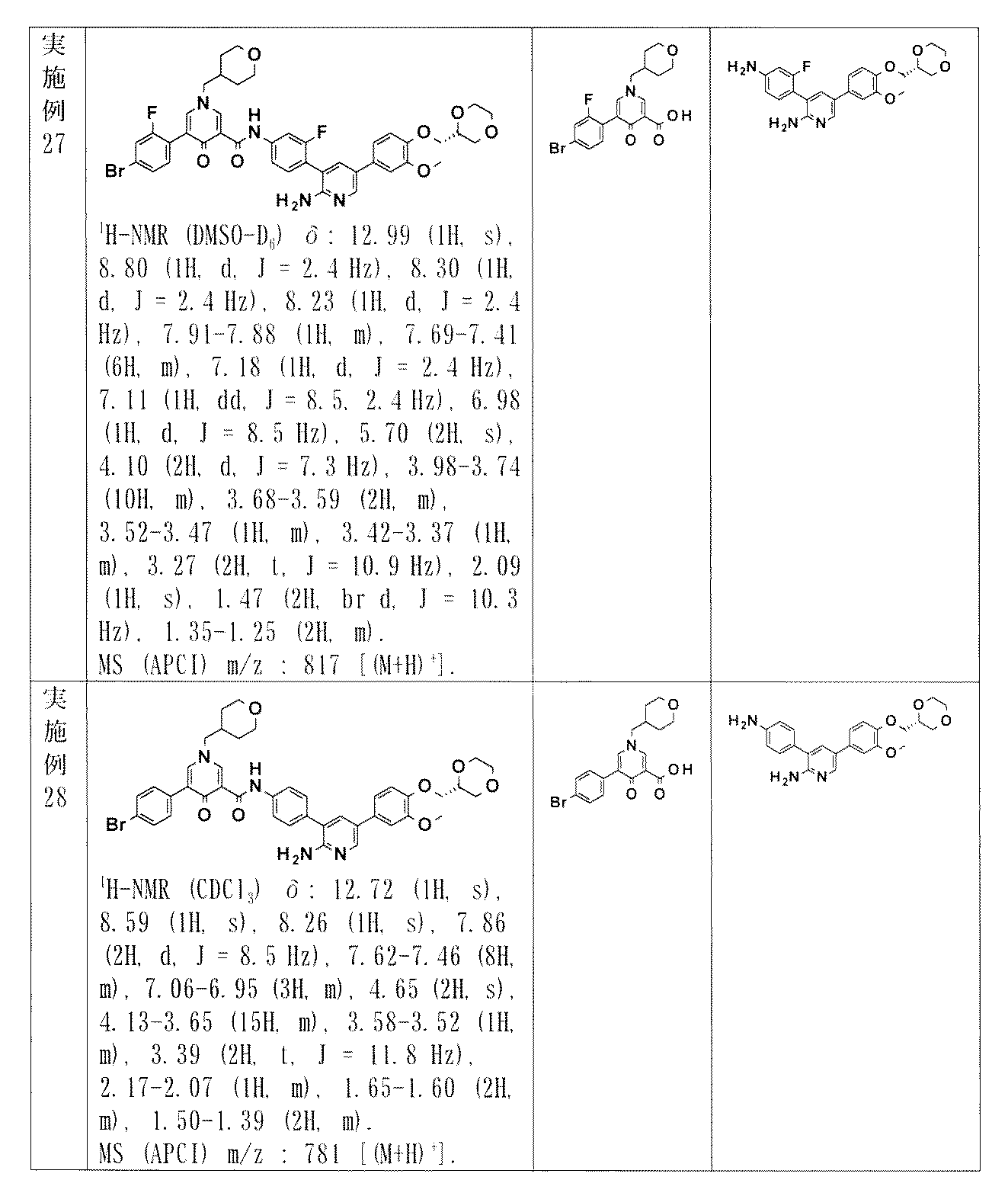




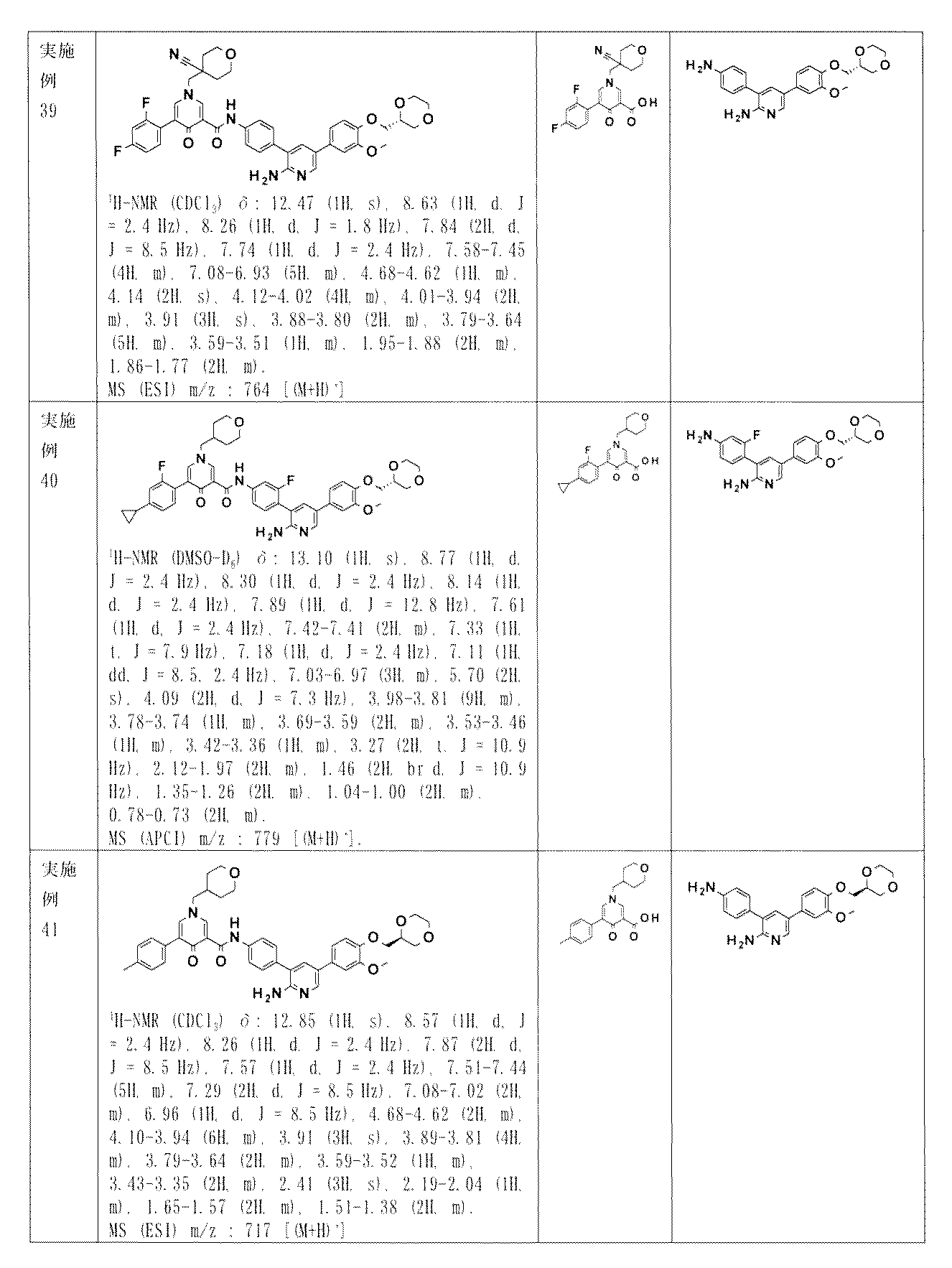









N−{4−[2−アミノ−5−(3,4−ジメトキシフェニル)ピリジン−3−イル]−3−フルオロフェニル}−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミド メタンスルホン酸塩
1H−NMR(CDCl3)δ:12.98(1H,s),8.56(1H,d,J=2.4Hz),8.31(1H,d,J=2.4Hz),7.92(1H,dd,J=12.1,1.8Hz),7.59(1H,d,J=2.4Hz),7.49−7.43(4H,m),7.36(1H,t,J=8.2Hz),7.29(2H,d,J=7.9Hz),7.08(1H,dd,J=8.2,2.1Hz),7.03(1H,d,J=1.8Hz),6.93(1H,d,J=7.9Hz),4.57−4.52(2H,m),4.07−4.00(2H,m),3.94(3H,s),3.92(3H,s),3.88(2H,d,J=7.3Hz),3.44−3.35(2H,m),2.41(3H,s),2.19−2.04(1H,m),1.65−1.57(2H,m),1.51−1.38(2H,m).
MS(ESI)m/z:649[(M+H)+]
同様にして対応する出発原料から表14の最終体を合成した。


出発原料として3−(4−アミノフェニル)−5−ブロモピリジン−2−アミンを用いて、実施例1と同様にして標記化合物を得た。
1H−NMR(CDCl3)δ:12.88−12.85(1H,m),8.56(1H,d,J=2.4Hz),8.08(1H,d,J=2.4Hz),7.87−7.84(2H,m),7.49−7.45(4H,m),7.43−7.39(2H,m),7.31−7.27(2H,m),4.63−4.58(2H,m),4.06−4.00(2H,m),3.89−3.86(2H,m),3.43−3.35(2H,m),2.41(3H,s),2.17−2.06(1H,m),1.65−1.38(4H,m).
MS(ES+APCI)m/z:573[(M+H)+].
(工程2)N−[4−(2−アミノ−5−{4−[(4−メチルピペラジン−1−イル)メチル]フェニル}ピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミド
出発原料としてN−[4−(2−アミノ−5−ブロモピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミドと1−メチル−4−[[4−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)フェニル]メチル]ピペラジンを用いて、溶媒として1,4−ジオキサンおよび水を用いて、触媒としてテトラキス(トリフェニルホスフィン)パラジウム(0)を用いて、塩基として炭酸カリウムを用いて、反応温度100℃で2時間加熱攪拌し中間体8と同様にして標記化合物を得た。
1H−NMR(CDCl3)δ:12.87−12.85(1H,m),8.57(1H,d,J=2.4Hz),8.31(1H,d,J=2.4Hz),7.87(2H,d,J=7.9Hz),7.61(1H,d,J=2.4Hz),7.52−7.45(7H,m),7.37(2H,d,J=7.9Hz),7.29(2H,d,J=7.9Hz),4.69−4.64(2H,m),4.06−4.00(2H,m),3.87(2H,d,J=7.3Hz),3.54(2H,s),3.43−3.35(2H,m),2.63−2.34(8H,m),2.41(3H,s),2.29(3H,s),2.15−2.07(1H,m),1.64−1.57(2H,m),1.50−1.38(2H,m).
MS(ES+APCI)m/z:683[(M+H)+].
(工程3)N−[4−(2−アミノ−5−{4−[(4−メチルピペラジン−1−イル)メチル]フェニル}ピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミド メタンスルホン酸塩
N−[4−(2−アミノ−5−{4−[(4−メチルピペラジン−1−イル)メチル]フェニル}ピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミド34.0mg(49.8μmol)をクロロホルム3.00mLに溶解し、室温で1mol/Lメタンスルホン酸/エタノール溶液49.8μL(49.8μmol)を加えた後溶媒を減圧下留去した。得られた残渣に、ジイソプロピルエーテルを加えて析出した固体を濾取して標記化合物25.7mg(収率66.3%)を得た。
1H−NMR(CDCl3)δ:12.92−12.90(1H,m),8.57(1H,d,J=2.4Hz),8.20(1H,d,J=2.4Hz),7.91−7.87(2H,m),7.69(1H,d,J=2.4Hz),7.52−7.44(7H,m),7.39−7.35(2H,m),7.31−7.27(2H,m),4.07−4.00(2H,m),3.88(2H,d,J=7.3Hz),3.69−3.63(2H,m),3.44−3.35(2H,m),3.08−2.61(8H,m),2.86(3H,s),2.41(3H,s),2.18(3H,s),2.16−2.06(1H,m),1.65−1.58(2H,m),1.51−1.39(2H,m).
MS(ES+APCI)m/z:683[(M+H)+].
同様にして対応する出発原料から表15の最終体を合成した。





実施例71の工程1で得たN−[4−(2−アミノ−5−ブロモピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミド及び3−フルオロ−4−ホルミルフェニルホウ酸を用いて、溶媒として1,4−ジオキサン及び水(比率10:1)を用いて、触媒としてテトラキス(トリフェニルホスフィン)パラジウム(0)を用いて、塩基として炭酸カリウムを用いて、反応温度100℃で7時間加熱攪拌し、中間体8と同様にして標記化合物を得た。
1H−NMR(CDCl3)δ:12.89(1H,s),10.37−10.35(1H,m),8.58−8.57(1H,m),8.37(1H,d,J=2.4Hz),7.94−7.87(3H,m),7.64(1H,d,J=2.4Hz),7.49−7.45(6H,m),7.36(1H,dd,J=12.1,1.8Hz),7.31−7.28(2H,m),4.86−4.81(2H,m),4.07−4.01(2H,m),3.88(2H,d,J=7.3Hz),3.43−3.35(2H,m),2.41(3H,s),2.17−2.07(1H,m),1.64−1.53(2H,m),1.51−1.39(2H,m).
MS(ES+APCI)m/z:617[(M+H)+].
(工程2)N−(4−{2−アミノ−5−[3−フルオロ−4−(モルホリン−4−イルメチル)フェニル]ピリジン−3−イル}フェニル)−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミド メタンスルホン酸塩
N−{4−[2−アミノ−5−(3−フルオロ−4−ホルミルフェニル)ピリジン−3−イル]フェニル}−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミド70.0mg(0.114mmol)をジクロロエタン2.27mlに懸濁し、モルホリン19.8μl(0.227mmol)を加え室温で5分攪拌した。反応液にトリアセトキシ水素化ホウ素ナトリウム48.1mg(0.227mmol)を加え室温で3時間攪拌した。反応液に水及び炭酸水素ナトリウムを加えクロロホルムで抽出し、無水硫酸ナトリウムで乾燥した。溶媒を減圧下留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(バイオタージ社、ジクロロメタン/エタノール=100/1~50/1)で精製し固体を得た。このものをクロロホルム8.00mlに溶解し、1Mメタンスルホン酸/エタノール溶液91.2μl(91.2μmol)を加え減圧下濃縮した。残渣にジエチルエーテルを加え析出した固体をろ取し、標記化合物30.5mg(34.7%)を固体として得た。
1H−NMR(DMSO−D6)δ:13.21−13.18(1H,m),10.01−9.83(1H,m),8.72(1H,d,J=2.4Hz),8.42(1H,d,J=2.4Hz),8.19(1H,d,J=2.4Hz),8.10−8.03(1H,m),7.91−7.81(3H,m),7.78−7.73(1H,m),7.69−7.63(1H,m),7.61−7.55(4H,m),7.27(2H,d,J=7.9Hz),4.49−4.36(2H,m),4.12(2H,d,J=7.3Hz),4.03−3.92(2H,m),3.90−3.84(2H,m),3.72−3.59(2H,m),3.47−3.10(8H,m),2.36(3H,s),2.32(3H,s),2.17−2.05(1H,m),1.50−1.42(2H,m),1.38−1.25(2H,m).
MS(ES+APCI)m/z:688[(M+H)+].
同様にして対応する出発原料から表16の最終体を合成した。


出発原料として特許(WO2013/115280 A1)記載の方法で合成した3−(4−アミノ−2−フルオロフェニル)−5−ブロモ−ピリジン−2−アミンを用いて、実施例1と同様にして標記化合物を得た。
1H−NMR(CDCl3)δ:13.01−12.98(1H,m),8.55(1H,d,J=2.4Hz),8.13(1H,d,J=2.4Hz),7.91(1H,dd,J=12.1,2.4Hz),7.51−7.41(5H,m),7.32−7.25(3H,m),4.54−4.50(2H,m),4.07−4.01(2H,m),3.88(2H,d,J=7.3Hz),3.43−3.35(2H,m),2.41(3H,s),2.16−2.05(1H,m),1.64−1.54(2H,m),1.51−1.38(2H,m).
MS(ES+APCI)m/z:591[(M+H)+].
(工程2)N−(4−{2−アミノ−5−[1−(テトラヒドロ−2H−ピラン−4−イル)−1H−ピラゾール−4−イル]ピリジン−3−イル}−3−フルオロフェニル)−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミド メタンスルホン酸塩
出発原料として工程1で得たN−[4−(2−アミノ−5−ブロモピリジン−3−イル)−3−フルオロフェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミド及び1−(テトラヒドロ−2H−ピラン−4−イル)−4−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)−1H−ピラゾールを用いて、溶媒として1,4−ジオキサン及び水(比率10:1)を用いて、触媒としてテトラキス(トリフェニルホスフィン)パラジウム(0)を用いて、塩基として炭酸カリウムを用いて、反応温度100℃で7時間加熱攪拌し、中間体8と同様にしてN−(4−{2−アミノ−5−[1−(テトラヒドロ−2H−ピラン−4−イル)−1H−ピラゾール−4−イル]ピリジン−3−イル}−3−フルオロフェニル)−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミドを得た。このものを用いて、実施例71工程3と同様の操作により標記化合物を得た。
1H−NMR(DMSO−D6)δ:13.38−13.36(1H,m),8.73(1H,d,J=2.4Hz),8.42−8.40(1H,m),8.26−8.19(3H,m),8.03−7.98(2H,m),7.61−7.44(6H,m),7.27(2H,d,J=7.9Hz),4.44−4.35(1H,m),4.12(2H,d,J=6.7Hz),3.99−3.93(2H,m),3.90−3.84(2H,m),3.52−3.43(2H,m),3.31−3.23(2H,m),2.36(3H,s),2.31(3H,s),2.17−2.06(1H,m),2.05−1.98(2H,m),1.97−1.84(2H,m),1.49−1.42(2H,m),1.37−1.25(2H,m).
MS(ES+APCI)m/z:663[(M+H)+].
同様にして対応する出発原料から表17の最終体を合成した。
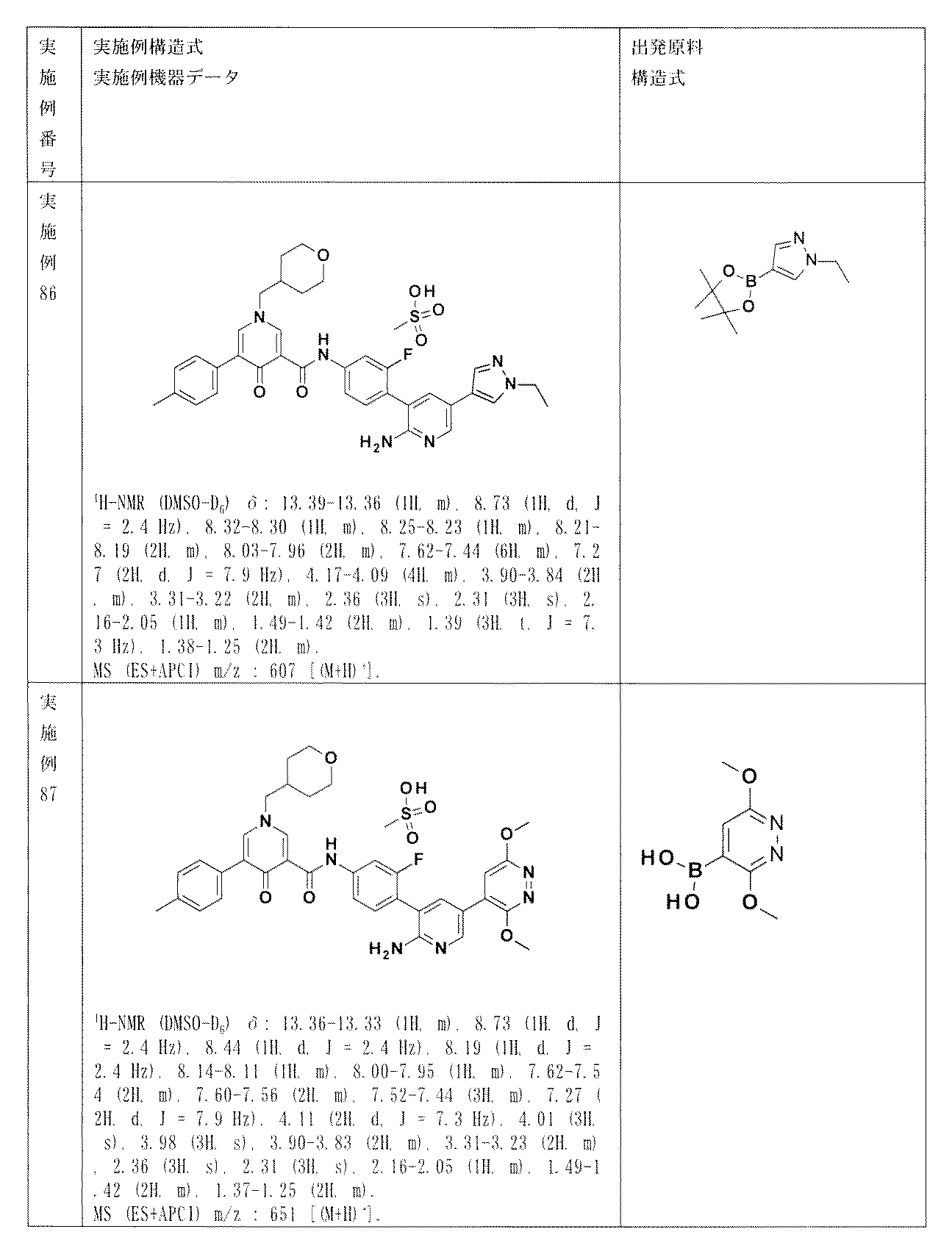

5−ブロモ−3−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)ピリジン−2−アミン200mg(0.669mmol)のジオキサン10.0ml、水1.00ml溶液に、N−(6−ヨードピリジン−3−イル)−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミド354mg(0.669mmol)、炭酸カリウム277mg(2.01mmol)、テトラキス(トリフェニルホスフィン)パラジウム(0)38.0mg(0.0344mmol)を加え、窒素雰囲気下、90℃にて3時間撹拌した。さらに5−ブロモ−3−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)ピリジン−2−アミン200mg(0.669mmol)を加えて90℃にて5時間撹拌した。
放冷後、水、塩化メチレン、メタノールを加え、分液操作を行った。有機層を無水硫酸ナトリウムにて乾燥後、ろ過し、溶液を濃縮した。残渣に酢酸エチルにてスラリーして、標記化合物352mg(91.6%)を固体として得た。
1H−NMR(CDCl3)δ:13.05(1H,s),8.94(1H,d,J=2.4Hz),8.56(1H,d,J=2.4Hz),8.32(1H,dd,J=8.5,2.4Hz),8.10(1H,d,J=1.8Hz),7.92(1H,d,J=2.4Hz),7.68(1H,d,J=9.2Hz),7.50−7.44(3H,m),7.29(2H,d,J=7.9Hz),6.76(2H,br s),4.04(2H,dd,J=11.9,3.4Hz),3.89(2H,d,J=7.3Hz),3.39(2H,t,J=11.0Hz),2.42(3H,s),2.16−2.06(1H,m),1.65−1.57(2H,m),1.51−1.38(2H,m).
MS(APCI)m/z:574[(M+H)+].
(工程2) N−[2’−アミノ−5’−(3,4−ジメトキシフェニル)−2,3’−ビピリジン−5−イル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミド メタンスルホン酸塩
N−(2’−アミノ−5’−ブロモ−2,3’−ビピリジン−5−イル)−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミド130mg(0.226mmol),3,4−ジメトキシフェニルボロン酸50mg(0.272mmol)のジオキサン10ml、水1ml懸濁液に炭酸カリウム94mg(0.679mmol)、テトラキス(トリフェニルホスフィン)パラジウム(0)13mg(0.0113mmol)を加え、窒素雰囲気下、100℃にて2時間撹拌した。反応液を塩化メチレンにて希釈後、水洗した。有機層を無水硫酸ナトリウムにて乾燥し、ろ過後、溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(塩化メチレン/メタノール=19/1、プレカラムとしてアミノシリカゲルカラムを使用)にて精製して、N−[2’−アミノ−5’−(3,4−ジメトキシフェニル)−2,3’−ビピリジン−5−イル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミド69.0mg(48.3%)を固体として得た。このものの塩化メチレン1.00ml、エタノール1.00ml溶液に、メタンスルホン酸10.5mg(0.192mmol)を加え、溶媒を留去した。残渣を塩化メチレン/酢酸エチル/イソプロピルエーテルに懸濁し、不溶物をろ取して、標記化合物65.0mg(81.8%)を固体として得た。
1H−NMR(CDCl3)δ:14.93(1H,s),13.27(1H,s),9.03(1H,d,J=2.4Hz),8.57(1H,s),8.41−8.36(2H,m),7.91−7.85(2H,m),7.52(1H,d,J=2.4Hz),7.46(2H,d,J=8.5Hz),7.30(2H,d,J=7.9Hz),7.05(1H,dd,J=8.2,2.1Hz),7.00−6.94(2H,m),4.04(2H,dd,J=11.6,3.7Hz),3.96(3H,s),3.95(3H,s),3.91(2H,d,J=7.3Hz),3.40(2H,t,J=11.0Hz),2.95(3H,s),2.42(3H,s),2.18−2.07(1H,m),1.65−1.55(2H,m),1.52−1.39(2H,m).
MS(ESI)m/z:632[(M+H)+].
同様にして対応する出発原料AおよびBから表18の最終体を合成した。






1H−NMR(DMSO−D6)δ:13.94(1H,s),8.78(1H,d,J=2.4Hz),8.64(1H,d,J=9.7Hz),8.52(1H,d,J=9.7Hz),8.42(1H,d,J=2.4Hz),8.25−8.21(2H,m),7.61(2H,d,J=7.9Hz),7.48(2H,s),7.28−7.23(4H,m),7.02(1H,d,J=8.5Hz),4.12(2H,d,J=7.3Hz),3.91−3.83(5H,m),3.79(3H,s),3.27(2H,t,J=10.9Hz),2.37(3H,s),2.16−2.07(1H,m),1.47(2H,br d,J=10.9Hz),1.37−1.26(2H,m).
MS(APCI)m/z:633[(M+H)+].
同様にして対応する出発原料AおよびBから表19の最終体を合成した。
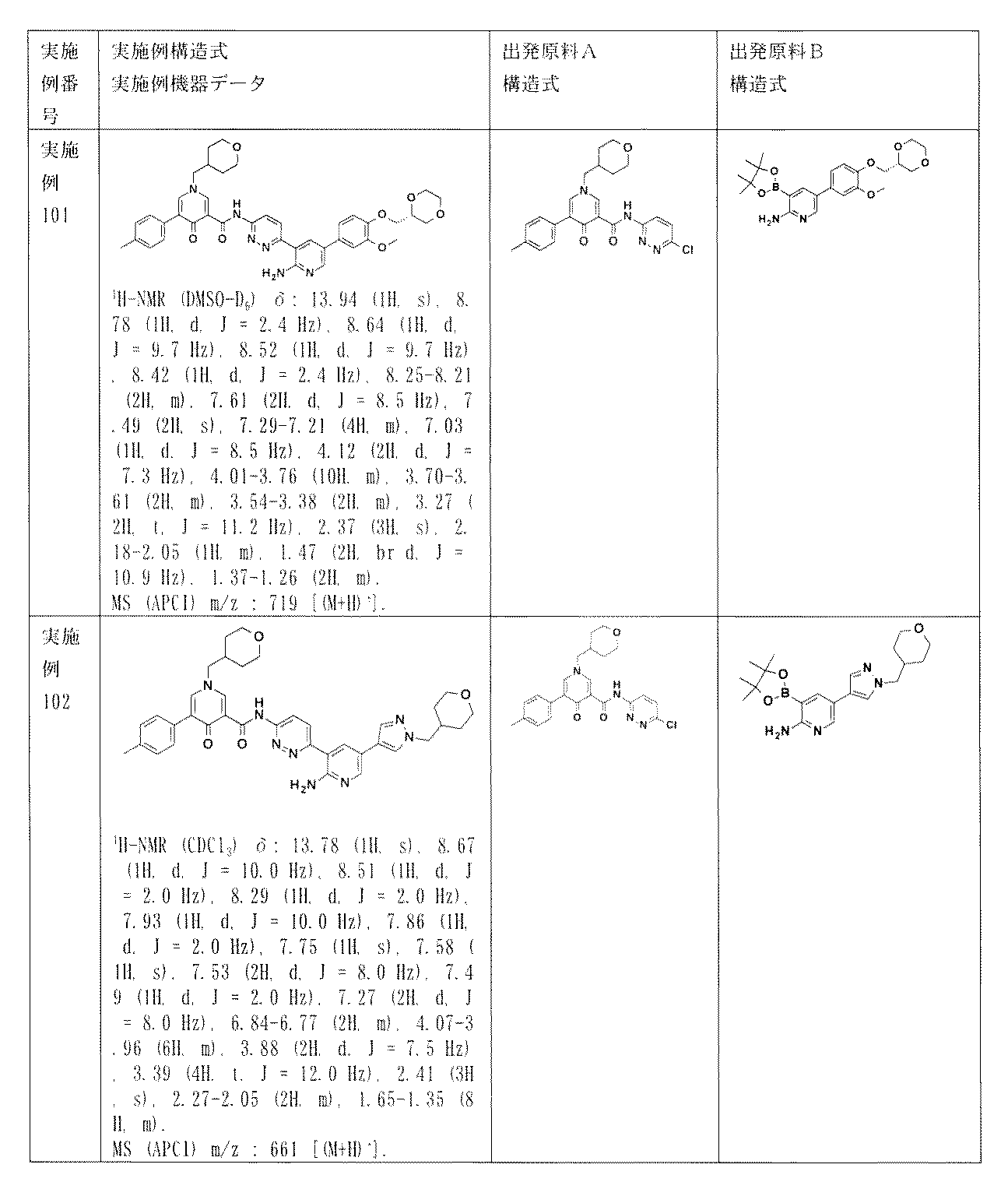




1H−NMR(CDCl3)δ:13.77(1H,s),8.67(1H,d,J=9.7Hz),8.51(1H,d,J=2.4Hz),8.35(1H,d,J=2.4Hz),7.96−7.94(2H,m),7.53−7.48(3H,m),7.27(2H,d,J=7.9Hz),7.08−7.05(2H,m),6.98(1H,d,J=7.9Hz),6.89(2H,s),4.21(2H,t,J=6.1Hz),4.03(2H,dd,J=11.2,3.3Hz),3.94(3H,s),3.87(2H,d,J=7.3Hz),3.76(4H,t,J=4.9Hz),3.39(2H,t,J=11.2Hz),2.88(2H,t,J=6.1Hz),2.63−2.61(4H,m),2.41(3H,s),2.15−2.07(1H,m),1.62−1.60(2H,br d、J=11.2Hz),1.49−1.39(2H,m).
MS(APCI)m/z:732[(M+H)+].
同様にして対応する出発原料から表20の最終体を合成した。


塩化メチレン体(1:1)0.0169g(0.0207mmol)、炭酸セシウム0.202g(0.621mmol)に1,4−ジオキサン2.00mLと水0.400mLを加え、80℃で1時間攪拌した。室温まで放冷後、反応液に水を加え、酢酸エチルで3回抽出し、有機層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥した。濾過、減圧濃縮後、得られた残渣をアミノシリカゲルカラムクロマトグラフィー(山善社、酢酸エチル/メタノール=100/0~90/10)で精製した。減圧濃縮後、残渣を酢酸エチルとジエチルエーテルを用いて固化し、固体を濾取、乾燥することで標記化合物0.111g(収率84.8%)を固体として得た。
1H−NMR(DMSO−D6)δ:13.61(1H,s),9.58(1H,d,J=1.2Hz),9.17(1H,d,J=2.4Hz),8.79(1H,d,J=2.4Hz),8.39(1H,d,J=2.4Hz),8.32(1H,d,J=2.4Hz),8.20(1H,d,J=2.4Hz),7.59(2H,d,J=8.5Hz),7.29−7.23(6H,m),7.02(1H,d,J=8.5Hz),4.13(2H,d,J=6.7Hz),3.89−3.85(5H,m),3.79(3H,s),3.27(2H,t,J=10.9Hz),2.36(3H,s),2.16−2.07(1H,m),1.49−1.44(2H,m),1.37−1.27(2H,m).
MS(APCI)m/z:633[(M+H)+].
(実施例113) N−{4−[2−アミノ−5−(3,4−ジメトキシフェニル)ピリジン−3−イル]フェニル}−5−(4−シクロプロピルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミド

1H−NMR(DMSO−D6)δ:13.11(1H,s),8.71(1H,d,J=2.4Hz),8.25(1H,d,J=2.4Hz),8.16(1H,d,J=2.4Hz),7.82(2H,d,J=8.5Hz),7.63(1H,d,J=2.4Hz),7.58−7.52(4H,m),7.20−7.14(4H,m),6.99(1H,d,J=8.5Hz),5.76(2H,s),4.10(2H,d,J=7.3Hz),3.88−3.83(5H,m),3.77(3H,s),3.27(2H,t,J=10.9Hz),2.16−2.04(1H,m),2.00−1.93(1H,m),1.46(2H,br d,J=10.9Hz),1.35−1.26(2H,m),1.02−0.97(2H,m),0.73−0.69(2H,m).
MS(APCI)m/z:657[(M+H)+].
(実施例114) N−{4−[2−アミノ−5−(3,4−ジメトキシフェニル)ピリジン−3−イル]フェニル}−5−メチル−4’−オキソ−1’−(テトラヒドロ−2H−ピラン−4−イルメチル)−1’,4’−ジヒドロ−2,3’−ビピリジン−5’−カルボキサミド

1H−NMR(DMSO−D6)δ:12.98(1H,s),8.75(1H,d,J=2.4Hz),8.71(1H,d,J=2.4Hz),8.30(1H,d,J=2.4Hz),8.25(1H,d,J=2.4Hz),8.02(1H,dd,J=8.5,2.4Hz),7.82(2H,d,J=8.5Hz),7.61(1H,d,J=2.4Hz),7.53(2H,d,J=8.5Hz),7.34(1H,d,J=8.5Hz),7.19−7.13(2H,m),6.98(1H,d,J=8.5Hz),5.69(2H,s),4.12(2H,d,J=7.3Hz),3.88−3.83(5H,m),3.77(3H,s),3.27(2H,t,J=11.0Hz),2.52(3H,s),2.17−2.07(1H,m),1.47(2H,br d,J=11.0Hz),1.37−1.26(2H,m).
MS(APCI)m/z:632[(M+H)+].
同様にして対応する出発原料から表21の最終体を合成した。


1H−NMR(DMSO−D6)δ:13.08(1H,s),8.76(1H,d,J=2.4Hz),8.69(1H,d,J=2.4Hz),8.51−8.47(2H,m),8.26(1H,d,J=2.4Hz),7.84(2H,d,J=8.5Hz),7.71(1H,dd,J=8.5,2.4Hz),7.62(1H,d,J=2.4Hz),7.55(2H,d,J=8.5Hz),7.20−7.14(2H,m),6.99(1H,d,J=8.5Hz),5.69(2H,s),4.20(2H,d,J=7.3Hz),3.88−3.83(5H,m),3.77(3H,s),3.27(2H,t,J=11.3Hz),2.36(3H,s),2.11−2.03(1H,m),1.47(2H,br d,J=11.0Hz),1.37−1.26(2H,m).
MS(APCI)m/z:632[(M+H)+].
同様にして対応する出発原料から表22の最終体を合成した。


1H−NMR(DMSO−D6)δ:12.90(1H,s),8.69−8.66(2H,m),8.26(1H,d,J=2.4Hz),7.84(2H,d,J=8.5Hz),7.62(1H,d,J=2.4Hz),7.57−7.54(3H,m),7.20−7.14(2H,m),6.99(1H,d,J=8.5Hz),6.87−6.85(1H,m),5.70(2H,s),4.14(2H,d,J=7.3Hz),3.88−3.83(5H,m),3.77(3H,s),3.27(2H,t,J=10.9Hz),2.49(3H,s),2.16−2.10(1H,m),1.45(2H,br d,J=10.9Hz),1.38−1.27(2H,m).
MS(APCI)m/z:637[(M+H)+].
同様にして対応する出発原料から表23の最終体を合成した。


1H−NMR(DMSO−D6)δ:13.07(1H,s),8.86(1H,d,J=2.4Hz),8.80(1H,d,J=2.4Hz),8.52−8.48(2H,m),8.31(1H,d,J=2.4Hz),7.94(1H,dd,J=12.1,2.4Hz),7.72(1H,dd,J=8.2,2.4Hz),7.63(1H,d,J=2.4Hz),7.50−7.42(2H,m),7.18(1H,d,J=2.4Hz),7.11(1H,dd,J=8.5,2.4Hz),6.99(1H,d,J=8.5Hz),5.71(2H,s),4.70(2H,s),3.97−3.74(10H,m),3.69−3.60(2H,m),3.52−3.32(4H,m),2.36(3H,s),1.88−1.77(4H,m).
MS(APCI)m/z:761[(M+H)+].
同様にして対応する出発原料から表24の最終体を合成した。


1H−NMR(DMSO−D6)δ:13.27(1H,s),8.79(1H,d,J=2.4Hz),8.72(1H,d,J=2.4Hz),8.54(1H,d,J=2.4Hz),8.46(1H,d,J=8.5Hz),8.34(1H,d,J=2.4Hz),8.29(1H,d,J=2.4Hz),8.03−7.99(1H,m),7.78(1H,d,J=8.5Hz),7.59−7.51(4H,m),7.31(1H,d,J=2.4Hz),7.25(1H,dd,J=8.5,2.4Hz),7.06(1H,d,J=8.5Hz),4.21(2H,d,J=7.3Hz),4.01−3.93(2H,m),3.90−3.74(8H,m),3.69−3.24(6H,m),2.37(3H,s),2.34(3H,s),2.13−2.02(1H,m),1.47(2H,br d,J=10.9Hz),1.37−1.28(2H,m).
MS(APCI)m/z:736[(M+H)+].
元素分析値C41H42FN5O7・1.0CH3SO3H・1.0H2Oとして
計算値:C,59.35;H,5.69;N,8.24;F,2.24;S,3.77.
実測値:C,59.38;H,5.80;N,8.10;F,2.23;S,3.71.
同様にして対応する出発原料から表25の各メシル酸塩を非晶性固体として得た。



(実施例130) N−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミド メタンスルホン酸塩 水和物
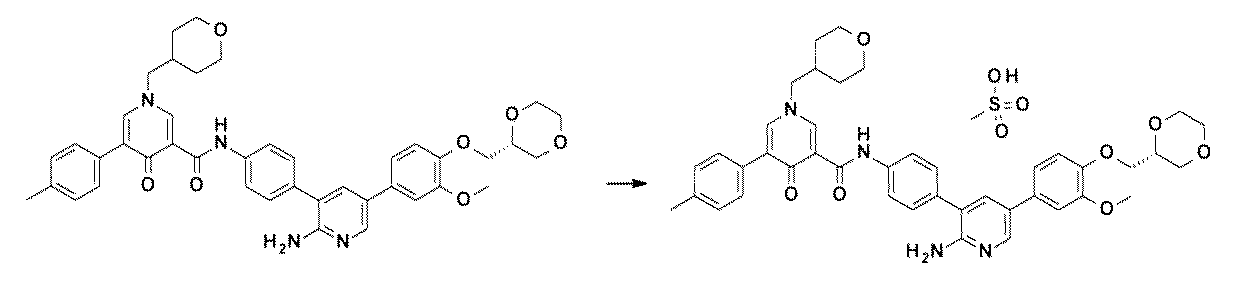
1H−NMR(DMSO−D6)δ:13.22(1H,s),8.72(1H,d,J=2.0Hz),8.28(1H,d,J=2.0Hz),8.21(1H,d,J=2.0Hz),8.19(1H,d,J=2.0Hz),7.90(2H,d,J=8.5Hz),7.60−7.52(6H,m),7.32(1H,d,J=2.0Hz),7.27(3H,d,J=8.5Hz),7.06(1H,d,J=8.5Hz),4.11(2H,d,J=7.5Hz),4.01−3.93(2H,m),3.89−3.22(14H,m),2.36(3H,s),2.32(3H,s),2.17−2.04(1H,m),1.46(2H,d,J=11.0Hz),1.37−1.25(2H,m).
MS(APCI)m/z:717[(M+H)+].
元素分析値C42H44N4O7・1.0CH3SO3H・3.0H2Oとして
計算値:C,59.57;H,6.28;N,6.46;S,3.70.
実測値:C,59.75;H,6.29;N,6.48;S,3.76.
粉末X線回折(CuKα、λ=1.54オングストローム、走査速度=20°/min)の回折パターンを図1において最大ピーク強度を100とした場合の相対強度22以上のピークを表26に示す。


1H−NMR(DMSO−D6)δ:13.22(1H,s),8.72(1H,d,J=2.5Hz),8.28(1H,d,J=2.5Hz),8.21(1H,d,J=2.5Hz),8.19(1H,d,J=2.5Hz),7.90(2H,d,J=8.8Hz),7.60−7.55(4H,m),7.50(2H,br s),7.31(1H,d,J=2.5Hz),7.29−7.23(3H,m),7.06(1H,d,J=8.8Hz),4.12(2H,d,J=7.5Hz),4.01−3.93(2H,m),3.89−3.24(14H,m),2.36(3H,s),2.16−2.05(1H,m),1.46(2H,d,J=12.5Hz),1.37−1.25(2H,m).
元素分析値C42H44N4O7・1HBr・3.3H2Oとして
計算値:C,58.85;H,6.07;N,6.53;Br,9.32.
実測値:C,58.91;H,5.98;N,6.58;Br,9.42.
粉末X線回折(CuKα、λ=1.54オングストローム、走査速度=20°/min)の回折パターンを図2において最大ピーク強度を100とした場合の相対強度19以上のピークを表27に示す。


1H−NMR(DMSO−D6)δ:13.22(1H,s),8.72(1H,d,J=2.5Hz),8.27(1H,d,J=2.5Hz),8.21(1H,d,J=2.5Hz),8.19(1H,d,J=2.5Hz),7.90(2H,d,J=8.8Hz),7.60−7.55(4H,m),7.49(2H,br s),7.31(1H,d,J=2.4Hz),7.29−7.24(3H,m),7.06(1H,d,J=8.8Hz),4.11(2H,d,J=7.5Hz),4.01−3.93(2H,m),3.90−3.22(14H,m),2.36(3H,s),2.17−2.04(1H,m),1.46(2H,d,J=12.5Hz),1.38−1.26(2H,m).
元素分析値C42H44N4O7・1HNO3・3H2Oとして
計算値:C,60.49;H,6.16;N,8.40.
実測値:C,60.58;H,6.15;N,8.43.
粉末X線回折(CuKα、λ=1.54オングストローム、走査速度=20°/min)の回折パターンを図3において最大ピーク強度を100とした場合の相対強度34以上のピークを表28に示す。


1H−NMR(DMSO−D6)δ:13.20(1H,s),8.72(1H,d,J=2.4Hz),8.27(1H,d,J=2.4Hz),8.18(1H,d,J=2.4Hz),8.08(1H,s),7.88(2H,d,J=8.5Hz),7.57(4H,t,J=8.5Hz),7.30−7.03(7H,m),4.11(2H,d,J=7.5Hz),4.01−3.92(2H,m),3.91−3.21(14H,m),2.36(3H,s),2.16−2.05(1H,m),1.46(2H,d,J=12.5Hz),1.37−1.25(2H,m).
元素分析値C42H44N4O7・0.75H2SO4・4H2Oとして
計算値:C,58.49;H,6.25;N,6.50;S,2.79.
実測値:C,58.53;H,6.13;N,6.52;S,2.80..
粉末X線回折(CuKα、λ=1.54オングストローム、走査速度=20°/min)の回折パターンを図4において最大ピーク強度を100とした場合の相対強度13以上のピークを表29に示す。


1H−NMR(DMSO−D6)δ:13.11(1H,s),8.72(1H,d,J=2.5Hz),8.25(1H,d,J=2.5Hz),8.17(1H,d,J=2.5Hz),7.82(2H,d,J=8.5Hz),7.62(1H,d,J=2.5Hz),7.58(2H,d,J=8.5Hz),7.53(2H,d,J=8.5Hz),7.26(2H,d,J=8.5Hz),7.20(1H,d,J=2.0Hz),7.13(1H,dd,J=8.5,2.0Hz),6.99(1H,d,J=8.5Hz),5.73(2H,br s),4.11(2H,d,J=7.5Hz),3.98−3.90(2H,m),3.89−3.22(14H,m),2.36(3H,s),2.16−2.04(1H,m),1.46(2H,d,J=11.5Hz),1.37−1.25(2H,m).
元素分析値C42H44N4O7・1.0H3PO4・3.5H2Oとして
計算値:C,57.46;H,6.20;N,6.38;P,3.53.
実測値:C,57.41;H,6.20;N,6.41;P,3.41.
粉末X線回折(CuKα、λ=1.54オングストローム、走査速度=20°/min)の回折パターンを図5において最大ピーク強度を100とした場合の相対強度20以上のピークを表30に示す。


種晶の取得方法
N−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミド10.4mg(14.5μmol)にメタノール41.6μL、水151μlを加えた。その後、1.00mol/L エタンスルホン酸水溶液15.2μl(15.2μmol)を加えた。混合液を40℃で約24時間、次いで、室温で約30分攪拌し、桐山ロートにより吸引ろ過し、析出した固体をろ取した。その後、風乾し、種晶として10.9mgを得た。
1H−NMR(DMSO−D6)δ:13.22(1H,s),8.72(1H,d,J=2.0Hz),8.28(1H,d,J=2.0Hz),8.20(2H,dd,J=8.5,2.0Hz),7.90(2H,d,J=8.5Hz),7.61−7.49(6H,m),7.32(1H,d,J=2.0Hz),7.29−7.24(3H,m),7.06(1H,d,J=8.5Hz),4.11(2H,d,J=7.5Hz),4.01−3.93(2H,m),3.87−3.24(14H,m),2.41−2.31(5H,m),2.16−2.04(1H,m),1.46(2H,d,J=11.5Hz),1.37−1.25(2H,m),1.06(3H,t,J=7.5Hz).
元素分析値C42H44N4O7・1.0C2H5SO3H・5.5H2Oとして
計算値:C,57.07;H,6.64;N,6.05;S,3.46.
実測値:C,57.13;H,6.78;N,6.08;S,3.60.
粉末X線回折(CuKα、λ=1.54オングストローム、走査速度=20°/min)の回折パターンを図6において最大ピーク強度を100とした場合の相対強度5以上のピークを表31に示す。

1H−NMR(DMSO−D6)δ:13.22(1H,s),8.72(1H,d,J=2.0Hz),8.27(1H,d,J=2.0Hz),8.19(2H,d,J=2.0Hz),7.90(2H,d,J=8.5Hz),7.61−7.55(6H,m),7.44(2H,br s),7.35−7.24(7H,m),7.06(1H,d,J=8.5Hz),4.11(2H,d,J=7.5Hz),4.01−3.93(2H,m),3.90−3.22(14H,m),2.36(3H,s),2.17−2.04(1H,m),1.46(2H,d,J=11.6Hz),1.31(2H,td,J=12.2,8.5Hz).
元素分析値C42H44N4O7・1.0C6H5SO3H・3H2Oとして
計算値:C,62.05;H,6.08;N,6.03;S,3.45.
実測値:C,62.18;H,6.07;N,6.08:S,3.46.
粉末X線回折(CuKα、λ=1.54オングストローム、走査速度=20°/min)の回折パターンを図7において最大ピーク強度を100とした場合の相対強度29以上のピークを表32に示す。

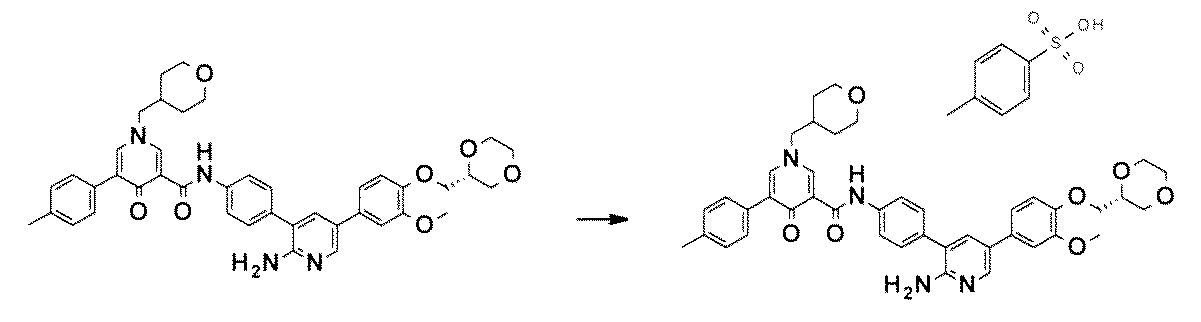
種晶の取得方法
N−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)フェニル]−5−(4−メチルフェニル)−4−オキソ−1−(テトラヒドロ−2H−ピラン−4−イルメチル)−1,4−ジヒドロピリジン−3−カルボキサミド 10.3mg(14.4μmol)にメタノール165μL、水26.2μlを加えた。その後、1.00mol/L p−トルエンスルホン酸水溶液15.0μl(15.0μmol)を加えた。混合液を40℃で約24時間、次いで、室温で約30分攪拌し、桐山ロートにより吸引ろ過し、析出した固体をろ取した。その後、風乾し、種晶として10.7mgを得た。
1H−NMR(DMSO−D6)δ:13.22(1H,s),8.73(1H,d,J=2.0Hz),8.28(1H,d,J=2.0Hz),8.21(1H,d,J=2.0Hz),8.19(1H,d,J=2.0Hz),7.90(2H,d,J=8.0Hz),7.60−7.45(8H,m),7.31(1H,d,J=2.0Hz),7.29−7.24(3H,m),7.11(2H,d,J=8.0Hz),7.06(1H,d,J=8.0Hz),4.11(2H,d,J=7.5Hz),4.02−3.93(2H,m),3.90−3.22(14H,m),2.36(3H,s),2.29(3H,s),2.16−2.04(1H,m),1.46(2H,d,J=11.5Hz),1.37−1.25(2H,m).
元素分析値C42H44N4O7・1.0C7H7SO3Hとして
計算値:C,66.19;H,5.89;N,6.30;S,3.61.
実測値:C,65.91;H,5.97;N,6.26;S,3.59.
粉末X線回折(CuKα、λ=1.54オングストローム、走査速度=20°/min)の回折パターンを図8において最大ピーク強度を100とした場合の相対強度23以上のピークを表33に示す。


1H−NMR(DMSO−D6)δ:13.22(1H,s),8.77(1H,d,J=2.0Hz),8.69(1H,d,J=2.0Hz),8.51(1H,d,J=2.0Hz),8.47(1H,d,J=8.5Hz),8.30(1H,d,J=2.0Hz),7.93(1H,dd,J=12.5,2.0Hz),7.71(1H,dd,J=8.5,2.0Hz),7.64(1H,d,J=2.0Hz),7.49−7.41(2H,m),7.19(1H,d,J=2.0Hz),7.12(1H,dd,J=8.5,2.0Hz),6.99(1H,d,J=8.5Hz),5.77(2H,br s),4.21(2H,d,J=7.5Hz),3.98−3.24(16H,m),2.36(3H,s),2.13−2.01(1H,m),1.46(2H,d,J=12.0Hz),1.38−1.26(2H,m).
MS(APCI)m/z:736[(M+H)+].
元素分析値C41H42FN5O7・1.0H3PO4・3.5H2Oとして
計算値:C,54.91;H,5.84;N,7.81;F,2.12;P,3.45.
実測値:C,54.95;H,5.54;N,7.86;F,2.20;P,3.19.
粉末X線回折(CuKα、λ=1.54オングストローム、走査速度=20°/min)の回折パターンを図9において最大ピーク強度を100とした場合の相対強度18以上のピークを表34に示す。


1H−NMR(DMSO−D6)δ:13.04(1H,br s),8.83(2H,d,J=9.5Hz),8.63(1H,s),8.44(1H,d,J=8.0Hz),8.34(2H,s),8.05−7.98(2H,m),7.72(2H,br s),7.59−7.52(2H,m),7.32(1H,d,J=2.0Hz),7.26(1H,dd,J=8.0,2.0Hz),7.07(1H,d,J=8.0Hz),4.22(2H,d,J=7.5Hz),4.01−3.93(2H,m),3.90−3.74(8H,m),3.70−3.59(2H,m),3.53−3.45(1H,m),3.40(1H,dd,J=11.0,10.0Hz),3.27(2H,t,J=11.0Hz),2.43(3H,s),2.16−2.05(1H,m),1.51−1.43(2H,m),1.39−1.26(2H,m).
MS(APCI)m/z:736[(M+H)+].
元素分析値C41H42FN5O7・1.8H2SO4・3.0H2Oとして
計算値:C,50.96;H,5.38;N,7.25;F,1.97;S,5.97.
実測値:C,50.98;H,5.36;N,7.23;F,1.97;S,6.19.
粉末X線回折(CuKα、λ=1.54オングストローム、走査速度=20°/min)の回折パターンを図10において最大ピーク強度を100とした場合の相対強度6以上のピークを表35に示す。

N−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)−3−フルオロフェニル]−5−メチル−4’−オキソ−1’−(テトラヒドロ−2H−ピラン−4−イルメチル)−1’,4’−ジヒドロ−2,3’−ビピリジン−5’−カルボキサミド 39.8mg(54.1μmol)にアセトン637μL、水78.3μlを加えた。その後、1.00mol/L硫酸水溶液80.9μl(80.9μmol)を加えた。混合液を40℃で約24時間、次いで、室温で約30分攪拌し、桐山ロートにより吸引ろ過し、析出した固体をろ取した。その後、風乾し、標記化合物36.7mg(収率71.6%)を結晶性固体として得た。
元素分析値C41H42FN5O7・1.60H2SO4・3.0H2Oとして
計算値:C,52.01;H,5.45;N,7.40;F,2.01;S,5.42.
実測値:C,52.07;H,5.24;N,7.25;F,2.09;S,5.50.
粉末X線回折(CuKα、λ=1.54オングストローム、走査速度=20°/min)の回折パターンを図11において最大ピーク強度を100とした場合の相対強度12以上のピークを表36に示す。

N−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)−3−フルオロフェニル]−5−メチル−4’−オキソ−1’−(テトラヒドロ−2H−ピラン−4−イルメチル)−1’,4’−ジヒドロ−2,3’−ビピリジン−5’−カルボキサミド 40.5mg(55.0μmol)にアセトン648μL、水24.8μlを加えた。その後、1.00mol/L硫酸水溶液137μl(137μmol)を加えた。混合液を40℃で約24時間、次いで、室温で約30分攪拌し、桐山ロートにより吸引ろ過し、析出した固体をろ取した。その後、風乾し、標記化合物47.2mg(収率88.8%)を結晶性固体として得た。
元素分析値C41H42FN5O7・1.80H2SO4・3.0H2Oとして
計算値:C,50.96;H,5.38;N,7.25;F,1.97;S,5.97.
実測値:C,50.85;H,5.20;N,7.06;F,2.09;S,5.99.
粉末X線回折(CuKα、λ=1.54オングストローム、走査速度=20°/min)の回折パターンを図12において最大ピーク強度を100とした場合の相対強度7以上のピークを表37に示す。

N−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)−3−フルオロフェニル]−5−メチル−4’−オキソ−1’−(テトラヒドロ−2H−ピラン−4−イルメチル)−1’,4’−ジヒドロ−2,3’−ビピリジン−5’−カルボキサミド 39.8mg(54.0μmol)にアセトン636μL、水112μlを加えた。その後、5.79mol/L硫酸水溶液46.7μl(270μmol)を加えた。混合液を40℃で約24時間、次いで、室温で約30分攪拌し、桐山ロートにより吸引ろ過し、析出した固体をろ取した。その後、風乾し、標記化合物47.5mg(収率89.2%)を結晶性固体として得た。
元素分析値C41H42FN5O7・2.00H2SO4・3.0H2Oとして
計算値:C,49.94;H,5.32;N,7.10;F,1.93;S,6.50.
実測値:C,49.75;H,5.08;N,6.91;F,2.20;S,6.69.
粉末X線回折(CuKα、λ=1.54オングストローム、走査速度=20°/min)の回折パターンを図13において最大ピーク強度を100とした場合の相対強度11以上のピークを表38に示す。

N−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)−3−フルオロフェニル]−5−メチル−4’−オキソ−1’−(テトラヒドロ−2H−ピラン−4−イルメチル)−1’,4’−ジヒドロ−2,3’−ビピリジン−5’−カルボキサミド 40.2mg(54.6μmol)にアセトン643μL、水51.8μlを加えた。その後、1.00mol/L硫酸水溶液109μl(109μmol)を加えた。混合液を40℃で約24時間、次いで、室温で約30分攪拌し、桐山ロートにより吸引ろ過し、析出した固体をろ取した。その後、風乾し、標記化合物44.4mg(収率84.6%)を結晶性固体として得た。
元素分析値C41H42FN5O7・1.75H2SO4・3.0H2Oとして
計算値:C,51.22;H,5.40;N,7.28;F,1.98;S,5.84.
実測値:C,50.92;H,5.19;N,7.11;F,2.25;S,5.81.
粉末X線回折(CuKα、λ=1.54オングストローム、走査速度=20□/min)の回折パターンを図14において最大ピーク強度を100とした場合の相対強度11以上のピークを表39に示す。


元素分析値C41H42N5O7F・1.75H2SO4・3.5H2Oとして
計算値:C,50.74;H,5.45;N,7.22;F,1.96;S,5.78.
実測値:C,50.70;H,5.33;N,7.13;F,2.01;S,5.82.
粉末X線回折(CuKα、λ=1.54オングストローム、走査速度=20°/min)の回折パターンを図15において最大ピーク強度を100とした場合の相対強度18以上のピークを表40に示す。


元素分析値C41H42N5O7F・1.5H2SO4・5.0H2Oとして
計算値:C,50.61;H,5.70;N,7.20;F,1.95;S,4.94.
実測値:C,50.59;H,5.55;N,7.24;F,2.04;S,5.09.
粉末X線回折(CuKα、λ=1.54オングストローム、走査速度=20°/min)の回折パターンを図16において最大ピーク強度を100とした場合の相対強度13以上のピークを表41に示す。
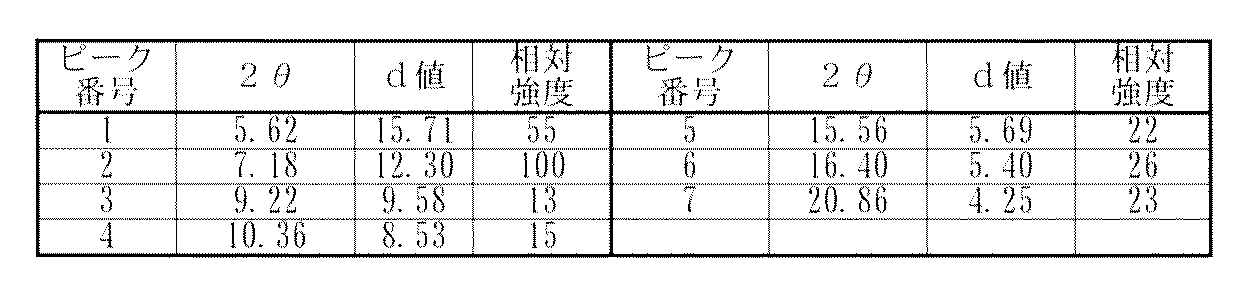

1H−NMR(DMSO−D6)δ:12.97(1H,br s),8.90−8.80(4H,m),8.64(1H,s),8.42(1H,d,J=8.5Hz),8.33(2H,s),8.11−7.97(2H,m),7.92(2H,d,J=7.0Hz),7.71(2H,br s),7.59−7.52(2H,m),7.40(2H,dd,J=8.5,7.0Hz),7.31(1H,d,J=2.5Hz),7.26(1H,dd,J=8.5,2.5Hz),7.06(1H,d,J=8.5Hz),4.22(2H,d,J=7.0Hz),3.99−3.24(16H,m),2.43(3H,s),2.18−2.05(1H,m),1.47(2H,d,J=10.5Hz),1.39−1.26(2H,m).
MS(APCI)m/z:736[(M+H)+].
元素分析値C41H42N5O7F・1.0C10H8O6S2・5.0H2Oとして
計算値:C,54.98;H,5.43;N,6.29;F,1.71;S,5.76.
実測値:C,54.74;H,5.37;N,6.24;F,1.92;S,5.82.
粉末X線回折(CuKα、λ=1.54オングストローム、走査速度=20°/min)の回折パターンを図17において最大ピーク強度を100とした場合の相対強度45以上のピークを表42に示す。

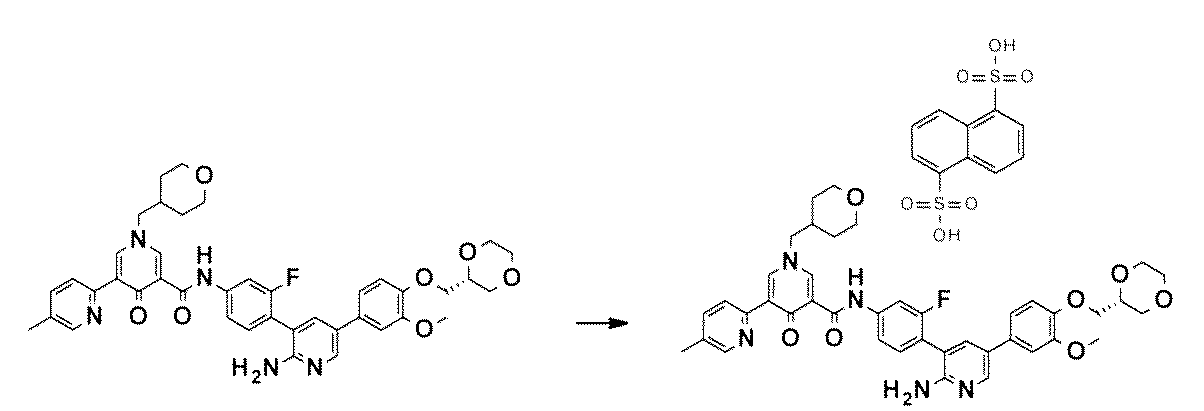
元素分析値C41H42N5O7F・1.0C10H8O6S2・4.0H2Oとして
計算値:C,55.88;H,5.33;N,6.39;F,1.73;S,5.85.
実測値:C,55.71;H,5.45;N,6.18;F,1.82;S,5.62.
粉末X線回折(CuKα、λ=1.54オングストローム、走査速度=20°/min)の回折パターンを図18において最大ピーク強度を100とした場合の相対強度42以上のピークを表43に示す。

キナーゼ反応緩衝液(100mM HEPES(pH7.4),0.003%Brij−35,0.004% Tween−20,1mM DTT,10mM MgCl2)を用いて、AXL(human AXLの細胞内ドメイン464~885番目のアミノ酸とグルタチオントランスフェラーゼとの融合タンパク質をバキュロウイルス発現システムで発現させ、グルタチオンセファロースクロマトグラフィーで精製したもの。カルナバイオ株式会社、カタログ番号08−107)を170ng/ml含むキナーゼ希釈溶液を作成し、384ウェルプレートの各ウェルに19μlずつ添加した。
次に、DMSOを用いて試験化合物を希釈し、この希釈液を各ウェルに1μlずつ添加した。
室温で30分間のプレインキュベーションを行った後、基質ペプチド(FL−Peptide 30(5FAM−KKKKEEIYFFF−CONH2)、Caliper Life Sciences、カタログ番号760430)およびATPをそれぞれ1.5μM、10μM含む溶液を作成し、これを各ウェルに5μlずつ添加することでキナーゼ反応を開始した。プレートを28℃で1.5時間インキュベートし、各ウェルに40μlのターミネーション緩衝液(100mM HEPES(pH7.4),0.015% Brij−35,40mM EDTA,0.1% Coating Reagent3)を添加して反応を停止させた。
反応溶液中の基質ペプチドとリン酸化ペプチドはEZ Reader II(Caliper Life Sciences)により分離、定量した。
キナーゼ反応は基質ペプチドピーク高さ(S)とリン酸化ペプチドピーク高さ(P)から計算される生成物比(P/(P+S))にて評価した。
阻害率(Inhibition)は、次の式により求めた(EZ Reader IIシステムのソフトウェアにより自動的に算出)。
Inhibition(%)=100×(1−Ci/C0)
ここでCiは試験化合物が添加された場合の生成物比を表し、C0は試験化合物の代わりにDMSOが添加された場合の生成物比を表す。
試験化合物濃度12点に対する阻害率のデータから、次の式を用いた非線形回帰(4パラメーターロジスティック回帰)によりIC50を求めた。
Inhibition(%)=Bottom+(Top−Bottom)/(1+([Compound]/IC50)slope)
(試験例2 セルフリーMerキナーゼ阻害活性)
キナーゼ反応緩衝液(100mM HEPES(pH7.4)、0.003% Brij−35,0.004% Tween−20,1mM DTT,10mM MgCl2)を用いて、Mer(human MERの細胞内ドメイン528~999番目のアミノ酸とグルタチオントランスフェラーゼとの融合タンパク質をバキュロウイルス発現システムで発現させ、グルタチオンセファロースクロマトグラフィーとイオン交換クロマトグラフィーで精製したもの。カルナバイオ株式会社、カタログ番号08−108)を20ng/ml含むキナーゼ希釈溶液を作成し、384ウェルプレートの各ウェルに19μlずつ添加した。
次に、DMSOを用いて試験化合物を希釈し、この希釈液を各ウェルに1μlずつ添加した。
室温で20分間のプレインキュベーションを行った後、基質ペプチド(FL−Peptide 27(5FAM−EFPIYDFLPAKKK−CONH2)、Caliper Life Sciences、カタログ番号760424)およびATP5mM含む溶液を作成し、これを各ウェルに5μlずつ添加することでキナーゼ反応を開始した。プレートを28℃で45分インキュベートし、各ウェルに40μlのターミネーション緩衝液(100mM HEPES(pH7.4),0.015% Brij−35,40mM EDTA,0.1% Coating Reagent3)を添加して反応を停止させた。
反応溶液中の基質ペプチドとリン酸化ペプチドはEZ Reader II(Caliper Life Sciences)により分離、定量した。
キナーゼ反応は基質ペプチドピーク高さ(S)とリン酸化ペプチドピーク高さ(P)から計算される生成物比(P/(P+S))にて評価した。
阻害率(Inhibition)は、次の式により求めた(EZ Reader IIシステムのソフトウェアにより自動的に算出)。
Inhibition(%)=100×(1−Ci/C0)
ここでCiは試験化合物が添加された場合の生成物比を表し、C0は試験化合物の代わりにDMSOが添加された場合の生成物比を表す。
試験化合物濃度12点に対する阻害率のデータから、次の式を用いた非線形回帰(4パラメーターロジスティック回帰)によりIC50を求めた。
Inhibition(%)=Bottom+(Top−Bottom)/(1+([Compound]/IC50)slope)
表44に、ATP濃度が1mMの条件でのAxlキナーゼ阻害活性をIC50(nM)値として、Merキナーゼ阻害IC50(nM)値として、さらにMerキナーゼに対するAxlキナーゼ選択性(倍)を示した。







ヒト非小細胞肺癌由来細胞株NCI−H1299を用いてリン酸化Axl(以下pAxl)阻害試験を実施した。
NCI−H1299細胞を、培地(10%牛胎児血清を含むRPMI1640培地)に懸濁し、96ウェルのマルチウェルプレートにそれぞれ15000細胞/100μl/ウェルで播種し、37℃、5% CO2存在で1日培養した。翌日に培地を除き、100μlの培地を添加し、37℃、5% CO2下で1日培養した。試験化合物をDMSOに溶解し、FBS非添加培地で希釈して検体溶液とした(DMSO濃度2%)。培地または検体添加培地をウェルに25μl添加し(DMSO濃度0.4%)、37℃、5% CO2存在下で1時間インキュベーションした。
GAS6(R&D、品番:885−GS)を6μg/mlとなるようにFBS非添加培地を用いて希釈し、各ウェルへ25μl添加し、攪拌後、37℃、5% CO2存在下で10分間インキュベーションした。
上清を捨て、37%ホルマリン液をリン酸緩衝液(PBS)で4%に希釈した溶液(以下、4%ホルマリン液)をウェルに0.1ml添加して室温で10分間静置した。次に4%ホルマリン液を捨て、TritonX−100をPBSで0.1%に希釈した溶液(以下wash buffer)を0.2ml添加し、デカンタで捨て、ペーパータオル上で余分な水分を除いた。
続いてwash buffer10.7mLに10%NaN3とH2O2110μlを加え(以下、quenching buffer)、ウェルへ0.1ml添加して室温で15分間静置した。
Quenching bufferを捨て、wash bufferを0.2ml添加し、デカンタで捨て、ペーパータオル上で余分な水分を除いた。Wash bufferにスキムミルク(WAKO #198−10605)を最終濃度5%で加え(blocking buffer)、ウェルへ0.25ml添加し、室温で1時間静置した。
Blocking bufferを捨て、Anti−phospho−Axl(Y702)(D12B2)rabbit monoclonal antibody(Cell Signaling、カタログ番号5724)を1/1000の濃度で反応させ、4℃で一晩静置した。Wash bufferで5回洗浄操作を繰り返し、Peroxidase AffiniPure Donkey Anti−Rabbit IgG(H+L)(Jackson ImmunoResearch、カタログ番号 711−035−152)を1/2000の濃度で室温で1時間反応させた。同様に洗浄操作を行い、Super Signal ELISA pico chemi luminescent substrate(Thermo Scientific、カタログ番号37069)を0.05ml添加して、軽く攪拌した後20分間インキュベーションした。その後、ARVO sx(Perkin ElMer)で発光を測定し、pAxl(Y702)レベルを測定した。
pAxl阻害活性は以下の式により求めた。
Inhibition %=100−(A−B)×100/(T−B)
A:被検化合物の測定値
B: ほぼ100%リン酸化を抑制する濃度のポジティブコントロール化合物が添加された反応液の発光値 (たとえばBMS−777607 1μMが添加された反応液の発光値)
T:化合物を添加していない反応液の発光値
複数濃度のpAxl阻害活性のデータから、GraphPad Prism4により50%阻害濃度(IC50)を求めた。
表45に細胞内Axlリン酸化阻害活性をIC50(nM)値として示した。


1.動物:NOG雌性マウス
2.薬物:
Compound A 1塩酸塩2水和物
実施例124
実施例128
実施例129
3.投与液調製:
Compound A、実施例124実施例128および実施例129は0.5%メチルセルロース溶液(0.5% MC,和光純薬工業)に懸濁した。
4.投与方法:
4連投、1日休薬、5連投、2日休薬、5連投、2日休薬、5連投、2日休薬、4連投。
5.病理学的検査
投与期間終了日の翌日にイソフルラン麻酔下で、放血安楽死後、眼球を採材し、ダビッドソン液で固定後、パラフィン包埋およびヘマトキシリン・エオジン染色標本を作製し、病理組織学的検査を実施した。
Compound A 1塩酸塩2水和物:100mg/kg投与群全例(8例)において軽度~中等度の網膜の外顆粒層及び桿錘体層の変性及び菲薄化が認められた。
実施例124、実施例128、実施例129:100および200mg/kg投与群全例(8例)において網膜に組織学的変化は認められなかった。
(試験例5:抗腫瘍試験)
NIH−3T3−Axl#7(全長Axl cDNAを挿入したpLXSNレトロウイルスをNIH3T3細胞にトランスフェクトして作製された細胞)をNOG雌性マウスにBlock移植し、推定腫瘍体積が約500mm3に達した時点で、Compound A 1塩酸塩2水和物(25、4.2、0.7mg/kg)、実施例124(50、12.5、3.1、0.8mg/kg)および実施例128(50、12.5、3.1、0.8mg/kg)を1日2回(bid)で5連投した(経口投与)。Compound Aは25及び4.2mg/kgで明確な腫瘍縮退効果を示し、実施例124及び実施例128は3.1mg/kgで投与期間中ほぼ完全に腫瘍増殖を抑制し、50および12.5mg/kgで明確な腫瘍縮退効果を示した。
(試験例6:erlotinibとのin vivo併用効果の検討)
EGFR遺伝子exon 19に欠失変異を有し、EGFR阻害薬に高感受性を示すHCC827肺癌細胞をリン酸緩衝生理食塩水を用いて5×107cells/mLになるよう懸濁し、調製した細胞懸濁液をヌードマウス(雌性、5週齢)の皮下に0.1mL移植した。大部分のマウスの推定腫瘍体積が約450mm3に達した時点(腫瘍移植後52日目)で腫瘍体積値による群分けを行い、erlotinib(LC Laboratories)を25mg/kg(1日1回:qd)もしくはN−[4−(2−アミノ−5−{4−[(2R)−1,4−ジオキサン−2−イルメトキシ]−3−メトキシフェニル}ピリジン−3−イル)−3−フルオロフェニル]−5−メチル−4’−オキソ−1’−(テトラヒドロ−2H−ピラン−4−イルメチル)−1’,4’−ジヒドロ−2,3’−ビピリジン−5’−カルボキサミド(Compound B)硫酸塩水和物を50mg/kg(1日2回:bid)を強制経口投与した(単独投与もしくは両剤の併用投与)。投与は群分け翌日(Day1)から週5日の割合(土日休薬)で行い、Vehicle及びCompound B硫酸塩水和物単独投与群はDay46、erlotinib単独投与群はDay86、併用投与群はDay106まで行なった。経時的に腫瘍の長径(mm)および短径(mm)を電子デジタルノギスで計測し、以下に示す計算式(1)により推定腫瘍体積を算出して図を作成した。また経時的に小動物用自動天秤を用いて体重を測定し、以下に示す計算式(2)により体重変化率(Body weight change %)を算出して薬剤投与の体重への影響を検討すると共に、直近の体重測定結果を投与量算出に用いた。
Estimated Tumor Volume (mm3) = 各個体の推定腫瘍体積の平均値・・・(1)
各個体の推定腫瘍体積 = An×Bn2/2
An:n日目の腫瘍の長径
Bn:n日目の腫瘍の短径
Body weight change(%) = 各個体の体重変化率の平均値・・・(2)
各個体の体重変化率 = (1−BWn/BWs)×100
BWn:n日目の体重
BWs:投与開始日の体重
erlotinib単剤では投与後直ちに腫瘍退縮効果が確認されたが、その後耐性を獲得し、Day77には投与開始時の大きさを上回った。しかしながら、Compound B硫酸塩水和物との併用群ではDay107においても投与開始時の大きさには達しなかった(図19−1)。erlotinibとCompound B硫酸塩水和物との併用群において、特に顕著な体重減少の低下は見られなかった(図19−2)。
(試験例7:erlotinib投与によるAXL発現上昇の検討)
HCC827肺癌細胞をリン酸緩衝生理食塩水を用いて5×107cells/mLになるよう懸濁し、調製した細胞懸濁液をヌードマウス(雌性、5週齢)の皮下に0.1mL移植した。大部分のマウスの推定腫瘍体積が約450mm3に達した時点(腫瘍移植後52日目)で腫瘍体積値による群分けを行い(Day0)、erlotinib(LC Laboratories)を25mg/kg、12.5mg/kg、或いは6.25mg/kgを週5日の割合(土日休薬)で強制経口投与を行った。Vehicle投与群はDay46、erlotinib投与群はDay86まで行なった。経時的に腫瘍の長径(mm)および短径(mm)を電子デジタルノギスで計測し、以下に示す計算式(1)により推定腫瘍体積を算出して図を作成した(図20−1)。各群2−6匹の腫瘍塊からCell lysateを調製し、ウエスタンブロットによりAXLの発現を確認し、個々のバンドをImageQuantLAS4000(GEヘルスケア)により定量化した(図20−2)。Cell lysateの調製、及びウエスタンブロットについては次のように行なった。100mg前後の腫瘍塊をフォスファターゼ阻害剤(ロシュアダイアグノスティックス、04 906 837 001)とプロテアーゼ阻害剤(ロシュアダイアグノスティックス、11 836 153 001)を含んだLysis Buffer(Cell signaling technology、9803)中でホモジナイズし、上清をLysateサンプルとした。このサンプルの蛋白質をバッファー(Life technologies、NP0008およびNP0009)と熱変性で固定しウエスタンブロットに供した。15μgのサンプルを電気泳動し、ニトロセルロース膜に転写し、1時間のブロッキングの後にウサギ抗AXL抗体(Cell signaling technology、9803、1/1000)またはウサギ抗Actin抗体(Santa Cruz Biotechnology、SC−1616、1/2000)と冷蔵下で一晩反応させた。トリス緩衝生理食塩水で洗浄後、HRP標識抗ウサギIgG抗体(Cell signaling technology、7074、1/2000)と室温下で1時間反応させ、トリス緩衝生理食塩水で洗浄した後にHRP基質(Merck Millipore、WBLUF0500)で発光させた。
ウエスタンブロットによりAXLの発現を確認し、個々のバンドをImageQuantLAS4000により定量化した結果、erlotinib投与群でAXLの発現が有意に上昇している事がWelch testにより確認された。
(試験例8:erlotinibとのin vivo併用効果の検討2)
HCC827肺癌細胞をリン酸緩衝生理食塩水を用いて4×107cells/mLになるよう懸濁し、調製した細胞懸濁液をヌードマウス(雌性、5週齢)の皮下に0.1mL移植した。腫瘍細胞移植後52日目に、推定腫瘍体積200mm3以上700mm3未満のマウス(86匹/129匹)にerlotinib(LC Laboratories)を25mg/kg(1日1回:qd)強制経口投与した。投与は群分け翌日(移植後53日目)から週5日の割合(土日休薬)で行い、腫瘍が一旦退縮し、明確に再増殖が確認された担癌マウスを用いて群分けを実施し(6匹/群、移植後115日目)、グループ1:erlotinib+vehicle投与群(平均推定腫瘍体積275mm3)、グループ2:erlotinib+Compound B硫酸塩水和物50mg/kg(平均推定腫瘍体積284mm3)、グループ3:erlotinib+Compound B硫酸塩水和物25mg/kg(平均推定腫瘍体積268mm3)の3群に分けてerlotinibで耐性化した腫瘍に対してCompound B硫酸塩水和物の併用によりerlotinibの効果が再び見られるようになるかを検討した。Compound B硫酸塩水和物の投与は移植後116日目から1日2回投与(bid)を週5日の割合(土日休薬)で継続した。グループ1では当初の移植時と同レベルの推定腫瘍体積まで腫瘍が増殖したが、グループ2およびグループ3ではerlotinibによる明確な増殖阻害効果が確認された。グループ2では移植後140日目においても併用投与を開始した移植後116日目の推定腫瘍体積が維持された。
Claims (53)
- 一般式(I)

W、X、Yは、それぞれ独立して窒素原子、C−H、C−FまたはC−Clを示し、Zは、窒素原子、C−H、C−F、C−ClまたはC−C1~C6アルキル基を示し、R1は、下記式(II−1)または(II−2)

QおよびTは、それぞれ独立して、窒素原子、C−HまたはC−Fを示し、
Tは、窒素原子またはC−Hを示し、
Uは、窒素原子またはC−Hを示し、
R6は、ハロゲン原子、C1~C6アルキル基、C3~C6シクロアルキル基、シアノ基またはトリフルオロメトキシ基を示し、
式(II−2)中、
Vは、硫黄原子または酸素原子を示し、
R7は、C1~C6アルキル基を示す。)を示し、
R2は、水素原子、ハロゲン原子、C1~C6アルキル基、C1~C6アルコキシ基またはシアノ基を示し、
R3は、水素原子またはC1~C6アルキル基を示し、
R4は、水素原子、ハロゲン原子またはC1~C6アルキル基を示し、
R5は、水素原子または下記式(III−1)、(III−2)、(III−3)もしくは(III−4)

R8およびR12は、それぞれ独立して水素原子または重水素原子を示し、
R9は、水素原子、ハロゲン原子またはC1~C6アルコキシ基を示し、
R10は、水素原子、ヘテロシクロアルキル基で置換されていてもよいC1~C6アルキル基またはC1~C6アルキル基で置換されていてもよいヘテロシクロアルキル基で置換されていてもよいC1~C6アルコキシ基を示し、
R11は、水素原子またはC1~C6アルコキシ基または重水素で置換されたC1~C6アルコキシ基を示し、
式(III−2)中、
R13は、ヘテロシクロアルキル基で置換されていてもよいC1~C6アルキル基またはヘテロシクロアルキル基を示し、
式(III−3)中、
R14は、水素原子またはC1~C6アルキル基を示し、
R15は、水素原子、C1~C6アルキル基またはC1~C6アルコキシ基を示し、
R16は、水素原子またはハロゲン原子を示し、
式(III−4)中、
R17は、C1~C6アルコキシ基を示し、
R18は、C1~C6アルコキシ基を示す。)を示す。]
で表される化合物またはその薬学的に許容される塩。 - 下記式

- 下記式
- 下記式
- 下記式

- 下記式

- 下記式

- 下記式

- 下記式

- 下記式

- 請求項2に記載の化合物の臭化水素酸塩、硝酸塩、硫酸塩、リン酸塩、メタンスルホン酸塩、エタンスルホン酸塩、ベンゼンスルホン酸塩またはp−トルエンスルホン酸塩。
- 請求項4に記載の化合物のメタンスルホン酸塩。
- 請求項5に記載の化合物のメタンスルホン酸塩、リン酸塩、ナフタレン−1,5−ジスルホン酸塩または硫酸塩。
- 銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.74、7.56、8.96、11.38、12.36、14.78、15.60、16.16、18.70および24.10に特徴的ピークを示す請求項11に記載のメタンスルホン酸塩の結晶。
- 銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.84、7.72、9.40、11.62、14.92、15.48、16.70、18.88、19.32および24.40に特徴的ピークを示す請求項11に記載の臭化水素酸塩の結晶。
- 銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.82、7.66、9.28、9.52、11.54、15.26、15.54、16.62、19.24および24.56に特徴的ピーク示す請求項11に記載の硝酸塩の結晶。
- 銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.74、7.56、8.92、9.58、11.36、12.38、14.68,15.64、16.06および24.38に特徴的ピーク示す請求項11に記載の硫酸塩の結晶。
- 銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.74、7.56、8.80、9.56、11.34、14.56、15.74、23.68、24.34および24.68に特徴的ピーク示す請求項11に記載のリン酸塩の結晶。
- 銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=6.72、7.90、12.02、13.40、16.90、17.88、19.00、19.80、21.26および24.18に特徴的ピーク示す請求項11に記載のエタンスルホン酸塩の結晶。
- 銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=9.22、10.60、10.82、11.10、13.40、15.78、17.50、18.66、21.02および26.10に特徴的ピーク示す請求項11に記載のベンゼンスルホン酸塩の結晶。
- 銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=4.18、5.12、13.44、14.98、16.96、17.44、18.92、19.72、20.16および23.04に特徴的ピーク示す請求項11に記載のp−トルエンスルホン酸塩の結晶。
- 銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=4.28、8.42、8.64、10.54、12.72、13.48、15.90、17.00.17.46および21.26に特徴的ピーク示す請求項13に記載のリン酸塩の結晶。
- 銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.66、6.42、7.32、9.76、11.00、12.88、18.42、19.62、20.54および24.22に特徴的ピーク示す請求項13に記載の硫酸塩の結晶。
- 銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.64、6.40、7.32、9.76、17.38、18.42、19.64、20.56、22.90および24.20に特徴的ピーク示す請求項13に記載の硫酸塩の結晶。
- 銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.64、6.40、7.30、9.76、17.34、18.38、19.34、20.56、21.52および22.94に特徴的ピーク示す請求項13に記載の硫酸塩の結晶。
- 銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.62、6.38、7.28、9.74、17.30、18.36、19.54、20.52、22.86および24.14に特徴的ピーク示す請求項13に記載の硫酸塩の結晶。
- 銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.64、6.40、7.30、9.76、12.86、18.40、19.62、20.54、22.92および24.20に特徴的ピーク示す請求項13に記載の硫酸塩の結晶。
- 銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=3.64、6.36、7.30、18.36、19.04、19.42、19.70、20.12、20.42および21.32に特徴的ピーク示す請求項13に記載の硫酸塩の結晶。
- 銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=5.62、7.18、9.22、10.36、15.56、16.40および20.86に特徴的ピーク示す請求項13に記載の硫酸塩の結晶。
- 銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=6.14、6.98、11.24、14.84、17.48、19.54、20.94、22.38、23.20および24.70に特徴的ピーク示す請求項13に記載のナフタレン−1,5−ジスルホン酸塩の結晶。
- 銅のKα線(波長λ=1.54オングストローム)の照射で得られる粉末X線回折図において、回折角度2θ=9.24、9.58、14.00、14.46、16.70、17.02、18.22、20.24、21.64および25.52に特徴的ピーク示す請求項13に記載のナフタレン−1,5−ジスルホン酸塩の結晶。
- 請求項1から10のいずれか1項に記載の化合物またはその薬学的に許容される塩を含む、Axl阻害剤。
- 請求項1から10のいずれか1項に記載の化合物またはその薬学的に許容される塩を有効成分とする医薬。
- 請求項1から10のいずれか1項に記載の化合物またはその薬学的に許容される塩を有効成分とするAxlキナーゼ機能亢進を原因とする疾患、Axlキナーゼ機能亢進と関連する疾患、および/またはAxlキナーゼ機能亢進を伴う疾患の治療のための医薬。
- 請求項1から10のいずれか1項に記載の化合物またはその薬学的に許容される塩を有効成分とする過剰増殖性疾患の治療のための医薬。
- 請求項1から10のいずれか1項に記載の化合物またはその薬学的に許容される塩を有効成分とする癌の治療のための医薬。
- 請求項1から10のいずれか1項に記載の化合物またはその薬学的に許容される塩を有効成分とする癌の転移予防のための医薬。
- 請求項1から10のいずれか1項に記載の化合物またはその薬学的に許容される塩を有効成分とする癌の薬剤耐性解除のための医薬。
- 請求項1から10のいずれか1項に記載の化合物またはその薬学的に許容される塩を有効成分とする癌の薬剤耐性獲得阻害のための医薬。
- 癌が、乳癌、結腸癌、前立腺癌、肺癌、胃癌、卵巣癌、子宮内膜癌、腎癌、肝細胞癌、甲状腺癌、子宮癌、食道癌、扁平上皮癌、白血病、骨肉腫、メラノーマ、膠芽細胞腫、神経芽細胞腫および膵臓癌から選択されるものである請求項36から39のいずれか1項に記載の医薬。
- 請求項1から10のいずれか1項に記載の化合物またはその薬学的に許容される塩および薬学的に許容し得る担体を含有する医薬組成物。
- 請求項1から10のいずれか1項に記載の化合物またはその薬学的に許容される塩を用いることを特徴とするAxlキナーゼ機能亢進を原因とする疾患、Axlキナーゼ機能亢進と関連する疾患、および/またはAxlキナーゼ機能亢進を伴う疾患の治療方法。
- 請求項1から10のいずれか1項に記載の化合物またはその薬学的に許容される塩を用いることを特徴とする過剰増殖性疾患の治療方法。
- 請求項1から10のいずれか1項に記載の化合物またはその薬学的に許容される塩を用いることを特徴とする癌の治療方法。
- 請求項1から10のいずれか1項に記載の化合物またはその薬学的に許容される塩を用いることを特徴とする癌の転移予防方法。
- 請求項1から10のいずれか1項に記載の化合物またはその薬学的に許容される塩を用いることを特徴とする癌の薬剤耐性解除方法。
- 請求項1から10のいずれか1項に記載の化合物またはその薬学的に許容される塩を用いることを特徴とする癌の薬剤耐性獲得の阻害方法。
- 癌が、乳癌、結腸癌、前立腺癌、肺癌、胃癌、卵巣癌、子宮内膜癌、腎癌、肝細胞癌、甲状腺癌、子宮癌、食道癌、扁平上皮癌、白血病、骨肉腫、メラノーマ、膠芽細胞腫および神経芽細胞腫から選択されるものである請求項44から47のいずれか1項に記載の方法。
- 請求項1から10のいずれか1項に記載の化合物またはその薬学的に許容される塩の、医薬製造のための使用。
- 請求項1から10のいずれか1項に記載の化合物またはその薬学的に許容される塩の、癌の治療のための使用。
- 請求項1から10のいずれか1項に記載の化合物またはその薬学的に許容される塩の、癌の転移予防のための使用。
- 請求項1から10のいずれか1項に記載の化合物またはその薬学的に許容される塩の、癌の薬剤耐性解除のための使用。
- 請求項1から10のいずれか1項に記載の化合物またはその薬学的に許容される塩の、癌の薬剤耐性獲得の阻害のための使用。
Priority Applications (30)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010427596.9A CN111467346B (zh) | 2014-07-07 | 2015-07-06 | 具有四氢吡喃基甲基的吡啶酮衍生物 |
| CA2954488A CA2954488C (en) | 2014-07-07 | 2015-07-06 | Pyridone derivative having tetrahydropyranylmethyl group |
| NZ728116A NZ728116A (en) | 2014-07-07 | 2015-07-06 | Pyridone derivative having tetrahydropyranylmethyl group |
| MX2017000330A MX2017000330A (es) | 2014-07-07 | 2015-07-06 | Derivado de piridona que tiene un grupo tetrahidropiranilmetilo. |
| ES15818440T ES2875360T3 (es) | 2014-07-07 | 2015-07-06 | Derivado de piridona que tiene un grupo tetrahidropiranil metilo |
| KR1020177000534A KR102348190B1 (ko) | 2014-07-07 | 2015-07-06 | 테트라하이드로피라닐메틸기를 갖는 피리돈 유도체 |
| JP2016532991A JP6086516B2 (ja) | 2014-07-07 | 2015-07-06 | テトラヒドロピラニルメチル基を有するピリドン誘導体 |
| BR112017000242-6A BR112017000242A2 (ja) | 2014-07-07 | 2015-07-06 | A pyridone derivative which has a tetrahydropyranyl methyl group |
| PL15818440T PL3168219T3 (pl) | 2014-07-07 | 2015-07-06 | Pochodna pirydonu z grupą tetrahydropiranylometylu |
| SI201531589T SI3168219T1 (sl) | 2014-07-07 | 2015-07-06 | Derivati piridona, ki imajo tetrahidropiranil metilno skupino |
| EP21166082.4A EP3868757A1 (en) | 2014-07-07 | 2015-07-06 | Pyridone derivative having tetrahydropyranylmethyl group |
| RU2017103753A RU2707953C2 (ru) | 2014-07-07 | 2015-07-06 | Производное пиридона, имеющее тетрагидропиранилметильную группу |
| DK15818440.8T DK3168219T3 (da) | 2014-07-07 | 2015-07-06 | Pyridonderivat med tetrahydropyranylmethylgruppe |
| MYPI2017000012A MY194307A (en) | 2014-07-07 | 2015-07-06 | Pyridone derivative having tetrahydropyranylmethyl group |
| RS20210707A RS61941B1 (sr) | 2014-07-07 | 2015-07-06 | Derivat piridona koji ima tetrahidropiranil metil grupu |
| US15/036,981 US10442797B2 (en) | 2014-07-07 | 2015-07-06 | Pyridone derivatives having tetrahydropyranylmethyl groups |
| AU2015288648A AU2015288648B2 (en) | 2014-07-07 | 2015-07-06 | Pyridone derivative having tetrahydropyranyl methyl group |
| SG11201700063RA SG11201700063RA (en) | 2014-07-07 | 2015-07-06 | Pyridone derivative having tetrahydropyranylmethyl group |
| CN201580047744.1A CN106604920B (zh) | 2014-07-07 | 2015-07-06 | 具有四氢吡喃基甲基的吡啶酮衍生物 |
| EP15818440.8A EP3168219B1 (en) | 2014-07-07 | 2015-07-06 | Pyridone derivative having tetrahydropyranyl methyl group |
| LTEP15818440.8T LT3168219T (lt) | 2014-07-07 | 2015-07-06 | Piridono dariniai, turintys tetrahidropiranilmetilo grupę |
| IL249948A IL249948B (en) | 2014-07-07 | 2017-01-05 | A pyridone derivative with a tetrahydropyranylmethyl group |
| PH12017500048A PH12017500048A1 (en) | 2014-07-07 | 2017-01-06 | Pyridone derivative having tetrahydropyranylmethyl group |
| ZA2017/00139A ZA201700139B (en) | 2014-07-07 | 2017-01-06 | Pyridone derivative having tetrahydropyranylmethyl group |
| CONC2017/0001178A CO2017001178A2 (es) | 2014-07-07 | 2017-02-07 | Derivado de piridona que tiene un grupo tetrahidropiranil metilo |
| US16/575,177 US11208403B2 (en) | 2014-07-07 | 2019-09-18 | Pyridone derivatives having tetrahydropyranylmethyl groups |
| IL278124A IL278124B (en) | 2014-07-07 | 2020-10-18 | Axl depressant pyridone derivative for use in the treatment of the disease |
| HRP20210899TT HRP20210899T1 (hr) | 2014-07-07 | 2021-06-07 | Derivat piridona koji ima tetrahidropiranil metil grupu |
| CY20211100586T CY1124537T1 (el) | 2014-07-07 | 2021-06-30 | Παραγωγο πυριδονης που εχει τετραϋδροπυρανυλ μεθυλ ομαδα |
| US17/400,795 US20220002277A1 (en) | 2014-07-07 | 2021-08-12 | Pyridone derivative having tetrahydropyranylmethyl group |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014139628 | 2014-07-07 | ||
| JP2014-139628 | 2014-07-07 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/036,981 A-371-Of-International US10442797B2 (en) | 2014-07-07 | 2015-07-06 | Pyridone derivatives having tetrahydropyranylmethyl groups |
| US16/575,177 Continuation US11208403B2 (en) | 2014-07-07 | 2019-09-18 | Pyridone derivatives having tetrahydropyranylmethyl groups |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016006706A1 true WO2016006706A1 (ja) | 2016-01-14 |
Family
ID=55064330
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/069976 WO2016006706A1 (ja) | 2014-07-07 | 2015-07-06 | テトラヒドロピラ二ルメチル基を有するピリドン誘導体 |
Country Status (29)
| Country | Link |
|---|---|
| US (3) | US10442797B2 (ja) |
| EP (2) | EP3168219B1 (ja) |
| JP (2) | JP6086516B2 (ja) |
| KR (1) | KR102348190B1 (ja) |
| CN (2) | CN106604920B (ja) |
| AU (1) | AU2015288648B2 (ja) |
| BR (1) | BR112017000242A2 (ja) |
| CA (1) | CA2954488C (ja) |
| CO (1) | CO2017001178A2 (ja) |
| CY (1) | CY1124537T1 (ja) |
| DK (1) | DK3168219T3 (ja) |
| ES (1) | ES2875360T3 (ja) |
| HR (1) | HRP20210899T1 (ja) |
| HU (1) | HUE054534T2 (ja) |
| IL (2) | IL249948B (ja) |
| LT (1) | LT3168219T (ja) |
| MX (1) | MX2017000330A (ja) |
| MY (1) | MY194307A (ja) |
| NZ (1) | NZ728116A (ja) |
| PH (1) | PH12017500048A1 (ja) |
| PL (1) | PL3168219T3 (ja) |
| PT (1) | PT3168219T (ja) |
| RS (1) | RS61941B1 (ja) |
| RU (1) | RU2707953C2 (ja) |
| SG (2) | SG11201700063RA (ja) |
| SI (1) | SI3168219T1 (ja) |
| TW (2) | TWI690525B (ja) |
| WO (1) | WO2016006706A1 (ja) |
| ZA (1) | ZA201700139B (ja) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017146236A1 (ja) | 2016-02-26 | 2017-08-31 | 小野薬品工業株式会社 | Axl阻害剤と免疫チェックポイント阻害剤とを組み合わせて投与することを特徴とする癌治療のための医薬 |
| WO2018121228A1 (zh) * | 2016-12-28 | 2018-07-05 | 中国科学院上海药物研究所 | 一种具有axl抑制活性的化合物及其制备和应用 |
| WO2018174219A1 (ja) | 2017-03-24 | 2018-09-27 | 第一三共株式会社 | Axl阻害剤とEGFRチロシンキナーゼ阻害薬との併用治療法 |
| WO2019039525A1 (ja) | 2017-08-23 | 2019-02-28 | 小野薬品工業株式会社 | Axl阻害剤を有効成分として含むがん治療剤 |
| WO2019074116A1 (ja) | 2017-10-13 | 2019-04-18 | 小野薬品工業株式会社 | Axl阻害剤を有効成分として含む固形がん治療剤 |
| WO2020009132A1 (ja) | 2018-07-04 | 2020-01-09 | 第一三共株式会社 | ジアリールピリジン誘導体の製造方法 |
| WO2020188015A1 (en) | 2019-03-21 | 2020-09-24 | Onxeo | A dbait molecule in combination with kinase inhibitor for the treatment of cancer |
| WO2021089791A1 (en) | 2019-11-08 | 2021-05-14 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for the treatment of cancers that have acquired resistance to kinase inhibitors |
| WO2021148581A1 (en) | 2020-01-22 | 2021-07-29 | Onxeo | Novel dbait molecule and its use |
| WO2022220227A1 (ja) * | 2021-04-14 | 2022-10-20 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | テトラヒドロピリドピリミジン化合物 |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI690525B (zh) | 2014-07-07 | 2020-04-11 | 日商第一三共股份有限公司 | 具有四氫吡喃基甲基之吡啶酮衍生物及其用途 |
| US11919902B2 (en) | 2017-12-22 | 2024-03-05 | Hibercell, Inc. | Aryl-bipyridine amine derivatives as phosphatidylinositol phosphate kinase inhibitors |
| CN110041316B (zh) * | 2018-01-17 | 2022-04-19 | 药捷安康(南京)科技股份有限公司 | Tam家族激酶/和csf1r激酶抑制剂及其用途 |
| KR20200128531A (ko) * | 2018-01-29 | 2020-11-13 | 카풀루스 테라퓨틱스, 엘엘씨 | 6원 중심 고리를 포함하는 srebp 억제제 |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001070726A1 (en) * | 2000-03-23 | 2001-09-27 | Sanofi-Synthelabo | Aminophenyl pyrimidone derivatives |
| WO2009047514A1 (en) * | 2007-10-10 | 2009-04-16 | Cancer Research Technology Limited | [1,2,4]triazolo[1,5-a]pyridine and [1,2,4]triazolo[1,5-c]pyrimidine compounds and their use |
| JP2010533159A (ja) * | 2007-07-09 | 2010-10-21 | アストラゼネカ アクチボラグ | 化合物947 |
| WO2010126914A1 (en) * | 2009-04-27 | 2010-11-04 | Elan Pharmaceuticals, Inc. | Pyridinone antagonists of alpha-4 integrins |
| JP2011500778A (ja) * | 2007-10-25 | 2011-01-06 | アストラゼネカ・アクチエボラーグ | ピリジン及びピラジン誘導体−083 |
| JP2011500801A (ja) * | 2007-10-26 | 2011-01-06 | ライジェル ファーマシューティカルズ, インコーポレイテッド | Axl阻害剤として有用な多環アリール置換トリアゾール及び多環ヘテロアリール置換トリアゾール |
| WO2013115280A1 (ja) * | 2012-01-31 | 2013-08-08 | 第一三共株式会社 | ピリドン誘導体 |
Family Cites Families (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0319241D0 (en) | 2003-08-15 | 2003-09-17 | Hotpods Ltd | Oven and a food delivery vehicle comprising said oven |
| WO2007030680A2 (en) | 2005-09-07 | 2007-03-15 | Rigel Pharmaceuticals, Inc. | Triazole derivatives useful as axl inhibitors |
| WO2007057399A2 (en) | 2005-11-15 | 2007-05-24 | Boehringer Ingelheim International Gmbh | Treatment of cancer with indole derivatives |
| US7825137B2 (en) | 2005-12-05 | 2010-11-02 | Pfizer Inc. | Method of treating abnormal cell growth |
| ES2562428T3 (es) | 2005-12-15 | 2016-03-04 | Rigel Pharmaceuticals, Inc. | Inhibidores de cinasa y sus usos |
| EP1900727A1 (en) | 2006-08-30 | 2008-03-19 | Cellzome Ag | Aminopyridine derivatives as kinase inhibitors |
| WO2008045978A1 (en) | 2006-10-10 | 2008-04-17 | Rigel Pharmaceuticals, Inc. | Pinane-substituted pyrimidinediamine derivatives useful as axl inhibitors |
| GB0625659D0 (en) | 2006-12-21 | 2007-01-31 | Cancer Rec Tech Ltd | Therapeutic compounds and their use |
| US7872000B2 (en) | 2006-12-29 | 2011-01-18 | Rigel Pharmaceuticals, Inc. | Bicyclic aryl and bicyclic heteroaryl substituted triazoles useful as Axl inhibitors |
| CA2946305C (en) | 2006-12-29 | 2019-09-17 | Rigel Pharmaceuticals, Inc. | Substituted triazoles useful as axl inhibitors |
| PL2114955T3 (pl) | 2006-12-29 | 2013-06-28 | Rigel Pharmaceuticals Inc | Triazole podstawione mostkowanym bicyklicznym arylem oraz mostkowanym bicyklicznym heteroarylem użyteczne jako inhibitory Axl |
| CA2710234C (en) | 2006-12-29 | 2015-10-20 | Rigel Pharmaceuticals, Inc. | Polycyclic heteroaryl substituted triazoles useful as axl inhibitors |
| ES2672172T3 (es) | 2006-12-29 | 2018-06-12 | Rigel Pharmaceuticals, Inc. | Triazoles N3-heteroarilsustituidos y triazoles N5-heteroarilsustituidos útiles como inhibidores de Axl |
| CN101679313A (zh) | 2007-04-13 | 2010-03-24 | 休普基因公司 | 用于治疗癌症或过度增殖性疾病的axl激酶抑制剂 |
| GB0713259D0 (en) | 2007-07-09 | 2007-08-15 | Astrazeneca Ab | Pyrazine derivatives 954 |
| WO2009024825A1 (en) | 2007-08-21 | 2009-02-26 | Astrazeneca Ab | 2-pyrazinylbenzimidazole derivatives as receptor tyrosine kinase inhibitors |
| RU2010120536A (ru) * | 2007-10-25 | 2011-11-27 | Астразенека Аб (Se) | Производные пиридина и пиразина, полезные для лечения клеточных проферативных расстройств |
| MX2010004967A (es) | 2007-11-02 | 2010-07-30 | Janssen Pharmaceutica Nv | Uso de inhibidor de cfms para el tratamiento y la prevención del cancer de hueso, así como la perdida osea y el dolor de hueso asociados con el cancer de hueso. |
| NZ586630A (en) | 2008-01-23 | 2011-12-22 | Bristol Myers Squibb Co | 4-Pyridinone compounds and their use for cancer |
| CN101977905B (zh) | 2008-01-23 | 2014-07-02 | 百时美施贵宝公司 | 4-吡啶酮化合物及其对于癌症的用途 |
| CN102131783A (zh) | 2008-04-16 | 2011-07-20 | 马克斯普朗克科学发展组织 | 作为axl激酶抑制剂的喹啉衍生物 |
| UY31800A (es) | 2008-05-05 | 2009-11-10 | Smithkline Beckman Corp | Metodo de tratamiento de cancer usando un inhibidor de cmet y axl y un inhibidor de erbb |
| WO2009138799A1 (en) | 2008-05-14 | 2009-11-19 | Astex Therapeutics Limited | Therapeutic uses of 1-cycl0pr0pyl-3 - [3- ( 5 -morpholin- 4 -ylmethyl- 1h-benz0imidaz0l- 2 -yl) -lh-pyrazol-4-yl] -urea |
| PL2328888T3 (pl) | 2008-07-09 | 2013-04-30 | Rigel Pharmaceuticals Inc | Triazole podstawione mostkowanym bicyklicznym heteroarylem użyteczne jako inhibitory AXL |
| EP2326641B1 (en) | 2008-07-09 | 2014-09-03 | Rigel Pharmaceuticals, Inc. | Polycyclic heteroaryl substituted triazoles useful as axl inhibitors |
| EP2393814A1 (en) | 2009-02-09 | 2011-12-14 | SuperGen, Inc. | Pyrrolopyrimidinyl axl kinase inhibitors |
| EP2311809A1 (en) | 2009-10-16 | 2011-04-20 | Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. | Quinolinyloxyphenylsulfonamides |
| AR080754A1 (es) | 2010-03-09 | 2012-05-09 | Janssen Pharmaceutica Nv | Derivados de imidazo (1,2-a) pirazina y su uso como inhibidores de pde10 |
| WO2012121939A2 (en) | 2011-03-04 | 2012-09-13 | Locus Pharmaceuticals, Inc. | Aminopyrazine compounds |
| WO2013162061A1 (ja) | 2012-04-26 | 2013-10-31 | 第一三共株式会社 | 二環性ピリミジン化合物 |
| TWI690525B (zh) | 2014-07-07 | 2020-04-11 | 日商第一三共股份有限公司 | 具有四氫吡喃基甲基之吡啶酮衍生物及其用途 |
-
2015
- 2015-07-03 TW TW104121592A patent/TWI690525B/zh active
- 2015-07-03 TW TW108136526A patent/TWI723572B/zh active
- 2015-07-06 WO PCT/JP2015/069976 patent/WO2016006706A1/ja active Application Filing
- 2015-07-06 ES ES15818440T patent/ES2875360T3/es active Active
- 2015-07-06 RS RS20210707A patent/RS61941B1/sr unknown
- 2015-07-06 EP EP15818440.8A patent/EP3168219B1/en active Active
- 2015-07-06 KR KR1020177000534A patent/KR102348190B1/ko active IP Right Grant
- 2015-07-06 LT LTEP15818440.8T patent/LT3168219T/lt unknown
- 2015-07-06 JP JP2016532991A patent/JP6086516B2/ja active Active
- 2015-07-06 MY MYPI2017000012A patent/MY194307A/en unknown
- 2015-07-06 PL PL15818440T patent/PL3168219T3/pl unknown
- 2015-07-06 HU HUE15818440A patent/HUE054534T2/hu unknown
- 2015-07-06 CA CA2954488A patent/CA2954488C/en active Active
- 2015-07-06 SI SI201531589T patent/SI3168219T1/sl unknown
- 2015-07-06 SG SG11201700063RA patent/SG11201700063RA/en unknown
- 2015-07-06 DK DK15818440.8T patent/DK3168219T3/da active
- 2015-07-06 BR BR112017000242-6A patent/BR112017000242A2/ja active Search and Examination
- 2015-07-06 MX MX2017000330A patent/MX2017000330A/es unknown
- 2015-07-06 CN CN201580047744.1A patent/CN106604920B/zh active Active
- 2015-07-06 SG SG10201900144TA patent/SG10201900144TA/en unknown
- 2015-07-06 NZ NZ728116A patent/NZ728116A/en unknown
- 2015-07-06 RU RU2017103753A patent/RU2707953C2/ru active
- 2015-07-06 AU AU2015288648A patent/AU2015288648B2/en active Active
- 2015-07-06 CN CN202010427596.9A patent/CN111467346B/zh active Active
- 2015-07-06 US US15/036,981 patent/US10442797B2/en active Active
- 2015-07-06 EP EP21166082.4A patent/EP3868757A1/en active Pending
- 2015-07-06 PT PT158184408T patent/PT3168219T/pt unknown
-
2017
- 2017-01-05 IL IL249948A patent/IL249948B/en active IP Right Grant
- 2017-01-06 ZA ZA2017/00139A patent/ZA201700139B/en unknown
- 2017-01-06 PH PH12017500048A patent/PH12017500048A1/en unknown
- 2017-02-01 JP JP2017016682A patent/JP6203439B2/ja active Active
- 2017-02-07 CO CONC2017/0001178A patent/CO2017001178A2/es unknown
-
2019
- 2019-09-18 US US16/575,177 patent/US11208403B2/en active Active
-
2020
- 2020-10-18 IL IL278124A patent/IL278124B/en unknown
-
2021
- 2021-06-07 HR HRP20210899TT patent/HRP20210899T1/hr unknown
- 2021-06-30 CY CY20211100586T patent/CY1124537T1/el unknown
- 2021-08-12 US US17/400,795 patent/US20220002277A1/en active Pending
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001070726A1 (en) * | 2000-03-23 | 2001-09-27 | Sanofi-Synthelabo | Aminophenyl pyrimidone derivatives |
| JP2010533159A (ja) * | 2007-07-09 | 2010-10-21 | アストラゼネカ アクチボラグ | 化合物947 |
| WO2009047514A1 (en) * | 2007-10-10 | 2009-04-16 | Cancer Research Technology Limited | [1,2,4]triazolo[1,5-a]pyridine and [1,2,4]triazolo[1,5-c]pyrimidine compounds and their use |
| JP2011500778A (ja) * | 2007-10-25 | 2011-01-06 | アストラゼネカ・アクチエボラーグ | ピリジン及びピラジン誘導体−083 |
| JP2011500801A (ja) * | 2007-10-26 | 2011-01-06 | ライジェル ファーマシューティカルズ, インコーポレイテッド | Axl阻害剤として有用な多環アリール置換トリアゾール及び多環ヘテロアリール置換トリアゾール |
| WO2010126914A1 (en) * | 2009-04-27 | 2010-11-04 | Elan Pharmaceuticals, Inc. | Pyridinone antagonists of alpha-4 integrins |
| WO2013115280A1 (ja) * | 2012-01-31 | 2013-08-08 | 第一三共株式会社 | ピリドン誘導体 |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017146236A1 (ja) | 2016-02-26 | 2017-08-31 | 小野薬品工業株式会社 | Axl阻害剤と免疫チェックポイント阻害剤とを組み合わせて投与することを特徴とする癌治療のための医薬 |
| WO2018121228A1 (zh) * | 2016-12-28 | 2018-07-05 | 中国科学院上海药物研究所 | 一种具有axl抑制活性的化合物及其制备和应用 |
| US11318124B2 (en) | 2017-03-24 | 2022-05-03 | Daiichi Sankyo Company, Limited | Combination therapy of Axl inhibitor and EGFR tyrosine kinase inhibitor |
| WO2018174219A1 (ja) | 2017-03-24 | 2018-09-27 | 第一三共株式会社 | Axl阻害剤とEGFRチロシンキナーゼ阻害薬との併用治療法 |
| CN110402141A (zh) * | 2017-03-24 | 2019-11-01 | 第一三共株式会社 | Axl抑制剂和EGFR酪氨酸激酶抑制剂的联合疗法 |
| KR20190128649A (ko) | 2017-03-24 | 2019-11-18 | 다이이찌 산쿄 가부시키가이샤 | Axl 저해제와 EGFR 티로신키나아제 저해약의 병용 치료법 |
| JPWO2018174219A1 (ja) * | 2017-03-24 | 2020-01-23 | 第一三共株式会社 | Axl阻害剤とEGFRチロシンキナーゼ阻害薬との併用治療法 |
| EP3603638A4 (en) * | 2017-03-24 | 2020-09-09 | Daiichi Sankyo Company, Limited | COMBINATION THERAPY TECHNIQUE FOR AXL INHIBITOR AND EGFR TYROSINKINASE INHIBITOR |
| US11806339B2 (en) | 2017-03-24 | 2023-11-07 | Daiichi Sankyo Company, Limited | Combination therapy of Axl inhibitor and EGFR tyrosine kinase inhibitor |
| JP7293108B2 (ja) | 2017-03-24 | 2023-06-19 | 第一三共株式会社 | Axl阻害剤とEGFRチロシンキナーゼ阻害薬との併用治療法 |
| TWI796326B (zh) * | 2017-03-24 | 2023-03-21 | 日商第一三共股份有限公司 | 含有axl抑制劑與egfr酪胺酸激酶抑制藥的醫藥品及其用途 |
| WO2019039525A1 (ja) | 2017-08-23 | 2019-02-28 | 小野薬品工業株式会社 | Axl阻害剤を有効成分として含むがん治療剤 |
| WO2019074116A1 (ja) | 2017-10-13 | 2019-04-18 | 小野薬品工業株式会社 | Axl阻害剤を有効成分として含む固形がん治療剤 |
| WO2020009132A1 (ja) | 2018-07-04 | 2020-01-09 | 第一三共株式会社 | ジアリールピリジン誘導体の製造方法 |
| JPWO2020009132A1 (ja) * | 2018-07-04 | 2021-07-08 | 第一三共株式会社 | ジアリールピリジン誘導体の製造方法 |
| US20210188823A1 (en) * | 2018-07-04 | 2021-06-24 | Daiichi Sankyo Company, Limited | Method for Producing Diarylpyridine Derivatives |
| JP7337058B2 (ja) | 2018-07-04 | 2023-09-01 | 第一三共株式会社 | ジアリールピリジン誘導体の製造方法 |
| WO2020188015A1 (en) | 2019-03-21 | 2020-09-24 | Onxeo | A dbait molecule in combination with kinase inhibitor for the treatment of cancer |
| WO2021089791A1 (en) | 2019-11-08 | 2021-05-14 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for the treatment of cancers that have acquired resistance to kinase inhibitors |
| WO2021148581A1 (en) | 2020-01-22 | 2021-07-29 | Onxeo | Novel dbait molecule and its use |
| WO2022220227A1 (ja) * | 2021-04-14 | 2022-10-20 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | テトラヒドロピリドピリミジン化合物 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6203439B2 (ja) | テトラヒドロピラニルメチル基を有するピリドン誘導体 | |
| AU2013216006B2 (en) | Pyridone derivatives | |
| AU2018282363B2 (en) | Compounds and methods of use | |
| WO2013162061A1 (ja) | 二環性ピリミジン化合物 | |
| TW202031653A (zh) | 新穎雜環芳香族醯胺衍生物及含有其之醫藥 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15818440 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15036981 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2016532991 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 249948 Country of ref document: IL |
|
| ENP | Entry into the national phase |
Ref document number: 20177000534 Country of ref document: KR Kind code of ref document: A Ref document number: 2954488 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12017500048 Country of ref document: PH Ref document number: MX/A/2017/000330 Country of ref document: MX Ref document number: 1020177000534 Country of ref document: KR |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015818440 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2015818440 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112017000242 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 2015288648 Country of ref document: AU Date of ref document: 20150706 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2017103753 Country of ref document: RU Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: NC2017/0001178 Country of ref document: CO |
|
| ENP | Entry into the national phase |
Ref document number: 112017000242 Country of ref document: BR Kind code of ref document: A2 Effective date: 20170105 |