ES2875360T3 - Derivado de piridona que tiene un grupo tetrahidropiranil metilo - Google Patents
Derivado de piridona que tiene un grupo tetrahidropiranil metilo Download PDFInfo
- Publication number
- ES2875360T3 ES2875360T3 ES15818440T ES15818440T ES2875360T3 ES 2875360 T3 ES2875360 T3 ES 2875360T3 ES 15818440 T ES15818440 T ES 15818440T ES 15818440 T ES15818440 T ES 15818440T ES 2875360 T3 ES2875360 T3 ES 2875360T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- crystal
- group
- cancer
- tetrahydro
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- UBQKCCHYAOITMY-UHFFFAOYSA-N pyridin-2-ol Chemical class OC1=CC=CC=N1 UBQKCCHYAOITMY-UHFFFAOYSA-N 0.000 title description 2
- 125000006173 tetrahydropyranylmethyl group Chemical group 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 491
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims abstract description 39
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 38
- 150000003839 salts Chemical class 0.000 claims abstract description 38
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims abstract description 27
- 229910052757 nitrogen Inorganic materials 0.000 claims abstract description 27
- 125000005843 halogen group Chemical group 0.000 claims abstract description 25
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims abstract description 17
- 125000004433 nitrogen atom Chemical group N* 0.000 claims abstract description 16
- 229910052805 deuterium Inorganic materials 0.000 claims abstract description 9
- 125000006414 CCl Chemical group ClC* 0.000 claims abstract description 6
- 125000004093 cyano group Chemical group *C#N 0.000 claims abstract description 6
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims abstract description 3
- 229910052717 sulfur Inorganic materials 0.000 claims abstract description 3
- 125000004434 sulfur atom Chemical group 0.000 claims abstract description 3
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims abstract description 3
- 239000013078 crystal Substances 0.000 claims description 141
- -1 2-Amino-5- {4 - [(2R) -1,4-dioxan-2-ylmethoxy] -3-methoxyphenyl} pyridin-3 -yl Chemical group 0.000 claims description 111
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 91
- 206010028980 Neoplasm Diseases 0.000 claims description 85
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 claims description 54
- 229910052802 copper Inorganic materials 0.000 claims description 54
- 239000010949 copper Substances 0.000 claims description 54
- 230000005855 radiation Effects 0.000 claims description 54
- 201000011510 cancer Diseases 0.000 claims description 32
- YCGFHHACUCVQPI-UUWRZZSWSA-N N-[4-[2-amino-5-[4-[[(2R)-1,4-dioxan-2-yl]methoxy]-3-methoxyphenyl]pyridin-3-yl]phenyl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)C1=CC(=C(C=C1)OC[C@@H]1OCCOC1)OC YCGFHHACUCVQPI-UUWRZZSWSA-N 0.000 claims description 30
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 26
- 201000010099 disease Diseases 0.000 claims description 25
- 230000002401 inhibitory effect Effects 0.000 claims description 25
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 claims description 24
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 22
- 239000003814 drug Substances 0.000 claims description 21
- 239000003112 inhibitor Substances 0.000 claims description 20
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 16
- 230000005764 inhibitory process Effects 0.000 claims description 16
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 claims description 16
- 229910019142 PO4 Inorganic materials 0.000 claims description 12
- 229940079593 drug Drugs 0.000 claims description 11
- 239000010452 phosphate Substances 0.000 claims description 11
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 claims description 10
- 206010059866 Drug resistance Diseases 0.000 claims description 9
- 206010027476 Metastases Diseases 0.000 claims description 8
- 230000009401 metastasis Effects 0.000 claims description 8
- 239000008194 pharmaceutical composition Substances 0.000 claims description 7
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical group [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 claims description 6
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 claims description 6
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 6
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 claims description 6
- 201000005202 lung cancer Diseases 0.000 claims description 6
- 208000020816 lung neoplasm Diseases 0.000 claims description 6
- 206010006187 Breast cancer Diseases 0.000 claims description 5
- 208000026310 Breast neoplasm Diseases 0.000 claims description 5
- 229910002651 NO3 Inorganic materials 0.000 claims description 5
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 claims description 5
- 206010033128 Ovarian cancer Diseases 0.000 claims description 5
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 5
- 229940077388 benzenesulfonate Drugs 0.000 claims description 5
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 claims description 5
- XTEGVFVZDVNBPF-UHFFFAOYSA-L naphthalene-1,5-disulfonate(2-) Chemical compound C1=CC=C2C(S(=O)(=O)[O-])=CC=CC2=C1S([O-])(=O)=O XTEGVFVZDVNBPF-UHFFFAOYSA-L 0.000 claims description 5
- 206010009944 Colon cancer Diseases 0.000 claims description 4
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 4
- 206010017758 gastric cancer Diseases 0.000 claims description 4
- 201000001441 melanoma Diseases 0.000 claims description 4
- 201000011549 stomach cancer Diseases 0.000 claims description 4
- 125000006415 CF Chemical group FC* 0.000 claims description 3
- 125000006417 CH Chemical group [H]C* 0.000 claims description 3
- 206010014733 Endometrial cancer Diseases 0.000 claims description 3
- 206010014759 Endometrial neoplasm Diseases 0.000 claims description 3
- 208000000461 Esophageal Neoplasms Diseases 0.000 claims description 3
- XYELEXOQONCGOW-UHFFFAOYSA-N N-[6-[2-amino-5-[3-methoxy-4-(2-morpholin-4-ylethoxy)phenyl]pyridin-3-yl]pyridazin-3-yl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=CC=C(N=N1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)C1=CC(=C(C=C1)OCCN1CCOCC1)OC XYELEXOQONCGOW-UHFFFAOYSA-N 0.000 claims description 3
- 206010030155 Oesophageal carcinoma Diseases 0.000 claims description 3
- 206010060862 Prostate cancer Diseases 0.000 claims description 3
- 125000003545 alkoxy group Chemical group 0.000 claims description 3
- 208000029742 colonic neoplasm Diseases 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- 201000004101 esophageal cancer Diseases 0.000 claims description 3
- 208000005017 glioblastoma Diseases 0.000 claims description 3
- 230000003463 hyperproliferative effect Effects 0.000 claims description 3
- 208000032839 leukemia Diseases 0.000 claims description 3
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 3
- 201000002510 thyroid cancer Diseases 0.000 claims description 3
- 208000013066 thyroid gland cancer Diseases 0.000 claims description 3
- 208000008839 Kidney Neoplasms Diseases 0.000 claims description 2
- RQAATFYANFZYNJ-UHFFFAOYSA-N N-[4-[2-amino-5-(1-ethylpyrazol-4-yl)pyridin-3-yl]-3-fluorophenyl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=C(C=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)F)C=1C=NN(C=1)CC RQAATFYANFZYNJ-UHFFFAOYSA-N 0.000 claims description 2
- MIJFYIHQEYKWGI-UHFFFAOYSA-N N-[4-[2-amino-5-(3,4-dimethoxyphenyl)pyridin-3-yl]phenyl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)C1=CC(=C(C=C1)OC)OC MIJFYIHQEYKWGI-UHFFFAOYSA-N 0.000 claims description 2
- YQGUQZWOHCDPTK-UHFFFAOYSA-N N-[4-[2-amino-5-[1-(oxan-4-yl)pyrazol-4-yl]pyridin-3-yl]-3-fluorophenyl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=C(C=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)F)C=1C=NN(C=1)C1CCOCC1 YQGUQZWOHCDPTK-UHFFFAOYSA-N 0.000 claims description 2
- YCGFHHACUCVQPI-UMSFTDKQSA-N N-[4-[2-amino-5-[4-[[(2S)-1,4-dioxan-2-yl]methoxy]-3-methoxyphenyl]pyridin-3-yl]phenyl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)C1=CC(=C(C=C1)OC[C@H]1OCCOC1)OC YCGFHHACUCVQPI-UMSFTDKQSA-N 0.000 claims description 2
- 206010029260 Neuroblastoma Diseases 0.000 claims description 2
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 2
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 2
- 206010038389 Renal cancer Diseases 0.000 claims description 2
- 239000004480 active ingredient Substances 0.000 claims description 2
- 150000001975 deuterium Chemical group 0.000 claims description 2
- 201000010982 kidney cancer Diseases 0.000 claims description 2
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 2
- 201000008968 osteosarcoma Diseases 0.000 claims description 2
- 201000002528 pancreatic cancer Diseases 0.000 claims description 2
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 2
- 125000002206 pyridazin-3-yl group Chemical group [H]C1=C([H])C([H])=C(*)N=N1 0.000 claims description 2
- 206010041823 squamous cell carcinoma Diseases 0.000 claims description 2
- 208000017572 squamous cell neoplasm Diseases 0.000 claims description 2
- 208000002495 Uterine Neoplasms Diseases 0.000 claims 1
- 239000003560 cancer drug Substances 0.000 claims 1
- 230000002265 prevention Effects 0.000 claims 1
- 238000002560 therapeutic procedure Methods 0.000 claims 1
- 206010046766 uterine cancer Diseases 0.000 claims 1
- 125000000217 alkyl group Chemical group 0.000 abstract description 6
- 125000004429 atom Chemical group 0.000 abstract description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 abstract 1
- 125000004431 deuterium atom Chemical group 0.000 abstract 1
- 229910052760 oxygen Inorganic materials 0.000 abstract 1
- 239000001301 oxygen Substances 0.000 abstract 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 212
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 150
- 239000007787 solid Substances 0.000 description 111
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 101
- 238000000034 method Methods 0.000 description 101
- 239000000203 mixture Substances 0.000 description 97
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 84
- 239000000243 solution Substances 0.000 description 74
- 238000006243 chemical reaction Methods 0.000 description 64
- 230000002829 reductive effect Effects 0.000 description 64
- 239000002904 solvent Substances 0.000 description 62
- 238000005160 1H NMR spectroscopy Methods 0.000 description 61
- 238000001914 filtration Methods 0.000 description 61
- 239000011541 reaction mixture Substances 0.000 description 60
- 238000004519 manufacturing process Methods 0.000 description 59
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 54
- 230000015572 biosynthetic process Effects 0.000 description 50
- 238000003786 synthesis reaction Methods 0.000 description 49
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 48
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 48
- 239000000543 intermediate Substances 0.000 description 40
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 38
- 239000012044 organic layer Substances 0.000 description 37
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 36
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 32
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 32
- 239000007858 starting material Substances 0.000 description 32
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 31
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical class CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 31
- 229960001433 erlotinib Drugs 0.000 description 31
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 31
- 238000010898 silica gel chromatography Methods 0.000 description 31
- 238000012360 testing method Methods 0.000 description 31
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 29
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 25
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 25
- 238000009835 boiling Methods 0.000 description 25
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 24
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 24
- 238000000605 extraction Methods 0.000 description 24
- 210000004027 cell Anatomy 0.000 description 23
- 239000012267 brine Substances 0.000 description 22
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 22
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 21
- 108091000080 Phosphotransferase Proteins 0.000 description 21
- 102000020233 phosphotransferase Human genes 0.000 description 21
- 241000699670 Mus sp. Species 0.000 description 20
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 20
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 19
- 238000000921 elemental analysis Methods 0.000 description 19
- 229910000027 potassium carbonate Inorganic materials 0.000 description 19
- 229910016523 CuKa Inorganic materials 0.000 description 18
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 18
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 18
- 239000002253 acid Substances 0.000 description 17
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 17
- 229910000024 caesium carbonate Inorganic materials 0.000 description 17
- 230000014509 gene expression Effects 0.000 description 17
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 16
- 239000012046 mixed solvent Substances 0.000 description 16
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 15
- 239000000725 suspension Substances 0.000 description 15
- 0 CCNC=C(C=*)C(/C=C1\C2C(F)=CC(*C(C3=CN(CC4CCOCC4)C=C(c4ccc(C)cc4)C3=O)=O)=CC2)=CCC/C(/C)=C1/N Chemical compound CCNC=C(C=*)C(/C=C1\C2C(F)=CC(*C(C3=CN(CC4CCOCC4)C=C(c4ccc(C)cc4)C3=O)=O)=CC2)=CCC/C(/C)=C1/N 0.000 description 14
- 239000002585 base Substances 0.000 description 14
- 230000037396 body weight Effects 0.000 description 14
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- 239000012156 elution solvent Substances 0.000 description 12
- FWFGVMYFCODZRD-UHFFFAOYSA-N oxidanium;hydrogen sulfate Chemical compound O.OS(O)(=O)=O FWFGVMYFCODZRD-UHFFFAOYSA-N 0.000 description 12
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 12
- 235000017557 sodium bicarbonate Nutrition 0.000 description 12
- 238000003756 stirring Methods 0.000 description 12
- 239000000706 filtrate Substances 0.000 description 11
- 238000009472 formulation Methods 0.000 description 11
- 229920006395 saturated elastomer Polymers 0.000 description 11
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 10
- 230000000694 effects Effects 0.000 description 10
- 108090000765 processed proteins & peptides Proteins 0.000 description 10
- 239000000047 product Substances 0.000 description 10
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 9
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 9
- 230000000259 anti-tumor effect Effects 0.000 description 9
- 239000000872 buffer Substances 0.000 description 9
- 239000003054 catalyst Substances 0.000 description 9
- 239000003153 chemical reaction reagent Substances 0.000 description 9
- 238000004440 column chromatography Methods 0.000 description 9
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 9
- 150000007530 organic bases Chemical class 0.000 description 9
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 9
- 238000002054 transplantation Methods 0.000 description 9
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 8
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 8
- 239000002246 antineoplastic agent Substances 0.000 description 8
- 239000007864 aqueous solution Substances 0.000 description 8
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 8
- 150000007529 inorganic bases Chemical class 0.000 description 8
- 239000012299 nitrogen atmosphere Substances 0.000 description 8
- 235000021317 phosphate Nutrition 0.000 description 8
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 8
- 239000012453 solvate Substances 0.000 description 8
- 239000000758 substrate Substances 0.000 description 8
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 8
- LVEYOSJUKRVCCF-UHFFFAOYSA-N 1,3-bis(diphenylphosphino)propane Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)CCCP(C=1C=CC=CC=1)C1=CC=CC=C1 LVEYOSJUKRVCCF-UHFFFAOYSA-N 0.000 description 7
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 7
- 125000000590 4-methylphenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])[H] 0.000 description 7
- 108700020796 Oncogene Proteins 0.000 description 7
- GPDHNZNLPKYHCN-DZOOLQPHSA-N [[(z)-(1-cyano-2-ethoxy-2-oxoethylidene)amino]oxy-morpholin-4-ylmethylidene]-dimethylazanium;hexafluorophosphate Chemical compound F[P-](F)(F)(F)(F)F.CCOC(=O)C(\C#N)=N/OC(=[N+](C)C)N1CCOCC1 GPDHNZNLPKYHCN-DZOOLQPHSA-N 0.000 description 7
- 125000001246 bromo group Chemical group Br* 0.000 description 7
- 230000008859 change Effects 0.000 description 7
- 239000003795 chemical substances by application Substances 0.000 description 7
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 7
- 239000010410 layer Substances 0.000 description 7
- 239000002609 medium Substances 0.000 description 7
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 7
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 7
- 239000000843 powder Substances 0.000 description 7
- 239000000741 silica gel Substances 0.000 description 7
- 229910002027 silica gel Inorganic materials 0.000 description 7
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 6
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 6
- 229940126062 Compound A Drugs 0.000 description 6
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 6
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 6
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 6
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 6
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 6
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 6
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 6
- 239000002953 phosphate buffered saline Substances 0.000 description 6
- 229940002612 prodrug Drugs 0.000 description 6
- 239000000651 prodrug Substances 0.000 description 6
- 108090000623 proteins and genes Proteins 0.000 description 6
- 125000001424 substituent group Chemical group 0.000 description 6
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 6
- WLPUWLXVBWGYMZ-UHFFFAOYSA-N tricyclohexylphosphine Chemical compound C1CCCCC1P(C1CCCCC1)C1CCCCC1 WLPUWLXVBWGYMZ-UHFFFAOYSA-N 0.000 description 6
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 6
- 239000003981 vehicle Substances 0.000 description 6
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 5
- 102100037236 Tyrosine-protein kinase receptor UFO Human genes 0.000 description 5
- 239000000654 additive Substances 0.000 description 5
- 229940100198 alkylating agent Drugs 0.000 description 5
- 239000002168 alkylating agent Substances 0.000 description 5
- 238000005516 engineering process Methods 0.000 description 5
- 230000012010 growth Effects 0.000 description 5
- 208000017169 kidney disease Diseases 0.000 description 5
- 239000003446 ligand Substances 0.000 description 5
- 229940098779 methanesulfonic acid Drugs 0.000 description 5
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 5
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 5
- 229910000029 sodium carbonate Inorganic materials 0.000 description 5
- 230000002194 synthesizing effect Effects 0.000 description 5
- 229910052723 transition metal Inorganic materials 0.000 description 5
- 150000003624 transition metals Chemical class 0.000 description 5
- 125000002827 triflate group Chemical class FC(S(=O)(=O)O*)(F)F 0.000 description 5
- 239000011534 wash buffer Substances 0.000 description 5
- IEQAICDLOKRSRL-UHFFFAOYSA-N 2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-(2-dodecoxyethoxy)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethanol Chemical compound CCCCCCCCCCCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCO IEQAICDLOKRSRL-UHFFFAOYSA-N 0.000 description 4
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 4
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 4
- VSXLNJTYRFDLNH-UHFFFAOYSA-N 5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxylic acid Chemical compound CC1=CC=C(C=C1)C=1C(C(=CN(C=1)CC1CCOCC1)C(=O)O)=O VSXLNJTYRFDLNH-UHFFFAOYSA-N 0.000 description 4
- JPVNIXANASYDQI-UHFFFAOYSA-N COC=1C=C(C=CC=1OC)C=1C=C(C(=NC=1)N)B1OC(C(O1)(C)C)(C)C Chemical compound COC=1C=C(C=CC=1OC)C=1C=C(C(=NC=1)N)B1OC(C(O1)(C)C)(C)C JPVNIXANASYDQI-UHFFFAOYSA-N 0.000 description 4
- 208000007342 Diabetic Nephropathies Diseases 0.000 description 4
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 4
- 101150022345 GAS6 gene Proteins 0.000 description 4
- 239000007995 HEPES buffer Substances 0.000 description 4
- 241001465754 Metazoa Species 0.000 description 4
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 4
- 125000003277 amino group Chemical group 0.000 description 4
- 125000004432 carbon atom Chemical group C* 0.000 description 4
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 4
- 230000010261 cell growth Effects 0.000 description 4
- 230000012292 cell migration Effects 0.000 description 4
- 230000005754 cellular signaling Effects 0.000 description 4
- 238000001816 cooling Methods 0.000 description 4
- 239000012043 crude product Substances 0.000 description 4
- 208000033679 diabetic kidney disease Diseases 0.000 description 4
- 238000010586 diagram Methods 0.000 description 4
- 238000010790 dilution Methods 0.000 description 4
- 239000012895 dilution Substances 0.000 description 4
- 238000004821 distillation Methods 0.000 description 4
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 4
- 239000000499 gel Substances 0.000 description 4
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 4
- 125000000623 heterocyclic group Chemical group 0.000 description 4
- 238000004128 high performance liquid chromatography Methods 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 230000002779 inactivation Effects 0.000 description 4
- 230000003834 intracellular effect Effects 0.000 description 4
- 238000005304 joining Methods 0.000 description 4
- 239000012528 membrane Substances 0.000 description 4
- 230000035772 mutation Effects 0.000 description 4
- XTEGVFVZDVNBPF-UHFFFAOYSA-N naphthalene-1,5-disulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1S(O)(=O)=O XTEGVFVZDVNBPF-UHFFFAOYSA-N 0.000 description 4
- 239000003960 organic solvent Substances 0.000 description 4
- 229910052763 palladium Inorganic materials 0.000 description 4
- IVDFJHOHABJVEH-UHFFFAOYSA-N pinacol Chemical compound CC(C)(O)C(C)(C)O IVDFJHOHABJVEH-UHFFFAOYSA-N 0.000 description 4
- 235000011056 potassium acetate Nutrition 0.000 description 4
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- 102000004169 proteins and genes Human genes 0.000 description 4
- 150000003222 pyridines Chemical class 0.000 description 4
- 239000000523 sample Substances 0.000 description 4
- 239000000377 silicon dioxide Substances 0.000 description 4
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 4
- 229940124597 therapeutic agent Drugs 0.000 description 4
- 210000001519 tissue Anatomy 0.000 description 4
- 230000004614 tumor growth Effects 0.000 description 4
- 125000005500 uronium group Chemical group 0.000 description 4
- 210000004509 vascular smooth muscle cell Anatomy 0.000 description 4
- 238000005406 washing Methods 0.000 description 4
- 238000001262 western blot Methods 0.000 description 4
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 description 4
- ABDDQTDRAHXHOC-QMMMGPOBSA-N 1-[(7s)-5,7-dihydro-4h-thieno[2,3-c]pyran-7-yl]-n-methylmethanamine Chemical compound CNC[C@@H]1OCCC2=C1SC=C2 ABDDQTDRAHXHOC-QMMMGPOBSA-N 0.000 description 3
- GXXXUZIRGXYDFP-UHFFFAOYSA-N 2-(4-methylphenyl)acetic acid Chemical compound CC1=CC=C(CC(O)=O)C=C1 GXXXUZIRGXYDFP-UHFFFAOYSA-N 0.000 description 3
- 125000003762 3,4-dimethoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C(OC([H])([H])[H])C([H])=C1* 0.000 description 3
- CYRIVXRQUKTHSD-WJOKGBTCSA-N 3-[1-[4-[2-amino-5-[4-[[(2R)-1,4-dioxan-2-yl]methoxy]-3-methoxyphenyl]pyridin-3-yl]-3-fluorophenyl]-3-methyl-2H-pyridin-6-yl]-1-(oxan-4-ylmethyl)pyridin-4-one Chemical compound NC1=NC=C(C=C1C1=C(C=C(C=C1)N1C(=CC=C(C1)C)C1=CN(C=CC1=O)CC1CCOCC1)F)C1=CC(=C(C=C1)OC[C@@H]1OCCOC1)OC CYRIVXRQUKTHSD-WJOKGBTCSA-N 0.000 description 3
- 125000004180 3-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(F)=C1[H] 0.000 description 3
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 3
- 101150018445 Axl gene Proteins 0.000 description 3
- 206010018366 Glomerulonephritis acute Diseases 0.000 description 3
- 206010018367 Glomerulonephritis chronic Diseases 0.000 description 3
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 3
- 241000699666 Mus <mouse, genus> Species 0.000 description 3
- SBQHEYGRLCNAMO-UHFFFAOYSA-N N-[4-[2-amino-5-(3,4-dimethoxyphenyl)pyridin-3-yl]phenyl]-5-bromo-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)Br)CC1CCOCC1)C1=CC(=C(C=C1)OC)OC SBQHEYGRLCNAMO-UHFFFAOYSA-N 0.000 description 3
- 241000283973 Oryctolagus cuniculus Species 0.000 description 3
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- 206010048955 Retinal toxicity Diseases 0.000 description 3
- 108091027967 Small hairpin RNA Proteins 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 230000001154 acute effect Effects 0.000 description 3
- 230000000340 anti-metabolite Effects 0.000 description 3
- 229940100197 antimetabolite Drugs 0.000 description 3
- 239000002256 antimetabolite Substances 0.000 description 3
- 230000000903 blocking effect Effects 0.000 description 3
- 230000004709 cell invasion Effects 0.000 description 3
- 239000006285 cell suspension Substances 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000002425 crystallisation Methods 0.000 description 3
- 230000008025 crystallization Effects 0.000 description 3
- 230000007850 degeneration Effects 0.000 description 3
- 238000012217 deletion Methods 0.000 description 3
- 230000037430 deletion Effects 0.000 description 3
- BSHICDXRSZQYBP-UHFFFAOYSA-N dichloromethane;palladium(2+) Chemical compound [Pd+2].ClCCl BSHICDXRSZQYBP-UHFFFAOYSA-N 0.000 description 3
- NSNHWTBQMQIDCF-UHFFFAOYSA-N dihydrate;hydrochloride Chemical compound O.O.Cl NSNHWTBQMQIDCF-UHFFFAOYSA-N 0.000 description 3
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 3
- GDXNSFVXXGAUMU-UHFFFAOYSA-N ethyl 4-(4-methylphenyl)-3-oxobutanoate Chemical compound CCOC(=O)CC(=O)CC1=CC=C(C)C=C1 GDXNSFVXXGAUMU-UHFFFAOYSA-N 0.000 description 3
- UIKUIBBGEJWAIP-UHFFFAOYSA-N ethyl 5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxylate Chemical compound CC1=CC=C(C=C1)C=1C(C(=CN(C=1)CC1CCOCC1)C(=O)OCC)=O UIKUIBBGEJWAIP-UHFFFAOYSA-N 0.000 description 3
- 235000019253 formic acid Nutrition 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 230000009545 invasion Effects 0.000 description 3
- 230000005012 migration Effects 0.000 description 3
- 238000013508 migration Methods 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 3
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 3
- 230000026731 phosphorylation Effects 0.000 description 3
- 238000006366 phosphorylation reaction Methods 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 125000006239 protecting group Chemical group 0.000 description 3
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 3
- 231100000385 retinal toxicity Toxicity 0.000 description 3
- 230000035945 sensitivity Effects 0.000 description 3
- 239000004055 small Interfering RNA Substances 0.000 description 3
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 3
- 210000004881 tumor cell Anatomy 0.000 description 3
- UGOMMVLRQDMAQQ-UHFFFAOYSA-N xphos Chemical group CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 UGOMMVLRQDMAQQ-UHFFFAOYSA-N 0.000 description 3
- MOWXJLUYGFNTAL-DEOSSOPVSA-N (s)-[2-chloro-4-fluoro-5-(7-morpholin-4-ylquinazolin-4-yl)phenyl]-(6-methoxypyridazin-3-yl)methanol Chemical compound N1=NC(OC)=CC=C1[C@@H](O)C1=CC(C=2C3=CC=C(C=C3N=CN=2)N2CCOCC2)=C(F)C=C1Cl MOWXJLUYGFNTAL-DEOSSOPVSA-N 0.000 description 2
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 2
- FPIRBHDGWMWJEP-UHFFFAOYSA-N 1-hydroxy-7-azabenzotriazole Chemical compound C1=CN=C2N(O)N=NC2=C1 FPIRBHDGWMWJEP-UHFFFAOYSA-N 0.000 description 2
- QVKJPKIMVKZJGM-UHFFFAOYSA-N 2-(4-cyclopropyl-2-fluorophenyl)acetic acid Chemical compound C1(CC1)C1=CC(=C(C=C1)CC(=O)O)F QVKJPKIMVKZJGM-UHFFFAOYSA-N 0.000 description 2
- SDTMFDGELKWGFT-UHFFFAOYSA-N 2-methylpropan-2-olate Chemical compound CC(C)(C)[O-] SDTMFDGELKWGFT-UHFFFAOYSA-N 0.000 description 2
- PYIVNGBQMPHEDY-QGZVFWFLSA-N 3-(4-amino-2-fluorophenyl)-5-[4-[[(2R)-1,4-dioxan-2-yl]methoxy]-3-methoxyphenyl]pyridin-2-amine Chemical compound NC1=CC(=C(C=C1)C=1C(=NC=C(C=1)C1=CC(=C(C=C1)OC[C@@H]1OCCOC1)OC)N)F PYIVNGBQMPHEDY-QGZVFWFLSA-N 0.000 description 2
- BYHQTRFJOGIQAO-GOSISDBHSA-N 3-(4-bromophenyl)-8-[(2R)-2-hydroxypropyl]-1-[(3-methoxyphenyl)methyl]-1,3,8-triazaspiro[4.5]decan-2-one Chemical compound C[C@H](CN1CCC2(CC1)CN(C(=O)N2CC3=CC(=CC=C3)OC)C4=CC=C(C=C4)Br)O BYHQTRFJOGIQAO-GOSISDBHSA-N 0.000 description 2
- WNEODWDFDXWOLU-QHCPKHFHSA-N 3-[3-(hydroxymethyl)-4-[1-methyl-5-[[5-[(2s)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl]amino]-6-oxopyridin-3-yl]pyridin-2-yl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one Chemical compound C([C@@H](N(CC1)C=2C=NC(NC=3C(N(C)C=C(C=3)C=3C(=C(N4C(C5=CC=6CC(C)(C)CC=6N5CC4)=O)N=CC=3)CO)=O)=CC=2)C)N1C1COC1 WNEODWDFDXWOLU-QHCPKHFHSA-N 0.000 description 2
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 2
- CCADMPWCDJNWMQ-UHFFFAOYSA-N 3-chloro-5-(3,4-dimethoxyphenyl)pyrazin-2-amine Chemical compound ClC=1C(=NC=C(N=1)C1=CC(=C(C=C1)OC)OC)N CCADMPWCDJNWMQ-UHFFFAOYSA-N 0.000 description 2
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 2
- LMOOYAKLEOGKJR-UHFFFAOYSA-N 4-(bromomethyl)oxane Chemical compound BrCC1CCOCC1 LMOOYAKLEOGKJR-UHFFFAOYSA-N 0.000 description 2
- 125000004195 4-methylpiperazin-1-yl group Chemical group [H]C([H])([H])N1C([H])([H])C([H])([H])N(*)C([H])([H])C1([H])[H] 0.000 description 2
- LLPFPLXIDMRGBA-UHFFFAOYSA-N 5-(4-bromophenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxylic acid Chemical compound BrC1=CC=C(C=C1)C=1C(C(=CN(C=1)CC1CCOCC1)C(=O)O)=O LLPFPLXIDMRGBA-UHFFFAOYSA-N 0.000 description 2
- WOVKYSAHUYNSMH-RRKCRQDMSA-N 5-bromodeoxyuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(Br)=C1 WOVKYSAHUYNSMH-RRKCRQDMSA-N 0.000 description 2
- KCBWAFJCKVKYHO-UHFFFAOYSA-N 6-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-1-[[4-[1-propan-2-yl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methyl]pyrazolo[3,4-d]pyrimidine Chemical compound C1(CC1)C1=NC=NC(=C1C1=NC=C2C(=N1)N(N=C2)CC1=CC=C(C=C1)C=1N(C=C(N=1)C(F)(F)F)C(C)C)OC KCBWAFJCKVKYHO-UHFFFAOYSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- 206010003210 Arteriosclerosis Diseases 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 201000001320 Atherosclerosis Diseases 0.000 description 2
- 208000037260 Atherosclerotic Plaque Diseases 0.000 description 2
- 208000031648 Body Weight Changes Diseases 0.000 description 2
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 2
- 201000009030 Carcinoma Diseases 0.000 description 2
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 2
- 206010012689 Diabetic retinopathy Diseases 0.000 description 2
- BUDQDWGNQVEFAC-UHFFFAOYSA-N Dihydropyran Chemical compound C1COC=CC1 BUDQDWGNQVEFAC-UHFFFAOYSA-N 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 101150039808 Egfr gene Proteins 0.000 description 2
- 241000196324 Embryophyta Species 0.000 description 2
- 108010024636 Glutathione Proteins 0.000 description 2
- 102000005720 Glutathione transferase Human genes 0.000 description 2
- 108010070675 Glutathione transferase Proteins 0.000 description 2
- WZUVPPKBWHMQCE-UHFFFAOYSA-N Haematoxylin Chemical compound C12=CC(O)=C(O)C=C2CC2(O)C1C1=CC=C(O)C(O)=C1OC2 WZUVPPKBWHMQCE-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 208000007766 Kaposi sarcoma Diseases 0.000 description 2
- 239000002136 L01XE07 - Lapatinib Substances 0.000 description 2
- XNRVGTHNYCNCFF-UHFFFAOYSA-N Lapatinib ditosylate monohydrate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1.CC1=CC=C(S(O)(=O)=O)C=C1.O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 XNRVGTHNYCNCFF-UHFFFAOYSA-N 0.000 description 2
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 2
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 2
- ZSXGLVDWWRXATF-UHFFFAOYSA-N N,N-dimethylformamide dimethyl acetal Chemical compound COC(OC)N(C)C ZSXGLVDWWRXATF-UHFFFAOYSA-N 0.000 description 2
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 2
- HVZYRAXHTSGBOE-UHFFFAOYSA-N N-[4-[2-amino-5-(3,4-dimethoxyphenyl)pyridin-3-yl]phenyl]-5-(4-cyclopropylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C1CC1)CC1CCOCC1)C1=CC(=C(C=C1)OC)OC HVZYRAXHTSGBOE-UHFFFAOYSA-N 0.000 description 2
- 150000001204 N-oxides Chemical group 0.000 description 2
- YNPNZTXNASCQKK-UHFFFAOYSA-N Phenanthrene Natural products C1=CC=C2C3=CC=CC=C3C=CC2=C1 YNPNZTXNASCQKK-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 229920001213 Polysorbate 20 Polymers 0.000 description 2
- 201000004681 Psoriasis Diseases 0.000 description 2
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 2
- 208000017442 Retinal disease Diseases 0.000 description 2
- 206010038923 Retinopathy Diseases 0.000 description 2
- 206010039491 Sarcoma Diseases 0.000 description 2
- 229920002684 Sepharose Polymers 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- 108090000190 Thrombin Proteins 0.000 description 2
- 208000007536 Thrombosis Diseases 0.000 description 2
- 208000005475 Vascular calcification Diseases 0.000 description 2
- LXRZVMYMQHNYJB-UNXOBOICSA-N [(1R,2S,4R)-4-[[5-[4-[(1R)-7-chloro-1,2,3,4-tetrahydroisoquinolin-1-yl]-5-methylthiophene-2-carbonyl]pyrimidin-4-yl]amino]-2-hydroxycyclopentyl]methyl sulfamate Chemical compound CC1=C(C=C(S1)C(=O)C1=C(N[C@H]2C[C@H](O)[C@@H](COS(N)(=O)=O)C2)N=CN=C1)[C@@H]1NCCC2=C1C=C(Cl)C=C2 LXRZVMYMQHNYJB-UNXOBOICSA-N 0.000 description 2
- DGEZNRSVGBDHLK-UHFFFAOYSA-N [1,10]phenanthroline Chemical compound C1=CN=C2C3=NC=CC=C3C=CC2=C1 DGEZNRSVGBDHLK-UHFFFAOYSA-N 0.000 description 2
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 2
- 229910052783 alkali metal Inorganic materials 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 230000033115 angiogenesis Effects 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- 230000004900 autophagic degradation Effects 0.000 description 2
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 2
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical class OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 2
- 230000004579 body weight change Effects 0.000 description 2
- 229910052796 boron Inorganic materials 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 229960004562 carboplatin Drugs 0.000 description 2
- 230000024245 cell differentiation Effects 0.000 description 2
- 230000032823 cell division Effects 0.000 description 2
- 239000013592 cell lysate Substances 0.000 description 2
- 230000036755 cellular response Effects 0.000 description 2
- 239000003638 chemical reducing agent Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- 230000001684 chronic effect Effects 0.000 description 2
- 229960004316 cisplatin Drugs 0.000 description 2
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 2
- 239000002131 composite material Substances 0.000 description 2
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- UKJLNMAFNRKWGR-UHFFFAOYSA-N cyclohexatrienamine Chemical group NC1=CC=C=C[CH]1 UKJLNMAFNRKWGR-UHFFFAOYSA-N 0.000 description 2
- WLVKDFJTYKELLQ-UHFFFAOYSA-N cyclopropylboronic acid Chemical compound OB(O)C1CC1 WLVKDFJTYKELLQ-UHFFFAOYSA-N 0.000 description 2
- 238000010908 decantation Methods 0.000 description 2
- WMKGGPCROCCUDY-PHEQNACWSA-N dibenzylideneacetone Chemical compound C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 WMKGGPCROCCUDY-PHEQNACWSA-N 0.000 description 2
- 125000000532 dioxanyl group Chemical group 0.000 description 2
- 229960004679 doxorubicin Drugs 0.000 description 2
- 229940121647 egfr inhibitor Drugs 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 108700021358 erbB-1 Genes Proteins 0.000 description 2
- 229940071111 erlotinib 25 mg Drugs 0.000 description 2
- BRYNITBKFYVFBV-UHFFFAOYSA-N ethyl 4-(4-bromophenyl)-3-oxobutanoate Chemical compound CCOC(=O)CC(=O)CC1=CC=C(Br)C=C1 BRYNITBKFYVFBV-UHFFFAOYSA-N 0.000 description 2
- KKAFIMYLMXUFKH-UHFFFAOYSA-N ethyl 5-(4-bromophenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxylate Chemical compound BrC1=CC=C(C=C1)C=1C(C(=CN(C=1)CC1CCOCC1)C(=O)OCC)=O KKAFIMYLMXUFKH-UHFFFAOYSA-N 0.000 description 2
- NHIDERHRUULCAN-UHFFFAOYSA-N ethyl 5-(4-bromophenyl)-4-oxo-1H-pyridine-3-carboxylate Chemical compound BrC1=CC=C(C=C1)C=1C(C(=CNC=1)C(=O)OCC)=O NHIDERHRUULCAN-UHFFFAOYSA-N 0.000 description 2
- KTWOOEGAPBSYNW-UHFFFAOYSA-N ferrocene Chemical compound [Fe+2].C=1C=C[CH-]C=1.C=1C=C[CH-]C=1 KTWOOEGAPBSYNW-UHFFFAOYSA-N 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 125000001153 fluoro group Chemical group F* 0.000 description 2
- 239000006260 foam Substances 0.000 description 2
- 108020001507 fusion proteins Proteins 0.000 description 2
- 102000037865 fusion proteins Human genes 0.000 description 2
- 210000004211 gastric acid Anatomy 0.000 description 2
- 229960003180 glutathione Drugs 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 230000009036 growth inhibition Effects 0.000 description 2
- 230000002140 halogenating effect Effects 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 239000005457 ice water Substances 0.000 description 2
- 230000036039 immunity Effects 0.000 description 2
- 239000000367 immunologic factor Substances 0.000 description 2
- 230000005917 in vivo anti-tumor Effects 0.000 description 2
- CGIGDMFJXJATDK-UHFFFAOYSA-N indomethacin Chemical compound CC1=C(CC(O)=O)C2=CC(OC)=CC=C2N1C(=O)C1=CC=C(Cl)C=C1 CGIGDMFJXJATDK-UHFFFAOYSA-N 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- 229960004891 lapatinib Drugs 0.000 description 2
- 239000012669 liquid formulation Substances 0.000 description 2
- 238000007477 logistic regression Methods 0.000 description 2
- 230000007774 longterm Effects 0.000 description 2
- 229910001629 magnesium chloride Inorganic materials 0.000 description 2
- 230000003211 malignant effect Effects 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 2
- 210000000822 natural killer cell Anatomy 0.000 description 2
- QMMRZOWCJAIUJA-UHFFFAOYSA-L nickel dichloride Chemical compound Cl[Ni]Cl QMMRZOWCJAIUJA-UHFFFAOYSA-L 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 2
- UQPUONNXJVWHRM-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 UQPUONNXJVWHRM-UHFFFAOYSA-N 0.000 description 2
- 230000004962 physiological condition Effects 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 125000004193 piperazinyl group Chemical group 0.000 description 2
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 2
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 2
- 229910000160 potassium phosphate Inorganic materials 0.000 description 2
- 235000011009 potassium phosphates Nutrition 0.000 description 2
- 238000011533 pre-incubation Methods 0.000 description 2
- 239000011535 reaction buffer Substances 0.000 description 2
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 2
- 208000037803 restenosis Diseases 0.000 description 2
- 210000001525 retina Anatomy 0.000 description 2
- 206010039073 rheumatoid arthritis Diseases 0.000 description 2
- 230000011664 signaling Effects 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 230000021595 spermatogenesis Effects 0.000 description 2
- 208000010110 spontaneous platelet aggregation Diseases 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 238000001308 synthesis method Methods 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 229960004072 thrombin Drugs 0.000 description 2
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 2
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 2
- 235000019798 tripotassium phosphate Nutrition 0.000 description 2
- 239000003656 tris buffered saline Substances 0.000 description 2
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 2
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 2
- 241000701447 unidentified baculovirus Species 0.000 description 2
- 208000019553 vascular disease Diseases 0.000 description 2
- 229960003862 vemurafenib Drugs 0.000 description 2
- GPXBXXGIAQBQNI-UHFFFAOYSA-N vemurafenib Chemical compound CCCS(=O)(=O)NC1=CC=C(F)C(C(=O)C=2C3=CC(=CN=C3NC=2)C=2C=CC(Cl)=CC=2)=C1F GPXBXXGIAQBQNI-UHFFFAOYSA-N 0.000 description 2
- 230000007998 vessel formation Effects 0.000 description 2
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 2
- 229960004528 vincristine Drugs 0.000 description 2
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 2
- 229930003231 vitamin Natural products 0.000 description 2
- 239000011782 vitamin Substances 0.000 description 2
- 235000013343 vitamin Nutrition 0.000 description 2
- 229940088594 vitamin Drugs 0.000 description 2
- VCGRFBXVSFAGGA-UHFFFAOYSA-N (1,1-dioxo-1,4-thiazinan-4-yl)-[6-[[3-(4-fluorophenyl)-5-methyl-1,2-oxazol-4-yl]methoxy]pyridin-3-yl]methanone Chemical compound CC=1ON=C(C=2C=CC(F)=CC=2)C=1COC(N=C1)=CC=C1C(=O)N1CCS(=O)(=O)CC1 VCGRFBXVSFAGGA-UHFFFAOYSA-N 0.000 description 1
- WDQLRUYAYXDIFW-RWKIJVEZSA-N (2r,3r,4s,5r,6r)-4-[(2s,3r,4s,5r,6r)-3,5-dihydroxy-4-[(2r,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-6-[[(2r,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxymethyl]oxan-2-yl]oxy-6-(hydroxymethyl)oxane-2,3,5-triol Chemical compound O[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)[C@H](O)[C@@H](CO[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)O1 WDQLRUYAYXDIFW-RWKIJVEZSA-N 0.000 description 1
- ATVDJKLZNBAVCC-UHFFFAOYSA-N (3,4-dimethoxyphenyl)boron Chemical compound [B]C1=CC=C(OC)C(OC)=C1 ATVDJKLZNBAVCC-UHFFFAOYSA-N 0.000 description 1
- RCVDPBFUMYUKPB-UHFFFAOYSA-N (3,4-dimethoxyphenyl)boronic acid Chemical compound COC1=CC=C(B(O)O)C=C1OC RCVDPBFUMYUKPB-UHFFFAOYSA-N 0.000 description 1
- NRIYPIBRPGAWDD-UHFFFAOYSA-N (5-methylthiophen-2-yl)boronic acid Chemical compound CC1=CC=C(B(O)O)S1 NRIYPIBRPGAWDD-UHFFFAOYSA-N 0.000 description 1
- KMSKQZKKOZQFFG-YXRRJAAWSA-N (7S,9S)-7-[[(2R,4S,5S,6S)-4-amino-6-methyl-5-[[(2R)-2-oxanyl]oxy]-2-oxanyl]oxy]-6,9,11-trihydroxy-9-(2-hydroxy-1-oxoethyl)-4-methoxy-8,10-dihydro-7H-tetracene-5,12-dione Chemical compound O([C@H]1[C@@H](N)C[C@@H](O[C@H]1C)O[C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@@H]1CCCCO1 KMSKQZKKOZQFFG-YXRRJAAWSA-N 0.000 description 1
- 125000004890 (C1-C6) alkylamino group Chemical group 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- JIHQDMXYYFUGFV-UHFFFAOYSA-N 1,3,5-triazine Chemical compound C1=NC=NC=N1 JIHQDMXYYFUGFV-UHFFFAOYSA-N 0.000 description 1
- FRASJONUBLZVQX-UHFFFAOYSA-N 1,4-naphthoquinone Chemical compound C1=CC=C2C(=O)C=CC(=O)C2=C1 FRASJONUBLZVQX-UHFFFAOYSA-N 0.000 description 1
- BOOVIFJKQGYEON-UHFFFAOYSA-N 1-(oxan-4-yl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole Chemical compound O1C(C)(C)C(C)(C)OB1C1=CN(C2CCOCC2)N=C1 BOOVIFJKQGYEON-UHFFFAOYSA-N 0.000 description 1
- OMRFOTIFDZORHU-UHFFFAOYSA-N 1-(oxan-4-ylmethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole Chemical compound O1C(C)(C)C(C)(C)OB1C1=CN(CC2CCOCC2)N=C1 OMRFOTIFDZORHU-UHFFFAOYSA-N 0.000 description 1
- GFTXALYVNPNJLF-UHFFFAOYSA-N 1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound O=C1C(=CN(C=C1)CC1CCOCC1)C(=O)N GFTXALYVNPNJLF-UHFFFAOYSA-N 0.000 description 1
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- 102100025573 1-alkyl-2-acetylglycerophosphocholine esterase Human genes 0.000 description 1
- CYEYLYGHCOHIDC-UHFFFAOYSA-N 1-methyl-4-[[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methyl]piperazine Chemical compound C1CN(C)CCN1CC1=CC=C(B2OC(C)(C)C(C)(C)O2)C=C1 CYEYLYGHCOHIDC-UHFFFAOYSA-N 0.000 description 1
- YYKBFGMYHQMXIL-UHFFFAOYSA-N 1-phenyl-2,3,4-tri(propan-2-yl)benzene Chemical group CC(C)C1=C(C(C)C)C(C(C)C)=CC=C1C1=CC=CC=C1 YYKBFGMYHQMXIL-UHFFFAOYSA-N 0.000 description 1
- 238000011714 129 mouse Methods 0.000 description 1
- FPIPGXGPPPQFEQ-UHFFFAOYSA-N 13-cis retinol Natural products OCC=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-UHFFFAOYSA-N 0.000 description 1
- BFPYWIDHMRZLRN-UHFFFAOYSA-N 17alpha-ethynyl estradiol Natural products OC1=CC=C2C3CCC(C)(C(CC4)(O)C#C)C4C3CCC2=C1 BFPYWIDHMRZLRN-UHFFFAOYSA-N 0.000 description 1
- ISWSIDIOOBJBQZ-KYKRLZBASA-N 2,3,4-trideuteriophenol Chemical compound [2H]c1ccc(O)c([2H])c1[2H] ISWSIDIOOBJBQZ-KYKRLZBASA-N 0.000 description 1
- GBBSAMQTQCPOBF-UHFFFAOYSA-N 2,4,6-trimethyl-1,3,5,2,4,6-trioxatriborinane Chemical compound CB1OB(C)OB(C)O1 GBBSAMQTQCPOBF-UHFFFAOYSA-N 0.000 description 1
- PNBIYFPZODYMOO-UHFFFAOYSA-N 2-(4-bromo-2-fluorophenyl)acetic acid Chemical compound OC(=O)CC1=CC=C(Br)C=C1F PNBIYFPZODYMOO-UHFFFAOYSA-N 0.000 description 1
- AVMWJACSYAQNDG-UHFFFAOYSA-N 2-(5-methylpyridin-2-yl)-6-phenyl-1,3,6,2-dioxazaborocane Chemical compound N1=CC(C)=CC=C1B1OCCN(C=2C=CC=CC=2)CCO1 AVMWJACSYAQNDG-UHFFFAOYSA-N 0.000 description 1
- TYHTUJMBDHMENY-UHFFFAOYSA-N 2-(5-methylpyridin-2-yl)acetic acid Chemical compound CC1=CC=C(CC(O)=O)N=C1 TYHTUJMBDHMENY-UHFFFAOYSA-N 0.000 description 1
- BZHDYBZLUCKBIA-ZCFIWIBFSA-N 2-[(2R)-1,4-dioxan-2-yl]ethanesulfonic acid Chemical compound O1[C@@H](COCC1)CCS(=O)(=O)O BZHDYBZLUCKBIA-ZCFIWIBFSA-N 0.000 description 1
- BZHDYBZLUCKBIA-LURJTMIESA-N 2-[(2S)-1,4-dioxan-2-yl]ethanesulfonic acid Chemical compound O1[C@H](COCC1)CCS(=O)(=O)O BZHDYBZLUCKBIA-LURJTMIESA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- AIXGNRNTXUKZLC-UHFFFAOYSA-N 2-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(N)C(F)=C1 AIXGNRNTXUKZLC-UHFFFAOYSA-N 0.000 description 1
- WFSJROCEOJANPD-UHFFFAOYSA-N 2-methoxy-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenol Chemical compound C1=C(O)C(OC)=CC(B2OC(C)(C)C(C)(C)O2)=C1 WFSJROCEOJANPD-UHFFFAOYSA-N 0.000 description 1
- JEECGGXGHJUYMN-UHFFFAOYSA-N 2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine Chemical compound C1=NC(C)=CC=C1B1OC(C)(C)C(C)(C)O1 JEECGGXGHJUYMN-UHFFFAOYSA-N 0.000 description 1
- 125000005975 2-phenylethyloxy group Chemical group 0.000 description 1
- RUDHSKFULPGCLM-UHFFFAOYSA-N 2-pyrazin-2-yl-1h-benzimidazole Chemical group N=1C2=CC=CC=C2NC=1C1=CN=CC=N1 RUDHSKFULPGCLM-UHFFFAOYSA-N 0.000 description 1
- DTXVKPOKPFWSFF-UHFFFAOYSA-N 3(S)-hydroxy-13-cis-eicosenoyl-CoA Chemical compound NC1=CC=C(Cl)N=N1 DTXVKPOKPFWSFF-UHFFFAOYSA-N 0.000 description 1
- YCIMNLLNPGFGHC-RHQRLBAQSA-N 3,4,5,6-tetradeuteriobenzene-1,2-diol Chemical compound [2H]C1=C([2H])C([2H])=C(O)C(O)=C1[2H] YCIMNLLNPGFGHC-RHQRLBAQSA-N 0.000 description 1
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 description 1
- RUDFGVCQAZXNAJ-UHFFFAOYSA-N 3-(4-amino-2-fluorophenyl)-5-(3,4-dimethoxyphenyl)pyridin-2-amine Chemical compound C1=C(OC)C(OC)=CC=C1C1=CN=C(N)C(C=2C(=CC(N)=CC=2)F)=C1 RUDFGVCQAZXNAJ-UHFFFAOYSA-N 0.000 description 1
- BLAHKHDCYYFYCE-UHFFFAOYSA-N 3-(4-amino-2-fluorophenyl)-5-bromopyridin-2-amine Chemical compound FC1=CC(N)=CC=C1C1=CC(Br)=CN=C1N BLAHKHDCYYFYCE-UHFFFAOYSA-N 0.000 description 1
- WOUBEGBESRQENP-UHFFFAOYSA-N 3-(4-amino-3-fluorophenyl)-4-methoxypyridin-2-amine Chemical compound NC1=C(C=C(C=C1)C=1C(=NC=CC=1OC)N)F WOUBEGBESRQENP-UHFFFAOYSA-N 0.000 description 1
- XQFAQBFIIPTPQF-LJQANCHMSA-N 3-(4-aminophenyl)-5-[4-[[(2r)-1,4-dioxan-2-yl]methoxy]-3-methoxyphenyl]pyridin-2-amine Chemical compound COC1=CC(C=2C=C(C(N)=NC=2)C=2C=CC(N)=CC=2)=CC=C1OC[C@H]1COCCO1 XQFAQBFIIPTPQF-LJQANCHMSA-N 0.000 description 1
- REZBRRWYFOIXJN-UHFFFAOYSA-N 3-(4-aminophenyl)-5-bromopyridin-2-amine Chemical compound C1=CC(N)=CC=C1C1=CC(Br)=CN=C1N REZBRRWYFOIXJN-UHFFFAOYSA-N 0.000 description 1
- HUTRLPYFPQSQBA-UHFFFAOYSA-N 3-bromo-5-(3,4-dimethoxyphenyl)pyridin-2-amine Chemical compound C1=C(OC)C(OC)=CC=C1C1=CN=C(N)C(Br)=C1 HUTRLPYFPQSQBA-UHFFFAOYSA-N 0.000 description 1
- GJITWRFPCHXBOP-UHFFFAOYSA-N 3-bromo-5-[3-methoxy-4-(2-morpholin-4-ylethoxy)phenyl]pyridin-2-amine Chemical compound BrC=1C(=NC=C(C=1)C1=CC(=C(C=C1)OCCN1CCOCC1)OC)N GJITWRFPCHXBOP-UHFFFAOYSA-N 0.000 description 1
- AAXRJLIGAKZDSS-CYBMUJFWSA-N 3-bromo-5-[4-[[(2r)-1,4-dioxan-2-yl]methoxy]-3-methoxyphenyl]pyridin-2-amine Chemical compound COC1=CC(C=2C=C(Br)C(N)=NC=2)=CC=C1OC[C@H]1COCCO1 AAXRJLIGAKZDSS-CYBMUJFWSA-N 0.000 description 1
- OYZOHRSCEASBER-UHFFFAOYSA-N 3-bromo-5-iodopyridin-2-amine Chemical compound NC1=NC=C(I)C=C1Br OYZOHRSCEASBER-UHFFFAOYSA-N 0.000 description 1
- KVGKDDCJSHJYCS-UHFFFAOYSA-N 3-chloro-5-iodopyrazin-2-amine Chemical compound NC1=NC=C(I)N=C1Cl KVGKDDCJSHJYCS-UHFFFAOYSA-N 0.000 description 1
- FLMNWVXAEGUVNY-UHFFFAOYSA-N 3-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(N)C=C1F FLMNWVXAEGUVNY-UHFFFAOYSA-N 0.000 description 1
- QBPYHICVLNIUPW-UHFFFAOYSA-N 3-iodo-4-methoxypyridin-2-amine Chemical compound COC1=CC=NC(N)=C1I QBPYHICVLNIUPW-UHFFFAOYSA-N 0.000 description 1
- TVOJIBGZFYMWDT-UHFFFAOYSA-N 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1h-pyrazole Chemical compound O1C(C)(C)C(C)(C)OB1C1=CNN=C1 TVOJIBGZFYMWDT-UHFFFAOYSA-N 0.000 description 1
- QOWSWEBLNVACCL-UHFFFAOYSA-N 4-Bromophenyl acetate Chemical compound OC(=O)CC1=CC=C(Br)C=C1 QOWSWEBLNVACCL-UHFFFAOYSA-N 0.000 description 1
- XWNSFEAWWGGSKJ-UHFFFAOYSA-N 4-acetyl-4-methylheptanedinitrile Chemical compound N#CCCC(C)(C(=O)C)CCC#N XWNSFEAWWGGSKJ-UHFFFAOYSA-N 0.000 description 1
- KVCQTKNUUQOELD-UHFFFAOYSA-N 4-amino-n-[1-(3-chloro-2-fluoroanilino)-6-methylisoquinolin-5-yl]thieno[3,2-d]pyrimidine-7-carboxamide Chemical compound N=1C=CC2=C(NC(=O)C=3C4=NC=NC(N)=C4SC=3)C(C)=CC=C2C=1NC1=CC=CC(Cl)=C1F KVCQTKNUUQOELD-UHFFFAOYSA-N 0.000 description 1
- WHSIIJQOEGXWSN-RLTMCGQMSA-N 4-bromo-2,3,5-trideuterio-6-(trideuteriomethoxy)phenol Chemical compound BrC1=C(C(=C(C(=C1[2H])OC([2H])([2H])[2H])O)[2H])[2H] WHSIIJQOEGXWSN-RLTMCGQMSA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- NRTSMZFIDWZGFJ-UHFFFAOYSA-N 5-(4-cyclopropyl-2-fluorophenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxylic acid Chemical compound C1(CC1)C1=CC(=C(C=C1)C=1C(C(=CN(C=1)CC1CCOCC1)C(=O)O)=O)F NRTSMZFIDWZGFJ-UHFFFAOYSA-N 0.000 description 1
- IVILGUFRMDBUEQ-UHFFFAOYSA-N 5-iodopyridin-2-amine Chemical compound NC1=CC=C(I)C=N1 IVILGUFRMDBUEQ-UHFFFAOYSA-N 0.000 description 1
- HAZMBQGWYYGRES-UHFFFAOYSA-N 6-chloro-5-fluoropyridin-3-amine Chemical compound NC1=CN=C(Cl)C(F)=C1 HAZMBQGWYYGRES-UHFFFAOYSA-N 0.000 description 1
- NKGPJODWTZCHGF-KQYNXXCUSA-N 6-thioinosinic acid Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(S)=C2N=C1 NKGPJODWTZCHGF-KQYNXXCUSA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- CYJRNFFLTBEQSQ-UHFFFAOYSA-N 8-(3-methyl-1-benzothiophen-5-yl)-N-(4-methylsulfonylpyridin-3-yl)quinoxalin-6-amine Chemical compound CS(=O)(=O)C1=C(C=NC=C1)NC=1C=C2N=CC=NC2=C(C=1)C=1C=CC2=C(C(=CS2)C)C=1 CYJRNFFLTBEQSQ-UHFFFAOYSA-N 0.000 description 1
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 1
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 108010024976 Asparaginase Proteins 0.000 description 1
- NOWKCMXCCJGMRR-UHFFFAOYSA-N Aziridine Chemical compound C1CN1 NOWKCMXCCJGMRR-UHFFFAOYSA-N 0.000 description 1
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 1
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 1
- WOVKYSAHUYNSMH-UHFFFAOYSA-N BROMODEOXYURIDINE Natural products C1C(O)C(CO)OC1N1C(=O)NC(=O)C(Br)=C1 WOVKYSAHUYNSMH-UHFFFAOYSA-N 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- 206010055113 Breast cancer metastatic Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- FVLVBPDQNARYJU-XAHDHGMMSA-N C[C@H]1CCC(CC1)NC(=O)N(CCCl)N=O Chemical compound C[C@H]1CCC(CC1)NC(=O)N(CCCl)N=O FVLVBPDQNARYJU-XAHDHGMMSA-N 0.000 description 1
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- SHHKQEUPHAENFK-UHFFFAOYSA-N Carboquone Chemical compound O=C1C(C)=C(N2CC2)C(=O)C(C(COC(N)=O)OC)=C1N1CC1 SHHKQEUPHAENFK-UHFFFAOYSA-N 0.000 description 1
- AOCCBINRVIKJHY-UHFFFAOYSA-N Carmofur Chemical compound CCCCCCNC(=O)N1C=C(F)C(=O)NC1=O AOCCBINRVIKJHY-UHFFFAOYSA-N 0.000 description 1
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 1
- 206010008342 Cervix carcinoma Diseases 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- QMBJSIBWORFWQT-DFXBJWIESA-N Chlormadinone acetate Chemical compound C1=C(Cl)C2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)C)[C@@]1(C)CC2 QMBJSIBWORFWQT-DFXBJWIESA-N 0.000 description 1
- MKQWTWSXVILIKJ-LXGUWJNJSA-N Chlorozotocin Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](C=O)NC(=O)N(N=O)CCCl MKQWTWSXVILIKJ-LXGUWJNJSA-N 0.000 description 1
- RURLVUZRUFHCJO-UHFFFAOYSA-N Chromomycin A3 Natural products COC(C1Cc2cc3cc(OC4CC(OC(=O)C)C(OC5CC(O)C(OC)C(C)O5)C(C)O4)c(C)c(O)c3c(O)c2C(=O)C1OC6CC(OC7CC(C)(O)C(OC(=O)C)C(C)O7)C(O)C(C)O6)C(=O)C(O)C(C)O RURLVUZRUFHCJO-UHFFFAOYSA-N 0.000 description 1
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 1
- 229910021591 Copper(I) chloride Inorganic materials 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 1
- ZZZCUOFIHGPKAK-UHFFFAOYSA-N D-erythro-ascorbic acid Natural products OCC1OC(=O)C(O)=C1O ZZZCUOFIHGPKAK-UHFFFAOYSA-N 0.000 description 1
- 108020004414 DNA Proteins 0.000 description 1
- 108010092160 Dactinomycin Proteins 0.000 description 1
- ZBNZXTGUTAYRHI-UHFFFAOYSA-N Dasatinib Chemical compound C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1Cl ZBNZXTGUTAYRHI-UHFFFAOYSA-N 0.000 description 1
- BWLUMTFWVZZZND-UHFFFAOYSA-N Dibenzylamine Chemical class C=1C=CC=CC=1CNCC1=CC=CC=C1 BWLUMTFWVZZZND-UHFFFAOYSA-N 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical class C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- 239000004593 Epoxy Substances 0.000 description 1
- 241000283074 Equus asinus Species 0.000 description 1
- BFPYWIDHMRZLRN-SLHNCBLASA-N Ethinyl estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@](CC4)(O)C#C)[C@@H]4[C@@H]3CCC2=C1 BFPYWIDHMRZLRN-SLHNCBLASA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical class NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- 102100031487 Growth arrest-specific protein 6 Human genes 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- 208000017891 HER2 positive breast carcinoma Diseases 0.000 description 1
- 108010068250 Herpes Simplex Virus Protein Vmw65 Proteins 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000923005 Homo sapiens Growth arrest-specific protein 6 Proteins 0.000 description 1
- 101000973901 Homo sapiens Tyrosine-protein kinase Mer Proteins 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 201000009794 Idiopathic Pulmonary Fibrosis Diseases 0.000 description 1
- PIWKPBJCKXDKJR-UHFFFAOYSA-N Isoflurane Chemical compound FC(F)OC(Cl)C(F)(F)F PIWKPBJCKXDKJR-UHFFFAOYSA-N 0.000 description 1
- AHLPHDHHMVZTML-BYPYZUCNSA-N L-Ornithine Chemical class NCCC[C@H](N)C(O)=O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 1
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 1
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 1
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 1
- 239000002067 L01XE06 - Dasatinib Substances 0.000 description 1
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 1
- 238000000134 MTT assay Methods 0.000 description 1
- 231100000002 MTT assay Toxicity 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- PGAUJQOPTMSERF-QWQRBHLCSA-N Methenolone acetate Chemical compound C([C@@H]1CC2)C(=O)C=C(C)[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H](OC(=O)C)[C@@]2(C)CC1 PGAUJQOPTMSERF-QWQRBHLCSA-N 0.000 description 1
- FQISKWAFAHGMGT-SGJOWKDISA-M Methylprednisolone sodium succinate Chemical compound [Na+].C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2[C@@H](O)C[C@]2(C)[C@@](O)(C(=O)COC(=O)CCC([O-])=O)CC[C@H]21 FQISKWAFAHGMGT-SGJOWKDISA-M 0.000 description 1
- VFKZTMPDYBFSTM-KVTDHHQDSA-N Mitobronitol Chemical compound BrC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CBr VFKZTMPDYBFSTM-KVTDHHQDSA-N 0.000 description 1
- HGINADPHJQTSKN-UHFFFAOYSA-N Monoethyl malonic acid Chemical compound CCOC(=O)CC(O)=O HGINADPHJQTSKN-UHFFFAOYSA-N 0.000 description 1
- 101100101585 Mus musculus Ubqln4 gene Proteins 0.000 description 1
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 1
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- FXBJJYIXVDHGJQ-UHFFFAOYSA-N N-(5-bromopyrazin-2-yl)-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound BrC=1N=CC(=NC=1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1 FXBJJYIXVDHGJQ-UHFFFAOYSA-N 0.000 description 1
- YYGLURGKDYINIT-UHFFFAOYSA-N N-(5-iodopyridin-2-yl)-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound IC=1C=CC(=NC=1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1 YYGLURGKDYINIT-UHFFFAOYSA-N 0.000 description 1
- DPUWYVAVBSHSDS-UHFFFAOYSA-N N-(6-chloropyridazin-3-yl)-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound ClC1=CC=C(N=N1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1 DPUWYVAVBSHSDS-UHFFFAOYSA-N 0.000 description 1
- AYCPARAPKDAOEN-LJQANCHMSA-N N-[(1S)-2-(dimethylamino)-1-phenylethyl]-6,6-dimethyl-3-[(2-methyl-4-thieno[3,2-d]pyrimidinyl)amino]-1,4-dihydropyrrolo[3,4-c]pyrazole-5-carboxamide Chemical compound C1([C@H](NC(=O)N2C(C=3NN=C(NC=4C=5SC=CC=5N=C(C)N=4)C=3C2)(C)C)CN(C)C)=CC=CC=C1 AYCPARAPKDAOEN-LJQANCHMSA-N 0.000 description 1
- NDHJRDQILUOWLR-UHFFFAOYSA-N N-[4-(2-amino-5-bromopyridin-3-yl)-3-fluorophenyl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=C(C=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)F)Br NDHJRDQILUOWLR-UHFFFAOYSA-N 0.000 description 1
- NATXQLRQRJHHRS-UHFFFAOYSA-N N-[4-(2-amino-5-bromopyridin-3-yl)phenyl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)Br NATXQLRQRJHHRS-UHFFFAOYSA-N 0.000 description 1
- VNBRGSXVFBYQNN-UHFFFAOYSA-N N-[4-[(2-amino-3-chloro-4-pyridinyl)oxy]-3-fluorophenyl]-4-ethoxy-1-(4-fluorophenyl)-2-oxo-3-pyridinecarboxamide Chemical compound O=C1C(C(=O)NC=2C=C(F)C(OC=3C(=C(N)N=CC=3)Cl)=CC=2)=C(OCC)C=CN1C1=CC=C(F)C=C1 VNBRGSXVFBYQNN-UHFFFAOYSA-N 0.000 description 1
- GEAJYTNNHUHUPD-UHFFFAOYSA-N N-[4-[2-amino-5-(3,4-dimethoxyphenyl)pyridin-3-yl]-3-fluorophenyl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound C1(C=2C=C(C3=CC=C(OC)C(OC)=C3)C=NC=2N)=CC=C(NC(=O)C=2C(=O)C(C3=CC=C(C=C3)C)=CN(C=2)CC2CCOCC2)C=C1F GEAJYTNNHUHUPD-UHFFFAOYSA-N 0.000 description 1
- YLKPPBUPVVBPDB-UHFFFAOYSA-N N-[4-[2-amino-5-(3,4-dimethoxyphenyl)pyridin-3-yl]phenyl]-5-(4-bromophenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)Br)CC1CCOCC1)C1=CC(=C(C=C1)OC)OC YLKPPBUPVVBPDB-UHFFFAOYSA-N 0.000 description 1
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical class CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- KCTZOTUQSGYWLV-UHFFFAOYSA-N N1C=NC=C2N=CC=C21 Chemical group N1C=NC=C2N=CC=C21 KCTZOTUQSGYWLV-UHFFFAOYSA-N 0.000 description 1
- 229910021586 Nickel(II) chloride Inorganic materials 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 239000000020 Nitrocellulose Substances 0.000 description 1
- AFLXUQUGROGEFA-UHFFFAOYSA-N Nitrogen mustard N-oxide Chemical compound ClCC[N+]([O-])(C)CCCl AFLXUQUGROGEFA-UHFFFAOYSA-N 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- 102000043276 Oncogene Human genes 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 108010057150 Peplomycin Proteins 0.000 description 1
- 102000003992 Peroxidases Human genes 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 1
- 229940122907 Phosphatase inhibitor Drugs 0.000 description 1
- 239000004153 Potassium bromate Substances 0.000 description 1
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 1
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 1
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- AHHFEZNOXOZZQA-ZEBDFXRSSA-N Ranimustine Chemical compound CO[C@H]1O[C@H](CNC(=O)N(CCCl)N=O)[C@@H](O)[C@H](O)[C@H]1O AHHFEZNOXOZZQA-ZEBDFXRSSA-N 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 208000007014 Retinitis pigmentosa Diseases 0.000 description 1
- 201000000582 Retinoblastoma Diseases 0.000 description 1
- 101100485158 Salmonella typhimurium (strain LT2 / SGSC1412 / ATCC 700720) wzzE gene Proteins 0.000 description 1
- 229920000519 Sizofiran Polymers 0.000 description 1
- 108020004459 Small interfering RNA Proteins 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical class [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 208000000277 Splenic Neoplasms Diseases 0.000 description 1
- 108091005729 TAM receptors Proteins 0.000 description 1
- 108091077436 Tam family Proteins 0.000 description 1
- 229940123237 Taxane Drugs 0.000 description 1
- WFWLQNSHRPWKFK-UHFFFAOYSA-N Tegafur Chemical compound O=C1NC(=O)C(F)=CN1C1OCCC1 WFWLQNSHRPWKFK-UHFFFAOYSA-N 0.000 description 1
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 1
- 208000024313 Testicular Neoplasms Diseases 0.000 description 1
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 1
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 1
- 201000009365 Thymic carcinoma Diseases 0.000 description 1
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 1
- 206010052779 Transplant rejections Diseases 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 239000013504 Triton X-100 Substances 0.000 description 1
- 229920004890 Triton X-100 Polymers 0.000 description 1
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 1
- 229910052770 Uranium Inorganic materials 0.000 description 1
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 229940122803 Vinca alkaloid Drugs 0.000 description 1
- FPIPGXGPPPQFEQ-BOOMUCAASA-N Vitamin A Natural products OC/C=C(/C)\C=C\C=C(\C)/C=C/C1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-BOOMUCAASA-N 0.000 description 1
- 229930003268 Vitamin C Natural products 0.000 description 1
- SPEUIVXLLWOEMJ-UHFFFAOYSA-N acetaldehyde dimethyl acetal Natural products COC(C)OC SPEUIVXLLWOEMJ-UHFFFAOYSA-N 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- 125000000218 acetic acid group Chemical group C(C)(=O)* 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- USZYSDMBJDPRIF-SVEJIMAYSA-N aclacinomycin A Chemical compound O([C@H]1[C@@H](O)C[C@@H](O[C@H]1C)O[C@H]1[C@H](C[C@@H](O[C@H]1C)O[C@H]1C[C@]([C@@H](C2=CC=3C(=O)C4=CC=CC(O)=C4C(=O)C=3C(O)=C21)C(=O)OC)(O)CC)N(C)C)[C@H]1CCC(=O)[C@H](C)O1 USZYSDMBJDPRIF-SVEJIMAYSA-N 0.000 description 1
- 229960004176 aclarubicin Drugs 0.000 description 1
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- FPIPGXGPPPQFEQ-OVSJKPMPSA-N all-trans-retinol Chemical compound OC\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-OVSJKPMPSA-N 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 239000003817 anthracycline antibiotic agent Substances 0.000 description 1
- 230000003432 anti-folate effect Effects 0.000 description 1
- 229940127074 antifolate Drugs 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000013011 aqueous formulation Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 125000005228 aryl sulfonate group Chemical group 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-L aspartate group Chemical class N[C@@H](CC(=O)[O-])C(=O)[O-] CKLJMWTZIZZHCS-REOHCLBHSA-L 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 108010023337 axl receptor tyrosine kinase Proteins 0.000 description 1
- 238000010533 azeotropic distillation Methods 0.000 description 1
- HONIICLYMWZJFZ-UHFFFAOYSA-N azetidine Chemical compound C1CNC1 HONIICLYMWZJFZ-UHFFFAOYSA-N 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 1
- 229960002537 betamethasone Drugs 0.000 description 1
- UREBDLICKHMUKA-DVTGEIKXSA-N betamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-DVTGEIKXSA-N 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- YNHIGQDRGKUECZ-UHFFFAOYSA-L bis(triphenylphosphine)palladium(ii) dichloride Chemical compound [Cl-].[Cl-].[Pd+2].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-L 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 230000031709 bromination Effects 0.000 description 1
- 238000005893 bromination reaction Methods 0.000 description 1
- 229950004398 broxuridine Drugs 0.000 description 1
- 210000005252 bulbus oculi Anatomy 0.000 description 1
- 239000004067 bulking agent Substances 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- 125000004106 butoxy group Chemical group [*]OC([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 1
- 229940127093 camptothecin Drugs 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 125000005587 carbonate group Chemical group 0.000 description 1
- JYYOBHFYCIDXHH-UHFFFAOYSA-N carbonic acid;hydrate Chemical compound O.OC(O)=O JYYOBHFYCIDXHH-UHFFFAOYSA-N 0.000 description 1
- 229960002115 carboquone Drugs 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 125000006244 carboxylic acid protecting group Chemical group 0.000 description 1
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 description 1
- 206010061592 cardiac fibrillation Diseases 0.000 description 1
- 229960003261 carmofur Drugs 0.000 description 1
- 229960005243 carmustine Drugs 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 210000003986 cell retinal photoreceptor Anatomy 0.000 description 1
- 201000010881 cervical cancer Diseases 0.000 description 1
- 229960005395 cetuximab Drugs 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 229960003996 chlormadinone Drugs 0.000 description 1
- 229960001480 chlorozotocin Drugs 0.000 description 1
- ZYVSOIYQKUDENJ-WKSBCEQHSA-N chromomycin A3 Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@@H]1OC(C)=O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@@H](O)[C@H](O[C@@H]3O[C@@H](C)[C@H](OC(C)=O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@@H]1C[C@@H](O)[C@@H](OC)[C@@H](C)O1 ZYVSOIYQKUDENJ-WKSBCEQHSA-N 0.000 description 1
- 150000001860 citric acid derivatives Chemical class 0.000 description 1
- 230000004186 co-expression Effects 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 201000006754 cone-rod dystrophy Diseases 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- OXBLHERUFWYNTN-UHFFFAOYSA-M copper(I) chloride Chemical compound [Cu]Cl OXBLHERUFWYNTN-UHFFFAOYSA-M 0.000 description 1
- 238000012258 culturing Methods 0.000 description 1
- 125000006165 cyclic alkyl group Chemical group 0.000 description 1
- 125000000392 cycloalkenyl group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 150000003946 cyclohexylamines Chemical class 0.000 description 1
- GCUVBACNBHGZRS-UHFFFAOYSA-N cyclopenta-1,3-diene cyclopenta-2,4-dien-1-yl(diphenyl)phosphane iron(2+) Chemical compound [Fe++].c1cc[cH-]c1.c1cc[c-](c1)P(c1ccccc1)c1ccccc1 GCUVBACNBHGZRS-UHFFFAOYSA-N 0.000 description 1
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 1
- HKJAEGDDNPSICL-UHFFFAOYSA-N cyclopenta-2,4-dien-1-yl(diphenyl)phosphane;dichloromethane;iron(2+) Chemical compound [Fe+2].ClCCl.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 HKJAEGDDNPSICL-UHFFFAOYSA-N 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 229960000684 cytarabine Drugs 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- 229960000640 dactinomycin Drugs 0.000 description 1
- 229960002448 dasatinib Drugs 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- FMGSKLZLMKYGDP-USOAJAOKSA-N dehydroepiandrosterone Chemical compound C1[C@@H](O)CC[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC=C21 FMGSKLZLMKYGDP-USOAJAOKSA-N 0.000 description 1
- 238000004925 denaturation Methods 0.000 description 1
- 230000036425 denaturation Effects 0.000 description 1
- 229960001251 denosumab Drugs 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 229960003957 dexamethasone Drugs 0.000 description 1
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 1
- 229940039227 diagnostic agent Drugs 0.000 description 1
- 239000000032 diagnostic agent Substances 0.000 description 1
- ZOCHARZZJNPSEU-UHFFFAOYSA-N diboron Chemical compound B#B ZOCHARZZJNPSEU-UHFFFAOYSA-N 0.000 description 1
- AGCOMFFHXJMNLN-UHFFFAOYSA-N dichloromethane;dihydrochloride Chemical compound Cl.Cl.ClCCl AGCOMFFHXJMNLN-UHFFFAOYSA-N 0.000 description 1
- 150000005332 diethylamines Chemical class 0.000 description 1
- NLORYLAYLIXTID-ISLYRVAYSA-N diethylstilbestrol diphosphate Chemical compound C=1C=C(OP(O)(O)=O)C=CC=1C(/CC)=C(\CC)C1=CC=C(OP(O)(O)=O)C=C1 NLORYLAYLIXTID-ISLYRVAYSA-N 0.000 description 1
- 238000009792 diffusion process Methods 0.000 description 1
- 125000005043 dihydropyranyl group Chemical group O1C(CCC=C1)* 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 125000006222 dimethylaminomethyl group Chemical group [H]C([H])([H])N(C([H])([H])[H])C([H])([H])* 0.000 description 1
- GPAYUJZHTULNBE-UHFFFAOYSA-N diphenylphosphine Chemical compound C=1C=CC=CC=1PC1=CC=CC=C1 GPAYUJZHTULNBE-UHFFFAOYSA-N 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 1
- 229960003668 docetaxel Drugs 0.000 description 1
- ZWAOHEXOSAUJHY-ZIYNGMLESA-N doxifluridine Chemical compound O[C@@H]1[C@H](O)[C@@H](C)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ZWAOHEXOSAUJHY-ZIYNGMLESA-N 0.000 description 1
- 229950005454 doxifluridine Drugs 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 201000006828 endometrial hyperplasia Diseases 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- YQGOJNYOYNNSMM-UHFFFAOYSA-N eosin Chemical compound [Na+].OC(=O)C1=CC=CC=C1C1=C2C=C(Br)C(=O)C(Br)=C2OC2=C(Br)C(O)=C(Br)C=C21 YQGOJNYOYNNSMM-UHFFFAOYSA-N 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-N ethanesulfonic acid Chemical class CCS(O)(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-N 0.000 description 1
- FXPHJTKVWZVEGA-UHFFFAOYSA-N ethenyl hydrogen carbonate Chemical group OC(=O)OC=C FXPHJTKVWZVEGA-UHFFFAOYSA-N 0.000 description 1
- 125000001033 ether group Chemical group 0.000 description 1
- 229960002568 ethinylestradiol Drugs 0.000 description 1
- NAIAJPLUTCZXKC-UHFFFAOYSA-N ethyl 2-(4-bromo-2-fluorophenyl)acetate Chemical compound CCOC(=O)CC1=CC=C(Br)C=C1F NAIAJPLUTCZXKC-UHFFFAOYSA-N 0.000 description 1
- PYUFYLOTFMNRSN-UHFFFAOYSA-N ethyl 4-(5-methylpyridin-2-yl)-3-oxobutanoate Chemical compound CC=1C=CC(=NC=1)CC(CC(=O)OCC)=O PYUFYLOTFMNRSN-UHFFFAOYSA-N 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 239000012894 fetal calf serum Substances 0.000 description 1
- 230000002600 fibrillogenic effect Effects 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- 239000004052 folic acid antagonist Substances 0.000 description 1
- 150000004675 formic acid derivatives Chemical class 0.000 description 1
- 229960000297 fosfestrol Drugs 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-L fumarate(2-) Chemical class [O-]C(=O)\C=C\C([O-])=O VZCYOOQTPOCHFL-OWOJBTEDSA-L 0.000 description 1
- 229960002584 gefitinib Drugs 0.000 description 1
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 150000002306 glutamic acid derivatives Chemical class 0.000 description 1
- 230000009422 growth inhibiting effect Effects 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 208000014829 head and neck neoplasm Diseases 0.000 description 1
- 201000005787 hematologic cancer Diseases 0.000 description 1
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 1
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 1
- 125000004366 heterocycloalkenyl group Chemical group 0.000 description 1
- 230000036732 histological change Effects 0.000 description 1
- 102000045684 human MERTK Human genes 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- 229960000890 hydrocortisone Drugs 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 1
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical class I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 1
- 229960002411 imatinib Drugs 0.000 description 1
- 229960003685 imatinib mesylate Drugs 0.000 description 1
- YLMAHDNUQAMNNX-UHFFFAOYSA-N imatinib methanesulfonate Chemical compound CS(O)(=O)=O.C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 YLMAHDNUQAMNNX-UHFFFAOYSA-N 0.000 description 1
- MTNDZQHUAFNZQY-UHFFFAOYSA-N imidazoline Chemical compound C1CN=CN1 MTNDZQHUAFNZQY-UHFFFAOYSA-N 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- DBIGHPPNXATHOF-UHFFFAOYSA-N improsulfan Chemical compound CS(=O)(=O)OCCCNCCCOS(C)(=O)=O DBIGHPPNXATHOF-UHFFFAOYSA-N 0.000 description 1
- 229950008097 improsulfan Drugs 0.000 description 1
- 238000011503 in vivo imaging Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- JYGFTBXVXVMTGB-UHFFFAOYSA-N indolin-2-one Chemical group C1=CC=C2NC(=O)CC2=C1 JYGFTBXVXVMTGB-UHFFFAOYSA-N 0.000 description 1
- 229960000905 indomethacin Drugs 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 229960000598 infliximab Drugs 0.000 description 1
- 208000036971 interstitial lung disease 2 Diseases 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- ZCYVEMRRCGMTRW-YPZZEJLDSA-N iodine-125 Chemical compound [125I] ZCYVEMRRCGMTRW-YPZZEJLDSA-N 0.000 description 1
- 229940044173 iodine-125 Drugs 0.000 description 1
- 238000004255 ion exchange chromatography Methods 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 229960002725 isoflurane Drugs 0.000 description 1
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000000155 isotopic effect Effects 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 229910003002 lithium salt Inorganic materials 0.000 description 1
- 159000000002 lithium salts Chemical class 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 229960002247 lomustine Drugs 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000001165 lymph node Anatomy 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 150000002688 maleic acid derivatives Chemical class 0.000 description 1
- 150000004701 malic acid derivatives Chemical class 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 229960004961 mechlorethamine Drugs 0.000 description 1
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical class ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 229960004616 medroxyprogesterone Drugs 0.000 description 1
- PSGAAPLEWMOORI-PEINSRQWSA-N medroxyprogesterone acetate Chemical compound C([C@@]12C)CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2CC[C@]2(C)[C@@](OC(C)=O)(C(C)=O)CC[C@H]21 PSGAAPLEWMOORI-PEINSRQWSA-N 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- 101150089110 metN gene Proteins 0.000 description 1
- 230000001394 metastastic effect Effects 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- 229960003578 metenolone Drugs 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-M methanesulfonate group Chemical class CS(=O)(=O)[O-] AFVFQIVMOAPDHO-UHFFFAOYSA-M 0.000 description 1
- 125000005948 methanesulfonyloxy group Chemical group 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- HRDXJKGNWSUIBT-UHFFFAOYSA-N methoxybenzene Chemical group [CH2]OC1=CC=CC=C1 HRDXJKGNWSUIBT-UHFFFAOYSA-N 0.000 description 1
- MOSJHTAFHHUNRB-UHFFFAOYSA-N methyl 5-bromo-4-oxo-1h-pyridine-3-carboxylate Chemical compound COC(=O)C1=CNC=C(Br)C1=O MOSJHTAFHHUNRB-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 229960004584 methylprednisolone Drugs 0.000 description 1
- 229960005485 mitobronitol Drugs 0.000 description 1
- VFKZTMPDYBFSTM-GUCUJZIJSA-N mitolactol Chemical compound BrC[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)CBr VFKZTMPDYBFSTM-GUCUJZIJSA-N 0.000 description 1
- 229950010913 mitolactol Drugs 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 150000002780 morpholines Chemical class 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- QLVWKBNYBAWIPW-UHFFFAOYSA-N n-quinolin-2-yloxybenzenesulfonamide Chemical group C=1C=C2C=CC=CC2=NC=1ONS(=O)(=O)C1=CC=CC=C1 QLVWKBNYBAWIPW-UHFFFAOYSA-N 0.000 description 1
- 229960004719 nandrolone Drugs 0.000 description 1
- NPAGDVCDWIYMMC-IZPLOLCNSA-N nandrolone Chemical compound O=C1CC[C@@H]2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 NPAGDVCDWIYMMC-IZPLOLCNSA-N 0.000 description 1
- 208000025189 neoplasm of testis Diseases 0.000 description 1
- 229950010203 nimotuzumab Drugs 0.000 description 1
- 229960001420 nimustine Drugs 0.000 description 1
- VFEDRRNHLBGPNN-UHFFFAOYSA-N nimustine Chemical compound CC1=NC=C(CNC(=O)N(CCCl)N=O)C(N)=N1 VFEDRRNHLBGPNN-UHFFFAOYSA-N 0.000 description 1
- 150000002823 nitrates Chemical class 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 229920001220 nitrocellulos Polymers 0.000 description 1
- OSTGTTZJOCZWJG-UHFFFAOYSA-N nitrosourea Chemical compound NC(=O)N=NO OSTGTTZJOCZWJG-UHFFFAOYSA-N 0.000 description 1
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- 150000003891 oxalate salts Chemical class 0.000 description 1
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 1
- 229960001756 oxaliplatin Drugs 0.000 description 1
- IPBPLHNLRKRLPJ-UHFFFAOYSA-N oxan-4-ylmethanamine Chemical compound NCC1CCOCC1 IPBPLHNLRKRLPJ-UHFFFAOYSA-N 0.000 description 1
- HJZXNSXKMBRTJZ-UHFFFAOYSA-N oxan-4-ylmethyl methanesulfonate Chemical compound CS(=O)(=O)OCC1CCOCC1 HJZXNSXKMBRTJZ-UHFFFAOYSA-N 0.000 description 1
- AHHWIHXENZJRFG-UHFFFAOYSA-N oxetane Chemical compound C1COC1 AHHWIHXENZJRFG-UHFFFAOYSA-N 0.000 description 1
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 229960005244 oxymetholone Drugs 0.000 description 1
- ICMWWNHDUZJFDW-DHODBPELSA-N oxymetholone Chemical compound C([C@@H]1CC2)C(=O)\C(=C/O)C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@](C)(O)[C@@]2(C)CC1 ICMWWNHDUZJFDW-DHODBPELSA-N 0.000 description 1
- ICMWWNHDUZJFDW-UHFFFAOYSA-N oxymetholone Natural products C1CC2CC(=O)C(=CO)CC2(C)C2C1C1CCC(C)(O)C1(C)CC2 ICMWWNHDUZJFDW-UHFFFAOYSA-N 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- MUJIDPITZJWBSW-UHFFFAOYSA-N palladium(2+) Chemical compound [Pd+2] MUJIDPITZJWBSW-UHFFFAOYSA-N 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 229950003180 peplomycin Drugs 0.000 description 1
- QIMGFXOHTOXMQP-GFAGFCTOSA-N peplomycin Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCCN[C@@H](C)C=1C=CC=CC=1)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1NC=NC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C QIMGFXOHTOXMQP-GFAGFCTOSA-N 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical class OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 1
- 108040007629 peroxidase activity proteins Proteins 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 1
- LJYVSUWJFYSYHT-UHFFFAOYSA-N phenyl ethanesulfonate Chemical compound CCS(=O)(=O)OC1=CC=CC=C1 LJYVSUWJFYSYHT-UHFFFAOYSA-N 0.000 description 1
- FPJQWFBQXIKMMP-UHFFFAOYSA-N phenyl nitrate Chemical compound [O-][N+](=O)OC1=CC=CC=C1 FPJQWFBQXIKMMP-UHFFFAOYSA-N 0.000 description 1
- CMPQUABWPXYYSH-UHFFFAOYSA-N phenyl phosphate Chemical compound OP(O)(=O)OC1=CC=CC=C1 CMPQUABWPXYYSH-UHFFFAOYSA-N 0.000 description 1
- NIXKBAZVOQAHGC-UHFFFAOYSA-M phenylmethanesulfonate Chemical compound [O-]S(=O)(=O)CC1=CC=CC=C1 NIXKBAZVOQAHGC-UHFFFAOYSA-M 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 230000000865 phosphorylative effect Effects 0.000 description 1
- 150000004885 piperazines Chemical class 0.000 description 1
- 229960000952 pipobroman Drugs 0.000 description 1
- NJBFOOCLYDNZJN-UHFFFAOYSA-N pipobroman Chemical compound BrCCC(=O)N1CCN(C(=O)CCBr)CC1 NJBFOOCLYDNZJN-UHFFFAOYSA-N 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 229940094037 potassium bromate Drugs 0.000 description 1
- 235000019396 potassium bromate Nutrition 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- WVUCPRGADMCTBN-UHFFFAOYSA-M potassium;3-ethoxy-3-oxopropanoate Chemical compound [K+].CCOC(=O)CC([O-])=O WVUCPRGADMCTBN-UHFFFAOYSA-M 0.000 description 1
- 229960002847 prasterone Drugs 0.000 description 1
- 229960005205 prednisolone Drugs 0.000 description 1
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 description 1
- 238000012746 preparative thin layer chromatography Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- YAAWASYJIRZXSZ-UHFFFAOYSA-N pyrimidine-2,4-diamine Chemical group NC1=CC=NC(N)=N1 YAAWASYJIRZXSZ-UHFFFAOYSA-N 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- SBYHFKPVCBCYGV-UHFFFAOYSA-N quinuclidine Chemical compound C1CC2CCN1CC2 SBYHFKPVCBCYGV-UHFFFAOYSA-N 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 229960002185 ranimustine Drugs 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 238000005057 refrigeration Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000013557 residual solvent Substances 0.000 description 1
- 230000008261 resistance mechanism Effects 0.000 description 1
- 230000002207 retinal effect Effects 0.000 description 1
- 229960004641 rituximab Drugs 0.000 description 1
- 239000012488 sample solution Substances 0.000 description 1
- 229960003440 semustine Drugs 0.000 description 1
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 1
- 229950001403 sizofiran Drugs 0.000 description 1
- 235000020183 skimmed milk Nutrition 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 1
- 229960003787 sorafenib Drugs 0.000 description 1
- 201000002471 spleen cancer Diseases 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 150000003431 steroids Chemical group 0.000 description 1
- 229960001052 streptozocin Drugs 0.000 description 1
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 150000003890 succinate salts Chemical class 0.000 description 1
- 125000000565 sulfonamide group Chemical group 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 1
- 229960001796 sunitinib Drugs 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- 150000003892 tartrate salts Chemical class 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 229960001674 tegafur Drugs 0.000 description 1
- WFWLQNSHRPWKFK-ZCFIWIBFSA-N tegafur Chemical compound O=C1NC(=O)C(F)=CN1[C@@H]1OCCC1 WFWLQNSHRPWKFK-ZCFIWIBFSA-N 0.000 description 1
- 229960004964 temozolomide Drugs 0.000 description 1
- 230000002123 temporal effect Effects 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- 201000003120 testicular cancer Diseases 0.000 description 1
- 238000012956 testing procedure Methods 0.000 description 1
- 125000004853 tetrahydropyridinyl group Chemical group N1(CCCC=C1)* 0.000 description 1
- QEMXHQIAXOOASZ-UHFFFAOYSA-N tetramethylammonium Chemical class C[N+](C)(C)C QEMXHQIAXOOASZ-UHFFFAOYSA-N 0.000 description 1
- 229960001196 thiotepa Drugs 0.000 description 1
- 208000008732 thymoma Diseases 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- 229960003087 tioguanine Drugs 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 229960000575 trastuzumab Drugs 0.000 description 1
- 229960005294 triamcinolone Drugs 0.000 description 1
- GFNANZIMVAIWHM-OBYCQNJPSA-N triamcinolone Chemical compound O=C1C=C[C@]2(C)[C@@]3(F)[C@@H](O)C[C@](C)([C@@]([C@H](O)C4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 GFNANZIMVAIWHM-OBYCQNJPSA-N 0.000 description 1
- 150000003852 triazoles Chemical group 0.000 description 1
- OVCXRBARSPBVMC-UHFFFAOYSA-N triazolopyridine Chemical compound C=1N2C(C(C)C)=NN=C2C=CC=1C=1OC=NC=1C1=CC=C(F)C=C1 OVCXRBARSPBVMC-UHFFFAOYSA-N 0.000 description 1
- YWBFPKPWMSWWEA-UHFFFAOYSA-O triazolopyrimidine Chemical group BrC1=CC=CC(C=2N=C3N=CN[N+]3=C(NCC=3C=CN=CC=3)C=2)=C1 YWBFPKPWMSWWEA-UHFFFAOYSA-O 0.000 description 1
- INQOMBQAUSQDDS-FIBGUPNXSA-N trideuterio(iodo)methane Chemical compound [2H]C([2H])([2H])I INQOMBQAUSQDDS-FIBGUPNXSA-N 0.000 description 1
- 125000005951 trifluoromethanesulfonyloxy group Chemical group 0.000 description 1
- NOYPYLRCIDNJJB-UHFFFAOYSA-N trimetrexate Chemical compound COC1=C(OC)C(OC)=CC(NCC=2C(=C3C(N)=NC(N)=NC3=CC=2)C)=C1 NOYPYLRCIDNJJB-UHFFFAOYSA-N 0.000 description 1
- 229960001099 trimetrexate Drugs 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical class OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 102000003390 tumor necrosis factor Human genes 0.000 description 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 1
- 229910052721 tungsten Inorganic materials 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- XSQUKJJJFZCRTK-UHFFFAOYSA-N urea group Chemical group NC(=O)N XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 1
- 201000009825 uterine corpus cancer Diseases 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- UGGWPQSBPIFKDZ-KOTLKJBCSA-N vindesine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(N)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 UGGWPQSBPIFKDZ-KOTLKJBCSA-N 0.000 description 1
- 229960004355 vindesine Drugs 0.000 description 1
- 235000019155 vitamin A Nutrition 0.000 description 1
- 239000011719 vitamin A Substances 0.000 description 1
- 235000019154 vitamin C Nutrition 0.000 description 1
- 239000011718 vitamin C Substances 0.000 description 1
- 229940045997 vitamin a Drugs 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/501—Pyridazines; Hydrogenated pyridazines not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B59/00—Introduction of isotopes of elements into organic compounds ; Labelled organic compounds per se
- C07B59/002—Heterocyclic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/01—Sulfonic acids
- C07C309/02—Sulfonic acids having sulfo groups bound to acyclic carbon atoms
- C07C309/03—Sulfonic acids having sulfo groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
- C07C309/04—Sulfonic acids having sulfo groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton containing only one sulfo group
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/01—Sulfonic acids
- C07C309/28—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C309/33—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton of six-membered aromatic rings being part of condensed ring systems
- C07C309/34—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton of six-membered aromatic rings being part of condensed ring systems formed by two rings
- C07C309/35—Naphthalene sulfonic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/05—Isotopically modified compounds, e.g. labelled
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Urology & Nephrology (AREA)
- Oncology (AREA)
- Physical Education & Sports Medicine (AREA)
- Endocrinology (AREA)
- Dermatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Gastroenterology & Hepatology (AREA)
- Pulmonology (AREA)
- Reproductive Health (AREA)
- Neurology (AREA)
- Diabetes (AREA)
- Rheumatology (AREA)
- Hematology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Un compuesto representado por la fórmula general (I) o una sal farmacéuticamente aceptable del mismo: **(Ver fórmula)** en la que W, X e Y representan cada uno independientemente un átomo de nitrógeno, C-H, C-F o C-Cl, Z representa un átomo de nitrógeno, C-H, C-F, C-Cl, alquilo C-C1-C6 o grupo alcoxi C-C1-C6, R1 representa un grupo representado por la siguiente fórmula (II-1) o (II-2): **(Ver fórmula)** en la que en la fórmula (II-1), Q representa un átomo de nitrógeno, C-H o C-F, T representa un átomo de nitrógeno o C-H, U representa un átomo de nitrógeno o C-H, y R6 representa un átomo de halógeno, un grupo alquilo C1-C6, un grupo cicloalquilo C3-C6, un grupo ciano o un grupo trifluorometoxi, y en la fórmula (II-2), V representa un átomo de azufre o un átomo de oxígeno, y R7 representa un grupo alquilo C1-C6, R2 representa un átomo de hidrógeno, un átomo de halógeno, un grupo alquilo C1-C6, un grupo alcoxi C1-C6 o un grupo ciano, R3 representa un átomo de hidrógeno o un grupo alquilo C1-C6, R4 representa un átomo de hidrógeno, un átomo de halógeno o un grupo alquilo C1-C6, y R5 representa un átomo de hidrógeno o un grupo representado por la siguiente fórmula (III-1), (III-2), (III-3) o (III- 4): **(Ver fórmula)** en la que en la fórmula (III-1), R8 y R12 representan cada uno independientemente un átomo de hidrógeno o un átomo de deuterio, R9 representa un átomo de hidrógeno, un átomo de halógeno o un grupo alcoxi C1-C6, R10 representa un átomo de hidrógeno, un grupo alquilo C1-C6 opcionalmente sustituido con un grupo heterocicloalquilo o un grupo alcoxi C1-C6 opcionalmente sustituido con un grupo heterocicloalquilo opcionalmente sustituido con un grupo alquilo C1-C6, y R11 representa un átomo de hidrógeno, un grupo alcoxi C1-C6 o un grupo alcoxi C1-C6 sustituido con deuterio, en la fórmula (III-2), R13 representa un grupo alquilo C1-C6 opcionalmente sustituido con un grupo heterocicloalquilo, o un grupo heterocicloalquilo, en la fórmula (III-3), R14 representa un átomo de hidrógeno o un grupo alquilo C1-C6, R15 representa un átomo de hidrógeno, un grupo alquilo C1-C6 o un grupo alcoxi C1-C6, y R16 representa un átomo de hidrógeno o un átomo de halógeno, y en la fórmula (III-4), R17 representa un grupo alcoxi C1-C6, y R18 representa un grupo alcoxi C1-C6.
Description
DESCRIPCIÓN
Derivado de piridona que tiene un grupo tetrahidropiranil metilo
[Campo técnico]
La presente invención se refiere a un compuesto o una sal del mismo que tiene actividad inhibidora de Axl.
[Técnica antecedente]
Axl es un receptor de tirosina quinasa que pertenece a la familia de receptores de tirosina quinasa Tyro3-Axl-Mer (TAM) cuyo ligando es una proteína codificada por el gen 6 específico de detención del crecimiento (Gas6). El gen de esta quinasa se identificó originalmente como un gen transformante en la leucemia mielógena crónica (Documento no de patente 1).
Se ha informado que el sistema de señalización Gas6/Axl regula diversas respuestas celulares tales como la supervivencia celular, la división celular, la autofagia, la migración celular, la angiogénesis, la agregación plaquetaria y la diferenciación de las células NK (Documento no de patente 2). Además, muchos informes muestran la sobreexpresión de Axl en tejidos de cánceres tales como cáncer de colon primario (Documento no de patente 3), cáncer gástrico (Documento no de patente 4), cáncer de esófago (Documento no de patente 5), melanoma (Documento no de patente 6), cáncer de ovario (Documento no de patente 7), cáncer de riñón (Documento no de patente 8), cáncer de endometrio (Documento no de patente 9) y cáncer de glándula tiroides (Documento no de patente 10) . Se ha descubierto que la presencia de Axl está estrechamente relacionada con la afectación de los ganglios linfáticos y el estadio del cáncer de pulmón y la expresión de ER en el cáncer de mama (Documento no de patente 11) .
Además, se ha descubierto que Axl desempeña un papel en la inmunidad (Documento no de patente 12), funciones plaquetarias (Documento no de patente 13), espermatogénesis (Documento no de patente 14), calcificación vascular (Documento no de patente 15), proliferación celular de músculo liso vascular inducido por trombina (VSMC) (Documento no de patente 16) y diversas enfermedades renales, por ejemplo, glomerulonefritis aguda y crónica, nefropatía diabética y rechazo crónico de aloinjertos (Documento no de patente 17). Se espera que los inhibidores de Axl proporcionen beneficios terapéuticos para muchas enfermedades, incluidos cánceres (incluidos tumores sólidos tales como carcinoma y sarcoma, leucemia y enfermedades linfoides malignas), así como enfermedades vasculares (incluidas, pero no limitadas a, trombosis, aterosclerosis y reestenosis), enfermedades renales (que incluyen, pero no se limitan a, glomerulonefritis aguda y crónica, nefropatía diabética y rechazo del injerto) y enfermedades en las que la formación desorganizada de los vasos sanguíneos tiene consecuencias graves (que incluyen, pero no se limitan a, retinopatía diabética, retinopatía, psoriasis, artritis reumatoide, ateroma, sarcoma de Kaposi y angioma).
Mientras tanto, se ha informado que Mer, otro miembro de la familia de tirosina quinasas del receptor TAM a la que pertenece Axl, causa retinitis pigmentosa autosómica recesiva a través de su mutación homocigótica (Documento no de patente 24). Se ha informado además de que una determinada mutación en Mer está relacionada con la distrofia de cono-bastón de inicio en la infancia (Documento no de patente 25).
Compuestos que tienen una estructura de sulfonamida (Documento de patente 3), compuestos que tienen una estructura de pirrolopirimidina (Documentos de patente 4 y 5), compuestos que tienen estructuras de piridina y pirazina (Documento de patente 6), compuestos que tienen una estructura de pirazina (Documento de patente 7), compuestos que tienen una estructura de pirazinilbencimidazol (Documento de patente 8), compuestos que tienen una estructura de indolinona (Documento de patente 9), compuestos que tienen estructuras de triazolopiridina y triazolopirimidina (Documento de patente 10), compuestos que tienen una estructura de imidazol (Documento de patente 11), compuestos que tienen una estructura de triazol (Documentos de patente 12, 13, 14, 15, 16, 17, 20, 24, 25, 26, 27 y 28), compuestos que tienen una estructura de pirimidindiamina (Documento de patente 18), compuestos que tienen una estructura de pirimidina (Documento de patente 19 y Documentos que no son de patente 18 y 22), compuestos que tienen una estructura de quinoliniloxifenilsulfonamida (Documento de patente 21), compuestos que tienen una estructura de quinolina (Documentos de patente 22 y 30 y Documentos que no son de patente 21), compuestos que tienen una estructura de piridina (Documentos de patente 23 y 33 y Documento no de patente 19), compuestos que tienen una estructura de urea (Documento de patente 29), compuestos que tienen una estructura de arilamida disustituida en 2,4 (Documento no de patente 20), compuestos que tienen una estructura secosteroide (Documento no de patente 23), compuestos que tienen una estructura de pirimidina bicíclica (Documentos de patente 31, 32 y 34) y similares se han informado como compuestos que inhiben Axl.
[Lista de citas]
[Documentos de Patente]
[Documento de Patente 1] Publicación Internacional No. WO2008/025820
[Documento de Patente 2] Publicación Internacional No. WO2008/074997
[Documento de Patente 3] Publicación Internacional No. WO2008/128072
[Documento de Patente 4] Publicación de Solicitud de Patente U.S. No. 20100204221 [Documento de Patente 5] Publicación Internacional No. WO2010/090764
[Documento de Patente 6] Publicación Internacional No. WO2009/053737
[Documento de Patente 7] Publicación Internacional No. WO2009/007390
[Documento de Patente 8] Publicación Internacional No. WO2009/024825
[Documento de Patente 9] Publicación Internacional No. WO2007/057399
[Documento de Patente 10] Pub icación Internacional No. WO2009/047514
[Documento de Patente 11] Pub icación Internacional No. WO2009/058801
[Documento de Patente 12] Pub icación Internacional No. WO2008/083367
[Documento de Patente 13] Pub icación Internacional No. WO2008/083353
[Documento de Patente 14] Pub icación Internacional No. WO2010/005879
[Documento de Patente 15] Pub icación Internacional No. WO2008/083357
[Documento de Patente 16] Pub icación Internacional No. WO2008/083356
[Documento de Patente 17] Pub icación Internacional No. WO2008/083354
[Documento de Patente 18] Pub icación Internacional No. WO2008/045978
[Documento de Patente 19] Pub icación Internacional No. WO2007/070872
[Documento de Patente 20] Pub icación Internacional No. WO2007/030680
[Documento de Patente 21] Pub icación Internacional No. WO2011/045084
[Documento de Patente 22] Pub icación Internacional No. WO2009/127417
[Documento de Patente 23] Pub icación Internacional No. WO2007/066187
[Documento de Patente 24] Pub icación Internacional No. WO2009/054864
[Documento de Patente 25] Pub icación Internacional No. WO2010/005876
[Documento de Patente 26] Pub icación Internacional No. WO2009/054864
[Documento de Patente 27] Pub icación de Solicitud de Patente U.S. No. 20090111816 [Documento de Patente 28] Pub icación de Solicitud de Patente U.S. No. 20100168416 [Documento de Patente 29] Pub icación Internacional No. WO2009/138799
[Documento de Patente 30] Pub icación de Solicitud de Patente U.S. No. 20090274693 [Documento de Patente 31] Pub icación de Solicitud de Patente U.S. No. 20100069369 [Documento de Patente 32] Pub icación de Solicitud de Patente U.S. No. 20070142402 [Documento de Patente 33] Pub icación Internacional No. WO2013/115280
[Documento de Patente 34] Publi icación Internacional No. WO2013/162061
[Documento no de Patentes]
[Documento no de Patente 1] O'Bryan et al., Mol. Cell. Biol., 11, 5031 (1991)
[Documento no de Patente 2] Rachel MA Linger et al., Expert Opin. Ther. Targets, 14, 1073 (2010) [Documento no de Patente 3] Craven et al., Int. J. Cancer., 60, 791 (1995)
[Documento no de Patente 4] Sawabu et al., Mol. Carcinog., 46, 155 (2007)
[Documento no de Patente 5] Nemoto et al., Pathobiology, 65, 195 (1997)
[Documento no de Patente 6] Quong et al., Melanoma Res., 4, 313 (1994)
[Documento no de Patente 7] Sun et al., Oncology, 66, 450 (2004)
[Documento no de Patente 8] Chung et al., DNA Cell Biol., 22, 533 (2003)
[Documento no de Patente 9] Sun et al., Ann. Oncol., 14, 898 (2003)
[Documento no de Patente 10] Ito et al., Thyroid, 9, 563 (1999)
[Documento no de Patente 11] Berclaz et al., Ann. Oncol., 12, 819 (2001)
[Documento no de Patente 12] Lu et al., Science, 293, 306 (2001)
[Documento no de Patente 13] Angelillo-Scherrer et al., Nat. Med., 7, 215 (2001)
[Documento no de Patente 14] Lu et al., Nature, 398, 723 (1999)
[Documento no de Patente 15] Son et al., Eur. J. Pharmacol., 556, 1 (2007)
[Documento no de Patente 16] Nakano et al., J. Biol. Chem., 270, 5702 (1995)
[Documento no de Patente 17] Yanagita et al., J. Clin. Invest., 110, 239 (2002)
[Documento no de Patente 18] AlexisMollard etal., Med. Chem. Lett., 2, 907 (2011)
[Documento no de Patente 19] Gretchen M. Schroeder et al., J. Med. Chem., 52, 1251 (2009)
[Documento no de Patente 20] Carl R. Illig et al., Bioorg. Med. Chem. Lett., 18, 1642 (2008)
[Documento no de Patente 21] Yi-Xiang Zhang et al., Cancer Res., 68, 1905 (2008)
[Documento no de Patente 22] D Mahadevan et al., Oncogene, 26, 3909 (2007)
[Documento no de Patente 23] Daowan Lai et al., Bioorg. Med. Chem., 19, 6873 (2011)
[Documento no de Patente 24] Ksantini M.,Eur J Ophthalmol., 22, 647 (2012)
[Documento no de Patente 25] Mackay et al., Molecular Vision., 16, 369 (2010)
[Sumario de la invención]
[Problema técnico]
Un objeto de la presente invención es proporcionar un compuesto inhibidor de Axl que tenga una alta especificidad inhibidora para Axl y una mayor seguridad. Otro objeto de la presente invención es proporcionar un agente terapéutico para una enfermedad causada por la hiperfunción de Axl, un agente terapéutico para una enfermedad asociada con la hiperfunción de Axl y/o un agente terapéutico para una enfermedad que involucra la hiperfunción de Axl, por ejemplo, un agente anticanceroso, que comprende un compuesto inhibidor de Axl.
[Solución al problema]
El compuesto del Ejemplo 9 del Documento 33 de patente (de aquí en adelante, denominado Compuesto A) tiene una alta actividad inhibidora de Axl, pero se ha confirmado que causa una degeneración irreversible de los fotorreceptores de la retina en una prueba de administración a largo plazo usando ratones. Se informa que la inactivación de Mer, un miembro de la familia TAM, como con Axl, está relacionada con la degeneración de las células retinianas, y el compuesto del Ejemplo 9 del Documento 33 de patente también tiene actividad inhibidora contra Mer. Teniendo en cuenta estos hechos, los presentes inventores han considerado que la inhibición de Mer por este compuesto conduce a toxicidad retiniana.
Como resultado de la realización de estudios diligentes, los presentes inventores han descubierto que un compuesto que tiene una estructura representada por la fórmula general (I) dada a continuación o una sal del mismo tiene una alta especificidad inhibidora de Axl y una baja actividad inhibidora contra Mer, y además encontraron que este el compuesto o sal del mismo no induce toxicidad retiniana en una prueba de administración a largo plazo usando ratones.
[Efectos ventajosos de la invención]
La presente invención proporciona un compuesto representado por la fórmula (I) que tiene actividad inhibidora de Axl. El compuesto es útil como agente terapéutico para una enfermedad causada por la hiperfunción de Axl, una enfermedad asociada con la hiperfunción de Axl y/o una enfermedad que implica la hiperfunción de Axl, por ejemplo, un agente anticanceroso.
[Breve descripción de los dibujos]
[Figura 1] La Figura 1 muestra un patrón de difracción de rayos X en polvo del cristal obtenido en el Ejemplo 130. En este dibujo, la ordenada indica la intensidad de difracción (Intensidad) por unidad de recuento/seg (cps), y la abscisa representa los valores del ángulo de difracción 20.
[Figura 2] La Figura 2 muestra un patrón de difracción de rayos X en polvo del cristal obtenido en el Ejemplo 131. En este dibujo, la ordenada indica la intensidad de difracción (Intensidad) por unidad de recuento/seg (cps), y la abscisa representa los valores del ángulo de difracción 20.
[Figura 3] La Figura 3 muestra un patrón de difracción de rayos X en polvo del cristal obtenido en el Ejemplo 132. En este dibujo, la ordenada indica la intensidad de difracción (Intensidad) por unidad de recuento/seg (cps), y la abscisa representa los valores del ángulo de difracción 20.
[Figura 4] La Figura 4 muestra un patrón de difracción de rayos X en polvo del cristal obtenido en el Ejemplo 133. En este dibujo, la ordenada indica la intensidad de difracción (Intensidad) por unidad de recuento/seg (cps), y la abscisa representa los valores del ángulo de difracción 20.
[Figura 5] La Figura 5 muestra un patrón de difracción de rayos X en polvo del cristal obtenido en el Ejemplo 134. En este dibujo, la ordenada indica la intensidad de difracción (Intensidad) por unidad de recuento/seg (cps), y la abscisa representa los valores del ángulo de difracción 20.
[Figura 6] La Figura 6 muestra un patrón de difracción de rayos X en polvo del cristal obtenido en el Ejemplo 135. En este dibujo, la ordenada indica la intensidad de difracción (Intensidad) por unidad de recuento/seg (cps), y la abscisa representa los valores del ángulo de difracción 20.
[Figura 7] La Figura 7 muestra un patrón de difracción de rayos X en polvo del cristal obtenido en el Ejemplo 136. En este dibujo, la ordenada indica la intensidad de difracción (Intensidad) por unidad de recuento/seg (cps), y la abscisa representa los valores del ángulo de difracción 20.
[Figura 8] La Figura 8 muestra un patrón de difracción de rayos X en polvo del cristal obtenido en el Ejemplo 137. En este dibujo, la ordenada indica la intensidad de difracción (Intensidad) por unidad de recuento/seg (cps), y la abscisa representa los valores del ángulo de difracción 20.
[Figura 9] La Figura 9 muestra un patrón de difracción de rayos X en polvo del cristal obtenido en el Ejemplo 138. En este dibujo, la ordenada indica la intensidad de difracción (Intensidad) por unidad de recuento/seg (cps), y la abscisa representa los valores del ángulo de difracción 20.
[Figura 10] La Figura 10 muestra un patrón de difracción de rayos X en polvo del cristal obtenido en el Ejemplo 139. En este dibujo, la ordenada indica la intensidad de difracción (Intensidad) por unidad de recuento/seg (cps), y la abscisa representa los valores del ángulo de difracción 20.
[Figura 11] La Figura 11 muestra un patrón de difracción de rayos X en polvo del cristal obtenido en el Ejemplo 140. En este dibujo, la ordenada indica la intensidad de difracción (Intensidad) por unidad de recuento/seg (cps), y la abscisa representa los valores del ángulo de difracción 20.
[Figura 12] La Figura 12 muestra un patrón de difracción de rayos X en polvo del cristal obtenido en el Ejemplo 141. En este dibujo, la ordenada indica la intensidad de difracción (Intensidad) por unidad de recuento/seg (cps), y la abscisa representa los valores del ángulo de difracción 20.
[Figura 13] La Figura 13 muestra un patrón de difracción de rayos X en polvo del cristal obtenido en el Ejemplo 142. En este dibujo, la ordenada indica la intensidad de difracción (Intensidad) por unidad de recuento/seg (cps), y la abscisa representa los valores del ángulo de difracción 20.
[Figura 14] La Figura 14 muestra un patrón de difracción de rayos X en polvo del cristal obtenido en el Ejemplo 143. En este dibujo, la ordenada indica la intensidad de difracción (Intensidad) por unidad de recuento/seg (cps), y la abscisa representa los valores del ángulo de difracción 20.
[Figura 15] La Figura 15 muestra un patrón de difracción de rayos X en polvo del cristal obtenido en el Ejemplo 144. En este dibujo, la ordenada indica la intensidad de difracción (Intensidad) por unidad de recuento/seg (cps), y la abscisa representa los valores del ángulo de difracción 20.
[Figura 16] La Figura 16 muestra un patrón de difracción de rayos X en polvo del cristal obtenido en el Ejemplo 145. En este dibujo, la ordenada indica la intensidad de difracción (Intensidad) por unidad de recuento/seg (cps), y la abscisa representa los valores del ángulo de difracción 20.
[Figura 17] La Figura 17 muestra un patrón de difracción de rayos X en polvo del cristal obtenido en el Ejemplo 146. En este dibujo, la ordenada indica la intensidad de difracción (Intensidad) por unidad de recuento/seg (cps), y la abscisa representa los valores del ángulo de difracción 20.
[Figura 18] La Figura 18 muestra un patrón de difracción de rayos X en polvo del cristal obtenido en el Ejemplo 147. En este dibujo, la ordenada indica la intensidad de difracción (Intensidad) por unidad de recuento/seg (cps), y la abscisa representa los valores del ángulo de difracción 20.
[Figura 19-1] La figura 19-1 es un diagrama que muestra un efecto provocado por el uso combinado del compuesto inhibidor de Axl de la presente solicitud con erlotinib en tumores derivados de HCC827 (que tiene una mutación por deleción en el gen EGFR exón 19 y muestra una alta sensibilidad a los inhibidores de EGFR) trasplantados por vía subcutánea en ratones. El símbolo x representa a un grupo que recibió un vehículo tres veces al día (Tid). El símbolo del círculo vacío representa un grupo que recibió 50 mg/kg de compuesto B sulfato hidrato dos veces al día (bid). El círculo lleno de símbolo representa a un grupo que recibió 25 mg/kg de erlotinib una vez al día (qd). El símbolo del triángulo abierto representa un grupo al que se le administran 50 mg/kg de compuesto B sulfato hidrato (dos veces al día) y 25 mg/kg de erlotinib (una vez al día). La abscisa representa el número de días después del inicio de la administración. La ordenada representa un volumen tumoral estimado calculado a partir de los ejes tumorales.
[Figura 19-2] La Figura 19-2 es un diagrama que muestra una tasa media de cambio en el peso corporal de los individuos en una prueba sobre el uso combinado del compuesto inhibidor de Axl de la presente solicitud con erlotinib (se utilizan las mismas leyendas como en la Figura 19-1).
[Figura 20-1] La Figura 20-1 es un diagrama que muestra el efecto antitumoral de erlotinib sobre tumores derivados de HCC827 trasplantado subcutáneamente en ratones. El símbolo x representa un grupo de administración de vehículos. El símbolo del círculo abierto representa un grupo al que se le administraron 25 mg/kg de erlotinib. El círculo lleno de símbolo representa un grupo que recibió 12,5 mg/kg de erlotinib. El símbolo del triángulo abierto representa un grupo al que se le han administrado 6,25 mg/kg de erlotinib. La abscisa representa el número de días después del inicio de la administración. La ordenada representa un volumen tumoral estimado calculado a partir de los ejes tumorales.
[Figura 20-2] La Figura 20-2 es un diagrama que muestra el cambio en el nivel de expresión de la proteína Axl cuando se administró erlotinib a tumores derivados de HCC827 trasplantados subcutáneamente en ratones.
[Descripción de realizaciones]
En la presente invención, Axl se refiere a una proteína codificada por un gen Axl. "Axl" incluye, por ejemplo, proteínas Axl codificadas por el gen Axl de longitud completa o proteínas Axl codificadas por una variante del gen Axl (que incluye una variante de deleción, una variante de sustitución o una variante de adición). En la presente invención, "Axl" también incluye homólogos derivados de diversas especies animales.
En la presente invención, "inhibidor de Axl" se refiere a un inhibidor de una función de Axl como una tirosina quinasa.
En la presente invención, los términos "tumor" y "cáncer" se usan indistintamente. En la presente invención, el tumor, tumor maligno, cáncer, neoplasma maligno, carcinoma, sarcoma, etc., también se denominan colectivamente "tumor" o "cáncer".
En la presente invención,
un "grupo alquilo C1-C6" significa un grupo alquilo lineal o ramificado que tiene de 1 a 6 átomos de carbono. Ejemplos de un "grupo alquilo C1-C6" incluyen un grupo metilo, un grupo etilo, un grupo propilo, un grupo isopropilo, un grupo butilo y un grupo tert-butilo.
Un "grupo alcoxi C1-C6" significa un grupo alcoxi que tiene un grupo alquilo lineal o ramificado que tiene de 1 a 6 átomos de carbono. Ejemplos de un "grupo alcoxi C1-C6" incluyen un grupo metoxi, un grupo etoxi, un grupo propoxi, un grupo isopropoxi y un grupo butoxi.
Ejemplos de un "átomo de halógeno" incluyen un átomo de flúor, un átomo de cloro, un átomo de bromo y un átomo de yodo.
Un "grupo cicloalquilo" significa un grupo alquilo cíclico que tiene de 3 a 8 átomos de carbono, a menos que se especifique otra cosa. Ejemplos del mismo incluyen un grupo ciclopropilo, un grupo ciclobutilo, un grupo ciclopentilo y un grupo ciclohexilo.
Un "grupo heterocicloalquilo" significa un grupo heterocíclico saturado monovalente. Ejemplos del mismo incluyen un grupo heterocíclico saturado que tiene un átomo de nitrógeno en el anillo y un grupo heterocíclico saturado que tiene un átomo de oxígeno en el anillo y específicamente incluyen grupos monovalentes derivados de pirrolidina, imidazolina, piperidina, piperazina, azetidina, morfolina, dioxano, oxetano, tetrahidropirano y quinuclidina.
Un "grupo cicloalquenilo" se refiere al "grupo cicloalquilo" mencionado anteriormente que tiene uno o más enlaces insaturados tales como dobles enlaces. Ejemplos del mismo incluyen un grupo ciclopentenilo y un grupo ciclohexenilo. Un "grupo heterocicloalquenilo" se refiere al "grupo heterocicloalquilo" mencionado anteriormente que tiene uno o más enlaces insaturados tales como dobles enlaces. Ejemplos del mismo incluyen un grupo tetrahidropiridinilo y un grupo dihidropiranilo. A continuación, se describirá cada sustituyente de la fórmula (I).
En la siguiente fórmula general (I),

W, X e Y representan cada uno independientemente un átomo de nitrógeno, C-H, C-F o C-Cl.
Z representa un átomo de nitrógeno, C-H, C-F, grupo alcoxi C-C1-C6, grupo alquilo C-C1-C6 o C-Cl.
R1 representa un grupo representado por la siguiente fórmula (II-1) o (II-2):

en la que en la fórmula (II-1), Q representa un átomo de nitrógeno, CH o CF, T representa un átomo de nitrógeno o C-H, U representa un átomo de nitrógeno o CH, y R6 representa un átomo de halógeno, un grupo alquilo C1-C6, un grupo cicloalquilo C3-C6, un grupo ciano o un grupo trifluorometoxi, y en la fórmula (II-2), V representa un átomo de azufre o un átomo de oxígeno, y R7 representa un grupo alquilo C1-C6.
R2 representa un átomo de hidrógeno, un átomo de halógeno, un grupo alquilo C1-C6, un grupo alcoxi C1-C6 o un grupo ciano. En este contexto, el grupo alquilo C1-C6 es preferentemente un grupo metilo, un grupo etilo o un grupo propilo. El grupo alcoxi C1-C6 es preferentemente un grupo metoxi o un grupo etoxi.
R3 representa un átomo de hidrógeno o un grupo alquilo C1-C6. El grupo alquilo C1-C6 es preferentemente un grupo metilo, un grupo etilo o un grupo propilo.
R4 representa un átomo de hidrógeno, un átomo de halógeno o un grupo alquilo C1-C6. R4 es preferentemente un átomo de hidrógeno, un átomo de flúor, un átomo de cloro, un grupo metilo o un grupo etilo.
R5 representa un átomo de hidrógeno o un grupo representado por la siguiente fórmula (III-1), (III-2), (III-3) o (III-4):
[Fórmula 15]

(¡lf-1) (in-2) {llf-3} (lil-4)
en la que
en la fórmula (III-1),
R8 y R12 representan cada uno independientemente un átomo de hidrógeno o un átomo de deuterio, Rg representa un átomo de hidrógeno, un átomo de halógeno o un grupo alcoxi C1-C6,
R10 representa un átomo de hidrógeno, un grupo alquilo C1-C6 opcionalmente sustituido con un grupo heterocicloalquilo o un grupo alcoxi C1-C6 opcionalmente sustituido con un grupo heterocicloalquilo opcionalmente sustituido con un grupo alquilo C1-C6, y
R11 representa un átomo de hidrógeno, un grupo alcoxi C1-C6 o un grupo alcoxi C1-C6 sustituido con deuterio, en la fórmula (III-2),
R13 representa un grupo alquilo C1-C6 opcionalmente sustituido con un grupo heterocicloalquilo o un grupo heterocicloalquilo
en la fórmula (III-3),
R14 representa un átomo de hidrógeno o un grupo alquilo C1-C6,
R15 representa un átomo de hidrógeno, un grupo alquilo C1-C6 o un grupo alcoxi C1-C6, y
R16 representa un átomo de hidrógeno o un átomo de halógeno, y
en la fórmula (III-4),
R17 representa un grupo alcoxi C1-C6, y
R18 representa un grupo alcoxi C1-C6.
Cuando R5 es un grupo representado por la fórmula (III-1), el grupo heterocicloalquilo de R10 es preferentemente un grupo dioxanilo, un grupo morfolino, un grupo piperazinilo o un grupo tetrahidropiranilo, y el grupo alquilo C1-C6 es preferentemente un grupo metoxi o un grupo etoxi. El grupo alcoxi C1-C6 de R11 es preferentemente un grupo metoxi o un grupo etoxi.
Cuando R5 es un grupo representado por la fórmula (III-2), el grupo heterocicloalquilo de R13 es preferentemente un grupo dioxanilo, un grupo morfolino, un grupo piperazinilo o un grupo tetrahidropiranilo, y el grupo alquilo C1-C6 de R13 es preferentemente un grupo metilo o un grupo etilo.
El compuesto representado por la fórmula general (I) o la sal farmacéuticamente aceptable del mismo es más preferentemente cualquiera de los siguientes compuestos o sales farmacéuticamente aceptables del mismo:
N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida representado por la siguiente fórmula:

N-[4-(2-amino-5-{4-[(2S)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida representado por la siguiente fórmula:

N-{4-[2-amino-5-(3,4-dimetoxifenil)piridin-3-il]fenil}-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida representado por la siguiente fórmula:

N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4l-dihidro-2,3l-bipiridin-5'-carboxamida representado por la siguiente fórmula:

N-[4-(2-amino-5-{4-[(2S)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1|,4’-dihidro-2,3|-bipiridin-5’-carboxamida representado por la siguiente fórmula:

N-{4-[2-amino-5-(1-etil-1H-pirazol-4-il)piridin-3-il]-3-fluorofenil}-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida representado por la siguiente fórmula:

N-(4-{2-amino-5-[1-(tetrahidro-2H-piran-4-il)-1H-pirazol-4-il]piridin-3-il}-3-fluorofenil)-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida representado por la siguiente fórmula:

N-[6-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)piridazin-3-il]-5-(5-metiltiofen-2-il)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida representado por la siguiente fórmula:

y
N-[6-(2-amino-5-{3-metoxi-4-[2-(morfolin4-il)etoxi]fenil}piridin-3-il)piridazin-3-il]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida representado por la siguiente fórmula:

Los compuestos representados por la fórmula (I) de la presente invención pueden tener estereoisómeros o isómeros ópticos derivados de átomos de carbono asimétricos. Todos estos estereoisómeros e isómeros ópticos y mezclas de los mismos están incluidos en la presente invención.
Los compuestos representados por la fórmula general (I) de la presente invención pueden formar una sal farmacéuticamente aceptable, si se desea, cuando tienen un grupo básico tal como un grupo amino. Ejemplos de tales sales pueden incluir: hidrohaluros tales como hidrocloruros, hidrobromuros e hidroyoduros; sales de ácidos inorgánicos tales como nitratos, percloratos, sulfatos y fosfatos; alcanosulfonatos inferiores tales como metanosulfonatos, trifluorometanosulfonatos y etanosulfonatos; arilsulfonatos tales como bencenosulfonatos y p-toluenosulfonatos; sales de ácidos orgánicos tales como formiatos, acetatos, malatos, fumaratos, succinatos, citratos, tartratos, oxalatos y maleatos; y sales de aminoácidos tales como sales de ornitina, glutamatos y aspartatos. Se prefieren los hidrohaluros y las sales de ácidos orgánicos.
Los compuestos representados por la fórmula general (I) de la presente invención generalmente pueden formar una sal de adición de base cuando tienen un grupo ácido tal como un grupo carboxi. Ejemplos de tales sales farmacéuticamente aceptables pueden incluir: sales de metales alcalinos tales como sales de sodio, sales de potasio y sales de litio; sales de metales alcalinotérreos tales como sales de calcio y sales de magnesio; sales inorgánicas tales como sales de amonio; y sales de aminas orgánicas tales como sales de dibencilamina, sales de morfolina, sales de éster de alquilo de fenilglicina, sales de etilendiamina, sales de N-metilglucamina, sales de dietilamina, sales de trietilamina, sales de ciclohexilamina, sales de diciclohexilamina, sales de N,N'-dibenciletilendiamina, sales de N-dibenciletilendiamina, sales de bencil-N-(2-feniletoxi)amina, sales de piperazina, sales de tetrametilamonio y sales de tris(hidroximetil)aminometano.
Los compuestos representados por la fórmula general (I) de la presente invención o las sales de los mismos pueden existir en forma libre o solvato. Los compuestos representados por la fórmula general (I) de la presente invención o sus sales pueden existir en forma de hidrato, por ejemplo, absorbiendo humedad en el aire. Los solvatos no están particularmente limitados siempre que el solvato sea farmacéuticamente aceptable. Específicamente, un solvato es preferentemente un hidrato, un solvato de etanol o similar. El compuesto representado por la fórmula general (I) de la presente invención puede estar en forma de N-óxido cuando contiene un átomo de nitrógeno. Estas formas de solvato y N-óxido también se incluyen en el ámbito de la presente invención. Los cristales de los compuestos de la presente invención o sus sales también se incluyen en el ámbito de la presente invención.
Más específicamente, Ejemplos de la sal incluyen un hidrobromuro, nitrato, sulfato, fosfato, etanosulfonato, bencenosulfonato y p-toluenosulfonato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida.
Ejemplos adicionales de los mismos incluyen un metanosulfonato de N-{4-[2-amino-5-(3,4-dimetoxifenil)piridin-3-il]fenil}-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida.
Ejemplos adicionales de los mismos incluyen un metanosulfonato, fosfato y sulfato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida.
En otro aspecto, la presente invención se refiere a cristales de los compuestos de la presente invención o las sales. En este contexto, los cristales se refieren a un sólido cuya estructura interna está formada por una repetición tridimensional regular de los átomos constituyentes (o un grupo de los mismos), y se distinguen de un sólido amorfo que no tiene una estructura interna tan regular.
Pueden generarse cristales del mismo compuesto que tienen una pluralidad de estructuras internas y propiedades fisicoquímicas diferentes (polimorfos de cristal) dependiendo de las condiciones de cristalización. Los cristales de la presente invención pueden ser cualquiera de estos polimorfos de cristal o pueden ser una mezcla de dos o más polimorfos de cristal.
Los cristales de la presente invención pueden absorber humedad o adsorber agua cuando se dejan reposar en el aire o pueden calentarse desde 25 hasta 150 °C bajo condiciones atmosféricas normales, por ejemplo, para formar un hidrato. Los cristales de la presente invención también pueden contener un disolvente de cristalización en el disolvente o solvato residual adjunto.
En la presente memoria descriptiva, los cristales de la presente invención se pueden representar sobre la base de datos de difracción de rayos X en polvo. La difracción de rayos X en polvo se puede medir y analizar mediante un enfoque usado convencionalmente en la técnica y se puede llevar a cabo, por ejemplo, mediante un procedimiento descrito en los Ejemplos. Generalmente, en los hidratos y deshidratados, la unión y desprendimiento del agua de cristalización puede cambiar sus constantes de red y así cambiar los ángulos de difracción (20) en la difracción de rayos X en polvo. Además, la intensidad del pico puede variar de acuerdo con, por ejemplo, la diferencia en la superficie de crecimiento de los cristales o similar (hábito de los cristales). Por consiguiente, cuando los cristales de la presente invención se representan sobre la base de datos de difracción de rayos X en polvo, los cristales cuyos ángulos de difracción máximos y patrones de difracción de rayos X en polvo en la difracción de rayos X en polvo son idénticos a los de los cristales de la presente invención así como los hidratos y deshidratados obtenidos a partir de estos cristales también se incluyen en el ámbito de la presente invención.
En un aspecto más específico, la presente invención se refiere a un cristal de un metanosulfonato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1.4- dihidropiridin-3-carboxamida. Más preferiblemente, la presente invención se refiere a un cristal de un metanosulfonato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 3,74, 7,56, 8,96 , 11,38, 12,36, 14,78, 15,60, 16,16, 18,70, y 24,10 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms). El cristal exhibe el patrón de difracción de rayos X en polvo de la Figura 1 por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms)
En un aspecto más específico, la presente invención se refiere a un cristal de un hidrobromuro de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida. Más preferiblemente, la presente invención se refiere a un cristal de un hidrobromuro de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 3,84, 7,72, 9,40, 11,62, 14,92, 15,48, 16,70, 18,88, 19,32, y 24,40 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms). El cristal exhibe el patrón de difracción de rayos X en polvo de la Figura 2 por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms)
En un aspecto más específico, la presente invención se refiere a un cristal de un nitrato de N-[4-(2-amino-5-{4-[(2R)-1.4- dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida. Más preferiblemente, la presente invención se refiere a un cristal de un nitrato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 3,82, 7,66, 9,28, 9,52, 11,54, 15,26, 15,54, 16,62, 19,24, y 24,56 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms). El cristal exhibe el patrón de difracción de rayos X en polvo de la Figura 3 por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms)
En un aspecto más específico, la presente invención se refiere a un cristal de un sulfato de N-[4-(2-amino-5-{4-[(2R)-1.4- dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida. Más preferiblemente, la presente invención se refiere a un cristal de un sulfato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 3,74, 7,56, 8,92, 9,58, 11,36, 12,38, 14,68, 15,64, 16,06, y 24,38 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms). El cristal exhibe el patrón de difracción de rayos X en polvo de la Figura 4 por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms)
En un aspecto más específico, la presente invención se refiere a un cristal de un fosfato de N-[4-(2-amino-5-{4-[(2R)-1.4- dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida. Más preferiblemente, la presente invención se refiere a un cristal de un fosfato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4- ilmetil)-1,4-dihidropiridin-3-carboxamida, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 3,74, 7,56, 8,80, 9,56, 11,34, 14,56, 15,74, 23,68, 24,34, y 24,68 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms). El cristal exhibe el patrón de difracción de rayos X en polvo de la Figura 5 por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms)
En un aspecto más específico, la presente invención se refiere a un cristal de un etanosulfonato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1.4- dihidropiridin-3-carboxamida. Más preferiblemente, la presente invención se refiere a un cristal de un etanosulfonato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 6,72, 7,90, 12,02, 13,40, 16,90, 17,88, 19,00, 19,80, 21,26, y 24,18 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms). El cristal exhibe el patrón de difracción de rayos X en polvo de la Figura 6 por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms)
En un aspecto más específico, la presente invención se refiere a un cristal de un bencenosulfonato de N-[4-(2-amino-5- {4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1.4- dihidropiridin-3-carboxamida. Más preferiblemente, la presente invención se refiere a un cristal de un bencenosulfonato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 9,22, 10,60, 10,82, 11,10, 13,40, 15,78, 17,50, 18,66, 21,02, y 26,10 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms). El cristal exhibe el patrón de difracción de rayos X en polvo de la Figura 7 por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms)
En un aspecto más específico, la presente invención se refiere a un cristal de un p-toluenosulfonato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1.4- dihidropiridin-3-carboxamida. Más preferiblemente, la presente invención se refiere a un cristal de un ptoluenosulfonato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 4,18, 5,12, 13,44, 14,98, 16,96, 17,44, 18,92, 19,72, 20,16, y 23,04 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms). El cristal exhibe el patrón de difracción de rayos X en polvo de la Figura 8 por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms)
En un aspecto más específico, la presente invención se refiere al cristal de un fosfato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida. Más preferiblemente, la presente invención se refiere a un cristal de un fosfato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 4,28, 8,42, 8,64, 10,54, 12,72, 13,48, 15,90, 17,00, 17,46, y 21,26 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms). El cristal exhibe el patrón de difracción de rayos X en polvo de la Figura 9 por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms)
En un aspecto más específico, la presente invención se refiere a un cristal de un sulfato de N-[4-(2-amino-5-{4-[(2R)-1.4- dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1',4-dihidro-2,3'-bipiridin-5'-carboxamida. Más preferiblemente, la presente invención se refiere a un cristal de un sulfato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 3,66, 6,42, 7,32, 9,76, 11,00, 12,88, 18,42, 19,62, 20,54, y 24,22 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms). El cristal exhibe
el patrón de difracción de rayos X en polvo de la Figura 10 por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms). También preferiblemente, la presente invención se refiere a un cristal de un sulfato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 3,64, 6,40, 7,32, 9,76, 17,38, 18,42, 19,64, 20,56, 22,90, y 24,20 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms). El cristal exhibe el patrón de difracción de rayos X en polvo de la Figura 11 por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms) También preferiblemente, la presente invención se refiere a un cristal de un sulfato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 3,64, 6,40, 7,30, 9,76, 17,34, 18,38, 19,34, 20,56, 21,52, y 22,94 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms). El cristal exhibe el patrón de difracción de rayos X en polvo de la Figura 12 por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms) También preferiblemente, la presente invención se refiere a un cristal de un sulfato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 3,62, 6,38, 7,28, 9,74, 17,30, 18,36, 19,54, 20,52, 22,86, y 24,14 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms). El cristal exhibe el patrón de difracción de rayos X en polvo de la Figura 13 por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms) También preferiblemente, la presente invención se refiere a un cristal de un sulfato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 3,64, 6,40, 7,30, 9,76, 12,86, 18,40, 19,62, 20,54, 22,92, y 24,20 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms). El cristal exhibe el patrón de difracción de rayos X en polvo de la Figura 14 por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms) También preferiblemente, la presente invención se refiere a un cristal de un sulfato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 3,64, 6,36, 7,30, 18,36, 19,04, 19,42, 19,70, 20,12, 20,42, y 21,32 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda X = 1,54 angstroms). El cristal exhibe el patrón de difracción de rayos X en polvo de la Figura 15 por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms) También preferiblemente, la presente invención se refiere a un cristal de un sulfato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 5,62, 7,18, 9,22, 10,36, 15,56, 16,40, y 20,86 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms). El cristal exhibe el patrón de difracción de rayos X en polvo de la Figura 16 por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms)
En un aspecto más específico, la presente invención se refiere a un cristal de un naftalen-1,5-disulfonato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida. Más preferiblemente, la presente invención se refiere a un cristal de un naftalen-1,5-disulfonato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1',4-dihidro-2,3-bipiridin-5-carboxamida, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 6,14, 6,98, 11,24, 14,84, 17,48, 19,54, 20,94, 22,38, 23,20,y24,70 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms). El cristal exhibe el patrón de difracción de rayos X en polvo de la Figura 17 por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms) También preferiblemente, la presente invención se refiere a un cristal de un naftalen-1,5-disulfonato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1',4-dihidro-2,3-bipiridin-5-carboxamida, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 9,24, 9,58, 14,00, 14,46, 16,70, 17,02, 18,22, 20,24, 21,64, y 25,52 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms). El cristal exhibe el patrón de difracción de rayos X en polvo de la Figura 18 por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms)
Los compuestos representados por la fórmula general (I) de la presente invención pueden existir como diversos isómeros que incluyen isómeros geométricos tales como isómeros cis y trans, tautómeros o isómeros ópticos tales como isómeros d y l, dependiendo de los tipos y combinaciones de los sustituyentes.. Los compuestos de la presente invención también incluyen todos estos isómeros y estereoisómeros, y mezclas de cualquier proporción de estos isómeros y estereoisómeros, a menos que se especifique otra cosa.
Los compuestos representados por la fórmula general (I) de la presente invención también pueden contener proporciones no naturales de isótopos atómicos en uno o más de los átomos que constituyen los compuestos. Ejemplos de isótopos atómicos incluyen deuterio (2H), también denominado D en la presente memoria descriptiva, tritio (3H), yodo-125 (125I) y carbono-14 (14C). Estos compuestos son útiles como agentes terapéuticos o profilácticos, reactivos de investigación (por ejemplo, reactivos de ensayo) y agentes de diagnóstico (por ejemplo, agentes de
diagnóstico por imagen in vivo). Todas las variantes isotópicas de los compuestos representados por la fórmula general (I) están incluidas en el ámbito de la presente invención, sean radiactivas o no.
La presente invención también abarca compuestos que se convierten en los compuestos representados por la fórmula general (I) que sirven como ingredientes activos en las composiciones farmacéuticas de la presente invención a través de reacciones con una enzima, ácido gástrico o similares bajo condiciones fisiológicas in vivo, es decir, "compuestos profármacos farmacéuticamente aceptables" que se convierten en los compuestos representados por la fórmula general (I) por oxidación enzimática, reducción, hidrólisis, etc., o se convierten en los compuestos representados por la fórmula general (I) por hidrólisis, etc., por ácido gástrico o similar.
Ejemplos de profármacos de los compuestos representados por la fórmula general (I) en los que está presente un grupo amino pueden incluir compuestos en los que el grupo amino está acilado, alquilado o fosforilado (por ejemplo, compuestos en los que el grupo amino está eicosanoilado, alanilado, pentilaminocarbonilado, (5-metil-2-oxo-1,3-dioxolen-4-il)metoxicarbonilado, tetrahidrofuranilado, pirrolidilmetilado, pivaloiloximetilado o tert-butilado). Ejemplos de profármacos de los compuestos representados por la fórmula general (I) en los que está presente un grupo hidroxi pueden incluir compuestos en los que el grupo hidroxi está acilado, alquilado, fosforilado o borado (por ejemplo, Compuestos en los que el grupo hidroxi está acetilado, palmitoilado, propanoilado, pivaloilado, succinilado, fumarilado, alanilado o dimetilaminometilcarbonilado). Ejemplos de profármacos de los compuestos representados por la fórmula general (I) en los que está presente un grupo carboxi incluyen compuestos en los que el grupo carboxi está esterificado o amidado (por ejemplo, compuestos en los que el grupo carboxi está esterificado con etilo, esterificado con fenilo, esterificado con carboximetilo, esterificado con dimetilaminometilo, esterificado con pivaloiloximetilo, esterificado con etoxicarboniloxietilo, amidado o metilamidado).
Los profármacos de los compuestos de la presente invención se pueden producir a partir de los compuestos representados por la fórmula general (I) mediante procedimientos conocidos en la técnica. Los profármacos de los compuestos de la presente invención también incluyen compuestos que se convierten en los compuestos representados por la fórmula general (I) bajo condiciones fisiológicas como se describe en "Iyakuhin No Kaihatsu" [Development of Pharmaceuticals], vol. 7, Bunshi Sekkei [Molecular Design], Hirokawa Shoten, 1990, págs. 163-198.
A continuación, se describirán los procedimientos de producción típicos de los compuestos representados por la fórmula general (I). Los compuestos de la presente invención se pueden producir mediante diversos procedimientos de producción. Los procedimientos de producción que se muestran a continuación se dan meramente con fines ilustrativos, y la presente invención no debe interpretarse como limitada por los mismos. Cada reacción se puede realizar con sustituyentes protegidos con grupos protectores apropiados según se requiera, y el tipo de grupo protector no está particularmente limitado.

Se puede suponer que los compuestos de fórmula general (I) están constituidos por sitios separados, es decir, el sitio A, el sitio B y el sitio C como se describió anteriormente. Ejemplos de los principales procedimientos de síntesis incluyen, pero no se limitan particularmente a, un procedimiento que implica un primer enlace del compuesto (B) al compuesto (CI) y el enlace del complejo al compuesto (AI), un procedimiento que implica un primer enlace del compuesto (AI) a compuesto (B) y unir el complejo al compuesto (CI), un procedimiento que implica primero unir el compuesto (B) al compuesto (CI), unir el complejo al compuesto (A-II) y luego convertir el grupo bromo en R1, un procedimiento que implica convertir el grupo bromo en el compuesto (BC) en R5 y luego unir el compuesto resultante al compuesto (AI) o al compuesto (A-II), un procedimiento que implica primero unir el compuesto (AI) al compuesto (BC) y luego convertir el grupo bromo en R5, y un procedimiento que implica unir un complejo de compuesto (A-I)-compuesto (B) al compuesto (C-II) y luego convertir el grupo bromo en R5.

en la que Ri a R5, W, X, Y y Z son como se definieron anteriormente; R31 es un sustituyente necesario para unir el compuesto (B) al compuesto (C-I) o al compuesto (C-II) y representa un grupo halógeno, un grupo triflato, ácido borónico o éster de ácido borónico; R32 es un sustituyente necesario para unir el compuesto (B) al compuesto (C-I) y representa un grupo halógeno, un grupo triflato, ácido borónico o éster de ácido borónico; y R33 es un sustituyente necesario para unir el compuesto (B) al compuesto (C-II) y representa ácido borónico o éster de ácido borónico. En primer lugar, se describirá el procedimiento para sintetizar el compuesto (A-I).
[Procedimiento de producción 1]
El compuesto (A-I) se puede sintetizar mediante, pero sin limitarse particularmente a, un procedimiento como se muestra en el siguiente esquema de reacción 1 :

en el que Ri a R3, Q, T y U son como se definieron anteriormente; R41 representa un grupo halógeno; R42 es un grupo protector del ácido carboxílico y representa un grupo alquilo C1-C6; R43 representa ácido borónico o éster de ácido borónico; y R44 representa un grupo alquilo C1-C6 o un grupo alquilo ciclo-C3-C6. El compuesto (1d) se incluye en el compuesto (1e).
Síntesis del compuesto (1c)
El compuesto (1a) disponible comercialmente o sintetizado apropiadamente puede someterse a una reacción de acoplamiento con el compuesto (1b) o el compuesto (1b) disponible comercialmente sintetizado mediante un
procedimiento mostrado en el procedimiento de producción 4, el procedimiento de producción 5 o el procedimiento de producción 6 en presencia de una base orgánica o inorgánica tal como carbonato de potasio, carbonato de sodio, carbonato de cesio, acetato de potasio, fosfato de potasio, tert-butóxido o trietilamina, y un catalizador de metal de transición tal como tris(dibencilidenacetona)-dipaladio (0), tetrakis(trifenilfosfina)paladio (0), cloruro de paladio (II) bis(trifenilfosfina), complejo de cloruro de paladio (II) [1,1'-bis(difenilfosfino)-ferroceno] diclorometano, acetato de paladio (II), yoduro de cobre (I) o cloruro de cobre (I) para obtener el compuesto (1c). La reacción también se puede llevar a cabo en presencia de un ligando tal como trifenilfosfina, triciclohexilfosfina, dibencilidenoacetona, 1,3-bis(difenilfosfino)propano, 2-diciclohexilfosfino-2',4',6'-triisopropilbifenilo o 4,5-bis(difenilfosfino)-9,9-dimetilxanteno. La reacción también se puede llevar a cabo usando dos o más tipos de catalizadores de metales de transición en combinación. Ejemplos del disolvente incluyen, pero no se limitan particularmente a, 1,4-dioxano, 1,2-dimetoxietano, metanol, tolueno, agua y disolventes mixtos de los mismos. La temperatura de reacción está usualmente en el intervalo de 0 °C hasta 200 °C o el punto de ebullición del disolvente, preferentemente en el intervalo de 0 °C al punto de ebullición del disolvente. El tratamiento mencionado anteriormente también se puede realizar con un microondas en un tubo sellado. La base orgánica o inorgánica se puede usar en 1 a un exceso de equivalentes molares, preferentemente de 1 a 5 equivalentes molares, con respecto al compuesto (1a). El catalizador de metal de transición o el ligando se puede usar en 0,001 a 1 equivalente molar, preferentemente 0,05 a 1 equivalente molar, con respecto al compuesto (1c). El éster de ácido borónico se puede usar de 1 a exceso de equivalentes molares, preferentemente de 1 a 5 equivalentes molares, con respecto al compuesto (1c).
Síntesis del compuesto (1d)
El compuesto (1c) se puede hidrolizar con un ácido tal como ácido sulfúrico o ácido clorhídrico, o un álcali tal como hidróxido de sodio o carbonato de potasio para obtener el compuesto (1d). La base o el ácido se pueden usar en 1 a equivalentes molares en exceso con respecto al compuesto (1c). Ejemplos del disolvente para su uso en la reacción incluyen, pero no se limitan particularmente a, metanol, etanol, propanol, agua y disolventes mixtos de los mismos. La temperatura de reacción está usualmente en el intervalo de 0 °C hasta 200 °C o el punto de ebullición del disolvente, preferentemente en el intervalo de 0 °C al punto de ebullición del disolvente.
Síntesis del compuesto (1f)
El compuesto (1e) comercialmente disponible o sintetizado apropiadamente puede activarse y hacerse reaccionar con monoéster de ácido malónico o una sal del mismo en presencia de cloruro de magnesio para obtener el compuesto (1f). Ejemplos del procedimiento para activar ácido carboxílico pueden incluir un procedimiento que usa 1,1'-carbonildiimidazol y un procedimiento mediado por cloruro de ácido. Se puede añadir una base, si es necesario, al sistema de reacción. Ejemplos de la base pueden incluir trietilamina. La base se puede utilizar en 1 a un exceso de equivalentes molares, preferentemente de 2 a 10 equivalentes molares, con respecto al compuesto (1e). Ejemplos del disolvente para su uso en la reacción incluyen, pero no se limitan particularmente a, tetrahidrofurano, dioxano, acetonitrilo, N,N-dimetilformamida, tolueno y disolventes mixtos de los mismos. La temperatura de reacción está usualmente en el intervalo de -78 °C hasta 100 °C o el punto de ebullición del disolvente, preferentemente en el intervalo de aproximadamente la temperatura ambiente hasta 100 °C.
Síntesis del compuesto (1g)
El compuesto (1f) se puede tratar con dimetil acetal de N,N-dimetilformamida para obtener el compuesto (1 g). El dimetil acetal de N,N-dimetilformamida se puede utilizar a 2 hasta un exceso de equivalentes molares con respecto al compuesto (1f). Ejemplos del disolvente para su uso en la reacción incluyen, pero no se limitan particularmente a, tolueno, xileno, diclorometano, etanol, N,N-dimetilformamida, acetato de etilo y disolventes mixtos de los mismos. Alternativamente, la reacción se lleva a cabo sin el disolvente. La temperatura de reacción está usualmente en el intervalo de 0 °C hasta 300 °C, preferentemente en el intervalo de aproximadamente la temperatura ambiente hasta 130 °C.
Síntesis del compuesto (1i)
El compuesto (1 g) se puede tratar con el compuesto (1h) disponible comercialmente o sintetizado apropiadamente para obtener el compuesto (1i). Ejemplos del disolvente para su uso en la reacción incluyen, pero no se limitan particularmente a, tolueno, acetato de etilo, etanol, 1,4-dioxano y disolventes mixtos de los mismos. La temperatura de reacción está usualmente en el intervalo de -78 °C hasta 130 °C o el punto de ebullición del disolvente, preferentemente en el intervalo de aproximadamente la temperatura ambiente al punto de ebullición del disolvente. La reacción se puede llevar a cabo, si es necesario, mediante la adición de una base orgánica o inorgánica como carbonato de potasio, carbonato de cesio, tert-butóxido de potasio o trietilamina, o un ácido como ácido acético, cloruro de hidrógeno, bromuro de hidrógeno o ácido sulfúrico. El compuesto (1h), la base o el ácido se pueden usar de 1 a un exceso de equivalentes molares, preferentemente de 1 a 5 equivalentes molares, con respecto al compuesto (1 g).
Síntesis del compuesto (A-I)
El compuesto (A-I) se puede obtener mediante la misma operación que en la síntesis del compuesto (1d).
[Procedimiento de producción 21
El compuesto (1i) también se puede sintetizar mediante, pero sin limitarse particularmente a, un procedimiento como se muestra en el siguiente esquema de reacción 2:

en donde R1 y R42 son como se definieron anteriormente; y R45 representa un grupo halógeno, un grupo metanosulfoniloxi, un grupo trifluorometanosulfoniloxi o un grupo p-toluenosulfoniloxi. El compuesto (2c) se incluye en el compuesto (1i).
Síntesis del compuesto (2a)
El compuesto (2a) está disponible comercialmente o puede sintetizarse a partir del compuesto (1f) con referencia a informes previos tales como J. Heterocyclic Chem. 17, 359 (1980) y Chem. Pharm. Toro. 43, 450 (1995).
Síntesis del compuesto (2c)
El compuesto (2a) se puede tratar con el compuesto (2b) en presencia de una base orgánica o inorgánica tal como carbonato de potasio, carbonato de cesio, tert-butóxido de potasio o trietilamina para obtener el compuesto (2c). Ejemplos del disolvente para su uso en la reacción incluyen, pero no se limitan particularmente a, N,N-dimetilformamida, dimetilsulfóxido, acetonitrilo y disolventes mixtos de los mismos. La temperatura de reacción está usualmente en el intervalo de 0 °C hasta 200 °C o el punto de ebullición del disolvente, preferentemente en el intervalo de 0 °C al punto de ebullición del disolvente. El compuesto (2b) o la base se puede usar en 1 a un exceso de equivalentes molares, preferentemente de 1 a 5 equivalentes molares, con respecto al compuesto (2a).
A continuación, se describirá el procedimiento para sintetizar el compuesto (A-II).
[Procedimiento de producción 31
El compuesto (A-II) se puede sintetizar mediante, pero sin limitarse particularmente a, un procedimiento como se muestra en el siguiente esquema de reacción 3:

Síntesis del compuesto (3b)
Compuesto (3a) o compuesto (3a) disponible comercialmente que se puede sintetizar fácilmente con referencia a informes previos tales como J. Med. Chem. 51, 5330 (2008) se puede tratar con el compuesto (2b) en presencia de una base orgánica o inorgánica tal como carbonato de potasio, carbonato de cesio, tert-butóxido de potasio o trietilamina para obtener el compuesto (3b). Ejemplos del disolvente para su uso en la reacción incluyen, pero no se
limitan particularmente a, N,N-dimetilformamida, dimetilsulfóxido, acetonitrilo y disolventes mixtos de los mismos. La temperatura de reacción está usualmente en el intervalo de 0 °C hasta 200 °C o el punto de ebullición del disolvente, preferentemente en el intervalo de 0 °C al punto de ebullición del disolvente. El compuesto (2b) o la base se puede utilizar en 1 a un exceso de equivalentes molares, preferentemente de 1 a 5 equivalentes molares, con respecto al compuesto (3a).
Síntesis del compuesto (A-II)
El compuesto (A-II) puede obtenerse mediante la misma operación que en la síntesis del compuesto (A-I) en el [Procedimiento de producción 1].
A continuación, se describirá el procedimiento para sintetizar el compuesto (C-I).
Cuando R5 es un átomo de hidrógeno, el compuesto (C-I) está disponible comercialmente o puede sintetizarse fácilmente a partir de un producto disponible comercialmente mediante un procedimiento conocido en la literatura.
Cuando R5 es un grupo distinto de un átomo de hidrógeno, el compuesto (C-I) se puede sintetizar mediante un procedimiento como se muestra en el procedimiento de producción 4 al procedimiento de producción 7 que se indica a continuación. Específicamente, el compuesto (4c), compuesto (5h) o compuesto (6 c) que constituye R5 se sintetiza seleccionando cualquiera de los procedimientos mostrados en el procedimiento de producción 4 al procedimiento de producción 6. Luego, el compuesto (CI) se puede sintetizar mediante el procedimiento de producción 7 utilizando el compuesto así sintetizado o un compuesto disponible comercialmente, aunque el procedimiento de síntesis no está particularmente limitado al mismo.
[Procedimiento de producción 4]

en el que R8, R9, R11, R12, R43 y R45 son como se definieron anteriormente; y R51 representa un grupo alquilo C1-C6 opcionalmente sustituido con un grupo heterocicloalquilo.
Síntesis del compuesto (4c)
El compuesto (4a) comercialmente disponible o sintetizado apropiadamente puede tratarse con el compuesto (4b) en presencia de una base orgánica o inorgánica tal como carbonato de potasio, carbonato de cesio, tert-butóxido de potasio o trietilamina para obtener el compuesto (4c). Ejemplos del disolvente para su uso en la reacción incluyen, pero no se limitan particularmente a, N,N-dimetilformamida, dimetilsulfóxido, acetonitrilo y disolventes mixtos de los mismos. La temperatura de reacción está usualmente en el intervalo de 0 °C hasta 200 °C o el punto de ebullición del disolvente, preferentemente en el intervalo de 0 °C al punto de ebullición del disolvente. El compuesto (4b) o la base se puede usar en 1 a un exceso de equivalentes molares, preferentemente de 1 a 5 equivalentes molares, con respecto al compuesto (4a).
[Procedimiento de producción 51

en el que R8, Rg, R12, R43, R45 y R51 son como se definieron anteriormente; R61 representa un átomo de deuterio o hidrógeno; R62 representa un grupo hidroxi o un grupo protector sustituido con deuterio para el grupo hidroxi, en el que los ejemplos del grupo protector incluyen grupos éter tipificados por un grupo tetrahidropiranilo, grupos sililo tipificados por un grupo t-butildifenilsililo, grupos éster tipificados por un grupo acetilo, y grupos carbonato tipificados por un grupo carbonato de vinilo; y R63 representa un grupo alquilo C1-C6 sustituido con deuterio.
Síntesis del compuesto (5b)
Solo uno de los dos grupos hidroxi en el compuesto (5a) disponible comercialmente o sintetizado apropiadamente puede protegerse para obtener el compuesto (5b). Por ejemplo, el compuesto (5a) se puede tratar con 3,4-dihidro-2H-pirano en presencia de un ácido tal como la sal de piridina del ácido p-toluensulfónico para obtener el compuesto (5b) con solo uno de los grupos hidroxi protegido con un grupo tetrahidropiranilo, aunque el procedimiento para proteger el grupo hidroxi no está particularmente limitado al mismo. Ejemplos del disolvente para su uso en la reacción incluyen, pero no se limitan particularmente a, diclorometano, cloroformo, tolueno y disolventes mixtos de los mismos. La temperatura de reacción está usualmente en el intervalo de 0 °C hasta 200 °C o el punto de ebullición del disolvente, preferentemente en el intervalo de 0 °C al punto de ebullición del disolvente. El compuesto (4b) o el ácido se pueden usar de 0,01 a equivalentes molares en exceso, preferentemente de 0,01 a 1 equivalentes molares, con respecto al compuesto (5a).
Síntesis del compuesto (5d)
El compuesto (5b) se puede tratar con el compuesto (5c) en presencia de una base orgánica o inorgánica tal como carbonato de potasio, carbonato de cesio, tert-butóxido de potasio o trietilamina para obtener el compuesto (5d). Ejemplos del disolvente para su uso en la reacción incluyen, pero no se limitan particularmente a, N,N-dimetilformamida, dimetilsulfóxido, acetonitrilo, acetona y disolventes mixtos de los mismos. La temperatura de reacción está usualmente en el intervalo de 0 °C hasta 200 °C o el punto de ebullición del disolvente, preferentemente en el intervalo de 0 °C al punto de ebullición del disolvente. El compuesto (5c) o la base se puede usar en 1 a un exceso de equivalentes molares, preferentemente de 1 a 5 equivalentes molares, con respecto al compuesto (5b).
Síntesis del compuesto (5e)
El compuesto (5d) se puede tratar con un ácido tal como ácido clorhídrico, ácido acético o sal de piridina del ácido ptoluenosulfónico para obtener el compuesto (5e). Ejemplos del disolvente para su uso en la reacción incluyen, pero no se limitan particularmente a, etanol, metanol, isopropanol, agua y disolventes mixtos de los mismos. La temperatura de reacción está usualmente en el intervalo de 0 °C hasta 200 °C o el punto de ebullición del disolvente, preferentemente en el intervalo de 0 °C al punto de ebullición del disolvente. El compuesto (4b) o el ácido se pueden usar en una cantidad de 0,01 a equivalentes molares en exceso, preferentemente de 0,01 a 1 equivalentes molares, con respecto al compuesto (5d).
Síntesis del compuesto (5f)
El compuesto (5e) se puede tratar con un reactivo bromante tal como N-bromosuccinimida o bromato de potasio para obtener el compuesto (5f). Ejemplos del disolvente para su uso en la reacción incluyen, pero no se limitan particularmente a, N,N-dimetilformamida, una solución al 30 % de ácido bromhídrico en ácido acético, ácido acético y disolventes mixtos de los mismos. La temperatura de reacción está usualmente en el intervalo de 0 °C hasta 200 °C o el punto de ebullición del disolvente, preferentemente en el intervalo de 0 °C al punto de ebullición del disolvente. El
reactivo de bromación se puede usar de 0,1 a equivalentes molares en exceso, preferentemente de 0,1 a 5 equivalentes molares, con respecto al compuesto (5e).
Síntesis del compuesto (5g)
El compuesto (5g) se puede obtener mediante la misma operación que en el [Procedimiento de producción 4].
Síntesis del compuesto (5h)
El compuesto (5 g) se puede tratar con éster de ácido borónico tal como bis(pinacolato)diboro o pinacol boro en presencia de una base orgánica o inorgánica tal como carbonato de potasio, carbonato de sodio, carbonato de cesio, acetato de potasio, fosfato de potasio, tert-butóxido, o trietilamina, un catalizador de metal de transición tal como tris(dibencilidenoacetona)-dipaladio (0), bis(trifenilfosfina)cloruro de paladio (II), complejo de [1,1'-bis(difenilfosfino)-ferroceno]cloruro de paladio (II)-diclorometano, complejo de [1,1'-bis(difenilfosfino)-ferroceno]cloruro de paladio (II)diclorometano, o 1,3-bis(difenilfosfino)propano]cloruro de níquel (II) y un ligando tal como trifenilfosfina, triciclohexilfosfina, dibencilidenacetona, 1,3-bis(difenilfosfino)propano, 2-diciclohexilfosfino-2',4',6'-triisopropilbifenilo o 4,5-bis(difenilfosfino)-9,9-dimetilxanteno para obtener el compuesto (5h). Ejemplos del disolvente incluyen, pero no se limitan particularmente a, 1,4-dioxano, 1,2-dimetoxietano, metanol, tolueno y disolventes mixtos de los mismos. La temperatura de reacción está usualmente en el intervalo de 0 °C hasta 200 °C o el punto de ebullición del disolvente, preferentemente en el intervalo de 0 °C al punto de ebullición del disolvente. El tratamiento mencionado anteriormente también se puede realizar con un microondas en un tubo sellado. La base orgánica o inorgánica se puede usar de 1 a un exceso de equivalentes molares, preferentemente de 1 a 5 equivalentes molares, con respecto al compuesto (5g). El catalizador de metal de transición o el ligando se puede usar en 0,001 a 1 equivalente molar, preferentemente 0,05 a 1 equivalente molar, con respecto al compuesto (5 g). El éster de ácido borónico se puede utilizar en 1 a un exceso de equivalentes molares, preferentemente de 1 a 5 equivalentes molares, con respecto al compuesto (5 g).
[Procedimiento de producción 6]

en el que R13, R43 y R45 son como se definieron anteriormente.
Síntesis del compuesto (6c)
El compuesto (6c) puede obtenerse mediante la misma operación que en el [Procedimiento de producción 4] utilizando compuestos (6a) y (6b) comercialmente disponibles o sintetizados apropiadamente.
[Procedimiento de producción 7]

en el que R5 y R43 son como se definieron anteriormente; y R81 representa un grupo halógeno. El compuesto (7a) es un compuesto de ácido borónico o éster de ácido borónico que contiene el compuesto (4c), el compuesto (5h) o el compuesto (6c). El compuesto (7c) y el compuesto (7d) se incluyen en el compuesto (C-I).
Síntesis del compuesto (7c)
El compuesto (7b) puede someterse a una reacción de acoplamiento con el compuesto (7a) disponible comercialmente o el compuesto (7a) sintetizado mediante un procedimiento que se muestra en el procedimiento de producción 4, el procedimiento de producción 5 o el procedimiento de producción 6 de la misma manera que en la síntesis del compuesto. (1c) en el [Procedimiento de producción 1] para obtener el compuesto (7c).
Síntesis del compuesto (7d)
El compuesto (7d) se puede obtener mediante la misma operación que en la síntesis del compuesto (5h) en el [Procedimiento de producción 5] usando el compuesto (7c) disponible comercialmente o sintetizado apropiadamente.
A continuación, se describirá el procedimiento para sintetizar un complejo de compuesto (B)-compuesto (C-I).
El complejo de compuesto (B)-compuesto (C-I) puede sintetizarse mediante un procedimiento para convertir el grupo bromo en el compuesto (BC) en R5 como se muestra en el [Procedimiento de producción 8], aunque el procedimiento de síntesis no está particularmente limitado al mismo.
[Procedimiento de producción 8]

en el qjue R4, R5, W, X, Y, Z y R43 son como se definieron anteriormente. El compuesto (8) se incluye en el complejo de compuesto (B)-compuesto (C-I).
El compuesto (8) se puede obtener mediante la misma operación que en la síntesis del compuesto (1c) en el [Procedimiento de producción 1] usando el compuesto (BC) o el compuesto (8) disponible comercialmente o sintetizado apropiadamente.
El compuesto (8) también se puede sintetizar mediante un procedimiento mostrado en el [Procedimiento de producción 9] o [Procedimiento de producción 10], aunque el procedimiento de síntesis no está particularmente limitado a los mismos.
[Procedimiento de producción 9]

en el que R4, R5, W, X, Y, Z y Rs1 son como se definieron anteriormente. El compuesto (8) se incluye en el complejo de compuesto (B)-compuesto (C-I).
El compuesto (8) se puede obtener mediante la misma operación que en la síntesis del compuesto (1c) en el [Procedimiento de producción 1] usando compuestos (9a) y (9b) disponibles comercialmente o sintetizados apropiadamente.
[Procedimiento de producción 10]

en el que R4, R5, W, X, Y, Z y Rs1 son como se definieron anteriormente. El compuesto (8) se incluye en el complejo de compuesto (B)-compuesto (C-I).
El compuesto (8) puede obtenerse mediante la misma operación que en la síntesis del compuesto (1c) en el [Procedimiento de producción 1] usando el compuesto (7d) y el compuesto (10) disponibles comercialmente o sintetizados apropiadamente.
A continuación, se describirá el procedimiento de síntesis para unir el complejo de compuesto (B)-compuesto (C-I) al compuesto (A-I).
El complejo de compuesto (B)-compuesto (C-I) se puede unir al compuesto (A-I) de acuerdo con el siguiente procedimiento de producción 11 o procedimiento de producción 12 :
[Procedimiento de producción 11]
El compuesto (11) se puede sintetizar mediante, pero sin limitarse particularmente a, un procedimiento como se muestra en el siguiente esquema de reacción 11 :

en el que R1 a R5, W, X, Y y Z son como se definieron anteriormente. El compuesto (8) se incluye en el complejo de compuesto (B)-compuesto (C-I).
El compuesto (A-I) se puede hacer reaccionar con el compuesto (8) en presencia de un agente de condensación para obtener el compuesto (11). Ejemplos del agente de condensación incluyen, pero no se limitan a, hexafluorofosfato de N-[1-(ciano-2-etoxi-2-oxoetilidenaminooxi)dimetilamino(morfolino)]uronio (COMU). Ejemplos del disolvente para su uso en la reacción incluyen, pero no se limitan particularmente a, N,N-dimetilformamida, diclorometano y disolventes mixtos de los mismos. La temperatura de reacción está usualmente en el intervalo de -78 °C hasta 200 °C o el punto de ebullición del disolvente, preferentemente en el intervalo de 0 °C hasta 100 °C. El agente de condensación se puede usar en 1 a un exceso de equivalentes molares, preferentemente de 1 a 5 equivalentes molares, con respecto al compuesto (A-I).
[Procedimiento de producción 121
El compuesto (11) se puede sintetizar mediante, pero sin limitarse particularmente a, un procedimiento como se muestra en el siguiente esquema de reacción 12:

en el que R1 a R5, W, X, Y y Z son como se definieron anteriormente. El compuesto (8) se incluye en el complejo de compuesto (B)-compuesto (C-I).
El compuesto (A-I) se puede tratar con un reactivo halogenante ácido tal como cloruro de tionilo o cloruro de oxalilo para obtener el compuesto (12), que luego se trata con el compuesto (8) para obtener el compuesto (11). Ejemplos del disolvente para su uso en la reacción incluyen, pero no se limitan particularmente a, diclorometano, dicloroetano y disolventes mixtos de los mismos. El reactivo de halogenación ácido se puede usar en 0,9 a equivalentes molares en exceso, preferentemente 0,9 a 2 equivalentes molares, con respecto al compuesto (A-I).
A continuación, se describirá el procedimiento para sintetizar el complejo de compuesto (A-I)-compuesto (B).
[Procedimiento de producción 131
El compuesto (13) se puede sintetizar mediante, pero sin limitarse particularmente a, un procedimiento como se muestra en el siguiente esquema de reacción 13.

en el que R1 a R4, W, X, Y y R31 son como se definieron anteriormente. El compuesto (13) se incluye en el complejo de compuesto (A-I)-compuesto (B).
Síntesis del compuesto (13)
El compuesto (B) está disponible comercialmente o puede sintetizarse fácilmente a partir de un producto disponible comercialmente mediante un procedimiento conocido en la literatura. El compuesto (13) puede obtenerse mediante la misma operación que en el [Procedimiento de producción 11] usando el compuesto (A-l) y el compuesto (B) disponible comercialmente o sintetizado apropiadamente.
A continuación, se describirá el procedimiento de síntesis para unir el complejo de compuesto (A-I)-compuesto (B) al compuesto (C-I).
[Procedimiento de producción 141
El compuesto (11) se puede sintetizar mediante, pero sin limitarse particularmente a, un procedimiento como se muestra en el siguiente esquema de reacción 14:

en el que R1 a R5, W, X, Y y Z son como se definieron anteriormente; cuando R31 es un grupo halógeno, un grupo triflato o similar, R32 representa ácido borónico o éster de ácido borónico; y cuando R31 es ácido borónico o éster de ácido borónico, R32 representa un grupo halógeno, un grupo triflato o similares. El compuesto (13) se incluye en el complejo de compuesto (A-I)-compuesto (B).
Síntesis del compuesto (11)
El compuesto (11) se puede obtener mediante la misma operación que en la síntesis del compuesto (1c) en el [Procedimiento de producción 1] usando el compuesto (13) y el compuesto (C-I) disponible comercialmente o sintetizado apropiadamente.
A continuación, se describirá el procedimiento de síntesis para unir el complejo de compuesto (B)-compuesto (C-I) al compuesto (A-II).
[Procedimiento de producción 15]
El compuesto (15) se puede sintetizar mediante, pero sin limitarse particularmente a, un procedimiento como se muestra en el siguiente esquema de reacción 15:

en el que R4 a R5, W, X, Y y Z son como se definieron anteriormente. El compuesto (8) se incluye en el complejo de compuesto (B)-compuesto (C-I).
Síntesis del compuesto (15)
El compuesto (15) se puede obtener mediante la misma operación que en el [Procedimiento de producción 11] usando el compuesto (A-II) y el compuesto (8).
A continuación, se describirá el procedimiento de síntesis para convertir el grupo bromo en el complejo de compuesto (A-II)-compuesto (B)-compuesto (C-I) en R1.
[Procedimiento de producción 161
El compuesto (16b) se puede sintetizar mediante, pero sin limitarse particularmente a, un procedimiento como se muestra en el siguiente esquema de reacción 16:

en el que R1, R4 a R5, W, X, Y, Z y R43 son como se definieron anteriormente. El compuesto (15) se incluye en el complejo de compuesto (A-I)-compuesto (B)-compuesto (C-I).
Síntesis del compuesto (16b)
El compuesto (16b) se puede obtener mediante la misma operación que en la síntesis del compuesto (1c) en el [Procedimiento de producción 1] usando el compuesto (15) y el compuesto (16a) disponible comercialmente o sintetizado apropiadamente.
A continuación, se describirá el procedimiento de síntesis para convertir el grupo halógeno en el complejo de compuesto (A-I)-compuesto (B)-compuesto (C-I) en un grupo alquilo.
[Procedimiento de producción 17]
El compuesto (17b) se puede sintetizar mediante, pero sin limitarse particularmente a, un procedimiento como se muestra en el siguiente esquema de reacción 17:

en el que R2 a R5, Q, T, U, W, X, Y, Z, R43 y R44 son como se definieron anteriormente. El compuesto (17a) y el compuesto (17b) se incluyen en el complejo de compuesto (A-I)-compuesto (B)-compuesto (C-I).
Síntesis del compuesto (17b)
El compuesto (17b) se puede obtener mediante la misma operación que en la síntesis del compuesto (1c) en el [Procedimiento de producción 1] usando el compuesto (17a) y el compuesto (1b) disponible comercialmente o sintetizado apropiadamente.
A continuación, se describirá el procedimiento de síntesis para unir el compuesto (A-I) al compuesto (BC) y luego convertir el grupo bromo en R5.
[Procedimiento de producción 181
El compuesto (11) se puede sintetizar mediante, pero sin limitarse particularmente a, un procedimiento como se muestra en el siguiente esquema de reacción 18:

en el que R1 a R5, W, X, Y, Z y R43 son como se definieron anteriormente.
Síntesis del compuesto (18)
El compuesto (18) se puede obtener mediante la misma operación que en el [Procedimiento de producción 11] usando el compuesto (A-I) y el compuesto (BC).
Síntesis del compuesto (11)
El compuesto (11) se puede obtener mediante la misma operación que en la síntesis del compuesto (1c) en el [Procedimiento de producción 1] usando el compuesto (18 ) y el compuesto (7a) disponible comercialmente o sintetizado apropiadamente.
El compuesto (18) también se puede sintetizar mediante un procedimiento como se muestra en [Procedimiento de producción 19].
[Procedimiento de producción 191

en el que Ri a R4, R33, R81, W, X, Y y Z son como se definieron anteriormente. El compuesto (19) se incluye en el compuesto (13).
El compuesto (C-II) está disponible comercialmente o puede sintetizarse fácilmente a partir de un producto disponible comercialmente mediante un procedimiento conocido en la literatura. El compuesto (18) se puede obtener mediante la misma operación que en la síntesis del compuesto (1c) en el [Procedimiento de producción 1] usando el compuesto (19) y el compuesto (C-II) disponible comercialmente o sintetizado apropiadamente.
El compuesto (11) también se puede producir de acuerdo con el procedimiento de producción 20.
[Procedimiento de producción 20]
en el que Ri a R4, R43, W, X, Y y Z son como se definieron anteriormente; R201 representa un átomo de hidrógeno o un grupo halógeno; y R202 representa un grupo alquilamino C1-C6 que forma opcionalmente un anillo o un grupo oxialquilamino C1-C6 que forma opcionalmente un anillo. El compuesto (20d) se incluye en el compuesto (11).
Síntesis del compuesto (20b)
El compuesto (20b) se puede obtener mediante la misma operación que en la síntesis del compuesto (1c) en el [Procedimiento de producción 1] usando el compuesto (18) y el compuesto (20a) disponible comercialmente o sintetizado apropiadamente.
Síntesis del compuesto (20d)
El compuesto (20b) se puede hacer reaccionar con el compuesto (20c) disponible comercialmente o sintetizado apropiadamente en presencia de un agente reductor para obtener el compuesto (20d). Ejemplos del agente reductor usado pueden incluir triacetoxiborohidruro de sodio, cianoborohidruro de sodio y borohidruro de sodio. Ejemplos del disolvente usado incluyen, pero no se limitan particularmente a, metanol, etanol, agua, tetrahidrofurano, dioxano, N,N-dimetilformamida, cloruro de metileno, ácido acético y disolventes mixtos de los mismos. La temperatura de reacción está usualmente en el intervalo de -78 °C hasta 100 °C o el punto de ebullición del disolvente, preferentemente en el intervalo de 0 °C hasta 100 °C.
Como se mencionó anteriormente, se ha informado que el sistema de señalización Gas6/Axl regula diversas respuestas celulares como la supervivencia celular, la división celular, la autofagia, la migración celular, la angiogénesis, la agregación plaquetaria y la diferenciación de células NK (Documento no de patente 2). Por lo tanto, los inhibidores de Axl son útiles para tratar una enfermedad causada por la hiperfunción de la quinasa Axl, una enfermedad asociada con la hiperfunción de la quinasa Axl y/o una enfermedad que implica la hiperfunción de la quinasa Axl.
Particularmente, los compuestos de la presente invención pueden usarse de manera más segura porque los compuestos de la presente invención tienen una alta especificidad inhibidora de Axl y no causan toxicidad retiniana.
Ejemplos de enfermedades causadas por la hiperfunción de la quinasa Axl, enfermedades asociadas con la hiperfunción de la quinasa Axl y enfermedades que implican la hiperfunción de la quinasa Axl incluyen enfermedades que tienen tejidos que contienen genes y/o proteínas Axl sobreexpresados, y enfermedades que tienen tejidos con una actividad fosforilante elevad. de Axl.
Ejemplos de las enfermedades mencionadas anteriormente incluyen enfermedades hiperproliferativas. Ejemplos de enfermedades hiperproliferativas incluyen, pero sin limitación, hiperplasia endometrial, proliferación de células de músculo liso vascular inducida por trombina (VSMC), tumor benigno, tumor maligno (cáncer), glomerulonefritis aguda y crónica y nefropatía diabética.
Además, se ha descubierto que Axl desempeña un papel en la inmunidad (Documento no de patente 12), funciones plaquetarias (Documento no de patente 13), espermatogénesis (Documento no de patente 14), calcificación vascular (Documento no de patente 15), (Documento no de patente 16) y diversas enfermedades renales y rechazo crónico de aloinjertos (Documento no de patente 17). Por lo tanto, los inhibidores de Axl son útiles en el tratamiento de muchas enfermedades que incluyen enfermedades vasculares (que incluyen, pero no se limitan a, trombosis, aterosclerosis y reestenosis) y enfermedades en las que la formación desorganizada de vasos sanguíneos tiene consecuencias graves (que incluyen, pero no se limitan a, retinopatía diabética, retinopatía, psoriasis, artritis reumatoide, ateroma, sarcoma de Kaposi y angioma).
Los compuestos de la presente invención inhiben Axl y, por tanto, son útiles en el tratamiento de las enfermedades descritas anteriormente.
Más preferentemente, los compuestos de la presente invención son útiles en el tratamiento de diversos cánceres. Ejemplos de cánceres incluyen, pero no se limitan a, cáncer de mama, cáncer de colon, cáncer de próstata, cáncer de pulmón, cáncer de estómago, cáncer de ovario, cáncer de cuello uterino, cáncer de endometrio, cáncer de cuerpo uterino, cáncer de riñón, cáncer hepatocelular, cáncer de glándula tiroides, cáncer de esófago, cáncer de células escamosas, leucemia, osteosarcoma, melanoma, glioblastoma, neuroblastoma, cáncer de ovario, cáncer de cabeza y cuello, tumor testicular, cáncer colorrectal, cáncer de sangre, retinoblastoma y cáncer de páncreas.
Se han realizado diversos informes sobre la relación de Axl con los cánceres desde el punto de vista de la inhibición del crecimiento, la inhibición de la metástasis, la migración o invasión y la superación de la resistencia a los fármacos.
Se ha informado que los mutantes negativos dominantes de Axl inhiben el crecimiento de tumores cerebrales (Vajkoczy et al., PNAS 2006, 103, 5799). Se ha informado que los tumores en tejidos derivados de pacientes con glioblastoma que tienen la expresión de Axl o la coexpresión de Axl/Gas6 crecen significativamente más rápidamente y acortan la vida de los pacientes (Hutterer et al., Clin Cancer Res 14, 130 (2008)). Se ha informado que el ARNhc de Ax1 inhibe el crecimiento de células de cáncer de mama (Yi-Xiang et al., Cancer Res 68, 1905 (2008)). Como es evidente a partir de estos informes, los inhibidores de Axl son útiles en la inhibición del crecimiento celular en cánceres.
Por otro lado, se ha informado que los mutantes negativos dominantes Axl inhiben la migración e invasión celular (Zhang et al., Cancer Res 68, 1905 (2008); Vajkoczy et al., PNAS 103, 5799 (2006); y Holland et al., Cancer Res 65, 9294 (2005)). Se ha informado que el ARNhc de Ax1 inhibe la metástasis in vivo (Li et al., Oncogene 28, 3442 (2009)). Se ha informado que los anticuerpos anti-Axl y el ARNip inhiben el crecimiento tumoral y la metástasis en un modelo de ratón (Li et al., Oncogene 28, 3442 (2009); Ye et al., Oncogene 29, 5254 (2010)). Se ha informado que Axl promueve la invasión celular (Tai et al., Oncogene 27, 4044 (2008)). Se ha informado que el R-428, un inhibidor de Axl, inhibe un modelo de difusión de cáncer de mama metastásico (Holland et al., Cancer Res 70, 1544 (2010)). Se ha informado que los anticuerpos Axl, el ARNhc de Axl y un inhibidor de Axl NA80x1 inhiben la migración y la invasión de las células de cáncer de mama (Yi-Xiang et al., Cancer Res 68, 1905 (2008)). Además, ha habido una pluralidad de informes sobre la participación de Axl en la metástasis y la progresión maligna del cáncer de próstata, cáncer de bazo, cáncer de ovario metastásico, carcinoma tímico y similares. Como resulta evidente a partir de estos informes, los inhibidores de Axl son útiles para, por ejemplo, inhibir, tratar y prevenir la metástasis del cáncer, la migración celular y la invasión celular.
Además, se ha informado que los inhibidores de Axl superan la resistencia al imatinib en el cáncer gástrico (Mahadevan et al., Oncogene 26, 3909 (2007)). Se ha encontrado que Axl se induce en la resistencia de la leucemia mieloide aguda a agentes quimioterapéuticos tales como doxorrubicina, VP16 y cisplatino (Hong et al., Cancer Letters 268, 314 (2008)). Según se informa, Axl se activa en la resistencia a lapatinib en células de cáncer de mama positivas para HER-2 (Liu et al., Cancer Res 69, 6871 (2009)). Se ha informado que Axl participa en el mecanismo de resistencia PLX4032 (vemurafenib) (Johannessen et al., Nature 468, 968 (2010)). Además, se ha informado que Axl participa en la resistencia a temozolomida, carboplatino y vincristina (AK Keeating et al., Mol Cancer Ther 9 (5), 1298 (2010)). Como es evidente a partir de estos informes, los inhibidores de Axl son útiles para superar la resistencia a los fármacos, por ejemplo, para superar la resistencia a diversos agentes anticancerígenos.
Los inhibidores de Axl de la presente solicitud, como se muestra en los Ejemplos de prueba de la presente solicitud, inhibieron la resistencia de los tumores a erlotinib mediante la administración en combinación con erlotinib. Se observó una inducción significativa de la proteína Axl por la administración de erlotinib, lo que sugiere que Axl está involucrado en la adquisición de resistencia a los fármacos por los cánceres.
El uso combinado con los inhibidores de Axl de la presente solicitud para tumores resistentes a erlotinib restauró la sensibilidad a erlotinib, lo que sugiere que los inhibidores de Axl son eficaces para superar la resistencia a fármacos de los cánceres.
Se ha informado además de que Axl participa en enfermedades renales tales como la formación de fibrillas en el riñón y la nefropatía diabética (Publicación Nacional de la Solicitud de Patente Internacional No. 2005-517412). Por tanto, los inhibidores de Axl son evidentemente útiles en el tratamiento de estas enfermedades renales así como enfermedades de fibrilización tales como fibrosis pulmonar idiopática.
La actividad inhibidora de Axl de los compuestos puede medirse mediante, pero sin limitarse a, los procedimientos descritos en, por ejemplo, los Ejemplos de prueba de la presente solicitud.
La actividad inhibidora contra el crecimiento celular puede examinarse mediante el uso de un procedimiento de prueba de inhibición del crecimiento usado convencionalmente por los expertos en la técnica. La actividad inhibidora contra el crecimiento celular se puede determinar comparando el grado de crecimiento de las células (por ejemplo, células tumorales) en presencia y en ausencia del compuesto de prueba. El grado de crecimiento se puede examinar usando, por ejemplo, un sistema de prueba para medir células vivas. Ejemplos del procedimiento para medir células vivas incluyen la prueba de captación de [3H]-timidina, el procedimiento BrdU y el ensayo MTT.
La actividad antitumoral in vivo puede examinarse mediante el uso de un procedimiento de prueba antitumoral convencionalmente utilizado por los expertos en la técnica. La actividad antitumoral in vivo de acuerdo con la presente invención puede confirmarse, por ejemplo: trasplantando diversas células tumorales en ratones, ratas o similares; después de la confirmación del injerto de las células trasplantadas, administrar un compuesto de la presente invención por vía oral, vía intravenosa o similar a los animales; unos pocos días a unas pocas semanas más tarde, comparando el crecimiento tumoral en un grupo de vehículo con el crecimiento tumoral en el grupo de administración del compuesto.
Además, la actividad inhibidora de la metástasis, la actividad inhibidora de la invasión, la actividad inhibidora de la migración y la actividad de superación de la resistencia al fármaco pueden medirse mediante procedimientos de ensayo descritos anteriormente en la literatura que informa la relación de Axl con cada actividad.
Las composiciones farmacéuticas de la presente invención comprenden un compuesto de la presente invención y un portador farmacéuticamente aceptable y se pueden administrar como diversas inyecciones tales como inyecciones intravenosas, inyecciones intramusculares o inyecciones subcutáneas, o mediante diversos procedimientos tales como administración oral o transdérmica. Un vehículo farmacéuticamente aceptable significa un material farmacéuticamente aceptable (por ejemplo, un excipiente, un diluyente, un aditivo o un disolvente) involucrado en el transporte del compuesto de la presente invención o la composición que comprende el compuesto de la presente invención de un órgano a otro órgano.
Las formulaciones (por ejemplo, formulaciones orales o inyecciones) se pueden seleccionar de manera apropiada de acuerdo con el procedimiento de administración y prepararse mediante procedimientos usados convencionalmente para preparar diversas formulaciones. Ejemplos de formulaciones orales pueden incluir comprimidos, polvos, gránulos, cápsulas, píldoras, trociscos, soluciones, jarabes, elixires, emulsiones y suspensiones oleosas o acuosas. Estas formulaciones se pueden administrar por vía oral en forma libre o en forma de sal. Las formulaciones acuosas se pueden preparar formando aductos ácidos con ácidos farmacéuticamente aceptables o formando sales de metales alcalinos tales como sodio. En el caso de inyecciones, se pueden usar estabilizantes, conservantes, agentes solubilizantes y similares en las formulaciones. Las soluciones que pueden contener estos adyuvantes, etc., pueden almacenarse en recipientes y luego liofilizarse, por ejemplo, para formar formulaciones sólidas que se prepararán antes de su uso. Se puede almacenar una dosis en un recipiente o se pueden almacenar múltiples dosis en un recipiente.
Ejemplos de formulaciones sólidas incluyen comprimidos, polvos, gránulos, cápsulas, píldoras y trociscos. Estas formulaciones sólidas pueden contener aditivos farmacéuticamente aceptables junto con los compuestos de la presente invención. Ejemplos de aditivos incluyen cargas, agentes de carga, aglutinantes, desintegrantes, promotores de disolución, agentes humectantes y lubricantes, que pueden seleccionarse y mezclarse según sea necesario para preparar formulaciones.
Ejemplos de formulaciones líquidas incluyen soluciones, jarabes, elixires, emulsiones y suspensiones. Estas formulaciones líquidas pueden contener aditivos farmacéuticamente aceptables junto con los compuestos de la presente invención. Ejemplos de aditivos incluyen agentes de suspensión y emulsionantes, que se pueden seleccionar y mezclar según sea necesario para preparar formulaciones.
Los compuestos de la presente invención pueden usarse para tratar cánceres en mamíferos, particularmente, humanos. La dosis y el intervalo de dosificación pueden seleccionarse apropiadamente a juicio del médico de acuerdo con el sitio de la enfermedad y la altura, peso corporal, sexo o historial médico del paciente. Cuando se administra un compuesto de la presente invención a un ser humano, la dosis está en el intervalo de aproximadamente 0,01 mg/kg de peso corporal a aproximadamente 500 mg/kg de peso corporal por día, preferentemente aproximadamente de 0,1 mg/kg de peso corporal a aproximadamente 100 mg./kg de peso corporal por día. En la administración a un ser humano, la dosis se administra preferentemente en una sola dosis o de dos a cuatro dosis separadas por día, y la administración se repite preferentemente a intervalos apropiados. La dosis diaria puede exceder la dosis antes mencionada según el criterio del médico, si es necesario.
Los compuestos de la presente invención pueden usarse en combinación con un agente antitumoral adicional. Ejemplos de los mismos incluyen inhibidores de tirosina quinasa (TKI) tales como erlotinib enumerados anteriormente, así como antibióticos antitumorales, componentes de plantas antitumorales, BRM (modificadores de la respuesta biológica), hormonas, vitaminas, anticuerpos antitumorales y otros fármacos diana moleculares y otros agentes antitumorales.
Más específicamente, ejemplos de agentes alquilantes incluyen: agentes alquilantes tales como mostaza nitrogenada, N-óxido de mostaza nitrogenada y clorambucilo; agentes alquilantes de aziridina tales como carbocuona y tiotepa; agentes alquilantes de epóxido tales como dibromomanitol y dibromodulcitol; agentes alquilantes de nitrosourea tales como carmustina, lomustina, semustina, clorhidrato de nimustina, estreptozocina, clorozotocina y ranimustina; y otros tales como busulfán, tosilato de improsulfán y dacarbazina.
Ejemplos de diversos antimetabolitos incluyen: antimetabolitos de purina tales como 6-mercaptopurina, 6-tioguanina y tioinosina; antimetabolitos de pirimidina tales como fluorouracilo, tegafur, tegafur uracilo, carmofur, doxifluridina, broxuridina, citarabina y enocitabina; y antifolatos tales como metotrexato y trimetrexato.
Ejemplos de antibióticos antitumorales incluyen: agentes antitumorales antibióticos de antraciclina tales como mitomicina C, bleomicina, peplomicina, daunorrubicina, aclarubicina, doxorrubicina, pirarubicina, THP-adriamicina,4'-epidoxorrubicina y epirrubicina; y otros tales como cromomicina A3 y actinomicina D.
Ejemplos de componentes de plantas antitumorales incluyen: alcaloides de la vinca tales como vindesina, vincristina y vinblastina; taxanos tales como paclitaxel y docetaxel; y epipodofilotoxinas tales como etopósido y tenipósido.
Ejemplos de BRM incluyen factores de necrosis tumoral e indometacina.
Ejemplos de hormonas incluyen hidrocortisona, dexametasona, metilprednisolona, prednisolona, prasterona, betametasona, triamcinolona, oximetolona, nandrolona, metenolona, fosfestrol, etinilestradiol, clormadinona y medroxiprogesterona.
Ejemplos de vitaminas incluyen vitamina C y vitamina A.
Ejemplos de anticuerpos antitumorales y fármacos objetivo moleculares incluyen trastuzumab, rituximab, cetuximab, nimotuzumab, denosumab, bevacizumab, infliximab, mesilato de imatinib, gefitinib, erlotinib, sunitinib, lapatinib, sorafenib, dasatinib, nilotinib, vemurafenib, y tivantinib.
Ejemplos de otros agentes antitumorales incluyen cisplatino, carboplatino, oxaliplatino, tamoxifeno, camptotecina, ifosfamida, ciclofosfamida, melfalán, L-asparaginasa, aceglatona, sizofiran, picibanil, procarbazina, pipobroman, neocarzinosatina, hidroxiurea, ubenimex y krestina.
La presente invención también incluye un procedimiento para prevenir y/o tratar un cáncer, que comprende administrar un compuesto de la presente invención o una sal del mismo.
La presente invención incluye además el uso de un compuesto de la presente invención o una sal del mismo, o un solvato del compuesto o la sal para la fabricación del medicamento mencionado anteriormente.
La presente invención se describirá específicamente con referencia a los Ejemplos ilustrados a continuación. Sin embargo, la presente invención no está limitada por estos Ejemplos, y estos Ejemplos no deben interpretarse como una limitación en ningún sentido. Los reactivos, disolventes y materiales de partida descritos en la presente memoria descriptiva están fácilmente disponibles en fuentes comerciales, a menos que se especifique otra cosa.
[Ejemplos]
Abreviaturas
DMF: N,N-dimetilformamida
THF: tetrahidrofurano
HATU: hexafluorofosfato de N,N,N',N'-tetrametil-O-(7-azabenzotriazol-1-il)uronio
HOAt: 1-hidroxi-7-azabenzotriazol
DIPEA: N,N-diisopropiletilamina
COMU: hexafluorofosfato de (1-ciano-2-etoxi-2-oxoetilidenaminooxi)dimetilamino-morfolino-carbenio TFA: ácido trifluoroacético
EDCHCl: clorhidrato de 1 -etil-3-(3-dimetilaminopropil)carbodiimida
HOBt: 1-hidroxibenzotriazol
DMAP: 4-dimetilaminopiridina
PLC: cromatografía preparativa en capa fina
HPLC: cromatografía líquida de alto rendimiento

(Etapa 1) 4-(4-metilfen¡l)-3-oxobutanoato de etilo
Se disolvió ácido (4-metilfenil)acético (75,0 g, 499 mmol) en tetrahidrofurano (1,00 l). A la solución, se le añadió carbonildiimidazol (105 g, 649 mmol) y la mezcla se agitó a temperatura ambiente durante 40 minutos. A la mezcla de reacción, se le añadieron sal potásica del éster monoetílico del ácido malónico (111 g, 649 mmol) y cloruro de magnesio anhidro (57,1 g, 599 mmol) y la mezcla se calentó con agitación a 60 °C durante 6 horas. La mezcla de reacción se enfrió hasta temperatura ambiente y se le añadió acetato de etilo (2,00 l), seguido de lavado con ácido clorhídrico 1 N (1,00 l). La capa orgánica se lavó con una solución acuosa saturada de bicarbonato de sodio y salmuera en este orden. La capa orgánica se secó sobre sulfato de sodio anhidro y luego el disolvente se eliminó por destilación bajo presión reducida. El aceite obtenido se sometió a destilación azeotrópica con tolueno bajo presión reducida para obtener el compuesto del título (86,8 g, rendimiento: 78,8 %) en forma de aceite. 1H-RMN (CDCh) g(47,18-7,07 (4H, m), 4,17 (2H, q, J = 7,5 Hz), 3,78 (2H, s), 3,43 (2H, s), 2,33 (3H, s), 1,26 (3H, t, J = 7,5 Hz).
MS (ESI) m/z : 221 [(M+H)+].
(Etapa 2) 5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1.4-dihidropiridin-3-carboxilato de etilo
Se disolvió 4-(4-metilfenil)-3-oxobutanoato de etilo (43.0 g. 195 mmol) en tolueno (390 ml). A la solución. se le añadió dimetil acetal de N.N-dimetilformamida (130 ml. 976 mmol) y la mezcla se calentó con agitación a 100 °C durante 5 horas mientras se atrapaba el metanol formado en un aparato Dean-Stark. La mezcla de reacción se enfrió hasta temperatura ambiente y se concentró bajo presión reducida. El residuo se disolvió en etanol (390 ml). A la solución. se le añadió tetrahidro-2H-piran-4-ilmetanamina (26.0 ml. 215 mmol) y la mezcla se calentó con agitación a 60 °C durante 9 horas. La mezcla de reacción se enfrió hasta temperatura ambiente y se concentró bajo presión reducida.
El residuo se purificó mediante cromatografía en columna de sílica gel (Shoko Scientific Co.. Ltd.. disolvente de elución: diclorometano/acetato de etilo = 75/25 -> 20/80) para obtener el compuesto del título (36.9 g. rendimiento: 53.2 %) como una sustancia parecida al caramelo.
1H-RMN (CDCls) 5: 8.10 (1H. d. J = 2.4 Hz). 7.48 (2H. d. J = 8.0 Hz). 7.32 (1H. d. J = 2.4 Hz). 7.20 (2H. d. J = 8.0 Hz).
4.38 (2H. q. J = 7.0 Hz). 4.02 (2H. dd. J = 11.5. 4.0 Hz). 3.74 (2H. d. J = 7.3 Hz). 3.38 (2H. td. J = 11.5. 2.0 Hz). 2.08 2.00 (1H. m). 1.63-1.56 (2H. m). 1.35-1.42 (5H. m).
MS (APCI) m/z : 356 [(M+H)+].
(Etapa 3) ácido 5-(4-Metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1.4-dihidropiridin-3-carboxílico
Se disolvió 5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1.4-dihidropiridin-3-carboxilato de etilo (36.9 g. 104 mmol) en metanol (519 ml).). A la solución. se le añadió una solución acuosa de hidróxido de sodio 1 N (311 ml. 311 mmol) y la mezcla se agitó a temperatura ambiente durante 1 hora. La reacción de la mezcla se concentró bajo presión reducida. Al residuo. se le añadió ácido clorhídrico 1 N (400 ml). seguido de extracción con diclorometano. La capa orgánica se secó sobre sulfato de sodio anhidro y luego el disolvente se eliminó por destilación bajo presión reducida.
El residuo se lavó con metanol mediante el procedimiento en suspensión para obtener el compuesto del título (31.6 g. rendimiento: 92.9 %) como un sólido.
1H-RMN (CDCls) 5: 8.46 (1H. d. J = 2.4 Hz). 7.56 (1H. d. J =2.4 Hz). 7.48 (2H. d. J = 8.5 Hz). 7.28 (2H. d. J = 8.5 Hz).
4.03 (2H. dd. J = 11.3. 4.0 Hz). 3.89 (2H. d. J = 7.5 Hz). 3.38 (2H. t. J = 11.3 Hz). 2.40 (3H. s). 2.16-2.04 (1H. m). 1.62 1.53 (3H. m). 1.49-1.37 (2H. m).
MS (APCI) m/z : 328 [(M+H)+].
De manera similar. los intermedios de la Tabla 1 se sintetizaron a partir de los correspondientes materiales de partida A y B.
[Tabla 1-1]


[Tabla 1-2]

continuación

[Tabla 1-3]

continuación

[Tabla 1-4]

(continuación)

[Tabla 1-5]

(Intermedio 2) ácido 5-(4-Ciclopropil-2-fluorofenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1.4-dihidropiridin-3-carboxílico

A una solución de ácido (4-bromo-2-fluorofenil)acético (2.00 g. 8.58 mmol) en cloruro de metileno (40.0 ml). etanol (0.969 ml. 0.791 g. 17.2 mmol). 4-dimetilaminopiridina (0.105 g. 0.858 mmol) y clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDACHCl) (1.97 g. 10.3 mmol) y la mezcla se agitó durante la noche a temperatura ambiente. A la mezcla de reacción se le añadió ácido clorhídrico 1 N. seguido de extracción con cloruro de metileno tres veces. La capa orgánica se lavó con una solución acuosa saturada de bicarbonato de sodio y salmuera en este orden y luego se secó sobre sulfato de sodio anhidro. Después de filtrar y concentrar bajo presión reducida. el residuo obtenido se purificó mediante cromatografía en columna de sílica gel (Yamazen Corp.. n-hexano/acetato de etilo = (100/0 -> 87/13) para obtener el compuesto del título (1.96 g. rendimiento: 87.5 %) como un sólido.
1H-RMN (CDCh) 5: 7.27-7.24 (2H. m). 7.18-7.14 (1H. m). 4.17 (2H. q. J = 7.0 Hz). 3.62 (2H. s). 1.26 (3H. t. J = 7.0 Hz). (Etapa 2) (4-Ciclopropil-2-fluorofenil)acetato de etilo
A una suspensión de (4-bromo-2-fluorofenil)acetato de etilo (1.95 g. 7.47 mmol) sintetizado en la etapa 1 en tolueno (30.0 ml). ácido ciclopropilborónico (0.962 g. 11.2 mmol). triciclohexilfosfina (0.419 g. 1.49 mmol)). se añadieron fosfato tripotásico (5.55 g. 26.1 mmol) y agua (6.00 ml) y la mezcla de reacción se purgó con nitrógeno. Luego. se añadió acetato de paladio (II) (0.168 g. 0.747 mmol) y la mezcla se agitó a 100 °C durante 2 horas. La mezcla de reacción se
dejó enfriar hasta temperatura ambiente y luego se le añadió agua, seguido de extracción con acetato de etilo tres veces. La capa orgánica se lavó con salmuera y luego se secó sobre sulfato de sodio anhidro. Después de filtrar y concentrar bajo presión reducida, el residuo obtenido se purificó mediante cromatografía en columna de sílica gel (Yamazen Corp., n-hexano/acetato de etilo = 100/0 -> 87/13) y se purificó adicionalmente mediante cromatografía en columna de sílica gel amino (Yamazen Corp., n-hexano/acetato de etilo = 100/0 -> 87/13) para obtener el compuesto del título (1,59 g, rendimiento: 95,8 %) como un aceite.
1H-RMN (CDCla) 8: 7,12 (1H, t, J = 7,9 Hz), 6,84-6,82 (1H, m), 6,76-6,72 (1H, m), 4,16 (2H, q, J = 7,0 Hz), 3,61 (2H, s), 1,90-1,83 (1H, m), 1,25 (3H, t, J = 7,0 Hz), 1,00-0,95 (2H, m), 0,70-0,66 (2H, m).
MS (APCI) m/z : 223 [(M+H)+].
(Etapa 3) ácido (4-C¡cloprop¡l-2-fluorofenil)acét¡co
A una solución de (4-ciclopropil-2-fluorofenil)acetato de etilo (1,59 g, 7,15 mmol) sintetizado en la etapa 2 en tetrahidrofurano (30,0 ml), metanol (15,0 ml) y una solución acuosa de hidróxido de sodio de 1 mol/l (14,3 ml, 14,3 mmol) y la mezcla se agitó durante la noche a temperatura ambiente. El disolvente orgánico se eliminó por destilación bajo presión reducida y luego, se añadió agua al residuo, seguido de lavado con éter dietílico. La capa acuosa se aciduló mediante la adición de ácido clorhídrico 1 N, seguido de extracción con acetato de etilo tres veces. La capa orgánica se lavó con salmuera y luego se secó sobre sulfato de sodio anhidro. Se llevaron a cabo filtración y concentración bajo presión reducida para obtener el compuesto del título (1,36 g, 7,00 mmol) como un sólido. 1H-RMN (CDCla) 8: 7,11 (1H, t, J = 7,9 Hz), 6,83 (1H, dd, J = 7,9, 1,8 Hz), 6,75 (1H, dd, J = 11,2, 1,8 Hz), 3,65 (2H, s), 1,90 1,83 (1H, m), 1,00-0,95 (2H, m), 0,70-0,66 (2H, m).
(Etapa 4) ácido 5-(4-C¡cloprop¡l-2-fluorofen¡l)-4-oxo-1-(tetrah¡dro-2H-p¡ran-4-¡lmet¡l)-1.4-d¡h¡drop¡r¡d¡n-3-carboxíl¡co
El compuesto del título se obtuvo como un sólido usando ácido (4-ciclopropil-2-fluorofenil)acético en lugar de ácido (4-metilfenil)acético en la etapa 1 del intermedio 1a y llevando a cabo las mismas reacciones subsecuentes que en la etapa 1 a la etapa 3 del intermedio 1a.
1H-RMN (DMSO-D6) 8: 8,80 (1H, d, J = 2,4 Hz), 8,31 (1H, d, J = 2,4 Hz), 7,34 (1H, t, J = 8,2 Hz), 7,03-6,99 (2H, m), 4,12 (2H, d, J = 7,3 Hz), 3,85 (2H, dd, J = 11,2, 3,3 Hz), 3,24 (2H, t, J = 11,2 Hz), 2,12-1,97 (2H, m), 1,43 (2H, brd, J = 10,9 Hz), 1,33-1,22 (2H, m), 1,04-0,99 (2H, m), 0,77-0,73 (2H, m).
MS (APCI) m/z : 372 [(M+H)+].

(Etapa 1) 4-(5-met¡lp¡r¡d¡n-2-¡l)-3-oxobutanoato de etilo
A una solución de ácido (5-metilpiridin-2-il)acético (0,500 g, 3,31 mmol) en tetrahidrofurano (15,0 ml), se añadió 1,1'-carbonilbis-1H-imidazol (0,805 g, 4,96 mmol) y la mezcla se agitó a temperatura ambiente durante 2,5 horas. En otro recipiente de reacción, se suspendieron malonato de monoetil potasio (1,69 g, 9,92 mmol) y cloruro de magnesio (0,945 g, 9,92 mmol) en tetrahidrofurano (25,0 ml). A la suspensión, se le añadió trietilamina (2,38 ml, 1,74 g, 17,2 mmol) enfriando con hielo y la mezcla se agitó a temperatura ambiente durante 2,5 horas y luego se enfrió con hielo de nuevo. Se le añadió la solución de éster activo preparada anteriormente y la mezcla se agitó durante la noche a temperatura ambiente. A la mezcla de reacción, se le añadió una solución acuosa de ácido cítrico al 10 %, seguido de extracción con acetato de etilo cinco veces. La capa orgánica se lavó con una solución acuosa saturada de bicarbonato de sodio y salmuera en este orden y luego se secó sobre sulfato de sodio anhidro. Después de filtrar y concentrar bajo presión reducida, el residuo obtenido se purificó mediante cromatografía en columna de sílica gel (Yamazen Corp., disolvente de elución: n-hexano/acetato de etilo = 57/43 -> 36/64) para obtener el compuesto del título (0,634 g)., rendimiento: 86,6 %) como un aceite. EM (APCI) m/z: 222 [(M+H)+].
(Etapas 2 y 3) ácido 5-Met¡l-4'-oxo-1'-(tetrah¡dro-2H-p¡ran-4-¡lmet¡l)-1'.4'-d¡h¡dro-2.3'-b¡p¡r¡d¡n-5'-carboxíl¡co
El compuesto del título se obtuvo como un sólido usando 4-(5-metilpiridin-2-il)-3-oxobutanoato de etilo en lugar de 4-(4-metilfenil)-3-oxobutanoato de etilo en la etapa 2 del intermedio 1a y llevando a cabo las mismas reacciones subsecuentes que en la etapa 2 y la etapa 3 del intermedio 1a.
1H-RMN (DMSO-D6) 5: 8,86 (1H, d, J = 2,4 Hz), 8,80 (1H, d, J = 2,4 Hz), 8,53 (1H, s), 8,38 (1H, d, J = 8,5 Hz), 7,72 (1H, dd, J = 8,5, 2,1 Hz), 4,24 (2H, d, J = 7,3 Hz), 3,84 (2H, dd, J = 11,2, 3,3 Hz), 3,24 (2H, t, J = 11,2 Hz), 2,35 (3H, s), 2,11-2,01 (1H, m), 1,44 (2H, brd, J = 10,9 Hz), 1,35-1,24 (2H, m).
MS (APCI) m/z : 329 [(M+H)+].
De manera similar, el intermedio de la tabla 2 se sintetizó a partir del material de partida correspondiente.
[Tabla 2]

(Intermedio 4a) ácido 5-(4-Bromofenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxílico

(Etapa 1) 4-(4-bromofenil)-3-oxobutanoato de etilo
El compuesto del título se obtuvo como un aceite usando ácido 2-(4-bromofenil)acético en lugar de ácido (4-metilfenil)acético en la etapa 1 del intermedio 1a y llevando a cabo las reacciones subsecuentes mediante la misma operación.1
1H-RMN (CDCls) 5: 7,47 (2H, d, J = 8,5 Hz), 7,09 (2H, d, J = 8,5 Hz), 4,18 (2H, q, J = 7,1 Hz), 3,81 (2H, s), 3,46 (2H, s), 1,27 (3H,t, J = 7,1 Hz).
(Etapa 2) 5-(4-bromofenil)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo
A una solución de 4-(4-bromofenil)-3-oxobutanoato de etilo (5,00 g, 17,5 mmol) sintetizado en la etapa 1 en etanol (50,0 ml), se añadieron 1,3,5-triazina (1,56 g, 19,3 mmol) y etóxido de sodio (1,43 g, 21,0 mmol) y la mezcla se agitó a 85 °C durante 4 horas. La mezcla de reacción se dejó enfriar hasta temperatura ambiente y luego se concentró bajo presión reducida. El residuo obtenido se aciduló mediante la adición de ácido clorhídrico 1 N, y luego, el sólido depositado se recolectó por filtración, luego se lavó con acetona y se secó para obtener un producto crudo del compuesto del título (3,73 g) como un sólido.
MS (APCI) m/z : 322 [(M+H)+].
(Etapa 3) 5-(4-bromofenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxilato de etilo
A una suspensión del producto crudo de 5-(4-bromofenil)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (0,322 g) sintetizado en la etapa 2 en N,N-dimetilformamida (45,0 ml), se añadió carbonato de cesio (0,977 g, 3,00 mmol) y la mezcla se agitó a 80 °C durante 15 minutos. A continuación, se le a la misma añadió tetrahidro-2H-piran-4-ilmetil éster del ácido metanosulfónico (0,583 g, 3,00 mmol) y la mezcla se agitó a 80 °C durante 4 horas. La mezcla de reacción se dejó enfriar hasta temperatura ambiente y luego se le añadió agua, seguido de extracción con acetato de etilo tres
veces. La capa orgánica se lavó con salmuera y luego se secó sobre sulfato de sodio anhidro. Después de filtrar y concentrar bajo presión reducida, el residuo obtenido se purificó mediante cromatografía en columna de sílica gel (Yamazen Corp., disolvente de elución: n-hexano/acetato de etilo = 57/43 -> 36/64) para obtener el compuesto del título (0,234 g)., rendimiento en 2 etapas: 36,7 %) como un sólido amorfo.
1H-RMN (DMSO-D6) 5: 8,30 (1H, d, J = 2,4 Hz), 8,02 (1H, d, J = 2,4 Hz), 7,59 (4H, s), 4,20 (2H, q, J = 7,0 Hz), 3,92 (2H, d, J = 7,3 Hz), 3,85 (2H, dd, J = 10,9, 3,0 Hz), 3,25 (2H, t, J = 10,9 Hz), 2,09-1,99 (1H, m), 1,43 (2H, br d, J = 10,9 Hz), 1,32-1,24 (5H, m).
MS (APCI) m/z : 420 [(M+H)+].
(Etapa 4) ácido 5-(4-Bromofenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxílico
El compuesto del título se obtuvo como un sólido usando 5-(4-bromofenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxilato de etilo en lugar de 5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxilato de etilo en la etapa 3 del intermedio 1a y llevando a cabo las reacciones subsecuentes mediante la misma operación. 1H-RMN (DMSO-D6) 5: 8,79 (1H, d, J = 1,8 Hz), 8,45 (1H, d, J = 1,8 Hz), 7,70-7,65 (4H, m), 4,14 (2H, d, J = 7,3 Hz), 3,85 (2H, dd, J = 11,2, 3,3 Hz), 3,24 (2H, t, J = 11,2 Hz), 2,16-2,06 (1H, m), 1,43 (2H, br d, J = 10,9 Hz), 1,34-1,24 (2H, m). MS (APCI) m/z : 392 [(M+H)+].
De manera similar, los intermedios de la Tabla 3 fueron sintetizados a partir de los correspondientes materiales de partida.
[Tabla 3]

(Intermedio 5a) (2S)-2-{[2-Metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenoxi1metil)-1,4-dioxano

Se disolvió 2-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenol (0,500 g, 1,99 mmol) en N,N-dimetilformamida (10,0 ml). A la solución se le añadió ácido (2S)-1,4-dioxan-2-ilmetilmetanosulfónico (0,401 g, 2,04 mmol) y la mezcla se agitó a 90 °C durante 10 horas. La mezcla de reacción se enfrió hasta temperatura ambiente y se separó en capas orgánica y acuosa mediante la adición de acetato de etilo y agua. La capa orgánica se lavó con salmuera y luego se secó sobre sulfato de magnesio anhidro. El disolvente se eliminó por destilación bajo presión reducida y el residuo se purificó mediante cromatografía en columna de sílica gel (Biotage Japan Ltd., disolvente de elución: hexano/acetato de etilo = 100/0 -> 50/50) para obtener el compuesto del título (0,451 g)., rendimiento: 64,4 %) como un aceite. 1H-RMN (CDCh) 5: 7,39 (1H, d, J = 7,9 Hz), 7,28 (1H, s), 6,89 (1H, d, J = 7,9 Hz), 4,09-4,03 (2H, m), 4,00-3,93 (2H, m), 3,89 (3H, s), 3,86-3,63 (4H, m), 3,57-3,50 (1H, m), 1,34 (12H, s).
De manera similar, el intermedio de la tabla 4 se sintetizó a partir del material de partida correspondiente.
[Tabla 4]

(Intermedio 6) 1-(Tetrahidro-2H-piran-4-ilmetil)-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol

El compuesto del título se obtuvo de la misma manera que en el intermedio 5a usando 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol y 4-(bromometil)tetrahidropirano como materiales de partida.1
1H-RMN (CDCls) 5: 7,79 (1H, s), 7,65 (1H, s), 4,00 (2H, d, J = 7,5 Hz), 3,96 (2H, dd, J = 11,5, 4,5 Hz), 3,35 (2H, td, J = 11,5, 2,0 Hz), 2,23-2,11 (1H, m), 1,48 (2H, d, J = 11,5 Hz), 1,40-1,28 (14H, m).
MS (APCI) m/z : 293 [(M+H)+1.
(Intermedio 7) (2R)-2-[({2-[(2H3)Metoxi1-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)(2H3)fenil}oxi)metil}-1,4-dioxano

(Etapa 1) 4-Bromo-2.3.5-trideuterio-6-(trideuteriometoxi)fenol 4-bromo-2-[(2H3)metoxi1(2H3)fenol
Se suspendió (2H4)benceno-1,2-diol (12,0 g, 105 mmol) en diclorometano (105 ml). A la suspensión, se le añadieron 3,4-dihidro-2H-pirano (9,53 ml, 105 mmol) y sal de piridina del ácido p-toluenosulfónico (396 mg, 1,58 mmol) y la mezcla se agitó a temperatura ambiente durante 3 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de bicarbonato de sodio y la mezcla se agitó durante 10 minutos. La capa orgánica se lavó con salmuera y se secó sobre sulfato de sodio anhidro y el disolvente se eliminó por destilación bajo presión reducida. El aceite obtenido se disolvió en acetona (207 ml). A la solución, se le añadieron carbonato de potasio (45,8 g, 330 mmol) y yoduro de trideuteriometilo (14,4 ml, 228 mmol) y la mezcla se calentó con agitación a 40 °C durante 24 horas bajo una corriente de nitrógeno. La mezcla de reacción se enfrió hasta temperatura ambiente y luego se filtró con un embudo Kiriyama y el filtrado se concentró bajo presión reducida. Al residuo se le añadió hexano y la mezcla se filtró de nuevo con un embudo Kiriyama. Luego, el filtrado se concentró bajo presión reducida. El aceite obtenido se disolvió en etanol (181 ml). A la solución se le añadió sal de piridina del ácido p-toluenosulfónico (341 mg, 1,36 mmol) y la mezcla se calentó con agitación a 65 °C durante 3 horas. La mezcla de reacción se enfrió hasta temperatura ambiente y luego se concentró bajo presión reducida. El residuo se disolvió en N,N-dimetilformamida (90,0 ml). A la solución, se le añadió N-bromosuccinimida (16,1 g, 90,6 mmol) en pequeñas porciones enfriando con hielo y la mezcla se agitó durante 1 hora enfriando con hielo. A la mezcla de reacción, se le añadió éter dietílico (500 ml) y luego, la reacción se terminó mediante la adición de una solución acuosa de tiosulfato de sodio de 8,85 mol/l (50 ml). La capa orgánica se secó sobre sulfato de magnesio anhidro y luego se concentró bajo presión reducida. El residuo se purificó mediante cromatografía en columna de sílica gel amino (Shoko Scientific Co., Ltd., disolvente de elución: hexano/acetato de etilo = 90/10 -> 55/45) para obtener el compuesto del título (6,97 g, rendimiento: 28,3 %). como un aceite.
1H-RMN(CDCl3) 6: 5,57(1H, s).
MS (CI) m/z: 207 [(M-H)+].
(Etapa 2) (2R)-2-[({4-Bromo-2-[(2H3)metoxi1(2H3)fenN}oxi)metN1-1,4-dioxano
Se disolvió 4-bromo-2-[(2H3)metoxi](2H3)fenol (2,00 g, 9,57 mmol) en N,N-dimetilformamida (48,0 ml). A la solución, se le añadieron carbonato de potasio (2,64 g, 19,1 mmol) y ácido (2R)-1,4-dioxan-2-ilmetil metanosulfónico (1,88 g, 9,57 mmol) y la mezcla se calentó con agitación a 90 °C. durante 4 horas. La mezcla de reacción se enfrió hasta temperatura ambiente y se le añadió acetato de etilo. La materia insoluble se separó por filtración. El filtrado se concentró bajo presión reducida y el residuo se purificó mediante cromatografía en columna de sílica gel (Shoko Scientific Co., Ltd., disolvente de elución: hexano/acetato de etilo = 100/0 -> 70/30) para obtener el compuesto del título (1,75 g, rendimiento: 59,2 %) como un sólido.
1H-RMN (CDCls) 6: 4,06-3,48 (9H, m).
MS (APCI) m/z : 309 [(M+H)+1.
(Etapa 3) (2R)-2-[({2-[(2H3)Metoxi1-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)(2H3)fenN}oxi)metil}-1,4-dioxano
Se suspendió (2R)-2-[({4-Bromo-2-[(2H3)metoxi1(2H3)fenil}oxi)metil1-1,4-dioxano (800 mg, 2,59 mmol) en tolueno (5,20 ml). A la suspensión, se añadieron cloruro de níquel (II) 1,3-bis(difenilfosfino)propano1 (140 mg, 259 pmol), 1,3-bis(difenilfosfino)propano (107 mg, 0,259 mmol), pinacol boro (0,744 ml, 5,17 mmol) y trietilamina (1,10 ml, 7,76 mmol) y la mezcla se calentó con agitación a 100°C durante 16 horas bajo una corriente de nitrógeno. La mezcla de reacción se enfrió hasta temperatura ambiente y se diluyó con acetato de etilo, y luego, la materia insoluble se filtró a través de celite. El filtrado se concentró bajo presión reducida y el residuo se purificó mediante cromatografía en columna de sílica gel (Shoko Scientific Co., Ltd., disolvente de elución: hexano/acetato de etilo = 100/0 -> 85/15) para obtener el compuesto del título (840 mg, 91,1 %) como un sólido.
1H-RMN (CDCl3) 6: 4,15-4,02 (2H, m), 4,00-3,93 (2H, m), 3,87-3,62 (4H, m), 3,57-3,49 (1H, m), 1,34 (12H, s).
MS (ESI) m/z : 357 [(M+H)+1.
(Intermedio 8)
3-(4-Am¡nofen¡l)-5-{4-[(2S)-1.4-d¡oxan-2-¡lmetox¡l-3-metox¡fen¡l}p¡r¡d¡n-2-am¡na

Se d¡solv¡ó 3-(4-Am¡nofen¡l)-5-bromop¡r¡d¡n-2-am¡na (0.510 g. 1.93 mmol) s¡ntet¡zada por el proced¡m¡ento descr¡to en la patente (WO2013/115280 A1) en 1.4-d¡oxano (5.00 ml). A la soluc¡ón, se agregaron agua (1.00 ml). (2S)-2-{[2-metox¡-4-(4.4.5.5-tetramet¡l-1.3.2-d¡oxaborolan-2-¡l)fenox¡lmet¡l}1.4-d¡oxano (0.451 g. 1.29 mmol). tetraqu¡s(tr¡fen¡lfosf¡na)palad¡o (0) (0.0744 g. 0.0644 mmol) y carbonato de potas¡o (0.356 g. 2.58 mmol) y la mezcla se ag¡tó a 100 °C durante 5 horas bajo atmósfera de n¡trógeno. La mezcla de reacc¡ón se separó en capas orgán¡ca y acuosa med¡ante la ad¡c¡ón de d¡clorometano y agua. La capa orgán¡ca se secó sobre sulfato de sod¡o anh¡dro y el d¡solvente se el¡m¡nó por dest¡lac¡ón bajo pres¡ón reduc¡da. El res¡duo se pur¡f¡có med¡ante cromatografía en columna de síl¡ca gel (B¡otage Japan Ltd.. d¡solvente de eluc¡ón: acetato de et¡lo/metanol = 99/1 -> 90/10) para obtener el compuesto del título (0.139 g. rend¡m¡ento: 26.5 %) como un ace¡te.
1H-RMN (CDCla) 8: 8.24-8.22 (1H. m). 7.53-7.51 (1H. m). 7.32-7.27 (2H. m). 7.07-6.99 (2H. m). 6.98-6.93 (1H. m).
6.81-6.76 (2H. m). 4.62-4.58 (2H. m). 4.14-4.01 (3H. m). 4.01-3.93 (3H. m). 3.92-3.63 (7H. m). 3.59-3.50 (1H. m).
MS (ESI) m/z : 408 [(M+H)+]
(Intermed¡o 9a) 3-Bromo-5-{4-[(2S)-1.4-d¡oxan-2-¡lmetox¡l-3-metox¡fen¡l}p¡r¡d¡n-2-am¡na

A 3-bromo-5-yodop¡r¡d¡n-2-am¡na (0.533 g. 1.79 mmol). (2S)-2-{[2-metox¡-4-(4.4.5.5-tetramet¡l-1.3.2-d¡oxaborolan-2-¡l) fenox¡] met¡l}-1.4-d¡oxano (0.625 g. 1.79 mmol). carbonato de sod¡o (0.378 g. 3.57 mmol) y tetraqu¡s(tr¡fen¡lfosf¡na)palad¡o (0) (0.103 g. 0.0892 mmol)). 1.4-d¡oxano (9.00 ml) y agua (1.80 ml) y la mezcla se agitó a 90 °C durante 7 horas bajo una atmósfera de n¡trógeno. La mezcla de reacc¡ón se dejó enfr¡ar hasta temperatura amb¡ente y luego se le añad¡ó agua. segu¡do de extracc¡ón con cloruro de met¡leno tres veces. La capa orgán¡ca se secó sobre sulfato de sod¡o anh¡dro. Después de f¡ltrar y concentrar bajo pres¡ón reduc¡da. el res¡duo obten¡do se pur¡f¡có med¡ante cromatografía en columna de síl¡ca gel (Yamazen Corp.. d¡solvente de eluc¡ón: cloroformo/acetato de etilo = 59/41 -> 38/62) para obtener el compuesto del título (0.388 g. rend¡m¡ento : 55.0 %) como un sól¡do. 1H-RMN. (DMSO-D6) 8: 8.28 (1H. d. J = 1.8 Hz). 8.05 (1H. d. J = 2.4 Hz). 7.17 (1H. d. J = 2.4 Hz). 7.10 (1H. dd. J = 8.5.
2.4 Hz). 6.99 (1H. d. J = 8.5 Hz). 6.28 (2H. s). 3.98-3.74 (8H. m). 3.69-3.60 (2H. m). 3.52-3.46 (1H. m). 3.42-3.36 (1H. m). MS (APCI) m/z : 395 [(M+H)+].
De manera s¡m¡lar. los ¡ntermed¡os de la Tabla 5 se s¡ntet¡zaron a part¡r de los mater¡ales de part¡da correspondentes.
[Tabla 5-1]

continuación

(Intermedio 10a)
3-(4-Amino-2-fluorofenil)-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-2-amina

El compuesto del título se obtuvo de la misma manera que en el intermedio 8 usando 3-fluoro-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)anilina y 3-bromo-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-2-amina sintetizada por el procedimiento descrito en la patente (WO2013/115280 A1) como materiales de partida, 1,4-dioxano y agua como disolvente, tetraquis(trifenilfosfina)paladio (0) como catalizador y carbonato de potasio como base y se calentó con agitación a una temperatura de reacción de 100 °C durante 5 horas.
1H-RMN (CDCls) 5: 8,27 (1H, d, J =2,4 Hz), 7,54 (1H, d, J =2,4 Hz), 7,17 (1H, t, J = 8,0 Hz), 7,06-7,01 (2H, m), 6,95 (1H, d, J = 8,0 Hz), 6,55 (1H, dd, J = 8,0, 2,4 Hz), 6,51 (1H, dd, J = 11,5, 2,4 Hz), 4,52 (2H, brs), 4,13-3,52 (12H, m). MS (APCI) m/z : 426 [(M+H)+].
De manera similar, los intermedios de la Tabla 6 se sintetizaron a partir de los materiales de partida correspondientes AandB.
[Tabla 6]

(Intermedio 11a) 3-(4-Amino-3-fluorofenil)-5-(3.4-dimetoxifenil)piridin-2-amina
A 3-bromo-5-(3,4-dimetoxifenil)piridin-2-amina (1,30 g, 4,20 mmol) sintetizada por el procedimiento descrito en la patente (WO2013/115280 A1), se agregaron 2-fluoro-4-(4, 4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)anilina (1,20 g, 5,06 mmol), complejo de dicloruro de [1,1'-bis(difenilfosfino)ferroceno]paladio (N)-didorometano (1:1) (0,340 g, 0,416 mmol) y carbonato de cesio (4,10 g, 12,6 mmol), 1,4-dioxano (25,0 ml) y agua (5,00 ml) y la mezcla se agitó a 80 °C. durante 5 horas. La mezcla de reacción se dejó enfriar hasta temperatura ambiente y luego se le añadieron agua y cloruro de metileno. La materia insoluble se separó por filtración a través de celite y el filtrado se sometió a extracción con cloruro de metileno tres veces. Luego, la capa orgánica se secó sobre sulfato de sodio anhidro. Después de filtrar y concentrar bajo presión reducida, el residuo obtenido se purificó mediante cromatografía en columna de sílica gel (Yamazen Corp., cloroformo/acetato de etilo = 50/50 -> 0/100) y luego se purificó mediante cromatografía en columna de sílica gel amino (Yamazen Corp., n-hexano/acetato de etilo = 25/75 -> 0/100 -> acetato de etilo/metanol = 100/0 -> 90/10). Después de la concentración bajo presión reducida, el sólido obtenido se suspendió en éter dietílico, se recolectó por filtración y se secó para obtener el compuesto del título (0,928 g, rendimiento: 65,1 %) como un sólido. 1H-RMN (DMSO-D6) 5: 8,20 (1H, d, J = 2,4 Hz), 7,52 (1H, d, J = 2,4 Hz), 7,18-7,11 (3H, m), 7,06 (1H, dd, J = 7,9, 2,4 Hz), 6,98 (1H, d, J = 8,5 Hz), 6,87-6,83 (1H, m), 5,61 (2H, s), 5,30 (2H, s), 3,82 (3H, s), 3,77 (3H, s).
MS (APCI) m/z : 340 [(M+H)+].
De manera similar, el intermedio de la tabla 7 se sintetizó a partir del material de partida correspondiente.
[Tabla 7]

(Intermedio 12a) 3-(4-Amino-3-fluorofenil)-4-metoxipiridin-2-amina

A 3-yodo-4-metoxipiridin-2-amina (0,500 g, 2,00 mmol), 2-fluoro-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)anilina (0,498 g, 2,10 mmol), tetraquis(trifenilfosfina)paladio (0) (0,231 g, 0,200 mmol) y carbonato de sodio (0,636 g, 6,00 mmol), 1,4-dioxano (10,0 ml) y agua (2,00 ml) se añadieron y la mezcla se agitó a 80 °C durante 12 horas bajo una atmósfera de nitrógeno. La mezcla de reacción se dejó enfriar hasta temperatura ambiente y luego se le añadió agua, seguido de extracción con acetato de etilo tres veces. La capa orgánica se lavó con salmuera y luego se secó sobre sulfato de sodio anhidro. Después de filtrar y concentrar bajo presión reducida, el residuo obtenido se purificó mediante cromatografía en columna de sílica gel (Yamazen Corp., acetato de etilo/metanol = 100/0 -> 90/10) y luego se purificó mediante cromatografía en columna de sílica gel amino (Yamazen Corp., acetato de etilo/metanol = 100/0 -> 90/10) para obtener el compuesto del título (0,177 g, rendimiento: 38,0 %) como un sólido.
1H-RMN (DMSO-D6) 5: 7,83 (1H, d, J = 5,5 Hz), 7,65-7,53 (1H, m), 6,83-6,72 (3H, m), 6,39 (1H, d, J = 6,1 Hz), 5,18 (2H, s), 5,10 (2H, s), 3,65 (3H, s).
MS (APCI) m/z : 234 [(M+H)+].
De manera similar, los intermedios de la Tabla 8 se sintetizaron a partir de los materiales de partida correspondientes A and B.
[Tabla 8]

(Intermedio 13) 3-(4-Am¡nofen¡l)-5-(3.4-d¡metox¡fen¡l)p¡razin-2-am¡na

(Etapa 1) 3-Cloro-5-(3.4-d¡metox¡fenil)p¡raz¡n-2-am¡na
A 3-cloro-5-yodop¡raz¡n-2-am¡na (0.510 g. 2.00 mmol) s¡ntet¡zada de acuerdo con el proced¡m¡ento descr¡to en la patente (WO2011/110545 A1). ác¡do (3,4-d¡metox¡fen¡l)borón¡co (0,363 g. 2,00 mmol)), complejo de d¡cloruro de [1.1'-b¡s(d¡fen¡lfosf¡no)ferroceno]palad¡o-d¡dorometano (II) (1:1) (0,163 g. 0,200 mmol) y carbonato de ces¡o (1,30 g. 4,00 mmol). se añad¡eron 1,4-d¡oxano (7.50 ml) y agua (2,50 ml) y la mezcla se agitó a 80 °C durante 4 horas. La mezcla de reacc¡ón se dejó enfr¡ar hasta temperatura amb¡ente y luego se le añad¡ó agua. segu¡do de extracc¡ón con cloruro de met¡leno tres veces. La capa orgán¡ca se secó sobre sulfato de sod¡o anh¡dro. Después de f¡ltrar y concentrar bajo pres¡ón reduc¡da, el res¡duo obten¡do se pur¡f¡có med¡ante cromatografía en columna de síl¡ca gel (Yamazen Corp., cloroformo/acetato de etilo = 100/0 -> 90/10) y se purificó ad¡c¡onalmente med¡ante cromatografía en columna de síl¡ca gel am¡no (Yamazen Corp., n-hexano/acetato de etilo = 25/75 -> 0/100) para obtener el compuesto del título (0,412 g. rend¡m¡ento: 77.7 %) como un sólido.
1H-RMN (DMSO-Da) 8: 8.57 (1H. s). 7,47-7,45 (2H. m). 7,01 (1H. d. J = 7.9 Hz). 6.85 (2H. s). 3.83 (3H. s). 3.79 (3H. s).
MS (APCI) m/z : 266 [(M+H)+].
(Etapa 2) 3-(4-Am¡nofenil)-5-(3.4-d¡metox¡fen¡l)p¡raz¡n-2-am¡na
A 3-cloro-5-(3,4-dimetox¡fen¡l)p¡raz¡n-2-am¡na (0,0800 g. 0,301 mmol) sintetizada en la etapa 1. 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)an¡l¡na (0,0990 g. 0,452 mmol). complejo de dicloruro de [1,1'-bis(difen¡lfosf¡no)ferroceno]palad¡o (II)-diclorometano (1:1) (0,0246 g. 0,030 mmol) y carbonato de cesio (0,294 g. 0,903 mmol). se agregaron 1,4-dioxano (2,00 ml) y agua (0,200 ml) y la mezcla se agitó a 100°C durante 4 horas. La mezcla de reacción se dejó enfriar hasta temperatura ambiente y luego se le añadió agua. seguido de extracción con cloruro de metileno tres veces. La capa orgánica se secó sobre sulfato de sodio anhidro. Después de filtrar y concentrar bajo presión reducida. el residuo obtenido se purificó mediante cromatografía en columna de gel de sílice (Yamazen Corp., acetato de etilo solo) para obtener el compuesto del título (0,0940 g. rendimiento: 96,8%) como un sólido amorfo.
1H-RMN (DMSO-D6) 8: 8,39 (1H. s). 7,54-7,50 (4H. m). 7,01 (1H. d. J = 8.5 Hz). 6,67 (2H. d. J = 8.5 Hz). 6,00 (2H. s).
5,44 (2H. s). 3,83 (3H. s). 3,79 (3H. s).
MS (APCI) m/z : 323 [(M+H)+].
(Intermedio 14a) N-(6-Clorop¡r¡daz¡n-3-¡l)-5-(4-met¡lfen¡l)-4-oxo-1-(tetrah¡dro-2H-p¡ran-4-ilmet¡l)-1.4-d¡h¡drop¡r¡d¡n-3-carboxamida

Se disolvió ácido 5-(4-met¡lfen¡l)-4-oxo-1-(tetrah¡dro-2H-p¡ran-4-¡lmetil)-1.4-d¡h¡drop¡r¡d¡n-3-carboxíl¡co (3,00 g. 9,16 mmol) en N.N-dimetilformamida (30,0 ml). A la solución. se le añadió hexafluorofosfato de N-[1-(ciano-2-etox¡-2-oxoetil¡denam¡noox¡)d¡met¡lam¡no(morfol¡no)]uron¡o (COMU) (4,32 g. 10,1 mmol) y la mezcla se agitó a temperatura ambiente durante 1 hora. A continuación. se le añadió 3-amino-6-cloropir¡daz¡na (1,31 g. 10,1 mmol) y la mezcla se agitó a temperatura ambiente durante 3 días. La mezcla de reacción se diluyó con acetato de etilo y luego se le añadió ácido clorhídrico 1 N. La capa orgánica se lavó con una solución acuosa saturada de bicarbonato de sodio y salmuera en este orden. La capa orgánica se secó sobre sulfato de sodio anhidro. El disolvente se concentró bajo presión
reducida y el residuo se purificó mediante cromatografía en columna de sílica gel (Shoko Scientific Co., Ltd., acetato de etilo/metanol = 100/0 -> 96/4) para obtener un sólido amorfo. Este sólido se lavó con metanol mediante el procedimiento de suspensión para obtener el compuesto del título (2,10 g, rendimiento: 52,2 %) como un sólido. 1H-RMN (CDCls) 5: 13,68 (1H, s), 8,59 (1H, d, J = 9,0 Hz), 8,48 (1H, d, J = 2,0 Hz), 7,51-7,46 (4H, m), 7,26 (2H, d, J = 8,0 Hz), 4,03 (2H, dd, J = 11,5, 4,0 Hz), 3,87 (2H, d, J = 7,5 Hz), 3,39 (2H, t, J = 11,5 Hz), 2,40 (3H, s), 2,16-2,06 (1H, m), 1,64-1,55 (2H, m), 1,50-1,38 (2H, m).
MS (APCI) m/z : 439 [(M+H)+].
De manera similar, los intermedios de la Tabla 9 se sintetizaron a partir de los materiales de partida correspondientes A and B.
[Tabla 9-1]

[Tabla 9-2]

(Intermedio 16a) N-(5-Yodopiridin-2-il)-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida

A una solución de ácido 5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxílico (300 mg, 0,916 mmol) en dimetilacetamida (1 ml), se añadió cloruro de tionilo (0,087 ml, 1,19 mmol) en un baño de agua helada. Después de agitar durante 0,5 horas, se le añadieron 5-yodopiridin-2-amina (201 mg, 0,916 mmol) y diisopropiletilamina (0,23 ml, 1,37 mmol) en un baño de agua helada, y la temperatura de la mezcla de reacción se elevó gradualmente hasta temperatura ambiente. La mezcla de reacción se agitó a temperatura ambiente durante 5 horas y luego se le añadió agua. La materia insoluble se recolectó por filtración. El producto obtenido se lavó con hexano y luego se secó a 50 °C durante 1 hora bajo presión reducida. El residuo se purificó mediante cromatografía en columna de sílica gel (cloroformo/metanol = 99/1 -> 95/5) para obtener el compuesto del título (302 mg, rendimiento: 62,3%) como un sólido.
1H-RMN (CDCls) 5: 13,19 (1H, s), 8,53 (1H, d, J = 2,4 Hz), 8,49 (1H, d, J = 2,4 Hz), 8,15 (1H, d, J = 8,5 Hz), 7,95 (1H, dd, J = 8,5, 2,4 Hz), 7,51 (2H, d, J = 7,9 Hz), 7,46 (1H, d, J = 2,4 Hz), 7,28-7,23 (2H, m), 4,03 (2H, dd, J = 11,0, 3,7 Hz), 3,85 (2H, d, J = 7,3 Hz), 3,42-3,34 (2H, m), 2,40 (3H, s), 2,15-2,03 (1H, m), 1,63-1,51 (2H, m), 1,49-1,37 (2H, m). MS (ESI/APCI) m/z : 530 [(M+H)+].
De manera similar, el intermedio de la tabla 10 se sintetizó a partir del material de partida correspondiente.
[Tabla 10]

(Intermedio 17a) 5-(3,4-Dimetoxifenil)-3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)piridin-2-amina
A 3-bromo-5-(3,4-dimetoxifenil)piridin-2-amina (2,48 g, 8,02 mmol) sintetizada por el procedimiento descrito en la patente (WO2013/115280 A1), bis(pinacolato)diboro (3,05 g, 12,0 mmol), tris(dibencilidenacetona)paladio (0) (0,441 g, 0,481 mmol), triciclohexilfosfina (0,315 g, 1,12 mmol) y acetato de potasio (1,18 g, 12,0 mmol), se añadió 1,4-dioxano (40,0 ml y la mezcla se agitó a 80 °C durante 6,5 horas bajo una atmósfera de nitrógeno. La mezcla de reacción se dejó enfriar hasta temperatura ambiente y luego se le añadió agua, seguido de extracción con acetato de etilo tres veces. La capa orgánica se lavó con salmuera. Después de filtrar y concentrar bajo presión reducida, el sólido obtenido se suspendió en éter dietílico, se recolectó por filtración y se secó para obtener el compuesto del título (2,20 g, rendimiento: 77,0 %) como un sólido amarillo.
1H-RMN (DMSO-Da) 5: 8,36 (1H, d, J = 3,0 Hz), 7,86 (1H, d, J = 3,0 Hz), 7,09-6,97 (3H, m), 6,18 (2H, s), 3,83 (3H, s), 3,77 (3H, s), 1,32 (12H, s).
De manera similar, los intermedios de la Tabla 11 se sintetizaron a partir de los materiales de partida correspondientes.
[Tabla 11-1]

[Tabla 11-2]

(Intermedio 18) 5'-(3,4-Dimetoxifenil)-3-fluoro-2,3'-bipiridin-2'.5-diamina

A 6-cloro-5-fluoropiridin-3-amina (0.0300 g. 0.205 mmol), 5-(3,4-dimetoxifenil)-3-(4.4.5.5-tetrametil-1,3.2-dioxaborolan-2-il)piridin-2-amina (0,109 g. 0,307 mmol). complejo de dicloruro de [1,1'-bis(difenilfosfino)ferroceno]paladio (II)-diclorometano (1:1) (0,0167 g. 0,0205 mmol) y se añadieron carbonato de cesio (0,200 g. 0,614 mmol). 1,4-dioxano (2,00 ml) y agua (0,200 ml) y la mezcla se agitó a 100 °C durante 3 horas. La mezcla de reacción se dejó enfriar hasta temperatura ambiente y luego se le añadió agua. seguido de extracción con cloruro de metileno tres veces. La capa orgánica se secó sobre sulfato de sodio anhidro. Después de filtrar y concentrar bajo presión reducida. el residuo obtenido se purificó mediante cromatografía en columna de sílica gel (Shoko Scientific Co., Ltd., acetato de etilo/metanol = 100/0 -> 85/15) para obtener el compuesto del título (0,0690 g. rendimiento: 99.0 %) como un aceite.
1H-RMN (CDCls) 5: 8.27 (1H. d. J = 2.4 Hz). 7,98-7,95 (2H. m). 7,10-7,04 (2H. m). 6.94 (1H. d. J = 7.9 Hz). 6.85 (1H. dd. J = 12,1, 2.4 Hz). 5,99 (2H. s). 3,99-3,92 (8H. m).
MS (APCI) m/z : 341 [(M+H)+].
(Intermedio 19) ácido 5-Bromo-4-oxo-1-(tetrah¡dro-2H-p¡ran-4-¡lmet¡l)-1.4-d¡h¡dropir¡d¡n-3-carboxíl¡co

(Etapa 1) 5-bromo-4-oxo-1-(tetrah¡dro-2H-p¡ran-4-¡lmet¡l)-1.4-d¡h¡drop¡r¡din-3-carbox¡lato de metilo
Al 5-bromo-4-hidrox¡p¡rid¡n-3-carbox¡lato de metilo (2.36 g. 10.2 mmol) sintetizado de acuerdo con el procedimiento descrito en la literatura (J. Med. Chem. 2008. 51. 5330-5341). se agregaron carbonato de cesio (9.94 g. 30.5 mmol) y N.N-dimetilformamida (30.0 ml) y la mezcla se agitó a 80 °C durante 15 minutos. Luego. se le añadió 4-(bromometil) tetrahidro-2H-pirano (5.46 g. 30.5 mmol) y la mezcla se agitó a 80 °C durante 11 horas y luego se dejó durante la noche a temperatura ambiente. A la mezcla de reacción se le añadieron agua y salmuera. seguido de extracción con acetato de etilo tres veces y luego extracción con cloruro de metileno cinco veces. Las capas orgánicas se combinaron y se secaron sobre sulfato de sodio anhidro. Después de filtrar y concentrar bajo presión reducida. el residuo obtenido se purificó mediante cromatografía en columna de sílica gel amino (Yamazen Corp.. disolvente de elución: acetato de etilo/metanol = 100/0 -> 85/15) para obtener un producto crudo del compuesto del título. (2.83 g) como aceite.
MS (APCI) m/z : 330 [(M+H)+].
(Etapa 2) ácido 5-Bromo-4-oxo-1-(tetrah¡dro-2H-p¡ran-4-¡lmet¡l)-1.4-dih¡drop¡r¡d¡n-3-carboxíl¡co
A una solución del producto crudo de 5-bromo-4-oxo-1-(tetrah¡dro-2H-piran-4-¡lmet¡l)-1.4-d¡h¡drop¡r¡d¡n-3-carbox¡lato de metilo (2.83 g) sintetizado en la etapa 1 en tetrahidrofurano (50.0 ml). se agregaron metanol (25.0 ml) y una solución acuosa de hidróxido de sodio 1 N (25.7 ml. 25.7 mmol) y la mezcla se agitó durante la noche a temperatura ambiente. El disolvente orgánico se eliminó por destilación bajo presión reducida y luego. se añadió agua al residuo. seguido de lavado con acetato de etilo dos veces. La capa acuosa se aciduló mediante la adición de ácido clorhídrico 1 N y luego se añadió acetato de etilo. El sólido depositado se recolectó por filtración y luego. el filtrado se sometió a extracción con acetato de etilo tres veces. La capa orgánica se lavó con salmuera y luego se secó sobre sulfato de sodio anhidro. Después de filtrar y concentrar bajo presión reducida. el sólido obtenido se suspendió en éter dietílico y se recolectó por filtración. Este sólido se combinó con el sólido obtenido preliminarmente y se secó para obtener el compuesto del título (1.93 g. rendimiento en 2 etapas: 60.0 %) como un sólido.
1H-RMN (DMSO-Da) 8: 8.79 (1H. d. J = 1.8 Hz). 8.75 (1H. d. J = 1.8 Hz). 4.08 (2H. d. J = 7.9 Hz). 3.84 (2H. dd. J = 11.6. 3.4 Hz). 3.23 (2H. t. J = 11.6 Hz). 2.10-2.00 (1H. m). 1.40 (2H. brd. J = 10.4 Hz). 1.30-1.20 (2H. m).
MS (APCI) m/z : 316 [(M+H)+].
(Intermedio 20) N-f4-[2-Am¡no-5-(3.4-d¡metox¡fen¡l)p¡r¡d¡n-3-¡l1fen¡l)-5-bromo-4-oxo-1-(tetrahidro-2H-p¡ran-4-¡lmet¡l)-1.4-dih¡drop¡r¡d¡n-3-carboxamida

A ácido 5-bromo-4-oxo-1-(tetrah¡dro-2H-piran-4-¡lmet¡l)-1.4-d¡h¡drop¡r¡d¡n-3-carboxíl¡co (0.754 g. 2.39 mmol). 3-(4-aminofen¡l)-5-(3.4-d¡metox¡fen¡l)p¡r¡d¡n-2-am¡na (0.730 g. 2.27 mmol) sintetizada por el procedimiento descrito en la patente (WO2013/115280 A1). y hexafluorofosfato de N-[1-(ciano-2-etox¡-2-oxoet¡l¡denam¡noox¡)d¡met¡lamino(morfol¡no)]uron¡o (COMU) (1.27 g. 2.95 mmol). se agregó N.N-dimetilformamida (12.0 ml). luego N.N-diisopropiletilamina (0.593 ml. 0.440 g. 3.41 mmol) y la mezcla se agitó durante la noche a temperatura ambiente. La mezcla de reacción se concentró bajo presión reducida y luego. se añadió al residuo una solución acuosa saturada de bicarbonato de sodio. seguido de extracción con acetato de etilo tres veces. La capa orgánica se lavó con salmuera y luego se secó sobre sulfato de sodio anhidro. Después de filtrar y concentrar bajo presión reducida. el residuo obtenido se purificó mediante cromatografía en columna de sílica gel (Yamazen Corp.. disolvente de elución: acetato de etilo/metanol = 100/0 -> 85/15). Después de concentrar bajo presión reducida. el
residuo se solidificó mediante la adición de éter dietílico y el sólido se recolectó por filtración y se secó para obtener el compuesto del título (0,724 g, rendimiento: 51,5 %) como un sólido.
1H-RMN (DMSO-D6) 5: 12,61 (1H, s), 8,74 (1H, d, J = 2,4 Hz), 8,63 (1H, d, J = 2,4 Hz), 8,26 (1H, d, J = 2,4 Hz), 7,82 (2H, d, J = 8,5 Hz), 7,64 (1H, d, J = 2,4 Hz), 7,55 (2H, d, J = 8,5 Hz), 7,20-7,14 (2H, m), 6,99 (1H, d, J = 8,5 Hz), 5,77 (2H, s), 4,07 (2H, d, J = 7,3 Hz), 3,87-3,83 (5H, m), 3,77 (3H, s), 3,26 (2H, t, J = 10,7 Hz), 2,11-2,02 (1H, m), 1,42 (2H, br d, J = 10,9 Hz), 1,33-1,23 (2H, m).
MS (APCI) m/z : 619 [(M+H)+].
De manera similar, el intermedio de la tabla 12 se sintetizó a partir del material de partida correspondiente.
[Tabla 12]

(Intermedio 21) N-[4-(2-Amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi1-3-metoxifenil)piridin-3-il)-3-fluorofenil1-5-bromo-1'-[(4-cianotetrahidro-2H-piran-4-il)metil1-4'-oxo-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida

A una suspensión de ácido 5-bromo-1'-[(4-cianotetrahidro-2H-piran-4-il)metil]-4'-oxo-1',4’-dihidro-2,3'-bipiridin-5'-carboxílico (0,168 g, 0,401 mmol) en N,N-dimetilformamida (5,00 ml), se agregaron hexafluorofosfato de N-[1-(ciano-2-etoxi-2-oxoetilidenaminooxi)dimetilamino(morfolino)]uronio (COMU) (0,172 g, 0,401 mmol) y N,N-diisopropiletilamina (0,0825 ml, 0,0612 g, 0,474 mmol) y la mezcla se agitó a temperatura ambiente durante 1 hora. En esta operación, la mezcla de reacción se volvió homogénea. Se le agregó a la misma 3-(4-Amino-2-fluorofenil)-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-2-amina (0,155 g, 0,364 mmol) y la mezcla se agitó durante la noche a temperatura ambiente. La mezcla de reacción se concentró bajo presión reducida y luego, se añadió al residuo una solución acuosa saturada de bicarbonato de sodio, seguido de extracción con acetato de etilo tres veces. La capa orgánica se lavó con salmuera y luego se secó sobre sulfato de sodio anhidro. Después de filtrar y concentrar bajo presión reducida, el residuo obtenido se purificó mediante cromatografía en columna de sílica gel (Yamazen Corp., acetato de etilo/metanol = 99/1 -> 85/15) y luego se purificó mediante cromatografía en columna de sílica gel amino (Yamazen Corp., acetato de etilo/metanol = 99/1 -> 93/7) para obtener el compuesto del título (0,238 g, rendimiento: 79,1 %) como un sólido amorfo. 1H-RMN (DMSO-D6) 5: 12,95 (1H, s), 8,89-8,80 (3H, m), 8,59 (1H, d, J = 8,5 Hz), 8,31 (1H, d, J = 2,4 Hz), 8,18 (1H, dd, J = 8,5, 2,4 Hz), 7,96-7,92 (1H, m), 7,63 (1H, d, J = 2,4 Hz), 7,51-7,42 (2H, m), 7,19 (1H, d, J = 2,4 Hz), 7,11 (1H, dd, J = 8,5, 2,4 Hz), 6,99 (1H, d, J = 8,5 Hz), 5,71 (2H, s), 4,71 (2H, s), 3,97-3,75 (10H, m), 3,69-3,59 (2H, m), 3,53-3,30 (4H, m), 1,88-1,78 (4H, m).
MS (APCI) m/z : 825 [(M+H)+].
(Ejemplo 1)
N-[4-(2-Amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida

Se disolvió ácido 5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxílico (0,121 g, 0,367 mmol) en N,N- dimetilformamida (3,00 ml). A la solución, se le añadió hexafluorofosfato de N-[1-(ciano-2-etoxi-2-oxoetilidenaminooxi)dimetilamino(morfolino)]uronio (COMU) (0,236 g, 0,367 mmol) y la mezcla se agitó a temperatura ambiente durante 30 minutos. Luego, 3-(4-aminofenil)-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-2-amina (0,149 g, 0,367 mmol) sintetizada por el procedimiento descrito en la patente (WO2013/115280 A1) se añadió a la misma y la mezcla se agitó a temperatura ambiente durante 4 horas. A la mezcla de reacción se le añadió agua y el sólido depositado se recolectó por filtración. Este sólido se purificó mediante cromatografía en columna de sílica gel (Biotage Japan Ltd., disolvente de elución: diclorometano/metanol = 99/1 -> 95/5) para obtener el compuesto del título (0,18 g, rendimiento: 69,8 %) como una espuma. 1H-RMN (CDCls) 5: 12,85 (1H, s), 8,57 (1H, d, J = 2,4 Hz), 8,25 (1H, d, J = 1,8 Hz), 7,87 (2H, d, J = 8,5 Hz), 7,57 (1H, d, J = 1,8 Hz), 7,47 (5H, dd, J = 8,2, 5,8 Hz), 7,29 (2H, d, J = 8,5 Hz), 7,08-7,02 (2H, m), 6,98-6,94 (1H, m), 4,71 (2H, s), 4,11-3,94 (6H, m), 3,93-3,79 (8H, m), 3,79-3,63 (1H, m), 3,59-3,51 (1H, m), 3,44-3,35 (2H, m), 2,41 (3H, s), 2,18-2,04 (1H, m), 1,64-1,58 (2H, m), 1,51-1,35 (2H, m).
MS (ESI) m/z: 717[(M+H)+]
De manera similar, los compuestos finales de la Tabla13 se sintetizaron a partir de los materiales de partida correspondientes A y B.
[Tabla 13-1]

continuación

[Tabla 13-2]


[Tabla 13-4]


[Tabla 13-6]


[Tabla 13-8]


[Tabla 13-10]





[Tabla 13-15]


[Tabla 13-17]

[Tabla 13-18]

[Tabla 13-19]


[Tabla 13-21]

[Tabla 13-22]

[Tabla 13-23]

[Tabla 13-24]


[Tabla 13-26]

(Ejemplo 68)
Metanosulfonato de N-{4-[2-Amino-5-(3,4-dimetoxifenil)piridin-3-il]-3-fluorofenil}-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida

Se disolvió ácido 5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxílico (0,127 g, 0,389 mmol) en N-metil- 2-pirrolidona (3,00 ml). A la solución, se le añadió cloruro de tionilo (0,0277 ml, 0,382 mmol) y la mezcla se agitó a temperatura ambiente durante 30 minutos. Luego, se le añadió 3-(4-amino-2-fluorofenil)-5-(3,4-dimetoxifenil)piridin-2-amina (0,121 g, 0,354 mmol) enfriando con hielo y la mezcla se agitó a temperatura ambiente durante 5 horas. La mezcla de reacción se enfrió de nuevo con hielo y se neutralizó con bicarbonato de sodio saturado, seguido de extracción con acetato de etilo. La capa orgánica se lavó con salmuera y luego se secó sobre sulfato de magnesio anhidro y el disolvente se eliminó por destilación bajo presión reducida. El residuo se purificó mediante cromatografía en columna de sílica gel (Biotage Japan Ltd., disolvente de elución: diclorometano/metanol = 99/1 -> 96/4) para obtener el compuesto del título (0,106 g, rendimiento: 46,2 %) como una espuma.
1H-RMN (CDCla) 5: 12,98 (1H, s), 8,56 (1H, d, J = 2,4 Hz), 8,31 (1H, d, J = 2,4 Hz), 7,92 (1H, dd, J = 12,1, 1,8 Hz), 7,59 (1H, d, J =2,4 Hz), 7,49-7,43 (4H, m), 7,36 (1H, t, J = 8,2 Hz), 7,29 (2H, d, J = 7,9 Hz), 7,08 (1H, dd, J = 8,2, 2,1 Hz), 7,03 (1H, d, J = 1,8 Hz), 6,93 (1H, d, J = 7,9 Hz), 4,57-4,52 (2H, m), 4,07-4,00 (2H, m), 3,94 (3H, s), 3,92 (3H, s), 3,88 (2H, d, J = 7,3 Hz), 3,44-3,35 (2H, m), 2,41 (3H, s), 2,19-2,04 (1H, m), 1,65-1,57 (2H, m), 1,51-1,38 (2H, m). MS (ESI) m/z : 649 [(M+H)+]
De manera similar, los compuestos finales de la Tabla14 se sintetizaron a partir de los materiales de partida correspondientes.
[Tabla 14]

continuación

(Ejemplo 71) Metanosulfonato de N-[4-(2-Amino-5-{4-[(4-metMpiperazm-1-M)metN]fenM}pmdm-3-M)fenM]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida
[Fórmula 69]

(Etapa_____ 1) N-r4-(2-Amino-5-bromopiridin-3-il)fenill-5-(4-metilfenil)-4-oxo-1 -(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida
El compuesto del título se obtuvo de la misma forma que en el Ejemplo 1 usando 3-(4-aminofenil)-5-bromopiridin-2-amina como material de partida.
1H-RMN (CDCls) 5: 12,88-12,85 (1H, m), 8,56 (1H, d, J = 2,4 Hz), 8,08 (1H, d, J = 2,4 Hz), 7,87-7,84 (2H, m), 7,49 7,45 (4H, m), 7,43-7,39 (2H, m), 7,31-7,27 (2H, m), 4,63-4,58 (2H, m), 4,06-4,00 (2H, m), 3,89-3,86 (2H, m), 3,43-3,35 (2H, m), 2,41 (3H, s), 2,17-2,06 (1H, m), 1,65-1,38 (4H, m).
MS (ES+APCI) m/z : 573 [(M+H)+].
(Etapa 2) N-[4-(2-Amino-5-{4-[(4-metilpiperazin-1-il)metinfenil)piridin-3-il)fenin-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida
El compuesto del título se obtuvo de la misma manera que en el intermedio 8 usando N-[4-(2-amino-5-bromopiridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida y 1-metil-4-[[4-(4, 4, 5, 5-tetrametil-1,3,2-dioxaborolan -2-il)fenil] metil] piperazina como materiales de partida, 1,4-dioxano y agua como disolvente, tetraquis(trifenilfosfina)paladio (0) como catalizador y carbonato de potasio como base y se calentó con agitación a una temperatura de reacción de 100 °C durante 2 horas.
1H-RMN (CDCls) 5: 12,87-12,85 (1H, m), 8,57 (1H, d, J = 2,4 Hz), 8,31 (1H, d, J = 2,4 Hz), 7,87 (2H, d, J = 7,9 Hz), 7,61 (1H, d, J = 2,4 Hz), 7,52-7,45 (7H, m), 7,37 (2H, d, J = 7,9 Hz), 7,29 (2H, d, J = 7,9 Hz), 4,69-4,64 (2H, m), 4,06 4,00 (2H, m), 3,87 (2H, d, J = 7,3 Hz), 3,54 (2H, s), 3,43-3,35 (2H, m), 2,63-2,34 (8H, m), 2,41 (3H, s), 2,29 (3H, s), 2,15-2,07 (1H, m), 1,64-1,57 (2H, m), 1,50-1,38 (2H, m).
MS (ES+APCI) m/z : 683 [(M+H)+].
(Etapa 3) Metanosulfonato de N-[4-(2-Amino-5-{4-[(4-metilpiperazin-1-il)metil1fenil)piridin-3-il)fenil1-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida
Se disolvió N-[4-(2-Amino-5-{4-[(4-metilpiperazin-1-il)metil]fenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida (34,0 mg, 49,8 pmol) en cloroformo (3,00 ml). A la solución, se le añadió una solución de 1 mol/l de ácido metanosulfónico en etanol (49,8 pl, 49,8 pmol) a temperatura ambiente y, a continuación, se destiló el disolvente bajo presión reducida. Al residuo obtenido, se le añadió éter diisopropílico y el sólido depositado se recolectó por filtración para obtener el compuesto del título (25,7 mg, rendimiento: 66,3 %).
1H-RMN (CDCls) 5: 12,92-12,90 (1H, m), 8,57 (1H, d, J = 2,4 Hz), 8,20 (1H, d, J = 2,4 Hz), 7,91-7,87 (2H, m), 7,69 (1H, d, J = 2,4 Hz), 7,52-7,44 (7H, m), 7,39-7,35 (2H, m), 7,31-7,27 (2H, m), 4,07-4,00 (2H, m), 3,88 (2H, d, J = 7,3 Hz), 3,69-3,63 (2H, m), 3,44-3,35 (2H, m), 3,08-2,61 (8 H, m), 2,86 (3H, s), 2,41 (3H, s), 2,18 (3H, s), 2,16-2,06 (1H, m), 1,65-1,58 (2H, m), 1,51-1,39 (2H, m).
MS (ES+APCI) m/z : 683 [(M+H)+].
De manera similar, los compuestos finales de la Tabla15 se sintetizaron a partir de los materiales de partida correspondientes.
[Tabla 15-1]

(continuación)

[Tabla 15-2]

(continuación)

[Tabla 15-3]

(continuación)

[Tabla 15-4]

(Ejemplo 83) Metanosulfonato de N-(4-{2-Ammo-5-[3-fluoro-4-(morfoMn4-NmetN)feml]piridm-3-N}fenM)-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida

(Etapa 1) N-(4-r2-Am¡no-5-í3-fluoro-4-form¡lfen¡l)p¡r¡d¡n-3-¡llfen¡l)-5-í4-met¡lfen¡lM-oxo-1-ítetrahidro-2H-p¡ran-4-¡lmet¡l)-1.4-d¡h¡drop¡r¡d¡n-3-carboxam¡da
El compuesto del título se obtuvo de la m¡sma manera que en el ¡ntermed¡o 8 usando N-[4-(2-am¡no-5-bromop¡r¡d¡n-3-¡l)fen¡l]-5-(4-met¡lfen¡l)-4-oxo-1-(tetrah¡dro-2H-p¡ran-4-¡lmet¡l)-1,4-d¡h¡drop¡r¡d¡n-3-carboxam¡da y ác¡do 3-fluoro-4-form¡lfen¡lbór¡co. 1,4-d¡oxano y agua (proporc¡ón: 10: 1) como d¡solvente, tetrak¡s(tr¡fen¡lfosf¡na)palad¡o (0) como catal¡zador y carbonato de potas¡o como base y se calentó con ag¡tac¡ón a una temperatura de reacc¡ón de 100 °C durante 7 horas.
1H-RMN (CDCla) 8: 12.89 (1H. s). 10,37-10,35 (1H. m). 8,58-8,57 (1H. m). 8.37 (1H. d. J = 2.4 Hz). 7,94-7,87 (3H. m).
7.64 (1H. d. J = 2.4 Hz). 7,49-7,45 (6 H. m). 7.36 (1H. dd. J = 12,1, 1.8 Hz). 7,31-7,28 (2H. m). 4,86-4,81 (2H. m). 4,07 4,01 (2H. m). 3,88 (2H. d. J = 7.3 Hz). 3,43-3,35 (2H. m). 2,41 (3H. s). 2,17-2,07 (1H. m). 1,64-1,53 (2H. m). 1,51-1,39 (2H. m).
MS (ES+APCI) m/z : 617 [(M+H)+].
(Etapa 2) Metanosulfonato de N-(4-(2-Am¡no-5-[3-fluoro-4-(morfol¡n4-¡lmet¡l)fen¡llp¡r¡d¡n-3-¡l)fen¡l)-5-(4-met¡lfen¡l)-4-oxo-1-(tetrah¡dro-2H-p¡ran-4-¡lmet¡l)-1.4-d¡h¡drop¡r¡d¡n-3-carboxam¡da
Se suspend¡ó N-{4-[2-Am¡no-5-(3-fluoro-4-form¡lfen¡l)p¡r¡d¡n-3-¡l]fen¡l}-5-(4-met¡lfen¡l)-4-oxo-1-(tetrah¡dro-2H-p¡rano-4-¡lmet¡l)-1,4-d¡h¡drop¡r¡d¡n-3-carboxam¡da (70,0 mg. 0,114 mmol) en d¡cloroetano (2,27 ml). A la suspens¡ón, se le añad¡ó morfol¡na (19,8 jl. 0,227 mmol) y la mezcla se agitó a temperatura amb¡ente durante 5 m¡nutos. A la mezcla de reacc¡ón se le añad¡ó tr¡acetox¡boroh¡druro de sod¡o (48,1 mg. 0,227 mmol) y la mezcla se ag¡tó a temperatura amb¡ente durante 3 horas. A la mezcla de reacc¡ón se le añad¡eron agua y b¡carbonato de sod¡o, segu¡do de extracc¡ón con cloroformo. Los extractos se secaron sobre sulfato de sod¡o anh¡dro. El d¡solvente se el¡m¡nó por dest¡lac¡ón bajo pres¡ón reduc¡da y el res¡duo obten¡do se pur¡f¡có med¡ante cromatografía en columna de síl¡ca gel (B¡otage Japan Ltd.. d¡clorometano/etanol = 100/1 -> 50/1) para obtener un sól¡do. Este sól¡do se d¡solv¡ó en cloroformo (8,00 ml). A la soluc¡ón, se le añad¡ó una soluc¡ón 1 M de ác¡do metanosulfón¡co en etanol (91,2 jl. 91,2 jm ol) y la mezcla se concentró bajo pres¡ón reduc¡da. Al res¡duo, se le añad¡ó éter d¡etíl¡co y el sól¡do depos¡tado se recolectó por f¡ltrac¡ón para obtener el compuesto del título (30,5 mg. 34,7 %) como un sól¡do.
1H-RMN (DMSO-D6) 8 : 13,21-13,18 (1H. m). 10,01-9,83 (1H. m). 8,72 (1H. d. J = 2.4 Hz). 8,42 (1H. d. J = 2.4 Hz). 8,19 (1H. d. J =2.4 Hz). 8,10-8,03 (1H. m). 7,91-7,81 (3H. m). 7,78-7,73 (1H. m). 7,69-7,63 (1H. m). 7,61-7,55 (4H. m). 7,27 (2H. d. J = 7.9 Hz). 4,49-4,36 (2H. m). 4,12 (2H. d. J = 7.3 Hz). 4,03-3,92 (2H. m). 3,90-3,84 (2H. m). 3,72-3,59 (2H. m). 3,47-3,10 (8H. m). 2,36 (3H. s). 2,32 (3H. s). 2,17-2,05 (1H. m). 1,50-1,42 (2H. m). 1,38-1,25 (2H. m).
MS (ES+APCI) m/z : 688 [(M+H)+].
De manera s¡m¡lar, el compuesto f¡nal de la Tabla 16 se s¡ntet¡zó a part¡r del mater¡al de part¡da correspondente.
[Tabla 16]

(Ejemplo 85) Metanosulfonato de N-(4-{2-Amino-5-[1-(tetrahidro-2H-piran-4-il)-1H-pirazol-4-il]piridin-3-il}-3-fluorofenil)-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida
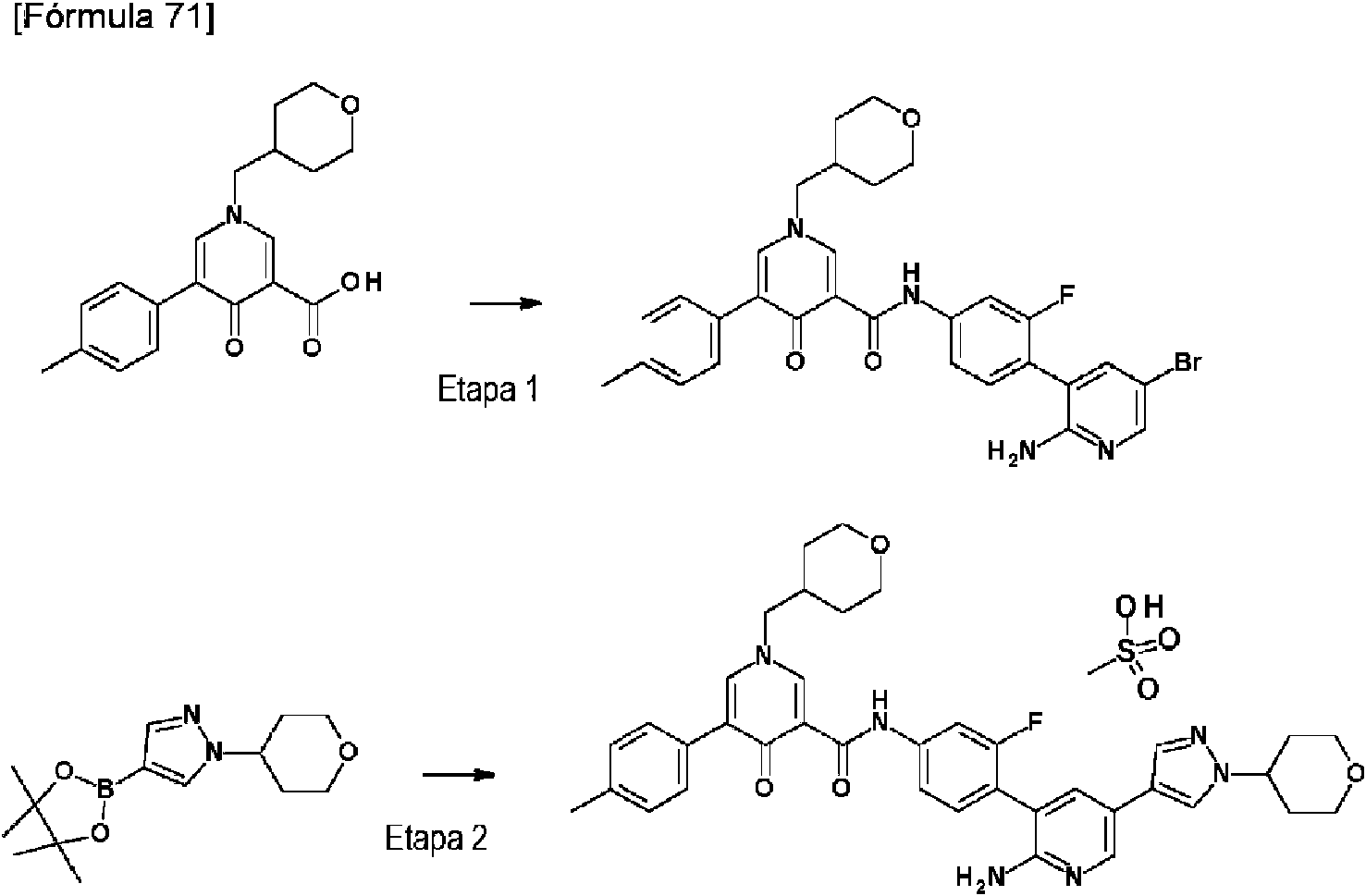
(Etapa 1) N-[4-(2-Amino-5-bromopiridin-3-ir)-3-fluorofenil1-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida
El compuesto del título se obtuvo de la misma manera que en el Ejemplo 1 usando 3-(4-amino-2-fluorofenil)-5-bromopiridin-2-amina sintetizada por el procedimiento descrito en la patente (WO2013/115280 A1) como material de partida.
1H-RMN (CDCh) 5: 13,01-12,98 (1H, m), 8,55 (1H, d, J = 2,4 Hz), 8,13 (1H, d, J = 2,4 Hz), 7,91 (1H, dd, J = 12,1, 2,4 Hz), 7,51-7,41 (5H, m), 7,32-7,25 (3H, m), 4,54-4,50 (2H, m), 4,07-4,01 (2H, m), 3,88 (2H, d, J = 7,3 Hz), 3,43-3,35 (2H, m), 2,41 (3H, s), 2,16-2,05 (1H, m), 1,64-1,54 (2H, m), 1,51-1,38 (2H, m).
MS (ES+APCI) m/z : 591 [(M+H)+].
(Etapa 2) Metanosulfonato de N-(4-{2-Amino-5-[1-(tetrahidro-2H-piran-4-ir)-1H-pirazol-4-il]piridin-3-il}-3-fluorofenil)-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida
Se obtuvo N-(4-{2-Amino-5-[1-(tetrahidro-2H-piran-4-il)-1H-pirazol-4-il] piridin-3-il}-3-fluorofenil)-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida de la misma manera que en el intermedio 8 usando N-[4-(2-amino-5-bromopiridin-3-il)-3-fluorofenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida obtenida en la etapa 1 y 1-(tetrahidro-2H-piran-4-il)-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol como materiales de partida, 1,4-dioxano y agua (proporción: 10: 1) como disolvente, tetraquis(trifenilfosfina)paladio (0) como catalizador y carbonato de potasio como base y se calentó con agitación a la temperatura de reacción. de 100 °C durante 7 horas. El compuesto del título se obtuvo mediante la misma operación que en la etapa 3 del Ejemplo 71 usando este compuesto.
1H-RMN (DMSO-D6) 5: 13,38-13,36 (1H, m), 8,73 (1H, d, J =2,4 Hz), 8,42-8,40 (1H, m), 8,26-8,19 (3H, m), 8,03-7,98 (2H, m), 7,61-7,44 (6 H, m), 7,27 (2H, d, J = 7,9 Hz), 4,44-4,35 (1H, m), 4,12 (2H, d, J = 6,7 Hz), 3,99-3,93 (2H, m), 3,90-3,84 (2H, m), 3,52-3,43 (2H, m), 3,31-3,23 (2H, m), 2,36 (3H, s), 2,31 (3H, s), 2,17-2,06 (1H, m), 2,05-1,98 (2H, m), 1,97-1,84 (2H, m), 1,49-1,42 (2H, m), 1,37-1,25 (2H, m).
MS (ES+APCI) m/z : 663 [(M+H)+].
De manera similar, los compuestos finales de la Tabla17 se sintetizaron a partir de los materiales de partida correspondientes.
[Tabla 17]

(Ejemplo 88) Metanosulfonato de N-[2'-Ammo-5'-(3,4-dimetoxifeml)-2,3'-bipiridm-5-N]-5-(4-metNfenM)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida

(Etapa_____1)_____N-(2'-Am¡no-5'-bromo-2.3'-b¡p¡r¡d¡n-5-¡l)-5-(4-met¡lfen¡l)-4-oxo-1-(tetrah¡dro-2H-p¡ran-4-¡lmet¡l)-1.4-dihidropiridin-3-carboxamida
A una solución de 5-bromo-3-(4.4.5.5-tetrametil-1.3.2-d¡oxaborolan-2-¡l)p¡r¡d¡n-2-amina (200 mg. 0.669 mmol) en dioxano (10.0 ml) y agua (1.00 ml). se agregaron N-(6-yodop¡rid¡n-3-¡l)-5-(4-met¡lfen¡l)-4-oxo-1-(tetrah¡dro-2H-p¡ran-4-ilmet¡l)-1.4-d¡h¡drop¡r¡d¡n-3-carboxamida (354 mg. 0.669 mmol). carbonato de potasio (277 mg. 2.01 mmol) y tetraquis(trifen¡lfosf¡na)palad¡o (0) (38.0 mg. 0.0344 mmol) y la mezcla se agitó a 90 °C durante 3 horas bajo una atmósfera de nitrógeno. Se añadió adicionalmente 5-bromo-3-(4.4.5.5-tetramet¡l-1.3.2-d¡oxaborolan-2-il)p¡r¡d¡n-2-amina (200 mg. 0.669 mmol) y la mezcla se se agita a 90 °C durante 5 horas. La mezcla de reacción se dejó enfriar y luego se separó en capas orgánica y acuosa mediante la adición de agua. cloruro de metileno y metanol. La capa orgánica se secó sobre sulfato de sodio anhidro y luego se filtró y el filtrado se concentró. El residuo se convirtió en una suspensión con acetato de etilo para obtener el compuesto del título (352 mg. 91.6 %) como un sólido.
1H-RMN (CDCla) 8 : 13. 05 (1H. s). 8. 94 (1H. d. J = 2.4 Hz). 8.56 (1H. d. J = 2.4 Hz). 8.32 (1H. dd. J = 8.5. 2.4 Hz).
8.10 (1H. d. J = 1.8 Hz). 7.92 (1H. d. J = 2.4 Hz). 7.68 (1H. d. J = 9.2 Hz). 7.50-7.44 (3H. m). 7.29 (2H. d. J = 7.9 Hz).
6.76 (2H. br s). 4.04 (2H. dd. J = 11.9. 3.4 Hz). 3.89 (2H. d. J = 7.3 Hz). 3.39 (2H. t. J = 11.0 Hz). 2.42 (3H. s). 2.16 2.06 (1H. m). 1.65-1.57 (2H. m). 1.51-1.38 (2H. m).
MS (APCI) m/z : 574 [(M+H)+].
(Etapa 2) Metanosulfonato de N-[2'-Am¡no-5'-(3.4-d¡metox¡fen¡l)-2.3'-b¡p¡r¡d¡n-5-¡l1-5-(4-met¡lfen¡l)-4-oxo-1-(tetrahidro-2H-p¡ran-4-¡lmetil)-1.4-d¡h¡drop¡r¡d¡n-3-carboxam¡da
A una suspensión de N-(2'-am¡no-5'-bromo-2.3'-b¡p¡rid¡n-5-¡l)-5-(4-met¡lfen¡l)-4-oxo-1-(tetrah¡dro-2H-p¡ran-4-¡lmet¡l)-1.4-dih¡drop¡rid¡n-3-carboxam¡da (130 mg. 0.226 mmol) y ácido 3.4-dimetoxifenilborónico (50 mg. 0.272 mmol) en dioxano (10 ml) y agua (1 ml). se añadieron carbonato de potasio (94 mg. 0.679 mmol) y tetraquis(trifen¡lfosf¡na)palad¡o (0) (13 mg. 0.0113 mmol) y la mezcla se agitó a 100 °C durante 2 horas bajo una atmósfera de nitrógeno. La mezcla de reacción se diluyó con cloruro de metileno y luego se lavó con agua. La capa orgánica se secó sobre sulfato de sodio anhidro. se filtró y luego se destiló el disolvente. El residuo se purificó mediante cromatografía en columna de sílica gel (cloruro de metileno/metanol = 19/1; se usó una columna de sílica gel amino como precolumna) para obtener N-[2'-am¡no-5'-(3.4-d¡metox¡fen¡l)-2.3'-b¡p¡rid¡n-5-¡l1-5-(4-met¡lfen¡l)-4-oxo-1-(tetrah¡dro-2H-p¡ran-4-¡lmet¡l)-1.4-dihidropiridin-3-carboxamida (69.0 mg. 48.3 %) como un sólido. A una solución de este compuesto en cloruro de metileno (1.00 ml) y etanol (1.00 ml). se añadió ácido metanosulfónico (10.5 mg. 0.192 mmol) y se destiló el disolvente.
El residuo se suspendió en cloruro de metileno/acetato de etilo/éter isopropílico y la materia insoluble se recolectó por filtración para obtener el compuesto del título (65,0 mg, 81,8 %) como un sólido.
1H-RMN (CDCh) 5: 14. 93 (1H, s), 13,27 (1H, s), 9,03 (1H, d, J = 2,4 Hz), 8,57 (1H, s), 8,41-8,36 (2H, m), 7,91-7,85 (2H, m), 7,52 (1H, d, J = 2,4 Hz), 7,46 (2H, d, J = 8,5 Hz), 7,30 (2H, d, J = 7,9 Hz), 7,05 (1H, dd, J = 8,2, 2,1 Hz), 7,00 6,94 (2H, m), 4,04 (2H, dd, J = 11,6, 3,7 Hz), 3,96 (3H, s), 3,95 (3H, s), 3,91 (2H, d, J = 7,3 Hz), 3,40 (2H, t, J = 11,0 Hz), 2,95 (3H, s), 2,42 (3H, s), 2,18-2,07 (1H, m), 1,65-1,55 (2H, m), 1,52-1,39 (2H, m).
MS (ESI) m/z : 632 [(M+H)+].
De manera similar, los compuestos finales de la Tabla18 se sintetizaron a partir de los materiales de partida correspondientes A and B.
[Tabla 18-1]

[Tabla 18-2]

[Tabla 18-3]
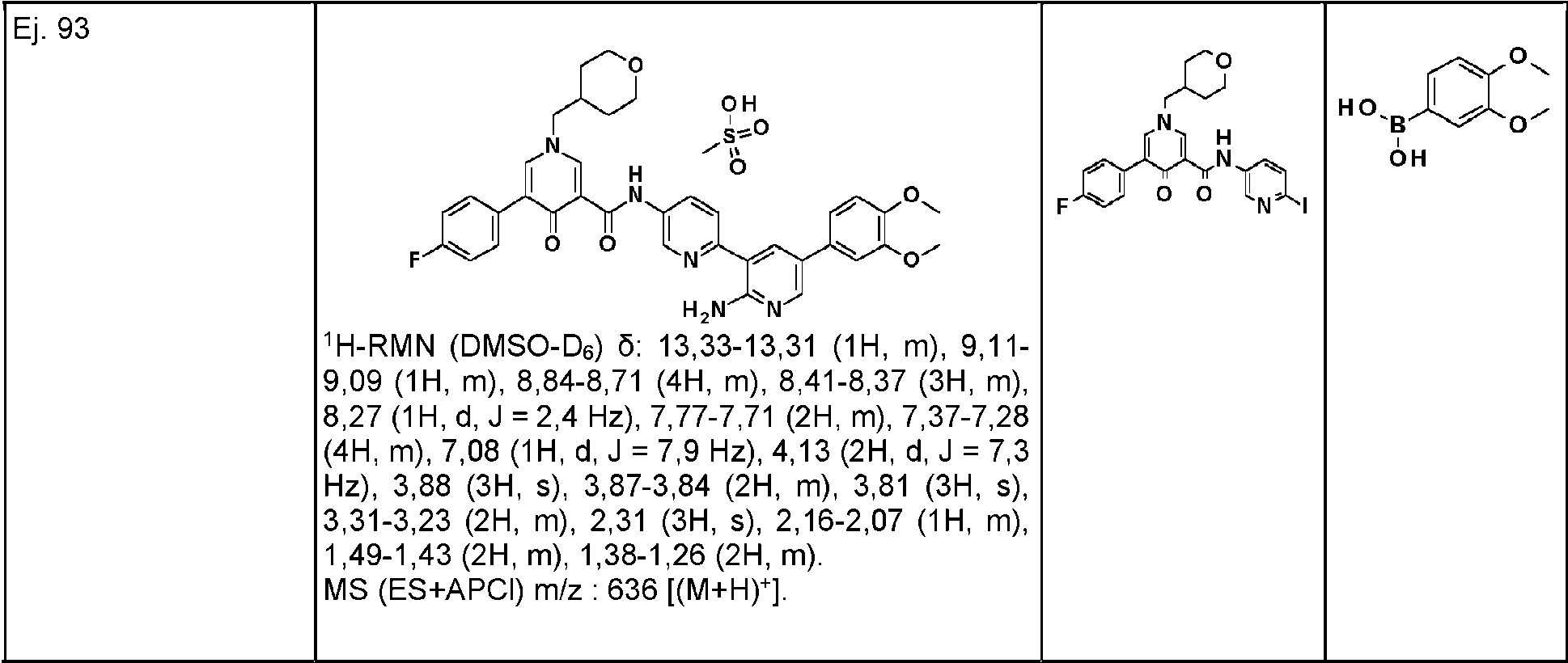
continuación

[Tabla 18-4]

[Tabla 18-5]

(Ejemplo 100) N-{6-[2-Amino-5-(3,4-dimetoxifenil)piridin-3-il] pmdazm-3-il}-5-(4-metilfeml)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridm-3-carboxamida

A 5-(3,4-dimetoxifenil)-3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)piridin-2-amina (0,0800 g, 0,182 mmol), N-(6-doropiridazin-3-il)-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida (0,0974 g, 0,273 mmol), tris(dibencilidenacetona)paladio (0) (0,0167 g, 0,0182 mmol), 2-diddohexilfosfino-2,4',6'
triisopropilbifenilo (XPhos) (0,0348 g, 0,0729 mmol) y carbonato de sodio (0,0229 g, 0,547 mmol), se agregaron 1,4-dioxano (2,00 ml) y agua (0,200 ml) y la mezcla se agitó a 100 °C durante 4 horas. La mezcla de reacción se dejó enfriar hasta temperatura ambiente y luego se le añadió agua, seguido de extracción con cloruro de metileno tres veces. La capa orgánica se secó sobre sulfato de sodio anhidro. Después de filtrar y concentrar bajo presión reducida, el residuo obtenido se purificó mediante cromatografía en columna de sílica gel (Yamazen Corp., acetato de etilo/metanol = 100/0 -> 80/20). Después de concentrar bajo presión reducida, el sólido obtenido se suspendió en etanol, se recolectó por filtración y se secó para obtener el compuesto del título (0,067 g, rendimiento: 58,1 %) como un sólido.
1H-RMN (DMSO-Da) 8: 13,94 (1H, s), 8,78 (1H, d, J = 2,4 Hz), 8,64 (1H, d, J = 9,7 Hz), 8,52 (1H, d, J = 9,7 Hz), 8,42 (1H, d, J = 2,4 Hz), 8,25-8,21 (2H, m), 7,61 (2H, d, J = 7,9 Hz), 7,48 (2H, s), 7,28-7,23 (4H, m), 7,02 (1H, d, J = 8,5 Hz), 4,12 (2H, d, J = 7,3 Hz), 3,91-3,83 (5H, m), 3,79 (3H, s), 3,27 (2H, t, J = 10,9 Hz), 2,37 (3H, s), 2,16-2,07 (1H, m), 1,47 (2H, brd, J = 10,9 Hz), 1,37-1,26 (2H, m).
MS (APCI) m/z : 633 [(M+H)+].
De manera similar, los compuestos finales de la Tabla19 se sintetizaron a partir de los materiales de partida correspondientes A and B.




(Ejemplo 110) N-[6-(2-Amino-5-{3-metoxi-4-[2-(morfolin4-il)etoxi]fenil}piridin-3-il)piridazin-3-il]-5-(4-metilfenil)-4-oxo-1 -(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida

A 3-bromo-5-{3-metoxi-4-[2-(morfolin-4-il) etoxi]fenil}piridin-2-amina (0,245 g, 0,600 mmol), bis(pinacolato)diboro (0,229 g, 0,900 mmol), tris(dibencilidenacetona) dipaladio (0) (0,0330 g, 0,0360 mmol), triciclohexilfosfina (0,0236 g, 0,0840 mmol) y acetato de potasio (0,0883 g, 0,900 mmol), se agregó 1,4-dioxano (3,00 ml) y la mezcla se agitó a 80 °C durante 6 horas bajo una atmósfera de nitrógeno. La mezcla de reacción se dejó enfriar hasta temperatura ambiente y luego se filtró y el filtrado se concentró bajo presión reducida. Al residuo obtenido, se agregaron N-(6-cloropiridazin-3-il)-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida (0,175 g, 0,399 mmol), complejo de dicloruro de [1,1'-bis(difenilfosfino)ferroceno]paladio (Il)-cloruro de metileno (1:1) (0,0326 g, 0,0399 mmol), carbonato de cesio (0,390 g, 1,20 mmol), 1,4-dioxano (4,00 ml) y agua (0,400 ml) y la mezcla se agitó a 100 °C durante 4 horas bajo una atmósfera de nitrógeno. La mezcla de reacción se dejó enfriar hasta temperatura ambiente y luego se le añadió una solución acuosa saturada de bicarbonato de sodio, seguido de extracción con cloruro de metileno tres veces. La capa orgánica se secó sobre sulfato de sodio anhidro. Después de filtrar y concentrar bajo presión reducida, el residuo obtenido se purificó mediante cromatografía en columna de sílica gel amino (Yamazen Corp., acetato de etilo/metanol = 100/0 -> 80/20). Después de concentrar bajo presión reducida, el sólido obtenido se suspendió en acetato de etilo, se recolectó por filtración y se secó para obtener el compuesto del título (0,153 g, rendimiento: 52,4 %) como un sólido.
1H-RMN (CDCls) 5: 13,77 (1H, s), 8,67 (1H, d, J = 9,7 Hz), 8,51 (1H, d, J = 2,4 Hz), 8,35 (1H, d, J = 2,4 Hz), 7,96-7,94 (2H, m), 7,53-7,48 (3H, m), 7,27 (2H, d, J = 7,9 Hz), 7,08-7,05 (2H, m), 6,98 (1H, d, J = 7,9 Hz), 6,89 (2H, s), 4,21 (2H, t, J = 6,1 Hz), 4,03 (2H, dd, J = 11,2, 3,3 Hz), 3,94 (3H, s), 3,87 (2H, d, J = 7,3 Hz), 3,76 (4H, t, J = 4,9 Hz), 3,39 (2H, t, J = 11,2 Hz), 2,88 (2H, t, J = 6,1 Hz), 2,63-2,61 (4H, m), 2,41 (3H, s), 2,15-2,07 (1H, m), 1,62-1,60 (2H, br d, J = 11,2 Hz), 1,49-1,39 (2H, m).
MS (APCI) m/z : 732 [(M+H)+].
De manera similar, el compuesto final de la Tabla 20 se sintetizó a partir del material de partida correspondiente.
[Tabla 20]

(Ejemplo 112) N-{5-[2-Amino-5-(3,4-dimetoxifenil)piridin-3-il]pirazin-2-il}-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida

A 5-(3,4-dimetoxifenil)-3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)piridin-2-amina (0,111 g, 0,310 mmol), N-(5-bromopirazin-2-il)-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida (0,100 g, 0,207 mmol), complejo de dicloruro de [1,1'-bis(difenilfosfino)ferroceno]paladio (N)-domro de metileno (1:1) (0,0169 g, 0,0207 mmol) y carbonato de cesio (0,202 g, 0,621 mmol), se agregaron 1,4-dioxano (2,00 ml) y agua (0,400 ml) y la mezcla se agitó a 80 °C durante 1 hora. La mezcla de reacción se dejó enfriar hasta temperatura ambiente y luego se le añadió agua, seguido de extracción con acetato de etilo tres veces. La capa orgánica se lavó con salmuera y luego se secó sobre sulfato de sodio anhidro. Después de filtrar y concentrar bajo presión reducida, el residuo obtenido se purificó mediante cromatografía en columna de sílica gel amino (Yamazen Corp., acetato de etilo/metanol = 100/0 -> 90/10). Después de concentrar bajo presión reducida, el residuo se solidificó usando acetato de etilo y éter dietílico, y el sólido se recolectó por filtración y se secó para obtener el compuesto del título (0,111 g, rendimiento: 84,8 %) como un sólido.
1H-RMN (DMSO-D6 ) 8: 13,61 (1H, s), 9,58 (1H, d, J = 1,2 Hz), 9,17 (1H, d, J = 2,4 Hz), 8,79 (1H, d, J = 2,4 Hz), 8,39 (1H, d, J = 2,4 Hz), 8,32 (1H, d, J = 2,4 Hz), 8,20 (1H, d, J = 2,4 Hz), 7,59 (2H, d, J = 8,5 Hz), 7,29-7,23 (6 H, m), 7,02 (1H, d, J = 8,5 Hz), 4,13 (2H, d, J = 6,7 Hz), 3,89-3,85 (5H, m), 3,79 (3H, s), 3,27 (2H, t, J = 10,9 Hz), 2,36 (3H, s), 2,16-2,07 (1H, m), 1,49-1,44 (2H, m), 1,37-1,27 (2H, m).
MS (APCI) m/z : 633 [(M+H)+].
(Ejemplo 113) N-{4-[2-Amino-5-(3,4-dimetoxifenil)piridin-3-il]fenil}-5-(4-ciclopropilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida

A una suspensión de N-{4-[2-amino-5-(3,4-dimetoxifenil)piridin-3-il]fenil}-5-(4-bromofenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida (0,350 g, 0,503 mmol) en tolueno (5,00 ml), se agregaron ácido ciclopropilborónico (0,0648 g, 0,755 mmol), triciclohexilfosfina (0,0282 g, 0,101 mmol), fosfato tripotásico (0,374 g, 1,76 mmol) y agua (1,00 ml) y la mezcla de reacción se purgó con nitrógeno. A continuación, se le añadió acetato de paladio (II) (0,0113 g, 0,0503 mmol) y la mezcla se calentó hasta 100 °C. Luego, se añadió a la misma 1,4-dioxano (2,00 ml) y la mezcla se agitó a la misma temperatura que antes durante 4 horas. La mezcla de reacción se dejó enfriar hasta temperatura ambiente y luego se agregó agua a la misma, seguido de extracción con cloruro de metileno tres veces. La capa orgánica se lavó con salmuera y luego se secó sobre sulfato de sodio anhidro. Después de la filtración y concentración bajo presión reducida, el residuo obtenido se purificó mediante cromatografía en columna de sílica gel (Yamazen Corp., acetato de etilo/metanol = 100/0 -> 85/15) y se purificó adicionalmente mediante cromatografía líquida de alto rendimiento (NOMURA Develosil Combi, acetonitrilo/agua/ácido fórmico al 0,1 %). El disolvente orgánico se eliminó por destilación bajo presión reducida y luego se añadió al residuo una solución acuosa saturada de bicarbonato de sodio. El sólido depositado se recolectó por filtración y se secó para obtener el compuesto del título (0,200 g, rendimiento: 60,5 %) como un sólido. 1H-RMN (DMSO-D6) 8: 13,11 (1H, s), 8,71 (1H, d, J = 2,4 Hz), 8,25 (1H, d, J = 2,4 Hz), 8,16 (1H, d, J = 2,4 Hz), 7,82 (2H, d, J = 8,5 Hz), 7,63 (1H, d, J = 2,4 Hz), 7,58-7,52 (4H, m), 7,20 7,14 (4H, m), 6,99 (1H, d, J = 8,5 Hz), 5,76 (2H, s), 4,10 (2H, d, J = 7,3 Hz), 3,88-3,83 (5H, m), 3,77 (3H, s), 3,27 (2H, t, J = 10,9 Hz), 2,16-2,04 (1H, m), 2,00-1,93 (1H, m), 1,46 (2H, br d, J = 10,9 Hz), 1,35-1,26 (2H, m), 1,02-0,97 (2H, m), 0,73-0,69 (2H, m).
MS (APCI) m/z : 657[(M+H)+],
(Ejemplo 114) N-{4-[2-Ammo-5-(3,4-dimetoxifeml)pmdm-3-M]feml}-5-metN-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida

A N-{4-[2-amino-5-(3,4-dimetoxifenil)piridin-3-il]fenil}-5-bromo-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida (0,620 g, 1,00 mmol), éster de N-fenildietanolamina del ácido 5-metilpiridin-2-borónico (0,480 g, 1,70 mmol) y yoduro de cobre (I) (0,0762 g, 0,400 mmol)), se añadieron tolueno (6,00 ml), metanol (1,85 ml) y una solución de carbonato de potasio (1,84 g, 13,3 mmol) en agua (1,20 ml) y la mezcla de reacción se purgó con nitrógeno. A continuación, se le añadió tetraquis(trifenilfosfina)paladio (0) (0,116 g, 0,100 mmol) y la mezcla se agitó a 100 °C durante 4 horas. La mezcla de reacción se dejó enfriar hasta temperatura ambiente y luego se le añadieron acetato de etilo y agua. La materia insoluble se separó por filtración. El filtrado se sometió a extracción con acetato de etilo tres veces. La capa orgánica se lavó con salmuera y luego se secó sobre sulfato de sodio anhidro. Después de filtrar y concentrar bajo presión reducida, el residuo obtenido se purificó mediante cromatografía en columna de sílica gel (Yamazen Corp., acetato de etilo/metanol = 99/1 -> 85/15). Después de concentrar bajo presión reducida, el residuo se solidificó mediante la adición de acetato de etilo y el sólido se recolectó por filtración y se secó para obtener el compuesto del título (0,229 g, rendimiento: 36,2 %) como un sólido.
1H-RMN (DMSO-D6) 5: 12,98 (1H, s), 8,75 (1H, d, J = 2,4 Hz), 8,71 (1H, d, J = 2,4 Hz), 8,30 (1H, d, J = 2,4 Hz), 8,25 (1H, d, J = 2,4 Hz), 8,02 (1H, dd, J = 8,5, 2,4 Hz), 7,82 (2H, d, J = 8,5 Hz), 7,61 (1H, d, J = 2,4 Hz), 7,53 (2H, d, J = 8,5 Hz), 7,34 (1H, d, J = 8,5 Hz), 7,19-7,13 (2H, m), 6,98 (1H, d, J = 8,5 Hz), 5,69 (2H, s), 4,12 (2H, d, J = 7,3 Hz), 3,88 3,83 (5H, m), 3,77 (3H, s), 3,27 (2H, t, J = 11,0 Hz), 2,52 (3H, s), 2,17-2,07 (1H, m), 1,47 (2H, brd, J = 11,0 Hz), 1,37 1,26 (2H, m).
MS (APCI) m/z: 632 [(M+H)+].
De manera similar, el compuesto final de la Tabla 21 se sintetizó a partir del material de partida correspondiente.
[Tabla 21]
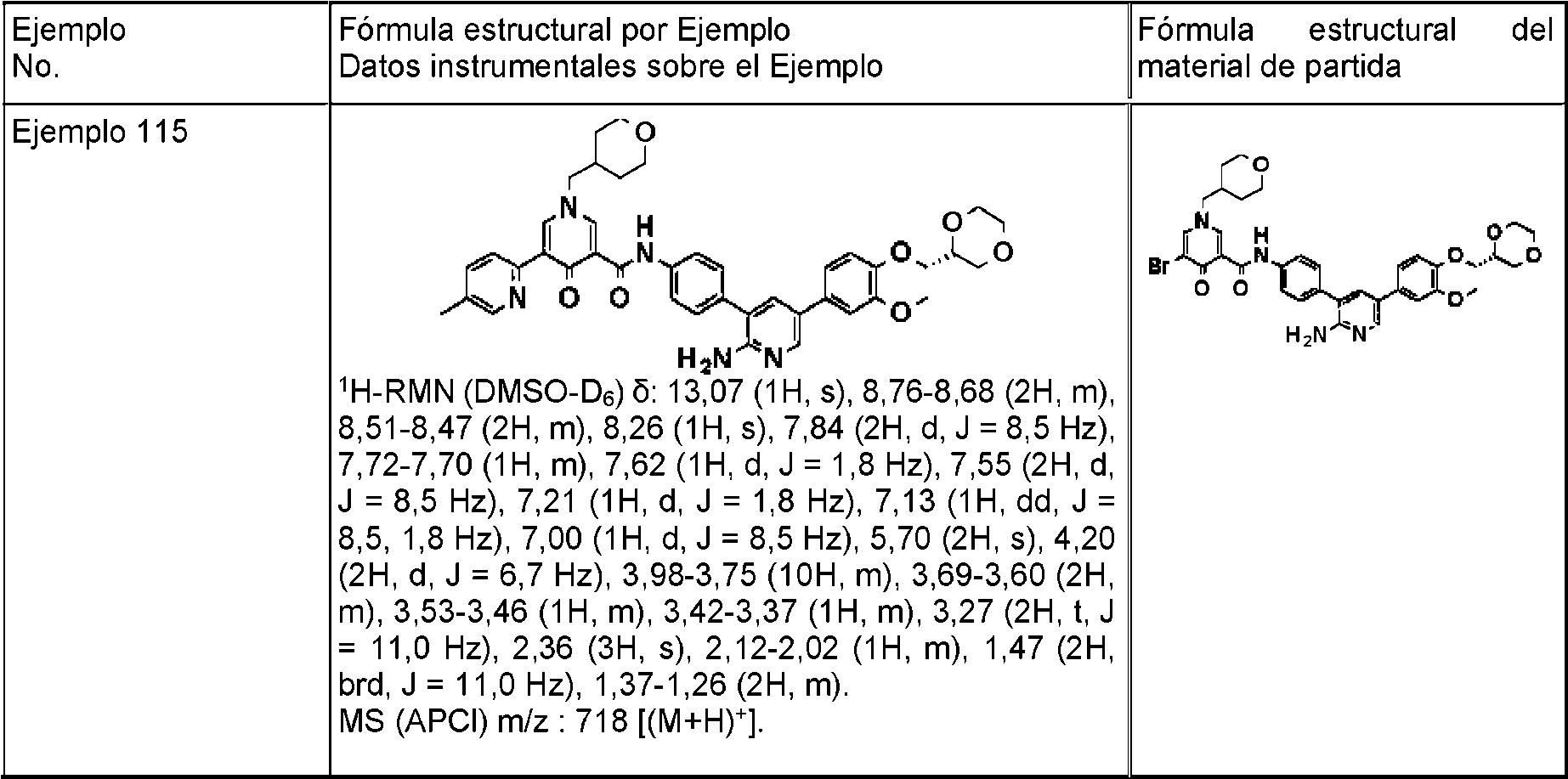
(Ejemplo 116) N-{4-[2-Ammo-5-(3,4-dimetoxifeml)piridm-3-N]feml}-6'-metN-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1, 4-dihidro-3,3'-bipiridin-5-carboxamida

A N-{4-[2-amino-5-(3,4-dimetoxifenil)piridin-3-il]fenil}-5-bromo-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida (0,400 g, 0,646 mmol), 2-metil-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)piridina (0,212 g, 0,969 mmol), carbonato de cesio (0,631 g, 1,94 mmol) y complejo de dicloruro de [1,1'-bis(difenilfosfino)ferroceno]paladio (N)-domro de metileno (1:1) (0,0527 g, 0,0646 mmol), se añadieron 1,4-dioxano (5,00 ml) y agua (1,00 ml) bajo una atmósfera de nitrógeno y la mezcla se agitó a 80 °C durante 4 horas. La mezcla de reacción se dejó enfriar hasta temperatura ambiente y luego se le añadieron agua y acetato de etilo. La materia insoluble se separó por filtración. El filtrado se sometió a extracción con acetato de etilo tres veces. La capa orgánica se lavó con salmuera y luego se secó sobre sulfato de sodio anhidro. Después de filtrar y concentrar bajo presión reducida, el residuo obtenido se purificó mediante cromatografía en columna de sílica gel amino (Yamazen Corp., acetato de etilo/metanol = 99/1 -> 90/10) y se purificó adicionalmente mediante cromatografía en columna de sílica gel (Yamazen Corp., acetato de etilo/metanol = 99/1 -> 5/95). Después de concentrar bajo presión reducida, el residuo se purificó mediante cromatografía líquida de alto rendimiento (NOMURA Develosil Combi, acetonitrilo/agua/ácido fórmico al 0,1 %). El disolvente orgánico se eliminó por destilación bajo presión reducida y luego se añadió al residuo una solución acuosa saturada de bicarbonato de sodio, seguido de extracción con acetato de etilo tres veces. La capa orgánica se lavó con salmuera y luego se secó sobre sulfato de sodio anhidro. Se llevaron a cabo filtración y concentración bajo presión reducida para obtener el compuesto del título (0,129 g, rendimiento: 31,6 %) como un sólido amorfo.
1H-RMN (DMSO-D6) 5: 13,08 (1H, s), 8,76 (1H, d, J = 2,4 Hz), 8,69 (1H, d, J = 2,4 Hz), 8,51-8,47 (2H, m), 8,26 (1H, d, J = 2,4 Hz), 7,84 (2H, d, J = 8,5 Hz), 7,71 (1H, dd, J = 8,5, 2,4 Hz), 7,62 (1H, d, J = 2,4 Hz), 7,55 (2H, d, J = 8,5 Hz), 7,20-7,14 (2H, m), 6,99 (1H, d, J = 8,5 Hz), 5,69 (2H, s), 4,20 (2H, d, J = 7,3 Hz), 3,88-3,83 (5H, m), 3,77 (3H, s), 3,27 (2H, t, J = 11,3 Hz), 2,36 (3H, s), 2,11-2,03 (1H, m), 1,47 (2H, brd, J = 11,0 Hz), 1,37-1,26 (2H, m).
MS (APCI) m/z: 632 [(M+H)+].
De manera similar, los compuestos finales de la Tabla 22 se sintetizaron a partir de los materiales de partida correspondientes.
[Tabla 22]


(Ejemplo 119) N-{4-[2-Ammo-5-(3,4-dimetoxifeml)pmdm-3-N]feml}-5-(5-metNtiofen-2-N)-4-oxo-1 -(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida

A N-{4-[2-amino-5-(3,4-dimetoxifenil)piridin-3-il]fenil}-5-bromo-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida (0,200 g, 0,323 mmol), ácido (5-metil-2-tienil)borónico (0,055 g, 0,387 mmol), carbonato de potasio (0,134 g, 0,969 mmol) y tetrakis(trifenilfosfina)paladio (0) (0,0373 g, 0,0323 mmol), se agregaron 1,2-dimetoxietano (5,00 ml) y agua (0,500 ml) y la mezcla se agitó a 90 °C durante 12 horas y luego se dejó enfriar. A la mezcla de reacción se le añadió agua, seguido de extracción con cloruro de metileno tres veces. La capa orgánica se secó sobre sulfato de sodio anhidro. Después de filtrar y concentrar bajo presión reducida, el residuo obtenido se purificó mediante cromatografía en columna de sílica gel (Yamazen Corp., acetato de etilo/metanol = 99/1 -> 80/20) y se purificó mediante cromatografía en columna de sílica gel amino (Yamazen Corp., acetato de etilo/metanol = 99/1 -> 93/7). El compuesto resultante se purificó adicionalmente mediante cromatografía líquida de alto rendimiento (NOMURA Develosil Combi, acetonitrilo/agua/ácido fórmico al 0,1 %) para obtener el compuesto del título (0,0860 g, rendimiento: 41,8 %) como un sólido. 1H-RMN (DMSO-D6) 5: 12,90 (1H, s), 8,69-8,66 (2H, m), 8,26 (1H, d, J = 2,4 Hz), 7,84 (2H, d, J = 8,5 Hz), 7,62 (1H, d, J = 2,4 Hz), 7,57-7,54 (3H, m), 7,20-7,14 (2H, m), 6,99 (1H, d, J = 8,5 Hz), 6,87 6,85 (1H, m), 5,70 (2H, s), 4,14 (2H, d, J = 7,3 Hz), 3,88-3,83 (5H, m), 3,77 (3H, s), 3,27 (2H, t, J = 10,9 Hz), 2,49 (3H, s), 2,16-2,10 (1H, m), 1,45 (2H, brd, J = 10,9 Hz), 1,38-1,27 (2H, m).
MS (APCI) m/z : 637 [(M+H)+].
De manera similar, el compuesto final de la Tabla 23 se sintetizó a partir del material de partida correspondiente.
[Tabla 23]

(Ejemplo 121) N-[4-(2-Amino-5-(4-[(2R)-1,4-dioxan-2-Nmetoxi]-3-metoxifenM}pmdm-3-M)-3-fluorofenN]-1'-[(4-cianotetrahidro-2H-piran-4-il)metil]-5-metil-4'-oxo-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida

A N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifeml}pindin-3-il)-3-fluorofeml]-5-bromo-1'-[(4-cianotetrahidro-2H-piran-4-il)metil]-4'-oxo-1',4’-dihidro-2,3'-bipiridin-5'-carboxamida (0,23o g, 0,279 mmol), complejo de dicloruro de [1,1'-bis(difenilfosfino)ferroceno]paladio (II)-doruro de metileno (1:1) (0,0228 g, 0,0279 mmol) y carbonato de cesio (0,272 g, 0,836 mmol), se añadieron 1,4-dioxano (5,00 ml), agua (0,500 ml) y una solución al 50 % de trimetilboroxina en tetrahidrofurano (3,5 mol/l, 0,0960 ml, 0,334 mmol) bajo una atmósfera de nitrógeno y la mezcla se agitó a 100 °C. °C durante 3,5 horas. La mezcla de reacción se dejó enfriar hasta temperatura ambiente y luego se le añadió una solución acuosa saturada de bicarbonato de sodio y acetato de etilo. La materia insoluble se separó por filtración. El filtrado se sometió a extracción con acetato de etilo tres veces. La capa orgánica se lavó con salmuera y luego se secó sobre sulfato de sodio anhidro. Después de filtrar y concentrar bajo presión reducida, el residuo obtenido se purificó mediante cromatografía en columna de sílica gel (Yamazen Corp., acetato de etilo/metanol = 99/1 -> 80/20) y luego se purificó adicionalmente mediante cromatografía en columna de sílica gel amino (Yamazen Corp.., acetato de etilo/metanol = 99/1 -> 93/7) para obtener el compuesto del título (0,145 g, rendimiento: 68,4 %) como un sólido amorfo.
1H-RMN (DMSO-Da) 5: 13,07 (1H, s), 8,86 (1H, d, J = 2,4 Hz), 8,80 (1H, d, J = 2,4 Hz), 8,52-8,48 (2H, m), 8,31 (1H, d, J = 2,4 Hz), 7,94 (1H, dd, J = 12,1,2,4 Hz), 7,72 (1H, dd, J = 8,2, 2,4 Hz), 7,63 (1H, d, J = 2,4 Hz), 7,50-7,42 (2H, m), 7,18 (1H, d, J = 2,4 Hz), 7,11 (1H, dd, J = 8,5, 2,4 Hz), 6,99 (1H, d, J = 8,5 Hz), 5,71 (2H, s), 4,70 (2H, s), 3,97-3,74 (10H, m), 3,69-3,60 (2H, m), 3,52-3,32 (4H, m), 2,36 (3H, s), 1,88-1,77 (4H, m).
MS (APCI) m/z : 761 [(M+H)+].
De manera similar, los compuestos finales de la Tabla 24 se sintetizaron a partir de los materiales de partida correspondientes.
[Tabla 24]

(Ejemplo 124) Metanosulfonato de N-[4-(2-Amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida (sólido amorfo)

A una solución de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4’-dihidro-2,3'-bipiridin-5'-carboxamida (23,9 g, 32,5 mmol) en cloruro de metileno (110 ml), se añadió una solución de ácido metanosulfónico (2 ,11 ml, 3,12 g, 32,5 mmol) en etanol (36,0 ml) y la mezcla se agitó a temperatura ambiente durante 15 minutos. La reacción de la mezcla se concentró bajo presión reducida. Luego, el sólido amorfo obtenido se suspendió en acetato de etilo, se recolectó por filtración y se secó para obtener el compuesto del título (25,7 g, rendimiento: 95,1 %) como un sólido amorfo.
1H-RMN (DMSO-D6) 5: 13,27 (1H, s), 8,79 (1H, d, J = 2,4 Hz), 8,72 (1H, d, J = 2,4 Hz), 8,54 (1H, d, J = 2,4 Hz), 8,46 (1H, d, J = 8,5 Hz), 8,34 (1H, d, J = 2,4 Hz), 8,29 (1H, d, J = 2,4 Hz), 8,03-7,99 (1H, m), 7,78 (1H, d, J = 8,5 Hz), 7,59 7,51 (4H, m), 7,31 (1H, d, J = 2,4 Hz), 7,25 (1H, dd, J = 8,5, 2,4 Hz), 7,06 (1H, d, J = 8,5 Hz), 4,21 (2H, d, J = 7,3 Hz), 4,01-3,93 (2H, m), 3,90-3,74 (8 H, m), 3,69-3,24 (6 H, m), 2,37 (3H, s), 2,34 (3H, s), 2,13-2,02 (1H, m), 1,47 (2H, brd, J = 10,9 Hz), 1,37-1,28 (2H, m).
MS (APCI) m/z : 736[(M+H)+].
Valores de análisis elemental para C41H42FN5Or1,0CH3SO3H1.OH2O
Calculado: C, 59,35; H, 5,69; N, 8,24; F, 2,24; S, 3,77.
Encontrado: C, 59,38; H, 5,80; N, 8,10; F, 2,23; S, 3,71.
De manera similar, cada mesilato de la Tabla 25 se obtuvo como un sólido amorfo a partir de los correspondientes materiales de partida.
[Tabla 25-1]

continuación

[Tabla 25-2]

[Tabla 25-3]

De aquí em adelante, N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida es el compuesto del Ejemplo 1, y N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida es el compuesto del Ejemplo 34.
(Ejemplo 130) Hidrato de metanosulfonato de N-[4-(2-Amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida
Se suspendió N-[4-(2-Amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenilo)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida (46,3 g, 62,6 mmol) en acetona (244 ml). A la suspensión se le añadió agua (27,0 ml). Se añadió ácido metanosulfónico (4,27 ml, 65,8 mmol) en pequeñas porciones a la mezcla. La mezcla de reacción se agitó a temperatura ambiente durante 30 horas y luego se filtró con succión con un embudo Buchner. El sólido recolectado por filtración se lavó con una mezcla de agua/acetona (agua/acetona = 1/9) para obtener el compuesto del título (47,5 g, rendimiento: 87,5 %) como un sólido cristalino. 1H-RMN (DMSO-D6) 8 : 13,22 (1H, s), 8,72 (1H, d, J = 2,0 Hz), 8,28 (1H, d, J = 2,0 Hz), 8,21 (1H, d, J = 2,0 Hz), 8,19 (1H, d, J = 2,0 Hz), 7,90 (2H, d, J = 8,5 Hz), 7,60-7,52 (6 H, m), 7,32 (1H, d, J =2,0 Hz), 7,27 (3H, d, J = 8,5 Hz), 7,06 (1H, d, J = 8,5 Hz), 4,11 (2H, d, J = 7,5 Hz), 4,01-3,93 (2H, m), 3. 89-3,22 (14H, m), 2,36 (3H, s), 2,32 (3H, s), 2,17-2,04 (1H, m), 1,46 (2H, d, J = 11,0 Hz), 1,37-1,25 (2H, m).
MS (APCI) m/z : 717 [(M+H)+].
Valores de análisis elemental para C42H44N4Or1,0CH3SO3H-3,0H2O
Calculado: C, 59,57; H, 6,28; N, 6,46; S, 3,70.
Encontrado: C, 59,75; H, 6,29; N, 6,48; S, 3,76.
El patrón de difracción de rayos X en polvo (CuKa, A = 1,54 angstroms, tasa de barrido = 20 /min) se muestra en la Figura 1, y los picos que tienen una intensidad relativa de 22 o más con la intensidad de pico máxima definida como 100 se muestran en la Tabla 26.
[Tabla 26]

(Ejemplo 131) hidrato de hidrobromuro de N-[4-(2-Ammo-5-{4-[(2R)-1,4-dioxan-2-Nmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida

A N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida (250 mg, 349 jmol), se agregaron metanol (3,99 ml) y agua (633 |jl). A continuación, se agregó a la misma una solución acuosa de ácido bromhídrico de 1,00 mol/l (365 jl, 365 jmol). La mezcla se agitó a 40 °C durante aproximadamente 21 horas y posteriormente a temperatura ambiente durante aproximadamente 30 minutos y se filtró con succión con un embudo Kiriyama, y el sólido depositado se recolectó por filtración. Luego, el sólido se secó al aire para obtener el compuesto del título (243 mg, rendimiento: 81,4 %) como un sólido cristalino.
1H-RMN (DMSO-D6) 8: 13,22 (1H, s), 8,72 (1H, d, J = 2,5 Hz), 8,28 (1H, d, J = 2,5 Hz), 8,21 (1H, d, J = 2,5 Hz), 8,19 (1H, d, J = 2,5 Hz), 7,90 (2H, d, J = 8,8 Hz), 7,60-7,55 (4H, m), 7,50 (2H, br s), 7,31 (1H, d, J = 2,5 Hz), 7,29-7,23 (3H, m), 7,06 (1H, d, J = 8,8 Hz), 4,12 (2H, d, J = 7,5 Hz), 4,01-3,93 (2H, m), 3,89-3,24 (14H, m), 2,36 (3H, s), 2,16-2,05 (1H, m), 1,46 (2H, d, J = 12,5 Hz), 1,37-1,25 (2H, m).
Valores de análisis elemental para C42H44N4O r 1 HBr-3 ,3 H2O
Calculado: C, 58,85; H, 6,07; N, 6,53; Br, 9,32.
Encontrado: C, 58,91; H, 5,98; N, 6,58; Br, 9,42.
El patrón de difracción de rayos X en polvo (CuKa, A = 1,54 angstroms, tasa de barrido = 20°/min) se muestra en la Figura 2, y los picos que tienen una intensidad relativa de 22 o más con la intensidad de pico máxima definida como 100 se muestran en la Tabla 27.
[Tabla 27]

(Ejemplo 132) hidrato de nitrato de N-[4-(2-Amino-5-(4-[(2R)-1,4-dioxan-2-Hmetoxi]-3-metoxifenN}pmdm-3-il)fenil]-5-(4-metilfenil)-4-oxo-1 -(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida

A N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida (249 mg, 347 pmol), se agregaron metanol (1,49 ml) y agua (9,10 pl). A continuación, se le añadió una solución acuosa de ácido nítrico de 1,00 mol/l (364 pl, 364 pmol). La mezcla se agitó a 40 °C durante aproximadamente 21 horas y posteriormente a temperatura ambiente durante aproximadamente 30 minutos y se filtró con succión con un embudo Kiriyama, y el sólido depositado se recolectó por filtración. Luego, el sólido se secó al aire para obtener el compuesto del título (252 mg, rendimiento: 87,0 %) como un sólido cristalino. 1H-RMN (DMSO-De) 6 : 13,22 (1H, s), 8,72 (1H, d ,J = 2,5 Hz), 8,27(1H, d ,J = 2,5 Hz), 8,21 (1H, d ,J = 2,5 Hz), 8,19 (1H ,d ,J = 2,5 Hz), 7,90 (2H, d ,J = 8,8 Hz), 7,60-7,55 (4H, m), 7,49 (2H, br s), 7,31 (1H ,d,J = 2,4 Hz), 7,29-7,24 (3H, m), 7,06 (1H, d, J = 8,8 Hz), 4,11 (2H, d ,J = 7,5 Hz), 4,01-3,93 (2H, m), 3,90-3,22 (14H, m), 2,36 (3H, s), 2,17-2,04 (1H, m), 1,46 (2H, d ,J = 12,5 Hz), 1,38-1,26(2H, m).
Valores de análisis elemental para C42H44N4O7 1 HNO33 H2O
Calculado: C, 60,49; H, 6,16; N, 8,40.
Encontrado: C, 60,58; H, 6,15; N, 8,43.
El patrón de difracción de rayos X en polvo (CuKa, A = 1,54 angstroms, tasa de barrido = 20°/min) se muestra en la Figura 3, y los picos que tienen una intensidad relativa de 34 o más con la intensidad de pico máxima definida como 100 se muestran en la Tabla 28.
[Tabla 28]

(Ejemplo 133) hidrato de sulfato de N-[4-(2-Amino-5-{4-[(2R)-1,4-dioxan-2-Nmetoxi]-3-metoxifenN}piridm-3-N)feml]-5-(4-metMfenM)-4-oxo-1 -(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida

A N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida (249 mg, 347 |jmol), se agregaron metanol (3,98 ml) y agua (632 jl). Luego, se le añadió una solución acuosa de ácido sulfúrico de 1,00 mol/l (363 jl, 363 jmol). La mezcla se agitó a 40 °C durante aproximadamente 21 horas y posteriormente a temperatura ambiente durante aproximadamente 30 minutos y se filtró con succión con un embudo Kiriyama, y el sólido depositado se recolectó por filtración. Luego, el sólido se secó al aire para obtener el compuesto del título (271 mg, rendimiento: 90,4 %) como un sólido cristalino.
1H-RMN (DMSO-Da) 5: 13,20 (1H, s), 8,72 (1H, d, J = 2,4 Hz), 8,27 (1H, d, J = 2,4 Hz), 8,18 (1H, d, J = 2,4 Hz), 8,08 (1H, s), 7,88 (2H, d, J = 8,5 Hz), 7,57 (4H, t, J = 8,5 Hz), 7,30-7,03 (7H, m), 4,11 (2H, d, J = 7,5 Hz), 4,01-3,92 (2H, m), 3,91-3,21 (14H, m), 2,36 (3H, s), 2,16-2,05 (1H, m), 1,46 (2H, d, J = 12,5 Hz), 1,37-1,25 (2H, m).
Valores de análisis elemental para C42H44N4O7-0,75H2SO4-4H2O
Calculado: C, 58,49; H, 6,25; N, 6,50; S, 2,79.
Encontrado: C, 58,53; H, 6,13; N, 6,52; S, 2,80.
El patrón de difracción de rayos X en polvo (CuKa, A = 1,54 angstroms, tasa de barrido = 20°/min) se muestra en la Figura 4, y los picos que tienen una intensidad relativa de 13 o más con la intensidad de pico máxima definida como 100 se muestran en la Tabla 29.
[Tabla 29]

(Ejemplo 134) hidrato de fosfato de N-[4-(2-Amino-5-{4-[(2R)-1,4-dioxan-2-Nmetoxi]-3-metoxifenN}piridm-3-il)fenil]-5-(4-metilfenil)-4-oxo-1 -(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida

A N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida (251 mg, 350 jmol), se agregaron metanol (501 j l) y agua (1,28
ml). Luego, se le añadió una solución acuosa de ácido fosfórico de 0,504 mol/l (729 jl, 367 |jmol). La mezcla se agitó a 40 °C durante aproximadamente 24 horas y posteriormente a temperatura ambiente durante aproximadamente 30 minutos y se filtró con succión con un embudo Kiriyama, y el sólido depositado se recolectó por filtración. Luego, el sólido se secó al aire para obtener el compuesto del título (254 mg, rendimiento: 82,8 %) como un sólido cristalino.
1H-RMN (DMSO-Da) 8: 13,11 (1H, s), 8,72 (1H, d, J = 2,5 Hz), 8,25 (1H, d, J = 2,5 Hz), 8,17 (1H, d, J = 2,5 Hz), 7,82 (2H, d, J = 8,5 Hz), 7,62 (1H, d, J = 2,5 Hz), 7,58 (2H, d, J = 8,5 Hz), 7,53 (2H, d, J = 8,5 Hz), 7,26 (2H, d, J = 8,5 Hz), 7,20 (1H, d, J = 2,0 Hz), 7,13 (1H, dd, J = 8,5, 2,0 Hz), 6,99 (1H, d, J = 8,5 Hz), 5,73 (2H, br s), 4,11 (2H, d, J = 7,5 Hz), 3,98-3,90 (2H, m), 3,89-3,22 (14H, m), 2,36 (3H, s), 2,16-2,04 (1H, m), 1,46 (2H, d, J = 11,5 Hz), 1,37-1,25 (2H, m).
Valores de análisis elemental para C42H44N4Or1,0HaPO4-3,5H2O
Calculado: C, 57,46; H, 6,20; N, 6,38; P, 3,53.
Encontrado: C, 57,41; H, 6,20; N, 6,41; P, 3,41.
El patrón de difracción de rayos X en polvo (CuKa, A = 1,54 angstroms, tasa de barrido = 20°/min) se muestra en la Figura 5, y los picos que tienen una intensidad relativa de 20 o más con la intensidad de pico máxima definida como 100 se muestran en la Tabla 30.
[Tabla 30]

(Ejemplo 135) hidrato de etanosulfonato de N-[4-(2-Amino-5-{4-[(2R)-1,4-dioxan-2-Nmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida

A N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida (251 mg, 350 jmol), se agregaron metanol (1,00 ml) y agua (3,65 ml). Luego, se le añadió una solución acuosa de ácido etanosulfónico 1,00 mol/l (368 jl, 368 jmol). Se añadió a la mezcla una pequeña cantidad de cristal semilla obtenido mediante el procedimiento que se indica a continuación. La mezcla se agitó a 40 °C durante aproximadamente 21 horas y posteriormente a temperatura ambiente durante aproximadamente 30 minutos y se filtró con succión con un embudo Kiriyama, y el sólido depositado se recolectó por filtración. Luego, el sólido se secó al aire para obtener el compuesto del título (265 mg, rendimiento: 81,8 %) como un sólido cristalino.
Procedimiento para obtener cristal semilla
A N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida (10,4 mg, 14,5 jmol), se agregaron metanol (41,6 j l ) y agua (151 jl). Luego, se le añadió una solución acuosa de ácido etanosulfónico 1,00 mol/l (15,2 jl, 15,2 jmol). La mezcla se agitó a 40 °C durante aproximadamente 24 horas y posteriormente a temperatura ambiente durante aproximadamente 30 minutos y se filtró con succión con un embudo Kiriyama, y el sólido depositado se recolectó por filtración. Luego, el sólido se secó al aire para obtener un cristal semilla (10,9 mg).
1H-RMN (DMSO-Da) 5: 13,22 (1H, s), 8,72 (1H, d, J = 2,0 Hz), 8,28 (1H, d, J = 2,0 Hz), 8,20 (2H, dd, J = 8,5, 2,0 Hz), 7,90 (2H, d, J = 8,5 Hz), 7,61-7,49 (6 H, m), 7,32 (1H, d, J = 2,0 Hz), 7,29-7,24 (3H, m), 7,06 (1H, d, J = 8,5 Hz), 4,11 (2H, d, J = 7,5 Hz), 4,01-3,93 (2H, m), 3,87-3,24 (14H, m), 2,41-2,31 (5H, m), 2,16-2,04 (1H, m), 1,46 (2H, d, J = 11,5 Hz), 1,37-1,25 (2H, m), 1,06 (3H, t, J = 7,5 Hz).
Valores de análisis elemental para C42H44N4Or1,0C2H5SOaH-5,5H2O
Calculado: C, 57,07; H, 6,64; N, 6,05; S, 3,46.
Encontrado: C, 57,13; H, 6,78; N, 6,08; S, 3,60.
El patrón de difracción de rayos X en polvo (CuKa, A = 1,54 angstroms, tasa de barrido = 20°/min) se muestra en la Figura 6 , y los picos que tienen una intensidad relativa de 5 o más con la intensidad de pico máxima definida como 100 se muestran en la Tabla 31.
[Tabla 31]

(Ejemplo 136) hidrato de bencenosulfonato de N-[4-(2-Amino-5-{4-[(2R)-1,4-dioxan-2-Nmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida

A N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida (250 mg, 349 pmol), se agregaron metanol (1,00 ml) y agua (3,64 ml). A continuación, se le añadió una solución acuosa de ácido bencenosulfónico de 1,00 mol/l (366 pl, 366 pmol). Se preparó una suspensión a partir de la misma usando una lavadora ultrasónica, luego se agitó a 40 °C durante aproximadamente 21 horas y posteriormente a temperatura ambiente durante aproximadamente 30 minutos, y se filtró con succión con un embudo Kiriyama, y el sólido depositado se recolectó por filtración. Luego, el sólido se secó al aire para obtener el compuesto del título (287 mg, rendimiento: 88,3 %) como un sólido cristalino.1
1H-RMN (DMSO-D6) 5: 13,22 (1H, s), 8,72 (1H, d, J = 2,0 Hz), 8,27 (1H, d, J = 2,0 Hz), 8,19 (2H, d, J = 2,0 Hz), 7,90 (2H, d, J = 8,5 Hz), 7,61-7,55 (6 H, m), 7,44 (2H, br s), 7,35-7,24 (7H, m), 7,06 (1H, d, J = 8,5 Hz), 4,11 (2H, d, J = 7,5 Hz), 4,01-3,93 (2H, m), 3,90-3,22 (14H, m), 2,36 (3H, s), 2,17-2,04 (1H, m), 1,46 (2H, d, J = 11,6 Hz), 1,31 (2H, td, J = 12,2, 8,5 Hz).
Valores de análisis elemental para C42H44N4O7-1 ,0C6H5SOaH-3 H2O
Calculado: C, 62,05; H, 6,08; N, 6,03; S, 3,45.
Encontrado: C, 62,18; H, 6,07; N, 6,08; S, 3,46.
El patrón de difracción de rayos X en polvo (CuKa, A = 1,54 angstroms, tasa de barrido = 20°/min) se muestra en la Figura 7, y los picos que tienen una intensidad relativa de 29 o más con la intensidad de pico máxima definida como 100 se muestran en la Tabla 32.
[Tabla 32]

(Ejemplo 137) p-toluenosulfonato de N-[4-(2-Ammo-5-{4-[(2R)-1,4-dioxan-2-Nmetoxi]-3-metoxifeml}piridm-3-M)feml]-5-(4-metMfenM)-4-oxo-1-(tetrahidro-2H-piran-4-MmetM)-1,4-dihidropiridm-3-carboxamida

A N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida (268 mg, 373 pmol), se agregaron metanol (2,14 ml) y agua (145 pl). A continuación, se le añadió una solución acuosa de ácido p-toluenosulfónico de 1,00 mol/l (390 pl, 390 |jmol). Se añadió a la mezcla una pequeña cantidad de cristal semilla obtenido mediante el procedimiento que se indica a continuación. La mezcla se agitó a 40 °C durante aproximadamente 21 horas y posteriormente a temperatura ambiente durante aproximadamente 30 minutos y se filtró con succión con un embudo Kiriyama, y el sólido depositado se recolectó por filtración. Luego, el sólido se secó al aire para obtener el compuesto del título (255 mg, rendimiento: 77,0 %) como un sólido cristalino.
Procedimiento para obtener cristal semilla
A N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida (10,3 mg, 14,4 pmol), se agregaron metanol (165 pl) y agua (26,2 pl). A continuación, se le añadió una solución acuosa de ácido p-toluenosulfónico de 1,00 mol/l (15,0 pl, 15,0 pmol). La mezcla se agitó a 40 °C durante aproximadamente 24 horas y posteriormente a temperatura ambiente durante aproximadamente 30 minutos y se filtró con succión con un embudo Kiriyama, y el sólido depositado se recolectó por filtración. Luego, el sólido se secó al aire para obtener un cristal semilla (10,7 mg).
1H-RMN (DMSO-Da) 8: 13,22 (1H, s), 8,73 (1H, d, J = 2,0 Hz), 8,28 (1H, d, J = 2,0 Hz), 8,21 (1H, d, J = 2,0 Hz), 8,19 (1H, d, J = 2,0 Hz), 7,90 (2H, d, J = 8,0 Hz), 7,60-7,45 (8H, m), 7,31 (1H, d, J = 2,0 Hz), 7,29-7,24 (3H, m), 7,11 (2H, d, J = 8,0 Hz), 7,06 (1H, d, J = 8,0 Hz), 4,11 (2H, d, J = 7,5 Hz), 4,02-3,93 (2H, m), 3,90-3,22 (14H, m), 2,36 (3H, s), 2,29 (3H, s), 2,16-2,04 (1H, m), 1,46 (2H, d, J = 11,5 Hz), 1,37-1,25 (2H, m).
Valores de análisis elemental para C42H44N4Or1,0C7HySO3H
Calculado: C, 66,19; H, 5,89; N, 6,30; S, 3,61.
Encontrado: C, 65,91; H, 5,97; N, 6,26; S, 3,59.
El patrón de difracción de rayos X en polvo (CuKa, A = 1,54 angstroms, tasa de barrido = 20°/min) se muestra en la Figura 8 , y los picos que tienen una intensidad relativa de 23 o más con la intensidad de pico máxima definida como 100 se muestran en la Tabla 33.
[Tabla 33]

(Ejemplo 138) hidrato de fosfato de N-[4-(2-Ammo-5-{4-[(2R)-1,4-dioxan-2-Nmetoxi]-3-metoxifeml}pindm-3-M)-3-fluorofenil]-5-metil-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida

Se suspendió N-[4-(2-Amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metilo-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida (50,0 mg, 67,9 |jmol) en acetona (400 |jl). A la suspensión se le añadió agua (64,4 jl). A continuación, se le añadió una solución acuosa de ácido fosfórico de 4,00 mol/l (35,7 j l , 143 jm ol) y la mezcla se agitó a 40 °C durante 3 horas. Luego, se le añadió acetona acuosa al 20 % (500 j l) y la mezcla se agitó adicionalmente durante aproximadamente 21 horas. Luego, la mezcla de reacción se agitó a temperatura ambiente durante aproximadamente 30 minutos y luego se filtró con succión con un embudo Kiriyama para obtener el compuesto del título (42,5 mg, rendimiento: 69,7 %) como un sólido cristalino.
1H-RMN (DMSO-Da) 8: 13,22 (1H, s), 8,77 (1H, d, J = 2,0 Hz), 8,69 (1H, d, J = 2,0 Hz), 8,51 (1H, d, J = 2,0 Hz), 8,47 (1H, d, J = 8,5 Hz), 8,30 (1H, d, J = 2,0 Hz), 7,93 (1H, dd, J = 12,5, 2,0 Hz), 7,71 (1H, dd, J = 8,5, 2,0 Hz), 7,64 (1H, d, J = 2,0 Hz), 7,49-7,41 (2H, m), 7,19 (1H, d, J = 2,0 Hz), 7,12 (1H, dd, J = 8,5, 2,0 Hz), 6,99 (1H, d, J = 8,5 Hz), 5,77 (2H, brs), 4,21 (2H, d, J = 7,5 Hz), 3,98-3,24 (16H, m), 2,36 (3H, s), 2,13-2,01 (1H, m), 1,46 (2H, d, J = 12,0 Hz), 1,38 1,26 (2H, m).
MS (APCI) m/z: 736[(M+H)+].
Valores de análisis elemental para C4-iH42FN5Or1,0H3PO4,3.5H2O
Calculado: C, 54,91; H, 5,84; N, 7,81; F, 2,12; P, 3,45.
Encontrado: C, 54,95; H, 5,54; N, 7,86; F, 2,20; P, 3,19.
El patrón de difracción de rayos X en polvo (CuKa, A = 1,54 angstroms, tasa de barrido = 20°/min) se muestra en la Figura 9, y los picos que tienen una intensidad relativa de 18 o más con la intensidad de pico máxima definida como 100 se muestran en la Tabla 34.
[Tabla 34]

(Ejemplo 139) hidrato de sulfato de N-[4-(2-Amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofeml]-5-metN-4'-oxo-1'-(tetrahidro-2H-piran-4-NmetN)-1',4'-dihidro-2,3'-bipindm-5'-carboxamida

Se suspendió N-[4-(2-Amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida (80,0 g, 109 mmol) en acetona (1,28 l). A la suspensión se le añadió agua (247 ml). Se le añadió gota a gota 6,00 mol/l de ácido sulfúrico (71,7 ml, 430 mmol) a 40 °C. La mezcla de reacción se agitó a 40 °C durante 3 días y luego se filtró con succión con un embudo Buchner y el sólido depositado se recolectó por filtración. El sólido se lavó con una mezcla de agua/acetona (agua/acetona= 1/9) para obtener el compuesto del título (92,3 g, rendimiento: 88,3 %) como un sólido cristalino.
H-RMN (DMSO-Da) 8: 13,04 (1H, br s), 8,83 (2H, d, J = 9,5 Hz), 8,63 (1H, s), 8,44 (1H, d, J = 8,0 Hz), 8,34 (2H, s), 8,05-7,98 (2H, m), 7,72 (2H, br s), 7,59-7,52 (2H, m), 7,32 (1H, d, J = 2,0 Hz), 7,26 (1H, dd, J = 8,0, 2,0 Hz), 7,07 (1H, d, J = 8,0 Hz), 4,22 (2H, d, J = 7,5 Hz), 4,01-3,93 (2H, m), 3,90-3,74 (8H, m), 3,70-3,59 (2H, m), 3,53-3,45 (1H, m), 3,40 (1H, dd, J = 11,0, 10,0 Hz), 3,27 (2H, t, J = 11,0 Hz), 2,43 (3H, s), 2,16-2,05 (1H, m), 1,51-1,43 (2H, m), 1,39-1,26 (2H, m).
MS (APCI) m/z : 736[(M+H)+].
Valores de análisis elemental para C4-iH42FN5Or1,8H2SO4-3,0H2O
Calculado: C, 50,96; H, 5,38; N, 7,25; F, 1,97; S, 5,97.
Encontrado: C, 50,98; H, 5,36; N, 7,23; F, 1,97; S, 6,19.
El patrón de difracción de rayos X en polvo (CuKa, A = 1,54 angstroms, tasa de barrido = 20°/min) se muestra en la Figura 10, y los picos que tienen una intensidad relativa de 6 o más con la intensidad de pico máxima definida como 100 se muestran en la Tabla 35.
[Tabla 35]

(Ejemplo 140) hidrato de sulfato de N-[4-(2-Amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida
A N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida (39,8 mg, 54,1 pmol), se agregaron acetona (637 pl) y agua (78,3 pl). A continuación, se le añadió una solución acuosa de ácido sulfúrico de 1,00 mol/l (80,9 pl, 80,9 pmol). La mezcla se agitó a 40 °C durante aproximadamente 24 horas y posteriormente a temperatura ambiente durante aproximadamente 30 minutos y se filtró con succión con un embudo Kiriyama, y el sólido depositado se recolectó por filtración. Luego, el sólido se secó al aire para obtener el compuesto del título (36,7 mg, rendimiento: 71,6 %) como un sólido cristalino.
Valores de análisis elemental para C4-iH42FN5Or1,60H2SO4-3,0H2O
Calculado: C, 52,01; H, 5,45; N, 7,40; F, 2,01; S, 5,42.
Encontrado: C, 52,07; H, 5,24; N, 7,25; F, 2,09; S, 5,50.
El patrón de difracción de rayos X en polvo (CuKa, A = 1,54 angstroms, tasa de barrido = 20°/min) se muestra en la Figura 11, y los picos que tienen una intensidad relativa de 12 o más con la intensidad de pico máxima definida como 100 se muestran en la Tabla 36.
[Tabla 36]
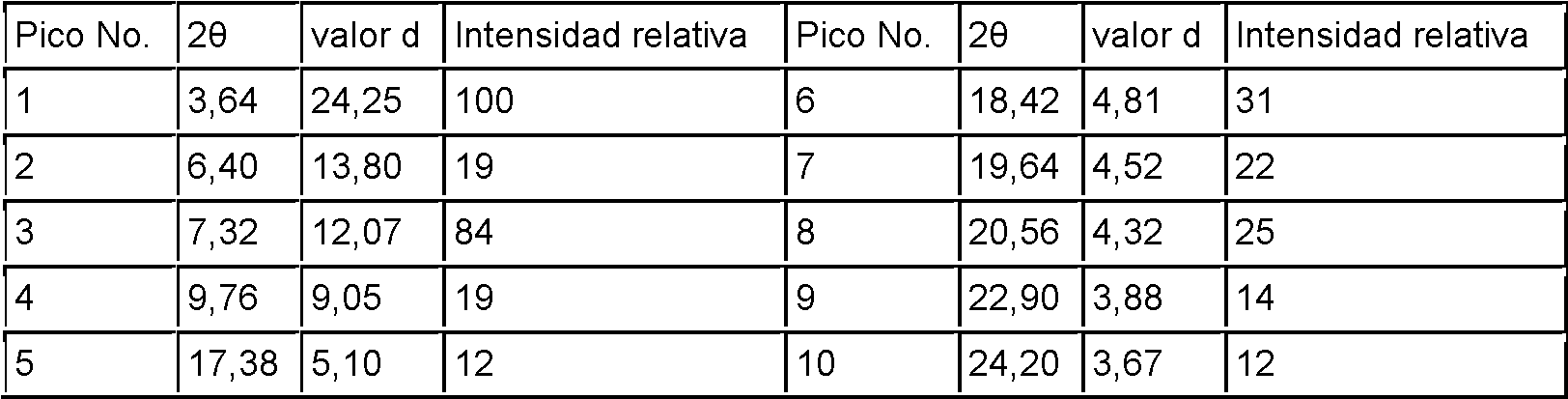
(Ejemplo 141) hidrato de sulfato de N-[4-(2-Amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofeml]-5-metN-4'-oxo-1'-(tetrahidro-2H-piran-4-NmetN)-1',4'-dihidro-2,3'-bipindm-5'-carboxamida
A N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5- metil-4'-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida (40,5 mg, 55,0 jmol), se agregaron acetona (648 j l) y agua (24,8 jl). A continuación, se le añadió una solución acuosa de ácido sulfúrico de 1,00 mol/l (137 jl, 137 jmol). La mezcla se agitó a 40 °C durante aproximadamente 24 horas y posteriormente a temperatura ambiente durante aproximadamente 30 minutos y se filtró con succión con un embudo Kiriyama, y el sólido depositado se recolectó por filtración. Luego, el sólido se secó al aire para obtener el compuesto del título (47,2 mg, rendimiento: 88,8 %) como un sólido cristalino.
Valores de análisis elemental para C4-iH42FN5O7-1,80H2SO4-3,0H2O
Calculado: C, 50,96; H, 5,38; N, 7,25; F, 1,97; S, 5,97.
Encontrado: C, 50,85; H, 5,20; N, 7,06; F, 2,09; S, 5,99.
El patrón de difracción de rayos X en polvo (CuKa, A = 1,54 angstroms, tasa de barrido = 20°/min) se muestra en la Figura 12, y los picos que tienen una intensidad relativa de 7 o más con la intensidad de pico máxima definida como 100 se muestran en la Tabla 37.
[Tabla 37]

(Ejemplo 142) hidrato de sulfato de N-[4-(2-Amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida
A N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5- metil-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida (39,8 mg, 54,0 jmol), se agregaron acetona (636 j l) y agua (112 jl). A continuación, se le añadió una solución acuosa de ácido sulfúrico de 5,79 mol/l (46,7 jl, 270 jmol). La mezcla se agitó a 40 °C durante aproximadamente 24 horas y posteriormente a temperatura ambiente durante aproximadamente 30 minutos y se filtró con succión con un embudo Kiriyama, y el sólido depositado se recolectó por filtración. Luego, el sólido se secó al aire para obtener el compuesto del título (47,5 mg, rendimiento: 89,2 %) como un sólido cristalino.
Valores de análisis elemental para C4-iH42FN5Or2,00H2SO4-3,0H2O
Calculado: C, 49,94; H, 5,32; N, 7,10; F, 1,93; S, 6,50.
Encontrado: C, 49,75; H, 5,08; N, 6,91; F, 2,20; S, 6,69.
El patrón de difracción de rayos X en polvo (CuKa, A = 1,54 angstroms, tasa de barrido = 20°/min) se muestra en la Figura 13, y los picos que tienen una intensidad relativa de 11 o más con la intensidad de pico máxima definida como 100 se muestran en la Tabla 38.
[Tabla 38]

(Ejemplo 143) hidrato de sulfato de N-[4-(2-Amino-5-{4-[(2R)-1,4-dioxan-2-Nmetoxi]-3-metoxifem l}piNdm -3-M)-3-fluorofem l]-5-metN-4'-oxo-1'-(tetrahidro-2H-piran-4-NmetN)-1',4'-dihidro-2,3'-bipindm -5'-carboxamida
A N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5- metil-4'-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida (40,2 mg, 54,6 jmol), se agregaron acetona (643 j l) y agua (51,8 jl). A continuación, se le añadió una solución acuosa de ácido sulfúrico de 1,00 mol/l (109 jl, 109 jmol). La mezcla se agitó a 40 °C durante aproximadamente 24 horas y posteriormente a temperatura ambiente durante aproximadamente 30 minutos y se filtró con succión con un embudo Kiriyama, y el sólido depositado se recolectó por filtración. Luego, el sólido se secó al aire para obtener el compuesto del título (44,4 mg, rendimiento: 84,6 %) como un sólido cristalino.
Valores de análisis elemental para C4-iH42FN5Or1,75H2SO4-3,0H2O
Calculado: C, 51,22; H, 5,40; N, 7,28; F, 1,98; S, 5,84.
Encontrado: C, 50,92; H, 5,19; N, 7,11; F, 2,25; S, 5,81.
El patrón de difracción de rayos X en polvo (CuKa, A = 1,54 angstroms, tasa de barrido = 20°/min) se muestra en la Figura 14, y los picos que tienen una intensidad relativa de 11 o más con la intensidad de pico máxima definida como 100 se muestran en la Tabla 39.
[Tabla 39]

(Ejemplo 144) hidrato de sulfato de N-{4-[2-Amino-5-(4-{[(2R)-1,4-dioxan-2-il]metoxi}-3-metoxifenil)-3-piridil]-3-fluorofenil}-5-(5-metil-2-piridil)-4-oxo-1-(tetrahidropiran-4-ilmetil)piridin-3-carboxamida
A N-{4-[2-amino-5-(4-{[(2R)-1,4-dioxan-2-il] metoxi}-3-metoxifenil)-3-piridil]-3-fluorofenil}- 5-(5-metil-2-piridil)-4-oxo-1 -(tetrahidropiran-4-ilmetil)piridin-3-carboxamida (2,l1 g, 2,88 mmol), se agregaron metanol (12,7 ml) y agua (2,72 ml) y se añadió gota a gota 1,00 mol/l de ácido sulfúrico (5,72 ml, 5,72 mmol). La mezcla de reacción se agitó a 25 °C durante aproximadamente 21 horas y luego, el sólido depositado se recolectó por filtración. El sólido se secó bajo presión reducida a temperatura ambiente durante aproximadamente 18 horas para obtener el compuesto del título (2,39 g, rendimiento: 85,8 %).
Valores de análisis elemental para C4-iH42N5O7F-1,75H2SO4-3,5H2O
Calculado: C, 50,74; H, 5,45; N, 7,22; F, 1,96; S, 5,78.
Encontrado: C, 50,70; H, 5,33; N, 7,13; F, 2,01; S, 5,82.
El patrón de difracción de rayos X en polvo (CuKa, A = 1,54 angstroms, tasa de barrido = 20°/min) se muestra en la Figura 15, y los picos que tienen una intensidad relativa de 18 o más con la intensidad de pico máxima definida como 100 se muestran en la Tabla 40.
[Tabla 40]

(Ejemplo 145) hidrato de sulfato de N-{4-[2-Ammo-5-(4-{[(2R)-1,4-dioxan-2-N]metoxi}-3-metoxifeml)-3-piridM]-3-fluorofenil}-5-(5-metil-2-piridil)-4-oxo-1-(tetrahidropiran-4-ilmetil)piridin-3-carboxamida

Al sulfato hidrato (2,44 g) descrito en el Ejemplo 139, se le añadió metanol acuoso al 20 % (24,4 ml) y la mezcla se agitó a 5 °C durante aproximadamente 27 horas. El sólido se recolectó por filtración y luego se secó bajo presión reducida a temperatura ambiente durante 4,5 horas para obtener el compuesto del título (2,30 g, rendimiento: 93,5 %). Valores de análisis elemental para C4-iH42N5O7F-1,5H2SO4,5.0H2O
Calculado: C, 50,61; H, 5,70; N, 7,20; F, 1,95; S, 4,94.
Encontrado: C, 50,59; H, 5,55; N, 7,24; F, 2,04; S, 5,09.
El patrón de difracción de rayos X en polvo (CuKa, A = 1,54 angstroms, tasa de barrido = 20°/min) se muestra en la Figura 16, y los picos que tienen una intensidad relativa de 13 o más con la intensidad de pico máxima definida como 100 se muestran en la Tabla 41.
[Tabla 41]

continuación

(Ejemplo 146) hidrato de naftalen-1,5-disulfonato de N-{4-[2-Ammo-5-(4-{[(2R)-1,4-dioxan-2-M]metoxi}-3-metoxifenil)-3-piridil]-3-fluorofenil}-5-(5-metil-2-piridil)-4-oxo-1-(tetrahidropiran-4-ilmetil)piridin-3-carboxamida

A N-{4-[2-amino-5-(4-{[(2R)-1,4-dioxan-2-il] metoxi}-3-metoxifenil)-3-piridil]-3-fluorofenil}- 5-(5-metil-2-piridil)-4-oxo-1 -(tetrahidropiran-4-ilmetil)piridin-3-carboxamida (199,34 mg, 271 |jmol), se agregaron metanol (3,19 ml), una solución acuosa de 1 mol/l de ácido naftalen-1,5-disulfónico (285 jl, 284 jmol) y agua (513 jl). La mezcla de reacción se agitó a 40 °C durante aproximadamente 26 horas y luego a temperatura ambiente durante aproximadamente 0,5 horas. El sólido se recolectó por filtración y luego se secó al aire durante la noche para obtener el compuesto del título (285,7 mg, rendimiento: 94,6 %).
1H-RMN (DMSO-Da) 8 : 12,97 (1H, br s), 8,90-8,80 (4H, m), 8,64 (1H, s), 8,42 (1H, d, J = 8,5 Hz), 8,33 (2H, s), 8,11 7,97 (2H, m), 7,92 (2H, d, J = 7,0 Hz), 7,71 (2H, br s), 7,59-7,52 (2H, m), 7,40 (2H, dd, J = 8,5, 7,0 Hz), 7,31 (1H, d, J = 2,5 Hz), 7,26 (1H, dd, J = 8,5, 2,5 Hz), 7,06 (1H, d, J = 8,5 Hz), 4,22 (2H, d, J = 7,0 Hz), 3,99-3,24 (16H, m), 2,43 (3H, s), 2,18-2,05 (1H, m), 1,47 (2H, d, J = 10,5 Hz), 1,39-1,26 (2H, m).
MS (APCI) m/z : 736 [(M+H)+].
Valores de análisis elemental para C41H42N5O7F-1,0C1üH8O6S2-5,0H2O
Calculado: C, 54,98; H, 5,43; N, 6,29; F, 1,71; S, 5,76.
Encontrado: C, 54,74; H, 5,37; N, 6,24; F, 1,92; S, 5,82.
El patrón de difracción de rayos X en polvo (CuKa, A = 1,54 angstroms, tasa de barrido = 20°/min) se muestra en la Figura 17, y los picos que tienen una intensidad relativa de 45 o más con la intensidad de pico máxima definida como 100 se muestran en la Tabla 42.
[Tabla 42]

(Ejemplo 147) hidrato de naftalen-1,5-disulfonato de N-{4-[2-Amino-5-(4-{[(2R)-1,4-dioxan-2-il]metoxi}-3-metoxifenil)-3-piridil]-3-fluorofenil}-5-(5-metil-2-piridil)-4-oxo-1-(tetrahidropiran-4-ilmetil)piridin-3-carboxamida

A N-{4-[2-amino-5-(4-{[(2R)-1,4-dioxan-2-il] metoxi}-3-metoxifenil)-3-piridil]-3-fluorofenil}- 5-(5-metil-2-piridil)-4-oxo-1 -(tetrahidropiran-4-ilmetil)piridin-3-carboxamida (199,50 mg, 271 pmol), se agregaron acetona (3,19 ml), una solución acuosa de 1 mol/l de ácido naftalen-1,5-disulfónico (285 pl, 284 pmol) y agua (513 pl). La mezcla de reacción se agitó a 40 °C durante aproximadamente 26 horas y luego a temperatura ambiente durante aproximadamente 0,5 horas. El sólido se recolectó por filtración y luego se secó al aire para obtener el compuesto del título (290,9 mg, rendimiento: 97,9 %).
Valores de análisis elemental para C41H42N5O7F-1,0C1üH8O6S2-4,0H2O
Calculado: C, 55,88; H, 5,33; N, 6,39; F, 1,73; S, 5,85.
Encontrado: C, 55,71; H, 5,45; N, 6,18; F, 1,82; S, 5,62.
El patrón de difracción de rayos X en polvo (CuKa, A = 1,54 angstroms, tasa de barrido = 20°/min) se muestra en la Figura 18, y los picos que tienen una intensidad relativa de 42 o más con la intensidad de pico máxima definida como 100 se muestran en la Tabla 43.
[Tabla 43]

(Ejemplo de prueba 1 Actividad inhibidora de la quinasa Axl libre de células)
Una solución de dilución de quinasa que contiene 170 ng/ml de AXL (los aminoácidos 464 a 885 del dominio intracelular de AXL humano expresados como una proteína de fusión con glutatión transferasa en un sistema de expresión de baculovirus y purificada por cromatografía de glutatión sefarosa; Carna Biosciences, Inc., No. de catálogo 08-107) se preparó usando una solución tampón de reacción de quinasa (HEPES 100 mM (pH 7,4), Brij-35 al 0,003 %, Tween-20 al 0,004 %, DTT 1 mM y MgCh 10 mM) y se añadió a 19 pl/pocillo a una placa de 384 pocillos.
A continuación, cada compuesto de prueba se diluyó con DMSO y esta solución de dilución se añadió a 1 pl/pocillo a la placa.
Después de la preincubación a temperatura ambiente durante 30 minutos, una solución que contenía un péptido sustrato (FL-Peptide 30 (5 FAM-KKKKEEIYFFF-CONH2), Caliper Life Sciences, número de catálogo 760430) y ATP a 1,5 pM y 10 pM, respectivamente, se preparó y se añadió a 5 pl/pocillo a la placa para iniciar la reacción de la quinasa. La placa se incubó a 28 °C durante 1,5 horas y la reacción se terminó mediante la adición de una solución tampón de terminación (HEPES 100 mM (pH 7,4), Brij-35 al 0,015 %, EDTA 40 mM y reactivo de revestimiento 3 al 0,1 %.) a 40 pl/pocillo.
El péptido sustrato y el péptido fosforilado en la mezcla de reacción se separaron y cuantificaron usando EZ Reader II (Caliper Life Sciences).
La reacción de la quinasa se evaluó sobre la base de una relación de producto (P/(P S)) calculada a partir de la altura pico (S) del péptido sustrato y la altura pico (P) del péptido fosforilado.
La tasa de inhibición (inhibición) se determinó de acuerdo con la siguiente expresión (calculada automáticamente usando el software del sistema EZ Reader II).
Inhibición (%) = loo x ( i - Ci / c0)
en la que Ci representa la relación de producto cuando se añadió el compuesto de prueba, y C0 representa la relación de producto cuando se añadió DMSO en lugar del compuesto de prueba.
La IC50 se determinó a partir de los datos sobre las tasas de inhibición a 12 concentraciones de compuesto de prueba mediante regresión no lineal (regresión logística de 4 parámetros) de acuerdo con la siguiente expresión:
Inhibición (%) = Inferior (Superior - Inferior) / (1+ ([Compuesto] / IC5o)pendiente)
(Ejemplo de prueba 2 Actividad inhibidora de la quinasa Mer libre de células)
Una solución de dilución de quinasa que contiene 20 ng/ml de Mer (los aminoácidos 528 a 999 del dominio intracelular de la MER humana expresada como una proteína de fusión con glutatión transferasa en un sistema de expresión de baculovirus y purificada por cromatografía de glutatión sefarosa y cromatografía de intercambio iónico; Carna Biosciences, Inc., catálogo No. 08-108) se preparó usando una solución tampón de reacción de quinasa (HEPES 100 mM (pH 7,4), Brij-35 al 0,003 %, Tween-20 al 0,004 %, DTT 1 mM y MgCh 10 mM) y se añadió a 19 pl/pocillo a una placa de 384 pocillos.
A continuación, cada compuesto de prueba se diluyó con DMSO y esta solución de dilución se añadió a 1 pl/pocillo a la placa.
Después de la preincubación a temperatura ambiente durante 20 minutos, se preparó una solución que contenía un péptido sustrato (FL-Peptide 27 (5FAM-EFPIYDFLPAKKK-CONH2), Caliper Life Sciences, número de catálogo 760424) y ATP a 5 mM y se añadió a 5 pl/bien a la placa para iniciar la reacción de quinasa. La placa se incubó a 28 °C durante 45 minutos y la reacción se terminó mediante la adición de una solución tampón de terminación (HEPES 100 mM (pH 7,4), Brij-35 al 0,015 %, EDTA 40 mM y Reactivo de recubrimiento 3 al 0,1 %.) a 40 pl/pocillo.
El péptido sustrato y el péptido fosforilado en la mezcla de reacción se separaron y cuantificaron usando EZ Reader II (Caliper Life Sciences).
La reacción de la quinasa se evaluó sobre la base de una relación de producto (P/(P S)) calculada a partir de la altura pico (S) del péptido sustrato y la altura pico (P) del péptido fosforilado.
La tasa de inhibición (inhibición) se determinó de acuerdo con la siguiente expresión (calculada automáticamente usando el software del sistema EZ Reader II).
Inhibición (%) = 100 x ( i - c± / c0)
en la que Ci representa la relación de producto cuando se añadió el compuesto de prueba, y C0 representa la relación de producto cuando se añadió DMSO en lugar del compuesto de prueba.
La IC50 se determinó a partir de los datos sobre las tasas de inhibición a 12 concentraciones de compuesto de prueba mediante regresión no lineal (regresión logística de 4 parámetros) de acuerdo con la siguiente expresión:
Inhibición (%) = Inferior (Superior - Inferior) / (1+ ([Compuesto] / IC5o)pend¡ente)
La Tabla 44 muestra la actividad inhibidora de la quinasa Axl como IC50 (nM) y la inhibición de la quinasa Mer IC50 (nM) en condiciones que implican una concentración de ATP de 1 mM y, además, la selectividad de la quinasa Axl (veces) con respecto a la quinasa Mer.
[Tabla 44-1]

[Tabla 44-2]

[Tabla 44-3]

[Tabla 44-4]

continuación

[Tabla 44-5]

[Tabla 44-6]

continuación

[Tabla 44-7]

(Ejemplo de prueba 3 Actividad inhibidora de la fosforilación de Axl intracelular)
Se llevó a cabo una prueba de inhibición de Axl fosforilada (de aquí en adelante, denominada como pAxI) usando una línea celular derivada de cáncer de pulmón de células no pequeñas humana NCI-H1299.
Las células NCI-H1299 se suspendieron en un medio (medio RPMI1640 que contenía suero bovino fetal al 10 %), luego se inocularon a 15000 células/100 pl/pocillo en cada placa de paredes múltiples de 96 pocillos y se cultivaron a 37 °C durante 1 día en presencia de 5 % de CO2. Al día siguiente, se descartó el medio y se añadió un medio a 100 pl/pocillo a la placa, seguido de cultivo a 37 °C durante 1 día en presencia de CO2 al 5 %. Cada compuesto de prueba se disolvió en DMSO y se diluyó con un medio libre de FBS para preparar una solución de muestra (concentración de DMSO: 2 %). Se añadió un medio o un medio complementado con la muestra a 25 pl/pocillo (concentración de DMSO: 0,4 %) a la placa y se incubó a 37 °C durante 1 hora en presencia de CO2 al 5 %.
GAS6 (R&D Systems Inc., modelo: 885-GS) se diluyó en 6 pg/ml con un medio libre de FBS, luego se añadió a 25 pl/pocillo a la placa y se incubó a 37 °C durante 10 minutos en presencia de CO2 al 5 % después de agitar.
Se descartó el sobrenadante, y se añadió a la placa una solución de formalina al 37 % diluida al 4 % con solución salina tamponada con fosfato (PBS) (en adelante, una solución de formalina al 4 %) a 0,1 ml/pocillo, que luego se dejó reposar a temperatura ambiente durante 10 minutos. A continuación, se descartó la solución de formalina al 4 % y se añadió una solución de Triton X-100 diluida al 0,1 % con PBS (en lo sucesivo, tampón de lavado) a 0,2 ml/pocillo a la placa y se descartó por decantación. Se eliminó el exceso de agua sobre una toalla de papel.
Posteriormente, se añadieron NaN3 al 10 % y 110 pl de H2O2 a 10,7 ml de un tampón de lavado (en adelante, denominado tampón de inactivación), y este tampón de inactivación se añadió a 0,1 ml/pocillo a la placa, que luego se dejó en reposo a temperatura ambiente durante 15 minutos.
Se descartó el tampón de inactivación y se añadió un tampón de lavado a 0,2 ml/pocillo a la placa y se descartó por decantación. Se eliminó el exceso de agua sobre una toalla de papel. Se añadió leche desnatada (WAKO # 198 10605) (concentración final 5 %) a un tampón de lavado (en adelante, denominado tampón de bloqueo), y este tampón de bloqueo se añadió a 0,25 ml/pocillo a la placa, que luego se se deja reposar a temperatura ambiente durante 1 hora.
Se descartó el tampón de bloqueo y se hizo reaccionar el anticuerpo monoclonal de conejo anti-fosfo-Axl (Y702) (D12B2) (Cell Signaling Technology, Inc., catálogo No. 5724) a una concentración de 1/1000 con la placa, que luego se deja reposar durante la noche a 4 °C. Cada pocillo se lavó repetidamente con un tampón de lavado cinco veces, y se hizo reaccionar Peroxidasa AffiniPure Burro Anti-Conejo IgG (H+L) (Jackson ImmunoResearch Inc., catálogo No.
711-035-152) a una concentración de 1/2000 con la placa a temperatura ambiente durante 1 hora. Se llevó a cabo
una operación de lavado similar, y se añadió sustrato picoquimioluminiscente ELISA Super Signal (Thermo Fisher Scientific, Inc., catálogo No. 37069) a 0,05 ml/pocillo a la placa y se agitó suavemente, seguido de incubación durante 20 minutos. Luego, se midió la luz desarrollada usando ARVO sx (PerkinElmer Inc.) para medir el nivel de pAxl (Y702). La actividad inhibidora de pAxl se determinó de acuerdo con la siguiente expresión:
Inhibición % = 100 - (a - b ) x 100 / (t - b )
A: valor de medición del compuesto de prueba
B: Intensidad de luz de la mezcla de reacción suplementada con un compuesto de control positivo que tiene una concentración que inhibe casi el 100% de la fosforilación (por ejemplo, intensidad de luz de la mezcla de reacción suplementada con 1 pM de BMS-777607)
T: Intensidad de luz de la mezcla de reacción no complementada con el compuesto.
La concentración de inhibición del 50 % (IC50) se determinó a partir de los datos sobre la actividad inhibidora de pAxl a una pluralidad de concentraciones usando GraphPad Prism 4.
La Tabla 45 muestra la actividad inhibidora de la fosforilación de Axl intracelular como IC50 (nM).
[Tabla 45-1]

[Tabla 45-2]

(Ejemplo de prueba 4 Examen histopatológico oftálmico)
1. Animal: ratones hembra NOG
2. Fármaco:
Compuesto A monohidrocloruro dihidrato
Compuesto del Ejemplo 124
Compuesto del Ejemplo 128
Compuesto del Ejemplo 129
3. Preparación de la solución de dosificación:
El compuesto A, el compuesto del Ejemplo 124, el compuesto del Ejemplo 128 y el compuesto del Ejemplo 129 se suspendieron cada uno en una solución de metilcelulosa al 0,5 % (Mc al 0,5 %, Wako Pure Chemical Industries, Ltd.).
4. Procedimiento de administración:
Cuatro dosis continuas, 1 descanso del fármaco, 5 dosis continuas, 2 perYodos de descanso del fármaco, 5 dosis continuas, 2 perYodos de descanso, 5 dosis continuas, 2 perYodos de descanso del fármaco y 4 dosis continuas.
5. Examen patológico
Al día siguiente de la fecha de finalización del período de administración, los ratones se sacrificaron mediante extracción de sangre bajo anestesia con isoflurano. Luego, se recolectaron sus globos oculares, se fijaron en soluciones de Davidson, luego se embebieron en parafina y se tiñeron con hematoxilina eosina para preparar preparaciones, que luego se examinaron histopatológicamente.
Se encontraron degeneración leve a moderada y adelgazamiento de la capa nuclear externa y la capa de células de bastón y cono en la retina en todos los ratones (n = 8) en el grupo que recibió 100 mg/kg del Compuesto A monohidrocloruro dihidrato.
No se encontró ningún cambio histológico en la retina en ninguno de los ratones (n = 8) en los grupos 100 o 200 mg/kg del compuesto del Ejemplo 124, Ejemplo 128 o Ejemplo 129.
(Ejemplo de prueba 5: Prueba antitumoral)
Se trasplantaron en bloque NIH-3T3-Axl # 7 (células preparadas mediante la transfección de células NIH3T3 con retrovirus pLXSN que tienen un inserto de ADNc de Ax1 de longitud completa) en ratones NOG hembra. Cuando sus volúmenes tumorales estimados alcanzaron aproximadamente 500 mm3, el compuesto A monohidrocloruro dihidrato (25, 4,2 o 0,7 mg/kg), el compuesto del Ejemplo 124 (50, 12,5, 3,1 o 0,8 mg/kg) y el compuesto del Ejemplo 128 (50, 12,5, 3,1 o 0,8 mg/kg) se administraron cada uno por vía oral dos veces al día (bid) en 5 dosis continuas. El compuesto A administrado a 25 y 4,2 mg/kg mostró un efecto de regresión tumoral distinto. Los compuestos del Ejemplo 124 y del Ejemplo 128 administrados a 3,1 mg/kg inhibieron casi completamente el crecimiento tumoral durante el período de administración, y estos compuestos administrados a 50 y 12,5 mg/kg exhibieron un efecto de regresión tumoral distinto.
(Ejemplo de prueba 6: Estudio sobre el efecto in vivo provocado por el uso combinado con erlotinib)
Se suspendieron células de cáncer de pulmón HCC827 que tenían una mutación por deleción en el exón 19 del gen EGFR y que presentaban una alta sensibilidad a los inhibidores de EGFR a 5 x 107 células/ml en solución salina tamponada con fosfato. Se trasplantaron subcutáneamente 0,1 ml de la suspensión celular preparada a cada ratón lampiño (hembra, 5 semanas de edad). Cuando los volúmenes tumorales estimados de la mayoría de los ratones alcanzaron aproximadamente 450 mm3 (52 días después del trasplante tumoral), los ratones se agruparon de acuerdo con sus volúmenes tumorales y se les obligó a recibir por vía oral 25 mg/kg de erlotinib (LC Laboratories) una vez al día (qd) o 50 mg/kg de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3-bipiridin-5-carboxamida (Compuesto B) sulfato hidrato dos veces al día (bid) (estos agentes se administraron solos o en combinación). La administración se inició el día (día 1) siguiente a la fecha de agrupación y se llevó a cabo cinco veces a la semana (el sábado y el domingo se establecieron como descansos del fármaco). La administración continuó hasta el día 46 para los grupos que recibieron un vehículo o el sulfato hidrato del compuesto B solo, el día 86 para el grupo que recibió erlotinib solo y el día 106 para el grupo que recibió estos agentes en combinación. El eje mayor (mm) y el eje menor (mm) de cada tumor se midieron a lo largo del tiempo utilizando un calibrador digital electrónico. El volumen tumoral estimado se calculó de acuerdo con la expresión (1) dada a continuación y se representó gráficamente. Además, el peso corporal se midió a lo largo del tiempo utilizando una balanza automática para animales pequeños. La tasa de cambio en el peso corporal (% de
cambio de peso corporal) se calculó de acuerdo con la expresión (2) dada a continuación. De esta forma, se estudió la influencia de la administración del fármaco sobre el peso corporal, mientras que los resultados de la última medición del peso corporal se utilizaron en el cálculo de la dosis.
Volumen tumoral estimado (mm3) = volumen tumoral estimado medio de los individuos ... (1)
Volumen tumoral estimado de cada individuo = An x Bn2 / 2
An: Eje mayor del tumor en el Día n
Bn: Eje menor del tumor en el Día n
Cambio de peso corporal (%) = Tasa media de cambio en pesos corporales
de los individuos ... (2)
Tasa de cambio en peso corporal de cada individuo = 1 (1 - BWn
/ BWs) x 100
BWn: peso corporal en el Día n
BWs: peso corporal el día inicial de la administración.
Se confirmó que erlotinib administrado solo tiene un efecto de regresión tumoral inmediatamente después de la administración. Sin embargo, el tumor adquirió resistencia y se volvió más grande que su tamaño al inicio de la administración en el día 77. Por el contrario, el tamaño del tumor en el grupo que recibió erlotinib y el sulfato hidrato del Compuesto B en combinación no alcanzó ese tamaño al comienzo de la administración, incluso en el día 107 (Figura 19-1). No se encontró una pérdida de peso significativa en el grupo que recibió erlotinib y el Compuesto B sulfato hidrato en combinación (Figura 19-2).
(Ejemplo de prueba 7: Estudio sobre la elevación de la expresión de AXL mediante la administración de erlotinib)
Se suspendieron células de cáncer de pulmón HCC827 a 5 x 107 células/ml en solución salina tamponada con fosfato. Se trasplantaron subcutáneamente 0,1 ml de la suspensión celular preparada a cada ratón lampiño (hembra, 5 semanas de edad). Cuando los volúmenes tumorales estimados de la mayoría de los ratones alcanzaron aproximadamente 450 mm3 (52 días después del trasplante tumoral), los ratones se agruparon de acuerdo con sus volúmenes tumorales (día 0) y se les obligó a recibir por vía oral 25 mg/kg, 12,5 mg/kg, o 6,25 mg/kg de erlotinib (LC Laboratories) cinco días a la semana (los sábados y domingos se establecieron como descansos de fármacos). La administración continuó hasta el día 46 para un grupo de administración de vehículo y el día 86 para los grupos de administración de erlotinib. El eje mayor (mm) y el eje menor (mm) de cada tumor se midieron a lo largo del tiempo utilizando un calibrador digital electrónico. El volumen tumoral estimado se calculó de acuerdo con la expresión (1) dada a continuación y se representó gráficamente (Figura 20-1). Se preparó un lisado celular a partir de masas tumorales de 2 a 6 ratones en cada grupo y se examinó la expresión de AXL mediante transferencia Western, y cada banda individual se cuantificó utilizando ImageQuant LAS4000 (GE Healthcare Japan Corp.) (Figura 20-2). La preparación del lisado celular y la transferencia Western se llevaron a cabo de la siguiente manera: se homogeneizaron aproximadamente 100 mg de las masas tumorales en tampón de lisis (Cell Signaling Technology, Inc., 9803) que contenía un inhibidor de fosfatasa (Roche Diagnostics., 04906837001) y un inhibidor de proteasa (Roche Diagnostics, 11 836 153 001), y el sobrenadante se utilizó como muestra de lisado. Las proteínas de esta muestra se fijaron utilizando tampones (Life Technologies Corp, NP0008 y NP0009) y desnaturalización térmica y se sometieron a transferencia Western. Se sometieron 15 pg de la muestra a electroforesis y se transfirieron a una membrana de nitrocelulosa. La membrana se bloqueó durante 1 hora y luego se hizo reaccionar durante la noche con un anticuerpo anti-AXL de conejo (Cell Signaling Technology, Inc., 9803, 1/1000) o un anticuerpo anti-actina de conejo (Santa Cruz Biotechnology, Inc., SC-1616)., 1/2000) bajo refrigeración. La membrana se lavó con solución salina tamponada con tris y luego se hizo reaccionar con un anticuerpo IgG anti-conejo marcado con HRP (Cell Signaling Technology, Inc., 7074, 1/2000) a temperatura ambiente durante 1 hora. La membrana se lavó con solución salina tamponada con tris y luego se dejó desarrollar luz con un sustrato de HRP (Merck Millipore Corp., WBLUF0500).
La expresión de AXL se confirmó mediante transferencia Western y cada banda individual se cuantificó usando ImageQuant LAS4000. Como resultado, la prueba de Welch confirmó la elevación significativa de la expresión de AXL en los grupos de administración de erlotinib.
(Ejemplo de prueba 8: Estudio sobre el efecto in vivo provocado por el uso combinado con erlotinib - 2)
Se suspendieron células de cáncer de pulmón HCC827 a 4 x 107 células/ml en solución salina tamponada con fosfato. Se trasplantaron subcutáneamente 0,1 ml de la suspensión celular preparada a cada ratón lampiño (hembra, 5 semanas de edad). 52 días después del trasplante de células tumorales, los ratones que tenían un volumen tumoral estimado de 200 mm3 o mayor y menor de 700 mm3 (86 ratones/129 ratones) se vieron obligados a recibir por vía oral 25 mg/kg de erlotinib (LC Laboratories) una vez al día (qd). La administración se inició el día (53 días después del trasplante) siguiente a la fecha de agrupación y se llevó a cabo cinco veces por semana (los sábados y domingos se establecieron como descansos del fármaco). Los ratones portadores de cáncer que se confirmó que tenían regresión temporal y un recrecimiento distinto de un tumor se agruparon (6 ratones/grupo, 115 días después del trasplante) en 3 grupos: grupo 1: grupo de administración de erlotinib vehículo (volumen tumoral medio estimado: 275 mm3), grupo 2: erlotinib 50 mg/kg de compuesto B sulfato hidrato (volumen tumoral medio estimado: 284 mm3) y grupo 3: erlotinib 25 mg/kg de compuesto B sulfato hidrato (volumen medio estimado del tumor: 268 mm3). Se realizó un estudio sobre si el uso combinado con el sulfato hidrato del compuesto B restablecería el efecto de erlotinib en los tumores resistentes a erlotinib. El compuesto B sulfato hidrato se administró dos veces al día (bid) a partir de los 116 días después del trasplante, y esta administración se continuó cinco veces a la semana (los sábados y domingos se establecieron como días de descanso del fármaco). El tumor creció hasta el volumen tumoral estimado al mismo nivel que el volumen original en el momento del trasplante en el grupo 1, mientras que el efecto inhibidor del crecimiento distintivo de erlotinib se confirmó en el grupo 2 y en el grupo 3. En el grupo 2, el volumen tumoral estimado a los 116 días después del trasplante cuando se inició la administración combinada se mantuvo incluso a los 140 días después del trasplante.
Claims (34)
1. Un compuesto representado por la fórmula general (I) o una sal farmacéuticamente aceptable del mismo:

en la que
W, X e Y representan cada uno independientemente un átomo de nitrógeno, C-H, C-F o C-Cl,
Z representa un átomo de nitrógeno, C-H, C-F, C-Cl, alquilo C-Ci-C6 o grupo alcoxi C-Ci-C6,
Ri representa un grupo representado por la siguiente fórmula (II-1) o (II-2):

en la que en la fórmula (II-1),
Q representa un átomo de nitrógeno, C-H o C-F,
T representa un átomo de nitrógeno o C-H,
U representa un átomo de nitrógeno o C-H, y
R6 representa un átomo de halógeno, un grupo alquilo C1-C6, un grupo cicloalquilo C3-C6, un grupo ciano o un grupo trifluorometoxi, y
en la fórmula (II-2),
V representa un átomo de azufre o un átomo de oxígeno, y
R7 representa un grupo alquilo C1-C6,
R2 representa un átomo de hidrógeno, un átomo de halógeno, un grupo alquilo C1-C6, un grupo alcoxi C1-C6 o un grupo ciano,
R3 representa un átomo de hidrógeno o un grupo alquilo C1-C6,
R4 representa un átomo de hidrógeno, un átomo de halógeno o un grupo alquilo C1-C6, y
R5 representa un átomo de hidrógeno o un grupo representado por la siguiente fórmula (III-1), (III-2), (III-3) o (III-4):

en la que en la fórmula (III-1),
R8 y Ri2 representan cada uno independientemente un átomo de hidrógeno o un átomo de deuterio,
R9 representa un átomo de hidrógeno, un átomo de halógeno o un grupo alcoxi Ci-C6,
R10 representa un átomo de hidrógeno, un grupo alquilo C1-C6 opcionalmente sustituido con un grupo heterocicloalquilo o un grupo alcoxi C1-C6 opcionalmente sustituido con un grupo heterocicloalquilo opcionalmente sustituido con un grupo alquilo C1-C6, y
R11 representa un átomo de hidrógeno, un grupo alcoxi C1-C6 o un grupo alcoxi C1-C6 sustituido con deuterio, en la fórmula (III-2),
R13 representa un grupo alquilo C1-C6 opcionalmente sustituido con un grupo heterocicloalquilo, o un grupo heterocicloalquilo,
en la fórmula (III-3),
R14 representa un átomo de hidrógeno o un grupo alquilo C1-C6,
R15 representa un átomo de hidrógeno, un grupo alquilo C1-C6 o un grupo alcoxi C1-C6, y
R16 representa un átomo de hidrógeno o un átomo de halógeno, y
en la fórmula (III-4),
R17 representa un grupo alcoxi C1-C6, y
R18 representa un grupo alcoxi C1-C6.
2. Un compuesto de acuerdo con la reivindicación 1 que es N-[4-(2-Amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida representado por la siguiente fórmula:

o una sal farmacéuticamente aceptable del mismo.
3. Un compuesto de acuerdo con la reivindicación 1 que es N-[4-(2-Amino-5-{4-[(2S)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)fenil]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida representado por la siguiente fórmula:

o una sal farmacéuticamente aceptable del mismo.
4. Un compuesto de acuerdo con la reivindicación 1 que es N-{4-[2-Amino-5-(3,4-dimetoxifenil)piridin-3-il]fenil}-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida representado por la siguiente fórmula:

o una sal farmacéuticamente aceptable del mismo.
5. Un compuesto de acuerdo con la reivindicación 1 que es N-[4-(2-Amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida representado por la siguiente fórmula:

o una sal farmacéuticamente aceptable del mismo.
6. Un compuesto de acuerdo con la reivindicación 1 que se selecciona del grupo que consiste en:
N-[4-(2-Amino-5-{4-[(2S)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4'-oxo-1'-(tetrahidro-2H-piran-4-ilmetil)-1',4'-dihidro-2,3'-bipiridin-5'-carboxamida representado por la siguiente fórmula:

o una sal farmacéuticamente aceptable del mismo;
N-{4-[2-Amino-5-(1-etil-1H-pirazol-4-il)piridin-3-il]-3-fluorofenil}-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida representado por la siguiente fórmula:

o una sal farmacéuticamente aceptable del mismo;
N-(4-{2-Amino-5-[1-(tetrahidro-2H-piran-4-il)-1H-pirazol-4-il]piridin-3-il}-3-fluorofenil)-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida representado por la siguiente fórmula:

o una sal farmacéuticamente aceptable del mismo;
N-[6-(2-Amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)piridazin-3-il]-5-(5-metiltiofen-2-il)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida representado por la siguiente fórmula:

o una sal farmacéuticamente aceptable del mismo; y
N-[6-(2-Amino-5-{3-metoxi-4-[2-(morfolin4-il)etoxi]fenil}piridin-3-il)piridazin-3-il]-5-(4-metilfenil)-4-oxo-1-(tetrahidro-2H-piran-4-ilmetil)-1,4-dihidropiridin-3-carboxamida representado por la siguiente fórmula:

o una sal farmacéuticamente aceptable del mismo.
7. Un hidrobromuro, nitrato, sulfato, fosfato, metanosulfonato, etanosulfonato, bencenosulfonato o p-toluensulfonato de un compuesto de acuerdo con la reivindicación 2.
8. Un metanosulfonato de un compuesto de acuerdo con la reivindicación 4.
9. Un metanosulfonato, fosfato, naftaleno-1,5-disulfonato o sulfato de un compuesto de acuerdo con la reivindicación 5.
10. Un cristal de un metanosulfonato de acuerdo con la reivindicación 7, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 3,74, 7,56, 8,96, 11,38, 12,36, 14,78, 15,60, 16,16, 18,70 y 24,10 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms).
11. Un cristal de un hidrobromuro de acuerdo con la reivindicación 7, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 3,84, 7,72, 9,40, 11,62, 14,92, 15,48, 16,70, 18,88, 19,32 y 24,40 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms).
12. Un cristal de un nitrato de acuerdo con la reivindicación 7, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 3,82, 7,66, 9,28, 9,52, 11,54, 15,26, 15,54, 16,62, 19,24 y 24,56 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms).
13. Un cristal de un sulfato de acuerdo con la reivindicación 7, en el que el cristal presenta picos característicos en ángulos de difracción 20 = 3,74, 7,56, 8,92, 9,58, 11,36, 12,38, 14,68, 15,64, 16,06 y 24,38 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms).
14. Un cristal de un fosfato de acuerdo con la reivindicación 7, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 3,74, 7,56, 8,80, 9,56, 11,34, 14,56, 15,74, 23,68, 24,34 y 24,68 en una difracción de rayos X en polvo patrón obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms).
15. Un cristal de un etanosulfonato de acuerdo con la reivindicación 7, en el que el cristal presenta picos característicos en ángulos de difracción 20 = 6,72, 7,90, 12,02, 13,40, 16,90, 17,88, 19,00, 19,80, 21,26 y 24,18 en una difracción de rayos X en polvo patrón obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms).
16. Un cristal de bencenosulfonato de acuerdo con la reivindicación 7, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 9,22, 10,60, 10,82, 11,10, 13,40, 15,78, 17,50, 18,66, 21,02 y 26,10 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms).
17. Un cristal de un p-toluenosulfonato de acuerdo con la reivindicación 7, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 4,18, 5,12, 13,44, 14,98, 16,96, 17,44, 18,92, 19,72, 20,16 y 23,04 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms).
18. Un cristal de un fosfato de acuerdo con la reivindicación 9, en el que el cristal presenta picos característicos en ángulos de difracción 20 = 4,28, 8,42, 8,64, 10,54, 12,72, 13,48, 15,90, 17,00. 17,46 y 21,26 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms).
19. Un cristal de un sulfato de acuerdo con la reivindicación 9, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 3,66, 6,42, 7,32, 9,76, 11,00, 12,88, 18,42, 19,62, 20,54 y 24,22 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms).
20. Un cristal de un sulfato de acuerdo con la reivindicación 9, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 3,64, 6,40, 7,32, 9,76, 17,38, 18,42, 19,64, 20,56, 22,90 y 24,20 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms).
21. Un cristal de un sulfato de acuerdo con la reivindicación 9, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 3,64, 6,40, 7,30, 9,76, 17,34, 18,38, 19,34, 20,56, 21,52 y 22,94 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms).
22. Un cristal de un sulfato de acuerdo con la reivindicación 9, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 3,62, 6,38, 7,28, 9,74, 17,30, 18,36, 19,54, 20,52, 22,86 y 24,14 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms).
23. Un cristal de un sulfato de acuerdo con la reivindicación 9, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 3,64, 6,40, 7,30, 9,76, 12,86, 18,40, 19,62, 20,54, 22,92 y 24,20 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms).
24. Un cristal de un sulfato de acuerdo con la reivindicación 9, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 3,64, 6,36, 7,30, 18,36, 19,04, 19,42, 19,70, 20,12, 20,42 y 21,32 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms).
25. Un cristal de un sulfato de acuerdo con la reivindicación 9, en el que el cristal presenta picos característicos en ángulos de difracción 20 = 5,62, 7,18, 9,22, 10,36, 15,56, 16,40 y 20,86 en un patrón de difracción de rayos X en polvo obtenido por irradiación con cobre. Radiación Ka (longitud de onda A = 1,54 angstroms).
26. Un cristal de un naftaleno-1,5-disulfonato de acuerdo con la reivindicación 9, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 6,14, 6,98, 11,24, 14,84, 17,48, 19,54, 20,94, 22,38, 23,20 y 24,70 en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms).
27. Un cristal de un naftaleno-1,5-disulfonato de acuerdo con la reivindicación 9, en el que el cristal exhibe picos característicos en ángulos de difracción 20 = 9,24, 9,58, 14,00, 14,46, 16,70, 17,02, 18,22, 20,24, 21,64 y 25,52en un patrón de difracción de rayos X en polvo obtenido por irradiación con radiación Ka de cobre (longitud de onda A = 1,54 angstroms).
28. Un inhibidor de Axl que comprende un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 6 o una sal farmacéuticamente aceptable del mismo.
29. Una composición farmacéutica que comprende un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 6 o una sal farmacéuticamente aceptable del mismo y un vehículo farmacéuticamente aceptable.
30. Un medicamento que comprende un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 6 o una sal farmacéuticamente aceptable del mismo como ingrediente activo.
31. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 6 o una sal farmacéuticamente aceptable del mismo, una composición farmacéutica de acuerdo con la reivindicación 29 o un medicamento de acuerdo con la reivindicación 30 para su uso en terapia.
32. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 6 o una sal farmacéuticamente aceptable del mismo, una composición farmacéutica de acuerdo con la reivindicación 29 o un medicamento de acuerdo con la reivindicación 30 para su uso en el tratamiento de una enfermedad hiperproliferativa.
33. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 6 o una sal farmacéuticamente aceptable del mismo, una composición farmacéutica de acuerdo con la reivindicación 29 o un medicamento de acuerdo con la reivindicación 30 para su uso en el tratamiento de un cáncer, la prevención de la metástasis del cáncer, la superación de la resistencia a fármacos de un cáncer, o la inhibición de la adquisición de resistencia a fármacos de un cáncer.
34. Un compuesto, una composición farmacéutica o un medicamento para su uso en el tratamiento de un cáncer, la prevención de la metástasis del cáncer, la superación de la resistencia a fármacos de un cáncer o la inhibición de la adquisición de resistencia a los fármacos de un cáncer de acuerdo con la reivindicación 33, en el que el cáncer se selecciona de cáncer de mama, cáncer de colon, cáncer de próstata, cáncer de pulmón, cáncer gástrico, cáncer de ovario, cáncer de endometrio, cáncer de riñón, cáncer hepatocelular, cáncer de glándula tiroides, cáncer de útero, cáncer de esófago, cáncer de células escamosas, leucemia, osteosarcoma, melanoma, glioblastoma, neuroblastoma y cáncer de páncreas.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014139628 | 2014-07-07 | ||
| PCT/JP2015/069976 WO2016006706A1 (ja) | 2014-07-07 | 2015-07-06 | テトラヒドロピラ二ルメチル基を有するピリドン誘導体 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2875360T3 true ES2875360T3 (es) | 2021-11-10 |
Family
ID=55064330
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15818440T Active ES2875360T3 (es) | 2014-07-07 | 2015-07-06 | Derivado de piridona que tiene un grupo tetrahidropiranil metilo |
Country Status (28)
| Country | Link |
|---|---|
| US (3) | US10442797B2 (es) |
| EP (2) | EP3168219B1 (es) |
| JP (2) | JP6086516B2 (es) |
| KR (1) | KR102348190B1 (es) |
| CN (2) | CN111467346B (es) |
| AU (1) | AU2015288648B2 (es) |
| BR (1) | BR112017000242A2 (es) |
| CA (1) | CA2954488C (es) |
| CO (1) | CO2017001178A2 (es) |
| CY (1) | CY1124537T1 (es) |
| DK (1) | DK3168219T3 (es) |
| ES (1) | ES2875360T3 (es) |
| HR (1) | HRP20210899T1 (es) |
| HU (1) | HUE054534T2 (es) |
| IL (2) | IL249948B (es) |
| LT (1) | LT3168219T (es) |
| MX (1) | MX2017000330A (es) |
| MY (1) | MY194307A (es) |
| PH (1) | PH12017500048A1 (es) |
| PL (1) | PL3168219T3 (es) |
| PT (1) | PT3168219T (es) |
| RS (1) | RS61941B1 (es) |
| RU (1) | RU2707953C2 (es) |
| SG (2) | SG10201900144TA (es) |
| SI (1) | SI3168219T1 (es) |
| TW (2) | TWI690525B (es) |
| WO (1) | WO2016006706A1 (es) |
| ZA (1) | ZA201700139B (es) |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI690525B (zh) | 2014-07-07 | 2020-04-11 | 日商第一三共股份有限公司 | 具有四氫吡喃基甲基之吡啶酮衍生物及其用途 |
| JP6885390B2 (ja) | 2016-02-26 | 2021-06-16 | 小野薬品工業株式会社 | Axl阻害剤と免疫チェックポイント阻害剤とを組み合わせて投与することを特徴とする癌治療のための医薬 |
| CN108250200A (zh) * | 2016-12-28 | 2018-07-06 | 中国科学院上海药物研究所 | 一种具有Axl抑制活性的化合物及其制备和应用 |
| TWI796326B (zh) | 2017-03-24 | 2023-03-21 | 日商第一三共股份有限公司 | 含有axl抑制劑與egfr酪胺酸激酶抑制藥的醫藥品及其用途 |
| JP7156287B2 (ja) | 2017-08-23 | 2022-10-19 | 小野薬品工業株式会社 | Axl阻害剤を有効成分として含むがん治療剤 |
| JP7223998B2 (ja) | 2017-10-13 | 2023-02-17 | 小野薬品工業株式会社 | Axl阻害剤を有効成分として含む固形がん治療剤 |
| RU2020124116A (ru) * | 2017-12-22 | 2022-01-25 | Хиберселл, Инк. | Производные арил-бипиридинамина в качестве ингибиторов фосфатидилинозитолфосфаткиназы |
| JP7282397B2 (ja) * | 2018-01-17 | 2023-05-29 | 薬捷安康(南京)科技股▲分▼有限公司 | Tamファミリーキナーゼ/及びcsf1rキナーゼ阻害剤及びその用途 |
| BR112020015375A2 (pt) * | 2018-01-29 | 2020-12-08 | Capulus Therapeutics, Llc | Inibidores de srebp compreendendo um anel central de seis membros |
| EP3819296B1 (en) * | 2018-07-04 | 2022-06-29 | Daiichi Sankyo Company, Limited | Method for producing diarylpyridine derivatives |
| EA202192575A1 (ru) | 2019-03-21 | 2022-01-14 | Онксео | Соединения dbait в сочетании с ингибиторами киназ для лечения рака |
| EP4054579A1 (en) | 2019-11-08 | 2022-09-14 | Institut National de la Santé et de la Recherche Médicale (INSERM) | Methods for the treatment of cancers that have acquired resistance to kinase inhibitors |
| WO2021148581A1 (en) | 2020-01-22 | 2021-07-29 | Onxeo | Novel dbait molecule and its use |
| WO2022220227A1 (ja) * | 2021-04-14 | 2022-10-20 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | テトラヒドロピリドピリミジン化合物 |
Family Cites Families (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1136485A1 (en) * | 2000-03-23 | 2001-09-26 | Sanofi-Synthelabo | Aminophenyl pyrimidone derivatives |
| GB0319241D0 (en) | 2003-08-15 | 2003-09-17 | Hotpods Ltd | Oven and a food delivery vehicle comprising said oven |
| US7884119B2 (en) | 2005-09-07 | 2011-02-08 | Rigel Pharmaceuticals, Inc. | Triazole derivatives useful as Axl inhibitors |
| WO2007057399A2 (en) | 2005-11-15 | 2007-05-24 | Boehringer Ingelheim International Gmbh | Treatment of cancer with indole derivatives |
| PL1959955T3 (pl) | 2005-12-05 | 2011-04-29 | Pfizer Prod Inc | Sposób traktowania nieprawidłowego wzrostu komórek |
| CA2633035C (en) | 2005-12-15 | 2016-05-10 | Rigel Pharmaceuticals, Inc. | Kinase inhibitors and their uses |
| EP1900727A1 (en) | 2006-08-30 | 2008-03-19 | Cellzome Ag | Aminopyridine derivatives as kinase inhibitors |
| US8097630B2 (en) | 2006-10-10 | 2012-01-17 | Rigel Pharmaceuticals, Inc. | Pinane-substituted pyrimidinediamine derivatives useful as Axl inhibitors |
| GB0625659D0 (en) | 2006-12-21 | 2007-01-31 | Cancer Rec Tech Ltd | Therapeutic compounds and their use |
| ES2607065T3 (es) | 2006-12-29 | 2017-03-29 | Rigel Pharmaceuticals, Inc. | Triazoles N3-heteroaril sustituidos y triazoles N5-heteroaril sustituidos útiles como inhibidores de axl |
| CN101622246B (zh) | 2006-12-29 | 2015-09-30 | 里格尔制药公司 | 可用作axl抑制剂的多环杂芳基取代的三唑 |
| ES2558477T3 (es) | 2006-12-29 | 2016-02-04 | Rigel Pharmaceuticals, Inc. | Triazoles sustituidos útiles como inhibidores de AXL |
| WO2008083353A1 (en) | 2006-12-29 | 2008-07-10 | Rigel Pharmaceuticals, Inc. | Bicyclic aryl and bicyclic heteroaryl substituted triazoles useful as axl inhibitors |
| WO2008083357A1 (en) | 2006-12-29 | 2008-07-10 | Rigel Pharmaceuticals, Inc. | Bridged bicyclic aryl and bridged bicyclic heteroaryl substituted triazoles useful as axl inhibitors |
| WO2008128072A2 (en) | 2007-04-13 | 2008-10-23 | Supergen, Inc. | Axl kinase inhibitors useful for the treatment of cancer or hyperproliferative disorders |
| GB0713259D0 (en) | 2007-07-09 | 2007-08-15 | Astrazeneca Ab | Pyrazine derivatives 954 |
| WO2009007749A2 (en) | 2007-07-09 | 2009-01-15 | Astrazeneca Ab | Trisubstituted pyrimidine derivatives for the treatment of proliferative diseases |
| WO2009024825A1 (en) | 2007-08-21 | 2009-02-26 | Astrazeneca Ab | 2-pyrazinylbenzimidazole derivatives as receptor tyrosine kinase inhibitors |
| GB0719803D0 (en) | 2007-10-10 | 2007-11-21 | Cancer Rec Tech Ltd | Therapeutic compounds and their use |
| RU2010120536A (ru) * | 2007-10-25 | 2011-11-27 | Астразенека Аб (Se) | Производные пиридина и пиразина, полезные для лечения клеточных проферативных расстройств |
| JP2011500778A (ja) | 2007-10-25 | 2011-01-06 | アストラゼネカ・アクチエボラーグ | ピリジン及びピラジン誘導体−083 |
| EP2205592B1 (en) * | 2007-10-26 | 2013-05-08 | Rigel Pharmaceuticals, Inc. | Polycyclic aryl substituted triazoles and polycyclic heteroaryl substituted triazoles useful as axl inhibitors |
| CN101917994A (zh) | 2007-11-02 | 2010-12-15 | 詹森药业有限公司 | Cfms抑制剂用于治疗或预防骨癌以及与骨癌相关的骨丢失和骨痛的用途 |
| WO2009094417A1 (en) | 2008-01-23 | 2009-07-30 | Bristol-Myers Squibb Company | 4-pyridinone compounds and their use for cancer |
| US8558000B2 (en) | 2008-01-23 | 2013-10-15 | Bristol-Myers Squibb Company | 4-pyridinone compounds and their use for cancer |
| WO2009127417A1 (en) | 2008-04-16 | 2009-10-22 | MAX-PLANCK-Gesellschaft zur Förderung der Wissenschaften e.V. | Quinoline derivatives as axl kinase inhibitors |
| UY31800A (es) | 2008-05-05 | 2009-11-10 | Smithkline Beckman Corp | Metodo de tratamiento de cancer usando un inhibidor de cmet y axl y un inhibidor de erbb |
| WO2009138799A1 (en) | 2008-05-14 | 2009-11-19 | Astex Therapeutics Limited | Therapeutic uses of 1-cycl0pr0pyl-3 - [3- ( 5 -morpholin- 4 -ylmethyl- 1h-benz0imidaz0l- 2 -yl) -lh-pyrazol-4-yl] -urea |
| DK2328888T3 (da) | 2008-07-09 | 2013-02-11 | Rigel Pharmaceuticals Inc | Broforbundne, bicykliske heteroarylsubstituerede triazoler, der er anvendelige som axl-inhitorer |
| CA2730231C (en) | 2008-07-09 | 2016-10-18 | Rigel Pharmaceuticals, Inc. | Polycyclic heteroaryl substituted triazoles useful as axl inhibitors |
| CA2748943A1 (en) | 2009-02-09 | 2010-08-12 | Supergen, Inc. | Pyrrolopyrimidinyl axl kinase inhibitors |
| SG175355A1 (en) * | 2009-04-27 | 2011-12-29 | Elan Pharm Inc | Pyridinone antagonists of alpha-4 integrins |
| EP2311809A1 (en) | 2009-10-16 | 2011-04-20 | Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. | Quinolinyloxyphenylsulfonamides |
| AR080754A1 (es) | 2010-03-09 | 2012-05-09 | Janssen Pharmaceutica Nv | Derivados de imidazo (1,2-a) pirazina y su uso como inhibidores de pde10 |
| US8962623B2 (en) | 2011-03-04 | 2015-02-24 | Locus Pharmaceuticals, Inc. | Aminopyrazine compounds |
| CA2863515C (en) * | 2012-01-31 | 2018-02-27 | Daiichi Sankyo Company, Limited | Pyridone derivatives |
| WO2013162061A1 (ja) | 2012-04-26 | 2013-10-31 | 第一三共株式会社 | 二環性ピリミジン化合物 |
| TWI690525B (zh) | 2014-07-07 | 2020-04-11 | 日商第一三共股份有限公司 | 具有四氫吡喃基甲基之吡啶酮衍生物及其用途 |
-
2015
- 2015-07-03 TW TW104121592A patent/TWI690525B/zh active
- 2015-07-03 TW TW108136526A patent/TWI723572B/zh active
- 2015-07-06 LT LTEP15818440.8T patent/LT3168219T/lt unknown
- 2015-07-06 BR BR112017000242-6A patent/BR112017000242A2/pt active Search and Examination
- 2015-07-06 AU AU2015288648A patent/AU2015288648B2/en active Active
- 2015-07-06 RU RU2017103753A patent/RU2707953C2/ru active
- 2015-07-06 SG SG10201900144TA patent/SG10201900144TA/en unknown
- 2015-07-06 DK DK15818440.8T patent/DK3168219T3/da active
- 2015-07-06 SG SG11201700063RA patent/SG11201700063RA/en unknown
- 2015-07-06 JP JP2016532991A patent/JP6086516B2/ja active Active
- 2015-07-06 PL PL15818440T patent/PL3168219T3/pl unknown
- 2015-07-06 SI SI201531589T patent/SI3168219T1/sl unknown
- 2015-07-06 CN CN202010427596.9A patent/CN111467346B/zh active Active
- 2015-07-06 EP EP15818440.8A patent/EP3168219B1/en active Active
- 2015-07-06 WO PCT/JP2015/069976 patent/WO2016006706A1/ja active Application Filing
- 2015-07-06 US US15/036,981 patent/US10442797B2/en active Active
- 2015-07-06 PT PT158184408T patent/PT3168219T/pt unknown
- 2015-07-06 MY MYPI2017000012A patent/MY194307A/en unknown
- 2015-07-06 RS RS20210707A patent/RS61941B1/sr unknown
- 2015-07-06 CA CA2954488A patent/CA2954488C/en active Active
- 2015-07-06 CN CN201580047744.1A patent/CN106604920B/zh active Active
- 2015-07-06 EP EP21166082.4A patent/EP3868757A1/en active Pending
- 2015-07-06 MX MX2017000330A patent/MX2017000330A/es unknown
- 2015-07-06 KR KR1020177000534A patent/KR102348190B1/ko active IP Right Grant
- 2015-07-06 HU HUE15818440A patent/HUE054534T2/hu unknown
- 2015-07-06 ES ES15818440T patent/ES2875360T3/es active Active
-
2017
- 2017-01-05 IL IL249948A patent/IL249948B/en active IP Right Grant
- 2017-01-06 PH PH12017500048A patent/PH12017500048A1/en unknown
- 2017-01-06 ZA ZA2017/00139A patent/ZA201700139B/en unknown
- 2017-02-01 JP JP2017016682A patent/JP6203439B2/ja active Active
- 2017-02-07 CO CONC2017/0001178A patent/CO2017001178A2/es unknown
-
2019
- 2019-09-18 US US16/575,177 patent/US11208403B2/en active Active
-
2020
- 2020-10-18 IL IL278124A patent/IL278124B/en unknown
-
2021
- 2021-06-07 HR HRP20210899TT patent/HRP20210899T1/hr unknown
- 2021-06-30 CY CY20211100586T patent/CY1124537T1/el unknown
- 2021-08-12 US US17/400,795 patent/US20220002277A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2875360T3 (es) | Derivado de piridona que tiene un grupo tetrahidropiranil metilo | |
| AU2013216006B2 (en) | Pyridone derivatives | |
| KR102534028B1 (ko) | 트리아졸로피리미딘 화합물 및 그의 용도 | |
| CA3113233A1 (en) | Fused tricyclic ring derivatives as src homology-2 phosphatase inhibitors | |
| KR102399891B1 (ko) | 항암 및 항-증식 활성을 나타내는 2-아미노피리미딘-6-온 및 유사체 | |
| ES2798424T3 (es) | Compuestos de triazolopiridina y usos de estos | |
| ES2813875T3 (es) | Compuestos y procedimientos de uso | |
| AU2014265957A1 (en) | Inhibitors of the kynurenine pathway | |
| TW200829594A (en) | Phosphoinositide 3-kinase inhibitor compounds and methods of use | |
| JP2016534038A (ja) | Rhoキナーゼ阻害剤 | |
| WO2020068867A1 (en) | Quinazoline derivatives as tyrosine kinase inhibitor, compositions, methods of making them and their use | |
| TW201733580A (zh) | Trk抑制劑抵抗性的癌治療劑 | |
| KR20170045748A (ko) | 증식성 질병의 치료를 위한 조성물 및 방법 |